/


























































































































































































































































































































Автор: Березовский В.М.
Теги: материальные основы жизни биохимия молекулярная биология биофизика пищевая промышленность химия витаминов
Год: 1973




Текст
В-М-БЕРЕЗОВСКИЙ
ХИМИЯ
ВИТАМИНОВ
ИЗДАНИЕ ВТОРОЕ,
ПЕРЕРАБОТАННОЕ И ДОПОЛНЕННОЕ
J Гнпн’нцМ ,
’ Шитамигнид 3..2ОД.
МОСКВА
ПИЩЕВАЯ ПРОМЫШЛЕННОСТЬ
1 973
УДК 577.16
Химия витаминов. Березовский В. М. Изд. 2-е, М.,
«Пищевая промышленность», 1973.
Изучение и получение витаминов — природных неза-
менимых пищевых веществ — имеет важное значение.
На основе предложенной химической классификации ви-
таминов детально изложены и обобщены вопросы химии
витаминов в ее современном состоянии, методы выделе-
ния из природных источников, различные методы синте-
за. Рассмотрена зависимость биологической активности
от структуры витаминов, коферментов и их химических
модификаций. Детально изложена химия провитаминов
и рассмотрены пути их превращения в витамины. Даны
представления о биологических свойствах витаминов,
их превращении в коферменты, о биокаталитических
функциях коферментов в обмене веществ животного ор-
ганизма, о роли витаминов в питании и путях их приме-
нения в пищевой промышленности, а также в животно-
водстве, о значении витаминов и коферментов в профи-
лактике и лечении различных заболеваний.
Таблиц 23. Иллюстраций 6. Список литературы —
3785 наименований.
Рецензент: академик И. Л. Кнунянц
(g) Издательство «Пищевая промышленность*, 1973 г.
Б
3177-052 R9_73
044(01)—73
ВЛАДИМИР МИРОНОВИЧ БЕРЕЗОВСКИЙ
ХИМИЯ ВИТАМИНОВ
Редактор Л. С. Беликова
Художник С. Р. Нак
Худож. редактор В. В. Водзинский
Технический редактор Т. С. Пронченкова
Корректор 3. В. Коршунова
Т-15236. Сдано в набор 29/1 1973 г. Подписано в печать 26/Х 1973 г.
Формат 70X108’Ле- Бумага книжно-журнальная № 2. Печ
л. 39.5—55,3 усл.-п. л. Уч.-изд. л. 55,0. Тираж 3400 экз. Зак. 69.
Цена 3 р. 10 к.
Издательство «Пищевая промышленность»
113035. Москва, М-35, 1-й Кадашевский, 12.
Ярославский поли граф комбинат «Союзполнграфпрома» прн Го-
сударственном комитете Совета Министров СССР по делам из-
дательств. полиграфии и книжной торговли. Ярославль, ул. Сво-
боды. 97.
ПРЕДИСЛОВИЕ
f—---------------- ,г
I Бийский |
’ витаминный завод. ;
Среди физиологически активных природных органических соединений —
алкалоидов, гормонов, антибиотиков и других — витамины занимают особое
место. Значение витаминов заключается не только в том, что они представ-
ляют собой постоянные составные части животного организма и незаменимы
для питания, нои в том, что они являются и лечебными средствами против
заболеваний витаминной недостаточностью и средствами, усиливающими
защитные функции организма в сопротивлении против других заболеваний.
Синтетические витамины полностью идентичны природным, они пред-
ставляют собой синтетические пищевые вещества. Витамины и витаминные
препараты перестали быть экзотическими веществами, они стали элементар-
ными продуктами питания первой необходимости для каждого человека и
каждой семьи наряду с другими продуктами полноценного рационального
питания. Значительно возросло применение витаминов для лечебных целей;
подавляющее большинство витаминов уже не является дефицитным. В на-
стоящее время осуществляется широкая витаминизация пищевых продуктов
к внедрение в лечебную практику таких ценных лечебных препаратов,
как коферменты. Широко применяются витамины в кормлении домашних
животных и птицы. Применение витаминов в животноводстве, можно ска-
зать без преувеличения, дает громадный экономический эффект и повышает
полноценность продуктов животноводства.
Изучение витаминов, их роли в обмене веществ, участия в фермента-
тивных реакциях, в функционировании органов чувств, в процессе размно-
жения и роста животного организма, применения в медицине и питании
производится многими разделами естествознания, в результате чего созда-
ны обширные обзорные монографии; в первую очередь это относится к моле-
кулярной биологии, биохимии, биологии и медицине. Однако в области
химического изучения витаминов обширный фактический материал оставал-
ся разрозненным и до последнего времени недостаточно обобщенным. В из-
вестной степени этот недостаток был восполнен изданием в 1959 г. книги
«Химия витаминов».
Химические исследования по витаминам продолжаются уже около со-
рока лет, эта область науки непрерывно пополняется новыми реакциями
открытиями и интересными данными.
За более чем десятилетний период, прошедший со времени первого из-
дания книги, в области химии витаминов появилось большое количество
научных исследований, посвященных уточнению химической структуры
витаминов, новым методам их синтеза, изучению физических, химических и
биологических свойств, определению конфигурации цис-транс-изомерных
3
форм или абсолютных конфигураций изомеров витаминов, имеющих асиммет-
рические центры, открытию новых коферментов, изучению реакционной
способности промежуточных соединений витаминов.
Однако список витаминов, принимающих участие в жизнедеятельности
живого организма, по-видимому, исчерпан, так как за последний период
времени открытия новых витаминов не было. Наоборот, некоторые соеди-
нения, относимые ранее к витаминам, должны быть исключены из их списка
как неудовлетворяющие общепринятому понятию «витамины».
Соответственно количественному и качественному скачку в области
научных изысканий, производства и потребления витаминов материал в
настоящей монографии по химии витаминов полностью переработан и зна-
чительно дополнен. Многие разделы, в том числе все разделы по кофермен-
там и их биохимическим функциям, написаны заново. Материал в книге
изложен применительно к предложенной рациональной химической клас-
сификации витаминов, что позволяет легче выявить зависимость между
химическим строением веществ и их физиологической активностью. В моно-
графию включены также краткие сведения по физиологии и биохимии ви-
таминов.
В книге использована патентная и периодическая литература, опублико-
ванная до 1972 г. и в отдельных случаях — в более позднее время.
ВВЕДЕНИЕ
Бийский £
1 битами и аый завод.
Со времени открытия витаминов были проведены многочисленные
исследования, определившие роль, которую витамины играют в обмене
веществ человека и животных; при этом открыты различные органиче-
ские соединения, отсутствие или недостаток которых приводит к специфи-
ческим нарушениям функций организма, однако химическая природа ни
одного из этих соединений к тридцатым годам нашего столетия еще не
была расшифрована.
Тем не менее Н. Д. Зелинский [1] в 1921 г. определил роль витами-
нов в обмене веществ животного организма следующим образом:
«Биологическое значение витаминов заключается не в том, что при
их помощи в организм вводятся большие запасы энергии или основные
кирпичи для постройки органического субстрата, а в том, что эти допол-
нительные вещества вызывают в клетках организма деятельность, подоб-
ную той, какая обусловливается ферментами и продуктами внутренней
секреции... Связь между ферментами и витаминами, возможно, и выра-
жается в том, что последние необходимы как строительный материал для
первых». Существо витаминов, раскрытое Н. Д. Зелинским, было блестя-
ще подтверждено в последующие годы.
Для жизнедеятельности организма человека и животных необходимы
белки, жиры и углеводы, являющиеся пластическими и энергетическими
материалами, а также минеральные соли и витамины. Среди жиров и
продуктов гидролиза белков имеются незаменимые органические веще-
ства, поступление которых должно обеспечиваться с пищей, так как они
не синтезируются организмом. По-видимому, по мере эволюционного
развития животного мира отдельные виды постепенно теряли способ-
ность к биосинтезу некоторых простых органических соединений, участву-
ющих в метаболических процессах, так как более эффективным для орга-
низма путем они могли получить их из окружающей органической приро-
ды — растений и микроорганизмов или с животной пищей. К таким
органическим соединениям относятся незаменимые /.-аминокислоты, не-
заменимые ненасыщенные жирные кислоты, а также витамины (термин
«витамины» предложен Функом [2]). На необходимость для питания та-
ких факторов («витаминов»), не синтезируемых животными, указывал
Лунин [3]. Дл-я человека незаменимыми оказались восемь /.-аминокислот
(из 20): валин, лейцин, изолейцин, лизин, треонин, метионин, фенилала-
нин и триптофан [4]. Для животных незаменимых аминокислот значитель-
но больше, например для крысы —11.
К незаменимым ненасыщенным жирным кислотам относятся лино-
левая, линоленовая и арахидоновая кислоты. Раньше их объединяли под
названием «витамин F», хотя отнести их к витаминам нельзя.
Витамины — незаменимые для жизни органические вещества
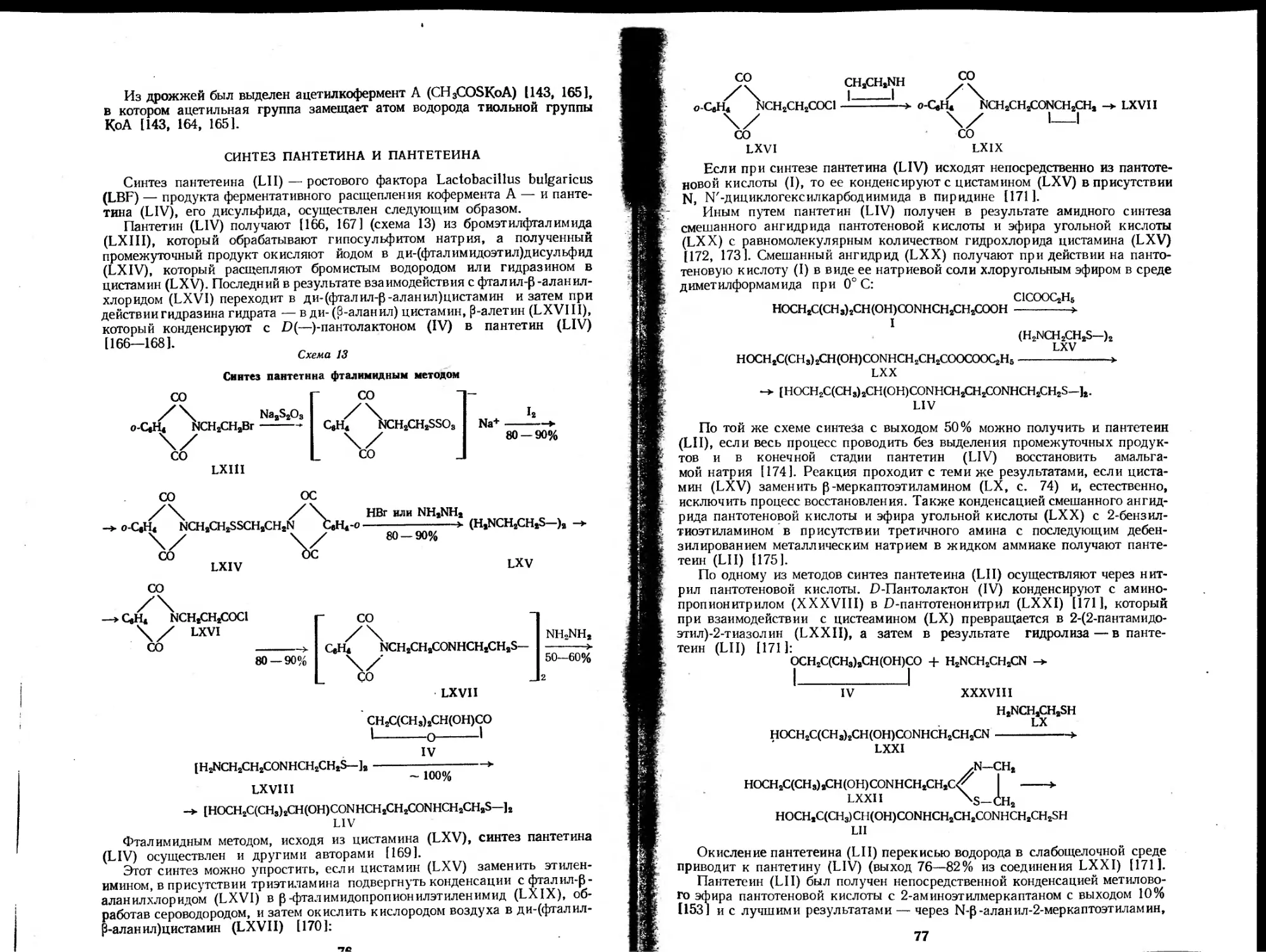
разнообразной структуры, являющиеся биологическими катализаторами
химических реакций или реагентами фотохимических процессов, протека-
ющих в живой клетке, и участвующие в обмене веществ преимуществен-
но в соединении со специфическими белками в составе ферментных
систем, причем в организме человека и животных не синтезирующиеся и
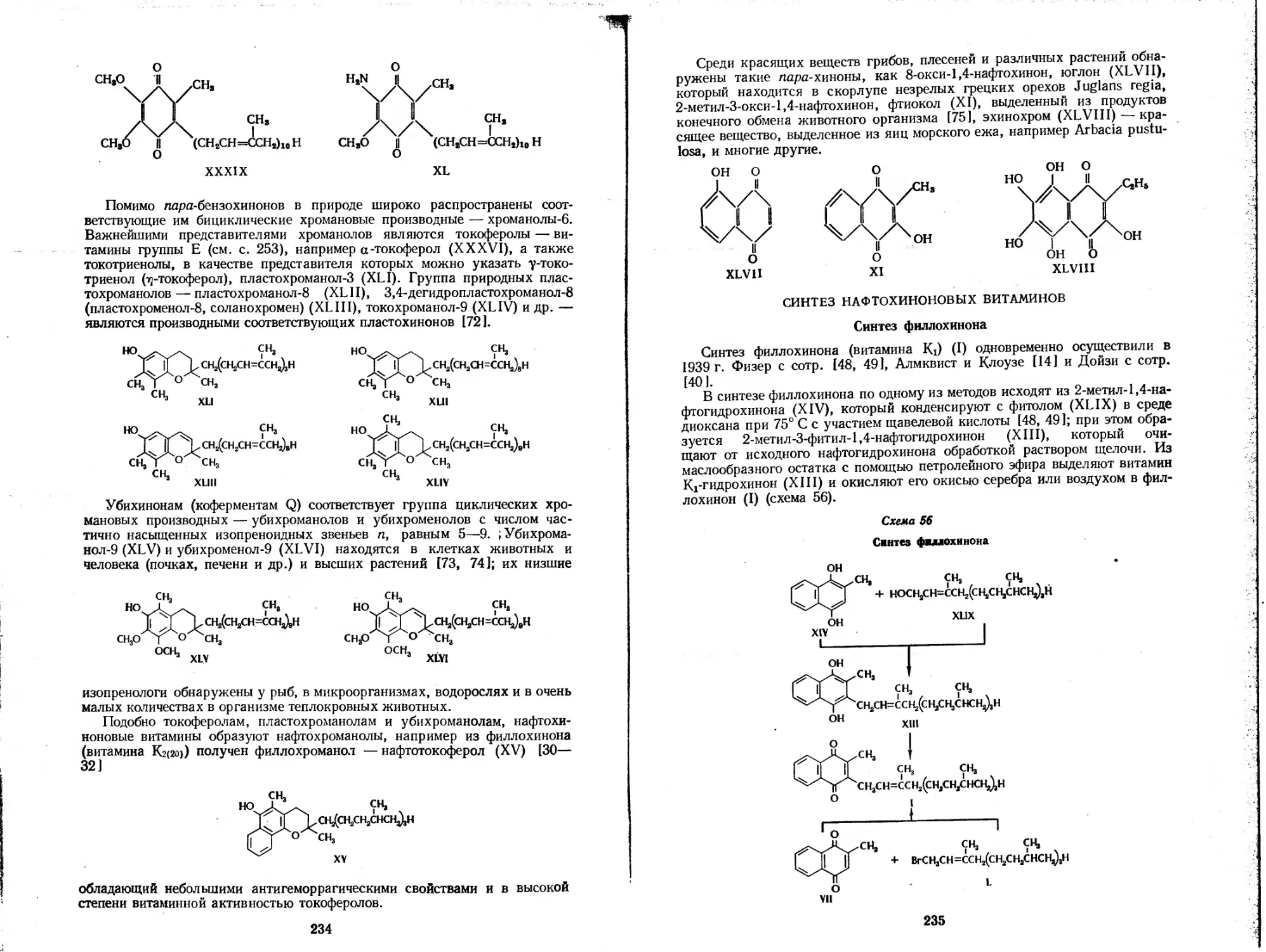
5
поступающие в него только из внешней среды (непосредственно; в соста-
ве ферментов и коферментов или в виде провитаминов).
Потребность человека и различных животных в витаминах неодинако-
ва. Некоторые витамины, такие, как тиамин, рибофлавин, пантотеновая
кислота, пиридоксаль и пиридоксамин и некоторые другие, необходимы
как катализаторы химических реакций для каждой живой клетки; они не
синтезируются тканями человека и животных и должны поступать из внеш-
ней среды. Другие витамины нужны не всем животным, так, например,
L-аскорбиновая кислота необходима для человека, обезьяны и морской
свинки, а остальные животные не нуждаются в поступлении ее из внешней
среды, так как способны к самостоятельному биосинтезу; поэтому для
этих видов животных L-аскорбиновая кислота не является витамином.
Непосредственно к витаминам примыкают ростовые вещества микро-
организмов, которые нуждаются в получении этих веществ из окружающей
среды. Многие витамины (пантотеновая кислота, биотин, фолиевая кислота,
тиамин, рибофлавин, пиридоксин и др.) одновременно являются и ростовы-
ми веществами для различных видов микроорганизмов.
Некоторые микроорганизмы обладают способностью к биосинтезу необ-
ходимых для них ростовых веществ, причем иногда в размерах, значительно
превышающих их собственную потребность. Излишек ростовых веществ
(в то же время являющихся и витаминами), вырабатываемых микрофлорой
кишечника некоторых животных, например жвачных (менахиноны, кобал-
амин, тиамин, рибофлавин и др.), усваивается этими животными, вследствие
чегс они не нуждаются в поступлении отдельных витаминов с пищей.
В природе биосинтез витаминов, как правило, осуществляется растения-
ми и микроорганизмами; многие витамины для самих растений являются
биокатализаторами химических реакций обмена веществ. Менахиноны и
кобаламин синтезируются только микроорганизмами.
Некоторые витамины представлены в природе не индивидуальными
веществами, а семействами родственных соединений; к ним относятся рети-
нолы, кальциферолы, токоферолы, нафтохиноновые витамины.
КЛАССИФИКАЦИЯ ВИТАМИНОВ
По мере открытия отдельных витаминов они обозначались буквами
латинского алфавита и назывались в зависимости от их биологической роли,
чапример витамин Е — токоферол (по гречески «токос»—деторождение,
«феро» — несущий), витамин А — аксерофтол (ксерофтальмия — глазное
заболевание) и т. п. В дальнейшем пришлось буквенные обозначения рас-
ширить, так как выделялись новые индивидуальные вещества близкого,
аналогичного или нового биологического характера; поэтому к буквам
были присоединены цифровые обозначения. В результате, например, вместо
одного наименования «витамин В» в настоящее время для обозначения раз-
личных «витаминов комплекса В» использованы наименования от «витами-
на Вр) до «витамина В15» и далее [2, 5—121. Буквенная классификация ви-
таминов представлена в табл. 1.
После установления химической структуры витаминов их тривиальные
наименования стали приобретать химический смысл, например тиамин,
рибофлавин, пиридоксаль, птероил-£-глутаминовая кислота и т. д. Затем
оказалось, что ряд давно известных органических веществ обладает свойст-
вами витаминов; к ним относятся никотиновая кислота, никотинамид,
т. е. химические соединения с давно установившимися наименованиями.
В настоящее время для обозначения витаминов широко пользуются
наименованиями витаминов биологического и химического смыслового
происхождения и в меньшей мере — буквенными обозначениями.
Помимо буквенной классификации, применяется физическая класси-
фикация витаминов, разделяющая их на две большие группы по признаку
растворимости в воде или жирах: водорастворимые витамины и жирораство-
Таблица 1
Буквенная классификация витаминов
Буквенное обозначение витамина Наименование1 Специфический биологический характер действия
А (АО Ретинол (аксерофтол) Противоксерофтальми- ческий, антиинфекцион-
Аа Дегидроретинол ный
В1 Тиамин (аневрин, фактор бери-бери) Противоневрический
в2 (G) Рибофлавин (лактофлавин, овофлавин)
В3 Пантотеновая кислота («универсальный витамин», противодерматитный фактор цыплят)
В5(?), (РР) Никотинамид, никотиновая кислота (ни- ацин, ниацинамид) Пиридоксин (пиридоксол), пиридоксаль, пиридоксамин (адермин, фактор R) Противопеллагрический
вв Противодерма тический
В7 Фактор пищеварения птиц
В8(?) мезо-Инозит (мышиный фактор)
Вю Фактор оперения
Вц Ростовой фактор цыплят
В12 Цианокобаламин, оксикобаламин (фактор животного протеина) Противопернициозиый
В15 Пангамовая кислота
вв, вс, м Фолиевая кислота (фактор U), птероил- Z.-глутаминовая кислота, ферментативный L. casei-фактор, витамин Вс-конъюгат Противоанемическнй
Вт Карнитин
с L-Аскорбиновая кислота Противоцинготный
Da Эргокальциферол (кальциферол, виосте- рол) Противорахитический
D3 Холекальциферол (кальциферол)
Е Токоферолы: а-, р-, у- и др. Противостерильный
F Линолевая, линоленовая, арахидоновая кислоты (незаменимые жирные кислоты)
Н Биотин (фактор W, коэнзим R)
Н' пара-Аминобензойная кислота (фактор, предохраняющий от поседения шерсть крыс)
Ki Филлохинон (а-филлохинон) Противогеморрагический
Ks Менахинон (8-филлохинон)
L Лактационный
Р d-Катехин, гесперидии, гесперетин, Противокапилляропрони-
эриодиктиол, рутин, кверцетин и др. цаемый
1 Названия витаминов, рекомендованные Международным союзом чистой н приклад-
ной химии (I U РАС), выделены курсивом.
римые витамины [5—7, 9, 10]. Такая классификация примитивна и не отра-
жает всего многообразия сложного химического строения органических
соединений, входящих в группу витаминов. Кроме того, она не верна,
так как характер отношения к воде каждого витамина, как правило, может
быть изменен на противоположный введением в витамин соответствующих
липофильных или липофобных групп, которые не изменяют уровня его
биологической активности. Например, липофобная L-аскорбиновая кисло-
та может быть превращена в жирорастворимый препарат этерификацией ее
какой-либо высшей жирной кислотой (пальмитиновой, олеиновой, стеари-
новой и т. д.). Гидрофобный витамин А при превращении его в фосфорный
эфир становится гидрофильным и т. д.
Как впоследствии оказалось, многие органические вещества, первона-
чально относимые к витаминам, на самом деле представляют собой или
пластический материал, используемый животными организмами для по-
7
строения тканей, или метаболиты биохимических процессов, синтезируемые
организмом, или природные соединения растительного происхождения,
обладающие терапевтическим эффектом. Поэтому нет оснований относить к
витаминам такие вещества: холин, инозит, карнитин (витамин Вт) панга-
мовую кислоту (витамин В15), линолевую, линоленовую, арахидоновую
кислоты (витамин F), рутин, гасперидин, гесперетин, катехин, эриодиктиол,
кверцетин и др. (витамин Р).
По своей химической структуре витамины многообразны. Они являются
производными ненасыщенных у-лактонов, р -аминокислот, амидов кислот,
циклогексана, нафтохинона, имидазола, пиролла, бензопирана, пиридина,
пиримидина, тиазола, изоаллоксазина и других циклических систем.
Следует отметить, что предложенный Функом [2] термин «витамины»,
включающий представление о жизненно необходимых аминах, не отражает
существа этих соединений, так как хотя многие витамины и содержат цикли-
ческий азот, но первичная аминогруппа входит в структуру только несколь-
ких витаминов — пиридоксамина, тиамина; аминогруппа птериновых
витаминов химически малоактивна. В то же время почти во всех витаминах
содержится гидроксильная или карбонильная группа, способная прев-
ращаться в гидроксильную. Только один витамин—никотинамид — не
содержит гидроксильной группы, но она содержится в молекуле кофер-
мента, в виде которого никотинамид участвует в обмене веществ.
Таким образом, можно считать установленным, что для витаминов
характерно наличие в молекуле гидроксильной функции.
Для многих витаминов характерна изомерия положения, геометриче-
ская и оптическая изомерии, причем биологической активностью обладают
только строго определенные изомерные формы.
Витамины нельзя рассматривать в отрыве от многочисленных физиоло-
гически активных природных и синтетических органических соединений.
Они находятся в тесной связи с гормонами, близки по химическому строению
к антибиотикам, алкалоидам и многочисленным лекарственным органиче-
ским веществам, а также к ростовым веществам растений. Для всех этих
соединений принята химическая классификация.
Поэтому наиболее рациональной следует признать химическую клас-
сификацию витаминов на основе классификации органических соединений.
Химическая классификация витаминов
А. Витамины алифатического ряда
I. Витамины — производные ненасыщенных полиокси-у-лактонов
L-A скорби нова я кислота (витамин С)
2. Витамины — производные р-аминокислот
Пантотеновая кислота (витамин В3)
Б. Витамины алициклического ряда
3. Циклогексанол-этиленгидриндановые витамины
Кальциферолы (витамины группы D)
Эргокальциферол (кальциферол, виостерол) (витамин D.-)
Холекальциферол (витамин D3) и др.
4. Циклогексенилизопреноидные витамины
Ретинолы (витамины группы А)
Ретинол (витамин Ai) и его стереоизомеры
Дегидроретинол (витамин А2) и его стереоизомеры
В. Витамины ароматического ряда
5. Нафтохиноновые витамины
Филлохинон (витамины Ki)
Менахиноны (витамины группы К2)
Г. Витамины гетероциклического ряда
6. Хромановые витамины
Токоферолы (витамины группы Е)
5,7,8- Триметилтокол (я-токоферол)
8
5,8-Диметилтокол (p-токоферол)
7,8-Диметилтокол (у-токоферол) и другие изомеры
7. Пиридинкарбоновые витамины
Никотинамид и никотиновая кислота (витамин РР)
8. Оксиметилпиридиновые витамины
Пиридоксин (витамины группы В«)
Пиридоксаль
Пиридоксамин
Пиридоксин
9. Пиримидилметилтиазолиевые витамины •
Тиамин (витамин Bi)
10. Гексагидроимидазолотиеновые витамины
Биотин (витамин Н)
11. Птериновые витамины
Фолиевая кислота — птероил Б-глутаминовая кислота (витамин Вс)
Птероилтри-Б-глутаминовая кислота (ферментативный «L. casei-фактор»)
12. Флавиновые витамины
Рибофлавин (витамин В2)
13. Корриновые витамины
Кобаламины (витамины группы В12)
Цианокобаламин
Оксикобаламин (гидроксокобаламин)
Терминология отдельных витаминов установлена комиссией по номен-
клатуре биологической химии Международного союза по чистой и приклад-
ной химии [13].
Структура витаминов приведена ниже.
Структура витаминов -
ОМОН
ОН С==^ СН3ОН
НОСН2 С-НСХ ZC=O HOCHfC-CHCONHC^CHgCOOH
н ° сн3
Z -Аскорбиновая кислота D -Пантотеновая кислота
ск,сн3 сн3 сн3 СН3( <YCH=CH-C“CH<H-CH-C-CH-CfijOH А- Ретинол сн3 сн3 _у13 1 1 рт—А т А СН3 1 и й i СН с / эргокальциферол / "S СИ, Н| НО"'2 нсг"-^ ОЛТСНз vH3 <?н3 4&XVCH2CH’CCH2 (сн2 сн2 снсн2)3 н О Филлохинои н°АСц ^Нз н° 5 Т ]Г Ъсн/СНгСНгСНСНДН снСго сн3 А '"^3 5,Т.8-Тримегилтокоп Q (oC-токоф еррл) <^н3 CH AJL J<CH2(CH2CH2CHCH2)3 н 3 СНд^СНз Z8 - д имет и л токол ( Г-тонсхрерол) :н3 сн3 сн, rCH^CH-i-CHCH-CH-C-CH-CHjOH ^СНз Дегидроретинол СНз?1* j—Р^^СНз ;Н V Холена льцисрерол ХН2 ^Хсн OjT 3 ?Нз %^Ya'(ch2ch=cch2)6h О Менахинон «3 9нз I Ксн/сн2сн2спсп2)3н ^О^СНз Но 5,8-Диметилтокол (р -токо<рерол) ^C°NH2 'n Никотинамид
9
но.
сн2он
As/Ch,oh
ch.Cn
Пиридоксин
Пиридоксаль
сно
но СН2ОН
Пиридонсаммн
HN-
__СН-ч.
ХХ^ГТСНз
снр-н nh2 Sx CI 12СНгОН
II
HN 'NH
Qc f 1,СН,СН2СН2 соон
К Биотин
СООН
СН-hn-AAcohnch
. . -X / !
сн,
СН2СООН
о „ Г с,х,,;
N%-CH2HN’Д)-С ОН хсн
Фолиевая
кислота
HN-
сн,
ШЫ
Птероилтри- 2-глутами-
новая кислота
CH..CO
соон
HNCH
<^Нг
СН2СООН
СН2ОН
носн
носн
носн
снг
N’ N^O
[ч^т-_NH
II
о
Рибофлавин
h.nocch2ch. сн
н,ыосн2с
Н3С
Н3С
Н2ЫОСН2С
к^хсн3
HN’OCCH.CH, СН3сн
сн2
СНСН-,
I
о
о
N
ОН
сн3
ch2conh2
CH;£H2CONH2i
сн3
сн3
ch,ch2conh2
•СН3
о
НгХ^И
ПРОВИТАМИНЫ
В отдельных случаях вместо витаминов организм животного может
удовлетворяться получением генетически с ними связанными органическими
веществами, которые также не синтезируются самим организмом, однако
в процессе обмена веществ или фотосинтеза способны переходить в витамин.
Такие вещества получили название провитаминов [5—11 ].
Важнейшие провитамины — каротиноиды, широко распространены в
растительном мире. Среди каротиноидов провитаминами являются только
соединения, содержащие в своей молекуле структурную часть ретинола,
в который они переходят при расщеплении в процессах метаболизма.
Другую большую группу провитаминов представляют стерины, содер-
жащие двойные связи, при расщеплении способные образовывать отдель-
ные циклы, соединенные полиеновой цепью. Эти стерины при облучении
кожи ультрафиолетовыми лучами солнечного или искусственного света
переходят в кальциферолы. Никотиновую кислоту, переходящую в никотин-
амид, также правильнее рассматривать как провитамин. Имеются основания
10
предполагать наличие провитаминной зависимости между никотинамидом и
триптофаном, являющимся незаменимой аминокислотой. Возможно, что
провитаминами являются и малоактивные ди- и гексапептиды птероил-
глутаминовой кислоты (ферментативный L. casei-фактор и витамин
Вс-конъюгат), под действием конъюгаз переходящие в птероилглутамино-
вую кислоту.
Не исключено, что природным провитаминам может принадлежать и
большая самостоятельная биологическая роль.
Ниже представлены важнейшие провитамины и их структура.
Витамины Провитамины
Кальциферолы Эргостерин (провитамин эргокальциферола)
7- Дегидрохолестерин (провитамин холекальциферола)
Ретинол а-Каротин
^-Каротин
7-Каротин
Нафтохиноновые витамины 2-Д\етил-1,4-нафтохинон (витамин К3)
2-Метил-1,4-нафтогидрохинон (витамин К«)
2-Метил-4-амино-1 -нафтол дигидрохлорид (витамин Кь)
2-Метил-1,4-диаминонафталин гидрохлорид (вита-
мин Ке)
З-Метил-4-амино-1-нафтол дигидрохлсрид (витамин К?)
Никотинамид Никотиновая кислота
Структура важнейших провитаминов
ПРОВИТАМИНЫ КАЛЬЦИФЕРОЛОВ (ПРОВИТАМИНЫ1))
ПРОВИТАМИНЫ РЕТИНОЛА (провитамины А)
СН3 СН3 7Нз СНз СН3 СН3 СН3
><Ссн=сн-с=сн-сн=сн-с=сн-сн=сн-сн=с-сн=сн-сн=с-сн=сНчгХ.
\Х^СН3 <<-Каротин СН3-^Ч/
СН3 СН3 71’ С”3 (ГНз СНз СНз
><-СН=СН-С=СН-СН=СН-С=СН-СН-СН-СН=С-С№СН-СН=С-СН=СНу>\
\/'СН3 р-Каротин’ СНз^4^
СН3 СН3 СН3 СН3 СН3 Cw СНз
>ССН=СН-С“СН-СН=СН-С“СН-СН=СН-СН=С-СН=СН-СН=С-СН=СН^ ч
Г-Каротин
провитамины нафтохиноновых витаминов (провитамины К)
О
2-Метил -1,4 - насртогид рохи-
нон (витамин К*) .
2-Метил-1,4-нафтохином (витамин К3) и натриевая
соль его бисульфитного производного „ викасоп •
он
NHyHCl
он
NH2 НС1
2-Метил‘4 - а миго-1 - нафтол
| гидрохлорид (витамин
NH2 НС1
2-Мети л -1- диам инонвфта л и и
дпгидроклорид (витамин
сн3
NHjHCl
3-Метил- 4 -амино-1-нафтол
гидрохлорид (витамин
ПРОВИТАМИН НИКОТИНАМИДА
Никотиновая кислота
11
БИО КАТАЛИТИЧЕСКИЕ ФУНКЦИИ ВИТАМИНОВ
Большинство витаминов в составе ферментных систем катализируют
реакции превращения аминокислот и белков, жиров, стероидов, углево-
дов и нуклеиновых кислот в животном организме; к таким химическим про-
цессам относятся реакции окисления и восстановления, переноса электро-
на, переаминирования, трансметилирования, изомеризации, карбоксили-
рования, декарбоксилирования, переноса ацильных и одноуглеродных групп,
реакции, в частности, связанные с кроветворением, с кальцификацией кос-
тей и др. При участии витаминов обеспечивается нормальное функциони-
рование всех животных тканей, органов и желез внутренней секреции,
нормальные процессы обмена веществ [11, 12, 14—21].
Витамины являются типичными биокатализаторами и, как правило,
осуществляют свои каталитические функции в составе ферментных систем,
находясь в животных тканях в весьма малых количествах.
Для некоторых витаминов (L-аскорбиновой кислоты, кальциферола,
ретинола, токоферола, нафтохиноновых витаминов) характер действия
в биохимических реакциях и возможного связывания с белком недостаточ-
но ясен.
Все химические реакции между различными химическими соединениями
в клетках животного организма протекают при невысокой температуре —
в интервале от 1 до 40° С — и идут с чрезвычайно высокой скоростью.
Ускорение биохимических процессов обусловливается дополнительным
участием в реакциях многообразных органических биокатализаторов —
ферментов, или энзимов.
Ферменты — высокомолекулярные (с молекулярной массой от 9000
до 1 000 000 и больше) органические вещества сложной структуры пептид-
ной природы, строение которых основано на строго последовательном
чередовании и определенном разветвлении различных L-аминокислот
точного состава.
Специфичность фермента к определенной химической реакции находится
в связи с природой функциональных групп и типом химических связей реаги-
рующего вещества (субстрата), его пространственной конфигурацией и
характерной белковой составной частью фермента (14—17, 19, 20].
Основной принцип действия каждого катализатора заключается в том,
что, участвуя в реакции в незначительных количествах, он образует с
субстратом промежуточную активную форму, в виде которой происходит
химическое превращение, причем в конце реакции катализатор регенериру-
ется в неизменном состоянии.
В состав многих ферментов, помимо полипептидных цепей из десятков,
сотен и тысяч молекул аминокислот, составляющих специфическую белко-
вую (протеиновую) его часть, входит одна или несколько молекул относи-
тельно низкомолекулярного органического соединения небелковой природы
(основания, кислоты, спирта, кетона и т. д. алифатического, алициклическо-
го или гетероциклического ряда) — так называемая простатическая груп-
па, или кофермент. В таком случае протеиновая часть фермента называется
апоферментом. В состав некоторых ферментов также входят неорганиче-
ские кофакторы — ионы металлов Fe, Со, Си, Мп и др.
В качестве коферментов к настоящему времени известны следующие
вещества [14, 16—19]:
>'
Убихинон (кофермент Q)
Липоевая кислота
Порфирины (протопорфирин, формилпорфирин, мезопорфирин, ди-
гидропорфирин)
D-Гл юкозо-1,6-дифосфат
D-Маннозо-1,6-дифосфат
Аденозин-5'-ди (три) фосфат
Гуанозин-5'-ди (три) фосфат
Уридин-5'-ди (три) фосфат
Цитидин-5'-ди(три) фосфат
Инозин-5'-ди (три) фосфат
Уридиндифосфатсахара (с углеводами: глюкозой, галактозой, араби-
нозой, ксилозой, глюкуроновой кислотой)
Гуанозиндифосфатсахара (с углеводами: фруктозой, маннозой)
Цитидиндифосфатспирты (со спиртами: рибитом, этаноламином, гли-
церином. холином)
S-Аденозилметионин
Глутатион (y-L-глутамин-А-цистеинилглицин)
Ациладенилаты
Различные производные витаминов и др.
Коферменты — производные витаминов [14, 16, 17, 19] —представ-
лены ниже.
Кофермент А (КоА)
Никотинамидадениндинуклеотид (НАД)
Никотинамидадениндинуклеотидфосфат (НАДФ)
Пиридоксаль-5а-фосфат
Пиридоксамин-5а-фосфат
Тиаминпирофосфат (тиаминдифосфат, ТДФ)
5, 6, 7, 8-Тетрагидрофолиевая кислота
/У5-Карбоксибиотин
Флавинмононуклеотид (рибофлавин-5'-фосфат, ФМН)
Флавинадениндинуклеотид (ФАД)
Гистидилфлавинадениндинуклеотид (8 а-гистидил-ФАД)
Цистеинилфлавинадениндинуклеотид (8 а-цистеинил-ФАД)
Кофермент-кобаламин (5'-дезоксиаденозилкобаламин)
Группа нуклеотидных коферментов производных витаминов (КоА,
НАД, НАДФ, ФАД и др.) представляет собой смешанный ангидрид Р1,
Р2-диэфиров пирофосфорной кислоты. В своей структуре они имеют вита-
мин, соединенный р -гликозидной связью с D-рибофуранозой (нуклеозид)
и по первичной гидроксильной группе этерифицированный ортофосфорной
кислотой (нуклеотид); эта часть молекулы пирофосфатной связью объеди-
няется с аденозин-5'-фосфатом. Вместо D-рибофуранозы в структуре ко-
фермента может находиться D-рибит или витамин может быть этерифици-
рован ортофосфорной кислотой по его гидроксильной группе, и тогда ко-
фермент, представляя собой смешанный фосфоангидрид, имеет только
один остаток' D-рибофуранозы.
Номенклатура коферментов установлена Международным союзом по
чистой и прикладной химии [13].
Кофермент обусловливает каталитические функции в химической реак-
ции.
Сам каталитический акт химической реакции происходит в фермент-
субстратном комлексе, состоящем из субстрата и активного центра фермен-
та, образованного из определенных функциональных групп аминокислот,
кофермента, иногда ионов металла, строго фиксированных в простран-
стве.
Химическая связь кофермента (простетической группы) с ферментным
белком осуществляется по-разному. Связь с апоферментом может быть
13
ковалентной, трудно расщепляемой, например в сукцинатдегидрогеназе —
с гистидил-ФАД; в этом случае простетическая группа в каталитическом
акте во все время реакции тесно связана с белком. Связь с ферментным
белком может быть ионной, водородной или иной, легко диализуемой,
например НАД и алкогольдегидрогеназы; в этом случае кофермент в ката-
литическом акте переходит от одного ферментного белка к другому.
В отсутствие белка фермента кофермент не проявляет или почти не
проявляет биологической каталитической активности.
Апофермент — протеиновая часть фермента — синтезируется из £-ами-
нокислот самим организмом. Витамин же поступает с пищей и превращается
в кофермент в печени, крови или других органах и тканях.
Возможно, что некоторые коферменты — производные витаминов —
к настоящему времени еще не обнаружены.
В ряде заболеваний коферменты оказывают по сравнению с витами-
нами дополнительный специфический, только им присущий лечебный эф-
фект.
Строение коферментов, производных витаминов, показано ниже:
Структура коферментов — производных витаминов
Н
Л'Н2
РО3Н2
н ОН он
&Н2ОР-О-Р -OCH2C(CH3)2CH(OH)CONHCH2CH2CONHCH2CH2 SH
II II
о о
Ко<рермеН7 А
,conh2
nh2
N—[| N
О
N н Н
II
о
он
Д——К с н2о-р-о-р он2с
/он но\1 2 I "
н\о/н
НАД (никотинамидадениндинукг.еотид)
Н
н он
,conh2
н О он
Д—\ СН,О-Р -о-p -он2с
А>нно/ 2 & ъ '
Н АДФ (никотина мидадеминдинуклеотидфоссрат;
'.N К
н
О , о
Н РО3Н2
Н
сно он ch2nh2 он
hoY:JVch2o-p-oh hoyavch2o-p-oh
1 “ II I X
СНзЧг ° СНз N* °
ПиридОксаль-5<£ -<рос<рат Пиридоксамин-5^— фосфат
: °
Сгь . л _ it
Гснг- ОН О НООСЬГ^МН
^<;Асн,сн2о-р-о-р-он ( •
13 N NH2- < /СНгС^СНгСНгСООТ
Тиамлндифосрат S S-N-карбоксибиотин
соон
2 N -CH-HN-/~VcOHNCH
"/ТУ ‘
H2N"^N^P} СНгСООН
• 5,6,7,8 -Тетрагидрофопиевая кислота
н
ФМН (флавинмононуклеотид,
рибофлавин-5-фосфат)
OH
H ),—
OH
4 H
h2nocch2ch2
h2nocch2
Cb3
. CH3
H2NOCCH2
снЛо<
ГЧНОССН2СН2
CH2
ch-ch3
Со +
N
ch3
H3
Снз CH3) ch2ch2conh2
5 NH2
CH2CONH2
ch2ch2conh2
O=p-O
HOCH2
<H3
CHj
Кофврмент-
новаламин
АНТИВИТАМИНЫ
Большая группа органических соединений обладает свойством подавлять
биологическую активность витаминов. Такие соединения относятся к анти-
витаминам [5—12, 14, 19].
По структуре антивитамины в своем большинстве сходны с витаминами
и отличаются от них отсутствием или дополнительным наличием в молекуле
алкильной или какой-либо функциональной группы. Иногда структурное
сходство является достаточно отдаленным, например среди антивитами-
нов к.
Как и антиметаболиты, антивитамины в биокаталитических реакциях
в ряде случаев подменяют истинные катализаторы — витамины, проявляю-
щие свои каталитические функции в составе ферментных систем. Возможно,
антивитамины могут входить в активный центр ферментных систем (конку-
рентные ингибиторы) или взаимодействовать с полипептидной цепью иным
путем, образуя псевдоферменты, не обладающие биокаталитическими функ-
циями, однако способные в результате конфигурации подавлять биоката-
литическую активность истинных ферментов, в состав которых входят
витамины, или вытеснять витамины из ферментных систем.
Для подавления витаминных свойств, как правило, требуется примене-
ние больших количеств антивитаминов.
Многие антивитамины имеют важное значение в медицинской практике и
применяются для лечения некоторых заболеваний. Антиростовые вещества
микроорганизмов широко используются для лечения заболеваний, вызы-
ваемых патогенными микроорганизмами.
15
Витамин Антивитамин
L-Аскорбиновая кислота Пантотеновая кислота D-Аскорбиновая кислота (I) и>-Метилпантотеновая кислота (II) Пантоилтаурин, (+)-сульфопаитотеновая кислота (111)
Нафтохиноны Никотинамид Дикумарин (3,3'-метилен-бцс-4-оксикумарин) (IV) Пиридин-З-сульфокислота (V) 3-Ацетопиридин (VI)
Пиридоксин 2-Этил-3-амино-4-этоксиметил-5-(аминометилпиридин) (VII) 5-Дезоксипиридоксаль (VIII)
Тиамин Пиритиамин (IX) ' Окситиамин (X)
Биотин Оксибиотинсульфокислота (XI) Авидин (протеид)
Фолиевая кислота Аминоптерин (4-амино(дезокси)птероил-Т-глутамино- вая кислота) (XII) Аметаптерин (4-амино(дезокси)-10-метилптероил-Т- глутаминовая кислота) (XIII)
Рибофлавин 7-Метил-8-хлор-10-(Г-О-рибитил) изоаллоксазин (XIV) 7-Метил-8-амино-10-(Г-О-рибитил) — изоаллоксазин (XV)
Цианокобаламин 2,5-Диметилбензимидазол (XVI)
Структура важнейших антивитаминов
CH3CH(OH)C(CH3)2CH(OH)CONHCH2CH2COOH
носн2с ich3)2ch(oh)conhch, ch2so3 н
ill
О NH.. СООН
nA>V
HN NH { J CH„
\--/ H2N NN XII CH,COOH
y-CH2CH2CH2CH2SO3H
о XI
CH,OH CH2OH
I 2 I i
HOCH HOCH
HOCH HOCH
HOCH HOCH
I I
CH, CH,
Антивитамины имеют большое значение для изучения специфического
действия витаминов на животных и микроорганизмах, так как создавать
искусственные питательные среды, лишенные какого-либо витамина, во
многих случаях трудно.
Выше приводится сводка важнейших антивитаминов и подавляемых
ими витаминов.
ОПРЕДЕЛЕНИЕ ВИТАМИНОВ
Для количественного определения витаминов в готовых препаратах,
в растительных и животных источниках, в продуктах питания, в кормах,
а также в биохимических системах разработаны химические, физические,
микробиологические и биологические методы анализа [22, 23].
Так как витамины в настоящее время вырабатываются в виде индиви-
дуальных, высокой степени чистоты веществ, то при их качественном и
количественном определении руководствуются требованиями и методами
Госфармакопеи [261 или ГОСТ.
При витаминизации пищевых продуктов витамины используются в
кристаллическом состоянии или в масляных растворах. В лечебной практи-
ке витамины обычно применяются в кристаллическом виде, в виде табле-
ток, капсул, масляных или спиртовых растворов или инъекционных раство-
ров в ампулах. Для витаминизации кормов витамины предварительно полу-
чают, как правило, в виде стабилизированной смеси с какими-либо
продуктами (премиксы), а затем уже смешивают с большим количеством кон-
центрированных кормов.
ПОТРЕБНОСТЬ В ВИТАМИНАХ
Потребность человека и сельскохозяйственных животных в витаминах
в настоящее время достаточно изучена. Однако исследования по уточнению
потребности в различных климатических условиях, при разных условиях
труда и питания продолжаются в довольно широких масштабах. Данные
о потребности взрослого человека (весящего в среднем 60—70 кг) в витами-
нах представлены ниже.
Витамин Потребность, мг/сут Витамин Потребность, мг/сут
L-Аскорбиновая кислота 70—100 Никотинамид (никотиновая
Пантотеновая кислота 5—10 кислота) 15—25
Эргокальциферол 0,01—о;о4 Пиридоксин 2.0—2,5
Ретинол 1,5—2,0 Тиамин 1,5—2,0
Филлохинон 2,0 Биотин 0,1—0,3
а-Токоферол 2-6 Фолиевая кислота Рибофлавин Цианокобаламин 0,1—0,5 2,0—2,5 0,005—0,050
СПИСОК ИСПОЛЬЗОВАННОЙ
ЛИТЕРАТУРЫ (К ВВЕДЕНИЮ)
1. К. Ф у н к. Витамины, предисло-
вие Н. Д. Зелинского. М., ГИЗ.
1922.
2. С. Funk. J.. Stat. Med., 20, 341
(1912).
3. Н. Лунин. Z. physiol. Chem.,
15, 93, 97. (1881).
4. К. М. Быков, Г. Е. В л а -
д и м и р о в, В. Е. Дело в,
Г. П. Конради, А. Д. С л о-
н и м. Учебник физиологии. М.,
Медгиз, 1954.
5. В. Н. Буки н, Витамины, М.,
Пищепромиздат, 1941.
6. Н. Rosenberg. Chemistry and
Physiology of the Vitamins, New
York, 1945.
7. Б. А. К у д p я ш о в. Биологи-
ческие основы учения о витаминах,
М., изд. «Сов. наука», 1948; Физио-
логическое и биохимическое значе-
ние витаминов, М., изд. Моск, о-ва
испыт. природы, 1953.
8. В. А. Девяти н н. Витамины.
. М., Пищепромиздат, 1948.
9. А. В. Т р у ф а н о в. Витамины и
антивитамины, М. Пищепромиздат,
1950.
10. Vogel. Chemie und Technik der
Vitamine, Bd. I, 1950; Bd. Il,
1955, Stuttgart.
11. The Vitamins, Chemistry, Physio-
logy, Pathology, Methods, Ed. W. Se-
brell, R. Harris, vol. 1. II, New
5 ork, 1967—1968.
12. Vitamine, Chemie und Biochemie,
Band 1, H под ред. J. Fragner,
.Jena, 1964 —1965.
13. I.U.P.A.C., Commission on the No,
menclature of Biological Chemistry-
J. Am. Chem. Soc., 82, 5581 (1960;
83, 563 (1961); Biochim. Biophys.
Acta, 107, 1 (1965); J. Biol. Chem.,
241, 2987 (1966); Z-physiol. chem.,
348, 266 (1967).
14. M. Диксон, Э. Уэбб. Фер-
менты. M., изд. «Мир», 1966.
15. Дж. Н е й л а ндс, П. Ш т у -
м п ф. Очерки по химии ферментов.
М., изд. ИЛ, 1958.
16. Ферменты, под ред. А. Е. Браун-
штейна, М„ изд. «Наука»,
1964.
17. Э. К о с о в е р. Молекулярная
биохимия, М., изд. «Мир , 1964.
18. В. Л. К р е т о в и ч. Введение в
энзимологию. М., Изд. «Наука», 1967.
19. Б. П ю л ь м а н, А. Июль-
май. Квантовая биохимия. М.,
изд. «Мир», 1965.
20. Механизм и кинетика фермента-
тивного катализа, под ред.
А. Е. Брауиштейна и
В. А. Яковлева. М., Изд.
АН СССР, 1964.
21. Номенклатура ферментов, М., Изд.
АН СССР, 1966.
22. В. А. Девятнин. Методы хи-
мического анализа в производстве
витаминов. М., изд-во «Медицина»,
1964.
23. The Vitamins, Chemistry, Physiolo-
gy, Pathology Methods, Ed. P. Gy-
orgy, W. Pearson, vol. VI, VII,
New York, 1967.
24. Методическое руководство цо опре-
делению витаминов, под ред.
Б. А. Лаврова, М., Медгиз,
1960.
25. Методы определения витаминов (хи-
мические и биологические), под
ред. В. А. Д е в я т и и н а, М.,
Пищепромиздат, 1954.
26. Госфармакопея СССР X; USP
Farmacopea.
27. ВОЗ — серия техн, докл., № 362;
изд. ВОЗ, Женева, 1968.
28. «Рекомендуемые величины физио-
логических потребностей в пище-
вых веществах и энергии», утве.ржд.
зам. министра здравоохранения
СССР, 16 апреля 1968 г. (№ 735—
68), М., изд. Минздрава СССР,
1968.
Часть первая
ВИТАМИНЫ АЛИФАТИЧЕСКОГО РЯДА
ГЛАВА I
ВИТАМИНЫ — ПРОИЗВОДНЫЕ НЕНАСЫЩЕННЫХ
ПОЛИОКСИ-1-ЛАКТОНОВ
L-АСКОРБИНОВАЯ КИСЛОТА
£-Аскорбиновая кислота — витамин С [1] (I) представляет собой ^-ла-
ктон 2,3-дегидро-£(-г-)-гулоновой кислоты (7-лактон £-трео-2,3,4,5,6-пен-
таокси гексен-2-овой кислоты).
НОН2С-С
6 5|
н
он он
?Н
1.н/"3 7
„—С л |.СЧ
о
I
£-Аскорбиновая кислота находится в тканях также и в виде окисленной
формы—дегидро-£-аскорбиновой кислоты (II).
По своему строению L-аскорбиновая кислота может быть отнесена'к
производным углеводов, в структуру которых включены 7-лактонное
кольцо системы Аа-3-бутенолидов и группировка «редуктона» с двумя сопря-
женнымидвойными связями—С=С—С=О. Важнейшее свойство такихсис-
I I
онон
тем — способность к обратимым окислительно-восстановительным превра-
щениям .
L-Аскорбиновая кислота имеет два асимметрических атома’углерода в
положениях 4 и 5 и образует четыре оптических изомера и два рацемата.
Она представлена в двух эпимерных формах, каждая из которых дает по
два оптических антипода: D- и L-аскорбиновые кислоты и их диастерео-
изомеры — D- и £-изоаскорбиновые кислоты (I, III, IV, V).
<0-Аскорбиновая кислота
носн2
Х-Изоаскорби новая кислота
IV
2?-Иэоаскорбимовая кислота
v
19
Природная биологически активная L-аскорбиновая кислота относится
к производным гулозы L-ряда. Нахождение среди биологически активных
веществ производных гексоз L-конфигурации является необычным, так как
природные моносахариды животного организма, как правило, имеют
D-конфигурацию. D-Аскорбиновая кислота витаминными свойствами не
обладает, а наоборот, является почти единственным антагонистом витами-
на С. D-Аскорбиновая и D- и L-изоаскорбиновые кислоты (III -—V) в при-
роде не встречаются и получены только синтетически.
L-А с к о р б и и о в а я кислота (I) представляет собой бес-
цветные призмы моноклинической системы, без запаха, с т. пл. 192° С
(с разл.); она имеет [а ]ь° + 23° (1,6%, Н2О) и [а g + 48° (0,85%, СН3ОН).
Молекула /.-аскорбиновой кислоты имеет плоскую конфигурацию, однако
атом С(5) находится вне этой плоскости, что доказано кристаллографически-
ми и другими измерениями [2, 3]. Аскорбиновая кислота хорошо раство-
рима в воде, значительно хуже — в спирте [4], что видно из данных табл. 2.
Таблица 2
Растворимость L-аскорбиновой кислоты в процентах при различных температурах
Растворитель Температура, СС
0 10 23 30 40 50 60 70 80 10G
Вода Спирт 96% об. 13,59 3,33 17,79 22,42 4,61 27,10 5,50 30,75 6,62 38,24 8,27 42,34 10,65 46,57 17,76 (78°) 50,47 57,51
В высших спиртах растворимость L-аскорбиновой кислоты сильно
уменьшается. L-Аскорбиновая кислота малорастворима в глицерине и в
ацетоне; нерастворима в неполярных растворителях, таких, как аромати-
ческие и алифатические углеводороды (бензол, петролейный эфир, бензин),
в галогенопроизводных (четыреххлористый углерод, хлороформ, дихлор-
этан, хлорбензол), в эфире.
В водных растворах L-аскорбиновая кислота обладает кислой реакцией
(для 0,1 н. раствора pH 2,2) и реагирует как одноосновная кислота. Так как
лактоны нейтральны, то кислые свойства вызываются главным образом
гидроксильной группой положения 3 и только частично гидроксильной
группой положения 2. Константы диссоциации L-аскорбиновой кислоты
следующие: по одним данным р/<\4,17 ирК2 И,57 [5], по другим [61 рК14,25
и рК2 11,79. Спектр поглощения L-аскорбиновой кислоты в ультрафиоле-
товом свете имеет подвижный максимум от 245 нм в кислой среде до 265 нм
в нейтральном водном или щелочном растворе (Ецм 3,98) [7], зависящий
от присутствия сопряженной системы двойных связей, а также небольшое
плечо между 350 и 400 нм [8]. Максимум поглощения в спиртовом растворе
245 нм. Следует отметить, что ультрафиолетовый свет вызывает расщепле-
ние L-аскорбиновой кислоты.
Окислительно-восстановительный потенциал при равномолекулярном
соотношении L-аскорбиновой и дегидро-L-аскорбиновой кислот в воде
следующий [9, 101:
рн е;, в рн е». В
2,04 4- 0,281
2,68 4-0,242 4,65 4-0.138
3,30 4- 0,204 5,31 4-0.119
4,01 4-0,166 5,75 4-0,106
Возможно, однако, что окислительно-восстановительные потенциалы
не характерны [111, так как система L-аскорбиновая кислота — дегидро-L-
аскорбиновая кислота обратима неполностью.
20
Полярографический окислительный потенциал L-аскорбиновой кислоты
[121 находится в зависимости от pH. Полуволновой потенциал Е ( окисления
составляет + 0,226 В при pH 2,19, -f- 0,090 В при pH 4,29 и — 0,074 В
при pH 8,32 [13].
L-Аскорбиновая кислота адсорбируется активированным углем.
L-Аскорбинат натрия C6H7O6Na — представляет собой бес-
цветные кристаллы,
ONa он
он
I ,
носн,-с— нс
Л
с=с
sA
(а ]о + Ю5° (Н2О). Он легко растворим в воде, трудно — в спирте
(95%-ном) и нерастворим в эфире. Выпускается как препарат для приготов-
ления ампульных растворов и для консервирования мяса и мясных изде-
лий.
L-А скорбинат кальция (С6Н7Ос)2Са-2Н2О — бледно-жел-
тые кристаллы, (а 1о + 96° (Н2О). Он легко растворим в воде, трудно —
в спирте и нерастворим в эфире.
L-А скорбинат железа — кристаллы ярко-фиолетового цвета;
используется в лечебных целях.
Пальмитат L-a скорбиновой кислоты — сложный
эфир по первичному гидроксилу положения 6,
он он
I ।
он н с=с
. | Н / \
сн3(снг)чсооснг—с—-'с Сх
I \ х <)
н о и
представляет собой бесцветные кристаллы со слабым желтоватым оттенком.
Почти нерастворим в воде, хорошо растворим во многих органических
растворителях, животных и растительных маслах.
Известны препараты аскорбиновой кислоты с сульфаниламидными
соединениями, например с норсульфазолом и др., с характерными химио-
терапевтическими свойствами этих соединений [14].
Дегидро-L-аскорбиновая кислота — у-лактон2,3-дике-
to-L(+)- гулоновой кислоты (II) — представляет собой бесцветные кристал-
лы ст. пл. 237—240°С (с разл.) [15], [а]о -{- 55° [16]. Константа диссоциа-
ции рК 9.
Безводная ,дегидро-А-аскорбиновая кислота, возможно, имеет диокса-
новую структуру и является димерным лактоидом, лишенным хромофор-
ных кетогрупп и первичной гидроксильной группы [17, 18]. В твердом
состоянии она, вероятно, имеет полукетальную структуру и связана меж-
молекулярны.ми водородными связями в форме полимерного агрегата [19].
В водном растворе дегидро-L-ac корбинов а я кислота находится в виде
мономера.
ХИМИЧЕСКИЕ СВОЙСТВА L-АСКОРБИНОВОЙ КИСЛОТЫ
Ненасыщенное у-лактонное кольцо L-аскорбиновой кислоты (I) при
действии сильных щелочей подвергается гидролитическому расщеплению,
превращаясь в соль кетокислоты (VI), но не в соль ненасыщенной окси-
кислоты
он он
?Н /С==СХ NaOH ОН Н ОН О
HOCII, С -нс. С ---'НОСИ,-с- с-с- C-COONa
' 1 х / О III
НО н онн
Кратная связь весьма сильно способствует стабилизации лактонного
кольца. Наличие двух гидроксильных групп у атомов углерода, соединен-
ных двойной связью, обусловливает, как это свойственно енольным формам,
кислый характер соединения. Сопряжение карбонильной группы с двой-
ной связью, находящейся между атомами углерода С(2) и С(з> I—С=С—С—О],
I I 1
также влияет на усиление кислого характера ендиольных групп. Поэтому
со слабыми щелочами L-аскорбиновая кислота легко образует нейтральные
монощелочные еноляты без расщепления лактонного кольца (см. с. 21).
Натриевые и аммонийные еноляты обладают хорошей растворимостью в
воде; нейтральный свинцовый енолят растворим в воде, но почти не раство-
рим в спирте; основной свинцовый енолят в воде и спирте нерастворим.
L-Аскорбиновая кислота может осаждаться из слабого аммиачного водного
раствора при pH 7,2—7,5 уксуснокислым свинцом.
Для L-аскорбиновой кислоты в растворах известны таутомерные формы.
В растворах она находится почти полностью в виде ненасыщенного «-гли-
коля (I), образующего ендиольную группировку, и в очень малом коли-
честве в кето-форме, причем 2-кето-форма (VII) достаточно стабильна и
может быть выделена, а 3-кето-форма (VIII), если таковая и существует б
действительности, лабильна и легко переходит в ендиольную форму [20]:
он о
он
он он
ОН I ।
носн,-с— с.
I
н
с -
ох о
носнг-с —с.
н
У*
о он
ОН П i
IH С— сн
,с=о
носн-с—с
I \ ,
н о
:—с
VII I VIII
L-Аскорбиновая кислота реагирует и в своей таутомерной кето-форме.
Она образует фенилозазоны и производные с о-фенилендиамином [21, 22).
По двойной связи L-аскорбиновой кислоты может происходить присоедине-
ние сульфгидрильных групп и галогенов.
Следует отметить, что реакционная способность карбоксильной группы
L-аскорбиновой кислоты характеризуется некоторыми особенностями; так,
она не реагирует с алюмогидридО1М лития, который обладает специфическим
свойством восстанавливать кислоты или лактоны в соответствующие спирты
[231.
L-Аскорбиновая кислота (I) в сухом состоянии в кристаллической форме
устойчива, но вследствие наличия одной ненасыщенной связи во влажном
состоянии или в растворах легко изменяется, особенно в присутствии
воздуха. В растворах L-аскорбиновая кислота аэробно окисляется, наиболее
легко при каталитическом влиянии некоторых металлов (например ионов
меди) в дегидроД-аскорбиновую кислоту (II) и перекись водорода [24, 25].
ДегидроД-аскорбиновая кислота обратимо превращается в L-аскорбино-
вую кислоту (I) при действии йодистого водорода или сероводорода при
pH 4—7.
сн он
।
он с=с ,
i / \ -Зе.~2Н
HOCH -С-НС С —. -а
\ / \ -2е.-2Н’
Н О О
° °
ОН /С—С
иосн..-с-нс ё
‘ н V о
ОО
С повышением щелочности скорость реакции окисления увеличивается.
При pH ниже 7 реакция протекает только при участии катализатора
[26, 27]. Активированный уголь способствует окислению L-аскорбиновой
кислоты [28].
L-Аскорбиновую кислоту дегидрируют, помимо кислорода воздуха
[29], селеновая кислота [30], перекись водорода, хлорное железо, хинон,
ацетат меди [31, 32], 2,4-дихлорфенолиндофенол в кислом растворе [33],
хлор, бром и йод в кислом и нейтральном растворах [34, 35], метиленовый
голубой [36, 37], йодноватая кислота, марганцовокислый калий, азотно-
кислое серебро, раствор фелинга, а также антибиотик террамицин [38]
и многие другие соединения. Изучена кинетика окисления L-аскорбиновой
кислоты перекисью водорода [39].
Окисление L-аскорбиновой кислоты помимо меди катализируют ионы маг-
ния [40], серебра. Следует отметить, что кальций, марганец, железо, никель
и кобальт почти не обладают каталитическими свойствами в реакциях
окисления аскорбиновой кислоты кислородом воздуха [26], а в безводном
спиртовом растворе или других неводных растворах йод и другие галогены
не реагируют с L-аскорбнновой кислотой. Влияние pH на кинетику окис-
ления L-аскорбиновой кислоты подвергалось подробному исследованию
[41 ]. В отсутствие катализаторов окисление кислородом воздуха не идет и
растворы L-аскорбиновой кислоты обладают стойкостью к умеренному
нагреванию. Двуокись углерода и сернистый ангидрид предохраняют L-ac-
корбиновую кислоту от окисления; они применяются для ее стабилизации.
Реакцию окисления L-аскорбиновой кислоты в растворах ингибируют
этилендиаминтетрауксусная кислота [42], флавоноиды 143], о-дифенолы
[33], метафосфорная кислота [44], тиоацетали [45], некоторые N-гетеро-
циклические соединения [46] и др.
Схема 1
Реакции окислительного расщепления L-аскорбиновой кислоты
ОН ОН о о
: Г II
ОН С=С -2е.-2Н" ОН С—С Н,О
I ! \ ... --Г I / \ —~
НОСН,-С-НС щ -2е*2Н* НОСН2-С~НС С.
н ох ° А V "о
1 - И
соон
I
с=о
с=о нго
неон
носн
I
СН2ОН
СНО !
соон+ неон ;
СООН носн
CH..OH
_ и' :
сн.,он
с-₽о
носн
CH.OH
соон
[oj неон [о]
носн
сн.он
]й х
соон
о; с=о
носн
СН..ОН
СНО
НОСН
СН.ОН
Lji :
Г сн.он!
I — I
I С=О I
I I
Г СН.ОН:
I - I
СООН
[О] НОСИ [О] СНО [О] СООН
СН,ОН СН.ОН соон
СН.ОН
СООН
jb] неон
носн
I
соон
— I
XIV
L-Аскорбиновая кислота легко подвергается окислительному расщепле-
нию (схема 1). Как указывалось выше, при действии на L-аскорбиновую
кислоту (I) кислорода образуется дегидроД-аскорбиновая кислота (II).
Затем лактонное кольцо дегидро^-аскорбиновой кислоты в отличие от
стабилизированного двойной связью лактонного кольца L-аскорбиновой
кислоты в водном растворе легко гидролизуется с образованием 2,3-дике-
TO-L-гулоновой кислоты (IX) [34, 47]. Эта реакция необратимого превра-
23
щения дегидро-Ь-аскорбиновой кислоты первого порядка [48], она протекает
в водном растворе со значительной скоростью [10], которая возрастает
с повышением температуры и pH растворов. Превращение половины коли-
чества вещества протекает при pH 5 и температуре 80°С в течение 5 мин
[27].
Следующей стадией превращения является переход 2,3-дикето-Ь-гу-
лоновой кислоты (IX) при дальнейшем окислении кислородом в L-треоно-
вую (X) и щавелевую кислоты. Вероятно, первоначально, в щелочных
условиях в результате гидролиза происходит расщепление молекулы и
образуется промежуточное соединение типа тетрозы, обладающее значи-
тельной восстановительной способностью, и только затем альдегидная
группа окисляется в карбоксильную.
Затем происходит дальнейшее окисление L-треоновой кислоты (X) в
£(+)-винную кислоту (XIV) и многочисленные другие хроматографически
обнаруженные соединения [49]. Среди продуктов окисления L-аскорбиновой
кислоты идентифицированы: 2,3-дикето-/>-гулоновая (IX), L-треоновая (X),
2-кето-Ь-треоновая (XI), глицериновая (XII), оксипировиноградная (XIII),
щавелевая кислоты и двуокись углерода [47, 50, 51 ]. Возможно, что обра-
зование кетокислот (XI, XIII) происходит не только в результате прямого
окисления оксикислот (X, XII), но и в результате предварительной изомери-
зации альдоз в кетозы, которые в последующем подвергаются окислению.
Для объяснения дальнейшей реакции расщепления 2,3-дикето-Ь-гу-
лоновой кислоты (IX) в растворе при pH 3,2 предложены два процесса:
1) декарбоксилирование с образованием L-ксилозона (XV7);
2) окисление таутомерной формы IX в 2, 3, 4-трикето-£-гулоновую кис-
лоту (XVI) с последующим образованием L-треозона (XVII) [48]:
соон соон ecu
1 1 +
сно с=о с=о со
с=о г° с=о сно
неон неон с=о с=о
носн носн носн носн
<Ь2ОН снгон снгон 6н2он
XV IX XVI XVII
L-Аскорбиновая кислота, помимо окислительного расщепления в опре-
деленных условиях, подобно углеводам, склонна к гидролитическому
расщеплению, связанному с декарбоксилированием и дегидратацией в фур-
фурол и дальнейшие продукты его глубокого расщепления. Известно, что
L-аскорбиновая кислота при кипячении с соляной кислотой количественно
образует фурфурол и двуокись углерода [21, 34, 52]; эта реакция может
применяться для аналитических целей. Образование фурфурола и двуоки-
си углерода наблюдалось и в других условиях [53]. Таким же путем, воз-
можно, происходит расщепление и при pH выше 7,6 с той разницей, что
реакция почти не задерживается на образовании фурфурола, а протекает
дальше. Скорость глубокого расщепления L-аскорбиновой кислоты повы-
шается с увеличением щелочности [26].
Реакция гидролитического расщепления L-аскорбиновой кислрты, воз-
можно, протекает по схеме 2.
L-Аскорбиновая кислота (I) в растворе, как лактон, находится в состоя-
нии подвижного равновесия с очень малыми количествами 2-кето-А-гуло-
новой кислоты (XVIII). Последняя под влиянием кислых или щелочных
реагентов или повышенных температурных условий медленно декарбокси-
лируется с образованием двуокиси углерода и L-ксилозы (XIX). Расщепле-
ние некоторой части 2-KeTo-L-rynoHOBoi“i кислоты сдвигает равновесие и
вызывает последующее превращение новой части L-аскорбиновой кислоты
24
в 2-кето-£-гулоновую кислоту. Затем L-ксилоза (XIX) гидролитически
расщепляется с образованием фурфурола (XX).
Схема 2
Реакции гидролитического расщепления L-аскорбиновой кислоты
СООН
сно
ОН он
он I I
। ч/H
носн2-с—С С
А
Н2О
с=о
носн —-
I со2
неон
носн
СН2ОН
носн
неон —-
НОСН
СН2ОН
XIX
Д И / \
О'СНО ХГ СООН
XX
XXI
XVIII
Реакция образования фурфурола изучена особенно тщательно именно
для ксилозы [54 ]. Следует отметить, что фурфурол легко вступает в реакции
присоединения, подвержен полимеризации и легко окисляется с раскрытием
цикла в янтарную, дегидроянтарную кислоты, а также многие другие орга-
нические кислоты [55] и смолистые продукты сложного строения.
Дегидратация L-ксилозы может идти и по другому направлению, в
результате чего образуется 2,5-дигидропирослизевая кислота (XXI), кото-
рая обнаружена среди продуктов расщепления L-аскорбиновой кислоты в
анаэробных кислых водных условиях [56], наряду с L-ксилозой (XIX)
[53].
Катализируемое соляной кислотой расщепление L-аскорбиновой кисло-
ты до фурфурола протекает через различные сложные промежуточные
соединения [57]
Образование дегидроД-аскорбиновой кислоты (II) при действии йода
на L-аскорбиновую кислоту (I) в растворе протекает через промежуточное
присоединение двух эквивалентов йода по двойной связи и последующее
отщепление йодистого водорода. Эта реакция применяется для количест-
венного определения L-аскорбиновой кислоты [34]. Широкое применение
имеет и йодатный метод анализа.
При окислении L-аскорбиновой кислоты формальдегидом образуется
дегидроД-аскорбиновая кислота, которая медленно декарбоксилируется
с выделением двуокиси углерода [581, причем скорости выделения двуокиси
углерода из дегидроД-аскорбиновой кислоты и из L-аекорбиновой кислоты
с раствором формальдегида приблизительно одинаковы. Выделение двуоки-
си углерода происходит за счет карбоксильного атома углерода L-аскорби-
новой кислоты, что доказано путем применения меченых атомов [59].
ДегидроД-аскорбиновая кислота образует разные формы редуктон-
производных [60] и различные другие производные, например с фенилгид-
разином—фенилозазон дегидро-Даскорбиновой кислоты в у-лактонной
форме (XXII) [34] или в o-форме (XXIII) [61], с о-фенилендиамином—
хиноксалиновое производное (XXIV7) [62] и с о-фенилендиамином ифенил-
гидразином — производное XXV [63]. ,
CeH5NHN NHNCtH5
„ Ч 11
он с—с
носнг-с-нс( к
1 xoz о
XXII
C6H5NHN nhnc6h5
И II
Н /С~С
но \ >
/с\ °
Н CHjOH
XXIII
N N
с-с
он
с=о носн,-с-нс С I
' 1 \ /У»
н о °
XXIV
C6HSNHN
ОН с—c^N
'N
XXV
25
* Для количественного определения малых количеств /.-аскорбиновой
кислоты используется ее реакция с 2,4-динитрофен илгидразином [64].
Вследствие легкой окисляемости /.-аскорбиновая кислота является
донором водорода; она количественно легко восстанавливает многочислен-
ные соединения. На способности этих соединений изменять окраску при
восстановлении основаны различные способы определения /.-аскорбиновой
кислоты. К таким соединениям относятся: синий 2,6-дихлорфенолиндо-
фенол, который переходит в бесцветный фенол имин 152, 65],
ci ct
метиленовый голубой, переходящий в бесцветное лейкосоединен ие [36, 66];
диазосульфаниловая кислота, образующая оранжевый азокраситель [67];
и др.
/.-Аскорбиновая кислота дает окраску с многочисленными соединениями:
с молибденовофосфорновольфрамовой кислотой — фиолетовую, с пяти-
окисыо ванадия в серной кислоте — синюю, с селенистой кислотой — оран-
жево-красную и др.
Дегидро-Ь-аскорбиновая кислота реагирует с аминокислотами, обра-
зуя продукт, имеющий розовую или оранжево-красную окраску [16, 68];
при этом выделяется двуокись углерода. Такая же окраска наблюдается
при соприкосновении дегидро-Ь-аскорбиновой кислоты с кожей животных,
L-Аскорбиновая кислота с хлора н гидр идам и высших жирных кислот
дает сложные эфиры [69—71 ], например З-О-па льмитоил- илиЗ-О-стеароил-
L-аскорбиновую кислоту [70], которые сохраняют витаминную активность
и применяются как жирорастворимые препараты L-аскорбиновой кислоты в
качестве антиоксидантов — стабилизаторов.
С никотиновой кислотой и ее амидом L-аскорбиновая кислота дает
устойчивый комплекс с т. пл. 185° С [72].
L-Аскорбиновая кислота образует многочисленные соли, например с
холином [73], аминокислотами 174, 75], 2-меркаптоэтиламином [76] и др.
Интересно отметить соединение L-аскорбиновой кислоты с тиамином, кото-
рому приписывают следующую структуру [77]:
ТСНз
СН2СН2ОН
он
I
с=с
о
ОН I
НОСН.СН-^о^О
СТРОЕНИЕ L-АСКОРБИНОВОЙ КИСЛОТЫ
В 1918—1925 гг. Цильва выделил из лимонов почти чистый антискор-
бутный фактор, установил его молекулярную массу, свойства (неустойчи-
вость к реакциям окисления и др.) [78—80]. Препарат L-аскорбиновой кис-
лоты был выделен в 1925 г. из капусты, а в 1928—1930 гг. под названием
«гексуроновая кислота» — из надпочечников быка [81 ] и затем из сока
апельсинов. Гексуроновая кислота впоследствии была названа L-аскорби-
новой кислотой [1 ].
В результате изучения свойств L-аскорбиновой кислоты и продуктов ее
расщепления в 1933 г. Херберт и Хирст с сотр. [34, 82, 83] и независимо
Эйлер с сотр. [84 ] предложили для L-аскорбиновой кислоты ее общеизвест-
hvio формулу (1), которая в том же году была подтверждена многими ис-
следователями 17, 32, 85—87].
L-Аскорбиновую кислоту впервые синтезировали в 1933 г. независимо
друг от друга Рейхштейн с corp. [85, 861 и Хэуорз и Хирст с сотр. [87, 881.
Было показано, что L-аскорбиновая кислота (I) имеет четыре активных
атома водорода, принадлежащих четырем гидроксильным группам, обра-
зующим тетраацетат. В определенных условиях L-аскорбиповая кислота
может ацетилироваться только по двум гидроксильным группам [89], кото-
рые. следовательно, отличаются по своей реакционной способности от двух
других. L-Аскорбиповая кислота способна метилироваться диазометаном
на холоду по гидроксильной группе при С(з) (более кислой, чем при С(о>)
с образованием 3-монометилового эфира аскорбиновой кислоты (XXVI)
[90, 91]. Эфир не обладает восстановительными свойствами, имеет только
слабую кислотность и дает с хлорным железом интенсивную фиолетово-
синюю окраску [90]. Диазометан при комнатной температуре этерифици-
рует и вторую гидроксильную группу при С(2), образуя 2,3-диметиловый
эфир L-аскорбиновой кислоты (XXVII), который также не обладает вос-
становительными свойствами [31, 32]. При дальнейшем метилировании
йодистым метилом образуется 2,3,5,6-тетраметиловый эфир L-аскорбиновой
кислоты (XXVIII) [34, 82]:
он
носн2-с-нс:
н
он он
с=с ch,n2
у, (на холоде)
осн3 он
I I
он с=с сн,ы2
I / \ -" . •
носнг-с-нс\ .с 20 25
н '"(У "°
осн3 осн3 осн3 осн3
он /Я==СЧ СНД. осн3 с=с
НОСН2-С- НС С СНоОСН^С-НС7 сч
А \ох V а 'о
XXVII
При ацетонировании L^acKop6nnoBofi кислоты (I) образуется только
5,б-моно-О-изопропилиден-L-аскорбиновая кислота (XXIX) по гидро-
ксильным группам при С(5) и С(б) [32], причем это производное имеет такие
же восстановительные свойства, как и L-аскорбиновая кислота [92, 93], но
обладает низкой витаминной активностью. При действии диазометана на
5,6-0-изопропил идеи-L-аскорбиновую кислоту (XXIX) образуется З-.моно-
метиловый [91 ], а затем 2,3-диметиловый эфиры 5,6-0-изопропилиден-
L-аскорбиновой кислоты (XXX и XXXI) [31 ], не обладающие восстано-
вительными свойствами.
Так как 2,3-диметиловый эфир L-аскорбиновой кислоты (XXVII) и
2,3-диметиловый эфир 5,б-О-изопропилиден-L-аскорбиновой кислоты
(XXXI) в отличие от L-аскорбиновой кислоты не обладают восстанови-
тельными свойствами, то этим определяется енольный характер двух
ГИДРОКСИЛЬНЫХ Групп при С(2) И С(3).
он
I /
носн2-с-нс,
н
он он
I i
<с С\ СН3СОСН3
С ------------
СНз сн3
о 'о
он он
с=с
н2с— с-НС
н
ch2n2
сн3/сн3 1 сн, сн,
\ осн, он 'с
6 О • - с CH..N, О О
I \ —-—~
сн,-с-н< ,СЧ НХ —С-1
н О
п f ь
27
Дальнейшее доказательство наличия двойной связи между атомами
углерода положений 2 и 3 молекулы L-аскорбиновой кислоты получено на
основании образования цветной реакции с тетранитрометаном [31,32], а
также того обстоятельства, что при каталитическом восстановлении L-ас-
корбиновой кислоты (I) 1 молем водорода была получена L-идснэвая кисло-
та [7|. При восстановлении двойной связи между С<2> и С(3) могут образо-
ваться четыре пентаоксикарбоновые кислоты (XXXII а—XXXII г) с двумя
2,3-эритро- и двумя 2,3-трео-гидроксильными группами. L-Идоновая
кислота представляет собой 2,3-трео-изомер (XXXIIв).
он н он он
iili
носн2-с—с—с—с-соон
1111
н онн н
х»хпв х. он
онс=
он н онн
1111
носн,-с—с—с—с-соон
II II
н он н он
Illi I
носн,-с-с-с-с-соон носн2-с-
llll I
Н (Ж он он н
н н он
I I I
с— с — с-соон
I I I
он он н
xxxfi г
В своей таутомерной кето-форме L-аскорбиновая кислота дает с фенил-
гидразином и его производными различные озазоны [21 ] в отличие от ее
диметилового эфира, не реагирующего с этими реагентами. Спектр погло-
щения ультрафиолетового света (максимум 245 нм — в кислом растворе и
265 нм — в нейтральном) указывает на присутствие в молекуле L-аскорби-
новой кислоты системы сопряженных двойных связей
—СН—С—С=О С=С—С=О
ОН О OR ОН ОН OR
Система сопряженных двойных связей, аналогичная таковой же у
L-аскорбиновой кислоты, имеется в диоксималеиновой кислоте, которая
ведет себя в окислительно-восстановительных реакциях так же, как и
L-аскорбиновая кислота [34].
Так как при окислении L-аскорбиновой кислоты перекисью водорода
образуется щавелевая кислота [32], то можно заключить, что в таутомер-
ной кето-форме кетогруппа находится во-положении к карбоксильной груп-
пе. Почти количественное образование при окислении йодной кислотой
наряду со щавелевой кислотой L-треоновой кислоты [83 ] указывает на поло-
жение двойной связи между атомами углерода положений 2 и 3, а также
на то, что молекула L-аскорбиновой кислоты содержит неразветвленную
цепь из 6 атомов углерода.
L-Треоновая кислота (X) идентифицирована путем метилирования в
1,2,3,4-тетраметиловый эфир L-треоновой кислоты (XXXIII) и выделения
кристаллического 2,3,4-триметил-О-/.-треонамида (XXXIV) [7]. При окис-
лении L-треоновой кислоты (X) выделена L(4-)-bhhh3H кислота (XIV),
строение которой установлено путем ее превращения в L(+)-AHMeTOKCH-
сукцинамид [83]:
СООН соон СООСНз conh2
НСОН ч - НСОН -> ► НСОСНз - -> НСОСНз
носн НОСН СН3ОСН СН3ОСН
с!оон СН2ОН 1 СН2ОСН3 CH2OCHS
XIV X XXXIII XXXIV
28
Присутствие первичной гидроксильной группы в молекуле L-аскорби-
новой кислоты было показано реакцией с трифенилметилхлоридом (94];
тритиловый эфир содержал ендиольную группировку и мог быть этерифи-
цирован диазометаном (32]. На наличие первичной гидроксильной группы
указывало также образование формальдегида при окислении L-аскорби-
новой кислоты тетраацетатом свинца (7].
Эти данные привели к утверждению, что L-аскорбиновая кислота яв-
ляется производным L-гулозы.
При дегидрировании L-аскорбиновой кислоты слабыми окислителями
образуется дегидро-L-аскорбиновая кислота (II), не обладающая кислыми
свойствами 129]. Дегидро-1-аскорбиновая кислота не обладает избиратель-
ным поглощением в ультрафиолетовом свете [34], чем доказывается отсут-
ствие в ней С=С двойных связей. При действии сероводорода она вновь
восстанавливается в L-аскорбиновую кислоту [34]. Однако если этот про-
цесс провести не быстро, то в водном растворе происходит гидролиз дегидро-
L-аскорбиновой кислоты с образованием ее нециклической формы — 2,3-
дикетоД-гулоновой кислоты, обладающей кислыми свойствами (см. схему 1).
Эта форма не восстанавливается сероводородом в L-аскорбиновую кислоту,
но если одновременно с реакцией восстановления производить выпаривание
раствора в присутствии йод истоводородной кислоты, то происходит регене-
рация L-аскорбиновой кислоты. Это указывает на то, что протекает реакция
образования лактонного кольца [34, 82] и что кислые свойства L-аскорби-
новой кислоты обусловливаются не карбоксильной группой, а енольными
гидроксильными группами [83].
Наличие лактонного кольца в молекуле L-аскорбиновой кислоты было
доказано путем окислительного расщепления озоном тетраметилового эфи-
ра L-аскорбиновой кислоты (XXVIII) (34, 82], который с разрывом двой-
ной связи переходил в метиловый эфир 2-О-(2'-метоксиоксалил)-3,4-ди-О-
метил-Д-треоновой кислоты (XXXV). При действии аммиака из него полу-
чен оксамид (XXXVII) и амид 3,4-ди-О-метил^-треоновой кислоты (XXXVI)
осн, осн
(^СНз
сн,осн,-с-нс
3 2 I
н
ОСН3 (рсНз
осн3 С-О С=О
сн3осн2-с- нс.
н
XXVIII
NH3
О'
XXXV
осн3 н
I I
сн3осн$-с—c-conh2+ conh2
н он соын2
XXXV»
XXXVII
Следовательно, только неэтерифицированный гидроксил в положении
4 L-аскорбиновой кислоты участвует в образовании у-лактонного кольца.
Эта же реакция указывает и место двойной связи в фурановом кольце
как Д’^-бутенолида.
Среди немногочисленных природных соединений, включающих в свою
молекулу характерное для аскорбиновой кислоты Да-?-ненасыщенное у-лак-
тонное кольцо, находится один из продуктов жизнедеятельности некоторых
видов плесени Penicillium, а именно пеницилловая кислота (XXXVIII)
195]:
соон сн3
с===с
сн/сн^н/ с
XXXIX
29
К таким же соединениям относятся лихостериновая кислота (XXXIX) и
сердечные гликозиды, основной скелет которых представлен следующей
формулой:
но
сн3
R=CH3 или СНО.
Из плесени Р. charlesii выделены различные производные у-лактонного
кольца системы А“^-бутенолидов, например кароловая (XL), карловая
(XLI), карлозовая (XLII) кислоты [96] и др.
он со(снг)гсн,он
он со(сн2)2сн2он
с=с
с=с
сн3нс
,с.
HOOCCHgHC
он <^о(сн2)гсн3
/с=с\
о'
С. НООССНоН
чо
О
АСКОРБИГЕН
В некоторых природных источниках /.-аскорбиновая кислота находится
в виде так называемой связанной формы [97], аскорбигена [98], —веще-
ства, почти не обладающего антискорбутными свойствами [99]. Чистый
аскорбиген выделил из капусты Прохазка [100]. Его [а ]д + 11° [99].
По своей структуре аскорбиген представляет индольное производное L-ас-
корбиновой кислоты [100]. Синтез его осуществили в 1961 г. Гмелин и Вир-
танен [101 ]. Он может быть получен из 3-оксиметилиндола и L-аскорбино-
вой кислоты при pH 4—5 и при 37° С с выходом 70—80% [102] или из
грамина и L-аскорбиновой кислоты [99]. При синтезе аскорбигена из L-ас-
корбиновой кислоты и 3-оксиметил индола образуются две диастереоизо-
мерные формы: A (XLIII) с выходом 55—60% и В (XLIV) с выходом 2%
[103]:
Природной форме отвечает аскорбиген A (XLIII) [104].
СИНТЕЗ L-АСКОРБИНОВОЙ КИСЛОТЫ
L-Аскорбиновую кислоту в виде концентратов получают из различных
растительных объектов: плодов шиповника [105], болгарского перца [92],
зеленого грецкого ореха и др. Однако значительно более эффективны синте-
тические методы получения аскорбиновой кислоты, которые по своей сущ-
ности направлены на создание определенной пространственной структуры
из соответствующих моносахаридов и их производных.
30
I. Синтез L-аскорбиновой кислоты конденсацией бензоинового типа
При конденсации двух альдегидов — этилового эфира глиоксиловой
кислоты (XLV) и L-треозы (XLVI) — под влиянием щелочного раство-
ра цианистого калия происходит бензоиновая (ацилоиновая) конден-
сация, по-видимому, через промежуточное получение двух возможных сме-
шанных бензоинов (XLVII) с образованием L-аскорбиновой кислоты (I)
[1061:
COOCZHS COOCjh 15 СООС2Н5 •н
О“СН о=с НОСН но9 А
+ «1* KCN(KOH) НОСН и о=А Il Y .нос |
о==сн 1 неон неон -ROH нс 1
неон носн ноАн 1 1 носн
носн 1 снгон снгон сн,он
снаон XWII I
XLVI
В качестве исходного вещества можно применить и ацетилированный
циангидрин L-треозы [106]. Однако конденсация бензоинового типа сопро-
вождается образованием бензоинов из одного и того же альдегида (а не из
смешанных), а также альдольной конденсацией альдегидов в щелочной среде
и, кроме того, реакциями изомеризации, которые свойственны моносахари-
дам, что приводит к небольшому выходу L-аскорбиновой кислоты.
Несколько лучшие результаты получаются, если предварительно из
альдозы, например L-ксилозы (XIX), через тетраацетил^-ксилозу (XLVIII),
получить нитрил тетраацетил^-ксилоновой кислоты (XLIX) и подверг-
нуть его конденсации с эфиром глиоксиловой кислоты (XLVa) [107] или его
полуацеталем (XLV6)[108L Реакция идет в присутствии метилата натрия с
отщеплением циангруппы и промежуточным образованием L-треозы (XLVI),
которая и конденсируется под влиянием цианистого натрия с этиловым
эфиром глиоксиловой кислоты (XLVa, R = СгН5):
>о
о
носн
неон
носн
снгон
Ас ОСН
— НСОАс-
АсОСН
СНгОАс
NH2OH
38%
я
с
Асобн
- 11 io Ас
Acoin
нбон
HoiH
ifijOH
COOR
COOR^u СН
<Lho но хос2н5
XLVa XLV 6
Na CM
80-90 У.
HOCH
СЦОАс
I
CH2OH
XLV1 Ac-COCH3
L-Ксилозу получают из D-глюкозы через лактон L-гулоновой кислоты
с последующим расщеплением по Руффу или Веерману [86].
2. Синтез L-аскорбиновой кислоты конденсацией эфиров замещенных
а-оксикислот
Бензиловый эфир бензоилгликолевой кислоты (L) конденсируют с этило-
вым эфиром трибензо ил^-треоновой кислоты (LI) [109, НО] в замещенный
эфир 3-кетогексоновой кислоты (LII), который подвергают омылению и
лактонизации в L-аскорбиновую кислоту (I); выход небольшой.
31
СООС2НЭ
нсооссвн3
г с=о _________
HCOOCC6HS
СвН5СООСН
СНгООСС6Н5
н°с I
НОС ]
но<~н
СЦЮН
LI1
?
COOCH8CeH5
сн2оосс6н5
4- L
соосгн5
НСООСС6Н3
СвН5СООСН
снгоосс6н5
LI
3. Синтез L-аскорбиновой кислоты циангидриновым (озон-цианидным)
методом
Этот синтез осуществляется через пентозон, соответствующий по кон-
: фигурации гидроксильных групп у С(4) и С(5> строению L-аскорбиновой
кислоты. Такой пентозон (XV) может быть получен через озазон (LIV) только
из L-ликсозы (LIII) или L-ксилозы (XIX) (схема 3). Из последней пентозы
были осуществлены первые синтезы аскорбиновой кислоты [85—87, 91, 111 ].
Схема 3
Синтез L-аскорбиновой кислоты циангидриновым методом
сно
неон
неон
НОСН
I X
сн2он '
L-Лимсоза
сно
сно
I
носн
неон
I
НОСН
I
снгон
CH-NNHC.H,
С—NNHC.H, £»HsCHO
неон
I
носн
^HjOH
I
с=о
I
неон
I
носн
I
СН2ОН
CN
НСХ^Н CN
I I
С=О нос
I II
неон-----НОС
I I
носн неон
I I
СН2ОН НОСН
LV снгон
L-Ксипоза
XV
ОН ОН
ОН С=С
носнг-с-н/ X
Н ХУ (
Для получения L-ксилозона (XV) L-ксилозу (XIX) или соответственно
L-ликсозу (LIII) при действии фенилгидразина превращают в озазон,
который разлагают реакцией с бензальдегидом [86, 87] или окислением
перекисью водорода при каталитическом участии солей двухвалентного
железа. По другому варианту L-ксилозу (XIX) подвергают непосредствен-
ному окислению ацетатом меди обычно в среде метилового спирта при
кратковременном кипячении [112, 113]. Выход L-ксилозона 50—55°6. Его
конденсируют с цианистым калием (или натрием) в присутствии хлористого
кальция в водном растворе в нитрил (LV), таутомерно превращающийся
в циклическую форму имино-Е-аскорбиновой кислоты (LVI).
Имино-Е-аскорбиновая кислота (LVI), содержащая реакционноспособ-
ную ендиольную систему, может быть выделена в кристаллическом виде,
но обычно производят непосредственное гидролитическое элиминирование
иминогруппы в разбавленной минеральной кислоте с образованием L-ac-
корбиновой кислоты (I). Синтез идет с невысокими выходами. Таким же
путем получена меченая Е(-г)-аскорбиновая-1-14О кислота [113].
L-Ликсоза (LIII) может быть получена из D-галактозы через D-галак-
туроновую кислоту и L-галактоновую кислоту, соль которой окисляют
перекисью водорода по методу укорачивания углеродной цепи по Руффу
[87]. Другим синтетическим путем L-ликсоза может быть получена из
D-глюкозы через L-сорбозу [20, 114—118].
32
4. Синтез. L-аскорбиновой кислоты изомеризацией и лактонизацией 2- или
3-кетогексоновых кислот
В этом синтезе исходят из шестиатомных моносахаридов L-ряда, которые
по конфигурации гидроксилов у С(4) и С(5) отвечают строению L-аскорбино-
вой кислоты. Можно применить 3-кетогексоновые кислоты, однако для их
синтеза нет приемлемого метода. Практическое значение имеют только мето-
ды синтеза через 2-кето-£-галактоновую или 2-кето-Ь-гулоновую кислоты,
которые рассматриваются ниже.
Получение L-аскорбиновой кислоты через 2-кето^-галактоновую кисло-
ту. В этом методе синтеза используют L-галактоновую кислоту (LVIII),
для получения которой синтетические методы не имеют практического зна-
чения из-за своей сложности. Однако метод ее получения из D-галактуроно-
вой кислоты (LVII), выделяемой ферментативным гидролизом из при-
родных пектинсодержащих веществ, уже представляет некоторый практи-
ческий интерес (схема 4). Так, натрий-кальциевая соль D-галактуроновой
кислоты получается с выходом 18% от сухого свекловичного жома, а ее
каталитическое гидрирование дает с выходом 93% L-галактоновую кислоту
(LVIII).
Схема 4
Синтез L-аскорбиновой кислоты из D-галактуроновой кислоты
СООН
I
НОСН
неон [Н]
неон 93z
।
носн
I
сно
соон СО—|
носн носн I
। । 9
неон ____неон
неон -Н«О нс—'
1 «* I
носн носн
I . I
снгон сн,он
соон соосн3
с=о с=о
I I
NaOCl неон сн,он неон
- .. I ....... — I ....... L-Аскорб,
** неон 55Х неон 707-
I I I
носн носн
I I
снгон СН2ОН
evil LVIII Lix
IXI
Дальнейший синтез состоит в удалении иона кальция и образовании
у-лактона L-галактоновой кислоты (LIX) почти с количественным выходом,
его окислении гипохлоритом натрия при каталитическом участии пятиоки-
си ванадия в среде безводного метилового спирта с добавлением фосфорного
ангидрида в 2-кето^-галактоновую кислоту (LX) с выходом 24%. Затем
получают метиловый эфир 2-кето^-галактоновой кислоты (LXI), нейтра-
лизуют кислый раствор углекислым серебром и после выпаривания фильтра-
та выделяют эфир (LX Невыходом около 55 % [1191. Лактонизацию и еноли-
зацию метилового эфира 2-кето^-галактоновой кислоты (LXI) в L-аскорби-
повую кислоту (I) производят в присутствии метилата натрия в безводном
спирте [119, 120] или при нагревании в спиртово-хлороформенной смеси
в присутствии хлористого водорода.
Из 100 кг сухого свекловичного жома получается 2,3 кг L-аскорбиновой
кислоты (около 10% теоретического выхода) [121].
Получение L-аскорбиновой кислоты через 2 кето-£-гулоновую кислоту.
Исходные моносахариды, применяемые в этом синтезе, — D-глюкоза,
L-сорбоза и L-гулоза — могут быть получены из доступных исходных
продуктов. Хотя 2-кето^-гулоновая кислота или изомерная ей по поло-
жению 3 2-кето^-галактоновая кислота (которая также может служить
полупродуктом для синтеза L-аскорбиновой кислоты) могла бы быть полу-
чена осторожным окислением L-идозы, L-тагатозы, L-талозы или L-галак-
тозы, однако этот путь синтеза может представить только теоретический
интерес из-за малой доступности исходных моносахаридов.
2—69
33
В одном из более ранних синтезов L-аскорбиновой кислоты (I) L-cop-
бозу (LXII) или L-гулозу (LXIII) при действии фенилгидразина превра-
щают в гулозазон (LXIV), который путем обработки бензальдегидом [1221
дает гулозон (LXV) (схема 5). Последнее соединение осторожным окислени-
ем водным раствором брома превращают в 2-кето-Ь-гулоновую кислоту
(XVIII) и затем енолизацией и лактонизацией — в L-аскорбиновую кисло-
ту (I)-
L-Гулозу (LXIII) получают довольно сложным синтезом из D-глюкозы
через лактон L-гулоновой кислоты [122].
Схема 5
Синтез L-аскорбииовой кислоты из L-сорбозы или L-гулозы через гулозон
(jHj.OH
г°
носн
н^он
носн
СН2ОН
1X11
сно ch=nnhc6h5
I I
НОСН С = NNHC6HS
носн c8hsnhnh2 носн с«н5сно
неон неон
I I
носн носн
I I
снгон снгон
LXIII LXIV
сно
j“° Вгг.НгО
НОСН --------
неон
I
НОСН
I
СН2ОН
I
носн
I
СН2ОН
СООН
Г_
С С Лаккжмэация,
1 «нопимции
НОСН ------------
I
неон
L-Сорбоза получается ферментативным окислением D-сорбита, встре-
чающегося в значительном количестве в ягодах рябины. Промышленным
источником D-сорбита служит D-глюкоза, переходящая в него при восста-
новлении. Эти методы синтеза описываются ниже.
Как видно из рассмотрения путей синтеза L-аскорбиновой кислоты,
основным исходным сырьем для нее является D-глюкоза, в чистом виде
получаемая гидролизом кукурузного крахмала. Наиболее простым и
экономичным техническим синтезом L-аскорбиновой кислоты (I) из D-глю-
козы (LXVI) является синтез через L-сорбозу (LXII) и 2,3;4,6-ди-О-изо-
пропилиден-2-кето^-гулоновую кислоту (LXIX) или 2-кето-Г-гулоновую
кислоту (XVIII).
Описываемый ниже метод синтеза L-аскорбиновой кислоты впервые
предложили Рейхштейн и Грюсснер в 1934 г. [201; его дополнил Эльгер
[123] и воспроизвели другие авторы [62, 124—127]. Этот синтез (схема 6)
проводится через следующие промежуточные соединения: D-глюкоза уз%
D-сорбит 88% L-сорбоза §2% 2,3; 4,6-ди-О-изопропилиден^-сорбоза &s% 2,3;
4,6-ди-О-изопропилиден-2-кето^-гулоновая кислота 88% L-аскорбино-
вая кислота; выход при перекристаллизации составляет около 94%. Общий
выход L-аскорбиновой кислоты свыше 55% от теоретического.
D-Сорбит (LXVII) получают восстановлением комплексными гид-
ридами металлов [128]. электролитическим восстановлением [129-131] или
каталитическим гидрированием [132] D-глюкозы (LXVI).
D-Глюкозу можно восстановить в D-сорбит боргидридом натрия, причем
восстановлению подвергается ациклическая форма, образующаяся из по-
луацетальной формы D-глюкозы при каталитическом участии гидроксил-
34
иона [133]. Из-за недостаточно высоких выходов этот метод получения
D-сорбита не имеет технического значения.
Для электролитического восстановления D-глюкозы применяют ванны
с диафрагмой; анод — свинцовый, катод — амальгамированный свинец
или сплав никеля с алюминием [131, 134]; выход 98—99%. При бездиафраг-
менном восстановлении выход составляет только 90% [135]. В качестве
католита применяют 30—35%-ный раствор D-глюкозы, содержащей суль-
фат аммония [136] или сульфат натрия, прибавляемые для увеличения
электропроводности. Электролиз проводят при 25—30° С и pH 10 [137].
Схема 6
Синтез /.-аскорбиновой кислоты через /.-сорбозу и 2,3:4,6-ди-0-изопропилидеи-2-кето-
L-гулоновую кислоту
СН2ОН
о н
н
но
н он
J) -Глюкоза
LXVI
н
ОН н
он
в СН2ОН
г, 5носн
2[н]
.. ’ 4
з
носн
неон
н о CttOH
(СН,УСО
ОН
[О]
н
он
н
н
НО
CHg-O
1.2-0- изопрол и л идея -
сорбопи раноза
lx VIII а
н
н
н
ОН н
.сн3
о—
носн
СН2ОН
D - Сорби г
LXVII
г
он н
I- Сорбоза
LXH
сн3 сн3
(сн3)гсо н
H9SO4
НгС
о
н
[О]
н2с
—I *
О н СН2ОН о
О
сн, сн3
Н
о н
о
н
~(СН3)2СО
соон
н2о
н
но оч он
н
о
н
снг—о
н
он н
1Л-0-ИЗОПРОПИ-
лидеи -<4- L- сорбо-
пираиоза
LXVIll <j
СН73 СН3
2Д4.6-ж*^Изопроп илиден-
-ос -£-сорбо<рураноза
1Х(Х
-(СНз)2СО||(СнДСО
сн3 сн3
\/
о
сн3 сн3
2 ЗЛ.6-ди-ОИзопропилмден-
2-кето-/.- гулоиовая
кислота (гидрат)
LXXI
сн3
•СН3
НОН/1
о
2 З-О-Изопропилиден-
-ot-£- сорбофураиоэа
LXX
о
н
сн2он
он н
ROH
-(СН3)гСО
ОН н
r-c2h5
он он
hoch2-c-c-c-co-coor
4 I I I
н I н
он
Эсрнр 2-нето~Д-гу-
ломовой кислоты
LXXII
Hf
-ROH
Электролитически получаемый D-сорбит содержит около 15% D-манни-
та, который образуется из продуктов частичной эпимеризации D-глюкозы
в щелочной среде. Поэтому применение такого сорбита для получения из
него L-сорбозы связано со значительными трудностями.
Каталитическое гидрирование углеводов впервые было осуществлено
в 1912 г. [132]. Гидрирование D-глюкозы в D-сорбит осуществляют со
скелетным никелевым, медно-хромовым или медно-никелево-марганцевым
катализатором [132,138—141 ]. Применяют 50%-ный водный раствор глюко-
2*
35
зы и гидрируют в автоклавах при 120° С и давлении свыше 10 мПа (100 ат)
в течение 1,5—2 ч. Можно применять и более концентрированные растворы
глюкозы — 70%-ный [139]. Во избежание слишком большого закисления
раствора при реакции в смесь прибавляют известковую воду и мел до
pH 8—8,4 [139], однако излишняя щелочность раствора приводит к увели-
чению побочных реакций. Для гидрирования применяют вертикальные
и горизонтальные автоклавы или автоклавы непрерывного действия [142],
работающие на сплавном алюминиево-никелевом катализаторе или на гра-
нулированном и порошкообразном скелетном никелевом катализаторе
[143].
Представляет интерес использование крахмала для получения D-сорбита.
Суспензию 1 части крахмала в 2*/3 частях воды с 0,7% хлористого магния
гидрируют с никелевым катализатором на диатомовой земле при давлении
10 мПа (100 ат) и температуре 200° С в течение 75 мин [144 ].
Раствор D-сорбита после гидрирования очищают от тяжелых металлов,
главным образом от никеля; он содержится в количестве 40—50 мг/л и яв-
ляется ядом для микроорганизмов, применяемых на следующей стадии
синтеза. Очистка основана на образовании нерастворимых углекислых
или фосфорнокислых солей тяжелых металлов при обменной реакции с
двузамещенным фосфорнокислым натрием при нагревании до 70—80°С
в присутствии углекислого кальция [145]. Возможно применение электро-
осаждения никеля [146] или ионнообменных смол.
При гидрировании D-глюкозы (LXVI), помимо D-сорбита (LXVII),
образуется небольшое количество D-маннита (LXXVI) и D-глюконовой
кислоты (LXXIII). Органическая кислота получается из £)-глюкозы
(LXVI) по реакции Канниццаро, которая протекает в присутствии скелет-
ного никелевого катализатора [147] с образованием D-сорбита (LXVII)
и D-глюконовой кислоты (LXXIII).
Схема 7
Реакции, протекающие при каталитическом гидрировании D-глюкозы
CH.OH
носн
I
носн
I
НСОН
I
НСОН
I
СН2ОН
LXXVI
|г[н]
сно
I
НОСН
I
носн____
НСОН
I
НСОН
I
СНгОН
сно
I
НСОН
I
НОСН
I
НСОН
I
НСОН
I
снгон
LXVI
II
CHOH
II
сон
I
носн
I •
неон.
НСОН
снгон
соон
н^он
но^н
неон
I
НСОН
снгон
LXXIII
снгон
НСОН
I
носн
I
НСОН
I
НСОН
I
снгон
LXXV
LXVII
Образование D-маннита (LXXVI) является следствием реакции еноли-
зации и изомеризации D-глюкозы (LXVI) в щелочном растворе в D-фрукто-
зу (LXXIV) и в D-маннозу (LXXV) [148—150], которая затем подвергается
восстановлению; D-фруктоза восстанавливается в D-сорбит и в D-маннит
(схема 7). Средн побочных продуктов найдены также 2-дезокси-О-сорбит,
1-дезокси-О-маннит, аллит и др. [151 1. Для подавления реакций изомерн-
36
зации ретроальдольного расщепления и процессов карамелизации реко-
мендуется быстрое проведение процесса, для чего применяют высокоак-
тивный катализатор гидрирования и повышают температуру.
L-С о р б о з a (LXII) не обладает мутаротацией и в кристаллическом
виде представляет собой P-форму пираноидной (S-окисной) структуры с
т. пл. 159—161° С и [ц]р —43,1° (Н2О). Она имеет такую же конфигура-
цию гидроксильных групп у С(4) и С(5), как и L-аскорбиновая кислота;
Химический путь синтеза L-сорбозы (LXII) [152] (схема 8) основан на
окислении гидроксильной группы положения 5 в производных
D-сорбита, у которых остальные гидроксильные группы защи-
щены. При действии паральдегида на D-сорбит (LXVII) получают 1,3;
2,4;5,6-три-О-этилиден7)-сорбит (LXXVII), частичный гидролиз которого
приводит к 1,3;2,4-ди-О-этилиден-О-сорбиту (LXXVIII). Свободная первич-
ная гидроксильная группа этого соединения защищается бензоилировани-
ем, и 6-бензоил-1,3; 2,4-ди-О-этилиден-П-сорбит (LXXIX) окисляется хро-
мовым ангидридом в уксусной кислоте в 1-бензоил-3,5;4,6-ди-О-этилиден
L-сорбозу (LXXX), из которой гидролизом в соединение LXXXI с после-
дующим омылением получается L-сорбоза (LXII) с общим выходом 0,75%.
Схема 8
Химический синтез £-сорбозы из D-сорбита
СН2ОН 1
I
НСОН 2
I
НОСН 3
I
НСОН 4
I
НСОН 5
I
СН2ОН 6
^ОСН2
CH..CH НСО.
\ I \
ХОСН /СНСНз
/ОСН,
СН3СН НСО
сносно
167.
нс о
HgO
337.
НСО^нгн
сн2ох 3
LXVII LX XVII
/ОСН, ,осн2 1 * 6
CHjCH НСО\ СН3СН НСО\ 1 \ 5
^ОСН СНСН, 1 / "ОСН ^снсн3
НСО сго3 нсо-^ з нго
НСОН с=о о (H2SO4) 2 857.
CH2OR ch2or 1
LXXIX LXXX
R=COC6HS
ОСН СНСН
НСОН
сн2он
LXXVIII
CHjOR i
НОС
НОСН
О I
НСОН
I
НОСН
I
--СНг
LXXXI
c^coa
S67.
СН2ОН
—
НОС
I
г носн
о I
з НСОН
4 НдО НОСН
5 ^.г) СН2
в I»»
С несколько большим выходом (около 3%) L-сорбозу ^ХП)'можно полу-
чить из 5-кето-Л-глюконовой кислоты (LXXXII) после ее метилирования
метиловым спиртом в присутствии хлористого водорода, восстановления
образующегося метил гликозида метилового эфира 5-кето-О-глюконовой кис-
лоты боргидридом натрия в метил-L-сорбофуранозу и ее последующего
гидролиза с н. серной кислотой при 100° С [1531:
соон соосн3
неон нс----1
НОСН носн I
I —— । о
НСОН НСОН [
С=О CHgOC——J
сн2он СН2ОН
LXXXII
СН2ОН
НС----]
НОСН I
‘ . I О /Сорбоза
НСОН I щи
СН3ОС-----’
СНгОН
R = ОССвНь
37
Однако техническое значение имеет стереохимически направленный
биохимический синтез L-сорбозы, экономически более выгодный.
Ферментативное окисление D-сорбита осуществляют по Бертрану [154]
с применением уксуснокислых бактерий [155—157] Acetobacter xylinum
[154, 156], Acetobacter melanoqenum или Acetobacter suboxydans. Эти
бактерии вызывают окисление вторичной гидроксильной группы в различ-
ных полиоксикарбоновых соединениях, преимущественно атомов углерода,
соседних с первичным гидроксилом; в случае возможности окисления одной
из двух вторичных гидроксильных групп, соседних с первичными, исклю-
чительный выбор падает на гидроксил, находящийся в D-эритро-положе-
нии к гидроксильной группе углеродных атомов положения 3 или 4.
Образование 7.-сорбозы при биохимическом окислении уксуснокислыми
микроорганизмами происходит в результате дегидрирования гидроксила
при С(5), находящегося в эритро-положении к соседней гидроксильной
группе положения 4 молекулы D-сорбита. Следует отметить, что при окис-
лении D-сорбита поглощается теоретическое количество кислорода.
Для успешного развития уксуснокислых бактерий на растворе D-сорбита
среда должна содержать витамины комплекса В. В качестве таких добавок
применяется водный экстракт хлебопекарных дрожжей, дрожжевой авто-
лизат, кукурузный экстракт или препарат витаминов группы В, получае-
мый из дрожжей. Для окисления с помощью Acetobacter melanogenum
в кюветах применяют 12—15%-ный раствор D-сорбита; образуется 80—93%
L-сорбозы [158], pH раствора 5,5—6,5; среда не должна иметь значитель-
ных количеств тяжелых металлов, так как, например, содержание никеля
5 мг/л или железа 150 мг/л уже сильно угнетает Acetobacter melanogenum.
Процесс длится 3—4 суток.
В технике L-сорбозу получают более эффективным глубинным способом
путем окисления D-сорбита в ферментаторах. В 15—25%-ные растворы
D-сорбита, содержащие необходимые для микроорганизмов ростовые фак-
торы, пропускают воздух через распылительные системы — 177—550 мл
в минуту на 1 л раствора. При температуре 30° С через 24 ч с применением
Acetobacter suboxydans L-сорбоза получается с выходом 93% в растворе
[159]. Повышение давления с 0,1 до 0,2 МПа (с 1 до 2 ат) ускоряет процесс
до 12—16 ч [157, 159]. Выяснены отдельные детали процесса, связанные с
ферментативным окислением сорбита в сорбозу [160—162 ]. В глубинном спо-
собе окисления D-сорбита применяют также культуру Acetobacter mela-
nogenum, однако реакция протекает более продолжительно.
Для ферментативного получения D-сорбита предложен непрерывный
процесс окисления растворов L-сорбозы в тонких пленках, т. е. в системе
жидкость — газ (в пене) [163]. Для окисления применяют обычно очи-
щенные от тяжелых металлов растворы D-сорбита, которые содержат не-
большие примеси D-маннита, D-глюконовой кислоты и D-глюкозы. Уксус-
нокислые бактерии способны окислять D-глюкозу (LXVI) в 2Э-глюконовую
кислоту (LXXIII); последняя также окисляется, например Acetobacter
suboxydans — в5-кето^-глюконовую кислоту (LXXXII, см. с 37). D-Маннит
(LXXVI) окисляется уксуснокислыми бактериями в D-фруктозу (LXXIV):
СН2ОН СН2ОН
I I
носн с=о
I I
НОСН Ферментация НОСН
I-------------------> I
неон неон
неон неон
I I
сн2он сн2он
LXXVI LXX1V
38
Таким образом, наряду с основной реакцией окисления D-сорбита в
L-сорбозу происходят побочные окислительные реакции с образованием
органических кислот и продуктов, обладающих восстановительными свой-
ствами и аналитически определяемых (реактивом Фелинга) как восстанав-
ливающие моносахариды.
При ферментативном окислении происходит в небольшом количестве
выделение двуокиси углерода, по-видимому, за счет полного окисления
части моносахаридов до двуокиси углерода и воды в результате биохими-
ческих реакций неустановленного характера, протекающих в живой клетке.
Тщательно изучена побочная микрофлора, встречающаяся в производствен-
ных условиях окисления D-сорбита в L-сорбозу [164].
2,3;4,6-Ди-О-и зоп роп и л идеи - L - с орбоза, 2,3;4,6-ди-О-изо-
пропилиден-а-Ь-сорбофураноза (LXIX), является третьим промежуточным
продуктом описываемого метода синтеза L-аскорбиновой кислоты (схема 6).
Кеталирование применяется для защиты в виде циклических кеталей гидрок-
сильных групп, которые желательно сохранить неизменными при дальнейших
превращениях L-сорбозы (LXII), и производится с целью последующего осу-
ществления реакции окисления остающейся свободной первичной гидро-
ксильной группы с превращением молекулы в 2-кетоД-гулоновую кислоту.
Реакция кеталирования сопровождается изомеризацией аномерной и
циклической конфигураций L-сорбозы. P-L-Сорбопираноза (LXII), по-ви-
димому, изомеризуется и кеталируется первоначально в промежуточную
ациклическую кето-форму — 1.3-О-изопропилиден-L-сорбозу,
находящуюся в равновесии с 1,2- и 1,3-О-изопропилиден-сеД-сорбопирано-
зами (LXVIIIa и LXVIII6); это первичная стадия кеталирования L-сорбо-
зы в ацетоне в присутствии серной кислоты [165]. Затем протекает бимо-
лекулярная реакция дальнейшего кеталирования 1,2- и 1,3-О-изопропилен-
a-L-сорбопиранозы в 2,3;4,6-ди-О-изопропилиден-аД-сорбофуранозу(...
(LXIX), сопровождающаяся перегруппировкой пиранозной структуры в
фуранозную (схема 6); реакция, по-видимому, идет через промежуточную
ациклическую кето-форму— 1,3;4,6-ди-О-изопропилиденД-сорбозу. Эта .
реакция необратима, она определяет скорость процесса. Промежуточная
ациклическая форма находится в равновесном состоянии с 1,3;4,6- и 1,2;
4,6-ди-О-изопропилиден-|3^-сорбофуранозами и 1,2;4,6-ди-О-изопропили-
ден-аД-сорбофуранозой [165]; эти соединения были выделены из смеси
продуктов реакции при непродолжительном ацетонировании L-сорбозы
в большом избытке ацетона в присутствии серной кислоты. Продукты кета-
лирования с пятичленным фуранозным циклом более стабильны. По окон-
чании процесса ацетонирования 1,2- и 1,3-О-изопропилиден-оД-сорбопи-
раноз (LXVIIIa и LXVIII6) остается малозаметное количество.
В позднейшей стадии процесса 2,3;4,6-ди-0-изопропилиден-аД-сорбофу-
раноза (LXIX) со значительной скоростью обратимо превращается в 2,3-0-
изопропилиден-аД-сорбофуранозу (LXX) [166—169] с т. пл. 92—93° С,
1а]д 4- 7° (Н20) [20] с одновременным отщеплением более лабильной 4,6-
О-изопропилиденовой группы, причем между этими соединениями устанав-
ливается подвижное равновесие. Такое течение реакции подтверждается
данными газожидкостной хроматографии.
Кеталирование циклических изомеров L-сорбозы первоначально затра-
гивает енольную форму кетогруппы положения 2; это следует из того, что
моно-О-изопропилиденД-сорбозы уже не обладают восстановительными
свойствами (с реактивом Фелинга). 2,3;4,6-Ди-О-изопропилиден-аД-сор-
39
бофураноза (LXIX) является главным продуктом реакции ацетонирования
£-сорбозы и образуется с выходом 78—80%. Если после ее отделения из
маточных растворов выделить 2,3-О-изопропилиден-Л-сорбозу (LXX) и
подвергнуть это вещество повторному ацетонированию, то можно поднять
выход до 85% 120, 170].
Сообщается, что при ацетонировании в присутствии 2,2-диэтоксипропа-
на и n-толуолсульфокислоты, помимо изопропилиденпроизводных LXIX
и LXX, образуется 1,2;4,6-ди-О-изопропилиден-а-А-сорбоза и немного
1,2-О-изопропилиден-а-А-сорбофуранозы [171 ].
Для определения 2,3-О-изопропилиден-а-£-сорбофуранозы (LXX) и
2,3;4,6-ди-О-изопропилиден-а-£-сорбофуранозы (LXIX) в смесях исполь-
зуют хроматографию на бумаге; вещество количественно определяют фото-
метрически по цветной реакции с дифениламином в кислом растворе и с са-
лициловым альдегидом соответственно [172].
Ацетонирование L-сорбозы проводится при взаимодействии суспензии
сахара с ацетоном при каталитическом участии различных водоотнимаю-
щих средств, в качестве которых применяются минеральные кислоты, их
ангидриды или соли. Наиболее широкое применение в качестве водоотни-
мающего средства имеет концентрированная серная кислота [20, 173], ее
моногидрат или олеум. Применяется также безводный хлористый цинк,
фосфорный ангидрид, эти же реагенты в смеси с ортофосфорной кислотой
[174]. Ацетонирование с этими катализаторами связано с рядом техниче-
ских трудностей из-за сильной гигроскопичности применяемых веществ
и в то же время не имеет преимуществ в выходах перед другими способами.
Применение хлористого водорода или безводной сернокислой меди
[20, 175] связано с малой скоростью реакции. Ацетонирование в присут-
ствии сернокислой меди и малых количеств серной кислоты [176]
протекает в течение 96—120 ч и менее эффективно, чем ацетонирование под
влиянием 3—5 об. % концентрированной серной кислоты (по отношению к
ацетону), которое при 22—26° С продолжается всего 1,5—2 ч. В этих же усло-
виях ацетонирования с последующим кратковременным охлаждением перед
нейтрализацией ниже 0° С образуется смесь из 30% 2,3-О-изопропилиден-
a-L-сорбозы и 70% 2,3;4,6-ди-О-изопропилиден-а-£-сорбозы [177].
Применение разбавленной серной кислоты сильно снижает выход;
так, ацетонирование с 7096-ной серной кислотой при 30° С (нейтрализация
при той же температуре) дает 2,3; 4,6-ди-О-изопропилиден-А-сорбозу только
с 16%-ным выходом [177]. Целесообразно для ацетонирования L-сорбозы
применять 12—4 4-кратное (по объему) количество ацетона по отношению
к L-сорбозе. Снижение количества ацетона до 10-кратного ведет к некоторо-
му уменьшению выхода 2,3; 4,6-ди-О-изопропилиден-а-£-сорбозы [126].
Так как при реакции ацетонирования отщепляется вода, то для полу-
чения оптимального выхода 2,3;4,6-ди-О-изопропилиден-а-£-сорбозы кон-
центрация серной кислоты к концу ацетонирования не должна быть ниже
81—82%. При увеличении количества концентрированной серной кислоты
в растворе ацетона свыше 4—596 об. возрастает уплотнение ацетона в
окись мезитила и форон.
Имеется сообщение, что если увеличить количество серной кислоты до
10% об. и ацетонирование вести в присутствиии ацетондиметилацеталя при
10° С 1 ч, то выход 2,3;4,6-ди-О-изопропилиден-а-£-сорбозы достигает
92% [178].
Так как использование многократно регенерированного ацетона ведет
к ослаблению реагирования [173, 179], то рекомендуется прибавление
0,5—1 % ацетальдегида [179]. Однако происходящее при этом некоторое
ускорение реакции сопровождается потемнением раствора и затруднением
выделения продуктов реакции [173].
При ацетонировании L-сорбозы под влиянием серной кислоты применим
широкий интервал температур: от —5 до +50° С [180]. Повышение тем-
пературы увеличивает скорость реакции, но одновременно происходит
40
сильное потемнение раствора и рост количества продуктов уплотнения ацето-
на, а следовательно, и воды, сдвигающей равновесие в сторону образования
2,3-О-изопропилиден-а-L-сорбозы. Между 2,3-О-изопропилиден-а-А-сорбо-
зой и 2,3;4,6-ди-О-изопропилиден-а-Л-сорбозой в кислом ацетоновом раст-
воре существует равновесие, которое сдвигается в сторону 2,3;4,6-ди-О-
изопропилиден-a-L-сорбозы при понижении температуры [180]. Оптималь-
ной температуру ацетонирования можно считать от + 2 до — 5° С (при
продолжительности реакции до 24 ч).
Попытка сдвинуть равновесие в сторону 2,3;4,6-ди-О-изопропилиден-а-
L-сорбозы путем проведения ацетонирования в среде, содержащей, помимо
ацетона, такие растворители, которые преимущественно растворяют 2,3;4,6-
ди-О-изопропилиден-а-Д-сорбозу и не растворяют 2,3-О-изопропилиден-а-£-
сорбозу, например бензол и хлороформ, не дала существенных резуль-
татов [181].
Следует отметить, что применение ультразвука при реакции ацетони-
рования сорбозы уменьшает продолжительность процесса [182].
После окончания ацетонирования кислый ацетоновый раствор смеси
моно- и ди-О-изопропилиден-Ь-сорбозы нейтрализуют углекислыми [20],
двууглекислыми [183] солями и основаниями щелочных металлов [126,
184] при температуре ниже 0° С [180, 184]. Нейтрализация значительно
ускоряется, если применить 10—50%-ные растворы едкого натра [126].
Осуществлено ацетонирование L-сорбозы ацетоном на ионообменных смо-
лах— сульфокатионитах типа амберлит-15 (сульфоспирт — бензальдегид,
сополимер макротипа, содержащий 6—55% дивинилбензола) [185, 186];
для проведения реакции перемешивают суспензию L-сорбозы с ацетоном
(1:2) с ионообменными смолами в течение нескольких часов или процесс
ацетонирования ведут в колоннах непрерывного действия [186].
Из продукта ацетонирования после удаления растворителя с водяным
паром-отгоняют окись мезитила и из остатка выделяют 2,3;4,6-дшО-изопро-
пилиден-а-/,-сорбозу высаливанием раствором едкого натра или экстрак-
цией органическими растворителями. В качестве растворителей для извле-
чения 2,3;4,6-ди-О-изопропилиден-а^-сорбозы известны бензин, петролей-
ный эфир, дихлорэтан, трихлорэтилен, хлороформ, бензол и другие,
практически не растворяющие изомерные моно-О-изопропилиден-L-сорбозы.
Для защиты гидроксильных групп L-сорбозы, помимо ацетона, приме-
няют многочисленные альдегиды и кетоны; к ним относятся: метилэтй л ке-
тон. бензальдегид, паральдегид, циклогексанон и др.
2,3;4,6-Ди-О-и зопропилиден -2-к е т o-L-r улоновая кис-
лота, 2,3;4,6-ди-0-изопропилиден-ксюго-гексулозоновая кислота (LXXI,
схема 6), получается окислением первичной гидроксильной группы 2,3;
4,6-ди-0-изопропилиден-а^-сорбозы (LXIX). Окислителями могут служить
марганцовокислый калий в слабощелочной среде [20], гипохлорит натрия
[187] или кислород [188] в присутствии катализаторов.
Целесообразно вносить в реакционную смесь марганцовокислый калий не
в растворе, а в твердом виде [126]. Температуру реакции поддерживают в
пределах 36—38° С.
Применение электролитического окисления 2,3;4,6-ди-О-изопропили-
fleH-a-L-cop6o3bi позволяет провести реакцию с каталитическими количест-
вами марганцовокислого калия, но с более низким выходом [189]. Электро-
лиз проводят на железном аноде в щелочном растворе при температуре
10—20° С; марганцовокислого калия расходуется около 25% стехиометри-
ческого количества.
Непосредственное электролитическое окисление 2,3;4,6-ди-О-изопропи-
лиден-а^-сорбозы в 2,3;4,6-ди-О-изопропилиден-2-кето^-гулоновую кисло-
ту осуществляют в 4%-ном растворе едкого натра с Ni(NO3)2 при 55° С;
конверсия составляет 89,4°6, выход 91,6% [190].
Каталитическое окисление гипохлоритом натрия требует более тщатель-
ного регулирования реакции; в качестве катализаторов реакции применя-
41
ют окисли никеля (например, получаемые из сернокислого или хлористого
никеля) или ванадия; окисление проводится при 50—60° С [187, 191, 192].
Эта реакция первого порядка [193]. Для окисления можно применить более
широкий температурный интервал (60—90° С), снизить количество катали-
затора до 2% и вести реакцию без избытка окислителя растворами гипохло-
рита с содержанием 180—210 г/л активного хлора [194]. Предложен
непрерывный способ проведения реакции окисления [195, 196]. Для окис-
ления с успехом можно применять хлор в щелочном растворе в присутствии
сернокислого никеля [197].
Каталитическое окисление 2,3;4,6-ди-О-изопропилиден-а-Л-сорбозы кис-
лородом воздуха проводится в водном растворе в среде бикарбоната натрия
при pH 8,5 в присутствии 10% Pt/C при 50—60° С (продолжительность
20 ч); выход 2,3;4,6-ди-О-изопропилиден-2-кето-А-гулоновой кислоты дости-
гает 98% [188].
Из щелочных растворов 2,3;4,6-ди-О-изопропилиден-2-кето-£-гулоно-
вую кислоту выделяют в кристаллическом виде с одной молекулой воды
путем нейтрализации соляной или серной кислотой при температуре около
0е С. Она почти не растворима в воде и после промывки холодной водой от
минеральных солей получается в чистом виде. Сопутствующая технической
2,3;4,6-О-изопропилиден-£-сорбозе как примесь моно-О-изопропилиден-L-
сорбоза подвергается деструктивному окислению; продукты ее расщепления
хорошо растворимы в щелочных и нейтральных растворах.
2,3;4,6-Ди-О-изопропилиден-2-кето-А-гулоновая кислота легко гидроли-
зуется при нагревании ее водных растворов в 2-кето-Л-гулоновую кислоту
(XVIII) с выходом свыше 90%, выделяемую в кристаллическом виде после
концентрации раствора; при этом происходит частичная енолизация и лакто-
низация с образованием небольших количеств L-аскорбиновой кис-
лоты.
Для получения 2-кето-£-гулоновой кислоты (XVIII) можно исходить из
L-гулоновой кислоты, которая подвергается избирательному окислению в
положении 2 хромовой кислотой или хроматами в присутствии пятиокиси
ванадия как катализатора [198], а также гипохлоритами с хлоридом никеля
[199]. Исходя из D-сорбита (LXVII), можно получить 1,3;2,4-ди-О-этили-
ден-П-сорбит (LXXVIII, с. 37), который окисляют кислородом при 50° С
в присутствии 15% Pt/C в 3,5;4,6-ди-О-этилиден-£-гулоновую кислоту
(выход 50—70%), а уже последнюю повторно окисляют гипохлоритом натрия
в присутствии никелевого катализатора в щелочной среде в 2-кето-Ь-гуло-
новую кислоту (выход 33%) [199].
Схема 9
Непосредственное окисление L-сорбозы в 2-кето-£-гулоновую кислоту
СН2ОН
---сон
I
носн ______
о неон •
носн
I
----сн2
сно
—<^он
носн
I
о неон
|но(^н
—СН2
соон
соон
1X11
—^он
носн
о неон
| носн
'—снг
с=о
носн
неон
носн
сн2 он
XVIII
Z-Аскорбиновая
кислота
соон
I
с=о
носн -
неон
нос^н
соон
ixxxni
он
ноос-с—НС
LXXXIV
н
42
г
Л
Интересным вариантом синтеза L-аскорбиновой кислоты является не-
посредственное окисление L-сорбозы (LXII) в тщательно регулируемых
условиях в 2-кето-Л-гулоновую кислоту (XVIII, схема 9); эта реакция
основана на большей реакционной способности а-водородных атомов поло-
; жения 1 вследствие активирования карбонильной группой. Окисление
сорбозы проводят разбавленной азотной кислотой при нагревании [200,2011
! или с лучшим выходом концентрированной азотной кислотой при низкой
температуре [202 ], а также окислами азота с выходом 58% [203 ]. Применяют
кислород воздуха при каталитическом участии платинового или палладие-
вого катализатора в нейтральной среде или в среде двууглекислых солей
щелочных металлов (выход 50%) [204—206]. Реакция окисления сорбозы,
по-видимому, протекает через промежуточное альдегидное производное —
L-гулозон (LXV).
Для непосредственного окисления L-сорбозы (LXII) в 2-кето-Т-гулоно-
вую кислоту (XVIII) предложены гипохлориты и хлораты [207], электро-
литический метод окисления [208], а также окисление диацетатом меди в
L-гулозон (LXV) с последующим окислением бромной водой в 2-KeTo-L-ry-
лоновую кислоту (XVIII) [209].
Реакция каталитического окисления L-сорбозы может сопровождаться
побочным образованием 2-кето-Е-гулосахарной кислоты (LXXXIII), которая
способна превращаться в 6-карбокси-Т-аскорбиновую кислоту (LXXXIV)
[204], с характерной ендиольной группировкой.
2-Кето-Е-гулоновую кислоту (XVIII) можно получить непосредственным
ферментативным окислением L-сорбозы (LXII) Pseudomonas viridifrava
[210].
L-A скорби новая кислота (I) получается в результате еноли-
зации и лактонизации 2-кето-Т-гулоновой кислоты (XVI11) или ее диизо-
пропил иденового производного LXXI (схема 6).
Превращение метилового эфира 2-кето-Е-гулоновой кислоты (LXXII,
R = СН3) в L-аскорбиновую кислоту может производиться в щелочной
среде в присутствии алкоголята натрия в метиловом спирте в токе инерт-
ного газа [211] с последующим подкислением для выделения свободной
L-аскорбиновой кислоты [20]. Реакцию катализируют также амины и их
соли [212], ацетат натрия, бикарбонат натрия, ионообменные смолы —
амберлит IR-45 ( в ОН-форме) [2131; выход небольшой.
Превращение под влиянием кислых катализаторов дает хорошие резуль-
таты [20, 214]. Из многочисленных вариантов заслуживает внимания не-
посредственная енолизация и лактонизация 2,3;4,6-ди-О-изопропилиден-2-
KeTo-L-гулоновой кислоты (LXXI) без растворителя в токе хлористого или
бромистого водорода при 120° С; реакция продолжается всего 4 мин, и выход
L-аскорбиновой кислоты составляет 70—80% [215]. Енолизация и лактони-
зация 2,3;4,6-ди-О-изопропилиден-2-кето^-гулоновой кислоты в 20%-ном
водном растворе в отсутствие кислых реагентов проходит при температуре
120—150° С под давлением в течение 30—60 мин; выход L-аскорбиновой
кислоты составляет 70—73% [216].
При нагревании 2,3;4,6-ди-О-изопропилиден-2-кето-А-гулоновой кисло-
ты в концентрированной соляной кислоте (при применении 0,5 • части
соляной кислоты) в течение 2—2,5 ч при 60—63° С превращение дости-
гает максимума и L-аскорбиновую кислоту можно выделить в кристалли-
ческом виде с выходом 70—71 % [217 ]; после сгущения маточных растворов
выход повышается до 78%. Однако если применять четырехкратное
количество соляной кислоты, то удается выделить всего 13% L-аскорбино-
вой кислоты [214]. Изучена кинетика превращения 2-кето-Е-гулоновой
кислоты в L-аскорбиновую кислоту в разбавленной соляной кислоте [218] и
2,3;4,6-ди-О-изопропилиден-2-кето-Т-гулоновой кислоты в концентрирован-
ной соляной кислоте при 60 и 70° С [219]; реакция в водной среде первона-
чально протекает с отщеплением ацетона. Так, если в соляную кислоту
внести 2,3;4,6-ди-О-изопропилиден-2-кето-Е-гулоновую кислоту при 50° С
43
и по растворении смесь охладить, то выделяется 2-кето-Е-гулоновая кислота.
Затем происходит енолизация и лактонизация образовавшейся 2-кето-£-
гулоновой кислоты, одновременно сопровождающаяся реакциями расщепле-
ния /.-аскорбиновой кислоты в фурфурол и смолистые продукты его поли-
мер изации.
В спиртовой среде превращение 2,3;4,6-ди-О-изопропилиден-2-кето-£-
гулоновой кислоты в L-аскорбиновую кислоту в присутствии хлористого
водорода достигает 78% (в растворе) [220].
Енолизация и лактонизация 2-кето-/_-гулоновой кислоты в кислой среде
при нагревании в безводных неполярных растворителях (хлороформ,
диоксан и другие) [221 ] дает L-аскорбиновую кислоту с выходом 78%,
загрязненную из-за неоднозначности реакции посторонними примесями
[214].
Важнейшим вариантом синтеза L-аскорбиновой кислоты является прев-
ращение 2-кето-/_-гулоновой кислоты или ее диизопропилиденового произ-
водного в неполярном растворителе в присутствии спирта под влиянием
кислых катализаторов. Эльгер [123] превращает 2-кето-Е-гулоновую кисло-
ту или 2,3;4,6-ди-О-изопропилиден-2-кето-Е-гулоновую кислоту в L-аскор-
биновую кислоту в хлороформе и 94%-ном спирте, содержащем хлористый
водород, нагреванием при 60° С в течение 55 ч. Выход L-аскорбиновой кис-
лоты, выделяющейся из реакционной смеси в кристаллическом виде, дости-
гает 88%.В таких же условиях в присутствии метилового спирта вместо этило-
вого происходит образование метилового эфира 2-кето-Е-гулоновой кислоты
уже в течение 1 ч [222]. Он мсжет быть получен в толуоле в присутствии
метилового спирта и серной кислоты [223]. Этот эфир далее превращают в
L-аскорбиновую кислоту [20, 224] с выходом свыше 70% [214]. Метиловый
эфир 2-кето-Е-гулоновой кислоты может быть получен также из 2-кето-Е-гу-
лоновой кислоты этерификацией метиловым спиртом в присутствии мине-
ральной кислоты [20, 201 ], на амберлите 1R-120 в присутствии метилового
спирта [225], этерификацией диазометаном [20], а также при действии
метилата натрия в метиловом спирте [62, 124].
Для получения L-аскорбиновой кислоты применяют этиловый и бутило-
вый эфиры 2-кето-Е-гулоновой кислоты [226]. В качестве катализатора
предложен хлористый тиснил (выход 86%) [227], для среды — муравьи-
ная кислота с катализатором хлористым водородом [228].
В качестве неполярного растворителя могут быть применены, помимо
хлороформа, дихлорэтан, трихлорэтилен, различные другие галогенза-
мещенные алифатические и ароматические углеводороды, а также сами
ароматические углеводороды; выходы с такими растворителями, как хло-
роформ, дихлорэтан, четыреххлористый углерод и бензол, однозначны
[214].
Реакция енолизации и лактонизации протекает при температуре кипе-
ния азеотропных смесей [214], например в присутствии дихлорэтана — при
70,1—70,6' С за 13 ч, четыреххлористого углерода—при 62,1—62,6° С
за 24 ч и т. д. Для получения промежуточных эфиров 2-кето-Е-гулоновой
кислоты с успехом можно применять и высшие спирты в смеси с индифферент-
ным растворителем. Скорость реакции превращения 2,3;4,6-ди-О-изопропи-
лиден-2-кето-Е-гулоновой кислоты в смесях с нормальным пропиловым и
бутиловым, а также изобутиловым спиртами незначительно отличается от
скорости реакции с этиловым спиртом, но замедляется с изоамиловым спир-
том [229]. При применении этилового и других спиртов выход L-аскор-
биновой кислоты зависит также от каталитического участия воды; выходы
в безводном этиловом и других спиртах ниже на 4—22%, чем при катали-
тического влиянии 0,3 моля воды [229, 230]. Енолизация и лактонизация
2,3;4,6-ди-О-изопропилиден-2-кето^-гулоновой кислоты в L-аскорбиновую
кислоту осуществлена также в смеси, состоящей из толуола, гексанола с
10% НС1 и ацетона при 70—75° С [231 ]. Изучались и соотношения состав-
ных частей среды для енолизации [232].
44
’ Реакция превращения 2,3;4,6-ди-О-изопропилиден-2-кето-Е-гулоновой
| кислоты (LXXI) в L-аскорбиновую кислоту (1) нагреванием в среде неполяр-
| ного растворителя в присутствии спирта при каталитическом влиянии кис-
Г лых реагентов протекает через несколько фаз [229] (см. схему 6). Первой
Г фазой превращения является гидролиз 2,3;4,6-ди-О-изопропилиден-2-кето-
Г £-гулоновой кислоты (LXXI) и образование эфира 2-кето-А-гулоновой
F кислоты (LXXII, R = С2Н5). При этой реакции происходит и гидролиз, и
Г этерификация карбоксильной группы при участии спирта в присутствии
минеральной кислоты. Изопропилиденовые группы отщепляются в виде
L двух молекул ацетона. Вода, потребная для образования ацетона, полу-
чается за счет одной молекулы кристаллизационной воды, содержащейся
в исходной 2,3;4,6-ди-О-изопропилиден-2-кето-Е-гулоновой кислоте
(LXXI), а вторая молекула воды получается за счет реакции этерификации.
Первая фаза реакции совер пенно необходима для образования L-аскор-
биновой кислоты. Во второй фазе полученный эфир 2-кето^-аскорбиновой
кислоты (LXXII, R = С2Н5) подвергается дальнейшему превращению под
влиянием минеральных и органических кислот (хотя в последнем случае
скорость реакции значительно меньше) в L-аскорбиновую кислоту в
результате отщепления молекулы спирта от эфира 2-кето^-гулоновой ки-
слоты с одновременной кетоенольной перегруппировкой и замыканием
у-лактонного цикла в систему Да-?-бутенолида.
Естественно, что вторая фаза реакции зависит от характера катализа-
L тора, концентрации водородных ионов и температуры. Скорость реакции
L при каталитическом участии хлористого водорода в 3—4 раза больше, чем
В под влиянием серной кислоты [229]. Скорость реакции зависит также от
[ количества катализатора [214]. Повышение температуры на 10,7° С увели-
| чивает скорость реакции более чем в три раза.
К Таким образом, 2,3;4,6-ди-О-изопропилиден-2-кето-Е-гулоновая кислота
* в среде индифферентного растворителя в присутствии спирта превращается
L в L-аскорбиновую кислоту через реакции гидролиза, этерификации, йзоме-
£ ризации и лактонизации [229].
L В бесспиртовых смесях (например, в дихлорэтане и дибутиловом эфире)
Ь выход L-аскорбиновой кислоты резко снижается [219].
Следует отметить, что реакция лактонизации 2-кето-Е-гулоновой кисло-
Г ты или ее эфира обладает небольшой скоростью в отличие от обычной реак-
[ ции лактонизации, вероятно, потому, что 2-кето-Е-гулоновая кислота на-
?. ходится в устойчивой циклической у-окисной форме (XVIПа). После того
как образуется открытая форма 2-кето-Е-гулоновой кислоты (XVIII),
происходит кетоенольная перегруппировка и получаются цис- и транс-
изомеры 2-кето-Е-гулоновой кислоты (XVI Пв и XVII16), из которых только
^пс-форма (XVII 1в) способна подвергаться лактонизации.
они он
носнг -с - с- с -со-соон
н они
он ?н?н
I *\/с=с\
носн2-с—с
н Х°-
45
Возможно, что первоначально происходит лактонизация 2-кето-L-ryлоно-
вой кислоты (XVIII) в лактон 2-кето-£-гулоновой кислоты (LXXXV), а
затем таутомерное его превращение в L-аскорбиновую кислоту (I) с обра-
зованием ненасыщенной связи.
В настоящее время нет бесспорных данных, которые говорили бы в поль-
зу той или другой схемы реакции. Двойная связь образующейся системы
Д“-Р-бутенолида сильно повышает прочность пятичленного цикла, причем
повышению прочности способствует также наличие алкильного заместителя
в цикле.
Техническая L-аскорбиновая кислота подвергается перекристаллиза-
ции из воды или водного спирта, что связано с незначительными ее потеря-
ми при многократном использовании маточных растворов путем их сгуще-
ния в вакууме. Для осветления растворов используется активированный
уголь, с гидросульфитом натрия или насыщенный сернистым газом. Пере-
кристаллизацию проводят в присутствии диоктилсульфосукцината натрия
как поверхностно активного вещества [233] или трилона Б как комплексо-
образователя [234]. Для уменьшения потерь при перекристаллизации ре-
комендуется ускорение технологических операций, так как L-аскорбиновая
кислота не стабильна к длительному нагреванию водных растворов [235].
Получение L-аскорбиновой кислоты через L-идоновую и 2-кето-£-гу-
лоновую кислоты. Интересный и перспективный метод синтеза L-аскорби-
новой кислоты из D-глюкозы через L-идоновую кислоту и 2-кето-£-гулоно-
вую кислоту разработан в последние годы. В нем применяются ферментатив-
ные методы избирательного окисления на двух стадиях синтеза. D-Глю-
козу (LXVI) первоначально подвергают электролитическому окислению в
присутствии бромистых солей в D-глюконовую кислоту (LXXIII), с высоким
выходом выделяемую в виде кальциевой соли. Затем D-глюконовую кислоту
окислительной ферментацией с Acetobacter suboxydans превращают в 5-кето-
D-глюконовую кислоту (LXXXII) также с высоким выходом [236—238].
Каталитическим гидрированием на никелевом катализаторе из5-kcto-D-глю-
коновой кислоты (LXXXII) с выходом 94% получают кальциевую соль
L-идоновой кислоты (ХХХНв) [236—239]. Одновременно образуется неко-
торое количество D-глюконовой кислоты (LXXIII) [236—2381:
Н он
V-соон
неон н^он
носн о^-ноАн т?-ия-
нсон| н<!:он
нс—' н^он
CHjOH . СН2ОН
LXVI LXXIII
соон
неон
hoJh
неон
с=о
(!:н2он
соон
НСОН ферыен-
НОСН
НСОН
НОСН
CHjOH
соон
I
с=о
НОСН —
н|он иис"ота
носн
СН2ОН-
LXXXII
хххнв
XVlil
L-Идоновую кислоту (XXXПв) ферментацией с Pseudomonas fluorescens
[236, 237] или Pseudomonas 2-ketogulonicum [240] в аэробных условиях
окисляют в 2-кето-£-гулоновую кислоту (XVIII); из кальциевой соли
L-идоновой кислоты выход составляет 64,7% [240], из натриевой соли —
почти количественный [241]. Образующаяся при восстановлении 5-кето-О-
глюконовой кислоты D-глюконовая кислота после отделения труднораство-
римого 2-кето-£-гулоната кальция может быть повторно подвергнута фермен-
тативному окислению Acetobacter suboxydans [236,237 ]. 2-Кето-£-гулоновую
кислоту реакцией енолизации и лактонизации превращают в L-аскорби-
новую кислоту с выходом 88% [236, 237].
Следует отметить, что, по-видимому, три стадии синтеза (ферментацию,
гидрирование, ферментацию) можно провести последовательно без выделе-
ния промежуточно образующихся веществ (5-кето-О-глюконовой и L-идо-
46
новой кислот). Если окисление смеси D-глюконовой и L-идоновой кислот
осуществить с Pseudomonas chroniospirans, то в первой фазе происходит
окисление D-глюконовой кислоты до двуокиси углерода (тем самым она
выводится из процесса), а во второй фазе L-идоновая кислота этим мик-
роорганизмом обычным образом окисляется в 2-кето-Е-гулоновую кислоту
[242]. Общий выход L-аскорбиновой кислоты из D-глюкозы по этому
методу составляет около 60%.
ПОЛУЧЕНИЕ ПРЕПАРАТОВ Е-АСКОРБИНОВОЙ КИСЛОТЫ
L-А скорбинат натрия получают из раствора L-аскорбиновой
кислоты в водно-метанольной смеси нейтрализацией двууглекислым натрием
при 55-70° С с последующим осаждением горячим метиловым спиртом [243].
L-А скорбинат кальция получают из водного раствора
L-аскорбиновой кислоты и углекислого кальция с последующим осаждением
спиртом или ацетоном [244].
L-А скорбинат железа образуется из L-аскорбиновой кислоты
и углекислого железа при осаждении спиртом [245, 246].
Пальмитат L-a скорбиновой кислоты получен из
L-аскорбиновой кислоты и пальмитоилхлорида в диметилформамиде [247].
Д е г и д р o-L-a скорбиновая кислота образуется из L-ac-
L корбиновой кислоты при окислении SeO2 в безводном метиловом спирте
(выход 65—70%) [248], при окислении марганцовокислым калием [249] и
азотистой кислотой при pH 1—5 [250], при окислении йодом в метиловом
L спирте (выход 23%) [251].
г При окислении L-аскорбиновой кислоты хлором в строго регулируемых
I условиях в безводном метиловом спирте в присутствии избытка углекислого
| свинца получен кристаллический метанольный аддукт дегидро-Е-аскорбино-
k вой кислоты с т. пл. 102—105° С [16].
БИОЛОГИЧЕСКОЕ ЗНАЧЕНИЕ L-АСКОРБИНОВОЙ КИСЛОТЫ. ЗАВИСИМОСТЬ
МЕЖДУ СТРОЕНИЕМ И БИОЛОГИЧЕСКОЙ АКТИВНОСТЬЮ
L-Аскорбиновая кислота является витамином для человека, обезьян и
некоторых других представителей теплокровных: морской свинки, так на-
зываемой фруктовой летучей мыши и индийского красного воробья. Осталь-
ные животные не нуждаются в получении витамина С с пищей и обладают
способностью к его биосинтезу. При недостатке в организме L-аскорбиновой
кислоты возникает цинга — болезнь, известная с древних времен, характе-
ризующаяся заболеванием десен, выпадением зубов, структурным измене-
нием хрящей и костей, в итоге приводящая к летальному исходу. L-Аскорби-
новая кислота способствует образованию соединительной ткани.
Эта кислота встречается в животных и растительных тканях как в свобод-
ной, так и в окисленной форме (дегидро-Е-аскорбиновая кислота); например,
в печени содержится 27% ив мышцах 50% дегидро-Е-аскорбиновой кислоты
от общего количества кислоты. В молоке и крови содержится только L-ас-
корбиновая кислота. В растениях L-аскорбиновой кислоты находится до
70% в связанном состоянии [252]. Дегидро-Е-аскорбиновая кислота обла-
дает такой же витаминной активностью, как и L-аскорбиновая кислота [52].
L-Аскорбиновая кислота (витамин С) является переносчиком водорода в
некоторых ферментативных реакциях, протекающих в живой клетке.
Она восстанавливает различные окисленные формы ферментов (например,
аскорбиназу), сама окисляясь при этом в дегидро-Е-аскорбиновую кислоту,
обратимо легко регенерируемую в L-аскорбиновую кислоту глутатионом
(у-глутамилцистеинилглицином) [253] за счет его сульфгидрильной группы
при воздействии редуктазы. Эту функцию L-аскорбиновой кислоты как
переносчика водорода можно представить следующей схемой:
47
1
L -Аскорбиновая кислота 4- — О>
Оксидаза L-аскор-
биновой кислоты
----------------► Дегидро-А-аскорбиновая 4- Н,О
кислота
Редуктаза дегидро-
L-аскорбиновой кислоты
Дегидро-А-аскорбиновая . 2CSH--------------------------
кислота
L -Аскорбиновая . qccq.
кислота ”’-
Дегидрогеназа
Субстрат + НАДФ+---------------> Окисленный субстрат 4- НАДФ-Н + Н+
Глутатион—редуктаза "
GSSG 4- НАДФ-Н 4- Н+------------------> 2GSH 4- НАДФ+ 1
NH2 CONHCHjjCOOH
GSH = НООССН (CH2)s conhchch2sh
Природная аскорбиназа — оксидаза L-аскорбиновой кислоты, фермент ]
с молекулярной массой 150 000, широко распространенный в растительных ]
тканях, — содержит около 0,26% меди, что соответствует 6 атомам Сп++ на
I моль [254].
L-Аскорбиновая кислота — самый сильный восстановитель живого орга-
низма, легко окисляемый различными ферментами, например пероксидазой в
присутствии перекиси водорода [81 ] и некоторых флаваноидов (флаванов,
флавонолов), фенолоксидазой, цитохромоксидазой в присутствии цитохрома
и др.
Способность к окислительно-восстановительным превращениям, связан-
ная с ендиольной группировкой, которая стабилизирована находящейся в
цикле соседней карбонильной группой, и сопровождающаяся перенесением
атомов водорода к акцепторам, является важнейшей каталитической функ-
цией L-аскорбиновой кислоты в живом организме.
L-Аскорбиновая кислота по своей биологической активности высоко
специфична. Витаминная активность находится в зависимости от наличия
системы Д“-₽-бутенолида, причем у-лактонное кольцо должно быть образо-
вано по гидроксильной группе положения 4 D-конфигурации. Аналоги L-ac-
корбиновой кислоты, имеющие у-лактонное кольцо, образованное по гидро-
ксильной группе при (^^-конфигурации, не обладают витаминной актив- а
ностью, например L-изоаскорбиновая (L-аробо-аскорбиновая) кислота (IV)
И др. |
Пространственное положение гидроксила при С<5) должно отвечать, ,
наоборот, L-конфигурации; для D-конфигурации активность уменьшается
в 20 раз, как это характерно для D-изоаскорбиновой (D-арабо-аскорбиновой)
кислоты (V) [62, 91 ]. Элиминирование гидроксильной группы в положенииб
снижает активность в три раза, как это наблюдается у б-дезокси-А-аскорби-
новой кислоты (LXXXVI) [255]. Увеличение числа углеродных атомов в .
молекуле снижает витаминную активность в пять раз для L-рамно-аскорби-
новой кислоты (LX XXVII), в 40 и 60 раз соответственно для L-глюко-
(LXXXVIII) [256] и L-галакто-аскорбиновой кислоты (LXXXIX) и в
100 раз для L-глюко-гептааскорбиновой кислоты (ХС) [257 ].
Уменьшение числа углеродных атомов по сравнению с шестью имеющими-
ся в молекуле L-аскорбиновой кислоты полностью лишает соединение вита-
минных свойств.
При сочетании в одной структуре L-конфигурации 4-гидроксильной груп-
пы иП-конфигурации 5-гидроксильной группы образуются сильнейшие анти-
витамины: энантиоморфная форма витамина С, D-аскорбиновая кислота
П1) и D-глкмсо-аскорбиновая кислота (XCI) [108, 258, 259].
48
он он он он
НОСН носн
I LXXAVIII | LaXXIX
сн2он сн2он
носн
носн
сн2он
хс
D-Аскорбиновая кислота может быть получена циангидриновым методом
из D-ксилозы [112].
Солеобразные соединения L-аскорбиновой кислоты с металлами, еноляты,
способные легко превращаться снова в L-аскорбиновую кислоту, например
L-аскорбинаты натрия, кальция, железа и т. д., обладают равнозначной
витаминной активностью. Витаминная активность проявляется только при
наличии свободных гидроксильных групп. Различные функциональные
производные по гидроксильным группам, такие, как метиловые эфиры,
изопропилиденовые производные и другие, лишают молекулу витаминных
свойств полностью или почти польностью, так же как и гидрирование не-
насыщенной связи лактонного кольца. Так, 3-метиловый эфир L-аскорбино-
вой кислоты (т. пл. 121° С, [ab + 29°) обладает всего 2—4% витаминной
активности [257].
Дегидро-Л-аскорбиновая кислота, 2,3-дикето-£-гулоно-у-лактон (II),
являющаяся окисленной формой L-аскорбиновой кислоты в организме, имеет
витаминную активность, равнозначную последней. При раскрытии лактон-
ного кольца образуется 2,3-дикето^-гулоновая кислота [260], которая
полностью лишена витаминной активности.
Интересно отметить, что среди нециклических соединений только мети-
ловый эфир 2-кето^-гулоновой кислоты (LXXII, R = СН3)
он о
он н с — с
। ।
носн,-с-нсон С'
А
LXXII
обладает 20%-ной активностью L-аскорбиновой кислоты [257], по-видимому,
вследствие его повышенной способности образовывать систему Дв1₽-бутено-
лида в организме, превращаясь в L-аскорбиновую кислоту.
Замена одной гидроксильной группы в положении 2 аминогруппой приво-
дит к образованию скорбаминовой кислоты (ХСП), обладающей незначитель-
ными витаминными свойствами [261 ], а замена двух гидроксильных групп
в положениях 2 и 3 — к диаминоаналогу L-аскорбиновой кислоты; оба
эти соединения обладают сильными восстановительными свойствами [262].
Аскорбиген (XLIII) — индольное производное L-аскорбиновой кисло-
ты — обладает менее чем 5%-ной активностью витамина С [99].
4$
L-Аскорбиновая кислота (I) синтезируется в растениях и организме неко-
торых животных из D-глюкозы (LXVI) через лактон D-глюкуроновой кис-
лоты (XCIII) и £-гулоно-7-лактон (XCIV) или их производные [263,264].
По-видимому, D-галактоза (ХСУ)также может переходить в L-аскорбиновую
кислоту (I) через соответствующие промежуточные соединения —D-галак-
туроновую кислоту (LVII) и L-галактоно-у-лактон (LIX, схема 10).
Схема 10
Синтез L-аскорбиновой кислоты в растениях из D-глюкозы и D-галактозы
СН2ОН
о неон
io
н I
I
носн
I
но н
сн
н
но н
хеш
СН2ОН
---сн
I
неон |О]
о неон
носн
нЛ
Xcv
СООН
I
—сн
I
неон
о неон
I
носн
I
L-i-C
/\
но н
LVU
'°~1
носн I
носн
I
сн2он
В этом биосинтезе происходит превращение соединений с конфигурацией
D-ряда в соединения с конфигурацией L-ряда, что доказано превращением в
организме крысы D-глюкозы с меченым атомом В * * * * * 14С в положении 1 в L-ас-
корбиновую кислоту с меченым атомом углерода в положении 6 [263].
у-Лактоны D-глюкуроновой (ХСШ) и L-гулоновой кислот. (XCIV) при
поступлении в ростки растений или при введении в организм крысы превра-
щаются в L-аскорбиновую кислоту. Также ведет себя метиловый эфир
D-галактуроновой кислоты и у-лактон L-галактоновой кислоты (LIX); но
другие у-лактоны альдоновых кислот, в том числе у-лактоны L-идоновой и
L-талоновой кислот, отличающиеся от у-лактонов L-гулоновой иL-галак-
тоновой кислот одной лишь конфигурацией заместителей у атома углерода
в положении 2, L-аскорбиновой кислоты не дают [263].
Доказательство превращения у-лактона L-гулоновой кислоты (XCIV) в
L-аскорбиновую кислоту получено и синтетическим путем — проведением
окисления с пергидролом и сернокислым железом в присутствии пирофос-
форной кислоты при температуре 80—90° С (выход 10%) [265].
Витамин С является одним из витаминов, потребность в котором не покры-
вается за счет пищевых продуктов. Это обстоятельство объясняется тем, что
L-аскорбиновая кислота довольно легко разрушается при нагревании во
время приготовления пищи, а также сезонной недостаточностью свежих
овощей и фруктов в весенние и первые летние месяцы, особенно у населения
северных районов. Применение для питания в больших количествах таких
продуктов, как консервированные и сухие овощи, фрукты, мясо и рыба, в
которых содержание витамина С понижено, также приводит к увеличению
недостаточности этого витамина. Поэтому L-аскорбиновая кислота приме-
няется для восполнения ее недостатка в различных пищевых продуктах
50
при их приготовлении, при получении пищевых продуктов в консервирован-
ном виде и специально для витаминизации, например, белого хлеба, молока
И др-
L-Аскорбиновая кислота имеет большое значение как антиоксидант для
сохранности пищевых продуктов и в разных странах широко используется
для стабилизации внешнего вида картофеля, мяса и мясных изделий в
охлажденном и расфасованном состоянии. Кроме того, L-аскорбиновая кис-
лота применяется для приготовления различных напитков.
Недостаток (гиповитаминоз) L-аскорбиновой кислоты в организме
проявляется в потере аппетита, анемии, быстрой утомляемости, весенней
усталости, неясных разлитых болях в различных частях тела, наклонностей
к кровотечениям, разрыхленности десен, склонности к инфекционным забо-
леваниям дыхательных путей и других симптомах.
Применение L-аскорбиновой кислоты для медицинских целей дает исклю-
чительно большой эффект при лечении различных заболеваний; в частности,
L-аскорбиновая кислота способствует заживлению ран и вылечиванию пере-
ломов, излечению язвы желудка и двенадцатиперстной кишки, а также
рекомендуется при артериосклерозе, остром инфекционном гепатите, стома-
тите, тромбозах, рвоте беременных, экземах и др.
Физиологическая потребность человека в L-аскорбиновой кислоте
70—100 мг в день; при очень тяжелом труде и для кормящих эта норма
возрастает до 200 мг в день.
Профилактическое применение больших доз L-аскорбиновой кислоты
(1—3 г в сутки) способствует предохранению от заболеваний простудного
характера.
L-Аскорбиновая кислота находит применение в качестве витаминной
кормовой добавки для домашней птицы во время носки яиц — около
10 мг на 100 г корма.
СПИСОК ИСПОЛЬЗОВАННОЙ
ЛИТЕРАТУРЫ (К ГЛАВЕ I)
1. A. Szent-Gyorgyi, W. Ha-
worth. Nature, 131, 23 (1933).
2. G. Co x. Nature, 130, 205 (1932).
3. G. С о x, T. Goodwin. J.
Chem. Soc., 769 (1936).
4. Я. M. Слободин, А. К. Ба-
сова, A. E. Гельме. ЖПХ,
19, 170 (1946).
5. W. В i r c h, L. H a r r i s. Bio-
chem. J„ 27, 595 (1933).
6. G. N e b b i a, E. M. Pizzo-
1 i. Acta Vitaminol., 13, 269 (1959);
Ann. chim. (Rome), 48, 695 (1958).
7. T. M i c h e 1, К. К г a f t. Z.
Physiol. Chem., 216, 233 (1933);
218, 280 (1933); 222, 235 (1933).
8. H. Mohler, H. L о h r. Helv.'
Chim. Acta, 21, 485 (1938).
9. E. В a 1 1. J. Biol. Chem., 118,
219 (1937).
10. H. В о r s о о k, H. Daven-
port, C. Jeffreys, R. War-
ne r. J. Biol. Chem., 117, 237 (1937).
И. P. Karr er, G. Schwarzen-
b a c h. Helv. Chim. Acta, 17, 58
(1934).
12. D. Kern. J. Am. Chem. Soc., 76,
1011 (1954).
13. Z. V a v r i n. Chem. listy, 41, 77
(1947).
14. S. Ruskin. Пат. США 2606903;
С. A. , 47, 5440 (1953).
15. H. S t a u d i n g e r, W. Weis.
Z. physiol. Chem., 337, 284 (1964).
16. В. P e c h e r e r. J. Am. Chem.
. Soc., 73, 3827 (1951).
17. H. A 1 b e r s, E. Muller. Na-
turwissensch., 46, 75 (1959).
18. H. A 1 b e r s, E. M fi 1 1 e r,
H. D i e t z. Z. physiol. Chem., 334,
243 (1963).
19. В. T e i c h m a n n, D. Z i e -
b a r t h. Acta Chim. Acad. Sci.
Hung., 49, 311 (1966); C. A. , 66,
37204 (1967).
• •20. T. Reichstein, A. Gruss-
n e r. Helv. Chim. Acta., 17, 311
(1934).
21. E. Cox, E. Hirst, R. Rey-
no 1 d s. Nature, 130, 888 (1932).
22. H. D a m, J. G 1 a v i n d. Bio-
chem. J„ 32, 1018 (1938).
23. F. Petyely, H. Bauer. Mo-
natsh., 83, 758 (1952).
24. R. Strohecker, H. Schmidt.
Z. Lebensm.-Untersuch.-Forsch.,
36, 370 (1943).
• 25. J. S c a i f e. Canad. J. Biochem.
Physiol., 37, 1049 (1959).
26. E. Barron, R. Al e i o,
F. К 1 e m p e r e r. J. Biol. Chem.,
112, 625 (1936).
27. В. А. Энгельгардт,
51
В. Н. Букин. Биохимия, 2,
587 (1937).
28. F. F о х, L. Levy. Biochem. J.,
30, 208 (1936).
29. J. A n t e n e r. Helv. Chim. Acta.,
20, 742 (1937).
30. G. De s h m u k h, M. В a p a t.
Chem. Ber., 88, 1121 (1955).
31. P. Karrer, H. Salomon,
K. Sch op p, R. M о r f. Helv.
Chim. Acta, 16, 181 (1933).
32. P. К a г г e r, G. Schwar-
zenbach, K. Schopp. Helv.
Chim. Acta, 16, 302 (1933).
33. E. Gero. Compt. rend., 240, 1818
(1955); Bull. Soc. Chim. BioL, 31,
817, 825 (1949).
34. R. Herbert, E. H i r s t,
E. Percival, R. Reynolds,
F. Smith. J. Chem. Soc., 1933,
1270.
35. С. A r c u s, S. Z i 1 v a. Biochem.
J., 34, 61 (1940).
36. H. L u n d, H. L i e c k. Nature,
137, 784 (1936).
37. L. C h a p о n, S. C h a p о n,
N. G e n t у - A i m ё, E. U r i о n.
Bull. Soc. Chim. France, 1958, 157,
1366; 1959, 81.
38. A. Dudani, C. Krishna-
murti. Biochim, Biophys. Acta.,
13, 505 (1954).
39. R. G r i n s t e a d. J. Am. Chem.
Soc., 82, 3464 (1960).
40. П. С. Романчук. ДАН СССР,
117, 665 (1957).
41. E. G e г о, P. G a 1 1 i c. Bull.
Soc. chim. biol., 34, 549, 557 (1952).
42. C. F 1 e s c h, W. Schuler,
R. Meier. Helv. Chim. Acta, 43,
2014 (1960).
43. H. В. Новотельной,
M. T. Головкина,
E. P. Ста в p о в a . Научи,
докл. высшей школы, биолог, нау-
ки, № 1, 137 (1959); С. А., 54,
8953 (1960).
44. R. Morse. Food Res., 18, 48
(1953).
45. М. A k a g i, I. А о k 1. Yakugaku
Zasshi, 77, 1314 (1957); C. A., 52,
6263 (1958).
46. E. C h e г b u 1 i e z, J. R a b i -
nowitz. Helv. Chim. Acta., 45,
1874 (1962).
47. S. К a m i у a, T. Nakabayas-
ki. Bitamin, 13, 250, 384 (1957);
C. A., 54, 7797 (1960).
48. C. Noire, A. C i e r, B. D r e -
von. Bull. Soc. chim. France,
1960, 245.
49. J. Herrmann, W. Andrae.
Nahrung., 7, 243 (1963).
50. Z. Csiiros, J. Petro. Acta
Chim. Acad. Sci. Hang., 14, 95
(1958).
51. S. К a m i у a, T. N a k a b a у a-
s h i. Bitamin, 13, 246 (1957); C. A.,
54, 7797 (1960).
52. J. Tillmans, P. H i r s c h et
al. Z. Untersuch. Lebensm., 63, 1,
21, 241, 267, 276 (1932); 65, 145
(1933).
53. A. C i e r, C. N о f r e, B. D r e -
v о n, A. L e f i e r. Bull. Soc. Chim.
France, 1959, 74.
54. С. H u r d, С. I s e n h о u r. J. Am.
Chem. Soc., 54, 317 (1932).
55. F. H u e 1 i n. Food Res., 18, 633
(1953).
56. I. M. С о g g i о 1 a. Nature, 200,
954 (1963).
57. K. Tokuyama, K. Goshi-
m a, N. M a e z о n о, T. Maeda.
Tetrahedron Lett., 1971, 2503.
58. F. R e i t h e 1, R. W i t h e r. J.
Am. Chem. Soc., 71, 1879 (1949).
59. E. Moslach, J. Burns,
С. К i n g. J. Am. Chem. Soc., 73,
1875 (1951).
60, D. Nomura, Y. U e h a r a,
H . H a k k o. Kogaku Zasshi, 36,
290 (1958); C. A., 53, 10047 (1959).
61. H. E 1 К h a d e m, S. E I Ash-
ry. J. Chem. Soc. ,,C“, 1968, 2247,
2248. 2251.
62. H. О h 1 e, H. Erlbach,
H. С a r i s. Ber., 67, 324, 555 (1934).
63. G. Henseke, K. Dittrich.
Chem. Ber., 92, 1550 (1959).
64. J. Roe. C. Kuether. J. BioL
Chem., 147, 399 (1943).
65. В. А. Де в я т н и н, В. M. Ио-
си ков a . ДАН СССР, 15, 85
(1937).
66. I. G a 1. Nature, 138, 799 (1936).
67. G. В a r a c. Compt. rend. Soc. biol.,
126, 61 (1937).
68. B. Wurtz, J. North. Compt.
Rend., 256, 1388 (1963).
69. D. Swern. J. Am. Chem. Soc.,
71, 3256 (1949).
70. H. Nomura, K. Sugimoto
Яп. пат. 9550 (1968); С. A., 70,
4545 ZIQfiQl
71. H. T a n a k a, Y. H i s t a n i. Яп.
пат. 20051 (1967); С. A., 68, 96104
(1968).
72. W. Wenner. J. org. Chem., 14, 23
(1949).
73. W. Hoffman. Пат. США
2823166; С. A., 52, 7623 (1958).
74. Франц, пат. 1173146, 1223224,
1273989; РЖХ, 1960, 78489; 1962
24345.
75. Н. Meyer-Doring. Arrnei-
mittel-Forsch., 10, 381 (1960); С. А„
54, 22388 (1960).
76. Ф. Я. Рачинский,
Н. М. Славачевская,
Д. В. Иоффе. ЖОХ, 28, 2998
(1958).
77. Англ. пат. 1088735; С. А., 69,
67415 (1968).
78. А. Н а г d е п, S. Z i 1 v a. Bio-
chem. J., 12, 259 (1918).
79. S. Z i 1 v a. Biochem. J„ 17, 410,
416 (1923); 18, 186, 632 (1924); 19,
589 (1925).
80. S. С о n n e 1 1, S. Z i 1 v a. Bio-
chem. J., 18, 638, 641 (1924).
81. A. Szent-Gyorgyi. Biochem.
52
J„ 22, 1387 (1928); 24, 1886 (1930).
Я2 E Hirst, E. Percival,
F.’ Smi t h. Nature, 131, 617 (1933).
83 R. Her bert, E. Perci val,
r. Reynolds, F. Smith,
E Hirst. J. Soc. Chem. Ind.,
52, 221 (1933).
Я4 H E u 1 e г, С. M a r t i u s. Lieb.
* Ann., 52, 221 (1933).
я.4 T R e i c h s t e i n. Nature, 132,
280 (1933).
86 T. R e i c h s t e i n. A. G г й ss-
n e r, A. Oppenauer. Helv.
Chim. Acta, 16, 561, 1019 (1933).
87 . R. A u 1 t, D. В a i г d, H. Car-
rin g t о n, W. Haworth,
R. Herbert, E. Hirst,
E. P e r c i v a 1, F. Smith,
N. S t a c e y. J. Chem. Soc., 1933,
1419.
88 W. H a w о r t, E. H i г s t. J.
' S о c. Chem. Ind., 52, 645 (1933).
89. At. С r e i g h t о n, W. W e n ner,
H. At. W и e s t. J. Org. Chem., 13,
613 (1948).
90. T. R e i c h s t e i n, R. Oppe-
nauer. Helv. Chim. Acta, 17,
390 (1934).
91. T. R e i c h s t e i n, A. G г й s s-
ner, R. Oppenauer. Helv.
Chim. Acta, 17, 510 (1934).
92. A. Szent-Gyorgyi, J.Stir-
b e 1 y. Biochem. J., 27, 279 (1933).
93 L. V a r g h a. Nature. 130, 846
(1932).
94 L. V a r g h a. Nature., 131, 363
(1933).
95. R. Raphael. J. Chem. Soc.
1947, 805; Nature, 160, 261 (1947).
96. Л. Ф и з e p, M. Ф и з e p. Орга-
ническая химия, At., ИЛ, 1949, с.
415, и др.
97. М. В е zssonof I. Bull. Soc.
chim. biol., 16, 1107 (1934).
98. В. G u h a , J. P a 1. Nature, 137,
946 (1936).
99. W. F e 1 d h e i m, Z. P г о c h a z-
k a. Intern. Z. Vitaminforsch., 32,
251 (1962).
100. 2 - Prochazka, V. Sand a.
F. Sorm. Chem. listy, 50, 167
(1956); 51, 1197 (1957); Coll. Czech-
osl. Chem. Comm. 22, 333 (1957).
101. R. G m e 1 i n, A. V i г t a n e n.
Ann. Acad. Sci. Fennicae, Ser. A,
107 (1961); Suomen Kemistilechti,
34B, 15 (1'961).
102. A. Virtanen, E. P i iro-
ne n, Suomen Kemistileht, 35B,
N 5—6, 104 (1962); Acta chem.
Scand., 16, 1286 (1962); РЖХ,
1963, 8 Ж 484, 485.
103. G. К i s s, H. N e u k о m. Helv.
Chim. Acta, 49, 989 (1966).
104. G. Kiss. Angew. Chem., 78, 1066
(1966).
105. А. А. Шмидт, К. 3. T у л ь-
ч и н с к а я. Витамины в теории
и практике, At., Пищепромиздат,
1937;
106. В. Helferich, О. Peters.
Вег., 70, 465 (1937).
107. J. Hamamura М. Otsuka,
М. Suzumoto. J. Agr. Chem.
Soc. Japan, 22, 24, 25; C. A., 46,
10108 (1952); Bull. Fac. Text.
Fibers. Kyoto Univ. Industr. Arts
and Text. Fibers, 1, 79 (1955); РЖХ,
1957, 32291.
108. P. Stedehouer. Rec. trav.
chim., 71,. 831 (1952).
109. F. M i c h e e 1, F. J u ng. Ber.,
66, 1291 (1933).
110. F. At i c h e e 1, H. H a a r h о f f.
Lieb. Ann., 545, 28 (1940).
111. W. H a w о r t h, E. H i r s t,
J. Jones., F. S m i t h. J.
Chem. Soc., 1934, 1192.
112. K. Hamilton, F. S m i t h. J.
Am. Chem. Soc., 74, 5162 (1952).
113. L. S a 1 о m о n, J. Burns,
С. К i n g. J. Am. Chem. Soc., 74,
5161 (1952).
114. T. R e i c h s t e i n. Helv. Chim.
Acta, 17, 324 (1934).
115. H. I s b e 1. J. Research National
Bureau Standards, 29, 227 (1942).
116. T. G a r d п e г, E. We nis. J.
Am. Chem. Soc., 73, 1855 (1951).
117. В. Березовский, E. Ро-
дионова, Л. Стрель-
ну н а с . ЖОХ, 24, 628 (1954).
118. В. Березовский,
Л. Стрельчунас. ЖОХ, 24,
856 (1954).
119. Н. I s b е 1 1. J. Research Natio-
nal Bureau Standards, 33, 45 (1944);
Chem. Age., 51, 35, 1305 (1944).
120. Д. Вернер. Укр. хим. ж. 18,
366 (1952).
121. Т. Kozakiewiez. Gaz. Cuk-
rownicza, 89, 394 (1949); C. A., 48,
7550 (1954).
122. P. S a h. Ber., 69, 158 (1936).
123. F. Eiger. Zbl., 1937, I, 2992;
пат. США 2179978; С. A., 34,
1823 (1940).
124. К. At a u г e г, В. S c h i e d t.
Ber., 66, 1054 (1933); 67, 1239
(1934).
125. A. E. Фаворский,
T. И. Темникова. Изв. АН
СССР, ОХН, 1936, 911.
126. И. Струков, Н. К о п ы л о -
в а. Фармация, 1947, № 3, 8.
127. Р. R u m р 1, S. At а г 1 i е г.
Bull. Soc. chim. France, 1959, 187.
128. At. Abdel-Akher. J. Ha-
milton, F. S m i t h. J. Am.
Chem. Soc., 73, 4696 (1951).
129. H. Creighton. Trans. Elect-
roch. Soc., 75, 389 (1939), C. A., 37,
1088 (1943).
130. R. Hales. Пат. США 2289189,
2300218; С. A., 37, 42, 1660 (1943).
131. H. Г. Беленькая,
Н. А. Белозерский, ЖОХ,
19, 164 (1949).
132. В. Н. Ипатьев, ЖРХО, 44,
1003, 1710 (1912); Вег., 45, 3225
(1912).
133. Р. В г a g g, L. Hough. J.
Chem. Soc., 1957, 4347.
53
134. W. S. F о d о г. Ind. Eng. Chem.,
52, 282 (1960).
135. H. H e f t i, W. К о 1 b. Пат.
США 2507973; С. А., 45, 2340
(1951).
136. К. S u g i п о, М. Y a m a s h i-
t а. Яп. пат. 175935; С. А., 44,
9836 (1950).
137. D. Killefer. Ind Eng. Chem.
News, 15, 489 (1937).
138. L. F 1 о у d, R. Con n or,
H. A d к i n s. J. Am. Chem.
Soc., 54, 1651 (1932).
139. С. Д. Борисоглебский.
ЖПХ, 13, 571 (1940).
140. P. В г a h m e, M. P a i, G. N a r-
s i m h a n. Brit. Chem. Eng., 9,
684 (1964).
141. K. S t i с к d о г n, E. К 6 n i g,
G. К о n e t z к e et al. Пат. ГДР
33788; РЖХ, 1966, 11 H 283.
142. I. Gomes, E. Hai de gger,
A. Monostory, I, Peter,
N. S z e i 1 e r n e. Kolorist ert.
6, №2, 70 (1964); РЖХ, 1965,
19 H 16.
143. E. M. А д а с к и н, H. И. Зи-
мин о в a, Б. Л. Лебедев,
С. В. Павлов. ЖПХ, 31, 595,
897 (1958).
144. L. Hartstr a, L. Bakker,
Н. Adrian van Westen.
Пат. США 2518235, С. А., 44,
10732 (1950); датск, пат. 64717,
С. А., 44, 1531 (1950).
145. А. Д. П о с и е е в, Е. А. Че-
бан. Авт. свид. СССР 64925;
Бюлл. изобрет., 1945, № 6, 12.
146. Н. И. Волынкин. Авт. свид.
СССР 66661; Бюлл. изобрет., 1946,
№ 7, 10.
147. М. Delepine, С.Напе-
g г a a f f. Bull. Soc. chim., 4,
2091 (1937).
148. C. Lobry de Bruyn e,
W. Alberdavan Ekens-
t e i n. Rev. trav. chim., 14, 203
(1895).
149. M. W о 1 f г о m, W. L e w i s.
J. Am. Chem. Soc., 50, 837 (1928).
150. J. Sowden, R. Shaffer. J.
Am. Chem. Soc., 74, 505 (1952).
151. M. W о 1 f г о m, M. К 6 n i g s-
b e r g, F. M о о d y, R. G о e p p.
J. Am. Chem. Soc., 68, 122, 578
(1946).
152. W. S u 1 1 i v a n. J. Am. Chem.
Soc., 67, 837 (1945).
153. J. J о n e s, W. Reid. Canad. J.
Chem., 33, 1682 (1955).
154. G. Bertrand. Bull. Soc. chim.,
(3), 15, 627 (1896); Lieb. Ann.
chim., (8) 3, 183, 227 (1904).
155. P. W e 1 1 s, L. L о c k w о о d,
J. S t u b b s, N. P о r g e s,
E. Gastrock. Ind. Eng. Chem.,
31, 1425 (1939).
156. 3. Г. Разумовская. Ар-
хив биол. наук, 43, 153 (1936).
157. Р. Wells, J. Stubbs,
L. Lock wood, E. R о 1,
N. P о r g e s, Gastrock. Ind. Eng.
Chem., 31, 1518 (1939).
158. E. Креслин г. Микробиоло-
гия, 6, 898 (1937); 11, 115 (1942).
159. P. W e 1 1 s, J. Stubbs,
L. L о c k w о о d, E. R о 1. Ind.
Eng. Chem., 29, 1385 (1937).
160. Э. Д. Михлин, M. Г. Го-
лышева. ДАН СССР, 82, 439
(1952); Биохимия, 17, 91 (1952);
Труды ВНИ витаминного ин-та,
4, 83, М., Пищепромиздат, 1953.
161. М. Г. Голышева, М. И. Л н-
б е р. Труды ВНИ витаминного
ин-та, 4, 79, М_, Пищепромиздат,
1953.
162. Э. Д. Михлин, М. Г. Го-
лышева, В. А. К е п п е н.
Микробиология, 21, 521 (1952).
163. Э. Д. Михлин, М. Г. Го-
лышева, И. С. Розен-
берг, Н. А. Крылов,
В. А. К е п п е и. Труды ВНИ
витаминного ин-та, 5, 66, М.,
Пищепромиздат, 1954.
164. 3. Г. Разумовская. Мик-
робиология, 11, 125 (1942).
165. Т. Maeda, Y. М i i с h i,
К. Takuyama. Bull. Chem.
Soc., Jap., 42, 2648 (1969).
166. В. M. Березовский,
Л. И. Стрельчунас. ЖОХ,
сборник, 1, 453 (1953).
167. Т. И. Темникова,
В. В. Склярова. ЖПХ, 27,
1131 (1954).
168. К. Т о k и у a m а, Е. Н о n d а,
N. Н о к i. J. org. Chem., 29, 133
(1964).
169. J. Р a t i 1, J. В о s e. J. Indian
Chem. Soc., 43, 161 (1966); РЖХ,
1967, 2Ж502.
170. F. C u i b a n, E. I s t г i c,
S. Morgan u, F. Serbes-
cu, S. Teodorescu, B. Be-
nes, E. К 1 e i n. Rec. Chim.
(Bucharest), 10, № 2, 71 (1959);
C. A., 57, 965 (1962).
171. К. T о к u у a m a, E. H о n -
d a. Bull. Chem. Soc. Japan., 37,
591 (1964); C. A., 61, 13394 (1964).
172. M. Sterescu, S. Ariza n,
S. P о p a. Ceskosl. farmac., 9,
398 (1960); РЖХ, 1961, 17Л274.
173. С. Д а н и л о в, И. Л и ш а н-
ский. ЖОХ, 21, 366 (1951).
174. Н. Grunenberg, С. Bredt,
W. Freudenberg. J. Am.
Chem. Soc., 60, 1507 (1938).
175. J. R a t i e, J. Rose. Indian J-
Chem., 5, 598 (1967).
176. В. M а к с и м о в, В. H и к о и о-
в а, А. Ла з а р е в а , Л. Зве-
рева, ЖОХ, 9, 936 (1939).
177. В. М. Березовский,
Г. Е. Цимаркина,
Л. Н. Стрельчунас. Тру-
ды ВНИ витаминного нн-та, 5,
21, М., Пищепромиздат, 1954.
178. Франц, пат. 1520243; С. А., 71,
13328 (1969).
54
179 Н. О h 1 е. Вег., 57, 403, 1566
' (1924).
180. Р- Кристаллинская. Ви-
тамины в теории и практике, 2,
78, 85, М„ Пищепромиздат, 1941.
181 Яп. пат. 3895 (1950); С. А., 47,
‘ 5962 (1953).
182. М. Hosokawa, Н. Y a g i,
К. N a i t о. Пат. ФРГ 921326;
РЖХ, 1957, 24588.
183 Я- Слободин, ЖОХ, 17, 485
(1947).
184. Н. О h 1 е. Вег., 71В, 562 (1938).
185. D. Hinkley, R. В е u t е 1.
Франц, пат. 1541849; С. А., 71,
102189 (1969).
186. Т. D г и s t г и г, J. G i 1 1 i s,
М. G о 1 1 a h е г. Франц, пат.
1403516, С. А., 63, 13554 (1965).
187. J. W е i j 1 а г d. J. Am. Chem.
Soc., 67, 1031 (1945).
188. В. G б г 1 i c h. Пат. ФРГ. 935968;
Zbl., 1956, 6243.
189. A. Verheyden. Пат. США
2559034; С. А. 45, 8926 (1951).
190. G. Frohlich, А. К г а 1 а -
v i 1, Е. L г i к е. Пат. США
3453191; РЖХ, 1970, 16Н19.
191. J. W е i j 1 а г d. J. Ziegler.
Пат. США 2367251; С. А., 39,
2766 (1945).
192. Н. Lehmann. Пат. ФРГ
1052395; РЖХ, 1961, 2Л191.
193. К. Накатав а, Р. Кона-
ка, Ю. Т а н и и, Т. Такана-
си. J. Chem. Soc. Japan, Industr.
Chem. Sec., 64, 1729 (1961); РЖХ,
1962, 13Ж51.
194. И. А. Рубцов, M. В. Б a -
лякина, Л. Г. Грызло-
ва, E. С. Жданович,
H. А. Преображенский.
Труды ВНИ витаминного ин-та,
5, 17, М., Пищепромиздат, 1954.
195. И. А. Рубцов, М. В. Ба-
лякина, Е. М. П о т а к,
Е. И. Ф и л л и п о в и ч, К- В. Л и-
п е ц, А. П. Н е ч а е в Н. А. Бе-
тел ьман. Авт. свид. СССР 106842
Бюлл. изобрет., 1957, № 6, 27.
196. К. Накатав а, Т. Така-
хаси. Яп. пат. 18491 (1958);
РЖХ, 1964, 2Н215.
197. J. М а у е г, J. С t v г t п i к,
J. Zak. Чехосл. пат. 104601;
РЖХ, 1964, 2Н216.
198. R. Pasternack, Р. Reg-
п а. Пат. США 2153311, 2188777;
Offiz. Gazette unit, states patent,
501, 210 (1939); 510, 1108 (1940).
199. A. D. ’A d d i e с о. Пат. США
2847421; С. A., 53, 3084 (1959).
200. W. Haworth. Nature, 134, 724
(1934); англ. пат. 443901; С. A.,
30, 5240 (1936).
201. J. Overhoff, W. Huyser.
Пат. США 2467442, С. A., 43, 6225
(1949).
202. Голл. пат. 59301; С. А., 41, 5895
(1947).
203. О. Braenden, О. Gis-
v о 1 d. J. Am. Pharm. Assoc.,
41, 382 (1942).
204. К. H e у n s. Lieb. Ann., 568, 117
(1947).
205. O. Dalmer, К. H e у n s.
Пат. США 2189778, 2190377; С. A.,
34, 4236, 4080 (1940); герм. пат.
692897, С. А., 35, 4396 (1941).
206. Т. Merck. Англ. пат. 495050,
франц, пат. 829236; С. А., 33,
2656, 1347 (1939).
207. S. Goldschmidt. Голл. пат.
57143; С. А., 41, 4168 (1947).
208. F. S m i d t h. Датск. пат. 68836
(1949); С. А., 43, 8919 (1949).
209. F. Boedecker, Н. Volk.
Пат. ФРГ 846846; С. А., 47,
11233 (1953).
210. Hsing Huang. Пат. США
3043749; РЖХ, 1964, 8Н272.
•211. Т. R е i с h s t е i п. Швейц,
пат. 174208; Zbl., 1936, I, 113.
212. F. Boedecker, Н. V о 1 k.
Пат. ФРГ 844294; С. А., 52, 101608
(1958); пат. ФРГ 892446; С. А., 50,
4203 (1956).
213. К. Мас у так и, К. Та к а -
сэ, С. У э н о , К. И т о. Яп. пат,
21767 (1965); РЖХ, 1968, 2Н435.
214. В. М. Березовский,
Л. И. Стрельчунас. ЖПХ,
22, 1113 (1949).
215. Р. Stedehouder. Голл. пат.
59710; С. А., 41, 6025 (1947).
216. В. М. Туре и н. Авт. свид.
СССР 65477; Бюлл. изобрет. 1945,
№ 11—12, 18.
217. Герм. пат. 676011; С. А., 33, 6530
(1939).
218. Р. R е g п а, В. С а 1 d w а 1 1.
J. Am. Chem. Soc., 66, 246 (1944).
219. В. И. Be к с лер, Г. Е. Ш а л-
тыков, ЖОХ, 24, 1422, 2150
(1954); 26, 1456 (1956).
220. Я. М. С л о б о д и н, А. К. Ба-
сова. ЖПХ, 19, 172 (1946).
221. Т. R е i с h s t е i п. Англ,
пат. 428815; Zbl., 1936, I, 113.
222. В. Березовский,
Л. Стрельчунас. Авт. свид.
СССР, 73447; Бюлл. Изобрет.,
1949, № 2, 19.
223. Н. Stopsack, G. Jun-
ghanns, М. Schmidt,
R. Ring, H. Koch. Пат. ГДР
33792, 39696; РЖХ, 1965, 19H36;
1966, 24H194. *
224. Т. Reichstein. Швейц, пат.
187933, 187934; англ. пат. 428815;
Zbl., 1937, II, 4471; 1936, 1, 113.
225. Shozo Wada. Яп. пат. 1406
(1964): С. А., 60, 14600_ (1964).
55
226 A. Ruys, J. Lem me ns.
' Пат. США 2491933; С. A., 44,
3013 (1950).
227 А В о g n a г. Венг. пат. 149668;
‘ C. A., 60, 9350 (1964).
228 Z. T о 1 d i. Венг. пат. 137778;
РЖХ, 1964, 10H218.
229. В. M. Березовский,
Л И. Стрельчунас. ЖОХ,
20, 2072 (1950).
230. В. М. Березовскн й,
Л. И. Стрельчунас. Авт.
свид. СССР, 75863; Бюлл. нзобрет.;
1949, № 7, 13.
231. Т. Tanaka, A. Matsu-
moto, М. Furukawa. Яп.
пат. 9217 (1968); С. А., 69, 107007
(1968).
232. Л. О. Шнайдман Труды
ВНИ витаминного ин-та. Б, 32, М.,
Пищепромиздат, 1954.
233. Т. Като. Яп. пат. 15772 (1961);
РЖХ, 1963, 15Н170.
234. Е. И. Григорашвили,
Н. С. Золотарев,
М. Д. М о с к в н и, 3. Г. Т хо-
ре в с к а я, А. Д. X а ф а з о-
в а. Хим. фарм. ж., 1971, № 9, 50.
235. Л. О. Шнайдман. Труды
ВНИ витаминного ии-та, 4, 47, 54,
М., Пищепромиздат, 1953.
236. М. Y a m a z a k i, Т. М i о к 1.
J. Ferment. Technol., 31, 39, 86,
126, 230, (1953).
237. М. Y a m a z а к i. J. Agr. Chem.
Soc. Japan, 27, 462, 633 (1953);
28, 748, 890 (1954).
238. В. Gray. Пат. США 2421611,
2421612; С. А., 41, 5683, 5684
(1947).
239. Т. Mioki, Т. Hasega-
wa. У. S a h a s h i. J. Vita-
minol., 6, 205 (1960); C. A., 55,
18607 (1961).
210. H. H а г а с а к a, M. X и c a-
така. Яп. пат. 2172 (1954); РЖХ,
1958, 47846.
241. R. Shoemaker. Пат. США
2948659; РЖХ, 1961, 17Л386.
242. G. Farber, О. Vondro-
v a, J. L i е b s t е г, В. L и к-
§ i к. Folia biologica, 4, 348 (1958).
243. Н. F о х, М. Creighton.
Пат. США 2495246; С. А., 44,
3220 (1950).
244. S. Ruskin. Пат. США 2596103;
С. А., 47, 275 (1953); пат. США
2631155, РЖХ, 1953, 3625.
245. A. Szent-Gyorgyi. Z.
Physiol. Chem., 225, 168 (1934)’
246. К. М а и г е г, В. S с h i 1 d t
Biochem. Z„ 285, 67 (1936).
247. I. Tanaka, S. Komatsu,
T. Sa da tome. Яп. пат. 21365
(1963); С. A., 60, 2726 (1964).
248. R. Pohloudek-Fabini,
W. F й r t i g. Arch. Pharmazie ,
292. 350 (1959); C. A., 54, 2183
(1960).
249. С. СтаннмировиЬ,
Д. И 6 p a j т e p. Гласник хем.
друштва, 23—24, № 7—10. 409,
415 (1958—1959); РЖХ, 1962,
11Ж 453.
250. Н. Dahn, L. Loewe et al.
Helv. Chim. Acta, 43, 287, 294,
303, 310, 317, 320 (1960); 46,
2431 (1963).
251. J. Kenyon, M. Munro. J.
Chem Soc., 1948, 158.
252. P. Z u m a n, Z. P г о c h a z-
k a. Chem. listy, 47, 357 (1953).
253. F. H о к i n s, J. Morgan.
Biochem. J„ 30, 1446 (1936).
254. T. Tadakoro, T. T a g a -
s u g i. J. Chem. Soc. (Japan),
60, 188 (1939).
255. H. Muller, T. R e i c fa-
st e i n. Helv. Chim. Acta, 21,
273 (1938).
256. T. Reichstein,
L. Schwarz, A. Grussner.
Helv. Chim. Acta, 18, 353(1935).
257. T. Reichstein, V. Demo-
le . Festschrift fur E. C. Bareli,
Basel, 1936, 136.
258. D. H e s 1 о p E. Salt,
F. Smith. J. Chem. Soc., 1944,
225.
259. E. H a w к i n s, E. Hirst,
J. J ones. J. Chem. Soc., 1939,
246.
260. J. P e n n e y, S. Z i 1 v a, Bio-
chem. J., 37, 403 (1943).
261. F. M i c h e e 1, R. M i t t a g.
Naturwissensch., 25, 158 (1937);
Z. physiol. Chem., 247, 34 (1937).
262. W. Haworth, E. Hirst.
Erg., Vitamin, Hormon. Forsch.,
2, 160 (1939).
263. F. Ischerwood, J. Chen,
L. M a p s о n. Nature, 171, 348
(1953); Biochem. J„ 56, 1 (1954).
264. J. В u r n s, С. E v a n s. J. Biol.
Chem., 223, 897 (1956).
265. W. Berends, J. Konings.
Rec. trav. chim., 74, 365 (1955).
ГЛАВА II
ВИТАМИНЫ И КОФЕРМЕНТЫ — ПРОИЗВОДНЫЕ ₽-АМИНОКИСЛОТ
ПАНТОТЕНОВАЯ КИСЛОТА
Пантотеновая кислота — витамин В3 — представляет собой производное
ft-аланина и является П(+)-а,у-диокси-р,р-диметилбутир ил-р'-аминопро-
пиондвой кислотой (I)
сн3
НОСНаС CH(OH)CONHCH2CH2COOH
7 | “ ₽'
сн3 I
Пантотеновая кислота [1 ] имеет один асимметрический атом углерода и,
следовательно, образует два оптических антипода и один рацемат. Природ-
ная D-пантотеновая кислота, обладающая витаминными свойствами, враща-
ет плоскость поляризации вправо: [а 1о + 37,5° (Н2О) [2]. Рацемат обла-
дает 50% активности природной кислоты. Синтетическая левовращающая
Ц—)-пантотеновая кислота биологически совершенно неактивна ни на
животных [3, 4], ни на бактериях [2].
П(+)-П антотеновая кислота (I) — светло-желтое вязкое мас-
лообразное вещество. Она гигроскопична, легко растворима в воде, этило-
вом спирте, уксусной кислоте, этилацетате и диоксане, мало растворима в
эфире и высших спиртах, например амиловом [5], и практически нерас-
творима в хлороформе, бензоле [6] и многих других органических
растворителях; адсорбируется на активированном угле [7]; фуллерова
земля не адсорбирует пантотеновой кислоты [7, 8].
Пантотеновая кислота образует кристаллические соли, в виде которых
она широко применяется. *
D-П антотенат натрия кристаллизуется в бесцветных, сильно
гигроскопических иглах с т. пл. 122 — 124° С (с разл.); [а 1о + 27° (Н2О).
D-Пантотенат кальция имеет т. пл. 193,5—195° С [9];
la lio + 26,8° (Н2О) [10]; он легко растворим в воде с нейтральной или сла-
бощелочной реакцией, хуже —в глицерине, мало — в спирте и почти не-
растворим в ацетоне и эфире. DL-Пантотенат кальция оптически неактивен,
сильно гигроскопичен.
О(+)-П антотенол (II) — соответствующий пантотеновой кислоте
спирт, HOCH2C(CH3)2CH(OH)CONHCH2CH2CH2OH, т. пл. 34—46° С (из
II
ацетона), [а ] р +31,8° (Н2О) [11], обладает равноценной пантотеновой
кислоте витаминной активностью на животных.
ХИМИЧЕСКИЕ СВОЙСТВА ПАНТОТЕНОВОЙ КИСЛОТЫ
Пантотеновой кислоте присущи амфотерные свойства с преобладанием
кислого характера [7, 12]. При нагревании с кислотами или щелочами
она гидролитически расщепляется по амидной связи на £ -аланин (III) и
пантоевую кислоту, которая в кислой среде легко превращается в а-окси-
p-диметил-у-бутиролактон, пантолактон (IV):
HOCH2C(CH3)2CH(OH)CONHCH2CH2COOH ->
I
-> H2NCH2CH2COOH + СН2С(СН3)2СН(ОН)СО
in I---------------------о—.—2d
IV
57
Пантотеновая кислота, в состав молекулы которой входят первичная и
вторичная гидроксильные группы и карбоксильная группа, способна обра-
зовывать простые и сложные эфиры, хлора н гидр иды, азиды и другие произ-
водные. Простые эфиры D (+)-пантотеновой кислоты — метиловый, этило-
вый, пропиловый, изобутиловый и другие—сиропообразные вещества
[13, 14]. Сложные эфиры: ацетат — маслообразное вещество, перегоняю-
щееся в высоком вакууме [6]; п-нитробензоат — т. пл. 137—138° С,
[a Id + 4,5°. Получен ряд эфиров пантотеновой кислоты одновременно по
окси- и карбоксильным группам, например этиловый эфира-О-ацетилпанто-
теновой кислоты и др. [15].
D-Пантотеновая кислота образует D-пантотенамид (V) [16, 17]
HOCH2C(CH3)2CH(OH)CONHCH2CH2CONH2,
V
маслообразное вещество; DL-пантотенамид имеет т. пл. 106—108° С [17].
Пантотеновая кислота образует соль с холином [18]; эта соль обладает
биологической активностью соединений, входящих в ее состав. Синтезиро-
ваны бетаинпантотенат [19], D-пантотеноил-Л-цистеин [20], ди-И-панто-
теноил-Л-цистеин [20, 211 и др.
Пантотеновая кислота в водном растворе способна окисляться марган-
цовокислым калием, но устойчива к кислороду воздуха.
Восстановление карбоксильной группы пантотеновой кислоты (I) в
оксиметильную группу дает пантотенол (II) [22].
СТРОЕНИЕ ПАНТОТЕНОВОЙ КИСЛОТЫ
Пантотеновая кислота (I) выделена из печени [7] посредством автолиза,
последующей адсорбции посторонних оснований на фуллеровой земле,
адсорбции витамина на активированном угле с последующей элюцией, по-
лучения бруциновой соли, экстракции хлороформом и последующего распре-
деления бруциновой соли между хлороформом и водой, превращения в
кальциевую соль и перекристаллизации из различных растворителей.
Строение D-пантотеновой кислоты (I) установили в 1938 г. Вильямс с
сотр. [23] на основании следующих данных.
В молекуле пантотеновой кислоты (I) содержится одна свободная кар-
боксильная группа, что установлено по образованию из пантотеновой кисло-
ты сложного эфира (VI) с диазометаном или метиловым спиртом; эфир
подвергается омылению в свободную пантотеновую кислоту [231. Таким
образом, из пяти атомов кислорода, содержащихся в молекуле, два отно-
сятся к карбоксильной группе, а два других — к свободным гидроксильным
группам [24], что доказано рядом реакций. Так, определением активного
водорода по Церевитинову в пантотеновой кислоте установлено наличие
трех активных атомов водорода (один из которых относится к гидроксилу
карбоксильной группы); при этерификации различными кислотами обра-
зуются сложные эфиры, например с уксусной кислотой — диацетилпанто-
теновая кислота (VII), легко подвергающаяся гидролизу обычным методом.
При взаимодействии с ацетальдегидом, ацетоном и бензальдегидом под
влиянием минеральных кислот образуются соответствующие ацетали или
кетали, например с ацетоном — О-изопропилиденпантотеновая кислота
(VIII); эти производные вновь переходят в исходную пантотеновую кислоту
в разбавленных водных кислотах. Последний тип реакций показывает на
присутствие гидроксильных групп в виде а, р-, а,у- или а.о-гликолей.
На это же указывают гидрофильные свойства пантотеновой кислоты: хоро-
шая растворимость в воде, так же как и ее сложного эфира, и несколько
худшая растворимость ее простого эфира (схема 11).
58
Схема 11
Установление строения пантотеновой кислоты реакциями ее превращений
и расщепления
ососн3 ососн3 оС5с^о3
ch2c(ch3)2chconhch/:h2cooh ch2c(ch3)2chconhch2ch2cooh
VIII
НОСН2С(СН3)2СН(ОН)СОННСН2СН2СООН =s=fc носн2с(снз)2сн (OHjcONHCHjCHjCOOCH,
7 ?n а vi
Пантотеновая кислота
I
I I SO^HCl^CH.COOH
носн2с(сн3)2сн(он)соон H2NCH2CH2COOH ---]Г J
X III
| ОСОСН3 IX
сн2с(сн3)2сн(он)со——сн2с(сн2)2снсо
I-----о-------1 I——О--------1
XI
I
СН3СОСН3
Слабая цветная реакция с хлорным железом свидетельствует о присут-
ствии гидроксильной группы в а-положен ии к карбоксильной группе, а
константа ионизации, равная 3,9-10-5, вызывает предположение, что вто-
рая гидроксильная группа находится вр- или у-положении.
При щелочном гидролизе пантотеновой кислоты выделен р-аланин (III)
[25], который идентифицирован как р-нафталинсульфо-р-аланин (IX)
[26]. Вторая часть молекулы выделена в виде кристаллического оксилактона
(IV) после нагревания подкисленного раствора [25] и последующей
экстракции [27,28]. Алифатическую диоксикислоту (X), из которой
образуется оксилактон, в свободном виде выделить не удается.
Отсюда вытекает, что имеющийся в молекуле пантотеновой кислоты
один атом азота входит в виде аминогруппы в состав р -аланина (III) и связан
с молекулой диоксикислоты (X) по ее карбоксильной группе амидной свя-
зью. Это следует из того, что азот пантотеновой кислоты не дает реакций
на амино-, имино- или четвертичную аминогруппу, что также указывает
на наличие остающегося неразмещенным пятого атома кислорода в карбок-
силе амидокислотной группы.
Естественно, что оксилактон (IV) при титровании его водного раствора
на холоду показывает отсутствие карбоксильной группы; при нагревании
он поглощает один эквивалент щелочи, что указывает на наличие одной
карбоксильной группы в виде лактона [29]. Оксилактон (IV) содержит
одну гидроксильную группу, связанную в виде внутреннего эфира (лакто-
на), а вторую — свободную, что определено по наличию одного активного
атома водорода и образованию моноацетата (XI) при ацетилировании,
а, Р-Положение двух гидроксильных групп в молекуле лактона (IV) исклю-
чается, так как пантотеновая кислота не окисляется тетраацетатом свинца и
йодной кислотой в водном растворе [5], т. е. не подвергается реакциям,
характерным для а-гликолей. Расположение одной гидроксильной группы
в a-положении к карбоксильной группе диоксикислоты (X) следует из того,
что диоксикислота дает положительную реакцию с хлорным железом [30]
и при разложении серной кислотой образует окись углерода [5]. Следова-
тельно, имеются все данные полагать, что образование лактона происходит
по второй гидроксильной группе, находящейся в у-положении.
59
Вместе с тем в молекуле оксилактона (IV) было обнаружено присутствие
диметильной группы, что следовало из образования ацетона при окислении
оксилактона 129]; реакцией Куна — Рота показано местоположение ме-
тильных групп в боковой цепи.
Окончательное строение оксилактона (IV) как а-окси-р,р-диметил-у-
бутиролактона [28] было установлено превращением его в ранее известную
а, а-диметил-р-оксипропионовую кислоту (XIV). При реакции с метилмаг-
нийиодидом оксилактон дает диоксидиметилкарбинол (XII), который при
окислении тетраацетатом свинца превращается в а,а-диметил-(3-оксипропио-
новый альдегид (XIII), переходящий при окислении щелочной гидроокисью
серебра в а ,а-диметил-р-оксипроп ионовую кислоту (XIV).
СН3 СН3
I CH3MgJ | уСН3
СНгССН(ОН)СО---------* НОСН2ССН(ОН)СХ ->
сн3 сн3 I СНз
он
----о-----
IV XII
сн3 сн3
I I
— НОСН2ССНО -> НОСН2ССООН
I I
СН3 СНз
XIII XIV
СИНТЕЗ ПАНТОТЕНОВОЙ КИСЛОТЫ
Конденсация пантолактона с р-аланином
Пантотеновую кислоту синтезировали в 1940 г. Вильямс с сотр. [28,
30, 31], а затем ее полный синтез одновременно произвели Рейхштейн,
и Грюсснер [32], Кун и Виланд [3], Стиллер, Харрис, Финкельштейн,
Керестези и Фолькерс [2].
Все пути синтеза пантотеновой кислоты в основном сводятся к конденса-
ции двух компонентов: а,у-диэкси-|3,р-диметилмасляной кислоты, ее эфи-
ров или производных ср-аланином, его эфирами и солями. Сущность реак-
ции состоит в образовании амидной связи между карбонильным атомом
углерода и аминогруппой, поэтому она может быть осуществлена обычными
методами органической химии, применяемыми для получения амидов кис-
лот. Первоначальный синтез пантотеновой кислоты проведен конденсаци-
ей синтетического этилового эфира р-аланина с хлорангидридом ацетилди-
оксикарбэновой кислоты, выделенной из гидролизата пантотеновой кисло-
ты, с последующим точным гидролизом продукта конденсации [25]; полу-
чен невысокий выход.
Значительно лучший синтез D(+) пантотеновой кислоты состоит в кон-
денсации при нагревании до 70° С D(—)-пантолактона — а-окси*р,р-ди-
метил-т-бутиролактона (IV) — с этиловым (или метиловым) эфиром Р-ала-
нина (XV) [30—32]; выход составляет 50%. Реакция протекает через про-
межуточное образование дегидратированной формы эфира пантотеновой
кислоты (XVI) и его гидролиз:
СН3
СНз I
I СНз-С------снон
СНз-С-----снон ._________________ 1 ] /ОН
, 0х ^NHCH^HjCOOCjHs ->
XVI
IV
НОСН2С(СНз)1СН(ОН)СОННСН2СН1СООН
. I
60
Если же применить бензиловый эфир Р-аланина, то образовавшийся
бензиловый эфир пантотеновой кислоты подвергают восстановительному
гидролизу [3]. Применение эфиров Р-аланина связано с относительно не-
высокими выходами пантотеновой кислоты из-за их способности полимери-
зоваться при стоянии [33].
Интересно отметить, что применение свободного р-аланина также дает
невысокий выход пантотеновой кислоты. P-Аланин конденсируют с D(—)-
пантолактоном при температуре 150° С с образованием пантотеновой кисло-
ты в виде маслообразного продукта с выходом 39% [34 ]; конденсацию можно
провести при более низкой температуре в среде метилового спирта в присут-
ствии метилата натрия [35].^
Однако если применить для конденсации с пантолактоном соли р-алани-
на или если после конденсации со свободным р-аланином образовавшуюся
пантотеновую кислоту выделять в виде соли, то выход значительно повы-
шается. Так, для конденсации с пантолактоном (IV) используют натриевую
соль р-аланина [30, 32, 36, 37]. Конденсацию ингридиентов проводят или
нагреванием их смеси, или кипячением в среде безводного спирта; после
удаления растворителя получают чистую соль пантотеновой кислоты [38t
39]. Конденсация сухой натриевой соли Р-аланина с пантолактоном в инерт-
"ном растворителе при температуре выше 90° С приводит к пантотенату натрия
с выходом 92 % [36 ].
Так как наиболее распространенной формой пантотеновой кислоты яв-
ляется ее кальциевая соль, то обычно конденсацию D(—)-пантолактона
проводят с кальциевой солью р-аланина или с основанием Р-аланина в при-
сутствии гидроокиси кальция, карбида кальция или мела нагреванием в
безводном метиловом спирте; пантотеновую кислоту получают с выходом
95% [40—44]. Этот путь синтеза представляет значительный технический
и нтере~ '
Удобно применение в качестве растворителя для конденсации кальциевой
соли р-аланина с £>(—)-пантолактоном смешанного метилэтилового эфира
этиленгликоля; реакция протекает при 25—30° С, и пантотенат кальция
выделяется из реакционной смеси в аналитически чистом состоянии с выхо-
дом 94% [9]^.
Для освобождения пантотената кальция от соли р-аланина можно при-
менить ионообменные смолы карбоксильного типа [45]. Для перекристал-
лизации в случае надобности применяют смесь вещь! и метилового спирта
[101,
* Следует отметить, что соли пантотеновой кислоты [1] получаются про-
стым методом синтеза с высоким выходом из пантолактона (IV) и свободного
Р-аланина [III], если конденсацию проводить в присутствии вторичных
или третичных аминов с последующим прибавлением окиси кальция или
этилата натрия [46]:
СНз
СНз-С1-----СНОН R2NH или RsN
| [ + H2NCH2CH2COOH------------>
HtC СО III ^Нз
\0/ -> HOCH2CCH(OH)CONHCHlCH2COOH
IV CHS
I
Конденсацию пантолактона (IV) и Р-аланина (III) в присутствии диэтил-
амина проводят в спиртовом растворе; по окончании реакции отгоняют
диэтиламин и прибавляют солеобразующий агент. Своеобразную роль
амина в этой реакции нельзя объяснить его каталитическим характером,
так как его требуется не менее, чем равномолекулярное количество. Выход
61
пантотената кальция составляет 89—91 % [46, 471, пантотената натрия —
75% [46L —-------•
Для получения пантотената кальция применяют также конденсацию
0-аланина (III) с а.у-диокси-Р.р-диметилбутиронитрилом (XVII) с одно-
временной обработкой гидратом окиси кальция [481:
HOCH2C(CH3)2CH(OH)CN + H2NCH2CH2COOH —► Пантотенат кальция.
XVII III
Представляет особый интерес возможность избежать процесса получе-
ния р-аланина и проводить конденсацию пантолактона (IV) непосредственно
с циануксусным эфиром (XVIII) в ледяной уксусной кислоте при одновре-
менном каталитическом восстановлении с палладиевым катализатором
при 40° С 149].
СНз
| СНз
СН3-С-----СНОН [HJ/Pd I
I | + СНСНгСООСгНь----> НОСН2 CCH(OH)CONHCH2CH2COOH
С° XVIII (!н3
ХУ
Среди других синтезов пантотеновой кислоты интересно остановиться
на ее получении через пантотеновый спирт (II) [22]. Первоначально полу-
чают p-аминопропанол (XXI) из этиленхлоргидрина (XIX) и цианистого
натрия с последующим каталитическим восстановлением нитрильной груп-
пы Р-оксипропионитрила (XX) в аминогрупп}' [22]. р-Аминопропанол
(XXI) конденсируют при нагревании с DL-пантолактоном (IV) в DL-панто-
тенол (т. пл. 60—61 ° С, т. кип. 118—120° С при 0,2 мм) [50]; для получения
П(+)-пантотенола (II) в конденсацию вводят £)(—)-пантолактон (IV)
[22, 50—52]. Кристаллический D-пантотенол получен с т. пл. 35—46° С,
[а]о+30,8° (Н2О) [52]. При окислении марганцовокислым калием или бари-
ем он образует природную D-пантотеновую кислоту (I).
С1СН2СН2ОН + KCN -> CNCH2CH2OH -*• H2NCH2CH2CH2OH
XIX XX XXI
I СНз
CHS-C-----СНОН I
I | + H2NCH2CHaCH2OH -> HOCH2CCH(OH)CONHCH1CH1CH2OH ->
H2C CO XXI jj
XX
IV
-> HOCHaC(CHs)2CH(OH)CONHCH2CH2COOH
I
Следует, однако, иметь в виду, что при окислении в щелочной среде
происходит частичная рацемизация П-пантотеновой кислоты.
При конденсации рацемического пантолактона с Р-аланином образуется
рацемическая оптически недеятельная ОЛ-пантотеновая кислота; она может
быть расщеплена на свои оптически активные компоненты кристаллиза-
цией ее хининовых [3, 4], стрихниновых или цинхониновых [53] солей;
для выделения природной (-г)-формы предпочтительнее применять цинхо-
ниновую соль как более легко кристаллизующуюся [54]. Хининовая соль
пантотеновой кислоты расщепляется с помощью гидрата окиси бария.
Пантотенат кальция можно получить конденсацией а.^-диокси-р.р-ди-
метилбутироамида (ХХ11) с пропиолактоном (XXIII) при 100—150°С в
62
отсутствие влаги с последующей обработкой мелом и осаждением метиловым
спиртом; реакция проходит с высоким выходом [55].
HOCH2C(CH,)2CH(OH)CONHa + СН2СН2СО -> Пантотеновая кислота
XXII I—O—I 1
XXIII
Помимо приведенных выше схем, для синтеза пантотеновой кислоты
использована реакция конденсации циангидрина, применяемого в виде
циклического изомерного соединения XXIV, с гидрохлоридом этилового
эфира 0-аланина (XV) [56]:
СН, СН,
СН,—А----СНОН СН,—С*----СНОН
II II
Н2С C=NH 4- HaNCH2CI I2COOC2HS -> Н2С C=NCH,CH,COOC,He->
\ / XV \ /
xoz xoz
XXIV XXV
-> HOCH2C(CH,)2CH(OH)CONHCH2CH2COOH
I
Реакция протекает в среде хлороформа в направлении образования
соли сильной кислоты с наиболее сильным основанием. Затем образующееся
соединение XXV в виде натриевой соли подвергают окислительному гидро-
лизу перекисью водорода, приводящему к получению пантотеновой кисло-
ты (I). Циклический изомерный циангидрин (XXIV) получают при конден-
сации бисульфитного соединения а,а-Диметил-0-оксипропионового альде-
гида (XIII, см. с. 60) с цианистым натрием с последующим насыщением
слоя, содержащего циангидрин, сухим хлористым водородом.
Оригинальный синтез пантотеновой кислоты (I) и £)(+)-пантотенола
(II) осуществлен конденсацией D(—)-пантолактона (IV) с 1,6-диамино-Д3-
гексеном (XXVI) при последующем нагревании в вакууме без раствори-
теля в 1,6-бис- (а,7-Диокси-0,0-диметилбутириламино)-Д3-гексен (XXVII),
который ацетилируют уксусным ангидридом, затем расщепляют озоном в
этилацетате при — 65° С и после окисления перекисью водорода прев-
ращают в диацетат £>(+)-пантотеновой кислоты (XXVIII) [57]:
сн,
। 3
сн,-с-снон
3 / \ .
н2с. .со +
о
IV
(снзсо)2о,оз. Н2Ог> СНзС00СНгС('снДсн|'0С0СНз)С0Ь|НСН2Сн^С00н Пантотеновая
XXVIII I
Если после озонирования вещества подвергнуть гидрированию, то
получается £>(+)-пантотенол.
Синтез пантолактона
В одном из важнейших методов синтеза а-окси-0,Р-диметил--[-бутиро-
лактона — пантолактона (IV) [2, 32, 58, 59] — исходят из изомасляного
альдегида (XXIX) с т. кип. 61° С, который пату чают окислением изобути-
лового спирта бихроматом калия (или натрия) в разбавленной серной кисло-
те при температуре 90° С с выходом 35% [60] или каталитическим дегидри-
63
рованием изобутилового спирта кислородом воздуха на медном или серебря-
ном катализаторе при 230—300° С с выходом 80—90% 161].
При проведении альдольной конденсации изобутилового альдегида
(XXIX) с формальдегидом в присутствии поташа образуется а,а-диметил-
Р-оксипропионовый альдегид (XIII) с т. пл. 96—97°С; выход 76% [62].
Циангидриновым синтезом конденсацией альдегида (XIII) с синильной
кислотой или лучше [63] путем конденсации с цианистым калием в присут- j
ствии хлористого кальция или с цианистым натрием, а также в результате
взаимодействия бисульфитного соединения а,а-Диметил-Р-оксипропионового
альдегида (XIII) с цианистым калием через циангидрин (XXX) получают g
а, 7-диокси-р, р-диметилмасляную кислоту (X); при ее лактонизации обра- н
зуется рацемический пантолактон (IV) с т. пл. около 80° С, с выходом
77—81%, считая на а,а-диметил-р-оксипропионовый альдегид (XIII):
СНз СНз
нсно + нссно -> несносно ->
СНз СНз
сн»
I
HOCH2CCH(OH)CN
I
СНз
XXIX XIII XXX
СНз
СНз
НОСН2ССН(ОН)СООН -> СН2ССН(ОН)СО
СНз
X
СНз
—о-
IV
Гидролиз промежуточного циангидрина (XXX) может быть проведен в
присутствии карбонатов щелочных металлов [§4 ]. Сделаны различные улуч-
шения и упрощения синтеза [65—67]. В качестве планирующего агента
предложен ацетонциангидрин — в этом случае выход пантолактона на
а,а-Диметил-Р-оксипропионовый альдегид составляет 61,6% [68].
Природную D (-(-)-пантотеновую кислоту лучше получать не расщепле-
нием ее рацемата, а непосредственно из оптически активной левовращаю-
щей формы пантолактона и Р-аланина. DL-Пантолактон предварительно
превращают в соответствующую кислоту; расщепление а, у-ди окси-р, р-ди-
метилмасляной кислоты (X) на оптические антиподы производят дробной
кристаллизацией ее солей с алкалоидами — с хинином [2, 321 хинидином,
цинхонином J69], бруцином [70—72j или эфедрином [73]. Так, натриевую
соль а, -jf-диокси-р, р-диметилмасляной кислоты (X) подвергают кристалли-
зации с половиной эквивалента гидрохлорида хинина при этом преимущест-
венно выкристаллизовывается соль хинина с (+)-формой кислоты, из
которой получают D(—)-пантолактон, имеющий т. пл. 92—93° С,
Ыс—49,8°, с выходом 71 %. Помимо оптически активных алкалоидов
для расщепления рацемического пантолактона на оптические антиподы
применяют оптически активные амины. D(—)-Пантолактон с выходом 59%
получают при использовании для разделения рацемата (—)-а-1-(п-нитро-
фенил)-2-аминопропандиола-1,3 в водной среде [51, 74—781. Для расщепле-
ния применяют также (-Ь)-а-фенилэтиламин Г79, 80] и 1-нафтилэтиламин
181.]. -------
Для рацемизации оптический антипод в виде натриевой соли а, у-диок-
си-р, р-дпметилмасляной кислоты нагревают при 100—150° С [2, 32, 82].
Водный раствор образующегося рацемического соединения подвергают
вновь расщеплению, что постепенно приводит к почти полному переведе-
нию рацемата в необходимый оптический изомер.
64
Синтез р-аланина
'> Второй компонент, необходимый для синтеза пантотеновой кислоты, —
Г 0-аланин, или p-аминопропионовую кислоту (III) — получают синтетически
по нескольким методам, которые рассматриваются ниже.
L I.Синтез p-а л а н и н а из имида янтарной кислоты
р-Аланин (III) получают [83] по реакции Гофмана в результате взаимо-
L действия сукцинимида (XXXI) с гипобромитом или гипохлоритом натрия
?' или калия в присутствии щелочи через промежуточный бромамид (XXXII).,
Г который при нагревании с избытком щелочи подвергается расщеплению и
перегруппировке в изоцианат (XXXIII): это соединение затем разлагают в
[ первичный амин (III) с отщеплением двуокиси углерода:
L КОВг КОН Н2О
L CHSCH2CO----> СН2СН2СООН —> СН2СНаС00Н ——>
L СО---NH О=С—NHBr O=C=N
£ XXXI XXXII XXXIII
-> [ HOC NCH2CH2COOH -> O=CNHCH2CHaCOOH’
OH
----> H2NCH2CH2COOH
-COa
III
OH
Выход р-аланина по этому методу получения составляет 41—45%;
|из реакционной смеси вещество лучше выделять в виде метилового или
этилового эфира [84].
2. Синтез Р-аланина окислением аминоспиртов
Р-Аланин (III) получают электрохимическим окислением 0-аминопропа-
нола (XXI) в электролизерах без диафрагмы на свинцовом электроде в
растворе серной кислоты при плотности тока 1 А/дм2; выход составляет
около 80% [85].
H2NCH2CH2CH2OH -> H2NCH2CH2COOH.
XXI III
3. Синтез Р-аланина из диоксима циклогекса н-
диона перегруппировкой Бекмана
Диоксим циклогександиона-1,4 (XXXIV), являющийся, по-видимому,
смесью син- и анлш-форм, при перегруппировке Бекмана дает смесь изо-
мерных лактамов XXXVa и XXXV6 [86]:
хн,-сн. сн-сн2 ch2-ch2-nh,
HOW=cf >C=NOH-*~O=Cf ^.С=О и О=С< ^С=О-—S-Алаиин
YHj-CH, NH-CH.-CH, г
£ XXXIV СН—СН2 XXXV б
XXXVa
из которых лактам XXXV6 при гидролизе образует р-аланин (III). Пере-
k группировке подвергается «диэфир», полученный из диоксима циклогек-
s сандиона-1,4 с n-толуолсульфохлоридом; р-аланин из продуктов перегруп-
I пировки выделен осаждением нитраниловой кислотой.
4. С и н т е з 0-а ланина аминированием аммиаком
производных пропионовой и акриловой кис-
лот
0-Аланин (III) получают аммонолизом 0-галогенпропионовой кислоты
187, 88]:
3-69
65
JCHSCH»COOH---
NH3
ClCHaCH2COOH—
H2NCH2CH2COOH
III
P-Аланин выделяется в смеси с иминодипропионовой кислотой, из которой
его получают с выходом 50%. Дополнительно 6% можно получить в резуль-
тате сплавления иминодипропионовой кислоты с фталевым ангидридом и
гидролизом Р-фталоиламинопропионовой кислоты [891; для очистки приме-
няют ионообменные смолы.
Важное практическое значение имеют методы, в которых исходят из
акрилового эфира или акрилонитрила. р-Аланин (III) получается при дей-
ствии концентрированного водного аммиака на акриловый эфир (XXXVI)
с последующим омылением эфира р-аланина (XV) кипячением с раство-
ром гидрата окиси бария [90]:
NH3 + СН2 = СНСООС2Н5 -> H2NCH2CH2COOCaH5—
XXXVI XV -> h2nch2ch2cooh
NH3 -1- СН2 = CHCN -> H2NCH2CH2CN-------- 111
XXXVII XXXVIII
Первоначальный выход 30—34% может быть повышен до 85% повторной
обработкой растворов после отделения Р-аланина водным аммиаком; очища-
ют Р-аланин кипячением с диизопропилаМином или триэтиламином [91 ].
Цианэтилирование аммиака осуществляют при взаимодействии акрило-
нитрила (XXXVII) с водным аммиаком под давлением при 75—150° С
192—96]. Реакция образования p-аминопропионитрила (XXXVIII) сопро-
вождается получением вторичного (XXXIX) и третичного (XL) аминов
[97, 981, находящихся в состоянии подвижного равновесия [99]:
—NH3 —NH3
HsNCHaCH2CN“zr± HN(CH.CH2CN)2 ~=± N(CH2CH2CN)S.
+NH3 +NH3
XXXVIII XXXIX XL
Образование бис- (Р-цианэтил) амина (XXXIX) в значительной степени
снижает выход Р-аминопропибнитрила (XXXVIII)—До 31—33%. Боль-
шой избыток аммиака, повышение температуры и введение акрилонитрила
под поверхность раствора аммиака способствуют повышению выхода Р-ами-
нопропионитрила (XXXVIII) [100] до 59% [96].
После фракционного отделения p-аминопропионитрил с выходом 93%
гидролизуют в р-аланин (III) соляной кислотой [92—96], серной кислотой
или щелочью [92, 101 ]. ' ’
Вторичный ’амин (XXXIX) может быть также использован для полу-
чения р-аланина (III) посредством конденсации с фталевым ангидридом
при 200е С в соответствующее Р-фталимидопроизводное XLI с последующим
расщеплением; реакция, по-видимому, протекает через промежуточный
продукт присоединения, из которого Р фталимидопропионитрил (XLI)
образуется при отщеплении акрилонитрила (XXXVII) [102]:
о-СвН4 О 4- HN
. 4CH2CH2CN
xxxix
CH2CH2CN
o-CeH4
CON
,ch2ch2cn~
ch,ch2cn
соон
66
соч
-> 0-с.н; \ch2ch2cn + СНа = CHCN + н2о
/ со/
XLI XXXVII:
При взаимодействии акрилонитрила с, ацетамидом (вместо аммиака)
также происходит его аминирование в присутствии каталитических коли-
честв натрия в бензоле. Образовавшийся р-ацетиламинопропионитрил
гидролизуют в р-аланин разбавленной серной кислотой [103].
5. Синтез Р-аланина аминированйем фталими-
дом производных пропионовой' и акриловой-
кислот
В синтезе Р-аланина (III) из p-фталимидопропиоальдегида (XLIII)
используется акролеин (XLII), для получения которого известны эффектив-
ные технические методы. Акролеин конденсируют с фталимидом в среде
безводного этилового спирта при каталитическом участии этилата натрия;
образующийся Р-фталимидопропиоальдегид (XLIII) окисляют марганцо-
вокислым калием в соответствующую кислоту (XL1V), которая при кипя-
чении с 20%-ной соляной кислотой дает р-аланин (III) [104]:
/СО. ,со.
/ \ (
o-CeH4 NH + СН2 = СНСНО -> о-С«Н4
NCH2CHaCHO ->
CO'
XLI I
"CO
XLIII
-> о-С«Н4 NCHaCH2COOH -> H2NCHaCH2COOH
CO
XLIV III
Через фталимидное производное протекает и синтез р-аланина (III)
из p-бромпропионового эфира (XLV), который конденсируют с фталимидом,
промежуточное соединение XLVI с водой превращают в Р-фталоиламидо-
пропионовый эфир (XLVII) и гидролизуют его с 30%-ной соляной кислотой
в Р-аланин (III) [105]:
со.
CO
0-CeH4 NH + BrCHaCH2COOC2H6 -> o-CeH4 ^СНаСНаСООС2Н5 -►
CO'
co
XLVI
XLV
^CONHCHaCHaCOOCjHe
н4 -> h2ncu2ch2cooh
СООН
XLVI I III
Использование фталимида для аминирования акрилонитрила имеет
большое практическое значение. При конденсации фталимида с акрило-
нитрилом (XXXVII) при каталитическом участии гидрата окиси триметил-
фениламмония или триметилбензиламмония образуется фталимидопропио-
нитрил (XLI), который гидролизуют соляной кислотой в хлоргидрат Р-ала-
нина, а затем при действии гидрата окиси лития или ионообменных смол
выделяют свободный Р-аланин (III) [106—1091:
3"
67
NH .+ CH2
CHCN---«,-C.H* NHCH2CH2CN-------►
73% \ / 89—92%
XC0
XXXVII
XLI
-> H2NCH2CH2C00H
III
В качестве катализатора можно применить и этилат натрия, но реакция
протекает несколько хуже и дает менее чистый продукт.
По одному из простых синтезов р-аланин получают методом присоеди-
нения фталимида к акриловому эфиру (XXXVI) под влиянием гидрата
окиси триметилфениламмония; реакция протекает с высоким выходом
[107]. Если производить непродолжительный гидролиз р-фталимидопро-
пионового эфира (XLVI) соляной кислотой, то можно в нем гидролизовать
только эфирную группу (соединение XLIV); в более жестких условиях
образуется гидрохлорид Р-аланина, который превращают в свободную
аминокислоту (III) обработкой окисью свинца и затем сероводородом:
н+ СН2 = СНСООС2Н6----> О-С.Н4
80% \
.СО.
1СН2СН2СООС2Н5
НС1
4 ч
О'
XXXVI
XLVI
/СО
о-СвН4 \сН2СН2СООН ---> H2NCH2CHiCOOH • НС1-> H2NCH2CH2COOH
\ / 83% 82%
XCOZ
XLIV III
Для выделения р-аланина из его гидрохлорида, помимо способов ука-
занных выше, хорошим является метод [107], основанный на взаимодействии
с. избытком окиси этилена на холоде; образующиеся при реакции этилен-
Хлоргидрин и этиленгликоль отгоняют с водяным паром. Выход кристалли-
ческого Р-аланина с т. пл. 192—197° С составляет 72%. Применение диэтил-
амина для этих же целей при pH 7,6 также дает хорошие результаты [89].
6. Синтез р-аланина гидрированием циануксус-
ной кислоты и ее эфиров
Простой метод получения Р-аланина (III) состоит в каталитическом
гидрировании циануксусного эфира (XVIII) [ПО] с последующим гидро-
лизом эфирной группы соединения XV водным раствором гидрата окиси
бария:
CNCH2COOC2H5 -> H2NCII2CH2CO(X^IL -► H2NCH2CH2COOH;
XVIII XV III
выход составляет 72%.
При гидрировании циануксусной кислоты [111] или ее соли [112 ] в спир-
товом растворе со скелетным никелевым катализатором реакция образова-
ния р-аланина при 90° С проходит за 15 мин, прибавление аммиака способ-
ствует более полному гидрированию нитрильной группы в первичную ами-
ногруппу за счет снижения количества вторичных аминов, всегда образую-
щихся при каталическом гидрировании нитрилов.
68
7. Синтез р-аланина из полимеров
Предложен метод получения Р-аланина (III) гидролизом разбавленной
минеральной кислотой полимеров р-аланина (молекулярная масса до 80 000),
образующихся из акриламида (катализатором может служить трет-бути-
лат натрия) 1113—115], или нагреванием этиленциангидрина при 70—250° С
со щелочным катализатором (аммиак, едкое кали) (116—119]:
СН2 = CHCONHg
Н8О
HOCH2CH2CN---> HOCH8CH2CONH8-----
— Н2О
-^ ... CH8CHjCD(NHCHsCH8CO)nNH ... -> H8NCH8CH8COOH
III
Выход Р-аланина 60—90 и 65% соответственно.
БИОЛОГИЧЕСКОЕ ЗНАЧЕНИЕ ПАНТОТЕНОВОЙ КИСЛОТЫ. ЗАВИСИМОСТЬ
МЕЖДУ СТРОЕНИЕМ И БИОЛОГИЧЕСКОЙ АКТИВНОСТЬЮ.
АНТИВИТАМИНЫ
Пантотеновая кислота — универсальный витамин; она содержится в
протоплазме клеток животных и растительных тканей и синтезируется не-
которыми плесенями, микроорганизмами и различными растениями.
Еще в 1933 г. было установлено, что природные соединения неизвест-
ного строения, названные Вильямсом «пантотеновой» (т. е. вездесущей)
кислотой, стимулируют развитие дрожжей [1 ]. В 1939 г. нашли, что панто-
теновая кислота и цыплячий антидерматический фактор идентичны (25, 120].
Недостаток пантотеновой кислоты в пище вызывает уменьшение выводи-
мости цыплят, понижение их жизнеспособности. Гиповитаминоз пантоте-
новой кислоты заключается в потере аппетита, вялости, огрубении волосяного
покрова, поседении волос у крыс и серебристо-черных лисиц (121], в рас-
стройстве движений, эксудатах вокруг глаз и в других местах. Пантотено-
вая кислота необходима для нормального воспроизведения животных,
например свиней; она способствует лучшему усвоению протеина и жиров,
повышает привес птицы и увеличивает яйценоскость [122]. В пантотеновой
кислоте нуждаются все позвоночные.
Пантотеновая кислота, входя в ферментные системы, является важней-
шим биокатализатором реакций ацилирования, протекающих в организме.
Она участвует в жировом и углеводном обмене. Характерно, что пантоте-^
новая кислота внутри клеток находится в связанном состоянии в виде ко-
фермента А; в свободном виде она содержится в плазме крови.
Природная D (Ч-)-пантотеновая кислота по своей биологической актив-
ности в высокой степени специфична. Как указывалось, ее оптический антит
под биологически неактивен; рацемат обладает 50%-ной активностью. Био-
логически активны только соли и сложные эфиры пантотеновой кислоты,
образованные по карбоксильной группе, например этиловый эфир (4] и др.
Сложные эфиры, образованные по гидроксильной группе, например ацетаты
(6], бензоаты, а- и 7-монофосфаты и дифосфаты (127], неактивны. Таким
образом, биологическая активность пантотеновой кислоты проявляется
только при наличии обеих свободных гидроксильных групп.
Введение в молекулу третьей гидроксильной группы в одну из боковых
метильных групп 0-положения дает окси пантотеновую кислоту, получае-
мую из 0-аланина (III) и а-окси-0-мегил-0-оксиметил--[-бутиролактона
(XLVIII) (1281
69
СН2ОН
НОСН2ССН(ОН) CONHCH2CH2COOH
I
СНз
XLVI 11
Биологическая активность оксипантотеновой кислоты составляет от
1,5 до 25% активности пантотеновой кислоты при испытании на различных
микроорганизмах.
Замещение карбоксильной группы на оксиметильную дает пантотенол
(II) 122]
HOCH2C(CH3)2CH(OH)CONHCH2CH.,CH2OH,
'< II
сохраняющий для животных полностью активность пантотеновой кислоты.
Биологическая активность пантотенового спирта объясняется его способ-
ностью переходить в организме в пантотеновую кислоту. Ацетилпантоте-
нол биологически неактивен {1291.
Замещение карбоксильной группы на альдегидную в виде ее ацеталя
дает пантотенальдиэтилацеталь (XLIX)
Н0СН2С(СН3)2С H(OH)CONHCH2CH2CH(OC2Hs)2 ,
! XLIX
витаминная активность которого составляет половину витаминной актив-
ности пантотеновой кислоты ИЗО]. Диэтилацеталь (XLIX) при действии
кислот образует межмолекулярный «дипантотеналь» (L) 1131]
Всякие другие структурные изменения молекулы пантотеновой кислоты,
кроме тех, которые связаны с карбоксильной группой, приводят к соеди-
нениям, лишенным витаминных свойств или обладающим ими в ничтожной
степени.
Замещение двух метильных групп на этильные в p-положении пантоте-
новой кислоты уменьшает витаминную активность до 1/1000. Удаление
одной боковой метильной группы в 0-положении уменьшает биологическую
активность уже более чем в 100 раз [132]. Изменение в окси карбоновой
части молекулы пантотеновой кислоты или замещение р-аланина а-аланином
или другими аминокислотами [5, 132] дает полностью или почти полностью
неактивные соединения. Были получены также различные аминоаналоги
пантотеновой кислоты, такие, как а-амино-р, Р-диметил-7-оксибутирил-Р'-
аминопропионовая кислота 1133]
HOCH2C(CH3)2CH(NH2)CONHCH2CH2COOH,
которая также не обладала активностью, а, ?-Диокси-0, р-диметилбутирил-
P'-N-метилаланид [1341
СН3
HOCH2C(CH3)2CH(OH)CONCH2CH2COOH
неактивен как ростовой фактор для Lactobacillus arabinosus и дрожжей и
обладает 5—10%-ной активностью для Acetobacter suboxydans.
Для некоторых микроорганизмов ростовым фактором является 0-ала-
нин, т. е. эти микроорганизмы могут синтезировать пантотеновую кислоту
при наличии Р-аланина, например дрожжи [26] и дифтеритные бактерии
[1231; другие микроорганизмы синтезируют пантотеновую кислоту при
наличии пантолактона, а третьи не могут осуществлять синтез и требуют в
качестве ростового фактора пантотеновую кислоту. Она может количествен-
но определяться при испытании на молочнокислых бактериях [124] и ге-
молитическом стрептококке [125].
Ряд аналогов пантотеновой кислоты получен при замене р-аланина дру-
гими соединениями, в том числе витаминами, например остатками 2-метил-
4-амино-5-аминометилпиримидина, п-аминобензойной кислоты или £-глу-
таминовой кислоты [135]; они оказались неактивными, хотя последнее
соединение и несколько благоприятствовало росту Bacillus typhosus.
Среди аналогов пантотеновой кислоты имеются многочисленные инги-
биторы ростовой активности различных микроорганизмов. К ним относятся
как аналоги со структурно измененной оксикарбоновой частью молекулы,
например а-дезоксипантотеновая кислота
HOCH2C(CI I3)2CH2CONHCH2CH2COOH,
так и аналоги, в молекуле которых Р-аланин замещен таурином. Наиболее
активный антагонист — сульфопантотеновая кислота (пантоилтаурин)
[131, 1361 HOCH2C(CH3)2CH(OH)CONHCH2CH2SO3H; она является росто-
вым ингибитором для микроорганизмов, например гемолитического стрепто-
кокка и дифтеритного микроба, но не блокирует пантотеновую кислоту у
некоторых животных, для которых является слабым витамином [136].
Однако антагонист пантотеновой кислоты — только (4~)-сульфопантоте-
новая кислота; ее оптический антипод, (—)-сульфопантотеновая кислота —
биологически неактивен [136].
Пантоилтауринамид HOCH2C(CH3)2CH(OH)CONHCH2CH2SO2NH2 и го-
мопантоилтаурин HOCH2C(CH3)2CH(OH)CH2CONHCH2CH2SO3H активны
против гемолитического стрептококка. бнс-Нордезоксипантотеновая кис-
лота — HOCH2CH2CH2CONHCH2CH2COOH — является сильным ингиби-
тором роста гемолитического стрептококка и некоторых других патогенных
микроорганизмов. со-Метилпантотеновая кислота
CH^H(OH)C(CH3)2CH(OH)CONHCH2CH2COOH [136, 1381, (о-алкилнор-
пантотеновая кислота [138], со-метилпантетин, ш-метилпантоилтаурин
[136] оказались сильными антиростовыми веществами для молочных бакте-
рий.
Производные пантетеина, в которых атом водорода сульфгидрильной
группы замещен метильным, этильным или фенильным радикалом
HOCH2C(CH3)2CH(OH)CONHCH2CH2CONHCH2CH2SR,
обладают антагонистическими свойствами для ряда микроорганизмов, по-
видимому, вследствие нарушения основного биологического синтеза кофер-
мента А [16]: пантетеин + адениловая кислота + фосфат->кофермент А.
Ингибитором пантотеновой кислоты для Leuconostoc mesenteroides
является пантотенол (II). Гидроксильный аналог пантетина (по SH-rpynne)
ингибирует L. helveticus [139], а различные производные пантотеновой
кислоты, у которых остаток р-аланина замещен различными ароматически-
ми аминами, ингибируют Lactobacillus arabinosus и др. [140].
Потребность человека в пантотеновой кислоте — около 10 мг в сутки
125]; для кормящих матерей и при очень тяжелом труде необходимо 20 мг
71
ее. Для лечебных целей применяется до 500 мг пантотената кальция.
Пантотеновая кислота применяется при различных полиневритах, аллер-
гиях, экземах, трофических язвах, стоматитах, колитах, токсикозе бере-
менных и др.
Токсичность пантотеновой кислоты при испытаниях на кроликах —
2 г/кг при внутривенном введении. Потребность в пантотеновой кислоте для
животных находится в пределах 0,1—2,5 мг на 1 кг например, для
собак около 0,1 мг на 1 кг [1261. Для нормального роста и развития цы-
плят необходимо 1—2 мг пантотеновой кислоты на 100 г корма.
В животноводстве применяют DL-пантотенат кальция или непосредствен-
но или в виде комплекса с СаС12- i
КОФЕРМЕНТ А И ДРУГИЕ КОФЕРМЕНТЫ ПАНТОТЕНОВОЙ КИСЛОТЫ
Пантотеновая кислота осуществляет свою биологическую функцию в
составе коферментов, которые в виде простетической группы в соединении
со специфическими белками — апоферментами входят в ферментные систе-
мы. Ферменты, включающие в свой состав пантотеновую кислоту, являются
важнейшими биокатализаторами реакций ацилирования, среди которых
находится реакция ацетилирования холина, связанная с возбудимостью
нервного волокна [141], реакции ацетилирования уксусной кислоты в
ацетоуксусную кислоту, ацетилирования аминов, спиртов и др. [142, 143].
Однако пантотеновая кислота проявляет свои би окатал ити чески е функции,
только входя в состав 2-меркаптоэтиламидных производных. Коферментом
ацилирования, переносящим ацетильную и другие ацильные группы по-
средством своей тиольной группы, является кофермент А [144]. Вся или
почти вся связанная пантотеновая кислота в клетках животного организма
представлена, вероятно, в виде этого кофермента.
Кофермент А — Р1-(3,-фосфоаденозин-5,)-Р2-(О-пантотенил-Р-
аминоэтилмеркаптан-4)дифосфат, пантетеинадениннуклеотиддифосфат, КоА,
HSKoA (LI) [1451 —
представляет собой бесцветное аморфное вещество, растворимое в воде и
нерастворимое в спирте, эфире, бензоле и многих других органических
растворителях. Препарат получен с чистотой 95%. Спектр поглощения ха-
рактеризуется максимумом Хмакс 260 нм, е 16,4-103. Кофермент А —
четырехосновная кислота.
В химическом отношении кофермент А представляет собой меркаптан,
способный при действии слабых окислителей переходить в дисульфидную
форму КоА. Окислителями могут служить кислород воздуха, йод, перекись
водорода и др. Восстановители осуществляют обратную реакцию.
При взаимодействии КоА с нитропруссидом натрия возникает характер-
ная цветная реакция. С гидроокисями щелочных металлов КоА образует
растворимые в воде меркаптиды, а с солями тяжелых металлов трудно-
растворимые меркаптиды.
72
Важнейшее свойство кофермента А — его способность к обратимому
образованию по тиольной группе с органическими карбоновыми кислотами
ацилтиоэфиров, таких, например, как ацетилкофермент А
8- 8+ 8+
СНз—С—S—КоА,
II
О
который характеризуется таким распределением электронной плотности,
при котором атом серы и атом углерода карбонильной группы имеют частич-
ный положительный заряд, а атом углерода метильной группы — отрица-
тельный.
В реакциях ацилирования активным центром является карбонильный
углерод, несущий положительный заряд.
Ацетил-, малонил- и бутирил-КоА способны к енолизации по типу
СН3С—SKoA СН2=С—SKoA
II I
О он
Более простыми производными пантотеновой кислоты, чем кофермент А,
является пантетеин, Р-тиоэтаноламид D-пантотеновой кислоты (LII)
[146, 1471, ростовой фактор Lactobacillus bulgaricus (LBF) [148—150]:
HOCH2C(CH3)2CH(OH)CONHCH2CH2CONHCH2CH2SH
LII
и 4-фосфат пантетеина (LIII), ростовой фактор Acetobacter
suboxydans [151 ]:
О
II
(HO)2POCH2C(CH3)2CH(OH)CONHCH2CH2CONHCH2CH2SH
LIII
4-Фосфат пантетеина (LIII), помимо того что он является составной
частью молекулы кофермента А, в качестве простетической группы входит
в состав так называемого «белкового переносчика ацильных групп» и свя-
зан, по-видимому, своей фосфорной группой с белком через гидроксил
серина [152].
Дисульфидными производными Р-тиоэтаноламида D-пантотеновой кис-
лоты являются пантетин (LIV)
[HOCH2C(CH3)2CH(OH)CONHCH2CH2CONHCH2CHaS—]а
LIV
и 4-ф осфат пантетина (LV)
О
II
(HO)jPOCH2C(CHs)2CH(OH)CONHCH2CH2CONHCH2CH2S—
2
LV
Этим соединениям свойственны обратимые окислительно-восстановитель-
ные превращения, так же как коферменту A (LI). Так, например, пан-
тетин (LIV) при действии восстановителей превращается в свою тиольную
форму — пантетеин (LII), который при действии слабых окислителей вновь
переходит в дисульфидную форму, пантетин (LIV) [146, 153].
Дисульфидными производными являются все неочищенные природные
препараты пантетеина и кофермента А, если во время их выделения не при-
менялись процессы восстановления. После восстановления препараты
дают характерную для кофермента А пурпурную окраску с нитропрусси-
дом натрия [145].
Интересно отметить, что высокая реакционная способность пантетеи-
на проявляется также в образовании кристаллического аддукта с юглоном
73
(5-окси-1,4-нафтохиноном) в виде 3-(5-пантетеил)-5-окси-1,4-нафтохинона
(LVI) [154], образующегося за счет окисления промежуточной гидрохино-
новой формы избытком юглона
II
он о
sch2ch2nhcoch2ch2nhcoch(oh)c(ch3)2ch2oh
LVI
Такой аддукт из 2-метил-1,4-нафтохинона (провитамина К) и пантетеи-
на обладает свойствами витамина К и одновременно свойствами ростового
фактора Lactobacillus bulqaricus.
Получены различные S-ацильные производные пантетеина, имеющие
ацильные радикалы: бутирил, капроил, стероил, бензоил, а-нафтоил,
Р-нафтоил и др. [155].
СТРОЕНИЕ КОФЕРМЕНТА А
Строение кофермента A (LI) установлено по результатам изучения про-
дуктов его гидролитического расщепления [146, 1561 (схема 12).
Схема 12
Расщепление кофермента А, подтверждающее его строение
О ОН
Ж
/° °х
ch2c(ch3)2chconhch2ch2conhch2ch2sh
4 LVIII
LIII
ch;c(ch3)2chconhch2ch2cooh-------H2NCH2CH2SH ----------1
4 2 LIX -------------------J
I hoch2c(ch3)2ch(oh)conhch2ch2conhch2ch2sh
о I Г" I
(ho)2poch2c(ch3)2ch(o^conhch2ch2cooh hoch2c(ch3)2ch(oh)conhch2ch2cooh
LXI 1
Кофермент А содержит 1 моль аденозина, 1 моль пантотеновой кис-
лоты и три остатка этерифицированной фосфорной кислоты [157], из которых
только один остаток отщепляется монофосфоэстеразой [158].
Первоначально при мягком щелочном гидролизе кофермента А проис-
ходит расщепление пирофосфатной связи с образованием аденозин-5'-фос-
фата (АМФ, LVII) и одной из пептидных связей с выделением 2-меркапто-
74
этиламина (LX), причем одновременно, по-видимому, образуется смесь
циклического 2,4-фосфорного эфира пантетеина (LVIII) и циклического
2,4-фосфорного эфира пантотеновой кислоты (LIX), а во второй стадии
гидролиза получаются 4-фосфорный эфир пантотеновой кислоты (LXI)
и 4-фосфорный эфир пантетеина (LIII) [156]. Строение этих веществ было
доказано синтезом.
Интересно отметить, что амидная связь в молекуле 4-фосфата пантоте-
новой кислоты (LXI) значительно более устойчива по отношению к щелочам,
чем у пантотеновой кислоты (I) [146].
При кислотном гидролизе кофермента А внутримолекулярной переэте-
рификации не может быть, и поэтому первичным продуктом гидролиза
является 4-фосфорный эфир пантетеина (LIII), затем гидролиз приводит
к 4-фосфорному эфиру пантотеновой кислоты (LXI). Конечными продук-
тами кислотного гидролиза, помимо 2-меркаптоэт илам ина (LX) и АМФ
(LVII), являются р-аланин (III) и пантолактон (IV) [156].
В молекуле кофермента А не может присутствовать циклический фос-
фат пантетеина (LVIII), так как он не гидролизуется кислотной или щелоч-
ной фосфатазой, в то время как осколки кофермента А таким путем легко
дефосфор ил провались.
Кофермент А имеетр-конфигурацию гликозидной связи. Гидролиз ко-
фермента А ферментами яда гремучей змеи приводит к аденозин-3,'5'-ди-
фосфату [159].
Кофермент А расщепляется пирофосфатазой на 4-фосфорный эфир панте-
теина (LIII) [153, 160, 161] и аденозиндифосфат [160], одна из фосфорно-
эфирных групп которого находится в положении 3' [161]. 4-Фосфорный
эфир пантетеина связан с молекулой D-рибозы пирофосфатной связью по
первичной гидроксильной группе положения 5 молекулы альдопентозы
[146, 154].
Подтверждение формулы кофермента A (LI) получено на основании его
биохимического синтеза. Пантотеновая кислота (I) конденсируется с цистеи-
ном в пантотенилцистеин (LXII)
HOCH2C(CH3)2CH(OH)CONHCHaCH2CONHCHCH!tSH,
I
СООН
LXII
который, возможно, является промежуточным веществом биосинтеза кофер-
мента А некоторыми микроорганизмами. После декарбоксилирования панто-
тенилцистеина (LXII) образуется пантетеин (LII), фосфорилирующийся
аденозинтрифосфатом в положении 4 при участии киназы I, затем пантетеин-
4-фосфат (LIII) конденсируется с аденозинтрифосфатом в 3'-дефосфокофер-
мент А при участии фермента с отщеплением неорганического фосфата [145]
и окончательно фосфорилируется в положенииЗ' D-рибозной части аденозин-
фосфата кофермента А при участии киназы II [145, 162]. З'-Дефосфокофер-
мент А является биологическим предшественником кофермента А [162].
ВЫДЕЛЕНИЕ КОФЕРМЕНТА А
Кофермент A (LI) первоначально выделили Липман, Каплан иНовелли
из свиной печени [144, 163]. Для его получения используют ферментатив-
ные процессы с культурой Streptomyces Iradiae [164]; КоА из кислого куль-
турального раствора адсорбируют на активированном угле, элюируют ацето-
ноаммиачной смесью, восстанавливают цинком и соляной кислотой для рас-
щепления дисульфидной связи, осаждают солями ртути [158] и после уда-
ления ионов ртути сероводородом очищают хроматографированием на
диолите-CS 100. Для получения КоА с чистотой 75% используют также дрож-
жи [157] с применением адсорбции на активированном угле [158, 164] и
переосаждения с восстановленным глутатионом.
75
Из дрожжей был выделен а цетил кофермент A (CH3COSKoA) 1143, 165],
в котором ацетильная группа замещает атом водорода тиольной группы
КоА [143, 164, 165].
СИНТЕЗ ПАНТЕТИНА И ПАНТЕТЕИНА
Синтез пантетеина (LII) — ростового фактора Lactobacillus bulgaricus
(LBF) — продукта ферментативного расщепления кофермента А — и панте-
тина (LIV), его дисульфида, осуществлен следующим образом.
Пантетин (L1V) получают [166, 167] (схема 13) из бромэтилфтал имида
(LXIII), который обрабатывают гипосульфитом натрия, а полученный
промежуточный продукт окисляют йодом в ди-(фталимидоэтил)дисульфид
(LXIV), который расщепляют бромистым водородом или гидразином в
цистамин (LXV). Последний в результате взаимодействия с фталил-0-аланил-
хлоридом (LXVI) переходит в ди-(фтал ил-0-алан ил)цистамин и затем при
действии гидразина гидрата — в ди-(8-аланил) цистамин, 0-алетин (LXVI II),
который конденсируют с D(—)-пантолактоном (IV) в пантетин (LIV)
[166—168].
Схема 13
Синтез пантетнна фталимидным методом
СО
NagSjOg
LXIII
СО
12
------>.
80 — 90%
NCHjCHjCOCl
' LXVI
CO
НВг или NHsNHa
-------------> (H2NCH2CH2S—)2 ->
80 — 90%
LXV
co
CeH4 NCH2CH2CONHCH2CH2S—
co
NHoNHj
50—60%
2
LX VII
CH2C(CHS)2CH(OH)CO
I-------о-------1
IV
fH2NCH2CH2CONHCH2CH2S—]2----------------->
~ 100%
LX VIII
-> [HOCH2C(CHs)2CH(OH)CONHCH2CH2CONHCH2CH2S—]2
LIV
Фталимидным методом, исходя из цистамина (LXV), синтез пантетина
(LIV) осуществлен и другими авторами [169].
Этот синтез можно упростить, если цистамин (LXV) заменить этилен-
имином, в присутствии триэтиламина подвергнуть конденсации с фтал ил-0 -
аланилхлоридом (LXVI) в 0-фталимидопропионилэтиленимид (LXlX), об-
работав сероводородом, и затем окислить кислородом воздуха в ди-(фталил-
₽-аланил)цистамин (LXVII) [170]:
CHaCHsNH /\
o CeHi NCH2CH2COC1 -- > 0-CeH* NCH2CH2CONCH2CH, -► LXVI I
co co
LXVI LXIX
Если при синтезе пантетина (LIV) исходят непосредственно из пантоте-
новой кислоты (I), то ее конденсируют с цистамином (LXV) в присутствии
N, N'-дициклогексилкарбодиимида в пиридине [171].
Иным путем пантетин (LIV) получен в результате амидного синтеза
смешанного ангидрида пантотеновой кислоты и эфира угольной кислоты
(LXX) с равномолекулярным количеством гидрохлорида цистамина (LXV)
[172, 1731. Смешанный ангидрид (LXX) получают при действии на панто-
теновую кислоту (I) в виде ее натриевой соли хлоругольным эфиром в среде
диметилформамида при 0° С:
С1СООС2Н6
HOCH2C(CHs)2CH(OH)CONHCH2CH2COOH -------->
I
(H2NCH2CH2S—)2
LXV
HOCH2C(CHs)2CH(OH)CONHCH2CH2COOCOOC2H5-------------->
LXX
[HOCH2C(CHs)2CH(OH)CONHCH2CH2CONHCH2CH2S—
LIV
По той же схеме синтеза с выходом 50% можно получить и пантетеин
(LII), если весь процесс проводить без выделения промежуточных продук-
тов и в конечной стадии пантетин (LIV) восстановить амальга-
мой натрия [174]. Реакция проходит с теми же результатами, если циста-
мин (LXV) заменить p-меркаптоэтиламином (LX, с. 74) и, естественно,
исключить процесс восстановления. Также конденсацией смешанного ангид-
рида пантотеновой кислоты и эфира угольной кислоты (LXX) с 2-бензил-
тиоэтиламином в присутствии третичного амина с последующим дебен-
зилированием металлическим натрием в жидком аммиаке получают панте-
теин (LII) [175].
По одному из методов синтез пантетеина (LII) осуществляют через нит-
рил пантотеновой кислоты. D-Пантолактон (IV) конденсируют с амино-
пропионитрилом (XXXVIII) в D-пантотенонитрил (LXXI) [171], который
при взаимодействии с цистеамином (LX) превращается в 2-(2-пантамидо-
этил)-2-тиазолин (LXXII), а затем в результате гидролиза — в панте-
теин (LII) [171]:
ОСН2С(СН3)аСН(ОН)СО + h2nch2ch2cn ->
IV XXXVIII
HjNCHjCHaSH
LX
HOCH2C(CHs)2CH(OH)CONHCH2CH2CN----------->
LXXI
,N-CHt
НОСН2С(СНз)2СН(О11)СОКНСН2СН2С<^ I ------->
LXXII \>-(1h2
HOCHsC(CH3)CH(OH)CONHCH2CH2CONHCH2CH2SH
LII
Окисление пантетеина (LII) перекисью водорода в слабощелочной среде
приводит к пантетину (LIV) (выход 76—82% из соединения LXXI) [171].
Пантетеин (LII) был получен непосредственной конденсацией метилово-
го эфира пантотеновой кислоты с 2-аминоэтилмеркаптаном с выходом 10%
1153] и с лучшими результатами — через №|3-аланил-2-меркаптоэтиламин,
77
р-алетеин (LXXIII) [146], получаемый пептидным синтезом (схема 14).
Из гидробромида 2-бромэт илам ина (LXXIV) и бензил-со-тиола (LXXV)
в присутствии этилата натрия получают 2-бензилтиоэтиламин (LXXVI), ко-
торый конденсируют с азидом N-карбобензокси-р-алан ина (LXXVII) в
хлороформе в М-(М-карбобензокси-р-аланил)-5-бензилтиоэтиламин
(LXXVIII) [146, 176, 177].
Схема 14
Синтез пантетеина из 2-бензилтиоэтиламина через р-алетеин
H2NCH2CH2Br 4- HSCH2C«H8 -► H2NCH2CH.2SCH2CeH5-->
LXXIV LXXV LXXVI
CeH8CH2OCONHCH2CHaCON3
LXXVII
-----------------------* C6HbCH/X2ONHCH2CH2C()NHCH2CH2SCH2C«H5 -►
LXXVIII
-> H2NCH2CH2CONHCH2CH2SH ->
LXXIII
CHaC(CH3)2CH(OH)CO
I-------о--------1
IV
----------- HOCH2C(CHs)2CH(OH)CONHCH2CH2CONHCH2CH2SH
LII
Вместо соединения LXXVII для конденсации также используют N-кар-
бобензокси-р-аланин в свободном виде [178]. В соединении LXXVIII
восстановлением натрием в жидком аммиаке удаляют карбобензилоксигруп-
пу и бензильный остаток и получаютр-алетеин (LXXIII) [146, 176—178].
При нагревании D (—)-пантолактона (IV)c р-алетеином (LXXIII) при 100° С
без растворителя получается чистый пантетеин (LII) [146, 176—178].
Весь синтез протекает с высокими выходами.
Известный интерес представляет синтез пантетеина (LII) через р-але-
теин (LXXIII) несколько иным методом [179], что видно из схемы 15.
Схема 15
Синтез пантетеина из цистамина через Р-алетеин
CeH8CH2OCOCl SOC12
H2NCHaCH2COOH-------------> C6H5CH2OCONHCH2CH2COOH---->
III LXXIX
h2nch2ch2ssch2ch2nh2
LXV
CeH5CH2OCONHCH2CH2COCl------------------->
LXXX
HOOCCOO-
Na,NH3 +
-> (C.H8CHaOCONHCH2CH2CONHCH2CHaS—)2-----> h8nch2ch2conhch2ch2sh->
LXXXI LXXXII
CH2C(CH3)2CH(OH)CO
I-----о--------1
IV
— HjNCH2CH2CONHCH2CH2SH ----------------->
LXXIII
-► HOCH2C(CH3)2CH(OH)CONHCHaCH2CONHCH2CH2SH
LII
78
Первоначально из р-аланина (III) получают N-карбобензокси-р-аланин
(LXXIX), который в виде его хлорангидрида (LXXX) конденсируют с
р-аминоэтилмеркаптаном (цистамином) (LXV) в соединение (LXXXI) с
последующим восстановлением и выделением щавелевокислого р-алетеина
(LXXXII). Из этого соединения обработкой этилатом натрия получают
алетеин (LXXIII), который конденсируют с пантолактоном (IV) в панте-
теин (LII) [154, 179].
С меньшими выходами (20—30%) пантетеин (LII) получен конденса-
цией метилового или этилового эфира пантотеновой кислоты (VI) или,
лучше, ее азида (LXXXIII) cp-меркаптоэтиламином (LX) [1541:
HOCH2C(CH3)2CH(OH)CONHCH2CH2COOR— h2nch2ch2sh
VI LX
|n2h4,hno2
HOCH2C(CH3) 2CII (OH)CONHCH2CH2CON3—
LXXXIII
> Пантетеин
LII
Таким же методом конденсация соединения VI или LXXXIII с цистами-
ном (LXV) приводит к пантетину (LIV).
В отличие от схемы 14 2-бензилтиоэтиламин (LXXVI) синтезирован из
Р-мерка птоэтиламина (LX) и бенз ил хлорида, что показано на схеме 16.
Схема 16
Синтез пантетеина из 2-бензилтиоэтиламина через S-бензилалетеии
H2NCH2CH2SH + С»Н8СН2С1 -> H2NCH2CH2SCH2CeH5 ->
LX LXXVI
C«H8CH2OCONHCH2CH2COC1
LXXX
СО
’0-CeH4 NCH2CH2COC1
LXVI
> СвНБСН2ОСОЫНСН2СН2СОКНСН2СН25СН2С«НБ
LXXXVIII
со
> о-С.На NCH2CH2CONHCH2CH2SCH2CeH8
СО
LXXXIV
CH2C(CH3)2CH(OH)CO
I-------о-------'
IV
H2NCH2CH2CONHCHgCH2SCH2CeH8 -----* 1 * LII---*
LXXXV
Na, NH3
-► HOCH2C(CH3)2CH(OH)CONHCH2CH2CONHCH2CH2SCH2CeH8------->
LXXXVI
HOCH2C(CH3)2CH(OH)CONHCH2CH2CONHCH2CH2SH
LII
Дальнейший синтез пантетеина (LII) проведен двумя путями: конденсаци-
ей 2-бензилтиоэтиламина (LXXVI) с х л оран гидридом карбобензокси-p-ала-
нина (LXXX) в соединение LXXVIII с последующим восстановлением нат-
рием в жидком аммиаке и элиминированием карбобензоксигруппы с обра-
79
зованием S-бенз илалете ина (LXXXV) или — более просто — конденса-
цией 2-бензилтиоэтиламина (LXXVI) с фтал ил-0-алан ил хлоридом (LXVI)
в М-фталил-5-бензилалетеин (LXXXIV), при действии на который гидра-
зином гидратом получают S-бензилалетеин (LXXXV) 1180].
Конденсацией S-бенз илалете ин a (LXXXV) с D(—)-пантолактоном
(IV) получают S-бенз ил пантетеин (LXXXVI) и затем—после восстановления
натрием в жидком аммиаке — пантетеин (LII) (180].
Известен и непосредственный синтез S-бенз ил пантете ина (LXXXVI),
идущий с высоким выходом, путем конденсации метилового эфира D-пан-
тотеновой кислоты (VI) с 2-бензилтиоэтиламином (LXXVI) в среде метано-
ла при 100—150° С с последующим восстановлением соединения LXXXVI
натрием в жидком аммиаке в пантетеин (LII) [181 ].
СИНТЕЗ КОФЕРМЕНТА А
На пути к синтезу кофермента А, помимо пантетеина, были получены
4-фосфорный эфир пантотеновой кислоты (LXI) и 4-фосфорный эфир панте-
теина (LIII). Чтобы избежать смеси 2- и 4-фосфорных эфиров [182], необ-
ходимо было защитить гидроксильную группу в положении 2 путем ее бен-
зилирования. На пантолактон (IV) действуют хлористым бензилом в горя-
чем ксилоле и полученный бензиловый эфир пантолактона (LXXXVII)
конденсируют с натриевой солью 0-аланина в 2-бензиловый эфир пантоте-:
новой кислоты (LXXXVIII), который затем этерифицируют дифенилхлор-
фосфатом в соединение LXXXIX; в результате последующего отщепле-
ния бензильной и фенильной групп восстановлением натрием в жидком
аммиаке получают 4-фосфорный эфир пантотеновой кислоты (LXI) [146]:
ОН ОСН2С6Н6
| | H2NCH2CH2COONa
СН2С(СН,)2СНСО -> СН2С(СН3)2СНСО---------------►
1—о—I 1—0—1
IV LXXXVI I
ОСН2С6Н5
-► HOCH2C(CH3)2CHCONHCH2CH2COOH ->
LXXXVIII
О ОСН2С6Н5
(CeH6O)2P0CH2C(CHs)2 CHCONHCH2CH2COOH ->
LXXXIX
о
II
(HO)2PCH2C(CH3)2CH(OH)CONHCH2CH2COOH
LXI
При получении 4-фосфата пантотеновой кислоты (LXI) также исходят
из бензилпантотената и применяют фенилдихлорфосфат [183] или на нит-
рил пантотеновой кислоты действуют фенилдихлорфосфатом в присутствии
третичных аминов [184] с последующим гидролизом гидратом окиси бария
образовавшегося фосфата D-пантотенонитрила [184, 185 ]. 4-Фосфат D-пан-
тотеновой кислоты синтезирован и другим путем—фосфорилированием
2-(2-П-пантамидоэтил)-2-тиазолина тетрахлорп ирофосфорной кислотой
[186].
Упрощенным методом, без защиты гидроксильной группы положения 2,
можно провести фосфорилирование D-пантотеновой кислоты (I) дибен-
зил хлорфосфатом в 4-фосфорный эфир пантотеновой кислоты (LXI), если
реакцию проводить в пиридине при —40°С [187].
Если на 2-бенз ил пантотеновую кислоту (LXXXVIII) подействовать хлор-
угольным эфиром в присутствии 4-метилморфолина и затем 2-бензилтио-
80
этиламином, то образуется дибензиловый эфир пантетеина (ХС) при одно-
временном выделении двуокиси углерода. Его фосфорилируют по свобод-
ной гидроксильной группе дибензил хлорфосфатом в пиридине и получен-
ное соединение XCI восстанавливают натрием в жидком аммиаке в панте-
теин-4-фосфат (LIII) [188]; реакция протекает с отщеплением четырех
бензильных групп:
ОСН2СвН5
| (СвНБСНгО)2РОС1
HOCH2C(CH3)2CHCONHCH2CH2CONHCH2CH2SCH2CeH5--------------->
ХС
О ОСН2С6НБ
II I
-> (CeH6CH2O)2PCX:H2C(CHs)2CHCONHCH2CH2CONHCH2CH2SCH2CeH5 ->
XCI
о
-> (HO)2POCH2C(CH3)2CH(OH)CONHCH2CH2CONHCH2CH2SH
LIII
Пантете ин-4-фосфат (LIII) синтезирован фосфорилированием S-бензо-
илпантетеина по первичной гидроксильной группе дихлорфосфоморфоли-
дом через S-бензоилпантетеин [189]. Для направленного синтеза пантетеин-
4-фосфата используют также 2-О-тоз ил пантотеновую кислоту с блокирован-
ной а-гидроксильной группой [190].
Синтез О-пантетеин-4-фосфата (LIII) осуществлен из D-пантотенонит-
рила и дибензилхлорфосфата с последующим восстановлением и конден-
сацией с цистеамином (LX) [191 ].
П-Пантетеин-4-фосфат (LIII) получен из D-пантетина (LIV) непосредст-
венным фосфорилированием дибензилхлорфосфатом по первичной гидро-
ксильной группе без блокирования гидроксила положения 2. Если проме-
жуточно образующийся бпс-(Р-дибензилфосфо)пантетин (ХСП) подвергнуть
восстановлению металлическим натрием в жидком аммиаке, то одновремен-
но происходит гидрогенолиз бенз ильных групп с образован ием П-пантетеин-
4-фосфата [LIII) [188,192]. Однако реакция сопровождается внутримолеку-
лярной циклизацией с образованием бнс-2,4-циклофосфата пантетина [193].
Поэтому первоначально одну бензильную группу фосфатного остатка бис-(Р-
дибензилфосфо) пантетина (ХСП) отщепляют нагреванием с 50%-ной уксус-
ной кислотой (100°С) в бпс-(Р-монобензил)пантетин (ХСШ), а его затем вос-
станавливают металлическим натрием в жидком аммиаке в П-пантетеин-4-
фосфат (LIII) [193]:
О
II
(СвН5СН2О)2РС1
[HOCH2C(CH3)2CH(OH)CONHCH2CH2CONHCH2CH2S—]2--------------
LIV
о
II
(QH6CH20)2POCH2C(CH3)2CH(OH)CONHCH2CH2NHCH2CH2S—]2 -►
ХСП
о
II
CeH5CH2OPOCH2C(CH3)2CH(OH)CONHCH2CH2NHCH2CH2S—
он
J2
хеш
о
II
-> (НО)2РОСН2С(СН8)2СН (OH)CONHCH2CH2CONHCH2CH2SH
LIII
Фосфорилирование 2, S-дибензилпантетеина дибензилхлорфосфатомс по-
следующим восстановлением металлическим натрием в жидком аммиаке
также приводит к пантетеин-4-фосфату [194].
Я1
Пантете ин-4-фосфат (LIII) получен и непосредственным фосфорилировани-
ем пантетина тетрахлорпирофосфатом с последующим восстановлением
(195]. Пантетин-4-фосфат получают из пантетина его фосфорилированием
дихлорфосфоморфолцдом или Р^дифенил-Р^морфол ил хлорпирофосфатом
(196].
Создание пирофосфатной связи несимметричного Р1, Р2-диэфира пиро-
фосфорной кислоты при синтезе кофермента А конденсацией аденозин-3', 5'-
дифосфата и 4-фосфата D-пантетеина (LIII) осуществляется фосфоамидным
методом, который заключается в конденсации фосфоамидного производ-
ного нуклеозида, имеющего незащищенные гидроксильные группы с другим,
также незащищенным фосфорным эфиром. Такие производные фосфорной
кислоты и ее эфиров доступны, реакционноспособны, устойчивы в условиях
опыта и обладают специфичностью в синтезе нуклеотидных коферментов.
Моноэфиры амида ортофосфорной кислоты могут реагировать с фосфат-
ными анионами с образованием пирофосфатной связи; при этом не происхо-
дит фосфорилирования спиртовых гидроксильных групп, фосфоамиды мо-
гут быть использованы для синтеза несимметричных динуклеотидов.
Незначительная активность фосфоамидов по отношению к спиртовым
гидроксильным группам позволяет использовать незащищенные нуклеоти-
ды и тем самым избежать реакций обмена, возможных при наличии пол-
ностью этерифицированных пирофосфатов, как это наблюдается при хлор-
фосфатном методе.
Реакция фосфорилирования гладко протекает в присутствии пиридина
или триал к илам инов. Исходные вещества чаще всего используются в виде
солей с металлами (калий, натрий) и органическими основаниями (пиридин,
триалкилам ины). Эти соли хорошо растворимы в формамиде, диметилфор-
мамиде, пиридине, о-хлорфеноле и смеси пиридина и о-хлорфенола — раст-
ворителях, наиболее часто употребляемых в этом методе.
Для реакции нуклеофильного замещения фосфоамидов был предложен
бимолекулярный механизм (197, 198]. Разрыв Р—N-связи обеспечивается
предварительным протонированием атома азота [197].
В сснову полного синтеза КоА (LI) Моффат и Хорана [193] был поло-
жен принцип фосфоамидного метода с применением для синтеза фосфомор-
фол идного производного (схема 17). Первоначально из аденозин-2'(3'),5'-ди-
фосфата и морфолина в присутствии N, N'-дициклогексилкарбодиимида
был получен аденозин-2', 3'-циклофосфат-5-дифосфоморфолид (XCIV) с
выходом 98%. Затем соединение XCIV конденсировали с Р-пантетёин-4-
фосфатом (LIII) в безводном пиридине при комнатной температуре в 2,'3'-
цикло-КоА (XCV), который при действии соляной кислоты, вызывающей
раскрытие циклической фосфорной связи, превращался в смесь изомерных
форм KoA((LI) и 2'-изо-КоА [193] с общим выходом 65%. Эти изомеры
разделяли на ионообменной эктеола-целлюлозе, и КоА получали с выхо-
дом 15%. Синтетический КоА обладал. 86 %-ной ферментной активностью,
2'-и-о-КоА — ничтожно малой, всего 4%.
Конденсация соединения XCIV проведена также с 4-фосфатом DL-пан-
тотеновой кислоты (LXI); продукт конденсации после выделения и очистки
методом ионообменной хроматографии обрабатывали 0,1 н. соляной кислотой,
после чэго вводили в реакцию с 2-меркаптоэтиламином (LX). Хроматогра-
фией на адсорбционной колонке была выделена смесь кофермента А и
2'-изо-КоА с общим выходом 50% [193]. Аналогично из 5'-морфолида АМФ
и Р£-пантетеин-4-фосфата синтезирован 3'-дефосфо-КоА [193].
Фосфоамидным методом осуществлен полный синтез КоА через тиазо-
линовое производное [199]. Из 4-морфолин-М, N'-дициклогексилкарбокс-
амидиниевой соли аденозин-2',3'-циклофосфат-5'-фосфоморфолида (XCIV)
иП-пантотенонитрил-4-фосфата в пиридине получен Р1-(аденозин-3'-фосфат-
5')-Р2-(П-пантотенонитрил-4) пирофосфат[2001, а в результате его последую-
щего взаимодействия с 2-меркаптоэтаноламином (LX) синтезирован Р1-(аде-
нозин-3'-фосфат-5')-Р2-Ы- [2- (2-тиазолин-2-ил)этил ]-Р-пантоамид-4-пирофос-
Схема 17
Синтез кофермента А фосфоамидным методом
О. он
о hsch4ch2nhcoch2ch2nhco(oh)ch(ch3)2cch2o-p-o9 + сГ ык он . N-P-OCHj О 1 1 Н IN OH 1 II xav
о о
hsch2ch2nhcoch2ch2nhco(oh)ch(ch3)2cch2o-p-o-p-
vr-w ОН он
Ок хон
о о
nh2
о,1н, на
о
-hsch2ch2nhcoch2ch2nhco(oh)ch(ch3)2cch2o-p-
, он
-2-изо-КоА
LI
Р°3Н2
МН2
фат, в котором нагреванием с нормальной соляной кислотой при 60° С
гидролизуется тиазолиновая конечная группировка молекулы
— с = nch8ch2s -> — CONHCH2CH2SH
и образуется кофермент А [200, 201 ]. Применение аденозин-5'-фосфоморфо-
лида приводит к З'-дефосфокоферменту А[199]. Таким же путем с примене-
нием тиазолинового производного 4-фосфата пантотенонитрила и его амида
были синтезированы кофермент A (LI) и 3'-дефосфокофермент А [200,201].
Фосфоморфолидным синтезом получены аналоги кофермента А: гуано-
зинкофермент А [202], DL-З'-дефосфоселеноаналог кофермента A, D- и
DL-селеноаналоги кофермента A, D- и DA-селеноаналоги 2'-изокофермен-
та А, бензоилселенокофермент А, бензо илселено-2'-изокофермент А [203],
кислородные аналоги окси-2'-изокси- и З'-дефосфооксикофермент А [204]
и другие аналоги КоА по меркаптоэтильной группе [205].
Со значительно лучшими результатами (выход 70—80%) кофермент А
синтезирован анионообменным методом.
Анионообменный метод получения несимметричных нуклеотидангидри-
дов заключается в обменной реакции неполностью этерифицированного
нуклеозидпирофосфата а с неэтерифицированным нуклеозидмонофосфатом б,
который представляет собой анион более слабой кислоты (так же как и
нуклеозидпирофосфат), чем анион отщепляющейся диарил- или диалкил-
фосфорной кислоты г [206]. В результате нуклеофильной атаки в пиридино-
вом растворе при комнатной температуре завязывается новая пирофосфат-
ная связь с образованием несимметричного нуклеотидангидрида в и отщеп-
лением устойчивого аниона г:
83
о о
II II
о
о о
II II
1 — - - -
Гон or"
О- a
I
HO—P—OR
II
О
c
При действии на аденозин-2'(3')»5'-дифосфат (XCVI) дифенилхлор-
фосфатом с количественным выходом получается Р1-(аденозин-2',3'-цикло-
фосфат-5')-Р2-Дифен ил пирофосфат (XCVII) [159]:
о=р(он)2
он о
I?
HO-P-OCH/Vr
он
N
nh2
XCVI
Р1-(Аденозин-2/,3,-циклофосфат-5/)-Р2- дифенил пирофосфат (XCVII) кон-
денсируют с 4-фосфатом пантетина (LV) в дисульфид 2,'3'-цикло-КоА
(XCVIII), который восстанавливают в смесь КоА (LI) и 2'-изо-КоА (вы-
ход 6,3%); КоА выделяют в виде литиевой соли (схема 18). Соединение
XCVIII превращают в КоА также ферментативным методом с рибонуклеа-
зой Т2 с одновременным раскрытием циклической фосфорной связи
исключительно в 3'-фосфат [159].
Схема 18
Синтез кофермента А анионообменным методом
О. „ОН
„Рх
О О
9 сО —
AO-P^b-P-OR —^AO-P-D-P-OR +~O-P-ORT
HI. II I „
OR"
он он
I
o^ ZOH
р
oz о
О
II
О
II
ОН
о
II
CSHSO-P-Ct—*-C6Hs-O-P-O -Р-ОСН^*о’
ОСвН5
О'-
OC6HjOH
NH2
XCVII
О
°
^-sch£ch2micoch2ch2nik:ochc(ch2)2ch2op(oh)J24C6h5op-o-poch2
ОС6Н5 ОН I
LV
он
1 / X
-sch2ch2nhcoch2ch2nhcochc(ch3)2i
о
II
.CHj.O-P-O-P-OCHj'O
он
Y N
XCVII nh2
о „он _
X
: О
OH
XCVIII
ОН
о
HSCH2CH2NHCOCH2CH2NHCOCHC (с На),СН2О- Р - О - Р
2'-изо-КоА ОН
N
Ь1Н2
о=р(он)2
о он
? I
-Р-ОСН.
он
О
Щ12 о
„Nx „N
2
U
NHj
84
Подобным образом при использовании в качестве аниона Рг-(адено-
зин-5')-Р2-Дифенилпирофосфата и в качестве нуклеофильного агента панте-
теин-4-фосфата синтезирован З'-дефосфо-КоА [206].
Аденозин-2'(3'),5'-дифосфат (XCVI) получен из аденозина (XCIX) фос-
форилированием дибензилхлорфосфатом в пиридине. Образовавшееся три-
и тетра бензилфосфопроизводное (Са и Сб) подвергали каталитическому
гидрогенолизу над палладиевым катализатором или анионному обмену с
щелочным гидролизом для отделения бензильных групп; полученная смесь
2',4'- и 3,'5'-дифосфатов аденозина (XCVIa и XCVI6) разделялась на ионо-
обменных смолах [193, 207, 208]:
(QHsCH^P-iH-
--------О
о о
—кн
о Кн
(сеН5СН2о)4Р-ОС|Сс>
J4 ,N
ОН ОН
о
NH,
XCIX
nh2
Са
О'" \>
О -
О
он о-р(он\
?о
НО—P-ОС н2 о
он
О1
nh2
XCVIa
NH,
Сб
(но)2р-о он
? н4гзИ
НО-Р-ОСН2 о
он
.N. Л
N.
ЫН2
XCY16
Фосфорилирование аденозина можно осуществить также 2-цианэтил-
фосфатом с N, N'-дицикло гекс ил кар боди имидом [209].
Для получения аденозин-3',5'-дифосфата из смеси изомеров очень эффек-
тивным оказался ферментативный метод — использование ферментов змеи-
ного яда или рибонуклеазы Т2 для расщепления 2',3'-циклофосфатной
связи промежуточно получаемого ангидрида аденозин-2',3'-циклофосфат-
5'-фосфорной и этилугольной кислот только в фосфат З'-положения [159].
Аденозин-3',5'-дифосфат является компонентом кофермента А, аденозин-
2', 5'-дифосфат— компонент никотинамидадениндинуклеотидфосфата
(НАДФ).
БИОХИМИЧЕСКИЕ ФУНКЦИИ КОФЕРМЕНТА А
Кофермент А является участником реакций, связанных с биосинтезом
аминокислот, высших жирных кислот, фосфолипидов, пуринов, терпенов,
стероидов, порфиринов, он участвует в синтезе лимонной, глутаминовой,
гиппуровой [142] кислот, аргинина, пролина и др.
Разнообразие функций кофермента А и ацилкофермента А определяется
апоферментом (белковой частью молекулы фермента), который общим хи-
мическим реакциям придает специфичность к определенным субстратам.
Кофермент А в виде своего высокореакционноспособного производно-
го — «активного ацетила» [165], ацетил-КоА, и соответствующих ему ациль-
ных производных других органических кислот является биологическим
ацилирующим реагентом в составе ацилтрансфераз. Он образует связи
С—С, С—N, С—О, С—S, ацилируя:
85
метильную группу уксусной кислоты с образованием 0-кетокислот;
оксигруппу аминоспиртов (холина, карнитина), сахаров, фосфорных
кислот;
аминогруппу аминокислот, аминосахаров, ароматических аминов;
циклический атом азота имидазола;
тиольную группу тиоэтанолам ина, сероводорода и др.
При участии кофермента А в виде его ацил-КоА осуществляются много-
численные биохимические реакции:
изомеризации D- и £-форм (например, 0-оксимасляной и метилмалоно-
вой кислот), карбоксильных групп (например, внутримолекулярная пере-
группировка метилмалоновой кислоты в янтарную кислоту при участии
кофермента витамина В12) и двойных связей (в ин ил уксусной и кротоновой :
кислот);
конденсации по а-углеродному атому карбоновых кислот; 1
дегидрирования (а, 0-дегидрирование кислот и 0-оксикислот в 0-кето-
кислоты) и обратимого окисления;
гидратации (по а, 0-двойной связи); з
расщеплен ия 0 -кеток ислот; 5
образования фосфоангидридов и др.
Эти реакции носят обратимый характер. 1
Кроме того, карбоксиацильные формы ацил-КоА являются переносчиками г
карбоксильных групп в реакциях, осуществляемых биотиновыми и другими
ферментами (декарбоксилирование щавелевоуксусной кислоты в пировино-
градную кислоту и образование метилмалоната из ацетата).
Ацил-КоА свойственна способность к обмену ацильных остатков с <
другими органическими кислотами над влиянием КоА-трансфераз. _•
Ацил-КоА осуществляет транспорт ацильных групп от одного субстрата ;
к другому, тем самым являясь их промежуточным акцептором и донором
в многочисленных химических реакциях распада пищевых продуктов и
синтеза жиров, аминокислот и других соединений в живой клетке. Однако '
он является не только переносчиком ацетила, но и участником его полного >
окисления в двуокись углерода и воду в цикле трикарбоновых кислот
(см. схему 74).
Ацетил-транспортная система КоА '
Углеводы Стероиды и др. j
_ / —Жиры
Гликолиз Х'Д \ з
/ Глицерин Ч. \ 1
/ / Жирные \
/ / ^-КИСЛОТЫ \
/ / Окисление х. \
Пфовино- / Аминокислоты х. \ 1 -1
градная / / \ \ I '
кислота / / \ \
ОкислительноеХ I
декарвоксилиА \ / '
рование \ \\/
\.CH3CO~SKoA
Окисление в цикле
трикарбоновых кислот
со2+нго
Ферментативные реакции, осуществляемые при участии кофермента А ;
[210], перечислены ниже
RR
Реакции карбоксилирования и ацилирования
Превращения субстрата
Пировиноградная кислота Щавелево-
уксусная кислота
L-Глутаминовая кислота 7~"~ N-Аце-
тил-£-глутаминовая кислота
Имидазол 721 N-Ацетилимидазол
2-Амино(дезокси)-О-глюкоза 7* 2-Аце-
тамино(дезокси)-£)-глюкоза
2-Амино(дезокси)-0-глюкозо-6-фос-
фат ~7~*~ 2-Ацетамино(дезокси)-О-глю-
козо-6-фосфат
Ароматический амин 2ZT Ароматический
N-ацетиламин
(Холин 7"* О-Ацетилхолин
Карнитин О-Ацетилкарнитин
иртофосфорная кислота т~~*~ Ацето-орто-
фосфорная кислота
Дцетил-КоА 7Z± Ацетоацетил-КоА
Ацил-КоА 717 Ацилацетил-КоА
Сероводород 7~*~ Тиоацетат
Тиоэтаноламин 7~*~ S-Ацетилтиоэтанол-
амин
6, 8-Дигидролипоевая кислота 7~*~ 6-S-
i Ацетил-6, 8-дигидролипоевая кислота
7,-Аспарагиновая кислота 7^~ N-Ацетил-
£-аспарагиновая кислота
J-D-Галактозид 7± 6-Ацетил-p-D-галак-
тозид
Глицин N-Ацилглицин
’£-Глицеро-3-фосфат 7~*~ Моноглицерид-
фосфат
11,2-Диглицерид 7~*~ Триглицерид
Ортофосфор на я кислота 7~*~ Бутироорто-
фосфорная кислота
L-Глутамин тг± a-N-Фенилацетил-Т-глу-
тамин
Превращения кофермента
Малонил- или метилмалонил-
КоА TZt Ацетил- или пропионил-КоА
Ацетил-КоА 2=7 КоА
>
> > 4
»
>
*
» »
» »
»
»
> э
Ацил-КоА 7^ КоА
»
» »
Бутирил-КоА 7Z± КоА
Фенилацетил-КоА тт± КоА
Реакции переацилировання
Ацетил-КоА 4- Пропионовая кислота 7~*~ Пропионил-КоА 4- Уксусная кислота
Ацетил-КоА 4- Малоновая кислота 7~*~ Малонил-КоА -f- Уксусная кислота
Сукцинил-КоА + Щавелевая кислота 7~*~ Оксалил-КоА -f- Янтарная кислота
Сукцинил-КоА + Ацилуксусная кислота 7~*~ Ацилацетил-КоА -f- Янтарная кислота
Сукцинил-КоА 4- З-Оксоадипиновая кислота 7~*~ 3-Оксоадипил-КоА 4- Янтарная кислота
Реакции изомеризации
L-3-Оксибутирил-КоА 227 D-3-Оксибутирил-КоА
О-Метилмалонил-КоА 7=7 £-Метилмалонил-КоА
Метилмалонил-КоА Сукцинил-КоА
Вииилацетил-КоА 2=7 Кротонил-КоА
Реакции карбоксилирования
Ацетил-КоА 4- СО2 4- АТФ 4- Н2О у~*~ Малонил-КоА 4- АДФ 4- Ортофосфориая кислота
Пропионил-КоА 4- СО2 4- АТФ 4- Н2О 2=7 Метилмалонил-КоА 4- АДФ 4- Ортофосфориая
кислота
3-Метилкротонил-КоА 4- СО2 4- АТФ 4- Н2О 2=7 3-Метилглутаконил-КоА 4- АДФ 4- Орто-
фосфорная кислота
87
Окислительно-восстановительные реакции ацил-КоА
прн участии НАД(Ф)+ НАД(Ф)-Н + Н+:
Мевалоновая кислота 7~*~ З-Окси-З-метилглутарил-КоА
3-Оксиацил-КоА 7~*~ 3-Оксоацил-КоА
Альдегид + КоА Ацил-КоА
Глиоксиловая кислота 4~ КоА Оксалил-КоА
Полуальдегид малоновой кислоты 4- КоА 7~*~ Ацетил-КоА 4- СО2
при участии Ф А Д ^=2 Ф А Д-Н2:
Бутирил-КоА у"* Кротонил-КоА
Ацил-КоА 2,3-Дегидроацил-КоА
Оксалил-КоА , Формил-КоА 4- СО2
Малонил-КоА 7~*~ Ацетил-КоА 4- СО2
L-Малеиновая кислота 4- КоА Ацетил-КоА 4- Глиоксиловая кислота 4- Н2О
З-Окси-З-метилглутарил-КоА Ацетил-КоА 4- Ацетоуксусная кислота
З-Окси-З-метилглутарил-КоА 4- КоА Ацетил-КоА 4* Ацетоацетил-КоА 4- Н2О
Лимонная кислота 4- КоА "Г* Ацетил-КоА 4- Щавелевоуксусная кислота 4- Н2О
Лимонная кислота 4- КоА 4- АТФ , Ацетил-КоА 4- Щавелевоуксусная кислота -J-
4- АДФ 4- Ортофосфорная кислота
2-Оксиглутаровая кислота 4- КоА т~*~ Пропионил-КоА 4- Глиоксиловая кислота 4- Н2О
w-Пропилмалоновая кислота 4- КоА yz± Бутирил-КоА 4- Глиоксиловая кислота 4- Н2О
L-3-Оксиацил-КоА т~* 2,3 (или 3,4)-пгранс-Еноил-КоА 4-Н2О
З-Окси-З-метилглутарил-КоА 7~*~ транс-З-Метилглутаконил-КоА -р Н2О
Р-Аланил-КоА 7~*~ Акри лил-КоА 4- NH3
Ацетил-КоА и другие ацил-КоА синтезируются при участии специфи-
ческих ферментов — синтетаз, которые образуют С—-S-связь между карбо-
нильным углеродом кислоты и тиольной группой кофермента А. Эти реак-
ции осуществляются за счет использования энергии макроэргического
аденозин-5'-трифосфата (АТФ) и протекают с выделением аденозин-5'-моно-
фосфата (АМФ) и неорганического пирофосфата.
Так, из кофермента А и уксусной кислоты под влиянием ацетил-КоА —
синтетазы образуется ацетил-КоА [211]. Промежуточным соединением в
этом синтезе является аденозин-5'-ацетофосфат, который уже затем реаги-
рует с коферментом А [212].
NH, NH.
АТФ ’ АМФ
Таким образом, реакция идет в две фазы:
СНзСООН 4- АТФ —> СНзСО-АМФ 4- Н4Р2О,,
СН3СО АМФ 4- HSKoA —> СН3СО - SKoA 4- АМФ-
88
Такое течение реакции установлено в ферментном синтезе ацетил-КоА
из КоА, АТФ и ацетата, меченного 18О; в этом синтезе изотоп кислорода
переносится на образующийся в реакции АМФ [213]. Образование ацил-КоА
с другими остатками кислот, например бутирил-КоА, бензоил-КоА и т. д.,
также связано с промежуточными неустойчивыми формами ацил-АМФ
(ациладен платами) [214].
В данной реакции происходит не только образование высоко реакцион-
носпособного ацетил-КоА, но и перенос энергии от АТФ (энергия макроэрги-
ческой связи АТФ составляет 7,65—8,25 ккал/моль) на макроэргический
ацетил-КоА, который становится аккумулятором энергии. Протекание
различных реакций ацетилирования обеспечивается энергией ацетил-КоА
при переносе ацетильной группы на субстрат.
Подобным образом из жирных кислот при участии ацил-КоА-синтетазы
образуется ацил-КоА [215, 216]:
HSKoA + СН3 (СН2)„ СООН + АТФ ->СН3 (СН2)„ СО ~ SKoA + АМФ + Н*Р2О7.
Из янтарной кислоты при участии сукцинил-КоА-синтетазы получается
сукцинил-КоА [2171:
HSKoA + НООССН2СН2СООН + АТФ -> НООССН2СН2СО ~ SKoA + АДФ + Н3РО4.
Вместо АТФ в подобной реакции участвует гуанозин-5'-трифосфат
<ГТФ), который переходит в гуанозин-5'-дифосфат (ГДФ) [217].
Соответственно из глутаровой кислоты под влиянием глутарил-КоА-
•синтетазы получается глутарил-КоА и т. п.
При действии ферментных систем [218] происходит включение органи-
ческих кислот в виде ацильных групп в молекулу кофермента А, в виде
ацил-КоА происходят их те или иные превращения, и затем под действием
специфических гидролаз сложноэфирная связь ацилтиолового эфира ко-
фермента А расщепляется и из ацил-КоА регенерируется КоА и выделяется
.соответствующая кислота:
ацил-КоА + Н2О —* КоА + ацил-ОН.
Из многочисленных реакций, в которых участвует ацил-КоА, интересно
остановиться на следующих.
Ацетил-КоА при участии ацетил-КоА-трансферазы [219] ацетилирует
холин в ацетилхолин:
[(CH,)sNCH2CH20h] ОН- + СН8СО ~ SKoA —* [(CHS)SNCH2CH2OCOCH8] он- +
+ HSKoA.
Эта реакция образования сложного эфира занимает важное место в
механизме нервного процесса.
При участии тиоэтаноламин-ацетилтрансферазы ацетил-КоА ацетилирует
тиоэтаноламин в S-ацетилтиоэтаноламин [220]:
H2NCH2CH2SH + СН3СО - SKoA —•> H2NCH2CH2SCOCH3 + HSKoA.
Из реакций образования амидов кислот можно отметить реакцию ацетили-
рования п-аминобензойной кислоты в n-ацетаминобензойную кислоту при
участии ацетил-КоА:
H2NCGH4COOH + СН3СО - SKoA —> CH3COHNC6H4COOH + HSKoA,
а сульфаниламидных соединений — в соответствующие ацетилпроизводные
[144]; эти реакции проходят при участии ариламин-ацетилтрансфераз [221 ].
При ацетилировании одной молекулы ацетил-КоА второй молекулой
ацетил-КоА под влиянием ацетил-КоА-ацетилтрансферазы происходит обра-
зование связанной р-кетокислоты—ацетоацетил-КоА [222]
89
о- н+
с- ||
СН3СО ~ SKoA + СН3СО ~ SKoA —> CHSCCH2CO ~ SKoA —»•
SKoA
—СН3СОСН2СО ~ SKoA 4- HSKoA
Углеродная цепь 0-кетокислоты может состоять из большего количества
атомов углерода, если в реакции будет участвовать одна из молекул кофер-
мента А, ацилированная не уксусной, а другой жирной кислотой.
Конденсации по а-углеродному атому органических кислот протекают
при участии ацетил-КоА, например в синтезе лимонной кислоты. Фаза
включения уксусной кислоты в виде «активного ацетила» в важнейший
биохимический цикл превращений трикарбоновых кислот (цикл Кребса,
см. с 324) заключается в электрофильной атаке карбонилом щавелевоуксус-
ной кислоты атома углерода метильной группы ацетил-КоА, имеющего
повышенную электронную плотность. В результате реакции, протекающей
под влиянием цитрат-синтазы, синтезируются лимонная кислота и кофер-
мент А [223]:
СООН
О+ I
НООССН2СОСООН + СН3СО - SKoA + Н2О —> НООССН2 С (ОН) СН2СООН + HSKoA
Уксусная кислота, включенная в цикл трикарбоновых кислот, полностью
окисляется в двуокись углерода и воду, а щавелевоуксусная кислота вновь
регенерируется. Уксусная кислота образуется при различных процессах
метаболизма и включается в ацетил-КоА при участии АТФ (см. выше).
Другими источниками образования ацетил-КоА является пировиноградная
кислота — важнейший продукт окислительного расщепления углеводов в
организме—или высшие жирные кислоты, подвергающиеся 0-окислительному
расщеплению.
Кофермент А участвует в сложной реакции дегидрирования и декарбокси-
лирования пировиноградной кислоты, при которой высвобождается ацетат,
акцептируемый КоА, аккумулируя при этом часть высвобождаемой энергии
в виде макроэргической связи. Эта реакция осуществляется ферментными
системами, состоящими из пируватдегидрогеназного комплекса и других
ферментов [224 ] при участии коферментов: никотинамидадениндинукле-
отида (НАД), тиаминдифосфата (ТДФ), флавинадениндинуклеотида (ФАД)
и липоевой кислоты (ЛК):
СН3СОСООН + HSKoA —> СН3СО ~ SKoA + 2 [Н] + СО2.
Течение реакции окислительного декарбоксилирования пировиноград-
ной кислоты и последующее превращение ацетильного остатка при рекомби-
нации со щавелевоуксусной кислотой в лимонную кислоту можно представить
следующей схемой:
Щавелевоуксусная СООН
кислота Лимонная кислота
90
Промежуточная биохимическая функция а-липоевой кислоты заключа-
ется в переносе ацетильного остатка от пировиноградной кислоты, акцеп-
тором которого она является, на кофермент А, в результате чего образуется
ацетил-КоА. При этом получается восстановленная форма — 6,8-дигидроли-
поевая кислота, которая регенерируется ва-липоевую кислоту при участии
НАД+ [225, 226].
Еще одной фазой превращения в цикле трикарбоновых кислот, проте-
кающей при участии КоА, является реакция дегидрирования и декарбокси-
лирования а-кетоглутаровой кислоты через сукцинил-КоА в янтарную
кислоту. Окислительное декарбоксилирование а-кетоглутаровой кислоты
в сукцинил-КоА протекает по сложному взаимодействию ферментных систем
включающих а-кето-глутаратдегидрогеназный комплекс, [227 ] и других,
катализирующих этот процесс, в состав которого входят коферменты:
липоевая кислота, НАД, ФАД, ТДФ
НООССН2СН2СОСООН + HSKoA —> НООССН2СН2СО ~ SKoA + 2Н+ + 2е -f- СО2
НООССН2СН2СО ~ SKoA + АДФ (или ГДФ) + Н3РО4 НООССН2СН2СООН +
+ HSKoA + АТФ (или ГТФ).
Превращение сукцинил-КоА в янтарную кислоту идет при участии АДФ
(или ГДФ) и ортофосфорной кислоты и протекает по реакции образования
фосфоангидридов (с выделением молекулы воды), при которой синтезирует-
ся макроэргический АТФ (ГТФ).
Р -Окислительное расщепление высших жирных кислот состоит в синхрон-
ном отщеплении двух атомов углерода, которые акцептируются КоА в виде
ацетил-КоА:
СН3 (СНг)л СНгСНгСО ~ SKoA -> СН3 (CH2)„ СН=СНСО - SKoA ->
-4- СН3 (СН2)Я СН (ОН) СН2СО ~ SKoA -> СН3 (СН2)„ СОСН2СО - SKoA ->
-► СН3 (СН2)„ СО ~ SKoA + СН3СО ~ SKoA.
Реакция идет через дегидрирование по а, р-атомам углерода ацил-КоА
при действии флавопротеида ацил-КоА-дегидрогеназы [228], последующую
гидратацию по двойной связи 2,3-дегидроацил-КоА, дегидрирование вто-
ричной спиртовой группы р-оксиацил-КоА 3-оксиацил-КоА-дегидрогена-
зой [215] в кето-группу ацилацетата-КоА с последующим включением в
процесс еще одной молекулы КоА и расщеплением р -кетоацил- КоА на
ацил-КоА и ацетил-КоА.
По мере распада высшие жирные кислоты превращаются в соединения
с укороченной углеродной цепью и, наконец, в бутирил-КоА, окончательное
Р-расщепление которого протекает в результате дегидрирования флаво-
протеидом бутирил-А-дегидрогеназой [228] в кротонил-КоА, и затем при
действии специфических ферментов в р -оксибутирил-КоА, ацетоацетил-КоА
и, наконец, в ацетил-КоА:
СН8СН2СН2СО ~ SKoA -> СН3СН=СНСО ~ SKoA -> СН3СН (ОН) СН2СО ~ SKoA ->
HSKoA
-► СН8СОСН2СО ~ SKoA-------*• 2СН3СО ~ SKoA.
Ацетильная группа ацетил-КоА окончательно окисляется до двуокиси угле-
рода и воды в цикле трнкарбоновых кислот.
Биосинтез жирных кислот протекает при участии ацильных форм ко-
фермента А, исходя из ацетил-КоА [229], в который при участии биотино-
вых ферментов включается двуокись углерода — при этой реакции образу-
ется малонил-КоА [230], превращающийся далее в соединения с увеличен-
ным количеством углеродных атомов [231 ]; среди них, вероятно, находятся
бутиромалонил-КоА и другие ацилмалонил-КоА
91
соон соон
I I
CH3CO ~ SKoA -> СН2СО - SKoA -> СН3СН2СН2СОСНСО ~ SKoA -> СН,(СН2)„СООН
Ингибитором реакций, катализируемых коферментов А, является п-хлор-
меркурбензоат. Оксикофермент А служит конкурентным ингибитором
КоА в фосфотрансацетилазной реакции [232].
СПИСОК ИСПОЛЬЗОВАННОЙ
1. R. Williams, С. Lyman,
G. Goodyear, J. Trues-
d a i 1, D. H a 1 a d a y. J. Am.
z- Chem. Soc., 55, 2912 (1933).
4 2. E. Stiller, S. Harris,
J. Finkelstein, J. Ke-
reszztesy, K. Folkers. J.
Am. Chem. Soc., 62, 1785 (1940).
3. R. К u h n, T. Wieland. Ber.,
73, 971, 1134 (1940); .пат. ФРГ
885848; Zbl., 1954, 11495.
4. A. Griissner, M. Gat zi-
Fichter, T. Reichstein.
Helv. Chim. Acta, 23, 1276 (1940).
5. H. Mitchell, H. Wein-
stock, E. Snell, S. S tan-
fa e r v, R. W i 1 1 i a m s. J. Am.
Chem. Soc., 62, 1776 (1940).
6. D. Woolley, H. Wais-
man, O. Michelson,
С. E 1 v e h i e m. J. Biol. Chem.,
125, 715 (1938).
7. R. Wil liams, J. Trues-
dail, H. Weinstock,
E. Rohrman n, C. Lyman,
C. Me Burney. J. Am. Chem.
Soc., 60, 2719 (1938).
8. S. L e p k о v s k у, T. Jukes,
M. Krause, J. Biol. Chem.,
115, 557 (1936).
9. F. Kogan, R. H e i n z e 1 -
man, D. W e i s b 1 a t,
W. G r e i n e r. J. Am. Chem. Soc.,
79, 3545 (1957).
10. К- Соя, С. T а д з а к и,
X. К у p и т а, А. К и т а д э,
И. С а к и я. Яп. пат. 2330 (1965);
РЖХ, 1968, ЗН440.
11. W. Braun, Н. G о t t h а г d t,
W. Bamberg, D. Schmidt,
H. Gruner t, D. Bartel.
Пат. ГДР 45705; С. A., 65, 3757
(1966).
12. R. Williams, S. Saun-
ders. Biochem. J., 28, 1887 (1934). \/
12. M. К о h i g a, S. К о j i m a,
К. M i z u h а г а, Яп. пат. 15822
(1964); С. A., 62, 7647 (1965).
14. G. E r 1 e m a n n, W. G u e x,
O. S c h i n d 1 e r, M. W a 1 t e r,
F. Weber. Parfuem. Kosmetik, 43,
№ 2, 38 (1962); C. A., 58, 1337 (1963).
15. S. H a r r i s, K. F о 1 k e г s, пат.
США 2557284; С. A., 46, 3071
(1952)..
16. T. Wi el a n d, E. Moller,
G. Dieckelmann. Chem. Ber.,
85, 1035 (1952).
ЛИТЕРАТУРЫ (К ГЛАВЕ II)
17. M. Vernsten, W. Braaten,
M. M о о r e. J. Am. Chem. Soc.,
76, 5811 (1954).
18. M. Freed. Пат. США 2653968;
С. A., 48, 8817 (1954).
19. Франц, пат. 1451182; РЖХ, 1968,
ЗН441.
20. G. О h t а, О. N a g a s е, Y. Н о-
sokawa, Н. Т a g a w а,
М. Shimizu. Chem. Pharm.
Bull. (Tokyo), 15, 644 (1967).
21. J. C a v a 1 1 a. J. Med. Chem.,
10, 743 (1967).
22. O. S c h n i d e r. Jubilee Vol. Emil
BarelL, 1946, 85; C. A., 41, 6199
(1947); англ. пат. 569083; С. A.,
41, 5146 (1947); пат. США 2413077; ’’
С. А., 41, 4802 (1947). ‘
23. R. W i 1 1 i a m s, Н. Wein- !
stock, E. R о h r m a n n,
J. T r u e s d a i 1, H. M i t -
c h e 1 1, С. M e у e r. J. Am. Chem.
Soc., 61, 454 (1939).
24. R. W i 1 1 i a m s, R, Moser. J.
Am. Chem. Soc., 56, 169 (1934).
25. D. W о о 1 1 e у, H. W a i s m a n,
С. E 1 v e h j e m. J. Am. Chem. ?
Soc., 61, 977 (1939); J. Biol. Chem., ’
129, 673 (1939). 1
26. H. W e i n s t о c k, H. M i t - ?
c h e 1 1, E. P r a t t , R. W i 1 - 1
1 i a m s. J. Am. Chem. Soc., 61, '•
1421 (1939).
27. D. Woolley. Science, 91, 245
(1940).
28. R. W i 1 1 i a m s, R. Major.
Science, 91, 246 (1940).
29. E. Stiller, J. Keresztesy,
J. F i n k e 1 s t e i n. J. Am. Chem.
Soc., 62, 1779 (1940).
30. R. W i 1 I i a m s, H. M i c h e 1 1,
H. W e i n s t о c k, E. S n e 1 1.
J. Am. Chem. Soc., 62, 1784 (1940). .
31. R. Williams. Science, 89, 486
(1939).
32. T. R e i c h s t e i n, A. Gruss-
n e r. Helv. Chim. Acta, 23, 650
(1940). <
33. E. A b d e r h a 1 d e n, A. T о -
d a r. Z. physiol. Chem., 85, 118
(1913). j
34. D. Rogers. Пат. США 2418902;
С. A., 41, 5145 (1947).
35. Швейц, пат. 215779; С. А., 42,
4606 (1948).
36. S. Babcock, Т. Jukes. J.
Am. Chem. Soc., 62, 1628 (1940).
37. M. Gatzi - Fichte г,
H. R e i c h, T. R e i c h s t e i n.
92
Helv. Chim. Acta, 24, 185 (1941).
38. F. Pickel. H. Weinstock.
Пат. США 2442143; С. A., 42, 5174
(1948).
39. Г. А. В а й д л и х. Усп. химии,
40, 804 (1941).
40. F. К a g а п. Пат. США 2845456,
2848489; С. А., 53, 3085 (1959).
^И 41. Пат. США 3150175; РЖХ, 1967,
ДВ 11Н374.
ВВ 42. Ван Ши-цинь, Мэн Ч ж э н-
фан, Чэнь Чжи-тун.
Яосюэ тунбао, 7, № 2, 67 (1959);
^В РЖХ, 1960, 9385.
43. W. В г а и п, D. Schmidt,
ПК. W. Bamberg. Пат. ГДР 44955;
5» ; С. А., 65, 15498 (1966).
BBt 44. Е. С. Ж Д а н о в и ч, Г. С. К оз-
лова, Т. Д. Мариева,
Т. В. Мельникова,
Н. А., Преображенский,
В ЖОрХ, 4, 1361 (1968).
^В.. 45. R. Карр, R. G г i f f i t h. Пат.
ВЁЛ США 2935528; РЖХ, 1961,
В 16Л383.
НВ- 46. Е. Wilson, J. W е j 1 а г d,
М. Т i S h 1 е г. Пат. США 2496363;
ИВ' Zbl., 1951, I, 366; J. Am. Chem.
|К Soc., 76, 5177 (1954).
|^В 47. R. L e k b e r g, R. Ho r n-
St beck. Пат. США 2809213; С. A.,
В»- 52, 2347 (1958).
Ии 48. Н. We hr mei st er. Пат. США
ДВ 2780645; РЖХ, 1959, 50811.
|^В 49. Saburo Funabashi, К i -
mi vo Michi. Bull. Inst.
KbS Phys. Chem. Research (Tokyo), 22,
KT 972 (1943); C. A., 43, 7903 (1949).
Еж 50. F. В о n a t i, D. P i t r e. Farma-
co Ed. Sci., 14 , 43 (1959).
^B V51. J. Jaworska, W. L e wen-
stein. Польск. пат. 47774; С. A.,
61 > 2977 (1964).
^H 52. Nett. Appl. 6504764; C. A., 66,
K- 55066 (1967).
JB 53. E. S t i 1 1 e r, P. W i 1 e y. J. Am.
UH Chem. Soc., 63, 1237 (1941).
F» 54. R. .Kuhn, T. Weiland.
I- Ber., 74, 257 (1941).
В 55. A. О p f e r m a n п. Пат. ФРГ
| 964498; РЖХ, 1958, 62013.
' £- 56. A. S a 1 a m о n. Miiegyetemi Koz-
Г lemenyek, 1949, 72; C. A., 45, 552
I (1951).
f 57, H. I n h о f f e n, G. H e i n e -
, < m a n n. Пат. ФРГ 1067443; РЖХ,
1961, 13Л470.
58. Е. Glaser. 'Monatsh., 25, 46
S (1904).
59. M. К о h n, V. N е u s t a d t е г.
Д Monatsh, 39, 293 (1918).
/г. 60. L. Vanino. Handbuch der Pra-
parativen Chemie, 2, 3, 61 (1923).
Д 61. И. Губен. Методы органической
<’? химии, т. II, 40—42, Госхимиздат,
А- 1941.
62. L. Wessely. Monatsh., 21, 231
(1900).
63. Н. С а г t е г, L. N е у. J. Ат.
Chem. Soc., 63, 312 (1941).
64. Англ. пат. 597648; С. А., 42, 4606
(1948).
65. J. Ford. Пат. США 2852530; С. А.,
53, 6089 (1959).
66. Р. J о u s s е t. Франц, пат. 1175516;
С. А., 54, 17272 (1960).
67. Н. Klein. Пат. США 3024250;
РЖХ, 1964, 2Н37.
68. Е. С. Жданович, Е. И. Бя-
лая, В. Н. Крылова,
Н. А. Преображенский,
Н. А. Стрельчунас. Авт.
свид. СССР, 150525; РЖХ, 1964,
13Н250.
69. R. Major, J. Finkelstein.
J. Am. Chem. Soc., 63, 1368 (1941).
70. R. В e u t e 1, M. Tishler. Пат.
США 2474719; С. A., 43, 7038
(1949).
71. С. В e c k m a n n, H. Klein,
R. Griffith. Пат. США 2967869;
РЖХ, 1962, 10Л219.
72. H. К 1 е i n, R. К а р р, R. G г i f-
f i t h. Пат. США 3009922; РЖХ,
1963, 9H115.
73. S. В a u e r. S. Of sz ag h. 4e-
хосл. пат. 88086; C. A., 54, 15250
(1960).
V 74. R. Rin g. Пат. ГДР. 16982; РЖХ,
1960, 27805.
75. W. В r a u n, D. Schmidt,
W. Bamberg. Пат. ГДР 32628,
37505; РЖХ, 1966, 1H125;
х/ 1967, 2Н402.
76. Е. С. Ж д а н о в и ч, В. Н. Кры-
лова, Г. С. Козло-
ва, Н. А. Преображен-
• с к и й, ЖОрХ, 3, 826 (1967); авт,
свид. СССР 147182; Бюлл. изобрет.
z 1962, № 10, 24.
77. Е. С. Ж д а н о в и ч, Г. С. К о з-
лова, Т. Д. Мариева,
Т. В. Мельникова,
Н. А. Преображенский.
Авт. свид. СССР 201426; Бюлл.
изобрет. 1967, № 44, 39; ЖОрХ,
4, 1359 (1968).
78. R. Ring. пат. ГДР 16982; С. А., 54,
. 19494 (1960).
79. W. Ozegowski, Н. Наег-
i п g. Pharmazie, 12, 254 (1957).
У/'80. М. Dunkel, I. Loter,
Н. Klein. Пат. США 3185710;
РЖХ, 1967, 12Н442.
81. R. W i n t е г b о t t о m, W. Zie-
gler. Пат. США 354.39; РЖХ,
1971, 16Н104.
v 82. Р. Hammond. Пат. США
2739157; РЖХ, 1958, 51312.
83. X. Кларк, Л. Бэр. Синтезы
органических препаратов, сб. 2, 20,
ИЛ, 1949.
84. А. Н. Парши н, ЖОХ, 20, 1826
(1950).
85. Швейц, пат. 244600; С. А., 43,
5682 (1949).
86. И. Л. К н у и я н ц, Б. П. Фаб-
ричный. ДАН СССР, 68, 701
(1949).
87. W. Heintz. Lieb. Ann., 156, 36
(1870).
88. Е. Mulder. Вег., 9, 1903 (1876).
89. С. И. Лурье, Е. С. Ч а м а и.
ЖОХ, 22, 1631 (1952).
93
90. К- М о г s с h. Monatsh., 63, 220
(1937).
91. F. Popplsdorf, R. Lemon.
J. Org. Chem., 26, 262 (1961).
92. J. Ford. J. Am. Chem. Soc., 67,
876 (1945).
93. G. Carlson, C. Hotchkiss.
Пат. США 2335997, С. A., 38, 2972
(1944).
94. S. В u c, J. F о r d, E. Wise. J.
Am. Chem. Soc., 67, 92 (1945).
95. D. Szlompek-Westeruk.
Przem. chem., 44, 85 (1965).
96. R. G r i 1 f i t h, W. D i S a 1 v o,
R. Kapp, L. Rosenberg. Пат.
США 2819303; С. A., 52, 14656
(1958).
97. A. H. Кост. Вести. Моск, ун-та,
1947, № 2, 141; Уч. зап. МГУ,
сер. хим., 1950, VI, вып. 131, 37.
98. О. Bayer. Angew. Chem., 61,
229 (1949).
99. А. Т е р е и т ь е в, К. Ч у р с и-
на, А. Кост. ЖОХ, 20, 1073
(1950).
100. J. Ford, S. Вис, J. Greiner. J.
Am. Chem. Soc., 69, 844 (1947).
101. R. Be и tel, P. Klem-
c h и k. Пат. США 2956080;
С. A., 55, 5365 (1961).
102. S. C h о d г о f f, R. Kapp,
C. Beckmann. J. Am. Chem.
Soc., 69, 256 (1947).
103. И. T а к а м а, К. Й о к а м a,
T. M а ц и д a. Bull. Fac. Engng.
Hokkaido Univ., 1962, № 31, 139;
РЖХ, 1963, 21, 373.
104. О. Moe, D. W a r n e r. J. Am.
Chem. Soc., 71, 1251 (1949).
105. P. F и s i e r. Ann. chim., (12) 5,
882 (1950); C. A., 45, 7010 (1951).
106. A. G a 1 a t. J. Am. Chem. Soc.,
67, 1414 (1945).
107. В. M. P о д и о и о в, Н. Г. Я р-
ц е в а. Изв. АН СССР, ОХН,
1948, 251, 1950, 108.
108. D. Pitre. Farmaco Ed. Sci., 14,
137 (1959).
109. E. С. Ж Д а н о в и ч, E. И. Б я-
лая, Н.А. Преображен-
ский. ЖОХ, 31, 446 (1961).
НО. F. W е у g a n d, Вег., 74 , 256
(1941).
111. Швейц, патент 226014; С. А., 43,
2225 (1949).
112. R. R u g g 1 i, А. В u s i n g е г.
Helv. Chim. Acta, 25, 35 (1942).
113. A. M a t 1 a с k. Пат. США
2672480; С. A., 49, 3244 (1955).
114. D. В r e s 1 о w, G. Hulse,
A. M a t 1 a c k. J. Am. Chem.
Soc., 79, 3760 (1957).
115. M. К i r c z, V. V о i n e s k u,
E. H e n d 1 e r. Rev. chim. (рум ),
10, № 2, 72 (1959).
116. L. В о a t r i g h t. Пат. США
2734081; С. A., 50, 15580
(1956).
117. R. Kleinschmidt, J. Ma-
han. Пат. США 2831890; С. A.,
52, 15573 (1958).
118. H. R a u c h - P u n t i g a m.
Пат. ФРГ 1098949; С. A., 55,
25783 (1961).
119. F. P о p p e 1 s d о r f. Пат. США
3105092; РЖХ, 1966, 17H221.
120. T. J u k e s. J. Am. Chem. Soc.,
61, 454 (1939).
121. В. M. Веселкина. Успехи
современной биологии, 24, 33
(1947).
122. R. Davey, J. Stevens on.
Animal Sci., 22, № 1, 9 (1963).
123. J. M u e 1 1 e г, А. К 1 о t z. J.
Am. Chem. Soc., 60, 3086 (1938).
124. D. Pennington, E. Snell,
R. Williams. J. Biol. Chem.
135, 213 (1940).
125. Y. S u b a г о w, L. R a n e. J.
Am. Chem. Soc., 61, 1616 (1939).
126. J. S t e r n, A. Campil lo,
M. Coon. J. Am. Chem. Soc.
75, 1517 , 2277 (1953).
127. D. W о 1 1 e y. J. Biol. Chem.,
134, 461 (1940).
128. H. Mitchell, E. Snell,
R. Williams. J. Am. Chem.
Soc., 62, 1791 (1940).
129. Швейц, пат. 282945; С. A., 48,
10063 (1954).
130. О p f e r m a n n. Англ. пат.
693801; С. A., 49, 1783 (1955).
131. F. В e r g e 1, N. Hi ndley,
A. Morrison, R. Moss.
Chem. Ber., 85, 711 (1952).
132. J. Barnett, F. Robinson.
Biochem. J., 36, 357, 364 (1942).
133. F. Holly, R. Barnes,
F. Koninszy, K. Folkers.
J. Am. Chem. Soc., 70, 3088 (1948);
пат. США 2571852; С. A., 46,
8676 (1952).
134. A. Hussey, I. W i e 1 k. J.
Am. Chem. Soc., 72, 828 (1950).
135. Ryozo Jujo. J. Pharm. Soc.
Japan, 63, 236 (1943); C. A., 45,
553 (1951).
136. H. A r i у a m a, Sh. Kimura.
J. Vitaminol., 6, 52 (1960); C. A.,
54, 24919 (1960).
137. M. M. Ш e м я к и н. Успехи,
химии, 16, 279 (1947).
138. W. D г е 1 1, М. D u n n. J. Ат.
Chem. Soc., 70, 2057 (1948); 76,
2804 (1954).
139. J. М о о г е, Е. W i t t 1 е. Пат.
США 2807644; РЖХ; 1959, 79538.
140. Е. Р а г k е г, Т. С о g d е 1 1 et
al. J. Med. Chem., 6, 73 (1963).
141. N. Kaplan, M. S о о d a k. Fe-
deration Proc., 8, 211 (1949).
142. А. Браунштейн, E. Ефи-
ме ч к и н а. ДАН СССР, 71,
347 (1950).
143. F. L у n е n, Е. Reichert. An-
gew. Chem., 63, 47 (1951).
144. F. L i р m a n n, N. Kaplan,
G. N о v e 1 1 i, L. T u t t 1 e,
B. G u i r a r d. J. Biol. Chem.,
167, 869 (1947); 186, 235 (1950).
145. M. H о a g 1 a n d, G. Novel-
1 i. J. Biol. Chem., 207, 767 (1954).
146. J. В a d d i 1 e у, E. T h a i n. J.
Chem. Soc., 1951, 246, 2253, 3425;
94
. 1952, 800; англ. пат. 716337; С. А.,
49, 12531 (1955).
147. J. Baddiley, Е. Thain,
G. N о v е 1 1 i, F. L i р m a n п.
Nature. 171, 76 (1953).
148. W. Williams, Е. Н o f f-
J г g е n s е п, Е. S n е 1 1. J. Biol.
Chem., 177, 933 (1949).
149. G. В г о w n, J. С г a i g,
Е. Snell. Arch. Biochem., 27,
473 (1950).
150. R . M c R о r i e, P. M a s 1 e у,
W. Williams, Arch. Biochem.,
27, 471 (1950).
151. T. К i n g, I. F e 1 s, V. C h el-
del i n. J. Am. Chem. Soc., 71,
131 (1949).
152. P. Majerus, A. Alberts,
P. V a g e 1 о s. Proc. Natl. Acad.
Sci, U. S„ 51, 123 (1964); 53, 410
(1965).
153. E. S n e 1 1, G. В г о w п, V. P e-
t e r s, J. Cr a i g, E. W i t t 1 e,
J. Moore, V. McGlohon,
О. В i r d. J. Am. Chem. Soc., 72,
5349 (1950).
154. E. W i t t 1 e. J. Moore,
R. S t i p о k, F. P e t e r s о n,
V. McGlohon, О. В i r d,
G. Bro wn, E. S n e 1 1. J. Am.
Chem. Soc., 75, 1694 (1953).
155. E. F e 1 d e r, D. Pitre. Angew.
chem., 68, 755 (1956); Gazz. chim.
ital., 88, 401 (1958); пат. США
2857408; С. A., 53, 6103 (1959).
156. J. Baddiley, E. Thain.
J. Chem. Soc., 1951, 3421; 1952,
3783
157. H. В e i n e r t, R. Van
К о r f f, D. G r e e n, D. В u y-
ske, R. Handschuma-
c h e r, H. Higgins, F.
Strong. J. Am. Chem. Soc., 74,
854 (1952).
158. J. Gregory, G. N о v e 1 1 i,
F. L i p m a n n. J. Am. Chem.
Soc., 74, 854 (1952).
159. A. Michelson. Biochim. Bio-
phys. Acta, 50, 605 (1961); 93,
71 (1964).
160. A. Cornberg, W. Pricer.
J. Biol. Chem., 182, 763 (1950).
161. L. S h u s t e r, N. Kaplan. Fe-
deration Proc., 11, 286 (1952).
162. G. N о v e 1 1 i. Tederation Proc.,
12, 675 (1953).
163. F. L i p m a n n, N. К a p 1 a n,
G. N о v e 1 1 i, Tederation Proc.,
6, 272 (1947).
164. W. Devries. W. G о v i e r,
J. Evans, J. Gregory,
G. N о v e 1 1 i, M. S о о d a k,
F. L i p m a n n. J. Am. Chem.
Soc., 72, 4838 (1950).
165. F. L у n e n, E. Reichert,
L. R u e f f. Lieb. Ann., 574, 1
(1951).
166. M. Viscont i ni, K. Adank,
N. M e г к 1 i n g, K. Ehr-
hardt, P. Karrer. Helv.
Chim. Acta, 36, 835 (1953); 37,
375 (1954).
167. Англ. пат. 747369; С. A- 51, 475
(1957).
168. P. Karrer, M. Viskonti-
n i. Швейц, пат. 312546; РЖХ,
1959, 39777.
169. R. В о wm an, J. Ca v al 1 a. J.
Chem. Soc., 1954, 1171.
170. D. F I es, A. Ma г ко v ас -
P r p i e. Arhiv. kem., 27, 211
(1955).
171. M. Shimizu, G. О h t a,
O. N a g a s e, S. Okada,
Y. Hosokawa. Chem. Pharm.
Bull. (Tokyo), 13, 180 (1965); яп.
пат. 28929 (1965); С. A., 64, 11093
(1966).
172. T. W i e 1 a n d, E. В о к e 1 -
m a n n. Naturwissensch., 38, 384
(1951).
173. E. С. Ж д а н о в и ч, В. M. Ко-
пелев ич, H. A. Преобра-
женский. ЖОХ, 37, 361 (1967).
174. A. Boehringer, E. Boeh-
ringer, I. Liebrecht,
J. Liebrecht. Англ. пат.
707709; С. A., 50, 4202 (1956).
175. J. О s b о n d. Англ. пат. 749715;
РЖХ, 1959, 75835.
176. E. Walton. Пат. США
2835704; С. А., 52, 17113 (1958).
177. J. Baddiley, Е. Т h a i n.
Пат. ФРГ 945090; РЖХ, 1958,
2285
178. Y. О s а п а, К. U е п о, N. Y а -
m a d а. Т. Wada. Яп. пат. 25418
(1965); С. А., 64, 9603 (1966).
179. Т. King, Ch. Stewart,
N. Cheldelin. J. Am. Chem.
Soc., 75, 1290 (1953).
180. E. Walton, A. W i 1 so n,
F. Holly, K. Fo 1 ke r s. J.
Am. Chem. Soc., 76, 1146
(1954).
181. M. M a t s u i , N. M i у a n o,
T. Sato. Яп- пат. 12111 (1961);
С. A., 58, 10089 (1963).
182. T . King, F. S t г о ng. J.
Biol. Chem., 191, 515 (1951).
183. J. В a d d i 1 e у, E. Thain.
Франц, пат. 1071149, РЖХ, 1956,
66410; англ. пат. 721950; С. А.,
.50, 2658 (1956).
184. F. Atherton. Англ. пат.
793017; С. А., 53, 2109 (1959).
185. S. О к a d а, О. N a g s е, М. S h i-
m i z u. Chem. Pharm. Bull. (To-
kyo), 15, 713 (1967).
186. Y. M i n o, Y. К a n a i. Яп.
пат. 2495 (1971); С. A., 75, 6337
(1971).
187. M. Yoshioka, К. Same-
j i m a, Z. Tamura. Chem.
Pharm. Bull., 17, 1265 (1969).
188. J. В a d d i 1 e у, E. Thain.
J. Chem. Soc., 1953, 1610.
189. В. M. Копелев и ч,
E. С. Жданович,
Н. А. Преображенский.
ЖОХ, 38, 1700 (1968).
190. В. М. К о п е л е в и ч.
С. Б. Сергиенко,
Е. С. Жданович,
95
Н. А. Преображенский.
ЖОХ, 38, 1135 (1968).
191. О. N agase. Chem. Pharm.
Bull. (Tokyo), 15, 648 (1967).
192. G. В a d d i 1 e у, E. T h a i n.
Англ. пат. 749512, 766536: РЖХ,
1959, 72363, 68854.
193. J. Moffatt, H. Khor ana.
J. Am. Chem. Soc., 80, 1265 (1959);
83, 663 (1961).
194. J. В a d d i 1 e у, E. Thai n.
Пат. ФРГ 1001996; С. A., 55,
1464 (1961).
195. Y. S а п n о, H. О s h i o,
Y. Kanai, H. H о n d а. Яп.
пат. 7213 (1968); С. A., 69,
76 675 (1968).
196. В. M. К о п е л е в и ч,
Е. С. Жданович,
Н. А. Преображенский.
Хим.-фарм. ж. 1,№11, 26 (1967).
197. V. Clark, S. Warren. J.
Chem. Soc., 1963, 178; 1965, 5509.
198. N. H a m e r. J. Chem. Soc. «С»,
1966, 404.
199. M. S h i m i z u, О. H a g a s e,
S. Okada, U. Hosokawa,
H- T a g a w a, U. A b i k o,
T. Suzuki. Chem. Pharm. Bull.
(Tokyo), 15, 655 (1967); C. A., 68,
13310 (1968).
200. M. Shimizu, О. H a g a s e,
S. Okada, T. Hosokawa,
H. T a g a w а. Яп. пат. 11078,
11079 (1967); С. A., 67, 108965,
108971 (1967).
201. M. Shimizu, О. Nagase,
S. О k a d a, Y. Hosokawa,
H. T a g a w a. Chem. Pharm.
Bull. (Tokyo), 13, 1142 (1965).
202. M. S h i m i z u, О. H a g a s e,
S. О k a d a, U. A b i k o,
T. Suzuki. Chem. Pharm.
Bull. (Tokyo), 14, 683 (1966).
203. W. Griither, H. Mautner.
J. Am. Chem. Soc., 87, 2708 (1965).
204. T. Miller, G. Rowley,
-Ch. Stewart. J. Am. Chem.
Soc., 88, 2299 (1966).
205. M. S h i m i z u, O. N a g a s e,
Y. Hosokawa, H. T a g a -
w a. Tetrahedron, 24, 5241 (1968).
Яп. пат. 24915, 24913 (1967); С. A.,
69, 97103, 97104 (1968).
206. A. Michelson. Biochim.
Biophys. Acta, 91, 1 (1964).
207. F. C r a m e r, G. Kenner,
N. H u g h e s, A. Todd. J.
Chem. Soc., 1957, 3297.
208. J. Baddiley, J. Bucha-
nan, R. Letters. J. Chem.
Soc., 1958, 1000.
209. L. F о g a r t y, W. R e e s. Na-
ture, 193, 1180 (1962).
210. M. Диксон, Э. У э б б. Фер-
менты, M., изд-во «Мир», 1966.
211. F. L i р m а п n, М. J о n е s,
S. В 1 а с k, R. F 1 у п n. J. Ат.
chem. Soc., 74 , 2384 (1952).
212. Р. В е г g. J. Am. Chem. Soc., 77,
3163 (1955); Science, 129, 895
(1959).
213. Р. В о у е г, О. К о е р р е,
W. L и с h s i п g е г, A. Fal-
cone. Federation Proc., 14, 185
(1955).
214. Р. Т а 1 b е г t, F. Н uen п е-
к е n s. J. Am. Chem. Soc., 78,
4671 (1956).
215. A. Lehninger, G. Grevil-
1 e. Biochim. Biophys. Acta, 12,
188 (1953).
216. B. Bergstrom, L. WheeJ-
1 о n. Biochim. Biophys. Acta, 50,
171 (1961).
217. R. Mazumder, D. Sa na di,
W. R о d w e I I. J. Biol. Chem.,
235, 2546 (1960).
218. J. Porter, R. Long. J. Biol.
Chem., 233, 20 (1958).
219. J. Berry, V. Whittaker.
Biochem. J., 73, 447 (1959).
220. R. Brady, E. Stadtman.
J. Biol. Chem., 211, 621 (1954).
221. H. W e i s s b a c h, B. Red-
field, J. A x e 1 г о d. Bio-
chim. Biophys. Acta, 54, 190 (1961).
222. F. L у n e n, S. О c h о a. Bio-
chim. Biophys. Acta, 12, 299
(1953).
223. J. Stern, B. Shapiro,
E. Stadtman, S. Ochoa.
J. Biol. Chem., 193, 703 (1951).
224. P. S с г i b a, H. H о 1 z e r. Bio-
chem. Z„ 334, 473 (1961).
225. J. G и n s a 1 и s. Feder Proc., 13,
715 (1954); The Mechanism of En-
zyme Action, John Hopkins Press,
1954, 545.
226. H. Grisebach. Angew. Chem.,
68, 554 (1956).
227. V. Massey. Biochim. Biophys.
Acta, 38, 447 (1960).
228. J. Hauge, F. Grane,
H. В e i n e r t. J. BioL Chem.,
219, 727 (1956).
229. F. L у n e n. Angew. Chem., 77,
929 (1965).
230. S. W a к i 1. J. Am. Chem. Soc.,.
80, 6465 (1958); J. Lipid Research,'
2, 1 (1961).
231. S. W a к i 1, R. Bressler. J.
Biol. Chem., 236, 1643 (1961).
232. T. Miller. G. Rowley,
C. S t e w a r t. J. Am. Chem. Soc.,
88, 2299 (1966).
Часть вторая
ВИТАМИНЫ АЛИЦИКЛИЧЕСКОГО РЯДА
ГЛАВА III
ЦИКЛОГЕКСАНОЛ-ЭТИЛЕНГИДРИНДАНОВЫЕ ВИТАМИНЫ И ИХ
ПРОВИТАМИНЫ
КАЛЬЦИФЕРОЛЫ
Кальциферолы — витамины группы D — являются производными
6-(3а-окси-10-метиленциклогексан- 5-илен)-7-(13р- метилгидриндан-8-илен)-
этана, замещенными в положении 17 алифатической разветвленной цепью
из 8—10 атомов углерода.
Углеродный скелет циклической части молекулы кальциферолов (по-
гречески «феро» — несущий) вследствие своей генетической связи со стери-
нами имеет своеобразную нумерацию атомов:
В свою молекулу кальциферолы включают циклогексановое кольцо А
и систему гидриндана из срощенных циклогексанового кольца С и цикло-
пентанового кольца D в транс-сочленении с ангулярной метильной группой
Р-конфигурации (кольцо В, находившееся в исходных стеринах, при превра-
щении в витамин D расщепляется и в молекуле витамина D отсутствует).
Кольца А и С соединены этиленовым мостиком с двумя экзоцикл ическими
двойными связями с вытянутой s-транс-конфигурацней1 [1,2]. Кольцо А
имеет гидроксильную группу (а-конфигурации) и метиленовую группу,
входящую в характерную ненасыщенную систему из трех сопряженных
двойных связей: Qis)—Сцо), С(5)—С(6> и С(7)—С(8) с 5-^нс-конфигурацией.
Поскольку молекула витамина D генетически связана со стеринами,
то ангулярную метильную группу, как и для стеринов, условно представля-
ют расположенной над плоскостью чертежа молекулы, изображая ее связь
с кольцом сплошной линией; заместители, находящиеся к ангулярной ме-
тильной группе в tjuc-положении, обозначают как р-заместители, а находя-
щиеся в транс-положении, т. е. за плоскостью чертежа молекулы, изобра-
жают пунктирной линией и обозначают как а-заместители [3].
1 s — означает вид цис-транс-пзомерин относительно простой (ординарной —
simple bond) связи, которая находится между двумя двойными связями.
4—69
97
К природным витаминам относятся: эргокальциферол — витамин D2 (I),
холекальциферол — витамин D3 (II) и дигидроэргокальциферол — вита-
мин D4 (III) 14, 5].
Природные и синтетические кальциферолы являются аналогами природ-
ного витамина D3, отличающимися от него боковой алифатической цепью
положения 17. Они характеризуются различными алкильными заместите-
лями при C(2t) у витаминов D2, D4, D5, De и D7 и двойной связью в алифати-
ческой части молекулы у витаминов D2 и D6 (табл. 3). Витамин представ-
ляет собой эквимолекулярное соединение эргокальциферола (витамина D2)
с лумистерином2 [61.
Заместители алифатической цепи, конфигурация асимметрических цен-
тров которой с достоверностью не связана с конфигурацией ядра, обозна-
чаются произвольно буквами «а» (связь заместителей с С<20> и С(24> изобра-
жается пунктирной линией) и «Ь» (связь изображается сплошной ли-
нией).
Насыщенная алифатическая цепь из шести атомов углерода с боковой
метильной группой b-конфигурации у С(20) и метильной группой у С(25)
содержится в молекуле холекальциферола (витамина D3). Все другие вита-
мины группы D имеют метильную группу при С(2о) также Ь-конфцгу-
рации.
Гомологами витамина D3, содержащими в алифатической цепи алкильные
заместители при С(24), являются дигидроэргокальциферол (витамин D4) с ме-
тильной группой b-конфигурации, ситокальциферол (витамин D(5)) с этиль-
ной группой b-конфигурации и кампекальциферол (витамин D7) с метильной
группой а-конфигурации, т. е. с противоположной конфигурацией, чем у
витамина D4 [81.
Алифатическая цепь витаминов D2 и D6 у C(i7) отличается от алифати-
ческой цепи витамина D3 присутствием двойной связи между С(22) и С(2з>-
Эти же витамины одновременно являются гомологами витамина D3, так
как содержат алкильные заместители при С(24>: витамин D2 — метильную
группу b-конфигурацип и витамин D6 — этильную группу Ь-конфигурации
[9].
Эргокальциферол — витамин D2 (I) — представляет собой бес-
цветные призмы (из ацетона) или иглы (из метилового спирта) без запаха,
с т. пл. 120—121° С [10] (в капилляре, запаянном в вакууме). Оптическое
вращение эргокальциферола [111: [а Ш + 106,25°, [а Шел +125,0°
(С2Н5ОН); [а Id + 83,5°, [а Швл + 99,5°(СН3СОСН3); [а $ + 88,75°;
(а ®6,1 + 105,5° (эфир); [а $+52,25°, [а Шел + 61,75°(СНС13);
[а $ + 87,5°, 1а $6,1 + 102,12° (С3Н3). Эргокальциферол имеет максимум
поглощения ультрафиолетового света 265 нм (в гексане или эфире),
е 1,94-10'* (А! с°м490) в спирте [10, 12].
Холекальциферол —- витамин D3 (II) —• кристаллизуется в
бесцветных тонких иглах с т. пл. 84—85° С (в капилляре, запаянном в
вакууме) [13,14]; [а $ + 105, + 112° (С2Н5ОН); [а Id + 83,3° (СН3СОСН3),
98
Кальциферолы (витамины группы D)
Т а блица 3
Наименование витамина* * Буквенное обозначение витамина Количество асимметричес- ких центров R—алифатический заместитель у Провитамины
Холекальциферол (кальциферол) D3 5 ^Нз хСНз -*CHCH2CH2CH2CH \н3 7-Дегидрохолесте- рин
Эргокальциферол (виостерол) D, 6 СИ, сн, ,сн3 1 3 ti 3/ж 3 -*снсн=снснсн 20 22 23 24 2^\ С Flo Эргостерин
СН, сн, .сн.
Диг идроэргокаль- циферол D* 6 -*СНСН£Н2СНСН \н3 22-Дигидроэрго- стерин
Ка мпека льцифе- рол D, 6 СНз # /СНз Л:нсн2сн2снсн сн3\н2 7-Дегидрокампес- терин
Ситокальциферол D6 6 сн3 с2н5/сн3 -*снсн2сн2снсн ХСНз 7-Дегидроситосте- рин
Стигмакальцифе- рол De 6 CH, C2HS/CH3 -*снсн=снснсн \н. 7-Дегидростиг- мастерин
1 Номенклатура установлена 18-й конференцией Международного союза по чис-
той и прикладной химии в 1955 г. [7].
Для витаминов группы D предложена [3] также номенклатура, генетически свя-
*5(6)-цисЛ, 10(19),22
зывающая их со стеринами [2J: для витамина D2 — 9,10-секо-Д -эрго-
статетраен-За-ол (приставка «секо» обозначает разорванную кольцевую связь); для
витамина D3—9,10-секо-Д -холестатриен-За-ол; для витамина D4 —
„ . 5(6)-цис,7> 10(19)
— 9,10-секо-Д -эргостатриен-За-ол.
[а]п + 51,9° (СНС13) [15]. Спектр поглощения имеет максимум при
265 нм; е 1,88-104 (Е1, см490) в спирте или гексане [16, 17].
Д и г и дроэргокальциферо л—витамин О4(Ш)—кристаллизу-
ется в листочках с т. пл. 106—108° С; [а ]о + 149°" (С2Н6ОН),
[а Id + 89,3° (СН3СОСН3) [18,19]. Максимум абсорбции 265 нм.
Ситокальциферол — витамин D5 (из 7-дегидроситостерина),
сти гма к а л ьцпфе рол — витамин DG (из 7-дегидростигмастерина)
и кампекальциферол — витамин D7 (из 7-дегидрокампестерина)
[8] — в кристаллическом виде не получены.
Кальциферолы растворимы во всех обычных органических растворите-
лях, а также в жирах; нерастворимы в воде. В 100 мл растворяется: при
7° С — в ацетоне 7 г эргокальциферола; при 26° С— в ацетоне 25 г, в
безводном спирте 28 г и в этилацетате 31 г эргокальциферола.
Эргокальциферол и холекальциферол образуют молекулярные продукты
присоединения: первый—с лумистерином (витамин D^, второй — с про-
дуктом облучения изодегндрохолекальциферола, вещества, сопровождаю-
щего синтетический 7-дегидрохолестерпн 120].
4
99
Холекальциферо л-х олестерин представляет собой ус-
тойчивый молекулярный продукт присоединения 1 моля холе кальциферола
и 1 моля холестерина; бесцветные кристаллы с т. пл. 117 — 121° С,
laic +24, 4-28° (СН3СОСН3), нерастворимые в воде, растворимые в
спирте, эфире, ацетоне, хлороформе, бензоле.
ХИМИЧЕСКИЕ СВОЙСТВА КАЛЬЦИФЕРОЛОВ
Кальциферолы по своей гидроксильной группе легко дают сложные
эфиры.
Важнейшие сложные эфиры эргокальциферола (D2) [10]: ацетат —
т. пл. 86° С, [al579 + 38° (СН3СОСН3); бензоат—т. пл. 92° С,
la ]д + 100° (СН3СОСН3); аллофанат—т. пл. 194— 195° С,
la Id 4- 50,4°(СНС13); п-нитробензоат — т. пл. 93° С, [а Id + 104° (СЫС13);
3,5-динитробензоат — т. пл. 148 — 149° С, [а Ь + 55° (CGH6),
(а Ь + 89° (СНС13) [21]; фосфат—натриевая соль (водорастворимая)
[22].
Важнейшие сложные эфиры холекальциферола (D3): аллофанат —
т. пл. 173° С, п-нитробензоат — т. пл. 127° С, [a Id + 114°(СНС13) [23, 241;
3,5-динитробензоат — т. пл. 128—129° С (из СН3ОН) и 133—134° С (из
эфира), [а 1о + 98° (СНС13); фосфат, натриевая соль С54 Н89-91О12-1зРз^а4
(водорастворимая)—т. пл. 215—216° С [25]; бутират [26].
Важнейшие сложные эфиры дигидроэргокальциферола (D4): 3,5-динитро-
бензоат — т. пл. 135—136°С, lab +95,4° (СН3СОСН3).
Кальциферолы не устойчивы к действию ультрафиолетового света;
происходящий при этом фотохимический процесс необратим. Реакция фото-
изомеризации кальциферолов приводит к образованию супрастерина-1,
супрастерина-Н и токе истер ина [27].
При фото изомеризации эргокальциферола вновь происходит замыкание
кольца В (отсутствующее в его молекуле), но образующаяся четырехколь-
цевая система супрастерина2-1 и супрастерина2-II (IV) [28] не имеет стероид-
ного строения [29, 30], а получаются спиросоединения (схема 19). При
фото изомеризации в течение 2,5 ч (возбуждение с л 260 — 280 нм) получа-
ются главным образом эти спиросоединения [31]. Супрастерин-I и супра-
стерин-П выделены в виде их эфиров: аллофаната супрастерина-1—
т. пл. 225—226° С, [a Id—58,4°; ацетата супрастер ина-1—т. пл. 73° С,
laic — 71° (СНС13); аллофаната супрастерина-II—т. пл. 224° С,
la Id 4* 80°.
При длительном облучении эргокальциферола ультрафиолетовым све-
том образуется токсистерин2; его строение не установлено. Он выделен в
виде вещества с т. пл. 50° С [32 ] и со спектром поглощения, имеющим макси-
мум около 248 нм и е 18 300 [33]; [a Id — 16° (СНС13) [32]. Токсистерин
обладает токсичными свойствами и как витамин неактивен [34]. В спиртовой
среде он образуется легче, чем при облучении в среде эфира [35]. После
облучения в течение 14,5 ч в спирте выделены токе истер ин 2-А, токсисте-
pHH2-Ax и токе истер ин 2-В [31].
Эргокальциферол (D2) устойчив к нагреванию и при 125° С в глубоком
вакууме может сублимироваться почти без разложения. При более высокой
температуре (160—180° С) он испытывает термическую циклизацию с обра-
зованием двух стереоизомеров: пирокальциферола2, имеющего 9a, 10a-кон-
фигурацию (V) [36], и изопирокальциферола2, имеющего 9|3, 10|3 -конфигу-
рацию (VI) [37], причем их получение сопровождается возникновением сте-
роидной структуры путем замыкания кольца В системы пергидроцикло-
пентанофенантрена вновь между атомами углерода положения 9 и 10,
но в ином пространственном направлении (схема 19). Термическая изоме-
ризация кальциферолов дает только два син-9, 10-изомера.
100
Схема 19
VII VIII
Холекальциферол (Ds) несколько более чувствителен к повышенной
температуре и менее устойчив при комнатной температуре, чем эргокаль-
циферол. При нагревании при 200° С без доступа воздуха он разлагается
с образованием пирокальциферола3 и изопирокальциферола3 [38].
В результате облучен ия пирокальциферола2 (V) и изопирокальциферола2
(VI) ультрафиолетовым светом происходит изомеризация двойных связей
в кольце В их молекулы и образование фотоизомеров [39]: фотопиро-
кальциферола2 (VII) и фотоизопирокальциферола2 (VIII). Фотопиросое-
динения получаются в кристаллическом виде и при нагревании до 190° С
вновь изомеризуются в исходные пиросоединения [39].
Эргокальциферол чувствителен к кислороду воздуха; его эмульсия с
водой почти полностью разрушается через пол года, однако его раствор в
оливковом масле при хранении в присутствии воздуха разрушается только
наполовину в течение 5 лет [40].
При действии кислорода воздуха на холекальциферол (II), адсорбиро-
ванный на активном глиноземе (флоридине), через 10-окси-10-метилпроиз-
водное происходит окисление его молекулы с образованием 9,10-секо-Д5-хо-
лестен-7-он-Зр, 10-диола, «кетона 2503» (IX), с д 250 нм и е 22 000 [41],
обладающего 10%-ной активностью витамина D3. Это соединение было вы-
делено также из жира печени тунца [42]; в жире печени трески оно нахо-
дится, в отличие от холекальциферола, в неэтерифицированном состоянии.
Окисление холекальциферола шрет-бутилатом аммония в смеси ацетона
с бензолом приводит к получению «изокетовитамина D3» (Х)ст. пл. 75° С
и выходом 30% [43].
Марганцовокислый калий в нейтральном или слабощелочном растворе
окисляет эргокальциферол [I] в 7,8-диокси-7,8-дигидроэргокальциферол
101
(XI) [44]. Этот же окислитель в более жестких условиях реакции наряду с
хромовым ангидридом или озоном расщепляет молекулу по С<з) —С(6) или
С(7) — С(8) двойной связи [43].
IX х XI хн
r=ch(ch^ch=ch(ch3)ch(ch3)2 или сн(сн3)сн2сн2сн2сн(сн3)2
При окислении эргокальциферола (I) тетраацетатом свинца идет присое-
динение гидроксильных или ацетильных групп в положение 5 и 6, и в ре-
зультате последующего гидролиза образуется эргокальциферол-5,6-диол
(XII) с выходом 25% (45], который присоединяет только 6 атомов водоро-
да, не имеет сопряженной системы двойных связей и при дальнейшем окис-
лении дает ненасыщенный альдегид С21 (XIII)
Пергидрол в бензонитриле окисляет эргокальциферол (I) в 5,6-эпокись
эргокальциферола [46].
Холекальциферол (II) при действии хлоранила подвергается дегидроге-
низации [47].
При частичном каталитическом гидрировании эргокальциферола со
скелетным никелевым катализатором в первую очередь происходит восста-
новление семициклической двойной связи [48]. Кристаллический 10,19-ди-
гидроэргокальциферол (XlVa, б) выделен в виде двух изомеров (схема 20),
различающихся по a-или В-конфигурации заместителя при С(19>:
дигидроэргокальциферола-IV (XlVa) [49]ст. пл. 63° С, [а Ь + 19,4°(СНС13)
и дигидроэргокальциферола-II (XIV6) [48, 49] с т. пл. 108° С,
[alo + 131,6° (СНС13). 10,19-Дигидроэргокальциферол не дает аддукта с
малеиновым ангидридом и имеет почти вдвое больший коэффициент экстинк-
ции максимума ультрафиолетового спектра поглощения, вследствие чего
этому соединению приписывается структура сД5>7-диеновой системой
Б-лгранс-конфигурации [48, 49].
Если дигидроэргокальциферол-II (XIV6) подвергнуть изомеризации
под действием лампы дневного света при каталитическом участии йода,
то образуется дигидротах истер ин (XV) с выходом 59% [50]. Это же соеди-
нение (XV) получается и при восстановлении эргокальциферола (I) натри-
ем в ксилоле [51 ]. Подобным образом восстанавливается и холекальцифе-
рол [51, 52]. Дигидротах истер ин (XV) имеет две сопряженные связи
С(5) —С(б) иС(7)—С(8) 153]; он поглощает в ультрафиолетовой области XMaKC
241 и 261 нм. Дигидротахистерин обладает слабой антирахитической актив-
ностью и применяется как противостолбнячный препарат (антитетанический
препарат № 10, АТ 10) [54], при этом он одновременно увеличивает и со-
держание кальция в крови.
102
Схема 20
Реакции восстановления эргокальциферола
Эргокальциферол (I) и холекальциферол (имеющие 5,6-^ас-конфигура-
цию) при каталитическом участии йода или эозина в нейтральной или
щелочной среде испытывают цис, транс-изомеризацию и превращаются в
5,6-транс-эргокальциферол (XVII) и 5,6-транс-холекальциферол [51, 55,
56]. Цис, транс-изомеризации способствует пространственная затруднен-
ность между атомами водорода в положениях 7 и 19. Образовавшийся в
результате изомеризации 5,6-транс-эргокальциферол (XVII) каталити-
ческим гидрированием с никелевым катализатором или восстановлением
натрием в ксилоле [51] превращают в дигидротахистерин (XV). Подобным
образом 5,6-транс-холекальциферол восстанавливается в дигидротахи-
стеринз [51, 55].
Частичное восстановление эргокальциферола (I) натрием в пропиловом
спирте приводит к 1,4-присоединению и образованию 6,19-дигидроэргокаль-
циферола (дигидроэргокальциферола-1, XVI) [57], который имеет две не-
сопряженные двойные связи С(ю)—С(5) и С<7)—С(8> и не поглощает ультра-
фиолетового света.
При исчерпывающем гидрировании все двойные связи эргокальцифе-
рола (I) восстанавливаются и образуется полностью насыщенный октагид-
роэргокальциферол (XX) [58] (см. с. 106).
В присутствии эфирата трехфтористого бора или фосфорной кислоты
эргокальциферол (I) изомеризуется в изотахистерин (XIX), характеризую-
щийся наличием двойной связи С(б)— С(7) и полной транс-Д10<5)-6>8<,4>-
триеновой конфигурацией [59]. Эта реакция протекает через промежуточ-
ное образование изоэргокальциферола (XVIII) [60].
Эргокальциферол 5 fi-транс -Эргокальциферол Изоэргокальцифероп Изотахистерин
XVIII XJX.
r= сн(сн3)сн=снсн(сн3)сн(сн3)2
103
Изотах истер ин (XIX) также образуется при изомеризации 5,6-транс-
эргокальциферола (XVII), изоэргокальциферола (XVIII) [59, 61], пре-
кальциферола и тахистерина [62]. При изомеризации, катализируемой
йодом, эргокальциферол (I) превращается в 5,6-транс-эргокальциферол
(XVII) с выходом 75% [51, 56, 63], затем он последовательно превращается
в изоэргокальциферол (XVIII) и изотахистсрин (XIX) [63]. По-видимому,
изотахистерин (XIX) является окончательной стабильной формой, образую-
щейся при всех изомеризациях. Эта форма имеет энергетически наиболее
благоприятную конфигурацию, т. е. конфигурацию с наименьшими про-
странственными помехами и с максимальной копланарностыо ненасыщен-
ной системы [59].
Образующийся из эргокальциферола (I) при цис, /пране-изомеризации
5,6-транс-эргокальциферол (XVII) обладает наименьшей энергией и более
стабилен, чем 5,6-/{нс-эргокальциферол (I), однако 5,6-транс-эргокальци-
ферол (XVII) может быть подвергнут транс, цис-фото изомеризации при
действии ультрафиолетового света с инверсией в эргокальциферол (I).
Подобным образом ведет себя и холекальциферол. Выход при фотоизоме-
ризации удается довести до 30% [64].
Кальциферолы устойчивы к щелочам, но чувствительны к действию кис-
лот. Эргокальциферол (как и его эфиры) по реакции Дильса — Альдера
образует продукт присоединения с малеиновым ангидридом по сопряжен-
ным ненасыщенным связям у атомов C^gj иС(б> [65]. Реакция Дильса— Аль-
дера дает возможность установить сопряжение диеновой системы; она осно-
вана на способности этиленовой связи, находящейся между двумя карбокси-
лами (например, в малеиновом и цитраконовом ангидриде), вступать в
реакцию с диенами с образованием шестичленных циклов. Присоединение
идет по концам диеновой сопряженной системы.
Реакция Дильса—Альдера имеет большое значение в химии витами-
нов D, А и др. и применяется не только для обнаружения кратных связей,
но и для установления их геометрической (цис, транс) конфигурации.
Витамин D2 дает различные цветные реакции. С треххлористой сурьмой
в хлороформе образуется оранжево-желтое окрашивание с максимумом
поглощения при5Э0 нм [66, 671, которое количественно измеряется в электро-
фотоколориметре. Эга реакция характерна и в присутствии витамина А,
и стер инов, которые мало мешают.
Известны разнообразные другие цветные реакции: с пирогалловой кис-
лотой, трихлоруксусной кислотой, сахарозой, дихлоргидрином глицерина
(зеленая окраска сХмакс 625 нм), ароматическими альдегидами и др.; они
предложены для качественного и количественного определения витамина D
в витаминных концентратах и пищевых продуктах [68]. Описан метод
количественного определения витаминов D2 и D3 хроматографией на бума-
ге с применением в качестве подвижной фазы петролейного эфира, насы-
щенного водой [69].
СТРОЕНИЕ ЭРГОКАЛЬЦИФЕРОЛА И ДРУГИХ ВИТАМИНОВ
Эргокальциферол образуется в результате фото изомеризации эргосте-
рина, которая активируется ультрафиолетовыми лучами. Его строение
полностью установили Винда ус с сотр. [70] и Хельброн с сотр. [71, 72]
в 1935—1936 гг.
Эргокальциферол (I) содержит одну вторичную гидроксильную группу.
Трициклическая молекула эргокальциферола содержит четыре двойные
связи, наличие которых установлено по поглощению восьми атомов водо-
рода при каталитическом гидрировании [581, протекающем с образованием
октагидроэргокальциферола (XX). Однако при титровании с пербензойной
кислотой обнаруживаются только три двойные связи, находящиеся в сопря-
жении. Четвертая двойная связь устанавливается следующими реакциями.
При восстановлении натрием в спирте или ксилоле происходит 1,4-присое-
104
динение (и частично также 1,2-присоединение) двух атомов водорода к
двойным связям С(б)—С(5) и С(1Э) —С(|9) с образованием 6,19-дигидроэрго-
кальциферола (дигидровитамина D2—I) (XVI)[73, 741 с т. пл. 65°С,
[а I/J + 12,4° (СНС13) 175], который уже реагирует с пербензойной кисло-
той с образованием кристаллического триоксида, что указывает на наличие
еще трех двойных связей [76] (схема 21).
Присутствие в молекуле витамина четырех двойных связей допускает
строение молекулы на основании эмпирического состава (С98Н44О) только
из трех гидрированных колец (двух шестичленных иодного конденсирован-
ного пятичленного) вместо четырех гидрированных колец (трех конденси-
рованных шестичленных и одного пятичленного), находящихся в исходном
эргостерине, содержащем три двойные связи. Следовательно, при фотоизо-
меризации должно произойти раскрытие одного из циклов. Отсутствие
стероидного цикла в молекуле эргокальциферола доказывается и теми
данными, что при дегидрировании эргокальциферола (I) селеном не обра-
зуется углеводорода Дильса — 3'’-метил-1,2-циклопентанофенантрена
(XXI) [57], характерного для реакции дегидрирования эргостерина и дру-
гих стеринов.
Одна из двойных связей находится в боковой цепи в транс-конфигура-
ции (как и у эргостерина [1 ]), так как при озонировании образуется метил и-
зопропилацетальдегид (XXIII) [77]. Две или три из остальных двойных
связей находятся в сопряжении, что следует из типичных для такихсистем
спектров поглощения в области 250—320 нм, а также из способности эрго-
кальциферола образовывать аддукт с малеиновым ангидридом (XXVIII)
[70].
Отсутствие кольца В в эргокальцифероле (I) вследствие происшедшего
при фото изомеризации эргостерина расщепления по связи С(9) — С(10) с
образованием новой двойной связи доказывается реакциями окисления.
При окислении эргокальциферола хромовой кислотой за счет расщепления
двойной связи С(5> —образуется бициклический а, (3-ненасыщенный
альдегид С,! (XIII), не содержащий гидроксильной группы [71 ]. При окис-
лении марганцовокислым калием получены муравьиная кислота и бици-
клический ненасыщенный кетон С19 (XXV), содержащий кольца С uD и
алифатическую цепь, такую же, как и у ненасыщенного альдегида С21
(XIII) [72]. Ненасыщенный кетон С19 (XXV) имеет карбонильную группу
в положении 8. Это указывает на то, что его образование произошло в ре-
зультате разрыва двойной связи С(7> —С(8>.
При озонировании эргокальциферола отщепляется метил изопропил-
ацетальдегид (XXIII) в результате расщепления двойной связи С(22>—С(23>,
затем формальдегид и бициклическая кетокислота С13 (XXII) [70] в резуль-
тате разрыва связи С(7) —С(8>.
Из этих данных следует, что кольцо А молекулы эргокальциферола свя-
зано с остальной частью молекулы (кольцо С) системой углеродных атомов,
имеющей две двойныесвязи С(5> —С(5> и С(7) —С(8>- При окислении эфира
эргокальциферола (XXIV) наряду с ненасыщенным кетоном С19 (XXV)
образуется левовращающая р-метоксиадипиновая кислота (XXVII) [78, 79]
в результате расщепления кольца А.
Строение эргокальциферола доказывается и реакциями окисления его
производных. Дигидропроизводное продукта присоединения эргокальци-
ферола (как него эфиров) и малеинового ангидрида (XXVIII) [65] при озо-
нировании дает бициклический кетон С19 (XXIX) с насыщенной алифати-
ческой боковой цепью, который идентичен продукту гидрирования нена-
сыщенного кетона С19 (XXV) [70]. Аддукт эргокальциферола и малеинового
ангидрида (XXVIII) при дегидрировании селеном дает 2,3-димет ил наф-
талин (XXX), причем, что интересно отметить, карбоксильные группы,
входящие в молекулу малеинового ангидрида, восстанавливаются в метиль-
ные [65].
105
Схема 21
Установление строения аргокальциферола реакциями его превращений и расщепления
Эфир дикарбоновой кислоты (XXXI), полученный в результате гидро-
лиза аддукта эргокальциферола и малеинового ангидрида (XXVIII), при
дегидрогенизации с платиной дает нафталин и р-нафтойную кислоту, вклю-
чающие в молекулу кольцо Я эргокальциферола, а при дегидрировании
106
селеном образует тот же самый 2,3-диметилнафталин (XXX) [65]. Окисле-
ние дикарбонового эфира (XXXI) приводит к насыщенному кетону Ci9
(XXIX).
Для кальциферолов, образующихся в результате фотохимического рас-
щепления кольца В стеринов по связи между Qgj и Сцоц остаются многие
характерные для провитаминов пространственные конфигурации, не затраги-
ваемые расщеплением молекулы.
Кольца С и D в молекуле эргокальциферола построены путем транс-соч-
ленения, а заместители: ангулярная метильная группа у С(|з> и алифати-
ческая боковая цепь (R) у С(17) пространственно представляют собой
Р-конфигурацию, как это характерно для стеринов (стр. 127). Эти данные
вытекают из рентгеноструктурного исследования эргостерина, а также
эргокальциферола [80, 81 ] и 4-йод-5-нитробензоата эргокальциферола
[1, 82] и подтверждаются изучением инфракрасных спектров [83].
В пользу транс-сочленения колец С и D свидетельствует и реакция изо-
меризации. При действии щелочью или кислотой ненасыщенный кетон С19
(XXV) 14а-транс-конфигурации подвергается необратимой изомеризации
в кетон с более низкой температурой плавления, имеющий более устойчивую
14р-форму (XXVI), т. е. ^ис-конфигурацию [84]. Такие же результаты по-
лучены и для продукта изомеризации кетона, образующегося при расщеп-
лении холекальциферола [85].
Сделана попытка установить абсолютную конфигурацию гидроксильной
группы у С(3) эргокальциферола [79, 86], однако она оказалась в противо-
речии с принятой для стеринов [87 ] и доказанной направленным асимметри-
ческим синтезом [88] и, следовательно, генетически переходящей на прост-
ранственную конфигурацию гидроксильной группы циклогексанового
кольца эргокальциферола.
Эргокальциферол, содержащий триеновую сопряженную систему, имеет
главный максимум поглощения при 265 нм, смещенный в коротковолновую
область спектра по сравнению с максимумом поглощения тахистерина
(280 нм), имеющего также триеновую сопряженную систему, эргостерина
(282 нм), с диеновой системой сопряженных связей (см. с. 119), или по срав-
нению с максимумом поглощения синтетических изомеров. Сдвигмаксиму-
ма поглощения говорит о наличии некоторых пространственных затрудне-
ний, которые вынуждают хромофорную систему выйти из плоскостного
положения и препятствуют свободному внутреннему вращению. Это сме-
щение полосы поглощения эргокальциферола может быть объяснено,
по крайней мере частично, наличием цис-диеновой структуры коль-
ца [5].
На основании кристаллографического анализа 4-йод-5-нитробензоата
эргокальциферола [1 ], а также дальнейших рентгеноструктурных измере-
ний [2] установлено, что кристаллический эргокальциферол имеет структу-
ру 1 с вытянутой диеновой системой s-транс-конфигурации, а не диеновой
системой 5-/{ас-конфигурации (XXXII), обладающей большими стерически-
ми помехами:
107
Предложенная для витамина D2 формула XXXIII также не подтверди-
лась [89].
Конфигурация Д19(10)-5чис’7-триеновой системы характерна для эрго-
кальциферола и других витаминов группы D. Из возможных геометрических
изомеров двойной связи алифатической боковой цепи витаминов D2 и De
ей приписывается транс-конфигурация, что вытекает из инфракрасных
спектров эргокальциферола (также и стигмакальциферола) и соответствую-
щих провитаминов эргостерина и стигмастерина, имеющих полосы при
964, 968 и 970 см4 [83]; для транс-конфигурации двойной связи характерна
полоса в инфракрасном спектре при 965 см-1 в отличие от грс-конфигурации,
для которой наблюдается полоса при 690 см-1 [83].
Строение холекальциферола (II) подтверждается тем, что он при окисле-
нии озоном образует бициклический ненасыщенный альдегид Сго (XXXIV),
насыщенный бициклический кетон С18 (XXXV) и формальдегид [23]. Соеди-
нения XXXIV и XXXV охарактеризованы по их семикарбазонам и 2,4-ди-
нитрофен ил гидразонам. Они получаются в результате расщепления двой-
ных связей С(5) —С(б) и С(7)—С(8>-
[ Строение холекальциферола (II) и дигидроэргокальциферола (III)
(витамина D4, получаемого при активировании 22-дигидроэргостерина)
установлено такими же реакциями превращения и расщепления, каки строе-
ние эргокальциферола [18].
Конформация циклогексановых колец А и С молекулы кальциферолов
представляет собой форму «кресла», энергетически более выгодную, чем
форма «ванны» [90, 91 ]. Конформация циклогексана связана с пространствен-
но направленными связями атомов углерода с атомами водорода или заме-
стителями, характеризующимися тремя северо-аксиальными, или полюс-
ными (са), тремя южно-аксиальными (юа) и шестью экваториальными (э)
связями (располагающимися радиально к главной оси симметрии, т. е.
в основной плоскости чертежа). Конформацию эргокальциферола можно
представить следующей формулой:
108
Необходимо отметить, что в превращении эргостерина в прекальциферол
(см. с. 113) под влиянием лучистой энергии происходит расщепление связи
С(ю) —С(9) и образование замещенного циклогексана при этом кольцо А
испытывает обращение и по своей конформации имеет экваториально свя-
занный гидроксил у Со, (т. е. расположенный в основной плоскости черте-
жа), а аксиально связанный атом водорода положения 3 — расположенный
перед плоскостью чертежа. Изомеризация триеновой системы связей пре-
кальциферола с С(б)—С(7)-^ис-конфигурацией приводит к эргокальциферолу
с С(б) —-С(7)-транс-конфигурацией, и так как при этом циклогексановое
кольцо не испытывает обращения, то в конформационной формуле располо-
жение гидроксила положения 3 остается в основной плоскости чертежа, а
аксиального водорода — перед плоскостью чертежа.
ВЫДЕЛЕНИЕ КАЛЬЦИФЕРОЛОВ (ВИТАМИНОВ ГРУППЫ D)
Для выделения холекальциферола (D3) из жира печени тунца или палтуса
применяют следующие операции [201: омыление и отделение неомыляемой
фракции жира, частичное отделение витамина D от витамина А путем разде-
ления между углеводородом и спиртом (на основании их различной раство-
римости), хроматографическую адсорбцию на окиси алюминия, удаление
стеринов кристаллизацией из метанола и осаждение дигитонином, этерифи-
кацию хлорангидридом 3,5-динитробензойной кислоты, хроматографическую
очистку сырого эфира, гидролиз очищенного эфира и кристаллизацию сво-
бодного витамина.
Холекальциферол по ранее разработанному методу может быть выделен
из природных источников через продукт присоединения с цитраконовым
альдегидом, чем достигается очистка от сопровождающих его примесей
[66].
Эргокальциферол (D2) и дигидроэргокальциферол (D4) в природных про-
дуктах содержатся в очень малых количествах, и методы их выделения не
имеют практического значения; остальные витамины (D5, De, D7) получены
только фотосинтезом.
ФОТОСИНТЕЗ КАЛЬЦИФЕРОЛОВ ИЗ ПРОВИТАМИНОВ
Фотоизомеризация эргостерина, 7-дегидрохолестерина и других
провитаминов в эргокальциферол, холекальциферол и другие витамины
Для получения кальциферолов важнейшее значение имеет метод фотоизо-
меризации провитаминовых стеринов (эргостерина, 7-дегидрохолестерина
И др.).
Витамины группы D получаются из А5’7-стеринов, содержащих сопряжен-
ные двойные связи в положениях С5—Сй и С7—С8 и соответствующую али-
фатическую боковую цепь приС([7), в результате возбуждения тс-электронной
системы молекулы и фотоизомеризации под влиянием лучистой энергии,
а также последующей термической цис, /пране-изомеризации; при этом
элементарный состав вещества остается неизменным (см. схему 22, с. 113).
Такие стерины с нормальной ^-конфигурацией метильной группы у
С(Ю), находящейся в транс-положении к а-конфигурации атома водорода
у С(9), способны к фотоактивированию и дальнейшему превращению в каль-
циферолы. Это характерно и для лумистерина с 10 а-метильной группой и
9£-атомом водорода. В то же время эпимерный эргостерину изопирокальци-
ферол2 с 9Р-конфигурацией и пнрокальциферол2 с 9а- и 10а-конфигурация-
ми, являющиеся стеринами с qnc-конфигурацией у С(9) и С(10), при фотоизо-
меризации не дают веществ с витаминной активностью.
Необходимая энергия для получения 1 мкг эргокальциферола оказалась
равной от 3-1015 [92] до 3,7-1015 квантов [93]. Для активирования провита-
минов необходимо ультрафиолетовое излучение с длиной волны от 230 до
109
300 нм, т. е. в тех пределах, в которых происходит поглощение света исход-
ными и промежуточными соединениями. Лучи с преимущественным содер-
жанием более длинных волн (в указанном пределе) поглощаются преиму-
щественно эргостерином, лумистерином и тахистерином; лучи с содержа-
нием волн средней длины поглощаются преимущественно прекальциферо-
лом и эргокальциферолом [27].
Однако лучший выход витамина D наблюдается, если применяются
ультрафиолетовые лучи с длиной волны 275—310 нм, и особенно 280,4 нм
[94], т. е. лучи, которые наиболее полно поглощаются веществом и отве-
чают главному максимуму поглощения эргостерина и 7-дегидрохолестерина
Рис. 1. Образование и расщепление витамина D2
(кальциферола) в различных растворителях в зависи-
мости от продолжительности фотоизомеризации эрго-
стерина ртутной лампой при комнатной температуре:
1 — в этиловом спирте; 2 — в циклогексане; 3 — в эфире.
никеля,
кислоты,
в растворе (в спирте).
Для активирования 7-
дегидрохолестерина оп-
тимальной найдена дли-
на волны 296 нм [95 ].
Для отделения лучей с X
ниже 275 нм применяют
светофильтры с аромати-
ческими углеводородами
(бензол, ксилол, дифе-
нил и др.) или раство-
ры азотнокислого калия,
уксуснокислого свинца,
сернокислого
салициловой
хлора, брома и т. д.
Применяют также стек-
лянные фильтры, изби-
рательно пропускающие
лучи с длиной волны
270—300 нм.
Следует отметить
что пропускание света
через фильтры (особен-
но жидкостные) сильно снижает его интенсивность; поэтому предпочита-
ют обходиться без фильтров и подбирать^источник света с более интенсив-
ным излучением в требуемой области.
В качестве источника ультрафиолетового света при фотоизомеризации
эргостерина и 7-дегидрохолестерина применяют ртутно-кварцевые (ПРК-2,
ПР К-7), дуговые, эритемные фосфоресцентные бактерицидные и другие
лампы.
Для активирования в растворах используют такие растворители, как
бензол, эфир, циклогексан, диоксан, спирт, растительные масла и др. Фото-
изомеризация в эфирной среде протекает более легко, чем в спиртовой [96],
что связано со специфическим эффектом растворителя [71 ]. При фотоизоме-
ризации эргостерина в спирте процесс протекает быстро, содержание эрго-
кальциферола достигает максимума в течение часа, однако с большой ско-
ростью проходит и последующий процесс дальнейшей необратимой изоме-
ризации эргокальциферола. Поэтому еще задолго до полного превращения
эргостерина в эргокальциферол начинает преобладать процесс фотоизомери-
зации и термических превращений эргокальциферола, и его содержание в
растворе резко снижается. Образование эргокальциферола в спирте дости-
гает 30% от эргостерина [97]. Фотоизомеризацию 7-дегидрохолестерина
[98, 99] осуществляют в спирте под двуокисью углерода [100], причем его
превращение в холекальцпферол составляет 47% [100].
В эфире и циклогексане процесс активирования эргостерина протекает
значительно медленнее, чем в спирте, однако медленнее проходят и фотохи-
мические и термические превращения образовавшегося эргокальциферола.
ПО
Максимум эргокальциферола в эфире достигает 70% от прореагировавшего
эргостерина [97]. Эфир, применяемый для фотоизомеризации, должен быть
свободен от перекисных соединений. Для фотоизомеризации эргостерина
используют его растворы малой концентрации (около 1%).
Специфический эффект растворителя иллюстрируется данными, пред-
ставленными на рис. 1, полученными в сравнимых условиях [96].
Существенное преимущество для процесса имеет применение жидкости
в подвижном состоянии (протекание, перемешивание, кипение), так как
это обеспечивает равномерное активирование содержащихся в ней провита-
минов и промежуточных фотоизомеров. Для активирования эргостерина
наиболее эффективны аппараты с непрерывным протеканием жидкости по
кварцевым трубкам. Реакцию фотоизомеризации проводят в специальных
аппаратах различных конструкций.
Повышение температуры очень мало влияет на увеличение выхода каль-
циферолов при реакции фотоизомеризации [101], так как способствует на-
коплению токсистерина, пирокальциферола и изопирокальциферола.
Реакция фотоизомеризации представляет собой сложный процесс, со-
провождающийся образованием многочисленных продуктов, и дает опти-
мальные результаты только при частичном, но не полном превращении про-
витамина.
Выход эргокальциферола составляет 30—60% на прореагировавший
эргостерин; для получения оптимального выхода конверсия провитамина
при облучении в спирте должна составить от 40 до 60%, а в среде эфира —
до 50% (определяется кристаллизацией эргостерина из спирта или, точ-
нее, осаждением дигитонином). Облучение 7-дегидрохолестерина заканчи-
вают, когда его конверсия составит40%. Для выделения непрореагировав-
шего эргостерина эфирный раствор с продуктами фотоизомеризации выпа-
ривают, растворяют остаток в метиловом спирте и подвергают кристал-
лизации при охлаждении (вымораживании).
Вследствие того что продукты фотоизомеризации чувствительны к кисло-
роду, особенно тахистерин, реакцию целесообразно проводить в отсутствие
воздуха (с инертным газом) [102]. Наличие значительного количества окис-
ленных продуктов затрудняет выделение кристаллического витамина.
Устойчивый выход кальциферолов получается при применении стабили-
заторов, в качестве которых могут применяться холестерин или стерины
моллюсков, не являющиеся провитаминами. При активировании 7-дегидро-
холестерина прямой выход холекальциферола в этом случае повышается
до 40% [103].
Если витамин D предназначается для целей животноводства, то фотоизоме-
ры без выделения из них индивидуального витамина используют в виде раст-
вора в растительном масле, спирте или в микрокапсулах с твердым покрытием.
Витамин D3 вырабатывается также в виде 0,5%-ного сухого казеиново-
го концентрата или другого продукта на твердой основе.
Провитамины, содержащиеся в природных продуктах, например дрож-
жах, могут активироваться в сухом состоянии [104]. Этот метод применя-
ют для витаминизации кормов. Следует отметить, что фотоизомеризания
протекает только на поверхности кристаллов, причем последующее активи-
рование разрушает в некотором количестве образовавшийся на поверхнос-
ти витамин. Для активирования используют кормовые дрожжи, выращивае-
мые на пентозных сахарах гидролизатов древесных отходов (опилки, стерж-
ни початков кукурузы, подсолнечная лузга) или на углеводородах нефти.
Из 1 т сырья получается около 235 кг сухих дрожжей. Содержание эрго-
кальциферола после фотоизомеризации в виде суспензии составляет 0,25—
0,50 мг в 1 кг сухих дрожжей.
Для изомеризации эргостерина, помимо активирования лучами (ультра-
фиолетовыми и др.), можно применить метод, основанный на перегонке эр-
гостерина в токе инертного газа при остаточном давлении 133 Па (1 мм рт.
столба); при этом образуется около 35% эргокальциферола наряду с други-
111
ми продуктами [105]. Получаемый активный препарат обладает рядом осо-
бенностей, отличающих его от препаратов эргокальциферола, полученных
в результате фотоизомеризации. Другим методом эргостерин может быть
превращен в эргокальциферол без облучения при действии ионизирующих
реагентов в среде хлороформа и дихлорэтана [106].
Механизм реакции фотоизомеризации стериновых провитаминов
в кальциферолы
При фотоизомеризации эргостерина и других провитаминов ультрафио-
летовыми лучами происходит поглощение световой энергии молекулой про-
витамина и превращение его в ряд изомерных продуктов, образование ко-
торых связано с пространственным обращением заместителя у атома угле-
рода в положении 10, расщеплением центрального фенантренового кольца
В между С(9) и С(Ю), перемещением двойных связей в сопряженную систему
и другими изомерными превращениями. Термические воздействия вызыва-
ют дальнейшую изомеризацию образовавшихся веществ. Алифатическая
боковая цепь молекулы эргостерина и содержащаяся в ней двойная связь
между С(22) и С(2з> в реакции фотоизомеризации остается неизменной, что
доказывается получением при озонировании из продуктов фотоизомериза-
ции метилизопропилацетальдегида (XXIII) [77].
Продукты изомеризации различных провитаминов обычно называются
по одной и той же номенклатуре [23 ] и различаются по цифровой пристав-
ке: из эргостерина — фотоизомеры2, из 7-дегидрохолестерина — фотоизо-
меры3 [23, 38, 107 ] и из 22-дигидроэргостерина — фотоизомеры4.
Механизм реакции фотоизомеризации Д5-7-стеринов заключается в рас-
крытии кольца В их молекулы с образованием 9,10-секостерина — в част-
ности, в результате реакций фотоизомеризации провитамин эргостерин
(XXXVI) превращается в прекальциферол (XXXVII), который в свою
очередь с перемещением сопряженной системы двойных связей термически
изомеризуется в кальциферол (I). Эти реакции фотоизомеризации не одно-
значны, сопровождаются фотопревращениями прекальциферола (схема 22)
в другие продукты облучения—лумистерин (XXXVIII) и тахистерин
(XXXIX) [108, 109] — и связаны с обратимостью реакций фотоизомериза-
ции. Ни лумистерин, ни тахистерин не являются необходимыми промежу-
точными соединениями при получении кальциферолов.
На основании работ по ультрафиолетовому облучению чистого прекаль-
циферола2 (XXXVII) в эфирном растворе, при котором образуется смесь
из прекальциферола (40%), тахистерина (30%), эргостерина (3%) и луми-
стерина (3%), высказана гипотеза, что в превращениях эргостерина пре-
кальциферол играет роль важнейшего промежуточного продукта, склон-
ного подвергаться фотоизомеризации в различных направлениях [НО, 111 ].
Это впоследствии было подтверждено другими исследованиями [108, 109].
Воздействие ультрафиолетовым светом на эргостерин (XXXVI) приво-
дит к раскрытию кольца В [28 ] по связи между асимметрическими центра-
ми С(9) и С(ю) [57], находящимися на концах возбужденной диеновой со-
пряженной системы. В результате поглощения световой энергии диеновой
системой происходит образование прекальциферола (XXXVII) с триеновой
сопряженной системой, с /{г/с-конфигурацией центральной двойной связи,
возникающей за счет новой двойной связи С(8>—С(9> и перемещения связей
С<5)—С(б) и С(7)—С(8) в положения С(Ю)—С(5> и С(б) —С(7). Прекальциферол
имеет в своей молекуле три цикла. Были предложены различные формулы
строения прекальциферола, предусматривающие далеко идущую изомери-
зацию двойных связей, однако наиболее достоверной следует считать форму-
лу XXXVII [108, 112—114]. Данными ПМР-спектроскопии установлено,
что строение молекулы прекальциферола отвечает tjuc-изомеру тахисте-
рина [115].
112
Схема 22
Реакции фотоизомеризации эргостерина
Кинетическими исследованиями показано, что при облучении эргостери-
на (XXXVI) ультрафиолетовым светом (к 254 нм) при комнатной темпера-
туре непосредственным продуктом фотоизомеризации является только пре-
кальциферол (XXXVII) — 26% [116—118] (85% всего количества продук-
тов фотоизомеризации) [108, 119], а уже из него побочно образуется тахи-
стерин (XXXIX) [117]—до 15% суммы продуктов фотоизомеризации [119].
Реакция образования прекальциферола не зависит от длины волны и ха-
рактера растворителя [119]. Такой же процесс идет при облучении твердых
растворов эргостерина в смеси эфира, спирта и изопентана (при 80° С) [120].
Возможно, что присутствие в смеси продуктов фотоизомеризации эрго-
стерина неустойчивого продукта неизвестного строения, так называемого
«протахистерина» (1931 г.) [121, 122], на основании последних данных сле-
дует отнести за счет прекальциферола2.
Прекальциферол (XXXVII) нестабилен и при последующей термической
изомеризации легко переходит в эргокальциферол (I), что связано с переме-
щением триеновой сопряженной системы в более стабильное C(i9>—С(Ю),
С(5>— С(б) и С(8)— С(7) положение с семициклической C(i9>—С(ю) двойной
связью и с образованием экзоциклической метиленовой группы. Эта реак-
ция изомеризации обратима, так как при нагревании в бензоле при 60° С
в течение 16 ч 3,5-динитробензоат эргокальциферола дает 20% 3,5-динитро-
бензоата прекальциферола2. Оба вещества находятся в растворе в опреде-
ленном равновесии.
В теплом нейтральном растворе (60° С) прекальциферол обратимо пре-
вращается в эргокальциферол [112] с содержанием 75% в равновесной
смеси [114].
При дальнейшем воздействии ультрафиолетовым светом прекальциферол
(XXXVII) в свою очередь подвергается цис, лгранс-фотоизомеризации в
более стабильную //гранс-форму— тахистерин (XXXIX) [117, 118]. Его
образование происходит именно из прекальциферола, а не из эргостерина
[117, 118]. Тахистерин в оптимальном количестве может быть получен без
промежуточного выделения прекальциферола при конверсии эргостерина
113
в пределах от 35 до 60% [38, 1231. Фотоизомеризация протекает особенно
благоприятно при Хмакс 254 нм [116] и ускоряется при каталитическом
участии йода [56, 113, 123].
Облучение ультрафиолетовым светом тахистерина (XXXIX) вызывает
и обратную транс, г{ас-изомеризацию, т. е. его превращение в прекальци-
ферол (XXXVII) [124, 125]. Таким образом, реакция фотоизомеризации
носит обратимый характер.
Другим направлением реакции фотоизомеризации тахистерина2
(XXXIX) является его превращение в лумистерин2 (XXXVIII) 1126].
Однако, по-видимому, образование лумистерина происходит не непосред-
ственно, а только через прекальциферол, который является единственным
продуктом фотоизомеризации тахистерина [127]. Прекальциферол
(XXXVII) при воздействии ультрафиолетовым светом испытывает в свою
очередь изомеризацию с завязыванием новой связи между C<9> и C(i0) и обра-
зованием кольца В изомерного стерина [66, 128], представляющего по сво-
ей структуре эргостерин, у которого произошло пространственное обраще-
ние ангулярной метильной группы у асимметрического центра С(ю).
Вследствие того что гидроксильная группа у С(з> и метильная группа у
С(10) становятся в транс-положение, лумистерин (XXXVIII) теряет спо-
собность осаждаться дигитонином (в отличие от эргостерина). Лумистерин
в отличие от эргостерина имеет 9Р-конфигурацию.
Образование лумистерина проходит при облучении ультрафиолетовым
светом с длиной волны 290—300 нм [128]. Конверсия эргостерина при фото-
изомеризации составляет 40—60%; лумистерин выделяется из маточных
растворов метилового спирта после удаления избытка непрореагировавше-
го эргостерина. Как указывалось, лумистерин2 образует аддукт с эргокаль-
циферолом с т. пл. 124—125° С, так называемый витамин D* [6], который
разрушается при ацетилировании, фракционной кристаллизации продук-
тов ацетилирования и последующем гидролизе ацетата лумистерина.
Характерно, что в отличие от лумистерина2 лумистерин3 и лумистерин*
не образуют продуктов присоединения с соответствующими витаминами,
из чего можно заключить, что образование аддукта связано с наличием
двойной связи в боковой алифатической цепи.
Реакция фотоизомеризации прекальциферола в лумистерин обратима.
При облучении УФ-светом лумистерин изомеризуется в прекальциферол
[129], который в свою очередь подвергается изомеризации в тахистерин.
Облучением эпилумистерина получен эпиэргокальциферол [130].
Возможно [131], фотоизомеризация эргостерина протекает с предвари-
тельным образованием промежуточного продукта неустановленного строе-
ния, который уже далее обратимо фотоизомеризуется в прекальциферол,
лумистерин и тахистерин:
Эргостерин ад Лромежуто«-ый , Прекальциферол, M 'Я , Эргокальциферол
h# продукт ну 2 Нагревание
XXXVI // \ , XXXVII I
$ \ У
Лумистерин^ Тахистерин
XXXVIII XXXIX
Все другие, помимо эргостерина, провитамины кальциферолов с харак-
терной сопряженной системой двойных связей в кольце В, как, например,
7-дегидрохолестерин [23] и 22-дигидроэргостерин [19], подвергаются фото-
изомеризации по той же реакции, что и эргостерин (схема 22).
При активировании 22-дигпдроэргостерина получены лумистерин*, та-
хистерин*, дигидроэргокальциферол (D*) [19].
Дальнейшее воздействие ультрафиолетовыми лучами на эргокальцифе-
рол приводит, по-видимому, к одновременному [27 ] образованию трех продук-
тов переоблучения — токсистерина, супрастерина-1 исупрастерина-II (IV).
114
Реакция фотоизомеризации (схема 19, с. 101, и схема 22, с. 113) изу-
чалась с установлением кинетических данных, определением квантового
выхода [119] и использованием меченых промежуточных соединений [131].
Выделение кристаллических кальциферолов из продуктов фотоизомеризации
Эргокальциферол из продуктов облучения выделяют в виде 3,5-динит-
робензоата (выход 78%), очищают перекристаллизацией и затем после
гидролиза эфира получают в чистом кристаллическом состоянии [21, 66].
Этот метод выделения имеет техническое значение. Из маточных эфирных
растворов после отделения 3,5-динитробензоата эргокальциферола выделен
3,5-динитробензоат тахистерина [132].
Другой путь выделения эргокальциферола из продуктов облучения сле-
дующий: предварительно отделяют тахистерин конденсацией с цитраконо-
вым ангидридом; после гидролиза аддукт тахистерин-цитраконовая кислота
остается в водном растворе, а эргокальциферол извлекается петролейным
эфиром. Витамин кристаллизуется из ацетона при низкой температуре.
При облучении 7-дегидрохолестерина был получен холекальциферол
(витамин D3) в кристаллическом виде [13, 14]. Кристаллический холекаль-
циферол выделяют из продуктов фотоизомеризации в виде эфиров с 3,5-ди-
нитробензойной [ПО, 133] или 3,5-динитро-4-метилбензойной кислотой с
последующим гидролизом и получением в свободном виде или в виде моле-
кулярного соединения с холестерином (содержание витамина D3 45%). Из
этого комплекса холекальциферол выделяют хроматографией на окиси
алюминия [134]. В кристаллическом виде витаминD3 можетбыть получен
из его бутирата путем гидролиза или восстановления гидридами металлов
с выходом 74% [135], а также из продуктов фотоизомеризации перекри-
сталлизацией из метилформиата.
Строение продуктов фотоизомеризации стериновых провитаминов
и кальциферолов и их физико-химическая характеристика
П р е к а л ь ц и ф е р о л2 (XXXVII) [ПО, 111, 113]. Первый продукт
фотоизомеризации эгростерина (XXXVI) — прекальциферол2 — имеет трие-
новую сопряженную систему с затрудненной ^wc-конфигурацией С(б}— С(7)-
двойной связью, образующуюся при расщеплении кольца В эргостерина.
Полоса поглощения ультрафиолетового света прекальциферола2 — лмакс
262 нм — значительно смещена в коротковолновую область по сравнению с
максимумами для эргостерина и тахистерина2 (см. табл. 4 и рис. 2) при одно-
временном сильном снижении экстинкции до s 9000 [113] по сравнению с
тахистерином2 (транс-конфигурация, г 29 550 при 280 нм).
Прекальциферол2 впервые выделил Веллуз с сотр. [136] в виде 3,5-ди-
нитробензоата, представляющего собой тонкие желтоватые иглы с т. пл.
103—104° С; lab +30° (С0Н6) и 4-45° (СНС13) [110—112, 136]. Прекальци-
ферол 3-3,5-динитробензоат имеет т. пл. 110—111° С [ПО, 111].
Л у м и с т е.р и н (лумистерин2) (XXXVIII) [36, 66, 128]. Так же как
и эргостерин, лумистерин содержит гидроксильную группу и три двойные
связи, а следовательно, и четыре кольца в молекуле, что установлено эте-
рификацией и по присоединению шести атомов водорода при каталитиче-
ском гидрировании, и по титрованию с пербензойной кислотой [128, 137].
Одна двойная связь находится в боковой цепи, в положении С(2?)— С(23),
как и в эргостерине, что доказывается образованием при озонировании ме-
тилизопропилацетальдегида (XXIII) [107]. Другие две двойные связи
находятся в сопряжении, как это вытекает из спектров поглощения; они
занимают положения С(5)—С(б> и С(7)—C(s> кольца В стероидного цикла.
Это следует из того, что при окислении лумистерина (XXXVIII) селеном
образуется углеводород Дильса — 3'-метил-1,2-циклопентанофенантрен
(XXI) [138], тот же самый, что и получаемый при окислении селеном эрго-
115
стерина, а при окислении азотной кислотой—толуолтетракарбоновая
кислота [139]. Изучались реакции восстановления лумистерина в ди-,
тетра- и гексагидропроизводные, его окисление пербензойной кислотой
[140, 141) и изомеризация при действии кислот [142].
При действии уксуснокислой ртути происходит дегидрирование луми-
стерина (XXXVIII) в дегидролумистерин (XL) [143], который является
10а-стереоизомером 9,11-дегидроэргостерина и подобен ему по спектру
поглощения [144]. Новая двойная связь в этих соединениях находится в
положении 9, 11 кольца С и образует сопряженную систему с двумя дру-
гими двойными связями. На основе дальнейших превращений [143, 144],
а также реакции образования дегидролумистерина (XL) при окислении пи-
рокальциферола (V) уксуснокислой ртутью [145] можно прийти к заключе-
нию, что лумистерин не отличается от эргостерина конфигурацией у асим-
метрического центра С(9), уничтожаемой при образовании двойной связи
С(9)—С(Ц); однако он отличается противоположной (а) пространственной
конфигурацией метильной группы у С(Ю).
. r= сн(сн3)сн=снсн(сна)сн(сн31)3
Для лумистерина (XXXVIII) установлена 90-конфигурация [36, 146],
а для пирокальциферола (V) — 9а-конфигурация [36]; оба соединения име-
ют одну и ту же 10а-конфигурацию.
Лумистерин кристаллизуется в тонких иглах с т. пл. 117—118° С (из
смеси ацетона и метилового спирта) [128]. Он легко растворим в эфире,
хлороформе, ацетоне, труднее — в метаноле. Ацетат лумистерина имеет
т. пл. 100° С, [а]р 4-130,5°; 3,5,4-динитробензоат лумистерина — т. пл.
140° С, Ыо +24°.
Тахистерин (тахистерин2) (XXXIX) [5, 38, 62, 122, 147]. Присут-
ствующая в молекуле тахистерина сопряженная система двойных связей
обнаруживается по характерному спектру поглощения, который имеет поч-
ти те же максимумы, что и эргостерин. Тахистерин (XXXIX) содержит че-
тыре двойные связи, т. е. на одну двойную связь больше, чем лумистерин
(XXXVIII). По реакции Дильса — Альдера он (в виде ацетата) легко дает
аддукт с цитраконовым ангидридом [57]; при этом из двух двойных связей
получается одна. Образование аддукта тахистерина с цитраконовым ангид-
ридом происходит с большой скоростью (название «тахистерин» происходит
от греческого «тахис»—быстрый [38]) в отличие от несколько пространст-
венно затрудненной реакции с цитраконовым ангидридом эргокальциферо-
ла, эргостерина и лумистерина, имеющих также диеновую сопряженную си-
стему (последние соединения более легко дают аддукты с малеиновым ан-
гидридом). При гидрировании аддукта тахистерина присоединяется только
четыре атома водорода, что указывает на наличие в молекуле двух двойных
связей, однако гидрированный аддукт показывает присутствие еще одной
связи, определяемой титрованием с пербензойной кислотой [57]. Наличие
в тахистерине (XXXIX) четырех двойных связей находится в соответствии
с тем, что его молекула на основании одного и того же эмпирического соста-
ва (С28, Н44О) может содержать только три кольца вместо четырех в луми-
стерине (XXXVIII). Это подтверждается тем, что тахистерин при дегидри-
ровании селеном не дает характерного для стеринов углеводорода Дильса
116
(XXI), но при восстановлении натрием в пропиловом спирте образует тот
же самый 6, 19-дигидроэргокалышферол (XVI), что и витамин D2, а также
дигидротахистерин (XV).
Триеновая сопряженная система в тахистерине (XXXIX) находится в
ином положении, чем у эргокальциферола (I). Это вытекает из реакции его
озонирования, не дающей кетона С19 (XXV) [148], обычно возникающего
при окислении эргокальциферола за счет расщепления связи С(7) — С<8).
Так как эта двойная связь в тахистеринеотсутствует, тотриеновая система
занимает положения С(ю) —С(5), С(6)—С(7) и С(8)—С(9).
Для тахистерина характерна Al0<5>’6<7)'mpaw-8 -триеновая система с бо-
лее высоким коэффициентом экстинкции максимума поглощения (г 24 600
при 281 нм) по сравнению с прекальциферолом и эргокальциферолом [62] и
с почти полным отсутствием пространственных помех [114]. Его конформа-
ция полностью установлена по данным ЯМР-спектроскопии [149].
Характерна для тахистерина его особенно легкая способность окислять-
ся кислородом воздуха с образованием перокисей, чем он отличается от дру-
гих продуктов фотоизомеризации эргостерина. Он легко растворим в обыч-
ных органических растворителях.
Тахистерин может быть получен из эргокальциферола реакцией йодиро-
вания и последующего дезйодирования [147]. В ионизированных растворах,
например в дихлорэтане, активированном 1% дихлоргидрина глицерина,
наблюдается обратный переход тахистерина (XXXIX) в эргокальциферол
[150].
Кристаллический тахистерин неизвестен: он получен в виде маслообраз-
ного продукта высокой степени чистоты термическим разложением аддукта
ацетата тахистерина с цитраконовым ангидридом (т. пл. 161° С, [alp+750
в хлороформе) или гидролизом кристаллического 3,5-динитро-4-метилбен-
зойнокислого эфира тахистерина2 (т. пл. 154—155°С) [38]. Сообщено [151]
о химическом синтезе тахистерина3. Тахистерин3-3,5-динитро-4-метилбен-
зоат имеет т. пл. 137° С, [ц]д +106,6° (СНС13).
Дигидротахистерин (XV) в отличие от тахистерина (XXXIX) легко по-
лучается в кристаллическом состоянии восстановлением 3,5-динитро-4-ме-
тилбензоата тахистерина натрием в спирте [152] или в жидком аммиаке
[74] с последующим гидролизом. С выходом 38% он получен непосредствен-
ным восстановлением тахистерина или прекальциферола литием в жидком
аммиаке [153, 154].
Из тахистерина3 синтезирован дигидротахистерин3; определены его свой-
ства [155].
Помимо тахистерина (XXXIX), синтетически получены два его изомера:
U-тахистерин (XLI) [5] и изотахистерин (XIX) [59]; для этих соединений
характерно наличие центральной С(б) — С(7) двойной связи /лрпнс-конфигу-
рации. Триеновая сопряженная система изотахистерина дает наибольший
максимум поглощения ультрафиолетового света и более высокую величину
экстинкции — г 41 800 при 290 нм.
117
Фотоизомеризация изотахистерина (XIX) при каталитическом участии
йода приводит к ^wc-изотахистерину (XLII), почти не обладающему анти-
рахитической активностью [156].
Супрастерины1иП (IV) [28]. Эти соединения не дают при окис-
лении селеном типичного для стероидных соединений углеводорода Дильса.
Супрастерины I и II содержат гидроксильную группу и такую же боковую
цепь, как и эргостерин. Они имеют три двойные связи, что установлено
каталитическим гидрированием и титрованием пербензойной кислотой
[1571; двойные связи не находятся в сопряжении, это следует из того, что
супрастерины не поглощают света в ультрафиолетовой области выше 240 нм.
Молекула супрастерина-П содержит циклопропановое кольцо в сопряже-
нии с тетразамещенной двойной связью [29].
Для супрастерина-П на основании УФ-, ПК- и ЯМР-спектроскопии
предложена формула спироциклопентана [157] и др. [158]. Строение и аб-
солютная конфигурация супрастерина-П — продукта фотоизомеризации
эргокальциферола — установлены в виде формулы IV (схем# 19, с. 101)
[29, 30].
Пирокальциферол2 (V) и изопирокальциферол2
(VI). Наличие стероидной системы из четырех циклов подтверждается тем,
что пирокальциферол2и изопирокальциферол2 при окислении селеном обра-
зуют характерный углеводород Дильса [138].
Пирокальциферол2 (9а-лумистерин) (V) [36, 159] обладает 9а-конфигу-
рацией, а не 90-конфигурацией, характерной для лумистерина (XXXVIII);
в остальной части молекулы его строение не отличается от строения соеди-
нения XXXVIII [36, 146]. Поэтому пирокальциферол2 является 9а-сте-
реоизомером лумистерина, имеющего, как и пирокальциферол2, а-ориента-
цию у С(Ю)- Так как при дегидрировании пербензойной кислотой или уксус-
нокислой ртутью пирокальциферола2 и лумистерина разрушается асиммет-
рический центр С(9) и образуется один и тот же дегидролумистерин (XL)
[143], то оба эти соединения имеют a-ориентацию у С(ю). Изопирокальци-
ферол2 (90-эргостерин) (VI) имеет 90-конфигурацию и является 90-стерео-
изомером эргостерина (XXXVI) [36] (с нормальной, как и у изопирокаль-
циферола2, 103-ориентацией).
При дегидрировании изопирокальциферола2 и эргостерина также разру-
шается асимметрический центр С(9) и образуется один и тот же дегидро-
эргостерин с двойной связью С(9)—С(Ц) [145], и, следовательно, оба эти
соединения имеют нормальную для стеринов 0-ориентацию у асимметриче-
ского центра С(Ю). Из трех фотоизомерных стеринов — лумистерина2, пи-
рокальциферола 2 и пзопирокальциферола2 — только последний, имеющий
конфигурацию метильной группы у С(ю), свойственную природным стери-
нам (ОН у С(з) и СН3 у С(ю) в zpc-положении), частично осаждается
дигитонином.
Пиросоединения имеют три двойные связи, что подтверждено каталити-
ческим гидрированием [157] и титрованием пербензойной кислотой. Одна
двойная связь находится между С(2г> и С(-23), Две другие — в сопряжении,
118
что доказывается реакцией с малеиновым ангидридом; то, что двойные свя-
зи находятся в одном ядре, следует из реакции окисления азотной кислотой,
приводящей к характерной для такого случая толил-2,3,4,5-тетракарбо-
новой кислоте (формулы пиросоединений V и VI см. на схеме 19, с.
101).
Фотопирокальциферол2 (VII) ифотоизопирокаль-
ц и ф е р о л2 (VIII). Наличие стероидной структуры этих соединений уста-
новлено на основании кристаллографического изучения [811. Фотопиро-
кальциферол 2 (VII) и фотоизопирокальциферол2 (VIII) имеют две несо-
пряженные двойные связи, так как отсутствует максимум поглощения в
области длин волн между 240—320 нм. Одна из этиленовых связей находится
в боковой цепи, а другая—в кольце в положении С(6)—С(7>. Так как
эмпирическому составу фотоизопирокальциферола2 (С28Н44О) должно было
бы отвечать наличие трех двойных связей, а количественным гидрирова-
нием обнаруживается присутствие только двух, то дополнительная связь
возникает между С(5) и С(8) с образованием циклобутенового кольца [160].
Формула VIП(с. 101) доказывается как изучением инфракрасных спектров,
так и реакциями превращений в соответствующий кетон [160, 161]. Ско-
рость реакции фотоизомеризации изопирокальциферола2 превышает тако-
вую для пирокальциферола2 в 20 раз.
Главнейшие физические характеристики фотоизомеров эргостерина и
веществ, образующихся при изомеризации эргокальциферола, приведены в
табл. 4.
Таблица 4
Фотоизомеры эргостерина и продукты изомеризации эргокальциферола
Вещество Т. пл., 'С [“I . град Реакция осаж- дения диги- тонином *макс в спирте,.нм е главного (г) максимума Литература
Эргостерин (XXXVI) 164—167 —135* + 262, 271, 282 г, 293,5 12 150 [5,162]
Лумистерин2 117—118 + 191** — 260,272 г, 9 450 [5,17,
(XXXVIII) 280 128]
Тахистерин2 Масло —70*** — 271, 280 г, 29550 [5, 17,
(XXXIX) 298 62]
Прекальциферол2 (XXXVII) — — 262 9000 [5, 113]
Эргокальциферол (1) 120—121 +52* — 265 19 400 [5, 10,12]
Токсистерин Около 50 —16* — 248 18 300 [5, 33]
Супрастерин-1 103-104 —76* — Не погло- щает до 240
Супрастерин-П (IV) 109—110 +63* — То же
Пирокальциферол2 (V) 93—95 +502**** — 274, 294
Изопи рока льцифе- рол2 (VI) 114—115 +332* + (час- тично) 280
Фотопирокальцифе- рол2 (VII) 103-105 +50* Нет
Ф отоизопирокальци- ферол2 (VIII) 78—80 -60* — >
• В хлороформе. •• В ацетоне. В лигроине. ***• В спирте.
Спектры поглощения продуктов фотоизомеризации эргостерина [131]
даны на рис. 2.
Продукты фотоизомеризации 7-дегидрохолестерина охарактеризованы
в табл. 5.
119
Рис. 2. Спектр поглощения продуктов
фотоизомеризации эргостерина в эфире:
/ — эргостерина; 2 — лум (стерина; 3 — тахис-
терина; 4 — прекальциферола; 6 — эргокаль-
циферола (витамина D,).
Таблица 5
Фотоизомеры 7-дегидрохолестеряна
Вещество Т. пл.. сс г л20 [°] D • град- Реакция осаж- дения дигито- нином макс в спирте, нм е главного (г) максимума Литература
7-Дег ндрохолесте- рин Лумистерииз Тахистерин3 Прекальциферол3 149—150 63—64 (из ацетона), 87—88 (из метанола) Масло Масло —120* + 197* —11,5** + 262, 271, 281,5 г, 293 281, 293 260 10900 9250 117] [110, 111] [17]
* В хлороформе. ♦* В лигроине.
СИНТЕЗ КАЛЬЦИФЕРОЛОВ И ИХ ИЗОМЕРОВ
Полный синтез природного холекальциферола (витамина D3) через хо-
лестерин формально можно считать осуществленным после того, как в 1951 —
1952 гг. Вудварт с сотр. [163, 164] из простых веществ синтезировал при-
родный холестерин. Синтез холекальциферола из холестерина через 7-де-
гидр охолестер ин и его фотоизомеризацию (с. 132) к тому времени был уже
известен.
Для частичного синтеза кальциферолов и их изомеров применен метод
построения молекулы из а,Р-ненасыщенного гидринданового альдегида
(XIII), являющегося продуктом окислительного расщепления кальциферо-
лов [45], и замещенного циклогексана (LIV). Были изучены методы полу-
чения гидринданов и замещенных циклогексанов [165—169], и эти соеди-
нения первоначально использованы в синтезе изоэргокалышферола (XVIII),
имеющего Хчанс 287 им и г 44 100 [5], обладающего полной /пракс-конфи-
гурацней д И‘и>-5>7-триеновой сопряженной системы [59, 62].
120
1 Частичный синтез 5,6-транс-эргокальциферола (XVII) [170] и эрго-
кальциферола (I) [165] осуществили Инхофен с сотрудниками в 1957 г. и
несколько позже Харрисон и Литго [171, 172]. Альдольная конденсация
а, 0-ненасыщенного альдегида (LIII, R=C9H17) с п-ацетоксициклогексано-
ном (LIV) под влиянием этилата натрия приводит к /нранс-диенолону (LV,
R=C9H17) в виде смеси эпимеров. Соединение LV применялось в реакции
в виде 2-тетрагидропиранилового [173] или уксусного эфира [170]. В по-
ложение 10 молекулы этого соединения по реакции Виттига [133] при
действии трифенилфосфинметилида вводят метиленовую группу на место кис-
лородного атома оксогруппы, что приводит к образованию эфира 5,6-транс-
эргокальциферола (XVII) в виде смеси а - и 0-эпимеров по оксигруппе по-
ложения 3 [170, 173]. Образуется вещество сД19(|0ь5(6)’"1₽снс‘7-триеновой со-
пряженной системой.
Схема 23
Синтез эргокальциферола и холекальциферола
Для соединений bill. LV. XVII. I R=C9H17 СН(СН3)СН=СНСН(СН3)СН(СН3)2:
для L-LIII, LV. LVI, II R=C8H17 СН (СН3)СН2СН2СН2СН(СН-
После гидролиза эфирной группы смесь эпимеров 5,6-транс-эргокаль-
циферола (XVII) при действии профильтрованного через стекло и бензол
ультрафиолетового света ртутной лампы подвергают изомеризации в
5,6-цас-изомер, эргокальциферол (I) и эпиэргокальциферол1 [64, 165, 171],
которые разделяют в виде их 3,5-динитробензоатов (схема 23).
1 Приставкой «эпи> обозначается гидроксильная группа с противоположной JJ-koh-
фигурацией у асимметрического центра С<з).
121
Полный синтез холекальциферола (витамина D3) осуществили Инхо-
фен с сотр. в 1958 г. [174]. Исходным веществом при синтезе служил за-
мещенный циклогексенон (XLIII) [175], который путем присоединения
нитрильной группы и последующего гидролиза превращают в циклогекса-
нондикарбоновую кислоту (XLIV) [176]. После расщепления на антиподы
транс-кислота (XL IV) метилируется с помощью диазометана и восстанавли-
вается в циклогексанолдикарбоновый эфир (XLV), который в виде ацетата
или бензоата под влиянием бутилата калия в толуоле циклизуют по Дикма-
ну с последующим омылением и декарбоксилированием в транс-8-метил-
гидринданол-4-он-1 (XLVI). Полученное соединение с [а Ь +109° идентич-
но веществу, образующемуся из альдегида (XIII) — продукта расщепления
эргокальциферола — в результате озонолиза, восстановительного расщеп-
ления озонида и окислительного отщепления боковой цепи (схема 23).
К соединению XLVI по кислороду оксогруппы присоединяют изоокти-
ловую боковую цепь с помощью целого ряда реакций. Первоначально гидрин-
данолон (XLVI) вводят в реакцию Гриньяра с кротилмагнийбромидом, в
результате чего получают изокротилкарбинол (XLVII) с концевой двойной
связью боковой цепи. Один атом углерода боковой цепи удаляют посредст-
вом образования гликола и последующим его расщеплением тетраацетатом
свинца, а затем в результате гидролиза промежуточного альдоля получают
ненасыщенный транс-альдегид (XLVIII) наряду с qwc-альдегидом, который
подвергают /пране-изомеризации. Альдегид (XLVIII) при действии лития
в жидком аммиаке превращают в литийенолят и после осторожного гидро-
лиза— в ненасыщенный нормальный альдегид (XLIX). Для получения
структуры боковой цепи, полностью соответствующей природному холекаль-
циферолу, альдегид (XL IX) вводят в реакцию Виттига с изоамилбромидом
и затем последующим гидрированием образовавшейся двойной связи пре-
вращают в гидриндановый карбинол (L, R=C8H17) [174, 176].
Для получения ненасыщенного альдегида (LIII, R=C8H17) карбинол
(L, R=C8H17) окисляют хромовым ангидридом в транс-гидринданон (LI,
R=C8H17) и затем присоединяют диеновую трехуглеродную цепочку по
реакции Виттига с аллилбромидом; образовавшееся транс-производное
(LII, R=C8H17) [166] после частичного озонолиза, восстановления алюмо-
гидридом лития и окисления двуокисью марганца дает а, 0-ненасыщенный
альдегид (LIII, R=C8H17), идентичный альдегиду, полученному при расщеп-
лении витамина D3.
Дальнейший путь синтеза холекальциферола (II) аналогичен описан-
ному для эргокальциферола (I). а, [3-Ненасыщенный альдегид (LIII,
R=C8H17) конденсирует с п-ацетоксициклогексаном (LIV) в эпимерную
смесь диенолона (LV, R=C8H17). Эпимеры успешно разделяют хроматогра-
фически и транс-изомер по реакции Виттига конденсируют с трифенилфос-
финметилидом в стабильный 5,6-транс-холекальциферол (LVI), а из него в
результате фото-транс, цае-пзомеризацни получают холекальциферол (II)
[61 174, 177, 178].
В полно,м синтезе прекальциферола3 [179, 180] в качестве исходных ве-
ществ послужили: 9а-хлор-дез-АВ-холестан-8-он, {5а-хлор-1р-[(17?)-1,5-ди-
метилгексил ]-7ар-метпл-транс-пергидриндан-4-он] (LVII) и (1Х)-3-эти-
нил-4-метилциклогекс-3-ен-1-ол (LVIII) в виде литиевого производного три-
метил сил илового эфира. В результате конденсации этих веществ получен
9а-хлор-9,10-секохолест-5(10)-ен-6-ин-3р ,8р-диол (LIX) с т. пл. 110,5° С,
в кольце С которого при действии бас-(этилендиамин)хромия создается
8,9-двойная связь. При этом образуется достаточно чистый диенин (LX).
Селективным гидрированием ацетиленовой связи до двойной с палладиевым
катализатором Линдлара [181 ] диенин (LX) превращается в прекальцифе-
рол3 (XXXVII), выделенный в виде 3,5-дшштробензоата с т. пл. 97,5—
98,5°С; [a Id 4-51,6° (СПС13). Общин выход из соединения LIX составляет
около 21%.
199
Обычным методом (термической изомеризацией в бензоле) прекальцифе-
рол3 превращают в холекальциферол (D3).
Хлоркетон (LVII) получен из 9а-хлор-дез-Ав-холестан-80-ола, который
в свою очередь синтезирован из дез-лв-холестан-8р-ола через дез-лв-хо-
лест-8-ен [182]. Дез-дв-холестан-8[}-ол получен полным синтезом [183].
БИОЛОГИЧЕСКОЕ ЗНАЧЕНИЕ КАЛЬЦИФЕРОЛОВ. ЗАВИСИМОСТЬ МЕЖДУ
СТРОЕНИЕМ И БИОЛОГИЧЕСКОЙ АКТИВНОСТЬЮ КАЛЬЦИФЕРОЛОВ
Витамин D принимает участие в регулировании обмена кальция в рас-
тущем организме. Он катализирует процессы, связанные с отложением фос-
форнокислого кальция на концах растущих костей, в костях молодых жи-
вотных, в скорлупе яиц; витамин D регулирует содержание солей кальция
в молоке. Возможно, что витамин D влияет на активность ферментов, от
которых зависят процессы обмена фосфора и кальция, связанные с осци-
фикацией [184]. Недостаток витамина D в раннем детском возрасте приво-
дит к развитию рахита, проявляющегося в недостаточном окостенении,
искривлении костей, неправильном развитии зубов и других явлениях.
В витамине D нуждаются и взрослые люди. Среди домашних животных ра-
хит наблюдается у кур и свиней. К излечению рахита приводит облучение
ультрафиолетовыми лучами солнечного света, производящими фотосинтез
витамина D из провитамина 7-дегидрохолестерина, содержащегося в коже
человека и животных, или питание рыбьим жиром, содержащим витамин D.
В настоящее время для лечебных и профилактических целей большое зна-
чение имеют синтетические препараты витаминов D2 и D3.
Первоначально полагали, что провитамином D является эргостерин,
кристаллический продукт облучения которого впоследствии (в 1931 г.)
был выделен как витамин D2 (эргокальциферол) [66, 185]. Однако этот ви-
тамин почти не обладал (всего около 3%) антирахитической активностью
для цыплят [186] и практически не имеет значения для защиты от рахита
совсем маленьких цыплят.
Природным витамином, активным для всех животных, является холе-
кальциферол (витамин D3), выделенный в 1936 г. Брокманом [187] из жира
печени тунца и палтуса в форме кристаллического 3,5-динитробензоата,
а затем и в свободном кристаллическом виде [20, 188]. Полагают, что в при-
родном витамине D3 содержится около 10% витамина D2 [4].
Так как все витамины группы D содержат одну и ту же циклическую
структурную часть молекулы из 19 атомов углерода с гидроксильной груп-
пой в положении 3 и специфической триеновой системой, но различаются
по строению боковой алифатической цепи при С(17) из 8—10 атомов угле-
рода, то их витаминная активность находится в прямой зависимости от
строения этой цепи или, точнее, от части этой цепи с атомами углерода
С(22) —С.,2!, .
123
Биологическая активность кальциферолов представлена в табл. 6.
Таблица 6
Сравнительная биологическая активность витаминов
группы D (1 и. е. = 0,025 мкг витамина D3)
Витамин Строение углеродной цепи С(22) — с(24) Активность [189]
для крыс, млн. и. е. в 1 г для цыплят, млн. и. е. в 1 г (%)
Холекальциферол (D3) —СН2СН2СН2— 22 23 24 СНз 40 (52,4) 40 (100)
Эргокальциферол (D2) —СН=СНСН— СНз 40 0,4—1,2 (1—3) [186]
Дигидроэргокальциферол (D*) —СН2СН2СН— 20-30 8(20)
Кампекальциферол (D7) —СН2СН2СН— 4 —
СНз СзНз
Ситокальциферол (Ds) -СН2СН2СН— с2н5 1.3 —
Стигмакальциферол (De) —сн=снсн— 0,1 *—*
Биологическая активность эргокальциферола (5,6-£{wc) и 5,6-щрпнс-
эргокальциферола на цыплятах оказалась практически одинаковой [56].
По некоторым данным, холекальциферол более активен для человека,
чем эргокальциферол. В опытах на крысах витамин D3 был активнее вита-
мина D2 [186] в 1,31 раза [190].’
Наибольшей активностью обладает холекальциферол — витамин,
имеющий насыщенную боковую цепь С(22>—С(24> без заместителей. Введе-
ние метильной группы в положение 24 уменьшает активность почти в 2 ра-
за для крыс и в 5 раз для цыплят (дигидроэргокальциферол). Изменение
конфигурации метильной группы в этом положении на противоположную,
т. е. на а-конфигурацпю, снижает активность еще больше — в 10 раз (кам-
пекальциферол). Этильная группа в положении 24 уменьшает активность
почти в 25 раз (ситокальциферол). Наличие кратной связи С(22>—С(23) в
молекуле витамина, хотя и связано с сохранением активности для крыс,
однако почти полностью лишает соединение витаминной активности для
цыплят (эргокальциферол) или резко снижает активность для всех живот-
ных (стигмакальциферол). Аналоги кальциферолов с боковой цепью желч-
ных КИСЛОТ при С(17) [191 ].
—СН (СНз) СН2 СН2 СООН,
или со спиртовой группой при С(17)—17-оксиэтиокальциферол г[192], или
без всякого заместителя при C(i7>—этиокальциферол [154] почти совсем
[87] или совсем биологически не активны [35].
Гомолог с СНСН3-группой при С(ю) по активности в 40 раз слабее на-
турального витамина D.
194
Изменение пространственного расположения гидроксильной группы у
асимметрического центра С(з> на противоположное уменьшает активность в
10—20 раз — эпиэргокальциферол [193] и эпихолекальциферол [194].
Изостер холекальциферола, в котором гидроксильная группа замещена
на тиольную (—SH), неактивен. Гидроксильная группа кальциферолов
должна быть свободна. Простые и сложные эфиры витамина D неактивны
(например, бензоат, пальмитат, аллофанат, циннамат, дифенилацетат, окса-
лат, нафтилуретан, фенилуретан); однако если сложные эфиры в организме
легко гидролизуются, то такие производные приобретают активность (на-
пример, ацетат, этилкарбонат, фосфат).
Продукт гидратации и окисления холекальциферола — «кетон 250» (IX)
отвечает 10%-ной, а его енолят кальция — полной активности природного
витамина D3. По-видимому, енолят кальция «кетона 250» (растворимый в
липидах) участвует в переносе ионов кальция в процессах обмена веществ
и способствует известкованию костей [195].
Среди природных продуктов обнаружены биологически активные окси-
производные кальциферолов, регулирующие обмен кальция. Из плазмы
крови выделен 25-окси холекальциферол (LXI) [196], обладающий более
высокой антирахитической активностью в классическом тесте на цыплятах,
чем холекальциферол (D3); это соединение образуется в печени из холе-
кальциферола. Осуществлен его синтез [197, 198]. Получена также другая
активная метаболическая форма витамина D2 — 25-оксиэргокальциферол
[199]. Из кишечника цыпленка выделена еще одна форма витамина D3,
катализирующая транспорт кальция, — 1,25-диоксихолекальццферол
(LXII) [200, 201]; эта форма образуется в почках в результате гидрокси-
лирования 25-оксихолекальциферола ферментами митохондрий. При испы-
тании на животных это соединение показало значительно более высокую
активность, чем холекальциферол [202].
Антирахитическая активность 7-дегидрохолестерина (провитамина D3)
в результате его метаболизма составляет 35% активности холекальциферола
[203].
Витамин D встречается только в животных организмах в очень малых
количествах. Больше всего его содержится в жире печени и внутренностей
различных рыб и морских животных—дельфинов, китов и др. [204].
Для определения витамина D3 в жире печени рыб и морских животных
применяется биологический метод анализа [205]. По своей биологической
активности 1 г витамина D3 принят равны.м 40 000 000 и. е., т. е. 1 и. е. =
= 0,025 мкг (USP-стандарт).
Витамин D находит широкое применение для витаминизации пищевых
продуктов: молока (в том числе сухого), маргарина и др.
Профилактическая доза витамина Пдля ребенка составляет 17,5—25 мкг
(700—1000 и. е.) в день. Суточная потребность для взрослых определяется
в 7—12 мкг (для жителей северных районов).
Для лечения рахита применяют по 10—50 мкг кальциферола или холе-
кальциферола (400—2000 и. е.) из расчета на 1 кг тела ребенка [189].
Ежедневные дозы 0,5 мг кальциферола (20 000 и. е.) на 1 кг тела или
дозы, превышающие в 3500 раз обычные терапевтические, могут вызвать
125
интоксикацию. Однако для лечения лейшманиоза (туберкулеза кожи) при-
меняют длительное лечение массивными дозами — по 5—25 мг кальцифе-
рола [189]. Витамины D2 и D3 для лечебных целей выпускают в виде спир-
товых или масляных растворов. Витамин D имеет важное значение в живот-
новодстве. Им витаминизируют корма для свиней, крупного рогатого скота
и птиц — 30—40 мг/т.
ПРОВИТАМИНЫ КАЛЬЦИФЕРОЛОВ (ПРОВИТАМИНЫ D)
Провитамины кальциферолов содержат в своей молекуле скелет пер-
гидро- 1,2-циклопентанофенантрена и относятся к стероидным соединениям,
к группе стеринов — полициклических одноатомных спиртов. Стерины ши-
роко распространены в животном и растительном мире, являясь неомыляе-
мой составной частью масел и жиров. Они встречаются как в виде эфиров
алифатических кислот, так и в свободном состоянии.
Помимо стеринов среди стероидных соединений находятся такие важней-
шие, биологически активные соединения, как половые гормоны, желчные
кислоты, гормоны надпочечников, сердечные гликозиды и многие другие
природные соединения.
Провитамины кальциферолов содержат систему сопряженных двойных
связей в положении С<5)—С(б>и С(7)—С(8> и боковую алифатическую цепь
в положении C(i7> из 8—9 атомов углерода. Наличие двойной связи в поло-
жении С(7)—С(8) ставит провитамины D в особую группу среди стероидных
соединений.
Провитамины имеют 6 асимметрических атомов углерода в четырехколь-
цевой системе и 1—2 асимметрических центра в боковой алифатической
К провитаминам кальцифе ролов относятся следующие соединения:
Эргостерин 21 СНз R=CHCH= СН, =сн*снсн< /СНз Провитамин эргокальциферо-
(XXXV!) 20 22 23 24 25 'СН3 ла (О2)
• СНз /СНз
7-Дегидрохолестерин (LXIII) r =снснгсн2сн2сн/ 'СНз Провитамин холекальциферо- ла (D3)
СНз СНз /СНз
22-Дигидроэргостерин (LXIV) R = снсн2сн2снсн<" 'СНз Провитамин дигидроэрго- кальциферола (D4)
СНз с2н5 /СНз
7-Дегидроситостерин r = снсн2сн2снсн/ СНз QHs 1 1 'СНз гСНз Провитамин снтокальциферо- ла (D5)
7-Дегидростигмастерин R = СНСНп СНз .1 =снснсн/ ЧСН3 /СНз Провитамин стигмакальцифе- рола (De)
7-Дегидрокампестерин R = CHCH2CH2CHCH<Q СН3 'СН3 Провитамин кампекальцифе- рола (D7)
126
Первые три провитамина обнаружены в природных продуктах, осталь-
ные провитамины получены только синтетически. Из важнейших провита-
минов эргостерин является фитостерином, а 7-дегидрохолестерин — жи-
вотным стерином.
Кроме этих провитаминов, из моллюска Mytilus edulis выделен малоизу-
ченный стерин, обладающий свойствами провитамина, — 22-дегидро-7-де-
гидрохолестерин (А5-7•22 -холестатриен-Зр -ол) [85 ]
СНз QIT
♦I /Нэ
R = —СНСН=СНСН2СН<'
х:н3
Провитамины различаются между собой по наличию или отсутствию двой-
ной связи у С22)—Q23) и по заместителям у асимметрического центра С(24).
По сравнению с эргостерином 7-дегидрохолестерин, 22-дигидроэргостерин,
7-дегидроситостерин и 7-дегидрокампестерин у С(22)—С(2з> не имеют двой-
ной связи. Эргостерин и 22-дигидроэргостерин у С<24) имеют метильную
группу b-конфигурации (связь изображается сплошной линией), 7-дегид-
роситостерин и 7-дегидростигмастерин — этильную группу той же Ь-кон-
фигурации, а 7-дегидрокампестерин является эпимером 22-дигидроэргосте-
рина и содержит у С(24) метильную группу противоположной а-конфигу-
рации (связь изображается пунктирной линией). 7-Дегидрохолестерин не
имеет асимметрического центра С<24>.
Структурную формулу эргостерина установили в 1934 г. Виндаус с сотр.
[206J. Эргостерин (XXXVI) по своей структуре представляет срощенную
из четырех ядер циклическую систему пергидропентанофенантрена. Он от-
носится к производным андростана.
Заместители в циклической части молекулы эргостерина и других про-
витаминов (вторичная гидроксильная группа положения 3, ангулярные
метильные группы при С;ю) и Сцз) и заместитель при C(i7)) имеют(3-конфи-
гурацию и все находятся в 7{пс-положении относительно друг друга [87].
Провитамины дают реакцию с дигитонином, характерную для стероидных
соединений с гидроксильной группой Зр -конфигурации, образующей более
устойчивую экваториальную связь [87 ].
Нормальная а-конфигурация у С(9> существенно необходима для’акти-
вирования. Так, эпимерный эргостерину изопирокальциферол2 (VI) с 9р-
конфигурацией и пирокальциферол2 (V) с 9а- и 10а-конфигурациями не да-
ют вещества с витаминной активностью. Однако перемещение метильной
группы из р-положения у С(1э) ва-положение (лумистерин, XXXVIII) не
мешает активированию.
Р-Конфигурация алифатического заместителя при C(i7) характерна для
всех природных стеринов и других стероидов; вещества с противоположной
а-конфигурацией не обладают биологической активностью.
Абсолютная конфигурация асимметрического центра С(2о) алифатиче-
ской цепи с метильной группой при нем в молекуле стериновых провитами-
нов отвечает принятым проекционным формулам в соответствии с данными
по пирролизу продукта озонирования Д14-холестенола-Зр в а, р-непредель-
ный альдегид и его генетической связи с (Ч-)-метилянтарной кислотой [207 ].
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА ПРОВИТАМИНОВ
КАЛЬЦИФЕРОЛОВ
Эргостерин1 —Д5,7.22 -эргостатриен-Зр-ол (XXXVI) кристалли-
зуется из 95%-ного спирта в бесцветных, блестящих листочках с одной
молекулой воды; температура плавления находится в пределах 164—167° С
[162], вещество трудно дегидратируется [208]. Т. кип. около 250°Спри
1 По-английски ergot — спорынья.
127
0,4 мм. УФ-спектр поглощения эргостерина имеет максимумы при к 262,
271, 282 и 293,5 нм (в спирте); молекулярная экстинкция при 282 нм е 12 150
[162].
Удельное вращение эргостерина: [a In —135° (СНС13) [209], —94°
(эфир), —93° (С2Н5ОН), —92° (СН3СОСН3) и —125° (QHJ; [а $6iI —171°
(СНС13). Наблюдаемое общее молекулярное вращение* 1 MD эргостерина
(и соответственно 7-дегидрохолестерина) составляется из отдельных частных
вращений асимметрических центров, оптической Экзальтации (вициналь-
ного эффекта) зависимой от двойных связей, алкильных заместителей и
гидроксилированных центров, направление и величину которых можно
представить следующей формулой2:
вычисленное -552 ±60
Эргостерин нерастворим в воде; растворимость в углеводородах, их хло-
рированных производных, в спиртах и других органических растворителях
приведена в табл. 7 и 8.
Таблица 7
Растворимость эргостерина в различных растворителях
Растворитель Температура, Число частей рас.творителя для растьоре- Ш1я 1 части Температура кипения, АС Число миллилитров растворителя для растворения 1 части
Метиловый спирт — 65 280
Изопропиловый спирт — — 82 10
Эфир 20 50 35 (40) 70
Ацетон 20 200 56 27 (40)
Хлороформ 18 50 61 Сильно растворим
Петролейный эфир (гексан) 16 94 65—70 24
Метилацетат —— — 54 35
Этилацетат — — 77 6,5
Сильные окислители (марганцовокислый калий и др.) вызывают рас-
щепление эргостерина; однако и кислород воздуха, особенно в присутствии
света, приводит к разложению с образованием перекисных соединений.
Эргостерин дает характерные цветные реакции с различными реактива-
ми: реакция Чугаева [211 ] — красное окрашивание при кипячении с хло-
ристым ацетилом и хлористым цинком в ледяной уксусной кислоте; красное
окрашивание с треххлористой сурьмой [212], с трихлоруксусвой кислотой
в хлороформе, с концентрированной серной кислотой [681; зеленое окраши-
ЫоХ Мол. масса
1 Молекулярное вращение определяют по формуле Md =----jqq----- .
8 Выведено на основании частных вращений, вычисленных Л. и М. Физера.ми
[87] для холестана и других стероидных соединений.
128
Таблица 8
Растворимость эргостерина при различной температуре
Растворитель Содержание, г в 100 г растворителя [210] при температуре, СС Число частей растворителя для растворе- ния I части при темпера- туре, °C
0 10 20 50 70 78 80
Дихлорэтан Бензол Этиловый спирт 0,040 0,020 0,083 0,260 0,077 0,308 0,521 0,110 2,57 4,01 0,74 9,04 16,16 0,79 36 3,68
вание с серной кислотой и уксусным ангидридом, с бромом в ледяной уксус-
ной кислоте, хлороформе и оливковом масле, сдиметилсульфатом и др. [68].
Эти реакции можно применить для качественного и количественного опре-
деления эргостерина. Для практического анализа эргостерина в дрожжах
и в мицелии применяют колориметрический метод определения с уксусным
ангидридом в присутствии серной кислоты, а в продуктах облучения —
метод осаждения дигитонином [213].
Производные эргостерина [68]: ацетат — т. пл. 179° С, [а Ь —90°; бен-
зоат—т. пл. 171° С, [a In —68°; п-нитробензоат— желтые листочки —
т. пл. 182° С, [а In—49,5° (СНС13); 3,5-динитробензоат—оранжевые
листочки — т. пл. 198—199° С, [а Ь —40,8° (СНС13); аллофанат,
C28H48OCONHCONH2, — иглы — т. пл. 250° С.
Производные эргостерина [68] труднорастворимы во многих органиче-
ских растворителях, за исключением пиридина.
7-Дегидрохолестерин —А5-7 -холестадиен-30 -ол (LXIII) крис-
таллизуется из смеси бензола с метиловым спиртом в присутствии следов
воды в виде моногидрата — тонкие иглы с т. пл. 149—150° С, Га ]п —120”
<СНС13) [214].
7-Дегидрохолестерин имеет те же максимумы поглощения, что и эргосте-
рин; е при 281,5 нм 10 900 (в спирте). Он легко растворим в эфире и труд-
но растворим в метаноле. 7-Дегидрохолестерин менее стабилен, чем
эргостерин, однако в смеси с холестерином обладает высокой устойчивостью
[215].
Производные 7-дегидрохолестерина [68]: ацетат — т. пл. 130° С,
[а Ь—85,3° (С6Не); бензоат — т. пл. 138—139° С, [а In —53,2° (СНС13);
п-нитробензоат — желтые иглы — т. пл. 153—154° С, [а ]п —49,8° (СНС13);
3,5-динитробензоат — оранжевые иглы — т. пл. 210—212° С, [а ]£> —45,7°
(СНС13); эпи-7-дегидрохолестерин —т. пл. 124—126° С, kin —70,5°
(СНС13) [1941; 7-бромхолестерилацетат—т. пл. 109—110° С, [а ]п
— 245° (СНС13); 7-хлорхолестерилбензоат —т. пл. 168° С,[а]©
— 122° (СНС13).
22-Д и г и д р о э р г о с т е р и н (LXIV) имеет т. пл. 152—153° С,
Га]п —109° (СНС13) Г216].
7-Дегидро-р-ситостерин —т. пл. 144—145°С, [а]п —116°
(СНС13).
7-Дегидростигмастерин — т. пл. 153—154°С, [а]п —113,1°
(СеНс) [175]. Производные 7-дегидростигмастерина: ацетат — т. пл. 172° С,
бензоат — т. пл. 180° С.
7-Дегидрокампестерин — пластинки с т. пл. 164—165° С,
Ь In —109° (СНС13). Он является стерическим изомером 22-дегидроэргосте-
5—69
129
рина по асимметрическому центру С(24>. Производное 7-дегидрокампесте-
рина: бензоат — тонкие иглы с т. пл. 156—157°С, [а]© —59,2° (СНС13).
ВЫДЕЛЕНИЕ И СИНТЕЗ ПРОВИТАМИНОВ КАЛЬЦИФЕРОЛОВ
Получение эргостерина (провитамина D2)
Эргостерин (XXXVI) содержится в тканях низших животных, в
растениях (в корнях скополии, в ростках пшеницы и др.), спорынье, грибах,
дрожжах, плесенях, а также в яичном желтке. Среди других стеринов его
содержание составляет: в дрожжах 98 —99%, в улитке Arion empiricori-
um 19—25%, в земляном черве 22%, в стеринах хлопкового масла 5%.
Эргостерин выделяют из пекарских дрожжей (содержание 0,29—1,17%
на сухую массу) или из плесеней. Технический эргостерин из плесеней мо-
жет содержать до 5% а-дигидроэргостерина, который обладает такой же
растворимостью, как и эргостерин. В качестве источника получения эрго-
стерина применяют мицелий плесени Penicillium notatum (содержаниеоко-
ло 1 %), остающийся после фильтрации ферментативной жидкости, направ-
ляемой на выделение пенициллина [217, 218].
Для выделения эргостерина из дрожжей необходимо первоначально раз-
рушить оболочку клеток, что обычно достигается при помощи раствора
едкого кали при нагревании; одновременно происходит омыление содержа-
щегося в клетках жира. Затем стерины, находящиеся в неомыляемой части
клеточного содержимого, экстрагируются спиртом или другим органическим
растворителем, например бензолом. Из небольшого остатка после удаления
основного количества растворителя выделяется эргостерин; всего, с обра-
боткой маточных растворов получается около 3,3 г эргостерина из 1 кг
прессованных дрожжей. Дополнительно из маточных растворов обработ-
кой петролейным эфиром можно получить еще около 0,07 г зимостерина
(LXV). Автолиз дрожжей в присутствии уксусноэтилового эфира способству-
ет повышенному извлечению эргостерина (на 30—50%) [219].
Эргостерин-сырец необходимо подвергнуть тщательной очистке, так
как в дрожжах, помимо эргостерина, содержатся в небольших количествах
следующие стерины: зимостерин (LXV)ct. пл. 110е С, [а]р 4-49° [220];
аскостерин (LXVI) с т. пл. 142° С, [a In +45° [220]; фекостерин (LXVII) с
т. пл. 162° С, [а]о +42° [220]; эпистерин (LXVIII) с т. пл. 151° С, [а]о
—5° [2091; 5-дигидроэргостерин [221]; анастерин с т. пл. 159° С, [а]о—8°
[2091; гипостерин с т. пл. 102° С, [а]о +12,5° [222]; церевистерин с т. пл.
254 и 265° С, [а]о —57°, —79°.
Очистку эргостерина от примесей можно осуществить и превращением
его в эфиры с последующим гидролизом [223].
130
Для получения эргостерина могут быть использованы пивные дрож-
жи [224], однако этот вариант не имеет практического значения.
Если до обработки дрожжей едким кали производят экстракцию их вод-
ным спиртом при 50° С, то после удаления растворителя получают концент-
рат комплекса витаминов группы В [225], который находит применение в
процессах ферментации, например при получении L-сорбозы из D-сорбита
в синтезе L-аскорбиновой кислоты.
Сводка обширной научной и патентной литературы о получении эргосте-
рина дана в обзорах [68, 210].
Синтез 7-дегидрохолестерина (провитамина D3)
7-Дегидрохолестерин (LXII1) находится в нервных и жировых тканях
человека и высших животных. В коже животных 7-дегидрохолестерин со-
держится в большем количестве (около 4% всего количества стеринов),
чем в других частях тела (от 0,1 до 0,5%). Среди различных стеринов 7-де-
гидрохолестерина содержится: в улитке Buccinum undatum 17—27%, В
коже свиньи 3—6%, в двустворчатых моллюсках Mytilus edulis 9—10% и
Modiolus demissus 40—50%. В стеринах моллюсков кроме 7-дегидрохоле-
стерина (3 части) найдены: эргостерин (1 часть), дз.7,22 -холестатриен-
3-ол (1—2 части) и провитамин Dx (1 часть) [85].
7-Дегидрохолестерин может быть выделен из природных источников,
например из моллюсков Modiolus demissus [226] или Mytilus edulis [227],
среди стеринов которых он находится в значительном количестве.
7-Дегидрохолестерин выделяют путем экстракции жира и стеринов орга-
ническими растворителями с последующим омылением жира щелочью, а
стеринов — экстракцией спиртом или ацетоном из реакционной смеси с
последующей кристаллизацией.
Однако важнейшее значение имеют методы синтеза 7-дегидрохолестери-
на путем введения в положение 7 и 8 холестерина двойной связи. Холестерин
(по-гречески «холе» — желчь) широко распространен почти во всех тканях
организма животных, особенно в нервных и мозговых, в надпочечниках; он
содержится в кожном жире, яичном желтке и т. д. Холестерин — т. пл.
149° С, [а Ь —39° (СНС13) — является главной составной частью желч-
ных камней. Он добывается из спинного мозга рогатого скота экстракцией
дихлорэтаном или другими растворителями (выход превышает 10%). Хо-
лестерин отличается от 7-дегидрохолестерина отсутствием двойной связи у
С-7) —С(8) и содержит всего одну двойную связь. Холестерин получен пол-
ным стерически направленным синтезом в 1951 г. [163, 228] из 5-метокси-
толухинона и бутадиена через 1{ис-2-метокси-1,4-дикето-10-метил-Д2>6-гек-
сагидронафталин, трициклический 2-кето-1,14-диметил-Д1<11)’6-'-гекса-
гидронафталин, Ш-Д4-9-Д-гомоандростадиен-16, 17-диол-З-он ацетонат, 3-ке-
тоэтиохолатриеновый эфир [228], 3-кетоэтиоаллохолановый эфир [163],
холестан-Зр-ол [163] и Д4-холестен-3-он [229, 230], превращенный затем в
холестерин [231 ]. Этот синтез осуществляется через многочисленные про-
межуточные соединения.
По методу Виндауса (схема 24, путь А) из холестерина (LXIX) при
действии уксусного ангидрида получают 3-ацетилхолестерин (LXX,
R'=COCH3), который окисляют хромовым ангидридом в ледяной уксусной
кислоте в З-ацетил-7-кетохолестерин (LXXI, R'=COCH3) [16, 87, 232].
Для получения этого кетона можно применить электролитическое окисле-
ние [233]. З-Ацетил-7-кетохолестерин (LXXI, R'=COCH3) восстанавлива-
ют по Меервейну — Понндорфу изопропилатом алюминия [234] или алко-
голятами магния, циркония [214] или натрия [233], или алюмогидридом
лития [87] в 7-оксихолестерин, причем образуются обе возможные а- и
Р-оксиконфигурации [235,236]: с алкоголятамн металлов — Зр,7р-диол
(LXXII) ст. пл. 178° С и [а Ь +7,2е с выходом 29 % [16]; с алюмогидридом
лития—также преимущественно Зр.7р-диол и одновременно около 5%
•’ -диола (LXXI11) с т. пл. 177е С и [а Ь — dh 1ь7].
131
Дальнейшими реакциями эпимеры 7-оксихолестерина (LXXII и
LXXIII) превращают в 7-дегидрохолестерин (LXIII). Предварительно
7-оксихолестеринбензоилируютизатем из дибензоата 7В- или 7а-холестери-
на (LXXIV и LXXV) термическим расщеплением [16], которое облегчает-
ся применением диметиланилина [87, 232], из положения 7 отщепляют бен-
зойную кислоту с одновременным введением С, — С8 двойной связи и
образованием З-бензоил-7-дегидрохолестерина (LXXVII). Большие преиму-
щества при термическом расщеплении с диметиланилином имеет примене-
ние в качестве среды органического растворителя [237, 238]. Следует от-
метить, что при этой реакции наблюдается образование З-бензоил-8-дегид-
роизохолестерина (LXXVI) [87, 232].
Гидролизом З-бензоил-7-дегидрохолестерина (LXXVII) получают 7-де-
гидрохолестерин (LXIII) [71, 232, 234]. Для получения 7-дегидрохоле-
стерина из эпимеров 7-оксихолестерина (LXXII иLXXIII) применяются
также эфиры серной кислоты [239]. 7-Окси холестерин может быть дегидри-
рован в 7-дегидрохолестерин непосредственным кипячением с концентри-
рованным раствором щавелевой кислоты [233] или нагреванием в вакууме
при 135° С [240]. Из кислого фталата холестерина окислением марганцово-
кислым калием в щелочной среде с небольшим выходом получают 7а-окси-
Схема 24
Синтез 7-дегидрохолестерина из холестерина
LXIII
132
холестерин (LXXIII) [241], который через дибензоат превращают в 7-де-
гидрохолестерин (LXIII).
Имеющий техническое значение более простой метод синтеза 7-дегидро-
холестерина (LXIII) (схема 24, путь Б) состоит в бромировании в аллиль-
ное положение (7-е) 3-бензоил- или 3-ацетилхолестерина (LXX, R'=COC6H6
или СОСН3) N-бромсукцинимидом при активировании на свету. Затем из
полученного 3-бензоил- или З-ацетил-7-бромхолестерина (LXXVIII, R'=
=СОС6Н5 или СОСН3) отщепляют бромистоводородную кислоту при взаи-
модействии с коллидином, диметиланилином, аммиаком, мочевиной или
карбонатами кальция и аммония в ксилоле или другом растворителе, при
этом образуется 7-дегидрохолестерин (LXIII) с общим выходом свыше
30% [242—250]. Эфир 7-бромхолестерина (LXXVIII) получают с выходом
55—65%, если производят непосредственное бромирование в среде четырех-
хлористого углерода на свету (с К 410 нм) [251]. Выход при дегидроброми-
ровании достигает 68% [249].
Общий выход 7-дегидрохолестерина повышен до 40% [252] и до 50%
применением коллидина для отщепления галогеноводорода [252, 253].
Если бромировать N-бромсукцинимидом в гексане в присутствии перокиси
лауриновой кислоты и после отгонки растворителя от сырого эфира 7-бром-
холестерина (LXXVIII) произвести отщепление бромистоводородной кис-
лоты с хинальдином в ксилоле, З-бензоил-7-дегидрохолестерин получают d
выходом 70%; его гидролиз с раствором едкого натра или со спиртовым
едким кали в свободный 7-дегидрохолестерин (LXIII) протекает с выходом
80% [98, 100, 254]. Еще более (до 52 % от холестерина) общий выход 7-де-
гидрохолестерина может быть увеличен, если бромирование 3-бензоилхо-
лестерина осуществлять П,№дибром-5, 5-диметилгидантоином, диброманти-
ном и полученный З-бензоил-7-бромхолестерин дегидробромировать при
125° С в ксилоле триметилфосфитом, который одновременно осуществляет
и защитные функции от окисления [100, 255].
При дегидробромировании З-бензоил-7-бромхолестерина (LXXVIII,
R'=COC6H5), помимо 7-дегидрохолестерина, обнаружены побочные продук-
ты: Д4>6-холестадиенол-3р (LXXIX) [256, 257] при применении в качестве
дегидробромирующего основания П,Ы-диэтилциклогексиламина, н-дибу-
тиламина или этиламина; Д2-4-6-холестатриен (LXXX)— с н-трибутил-
амином [257 ].
LXXIX LXXX
сн3
R = СНСН2СН2СН2СНСН3)2
При дегидробромировании в присутствии 50 %-ного раствора едкого нат-
ра, помимо 7-дегидрохолестерина, выделены: Д6-8(14)-холестадиенол-Зр и
Д4,б,8(14).холестаТриен [257].
Кроме указанных выше химических методов дегидрирования холестери-
на, имеющих техническое значение, известны методы непосредственного
биохимического дегидрирования холестерина хиноном, хлоранилом, серой,
метиленовым голубым, бензальдегидом и другими окислителями в присут-
ствии света и сукцпнатдегидрогеназы сердечной мышцы, дающие до 20%
провитамина [258].
Синтез 22-дигидроэргостерина (провитамина D4)
22-Дигидроэргостерин (LXIV) получают из 3-ацетилэргостерина
(LXXXI), который конденсируют с малеиновым ангидридом в среде ксило-
133
ла при 130°С [216, 259, 260]; в аддукте LXXXII каталитическим путем
восстанавливают двойную связь боковой цепи и затем из соединения
LXXXIII в результате термического расщепления в глубоком вакууме
и гидролиза получают 22-дигидроэргостерин (LXIV) [216].
Синтез других стериновых
провитаминов
Синтезы 7-дегидроситостерина (провитамина D5), 7-дегидростигмастери-
на (провитамина D6) и 7-дегидрокампестерина (провитамина D7) осуществля-
ют по методу Виндауса [16], описанному выше (с. 131).
В синтезе 7-дегидроситостерина [191] исходят из р-ситостерина [261]
(по-гречески «сито» — зерно), наиболее распространенного фитостерина,
встречающегося в хлопковом масле и масле из ростков пшеницы, ржи и
кукурузы, р -Ситостерин имеет т. пл. 140° С, [а]о —36°. Он выделен также
из смоляных мыл сосновой древесины [262, 263 ].
Исходным веществом для синтетического получения 7-дегидростигма-
стерина [175]служит стигмастерин, содержащийся в соевых и калабарских
бобах. Стигмастерин имеет т. пл. 170° С, [а]д —49° (СНС13).
Для получения 7-дегидрокампестерина [8, 264 ] исходят из кампестери-.
на, содержащегося в рапсовом, соевом масле и масле ростков пшеницы и
ржи. Т. пл. кампестерина 158° С.
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ (К ГЛАВЕ III)
1. D. Crowfoot, J. Dunitz.
Nature, 162, 608 (1948).
2. D. Н о d g k i п, M. W e b s t e г,
J. Dunitz. Chem. and Ind., 1957,
1148.
3. И. H. Назаров, Л. Д. Б ер-
гель с о н. Успехи химии, 19,
88 (1950).
4. Н. В г о с k m а п п, A. Busse.
Z. physiol. Chem., 256, 252 (1938).
5. Н. Inhoffen, К. Bruck-
ner, К. I г m s с h е г, G. Q u i n-
к e г t. Chem. Ber., 88, 1424 (1955).
6. A. W i n d a u s, A. Liittrin-
g h a u s. Z. physiol. Chem., 203,
70 (1931).
7. J. Courtois. Bull. Soc. chim.
biol., 38, 295 (1956).
8. W. R uigh. J. Am. Chem. Soc.,
64, 1900 (1942).
9. W. В e r g m a n n. E. L о w. J.
Org. Chem., 12, 67 (1947).
10. H. P e n a u, G. H a g e m a n n.
Helv. Chim. Acta, 29, 1366 (1946).
11. A. Bacharach, E. Smith,
S. Stevenson. Analyst, 58, 128
(1933)
12. S. C r e w s, E. S m i t h. Ana-
lyst, 64, 568 (1939).
13. A. Windaus, F. S c h e n c k,
F. v. Werder. Z. physiol. Chem.,
241 , 100 (1936).
134
14. F. Schenck. Naturwissensch, 25,
159 (1937).
15. W. H u ber, О. В ar 1 о w. J.
Biol. Chem., 149, 125 (1943).
16. A. Windaus, H. L e t t r e,
F. Schenck. Lieb. Ann., 520,
98 (1935).
17. D. Ewing, T. S 1 a b a c h,
H. Powell, J. V a i t к u s,
O. Bird. Analyt. Chem., 24,
1406 (1954).
18. A. W i n d a u s, G. T r a u t -
m a n n. Z. physiol. Chem., 247,
185 (1937).
19. A. W i n d a u s, B. G ii n t z e I.
Lieb. Ann., 538, 120 (1939).
20. H. В г о с к m a n n, A. Busse.
Z. physiol. Chem., 249, 176 (1937);
Naturwissensch, 26, 122 (1938).
21. J. T u 1 e с к i, I. Kazakie-
w i e r. Acta polon. pharmac., 17,
Ke 1, 81 (1960).
22. R. Z e t t e r s t r 6 m. Nature, 167,
409 (1951).
23. A. W i n d a u s, M. D e p p e,
W. Wunderlich. Lieb. Ann.,
533, 118 (1937).
24. К. В h a r u c h a, F. M a г t i n.
Пат. США 3168535; РЖХ, 1966,
17H322
25. N. M i 1 a s, P. Davis, L i -
Chin Chiang. J. Am. Chem.
Soc., 77, 1640 (1955).
26. H. Sc h a 1 t egg er, F. M ii I 1-
n e г. Швейц, пат. 314148; РЖХ,
1958, 74998.
27. P. S e t z. Z. physiol. Chem., 215,
183 (1933).
28. A. W i n d a u s, J. G a e d e,
J. К 6 s e r, G. S t e i n. Lieb.
Ann., 483, 17 (1930).
29. W. D a u b e n, J. В e 1 1, T. Hut-
ton, G. L a w s, A. R e i n e r,
H. Urschler. J. Am. Chem.
Soc., 80, 4116 (1958).
30. W. D a u b e n, P. Baumann.
Tetrahedron Leters, 1961, 565.
31. P. W e s t e r h о f. J. Buis-
man. Rec. trav. chim., 75, 1243
(1956).
32. O. L i n s e r t. Пат. США
2030377; С. A., 30, 2326 (1936).
33. A. W i n d a u s, A. Lu 11 r i n-
g h a u s, P. В u s s e. Nachr. Ges.
Wiss. Gottingen. Math. physik.
KL, Fachgruppe, III, 150 (1932).
34. F. L a q u e r, O. L i n s e г t.
Klin. Wochschr., 12, 753 (1933).
35. A. H a r r i s, T. Mo о r e . Bioch.
J., 23 , 261 (1929).
36. J. C a s t e 1 1 s, E. Jones,
G. M e a к i n s, R. Willi-
ams. J. Chem. Soc., 1959, 1159.
37. P. Buss e. Z. physiol. Chem.,214,
211 (1933).
38. A. Wi n d a us, F. v. Werder,
A. Liittringhaus. Lieb.
Ann., 499, 188 (1932).
39. K. D i m г о t h. Ber., 70, 1631
(1937).
40. H. Lindholm. Arch. Pharm.
org. Chem , 51, 715 (1944).
41. Y. Raoul, N. L e Boulch,
A. Guerillot-Vinet,
R. D u 1 о u, С. В a г о n. Comp,
rend., 241, 1882 (1955).
42. Y. R a о u 1, N. Le Bou 1 ch.
Bull. Soc. chim. biol., 45, 145 (1963).
43. C. Baron, J. В i d a 1 1 i e r.
Bull. Soc. chim. France, 1959, 1330.
44. Wang Yu, Ting Hung-
s h i u n, H u a n g Jing-jain,
Chow Yung-chi, Huang
Yao-tseng. Acta chim. sinica,
24, 126 (1958); C. A., 53, 5331 (1959).
45. A. W i n d a u s, U. R i e m a n n.
Z. physiol. Chem., 274, 206 (1942).
46. N. L e Boulch, Y. R a о u 1.
G. О u r i s s о n. Bull. Soc. chim.
Franc., 1967, 2413.
47. H. D a n n e n b e r g, К. H e -
benbrock. Lieb. Ann., 662, 21
(1963).
48. K. Schubert. Biochem. Z., 326,
132 (1954); Naturwissensch., 41, 231
(1954).
49. P- West er h of, J. В u i s-
m a n. Rec. trav. chim., 76, 679
(1957).
50. К. 1 r m s c h e r. Naturwissensch.
47, 425 (1960); пат. ФРГ 1108215;
С. A., 56, 531 (1962).
51. Англ. пат. 790448; С. А., 54, 9995
(1960).
52. F. Schenck. Пат. ФРГ 1077660;
С. А., 55, 11469 (1961).
53. A. W i n d a u s, О. L i n s е г t,
A. Liittringhaus, G. Wei ri-
lle h. Lieb. Ann., 492, 226
(1932).
54. E. Merc к. Герм. пат. 624231;
С. A., 30, 2708 (1936).
55. A. Koevoet, A. Verloop,
J. В u i s m a n. Пат. США 2840575;
С. A., 52, 20262 (1958); гол. пат.
92829; РЖХ, 1961, 22Л249.
56. A. Verloop, A. Koevoet,
R. van Moo г se 1 a ar,
Е. Н a v i п g а . Rec. trav. chim.,
78,. 1004 (1959).
57. Н. L е t t г ё. Lieb. Ann., 511, 280
(1934).
58. R. К u h n, E. M 6 1 1 e r. AngeW.
Chem., 47, 145 (1934).
59. H. I n h о f f e п, K. Bruckner,
R. G г й n d e 1. Chem. Ber., 87, 1
(1954).
60. T. Kobayashi. Bitamin, 35,
362 (1967); C. A., 67, 44013 (1967).
61. H. Inhoffen, G. Quin-
k e г t, H.-I. Hess, H.-M. Erd-
mann. Chem. Ber., 89, 2273 (1956).
62. H. 1 n h о f f e n, К. В r ii с к -
пег, R. Griindel, G. Quin-
k e r t. Chem. Ber., 87, 1407 (1954).
63. T. Kobayashi. Bitamin, 35,
366 (1967); C. A., 67, 44014 (1967).
64. H. Inhoffen, G. Q и i n -
к e r t, H.-I. Hess, H.Hir-
s c h f e 1 d. Chem. Ber., 90 , 2544
(1957).
65. W. Thiele, G. Trautman n.
Ber., 68, 2245 (1935).
66. F. A s к e w, R. В с и r d i : ! / n
135
Н. В r u ce, R. Callow,
J. Philpot, Т. Webster.
Proc. Roy. Soc. (London), B107, 76,
91 (1930); B108, 340 (1931); B109,
488 (1932).
67. D. W i t t, M. S u 1 1 i v a n. Ind.
Eng. Chem., 18, 117 (1946).
68. H. Vogel-Knobloch. Che-
mie und Technik der Vitamine. II,
Stuttgart, 1950.
69. H. L e e m a n n, K- S t i c h. Helv.
Chim. Acta, 45, 1275 (1962).
70. A. W i n d a u s, W. G r u n ri-
ma n n. Lieb. Ann., 521, 160
(1935); 524 , 295 (1936).
71. J. H e i 1 b г о n, R. Jones,
K. S a m a n t, F. S p r i n g. J.
Chem. Soc., 1936, 905.
72. J. H e i 1 b г о n, F. Spring.
Chem. and Ind., 54, 795 (1935).
73. K. S c h u b e г t, K. Wehr -
b e r g e r. Biochem. Z., 328, 199
(1955).
74. P. W e s t e r h о f, J. Buis-
man. Rec. trav. chim., 75, 453
(1956).
75. W. Werder. Lieb. Ann., 603, 15
(1957).
76. S. v. R e i c h e 1, M. D e p p e.
Z. physiol. Chem., 239, 143 (1936).
77. A. G u i t e r a s, Z. N a к a-
m i у a, H. I n h о f f e n. Lieb.
Ann., 494, 116 (1932).
78. S. Bergstrom. Helv. Chim.
Acta, 32, 1 (1949).
79. S. Bergstrom, A. Larden,
T. Reichstein. Helv. Chim.
Acta, 32, 1617 (1949).
80. J. Bernal. Nature. 129, 277
(1932).
81. J. Bernal, D. Crowfoot,
I. Fankuchen. Trans. Roy.
Soc. (London), A239, 135 (1940).
82. C. Carlisle, D. Crowfoot.
Proc. Roy, Soc., A184, 64 (1945).
83. R. J о n e s. J. Am. Chem. Soc.,
72, 5322 (1950).
84. K- Dimroth, H. Johnson.
Ber., 74, 520 (1941).
85. J. V 1 i e t. Rec. trav. chim., 67,
246, 265, 343 (1948).
86. A. Lardon,!. Reichstein.
Helv. Chim. Acta, 32, 2003 (1949).
87. Л. Ф и з e p, M. Ф и з e p. Химия
природных соединений фенантре-
нового ряда, AL, Госхимиздат,
1953.
88. V. Р г е 1 о g a. et Helv. Chim.
Acta, 36, 308, 320, 325 (1953).
89. L. V e 1 1 u z, G. A m i a r d. Bull,
soc. chim. France, 1955, 205.
90. О. H a s s e 1, В. О t t a r. Acta
chem. scand., 1, 929 (1947).
91. O. Hassel. H. V i e r v о 1 1.
Acta chem. scand., 1, 149 (1947).
92. R. Harr i s, J. Bunker,
L. Mosier. J. Am. Chem. Soc.,
60, 2579 (1938).
93. A. Arnold. Proc. Soc. Exptl.
Biol. Ated., 63, 230 (1946).
94 A. К n u d s о n, F. В e n f о r d.
Biol. Chem., 124 , 287 (1938).
95. J. Bunker, R. Harris,
L. Mosier. J. Am. Chem. Soc.,
62, 508, 1760 (1940).
96. С. В i 1 1 s, E. H о n e у w e 1 1,
W. Cox, J. Biol. Chem., 92, 601
(1931).
97. C. Bills. Глава «Витамины груп-
пы D» в kh. “The Vitamins”,
m. 11, New York, 1954.
98. В. П. Вендт. Витамины, 2, 14
(1956), изд. АН УССР, Киев.
99. I. Hercsel, Z. Е с s е г у,
М. Muller. Венг. пат. 148480;
РЖХ, 1963 , 6Н212.
100. I. Hercsel, Z. Ecsery,
Z. Laurencsik, J. Sume-
g i, F. К о m 1 о s. Венг. пат.
151690; С. А., 62, 7846 (1965).
101. С. В i 1 1 s, F. Brickwedde.
Nature, 121, 452 (1928).
102. С. Bills, Е. Honeywell,
W. Сох. J. Biol. Chem., 80, 557
(1928).
103. J. W add el 1, W. W о e s s-
n e г. Пат. США 2410254; С. A.,
41, 1815 (1947).
104. H. Beard, R. Bark,
H. Thompson, H. Gold-
blatt. J. Biol. Chem., 96, 307
(1932).
105. W. D a s 1 e r, C. Bauer. J.
Biol. Chem., 167, 581 (1947).
106. Y. Raoul. Франц, пат. 984809;
С. A., 50, 5784 (1956).
107. A. Wi nd a us, M. D e p p e,
C. Roosen - Runge. Leib.
Ann., 537, 1 (1938).
108. L. V e 1 1 u z, G. A m i a r d,
B. G о f f i n e t. Compt. rend.,
240, 2076, 2326 (1955).
109. L. V e 1 1 u z, A. Gaston,
B. G о f f i n e t. Bull. soc. chim.
France, 1955, 1341.
110. L. V e 1 1 u z, A. Petit, G. Mi-
chel, G. Rousseau. Compt.
rend., 226, 1287 (1948).
111. L. V e 1 1 u z, A. Petit,
G. A mi a rd. Bull. soc. chim.
France, 15, 1115 (1948).
112. L. V e 1 1 u z, G. A m i a r d,
A. Petit. Bull. soc. chim. Fran-
ce, 16, 501 (1949).
113. А. К о e у о e t, A. Ver loo p,
E. H a v i n g a. Rec. trav. chim.,
74, 788 (1955).
114. A. V e r 1 о о p, А. Koevoet,
E. H a v i n g a. Rec. trav. chim.,
76, 689 (1957).
115. V. D e 1 a г о f f, P. R a t h 1 e,
M. Legrand. Bull. soc. chim.
France, 1963, 1739.
116. M. Rappoldt, P. Wester-
h о f, K. Hanewald, J. В u i s-
m a n. Rec. trav. chim., 77,
241 (1958).
117. M. R a p p о 1 d t, E. Havin-
g a. Rec. trav. chim., 79, 369
(1960).
118. M. Rappoldt. Rec. trav.
chim., 79, 1012 (1960).
119. M. Rappoldt, J. Buis-
r ;i. Havin:. •. Rec.
136
trav. chim., 77, 327 (1958).
120. R. de Kock, G. van der
К u i p, A. V e r 1 о о p, E. Ha-
vin g a. Rec. trav. chim., 80, 20
(1961).
121. A. Windaus, E. Auhagen.
Z. physiol. Chem., 196, 108 (1931).
122. A. Windaus, F. Werder,
A. Liittringhaus. Lieb.
Ann., 499, 188 (1932).
123. Англ. пат. 790956; С. A., 54,
11085 (1960).
124. H. Inhoffen, H. Schae-
fer. Chem. Ber., 92, 1126 (1959).
125. L. V e 1 1 u z, G. A m i a r d,
B. G о f f i n e t. Франц. пат.
1186124; РЖХ, 1960, 66560.
126. E. Having a, J.Schlat-
m a n n. Tetrahedron, 16, 146
(1961).
127. G. Sanders, E. Having a.
Rec. trav. chim., 83, 665 (1964).
128. A. W i n d a u s, K. D i t h m ar,
E. F e r n h о 1 t z. Lieb. Ann.,
493, 259, 265 (1932).
129. M. Rappoldt. Rec. trav.
chim., 79, 392 (1960).
130. I. Harrison, R. Hurst,
B. Lythgoe, D. Willi-
ams. J. Chem. Soc., 1960, 5176.
131. E. Having a, A. Koevoet,
A. V e r 1 о о p. Rec. trav. chim.,
74, 1230 (1955). 75, 371 (1956).
132. G. A m i a r d. Naturwissensch.,
47, 252 (1960).
133. L. V e 1 1 u z, C. A mi ar d. Пат.
США 2707710; С. A., 50, 6517
(1956).
134. R. J a wor ska, H. S a 1 w a.
Acta Polon. Pharm., 20, 405
(1963); Neth. Appl., 6516485; C. A.,
67, 73784 (1967).
135. M. Rappoldt. Пат. ФРГ
1156406; С. A., 60, 3072 (1964);
гол. пат. 102019; РЖХ, 1964,
7Н215.
136. L. V е 1 1 u z, G. A m i а г d.
Compt. rend., 228, 692, 853, 1037
(1949); пат. США 2693475; С. А.,
49, 14818 (1955).
137. К. D i m г о t h. Вег., 68, 539
(1935).
138. Н. Inhoffen. Lieb. Ann., 494,
116, 122 (1932).
139. G. A h г е n s, E. F e r n h о 1 t z,
W. Stoll. Lieb. Ann., 500, 109
(1932).
140. J. Castells, G. Fletcher,
E. J о n e s, G. M e a к i n s,
R. Swindells. J. Chem. Soc.,
1960, 2627, 2785.
141. P. M а у о r, G. M e с к i n s. J.
Chem. Soc., 1960, 2792, 2800.
142. J. C a s t e 1 1 s, G. Mea ki ns,
R. Swindells. J. Chem. Soc.,
1962, 2917.
143. J. H e i 1 b г о n, F. S p r i n g,
P. S t e w a r t. J. Chem. Soc.,
1935, 1221.
144. K. D i m г о t h. Ber., 69, 1123
145. A. Windaus, K. Dimroih.
Ber., 70, 376 (1937).
146. J. Cast el 1 s, E. Jones,
R. Williams, G. Mea-
kins. Proc. Chem. Soc., 1958, 7.
147. P. Meunier, G. Thibau-
d e t. Compt. rend., 223, 172 (1946).
148. W. G г и n d m a n n. Z. physiol.
Chem., 252, 151 (1938).
149. J. Lugtenburg. E. Havin-
ga. Tetrahedron. Lett. 1969, 2391.
150. Y. R а о и 1, I. Chopin,
P. M e и n i e r. N. Boulch. Compt.
rend., 228, 1064 (1949).
151. R. Davidson, Sh. Wad-
dington - Feather,
D. Williams, B. Lythgoe.
J. Chem. Soc. «С», 1967, 2534.
152. F. v. W e r d e r. Z. physiol.
Chem., 260, 119 (1939).
153. P. Westerhof. Гол. пат.
93270; РЖХ, 1961, 22Л250.
154. L. Veil uz, G. A m i a r d,
B. G о f f i n e t. Пат. ФРГ
1040025; С. A., 55, 4587 (1961).
155. J. van de VI iervoet,
P. Westerhof, I. Buis-
m a n, E. H a v i n g a. Rec.
trav. chim., 75, 453 (1956).
156. A. V e r 1 о о p, G. Corts,
E. H a v i n g a. Rec. trav. chim.,
79, 164 (1960).
157. M. M u 1 1 e r. Z. physiol. Chem.,
233, 223 (1935).
158. O. Rosenheim, King,
Chem. and Ind., 54 , 699 (1935).
159. J. C a s t e 1 1 s, E. Jones,
G. M e a к i n s, S. P a 1 m e r,
R. Swindells. J. Chem. Soc.,
1962, 2907.
160. W. D a и b e n, G. F о n к e n. J.
Am. Chem. Soc., 79, 29?! (1957).
161. W. D a и b e n, G. F о n к e n. J.
Am. Chem. Soc., 81, 4060 (1959).
162. W. Huber, G. Ewing,
J. К r i g e r. J. Amer. Chem. Soc.,
67, 609 (1945).
163. R. W о о d w a r d, F. So nd -
h e i m e r, D. T a и b. J. Am.
Chem. Soc., 73, 3547, 3548 (1951).
164. R. Woodward, F. Sond-
heimer, D. Taub, Heus-
ler, McLamore. J. Am. Chem.
Soc., 74, 4223 (1952).
165. H. Inhoffen, G. Quin-
к e r t. H.-J. Hess. Naturwis-
sensch., 44, 11 (1957).
166. H. Inhoffen, G. Quin-
k e r t, S. S c h u t z. Chem. Ber.,
90, 1283 (1957).
167. H. Inhoffen, G. Quin-
k e r t, S. S c h u t z, D. К a m-
p e, G. D о m a g k. Chem. Ber.,
90, 664 (1957).
168. H. Inhoffen, E. Prinz.
Chem. Ber., 87, 684 (1954).
169. H. Inhoffen, H. Kramer.
Chem. Ber., 87, 488 (1954).
170. H. Inhoffen, J. K. a t h,
W. S t i e h e r 1 i n g, К. В г и v-
k n e r. Lieb. Ann., 603, 25 (1957).
71. I. Harrison, B. L у t h g о
137
Proc. Chem. Soc., 1957, 261; J.
Chem. Soc., 1958, 837.
172. B. L у t h g о e. Proc. Chem.
Soc., 1959, 141.
173. H. I n h о f f e n, J. К a t h,
K. Bruckner. Angew. Chem.,
67, 276 (1955).
174. H. Inhoffen. Angew Chem.,
70, 576 (1958).
175. O. L i n s e r t. Z. physiol. Chem.,
241, 125 (1936).
176. H. Inhoffen. Angew. Chem.,
72, 875, (1960).
177. H. Inhoffen, R. I r m -
scher, H. Hirschfeld,
U. Stache, A. Kreutzer.
Chem. Ber., 91, 2309 (1958).
178. H. I n h о f f e n, К. I r m s c h e r,
G. F r i e d r i c h. D. К a m p e,
O. Berges. Chem. Ber., 92,
1779
179. J. Dixon, P. Littlewood,
B. Lythgoe, A. Sak Sena,
Chem. Communs., 1970, 993.
180. T. Dawson, J. Dixon,
P. Littlewood, B. Lyth-
goe. A. Sak Sena. J. Chem.
Soc., «С», 1971, 2960.
181. H. Lindlar. Helv. Chim. Acta,
35, 446 (1952).
182. H. I n h о f f e n, G. Qui n-
k e r t, S. S c h й t z, D. К a m-
p e, G. D о m a g k. Chem. Ber.
90, 664 (1957).
183. H. I n h о f f e n, G. Fried-
r i c h., D. К a m p e, O. Ber-
ges. Chem. Ber., 92, 1772 (1959).
184. E. R о m i n g e r. Ergebn. Vi-
tamin. Hormon-Forschung. 2, 104
(1939).
185. A. Windaus, O. Linser t.
Lieb. Ann., 489, 269 (1931).
186. W. G r a b. Z. physiol. Chem., 243,
63 (1936).
187. H. В г о с к m a n n. Z. physiol.
Chem., 241, 104 (1936).
188. E. S i m о n s, T. Z u с к e r. J.
Am. Chem. Soc., 58, 2655 (1936).
189. H. Rosenberg. Chemistry
and Physiology of the Vitamins,
Ne'w York, 1945.
190. N. Gridgeman. Quart. J.
Pharm. Pharmacol., 18, 24 (1945).
191. W. W u n d e r 1 i c h. Z. phy-
siol. Chem., 241, 116 (1936).
192. L. V e 1 1 u z, G. A m i a r d,
B. G о f f i n e t. Франц, пат.
1192180; С. A., 56, 6060 (1962).
193. A. Windaus, K. Buch-
holz. Ber., 71, 576 (1938); 72
597 (1939).
194. A. Windaus, J. Naggatz.
Lieb. Ann., 542, 204 (1939).
195. Л. Ф и з e p, M. Ф и з e p. Сте-
роиды, M., пзд-во «Мир», 1964,
c. 171.
196. J. В 1 u n t, H. D e L u c a,
H. S c h n о e s. Chem. Communs,
1968, 801.
197. J. Campbell, D. Squires,
J. Babcock. Steroids, 13, 567
(1969).
198. S. H a 1 к e s, N. V 1 i e t. Rec.
trav. chim. 88, 1080 (1969).
199. T. S u d a, H. De Luca,
H. Schnoes, J. Blunt. Bio-
chem. Biophys, Res. Communs,
1969, 35, 182.
200. D. L a w s о n, D. Fraser,
E. К о d i с e к, H. M о r r i s,
D. Williams. Nature, 230,
(5291), 228 (1971).
201. M. Holick, H. Schnoes,
H. D e L u с a, T. Suda,
R. Cousins. Biochemistry, 10,
2799 (1971).
202. H. DeLuca, J. Omdahl,
M. Holick, T. Suda, Y. Ta-
naka. Biochemistry, 10, 2935
(1971).
203. К- H a nqwa I d, M. R a p -
poldt, J. Roborgh. Rec.
trav. chim., 80, 1003 (1961).
204. В. H. Б у к и н, Е. П. С к о р о-
богатова. Сборн. Витамин-
ные ресурсы и их использование,
I, стр. 207, М., Изд. АН СССР,
1951.
205. В. Н. Букин, Н. Ерофее-
в а. Сборн. Витаминные ресурсы
и их использование, I, стр. 250,
М., Изд. АН СССР, 1951.
206. A. W i n d a u s, Н. Inhof-
fen, S. V. R е i с h е 1. Lieb.
Ann., 510, 248 (1934).
207. J. Cornfort h, J. Youhot-
s k y, G. P о p j a k. Nature,
173, 536 (1954).
208. C. Bills, E. H о n e у w e 1 1.
J. Biol. Chem., 80, 15 (1928).
209. H. W i e 1 a n d, G. G о u g h.
Lieb. Ann., 482, 36 (1930).
210. A. P. Татьянин. Производ-
ство витамина D.M., Пищепром-
издат, 1943.
211. Л. Чугаев. Angew. Chem., 13,
618 (1900).
212. Н. Brockman n, Y. Chen.
Z. physiol. Chem., 241, 104 (1936).
213. Методы определения витаминов,
ВНИ витаминный институт, М.,
Пищепромиздат, 1954.
214. A. Windaus, F. Schenck.
Пат. США 2098984. 2098985;
С. А., 32, 196 (1938).
215. Н. Rosenberg. W. Woess-
ner. Пат. США 2434015; С. А.,
42, 1710 (1948).
216. A. W i п d a u s, R. L а п g е г.
Lieb. Ann., 508, 105 (1933).
217. R. N i 1 s s о п, N. Olsen,
P. Nilsson. The Svedberg,
1944, 484.
218. G. T a p p i. Gaz. chim. itaL, 78,
311 (1948).
219. А. В. T p у ф а и о в,
В. А. Кирсанова. Биохи-
мия, 4, 377 (1939).
138
220. H. W i e 1 a n d, Y. К a n а о к a.
Lieb. Ann., 530, 146 (1937).
221. D. В a r t о n, J. С о x. J. Chem.
Soc., 1948, 1357.
222. H. Wi el a nd, R. Rath,
H. Hess. Lieb. Ann., 548, 34
(1941).
223. В. H. Букин, И. H. Га p -
кина, И. H. Кущинская,
Л. Б. Л и хо шва, Е. П. Ско-
робогатова, Б. В. Ш е-
« стаков. Авт. свид. СССР 97222;
Бюлл. изобрет., 1954, № 2, 15.
224 R. F i s с h е г. Пат. США 2865934;
РЖХ, I960, 70684.
225. В. А. В а д о в а, М. В. П е р е-
пёлкина, 3. Е. Ге лео-
новская, И. В. Бурут-
т о, П. В. К о т л я р е в с к н й,
Т. И. Бедер, В. И. Исаев.
Авт. свид. СССР 134389; С. А., 55,
12783 (1961).
226. W. С а 1 с о t t, J. W a d d е 1 1»
H. Rosenberg. Пат. США
2383446; С. A., 39, 5413 (1945),
227. A. Boer. J. van Niekerk,
E. ReerinkA. van Wijk.
Пат. США 2163659; С. A., 33,
7964 (1939).
228. R. Woodward, F. So n d-
h e i m e r, D. T a u b, К. H e u s-
1 e r, W. McLamore. J. Am.
Chem. Soc., 73, 2403 (1951).
229. A. Butenandt, A. Woff.
Ber., 68, 2091 (1935).
230. L. Ruzicka, P. Plattner,
R. Aeschbacher. Helv.
Chim. Acta, 21, 866
(1938).
231. W. D a u ben, J. Eastham.
J. Am. Chem. Soc., 72, 2305 (1950).
232. W. В u s e r . Helv. Chim. Acta»
30, 1385 (1947).
233. A. К r a m 1 i. Arch. Biol.
Hung., 17, 337, 343 (1947).
234. O. Wintersteiner,
W. R u i g h. J. Am. Chem. Soc.,’
64, 1177, 2453 (1942); пат. США
2411177; С. A., 41, 1398 (1947).
235. A. В i d e, H. H e n b e s t,
E. J о n e s, P. Wilkinson.
J. Chem. Soc., 1948, 1783,
1788.
236. H. Henbest, E. Jones. J.
Chem. Soc.', 1948, 1792.
237. G. H a s 1 e w о о d. J. Chem.
Soc., 1938, 224; Biochem. J., 33,
454 (1939).
238. H. Rosenberg. Пат. США
2209934; С. A., 35, 138
(1940).
239. A. S о b e 1, P. О w a d e s>
J. О w a d e s. J. Am. Chem. Soc.,
71, 1487 (1949).
240. Яп. пат. 8937 (1955); С. A., 52,
1296 (1958).
241. T. В a r r, J. Н е i 1 b г о п,
Е. Р а г г у, Т. S р г i п g. J.
Chem. Soc., 1936, 1437.
242. S. Bernstein, L. Binovi,
L. D о r e m a n, K. Sax,
Y. Subbarow. J. Org. Chem.,
14, 433 (1949).'
243. H. H e n b e s t, E. Jones,
A. Bide, R. P e e v e r s,
P. Wilkinson. Nature, 158,
169, (1946).
244. J. Bui sm an, W. Stevens,
J. V 1 i e t. Rec. trav. chim.,
66, 83, 87 (1947).
245. J. Rede 1, B. Gauthier.
Bull. soc. chim. France., 15, 607
(1948).
246. A. Bide, H. H e n b e s t,—
E. Jones, R. P e e v e r s,
P. W i 1 k i n s о n. J. Chem. Soc.,
1948, 1783.
247. S. В e r n s t e i n, K. S a x. Пат.
США 2498390; С. A., 44, 5401
(1950); J. Am. Chem. Soc., 73,
846 (1951).
248. A. Wander. Швейц. пат.
305111; англ. пат. 760460; С. А.,
51, 2067, 9722 (1957).
249. Н. Schaltegger. Пат. ФРГ
956509; РЖХ, 1958, 33596; швейц,
пат. 314796; РЖХ, 1959,
5789.
250. G. Handley. Austral. J.
Charm., 40, 1079 (1959); С. A.,
54, 11385 (1960).
251. H. Schaltegger. Experien-
ce, 5, 321 (1949); Helv. Chim.
Acta, 33, 2101 (1950).
252. K. Schaaf. Пат. США 2542291,
2546788; С. A., 45, 7606, 7608
(1951).
253. W. R u i g h, D. G о u I d. Пат.
США 2546787; С. A., 45, 7607
(1951).
254.
255.
256.
257.
258.
259.
260.
261.
262.
263.
264.
R. Kapp, S. Chodroff, Le
S о f i e 1 d. Пат. США 2642446;
С. A., 48, 7647 (1954).
’.Hunziker, F. M ii 1 1 n e r.
-lelv. Chim. Acta, 41, 70
(1958).
). G о u I d, W. R u i g h. Пат.
США 2563235; С. A., 46, 1597
(1952).
F. Hunziker. Helv. Chim.
Acta, 38, 1316 (1955).
N. M i 1 a s. R. H e g g i e. J. Am.
Chem. Soc., 60, 984 (1938).
A. Windaus, A. L u t t r i n g-
h a u s. Ber., 64 , 850 (1931).
K. Schubert, K.-H. В 6 h -
m e. Chem. Ber., 93, 1878, 1884
(1960).
W. D i r s c h e r 1, H. N a h m.
Lieb. Ann., 558, 231, (1947).
Ф. T. С о л о д к и й, А. А. Но-
вак. ЖПХ, 20, 864 (1947).
А. М. X а л е ц к и й, Н. Н. С о-
л о м о н и к. ЖОХ, 17, 1171
(1947).
W. R u i g h. Пат. США 2378435;
С. А., 39, 5413 (1945).
139
ГЛАВА IV
ЦИКЛОГЕКСЕНИЛИЗОПРЕНОИДНЫЕ ВИТАМИНЫ И
ПРОВИТАМИНЫ
РЕТИНОЛ, ДЕГИДРОРЕТИНОЛ
К ретинолам — витаминам группы А — относятся соединения из 20
атомов углерода, включающие в свою структуру триметилциклогексеновое
кольцо, связанное с тетраеновой сопряженной цепью изопреноидного типа,
оканчивающейся спиртовой или альдегидной группой.
Ретинол—витамин Ах (аксерофтол), 9,13-диметил-7-(1,2,5-триметил-
циклогексен-5-ил-6)нонатетраен-7,9,11,13-ол-15 (I) — представляет собой
соединение с полной транс-конфигурацией всех четырех двойных связей
боковой цепи [1, 2] и s-грс-конфигурацией С(б>—С(7)-связи
19 20
сн3 СН,
I 3 I
с=сн-сн=сн-с=с н-сн2он
9 10 11 12 13 14 15
I
СН3 СН3 VH3 VH3
S<\/CH=CH-C=CH-CH=CH-C=CH-CH2OH
^сн3 yin
Из природных источников и синтетически витамин Ах получен в виде
шести стереоизомерных форм.
Неоретинол — неовитамин Ах (II) — представляет собой стереоизомер
витамина Ах с zpc-конфигурацией конечной (пятой) двойной связи боко-
вой цепи — 13-1{ос-ретинол; он выделен из природных источников. Осталь-
ные стереоизомеры; 11-1{ис-ретинол (III) [5], 9-1{ас-ретинол (IV) [3,4], 11,
13-ди-1{ас-ретинол (V) [5] и 9,13-ди-^нс-ретинол (VI) [4] впервые получены
синтетически.
Природный витамин Ах из жира печени морских рыб в основной своей
части состоит из полного транс-изомера (I); неовитамина А (II) в природ-
ном продукте содержится около 35% всего количества витамина [61.
Ч, СН.Н н V'- _ц
.С X. X. ХН.-ОН
'"С ''С '"С 'х
н н н н
сн3
Полный гране -Ретинол
I *
СН3 СНзН Н
>< _с с с
Г >сн
I Н н н 1
""СНЭ С|ф "СИ
СН2ОН
11-4W-Ретинол (нео-4
111
СНз СНз
СНз JHjH I Н I
Сх /Сх /С.
"С "с "с сн
С1£ Н Н СН2ОН
13-^г-Ретинол (нео-неоретинол
3-иис -Ретинол (изо-г)
IV
140
СНз «НН l”3 М
>< ^с. XL AL
< "С "С ''СН
НСНАС-С^н
3 н
11,13-ди-ссг-Ретинол (нео-с)
V
снГ^с-^и
’ н
ЭДЗ-ди-сда -Ретинол (изо-i)
VI
Дегидроретинол — 3,4-дегидроретинол, витамин А2 (VII) — отличает-
ся от ретинола наличием дополнительной С(3)—С(4) двойной связи в цикло-
гексеновом ядре {7, 8]
сна сн8
сн=сн—с=сн—сн=сн-с=сн— СН2ОН
Строение дегидроретинола (VII) характеризуется полной транс-конфи-
гурацией всех двойных связей изопреноидной боковой цепи.
Синтетически дегидроретинол получен в виде шести Zjoc-mpawc-стерео-
изомеров: полный транс-изомер (VII), 13-цис-дегидроретинол (нео-а)
(VIII), 11-цис-дегидроретинол (нео-Z?) (IX), 9-^ас-дегидроретинол (изо-а)
(X), 11,13-ди-цис-дегидроретинол (нео-с) (XI) и 9,13-ди-/{ас-дегидрорети-
нол (изо-/?) (XII) [9, 10].
сн3 C1L
СНз Я'эН I Н I
/L хСч ХД /L /СШОН
*с Ч Ч
н н н н
СНз
Полный транс—Дегидроретинол
VII
СН. СЦн- н
><
Г Y ^С ''С "с "СН
ЧАсн,Н Н ". «юн,
13 -цис-Дегидроретинол (нео-с)
VIII
СНз СНзН V”3 н
>< XL с^
< Y С ''С ''СН
к JL Н Н т
^ СН3 СНз^СН
СНзОН
И- цис -Дегидроретинал (нео-i)
IX
CI4. О4зН Р1»
с^ .С.
^с ''сн
н НС
СНз нс^сн
I
^с^
сн3 ^сн
CHjOH
9-цис-Дегидроретинол (изо-с)
X
11,13-ди-ссс-Дегидроретинол (нео-с)
XI
сн, СНзН
-С^ ,
"с
Н
СН3
СН,
,с.
''СН
I
НС^
*сн
/СН.ОН
СН3 ^С*
3 Н
9,13-ди-ссс -Дегидроретинол (иэо-Z)
XII
Для витамина А характерна s-^ac-конфигурация С(6)—С(7)-связи, как
это следует из рентгеноструктурного изучения витамин А-кислоты (рети-
141
новой кислоты) [11], 15,15'-дегидро-0-каротина [11, 12] и 0-каротина [13].
Циклогексеновое кольцо витамина А по своей конформации в наиболее энер-
гетически выгодной форме может быть представлено в виде полукресла
[14]
(обозначение связей: э — экваториальная; а — аксиальная, «осевая» или
«полюсная»; э' — квазиэкваториальная и а' — квазиаксиальная).
Для ретинола в соответствии с количеством двойных связей полиеновой
алифатической цепи возможно 16 (24) геометрических изомеров. Как уста-
новлено для каротиноидных полиенов [1, 2], содержащих такие же изо-
преноидные группы, как и ретинол, транс-конфигурация двойных связей
свободна от пространственных помех. Однако ^ас-изомеризация двойных
связей полиеновой цепи, содержащей метильные заместители, протекает не
во всех случаях с одинаковой легкостью.
Метильные группы обладают радиусом помех около 2 А, который пре-
пятствует дальнейшему сближению атомов. Поэтому ^ас-превращения от-
дельных этиленовых связей, приводящие к искривлению зигзагообразной
молекулы полной транс-конфигурации, легко возникают без пространствен-
ных затруднений около центральной двойной связи только в триеновых си-
стемах с незамещенными концевыми членами типа I,
( СЙ ®
=с с=
Тип I
с=с
к—X
Тип II
но становятся заторможенными из-за пространственных помех, вызывае-
мых метильными группами в триеновых системах с монозамещенными
концевыми членами типа II. Следовательно, для молекулы витами-
на Аг свободное превращение с образованием стабильных ^пс-конфигу-
раций возможно для С(9>—С(ю) и С(|3)—C(i4) двойных связей, один из ато-
мов углерода которых связан с метильной группой. У двойной связи С(1() —
С(12), атомы углерода которой не содержат заместителей, ^ас-изомеризация
будет заторможена метильной группой концевого члена триеновой системы,
а для С(7)—С(8) двойной связи цис-конфигурация невозможна. 7-цис-Рети-
нол и соответствующие каротиноиды неизвестны.
Таким образом, из 16 теоретически возможных стереоизомеров витами-
на At число изомеров, не испытывающих пространственных затруднений,
ограничивается четырьмя (формулы I, 11, IV, VI) [2]. Все они получены
из природных источников или синтетически. Остальные 12 изомеров про-
странственно затруднены, однако среди них в природе обнаружен 11-цис-
ретинол (III) и синтезом получен еще один изомер— 11,13-ди-цас-ретинол
(V). Стерическпе затруднения возникают и между 5-метильной группой и
атомом водорода положения 8, однако представленная на формуле s-транс-
конфигурання С(й)—С(7)-связи менее эффективна, чем s-цас-конфигура-
ция [11—13].
142
При расчете внутренней энергии s-цис- и s-транс-изомеров ретиналя
как функции угла вращения заместителей вокруг С(б>—С(7)-связи показано,
что минимальная энергия соответствует s-^uc-изомеру, при этом циклогек-
сеновое кольцо и полиеновая цепь расположены в разных плоскостях —
для витамина А под углом 35° [15]. Природный витамин А, ретиналь и каро-
тиноиды существуют в виде s-^ос-изомеров.
Полиеновая сопряженная система витамина А и каротиноидов представ-
ляет собой хромофор, обусловливающий окраску этих соединений. В транс-
конфигурациях вся система двойных связей выступает как общий хромо-
фор, свободно проводящий электронные колебания от конца к концу
сопряженной системы; такие соединения показывают наиболее длинноволно-
вый максимум поглощения видимого (для каротиноидов) и ультрафиолето-
вого света (для каротиноидов и витамина А).
Изгибание зигзагообразной молекулы при ^uc-превращениях связа-
но с укорачиванием общего хромофора или с разрывом его на частичные
хромофоры с укороченными сопряженными полиеновыми системами; полосы
поглощения таких соединений смещены в коротковолновую часть с ослаб-
лением их интенсивности. Изомеры ретинола с цепью искривленных ^ре-
конфигурациями двойных связей обладают меньшей биологической актив-
ностью, чем полный транс-ретинол (см. табл. 9).
Ретинол и его стереоизомеры, дегидроретинол и его стер иоизомеры [9,
10] представляют собой желтоватого цвета маслообразные или низкопла-
вящиеся кристаллические вещества. Ретинол перегоняется при 120—125° С и
0,005 мм [28].
Для витаминов группы А характерна растворимость во многих орга-
нических растворителях (в ацетоне, хлороформе, бензоле и др.); они легко-
растворимы в жирах и совершенно нерастворимы в воде.
Благодаря наличию сопряженной системы двойных связей витамины
группы А поглощают в ультрафиолетовой области, давая спектр с одним
интенсивным максимумом поглощения — для стереоизомеров ретинола
143
Хмакс 311—328 нм [4, 29] (рис. 3), для стереоизомеров дегидроретинола
Хмакс 337—352 нм. Дегидроретинол и некоторые стереоизомеры ретинола и
дегидроретинола имеют еще 1—2 максимума гораздо меньшей интенсив-
ности (табл. 9 и 10).
Физические свойства стереоизомеров ретинола
Таблица 9
Изомеры витаминов группы А, т. пл., °C Спектр поглощения А g T. пл. п-фенил- азобензоата, °C Литература
максимум в спирте, X нм
витаминная ан ность, %
Ретинол, витамин Ах, полный транс-ретинол (I) 62—64 325 1835 100 79—80 [16—20]
Неоретинол, 13-цис- ретинол (нео-а) (II) 58—60 328 1686 75,3 94—96 [4, 6, 21, 22]
11-час-Ретинол (нео-6) (III) Масло 319 233 1220 370 23,4 66—67 (5 , 22—24]
9-цас-Ретинол (изо-а) (IV) 81,5—82,5 323 258 1477 382 21,8 79—80 [4, 22, 25]
11,13-ди-час- Ретинол (нео-с) (V) 86—88 311 1024 5 99—100 [5, 22, 25—271
9,13-ди-час-Ретинол (изо-6) (VI) 58—59 324 263 1379 330 23,7 91,5—92,5 [4, 22, 25]
Физические свойства витаминов группы Аг
Таблица 10
Изомеры витаминов группы А, Т. пл., °C Спектр поглощения Витаминная актив- ность, % (ретинол» = 100) Т. пл. п-фенилазо- бензоата, °C Литература
максимум в спирте, X нм е'% 1см
Дегидрорет инол, витамин А2, пол- ный транс-3,4-де- гидроретинол (VII) 63-65 17—19 350 286 276 1455 715 555 40 или 51 94—95 [8, 25, 30—32]
13-час-Дегидроре- тинол (нео-а) (VIII) 73—74 352 288 277 1375 649 493 35 или 26 96—98 [25, 32]
11-Час-Дегидроре- тинол (нео-fe) (IX) Масло 344 286 990 566 15* 38—40 [25]
9-фгс-Дегидроре- тинол (изо-а) (X) 77—79 348 287 277 1143 919 767 14 83—85 [25 , 32]
И, 13-ди-час-Де- гидроретинол (нео-с) (XI) 91—93 337 277 905 470 14* 82—84 [25]
9,13-ди-час-Дегид- роретинол (изо-fc) (ХП) <—30 350 288 1030 761 8* 81—83 [25]
* Предварительные данные.
144
13-цис-Конфигурация (конечной) двойной связи неоретинола оказывает не-
большое влияние на состояние хромофорной системы, что мало отражается на
изменении полосы поглощения ультрафиолетового света [29]. Однако в от-
личие от общего правила для неоретинола (II) наблюдается смещение мак-
симума с 325 нм для полного транс-ретинола не в сторону коротких волн,
а в длинноволновую область до 328 нм, что находится в соответствии с та-
ким же явлением для ^нс-Р-ионилиденэтилового спирта, у которого (так же
как и у неоретинола) концевая г{ас-конфигурация двойной связи находится
в аллильном положении к гидроксильной группе [4, 29]. Полоса поглоще-
ния 9-цис- и 9,13-ди-^нс-ретинолов содержит цис-пик при 258—263 нм, ха-
рактерный для диеновых и триеновых сопряженных систем.
Состояние 13-ф/с-конфигурации двойной связи сказывается на физи-
ческих свойствах, что проявляется в более слабой адсорбции на натриево-
алюминиевой соли кремневой кислоты, окиси алюминия и магния по срав-
нению с ретинолом.
Ретинол, дегидроретинол и их стереоизомеры значительно различаются
по своей витаминной активности (табл. 9 и 10).
ХИМИЧЕСКИЕ СВОЙСТВА РЕТИНОЛОВ
С(1з>—С(и) -Двойная связь ретинолов легко подвергается изомеризации.
Ретинол (I) и 9,13-ди-^нс-ретинол (VI) в виде своих эфиров под влиянием
следов йода в петролейном эфире или бензоле на свету или при термиче-
ском воздействии изомеризуются в неоретинол (II) или 9-1{нс-ретинол (IV)
[6, 33]. Изомеризация полного транс-ретинола (I) в пиридине в присутст-
вии йода приводит к смеси: 67% ретинола, 30,5% неоретинола (\3-цис) (II)
и менее 3% 9-ф/с-ретинола (IV) [34]. Неоретинол в определенных природ-
ных условиях, например в организме крысы, способен подвергаться полной
транс-стереоизомеризации. При изомеризации неоретинола (II) под влия-
нием следов йода устанавливается равновесие на уровне 70% ретинола и
30% неоретинола при 25° С за 2 ч [6]. Неоретинол-6 (III), стереоизомер с
заторможенной 11-цнс-конфигурацией, лабилен и под влиянием следов
йода также легко изомеризуется в полный транс-ретинол (I) [5]. Подоб-
ным образом смесь 9-цис-, 11-цис-, 9,13-дн-цис- и 11,13-ди-цнс-ретинолов
под влиянием йода в пиридине изомеризуется в полный транс-резинол и
неоретинол [35].
Для витамина А характерна реакция присоединения с треххлористой
сурьмой в хлороформе (реакция Карр — Прайса), протекающая с обра-
зованием синего окрашивания [36 ], характеризующегося четким максимумом
поглощения (Хмакс 620 нм, Е\ 5070). Эта окраска, в действительности за-
висящая от следов пятихлористой сурьмы, находящихся в SbCl3 [37], не-
устойчива и довольно быстро изменяется (для ретинола до Хмакс 580 нм).
Реакция с треххлористой сурьмой, другими хлоридами металлов и цвет-
ные реакции с различными кислотами используются в методах колориметри-
ческого определения ретинола [38, 39]. Достаточно надежные данные дает
метод определения ретинола по интенсивности максимума поглощения при
325 нм [40, 41].
Как ретинол в его полной транс-форме (I), так и 9-^нс-ретинол (IV)
вступают в реакцию присоединения малеинового ангидрида [4] и быстро
взаимодействуют по реакции Дильса — Альдера по конечной, пространст-
венно не затрудненной диеновой системе цепи с образованием соответствую-
щего аддукта
СН3 CHS
—CH=CH-i=CH—СН2ОН -> — сн—сн=с СН—СН2ОН
11 13 \ /
СН-СН
ОС—О-^СО
145
Эта реакция определяет транс-конфигурацию С(ц) —С(12) и С(13) —С(М)
двойных связей для обоих этих изомеров [4, 42]. Диеновая система, в ко-
торой имеется ^ас-конфигурация двойных связей, затрудняет реагирование,
и поэтому 11-цис-, 13-цис-, 9,13-ди-цас- и ПДЗ-ди-цос-ретинолы (III, II,
VI, V) с малеиновым ангидридом взаимодействуют очень медленно.
Ретинол по своей гидроксильной группе образует как простые, так и
сложные эфиры [43], например ретинол-ацетат (витамин А-ацетат) (XIII).
СН8 СН8
СНз СНз
-i =СН-СН =СН—С=СН-СН2ОСОСН8
XIII
Свойства эфиров группы витамина Ах приведены в табл. 11.
Таблица 11
Физические свойства эфиров ретинола
Наименование эфиров Т. пл., °C 20 "ЛИ Яд Спектр поглощения Биологическая актив- ность, % (витамин А1=100. молярные соотношения) Литература
максимум в спирте, д нм
Простые эфиры метиловый 34—35 326* 1660 НО [44]
фениловый Сложные эфиры 90—92 327 1460 4 [45]
ацетат 58-59 326 1550 100 [18. 19]
бутират 1,5986 325—328 1345 82 [45, 46]
лауриат 1,5668 325—328 1035 84 [45, 46]
пальмитат 28—29 325—328 975 88 [45, 46]
стеарат 1,5548 325—328 940 87 [45, 46]
олеат 1,5559- 325—328 870 115 [45, 46]
бензоат 1,6305 325—328 1240 73 [45, 46]
сукцинат 77—78 325—328 1480 86 [45, 46]
р-нафтоат 74—75 328 1090 97 [20, 45]
антрахинон ф -карбоксилат 122—123 330 1065 98 [19, 46]
* В изопропаноле.
Для изомеров ретинола и дегидроретинола характерны хорошо кристал-
лизующиеся эфиры с /z-фенилазобензойной кислотой, которые используют-
ся для их выделения и идентификации (табл. 9 и 10). Так, неоретинол (II)
выделен из некристаллизующейся фракции природного витамина А через
п-фенилазобензоат [6]. Интересно отметить, что 13-1{ш>изомеризация ан-
трахинон-р-карбонового эфира ретинола вызывает гипсохромный сдвиг
от красного до желтого цвета (для эфира неоретинола).
Сложные эфиры витамина А получают или непосредственно в результа-
те синтеза, или при действии на витамин А хлорангидридами кислот в пи-
ридине [20, 45]. Ретинол-пальмитат получают путем переэтерификации
ретинол-ацетата с метилпальмитатом — СН3(СН2)14СООСН3 [47]. Некото-
рые сложные эфиры водорастворимы, например витамин А-сукцинатсаха-
роза (—СН2ОСОСН2СН2СООС12Н21О10) [48].
15
146
За стандартную интернациональную единицу витаминной активности
(и. е.) принято 0,344 мкг кристаллического витамина А-ацетата с т. пл.
57,8—59° С, что соответствует 0,300 мкг кристаллического витамина
Ai-спирта [49]. Таким образом, 1 г ретинола (витамина Агспирта) соответст-
вует 3 300 000 и. е., 1 г ретинол-ацетата — 2 900 000 и. е. и 1 г ретинол-
пальмитата — 1 800 000 и. е.
Кристаллический ретинол и его кристаллические эфиры с уксусной, ян-
тарной, пальмитиновой и другими кислотами обладают значительно боль-
шей устойчивостью к умеренному нагреванию в инертном газе в отсутствие
света, а также при хранении в вакууме, чем их растворы [20].
Ретинол (витамин A-спирт) неустойчив, при стоянии его раствора в
60—100%-ном спирте при повышенной температуре были обнаружены
этиловый эфир ретинола, ретиналь, ретиновая кислота, ангидроретинол
(см. ниже) [49а].
Ретинолы легко подвергаются окислению кислородом воздуха. Сле-
дует отметить, что неоретинол (II) (витамин А с 13-^ис-конфигурацией)
несколько более устойчив, чем ретинол (I) (витамин с полной /пра«с-кон-
фигурацией двойных связей).
Первоначальным продуктом окисления ретинола кислородом воздуха
являются его эпокиси — ретинол-5,6-эпокись. (гепаксантин) (XIV) и ре-
тинол-7,8-эпокись (XV) [50, 51 ]
//Х^/СН=СН—с=сн—сн=сн-с=сн-сн,он
I |/°
СН8 СНз СНз СНз
XV
Эти эпокиси могут быть получены из ретинола или его ацетата при их
окислении перфталевой кислотой [52]. Под влиянием хлористого водорода
в хлороформе происходит изомеризация в так называемые фуранокиси,
например в ретинол-5,8-эпокись (XVI) [53]
СНз СНз
___СН СНз СНз
I । ।
•сн—С=СН—СН =СН—С=СН—СНзОН
о
СНз
XVI
Для увеличения устойчивости масляных и других растворов ретинола
к окислению к ним прибавляют антиоксиданты, такие, как а-токоферол,
гидрохинон, октил галлат, децил галлат, бутилоксианизол и др. Жирные
масла должны быть свободны от перекисей. Эфиры ретинола (ацетат, паль-
митат и другие) обладают большей стойкостью, чем свободный витамин
А-спирт [20]. Неовитамин А также обладает большей устойчивостью к
окислению, чем полный транс-витамин Ах.
Дегидро ретинол более чувствителен к кислороду, чем ретинол [10].
147
При энергичном окислении ретинола озоном происходит расщепление
всей молекулы и образуется героновая кислота [54].
Производным ретинола с полностью окисленной спиртовой группой
является ретиновая кислота (полная /пранс-витамин А-кислота) (XVII)
СН8 СН,
СНз СНз
I I
сн=сн—с=сн-сн=сн-с=сн-соон
сн
3
XVII
сд. пл. 180—181°С, Хмакс (в спирте) 350 нм; Е\сы 1510 [4]. Ретиновая кис-
лота может быть получена из ретинола при его окислении окисью серебра.
Соответствующие стереоизомерные ретиновые кислоты известны для произ-
водных всех стереоизомеров витамина А [4, 25]. Известны сложные эфи-
ры изомерных ретиновых кислот [4], например метиловый эфир полной
трпнс-ретиновой кислоты (т. пл. 56—56,5 и 72—73° С) [4] и др.
Дегидроретиновая кислота (витамин А2-кислота) имеет т. пл. 183—
184° С, ХМ2КС (в спирте) 370 нм; Е\ % 1395; метиловый эфир дегидрорети-
новой кислоты — т. пл. 44—46° С [9, 10].
При селективном окислении ретинола марганцовокислым калием [55]
или лучше специально активированной двуокисью марганца в петролейном
эфире при комнатной температуре образуется ретиналь — витамин А-аль-
дегид (ретинен) (XVIII) [7, 33, 56, 57]. В качестве окислителя ретинола
можно использовать тетрахлор-п-бензохинон [58]. Каталитическое окисле-
ние ретинола в уксусной кислоте с платиновым катализатором также при-
водит к ретиналю [59].
Витамин А в виде его альдегида (ретинена, ретиналя) является пигмен-
том зрительного пурпура сетчатки глаза. Ретиналь образует несколько гео-
метрических изомеров [31, 60]: полный тра«с-ретиналь (ретиненг) (XVIII),
13-грс-ретиналь (неоретиналь-fl) (XIX), 9-1{ис-ретиналь (изоретиналь-а)
(XXI), 9,13-ди-ф/с-ретиналь (изоретина^ь-5) (XXIII). Помимо этих четы-
рех пространственно незатрудненных форм альдегида, отвечающих четы-
рем природным и синтетическим формам витамина А, из природных источ-
ников выделен пространственно затрудненный 11-цис-ретиналь (неорети-
наль-6) (XX) [5, 61] и синтезирован 11,13-ди-цнс-ретиналь (неоретиналь-с)
(XXII) [5].
11-дол-Ретиналь (неоретиналь-г)
XX
сн, сцН СНз н
сн.
н
н н н сно
СНз
13—дол—Ретиналь (неоретиналь-в)
XIX
148
11,13-ди-до-Ретиналь (неорегиналь-f)
XXI!
9,13-ди-до -Ретиналь (иэоретиналь-i)
ХХШ
В качестве пигмента зрительного пурпура (родопсина) в механизме
зрения участвует, помимо полного /праяс-ретиналя, также 11-^нс-ретиналь
(XX), который может быть получен при окислении ll-qac-ретинола (III)
двуокисью марганца.
Рис. 4. Спектр погло-
щения стереоизомеров
ретиналя (витамин А-
альдегида) в спирте:
1— полного транс; 2 —
нео-а {13-цис); 3— нео-Ь
(11-цис); 4 — изо-а (9-
цису, 5 — изо-i (9,13-ди-
Ч«с).
Физические свойства стереоизомеров ретиналя представлены на рис. 4
[62] и в табл. 12.
Витамин А-альдегид и 13-^нс-ретиналь обладают высокой витаминной
активностью, другие ^ос-стереоизомеры — значительно меньшей (табл. 12).
Как характерно для альдегидов, витамин А-альдегид образует ацетали
[67], вступает в реакцию Канниццаро [68] и др. Ретиналь с С(о)—С(ю) и
С(1з> —С(14) транс-конфигурациями двойных связей дает с гидрохиноном
или пирокатехином кристаллический аддукт [69].
При окислении дегидроретинола двуокисью марганца образуется дегид-
роретиналь (витамин А2-альдегид) (XXIV) без стереоизомеризации, если
реакция проводится без нагревания в отсутствие света [8—10]. Дегидроре-.
тиналь вместе с 11-^ос-дегидроретиналем (XXV) является пигментом зри-
тельного пурпура сетчатки глаз пресноводных рыб; обе эти стереоизомер-
ные формы участвуют в механизме зрения [70].
Ретиналь и дегидроретиналь способны изомеризоваться под влиянием
света. Под действием разбавленной соляной кислоты полный траяс-рети-
наль изомеризуется в 9-цос- и 9,13-ди-цос-ретиналь [71]. Йод в темноте
149
Таблица 12
Физические свойства стереоизомеров ретииаля
Изомеры витамин А-альдегида Т. пл., °C Спектр поглощения Биологическая активность, % (активность ретинол-аце- тата-100 %) Литература
хмакс в спи₽™. нм £1% £1см SbCIj-реак- ция
Ж X Ъ со X 0s*
Ретиналь, полный 57 и 64—65* 381 1530 664 3470 90,9 [8, 22, 57,
транс-витамин А-альдегид (XVIII) Неоретиналь, 77 375 1250 664 3490 92,9 63—65] [22, 33, 63]
13-цис- ретина ль (неоретиналь-а) 257 336
(XIX) И-ф/с-Ретиналь 64,5 376 878 664 3498 48 [5 , 22 , 24,
(нео ретина лъ-Ь) 290 412 63]
(XX) 254 614
9-цис-Ретиналь (изоретиналь-а) (XXI) 64 Масло 373 1270 664 3470 19 [3, 22, 63]
11,13-ди-4/ис-Рети- наль (неорети- 373 700 31 [5 , 22 , 66]
наль-с) (XXII) 9,13-ди-чис-Рети- 49 и 85* 368 1140 664 3490 17,3 [22, 63]
наль (изорети-
наль-й)(ХХ1Н) Дегидроретиналь, 77—78 401, 314 1470, 395 735 4000 32—36 [8 , 22, 25]
полный транс-ви- тамин А2-альдегид (XXIV) 13-цис- Дегид pope - тиналь (неодегид- роретиналь-а) Масло 395, 314 1180, 412 [25]
11-Час-Дегидроре- Масло 393, 352 882, 452 [25]
тиналь (неодегид- роретиналь-6) (XXV) 9-цис-Дегидроре- 54—56 391, 315 1208, 672 [25]
тиналь (изодегид- роретиналь-а) 11,13-ди-цис- Де- гидроретиналь (неодегид рорети- наль-с) Масло 386, 269 963, 392 [25]
* Диморфные формы.
катализирует изомеризацию ll-tpc-неоретиналя (неоретиналя-6) и неорети-
наля (13-ф/с-ретиналя), но не 9-ф/с-ретиналя (изоретиналя-а) в полный
mpawc-ретиналь.
При восстановлении витамин А-альдегида (XVIII) образуется ретинол (I).
Восстановление может быть проведено алюмогидридом лития [4], бор-
гидридом калия [72 , 73], этилэтоксиалюмогидридом натрия и диэтоксиалю-
могидридом натрия [74]. Подобным образом ведет себя ретиновая кислота
(XVII) (см. с. 148), а также витамин А2-альдегид [75] и дегидроретиновая
кислота [10].
150
Восстановлением природного 1 l-ijuc-ретиналя (неоретиналя-6) (XX)
впервые получен 11-1{«с-ретинол (III) [61].
При каталитическом восстановлении в присутствии платинового катали-
затора в уксусной кислоте витамин А восстанавливается в пергидровитамин
А (XXVI) [76]:
СНз СНз СН, СН8
СНзСНзАнСНзСНзСНзСНСНзСНзОН
Следует отметить, что витамин А устойчив к восстановлению в присутствии
пассивированного катализатора Линдлара [77].
Продуктом восстановления спиртовой группы ретинола (I) является
углеводород аксерофтен (XXVII) [78 , 79], полученный синтетически
из ретинилтрифенилфосфониевой соли путем ее гидролиза в водно-спирто-
вом растворе едкого кали [80],
СНз
СН8
СНз СНз
СН=СН-С=СН-СН=СН-С=СН-СНз
XXVII
т. пл. 75—76° С, Хмакс (в гексане) 326 нм; Е\& 1850. При окислении аксе-
рофтена (XXVII) двуокисью селена образуется ретинол [81].
Ретинол способен к фотополимеризации [82, 83], Его димер — китол
(XXVH4) состоит из связанных по С(14)-атомам обеих молекул ретинола и
одновременно связанных по атому С(ц) одной молекулы и атому C(i3) дру-
гой молекулы, при этом происходит образование третьего циклогексано-
вого цикла [82—87]:
сн2он сн,
НОСН,—<14 ')
СН СН3
C=CH-CH=CHZ сн? сн=с-сн=сн
12 ’ *
ХХУ1П СН’
Его структура подтверждена данными ПК- и ЯМР-спектроскопии [82, 84].
Китол имеет т. пл. 88—90° С, Хмакс 290 нм; Е\ % 707. Впервые он выделен
из китовой печени; не обладает витаминной активностью.
При фотополимеризации наблюдается образование и других соедине-
ний, содержащих, в частности, в центральной части димера вместо цикло-
гексана циклобутан [88].
При длительном облучении дегидроретинола-пальмитата образуется
китол2-дипальмитат [89].
Ретинол и его изомеры неустойчивы к минеральным кислотам [90]. Так,
при действии концентрированной соляной кислоты в спирте происходит
дегидратация витамина А и его изомеров, сопровождающаяся изомериза-
цией полиеновой системы с образованием ангидроретинола (XXIX) (91,
151
сн, сн, сн,
сн,
XXIX
Дегидратацию витамина А также вызывают серная кислота в смеси с
уксусной кислотой и п-тулуолсульфокислота [93]. При образовании ангид-
ровитамина А возникает еще одна ненасыщенная (шестая) связь и проис-
ходит перемещение всей сопряженной полиеновой системы [80 ], в том числе
и в циклогексеновом кольце, в так называемую ретроизомерную систему
[94]. Строение ангидроретинола как 4,15-дегидроаксерофтена с достовер-
ностью следует из его синтеза из Р-ионона и 3-метилгексадиен-1,3-ина-5
и соответствующего промежуточного оксипроизводного [95]. Его витамин-
ная активность незначительна. Ангидроретинол выделен в оранжевых иг-
лах ст. пл. 76—77° С, Хмакс (в спирте) 392, 371, 351 нм; £} 7» 3180, 3650,
2500 [91, 94, 93].
13-1{мс-Конфигурация двойной связи влияет на замедление дегидрата-
ции при образовании ангидроретинола из неоретинола.
Ангидроретинолу отвечает соединение с гидроксильной функцией —
ретроретинол (ретровитамин А, регидровитамин А) (XXX), в виде ацетата
легко образующийся из витамина А-ацетата в метиленхлориде с концентри-
рованной бромистоводородной кислотой [96],
СНз
сн, сн.
сн,
сн-сн=с^сн=сн-сн=с—сн,—сн,он
Н,
XXX
Хмакс (в спирте) 369, 351, 330 нм. Ретроретинол выделен из печени А-авита-
минозных крыс при даче им ангидроретинола [94 ]. Его биологическая ак-
тивность отвечает около 10% активности ретинола [94].
При кислотной обработке дегидроретинола (VII) в спиртовом растворе
происходит дегидратация и изомеризация, в результате чего получается
ангидродегидроретинол (ангидровитамин А2), который представляет собой
3-этоксиангидроретинол (XXXI)
С,Н6О
СН,
СНз СНз
сн,
:-сн=сн.
XXXI
с т. пл. 86—88° С, Хиакс (в спирте) 391, 370, 352 нм; Е\ ?м 2620, 2980, 2040
[97].
Регидродегидроретинол (регидровитамин А2) (XXXII) является 3-это-
ксиретроретинолом [98 ]
152
сн8
СН,
сн, сн,
сн-сн=с-сн=сн—сн=с-сн,-сн,он
С,Н,0
XXXII
с Хмажс (в спирте) 365, 348, 330 нм.
а-Витамин А (XXXIII) испытывает под влиянием метилата натрия изо-
меризацию в ретинол (I) с ^-структурой замещенного циклогексенового
цикла (выход 75%) [99].
СТРОЕНИЕ РЕТИНОЛОВ
Ретинол
Строение ретинола (витамина AJ установили Каррер, Морф и Шопп
[54] в 1931 г. реакциями окислительного расщепления, подтвердили Хейль-
брон, Мортон и Вебстер [90] в 1932 г. и доказали Каррер и Морф [76] в
1933 г. синтезом пергидровитамина А.
Присутствие в молекуле ретинола (I) первичной гидроксильной группы
установлено как реакциями получения сложных эфиров (ацетата и нитро-
бензоата), так и мягким окислением витамина А в ретиналь (XVIII) [54,
55]. Так как при озонировании ретинола (I) и р-ионона образуется одна и
та же героновая кислота (XXXIV), то это является доказательством общ-
ности строения циклической части молекулы данных соединений. Наличие
в молекуле ретинола шестичленного цикла и метильной группы при нем
следует также из реакции образования 1,6-диметилнафталина (XXXV) при
дегидрировании селеном [90], что одновременно указывает и на положе-
ние в цикле алифатической боковой цепи и первой метильной группы в ней
(схема 25).
Схема 25
Установление строения ретинола реакциями окисления и восстановления
сн. сн,
СН. СН, I ’ I 3
" " CH2CH^HCH2CHjCH2CHCH,CH2OH
сн3
XXVI
|[Н]
СН, СН, 1
>< / сн=сн-с=сн-сн=сн-с=сн-сн,он
Ъ сн. Г3 I 3
></СН=СН-С=СН-СН=СН-С=СН-СНО
СI
СООН
153
В результате жесткого окисления марганцовокислым калием боковая
часть молекулы расщепляется с образованием двух молекул уксусной кис-
лоты, что говорит о наличии разветвленной цепи, содержащей две метиль-
ные группы. Еще более жесткое окисление при нагревании с хромовой кис-
лотой указывает на наличие и третьей метильной группы (в цикле).
Так как при каталитическом гидрировании присоединяется 5 молей во-
дорода с образованием пергидровитамина А (XXVI), то это дает основание
предполагать присутствие пяти двойных связей. Способность к присоедине-
нию малеинового или цитраконового ангидрида приводит к заключению о
наличии сопряженной системы двойных связей. Все эти данные дают осно-
вание предположить для строения боковой цепи изопреноидную структуру.
Окончательное доказательство строения ретинола получено превраще-
нием его в пергидровитамин А (XXVI) [76] и идентификацией этого вещест-
ва с синтетическим продуктом.
Для синтеза пергидровитамина А (XXVI) [76] 0-ионон (XXXVI)
и бромуксусный эфир подвергают конденсации по реакции Реформатского
в присутствии цинка в 0-ионилиденуксусный эфир (XXXVII), который ка-
талитическим гидрированием переводят в насыщенное соединение, карбал-
коксильную группу этого вещества восстанавливают натрием в спирте в
оксиметильную группу и из полученного спирта С15 получают бромид С1&
(XXXVIII, схема 26). В результате конденсации бромида С15 (XXXVIII)
с натриймалоновым эфиром с последующим декарбоксилированием получа-
ют кислоту С17 (XXXIX), которую превращают в хлорангидрид кислоты
С17 и конденсируют с метилцинкйодидом — образуется насыщенный ке-
тон С18 (XL). Его конденсируют с бромуксусным эфиром по реакции Рефор-
матского в эфир насыщенной оксикислоты Сго, в котором гидроксильную
группу замещают на галоген и затем полученное соединение восстанавли-
вают цинком в уксусной кислоте в эфир пергидровитамина А-кислоты
(XLI).
Схема 26
Синтез пергидровнтамнна А
CHj снз
СН,
I 3
сн=сн-с=о
сн3
.CH=CH-C=CH-COOR
+ BrCHCOOR'
сн, сн3
XXXVI • XXXVII
сн, сн3 сн3
I 3 I I
RCH2CH2CHCH2COOR' —RCH2CH2CHCH2CH2OH ——RCH2CH2CHCHaCH2Br -
XXXVIII
сн3 сн3 ' СН3 СНз
RCH2CH2CHCH2CH2CH(COOh)2----RCH2CH2CHCH2CH2CH2COOH—-RCH2CH2CHCH2CH2CH2CO-
XXXIX XL
CH3 CH3 CH3 CH3
RCHj.CH^HCHjCHjCHjCCHj.COOR’-----RCH2CH2CHCH2CH2CH2CCH2COOR’ ---------—
Oil Br
CH3 CH3 I 3 | 3
CH3 CH3 X CH2CH2CHCH2CH2CH2CHCH2CH2OH
RCH2CH2CHCH2CH2CH,CHCH2COOR’------ Г |
XLI \^CH3
Пергидровитамин A
XXVI
R’=C,HS
154
Из полученного соединения восстановлением натрием в спирте синтези-
руют тот же самый пергидровитамин А (XXVI), который является продуктом
восстановления природного витамина А (что установлено переведением обо-
их соединений в кетон С2з и идентификацией по отсутствию депрессии сме-
шанной пробы их семикарбазонов с т. пл. 67° С) [76].
Кристаллический синтетический витамин А получен только в 1947 г.
[1001; по спектрам поглощения его эфиров, по смешанной пробе и по физио-
логической активности он был идентичен натуральному витамину.
Установление геометрической конфигурации изомеров ретинола осно-
вано на генетической связи с исходными для их синтеза соединениями, на
интерпретации спектров поглощения (по величинам экстинкций, появлению
zfuc-пика для соединений IV и VI), а также на отношении их п-фенилазобен-
зойных эфиров к малеиновому ангидриду.
Так, строение 1 l-ijac-ретинола (III) установлено по ультрафиолетовому
и инфракрасному спектрам поглощения [5], подтверждено [101] и дока-
зано синтезом из 11,12-дегидро-13-тракс-ретинола (XLII) [5] путем час-
тичного каталитического гидрирования.
Таким же путем установлено строение 11,13-ди-^нс-ретинола (V)—в
результате частичного гидрирования 11,12-дегидро-13-ф/с-ретинола (XLIII)
[5]. Такой метод установления цис,транс-конфигурации основан на том,
что гидрирование ацетиленовой связи до этиленовой водородом в присутст-
вии окиси платины или скелетного никелевого катализатора однозначно
приводит к 1{нс-конфигурации образующейся двойной связи так же, как
и химическое восстановление цинком и кислотой, в отличие от химического
восстановления другими реагентами, дающими транс-конфигурацию [102].
Дегидроретинол
•
Дегидроретинол — витамин А2 (VII) — открыт во Всесоюзном научно-
исследовательском витаминном институте в 1937 г. Ледерером и Розановой
[103, 104]. Ими было показано, что в неомыляемой фракции жира печени
пресноводных рыб, выловленных в реках Дон, Волга и в реках Ленинград-
ской области, помимо витамина А1? содержится хромоген, дающий с трех-
хлористой сурьмой синие растворы, обладающие спектром поглощения с
сильным максимумом около 690 нм и более слабым — при 645 нм (в отли-
чие от витамина Аь дающего окраску с Хмакс 620 нм). В некоторых жирах
хромоген с максимумом поглощения около 690 нм составлял подавляющую
часть спектра. Эти данные были получены для жира севрюги, судака, леща
и других пресноводных рыб. Новый хромоген по своим свойствам очень
близок в витамину Alt он показал высокую биологическую активность.
Впоследствии этими же авторами хромогену было присвоено наимено-
вание «витамин А2» [105, 106]. В 1939 г. витамин А2 был выделен в виде
отдельной фракции в результате применения хроматографического разде-
ления; он охарактеризован как кристаллический фенилазобензоат с т. пл.
76-77° С, Хмажс 341 нм; Е\ * 1190 [30].
Для строения витамина А2 (VII) Грей и Каулей [107] предложили в
1939 г. формулу 3,4-дегидроретинола, которая была поддержана другими
авторами [7 ]; Фаррар, Хамлет, Хенбест и Джонс [8] доказали эту структу-
ру синтезом только в 1952 г.
Дегидроретинол (VII) содержит шесть двойных связей, что доказывает-
ся присоединением шести молекул водорода при каталитическом гидриро-
155
вании с образованием пергидровитамина А (XXVI), т. е. он содержит на
одну двойную связь больше, чем ретинол. При окислении дегидроретинола
активированной двуокисью марганца образуется дегидроретина ль — ви-
тамин А2-альдегид (XXIV) [7]. При озонировании дегидроретинол дает
героновую кислоту (XXXIV) [14], ацетон и формальдегид [62, 108] (схе-
ма 27).
Схема 27
Установление строения дегидроретниола реакциями окисления восстановления
СИ, CH3 9^3
^Х'.СЦ- C^-CH-CHj-CHj-C^-CH-CH,- сн2он
СНз XXVI
|[н]
СИ., СН3 СИз ‘ СИа
><\,CH=CH-C=CH-CH=CFi-C=CH-CH2OH
to]
СИ, СН3 «(И» I
5<,сн=сн-с=сн-сн=сн-с=сн-сно
СНз XXIV
СДСОСН/.
н
сн3 ^сн3
chScooh
сн^сн,
XXXIV
сш ^сн3
сн2 хсоон
соон
Однако дегидроретинол не образует а,а-диметилянтарной кислоты, ко-
торая должна была получиться, если бы расщепление происходило не толь-
ко по связи С(5)—С(б), но и по связи С(з)—С(4> • Можно полагать, что в цик-
логексадиеновом кольце дегидроретинола первоначально по связи С(5) —С(6)
образуется озонид, который расщепляется в дикетон, и только затем посте-
пенно расщепляется полиеновая цепь:
"з
.сн=сн--
сн3
сн
сн3
‘3
сн=сн----
о
сн3
Этот механизм реакции- находится в соответствии с последовательным
образованием 0-семикаротннона (дикетона) и 0-каротинона (тетракетона) в
условиях мягкого окисления Р-каротина хромовой кислотой (см. с. 196).
По-видимому, в присутствии образующегося при озонировании формаль-
дегида происходит восстановление двойной связи С(з)—С(4) и в итоге обра-
зование героновой кислоты.
ВЫДЕЛЕНИЕ РЕТИНОЛА (ВИТАМИНА А) ГИЗ ПРИРОДНЫХ ИСТОЧНИКОВ
Одним из богатых по содержанию источников витамина А является жир
печени морских животных и рыб, например палтуса [109]. В технике кон-
центраты витамина А получаются методом молекулярной дистилляции
[НО]. Для выделения витамина А применяется метод омыления жиров рас-
твором едкого кали при 60° С в отсутствие воздуха, извлечение эфиром,
удаление растворителя, отделение стеринов от неомыляемого остатка из
раствора метилового спирта при большом охлаждении, затем или хромато-
графическое разделение на окиси алюминия по методу Цвета [111], или
кристаллизация из 10%-ного этилформиата при —35е С [20], а также мно-
гократная молекулярная дистилляция. Природный витамин А получен с
156
т. пл. 63—64° С [201; из него синтезированы: ацетат (т. пл. 57—58° С), паль-
митат (т. пл. 27—28° С), антрахинон-₽-карбоксилат и другие эфиры.
Природный витамин A-эфир перегоняется при молекулярной дистилля-
ции при температуре около 200° С, полученный из него витамин А — при
123° С [28], витамин А-ацетат— при 132,5° С и витамин А-пальмитат —
при 213° С [20].
I I ;СИНТЕЗ РЕТИНОЛОВ
В настоящее время важнейшим методом получения ретинола, дегидро-
ретинола и их стереоизомеров (витаминов группы А) является их синтез.
Долгое время попытки синтетического получения витамина А не при-
водили к удовлетворительным результатам, и с достоверностью витамин А,
его метиловый эфир и ретиновая кислота (витамин А-кислота) были полу-
чены лишь в 1946—1947 гг. [100, 112—116]. Ислер, Губер, Ронко и Коф-
лер [100, 112] осуществили первый полный синтез витамина А в кристал-
лическом виде и синтез его метилового эфира; Аренс и ван-Дорп [114, 116]
синтезировали витамин А и витамин А-кислоту; Майлас и др. [113] — ме-
тиловый эфир витамина А; Каррер, Джуккер и Шик [115] — витамин А-
кислоту; в 1948 г. Коули, Робесон и др. [16] — витамин А. В последующие
годы были опубликованы новые схемы синтеза и многочисленные патенты.
Витамин А вырабатывают в виде сложных эфиров — ретинол-ацетата
или-ретинол-пальмитата, обычно в виде стабилизированных масляных пре-
паратов, содержащих от 200 000 до 1 800 000 и. е. в 1 г.
1. Синтез исходных веществ для получения ретинола—цитраля,
псевдоионона и р-ионона
В качестве важнейшего исходного вещества во многих синтезах ретино-
ла применяют Р-ионон (XXXVII), функциональная группа которого служит
мостиком для структурного преобразования в направлении наращивания
полиеновой углеродной цепи молекулы. P-Ионон получают или из при-
родного цитраля (XLIV) через псевдоионон (XLV), или синтетически.
Цитраль (XLIV) получают в виде смеси цис и /пранс-стереоизомеров из
лемонграссового масла (содержание в нем цитраля около 75%), выделяемо-
го из травы Cymbogon flexuosus (Индия, Гватемала), или из кориандрового
масла растения Coriandrum Sativum L. окислением содержащегося в нем
линалоола.
Псевдоионон (XLV) представляет собой продукт конденсации цитраля
(XLIV) и ацетона в результате реакции, протекающей под влиянием ос-
новных катализаторов, например раствора щелочи [117—119] или лучше
фенолята натрия в бензольном растворе [120] с выходом свыше 75%:
(H2SO<)
157
Р-Ионон (XXXVII) наряду с а-иононом, являющимся ценным душис-
тым веществом с запахом фиалки, получается при циклизации псевдоионо-
на (XLV) под влиянием кислот. При действии на псевдоионон смеси уксус-
ной и концентрированной серной кислот при 10—15° С [117, 121] или
одной 98%-ной серной кислоты в среде эфира при—5,—7° С [122 ] образует-
ся преимущественно р-ионон с выходом 70—80%. Изучен механизм цикли-
зации псевдоионона [123, 1241.
С и н.т ез цитраля
Цитраль (XL IV) синтезирован из природного р-пинена (основной части
скипидара, получаемого из смолы хвойных) посредством его пиролиза в
мирцен (XLVI) [125] с последующей обработкой бромистым водородом, а
затем нитропропаном в едком кали [126, 1271:
Один из синтезов цитраля (XLIV) состоит в получении его из изопрена
через геранилхлорид [128]. Для этого первоначально на изопрен (XLVII)
действуют хлористым водородом и получают смесь а,а- и у.у-диметилал-
лилгалогенидов (XLVIII и XLIX) [129], которую совместно с изопреном
подвергают теломеризации в присутствии четыреххлористого олова в гера-
нилхлорид, образующийся совместно с терпенилхлоридом [128, 130]. Ге-
ранилхлорид выделяют в виде уротропиновой соли четвертичного основа-
ния и затем превращают в цитраль (XL IV) нагреванием с избытком фор-
мальдегида [128]:
СН, СН, СНзСЦ,
НЦ р
СН, сн,а
XLVI! XLYIH
CH, CH, CH,
[[Cl CHj=C-CH=CH,
CH, SnCl4; 50%
HCHO
75%
CH3 CH,
CHO
XLJY
интез псевдоионона
Синтез псевдоионона (XLV) осуществлен из 2-метилгептен-2-она-6
(LIII) через дегидролиналоол (LIV). Для получения 2-метилгептен-2-
она-6 разработаны следующие методы.
По первому методу а,а-диметилаллилгалогенид (XLVIII) легко кон-
денсируется с натрийацетоуксусным эфиром и в результате последующе-
го гидролиза превращается в метилгептенон (LIII) [131]. В синтезе а,а-
диметилаллилгалогенида можно исходить из изопрена (XLVII) [131] или
диметилвинилкарбинола (LI) [132] и действовать на них галогеноводоро-
дом. При использовании диметилвинилкарбинола (LI) реакцию получе-
ния 2-метилгептен-2-она-6 (ЫП) проводят в одну стадию, минуя выделе-
ние соединения XLVIII, причем 2-метилгептен-2-он-6 получается с выхо-
дом 60% через хлорпроизводное и 75% через бромпроизводное [133—
135] (схема 28).
Диметилвинилкарбинол (З-метилбутен-1-ол-З) (LI) получают [136]
из ацетона конденсацией с ацетиленидом натрия в диметилэтинилкарби-
нол, который подвергают селективному гидрированию в присутствии пал-
ладия на углекислом кальции с выходом около 95%.
Второй метод заключается в непосредственной конденсации диметил-
винилкарбинола (LI) с ацетоуксусным эфиром в ацетоацетат диметил-
винилкарбинола (LII) и последующем его пиролизе в 2-метилгептен-2-
он-6 (LIII)- Еще в 1940 г. [137] была показана реакция между третичны-
158
ми виниловыми спиртами и ацетоуксусным эфиром под воздействием ката-
лизаторов основного характера, приводящая при пиролизе ацетоуксусных
эфиров непредельных спиртов к внутримолекулярной перегруппировке и
отщеплению двуокиси углерода с образованием у, 8-ненасыщенных кетонов
[137—139] (схема 28). -
Схема 28
Синтез 2-метилгептен-2-она-6
Конденсация диметилвинилкарбинола (LI) с ацетоуксусным эфиром
проводится при температуре 160—170° С под давлением или в присутствии
вазелинового масла; выход 2-метилгептен-2-она-6 (ЫП) составляет свыше
70% [133]. Применение металлического натрия [137] или других катализа-
торов в реакции не вызывается необходимостью [133]. Промежуточным про-
дуктом в этом синтезе является ацетоуксусный эфир диметилвинилкарби-
нола (LII), который может быть выделен [133].
Для получения ацетоуксусного эфира диметилвинилкарбинола (LII)
предложен метод этерификации диметилвинилкарбинола (LI) дикетеном
[138, 140, 141]; реакцию проводят в присутствии металлического натрия
(выход 60%) [138] или лучше пиридина (выход свыше 92%) [133].
Третий метод синтеза 2-метилгептен-2-она-6 (LIII) состоит в конденса-
ции диметилвинилкарбинола (LI) с избытком изопропенилового эфира,
причем образующийся лабильный кеталь в кислой среде отщепляет метило-
вый спирт и изомеризуется в 2-метилгептен-2-он-6 (LIH) с общим выходом
41% [142] (схема 28).
2-Метилгептен-2-он-6 (LIII) превращается далее в псевдоионон (XLV)
(схема 29). Для этого его конденсируют с ацетиленом в 3,7-диметилоктаен-
б-ин-1-ол-З, дегидролиналоол (LIV) в среде серного эфира в присутствии
амида натрия (выход 80%) [143], в жидком аммиаке при —50, —60° С и
при воздействии металлического натрия (выход 74%) [144, 145], по методу
Назарова [146] в среде эфира под влиянием порошкообразного едкого кали
при температуре 0—20° С и давлении 0,5—1 МПа (5—10 ат)—выход
92% [135, 148] — или в среде N-метилпирролидона в присутствии едкого
натра [147]. Селективным гидрированием дегидролиналоола (LIV) получен
линалоол (LV) с выходом 96% [143, 148].
Если линалоол (LV) нагревать с а-хлорацетоуксусным эфиром, то, по-
видимому, через промежуточный эфир хлорацетоуксусной кислоты образу-
ется а-хлордигидропсевдоионон (LVI) (частный случай реакции третичных
виниловых спиртов с ацетоуксусным эфиром и последующим пиролизом)
[137], который при действии пиридина отщепляет хлористый водород с об-
разованием псевдоионона (XLV) [149] (схема 29).
159
Схема 29
Синтез псевдоионона нз 2-метилгептен-2-ола-6
LVIII
LIX
XLV
Псевдоионон (XLV) может быть получен непосредственно из дегидроли-
налоола (XL IV) конденсацией с изопропениловым эфиром [150—153 J в
кислой среде через промежуточные кеталь и алленкетон, который далее под-
вергается перегруппировке в щелочной среде (схема 29). Этот вариант син-
теза имеет достаточно высокую эффективность.
Однако дегидролиналоол (LIV) с успехом конденсируют с дикетеном и
через ацетоацетат (LVII) непосредственно превращают в псевдоионон
(XLV) практически в одну стадию по Каймелу [140] (схема 29). Реакция
получения диенонов из третичных этинилкарбинолов и дикетена с после-
дующим пиролизом ацетоацетатов третичных этинилкарбинолов сопровож-
дается отщеплением углекислоты и внутримолекулярной перегруппировкой.
Такая же реакция осуществлена с диметилэтинилкарбинолом [154]. Дегид-
ролиналоол ацилируют дикетеном в сложный эфир ацетоуксусной кислоты
с дегидролиналоолом (LVII) [140] в присутствии металлического натрия
(выход 72%) [144, 145] или, лучше, пиридина или триэтиламина (выход
90%) при температуре 70—80° С [155]. Ацетоацетат дегидролиналоола
(LVII) превращают в псевдоионон (XLV) через алленкетон пиролизом при
170—180°С [140, 145, 155, 156], выход на дегидролиналоол составляет
50—55%. Общий выход псевдопонона на ацетон —35% [140].
Применение ацетоуксусного эфира для ацилирования третичных эти-
нилкарбинолов [157, 158], например дегидролиналоола (LIV), несколько
упрощает синтез псевдоионона (XLV) [157]. Эта реакция лучше всего про-
ходит в одну стадию одновременно с пиролизом в отсутствие катализаторов
при нагревании в токе азота при 170—180° С (выход псевдоионона 55%)
[155]. Пиролиз ацетоацетата дегидролиналоола сопровождается образова-
нием побочных продуктов [140, 159].
Интересный вариант синтеза псевдоионона (XLV) через цитраль осу-
ществляется по способу Соси—Марбета [160]. Конденсация 2-метилгеп-
тен-2-она-6 (ЫП) с ацетиленом через дегидролиналоол (LIV) с последую-
щим ацетилированием приводит к ацетату дегпдролиналоола (LVI1I), ко-
торый при каталитическом участии солей серебра перегруппировывается
в цитраль-алленацетат (LIX). Он легко гидролизуется в а, р -ненасыщенный
160
альдегид — цитраль (XL IV) с выходом 61 % из дегидролиналоола; цитраль
по известной реакции конденсируется с ацетоном в псевдоионон (XLV) (схе-
ма 29).
К цитралю (XLIV) от 2-метилгептен-2-она-6 (LIII) можно перейти через
его ацеталь, который вводят в конденсацию с виниловым эфиром [161].
Менее эффективным путем псевдоионон был получен конденсацией 2-ме-
тилгептен-2-она-6 (LIII) с эфиром у-бромкротоновой кислоты по Реформатско-
му с последующей дегидратацией образующегося 3-оксиэфира и дальнейшим
наращиванием углеродного скелета при действии литийметила в безводном
эфире [134].
2. Синтез ретинола из замещенного циклогексанона
В многочисленных исследованиях Хейльброн с сотр. [162, 163] и другие
[164, 165] первоначально на модельных соединениях циклогексана разра-
ботали основы для синтеза ретинола, которые впоследствии привели к его
полному синтезу [166] (схема 30).
В этих синтезах применяют алкилзамещенные циклогексаноны, которые
подвергают этинилированию в этинилкарбинолы — соединения, пригодные
для построения на их основе молекулы ретинола с полиеновой разветвлен-
ной углеродной цепью и конечной гидроксильной группой. 1
Так, из 1,1,3-триметилциклогексанона-2 (LX) [167] при действии аце-
тиленида натрия в жидком аммиаке был получен 1,1,3-триметил-2-этинил-
циклогексанол-2 (LXI) [165, 168]. Это соединение, молекула которого со-
стоит из 11 атомов углерода, конденсировалось с полиеновым кетоном C#
(LXII) [169] в условиях реакции Гриньяра с образованием полного угле-
родного скелета витамина А в виде гликоля Сго (LXIII). В результате анио-
нотропной перегруппировки под влиянием кислых реагентов образуется
сопряженная система ненасыщенных связей с гидроксильной функцией на
конце алифатической цепи соединения LXIV. Тройная связь соединения
LXIV была частично восстановлена в двойную [170] с применением алюмо-
гидрида лития [171], и полученное вещество частично ацетилировано в
моноацетат (LXV). В результате дегидратации под влиянием п-толуол-
сульфокислоты в толуоле или бензоле при кипячении из этого соединения
получен ретинол-ацетат (XIII) с примесью ангидроретинола [1661 с не-
высоким выходом.
Схема 30
Синтез ретинол-ацетата из замещенного циклогексанона
СН3 СН.
сн3 сн, сн, СН, <1 I
Ж с=сн ОС-СН=СН-СН=С-СН=СН,
rY NaC='CH УЖ IX»
^-^СНз ^^ХН, LX LXI СН3 СН3 СИ* *[Нз У<УС5С-С-СН=СН-СН=С-СН=СН2 Г 7ой бн — LXII1 СН3 СН3 ^3 <fH3 ><.СН=С1ЬС=СН-СНСН-С=СНСН2ОССХ:Н3 [ ?он — • d 1 VV сн3 сн3 V"3 73 3></С5С-С=СН-СН=СН-С=СН-СН2ОН f 'гон — Чч^хн, LXIV сн3 сн3 5Нз С”3 ><жсн=сн-с=сн-сн=сн-с=сн-снрсосн3 '''"'''ЖН, XIII
6—69
161
3. Синтез ретинола из цитраля
В синтезе ретинола по схеме С1о+С1о=С2о (схема 31) в качестве исходно-
го вещества служит цитраль (XLIV), который через В-циклоцитраль (LXVI)
превращают в 0-циклогераниол (LXVII) [172, 173] и затем при действии
трифенилфосфоний-бромида в среде диметилформамида—в 0-циклоге-
ранилтрифенилфосфоний-бромид (LXVIII) [1741. Полученный из него фос-
форан конденсируют с триеновым альдегидоэфиром С1о (LXIX) [175] в
этиловый эфир полной транс-ретиновой кислоты (LXX, R=C2H5) [176],
который восстанавливают алюмогидридом лития [4] или диэтоксиалюмо-
гид ридом натрия [72, 741 в ретинол (I).
Схема 31
Синтез ретинола из цитраля через Р-циклогераниол
XLIV LXVI
СНз СН3
ohc-c=ch-ch=ch-c=ch-coor
LXIX
сн3 сн3
СН2ОН
СН;
LXVII
сн3 сн3 +
" -СН2 Р(с6Н5)3
Вт"
сн3
LXYII1
(CeHs^P-HBr
сн3
I 3
сн3
СНз
СН3
сн=сн -c=ch-ch=ch-c=ch-coor
сн3
* Ретинол
I
LXX
Интересно отметить, что, если обычно реакция Виттига приводит к по-
лучению смеси цис-транс-изомеров, в данном синтезе вследствие стериче-
ских затруднений образования г{цс-конфигурации по С(7>—С(8) двойной
.связи не происходит.
Исходя из 0-циклоцитраля (LXVI), осуществлен синтез витамина А по
схеме С1о+С54-С5=С2о через 0-ионилиденуксусный альдегид и витамин А-
альдегид (схема 32). Ацеталь 0-циклоцитраля (LXXI) конденси-
руют с 1-этоксиизопреном в присутствии хлористого цинка в 7-эток-
сиацеталь С15 (LXXII) и затем после гидролиза и элиминирования
этоксигруппы л-толуолсульфокислотой в толуоле получают 0-ионилиденук-
сусный альдегид, или альдегид С15 (LXXIII) [177]. Синтез альдегида
осуществлен и другими, более доступными методами (с. 166). Из него полу-
чают ацеталь альдегида С16 (LXXIV), который подобной конденсацией с
Цитраль
XLIV
Схема 32
) .
Синтез ретинола нз цитраля через альдегид С15
, OR СН,
э! 1 ' / \
ch-ch2-c=ch-ch(or)2
СН,
LXXI LXXII
сн3
сн, сн,
СН=СН-СзСН-СНО
СНз>Нз- СИз СН2
ch=ch-c=ch-ch(or)2 1
сн3
CHj=C~CH-=CHOR
сн3 сн.
LXXIII
сн.
OR
СНз LXXIV
С|,з сн, сн.
3 I I I / X
сн=сн-с=сн-сн^сн2-с=сн-сн(рк^
сн3
3 LXXV
сн,
3 XVIII
r=c2hs
сн3 сн3
сн=сн-с=сн-сн=сн-с=сн-сно
------------------------— Рет*юл
I
162
1-этоксиизопреном превращают в II-этоксидигидроацеталь (LXXV),.
из этого соединения в результате гидролиза образуется 11 -этоксидигидро-
ретиналь, а из него посредством отщепления этоксигруппы на окиси алюми-
ния— витамин А-альдегид (XVIII) [178]. Восстановление ретиналя (XVIII)
алюмогидридом лития приводит к витамину А (I).
4. Синтез ретинола из ₽-ионона через углеводороды С15 и С16 или
соединением с цепочкой С7
Известный интерес представляет применение для синтеза витамина А,
пропаргилбромида (ВгСН2С=СН; пропаргиловый спирт получают кон-,
денсацией параформа и ацетилена в присутствии медного катализатора);
или его производных, реакционная способность которых к карбонильным,
соединениям связана с высокоподвижным атомом брома, активированным
ацетиленовой связью. Этот метод позволяет удлинить углеродную цепь
Р-ионона на 3 или 7 атомов углерода [46, 113 J, однако он связан с побочным
образованием соединений с искаженной полиеновой системой.
Витамин А-метиловый эфир (LXXXII, R=CH3) по одному из вариантов
синтеза (схема 33) получают из р-ионона (XXXVI) и замещенного пропар-
гилбромида — 1-алкокси-6-бром-3-метилгексен-2-ина-4 (LXXVI), которые
конденсируют под влиянием цинка в ацетиленовый гликоль (L XXVII),
в результате частичного гидрирования превращают в этиленовый гликоль
С20 (LXXVIII) [179] изатем в метиловый эфир ретинола (LXXXII, R=CH3).
Конечный продукт — ретинол (I) — получается с очень малой витамин- к
ной активностью (всего */х— i/so активности природного витамина А).
Схема 33
Синтез ретинола из ^-ионона через углеводород С1в или соединением с цепочкой С7
сн2
СН3 СН3 *“Н3
сн=сн-с=о
сн3
СН3 XXXVI
BrCH2CsC-C=CH-CH2OR;Zn HC=C-CHrC=CH-CH3OR
LXXVI | LXXIX
CH3
сн3
ch=ch-c-ch2-c=c-c=ch-ch2or
1[ ' он
CH, CH3 I
CH=CH-C- C=C-CH3- c=ch-ch2or
OH
сн2
сн3
LXXVII
сн3
LXXX
СН3
€Н3
сн3
Cll3 CH3
CH=CH-C-CH2-CH=CH-C=CH-CH2OR
OH
СН,
сн3 сн3
LXXVIII
сн3
СН=СН-С-СН=СН-СН2- C^CH-CHjOR,
он
сн3
LXXXI
CH3 CH3 VH’ СНз
^><XH=CH-C=CH-CH=CH-C=CH-CH2OR
^^CH,
3 LXXXII
CH3 CH3 си*
5<хн=сн-с=сн-сн=сн-с=а i-ch2oh
По другому варианту синтеза ft-ионон (XXXVI) конденсируют с 1-ал-
кокси-3-метилгексен-2-ином-5 (LXXIX) по реакции Иоцича в замещен-
ный ацетиленовый гликоль (LXXX), который частичным гидрированием
6*
163
превращают в этиленовый гликоль (LXXXI); дегидратация последнего
соединения (схема 33) дает витамин А-метиловый эфир (LXXXII, R=CH3),
а затем витамин А (1)1180].
Ацетиленовый гликоль С20 (LXXX) в присутствии щелочи претерпевает
прототропную перегруппировку с перемещением изолированной двойной
связи [181 ] в сопряжение с триениновой системой и образованием веществ
с ретроионилиденовой системой [182, 1831. Этиленовый гликоль
(LXXXI) под влиянием кислот, помимо нормального превращения в эфир
ретинола, подвергается аллильной перегруппировке гидроксильной группы
к наиболее алкилированному углероду на конце сопряженной системы с де-
гидратацией и образованием в результате смещения всех двойных связей
ретроионилиденовой системы [182—185] эфира ретроретинола (LXXXIII)
317 нм):
СН, СНз сн, сн
сн-сн=с-сн=сн-сн=с-сн2—ch2or
и,
LXXXIII
Метиловый эфир ретроретинола показывает всего 1—3% активности ви-
тамина А. Эта побочная реакция приводит к низкому выходу ретинола.
I* 1-Алкокси-3-метилгексен-2-ин-5 (LXXIX) получают конденсацией ме-
тилвинилкетона и пропаргилбромида под воздействием цинка с последую-
щей этерификацией.
Синтез ретинола также осуществлен через витамин А-альдегид, получае-
мый взаимодействием 0 -ионона с 1-метокси-3-метилгекса-1,3-диен-5-ином
через промежуточный метоксиацетиленовый карбинол Сго, тройную связь
которого гидрируют до двойной и гидролизуют в альдегид [186, 187].
При конденсации р-ионона с комплексом Иоцича, полученным из 1-эток-
си-3-метилгексен-2-ин-5-ола-4, образуется 1-этокси-9-(2',6',6'-триметил-
циклогексен-1'-ил)-3,7-диметилнонадиен-2,8-ин-5-диол-4,7, а из него в
результате восстановления алюмогидридом лития — эфир витамина А
[188].
Схема 34
Синтез ретинола из Р-ионона через 8-ионол
СН3 СНз 5-Нз
_Х.СН=СН-СНОН
3~Монон— > Г |Т —
XXXYI
LXXXIV
СН, сн, сн,
СН, СН, I 3 I 3 снз СН, 1^
6Ли С ^COOR tH С^
"сн "сн *сн ^сн . г п "СН "VH
ч I JL сн
"сн ^СН3 4сн
LXXXYII с
сн3- *сн
|_________________________LXXXY1U ^о,
Смесь полного /|м»г-ретигюла(1) и
9-цис-ретинола (iv)
R=C2Hs
164
Ретинол можно синтезировать, исходя из р-ионона с применением реак-
ции Виттита (схема 34). Первоначальнор-ионон (XXXVI) восстанавливают
алюмогидридом лития вр-ионол (LXXXIV) [189], который с трифенилфос-
фоний-хлоридом превращают вp-ионилтрифенилфосфоний-хлорид (LXXXV)
[190] и вводят в реакцию с диеновым альдегидоэфиром С, (LXXXVI). По-
лучают смесь этиловых эфиров полной транс- (LXXXVII) и 9-цис-
(LXXXVIII) ретиновых кислот [190], которые восстановлением алюмогид-
ридом лития превращают в смесь ретинола (I) и 9-цис- ретинола (IV).
Следует отметить, что 9-^ис-ретинол в полный транс-ретинол не изоме-
ризуется.
Технический синтез витамина А-ацетата с применением реакции Виттита
разработан (способ фирмы BASF) по схеме: С13+С2+С5=С2о[74] (схема 35).
При присоединении к р-ионону (XXXVI) двух атомов углерода конден-
сацией с ацетиленидом лития образуется ацетиленовый карбинол С16
(LXXXIX). После его селективного восстановления получают этиленовый
карбинол С15 (ХС) [184], который вводят в реакцию с трифен илфосфон и й-
хлоридом, образовавшийся р -ионилиденэтилтрифенилфосфоний-хлорид С15
(XCI) превращают в С15-фосфоран (ХСП) и затем конденсируют [78] с
Р-формилбутенолацетатом (ХСШ) [191—193]. При этой реакции образу-
ется смесь ретинол-ацетата и его 13-цис- и 1 l-^uc-изомеров, которые изоме-
ризацией превращают в полный троне-ретинол-ацетат (XIII).
Схема 35
Синтез ретинол-ацетата из f-ионона через ацетиленовый карбинол Сде
СИ, СН,
"><,снсн-со
CH3
сн=сн-с-с=сн
ОН
сн3
СН3 СН, fh3
|1ГСН=СН1нСН=СЦ>(СбН^Р>-
\ НС1
ХС
LiC=CH
XXXVI LXXX1X
сно
сн,-с=сн-сн,ососн3
хеш
сн3 сн, V14®
>< / сн=сн-с=сн-сн=р(свн5),
СН’ XCII
СН, СН3 V”3
5<.сн=сн-с=сн-сн=сн-с=сн-сн,ососн3
СНз XIII
Ацетиленовый карбинол (LXXXIX) конденсацией с эфиром р-метил-
у-бромкротонового спирта может быть превращен в этиленовый гликоль
С20 (LXXXI) [194] (схема 33), конденсацией с у-этокситиглиновым альде-
гидом ROCH2CH=C(CH3)CHO—в эфир диоксидегидровитамина А, а после
его восстановления — в эфир витамина A (LXXXII) [188].
Техническое значение имеет синтез из ацетиленового карбинола Cje
(XCIV) (применяемый фирмой Eastman Kodak, разработанный в Glaxo
Laboratories), который получают из р-ионона (XXXVI) конденсацией с
пропаргилбромидом по реакции Реформатского с выходом 85% [195—198J
(схема 36). Ацетиленовый карбинол С1е (XCIV) конденсируют с ацеталем
ацетоуксусного альдегида (XCV) [197, 199—201 ] по реакции Гриньяра в
ацеталь ацетиленового гликоля С2о (XCVI), затем избирательно восстанав-
ливают в ацеталь этиленового гликоля (XCVII). Это соединение дегидра-
тацией и деацетализацией в присутствии хлоргидрата хинолина в этилме-
тилкетоне превращают в ретиналь (XVIII) [197, 200—202].
165
сн3 сн.
1 '• • Схема 36
Синтез ретинол а нз р-Йоно и а через ацетиленовый карбинол Cie
СПз
CH=CH-C-CHj-CsCII
он
сн3
XCIV
СН3 ^сн3
• BrCHjCsCH
I - В -Ионон ' '• -—=-—
XXXVI
сн3
О^СНгСнГоСнХ • ..
АСУ ___________
, СН3 1 СК
13 1 ’ 1 X
СН=СН-ССН2-С=СС-СН2-СН(ОСН3)2
Zn
СН3 сн3
..сн=сн-с=сн-с=сн
ОН
ОН
сн3
XCVI
сн3
XCVIII
сн3
сн3
сн=сн-с-снг-сн
он
• сн3
|=сн-с-снгсн(оснД
он
СН3СОСН=СН2
сн3 сн3
СН3 CHj
' CH=CH-C=CH-fCsC-C-CH=CH2
он
сн3
> Лсуп
сн3
3 ХОХ
СИз
сн3
СН, сн,
' " хсн=сн-с=сн-сн=сн-с=сн-сно
сн3
XVIII
L—
СК СН,
сн3
f сн3
I I
СН=СН-С=О1-СН=СН-С-СН=СН2
он
сн3 с
. сн.
сн8
ей, сн3
СН3
сн=сн-с=сн-сн=сн-с=сн-сн2он
сн3
I
После очистки ретиналя через кристаллический аддукт с гидрохиноном
[203] его восстанавливает в ретинол (I) боргидридом натрия [200, 201,
2041.
Ацетиленовый карбинол С16 (XCIV) можно использовать для другого
варианта синтеза витамина А (схема 36). При дегидратации под влиянием
n-толуолсульфокислоты образуется ацетиленовый углеводород С16 (XCVIII)
наряду с его ретроизомером. Лучше проводить реакцию при участии хлор-
окиси фосфора в пиридине [205[. Ацетиленовый углеводород С16 (XCVIII)
конденсируют с метилвинилкетоном по реакции Гриньяра в ацетиленовый
карбинол Сго (XCIX). Частичное восстановление тройной связи до двойной,
в соединение С, и аллильная перегруппировка приводят к витамину А
[179, 195, 205].
5. Синтез ретинола из Р-ионона через кетон С18 или альдегид С15
Синтез альдегида С15
р-Ионилиденуксусный альдегид, или альдегид С15 (LXXIII), получают
' из р-ионона путем удлинения его углеродной цепи на группу =СН—СНО
по методу Преображенского [206], в котором применялся алкоксиацетилен
CH=COR [207]. Позднее было найдено лучшим применять литиевые про-
изводные этого соединения [208].
Р-Ионон (XXXVI) конденсируют с броммагнийэтоксиацетиленом по
реакции Гриньяра в алкоксиацетиленовый эфир С15 (CI), который частич-
ным гидрированием с палладиевым катализатором переводят в оксивини-
166
ловый эфир С15 (СН). Последний под влиянием разбавленной соляной кис-]
лоты гидролизуется в оксиальдегид (СШ), который, .дегидратируется,
и преобразовывается в альдегид С15 (LXXIII) [209]. Эти реакции могут-
протекать в одну стадию без выделения промежуточных соединений
(схема 37). Альдегид С15 этим методом синтезирован и другими автора-
ми [210, 211].
Схема 37
Синтез альдегида Си из р-ионона
BrMgCsCOR XXfVI
ВгСНгСООК
сн3
сн3
ОН
CI
сн3
Н2О
СИ3 СМз
" ' .ch=ch-c-ch2coor
он
сна
3 CIV
сн. сн3
CH=CH-C-CsCOR
LXXIII
Для получения альдегида С16 применена реакция Реформатского. Син-
тез проводится через р-ионилиденэтиловый спирт (CV). По этому методу
(схема 37)0-ионон (XXXVI) конденсируют с бромуксусным эфиром в при-
сутствии цинка в р-ионилиденоксиуксусный эфир (CIV), который при по-
следующем отщеплении элементов воды дает 0-ион и л идеи уксусный эфир
(XXXVII) [19, 212—216] с выходом около 70% [215]. Вместо бромуксус-
ного эфира можно применить хлоруксусный эфир [217]. 0-Ионилиденук-
сусный эфир (XXXVII) также может быть получен в результате гидратации
алкоксиацетиленового эфира С15 (CI) в кислой среде через промежуточный
0 -ион и л иденокси уксусный эфир (CIV, R—С2Н5) [209,218] или непосредствен-
но. р-Ионилиденуксусный эфир (XXXVII) получается в виде смеси геомет-
рических изомеров [219]. После гидролиза смеси tfoc-транс-изомерных
эфиров и разделения смеси кислот транс-p-ионилиденуксусная кислота
получена с т. пл. 126—127° С и Хыакс 297 нм [214], а цас-кислота с т. пл.
98,5—99,5° С и Хмакс 306 нм с более низкой экстинкцией, чем транс-кисло-
та [4].
167
0-Иоилиденуксусный эфир (XXXVII) в виде трудно разделяемой смеси
цис-транс-изомеров переводится восстановлением алюмогидридом ли-
тия в 0 -ионилиденэтиловый спирт (CV); эта реакция дает возможность про-
ведения избирательного восстановления карбоксильной группы, не затра-
гивая двойных связей [220].
Затем р-ионилиденэтиловый спирт (CV) окисляют высокоактивной дву-
окисью марганца в альдегид С15 [4, 213, 214, 221 ] с выходом 60%. Однако
альдегид С15 получается также в виде смеси 9- цис-транс-изомеров [167, 214],
и его применение для синтеза приводит к ретинолу, обладающему малой
биологической активностью и содержащему, кроме того, примесь больших
количеств побочных ретроизомерных продуктов (см. ниже). Следует ука-
зать, что для синтеза ретинола (имеющего полную транс-конфигурацию
двойных связей) нужен только 9-транс-альдегид С15 [4, 167, 214].
Необходимо отметить, что при дегидратации р-ионилиденоксиуксусного
эфира (CIV) образуется не только эфир (XXXVII) с требующимся располо-
жением двойной связи в а, р -положении к карбоксильной группе, но и
0,у-изомер (CVI) [222], у которого в результате анионотропной перегруппи-
ровки гидроксильной группы в аллильное положение вся полиеновая си-
стема смещена на один атом углерода с образованием в циклогексеновом
кольце структуры кольца а-ионона и семициклической двойной связи,
так называемой ретроионилиденовой системы.
СН, СИ,
><-CH=CH-C-CH.COOR
-^^сн3
CIV
сн
сн, сн,
f^CH-CH=C-CH2COOR
Г он
XH,
сн3 сн,
^CH-CH^C-CHjCOOR.
Г т F
<н,
CVI
Аллильную перегруппировку гидроксильной группы, протекающую
при дегидратации 0-ионилиденовых карбиноловувпервые обнаружили Гуис-
ман с сотр. [167] и Орошник с сотр. [184]. Одна из первых попыток синте-
за витамина А потерпела неудачу главным образом из-за того, что получае-
мый по реакции Реформатского р-ионилиденуксусный эфир представлял
собой смесь геометрических изомеров [219] с преимущественным содержа-
нием изомера (CVI).
Смесь изомерных эфиров (CIV иСУ1)может быть разделена фракционной
кристаллизацией, и изомер (CVI) со смещенной сопряженной системой двой-
ных связей после гидролиза может быть подвергнут изомеризации при ка-
талитическом действии хлорокиси фосфора [223] или специально очищен-
ного треххлористого фосфора [222] в хлорангидрид кислоты С15, соответ-
ствующей нормальному эфиру (XXXVII) с выходом 99%. Восстановлению
алюмогидридом лития в 0-ионилиденэтиловый спирт (CV) с выходом 99%
может быть подвергнут и хлорангидрид кислоты С15 [222], образующейся
в результате гидролиза и изомеризации ретроионилиденуксусного эфира '
(CV!).
Разработан интересный синтез альдегида С15 (LXXIII), позволяющий
обойти ретроионилиденовую перегруппировку промежуточных соединений.
По этому синтезу [167, 181, 224, 225], имеющему техническое значение для
получения ретинола (способфирмы Philips Gloeilampenfabriken) (см. с. 172),
0-ионон (XXXVI) конденсируют с циануксусной кислотой или ее эфиром по
Кневенагелю в 0-ионилиденциануксусную кислоту (CVII) [224, 225] или ее
эфир, затем декарбоксилированием в процессе конденсации или полным
168
диизобутилалюминийгидридом [225, 226] c
и отщеплением аммиака
декарбоксилированием нагреванием в пиридине [224 ] или с медью в толуоле
[225] получают 0-ионилиденацетонитрил (СУШ), который восстанавливают
алюмогидридом лития или ' ’
одновременной гидратацией
СН, CH, VH3
>< /СН=СН-СО
СН3
XXXVI
сн, сн,
Х^сн-ган-CN М; НзО
Т 1 -NH,
ru ru CHjCOOR
COOR ко
-СО,;-ROH
CH.
сн,
CVII
СНа СН,
2></СН=<
сн,
сн-с=сн-сно
ch,
cvni
r=h; csh5
сн3
-lxxiii
p-Ионилиденацетонитрил (CVIII) может быть получен из р-ионона и ди-
этилфосфонийацетонитрила [227] или через циангидрин альдегида С14
1228].
Выше указывалось (см. с. 162), что для синтеза альдегида С15 (LXXIII)
был предложен метод, в основу которого положена конденсация ацеталя
Р-циклоцитраля (LXVI) с 1-этоксиизопреном [177].
Альдегид С15 (LXXIII) синтезирован также путем конденсации кеталя
Р-ионона с винилэтиловым эфиром с последующим отщеплением этокси-
группы от промежуточного соединения и гидролиза [229].
Альдегид С15 успешно получен из р-ионона конденсацией с этилформиа-
том, метилированием енолята с последующей реакцией Гриньяра с метил-
магнийгалогенидом, гидролизом и дегидратацией (способ фирмы АЕС)
[230].
Еще два варианта синтеза р-ионилиденуксусного альдегида (альдеги-
да С15) заключаются в конденсации р-ионона с (RO)2P(O)CH2CH(OR')2
* в присутствии амида натрия [231 ] и в конденсации 2,6,6-триметилциклогек-
| сен-1-ила-1 лития с 3-метил-5-диметиламинопентадиен-2,4-ален-1-ом»
£
| Синтез кетона С18
| Первоначально кетон С18 — 9-метил-7-(1,1,5-триметилциклогексен-5-ил-
-6)октатриен-7,9,1 Loh-13 (CXIII) — был получен из Р-ионона (XXXVI)
I и метилового эфира Р-бромкротоновой кислоты (CIX) по реакции Рефор-
матского (в присутствии цинка) с последующими превращениями образую-
щегося оксиэфира CI7 (СХ) через кислоту С17 (СХП) [116, 232]. Отщепление
воды от оксиэфира С17 (СХ) производилось с помощью щавелевой кислоты.
Полученный с выходом 30% щранс-Р-ионилиденкротоновый эфир (CXI)
[114, 233, 244] подвергался гидролизу едким кали в транс-Р-ионилиденкро-
тоновую кислоту (СХП), которая при действии эфирного раствора литиймети-
ла [114] превращалась в кетон С18 (СХШ). Последнюю стадию можно про-
вести через хлорангидрид кислоты С17 (СХП) с диметилкадмием [235] или
метилцинкйодидом [115] (схема 38).
Необходимый для синтеза кетона С18 у-бромкротоновый эфир (CIX) по-
лучают из малонового эфира [236 ] или лучше по более удобному методу из
ацетоуксусного эфира его гидрированием с последующей дегидратацией
образующегося эфира Р-оксимасляной кислоты [162] и бромированием эфи-
ра кротоновой кислоты N-бромсукцинимидом.
Как впоследствии оказалось, невысокий выход гпранс-Р-ионилиденкро-
тонового эфира (CXI) с 1маис 325 нм, характерным для карбонильных
сопряженных пентаенов, не может быть улучшен, так как, помимо образо-
вания побочного tyizc-изомера, реакция Реформатского сопровождается ани-
169
Схема 38
Синтез кетона С18 нз [3-ионона через кислоту Cj7
СН3 СН, ^Н,
' сн=сн-с=о
* BrCH„CH=CHCOOR
С1Х
С^з
CH=CH-C-CHrCH=CH-COOR
Ан -н-°
сн3
XXXVI
СИ, СН, СН3 СН, СНа
' ' ch=ch-c=ch-ch=ch-coor '
HQ
сн,
3 СХ1
СН3
сх
сн,
I 3
CH=CH.-C=CH-CH=CH-COOR сн L.
сн,
3 схп
сн, сна СНз сн,
,СН=СН-С=СН-СН=СН-СО
СН3 схш
R=CH3
онотропной (аллильной) перегруппировкой гидроксильной группы окси-
эфира С17 (СХ) к концу сопряженной системы в циклогексеновое ядро [167],
перемещением двойных связей боковой цепи, последующей дегидратацией и
смещением ненасыщенной связи в циклогексеновом ядре с образованием ре-
троионилиденовой системы — ретроионилиденкротонового эфира (CXIV) с
Хмакс 285 нм, характерным для сопряженных триенов. Аллильная перегруп-
пировка с образованием ретроионилиденового изомера (CXIV) протекает
под влиянием минеральных и органических кислот даже при низкой тем-
пературе.
сн, сн3 сн3
£Н=С Н-С-С Н2—С Н=С H-COOR
""—ОН
сн3
сх
сн, сн3
" " ^CH-CH^-CH^CH-CHj-COOR
СН, СН, । 3
'Х^.сн-с н=с-сн2-с Н=С H-COOR
^''^^СН,
CXIV
сн3
CXV
Исследованиями инфракрасных спектров поглощения и другими по-
казано, что ретроионилиденкротоновый эфир (CXIV) образуется в значитель-
ном количестве [167, 182—184, 237—239 J.
Помимо этой изомеризации, наблюдается прототропная перегруппиров-
ка под влиянием щелочи или при нагревании с перемещением водорода ме-
тиленовой группы и удлинением сопряженной системы двойных связей
ретроионилиденкротонового эфира (СХ IV) [239 ] в нормальный р-ионилиден-
кротоновый эфир (CXI). Однако прототропная перегруппировка сопрово-
ждается перемещением атома водорода метиленовой группы и в направле-
нии свободного конца боковой цепи, что приводит к образованию второго рет-
роионилиденового эфира (CXV) [239]. Такая неоднозначность реакций и
является причиной того, что этот метод получения р-ионилиденкротонового
эфира (CXI) неэффективен.
Кетон С18 (СХIII) из альдегида С15 (LXXIII) получен путем конденсации
с ацетоном под влиянием mpem-изобутилата алюминия с выходом 80—85%
[213, 214] или с несколько большим в присутствии раствора едкого натра
[222].
170
сн, сн,
сн,
сн =сн—с=сн—сно
СН.СОСН,
сн,
LXXIII
сн, сн.
сн,
СНз
сн=сн с=сн—сн=сн—со
сн,
схш
цис- и транс-Кетон С18 получают из соответствующих изомеров альдегида
С15 [214, 240 J.
Если использовать для получения кетона С18 (СХШ) схему конденсации
р-ионона (XXXVI) с бромуксусным эфиром и последующими превращениями
через соединения CIV, XXXVII, CV и альдегид С15 (LXXIII) (схема 37)
с применением изомеризации ретросоединений по Гуисману [222 ], то выход
кетона С18 из 0-ионона можно довести до 85%.
Для получения кетона С18 (СХШ) можно также подвергнуть окислению
и последующей конденсации с ацетоном непосредственно [J-ионилиденэтило-
вый спирт (CV) (схема 37). Если применить альдегид С15 в виде альдимина
и конденсировать его с ацетоном в аминокетон, то при обработке хлористым
кальцием в спирте происходит дезаминирование в кетон С18 [241 ].
Предложен метод синтеза кетона С18 (СХШ) через ацетиленовый кетон
С18 (CXVIII), получаемый конденсацией альдегида С14 (CXVI) и литиевого
производного 7-алкоксивинилацетилена (CXIX) с последующим гидроли-
зом эфирной группы ацетиленового гликоля С18 (CXVII); при этом проис-
ходит перегруппировка с образованием триеновой сопряженной системы
[242]. В результате частичного каталитического гидрирования соединения
CXVIII получается кетон С18 (СХШ). * .
сн3 сн, । 3
/ сн2-сн=с-сно
+ ЫС=С-С=СНа
ОС2Н5
CXIX
к 1
CH2-CH=C-CH-CSC-C=сн2
сн,
CXVII
ОС2Н5
СН3 CH, VH’ СН, сн СНз сн3
X/СН=СН-С=СН-С2С-СО X <н=сн-с=сн-сн=сн-со
С и н т е з р е т и н о л а из альдегида Сц и кетона С18
Одна из первых попыток синтеза витамина А, в которой неочищенный
альдегид С15 (LXXIII) конденсировали с метилкротоновым альдегидом
под влиянием пиперидилацетата в ретиналь (XVIII) с последующим его
восстановлением по Меервейну—Пондорфу, привела к получению продукта
с малым содержанием витамина А. С несколько лучшими результатами ре-
тинол (I) синтезирован из альдегида С15 (LXXIII) через этиловый или мети-
ловый эфир ретиновой кислоты (LXX, R=CH3 или С2Н5) из р-ионилиденук-
сусного альдегида (LXXIII) и р-метил-7-бромкротонового эфира (СХХ)
конденсацией по Рефюрматскому в эфир полиеновой оксикислоты С20 (CXXI)
171
[167, 243 ], дегидратация которого привела к получению эфира ретиновой кис-
лоты (LXX), а последующее восстановление — к сложной смеси изомеров
ретинола. В этом направлении проводились и другие исследования [181,
221, 243].
СН, СН3 | 3
><1^ сн=сн-с=сн-сно
СНз LXXIII
сн, сн, сн,
I СН, CH, I 3 I 3
BrCH/X=CHCOOR СН=СН-С=СН-СН-СН,- C=CH-COOR
СХХ । | __
а он
СНз CXXI
СН,
СН3 СНз
СН,
CH=CH-C=CH-CH=CH-C=CH-COOR
Смесь изомеров ретинола
•СН,
LXX
Однако если исходят из транс-альдегида С15 (LXXIII) и конденсируют
его с Р-метилкротоновым эфиром (СХХП) в присутствии амида калия (по
реакции Кневенагеля) в среде жидкого аммиака [244 ] или безводного эфира
[245], то получают этиловый эфир полной транс-ретиновой кислоты (LXX,
Й=С2Н5) [244], который восстанавливают алюмогидридом лития в рети-
нол (I) (способ фирмы «Sumitomo»)
сн, сн,
сн
СН,
=сн-с=сн-сно
сн3
CH,C=CHCOOR
СХХП.
СН, СН, 1Нз
'><,CH=CH“C=CH-CH=CH-C=CHCOOR
Ретинол
I
-LXXIII
R=C,HS
Важно отметить, что если для конденсации соединений LXXIII и CXXI
применить амид натрия или лития, то будет образовываться 13-цос-ретино-
вая кислота и соответственно 13-цис-ретинол. При использовании для кон-
денсации цис-альдегида С15 синтез приводит к 9-цис- и 9,13-ди-цис-ретинолу
[244].
Можно непосредственно от альдегида С16 (LXXIII) перейти к эфиру ре-
тиновой кислоты (LXX), если p-метил-у-бромкротоновый эфир (CXIX) по
реакции Виттига предварительно с трифенилфосфином превратить в фосфо-
ниевую соль (CXXIII) [246]:
СН3
+ | _ Эфир ретино-
Альдегид Cj8 -}- L(CeHs)3P—CH2C=CHCOORJ Br -► вой кислоты —> Ретинол
LXXIII CXXIII LXX I
С применением реакции Виттига из альдегида С15 и (4,4-диэтокси-2-ме-
тилбутен-2-ил)трифенилфосфина (получаемого из диэтилацеталя Р-метил-у-
бромкротонового альдегида) синтезирован диэтилретиналь, гидролизуе-
мый в ретиналь [247], а затем восстанавливаемый в ретинол.
В техническом синтезе ретинола (способ фирмы Philips Gloeilampenfab-
riken) [225] альдегид С1Б (LXXIII) получают из Р-ионона (XXXVI) конден-
сацией с циануксусной кислотой в p-ионилиденацетонитрил (CVIII) с пос-
ледующим восстановлением (см. с. 168), затем альдегид С15 превращают в
кетон С18 и подобным повторением реакции кетон С18 (СХШ) или его шиф-
фово основание [248 ] конденсируют с циануксусной кислотой в циановита-
мин А-кислоту (CXXIV), затем декарбоксилируемую в нитрил ретиновой
кислоты (CXXV) [224, 249], который восстанавливают диизобутилалюми-
нийгидридом в альдимин и гидролизуют его в ретиналь (XVIII) [197];
из него получают витамин А (I) и затем витамин А-пальмитат (схема 39).
172
н
Схема 39
Синтез ретинола из кетона Си через нитрил ретиновой кислоты
СН, СН3 VHa СИз
СН=СН-С=СН-СН=СН-СО
соон сн3
™ /СН=СН-С=СН-СН=СН-С=С
CHjCN
сн,
СИ,
1 .CN
соон
сн3 сн3
СХШ
сн3
•СН=СН-С=СН-СН=СН—С=СН-CN
сн3
СН3
CXXIY
сн,
сн,
сн, сн,
сн=сн-с=сн-сн=сн-с=сн-сно
сн,
CXXV
сн3
XVIII
сн3 сн,
сн3 сн3
сн=сн-с=сн-сн=сн-с=сн-сн2он
сн3
Нитрил ретиновой кислоты (CXXV) может быть получен из альдегида
С18 (см. с. 206) при действии на него ацетонциангидрина или цианистого
калия и последующей дегидратации промежуточного циангидрина хлор-
окисью фосфора в пиридине [2501.
Для синтеза ретиналя (XVIII), превращаемого затем в ретинол (I),
предложен метод его получения из кетона С18 (СХШ) и ортомуравьиного
эфира через ацеталь и конденсацию с виниловым эфиром под влиянием
эфирата трехфтористого бора с последующим гидролизом [251, 252].
Кетон С18 (СХШ) превращают в ретинол (I) через ретиналь (XVIII)
[116] и другими методами (схема 40). Первоначально кетон С18 (СХШ)
по реакции Гриньяра конденсируют с броммагнийэтоксиацетиленом [253]
в оксиацетиленовый эфир Сго (CXXVI), который подвергают частичному
восстановлению в оксиполиеновый эфир (CXXVII); последнее соедине-
ние при действии разбавленной соляной кислоты подвергается гидролизу
и преобразованию в ретиналь (XVIII). Превращение альдегидной’группы
соединения XVIII в спиртовую такими реагентами, которые не восстанавли-
вают этиленовых связей, например изопропилатом алюминия по Меервей-
ну — Пондорфу, приводит к ретинолу (I) в виде 35%-ного концентрата
[116]; применение алюмогидрида лития для восстановления дает лучший
выход витамина А [64 ].
Другим путем кетон С18 (СХШ) с помощью реакции Реформатского
превращают в эфир ретиновой кислоты (LXX) [114—116, 232], который
восстанавливают алюмогидридом лития в ретинол (I) [254]. Первоначально
конденсацией кетона С18 с бромуксусным эфиром в присутствии активного
цинка получают эфир ненасыщенной оксикислоты Сго (CXXVII), который
подвергают дегидратации безводной уксусной кислотой и йодом или с луч-
шими результатами п-толуолсульфокислотой [254, 255] в эфир ретиновой
кислоты (LXX) (схема 40).
Для получения витамина A-спирта (I) карбоксильную группу эфира ре-
тиновой кислоты (LXX) избирательно восстанавливают алюмогидридом
лития [254, 255] в витамин A-спирт, который в результате очистки получен
с хорошим выходом в виде к ристал лизатов. Синтезированное вещество на-
ряду с транс-изомером витамина Аг содержит 36—40% стереоизомерного
13-цис-ретинола (II) [254].
Эфир ретиновой кислоты (LXX) предварительно отделяют от примеси
изомерного эфира ретроретиновой кислоты (CXXIX); последний с неболь-
шим выходом может быть изомеризован в эфир (LXX) при каталитическом
действии хлорокиси фосфора [223]. После гидролиза получается кристал-
173
Схема 40
Синтез ретинола нз кетона С18 через ретиналь или эфир ретиновой кислоты
СН СН V^*3
Ч^з ^-пз I I
СН=СН-С=СН-СН=СН-С=О
BrMgC^COR
СН,
схш
сн, сн,
1з сн3 I сщ
CH=CH-C=CH-CH=CH-C-ChCOR
ОН
СН3 СНз
BrCH3COOR
СНз I
СНз
СН=СН-С=СН-СН=С1 |-C-CH3COOR
он
СН.
CXXVI
1[H}/pd(BaSO4)+PbCl2
СН3
сн, сн.
СН3
CH=CH-C=CH-CH=CH-C-CII=CHOR
OH
сн3
CXXVIII
сн3
сн3 сн.
-HjO
1 сн3
СН=С1l-C=CI l-CI l=CH-C=CH-COOR
СН3
сн, сн.
CXXYII
н2о(н+)
сн3
сн3
сн=сн-с=сн-сн=сн-с=сн-сно
СН3
[Н]
сн3 сн.
XVIII
R=CjHs
СН,
СН3
LXX
сн3
,сн=сн-с=сн-сн=сн-с=сн-сн2он
СН,
лическая ретиновая кислота с т. пл. 180—182° С [114, 115,232,255] и выхо-
дом 20—25% (на кетон С18) [256]. Получение ее исследовано многими авто-
рами [181, 235].
По некоторым данным [222], изомерный эфир ретроретиновой кислоты
(CXXIX) после гидролиза может быть почти количественно перегруппиро-
ван под влиянием треххлористого фосфора в хлорангидрид ретиновой кис-
лоты (СХХХ) с нормальной системой сопряженных двойных связей; послед-
ний восстанавливают алюмогидридом лития почти с количественным вы-
ходом в ретинол (I), при очистке которого удается получить около 75% крис-
таллического вещества.
СНз СНз
СНз
СН3
СН—СН=С—СН=СН—СН=С-CH2COOR
СН3
CXXIX
СНз СНз СНз
СНз
СН =СН—С=СН-СН =СН—С=СН-СОС 1
—> Ретинол
СХХХ
Полный синтез ретинола через эфир ретиновой кислоты проведен и дру-
гими авторами [214, 257]. Синтез ретинола через альдегид С15 и кетон С18
протекает через ряд кристаллических промежуточных соединений, которые
могут быть очищены от изомеров.
174
В основу описываемого ниже метода синтеза ретинола положен принцип
постепенного наращивания молекулы по схеме: С13 + С1+С1+С34-С14-
4-Сх — С20 (схема 41). Этот метод дает возможность использовать простей-
шие соединения (этилформиат и ацетон) вместо сложных цепочек, получе-
ние которых обычно представляет сложную химическую задачу. В синтезе
витамина А (способ фирмы Alimentation Equilibree de Commentry) [230]
в качестве исходного вещества используют р-ионон (XXXVI), а в качестве
промежуточных соединений — альдегид С15, кетон С18 и альдегид С2о (ре-
тиналь), причем при наращивании на кетоне С13 (р-иононе) и кетоне С18
двух атомов углерода производятся однотипные химические реакции про-
межуточного образования кстоенолов и кетоацеталей.
P-Ионон (XXXVI) по реакции Кляйзена конденсируют с этилформиатом
в присутствии сухого метилата натрия в гексане и образовавшийся кетоенолят
С14 (СХ XXI) подвергают ацетализации метиловым спиртом в присутствии
серной кислоты в кетоацеталь С14 (СХХХII); очистку осуществляют
перегонкой в вакууме. Реакцией Гриньяра с метилмагнийхлоридом в без-
водном эфире по карбонилу кетоацеталя С14 (СХХХII) присоединяют ме-
тильную группу и полученный оксиацеталь С15 (CXXXIII) дегидратируют
и гидролизуют при действии хлористого водорода в альдегид С15 (LXXIII).
Его конденсируют с ацетоном в щелочной среде в кетон С18 (СХШ) (см.
с. 170), который после молекулярной дистилляции однотипными реакция-
ми через кетоенолятС19 (СХХХIV), кетоацеталь C19(CXXXV) и оксиацеталь
С^ (CXXXVI) превращают в ретиналь (XVIII) [2301. Ретиналь получается
с примесью около 30% цис-изомеров, которые подвергаются цис-транс-изо-
меризации в присутствии йодистого водорода или йода в изопропиловом
спирте; продукт изомеризации выделяют в виде комплекса с гидрохиноном
или пирокатехином [258]. Ретиналь восстанавливают боргидридом натрия
в витамин А (I). Этот синтез имеет техническое значение.
Схема 41
Синтез ретинола из р-нонона через альдегид и кетон С18 .
CXXXIV CXXXV
CXXXVI XVIII
175
6. Синтез ретинола из р-ионона через альдегид С14
Эта группа синтезов [100, 112, 113, 259] осуществляется по схеме С13-[-
-}-Cj~pCe — С^. Основная характерная особенность этих синтезов — отсут-
ствие ограничений, связанных с образованием побочных продуктов в ре-
зультате реакции изомеризации. Все превращения производятся с проме-
жуточными продуктами синтеза, у которых прервано сопряжение двойной
связи цикла с непредельными связями цепочки и образующаяся в резуль-
тате последующих конденсаций гидроксильная группа не находится в
аллильном положении к двойной связи ядра, вследствие чего не может
произойти анионотропной перегруппировки с перемещением полиеновой
системы и образованием ретроионилиденовой системы.
Синтез альдегида С14
Альдегид С14 — 9-метил-7-(1,1,3-триметилциклогексен-5-ил-6)бутен-8-
аль-10 (CXVI) — впервые получили Ишикава и Матсуура [260] из Р-ионона
через глицидный эфир.
P-Ионон (XXXVI) конденсируют с хлоруксусным эфиром по реакции
Дарзана под влиянием метилата натрия в глицидный эфир С15 (CXXXVII)
[260], который вследствие своей чувствительности к нагреванию [100] по-
лучается с высоким выходом только при проведении реакции при темпера-
туре ниже 5° С [46]. При гидролизе едким натром в метиловом спирте гли-
цидный эфир С15 (CXXXVII) превращается в неустойчивую натриевую соль
глицидной кислоты С15, которая декарбоксилируется с образованием
₽-альдегида С14 (CXVI) (т. пл. 2° С) с общим выходом из Р-ионона 80—90%
(4*6, 2611. Для получения р-альдегида С14 в чистом виде, что необходимо для
последующей реакции, применяют высоковакуумную перегонку и ректи-
фикацию или с большим эффектом кристаллизацию при температуре
— 30° С [262].
Альдегид С14 (CXVI) использован для технического синтеза ретинола
[100].
Синтезу Р-альдегида CJ4 было посвящено много исследований Хейльбро-
на, причем для этого соединения была установлена формула CXVI [263—
267 ], как а, P-ненасыщенного альдегида; для него характерны полосы с час-
тотами 1630—1680 см-1 в спектре комбинационного рассеяния [266, 268],
которые типичны для а, Р-непредельных карбонильных соединений. Полу-
ченный из этого вещества а, р-непредельный спирт окисляется активирован-
ной двуокисью марганца в растворе гексана снова в альдегид С14 в отличие
от неспособных к этой реакции р, -[--непредельных спиртов [269]. Альдегид
С14 при озонировании почти не дает героновой кислоты [265 ], образование
которой характерно для Р-ионона. Получены и дополнительные доказатель-
ства в пользу такой структуры Р-альдегида С14 (CXVI) [270].
176
Однако формула CXVI оспаривалась [271 ]; для Р-альдегида С14 предла-
галась формула CXXXVIII с сопряженной системой двойных связей,
CXXXVIII
но соображения в ее пользу оказались несостоятельными и были отверг-
нуты. р, у-Ненасыщенный альдегид С14, или алло-альдегид С14 (CXXXVIII),
был получен [217, 272, 273] и использован для синтеза витамина А, при
этом получился не полный транс-ретинол (I), а 9-4{пс-ретинол (IV) [2741.
Получение ретинола из альдегида С14
Альдегид С14 для синтеза ретинола был предложен еще в 1942 г. [263,
267 ]. В одном из первых синтезов витамина А (схема 42), в котором Майлас
[259] применил альдегид С14 (CXVI), его конденсацию с ацетиленовым кар-
бинолом, 3-метилпентен-1-ин-4-олом-3 (CXXXIX) в ацетиленовый гликоль
Сзд (CXL) проводят по реакции Гриньяра.
Схема 42
Синтез ретинол-ацетата из альдегида Ci<
СН, СН3 Q
СНа-СН=С-С^
Н
сн3
нс=с-с-сн=сн.
он
CXXXIX
СНз
CXVI
LiC=CH
СН, Упз СН,
' СНгСН=ССН-Сг€-ССН=СН3
он он
сн3'
СН3>Н3 , СИз
сн2-сН=с-сн=сн
сн,
он
CXL
сн,
CXLII
сн3
СПз
сн, сн,
’><Г,сн2-сн==с-сн-сн==сн-с-сн=сн2
сн,
|сн,сосн,сцососнэ
СН, ’ сн,
он
СН, СН, Г’3 т *
'><.сн2-сн=с-сн-с=с-с-сн2-сн,ососн3
сн3
он он
CXLIII
сн, сн3
он
CXLI
сн3
с н=с н-с=с н-с н=с н-с=с н-с Н2ОН
сн.
СН3 сц
сн3
сн,
. - з СН,
ia I I
.сн,-сн=с-сн-сн=сн-с-сн,-сн,ососн,
I I
сн,
он
CXLIV
сн,
сн, сн, Vй»
СН=СН-С=СН-СН=СН-С=СН-СН2ОСОСНз
СН3
XIII
177
Тройная связь этого соединения была частично восстановлена до двой-
ной в пиридиново-этилацетатной смеси с палладиевым катализатором на
угле. Получающийся этиленовый гликоль (CXLI) подвергался дегидратации
бромистым водородом в пиридине и в ледяной уксусной кислоте с одновре-
менной аллильной перегруппировкой гидроксила к конечной метиленовой
группе и ацетилированием в витамин А-ацетат (XIII) [2751.
По другому принципу [113, 259] ретинол получают путем постепенного
наращивания углеродного скелета, для чего из альдегида С14 с ацетиленидом
лития в жидком аммиаке первоначально получают ацетиленовый карбинол
С16 (CXLII), который в виде броммагниевого производного конденсируют
с 1-ацетоксибутаноном-З в моноацетат ацетиленового триола (CXLIII).
Таким же путем, как и в предыдущем методе синтеза, моноацетат ацетиле-
нового триола Сго частично восстанавливают в моноацетат этиленового трио-
ла Сго (CXLIV), который с хорошим выходом дегидратируют п-толуолсуль-
фокислотой, йодом или бромистым водородом в пиридине в ретинол-ацетат
(XIII). Общий выход по этой схеме получается несколько ниже, чем в дру-
гих синтезах ретинола, в которых применяется альдегид С14.
Следует отметить, что из альдегида С14 (CXVI) через ацетиленовый кар-
бинол С16 путем его дегидратации можно получить активный к последующим
превращениям углеводород С16 [259, 263, 267 ].
Альдегид CJ4 (CXVI) использован для синтеза витамина А через витамин
А-кислоту. Для этой цели его конденсируют с 1-этокси-3-метил пента диин-
-1,4-олом-З в условиях реакции Гриньяра, образовавшийся эфир диинтрио-
ла Сгр изомеризуют в 10%-ной серной кислоте в 7,8-дигидро-8-окси-11,12-де-
гидроретиновый эфир, который дегидратацией и частичным каталитическим
гидрированием превращают в эфир ретиновой кислоты (LXX, R=C2H5)
[276].
Важнейшая схема синтеза ретинола, которую разработали Ислер с corp.
[45, 100, 112, 277], была внедрена фирмой «Гоффман-ла-Роше» (Швейцария)
в производство (схема 43); в ней применяются альдегид С14 (CXVI) и аце-
тиленвиниловый спирт—3-метилпентен-2-ин-4-ол-1 (CXLV). Синтез витамина
А из альдегида С14 осуществлен также фирмами «Мерк», «Пфитцер» и др.
Соединения CXVI и CXLV вводятся в реакцию Гриньяра, в результате
которой однозначно образуется диолин С^ (CXLVI) в виде кристаллическо-
го продукта ст. пл. 58—59°С (из петролейного эфира) и выходом около
80%. Посредством частичного каталитического гидрирования тройной
связи до двойной диолин С^ (CXLVI) в среде петролейного эфира или мета-
нола превращается в этиленовый гликоль C^ (CXLVI I) почти с количест-
венным выходом. Реакция частичного восстановления высокоспецифична;
она осуществляется под влиянием отравленного солями свинца и хиноли-
ном палладиевого катализатора Линдлара (Pd/CaCO3) [77], что позволяет
избирательно гидрировать ацетиленовую связь до этиленовой. Этиленовый
гликоль Сго (CXLVII) частично ацетилируют по первичной гидроксильной
группе для ее защиты при последующих реакциях одним эквивалентом
уксусного ангидрида или хлористого ацетила в присутствии пиридина
при 0° С и получают моноацетат этиленового гликоля С2о (CXLVIII)
с т. пл. 73—74° С и выходом 97%.
Для аллильной перегруппировки вторичной гидроксильной группы сое-
динения CXLVIII и дегидратации с образованием новой двойной связи,
приводящей к сопряженной с ядром пентаеновой системы витамин А-аце-
тата (XIII), можно применить йод в петролейном эфире [100]; однако найде-
но лучшим эту реакцию проводить с хлорокисью фосфора в пиридине (для
связывания выделяющегося при реакции хлористого водорода) [278], при-
чем ретинол-ацетат получают с т. пл. 60° С и выходом 45%. При возвраще-
нии вновь в реакцию непрореагировавшего ацетата гликоля С^ (CXLVIII),
проведении реакции дегидратации бромистоводородной кислотой [275,
2791 в пиридине и уксусной кислоте (в этом случае стадию специального аце-
тилирования первичной гидроксильной группы можно исключить) или п-то-
178
Схема 43
Технический синтез ретинол-ацетата из альдегида С14
СН, СН3 । ’
аСН,-СН=С-СНО
сн,
3 CXYI
СПз
+ нс=с-с=сн-сн2он
CXLV
сн, СН.
' .а^-сн=с-сн-с=с-с=сн-снрн
он
сн,
CXLVI
СН3 СН,
СН=С-СН-СН=СН-С=СН-СН3ОН
ОН
CXLVII
СН3 СН,
" ' .ch2-ch=c-ch-ch=ch-c=ch-ch2or
ОН —
сн3
CXLVIII
СН, СН, р | 3
аСН=СН-С=СН-СН=СН-С=СН-СН2ОСОСН3
см.
R=COCH3
луолсульфокислотой в пиридине, или уксусной кислотой с применением а-то-
коферола для предохранения витамина А-ацетата от окисления во время
реакции выход равен 70% [275].
По этому синтезу общий выход ретинола из р -ионона составляет свыше.
45%. Таким путем, через альдегид С14, кристаллический синтетический
витамин А был впервые получен в 1947 г. [100].
Важно отметить, что при конденсации альдегида С14 (CXVI) используют
смесь цос-траяс-изомеров 3-метилпентен-2-ин-4-ола-1 (CXLV):
W -CXLV
сн2он
дос-CXLV
которые могут быть разделены дистилляцией; /лраяс-CXLV имеет более вы-
сокую температуру кипения [5, 25]. Из цис-CXLV образуется транс-дио-
лин-CXLVI, который в результате частичного восстановления превращается
в этиленовый гликоль С20 (CXLVII) с И-цис-конфигурацией. Из транс-
CXLV получается 13-цuc-диoлин-CXLVI и затем этиленовый гликоль Сго
(CXLVII), имеющий 11,13-ди-цис-конфигурацию. Оба изомерных этилено-
вых гликоля Сго с И-Цис-и 11,13-ди-цис-двойными связями после ацетили-
рования в соединение CXLVIII и последующей дегидратации, при которой
происходит перегруппировка цис-двойных связей, образуют в преобладаю-
щем количестве-полный /пранс-ретинол-ацетат (XIII).
Если изменить ход реакции, т. е. сначала провести дегидратацию, а за-
тем частичное гидрирование, то из цис-CXLV получается 11,13-ди-цис-ре-
тинол, а из mpauc-CXLV—11-цис-ретинол [5].
Если для моноацилирования этиленового гликоля Сго (CXLVII), помимо
уксусного ангидрида или хлористого ацетила, применить ацилирующие реа-
генты из масляной, пальмитиновой, янтарной и других кислот, то можно
получить соответствующие эфиры ретинола [45].
Из альдегида С14 (CXVI) с меньшим выходом, чем витамин А-ацетат, по-
лучен метиловый эфир витамина А 1112, 113, 259, 280].
Необходимую для конденсации с альдегидом С14 (по схеме 43) цепочку
Св—3-метилпентен-2-ин-4-ол-1 (CXLV) получают из метплвинилкетона.
В синтезе метплвинилкетона (CL) исходят или из ацетона, который конден-
179
сируют с формальдегидом в присутствии диэтиламина по реакции Манниха
с последующим отщеплением диэтиламина от продукта конденсации
(CXLIX) [281 ] (технический способ), или из винилацетилена, подвергаемого
гидратации под влиянием ртутного или другого катализатора:
СН3 (QH^NH-HCl СН3
| +НСНО-------------» |
СОСН3 COCH2CHsN(CjH8)2-HC1—
CXLIX СН3
Н2О сосн=сн2
2СН=СН СН=С—СН=СН2 ------------ CL
Метилвинилкетон с хорошими результатами получают исходя из ацети-
лена с последующим гидролизом промежуточного 1,3-дихлорбутена-2 при
температуре кипения (выход 50—60%) [282].
В дальнейшем синтезе для превращения метилвинилкетона (CL) в сое-
динение CXLV необходимо удлинить углеродный скелет молекулы на два
атома. Реакция этинилирования непредельных кетонов ацетиленом в со-
ответствующие карбинолы изучалась во многих работах [283—285 ]. Исполь-
зование метода Фаворского [2861 для этой реакции (в присутствии едкого
кали) сопровождается реакцией полимеризации, поэтому применяют кон-
денсацию при низких температурах в жидком аммиаке. Метилвинилкетон
(CL) конденсируют с ацетиленидами щелочных и щелочноземельных метал-
лов: натрия [284], лития или кальция [194] — в жидком аммиаке под не-
большим давлением в избытке ацетилена в третичный З-метилпентен-1-ин-
4-ол-З (СХХХIX) с выходом около 85%, который подвергают аллильной
перегруппировке в присутствии разбавленных минеральных кислот в пер-
вичный 3-метилпентен-2-ин-4-ол-1 (CXLV) с выходом около 90%:
СНз СН3 СН3
I I (Н+) I
COCH=CHt + LiC=CH -> СН^С—С—СН=СН2----------> СН=С—С СН—СН2ОН
I
он
CL CXXXIX CXLV
Соединение CXLV представляет собой смесь цис- и /иромс-изомеров [284].
7. Направленный стерический синтез геометрических изомеров ретинола
Для получения геометрических изомеров витамина А [7 ] в качестве исхо-
дного ключевого вещества применяют р-ионилиденуксусный альдегид, альде-
гид С16 (LXXIII), в виде его 9-цис- и 9-/ирп«с-изомерных форм (см. с. 168).
В качестве второго компонента необходимого для создания полного углерод-
ного скелета ретинола, используют метиловый эфир 0 -метилглутаконовой
кислоты (CLI). Конденсацию соединений цис- и транс-LXXIII и CLI в
12-карбоксиретиновые кислоты (11 -цис-CL 11 и 9,11-ди-^«с-(ХП) осуществ-
ляют под влиянием спиртового раствора едкого кали, причем во время реак-
ции происходит гидролиз промежуточного соединения. Для геометрической
формы конечного продукта синтеза не имеет никакого значения, с какими
(цис или транс) конфигурациями р-метилглутаконовый эфир вводят в реак-
цию с альдегидом С15 (схема 44).
Образующиеся с высоким выходом 12-карбоксиретиновые кислоты (CLII)
имеют пространственно менее затрудненную 11 -^ис-конфигурацию (с кар-
боксильной группой при С12) независимо от того, имеется ли ^пс-конфигу-
рация у Со»—-С(!0) двойной связи или нет [287].
Отщепление С12-карбоксила от 12-карбоксиретиновых кислот (CLII)
производят при нагревании раствора в 2,4-лутидине в присутствии уксусно-
кислой меди в течение 2 ч при 120—125° С. Одновременно с отщеплением
углекислоты происходит изомеризация с образованием пространственно не-
180
Схема 44
Стерический синтез геометрических изомеров ретинола
транс - LXXIII
затрудненных 11-траяс-ретиновых кислот. При этом, если в качестве исход-
ного продукта применяют 9-транс-альдегид С15 (транс-LXXIII), образует-
ся неоретиновая кислота (13-цис) (CLIII), а если 9-^нс-альдегид С15 (цис-
LXXIII), то 9,13-ди-цис-ретиновая кислота (CLIV).
Под влиянием следов йода ретиновые кислоты (СЫН и CLIV) в виде их
метиловых эфиров изомеризуются в среде бензола и этилового эфира или в
изопропиловом спирте в транс-ретиновую кислоту (XVII) и 9-1/ис-ретиновую
кислоту (CLV) соответственно.
Превращение изомерных ретиновых кислот (СЫН, XVII, CLIV и CLV)
в виде их эфиров в неоретинол (II), полный транс-ретинол (I), 9,13-ди-
^ас-ретинол (VI) и 9-^ас-ретинол (IV) осуществляют восстановлением алю-
могидридом лития в среде эфира.
Ретинол (I) может быть получен также изомеризацией неоретинола
(II) через его ацетат, а 9-^пс-ретинол (IV) —изомеризацией 9,13-ди-д{ас-ре-
тинола (VI) в виде его n-фенилбензоата под влиянием следов йода в петро-
лейном эфире или бензоле.
181
Помимо природного полного транс- и 11-цис-ретинолов, получены и дру-
гие 9-цис-, 13-цис-, 9,13-ди-цис- и 11,13-ди-ц«с-изомеры витамина А, а так-
же соответствующие альдегиды, кислоты и эфиры [25, 288—290].
8. Синтез дегидроретинола
Дегидроретинол получают из полной трояс-ретиновой кислоты, в цик-
логексеновом ядре которой создают вторую двойную связь. Для этого в
положение 4 метилового эфира ретиновой кислоты(ЬХХ, R=CH3) с т. пл.
55—56° С (полученного одним из методов, описанных выше) вводят атом бро-
ма при взаимодействии с N-бромсукцинимидом в хлороформе при 0° С (схе-
ма 45). Из полученного 4-бромпроизводного CLVI нагреванием с 4-фенил-
морфолином отщепляют бромистый водород и затем после хроматографи-
ческой очистки выделяют метиловый эфир витамина А2-кислоты (CLVII)
ст. пл. 45—47° С. Его восстанавливают в дегидроретинол (VII) алюмогид-
ридом лития с общим выходом 25% [8].
Схема 45
Синтез дегидроретинола из эфира ретииовой кислоты
СН3 СН, ^Н’
ch=ch-c=ch-ch=ch-c=ch-coor
СН, CH,
" ' ,ch=ch-c=ch-ch=ch-c=ch-coor
сн3
LXX
сн3
Вг
CLVI
сн, сн3 ?Нз
'><XCH=CH-C=CH-CH=CH-C=CH-COOR
L 1 —*
^^сн
3 CLVII
R=CH3
сн, СН,
гХ_СН=СН-С=СН-СН=СН-С=СН-СН2ОН
¥|1
В синтезе дегидроретинола можно исходить и непосредственно из дегид-
po-р-ионона [291 ], а также из 3,4-дегидроальдегида С14, подвергая его кон-
денсации с 3-метилпентен-2-ин-4-олом-1 (CXLV) с последующими превраще-
ниями [9].
Из 3,4-дегидроальдегида С15 с помощью реакции Виттига синтезирован
эфир витамина А2-кислоты (CLVII), который алюмогидридом лития вос-
становлен в дегидроретинол (VII) [101.
Исходя непосредственно из ретинола, окислением его в контролируе-
мых условиях в колонке двуокисью марганца получают 3-оксиретиналь, ко-
торый количественно восстанавливают алюмогидридом лития в 3-оксирети-
нол. После селективного ацетилирования и дегидратации n-толуолсульфо-
кислотой из него получают 3,4-дегидроретинол (VII) [292].
Еще в одном синтезе дегидроретинола исходят из ретиналя (XVIII),
который при взаимодействии с N-бромсукцинимидом образует 4-бромрети-
наль, а после отщепления бромистого водорода — 3,4-дегидроретиналь. Его
восстановление обычным способом с алюмогидридом лития дает дегидроре-
тинол (VII) [891.
Витамин А2-альдегид синтезирован еще в 1939 г. [293] дегидрированием
витамина А, дпэтилкетоном в присутствии mpem-бутилата алюминия. В кри-
сталлическом виде он был получен в 1962 г. [9].
Синтезированы кристаллические полный транс-, 9-цис-, 13-цис-, 9,13-
ди- г{пс-3,4-дегпдроретинолы [10] и 11-цис-, 11,13-ди-цис-3,4-дегидроретн-
нолы [9], т. е. все шесть известных изомерных дегидроретинолов.
182
БИОЛОГИЧЕСКОЕ ЗНАЧЕНИЕ РЕТИНОЛОВ. ЗАВИСИМОСТЬ МЕЖДУ
СТРОЕНИЕМ И А-ВИТАМИННОЙ АКТИВНОСТЬЮ
Витамин А — важнейший витамин роста, необходимый для всех млеко-
питающих животных, птиц и человека. При его недостатке в организме при-
останавливается рост (молодых животных), а затем наступает смерть. Еще
в 1913 г. было установлено, что для развития молодых животных (крыс)
необходим эфирный экстракт яичного желтка или любой животный жир (кро-
ме свиного сала) [294, 295]. Впоследствии было доказано, что активным ве-
ществом этих пищевых продуктов является жирорастворимый ростовой фак-
тор, который в 1915 г. выделила Макколлум и Девис [296] из жира живот-
ных и рыбьего жира. Оказалось, что недостаток витамина А вызывает
заболевание глаз, так называемую куриную слепоту, более тяжелое забо-
левание — ксерофтальмию, перерождение эпителиальных тканей (кожи, ды-
хательных и мочеполовых органов, слизистой кишечника), которые подвер-
гаются керотинизации (ороговению, шероховатости), и общую неустойчи-
вость организма к инфекционным заболеваниям. Витамин А необходим для
нормального размножения; он выступает в качестве регулятора желез вну-
тренней секреции (яичников, щитовидной железы).
Витамин А выполняет в организме биохимические функции, которые за-
ключаются в специфическом участии в различных реакциях обмена веществ;
возможно, его основная роль заключается в регулировании прохождения ме-
таболитов через мембраны. Обзор по биохимии витамина А дан в литературе
[297].
Витамин А осуществляет в механизме сумеречного зрения важнейшие
процессы, участвуя в адаптации глаз позвоночных животных к темноте.
Из обесцвеченной сетчатки глаз в 1935 г. был выделен ретиналь [298],
а в 1944 г. Мортон показал, что зрительный пигмент (родопсин) содержит
ретиналь (витамин А-альдегид) [299 ]. Витамин А-альдегид, являясь просте-
тической группой, совместно с протеином — опсином — образует зритель-
ный пигмент сетчатки глаз: родопсин и порфиропсин. В состав родопсина
входит ретиналь (витамин Aj-альдегид, ретинен^ (XVIII) [298, 299], в
состав порфиропсина—дегидроретиналь (витамин А2-альдегид, ретинен2)
(XXIV) [300, ЗОН.
В организме человека и животных полный транс-ретинол (I) и 13-^ас-ре-
тинол (неовитамин А) (И) эффективны в зрительном процессе в связи с про-
текающими реакциями их превращений в ретиналь.
Зрительный импульс продуцируется в результате реакции фотоизомери-
зации 11-цис-ретиналя (и соответственно 1 l-^wc-дегидроретиналя) в полный
транс-ретиналь (соответственно транс-дегидротиналь).
При фотохимической реакции происходит поглощение квантов световой
энергии зрительным пигментом сетчатки глаз — родопсином. Родопсин,
красный пигмент (Хмакс 498 нм), содержит в качестве хромофора 11-цис-ре-
тиналь (XX), первоначально под действием света превращается в неста-
бильный лумиродопсин оранжевого цвета [70], в котором в результате
фотоизомеризации образуется полный транс-ретиналь (XVIII). Затем проис-
ходит превращение в желтый метародопсин (/.макс 478 нм) и наконец обес-
цвечивание и расщепление родопсина на полный транс-ретиналь (XVIII)
с 'макс 381 нм и протеин —опсин [70, 302].
В дальнейшем процессе под влиянием ретинальизомеразы полный
транс-ретиналь (XVIII) изомеризуется в 11 -г{ас-ретиналь (неоретиналь Ь)
(XX), который в темноте взаимодействует с опсином и вновь регенерирует
родопсин. В экспериментальных условиях этот процесс почти полностью
заканчивается через 20 мин. По-видимому, превращение полного транс-ре-
тиналя в 11-1{нс-ретиналь под влиянием ретинальизомеразы происходит в
темноте [303, 304]. Цикл 11-цис-транс- стереоизомерии, возможно, проте-
кает через стадию изомеризации ретинола (I) в 1 l-tjuc-ретпнол (неоретинол
b) (III) [61 ]. Эти превращения могут быть изображены следующим образом:
183
Родопсин-
Темнота зипельный пигмент
(содержит 11-цис -
ретиналь)
Свет
Лумиродопсин
(содержит полный
транс-ре тиналь)
Метародопсин
(содержит полный
транс - ретиналь)
Ретинальизомераза
11 -цис -ретиналь ------------------
Спейн Ж pgQ
Алкоголь-
дегидроге-
наза
НАДН+Н*
П-цис -Ретинол
НАД
Полный транс - _
ретиналь(ХУШ) +Опсин
НАД
Алкоголь-
дегидроге-
наза
НАДНЩ
Изомераза (?) Полный
транс -ретинол (1)
Источником ретиналя, покрывающим его убыль, является ретинол, ко-
торый с высоким выходом окисляется в ретиналь под влиянием ферментных
систем типа ретинальредуктазы (алкогольдегидрогеназы). Ретиналь может
обратимо восстанавливаться в ретинол под влиянием восстановленной формы
рети на л ьредуктазы.
Следует отметить, что родопсин не может синтезироваться in vitro из опси-
на, ретинальредуктазы и чистого витамина Аъ обладающего полной
тра«с-конфигурацией. Однако из кристаллического ретинола (I) и неовита-
мина А (II) под влиянием следов йода на свету образуется 11-^пс-ретинол
(III), из которого родопсин уже может синтезироваться.
Происходящие в сетчатке глаз процессы, лежащие в основе зрения,
связанные с фотопревращениями ретиналя, в значительной степени изуче-
ны, однако многое еще остается неясным, что видно из обзора [302].
П-гфс-Дегидроретиналь (XXV) является хромофором порфиропсина —
зрительного пигмента пурпурного цвета (Хмакс522 нм) сетчатки глаз пресно-
водных рыб. Он участвует в зрительном процессе по такому же циклу, что и
11 -цис-ретиналь в системе родопсина [70].
Витамин А встречается только в продуктах животного происхождения в
противоположность каротиноидным провитаминам, которыцнаходится глав-
ным образом в растительных источниках.
Молекула витамина А по своей биологической активности высокоспеци-
фична. В ее структуру должны входить цикл 1,1,5-триметилциклогексена-5
(кольцо 3-ионона) и боковая алифатическая цепь из 9 атомов углерода с че-
тырьмя сопряженными двойными связями, соединенная с ядром в положе-
нии 6; алифатическая цепь должна содержать две метильные группы в по-
ложении 9 и 13 и оканчиваться гидроксильной группой или другими функ-
циональными группами (карбоксильной, альдегидной), способными в орга-
низме превращаться в гидроксильную группу.
Только полная транс-конфигурация двойных связей боковой цепи обу-
словливает полную биологическую активность молекулы витамина А. Одна
ф/с-конфигурация, например у пятой двойной связи, снижает активность
почти на 13% [21 ] (или на 25%) [22], как это установлено для неоретинола
(II), а у третьей двойной связи —почти в 5 раз; две ^нс-конфигурации (у '
третьей и пятой двойных связей) снижают активность более чем в 4 раза [22 ]
(см. табл. 9, с. 144).
Введение второй двойной связи в ядро р-ионона, как это происходит в
молекуле витамина А2, снижает витаминную активность до 40% [8, 30],
а его час-стереоизомеров—еще больше (см. табл. 10).
Полное или частичное гидрирование молекулы дает неактивные соеди-
нения— дигидро-, тетрагидро- и пергидровитамины А [76, 305, 306]. За-
мещение одной двойной связи на тройную сильно снижает активность сое-
184
динения — 7,8-дегидровитамин A (CLVIII) имеет 40% активности витамина
А [116L
СН, СН,
СН,
CH,
:=сн—сн,он
CLVIII
Удлинение алифатической цепи на одну метиленовую группу почти пол-
ностью дезактивирует соединение: этиловый эфир гомовитамина А
сн,
сн, сн,
Н,
сн,
СН =СН—С -СН—СН =СН—с=сн—сн,—сн,ос2н6
9 13
обладает 15% активности витамина А 1307].
Высшие гомологи витамина А, такие, как р -апо-8'-(или 12' и 14')кароти-
нолы, могут быть получены восстановлением продуктов окисления р -каро-
тина, например р-апо-8'-, р-апо-12'-, р-апо-14'-каротиналей; эти гомологи
обладают значительной активностью витамина А [308], по-видимому, вслед-
ствие их способности подвергаться в организме дальнейшему окислению до
структуры витамина А.
СН, СН, СН3 СН, СН, СН,
\/ II II
сн =сн-с=сн-сн=сн-с=сн-сн=сн-сн=с—сн=сн-сн=с—СН2ОН
। Y
Ych,
₽-апо-8'-Каротинол •
Удаление метильной группы, например из положения 13, также почти
полностью лишает соединение витаминной активности — метиловый эфир
норвитамина А
СН, СН, СН,
,СН=СН—i=CH—СН=СН—СН=СН—СН2ОН
I II ‘ 13
^/^СНз
обладает всего около 3 % активности витамина [264 ]. Также почти лишается
витаминной активности соединение, в котором удаляются метильные груп-
пы ядра, например тринорвитамин А.
Превращение гидроксильной группы в сложноэфирную группу почти не
изменяет витаминной активности образующегося соединения.
Из простых эфиров высокой активностью обладают те, которые могут
расщепляться в организме, например метиловый эфир витамина А (см.
табл. 11).
Соответствующий витамину А углеводород — аксерофтен (XXVII)
(с. 151) обладает 10% витаминной активности [309], а димер ретинола —
китол (XXVIII) биологически неактивен. Продукт дегидратации витами-
на А —ангидроретинол (XXIX)
185
CH3 CHS СНз СНз
\/ । ।
/><\^СН—СН =С-СН=СН—сн=с—сн =СН,
\^\сн3
XXIX
обладает всего 0,4%-ной активностью [80, 91], а ангидровитамин А2 неак-
тивен [30].
Витамин А-кислота, ретиновая кислота (XVII) (с. 148) в виде ее водно-
растворимой соли, отвечает 2/3 активности витамина A-спирта, а свободная
кислота — ‘/10.
При замещении оксиметильной группы на альдегидную образуется ре-
тиналь (XVIII) (полная транс-конфигурация, с. 148), сохраняющий почти
полную активность ретинола [22, 64 , 310]. Дегидроретиналь (XXIV) рав-
нозначен по активности витамину А2 (см. табл. 10). Эфиры и нитрилы ви-
тамина А2-кислоты также обладают витаминной активностью [311 ].
Ретиналь с одной tfac-конфигурацией у двойной связи положения 9,
изоретиналь-а (XXI), обладает почти такой же активностью, как и ретиналь
(XVIII) [3]. Однако, по другим данным, его активность, как и активность
ретиналя с двумя quc-конфигурациями (у третьей и пятой двойных связей),
изоретиналя-6 (XXIII) (см. с. 61), составляет всего */5 активности вита-
мина А [22]. В организме крыс витамин А-альдегид превращается в витамин
А [461, так же как и неовитамин А.
Интересно отметить, что окисление по двойной связи циклогексенового
ядра витамина А-альдегида не снижает витаминной активности — 5,6-эпо-
кись витамина А-альдегида имеет 108% активности полного транс-витамина
А-ацетата [312], в то время как 5,6-эпокиси ретинола — гепаксантину
(XIV)—свойственна незначительная активность [313].
Потребность человека в витамине А составляет около 1,5—2,0 мг в
сутки и увеличивается в зависимости от физической нагрузки до 3 мг при
очень тяжелом труде; она возрастает также у беременных и кормящих
женщин —до 2,5—Змг в сутки. Только для предупреждения зрительной
недостаточности человека необходимо 0,39 мг витамина А в сутки.
Витамин А в виде своих стабилизированных препаратов имеет большое
значение для лечебных целей, косметики и витаминизации пищевых продук-
тов. В частности, витамином А производится обогащение молока и маргари-
на: на 1 кг маргарина вносится 7—10 мг ретинола. Витамин А широко при-
меняется в животноводстве и птицеводстве. Телята, поросята и цыплята
нуждаются в специальном витаминном рационе или в дополнительном скар-
мливании концентратов витамина А и его провитамина —р-каротина, обла-
дающего частичной (V4—1/в) биологической активностью витамина А
[3141.
Помимо ряда полиеновых соединений, обладающих стимулирующей рост
биологической активностью, известны вещества, подавляющие витаминные
свойства. Из ретиналя получен антивитамин А [315] следующего строения:
СНз СНз СНз СНз
,CH=CH—С—СН—СН=СН—(L=<
Z \/ I I
1 ОН он
\/\сн3
Антивитамины А образуются также при гидроксилировании двойной свя-
зи витамина А или каротина.
186
ПРОВИТАМИНЫ РЕТИНОЛА И ДЕГИДРОРЕТИНОЛА
Многочисленные природные красящие вещества, обусловливающие жел-
тую, оранжевую и красную окраску плодов, цветов и осенних листьев рас-
тений, относимые к каротиноидам, обладают биологической активностью ви-
тамина А [316].
Молекула каротиноидов, обычно состоящая из 40 атомов углерода,
включает сопряженную полиеновую систему, находящуюся в ряде соедине-
ний между двумя циклогексеновыми кольцами. Отдельные представители
этих соединений являются как провитаминами ретинола, так и провитамина-
ми дегидроретинола [317].
Биологическая активность таких каротиноидов связана с их способностью
в печени животных расщепляться с образованием витамина А. Поэтому
активностью витамина А обладают только такие каротиноиды (провитами-
ны А), в молекулу которых входит кольцо 0-ионона (или 3,4-дигидро-
0-ионона), связанное с алифатической цепью, содержащей систему сопря-
женных двойных связей изопреноидного характера, которые при расщеп-
лении могут образовать структуру витамина А.
К важнейшим провитаминам А относятся природные каротиноидные угле-
водороды: а-каротин (CLIX),0-каротин— 1,Т-бис- (1,1,5-триметилциклогек-
сен-5-ил-6)-9,13,13',9'-тетраметилоктадеканонаен-7,9,11,13,15,14',12', 10',8'
(CLX) [1] и у-каротин'(СБХ1).
19 20 20' 18' .
•» 17 сн, сн, СН, СН, 1/ 16
сн3 сн3 . у"9 сн, сн3
" >сн=сн-с=сн-сн=сн-с=сн-сн=сн-сн=с-сн=сн-сн=с-сн=сн
7 8 8 10 II 12 13 14 1S 15'
сн,
18 3
Ci-Каротин
CUX
Н’ 13’ И’ Ю’ 9’ 8' 7*
сн3
«г 3
СНз СН3 СНз СНз
сн=сн-с=сн-сн=сн~с=сн-сн=сн-
СНз СНз
сн=с-сн=сн-сн=с-сн=сн
сн3
СН3
Сн, ей.
"снэ
В-Каротин
CLX
СН3 СНз
сн=сн-с=сн-сн=сн-с=сн-сн=сн-сн=с-сн=сн-сн=с-сн=сн
у-Каротин
CLXI
СНз
сн3
сн3'
сн, <Х
Известны синтетические каротиноидные углеводороды — провитамины
Аг, такие, как 3,4;3',4'-бпс-дегидро-0 -каротин и 3,4-монодегидро-0-каротин.
Помимо каротиноидных углеводородов (а-, 0- и у-каротинов), к провита-
минам А относятся природные окси- и оксо каротиноиды с кольцом 0-ионона
в молекуле (табл. 13). Все они обладают витаминной активностью.
Двойные связи алифатической цепи каротиноидов образуют многочис-
ленные геометрические изомеры. Теоретически для 0-каротина, имеющего
девять сопряженных двойных связей в алифатической цепи, возможно пред-
ставить 512 ^«с-транс-изомеров. Фактически tyuc-изомеризация сильно огра-
ничена пространственными помехами [1,2] из-за разветвления боковой цепи,
имеющей в наличии четыре метильные группы. Оказывается, что свобод-
ное ^uc-превращение возможно только у двойных связей тех атомов углеро-
да каротиноидной полиеновой цепи,которые имеют при себе метильные груп-
пы, и, кроме того, у центральной двойной связи. Для остальных двойных
связей с обозначениями 2, 4, 8, 10 возможна только заторможенная zpc-изо-
меризация.
Поэтому наличие пространственных затруднений делает возможным для
0-каротина только 20 незатрудненных стереоизомеров, для а-каротина — 32
и для у-каротина—64 изомера [2].
187
Таблица 13
Провитаминовые каротиноиды1
CHS СНз СН, СН.
I । । I
К=—С=СН—СН=СН—С=СН—СН=СН—СН=С—СН=СН—сн=с—
В формулах
Название Структурная формула Т. пл., °C Спектр погло- щения* Лите- ратура
^макс* нм ₽|% 1см
Каротиноидные углеводороды
tU-a-Каротин — Сц>Нзв — темно- красные призмы СН, СН3 СН, «^.СН^СН-^-СН^Н^Ъ- '"CH, CHf^ СН, 187—188 160—162 (синтетичес- кий) а) 509, 477 б) 485. 454 в) 473, 444, 422 2520, 2800, 1900 (318, 319]
P-Каротин (пол- СНз сн» . . <Н сн, 183 (в ва- а) 520, [319]
ный транс- изо- ^СН=СН-(|^-СН=СН кууме) 484,
мер) — С40Н56— II 11 452
темно-красные б) 497,
ромбические Vltg С Hj 466
пластинки 180 в) 481, 2268,
453 2592
т-Каротин — С^ СН3 СН, х СН=СН-(к)~СН=С1< у >сн /Н 178 (в ва- а) 533. [318,
С40Н5в — темно- кууме) 496, 319]
красные призмы i 463
"TH, CHfXz 152—154 (синтетичес- б) 509, 475,
кий) • 466 в) 494, 2720,
462, 3100, •
437 2055
3,4-Монодегид- ро-р-каротин (синтетический СЛ сн, сн, ^CH=C11-(k)-CH=CH^S< сн, 186 в) 461 2330 [320]
провитамин А2)—С4оН,54 — 'сн, сн,"^
сине-фиолето- вые кристаллы 3,4; 3',4'-бис- Дегидро-р-каро- СН, г» сн, сн, ' сн=сн-^к)-сн=сн S< сн» 190-191 в) 47Г 2400 (320)
тин (синтетиче- ский провита- мин А2) —
11 • J]
С40 Н52 — темно- фиолетовые ли- хн, сн/^
сточки сн, сн. сн» 183—184 в) 518, 2680, [318]
Торулнн (3', 4'- сн. 484, 3240,
дегидро-7-каро- - сн=сн-(19-сн=сн 460 2315
тин) (нз микро- организмов) II п ^СН, СНз^4^
З-Зеакаротин (7', 8'-днгидро- 7-каротин) (пиг- СНз СН, СН. ' ci+=CH-(^~CH,-ctu 7; [СН, 96—98 в) 454, 428, 406 2300, 2520, 1660 [318]
мент желтой ку- курузы) Н 11 ХИ, Cllf^
Ок сипро изводные каротины
а-Криптоксан-
тин (З'-окси-а-
каротин)—
С40Н6вО
СНз СН, СН, СН,
' ,сн=сн-(к)-сн=сн
Нз
СН, - ОН
182
175—176
а) 509,
477
446
б) 490,
457,
431
[321]
188
Продолжение табл. 13
Название Структурная формула Т. пл., °C Спектр по- глощения1 Лите- ратура
^макс* нм £1% с1см
Р-Криптоксан- тин (З-окси-Р- каротин)— С40Н58О (пиг- мент желтой ку- курузы, фрук- тов и цветов)— блестящие приз- мы ; ! 1 СН3 СН, СН, ОД >С/С Н =сн—(к}-СН=О1 С>\ L I JL 169 (в ва- кууме), 158—159 а) 519, 483, 452 б) 497, 463, 433 в) 480, 452 1986, 2370 [322]
Кетопроизводные каротины
Эхиненон (мий- соксантин, афа- нин) (4-кето-Р- СН3 СН, % СН, СН, S<^h^h-(k)^h=ch 178—179 а) 520, 488 г) 472 2040 [323, 324]
каротин) — С40НБ8О (из во- дорослей и по- ловых же- лез морских ежей) — фиоле- товые иглы ^"XH, CHf о -
Криптокапсин (пигмент крас- ного перца) сн, сн, сн=сн-(к/-сн=сн-с -t 'О X 160—161 г) 486 1968 [325]
Цитранаксантин (из цитрусовых) СИз ОД V '><^сн=сн-(к)-сн=сн-с-од ^сн, [326]
Синтаксантив (из цитрусовых) ОД ОД 9 Х<,сн=с.н-(к)-с -ОД [326]
Каротиноидные альдегиды
1', 2'; 3', 4'- бис-Дегидро-7- каротин-1-аль- дегид — С40 (из микроорганиз- мов) сн3 СН3 сн, сно ХСН=СН-(к)-СН=СНхХ^< '(SH, сн^^ 167—168 в) 540, 508 2120, 2865
Р-Апо-2'-каро- тиналь — Сз7 (из плодов цит- русовых) ОД ^5н=сн-(к)-сн=очсн 160—161 в) 498 2730
Р-Апо-8'-каро- тиналь —С30 (из плодов цит- русовых, пече- ни) Р-Апо-Ю'-каро- тиналь — Сг7 СН, CHf^ сн3 од XL-CH=CH-(K)-CHO ^"сн, 138—139 97—99 в) 457 в) 437 2640 2550 [327] [328]
189
Продолжение табл. 13
- Название
1 Структурная формула Т. пл.. °C Спектр погло- щения1 Лите- ратура
*макс» нм pi % Нем
Каротиноидные кислоты
Торуларод ин —
СзтН^Ог (из
микроорганиз-
мов) — бле стя-
щие крас-
ные иглы
Нейроспорак-
сантин (из мик-
роорганизмов)
CH3 СНз сн3 соон
" " ,сн=сн-(к)-сн=сн.
'сн3
сн3 сн3
><J:h=ch-(k)-ch=ch
VH
II
CHfCxCOOH
Нз
210—212
а) 582, 541, 502 б) 554, 515 в) 535, 2040 [3291
507
[3301
СН, .
1 а) в сероуглероде; б) в хлороформе; в) в петролейном эфире; г) в бензоле.
Выделенные из природных источников каротиноиды, являющиеся про-
витаминами ретинола, как правило, представляют собой полную транс-
конфигурацию, т. е. имеют общую хромофорную систему всей полиеновой
цепи, что связано с максимумом поглощения в наиболее длинноволновой
части спектра по сравнению с максимумами их стереоизомеров, имеющими
одну или несколько двойных связей в ^пс-форме [2]. Природный 0 -каротин
(CLX): также является полным траяс-изомером.
г
* *. г
Возможные ^пс-траяс-изомерные пространственно незатрудненные формы
0-каротина представлены ниже.
quc-транс-Изомеры р-каротина
Полный транс-
9-цис (нео-з-каротин U)
13-цис-
15-цис-
9,13-ди-цис-
9,15-ди-чис- (нео-Р-каротин В)
9,13'-ди-цис- (нео-р-каротин В)
9,9'-ди-цис- (нео-Р-каротин V)
13,15-ди-чнс-
13,13'-ди-ч«с-
9,13,15-три-чис-
9,13,13'-три-чис-
9,13,9'-три-чнс-
9,15,13'-три-чис-
9,15,9'-три-чис-
13,15,13'-три-чис-
9,13,15,13'-тетра-цис-
9,13,15.9'-тетра-ч«с-
9,13,13',9'-тетра-чис-
9,13,15,13'9'-пента-1/ис-
Из природных источников выделена и получена искусственными превра-
щениями [331] большая часть стереоизомеров, в том числе 15,15'-цис-[} -ка-
ротин (CLXII) [332], нео-В-каротин В (9,15-ди-г{нс) (CLXIII), нео-0-ка-
ротины U, V (9-цис и 9,9--дм-цис) и др.
190
CLXIU
Помимо пространственно незатрудненных каротинов, из 11,12; 11',
12'-бпс-дегидро-0 -каротина частичным каталитическим гидрированием полу-
чены пространственно заторможенные ll-tjuc-p-каротин В и 11-цисф -каро-
тин С [333 ]. Другим синтетическим путем пространственно заторможенный
11,11'-ди-г/uc-p -каротин получен из альдегида С14 и 3,8-диметилдекатриен-
3,5,7-диина-1,9 [3341.
Для p-каротина (CLX) и 15,15'-дегидро-p-каротина С(б)—С<7>-связь иони-
лиденового кольца с полиеновой цепью имеет s-t/uc-конфигурацию, как это
установлено рентгеноструктурным анализом [11—13], а не транс-конфи-
гурацию.
Среди приведенных в табл. 13 каротиноидов только природный (+)-а-ка-
ротин (CLIX) имеет асимметрический атом углерода и обладает оптической
активностью: (a law+385°, [а Id +538° (С6Н6). Синтетически получена энан-
тиомерная форма — (—)-а -каротин с [а.1з4з —400°, [а Id—556°. (С6Н6)
[335]. Каротиноиды — высокоплавкие вещества (табл. 13). а-, р- и у-Ка-
ротины легко растворимы во многих органических растворителях: сероугле-
роде, дихлорэтане, хлороформе, треххлористом углероде, бензоле, толуоле и
кипящем петролейном эфире; в гексане (при 0°С) а-каротин лучше растворим
(1 : 100), чем р-каротин (1 : 300). Они малорастворимы в эфире (1 : 1000),
в холодном петролейном эфире (1 : 1500), почти нерастворимы в кипящем
абсолютном спирте, растворимы в жирах, нерастворимы в воде.
Каротин легко адсорбируется окисью алюминия и другими адсорбентами;
в верхней части адсорбционной колонки располагается у-каротин (12 двой-
ных связей), ниже —р-каротин (11 двойных связей) и еще ниже —а-каро-
тин (10 сопряженных двойных связей и 1 несопряженная).
В растворах каротин обладает окраской от желтой до оранжево-красной
со слабой желто-зеленой флуоресценцией. Для каротиноидов вследствие
наличия длинной полиеновой цепи сопряженных двойных связей характер-
но интенсивное поглощение как в ультрафиолетовой, так и в видимой обла-
стях света. Главный максимум поглощения .важнейших провитаминовых ка-_
ретиноидов лежит в области 461—509 нм (в хлороформе или бензоле), что
видно из табл. 13 и рис. 5.
г/пс-Изомеризация пространственно незатрудненной двойной связи ка-
ротиноидов вызывает гипсохромное смещение максимума поглощения ви-
димого света на 3—7 нм со значительным ослаблением интенсивности по-
глощения [2]. Одновременно весьма незначительная полоса поглощения
ультрафиолетового света в области 320—380 нм усиливается и сопрово-
191
ждается возникновением нового максимума, так называемого цис-пика, дос-
тигающего наибольшей интенсивности при образовании цис-конфигурации
центральной 15,15'-двойной связи соединения CLXII. Характерное изме-
нение спектра поглощения для этого 15,15'-цис-$-каротина (CLXII) пока-
зано на рис. 5 [3361.
Рис. 5. Спектр поглоще-
ния в петролейном эфире:
/ — полного транс -а-каготи-
на; 2 — полно-о транс-^-ка-
ротина: 3 — полного транс-у.
каротина; 4 — 16. 15'-цис-р-Ка.
ротина.
Из большого числа провитаминовых каротиноидов производят главным
образом р-каротин —в виде его масляных растворов. В последние годы в
ряде стран стали получать р-апо-8'-каротиналь, р-апо-12'-каротиналь и
этиловый эфир р-апо-8'-каротиновой кислоты как пигменты для пищевых
продуктов.
Для условной биологической оценки препаратов используют интерна-
циональную стандартную единицу (и. е.) активности. Принято, что актив-
ность р -каротина в 2 раза ниже активности витамина А, и 1 и. е. отвечает
0,6 мг р-каротина. 1 г полного транс-R -каротина соответствует 1 650 000
и. е. [49 ].
Каротин легко расщепляется под воздействием ультрафиолетового све-
та. Провитамины А способны к легкой цис-транс-изомеризации [ 2, 331].
Эта изомеризация происходит в растворах, особенно легко на свету [331],
при нагревании в темноте под влиянием галогеноводородных и других кис-
лот или в петролейном эфире при каталитическом участии’'йода на свету
[3371, а также в расплавах. В кристаллическом состоянии вещество изоме-
ризации не испытывает [21. Длительное термическое воздействие вызывает
tytzc-изомеризацию с последующим расщеплением молекулы каротиноидов.
Одним из первоначальных продуктов термической изомеризации полного
транс-$-каротина (CLX) является нео -р -каротин В (9,15-ди-^ис-изомер)
(CLXIII) [2, 338]. Разделение стереоизомеров производят хроматографи-
ческим методом Цвета [264]. 5,15'-t{uc-p-Каротин (CLXII) лабилен и под
влиянием следов йода при нагревании легко превращается в полный транс-
-р-каротин (CLX) [332].
а-Каротин перегруппировывается вр-каротин под влиянием этилата нат-
рия при нагревании [339]. а-, р- и y-Каротины очень чувствительны к раз-
личным химическим воздействиям. Однако они устойчивы к сильным щело-
чам и почти не изменяются при нагревании в инертной атмосфере.
Элементарный бром присоединяется к кратным связям всей полиеновой
системы [340]. При воздействии йодом на а- и р-каротины происходит при-
192
соединение по двойным связям циклогексенового ядра с образованием
дийод- и тетрайодкаротинов [341]. При бромировании Р-каротина N-бром-
сукцинимидом бром идет в ядро (в положение 4), не затрагивая двойных
связей.
При восстановлении каротинов над платиновым катализатором в цикло-
гексане или уксусной кислоте водород постепенно присоединяется ко
всем двойным связям с образованием смеси продуктов частичного восстанов-
ления или конечного бесцветного пергидрокаротина [342].
Йодистоводородная кислота восстанавливает а- и 0 -каротины с образо-
ванием соответствующих 5,6-дигидро-а- и 5,6-дигидро-0 -каротинов [343,
344]; реакция протекает по наименее устойчивой непредельной связи
ядра.
Амальгама алюминия производит восстановление 0 -каротина с а, <о-при-
соединением водорода и одновременной перегрупппировкой двойных связей
с образованием 7,7'-дигидро-0 -каротина (CLXIV) [345]:
Все каротиноиды очень чувствительны к кислороду и претерпевают в
различной степени окисление, превращаясь в эпокиси — продукты присое-
динения одного атома кислорода по двойной связи цикла, так называемые
«фуранокиси»—продукты присоединения одного атома кислорода по двум
двойным связям (циклической и нециклической), альдегиды — с отщепле-
нием части молекулы главным образом по пространственно затрудненным
двойным связям, т. е. по 8-й и 10-й непредельным связям, кетоны — с рас-
щеплением цикла по двойной связи, кетали — с расщеплением цикла по
двойной связи и полиеновой цепи по двойным связям, оксикаротийы — с
присоединением двух гидроксильных групп по двойной связи цикла и каро-
тиноиды с различной степенью дегидрирования циклической части моле-
кулы.
Необходимо отметить, что способностью к образованию эпокисей обла-
дают лишь те двойные связи, которые находятся в сопряжении. Изолирован-
ная двойная связь а-каротина не образует эпокисей.
Окисление каротина начинается с наименее устойчивой двойной связи
ядра и постепенно распространяется по пространственно наиболее затруд-
ненным непредельным связям. Центральная двойная связь каротина, сво-
бодная от всяких помех благодаря двустороннему электронному взаимо-
действию полиеновой сопряженной цепи, по своему характеру приближает-
ся к одинарной связи и наиболее устойчива к окислению.
Кислоты не оказывают влияния в отсутствие воздуха, однако в присут-
ствии кислорода сильно ускоряют окисление и распад каротиноидов; пред-
варительно происходит изомеризация образующихся промежуточных сое-
динений [3461.
В синтетических условиях эпокиси и «фуранокиси» получаются при
обработке провитаминов моноперфталевой кислотой или другими органичес-
кими перекисями [347 ]. Эпокиси изомеризуются в фуранокиси под влиянием
следов хлористого водорода в хлороформе. Марганцовокислый калий пер-
воначально окисляет одно циклогексеновое кольцо с постепенным расщеп-
лением молекулы по пространственно затрудненным двойным связям с
образованием (из 0 -каротина) 0-апокаротиналей [328] (схема 46).
7—69
193
Схема 46
Окислительное расщепление 0-каротина
Р-.Каротин
CLX
СН,
I 3
СН,
I 3
сн3
•Н г--” сн3
н3 II . .
>сн=сн-с=сн-сн=сн-с=сн-сн=сн-сн=с-сн=сн-сн=с-сн=сн
СНз
СН3
Перокись р-каротииа
Ч^СИз
сн3сн3
СН3
__т —„ СН3
СН3 СНо 1
£н=сн-с=сн
СН3
[-СН =сн - с=сн-сн =сн-сн=с-сн=сн-сн=с
•СН3
фуранокись р-каротина
сн3
СНз СН3 сн, сн3 _
СН3 СНз I 3 ! 3 I 3 I 3
" ЯН=СН-С=СН-СН=СН-С=СН-СН=СН-СН=С-СН =сн-сн=с - cf
н
СН3
р-апо-8-Каротиналь (С3о)
сн3
СНз
сн3
I 3
СН3СН3
,СН=СН-С=СН-СН=СН-С=СН-СН=СН-СН=С-СН=СН-С'
СНз
р-апо-10-Каротиналь (С27)
сн3
?Нз.о
№ сн3 7Нз
сн=сн-с=сн-сн=сн-с=сн-сн=сн-сн=с-сС
н
•сн3
р-апб-12-Каротииаль (С25)
сн3
CH3JZH3 сНз
1H=CH-C=CH-CH=CH-C=CH-CHO
сн3
₽-апо-15-КаротиниЛь (Cjq)^
витамин А-альдегид
XVIII
Кроме представленных на схеме 46 каротиналей при окислений об-
разуется небольшое количество р-апо-14'-каротиналя (укорочение полие-
новой цепи на две, четыре п т. д. двойные связи обозначается приставкой
«апо» и цифрой, соответствующей нумерацииатомов углерода).
Из 0-каротина при окислении воздухом в присутствии двуокиси марган-
ца образуется ретиналь (XVIII) [348, 349].
о
Н
194
В животном организме р -каротин превращается отчасти в слизистой ки-
шечника через р-апо-8'-каротиналь (С^) и р-апо-12'-каротиналь (С25) в р-
-апо-15-каротиналь (ретиналь, XVIII) и затем с последующим восстановле-
нием — в витамин А (I) [310].
При мягком окислении р-каротина (CLX) в смеси петролейного эфира,
спирта и эфира взбалтыванием с двуокисью свинца (или двуокисью марган-
ца) при 26° С в течение 5 мин образуется витамин А (I) с выходом несколько
более 50% [350].
При каталитическом окислении р-каротина (CLX) перекисью водорода
в эфире в присутствии OsO4 [351, 352] происходит более глубокое окисление
с образованием: ретиналя (XVIII) с выходом 30%, р-ионилиденуксусного
альдегида (альдегида С15) (LXXIII) и 2,7-диметилоктатриендиаля (CLXV)
[310] из центральной части молекулы р-каротина.
СН3 СН3 ?Нз О
хн=сн-осн-с
СХ н
Хчх"сН3 LXXIII
СН3 сн3 л
С) I 3 1 3 °
^С-С=СН-С‘Н=СН-€Н= с-с '
н н
CLXV
В тех же условиях 100 мг 5,6-моноэпокиси р-каротина дает: 22 мг рети-
наля (XVIII), 10 мг 5,6-эпокиси ретиналя, 12 мгр-апо-10'-каротиналя и др.
[353].
При окислении p-каротина первоначально хромовой кислотой, а затем
тетраацетатом свинца [354] образуется биологически активный Р-семикаро-
тинон, в свою очередь окисляющийся под влиянием тех же реагентов в тет-
ракетон—p-каротинон. При окислении p-каротинона хромовой кислотой
/получается дикетоальдегид — р-каротинональдегид [328].
СНз сн, СНз сн,
СН3j;h3 1 1 3 1 1 3 СН3 сн,
н=сн-с=сн-сн-сн-с-сн-сн=сн~сн=с-сн=сн-сн=с-сн=сн >< *
р-Семикаротинон
О=<
СНз
сн3 сн3
Сн3
сн3
сн3
сн3
сн=сн-с=сн-сн=сн-с=сн-сн=сн-сн=с-сн=сн-сн=с-сн=сн
сн3
;р-Каротинов
СНосн3
O-C>S
О=С^>
СН3
сн3
сн3
сн3
СНз СНз
><_^СН=СН-С=СН-СН=СН-С=СН-СН=СН-СН=С-СН = СН-С'
Г
сн3
-Каротинональдегид
о
н
Каротиноиды кетоны легко образуют оксимы с гидроксиламином.
Окисление разбавленными растворами хромовой кислоты приводит к
образованию оксикаротинов, у которых по двойной связи циклогексенового
ядра присоединены две гидроксильные группы, как, например, у окси-р-ка-
ротина (CLXVI).
СИ СН, СН3 CH3 СНд
сйз£н3 I J I 3 I I СН3 СН3
^CH=CH-.C=CH-CH=CH-;C=CH-CH=CH-CH=C-CH=CH-CH=C-CH=Crj
"ЮН
>он
сн3
-CLXVI
СНз
7*
195
При окислении каротиноидов хромовой кислотой выделено и много дру-
гих веществ, в том числе и неизвестной структуры [355].
Фотосенсибилизированное окисление £-каротина кислородом дает
5-окси-8-оксо-, 5-окси-7,8-дегидро-,5,8-эпокси-8-оксикаротиноиды и др. с не-
изменной остальной частью молекулы [356}.
При мягком дегидрировании £ -каротина (CLX) образуются каротины
с дополнительными двойными связями: изокаротин (дегидро-£-каротин)
(CL XVII) [357 ] с 12 двойными связями и со смещенной полиеновой системой
типа ретроионилиденовой системы, 3,4; 3',4'-бпс-дегидро-£ -каротин
(CLXVIII) с 13 двойными связями и бис-дегид роизокаротин с 14 двойными
связями. Эти вещества могут быть получены при дегидрировании £-каротина
N-бромсукцин имидом в четыреххлористом углероде [358].
и сн 9нз 9нз сн3 сн3 гн гн
(Н3 СНз • 1 I I СН3 сн3
,СН=СНтС=СН-СН=СН-С=СН-СН=СН-СН=С-СН=СН-^СН=С-СН=СН.
сн3
CLXVIII
сн3
Формулы, температуры плавления, максимумы спектра поглощения
[53] окисей а- и £-каротина и окисей криптоксантина [347, 359], многие из
которых встречаются в природе, приведены в табл. 14.
Таблица 14
Окиси а- и ^-каротинов и криптоксантина
СН3 СН3 СНа CHS
В формулах К=—i=CH—СН=СН—С=СН—СН=СН—СН =С СН=СН—СН=С—
9 15 15' 9'
196
Название
Аурохром —
С^НзвОг —
желто-оранже-
вые листочки
а-Каротин-5,6-
моноэпокнсь—
С40Н5вО —крас-
новато-желтые
пластинк и
Р-Каротин-мо-
ноэпокись —
С40Н56О —оран-
жевые листочки
Р-Каротин-диэ-
покись —
С40Н5вО2 (син-
тетический) —
желто-оранже-
вые листочки
Криптоксантин-
эпокись I —
С4оНбв08
Криптоксантин-
эпокись II —
С4оН5602
1
Структурная формула
СН3 CHS
СН
СН
СНз СНз
СН3
СНз^СНз СНз СН3
П- СН=СН—(К)-СН =СНу / \
ги О I I
>3
СН3
СН3 СН»
СН3СН3
Окиси криптоксантина
СНз СН3
CHS СН3
СН3 СНа
Спектр
поглощеяия‘к
^макс, нм
185 а) 457, 428
175 (в
вакууме)
а) 503, 471
б) 483, 454
в) 471, 442
160
а) 511, 479
в) 478, 447
184 в) 470, 443
а) 512, 479
197
Напакие
Структурная формула
Спектр
поглощения*.
хмакс .им
Криптоксантин-
диэпокись I —
С40НБвО2
Криптофла-
вин 1 —
CaoHsjOj
СН3 СН3
СН3 СН3
СН;
194
171
а) 503, 473
а) 490, 453
Криптофла-
вин II —
СаоН&вОа
1-СН3
СН3
а) 456, 424
Криптохром —
CaoHsaOj
1 а) в сероуглероде, б) в хлороформе, в) в петролейном эфире.
СТРОЕНИЕ ПРОВИТАМИНОВ
Впервые каротин выделил из моркови (Daucus carota) Вакенродер в
1831 г. Эмпирическую формулу ^-каротина определил Вальштеттер в 1906 г.,
а его строение установили Цехмейстер [360], Каррер [361] и Кун [362] с
сотр. только в 1928—1930 гг.
Присутствие ва-,0- и у-каротинах (CLIX, CLX, CLXI) полиеновой угле-
родной системы двойных связей установлено реакцией исчерпывающего ка-
талитического гидрирования, протекающей с образованием пергидрокаро-
тинов. По количеству присоединенного водорода в молекуле а-каротина
найдено 11 двойных связей, в молекуле 0 -каротина — также 11 двойных
связей [360] и у-каротина — 12 двойных связей [342]. Иза- и0-каротинов
образуется один и тот же пергидрокаротин.
При озонировании 0-каротина (CLX) образуются 2 молекулы героновой
кислоты (XXXIV), что соответствует двум остаткам цикла 0-ионона; на
остатки цикла 0-ионона приходятся две двойные связи (схема 46 а).
198
Схема 46 а
Установление строения
{3-каротина реакциями расщепления
СН, СН3 о
О Н
О
СН3
СН3 ^СНз
eft ^СООН
JL, _-*О
СН3 СН,
О,
сн3
СН3 CHj
СН2 СН3
Героновая кислота
XXXIV
СН,
СООН
CLXIX
СН, ^СН3
С(£ "'СООН
соон
СН3 СН,
- ^с^
соон соон
CLXX
СН,
СНз^Ив
,СН=СН-С=СН-СН=СН-С=СН-СН=СН-СН=С-СН=СН-СН=С-СН=СН
сн,
р-Каротин
CLX
сн;
Изопреноидные группы
cit |-----сн3-|
=СН-С=СН-СН=СН-С=СН-СН=СН-СН=С-СН=СН-СН=С-СН=
{ее ! 11 13 I 15 15' | и' if i 9* 8'!
‘з
'з
у Центральная часть; молекулы каротина
Термическое : расщепление
t I
,сн сн
С^ ^CHlf*CH
I >
СН СН
исн сн,3сн3
CLXXI
9* 8'
I 8 8
Это подтверждается деструктивным окислением молекулы р-каротина
марганцовокислым калием в щелочном растворе, которая в результате пос-
тепенного укорачивания полиеновой цепи с промежуточным образованием
каротинональдегидов (см. с. 194), помимо героновой кислоты (XXXIV),
даеттакжеа, а-диметилглутаровую кислоту (СЕХ1Х),а,а-диметилянтарную
кислоту (CLXX) и диметилмалоновую кислоту; эти кислоты образуются из
циклогексановых циклов. Разделением хроматографией на бумаге продук-
тов окисления В-каротина от исходного соединения найдено: соедине-
ния CLXIX 27%, CLXX 26% и диметилмалоновой кислоты —следы [363].
Остальные девять двойных связей находятся в сопряженной системе
между 18 атомами углерода, составляющими симметричную центральную
часть молекулы, по концам которой расположены кольца р-ионона. Цент-
ральная часть молекулы имеет ответвления в виде четырех метильных групп,
что устанавливается по образованию четырех молекул уксусной кислоты при
окислении р-каротина марганцовокислым калием. Получение уксусной
кислоты указывает также на наличие ненасыщенной группировки типа.
=С(СН3)—СН= [364]. Еще две метильные группы обнаруживаются в
Р -иононовых циклах при более жестком окислении хромовой кислотой
[361, 365]. В центральной части молекулы метильные группы находятся
в положении 1,6, что следует из получения 2,6-диметилнафталина (CLXXI)
при реакции термического расщепления [3661, при этом в образовании аро-
матического цикла участвует цепь из 10 атомов углерода. Метильные груп-
пы в боковой части алифатической цепи находятся в положении 1,5, что до-
199
называется образованием jw-ксилола при реакции термического расщепле-
ния алифатической части каротиноида ликопина [362]. Таким образом,
центральная часть молекулы 0-каротина (CLX) состоит из четырех конден-
сированных попарно симметрично расположенных изопреноидных групп.
При расщеплении молекулы а-каротина (CLIX) озоном обнаруживается
присутствие одного кольца 0-ионона и одного кольца а-ионона (CLXXII),
что установлено по образованию героновой (XXXIV) и изогероновой кис-
лот (CLXXIII). Наличие одной изолированной двойной связи в триметил-
циклогексановом кольце, отвечающее структуре кольца а-ионона, подтвер-
ждается также оптической активностью и положением максимума поглоще-
ния ультрафиолетового света. В остальной своей части строение а-каротина
соответствует р -каротину и доказывается подобными же реакциями [360,
365, 367].
CLXXIII
•у-Каротин (CLXI) при расщеплении озоном образует одну молекулу ге-
роновой кислоты (XXXIV), что указывает на присутствие кольца р-ионона.
Вместо второго кольца находится открытая алифатическая часть молекулы
с тем же количеством атомов углерода, отвечающая структуре псевдоионона
(имеющейся также в каротиноиде ликопине); это подтверждается образо-
ванием одной молекулы ацетона при озонировании и наличием 12-й изоли-
рованной двойной связи. Изолированная двойная связь отделяется от со-
пряженной системы кратных связей остатком с двумя насыщенными атомами
углерода, который при расщеплении марганцовокислым калием в щелочном
растворе образует одну молекулу янтарной кислоты. Остальная часть мо-
лекулы содержит полиеновую сопряженную систему из 12 двойных связей
(и одной в алифатическом остатке структуры псевдоионона), отвечающую
строению 0-каротина [342, 365].
ВЫДЕЛЕНИЕ КАРОТИНОИДНЫХ ПРОВИТАМИНОВ
Принцип извлечения каротиноидов из растительных или животных
источников основан на экстракции сухого измельченного сырья органичес-
ким растворителем с последующей отгонкой избытка растворителя из
экстракта; остаток подвергают обработке едкой щелочью с целью омыления
липоидных веществ и каротиноиды извлекают петролейным эфиром или гек-
саном. Экстракт смешивают с метиловым спиртом и после расслаивания по-
лучают два слоя: углеводородный, содержащий каротиноидные углеводоро-
ды, в том числе а-, 0- и -(-каротины, и метанольный, в котором заключаются
кислородсодержащие каротиноиды. Дальнейшее разделение каротиноидов
производят хроматографически по методу Цвета [111] на окиси алюминия
[368] или других адсорбентах с последующим избирательным вымыванием
смесью бензола и метанола или другими растворителями (см. с. 191).
0-Каротин в виде его концентратов или в кристаллическом виде полу-
чают из люцерны [369], моркови [3701 или тыквы.
В растениях каротиноиды находятся в смесях различного состава. Среди
каротиноидов моркови, люцерны и тыквы 0-каротин содержится в наиболь-
200
шем количестве наряду с небольшим количеством а-каротина (около 15%)
и ничтожным — 7-каротина (около 0,1%). Много а-каротина среди кароти-
ноидов красного пальмового масла, он составляет основную часть кароти-
ноидов листьев некоторых сортов чая.
В технике при извлечении каротина из люцерны одновременно исполь-
зуют ксантофилл и хлорофилл [369], который может быть подвергнут гид-
ролизу в фитол, необходимый для синтеза а-токоферола (см. с. 271) и фил-
лохинона (см. с. 235). Люцерну сушат, экстрагируют гексаном, избыток
растворителя отгоняют, а остаток в растворителе пропускают через актив-
ный уголь, из которого адсорбированные природные красящие вещества
фракционно вымываются.
Технология каротина из моркови заключается в измельчении ее, отжима-
нии сока, коагуляции белковых веществ морковного сока подкислением до
pH 4,2 или нагреванием до 80—90° С и адсорбции каротина образующимся
белковым осадком; из обезвоженного коагулята каротин экстрагируют мас-
лом или бензином [370]. Для получения 1 г каротинов (с содержанием око-
ло 15% а-каротина, 85% 0-каротина и 0,1% 7-каротина) необходимо около
15 кг моркови.
Для выделения а-каротина используют реакцию йодирования, приво-
дящую к получению растворимого дийод-а-каротина, в то время как дийод-
0-каротин выделяется в осадок. Из раствора а-каротин регенерируют обра-
боткой гипосульфитом натрия [367].
ПОЛУЧЕНИЕ р-КАРОТИНА ФЕРМЕНТАТИВНЫМ ПУТЕМ
Многие микроорганизмы могут продуцировать различные каротиноиды,
в частности двуполая культура Blakeslea trispora [371 ] продуцирует 0-ка-
ротин в значительном количестве — 18—20 мг в 1 г сухой биомассы.
СИНТЕЗ КАРОТИНОИДНЫХ ПРОВИТАМИНОВ
Синтез a-к а р о т и н а
dl-a-Каротин впервые синтезирован в 1950 г. [3721 из ацетиленовых кар-
бинолов С1в, полученных из а- и 0-иононов и октен-4-диона-2,7.
Схема 47
Синтез а-каротина
CLXXVIII
II-d-Каротин
СИХ
201
Для синтеза полного /npawc-dZ-a-каротина (CLIX) предложен метод [373]
(схема 47), в котором исходят из a-ионона (CLXXII), глицидным синтезом
его превращают в a-альдегид С\4 (CLXXVI) и затем реакцией Реформатского
с метиловым эфиром 7-бромтиглиновой кислоты — в эфир а-кислоты С19,
восстанавливаемый алюмогидридом лития в a-спирт С19 и затем окисляемый
двуокисью марганца в a-альдегид С19 (CLXXVII). Р-Альдегид С19 (CLXXIV)
(см. с. 204) конденсируют с броммагнийацетиленом в Р-этиниловый спирт
С21 (CLXXV), соединяемый с a-альдегидом С19 (CLXXVII) в a-диол С40
(CLXXVIII), дегидратация которого приводит к 15,15'-дегидро-^/-а-каро-
тину, затем частичное восстановление — к цис- и изомеризация под влия-
нием йода — к транс-Ш-а-каротину (CLIX).
dZ-a-Каротин синтезирован из 15,15'-дегидро-Р-апо-12'-каротиналя (С^)
(см. с. 212) конденсацией егос фосфораном, полученным из а-ионилиден-
этилтрифенилфосфоний-хлорида (С15) в 15,15'-дегидропроизводное а-каро-
тина, которое подвергают частичному гидрированию и изомеризации в пол-
ный mpa«c-a-каротин (CLIX) [318].
CLIX
Синтез Р-каротина
Структура Р-каротина (CLX) была известна еще в 1930 г., однако его пол-
ный синтез осуществили только в 1950 г. Каррер [374], Инхоффен [375]
и Майлас [376] с corp.
P-Каротин синтезирован по схеме; С10+С2о1-С10 = С40 из 2 молей фосфо-
рана (полученного из p-циклогеранилтрифенилфосфоний-бромида —
LXVIII, с. 162) и 1 моля кроцеиндиальдегида в среде диметилформамида
[377].
Если исходят из ацетиленового карбинола С16 (XCIV), получаемого из
0-ионона и пропаргилбромида (см. с. 166), то применяют его конденсацию
по реакции Гриньяра с октен-4-дионом-2,7 (CLXXIX) (см. с. 203) в диаце-
тиленовый тетрол с углеродным скелетом p-каротина, который подвергают
частичному гидрированию и дегидратируют n-толуолсульфокислотой. Про-
межуточные соединения получаются с гидроксильной группой в аллильном
положении к двойной связи ядра, что связано с ретроионилиденовой пере-
группировкой и образованием Р-каротина с небольшим выходом (меньше
3%) [374].
Для получения Р-каротина в качестве исходного вещества применяют
ацетиленовый углеводород Cln (XCVIII, с. 166), который конденсируют
с октен-4-дионом-2,7 (CLXXIX), что несколько упрощает синтез [375, 378—
380].
Показана возможность применить для синтеза Р-каротина кетон С18
(СХШ, с. 170); этот кетон конденсируют с димагнийдибромдиацетиленом
(BrMgC=C—C=CMgBr) в диацетиленовый гликоль Р-каротина, частичным
гидрированием переводят в гликоль и затем дегидратируют в р-каротин
[381 ] с небольшим выходом.
202
По одному из методов [376 ], дающих лучший результат, чем предыдущие,
0-каротин синтезируют из альдегида С14 (CXVI) и ацетиленового комплекса
Иоцича через ацетиленовый карбинол С16 (CXLII, с. 178), в котором гидрок-
сильная группа не находится в аллильном положении к двойной связи ядра,
чем избегается образование изомерных соединений (схема 48).
Схема 48
Синтез p-каротина из альдегида С14 через ацетиленовый карбинол Сц
СИ,
сн=с-сно
BrMgC=CMgBr
CXVI
I— _____________________I
СИ, CH, T \r,3
JX. CH,-CH=C-CH-C=CH
Cl^ CXLII
CH, сн,
OCCH2CH=CHCH2CO
CLXXIX CXLII
СИ, СН
, сн, сн,J “ghT
CH2-CH=C-CH-C=C-C-CH,-CH=CH-CH2-C-CSC-CH-C=CH
он OH OH OH
CH,
CLXXX
CH,
CH, CH,
5<,ch,-ch=c
CH,
I 3
CH,
CH,
CH,
1 3
:-CH-CH=CH-C-CH,-CH=CH-CH,-C-CH=CH-CH-C=CH-CH,
OH
OH
OH
OH
CLXXXI
CH,
CH, CH,
р-Каротин
CLX
Ацетиленовый карбинол Cie (CXLII) в виде магнийорганического сое-
динения конденсируют с ненасыщенным дикетоном С8 октен-4-дионом-2,7
(CLXXIX) в диацетиленовый тетрол С4() (CLXXX), ацетиленовые связи его
подвергают частичному каталитическому гидрированию в присутствии дез-
активированного палладиевого на углекислом кальцие катализатора в
полный полиеновый тетрол С40 (CLXXXI); дегидратация последнего с бро-
мистоводородной кислотой в кипящем пиридине [2801 приводит к получению
0-каротина (CLX). Вещество выделяется в кристаллическом виде в резуль-
тате хроматографической очистки на гидроокиси кальция [376].
Для получения ненасыщенного дикетона С8 (CLXXIX) исходят из транс-
дигидрокумонилхлорида, ацетоуксусной кислоты и глиоксаля или метил-
этинилкарбинола [382—385 ].
0-Каротин (CLX) синтезирован из 0-альдегида С19 (CLXXIV) конденса-
цией двух его молекул с ацетиленом [332, 386].
Необходимый для синтеза каротина кристаллический 9,13-ди мети л-7-
(1,1,5-триметилциклогексен-5-ил-6)октатриен-8,10,12-аль-14, 0-альдегид
С19 (CLXXIV), получают [387, 388] (схема 49) из альдегида С14 (CXVI),
который конденсируют с магнийорганическим производным метоксиметил-
этинилкарбинола (CLXXXII) в ацетиленовый триол С19 (CLXXX1II), затем
это соединение в результате аллильной перегруппировки, частичного ката-
литического гидрирования и дегидратации с помощью п-толуолсульфокис-
203
лоты через соединение CLXXXIV превращают в производное винилового
карбинола (CLXXXV), которое при действии разбавленной серной кислоты
перегруппировывается с небольшим выходом в альдегид С19 (CLXXIV).
Схема 49
Синтез альдегида С1в
ск . сн3
СН3
CHj-CH-C-CHO СН3
+ НС=С-С-СН2ОСН3 -------—
он
CLXXXK
СК снз СН3
сн2-сн-с=сн-сн=<
он
CH’cxvi
сн3 ^сн3 Vм’ Vм’
сн2-сн=с-сн-с®с-с-сн2осн3 йНк а)од
ОН ---------------------------:---
он
сн3
сн3
СН-С-СЦОСНз
он
-ир
CLXXXIII
сн3
сн3
СН3 СН3
СПз сн. сн3
СН=СН-С=СН-СН=СН-С=СНОСК ,, г,
3 Н2О
‘Г LI
3 CLXXXV
CLXXXIY
сн3
CHj-CH=C-CH=CH-CH=C-CHO
р- Альдегид Св
Нз CLXX1V
сн3
Для синтеза альдегида С19 можно исходить из ацеталя альдегида С14
и конденсировать его с 1-этокси-2-метилбутадиеном-1,3 [389].
По другому варианту исходят из 1,2-диэтокси-4-бром-2-метилбутена-2
(ацеталя -рбромтиглинового альдегида), получают из него по реакции Вит-
тига соответствующий фосфоран (CLXXXVI), который вводят в конденса-
цию с альдегидом С14 (CXVI), и образовавшийся ацеталь С19 (CLXXXVII)
разбавленной фосфорной кислотой количественно гидролизуют в альдегид
С19 (CLXXIV) [3901:
CHS
+ - I
+ (CjHjJsP—СН—СН=С—CH(OR,) -►
CLXXXVI
CHs CH, CHS СНз
Н8-СН=С-СН-=СН—СН=С—CH(OR), ->
СНз
CLXXXVII
CLXXIV
Имеющий техническое значение синтез р-каротина [391 ] основан на спо-
собе наращивания полиеновой цепи путем конденсации ацеталей с винило-
выми эфирами, приводящем к получению альдегида С19, который конденси-
руют затем с ацетиленом [3321.
204
Первоначально из Р-ионона (XXXVI) глицидным синтезом и перегруппи-
ровкой получают альдегид С14 (CXVI, с. 176), который в две ступени одно-
типных реакций превращают в альдегид С16 (СХС) и затем в р-альдегид С19
(CLXXIV, схема 50).
Альдегид С14 (CXVI) при действии ортомуравьиным эфиром в присутст-
вии каталитических количеств n-толуолсульфокислоты превращают в а-
Р-ненасыщенный ацеталь альдегида С14 (CLXXXVIII), который конденси-
руют с виниловым эфиром в присутствии 10%-ного раствора хлористого
цинка или эфирата трехфтористого бора в эфироацеталь С16 (CLXXXIX),
затем кипячением с уксусной или фосфорной кислотой этот эфироацеталь
гидролизуют и получают кристаллический альдегид С1в (СХС) с т. пл. 78—
79° С. Все эти превращения (ацетилирование, конденсация и гидролиз)
с хорошим выходом протекают также без выделения промежуточных соеди-
нений в одну стадию [336].
Схема 50
Технический синтез ^-каротина из альдегида С14 через альдегид С18
СН, СЦ,
" ' СН,-СН=С-СНО
92%
CXVI
65% I CH^CHOR
I сн3
CH, CH, CH3OR
CHfCHOR
’ OR 91%
CH,
’ CLXXXVIII
СН, СН, СНз OR
' ' cil-ch=c-chcil-ch
Pi сС I .
OR OR
СН,
3 CLXXXIX
СН3 CHj
сн,
I, '
сн,-сн=с-сн=сн-сно
сн3
<!:h=chor
CH3 CXC
CH,
CH=CHOR
CH, CH3 ' ^н»
" ' CH2-CH=C-CH=CH-CH=C-CHO
CH3 CH, CH, CHjOR
" ' ch,-ch=c-ch=ch-ch-ch-ch
OR OR
CHj CXCI
68 - 73%
+ сн=сн
Альдегид С19
+ Альдегид Си
CLXX1V
CLXXIV 99%
сн,
CH,
J 3
CH,
сн=сн-сн=с-сн-с=с-сн-с=сн-сн=сн-с=сн-сц1.
сн;
ch, ch,
OH OH
CXCII
CH CH CH, |85%
'-”3 .ч-г,з I I '
><>CH=CH-C=CH-CH=CH-C=CH-C=C-CH
^Nh,
CXC1IL"
CH,
=c-ch=ch-ch=c-ch=ch
CH,
сн,
CH, CH
CH3 CH.
CH,
CH,
CH,
3 . . .
CH=CH-C=CH-CH=CH-C=CH-CH=CH-CH=C-CH=CH-CH=C-CH=CH
CH.
в-Каротин
CLX
CH,"
CH3 CH,
|i»* CH,
При присоединении виниловых эфиров к ацеталям а, t3-непредельных кар-
бонильных соединений происходит внутримолекулярная перегруппировка
одной алкоксигруппы от ацеталя к двойной связи винилового эфира с одно-
временным присоединением остатка ацеталя к метиленовой группе вини-
205
левого эфира [391 ]. В то же время образующиеся а, насыщенные ацетали
в условиях реакции к взаимодействию с виниловыми эфирами не способны,
что обеспечивает однозначность реакции и высокий выход конечного про-
дукта.
Таким же путем через а, p-ненасыщенный ацеталь альдегида С16 и про-
дукт его конденсации с пропениловым эфиром—эфироацеталь С19 (CXCI)
или непосредственно из альдегида Clfi (СХС) с применением пропенилового
эфира получают альдегид С19 (CLXXIV) с т. пл. 66—68° С.
Альдегид С19 (CLXXIV) конденсируют с ацетилендимагнийдибромидом
в диол С49 (СХСП). В конденсацию с ацетиленом по реакции Гриньяра вво-
дят две молекулы альдегида С19 или первоначально одну молекулу альдегида
С19 конденсируют с ацетиленидом лития в жидком аммиаке в ацетиленкарби-
нол С21, который затем по реакции Гриньяра со второй молекулой альдегида
С19 превращают в диол С40 (СХСП) [336]. Затем производят дегидратацию
диола С40 (СХСП), причем одновременно с отщеплением двух молекул воды
от диола С40 в присутствии галогеноводородной кислоты происходит аллиль-
ная перегруппировка с установлением цепи сопряженных двойных связей
и образованием 15,15'-дегидро-Р-каротина (CXCIII) в виде оранжево-жел-
тых листочков ст. пл. 150° С. Промежуточным соединением при обработке
60%-ной бромистоводородной кислотой в течение 1,5 мин при —30° С явля-
ется неустойчивый бромид 15,15'-дегидро-Р-каротина, теряющий элементы
бромистого водорода при воздействии воды. У соединения CXCIII частич-
ному каталитическому гидрированию с ослабленным катализатором Линд-
лара [77 ] подвергают центральную тройную связь. При этом образуется ла-
бильный 15,15'-г{ис-Р-каротин (см. формулу CLXII на с. 191) в виде длин-
ных оранжево-красных призм с характерным цис-пиком при 338 нм. Его
подвергают изомеризации в стабильный полный /7гранс-0-каротин (CLX)
прй нагревании суспензии в петролейном эфире при 90—100° С в присутст-
вии следов йода на свету в течение 10 ч, выделяемый в виде темно-красных
кристаллов с т. пл. 179—180°С [336]. Полностью и с хорошим выходом
изомеризация протекает также в растворах на свету [392]. Для получения
15,15'-дегидро-|3-каротина (CXCIII) известны и методы, использующие фос-
форорганические производные [393].
0-Каротин (CLX) получен также, исходя из ацеталя альдегида С14 и
симметричного диенолина С12 [394]. Конденсацией двух молекул тиглино-
вого альдегида (CXCIV) с ацетилендимагнийдибромидом получен 4,7-ди-
окси-3,8-диметилдекадиен-2,8-ин-5, диенолин С12 (CXCV), который в резуль-
тате аллильной перегруппировки под влиянием кислот и последующего оки-
сления двуокисью марганца дает 3,8-диметилдекадиен-3,7-ин-5-дион-2,9,
дикетон С12 (CXCVI), с т. пл. 108—109° С. Диенолин С12 (CXCV) с лучшими
результатами можно превратить в дикетон С12 (CXCVI) через нитроновый
эфир [127]. При действии на дикетон С12 (CXCVI) ортомуравьиного эфира
получен дикеталь, переходящий под влиянием хлорокиси фосфора или смеси
уксусного ангидрида и пиридина в эфир 2,9-диокси-3,8-диметилдекатетра-
ен-1,3,7,9-ина-5, тетраенолина С12 (CXCVII), с т. пл. 60° С (схема 51).
Эфир тетраенолина С12 (CXCVII) конденсируют с двумя молекулами
а, P-ненасыщенного ацеталя альдегида С14 (CLXXXVIII) под влиянием хло-
ристого цинка по методу конденсации с виниловыми эфирами, изложенному
выше. Диэфир р-дикеталя С40 (CXCVI II) подвергают гидролизу; при отще-
плении спирта по а, 0-положению возникает новая двойная связь, в резуль.
тате чего образуется Р-дикетон С40 (CXCIX) с т. пл. 172—173° С. Восста-
новление последнего соединения изопропилатом алюминия приводит к
Р-диолу С40 (СС), затем в результате аллильной перегруппировки и отщепле-
ния воды под влиянием спиртового раствора хлористого водорода — к
15,15'-дегидро-р-каротину (CXCIII) и в результате изомеризации — к 0-ка-
ротину (CLX, схема 51) [394]. Выход 0-каротина около 60%, считая на
ацеталь альдегида C14(CLXXXVIII).
206
' Схема 51
Синтез 0-каротина из альдегида С14 и диеиолина Сц
CXCV1
сс
схсш
транс —0-Каротин
CLX
Реакция Виттига была применена для синтеза 0-каротина по схеме:
Ci54- Ci0+C15 = С40. Этиленовый карбинол (ХС) вступает в реакцию с три-
фенилфосфином в метанольном растворе хлористого водорода и диметилфор-
мамида, образовавшийся 0-ионилиденэтилтрифенилфосфоний-хлорид С15
(XCI) при действии метилата натрия дает соответствующий С15-фосфоран
(ХСП); эти превращения указаны на схеме 35; С15-фосфоран (ХСП) и 2,7-ди-
метилоктатриен-2,4,6-диален-1,8 (СО) конденсируют в полный /пранс-0-ка-
ротин (CLX) с выходом около 60% [395].
СН3 СНз СНз СН3 СНз
/Сн=сн—с=сн—сн^(СвН6)3 + онс-с=сн—сн=сн—сн=с—сно-
ха I
CCI
Полный транс-
0-каротин
CLX
207
По схеме C^+C^ = С40 полный транс-0-каротин получен по реакции
Виттига из 15,15'-дегидро-0-апо-12'-каротиналя и 0-ионилиденэтилтрифенил-
фосфоний-хлорида С15 (XCI) с последующим гидрированием образовавшего-
ся 15,15'-дегидро-0-каротина и термической изомеризацией [318].
Интересный синтез 0-каротина состоит в конденсации ретинола и ретина-
ля по реакции Виттига [395—397]. Первоначально из ретинол-ацетата
(XIII) с трифенилфосфоний-сульфатом в метанольном растворе получают
0-ретинилтрифенилфосфоний-сульфат (GCII), который после обработки спир-
товым едким кали вводят в реакцию с ретиналем (XVIII); выход 70%.
сн, сн, Vм» f”3
г></СН=СН-С=СН-СН=СН-С=СНСН2ОСОСН3
Витамин А(1) превращают в 0-каротин в результате димеризации двух
молекул ретинилтриметилфосфорана (получаемого из 0-ретинилтрифенил-
фосфоний-сульфата) под влиянием кислорода [398].
Следует остановиться на синтезе 0-каротина [399], в котором исполь-
зуется промежуточный продукт технического синтеза ретинол-ацетата (XIII)
из альдегида С14 (CXVI) и 3-метилпентен-2-ин-4-ола-1 (CXLV) — диолин С^
(CXLVI) [45] — по схеме 42. Из этого соединения с помощью трехбромис-
того фосфора получают дибромид (ССШ) с одновременной перегруппиров-
кой двойной связи, затем обработкой триэтилфосфитом его превращают в
соответствующий замещенный диэтилфосфонат (CCIV) с выходом 70% на
исходный диолин С2о. В результате селективного гидрирования ацетилено-
вой связи по двойной с катализатором Линдлара из соединения CCIV по-
лучают ll-quc-ретинилфосфонат (CCV), который в результате Михаэль
Арбузовской реакции конденсируется с ретиналем (XVIII) в присутствии ме-
тилата натрия в пиридине при 0°С с образованием /71рапс-0-каротина (CLX)
с выходом 61% на соединение XVIII [399].
Следует отметить также и другие синтезы 0-каротина, в которых приме-
няют витамин А-альдегид (XVIII). По одному из них первоначально из ке-
тона С1е (СХШ) при действии ацетиленида лития в среде жидкого аммиака
получают ацетиленовый карбинол С2о, который по реакции Гриньяра кон-
денсируют с ретиналем (XVIII) в диолин С40, подвергаемый затем восста-
новлению алюмогидридом лития в присутствии ди метила нил и на в 0-каротин
(CLX) с выходом 10% [400].
208
При действии сероводорода ретиналь (XVIII) в растворе амина замещает
атом кислорода на атом серы и дает тиопирановое производное, которое пос-
ле обессеривания металлами превращается в 0-каротин [401 ].
Синтез 7-к аротина
Полный синтез 7-каротина (CLXI) осуществлен [402] одновременной кон-
денсацией диброммагниевых производных ацетиленового карбинола С16
(XCIV и ациклического ацетиленового карбинола С16 (CCVI) с ненасыщен-
ным дикетоном С8, октен-4-дионом-2,7 (CLXXIX), в диацетиленовый тет-
рол С40 (CCVII), образующийся наряду с тетролом 0-каротиноидного ря-
да (схема 52/.
В соответствии с общим правилом, что при частичном каталитическом
гидрировании ацетиленовой связи образуется двойная связь tjuc-конфигу-
рации, в результате частичного восстановления диацетиленового тетрола
(CCVII) в соединение CCVIII и отщепления воды образуется 11,12; 11',
12'-ди-1{ис-'[-каротин (наряду с ди-г{ис-0-каротином). Под влиянием следов
йода происходит изомеризация с образованием полного транс-7-каротина
(CLXI), который с небольшим выходом отделяют хроматографическим мето-
дом от примеси 0-каротина.
Полный транс-7-каротин синтезирован с применением реакции Виттига
из 15,15'-дегидро-0-апо-12'-каротиналя (С25) и псевдоионилиденэтилтри-
фенилфосфоний-хлорида (С15) с последующим частичным гидрированием
образовавшегося 15,15'-дегидрокаротина и изомеризацией [395] или из 0-апо-
8'-каротиналя (С^) и геранилтрифенилфосфоний-бромида [327].
Синтез 3,4-м о н о д е г и д р о- и 3,4;3',4'-б и с -д е г и д р 0-0-ка-
ротинов
3,4-Монодегидро- и 3,4; 3',4'-бнс-дегидро-0-каротин (CLXVIII, схема 53)
[320, 403], являющиеся провитаминами А2, синтезированы из бнс-дегидро-
альдегида Clg (CCXIV) [320], который получают из альдегида С14 (CXVI)
[208]. Для введения в циклогексеновое кольцо альдегида С14 второй двойной
связи использована реакция образования ретроизомерной полиеновой сис-
209
Схема 52
Синтез 7-каротина
сн.
сна сн,
сн=сн-с-сн,-с=сн
он
сн,
3 XCIV
сн3
СН,
нс=с-сн.-с-сн=сн -сн
л а It
сн,
он
О=С-СН2-СН=СН-СН2-С=О
CLXX1X
CCV1
сн, сн,
сн
сн, сн,
СН, сн, СНз I СЦ. сн,
5<г,сн=сн-с-сн2-с=с-с-сн2-сн=сн-сн2-с-с=с-сн,-с-сн=сн-сн ^сн
[ JL ОН ОН ОН ОН ХСН ^СН,
^-'''сн, сн, СН2
CCV11
сн, сн,
сн,
I d
сн,
СН,
СН, J7I1,
... _.( *сн
J2H J2H,
СН, ''сн,
ч.сн=сн-с-сн2-сн=сн-с-сн,-сн=сн-сн2-с-сн=сн-сн,-с-сн=сн-сн
11 ОН ОН ОН ОН ^СН
СН,
ccvm
сн,
СНз
СН,
сн.
СН, сн,
" " СН=СН-С=СН-СН=СН-С=СН-СН=СН-СН=С-СН=СН-СН=С-СН=СН-СН
сн,
-у-Карелии
С.
______ СН
II I
J7.ll ^СН,
сн, "сн,
CLX1
темы в результате отщепления элементов бромистого водорода и последую-
щей перегруппировки семициклической двойной связи в ядро. Для броми-
рования альдегида С14 (CXVI) N-бромсукцинимидом используют повышен-
ную подвижность водорода метиленовой группы положения 7, активиро-
ванной двумя двойными связями, по сравнению с подвижностью атома
водорода положения 4 циклогексенового кольца, используемой в синтезе ви-
тамина А2 из эфира ретиновой кислоты. Реакцию бромирования проводят
в среде метиленхлорида при 5—10° С. Отщепление бромистого водорода
происходит при обработке соединения CCIX третичным основанием, напри-
мер хинолином, в вакууме. Образующийся дегидроретроальдегид С14 (ССХ)
превращают в енолацетат (CCXI) при действии изопропилинилацетата в
присутствии следов n-толуолсульфокислоты и затем в результате обработки
бикарбонатом натрия получают 3,4-дегидроальдегид С14 (ССХ II) [208].
Реакции превращения 3,4-дегидроальдегида С14 (ССХ II) в дегидроальде-
гид С19 (ССХIV) через дегидроальдегид С16 (ССХ III) протекают аналогично
превращениям ацеталей с виниловыми эфирами в ряду р-ионилиденовых про-
изводных (см. с. 205) [208].
и
Синтез3,4;3',4'-бис-дегидро-Р-каротина (CLXVIII, схема 53) осуществлен
и методом введения двойной связи в ядро через ретроизомерные произ-
водные; в этом методе используется анионотропная перегруппировка гид-
роксильной группы с последующим отщеплением элементов воды, применяе-
мая дважды [403]. Из ацетиленового карбинола С15 (LXXXIX) (полу-
чение см. на схеме 35) и а-метилакролеина (CCXV) магнийорганическим син-
тезом получают диол С19 (CCXVI), который подвергают анионотропной пере-
группировке и дегидратации под действием серной кислоты в дегидроретро-
ацетиленовый спирт С19 (CCXVII); его окисляют активированной двуокисью
марганца в ацетиленовый альдегид С19 (CCXVIII),b котором с частично дез-
активированным палладиевым катализатором восстанавливают тройную
связь до двойной с образованием 5,6-дегидроретроальдегида С19 (ССХ IX).
Конденсацией двух молекул альдегида ССХIX с броммагнийацетиленом по-
лучают дегидроретродиол С40 (ССХХ), который в результате анионотропной
перегруппировки и дегидратации под влиянием п-толуолсульфокислоты
дает 3,4;3',4'; 15,15'-щрнс-дегидро-Р-каротин (CCXXI) с выходом около 5%.
Восстановление центральной ацетиленовой связи соединения CCXXI до
этиленовой с последующей изомеризацией приводит к 3,4;3',4'-бпс-дегидро-
p-каротину (CLXVIII) [403].
Схема 53
Синтез 3,4; 3',4'-бис-дегидроф-каротина через дегидроретроальдегид Ci9
сн3
сн, сн3
сн=сн-с-с=сн
сн3
I
он
LXXXIX
сн3
СНЭ
+ онс-с=сн2
CCXY
сн3
сн
СН3 СН3 Is
,сн=сн-с-с=с
сн3
он
сн3
-сн-с=сн2
он
сн, сн.
сн3
'з
CH-CH=C-C=C-CH=C-CHjOH
CCXVI
СН3
СН,
I л
3
сн-сн=с-ос-сн=с-сно
ССХ VII
сн3
сн3
сн, СНЭ
Х^сн-сн=с-сн=сн-сн=с-сно
сн3
3 CCXVIII
СН3. СН3
ОНС-С=СН-СН=СН-С=СН-СН
й
5ИГ
: I-
I
снэ
CCXIX
CCXIX
сн;
СН3
нс=сн
СН3
СНз
CHS
СН3 сн3
X^CH-CH=C-CH=CH-CH=C-CH-Csc-CH-C=CH-CH=CH-C=CH-CH
СН3 сн3
он он
сн3
ссхх
сн3
сн, сн, S43
СК*
сн=сн-с=сн-сн=сн-с=сн-ос-сн=с-сн=сн-<
СН, СН,
сн=с-сн=сн
сн3
CCXXI
сн3
сн3 сна
5</СН=СН-
СН3 СН3 | СН3 СН3 сн, сн^
с=сн-сн=сн-с=сн-сн=сн-сн=с-сн=сн-сн=с-сн=сн
сн3
сн3
CLXVIII
Этот же каротин был получен из дегидро-0-С14-ацеталя и эфира тетрадено-
лина С12 (CXCVII) 1394].
211
Синтез криптоксантина
Криптоксантин — З-окси-0-каротин (CCXXIV) — получен конденсацией
Р-альдегида С19 (CLXXIV; получение см. на с. 204) с ацетиленом по реакции
Гриньяра в Р-этиниловый спирт C2i (CLXXV) с последующим соединением
с З'-ацетокси-р-альдегидом С19 (ССХХП) в З'-ацетоксидиол С40 (ССХХШ).
Последующее частичное гидрирование, аллильная перегруппировка, дегид-
ратация и деацетилирование приводят к получению криптоксантина [404].
ОН
CCXXIV
Синтез P-а п о-8'-и p-а п о-12'-к а р о т и н а л е й и
этилового эфира p-а п о-8'-к аротиновой кислоты
Синтез щра«с-р-апо-8'-каротиналя осуществлен из Р-альдегида С19
(CLXXIV), который вводился в реакцию Гриньяра с ацеталем метилпенте-
нина С6; после дегидратации образовавшийся 15,15'-дегидро-апо-12'-каро-
тиналь конденсировался сначала с виниловым эфиром в 15,15'-дегидро-апо-
Ю'-каротиналь (С27) и затем с пропениловым эфиром, и после частичного вос-
становления и изомеризации получался полный пгранс-р-апо-8'-каротиналь
(С30) (3271.
Р-апо-8'- и Р-апо-12'-Каротинали синтезированы из ретинола или рети-
наля различными другими методами [311, 405, 406].
Из 15,15'-дегидро-апо-10'-каротиналя (С27) по реакции Виттига с фос-
фораном, полученным из трифенилфосфина и а-бромпропионового эфира с
последующим частичным восстановлением и изомеризацией, синтезирован
этиловый эфир полной транс-^-апо-8'-каротиновой кислоты [329J.
♦ * *
По химии витамина А и каротиноидов сделаны многочисленные обзоры
[2, 19, 46, 74 , 316, 393, 407—^10].
Каротиноиды являются провитаминами только в том случае, если при
расщеплении их молекулы может образоваться витамин А; в молекулу про-
витаминов включаются триметилциклогексеновое кольцо витамина А и
212
алифатическая цепь из 22 атомов углерода с девятью сопряженными двой-
ными связями изопреноидного характера.
Провитамины с двумя триметилциклогексеновыми кольцами витамина
А ф-каротин) обладают вдвое большей активностью по сравнению с прови-
таминами с одним кольцом (например, а-каротином).
Продукты окисления провитаминов — моно- и диэпокиси-, моно- и
дифуранокиси-, моноэпоксимонофуранокиси-5,6-диокси-, 5,6-дигидро-Р-ка-
ротины, моноэпокись а-каротина, 5,6-дигидро-а-каротин, р-апо-12'- и Р-апо-
8'-каротинали, р-апо-12' -и Р-апо-8'-каротинолы, Р-семикаротинон, содер-
жащие одно или два незамещенных триметилциклогексеновых кольца рети-
нола и боковую цепь большей длины, чем у витамина А, а также кетоны,
образующиеся при расщеплении одного кольца ретинола, обладают в 2—8
раза меньшей биологической активностью, однако при том условии, что одно
триметилциклогексеновое кольцо не подверглось изменениям.
P-Каротин с боковой цепью, в которой двойные связи находятся в транс-
конфигурации, обладает большей витаминной активностью, чем каротинои-
ды, имеющие отдельные двойные связи в qwc-конфигурации [334 , 411]
[табл. 15].
Таблица 15
Биологическая активность стереоизомерных каротиноидов
Каротиноид Конфигурация стереоизомера Витаминная активность при и< пытан in на крысах, % от активности полного транс-3-каротина
а-Каротин Полный транс 53
нео-а-Каротин U 9- или 9'-moho-z{uc- 13
нео-а-Каротин В 9,15- или 9,13'-ди-ч«с 16
3-Каротин Полный транс- 100
нео-р-Каротин U 9-MOHO-ijuc- 38
нео-р-Каротин В 9,15- или 9,13'-ди-цис 53
11,1 Г-ди-чнс-р-Каротин 30
р-Каротин-диэпокись Полный транс- 70
3,4-Монодегидро-р-каротин Полный транс- 75
3,4; 3',4'-бис-Дегидро-р-каротин Полный транс- 83.
7-Каротин Полный транс- 27
нео-7-Каротин Р 9-моно-чнс- 19
Смешанный нео-ркаротин Центр.-моно-чис- 16
про-7-Каротин Пента-чис 44
З-Криптоксантин Полный транс- 57
нео-Р-Криптоксантин U 9-моно-чис- 27
нео-Р-Криптоксантин А Центр.-моно-чис- 42
Ликопин, 6- и е-каротины, не содержащие в своей структуре триметил-
циклогексенового кольца ретинола, активности витамина А не показывают.
На основании многочисленных исследований можно считать, что живот-
ный организм усваивает около ^Р-каротина, имеющегося в пище или кормах;
по-видимому, только половина p-каротина превращается в организме в ви-
тамин А и, следовательно, только V6 Р-каротина, по равнозначности соответ-
ствующая 0,167 мг ретинола на 1 мг p-каротина, утилизируется организмом
в виде витамина.
P-Каротин и каротиноиды, содержащиеся в кормах природного проис-
хождения (ряда ксантофилла и особенно непровитаминные каротиноиды —
зеаксантин и лютеин), используются в кормлении для усиления окраски
желтка яиц и пигментации кожи и ног тушек цыплят. В корма добавляют
каротиноиды — 6 г/т.
Р-Каротин находит широкое применение в качестве красителя пищевых
жиров, например маргарина [412]. Как пигменты, имитирующие натураль-
213
ные цвета, для окраски пищевых продуктов используют 0-апо-8'-каротиналь,
этиловый эфир 0-апо-8'-каротиновой кислоты, а также другие каротиноиды.
Каротиноиды широко распространены в растениях, особенно в листьях
и плодах 1316]; они играют важную роль в обмене веществ в эпителиальных
и других растительных клетках. Возможно, что каротиноиды выполняют
роль светофильтра в механизме поглощения поверхностью листа лучистой
энергии определенной длины волны, необходимой для фотосинтеза орга-
нического вещества, и защищают хлорофил от фотоокисления [413, 414].
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ (К ГЛАВЕ IV)
1. L. Pauling. Fortschr.
Chem. org. Naturstoffe, 3, 203
(1939).
2. L. Zechmeister. Chem. Revs.,
34, 267 (1944); Vitamins and Hor-
mones, 7, 57 (1949).
3. W. G r a h a m, D. v a n Dorp,
J. Arens. Rec. trav. chim., 68,
609 (1949).
4. C. Robeson, J. Cawley,
L. W e i s 1 e r, M. Stern,
C. Eddinger, A. Chechak.
J. Am. Chem. Soc., 77, 4111 (1955).
5. W. О г о s h n i k. J. Am. Chem.
Soc., 78, 2651 (1956).
6. C. R о b e s о n, J. В a x t e r. J.
Am. Chem. Soc., 69, 136 (1947).
7. R. Morton, M. Salah,
A. Stubbs. Nature, 159, 744
(1947).
8. K. Farrar, I. Hamlet,
H. H e n b e s t, E. J о n e s. J.
Chem. Soc., 1952, 2657; Chem. and
Ind., 1952, 49.
9. O. S c h w i e t e r, G. S a u c y,
M. Montavon, C. von Plan-
ta, R. R ii e g g, О. I s 1 e r.
Helv. chim. Acta, 45, 517 (1962).
10. U. Schwieter, C. vonPlan-
t a, R. R ii e g g, О. I s 1 e r. Helv.
Chim. Acta, 45, 528, 541 (1962)-
11. C. Stam, C. McGillavry.
Acta, Cryst., 16, 62 (1963).
12. W. S 1 y. Acta Cryst., 8, 115 (1955).
13. C. Sterling. Acta Cryst., 17,
1224 (1964).
14. D. Barton, R. Cookson,
W. К 1 у n e, S. S h о p p e e .
Chem. and Ind., 1954 , 21.
15. B. Pullman, J. Langle t,
H. В e г h о d. J. Theor. BioL, 23,
492 (1969).
16. J. C a w 1 e у, C. R о b e s о n,
L. W e i s 1 e r, E. Schantz,
N. E m b r e e, J. Baxter. Scie-
nce, 107, 346 (1948).
17. H. C a m a, F. Collins,
R. Morton. Biochem. J., 50,
48 (1951).
18. J. В о 1 d i n g h, H. C a m a,
F. С о 1 1 i n s, R. Morton,
N. G r i d g e m a n, О. I s 1 e r,
M. К о f 1 e r, R. Taylor,
A. Welland, T. В 1 a d b u r y.
Nature, 168, 598 (1951).
19. J. Baxter. Fortschr. Chem. org.
Naturstoffe, 9, 41 (1952).
20. J. В a x t e r, C. Robeson. J.
Am. Chem. Soc., 64, 2407, 2411
(1942).
21. P. H a r r i s, S. Ames,
L Brinkman. J. Am. Chem.
Soc., 73, 1252 (1951).
22. S. A m e s, W. Swanson,
P. Harris. J. Am. Chem. Soc.,
77, 4134, 4136 (1955).
23. W. О г о s h n i k, G. W a I d.
Abstracts 14-th International con-
gress of I UP AC. (Zurich), July
1955.
24. P. В г о w n, G. W a 1 d. J. BioL
Chem., 222, 865 (1956).
25. C. vonPlanta, U. Schwie-
ter, L. Chopard-dit-Jean,
R. Riiegg, M. Kofler,
O. Isler. Helv. Chim. Acta, 45,
548 (1962).
26. S. Ames. Ann. Rev. Biochem.,
27, 375 (1958).
27. G. Wald, P. Brown, R. Hub-
bard, W. Oroshnik. Proc.
Nat. Acad. Sci., 41, 438 (1955).
28. K. Hickman. Ind. Eng. Chem.,
29, 968, 1107 (1937).
29. W. О г о s h n i k, A. M e b a n e.
J. Am. Chem. Soc., 76, 5719 (1954).
30. E. Schantz. Science. 108, 417
(1948).
31. Meunier, I.Jouanneteau.
Bull. Soc. chim. bioL, 30, 185, 260
(1948).
32. H. W e i s e r. Biochem. Biophys.
Res. CommunS, 14, 183 (1964).
33. R. Hubbard, G. Wald. Scien-
ce. 114, 60 (1952).
34. Neth. appl. 6504287; C. A., 64,
11262 (1966).
35. J. К a r d у s. Пат. США 3384633;
С. A., 69, 77548 (1968).
36. F. С a r r, E. P r i c e. Biochem.
J., 20, 498 (1926).
37. J. В r ii g g e m a n, W. Kraus,
J. T i e w s. Chem. Ber., 85, 315
(1952).
38. N. G r i d g e m a n. Chem. and
Ind., 38, 574 (1947).
39. M. К о f 1 e r, S. R u b i n. Vi-
tamins and Hormones, 18, 315 (1960).
40. A. G i 1 1 a m, R. Mor ton. Bio-
chem. J., 25, 1346 (1931).
41. R. Morton,' J. H e i 1 b г о n.
Biochem. J., 22, 987 (1928).
214
42. C. R о b e s о n, W. Blum,
J. D i e t e г 1 e, J. Cawley,
J. Baxter. J. Am. Chem. Soc.,
77, 4120 (1955).
43. T. Mead. Biochem. J., 33, 589
(1939).
44. A. H a n z e, T. С о n g e r, E. W i-
s e, D. We is b I a t. J. Am. Chem.
Soc., 70, 1253, (1948).
45. О. I s 1 e r, A. Ronco, W.
G u e x, N. H i n d 1 e y, W. H u -
ber, K. D i a 1 e r, M. Kofi er.
Helv. Chim. Acta, 32, 489 (1949).
46. O. Isler. Chimia, 4, 103 (1950).
47. A. В u s i n g e r, O. Isler,
R. R ii e g g, P. Z e 1 1 e r. J. Sci.
Ind. Res. (India) 17A, 502 (1958).
48. M. M a t s u i, T. О к a m о t o.
Яп. пат. 11251 (1966); С. A., 65,
1549 (1966).
49. Expert Committee on Biological
Standardization, World Health Or-
gan. Tech. Rept. Ser., 187, 10 (1960);
222, 10 (1961).
49a.Г. Анмо, M. Васитакэ. Вита-
мин, 46, 193 (1972).
50. P. К a r r e r, E. J u с к e r. Helv.
Chim. Acta, 28, 717 (1945); 30, 559
(1947).
51. Г. В. Троицкий. Биохимия,
13, 7 (1948).
52. F. J u n g a 1 w a 1 a, H. C a m a.
Biochem. J., 95, 17 (1965).
53. R. M a 1 1 e i n. Compt. rend., 234,
143 (1952).
54. P. К a r r e r, R. M о r f,
K. S h о p p. Helv. Chim. Acta,
14, 1036, 1431 (1931).
55. R. Hunter, E. Hawkins.
J. Chem. Soc., 1944 , 411.
56. R. M о r t о n, T. G о о d w i n.
Nature, 153, 405 (1944).
57. S. В a 1 1, T. Goodwin,
R. Morton. Biochem. J., 42,
516 (1948).
58. H. К о e n i g. W. R e i f, H. P om-
m e г. Пат. ФРГ 1254613; С. A.,
68, 49818 (1968).
59. P. Karrer, W. Hess. Helv.
Chim. Acta, 40, 265 (1957).
60. H. H u b b a r d. R. Greger-
m a n, G. W a 1 d. J. Gen. Phy-
siol., 36, 415 (1953).
61. R. Hubbard, G. Wald. J.
Gen. Physiol., 36, 269 (1952—1953).
62. P. К a r r e r, A. Geiger,
E. В r e t s c h e r. Helv. Chim.
Acta, 24, 161E (1941).
63. C. R о b e s о n, W. Blum,
J. D i e t e r 1 e, J. Cawley,
J. Baxter. J. Am. Chem. Soc.,
77, 4120 (1955).
64. J. A r e n s, D. v a n Dorp. Rec.
trav. chim., 68, 604 (1949).
65. G. W a 1 d, P. В г о w n. J. Gen.
Physiol., 37, 189 (1953—1954).
66. G. Wald, Proc. Natl. Acad. Sci.
U. S., 41, 438 (1955).
67. H. H e n b e s t, E. Jones,
T. Owen. J. Chem. Soc., 1957,
4909.
68. 1. Koizumi, T. Suzuki,
Y. S a h a s h i. J. Vitaminol. (Kyo-
to), 9, 154 (1963).
69. J. Redel, G. Nicolaux, Франц,
пат. 1288975; С. A., 57, 16672
(1962).
70. G. Wald, I. В u г e 11, R. G e о r-
g e. Science, 111, 179 (1950).
71. P. Brown, W. Blum,
M. Stern. Nature, 184, 1377
(1959).
72. W. Sarnecki, M. Schwa r-
zmann, G. Hamprecht,
G. Hummel, H. P о m m e r.
Пат. ФРГ 1126872; С. A., 57, 7172
(1962).
73. P. В г о w n, G. W a 1 d, J. Biol.
Chem., 222, 865 (1956).
74. H. P о m m e r. Angew. Chem., 72,
811 (1960).
75. H.Cama, A. Field, J. Glover,
R. M о r t о n, M. S a 1 a h. Bio-
chem. J., 52, 548 (1952).
76. P. К a r r e r. R. M о r f. Helv.
Chim. Acta, 16, 557, 625 (1933).
77. H. Lindlar. Helv. Chim. Acta,
35, 446 (1952).
78. H. Pommer, W. Sarnecki.
Пат. ФРГ 1059900, 1068710; С. A.,
55, 14511, 12446 (1961).
79. C. Robeson. Пат. США
2835713; англ. пат. 786104; С. А.,
52, 13798, 10191 (1958).
80. Р. Karrer, R. S с h w у z е г.
Helv. Chim. Acta, 31, 1055 (1948).
81. Н. Pommer. Пат. США 2898368;
С. А., 54, 1596 (1960).
82. М. Mousseron - Canet,
J. М а п i, D. L е г п е г. Bull,
soc. chim. Frans., 1966, 3043.
83. P e x э й К а н э к о. Tokyo Koguo
Shikensho hokoku, 57, № 4, 194,
203 (1962); РЖХ, 1963, 13Ж398,
399.
84. В. Burger, С. Garbers,
К. Pachler, R. Bonnett,
В. Weedon. Chem, Communs,
1965, 588.
85. R. К a n e k o. RepL Gov. Chem.
Ind. Res. Inst., Tokyo, 57, 23
(1962).
86. C. Giannotti, B. Das,
E. Lederer. Chem. Communs,
1966, 28; Bull. soc. chim. Frans,
1966, 3299.
87. M. Monsseron - Canet,
J. Mani, D. Lerner. Bull. soc.
chim. Frans., 1966, 30433.
88. C. G i a n n о t t i. Can. J. Chem.,
46, 3025 (1968).
89. R. Kaneko, T. Motohashi.
Nippon Kagaku Zasshi, 85, 65
(1964); C. A., 61, 6859 (1964).
90. J. H e i 1 b г о n, R. Morton,
E. Webster. Biochem. J., 26,
1194 (1932).
91. E. Schantz, J. Cawley,
N. E m b r e e. J. Am. Chem. Soc.,
65, 901 (1943).
92. P. Meunier, R. D u 1 о u,
A. V i n e t. Bull. Soc. chim. biol .
25, 371 (1943).
215
93. В. В а г п h о 1 d t. Acta chem.
scand., 11, 909 (1957).
94. E. Schantz. J. Biol. Chem.,
182, 515 (1950).
95. Англ. пат. 753157; С. A., 51, 7419
(1957).
96. R. В e u t e 1, D. Hinkley,
P. Pollak. J. Am. Chem. Soc.,
77, 5166 (1955).
97. H. H e n b e s t, E. Jones,
T. Owen, V. Thaller. J.
Chem. Soc., 1955, 2763.
98. S. В a 1 as и n dar a tn, M. В a m-
j i, H. C a m a ., P. S u n da-
re s a n, T. Varma. J. Biol.
Chem., 233, 827 (1958).
99. С. И с и к а в а, К. Суга. Яп.
пат. 6109 (1959); РЖХ, 1963,
7Н185.
100. О. I s 1 е г, W. Н u b е г, A. R о п-
с о, М. К о е f 1 е г. Helv. Chim.
Acta, 30, 1911 (1947).
101. R. Hubbard. J. Am. Chem.
Soc., 78, 4662 (1956).
102. B. Rabinovitch, F. Loo-
ney. J. Am. Chem. Soc., 75, 2652
(1953).
103. E. А. Ледерер, В. А. Ро-
занова. Биохимия, 2, 293
(1937).
104. A. G i 1 1 a m, J. H e i 1 b г о n,
E. Jones, E. Lederer,
В. Розанова. Biochem. J.,
32, 405 (1938).
105. E. Lederer, В. А. Роза-
нова, A. Gillam, J. Heil-
b г о n. Nature, 140, 233 (1937).
106. J. Edisbury, R. Morton,
G. Simpkins. Nature, 140,
234 (1937).
107. E. Gray, J. Cawley. J. Biol.
Chem., 131, 317 (1939); 134, 397
(1940).
108. P. Karrer, E. Bretscher.
Helv. Chim. Acta, 25, 1650 (1942);
26, 1758 (143).
109. E. Poulson. Strahlentherapie,
34, 648 (1929).
110. I. H e i 1 b г о n, R. H e s 1 о p,
R. Morton, E. Webster,
J. Rea, J. Drummond.
Biochem. J., 26, 1178 (1932).
111. AL С. Цвет. Труды Варшав-
ского общества естествоиспытате-
лей, отд. биолог., 14, 20 (1903);
Хроматографический адсорбцион-
ный анализ, М., 1946.
112. О. I s 1 е г, W. Н u b е г, A. R о п-
с о, М. Koeller. Experientia,
2, 31 (1946).
113. N. Milas. Science, 103, 581
(1946).
114. J. Arens, D. Van Dorp.
Rec. trav. chim., 65, 338 (1946).
115. P. К a г г e г, E. J и c k e r,
K. Schick. Helv. Chim. Acta,
29, 704 (1946).
116. J. A r e n s, D. Van Dorp.
Nature, 160, 189 (1947).
117. E. R о v a 1 s. Ind. Eng. Chem.,
38, 546'(1946).
118. В. H. Белов, T. А. Диль-
м а н, H. Г. Крохин, Л. H.
Петрова,
H. И. Скворцова. Химия и
технология душистых веществ,
с. 176, М., Гизлегпищепром, 1953.
119. I. S a u s a, L. G a z о, J. Мог-
v i с. Чехосл. пат. 129547; С. А.-,
71, 38354 (1969).
120. Z. Arnold, К. Н е i п о. Че-
хосл. пат. 85207; С. А., 50,
10781 (1956).
121. Н. Krishna, В. Joshi, J.
Org. Chem., 22, 224 (1957).
122. Z. А г п о 1 d, К. Н е i п о. Чехосл.
пат. 85520; С. А., 51, 479 (1957).
123. В. А. Смит, А. В. Семе-
новский, В. М. Медведе-
ва, В. Ф. Кучеров, ДАН
СССР, 124, 1080 (1959).
124. А. В. Семеновский,
В. А. С м и т, В. Ф. К У ч е р о в,
ДАН СССР, 132, 1107 (1960).
125. Т. S a v i с h, L. G о 1 d b 1 a t t.
Пат. США 2507546; С. А., 44,
7341 (1950).
126. R. Webb. Пат. США 3016408;
С. А., 56, 10197 (1962).
127. М. Montavon, Н. Lind-
lar, R. М а г b е t, R. R й е g g,
G. R у s e r, G. S а и с у, P. Z e 1-
1 e г, О. I s 1 e r. Helv. Chim. Acta,
40, 1250 (1957).
128. К- В. Л э э т с, А. К. Ш у м e fl-
ко, A. A. P о з e н о e p,
H. В. К у д p я ш о в а, А. И. П и-
л я в с к а я. ЖОХ, 27, 1510
(1957).
129. Н. U 1 t е е , J. Chem. Soc., 1948,
530.
130. К. В. Л э э т с. Авт. свид. СССР
105428; Бюлл. изобрет., 1957,
№ 3, 17.
131. L. R u z i с k а, Н. S с h i n z.
Helv. Chim. Acta, 23, 960 (1940).
132. И. H. H а з a p о в, И. H. А з e p-
баев. ЖОХ, 18, 414 (1948).
133. И. H. H а з a p о в, Л. А. Я н о в-
с к а я, Б. П. Гусев,
С. С. Ю ф и т, В. И. Г у н а р,
В. А. Смит. ДАН СССР, 114,
331 (1957).
134. Г. И. Самохвалов,
М. А. Миропольская,
Л. А. В а к у л о в а, Н. А. П р е-
ображенский, ДАН СССР,
84, 1179 (1952); ЖОХ, 25, 545
(1955).
135 . И. Н. Н а з а р о в, Б. П. Г у-
с е в, В. И. Г у н а р. ЖОХ, 28,
1444 (1958).
136. И. Н. Назаров, В. Н. Рак-
чеева, В. Я. Райгород-
ская, И. Н. Азербаев.
Изв. АН СССР, ОХН, 1946, 305.
137. М. Carrol. J. Chem. Soc.,
1940, 704.
138. W. К i m e 1, Пат. США 2638484;
С. A., 48, 2763 (1954).
139. W. К i m e 1, А. С о p e. J. Am.
Chem. Soc., 65, 1992 (1943).
140. W. К i m e 1, N. S a x, Пат. США
2661368; С.' A., 49, № 3, 1784
216
(1955); Angew. Chem., 69, 518
(1957).
141. W. К i m e 1, J. S u г m a t i s,
J. W e b e r, G. C h a s e, N. S a x,
A. О f n e г s. J. Org. Chem., 22,
1611 (1957).
142. G. Saucy, R. Marbet. Helv.
Chim. Acta, 50, 2091 (1967).
143. L. Ruzicka, V. For n as i r.
Helv. Chim. Acta, 2, 182 (1919).
144. Г. И. Самохвалов,
M. А. Миропольская,
H. А. Преображенский.
ДАН СССР, 107, 103 (1956); авт.
свид. СССР 103776 (заявка от 29 де-
кабря 1955); Бюлл. изобрет., 1956,
№ 7, 10.
145. Г. И. Самохвалов,
М. А. М и р о п о л ь с к а я,
Л. В. Лукьянова,
Н. А. Преображенский.
ЖОХ, 27, 2501 (1957).
146. И. Н. Н а з а р о в, И. Л. К о т -
ляревский, В. Ф. Р я б -
ченко. ЖОХ, 23, 1900 (1953;)
Изв. АН СССР, ОХН, 1956, 960.
147. Франц, пат. 2031799; РЖХ, 1971,
23Н27.
148. И. Н. Н а з а р о в, Б. П. Г у -
сев, С. М. Маркин,
В. Б. М о ч а л и н, И. И. Н а з а-
рова, В. П. Виноградов,
Б. К- К р У и ц о в, О. А. Ш а -
врыгина, Д. В. Назаро-
в а. ДАН СССР, 114, 796 (1957).
149. С. Т a v е 1. Helv. Chim. Acta,
33, 1266 (1950).
150. R. Marbet, G. Saucy. Chi-
mia, 14, 362 (1960).
151. G. Saucy, R. Marbet. Helv.
Chim. Acta, 50, 1158 (1967).
152. H. И. 3 a x a p о в a, M. А. Ми-
ропольская, И. M. К ус-
та н о в и ч, Т. М. Филиппо-
ва, Л. И. Казакова,
О. Т. Юркина, О. В. Ко-
вальчук, Г. И. Самохва-
лов. ДАН СССР, 181, 603 (1968).
153. Н. И. Захарова, М. А. Ми-
ропольская, Т. М. Фи-
липпова, О. Т. Юркина,
И. М. Кустанович,
Г. И. Самохвалов, ЖОХ,
40, 1657 (1970).
154. R. Lacey. J. Chem. Soc., 1954,
827.
155. И. Н. Наз аров, Л. Я. Я нев-
ская. Б. П. Гусев,
С. М. М а к и н, И. И. Наза-
рова. ДАН СССР, 114, 1029
(1957).
156. W. К i m е 1, N. Sax, S. Kai-
ser, G. Eichmann, G. Cha-
se, A. О f n e r. J. Org. Chem.,
23, 153 (1958).
157. Y.-R. Naves. Compt. rend., 240,
1437 (1955).
158. Y.-R. Naves, P. A r d i z i o.
Bull. Soc. chim. France, 1955,
1479; 1956, 672.
159. Y.-R. Naves, P. A r d i z i_o,
B. Wolf. Bull. Soc. chim. Fran-
ce, 1957, 1213.
160. G. Saucy, R. Marbet,
H. L i n d 1 а г, O. Isler. Helv.
chim. Acta, 42, 1945 (1959).
161. Г. И. Самохвалов,
Л. А. В а к у л о в a, T. В. M e h,
Л. T. Ж и x a p e в а, В. И. К о л-
тунова, H. А. Преобра-
женский. ЖОХ, 29, 2575
(1959).
162. I. Н е i 1 b г о n. J. Chem.
Soc., 1948, 386.
163. I. Н е i 1 b г о n. е. a., J. Chem.
Soc., 1949, 287, 742, 2023, 3120,
3123.
164. N. М i 1 a s, F. G г о s s i, S. Р е n-
n e z, S. Kahn. J. Am. Chem.
Soc., 70, 1292 (1948).
165. N. Mi 1 as, N. McDonald,
D. В 1 a c k. J. Am. Chem. Soc.,
70, 1829 (1948)
166. I. Attenburrow, A. Came-
ron, J. Chapman,
R. E v a ns, В. H e ms, A. J a n-
s e г, T. Walker. J. Chem.
Soc., 1952, 1094.
167. H. Huisman, A. Smit,
S. V г о m e r, L. Fischer.
Rec. trav. chim., 71, 899 (1952).
168. H. Sobotka, J. Chanlev.
J. Am. Chem. Soc., 71, 4136 (1949).
169. G. Cheeseman, I. Heil-
b г о n, E. Jones, F. So n d-
h e i m e r, B. Weedon. J.
Chem. Soc., 1949 , 2031.
170. J. Chanley, H. Sobotka.
J. Am. Chem. Soc., 71, 4140 (1949).
171. R. N у s t г о m, W. Brown.
J. Am. Chem. Soc., 69, 1197 (1947).
172. R. Kuhn, M. Hoffer. Ber.,
67, 357 (1934).
173. L. Colombi, A. Bosshard
H. Sc h i n z, C. Seidel. Helv.
Chim. Acta, 34 , 265 (1951).
174. H. Pommer, W. Sarnecki.
Пат. ФРГ 1068707; РЖХ, 1961,
12Л433; пат. США 3006939; С. А.,
56, 12960 (1962).
175. Н. Р о m m е г, W. Arend. Пат.
ФРГ 1008729; С. А., 53, 15989
(1959).
176. Н. Р о m m е г, G. Wittig.
Пат. ФРГ 951212; С. А., 53, 437
(1959)
177. И. Н. Н а з а р о в, Ж. А. К Р а с-
н а я. ДАН СССР, 118, 716 (1958).
178. Ж- А. К Р а с н а я, В. Ф. К v -
ч е р о в. Изв. АН СССР, ОХН,
1961, 1160; ЖОХ, 32, 64 (1962).
179. О. Isler. Chimia, 3, 150 (1949).
180. Швейц, пат. 254948; С. А., 44,
3025 (1950).
181. Н. Huisman, Rec. trav. chim.,
69, 851 (1950).
182. W. Oroshnik, G. Karma s,
A. M e b a n e. J. Am. Chem. Soc.,
74, 3807 (1952).
183. W. Oroshnik, A. Mebane,
G. К a r m a s. J. Am. Chem. Soc.,
75, 1050 (1953).
184. W. О г о s h n i k, G. К a r m a s,
217
A. Me b a ne. J. Am, Chem. Soc.,
74, 295 (1952).
185. W. Orosh ni k, J. Am. Chem.
Soc., 76, 5499 (1954).
186. Neth. Appl. 6404175; C. A., 64,
19693 (1966).
187. H. J acobs, M. Berg, L. В r and-
sma, J. Arens. Rec. trav.
chim. 84, 1113 (1965).
188. О. И. Ш а в p ы г и н а, Д. В. На-
зар о в a, С. M. Макин.
ЖОрХ, 2, 1586 (1966).
189. Н. I n h о f f е п, F. Bohl-
mann. MBohlmann Lieb.
Ann., 565, 35 (1949).
190. Н. Pommer, W. Sarnec-
ki. Пат. ФРГ 1050763, 1068704;
С. А., 55, 5572, 13473 (1961).
191. С. М. Макин, Д. В. Наза-
рова, Е. А. Кирсанова,
Л. Н. Смирнова. ЖОХ, 32,
1111 (1962).
192. W. Orosh п i k, R. М а 1 1 о -
г у. J. Am. Chem. Soc., 72, 4608
(1950).
193. А. А 1 1 a i s, D. Berlin,
G. Nomine. Bull. soc. chim.
France, 1955, 209.
194. W. Oroshnik, A. Mebane.
J. Am. Chem. Soc., 71, 2062 (1949).
195. Швейц, пат. 258514; С. A., 44,
3026 (1950).
196. К. E i t e r. E. T r u s c h e i t,
H. О e d i g e г. Пат. ФРГ
1110158; С. A., 56, 9999 (1962).
197. Y. Abe, T. M i к i, К. T a r u i,
T. M a r u у a m a, T. Toda,
К- H i r a g а. Яп. пат. 4876
(1972); С. A., 59, 478 (1963).
198. Я- Абэ, Т. М и к и, Д. X а р а о.
Яп. пат. 12117 (1961); РЖХ, 1962,
22Л42.
199. G. Fletc her, J. Н u 1 1. Пат
США 2760986; С. А., 51,2854 (1957)'
200. Я. А б э, Т. М и к и, К- Та р у и,
Т. М а р у я м а и др. Яп. пат
4876 (1962); РЖХ, 1964, 24Н262.
201. Я. Абэ, Т. Мики, М. Тога.
Яп. пат. 17461 (1961); РЖХ, 1964,
8Н267.
202. W. Н u m р h 1 е t t, D. В u г -
ness, Пат. США 2676990; С. А.,
50, 408 (1956).
203. С. Benton, С. Robeson.
Пат. США 2683746, 2683747; С. А.,
49, 10375 (1955).
204. О. Hawks. Пат. США 2839585;
С. А., 52, 17627 (1958).
205. К- Е i t е г, Е. Т г u s с h е i t.
Пат. ФРГ 1110156; С. А., 57, 4567
. (1962); пат. США 3060229; С. А„
58, 5732 (1963).
206. Н. А. Преображенский,
В. Ш о к и н а. ЖОХ, 15, 65
(1945).
207. А. Е. Фаворский,
М. Н. Щукин а. Информацион-
ный бюллетень Всесоюзного Мен-
делеевского общества, 5, 7 (1941);
ЖОХ, 15, 394 (1945).
208. О. Isler, М. М о п t a v о п,
R. R fl е g g, Р. Zeller. Helv.
Chim. Acta, 39, 259 (1956).
209. H. А. Преображенский,
И. А. Рубцов. ЖОХ, 18, 1719
(1948).
210. J. A r e n s, D. van Dorp,
G. v a n Dijk, B. Brandt.
Rec trav. chim., 67, 973 (1948).
211. I. H e i 1 b г о n, E. Jones,
B. Weedon. J. Chem. Soc.,
1949, 1823.
212. W. Y о u n g, L. Andrews,
S. С r i s t о 1. J. Am. Chem. Soc.,
66,. 520 (1944).
213. N. M i 1 a s, T. H а г г i n g t о n.
J. Am. Chem. Soc., 69, 2247
(1947); 70, 4275 (1948).
214. N. Wendler, H. Slates,
N. T г e n n e r, M. T i s h 1 e r.
J. Am. Chem. Soc., 71, 3267 (1949);
73, 719 (1951).
215. P. Karrer, H. Salomon,
R. M о r f, O. Walker. Helv.
Chim. Acta, 15, 878(1932).
216. H. Sobotka, E. Bloch,
D. G 1 i c k. J. Am. Chem. Soc.,
65, 1961 (1943).
217. К. E i t e г, H. О e d i g e r,
R. Lorenz, E. Stein. Бельг,
пат. 617181 (пат. ФРГ 1142595);
С. А., 58, 11238 (1963).
218. М. Н. Щ у к и н а, И. А. Руб-
цов. ЖОХ, 18, 1716 (1948).
219. W. Young, S. Linder. J.
Am. Chem. Soc., 69, 2042 (1947).
220. A. F i n h о 1 t, A. Bond,
H. S c h 1 e s i n g e r. J. Am.
Chem. Soc., 69, 1199 (1947).
221. C. Robeson. Англ. пат.
668604; С. A., 47, 2212 (1953).
222. H. Huisman, A. Smit,
P. van Leenwen, J. van
R i j. Rec. trav. chim., 75, 977
(1956).
223. F. Shantz, C. Robeson,
H. К a s c h e г. Пат. США
2576104; С. A., 46, 6155 (1952).
- 224. A. Smit. Rec. trav. chim., 80,
891 (1961).
225. H. H u i s m a n, A. S m i t. Пат.
ФРГ 1041950; С. A., 54, 10755
(1969).
226. H. Huisman, A. Smit,
< L. Fisscher. Гол. пат. 83143;
С. A., 52, 2919 (1958).
227. W. S t i 1 z, H. Pommer. Пат.
ФРГ 1108208; С. A., 56, 11422
(1962).
228. Г. И. Самохвалов,
Л. И. 3 а х а р к и н, Л. П. Да-
выд о в а, И. М. X о р л и н а.
ДАН СССР, 126, 1013 (1959).
229. Б. М. Михайлов, Л. С. По-
варов. Изв. АН СССР, ОХН,
1963, 1144.
230. G. N i с о 1 а и х, Е. G а у, J. М а -
tet, R. Mauge, С. San-
devoir, A. Wasmer. Франц,
пат. 1243824; С. А., 57, 16671
(1962).
218
231. С. Сайдзе, С. О к а и о,
М. А д а т и. Яп. пат. 4783 (1966);
РЖХ, 1968, 2Н428.
232. J. Arens, D. van Dorp. Na-
ture, 157, 190 (1946); Rec. trav.
chim., 66, 759 (1947).
233. H. Inhoffen, F. Bohl-
mann, M. Bohlmann. Lieb.
Ann., 561, 13 (1948).
234. Г. И. Самохвалов, Г. Гу-
са ко в a, M. А. Мирополь-
ская, H. А. Преображен-
ский. Авт. свид. СССР 93422;
Бюлл. изобрет., 1952, № 2—3, 7.
235. I. H e i 1 b г о n, E. Jones,
D. O’ S u 1 1 i v a n. J. Chem.
Soc., 1946, 866.
236. K. Ziegler, A. Spath,
E. S c h a a f et al. Lieb. Ann.,
551, 80 (1942).
237. Г. И. Самохвалов,
M. А. Миропольская,
Л. А. В а к у л о в а. Н. А. Пре-
ображенский, Труды
ВНИ витаминного ин-та, IV,
5 (1953); V, 5 (1954).
238. Н. Словохотова,
Г. И. Самохвалов, Г. К у-
ницкая, М. А. Мирополь-
ская, ЖОХ, 24, 2222 (1954).
239. Г. И. С а м о х в а л о в, М. А. М и-
ропольская, Л. А. В а к у-
л о в а, Л. П. Жукова,
Н. Словохотова, В. Ма-
люсов, Н. А. Преобра-
женский. ДАН СССР, 99, 273
(1954).
240. Н. Н u i s m а п, A. Smit,
Р. van Leeuwen, J. van
R i j. Rec. trav. chim., 75, 977
(1956).
241. H. van Geelen. Пат. ФРГ
1139835 (1963); С. A., 58, 10102
(1963).
242. Г. И. Самохвалов,
И. А. Рубцов, М. А. Миро-
польская, Н. А. Пре-
ображенский. Труды
ВНИ витаминного ин-та, IV,
10 (1953); авт. свид. СССР 93902;
Бюлл. изобрет., 1952, № 6, 9.
243. К age h i г a Ueno. Яп. пат.
2225 (1953); С. А., 49, 377 (1955).
•244. М. Matsui, S. Oka по,
К. Yamashita, М. М i у а -
mo, S. К i t а т u г a, A. Ko-
bayashi,,!. Sato, R. М i -
kami. J. Vitaminol. (Kyoto), 4.
178, (1958).
245. Пат. США 2951853; англ, пат-
835526; С. А., 54, 24557 (1960).
246. G. Wittig, Н. Pommer. DPB"
Anm. В., 32741 IVb/120 V. 25 sept-
1954, der BASF-Ludwigshafen; An"
gew. Chem., 68, 508 (1956).
247. С. M. Макин. ЖОХ, 32, 3159
(1962).
248. Бельг, пат. 603425 (1963).
249. Н. Huisman, A. Smit.
Гол. пат. 99527; РЖХ, 1962,
22Л150.
250. Г. И. Самохвалов,
Л. П. Д а в ы д о в а, Л. И. За-
хар к и н, И. М. Хорлина,
Л. А. В а к у л о в а,
Л. Т. Ж и х а р е в а, Н. А. П р е-
ображе некий. ЖОХ, 30,
1823 (1960).
251. J. Copenhaver. Пат. США
2586305; С. А., 47, 2212 (1953).
252. A. Starke. Пат. США 2615922;
С. А., 48, 7638 (1954).
253. Т. Jacobs, R. Cramer,
J. Hanson. J. Am. Chem. Soc.,
64, 223 (1942).
254. O. Schwarzkopf, H. C a h n-
m a n n, A. Lewis, I. S w i n -
dinsky. H. Wuest. Helv.
Chim. Acta, 32, 443 (1949).
255. H. Huisman, A. Smit. Пат.
США 3056834; С. A., 58, 11410
(1963).
256. H. Inhoffen, F. Bohl-
mann, M. В о h 1 m a n n. Lieb.
Ann., 568, 47 (1950).
257. J. Cawley, C. Robeson,
E. S h a n t z, L. W e i s 1 e r,
J. Baxter, Пат. США 2576103;
С. A., 46, 6154 (1952).
258. J. Redel, G. Nicolaux. Франц,
пат. 1291622; С. A., 57, 14966
(1962).
259. N. Milas. Пат. США 2369156,
2369163—2369166; С. A., 39, 5043,
5046 (1945); пат. США 2382085,
2382086; С. А., 40, 681 (1946).
260. S. Ishikawa, Т. Matsuu-
r а . Sci. Repts, Tokyo Burnika
Daigaku, ЗА, 173 (1937); Zbl.
1937, II, 3452.
261. H. L i n d 1 a г. Пат. США 2451740;
С. A., 43, 1800 (1949).
262. R. Posternack, A. В a w-
ley. Пат. США 2700058;.С. A.,
49, 15970 (1955).
263. J.Cymerman, I. Heilbron,
E. J о n e s, R. L а с e у . J. Chem.
Soc., 1946, 500, 727.
264. G. Cheeseman, I. Heil-
bron, E. Jones, F. Sond-
heim e r, B. Weedon. J.
Chem. Soc., 1949, 1516.
265. H.Inhoffen, F. Bohl ma nn,
G. L i n h о f f. Lieb. Ann., 570,
73 (1950).
266. Y. Naves, H. Barbier,
P. A r d i z i o. Bull. Soc. chim.
France, 1950, 1188.
267. I. Heilbron, A. Johnson,
E. J о n n e s, A. Spinks. J.
Chem. Soc., 1942, 727.
268. H. Inhoffen, H. Pommer,
F. Bohlmann. Lieb. Ann., 561,
26 (1948).
269. M. Harfenist, A. Bavley,
W. L a z i e v. J. Org. Chem., 19,
1608 (1954).
270. Г. И. Самохвалов,
Л. П. Ж У к о в a, H. А. Пре-
ображенский. ЖОХ, 26,
3105 (1956).
271. N. М i 1 a s, S. L е е, Е. S a k а 1,
Н. Wohlers, N. McDonald,
219
F. G г о s s i, H. Wright. J.
Am. Chem. Soc., 70, 1584 (1948).
272. H. О e d i g e г, К. E i t e r. Chem.
Ber., 97, 549 (1964).
273. H. О e d i g e г, К. E i t e r,
R. Lorenz, E. Stein. Chem.
Ber., 97, 549 (1964).
274. H. О e d i g e г, К. E i t e r.
Пат. ФРГ 1157606; С. A., 60,
5568 (1964).
275. N. Milas. Пат. США 2577538;
С. A., 46, 9604 (1952); англ. пат.
664815; С. А., 46, 6671 (1952).
276. И. А. Рубцов, Н. В. 3 о т -
чик, М. Баткибекова.
Биол. акт. соед. (ЖОрХ). Л.,
«Наука», 1968, с. 11.
277. О. Isler. Chem. Eng. Nejvs 29,
3962 (1951); пат. США 2451735—
2451739; С. А., 43, 1801 (1949).
278. Н. Luther. Ind. Eng. News.,
30, 539 (1952).
279. L. S u r m a t i s. Пат. США
2610208; С. A., 47, 8777 (1953).
280. N. Milas, E. Sakai, I. Pla-
t i, I. Rivers, I. Gladding,
F. Grossi, Z. W e i s s,
M. Campbell, H. Wright.
J. Am. Chem. Soc., 70, 1597 (1948).
281. Г. С. M и p о н о в, Г. H. X p и c -
товская, M. И. Фарбе-
ров, T. H. А н т о н о в а. ЖПХ,
40, 1777 (1967).
282. К. Kalina. Чехосл. пат.
102024; РЖХ, 1963, 15Н19.
283. Е. Jones, McCombie. J.
Chem. Soc., 1943, 261.
284. J. Cymerman, I. Heilb-
r о n, E. Jones. J. Chem.
Soc., 1945 , 90.
285. I. Heilbron, E. Jones,
Lacey, McCombie, Ra-
phael. J. Chem. Soc., 1945,
77.
286. A. E. Фаворский, Скоса-
ре в с к и й. ЖРФХО, 32, 652
(1900).
287. J. Cawley. J. Am. Chem. Soc.,
77, 4125 (1955);
288. G. Pattenden, B. Weed on,
C. Garbers, D. Schneider,
J. van der Merwe. Chem.
Communs, 1965, 347.
289. Sh. О k a n o, R. M i k a m i,
Sh. S a i j о, M. M a t s u i. J.
VitaminoL 7, 304 (1961); РЖХ,
1963, ЗЖ402.
290. К. Eiter, E. T r u s c h e i t.
Пат. ФРГ 1075598; С. A., 57, 4706
(1962).
291. H. Henbest. J. Chem. Soc.,
1951, 1074.
292. R. В a r u a, M. Nair. Nature,
193, 165 (1962).
293. E. Haworth, J. Heilbron.
J. Chem. Soc., 1939, 128.
294. E. McCollum, Davis. J.
Biol. Chem., 15, 167 (1913).
295. T. Osborne, L. Mendel. J.
Biol., Chem., 15, 311 (1913).
296. E. McCollum, M. Davis.
J. Biol. Chem., 23, 181 (1915).
297. T. Moore. Proc. Nutr. Dieta, 3
102 (1962).
298. G. Wald. J. Gen. Physiol., 19
351 (1935).
299. R. Morton. Nature, 153, 69
(1944); Biochem. J., 42, 516 (1948).
300. G. Wald. Nature, 139, 1017
(1937).
301. R. Morton, Salah, Stubbs
Nature, 159, 744 (1947).
302. G. Wald. Vitamins and Hormo-
nes, 18, 417 (1960).
303. R. Hubbard, A. Colman
Science, 130, 977 (1959).
304. J. Dowling. Nature, 188, 114
(1960).
305. L. Ruzick a, Fischer. Helv.
Chim. Acta, 17, 633 (1934).
306. R. Gould, A. Thompson.
J. Am. Chem. Soc., 57, 340 (1935);
J. Biol. Chem., 114, Xli (1936).
307. N. Milas, S. Lee, C. Schu-
' e r c h. R. Edgerton, 1. P 1 a-
t i, F. Grossi, Z. Weiss,
M. C a m p b e 1 1. J. Am. Chem.
Soc., 70, 1591 (1948).
308. H. E u 1 e r, P. Karrer,
LJ. S о 1 m s s e n. Helv. Chim. Acta, ’
21, 211 (1938).
309. P. Karrer, J. Benz. Helv.
Chim. Acta, 31, 1048 (1948);
32, 232 (1949).
310. N. Wendler, C. Rosen-
blum, M. T i s h 1 e r. J. Am.
Chem. Soc., 72, 234 (1950).
311. К. E i t e г, H. О e d i g e r,
E. Truscheit. Пат. ФРГ
1110633; С. A., 56, 3522 (1962).
312. M. Lakshmanan, F.Jun-
g a 1 w a 1 a, H. C a m ,a. Bio-
chem. J., 95, 27 (1965).
313. P. Karrer. E. Jucker. Helv.
Chim. Acta,28, 717 (1945); 30, 559
(1947).
314. В. E u 1 er, H. Euler,
P. Karrer. Helv. Chim. Acta,
12, 278 (1929).
315. P. Meunier. Fortsch. Chem.
org. Naturstoffe, 9, 112 (1952).
i/316. P. Karrer, E. Jucker. Ca-
rotenoids, Amsterdam, 1950.
317. R. M о r t о n, R. Creed. Bio-
chem., J., 33, 318 (1939).
318. R. Rflegg. U. Schwieter,
G. R у s e r, P. S c h u d e 1,
O. Isler. Helv. chim. Acta, 44,
985, 994 (1961).
319. U. Schwieter, H. Bolli-
ger, L. Chopard-did-
Je a n, E. E n g 1 e r t, M. К о f -
1 e г, К. К 6 n i g, C. v о п P 1 an-
t a, R. Rflegg, W. Vetter,
O. Isler. Chimia, 19, 294 (1965).
320. O. Isler, H. Lindlar,
M. Montavon, R. Rflegg,
P. Zeller. Helv. Chim. Acta,
39, 274 (1956).
321. L. Cholnoky, J. Szabolcs,
E. Nagy. Lieb. Ann., 616, 207
(1958).
322. R. Kuhn, C. G r u n d m a n n.
Ber., 66, 1746 (1933).
220
323. T. Goodwin, М. Taha. Bio-
chem. J., 47, 244 (1950); 48, 513
(1951).
324. A. Left wick, B. Wee do n.
Chem. Communs, 1967, 49.
325. M. Barber, L. Jackman,
C. W a г г er, B. Weedon.
Proc. Chem. Soc., 1960, 19.
326. H. Yokoyama, W. Micha-
el. J. Org. Chem., 30, 2481, 3994
(1965).
327. R. R ii e g g, M. Mont a von,
G. R у s e r, G. Saucy,
U. Schwieter, O. Isler.
Helv. Chim. Acta, 42, 854 (1959).
328. P. Karrer, U. Solmssen.
Helv. Chim. Acta, 20, 682, 1020
(1937).
329. O. Isler, W. G u e x,
R. R ii e g g, G. R у s e r, G. S a u-
cy, U. Schwieter, M. W a 1-
ter, A. Winterstein. Helv.
Chim. Acta, 42, 864 (1959).
330. A. A a s e n, S. J e nse n. Acta,
chem. scand., 19, 1843 (1965).
331. L. Zechmeisterc сотр. Bio-
chem. J., 32, 1305 (1938); Ber.,
72, 1340 (1939); J. Am. Chem.
Soc., 64, 1856 (1942); 66, 137
(1944); Arch. Biochem., 5, 107
(1944); 7, 247 (1945).
332. H. Inhoffen, F. В о h 1 -
m a n n, К. В a r t r a m, G. Rum-
rn e r t, H. Po m m er. Lieb. Ann.,
570, 54 (1950).
333. C. Eugster, C. Garber,
P. Karrer, Helv. Chim. Acta,
36, 1378 (1953).
334. O. Isler, L. Choppard-
dit-Jean, M. Montavon,
R. R ii e g g, P. Z e 1 1 e r. Helv.
Chim. Acta, 40, 1256 (1957).
335. C. Euguster, R. Buche-
cker, C. Tscharner,
G. U h d e, G. О h 1 о f f. Helv.
Chim. Acta, 52, 1729 (1969).
336. О. I s 1 e r, H. Lindlar,
M. Montavon, R. Riiegg,
P. Zeller. Helv. Chim. Acta,
39, 249 (1956).
337. P. Karrer, A. Helfen-
stein. Helv. Chim. Acta, 12,
741 (1929).
338. Б. Г. С а в и н о в, A. M и хай-
ло в н и н а . ДАН СССР, 88, 887
(1953).
339. Р. Karrer, Е. Jucker.
Helv. Chim. Acta, 30, 266 (1947).
340. Б. Г. С а в и н о в, Г. Третья-
кова. ДАН СССР, 78, 553 (1951);
Укр. хим. журнал, 17, 502 (1951).
341. Р. Karrer, Н. von Euler,
Н. Solemssen, О. Wal-
ker. Helv. Chem. Acta, 17, 417
1169 (1934).
342. R. Kuhn, H. Brockman n.
Ber., 66, 407 (1933).
343. L. Z e c h m e i s t e г, A. P о 1 -
gar. J. Am. Chem. Soc., 65,
1524 (1943).
344. A. P о 1 g a 1, L. Z e c h m e i s-
t e r. J. Am. Chem. Soc., 65, 1528
(1943).
345. P. Karrer, A. Riiegger.
Helv. Chim. Acta, 23, 955 (1940).
346. Б. Савинове сотр. ДАН СССР,
72, 1087 (1950); Укр. хим. журн.;
15, 41, 93 (1949); 16, 57, 358 (1950);
17, 496 (1951); 18, 361 (1952).
347. Р. Karrer, Е. Jucker.
Helv. Chim. Acta, 28, 427, 471
(1945).
348. P. Meunier, J. Jouanne-
tean, G. Zwingelstein.
Compt. rend., 231, 1170 (1950).
349. A. Winterstein. Angew.
Chem., 72, 907 (1960).
350. Y. Mura, Y. Mose, Яп. пат.
4784 (1962); С. A., 48, 14008 (1963).
351. N. Milas, S. Sussman. J.
Am. Chem. Soc., 58, 1302 (1936);
59, 2545 (1937).
352. E. Grob, R. Buttle r. Helv.
Chim. Acta, 38, 737 (1955).
353. R. В a r u a, A. В a r u a. Indian J.
Chem., 7, 1017 (1969).
354. R. Kuhn, H. Brockman n.
Ber., 65, 894 (1932); Lieb. Ann.,
516, 95 (1935).
355. U. Schwieter, W. Arnold,
W. О b e r ha n sei, N. Ri-
ga ssi, W. Vetter. Helv.
Chem. Acta, 54, 2447 (1971).
356. K. Tsukida, Cho Shuku-
C h i n, M. Y о k о t a, Chem.
Tharm. Bull., 17, 1755 (1969).
357. P. Karrer, G. Schwab.
Helv. Chim. Acta, 23, 578 (1940).
358. L. Zechmeister, L. Wail-
ea v e. J. Am. Chem. Soc., 75,
4493 flQSTi
359. P. К a r r er, E. J u c k e r. Helv.
Chim. Acta, 29, 229 (1946).
360. L. Zechmeister, L. Q h о 1
n о k у, V. V r a b e 1 у. Ber., 61,
566, 1534 (1928); 66, 123 (1935).
361. P. Karrer, e. a. Helv. Chim.
Acta, 11, 751 , 1201 (1928).; 12,
185, 1142 (1929); 13, 1084 (1930);
14, 43 (1931).
362. R. Kuhn, A. Winterstein.
Helv. Chim. Acta, 11, 427 (1928).
363. N. H о 1 у e r, B. Weedon.
Chemistry and Industry, 1955, 1219.
364. R. К u h и, H. R о t h. Ber., 66,
1274 (1933).
365. P. Karrer, e. a. Helv. Chim.
Acta, 14, 614, 833, 1033 (1931);
15, 490, 1153 (1932); 16, 975 (1933);
17, 417, 1169 (1934); 18, 25 (1935).
366. R. Kuhn, A. Winterstein.
Ber., 65, 1873 (1932); 66, 429,
1733 (1933).
367. R. Kuhn, E. Lederer. Ber.,
64, 1349 (1931); 65, 637 (1932).
368. R. Kuhn, E. Lederer. Z.
physiol. Chem., 200, 246, 255
(1931).
369. W. Shearon, J.Gee, O. Gee.
Ind. Eng. Chem., 41, 218 (1949).
370. Л. О. Шнайдман. Производ-
ство витаминов из растительного
221
и животного сырья. М., Пище-
промиздат, 1950.
371. А. С i е g 1 е г, G. Nelson,
Н. Hal. Appl. Microbiol., 11,
128 (1963).
372. Р. Karrer. С. Eugster. Helv.
Chim. Acta, 33, 1952 (1950).
373. H. Inhoffen, U. Schwie-
t e r, G. R asp e. Lieb. Ann.,
588, 117 (1954).
374. P. Karrer, C. Eugster.
Compt. rend., 230, 1920 (1950);
Helv. Chim. Acta, 33, 1172 (1950).
375. H. Inhoffen, F. Bohl-
mann, K. Bartram,
H. P о m m e r. Chem. Ztg., 74,
_ 285 (1950).
376. N. M i 1 a s, P. Davis, I. В e -
lie, D. F1 e § . J. Am. Chem.
Soc., 72, 4844 (1950).
377. H. Pommer, W. Sarnecki.
Пат. ФРГ 1068707; С A., 55,
10499 (1961).
378. H-J. Kabbe, E. Truscheit,
К. E i t e r. Lieb. Ann., 684, 14
(1965).
379. H. К л ю e в а, И. A. P у б ц о в.
Сб. Биол. акт. соед. АН СССР,
1965, 228.
380. К. Е i t е г, Н. Kabbe,
Е. Truscheit. Пат. ФРГ
1114812; РЖХ, 1963, 7Н184.
381. Н. Inhoffen, F. Bohl-
m a n n, H. A 1 d a g, S. Bock,
G. L e i b n e r. Lieb. Ann., 573,
1 (1951).
382. P. Karrer, C. Eugster,
S. Perl. Helv. Chim. Acta, 32,
1013 (1949).
383. P. Karrer, C. Eugs-
ter. Helv. Chim. Acta, 32, 1934
(1949).
384. H. Inhoffen, H. Pommer,
K. W i n k e 1 m a n n, H. A 1 -
day. Chem. Ber., 84, 87 (1951).
385. R. Ahmad, F. Sondheimer,
B. Weedon, R. Woods.
J. Chem. Soc., 1952, 4089.
386. H. Inhoffen, H. S i em er.
Fortsch. Chem. org. Naturstoffe,
9, 1 (1952).
387. H. Inhoffen, G. Leibner.
Lieb. Ann., 575, 105 (1952).
388. H. Inhoffen, H. Si e m er,
K. Mohle. Lieb. Ann., 585, 126
(1954).
389. С. M. M а к и н, И. Н. Р о ж к о в.
ЖОХ, 31, 3214 (1961).
390. С. М. Макин. ДАН СССР, 138,
387 (1961).
391. R. Н о a g 1 i n, D. Н I г s h. J.
Am. Chem. Soc., 71, 3468 (1949).
392. H. Inhoffen, F. Bohlmann,
G. R u m m e r t. Lieb. Ann., 571,
75 (1951).
J393. G. Wittig. Experientia, 12,
* 41 (1956).
394. O. Isler, M. M о n t a v о n,
R. Riiegg, P. Zeller. Lieb.
Ann., 603, 129 (1957).
395. H. Pommer. Angew. Chem., 72,
911 (1960).
396. W. Sarnecki, A. Niirren-
b a c h, W. Reif. Пат. ФРГ
1158505; С. A„ 60, 5570 (1964).
397. H. Pommer, W. Sarnecki.
Пат. ФРГ 1068709; С. A., 55,
13472 (1961).
398. H. Bestman n. Angew. Chem.,
76, 754 (1964).
399. J. Surmatis, R. Thorn-
men. J. Org. Chem., 34 , 559(1969).
400. O. Isler, M. Montavon,
R. R fl e g g, P. Z e 1 1 er, Helv.
Chim. Acta, 39, 454 (1956).
401. A. C h e c h a k, M. Stern,
C. Robeson. J. Org. Chem.,
29, 187 (1964).
402. C. Garbers, C. Eugster,
P. Karrer. Helv. Chim. Acta,
36, 1783 (1953).
403. H. Inhoffen, G. Raspe.
Lieb. Ann., 594, 165 (1955).
404. O. Isler, H. Lindlar,
M. Montavon, R. Riiegg,
G. S a u c v, P. Zeller. Helv.
Chim. Acta, 39, 456 (1956).
405. J. Redel. J. Boch. Compt.
rend., 258, 1840 (1964).
406. H. Pommer. Angdw. Chem.,
76, 754 (1964).
407. O. Isler, R. R fl e g g,
U. Schwieter, J. Wflrsch.
Vitamins and Hormones. 18, 295
(1960).
408. O. Isler, M. Montavon.
“Chimia, 12, 1 (1958).
409. O. Isler, P. Zeller, Vita-
mins and Hormones, 15, 31 (1957).
410. L. Zechmeister “Cis-trans
Isomeric Carotenoids, vitamins A
and Arylpolyenes”, Vianna, 1962.
411. L. Zechmeister. Vitamins
and Hormones, 7, 73 (1949).
412. W. Schuchardt. Fette and
Seifen, 53, 986 (1954).
413. T. Goodwin. Ann. Rev. Plant.
Physiol., 12, 219 (1961).
414. R. S t a n i e r. Harvey Lectures,
54, 219 (1958—1959).
Часть третья
ВИТАМИНЫ АРОМАТИЧЕСКОГО РЯДА
ГЛАВА V
НАФТОХИНОНОВЫЕ ВИТАМИНЫ И ПРОВИТАМИНЫ
ФИЛЛОХИНОН, МЕНАХИНОНЫ
К витаминам группы 2-метил-1,4-нафтохинона (витамины группы К)
относятся его производные, имеющие в качестве заместителя в положении
3 фитил, дигеранил, дифарнезил, фарнезилдигеранил, дифарнезилгеранил
или соланезил.
Филлохинон [1]—а-филлохинон [2] — представляет собой 2-мегил-З-
фитил-1,4-нафтохинон (витамин Кх или витамин Ккгл) (I) и имеет в боко-
вой цепи одно частично насыщенное изопреноидное звено.
о
¥ сн3 сн3 сн3 сн,
йх^х'СНгСН:=ССН2СН2СН2СНСН2СНгСН2СНСН;(СН2СН2СНСНэ
О 3' У
I
Менахиноны — витамины группы К2 — в структуре своей молекулы
имеют тот же 2-метил-1,4-нафтохинон и в положении 3 алифатическую
боковую цепь, состоящую из различного числа частично насыщейных
изопреноидных звеньев.
Менахинон-4—2-метил-3-дигеранил-1,4-нафтохинон (витамин К2(2;)) (И)
[3]; менахинон-6—2-метил-3-дифарнезил-1,4-нафтохинон (витамин К2(зэ))(1П)
[4], менахинон-7 —0-филлохинон, 2-метил-3-фарнезилдигеранил-1,4-наф-
тохинон (витамин К2(35)) (IV) [4], первоначально открыт Дойзи с сотр. как
витамин К2 [5], ранее ему приписывалась структура соединения III, мена-
хинон-8—2-метил-3-дифарнезилгеранил-1,4-нафтохинон (витамин K2(43)) (V)
и менахинон-9—2-метил-3-соланезил-1,4-нафтохинон (витамин Кгцб))
(VI) [6—81, обнаруженные в микроорганизмах, —группа природных изо-
мерных соединений, производных 2-метил-1,4-нафтохинона с боковой цепью
223
о
в положении 3, состоящей из различного количества частично насыщен-
ных изопреноидных звеньев. Так, менахинон-7 содержит семь изопреноид-
ных остатков из 35 атомов углерода с семью двойными связями.
Филлохинон (I) имеет два асимметрических атома углерода. Так как
у оптически активных фитолов асимметрические атомы углерода в поло-
жениях 7 и 11, как правило, по знаку удельного вращения однозначны, то,
следовательно, для витамина Кх возможны два оптических антипода и один
рацемат. Природный фитол, который входитвмолекулуфиллохинона, имеет
транс-конфигурацию двойной связи и ^-конфигурацию; значит, и филлохи-
нон (I) должен быть отнесен к /?-конфигурации, т. е. С* 7'/?, 11'/? [9, 10]
о
Оптическая активность филлохинона [а й> —0,4° (57,5%, СвНв) [11].
Помимо филлохинона с природной 2',З'-транс-конфигурацией [10]
синтетически получен из цис-фитола [10] 2',3'-цис-филлохинон
Опубликованы правила определения /?- и S-конфигураций [12].
Мена хиноны (II—VI) оптической активностью не обладают. Все изо-
пренологи менахинонов имеют полную транс-конфигурацию двойных связей
боковой цепи
Филлохинон — витамин Kr (I) — представляет собой вязкое масло-
образное вещество желтого цвета с т. пл. —20° С и т. кип. 115—145° С
при 0,0002 мм. Спектр поглощения характеризуется максимумами при 243,
‘248, 261, 270 и 328 нм [131; Е\ %, 328 при 248 нм. Для филлохинона харак-
терна способность флуоресцировать при освещении аргоновой лампой [14],
что, однако, свойственно и нафтохинонам, лишенным заместителя в поло-
жении 3. Филлохинон нерастворим в воде, мало растворим в метиловом
спирте, легко растворим в петролейном эфире, бензоле, эфире, ацетоне и
многих других органических растворителях. Из растворов он адсорбируется
сернокислым магнием, пермутитом и активированным углем. Филлохинон
обладает способностью образовывать дисперсные системы с жидкостями,
в которых нерастворим.
224
Meна хин он-7 — витамин К2(35) (IV) — представляет собой кристалли-
ческое вещество желтого цвета с т. пл. 53,5—54,5° С [2, 41 и т. кип. 200° С
(с разл. при 0,0002 мм; кристаллизуется из петролейного эфира или
метанола. Спектр поглощения мена хинона-7 имеет те же максимумы, что
и для филлохинона: 243, 249, 260, 270, 320 нм; Е{ см 350 при 249 нм. Мена-
хинон-7 нерастворим в воде, хорошо растворим в эфире, бензоле, ацетоне,
петролейном эфире, а также в безводном этиловом спирте.
Другие менахиноны также являются кристаллическими веществами
желтого цвета : мен а хинон-4 (витамин Кг(20)) с т.пл. 35 °C [4], ме-
на хинон-6 (витамин Кг(зо)) с т. пл. 50 СС [41, мен а хи нон-9 (витамин
Кг(45)) с т. пл. 58—59 СС [31. Спектры поглощения менахинонов характе-
ризуются теми же максимумами, что и для менахинона-7.
Филлохинон (I) широко распространен в природе, главным образом в
зеленых частях растений. Он выделен из люцерны [15, 16 J и других рас-
тений. Менахиноны являются продуктами жизнедеятельности бактерий. Их
источниками служат, например, Escherichia coli, обитающие в кишечнике
животных, или другие микроорганизмы. Менахинон-6 (III) и менахинон-7
(IV) выделены из гниющей рыбной муки [4, 51, менахинон-8 (V) — из Pro-
teus Vulgaris и др. [6—81, менахинон-9 (VI) — из Mycobacterium phlei и др.
[7, 81, менахинон-4 (II) — из разных микроорганизмов [5, 17].
ХИМИЧЕСКИЕ СВОЙСТВА НАФТОХИНОНОВЫХ ВИТАМИНОВ
Нафтохиноны не устойчивы к окислителям. При действии света и кисло-
рода воздуха 2-метил-1,4-нафтохинон (менадион, витамин Л3) (VII) окисля-
ется и фотополимеризуется в «димер» (VIII). Особый интерес вызывает
свойство нафтохинонов легко окисляться с присоединением по двойной свя-
зи хинона атома кислорода с образованием эпокисей. Так, 2-метил-1,4-наф-
тохинон (VII) при действии перекиси водорода дает 2,3-перокись 2-метил-
1,4-нафтохинона (IX),
представляющую собой бесцветные кристаллы с т. пл. 95,5—96,5° С; это
соединение имеет достаточно высокую биологическую активность.
Филлохинон (I) в результате окисления перекисью водорода при 75° С
дает устойчивую к свету 2,3-эпокись 2-метил-3-фитил-1,4-нафтохинона (X) *
с выходом 82% [18].
о
о
СН,
о СНз СНз
сн2сн=ссн2(сн2сн2снснДн
X
Это почти бесцветное маслообразное вещество с Пр 1,5132, обладающее
почти такой же активностью, как и неокпсленный витамин.
При умеренном окислении по двойной связи молекулы филлохинона
отщепляется боковая алифатическая цепь, а при действии энергичных окис-
лителей образуется фталевая кислота. Под влиянием разбавленной азотной
кислоты 2-метил-1,4-нафтохинон окисляется во фталевый ангидрид. В свя-
8—69
225
зи с чувствительностью к окислению нафтохиноновые витамины, включа-
ющие в свою структуру пара-хиноидную группировку и ненасыщенные свя-
зи, неустойчивы к действию прямого солнечного света [2], к ультрафиоле-
товому излучению и к высокой температуре [19]; инфракрасные лучи не
расщепляют филлохинон (I), это относится и к 2-метил-1,4-нафтохинону.
При фотолитическом окислении кислородом в циклогексане филлохи-
нон и менахиноны расщепляются по первой двойной связи боковой цепи
с образованием кетопроизводного [20].
Нафтохиноны легко претерпевают окислительно-гидролитические пре-
вращения [21—23]. Так, длительное нагревание водных растворов 2-метил-
1,4-нафтохинона при pH выше или ниже 7 протекает с образованием фтале-
вой кислоты и фтиокола( XI), а также с образованием хингидронов и различ-
ных продуктов уплотнения его молекулы [21]. Если реакцию проводить точ-
но при pH 7, то нафтохинон изменениям не подвергается. Так как окисли-
тельная реакция протекает и в отсутствие кислорода, то, следовательно,
окисляющим реагентом служит сам нафтохинон, превращающийся при этом
в нафтогидрохинон.
Окислительно-гидролитическое превращение 2,3-перокиси 2-метил-1,4-
нафтохинона (IX) протекает при простом кипячении в воде с образованием
2-метил-1,4-нафтохинона (VII), фтиокола (XI) и о-лактилфенилглиоксило-
вой кислоты (XII), возникающей в результате окислительно-гидролитичес-
кого расщепления нафтохинона [22, 23]. Окислителем в этой реакции слу-
жит или исходная перокись IX, или образующийся при реакции нафтохи-
нон, который, как и при описанных ниже реакциях присоединения аминов
или тиокислот, восстанавливается при этом в нафгогидрохинон.
Окислительно-гидролитическое расщепление филлохинона приводит к
образованию (2-метил-1,4-нафтохинонил-3)уксусной кислоты и соответст-
вующего кетона С18.
При действии щелочей нафтохиноновые вита мины испытывают гидроли-
тическое расщепление. Так, филлохинон при этом дает фтиокол (XI) и
фитол.
Нафтохинонам свойственны все реакции, типичные для карбонильной
функции. Характерной особенностью нафтохинонов и нафтохиноновых ви-
таминов является их способность к окислительно-восстановительным пре-
вращениям. Так, при действии двух протонов и двух электронов они очень
легко и количественно восстанавливаются в бесцветные нафтогидрохиноны,
которые уже под влиянием кислорода воздуха вновь окисляются в окра-
шенные хиноны.
Реакция восстановления в водном растворе носит обратимый характер.
По-видимому, первоначально к окрашенному 2-метил-1,4-нафтохинону
(VII) или филлохинону (I) присоединяется два электрона, а затем к обра-
зовавшемуся дианиону присоединяется два протона и образуется бесцвет-
ный 2-метил-1,4-нафтогидрохинон (XIV) пли филлогидрохинон.
226
Окислительно-восстановительный потенциал хинон-гидрохиноновой си-
стемы фнллохинона (I), характеризующий окислительную способность хи-
нона, составляет Ео = 0,363 В при 20° С или 0,328 В при 22° С [24]. Для
фтиокола (XI), обладающего 1/500 активности витамина окислительно-
восстановительный потенциал понижается: Ео = 0,256 В при 22° С и
0,300 В при 30° С, а для 2-метил-1,4-нафтохинона (VII), более активного,
чем витамин Ki, повышается: Ео — 0,428 В при 25° С и 0,458 В при 28° С.
При восстановительном ацетилировании витамины К образуют диаце-
тилдигидровитамины: филлохинон (I) — филлогидрохинондиацетат с т. пл.
61—62° С [25], обладающий 50%-ной активностью фнллохинона; менахи-
нон-6 (III) — гидрохинондиацетат менахинона-6 с т. пл. 56,5—57,5° С
[4 ], обладающий 50%-ной активностью исходного менахинона; менахинон-7
(IV) —гидрохинондиацетат менахинона-7 с т. пл. 57° С [4]. Филлогидро-
хинондибензоат имеет т. пл. 86° С, филлогидрохинондисукцинат — 182—
184° С [25]. Известны и другие ацилгидрохиноны витамина Кх [26, 27].
Нафтохиноновые витамины, например филлохинон (I), под влиянием
перхлорной кислоты в дихлорэтане испытывают димеризацию с образова-
нием хроманового спиродимера (XVII) [28, 29]. Реакция протекает через
филлохроманол — нафтотокоферол (XV), образующийся в результате вос-
становительной внутримолекулярной циклизации фнллохинона [30—32]
с последующим окислением феррицианидом калия в щелочной среде [29]
8*
227
или кислородом воздуха [33], или реакция протекает непосредственно под
влиянием кислоты через хинонметид (XVI) [28, 291.
Восстановление спиродимера (XVII) цинком в уксусном ангидриде при-
водит к бис-(нафтотокоферил-5)этану (XIX), который в результате деаце-
тилирования с помощью алюмогидрида лития и окисления хлорным желе-
зом превращается в димер (XVIII) [29]; этот димер получается также
непосредственным окислением спиродимера (XVII) [29, 34].
При фотолизе филлохинона (I) в этиловом спирте или бензоле в ана-
эробных условиях происходит его расщепление с образованием димера
нафтохроменола (XX) [35].
В результате обработки филлохинона (I) концентрированной серной кис-
лотой с последующим разложением водой происходит его гидратация и по-
лучается у-оксипроизводное нафтохинона (XXI) [31, 36].
Для нафтохинонов характерны различные реакции присоединения, про-
текающие по двойной связи, находящейся между двумя карбонилами, и
по атому кислорода карбонила и 0-углеродному атому —1,4-присоедине-
ние, сопровождающееся окислением промежуточной енольной формы вто-
рой молекулой нафтохинона или специально применяемым окислителем.
Для 2-метил-1,4-нафтохинона (VII) реакции присоединения протекают
труднее, чем для неалкилированных хинонов. Однако соединение VII при-
соединяет бром (или хлор) по двойной связи с последующим отщеплением
бромистого водорода и образованием 2-метил-3-бром-1,4-нафтохинона (XXII)
О
VII XXII
Присоединение аминосоединений, например анилина, к 2-метил-1,4-наф-
тохинону (VII) с образованием промежуточного аддукта и получением за-
мещенного аминонафтохинона (XXIII) сопровождается реакцией восста-
новления второй молекулы нафтохпнона (VII), переходящей в 2-метил-1,4-
нафтогидрохинон (XIV) [37].
о
VII
Oi
2 СИ
3 WH»
он
он
XIV
Получен ряд продуктов присоединения 2-метил-1,4-нафтохинона с дру-
гими аминами — с толуидином, бензидином и нафтиламином — в резуль-
228
тате одновременного окисления промежуточных соединений в ходе реак-
ции второй молекулой нафтохинона [37].
Интересно отметить, что реакции присоединения к нафтохинонам про-
текают значительно легче для меркаптопроизводных, чем для аминосое-
динений; так, реакция присоединения к 2-метил-1,4-нафтохинону (VII)
тиогликолевой кислоты проводится в присутствии его избытка, который
используется для окисления промежуточной нафтогидрохиноновой формы
продукта присоединения: при этом образуется (2-метил-1,4-нафтохинонил-3)-
тиогликолевая кислота (XXIV), обладающая высокой анти геморрагичес-
кой активностью [38].
Введение алкильных групп в 3-е положение 2-метил-1,4-нафтохинона
может быть осуществлено при участии окислов свинца или других ката-
лизаторов, причем удлинение углеродной цепи алкильных групп облег-
чает протекание реакции.
Большая способность к реакциям присоединения и к конденсациям с али-
фатическими соединениями объясняет возможность 2-метил-1,4-нафтохино-
на как провитамина превращаться в животном организме в витамин К или
соединения подобного типа [39].
СТРОЕНИЕ НАФТОХИНОНОВЫХ ВИТАМИНОВ
Филлохинон
О наличии в молекуле филлохинона пара-хиноидной структуры можно
заключить на основании его желтой окраски (орто-хиноны обладают оран-
жевой или красной окраской), близкому подобию его восстановительного
потенциала к 1,4-хинонамтипа антрахинон-гидроантрахинон [15] и сходству
его спектра поглощения со спектром 1,4-нафтохинонов [15]. При восстано-
вительном ацетилировании филлохинона в присутствии уксусного ангид-
рида с поглощением двух атомов водорода (схема 54) образуется бесцвет-
ный дигидровитамин Кгдиацетат (XXVII), который при окислении озо-
ном дает кетон — 2,6,10-триметилпентадеканон-14 (XXX), идентичный
[40] с синтетическим кетоном, полученным из фитола, и (2-метил-1,4-ди-
ацетоксинафтил-3)ацетальдегид (XXIX), охарактеризованный по семикар-
базону [41]. Соединение XXVII при мягком окислении хромовой кис-
лотой образует (2-метил-1,4-диацетоксинафтил-3)уксусную кислоту (XXVIII)
[40], метиловый эфир которой идентичен с синтетическим веществом.
Подобным же образом филлохинон при осторожном действии хромовой
кислоты дает (2-метил-1,4-нафтохинонил-3)уксусную кислоту (XXV) [42,
43 ], идентичную синтетической, а при более энергичном окислении — фта-
левую кислоту (XXVI) [43]. Образование фталевой кислоты при деструк-
тивном окислении филлохинона свидетельствует об отсутствии заместите-
лей в бензольном цикле нафтохиноновой части молекулы.
Дигидровитамин Кх-диацетат (XXVII) гидролизом и последующим окис-
лением кислородом воздуха превращается вновь в филлохинон (I) [44].
Из восьми атомов водорода, которые присоединяются к молекуле филло-
хинона при каталитическом гидрировании с образованием бесцветного за-
мещенного тетрагидронафтогидрохинона (XXXI) [15], шесть атомов не-
обходимы для гидрирования бензольного ядра и оксогрупп нафтохинона;
229
Схема 54
Установление строения фнллохинона реакциями его превращений и расщепления
ососн,
сн.
CHjCOOH
ососн,
XXVIII
£н3со)2о| [о]
ососн,
СНз
СН3 СН,
ОСО CH^^CCH^CHjCHjCHCHj^H
{ососн,
СН3
Н2О
си,
CHjCHO
ососн,
XXIX
СН3 СН,
OCCHj^CHjCHCH^H
XXX
присоединение еще двух атомов водорода указывает на присутствие одной
двойной связи в боковой цепи. Замещенный тетрагидронафтогидрохинон
(XXXI) на свету в результате частичного окисления превращается в сое-
динение XXXII желтого цвета, обладающее структурой хинона
Филло-
хинон —►
I
сн,
XXXII
(СН2СН2СНСН2)4Н
XXXI
На основании изучения продуктов расщепления фнллохинона Физер
С сотр. [45—47 ] и Дойзи с сотр. [40, 41, 43] установили, что в состав моле-
кулы витамина включается остаток природного изопреноидного спирта фи-
тола и что строение его отвечает 2-метил-3-фитил-1,4-нафтохинону [47].
Строение филлохинона окончательно было доказано его синтезом [14, 40,
48, 49].
Менахиноны
Для определения строения менахинона-6 — витамина Кг(зо) (Ш)—
Дойзи с сотр. [41, 42] применил те же методы окислительного расщепле-
ния, которые были использованы для установления строения витамина Кх.
Наличие структуры нафтохинона в молекуле менахинона (III) установ-
лено по подобию его ультрафиолетового спектра со спектром поглощения
филлохинона, а также присутствию двух атомов кислорода. При восстано-
вительном ацетилировании с поглощением двух атомов водорода был по-
лучен диацетат дигидровитамина Кг(зэ> (XXXIII) [42], который при окис-
лении озоном [41] образует (2-метил-1,4-диацетоксинафтил-3)ацеталь-
дегид (XXIX), охарактеризованный через семикарбазон, и левулиновый
альдегид (XXXIV), идентифицированный в виде бис-2,4-динитрофенилгид-
разона; выделено около 5 молей левулинового альдегида из каждого моля
соединения XXXIII наряду с 1 молем ацетона (схема 55).
При окислении диацетата дигидровитамина К2(зэ> (XXXIII) марганцо-
вокислым калием была выделена фталевая кислота (XXVI), что свидетель-
230
ствует об отсутствии заместителей в бензольном ядре молекулы нафгохи-
нона.
Схема 55
Установление строения менахииона-6 реакциями его превращении и расщепления
О
f/l"1’ ?<•
Ч^Х^^СИгСН=ССНХСН3СН=ССН!)/::Н2СН = ССН3
О Мвнахинон-6
III
ОСОСН,
сн3
сн, сн, сн,
сн2сн=ссн2(сн2сн=ссн2)4сн2сн=ссн,
ОСОСНз XXXIII
СООН
соон
XXVI
ососн..
СНз
СН2СНО
ОСОСН3
XXIX
сн3
5ОССН2СН,СНО
XXXIV
сн,
соси,
Так как при каталитическом восстановлении менахинона-6 поглощается
18 атомов водорода, из которых шесть необходимы для гидрирования наф-
тохинона, то поглощение остальных 12 атомов водорода доказывает наличие
шести двойных связей в боковой цепи. Это подтверждается также способ-
ностью молекулы хинона присоединять 12 атомов брома. Установлено, что
двойные связи не находятся в сопряжении, так как витамин не образует
продуктов присоединения с малеиновым ангидридом. Строение менахино-
на-6 (III) отвечает 2-метил-3-дифарнезил-1,4-нафтохинону [41, 50].
Из гниющей рыбной муки выделен 2-метил-3-фарнезилдигеранил-1,4-
нафтохинон—менахинон-7 (IV)— полной транс-конфигурации, изопреноид-
ная боковая цепь которого состоит из 35 атомов углерода с семью двойными
связями [4].
Строение этого соединения доказано полным синтезом из 2-метил-1,4-на-,
фтогидрохинона и полного /пра/охД-фарнезилгераниллиналоола. Соеди-
нение IV имеет т. пл. 54° С, т. е. ту температуру плавления, которая все
время относилась к 2-метил-3-дифарнезил-1,4-нафтохинону (III). Таким
образом, витамину Кг с т. пл. 54° С, открытому Дойзи с сотр. [5], на самом
деле соответствует формула IV.
Витамин с формулой III —изопренолог с боковой цепью из 30 атомов
углерода, менахинон-6 —также выделен [4] из гниющей рыбы, но он имеет
т. пл. 50° С. Его строение как 2-метил-3-дифарнезил-1,4-нафтохинона пол-
ной mpawc-конфигурации доказано синтезом из 2-метил-1,4-нафтогидрохино-
на и полного шроас-Л-фарнезилнеролидола.
ВЕЩЕСТВА, РОДСТВЕННЫЕ НАФТОХИНОНОВЫМ ВИТАМИНАМ
Нафтохиноновые витамины, обладающие кровеостанавливающим дей-
ствием, принадлежат к группе пара-хинонов, широко распространенных
в растительном мире среди желтых красящих веществ, в животных орга-
низмах и микроорганизмах.
Помимо витаминов группы менахинона, в микроорганизмах обнаружены
соединения, построенные по типу менахинона-8 и менахинона-9 (витаминов
К«(40) и Кг(45>), с изопреноидными частично насыщенными звеньями в боковой
231
цепи из 40 или 45 атомов углерода, кроме того, одно из этих звеньев полно-
стью насыщено, т. е. соединения имеют на одну двойную связь меньше, чем
у соответствующих менахинонов. Эти соединения обозначаются как 6',7'-
дигидроменахинон-8 или витамин К2(4о>Н (п = 6), выделенный из Myco-
bacterium phlei [511 и Corynebacterium diphteriae [52], и 6',7'-дигидро-
менахинон-9 или витамин К2(45)Н (л = 7), выделенный из Mycobacterium
tuberculesis и др. [53, 54].
Синтезирован аналог — витамин K2(i5)H [54].
Хлоробиумхинон — пигмент нехлорофильной природы, выделенный из
Chlorobium thiosulfatophilum наряду с менахиноном-7 (витамином Кг(зб)) —
является производным винилметилнафтохинона [55], т. е. он имеет в со-
пряжении с хиноном двойную связь и не содержит в первом изопреновом
звене одной метиленовой группы, т. е. является как бы витамином Кг(34)
(XXXV).
о
о
XXXV
Из микроорганизмов выделено вещество нафтохиноновой природы, ли-
шенное метильной группы в положении 2 [56].
К биологически активным пара-хинонам относится окисленная форма
природных 6-оксихроманов — токоферолов, или витаминов группы Е
(см. с. 253), осуществляющих антистерильные и антиокислительные функ-
ции в животном организме. Для важнейшего витамина этой группы —
а-токоферола (XXXVI) —такой окисленной формой является а -токоферол-
хинон (XXXVII), производное 2,3,5-триметилбензохинона, который полу-
чается в результате реакции мягкого окисления а-токоферола хлорным же-
лезом или хлорным золотом.
232
К растительным пара-бензохинонам относится большая разнообразная
группа соединений. 2,3,5-Триметил-6-декапренил-1,4-бензохинон, токохи-
нон-10 (XXXVIII), имеет боковую цепь из десяти частично насыщенных
изопреноидных звеньев с десятью двойными связями в положении 6
Пластохиноны — производные 2,3-диметил-1,4-бензохинона — имеют в
положении 6 боковую цепь из 4—10 частично насыщенных изопреноидных
звеньев с таким же количеством двойных связей
О
СНз II
CHS
(СН2СН = ССН2)„ н
п = 4 4- 10
Пластохиноны содержатся в хлоропластах листа и связаны с процессами фо-
тосинтеза. 2,3-Диметил-6-тетрапренил-1,4-бензохинон, пластохинон-4 (п =4),
имеет такую же боковую цепь, что и менахинон-4 (витамин Касад)- Доказано
строение и осуществлен синтез натурального 2,3-диметил-6-нонапренил-
1,4-бензохинона, пластохинона-9 (п = 9) [57], имеющего ту же соланезиль-
ную боковую цепь, что и менахинон-9 (витамин К2(45>); выделен и иденти-
фицирован пластохинон-10 [58—60] с частично насыщенной изопреноидной
боковой цепью, аналогичной убихинону-10.
К нафтохиноновым витаминам близко примыкает группа природных
пара-бензохинонов—убихинонов (коферментов Q), соединений следующего
строения:
Убихиноны — производные 2-метил-5,6-диметокси-1,4-бензохинона с бо-
ковой цепью в,положении 3, состоящей из 5—10 частично насыщенных
изопреноидных звеньев и такого же количества двойных связей.
Убихинон-10 (кофермент Qlo) (XXXIX) входит в состав митохондрий
клеток печени, сердца, а также других тканей [61—63]. Он участвует в ре-
акциях окислительного фосфорилирования, осуществляя функцию перено-
са электронов по цепи дыхания между флавопротеидами и цитохромом с
[64, 65]. Коферменты Qcn = 5, 6, 7и8 входят главным образом в клетки
микроорганизмов [66—69]. В животных организмах распространены уби-
хинон-10 и убихинон-9 [681; они встречаются также в высших растениях
[63]. Вопросам химии убихинонов посвящены обзорные работы [8, 70].
Из Rhodospirillum rubrum выделен и затем синтезирован аминоаналог
убихинона-10 — родохинон (XL), которому свойственны фотосинтетические
функции [71].
233
XL
Помимо пара-бензохинонов в природе широко распространены соот-
ветствующие им бициклические хромановые производные — хроманолы-6.
Важнейшими представителями хроманолов являются токоферолы — ви-
тамины группы Е (см. с. 253), например а-токоферол (XXXVI), а также
токотриенолы, в качестве представителя которых можно указать у-токо-
триенол (^-токоферол), пластохроманол-3 (XLI). Группа природных плас-
тохроманолов — пластохроманол-8 (XLII), 3,4-дегидропластохроманол-8
(пластохроменол-8, соланохромен) (XLIII), токохроманол-9 (XLIV) и др. —
являются производными соответствующих пластохинонов [721.
сн3
Y" jT jLCH2(CH2CH=CCH2)eH
СНД ° CHj
сн,
jYT XcH^CHjCH^CCHj^H
сн/т 0 ch3
CHa
CH,
HO YY. CH,
Ych2(ch2ch=cch2)8h
ch/T 0 CH3
CH,
XLIV
Убихинонам (коферментам Q) соответствует группа циклических хро-
мановых производных — убихроманолов и убихроменолов с числом час-
тично насыщенных изопреноидных звеньев п, равным 5—9. ;Убихрома-
нол-9 (XLV) и убихроменол-9 (XLVI) находятся в клетках животных и
человека (почках, печени и др.) и высших растений [73, 74]; их низшие
изопренологи обнаружены у рыб, в микроорганизмах, водорослях и в очень
малых количествах в организме теплокровных животных.
Подобно токоферолам, пластохроманолам и убихроманолам, нафтохи-
ноновые витамины образуют нафтохроманолы, например из филлохинона
(витамина K2<2oj) получен филлохроманол — нафтотокоферол (XV) [30—
32]
обладающий небольшими антигеморрагическими свойствами и в высокой
степени витаминной активностью токоферолов.
234
Среди красящих веществ грибов, плесеней и различных растений обна-
ружены такие пара-хиноны, как 8-окси-1,4-нафтохинон, юглон (XLVII),
который находится в скорлупе незрелых грецких орехов Juglans regia,
2-метил-3-окси-1,4-нафтохинон, фтиокол (XI), выделенный из продуктов
конечного обмена животного организма (75J, эхинохром (XLVIII) — кра-
сящее вещество, выделенное из яиц морского ежа, например Arbacia pustu-
losa, и многие другие.
XLV1I
XLVIII
СИНТЕЗ НАФТОХИНОНОВЫХ ВИТАМИНОВ
Синтез филлохинона
Синтез филлохинона (витамина Ki) (I) одновременно осуществили в
1939 г. Физер с сотр. [48, 49], Алмквист и Клоузе [14] и Дойзи с сотр.
[40].
В синтезе филлохинона по одному из методов исходят из 2-метил-1,4-на-
фтогидрохинона (XIV), который конденсируют с фитолом (XLIX) в среде
диоксана при 75° С с участием щавелевой кислоты [48, 49]; при этом обра-
зуется 2-метил-3-фитил-1,4-нафтогидрохинон (XIII), который очи-
щают от исходного нафтогидрохинона обработкой раствором щелочи. Из
маслообразного остатка с помощью петролейного эфира выделяют витамин
Ki-гидрохинон (XIII) и окисляют его окисью серебра или воздухом в фил-
Схема 56
Синтез филлохинона
СН, СИ,
+ носн2сн=ссн2(сн2сн2снснДн
XL1X
I________
СН,
сн, сн,
сн2сн=ссн2(сн2сн3снсн1!),н
XIII
-СН,
СН, сн,
сн3сн=ссн2(сн,сн,снснДн
лохинон (I) (схема 56).
он
он
XIV
сн, СИ,
+ Вгсн,сн=ссн2(сн2сн,снсн,),н
L
VII
235
Важное практическое значение имеет путь синтезаХфиллохинона (I),
заключающийся в конденсации 2-метил-1,4-нафтохинона (VII) с фитил-
бромидом (L) в среде петролейного эфира (схема 56) в присутствии цинко-
вой пыли [14].
Реакция конденсации 2-метил-1,4-нафтогидрохинона (XIV) с фитолом
(XLIX) сопровождается образованием фитилоксалата и изомерного 2-ме-
тил-2-фитил-2,3-дигидро-1,4-нафтохинона (LI) [76]
О
LI
в результате фитилирования 2-метил-1,4-нафтогидрохинона (XIV) в поло-
жении 2.
Помимо щавелевой кислоты, при взаимодействии соединений XIV и
XLIX в качестве конденсирующих средств применяют кислый сульфат ка-
лия [76, 77], этилат натрия в присутствии воздуха [78], эфират трехфто-
ристого бора [10, 25, 76], хлоруксусную кислоту или хлористый цинк [79].
В более мягких условиях конденсации (с несильными кислотами) получа-
ется лучший выход витамина Ki-гидрохинона (XIII). Применение мине-
ральных кислот или сильных конденсирующих средств (типа хлористого
цинка) в более жестких условиях конденсации ведет к получению значитель-
ного количества побочного нафтотокоферола (XV, см. с. 229) [47, 80].
Выход филлохинона при конденсации фитола (XLIX) с 2-метил-1,4-наф-
тогидрохиноном (XIV) повышается до 42%, если в качестве катализатора
применить кислый сульфат калия, причем продолжительность реакции сни-
жается в 27 раз [76].
Если вместо 2-метил-1,4-нафтогидрохииона в реакцию взять его 1-моно-
ацетат, то получается 1-моноацетил-2-метил-3-фитил-1,4-нафтогидрохинон
и изомерный продукт не образуется; при этом выход филлохинона (I) по-
вышается при применении кислого сульфата калия до 50% [81 ], а в случае
эфирата трехфтористого бора —до 73% [76, 82]. Хорошие результаты дает
применение в качестве конденсирующего средства безводного хлористого
алюминия [82, 83], а для окисления промежуточного витамина Кргидро-
хинона—окиси свинца [83]. Щелочные конденсирующие реагенты дают
малый выход конечного вещества.
Конденсация 2-метил-1,4-нафтогидрохинона с синтетическим изофито-
лом — 3,7,11,15-тетраметилгексадекан-1-олом-З (LII)
СН3 СН3 СН3
СН2=СНССН2(СН2СН2СНСН2)2СН1СН2СНСН3
I
он
LH
при каталитическом участии эфирата трехфтористого бора в среде диоксана
при 50° С приводит с высоким выходом к получению витамина Кргидро-
хинона с рацемической боковой цепью, при окислении переходящего в ра-
цемический филлохинон [25, 84]. В качестве конденсирующего средства в
конденсации применяют метансульфокислоту и хлористое олово [85].
Из 1-моноацетата 2-метил-1,4-гидрохинона и изофитола в присутствии без-
водного хлористого алюминия выход филлохинона (I) достигает 70 % [86].
Филлохинон также получен из мононатриевого производного 2-метил- 1,4-на-
фтогидрохинона и фитилбромида [40]. После окисления продукта конден-
23G
сации кислородом воздуха производится хроматографическая очистка и
вакуум-перегонка.
По одному из вариантов витамин Ki-гидрохинон (XIV) синтезирован с
невысоким выходом в результате конденсации фитола (XLIX) с восстанов-
ленной формой фтиокола (XI) [80]. Для конденсации с 2-метил-1,4-нафтогид-
рохиноном применен 3, 7, 11, 15-тетраметилгексадека-1,3-диол [87].
Из 2-метил-1,4-нафтохинона-2-14С получен соответствующий ему филло-
хинон с меченым 14С [88 ].
Синтез менахинонов
Менахиноны с различным количеством изопреноидных звеньев синте-
зированы по тому же принципу, который применен для синтеза филлохи-
нона, в частности посредством конденсации 1-моноацетата 2-метил-1,4-наф-
тогидрохинона (LIII) и соответствующего ненасыщенного изопреноидного
спирта (LIV) в среде циклогексана, эфира или другого растворителя в при-
сутствии катализатора—эфирата трехфтористого бора, хлористого алю-
миния или хлористого цинка; образовавшееся в результате конденсации
промежуточное соединение LV окисляют окисью серебра или другими окис-
лителями в менахиноны (II—VI).
Для синтеза менахинона-4 (витамина Кг(20)) (II) применяют геранилли-
налоол (Сго) (LIV, п = 4) [89], для синтеза менахинона-6 (витамина Кг(зо))
(III) —фарнезилнеролидол (С3о) (LIV, п = 6) [4, 89, 90], для менахино-
на-7 (витамина Кг<з5)) (IV)—фарнезилгераниллиналоол (С(з5>) (LIV, п =7)
[4, 89]. Синтез менахинона-9 (витамина Кг (45>) (VI) осуществлен из 2-ме-
тил-1,4-нафтогидрохинона (XIV) и соланезола (С45), выделенного из таба-
ка [3].
Получение исходных веществ для синтеза нафтохиноновых витаминов
Методы получения 2-метил-1,4-нафтохинона (менадиона, витамина К3),
необходимого для синтеза нафтохиноновых витаминов, рассматриваются
ниже.
2-Метил- 1,4-нафто хинон (VII) синтезируют из легкодоступного 2-метил-
нафталина, являющегося одним из продуктов каменноугольного дегтя,
путем окисления хромовой кислотой в среде уксусной кислоты при 50—90° С
[48, 91—93] с выходом до 60% [94]. В качестве окислительных реагентов
могут служить также кислород воздуха, перекись водорода [95], хромовый
ангидрид [961 (выход 30%) и др.
Кроме того, 2-метил-1,4-нафтохинон ^П)может быть получен диеновым
синтезом Дильса—Альдера из толухинона. По одному из методов для кон-
денсации с толухиноном (LVI) применяют 1,3-бутадпен [97, 98]
237
Конденсацию, как правило, проводят под давлением; образуется 2-метил-
4а,5,8,8а-тетрагидро-1,4-нафтохинон (LVII), который под влиянием хло-
ристого водорода и хлористого олова [97] или бромистого водорода в ле-
дяной уксусной кислоте [99] изомеризуется в 2-метил-5,8-дигидро-1,4-наф-
тогидрохинон (LVIII). Это соединение окисляют хромовым ангидридом или
окисью серебра и затем тетраацетатом свинца с 58%-ным выходом в 2-ме-
тил-1,4-нафтохинон (VII). Конденсация осуществлена также в ледяной
уксусной кислоте при 110° С [98], причем образовавшееся вещество LVII
одновременно изомеризуется в соединение LVIII с выходом 95%; его без
выделения из раствора окисляют в 2-метил-1,4-нафтохинон (VII) в той же
среде [98]. Если конденсацию проводить при комнатной температуре, то
первоначально получается вещество LVII, которое отделяют от раствора и
изомеризуют кипячением с водой в соединение LVIII. В качестве среды для
конденсации применяется также нитробензол.
По второму варианту диенового синтеза толухинон (LVI) конденсируют
с циклогексадиеном [100] в 5,8-этилен-4а,5,8,8а-тетрагидро-2-метил-1,4-на-
фтохинон (LIX), который в результате каталитической изомеризации при
действии бромистоводородной кислоты переходит в 5,8-этилен-5,8-дигидро-
2-метил-1,4-нафтогидрохинон (LX). Окислением хлорным железом его
переводят в 5,8-этилен-5,8-дигидро-2-метил-1,4-нафтохинон (LXI), а затем
термической деэтиленизацией — в 2-метил-1,4-нафтохинон (VII).
Установлено, что 2-метил-1,4-нафтохинон может быть получен из 1,4-наф-
тогидрохинондиацетата хлорметилированием формалином и соляной кис-
лотой с последующим восстановлением цинком и соляной кислотой [101].
Описан синтез 2-метил-1,4-нафтохинона из итаконовой кислоты [102].
Для очистки сырой 2-метил-1,4-нафтохинон промывают четыреххлорис-
тым углеродом [103].
2-Метил-1,4-нафтохинон с радиоактивным 14С получен в виде ряда сое-
динений, отличающихся положением и количеством радиоактивных ато-
мов углерода: из 0 -бромнафталина через £ -нафталинкарбоновую кислоту
[104], из р-нафтилмагниййодпроизводного и меченого йодистого метила, из
меченой метилянтарной кислоты и бензола, из меченой янтарной кислоты
и толуола [93]. Синтез 2-метил-1,4-нафтохпнона-4-14С осуществлен из бенз-
альдегида с применением изотопной углекислоты [105].
Природным источником фитола, необходимого для синтеза филлохино-
на, служит хлорофилл, получаемый из зеленых частей растений. Для син-
238
теза фитола известны методы, в которых исходят из псевдоионона и фарне-
зола (106, 107], и др. (см. с. 278).
Изофитол получают из псевдоионона через гекса гидропсевдоионон [84,
108] или из линалоола через геранилацетон [109] и др. (см. с. 282).
Синтез гераниллиналоола (С^) (LXIII), фарнезилнеролидола (С3о)
(LXIV) и фарнезилгераниллиналоола (С35) (LXV), необходимых для полу-
чения менахинона-4, менахинона-6 и менахинона-7, осуществлен из не-
ролидола (LXII) путем последовательного наращивания изопреноидных
звеньев, что достигалось при использовании трех однотипных методов,
включающих синтез кетонов из винилкарбинолов с помощью ацетоуксусного
эфира или дикетена, конденсации кетонов с ацетиленом в ацетиленкарбино-
лы и их избирательного гидрирования на катализаторе Линдлара в винил-
карбинолы [89, 90, НО].
Описан синтез фарнезилнеролидола (LXIV) из 2-метил-2-гептен-6-она
через линалоол [89]. Полный транс, транс-герани л линалоол синтезирован
из псевдоионона с использованием метода гидролиза силикоэфиров, обра-
зующихся присоединением триэтоксисилангидрида к а, 0-непредельным
карбонильным соединениям [111].
БИОЛОГИЧЕСКОЕ ЗНАЧЕНИЕ НАФТОХИНОНОВЫХ ВИТАМИНОВ.
ЗАВИСИМОСТЬ МЕЖДУ СТРОЕНИЕМ И БИОЛОГИЧЕСКОЙ АКТИВНОСТЬЮ,
АНТИВИТАМИНЫ
Недостаток в животном организме нафтохиноновых витаминов лишает
кровь способности свертываться при повреждении ткани — возникает кро-
вотечение (геморрагия).
Авитаминоз К проявляется у птиц в виде геморрагического диатеза.
Авитаминоз К у млекопитающих и человека почти не наблюдается, по-ви-
димому, из-за синтеза витамина К микрофлорой кишечника. Однако в не-
которых случаях склонность к кровотечениям вызывается недостаточностью
всасывающей способности кишечника из-за малого поступления в кишечник
желчи, которая служит эмульгатором витамина К. Лучшая всасываемость
витамина К находится в зависимости от наибольшей его дисперсности.
Механизм действия витамина К в регулировании процессов свертывания
крови, вероятно, состоит в том, что он, обладая окислптельно-восстановп-
239
тельной хинон-гидрохиноновой системой, образует с белковым апофермен-
том фермент, который катализирует синтез протромбина. При повреждении
клеточных тканей из протромбина плазмы крови в присутствии ионов каль-
ция под влиянием фермента тромбокиназы образуется специфический бел-
ковый фермент тромбин (не содержащийся в крови); тромбин вызывает свер-
тывание крови путем полимеризации растворимого протеинообразного
вещества фибриногена (молекулярная масса 330 000) в нерастворимый про-
теин фибрин (молекулярная масса около 5 000 000), образующий сгустки
и нити, вызывающие тромб.
Возможно, что в процессе свертывания крови принимает участие также
тромботропин, получающийся при участии витамина К [112]. Биосинтез
протромбина происходит в печени. В случае заболевания печени или за-
купорки желчных протоков (обтурационная желтуха) поступления вита-
мина К в организм не происходит и наблюдается недостаток протромбина.
Менадион (витамин Кз) имеет значение в птицеводстве, в борьбе против
так называемого каннибализма, когда при случайном ранении другие куры
расклевывают кровоточащие раны до полной гибели птицы.
Витаминная активность определяется по ускорению свертывания крови
цыплят — до 4—5 мин вместо 1 ч и более при авитаминозе — и мышей.
Дневная потребность авитаминозных цыплят составляет 1 мкг витамина
Ki или 1,6 мкг витамина К2<35>-
Витамин К имеет значение для предупреждения послеоперационных кро-
вотечений и геморрагии новорожденных.
В настоящее время имеется много данных в пользу того, что нафтохино-
новые витамины принимают участие в переносе электронов и протона в био-
химических реакциях окислительного фосфорилирования, превращаясь
при этом в циклическую хромановую форму замещенного 6-нафтохроманола
[30, 113, 114].
Нафтохиноновые витамины не обладают высокой специфичностью. Анти-
геморрагическая активность свойственна не только природным производ-
ным 2-метил-1,4-нафтохинона с боковой цепью в положении 3, состоящей
из различного количества насыщенных и частично насыщенных изопре-
ноидных звеньев, но и отдаленным природным и синтетическим аналогам.
Гидрирование витамина Ki двумя атомами водорода, при котором хи-
ноидная группа превращается в гидрохиноновую, почти не связано с по-
терей антигеморрагической активности [115]. Гидрирование бензольного
ядра нафтохиноновой части молекулы витамина снижает активность в 4—
125 раз. Метилирование этого же ядра (кроме положения 5) ведет к полной
потере активности; введение метильной группы в положении 5 еще сохра-
няет активность, понижая ее в 170 раз. Чрезвычайно важное значение имеет
метильная группа в положении 2. Удлинение ее на метиленовый остаток,
т. е. замена на этильную группу, приводит к уменьшению активности в
1000 раз; также влияет и ее замещение на аллильную или ацетоксиме-
тильную группу. Удаление метильной группы снижает активность в 50 раз.
Дальнейшее удлинение алкильного остатка в положении 3, имеющего
четыре почти полностью насыщенных изопреноидных звена (20 атомов
углерода), на одну или две насыщенные изопреноидные группы, т. е. на
5 или 10 атомов углерода (С25 С3о), приводит к уменьшению антигеморра-
гической активности на 20 и 50% соответственно. Укорачивание боковой
цепи на одну, две или три насыщенные изопреноидные группы уменьшает
активность до 30, 10 или 5% по сравнению с активностью филлохинона.
Удаление метильных групп в положении 11' и 15' связано с уменьшени-
ем активности в 3 и 9 раз, а в положении 3' и 7' с 8-кратным уменьшени-
ем активности, так же как и насыщение р, у-двойной связи.
Синтезированы и испытаны на витаминную активность аналоги филлохино-
на с одной частично насыщенной изопреноидной группой и остальной нераз-
ветвленной цепью с 11—20 атомами углерода 1116, 117], с дважды разветв-
ленной боковой цепью [118], 6,7-дпметил-и 6,7-диметокси- [119], а также
240
со-карбокси- и со-оксианалоги филлохинона [120—122], аналоги без метиль-
ной группы в положении 2 [123] и с винильной группой в положении 3
[124, 125], оказавшиеся неактивными или малоактивными соединениями.
О влиянии на активность филлохинона различных модификаций его
молекулы можно судить по следующей схеме (за 100% принята активность
витамина Кг):
03-25%
I
Г идрировдние
Удаление группы СН3
।—।---------:—।---------------1------------1
2% 12,5% 12,5% 11-33% 11-33%
J . . I ।
сн3 сн3 сн3 сн3
СН2СН=ССН2СН2СН2СНСН2СН2СН2СНСН2СН2СН2СНСН3
Z । 4 Z 2 ** * Л. it Л
Р 11 I у ♦ ... t 1S.
12,э% 5% Ю% 30%
II I I
Гидрирование Укорачивание боковой цепи
Рацемизация двух асимметрических центров витамина Ki не отражается
на его биологической активности [25].
Уменьшение количества частично насыщенных изопреноидных звеньев в
боковой цепи положения 3 по сравнению с молекулой менахинона-4 (витами-
на Кг(2Э)) ведет к многократному уменьшению активности [126] (табл. 16).
Таблица 16
Влияние заместителя в положении 3 молекулы
2-метил-1,4-нафтохинона иа антигеморрагическую активность
Заместитель в положении 3 2-метил-1,4-нафтохинона Число атомов углеро- да в боковой цепи Количество ненасыщен- ных групп СН3 1 (СН,СН=ССН,) в боковой цепи Антигеморрагнчеосая активность, %
Геранил 10 2 Около 15
Фарнезил 15 3 Около 40
Дигеранил 20 4 100
Фарнезилгеранил 25 5 Около 120
Дифар незил 30 6 100
Фарнезилдигеранил 35 7 Около 70
2-Метил-1,4-нафтохинон (менадион), не имеющий алкильного замести-
теля в положении 3, обладает высокой витаминной активностью (0,3 мкг)1
[127], превышающей активность витамина Ki более чем в 3 раза (при отне-
сении к единице массы, а на моль вещества оказывается почти равной),
по-видимому, вследствие его способности замещаться в живом организме
в положении 3 группой фитила с образованием филлохинона. Однако
и токсичность 2-метил-1,4-нафтохинона значительно превышает токсич-
ность витамина Ki- При испытании на мышах 2-метил-1,4-нафтохинон ток-
сичен при дозировке 0,8—1 г на 1 кг, в то время как витамин Ki не ток-
сичен в дозах 25 г на 1 кг (при пероральном применении). Отсутствие
также и метильной группы в положении 2 почти полностью лишает это со-
единение (1,4-нафтохинон) активности [14].
Введение гидроксильной группы в любое положение почти полностью
инактивирует молекулу витамина К; активностьфтиокола (XI) (гидроксиль-
ная группа замещает положение 3) составляет 1/500 активности вита-
мина Ki-
1 Активность выражена в дневной потребности авитаминозных цыплят.
241
Введение в положение 3 молекулы 2-метил-1,4-нафтохинона некоторых
заместителей сохраняет биологическую активность соединения на высоком
уровне. Это относится к 2-метил-3-диметиламиноэтиламино-1,4-нафтохино-
ну (LXVI) [128]
XXIY
и к (2-метил-1,4-нафтохинонил-3) тиогликолевой кислоте (XXIV).
Замена метильной группы в положении 2 молекулы 2-метил-1,4-нафто-
хинона на аллильную или ацетоксиметильную приводит к соединениям с
очень низкой активностью.
При изучении строения витаминов К было установлено [127], что не-
которые нафтохиноны обладают высокой антигеморрагической активностью.
При заболевании желтухой лечебная доза 2-метил-1,4-нафтохинона или
натриевой соли его бисульфитного производного для человека составляет
10—15 мг в течение 3 дней, однако активность калиевой соли бисульфит-
ного производного [129—131 ] составляет только 1,25% активности 2-метил-
1,4-нафтохинона [128, 132].
Биологической активностью характеризуются не только нафтохиноны,
но и нафтогидрохиноны — почти в той же степени (табл. 17).
Сильно измененные структуры — нафтиламинопроизводные и нафтолы
с метильной группой в положении 2 (или 3) — также высокоактивны, что
может быть объяснено их способностью легко переходить в процессе обмена
веществ в производные 2-метил-1,4-нафтохинона. В табл. 17 приведен ряд
таких соединений, активность которых составляет.не менее 12% активности
филлохинона [115].
Некоторые соединения, сильно отличающиеся по своей структуре от
2-метил-1,4-нафтохинона, все же показывают, хотя и незначительную,
антигеморрагическую активность. Так, 3-метилхромон (LXVII) [76] обла-
дает свойствами нафтохиноновых витаминов, но 3,6-диалкилхромон мало-
активен. Замена бензольного цикла 2-метил-1,4-нафтохинона на тиофеновый
дает5-метилбензотиофен-4,7-хинон (LXVIII) [143,144], обладающий 3 %-ной
активностью 2-метил-1,4-нафтохинона [144]; так как тиофен, являясь изо-
стером бензола, по своим физико-химическим свойствам, а также по своему
химико-терапевтическому действию в виде его производных почти анало-
гичен бензолу, то этим можно объяснить относительно высокую активность
тиофенового аналога. Однако замещение бензольного ядра на пятичленное
ядро, включающее атом селена, а также на пиридиновое ядро приводит к
неактивным 3,5-диметилбензоселенофен-4,7-хинону [145] и 6-метилхинолин-
5,8-хинону (LXIX) [146].
LXVII
LXVIII
LXIX
Еще более сильное упрощение молекулы витамина Ki за счет удаления
хиноновой или гидрохиноновой группировки и сведения циклической части
молекулы только к бензольному кольцу все же оказывается возможным для
сохранения у некоторых соединений небольшой антигеморрагической актив-
ности. Так, фталевая кислота, являющаяся продуктом метаболизма впта-
242
Таблица 17
Важнейшие соединения, обладающие активностью иафтохиноновых витаминов
Соединение Минимальная активная доза, мкг Литературная ссылка
2,3 -Д налкил-1,4- нафтохиноны
2-Метил-3-фитил-1,4-нафтохинон, филлохинон (ни- 1 [48]
тамин К1(2э))
Витамин Кх-гидрохинондиацетат 2 42]
5,8-Дигидровитамин Ki 4 97, 115]
2-Метил-З-дигидрофитил-1,4-нафтохинон 8 18, 50, 133]
Витамин Ki-перокись 1,2 18, 133]
2-Метил-3-дифарнезил-1,4-нафтохинон, менахи- 1,6 15]
нон-6 (витамин К2(30))
Витамин К2(з5) -гидрохинондиацетат 3,2 [42]
2-Метил-3-фарнезил-1,4-нафтохннон 5 [18, 97]
2-Алкил-1,4-нафтохиноны
2-Метил-1,4-иафтохинон (витамин Кз) 0,3 [127]
2-Мети л-1,4-нафтохинон -2,3-перокись 5 [48]
2-Метил-5,8,9,10-тетраг идро-1,4-нафтохинон 8 [97]
2-Метил-1,4-нафтохинонмонооксим Около 2 [134]
2-Алкил-1,4- нафтогидрохиноны
2-Метил-1,4-нафтогидрохинон (витамин К<) 0,5 [135]
2-Метил-1,4-нафтогидрохиноидиацетат I 48, 136]
2-Метил-1,4-нафтогидрохинондифосфат 0,5 137, 138]
2-Метил-1,4-нафтогидрохинон (дибензоат, дисук- 1 48 , 80, 134, 136]
цинат, моноэтилат)
2-Метил-1,4-нафтогидрохинондиметилат 5 [136]
2-Метил-1,4-нафтогидрохиноидибензилат 7 [48]
2-Метил-1,4- нафтог идрох инондисульфат 2 [97, 138]
2-Метил-5,8-дигидро-1,4-нафтогидрохинон 6 [97]
Нафтолы •
2-Метил-1 -нафтол 1 [115, 139]
З-Метил-1 -нафтол 0,6 [115, 139]
2-Метил-1 -тетралон 0,6 [115, 139]
З-Метил-1 -тетралон 1 [115, 139]
Нафтиламинопроизводные
2-Метил-4-амино-1-нафтол НС1 (витамин К5) 1 [140, 141]
3-Метил-4-амино-1-нафтол НС1 (витамин Кт) 1 [140]
2-Метил-3-амино-1-н'афтол НС1 2 [142]
2-Метил-1 -нафтиламин 5 [133, 139]
2-Метил-1,4-нафтилдиамин 2НС1 (витамин Кв) —
S-Производные 2-мети л-1,4-
нафтохииона
8-(2-Метил-1,4-нафтохинонил-З) тиогликолевая 2 [38]
кислота
5-(Метил-1,4-иафтохинонил-З) тиогликолилглицин 5—50 [38]
мина К [147], при испытании на крысах [1481 проявила слабую актив-
ность, а ее диэтиловый эфир был в этом отношении несколько более эффек-
тивен.
Ряд химических соединений обладает способностью приостанавливать
процесс свертывания крови. Эти соединения являются антагонистами вита-
мина К.
3,3,-Метилен-бнг-(4-оксикумарин), дикумарин (дикумарол) (LXX), явля-
ется сильным геморрагическим средством [149]. Он вызывает кровотечение
у крупного рогатого скота при кормлении испорченным донником, из ко-
торого он выделен.
он он он
LXX LXXI
Являясь антивитамином, дикумарин (LXX) понижает содержание
протромбина в крови; применение витамина К устраняет этот недостаток.
В медицине дикумарин применяется при лечении внутренних кровоизли-
яний, тромбофлебитов, инфаркта миокарда и других заболеваний. Он
имеет значение как заменитель гепарина, используемого для консервиро-
вания крови. Дикумарин синтетически получают из ацетилированного ме-
тилсалицилата, который под влиянием натрия циклизуют в 4-оксикума-
рин и затем с формальдегидом переводят в дикумарин. В отличие от ди-
кумарина З-метил-4-оксикумарин (LXXI) обладает слабым антигемор-
рагическим действием [150].
В медицине находит применение неодикумарин, или пелентан
(LXXII), обладающий теми же геморрагическими свойствами, что и ди-
кумарин, но более мягко выраженными. Много антикоагулянтов оказа-
лось среди эфиров производных 3,3'-метилен-бпс-(4-оксикумарина), по-
строенных по типу соединения LXXIII [151].
LXXII LXXIII
Гомолог дикумарина 3,3'-этилен-бис-(4-оксикумарин) также относится к
группе антивитаминов.
Высокоактивное производное монодикумарина, каким является 3-(а-фе-
нил)пропил-4-оксикумарин (LXXIV), имеет значение для лечения и профи-
лактики тромбозов.
Для борьбы с грызунами применяется 3-(а-фенил-р-ацетил)этил-4-окси-
кумарин, вызывающий внутреннее кровотечение [152 ], а его нитропроизвод-
ное — 3-(а-м-нитрофенил-р-ацетил)этил-4-оксикумарин (LXXV) — имеет
применение в медицине вследствие малой токсичности и слабых кумуля-
тивных свойств.
он
LXXIV
Антивитамином является также 2,2'-метилен-бис-3-окси-1,4-нафтохинон
(LXXVI) [150],
244
о
о
LXXVI
в то время как его структурный элемент — фтиокол (XI) обладает слабыми
витаминными свойствами.
Вместе с тем антивитаминную активность проявляет и ряд других про-
изводных 1,4-нафтохинона, как, например, 2-хлор-1,4-нафтохинон и про-
изводные 3-окси-1,4-нафтохинона, такие, как 2-хлор-З-окси-1,4-нафтохи-
нон [150], 2-(2-метилоктил)-3-окси-1,4-нафтохинон (LXXVII), 2-(3'-цикло-
гексилпропил)-3-окси-1,4-нафтохинон (LXXVIII) [153],
LXXVII
а также 2-[3'-(декагидро-0 -нафтил)пропил]-3-окси-[153], 3-(1-фенил-2- аце-
тилэтил)-2-окси- и 3-(1-фенилбензоилэтил)-2-окси-1,4-нафтохиноны [154].
Синтезировано большое количество нафтохинонов с 3-тиоалкильными
и 6-ацильными и другими группами, а также галогензамещенные с целью
изучения их антивитаминной активности [155].
Провитамины (например, 2-метил-1,4-нафтохинон) малоэффективны для
противодействия антивитаминам (антикоагулянтам) в отличие от витамина
Ki [153].
ПРОВИТАМИНЫ
В основе строения нафтохиноновых витаминов лежит молекула 2-ме-
тил-1,4-нафтохинона. В настоящее время синтетически получены много-
численные производные нафтохинона и нафталина, структурно отличаю-
щиеся от нафтохиноновых витаминов, но обладающие высокой биологичес-
кой активностью. Наиболее активные из этих соединений, обладающие
относительно невысокой токсичностью, следует рассматривать как прови-
тамины нафтохиноновых витаминов.
2-М е т и л-1,4-н афтохинон, менадион (витамин Кз) (VII)
[98, 156, 157] обладает тонким характерным запахом и жгучим вкусом;
он раздражающе действует на кожу и верхние дыхательные пути. Менадион
кристаллизуется в иглах лимонно-желтого цвета с т. пл. 106—106,5° С.
Он малорастворпм в воде (0,013%), хорошо растворим в спиртах (1,7 г
в 100 мл этилового спирта), маслах (1,3 г в 100 мл сезамового масла), бен-
золе и других органических растворителях; плохо растворим в петролейном
эфире. Его минимальная активная доза —0,3 мкг*.
В и касол (LXXIX) [158] — натриевая соль бисульфитного произ-
водного 2-метил-1,4-нафтохинона, 2-метил-1,4-диоксо-1,2,3,4-тетрагидро-2-
нафталинсульфонат натрия [129, 130, 158—161] — бесцветные кристаллы
с т. пл. 154—157° С; имеет горький вкус. Он хорошо растворим в воде,
трудно в спирте, почти нерастворим в эфире, малотоксичен и поэтому имеет
большие преимущества для терапевтического применения [129, 130, 162,
163]; по активности равнозначен менадиону.
• См. примечание на с. 243.
245
LXXIX
Получена и кальциевая соль бисульфитного производного 2-метил-1,4-
нафтохинона [1641 — кристаллы с т. пл. 97—98° С; для безводной соли
т. пл. 115—117° С.
2-М е т и л-1,4-н а ф т о г и д р о х и н о н (витамин К4) (XIV) пред-
ставляет собой бесцветные кристаллы. Его диацетат имеет т. пл. 115° С;
к-дибутират 2-метил-1,4-нафтогидрохинона (LXXX)—т. пл. 50—53° С;
дифосфат 2-метил-1,4-нафтогидрохинона, тетранатриевая соль (LXXXI),
кристаллизуется в бесцветных кристаллах. Минимальная активная доза
витамина К4 — 0,5 мкг, его солей 0,5 —1 мкг.
XIY LXXX LXXXI
2-М е т и л-4-а мино-1-нафтол гидрохлорид — (витамин IQ
(LXXXII) — иглы розового цвета, т. пл. 280 ° С (с разл.); он хорошо
растворим в воде и вследствие малой токсичности рекомендован для приме-
нения [165]. Его N-ацетат — бесцветные иглы с т. пл. 209° С.
2- М е т и л-1,4-д иаминонафталин дигидрохлорид
(витамин Кс) (LXXXIII) —бесцветные кристаллы ст. пл.около300°С; он
хорошо растворим в воде и обладает большей стабильностью, чем витамин
Кх. Его диацетат плавится при 310° С.
3-М е т и л-4-а м и н о-1-н а ф т о л гидрохлорид (витамин К7)
(LXXXIV) — бесцветные кристаллы.
LXXXIII LXXXIV
Для витаминов К5, К6 и К7 минимальная активная доза 1 мкг.
Метод анализа менадиона (VII) и викасола (LXXIX) основан на реакции
восстановления хинона в гидрохинон йодистым водородом; выделяющийся
при реакции йод титруется гипосульфитом натрия [166].
СИНТЕЗ ПРОВИТАМИНОВ
Методы синтеза 2-метил-1,4-нафтохинона — менадиона (витамина Кз)
(VII) — приведены выше (см. с. 239). Кальциевая соль бисульфитногопро-
изводного 2-метил-1,4-нафтохинона получается из суспензии 2-метил-1,4-
нафтохинона и углекислого кальция в воде при пропускании сернистого
ангидрида или при действии бисульфита кальция в метиловом спирте [167].
Натриевая соль бисульфитного производного 2-метил-1,4-нафтохинона
(LXXIX) получается из водного раствора бисульфита натрия и 2-метил-
1,4-нафтохинона (VII) [128, 129, 157, 158, 168—170]. Эта реакция протекает
с образованием хорошо растворимого бисульфитного производного 2-ме-
тил-1,4-нафтохинона, которое при длительном кипячении в водном растворе
246
переходит в соль нормальной сульфокислоты, т. е. в натриевую соль 2-ме-
тил-1,4-нафтогидрохинон-З-сульфокислоты (LXXXV).
LXXXV
Реакция образования бисульфитного производного 2-метил-1,4-нафто-
хинона изучалась многими авторами [130, 157, 158, 163, 171, 172]. Это сое-
динение способно давать соответствующие гидразины [172], что указывает
на присутствие карбонильных групп, однако они не образуют характерной
сопряженной хиноидной системы. Это подтверждается тем, что ультрафио-
летовый спектр поглощения бисульфитного производного 2-метил-1,4-наф-
тохинона (LXXIX) отличается от спектра поглощения типичных нафтохи-
нонов, для которых характерно сопряжение карбонильных групп хиноидной
системы. Однако спектр поглощения соединения LXXIX подобен спектру
2,3-перокиси 2-метил-1,4-нафтохинона, в котором окисное кольцо прерывает
сопряжение двойных связей хиноидной системы [160]. В инфракрасном
спектре бисульфитного производного 2-метил-1,4-нафтохинона обнаружи-
вается отсутствие гидроксильной группы, что наряду с данными ЯМР под-
тверждает его строение, выраженное формулой LXXIX [163, 173].
2-Метил-1,4-нафтогидрохинон (витамин К4) получают восстановлением
2-метил-1,4-нафтохинона (VII). Его диацетат образуется в результате вос-
становительного ацетилирования 2-метил-1,4-нафтохинона. Дифосфат 2-ме-
тил-1,4-нафтогидрохинона синтезируют при действии хлорокиси фосфора
на 2-метил-1,4-нафтогидрохинон (XIV) [154, 174].
Для получения гидрохлорида 2-метил-4-амино-1-нафтола (витамина К5)
(LXXXII) 2-метил-1,4-нафтохинон (VII) превращают в его монооксим
(LXXXVI), т. пл. 168—170° С, который при действии едкого натра пере-
ходит в 4-нитрозо-2-метил-1-нафтол (LXXXVII) в виде натриевого произ-
водного, далее подвергаемого восстановлению в гидрохлорид 2-метил-4-ами-
но-1-нафтола (LXXXII) при действии хлористого олова [175] или гидросуль-
фита натрия [176]
Исходя из а-нитронафталина, путем его аминирования в 1-нитро-4-ами-
нонафталин с последующим гидролизом синтезируют 1-нитро-4-нафтол, ко-
торый подвергают хлорметилированию; после восстановления получают
гидрохлорид 2-метил-4-амино-1-нафтола (витамин К5) (LXXXII) [177].
Еще один путь синтеза гидрохлорида 2-метил-4-амино-1-нафтола
(XLVI)—иза-нафтола [178]—осуществляется по схеме 57. Метильную
группу положения 2 молекулы а-нафтола получают из альдегидной, ко-
торую вводят конденсацией а-нафтола (LXXXVIII) с изатин-а-анилидом
(LXXXIX) при нагревании в уксусном ангидриде с последующим расщеп-
лением образующегося 2-нафталин-2-индолиндиго (ХС). Полученный 1-окси-
2-нафтальдегид (XCI) восстанавливают по Клеменсену в спиртовой среде
гранулированным цинком, активированным сулемой и соляной кислотой в
2-метил-1-нафтол (ХСП); после нитрозирования амилнитрнтом в положении
4 образовавшийся 2-метил-4-нитрозо-1-нафтол (LXXXVI) восстанавливают
хлористым оловом в гидрохлорид 2-метил-4-амино-1-нафтола (LXXXII).
247
Схема 57
к
Синтез 2-метил-4-амино-1-нафтола из а-иафтола
Можно первоначально получить 4-семикарбазон или 2,4-дигидрофенил-
гидразон 2-метил-1,4-нафтохинона, который затем подвергнуть восстанов-
лению в 2-метил-4-амино-1-нафтол [93]. При непосредственном получении
этого вещества из 2-метилнафталина (схема 58) его нитруют в соединение
XCIII, после чего восстанавливают в соединение XCIV, диазотируют, а за-
тем разлагают диазосоединение в 1-окси-2-метилнафталин (ХСП), который
сочетают с диазосульфаниловой кислотой, затем азокраситель (XCV) вос-
станавливают в гидрохлорид 2-метил-4-амино-1-нафтола (LXXXII) [179,
180].
Схема 58
Синтез 2-метил-4-амино-1-нафтола из 2-метилиафталина
Следует отметить, что гидрохлорид 2-метил-4-амино-1-нафтола (LXXXII)
легко окисляется через промежуточный нафтохиноноимин (XCVI) с пос-
ледующим гидролизом в 2-метил-1,4-нафтохинон (VII)
Высокую витаминную активность аминометилнафтолов, а также диа-
мннометилнафталинов можно объяснить их легким переходом в организме
в 2-метил-1,4-нафтохинон, возможно, в результате аналогичной реакции.
Для получения 2-метил-1,4-диаминонафталина дигидрохлорида (вита-
мина Kfi) (LXXXIII) 2-метил-1-нафтиламин (XCIV) сочетают с диазобензо-
лом, диазобензолсульфокислотой или диазо- n-нитробензолом; получаю-
щийся азокраситель (XCVII) восстанавливают и выделяют дигидрохлорид
2-метил-1,4-диаминонафталина (LXXXIII) [180—183].
248
XCIV
nh2-hci
LXXXIII
Для практического применения, помимо филлохинона, синтетическим
путем получают 2-метил-1,4-нафтохинон (витамин Кз) (VII) и водораствори-
мые препараты — натриевую соль бисульфитного производного 2-метил-
1,4-нафтохинона (LXXIX), тетранатриевую соль дифосфорного эфира 2-ме-
тил-1,4-нафтогидрохинона (витамин К4) (LXXX) и 2-метил-4-амино-1-наф-
тол гидрохлорид (витамин К5) (LXXXII).
СПИСОК ИСПОЛЬЗОВАННОЙ
1. Номенклатура нафтохинонов, Bio-
chim. Biophys, Acta, 107, 5 (1965).
2. P. Karrer, A. Geiger. Helv.
Chim. Acta, 22, 945 (1935).
3. H. N о 1 1, R. R fl e g g, U. G 1 о or,
G. Ryser, O. Isler. Helv.
Chim. Acta, 43, 433 (1960).
4. 0. Isler, R. R fl egg, L. C h o-
pard-di t-Jean, A. Win-
ter s t e i n, O. W i s s. Helv.
Chim. Acta, 41, 786 (1958).
5. R. McKee, S. Binkley,
S. Thayer, D. Me Co r q 11 о -
dale, E. D о i s y. J. Biol. Chem.,
131, 327 (1939).
6. O. Isler. Vitamins and Hormo-
nes, 17, 53 (1959).
7. H. Holl. J. Biol. Chem., 232,
919 (1958).
8. A. Wagner, K. F о 1 k e r s.
Vitamins and Coenzymes, New
York, 1964 . 407, 435.
9. H. M a у e r, U. G 1 о о г, О. Is-
ler, R. Riiegg, О. W i s s.
Helv. Chim. Acta, 47, 221 (1964).
10. L. J a c k m a n, R. R fl e g g,
O. Isler. Helv. Chim. Acta, 48,
1332 (1965).
11. P. Karrer, H. Simon,
F. Z b i n d e n. Helv. Chim. Acta,
27, 317 (1944).
12. R. Cahn, C. Ingold,
V. P r e 1 о g. Experientia, 12, 81
(1956).
13. D. E w i n g, J. V a n d e r b e 1 t,
О. К a m m. J. Biol. Chem., 131,
345 (1939).
14. H. A 1 m q u i st, A. Klose.
► ' J. Am. Chem. Soc., 61, 2557 (1939).
15. R. McKee, S. Binkley,
D. M с С о r q u о d a 1 e, S. T a y-
1 о r, E. D о i s y. J. Am. Chem.
Soc., 61, 1295 (1939).
16. P. Karrer,, A. Geiger,
A. Ruegge r, H. Salomon.
Helv. Chim. Acta, 22, 1464, 1513
(1939).
17. С. M a r t i n s, H. Esser. Bio-
chem. Z., 331, 1 (1958).
18. L. F i e s e г, M. T i s h 1 e r,
W. Sampson. J. Am. Chem.
Soc., 62, 1628 (1940).
ЛИТЕРАТУРЫ (К ГЛАВЕ V)
19. H. A 1 m q u i s t, E. Stock-
s t a d. J. Nutrition, 14, 235 (1937).
20. C. S и у d e r, H. Rapoport.
J. Am. Chem. Soc., 91, 731 (1969).
21. Л. А. Щукина, А. П. К о н д-
ратьева, М. М. Шемякин.
ЖОХ, 18, 1945 (1948).
22. Л. А. Щукина, А. П. Кон д-
ратьева, М. М. Шемякин.
ЖОХ, 19, 183 (1949).
23. Л. А. Щукина, М. М. Ш е м я-
к и н. ЖОХ, 19, 193 (1949).
24. В. Riegel, Р. Smith,
С. S с h w е i t z е г. J. Am. Chem.
Soc., 62, 992 (1940).
25. О. Isler, К. D о е b е 1, Helv.
Chim. Acta, 37, 227 (1954); пат.
США 2683176; С. А., 49, 10377
(1955).
26. R. Н i г s с h m а п п, N. Wend-
ler, R. Miller. Пат. США
2906780; РЖХ, 1961, 11Л383.
27. R. Н i г s с h m а п п, A. Bas-
so. Пат. США 3065255; С. А., 58,
6762 (1963).
28. Е. Lederer. Compt. rend., 257,
706 (1963).
29. Р. М a m о п t,- Р. С о h еп,
R. A z е г a d, М. V i 1 k a s. Bull.
Soc. chim. France, 1965, 2513.
30. A. Wagner, P. W i t t r e i c h,
B. A r i s о n, N. T r e n n e r,
K. F о 1 к e r s. J. Am. Chem.
Soc., 85, 1178 (1963).
31. A. Wagner, K. F о 1 к e r s. J.
Am. Chem. Soc., 85, 3793 (1963).
32. Э. И. Козлов, Г. И. Само-
хвалов. ЖОХ, 36, 2120 (1966).
33. Э. И. К о з л о в, Е. А. О б о л ь-
никова, Г. И. Самохва-
лов. ЖОХ, 37, 544 (1967).
34. Р. Со hen, Р. М a m о п t,
R. A z е г a d, М. V i 1 к a s. Compt.
rend., 261, 5144 (1965).
35. Sh. F u j i s a m a, Sh. К a w a -
b a t a, Y. R у u i c h i. Yaku-
gaku Zasshi, 87, 1451 (1967); C. A.,
69, 35874 (1968).
36. Neth. Appl. 6402953; C. A., 62,
9068 (1965).
249
37. М. А. Голованова. ЖОХ,
18, 835 (1948).
38. L. F i е s е г, R. Т u г n е г. J. Ат.
Chem. Soc., 69, 2335 (1947).
39. L. F i е s е г. J. Am. Chem. Soc.,
61, 3467 (1939).
40. S. В i п к 1 e y, L. Cheney,
W. Ho 1 co m b, R. McKee,
S. T h а у 1 о r, D. McCorquo-
d a 1 e, E. D о i s y. J. Am. Chem.
Soc., 61, 2558 (1939).
41. S. Binkley, W. Me К e e,
S. T h а у 1 о r, E. D о i s y. J.
BioL Chem., 133, 721 (1940).
42. S. Binkley, D. McCorquo-
dale, L. Cheney, S. Tay-
lor, R. McKee, E. D о i s y.
J. Am. Chem., Soc., 61, 1612 (1939).
43. D. McCorquodale, S. Bin-
kley, S. T h а у I о r, E. D о i -
s y. J. Am. Chem. Soc., 61, 1928
(1939).
44. S. Binkley, D. McCorquo-
dale, J. Cheney, S. Tay-
lor, R. McKee, E. D о i s y.
J. Am. Chem., Soc., 61, 1621 (1939).
45. L. F i e s e r, D. Bowen,
W. Campbell, M. F i e s e r,
E. F r e i, R. Jones, B. R i -
gel, C. Schweitzer,
P. Smith. J. Am. Chem. Soc.,
61, 1925 (1939).
46. L. F i e s e r, D. Bowen,
W. Campbell, E. Frei,
M. Gates. J. Am. Chem. Soc.,
61, 1926 (1939).
47. L. F i e s e r, W. Campbell,
E. Frei. J. Am. Chem. Soc., 61,
2206 (1939).
48. L. F i e s e r, W. Campbell,
E. Frei, M. Gates. J. Am.
Chem. Soc., 61, 2559 (1939).
49. L. F i e s e r. J. Am. Chem. Soc.,
61, 2561 (1939).
50. P. Karrer, A. E p p r e c h t.
Helv. Chim. Acta, 23, 272 (1940).
.51. P. G al e, B. A r i son, N. Tren-
n e r, A. Page, K. Folkers.
Biochem., 2, 200 (1963).
52. P. Scholes, H. King. Bio-
chem. J., 91, 9P (1964).
53. R. Azerad, M. С у г о t,
E. Lederer. Biochem. Bio-
phys. Res. Commun., 27, 249 (1967).
54. R. Azerad, M.-O. С у г о t.
Bull. soc. chim. France, 1965, 3740.
55. B. Frydman, H. Rapoport.
J. Am. Chem. Soc., 85, 823 (1963).
56. R. В a u m, M. D о 1 i n. J. Biol.
Chem., 238, PC 4109 (1963).
57. M. К о f 1 e r, O. Isler. Helv.
Chim. Acta, 42, 2252 (1959).
58. С. P 1 a n t a, E. В i 1 1 e t e r,
M. К о f 1 e r. Helv. Chim. Acta,
42, 1278 (1959).
59. M. К о f 1 e r, A. Langemann,
R. Rflegg, L. Chopard-dit-
Jean, A. Rayroud, O. I s -
1 e r. Helv. chim. Acta, 42, 1283
(1959).
60. M. К о f 1 e r, A. Langemann,
R. Rflegg. Chimia, 13, 115 (1959).
61. D. W о 1 f, C. Hoffman,
N. F r e n n e r, B. Arison,
C. S h u n к, B. Linn,
J. McPherson, K. Folkers.
J. Am. Chem. Soc., 80, 4752 (1958).
62. R. M о r t о n, U. G 1 о о г,
О. S c h i n d 1 e r, G. Wilson,
L. Chopard - dit-Jean,
F. Hemming, O. Isler,
W. L e a t, J. Pennock,
P. R u e g g, U. S c h w i e t e r,
O. W i s s. Helv. chim. Acta, 41,
2343 (1958).
63. R. L e s t e r, F. С r a n e. J. BioL
Chem., 234, 2169 (1959).
64. Y. H a t e f i. Biochim. Biophys.
Acta, 34, 183 (1959); 31, 502 (1961).
65. K. D о e g, S. Krueger,
D. Ziegler. Arch. Biochem.
Biophys., 85, 282 (1959); Biochim.
Biophys Acta, 41, 491 (1960).
66. U. G 1 о о г, О. Isler, R. M о r-
t о n, R. Rflegg, O. W i s s.
Helv. Chim. Acta, 41, 2357 (1958).
67. P. F г i i s, G. Daves, K. Fol-
kers. Biochem. Biophys. Res.
Comm., 24, 252 (1966).
68. A. Page., P. Gale, H. W a 1-
1 i c k, R. W a 1 t о n, L. M c D a n i-
el, H. Woodruff, K. Fol-
kers. Arch. Biochem. Biophys., 89,
318 (1960).
69. R. L e s t e r, F. С r a n e, Y. H a-
t e f i. J. Am. Chem. Soc., 80, 4751
(1958).
70. Г. И. Самохвалов,
E. А. Обольникова, Успе-
хи химии, 36, 1012 (1967).
71. H. Moore, К. Folkers. J.
Am. Chem. Soc., 87, 1409 (1965);
88, 567 (1966).
72. R. Ericson, C. S h u n k,
N. T г e п n e r, B. Anson,
K. Folkers. J. Am. Chem.
Soc., 81, 4999, 5000 (1959).
73. E. Edwin, A. D i p 1 о c k,
J. Bunyan, J. Green. Bio-
chem. J., 79, 91 (1960).
74. J. Links. Biochim. Biophys. Ac-
ta, 38, 193 (1960).
75. R. A n d e r so п, M. N e w m a n n.
J. Bi,ol. Chem., 101, 773; 103, 197,
405 (1933).
76. R..H i r s c h m a n n, R. M i 1 -
1 e r, N. W e n d 1 e r. J. Am. Chem.
Soc., 76, 4592 (1954).
77. N. W e n d 1 e г. Пат. США 2831899;
РЖХ, 1959, 87674.
78. S. T h а у 1 о r, S. Binkley,
R. McKee, D. McCorquo-
dale, E. D о i s у. Пат. США
2348037; С. A., 39, 159 (1945).
79. 0, Isler. Пат. США 2325681;
С. А., 38, 1077 (1944).
80. М. Т i s h 1 е г, L. Fies.er,
N. W е n d 1 е г. J. Am. Chem.
Soc., 62, 1982 (1940).
81. R. Н i г s с h т а п и. Пат. США
3076004; С. А., 59, 2743 (1963).
250
82. J. W e i c h e t, 'V. К v i t a . Че-
хосл. пат. 86621; C. A., 54, 2294
(1960).
83. V. К v i t a, J. Wei che t,
V. Trcka. Chem. listy, 50, 1979
(1956); Coll. Czech. Chem. Comm.,
22, 583 (1957).
84. И. К. С a p ы ч e в а, Ю. Г. M о -
л о т к о в с к и й, Г. А. Воро-
бьева, Н. А. Преображен-
ский. ЖОХ, 29, 1123 (1959).
85. С. К и т а м у р а, С. М а ц у я м а,
М. М о р и о к а. Яп. пат. 28771
(1970); РЖХ, 1971, 19Н586.
86. J. Wei с h е t, V. Kvita. Пат.
ГДР 17296: С. А., 55, 1561 (1961).
87. М. М a t s u i, S. Ki t am ur а.
Agr. Biol. Chem. (Tokyo), 29, 978
(1965); C. A., 64, 6603 (1966).
88. C. Lee, F. H о s k i n, L. T r e -
v о y, L. laques, I. Spinks.
Canad. J. Chem., 31, 709 (1953).
89. J. Wei che t, L. Blaha,
V. К v i t a. Coll. Czech. Chem.
Comm., 25, 1914 (1960).
90. J. Weichet, L. Blaha,
V. Kvita. Chem. Ind., 1958,
227.
91. V. V e s e 1 у, I. К a p p. J. Chem.
listy, 18, 201 (1924); Rec. trav.
chim., 44, 360 (1925).
92. G. Martin, V. Swayne.
Science, 109, 201 (1949).
93. Nai-Hsuang Chang,
J. О n e t о, P. S a h. Ztsch. Vita-
mins-Hormon.-Fermentforsch., 3
61 (1949/50).
94. Яп. пат. 162647; С. A., 42, 4201
(1948).
95. R. Arnold, R. Larson. J.
Org. Chem., 5, 250 (1940).
96. N. Lundin. Пат. США 2413253;
С. A., 41, 2085 (1947).
97. L. F i e s e r, M- T i s h 1 e r,
N. We nd 1 er. J. Am. Chem. Soc.,
62, 2861 (1940).
98. Г. И. О с т p о ж и н с к а я.
ЖОХ, 16, 1053 (1946).
99. Ch. К. Chun g, Ch. Ts. Han.
Ber., 68, 876 (1935).
100. M. G r d i n i с, V. J u g о v i c.
Archiv. Kern., 23, 73 (1951).
101. M. Wakae, К. К о n i s h i.
Repts. Ind. Research. Inst., Osaka
Prefect. 7/2, 59 (1955); C. A., 50,
12005 (1956).
102. Z. H о r i i, T. Tanaka,
Y. Murakami. Chem. Pharm.
Bull. (Tokyo), 5, 82 (1957). C. A.
51, 17858 (1957).
103. W. G a 1 1 e г. Пат. США 3079405;
С. A., 59. 6336 (1963).
104. R. Phillips, L. T r e v о y,
L. I a q u e s, I. S p i n k s. Canad.
J. Chem., 30, 844 (1952).
105. Liang Li, W. Elliott, J.
Am. Chem. Soc., 74, 4089 (1952).
106. F. F i s c h e r, K. L о w e n b e r g.
Lieb. Ann., 475, 183 (1929).
107. P. Karrer, A. Geiger,
H. Rentschler, E. Z b i n-
d e n, A. Kugler. Helv. Chim.
Acta, 26, 1741 (1943).
108. J. Weichet, J. Hodrova,
V. Kvita. Chem. listy, 51, 568
(1957); Coll. Czech. Chem. Comm.,
22, 1180 (1957).
109. И. К- С a p ы ч e в а, Г. А. В о -
робьева, H. А. Кузне-
цова, Н. А. П р.е о б ра-
же и с к и й. ЖОХ, 28, 647
(1958).
110. Г. А. В о р о б ь е в а, И. К- Са-
ры ч е в а, Н. А. Преобра-
женский. ЖОХ, 29, 2314
(1959).
111. Э. И. Козлов, М. Ц. Я но-
те вс ки й, Г. И. Самохва-
лов. ЖОХ, 34, 2748 (1964).
112. Б. А. Кудряшов. ДАН СССР,
60, 1469 (1948).
113. A. Asano, Т. Kaneshiro,
А. В г о d е. J. BioL Chem., 240,
895 (1965).
114. М. V i 1 k a s, Е. Lederer. Ех-
perientia. 18, 546 (1962).
115. L. F i e s e r. M. T i s h 1 e r,
W. S a m p s о n. J. Biol. Chem.,
137, 659 (1941).
116. J. Weichet, V. Kvita,:
V. Trcka. Chem. listy, 51, 127
(1957).
117. S. § m о 1 i k, V. Kvita,
J. Wei chet, V. Trcka. Coll.
Czech. Chem. Comm. 25, 259
(1960).
118. J. Weichet, V. Kvita,
L. Blaha, V. Trcka. Coll.
5»-'. Czech. Chem. Comm., 24, 2754
. '• (1959).
119. J. Weichet, J. Hodrova,
L. Blaha. Coll. Czech. Chem.
Comm., 29, 197 (1964).
120. J. Weichet, L. Blaha,
J. Hodrova, В. К a k a c,
V. Trcka. Coll. Czech. Chem.
Comm., 31, 3607 (1966).
121. J. Weichet, L. Blaha,
J. Hodrova. Чехосл. пат.
118619; С. A., 67, 21735 (1967).
122. L. Blaha, J. Weichet,
J. Hodrova. Чехосл. пат.
124017; С. A., 69, 18914 (1968).
123. D. M i s i t i. H. Moore,
K. Folkers. Biochemistry, 4,
1156 (1965).
124. G. Manecke, W. Storck.
Chem. Ber., 94, 300 (1961).
125. W. В о n d i n e 1 1, S. D i M а г I,
B. Frydman, K. Matsu-
moto, H. R о p о p о r t, J. Org.
Chem., 33, 4351 (1968).
126. O. Isler. Angew. Chem., 71, 7
(1959).
127. S. Ansbacher, E. Fern-
fa о 1 z. J. Am. Chem. Soc., 61,
1924 (1939); J. Biol. Chem., 131,
399 (1939).
128. M. Moore. J. Am. Chem. Soc.,
63, 2049 (1941).
129. В. В a k e r, T. Davis,
L. M с E r 1 о v, G. Carlton.
J. Am. Chem., Soc., 66, 1096
(1942).
251
130. Д. А. Б о ч в а р, М. М. Шемя-
кин. ЖОХ, 13, 467 (1943); 16,
2033 (1946).
131. Menotti. J. Am. Chem. Soc.,
65, 1209 (1943).
132. П. Д. У л и т и н а, Б. А. Куд-
ряшов. ДАН СССР, 63, 465
(1948).
133. М. Т i s h 1 е г, L. F i е s е г,
N. Wendler. J. Am. Chem.
Soc., 62, 2866 (1940).
134. H. Dam, J.G1 avi nd. P. Kar-
r e r. Helv. Chim. Acta, 23, 224
(1940).
135. L. Fosdick, O. Fancher,
J. Calandra. Science, 96, 45
(1942).
136. S. Ansbacher, E. Fern-
h о 1 z, M. D о 1 1 i v e r. J. Am.
Chem. Soc., 62, 155 (1940).
137. L. F i e s e r, E. Frei. J. Am.
Chem. Soc., 62, 228 (1940).
138. R.Foster, J. Lee, U. Solms-
s e n. J. Am. Chem. Soc., 62, 453
(1940).
139. M. fishier, L. F i e s e r,
W. Sampson. J. Am. Chem.
Soc., 62, 1881 (1940).
140. H. A 1 m q u i s t, A. Klose.
Proc. Soc. Exptl. biol. Med., 45,
55 (1940).
141. E. D о i s y, D. M с С о r q u o-
d a 1 e, S. T а у 1 о r, S. В i n k-
1 e y, R. M с К e e. Science, 90,
407 (1939).
142. S. Ansbacher. Proc. Soc. Exp.
Biol. Med., 46, 421 (1941).
143. R. Kitchen, R. Sandin.
J. Am. Chem. Soc., 67, 1645 (1945).
144. D. Tarbell, D. Fukushi-
ma, H. Dam. J. Am. Chem. Soc.
67, 197, 1643 (1945).
145. I. Schmitt, I. S e i 1 e r t.
Lieb. Ann., 562, 15 (1949).
146. W. Christiansen, M. Dol-
liver. J. Am. Chem. Soc., 63,
1470 (1941).
147. M. M. Ш e м я к н н, Л. А. Щ y-
к и н а. ДАН СССР, 45, 167 (1944).
148. К- Д- Пакендорф,
Б. А. Кудряшов, Е. Н. Л а •
зарева. ДАН СССР, 31, 484
(1941).
149. Н. Campbell, К. Link. J.
Biol. Chem., 138, 21 (1941).
150. Р. Meunier, С. М е п t г е г,
В. Н о i, Р. С a g m i а и t. Bull.
Soc. chim. biol., 25, 384 (1943);
27, 191 (1945).
151. H. V e 1 d s t r a, P. Wiardi,
G. A 1 b e r d a. Rec. trav. chim.,
72, 358 (1953). .
152. F. Beran. Osterr. Chem. Ztg.,
54, 68 (1953).
153. C. Smith, R. F r a d k i n,
M. Lackey. Proc. Soc. Exp.
Med. Biol., 61, 398 (1946).
154. J.Chmielevska, H. Kavar-
zyk, B.J urecka, A. Pachec-
k a, Prace Jugoclav. Tovarz. Naux.
Ser. B., 40, 5 (1951).
155. L. F i e s e r, R. Brown. J. Am.
Chem. Soc., 71, 3609, 3615 (1949).
156. L. F i e s e r. J. Biol. Chem., 133,
391 (1940).
157. А. Ш м у к, А. Гусева. ЖПХ,
21, 1180 (1948).
158. А. В. Палладии. ДАН СССР,
41, 85, 258 (1943).
159. Д. А. Б 6 ч в а р, Л. А. Щу-
кина, А. С. Чернышев,
Н. Г. С е м е н о в, М. М. Ше-
мякин. J. Am. Chem. Soc., 65,
2162 (1943).
160. М. Carmack, М. Moore,
М. В а 1 i s. J. Am. Chem. Soc., 72,
844 (1950).
‘ Л61. Y. Asahi. Chem. Pharm. Bull,
fev . (Tokyo), 11, 813 (1963).
162. Д. А. Б о ч в a p, M. M. Шемя-
кин. ЖОХ, 16, 2033 (1946).
163. Д. А. Б оч в a p, А. С. Ч ер н ы -
ш е в, М. М. Ш е м я к и н. ЖОХ,
15, 844 (1945); 20, 2118 (1950).
164. F. A b 1 о и i, R. Р г i с е, В. В а -
к е г, G. С а г 1 s о n. J. Am. Chem.
Soc., 65, 1776 (1943).
165. D. Richert. J. Biol. Chem.,
154, 1 (1944).
z;;166. M. П. 3 a x a p о в а, В. А. Де-
вятнин. Биохимия, 9, 256 (1944).
167. Франц, пат. 996080; С. А., 52,
2079 (1958).
168. А. Ш м у к, А. Гусева. Авт.
свид. СССР 63828; Бюлл. изобрет.,
1944, № 6, 11.
169. М. Moore, F. Kirchmeyr.
Пат. США 2367302; С. А., 39,
2849 (1945).
170. Англ. пат. 771180; С. А., 51, 15584
(1957).
171. В. Н. Уфимцев. ДАН СССР,
44, 353 (1944); ЖОХ, 14, 1064
(1944).
172. Д. А. Б о ч в а р, Е. И. Вино-
градова, Ю. Б. Швецов,
М. М. Шемякин. ЖОХ, 18,
87 (1948).
173. М. Moore, W. Washburn.
J. Am. Chem. Soc., 77, 6384 (1955).
174. H. Ishikawa. Яп. пат. 6113
(1960); С. A., 55, 6456 (1961).
175. P. S a h, W. В r u 1 1. Ber.,
74, 552 (1941).
176. Голл. пат. 60933; С. A., 42, 4311
(1948).
177. L. Szotyori, J. Vodnar,
M. A 1 m a s i, C. A., 54, 18449
(1960).
178. P. S a h. Rec. trav. chim., 60,
373 (1941).
179. P. S a h, T. Daniels. Ztsch.
Vitamins-Normon.- Fermentforsch,
3, 81 (1949).
180. H. V e 1 d s t r a. P. Wiardi.
Rec. trav. chim., 62, 75 (1943).
181. P. Sah, Gokaraju Sub-
fa a r a j u, T. D e n i e 1 s. Ztsch.
Vitamin-, Hormon-, Fermentforsch.,
3, 87 (1949).
182. G. Car 1 so n, В. В a ker. Англ,
пат. 559522; С. A., 39, 5411 (1945).
183. Голл. пат. 60934; С. А., 42, 4312
(1948).
252
Часть четвертая
ВИТАМИНЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
ГЛАВА VI
ХРОМАНОВЫЕ ВИТАМИНЫ
ТОКОФЕРОЛЫ
Токоферолы [1, 2] — витамины группы Е — относятся к природным
производным хромана (бензо-у-дигидропирана). В основе молекулы токо-
феролов [3] лежит токол [4, 5], представляющий собой 2-метил-2-(4',8',
12 '-триметилтридека)-6-оксихроман
сн,
а-Токоферол — 5,7,8-триметилтокол, 2,5,7,8-тетраметил-2-(4',8',12'-
триметилтридека)-б-оксихроман (I) представляет собой токол, в котором
бензольное ядро полностью замещено метильными группами
сн, сн, сн,
CHjC^CHjCHCHjCHjC^CHCHjCHjCHjCHCH,
ск
но
сн
сн, сн>
I
Р -Токоферол—5,8-диметилтокол (II), у-токоферол—7,8-диметилтокол (III)
НО^^гх^х^ СН,
jk ST 'Ъсн,(сн2сн2снс^н
СНГт'чО'ТН,
сн,
III
Сз-токоферол—5,7-диметилтокол (IV) (из риса), — три природных изомера
диметилтокола. 8-Токоферол, 8-метилтокол (V) — природный изомер моно-
метилтокола,
253
Y J JLch2(ch2ch2chch;)3h T )XCWWH
CHa CH’ CH, CH*
5-метилтокол (VI), ранее называвшийся е-токоферолом, и 7-метилтокол
(VII), ранее называвшийся ^-токоферолом, — два других теоретически воз-
можных изомера монометилтокола
— сн, ; .
• НО. к’ СНз ; сн,
YY Ъсн2(сн2сн2снснДн f, Jj jT >н2(сн2сн2снсн>
- ^/"осна 5 снУ'о С11,
VI VII
в природных источниках не обнаружены и получены только синтетически.
Номенклатура токоферолов опубликована [5].
а-Токоферол, в котором бензольное ядро молекулы токола содержит
три метильные группы, имеет наиболее высокую биологическую
активность. При двух метильных группах в молекуле токола активность
снижается; для синтетического 5,7-диметилтокола (С2-токоферола) она со-
ставляет 80% (по отношению к а-токоферол у), а для его изомеров —природ-
ных 5,8-диметилтокола (р-токоферола) — 40—50% и 7,8-диметилтокола
(у-токоферола) —только 8% [6]. Активность 8-метилтокола (о-токоферола)
составляет менее 1 % 17 ].
В молекуле токола имеется три асимметрических атома углерода р по-
ложениях 2, 4 х и 8х. Следовательно, для токола (и, таким образом, для каж-
дого из а-, р-, у-,С2-, S-токоферолов) возможны восемь оптических изомеров
и четыре рацемата. Из этого количества оптических изомеров дляа-токофе-
рола известно лишь четыре.
Природный а-токоферол (I) имеет /?-конфигу рацию [8] у асимметрических
атомов углерода в положении 2 ядра хромана и в положениях 4х и 8х али-
фатической цепи, т. е. С*2/?, 4 х/?, 8х/? [9, 10]:
I
Природные ₽- и у-токоферолы также имеют абсолютную 2/?-конфигурацию
[111. Все природные токоферолы правовращающие (d); синтетические токо-
феролы являются рацематами по оптическому центру положения 2 ядра
хромана.
Природный токоферол по физическим свойствам, главным образом по
температуре плавления его аллофанатов и других его эфиров, и по несколько
большей биологической активности отличается от синтетического токоферо-
ла, полученного из триметил-и-гидрохинона и природного d-фитола, ко-
торый по отношению к атому углерода в положении 2 рацемичен [12], т. е.
С* 25/?, 4х/?, 8х/?. Из синтетических I- и dZ-фитолов [13, 14] были по-
лучены С* 25/?, 4XS, 8XS- и С *25/?, 4Х5/?, 8хХ/?-а-токоферолы [9, 15];
в последнем соединении все три асимметрических центра рацемические.
Токоферолы представляют собой прозрачную маслянистую жидкость
светло-желтого цвета, нерастворимую в воде, хорошо растворимую в хло-
роформе, эфире, петролейном эфире, хуже — в спирте и ацетоне. Физичес-
кие свойства токоферолов представлены в табл. 18.
254
Таблица 18
Физические свойства токоферолов
Изомеры токофе- ролов Относительная плотность Т. пл., °C Т. кип., “С Йодное число [4] [®]346,1 и спирте, град. [7] Спектр поглощения
X ЪА 5?- Тц"
d-a- 0,953 при d-ц, 1,5052 2,5—3,5 [17] 225—230 при 0,1 мм 143 +0,32 (с 14,2) 292 75,8 [18]
dl-a- 0,947—0,958 при dg 1,5030— 1,5070 0 — 292 74,7 [16]
4-₽- 0,953 при du 1,5052 138 +2,9 (с 7,15) 296 89,4 [18]
d-7- 152 +2,2 (с 9,32) 298 92,8 [16]
d-8- —2, —3 [17] +3,4 (с 15,5) 298 91,2 [7]
Для С* 27?, 4'7?, 8'Rd -токоферола (I) оптическое вращение в изооктане
составляет гораздо большую величину — [а от +24,5 до 28,4? (1—3%,
изооктан) [19].
ХИМИЧЕСКИЕ СВОЙСТВА ТОКОФЕРОЛОВ
а-Токоферол чувствителен к ультрафиолетовому свету [20], устойчив
к нагреванию до 200° С в присутствии кислорода и к действию минеральных
кислот даже при 100° С; с едкими щелочами реагирует очень медленно,
вследствие чего токоферол может быть получен из содержащих его расти-
тельных масел методом омыления.
‘Токоферолы легко образуют сложные эфиры с такими кислотами, как
фосфорная [21, 22], уксусная, пропионовая, янтарная, капроновая, стеа-
риновая, бензойная [23, 24], пальмитиновая [16, 24 ], олеиновая, линоле-
вая, линоленовая [24 ], никотиновая [25 ] и др.
d - а - Токоферилацетат имеет т. пл. 26,5—27,5° С (из муравьиного
эфира при —30°С) [17], [а Ь+0,25° (10%, СНС13), df 0,950-0,964, ng
1,4940—1,4985, fj'^40—44 при 284 нм (в спирте), dl-а- Токоферилаце-
тат (VIII)—светло-желтое вязкое масло с dg 0,9545—0,9665, ng 1,4958—
1,4972, Д[^м42,5 + 1 при 285,5 нм
VIII
255
Другие изомерные токоферолы не дают кристаллических ацетатов.
Пальмитат d-a -токоферола имеет т. пл. 42—43° С (из ацетона) [16],
пальмитат dl-a-токоферола —т. пл. 36—38° С (из ацетона).
Водорастворимыми формами а-токоферола являются кристаллические
высокоактивные эфиры, такие, как кислый эфир а-токоферола и янтарной
кислоты с т. пл. 76—77° С [26,27 ], его натриевая и кальциевая соли [27 ],
а также фосфорный эфир о-токоферола (IX)
в виде диэтаноламиновой соли [28] или полиэтиленгликолевого производ-
ного [29].
Для идентификации токоферолов могут служить хорошо кристаллизую-
щиеся сложные эфиры токоферолов [21, 23, 301: n-аминобензоат dl-a -токо-
ферола с т. пл. 158° С; n-фен ил бензоат d-[J -токоферола с т. пл. 70—71 ° С
[71; n-фенилбензоат d-o-токоферола с т. пл. 41—42° С; 2,4-динитробензоат
d-a-токоферола с т. пл. 86—87° С; 2,4-динитробензоат dl-a-токоферола с т.
пл. 63° С; аллофанат d-a-токоферола с т. пл. 161—162° С [9]; аллофанат
dl-a-токоферола ст. пл. 172° С [9]; аллофанат d-[J-токоферола ст. пл. 138—
139° С, [а Ь +5,7°; аллофанат d-y-токоферола с т. пл. 136—138° С, [а Ь +
+3,4°.
Аллофанаты образуются из токоферола (ROH) и циановой кислоты по
следующей реакции:
HN=C=O
HN=C=O + HOR [H2NCOOR1 ---------—> H2NCOHNCOOR.
Изомерные токоферолы неустойчивы к окислителям. Сильные окисли-
тели — озон, марганцовокислый калий, пербензойная кислота и др. —
производят глубокие изменения молекулы токоферола [31 ].
При действии азотной кислотой в спиртовом растворе происходит окис-
ление токоферолов в положении 5 даже и в том случае, если оно замещено
метильной группой, с образованием о-хинонат-хроман-5,6-хинона, напри-
мер для а- и -у-токоферолов— тококрасного (X) [32—35]:
но ^Нз
[ |[ ]/На(сНаСН,СНСнДн
СН3х'те’ СЦ,
сн.
I, R=CH,; III, R=H
сн.
сн,
сн2(сн,сн/:нснДн
СН,
X
Подобного типа о-хинон образуется и при окислении 8-токоферола азот-
ной кислотой [35]. Реакция окисления токоферолов в тококрасный проте-
кает количественно и может применяться для колориметрического опреде-
ления токоферолов по интенсивности образующейся красной окраски со
256
светофильтром 4б0—520 нм [36—38]. Имеются только немногие ограниче-
ния для этого метода, зависящие от присутствия посторонних веществ [39,
40]. Образование тококрасиого (X) при окислении у-токоферола (III) азот-
ной кислотой в спиртовом растворе из-за отсутствия метильной группы
в положении 5 происходит с большей скоростью, чем при окислении а-то-
коферола.
Одним из продуктов окисления а-токоферола хлорным железом явля-
ется п-хинон —токопурпурный, 2,7-диметил-2-(4',8',12'-триметилтридека)-
6-оксихроман-5,8-хинон (XI) [41 ],
XI
При мягком окислении изомерных токоферолов расщепляется хромано-
вый цикл и образуются токоферол-и-хиноны. Так, окисление а-токоферола
хлорным железом приводит к желтому а-токоферолхинону (XII) [42, 43]:
Промежуточным продуктом при окислении а-токоферола хлорным же-
лезом [44], хлорным железом в присутствии а, а'-дипиридила [45], а также
N-бромсукцинимидом или тетрахлор-о-хиноном [46] является бесцветный
8а-окси-а-токоферон (VIII)—«а-токоферолперекись» [45] <<
. СИ* •*
СН^СНзСНСнДн
сна
циклический полуацеталь таутомерного а-токоферол хинона (ХП).8а-Окси-
а-токоферон может быть вновь восстановлен в а-токоферол [45].
Следует отметить, что скорость реакции токоферолов с хлорным желе-
зом различна и уменьшается в таком порядке: а>|3>у>о [47, 48].
Для мягкого окисления а-токоферола используют молекулярный кис-
лород, хлорное золото, сернокислый церий, азотнокислое серебро [42, 49],
активированную двуокись марганца (выход 27%) [50], соли трехвалентного
железа, в частности хлорное железо [42, 43, 51—54]; при окислении FeCl3
в метиловом спирте выход индивидуального а-токоферол хинона (XII) сос-
тавляет 88% [52]. Окисление а-токоферола осуществляют и электрохими-
ческим методом [42].
При восстановлении а-токоферолхинона (XII) бисульфитом калия или
натрия или каталитическим гидрированием образуется а-токоферолгидро-
хинон (XIV) [43, 52, 55]:
9—69
257
сн, II
о
СН2СН2ССН2(СН2СН2СНСН2), н ->
I
он
XII
он
сн, I
сн.
сн,
сн3
I
СН» I CH2CHtCCH2(CH2CH2CHCHt), н
он он
XIV
Образование а-токоферолхинона(ХII) при окислении природного а-то-
коферола (I) хлорным железом происходит без изменения конфигурации
цри оптическом центре атома углерода положения 2 хроманового цикла. Под
йлиянием концентрированной серной кислоты в метиловом спирте а -токо-
феролхинон (XII) подвергается стереоспецифической рециклизации с сохра-
нением 3'^-конфигурации и образованием природного С* 2R, 4'R, 8'R-a-m-
коферола (I) [54], [a J£5 + 0,75° (спирт); а-токоферилацетат— [а ]о +3,55°
(спирт).
При каталитическом гидрировании а-токоферол хинона (XII) происхо-
дит инверсия асимметрического центра при атоме углерода положения 3',
и образовавшийся а-токоферолгидрохинон (XIV) при последующей рецик-
лизации под влиянием хлористого цинка в петролейном эфире образует
не природный С* 2R, 4'R, 8'R-а-токоферол, а его антипод С* 2S, 4'R, 8'R-a-
токоферол (XV) с неприродной конфигурацией [51, 54], [а 1 о +0,19°
(спирт); а-токоферилацетат— [« ]д —2,14° (спирт)
При окислении изомерных токоферолов щелочным феррицианидом ка-
лия или л-бензохиноном образуются различные димерные, спиродимерные,
тримерные и спиротримерные продукты окисления. При окислении а-токо-
ферола (I) КзРе(СМ)6 в щелочном растворе образуется спиродимер (XVI)
[56, 57 ], который при испытании на животных (на крысах) соответствует 2%
активности ацетата токоферола [57]. Выход спиродимера (XVI) составляет
65—75% [58]; предложенная для него структура была подтверждена [59].
Одновременно при окислении а-токоферола образуется бис-(у-токоферил-5)-
этан (XVIII) с выходом 2—3% и спиротример (XVII) (с выходом около 2—
3%) [58, 60]. В качестве побочного (или промежуточного) продукта при
окислении получается некоторое количество а-токоферолхинона.
258
Промежуточным соединением при окислении а-токоферола (I) в спиро-
' димер (XVI) является хинонметид (XIX, R=CH2[CH2CH2CH(CHJCH2]3H),
как это установлено на модельном соединении (XIX, R=CH3) [611:
XIX
При восстановлении спиродимера а-токоферола (XVI) водородом на
Рд/Ва5О4-катализаторе [19] или алюмогидридом лития получен бис-(у-то-
коферил-5)этан (XVIII) [19, 58]; его окисление вновь приводит к тому же
самому спиродимеру (XVI) [19]. Спиродимер (XVI) был получен и непо-
средственным синтезом и разделен на цис- и транс-изомеры [62 ].
Образование спиротримера (XVII) характерно и в случае применения
для окисления а-токоферола «-бензохинона [63] наряду с другими димерами
XVI, XVIII, как это следует из реакций, осуществленных на модельном сое-
динении (где R=CH3) [64].
Окисление а-токоферола свободнорадикальными инициаторами реакции,
например азо-бос-изобутиронитрилом, приводит к бис-(ч-токоферил-5)этану
(XVIII) [65].
Подобное течение реакции окислительной ди- и тримеризации наблю-
дается и для p-токоферола (II) — происходит образование спиротримера ти-
па XVII [63], димеров типа XVI и XVIII [64], как это следует из реакций,
проведенных на модельных соединениях. Кроме того, в этой реакции окис-
ления [^токоферола щелочным феррицианидом калия происходит образо-
вание димеров XX и XXI [63].
Окислительная ди- и тримеризация -^-токоферола (III) п-бензохиноном
приводит к образованию димеров —5-(7-токоферил-5)-ртокоферола (XXII)
9*
259
и 5-(7-токоферилокси)-7-токоферола (XXIII) [63]. Один из этих продуктов
окисления 7-токоферола—5-(у-токоферилокси)-т-токоферол (XXIII) — вы-
делен из накипи дезодоратора хлопкового масла [66].
Наряду с димерами XXII и XXIII образуется тример XXIV, как это
следует из реакций, осуществленных на модельном соединении (R =
СН3) [64].
Подобным образом протекает окислительная ди- и тримеризация о-то-
коферола (V) п-бензохиноном— образуются соединения типа XXIII и
XXIV [63, 64].
Иначе идет реакция окисления а-токоферола (I), если в качестве окис-
лителя применить перекись бензоила (в среде углеводородов под азотом);
продуктом окисления является 5-бензоилоксиметил-7-токоферол (XXV)
[67, 68].
СН2ОСОС6Н5 „„
НО ।
\^\/\ CHHCHsCH^HCH-OgH
I I I/
/W\
сн8 сн8
XXV
Кроме того в реакции образуется а-токоферолхинон (XII), спиродимер (XVI)
и небольшое количество спиротримера (XVII).
Токоферолы — сильные восстановители. Способность токоферолов к
легкому окислению, особенно 3- и -[--изомеров, лежит в основе их эффектив-
ности как антиоксидантов растительных масел, витамина А (ретинола) и
других веществ; по антиокислительной способности токоферолы распола-
гаются в следующий ряд:
Токоферолы способны восстанавливаться; при гидрировании поглощает-
ся 4 моля водорода [69]. В качестве восстановительного агента можно при-
менять гидросульфит [70].
Реакция мягкого окисления изомерных токоферолов в токоферолхино-
ны, количественно протекающая с раскрытием пиранового цикла, лежит
в основе важнейших методов их аналитического определения в природных
продуктах. В данном случае окисление хлорным железом ведут в присут-
ствии о-фенантролина или а, а'-дипиридила [45], являющихся индикаторами.
Интенсивную красную окраску комплекса а, а'-дипиридила с образовав-
260
шимся при реакции двухвалентным железом измеряют фотометрически
[33,71]. ' ..... .
Количественное определение токоферолов при использовании для окис-
ления хлорного золота производится потенциометрически [72] или ампе-
рометрически [73]. Этот метод находит ограниченное применение, так как
хлорное золото не обладает способностью окислять эфиры токоферолов и
другие производные. Окисление сульфатом церия дает возможность исполь-
зовать объемный метод с применением в качестве индикатора п-этоксихри-
зоидина [74].
Для качественного и количественного определения токоферолов исполь-
зуют различные реакции. Продукт окисления а-токоферола — тококрасный
(X) — при конденсации с о-фенилендиамином дает феназиновый флуорес-
цирующий краситель (XXVI) [75].
Эта реакция служит для флуореметрического количественного определе-
ния а-токоферола [76 ]. Для 7- и 8-токоферолов характерно образование крас-
ного азокрасителя при сочетании с диазосоединениями (например, с диазо-
тированным о-дианизидином) [77, 78]. Азогруппа вступает в положение
5 ароматического ядра хромана, активированного оксигруппой; поэтому
а-, Р- и с2-токоферолы, у которых положение 5 замещено, азокрасителей не
образуют. Эту реакцию используют для количественного определения 7-
и 8-изомеров токоферола в смесях [77, 79]. Подобные реакции изомерных
токоферолов могут применяться для их качественного и количественного рас-
познавания (табл. 19).
Таблица 1ft
Реакции изомерных токоферолов
Метод определения Токоферолы
а Р 7 &
Флуоресцентный (с о-фенилендиами- ном) [76] Образование флуоресцирую- щего фаназина Не применим Образование флуоресцирую- щего фенази- на Не применим
Полярографический (электролитическое окисление) [73] Наблюдается характерная полярографи- ческая волна Полярографи ческой волны не наблюдается
Азосочетанием [77— 79] Реакция с уксусной и азотной кислотами (для определения ₽- и 7-изомеров в при- сутствии а-изомера) Ьнролиз при 350°С Не применим — Образование азокрасителя
Желтое окра- шивание Красное ок )ашнваиие
Образование тетраметил-п- гидрохинона [81] Образование триметил-п- гидрохинона [69 , 82 , 83] Образование диметил-п- гидрохииона [7] —
Восстановление [82] Образование оксипсевдоку- мола Образование п-ксиленола —
261
СТРОЕНИЕ ТОКОФЕРОЛОВ
Строение а-токоферола в 1938 г. установил Фернгольц [811 и доказали
путем синтеза Каррер с сотр. [4, 12, 15], а затем Смит с сотр. [84], Бергель
и Тодд с сотр. [85].
а-Токоферол, 5,7,8-триметилтокол
Образование сложных [86, 87 ] и простых эфиров токоферола указывает
на присутствие в его молекуле свободной гидроксильной группы фенольно-
го характера, что установлено также на основании смещения полосы пог-
лощения ультрафиолетового света в коротковолновую область при этерифи-
кации [82 ]. Строение алифатической части молекулы а-токоферола было
выяснено в результате изучения продуктов реакции деструктивного окисле-
ния хромовой кислотой (схема 59).
а-Токоферол (I) в бензольном ядре дигидробензопирановой части мо-
лекулы содержит по крайней мере три метильные группы и одну гид-
роксильную группу, что следует из образования при пиролизе дурогидрохи-
нона (XXVII) [81 ] и при деструктивном восстановлении йодистоводородной
кислотой оксипсевдокумола (XXVIII) [69]. Продукты, полученные при
окислении а-токоферола, такие, как диметилмалеиновый ангидрид (XXIX)
и диацетил [81 ], полностью объясняют строение фенильного остатка.
Схема 59
Установление строении а-токоферола реакциями его превращений и расщеплении
СН, сн, сн,
Г ' I 3 I 3
HOOC^HjCHjCHCH^H -«----COCK^HjCHjCHCH^H
XXXIII XXXII
Образующийся при окислении а-токоферола (1) лактон C2i (XXXI)
[81] получается через промежуточную оксикислоту С21 (XXX), которая легко
лактонизируется с образованием 7-лактонного кольца. Неспособность
метилового эфира оксикислоты окисляться в кетокислоту указывает на тре-
тичный характер оксигруппы, имеющейся в ее молекуле. При окислении
а-токоферола образуются также кетон С18 (XXXII) и кислота C1G (XXXIII);
происхождение последней может быть объяснено образованием ее из лакто-
на С21 (XXXI) через кетон С18 (XXXII).
262
На основании эмпирического состава С1сН32О2 для кислоты С16 (XXXIII)
была предложена насыщенная изопреноидная структура; соединения это-
го ряда широко распространены в природе. Отсюда следует, что третья бо-
ковая метильная группа будет находиться у атома углерода с третичной
гидроксильной группой (образуя еще одно насыщенное изопреноидное зве-
но), и строение лактона C2I (XXXI) выразится вышеприведенной формулой.
Доказательством наличия в молекуле токоферола ядра хромана [88],
а не кумарана 182 ] (оба эти соединения могут образоваться при циклиза-
ции одного и того же исходного аллилфенола) служит реакция окисления
а-токоферола хлорным золотом, хлорным железом или азотнокислым се-
ребром, при которой происходит раскрытие пиранового цикла и образование
у-оксиалкил-п-хинона (XII) желтого цвета [55]. Восстановление этого
а-токоферолхинона (XII)
легко приводит к получению производного оксигидрохинона — а-токоферол-
гидрохинона (XIV). При действии минеральной, например соляной, кислоты
этот гидрохинон с третичной гидроксильной группой в боковой цепи цик-
лизуется в а-токоферол (I) [33]. Третичный характер алифатической гид-
роксильной группы хинона был установлен на основании устойчивости
этого гидроксила к окислению и по трудности этерификации.
Образование дурогидрохинона (XXVII) (схема 59) и а-токоферол хи но-
на (XII) указывает на то, что гидроксильная группа находится .в положении
6 ядра хромана.
Таким образом, при расщеплении ядра хромана насыщенные углерод-
ные атомы пиранового цикла вместе с боковой алифатической цепью а-то-
коферола переходят в оксикислоту. Строение а-токоферола было подтверж-
дено его синтезом [4, 12, 15, 84, 85]. Таким образом а-токоферол является
2,5,7,8-тетраметил-2-(4',8',12'-триметилтридека)-6-оксихроманом (I).
Конфигурация асимметрических центров в положении 2 дигидропирано-
вого цикла хромана и алифатической цепи природного а-токоферола (I)
определена как С* 2R, 4/R, 8'R на основании сравнения кривых дисперсии
оптического вращения алифатических кислот, альдегидов и кетонов, обра-
зующихся при расщеплении а-токоферола, а также синтеза природного а-то-
коферола (I) [51] из природного транс-фитола [9, 10] и изомерного ему С*
2S, 4'/?, 8'/?-а-токоферола (XV) [54].
р-Токоферол—5,8-диметилтокол
При деструктивном восстановлении йодистоводородной кислотой [J-то-
коферол (11) дает n-ксиленол (XXXIV), а при пиролизе — триметил-п-гид--
рохинон (псевдокумогидрохинон) (XXXV) [82, 89], что показано на схеме
60. Окисление Р-токоферола хромовой кислотой приводит к различным про-
дуктам распада, в том числе к лактону C2i (XXXI) [83] такого же строения,
который образуется и при окислении а-токоферола.
263
Схема 60
Установление строения ^-токоферола реакциями его расщепления
Две метильные группы в бензольном ядре 0-токоферола находятся в па-
ра-положении; одно положение не замещено, что доказано введением аллиль-
ной группы в молекулу p-токоферола при действии аллилбромида и хлорис-
того цинка [551. Все эти данные дают основание отнести fl-токоферол к низ-
шему гомологу а-токоферола, имеющему на одну метильную группу мень-
ше. Строение 0-токоферола было подтверждено его синтезом из диметил-
гидрохинона и бромпроизводного фитола [4, 15].
Y-Токоферол—7,8-диметилтокол
При пиролизе этот изомер дает тот же триметил-п-гидрохинон (XXXV),
что и Р-токоферол (II), а при деструктивном окислении —диметилмалеино-
вый ангидрид (XXIX) и другие продукты окисления [90], такие же, какие
образуются при окислении а-токоферола.
Так как -(-токоферол (III) имеет такую же эмпирическую формулу, как
и.Р-токоферол (II), то он является его изомером. Строение у-токоферола до-
казано синтезом [4, 15].
S-То кофер о л—8 метилтокол
Этот изомер первоначально был получен синтетическим путем в 1940 г.
[91 ] и только в 1957 г. был выделен из соевого масла [7]. Строение природ-
ного S-токофероЛа было доказано по идентичности продуктов его пиролиза
е продуктами пиролиза синтетического 8-метилтокола (V).
С2-Токоферол—5,7-диметилтокол
Этот токоферол первоначально получен синтетическим путем в 1938 г.
[4]. Позднее, в 1955 г., впервые выделенному из пшеничных отрубей ве-
ществу, обозначенному как с-токоферол [18, 92], была приписана структура
5,7-диметилтокола [93]. Однако в 1960 г. повторно выделенный из масла
пшеничных отрубей, а также из пальмового масла природный с-токоферол
не был идентичен 5,7-диметилтоколу; он отличался также от с-токоферола,
выделенного из риса [94 ]. На самом деле оказалось, что природный, так на-
зываемый с-токоферол из пшеничных отрубей и пальмового масла не явля-
•ется токоферолом: он соответствует по структуре триметил-6-оксихроману
£ ненасыщенной боковой алифатической цепью в положении 2 и является
изопренологом а-токоферола—2,5,7,8-тетраметил-2-(4',8'12'-триметилтриде-
ка-3’,7'11'-триенил)-6-оксихроманом (XXXVII, с. 269) [94]. Это соединение
теперь стало обозначаться как ^-токоферол или а-токотриенол (5,7,8-три-
метилтокотриенол, токохроманол-3).
При восстановлении а-токотриенол (^-токоферол) (XXXVII) переходит
в а-токоферол (I). Он может быть получен в результате хдорметилирования
или оксиметилирования природного 0-токотриенола, так называемого е-то-
264
коферола — 2,5,8-триметил-2-(4',8',12'-триметилтридека-3',7',11' -триенил)-
6-оксихромана (XXXVIII) 194, 95]. а-Токотриенол (XXXVII) синте-
зирован из 2,3,5-триметилгидрохинона и гераниллиналоола [96].
C-Токоферол, выделенный из риса, по своей структуре является 5,7-ди-
метилтоколом и обозначается как С2-токоферол (IV) [94, 95]; этот токоферол
идентичен ранее полученному синтетическому 5,7-диметилтоколу [4].
е-Токоферол—р-токотриенол
Так называемый е-токоферол (XXXVIII, с. 269), первоначально выде-
ленный из пшеничных отрубей [92 ], по своей структуре относился к 5-ме-
тилтоколу. Однако позднее было установлено [95], что это природное ве-
щество отличается от 5-метилтокола и не является токоферолом, а представ-
ляет собой изомер Р-токоферола с ненасыщенной боковой алифатической
л.- цепью и при восстановлении переходит в p-токоферол (II) [96]. Для е-то-
•X7 коферола было установлено строение 2,5,8-триметил-2-(4',8',12'-триметил-
тридека-3',7',1Г-триенил)-6-оксихромана (XXXVIII), т. е. изопренолога
0-токоферола [94, 96]. Р-Токотриенол—5,8-диметилтокотриенол (е-токо-
. ферол) (XXXVIII)—синтезирован из 2,5-диметилгидрохинона и трансге-
раниллиналоола [96]. Р-Токотриенол (e-токоферол) (XXXVIII) при хлор-
метилировании или оксиметилировании с последующим восстановлением
превращается в а-токотриенол (^-токоферол) (XXXVII) [94, 95].
ВЫДЕЛЕНИЕ ТОКОФЕРОЛОВ
Впервые витамины Е (а- и p-токоферолы) выделили из природных источ-
ников в виде кристаллических эфиров Эванс с сотр. [3]. а-Токоферол изоли-
рован в виде аллофаната из неомыляемой части зародышей пшеницы [3].
а- и p-Токоферолы и небольшое количество 8-изомера содержатся в мас-
ле зародышей пшеницы, причем в масле американской пшеницы больше
а-изомера, чем p-изомера, а в масле европейской пшеницы больше р-токо-
ферола, чем а-изомера [97]. p-Токоферол находится в кукурузном и в хлоп-
ковом маслах. В хлопковом, кукурузном и пальмовом маслах содержится
также -[-токоферол и небольшое количество а-токоферола.
Для получения витамина Е из растительных масел хлопковых семян,
зародышей кукурузы и пшеницы существует два основных метода — омы-
ление и молекулярная дистилляция. По первому методу растительное масло
подвергают омылению при невысокой температуре и неомыляемый остаток
(около 5%) переводят в органический растворитель (спирт, хлороформ, ди-
хлорэтан и др.). После удаления растворителя из неомыляемой части масла
в растворе ацетона или метилового спирта при температуре — 10° С вы-;
кристаллизовываюг стерины (около 90% неомыляемой части), остаток сте-
ринов осаждают дигитонином (или выделяют при температуре —15, —18° С) j
и концентрат витамина Е очищают высоковакуумной перегонкой [98].
По другому методу растительное масло подвергают молекулярной дистилля-
ции при 0,004 мм рт. ст. и температуре 200—240° С и отбирают фракцию
концентрированного витамина Е [99]; при двукратной дистилляции концент-
рат содержит около 60% токоферолов.
Для получения более чистых концентратов витамина Е применяют ад-
сорбционную хроматографию. Для стабилизации препаратов прибавляют в
качестве антиоксидантов гидрохинон или L-аскорбиновую кислоту [30].
В совершенно чистом виде токоферолы получают через их кисталли-
ческие эфиры, главным образом аллофанаты [9]. Токоферолы могут быть
разделены методом кристаллизации их аллофанатов:труднорастворимая
фракция содержит эфир а-токоферола, легкорастворимая —эфир р-токофе-
рола. Для получения эфира 7-токоферола применяют масло зародышей ку-
курузы [100].
265
, Для разделения токоферолов используют также хроматографическую
адсорбцию.
ПРИРОДНЫЕ ВЕЩЕСТВА, РОДСТВЕННЫЕ ТОКОФЕРОЛАМ
Помимо токоферолов, из растительных природных источников выделены
их изопреноидные аналоги, представляющие собой хромаполы с боковой
ненасыщенной цепью, состоящей из трех или более частично гидрированных
изопреноидных остатков.
В основе таких хроманолов лежит токотриенол, т. е. молекула токола с
боковой, частично насыщенной изопреноидной цепью, 2-метил-2-(4', 8'
12'-триметилтридека-3',7',11 -триенил)-6-оксихроман (XXXVI) .
сн3 сн3 сн,
17 П J/CH2CHJ,CH=CCH2CH,Ctt=CCH,Cfi,CH=CCH3
^ХоЖсн3 *'
XXXVI
Так, а-токотриенол — 5,7,8-триметилтокотриенол, токохроманол-3 (или
Cj-токоферол из пшеницы) (XXXVII) — представляет собой 2,5,7,8-тет-
раметил-2-(4',8',12'-триметилтридека-3',7',11 '-триенил)-6-оксихроман [94 J
или 2,5,7,8-тетраметил-2-трипренилметил-6-оксихроман и является
изопренологом а-токоферола.
Так же построены p-токотриенол, 5,8-диметилтокотриенол (или е-токо-
ферол из пшеницы) (XXXVIII) [94,95] — изопренолог р-токоферола,
СНз '
XXXVII
XXXVIII
у-токотриенол, 7,8-диметилтокотриенол, пластохроманол-3 (или ^-токоферол
(XXXIX) — изопренолог f-токоферола и o-токотриенол, 8-метилтокотрие-
нол (XL) — изопренолог S-токоферола (см. номенклатуру— [5]).
НО.
СН3
I
СН2(СН4СН=ССН»)»Н
СН8‘
СН,
‘СН8
XXXIX
266
CHj
сн8
,Н
а- и Р-Токотриенолы выделены из пшеничных отрубей [95, 1011, а- и
ртокотриенолы —из риса [102]; все четыре а-, Р-, у- и S-токотриенола обна-
ружены в латексе Hevea brasiliensis [103, 104].
Среди различных изопренологов пластохроманола-3 (7-токотриенола)
в природных источниках обнаружены вещества с большим количеством изо-
преноидных звеньев в боковой алифатической цепи. Из листьев Hevea bra-
siliensis выделен [105] пластохроманол-8 (XLI), а из листьев табака [106]—
пластохроменол-8, 3,4-дегидропластохроманол-8 (соланохромен) (XL II)
[107]
CHS
I
CHt (CH»CH=CCH2)8H
сн8
XLI
сн3
CH2(CH2CH=CCHt)8H
сн8
-сн8
XLII
Для токотриенолов установлена абсолютная пространственная конфигу-
рация боковой цепи положения 2. а-Токотриенол (XXXVII) и р-токотриенол
(XXXVIII) имеют 2 ^-конфигурацию боковой цепи и полную транс-конфи-
гурацию двойных связей [11]. Установлено, что у-токотриенол (XXXIX)
[102] и пластохроманол-8 (XLI) из листьев Hevea brasiliensis [102] также
имеют конфигурацию асимметрического центра, как 2R, и полную транс-кон-
фигурацию двойных связей боковой цепи [108],
XXXVII
однако боковая цепь пластохроманола-8 из табака имеет С*2 рацемический
[108].
Известны еще две группы природных и синтетических хромановых про-
изводных с боковой алифатической цепью, состоящей из частично или пол-
ностью насыщенных изопреноидных звеньев, родственных токоферолам.
К ним относятся филлохроманол, нафтотокоферол (XLIII) и производные
убихинона (коэнзима Q) [109—111], например убихроманол-9 (XLIV) и
убихроменол-9 (XLV).
267
сн.
сир
осн
но
сн3
сн2(сн2сн=ссн>)8н
СНз
XLIV
сир
СНз
CHj^HjCH^CCHjsH
сн,
осн,
XLY
Филлохроманол — 5-метил-7,8-бензотокол (XLIII)—синтезирован из
2-метил-3-фитил- 1,4-нафтохинона (витамина КР [112, 1131; он показывает
10%-ную активность а-токоферола и одновременно около 0,5% активности
филлохинона (витамина Kj) [112].
Если токоферолам, как указывалось выше, соответствует продукт их
мягкого окисления — n-бензохинон с раскрытым хромановым циклом, на-
пример а-токоферолу (I) соответствует а-токоферолхинон (XII), то а-токо-
триенолу, токохроманолу-3 (XXXVII) соответствует а-токотриенолхинон
(XLVI),
о
II
сн,-./4!
СН3-Л ’•
II
о
__СНз СНз СН,
I—СН,СН2ССН2 (Снрн Л1СН,),Н
он
XII
II
гп гн СН, СН,
П Г 8 1 1
СН,—I СН2СН/ХН,(СН£Н=ССН,),Н
II ОН
о
XLVI
7-токотриенолу, пластохроманолу-3 (XXXIX), — пластохинон-4 (XLVII),
а пластохроманолу-8 (XLI) — пластохинон-9 (XLVIII)
О
СН,-/ \ СН,
сн,-\ /-(CHjChUchj.h
&
XLVII
XLVIII
268
филлохроманолу, иафготокоферолу (XLIII),—филлохинон, витамин Ki
(XLIX), а убихроманолу-9 (XLIV) — убцхинон-10, коэнзим Qw (L).
О
СНз
!—СН2СН=ССН2(СН2СН2СНСН2)8Н
о
XLIX
Следует отметить, что пластохиноны в связанном с хлорофиллом сос-
тоянии участвуют в фотосинтезе, а также принимают участие в окислитель-
но-восстановительных реакциях, происходящих в растениях. Убихиноны
участвуют в переносе электронов и протона в окислительно-восстанови-
тельных реакциях, а также реакциях окислительного фосфорилирования
в животных тканях и микроорганизмах [109—111, 114].
Помимо токоферолов и токотриенолов, в кукурузном масле обнаруже-
ны продукты окислительной димеризации 7- и S-токоферолов и 7-токо-
триенола — б-^-токоферил-бфртокоферол (XXII, с. 263), 5-(-у-токотри-
енил)-7-токоферол (LI), 5-(7-токоферилокси)-у-токоферол (XXIII, с. 263)
и 5- (о-токс)ферилокси)-7-токоферол (LII) [115].
СИНТЕЗ ТОКОФЕРОЛОВ
Важнейшие синтезы токоферолов заключаются в конденсации алкилза-
мещенных n-гидрохинонов соответствующего (для а-, 0-, 7-, Сг- и 8-изомеров)
строения с аллильной алифатической цепью, представляющей собой по
строению углеродного скелета четыре конденсированных изопреноидных зве-
на. В качестве такой цепи применяют фитол, изофитол и их производные.
Преимущественное образование производных хромана находится
в связи с наличием одного или двух алкильных заместителей у 7-углерод-
ного атома алифатической цепи. В отсутствие алкильных заместителей про-
исходит циклизация с образованием производных кумарана [88].
269
Синтез а-токоферола
а-Токоферол синтезировали Каррер с сотр. [12] из триметил-п-гидро-
хинона (XXXV) и фитилбромида (LIII) в среде бензола или петролейного
эфира в присутствии хлористого цинка при 80° С в токе азота почти с ко-
личественным выходом.
Катализаторами реакции конденсации, помимо хлористого цинка [12,
1161, могут служить: муравьиная кислота [85, 117], уксусная кислота
[121, фосфорный ангидрид [118], эфират трехфтористого бора [119]; одна-
ко конденсация может протекать при 180—190° С и без катализатора
[85, 1201.
В качестве среды известно применение также сероуглерода [12], дека-
лина 185] и других растворителей.
Реакция конденсации протекает через ряд промежуточных соединений.
Первоначально образуется аллиловый эфир фенола (LIV) по одной из гид-
роксильных групп триметил-п-гидрохинона с фитилбромидом.
Затем, по-видимому, происходит перегруппировка аллилового эфира
в бензольное ядро соединения LV, в ор/по-положение к гидроксильной
группе и циклизация с образованием а-токоферола (I) [121], что пока-
зано на схеме 61.
Схема 61
Синтез а-токоферола из триметил-п-гидрохинола и фитилбромида
СН,
НОА
СНз Y ОН -ус.
СНз '
XXXV
ВгСН
oil СНз ZnCl3
^B/CH/cHsCHjCHCH^H -HBr
'СНЭ
LUI
СН,
НО А
СН Т
CHS
СНз
НО А
СН
СНз I ОН
СНз LV
L1Y
СН.
НО
сн,
СН,
СН8
.СН^СНзСНгСНСН^Н
СНа
CH^CHjCHjCHCH^H
СН3
I
Если конденсацию проводить в присутствии поташа, то можно выделить
аллиловый эфир фенола (LIV) [22].
Фитилбромид получают при действии концентрированной бромистово-
дородной кислоты [122] или трехбромистого фосфора на фитол. Для полу-
чения фитилхлорида, также применяемого в синтезе токоферола, исходят
из изофитола и концентрированной соляной кислоты [119].
Если для конденсации с триметил-n-гидрохиноном (XXXV) применить
дигидрофитилдибромид (LVI) в присутствии хлористого цинка [123], то
реакция образования а-токоферола (I) протекает также через промежуточ-
ную форму замещения LVII. В случае применения 2,6,10-триметил-14-эти-
нилпентадеканола-14 (LVIII) в присутствии хлористого цинка в среде де-
кагидронафталина [124] промежуточно получается 3,4-дегидро-а-токоферол
(LIX), который должен быть подвергнут восстановлению. При использова-
нии ацетилфитола [125] реакция идет через.подобную промежуточную фор-
му замещения.
270
СН, БгС'ЧЦ1
LYI
civr он
сна сн
XXXV ' с —
.... I^CH^CHCHjCHCH^H
•HOZ \нз
LVIII
При конденсации триметил-п-гидрохинона (XXXV) с фитолом (LX)
[85, 121, 126—1291
СН3 СНз
НОСН2СН==ССН2(СН2СН2СНСН2)8Н
. LX
реакция протекает непосредственно в ядро, затем происходит циклизация
полученного промежуточного соединения с образованием хроманового цик-
ла и получением а-токоферола [85]. Конденсация проводится при 140° С
с фосфорным ангидридом как катализатором; dZ-a-токоферол (I) получается
почти с количественным выходом [130].
Для конденсации с триметил-и-гидрохиноном (XXXV), приводящей
к получению-а-токоферола с высоким выходом, применяют изофитол (LXI)
[131, 132].
СНз СНз
I I
СН2=СНССН2(СН2СН2СНСН2)8Н
I '
, ОН
LXI
В качестве катализаторов реакции взаимодействия триметил-п-гидро-
хинона с фитолом или изофитолом, помимо фосфорного ангидрида, приме-
няют хлористый цинк [127 129], эфират трехфтористого бора [126, 129],
уксусную кислоту [126, 128], муравьиную кислоту [85], ионообменные смо-
лы — амберлит-15 [133] и реакцию проводят при более низкой температуре
в присутствии растворителя.
Каталитическое действие трехфтористого бора заключается, по-видимо-
му, в том, что он первоначально образует с триметил-n-гидрохиноном ком-
плекс, который далее уже реагирует с третичным виниловым или первичным
аллиловым спиртом [134].
Метод конденсации триметил-п-гидрохинона с изофитолом имеет тех-
ническое значение.
Известны синтезы a-токоферола, в которых триметил-п-гидрохинон кон-
денсирует сфитадиеном [84], дигидро-3-оксифитилхлоридом [135], 3,7,11,
15-тетраметил гексадекадиол ом-1,3 [136—138], а также с фитоевой кислотой
(LXII) [1391 ’
СН8 СН8
I I
НООС£Н=ССН2(СН2СН2СНСН2)8Н
LXII
Реакцию проводят в присутствии хлористого цинка или эфирата трехфто-
ристого бора.
271
Монобензоаты гидрохинонов дают лучшие выходы токолов, чем свобод-
ные гидрохиноны [140]; в полученных соединениях эфирные группы под-
вергают гидролизу.
Наряду с применением для синтеза а-токоферола (I) триметил-«-гидрохи-
нона используют также 3-формил- или 3-ацетиламино-6-окси-1,2,4-триметил-
бензол (LXIII), который конденсируют в кислой среде с фитилбромидом
(LIII) или фитолом (LX) в хромановое производное LXIV, и затем деацили-
руют в 6-амино(дезокси)-а-токоферол (LXV) (схема 62). Для получения
а-токоферола аминогруппу соединения LXV превращают в гидроксильную
через диазосоединение или окислением через хинон с последующим восста-
новлением в гидрохиноновое производное и его циклизацию [141 ].
u Схема 62 •
Синтез а-токоферола из 3-амино-6-окси-1.2.4-триметилбензола и фитола или
фитилбромида
V’ сн3
ь CH , I .
II ^CH^CHjCHjCHCH^jH
ХН3
LIII или LX
СНд
^CHjfCHjCHjCHCH^H -------
хсна
LXV
СН,
СН3
----- jf If jLcHj^HjC^chch^h——
снГХ^сн,
<4 LXIV
к‘Г
. С LL
но <^3
- Т If Ъсн^снснДн
сн?то^сна
Ас=НСО: СОСН/, X=Br(Llll) он (lx)
Синтез может быть произведен и из 3-нитро-6-окси-1,2,4-триметилбен-
зола [142] с последующим восстановлением 6-нитрохромана в 6-аминохро-
мановое производное LXV.
Синтез а-токоферола (I) осуществлен из 2,4,5-триметил-3,6-диметоксифе-
нил-2,6,10,14-тетраметилпептадекаен-1-кетона (LXVI), который нагреванием
с 40%-ной бромистоводородной кислотой превращают в 2,5,7,8-тетраме-
тил-6-окси-(4',8',12'-триметилтридека) хроманон-4 (LXVII) с последующим
восстановлением амальгамой цинка в толуоле и обработкой соляной кисло-
той с выходом 88% [143].
Реакция получения а-токоферола (I) может быть осуществлена и не-
посредственно из кетона (LXVI) при действии соляной кислоты и амальга-
мы натрия [143].
Принципиально иной метод синтеза а-токоферола, в котором не при-
меняется фитол, изофитол или их производные, состоит в конденсации
272
4,8,12-триметилтридекамагнийбромида (LXIX) по реакции Гриньяра с три-
метил-0-бутанон-п-гидрохиноном (LXVIII). Последующая внутримолеку-
лярная циклизация образующегося а-токоферолгидрохинона (XIV) по тре-
тичной гидроксильной группе алифатической цепи при обработке минераль-
ной кислотой приводит к получению рацемического а-токоферола {1441:
сн, сн,
HO^A^CHpijCOCH, сн, сн,ч сц,
Т If +BrMfCH(CH,CH,CHCH^,H-— Т jf £%H,(CH,CH,CHCI^),H —-сМоиофары
CHj^T^OH LXIX CHfV'^OH l"CH, Д
СН, СН, ОН
LXVIII XIV
Этот метод применяется также для получения некоторых аналогов ви-
тамина Е [145, 146}.
Из других синтезов природного С* 2R, 4'R, 8'7?-а-токоферола (схема 63)
и его антипода — С* 2S, 47?, 8'7?-а-токоферола —следует остановиться на
синтезе из 2-формил-2,5,7,8-тетраметил-6-ацетоксихромана (LXXIII) с при-
менением реакции Виттига [147]. Для получения этого замещенного 2-фор-
мил-2-метилхромана (LXXIII) триметил-п-гидрохинон (XXXV) с защищен-
ными гидроксильными группами формилируют, затем из соединения LXX по-
лучают замещенный кетон (LXXI), вводят его в реакцию Гриньяра с ацети-
леном и получают этиниЛхроман (LXXII), который посредством частичного
гидрирования и озонирования превращают в 2-формил-2,5,7,8-тетраме-
Схема 63
Синтез а-токоферилацетата через замещенный 2-формнл-2-метилхромаи
тил-6-ацетоксихроман (LXXIII). Его конденсируют с гексагидрофарне-
зилтрифенилфосфоний-бромидом (LXXIV) в Д*'-дегидро-а-токоферол (LXXV)
и в результате восстановления получают а-токоферилацетат [147—1491.
Гексагидрофарнезилтрифосфоний-бромид (LXXIV) синтезируют из при-
родного фитола, который дегидратируют и озонируют; на полученный
3,7,11-триметилдодеканаль последовательно действуют алюмогидридом ли-
тия, трехбромистым фосфором и трифенилфосфором [147].
Реакция Виттига была применена и для следующего синтеза а-токоферо-
ла [1501 (схема 64). Триметил-п-гидрохинон (XXXV) и гераниол (LXXVI)
273
конденсируют в присутствии эфирата трехфтористого бора в бензоле в хро-
меновое производное (LXXVII), его ацетилируют и подвергают озонирова-
нию и получают 2,5,7,8-тетраметил-2-(2'-формилэтнл)-6-ацетоксихроман
(LXXVIII). После конденсации замещенного 2-(2'-формилэтил)-2-метилхро-
мана (LXXVIII) с псевдоионолом (продукт восстановления псевдоионона бор-
гидридом натрия) по Виттигу получают 2,5,7,8-тетраметил-2-(4',8',12'-три-
метил-3', 5', 7', 11'-тридекатетраен-1-ил)-6-ацетоксихроман (LXXIX); его
восстановление приводит к получению а-токоферилацетата (VIII) [150]:
Схема 64
Рацемический а-токоферол может быть расщеплен на оптические анти-
поды через 3-бромкамфарсульфокислые эфиры, получаемые из токоферола
и 3:бромкамфарсульфохлорида нагреванием в пиридине [15, 151]; эфир
d-a-токоферола идентичен эфиру природного токоферола [15, 140]. Опти-
ческие антиподы а-токоферола выделены также путем раскристаллизации
рацемата [152].
Синтезирован рацемический а-токоферол с меченым 14С [153].
Полусинтез а-токоферола из у- и 6-токоферолов
Низкоактивныс Р-, у- и о-токоферолы (П, III, V) могут быть превращены
в а-токоферол (I) хлорметилированием с последующим восстановлением
[154—156], что показано на схеме 65. Так, на [}- или -у-токоферолы (II и
Ш) действуют монохлорметиловым эфиром, образующимся из формальде-
гида и хлористого водорода [154], ди хлор диметил овым эфиром [156], па-
раформом и хлорокисью фосфора [155] или алкиламином[154 I; при этом в
ядро (положения 5 и 7) вступает хлорметильная группа (соединения LXXX
и LXXXI), которая восстанавливается амальгамированным цинком или
цинком и соляной кислотой в метильную группу с образованием а-токоферо-
ла (I); выход составляет 75% [154 — 156].
Методом хлорметилирования и последующего восстановления 5-метил-
токол (VI) можно превратить в высокоактивный 5,7-диметилтокол—^-то-
коферол (IV) [157].
Таким методом могут быть получены и эфиры токоферолов.
274
Схема 65
Превращение р- и у-токоферолов в а-токоферол через хлорметильные производные
СН,
СИ,
Т 7Г 'Lcii2(ch2ch2ciicii2)jh
CI1,
СН, В -Токоферол
г II
сн,
Т |Г V а^СН/ЖН^
СН^"1Г о сн3
СН, "у-Токоферол
III
сн,а
сн.
ai2(cit,cH2cncn,j3H -
сц,
сн,
LXXXI
Превращения 0-, 7- и о -токоферолов в а-токоферол можно достигнуть так-
же посредством форматирования формальдегидом в присутствии щелочного
катализатора и восстановления полученного метильного производного
[158] или действием гексаметилентетрамином в присутствии кислого ка-
тализатора с последующим гидролизом и восстановлением образовавшегося
формильного производного [159].
Еще одним методом — аминоалкилированием в свободные положения
ароматического цикла — 0-, у- и S-токоферолы (II, III, V) могут быть пре-
вращены в высокоактивный а-токоферол [154, 160] (схема 66). Так, на-
пример, 0-токоферол (II) аминоалкилируют диметиламином и формальде-
гидом по реакции Манниха в 7-диметиламинометил-0-токоферол (LXXXII);
его подвергают восстановительному расщеплению при действии цинковой
пыли в уксусной кислоте с одновременным ацетолизом уксусным ангидри-
дом в 7-ацетоксиметил-0-токоферилацетат (LXXXIII), который деацетили-
руют и полученное соединение LXXXIV восстанавливают в а-токоферол
(I) с общим выходом 27% [160].
Схема 66
Превращение ^-токоферола в а-токоферол через амниоалкильное производное
СН, СН,
НО Г/v СН, НО СН,
[ ХсН^СН/Т^СНСнДн------- Тд J^CHj^HjCHjCHCH^H-------
y'TT CIL, ^ГЦСТ4 О СН,
СН» ц СН, LXXXII
НОН2С
сн,
но
сн,
сн,
CH^CHjCH^HCH^H
сн,
LXXXIV
-----»-с( -Токоферол
I
Интересен также метод образования а-токоферола (II) из 0-, у и 8-токо-
феролов (II, III, V) путем их предварительного превращения в токоферол-
хиноны окислением хлорным железом, хлорметилирования, последующего
восстановления и циклизации [161, 162].
Синтез других токоферолов
Синтез 0-, у- и С2-токоферолов выполнен теми же методами из фитола и
соответственно из п-, о- и м-ксилил гидрохинонов в присутствии муравьиной
кислоты [4, 15, 911. При получении 0-токоферола (II) конденсация п-ксилил-
275
I-
V*
I
гидрохинона (LXXXV) с фитилбромидом (LIII) протекает более сложно
(схема 67) с образованием 7-фитил-Р-токоферола (LXXXVI) или трицикли-
ческого соединения LXXXVII, что происходит при применении избытка
фитил бр омида [4 ].
Схема 67
Синтез f-токоферола из п-ксилилгидрохинона и фитилбромида
«и,
сн
II ^СН^СНСНзСНСнДи
с\н,
UII
сн,
CH^CHjCHjCHCH^H
СН,
СНз сн,
Н^НзСНСНзСнДсН,^ но
сн"С
II
с
'сн.
сн,
LXXXVI
сн,
сн, сн,
H^h/hCHjjCH^jCH,
сн,
сн,
CH/CHjCHjCHCH^H
СНз
LXXXVII
При конденсации о-ксилил-п-гидрохинона (LXXXVIII) с фитолом (LX)
в присутствии муравьиной кислоты образуется у-токоферол (III) [15]; если
конденсацию проводить с фитилбромидом (LIII) в присутствии хлористого
цинка в бензоле, реакция идет дальше, и получается трициклическое сое-
динение LXXXIX [4].
сн3
r=ch,(ch2ch2chch.),h; х=Вг(ьш), oh(lx)
Разделение продуктов реакции производят хроматографией на окиси алю-
миния.
Аналогично протекает реакция и с х-ксйлил-п-гидрохиноном (ХС) и
фитолом (LX), при этом получается 5,7-диметилтокол (IV) [4], впоследст-
вии выделенный из природных источников под названием Сг-токоферола;
конденсация соединения ХС с фитилбромидом (LIII) приводит к получению
Ч-фитилзамещенного 5,7-диметилтокола (XCD [4]:
ХС
хсн2.
СН„
сн.
LI1I или LX
СН,
r=ch/ch2ch,chch^h; x=Br(un) oh(lx)
276
--------ггйяМВТ"
При образовании этих более сложных продуктов конденсации [4, 15]
хлористый цинк является более активным катализатором реакции, чем му-
равьиная кислота.
Три возможных монометилтокола—8-токоферол, 5- и 7-метилтоколы (V,
VI и VII) получают в виде их смеси конденсацией толил-п-гидрохинона
(ХСП) и фитола (LX) под влиянием муравьиной кислоты [4, 163, 164 ]
8-Метилтокол (8-токоферол) (V) в индивидуальном виде получен из мо-
нобензоата толил-п-гидрохинона и фитола [91 ] или из 4-ацетокси-о-крезола
и изофитола при действии хлористого цинка или эфирата трехфтористого
бора [165]. 8-Метилтокол (V) синтезирован также йз 5-бром-2-метил-п-ги-
дрохинона (ХСШ) через 5-меркаптопроизводное (XCIV) конденсацией с
хлоруксусной кислотой в 5-карбоксиметилтио-2-метил-п-гидрохинон (XCV)
и превращением его при 180° С в 7-метил-6-окси-2,3-дигидробензо-1,4-окса-
тиин-2-он (XCVI):
Соединение XCVI конденсируют с фитолом, десульфируют с никелем Ренея
и гидролизуют [166].
7-Метнлтокол (VII) получают конденсацией диметилового эфира 5-(2'-
бромэтил)-2-метил-п-гидрохинона с кетоном С18(XXXII) по реакции Гринь-
яра с последующим гидролизом и циклизацией [167].
5-Метилтокол (VI) синтезирован из 2-метил-З-бромэтил-п-диметоксибен-
зола и кетона Ci8 (XXXII) (см. с. 265) по реакции Гриньяра с последую-
щим гидролизом и циклизацией [168].
Аналог а-токоферола—Сх-токоферол, а-токотриенол (XXXVII),—син-
тезирован (схема 68) в виде природной формы с полной транс-конфигура-
цией боковой углеродной цепи из триметил-п-гидрохинона (XXXV) и
полного тране-гераниллиналоола (ХСУ11) в присутствии эфирата трехфторис-
того бора в диоксане [101] через промежуточные геранилгеранилтриме-
тил-п-гидрохинон (XCVIII), замещенный хинон (XCIX) и 3,4-дегидро-а-токо-
триенол (С).
₽-Токотриенол — e-токоферол (XXXVIII)—получен из 2,5-диметил-
гидрохинона тем же путем [101 ].
Для получения некоторых аналогов витамина Е с различными группами
в положении 2 известен метод введения алкильной группы в ядро хромана
по реакции Гриньяра конденсацией замещенного дигидрокумарина со
смесью галогенмагнийметила и галогенмагнийалкила [169].
277
Схема 68
Синтез а-токотриенола (Ci-токоферола) из тримеиил-л-гидрохиноиа
и траяс-гераииллниалоола
Получение исходных веществ для синтеза токоферолов —фитола,
изофитола и триметил-п-гидрохинона
Синтез фитола и изофитола
Наиболее распространенным источником фитола (LX), применяемого
для синтеза токоферолов, служит хлорофилл, подвергаемый гидролизу.
Фитол можно получить непосредственно из зеленой массы растений —
из люцерны, крапивы [170], из перечной мяты после отгонки из нее
мятного масла [171], из экскрементов гусеницы тутового шелкопряда
(после их гидролиза уксусной или серной кислотой) [172, 173] и др.
Один из методов получения фитола из хлорофилла состоит в экстракции
из крапивы сухим ацетоном липоидной фракции с последующим гидроли-
зом заключающегося в ней хлорофилла твердым едким натром; при этом
исключается образование стойких эмульсий [174]. Из 1 кг муки сухой кра-
пивы выделено 2,8—3,0 г фитола. Фитол —вязкое масло с т. кип. 145° С
при 0,03 мм.
Важное практическое значение имеют синтетические методы получения
фитола [14, 175—178] и изофитола [129, 179]? Во многих методах фигол
(LX) и изофитол (LXI) синтезируются через 2,6,10-триметилпентадекан-
15-он — кетон CJg, фитон (XXXII)—для получения которого известны раз-
личные пути.
Исходя из псевдоинона (CI), через гексагидропсевдоионон (СИ) и ряд
превращений можно получить гексагидрофарнезол (СШ), а из него синте-
зировать кетон C1S (XXXII); весь путь занимает семь стадий [175 J (схема 69,
путь А). Гекса гидрофар незол (CII1) может быть получен из псевдоионона
(CI) и другим вариантом, в результате его конденсации по Реформатскому
с бромуксусным эфиром, дегидратации, гидрирования двойных связей и
восстановления эфирной группы в спиртовую [180].
Гексагидрофарнезол можно также получить и непосредственно из при-
родного душистого вещества фарнезола посредством его гидрирования
[14], однако экономически этот вариант невыгоден. Рациональнее гекса-
гидропсевдоионон (СП) в четыре стадии превратить в кетон С18 (XXXII)
через ацетиленовый спирт С15 (CIV) (схема 69, путь Б) [178]. Преимущест-
278
Схема 69
Синтез кетона С18 (фитона)
Путь А
СН, СН, СН,
сосн=СНСН = А-СН 2СН2СН=JcH,—1
Псевдоионон
CI
СН3 СН3 СН,
| I I BrCHsCOOR
сосн^сн,сн2снсн2Сн2сн/знсн,------------.
4
Г екса гидропсевдоионон
СП
СН, СН,
ROOCCH2CCH2(CH2CH2CHCH2)2H----
I -н,0
он
сн, сн3
| | Na.QHbOH
ROOCCH =ССН2(СН,СН2СНСН2)2Н------->
сн, сн,
I I
НОСН2СН2СНСН2(СН2СН2СНСН2)2Н ->
С III
сосн,
СН, СН3 I
I I CH2COOR
ВгСН2СН2СНСН2(СН2СН2СНСН,)2Н-------
СООН СН3 СН,
II I
СН,СОСНСН2СН2СНСН2(СН2СН2СНСН2)2Н --->
СН, сн,- СН,
<!осн2сн2сн2(!:нсн2(сн2сн2(!нсн2)2н
Кетон С18
XXXII
Путь Б
СН, СН, СН,
[Н] | I I НС=СН; КОН
Псевдоионон---> СОСН2СН2СН2СНСН2СН2СН2СНСН,------------*
89%
CI Гексагидропсевдоионон
СИ
I I (H]/Pd
CHsCCCH2(CH2CH2CHCH2)2H----------»
I 94%
ОН CIV
сн, сн,
I I ch,coch2coor
СН2=СНССН2 (СН2СН2СНСН,)2Н--------------
I 65%
ОН
279
CHS CHS CHs CHs CHs
СОСН2СН2СН^^СНг(СН2СН2СНСН2)2Н(1^(1:ОСН2(СН2СН1СНСН2)3Н
95%
Кетон C18
XXXII
CHS СН3
I I
COCH2CHoCH2CHCH,
Путь В
CHS
2СН2СН,СНСН, + QHsOOCCH = CHCHgP (ОСгНв)2 ->
О
Гексагидропсевдононон
СП
CV
A
I
J 4
7
CHS CHj
I I (Hl
CsHoOOCCH=CHCH=CCHs (CH2CH2CHCH2)2H -*
CH,
CgHjOOCCHs (CH2CH2CHCHS)SH ->
CVI
CH8S CH2 CH,
|| I | [H]/A1—Hg
OCOCHs (CH2CH2CHCHg)sH----------
CHs CH,
COCH, (CHsCHsCHCHgJsH
Кетон Cj8
XXXII
Путь !'
CHs / CH3 CH,
I I | CH=CH; KOH
COCH2CH2CH2CHCH.CHXH2CHCH3--------------->
Гексагидропсевдоионон
СП
CH8 CH8
CH= CCCHj (CHaCHj^HCH^aH ->
Лн
civ i
CH8 CHs CHs CHS CH8
I I I [H] | |
COCH=CHCH=CCH2'(CH2CH2CHCH2)2H-----♦ COCH2 (CH2CH2CHCH2)SH
Кетон C18
CVII XXXII
i
во этого варианта заключается в повторении однотипных реакций: конден- s
сации кетонов с ацетиленом, частичном гидрировании тройных связей аце- '
тиленовых спиртов до двойных в присутствии ослабленного катализатора
Линдлара [181], в конденсации третичного ненасыщенного спирта с ацето- is
уксусным эфиром и его гидрировании в кетон С18 (XXXII); частичное про-
должение этих реакций приводит к получению изофитола (LXI) и фитола
(LX) [178] (см. схему 70). <
280 |
1
Еще один путь синтеза кетона Ci8 (XXXII) состоит в конденсации гекса-
гидропсевдоионона (СП) с триэтилфосфонокротонатом (CV) с последующим
восстановлением в эфир кислоты С17 (CVI), которая вводится в реакцию
с метилсульфинилкарбанионом, в результате чего образуется промежуточ-
ный 0-кетосульфоксид, восстанавливаемый амальгамой алюминия в кетон
С16 (XXXII) [182] (схема 69, путь В).
И еще короче путь синтеза кетона С18 из гексагидропсевдоионона [129,
183, 184] — всего в три стадии (схема 69, путь Г), если гекса гидропсевдо-
ионон (СП) конденсировать с ацетиленом в присутствии порошкообразного
едкого кали (или натра) в среде жидкого аммиака в ацетиленовый спирт
С15 (CIV) [178]. Это соединение затем вводят в реакцию с ацетоуксусным
эфиром [184] или превращают в ацетат и диацетат ацеталя, конденсируют
с ацетоном [183] в диеновый кетон С18 (CVII) и восстанавливают в кетон C^g
(XXXII).
Для синтеза псевдоинона (CI) известен двухстадийный путь из метил-
гептенона (СУ1Ц) через дегидролиналоол (см. с. 160)
СН3 CHS
СХХЗН„СН2СН=ССН3
CVIII
Гексагидропсевдоионон (СП) можно получить из метилгептенона (CVIII),
минуя псевдоионон [178], однако этот путь многостадиен и дает меньший
выход (36% вместо 49%).
Описан синтез кетона CJ8 из линалоола [179] и из цитронеллола [185].
Для синтеза фитола или изофитола кетон С18 (XXXII) наращивают на
два атома углерода конденсацией его с ацетиленом и получают ацетиленовый
карбинол С20 (CIX), каталитическое гидрирование которого приводит к по-
лучению изофитола (LXI) [14,129, 178, 179,1831. В результате последующей
аллильной перегруппировки и изомеризации третичного винилового спирта
изофитола образуется первичный аллиловый спирт — фитол (LX), что по-
казано на схеме 70 [14, 178].
Схема 70
Синтез фитола и изофитола из -кетона С18
СН, СН3 СНз
| | | НС=СН; КОН
COCHjCHgC^CHCH, (CH.CHpiCH^H---------------
90%
Кетон С18
XXXII
I I [H]/Pd
СНнССН, (CH^HgCHCHJgH--------------
I 94%
ОН
CIX
СН8 СН3 Аллильная
| j перегруппировка
СН2-=СНССН2 (СН.СН2СНСН2)3Н---------------
Ьн
Изофитол
LXI
281
CHa CHS
. -> НОСНгСН^ССН„ (CHjCH/iHCH^aH
Фитол
LX
Кетон CJ8 (XXXII) через эфир фитоевой кислоты можно превратить в
фитол (LX) с природной транс-конфигурацией двойной связи и /?-конфигу-
рацией по обоим асимметрическим центрам [182, 185]. Синтезирован опти-
чески активный фитол [186]. Для синтеза изофитола (LXI) из псевдо-
ионона (CI) предложен метод конденсации гекса гидропсевдоионона (СП)
с пропаргиловым спиртом с последующими превращениями и заключитель-
ной конденсацией с метилвинилкетоном (шесть стадий) [187, 188].
СН3 сн3 сн3
| | | НС = ССН2ОН; КОН
СОСН2СН2СН2СНСН2СН2СН2СНСН, ---------------*
Гексагидропсевдоионон
СП
СН, сн,
НОСН2С=СССН,(СН2СН2(!нСН2)гН----►
I —н,о
он
сн, сн, сн, сн,
I I [Н] II
НОСН2С=СС=СН (СН2СН2СНСН2)2Н----> HOCH2CH,CH2CHCH2 (CH2CH2CHCH2)2H->
сн3 сн3 сн,
| СН3СОСН=СН2 | I
ХСН2 (CH2CH2CHCHa)sH-------------> СН2=СНССН2 (СНгСН2СНСН2),Н
ОН
Изофитол
Х=Вг или Cl LXI
Для получения фитола (LX), минуя кетон Ci8, предложена конденса-
ция гекса гидропсевдоионона (СП) с у-бромкротоновым эфиром с последую-
щими превращениями через 5,9,13-триметилтетрадекановую кислоту [189].
Синтез триметил-п-гидрохинона
2,3,5-Триметил-п-гидрохинон (псевдокумогидрохинон) (XXXV), необ-
ходимый для синтеза а-токоферола, может быть получен несколькими пу-
тями. Один из относительно сложных синтезов 2,3,5-триметил-п-гидрохинона
(XXXV) заключается в ряде химических превращений 3,5-диметилфенола
(л-ксиленола), продукта каменноугольного дегтя, через основание Манниха
[190, 191].
Для синтеза триметил-п-гидрохинона (XX XV') можно исходить из моно-
бензил-п-гидрохинона (СХ) [192], который по реакции Манниха конденса-
цией с формальдегидом и морфолином превращают в производноеCXI; в ре-
зультате последующей кислой обработки, приводящей к удалению бензиль-
ной группы, получают 2,6-ди-(морфолинометил)-п-гидрохинон, и затем после
повторного проведения реакции с формальдегидом и морфолином —
2,3,6-три-(морфолинометил)-п-гидрохинон (СХП). Каталитическое восста-
новление этого соединения приводит к получению 2,3,5-триметил-п-гидро-
хинона (XXXV) с общим выходом 36%.
Продукт уплотнения ацетона, изофорон (СХШ), с успехом используют
для синтеза триметил-п-гидрохинона. Изофорон подвергают метилированию
в производное циклогексенона и в результате обработки получают 2,3,5-три-
282
метилфенол (CXIV), который окисляют нитрозодисульфонатом калия в псев-
докумохинон (CXV) [193], а затем восстановлением превращают в три-
метил-л-гидрохинон (XXXV):
схш CXIV
сн3
XXXV
Значительный интерес представляет синтез триметил-п-гидрохинона
(XXXV) с использованием мезитилена (CXVI) — продукта каменноугольно-
го дегтя [194 ]. В этом синтезе применяется перегруппировка хинолов в гидг
рохиноны по Бамбергеру [195, 196]
Первоначально мезитилен (CXVI) нитруютв мононитромезитил ен (CXVII),
затем осторожным восстановлением превращают в 2,4,6-триметилфенилгид-
роксиламин (CXVIII) и при действии серной кислоты — в промежуточный
хинол (СХ1Х); из этого соединения в результате последующей перегруппи-
ровки получают триметил-п-гидрохинон (XXXV) с выходом 67%.
Описан синтез триметил-п-гидрохинона из п-ксилола через ряд произ-
водных, получаемых в результате реакций формилирования, нитрования,
восстановления, окисления и др. [197].
Для получения триметил-п-гидрохинона (XXXV) можно исходить из
изодуридина (CXXXI) [198], который окисляют в псевдокумохинон (СХУ)
и затем восстанавливают в соединение XXXV, что показано на схеме 71;
однако получаемый таким путем хинон сильно загрязнен побочными про-
дуктами.
Улучшенным вариантом является синтез триметил-п-гидрохинона из псев-
докумола (СХХ). Через соединения CXXI—CXXIV его превращают в п-ди-
нитрокумол (CXXV), который восстанавливают в п-диаминопсевдокумол
(СХХХ), и в результате последующего окисления хромовой кислотой, суль-
фатом трехвалентного железа или хлорным железом получают псевдокумо-
хинон (CXV) [198 —202], затем путем восстановления сернистой кислотой
или другими восстановителями превращают в триметил-п-гидрохинон
(XXXV) [2031. Однако путь синтеза п-диаминопсевдокумола (СХХХ) через
n-динитрокумол (CXXV) многостадиен (схема 71).
Более рациональные методы синтеза триметил-п-гидрохинона (XXXV)
из псевдокумола (СХХ) через n-диаминопсевдокумол (СХХХ) состоят в
предварительной защите реакционноспособного положения 5 введением
брома [204—207] или сульфогруппы [136, 208] (соединения CXXVI и
CXXVIII), в последующем нитровании (соединения CXXVIII и CXXIX).
283
химическом или каталитическом (со скелетным никелем) восстановлении
двух нитрогрупп и элиминировании атома брома [116, 207, 209—212] или
сульфогруппы [199—202,208] с образованием диаминопсевдокумола (СХХХ)
(схема 71). Эти методы дают высокие выходы п-диаминопсевдокумо-
ла; синтез триметил-п-гидрохинона (XXXV) из него описан выше. Псев-
докумол (СХХ)—легкодоступный продукт перегонки каменноугольного
дегтя.
Схема 71
Синтез триметнл-п-гидрохинона из псевдокумола
Сульфирование псевдокумола позволяет выделить его из соответствую-
щей фракции в виде чистой псевдокумол-5-сульфокислоты (CXXVIII),
которую можно превратить в 5-бромпсевдокумол (CXXVI) [116].
- Хорошие результаты дает синтез триметил-п-гидрохинона (XXXV) пу-
тем конденсации диэтилкетона с метилвинилкртоном в присутствии едко-
го натра в 2,5,6-триметилциклогексен-2-он-1 (выход 77%) с последующим
термическим (300° С) дегидрированием на катализаторе Pd на силикаге-
ле [212а].
БИОЛОГИЧЕСКОЕ ЗНАЧЕНИЕ ТОКОФЕРОЛОВ. ЗАВИСИМОСТЬ МЕЖДУ
СТРОЕНИЕМ И БИОЛОГИЧЕСКОЙ АКТИВНОСТЬЮ
Токоферолы участвуют в переносе водорода в окислительно-восстано-
вительных реакциях, протекающих в мышечной, сосудистой, соединитель-
ной и других тканях организма, по-видимому, входя в состав ферментных
систем. Механизм действия токоферолов точно не установлен; возможно,
они принимают участие в окислительном фосфорилировании [2131.
Участие токоферолов в реакциях обмена веществ, по-видимому, заклю-
чается в обратимом превращении в токохиноны, например а-токоферола
(I) в а-токохинон (СХХХII)
284
Витамин Е является антистерильным витамином. При его отсутствии
или недостатке у самок возникает бесплодие или самопроизвольный аборт,
а у самцов перерождаются сперматозоиды; у животных наблюдается дистро-
фия мышц и дегенерация нервных клеток.
Возможно, что механизм физиологического действия витамина
Е состоит в том, что токоферол предупреждает образование токси-
ческих веществ, возникающих при неполном расщеплении жиров (жирных
кислот) в организме животного; токсические вещества вызывают торможение
высокой активности деления клеток эмбриона или зародышевого эпителия
семенников, что приводит к их гибели. Это подтверждается различными
экспериментами, а также тем, что токоферол является специфическим анти-
оксидантом при обмене ненасыщенных жирных кислот, образующих жир
с нормальным уровнем устойчивости к окислению [216]. Продукты окис-
ления жиров вызывают бесплодие у самок крыс, подобное тому, которое
возникает при отсутствии в диете витамина Е [217].
Метаболитом а-токоферола является 4-метил-4-окси-6-(3,5,6-триметил-
бензохинонил) гексакарбоновая кислота, выделенная из мочи в виде лак-
тона [214].
Потребность человека в витамине Е составляет от 10 до 25 мг в сутки
(высшая дозировка — при очень тяжелом труде и для беременных и кор.-,
мящих). В лечебных целях препарат витамина Е применяют в дозах до 1 г
для лечения стерильности, мышечной дистрофии и в некоторых случаях
для предупреждения самопроизвольных абортов у женщин [215]. Токсич-
ность а-токоферола еще не наблюдается при 4 г на 1 кг (испытание на кры-
сах) [220].
Витамин Е нужен для нормального размножения собак, кроликов, крыс,
морских свинок и кур; он необходим для нормальной жизнедеятельности
ряда мелких домашних животных (овец, коз, свиней) и крупного рогатого
скота. Из насекомых в токоферолах как ростовом факторе нуждаются пче-
лы; в отсутствие токоферолов личинки развиваются в рабочие пчелы, для
развития личинки в матку в пище должно быть большое количество токофе-
ролов.
В природе токоферолы синтезируются только растениями.
Витамин Е преимущественно содержится в растительных маслах и мас-
лах ростков семян злаков и других растений; в животных продуктах, кроме
рыбьего жира, витамин Е содержится в незначительном количестве. Он
встречается в маслах главным образом в свободной форме [2181 наряду с
другими соединениями, более сильными антиоксидантами.
285
Токоферолы широко применяются как антиоксиданты для предохранения
от прогоркания масел и для стабилизации препаратов витаминов А и D
[2191.
По своей химической структуре токоферолы характеризуются относи-
тельно высокой специфичностью. Пространственная конфигурация метиль-
ных групп в положениях 2, 4' и 8' влияет на биологические свойства токо-
феролов [221 ]. Витаминная активность Л-а-токоферилацетата, который
более устойчив к окислению, чем свободный токоферол, в 1,47 раза превы-
шает активность свободного dAa-токоферола. Активность d/a-токоферола
составляет 0,68 и. е., для природного d-a-токоферола она значительно вы-
ше — 0,92 и. е., а для J-a-токоферилацетата — 1,36 и. е. по сравнению с
витаминной активностью Ш-а-токоферилацетата, активность 1 мг которого
принята за 1 и. е. [222]. Таким образом, активность природного а-токофе-
рола с /^-конфигурацией в положении 2 выше активности синтетического
a-токоферола (рацемического) в положении 2 всего на 30%. a-Токоферол с
S-конфигурацией в положениях 4' и 8' имеет такую же активность, как и
его рацемат [9].
Наличие гидроксильной группы в положении 6 необходимо для про-
явления Е-витаминной активности соединений. Однако некоторые сложные
эфиры (ацетат, пропионат, бутират, сукцинат, фосфат) a-токоферола по-
казыват большую активность, чем свободный витамин [21, 23, 30, 221],
в частности d-a-токоферилсукцинат — 1, 21 и. е., по-видимому, вследствие
более легкого усвоения организмом. Простые эфиры обладают низкой акти-
вностью; аллофанаты совсем неактивны [223].
Зависимость биологической активности от количества и расположения ме-
тильных групп в ароматическом цикле молекулы токоферолов освещена
на с. 257.
Введение иных заместителей вместо метильных групп в бензольное ядро
в сильнейшей степени снижает витаминную активность токолов. Так, го-
молог 5, 7-диметилтокола с этильными группами в положениях 5 и 7 обла-
дает примерно 5% активности [4, 91 ]; однако введение одной или двух ме-
тильных групп наряду с этильными делает токолы более активными. Акти-
вность 5, 7-диметил-8-этилтокола [15, 2241 и диэтилметилтокола [225] сос-
тавляет уже 25%. Токол, не имеющий метильных групп, и 6-дезокситокол
[226] не показывают витаминной активности в 40-кратных дозировках по
отношению к a-токоферолу. Замещение положений 7 и 8 трех- и четырех-
членными циклическими системами —(СН2)3— и —(СН2)4— дает соедине-
ния, не активные в 20-кратных дозировках [2271.
Введение метоксильных групп в бензольное ядро токола сильно понижает
активность соединений [125]. Дегидрирование ядра хромана приводит к
некоторому понижению активности — до 50%, например для d/-3, 4-дегид-
ро-а-токоферола (LIX) [228].
СНа
СН8
-СН2 (СН»СН2СНСН2)8Н
CHS
LIX
Однако значительно большая специфичность наблюдается при измене-
ний заместителей в положении 2 цикла хромана. Укорачивание алифати-
ческой цепи на одну изопреноидную группу (из 5 атомов углерода) [55, 229,
230] уменьшает активность до 10%, а укорачивание на две или три изопре-
286
ноидные группы или удаление метильных заместителей алифатической цепи
приводит к полной или почти полной потере активности, так же как и вве-
дение в алифатическую цепь ненасыщенных связей [55, 2311. 2-Додецил-2,
5, 7, 8-тетраметил-6-оксихроман все же имеет около 5% активности [1691.
Аналоги токоферолов, у которых гидроксильная группа заменена на
другие группы, например аналоги а-, Р-, у- и С2-токоферолов с
—CH2N(CH3)2-rpynnaMH [2321, аналоги у-токоферола с группами
—СН2ОСН3, —СН2ОС2Н3, —CH2N(CH3)2 обнаруживают некоторую Е-ви-
таминную активность.
Аналоги и низшие изопренологи а-токоферола с количеством гидриро-
ванных изопреноидных групп п от 1 до 3, боковая цепь которых оканчивает-
ся спиртовой [233 J или карбоксильной группой [234]
являются антиоксидантами и обнаруживают некоторую активность .вита-
мина Е.
Раскрытие ядра хромана в молекуле токоферолов с образованием п-хи-
нона, например а-токоферолхинона (XII) из а-токоферола, приводит к ина-
ктивированию молекулы [33, 223].
Интересно отметить, что кумариновое производное CXXXIII
СН3
1 , СООС2Н
°Ц I
I О
CHS
схххш
имеет около 10% биологической активности витамина Е, однако другие ку-
мариновые производные неактивны или почти неактивны [235].
Вместе с тем оказалось, что многочисленные упрощенные по своему
строению соединения, имеющие фенольную группу или о- и «-гидрохино-
новое строение, обладали все же небольшой (до2%) Е-витаминной актив-
ностью при испытаниях на крысах. К ним относятся: о-аллилфенол,
ди-о-гексенилфенол, п-амино-о-алил-фенол [236], о- и п-ксилилгидрохинон
[226], триметил-л-гидрохинон, дурогидрохинон, некоторые нафтохиноны
[226, 237]. По другим данным [238], многие из этих соединений все же неак-
тивны.
Антагонистами токоферолов являются продукты, содержащие альде-
гидные и кетонные функции, образующиеся при прогоркании жиров (жир-
ных кислот или стеринов), независимо от того, присутствует ли токоферол
в пище или нет. Антогонистом витамина Е является продукт мягкого окис-
ления а-токрферола — а-токоферолхинон (XII) [2391
287
сн3
\Az\
I | 7f/CH2(CH2CU2CHCH2)sH
cZ
ch3 ° in CH
XII
причем большие количества а-токоферола не могут снять его антагонисти-
ческого действия. Это соединение и продукт его дальнейшего восстанов-
ления — а-токоферолгидрохинон (XIV) — применяются для лечения ат-
рофии и дистрофии мышц [43] наряду с препаратами витамина Е.
Физиологическое действие гомологов токоферола и их производных
рассмотрено в обзоре [240].
&: СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ (К ГЛАВЕ VI)
V 1. Н. Е v a n s, К. Bishop. Scien-
се, 56, 650 (1922).
"'•;-2. В. Sure. J. Biol. Chem., 59, 19
(1924).
3. Н. Evans, О. Emerson,
G. Emerson. J. Biol. Chem.,
113, 319 (1936).
4. P. К a r r e r, H. F r i t zsc he.
Helv. Chim. Acta, 21, 1234 (1938);
22, 260 (1939).
, 5. IUPAC, J. Am. Chem. Soc., 82,
5581 (1960); Biochem. Biophys.
Acta, 107, 1 (1965); J. Biol. Chem.,
,«.g 241, 2987 (1966).
. .7. 6. M. J о f f e, P. Harris, J. Am.
5?’ Chem. Soc., 65, 925 (1943).
>4 7. M. Stern, C. Robeson,
?•L. Weisler, J. Baxter. J.
Am. Chem. Soc., 69, 869 (1947).
8. R. C a h п, С. I n g о 1 d, V. P r e -
log. Experientia, 12, 81 (1956).
9. P. Karrer, H. Rentschler.
Helv. Chim. Acta, 26, 1750 (1943).
10. H. Mayer, P. S c h u d e 1,
R. R ii e g g, O. Isler. Helv.
Chim. Acta, 46, 963 (1963).
II. О. I s 1 e г, H. M a у e r, J. M e t z-
g e r, R. R ii e g g, P. S c h u d e 1.
Angew. Chem., 75, 1030 (1963).
12. P. Karrer, H. Fritzsche,
B. R i n g i e r, H. Salomon.
Helv. Chim. Acta, 21, 520, 820
(1938); Nature, 141, 1057 (1937).
13. P. Karrer. Helv. Chim. Acta, 26,
1741 (1943).
14. F. Fischer, K. Lowenberg.
Lieb. Ann., 475, 183 (1929).
15. P. Karrer, H. Koenig,
B. R i n g i e r, H. Salomon.
Helv. Chim. Acta, 22, 1139 (1939).
16. J. Baxter, C. Robeson,
J. Taylor, R. Lehman. J.
Am. Chem. Soc., 65, 918 (1943).
17. C. Robeson, J. Am. Chem.
Soc., 65, 1660 (1943).
18. P. E g g i t, F. N о r r i s, J. Sci.
Food. Agric., 6, 689 (1955).
19. P. Schudel, H. Mayer,
J. M e t z g e r, R. R ii e g g, О. I s-
1 e r. Helv.Chim. Acta, 46, 636 (1963).
20. J. Drummond, E. S i n g e r,
R. M c W a 1 t e r. Biochem. J., 29,
456, 2510 (1935).
21. P. Karrer, G. В ussm a n n.
Helv. Chim. Acta, 23, 1137 (1940).
22. Швейц, пат. 213146; С. A., 36,
4673 (1942).
23. V. D e m о 1 e, О. Isler, B. R i n-
g i er, H. Salomon, P. Kar-
rer. Helv. Chim. Acta, 22, 65 (1939).
24. P. P r a i 1 1, J. Chem. Soc., 1959,
3100.
25. Франц, пат. 6320M; РЖХ, 1970,
17H439.
26. J. Cawley, M. Stern. Пат.
США 2680749; С. A., 48, 11009
(1954).
27. L. Smith, W. Renfrow,
J. О p i e. J. Am. Chem. Soc., 64,
1084 (1942).
28. О. I s h i s a k а, К. M u r a k a -
m i. Яп. пат. 1737 (1962); С. A., 58,
3400 (1963).
29. T. N a k a g a w a, Y. Mori. Яп.
пат. 35 (1958); С. A., 52, 20203
(1958).
30. О. Isle r. Helv. Chim. Acta, 21,
1756 (1938).
31. R. R i d eway, J. Drummond,
M. Wright. Biochem. J., 34,
1369 (1940).
32. W. J о h n, W. E m t e. Z. Physiol
Chem., 261, 24 (1939).
33. W. John, E. Dietz el,
W. E m t e. Z. Physiol. Chem.,
257, 180 (1939).
34. L. S m i t h, W. Irvin,
H. Ungnade. J. Am. Chem.
Soc., 61, 2424 (1939).
35. C. Swift, G. Mann, G. Fi-
sher. Oil and Soap, 21, 37 (1944).
36. P. Karrer, H. Rentschler.
Helv. Chem., Acta, 24, 302 (1941).
37. M. F u r t e r, R. Meyer. Helv.
Chim. Acta, 22, 240 (1939).
38. H. Ungnade, L. S m i t h. J.
Org. Chem., 4, 397 (1939).
39. F. Quackenbush, H. Got-
tlieb, H. Steenbock. Ind.
Eng. Chem., 33, 1276 (1941).
288
, 40. M. Wall, Е. Kelley. Ind.
End. Chem. Anal. Ed., 18, 198
(1946).
41. V. Frampton, W. S к i n n e r,
P. C a m boor, P. Bailey. J.
Am. Chem. Soc., 82, 4632 (1960).
42. J. D i e t z e 1. E. Dietzel,
W. Emle. Z. Physiol. Chem., 257,
173 (1939).
43. D. C h a r 1 e s, D. N e I a n. Пат.
США 2856414; РЖХ, 1961, 4Л293.
44. С. М а г t i ns, Н. Е i I i n g s I е Id.
Biochem. Z„ 328, 507 (1957).
45. P. В о у e r. J. Am. Chem. Soc., 73,
733 (1951).
46. W. D ii г с к h e i m e r. L. Co-
hen. J. Am. Chem. Soc., 86, 4388
(1964).
47. A. E m m e r i e, C. Eng Л Rec.
trav. chim., 57, 1351 (1938); 58,
283 (1939).
48. M. Stern, J. В a x t e r. Anal.
Chem., 19, 902 (1947).
49. S. S о g e s p a r. Fr. Addn. 75631;
С. A., 58, 2438 (1963).
50. Neth. Appl. 6503840; C. A., 64,
11180 (1966).
51. P. S c h u d e 1, H. Mayer,
J. Metzger, R. Rflegg,
O. Isler. Helv. Chim. Acta, 46,
333 (1963).
52. T. Фудзисава, К- К a p и ё-
н э. Yakugaku Kenkyu, 34, 354
(1962).
53. С. R о b е s о n, D. N е 1 а п. Пат.
США 2856414; С. А.» 53, 5254 (1959).
54. Н. М а у е г, W. V е t t е г,
J. Metzger, R. Rflegg,
О. Isler. Helv. Chim. Acta, 50,
1168 (1967).
55. P. К a r r e r, R. Escher,
H. Fritzsche, H. Keller,
B. R i n g i e r, H. Salomon.
Helv. Chim. Acta, 21, 939 (1938).
56. C. Martins, H. Eilings-
f e 1 d. Lieb. Ann., 607, 159 (1957).
57. D. N e 1 a n, C. Robeson, J.
Am. Chem. Soc., 84, 2963 (1962).
58. W. Skinner, P. Al anpovic.
Science, 140, 803 (1963); J. Ogr.
Chem., 28, 2854 (1963).
59. D. M с H a 1 e, J. G r e e n. Chem.
Ind., 1964, 366.
60. W. Skinner, R. Parkhurst.
J. Org. Chim., 29, 3601 (1964).
61. J. Nilsson, J. Branstad,
H. Sievertsson. Acta. Pharm.
Suecica, 5, 509 (1968).
62. A. C s a 1 1 a n у, H. D r a p e r.
J. Biol. Chem., 239, 574 (1964).
63. J. N i 1 1 s о n, C. D a v e s,
K. F о 1 к e г s. Acta chem. scand.,
22, 207 (1968).
64. J. N i 1 s s о n, G. L a r s,
H. S i e v e r t s s о n, H. Se-
la n d e r. Tetrahedron Lett., 1968,
5023; Acta chem. scund, 23, 268
(1969).
65. W. Skinner. Biochem. Biophys.
Res. Communs, 15, 469 (1964).
66. D. Me H a 1 e. J. Green, Chem.
Ind., 1963, 982.
67. C. G о о d h u e, H. R i s 1 e y.
Biochem. Biophys. Res. Communs,
17, 549 (1964).
68. W. Skinner, R. Park,
hurst. J. Org. Chem., 31, 1248
(1966).
69. F. В e r g e 1, A. T о d d,
T. W о r k. J. Chem. Soc., 1938,
253.
70. J. Scud i, R. Bush. J.
Biol. Chem., 146, 1 (1942).
71. M. T i s h 1 e r, N. W e n d 1 e r.
J. Am. Chem. Soc., 63, 1532
(1941).
72. P. К a г г e г, H. К e I 1 e r.
Helv. Chim. Acta, 22, 253, 617 (1939).
73. L. S m i t h, К о 1 t h о f f,
Spillane. J. Am. Chem. Soc.,
447, 646 (1942).
74. E. S c h u I e к, P. R 6 z s a.
Z. Anal, chem., 126, 253 (1943).
75. W. J о h n, W. E m t e. Z.
Physiol. Chem., 268, 85 (1941).
76. M. Koller. Helv. Chim. Acta, 25,
1469 (1942); 26, 2166 (1943); 28, 26
(1945); 30, 1053 (1947).
77. M. Q u a i I e. J. Am. Chem. Soc.,
66, 308 (1944).
78. J. G r e e n, S. Marti n kle-
wi cz. Nature, 177, 86 (1956).
79. L. W e i s 1 e r, C. R о b e son,
J. Baxter. Anal. Chem., 19,
906 (1947).
80. G. Fisher. Ind. End. Chem.
Anal. Ed., 17, 224 (1945).
81. E. F e r n h о 1 z. J. Am. Chem.
Soc., 59, 1154 (1937); 60, 700 (1938).
82. W. J о h n. Z. Physiol. Chem., 250,
11 (1937); 252, 208, 222 (1938).
83. О. Emerson.!. Am. Chem. Soc.,
60, 1741 (1938).
84. L. S m i t h, H. U ng n a d e,
W. Prichard. Science, 88, 37
(1938).
85. F. В e r g e 1, A. Copping,
A. J а с о b, A. T о d d, T. Work.
Nature, 142, 36 (1938); J. Chem. Soc.;
1938, 1382-
86. H. Olcott. J. Biol. Chem., 107,
471 (1934); 110, 695 (1935).
87. H. О 1 с о t t, H. M a t i 1. J.
Biol. Chem., 104, 423 (1934).
88. P. К a r r e r, R. E s c h e r,
H. Rentschler. Helv. Chim.
Acta, 22, 1287 (1939).
89. F. В e г g e 1, A. Todd,
T. W о r k. J. Soc. Chem. Ind., 56,
1054 (1937).
90. О. E m e r s о n, L. S m i t h.
J. Am. Chem. Soc., 62, 1869 (1940).
91. A. J а с о b, F. S u t h c h i f-
f e, A. T о d d. J. Chem. Soc.,
1940, 327.
92. P. E g g i t, L. W a r d. J. Sci.
Food Agric., 4, 569 (1953).
93. J. Green, S. Marcinkie-
wi cz, P. W a r d. J. Sci. Food
Agric., 6, 274 (1955).
94. J. Green, P. Mamalis,
S. Marcinkiewicz,
D. McHale. Chem. Ind.,
1960, 73.
10—69
289
95. J. G r een, D. McHale,
S. M а г c i n к i e.w i c z, P. M a-
m a I i s, P. Wa 11. J. Chem.
. Soc., 1959, 3362.
96. D. M с H a 1 e, J. G r e e n,
S. Marci n ki ewi cz, J. Fee-
ney, L. S u t с 1 i f f e. J. Chem.
Soc., 1963, 784.
' 97. A. Tod d, F. В e г g e 1,
T. Work. Biochem. J., 31, 2257
•' (1937).
.„,98. В. А. Девятнин. Пищевая
V промышленность СССР, 1944, № 9,
- - 8
99. N. E m b r e e. Chem. Revs., 29, 317
(1941).
100. О. E m e r s о n, G. E m e r-
s о n, H. E v a n s. Science, 83,
421 (1936).
101. P. S c h u d e 1, H. Mayer,
J. M e t z g e r, R. R fl e g g,
O. Isler. Helv. Chim. Acta, 46,
2517 (1963).
4 102. J. P e n n о c k, F. H e гп-
rn i n g, J. К e r r. Biochem. Bio-
phys. Res. Communs, 17, 542 (1964).
103. P. D u n p h у, K. W h i 111 e,
J. Pennock, R. Morton.
Nature, 207, 521 (1965).
104. K. W h i t t 1 e, P. D u n p h y,
J. Pennock. Biochem. J., 100,
138 (1966).
105. K- Whittle, P. Dunphy,
J. Pennock. Biochem. J., 96,
17C (1965). '
106. R. R owl and. J. Am. Chem.
Soc., 80, 6130 (1958).
107. R. E r i c s о n, C. S h u n k,
N. T r e n n e r, B. A n s о n,
K. F о 1 к e r s. J. Am. Chem. Soc.,
81, 4999, 5000 (1959).
108. H. M a у e r, J. Me t zger,
O. Isler. Helv. Chim. Acta, 50,
1376 (1967).
109. F. C r a n e, R. Lester,
C. W i d m er, Y. H a t e f i.
Biochem. Biophys. Acta, 32, 73
(1959).
110. R. Lester, F. G r a n e,
Y. Hatefi. J. Am. Chem. Soc.,
80, 4751 (1958).
111. R. M о r t о n, U. G 1 о о r,
О. S c h i n d I e r, G. W i 1-
son, L. Chopar d-d i t-
J e a n, F. Hemming,
O. Isler, W. L e a t, J. Pen-
nock, R. Rflegg, U.
Schwieter, O.’Wiss. Helv.
Chim. Acta, 31, 2343 (1958).
112. M. T i s h 1 e r, L. F i e s e r,
N. W e n d 1 e r. J. Am. Chem.
Soc., 62, 1982 (1940).
113. L. C h e n, R. D a 1 1 a m. Na-
ture, 198, 386 (1963).
114. F. H e m m i n g, J. Pen-
n о c k, R. Morton. Biochem.
J., 68, 29P (1958).
115. J. N i 1 s s о n, C. D a v e s,
K. Folkers. Acta chem. scand.,
22, 200 (1968).
116. Cheng Kuo Hui. J. Vita-
minol. (Japan) 1, 8 (1954); C. A.,
50, 320 (1956).
117. Швейц, пат. 205896; С. A., 35,
2905 (1941).
118. F. Werder. Пат. США 2230659;
С. A., 35, 3270 (1941).
119. J. S u r m a t i s, J. W e b e r.
Пат. США 2723278; С. A., 50,
10794 (1956).
120. L Sm i th, H. U n g n a d e.
J. Org. Chem., 4, 298 (1939).
121. L. Smith. J. Am. Chem. Soc.,
61, 2615 (1939).
122. R. В e h n i s h. Канад. пат.
494731; РЖХ, 1955, 32802.
123. Швейц, пат. 205895; С. А., 35,
6067 (1941).
124. Герм. пат. 719438; С. А., 37, 4747
(1943).
125. Р. К а г г е г, К. D ц г г. Helv.
Chim. Acta, 32, 1361 (1949).
126. О. Ehrmann. Пат. ФРГ
1015446; РЖХ, 1959, 61916.
127. R. Р а 1 а п о v s к i, Е. Wa-
1 е d z i а к. Польск. пат. 45483;
РЖХ, 1962, 24Л343.
128. К. Nakagawa, S. М и г а-
к i. Яп. пат. 11993 (1963); С. А.,
59, 13953 (1963).
129. М. Е. М а у р и т, Г. В. С м и р-
н о в а, Э. А. П а р ф е н о в,
И. К- С а р ы ч е в а, Н. А. Пре-
ображенский. ДАН СССР,
140, 1330 (1961).
130. F. Werder. Пат. США 2230970,
С. А., 35 , 3270 (1941);
131. Швейц, пат. 208446; С. А., 35,
3774 (1941). .
132. Англ. пат. 867166; С. А., 55,
23564 (1961).
133. Т. Мо г о е, S. Н a 11 о г i.
A. Komatsu, Т. М a t s u i,
H. К u r i h a г а, пат. США
3459773; С. А., 71, 91307 (1969).
134. A. W е h г 1 i, R. F г у е г,
W. Metlesics. J. Org. Chem.,
36, 2910 (1971).
135. L. S m i t h, J. S p r u n g. J.
Am. Chem. Soc., 65, 1276 (1943).
136. L. В 1 a h a, J. H о d г о v a,
J. Weichet. Coll. Czechoslov.
Chem. Comm., 24, 2023 (1959).
137. Li Blaha,. J. W e i c h e t.
Чехосл. пат. 88904; С. A., 54,
.2362 (1960).
138. M. M a t s u i, S. Ki t am u-
r a. Agric. and Biol. Chem., 29,
978 (1965).
139. Яп. пат. 964 (1962); РЖХ, 1963,
6H213.
140. A. J а с о b, Al. Steiger,
A. Todd. J. Soc. Chem". Ind. 57,
1188 (1938).
141. О. H г о m о t к а. Герм. пат.
703957; С. A., 36, 1144 (1942).
142. О. Hromotka. Герм. пат.
741688; С. А., 39, 2768 (1945).
143. М. М a t s u i, S. К i t a m u г а,
Н. F и к a w а, Н. К u г i h а-
г а. Яп. пат. 2621—2624 (1962);
С. А., 58, 7911 (1963).
144. W. J о h n, Н. Р i n i. 2. phy-
290
siol. Chem., 273, 225 (1942).
145. W. J ohn. Chemie, 55, 98, 110
(1942).
146. L. S m i t h, H. U n g n a d e,
J. О p i e, W. Prichard,
R. Car I i n, E. К a i s e r.
J. Org. Chem., 4, 323 (1939).
147. H. M а у e r, P. Sc hu d e I,
R. R ii e g g, О. I s 1 e r. Chi-
mia, 16, 367 (1962).
;4148. H. Mayer, P. Schudel,
< R. R ii e g g, О. I s 1 e r. Helv.
Kv Chim. Acta, 46, 650 (1963).
^j49. P. S c h u d e 1, H. Mayer,
'•fe R. R й e g g, О. 1 s I e r. Chi-
mia, 16, 368 (1962).
150. T. I c h i к a w a, T. К a t o.
' - Bull. Chem. Soc. Japan, 41, 1224
(1968); яп. пат. 11065 (1967); С. A.,
67, 100001 (1967).
151. Швейц, пат. 205362; С. А., 35,
2533 (1941).
152. D. N е 1 а п. Пат. США 3344151;
С. А., 69, 10362 (1968).
153. А. А. Св ищу к, Е. А. Т и-
хомирова. Укр., хим. ж, 29,
1070 (1969).
154. L. W е i s 1 е г. Пат. США
2519863; С. А., 45, 669 (1951).
155. J. G г е е п, S. М а г с i п к i е-
w i с z. Англ. пат. 827391; РЖХ,
1961, 13Л469.
156. Англ. пат. 1000517; С. А., 63,
13219 (1965).
157. Р. Е g g i t t, F. H о r r i s.
J. Sci. Food. Agric., 7, 493 (1956).
158. L. W i e s I e г. Пат. США 2673858;
РЖХ, 1956, 26958.
159. J. Baxter. Канад, пат. 513700;
РЖХ, 1958, 26225.
160. Т. Nakamura, S. К i j i-
m a.. Chem. Pharm. Bull., 19, 2318
(1971).
161. F. S e v i g n e. Пат. США 2998430;
С. A., 56, 3462 (1962).
162. F. S e v i g n e. Пат. США 3187011;
С. A., 63, 8322 (1965).
163. H. P e n d s e, P. К a r r e r.
Helv. Chim. Acta, 41, 396 (1958).
164. S. Marcinkiewicz,
D. M с H a 11, P. M a m a I i s.
J. Green, J. Chem. Soc., 1959,
3377.
165. Англ. пат. 900217; С. A., 57,
13730 (1962).
166. J. Green, D. McHale,
P. M a m a 1 i s, S. M a r c i n-
kiewicr. J. Chem. Soc., 1959, 3374-
167. D. M с H a I e, P.Mamalis-
J. G r e e n, S. M a r c i n к i e-
wicz. J. Chem. Soc., 1958, 1600.
168. D. M с H a 1 e, P. M a m a 1 i s,
S. Marcinkiewicz,
J. Green. J. Chem. Soc., 1959,
3358.
169. W. J о h n, P. G й n t h e r,
M. Schmeil. Ber., 71, 2637
(1938).
170. А. А. Св ищу к, С. И. X a*
нин, M. M. Гольдберг,
E. Д. О в e p ч у к. Авт. свид.
СССР 137506; РЖХ, 1962, 4Л339.
171. П. И. К а л у г и н, М. Ф. Ш а-
х о в а. Авт. свид. СССР 136855;
Бюлл. изобрет., 1961, № 6, 42-
172. Т. А к a g i. Яп. пат. 6121 (I960)!
РЖХ, 1961, 15Л415.
173. О. И. Великославинс-
к а я. Авт. свид. СССР 160801;
С. А., 61, 2912 (1964).
174. А. А. С в и щ у к, Б. Г. Са-
ви н о в. Укр. хим. ж., 22, 518
(1956).
175. Р. К а г г е г, В. R i п g 1 е г.
Helv: Chim. Acta, 22, 610 (1939).
176. R. Lukes, A . Zobacova.
Chem. listy, 51, 330 (1957); Coll.
Czechoslov. Chem. Comm., 22, 1649
(1957).
177. Англ. пат. 633711; С. A., 44, 4930
(1950)
178. И. H. H а з a p о в, Б. П. Г у-
с е в, В. И. Г у н а р. Изв. АН
СССР, ОХН, 1957, 1267; ЖОХ, 28,
1444 (1958).
179. И. К. С а р ы ч е в а, Г. А. Во-
роб ь е в а, Н. А. К У з н е-
ц о в а, Н. А. П р е о б р а-
женский. ЖОХ, 28, 647 (1958).
180. W. Fruhstorfer, К. Schulte.
Пат. ФРГ 1013275; С. А., 54, 1596
(1960).
181. Н. Lindlar. Helv. Chim. Acta,
35, 446 (1952). .
182. К. S a t о, S h. M i z u n o,
M. Hi г a у a m a. J. Org. Chem.,
32, 177 (1967).
183. M. E. M а у p и т, Г. В. С м и p-
и о в а, Э. А. Парфенов,’
T. M. В и н к о в с к а я, Н. А.
Преображенский. ЖОХ,
32, 2483 (1962). .
184. И. К. С а р ы ч е в а, Ю. Г.
М о л о т к о в с к н й, Г. А.
Воробьева, Н. А. П р е-
о б р а ж е некий. ЖОХ, 29,
1123 (1959).
185. J. Burrell, R. Garwood,
L. J а с k m a n, E. О s k а у,
В. Weed о n. J. Chem. Soc.
<C», 1966, 2144.
186. N. Beredjick, C. Schu-
e r c h. J. Org. Chem., 22, 469
(1957).
187. K. Sa to, Y. К u r i h a r a,
S h. A b e. J. Org. Chem., 28, 45
(1963).
188. K. Sa t o, Y. 1 s h i b i k i,
S h. Abe. Яп. пат. 16868 (1963);
С. A., 60, 572 (1964).
189. J. W e i c h e t, J. H о d r o-
v a, V. К v i t a. Chem. listy,
51, 568 (1957).
190. W. С a 1 d w e 1 1, T. T h о m p-
s о n. J. Am. Chem. Soc., 61, 765
(1939).
191. L. S m i th, J. К i n g. J. Am.
Chem. Soc., 63, 1887 (1941).
192. W. Burke, J. Warburton,
J. Bishop. J. Bi 1 is. J. Org.
Chem., 26, 4669 (1961).
193. И. К. С a p ы ч e в а, Г. A. Ce-
10*
291
ребренннкова, Л. И. Мит-
ру ш к и и а, Н. А. П р е о б-
раже некий. ЖОХ, 31, 2190
(1961).
194. М. Я. К р а ф т, А. М. Ц ыг а-
н о в а. Авт. свид. СССР 122152;
Бюлл. изобрет., 1959, N 17, 16;
Мед. пром. СССР, 1960, № 10, 27.
195. Е. Bamberger. Вег., 33, 3600
(1900); 59, 418 (1926).
196. Е. Bamberger, A. Ri-
sing. Вег., 33, 3636 (1900).
197. К. S a t о, V. F i j i m a,
A. Y ama de. Bull. Chem. Soc.
Japan, 41, 442 (1968).
198. H. H о e 1 t i n g, T. В a u-
m a n n. Ber., 18, 1152 (1885).
199. R. Jaworska, W. L e wen-
stein, B. Zyczynska.
Польск. пат. 37288; РЖХ, 1958,
29775.
200. P. В i t e, P. T u z s о n. Ma-
gyar Kem. folyoirat., 64, 396 (1958);
C. A., 54, 16416 (1960).
201. G. Leuschner, K. Pford-
t e. J. prakt. chem., 10, 102 (1960).
202. А. А. С в и щ у к, Н. М а х-
новский, Р. Ковтун.
Авт. свид. СССР 158873; РЖХ,
1965, 1Н55.
203. L. Smith. J. Am. Chem. Soc.,
56, 473 (1934).
204. F. В e i 1 s t e i n, А. К 6 g 1 e r.
Lieb. Ann., 137, 317 (1866).
205. M. H u e n d e r. Rec. trav. chim.,
34, 1 (1915).
206. R. M e у e r, W. M e у e r. Ber.,
51, 1571 (1918).
207. А. А. С в и щ у к, Ф. Л. Г р и н-
б е р г, Е. Д. Б а с а л к е-
в и ч, Е. Д. О в е р ч у к. Укр.
хим. ж., 29, 411 (1963).
208. А. Р о n g г a t z, К. Z i г m.
Monatsh., 83, 13 (1952).
209. R. F i t t i g. Lieb Ann., 147, 11
(1868).
210. W. К e 1 b e, K. Pa t he. Ber.,
19, 1548 (1886).
211. L. S m i t h, K. J о h n s о n.
J. Am. Chem. Soc., 59, 673 (1937).
212. Ф. Л. Г p и н б e p г, А. А. С в и-
щу к. Укр. хим. ж., 23, 79
(1957).
212а. Fr. Demande 2003508; С. А., 72,
100274 (1970).
213. F. V a s i n g t о n, S. Rei-
ch а г d, A. N a s о n. Vitamins
and Hormones, 18, 43 (1960).
214. Н. Mayer, Р. Schude 1,;<
R. R й е g g, О. 1 s 1 e r. Helv. '
Chim. Acta, 47, 229 (1964).
215. Ю. А. Арбузов. Успехи хи-
мии, 8, 11 (1939).
216. W. L u n d b e r g, R. В a r-
n e s, M. С 1 a u s e n, G. В u r r.
J. Biol. Chem., 153, 265 (1944).
- 217. O. F i t z h u g h, A. H e 1 s о n,
H. С a 1 v e r y. Proc. Soc. Exp.
Biol. Med., 56, 129 (1944).
218. A. M о s s, J. D r u m m о n d.
Biochem. J., 32, 1953 (1938).
219. H. О 1 с о t t, A. E m e r s о n.
J. Am. Chem. Soc., 59, 1008 (1937).
220. V. D e m о 1 e. Intern. Z. Vita-
minforsch., 8, 338 (1939).
221. P. H a r r i s, M. L u d w i g.
J. Biol. Chem., 179, 1111; 180, 611
(1949).
222. E. Hum e. Nature, 148, 478
(1941).
223. P. К a r r e r, A. G e i g e r.
Helv. Chim. Acta, 23, 455 (1940).
224. P. Karrer, O. Hoffmann.
Helv. Chim. Acta, 22, 654 (1939).
225. P. Karrer, O. Hoffmann.
Helv. Chim. Acta, 23, 1126 (1940).
226. F. W e r d e r, T. Moll,
F. J u n g. Z. Physiol. Chem., 257,
129 (1939).
227. P. К a г г e г, A. К u g 1 e r.
Helv. Chim. Acta, 28, 436 (1945).
228. P. К a r r e r, R. Leg 1 er,
G. Schwab. Helv. Chim. Acta,
23, 1132 (1940).
229. P. К a r r e r, K. J e n s e n.
Helv. Chim. Acta, 21, 1622 (1938).
230. P. К a г г e r, K. Y a p. Helv.
Chim. Acta, 23, 581 (1940).
231. J. We i c h e t, J. H о d r o-
v a. Chem. listy, 51, 595 (1957).
232. U. Schwieter, R. Tamm,
H. W e i s e r, O. Wi ss. Helv.
Chim. Acta, 49, 2297 (1966).
233. J. We i c he t, L. В 1 a h a,
В. К a k a c. Coll. Czechoslov.
Chem. Comm., 31, 4598 (1966).
234. J. W e i c h e t, L. В 1 a h a,
В. К a k a 6, J. H о d г о v a.
Coll. Czechoclov., Chem. Comm.,
31, 2434 (1966).
235. P. Karrer, M. Favarger,
A. M e r z, G. M i 1 h a n d.
Helv. Chim. Acta, 31, 1505 (1948).
236. Ц. E v a n s, О. E m e r s о n,
G. E m e r s о n, L. S m i t h,
. H. U n g n a d e, W. P ri-
ch a r d, F. A u s t i n, H. H o-
e h n, J. О p i e, S. Wawzo-
n e k. J. Org. Chem., 4, 376 (1939).
237. E. F e r n h о I z, J. F i n k el-
stein. J. Am. Chem. Soc., 60,
2402 (1938).
238. P. В о у e r, M. R a b i no-
wit z, E. L i e b e. J. Biol.
Chem., 192, 95 (1951).
239. D. Wooley. Science, 100, 749
(1944); J. Biol. Chem., 159, 59
(1945).
240. L. Smith. Chem. Revs., 27, 287
(1940).
292
ГЛАВА VII
ВИТАМИНЫ И КОФЕРМЕНТЫ — ПРОИЗВОДНЫЕ
ПИРИДИНКАРБОНОВЫХ КИСЛОТ
НИКОТИНАМИД И НИКОТИНОВАЯ КИСЛОТА
Никотиновая кислота (ниацин, витамин РР) (I), являющаяся ₽-пири-
динкарбоновой кислотой, и никотинамид (II)
были известны еще в прошлом столетии, но к витаминам они были отнесе-
ны только в 1935—1937 гг. [1—4].
Собственно противопеллагрическим витамином является никотинамид
[41, входящий в виде простетической группы в ферментные системы; нико-
тиновую кислоту следует рассматривать как провитамин никотинамида.
Никотиновая кислота превращается в никотинамид в процессе обмена
веществ в организме. !
Никотиновая кислота (I) представляет собой бесцветные
иглы (из воды и спирта) с т. пл. 235,5—236,5° С [5]. Она способна субли-
мироваться. Спектр поглощения имеет максимум при 261,5 нм [61;
е 3,1 - 103 при pH 5,6 и 4,6 103 при pH 3,85. Никотиновая кислота,
умеренно растворима в воде — в 100 мл при 0°С — 86 г, при 15° С — 1,30 г;
38° С — 2,47 г, 61° С — 4,06 г, 78° С — 6,00 г, 100° С — 9,76 г, в 96%.-ном
спирте в 100 мл при 0°С растворяется 0,57 г, при 15° С — 0,92 г, 38°С—:
2,10 г, 61° С — 4,20 г, 78° С — 7,06 г [7]. Она малорастворима в обычных
органических растворителях. г
Константа диссоциации никотиновой кислоты Кв= 1,4-10-5 при 25° С.'
Изоэлектрическая точка находится при pH 4,23—4,25. В 1%-ном вод-
ном растворе никотиновая кислота имеет pH около 3.
Никотиновая кислота вследствие своего амфотерного характера обра-
зует с щелочами нормальные и с кислотами четвертичные соли. Соли нико-
тиновой кислоты легко получаются в кристаллическом виде как с кисло-
тами, так и с основаниями, причем для серебряной, медной и кальциевой
солей характерна очень трудная растворимость в воде.
Никотинат гидрохлорид — C6H5O2N • НС1 - бесцветные призмы с т.
пл. 274° С (с разл.) 18], хорошо растворимые в воде— в 100 мл при 0°С
растворяется 1,73 г, при 15° С — 8,50 г, 38° С — 14,23 г, 61° С — 17,01 г,
78° С — 19,50 г, 100° С —21,48 г [7].
Никотинат натрия — C6H4O2NNa — кристаллы, хорошо растворимые
в воде — в 100 мл при 0° С растворяется 9,5 г, при 15° С— 23,11 г,
30° С — 31, 16 г, 61° С — 39,11 г, 78° С — 43,20 г, 100° С— 49,78 г [7].
Никотинат меди — (CeH4O2N)2Cu — кристаллы синего цвета [9], ве-
щество практически нерастворимо в воде.
Никотинат нитрат — СбН5О2Ы-НМО3-Н2О— бесцветные листочки или
призмы с т. пл. 190—192° С [10].
Пикрат никотиновой кислоты — ромбические призмы с т. пл. 225—
227° С.
Н икотинамид (II) — бесцветные иглы с т. пл. 131—132° С [11];
он может кристаллизоваться в четырех полиморфных формах [12,13]. Нико-
тинамид сублимируется в вакууме при 150—160° С и 0,0005 мм [141. Спектр
поглощения никотинамида имеет Хманс 261,5 нм; е 2,85-10а при pH 5,72,
293
3,70* 103 при pH 3,48 и 4,55-103 при pH 2,75 [151. Водные растворы нико-
тинамида нейтральны на лакмус. Он растворим в воде (1:1) и экстраги-
руется из водных растворов хлороформом, этилацетатом и бутиловым
спиртом [16]; растворим в спирте (1 : 1,5), ацетоне, этиленгликоле, глице-
рине (1 : 10), хлороформе и многих других органических растворителях;
практически нерастворим в эфире и бензоле. Никотинамид в водном ра-
створе при pH 6 обладает светло-желтой флуоресценцией в ультрафиоле-
товом свете [171.
Гидрохлорид никотинамида имеет т. пл. 227—228° С, пикрат нико-d
тинамида — т. пл. 193° С.
ХИМИЧЕСКИЕ СВОЙСТВА НИКОГИНОВОЙ КИСЛОТЫ И НИКОТИНАМИДА
Для пиридинмонокарбоновых кислот известны три изомерные кислоты:
пиколиновая (а-) (III), никотиновая (0-) (I) и изоникотиновая (7-пиридин-
карбоновая) (IV)
СООН
Различаются они по своим физическим и химическим свойствам. Так,
растворимость изомерных кислот в воде понижается от пиколиновой к ни-
котиновой и далее к изоникотиновой кислоте, растворимость которой в
100 раз меньше, чем у никотиновой.
Пиридинмонокарбоновые кислоты при повышенной температуре де- ’
карбоксилируются с образованием пиридина; никотиновая кислота де-
карбоксилируется наиболее трудно (260° С) по сравнению с изоникотино-
вой и особенно с пиколиновой кислотой. В устойчивости карбоксильных
групп наблюдается следующая зависимость от изомерии их положения:
Р >7 >а. Эта зависимость распространяется и на пиридинди- и пири-
динтрикарбоновые кислоты. При декарбоксилировании а, 6, 7-пиридин-
трикарбоновой кислоты (V) первоначально отщепляется а-карбоксильная
группа с образованием цинхомероновой кислоты (VI), затем при более по-
вышенной температуре — 7-карбоксильная группа с образованием нико-
тиновой кислоты (I). Хинолиновая кислота (VII) также декарбоксили-
руется с отщеплением а-карбоксилыюй группы:
Ацетильная группа, замещающая ^-положение молекулы пиридина, в
отличие от заместителей в а- и у-положениях обладает характером алифа-
тических групп, замещающих атомы водорода ароматических соединений.
Водородные атомы метильных групп в а- или у-положении пиридинового
ядра обладают повышенной подвижностью и сравнительно легко реаги-
руют с альдегидами, нитрозо-соединениями и др.
Как метильные группы, так и атомы галогена в а- и у-положениях пи-
ридинового ядра значительно легче вступают в обменные реакции, напри-
294
мер с аминами, чем те же заместители в 0-положении. Неравноценность'
а-, 7- и 0-положений пиридинового ядра характеризуется также разли-
чием аминопиридинов по своим реакциям. 0-Аминопи ридин подобен арома-.
тическим аминам, в то время как а- и 7-аминопиридины отличаются как
от ароматических аминов, так и от алифатических аминосоединений.
По химическим свойствам пиридинмонокарбоновые кислоты во многом
подобны ароматическим кислотам. Они образуют ангидриды, хлорангид-
риды, амиды, эфиры, нитрилы и Др. Однако этерификация никотиновой
кислоты протекает несколько труднее, чем ароматических кислот; при
получении сложных эфиров хлорангидрид дает лучшие результаты. Эфиры
никотиновой кислоты хорошо очищаются перегонкой. Метиловый эфир
никотиновой кислоты имеет т. пл. 37—38° С; этиловый эфир — жидкость
с т. кип. 88° С при 3,5 мм, Лд 1,5008; в 100 мл воды при 25° С растворяется
5,6 г этилового эфира никотиновой кислоты.
Из никотинамида (II) при действии пятиокиси фосфора или тионил-
хлорида при 100° С образуется 3-цианпиридин, никотинонитрил (VIII).
При .действии гипохлорита натрия в щелочной среде никотинамид (II)
подвергается гоффмановской перегруппировке, в результате которой полу-
чается 3-аминопиридин (IX) [18]:
Изучен гидролиз никотинамида в солянокислых растворах [19].
Известно несколько природных производных никотиновой кислоты
и никотинамида. Важнейшие из них — никотинамидадениндинуклеотид
(НАД) и никотинамидадениндинуклеотидфосфат (НАДФ) — коферменты,
соединения, образованные из никотинамида, Д-рибозы, аденина и фосфор-
ной кислоты (см. с. 309), участвующие в переносе водорода в многочис-
ленных окислительно-восстановительных реакциях обмена веществ.
Продукт конденсации глицина и никотиновой кислоты — никотино-
илглицин, никотинуровая кислота (X), с т. пл. 240—242° С (пикрат имеет
т. пл. 160—162° С) [20] — является одним из продуктов расщепления ни-
котинамидных коферментов, в виде которого наряду с другими соедине-
ниями, образующимися в результате метаболизма, они выводятся из ор-
ганизма. Никотинуровая кислота обнаружена в моче [201.
2,5-Диникотиноилорнитин (XI) имеет т. пл. 262—263° С, также яв-
ляется продуктом обмена веществ и выделяется из организма [21].
CONHCHjCOOH
X
Для никотиновой кислоты и никотинамида характерна способность к
образованию четвертичных аммониевых солей [22, 23]. Никотиновая ки-
слота образует, подобно ^-аминокислотам, бетаины, например тригонеллин
(XII), который может быть получен при действии на никотиновую кислоту
йодистым метилом в щелочном растворе. Тригонеллин выделяется с мочой
[20]; кроме того, он широко распространен в растительном мире. Фармако-
логическим действием это соединение, по-видимому, не обладает.
При действии галогеналкилов никотинамид образует четвертичные соли,
например М-метил-3-карбоксамидпиридинийхлорид(ХШ)ст. пл. 240° С [22];
это соединение является одним из продуктов метаболизма и выделено из
295
мочи [24]. Метилирование никотинамида в процессах метаболизма осу-
ществляется в результате переноса метильной группы метионина от мета-
болита-переносчика (косубстрата) S-аденозилметионина при каталитичес-
ком участии никотинамидметилтрансферазы. Еще один из продуктов рас-
щепления— 6-оксо-Ы-метилникотинамнд (XIV) с т. пл. 212—214° С —
также выделен из мочи [25].
Четвертичные соли никотиновой кислоты и никотинамида, например
тригонеллин (XII) и галогенметилат никотинамида, легко подвергаются
частичному восстановлению (например, гидросульфитом натрия) с обра-
зованием 1,2 (или 1,4)-дигидропиридинпроизводных [22, 26]. Эта реакция
обратима. Восстановительно-окислительные свойства четвертичных солей
никотинамида лежат в основе механизма ферментативного действия нико-
тинамидных коферментов. Следует отметить, что сам никотинамид (II) спо-
собностью к обратимому частичному восстановлению не обладает.
Многие производные никотиновой кислоты и ее амида обладают цен-
ными физиологическими свойствами. Так, кордиамин (XV) представляет
собой препарат, стимулирующий сердечную деятельность и дыхательные
центры; изопропилату никотиновой кислоты (XVI) свойственны анальге-
тические и местноанестезирующие свойства [27]. Никотиновый эфир холи-
на (XVII) обладает в некоторой степени свойствами холина [28].
XV
XVI
XVII
CCX3CHsCH4N (СНв)з-С1-
I
Интересно отметить соединения никотиновой кислоты с другими вита-
минами.
• Из гидразида никотиновой кислоты и пиридоксаля получен никотинил-
гидразон пиридоксаля (XVIII), обладающий некоторой противотуберку-
лезной активностью in vitro и проявляющий активность против рака мо-
лочной железы у мышей [29]. Никотинамид с L-аскорбиновой кислотой
при кипячении в среде ацетона образует аддукт с т. пл. 152—153° С воз-
можного строения (XIX) [30, 31].
Хлорангидрид никотиновой кислоты с пантотенолом образует пантоте-
нилникотинат (XX) [32].
296
XIX
hoch2c(ch^sch(oh)co
^^COOCHjCHjNH
Sr
XX
Для анализа никотиновой кислоты известны методы осаждения (по не-
растворимой медной соли) или колориметрические методы, основанные на
образовании окрашенных растворов с цианистым калием и хлорамином
[33] или роданистым калием, бромом и анилином [341; в последнем случае
предварительно получается роданистый бромид (BrCNS). В этих реакци-
ях происходит расщепление а-незамещенного пиридинового цикла с об-
разованием анилида замещенного глутаконового альдегида типа
СН
СН С—СООН
11 1
QHsNHCH CH=NCSH&
представляющего собой полиметиновый краситель.
СТРОЕНИЕ НИКОТИНОВОЙ КИСЛОТЫ
Никотиновая кислота является пиридинкарбоновой-3 кислотой [35].
Наличие в ее молекуле пиридинового ядра установлено реакцией декарбок-
силирования с гидроокисью кальция, при которой образуется незамещенный
пиридин; это основание дает четвертичные соли с кислотами и с галогено-
алкилами. Присутствие карбоксильной группы подтверждается реакцией
этерификации со спиртами и образованием солей с щелочными металлами.
0-Положение карбоксильной группы было установлено путем окис-
ления марганцовокислым калием синтетического 3-фенилпиридина (XXIII)
в 0-пиридинкарбоновую кислоту (I) [36]); соединение XXIII получают
из 0-нафтиламина (XXI), который по реакции Скраупа превращают в 0-
нафтохинолин (XXII), затем в 0-фенилпиридинкарбоновую кислоту,
подвергаемую последующему декарбоксилированию в 3-фенилпиридин
(XXIII):
По своей структуре пиридиновое кольцо никотиновой кислоты плос-
кое, а все атомы карбоксильной группы выведены из плоскости кольца;
молекулы никотиновой кислоты объединены водородными связями
—О—H...N в зигзагообразные цепи. У никотинамида амидная группа
лежит в плоскости кольца [37].
СИНТЕЗ НИКОТИНОВОЙ КИСЛОТЫ И НИКОТИНАМИДА
Никотиновая кислота' была выделена из дрожжей, рисовых отрубей
и риса еще в 1911—1912 гг. [38, 39], но только в 1934—1935 гг. установлена
ее связь с коферментами [1—3]. Однако впервые никотиновую кислоту полу-
297
чил еще в 1867 г. Губер [35] окислением никотина и определил ее строение
как пиридинмонокарбоновой кислоты. Окислением гомологов пиридина
(p-этилпиридина) никотиновая кислота получена Вышнеградским в 1879 г.
[40]. Никотинамид впервые синтезировал в 1894 г. Энглер [41].
Витаминные свойства никотиновой кислоты и никотинамида были уста-
новлены только в 1937 г. 141. Никотинамид был выделен из печени [4], из
сердечной мышцы [14], из коферментов — никотинамидадениндинуклеотид-
фосфата, коэнзима II [42, 431, и никотинамидадениндинуклеотида, коэн-
зима I [3]. Никотинамид оказался активным как ростовой фактор для мик-
роорганизмов [44].
Выделение никотиновой кислоты или никотинамида из дрожжей,
печени и других природных источников или продуктов микробиологичес-
кого синтеза не представляет никакого практического интереса. Для
получения этих соединений имеют значение только синтетические методы..
Существует несколько основных методов синтеза никотиновой кислоты.
1. Окисление алкалоидов никотина и анабазина.
2. Превращение различных производных пиридина с 0-замещающими
функциональными группами, например цианпиридина.
3. Окисление различных (3-алкилзамещенных пиридина.
4. Окисление хинолина и его производных.
Важное практическое значение имеют методы 2, 3 и 4.
1. Синтез никотиновой кислоты из никотина и анабазина
Природным источником сырья для получения никотиновой кислоты
служат алкалоиды — никотин и анабазин. Никотин — ^-(N-метил-а-
пирролидил)пиридин (XXIV) (т. кип. 246° С, d23 1,0099), главный алка-
лоид растения Nicotiana tobacum,— является отходом производства та-
бака. Анабазин, (а-пиперидил)пиридин (XXV) — т. кип. 276° С, d2°
1,0455, открытый Ореховым [45, 46] в среднеазиатском растении Anabasis
aphylla, вырабатывается как ядохимикат для сельскохозяйственных вре-
дителей.
Пиридиновое ядро молекулы алкалоидов значительно более устойчиво,
и при окислении преимущественно расщепляется пирролидиновое и пипе-
ридиновое ядро с образованием никотиновой кислоты. Марганцовокислый
калий в щелочной среде при температуре 70° С является хорошим окисли-
телем никотина (XXIV) [47, 48] и анабазина (XXV) [46, 491; никотиновую
кислоту получают с выходом 80% [48]. Двуокись марганца в 35%-ной
серной кислоте окисляет никотин [48] и анабазин [49]; в последнем случае
с выходом 45%.
Кроме того, в качестве окислителей никотина применяют хромовую
кислоту [35, 49] или азотную кислоту [50—521. С азотной кислотой реакцию
проводят при температуре кипения и получают никотиновую кислоту с
выходом 63—74% [52], причем расход 70%-ной азотной кислоты (d2<
1,42) достигает 20 частей, а 27%-ной — 40 частей на 1 часть никотина
[481; трехкратного теоретического количества азотной кислоты для про-
ведения реакции явно недостаточно [50]. Анабазин также успешно окис-
ляют в никотиновую кислоту азотной кислотой [46, 53—55].
Метод каталитического окисления никотина [56] и анабазина [57—
59] серной кислотой в присутствии металлического селена при температуре
298
около 300° С может применяться для получения никотиновой кислоты с
выходом до 77%.
Описано электрохимическое окисление никотина в никотиновую кис-
лоту в щелочной среде на никелевом аноде с применением в качестве пере-
носчика кислорода марганцовокислого калия; выход при 80° С достигает
82% (601, на платиновом аноде в кислой среде выход составляет только
15% 161]. При окислении анабазина на вращающемся платиновом аноде
в диафрагменном электролизе в кислой среде никотиновая кислота полу-
чена с выходом 69% (62].
2. Синтез никотиновой кислоты из пиридина
Один из важнейших продуктов коксования каменного угля — пиридин —
’является избыточным продуктом коксобензольной промышленности, и
методы его использования для синтеза никотиновой кислоты через 0-циан-
пиридин (никотинонитрил) представляют значительный интерес (схема 72).
По одному из вариантов 0-цианпиридин (XXVIII) получают путем
сульфирования пиридина (XXVI) олеумом при 230° С при каталитическом
участии сернокислой ртути и сплавления натриевой соли образовавшейся
0-пиридинсульфокислоты (XXVII) с цианистым калием или натрием при
350—400° С (выход 70—80%) (63]. Более просто проводить синтез 0-циан-
пиридина (XXVIII) бромированием пиридина при 300° С в 0-бромпири-
дин (XXIX) с выходом 38% с последующим замещением галогена циан-,
группой при действии цианистой меди — выход 50% (64]. Парофазный ме-
тод каталитического окисления никотина при 400° С также приводит к
получению 0-цианпиридина с выходом 40% [65, 66]; образования никоти-
новой кислоты в этой реакции не наблюдается.
0-Цианпиридин (XXVIII) в результате гидролиза с раствором едкого
натра превращается в никотиновую кислоту (I) с выходом 90% [67]; гид-
ролиз может быть осуществлен или с водным аммиаком при 260° (68] или
в присутствии ионообменной смолы АВ-17 при 60° с выходом 85% [69].
0-Цианпиридин может, быть непосредственно использован для синтеза
никотинамида (II).
Схема 72
Синтез никотиновой кислоты из пиридина
Никотиновую кислоту с меченым атомом углерода получают из 0-бром-
пиридина (XXIX) с я-бутиллитием и последующим карбоксилированием
изотопной углекислотой с общим выходом 60—80% [701.
3. Синтез никотиновой кислоты из р-алкилзамещенных пиридина
В качестве 0-ал кил замещенных пиридина практическое значение имеют
0-пиколин (XXX) и а-метил-0'-этилпиридин (2-метил-5-этилпиридин)
(XXXI).
299
р-Пиколин (т. кип. 143° С) содержится в летучих продуктах коксования
каменного угля наряду с близкокипящими трудно отделимыми пиридино-
выми основаниями: f-пиколином (т. кип. 144° С) и а, а'-лутидином (т.
кип. 142° С). Применение фракционной перегонки легких пиридиновых ос-
нований каменноугольного дегтя не дает возможности получить Р-пико-
лин в чистом виде. Однако методом распределительного разделения ком-
понентов легких пиридиновых оснований, освобожденных от пиридина
(т. кип. 115° С), между органическими растворителями и водой можно полу- -
чить Р-пиколин с 95%-ным содержанием [711; дальнейшая его очистка orf
примесей в случае надобности производится фракционным вымораживанием.
Так как коэффициенты распределения Р-пиколина, f-пиколина и а,
с'-лутидина между органическим растворителем и водой уменьшаются в
той же последовательности, как и растворимость воды в тех же раствори-
телях, то для отделения а, а'-лутидина от изомерных пиколинов приме-
няют н-гексан и воду. Из водного экстракта пиколины отгоняют, смеши-
вают с бензолом и отделяют f-пиколин от Р-пиколина, распределяя их
между водным раствором одноосновного фосфорнокислого натрия и бен-
золом [71J.
Химические методы выделения Р-пиколина основаны на образовании
различных производных, которые дают Р-пиколин или преимуществен-
но сопутствующие ему пиридиновые основания. Для отделения а, а'-лу-
тидина в виде кристаллического фталата [72] применяют конденсацию с
фталевым ангидридом [73—75] или с фталевым и уксусным ангидридом [76],
Р- и f-пиколины отделяют от а, а'-лутидина также в виде нерастворимых в
воде комплексов с хлористым цинком, которые затем разлагают щелочью
с выделением смеси свободных пиколинов [77] и др. .
Для отделения Р-пиколина от f-пиколина используют повышенную
подвижность атомов водорода f-метильной группы, для чего применяют
конденсацию f-пиколина с избытком формальдегида при нагревании в авто-
клаве [74] — при этом образуется смесь f-моно-, f-ди- и f-триме-
тилолпиколинов [78], f-NC5H4=CH2CH2OH, f-NCsH4-CH(CH2OH)2;
f-NC5H4-C(CH2OH)3, из которой свободный Р-пиколин легко отделяют от-
гонкой с паром [77]. Известны также методы фракционного осаждения гид-
рохлоридов — компонентов легких пиридиновых оснований [79], методы
конденсации с муравьиной [80] и бензойной [81] кислотами, с мочевиной
[82], полимеризации с серой [73], осаждения Р-пиколина в виде соли с од-
нохлористой медью [83, 84] и др.
Содержание Р-пиколина в легких пиридиновых основаниях можно
повысить до 35% и больше в результате каталитического (над пятиокисью
ванадия с трехокисью молибдена и кобальта) парофазного окисления его
спутников, которые окисляются до двуокиси углерода легче (при 350° С),
чем Р-пиколин [74, 85]. Однако в настоящее время в связи с применением
f-пиколина для синтеза производных изоникоршилгидразонов, активных
противотуберкулезных препаратов (фтивазид и др.) [86], практическое зна-
чение могут иметь только такие методы, которые дают возможность пол-
ностью использовать изомерные пиколины. Из отходов пиридиновых осно-
ваний от производства фтивазида, содержащих Р-пиколин и а, а'-лути-
дин (после выделения f-пиколина), р-пиколан получают с выходом 40%
через комплекс с хлористым цинком и используют для синтеза никотино-
вой кислоты [87].
Можно применять для окисления смесь пиридиновых оснований (Р-
и f-пиколины) и разделять конечные продукты окисления, выделяя нико-
тиновую и изоникотиновую кислоты [88], используя различную раствори-
мость их гидрохлоридов; никотиновую кислоту осаждают при pH 3,2—
4,0, изоникотиновую — при pH 1,5—2,2 [89].
P-Пиколин получают также синтетическим путем высокотемпературной
конденсацией акролеина и аммиака в присутствии фосфорнокислого алю-
миния с высоким выходом [90],
300
XXX
или из акролеина, пропионового альдегида и аммиака при 350—450°С
на катализаторе селикагель — окись алюминия [91] или из формалина
(параформа), ацетальдегида и аммиака при 425° С на катализаторе окиси
алюминия, силикагеле или их смеси с выходом около 60% [921; реакция
сопровождается одновременным образованием пиридина.
Для окисления Р-пиколина (XXX) в никотиновую кислоту (I) известно
применение различных реагентов. Его можно окислять в щелочных, ней-
тральных или кислых растворах. При окислении Р-пиколина марганцово-
кислым калием в щелочной среде выход никотиновой кислоты составля-
ет 77% [65, 87, 93]. Реакцию проводят при 70°С с постепенным прибав-
лением кристаллического марганцовокислого калия в водную реакцион-
ную смесь [93].
Так как пиридиновая молекула недостаточно устойчива к окислителям,
особенно в щелочной среде, то температуру реакции не поднимают выше
80° С [87]; для повышения выхода рекомендуется пропускать в реакцион-
ную смесь ток углекислоты [94].
1) 2КМпО4 или
3 [О] 2) 3H2SO4,
3) 2HNO3
Выделяется:
1) 2МпО2,2КОН
2) 3SO2, 4НгО
3) 2NO; 2Н2О
Метод окисления Р-пиколина марганцовокислым калием технологичес-
ки очень удобен, однако экономически мало эффективен.
Применение для окисления Р-пиколина в никотиновую кислоту ман-
ганата калия приводит к сильному снижению выхода —до 40%.
Суспензия двуокиси марганца в концентрированной серной кислоте
окисляет Р-пиколин при 140° С [95]: никотиновую кислоту выделяют в
виде медной соли [87, 95], которую разлагают кипячением с раствором
едкого натра — общий выход составляет 53—60% [87].
В качестве окислителя Р-пиколина можно применять хлорную кислоту
в присутствии селена и серной кислоты [96].
Метод жидкофазного окисления р-пиколина серной кислотой при
участии селена как катализатора осуществляют при 300° С [56]; реакция
сопровождается выделением сернистого ангидрида и воды.
При применении в качестве окислителя Р-пиколина двухромовокислого
натрия в нейтральном или щелочном растворе реакцию проводят при
200—250° С под давлением; выход никотиновой кислоты 83% [97, 98].
Если эту реакцию проводить в присутствии борной кислоты при 450° С
под давлением, то выход достигает 92% [99].
Окисление Р-пиколина азотной кислотой проводят при температуре
около 200° С [100, 101]; процесс сопровождается выделением окислов азота
и воды, а также отгонкой азотной кислоты, количество которой в реакции
необходимо все время восполнять. Если применить давление 15—20 ат
(1,5—2 МПа), то окисление 30%-ной азотной кислотой можно проводить
при более низкой температуре (130—150° С) [102]. В качестве материала
301
для аппаратуры оказалась пригодной нержавеющая сталь [101]; ингиби-
тором коррозии служит ортофосфорная кислота [100, 101]. Применение
катализаторов, таких, как серная кислота, соли ванадия, марганца [103]
и ртути [1021, позволяет несколько снизить температуру реакции. Выход
никотиновой кислоты достигает 60% и выше [102]. Этот метод связан с
трудностями технического оформления.
Были сделаны попытки применить в качестве окислителя |3-пиколина
хлор на свету в сернокислом растворе [90].
P-Пиколин способен окисляться в никотиновую кислоту электрохимичес-
ким путем в 30%-ной серной кислоте на свинцовом катоде с выходом 60%
[1041 Эта реакция изучена в зависимости от состава раствора и режима
электролиза; оптимальная температура окисления составляет 40° С при
плотности тока 5 А/дм2 [105, 1061.
Парофазное окисление ₽-пиколина кислородом над ванадиевым ката-
лизатором при 400° С [107] не дает хороших результатов, по-видимому, из-
за большой активности катализатора. При парофазном каталитическом
окислении кислородом воздуха выход никотиновой кислоты составляет
всего 17% [108]. Если эту реакцию проводить в присутствии аммиака, то
образуется нитрил никотиновой кислоты (XXVIII) с выходом свыше 60%
[108], гидролизуемый в никотиновую кислоту или ее амид (см. 307).
Окислительный аммонолиз ^-пиколина (XXX) кислородом воздуха
при 320—350° С на пятиокиси ванадия или У2О5/А12О3 дает выход до 82%
никотинонитрила (XXVIII) [109—112]. В качестве катализатора приме-
няют также ванадат олова [68, 113, 114] или смесь окисей титана и ванадия
[115].
XXX
XXVIII
Методы окисления ₽-пиколина азотной кислотой в никотиновую кисло-
ту и парофазное окисление воздухом через ее нитрил являются одними из
наиболее экономически выгодных.
Значительный интерес представляют методы, восполняющие дефицит-
ный Р-пиколин синтетическими алкилзамещенными пиридинами. Так, раз-
работаны высокоэффективные методы получения никотиновой кислоты из
альдегид-коллидина (2-метил-5-этилпиридина). Во многих странах рабо-
тают заводы по синтезу 2-метил-5-этилпиридина и дегидрирования его
в 2-метил-5-винилпиридин; последнее соединение имеет большое значение
для получения специальных видов синтетического каучука.
Для получения 2-метил-5-этилпиридина (XXXI) (т. кип. 178,3е С
при 160 мм, 66° С при 17 мм; растворимость в воде 1,22%) по методу Чичи-
бабина аммиак конденсируют с ацетиленом [116] в присутствии катализа-
торов-солей кобальта [117], кадмия, ртути и меди [118] или конденси-
руют с ацетальдегидом [119] в паровой фазе (в присутствии окиси алю-
миния).
При жидкофазной конденсации ацетальдегид первоначально полимери-
зуют в жидкий тример под влиянием кислых катализаторов, а затем после
нейтрализации конденсируют паральдегид с 25—30%-ным аммиаком в
присутствии уксуснокислого аммония как катализатора при 200—260° С и
55—210 ат (5,5—21 МПа); после фракционирования отделяют а-пиколин
(5—10%) и получают 2-метил-5-этилпиридин (XXXI) с выходом 70—80%
[120—124]
302
CHjCH CHCH,
CL /О
сн
+ сн3
сн
II
о
Нз
о
II
СН-СНз
3NH4OH
CH3COONH4
сн
сн сн
СН III
3 CHSCH * СН
мнэ
CHSCH
(А1»О3)
В качестве побочных продуктов, помимо а-пиколина, наблюдается об-
разование -у-пиколина, P-коллидина, 2-пропенил-5-этилпиридина и 2-ме-
тил-5-(2-бутенил)пириднна [ 122].
С выходом 73% 2-метил-5-этилпиридин (XXXI) получают, если в ка-
честве растворителя при конденсации 176 частей паральдегида с 420 час-
тями концентрированного аммиака и 60 частями уксусной кислоты приме-
няют 45 частей высококипящего основания (3-й фракции от перегонки по-
лученного продукта с т. кип. 50—180° С при 1—2 мм); реакцию проводят
в автоклаве при температуре 220° С и давлении около 50 ат (5 МПа) [125].
Уксуснокислого аммония как катализатора реакции прибавляют 0,2
моля на моль паральдегида [122]. Для реакции используют 50%-ный ам-
миак [126] и катализаторы — окислы цинка, хрома, железа, алюминия
[126], молибденат аммония [127], соли фторсодержащих кислот [128] и
др. Разработаны непрерывные синтезы 2-метил-5-этилпиридина [129, 130].
Для синтеза 2-метил-5-этилпиридина применяют также виниловые
эфиры, на которые действуют аммиаком при температуре 155—250° С
и давлении до 250 ат; реакция протекает под влиянием уксуснокислого
хлористого или фтористого аммония и однохлористой меди как катали-
заторов [131—133].
Окисление 2-метил-5-этилпиридина в пиридиндикарбоновую-2,5 (изо-
цинхомероновую) кислоту (XXXIII) осуществляют при помощи сус-
пензии марганцовокислого калия при 90—95° С (выход 76%) [134], а также
серной кислоты при каталитическом участии металлического селена при
температуре около 300° С [135].
Если для окисления применить водный раствор азотнокислой меди
при 210° С и давлении 60—250 ат (6—25 МПа), то пиридиндикарбоновую-2,5
кислоту можно получить с выходом 84—95% [134, 136, 137]. Одновремен-
ное применение в реакции окисления избытка кислорода дает возмож-
ность снизить необходимое количество азотнокислой меди в несколько раз
[138—140].
Применение для окисления 2-метил-5-этилпиридина азотной кислоты
дает хорошие результаты; 20—50%-ную азотную кислоту применяют од-
новременно с азотнокислой медью [141].
В окислении 2-метил-5-этилпиридина с успехом используют и одну азот-
ную кислоту в концентрации 10—30%. Процесс окисления осуществляют
под давлением в непрерывно действующей колонне с 20%-ной азотной
кислотой при 210°С и получают изоцинхомероновую кислоту (XXXIII)
с выходом 92%; при этом верхняя часть аппарата нагревается, а нижняя
охлаждается [141]. Реакция с 25%-ной азотной кислотой при 285° С и 80 ат
(8 МПа) с одновременным декарбоксилированием промежуточно образую-
щейся изоцинхомероновой кислоты приводит к получению никотиновой
кислоты (I) с выходом 64% [142]. При более низкой температуре (160—
180° С) и применении 10—30%-ной азотной кислоты выход изоцинхомеро-
новой кислоты достигает 70—80%; одновременно образуется более 8%
303
никотиновой кислоты [143, 144]. При той же температуре используют по-
вышенную концентрацию азотной кислоты — 40% 11451.
Реакция окисления 2-метил-5-этилпиридина (XXXI) азотной кислотой
или азотнокислой медью в пиридиндикарбоновую-2,5 кислоту (XXXIII)
протекает через промежуточное образование 2-метилпиридинкарбоновой-5
кислоты (XXXII) [134, 144]:
5HNO3,
У Выделяется. s
СН- N 2NO+l,5NsO+CO2+S5H,O LCH.
XXXI
соон
хххш
13
XXXII
Интересно отметить, что Р-пиколин окисляется азотной кислотой в 29
раз медленнее, чем 2-метил-5-этилпиридин [144].
Парофазное окисление 2-метил-5-этилпиридина кислородом при 180—
230° С под давлением 10—200 ат (1—20 МПа) на окиси свинца приводит К ;,’
получению изоцинхомероновой кислоты с выходом 79,5% [146].
Пиридиндикарбоновую-2,5 кислоту (ХХХШ) декарбоксилируют в ни->7*
котиновую кислоту (I) нагреванием в водной суспензии при 180—195° Cfe?
и давлении около 28 ат с выходом 95,6—98,5% [134] или в толуоле прик^ч
175—180° С [147], а также в минеральном масле при 200° С [148]. Для де-^й
карбоксилирования используют также диаммонийную соль пиридинди-
карбоновой-2,5 кислоты в воде при 225° С и 60 ат (6 МПа) [149]. Таким об-ij;
разом, общий выход никотиновой кислоты из паральдегида достигает^
70%. Этот метод синтеза высоко экономичен; он имеет важное техниче-^
ское значение. Sf'
Никотиновую кислоту можно выделить в чистом виде из растворов ГгУ
ее солей, загрязненных изоникотиновой кислотой, фракционным осажде-^?
нием соляной кислотой в изоэлектрической точке; первоначально вы->у
кристаллизовывается изоникотиновая кислота при pH 2,8—3,1, затем
никотиновая кислота при pH 4—4,5 [150].
4. Синтез никотиновой кислоты из хинолина
В гетероциклических конденсированных системах из пиридинового и
ароматических ядер бензольное кольцо менее устойчиво к окислителям,
чем гетероциклическое, вследствие чего такие системы расщепляются с
образованием пиридинполикарбоновых кислот. Так, например, хино-
лин (т. кип. 238,5° С) и 8-метилхинолин (т. кип. 247,75° С) образуют хи-
нолиновую кислоту (а, 6-пиридиндикарбоновую), изохинолин (т. кип.
243,25° С) — цинхомероновую кислоту (6, у-пиридиндикарбоновую), хи-
нальдин (2-метилхинолин) с т. кип. 247,5° С — а, а , ₽-пиридинтрикарбо-
новую кислоту. Различные заместители в бензольном кольце хинолина
делают его еще менее устойчивым к окислителям, и хотя, например, хро-
мовая кислота не действует на хинолин, его гомологи, замещенные в аро-
матическом кольце, окисляются ею в хинолиновую кислоту. Так как при
декарбоксилировании пиридинполикарбоновых кислот заместители в по-
ложениях а и у удаляются при значительно более низкой температуре, чем
Р-карбоксильная группа, то метод окисления конденсированных пири-
диновых соединений с последующим частичным декарбоксилированием
образовавшихся кислот широко применяется для синтеза никотиновой
кислоты. Важнейшим исходным продуктом в этом методе синтеза являет-
ся хинолин, содержащийся наряду с изохинолином, хинальдином, 8-ме-
тилхинолином и другими соединениями в тяжелых пиридиновых осно-
ваниях каменноугольного дегтя.
Никотиновую кислоту можно получить с выходом 25—35% непосред-
ственным окислением фракции тяжелых пиридиновых оснований с т. кип.
236—247° С серной кислотой в присутствии селена [57], однако значитель-
304
но рациональнее применять более обогащенные хинолином основания.
При обогащении хинолином тяжелых пиридиновых оснований паро-
фазным окислением кислородом воздуха над пятиокисью ванадия при
температуре 370° С преимущественно окисляются гомологи хинолина и
другие основания [1521; сам хинолин подвергается окислению только при
420—450° С с образованием углекислого аммония и муравьиной кислоты
[153]. Однако обогащенную хинолиновую фракцию можно получить и путем
тщательного фракционирования.
Хинолин (XXXIV) окисляют марганцовокислым калием в щелочном
растворе в хинолиновую кислоту (VII) свыходом 60—70% [151]. Перекись
водорода в сернокислой [154] или уксуснокислой среде [155] при ката-
литическом участии медных солей является хорошим окислителем хи-
нолина; хинолиновую кислоту (VII) выделяют в виде медной соли с выхо-
дом 65—70%.
XXXIV
Я Го] • ^2^4*
9l°J' ^H^C^Se
Выделяется ••
»)9NO, Н2О, 2СО2
s)9SO2,10H2O, 2СО2
VII
СООН
соон
Электрохимическим путем в среде 50%-ной [56, 105] или 75%-ной сер-
ной кислоты [104, 156] хинолин окисляется на аноде из платины или окиси
свинца при температуре около 70° С; пятиокись ванадия, двуокись селена,
трехокись хрома катализируют реакцию и увеличивают выход на 6—18%;
в оптимальных условиях выход хинолиновой кислоты достигает 77%
[104]. В качестве среды применяют водный раствор сернокислого аммония
и серной кислоты [157].
Декарбоксилирование хинолиновой кислоты (VII) в никотиновую ки-
слоту (1) проводят нагреванием при 180° С; выход составляет свыше 90%.
Если после монодекарбоксилирования продукт подвергать азеотропной
перегонке с инертными растворителями, такими, как тетрагидронафталин,
дифенил или дифениловый эфир при 250° С, то никотиновая кислота полу-
чается в чистом виде в бесцветных кристаллах [158].
Более рациональны методы, по которым хинолин (XXXIV) окисляется
и непосредственно декарбоксилируется в никотиновую кислоту (I), минуя
хинолиновую кислоту (VII). Окислителем хинолина может служить дву-
окись марганца в концентрированной серной кислоте при 165—175° С
[159]. При окислении 65%-ной азотной кислотой при 150—180° С выход
никотиновой кислоты составляет всего 30—35% [102]; однако с повыше-
нием температуры до 200—250° С и проведением реакции под давлением
он увеличивается [101]. Так, с 32%-ной азотной кислотой при 260° С
выход составляет 64%, а с использованием маточных растворов — до 86%
[160]. Осуществлено также окисление хинолина азотной кислотой с ее
постепенным прибавлением при кипячении (без давления) с катализатором
пятиокисью ванадия и последующим выдерживанием при 220—230° С
[161].
Сульфирование хинолина в ароматический цикл его молекулы (в по-
ложение 6) резко ослабляет устойчивость получаемого соединения по от-
ношению к окислительным агентам [162]. Это дает возможность после
предварительного сульфирования моногидратом серной кислоты при
240—270° С [17, 1631 или 65%-ным олеумом [164] провести окисление
хинолина 50—70%-ной азотной кислотой при 220° С или 86—90%-ной
азотной кислотой в присутствии катализаторов марганцовокислого калия
или двуокиси марганца [165].
Окисляя хинолин смесью азотной и серной кислот при 300° С [103,
166] с окисью ртути в качестве катализатора, никотиновую кислоту полу-
чают с выходом 88% [166]. Окислы азота (N2O4) в присутствии двуокиси
305
селена окисляют хинолин в среде серной кислоты при 200—300° С с вы-
ходом никотиновой кислоты около 89 % [167]; без катализатора выход до-
стигает только 63%. Катализатором может служить также молибденат
аммония [157].
Для окисления хинолина можно использовать в качестве окисли-
тельного агента непосредственно серную кислоту при участии металличес-
кого селена как катализатора. В этом методе Вудварда реакция проте-
кает при 300° С [56] или ниже [168, 169] и сопровождается выделением
сернистого и углекислого газов и отгонкой воды. Спедует отметить, что
в данной реакции предварительно происходит сульфирование хинолина.
В качестве катализатора реакции применяют также двуокись селена
(167, 170).
Серный ангидрид в присутствии селена является активным окислите-
лем хинолина при 220—230е С; выход никотиновой кислоты достигает 70%
[171, 172]. Озонолиз хинолина в 90%-ной уксусной кислоте приводит к
получению никотиновой кислоты [173].
Если, не выделяя никотиновой кислоты реакционную смесь по окон-
чании реакции окисления подвергнуть кипячению с метанолом, то обра-
зуется метиловый эфир никотиновой кислоты, который извлекают органи-
ческим растворителем, перегоняют в вакууме и используют для получения
амида никотиновой кислоты (II) [163, 174].
5. Биосинтез никотиновой кислоты из незаменимых аминокислот
При наличии в пище значительного количества триптофана (а-амино-
p-индолилпропионовой кислоты) (XXXIV), являющегося незаменимой
аминокислотой для человека и многих животных, в организме может со-
вершаться биосинтез никотиновой кислоты (I) при участии пиридоксале-
вых ферментов (схема 73). Триптофан (XXXIV) способен превращаться
через кинуренин (XXXV) в 3-оксикинуренин. Предшественником никоти-
новой кислоты является 3-оксиантраниловая кислота (XXXVII) [175],
которая образуется из 3-оксикинуренина (XXXVI) при действии кинуре-
ниназы [176, 177].
З-Оксиантраниловая кислота (XXXVII), по-видимому, под влиянием
ферментных систем способна окисляться с расщеплением цикла и образо-
ванием новых карбоксильных и альдегидных групп [178]. В результате
декарбоксилирования промежуточного соединения и замыкания в гете-
роцикл образуется никотиновая кислота (I).
Схема 73
Биосинтез никотиновой кислоты
306
Триптофан обычно рассматривается как биологический предшествен-
ник никотиновой кислоты.
6. Синтез никотинамида
Никотинамид (II) получают при пропускании аммиака в никотиновую
кислоту (I) при 180—230° С [179—1811 или при ее взаимодействии с моче-
виной в расплаве [179, 1821, преимущественно при каталитическом учас-
тии молибдената аммония; при температуре 160° С выход достигает 85%
[183]. Никотинамид получают также при действии водного или спиртового
раствора аммиака на метиловый (или этиловый) эфир никотиновой кислоты
(XXXVIII) [II, 41, 174, 184] или при взаимодействии изопропилового эфира
никотиновой кислоты с водным концентрированным раствором аммиака
в присутствии гранулированного алюминия почти с количественным вы-
ходом [1851.
Частичным гидролизом fj-цианпиридина (XXVIII) при действии пере-
киси водорода и едкого натра никотинамид (II) получен почти с количе-
ственным выходом [63, 186]. Гидратация Р-цианпиридина может быть
проведена с водой или водным аммиаком при 100—250° С (давление 4—
45 ат) с выходом 95% на прореагировавший нитрил [187, 1881 или нагре-
ванием до кипения водного раствора с основным высокомолекулярным
катализатором четвертичного характера (выход 86—90%) [189], а также
в присутствии карбонатов щелочных металлов [190].
Известен способ получения никотинамида из никотиновой кислоты
через ее хлорангидрид при действии аммиака, однако он не представляет
практического интереса.
Для очистки никотинамида от примеси никотиновой кислоты или
других соединений используют ионообменные смолы — амберлит IRA-410
11401, амберлит IR-4B [134], КБ-1, АВ-17 [191].
БИОЛОГИЧЕСКОЕ ЗНАЧЕНИЕ НИКОТИНОВОЙ КИСЛОТЫ
И НИКОТИНАМИДА. ЗАВИСИМОСТЬ МЕЖДУ СТРОЕНИЕМ
И БИОЛОГИЧЕСКОЙ АКТИВНОСТЬЮ. АНТИВИТАМИНЫ
Никотинамид (витамин) и никотиновая кислота (провитамин) яв-
ляются веществами, предохраняющими человека и животных от заболе-
вания пеллагрой (от латинского pelle agra, что означает «грубая кожа») —
болезнью, выражающейся в своеобразной шершавости кожи и изъязв-
лении языка [192]; в тяжелых случаях авитаминоза поражается
центральная нервная система. У животных пеллагра внешне про-
является следующим образом: у свиней -— изъязвление слизистых оболо-
чек вокруг рта и языка, шершавость кожи, огрубение волосяного покрова,
потеря аппетита; у птицы — плохое оперение, слабость ног, поносы.
307
Никотинамид проявляет свое биологическое действие в виде своей
коферментной формы — НАД и НАДФ (см. с. 309) в системах с фермен-
тами— дегидрогеназами.
В животный организм никотиновая кислота и никотинамид поступают
с пищей. Возможно, что надпочечники крысы способны синтезировать
никотиновую кислоту [193]. Триптофан в организме способен превращать-
ся в никотиновую кислоту, но с небольшим выходом — 60 мг триптофана
дает 1 мг никотиновой кислоты. Следует отметить, что способностью к би-
осинтезу никотиновой кислоты обладают и некоторые микроорганизмы.
Никотиновая кислота и никотинамид является ростовыми факторами
для многих видов микроорганизмов.
Для человека в сутки требуется 16—22 мг никотинамида (или никоти-
новой кислоты).
При приеме внутрь никотиновой кислоты наблюдается прилив крови
к лицу и конечностям, чувство жжения и покалывания в пальцах. Этих
явлений нет при приеме никотинамида.
Помимо специфической витаминной активности, никотинамид обла-
дает слабо выраженным противотуберкулезным действием [194].
Никотиновая кислота находит широкое применение для витами-
низации хлеба, муки, макаронных изделий и других пищевых продуктов
с целью повышения их питательной ценности.
Следует отметить, что в зерне кукурузы и других зерновых никотино-
вая кислота частично находится в неусвояемой связанной форме.
Никотиновая кислота имеет важное значение в кормлении сельско-
хозяйственных животных. В комбикорма ее добавляют 40—60 г/т.
Молекула амида никотиновой кислоты по своему витаминному дей-
ствию специфична. В полной мере витаминной активностью обладает толь-
ко его провитамин — никотиновая кислота. ^Изомерные пиколиновая и
изоникотиновая кислоты неактивны [195].
Слабой витаминной активностью (10% и ниже) обладают некоторые со-
единения, как, например, кордиамин — диэтиламид никотиновой кислоты
(XV, с. 296), диникотиновая кислота и ₽-пиколин, способные в организме
превращаться в никотинамид в результате реакций окисления, декарбок-
силирования, амидирования и др. Кроме того, кордиамин является возбу-
дителем сердечной деятельности (обладает физиологическим действием,
сходным с действием камфоры и кофеина) и имеет некоторое применение
при лечении шизофрении.
Более существенные изменения молекулы, сводящиеся к замене карбо-
ксильной группы в ^-положении пиридинового ядра на сульфогруппу —
Р-пиридинсульфокислота (XXVII) или ацетогруппу — 0-ацетопиридин
(XXXIX), приводят к получению соединений, обладающих антивитамин-
ными свойствами; сильным антагонистом является 6-аминоникотинамид
(XL) [196].
₽-Пиридинсульфокислота (XXVII) токсична для животных и служит
фактором задерживающим рост микроорганизмов. fl-Ацетопиридин
(XXXIX) — антивитамин для животных, хотя он и не влияет на рост
микроор ганизмов.
308
НИКОТИНАМИДНЫЕ КОФЕРМЕНТЫ
Никотинамид осуществляет свою биокаталитическую функцию в виде
коферментов — соединений с D-рибозой, фосфорной кислотой и аденози-
ном,— связываясь при этом со специфическим белком многочисленных
окислительно-восстановительных ферментов [197, 198] класса оксидоре-
дуктаз. Никотинамидные коферменты состоят из никотинамиднуклеози-
да — Ы-никотинамид-р Р-рибофуранозида, четвертичного аммониевого ос-
нования, и N(9(-аденил-[З-О-рибофуранозида, соединенных по первичным
гидроксильным группам 5'-положений D-рибозы пирофосфатной связью.
К никотинамидным коферментам относятся: никотинамидадениндинуклео-
тид (XLI) и никотинамидадениндинуклеотидфосфат (XLIV), а также их
восстановленные формы (XL1II и XLV). Из природных источников выде-
лен биологически неактивный я-никотинамидадениндинуклеотид (XLII).
Никотинам и дадениндинуклеотид — P*-(N- p-D-нико-
тинамидрибофуранозил-5')-Р2-(аденозил-5')пирофосфат (дифосфопиридинну-
клеотид, ДПН, НАД+, НАД) (XLI) — характеризуется наличием р-гли-
козидных связей адениновой и никотинамидрибофуранозидных частей
молекулы.
По своему строению НАД имеет четвертичный пиридиниевый ион,
который с одной из гидроксильных групп остатка фосфорной кислоты
образует внутреннюю соль. НАД представляет собой бесцветное вещество,
растворимое в воде, спирте и ацетоне и нерастворимое в большинстве дру-
гих органических растворителей. Он оптически активен: [(Дй.д —20°,
[aJiie — 70° (Н2О). Спектр поглощения имеет максимум при X 260 нм,
е 16,5-103. НАД мало устойчив в слабокислых растворах и неустой-
чив в слабощелочных растворах; так, в 0,1 н. NaOH при 20° С за 17 мин
он расщепляется на 50% [199].
НАД является одноосновной кислотой [200]. Соли НАД с тяжелыми
металлами в воде нерастворимы. НАД (2 мол.) образует с хинином (3 мол.)
хининовую соль с т. пл. 162—170° С, [ale —34,8° (Н2О).
a-Н икотинамидаденинд и нуклеотид — P*-(N-a-D-HH-
котинамидрибофуранозил-5')-Р2-(аденозил-5')пирофосфат (а-НАД)(ХЫ1) —
по своему строению имеет p-анамерную конфигурацию гликозидной свя-
зи аденозиновой части и a-гликозидную связь никотинамидрибофуранозид-
ной части молекулы нуклеотида.
309
а-НАД имеет Х„акс 260 нм, е 17,0-103. а-НАД биологически не активен
(2011. Наряду с биологически активным НАД ^-конфигурации (XLI)
(202] а-НАД находится, например, в дрожжах (10—15% общего содержа-
ния никотинамидных коферментов) [202]. а-НАД образует хининовую соль
с (а]о +14,3° (Н2О).
Никотинамидные коферменты осуществляют свои каталитические функ-
ции переноса водорода как в окисленной, так и в восстановленной
формах.
Дигидроникотинамидадениндинуклеотид
(НАД-Н) (XLI1I), восстановленный НАД
в отличие от окисленной формы НАД+ (XLI) вследствие наличия пара-хи-
ноидной структуры N-замещенного пиридинового цикла [203] имеет не толь-
ко Хмакс 260 нм, е 14,5-103, но и характерный дополнительный максимум
поглощения при А 340 нм, е 6,3-103 [199]. Этот второй максимум обычно
используют для количественного определения вещества, а также при
изучении окислительно-восстановительных превращений НАД. При воз-
буждении ультрафиолетовым светом с X 260 и 340 нм НАД-Н обладает го-
лубоватой флуоресценцией с максимумом излучения при X 460 нм. Сле-
дует отметить, что квантовый выход флуоресценции при возбуждении све-
том с X 260 нм для ₽ -НАД-Н гораздо больше, чем для а-НАД-Н [204].
НАД-Н является двухосновной кислотой. Он устойчив в слабощелоч-
ных и неустойчив в слабокислых растворах. Возможно, что при расщеп-
лении в кислой среде образуется промежуточный продукт присоединения
кислоты по одной из двойных связей пиридинового ядра [205].
Никотина мидадениндинуклеотидфосфат —
Р1-(Н-₽-Р-никотинамидрибофуранозил-5')-Ра-(2'-фосфоаденозил-5') пирофос-
фат (трифосфопиридиннуклеотид, ТПН, НАДФ+, НАДФ) (XLIV) — по
сравнению с НАД имеет дополнительную третью фосфорноэфирную
группу в положении 2 остатка £)-рибозы аденозиновой части молекулы.
Он представляет собой бесцветное вещество, растворимое в воде и
метаноле, трудно растворимое в этиловом спирте и этилацетате и нера-
створимое во многих других органических растворителях. Спектр погло-
щения имеет Хыакс 260 нм, е 16,5* 103, [а]^—24,6° и [аШв— 29,4° (Н^Э).
310
НАДФ мало устойчив в кислых растворах и неустойчив в слабощелочных —
при 23° С за 12 мин в 0,1 н. NaOH расщепляется 50% НАДФ 1199].
НАДФ является трехосновной кислотой; с тяжелыми металлами об-
разует нерастворимые в воде соли.
Дигидроникотинамидадениндинуклеотидфос-
ф а т (НАДФ-Н) (XLV), восстановленный НАДФ
XLV
имеет Хмакс 260 нм, е 14,5 • 103 и характеризуется появлением дополни-
тельного максимума поглощения при Хмакс 340 нм, в 6,3 • 10s [199]. НАДФ-Н
флуоресцирует с лыакс 460 нм при возбуждении УФ-светом с Хыакс 260
и 340 нм.
НАДФ-Н является четырехосновной кислотой. Он устойчив в слабо-
щелочных растворах и неустойчив в слабокислых.
Основной каталитически активной группировкой никотинамидных ко-
ферментов служит пиридиниевый цикл амида никотиновой кислоты, яв-
ляющийся акцептором водорода. Амид никотиновой кислоты в биохи-
мических реакциях обмена веществ превращается из четвертичного пири-
диниевого иона, в виде которого он находится в молекуле кофермента, в
третичный гетероциклический амин за счет присоединения электронов и
частичного гидрирования ядра; образующийся дигидроникотинамидный
кофермент является донором водорода. Эта реакция обратима:
Rib-P-O-P-Rib-Ad
II II
о о
XU
2е+2Н*
Rib-P-O-P-Rib-Ad
II II
Механизм реакции дегидрирования субстрата установлен путем при-
менения изотопного метода на примере превращения этилового спирта с
тяжелым водородом в ацетальдегид, причем дейтерий переходит в кофер-
мент [206]
D
_CONH2
aCONH,
-
п О' ОН
I I I
Rib-P-O-P-Rib-Ad
II II
О О
Н
CH,CDO + H+ + £
чьГ о" он
1 1 I
Rib-P-O-P-Rib-Ad
II II
О О
Присоединение дейтерия идет в положение 4 N-замещенного пиридиние-
вого цикла [2031. В качестве простой модели окислительно-восстановительной
никотинамидной системы был изучен N-метилдигидроникотинамид [207].
311
В изображении окислительно-восстановительных реакций окисленная
форма НАД может быть представлена символом НАД* (а НАДФ —
НАДФ*) тогда уравнение реакции будет следующим:
НАД+ + 2Н (2Н+ + 2г)=НАД-Н + Н*.
НАД-Н + Н* в этом уравнении является восстановленной формой НАД.
Согласно уравнению, к молекуле окисленной формы НАД (НАД*) проис-
ходит присоединение одного протона и двух электронов, т. е. в сумме
гидридиона Н“; второй протон обычно переходит в среду.
Реакции восстановления НАД и НАДФ имеют обратимый характер.
При дегидрировании молекула восстановленной формы НАД отдает про-
тон и два электрона и превращается в окисленную форму НАД.
НАД* легко восстанавливается в НАД-Н + Н* гидросульфитом нат-
рия [43] и другими восстановителями. НАД-Н может быть получен элек-
тролитическим восстановлением на платиновом, свинцовом и других като-
дах при pH 7,5 в присутствии (CH2OH)3CNH2 [208J. Также ведет себя и
НАДФ*, превращаясь в НАДФ-Н + Н*. При химическом восстановлении
образуется смесь двух стереоизомеров — по атому углерода положения
4 (см. с. 320).
Эта реакция восстановления обратима. Восстановленные НАД и НАДФ
ие подвергаются реокислению кислородом воздуха, но окисляются ферри-
цианидом калия и другими окислителями или ферментным путем.
При каталитическом гидрировании пиридиниевый цикл НАД и НАДФ
полностью восстанавливается и получается гексагидропиридиновое (пи-
перидиновое) производное, не обладающее способностью к обратимому
окислению.
НАД и НАДФ устойчивы к окислителям — брому, перекиси водорода
и марганцовокислому калию в кислых условиях [43].
При действии азотистой кислоты НАД дезаминируется в производное
гипоксантина — 6-окси(дезамино)-НАД 1209].
СТРОЕНИЕ НИКОТИНАМИДНЫХ КОФЕРМЕНТОВ
Структура никотинамидадениндинуклеотида (НАД) была установлена
на основании данных его химического и ферментативного расщепления
и синтеза отдельных фрагментов молекулы.
В молекуле НАД были найдены адениннуклеотид и никотинамид [43].
Гидролиз НАД в кислых условиях привел к расщеплению его молекулы
по ^-гликозидным связям и по фосфоангидридной связи с образованием
никотинамида [3, 43], аденина и двух молекул D-рибозо-5'-фосфата [210].
Наличие фосфоэфирной связи по первичной гидроксильной группе рибозы
(положение 5') следует из того, что при окислении перйодатом D-рибозо-
5-фосфата не образуется формальдегида [210, 211]. Пентоза, выделяющаяся
из никотинамид нуклеозида, была идентифицирована как D-рибоза
в виде ее п-бромфенилгидразона и другими реакциями [210].
При гидролизе НАД в слабощелочных условиях образовались нико-
тинамид и адепозиндифосфат-П-рибоза [212], а при повышенной темпера-
туре в результате дальнейшего расщепления — аденозин-5'-дифосфат (АДФ)
[213]. Следует отметить, что [3-гликозидная связь N-P-D-никотинамид-
рибофуранозид-5'-фосфата никотинамидмононуклеотида (НМН) в моле-
куле НАД оказалась значительно более лабильной, чем фосфоангидридная
связь пирофосфатной группы или ₽-гликозидная связь аденозин-5'-моно-
фосфата (АМФ).
Гидролиз НАД ферментом НАД-азой приводит к отщеплению пирофос-
фатной группы и расщеплению молекулы на два фрагмента — аденозин
и N-fJ-D-рибофуранозилникотинамид [214]. При действии пирофосфатазы
312
[>асщепление идет 'по фосфоангидридной связи и образуется НМН и АМФ
215], а в результате обратной реакции под действием ферментов из этих
фрагментов был ресинтезирован НАД [216].
Так как НАД титруется как одноосновная кислота [200], то, следо-
вательно, он имеет два этерифицированных гидроксила пирофосфатной
группы и представляет собой структуру цвиттер-иона, являясь одновре-
менно четвертичным пиридиниевым основанием [200].
В результате исследований большой группы авторов для строения НАД
в 1936 г. была предложена его формула XLI [200, 217].
В молекуле никотинамидадениндинуклеотидфосфата (НАДФ) установ-
лено наличие никотинамида [1, 11, 43]. Гидролиз в слабокислых условиях
расщепляет НАДФ с образованием никотинамида, аденина, двух моле-
кул пентозы и трех молекул фосфорной кислоты [42, 43]. При фермента-
тивном гидролизе с нуклеотидпирофосфатазой из НАДФ получается НМН
и аденозиндифосфат, в котором, однако, отсутствует пирофосфатная группа
[215]. В результате частичного дефосфорилирования одной фосфорноэфир-
ной группы из аденозиндифосфата образуется аденозинмонофосфат.
Три остатка фосфорной кислоты [200] в молекуле НАДФ соединены не
в виде линейной цепи, а так, что два остатка образуют пирофосфатную
связь, такую же, как и в никотинамидадениндинуклеотиде (XLI), а тре-
тий остаток соединен сложноэфирной связью с гидроксильной группой 2'
положения аденозиновой части молекулы [218]. В пользу такого строения
НАДФ (XLIV) говорит то, что он образуется из НАД (XLI) при взаимодей-
ствии с хлорокисью фосфора [219], т. е. в условиях, не требующих разрыва
существующей пирофосфатной связи. К тому же, если при щелочном ги-
дролизе расщепление НАД протекает первоначально по связи цикличес-
кого атома азота пиридиниевого ядра с D-рибозой, а затем с образованием
аденозинпирофосфата и адениловой кислоты [220], то из НАДФ эти сое-
динения не образуются, что говорит против присоединения третьего
остатка фосфорной кислоты к никотинамиднуклеозиду или к пирофос-
фатной группе.
ВЫДЕЛЕНИЕ НИКОТИНАМИДНЫХ КОФЕРМЕНТОВ
Основным методом получения коферментов НАД и НАДФ до послед-
него времени был метод выделения из природных источников, в качестве
которых могут служить дрожжи и животные ткани. Содержание НАД в
свежих дрожжах составляет около 0,5 г в 1 кг [221], в них очень мало НАДФ.
Сердечная мышца кролика содержит около 0,4 г НАД в 1 кг [16]. Содер-
жание НАДФ в печени, мышцах, эритроцитах составляет от 0,04 до 0,08г
в 1 кг [222].
Для получения НАД обычно используют хлебопекарные дрожжи, сво-
бодные от крахмала. Дрожжи экстрагируют горячей водой, экстракт
обрабатывают для удаления примесей основным ацетатом свинца и из
фильтрата НАД осаждают азотнокислым серебром. После удаления иона
металла сероводородом НАД из водного раствора осаждают ацетоном,
при этом его содержание достигает 76—83%. Окончательная очистка НАД
до содержания 95% производится на ионообменной смоле дауэкс-1 с по-
следующей элюцией 0,1 н. муравьиной кислоты и осаждением ацетоном
[2, 223, 224]. Для получения НАДФ по одному из методов свежую
печень овцы измельчают и экстрагируют теплой водой, осаждением три-
хлоруксусной кислотой удаляют белки и нуклеиновые кислоты, из филь-
трата НАДФ осаждают ацетатом ртути, удаляют ион металла сероводоро-
дом и осаждением ацетоном из водного раствора получают НАДФ с содер-
жанием 8—13%. Для последующей очистки используют ионообменную
смолу дауэкс-1, элюируют НАДФ формиатным раствором и после осажде-
ния ацетоном получают НАДФ с содержанием 92% [42, 43].
313
СИНТЕЗ НИКОТИНАМИДНЫХ КОФЕРМЕНТОВ
Для синтеза никотинамидадениндинуклеотида, НАД (XLI), исходят
из отдельных фрагментов его молекулы. Никотинамидный фрагмент полу-
чен из аморфного никотинамид-М-£)-рибофуранозида (XLVIII), кото-
рый синтезирован путем конденсации никотинамида (II), с 2, 3, 5-
триацстил-Р-бромрибофуранозой (XLVI) в растворе ацетонитрила в чет-
вертичную соль (XLVII) с последующим ее деацетилированием (225]
Соединение XLVI 11 фосфорилируют по первичной оксигруппе хлор-
окисью фосфора в нитрометане и получают N-p-D-никотинамидрибофу-
ранозид-5'-фосфат (XLIX) [226].
В качестве второго фрагмента молекулы НАД используют аденозин-
5'-монофосфат (L) [227].
Аденозин-5'-монофосфат (АМФ) синтетически получен прямым фосфор-
илированием аденозина хлорокисью фосфора в пиридине [228], при вза-
имодействии 2,'3'-диацетиладенозина с дифенил хлорфосфатом с последую-
щим удалением защитных групп [229]. Для синтеза АМФ широко ис-
пользовали аденозин с защищенными вторичными гидроксильными груп-
пами — 2',3'-О-изопропилиденаденозин. Для его фосфорилирования по
первичной гидроксильной группе положения 5' применялись хлорокись
фосфора [230], фосфорная кислота в присутствии трихлораиетонитрила
[231], тетрахлорпирофосфорная кислота с последующим мягким гидро-
лизом [232], дибензил хлорфосфат с последующим каталитическим гидро-
генолизом для удаления бензильных групп и кислотным гидролизом для
отщепления изопропилиденовой защиты [233] или гидролизом изопропи-
лиденовой группы и одной бензильной группы при действии уксусной ки-
слоты и каталитическим гидрогенолизом второй бензильной группы [2331.
Синтез АМФ осуществлен с использованием фенилборной защиты — фос-
форилированием 2',3'-О-фенилбороната аденозина Ридифенил-Р2-морфо-
лидофосфохлоридом, морфолидофосфодихлоридом или ₽-цианэтилфосфатом
[234, 235].
Среди различных методов синтеза несимметричных нуклеотидных ко-
ферментов НАД и НАДФ лучшим оказался карбодиимидный метод, сос-
тоящий в непосредственной конденсации двух монофосфатов нуклеозидов,
содержащих незащищенные гидроксильные группы, под влиянием конден-
сирующих агентов — М,1\т'-дициклогексилкарбодиимида, ди-;г-толил-
карбодиимида и др. Этот метод был проверен сначала на синтезе бензи-
ловых эфиров пирофосфорной кислоты [236]. Реакция между дибензилфос-
фатом и М,М'-дициклогексилкарбодиимидом в безводном пиридине идет
мгновенно с образованием тетрабензилпирофосфата и N.N'-дициклогексил-
мочевины. Оказалось, что даже значительный избыток воды не мешает
образованию смешанного ангидрида фосфорной кислоты. Реакция образо-
вания фосфоангидридов, по-видимому, заключается в первоначальном про-
тонировании N,N'-дициклогексилкарбодиимида [237,238] с последующим
314
присоединением фосфорного эфира и получением аддукта [237, 239], кото-
рый далее атакуется фосфор ноэфирным анионом с образованием пирофос-
фата и замещенной мочевины.
Синтез НАД (XLI) осуществлен из аденозин-5'-монофосфата (L) и
М-Р-£>-никотинамидрибофуранозид-5'-фосфата (XLIX) в водном пиридине
в присутствии избытка N,N'-дициклогексил карбодиимида [240]; реакция
проводилась при 0° С. После отделения образовавшейся дициклогексилмо-
чевины снова добавляют М,М'-дициклогексикарбодиимид и реакцию про-
должают при той же температуре; эту операцию повторяют трижды.
Около 50% никотинамидмононуклеотида превращалось в НАД и толь-
ко примерно десятая часть — в симметричный Р1, Р2-диникотинамиднукле-
озид-5'-пирофосфат; помимо него, в реакционной смеси обнаружен сим-
метричный Р1, Р2-диаденозин-5'-пирофосфат.
Разделение полученных веществ проведено на ионообменной смоле
дауэкс-2. Водные фракции содержали никотинамиднуклеотид и симметрич-
ный Р1, Р2-диникотинамиднуклеозид-5'-пирофосфат, а НАД удерживался
на смоле вместе с АМФ и Р1, Р2-диаденозин-5'-пирофосфатом. НАД вымы-
вался со смолы 0,01 н. муравьиной кислотой, АМФ — 0,1 н. и Р1, Р2-диа-
денозин-5'-пирофосфат — 1 н. муравьиной кислотой. В результате ионооб-
менной хроматографии НАД выделяли в виде аморфного порошка с содер-
жанием 70% кофермента.
В реакцию были взяты а- и p-анамеры НМН (приблизительно в соот-
ношении 80 : 20), поэтому в результате конденсации получилась смесь
а- и p-анамерных форм кофермента. Для их разделения использовали
изящный способ восстановления НАД алкоголь дегидрогеназой в НАД-Н
с последующей щелочной обработкой с целью расщепления а-анамера.
Полученный таким путем НАД был идентичен природному. По энзиматиче-
ской активности образец имел 90%-ную чистоту.
Подобным же методом синтезирован никотинамидадениндинуклеотид-
фосфат (НАДФ) (XLIV) из аденозин-2'(3'),5'-дифосфата и никотин-
амидмононуклеотида в присутствии М,М'-дициклогексилкарбодиимида в
водном пиридине
315
НАДФ был выделен на колонке с ионообменной смолой дауэкс-50
и содержал небольшие примеси никотинамидмононуклеотида и аденозин-
s',5'- и аденозин-3',5'-дифосфатов.
С невысоким выходом НАД был получен из АМФ и НМН в присутствии
трифторуксусного ангидрида [241]. Синтез НАД осуществлен также из
НМН и аденозин-5'-фосфоморфолида [242].
Карбодиимидным методом синтезированы различные аналоги НАД
[242—250].
Биологический синтез никотинамидмононуклеотида (XLIX) катали-
зирует трансфераза — рибозилникотинамидкиназа, исходным соединением
служит М-никотинамид-(3-£)-рибофуранозид, донором фосфатной группы —
' АТФ [251]:
Н-Никотинамидф-О-рибофуранозид + АТФ -> НМН + АДФ.
Биосинтез НАД и НМН и АТФ катализирует НМН-аденилилтрансфе-
эаза; реакция сопровождается выделением неорганического пирофосфата
.215, 216].
НМН + АТФ -> НАД + H*PtO7.
НАД без амидной группы может быть синтезирован при участии той же
никотинатмононуклеотид-аденилилтрансферазы:
Дезамидо-НМН + АТФ —* Дезакидо-НАД + Н4Р2ОТ.
НАДФ в организме образуется в результате строго специфического
фосфорилирования по 2-гидроксилу аденозиновой части молекулы НАД
НАД-киназой [252].
НАД 4- АТФ -> НАДФ + АДФ.
БИОХИМИЧЕСКИЕ ФУНКЦИИ НИКОТИНАМИДНЫХ КОФЕРМЕНТОВ
Никотинамидадениндинуклеотид (XLI) и никотинамидадениндинуклео-
тидфосфат (XLIV), как указывалось выше, представляют собой коферменты,
которые со специфическим белком многочисленных ферментов, относимых
преимущественно к классу оксидоредуктаз, образуют биокаталитические
комплексы. В процессе обмена веществ в клетках живого организма они
катализируют реакции дегидрирования различных органических соединений
и передают водород, как правило, промежуточному акцептору — флавино-
вым ферментам и в итоге -— на кислород.
Никотинамидные коферменты принимают участие в катализе свыше
130 различных биохимических обратимых реакций дегидрирования или
окисления субстратов с образованием химических связей: С=О, С—ОН,
CH = NH,CH—N,C—N,C—S, С—Си расщеплением связи С—С и др. При
этом они дегидрируют или окисляют следующие вещества:
первичные и вторичные спирты в альдегиды и кетоны;
альдегиды и кетоны в органические кислоты;
аминосоединения в иминосоединения с последующим образованием ок-
сосоединений;
316
) частично гидрированные циклические соединения в менее гидриро-
ванные или ароматизированные системы;
сероводород в сульфит и затем в сульфат и др.
Они осуществляют восстановительное внутримолекулярное N-алки-
। лирование. Кроме того, известен один никотинамидный фермент из класса
( изомераз, который принимает участие в реакции эпимеризации сахаров.
Плоскость пиридинового цикла никотинамидных коферментов не коп-
ланарна с почти плоским рибофуранозным кольцом, которое имеет почти
перпендикулярное расположение, причем гликозидная связь находится
в плоскости г_терэцикла [2531. Пиридиновый и пуриновый циклы про-
странственно сближены и, по-видимому, находятся на близких к парал-
лельным плоскостях, что облегчает взаимодействие между атомами азота
аминогруппы и других гетероциклов, возможно, за счет водородных свя-
) зей, с активными центрами ферментного белка. О возможности внутри-
। молекулярного взаимодействия пиридиновой и адениновой части в молекуле
j НАД получены некоторые данные электронных спектров [254], ПМР-
спектров [255] и спектров флуоресценции [256], из которых следует, что
НАД в водном растворе находится в свернутой конформации. Однако
по данным спектров ПМР высокого разрешения такая конформация НАД
быстро превращается в развернутую [257, 258].
Никотинамидные коферменты — НАД и НАДФ — в высокой степени
стереоспецифичны и в различных биохимических реакциях, в зависимости
от структуры ферментного белка, реагируют с субстратом путем присоеди-
нения или отщепления атома водорода в положении 4 пиридинового цик-
ла только той или иной (как правило, одной) стороной планарного пири-
динового (дигидропиридинового) кольца — формы А или В [259, 260].
Атом углерода в положении 4 при восстановлении становится асимме-
тричным.
При ферментативном восстановлении образуется только один стереоизомер.
При химическом восстановлении получается смесь стереоизомеров. Такая
стереоспецифичность дегидрогеназ установлена реакциями с применением
дейтерия. Дейтерированная молекула, относимая к форме А, будет иметь
атом дейтерия, расположенный над плоскостью молекулы, изображенной
на чертеже, и будет реагировать с субстратом этой стороной молекулы.
Форма В НАД будет иметь атом дейтерия со связью, направленной под
плоскость чертежа.
К ферментам, образующим форму В НАД, относятся алкогольдегидро-
геназа, L- и D-лактатдегидрогеназа, малатдегидрогеназа и др.; форму В
НАД — глюкозодегидрогеназа, глутаматдегидрогеназа, НАД-Н-цитохром-
с-редуктаза и др. [259].
Все разнообразные обратимые окислительно-восстановительные реакции
субстратов осуществляются почти исключительно при участии двух кофакто-
ров — НАД и НАДФ. Высокая специфичность фермента к субстрату обус-
ловливается почти исключительно структурой белка —последовательностью
соединения /.-аминокислот и их пространственного расположения. Моле-
кулярная масса ферментов составляет от 84 000 для алкогольдегидрогеназы
из печени до 1 000 000 для глутаматдегидрогеназы из митохондрий печени
быка.
317
Связь НАД и НАДФ с ферментами лабильна, и в процессе биокатали-
тического переноса водорода от донора к акцептору они могут связываться
с другим ферментом.
Ферменты, участвующие в переносе водорода, обычно в виде общей
цепи дыхания находятся в митохондриях клеток.
НАД и НАДФ есть во всех животных и растительных клетках, причем
НАДФ сравнительно в большем количестве содержится в животных клет-
ках, чем в растительных. Дегидрогеназа при участии НАД катализирует
преимущественно окислительные реакции субстратов — в живой клетке
кофермент находится главным образом в окисленной, а не в восстановлен-
ной форме. Дегидрогеназы при участии НАДФ катализируют в основнем
восстановительные реакции субстратов: в клетке кофермент представлен в
большем количестве в форме НАДФ-Н и в небольшом — в окисленной
форме.
В клетке и в клеточных структурах, например в митохондриях, под-
держивается постоянство соотношений между окисленными и восстанов-
ленными формами никотинамидных коферментов.
Каталитические функции, осуществляемые при участии восстановлен-
ных форм никотинамидных коферментов (НАДФ-Н), лежат в основе жиз-
ненных процессов — в синтезе первичного органического вещества из дву-
окиси углерода, воды, минеральных солей, фосфора, азота с поглощением
квантов света солнечной энергии. Процесс фотосинтеза осуществляется в
клетках зеленых частей растений и сопровождается выделением молеку-
лярного кислорода в атмосферу. Возможно, и к этому имеются серьезные
основания, весь или почти весь кислород атмосферы Земли образовался за
счет реакции фотосинтеза.
В сопряженных фотохимических и ферментативных реакциях проис-
ходит перенос электрона и протона от воды на НАДФ; образовавшийся
НАДФ-Н участвует в одной из реакций цикла восстановления двуокиси
углерода до D-фруктозы с последующим ее превращением в сахарозу.
НАДФ-Н в ферментной системе принимает участие в каталитическом вос-
становлении 3-фосфорглицериновой кислоты в З-фосфо-О-глицериновый
альдегид при участии АТФ [261 ]:
СООН СНО
| НАДФ-Н + Н+ |
нсон у- НСОН + Н,0
I НАДФ+ |
СН2ОРО3Н2 СН2ОРО»Н2
Эта реакция обратима, и превращение З-форфо-Р-глицеринового альдеги-
да в З-фосфо-О-глицериновую кислоту происходит при расщеплении гексоз
в процессах обмена веществ (см. ниже).
Восстановленные формы никотинамидных коферментов участвуют в
восстановлении нитрата в нитрит и затем в аммиак, 2,3-дегидро-А-аскор-
биновой кислоты в /.-аскорбиновую кислоту, L-цистина в £-цистеин, липо-
амида в дпгидролипоамид, а совместно с кислородом участвуют в образо-
вании оксисоединений, например 4-оксианилпна из анилина и т. п.
Никотинамидные коферменты принимают участие в отдельных реакциях
углеводного, липидного и аминокислотного обмена в процессах фотосин-
теза в растениях. Дегидрогеназы катализируют отдельные этапы реакций
анаэробного расщепления моносахаридов с высвобождением свободной
энергии и накоплением ее в аденозин-5'-трифосфате (АТФ), который яв-
ляется основным аккумулятором и затем генератором энергии в живой
клетке. В этой метаболической реакции происходит образование макроэр-
гической связи с превращением АДФ в АТФ, которые являются ключевы-
ми энергетическими переносчиками.
318
В реакциях анаэробных превращений углеводов (P-D-глюкозы, глико-
гена) до пировиноградной кислоты из 11 стадий этого гликолитического
процесса НАД участвует в катализе только одной стадии — дегидриро-
вании З-фосфо-Д-глицеринового альдегида (LI) в З-фосфо-Д-глицериповую
кислоту (LIII) через 1,3-дифосфат D-глицериновой кислоты (LII), имеющей
макроэргическую связь, при участии глицеральдегидфосфатдегидрогеназы
(молекулярная масса 137 000) [262]:
СНО
СНОН 4- Н8РО4
СН.2ОРО8Н2
L1
>он
сн/
I \оро3н.
СНОН
+ НАД+
—НАД-Н;
—Н+
СНОН
I
СН2ОРО8Н2
LII
4-АТФ
:н»оро,н
LIII
Последняя фаза этого процесса протекает при взаимодействии с адено-
зин-5'-дифосфатом (АДФ), который акцептирует фосфорную кислоту с
образованием АТФ.
Процесс гликолиза, протекающий в мышечных и других тканях, за-
вершается восстановлением пировиноградной кислоты в А-молочную
кислоту восстановленной формой НАД, образовавшейся на одной из
предыдущих стадий — при дегидрировании З-фосфо-О-глицеринового
альдегида. Эта реакция осуществляется при участии лактатдегидрогеназы
(особенно распространенной в скелетных и сердечной мышцах), при этом
заканчивается цикл регенерации НАД в исходной окисленной форме
[263—265], что обусловливает непрерывность гликолитического цикла
НАД-Н 4- Н+
СНзСОСООН----------------f СН8СН (ОН) соон.
-НАД+
Дрожжевая лактатдегидрогеназа участвует в осуществлении обратимой
реакции дегидрирования L-молочной кислоты в пировиноградную кислоту.
В заключительной фазе процесса спиртового брожения углеводов, проте-
кающего через пировиноградную кислоту, НАД-Н осуществляет гидриро-
вание ацетальдегида в этиловый спирт при участии алкогольдегидрогеназы
[265]:
НАД-Н 4- Н+
СН8СНО т............ СН3СН2ОН.
НАД+
Так как спиртовое брожение представляет собой анаэробный процесс,
то эта реакция протекает одновременно с окислением З-фосфо-О-глице-
ринового альдегида (LI) в 1,3-дифосфат Д-глицериновои кислоты (LII)
В процессе получения пировиноградной кислоты из углеводов при участии
НАД, вследствие чего носит характер перекрестной реакции Канниццаро.
Алкогольдегидрогеназа дрожжей связывается с 4 молекулами НАД и
4 молекулами Zn^ на молекулу фермента; она окисляет первичные и вто-
ричные спирты в соответствующие альдегиды и кетоны.
319
Биологическое окисление — источник энергии живых организмов. Окис-
лительные превращения охватывают все виды питательных веществ: бел-
ки, углеводы и жиры, которые распадаются под влиянием ферментов пище-
варительного тракта на аминокислоты, моносахариды, глицерин и жир-
ные кислоты. Продукты расщепления образуют метаболический фонд био-
синтеза и получения энергии.
Центральную роль в процессе обмена веществ играют ди- и трикарбо-
новые кислоты, образующиеся при дальнейших превращениях аминокис-
лот и жирных кислот и входящие в так называемый цикл трикарбоновых
кислот: лимонная, изолимонная, щавелевоянтарная, а-кетоглутаровая,
янтарная, фумаровая, А-яблочная, щавелевоуксусная и пировиноградная
(схема 74). Последняя из них не входит непосредственно в цикл трикарбо-
новых кислот, но является продуктом расщепления D-глюкозы и предшес-
твенником активной ацетильной группы в ацетил коферменте А. Реакции
превращений органических кислот приводят к полному расщеплению пиро-
виноградной кислоты до двуокиси углерода и водорода при последователь-
ном катализе 10 различными ферментами при участии коферментов: НАД,
НАДФ, флавинадениндинуклеотида (ФАД), 8я-гистидил-ФАД, пантетеи-
надениннуклеозиддифосфата (коэнзима А, КоА, KoASH), тиаминдифосфа-
та (ТДФ) и липоевой кислоты (ЛК).
Схема 74
НАД-Н+И
фап-н2 фа,д
НООСН2СОСООН
нооссн2снсоон
Яблочная кислота
нооссн=снсоон
Фумаровая кислота
Гистидил- А
,фад-н2 /
Гистидилу
2Н + 2е ФАД
HOOCCII2CII/OOI1
Янтарная кислота
атф(гтф) V
HOOCCH2CO~SKoA
Сукцинил-КоА
Щавелевоуксусная
кислота
CH,COCOOH+W2O
адф(гдф)+н3ро4
нооссн,с=снсоон
соон
Н О —ifcf-Анонитоаая кислота
-3CO2+ri2OH0HMte-^l
'ОН
HOOCCHjCHCHCOOH
соон
Изолимонная кислота
нооссцснсосода
Щавелевоянтарнаялислота НАДФ+Н
ФАД ФДД-Н2
HOOCCHCHjCOCOOHVq
а-Кетоглутароаая кислота
2Н+2е
Превращения в цикле трикарбоновых кислот — важный источник
энергии организма. Превращения питательных веществ в энергетическом
аспекте могут быть разделены на три стадии: а) расщепление макромолекул
биополимеров — углеводов до гексоз, белков до аминокислот и жиров до
320
глицерина и жирйых кислот — высвобождает 0,1—0,5% энергии; б) пре1
вращение образовавшихся мономеров в ацетилкоэнзим А и другие компо-
ненты цикла три карбоновых кислот высвобождает 15—30% энергии; в) окис-
ление по циклу трикарбоновых кислот высвобождает яри переносе элект-
ронов (водорода) на О2 70—80% энергии.
Дегидрогеназы катализируют три реакции в аэробных процессах пре-
вращений органических кислот по циклу трикарбоновых кислот: синтез
сукцинвл-КоА из ц-кетоглутаровой кислоты при участии НАД, ФАД,
ТДФ и липоевой кислоты (см. схему 74), дегидрирование изолимонной
кислоты в щавелевоянтарную кислоту НАДФ при участии изоцитратдегиД-
рогеназы 1266]
ОН
I + НАДФ4
НООССНгСНСНСООН --------------«• НООССН2СНСОСООН
| — НАДФ-Н — Н+ |
СООН соон
и обратимое дегидрирование £-яблочной кислоты в щавелевоуксусную
кислоту НАД под влиянием малатдегидрогеназы (см. схему 69) [2671
ОН
I +НАД+
НООССН2СНСООН-------------f НООССН2СОСООН
— НАД-Н —Н+
Тканевое дыхание — завершающий этап биологического окисления, в
результате которого происходит перенос водорода (протонов, электронов)
от НАД-Н и НАДФ-Н (возникающих при первичном акцептировании водо-
рода субстратов — кислот, компонентов цикла три карбоновых кислот и
других реакций) на молекулярный кислород. Процесс переноса водорода
осуществляется при каталитическом участии ферментов цепи дыхания, в
которой НАД является первичным звеном
Флавопротеиды акцептируют водород НАД-Н (НАДФ-Н) и передают
его на систем}' цитохромов или (возможно, как правило) предварительно
на убихинон (коэнзим Q). Протоны переносятся в раствор окружающей
среды, а электроны транспортируются системой цитохромов на молекуляр-
ный кислород. Образующийся при этом отрицательно заряженный ион ки-
слорода (О--) соединяется с протонами буферной среды митохондрий, об-
разуя воду. Следует отметить, что цитохромные ферменты осуществляют
только функцию исключительно переноса электронов.
В окислительно-восстановительных ферментных системах никотин-
амидные ферменты имеют более низкий потенциал, чем системы, образо-
ванные из флавиновых или порфириновых коферментов.
Оксидоредуктазы при участии НАДФ катализируют отдельные реак-
ции прямого окисления P-D-глюкозы в двуокись углерода по пентозному
циклу, окисляя при действии глюкозо-6-фосфатдегидрогеназы £)-глюкозо-
6-фосфат (LIV) в D-глюконо-б-лактоно-6-фосфат (LV) [2681, который гидро-
лизуется при участии О-глюконо-6-лактонгидролазы (или без нее) в 6-
фосфо-£)-глюконовую кислоту (LVI), в свою очередь подвергающуюся
дегидрированию и декарбоксилированию, по-видимому, через промежу-
точную 6-фосфо-£)-3-кетоглюконовую кислоту, при действии фосфоглюконат-
дегидрогеназы в П-рибулозо-5-фосфат (LVII) [2681
11—69
321
н н о н
H3o,POCHj-c—соон
он он Ьн
4 НАДФ*
- НАДФ-Н;-Н+’
н н
I I
HjOjPOCHs-C-C-CO-CHpH + со„
он он
LV1I
Следует отметить, что D-глюкозо-б-фосфат (LIV) является общим проме-
жуточным соединением как для пентозного цикла, так и для гликолити-
ческого расщепления углеводов. D-Рибулозо-б-фосфат (LVII) далее окис-
ляется в двуокись углерода или используется для синтеза D-рибозы, вхо-
дящей в структуру рибонуклеиновых кислот (РНК), НАД, НАДФ, ФАД,
КоА, АМФ, АДФ, АТФ и др.
Среди реакций белкового обмена интересно отметить обратимую реак-
цию окисления L-глутаминовой кислоты (LVIII) в а-кетоглутаровую кис-
лоту (LIX) с участием НАДФ или НАД через 2-иминоглутаровую кисло-
ту, протекающую при участии глутаматдегидрогеназы [269] с выделени-
ем аммиака:
+ НАДФ*
HOOCCH2CH2CH (NH2) СООН-------------► НООССН2СН2С (=NH) СООН
—НАДФ—Н;—Н+
LVIII
НООССН2СН2С (=NH) СООН + Н2О----> НООССН2СН2СОСООН 4- NH„
LIX
а также реакцию окисления бетаинальдегида в бетаин НАД катализируе-
мую бетаинальдегиддегидрогеназой [2701:
+ + НАД* +
(СНэ)з NCH2CHO + Н2О------------► (СН3)з nch2coo~.
— НАД—НН+
В ^-окислительном расщеплении жирных кислот до ацетилкофермента
А, включаемого для дальнейшего окисления в аэробный окислительный
цикл трикарбоновых кислот, в качестве одного из звеньев этого расщепле-
ния жирных кислот имеет место реакция дегидрирования 3-оксиацнл-КоА
в 3-оксоацил-КоА. Эта реакция катализируется 3-оксиацил-КоА-дегидро-
геназой (кристаллический фермент) при участии НАД [271, 272]
НАД* НАД-Н+Н'
/ i
RCH(OH)CH2CO~SKoA S-----------—
RCOCKCO'SKoA
Некоторые реакции обмена витаминов протекают при участии НАД.
К таким реакциям относятся следующие.
Восстановление дегидро-А-аскорбиновой кислоты (LX) в /.-аскорби-
новую кислоту (LXI) НАДФ-Н при участии восстановленной дегидроас-
корбатредуктазы [273]
ОН ОН
+ НАДФ-Н; +Н+
— НАДФ*
ОН Н | |
| |/С=С\
-С—с с
I ^О
н хох
LXI
322
Дегидрирование пиридоксина (LXII) в пиридоксаль (LXIII) (и соответ-
ственно их фосфатов) НАДФ при участии пиридоксиндегидрогеназы
+ НАДФ+
— НАДФ-Н; —Н+
LXIII
Дегидрирование в механизме зрительного процесса ретинола, витамина
A (LXIV) в ретиналь, витамин А-альдегид (LXV) НАД под влиянием альдегид
дегидрогеназы [265, 269]
СНз СНз СНз СН3
£Н=СН-С=СН-сн=сн—<1=сн-сн,он
+ НАД+
»НС СНз
СНз СНз
сн=сн—с=сн—-СН=СН—А=СН—<СНО
LXV
Восстановление фолиевой кислоты (LXVI) в 7,8-дигидрофолиевую кисло-
ту (LXVII) НАДФ-Н при участии восстановленной дегидрофолеатдегидро-
геназы 1274]
СООН
HNCeH4COHNCH
с!н
CHgCOOH
LXVI
+ НАДФ-Н; + Н+
—НАДФ+
н
LXVII
СООН
I
CH2HNCeH*COHNCH
I
СН,
I
СН,СООН
и дальнейшее восстановление 7,8-дигидрофолиевой кислоты (LXVII) в
5, 6, 7, 8-тетрагидрофолиевую кислоту также НАДФ-Н при участии восста-
новленной тетрагидрофол еатдегидрогеназы.
Уридиндифосфатглюкозоэиимераза (УДФ-глюкозо-4-эпимераза) при
участии НАД осуществляет в животном и растительном организме, а также
у микроорганизмов [275, 2761, реакцию взаимного превращения О-глю-
11*
323
козы и £)-галактозы путем эпимеризации гидроксильной группы поло-
жения 4 УДФ-глюкозы (LXVIII) и УДФ-галактозы (LXIX)
НАД и НАДФ при действии НАД-иуклеозидазы расщепляются в клетках
на никотинамид и соответствующий остаток. При действии нуклеотидпи-
рофосфатазы (из змеиного яда и др.) (2151 НАД и НАДФ расщепляются по
пирофосфорной связи на два мононуклеотида — НМН и АМФ.
Высокая специфичность структуры НАД в ферментативных реакциях
была установлена на основании сравнительного изучения многочисленных
аналогов коферментов [204, 2771).
СПИСОК ИСПОЛЬЗОВАННОЙ
ЛИТЕРАТУРЫ (К ГЛАВЕ VII)
1. О. Warburg, W. Chris-
tian. Biochem. Z., 274, 112(1934);
275, 112, 464 (1935).
2. О. W a r b u r g, W. Chris-
tian, A. G r i e s e. Biochem.
Z., 279, 143 (1935).
3- H. v о n Euler, H. A 1 b e r s,
F. Sc h I e n k. Z. physiol. Chem.,
237. 1 (1935); 240, 113 (1936).
4. С. E 1 v e h i e m, R. Madden,
F. S t г о n g, D. Woolley.
J. Am. Chem. Soc., 59, 1767 (1937);
J. Biol. Chem., 123, 137 (1938).
5. R. Go r d i ng, L. F 1 e x e r.
J. Am. Pharm. Assoc., 29, 230
(1940).
6. E. H у g h e s, H. J e 1 1 i n e k,
B. A m b г о s e. J. Phys. Col-
loid. Chem., 53, 414 (1949).
7. Я. M. Слободин, M. M.
Голь д м а н. ЖПХ, 21, 859
(1948).
8. H. V i c k e г v. J. Biol. Chem., 68,
585 (1926).
9. A. Pinner. Ber., 33, 1227 (1900).
10. A. W i c k s t г о m. J. Pharmac.
Pharmacol., 2, 444 (1950).
11. P. Karrer, B. Ringier,
J. В u c It i, H. Fritzsche,
C. S о 1 in s s e n. Helv. Chim.
Acta, 20, 55 (1937).
12. W. M с С г о n e, J. С о о k. The
Frontier, 10, 12 (1947).
13. W. W r i g h t, G. К i n g. Acta
Cryst., 3, 31 (1950).
14. R. К u h n, H. V e t t e r. Ber.,
68, 2374 (1935).
15. H. J e 1 1 i n e k, M. W a у n e.
J. Phys. Colloid. Chem., 55, 173
(1951).
16. P. К a г г e г, H. К e 1 1 e r. Helv.
Chim. Acta, 22, 1292 (1939).
17. A. H i r t, K. W i m m e r. Klin.
Wschr., 18, 765 (1939).
18. A. Philips. Lieb. Ann., 288,
253 (1895).
19. H. J e 1 I i n e k, A. Gor d о n.
J. Phys. Colloid. Chem., 53, 996
(1949).
20. D. Ackerman. Z. Biol., 59, 17
(1912).
21. W. Dann, J. HuH. J. Biol.
Chem., 168, 121 (1947).
22. P. К a r r e r, G. Schwarzen-
b a c h, F. В e n z, U. S о 1 m s-
s’e n. Helv. Chim. Acta, 19, 811,
826 (1936).
23: W. C i u s a, G. Neb bi a. Gazz.
chim. ital., 79, 521 (1949).
24. J. H u f f, W. Perlzweig.
J. Biol. Chem., 150, 395 (1943).
25. W. Knox, W. Grossman.
J. Biol. Chem., 166, 391 (1946);
168, 363 (1947).
26. O. W a r b u r g, W. C h r i s-
t i a ii. Biochem. Z., 285, 297 (1936).
27. Бельг, пат. 558779; РЖХ, 1961,
ЮЛ383.
28. J. Stepan, V. Kral, M. Ju-
re c e k. Chem. listy, 49, 144 (1955).
29. P. S a h. J. Am. Chem. Soc., 76, 300
(1954).
30. H. N a j e r, R. G u e p e t. Ann.
pharmac. Franc., 12, 712 (1954).
324
31. C. G е п о t. Англ. пат. 771317;
С. А., 51, 13938 (1957).
32. Т. I г i к u г a, S. S a t о. Яп. пат.
18109 (1965); С. А., 63, 18037 (1965).
33. Ю. Ф. Рачинский, Я. М.
Слободин, И. Н. Ш о х о р.
ЖПХ, 19, 176 (1946).
34. В. А. Д е в я т к и н, В. И о с и-
к о в а. Авт. свид. СССР 69191;
Бюлл. изобрет., 1947, № 8, 42.
35. С. Huber. Вег., 3. 849 (1870);
Lieb. Ann. 141, 271 (1867).
36. Z. Skraup, A. Cobenzl.
Monatsh., 4, 436 (1883).
37. W. Wright, G. К i n g. Acta
cryst al logr., 6, 305 (1953); 7, 283
(1954).
38. C. F u n k, J. Physiol., 43, 395
(1911); 46, 173 (1913).
39. L). S u z u k i, T. Shimamura,
S. О d a k e. Biochem, Z., 43, 89,
99 (1912).
40. А. Вышнеградский. Ber., 12,
1480 (1879).
41. E. Engler. Ber,, 27, 1787 (1894).
42. O. W a r b u r g, W. C h r i s-
tian, W. C r i e s e. Biochem.
Z., 282, 157 (1935).
43. O. Warburg, W. Chris-
ti a n. Biochem. Z., 287, 291 (1936).
44. J. Muel 1 er. J. Bact., 34, 429
(1937); J. Biol. Chem., 120, 219
(1937).
45. А. П. О p e x о в. Compt. rend.,
189, 945 (1929).
46. А. П. О p e x о в, Г. П. M e н ь-
шиков. Ber., 64, 266 (1931); 65,
232 (1932).
47. R. L a i b 1 i n. Ber., 10, 2136 (1877);
Lieb. Ann., 196, 135 (1879).
48. H. А. Васюнина, A. A.
Беэр, H. А. П p e о 6 p a-
женский. ЖПХ, 16, 206 (1943).
49. А. С. С а д ы к о в, ЖОХ, 17, 1710
(1947).
50. А. Шм у к, ЖПХ, 20, 245 (1947).
51. Ю. П. Ч у м а к о в, А. И. М ед-
ин к о в, Р. И. В и р н и к.
ЖПХ, 35, 602 (1962).
52. М а к-Э л ь в е н. В сборнике «Син-
тезы органических препаратов>, сб.
I, 288, М., ИЛ, 1949.
53. В. В. Вилья м с, И. М. М а-
влянов. ЖПХ, 17, 228 (1944).
54. А. С. Садыков, ЖОХ, 15, 252
(1945).
55. А. М. X а л е ц к и й, Ю. Н. Р о-
з е н б л ю м, А. А. Аносо-
ва. Фармация, 1944, № 1, 19.
56. С. Woo d war d, С. Bad-
gett, G. Kau 1 man. Ind.
Eng. Chem., 36, 544 (1944).
57. Т. М а р к а ч е в а, Г. Л е б е-
дева. ЖПХ, 23, 299 (1950).
58. И. Б. С и м о н. Авт. свид. СССР
69938; Бюлл. изобрет., 1947, № 12,
11.
59. Б. В. Матвеев, ЖОХ, 17, 482
(1947).
60. В. Lovrecek. Radovi Jugos-
lav. Acad. Znanosti i umjetnosti,
296, 65 (1953).
61. F. F i c h t e r, H. Stenzl.
Helv. Chim. Acta, 19, 1171 (1936).
62. И. В. С и м о н. Авт. свид. СССР
70302; Бюлл. изобрет., 1948, № 1,
11.
63. W. К г о h s. Пат. ФРГ 828246,
828247; С. А., 50, 3504 (1956).
64. S. М с Е 1 v a i n, М. G о е s е.
J. Am. Chem. Soc., 63, 2283 (1941).
65. С. Wood ward, С. В a d-
g е t t, J. W i J 1 a in a n. Ind.
Eng. Chem., 36, 540 (1944).
66. F. C i s 1 a k, W. Wheeler.
Пат. США 2456380; С. A., 43,
1811 (1949).
67. О. Fischer. Ber., 15, 63 (1882).
68. Б. В. Суворов, P. С. Раф и-
к о в, А. Д. Кагарлицкий,
Д. X. С е м б а е в, Ю. Н. С о л н-
ц е в, и др. Авт. свид. СССР
235764; РЖХ, 1970, 2Н253.
69. Е. Ф. К о з л о в а, И. М. К у с-
т а н о в и ч, М. М. Я н и н а,
И. Б. Чекмарева. Хим. фарм.
ж., 2, № 7, 28 (1968).
70. А. М у г г а у, W. F о г е m а п,
W. Langham. Science, 106,
277 (1947).
71. A. Karr, Е. Scheibel.
Ind. Eng. Chem., 46, 1583 (1954).
72. A. M. Г p и г о p о в с к и й, 3. М.
К н м е н. ЖПХ, 18, 259 (1945).
73. F. С i s 1 а к. Пат. США 2272159;
С. А., 36, 3514 (1942).
74. И. Поляков, ЖПХ, 20, 845
(1947).
75. F. С i s 1 a k, Е. W h е е 1 е г.
Пат. США 2430804; С. А., 42, 1322
(1948).
76. G. R i е t h о f. Пат. США 2443479;
С. А., 42, 6860 (1948).
77. М- В. Р у б ц о в, Е. Е. Мих-
ли и а, В. Я. Фурштат о-
в а. ЖПХ, 29, 949 (1956).
78. М. В. Р у б ц о в, Е. Е. Ми-
хлина, В. Я. Фурштат о-
в а. Авт. свид. СССР 97408; Бюлл.
изобрет., 1954, № 3, 15.
79. J. Jones. Англ. пат. 598036;С. А.,
42, 4614 (1948).
80. R. Bowman. Пат. США 2459359;
С. А., 43, 3043 (1949).
81. К. S 1 a g 1 е, R. В о w m а п.
Пат. США 2459191; С. А., 43,
3043 (1949).
82. G. R i е t h о f. Пат. США 2295606;
С. А., 37, 1131 (1943).
83. L. Rusek. Пат. ГДР6013;РЖХ,
1955, 19865.
84. IO. И. Ч у м а к о в, П. А. Г а н-
греки й. Авт. свид. СССР
128021; РЖХ, 1961, 13Л183.
85. F. С i s 1 a k, W. Wheeler
Пат. США 2300741; С. А., 37, 2019
(1943).
86. М. Н. Щ у к и и а, Г. Н. П е р-
ш п н, О. О. М а кее в а, Е. Д.
Сазонова, Е. С. Никитин-
ская, А. Д. Янина;
А. И. Яковлева. ДАН СССР,
84 , 981 (1952).
325
87. М. В. Р у б ц о в, Л. Н. Я х о н-
т о в, С. В. Я Ц е н к о. ЖПХ,
30, 315 (1957).
88. W. S wietosl a wski, А. By-
lick i, D. Pastafinska,
Z. L i s i с k i, W. L e wen-
stein, T. Sikorska,
E. Wardzinskj. Польск. пат.
39997; РЖХ, 1960, 43850.
89. В. L i p k а, Е. Treszczno-
wi cz. Пат. ПНР 50077; РЖХ,
1968, 8Н306.
90. F. S t i t z. Osterr. Chem. Ztg.,
45, 159 (1942).
91. K. Hargrave Англ. пат.
887688, 896049—896051, 929694;
С. A., 56, 12859; 57, 13736—13737
\{1962); РЖХ, 1964, 24H146.
. 92. R. Aries. Англ. пат. 790994;
РЖХ, 1960, 62445.
93. G. В 1 a c k e, E. D e p p,
B. Corson. J. org. Chem. 14, 14
. (1949).
94. J.. H e a p, W. Jones,
J. Speakman. J. Amer. Chem.
Soc., 43, 1936 (1921).
95. С. В i s w e 1 I, W. W i r t h.
Пат. США 2109954; Zbl., 1938, I,
4382.
96. M. M u e 1 1 e г. Пат. США 2586555;
С. A., 46, 9127 (1952).
97. J. О g i I v i e, A. Sweet. Пат.
США 2415147; С. A., 41, 2754 (1947).
98. Франц, пат. 976594; С. А., 47,
9367 (1953).
99. R. Lekberg, R. Jensen,
W. Bui ter. Пат. США 3313821;
. С. А., 67, 90685 (1967).
100. F. С i s 1 a k, W. Wheeler.
Пат. США 2396457; С. А., 40,
3142 (1946).
101. Англ. пат. 718007; J. Appl. Chem.,
5, 1781 (1955).
102. Е. Р 1 a z е к, H.Kozdro-
j о w n a. Roczniki Chem., 25,
509 (1951); С. А., 48, 5863 (1954).
103. F. С i s I а к, W. W h е е 1 е г.
Пат. США 2522163; С. А., 45, 8048
(1951).
104. М. К й 1 к a. J. Am. Chem. Soc.,
68, 2472 (1946).
105. В. Г. X о м я к о в, С. С. Кру-
гл и ко в, В. М. Б ере-
з о в с к и й. ЖОХ, 28, 2898 (1958).
106. В. Г. X о м я к о в, С. С. Кру-
гл и к о в, Н. А. И з г а р ы-
шев. ДАН СССР, 115, 557
(1957).
107. F. С i s I а к, W. W h е е 1 е г.
Пат. США 2437938; С. А., 42, 4204
(1948).
108. G. М а у и г п i к, A. Mos-
ch et to, Н. Bloch, J. S c u-
d i. Ind. Eng. Chem., 44, 1630
(1952).
109. D. Hadley. Англ. пат. 790937;
РЖХ, 1960, 70536.
110. D. Н a d 1 е у, В. W о о d. Англ,
пат. 777746; РЖХ, 1959, 39756.
111. Н. К У х а р ч и к, А. Ж в а к о-
в a. Collect. Czechosl. Chem.
Comm., 28, 55 (1963).
112. E. С. Ж Д а и о в и ч, И. Б. Че к-
марева, Н. А. Пре-
ображенский. ЖОХ, 81,
3272 (1961).
113. С. Р. Р а ф и к о в, Б. В. С у-
воров, Б. А. Ж у б а н о в,
М. И. Хмур а, М. В. П р о-
к о ф ь е в а, ДАН СССР, 126,
1286 (1959).
114. Б. В. С у в о р о в, С. Р. Р а-
ф и ко в, Б. А. Ж у б а н о в,
М. И. Хмура. Авт. свид. СССР
119878; РЖХ, 1960, 35817.
115. Б. В. С у в о р о в, А. Д. Ka-
ra р л л ц к и й, Т. А. А ф а-
насьев, О. Б. Лебедева,
А. И. Л о й ко, В. А. Сер а-
зетдинова. ХГС, 1969, 1024.
116. А. Е. Ч и ч и б а б и и, П. А.
; Мошкин. ЖРФХО, 54, 611
(1922—1923).
.7i;\117. W. R е р р е, Н. К г z i k а 1-
la, Е. W о е d а п. Пат. ФРГ
858399; С. А., 47, 12810 (1954).
'•5'418. К. Yamomoto. Яп. пат. 503
(1951); С. А., 47, 3883 (1953).
•’‘J119. А. Е. Ч и ч и б а б и и, П. А.
£<-• Мошкин, Л. С. Тяжело-
гч’’ ' В а. ЖРФХО, 54, 413 (1920); J.
prakt. Chem., [2], 107, 132 (1924).
‘bS-420. R. F r a n k, I. В 1 e g e n,
; - R. D e a r b о r n, R. N e у e r s-
' 'V; F. W о о d w а г d. J. Am. Chem.
Soc., 68, 1368 (1946).
-..'>121. J. Dunn. Англ. пат. 706816,
-ф-.‘ 742268; J. Appl. Chem., 4, 635
-W’ (1954); C. A., 50, 16874 (1956).
Ж 122. H. Narasaki, H. Suzuki.
ч,:-' Tokio Kogyo Shikensho Hokoku,
A- 52, 8 (1957); C. A., 51, 12089 (1957).
123. S. Kudo. Statist. Quality cont-
’•..t rol, 9, 180 (1958); РЖХ, 1959,
11834, см. также 1960, 42743.
124. W. Bamford. Англ. пат.
758076; РЖХ, 1960, 10331.
125. Англ. пат. 749718; С. А., 51,
11396 (1957).
126. М. И. Ф а р б е р о в, Б. Ф.
Уставщиков, А. М. К у-
т ь и н, Т. П. В е р н о в а,
Е. В. Ярош. Изв. высш, учебн.
завед., хим. и хим. техн., 1958,
г№ 5, 92; РЖХ, 1960, 58065.
127. К- Т а к э б а, Т. С а т о. Яп. пат.
6118 (1959); С. А., 54, 14274
(1960).
128. С. S t о о р s, С. В е с к е г.
Пат. США 2745833; РЖХ, 1959,
68715.
129. Р. Т э р а д а, К. Т а к э д а.
Яп. пат. 3071 (1959); РЖХ, 1961,
5Л182.
130. К. Т a k е b а, Т. Т е г a d а,
Т. Sato. Пат. США 2935513;
С. А., 55, 573 (1961).
131. J. Mahan. Пат. США 2706730;
С. А., 50, 2683 (1956).
132. Н. К г z i к а 1 1 а, К. Мег-
к е 1. Пат. ФРГ 918030; С. А., 52,
12931 (1958).
326
133. X. К о б о я с и. Яп. пат. 1134
(1956); РЖХ, 1959, 12886.
134. К. А г a i, Н. О s u к а, К- Т а-
nabe, К. Teramoto,
1. Ichikizaki. J. Chem.
Soc. Japan, 57, 495 (1954); РЖХ,
1955, 45897.
135. T. Jordon. Ind. End. Chem.,
44, 332 (1952).
136. Б. Ф. Уставщиков, M. И.
Фарберов, A. M. Ку-
тьин, Г. С. Левека я.
Учен, записки Ярослав, техн, инет.,
1960, № 5, 71; РЖХ, 1961,
13Л58.
137. X. С о б у э, К- Т о м и т а,
Я. С v м и д а. Яп. пат. 13081
(1963)'; РЖХ, 1965, 17Н169.
138. Т. Kato. Bull. Chem. Soc. Ja-
pan, 34, 636 (1961); C. A., 56,
10088 (1962).
139. T. Kato, Y. Tsu no d a. Ko-
gyo Kagaku Zasshi, 63, 1278 (1960);
C. A., 56, 10091 (1962).
140. W. Schwarz e. Пат. ФРГ
1010524; РЖХ, 1959, 36086.
141. W. Schwarz e. Пат. ФРГ
936447; РЖХ, 1957, 49314.
142. G. I 1 1 i c h. Пат. США. 2905688;
С. A., 54, 2369 (1960).
143. Б. Ф. У с т а в щ и к о в, Т. С.
Титова, Е. В. Дегтя-
рев, М. И. Фарберов.
ЖПХ, 39, 1388 (1966).
144. Т. С. Т и т о в а, Б. Ф. У с т а в-
щ и к о в, М. И. Фарберов,
Е. В. Дегтярев. ЖПХ, 42,
910 (1969).
145. J. Zundel. Франц. пат.
1509120; РЖХ, 1969, 1IH352.
146. W. S с h w а г z е. Пат. ФРГ
1071085; РЖХ, 1961, 13Л187.
147. К. U d a, A. S a k u г a i,
К. Sakabibara. Яп. пат.
20555 (1965); С. А., 64 , 2069 (1966).
148. R. Benner. Пат. США 2829144;
РЖХ, 1960, 2184.
149. М. Mueller. Пат. США
2834786; С. А., 52, 16377 (1958).
150. R. Gebauer. Пат. ГДР 9462;
РЖХ, 1957, 35696.
151. S. Heogewerff, W. v а п
Dorp. Lieb. Ann., 204, 117
(1880); 207, 219, 226 (1881); Ber.,
12, 747 (1879); 14, 974, (1881).
152. И. П о л я к о в. ЖПХ, 22, 771
(1949).
153. Ю. С. 3 а л ь к и н д, В. В. Ко-
са р е в. ЖОХ, 5, 879 (1937).
154. W. S t i х, S. В u 1 g a t s с h.
Ber., 65, 11 (1932).
155. A. W. Hawkinson, A. E I-
ston. Пат. США 2371691;
• С. A., 39, 4336 (1945).
156. С. С. К P у г л и к о в, В. Г.
Хомяков, Н. Г. Б а х ч и-
сарайцьян. Авт. свид. СССР
140062; РЖХ, 1962, 10Л117.
157. В. Г. X о м я к о в, Н. А.
Дзбаиовский, Л. Д. Б о р-
х и. Труды Вс. н. и. хим. реакт.
особо ч. х. в-в, 29, 304 (1966); С. А.,
67, 116796 (1967).
158. W. W i г z. Пат. ФРГ 923914;
РЖХ, 1957, 49313.
159. VendeKamp, Sletzin-
g е г. Пат. США 2392437; С. А.,
40, 2473 (1946).
160. A. S t о с к е г, О. М а г t 1,
G. S с h г е i пег. Швейц. пат.
504437; РЖХ, 1971, 23Н332.
161. А. А. Кондрашова, Л. О.
Ш н а й д м а н, С.В.Бала-
цепко, Е. С. Ж Д а н о в и ч.
Авт. свид. СССР 132225; РЖХ,
1961, 15Л385.
162. В. И. X м е л е в с к и й, В. Д.
Н и к и т и н. Медиц. пром. 1951,
№ 4, 23.
163. А. Н е у m о n s. Пат. ФРГ
912216; С. А., 52, 10213 (1958).
164. J. О’В г о с h t а. Пат. США
2999094; С. А., 56, 2432 (1962).
165. S. К- Saha. J. Indian Chem.
Soc., Ind. & News Ed., 19, 95, 100
(1956); C. A., 52, 18407 (1958).
166. M. L а г r i s о n. Пат. США
2475969; С. A., 44, 2038 (1950).
167. F. P о r t e г, M. В u m p us,
J. Cosby. Пат. США 2513251;
С. A., 44, 8380 (1950).
168. W. Schwarz e. Пат. ФРГ
901648; С. A., 52, 12931 (1958).
169. Бельг, пат. 473726; С. А., 43,
3979 (1949).
170. М. Mueller. Пат. США
2436660; С. А., 42, 4203 (1948).
171. W. Swoetosl awski,
J. В i а 1 е к, А. В у 1 i с k i,
A. Kotarski, J. Malczyn-
s k i, K. D e b n i c k i. Польск.
пат. 44195; англ. пат. 979761;
РЖХ, 1965, 1Н78; С. А., 62, 16207
(1965).
172. J. В i а 1 е k, J. Malczy п-
s к 1. Chim. et ind., 91, № 1, 69
(1966).
173. Л. П. Юрки и а, Н. Д. Р у-
с я н о в а, Н. В. Мал ыше-
в а. Сб. «Хим. прод. коксования
углей востока СССР», ВНИУИ,
1967, № 4, 251; авт. свид. СССР
191562; С. А., 68, 104997 (1968).
174. J. Kaufman. J. Am. Chem.
Soc., 37, 497 (1945).
175. H. Mitchel 1. J. Nutl. Proc.
Acad. Sci. U. S., 34, 1 (1948).
176. А. Браунштейн. ДАН СССР,
65, 715 (1949).
177. A. В u t e n a n d t, W. W e i-
d el, H. S c h 1 ossb erger.
Naturforschung, 48, 242 (1949).
178. J. H a г r i s, F. В i n n s. Na-
ture, 179, 475 (1957).
179. S. К e i m a t s u, K. Yoka-
t a, I. S a t о d a. J. Pharm.
Soc. Japan, 53, 994 (1933).
180. A. T r u c h a n, J. D avid-
son. Пат. США 2993051; РЖХ,
1962, 13Л296.
181. E. С. Ж Д а и о в и ч, И. Б.
Чекмарева, Т. С. Но во-
327
покровская, Н. А. Пре-
ображенский, ЖОХ, 32,
2828 (1962).
182. Е. С h е г b u 1 i е z, F. L а п-
d о 1 t. Helv. Chim. Acta, 29, 1438
(1946).
183. W. Hoefl ing, D. E i 1 h a-
u e r, G. R ec k 1 i n g. Франц,
пат. 1189995; С. A., 63, 583
(1965).
184. И. Б. Чекмарева, Е. С.
Жданович, Т. С. Н о в о-
покровская, Н. А. Прео-
браженский. ЖПХ, 35, 1157
(1962).
185. О. Lusting. Пат. США 2752355;
РЖХ, 1958, 33593.
186. J. Couch, Ch. Krewso n.
Пат. США 2453496; С. А., 43,
1811 (1949).
187. Н. Rosenberg. Пат. США
2446957; С. А., 43, 694 (1949).
188. И. А. А р х и п о в а, С. Р. Ра-
фи к о в, Б. В. Суворов.
ЖПХ, 35 , 389 (1962).
189. A. G а 1 a t. J. Am. Chem. Soc.,
70, 3945 (1948).
190. E. G a s s о n, D. H a d I e y.
Англ. пат. 777517; С. A., 52,
7361 (1958).
191. И. Б. Ч e к м a p e в а, Е. С.
Жданович, Т. А. Л у щ и к,
Н. А. Преображенский,
ЖОрХ, 1, 375 (1965).
192. В. В. Ефр ем о в. Пеллагра. М.
Биомедгиз, 1934.
193. L. L a s z t. Witaminforsch., 11,
76 (1941).
194. D. М с К е n z i е, L. М а 1 е-
n е, S. К u s h е г, J. О 1 е-
s о п, Y. S u b а г о w. J. Lab.
Ciin. Med., 33, 1249.(1949).
195. С. Е 1 v е h j е m. Physiol. Rev.,
20, 249 (1940).
196. R. W i 1 1 i a m s, R. Ea ki n,
E. Beerstecher, W. Shi-
v e. The Biochemistry of B-Vita-
mins, New York, 1950.
197. О. M e у e r h о f, P. О h 1 m e-
y e r. Biochem. Z., 290, 334 (1937).
198. H. von Euler. Angew. Chem.,
50, 831 (1937).
199. F. S c h I e n k. In the Enzymes,
vol. 2 part 1, p. 263, New York,
1951.
200. H. v о n Euler, F. Schlenk.
Z. physiol. Chem., 246, 64
(1937).
201. N. К a p 1 a n, S. С о 1 о w i c k,
A. Nason. J. Biol. Chem., 191,
473 (1951).
202. N. К a p 1 a n, M. C i о t t i,
F. S t о 1 z e n b a c h, N. Ba-
ch u r. J. Am. Chem. Soc., 77, 815
(1955).
203. M. P U 1 I m a n, A. S a n P i e t-
r o, S. С о 1 о w i c k. J. Biol.
Chem., 206, 129 (1954).
204. N. К a p 1 a n. V Межд. биохим.
конгресс, симпоз. IV, 5, Москва,
1961.
205. Е. Haas. Bioch. Z., 311, 209
(1942).
206. F. West hei mer, H. F i s-
c h e г, E. С о n n, B.Ven-
n e s 1 a n d. J. Am. Chem. Soc.,
73, 2403 (1951).
207. P. Karrer, Blume r. Helv.
Chim. Acta. 30, 1157 (1947).
208. B. Ke. J. Am. Chem. Soc., 78,
3649 (1956).
209. F. S c h 1 e n k, H. H e 1 1 s t-
r о m, H. von Euler, Ber.,
71, 1471 (1938).
210. F. Schlenk. Arkiv Kemi, Mi-
neral. Geol., 12B, 20 (1936); J.
Biol. Chem., 146, 619 (1942).
211. H. von E ii 1 e г, P. К a r-
r e г, В. В e c k e r. Helv. Chim.
Acta, 19, 1060 (1936).
212. F. S c h 1 e n k, H. von Eu-
ler, H. H e i w i n k e 1,
W. G 1 e i n, H. Ny st r om, Z.
physiol. Chem., 247, 23 (1937).
213. R. V e s t i n, F. Sc hl e n k,
H. von Euler. Ber., 70, 1369
(1937).
214. P. К a r r e r, B. R i n g i e r,
J. Buch i, H. Fritzsche,
U. S о 1 m s s e n. Helv. Chim.
Acta, 20, 55 (1937).
215. A. Kornberg, W. Pricer.
J. Biol. Chem., 182, 763 (1950).
216. A. Kornberg. J. Biol. Chem.,
182, 779 (1950).
217. F. Schlenk, H. von E u-
1 e r. Naturwissensch., 24, 794
(1936).
218. D. В г a w n, О. M a g r a t h,
A. Todd. J. Chem. Soc., 1952,
2708.
219. F. Schlenk, Naturwissensch.,
25, 688 (1937).
220. H. von Euler, F. Schlenk,
R. V e s t i n. Naturwissensch.,
25, 318 (1937).
221. О. M e у e r h о f, P. О h I m e-
y e r. Biochem. Z., 290, 334 (1937).
222. H. von Euler, F. Schlenk,
H. H e i w i n k e I, B. Hog-
berg. Z. physiol. Chem.,
256, 208 (1938).
223. S. Williamson, D. Green.
»J. Biol. Chem., 135, 347 (1940).
224. J. N e i 1 a n d s, A. A c e s о n.
J. Biol. Chem., 188, 307 (1951).
225. L. H a у n e s, A. T о d d. J.
Chem. Soc., 1950, 303.
226. L. Haynes, N. H u g h e s,
G. К e n n e г, A. Todd. J.
Chem. Soc., 1957, 3727.
227. J. D a v о 1 1, В. L о. w у. J.
Am. Chem. Soc., 73, 1650 (1951).
228. J. Gul land, E. Holi-
day. J. Chem. Soc., 1940, 746.
229. H. В r e d e r e c k, E. В e r-
ger, J. Ehrenberg, Ber.,
73, 269 (1940).
230. P. Levene, R.Tipson.
J. Biol. Chem., 121, 131 (1937).
231. F. C r a m e r, G. Wei mann-
Chem. Ind., 1960, 46.
328
232. H. G r u nze, W. К о г а п-
s к у. Angew. Chem., 71, 407
(1959).
233. J. В a d d i 1 е у, A. T о d d.
J. Chem. Soc., 1947, 648.
234. A. M. Ю p к e в и ч, И. И. К о-
л о д к и и а, Г. С. Евдоки-
мова, Е. Т. Б а ж е и о в а,
Н. А. Преображенский.
Сб. «Хим. орг. соед. фосфора»
(ЖОХ), 1967, 215.
235. А. М. Y u г k е v i с h, 1. I. Ko-
lodkina, L.S.Varshav-
s к a j а, V. I. Borodulina-
S h v e t z, 1. P. R u d a к o-
va, N. A. Preo bra z hen-
ski, Tetrahedron, 25, 477 (1969).
236. H. Khorana, A. Todd.
J. Chem. Soc., 1953, 2257.
237. M. Smith, J. Mo f f a 11,
H. Khorana. J. Am. Chem.
Soc., 80, 6204 (1958).
238. F. Cramer. Angew. Chem., 72,
236 (I960).
239. A. Todd. Gazz. chim. ital., 89,
126 (1954); Chem. Ind., 1958,
170.
240. N. H u g h e s, G. К e n n e r,
A. T о d d. J. Am. Chem. Soc.,
1957, 3733.
241. L. Shuster, N. Kaplan,
F. Stolzenbach. J. Biol.
Chem., 215, 195 (1955).
242. JI. M. Мельникова, В, M.
Березовский. ЖОХ, 40,
918 (1970).
243. М. Н о n j о, U. Furukawa,
N. М о г i у a m а, К. Т а п а-
k a. Chem. Pharm. Bull. Tokyo,
10, 73 (1962); 11, 712 (1963).
244. G. P f e i d e г e r, C.Woen-
c k h a u s, K. S c ii о 1 z,
H. Feller. Lieb. Ann., 675, 205
(1964).
245. G. Woenckhaus. Chem. Ber.,
97, 2439 (1964).
246. I. Gohri ng, A. W a c k e r,
G. Pfleiderer. Biochem. Z.,
339, 514 (1964).
247. C. \V о e n c k h a u s, M. V о 1 z,
G. P f 1 e i d e г e r. Z. Natur-
forsch., 197, 467 (1964).
248. C. Woenckhaus, G. Pflei-
derer. Biochem. Z., 341, 4951
(1965).
249. C. W о e n c k h a u s, M. V о 1 z.
Chem. Ber., 99, 1712 (1966).
250. K. G о b be I er, C. Woen-
ckhaus. Lieb. Ann., 700, 180
(1966).
251. J. Rowen, A. Kornberg.
J. Biol. Chem., 193, 497 (1951).
252. T. W a n g, N. К a p 1 a n. J.
Biol. Chem., 206, 311 (1954).
253. S. F u r b e r g, Acta chem. scand.,
4, 751 (1950).
254. J. Siegel, G. Montgome-
ry, R. В о c k. Arch. Biochem.
Biophys., 82, 288 (1959).
255. O. J a r d e t z k y, N. W a d e-
Jardetzky. J. BioL Chem.,
241, 85 (1966).
256. S. F r e e d, E. N e у f a c h,
L. T u m e г m a n. Biochim. Bio-
phys. Acta, 143, 432 (1967).
257. J. J acobus. Biochemistry, 10,
161 (1971).
258. N. Oppenheimer,
L. А г n о I d, N. К a p 1 a n.
Proc. Natl. Acad. Sci., 68, 3200
(1971).
259. T. N a k a m о t o, B.Venne-
s 1 a n d. J. Biol. Chem., 235, 202
(1960).
260. J. Cornfort h, G. Ry-
back, G. P о p j a k, C. D о n-
n i n ger, G. Schroepfer.
Biochem. Biophys. Res. Communs,
9, 371 (1962).
261. L. Rosenberg, D. Arnon.
J. Biol. Chem., 217, 361 (1955).
262. E. К r e b s, G. R a f t e r,
J. Junge. J. Biol. Chem., 200,
479 (1953).
263. D. D e n n i s, N. К a p 1 a n.
J. Biol. Chem., 235, 810 (1960).
264. В. V a 1 1 e e, W. W a c k e r.
J. Am. Chem. Soc., 78, 1771 (1956).
265. K. D a 1 z i e 1. Acta chem. scand.,
12, 459 (1958); Biochem. J., 80,
440 (1961).
266. J. M о у 1 e, M. D i x о n. Bio-
chim. Biophys. Acta, 16, 434 (1955);
Biochim. J., 63, 548 (1956).
267. E. E n g 1 a r d, H. В r e i g e r.
Biochim. Biophys. Acta, 56, 571
(1962).
268. D. S с о t t, S. С о h e n. Bio-
chem. J., 55, 23 (1953).
269. J. W a c h s m a n. J. Biol. Chem.,
223, 19 (1956).
270. H. R о t h s c h i 1 d, E. В a r-
r о n. J. Biol. Chem., 209, 511
(1954).
271. F. L у n e n, L. Wessely,
O. W i e 1 a n d, L. R u e f f.
Angew. Chem., 64, 687 (1952).
272. J. Stern. Biochim. Biophys.
Acta, 26, 448 (1957).
273. M. Kern, E. R a c k e r. Arch.
Biochem. Biophys., 48, 235 (1954).
274. R. В 1 a k 1 e у, В. M c D о u-
g a 1 1. J. Biol. Chem., 236, 1163
(1961).
275. T. S t a d t m a n, P. E 1 1 i-
o t t. J. Biol. Chem., 228, 983
(1957).
276. I. S t e r n, A. del Cam-
pi 1 1 o, A. L e h n i n g e r. J.
Am. Chem. Soc., 77, 1073 (1955).
277. M. Д и к co н, Э. У э б б. Фер-
менты. M., «Мир», 1966.
329
ГЛАВА VIII
ОКСИМЕТИЛПИРИДИНОВЫЕ ВИТАМИНЫ И КОФЕРМЕНТЫ
ПИРИДОКСИН, ПИРИДОКСАЛЬ, ПИРИДОКСАМИН
К оксиметилплридиновым витаминам относятся пиридоксин (I), пири-
доксаль (II) и пиридоксамин (III) — витамины группы Вс—соединения,
являющиеся производными р -окси-р '-оксиметил-а-пиколина.
СН,ОН
НО. J4..CH.OH
СН3 N
СН3 N
ш
CHjNH,
но Тщон
Эти витамины различаются функциональными группами в у-положении
пиридинового ядра — оксиметильной, формильной или аминометильной.
К оксиметилпиридиновым витаминам относятся пиридоксаль и пири-
доксамин; пиридоксин следует рассматривать как провитамин, так как он
проявляет свои витаминные свойства не непосредственно, а превращаясь
в организме в пиридоксаль или пиридоксамин.
Пиридоксин — пиридоксол, адермин, 2-метил-3-окси-4,5-бис-ок-
симетилпиридин (I) — представляет собой бесцветные призматические
кристаллы с т. пл. 160° С (с разл.), обладающие слабым горьким вкусом
[1, 2]. Он хорошо растворим в воде, спирте, хуже — в ацетоне, плохо раст-
ворим в эфире и хлороформе; легко сублимируется; образует пикрат с
т. пл. 156° С [3].
£70*
220 260 300 340
Рис. 6. Спектр поглоще-
ния пиридоксина в воде
при различных pH.
Гидрохлорид пиридоксина кристаллизуется в бесцветных призмах
с т. пл. 204—208° С (с разл). Он хорошо растворим в воде (1:4,5), значитель-
но хуже — в этиловом спирте (1:90 мл), отчасти растворим в ацетоне. Так
же как и основание, он хорошо сублимируется [1,2]. Водные растворы этого
соединения имеют pH 3,2. Спектр поглощения характеризуется максимума-
ми, интенсивность которых находится в зависимости от концентрации
водородных ионов (рис. 6). Так, при pH 5,1 максимумы поглощения пири-
доксина следующие: I—256 нм, II—292,5 нм и III—327,5 нм. При
pH 4 I максимума нет, интенсивность максимума II возрастает, а III —
уменьшается. При pH 6,75 максимум I по интенсивности возрастает, II
максимума нет, III максимум также возрастает [1, 4], что видно из рис. 6.
330
При pH 7,0 максимум I имеет 253 нм, III — 325 нм; в 0,1 н. NaOH макси-
мум I — 245 нм и III — 308 нм (е 7,0-103).
Из растворов пиридоксин хорошо осаждается фосфорновольфрамовой,
кремневольфрамовой и серной кислотами и не осаждается солями свинца и
ртути. Из нейтральных или кислых водных растворов пиридоксин адсорби-
руется на древесном угле, фуллеровой земле [5], цеолите, из которых вы-
мывается раствором гидрата окиси бария или бутиловым спиртом.
Пиридоксаль — 2-метил-3-окси-4-формил-5-оксиметилпиридпн
(И) — кристаллизуется в виде гидрохлорида с т. пл. 173—174° С (с разл.)
[61 и в виде оксима с т. пл. 225—226° С [7]. Растворимость гидрохлорида
пиридоксаля: 1 г в 2 мл воды, 1,7 г в 100 мл 95%-ного спирта. pH 1 %-ного
водного раствора 2,65.
Пиридоксамин — 2-метил-3-окси-4-аминометил-5-оксиметилпи-
ридин (III) — бесцветные кристаллы с т. пл. 193—193,5°С; гидрохлорид
пиридоксамина имеет т. пл. 226—227° С (с разл.) [7]. Растворимость
гидрохлорида пиридоксамина: 1 г в 2 мл воды, 0,65 г в 100 мл 95%-ного
спирта. pH 1 %-ного водного раствора 2,4.
ХИМИЧЕСКИЕ СВОЙСТВА ОКСИМЕТИЛПИРИДИНОВЫХ ВИТАМИНОВ
Пиридиксин (I), пиридоксаль (II) и пиридоксамин (III) устойчивы к
концентрированной соляной кислоте и к нагреванию в щелочных растворах.
Они не стабильны к окислителям (перекись водорода, марганцовокислый
калий) [8]; по отношению к разбавленной азотной кислоте пиридоксаль более
устойчив, чем пиридоксин. Растворы пиридоксина, пиридоксаля и пири-
доксамина не устойчивы к свету 19].
С хлорным железом растворы пиридоксина дают оранжево-красную ок-
раску. Ацетильное производное пиридоксина способно перегоняться в
высоком вакууме (при 85—90° С и 10~4 мм).
В молекуле пиридоксина первичный гидроксил оксиметильной группы
в 4а-положении обладает значительно большей реакционной способно-
стью, чем такой же гидроксил в 5а-положении. Это обеспечивает способ-
ность пиридоксина образовывать димер и возможность перехода оксиме-
тильной группы пиридоксина в положении 4 в формильную (пиридоксаль)
или аминометильную (пиридоксамин) группу при неизменности первичного
гидроксила в 5а-положении, чем обусловливается способность витаминов
группы В6 вступать в разнообразные обратимые реакции.
Пиридоксин полимеризуется в нейтральных водных растворах при
нагревании до 120° С с выделением одной молекулы воды, получающейся
за счет гидроксила 4-оксиметильной группы одной молекулы пиридоксина
и атома водорода четвертичного атома азота промежуточной формы другой
молекулы, образуя димер IV [10] с т. пл. 205—109° С и при дальнейшем
нагревании — тример. Пиридоксин с йодистым метилом образует йодмети-
лат, который при обработке углекислым серебром дает соединение бетаи-
нового типа — N-метилпиридоксин (V) [11].
IV
331
По своим гидроксильным группам окспметилпиридиновые витамины
образуют простые и сложные эфиры. С хлорапгидридами или ангидридами
органических кислот получены многочисленные производные — ацетаты,
бензоаты 112, 13] и др., а например, с высшими жирными кислотами —
жирорастворимые сложные эфиры: трилинолеил- [14, 15], тристеаринил
[15], 5а-моно-[16], 3,4а-ди- [13] и трипальмитинилпиридоксин (т.
пл. 73—74° С) [14, 15, 17], 5а-моно- (т. пл. 74° С) и 3,5а-дипальмитииил-
пиридоксаль (т. пл. 48—51 °C) [14, 16], трипальмнтинилпиридоксамин
(т. пл. 102—103° С) [14] и др.
Были получены различные сложные эфиры с никотиновой кислотой
(некоторые из них оказались гипотензивными, сосудорасширяющими и
активными для предупреждения атеросклероза и ожирения) [18, 19]:
5а-О- (VI) [18—22], 4а,5а-ди-О-[21 ] и три-О-никотиноилпиридоксин [22,
23], 5а-О-моно-, Ы-моно-[21], 5а-О, N-ди- [21] и триникотиноилпиридо-
ксамин (VII) [22, 23], 5а-О-никотиноилппридоксаль [21] и др.
Из сложных эфиров с минеральными кислотами следует отметить фос-
форные эфиры.
Пиридокси н-5а-ф о с ф а т однозамещенный, кристаллизуется с
одной молекулой воды; т. пл. 123—126° С. Вещество представляет собой
бесцветные кристаллы, хорошо растворимые в воде, трудно в спирте, почти
нерастворимые в эфире, хлороформе и ацетоне. Получение его описано на
с. 360.
П и р и д о к са л ь-5а-ф ос ф а т и п и р и до к с а м и н-5а-ф о сфа т
являются коферментами; они описаны на с. 360, 361.
Гидроксил оксиметильной группы положения 5 оксиметилпиридиновых
витаминов при действии хлорсульфоновой кислоты дает сернокислые эфиры—
образуются пиридоксин- (VIII), пиридоксамин- (IX) и пиридоксаль-5а-
сульфаты (X) [24, 25]
Для этих соединений характерна антивитаминная активность.
Гидроксильные группы оксиметилпиридиновых витаминов способны
обменивать свой кислород на серу. При действии на пиридоксин (I) сероуг-
леродом в щелочном растворе получается 4-ппридоксинтпол (XI) [26]. Реак-
ция 2-метил-3-окси-4,5-бис-бромметилпиридина с сероводородом приводит
к тиоэфиру бис-пиридоксина (XII) [27, 28]. бис-Пиридоксиндпсульфид—
бис-(2-метил-3-окси-4-оксиметил-5-пиридилметил) дисульфид, «пиритиоксин»
(XIII) с т. пл. 221 —222° С [29—32 ]—обладает противоэпилептическим
действием [33], ослабляет некоторые симптомы мозговой недостаточности
и др.
332
Для однозначного получения этого соединения с замещением атома
кислорода по гидроксильной группе положения 5а реакцию проводят
при взаимодействии полисульфида [34, 35] или ксантогената калия [29,
33] с 2-метил-3-окси-4,5-бас-бромметилпиридином [29, 35] или с 3,4а-О-
изопропилиденпиридоксином (XVI) [34] (с последующей обработкой аммиа-
ком). Получен также бпс-пиридоксаминдисульфид [31 ] и бис-пиридоксаль-
дисульфид (XIV) [36] с характерными для него антиконвульсивным,
анальгетическим и антирвотным действием.
Интересно отметить тиоэфиры пиридоксина с липоевой кислотой [37,
38] и с открытой тиольной формой тиамина [39], в частности дисульфидно-
го типа — S-пиридоксинтиаминдисульфид (XV) [40].
СН2ОН 7'3 . NHj
но J^ch2ssc=cn(cho)chi
L Jj CIlXH.OH L if
CH3 N TN CH3
XV
Пиридоксин дает циклические кетали двух типов, например с ацетоном,
стабильный в щелочной среде 3,4а-О-изопропилиденпиридоксин (XVI) с
шестичленным циклом, образованный по фенольной и оксиметильной груп-
пе положения 4 [41—44], и 4а, 5а-О-изопропилиденпиридоксин (XVII)
с семичленным циклом с т. пл. 184—185°С [43].
Циклический кеталь (XVI) образуется в ацетоне с 14% хлористого во-
дорода, а циклический кеталь (XVII) —в ацетоне с 4% хлористого водо-
рода. Строение соединения XVI доказано окислением марганцовокислым
калием с последующим кислотным гидролизом в лактон 2-метил-3-окси-4-ок-
симетилппридин-5-карбоновой кислоты [42].
Пиридоксин (через свой триацетат) при действии 30%-ной перекиси
водорода образует N-окись пиридоксина (XVIII) с т. пл. 145—146° С [45,
46], которая обладает 15%-ной витаминной активностью. N-Окись пиридо-
ксина (XVIII) при окислении двуокисью марганца превращается в N-окись
пиридоксаля (XIX) [46].
333
Эти N-окиси легко восстанавливаются в соответствующие пиридоксин и
пиридоксаль.
Пиридоксаль, как и всякое соединение с альдегидной функцией, спосо-
бен реагировать с бисульфитом натрия, цианидами, аминами, гидроксил-
амином, гидразином и др.
Никотиноилгидразон пиридоксаля (XX) обладает биологической актив-
ностью, проявляя противотуберкулезные свойства in vitro и активность
против рака молочной железы у мышей [47 J. Азометины (шиффовы основа-
ния, XXI) являются первичным продуктом конденсации пиридоксаля с
аминами. Производные пиридоксаля типа XXII обладают пролонгирован-
ным витаминным действием [481.
CH=NR
XXI
Изучена реакция пиридоксаль-5а -фосфата с аминотиолами [49 L
Пиридоксаль способен вступать в реакции конденсации с различными
аминокислотами, образуя многочисленные производные. Реакции конден-
сации пиридоксаля с аминокислотами лежат в основе неферментативного
переаминироваиия, причем в качестве промежуточного акцептора амино-
группы выступает пиридоксамин.
Превращения пиридоксаля (II) и пиридоксамина (III) протекают в
присутствии аминокислот (белковых гидролизатов, например казеина)
при нагревании до 115—120° С. Эти реакции небиологического переамини-
рования [50 J были воспроизведены также с водными растворами /.-глута-
миновой кислоты (XXIII) и пиридоксаля (II) и соответственно са-кетоглу-
таровой кислотой (XXIV) и пиридоксамином (III)
сно соон CHjNHj соон
СНМНз НО 1 СДОН с=о 1
" CHS ^Н,
СНз N СНзСООН снз N CHjCOOH
II XXIII III XXIV
Реакции переаминироваиия протекают с большой скоростью.
Реакция между пиридоксалем, аминокислотами и их имииами подвер-
галась детальному изучению [511.
Обратимые реакции неферментативного переаминироваиия между пи-
ридоксалем и многими аминокислотами, протекающие при 100° С, катали-
зируются солями меди, железа и алюминия. Скорость реакции в зависимо-
сти от применяемой аминокислоты уменьшается в такой последовательности:
большинство аминокислот > фениламии и тирозин > изолейцин > валин > треонин >
> глицин [52].
Азометины пиридоксаля с аминокислотами дают хелатные металличе-
ские комплексы пиридоксилиденаминокислот, для которых предложены
следующие структуры типа XXV и XXVI [531:
334
XXV
Изучено образование металлических хелатов соединений пиридоксаля
с аминокислотами в водном растворе с Zn, Си, Ni типа XXVII [54] и из
пиридоксаля с полиметилендиаминами с Zn, Си, Ni, Со, Fe, Mg типа XXVIII
155]
XXVII
Пиридоксиламины общей формулы типа XXIX:
XXX
где R—остатки 0 -фенилэтиламина, изобутиламина, бензиламина, гиста-
мина, триптамина, тирамина и другие получены конденсацией пиридок-
саля с аминосоединениями при одновременном восстановлении [56]. Пири-
доксиламины типа XXX, где R-остаток аминокислоты, получены из пири-
доксаля и аминокислот через соединения типа шиффовых оснований, кото-
рые затем подвергались гидрированию [57]. Пиридоксиламинокислоты
проявили витаминную активность на крысах в пределах активности вита-
мина В,; и как ростовые факторы оказались не активными для многих микро-
организмов.
Пиридоксин, пиридоксамин и пиридоксаль выводятся из организма
преимущественно в виде продукта дальнейшего окисления пиридоксаля —
ппридокспловой кислоты (XXXI), обладающей малой ростовой актив-
ностью для некоторых микроорганизмов. Пирпдоксиловая кислота (XXXI)
синтетически получается из оксима пиридоксаля [581 или окислением пи-
ридоксаля (II) марганцовокислым калием в нейтральной, а также в слабо-
щелочной среде. Если окисление проводить в кислой среде, то в качестве
продукта реакции образуется пиридоксилактон (ХХХШ). Он обладает
сильной голубой флуоресценцией, по интенсивности превышающей флуо-
ресценцию пиридоксиловой кислоты в 25 раз. При окислении пиридоксина
перекисью водорода при повышенной температуре образуется лактон изо-
пиридоксиловой кислоты (5-пиридоксиловой кислоты) (XXXII); он может
быть получен окислением 3,4а -О-изопропилцденпиридоксина (XVI)
335
Аминометильная группа пиридоксамина (III) способна при продолжи-
тельном нагревании с двуокисью марганца в водном растворе серной или
фосфорной кислоты превращаться в карбонильную, и из пиридоксамина
при этом образуется пиридоксиловая кислота (XXXI) [59].
Пиридоксин, пиридоксаль и пиридоксамин способны взаимно превра-
щаться один в другой по следующей схеме:
XXXIV
XXXV ш
Пиридоксин (I) окисляется в пиридоксаль (II), затем с аммиаком при от-
щеплении воды переходит в альдегидимин (XXXV) и далее восстанавливает-
ся в пиридоксамин (III). В обратном направлении превращение будет
проходить через реакции окисления, гидратации, отщепления аммиака и
восстановления. В превращениях пиридоксин — пиридоксаль реакции
гидрирования и дегидрирования, возможно, протекают через промежуточ-
ный полуацеталь пиридоксаля (XXXIV). То, что пиридоксаль находится
в циклической полуацетальной форме, следует из тех данных, что 5-окси-
метильная группа пиридоксаля остается инертной в отношении реагиро-
вания с хлоридом дибензилового эфира фосфорной кислоты [41 ].
Для химического определения витамина В6 был применен метод сочета-
ния с диазотированной сульфаниловой кислотой [60]. Получаемые произ-
водные пиридоксина, пиридоксаля и пиридоксамина отличаются по цвету
и могут быть дифференцированы спектроскопически [61 ]. Пиридоксин
определяется в присутствии пиридоксаля и пиридоксамина [62]; этот метод
основан на реакции пиридоксина с 2,6-дихлорхинонхлоримидом [1 ] — реак-
ции, характерной для фенолов со свободным /шрц-положением [63]. Йодме-
тилат пиридоксина не дает окраски с этим реагентом.
СТРОЕНИЕ ПИРИДОКСИНА, ПИРИДОКСАЛЯ И ПИРИДОКСАМИНА
Пиридоксин в виде гидрохлорида (C8HnO3N-HCl) впервые был выделен
в 1932 г. Одаке [64 ] из полированного риса, но витаминные свойства этого
соединения установлены не были. Затем в кристаллическом виде пиридоксин
в 1938 г. выделили из дрожжей, рисовых отрубей и других пищевых про-
дуктов [3, 4, 65—67]. В 1939 г. две группы исследователей—Стиллер,
336
Керештези, Стивенс [1] и Кун, Вендт и Вестфаль [68—701 — установили
строение пиридоксина как 2-метил-3-окси-4,5-бас-оксиметилпиридина. Та-
кое строение доказывается тем, что молекула пиридоксина содержит три
активных атома водорода, определенных по методу Церевитинова, причем
в качестве производных пиридоксина образуются его сложные эфиры —
триацетат [71 ] и трибензоат [11, из чего следует, что все три имеющиеся
атома кислорода находятся в форме гидроксильных групп. Отсутствие
активного атома водорода у азота и невозможность образования ацильных
производных по азоту доказывает его третичный характер и нахождение в
ядре [2).
Характер гидроксильных групп определяется тем, что диазометан обра-
зует монометиловый эфир пиридоксина по фенольному гидроксилу, имею-
щему особенно подвижный атом водорода, не затрагивая двух других гид-
роксильных групп. В отличие от пиридоксина (I) его метиловый эфир-
ах XXVI) не дает цветной реакции с хлорным железом и не сочетается с
диазотированной сульфаниловой кислотой; эта реакция характерна для:
свободных фенольных или енольных групп. При ацетилировании уксусным:
ангидридом в присутствии пиридина метиловый эфир образует диацетил-
производное метилового эфира пиридоксина (XXXVII) по двум другим
гидроксильным группам [11:
СИ,ОН
СН3 N
СН3 N
XXXVI
СН2ОСОСНз
(СН,СО),О СН.О Г.СН.ОСОСН,
СН3 N
XXXY1I
При обработке метилового эфира пиридоксина (XXXVI) бромистоводо-
родной кислотой происходит гидролиз эфира и замещение двух гидроксиль-
ных групп галогеном. Эти данные указывают на то, что одна гидроксильная
группа имеет фенольный характер, а две другие — алифатический.
Фенольная гидроксильная группа замещает р-положение пиридинового
ядра, что было установлено на основании подобия максимумов спектра
поглощения молекулы по сравнению с р-оксипиридином 11, 68), а также
по цветной реакции (синяя окраска) с реактивом на фенольные и на р -окси-
пиридиновые группы, не дающим реакции с а- или у-оксппиридинохМ [681
(реактив, состоящий из смеси молибденовокислого натрия, фосфорномолиб-
деновой кислоты, фосфорной кислоты и гидроокиси лития [72]).
В молекуле пиридоксина (I) атом водорода а-положения (пара-положе-
ние к гидроксильной группе) не замещен, что установлено по образованию
синей окраски с 2,6-дихлорхинонхлоримидом — специфическим реактивом
для открытия подобного рода соединений [73].
При окислении метилового эфира пиридоксина (XXXVI) двумя атомами
кислорода—действием марганцовокислым калием в нейтральном растворе—
образуется лактон (XXXVIII), чем устанавливается расположение алифа-
тических гидроксильных групп в p-и у-положениях. При окислении метило-
вого эфира пиридоксина (XXXVI) четырьмя атомами кислорода —действи-
ем марганцовокислым барием — образуется 5-метоксп-6-метплцпнхомероно-
вая кислота (XXXIX) [1, 69, 70, 741, переходящая при обработке уксусным
ангидридом в ангидрид замещенной пинхомероновой кислоты (XLIII).
Эта реакция, а также способность образовывать с резорцином краситель
фталеинового типа показывает, что карбоксильные группы, ведущие свое
происхождение от оксиметил ьных групп, расположены рядом. Декарбокси-
лирование натриевой соли 5-метокси-6-метилцинхомероновой кислоты
(XXXIX), которая была получена встречным синтезом [75], приводит к.
получению ₽-метокси-а-пиколина (XL).
337
сн3<э
^-o
CH, I
^G°KMnO< CH^
pH7
CHOH COOH
-С-сн»°н в^мпоД снр^соон
снд
СН, N
XXXYII1
CH, N
XXXYI
|КМпО4, KOH
,-o
CO \
ch,oAxo
COOH
CHjOA. cooh
CH, N
XXXIX
lfclL,CO)p
Lo
CO \
co
-cq
CH, N'
XL
N
XU!
-co,,-i^o X <
* 2HOOC N
XLI
XLIII
При окислении метилового эфира пиридоксина (XXXVI) семью
атомами кислорода — действием марганцовокислым калием в щелоч-
ном растворе — образуется 3-метокси-2,4,5-пиридиитрикарбоновая кис-
лота (XLI) [681, дающая характерную для пиридин-а-карбоновых
кислот красную окраску с сульфатом железа. Эта третья карбоксильная
группа ва-положении происходит от метильной группы и легко декарбокси-
лируется; так, при ангидридизации соединения XLI был получен ангидрид
З-метокси-4,5-пиридиндикарбоновой кислоты (XLII) [68, 691.
Окончательно строение пиридоксина как 2-метил-3-окси-4,5-быс-окси-
метилпиридина (I) было установлено по идентичности синтетически полу-
ченного пиридоксилактона (XXXII), его метилового эфира (XXXVIII)
и 2-карбокси-З-метоксицинхомероиовой кислоты (XLI) с такими же соеди-
нениями, полученными окислением пиридоксина. Синтез пиридоксина осу-
ществили в 1939 г. Харрис, Стиллер и Фолькерс [751.
Строение пиридоксамина и пиридоксаля вытекает, естественно, из реак-
ций их получения из пиридоксина и доказывается их синтезом из метилово-
го эфира пиридоксина.
СИНТЕЗ ПИРИДОКСИНА, ПИРИДОКСАЛЯ И ПИРИДОКСАМИНА
Многочисленные синтезы пиридоксина основаны главным образом или
на непосредственном получении пиридинового цикла из алифатиче-
ских соединений с заместителями, которые образуют функциональные
группы в положениях 4 и 5, метильную группу в положении 2 молекулы
пиридоксина или могут быть в них преобразованы, или на получении гетеро-
циклов и системы конденсированных циклов, которые способны превращать-
ся в пиридиновый цикл, при этом имевшиеся у них заместители пригодны
для дальнейших преобразований в заместители молекулы пиридоксина.
Построение молекулы пиридоксина из Простых пиридиновых соедине-
ний трудно осуществимо из-за сложности введения углеродсодержащих
заместителей ври у-положения молекулы пиридина.
Синтезы пиридоксамина и пиридоксаля основаны на превращениях уже
готовой молекулы пиридоксина.
1. Синтез пиридоксина через производные нитрила никотиновой кислоты
В этом синтезе гетероциклическое ядро с заместителями, которые затем
преобразовываются в необходимые функциональные группы, отвечающие
молекуле пиридоксина, создается посредством конденсации р-дикетона—
алкоксиацетилацетона (XLIV) и цианацетамида (XLV). Направление реак-
ции, которая может идти в двух направлениях [761, зависит от условий ее
проведения и от относительно большей активности одной из карбонильных
групп, которая вступает в конденсацию предпочтительно с активированной
метиленовой группой цианацетамида. При конденсации алкоксиацетилаце-
338
тона (XLIV) с цианацетамидом (XLV) в спирте при каталитическом действии
пиперидина (или диэтиламина, аммиака) образуется необходимый для после-
дующего синтеза 2-метпл-4-алкоксиметил-5-циан-6-пиридон (XLVI,
R = С2Н5) с выходом 75—81 % (для XLVI, R == С2Н5, т. пл. 209—210° С)
[75]; его изомер (XLVII) при этой реакции получается с выходом 15% [77]:
XLVH
R=CHS или CjH5
CH2OR RCN
1 1 1
с=о сн2
CHjCOCH, 1 CONHt
XLIV XLV
CH.OR
XLVI
Таким же путем были получены метильный (т. пл. 242° С) [78—80],
пропильный и фенильный гомологи эфира (XLVI, R = СН3, С3Н7, СвН5)
[81, 82].
Применяемый в этом синтезе этоксиацетилацетон (XLIV, R = СгН5)
может быть получен из хлоруксусной кислоты с общим выходом около 20%
[83, 841:
C2HsONa СгН5ОН
С1СН2СООН--------> С2Н6ОСН2СООН-------->
(CHS)2CO
С2Н5ОСН2СООС2Н5--------> QH5(XH2COCH2COCH3.
Цианацетамид (XLV) синтезируют из циануксусного эфира с выходом
86% [85].
2-Метил-4-алкоксиметил-5-циан-6-пиридон (XLVI) получают также
при конденсировании 2-амино-4-кето-5-алкоксиметил-2-пентена (XLVIII)
с малоновым динитрилом [86]. Дальнейшее превращение 2-метил-4-алкокси-
метил-5-циан-6-пиридона (XLVI) в пиридоксин (I) представлено серией
реакций на схеме 75.
Основным промежуточным соединением в различных вариантах синтеза
пиридоксина является 3-амино-4-алкоксиметил-5-аминометил-2-пиколин
(LIII).
В положение 3 молекулы 2-метил-4-этоксиметил-5-циан-6-пиридона
(XLVI, R = С2Н5) в одном из первых синтезов Гаррис и Фолькерс [87,88]
вводили оксигруппу рядом последовательных превращений:
NO2->NH2->OH. Нитрование протекает относительно легко в пара-поло-
жение к оксогруппе, т. е. в положение. 3.
Нитрование пиридона (XLVI) производят дымящейся азотной кислотой
в смеси с уксусным ангидридом при температуре около 50° С. Нитропири-
дон (XLIX, R — С2Н5. т. пл. 164—165°С) образуется с выходом около 64%;
метильный гомолог эфира (т. пл. 210° С) получается с несколько лучшим ре-
зультатом — до 75%. Описано 189] нитрование при температуре 70—80°С
смесью уксусного ангидрида и окисного азотнокислого железа.
Соединение XL IX каталитическим восстановлением превращают в
2-метил-3-амино-4-алкокспметпл-5-циан-6-пиридон (L) [87 ], однако с не-
достаточно высоким выходом. Восстановление нптрогруппы протекает легко
и в присутствии скелетного никелевого катализатора. В жестких условиях
гидрирования одновременно восстанавливаются двойные связи пириди-
нового ядра.
Из этого соединения L дальше получают аминохлорпиридин (LII), из
которого при последующем восстановлении образуется дигидрохлорид
3-амино-4-алкоксиметил-5-аминометил-2-пиколина (LIII); при этомодновре-
менно элиминируется атом хлора, а нитрильная группа восстанавливается
в аминометильную в присутствии 5%-ного палладиевого катализатора на
339
Схема 75
•Синтез пиридоксина через производные нитрила никотиновой кислоты
CHjOR
ЛМ CH.COHN
= - СН О J
CHjOR
СН О
II +
'С'.
сн, nh2
XLVIII
CH,OR
сСО+ $
СН, ° °
СН?0
,CN
СН,
нХ<О
XLV
LVIII
XLVI
R=CH,; C,HS
угле ( т. пл. 195° С для этокси- и 147° С для метоксипроизводного) [79, 881
или никелевого катализатора (в присутствии аммиака) [90].
По другому варианту 2-метил-3-нитро-4-алкоксиметил-5-циан-6-пири-
дон (XLIX) превращают в замещенный 2-хлорнитропиридин (LI) действием
пятихлористым фосфором при нагревании в среде хлорбензола или дихлор-
этана [88, 91 ] или, что значительно удобнее, хлорокисью фосфора в среде
хлорбензола или бензола в присутствии пиридина или диметилформамида
(выход 65—80%) [92—95].
Для 2-хлорнитропиридина (LI, R = CHS) описан одноступенчатый пере-
ход непосредственно к дигидрохлориду 3-амино-4-метоксиметил-5-амино-
метил-2-пиколина (LIII, R = СН3) с лучшими результатами, чем путь синте-
за через промежуточное восстановление нитрогруппы (LI-»-LII-»-LIII)
340
[80, 94,961. Для восстановления, помимо палладиевого катализатора на угле,
показано применение скелетного никелевого катализатора в спиртовок
среде в присутствии аммиака (для гидрирования нитрильной группы) [971.
Дальнейшее превращение соединения LIII в пиридоксин (I) было осу-
ществлено следующими путями.
Замещение аминогруппы в положении 3 на оксигруппу и аминометиль-
ной группы в положении 5 на окспметильную в соединении LIII производится
диазотированием и разложением соли диазония в горячем растворе смеси
серной кислоты и нитрита натрия. Получают эфир пиридоксина (LIV)
[78, 88, 94, 96], который гидролизом соляной кислотой под давлением при
температуре 50° С непосредственно превращают в пиридоксин (I) [78, 88,
94]. Разработан метод очистки соединения LIV на ионообменных смолах
[981. В маточных растворах находится, а при обработке эфира LIV 50 %-ной.
серной кислотой образуется внутренний эфир пиридоксина, который мо-
жет быть гидролизовав в пиридоксин разбавленной соляной кислотой при
175° С [87]. Это один из лучших методов получения пиридоксина.
По другому варианту пиридоксин (I) можно получить из его эфира
LIV кипячением с азеотропной бромистоводородной кислотой с последую-
щим гидролизом 4,5-бцс-бромметилпроизводного пиридоксина (LV) кипяче-
нием в водном растворе и удалением ионов брома обменной реакцией с соля-
ми серебра [88, 99]. Можно диамин (LIII) превратить в монобромметил-
производное (LVI), которое затем подвергнуть гидролизу кипячением с
водой в 3-амийо-4-оксиметил-5-аминометил-2-пиколин (LVII) [100]. Короче
будет путь, если диамин (LIII) непосредственно подвергнуть гидролизу
нагреванием с разбавленной соляной кислотой под давлением при 175° С в
соединение LVII, которое превратить в пиридоксин (I) обработкой нитри-
том натрия в кислом растворе при нагревании (выход 45%) [87, 101 ]. Была
найдено, что при обработке соединения LVII нитритом натрия с серной
кислотой при 80° С в присутствии 2 молей бромистого натрия выход пири-
доксина (I) значительно повышается—до 70% [102, 103]. Если взаимо-
действие соединения LIII с нитритом натрия и соляной кислотой произ-
водить под давлением при 80° С, то происходит непосредственное образо-
вание пиридоксина (I) [104]. Обычно, после выпаривания реакционной смеси,
досуха, экстракции этиловым спиртом и удаления большей части раство-
рителя пиридоксин в виде хлоргидрата осаждается ацетоном [105].
Для получения аминопиридона (L) представляет интерес метод конден-
сации у-ацетамида этоксиацетилацетона (LVIII, R — С2Н5) с цианацета-
мидом (XLV), приводящей к получению ацетиламино-2-пиридона (LIX)
[106], деацетилирование которого и дает аминопиридон (L).
Общий выход пиридоксина через соединения XLVI, XLIX, LI—LIII,
LVIII (схема 75) составляет 1,9% для этоксиметилпроизводных и 3,9%
для метоксиметилпроизводных и через соединения XLVI, XLIX, LI—LIV
8,5% для метоксиметилпроизводных (исходя из алкоксиацетилаце-
тона).
Синтез пиридоксина через производные нитрила никотиновой кислоты
по последнему варианту представляет собой один из наиболее рациональных
путей его технического получения как по выходам, так и по достаточно
четкой воспроизводимости процесса.
Для получения 3-амино-4-метоксиметил-5-аминометил-2-пиколина (LIII,
R = СН3) предложен путь, по которому в качестве исходного вещества для
конденсации с цианацетамидом (XLV) используют 3-фенилгидразон метокси-
ацетилацетона; далее синтез идет через 2-метил-3-фенилазо-4-метоксиметил-5-
циан-6-оксипиридин и его тозильное производное [107].
По одному из вариантов синтез пиридоксина осуществляют исходя из
циантиоацетамида и производного метоксиацетилацетона [108].
Первый синтез пиридоксина (I) осуществлен из 2-метил-4-алкоксиметил-5-
циан-6-пиридона (XLVI) через лактон 5-пиридоксиловой кислоты (XXII)
(схема 76) [75, 109—1111.
341*
Схема 76
Синтез пиридоксина из 2-метил-4-алкоксиметил-5-циан-6-пнридона через лактон
5-пиридоксиловой кислоты
Лактон (XXXII) может быть получен также из диметилового эфира
2-метил-3-окси-4,5-пиридиндикарбоновой кислоты при восстановлении бор* .
гидридом натрия в присутствии хлористого алюминия [111].
Синтезирован пиридоксин гидрохлорид, меченный 14С по оксиметильной
группе положения 5 [112].
2. Синтез пиридоксина через производные азида никотиновой кислоты
Этот путь синтеза пиридоксина разработан в ряде патентов [82, 113,
114]; в них исходят из 2-метил-4-феноксиметил-5-циан-6-оксиникотиновой
кислоты (LX), превращая карбоксильную группу этого соединения по реак-
ции Курциуса через азид в аминогруппу и затем в оксигруппу, получая i
соединение I. Первоначально из соединения LX (схема 77) получают егоЛ$
хлоран гидрид, одновременно замещая оксо группу в положении 6 на хлор.*’;.
Действуя на соединение LXI азидом натрия или гидразином в щелочном
растворе и затем азотистой кислотой, получают 2-метил-4-феноксиметил-5-
циан-6-хлоразид никотиновой кислоты (LXII). Его прибавляют к спирту,
причем происходит выделение азота и образование промежуточного эфира
изоциановой кислоты, который под действием спирта переходит в урета-
новое производное LXIII.
Схема 77
Синтез пиридоксина через производные азида никотиновой кислоты
C2H5OOCIIN
СН.
'N С1
LX1
СН2ОС6Н3
CWTC JL CN INaN, или
ClOt CN NHJMHg, NaNO2
h2n
CH, N
LXIV
CH,OC6HS
qiljOOCHN
CH, IN
LXV
CH, N
LXVI
Образовавшийся 3-карбоэтоксиамино-4-феноксиметил-5-циан-6-хлор-2-
пиколин (LXIII) гидрируют с никелевым катализатором под давлением в
метиловом спирте в присутствии аммиака в 5-аминометилпроизводное
(LXIV), аминогруппу которого превращают в оксигруппу диазореакцией
с нитритом натрия, и затем из соединения LXV переходят к пиридоксину.
Для этого фениловый эфир и уретановую группу гидролизуют нагреванием
342
с азеотропной бромистоводородной кислотой, одновременно получая дибром-
метнлзамещенный аминопиколин (LXVI), который с нитритом серебра прев-
ращают в пиридоксол (I). По другому варианту соединение L XV гидролизуют
соляной кислотой при 170° С под давлением и обработкой нитритом серебра
непосредственно получают пиридоксин (I).
3. Синтез пиридоксина через производные моно- и динитрила
цинхомероновой кислоты
В этом методе используют конденсацию амида ацетопировиноградной
кислоты (LXVII) с цианацетамндом (XLV) в спиртовой среде при катали-
тическом действии диэтиламина в 2-метил-4-амидокарбокси-5-цнан-6-пи-
ридон (LXVIII), который рядом превращений через динитрил цинхомеро-
новой кислоты переводят в пиридоксин (I) [115, 116], что показано на схеме
78. Первоначально конденсацией этилового эфира щавелевой кислоты и
ацетона получают этиловый эфир ацетопировиноградной кислоты (LXIX) с
выходом 61 % [117], затем его с раствором аммиака превращают в со-
единение LXVII.
Однако проще из этилового эфира ацетопировиноградной кислоты
(LXIX) и цианацетамида (XLV) получить эфир 2-метил-4-карбокси-5-циан-
6-пиридона (LXX), а затем его с аммиаком превратить в пиридонамид
(LXVIII). Конденсация цианацетамида с 0-дикетоном — с карбонильной
группой ацетопировиноградного эфира — протекает первоначально по его
метиленовой группе, активированной карбоалкоксильной группой [76].
Реакция проходит в присутствии органических катализаторов — диэтил-
амина или пиперидина или с неорганическим катализатором — аммиаком
в спиртовом растворе (выход 55%) [118, 119]; преимущество имеет синтез
с применением метиловых эфиров — щавелевой кислоты, ацетопировиног-
радной кислоты, производного пиридона [120].
Значительное упрощение в синтез пиридонамида (LXVIII) внесено при-
менением водного аммиака как катализатора реакции конденсации циана-
цетампда (XLV)c ацетопировиноградным эфиром (LXIX), причем одно-
временно происходит и образование карбамидной группы; процесс
приводит непосредственно к получению 2-метил-4-амидокарбокси-5-циан-6-
пиридона (LXVIII) [121].
Для синтеза 2-метил-4-карбоэтокси-5-циан-6-пиридона (LXX) можно
исходить из дегидролевулинового эфира и цианацетамида и подвергнуть их
конденсации в присутствии пиперидина как катализатора [122].
В дальнейшем синтезе пиридоксина 2-метил-4-амидокарбокси-5-циан-6-
пиридон (LXVIII) превращают в динитрил 6-метил-2-пиридонцинхомероно-
вой кислоты (LXXI) (т. пл. 242° С) дегидратацией хлорокисью фосфора в
среде пиридина. Можно также дегидратацию пиридонамида (LXVIII) в
динитрил (LXXI) осуществить кипячением с уксусным ангидридом при
участии молибденового катализатора [1231.
Затем динптрил (LXXI) нитруют дымящей азотной кислотой в смеси
с уксусным ангидридом в присутствии небольшого количества мочевины для
связывания окнслов азота и полученный 2-метил-3-нитро-4,5-дициан-6-пи-
ридон (LXXII) превращают в хлорнптропроизводное (LXXIII) нагревани-
ем с пятихлористым фосфором в хлорбензоле. Это соединение превращают в
соединение LXXIV различными путями. Один из вариантов заключается в
последовательном гидрировании нитрогруппы в аминогруппу соединения
LXXV, а затем в элиминировании атома хлора и восстановлении двух
нитрильных групп [124] с катализаторами различной активности: для
гидрирования нитрогруппы — окись платины, треххлористый титан или
скелетный никелевый катализатор, для элиминирования атома хлора и
восстановления нитрильных групп — 5?^-ный палладий на сернокислом
барии или угле, промотированный платиной. По другому варианту хлор-
нитропроизводное (LXXIII) превращают в соединение LXXIV в одну ста-
343
Схема 78
Синтез пиридоксина через производные моно- и ди нитрила цинхомероновой кислоты
CONHj
82%
Ao
CH, °
LXVII
CN
+ ffl, —.
^С_ 73%
H,N 4°
XLV
CONH2
1 CN C=O
Nil,. 11,0
CH, NH О
LXVIII
(СНэ)
,COOC2HS
_CN
CH2
CN
O,N A,
CN
CN , x A CN
HNO3,(cH3CO)2O
92% РОСТ,
"О
LXIX
нХ ''о
I XLV
NH,,
спирт
:ooc,hs
CN
CH, NH О
LXXII
68%
CH, NH О
LXXI
96%|PCls
CN
ОД Л CN
cooai,
O^JxCN
CH, NH "O
LXX
COOCjHs
A AN
CHjNH,
HjN
СН3 N
LXXIV
45%|NaNO2
СН, N
LXX1I1
СИ, N О
LXXVI
CN
A AN
LXXVII
Ihno,
♦Jfe
QN CN
CH, N a
CH2OH
HO JL CH,OH
CH3 N
CH, N Cl
LXXVIII
51% A <A
CH, N Ct
LXXY
LXXIX
дню — в присутствии 10%-ного палладия на угле под давлением [125].
3-Амино-4,5-бис-амшюметил-2-пиколин (LXXIV) получается в виде
тригидрохлорида или дпсульфата; его переводят в пиридоксин (I) диазо-
реакцией с нитритом натрия в горячем кислом растворе. Пиридоксин выде-
ляют концентрированием реакционного раствора в вакууме, растворением
остатка в безводном спирте и кристаллизацией в виде гидрохлорида [123,
1251. Синтез пиридоксина через соединения LXVIII, LXXI—LXXIV
осуществлен с общим выходом 6,8%.
Заслуживает внимания вариант синтеза пиридоксина через эфир заме-
щенного ппрпдопа (L.XX), из которого рядом превращении (схема 76) че-
рез соединения LXXVII—LXXVIII получают 2-метил-3-амино-4-карбо-
этокси-5-циапппридин (LXX1X) [126—128], восстанавливаемый алюмо-
гидридом лития в 3-ампно-4-оксиметил-5-аминометил-2-пнколин (LVII)
[1291; из него пиридоксин (I) получают обычным методом дпазореакцией с
нитритом натрия. Из соединения LXX можно перейти к соединению LXXVIII
через 2-метил-3-нитро-4-карбоалкокси-5-циан-6-пирпдон (LXXVI) [119].
4. Синтез пиридоксина через производные эфира цинхомероновой кислоты
В одном из вариантов синтеза пиридоксина через производные эфира
цинхомероновой кислоты используются производные а-аланина и янтарной
кислоты [130] (схема 79).
344
При конденсации N-алкил (или бензил) аланиновых эфиров (LXXX) с
эфирами а-формилянтарной кислоты (LXXXI) образуются N-алкил (или
бензил) замещенные аминометилен янтарные эфиры (LXXXII), которые под
влиянием металлического натрия, амида натрия или алкоголята натрия
(реакция Дикмана) подвергаются циклизации в производные эфира дигидро-
пиридона с карбоалкоксильными группами в положениях 4 и 5 (LXXXIII);
циклизация протекает только с соединением, содержащим третичный атом
азота. Затем оксоэфир (LXXXIII) в виде его гидрохлорида дегидрируется
хлористым сульфурилом в пиридиниевый оксидикарбоновый эфир
(LXXXIV), причем реакция, по-видимому, идет через промежуточное хло-
рирование.
Схема 79
Синтез пиридоксина через производные эфира цинхомероновой кислоты
COOR
I
.СН +
СН, ''NH
R’
LXXX
COOR
I
CH,
Sjj-COOR
,CH
HO
COOR
CH,
ROOC "<j-COOR
CH CH
CH, "N
R’
LXXXII
LXXXIII
If-CH
II +
CH, NH,
LXXXVI
cooqh5
,C
0х" C-COOCjHs
CHOH -W-60%
LXXXVII
LXXXIY
СООСЛ
O^_______
COOQHs
lf-СН, ^CCOOC^S^R’_X COCK^H5
/ч CH 11
CH,
LXXXVIII
80-85%
CH, N
LXXXIX
R=CH„ C,HS; R'=CH,. CH,^;
R’COCH,: COOC,HS
В результате гидрирования соединения LXXXIV получается замещен-
ный диэфир производного цинхомероновой кислоты (LXXXY). Его превра-
щение в пиридоксин (I) осуществляют восстановлением алюмогидридом
лития [131 1.
В другом варианте пиридиновый цикл создается из двух частей:
из аминометилвинилметилкетона (LXXXVI, R"= СОСН3) или эфира0-ами-
нокротоиовой кислоты (LXXXVI, R"= СОЭС2Н5) и эфира оксиметилен-
оксал ил уксусной кислоты (LXXXVII) [132]. Первоначально в безводном
растворе образуется ациклический эфир (LXXXVIII), причем выделяющая-
ся при реакции вода в присутствии кислоты ведет к частичному гидролизу
исходного ампносоединения (LXXXVI), затем происходит циклизация
ациклического эфира (LXXXVIII) в соединение LXXXIX при действии
концентрированной серной кислоты. Частичного гидролиза эфиров можно
избежать, если по окончании экзотермической реакции вылить реакцион-
ный раствор в безводный спирт, а затем разбавить водой.
З-Оксометильную группу соединения LXXXIX превращают в амино-
группу соединения ХС по реакции Шмидта с азотистоводородной кислотой
в присутствии серной кислоты и последующим гидролизом реакционной
смеси [132]. Если циклическое соединение LXXXIX из реакционной смеси
не выделять, а непосредственно на ациклический эфир (LXXXVIII) подей-
ствовать серной кислотой и через некоторый промежуток времени приба-
вить измельченный азид натрия, то выход 2-метил-З-аминоцинхомероновой
345
кислоты (ХС) будет меньше [132]. Соединение ХС после его этерификации
восстанавливают алюмогидридом лития в безводном эфире или тетрагидро-
фуране в амино (дезокси)пиридоксин [131—133], а из него диазореакцией
получают пиридоксин (I).
Синтез пиридоксина можно осуществить по той же принципиальной
схеме, исходя из р-ампнокротононнтрила (L.XXXVI, R"= CN), через
ппридиитрикарбоновые эфиры с последующим декарбоксилированием а-кар-
боксильной группы [134, 135].
Для синтеза пиридоксина предложен вариант, в котором исходят из
2-метил-3-нитро-4-карбометокси-5-цпан-6-хлорпиридина (ЬХХУШ) (схема
78, с. 344), нитрогруппу восстанавливают в аминогруппу, сложноэфирную
и нитрильную группу гидролизуют в карбоксильные — соединение XCI
[119, 136, 137]. В результате последующего восстановления эфира этого
соединения гидридами металлов получают 2-метил-3-амино-4,5-бпс-окси-
метилпиридин (ХСП) [136], а затем диазореакцией — пиридоксин (I)
[119, 136].
LXXVIII
5. Синтез пиридоксина через производные хинолина и изохинолина
Принципиально отличающимися от описанных методов синтеза пири-
доксина являются методы, основанные на расщеплении более сложных
молекул, в гетероциклическое ядро которых введен ряд функциональных
групп, отвечающих по своему расположению молекуле пиридоксина. Эти
синтезы ведутся через производные цинхомероновой кислоты (схема 80).
В одном из методов применяют З-метил-4-метоксиизохинолин (ХСШ),
который получают конденсацией фталимида калия и а-бромпропнонового
эфира с последующими сложными превращениями [138—140]. Для синте-
за соединения ХСШ можно исходить из 4-оксиизохинолпна, в положение 3
которого вводят метильную группу через основание Манниха при действии
формальдегида и диэтиламина с последующим каталитическим восстанов-
лением образовавшегося З-диметиламинометил-4-оксиизохинолина [141].
Окислением марганцовокислым калием в щелочной среде непосредствен-
но замещенного изохинолпна (ХСШ) [139] или, лучше, его аминопроиз-
водного (XCV) [1401 получают 2-метил-З-метоксицинхомероновую кислоту
(XXXIX); введение аминогруппы (через нитрогруппу —соединение XCIV)
или сульфогруппы в бензольное ядро изохинолиновой молекулы сильно
его ослабляет и делает мало устойчивым к окислителям.
2-Метил-З-метокспцннхомероновую кислоту (XXXIX) можно получить
также из 2-метил-З-метоксицинхониновой кислоты (XCVI) [142]. Перво-
начально путем ее нитрования и последующего восстановления получают
соединение XCVII, которое окислением марганцовокислым калием в
щелочном растворе превращают в 2-метил-3-метоксипиридин-4,5,6-трикар-
боновую кислоту (XCVIII); она легко декарбоксилируется в положении 6
нагреванием с уксусным ангидридом, и образовавшийся ангидрид гидроли-
зуется в замещенную цинхомероновую кислоту (XXXIX).
2-Метил-З-метоксицинхомероновую кислоту (XXXIX) в виде ее диэти-
лового эфира (XCIX) при действии аммиака переводят в диамид замещен-
ной цинхомероновой кислоты (С), из которого дегидратацией с хлорокисью
фосфора, пятихлористым фосфором, тионилхлоридом или уксусным ангид-
ридом получают динитрил 2-метил-З-метоксицинхомероновой кислоты (CI)
[140, 1431. Из этого соединения гидрированием с 5%-вым палладием на угле,
346
промотированном ' платиной, получают бпс-аминометнл производное (СП)
[109, 140, 143, 144], а из него диазореакцией —эфир пиридоксина (XXXVI),
затем при действии концентрированной бромистоводородной кислоты гидро-
лизуют метоксигруппу и получают пиридоксин (I) [140, 144]. Весь синтез
по схеме 80 может быть проведен также с этокси- [144] или бензилоксипро-
изводными [145].
По другому варианту бис-аминометплпроизводное (СП) подвергают
гидролизу бромистоводородной кислотой через соединение CIV и после
замещения аминогрупп диазореакцией на оксигруппы получают пиридок-
син (I). Можно от динитрила 2-метил-З-метоксицинхомероновой кислоты (CI)
перейти к 2-метил-3-окси-4,5-бщ>аминометилпиридину (CIV) первоначаль-
но через гидролиз эфира в оксидинитрил (СШ), затем в полученном соеди-
нении восстановить нитрильные группы [146].
Схема 80
Синтез пиридоксина через производные изохннолнна и хинолина
COOC2HS
CH,О JL COOC-Hs
gHgOH ’ yV
CONH,
1 CONH,
СН2К
С
СН, N
XCIX
|[n]/LiAIH4
CH,OH
HBr
A J CHCOOAg-
CH, N Ъ CH3 N
XXXY1 I
CN
л СН3О JI CN
роа. •
сцХ
CI
CHjNHj
[h]
ch, n"
Cll
CHjOH
CH.NH, CN
zUcMjNHj эд НО J. CN
СН, N
СIV
CH, N
СНГ
Широко применяемый в последние годы метод восстановления карбо-
новых кислот в спирты алюмогидридом лития использован и для получе-
ния пиридоксина. Дикарбоновый эфир (XCIX), в котором вместо 3-метокси-
группы могут содержаться 3-этокси- или 3-бензилоксигруппы, восстанав-
ливают в эфирном растворе в токе азота эфирным раствором алюмогидрида
лития при 0°С и после гидролиза эфира (XXXVI) получают пиридоксин (I)
[131, 147].
В случае применения 3-бензилоксипроизводного удаление защитной
группы производится восстановлением с катализатором — палладий на
угле [130].
6. Синтез пиридоксина через производные фурана
Этот метод синтеза основан на реакции гидролитического расщепления
аминометил замещенных фурана и перегруппировки в замещенные пиридина
(схема 81) [148—150].
Для получения пиридоксина исходным веществом служит 2-(а-ацетил-
аминоэтил)-3,4-бос-ацетоксиметилфуран (CXVII), который синтезируют из
347
3,4-бис-карбоэтоксифурана (CXIII) [150]. Для получения соединения
СХШ янтарный эфир (CV) формилируют этилформнатом под действием
натрия (соединение CVI), затем формильную группу защищают ортомуравьи-
ным эфиром и в диэтоксиметилянтарнын эфир (CVI 1) вводят вторую формиль-
ную группу. При действии концентрированной серной кислоты при 50° С
из полученного соединения CVIII получают 3,4-бнс-карбоэтоксифуран
(СХШ) [1511. Со значительно лучшим выходом это вещество получают
диеновым синтезом из фурана (CIX) при взаимодействии с ацетилендикар-
боновым эфиром (СХ) при 100° С; в результате присоединения к концам
сопряженной диеновой системы фурана образуется соответствующий аддукт
(CXI), который при частичном гидрировании в ацетоне дает аддукт (СХП)
с одной двойной связью. При нагревании 3,6-эпокси-3,4.5,6-тетра-гидро-
фталевого эфира (СХП) до 200° С с одновременным отщеплением этилена
получается соединение СХШ [152].
При восстановлении карбоэтокси групп алюмогидридом лития в эфирном
растворе в присутствии уксусного ангидрида из соединения СХШ полу-?:,
чают 3,4-бис-ацетоксиметилфуран (CXIV) [150], в положении 2 которого
действием уксусного ангидрида с эфиратом трехфтористого бора в качестве ел
катализатора вводят ацетильную группу, затем из соединения CXV полу-;>=.
чают его оксим (CXVII); гидрирование оксима со скелетным никелевым
катализатором под давлением 100 ат при 80° С в присутствии уксусного
ангидрида приводит к получению исходного для синтеза пиридоксина
2-(а-ацетиламиноэтил)-3,4-биоацетоксиметилфурану (CXVII) [150, 153].
Схема 81
Синтез пиридоксина через производные фурана
ССЮС,Н5 COOCjHj
СНг окно
СН2 65% СН,
COOC,HS COOC2HS
CV CVI
COOCjHj
COOCjHj
ex
COOCjHj COOCjHg
chch(or)3 chch(or)2
82% - CH, 87%' CHCHO
I I
COOCjHg cooc2h5
evil CYUI
^^/СООСгН5 [Hl/Pd . ^rCOOqHs______________I
К%(изфура^)кдА -C2H4;200o
^ЧООС2Н5 k >'^^COOCaHs
CXI CXII
CjHgOOC COOC,H5 AcOHjC C
: W [h](uaihQ; (CHjCQ)p W
83%
CXIII
83%
o*
CXIV
CHjOAc
(CHjCoXo, BF3
1 72%
AcOHjC CH2OAc
/ NH2OH
^c> coch, 900/0
CXV
AcOHjC CH2OAc
AcOH2C
CHjOAc
/ Электролитическое
\ окисление в СЦОН
CXVI NOH
о снен.
CXVII NHAc
АсОНС CHjOAc
лхОСНэ
CHaO'Y) СНСН,
CXVill NHAc"
H2O; NaOH
AcOH„C CHjOAc
гЧ/ОСН, HQ; H2O
«уАЛсНСнДЖ
CXIX nh2 83%
95%
CHjOH
oe'^-cHjOH
jw сно
nh2
Ac=CH)CO
348
Для получения соединения CXVIII вместо фурана в диеновом синтезе
с ацетилендикарбоиовым эфиром (СХ) используют 2-(а-ацетиламиноэтил)-
фуран и в результате восстановления образовавшегося 2-(а-ацетиламино-
этил)-3,4-бис-карбоэтоксифурана алюмогидридом натрия получают 2-(а-аце-
тиламиноэтил)-3,4-бис-ацетоксиметилфураи (CXVII) [154]. Известны и
другие усовершенствования процесса [155, 156].
С выходом около 1 % нагреванием в растворе хлористого аммония или
аммиака пиридоксин можно получить и из2-ацето-3,4-бис-ацетоксиметилфу-
рана (CXV) [157]. Однако выход пиридоксина достигает 76%, если перво-
начально из соединения CXV получить его ацетиламинопроизводное CXVII,
которое подвергнуть электролитическому окислительному метоксилирова-
нию [150, 158], и образующееся 2,5-диметокси-2,5-дигидрофурановое
производное (CXVIII) подвергнуть N-деацетилированиюваминоэтилпроиз-
водное (CXIX); последнее соединение с предварительным выделением [159]
или без выделения [150] при действии н. соляной кислоты подвергают гидро-
литическому расщеплению и внутримолекулярной конденсации продукта
гидролиза в пиридоксин (I). Предварительная защита аминогруппы ацети-
лированием необходима для повышения выхода при электролитическом
метоксилированни [148]; последнее проводят на платиновом аноде при
температуре —18, —22° С и плотности тока 2—4 А/дм2 [160]. Общий выход
пиридоксина из фурана составляет около 25%, а из янтарного эфира — око-
ло 12%.
7. Синтез пиридоксина через конденсированные пиримвдо-фурановые
системы
В этой группе синтезов пиридоксин (I) получают созданием и расщепле-
нием гетероциклов, представляющих собой срощенные пиридинофурановые
кольцевые системы. Так, производное 2-метилникотиновой кислоты (СХХП)
получают конденсацией 8-аминомасляного метилового эфира (СХХ) с 4-фор-
мил-3-оксотетрагидрофураном (CXXI) [161]. После окисления 2-метил-З-
карбометокси-4,5-оксидодиметилдигидропиридина (СХХП) перекисью водо-
рода в соединение CXXIII его карбометоксигруппу замещают при действии
концентрированной серной кислоты и азида натрия на 3-аминогруппу
2-метил-3-амино-4,5-оксидодиметилпиридина (CXXIV). Соединение CXXIV
далёе превращают в пиридоксин (I) через соединения CXXV и LV (схема 82)
Схема 82
Синтез пиридоксина через соединения с конденсированными пиридино-фурановыми
кольцевыми системами
349
по известным реакциям (замещением аминогруппы иа оксигруппу диазо-
реакцией, расщеплением циклического эфира бромистоводородной кислотой
и последующим гидролизом бромметильных групп).
Значительно проще исходить из нитрила пировиноградной кислоты
(CXXVI), конденсировать его с метиламином и на полученное соединение
подействовать 3-оксотетрагидрофураном (CXXVII) в присутствии ацетата
пиперидина. Образующийся 2-метил-3-амино-4,5-оксидодиметплпирндин
(CXXIV) превращают затем в пиридоксин (I), как указано выше 1162].
Еще один вариант синтеза пиридоксина (I) заключается в конденсации
3-оксотетрагидрофурана (CXXVII) с этиловым эфиром а-карбоэтоксиметил-
иминопировпноградной кислоты (СХXVIII) в метиловом спирте в присут-
ствии оснований. Соединение CXXVIII получают из пировиноградной кис-
лоты и глицинового эфира в безводном метаноле, насыщенном хлористым
водородом. Образовавшийся 2-метил-3-окси-4,5-оксидодиметил-6-карбоэто-
ксипиридин (CXXIX) декарбоксилированием и гидролитическим расщепле-
нием циклического эфира при действии кислоты превращают в пиридоксол
(I) [1631.
CHjCOCOOH
+
HaNCH-COOCaHj
CXXVIII
Для конденсации с соединением CXXVIII применяют и такие вещества,
которые можно рассматривать как производные 3-оксотетрагидрофурана
с раскрытым циклом: 1,4-диметоксибутанон-2 [1631, 1,4-диэтоксибутанон-2
и 1,4-бцс-бензилоксибутанон-2 [1641. С последним веществом и соединением
CXXVIII реакцию ведут в метаноле в присутствии карбоната натрия; об-
разовавшийся замещенный пиридин декарбоксилируют и освобождают от
защитных групп гидролизом в разбавленной соляной кислоте под давлением
при 155° С [164].
8. Диеновый синтез пиридоксина из замещенных оксазолов и тиазолов
Эта важная группа синтезов, имеющая техническое значение, основана
на реакции Дильса — Альдера между замещенными оксазолами или тиазо-
лами и различными диенофилами, в результате чего создается основной
углеродный скелет молекулы пиридоксина. В качестве диенофилов исполь-
зуются различные дикарбоновые кислоты, их ангидриды, дикарбоновые
эфиры, динитрилы, оксиэфиры и др. '
Ранее было установлено, что оксазолы реагируют по концам сопряжен-
ной диеновой системы с двойной связью малеиновой кислоты или ее ангид-
рида по методу диенового синтеза с образованием соответствующего аддукта,
который после раскрытия окисного мостика ароматизируется в 3,4-пи-
ридиндикарбоновую кислоту (цинхомероновую кислоту) [165], а при при-
менении 5-алкоксиоксазола—в 5-окспцпнхомероновую кислоту [166].
В ряде синтезов пиридоксина (I) в качестве исходного вещества Хар-
рис, Фирестон, Пфистер и др. [167] применили 4-метил-5-этоксиоксазол
(СХХХП), который получают следующим путем. Этиловый эфир /ЭЕ-п-ала-
нина (СХХХ), полученный с выходом 90% из DL-a-аланина, формилнруют
формплуксусным ангидридом в этиловый эфир N-формил-DL-a -аланина
(CXXXI) с выходом 78% [167 ]; эту же реакцию осуществляют при действии
формамида и пятиокиси фосфора при 105° С [168]. Внутримолекулярную
циклизацию этилового эфира М-формил-D L-a-аланина проводят под действи-
ем пятиокиси фосфора в среде хлороформа при нагревании в течение 5 ч.
350
После щелочной обработки и отгонки органического слоя получают 4-метил-
5-этоксиоксазол (CXXXII) с выходом 60% 1167—169].
COOCiHs
ch3chnh2-hci
сххх
COOQHs
I
CHgCHNHCHO
CXXXI
CXXXII
По одному из вариантов для синтеза DL-a -аланина исходят из пропио-
новой кислоты, которую при действии тионилхлорида превращают в ее
хлорангидрид, подвергаемый затем бромированию элементарным бромом в
«-положение. После нейтрализации полученную и -бромпропионовую кисло-
ту (выход 88%) подвергают аммонолизу реакцией с водным аммиаком и
через уротропиновый комплекс (образующийся при действии гексаметилен-
тетрамина) выделяют DL-a-аланин (выход 68%).
Диеновая конденсация 4-метил-5-этоксиоксазола (CXXXII) с 2 молями
диэтилового эфира малеиновой кислоты (СХХХIII) при 110° С (2 ч) по
Дильсу — Альдеру приводит к образованию соответствующего аддукта
(CXXXIV), который при нагревании с соляной кислотой отщепляет молеку-
лу спирта, раскрывает окисный мостик и превращается с выходом 85% в
2-метил-3-окси-4,5-б«с-карбоэтоксипиридин (CXXXV) [167]. Это соедине-
ние, как описано ранее (см. с. 345.), при восстановлении карбоэтоксильных
групп в оксиметильные группы алюмогидридом лития дает пиридоксин
(I) [131, 132, 167].
COOQHs
11 —
НС
СООС2Н5
CH3 N
HC1
CXXXIV
COOCjHj
A COOQHs HO
CHjOH
jKL CHjOH
CH, N
CXXXII CXXXII!
CXXXV
Для гетеродпенового синтеза пиридоксина в качестве исходного вещества
с успехом используют 4-метил-5-пропоксиоксазол (CXLV), который синтези-
руют аналогично описанному выше. Первоначально DL-a-аланин подверга-
ют азеотропной этерификации «-пропиловым спиртом с отделением воды в
виде азеотропа с пропанол-толуолом с последующим формилированием
образовавшегося «-пропилового эфира DL-a-аланина этилформнатом в
«-пропиловый эфир N-формил-DL-a-аланина (выход 68%). Затем осуществ-
ляют его внутримолекулярную конденсацию в 4-метил-5-пропоксиоксазол
(CXLV), которая протекает при нагревании с отщеплением воды при дей-
ствии пятиокиси фосфора в среде хлороформа с последующей нейтрализа-
цией образовавшейся фосфорной кислоты (выход 60%); для облегчения
условий перемешивания используют аэросил [1701.
Диеновая конденсация 4-метил-5-пропоксиоксазола (CXLV) с диметило-
вым эфиром малеиновой кислоты в толуоле в присутствии гидрохинона при
110°С (8 ч) дает 2-метил-3-окси-4,5-бис-карбометокспппридин (CLI) [170].
Его восстанавливают алюмогидридом лития в пиридоксин (I) с выходом
70%, как указано выше.
В качестве диенофила предложен малеиновый ангидрид; превращение
аддукта (CXXXIV) в соединение CXXXV осуществляют при кипячении в
спирте, насыщенном хлористым водородом, при этом одновременно проис-
ходит этерификация карбонильных групп [168].
Конденсация 4-метил-5-этоксиоксазола (CXXXII) с 1 молем фумаронит-
рила (CXXXV1) при кипячении в течение 5 ч в спирте дает аддукт
(CXXXVII), который при обработке концентрированной соляной кислотой
351
при нагревании отщепляет молекулу спирта и превращается в 2-метил-З-
окси-4,5-дицианпиридин (CIII) с выходом 75% 1167 J. Динитрил (СШ) гид-
рируют в метаноле и соляной кислоте с 5% Pd на угле с выходом 70% в
2-метил-3-окси-4,5-бпс-аминометилпиридин (CIV), превращаемый диазореак-
цией при 85° С по [1301 в пиридоксин (I) с общим выходом свыше 41 % [167 ].
схххи CXXXVI
Пири-
доксин
I
4-Метил-5-этоксиоксазол (СХХХП) и избыток 2,5-дигидрофунара
(CXXXVIII) в результате конденсации по Дильсу — Альдеру при 175е С
в течение 3 ч в присутствии трихлоруксусной кислоты как катализатора
дают аддукт (CXXXIX), который в среде дигидрофурана с минеральной
кислотой отщепляет молекулу спирта, раскрывает окисный мостик и пере-
ходит в 2 метил-3-окси-4,5-оксидодиметилпиридин (CXXV) с выходом 58%
[167, 171]
СХХХП
Пиридоксин
I
При нагревании с 48 %-ной бромистоводородной кислотой циклический
эфир (CXXV) образует 2-метил-3-окси-4,5-бпс-бромметилпиридин (LV),
который гидролизуют в пиридоксин (I) [167].
Если применить в качестве диенофила 2,5-диметокси-2,5-ди гидрофуран,
то диеновая конденсация с соединением СХХХП с последующей обработ-
кой соляной кислотой приводит к получению 2-метил-3-окси-4,5-диформил-
пиридина, а его восстановление с алюмогидридом лития или гидрирование
с никелевым катализатором дает пиридоксин (I) [172]. Использование в
реакции 1,4-диметоксибутена-2 с последующим нагреванием в соляной кис-
лоте в результате расщепления окисного мостика и гидролиза эфирных
групп дает непосредственно пиридоксин (I) [168].
В приведенных выше примерах диенового синтеза промежуточный ад-
дукт из реакционной смеси обычно не выделяется. Однако при взаимодей-
ствии 4-метил-5-этоксиоксазола (СХХХП) ,с 2-диметил-4,7-дигидро-1,3-ди-
оксеппном (CXL) при 190° С в присутствии трихлоруксусной кислоты был
изолирован аддукт (CXL1) [173, 174]. Превращение аддукта (CXLI) в за-
мещенный пиридин (CXLII) и затем без его выделения — в пиридоксин (I)
происходит при действии соляной кислоты
2-Диметил-4,7-дигидро-1,3-диоксепин (CXL) синтезируют из цис-бутен-
2-диола-1,4 и ацетона [173, 174]. Бутен-2-д иол-1,4 (CXLIII) получают
352
каталитическим гидрированием бутиндиола-1,4 на палладиевом катализа-
торе, пассивированном пиридином (выход 92%). Бутиндиол-1,4 является
продуктом синтеза на основе ацетилена и формальдегида. -• <
Удобным диенофилом оказался циклический ацеталь ацетальдегида с
цис-бутен-2-диолом-1,4 (CXLIV), который получают ацетализацией цис-
бутен-2-диола-1,4 (CXLIII) ацетальдегидом в присутствии каталитических
количеств соляной кислоты. Его вводят в гетеродиеновую конденсацию с
4-метил-5-н-пропоксиоксазолом(СХЬУ). Конденсация протекает при нагре-
вании в присутствии гидрохинона с образованием неустойчивого бицикли-
ческого аддукта (CXLVI), расщепляющегося с ароматизацией в 2-метил-З-
пропокси-4,5-циклоацетацеталь-бис-оксиметилпиридин (CXLVII) с выхо-
дом 64%.
Циклоацеталь (CXLVII) подвергают гидролитическому расщеплению по
эфирной и ацетальной группам при действии концентрированной соляной
кислоты в пиридоксин (I) с выходом 85%.
В качестве диенофила используют и циклический ацеталь формальде-
гида с бутен-2-диолом-1,4—4,7-дигидро-1,3-диоксепин; реакцию с соеди-
нением СХХХП проводят при 165—175° С в присутствии гидрохинона под
азотом с последующей обработкой соляной кислотой [174, 175]. 3,6-Дигид-
ро-1,2-диоксан также может служить диенофилом в реакции с соединением
СХХХП; полученное после обработки метанольным раствором хлористого
водорода производное пиридина превращается в пиридоксин (I) в результате
каталитического гидрирования с палладиевым на угле катализатором [1761.
Известен и ряд других диеновых синтезов пиридоксина, в которых
применяют 4-карбоксиметил- или 4-карбоэтоксиметил-5-этоксиоксазол и
малеиновый эфир, малеиновый ангидрид, 4-оксикротононитрил, 4,7-ди-
гидро-1,3-диоксепин [177, 178]. При последующей обработке карбоксиме-
тильная группа, занимающая положение 2, декарбоксилируется и превра-
щается в необходимую для структуры пиридоксина метильную группу.
Несколько иначе идет диеновый синтез пиридоксина в том случае, когда
применяется 4-метилоксазол (CXLVIII). Его конденсация с диметиловым
эфиром малеиновой или фумаровой кислоты (CXLIX) приводит к получе-
нию соответствующего аддукта (CL), последующее превращение которого в
2-метил-3-окси-4,5-бпс-карбометоксипиридин (CLI) сопровождается раскры-
тием окисного мостика и дегидрированием (а не отщеплением спирта [167])
при нагревании в нитробензоле [179]. Соединение СЫ превращается в
пиридоксин (I) в результате восстановления гидридами металлов
CXLVIII
соосн,
НС ’
нс\,
COOCHj
CXUX
12—69
353
. Реакция идет с отщеплением цианистого водорода, если для конденса-
ции с 4-метилоксазолом (CXLVIII) в качестве диенофила применить 1,4-ди-
метокси-2-цианбутен-2 [180]; пиридоксин образуется через промежуточ-
ный 2-метил-3-окси-4,5-бис-метоксиметнлпиридин с невысоким выходом.
Описаны и другие варианты гетеродиенового синтеза пиридоксина из
замещенного оксазола [172, 181, 1821.
Вместо оксазолов для диенового синтеза пиридоксина (I) предложен
4-метил-5-этокситиазол (CLII), который вводят в реакцию при 200° С с
диэтиловым эфиром малеиновой (СХХХIII) или фумаровой кислоты. Про-
межуточный аддукт (CLIII) при нагревании с концентрированной соляной
кислотой отщепляет сероводород и с выходом 63,5 % дает 2-метнл-З-этокси-
4,5-бис-карбоксипиридин (CLVI) [183], который по известным рэакциям
превращается в пиридоксин (I).
CUI
/COOQHg
Чг
нс..
^COOCjHe
схххш
COOQHs
- JLSJ
CU1I
9. Синтез пиридоксамина и пиридоксаля
Пиридоксамин (III) получен из 4а-метилового эфира пиридоксина (LIV,
R = СН3) аминированием в автоклаве раствором аммиака в метиловом
спирте при температуре 140° С в течение 16 ч с выходом 71 % [184].
сн, N
ш
CH,OR
но.>чхцон
Пиридоксамин можно получить из 3,4а-диацетилпиридоксина таким же
путем [185]. Описаны методы восстановления оксима пиридоксаля (CLV)
в метаноле водородом с платиновым катализатором под давлением 2 ат
[1861 или с палладием на угле в разбавленной соляной кислоте с выходом
98% [187 ], а также при действии цинковой пылью и ледяной уксусной кис-
лотой с выходом 87% [188, 189] и, наконец, электролитическим путем в
1—2 н. соляной кислоте с выходом 83—98% [190, 191 ].
Пиридоксаль, являющийся 2-метил-3-окси-4-формил-5-оксиметилпири-
дином (II), получают из гидрохлорида пиридоксина (I) осторожным окисле-
нием марганцовокислым калием в присутствии бикарбоната натрия с одно-
временным превращением полученного альдегида в оксим (CLV) с гидро-
хлоридом гидроксиламина и ацетатом натрия [184]; выход оксима 22%.
Окисление гидрохлорида пиридоксина также проводят двухромовокислым
калием [185] или нагреванием с суспензией двуокиси марганца, прибавляе-
мой в раствор пиридоксина с серной кислотой при 60—70° С до pH 6 с выхо-
дом 59% [6].
Из оксима свободный пиридоксаль выделяют обработкой нитритом натрия
или серебра в растворе соляной кислоты [184, 185, 192].
354
БИОЛОГИЧЕСКОЕ ЗНАЧЕНИЕ ОКСИМЕТИЛПИРИДИНОВЫХ ВИТАМИНОВ.
ЗАВИСИМОСТЬ МЕЖДУ СТРОЕНИЕМ И БИОЛОГИЧЕСКОЙ АКТИВНОСТЬЮ.
АНТИВИТАМИНЫ
Широкая и разнообразная биологическая активность оксиметилпири-
диновых витаминов связана со способностью пиридоксаль-5а-фосфорного
эфира, являющегося коферментом, в соединении с апоферментом, обеспе-
чивать многочисленные биокаталитические реакции, протекающие в живой
клетке.
Для животного организма витамин Вс является важнейшим витамином,
входящим в состав ферментов, катализирующих белковый обмен; он выпол-
няет важную функцию в превращениях аминокислот. Для каждого живот-
ного организма необходимо получать с пищей некоторые аминокислоты
(например, для человека незаменимы валин, лейцин, изолейцин, лизин,
треонин, метионин, фенилаланин, триптофан), которые он не в состоянии
синтезировать; все же другие необходимые аминокислоты синтезируются
организмом из продуктов расщепления белков или из а-кетокислот.
Специфическое авитаминное действие на животных проявляется в ряде
физиологических аномалий, происходящих при недостатке оксиметилпири-
диновых витаминов. Характерный авитаминоз выявлен у крыс — у них
наблюдается своеобразный симметрический дерматит — акродиния [9].
Почти полный недостаток витамина В6 приводит животных к гибели.
Витамины группы В6 необходимы для нормального кровеобразования.
Так, у собак и свиней при В6-авитаминозе развивается анемия, сопровож-
дающаяся ненормально повышенным содержанием железа в сыворотке
крови [1931. У людей этого явления обнаружено не было. При авитаминозе
у цыплят наблюдается ускорение свертывания крови. Витамины группы Вв
принимают участие в жировом обмене и способствуют утилизации ненасы-
щенных жирных кислот [194 J. Витамин В6 применяется в послелучевой
терапии.
При недостатке пиридоксина в организме животных происходит нару-
шение обмена триптофана [195—1981. Тесная взаимная связь между витами-
нами и аминокислотами проявляется, в частности, в участии витамина В6
в реакциях превращения L-триптофана (под влиянием кинурениназы), при-
водящих к биосинтезу никотиновой кислоты (витамина РР) [196, 199J.
Оксиметилпиридиновые витамины синтезируются многочисленными ви-
дами микроорганизмов и растениями. ЛДжрофлора желудка жвачных
животных также продуцирует витамин В6, вследствие чего эти животные
не нуждаются в получении витамина с пищей.
Пиридоксин является важнейшим ростовым фактором для жизнедея-
тельности различных бактерий [200], дрожжей и плесневых грибов. Пири-
доксаль и пиридоксамин показывают ростовую активность на микроорга-
низмах, причем для некоторых видов активность превышает в
1000—9000 раз активность пиридоксина [201 ].
Способность к биосинтезу незаменимых аминокислот у некоторых микро-
организмов (например, молочнокислых бактерий) зависит от участия пири-
доксина как ростового фактора, так же как синтез L-аминокислотиз их оп-
тических антиподов [202, 203]. Пиридоксин необходим для роста некоторых
корнеплодов (например, моркови) и корней томата, подсолнечника, хлоп-
чатника и др.
Из трех витаминов группы В6, находящихся в животных и расти-
тельных организмах на долю пиридоксаля и пиридоксамина приходится
более 80%; они встречаются в связанном высокомолекулярном соединении
с белком. Пиридоксин составляет меньшую часть—до 20%.
Для биологического определения оксиметилпиридиновых витаминов
применяют различные микроорганизмы, которые избирательно использу-
ют витамины группы В6 в качестве ростовых факторов. Так, для определе-
ния пиридоксаля применяется Lactobacillus casei, для определения суммы
12*
355
•пиридоксаля и пиридоксамина — Streptococcus faecalis и для всех трех
витаминов (пиридоксина, пиридоксаля и пиридоксамина) — Saccharomy-
ces carlsbergensis [2041 или S. ludwiqii [205].
Потребность в пиридоксине (витамине Вв) для человека составляет
от 1,5 [206] до 3 мг в сутки. При очень тяжелом труде, для беременных и
кормящих оптимальная потребность возрастает до 5 мг в сутки. Токсичность
пиридоксина определена в 5,5 г/кг при испытании на крысах.
Пиридоксин используют для витаминизации кормов в животноводст-
ве — 5—6 г/т.
Так как две формы витамина В6 — пиридоксаль и пиридоксамин —
являются продуктами взаимного превращения в процессах метаболизма,
следует признать, что молекула витамина специфична. Ацетилирование
двух или трех гидроксилов пиридоксина, который является наиболее
устойчивой формой витамина В6, сохраняет витаминные свойства, вероят-
но, вследствие того, что эти эфиры легко гидролизуются в организме.
Характерна высокая специфическая активность метильной группы
положения 2 молекулы витамина В6; ее замещение почти полностью инак-
тивирует несущую ее молекулу.
Гомолог пиридоксина с этильной группой в положении 2 обладает ни-
чтожной активностью — 2% [207] или 1—34% [2081, а в отношении дрож-
жей— даже антиростовыми свойствами [208]. Однако при испытании
в качестве субстрата пиридоксальдегидрогеназного фермента он показал
95%-ную активность [209]. Лишены или почти лишены витаминной актив-
ности гомологи с изопропильной (активность 0,25%), фенильной [210],
бензильной (0,02%) [211], изобутильной [210, 212], н-амильной [212]
и другими группами в положении 2.
В биокаталитических реакциях участвуют только 4- и 5-оксиметильные
группы; первая превращается в альдегидную группу пиридоксаля или в ами-
нометильную группу пиридоксамина, а вторая этерифицируется фосфорной
кислотой в пирндоксаль-5а-фосфат или пиридоксамин-5а-фосфат. Замеще-
ние гидроксильной группы в положении 5а водородом [11 ] или аминогруп-
пой, метилирование фенольного гидроксила [2, 200], образование N-метил-
производного [11 ], замещение гидроксила в положении 4а водородом (4-де-
зоксипиридоксин) [2001 или одновременное замещение гидроксила в
положениях 4а и 5а (4,5-бпс-дезокснпиридоксин, CLVI) [200], перенесение
гидроксильной группы из положения 3 в положение 6 (при наличии в поло-
жении 5 аминометильной группы) [213] устраняют полностью или почти пол-
ностью витаминную активность, характерную для пиридоксина. Переме-
щение заместителей между различными положениями молекулы витаминов
группы В6 лишает эти соединения физиологических свойств. Изопиридок-
син (CLVII), изопиридоксаль (CLVIII) [214] и изопиридоксамин (CLIX)
не активны для высших организмов.
CLV1
СН2ОН
CLIX
356
Однако изопиридоксаль (2-метил-3-окси-4-оксиметил-5-формилпиридин)
обладает достаточно большой ростовой активностью для дрожжей. 2-Метил-
3-окси-4,5-бнс-бромметилпириднн (LV) является ростовым фактором для
молочнокислых бактерий.
Некоторые производные 2-алкилпиридинов являются антагонистами
витаминных свойств пиридоксаля и пиридоксамина. 4-Дезоксипиридоксин
(2,4-димстил-3-окси-5-окснметилпиридин,СЕХ) — очень сильный ингибитор
(в отношении 2:1) при испытании на цыплятах [215]; сильными инги-
биторами являются также гомологи 4-дезоксипиридоксина [167] — 4а-ме-
тиловый эфир пиридоксина (CLXI) и 5-дезоксипиридоксин [215]. Но наибо-
лее сильный из всех известных антивитаминов пиридоксаля — 2-этил-З-ами-
но-4-этоксиметил-5-аминометилпиридин (CLXH) [216].
СН3
CLXII
5-Дезоксипиридоксин (CLXIII), 5-дезоксипиридоксаль (CLXIV) и 5-де-
зоксипиридоксамин (CLXV) представляют химический и биологический ин-
терес. С потерей 5-оксиметильной группы эти соединения ..........
CLXIII
не могут быть превращены в 5а-фосфорные эфиры, но функциональная группа
в положении 4, по-видимому, еще способна нормально участвовать в био-
химических реакциях [2171. Вследствие отсутствия 5-оксиметильной груп-
пы у 5-дезоксипиридоксаля (CLXIV) это соединение не может образовать
полуацетальной формы пиридоксаля. 5-Дезоксипиридоксаль и 5-дезокси-
пиридоксамин оказались сильными ингибиторами витамина Ве. Ингиби-
тором является также пиридоксаль-5а-сульфат [24].
Из соединений непиридинового ряда, которые ингибируют реакции,
катализируемые пиридоксалевыми ферментами, следует указать алкалоид
берберин [218] и антибиотик циклосерин, £>-4-аминоизоксазолидон-3
(CLXVI) [219,220]
н
CLXVI
... . . . Моль антагониста
АИ (антивитаминный индекс) - Модь пиридоксина гидрОхлБрйда '
357
Рассмотрены реакции взаимодействия пиридоксаля и пиридоксаль-5а-
фосфата с циклосерином, объясняющие его ингибирующие свойства на мо-
лекулярном уровне [221J.
ПИРИДОКСАЛЕВЫЕ КОФЕРМЕНТЫ
Пиридоксаль и пиридоксамин в виде своих 5а-фосфорных эфиров,
находясь в качестве простетической группы совместно со специфическими
белками в составе аминотрансфераз (трансаминаз), декарбоксилаз аминокис-
лот и других пнридоксальфосфатных ферментов, принимают биокаталити-
ческое участие в синтезе и расщеплении аминокислот. Пирпдоксаль-5а-
фосфат и пиридоксамин-5а-фосфат являются коферментными формами ви-
тамина В6.
Пиридоксаль-5а-фосфат — 2-метил-3-окси-4-формил-5-окси-
метил пиридинфосфат (CLXVII) —
представляет собой желтый кристаллический порошок, растворимый в
воде, неустойчивый на свету, в водных нейтральных или щелочных раство-
рах. Спектр поглощения имеет максимум: в 0,1 н. НС1 —295 нм, при pH
7,0—330 и 388 нм, в 0,1 н. NaOH —305 и 388 нм (е 6,6-103). Растворы
флуоресцируют в УФ-свете.
Пир и доксам и н - 5а -фосфат — 2-метил-3-окси-4-аминометил-
5-оксиметилпиридинфосфат (CLXVI11)
вторая, взаимно с пнридоксаль-5а-фосфатом'обратимая форма кофермента.
Пнридоксаль-5а-фосфат в присутствии металлических солей является
катализатором многих химических реакций, протекающих без участия
ферментных систем [222 ]. Он катализирует неферментные реакции переами-
нирования аминокислот и дегидратирования серина. Каталитическая спо-
собность пиридоксаля связана с большей реакционной способностью функ-
циональной группы, замещающей ^-положение пиридиновой молекулы.
Взаимодействие альдегидной группы пиридоксаля с аминогруппой амино-
кислот или первичных аминов и аминогруппы пиридоксамина с карбо-
нильной группой а-кетокарбоновых кислот приводит к образованию аль-
диминов (шиффовых оснований) и кетиминов (представленных в форме
замещенных а-иминокислот). Образующиеся промежуточные соединения в
определенной степени стабилизируются путем образования «клешневидных»
или хелатных внутренних комплексов с ионом металла по азометиновой
связи, фенольной группе и, вероятно, карбоксильной группе аминокислот-
ного остатка, например, соединения типа CLXIX и CLXX.
358
Как и в ферментных реакциях, основной структурной частью молекулы
пиридоксаля, участвующей в таутомерных превращениях, является 4-фор-
мильная группа. Присутствие 5-оксиметильной группы (или ее фосфорного
эфира), которая участвует в биологических процессах, по-видимому, не
вызывается необходимостью. Однако для проявления каталитического
действия существенно необходимо присутствие свободного фенольного гид-
роксила в орто-положении к формильной группе [222].
СТРОЕНИЕ ПИРИДОКСАЛЬ-5а-ФОСФАТА
Синтетический пиридоксаль-5а-фосфат (CLXVII) обладает всеми свой-
ствами природного кофермента. Образование эфирной связи оксиме-
тильной группы положения 5 этого соединения с фосфорной кислотой сле-
дует из того, что при окислении перекисью водорода образуется 2-метил-
3,4-диокси-5-оксиметилпиридилфосфорный эфир (CLXXI) [223], т. е.
диоксипроизводное, образование которого характерно для продуктов окисле-
ния о-оксиальдегидов перекисью водорода
Н»О2
OCsH5
I
СН—о
CLXXII
Соединение CLXVII отличается от пиридоксаль-3-фосфата, полученного
в виде полуацеталя (CLXXII) [41, 186, 224], оксима или бариевой соли
[225]; полуацеталь не дает окраски с хлорным железом, что указывает на
замещение гидроксильной группы положения 3 и обладает только ни-
чтожной коферментной активностью, так же как и пиридоксаль-3- пиро-
фосфор ны й эфи р [226 — 228 ].
Строение пиридоксаль-5я-фосфата (CLXVII) подтверждено различными
синтеза ми [41, 228 ].
СИНТЕЗ ПИРИД0КСАЛЬ-5а-Ф0СФАТА
Фосфорный эфир пиридоксаля по оксиметильной группе положения
5 (CLXVII) в свободном виде получается с трудом. Значительно
легче получить его кальциевые или бариевые соли или выделить
359
в виде оксима (т. пл. 229-230° С). Впервые пиридоксаль-5а-фосфат полу-
чили в 1951 г. Вильсон и Харрис [59] и Висконтини, Эбнозер и Каррер
[229]. Пути синтеза пиридоксаль-5а-фосфата (CLXVII) представлены на
схеме 83.
Схема 83
Синтез пнрид<жсаль-5а-фосфата
XXXJV.
OXXV
Синтез пиридоксаль-5а-фосфата (CLXVII) [4] осуществлен из пири-
доксина (I) через пиридоксин-5а-фосфат (CLXXIV) с т. пл. 210—211° С.
В пиридоксине (I) подвергают предварительной изопропилиденовой защите
гидроксильную (в положении 3) и оксиметильную (в положении 4) группы,
затем 3,4а-О-изопропилиденпиридоксин (XVI) фосфорилируют метафос-
форной кислотой [192], смесью 85%-ной ортофосфорной кислоты и пяти-
окисью фосфора [41 ] или тетрахлорфосфатом [230, 2311 с последующим
гидролизом в пиридоксин-5а-фосфат (CLXXIV). Для синтеза этого соеди-
нения исходят также непосредственно из пиридоксина (I), который фосфо-
рилируют гидратированной хлорокисью фосфора [228] или полифосфорной
кислотой — смесью ортофосфорной кислоты и пятиокиси фосфора [232,
233]. Пиридоксин-5а-фосфат (CLXXIV) окислением оксиметильной группы
в альдегидную двуокисью марганца превращают в пиридоксаль-5а-фосфат
(CLXVII) [41, 228]. Фосфорилированием бензилиденпиридоксина дифенил-
хлорфосфитом через пиридоксин-5а-фосфатf с последующим окислением
марганцовокислым калием также получен пиридоксаль-5а-фосфат [234].
Лучше для получения пиридоксаль-5а-фосфата (CLXVII) исходить из
пиридоксамина (III), который фосфорилируют в пиридоксамин-5а-фосфат
(CLXVIII) гидратированной хлорокисью фосфора [235]. смесью ортофос-
форной кислоты и фосфорного ангидрида [59, 232, 236, 237] или метафос-
форной кислотой [238]. В последних двух случаях при фосфорилировании
первоначально образуется полифосфорный эфир пиридоксамина, который
гидролизом переводится в монофосфорный эфир [237, 238]. Затем аминоме-
тильную группу пиридоксамин-5а-фосфата (CLXVIII) окисляют двуокисью
марганца в альдегидную и получают пиридоксаль-5а-фосфат (CLXVII)
[59, 236, 239, 240]. Пиридоксамин-5а-фосфат может быть превращен в пи-
ридоксаль-5а-фосфат в результате окислительного дезаминирования в
присутствии медных солей (например, ацетата меди) при действии пиро-
виноградной [241] или глиоксиловой кислоты с выходом 72% [232, 237].
С меньшим выходом пиридоксаль-5а-фосфат синтезирован из пиридок-
360
саля — его полуацетальной формы (XXXIV) — через оксим пиридоксаля
(CLV), фосфорилированием его хлорокисью фосфора 12421 или гидра-
тированной хлорокисью фосфора [243] в оксим пиридоксаль-5а-фосфа-
та (CLXXX7) с последующей обработкой нитритом натрия и соляной кис-
лотой [242, 2431.
Вместо оксима пиридоксаля для синтеза пиридоксаль-5а-фосфата го-
раздо лучше использовать шиффово основание пиридоксаля [244 J. Поэто-
му исходят из пиридоксина (I), который окисляют в пиридоксаль
(см. с. 354), и выделяют в виде основания Шиффа с каким-либо аромати-
ческим амином, например n-анизидином, затем это соединение фосфорили-
руют полифосфорной кислотой или другим фосфорилирующим агентом,
полученный фосфат шиффова основания пиридоксаля разлагают раствором
щелочи и превращают в пиридоксаль-5а-фосфат (CLXVII) с выходом свы-
ше 55% на пиридоксин.
Пиридоксаль-5а-фосфат также получен в виде магниевой или кальциевой
соли с выходом 50% из пиридоксаль-4-М-диметилглицилгидразона и мета-
фосфорной кислоты с последующим частичным гидролизом образовавшегося
полифосфата и диазорасщеплением при помощи азотистой кислоты [229].
Гидразон пиридоксаля фосфорилируют и полифосфорной кислотой—смесью
85 %-ной ортофосфорной кислоты и пятиокиси фосфора [245—247]. Очи-
стку производят через соль с акридином равномолекулярного состава
[248].
Помимо изложенных выше вариантов синтеза пиридоксаль-5а-фосфата,
описаны другие пути его получения: фосфорилированием пиридоксаля
[232], пиридоксальазина [245], пиридоксальоксазолидона [249] и
Ь1,М-бис-(2-метил-3-окси-5-оксиметил-4-пиридилметилен)гидразида [ 181 ],
окислительным дезаминированием изоникотинилгидразона пиридоксаль-5а-
фосфата [237] и др.
В качестве производных фосфорных эфиров оксиметилпиридиновых
витаминов интересно отметить синтез несимметричных соединений: Р*-(пи-
ридоксаль-5а)-Р2-(аденозин-5')дифосфата, Р1-(пиридоксамин-5а)-Р2-(адено-
зин-5')дифосфата и Р1-(пиридоксин-5а)-Р1-(аденозин-5')дифосфата, которые
получены фосфоамидным методом из аденозин-5'-фосфата и соответствую-
щих фосфатов другого компонента [250].
Биологический синтез пиридоксаль-5а-фосфата может быть осуществлен
из пиридоксина при инкубации его с животными тканями [251 ] и с дрожжа-
ми [252]; этот синтез осуществлен также с ферментными системами
[251 ] и при добавлении пиридоксаля вместе с АТФ к суспензии высу-
шенных клеток Streptococcus faecalis [253].
Пиридоксин, пиридоксаль и пиридоксамин подвергаются фосфорилиро-
ванию в соответствующие 5а-фосфаты при действии пиридоксалькиназы
в присутствии АТФ [254, 255].
БИОХИМИЧЕСКИЕ ФУНКЦИИ ПИРИДОКСАЛЕВЫХ КОФЕРМЕНТОВ
Основная и важнейшая биологическая функция окспметилппридиновых
витаминов в животном организме состоит в том, что они в составе различ-
ных ферментов выполняют роль биокатализаторов. Они участвуют в мета-
болизме, главным образом в виде коферментов — пирпдоксаль-5а.-фосфор-
ного эфира и его взаимно-обратимой формы — пиридоксамин-5а-фосфор-
ного эфира; являясь простетической группой, эти коферменты про-
являют биокаталитические свойства только в соединении с «носителем»—
белком фермента.
Пиридоксаль-5а-фосфат входит в состав различных ферментов амино-
кислотного обмена: декарбоксилаз, аминотрансфераз (трансаминаз), ки-
нурениназы, триптофансинтетазы, цистеиндесульфгидразы, а также в
состав ферментов, осуществляющих пересульфирование аминокислот,
361
и многих других. Содержание пиридоксаль-5а-фосфата, например
в аланинаминотрансферазе, составляет 0,18 мкг в 1 мг фермента
[256].
Активность фермента как катализатора одной определенной химиче-
ской реакции находится в зависимости от специфического белка, свя-
занного с пиридоксаль-5а-фосфатом, а также от структуры компонентов, в
частности аминокислоты, участвующих в реакции [196].
Установлены функции свыше 50 различных пиридоксальфосфатных фер-
ментов. Так, они осуществляют переаминирование а-аминокислот с а-ке-
токислотами [257—2591, декарбоксилирование с отщеплением а- или р-кар-
боксила аминокислот [260—261], отщепление окси-, амино- и сульфгид-
рильных групп, расщепление 0-окси- и у-кето-а-аминокислот, замещение
Р-оксигрупп а-аминокислот, рацемизацию аминокислот [262] и другие
реакции (табл. 20).
Пиридоксаль-5а-фосфат в составе ферментых систем катализирует сле-
дующие химические реакции,
Таблица 20
Химические реакции, катализируемые пиридоксаль-ба-фосфатом
Тип реакции Разрыв связей Образование связей
Переаминирование Рацемизация ^-Замещение Дегидротирование, дегидроксилирова- ние, детиолирование Декарбоксилирование а-, [1-Расщепление Гидролитическое расщепление (Р, расщепление) Са—N; Са=О; Са—Н Са—Н Qj-o С-О; С—S; C«-N; Са—Н Са—С: Ср—С Са—Ср с₽-с7 Са—N: Са—О; Са—Н Са—Н Ср—С: С₽—S Са=О; С—Н с-н с—н С—н
Переаминирование
Важнейшую реакцию обмена аминокислот, а именно их переаминиро-
вание, открыли Браунштейн и Крицман в 1937 г. [257, 258]. В основе
механизма биокаталитической функции пиридоксальфосфатпротеида, пред-
ложенного Браунштейном [195—197, 257, 258, 263—267], лежит подвиж-
ность а-водородного атома остатка аминокислоты в образующихся азоме-
тинах (шиффовых основаниях) и способность последних к обратимым тау-
томерным перегруппировкам, к отщеплению смежного с двойной связью
карбоксила а-иминокислот, к гидролитическому расщеплению по иминной
связи и другим реакциям. Подвижность а-водородного атома остатка ами-
нокислоты установлена опытами с радиоактивными изотопами 1264,
265].
В реакциях переаминирования пиридоксаль-5а-фосфат (CLXVII) яв-
ляется переносчиком аминогруппы от одного компонента реакции к другому,
но сам остается в конце реакции в неизмененном состоянии [265]. Реакцию
переаминирования катализируют аминотрансферазы, простетической груп-
пой которых является пиридоксаль-5а-фосфат [256, 268, 269].
Под влиянием пиридоксальфосфатпротеида —аспартатаминотрансфера-
зы (молекулярная масса 112 000) пррисходит обратный перенос аминной
группы L-аспарагиновой кислоты на а-кетоглутаровую кислоту с образова-
нием £-глутаминовой кислоты 12701 и щавелевоуксусной кислоты
362
СООН 1 chnh2 + соон 1 , С=О Z2 соон chnh2 + СООН 1 с=о
СН2СООН 1 СН2СН2СООН СН2СН2СООН CHsCOOH
(этот биохимический тест применяется для диагностики инфаркта миокарда),
а-аланина (в присутствии а-кетоглутаровой кислоты) в L-глутаминовую
кислоту {257, 258, 268, 270] с образованием пировиноградной кислоты при
каталитическом участии аланинаминотрансферазы
соон соон 1 соон соон
CHNH, + со chnh2 4- СО
сн3 СН2СН2СООН СН2СН2СООН СН3
L-тирозина 1271, 272] в результате переаминирования той же а-кетоглута-
ровой кислоты—в L-глутаминовую кислоту с образованием п-оксифенил-
пировиноградной кислоты при действии тирозинаминотрансферазы
СООН СООН
НОСеН4СН2СНСООН + СО ' CHNH2 + HOC^CH2COCOOH
I I I
nh2 CH2CH2COOH ch2ch2cooh
а также L-аспарагиновой кислоты за счет аминирования пировиноградной
кислоты — в а-аланин [273 ]
СООН
I
chnh2 +
I
СН2СООН
соон
со
I
сн,
соон соон
I I
CHNHg + СО
СН3 СН2СООН
Подобным путем многие другие аминокислоты, например глицин, цисте-
ин, лейцин, кинуренин, при участии а-кетоглутаровой кислоты и соответ-
ствующих пиридоксальфосфатпротеидов обратимо переаминируются в L-глу-
таминовую кислоту. Переаминирование аминокислот происходит также с
другими а-кетокислотами.
Механизм реакции переаминирования изучался с применением дейте-
рированных соединений [265, 274] и метилированных аминокислот, исклю-
чающих образование шиффовых оснований, в результате чего аминокислота
лишалась сродства к аминотрансферазе [274 ].
Механизм реакции переаминирования в соответствии с теорией Браун-
штейна — Шемякина — Снелла [222, 266, 275—277] состоит в следующем.
Первоначально в первой фазе реакции происходит конденсация аминогруп-
пы а-аминокислоты с альдегидной (формильной) группой пиридоксаль-5а-фос-
фата (CLXVI I), связанного с белком, с образованием альдимина—промежу-
точного шиффова основания I (CLXXVI) с двойной связью между атомом
азота и формильным атомом углерода. При этом вследствие происходящего
смещения электронной плотности от а-углерода по системе сопряжения к
атому азота пиридинового цикла происходит ослабление связей Са и Ср
с атомом углерода карбоксильной группы и с атомом водорода. Необ-
ходимо отметить, что атом азота пиридинового кольца шиффова основания
протонирован [51, 53, 278].
В следующей фазе реакции это шиффово основание претерпевает тауто-
мерную перегруппировку с отщеплением протона и перемещением двойной
связи от формильного атома углерода к а-атому углерода аминокислотного
остатка с образованием переходной формы шиффова основания.
Третья фаза реакции заключается в поляризации переходного шиффова
основания с созданием активного электрофильного центра на формильном
363
атоме углерода, по которому и происходит атака протона, приводящая к
образованию шиффова основания II (CLXXVII), у которого между а-уг-
леродным атомом и атомом азота имеется иминная форма связи.
но
RCHCOOH
NH,
CLXYII
RCCOOH
II
Y
~CH
CHO
Н,ОРО3На-нгО;Ж-НО.
ClijOPOJT
H+ HO.
[CH3N
Н
RC)COOH
N
„CHjOPQjH, ноч
RCCOOH
N
I
СН
A-CHaOPOjHj
CH^N
CLXXVI
RCCOOH
II
Y
CH2 CH,NH,
ACHpPO3H, HO J. C^OPOjHj
Г JJ Y¥ + RCOCOOH
•сн3к
ей,"
СНз N'
C XXVII
CLXVIII
В заключительной фазе реакции происходит гидролитическое расщепле-
ние шиффова основания II по иминной связи Ca = N, в результате чего полу-
чается а-кетокислота и пиридоксамин-5а-фосфат (CLXVIII).
Таким образом, в реакции переаминироваиия одна коферментная фор-
ма — пиридоксаль-5а-фосфат переходит в другую форму — пиридоксамин-
5а-фосфат. Эта другая коферментная форма вступает в конденсацию с новой
а-кетокислотой, на которую и переносит аминогруппу, проходя последо-
вательно в обратном порядке все фазы реакции (через шиффовы основания II
и I). Реакция оканчивается образованием новой а-аминокислоты и пиридо-
ксаль-5а-фосфата в первоначальном неизмененном состоянии.
Рацемизация
Аланин-, метионин- и глутаматрацемазы, содержащие в качестве про-
статической группы пиридоксаль-5а-фосфат, катализируют обратимое прев-
ращение L^D-форм соответствующих аминокислот. Диаминопимелат-
эпимераза катализирует превращение Ь,£-2,6-диаминопимелиновой кис-
лоты в жзо-диаминопимелиновую кислоту.
Механизм реакции рацемизации основан на подвижности а-водородного
атома продукта конденсации аминокислоты и пиридоксаль-5а-фосфата —
шиффова основания I (CLXXVI). Образовавшееся шиффово основание I
испытывает таутомерную перегруппировку в. промежуточную форму шиф-
фова основания, которая теряет центр асимметрии на а-углеродном атоме
вследствие отщепления протона и образования Са = Ь1-связи.
Последующее присоединение протона приводит к шиффову основанию
типа I (CLXXVIII), но уже рацемизированной по Са структуры. Его гид-
ролиз связан с расщеплением на DL-a-аминокпслоту и пиридоксаль-5а-
фосфат.
н
।
R—С—СООН
Ан, +
Пиридоксаль-
фосфат
CLXVII
-НоО;+Н+
CLXXVI
364
HOOC-C-R
I
N
il
CH
HOOC-C-R
1
N
II
CH
CLXXVIII
NH,
R-C-COOH
+ H
Пиридоксаль-
фосфат
CLXVII
Так как рацемизация более легко протекает в условиях, когда среда име-
ет pH выше 7, то, возможно [279], протонизации атома азота пиридинового
цикла не происходит и механизм реакции связан с непротонированной фор-
мой шиффова основания (CLXXIX). Депротонизация этого соединения
приводит к аниону, не имеющему центра асимметрии, а дальше обычным
путем присоединяется протон и вновь возникает асимметрический центр в
шиффовом основании (CLXXX)
НООС-С—R
N.
II
I
HOOC-C-R
CLXXX
^-Замещение
Пиридоксалевые ферменты — цистеинсинтетаза, метилцистеинсинтетаза
и триптофансинтетаза—катализируют замещение Р-оксигруппы L-серина ти-
ольной, тиометильной и индольной группами с образованием L-цистеина,
5-метил-£-цистеина и L-триптофана [280, 281 ] соответственно
h2s
-н2о
HOCHjCHCOOH
nh2
CH3SH
-Н2О
- Н2О
HSCHjCHCOOH
NH,
CHjSCH2CHCOOH
NH,
,CH2CHCOOH
V* NH,
H
Механизм реакции на примере синтеза L-триптофана может быть пред-
ставлен как протекающий через шиффово основание I (CLXXXI), переход-
ную форму шиффова основания, затем отщепление заместителя в р-положе-
нии и образование активного переходного основания (CLXXXII), р-углерод
которого подвергается нуклеофильной атаке анионом индола с переходом
в промежуточную форму с последующим протонированием в шиффово осно-
вание (CLXXXIII), которое уже гидролитически, обычным путем, расщепля-
ется на L-триптофан и пиридоксаль-5а-фосфат.
365
HOCHgCHCOOH
' if
CH
СН3ОРОзН2
CLXXXI
Реакция ферментативного пересульфирования L-гомоцистеина при уча-
стии L-серина в L-цистеин с одновременным образованием L-гомосерина
протекает в присутствии пиридоксаль-5а-фосфата [282] в две фазы: перво-
начально происходит конденсация в цистатионин, который затем расще-
пляется на L-гомосерин и L-цистеин
CH,SH СН,ОН СН2—ь—CHg сн,он CH,SH
сн, CHNH, + CHNH, I сн, 1 CHNH, + н,0 1 сн, CHNH, + CHNH, 1 соон
СООН -н,о’ CHNH, 1 соон
СООН СООН соон
Промежуточно образующийся пиридоксальфосфатгомоцистеин показы-
вает эквивалентную активность пиридоксаль-5я-фосфата [283].
Дегидратирование, дегидроксилирование и детиолирование
Для а-аминокислот, замещенных в 0- или ^-положении гидроксильной,
сульфгидрильной или иной сильной электрофильной группой, характерна
способность при катализе пиридоксальфосфатными ферментами в при-
сутствии воды отщеплять эти функциональные группы одновременно с а-
аминогруппой и с образованием а-кетокислот.
Так, L-серин при участии сериндегидратазы дегидратируется в а-ами-
ноакриловую кислоту, которая затем не ферментативно присоединяет мо-
лекулу воды, в результате чего получается пировиноградная кислота и
аммиак
Н,0
HOCH.CI 1СООН------1- СН2 = ССООН-----> СНзСОСООН + NH40H
Также ведет себя и L-гомосерин, образуя при действии гомосеринде-
гидратазы а-аминокротоновую кислоту, затем не ферментативно переходя-
щую в а-кетомасляную кислоту и аммиак
Н2О
НОСН, СН, СНСООН-----> СН3СН=ССООН ------► СН, СН, СОСООН + NH,
I -Н,О I
NH, NH,
366
Та же самая а-кетомасляная кислота образуется из L-треонина при
каталитическом участии треониндегидратазы
Н2О
сн3 сн—снсоон---------♦ сн3 сн2 сосоон + NH3
I I
он nh2
Цистеиндесульфгидраза катализирует отщепление меркапто- и амино-
группы от L-цистеина, который при этом превращается в пировиноград-
ную кислоту [284]
Н2О
HSCH2CHCOOH--------> СНз СОСООН + NH*SH
I
nh2
Такая же реакция отщепления HS- и NH2-rpynn, расположенных в
а- и у-положениях L-гомоцистеина, идет с образованием а-кетомасляной
кислоты при каталитическом участии гомоцистеиндесульфгидразы
Н2О
HSCH2CH2CHCOOH--------> СНз сн» сосоон +nh4sh
nh2
8-Метил-£-цистеин отщепляет метилмеркаптан и аммиак с выделением
пировиноградной кислоты под влиянием S-алкилцистеинлиазы
HjP
СН3 SCH» СНСООН-------» СН3 СО СООН + СНз SH + NH,
I
NH2
и т. д.
Эти реакции на примере L-гомоцистеина протекают в результате взаимо-
действия аминокислоты и пиридоксаль-5а-фосфата, депротонирования обра-
зовавшегося шиффова основания I (CLXXXIV) в переходную форму осно-
вания, от которой отщепляется заместитель в виде аниона (HS~), причем
образующаяся новая форма шиффова основания (CLXXXV) с удлиненной
цепью сопряжения подвергается гидролизу с отщеплением аммиака и отде-
лением а-кетомасляной кислоты и пиридоксаль-5а-фосфата
HSCHXH/SHCOOH
N
II
СН
№ X снрро^ „+ НО CH.Ol’OjH,
In I! ~П Tl If ~TI3
HSCHjCHjCCOOH
V
сн
сн, и
3 н
CLXXXIV
CHjCH-CCOOH
N
II
CH
НО.Х.С^ОРОзН.
NHj
сн3 й
CLXXXY
CHjCHjCOCOOH
* Пфидоксал-
фосфат
CLVII
СН3 N
Декарбоксилирование
Пиридоксаль-5а-фосфат в соединении со специфическими белками
катализирует необратимое декарбоксилирование L-аминокислот и аромати-
ческих кислот. Так, моноаминомонокарбоновые аминокислоты декарбокси-
лируются в первичные амины [253, 263, 285, 286] с выделением двуокиси
углерода.
L-Валин под влиянием валиндекарбоксилазы превращается в изобутил-
амин
СНзСН-СНСООН
JsHs NH»
----> СНз снсн2 nh2 + со»
I
сн»
367
L-Тирозин при участии тирозиндекарбоксилазы переходят в тирании
n-НОС» н4 СН2 СНСООН----------> n-НОС. н4 СН2 СН, NH, + со»
nh2
Подобное декарбоксилирование испытывает L-гистидин под влиянием
гистидиндекарбоксилазы, превращаясь при этом в гистамин
N—п—СНзСНСООН N—тг—CHjCH2NH2
кн2 * +с°2
Диаминомонокарбоновые аминокислоты при катализе пиридокса-
левыми ферментами превращаются в диамины с выделением двуокиси
углерода.
L-Лизин при участии лизиндекарбоксилазы переходит в кадаверин
СН2СН,СН,СН2 СНСООН --< сн2сн,сн,сн2сн, + со8
1111
nh2 nh, nh2 nh,
Подобным образом орнитин декарбоксилируется под влиянием орни-
тиндекарбоксилазы в путресцин
СН, СН2 СН, СНСООН--► СН, СН, СН, СН2 + СО,
fki2 nh2 NH, nh2
Гуаниноаминокислота —L-аргинин декарбоксилируется при участии ар-
гининдекарбоксилазы в агматин
hn=cnhch2ch2 СН, СНСООН--* HN=CNHCH, СН,СН,СН, +СО,
NH2 NH2 NH, NH,
И T. д.
Пиридоксалевые ферменты также декарбоксилируют дикарбоновые ами-
нокислоты в монокарбоновые, например L-глутаминовую кислоту в у-ами-
номасляную:
НООССН, СН2 СНСООН--> НООС (СН,), nh2 + СО2
jIh,
а L-аспарагиновую кислоту в а-аланин и Р-аланин [287 ]:
НООССН, СНСООН НООССН, ch2nh2
NH, — СО2Х СН;’ <NH2> С00Н
при каталитическом участии глутаматдекарбоксилазы, аспартат-4-декар-
боксилазы и аспартат-1-декарбоксилазы соответственно. Следует отметить,
что р-аланин является составной частью витамина — пантотеновой кислоты
(CLXX XVI)
НОСН2 С (СН3), СН (ОН) CONHCH, СН, СООН.
CLXXXVI
Так как животный организм использует для метаболизма а-аминокислоты,
то настоящая реакция интересна тем, что приводит к образованию не ис-
пользуемой для построения белков Р-аминокислоты, однако необходимой
для биосинтеза кофермента А.
Реакция декарбоксилирования происходит только в отношении природ-
ных аминокислот, принадлежащих по своей стереохимической конфигу-
рации к L-ряду, т. е. в отношении аминокислот, из которых построены
368
почти все белки. При этой реакции все функциональные группы реагирую-
щих аминокислот должны находиться в свободном (незамещенном) состоя-
нии (285].
Дикарбоновые аминокислоты, помимо а-декарбоксилирования, могут
подвергаться реакции со-декарбоксилирования [285, 288], т. е. реакции,
при которой отщепляется карбоксил, наиболее отдаленный от атома угле-
рода, несущего аминогруппу. В частности, выше указывалось, что /.-аспа-
рагиновая кислота может подвергаться кака-, так и ^-декарбоксилированию.
Помимо а-аминокислот, ферментативному декарбоксилированию под-
вергаются п- и о-аминобензойные кислоты, которые при участии аминобен-
зоатдекарбоксилазы превращаются в анилин и двуокись углерода.
Механизм реакции декарбоксилирования аминокислот также основан на
свойстве пиридоксаль-5а-фосфата, входящего в состав фермента, образо-
вывать с аминогруппой аминокислоты шиффовы основания, способные к
обратимым таутомерным превращениям [2891.
В реакции декарбоксилирования первая фаза аналогична реакции пере-
аминирования. Во второй фазе реакции шиффово основание I (CLXXVI)
подвергается таутомерной перегруппировке с отщеплением протона и дву-
окиси углерода, сопровождающейся перемещением двойной связи
от формильного атома углерода к а-углеродному атому с образованием пе-
реходной формы шиффова основания. В результате поляризации этого
азометина вновь с перегруппировкой двойных связей и сосредоточением на
а-углеродном атоме электронной плотности по этому центру присоединяется
протон и возникает новое шиффово основание II (CLXXXVII)
R СНСООН
NH, +
Пиридомяиъфосфат
CLXV1I
RCH,
N
СН
HO^k^CHjOPOjH,
CH?N
RCHCOOII RCH
N N
сн СН
НО X HO^yCHpPQiH,
с£у СН, N
CLXXVI
RCHjbiH,
+
Пиридоксаль фосфат
CLXVIl
CLXXXVII
В последней фазе реакции происходит гидролитическое расщепление
соединения CLXXXVII по двойной связи, при этом получается амин и не-
измененный пиридоксаль-5а-фосфат (CLXVII).
а, 3-Расщепление
Треонинальдолаза и серинальдолаза, включающие в свой состав пири-
доксаль-5а-фосфат, вызывают каталитическое расщепление L-треонина по
Са —Ср - связи, при этом образуются глицин и ацетальдегид
СН, СН—СНСООН zr h,nch2 СООН + СН, сно
i>H nh2
Подобным образом идет расщепление L-серина на глицин и формальде-
гид.
369
I
НОСН2СНСООН H,NCH,COOH-f-HCHO
nh2
Обе реакции обратимы. Формальдегид переносится на тетрагидро-
фолиевую кислоту (ТГФК), которая принимает участие во второй реакции.
Расщепление L-треонила идет по общей принципиальной схеме через
шиффово основание (CLXXVI), которая предложена для декарбоксилиро-
вания аминокислот, с тем отличием, что переходная форма шиффова осно-
вания образуется не в результате отщепления двуокиси углерода, а путем
разрыва Са —С-связи с атомом углерода, несущим гидроксил
RCHCOOH
I
м
II
сн
HO.k^CHjOPOaHa
снсоон
II
N
I
CH.N
CLXXVI
CHOPOjHj
CLXXXYIU
В результате протонирования переходной формы шиффова основания
образуется шиффово основание II (CLXXXVIII), гидролитическое расщеп-
ление которого приводит к аминокислоте с укороченной цепью атомов
углерода и пиридоксаль-5я-фосфату.
Ниже приведен механизм реакции образования L-серина из глицина и
формальдегида, донором которого служит 5,10-метилен-ТГФК серинальдо-
лазы [290]
HjNcHjCooh^ -Н;О;+н*
Пфидоксальфосфат
CLXVII
С IL.С ООН снсоон
A N
сн -н* сн _______________
HO^Js-CHsOPOaHj-’---HO^AcHjOPOjH,
X J ХУ
CH.N с£*
CLXXXVIII
^-сн2..
-N-CH-CHj—N—
б,Ю-метилен- ТГФК
+ нри-н+
н н
----N-CH-CH3—N—
ТГФК
носн2снсоон
N HOCHjCHCOOH
СН HiOl-tf . NH,
HOL X XHjOPOjH, n . .
34 Пиридоксальфосфат
CLXXXI
В данной реакции к электрофильному центру Си присоединяется оксиме-
тильная группа.
Гидролитическое расщепление
Каталитическое гидролитическое расщепление по связи Ср—Ст харак-
терно для ркето-а-ампнокислоты — L-кинуренина. Оно протекает с обра-
зованием антраниловой кислоты и а-аланина при участии кинурениназы
.?о£на<~н соон
NH,
‘NH,
С соон
if + СНаСНСООН
NH, NH,
370
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ (К ГЛАВЕ VIII)
1. Е. Stiller, J. Keresztesy,
J. Stevens. J. Am. Chem. Soc.,
61, 1237 (1939).
2. R. К u h n, G. Wend t. Ber.,
71, 1118, 1534 (1938).
3. A. I c h i b а, К. M i c .h i. Sci.
Papers Inst. Phys. Chem. Res. (To-
kyo), 34, 623, 1014 (1938); Zbl., 1938,
II, 1782; 1939, II, 3999.
4. J. К e r e s z t e s y, J. Ste-
vens. J. Am. Chem. Soc., 60, 1267
(1938).
5. T. В i г c h, P. G у о r g y. Bio-
chem. J., 30, 304 (1936).
6. Y. N i 11 а, K. Takamura,
H. A s a d а. Яп. пат. 26728
(1964); С. A., 62, 11788 (1965).
7. S. H a r r i s, D. H e у 1, К. F о 1-
k e r s. J. Am. Chem. Soc., 66,
2088 (1944).
8. M. H о c h b e r g, D. Me 1 n i c k,
В. О s e r. J. Biol. Chem., 155,
129 (1944).
9. P. Gyorgy. Biochem. J., 29,
741, 760, 767 (1935).
10. S. Harris. J. Am. Chem. Soc.,
63, 3363 (1941).
II. S. H a r r i s, Webb, K.Fol-
k e r s. J. Am. Chem. Soc., 62, 3198,
3203 (1940).
12. T. Matsukawa. J. Pharm. Soc.
Japan, 60, 216 (1940).
13. M. U c h i b a у a s c h i. Chem.
Pharm. Bull. Japan, 9, 182 (1961).
14. F. К u m m e г о w, T. S a k u-
r a g i. Пат. США 2955155; С. A.,
55, 5539 (1961).
15. M. В. Балякин а, Е. С.
Жданович, А. Г. 3 е м с-
к о в а, Н. А. П р е о б р а-
женский. ЖОХ, 32, 1172
(1962).
16. L. S е n n е 1 1 о, С. А г g о u de-
li s. J. Org. Chem., 33, 3983 (1968).
17. Т. М о г i, А. М i t a b i у а.
Яп. пат. 23945 (1968); С. А., 70,
57651 (1969).
18. Франц, пат. 1479985; С. А., 68,
114447 (1968).
19. N. S и g i m о t о , S. I m a d а.
Англ. пат. 1070120; С. А., 68,
78148 (1968).
20. N. S и g i ni о t о, S. I m a d а.
Яп. пат. 9348, 9341 (1967); С. А.,
68, 68892, 68893 (1968).
21. Т. Kuroda, R. Tanaka,
М. М а е d a. Bitamin, 35, 20 (1967).
22. К. О к и m и г a, S. I m a d о,
Т. Oda. Bitamin, 35, 375 (1967).
23. Англ. пат. 1101369; С. А., 69, 10374
(1968).
24. Т. Kuroda, Vitamins, 28, 21
(1963); РЖХ, 1964, 7Ж423.
25. К. О i к е, Т. К и г о d а. Яп.
пат. 5034 (1963); С. А., 61, 6997
(1964).
26. U. Schmidt, G. Giessel-
m а п n. Lieb. Ann., 657, 162 (1962).
AngeV. Chem., 72, 709 (1960);
27. G. S c h о г г e. Пат. США 3086023
С. A., 59, 9995 (1963); пат. ФРГ
1197455; РЖХ, 1968, 15Н426.
28. Бельг, пат. 640118; С. А., 63, 586
(1965).
29. Франц, пат. М948; С. А., 58, 9032
(1963).
30. Бельг, пат. 659401; С. А., 64, 3500
(1966).
31. М. I w a n a m i, I. О s a w а,
М. Murakami. Bitamin, 36,
122 (1967); Яп. пал 11369 (1969);
С. А., 71, 70497 (1969).
32. Л. А. Петрова, Н. Н. Бел ь-
ц о в а. ЖОХ, 32, 274 (1962).
33. О. Z i m a, G. S с h о г г е. Пат.
ФРГ 1135460; РЖХ, 1964, 7Н212.
34. Т. Kuroda. Vitamins, 30, 431
(1964).
35. Англ. пат. 927666; С. А., 60, 5467
(1964).
36. G. S с h о г г е, пат. ФРГ 1193049;
РЖХ, 1967, 19Н337.
37. X. Я м а д а, И. X и р а д а,
А. Татэмацу. Яп. пат. 2888
(1963); РЖХ, 1965, 8Н262.
38. Neth. Appl. 6408315; С. А., 63,
2977 (1965).
39. Y. D е g и с h i. Яп. пат. 6790
(1968); С. А., 69, 87010 (1968).
40. М. I wanami, I. О s a w а,
М. М и г а к a m i. Bitamin, 36,
119 (1967).
41. J. Baddiley, A. Mathias.
J. Chem. Soc., 1952, 2583.
42. А. С о h e n, E. H и g h e s. J.
Chem. Soc., 1952, 4384.
43. W. К о г у t п у к. J. Org. Chem.,
27, 3724 (1962).
44. W. К о г у t h у к, W. W i е d е-
m а п. J. Chem. Soc., 1962, 2531.
45. Т. Sakuragi, F. К и m m е-
г о w. J. Org. Chem., 24, 1032 (1959).
46. Y. N a к a i, N. О h i s h i,
S. S h i m i z u, S. F и к и i. Bi-
tamin, 35, 213 (1967).
47. P. S a h. J. Am. Chem. Soc., 76,
300 (1954).
48. К- О к и m и г а. Яп. пат. 2716
(1968); С. А., 69, 59138 (1968).
49. М. В и е 1 1, R. Н а п s е п. J.
Am. Chem. Soc., 82, 6042 (1960).
50. Е. Snell. J. Biol. Chem., 157,
491 (1945); J. Am. Chem. Soc.. 67,
194 (1945).
51. D. M e t z 1 e r. J. Am. Chem. Soc.,
79, 485 (1957).
52. D. M e t z 1 e г, E. S n e 1 1. J.
Am. Chem. Soc., 74, 979 (1952).
53. H. C h r i s t e n s e n. J. Am. Chem.
Soc., 79, 4073 (1957); 80, 99 (1958);
81, 6495 (1959).
54. L. D a v i s, F. R о d d y,
D. M e t z 1 e r. J. Am. Chem. Soc.,
83, 127 (1961).
55. K. N a к a g a w a, M. M a t sui-
Nippon Nogei Kagaku Kaishi, 42,
300 (1968); C. A., 70, 57582 (1969).
56. D. H e у E. L и z, S. H a r-
371
г i s, К. F о 1 к e r s. J. Am.
Chem. Soc., 70, 1670, 3669 (1948);
74, 414 (1952).
57. D. H e у I, S. H a r r i s,
K. F о i к e г s. J. Am. Chem. Soc.,
70, 3429 (1948).
58. D. H e у 1. J. Am. Chem. Soc., 70,
2434 (1948).
59. A. W i 1 s о n, S. H arr is. J.
Am. Chem. Soc., 73, 4693 (1951).
60. R. Kuhn, J. Low. Ber., 72,
1453 (1939).
61. A. Ormsby, A. Fischer,
F. Schlenk. Arch. Biochem., 12,
79 (1947).
62. D. Xi e 1 n i с к, M. H о ch-
b e r g, H. H i m e s, B. Oser.
J. Biol. Chem., 160, 1 (1945).
63. H. Gibbs. J. Biol. Chem., 72,
649 (1927).
64. S. О h d a к e. Bull. Agr. Chem.
Soc. Japan, 8, 11 (1932).
65. P. Gyorgy. J. Am. Chem. Soc.,
60, 983 (1938).
66. R. Kuhn. G. Wendt. Ber., 71.
780 (1938).
67. S. L e p к о v s к y. Science, 87, 169
(1938).
68. R. К h u n, G. W e n d t. Ber.,
72, 305 (1939).
69. R. К u h n, H. A n d er sag,
K. Westphal, G. Wendt.
Ber., 72, 309 (1939).
70. R. Kuhn, G. W e n d t, K. W es t-
p h a 1. Ber., 72, 310 (1939).
71. R. К u h n, G. Wendt. Ber.,
71, 78 (1938).
72. O. F о 1 i n, W. D enis. J.
Biol. Chem., 12, 239 (1912); 22, 305
(1915).
73. H. G i b b s. J. Biol. Chem., 72,
649 (1927).
74. A. I c h i b а, К. M i c h i. Sci.
Papers Inst. Phys. Chem. Res. (To-
kyo), 35, 73 (1938); 36, 1 (1939).
75. S. H а г г i s, E. S t i 1 1 e r,
K. F о 1 к e г s. J. Am. Chem. Soc.,
61, 1242 (1939).
76. J. В a r d h a n. J. Chem. Soc., 1929,
2223.
77. W. Wenner, J. Pl at i. J.
Org. Chem., 11, 751 (1946).
78. S. M о r i i, К. M a к i no. Enzy-
mologia, 7, 385 (1939).
79. S. Harris. Англ. пат. 593796;
С. A., 42, 1710 (1948).
80. W. В r u с e, H. С о о v e r. J.
Am. Chem. Soc., 66. 2092 (1944).
81. S. Harris, пат. США 2422627;
С. A., 41, 5554 (1947).
82. Швейц, пат. 217228 (1942); С. А.,
42, 5472 (1948).
83. Синтезы органических препаратов.
Сборн. 2, ИЛ, 1949, стр. 607.
84. Г. С. Михайлов, В. Кон ь-
к о в а, А. К- Б а с о в а. ЖПХ,
25, 1329 (1952).
85. Синтезы органических препаратов.
Сборн. 1, ИЛ, 1949, стр. 498.
86. Швейц, пат. 217231; С. А., 42, 5472
(1948).
87. S. Н а г г i s. К- F о 1 k е г s.
J. Am. Chem. Soc., 61, 3307 (1939);
пат. США 2226754; С. А., 36, 2378
(1942); Англ. пат. 557805; С. А., 39,
3398 (1945).
88. S. Н а г г i s, К. F о 1 к е г s.
J. Am. Chem. Soc., 61, 1245 (1939);
англ. пат. 543615; С. А., 36, 5957
(1942).
89. S. Н а г г i s. Пат. США 2382876;
С. А., 40, 682 (1946).
90. Д. И с и к а в а, К- С и р а к а-
в а, И. У с v и. Яп. пат. 6523
(1956); РЖХ, 1959, 46917.
91. К. Р a t z е 1 t, М. L i 2 к а,
V. К 1 е i п е г о v а. Чехосл. пат.
106515; РЖХ, 1964, 9Н231.
92. Е. Т е s t а, А. V е с с h i. Gazz.
chim. ital., 87, 467 (1957).
93. Y. Murakami, A. К u r i t a,
’у/, O. Y о n e у а. Яп. пат. 22886
•yfV (1963); C. A., 60, 5466 (1964).
J ®4. F. C u i b a n, S. C i 1 i a n n.
•j.Rec. chim. (Bucharest), 10, № 2, 74
•».S (1959).
£?;S5. M. В. Б а л я к и и а, Е. С.
Жданович, Н. А. Преоб-
раженский. ЖПХ, 35, 1864
(1962).
96. М. В. Балякина, Е. С.
Жданович, Н. А. Преоб-
раженский. ЖОХ, 31, 542
(1961).
97. М. М. Гольдман, Р. А.
Ш т е р н е р. Авт. свид. СССР
94345; Бюлл. изобрет., 1952, № 10,
9.
98. А. Н о w s h i р, G. Р h i 1-
I i р s. Англ. пат. 844883; РЖХ,
1961, 16Л380.
99. S. Н а г г i s. пат. США 2422616,
2422618, 2422620, 2422621; С. А.,
41, 5554 (1947); пат. США 2422195;
С. А., 41, 5555 (1947); пат. США
2483137; С. А., 44, 1545 (1950).
100. Пат. США 2272198; С. А., 36,
3634 (1942); англ. пат. 557804;
С. А., 39, 3398 (1945).
101. S. Harris. Пат. США 2422619,
2422628; С. А., 41, 5554, 5555
(1947).
102. G. С а г г а г а, Е. Testa.
Англ. пат. 765526; С. А., 51, 12152
(1957).
103. Е. Т е s t a, F. F a v a. Chi-
mia (Switz), 11, 307 (1957).
104. Англ. пат. 834451; С. А., 55,
17657 (1961).
105. S. Harris. Пат. США 2399347;
С. А., 40, 4181 (1946).
106. Е. Stiller. Пат. США 2372690;
С. А., 39, 4199 (1945); пат. США
2476464; С. А., 44, 1145 (1950).
107. U. Schmidt. Lieb. Ann., 657,
156 (1962).
108. U. Schmidt. Angew. Chem.,
69, 138 (1957).
109. S. Harris. Пат. США 2248078;
С.A., 35, 6741 (1941); пат. США
2325053; С. А., 38, 221 (1944).
110. Англ. пат. 534916; С. А., 36, 1739
(1942).
111. Н. Wuest, J. Bigot, Т. de
372
Boer, В. Wai, J. W i b a u t.
Rec. trav. chim., 78, 244 (1959).
112. H. E. Альперович. Ме-
тоды получ. радиоакт. препар., сб.
статен М., изд. «Наука», (1962, 37).
113. Герм. пат. 732238; С. А., 38,
1251 (1944); Герм. пат. 742428;
С. А., 39, 2624 (1945); Бельг, пат.
447824; С. А., 39, 1510 (1945); пат.
США 2389054 С. А., 40, 905
(1946); Швейц. пат. 224069,
230439, 236162; С. А., 43, 1917,
2741, 7051 (1949).
114. М. Hoffer. Пат. США
2410938—2410941; С. А., 41, 1395
(1947).
115. J. М о w a t, F. Pilgrim,
G. Carlson. J. Chem. Soc., 65,
954 (1943).
116. Англ. пат. 567611; С. A., 41, 2755
(1947).
117. Синтезы органических препара-
тов, сборн. I, М., ИЛ, 1949, с. 534.
118. В. М. Березовский. Авт.
свид. СССР 94217; Бюлл. изобрет.,
1952, № 9, 7.
119. R. В 1 а с k w о о d, G. Н е s s,
С. L а г г a b е е, F. Р i 1 g г i т.
J. Am. Chem. Soc., 80, 6244 (1958).
120. В. М. Березовский. Авт.
свид. СССР 93723; .Бюлл. изобрет.,
1952, № 5, 8.
121. В. М. Березовский. . Авт.
свид. СССР 97249; Бюлл. изобрет.,
1954, № 2, 15.
122. М. М a t s u i, А. К о b а у а-
shi, S. Watanabe. Agr.
Biol. Chem., 25, 240 (1961); C. A.,
55, 13424 (1961); Яп. пат. 8713
(1963); С. A., 59, 11442 (1963).
123. A. I c h i b a, S. Emoto. J.
Sci. Research Inst. (Tokyo), 43, 30
(1948); C. A., 43, 4670 (1949).
124. A. 1 c h i b a, S. E m о t o,
M. N a g a i. J. Sci. Research, Inst.
(Tokyo), 43, 23 (1948); C. A., 43,
4673 (1949); Яп. пат. 158276; С. A.,
44, 1134 (1950).
125. Англ. пат. 626368; С. А., 44, 2570
(1950).
126. М. S с о t t, L. N о г г i s,
G. Н е u s е г, W. В г u с е. J.
Am. Chem. Soc., 67, 157 (1945).
127. J. M о w a t. Пат. США 2475569;
С. A., 44, 2570 (1950).
128. К. V e r r i I 1, A. S c h n e i-
der. Англ. пат. 686012; С. A., 48,
2784 (1954).
129. T. M a t s u k a w a, K. Shi-
ra k a w a. J. Pharm. Soc. Japan,
71. 1408 (1951); C. A., 46, 8113
(1952).
130. A. Cohen, J. Haworth,
E. H u ghes. J. Chem. Soc.,
1952, 4374.
131. R. Jones, E. К о r n f e 1 d.
J. Am. Chem. Soc., 73, 107 (1951).
132. R. J о n e s. J. Am. Chem. Soc.,
73, 5244 (1951).
133. R. Jones, пат. США 2744114;
С. A., 51, 2056 (1957).
134. R. Jones. J. Am. Chem. Soc.,
73, 5610 (1951).
135. R. J ones. пат. США 2748135;
С. A., 51, 2877 (1957).
136. В. van d e r Wai, T. de
Boer, H. Huisman. Rec.
grav, chim., 80, 228 (1961).
137. C. Larrabee. Пат. США
2860141; С. A., 53, 7205 (1959).
138. S. Gabriel, J. Colman.
Ber., 33, 988 (1900).
139. A. I c h i b а, К. M ich i. Sci.
Papers Inst. Phys. Chem. Res.
(Tokyo), 36, 173 (1939); C. A., 33,
8201 (1939).
140. R. К u h n, K. Westphal,
G. Wendt, O. Westphal.
Naturwiss., 27, 469 (1939).
141. Y. Suzuki. Yakugaku Zasshi,
81, 792 (1961).
142. L. Szabo. Пат. США 2410531;
С. A., 41, 1714 (1947).
143. L. Szabo. Пат. США 2359260;
С. A., 39, 1965 (1945).
144. К. Westphal. Пат. США
2349318; С. А., 39, 1513(1945).
145. А. С о h е n, Е. Hughes.
Англ. пат. 625997, 629423; С. А.,
44, 3533, 7353 (1950).
146. К. М a k i п о, S с h. М о г i i>
F. S h о n g, W. T a g a m i. Bull.
Chem. Soc. Japan, 19, 1 (1944);
C. A., 41, 4493 (1947).
147. A. Cohen. Англ. пат. 629450;
С. A., 44, 7354 (1950).
148. N. С 1 a u s о п-К a a s, N. E 1-
m i n g, Z. T у 1 e. Acta
chim. scand., 9, 1 (1955).
149. N. С 1 a u s о n-K a a s, P.Ne-
b e n s k о v. Acta chim. scand.,
9, 14 (1955).
150. N. E 1 m i n g, N. С 1 a u son-
K a a s. Acta chim. scand., 9, 23
(1955).
151. E. К о г n f e 1 d, R. Jones.
J. Org. Chem., 19, 1671 (1934).
152. H. W i 1 1 i a m s, P. Kauf-
mann, H. M о s her. J. Org.
Chem., 20, 1139 (1955).
153. N. Clause n-K a a s, N. E 1-
m i n g. Пат. ФРГ 1134379; С. A.,
58, 1436 (1963).
154. J. N i e 1 s о n, N. E 1 m i n g,
N. С 1 a u s о n-K a a s. Acta
chem. scand., 14, 938 (1960).
155. N. С I a u s о n-K a a s, N. El-
mi n g. Пат. ГДР 14043; РЖХ,
1960, 6081; пат. США 2875207;
.С. А., 54, 569 (1960).
156. Т. N i е 1 s е п, N. Е 1 m i n g,
N. С 1 a u s о n-K a a s. Acta
chem. scand., 14. 938 (1960).
157. E. Kornleld. J. Org. Chem.,
20, H35 (1955).
158. N. С I a u s о n-K a a s, F. Lim-
bo r g, K. G I e n s. Acta chim.
scand., 6, 531 (1952).
159. Датск. пат. 80973, 80971; С. А.»
50, 14811 (1956).
160. F. L i m b о г g, N. С 1 a u s о n-
K a a s. Acta chim. scand., 7, 234
(1953).
373
161. P. Stevens. Пат. США
2734063; С. А., 50, 13099 (1956).
162. Р. Stevens. Пат. США
2680743; С. А., 49, 6315 (1955).
163. Р. Pollak. Пат. США 3024245;
РЖХ, 1963, 16Н223.
164. Р. Pollak. Пат. США 2904551;
С. А., 55, 572 (1961).
165. Г. Я. Кондратьева. Хим.
наука пром., 2, 666 (1957); Изв.
АН СССР, ОХН, 1959, 484.
166. Г. Я. Кондратьева
С. Huang, ДАН СССР, 141,
628, 861 (1961)..
167. Е. Harris. R. Firestone,
К. Pfister, R. Boett-
cher, T. Cross, R.Currie,
M. Monaco, E. Peterson,
W. Reuter. J. Org. Chem., 27,
2705 (1962).
168. К. P f i s t e r, E. Harris,
R. Firestone. Пат. США
3227724, 3227721; РЖХ, 1967,
17H390; 1968, 1H477.
169. M. В. Б а л я к и н а, 3. Н. Ж у-
к о в а, Е. С. Жданович.
ЖПХ, 41, 2324 (1968).
170. Г. Я. Кондратьева,
Б. А. К а з а н с к и й, Г. Н.
Прошина, Н. А. Ошуева.
Авт. свид. СССР 213879; РЖХ,
1969, 3H433.
171. R. F i г е s t о n е, Е. Harris,
W. Reuter. Tetrahedron, 23,
943 (1967).
172. Е. Harris, D. Rosen-
burg, E. Chamberlin.
Пат. США 3381014; С. A., «9,
52023 (1968).
173. W. К i m e 1, W. L e i m g r u-
b e г. Франц, пат. 1384099; С. A.,
63, 4263 (1965).
174. Neth. Appl. 6506703; C. A., 64,
15851 (1966).
175. Г. Я. Кондратьева. Авт.
свид. СССР 196854; С. А., 69, 67240
(1968).
176. Р. Pollak. Пат. США 3365461;
С. А., 69, 35963 (1968).
177. М. К a w a z и. Яп. пат. 18627
(1967); С. А., 69, 10366 (1968).
178. Т. М i k i, Т. Matsuo. Yaku-
gaku Zasshi, 87, 323 (1967).
179. Франц, пат. 1343270; С. А., 60,
11991 (1964).
180. Т. Yoshikawa, F. I s h i-
k a w a, T. N a i t o. Chem.
Pharm. Bull. Japan. 13, 878 (1965).
181. Neth. Appl. 6614801, 6614802;
C. A., 68, 87190, 87191 (1968).
182. Бельг, пат. 671385; С. A., 65,
15318 (1966).
183. Франц, пат. 1400843; С. А., 63,
9922 (1965).
184. S. Н а г г i s, D. Н е у 1,
К. F о 1 к е г s. J. Am. Chem.
Soc., 66, 2088 (1944); J. Biol.
Chem., 154, 315 (1944); пат. США
2497730; С. А., 44, 5923 (1950).
185. Е. Snell. Англ. цат. 603289,
603290; С. А., 43, 725 (1949).
186. Р. Karrer, М. Visconti-
n i, О. F о s t e г. Helv. Chim.
Acta, 31, 1004 (1948).
187. M. В. Б а л я к и и a, E. С. Ж да-
но в и ч, Н. А. Преоб-
раженский. ЖОХ, 31, 2983
(1961); авт. свид. СССР 133885;
РЖХ, 1961, 16 Л 381.
188. Е. Т е s t a, F. F a v a. Chimia,
11, 310 (1957).
189. Англ. пат. 773354; С. А., 51, 14833
(1957).
190. О. М а п о u s е к. Coll. Czechosl.
Chem. Comm., 25, 2250 (1960).
191. К. О к у м у p a. Vitamins, 23,
236 (1961); РЖХ, 1962, 23Ж412.
192. Т. Kuroda. Яп. пат. 9090,
9491 (1962); С. А., 59, 5140, 10005
(1963).
193. G. Cartwright, М. Wint-
robe, S. Humphreys. J.
Biol. Chem., 153, 171 (1944).
< 7: 194. E. M с H e n r y, G. Ga v i n. J.
Biol. Chem., 138, 471 (1941).
195. A. E. Браунштейн. Био-
' ‘n?. химия аминокислотного обмена,
Изв. АМН СССР, 1949.
196. А. Е. Браунштейн. ДАН
СССР, 65, 715 (1949).
4:.^197. А. Е. Б р а у н ш т е й н, Е. Г о-
рячеикова. Биохимия, 14,
1631 (1949).
198. D. Reid, S. L е р к о v s к у.
J. Biol. Chem., 155, 299 (1944).
199. О. W i s s, H. F u c h s. Expe-
rientia, 6, 472 (1950).
200. E. M о 1 1 e r. Z. physiol. Chem.,
260, 246 (1939); Angew. Chem.,
53, 204 (1949).
201. E. S n e 1 1. J. Am. Chem. Soc.,
66, 2082 (1944); J. Biol. Chem.,
154, 313 (1944); 158, 497 (1945).
202. A. E. Браунштейн.
Украин. биохим. журн., 22, 273
(1950).
203. W. М с N u t t, S. S пе 1 1. J.
Biol. Chem., 182, 557 (1950).
204. I. R a b i п о w i t z, E. S n e 1 1.
J. Biol. Chem., 176, 1157 (1948).
205. M. M e й с e л ь, H. Помощ-
ник ob а. Биохимия, 17, 593
(1952).
206. R. Williams. J. Am. Med.
Assoc., 119, 1 (1942).
207. iS. Harris, A. Wilson. J.
Am. Chem. Soc., 63, 2526 (1941).
208. M i r i s h i I k a w a, E. S n e 1 1.
J. Am. Chem. Soc., 76, 637 (1954).
209. P. M e 1 i n s, D. M a r s c h a 1 1.
J. Med. Chem., 10, 1157 (1967).
210. H. D e v о 1 1, F. Kipping.
J. Chem. Soc., 1953, 1935.
211. А. С о h e ii, J. S i 1 k. J. Chem.
Soc., 1952, 4386.
212. D. He у I, E. L u z, S. H a r-
r i s, K. F о 1 k e r s. J. Am.
Chem. Soc., 75, 4079, 4080, (1953).
213. R. M a г i e 1 1 a, E. В e 1 c h e r.
J. Am. Chem. Soc., 74, 4049 (1952).
214. W. Ко г у t n у k, E. Kris,
R. S i n g h. J. Org. Chem., 29,
574 (1964).
215. W. О t t. Proc. Soc. Exp. Biol.
374
Med., 61, 125 (1946); 66, 215 (1947).
216. R. Mariella, J. Leech.
J. Am. Chem. Soc., 71, 331 (1949).
217. D. He у 1, S. Harris,
K. Fol ke r s. J. Am. Chem.
Soc., 75, 663 (1953).
218. S. К u w a n a, K. Y a m a u c h i.
Chem. Pharm. Bull. (Japan), 8,
491, 497 (1960).
219. K. Y a m a d a, S. S a n a к i,
S. H a у m i. J. . Vitaminol. (Kyo-
to), 3, 68 (1957).
220. H. К. К о ч e т к о в, P. M. X o-
мутов, M. Я. К a p n e й-
c к и й, Э. И. Б у д о в с к и й,
Е. С. С е в е р и и. ДАН СССР,
126, 1132 (1959).
221. Р. М. Хомутов, М. Я. Кар-
пе й си и й, Е. С. С е в е р и н.
Биохимия, 26, 772 (1961).
222. D. М е t z 1 е г, М. J к a w а,
Е. S n е 1 1. J. Am. Chem. Soc.,
76, 648 (1954).
223. D. Н е у I, Е. L u z, S. Н а г-
г i s. J. Am. Chem. Soc., 73, 3437
(1951).
224. Р. Karrer, М. Visconti-
n i. Helv. Chim. Acta, 30, 52, 524
(1947).
225. D. H e у 1, S. H a r r i s. J. Am.
Chem. Soc., 73, 3434 (1951).
226. W. Um brei t, I. G и n s a-
1 и s. J. Biol. Chem., 179, 279
(1949).
227. M. Viscontini, C. Bonet-
t i. Helv. Chim. Acta, 34, 2438
(1951).
228. D. H e у 1, E. L и z, S. H a r-
r i s, K. F о I к e r s. J. Am.
Chem. Soc., 73, 3430 (1951).
229. M. V i s с о n t i n i, С. E b n o-
t h e r, P. Karrer. Helv. Chim.
Acta, 34, 1834, 2198 (1951).
230. Neth. Appl. 6514131; C. A., 65,
12176 (1966).
231. M. Yosh ika wa, T. К a t o,
T. Takenishi. Пат. США
3365460; С. A., 69, 27257 (1968).
232. T. Kuroda. Vitamins, 28, 211
(1963); Яп. пат. 19739 (1963);
С. А., 60, 4116 (1964).
233. А. Т г i е, J. U п о, Y. Yoshimu-
га. Яп. пат. 39267 (1970); С. А.,
74, 87842 (1971).
234. Т. Tanaka. Yakugaku Zasshi,
79, 1301 (1959).
235. D. Н е у 1, Е. L и z, S. Н а г-
r i s, К. F о 1 к е г s. J. Ат.
Chem. Soc., 73, 3436 (1951).
236. Е. Peterson, Н. Sober,
A. Meister. J. Am. Chem. Soc.,
74. 570 (1952).
237. T. К и г о d a. J. Vitaminol., 10,
252 (1964).
238. M. V i s с о n t i n i, С . E b ne-
ther, P. Karrer. Helv. Chim.
Acta, 34, 2199 (1951).
239. E. P e t e r s о n, H. Sob e r. J.
Am. Chem. Soc., 76, 169 (1954).
240. S. H a r r i s, A. Wilson.
Пат. США 2660061; С. A., 48,
12811 (1954).
241. R. Long. Англ. пат. 749800;
С. A., 51, 1297 (1957).
242. I. G и n s a 1 и s, W. Um-
bre i t, W. Bellamy, C.
Foust. J. Biol. Chem., 161, 743
(1945).
243. A. M i t a b i у а. Яп. пат. 2533
(1963); С. A. 59, 11445 (1963).
244. M. I w a n a m i, T. N и m at a,
M. M и г a к a m i. Bull. Chem.
Soc. Japan, 41, 161 (1968).
245. К. О к и m и г а, Т. Oda,
Т. N i s h i h a r a. Bitamin, 35,
380, 384 (1967).
246. К. О к и m и r a, T. Morita-
n i. Англ. пат. 1076910; С. A., 68,
59443 (1968).
247. Франц, пат. 1477940; С. А., 68,
29610 (1968).
248. М. V i s с о n t i n i, P. Kar-
rer. Helv. Chim. Acta, 35, 1924
(1952).
249. G. S c h о г г e. Пат. США
3124587; С. A., 61, 3078 (1964).
250. T. Kuroda. Vitamins, 28, 354,
362 (1963); РЖХ, 1965, ЗЖ445,
446; Яп. пат. 12915 (1965); С. А.,
63, 13393 (1965).
251. А. Труфанов, В. Кирса-
нова, 3. Соловьева. Био-
химия, 12, 482 (1947).
252. А. Труфанов, 3. Со-
ловьева. Биохимия, 14, 327
(1949).
253. 1. Gunsalus, W. Bellamy,
W. U m b г е i t. J. Biol.
Chem., 155, 357, 685 (1944); 160,
461 (1945); Arch. Biochem., 7, 185
(1945).
254. J. H u r w i z. J. Biol. Chem., 205,
935 (1953); Biochim. Biophys. Acta,
9, 496 (1952).
255. D. McCormick, M. Grego-
ry, E. S n e 1 1. J. Biol. Chem.,
236, 2076 (1961).
’256. A. E. Браунштейн,
M. К p и п к а я, О. С а м a p и-
н a, Е. G а 1 е, Н. Tomlin-
son. Биохимия, 11, 423 (1946).
257. А. Е. Браунштейн,
М. К р и ц м а н. Биохимия, 2,
242, 859 (1937).
258. А. Е. Браунштейн. Сборн.
Усп. совр. биохимии, 1, 40 (1947).
259. Е. S n е 1 1. J. Am. Chem. Soc.,
67, 194 (1949).
260. W. U т b г е i t, I. G u n s а 1 u s.
J. Biol. Chem., 159, 333 (1945).
261. S. R о t h b e г g, D. Stein-
berg. J. Am. Chem. Soc., 79,
3274 (1957).
262. W. W о о d, I. Gunsalus.
J. Biol. Chem., 190, 403 (1951).
263. Б. П ю л ь м а и, А. П ю л ь-
м а н. Квантовая биохимия, М.,
изд-во «Мир», 1965.
264. А. Е. К о н и к о в а, М. Кри ц-
м а н, Р. Тейс. Биохимия, 7, 86
(1942).' .1
265. А. Е. К о и и к о в а, Н. Д о б- ,
берт, А. Браунштейн.
375
Биохимия, 12, 465, 556 (1947).
266. А, Е. Б р а у н ш т е й н, М. М. Ше"
мякни. ДАН СССР, 85, 1115
(1952); Биохимия, 18, 393 (1953).
267. М. М. Шемякин, Л. Щуки-
и а. Успехи химии, 26, 528 (1957);
Биохимия, 22, 214 (1957).
268. Н. Lichtstein, I. Gunsa-
1 u s, W. Umbreit. J. Biol.
Chem., 161, 311 (1945).
269. D. Green, L. L e 1 о i г, V. N o-
ci to. J. Biol. Chem., 161,
559 (1945).
270. F. S c h 1 e n k, E. S n e 11. J.
Biol. Chem., 157, 425 (1945).
271. L. Feldman, I. G unsal us.
J. Biol. Chem., 187, 821 (1950).
272. J. H i r d, E. R owsell. Na-
ture, 166, 517 (1950).
273. M. Карягина, Биохимия, 4,
168 (1939).
274. Ц. Осипенко. ДАН СССР, 75,
91 (1950).
275. А. Е. Браунштейи. В кн.
The Enzymes, vol. II, р. 113,
New York, 1960.
276. E. Snell. Vitamins and Hor-
mones, 16, 77 (1958).
277. E. S n e 1 1, W. J e n kins. J.
Cellular Comp. Phys., 54, 161 (1959).
278. D. Me tzler, E. S n e 11. J.
Am. Chem. Soc., 77, 2431 (1955).
279. J. О 1 i v а г d, D. M e t z 1 e r,
E. S n e 1 1. J. Biol. Chem., 199,
669 (1952).
280. C. Yanofsky. J. Biol. Chem.,
224, 783 (1957).
281. D. Metzler, J. Longene-
c k e r, E. S n e 1 1. J. Am. Chem.
Soc., 76, 639 (1954).
282. A. E. Б p а у и ш т e й и, E. Г o-
ряченкова. ДАН СССР, 74,
529 (1950).
283. К. Okumura, К. Kotera,
К. Masukawa, Т. Danno
et al. Bitamin, 35, 301, 308, 322
(1967).
284. A. E. Браунштейи,
P. Азарх. ДАН СССР, 71, 93
(1950).
285. M. Мардашев. Успехи сов-• £,
ременной биологии, 28, 365 (1949).
286. J. В a d d i 1 е у, Е. Gale. Na- i‘
ture, 155, 727 (1945). r
287. A. V i r t a n e n, T. L a i n e.
Enzymologia, 3, 266 (1937).
288. С. Мардашев, Л. Семина,
P. Э т и н г о ф, А. Баляс-
н а я. Биохимия, 14, 44 (1949).
289. Ц. Осипенко. ДАН СССР, 75,
225 (1950).
290. D. Sanadi, М. Bennett.
Biochim. Biophys. Acta, 39, 367
(1960).
ГЛАВА IX
ПИРИМИДИЛМЕТИЛТИАЗОЛИЕВЫЕ ВИТАМИНЫ И КОФЕРМЕНТЫ
ТИАМИН
Тиамин — аневрин, витамин Bj — является хлористоводородной солью
4-метил-5-^-оксиэтил-М-(2'-метил-4/-аминопиримидил-5,-метил)тиазолийхло-
рида следующей структурной формулы:
Пиримидиновый и тиазолиевый циклы тиамин-хлорида, по данным
реитгеноструктурного анализа, расположены под углом 76°, а не в одной
плоскости [1].
Тиампн-основание представляет собой аморфное бесцветное гигроско-
пическое вещество. Он получается из галогеноводородных солей при обра-
ботке их окисью серебра.
Т и а м и н-х лорид гидрохлорид (C12H17ON4S)+ СГ • НС1 • ^Н-О)
кристаллизуется с половиной молекулы воды в бесцветных моноклини-
ческих иглах. Вещество обладает диморфизмом и имеет т. пл.
240—-244° С [2, 3] (преобладающая a-форма) и 250—252° С (с разл.)
376
[4,5] (0-форма). 0*-Кристаллическая форма превращается в a-форму при
193° С 16].
К четвертичным солям тиамина также относятся: тиамин-бро-
мид гидробромид (C12H17ON4S)+ Вг НВг-*/2Н2О — бесцветные
иглы с т. пл. 229—231 ° С [5] и 220° С [4] (диморфизм). Другие соли имеют
следующую температуру плавления: тиамин-сульфат 203 и 276—278° С
[3 ]; тиа м и н-м ононитрат — (C12H17ON4S)+NO3 164 — 165° С [3 ] и
196—200е С 17]; тиа мн н-й од и д г и д р о й о д и д 238—244° С [8, 9];
тиамин-п икролонат 165 и 229° С; тиамин-п и к р а т 208° С; ти-
а м и н-х лораурат 189° С (с разл.) [10].
Тиамин-хлорид и тиамин-бромид растворимы в спиртах, глицерине,
ледяной уксусной кислоте; нерастворимы в эфире, хлороформе, бензоле и
ацетоне [11]. В 1 мл воды растворяется 1 г тиамина-хлорида, в 95%-ном
спирте — около 1 г и в безводном спирте — около 0,3 г в 100 мл; в глице-
рине — около 4,5 г в 100 мл; pH 1 %-ного водного раствора 3,58.
Тиамин-мононитрат вследствие своей малой гигроскопичности и большой
устойчивости применяется для витаминизации пищевых продуктов. Его
растворимость в воде: 1 г в 37 мл при 25° Сив 3,4 мл при 100° С.
Тиамин-хлорид и тиамин-бромид обладают слабым специфическим за-
пахом, подобным запаху дрожжей или орехов.
Тиамин образует соли с органическими кислотами — сахариновой
[12], тетрафенил бор ной [13], циклогексилсульфаминовой [14], нафталин-
1,5-дисульфоновой [15] и др.
Характер спектра абсорбции тиамина зависит от концентрации водород-
ных ионов [16]. Спектр поглощения тиамин-хлорида имеет два
максимума при pH 7 — 235 и 267 нм и только один максимум при pH
5,5 и в более кислом растворе — 245 — 247 нм (Е}^м 442) [17, 18].
Из растворов тиамин количественно осаждается фосфорновольфрамовой
кислотой (pH 4,5—5,5), пикролоновой и золотохлористоводородной кислота-
ми, но не осаждается сернокислой ртутью и уксуснокислым свинцом. Тиамин
адсорбируется на фуллеровой земле, каолине, древесном угле и вымывает-
ся растворами гидроокиси бария. Степень адсорбции сильно зависит от
pH среды.
ХИМИЧЕСКИЕ СВОЙСТВА ТИАМИНА
В сильнокислой водной среде тиамин-хлорид (I) обладает высокой ус-
тойчивостью и не разрушается под действием таких энергичных окислите-
лей, как перекись водорода, марганцовокислый калий и озон. При pH 3,5
тиамин может нагреваться до 120° С без заметных признаков разложения
[19]. В результате нагревания водного раствора тиамина при температуре
кипения в течение 30 ч образуются муравьиная кислота, 4-метил-5-0-окси-
этилтиазол,2-метил-4-амино-5-аминометилпиримидпн, 2-метил-4-амино-5-ок-
симетилпиримидин и у-ацето-у-тиопропиловый спирт [20]. В сильнокислой
среде при 150° С под давлением происходит отщепление аминогруппы
[21].
В щелочной среде тиазолиевый цикл тиамина становится неустойчивым
и легко расщепляется с образованием открытой тиольной формы тиамина —
«тиамин-тиола» [II)
-----------------7Т-СН3 О1Г г ---С-СН3
xU k. Jk /к- Jk СНОI CCHjCHjOH
СНз ™ NHa s CHjCHjOH СН3 N NHj $Й
I II
377
Эта форма тиамина обладает высокой реакционной способностью и склон-
на к различным химическим превращениям — легкому окислению и др.
(см. раздел «Реакции и производные тиольной формы тиамина»).
Тиамин-хлорид (I) при осторожном восстановлении поД2-связи тиазо-
лиевого цикла алюмогидридом лития в тетрагидрофуране [22 —24],
NaBH(OCH3)3 [24], боргидридом натрия в безводном бутаноле [25] или
гидросульфитом [26] превращается в биологически неактивный дпгидро-
тиамин (V) с т. пл. 150е С. Его нагревание в воде [23] приводит к получе-
нию более стабильного изомерного соединения с циклической пергидрофу-
ротиазольной формой — изодигидротиамину (III) с т. пл. 160° С [27].
Это же соединение получается непосредственно при восстановлении тиамина
боргидридом натрия наряду с тетрагидротиамином (IV) [24].
По некоторым данным [27, 28] дигидротиамин (V) имеет также пергидро-
фуротиазольную структуру, которая находится в транс-конфигурации
(соединение III — в цис-конфигурации).
Дигидротиамин (V) при окислении хлорным железом в 10%-ной уксус-
ной кислоте или кислородом воздуха в слабом солянокислом растворе в
присутствии активного угля вновь образует тиамин (I) [29, 30], а при даль-
нейшем восстановлении боргидридом натрия, так же как и изодигидротиа-
мин,— тетрагидротиамин (IV) [25]. Тетрагидротиамин (IV) легко десуль-
фируется при действии активного никеля с образованием соединения VI
[24 I.
При восстановлении тиамина образуется еще одно вещество — псевдо-
дигидротиамин с т. пл. 175° С [24]. Это соединение не восстанавливается
боргидридом натрия в тетрагидротиамин [25], но способно окисляться в
тиамин [30]. Псевдодигидротиамину приписывается структура производ-
ного пиримидо[4,5-<2] тиазоло[3,4-а[пиримидина (VII) [31]
При взаимодействии с сульфитом натрия в водном растворе тиамин-
хлорид (I) подвергается реакции сульфитного расщепления, которая при-
водит к образованию 2-метил-4-амино-5-пиримидилметилсульфокислоты
(VIII) и 4-метил-5ф-оксиэтилтиазола (IX) [19, 32]
сг
378
Реакция сульфитного расщепления характерна для четвертичной тиазо-
лиевой формы тиамина; ни открытая тиольная форма тиамина, ни произ-
водные тиамина с третичным атомом азота тиазолинового цикла реакции
сульфитного расщепления не подвергаются [33]. Тиамин-тион-2 (тиотиамин)
в концентрированных водных растворах устойчив к действию сульфита нат-
рия, однако в разбавленных растворах его расщепление идет при аэрации,
так как по-видимому, он первоначально окисляется в тиамин [33].
Тиогликолевая кислота вызывает аналогичное расщепление молекулы
тиамина при нагревании, причем наряду с 4-метил-5-р-оксиэтилтиазолом
(IX), выход которого составляет 70%, образуется 2-метил-4-амино-5-пири-
мидилметилтиоуксусная кислота (X) с выходом 75% [34].
/ОДБОДСООН
од
xNH2
X
Тиамин по своей оксиэтильной группе образует простые и сложные эфи-
ры, например ацетат, бензоат [35, 36], пальмитат, стеарат [35—38] и др.
Эфиры тиамина с фосфорной кислотой образуются в виде моно-, ди-, три-
и полифосфатов. Тиаминдифосфат (кокарбоксилаза) представляет собой
коферментную форму тиамина, в виде которой он принимает участие в биохи-
мических реакциях (см. с. 417).
Ацилирование аминогруппы положения 4 пиримидинового цикла тиами-
на протекает с трудом. Аминогруппа тиамина (так же как и 2-метил-4-амино-
5-аминометилпиримидина) слабо реагирует с азотистой кислотой [39].
Протонирование тиамина при солеобразовании с минеральными кисло-
тами идет не по аминогруппе, а по циклическому атому азота положения 1
[40].
С гидратом окиси меди в щелочном растворе тиамин дает комплекс типа:
тиамин—Си—тиамин [41].
При взаимодействии тиамина с карбонильными соединениями идет реак-
ция электрофильного замещения по атому углерода положения 2 тиазолие-
вого цикла с образованием соответствующих производных. Так, в водном
или водно-спиртовом растворе с 2 молями едкого натра из тиамина и аце-
тальдегида образуется 2-а-оксиэтилтиамин (XI), а из тиамина и пировиног-
радной кислоты—соединение XII [42].
CH.'Tf NH, rs"XH2CHpH CH3 N'NI. Г''5ХСН3СН2ОН
Cl l3CHOH c h3c(oh)cooh
XI XII
Подобная реакция осуществлена с изо-бутиральдегидом (с выходом 60%)
/«-оксибензальдегидом (с выходом 35%), формальдегидом [42], бензальде-
гидом— получается 2-оксибензилтиамин [43] — и другими соединениями
[42]. Эта реакция лежит в основе механизма биокаталитической функции
тиамина в тканях организма.
Если же на тиамин-хлорид (I) первоначально подействовать триэтил-
амином, а затем бензальдегидом, то по положению 2 тиазолиевого цикла
происходит реакция присоединения, по-видимому, первоначально триэтил-
аминной группы с одновременной циклизацией оксиэтильной группы в сро-
щенный с тиазолиевым тетрагидрофурановый цикл с образованием соедине-
ния XIII [43]
379
Тиамин-
хлорид
1
Интересно отметить, что в условиях реакции происходит частичное расщеп-
ление соединения XIII на 4-амино-2,5-диметилпиримидин и 2-бензоил-5-
(2/-оксиэтил)-4-метилтиазол [44]. Соединение XIII боргидридом натрия
восстанавливается в дигидропроизводное (XIV) [43], а в кислой среде в
присутствии диметилформамида легко количественно гидролизуется вновь в
тиамин (I).
а-Кетоальдегиды в реакции с тиамином также присоединяются в поло-
жение 2 тиазолиевого цикла [45]. Так, тиамин-хлорид (I) и фенилглиоксаль
в присутствии триэтиламина и двуокиси углерода дают фенилоксалилтиа-
мин (XV), который кислородом воздуха легко окисляется в тиамин-тиа-
золон (XVI)
Тиамин-
хлорид
1
о., —1ГСНз
CHj ЬГ NH, C^S'XHjCHjOH
XVI
Такая же реакция присоединения проходит между тиамином и гетеро-
циклическими основаниями — морфолином, пиперазином и др. [46, 47].
Диалкилфосфиты также присоединяются к тиамину — образуется соеди-
нение XVII, которое при нагревании в спирте изомеризуется в производ-
ное пиримидо[4,5-^]тиазоло[3,4-а]пиримидина (XVIII) [46, 48]
Реакция тиамина с фосфитами детально исследована [49]. Изучен
масс-спектр тиамина [50 ].
Количественно тиамин определяется по интенсивности флуоресценции
тиохрома (см. с. 381), в который он окисляется [51—53]. Связанные формы
природного витамина перед определением подвергаются предварительному
ферментативному расщеплению [54 ]. При взаимодействии с диазотированной
сульфаниловой кислотой в присутствии формальдегида тиамин образует
красное окрашивание, интенсивность которого зависит от концентрации.
Это свойство тиамина также находит применение для его количественного
определения [55]. В синтетическом кристаллическом препарате тиамин
определяется последовательным титрованием азотнокислым серебром и
щелочью в присутствии бромтимолового синего; разность между этими
определениями соответствует количеству брома, связанного в виде четвер-
тичной соли.
РЕАКЦИИ И ПРОИЗВОДНЫЕ ТИОЛЬНОЙ ФОРМЫ ТИАМИНА
Тиольная форма тиамина (II) с раскрытым тиазолиевым циклом обла-
дает исключительно высокой реакционной способностью. Являясь солью
четвертичного основания, тиамин под воздействием трех эквивалентов щело-
880
чи претерпевает превращение в открытую тиольную форму с размыканием
тиазолиевого цикла. Эти превращения, идущие при постепенной нейтрали-
зации, можно изобразить следующей схемой 156]:
ок Tj^YCHS<—ij-сн, он~
,Г СН2hTNIL, ^'''хНгСЦ.ОН н+
XIX
ХХсн"йсСНз
CH, N NH2 HS CHjCHjOH
II
Промежуточной формой между четвертичным аммониевым основанием
(XX) и открытой тиольной формой тиамина —тиамин-тиолом (II) служит
псевдооснование (XXI) [56]. Количественное обратимое превращение ти-
амин-тиола (II) в тиамин (I) возможно только в том случае, если после-
дующая обработка кислотой следует тотчас же за обработкой щелочью.
Из четвертичного аммониевого основания (тиамин-основания) (XX) бы-
ли получены различные соли тиамина [57].
Тиамин (I) в щелочной среде легко подвергается окислению. Эта реак-
ция протекает через открытую тиольную форму тиамина (II) с образованием
трициклического соединения — тиохрома (XXIII)
т— хт“3'—rcHi
XXII
Открытая тиольная форма тиамина — бесцветное вещество состава
C12H17O2N4SNa-4H2O [58] — находится в равновесном состоянии со сво-
ей циклической формой — XXII (темно-желтые нестабильные кристаллы
состава C12Hl5ON4SNa-ЗН2О, получаемые’ при обработке суспензии тиамина
в безводном спирте этилатом натрия [58]), которая дегидрируется различ-
ными окислительными агентами. Эта схема получения тиохрома через
тиольную форму подтверждается тем, что тиохрома при использовании
трех эквивалентов щелочи (I, XIX, XX, II) образуется значительно больше,
чем с двумя эквивалентами, когда раскрытия тиазолиевого цикла не проис-
ходит. Для окисления тиамина в тиохром применяют йод [59], красную
кровяную соль КзРе(СМ)6, двуокись селена, окись ртути, перекись водо-
рода, марганцовокислый калий и кислород [51, 60, 611. Превращение тиами-
на в тиохром в щелочной среде протекает под влиянием только сильных
окислителей или в водной среде при действии цианбромида [62].
Тиохром (XXIII) — желтое кристаллическое вещество с т. пл. 227°С
(с разл.); он легко растворим в воде и спирте. Для тиохрома характерны
два максимума поглощения — при 358 и 375 нм. В ультрафиолетовом све-
те его водные растворы имеют интенсивную синюю флуоресценцию с 1ма1С в
381
пределах 460—470 нм. Это свойство используется для качественного и коли-
чественного определения малых количеств тиамина. Тиохром не обладает
витаминной активностью [51, 60]; он выделен из дрожжей (20 мг из 1,2 кг)
[63] и получен встречным синтезом [64, 65].
Открытая тиольная форма тиамина (II) неустойчива к окислителям и
легко превращается в симметричный тиаминдисульфид (XXIV) с т. пл.
177° С (для его гидрохлорида т. пл. 231 ° С)
СЦСН.ОН
—Гсн’ ® г r>^rC^rj‘c(CH^=asc=c(cH^CH2'Y^N
снАЛн, СНО НЛ'Н нААн,
IV XXIV
Эту первую стабильную тиольную форму тиамина получили Цима и Виль-
ямс в 1940 г. [58].
Окисление тиамина или его производных в щелочной среде в дисуль-
фидные соединения происходит при действии гипойодита [58], гипохлори-
та [66], перекиси водорода [67], феррицианида [68] и других окислителей.
Окисление феррицианидом калия происходит почти количественно, если
тиамину в щелочном растворе (pH 11—12) предварительно дают постоять
некоторое время. Образование дисульфида тиамина ускоряется растворен-
ным кислородом [69, 70].
Тиаминдисульфид (XXIV) также образуется из тиамина при его окисле-
нии йодом [71 ] или разбавленными растворами перекиси водорода при
pH 7,5, т. е. в условиях, близких к биологическим. Он обладает активностью,
равноценной активности тиамина, но для мелких животных почти в 5 раз
менее токсичен [67]. Тиаминдисульфид (XXIV) восстанавливается актив-
ным водородом, сероводородом и другими восстановителями вновь в откры-
тую форму тиамина (II), которая при обработке кислотами превращается у
в тиамин [67].
Тиаминдисульфид (XXIV) в результате окислительного диспропорцио-
нирования превращается в тиамин-тиазолон, тиаминон-2 (XVI) и тио- !
хром (XXIII) [72]
Образование тиохрома из тиаминдисульфида наблюдается под влиянием
кислорода воздуха [58, 73]. Тиаминон-2 является устойчивым бесцветным
кристаллическим соединением п не превращается в тиохром [58].
При окислении тиаминдисульфида 30%-ной перекисью водорода в среде
уксусной кислоты образуется сульфокислота открытой тиольной формы
тиамина—тиаминовая кислота (XXV) [74]
СН3
уХ^/СНг—1 NcLcCH2CH2OH
N СНО SO3H
ch//^n \nhs
XXV
382
Метильная и сульфогруппы, помимо /пра«с-конфигурации по отноше-
нию к двойной связи, образуют и ^ис-изомер тиаминовой кислоты [75].
Многие производные тиаминовой кислоты обладают анальгетическим и про-
тивовоспалительным действием.
Синтезированы разнообразные О-замещенные производные тиаминди-
сульфида и асимметричные дисульфидные производные тиамина. Как пра-
вило, соединения такого типа показали значительные преимущества перед
тиамином — они обладали пролонгированным действием и не разрушались
тиаминазой. Легкая проницаемость через клеточные мембраны [76], сродство
к тканям [77] и быстрая рециклизация в тиамин [76, 78] определяют высо-
кую физиологическую активность многих из этих соединений.
При окислении тиамин-О-монофосфата в щелочном растворе получен
быс-(тиамин-О-монофосфат)дисульфид (XXVI) [79, 801, который обладает
активностью тиамин-хлорида, однако легче усваивается организмом и
дольше удерживается в крови [80]
сн2сн2ор(он)2
nc(ch^=cssc=c(chJn-ch3
сно CKCHOPtoHlcHo
XXVI
Нз
Среди многочисленных симметричных дисульфидных производных тиами-
на для применения в лечебной практике были отобраны высокоэффектив-
ные соединения, зарекомендовавшие себя с лучшей стороны. К таким симмет-
ричным дисульфидным производным относятся: тиаминдисульфид (XXIV),
описанный выше [81, 82], и О-бензоилтиаминдисульфид, бестон (XXVII)
[83—85]
CHjCP^OCOCgH/
„^Ya^ijc(cHj=cssc=c(cii,)r|xCB’x|«,!4i
AAnh сно “*н*хос'н?я^ААсн.
XXVII
Как оказалось, в присутствии экстракта чеснока тиамин лучше усваива-
ется вследствие образования с активной составной частью луковиц расте-
ний вида Allium асимметричного дисульфидного производного — тиамин-
аллилдисульфида, аллитиамина, алинамина (XXVIII) [86].
СН/
СН2\ 9Hs
nc=cch2ch2oh
:hossch2ch=ch2
4nh2
XXVIII
CHs
СНО SSC3H7
н CH3
I
'NC=CCH2CH2OH
nh2
XXIX
Аллитиамин выделен в чистом виде из чеснока [87, 88]. При реакции
тиамина с ингредиентами экстракта луковиц растений вида Allium в щелоч-
ной среде при нагревании, помимо тиаминаллилдисульфида (XXVIII)
[89—91], был выделен тиаминпропилдисульфид (XXIX); эти соединения в
отличие от тиамина не разрушаются тиаминазой.
Оба эти соединения были получены синтетически [78, 82, 90, 92—97] из
тиольной формы тиамина при действии соответствующих тиолирующих
агентов при pH 7—9. Следует отметить, что более щелочная среда расщёпля-
383
ет не только сам тиамин, но и возникшую S—S-связь; в качестве раствори-
теля в реакции применяют воду, спирт, ацетон и др.
Среди многочисленной группы синтетических асимметричных дисульфид-
ных производных, большинство из которых имеет неприятный меркапта-
ноподобный запах, нашел широкое терапевтическое применение препарат,
более эффективный, чем тиаминпропилдисульфид, и к тому же лишенный
неприятного запаха, — тиаминтетрагидрофурфурилдисульфид (XXX) [96,
981.
СН,
Ь1<:ГхуСНа"МС =ССН2СН2ОН
снАЛгчн>° SSCHCHsCH2CH2
’ l-.o-----1
XXX
Разнообразное количество асимметричных тиаминдисульфидов было
получено в результате ацилирования по ОН-группе тиаминдисульфидов
или тиолирования О-ацилпроизводных тиамина (см. обзор [99]).
Тиамин и (+)-а-липоевая кислота (тиоктовая кислота) [100] выполня-
ют специфическую роль в процессах метаболизма, и поэтому были получе-
ны различные соединения этих веществ, связанных дисульфидной связью
по трем типам: а) тиамин присоединен к обоим атомам серы липоевой кисло-
ты [101, 102], б) тиамин присоединен к атому серы в положении 6 липоевой
кислоты и в) тиамин присоединен к атому серы в положении 8 [101—104].
В зависимости от различия в построении молекулы S-липоилтиамина в той
или иной степени проявляются свойства тиамина и липоевой кислоты [105].
Практическое применение нашел S-липоилтиамин, неовитан (XXXI),
СН3 СН2 СН2 СН (СН2)4 СООСНз
NC=CSS SCOC Н
I I
СНО СН2СН2ОН
XXXI
который легко всасывается при оральном введении; кроме того, у него хо-
рошая проницаемость тиамина (65,5%) и высокий переход липоевой части
молекулы (96,5%) во все ткани организма [105 I. При его инъекциях в боль-
ших дозах длительное время сохраняется высокий уровень тиамина в крови
[106, 107].
Синтезированы различные асимметричные дисульфидные производные
тиамина в комбинации с другими витаминами. К ним относятся тиаминпи-
ридоксиндисульфид (XXXII) [108], тиаминпантетеиндисульфид (XXXIII)
[109]
СНа СН2ОН
^NC=CSSCH2 он
chocHjCHjOh Ъ X
СН3 N Щ СН.
СН, ОНСНз
I I I
NC=CSSCH, СН, NHCOCH, СН2 NHCOCHCCH, ОН
II I
СНОСН,СН2ОН СН3
XXXIII
384
а также его многочисленные производные по гидроксильным группам.
Тиамин-тиол (II) легко реагирует в щелочной среде с галогеналкилами
с образованием S-ал кил производных тиамина моносульфидного типа,
NC (СН)3=ССН2 СН. OR
СНО SR'
где R' = СН=СНСОСН3, СН—СНСОС6Н5 и другие винильные производные,
a R"=H [НО]; синтезированы и многочисленные другие соединения с заме-
стителями по R' и R" [111, 112].
При действии хлористого бензоила на тиамин в открытой тиольной форме
(II) в щелочной среде образуется [113] О, S-дибензоилтиамин (XXXIV)
Тиамин-
тиол
II
С.Н5СОС1
NC (СН8)=ССН2 СН, OCOQHs
I I
СНО SCOC,H6
XXXIV
При использовании соответствующего ацилирующего агента и определен-
ных условий можно осуществить ацилирование только по SH- или ОН-груп-
пе. Было синтезировано большое количество S-моноацилпроизводных тиами-
на, однако из-за склонности к внутримолекулярной перегруппировке
ацильного радикала от S к атому О в кислой или щелочной среде [114, 115]
многие S-ацилпроизводные тиамина оказались нестабильными. Для пре-
дотвращения такой перегруппировки ацильного радикала применяют за-
щиту гидроксильной группы тиамина ацилированием или этерификацией.
Таким путем была получена большая группа О-ацил- или О, S-диацил-
производных. Некоторые S-алкоксикарбонилпроизводные тиамина обла-
дают высокой тиаминовой активностью и лучше, чем тиаминпропилдисуль-
фид, усваиваются в кишечнике [116].
Следует отметить, что соединение XXXV, содержащее S-карбомоиль-
ную группу при атоме серы, в присутствии щелочей или кислот испытыва-
ет перегруппировку карбомоильной группы к атому кислорода гидроксиль-
ной группы в результате чего получается соответствующее О-карбомоил-
замещенное циклической формы тиамина (XXXVI) [114]
ыс(сн^=ссн2сн2он
СНО SCOR’
'г
C1U+
N---------Т-СН3
CHjCHjOCOR,'
СН3 N NHj
ССН2СН2'
sx хо
RI''C''OH
XXXVI
При взаимодействии S-бензоилтиамина (XXXVII) с диметилкарбомо-
илхлоридом (CH3)2NCOC1 в пиридине при нагревании происходит перегруп-
пировка бензоильной группы от серы к кислороду с образованием S-диметил -
карбомоил-О-бензоилтиамина (XXXVIII) [117]
13—69
385
сн,
СН 1
^7С=^н2он (ch3)2ncoci
CHfS'J Nil, С1Ю scocell5
СН3
N<=fxV'al2^NC = CCU2Cll2OCOq.Hs
CH,AAnH, Cll° SCON(CH3),
xxxvu
XXXVIII
Среди большого количества S-ацилпроизводных тиамина с блокирован-
ной оксиэтильной группировкой следует отметить сложные эфиры с фосфор-
ными кислотами; такие соединения создают более высокую концентра-
цию тиамина в крови, чем соответствующие S-ацил- или О-ацилпроизводные
[118], Особенно хорошо выражены эти свойства у S-бензоилтиамин-О-мо-
нофосфата—биотамина, бенфотиамина (XXXIX),
CHS
МН2
CHS
NG=CCH2 СН2 ОР (ОН)з
СНО SCOCeHe
О
XXXIX
который проявил себя в лечебной практике как очень эффективный пре-
парат пролонгированного действия [119]. Он получается при взаимодей-
ствии тиамин-О-монофосфата с бензоилхлоридом [120], дибензоилдисуль-
фидом [121] или бензоилтиосульфатом натрия [120, 121] в щелочном
растворе при охлаждении.
Из производных никотиновой кислоты и открытой тиольной формы тиа-
мина следует отметить О-бензоил-8-никотиноилтиамин (XL) и О-никоти-
ноил-8-бензоилтиамин (XLI) [122]
сн
сн;
сн^
NH3
сн3
NC=CCH3CH2OOC
СНО SCOC6Hs
XU
которые обладают пролонгированным действием.
О, S-Диацилпроизводные тиамина плохо растворимы в воде и обычных
органических растворителях и хорошо — в подкисленных соляной кисло-
той растворителях. Они образуют устойчивые гидрохлорид, не дают тиохром-
ной реакции и рециклизуются в тиамин в животных тканях in vivo, чем и
определяется их биологическая активность [78].
Некоторые О, S-диацилпроизводные тиамина в кислых и щелочных сре-
дах оказались неустойчивыми. Выяснено, что стабильность зависит от при-
роды ацильного радикала: S-ацилтиамины, содержащие алифатические ра-
дикалы, менее стабильны, чем имеющие ароматический или гетероцикличес-
кий радикалы.
Тнамин-тиол (II) при действии n-толуол- или n-бензолсульфохлорида с
последующим нагреванием в 30 %-ном растворе едкого натра превращается
в биологически почти неактивный тиамин-ангидрид (XLII) [123, 124]
с т. пл. 136°С.
XUI
386
Реакция протекает ступенчато; сначала при действии л-бензолсульфохлори-
да идет образование сложного эфира и получается О,О-бис-фенилсульфонил-
тиаминдисульфид [123] или О-фенилсульфонилтиамин-тиол [125], которые
при нагревании в спиртовом растворе едкого натра отщепляют О-бензолсуль-
фокислоту и количественно превращаются в тиамин-ангидрид. При окис-
лении перекисью водорода или марганцовокислым калием тиамин-ангид-
рид (XL1I) образует тиамин-ангидридсульфон (XL1I1) 1126, 127].
При действии сероводорода в диметилформамиде тиамин-ангидрид
(XLII) дает 1,2-тиолановое производное — тиамин-тиолан (XLIV) с выхо-
дом 91 % 11281
Иначе идет реакция тиамин-ангидрида с тиофенолом — образуется сое-
динение XLV, причем реакция сопровождается одновременным десульфиро-
ванием тиамин-ангидрида с образованием замещенного 1,4-диазепина (XLVI)
[129]
Подобно протекает реакция с п-тиокрезолом.
Тиамин-ангидрид восстановлению цистеином в тиамин не подвергается
[124]; при действии соляной кислоты он легко расщепляется на 2-метил-4-
амино-5-аминометилпиримидин н 2-ацетилтиациклобутан [124, 126].
Тиамин-тиол (II) взаимодействует с цианбромидом в щелочной среде с
образованием N-(l-метил-2-тиоциан-4-оксобутен-1-ил)-М-(2-метил-4-ами-
но-5-пиримидилметил)формамида, S-цианотиамина (XLVII) [126, 130, 131],
с т. пл. 80°С
,СН2Ч . 9Нз
N
Ж=ССН2СН2ОН
СНО SH
сн/ N /н2 1
сн;1
сн2Ч
NC=CCH2CH2OH
СНО SCN
nh2
XLVII
Для нитрильной группы S-цианотиамина характерна очень высокая под-
вижность; эта группа легко гидролизуется и замещается. •
Так, при нагревании S-цианотиамина (XLVII) в бутиловом спирте [132]
или при щелочном гидролизе происходит образование тиамин-ангидрида
(XLII) [126, 130, 133], а при действии на него 30%-ной перекиси водорода
в уксусной кислоте — тиаминовой кислоты (XXV) [112].
13*
387
: В присутствии метилового спирта в растворе едкого натра S-циансггиа-
мин (XLVII) образует S-метилтиамин (XLVIII) наряду с тиамин-ангидри-
дом (XLII) 1133]; то же соединение получается при обработке метаноль-
ного раствора тиамин-ан гидрида щелочью [133].
СН,
СН3
NC=CCH2CH2 ОСОСНз
I I
СНО SCOCH,
XLIX
При действии на S-цианотиамин (XLVII) хлорангидридами органичес-
ких кислот, например хлористого ацетила, циан роданистой группы наряду
с атомом водорода оксигруппы замещается ацильным остатком и образует-
ся О,5-диацетилтиамин (ХЫХ) [134].
S-Цианотиамин (XLVII) при действии 4—5%-ной соляной кислоты при
80°С превращается в тиамлн-имин-2 (L) [126], а при 100° С—в производное
пиримидилметилтетрагидрофуротиазолинимина (LI) [126]
ГХс"Ттсн’
CHj N NHjHN^S CHjCHjOH
L
При нагревании со слабыми кислотами, например с 50%-ной уксусной
кислотой, S-цианотиамин (XLVII) циклизуется в тиохром (XXIII) [126,
132], а при восстановлении сероводородом или цистеином он превращается
в тиамин (I) [130].
Реакция S-цианотиамина (XLVII) с алкил-, арил- или аралкилмеркапта-
нами (RSH) приводит к замещению циангруппы и образованию замещенного
несимметричного тиаминдисульфида (LII) [135]
CHS-
Н2-
СН3
NC=CpH2CH2OH
СНО SSR
nh2
LII
R=G,H5, С3Н7. CeH5, C5HU, C3H6COC8H5 И др.
S-Цианотиамин концентрированной соляной кислотой расщепляется на
2-мети.т-4-амино-5-аминометилпиримидин и муравьиную кислоту [130].
Из других реакций тиамин-тиола необходимо отметить следующие.
При действии фосгена тиольная форма тиамина (II) в щелочном водном
растворе дает циклокарботиамин (LIII), а в водно-спиртовом растворе
при —20° С — S, S'-карбодитиамин [136]. Нагревание тиамин-тиола (II) с
серой в щеточном растворе приводит к циклизации ациклической части
молекулы в тиазолиновый цикл с образованием тиамин-тиона-2 (LIV)
[137].'
388
Тиамин-тион-2 при действии 30%-ной перекиси водорода легко окисля-
ется в тиамин (I) [138, 139].
Тиамин-тиол (II) в виде таутомерного тиокетона реагируете гидроксил-
амином в щелочном растворе; при этом отщепляется сероводород и образует-
ся оксим дезтиотиамина (LV). Подобным образом протекает реакция тиамин-
тиола с гидразином, фенилгидразином и тиосемикарбазидом с образованием
соответствующих гидразона, фенилгидразона и тиосеми карбазона, однако
с гидразин-гидратом реакция сопровождается одновременным элиминиро-
ванием одной молекулы воды и циклизацией в соединение, содержащее
1,2,4-триазиновый цикл (LVI) [140]
сн
сн3
I
NCHCCH,CHaOH
СНО NOH
СН,
fvCHySrCH,CHfOH
СНз'ТГЛЧН,
LVI
Аммиак в растворе едкого натра десульфирует тиамин-тиол (II) с одно-
временным замещением на аминогруппу; однако реакция на этом не закан-
чивается и сопровождается замыканием ациклической части молекулы в
имидазольный цикл с образованием Ы(3)-(2-метил-4-амино-5-пиримидилме-
тил)-4-метил-5-0-оксиэтил имидазола (LVII) [140]
LVII
Тиамин-тиол вступает в реакцию с первичными алифа тическими аминами
в щелочной среде, при этом происходит замещение тиольной группы на
NHR и получаются производные аминодезтиотиамина (LVIII) [140]. При
действии на такие соединения соляной кислотой в спирте происходит
гидролитическое отщепление R, замыкание имидазольного цикла и обра-
зуется соединение LVII. Со вторичными и третичными алифатическими
аминами и с ароматическими аминами протекает реакция десульфирования
тиамин-тиола в 2,7-диметил-5, 6-дигидро-6-формил-8-0-оксиэтил-9Н-пирими-
до[4,5-е]-1,4-диазепин (LIX) [46, 140]
сн,
СНл I ”
qa'4NC=CCH,CH,OH
chAAnh,
LVIII
и одновременного образования тиамин-тиона-2 (LIV).
Ацетамидин в сильнсщелочном растворе также вызывает замещение
меркаптогруппы тиамин-тиола (II) на остаток ацетамидина с образованием
1-иминоэтиламинодезтиотиамина [140]. При действии гуанидина тиамин-
тиол (II) превращается в тиохром (XXIII), тиаминдисульфид (XXIV),
замещенный 1,4-диазепин (LIX) и вещество, содержащее в своей молекуле
1,4-диазин [140].
380
Тиамин-тиол (II) вступает в реакцию с альдегидами (ацетальдегидом,
бензальдегидом, фурфуролом и др.), т. е. в аналогичную реакцию, протекаю-
щую между тиамином и альдегидами в присутствии триэтиламина (с. 379);
реакция сопровождается замыканием ациклической части молекулы в
пергидрофуротиазоловый цикл и вступлением заместителя в положение 2
этого цикла [43] — см. формулы XIII и XVII. Первоначально перед при-
бавлением бензальдегида или другого альдегида к тиамин-тиолу в виде
натриевой соли в среде толуола или спирта в раствор пропускают двуокись
углерода [43]. Подобная реакция имеет место для тиамин-тиола (II) с
морфолином, пиперидином или диалкилфосфитами [46].
Использование высокой реакционной способности тиамин-тиола дало
возможность получить среди различных производных тиамина тиольного
типа свыше 700 стабильных соединений, большинству из которых свойст-
венна высокая витаминная активность и пролонгированное действие —
см. в обзоре [99].
СТРОЕНИЕ ТИАМИНА
Впервые препарат тиамина выделил Функ в 1912 г. [141 ] из рисовых
отрубей, а еще более чистый препарат — Янсен и Донат в 1927 г. [142].
В кристаллическом виде витамин Вг выделили из дрожжей в 1931 г. Виндаус
с сотр. [143] (1 мг из 1 кг дрожжей).
Строение тиамина установили независимо друг от друга Вильямс [171
и Греве [19] в 1936 г. в результате расшифровки строения его составных
частей — пиримидинового и тиазолового компонентов и синтеза многочис-
ленных веществ, строение которых, казалось бы, отвечало тиамину. В то же
время строение тиамина окончательно подтвердили его синтезом в 1936 г.
Вильямс и Клин [2] и Андерзаг и Вестфаль [4].
В установлении структуры тиамина большое значение имела реакция
его сульфитного расщепления [32 ]. При обработке водного раствора тиамин-
хлорида (I) сульфитом натрия при pH 4,8—5 и 20—25° С вещество почти
количественно распадается на третичное тиазоловое основание (IX) состава
C6HsONS, растворимое в воде, и пиримидиламиносульфокислоту (VIII)
состава CeHgO3N3S, нерастворимую в воде.
^Гт сн- •
CHj гХмН, 4‘S"'CH2CH3OH \S'CH2CH:OH
CH, IVNH,
LX LXI
Возможно, что в результате ионного обмена первоначально образуется
четвертичная соль тиамин-сульфата, которая подвергается расщеплению на
соединения VIII и IX [19].
Тиазоловое основание (IX) содержит третичный атом азота, что было
доказано образованием с йодистым метилом четвертичной соли. Наличие в
этом основании одной гидроксильной группы установлено по образованию
сложного эфира с n-нитробензойной кислотой [51].
Если тиазоловое основание (IX) подвергнуть окислению азотной кисло-
той, то образуется 4-метилтиазолкарбоновая-5 кислота (LX), содержащая
пять атомов углерода; эта же кислота получается при непосредственном
окислении тиамин-нитрата азотной кислотой [144], она идентична ранее
синтезированному соединению LX [145, 146].
390
Происхожденйе карбоновой кислоты (LX) из соединения IX с потерей
одного атома углерода, а также отсутствие положительной реакции с йодо-’
форменной пробой и оптической активности у соединения IX указывают на
то, что гидроксильная группа имеет первичный, а не вторичный характер:
она должна находиться у крайнего атома углерода боковой цепи и, следо-
вательно, расположена в положении 5 тиазолового кольца в виде [5-оксиэтиль-
ной группы. Все эти данные приводят к структуре 4-метил-5-Р-оксиэтилтиа-
зола (IX), что было подтверждено его синтезом [145].
Значительно сложнее было установить структуру пиримидинового ком-
понента и самого тиамина. Второй продукт сульфитного расщепления —
пиримидиламиносульфокислота (VIII) содержит атом серы в виде сульфо-
группы, так как при сплавлении соединения VIII со щелочью образуется
сернистокислый натрий; очевидно, что сульфогруппа вводится в это веще-
ство при реакции сульфитного расщепления и, вероятно, занимает то поло-
жение, по которому осуществляется связь между составными частями
молекулы тиамина. Пиримидиламиносульфокислота (VIII) при нагревании
с соляной кислотой отщепляет аммиак с образованием оксисульфоновой
кислоты (LXI), чем доказывается наличие в соединении VIII первичной
аминогруппы [51, 147, 148].
Пиримидиламиносульфокислота (VIII), как установлено подобием спек-
тров поглощения ультрафиолетового света, является производным 6-ами-
нопиримидина [21 ]. Таким образом, устанавливалась основная структура
соединения VIII. Невыясненным оставалось место фиксации сульфогруппы
и двух атомов углерода алифатического характера.
По результатам определения структуры тиазолового основания как 4-ме-
тил-5-р-оксиэтилтиазола (IX), по предположению, что тиамин является
четвертичной солью аммониевого основания [145, 149], и данным потенцио-
метрического титрования [150—152] для структуры тиамина было предло-
жено несколько формул с различными расположениями алкильных замести-
телей пиримидинового цикла и местом взаимосвязи пиримидинового и тиа-
золового компонентов — LXII [10, 153, 154], LXIII [155], LXIV [156]
LXII
LXIII
LXIV
R=CH2CH2OH
Однако все эти формулы оказались ошибочными, и полученные соединения'
отличающимися от природного тиамина.
Следовательно, пиримидиновый цикл тиамина должен иметь или одну ’
свободную метильную группу, если тиазоловый и пиримидиновый циклы
связаны через метиленовый мостик, или две метильные группы.
Положение метильных групп было выяснено в следующем исследовании.
При восстановлении пиримидиламиносульфокислоты (VIII) натрием в
жидком аммиаке [16] было получено основание (LXXI), которое при срав-
нении пикратов оказалось идентичным синтетическому 2,5-диметил-6-ами-
нопиримидину (LXXI), полученному конденсацией ацетамидина с а-фор-
милпроппоповым эфиром и последующим превращением соединения LXIX
в хлорпроизводное (LXX), а затем в основание (LXXI) [157] (схема 84).
Оставался еще неясным вопрос о месте связи пиримидинового и тиазоло-
вого циклов. При расщеплении тиамина жидким аммиаком [16, 17, 157] или
при окислении его марганцовокислым барием был выделен гетероцикличе-
ский диамин (LXV). По спектру поглощения этого диамина можно было
заключить, что он относится к аминопроизводному пиримидина. Этот же
диамин (LXV) был получен синтетически [19] и, что особенно важно для
установления места связи компонентов, из него была получена пиримидил-
391
аминосульфокислота (VIII) через 2-метил-4-амино-5-бромметилпиримидин
(LXVI), обработанный бисульфитом натрия. Из полученных данных сле-
дует, что пиримидиновый и тиазоловый циклы в молекуле тиамина связаны
метиленовым мостиком по положению 5 пиримидина и по атому азота тиа-
зола в виде соли четвертичного аммониевого основания.
Схема 84
Установление строения тиамина реакциями его превращения и расщепления
LXIX LXX LXXI R=CH,CH,OH
Подтверждение структурной формулы тиамина было получено также в
расшифровке и синтезе его производного — тиохрома (XXIII), количест-
венно получаемого окислением тиамин-хлорида щелочным раствором крас-
ной кровяной соли [51 ]. Этот синтез осуществлен из 2-амино-4-метил-5-Р-
оксиэтилтиазола (LXVIII) и 2-метил-4-хлор-5-хлорметилпиримидина
(LXVII) [64].
СИНТЕЗ ТИАМИНА
Молекула тиамина представляет собой соединенные метиленовым мости-
ком замещенные пиримидиновый и тиазолиевый циклы и является солью
четвертичного аммониевого основания. С учетом такого строения молекулы
синтез тиамина может быть осуществлен тремя принципиально различными
путями: конденсацией пиримидинового и тиазолового компонентов, построе-
нием молекулы на пиримидиновом цикле и построением молекулы на тиазо-
ловом цикле.
I. Синтез тиамина конденсацией пиримидинового и тиазолового
компонентов
Этот синтез тиамина заключается в параллельном построении основных
компонентов его молекулы и последующем соединении их в молекулу тиами-
на. Он реализуется посредством взаимодействия 2-метил-4-амино-5-хлор-
метилпиримидина (LXXI1) и 4-метил-5-оксиэтилтиазола (IX)
392:
причем при конденсации происходит образование четвертичной тиазолиевой
соли.
Конденсацию соединений LXXII и IX в тиамин проводят сплавлением
при температуре 100—120° С. Однако лучше проводить конденсацию ком-
понентов нагреванием в растворителях, в качестве которых применяют
толуол и высококнпящие спирты, например бутиловый [2,3,4,51. или
полярные органические растворители, такие, как нитрометан, ацетонитрил
[158], бромоформ [159], Р-этоксипропионитрил [160], а также керосин. Выход
тиамина находится в пределах 6о—65%. Конденсацию можно осуществить
в две стадии — первоначально в толуоле при 20° С компоненты образуют
аддукт (т. пл. 125—130° С), который затем нагревают в этом же растворите-
ле с дополнительным количеством тиазолового компонента при 125° С;
выход составляет 60% [161 ].
Недостаточно высокий выход тиамина при конденсации в спиртовой
среде можно объяснить побочной реакцией, протекающей между галоген-
метилпиримидиновым производным (LXXII) и спиртом, в результате чего
происходит алкоголиз с образованием 2-метил-4-амино-5-этоксиметилпири-
мидина (LXXIII). Вместе с тем указание о том, что тиамин (I) можно полу-
чать сплавлением гидрохлорида 2-метил-4-амино-5-этоксиметилпиримидина
(LXXIII) и гидрохлорида 4-метил-5-р-оксиэтилтиазола (IX) при 165—170°С
1162],
СИ,
LXXIII
+trCHs
^S^CILCHjOH
IX
не противоречит высказанному выше положению о побочной реакции, так
как конденсация соединений LXXIII и IX протекает в жестких условиях.
Для конденсации с 2-метил-4-амино-5-хлорметилпиримидином (LXXII)
вместо 4-метил-5-р-оксиэтилтиазола (IX) можно применять азометиновые
производные 4-метил-5-[3-аминоэтилтиазола, и так как азометиновая груп-
пировка легко расщепляется галогеноводородными кислотами в присутствии
воды и спирта, то в результате конденсации получается 50-аминоаналог
тиамина, который переводится в тиамин вычисленным количеством азоти-
стой кислоты [1631.
Вследствие того что получение бромпроизводных пиримидина, например
2-метил-4-амино-5-бромметилпиримидина, осуществляется несколько легче,
чем хлорпроизводных, то иногда синтезируют бромистоводородную соль
тиамин-бромида, обладающего соответствующей эквивалентной биологи-
ческой активностью, и, если нужно, переводят ее в тиамин-хлорид.
Технический тиамин выделяют из реакционной смеси посредством его
осаждения ацетоном [1641 и очищают перекристаллизацией из метанола
или из 75—90%-ного этилового спирта; очистку осуществляют и растворе-
нием тиамина в воде с последующим сгущением раствора и осаждением
спиртом.
Представляет интерес рассмотрение обменных реакций для получения
различных форм тиамина. Так, тиамин-хлорид с количественным выходом
можно получить из тиамин-бромида встряхиванием с хлористым серебром
[4], при действии хлора в присутствии органических адсорбентов брома
(пиридина, фенола) с выходом 87% [165], при действии хлористого кальция
в среде метилового спирта [166], путем обработки безводным хлористым
водородом в растворе алифатического спирта (при этом происходит удаление
легколетучего бромистого алкила) [167].
С хорошими результатами превращают тиамин-бромид в тиамин-хлорид
через роданат [168, 169]. Так, тиамин-бромид при взаимодействии с рода-
нистым аммонием в водном растворе аммиака легко осаждается в виде
393
тиамин-роданата, который на анионообменной смоле в растворе слабой
соляной кислоты обменивает CSN"-hoh на С1"-ион. Таким же путем из тиамин-
бромида через тиамин-роданат синтезируют тиамин-мононитрат и другие
соли. Для обменной реакции применяют вофатит L-150, аниониты ЭДЭ-10П
и АВ-17.
Тиамин-хлорид получают из тиамин-бромида и непосредственным обме-
ном ионов на анионообменной смоле из полиаминоформальдегндной смолы
[170] или других анионитов [171, 172]. С использованием этого же метода
образуются и другие соли тиамина [173]. Из тиамин-сульфата при действии
хлористого бария получают тиамин-хлорид [174, 175],а с азотной кислотой
[176] или, лучше, с азотнокислым калием — тиамин-мононитрат (выход
91%) [177]. Тиамин-сульфат может быть получен из тиамин-хлорида
при действии хлорсульфоновой кислоты [178].
Синтезирован тиамин, меченный 14С и 35S, по положению 6 пиримидино-
вого цикла [179].
Одна из схем полного технического синтеза тиамина представлена ниже
(схема 85).
Схема 85
Полный технический синтез тиамина из ацетонитрила, акрилонитрила, формамида
н 7-ацетобутиролактоиа .
CHjCOOH+NH,
CH,=CH-CN
Акрилонитрил
НСООН
CH3CN
Ацетонитрил
C2HSOCH2CH2CN
р-Этокси-
пропионитрил
нсоос2н5-
Этилформиат
HCONHj
Формамид
NH
II
сш-с.
ОС2Н£
Ацетиминоэфир
NH
II
НС
СН3ОСН
CN
Of-Метоксиметилен-
O=<^CHj
'СН-СН,
CO CH2
y-АцетоБутиро-
лактон
Тиоформамид
NH
CHj-C.
nh2-hci
о=ссн3
I 3
CtC-CH2
CO CH2
•у-Хлор=у-ацетобути-
ролактон
Ацетамидин Этоксипропионитрил
^jyC^oqHs
ciO^NH,
2-Метил-4-амино-
5-этоксиметилпиримидин
ГГ'" ♦ гг —
СН3 N NHj-HCl s СН2СН2ОН
LXXII IX
о=ссн3
CICHCHjCHjOH
у-Хлору-ацето-
пропиловый спирт
2. Синтез тиамина построением его молекулы на пиримидиновом цикле
В этом методе получения тиамина исходят из 2-метил-4-амино-
5-аминометилпиримидина (LXV), который превращают (схема 86) в 2-метил-
4-амино-5-тиоформаминометилпиримидин (LXXV) или непосредственно, пу-
394
тем конденсации с дитиоформиатом калия в водном растворе [4, 180—182],
или через формильное производное (LXXIV), получаемое при взаимодейст-
вии соединения LXV с муравьиной кислотой или ее эфиром [183], которое
затем обработкой пятисернистым фосфором переводят в соединение LXXV
[4, 18, 183—185]. После нагревания с енольной формой эфира у-хлор-у-
ацетопропилового спирта (LXXVI, R = СОСН3) 2-метил-4-амино-5-тио-
формаминометилпиримидин (LXXV) образует эфир тиамина (LXXVII,
R = СОСН3); конденсация проводится в присутствии минеральной или
муравьиной кислоты [3, 4, 180, 182—186]. Эфир тиамина в результате гидро-
лиза превращают в тиамин (I).
Схема 86
Синтез тиамина методом построения его молекулы на пиримидиновом цикле
LXV LXXV
Для синтеза тиамина из соединения LXXV вместо эфира у-хлор-у-аце-
топропилового спирта (LXXVI) можно применять его циклические тауто-
мерные соединения: 2-метил-З-алкокси-З-хлортетрагидрофуран (LXXVIII)
в среде 95 %-ной муравьиной или 80 %-ной уксусной кислоты с выходом
54% [187], 2-метил-4-дигидро-2,3-дихлорфуран (LXXIX) в среде муравьи-
ной кислоты и пиридина с выходом 57% [188—190], у-хлор-у, у-диацето-
пропиловый спирт (LXXX) [185] или соединение LXXXI в 91%-ной му-
равьиной кислоте с выходом 56—62% [190]
LXXVIII
LXXXI
В некоторых способах создание тиоформильной группы в аминопирими-
диновом производном (LXXIV) достигается не с помощью пятпссрнпстого
фосфора, а при действии сероводорода (схема 86). Так, при непосредствен-
ной конденсации соединения LXXIV с у-хлор-у-ацетопропилацетатом
(LXXVI, R — СОСН3) в токе сероводорода в присутствии муравьиной кис-
лоты образуется эфир тиамина (LXXVII) [186, 191 ].
Следует отметить, что описанные выше синтезы тиамина имеют недо-
статочно высокий выход на стадии конденсации с производными у-ацетопро-
пилового спирта; реакция, по-видимому, сопровождается отщеплением
тиоформильной группы от малостабильного 2-метил-4-амино-5-тиоформами-
395
пометил пиримидина (LXXV) с образованием 2-метил-4-амино-5-аминометил-
-пиримидина (LXV).
Для построения молекулы тиамина на пиримидиновом цикле, помимо
пятисернистого фосфора, дитиоформиата калия или сероводорода, можно
применять сероуглерод [138] (схема 87). На соединение LXV действуют
аммиаком и сероуглеродом; образующийся 2-метил-4-амино-5-пиримидил-
метилдитиокарбамат (LXXXIII), конденсируют с у-хлор-^-ацетопропил-
ацетатом (LXXVI, R = СОСН3) в тиотиазоли новое производное -— тиамин-
тион-2 (LXXXVI), который окисляют перекисью водорода в тиамин-суль-
фат (I).
Этот высокоэкономический метод синтеза, имеющий важное техническое
значение, детально разработан японскими авторами; он протекает через
ряд промежуточных соединений (схема 87). Так, при действии аммиака и
сероуглерода в присутствии едкого натра из соединения LXV первоначально
образуется 2-метил-4-амино-5-пиримидилметилдитиокарбамат аммония
(LXXXII) [174], который в избытке раствора едкого натра переходит в
меркаптопроизводное (LXXXIII) [192]. Конденсация этого соединения с
•рхлор-т-ацетопропилацетатом (LXXVI, R — СОСН3) приводит к получе-
нию тиотиона (LXXXV) [174, 1931; одновременно получается около 10—12%
побочного 2-меркапто-7-метил-1,2,3,4-тетрагидропиримидо [4,5-d] пири-
мидина (LXXXIV) [192]. Однако можно не выделять промежуточных сое-
динений LXXXII и LXXXIII в кристаллическом виде и тиотион (LXXXV)
получить непосредственно конденсацией 2-метил-4-амино-5-аминометилпи-
римидина (LXV) с у-хлор-у-ацетопропиловым спиртом (LXXVI, R = Н)
или его ацетатом и сероуглеродом с избытком аммиака и едкого кали или
натра, при этом также образуется побочный бициклический продукт
(LXXXIV) [174, 194, 195].
Схема 87
Синтез тиамина через тиамин-тиои-2
-2НС1 148011
82%
75%
сн^н
s=c
CH nh2 ^snh4
LXXXII
NaOH
носсн,
CCL
CH£H,OR
^ГЧН LXXVI
S=C^ *
NH, SH
LXXXIII
CH, N NH,
LXV
CS2,NH„
NaOH,
C2HSOH
HOCCH,
ll 3
ClCCHjCHPR
LXXXVI
396
Неустойчивый тиотион (LXXXV) или его О-эфиры перегруппировыва-
ются в 2-метил-2-окси-3-тетрагидрсфурил-М-(2'-метил-4'-аминопиримидил-
5'-метил)дитиокарбаминовую кислоту (LXXXVII) [196], которая при обра-
ботке разбавленной соляной кислотой превращается в 3-М-(2'-метил-4'-ами-
нопиримидил-5'-метил)-4-метил-5-[3-оксиэтилтиазолинтион-2, тиамин-тион-2
(LXXXVI) [196]. При действии 1 %-ной соляной кислотой на соедине-
ние LXXXV первоначально образуется ерошенный пергидрофура нотиазоло-
вый цикл (LXXXVIII), который с 10%-ной соляной кислотой затем пере-
ходит в тиамин-тион-2 (LXXXVI) [197]. Однако при действии 10%-ной
соляной кислоты на соединение LXXXV происходит и непосредственная
циклизация в тиазолиновый цикл с образованием тиамин-тиона-2 (LXXXVI)
[194, 195], минуя промежуточные соединения LXXXVII и LXXXVIII.
Еще более упрощенным методом синтеза по способу Мацукава — конденса-
цией соединения LXV с сероуглеродом и у-хлор-у-ацетопропилацетатом
(LXXVI, R = СОСН3) в присутствии аммиака и едкого натра с последую-
щей обработкой соляной кислотой без выделения промежуточных продук-
тов — получают тиамин-тион-2 (LXXXVI) [175]. Последний вариант синте-
за получил дальнейшее улучшение [198, 199].
При действии на тиамин-тион-2 (LXXXVI) пергидролом при 25—30° С
образуется тиамин-сульфат (I), ацетильное производное предварительно
гидролизуется 30%-ной соляной кислотой [139, 174]. Из соединения
LXXXVI также получают тиамин-хлорид. Известен и такой вариант, когда
соединение LXXXV путем окисления пергидролом в присутствии соляной
кислоты непосредственно превращают в тиамин-сульфат [174]. Тиамин-
тион-2 (LXXXVI) при окислении перекисью водорода с последующим при-
бавлением нитрата аммония дает тиамин-мононитрат [200]. Общий выход
тиамина по этому методу из 2-метил-4-амино-5-аминометилпиримидина
(LXV) превышает 46%. Обработка тиамин-тиона-2 (LXXXVI) перекисью
водорода в присутствии карбонатов щелочноземельных металлов приводит
к получению солей тиамина—монохлорида, мононитрата, монороданата
и др. [201 ].
Для синтеза тиамина этим методом известно и применение у-хлор-у-аце-
топропилхлорида [202, 203] и 1-ацетокси-4-хлорпентанона-3 [140]. у-Хлор-
у-ацетопропиловый спирт также может быть заменен его циклическими
таутомерными производными: 2-метил-2-окси-3-ацетотетрагидрофураном
(LXXXIX) и 2-метил-2-ацетокси-3-хлортетрагидрофураном (ХС) [138]
сосн,
ососн,
CHj
LXXXIX
3. Синтез гомотиамина построением его молекулы на тиазоловом цикле
Этот метод синтеза экспериментально осуществлен только для уксусно-
кислого эфира гомолога тиамина (XCIV) с двумя метиленовыми группами
между пиримидиновым и тиазоловым циклами [184]. 4-Метил-5-|3-оксиэтил-
тиазол (IX) конденсируют с а-циан-у-броммасляным эфиром (XCI) в соеди-
нение XCII, которое затем при действии ацетамидина превращают в окси-
производное гомолога тиамина (ХСШ) (схема 88).
Последующая замена гидроксильной группы на водород достигается
через хлорпроизводное, восстановлением которого получают гомотиамин
(XCIV). Для синтеза тиамина этот вариант^не нашел^развития в последую-
щих исследованиях.
397
Схема 88
Синтез гомотиамина
ROOC -СЦ^НЛг
СН
I
CN
CHjCHjOH
XCI IX
ROOC^ ^.NH
CHCHjCHJS—т-СН, CH/C"NH.
- <!n Cx ’ 4
SX CHjCHjOH
XCII
PCI,
Ct
CH,CH,\ +
CH,
CH/SHjOH
OH
XX oz
CH,NNH, S Cli^OH
XCHl
CH, N NH,
4. Синтез важнейших структурных частей молекулы тиамина
Синтез 2-метил-4-амино-5-аминометилпиримидина и 2-
метил-4-амино-5-хлор(бром)метилпиримидина
В качестве пиримидинового компонента во всех синтезах тиамина при-
меняют 2-метил-4-амино-5-хлорметилпиримидин (LXXII), 2-метил-4-амино-
5-бромметилпиримидин или 2-метил-4-амино-5-аминометилпиримидин
(LXV). Во многих синтезах обычно путем конденсации ацетамидина (XCV) с
соединениями XCVI, в структуру которых включается трехуглеродная
система с оксиметиленовой группой, получают замещенные 2-метилпирими-
дина (XCV1I),
NH R'OCH R'
СН3 NH2 R"
XCV XCVI
R=CHS,CH2CHS; R'=CH2OR, CH2COOR, COOR, CN; R"=CN, COOR;
R"'=H, CH„ CH2CH3, COCH5, CH2OCHS
затем различными реакциями их превращают в пиримидиновые компоненты.
В конденсацию с ацетамидином вместо оксиметиленовой группы можно
ввести карбалкоксигруппу. Если вместо ацетамидина для конденсации
применять ацетаминоэфир или тиоацетамид, то оксиметиленовая группа
должна быть заменена аминометильной.
Для конденсации ацетамидина (XCV) с чнс-оксиметиленовыми произ-
водными, например алкоксиметиленциануксусным эфиром (XCVIII), реак-
ция первоначально протекает с образованием промежуточного ацетамиди-
нометиленового соединения (XCIX) [181], которое под влиянием следов
оснований (этилата натрия, едкого натра или ацетамидина) или кислот
циклизуется. Если формильное производное (XCVIII) имеет в положениях
1 и 3 группы, способные конденсироваться с ацетамидином или ацетимино-
эфиром в пиримидиновый цикл, то направление реакции зависит как от
pH среды, так и от характера функциональных групп [181,204 ]. При цикли-
398
ft
зации в кислой среде (например, при нагревании в водной уксусной кислоте)
j реакция протекает преимущественно с образованием аминопиримидина (С),
а в щелочной среде — оксипиримидина (CI)
NH
и
СНз'' '"ЫН.
XCY
ROCH^ COOR
'"С
I
CN
XCVIII
Ацетамидин, употребляемый в подавляющем числе синтезов пиримиди-
нового компонента тиамина, может быть получен из ацетамида через ацето-
иминоэфир. Ацетамид, с прекрасным выходом получаемый насыщением
уксусного ангидрида или уксусной кислоты аммиаком или при отгонке
воды из смеси уксусной кислоты и углекислого аммония [205], дегидратиру-
ется при взаимодействии с хлорокисью фосфора при 100—150° С, образуя
ацетонитрил. Его также получают непосредственно из уксусной кислоты и
аммиака при пропускании смеси их паров над окисью алюминия или окисью
тория при температуре 400—500° С [206 ], над селикагелем при 500° С с
выходом 95% [207] или над смесью селикагеля и фосфорной кислоты при
280—300° С с выходом 87% [208]. Для получения ацетонитрила можно
подвергнуть парофазной конденсации пентан и аммиак при 520° С над алю-
момолибденовым катализатором (выход 43,8%) [209] или этилен и аммиак
над окислами металлов, нанесенных на окись алюминия [210].
Действуя на безводный ацетонитрил сухим хлористым водородом в
присутствии этилового спирта, получают ацетоиминоэфир, который со
спиртовым раствором аммиака в отсутствие воды дает гидрохлорид ацетами-
дина с выходом 91—95% [211 ]. Так как в большинстве синтезов ацетамидин
применяют в свободном виде, то его получают из солей в результате обработ-
ки алкоголятом натрия.
В качестве трехуглеродной системы с оксиметиленовой группой (или
карбалкоксигруппой) для конденсации с ацетамидином в производные
2-метилпиримидина применяют замещенные янтарной, малоновой и пропио-
новой кислот. Далее описываются синтезы пиримидиновых компонентов,
используемых в различных путях построения молекулы тиамина.
1) Синтез из производных янтарной кислоты
Замещенный пиримидин по методу Андерзага и Вестфаля создается пу-
тем конденсации ацетамидина (XCV) с суспензией натриевого производного
оксиметиленянтарного эфира (CVI) в ксилоле [4, 184]. При нагревании с
хлорокисью фосфора гидроксил образовавшегося 2-метил-4-окси-5-пприми-
днлуксусного эфира (CVII) замещается на хлор и полученный хлорпирими-
днн (CV1II) переводится в 2-метил-4-амино-5-пиримидилацетамид (CIX)
действием жидкого аммиака пли спиртового раствора аммиака под давлением
при 120—130° С (схема 89).
Соединение CIX при расщеплении по Гофману с применением гипобро-
мита натрия дает 2-метил-4-амино-5-аминометнлпиримидин (LXV) с выходом,
который может быть повышен до 70% при применении гипохлорита натрия
(реакцию проводят при 0°С и затем в течение 3 ч при 100° С) [164].
2-Метил-4-амино-5-галогенметилпиримидин (LXXII) получают из
2-метил-4-амино-5-аминометилпиримидина (LXV) через 5-оксиметилпроиз-
водное (СХ) [164]. В этом способе синтеза общий выход соединения LXXII
составляет около 22% на формилянтарный эфир [164].
399
х
Схема 89
Синтез 2-метил-4-амино-5-галогенметилпиримидииа из производных янтарной
кислоты
NH
п
СН, NH,
ХС¥
COOR
I
СН,
ROOC-CH •
I
CN
СИ
ОН С!
jRc^coor Ach^oor
I -^11
СН3'Г* ГН CIVN'NH,
CHI CIV
N'
[н]
снЛ nh,
NH
II
zcx
CH, NH,
xcv
CHjCOOR_____
' ’ POCl;
80-90%
£H,OOOR
NH3
CHjCONH,
CH,OH
COOR
CH,
HOCH=C ------
I 63% ,
COOR CH, N OH
CVI CVII
N»OCl N^A^^NaNO^H4) гА
»% ’ A- A 67% A-,
CH, N NH, CH, N NH,
LXV CX
HC1
CH, N Cl
CVIII
(Br)
CH,Cl
NH,
87% , . , .
CH, N NH,
CIX
CH, N NH,
LXXII
R-c,h5
В другом варианте [18] с ацетамидином (XCV) конденсируют цианянтар-
ный эфир (СН); гидроксильная группа в образовавшемся 2-метил-4-амино-6-
окси-5-карбалкоксиметилпиримидине (СШ) замещается на хлор (соедине-
ние CIV), который элиминируется с помощью цинковой пыли в уксусной
кислоте; полученное соединение CV переводится в 2-метил-4-амино-5-пири-
мидилацетамид (CIX) обработкой аммиаком. Дальнейшее превращение в
пиримидиновый компонент производится по той же схеме. Синтез пирими-
динового компонента LXV или LXXII по схеме 89 недостаточно эффективен.
Формилянтарный эфир (CVI) получают из янтарного эфира с выходом
60—70% [212]. Для янтарной кислоты известен метод каталитического
гидрирования малеиновой кислоты, синтезируемой из бензола [213 ]. Описан
метод получения динитрила янтарной кислоты из акрилонитрила при
действии водного раствора цианистого калия и сернокислого магния с
выходом 80—85% [214]; при гидролизе нитрила легко получается янтарная
кислота.
2) Синтез из производных малоновой кислоты
Для конденсации с ацетамидином (XCV) применяется этоксиметиленма-
лоновой эфир (CXI) в присутствии этилата натрия [18, 181,215]. Гидроксиль-
ная группа в образующемся 2-метнл-4-окси-5-карбалкоксипиримидине
(СХП) превращается в 4-аминогруппу соединения С через хлорпроизводное
(СХШ) описанным выше способом (схема 90). В соединении С [181 ] карбок-
сильная группа превращается в амннометилрную через амид (CXVI), кото-
рый затем дегидратацией хлорокисью фосфора дает нитрил (CXV), вос-
станавливаемый в 2-метил-4-амино-5-аминометилпиримидин (LXV) [19].
Для получения пиримидинового компонента по методу Тодда и Бергеля
[181] можно исходить из производного циануксусного эфира (XCVIII) и
ацетамидина (XCV); этот путь синтеза сокращает две стадии и дает непосред-
ственно 2-метил-4-амино-5-карбалкоксипиримидин (С) с выходом около
70% [204]. Можно также получить соединение С исходя из аминометилен-
циануксусного эфира и ацетаминоэфира и тиоацетамида [216].
По методу Греве [19] ацетамидин (XCV) конденсируют с этокси мет ил ен-
малонилдинитрилом (CXIV, R = C2HS), что сокращает синтез на четыре
стадии (схема 91). Хотя производные динитрила малоновой кислоты и более
труднодоступны, чем производные малонового эфира, однако этот способ
имеет несомненные преимущества перед ранее описанным. При конденсации
ацетамидина (XCV) с этоксиметиленмалонилдинитрилом (CXIV, R = С2Н6)
непосредственно образуется 2-метил-4-амино-5-цианпиримидин (CXV), циан-
400
группу которого подвергают дальнейшему преобразованию в аминометиль-
ную группу; восстановление проводят алюмогидридом лития [217] или
каталитически с палладиевым или никелевым катализаторами [56] (в послед-
нем случае при 120° Си 113 ат в метиловом спирте и аммиаке); выход соеди-
нения LXV составляет 98% [2181. Получены выходы свыше 80%, в частно-
сти при гидрировании в уксусной кислоте с палладиевым катализатором в
присутствии хлористого водорода [191, 219, 220] и при электролитическом
восстановлении в присутствии минеральных кислот [219, 220].
Схема 90
Синтез 2-метнл-4-амиио-5-галогенметилпиримидина из производных малоновой
кислоты
NH
H
CH3 1
xcv
nh2
ROCH COOR
V
I
COOR
CXI
I, N OH
CXII
.COOR
COOR poet,
65%
CHa N Ct
CXIII
NH
II
XCX -
CH3 NHj
XCV
ROCH COOR
V
CN
XCVIII
45%
H
2И % раствор NaOH; 37%
или CHjCOOH; около 70%
CH3
NH
11
XCX и
CH3 NHj
XCV
ROCH CN
V
I
CN
CXIV
<^H5ONa
69%
^C>. CN
CH, NH2
XCIX
CN
Cl
CN
CH3 N NH,
CXV1I
(NH3,ctiMpT
5o°
Cl
POCla
. JL 76%
85%
ch;
ci
N'
ch
OH
.PCU
x„, 50%(«C)
CHj N NHa A 7
CXV
[H]/Ni
CHjOH,NH3
80-98%
Ny^NaNQ/H-
Jo JL 67%
COOR
NH,
.CONHj
CHj N NH2
CXVI
,СНд°ННС1(нВг) N
CH3 N NH3
LXV
95%
CH3 N NH,
LXXII
oh , '
jC CONH, ccXnh,)
N NH3
ex
44%
NH2
COOC2H5
CHj
cooc2hs
CXX CXIX CXVI1I XCV
R=c2Hs
2-Метил-4-амино-5-аминометилпиримидин (LXV) по методу Греве полу-
чают с общим выходом 67,5%, исходя из этоксиметиленмалонилдинитрила
(CXIV). т. е. 30% — из малонового эфира, что ставит этот метод синтеза
пиримидинового компонента в разряд лучших.
С худшим выходом — около 15% — и с большей затратой ацетамидина
соединение LXV можно получить конденсацией ацетамидина с малоновым
эфиром в 2-метил-4,6-диоксипиримидин (CXVIII) и с последующим его
преобразованием через соединения CXIX, CXX, CXVII [221 ] (схема 90).
Образующийся при восстановлении 2-метнл-4-амино-5-аминометилпири-
мидин (LXV) превращают первоначально в 2-метнл-4-амино-5-оксиметилпи-
римидин (СХ), причем с одним эквивалентом азотистой кислоты при 60° С
происходит преобразование только алифатической аминогруппы; затем
нагреванием оксиметиламинопроизводного СХ с бромистым водородом в сре-
де уксусной кислоты (60° С) [164 ], или с хлористым водородом в спиртовой
401
среде, или с тионилхлоридом [217, 2221 получают 2-метил-4-амино-5-гало-
генметилпиримидин (LXXII). Общий выход соединения LXXII на этоксиме-
тиленмалонилдинитрил (CXIV) составляет около 34%.
Для непосредственного получения 2-метил-4-амино-5-оксиметилпирими-
дина (СХ) из 2-метил-4-амино-5-карбоалкоксипиримидина (С) применяют
восстановление карбоалкоксильной группы алюмогидридом лития в эфир-
ной среде [216, 217].
2-Метпл-4-амино-5-цианпиримидин (CXV) может быть получен в ре-
зультате конденсации ацетоксиметиленмалонилдинитрила с ацетамидином
[216] или ацетиминоэфира в присутствии аммиака с производными нитрила
или динитрила малоновой кислоты; в последнем случае образование пири-
мидинового цикла протекает с высоким выходом, но для этого необходимо
применить аминометиленциануксусный эфир [223], аминометиленмало-
нилдинитрил или этоксиметиленмалонилдинитрил (CXIV) в присутствии
аммиака [224].
Для получения 2-метил-4-амино-5-цианпиримидина (CXV) предложен
метод, основанный на взаимодействии тиоацетамида с аминометиленмало-
нилдинитрилом [225] в спиртовом растворе алкоголята натрия с образова-
нием таутомерной открытой формы ацетамидинометиленмалонилдинитрила
с последующей циклизацией.
Создание оксиметиленовой группы в производных с высокоподвижным
а-водородным атомом метиленовой группы возможно методами сложноэфир-
ной конденсации или цианэтилирования муравьиного эфира, а также мето-
дом конденсации таких соединений с эфирами ортокарбоновых кислот.
В этом способе синтеза применяют в качестве исходных веществ производ-
ные малоновой и циануксусной кислот [226 ]. Кроме простоты осуществления,
этот способ обладает еще и тем удобством, что в одну стадию с высоким вы-
ходом получаются устойчивые защищенные оксиметиленовые производные.
Получение такого алкоксиметиленового производного (CXIV) для синтеза
пиримидинового компонента по Греве [19] (по схеме 90) из малонилдинит-
рила (образующегося при вакуум-перегонке цианацетамидина с пятихло-
ристым фосфором с выходом 60%) и ортомуравьинового эфира в уксусном
ангидриде иллюстрируется следующей схемой:
(СНзСО)2О
NCCH8CN -f- НС (OR)S-----> NCCCN -f- 2ROH
75% |
HCOR
CXIV
3)Синтез из производных пропионовой кислоты
Более совершенным является метод, предложенный Вильямсом, Клином и
Финкельштейном [2, 5, 227, 228], в котором-в качестве оксиметиленового
производного применен а-оксиметилен-р-этоксипропионовый эфир (CXXI)
в виде натриевой соли. При его конденсации с ацетамидином (XCV) образу-
ется 2-метил4-окси-5-этоксиметнлпиримидин (СХХП) (схема 91), в котором
гидроксил положения 4 при действии хлорокиси фосфата замещается на
хлор, а затем с помощью насыщенного спиртного раствора аммиака под
давлением при 140°С соединение СХХIII превращается в 2-метил-4-амино-5-
этоксиметилпиримидин (LXXIII). В этом соединении 5а-этоксигруппа
замещается на хлор при нагревании в 10%-ном растворе хлористого водоро-
да в уксусной кислоте при 120° С под давлением или при воздействии спирт-
ного раствора хлористого водорода [217, 229]; при этом образуется 2-метил-
4-амино-5-хлорметилпиримидин (LXXII).
Превращение 2-метил-4-окси-5-этоксиметилпиримидина (СХХП) в ко-
нечное соединение LXXII протекает с хорошим выходом в три стадии.
Этот метод после улучшения нашел применение на заводах фирмы «Мерк»,
возможно, благодаря стабилизации малоустойчивого а-оксиметилен-р-эт-
402
Схема SI
Синтез 2-метнл-4-амино-5-галогеиметилпиримидииа из производных пропионовой
кислоты
COOR
СХХ!
NH AUOCH СЦОС.Н,
С,Н5
СХХП
роа,
70%
сххш
XCV
ROCH ^CHjOQHs
I
CN
CXXIV, «г
.^снрсА
CH3 NH,
CXXV
70% NH,, C,HSOH
оксипропионового эфира (CXXI) защитными алкильными группами; в этом
случае выход метокси- или этоксипроизводных цис- или транс-форм дости-
гает 70% [2301. Для образования пиримидинового производного (СХХП) в
конденсацию с ацетамидином вводят тольдо цис-форму а-алкоксиметилен-р-
этоксипропионового эфира (CXXI)
AIkOCH CFUOCjHj
AIkOCH COOR
OOR
CXXI -цис
СНгОСгНб
CXXI-транс
Рациональный синтез пиримидинового компонента ведется по методу
Челинцева и Беневоленской [231 ]; в этом синтезе для конденсации с ацета-
мидином (XCV) применяют а-ацетоксиметилен-₽-этоксипропионитрил
(CXXIV, R = СОСН3), в результате чего образующийся замещенный пири-
мидин (LXXIII) требует всего только одного преобразования (схема 91) в
2-метил-4-амино-5-галогенметилпиримидин (LXXII). В реакцию с ацетами-
дином вводится защищенный ацилированием цос-а-оксиметилен-₽-этокси-
пропионитрил (CXXIV, R = СОСН3), получаемый из его натриевой соли
при обработке хлористым ацетилом. Следует отметить, что оксиметиленовые
производные нитрила пропионовой кислоты существуют в форме цис-транс-
изомеров [2291
ROCH2CCN
HCOR
CXXIV-цис
ROCH2CCN
II
ROCH
CXXW-транс
При конденсации соединений XCV и CXXIV в спирто-бензольной среде
первоначально, по-видимому, образуется промежуточный а-ацетамидино-
метилен-3-этоксипропионитрил (CXXV) [2291; это соединение затем цикли-
зуется и перегруппировывается при нагревании со щелочью в 2-метил-4-
амино-5-этоксиметилпиримидин (LXXIII) с выходом 50—55%. При реакции
выделяется уксусная кислота, которая дополнительно связывает одну моле-
кулу ацетамидина. Следует указать, что из ряда изученных ацилпроизвод-
ных [231,232 ] лишь ацетильное производное а-окспметилен-Р-этоксипропио-
нитрила обладало реакционной способностью к циклизации с ацетамидином.
Этот синтез значительно улучшили Федор с corp. — оказалось, что
с более высоким и устойчивым выходом с ацетамидином реагируют простые
эфиры а-оксиметилен-^-этоксипропионитрила, например метиловый
(CXXIV, R = СН3) или этиловый [2291; в этом случае для реакции
достаточно брать только один эквивалент ацетамидина.
При конденсации 1 моля ацетамидина с цис-а-метоксиметилен-р-этокси-
пропионитрилом (CXXIV, R = СН3) с последующим нагреванием до 40°С
403
и обработкой раствором едкого натра получают 2-метил-4-амино-5-этоксиме-
тилпиримидин (LXXIII) с выходом 81% [233, 2341. Для реакции с тем же
успехом используют /{uc-a-этоксиметилен-^-этоксипропионитрил [234 ]. Ана-
логично получают соединение LXXIII из транс-а-метокси- и л//?анс-а-этокси-
метилен-°-этоксипропионитрила с тем же выходом [234].
Из 2-метил-4-амино-5-этоксиметилпиримидина (LXXIII) действием бро-
мистоводородной кислоты или пропусканием хлористого водорода в спирто-
вой раствор (при 70° С, выход 92%), а также обработкой соляной кислотой
под давлением [217] или 10%-ным раствором хлористого водорода в уксус-
ной кислоте под давлением при 120°С [229] получают 2-метил-4-амино-5-
галогенметилпиримидин (LXXII) с выходом 90%. Общий выход 2-метил-4-
амино-5-галогенметилпиримидина (LXXII) по этому методу достигает
56% на Р-этоксипропионитрил, применяемый для получения а-метоксиме-
тилен-Р-этоксипропионитрила (CXXIV, R = СН3).
Синтез пиримидинового компонента по методу Челинцева и Федора (схе-
ма 91) имеет известные преимущества по сравнению с другими синтеза-
ми в связи с применением для конденсации с ацетамидином (XCV) а-мет-
оксиметилен-р-этоксипропионитрила (СХХIV), получаемого достаточно прос-
то с устойчивыми и высокими выходами (см. схему 85, с. 394). По этому
методу осуществляется синтез пиримидинового компонента тиамина на
заводах СССР и некоторых других стран.
Для получения соединения LXXIII реакцию ацетамидина с р-этокси-
пропионитрилом и этилформиатом осуществляют и непосредственно в
присутствии спирта в среде ксилола с распыленным в нем металлическим нат-
рием с последующей обработкой диметилсульфатом [235 ]._
Для получения а-оксиметилен-р-этоксипропионового эфира (CXXI) в
виде его метокси-или этоксипроизводных (метод Вильямса, Клина и Финкель-
штейна) исходят из p-этоксипропионитрила (CXXVI), который гидролизом
переводят в Р-этоксипропионовую кислоту, этерифицируют ее, затем Р-эт-
оксипропионовый эфир конденсируют по реакции Кляйзена с эфиром му-
равьиной кислоты в присутствии алкоголята натрия, свободного от спирта
[2, 5], или металлического натрия в бензоле и алкилируют при действии
диметилсульфата (выход цис-формы 52%) или диэтилсульфата (выход
цис-формы этоксипроизводного 70%) [230]. Едкий натр вызывает изомери-
зацию цис-формы в /прайс-форму.
Н2О СНаОН
QHbOCHjCHiCN —> CjHsOCHjCHtCOOH-------->
CXXVI
НСООС2Н5; (A1k)2SO«
—> CjHsOCHjCHtCOOCjHs----------------> CJLCXZILCCOOQHs
C»HsONa; 52—70% p
HCOR
' CXXI
Для получения соединения CXXI использован акриловый эфир, который
форматируют муравьиным эфиром [236].
Помимо соединения CXXI. для конденсации с ацетамидином применяют
а-диметокси- и а-метоксиэтоксиметил-Р-алкоксипропионовый эфир [34, 237],
а также а-дибензилоксиметил-р-этоксипропиобензилат [237].
а-Ацетокси- или а алкоксиметплен-р-этоксипропионитрил (CXXIV) по-
лучают из соответствующего эфира и формиата методом сложноэфирной
конденсации в виде натриевых производных.
В синтезе а-ацетоксиметилен-Р-этоксипропионитрила (CXXIV, R =
= СОСН3) [231] исходят из этиленциангидрина, который алкилируют с
помощью муравьиного эфира в присутствии алкоголята натрия в р-этокси-
пропионитрил (CXXVI); последний подвергают конденсации Кляйзена с
муравьиным эфиром в бензольной среде в присутствии алкоголята натрия,
натриевое производное а-оксиметилен-р-этоксипропионитрила (CXXVII)
404
выделяют в виде желтоватого осадка, свободного от спирта, и затем обраба-
тывают хлористым ацетилом.
NaOH
CH2=CHCN + СгНьОН --------
83—85%
НСООС2Н5
HOCH2CH2CN----------------
QHeONa; 72—75%
QHsOCHjCHjCN ->
CXXVI
HCOOQHs CHjCOCl
> QHbOCHjCCN-> CaHsOCHssCCN
CjHsONa; 65—68%--------------------il 30%-|j
HCONa HCOR
CXXVII CXXIV
0-Эгоксипропионитрил (CXXVI) с более высоким выходом и значитель-
но проще синтезируют путем цианэтилирования этилового спирта акрило-
нитрилом под влиянием каталитических количеств едких щелочей [238,
239].
Для получения а-этоксиметилен-Р-этоксипропионитрила (CXXIV,
R = С2Н5) была предложена схема, в которой исходят непосредственно
из акрилонитрила, минуя Р-этоксипропионитрил (CXXVI) [240]. Изучено
взаимодействие акрилонитрила с муравьиным эфиром [241].
Натрийенолят а-оксиметилен-р-этоксипропионитрила (CXXVII) мо-
жет образовывать алкиленолы (CXXIV, R = алкил) при обработке его
такими алкилирующими средствами, как монохлорметиловый эфир или
диметилсульфат. В этом случае для алкилирования нет надобности выделять
натриевое производное енола из реакционной среды [229].
Для получения а-метоксиметилен-0-этоксипропионитрила (CXXIV,
R = CHS) Р-этоксипропионитрил (CXXVI) конденсируют с муравьиным
эфиром в бензоле, ксилоле или керосине в присутствии металлического
натрия и спирта, затем образующийся натрийенолят а-оксиметилен-Р-этокси-
пропионитрила (CXXVII) метилируют диметилсульфатом в той же реакцион-
ной смеси при температуре 55° С. 1{нс-а-Метоксиметилен-Р-этоксипропио-
нитрил (т. кип. 103—104° С при 3 мм) выделяют с выходом 78% [242].
Для получения транс-формы этого соединения (т. кип. 94—96° С при 3 мм)
реакционную смесь после метилирования обрабатывают 50%-ным раство-
ром едкого натра; выход составляет 92% [173].
цнс-а-Алкоксиметилен-Р-алкоксипропионитрил (CXXIV) применяется в
синтезе 2-метил-4-амино-5-этоксиметилпиримидина (LXXIII) (схема 91).
щранс-а-Метоксиметилен-Р-алкоксипропионитрил (CXXIV, R = С2Н5) ис-
пользуется для синтеза 2-метнл-4-амино-5-аминометилпиримидина (LXV)
через бициклический пиримпдопиримидин (схема 92) в методе построения
молекулы тиамина на пиримидиновом компоненте.
Еще один важный метод синтеза пиримидинового компонента представ-
лен на схеме 92. По реакции Такамицава [242, 243] шранс-а-метоксиметилен-
0-этоксипропионитрил (CXXIV, R = СН3) вступает в конденсацию в при-
сутствии щелочи с избытком ацетамидина (XCV) с образованием бицикли-
ческого 2,7-диметил-5,6-дигидропиримидо [4,5-d) пиримидина (CXXIX),
имеющего т. пл. 172—173°С с выходом 60%, который после кипячения с
разбавленным раствором едкого натра почти количественно превращается
в 2-метил-4-амино-5-ацетамидометилпиримидин (СХХХ) [244]. цпс-Форма
CXXIV7 под влиянием щелочи легко изомеризуется в транс-форму CXXIV,
поэтому в конденсации с ацетамидином применяют без разделения их смесь,
образующуюся при взаимодействии соответствующего эфира и формиата
(см. выше), что сильно повышает экономичность этого метода.
405
Схема 92
Синтез 2-метил-4~амнио-5-аминометилпиримидииа из производных пропионовой
кислоты
XCV
И'<Э н RO
V ROH R O "сн
CH2OR CH2OR
CXXIV, трак CXXVIII
NH
CH „CN CHjCNH.
I? 'V .jcv t
Cx CHjOR
CXXV
„СН „CN
N *C NH2
н Нз°- N^yCH=>NHCOCH2 20%Ha
снААгАа^’0^ CH, nAvh, m%
CXXIX CXXX
R=CH3,<:2H5; R'=CH,, CsHs, COCHj
CH^bi NHj
LXV
При проведении конденсации а-алкоксиметилен-Р-алкоксипропионитри-
ла с 2 молями ацетамидина (XCV) в растворе этилата натрия в спирте с
последующей обработкой едким натром выход повышается до 80% [234,
235 J. Такое повышение выхода связано с тем, что предварительно прохо-
дит диацетализация а-алкоксиметилен-Р-алкоксипропионитрила под дей-
ствием спирта в а-диалкоксиметил-р-алкоксипропионитрил (CXXVIII),
который реагирует с одной молекулой ацетамидина и образует а-ацетами-
динометилен-Р-алкоксипропионитрил (CXXV) [245 J, а затем при действии
второй молекулы ацетамидина циклизуется в пиримидопиримидин (CXXIX)
[234]. Следует отметить, что соединение CXXV при действии газообразного
хлористого водорода согласно схеме 91 превращается в 2-метил-4-амино-5-
алкоксиметилпиримидин (LXXIII).
В реакцию с ацетамидином поэтому стали вводить уже готовый а-диалк-
оксиметил-Р-алкоксипропионитрил (CXXVIII), в качестве которого приме-
няют а-диметокси- [234], а-диэтокси- [234, 246, 247 ], а-метоксиэтоксиметил-
Р-этоксипропионитрил [234 ], а-диметоксиметил-Р-метоксипропионитрил
[234], а также а-метоксиэтоксиметилакрилонитрил, который присоединяет
спирт, находящийся в реакционной смеси, и превращается в а-метоксиэт-
оксиметил-Р-этоксипропионитрил [234, 248].
В результате гидролитического дезацилирования 2-метил-4-амино-5-
ацетамидометилпиримидин (CXXIX) при действии 25%-ного раствора
серной кислоты [2491, спиртового раствора хлористого водорода или щелочи
с выходом 85% получают 2-метил-4-амино-5-аминометилпиримидин
(LXV) [244, 250]; общий выход из p-этоксипропионитрила составляет
около 62%.
Соединение LXV используют для синтеза тиамина по методу построения
его молекулы на пиримидиновом цикле (см. с. 394) или по методу конденса-
ции пиримидинового и тиазолового компонентов (см. с. 392) с предваритель-
ным превращением в 2-метил-4-ампно 5-хлор (бром) метилпиримидин (см.
с. 400). а-Диалкоксиметил-Р-алкоксипропионитрил (СХХХ) получают реак-
цией а-диалкоксиметилакрплонитрила с алифатическими спиртами в при-
сутствии кислот или оснований [251 ].
Рассматриваемый метод синтеза пиримидинового компонента LXV
имеет важное техническое значение и используется для получения тиамина
на заводах Японии и других стран.
Для конденсации с ацетамидином предложено применение а-этоксимети-
лен-Р-ацетоксипропионитрила [252 ] и а-диэтоксиметил-р-аминопропионит-
рила [253].
406
Си нтез у-га л оген-у-а цетоп роп и л ового спирта, его
ацетата и других производных
Для синтеза тиамина по методу построения его молекулы на пиримиди-
новом компоненте, а также для получения тиазолового компонента (4-метил-
5-р-оксиэтилтиазола) при двухкомпонентном синтезе тиамина в качестве
структурной части молекулы применяют у-галоген-т-ацетопропиловый
спирт, или его сложные эфиры, или таутомерные циклические его формы.
Исходный у-ацетопропиловый спирт (СХХXIV) можег быть получен дейст-
вием эфиров р-галогенэтилового спирта на натрийацетоуксусный эфир
(CXXXI)
CH2COCHNa + HalCH2CH»OR -> CH3COCHCH2CHaOR ->
I ' I
COOQHs COOC2H5
CXXXI
СХХХП
-> CHjCOCHCHsCHjOR -> CH3COCH2CH2CH2OR ->
COOH CXXXIII
-> СН*СОСН2СН2СН2ОН
CXXXIV
Продукт конденсации СХХХП подвергают гидролизу в кислой среде,
причем одновременно происходит отщепление углекислоты и получается
соединение СХХХШ; лучшие выходы наблюдаются при применении йоди-
стых производных, однако предпочитают работать с более дешевыми и до-
ступными эфирами бромзамещенных спиртов. В результате применения
простых эфиров Р-бромэтилового спирта удалось достигнуть выхода 55%
[145]; при использовании ацетата Р-бромэтилового спирта выход едва дости-
гает 25% [181].
Важнейший общепринятый метод синтеза у-ацетопропилового спирта
(CXXXIV) [254] состоит в конденсации натрийацетоуксусного эфира
(CXXXI) с окисью этилена в присутствии этилата натрия в промежуточный
у-ацетобутиролактон (CXXXV), который одновременно гидролизуют и
декарбоксилируют в соединение CXXXIV с выходом 77—80%:
G,H6ONa Н2О(НС1)
CHjCOCHNa + Н2С---СН2 —------> СН3СОСН----СН2 --------->
I \/ II —СО2;80%
СООС2Н5 о со сн2
CXXXI
CXXXV
-* CHSCOCH2CH2CH2OH
CXXXIV
у-Ацетопропиловый спирт получают также из лесохимического фурфу-
рола (CXXXVI), который первоначально гидрируют в 2-метилфуран,
сильван (CXXXVII) с выходом 92% [255, 256], подвергают очистке [257],
а затем в присутствии палладиевой черни или никелевого катализатора в
водной среде частично гидрируют в 2-метил-4,5-дигидрофуран (CXXXVIII)
[258, 259]; последний в реакционной смеси в результате гидролиза дает
у-ацетопропиловый спирт (CXXXIV).
407
CXXXVI CXXXVH
/ V'OCHjCHsCHjCOC 1I3
V£h3
CXL
H»°, / V-OH
"HP S/Vh,
CXXXVIH CXXXIX
HOCHjCHjCHjCOCH,
CXXXIV
Этот метод имеет практическое значение.
Стедует, однако, отметить, что для получения у-галоген-у-ацетопропи-
лового спирта применение свободного у-ацетопропиловсго спирта
(CXXXIV) нецелесообразно, так как, подобно моносахаридам, он
подвержен таутомерным превращениям в циклический полуацеталь
(CXXXIX) [260], который легко претерпевает дальнейшие изменения,
или отщепляя воду с образованием 2-метил-4,5-дигидрофурана (CXXXVIH),
или этерифицируясь второй открытой формой у-ацетопропилового спирта
(CXXXIV) в димерный эфир (CXL).
Необходимо указать, что применение свободного у-ацетопропилового
спирта оказалось нецелесообразным также и потому, что при последующем
бромировании в водной среде [261 ], по-видимому, при действии образовав-
шегося бромистого водорода происходит изомеризация первичного у-ацето-
пропилового спирта (CXXXIV) во вторичный пентанол-2-он-4. Хотя
свободный у-ацетопропиловый спирт и легко образует циклический полуаце-
таль (CXXXIX), однако это не может предотвратить процесса изомериза-
ции первичного спирта во вторичный, так как в кислой среде равновесие
между у-ацетопропиловым спиртом (CXXXIV) и его полуацеталем
(CXXXIX) сдвигается в сторону ацетопропилового спирта.
Вследствие того что галогенирование свободного ацетопропилового
спирта проходит неудовлетворительно, была предпринята попытка [262]
провести галогенирование в процессе синтеза ацетопропилового спирта.
Конденсация натрийацетоуксусного эфира (CXXXI) проводилась или с
этиленбромгидрином, или с симметричным дибромэтаном. Образующийся
у-карбэтокси-у-ацетопропиловый спирт или его бромид хлорируют хло-
ристым сульфурилом в соединения CXLI и CXLII, затем их декарбоксили-
руют и гидролизуют в у-хлор-у-ацетопропиловый спирт (CXLIII):
СН3СОСНСН2СН2ОН
COOCjHg
SO2CI2
CH,COCCH„CH„OH
Cl COOCjH-
CXLI
CH3COCHNa
COOC2H5
CXXXI
СН3СОСНСН2СН2Вг
COOC2HS
СН1СОСНС1СН2СН„ОН •
CXLIII * *
CH3COCCH2CH2Br
Cl COOC2H.
CXLII
SO2Cl2
Промежуточный продукт синтеза ацетопропилового спирта из ацетоук-
сусного эфира и окиси этилена —у-ацетобутиролактон (CXXXV) — [254]
использован для непосредственного получения у-хлор-у-ацетопропилового
спирта (CXLIII) [11].
у-Ацетобутиролактон (CXXXV) хлорируют при действии хлора в вод-
ном растворе или хлористого сульфурила, используя повышенную под-
вижность а-водородного атома, находящегося между двух карбонильных
групп. Отсутствие склонности к изомеризации ацетобутиролактона связано,
408
по-видимому, с тем, что гидроксил защищен внутримолекулярной этерифи-
кацией карбоксильной группой; у-хлор-у-ацетобутиролактон (CXLIV) по-
лучается с выходом 83%. Его гидролиз при действии слабой соляной кисло-
ты и последующее декарбоксилирование приводят к получению у-хлор-
у-ацетопроп илового спирта (CXLIII) с выходом 73—75%
Cl
CHjCOCH----СН, SO2Cl2MH Clg+HaO CHgCoj:------ch2 h2o(hci)
CO CH. ” CO -CHa -CO2
"O 100°
CXLIV
CXXXV
CH2COCHClCH2CHjOH
CXLIII
Cl
-ОСН2СН2СНСОСНз
'СН3
CXLV
Однако значительная часть у-хлор-у-ацетопропилового спирта образует-
ся в виде димерного эфира (CXLV), который в реакции с тиоформамидом
также приводит к образованию 4-метил-5ф-оксиэтилтиазола [263]; димер-
ный эфир (CXLV) обладает меньшей реакционной способностью. Если
у-хлор-у-ацетопропиловый спирт выделять в виде ацетата — у-хлор-у-аце-
топропилацетата (CXLVII)—при гидролизе и декарбоксилировании хло-
ристым водородом в ледяной уксусной кислоте с дозированным количеством
воды и в последующем прибавлять уксусный ангидрид, то выход повышается
до 93—95% [264].
Этот наиболее рациональный метод получения у-хлор-у-ацетопропилово-
го спирта (CXLIII) имеет важное техническое значение вследствие того что
В нем реакция галогенирования протекает в благоприятных условиях,
исключающих размыкание у-ацетобутиролактона (CXXXV) в ацетопропило-
вый спирт.
Известное практическое значение для синтеза у-галоген-у-ацетопропил-
ацетата (CXLVII) имеет применение уксусного эфира у-ацетопропилового
спирта (CXLVI), который получают из соединения CXXXIV с выходом 80%;
галогенирование у-ацетопропилацетата проходит с лучшими результатами,
если выделяющийся при реакции галогеноводород нейтрализуется угле-
кислым кальцием [4, 261 ]:
(СН3СО)2О
CHsCOCH2CHtCH2OH----------> СНзСОСН2СН2СН2ОСООН, ->
80%
CXXXIV CXLVI
—> CHSCOCHCICH2CH?,ОСОСНз-
CXLVII
Синтез 4-мети л-5-^-оксиэтилтиазол а
В основе синтезов 4-метил-5-р-оксиэтилтиазола лежит общий метод
получения производных тиазола [265], основанный на конденсации а-га-
логенозамещенных кетонов с азотсодержащими тиосоединениями, в качестве
которых применяют тиоамид и его производные или тиомочевину, реагирую-
щие в своих таутомерных формах. Тиоамиды кислот легко приготовляются
из амидов алифатических кислот при взаимодействии с пятисернистым
фосфором. Часто получаемые в этой реакции плохие выходы тиоамида
объясняются тем, что образующийся фосфорный ангидрид переводит часть
амида в нитрил. При взаимодействии амида с пятисернистым фосфором в
среде органического растворителя (например, эфира), который извлекает
образующийся тиоамид и тем выводит его из сферы реакции, можно зна-
чительно повысить выход.
Синтез тиазолового компонента осуществляется путем взаимодействия
а-галогенокарбонильного соединения с одной из таутомерных форм тиосое-
409
динения, причем образование связи между атомом серы сульфгидрильной
группы и атомом углерода метиленовой группы сопровождается выделением
элементов галогеноводорода; вторая циклическая связь возникает между
углеродным атомом карбонила, реагирующего в енольной форме, и атомом
азота аминогруппы с выделением одной молекулы воды.
Почти все многочисленные варианты синтеза 4-метил-5-£ -оксиэтилтиазо-
ла (IX) можно объединить в две основные группы. К первой группе отно-
сятся синтезы через производные тиазола, замещенные в положении 2
легко удаляемыми посторонними заместителями; выходы в этой группе
недостаточно высокие. Вторая группа объединяет синтезы, основанные на
применении тиоформамида, непосредственно приводящие к построению
гетероцикла с необходимыми заместителями в одну стадию.
Следует отметить, что выбор способа получения 4-метил-5-р -оксиэтилтиа-
зола (IX) не только связан с преимуществами того или иного варианта по-
строения молекулы тиазола с необходимыми заместителями, но и находится
в зависимости от метода получения и доступности основного исходного
вещества — у-галоген -у-ацетопропилацетата.
Синтез через п р о и з в о д н ы е тиазол а, замещенные
в положении 2
В этих синтезах (схема 93) применяются замещенные тиоформамиды—
RCSNH2, где R — ОН, SH, NH2, OR', SR', a R'=алкил, в частности, дитио-
карбамат аммония, тиомочевина, ксантогенамид и роданиды металлов.
1) Синтез через 2-оксипроизводные тиазола.
При конденсации у-бром-у-ацетопропилацетата (CXLVIII) с роданидом
бария или свинца [4, 266] образуется у-тиоциан-у-ацетопропилацетат
(CXLIX), который циклизуется в уксусной кислоте в 2-окси-4-метил-5ф-
ацетоксиэтилтиазол (CL) с малым выходом — 10%. Это соединение с хлор-
окисью фосфора переводят в 2-хлорпроизводное, затем при действии цинковой
пыли в ледяной уксусной кислоте удаляют хлор и 4-метил-5-р -ацетокси-
этилтиазол (CLI) подвергают гидролизу в щелочной среде — получают 4-ме-
тил-5-р-оксиэтилтиазол (IX).
2) Синтез через 2-этоксипроизводное тиазола.
При взаимодействии у-хлор-у-ацетопропилового спирта (CXLIII) с ксанто-
генамидом образуется 2-этокси-4-метил-5ф-оксиэтилтиазол (CLII), который
после гидролиза, замещения гидроксила на хлор и последующего его
удаления, как описано выше, превращается в 4-метил-5-0-оксиэтилтиазол
(IX) [266]. Этот синтез идет с недостаточно высоким выходом.
3) Синтез через 2-аминопроизводное тиазола.
Конденсация тиомочевины с у-хлор-у-ацетопропиловым спиртом (CXLIII)-
[157, 267—269] или его эфиром [270] приводит к получению 2-амино-4-
метил-5-р-оксиэтилтиазола (LXVIII). Замещение аминогруппы этого соеди-
нения на водород достигается диазотированием и последующим расщеплени-
ем соли диазония нагреванием со спиртом или восстановлением цинком или
железом при температуре не выше — 10° С [271 ]. Выход 4-метил-5-0-окси-
этилтпазола (IX) достигает 30%, если разложение соли диазония проводить в
30%-ной ортофосфорной кислоте [270]. Однако лучше соль диазония под-
вергнуть бромированию и элиминировать диазогруппу через 2-пербромид
(CLIII); если его подвергнуть восстановлению цинковой пылью в ледяной
уксусной кислоте, то образуется соединение IX с выходом до 90% [272,
273]. Этот метод сложен в осуществлении.
4) Синтез через 2-меркаптопроизводное тиазо-
л а. При конденсации у-хлор-у-ацетопропплацетата (CXLVII) с дитиокар-
баматом аммония в спиртовой или водной среде при 50° С получают 2-мер-
капто-4-метил-5-ацетоксиэтилтиазол (CLIV), в котором окислением перекисью
водорода в кислой среде меркаптогруппу замещают на водород [274].
Окисление меркаптогруппы 10%-ной азотной кислотой или пергидролом в
концентрированной соляной кислоте в присутствии хлористого бария по-
410
Схема 93
Синтез 4-метял-5-р-оксиэтилтиазола через его производные, замещенные
в положении 2
Путь 1
о=с-сн3
ВгСНСН2С1 IUOCOCH,
CXLVI11
Ba(CNS)j
й о=с-сн,
C^ CHCH3CH,OCOCH3
CXLIX
Путь 2
Путь 3
CH,CHjOCOCH3
сн.
CH^CHjOCOCH)
-сн,
сн,
"S CHCHjOCOCH,
CLI
л~тгСНз
'Xs -^CHjCHjOCOCH,
h2n o=c-ch3
S^S^CHiiCHjOCOCH,
HOjS
•сн2
CHjCHjOCOCH,
CLIV
Путь 4
NH
II
xc
NH«S SH
O=C-CH3
C1CHCH2CH2OCOCH;
CXLVII
вышает выход 4-метил-5-0-ацетокситиазола (CL I) до 58—62% [1641. Для
окисления предложена смесь азотной кислоты и нитрита натрия при
90—100° С под давлением, дающая более высокий выход [275]. Из соедине-
ния CLI гидролизом получают 4-метил-5-р-оксиэтилтиазол (IX) с общим
выходом на у-хлор-у-ацетопропилацетат свыше 32%. Синтез по этому ва-
рианту представляет практический интерес.
Синтез из незамещенного тиоформамида
5) Синтез через эфиры у-г алоген-у-а ц е топ р on ило-
вого спирта. При конденсации тиоформамида с у-хлор-у-ацетопропил-
ацетатом (CXLV11) (схема 94) образуется 4-метил-5-р-ацетоксиэтилтиазол
(CLI), который затем при действии щелочей гидролизуют в 4-метил-5-0 -ок-
сиэтилтиазол (IX) [3, 51, 276, 277]. Применение у-бром-у-ацетопропилаце-
тата (CXLV1II) дает 4-метил-5-р-оксиэтилтиазол также с хорошим выходом
[261 ].
При замене сложного эфира у-хлор-у-ацетопропилового спирта на
простой, например на этиловый, продукт его конденсации с тиоформамидом—
этиловый эфир 4-метил-5-р -оксиэтилтиазола — получается с малым выходом
[145].
Для получения тиоформамида можно исходить из дитиомуравьиной
кислоты и аммиака [277 J. Вследствие лабильности тиоформамида в кристал-
411
лическом состоянии предпочитают получать его одновременно с конден-
сацией, т. е. проводить реакцию у-галогеп-у-ацетопропилового спирта или
его ацетата с формамидом в присутствии пятисернистого фосфора в одну
стадию [278] (см. схему 85).
Благодаря проведению различных улучшений вариант получения 4-ме-
тил-5-р-оксиэтилтиазола через сложный эфир у-хлор (или бром)-у-ацето-
пропилового спирта и тиоформамид имеет существенные технические пре-
имущества перед многими другими способами.
6) Синтез через свободный у-г а л о г е н -у-a ц е т о п ро-
ли левый спирт. При конденсации тиоформамида с у-галоген-у-аце-
топропиловьш спиртом (CXLIII) получают 4-метил-5-£-оксиэтилтиазол (IX)
в одну стадию в спиртовой среде с выходом 50—55% [11], а в среде эфира —
до 70% [263] (схема 94). Этот синтез имеет важное техническое значение.
Однако он применим только при методе, в котором для получения у-хлор-
у-ацетопропилового спирта пользуются ацетоуксусным эфиром (см. с. 407),
так как в случае, если исходным сырьем служит сильван (см. с. 408), ко-
нечным продуктом синтеза является у-галоген-у-ацетопропилацетат.
Схема 94
Синтез 4-метил-5-3-оксиэтилтназола из тиоформамида
Путь 5
Путь 6
Путь ?
CH,
co
I
etc--CH,
C^CH,
СНа СН,
со со
I I
zc« Л?
Cl СН,СН2ОСОСН, Cl CHjCHjOH
CXLVII CXUU
CXL1V
C1CH— CH,
CLV
LXXIX
CH,
CHjOOOR
CLV111
cxxxvm
Путь 10
CLX
^NH
HC^
SH
CH,COCHCH,COOR
a
CLIX
Путь 9
N---П-С1Ц HCCS11
JI —-------CH3COCHCH,CH,COOC,HS
'S^CH,CIIXOOC,II5 Cl
CLVII Путь 8 CLV1
R=C,HS, COCH,
7) Синтез из 2-м e т и л-4-эт о к с и - 3 - т етр а ги д p о ф у -
рана. Была сделана попытка применить для конденсации с тиоформами-
дом стабилизированную таутомерную гетероциклическую форму у-хлор-
412
у-ацетопропилового спирта [2791, а именно 2-метил-2-этокси-3-хлортетра-
гидрофуран (CLV), получаемый из у-хлор-у-ацетобутиролактона (CXLIV)
при действии спирта и серной кислоты (схема 94). Однако синтез 4-метил-5-
Р -оксиэтилтиазола этим способом не имеет существенных преимуществ
перед другими.
8) Синтез из у-хлорацето масляной кислоты.
В конденсацию с тиоформамидом вводят этиловый эфир у-хлорацетомасля-
ной кислоты (CLVI). Образующийся эфир (CLVII) обычным образом пере-
водят в амид и затем по реакции Гофмана в 4-метил-5-£-аминоэтилтиазол
(CLVIII). Это соединение диазотируют, затем диазогруппу замещают на
оксигруппу и получают 4-метил-5-р-оксиэтилтиазол (IX) [2 ^.Осуществле-
ние этого синтеза достаточно сложно (схема 94).
9) Синтез из р- хлорлевулиновой кислоты. Пред-
ложена схема синтеза 4-метил-5-тиазолилуксусного эфира (CLX) [280]
конденсацией тиоформамида и эфира р-хлорлевулиновой кислоты (CLIX)
или р-бромлевулиновой кислоты [281] (схема 94). Полученное соединение
CLX восстанавливают в 4-метил-5-0-оксиэтилтиазол (IX) алюмогидридом
лития с выходом 60% [282].
10) Синтез из 2-метил -4,5- дигидрофуран а. Для
конденсации с тиоформамидом в растворе 88—90 %-ной муравьиной кислоты
применяют 2-метил-4,5-дигидро-2,3-дихлорфуран (LXXIX), при этом полу-
чают 4-метил-5-р-оксиэтилтиазол (IX) с выходом 76% [283] (схема 94).
Соединение LXXIX образуется при хлорировании 2-метил-4,5-дигидрофура-
на (CXXVIII), являющегося промежуточным продуктом в синтезе ацето-
пропилового спирта из сильвана [258, 284], при температуре ниже — 50° С
с выходом 75% [189, 283].
О возможности применения соединения LXXIX для построения моле-
кулы тиамина на основе 2-метил-4-амино-5-тиоформиламинометилпирими-
дина указано ранее.
Другие синтезы
При взаимодействии у-хлор-у-ацетопропилацетата (CXLVII) с сернистым
-натрием в спиртовом растворе получают у-меркапто-у-ацетопропилацетат
(CLXI); без выделения из реакционной смеси на него действуют аммиаком
и формальдегидом, в результате чего образуется 4-метил-5 р -оксиэтилтиа-
золин-Д3 (CLXII). В качестве побочного продукта при реакции получается
тетрагидрофуриловый эфир тиазолина. Соединение CLXII дегидрируют
феррицианидом калия и гидролизуют в тиазоловый компонент IX; общий
выход составляет 34% [285].
соси,
с^н
ХСН2СН3ОСОСН3
CXLVII
сосн3
Т NHj+HCHO IN
- хн —--------
HSX XCH2CH1OCOCH,
CLXI
сн,
S сн.сн2ососн2
CLXII
N—|TCH2
S'CHjCHpH
IX
Принципиально иным путем 4-метил-5-0-этоксиэтилтиазол (IX) может
быть получен из 4-метил-5-ацетилтиазола, который нагреванием с водным
раствором многосернистого аммония (реакция Вильгеродта) переводят в
амид, затем в сложный эфир и восстанавливают алюмогидридом лития [217 ].
БИОЛОГИЧЕСКОЕ ЗНАЧЕНИЕ ТИАМИНА. ЗАВИСИМОСТЬ МЕЖДУ
СТРОЕНИЕМ И БИОЛОГИЧЕСКОЙ АКТИВНОСТЬЮ. АНТИВИТАМИНЫ
Тиамин в виде фосфорного эфира (кокарбоксилазы) входит как про-
стетическая группа в состав тиаминовых ферментов, которые принимают
участие в углеводном обмене, в осуществлении реакции декарбоксилирова-
ния пировиноградной кислоты и других а-кетокислот, наприхмера-кетоглу-
413
таровой. Излишнее накопление в животном организме пировиноградной
кислоты — продукта метаболизма углеводов — является токсичным для
нервных клеток и сердечной мышцы и в результате приводит к заболеванию
полиневритом. Тиамин оказывает специфическое витаминное действие, из-
лечивая тяжелое заболевание «бери-бери» — форму периферического поли-
неврита, связанного с поражением двигательных и чувствительных нейро-
нов. Витамин В] имеет важное значение для функции нервной системы.
Он принимает также участие в регулировании водного обмена и связан с
функцией органов кроветворения.
Тиамин-хлорид и тиамин-бромид, а также различные препараты откры-
той тиольной формы тиамина, такие, как тиаминаллилдисульфид, амина-
мин (XXVIII), О-бензоилтиаминдисульфид, бестон (XXVII), S-липоилтиа-
мин, неовитан (XXXI), S-бензоилтиамин-О-монофосфат, бенфотиамин,
биотамин (XXXIX), тиаминдисульфид, дайолин (XXIV) и др., широко
применяются в медицинской практике при лечении различных нервных
заболеваний (неврозов, радикулитов, люмбаго, мышечной дистрофии,
полиневрита, полиомиелита), сердечно-сосудистых расстройств (гипертонии,
атеросклероза, склероза коронарных сосудов) и др. Изучено фармакодина-
мическое превращение S-бензоилтиамин-О-монофосфата в тиамин в крови
животных [286].
Суточная потребность человека составляет 1,5—2 мгтиамина. Токсич-
ность тиамина при внутривенном введении составляет 300 мг/кг при испы-
тании на кроликах и 350 мг/кг — на собаках [287 ].
Тиамин, обычно с рибофлавином и никотиновой кислотой, применяется
для витаминизации белого хлеба и изделий из пшеничной муки.
Тиамин применяют для витаминизации концентрированных кормов в
животноводстве — 2—4 г/т.
Тиамин широко распространен в природе в растительных и животных
клетках. Он синтезируется растительной клеткой и некоторыми микроор-
ганизмами. Микрофлора желудка жвачных животных также синтезирует
тиамин, вследствие чего только эти животные не нуждаются в получении
витамина с пищей.
Молекула тиамина в высокой степени специфична. Однако раскрытие
тиазолового цикла с образованием различных стабильных производных
по атому серы полностью сохраняет биологическую активность и часто
придает новым соединениям пролонгированное или другое дополнительное
действие. В качестве примера тиольных форм тиамина можно привести
S-бензоилтиамин (XXXVII) и природный аллитиамин (XXIII) [86]
:но scoCeH6
/ N \
СН3 NHS
XXXVII
сн2. сн3
NC=CCH2CH2OH
:но ssch2ch==ch2
nh2
XXVIII
Высокой витаминной активностью обладает продукт окисления тиамина —
тиаминдисульфид (XXIV) [67]—в результате его превращения в орга-
низме в тиамин
414
CHjCHaOH
МС(СН3)=С$5С=(СН^Г^Н’Х^ХЧМ
СНО Cl^CHjOH OHC HjN^N^CHj
XXIV
Активны и многие другие стабильные производные тиамин-тиола [99].
У ряда производных тиамин-тиола активность определяется химической
структурой и проницаемостью через биомембраны. Так, S-беизоилтиамин
отличается хорошей проницаемостью через клеточные мембраны форменных
элементов крови, тогда как О-бензоилтиамин, S-бензоилтиамип-О-моно-
фосфат, тиамин-О-дифосфат (кокарбоксилаза) не обладают этим свойством.
Сравнительно медленное проникновение S-бензоилтиамин-О-монофосфата
через клеточные мембраны обусловлено его дефосфорилированием в S-бен-
зоилтиамин 1288]. В клетках крови S-бензоилтиамин накапливается в кон-
центрации, значительно превышающей его концентрацию в окружающей
среде, и быстро рециклизуется в тиамин [2891. Дебензоилирование в тиамину
происходит главным образом через обмен с SH-группой глутатиона [289].
Замещение метильной группы положения 2 пиримидинового цикла на
этильную группу — соединение CLXIII [290]
qh/tm nh^ s снснрн
CLXIIl
даже несколько усиливает (в 1,2 раза) биологическую активность. Однако
замещение метильной группы положения 2 на изо- или н-пропильную груп-
пу приводит к уменьшению активности в 3 раза. Введение этильной группы
вместо метильной в положение 4 тиазолиевого цикла уменьшает активность
вдвое, а при одновременном введении в положении 2 пиримидинового
цикла — в 6 раз. Такие изменения молекулы тиамина, как удаление за-
местителей (например, аминогруппы в положении 4 пиримидинового цикла)
или замена их на другие, а также введение заместителей в ядро (например, в
положение 2 тиазолиевого цикла), приводят к уменьшению активности в
десятки и сотни раз или даже к полной потере биологической активности
[55, 148, 290].
Окситиамин (CLXIV) — аналог тиамина, в котором вместо аминогруп-
пы пиримидинового цикла находится гидроксильная группа, — как вита-
мин, неактивен. Метилирование или фенилирование аминогруппы тиамина
приводит к получению неактивных производных. При бензилировании
сохраняется некоторая активность. Следует отметить, что тиамин-альде-
гид (CLXV) обладает некоторой витаминной активностью [291 ].
Неактивные дигидро- (V) и тетрагидротиамины (IV) [292 ] уже не явля-
ются четвертичными солями.
сн
СПз
CHjCH-OH
—ГСНг
CH3 ^S'<CH3CH!OH
V IV
Введение дополнительной метиленовой группы в соединяющий оба
цикла тиамина метиленовый мостик — гомотиамин (XCIV) [293], замена
415
атома серы в тиазолиевом цикле на другие гетероатомы: селен, кислород —
оксазольный аналог (CLXVI) [2941, азот — имидазолиевый аналог (CLXVII)
[2951 — или на винильную группу — пиритиамин, неопиритиамин
(CLXVIII) [296, 297 ] (синтезы приведены в работах [296—2991) -—дают сое-
динения с сильно пониженной активностью (для XCIV) или лишенные
витаминных свойств.
CLXYII
СНГКДД ОЛМОНД!
CLXVI
Пиридиниевый аналог тиамина, отличающийся от пиритиамина отсут-
ствием метильной группы в пиридиновом компоненте, обладает слабыми
биологическими свойствами [299]. Аналог тиамина (CLXIX), содержащий
ту же самую тиазолиевую часть молекулы, что и тиамин, но в отличие от
последнего имеющий а-аминопиридиновое кольцо, почти полностью лишен
витаминной активности [300]. Не обладает витаминной активностью и
трициклический продукт окисления тиамина —тиохром (XXIII).
jOCOz™3
С f C H2C HjOH
XXIII
В качестве ростовых факторов для некоторых микроорганизмов актив-
ны отдельные компоненты тиамина, которые используются для построе-
ния его молекулы [3011. Лишь немногие соединения, главным образом
некоторые аналоги тиамина, являются антагонистами его витаминных
свойств. Сильными антивитаминами являются такие соединения, как го-
мотиамин гликоль, в котором 2-оксиэтпльная группа тиазолиевого цикла
заменена 2-оксипропильной группой[2911, и 2-я-бутильный гомолог [302].
Антиметаболитом является также 2-тиоаналог тиамина. 2-Трифторметиль-
ный аналог тиамина подавляет развитие лейкемии у мышей [3031.
Изостерическими антивитаминами тиамина являются его имидазолиевый
аналог (CLXVII) [295, 304] и более сильный антагонист — пиритиамин
(CLXVIII) [296, 297]. Для многих микроорганизмов пиритиамин как инги-
битор более активен, чем окситиамин (CLXVI) [305, 306]. Как антимикроб-
ный претарат широко используется 2-мегпл-1чт-(2'-я-пропнл-4'-амино.тири-
мидил-5'-метил)пиридиний-бромид, активный против кокцидий —Eimeria
Tenella, поражающих слизистую кишечника цыплят и индюшат.
Отдельные группировки молекулы тиамина входят в состав некоторых
природных веществ. Ихтиамин —2-метил-4-амино-5-(0-аминоэтансульфонил-
метил)пиримидин (CLXX) —
CLXX
416
является продуктом инактивации тиамина тканью молюсков [307] и рыб
[308].
В молекулу скординина A (CLXXI), выделенного из японского чеснока,
входит группировка 2-метил-5-р-оксиэтилтиазолия [309]
нс--->
I.
неон I
I о
СНСООН СООН ГЧНг НСОН I
СН,—т—N-CHjNCNHSCHjCH2CHN НСОС НС I4,CH2COCH=C HCH.SC—
HOCHjCH/'V ____ CHjOH
CLXXI
Часть этой молекулы — 3-креатинил-М-4-метил-5-£ -оксиэтилтиазолий — и
его дифосфат, синтезированы [310].
Для количественного определения тиамина в естественных источниках
используются биологические методы [311]. Так, применяется метод его
определения по активности процесса брожения, вызываемого хлебопекар-
ными дрожжами или грибком Endomyces magnusii [312].
ТИАМИНОВЫЕ КОФЕРМЕНТЫ
Тиамин обладает способностью образовывать моно-, ди- и трифосфор-
ные эфиры, причем тиаминдифосфат в качестве простетической группы в
соединении со специфическими белками и металлами образует тиамино-
вые ферменты; их в настоящее время известно около десяти. Биокаталити-
ческая функция тиаминовых ферментов заключается в ферментативном
расщеплении а-кетокислот и кетосахаров и в ацилоиновом синтезе, т. е.
в реакциях, связанных с обменом углеводов.
Т иаминдифосфат — О-дифосфорный эфир 4-метил-5ф -оксиэтилг
М-(2'-метил-4'-аминопиримидил-5'-метил)тиазолий-хлорид, тиаминпирофос-
фат, кофермент, называемый кокарбоксилазой (ТДФ) CLXXII,
N—п-СН,
сн,
NH, S CHjCHjOPOPOH
CLXXII ОН ОН
представляет собой бесцветные иглы с т. пл. 241—243° С (с разл.), при
кристаллизации с одной молекулой воды т. пл. 215—216° С (с разл.) [313];
существует также в виде внутренней четвертичной соли, кристаллизуется
с четырьмя молекулами воды — бесцветные иглы с т. пл. 220—222° С (с
разл.) [314]. ТДФ очень хорошо растворим в воде (ТДФ-хлорид — 22 г
в 100 мл), нерастворим в безводном спирте, эфире, ацетоне, бензоле и мно-
гих других органических растворителях. Спектр поглощения (в воде)
Хмакс 250 нм при pH < 5 и два максимума — 235 и 264 нм при pH > 5
[315—317]. 0,3%-ный водный раствор ТДФ-хлорида имеет pH 2,23 при
25° С.
ТДФ хорошо выявляется при хроматографировании на бумаге: его
Rf0,30 (в системе н-пропанол — вода—0,1 М фосфатный буфер pH 5,
3:1:1) и 0,10 (в системе н-бутанол— уксусная кислота — вода, 4:1:5) в
восходящем потоке.
Помимо кокарбоксилазы (CLXXII), коферментной активностью обла-
дает открытая тиольная форма тиаминдифосфорного эфира (CLXXIII),
получаемая из кокарбоксилазы при действии этилата натрия,
14—69
417
----------JT-CHg
II ।------ONa ONa
A HC Jk. i I
CH, N NH,^ SNaCH2CH2O~P-O-P~<)H
CLXXIII ° °
в то время как дисульфидная форма тиаминдифосфорного эфира имеет
60% активности ТДФ.
Дифосфорный эфир тиамина широко распространен в животном и расти-
тельном мире [3181. Помимо него, в различных растительных и животных
тканях обнаружены тиаминтрифосфат [319—321 ] и тиаминмонофосфат
[320—-321 ]. Трифосфорный эфир тиамина не обнаруживает коферментных
свойств, однако он быстро приобретает каталитическую активность благо-
даря легкости ферментативного гидролиза в дифосфат [322].
В организме тиаминдифосфат образуется в результате этерификации
тиамина универсальным донором фосфатных групп — аденозин-5'-трифос-
фатом [323] при участии тиаминпирофосфокиназы, возможно, через тиамин-
монофосфат. Образование кокарбоксилазы из тиаминмонофосфата происхо-
дит быстрее, чем из тиамина [321 ].
ХИМИЧЕСКИЕ СВОЙСТВА ФОСФОРНЫХ ЭФИРОВ ТИАМИНА
Тиаминдифосфат по своему химическому поведению во многом подобен
тиамину. Он стабилен при хранении на холоду, однако менее устойчив в
водных растворах, постепенно превращаясь в тиаминмонофосфат. ТДФ при
окислении феррицианидом калия в щелочном растворе образует тиохром-
дифосфорный эфир, флуоресцирующий в УФ-свете интенсивным синим све-
том, эта реакция обычно используется для количественного определения
малых количеств ТДФ.
Фосфорноэфирные связи у моно-, ди- и трифосфорных эфиров тиамина
имеют различную устойчивость к гидролитическому расщеплению в кислой
и щелочной средах [324]. При гидролизе в кислой среде отщепляются ато-
мы фосфора положений р и у. В щелочной среде отщепляется атом фосфора
а-положения, связанный с органическим остатком молекулы тиамина через
атом кислорода.
По своей устойчивости в растворах моно-, ди- и трифосфорные эфиры
тиамина располагаются в следующий ряд: при pH 1(100° С)
ТТФ < ТДФ < ТМФ, при pH 8,7 (100° С) ТТФ > ТДФ > ТМФ [324].
Тиаминдифосфат наиболее устойчив в растворах с pH 5—5,5 и 4,0 [317,
325]. Водный раствор тиаминтрифэсфорного эфира (CLXXIV) довольно
устойчив при комнатной температуре, только через несколько часов можно
обнаружить ТДФ и свободную ортофосфорную кислоту. При 100° С гидро-
лиз протекает с большой скоростью в сторону образования тиаминдифос-
форного эфира (CLXXII); фосфэангпдридная связь ТДФ почти с такой
же скоростью гидролизуется с образованием тиаминмонофосфорного эфира
(CLXXV) с т. пл. 194—199° С [326]. Дальнейший гидролиз протекает в
значительно более жестких условиях [326, 327 ].
418
Распад кокарбоксилазы при комнатной температуре идет при pH 8
и даже в нейтральной среде, но при pH 4,6—4,7он незначителен; фосфор-
но-эфирная связь ТМФ не гидролизуется при pH 4,6—7,8 [326].
Тиаминдифосфат в гидратной форме более устойчив, чем ТДФ-хлорид в
водном растворе при различной температуре [328]. То же самое относится
и к кристаллическому состоянию этих двух форм ТДФ [328]; следует отме-
тить, что на холоду расщеплению подвергается менее 1 % тиаминдифосфата
в обеих формах за 6 месяцев хранения.
СИНТЕЗ ТИАМИНДИФОСФОРНОГО ЭФИРА И ЕГО ПРОИЗВОДНЫХ
Тиаминдифосфат (кокарбоксилазу) в кристаллическом виде впервые
выделили из дрожжей в 1937 г. Ломан и Шустер [316]; его строение уста-
новлено в виде 50-пирофосфорного эфира тиамина (CLXXII) [316, 329].
Для синтеза тиаминдифосфата в качестве фосфорилирующего агента
используют смесь ортофосфорной кислоты и фосфорного ангидрида [330]
в различных вариантах [313, 328, 331—335]. 50 -Гидроксильную группу тиа-
золиевого цикла тиамина этерифицируют при действии хлорокиси фосфора
с низким выходом [336] или с лучшими результатами при действии ортофос-
форной кислоты в присутствии пирофосфата натрия [337, 338, 339], мета-
фосфорной кислоты и пирофосфата натрия при 150° С [330, 337, 339, 340],
«юсфорного ангидрида [341, 342] или его смеси с пирофосфатом натрия
343] или пирофосфорной кислотой [344], хлорангидрида пирофосфорной
кислоты, ортофосфорной кислоты [345, 346], гидратированной хлорокиси
фосфора [347], монохлорангидрида тетрафосфорной кислоты [348], диме-
тилового эфира монохлорангидрида ортофосфорной кислоты [349] и др.
В 1941 г. Вейлард использовал для получения тиаминдифосфата фосфо-
рилирование тиамина полифосфорной кислотой — смесью ортофосфорной
кислоты и фосфорного ангидрида [330]; этот метод в виде различных ва-
риантов широко применяется как технический [313, 328, 332,333, 335].
Выход ТДФ составляет 67% [344]. При действии метафосфорной кислоты
(100° С) [345, 350] преимущественно получается трифосфорный эфир тиами-
на (т. пл. 228—232° С), который от других эфиров может быть отделен в
виде стифниновой соли или методом ионного обмена.
Из сравнительного изучения различных фосфорилирующих агентов
следует, что ортофосфориая кислота почти не реагирует с тиамином, пиро-
фосфорная кислота реагирует слабо и образует преимущественно монофюс-
фат тиамина, метафосфорная кислота приводит к образованию ди-, три- и
полифосфатов тиамина [351 ]. Во всех методах реакция фосфорилирования
тиамина протекает неоднозначно с образованием смеси моно-, ди- и трифос-
форных эфиров в различных соотношениях.
Из реакционного раствора смесь моно-, ди- и трифосфатов тиамина вы-
деляют ацетоном или спиртом. В небольших количествах это осуществляется
хроматографией на бумаге [352]; ТДФ выделяется через фосфорновольфра-
мовую соль [337, 3401. Для больших количеств предложены методы разде-
ления хроматографией на крахмале, электрофорезом на колонках с порош-
кообразной целлюлозой или на ионообменных смолах [313, 314, 353—355].
В качестве исходного продукта для получения ТДФ известные преиму-
щества имеет применение тиампн-фосфата [356] вследствие уменьшения в
реакционной среде содержания соляной кислоты, которая способствует
гидролизу кокарбоксилазы в тиаминмонофосфат. Предложен метод полу-
чения ТДФ фосфорилированием монофосфорного эфира тиамина [344].
Легкость гидролиза ТДФ в тиаминмонофосфорный эфир [316, 326]
является одной из причин недостаточно высоких выходов при его синтети-
ческом получении. Синтез тиаминмонофосфата описан [340, 351, 357].
Осуществлен синтез ТДФ путем конденсации пирофосфорного эфира
4-метил-5-0 -оксиэтилтиазола и 2-метил-4-амино-5-галогенметилпиримидина
[358—3601.
14*
419
Производные тиаминдифосфата (кокарбоксилазы) с раскрытым тиазолие-
вым циклом —общей формулы CLXXVI
СН
СНЯ
NC=CCH2CH2OP(OH)OP(OH)2
I I II II
СНО SR О О
CLXXVI
R = СОСвН4СН3-м, С0СН3, циннамоил, COCeH5, COQH.Cl
были получены либо ацилированием тиаминдифосфата в щелочной среде
[361—-364], либо фосфорилированием S-ацилтиамин-О-монофосфата [365].
S-Бензоилтиамин-О-дифосфат (CLXXVI, R = СОС6Н5) по витаминной
активности равнозначен кокарбоксилазе, а по действию на восстановление
активности транскетолазы в сердечной мышце и крови обнаружил значи-
тельно лучший эффект, чем кокарбоксилаза.
Получен ряд N, S-диацилпроизводных фосфорных эфиров тиамина при
действии уксусного ангидрида на S-ацилтиамин-О-фосфаты [364]; из них
наиболее интересным является М-ацетил-8-бензоилтиамин-О-дифосфат
(CL XXVII) '
СН, О О
I П II
^СНг—NC— O2H2CH20P(0H)OP(0H)2
n j с:но scoceH,
/^N / \NHCOCH,
СН,
CLXXVI I
Следует отметить, что реакция N-ацилирования S-ацилтиамин-О-ди-
фосфата сопровождается частичным расщеплением фосфоангидридной свя-
зи, приводящим к образованию М-ацетил-8-бензоилтиамин-0-монофосфата.
Как Р1-(8-бензоилтиамин)-Р2-(аденозин-5')дифосфат, так и Р1-(8-л-хлор-
бензоилтиамин)-Р2(аденозин-5 ')дифосфат (CL X XVI11)
СИ, о о
СН— I 2 II. , II,
V if ^c=5CH4CHj>0P(0H)0P(°h)0CH2
СН^ N NH, CH°SCOCeH4C1
CLXXVIII
О
Сн Ц
н Г
он
f Н
он
были получены непосредственной конденсацией аденозин-5'-фосфата с
соответствующими S-ацилтиамин-О-монофосфатом в присутствии N, N'-ди-
циклогексилкарбодиимида в среде водного пиридина [366].
Р1-(8-Бензоилтиамин)-Р2-(аденозин-5')дифосфат при дебензоилировании
цистеином замыкает тиазолиевый цикл и превращается в Р1-(тиамин)-Р2-
(аденозин-5')дифосфат [365].
Карбодпимидным методом также синтезированы тиаминаденозинди-
фосфат, тиамининозиндифосфат [367] и триацил-О-рибофлавинтиамин-
дифосфат [368]. Синтезирован также биологически неактивный тиохром-
дифосфат [369].
420
БИОХИМИЧЕСКИЕ ФУНКЦИИ ТИАМИНДИФОСФАТА
Тиаминдифосфат, соединяясь со специфическими белками и металлами,
образует тиаминпротеиды (тиаминовые ферменты), которые и осуществляют
биокаталитические функции. Связь кофермента с апоферментом достаточно
прочна. При воздействии тиаминовых ферментов на субстраты происходит
разрыв и образование С—С-связей в различных группах: между карбони-
лом и карбоксилом —СО—СООН, между карбонилом и вторичной гидрокси-
льной группой —СО—СНОН— (разрыв и синтез а-кетола).
Тиаминовые ферменты и их системы принимают участие в углеводном
обмене, который в свою очередь через низкомолекулярные органические
кислоты (окислительный цикл трикарбоновых кислот и другие обменные
реакции) находится во взаимосвязи с обменом жиров и аминокислот в
животном организме.
Тиаминдифосфат в составе ферментных систем осуществляет следующие
реакции:
декарбоксилирование а-кетокислот с образованием альдегидов (реакция
а-расщепления);
окислительное декарбоксилирование а-кетокислот с образованием кислот
(реакция а-расщепления);
межмолекулярное перенесение гликольальдегидного остатка между ке-
тосахарами (р-расщепление) и альдозами с образованием (ацилоиновый син-
тез) новых альдозы и кетозы (и новой в ней группировки а-кетола с новым
асимметрическим центром).
Декарбоксилирование пировиноградной кислоты в ацетальдегид
СН3СОСООН —»• CHSCHO + СО2
происходит при каталитическом участии пируватдекарбоксилазы (карбо-
ксилазы). Являясь простетической группой, дифосфорный эфир тиамина
совместно с белком и магнием входит в состав карбоксилазы [370, 371]—
фермента с молекулярной массой около 75 000 [372—373 ]. Реакция декар-
боксилирования имеет место в гликолизе и при спиртовом брожении [374].
Пируватдекарбоксилаза — довольно специфический по отношению к
субстрату фермент. Например, а-кетоглутаровая кислота декарбоксилиру-
ется другим ферментом — а-кетоглутаратдекарбоксилазой. Изучались струк-
тура и функции активного центра пируватдекарбоксилазы [375].
Окислительное декарбоксилирование а-кетокислот в карбоновые кислоты
с уменьшенной на один атом цепью углеродных атомов осуществляется
при участии системы ферментов. Через ряд каталитических превращений
пировиноградная кислота, являющаяся одним из продуктов углеводного
обмена (в частности гликолиза), в виде продукта ее декарбоксилирова-
ния и дегидрирования — высоко макроэргического ацетил-КоА (схе-
ма 95) — вводится в цикл трикарбоновых кислот в звене превращений
щавелевоуксусной кислоты в лимонную кислоту и в конечном счете окис-
ляется в двуокись углерода и воду. Первичное расщепление пировиноград-
ной кислоты с отделением двуокиси углерода осуществляет ТДФ. В после-
дующих превращениях образовавшегося ацильного остатка окислительным
агентом служит (+) а-липоевая кислота (ЛК, тиоктовая кислота) [376], ко1
торая сама при этом подвергается восстановительному ацилированию при
каталитическом действии пируватдегидрогеназы в 6-ацетилдигидролипоевую
кислоту.
Помимо ТДФ и липоевой кислоты, связанной с белком, в декарбо-
ксилировании и дегидрировании пировиноградной кислоты участвуют ко-
ферменты: пантетеинадениннуклеотиддифосфат — кофермент A (HSKoA),
никотинамидадениндинуклеотид (НАД), флавинадениндинуклеотид (ФАД)
и др. — в виде системы ферментов. Возможно, что ферментативные реакции
пировиноградной кислоты начинаются через промежуточное превращение
кофермента в 2-(р-оксиэтил)ТДФ.
421
Течение реакции окислительного декарбоксилирования представлено на
схеме 95 [377 ]. Пировиноградная кислота вступает в реакцию с тиаминди-
фосфатом и в результате отщепления двуокиси углерода образует «активный
ацетальдегид», который затем передает ацетильную группу липоевой ки-
слоте.
Схема 95
Окислительное декарбоксилирование пировиноградной кислоты
NH,
соон
I
сосн,
сн, N'
СН,
NH,
сн.
ТДФ
н
।
—с—сн,
• °н °
° CH., N
CHjCHjO-P-O-P-OH
он он
NH, СООН
=|-С-СН,
chjYS °и °
о “СО,
СНРУЭ-Р-О-Р-ОН
ОН ОН
о
. II
CHjCHjO-P-O-P-OH
он он
^сн,
СН, СН^нД/ХЮН
S----S
сн,
CH, CHjCHjSH
=J- C-SCH^H^COOH
ОН о о
Т и и
сц^нр-р-о-р-он
он он
о
к
лк
^CH^CHfCH^pOOH-^^-CHjCH/ZH^H^OOH * CH,CO~SKoA
SH scoch, sh sh
H*
ЛК+НАД-Н+Н*
Промежуточно образующаяся 6-ацетилдигидролипоевая кислота пере-
дает далее ацетильную группу коферменту А, а сама вновь регенерируется
в липоевую кислоту окислением НАД [378, 3791. Акцептором водорода от
НАД-Н является ФАД.
Окислительное декарбоксилирование а -кетоглутаровой кислоты через
сукцинил-КоА в янтарную кислоту — это еще одно звено в цикле трикарбо-
новых кислот, осуществляемое ферментными системами, содержащими ТДФ,
HSKoA, НАД, ФАД, липоевую кислоту и другие коферменты.
HSKoA Н2О
НООССН2СН^СОСООН------------> НООССН2СН2СО ~ SKoA-------->
— СО2, — Н2 ' —HSKoA
—> НООССН2СН2СООН.
В основе механизма каталитического действия ТДФ в реакциях простого
и окислительного декарбоксилирования пировиноградной кислоты и других
а-кетокислот в соответствии с теорией Бреслау [380—383) лежит способность
ТДФ легко диссоциировать в нейтральных водных растворах с отщеплением
протона при атоме углерода положения 2 тиазолиевого цикла, в результате
чего ТДФ приобретает структуру биполярного иона (а). Ион ТДФ является
каталитически активной формой, которая непосредственно взаимодействует
с молекулой субстрата, обеспечивая тем самым осуществление ферментати-
вной реакции. Механизм каталитического действия ТДФ принципиально
одинаков во всех катализируемых им реакциях.
При декарбоксилировании пировиноградной кислоты ион ТДФ (а)
взаимодействует с пировиноградной кислотой с образованием неустойчивого
422
промежуточного Соединения (б). Это соединение легко декарбоксилируется
и превращается в так называемый активный альдегид (в), представляющий
собой ацилкарбанион, в котором свободная электронная пара стабилизиро-
вана за счет структуры (г). Дальнейшие превращения «активного альдеги-.
да» зависят от того, в составе какой ферментной системы функционирует
ТДФ. В реакции простого декарбоксилирования 2-(а-окснэтил)-ТДФ_(в)
расщепляется с образованием свободного ацетальдегида
“ООС
•W1
I
R
“ООС гм—
Тиаминдифосфат в составе ферментных систем катализирует ацилоино-
вый синтез. Это реакция конденсации двух альдегидов, приводящая к груп-
пировке а-кетола; так, например, продуктом конденсации двух молекул
ацетальдегида является ацетоин СНзСОСН(ОН)СН3, который образуется
при брожении сахара. У многих бактерий, накапливающих в среде аце-
тоин, он образуется в результате декарбоксилирования а-ацетомолочной
кислоты.
В своей простейшей форме транскетолазная реакция [383, 384], катали-
зируемая транскетолазой, содержащей ТДФ, состоит из реакции а-расщеп-
ления оксипировиноградной кислоты (илир -расщепления, если отсчет вести,
как в сахарах, от первичной гидроксильной группы), сопровождающегося
декарбоксилированием с последующим ацетоиновым ресинтезом остатка
гликолевого альдегида с другим альдегидом в а-кето-р -оксисоединения
сн2он
со
соон
RCHO СО2 R
Окислительный пентозный цикл распада моносахаров до двуокиси угле-
рода, протекающий при участии окисленной формы НАДФ (НАДФ+), со-
провождается генерированием восстановленной формы — НАДФ-Н. Эта
форма используется в синтетических реакциях получения макроэргического
АТФ (при окислительном фосфорилировании и др.). В пентозном цикле из
молекулы D-глюкозо-б-фосфата (CLXXIX) получается 12 молекул НАДФ-Н
С6НПО6РО3Н2 + 12НАДФ* + 7Н»О 6СО2 + 12НАДФ-Н + 12Н+ + Н3РО*.
CLXXIX
В промежуточных фазах окислительного пентозного цикла (схема 96),
реакции которого строго стереоспецифичны для сахаров D-конфигурации,
происходят различные ферментативные превращения, сопровождающиеся
реакциями окисления, укорачивания и удлинения углеродной цепи, изоме-
ризации, эпимеризации, а также альдольного расщепления и синтеза.
В двух реакциях пентозного цикла осуществляется межмолекулярный
перенос остатка гликолевого альдегида, катализируемый ТДФ транскетола-
зы (молекулярная масса 140 000) [385]. D-Глюкозо-б-фосфат (CLXXIX) в ре-
зультате окислительного превращения с отщеплением двуокиси углерода
при участии НАД превращается в О-рибулозо-5-фосфат (CLXXX), который
при участии рибозофосфатизомеразы переходит в альдозу — D-рибозо-б-фо-
сфат (CLXXXI) или при каталитическом действии рибулозофосфатэпимера-
зы в кетозу — D-кcилyлoзo-5-фocфaт (CLXXXII). Оба эти пентозофосфата
подвергаются транскетолазному расщеплению и ресинтезу в углеводы с
семью и тремя атомами углерода — П-седо'гептулозо-7-фосфат (CLXXXIII)
и D-глицеральдегид-З-фосфат (CLXXXIV).
423
Первоначально Р-ксилулозо-5-фосфат (CLXXXII) испытывает 0-рас-
щепление связи между карбонилом и атомом углерода, несущим вторичный
гидроксил; отщепляется остаток гликолевого альдегида и образуется трех-
атомная альдоза —D-глицеральдегид-З-фосфат (CLXXXIV)
Схема 96
Окислительный пентозный цикл превращений D-глюкозо-б-фосфата
сно
неон
неон
неон
I
ru о Н2ОРО3Н.
СН2ОН РиБОЗО— 2 4 1
' фосфат-XCLXXXI
CU изомера-
I за
неон
I РиБуЛО-
НСОН эофосфат-
I эпимераза ''ru ruj
СН2ОРО3Н2 уМ*
CLXXX
CHjOH
со
носн
неон
Транскетолаза
СО_
НОСН
неон
CHjOPOjHj
CLXXXII
Транскетолаза
неон
I
неон
СН2ОРО3Н2
CLXXXIII
HOCHjCHO)
сно
неон
носн
неон
неон Сно
CHjOPOjHj неон
CLXX,X неон
СН„ОРО3Н2
CLXXXV
сно
I
неон
СН2ОРОаН,
CLXXXIY
HOCHjCHO) Трансальдолаза
СН2ОН
СО
НОСН
неон
неон
СН„ОРО3Н2
CLXXXY1
Затем остаток гликолевого альдегида вступает в ацетоиновую конденсацию
с альдозой — D-рибозо-б-фосфатом (CLXXXI), образуя кетозу — О-седо-
гептулозо-7-фосфат (CL X X X111).
£>-Седогептулозо-7-фосфат (CLXXXIII) при каталитическом участии
трансальдолазы взаимодействует с D-глицеральдегид-З-фосфатом
(CLXXXIV), образуя альдозу — О-эритрозо-4-фосфат (CLXXXV) и кето-
зу— D-фруктозо-б-фосфат (CLXXXVI), которая превращается в D-глю-
козо-6-фосфат (CLXXIX), замыкая пентозный цикл.
Транскетолазную реакцию испытывает вторая пара —альдоза и кето-
за — окислительного пентозного цикла. Взаимодействует образовавшийся
П-эритрозо-4-фосфат (CLXXXV) с D-ксилулозо-б-фосфатом (CLXXXII) при
каталитическом участии ТДФ-транскетолазы, в результате реакции приводя
к получению D-глицеральдегид-З-фосфата (CLXXXIV) и D-фруктозо-б-фос-
фата (CLXXXVI). Здесь также происходит ацетоиновый распад и ресинтез.
Все реакции носят обратимый характер.
ТДФ в составе транскетолазы является одним из катализаторов реакций
фотосинтеза у растений. Синтезируемый в пентозном цикле £>-рибозо-5-фос-
фат (CLXXXI) является источником D-рибозы в реакциях биосинтеза рибо-
нуклеиновых кислот (РНК), АТФ, НАД, ФАД и других соединений.
Ингибитором транскетолазы может служить окситиаминдифосфат.
ТДФ является также коферментом синтетазы а-ацето-а-оксикислот, ко-
торая участвует в биосинтезе у растений и микроорганизмов А-валина и
424
£-изолейцина, катализируя альдольную конденсацию пировиноградной и
а-кетомасляной кислот с «активным альдегидом», т. е. с 2-р-оксиэтиль-
ной группой связанного с белком ТДФ с последующим образованием
а-ацетомолочной и а-ането-а-оксимасляной кислот.
Кокарбоксилазу (ТДФ) с лечебной целью, обычно в виде сухого препа-
рата в ампулах, сопровождаемых отдельно буферным растворителем, в до-
зировке 50—100 мг в сутки применяют при сахарном диабете, при ацидо-
зах различного другого происхождения, инсулиновом шоке, заболевани-
ях сердца, при недостаточности коронарного кровообращения, инфаркте
миокарда, легких формах рассеянного склероза.
СПИСОК
ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ (К ГЛАВЕ IX)
1. J. Kraut, Н. Reed. Actacryst.,
15, 747 (1962).
2. R. W i 1 1 i a m s, J. С 1 i n e. J.
Am. Chem. Soc., 58, 1504 (1936).
3. A. T о d d, F. В e r ge 1, F. Ka-
rim u 1 1 a h, R. К e 1 1 e r. J.
Chem. Soc., 1937, 361.
4. H. A n d e r s a g, K. West-
phal. Ber., 70, 2035 (1937); Англ,
пат. 456751; Zbl., 1937, 1, 2868.
5. J. С 1 i n e, R. Williams,
J. Finkelstein. J. Am.
Chem. Soc., 59, 1052 (1937).
6. A. Watanabe, T. Kanzawa,
H. О k u t o. Yakugaku Zasshi,
79, 883, 887 (1959); C. A., 54, 549
(1960).
7. T. M ace k, В. T e 1 1 e r, E. H a-
n u s. J. Am. Pharm. Assoc., 39,
365 (1950).
8. Швейц, пат. 206628; Zbl., 1940, 1,
3685.
9. G. Leichssenring,
J. S m i d t. Пат. ГДР 55022;
С. A„ 68, 12994 (1968).
10. A. Todd, F. В e r ge 1. J.Chem.
Soc., 1936, 1559.
11. E. Buchman. J. Am. Chem.
Soc., 58, 1803 (1936): герм. пат.
673174, Zbl., 1939, 1, 4561.
12. R. Y a m a m о t о, T. T a k a h a-
s h i. Яп. пат. 3879, 4328 (1957);
С. A., 52, 5757, 5758 (1958).
13. I. U c h i m i, N. Tanaka,
К. H a r a d а. Яп. пат. 3995
(1959): С. A., 54, 1568 (1960).
• 4. К. К a w a h a r a, Y. H о c h i,
К. Ta ni. Яп. пат. 3347 (1959);
С. A., 54, 13152 (1960).
15. A. I t о, Tokutake, К u n i o.
Яп. пат. 7889 (1955); РЖХ, 1958,
54954.
16. J. Cline, R. Williams,
A. R u e h 1 e, R. Waterman.
J. Am. Chem. Soc., 59, 530 (1937).
17. R. W i 1 1 i a m s. J. Am. Chem.
Soc., 58, 1063 (1936).
18. H. A n d e r s a g, K. West-
phal. Герм. пат. 671787; франц,
пат. 819595; норв. пат. 59015; Zbl.,
1938, 1, 3800.
19. R. Gre we. Z. Physiol. Chem.,
242, 89 (1936); Natuhviss., 24,
657 (1936).
20. T. Ma t suk a wa, T. I w a t s u,
S. Yurugi. J. Pharm. Soc. Ja-
pan, 71, 369 (1951); C. A., 46,
4550 (1952).
21. R. Williams, E. В u c fa-
man, A. R u e h 1 e. J. Am.
Chem. Soc., 57, 1093 (1935).
22. T. Iwa t zu. J. Pharm. Soc. Ja-
pan, 75, 677 (1955); C. A., 50, 3459
(1956).
23. P. Karrer, H. Krishna.
Helv. Chim. Acta, 33, 555 (1950).
24. G. В on v i ci no, D. H e n n e s-
s y. J. Am. Chem. Soc., 79, 6325
(1957).
25. H. H i r a n o. J. Pharm. Soc. Ja-
pan, 78, 1387 (1958); C. A., 53,
8145 (1959).
26. F. Lipman, G. P e r 1 m a n n.
J. Am. Chem. Soc., 60, 2574 (1938).
27. T. Ma ts uka wa, H. H i r a n o,
T. I w a t s u, S h. Y u r u-
g i. J. Vitaminol., 3, 213 (1957);
C. A., 52, 5423 (1958).
28. S h. Yoshida, M. К a t a e k a.
Chem. Pharm. Bull. (Tokyo), 5, 176
(1957); C. A., 51, 17932 (1957).
29. T. I w a t s u. Яп. пат. 1881 (1958);
С. A., 53, 5297 (1959).
30. H. H i г a n о, H. Y о n e m о t о,
Y. H a r a. Chem. Pharmac. Bull.
(Tokyo), 7, 545 (1959).
31. T. Ma t suka wa, H. H i r a-
n о, T. I w a t s u, S h . Y u r u-
g i. J. Vitaminol. (Osaka), 3, 218
(1957); C. A., 52, 5423 (1958).
32. R. W i 1 1 i a m s, R. Water-
man, J. Keretztesy,
E. В u c h m a n. J. Am. Chem
Soc., 57, 536 (1935);
33. C h. Kawasaki, T. S u h a-
r a, T. H о г i o. Yakugaku Zas
shi, 78. 65, 69 (1958); C. A., 58,
11859 (1958).
34. G. В о n v i c i n o, D. H e n n e s-
s y. J. Org. Chem., 24, 451 (1959)
35. M. U п о k i, T. M i t s u g i. Ya-
kugaku Zasshi, 84, 780 (1964); C. A.,
61, 11992 (1964).
36. K. L'e m a t s u. Annual Rept. Ta-
kamine Lab., 1959, № 11, 42.
37. S h. J о s h i d a, K. N a ga w a,
M. К a t а о k а. Яп. пат. 1728
(1962); РЖХ, 1963, I6H222; яп.
пат. 9496, 9593 (1962); С. А., 59,
2836, 5176 (1963).
38. М. К a t а о k a, S. loshida,
М. N a g a w a. Takamine Кеп-
425
kyusho Nempo, 13, 31 (1961); C. A.,
55, 6488 (1961).
39. Ути да, Ф у д з у к и. J. Japan.
Biochem. Soc., 25, 35 (1953); РЖХ,
1955, 21247.
40. J. К г a u t, Н. R е е d. Acta
Crist., 15, 747 (1962).
41. A. Tomaka. Shigakenritsu Tan-
ki Daigaku Gakufutsu Zasshi, 6, 5
(1965); Bitamin, 33, 19 (1966);
C. A., 64, 9719, 8178 (1966).
42. С. M i 1 1 e r, J. Sprague,
L. К r a m p i t z. Ann. N. J.
Acad. Sci, 98. 401 (1962); C. A„
60, 6832 (1964)
43. A. T a k a mi z aw a, K. Hirai,
Y. H a m a s h ira a, S. Ma t-
s u m о t o. Tetrahedron Letters,
1967, 5071.
44. Y. Oka, S. К i s h i m о t o,
H. H i r a n o. Chem. Pharm. Bull.
18, 527 (1970).
45. A. T a k a m i z a w a, S. M a t s u-
moto, S. Sakai. Tetrahedron
Lett., 1968, 2189; Chem. Pharm.
Bull., 17, 128 (1969).
46. A. Takamizawa, K. Hirai,
Y. H a m a s h i m a, S. Mat-
sumoto. Tetrahedron Letters,
1967, 5077, 5081.
47. A. Takamizawa, K. Hi-
rai. Y. Hamashima, Chem.
Pharm. Bull, 16, 1758(1968).
48. A. T a k a m i z a w a, Y. S a t o.
Chem. Pharm. Bull., 15, 1183 (1967).
49. A. T a k a m i z a w а, К. H i-
rai, Y. Matsumoto,
S. T a n a k a. Chem. Pharm.
Bull., 16, 1764 (1968).
50. M. Hesse, H. Bild,
H. Sc hm i d. Helv. Chim. Acta,
50, 808 (1967).
51. G. В a r g e r, F. В e r g e I,
A. Todd. Nature, 136, 259
(1935); Ber., 68, 2257 (1935).
52. W. Karrer, U. К u b 1 i. Helv.
Chim. Acta, 20, 369 (1937).
53. B. Jansen. Rec. trav. chim.,
55, 1046 (1936).
54. M. P у k e. J. Soc. Chem. Ind., 58,
trans, 338 (1939).
55. F. В e r g e 1, A. T о d d. J. Chem.
Soc., 1937, 1504; 1938, 26.
56. R. W i 1 1 i a m s, T. S p i e s. Vi-
tamin Bj. New York, 1938.
57. A. T a k a m i z a w a, S. N a k a-
r i m a, H. S a t о h. J. Pharm.
Soc. Japan, 79, 399 (1959); РЖХ,
1960, 47793.
58. O. Z i m a, R. Williams. Ber.,
73, 941 (1940).
59. G. R i s i n g e r, P. P a г k e r.
Experientia, 21, 305 (1965).
60. R. К u h n, H. V e t t e r. Ber.,
68, 2375 (1935).
61. H. К i n n e r s 1 e y, J. О’ В r i -
en, R. Peters. Biochem. J.,
29, 2369 (1935).
62. C h. Kasahara. Yakugaku
Zasshi, 79, 963 (1959); C. A., 54,
559 (1960).
63. R. К u h n, T. W a g n e r-J a u-
regg, van Klaveren,
H. V e t t e r. Z. physiol. Chem.,
234, 196 (1935).
64. A. Todd, F. В e r ge 1, H.
F r a e n k e 1-C о n r a t, J. J a-
c о b. J. Chem. Soc., 1936, 1601.
65. К. M и я т а к э, T. О т а. Яп.
пат. 8287 (1956); РЖХ, 1960,
58352.
66. О. Z i m a, G. G о t t m а п,
A. Hoffman, L. Hep-
ding, G. H о t о v y. Natur-
wiss. 41, 214 (1954).
67. O. Z i m a, K. R i e t s e r t,
T. Mai 1. Z. physiol. Chem., 267,
210 (1941).
68. C h. К a w a s a k i, T. H о r i o,
I. D a i r a. J. Vitaminol., 9, 317
(1963); C. A., 61, 14669 (1964).
69. Ch. Kawasaki. T. H о r i o.
Bitamin, 19, 48 (1960).
70. T. H о r i o. Bitamin, 24, 79 (1961).
71. T. M a t s u k a w a, T. I w a t s u.
J. Pharm. Soc. Japan, 69, 550
(1949); 70, 28 (1950); C. A., 44,
4475 (1950).
72. P. N e s b i 11, P. S yke s. J.
Chem. Soc., 1954, 4581.
73. P. S у k e s, A. Todd . J. Chem.
Soc., 1951, 534.
74. I. U t su mi, К. H a r a d a,
G. Tsukamoto et al., Bita-
min, 31, 119, 126, 130, 137 (1965);
C. A., 62, 10433, 10434 (1965); J.
vitaminol., 11, 225 (1965).
75. G. Tsukamoto, K. Hara-
da, I. U t s u m i. Chem. Pharm.
Bull., 14, 823 (1966); C. A., 65,
18579 (1966).
76. R. Yamamoto, T. Kubo-
ta, К. I n a z u, Bitamin, 25,
483 (1962); C. A., 60, 9773 (1964).
77. H. Fukutomi. Bitamin, 33, 144
(1966); C. A., 64, 13144 (1966).
78. B. G a u t h i e r, R. T i x i e r,
A. U z a n. Ann. Fharm. Franc.,
21, 655 (1963).
79. T. F u j i t a, Y. M u t s h i k a.
J. Vitaminol., 12, 18 (1966); РЖХ,
1967, 12Ж568.
80. T. F u j i t a, Y. M u t s h i k a,
T. Kobayashi. Англ. пат.
1038920, РЖХ, 1967, 20H298.
81. G. • R i s i n g e r, P. P a r k e r,
H. Hsieh. Experientia, 21, 434
(1965).
82. A. T a k a m i z a w а, К. H i-
r a i, Y. S a t o. J. Pharm. Soc.
Japan, 82, 1202 (1962); C. A., 58,
6824 (1963).
83. T. S e i у a k u. Англ. пат. 922444;
С. A., 59, 10075 (1963).
84. Neth. AppL 300740; C. A., 64,
8204 (1966).
85. Z. I m a i, Y. M i у a j i, К. Ta-
d о k i г о, M. К u b о, T. H a-
v a s h i, M. S a k о. Яп. пат.
15111 (1966); С. A., 66, 37947
(1967).
86. V. L i 1 1 у, H. В a r n e tt,
G. Anderson. Science, 118,
548 (1952).
426
87. T. M a t s u к a w a, S. Y u r u-
gi. J. Pharm. Soc. Japan, 72,
1602, 1616 (1952); C. A„ 47, 9331
(1953).
88. T. M a t s u к a xv a, S. Y u r u g i.
J. Pharm. Soc. Japan, 73, 73
(1953); C. A., 47, 11198 (1953).
89. Sh. Y ur ugi. J. Pharm. Soc.
Japan, 74, 502 (1954); C. A., 49,
8300 (1955).
90. Англ. пат. 749307; С. A., 51, 5126
(1957).
91. H. Watanabe с сотр. Яп. пат.
1287 (1954); С. А., 49, 573 (1955).
92. Т. М a t s и к a w a, S h. Y и г и-
g i. Яп. пат^ 1484, 1840 (1954);
С. А., 49, 11727, 573 (1955).
93. Т. Mat sukawa, Н. Kawa-
saki, Т. I w a t s и, S h. Y и-
r и g i. J. Vitaminol., 1, 131
(1954); C. A., 49, 7572 (1955).
94. S h. Y m a d a, T. F и j i t a,
T. Mizoguchi. J. Pharm.
Soc. Japan, 74, 963 (1954); C. A.,
49, 10306 (1955).
95. T. M a t s и к a w a, T. I w a t s u,
H. Kawasaki. J. Pharm. Soc.
Japan, 73, 497 (1953); C. A., 48,
3372 (1954).
96. H. О к и d a. Bitamin, 36, 317
(1967); C. A., 68, 68952 (1968).
97. В. M. T у p с и н, E. А. Ивано-
ва. ЖОрХ, 1, 1151 (1965).
98. S h. Y и г и g i, T. F и s h i m i.
Пат. ФРГ 1110648; С. A., 56, 11602
(1962).
99. В. И. Колтунова,
В. М. Березовский. Вита-
мины и витаминные препараты, сб.
М., изд-во «Медицина», 1973, с. 6.
100. М. Д. Д н к с о н, Э. У э б б.
Ферменты, изд, «Мир», М.,
стр. 327, 364, 3471 (1966).
101. Y. Deguchi, К. Nakani-
s h i. J. Pharm. Soc. Japan, 83,
701, 704, 708 (1963); C. A., 59,
12913 (1963).
102. M. Ohara, K. Yomamoto,
K. Nakanishi. Яп. пат.
3143 (1963); С. A., 59, 11522 (1963).
103. M. Ohara. Bitamin. 23, 164
(1961).
104. Y. D e g u c h i, H. Miura,
M. I t o. J. Pharm. Soc. Japan,
83, 713 (1963); C. A., 59, 12913
(1963).
105. Y. Deguchi, К- M i u r a. J.
Pharm. Soc. Japan, 83, 717 (1963);
C. A., 59, 12913 (1963).
106. A. Hosino. Bitamin, 23, 424
(1961); 27, 402 (1963).
107. M. A k i a j i m a. Bitamin, 24,
50 (1961).
108. M. I w a n a m i, I. О s a w a,
M. M u r a k a m i. Bitamin, 36,
119 (1967); C. A., 68, 121817 (1968).
109. Франц, пат. 1418664; С. A., 65,
5470 (1966).
110. R. F u s с о, F. T e п с о n i.
Франц, пат. 1442289; РЖХ, 1967,
23H485.
111. G. Sunagawa, N. Y oshi-
da, K- N a k a m u г а. Яп. пат.
13997 (1962); С. A., 59, 10078
(1963).
112. I. Utsumi, K. Harada,
G. Tsukamoto. Яп. пат.
17262 (1967); С. A., 68, 105231
(1968).
113. T. M a t s u k a w a, H. Kawa-
saki. J. Pharm. Soc. Japan, 73,
705 (1953).
114. A. T a k a m i z a w a, K. Hi-
rai, Y. H a m a s h i m a. Chem.
Pharm. Bull., 10, 1107 (1962); 11,
882 (1963); РЖХ, 1964, 14Ж294,
14Ж289.
115. M. M u r a k a m i, K. Icfaika-
wa, K. Takahashi. Яп. пат.
17265 (1967); С. A., 68, 49637
(1968).
116. A. T a k a m i z a w а, К. H i-
r a i. Chem. Pharm. Bull., 10,
1102 (1962); C. A., 59, 5162 (1963).
117. M. Yoshida. Takamine Ken-
kyusho Nempo, 14, 57 (1962); РЖХ,
1963, 20Ж348.
118. A. 11 o, Y. N a g a w a, W. H a-
manaka, K. Kawada,
M. К a t а о k a, T. Wada,
T. Y о s h i о k а. Яп. пат.
21526 (1964); С. A., 62, 11827 (1965).
119. T. Wa d a, H. T a k a g i,
Sh. Miyazawa, Y. Suzu-
ki, H. M i n a k a m i. Bita-
min, 22, 342 (1961); C. A., 62,
2956 (1965).
120. W. H a m a n a k a, A. I t o,
H. T a k a g i, T. W a d а. Яп.
пат. 11037 (1962); С. A., 59, 10077
(1963).
121. A. I t о, W. H a m a n a k a,
K. Kawada. Яп. пат. 11038,
11039 (1966); РЖХ, 1966, 13H323.
122. H. N i s h i m u г a, H. К i n u-
g a s а, К. О n i s h i. Яп. пат.
24906—24910(1967); С. A., 69,
43928, 52159—52162 (1968).
123. C h. К a w a s a k i, I. Tomi-
t a. Yakugaku Zasshi, 78, 1160,
' 1163 (1958); C. A., 53, 5273 (1959).
124. Ch. Kawasaki, I. To m i-
t a, T. M о t о у a m a. Vitamins,
13, 57 (1957); C. A., 54, 4595
(1960).
125. Ch. Kawasaki, I. T о m i-
t a. Yakugaku Zasshi, 79, 295
(1959); C. A., 53, 15090 (1959).
126. H. Yonemoto. Yakugaku Zas-
shi, 77, 1124, 1128 (1957); 78, 472
(1958); C. A., 52, 5420, 17278
(1958).
127. T. M о t о v a m a. Yakugaku Zas-
shi, 79, 115 (1959).
128. A. Takamizawa, K. Hi-
rai, T. I s h i b a. Tetrahedron,
Lett., 1970, 441; Chem. Pharm.
Bull, 19, 1022 (1971).
129. A. T a k a m i z a w а, К. H i-
r a i, T. I s h i b a. Tetrahedron
Lett., 1970, 437; Chem. Pharm.
Bull., 19, 2009 (1971).
130. Sh. Kasahara. Yakugaku
Zasshi, 77, 1133 (1957); С. A-, 52,
427
5422 (1958); J. VitaminoL, 4, 100
(1958); РЖХ, 1959, 49688.
131. H. Y о ne m o t о. Яп. пат.
9268 (1959); РЖХ, 1963, 7H188.
132. H. Yonemoto. J. Pharm. Soc.
Japan, 78, 1391 (1958); С. А., 53,
8146 (1959).
133. Н. Yonemoto. Yakugaku
Zasshi, 79, 143 (1959); С. А„ 53,
13168 (1959).
134. М. A s a i, Т. Konotsune.
Takamine Kenkyusho Nempo, 13,
45 (1961).
135. H. Y о n e ni о t о. Яп. пат. 13023
(1960); С. А., 55, 3629 (1961).
136. М. М ига ка mi, К. Taka-
hashi, Y. Н i г a t а, Н. I w а-
m о t о. Bitamin, 34, 71 (1966),
С. А., 65, 8905 (1966); J. Vitami-
nol., 13, 122 (1967).
137. Н. Н i г а п о. Yakugaku Zasshi,
77, 1004 (1957); С. А., 52, 3828
(1958).
138. К. Y о s h i m i. J. Pharm. Soc.
Japan. 66, 62 (1946); C. A., 45,
6207 (1951).
139. M T о m i t a, S. U у e o,
H. Jnouye, H. Sakurai,
S. Moriguchi. J. Pharm.
Soc. Japan, 68, 151, 154 (1948);
C. A,. 48, 3984 (1954).
140. К. M a s u d a. Yakugaku Zasshi,
81, 533, 536, 540, 544, 549 (1961);
C. A., 55, 21131—21133 (1961).
141. C. F u n k. J. Physiol., 45, 75
(1912).
142. B. J a n s e n, W. Donath.
Chem. Weckblad, 23, 201; Zbl.,
1926, II, 607; 1927, 1, 1850.
143. A. Wi nda us, R. T s c h e s-
c h e, H. R uhkopl, F. La-
q u e r, F. S c h u 1 t z. Z. Phy-
siol. Chem., 204, 123 (1932).
144. A. W i n d a u s, R. Tsches-
c h e, R. Grewe. Z. physiol.
Chem., 228, 27 (1934).
145. H. Cl ar ke, S. G u r i n. J.
Am. Chem. Soc., 57, 1876 (1935).
146. M. Tomlinson. J. Chem.
Soc., 1935, 1030.
147. A. Wi n d a us, R. T s c h e s-
c h e, H. R u h к о p f. Nachr.
Ges. Wiss. Gottingen, 1932, 342
148. E. В u c h m a n, R. Willi-
ams. J. Am. Chem. Soc., 57, 1751
(1935).
149. E. В u c h m a n, R. W i 1 I i-
ams, J. Keresztesy, J. Am.
Chem. Soc., 57, 1849 (1935).
150. R. Williams, A. R u e h 1 e.
J. Am. Chem. Soc., 57, 1856 (1935).
151. T. В i r c h, L. H a r r i s. Na-
ture, 135, 654 (1935).
152. R. Mo ggridge, A. Ogston-
Biochem. J„ 29, 866 (1935).
153. R. Williams. J. Am. Chem.
Soc., 57, 229 (1935).
154. A. Todd, F. В e r g e 1, F. Ka-
rim u 1 1 a h. J. Chem. Soc.,
1936, 1557.
155. A, Windaus, R. Tsches-
c h e, R. G r e we. Z. physiol.
Chem. 237, 98 (1935).
156. К. M a к i n о, T. I m a i. Z.
physiol. Chem., 239, 1 (1936).
157. R. Williams, A. R u e h 1 e,
J.Finkelstein. J. Am. Chem.
Soc., 59, 526 (1937).
158. P. Baumgarten, A. D or-
n о w. Ber., 73, 44 (1940); герм,
пат. 730576; С. A., 38, 457 (1944).
159. О. Ю. M а г и д с о и, А. И.
Травин. Авт. свид. СССР
59808; Бюлл. изобрет., 1941,
N 4, 19.
160. И. А. Рубцов, Е. С. Жда-
но в и ч, М. В. Б а л я к и н а,
Е. И. Косарева, М. К. Ша-
хова. Авт. свид. СССР 99479;
Бюлл. изобрет., 1954, № 12, 13.
161. В. Szczycinski. Польск.
пат. 41840; С. А., 55, 18782 (1961).
162. Датск. пат. 55976; Zbl., 1939,
II, 4032.
163. Герм. пат. 703775: Zbl., 1941, I,
3551.
164. А. Травин. ЖПХ, 16, 105
(1943).
165. В. Carnahan. Пат. США
2678312; С. А., 49, 6323 (1955);
англ. пат. 717445; РЖХ, 1956,
69768; J. Appl. Chem., S, 781
(1955).
166. Англ. пат. 768773; С. А., 51, 15590
(1957).
167. W. S t i е g. Пат. США 2833769
С. А., 53, 1388 (1959).
168. G. Leichssenring. Пат.
ГДР 11163; С. А., 53,1389 (1959).
169. В. М. Березовский,
В. И. Колтунова,
Н. Д. П е к е л ь. Мед. пром.,
1962, № 12, 20.
170. Е. Howe, М. Т i shier. Ка-
над. пат. 501311; РЖХ, 1956,
62964.
171. Пат. ФРГ 1004613; С. А., 54, 8866
(1960).
172. В. И. Колтунова, В. М.
Березовский. Сб. Ионо-
обменная технология, М., изд.
«Наука», 1964, 202.
173. К. О k u m и г a, Y. Sa к’и-
r a i J. Vitaminol., 2, 276 (1956);
С. А., 51, 7331 (1957).
174. S. Y о s h i d а, К. U п о к i. J.
Pharm. Soc. Japan, 72, 493, 960,
964, 966, 968 (1952); C. A., 47,
2181 (1953).
175. T. M a t s и к a w a, T. J w a t-
s и. Пат. США 2592930; С. A.,
47, 4379 (1953).
176. К. U n о к i, Ка t or i. J.
Pharm. Soc. Japan, 74, 423 (1954);
РЖХ, 1956, 19379.
177. J. К о к и r a, S. W a к i. Яп.
пат. 2778 (1955); С. A., 51, 13323
(1957).
178. Яп. пат. 6726 (1959); С. А., 54,
15405 (1960).
179. И. X. Фе л ь дм а н, Л. А.
Петрова, 3. Я- Дрок,
428
Г. С. Семичева. Мечен, биол.
акт. в-ва, сборн. 1962, 20.
180. Т. I m a i, К. М a kino. Z.
physiol. Chem., 252, 76 (1938).
181. А. Т о d d , F. Be г gel. J.
Chem. Soc., 1937, 364.
182. Франц, пат. 831110, Zbl., 1939,
I, 729.
183. T. Matsuka wa. J. Pharm.
Soc. Japan, 62, 417 (1942); C. A.,
45, 4723 (1951).
184. H. A n d e r s a g, K. West-
phal. Франц, пат. 816432;
Zbl., 1938, I, 937.
185. T. M a t s u k a w a, M. О h t a.
Пат. США 2184720; инд. пат.
25808; Zbl., 1939, I, 3930.
186. T. Matsukawa, S. Y u г u-
g i. J. Pharm. Soc. Japan, 69, 508
(1949); C. A., 44, 4476 (1950).
187. M. Klingefuss. Пат. США
2127446; Zbl., 1938, II, 3573.
188. Англ. пат. 609803; С. А., 44,
4516 (1950).
189. R. Paul, S. Т с h е 1 i t -
с h е f f. Bull. Soc. chim. France,
1950, 520; C. A., 45, 602 (1951).
190. Австр. пат. 164546; С. A., 47,
7554 (1953).
191. T. Matsuka wa, S. Yuru-
g i. J. Pharm. Soc. Japan, 70,
331 (1950); C. A., 45, 2952 (1951).
192. T. Matsukawa, T. J w a t-
s u. J. Pharm. Soc. Japan, 72, 1203
(1952); C. A., 47 6950 (1953).
193. S. Y osh i da, K. Unoki.
J. Pharm. Soc. Japan, 73, 174
(1953); C. A., 47, 11199 (1953).
194. T. I w a t s u, J. Pharm. Soc.
Japan, 72, 354 (1952); C. A., 47,
2178 (1953).
195. В. M. T у p с и н, Л. Г. 4 e 6 o-
т a p e в a, H. Д. Кол от и-
лова, Л. Н. Б е л о у с о в а.
Авт. свид. СССР 118502; РЖХ,
1960, 70670.
196. S. Y о s h i da, К. U no ki. J.
Pharm. Soc. Japan, 73, 261, 627,
631 (1953); C. A., 48, 5866 (1954).
197. H. H i r a n o. J. Pharm. Soc.
Japan, 74, 56, 59 (1954); C. A., 49,
1730 (1955).
198. R. Horton. Пат. США
2770623; С. A., 51, 7441 (1957).
199. E. M a x i о n. Пат. США
2799676; С. A., 52, 1284 (1958).
200. R. T u r n e r, G. Schmit t.
Пат. США 2844579; С. A., 53,
3230 (1959).
201. О. S h i m u г а. Яп. пат. 18770
(1961); С. A., 57, 13763 (1962).
202. S. Y о s h i d a, K. Un о k i-
J. Pharm. Soc. Japan, 72, 1431
(1952); C.A ., 47, 8078 (1953); пат.
США 2676175; С. A., 49, 11028
(1955).
203. Sh. Yoshiga, W. I s h i z u-
k a, M. К a t а о k а. Яп. пат. 10232
(1956); С. A., 52, 15599 (1958).
204. Z. F б 1 d i, A. Salomon-
Ber„ 74, 1126 (1941).
205. Синтезы органических препаратов,
I, 63; M„ ИЛ, 1949.
206. V a n Е р р s, Е. R е i d. J.
Am. Chem. Soc., 38, 2130 (1916).
207. J. M i t c h e 1 1, E. R e i d. J.
Am. Chem. Soc., 53, 321 (1931).
208. И. M. Лиснянски й,
E. С. Жданович. Труды
ВНИвитаминного института, 6,
23 (1959); РЖХ, 1960, ' 62331.
209. Я. П а у ш к и н, Л. О с и п о-
в а. ДАН СССР, 111, 117 (1956).
210. Т. П л а т э, М. В о л ь п и н.
ДАН СССР, 89, 317, 491 (1953).
211. Синтезы органических препара-
тов. 1, 66; М., ИЛ, 1949.
212. W. W i s 1 i се n us, Е. В 6 k-
1 е n, F. R е u t h е. Lieb. Ann.,
363, 347 (1908).
213. Н. Н. Ворожцов. Основы
синтеза промежуточных продук-
тов и красителей, М., Госхимиздат,
1955, 643.
214. А. П. Терентьев,
А. Н. Кост. ЖОХ. 21, 1867
(1951).
215. Швейц, пат. 190718; Zbl., 1938, I,
3799.
216. И. А. Р у б ц о в, М. В. Б а-
лякина, Е. В. Зайцева,
Н. А. Преображенский.
Труды ВНИ витаминного институ-
та, 4, 20, М., Пищепромиздат, 1953.
217. A. D о г п о w, G. Р е t s с h.
Chem. Ber., 86, 1404 (1953).
218. J. Jchika wa, K. A k i t a.
Яп. пат. 1839 (1954); С. A., 49,
11726 (1955).
219. Англ. пат. 456751; С. А., 31,
2232 (1937)
220. L. R u z i с k a. Helv. Chim.
Аста, 2, 144 (1919).
221. Z. В u d е s i п s к у, J. Ко-
ре s к у. Chem. listy, 48, 1364
(1954).
222. L. S и г а п у i, Н. Wilk. Пат.
ФРГ 1150987; С. А., 60, 2971
(1964).
223. Швейц, пат. 195951, 195952; Zbl.,
1938, II, 2792.
224. О. Н го ma t к а. Герм. пат.
670635; 667990; Zbl., 1939, I, 1449.
225. Герм. пат. 731562; Zbl., 1943, II,
1210.
226. Р. Passalacgua. Gaz. chim.
Ital., 43, II—566 (1913).
227. R. Williams, J. Cline.
J. Am. Chem. Soc., 59, 216 (1937).
228. R. Williams. Ind. Eng.
Chem., 29, 980 (1937).
229. Г. Ф о д о p, А. Г e p e ч,
И. К и ш ш, Я- К о л о н и ч,
Я. В е й н, Э. К о в а ч. ЖОХ,
21, 1897 (1951).
230. A. Takamizawa. J. Pharm.
Soc. Japan, 74, 752, 756 (1954),
С. A., 49, 11663 (1955).
231. Г. В. Ч e л и н ц e в, 3. А. Б е-
неволенская. ЖОХ, 14,
1142 (1944).
232. Г. В. Ч е л и н ц е в, 3. А. Б е-
429
и е в о ленская, Б. Дуби-
ни н. ЖОХ, .17, 275 (1947).
233. A. Takamizawa, R. Mae-
da. J. Pharm. Soc., Japan, 74,
746 (1954), С. A., 49, 11662 (1955).
234. A. Takamizawa, К. I к a-
w a, K. Tor i. Yakugaku Zas-
shi, 78, 647 (1958); C. A., 52, 18411
(1958).
235. G. Leichssenring. Пат.
ГДР 11119; С. A„ 52, 17295
(1958).
236. G. Stei n, J. S t e v e n s. Пат.
США 2516158; С. A., 45, 312
(1951).
237. A. Takamizawa. Яп. пат.
1126 (1958); С. A., 53, 1388 (1959).
238. J. Ma c-G r e g о г, C. Pugh.
J. Chem. Soc., 1945, 535.
239. А. А. Б e э p, В. А. Ку p дю-
нова. Авт. свид. СССР 70497;
Бюлл. изобрет., 1948, № 2, 9.
240. Н. А. Преображенский,
И. А. Р у б ц о в, И.М.Лис-
н я н с к и й. Авт. свид. СССР
93885; Бюлл. изобрет., 1952, № 6,
стр. 9.
241. И. А. Р у б ц о в, М. В. Б а л я-
к и н а, Е. С. Ж д а н о в и ч,
Н. А. Преображенский.
Труды ВНИ витаминного инсти-
тута, 4, 23, М., Пищепромиздат,
1953.
242. М. Т о m j t a, S. U у е о, A. Ta-
kamizawa, R. М а е d a. J.
Pharm. Soc. Japan, 74, 742 (1954);
С. А., 49, 11661 (1955).
243. A. Takamizawa. Яп. пат.
7025 (1954); РЖХ, 1957, 49300;
пат. США 2775592; РЖХ, 1959,
61903.
244. A. Takamizawa. J. Pharm.
Soc. Japan, 74, 748 (1954); С. A.,
49, 11662 (1955).
245. A. T a k a m i z a w a, K.To-
k u у a m а, К. T о r i. Bull.
Chem. Soc. Japan, 32, 188 (1959);
РЖХ, 1960, 26501.
246. A. T a k a m i z a w а, К. I k a-
wa. Яп. пат. 3976 (1957); С. A.,
52, 10220 (1958).
247. M. T о m i t a, S. U у e o,
K. Takeda, A. Takamiza-
wa, R. M a e d а. Англ. пат.
772256; С. A., 51, 14835 (1957).
248. H. T a k a m i z a w а, К. I k a-
w a, S. H a у a s h i, M. T a n i i,
M. Narisa da. Яп. пат. 8673
(1958); С. A., 54, 5714 (1960).
249. Z. В u d ё s i n s k у. Чехослов
пат. 86866; С. А., 54, 2380 (1960).
250. М. Т о m i t a, S. U у е о,
К. Takeda, A. Takamiza-
w a, R. М а е d а. Пат. ФРГ
937057; РЖХ, 1958, 33564.
251. A. Takamizawa, К. I к а'
w a, S. Н а у a s h i, М. Т a n i i>
М. N а г i s a d а. Яп. пат. 770,
771 (1959); РЖХ, 1960, 14528,
35783.
252. A. Takamizawa, К. Т о к и-
у a m а. Яп. пат. 6122 (1959);
С. А., 54, 14284 (1960).
253. A. Takamizawa, К. То-
ки у a ma. Яп. пат. 3325
(1959); С. А., 54, 13152 (1960).
254. И. Л. К н у н я н ц, Г. В. Ч е-
л и п не в, Е. Осетрова
ДАН СССР, 1, 312 (1934).
255. L. S с h n i е р р, Н. Geller,
R. Vonrorff. J. Am. Chem.
Soc., 69, 672 (1947).
256. А. С. Султанов, В. A. Ma c-
леннпкова. Авт. свнд. СССР
107765; Бюлл. изобрет., 1957 № 8
17.
257. А. Г. Н а т р а д з е, С. Коган,
К. Новикова. Меднц. пром.
СССР, 1950, № 3, 15.
258. К. С. Т о п ч и е в, Л. П а в-
л о в. Авт. свид. СССР 48104;
Бюлл. изобрет., 1937, № 2, 4.
259. Н. И. Ш у й к и н, И. Ф. Бель-
ский, О. Н. С а в е к и н а.
ЖОХ, 29, 869 (1959).
260. В. Н е 1 f е г i ch . Ber., 52, 1123
(1919); 55, 702 (1922); 56, 759
(1923); 57, 1911 (1924).
261. Я. Слободин, Е. Гельме.
ДАН СССР, 39, 152 (1943).
262. А. П е с и н а. ЖОХ, 9, 804
(1939).
263. J. Stevens, G. Stein. J.
Am. Chem. Soc., 62, 1045 (1940).
264. J. L о w, R. S m i t h. Англ. пат.
606026; С. A., 43, 677 (1949).
265. A. Hantzsch с сотр., Ber.,
20, 3118 (1887); Lieb. Ann., 249, 1
(1888); 250, 257, 262 (1889).
266. Англ. пат. 456751; Zbl., 1937, I,
2868.
267. A. T о d d, F. В e r g e 1. Natu-
re, 138, 76, 406 (1936).
268. A. W e n z. Герм. пат. 664789;
Zbl., 1938, 11, 3573.
269. Гол. пат. 44419; швейц. пат.
199089; Zbl., 1939, I, 1411.
270. J. Н a t h е г. J. Am. Chem. Soc.,
69, 465 (1947).
271. L. S u г a n у i, H. W i 1 k.
Пат. ФРГ 1159451; РЖХ, 1966,
3H272.
272. К. Takeda, Kazuhika;
T a z a k i. Яп. пат. 175910, С. A.,
44, 8960 (1950).
273. К. Takeda, Tamamura,
Toni. J. Pharm. Soc. Japan, 71,
84 (1951); 74, 290 (1954); РЖХ,
1956, 47010.
274. Англ. пат. 492637; франц, пат.
883717; Zbl., 1939, I, 1064.
275. G. Lie c hsse nr i ng. Франц,
пат. 1310062; пат. ГДР 35205;
С. А., 58, 13963 (1963); 63, 1791
(1965).
276. А. Т о d d, F. В е г ge 1,
J. J а с о b. J. Chem. Soc., 1936,
1555.
277. А. Т о d d, F. В е г g е 1, F. К а-
r i m u 1 1 a h. Вег., 69, 217 (1936).
278. О. Hromatka. Пат. США
2160867; герм. пат. 670131; Zbl.,
1939, I, 2296.
430
279. Герм. пат. 663305; Zbl., 1938, II,
3007.
280. L. Cerecedo, J. Т о I р i п.
J. Am. Chem. Soc., 58, 1660 (1937).
281. С. R а р р е. Arkiv kemi, 13, 425
(1959).
282. А. Е и г е b i, Е. А г о w п,
L. Cerecedo. J. Am. Chem.
Soc., 71, 2931 (1949).
283. Т. Londergan, N. Н a u s е,
W. S c h m i t. J. Am. Chem. Soc.,
75, 4456 (1953).
284. К. С. Топчиев. ДАН СССР,
19, 497 (1938).
285. M. T h i e 1, F. A s i n g e г,
W. S t e n g 1 e r. Lieb. Ann.,
619, 161 (1958).
286. H. N о g a mi, M. N a n a n o,
S. A w a z u, T. T u w a. Chem.
Pharm. Bull., 18, 1937 (1970).
287. E. M о 1 i t о r, G. Emerson.
Vitamins and Hormones, 6, 69;
New York, 1948.
288. M. Y a m a z a к i, Bitamin, 38,
12 (1968); C. A., 69, 50697 (1968).
289. H. S h i n d о, К. О к a m о t o,
J. Tohtsu, I. Takahashi.
Bitamin, 38, 21 (1968); C. A., 69,
59698 (1968).
290. F. S c h u 1 t z e. Z. physiol.
Chem., 265, 113 (1939); 272, 29
(1941).
291. P. Karrer, M. Schoeller.
Helv. Chim. Acta, 34, 826 (1951).
292. E. Rogers. Ann. N. Y. Acad.
Sci., 98, 412 (1962).
293. J. В i g g s, P. S у к e s. J. Chem.
Soc., 1959, 1849.
294. A. Dor now, H. H e 1 1. Chem.
Ber., 94, 1248 (1961).
295. H. Erlenmeyer, D. Wald,
E. S о г к i n. Helv. Chim. Acta,
31, 32 (1948).
296. P. Baumgarten, A. D о r-
n о w. Ber., 72, 563 (1939); 73, 44
(1940).
297. A. Tracv, R. Elderfield.
J. Org. Chem., 6, 54 (1941).
298. A. W i 1 s о n, S. H a r r i s. J.
Am. Chem. Soc., 71, 2231 (1949);
73, 2388 (1951).
299. A. D о r n о w, W. Schacht.
Chem. Ber., 80, 502 (1947); 82, 117
(1949).
300. A. Dor now, A.Harge-
s h e i m e r. Chem. Ber., 86, 461
(1953).
301. А. Кузин. Химия и биология
патогенных микробов, 133—139,
Медгиз, 1946.
302. G. Е m е г s о n, Р h. S о u t h-
w i с h. J. Biol. Chem., 160, 169
(1945).
303. J. В a г о n e, H. T i e с к e 1-
m a n n, R. Guthrie,
J. H о 1 1 a n d. J. Org. Chem., 25,
24 (1960).
304. H. S t a a b, G. Schwab
bach. Sueb. Ann., 715, 128
305. L. Cerecedo, A. Eusebi,
M. S о о d a n. Chem. Ber., 85,
892 (1952).
306. T. Matsukawa, S. Yuru-
g i. J. Pharm. Soc. Japan, 71,
827 (1951); C. A., 46, 8125 (1952).
307. J. Barnhurst, D. Hennes-
sy. J. Am. Chem. Soc., 74, 353,
356 (1952).
308. E. Kupstas, D. Hennes-
sy. J. Am. Chem. Soc., 79, 5217,
5220, 5222 (1957).
309. К. К о m i n a t о. Яп. пат.
12520 (1965); С. A., 63, 13287 (1965).
310. К. К о m i n a t о. Яп. пат.
9078 (1957); РЖХ, 1961, УЛ377;
пат. ФРГ 1063334; С. А., 55,
18025 (1961).
311. Е. О д и и ц е в а. Успехи совре-
менной биологии., 27, 63 (1949).
312. Е. О д и н ц е в а, М. Me й-
сель, А. Г у с е в а. Микробио-
логия, 20, 273 (1951).
313. Т. Т а п а к a, J. Pharm. Soc. Ja-
pan, 76, 1314 (1956); С. А., 51,
3607 (1957).
314. A. W е п z, G. G б t t m а п и,
Н. Koop. Lieb. Ann., 618, 210
(1958).
315. A. W a t а п a b е, Y. A s a h i,
М. Н о г i. Yakugaku Zasshi, 77,
157 (1957); С. А., 51, 10493 (1957).
316. К. Loh ma п п, Р. S с h u-
s t е г. Biochem. Z., 294, 188
(1937); Naturwissensch., 25, 26
(1937).
317. A. R о s s i-F a n e 1 1 i, A. S e -
g г e, D. S i 1 i p r a n d i,
N. S i 1 i p r a n d i. Farmaco Ed.
Sci., 10, 173 (1955).
318. D. S i 1 i p r a n d i, N. Sili-
p r a n d i. Biochim. Biophys.
Acta, 14, 52 (1954).
319. A. Ross i-F a n e 1 1 i, N. Si-
li p. r a n d i, P. F a s e 11 a.
Science, 116, 711 (1952).
320. H. G r e i 1 i n g, L. К i e s о w.
Naturforsch., 13, 251 (1958).
321. A. R о s s i-F a n e 1 1 i, N. S i-
liprandi, P. Fasella et
al. Experientia, 10, 70 (1954).
322. H. I r e i 1 i n g, L. К i e s о w.
Naturforsch, 12, 672 (1957).
323. F. L e u t h a r d t, H. N i e 1-
sen. Helv. Chim. Acta, 35, 1196
(1952).
324. H.Greiling, L. Kiesow.
Naturforsch., 12, 606 (1957); 13b,
152 (1958).
325. F. R i v a, D. L i a r me r i,
T. H a m m a d y. Ital. J. Bio-
chem., 11, 346 (1962).
326. M. Viscontini, G. Bonet-
t i, С. E b n 6 t h e r, P. Kar-
rer. Helv. Chim. Acta, 34, 1384,
1388 (1951).
327. Z. Velluz, J. Bartos. Bull.
Soc. Chim France, 1952, 115.
328. В. M. Березовский,
В. И. К о л т у н о в а, Н. Д.
П е к е л ь, Е, А. Ш л и м о-
в и ч. ЖОХ, 33, 49 (1963).
329. В. J а п s е п. Vitamins and Ног-
431
mones, 7, 83; New York, 1949.
330. J. Weyl ar d. J. Am. Chem.
Soc., 63 1160 (1941).
331. B. Mat kovics, P. P e n-
z e s, S. F о 1 d e a k. Acta
Phys. Chem., 7, 55 (1961).
332. J. В i g 1 i n o, G. S e g г e. Far-
maco Ed. Sci., 8, 455 (1953); C. A.,
47, 12485 (1953).
333. T. Y u s a. J. Biochem., 46, 391
(1959).
334. A. We n z, G. G 6 t t m a n n,
H. Koop. Lieb. Ann., 618, 210
(1958); пат. США 2991284; Offic.
Gazette U.S., 768, 211 (1961).
335. Польск. пат. 45734; РЖХ, 1963,
3H289.
336. К. S t e r n, J. H of er. Science,
85, 483 (1937).
337. J. W e у 1 a r d, H. Tauber.
J. Am. Chem. Soc., 60, 2263 (1938).
338. K. Tarui, K. Matsumoto,
K. Takahashi. Яп. пат.
7676 (1960); РЖХ, 1962, 10Л276.
339. H. Ta uber. J. Am. Chem.
Soc., 60, 730 (1938); J. Biol. Chem.,
125, 191 (1938).
340. P. Karrer, M. Viscont i-
n i. Helv. Chim. Acta, 29, 711
(1946).
341. K. U c h a г а. Яп. пат. 3977
(1957); С. A., 52, 5758 (1958).
342. L. Tarui, K. Matsumoto.
Яп. пат. 10872 11962); С. A., 55,
8774 (1961).
343. К. U c h а г а, К- N а г a i,
К. Matsumoto. Яп. пат.
6881 (1957); С. А., 52, 10260 (1958).
344. G. S е г с h 1 Chimica, 30, 39
(1954).
345. М. Viscontini, G. Bonet-
t i, P. К a r r e r. Helv. Chim.
Acta, 32, 1478 (1949).
346. Y. W a k i s a k a, T. Ishida.
J. Vitaminol. 5, 44 (1959); C. A.,
53, 18145 (1959).
347. H. Roux, Y. Teysseire,
G. Duchesne. Compt. rend,
soc. biol., 142, 368 (1948).
348. H. 11 о, T. Hazumi, К. T a-
n a b e. Яп. пат. 10326 (I960);
С. A., 55, 9440 (1961).
349. В. И. Колтунова,
В. М. Березовский. Хим.
фарм. ж., 1967, № 2, 10.
350. L. V е 1 1 u z, G. A m i а г d,
J. Bartos. Bull. soc. chim.
France. 1948, 871; франц, пат.
1006637; С. A., 51, 12157 (1957).
351. В. M. Б e p e з о в с к и й, В. И.
Колтунова, Е. А. Ш л и-
м о в и ч, В. А. Д е в я т и и и.
ЖОХ, 32, 3890 (1962).
352. М. Viscontini, G.Bonet-
t i, С. Е b п о t h е г, P. К a r-
r e r. Helv. Chim. Acta, 34, 1384
(1951).
353. P. N e s b i t t, P. S у k e s.
J. Chem. Soc., 1954, 3057.
354. D. S i 1 i p r a n d i, M. S i 1 ip-
rand i. Biochim. Biophys. Acta,
14, 52 (1954).
355. В. M. Березовский,
В. И. Колтунова,
Н. С. Г е н о х о в а. Хим. фарм.
ж., 1969, № 8, 40.
356. J. Bartos. Bull. soc. chim.
biol., 38, 429 (1956).
357. В. M. Березовский,
В. И. Колтунов а, Е. И.
Ш л н м о в и ч. авт. свид. СССР
137920; Бюлл. изобрет., 1961, № 9,
25.
358. G. Leichssenring,
J. Schmidt. Chem. Ber., 95,
767 (1962); пат. ГДР 36516; С. А.,
63, 18111 (1965). ‘
359. Т. G а 1 a m о п, В. F i 1 i р о-
w i с z. Roczniki chem., 41, 1513
(1967).
360. Y. N о s е, К. N e d а. Яп. пат.
2292 (1963); С. A., 59, 11502 (1963).
361. A. I t о. J. Pharm. Soc. Japan,
82, 883 (1962); C. A., 58, 6732
(1963).
362. W. Ha m a n a k а, К. К a w a-
d а. Яп. пат. 1957 (1965); С. A.,
62, 14692 (1965).
363. К. Uchara, I. Muramat-
s u, К. К a b a s h i, T. Oka-
d a. Bitamin, 10, 353 (1956);
C. A., 51, 16488 (1957).
364. В. И. К о л т у н о в а, В. М.
Березовский. ЖОХ, 39, 102
(1969).
365. Яп. пат. 4410 (1965); С. А., 64,
6740 (1966).
366. В. М. Березовский,
В. И. К о л т у и о в а. ДАН
СССР, 174, 1325 (1967).
367. Т. Kuroda, М. М a s a k i.
Bitamin, 29, 109 (1964); С. А., 62,
7851 (1965).
368. К. Takamatsu, М. Susai.
Яп. пат. 3180 (1966); С. А., 64,
19750 (1966).
369. W. Hamanaka, J. Vitami-
nol., 12, 231 (1966).
370. R. Peters. Biochem. J. 31,
2240 (1937).
371. I. В a n g a, S. О c h о a, R. P e-
t e r s. Biochem. J., 33, 1109 (1939).
372. D. G r e e n, D. H e r b e r t,
V. Subrahmanvan. J. Biol.
Chem., 135, 785 (1940); 138,
327 (1941).
373. F. К u b о w i t z, W. L ii t t.
Biochem. Z., 307, 170 (1941).
374. E. A u h a ge n. Z. Physiol.Chem.,
204, 1 19; 209, 20 (1932).
375. A. Schellenberger. Angew.
Chem., 79, 1050 (1967).
376. M. Das, M. К о i к e, L. R ее d.
Proc. Natl. Acad. Sci. U. S„ 47,
753 (1961).
377. Э. Косове p. Молекулярная
биохимия. M., «Мир», 1964.
378. Н. Grisebach. Angew. Chem.,
68, 554 (1956).
379. J. Gunsalus. Feder. Proc.,
13, 715 (1954); The Mechanism of
Enzyme Action, John Hopkins
Ргрчч 1Q44 ^4H
380. R. B r e s l’o w. ’ Chem. and Ind.
432
(1957) 893; J. Am. Chem. Soc., 79,
1762 (1957); 80, 3719 (1958); Ann.
N. Y„ Acad. Sciences, 98, 445
(1962).
381. R. Breslow, E. McNeils.
J. Am. Chem. Soc., 81, 3080 (1959).
382. H. H о 1 z e r, F.Fonseca-
W о I 1 he i m, G. К о h 1 h a w,
Ch. Weenckhaus. Proc.
N. Y„ Acad. Sci., 98, 458 (1962).
383. L. К r a m p i t z, J. S u z u к i,
G. G r e u 1 1. Federal. Proc., 20,
971 (1961). Труды V Междуна-
родного биохимического конгрес-
са, симпозиум IV, Изд. АН СССР,
1962. стр. 343.
384. G. D a t t а, Е. R а с k е г. J.
Biol. Chem., 236, 624 (1961).
385. В. L. Но re скег, in Compre-
hensive Biochemistry, vol. 15, Am-
sterdam, 1964.
ГЛАВА X
ГЕКСАГИДРОИМИДАЗОЛОТИЕНОВЫЕ ВИТАМИНЫ И КОФЕРМЕНТЫ
БИОТИН
Биотин относится к производным конденсированных гекса гид роимидазо-
лотиеновых кольцевых систем; по своей структуре имидазольное кольцо
молекулы биотина относится к глиоксалидону. Биотин (витамин Н) явля-
ется (+)-цис-гексагидро-2-(В-карбоксибутил)-ЗН-4-оксоимидазоло (3,4-dJ-
тиеном (1).
О
II
X
HNf4^NH
НС—*СН
/ \*
Н3С6 t;СНСН2СН2СН2СН2СООН
S
I
Благодаря наличию трех асимметрических атомов углерода биотин обра-
зует восемь оптических антиподов и четыре рацемата. Его пространствен-
ная конфигурация может быть изображена следующими четырьмя цис-
транс-стереоизомерными формами I—IV.
R= (сн^соон
Форма I отвечает природному биотину, выделенному из молока и печени
[1, 2]. Формы II, III и IV получены только синтетическим путем. Рацеми-
ческий биотин обладает в 2 раза меньшей витаминной активностью, чем при-
родный d(-|-)-биотин. /(—)-Биотин и рацематы аллобиотина (III), эпиалло-
биотина (IV) [3] и эпибиотина (II) биологически неактивны.
d-Биотин представляет собой бесцветные тонкие иглы (из воды) с т. пл.
232,5е С (с разл.); он обнаруживает полиморфизм, и его вторая форма
кристаллизуется в призмах (из 80%-ного спирта) с т. пл. 230° С [4]. Его
оптическое вращение la Id+92° (0,1 н. NaOH) [5]. Биотин трудно раст-
433
ворим в воде — 0,022 г в 100 мл при 25° С, легче растворим в воде при наг-
ревании, хорошо растворим в слабых растворах щелочей. Он трудно раство-
рим в спиртах — 0,080 г в 100 мл спирта при 25° С, практически нераст-
ворим в эфире, хлороформе и других органических растворителях.
dZ-Биотин имеетт. пл. 231—233° С (с разл.) (3]. Температура плавления
его стереоизомеров: dZ-эпибиотина 190—191° С (с разл.) [6], dZ-аллобиотина
194—196° С (с разл.) и dZ-эпиаллобиотина—выше 195° С (с разл.) [3].
ХИМИЧЕСКИЕ СВОЙСТВА БИОТИНА
Биотин —слабая одноосновная кислота с Ка 6,3-10"®. Он адсорбируется
на угле и вымывается аммиачным раствором ацетона.
Биотин стабилен в кристаллическом виде при температуре 100° С, в
растворах устойчив к действию разбавленных кислот и щелочей, а также
нагреванию, однако он расщепляется при обработке с нагреванием концен-
трированными кислотами, щелочами и растворами перекиси водорода Г7 ]. “
В отличие от биотина (I) аллобиотин (III) и эпиаллобиотин (IV) не устойчивы
при кипячении в разбавленной серной кислоте и быстро гидролизуются, л
Для биотина свойственны все реакции, характерные для соединений с
карбоксильной группой, — образование сложных эфиров, амидов кислот,
гидразидов, азидов, нитрилов, ангидридов и т. д.
Метиловый эфир d-биотина кристаллизуется в бесцветных пластинках
с т. пл. 166—167° С, [а +57° (СНС13); он почти нерастворим в воде и
эфире, но хорошо растворим в спирте, ацетоне и хлороформе, в вакууме под-
вергается молекулярной дистилляции [8] и легко гидролизуется разбавлен-
ным раствором щелочи при комнатной температуре в биотин.
Биотин или, лучше, его метиловый эфир при действии аммиака или орга-
нических оснований образует амиды, отдельные представители которых
обладают биологической активностью. Особенный интерес представляет
амид биотина с аминокислотой £-лизином — E-N-биотинил-А-лизин (V)
19], выделенный из дрожжей. С одним из фрагментов D-пантотеновой кис-
лоты— р-аланином — биотин образует N-биотинил-р-аланин (VI) [10].
nh2
(CHj)4COHN(€iy4CHCOOH
V
ЮгТчн
^Z^CHj^COHNCHjCHjCOOH
vi
Биотин может быть превращен в его спиртовой аналог dZ-биотинол (VII)
ст. пл. 160—161° С [11]. Десульфирование биотина с никелевым катализа-
тором приводит к дезтиобиотнну (VIII).
о
HN^NH
( Ы
^"•(СЦ^СЦОН < Нз CH/CHsXcOOH
Из природных продуктов выделены S-окиси биотина, например из моло-
ка — S-о'кись d-биотина (IX), а ее оптический антипод — S-окись Z-биоти-
на — продуцируется плесенью Aspergillus niger [12, 13]. Изучена эпимери-
зация S-окиси d-биотина, синтез [14] и конфигурация [15]. Окисление био-
434
тина марганцовокислым калием приводит к образованию биотинсульфона
(X).
При жестком окислении биотин первоначально дегидрируется в своей
тиофановой части в дегидробиотин—2-(о-карбоксибутил)-3,4-уреилен-2,5-
дигидротиофен (XI), а затем в полный тиофеновый аналог — 2,3,4,5-тетра-
гидробиотин (XII) [16].
хп
СТРОЕНИЕ БИОТИНА
Впервые биотин в кристаллическом состоянии выделил Кегль [17] из
биоса II в 1935 г., а в следующем году Кегль и Тэннис [18] выделили био-
тин в виде метилового эфира из яичного желтка.
Биотин не содержит в своей молекуле ненасыщенных связей, что уста-
новлено по его неспособности присоединять водород. Отсутствие характер-
ного спектра поглощения в ульрафиолетовом свете указывает на отсутствие
ароматической системы в молекуле биотина.
Он имеет одну карбоксильную группу, определенную реакцией этери-
фикации и ацидиметрическим анализом [19]. Свободной или замещенной ами-
ногруппы в его молекуле не обнаружено.
Образование диаминокарбоновой кислоты (XIII) при гидролизе биотина
(I) с соляной кислотой при одновременном отщеплении двуокиси углерода
[8] или с окисью бария под давлением [19] и ресинтез из нее биотина под
действием фосгена [20] указывают на наличие в молекуле биотина группи-
ровки мочевины (схема 97). Почти количественный выход при ресинтезе
дает основание полагать, что свободные аминогруппы находятся при сосед-
них атомах углерода и при конденсации с фосгеном замыкаются в пятичлен-
ное кольцо.
Наличие свободной карбоксильной группы установлено на основании
образования с диазометаном метилового эфира биотина (XIV) [5]. Далее
было выяснено, что при окислении диаминокарбоновой кислоты (XIII)
марганцовокислым калием или азотной кислотой образуется адипиновая
кислота [21 ]; это дает основание предполагать присутствие в молекуле био-
тина боковой цепи из валериановой кислоты (одна из карбоксильных групп
получается при окислении атома углерода, несущего цепь валериановой кис-
лоты). Доказательство того, что вторая карбоксильная группа адипиновой
кислоты происходит от свободной карбоксильной группы биотина, было по-
лучено на основании изучения превращений метилового эфира биотина (XIV)
через гидразид в азид биотина (XV) и затем по реакции Курциуса через про-
межуточные соединения в 2-аминобутил-3,4-диаминотиофан (XVI), в кото-
ром одна из аминогрупп получена из карбоксильной группы биотина. Сое-
435
динение XVI при окислении марганцовокислым калием уже не дает адипи-
новой кислоты [22, 23]. Вопрос о том, входит ли группировка мочевины
в пяти-(имидазолидоновое) или шестичленное кольцо, выяснен в результа-
те обнаруженной способности диаминокарбоновой кислоты (XIII) образо-
вывать хиноксалиновое производное (XVIII) с фенантренхиноном (являю-
щимся реактивом на 1,2-диамины) [24] через соединение XVII, что доказы-
вает наличие имидазолидонового кольца в биотине [25]. Эта реакция
указывает и на присутствие у двух циклических атомов углерода, несущих
аминогруппы, также атомов водорода, что исключает боковую цепь при
них [251.
Схема 97
Установление строения р-биотнна реакциями его расщепления
c8h1Son8sn=c=o
I
CeHtfOr^SNHCOOCjHs
NH, NH,
XVI
h,)4nh,
Атом серы прочно связан в молекуле биотина (I); он не удаляется при дей-
ствии щелочи, йодистоводородной кислоты, цинка и соляной кислоты или
бромной воды и не образует меркаптогруппы. Ацетиловый эфир биотина
(XIV) способен при окислении марганцовокислым калием или перекисью
водорода в уксусной кислоте образовывать биотинсульфон (выход 90%),
гидролиз которого приводит к получению биотинсульфокислоты [26]. Так
как три атома кислорода распределяются в молекуле биотина между кар-
боксильной и карбонильной группами, то атом серы не может быть связан
с атомом кислорода в виде сульфоновой или других групп.
Вместе с тем, помимо приведенных выше доказательств, эфирный харак-
тер атома серы подтверждается образованием сульфона (XIX) при окисле-
нии диаминокарбоновой кислоты (XIII). Однако для установления точного
характера связи двух атомов углерода с атомом серы и имидазоидоновым
кольцом необходимо было провести дополнительные исследования.
436
При действии скелетного никелевого катализатора как десульфирующего
агента [27] на метиловый эфир биотина происходит десульфирование мо-
лекулы с образованием метилового эфира дезтиобиотина (VIII). При гидро-
лизе с окисью бария он образует дезтиодиамннокарбоновую кислоту, ока-
завшуюся С, Tj-диаминопелларгоновой кислотой (XX) [28], так как при
окислении щелочным раствором марганцовокислого калия получается пиме-
линовая кислота. Строение соединения XX доказано синтезом [28]. При дей-
ствии фосгена дезтиодиаминокарбоновая кислота (XX) превращается вновь
в дезтиобиотин [29]. Все эти данные говорят в пользу структуры биотина,
изображенной формулой 1 [2].
Дальнейшее подтверждение формулы биотина было получено в резуль-
тате исчерпывающего метилирования диамикокарбоновой кислоты (XIII)
диметилсульфатом с последующим гофмановским распадом образовавшегося
четвертичного аммониевого основания, при котором под влиянием концен-
трированной соляной кислоты отщепляются а-водородный атом и аминогруп-
пы, в результате чего происходило образование о-(тиофен-2)валериановой
кислоты (XXI) [30].
Кислота (XXI) оказалась идентичной с синтетической, полученной в резуль-
тате взаимодействия тиофена и ангидрида глутаровой кислоты с последую-
щим восстановлением продукта конденсации. Этим окончательно доказы-
валось наличие в молекуле биотина тиофанового кольца.
СТЕРЕОИЗОМЕРИЯ БИОТИНА
Так как молекула биотина имеет три асимметрических атома углерода:
С(2), С(з) и С(4), то в ней, так же как и в 2,3,4-замещенных тиофана, возмож-
на цис-и транс-конфигурация заместителей относительно основной плоскости
пятичленного кольца.
Полная
цис -конфигурация
„Эпи" цис -конфигурация
атомов азота
атомов азота
„Эпиалло7 транс -конфи-
гурация атомов азота
В биотине (I) и эпибиотине (II) оба атома азота находятся в i/uc-положе-
нии, а в аллобиотине (III) и эпиаллобиотине (IV) — в транс-положении. За-
местители в положении 2 тиофанового кольца в биотине (I) и эпибиотине (II)
эпимерны, так же как и в аллобиотине (III) и эпиаллобиотине (IV). Отсюда
следует, что биотин и эпиаллобиотин различаются положением заместителей
при атоме углерода положения 3, а эпибиотин и эпиаллобиотин — при ато-
ме углерода положения 4 тиофанового кольца. цис-Положение атомов азо-
та для Л(±)-биотина, следовательно, отвечает и t^ac-положению для при-
родного с[(+)-биотина. Для биотина принимается полная quc-конфигура-
ция всех заместителей молекулы (двух атомов азота и заместителя у атома
углерода в положении 2).
Важные доказательства относительной конфигурации четырех рацеми-
ческих форм биотипа были получены на основании превращения их в дез-
тиобиотины при нагревании со скелетным никелевым катализатором. Один
и тот же дезтиобиотин (VIII), имеющий только два асимметрических атома.
437
получен из биотина (I) [31 ] и эпиобиотина (II) [321, что говорит об одина-
ковом пространственном расположении атомов азота в молекуле этих сте-
реоизомеров биотина. Дезтиоаллобиотин (XXII) был получен из эпиалло-
биотина (IV) 131, 32) и аллобиотина (III) [311, которые имеют, следователь-
но, иное расположение атомов азота в пространстве.
цис-Конфигурация атомов азота приписывается дезтиобиотину (VIII)
и, следовательно, биотину и эпибиотину на основании легкости замыкания
кольца с фосгеном и почти количественного выхода в отличие от худшего
выхода для дезтиоаллобпотина (XXII). Это подтверждается большей ста-
бильностью биотина при нагревании его водного раствора, из которого он
легко перекристаллизовывается; в этих же условиях у аллобиотина наблю-
дается раскрытие имидазолидонового кольца, по-видимому, из-за внутрен-
них напряжений /пране-конфигурации. Биотин также устойчив к нагре-
ванию с разбавленной серной кислотой, в то время как <2/-аллобиотин легко
гидролизуется с образованием соответствующей диаминокарбоновой кисло-
ты.
Схема 98
Инверсия
стереоизомерных траяс-дикарбоиовых кислот
438
У замещенных тнофана возможно избирательное обращение заместителей
у атомов углерода в положениях 3 и 4 [321. Так, транс-3,4-дикарбокси-2-
(S-карбоксибутил)тиофан может иметь конфигурации XXIII и XXIV (схе-
ма 98). Путем избирательного обращения из этих стереоизомерных транс-
дикарбоновых кислот могут быть получены оба возможных цис-рацемата
биотина (биотин и эпибпотин) независимо от того, какая из этих конфигу-
раций /пранс-дикарбоновых кислот будет применена как исходная. Так, на-
пример, при инверсии заместителей у С(3) из дикарбоновой кислоты (XXIII)
будет получен в результате дальнейших синтетических превращений dZ-би-
отин, а при обращении уС(4) из той же кислоты —- d/эпибиотин. Теоретичес-
ки возможные превращения представлены в схеме 98.
В этой схеме приведены асимметрические центры с их нумерацией в тио-
фановом ядре. Знаком b отмечены атомы водорода, знаком х — заместители;
вертикальная черта, разделяющая эти знаки, условно представляет собой
главную плоскость молекулы.
ВЫДЕЛЕНИЕ БИОТИНА
Выделенный из природных источников «биос», наряду с другими вита-
минами содержащий биотин, является ростовым фактором дрожжей и мно-
гих других микроорганизмов. В 1936 г. Кегль и Тэннис выделили из желтка
яиц биологически активное вещество, названное ими биотином [181. Из
250 кг высушенного яичного желтка было получено 1,1 мг метилового эфи-
ра биотина [81. В 1937 г. Кегль установил эмпирическую формулу метило-
вого эфира биотина [331, которая затем была подтверждена [51. В 1939 г.
Дьёрги и др. было выделено из печени активное вещество, названное вита-
мином Н [34]. В следующем году была установлена идентичность витами-
на Н из печени с биотином из яичного желтка [35]. В 1941 г. дю-Виньо
с сотрудниками из печени был выделен метиловый эфир биотина [1].
В 1942 г. чистый биотин был выделен из концентратов свежего молока [36].
Установлению структуры биотина были посвящены многочисленные
исследования. Окончательно его структуру подтвердили полным синтезом
Гаррис, Фолькерс и др. в 1944—1945 гг. [3, 37].
СИНТЕЗ БИОТИНА И ЕГО СТЕРЕОИЗОМЕРОВ
Биотин, имеющий три асимметрических атома углерода, и его стереоизо-
меры синтезированы несколькими методами, в которых применяют прин-
цип первоначального построения тпофанового кольца с последующим на-
ращиванием на его основе имидазолидонового цикла или построения имида-
золидонового цикла с последующим наращиванием тиофанового кольца и
остальной части молекулы. Все эти синтезы сложны и сопровождаются обра-
зованием стереоизомерных продуктов.
1. Синтез биотина, аллобиотина и эпиаллобиотина из L-цистеина через
3-оксотиофан без заместителя в положении 2
В этом синтезе, который предложили Гаррис с сотр. [37, 38], использо-
вана подвижность атомов водорода положения 2 тиофановой молекулы для
конденсации ее с альдегидсодержащей боковой цепочкой, впоследствии пре-
вращаемой в необходимую о-карбоксибутильную группу (схема 99).
L-Цистеин в виде натриевой соли (XXVI), получаемый из L-цистина
(XXV) восстановлением натрием в жидком аммиаке, конденсируют с моно-
хлоруксусной кислотой в присутствии едкого натра в 5-карбоксиметил-£-
цистеин (XXVII). Аминогруппу этого соединения защищают бензоилиро-
ванием, карбоксильные группы этерифицируют метиловым спиртом и по-
лучают диметиловый эфир М-бензоил-5-карбоксиметил-£-цистеина (XXVIII)
[38—401. По другому варианту [371 исходят из L-серина (XXIX), амино-
группу которого бензоилируют, карбоксильную — этерифицируют, а гид-
роксильную — замещают атомом хлора; промежуточное соединение кон-
денсируют с натриевой солью тиогликолевого эфира в соединение XXVIII.
439
Диметиловый эфир М-бензоил-Б-карбоксиметил-А-цистеина (XXVIII)
подвергают далее внутримолекулярной конденсации в присутствии мети-
лата натрия (реакция Дикмана) и легко превращают в 0 -кетоэфир с образо-
ванием натриевого производного енольной формы метилового эфира 4-бен-
зоиламино-3-оксо-2-карбокси-4,5-дигпдротиофена (XXX); циклизация одно-
временно сопровождается рацемизацией [38—40]. После гидролиза и декар-
боксилирования соединения XXX при 125° С в смеси уксусной и соляной
кислот получают 4-бензоиламино-З-оксотиофан (XXXI); это соединение до-
статочно лабильно и с метиловым спиртом может расщепляться по связи
S—С с образованием ациклического дисульфида [41 ].
В положение 2 4-бензоиламино-З-оксотиофана (XXXI) вводят алифати-
ческий заместитель конденсацией с метиловым эфиром моноальдегида глута-
ровой кислоты при каталитическом участии уксуснокислого пиперидина.
Образующийся при конденсации 4-бензоиламино-3-оксо-2-(о-карбометокси-
бутилиден)тиофан (XXXII) при действии гидроксиламина в пиридине [3]
превращают в оксим (XXXIII), получаемый в син- и анти-формах. Однако
в водно-спиртовой среде в присутствии углекислого натрия гидроксиламин
присоединяется не по оксогруппе, а по кратной связи с образованием 4-бен-
зоиламино-3-оксо-2-(а-гидроксиламино-о-карбометоксибутил)тиофана [42 ].
Из обеих форм оксима (XXXIII) при избирательном восстановлении цинко-
вой пылью в уксусной кислоте и ацетилировании уксусным ангидридом обра-
зуется два изомерных соединения: 3-ацетиламино-4-бензоиламино-2-(о-кар-
бометоксибутил)-4,5-д и гидротиофен с т. пл. 185—186° С, «изодегидроэфир»
(XXXIV) с одним асимметрическим атомом углерода, и З-ацетиламино-4-
бензоиламино-2-(8-карбометоксибутилиден) тиофан с т. пл. 162—163° С,
«аллодегидроэфир» (XXXV) в двумя асимметрическими атомами углерода.
При каталитическом восстановлении двойной связи соединения XXXIV
получаются соединения, в которых атомы водорода положений 2 и 3 при-
соединяются в 4{ис-положении. Это положение само по себе может явиться
цис- или транс-положением в отношении какого-либо заместителя, уже име-
ющегося в молекуле. В результате исчерпывающего гидрирования на Pd-ка-
тализаторе изомерных соединений XXXIV и XXXV, различающихся по-
ложением двойной связи, образуется три различных рацемических сте-
реоизомерных 3-ацетиламино-4-бензоиламино-2-(о-карбоксибутил)тиофана
(XXXVI, XXXVII, XXXVIII). Из них в результате отщепления бензо-
ильного и ацетильного остатков при действии гидрата окиси бария при
140° С и гидролиза эфирной группы были получены три стереоизомерные
диаминокарбоновые кислоты (XIII, XXXIX, XL).
В заключение диаминокарбоновые кислоты (XIII, XXXIX, XL) при
взаимодействии с фосгеном подвергаются циклизации [19] в стереоизомер-
ные биотины (I, III, IV) [3, 37]. Из изодегидроэфира (XXXIV) при гид-
рировании получены два рацемата, из которых один — ст. пл. 153—154° С
(XXXVI)—дал dZ-биотин (I), а другой — с т. пл. 172 — 173° С
(XXXVII) —образовал dZ-аллобиотин (III). Соответственно из аллодегид-
роэфира (XXXV) было получено также два рацемата, из которых соедине-
ние XXXVII с т. пл. 172—173° С дало dZ-аллобиотин (III), а соединение
XXXVIII образовало с!/-эпиаллобиотин (IV).
Обычные оптически активные алкалоиды не дают кристаллических солей
с биотином. Разделение рацемического биотина на оптические антиподы бы-
ло осуществлено [43] превращением биотина в его хлорангидрид и полу-
чением производного с D(—)-миндальной (оксифенилуксусной) кислотой
или же в результате взаимодействия биотина с Ц+)-аргинином и превра-
щения в его диастереоизомерные соли. При фракционированной кристал-
лизации из изопропилового спирта был выделен Н-биотинил-£(-|-)-арги-
нин, из которого при действии разбавленной соляной кислотой получен
^(+)-биотин, идентичный с природным.
Метиловый эфир моноальдегида глутаровой кислоты получают из глута-
ровой кислоты [44 ] или лучше из акролеина и малонового эфира, которые
440
Схема 99
Синтез биотина, аллобиотнна и эпиаллобиотина из £-цистеииа через 3-оксотиофаи
без заместителя в положении 2
i^h,
снсоон
lyn,
снсоон
I
CH,
S------
XXV
ТЧН NH,
[11] CHCOONa ClCl tCOOH^ СНСООН
Cll2
SCH,COOH
XXVII
^CeHgCOCl,
з) этерификация
NHCOCgHj
\---<-ONa
(Na,NH3; Cl I,
SNa
XXVI
i) Бензоилирование,
2) этерификация,
a) SOCls_________
1
l\'HCOCeHs
yiCOOCH, RONa
CH2CH2COOCH,
1^н3
снсоон
I^HCOQHs
CIICOOCH,
j XXV1H
CH2OH
XXIX
СП2С1
CH,COOH,
HCl, H,O
x x -CO,,-ROH
S COOR
XXX
<jHO
NHCOC6HS (CH,), NHCOCeHs
f ° COOR / ~f°
nh2oh
NHCOC6HS
. \---. -NOH
S
XXXI
CjHgCO COCH,
NHH NHh
'H-h
4s'xh(ch2)3coor
XXXII
4s xh(ch2)3coor
XXXUl
N^HNH?H
Ва^ОНХУЛ" H
HN< H"NHH
COCt, H
COC(.H,, COCH,
NHh NH
s (ch2)4coor
XXXVI
* (CH^COOH
XIII
S' ItH^COOH
(fZ-Биотин
I
xs^[ch,)4coor
XXXIV
H j
COC6HS COCH,
NHHH NH
COCJIg COCH,
nhhh Ah
XXXVII
NH,HH NH,
ГО
HNV*1
(CHj^COOH COCI, /
( (СН^СООН
S H
XXXIX
т H
ill -Аллобиотмк
III
< A, , . COC6HS coch,
\h CH^COOR I I
* , NHh H NH
xxxy - -
NHaHH NH,
Ba(OH^
R=CH,
s (ch,)4goor
XXXVIII
\ch,)4cooh
XL
/C°\
Hl/н H NH
COCIj, 7~ \ н
'S^CH^OOOH
Л-Эпиалловиотин
IV
н
конденсируют в присутствии метилата натрия по реакции Михаэля в у,
Т-дикарбээтоксимасляный альдегид [45] с последующим его гидролизом
и декарбоксилированием в моноальдегпд глутаровой кислоты, подвергае-
мый затем этерификации [45]. Моноальдегид глутаровой кислоты сущест-
вует в форме мономерной альдегидокислоты, а также в форме циклического
лактола; кроме того, при стоянии он легко полимеризуется с образованием
тримера, который вновь может бытьпревращен в мономерную альдегидокис-
лоту при нагревании водного раствора [46].
2. Синтез биотина через 3-оксотиофан с ю-метоксибутильным заместителем
в положении 2
Для получения биотина Грюсснер, Бурквин и Шнидер предложили ме-
тод [47, 48], в последних стадиях которого метоксигруппа боковой цепи гек-
сагидроимидазолтиенового цикла должна быть превращена в карбоксиль-
ную группу.
441
В качестве исходного вещества применяют этиловый эфир 2-бром-6-ме-
токсикапроновой кислоты (XLI), который конденсируют с натриевым про-
изводным р-меркаптопропионового эфира в соединение XLII, подвергае-
мое затем дальнейшей циклизации в 2-(б-метоксибутил)-3-оксо-4-карбоэто-
кситиофан (ХЕШ, схема 100) [49]. Карбоксильную группу в положение 3
этого соединения вводят циангидрнновым синтезом и получают циангидрин
тиофана (XLIV).
В соединении XLIV нитрильную группу последовательно превращают
в амидную, карбоксильную, карбоалкоксильную, а гидроксильную груп-
пу положения 3 заменяют на хлор; после восстановления цинком и уксус-
ной кислотой получают 2-(о-метоксибутил)-3,4-дикарбоалкокситиофан (XLV).
Из соединения XLV при действии гидразин-гидрата получают дигидра-
зид (XLVI), некристаллизующаяся часть которого была переведена по ре-
акции Курциуса в 3,4-диуретан (XLVII). Это соединение подвергнуто хро-
матографическому разделению на окиси алюминия с последующей хлоро-
формной элюцией некристаллизующейся части, которая при нагревании
с 48 %-ной бромистоводородной кислотой с одновременным замещением ме-
токсильной группы боковой цепи на атом брома превращается в диамин
(XLVIII).
Схема 100
Синтез биотина через 3-оксотиофан с ю-метоксибутильным заместителем в положении 2
COOR
CHa
CH3
SNa
COOC2H5
Oi(cH2)4OCHj
Br
COOR-
CH2 COOCjHs
COOR
XU
XLII
COOR CONHj
’а
на
kcn, на
XS fHj^OCHj
xun
COOR COOH
XSX (CH2)4OCH3
XLIV
COOR COOR
VOH
"SZ|CH2)4OCH3
VOH
S^CI^OCH,
ROH
VOH _____
S'(CHAlOCH3
COOR COOR
soa2
COOR COOR
[H](Zn,CH3COOn]
MlgNHa
INHNHj 1NHNH2
CO CO
COOQHg
INH NHCOOQHg
HNQfrROH
НВг
XLVI
Hn hNH„
r \ h
KCN
(СнЛВг
XL1X
^^(cf^OCHj
XLVil
CO
ry
V^CH^^N
на
R- СН3, QHg
"S (СНд)4ОСН3
XLV
HH соси, NaOH
XSX (СН2)4Вг
XLVI11
CO
П H
S (CH2)4COOH
dl~ Биотин
I
При взаимодействии диамина (XLVIII) с фосгеном происходит образо-
вание имидазолидонового кольца, и получается соединение XLIX. Введение
карбоксильной группы в боковую цепь этого соединения осуществлено че-
рез биотинонитрил (L), который при нагревании со щелочью образует dZ-би-
отин (I) с т. пл. 231—232° С. Эти реакции протекают с малым выходом; сое-
442
динения XLVII—L в процессе синтеза не выделялись. Следует отметить, что
рассматриваемый метод получения биотина сопровождается образованием
ряда побочных продуктов [50].
3. Синтез биотина, эпибиотина и эпиаллобиотина через
транс-3,4-дикарбокси-2-(о-карбоксибутил)тиофан
Этот метод синтеза, который разработал Бакер с сотр. [51 ], характери-
зуется стереохимической направленностью получения отдельных стереоизо-
меров: с[/-биотина, dZ-эпибиотина или Н/-эпиаллобиотина, что достигается
обращением заместителей у С(з> и С(4) транс-3,'4-дикарбокси-2-(о-кар-
боксибутил)тиофана (XXIII). Обращение заместителей первоначально
было изучено на тиофандикарбоновых кислотах, не содержащих замести-
теля в положении 2 [52].
Для получения транс-3,4-дикарбоновой кислоты (XXIII) исходят из пи-
мелиновон кислоты [53, 54], которую превращают в а-бромпимелиновый
эфир (LI), его конденсируют с натриевым производным р-меркаптопропионо-
вого эфира и получают соединение LII. Это соединение внутримолекулярной
циклизацией в присутствии этилата натрия переводят в 4-карбометокси-З-
-оксо-2-(о-карбометоксибутил)тиофан (LIII) и затем оксогруппу посредст-
вом циангидринового синтеза через промежуточные соединения замещают
карбоксильной группой — после гидролиза образуется 3,4-дикарбоновая
кислота в виде нескольких стереоизомерных форм. Для дальнейшего син-
теза применяется транс-3,4-дикарбоновая кислота (XXIII).
?оосн3 соосн3
М + СООСН, СНг СООСН,
-Н, ВгСН^Н^СООСЦ, СН, ^(Цсц^соосн,
U LU
СООСНа о
—X HCN
UH
COOCHj CN
POCL, Х)==\
HCl;
S '(СН2)4СООСН3
C4°hV-cooh
-О<н
x'sX4(ch2\ccm3H
XXIU
Следует отметить, что обращение заместителей у С(з) и С<4) легче проис-
ходит при их меньшем объеме. Соединения с /нранс-расположенными замес-
тителями более стабильны, по-видимому, из-за меньших пространственных
препятствий, транс-3,4-Дикарбоновая кислота (XXIII) легче выкристалли-
зовывается из растворов [53—55], чем ее цис-форма. в то же время остаю-
щаяся в маточном растворе цис-дикарбоновая кислота может быть подверг-
нута обращению в транс-форму обработкой метилатом натрия [53, 54].
Вместе с тем цис-3,4-дикарбоновые эфиры вследствие пространственных
помех трудно образуют гидразиды, поэтому при одновременном расщепле-
нии обоих карбоксилов (по реакции Курциуса) цис-диамины получаются
только в незначительном количестве [53, 56], что было показано последую-
щим превращением в биотин [47].
В дальнейшем синтезе транс-3,4-дикарбоновую кислоту (XXIII) (схе-
ма 101) через полный трикарбоновый эфир с одним эквивалентом щелочи
превращают в дикарбоновый эфир /пранс-3,4-дикарбокси-2-(о-карбокспбу-
тил)тиофана (LIV) со свободной карбоксильной группой в положении 4.
Из него вальденовским обращением заместителей, находящихся в разных
положениях, и их последующими превращениями проведено три синтеза
(пути А, Б и В), приведших к получению трех различных стереоизомеров био-
тина.
В соединении LIV по реакции Курциуса с последующей обработкой ани-
лином карбоксильная группа положения 4 замещается уретананилидной
443
Схема 101
Синтез биотина, эЬибиотина и епиаллобиотина через транс-3,4-дикарбокси-2-
(о-карбоксибутил)ткофан
сХсоон
и
XXIII
’(СН^СООН
Whotoh
0/н -
5/4(cH2\C00R
L1V
J Реакция
Курциуса
CONHCgHj
NHHH COOR
П'и
N1C6HS
СО НН COOR
Пи
'S (CH^GOOR
со
S (CHXCOOR
LY
(Обращение у С3
(сн,со)р
CO-ljFQHs
IJHQlig
СО н Н CONj
“ с<н
k SMCH^OOR
LIX
(ОБращение у С4
(реакция Курциуса
QHg-N—СО
I
Путь А
CONHC6Hs
NHH н CONHNHj
И
Xй
^'XadiCOOR
LVI
'$ '^FlJtCOOH
LX
CONHQH,
NlIpCONHlNH,
гА H
s/\ch2Ycooh
LVII
| Реакция
1Курциуса
CONHC6HS
AhHHJNHCOOR
/\н
i/4(CH2)4COOH
|ва(ОН)2
МН2нн NHj
&н
S (сн^соон
XL
СОС12
s (CHlcONHQH;
I.VIII
Реакция
, Курциуса
NHNH,
Н СОн NHCONHCeHs
О"
i^^CHA^ONHCeHs
LXI
1 Реакция
Курциуса
QHSNHCO СО
IJ 1
со
н
н NHH;NcoNHceHs
/\ л
/СО,
И
s (сн^соон
dt -Эпиалловиотин
j (CH^CONHCeH,
|Ва(0Н)»
NHiHNHj
Мн
s (сн2)4соон
I Ва(он)2
H^H.NHs
Г \ ’i
со
/Л Н
(а^соон
XIII
s (сн2)4соон
Л-Биотин
5.
(СНСООН
444
группой с образованием соединения LV, которое под влиянием уксусного
ангидрида с обращением транс-заместителя у С(3)в цис-положение дает цик-
лический анилид (LVI). Этот цис-ряд (LVI, LVIII->XIII) (путь Б) ведет к
получению биотина (I).
При размыкании цикла у анилида (LVI) под влиянием метилата натрия
цис-заместитель у С(3) вновь подвергается обращению в транс-положение и
при последующем воздействии гидразин-гидрата дает гидразид (LVII). Этот
транс-ряд (LVII->XL) ведет к получению эпиаллобиотина (IV) (путь А)
151].
Третий цис-ряд (LIX—LXI->LXII) ведет к эпибиотину (путь В); для
этого исходят из транс-дикарбонового эфира (LIV), из которого получают
анилид по карбоксильной группе положения 4, гидролизуют эфирную груп-
пу положения 3 и переходят к азиду карбоновой кислоты (LlX). У этого
соединения после перегруппировки Курциуса в присутствии уксусного анги-
дрида и ацетата натрия происходит обращение транс-заместителя у С{4)
в цис-положение с образованием циклического анилида (LX).
Из циклических анилидов (LVI и LX) при действии гидразин-гидрата
получают гидразиды (LVIII и LXI).
Аналогичным образом из гидразидов (LVII, LVIII и LXI) трех различ-
ных цис-транс-рядов (пути А, Б, и В) второй карбоксил уС(3)ИЛИ С{4> за-
мещается аминогруппой (реакция Курциуса) с образованием диаминов. За-
тем после гидролиза уретановой или анилидуретановой группы при дейст-
вии гидрата окиси бария во всех трех рядах получены диаминокарбоновые
кислоты (XL, XIII и LXII), которые с фосгеном образовали dZ-эпиаллобио-
тин (IV), dZ-биотнн (I) и dZ-эпибиотин (II). Этим методом из 100 г пимели-
новой кислоты получают 2,7 г dl-биотина (выход 1,7%).
4. Синтез эпибиотина и эпиаллобиотина через 3-нитротиофан
Этот синтез предложили Гроб иШпрехер[57] (схема 102); в нем исходят
из метилового эфира ю-карбоксивалеральдегида (LXIII, R—СН3), легко кон-
денсирующегося с нитрометаном в присутствии щелочи в метиловый эфир
6-окси-7-нитрогептановой кислоты (LXIV, R=CH3); при действии пятихло-
ристого фосфора в положение 6 вводится атом хлора, и полученное соеди-
нение с помощью меркаптоацетальдегидацеталя превращается в ацеталь
тиоэфира (LX V). Этот ацеталь гидролизуется в альдегид, который в присутст-
вии алькоголята натрия дает смесь стереоизомерных эфиров З-нитро-4-окси-
2-(о-карбоксибутил)тиофана (LXVI).
Смесь рацемических нитросппртов (LXVI) ацетилирована в эфиры 3-ни-
тро-4-ацетокси-2-(о-карбоксибутил)тиофана (LXVII). При действии на них
аммиаком в растворе диоксана получается смесь двух З-нитро-4-аминотио-
фановых производных, при ацетилировании которых образуется два изомера
эфиров 3-нитро-4-ацетиламино-2-(о-карбоксибутил)тиофана: цис-изомер с
т. пл. 78—80е С (LXVIII) — около 70% и транс-изомер с т. пл. 114° С
(LXIX) — около 30%.
В дальнейшем синтезе при восстановлении амальгамированным алюми-
нием из изомеров LXVIII и LXIX получены два моноацетилдиамина, кото-
рые после гидролиза дали эфиры эпи-3,4-диамино-2-(о-карбоксибутил)тио-
фана (LXX) и его эпиаллопзомера (LXXI). Замыкание имидазолидонового
кольца у обоих этих соединений фосгеном приводило соответственно к dl-
эпибиотину (II) и к dZ-эпиаллобиотину (IV).
5. Синтез биотина через мезо-а,р-дибензиламиноянтарную кислоту
Стереоспецифичный синтез биотина, приводящий к получению только
его полной цис-стереоизомерной формы, описали Гольдберг и Штернбах
[11, 58]. Синтез, имеющий техническое значение, основан на применении
445
Схема 102
Синтез эпибиотнна и эпналлобиотииа через 3-нитротнофан
ROOC(CHj)4CHO
LXUI
rooc(ch2)4chch2no2
OH
LXIV
ROOC(CH,)^HCH2NO2
70,
(ro)2ch ch,
CH2 CHfCH^COOR
NH,
CH2
ОН NO:
LXV
?"i' г 7 .
CHj CH(CH2)4COOR
—ex
S (CH^OOR
LXVI
COCH,
NH h NO?
Ряд А
OCOCHj ЬЮ,
NHj
'2
\CH2)4COOR
LXVII
s/\chj)4coor
S H
LXVIII
COCH,
I
NHHH J\IOj
Ряд Б
COCHj
NHHNH.
Ряда / \
"S н
LXIX
Ряд Б
)4COOR
СОС12
(CH,)4COOH
LXX
COCH,
NH|]H NHj
Y’AfcH^COOR
V'H
S' H
dl -Эпивмотин
II
NH,HHNH, HN^ hH NH-
/\ (ch2)4coor COCl2 (CH,
г)4СООН
R=CH3
s' н
LXXI
s н
dl - Эпиаллобиотин
IV
s н
мезо-а, 0-дибензиламиноянтарной кислоты (LXXIII); принципиальное его
отличие от предыдущих состоит в том, что первоначально на углеродном
скелете фумаровой кислоты (LXXII) строят имидазолидоновое кольцо (схе-
ма 103), а только затем тиофановое, после чего в молекулу вводят боковую
углеродную цепь. Характерно, что образующаяся в этом синтезе полная цис-
стереоизомерная форма биотина легко отделяется от эпибиотина с цос-кон-
фигурацией атомов азота, в то время как эпиаллобиотин и аллобиотин с
транс-конфигурацией атомов азота не обнаружены.
Фумаровая кислота (LXXII) в результате присоединения брома, испыты-
вая при этом изомеризацию, превращается в л<езо-дибромянтарную кислоту,
затем при действии бензиламина получается мезо-а, 0-дибензиламиноянтар-
ная кислота (LXXIII). С фосгеном в ней происходит замыкание имида-
зольного цикла, а затем из цис-1,3-дибензил-4,5-дикарбокси-2-оксоимидазола
(LXXIV) при восстановлении цинком в смеси уксусного ангидрида и уксус-
ной кислоты получается ангидрид, переходящий в циклическую альдегидо-
кислоту — имидазолидоновое производное 2-оксо-5-ацетокситетрагидрофу-
рана (LXXV). При действии сероводорода в присутствии хлористого водород
да соединение LXXV через промежуточную альдегиДокислоту превращает-
446
Схема 103
Синтез биотина через лезо-аф-дибензиламииоянтарную кислоту
СООН
СН=СН ---
СООН
LXXII
СО
R<^H
гс;
ср со
Вт
^н—СН
соон соон
Т Т СОС1,
сн сн---------------
I I
соон соон
LXXIU
со
RI\ Н^Н
\ НС1, HjjS
сн----сн ——
I I ~Н2О
СООН соон
LXXIY
сн соон
и
о
[H][Zn,(cH3Co)sO]
’"'CHjCOOCH со
о
LXXV
LXXY1
LXXX
R—CGHgCH2; R'=CsHs; Я'~остаток d-камфарсульфоновой кислоты
ся в производное тиофана, которое при последующем восстановлении дает тио-
лактон — 3,4-(Г,3'-дибензил-2'-оксоимидазолидо)-2-оксотиофан (LXXVI).
Реакция превращения 3,4-(Г,3'-дибензил-2'-оксоимидазолидо)-5-аце-
токси-2-оксотетрагидрофурана (LXXV) в гиолактон (LXXVI) подробно изу-
чена и значительно усовершенствована [591. Если раствор соединения LXXV
в диоксане периодически насыщать хлористым водородом и сероводородом на
холоду, то через 96 ч образуется 5-галогенопроизводное, которое восстанав-
ливают в 3,4-(1',3'-дибензил-2'-оксоимидазолидо)-2-оксотиофан (LXXVI);
выход составляет 75—80%. Предложены и другие варианты синтеза соеди-
нения LXXVI [60]. Затем в положение 2 этого соединения вводят боковую
алифатическую цепь.
По одному из вариантов [11, 58] соединение LXXVI конденсируют с
3-этоксипропилмагнийбромидом в со-этоксипропилтиофановое производное,
которое дегидратируют в тиофан с экзоциклической двойной связью, вос-
станавливаемый затем в соединение LXXVII. При обработке бромистым во-
дородом в уксусной кислоте оно дает циклический триметилентиофаний-бро-
мид (LXXVIII), в виде которого лучше всего произвести разделение опти-
ческих изомеров. Это достигается через производные £)-камфарсульфоно-
вой кислоты; полученные две диастериоизомерные формы разделяются крис-
таллизацией из изопренилового спирта; конфигурации биотина отвечает
(—)-3,4-( 1 ',3'-дибензил-2 '-оксоимидазолидо)-! ,2-триметилентиофаний-П-кам-
447
фарсульфонат (LXXIX), который при обработке натриймалоновым эфиром
в толуоле образует производное 2-(о>,<в-дикарбоэтоксибутил)тиофана (LXXX),
обладающее левым вращением. В результате кипячения соединения LXXX
с концентрированной бромистоводородной кислотой происходит гидролиз
эфирных групп и монодекарбоксилирование в боковой цепи, а также одно-
временное удаление двух защитных бензильных групп в виде бензилбро-
мида; d-биотин (I) образуется с высоким выходом.
Выход рацемического биотина можно значительно повысить, до 28%,
если в положение 2 3,4-(1',3'-дибензил-2-оксоимидазолидо)-2-оксотиофана
(LXXVI) ввести тетрауглеродную боковую цепь конденсацией с 4-бензил-
оксибутмлмагнийхлоридом [611 в среде бензола. Полученное соединение
LXXXI дегидратируют нагреванием с уксусной кислотой и последующим
каталитическим гидрированием с Pd на угле превращают в 3,4-(Г,3'-ди-
бензил-2'-оксоимидазолидо)-2-(о-бензилоксибутил)тиофан (LXXXII). При
нагревании в растворе 70%-ногоспирта, насыщенного хлористым водородом,
снимается только бензильная защита спиртовой группы алифатической бо-
ковой цепи и не затрагивается защитная группировка циклических имино-
групп; в результате реакции получается 3,4-(Г,3'-дибензил-2 '-оксоимидазо-
лидо)-2-(й-оксибутил)тиофан (LXXXIII). Дальнейшие его превращения в
биотин (I) осуществляют путем введения в боковую цепь карбоксильной
группы через бромид и нитрил (LXXXIV). Превращение дибензилбиотино-
нитрила (LXXXIV) в dZ-биотнн (I), при котором происходит гидролиз нит-
рильной группы и N-дебензилирование, осуществляют нагреванием с 48%-
ной бромистоводородной кислотой; выход на этой стадии составляет около
60% [611.
RN
co
/ , (CH,)4OR СНдСООН
<hH
LXXVI
CO
Г~\ H
на,
s oh
LXXXI
co
7 \ н
РВГз
. [H]/Pd
52=сн(сн^ *
(cHj)4OR
LXXXII
SX (СНг)4ОН
LXXXIII
Г \ H NaCN
Ч^СН^^г
роК
-н
ъ'^сн^см
LXXXIV
48%НВг
НМ н
-f?
з (спг)4соон
I
r=c6h5ch2
Расщепление d/ биотина на оптические антиподы и выделение d-биотина
[61 ] осуществляют с £<+>-аргинином по [43].
Другой вариант синтеза биотина, в котором исходят из 3,4-(Г,3'-ди-
бензил-2'-оксоимидазолидо)-2-оксотиофана (LXXVI), заключается в при-
менении для конденсации с этим веществом 1,4-бутилендимагнийгалогенида
[621.
К образующемуся по реакции Гриньяра первичному промежуточному
соединению присоединяется молекула двуокиси углерода, и после разло-
жения вторичного промежуточного соединения разбавленной соляной кис-
лотой с выходом 74% образуется 2-окси-Г1,М-дибензилбиотин (LXXXV).
Его дегидратируют нагреванием с уксусной кислотой в соединение LXXXVI
и затем восстанавливают в метиловый эфир дибензилбиотина (LXXXVII).
Следует отметить, что при гидрировании соединения LXXXVI на катализа-
448
торе Pd/C происходит десульфирование молекулы. Успешное гидрирование
удалось осуществить на кислотоустойчивом катализаторе — никель на ки-
зельгуре — в среде метилового спирта при температуре 160—180° С и дав-
лении 100 ат (10 МПа). Метиловый эфирдибензилбиотина (LXXXVII) гид-
ролизуют и дебензилируют нагреванием в 48%-ной бромистоводородной
кислоте в dZ-биотин [62] с выходом 49%, исходя из соединения LXXXV.
Проводились дальнейшие усовершенствования синтеза биотина на основе
соединения LXXVI [63].
со
уЛ;сн?\мевг СО,
5х OMgBr
rn^nr
со
(ciij'uCooMgBr
S'tMgBr
rn' №h
CO
7 \ и
48%HBr
-------«-Биотин
s
V OH
S (CHs^OOCHj
I
LXXXV
LXXXVI
LXXXVII
R- C^HgCH, !
БИОЛОГИЧЕСКОЕ ЗНАЧЕНИЕ БИОТИНА. ЗАВИСИМОСТЬ МЕЖДУ
СТРОЕНИЕМ И БИОЛОГИЧЕСКОЙ АКТИВНОСТЬЮ
Биотин содержится во всех животных и растительных тканях [64]
главным образом в связанном с белком состоянии; белковый комплекс био-
тина расщепляется папаином или кислотным гидролизом.
Биотин, витамин Н [34 , 35, 65], необходим человеку и животным как
биокатализатор реакций переноса двуокиси углерода в процессах метабо-
лизма. Он осуществляет свои биокаталитические функции в виде кофер-
мента.
Кроме того, биотин необходим для нейтрализации токсичности авидина,
кристаллического протеина сырого яичного белка, вытесняющего биотин
из ферментных систем и образующего с ним нерастворимый комплекс по
имидазолидоновой части молекулы; авидин является сильнейшим антивита-
мином биотина [66].
При воспроизведении искусственной недостаточности биотина на жи-
вотных наблюдается депигментация кожи, развитие дерматитов, выпадение
волос, нервные расстройства, торможение роста. У птиц наблюдается плохое
оперение и снижение яйценоскости. Одновременно авитаминоз сопровожда-
ется нарушением обмена белков, жиров и углеводов в организме.
Потребность человека в биотине составляет 0,1-—0,3 мг в сутки. Чело-
век и животные в условиях нормального питания не испытывают недоста-
точности биотина, так как микрофлора кишечника продуцирует его в ко-
личествах, необходимых для восполнения потерь макроорганизмом. Однако
в условиях интенсивного воспроизводства его нужно прибавлять в корм
птицы. Биотин применяют для лечения кожных заболеваний.
Как фактор, способствующий клеточному делению, биотин необходим
для высших растений [67, 681; он является ростовым фактором для многих
рас дрожжей, а также других микроорганизмов.
Биотин характеризуется высокой специфичностью. Небольшие измене-
ния его молекулы в сильнейшей степени снижают витаминную активность,
или полностью лишают соединение биологических свойств, или вызывают
появление антивитаминной активности.
Витаминной активностью обладает только одна оптически активная фор-
ма биотина, именно d(4~)-форма; как отмечалось выше, оптический антипод
15-69
449
природного биотина неактивен, так же как и другие стереоизомеры. Этери-
фикация карбоксильной группы с образованием метилового эфира не снижает
витаминных свойств биотина [71.
Однако ацилирование биотина вызывает понижение, а бензоилирова-
ние— полное лишение соединения биологической активности [18]. N,N'-
Дибензилбиотин также не обладает биологической активностью [69].
Соединения XI и XII, частично или полностью дегидрированные в тио-
фановом кольце молекулы биотина как ростовые факторы биологически не-
активны для Lactobacillus casei и некоторых других микроорганизмов [16,
70].
При окислении серы в молекуле биотина образуется биотинсульфон,
который проявляет ростовую активность для одних микроорганизмов; для
других же имеет антагонистические свойства, в том числе и по отношению
стимулирования оксибиотином. S-Окись биотина не обладает витаминной
активностью на животных [12, 13]. Интересно отметить, что при замене
в молекуле биотина атома серы кислородом, т. е. тиофанового кольца тет-
рагидрофурановым, получается dl-оксибиотин—2-(5-карбоксибутил)-3,4-уре-
илентетрагидрофуран (LXXX VIII) [71],
который сохраняет в определенной степени активность ростового фактора
для некоторых микроорганизмов (25—40%) и витаминную активность для
животных (5% для крыс и 17% для цыплят), так же как и некоторые произ-
водные оксибиотина [72, 73]. В организме оксибиотин, по-видимому, пре-
вращается в биотин [118].
Замена тиофанового кольца циклогексановым — соединение LXXXIX—
или бензольным — соединение ХС — дает аналоги биотина [74], лишенные
витаминной активности и даже обнаруживающие антагонистические свойст-
ва; это особенно относится к циклогексановому аналогу.
о о
А А
HN NH HN NH
LXXXIX
Характерно, что некоторые микроорганизмы могут удовлетворяться
осколками молекулы биотина и упрощенными соединениями, близкими по
строению к этим осколкам, обладая, по-видимому, способностью достраи-
вать молекулу биотина в процессе своей жизнедеятельности, например из
дезтиобиотина [75], или ограничиться этими упрощенными соединениями
как ростовыми факторами. Так, раскрытие имидазолонового кольца био-
тина с удалением одного атома углерода дает диаминокислоту (XIII), обла-
дающую 10% активности биотина. Раскрытие тиофанового кольца с удале-
нием атома серы дает дезтиобиотин (VIII), который на крысах проявляет
от 0,01 до 0,1% витаминной активности биотина [76]; он также обладает
частичной ростовой активностью биотина для некоторых микроорганизмов,
в. том числе для дрожжей.
450,
h,n NH,
Ч$/~(СН2)4СООН
о
HN^NH
CH, СН,(снД,СООН
XIII VIII
Однако дезтиобиотин является ингибитором роста Lactobacillus casei.
Синтезы дезтиобиотина (VIII) [31] и его производных описаны многими
авторами [77—791.
Следует отметить, что небольшой антибиотиновой активностью обладают
некоторые производные имидазолидона типа
о
HN NH
(сн^соон
п = 4 (цепь валериановой кислоты);
п — 5 (цепь капроновой кислоты);
п = 6 (цепь этантовой кислоты);
л — 7 (цепь каприловой кислоты).
В этом ряду слабая ростовая активность имидазолидонкапроновой кис-
лоты снижается еще более с увеличением или уменьшением числа метиле-
новых групп в боковой цепи при испытании на Saccharomyces cerevisiae и
Lactobacillus casei; в такой же последовательности уменьшается и способ-
ность связываться с авидином.
Вместе с тем некоторой ростовой активностью на микроорганизмах
[80] обладает еще более упрощенный дезтиобиотин, одновременно лишен-
ный карбоксильной группы, — гексаметилглиоксилидон (XCI) [81, 82].
Раскрытие обоих колец, имидазольного и тиофанового, с отщеплением одно-
го атома углерода и атома серы приводит к диаминопелларгоновой кислоте
(XX), сохраняющей в небольшой степени ростовую активность для неко-
торых микроорганизмов: ; '
Н(СН2)8СН8
XCI
HjN NH2
----СН
СН, СН2(СН2)4СООН
XX
Для Corynobacter diphtheric ростовым фактором служит даже осколок
тиофановой и боковой части молекулы биотина — пимелиновая кислота
[83] НООС(СН2)5СООН.
Биологическая активность некоторых аналогов биотина для S. cerevi-
siae (в % от активности d-биотина) составляет: (+)-дезтиобиотин — 100
[75], <1/-дезтиобиотин— 50—60 [77], метиловый эфир dZ-биотина— 100
[18], метиловый эфир dZ-оксибиотина — 10—16 [84], метиловый эфир S-оки-
си биотина ~100 [85], ^нс-3,4-диамино-2-карбоксибутилтиофан— 10 [5],
диаминопелларгоновая кислота — 10 [28]; эти же соединения для Lacto-
bacillus casei факторами роста не являются.
К антагонистам биотина относятся биотинсульфон, имидазолидонкар-
боновые кислоты, а также сульфокислые аналоги оксибиотина (ХСП) [86]
и дезтиобиотина (ХСШ) [87].
хси
СН, CH^CH^SCyi
хеш
15*
451
В последние годы из микроорганизмов выделены наиболее эффективные
природные антиметаболиты кофактора биотина —2уо-дегидробпотин(^-а-
дегидробиотин, XCIV), синтез которого осуществлен [88], и 2о-метил-
биотин(а-метилбиотин, XCV) [89]
XC1V
XCV
БИОТИНОВЫЕ КОФЕРМЕНТЫ
Биотин в составе ферментных систем принимает биокаталитическое учас-
тие в химических реакциях обмена веществ, протекающих в живом организм
ме [90, 91 ].
Коферментной формой биотина, в соединении со специфическими белка-?
ми образующей биотиновые протеиды, является N5-карбокси биотин-
(XCVI).
? »
НО Ns 3NH
CHjCHjjCHjCHjCCXJH
XCY1
По своему строению он представляет собой биотин, замещенный карбок-
сильной группой по 5-М-положению. Диметиловый эфир Ы5-карбоксибиотина
имеет т. пл. 131—132° С [92].
Биотин во многих природных продуктах, особенно в автолизате дрож-
жей, содержится в виде е- N-биотинил- £-лизина, биоцитина (V) [9, 64,
93], который обладает ростовой активностью по отношению ко многим
микроорганизмам [94]. Возможно, что это соединение является оско-
лком молекулы биотинового фермента [95]. Если предположить, что карбок-
сильная группа боковой углеродной цепи М5-карбоксибиотина связана пеп-
тидной связью с е-аминогруппой одного из лизиновых аминокислотных
остатков полипептидной цепи апофермента, то структуру Ь[5-карбоксибио-
тинфермента можно изобразить следующей формулой [96, 97 ]:
NHj
(сн^)4ССЙ4М(сН^4СНСО-фермею
(остаток)
Из этого Ng-карбоксибиотинфермента был выделен К5-карбоксибиоцитин
[98].
Простетическая группа биотиновых ферментов образует с апоферментом
прочные, недиссоциирующие связи.
СТРОЕНИЕ И СИНТЕЗ КОФЕРМЕНТА БИОТИНА
При действии на метиловый эфир биотина (XIV) хлоругольным эфиром
были выделены два изомерных эфира — диметиловый эфир М5-карбокси-
биотина (XCVII) с т. пл. 131—132° С и диметиловый эфир Ы3-карбоксибио-
тина (XCVIII) с т. пл. 141°С в соотношении 14 : 1 [92].
452
Различие в выходах диметиловых эфиров (XCVII и XCVIII) можно объя-
снить влиянием экранирования углеродной цепи положения 2 замещающей
группой при N(3>, поэтому выход диэфира(ХСУШ) мал. Это давало осно-
вание приписать природному соединению структуру эфира (XCVII). Он был
идентифицирован с дианилидом известной структуры (т. пл. 214—216° С)
путем действия на Ы5-карбоксибиотин анилинмагнийиодидом.
СИНТЕЗ БИОЦИТИНА
Биоцитин (V) синтезируют из хлорангидрида биотина (XCIX) посредст-
вом конденсации с «хелатным» медным комплексом Z.-лизина (СП) или с
a-N-формил-А-лизином (С) с последующим избирательным гидролизом по-
лученного соединения CI в биоцитин (V) [99].
NHCHO
HjN^Hj^CHCOOH
О
NHCHO
СН,)4СОНМ(сЦ>)р1СООН
CI
о
БИОХИМИЧЕСКИЕ ФУНКЦИИ КОФЕРМЕНТА БИОТИНА
Биотин как кофактор входит в состав ферментов: метилмалонил-КоА-
транскарбоксилазы, пируваткарбоксилазы, ацетил-, пропионил-, метил-,
кротонил-КоА-карбоксилаз [ 100 ].
Биотиновые ферменты катализируют обратимые реакции карбоксилиро-
вания, транскарбоксилирования, монодекарбоксилирования многооснов-
ных кислот, участвуя тем самым в биосинтезе липидов, аминокислот, угле-
водов, нуклеиновых кислот и в других реакциях обмена веществ. Однако
биотиновые ферменты катализируют реакции переноса двуокиси углерода
не на самом субстрате, а главным образом на стадиях промежуточных пре-
вращений коферментов, так называемых «активных ацетатов» — ацилпро-
изводных кофермента А (кофермента пантотеновой кислоты) [101, 102].
453
Изучение каталитических реакций, зависимых от биотиновых ферментов,
осуществлялось с использованием меченых атомов и применением авидина
для ингибирования активности биотина [103, 104].
Перенос двуокиси углерода протекает постадийно с предварительным
активированием Ы5-карбоксибиотинового фермента в присутствии ионов
Mg++ и Мп++ и образованием макроэргической «активной двуокиси углерода».
Необходимая для осуществления этой реакции энергия черпается из АТФ,
который при этом превращается в АДФ и ортофосфорную кислоту [105,
106].
Во второй фазе процесса Ы5-карбоксибиотин (XCVI) переносит на проме-
жуточный кофермент двуокись углерода, регенерируя при этом биотин (I).
Так, например, ацетил-КоА-карбоксилаза катализирует карбоксилирова-
ние ацетил-КоА (СШ) в малонил-КоА (CIV). Эта реакция является одной
из первых стадий биосинтеза жирных кислот [107—ПО].
о
+ со,
-^сн^соон
он
OH OH
+ ho-p-o-p-o-p-oh2c
II II
о о
II
о
он он
J—кн
+ н,о
IH
АТФ
он
о
< /-(СЦ^СООН
XCVI
он
I
НО—Р—О—Р—ОН,С
II
о
II
о
АДФ
N
MH,
OH OH
hJ-----kH
+ H,PO4
ОН
он
NH2, .
о=р(он),
о он
W i >, ?н
)--( + CHjCO-SCHjC^NHCOCHjCH^HCOCHC^H^CHjO-P-O-P-OCH.
^/ДСН^ООН
XCVI
СШ
о
он о
+ HOOCCHj.CO-SCHjCH^HCOCHiCHsNHCCJCHC^CH^jCH^y-P-O-P
н^оон ' ° °
I
он
CIV
МН,
‘2
('
14
мн
hcA'n'^Nh
Возможно, что активная форма биотина представляет собой биотин-аде-
нилат [111].
Дальнейшее включение малонил-КоА в цикл синтеза жирных кислот
происходит при каталитическом участии группы из 7 различных ферментов,
которые организованы в полиферментный комплекс — синтетазу жирных
кислот; синтетаза из дрожжей синтезирует в основном пальмитиновую и
стеариновую кислоты, а синтетаза из животных тканей—преимуществен-
но пальмитиновую кислоту. Реакция заключается в конденсации мало-
нил-КоА с ацетил-КоА в ацетомалонил-КоА [112]
454
• соон
| 2Н+; ге'
НООССНгСО ~ SKoA 4- СН,СО ~ SKoA----> СН3СОСНСО ~ SKoA ----->
—HSKoA
СООН] СООН
| 1 2Н+; 2е~
— СН3СН(ОН)СНСО ~ SKoA-----> СН3СН=ССО - SKoA----->
—Н2О
СООН
-> СНзСН^НСО ~ SKoA-----> CHSCH2CH2CO - SKoA
-со,
В последней фазе этих превращений образование бутирил-КоА проис-
ходит в результате отщепления двуокиси углерода (ранее введенной в мо-
лекулу ацетил-КоА). Декарбоксилирование этилмалонил-КоА происходит
при каталитическом участии биотиновых ферментов (биотин превращается
в Ы5-карбоксибиотин) с выделением энергии, которая аккумулируется в
АТФ (113J
СООН
СН,СН2^НСО - SKoA + АМФ + HSPO4 CH/H2CH2CO ~ SKoA +
+ СО2 + АТФ 4- Н2О
л Далее бутирил-КоА подвергается циклу превращений, подобному тому,
который ранее был изображен для ацетил-КоА и малонил-КоА. Так, посте-
пенно происходит наращивание углеродной цепи по два углеродных звена,
ф в конечном счете путем включения в синтез жирных кислот (содержащих С1в
и С18) уксусной кислоты в виде ацетил-КоА. Биотин участвует в образовании
насыщенных и ненасыщенных жирных кислот с 18 атомами углерода.
Пропионовая кислота, участвующая в реакциях, которые связывают жи-
ровой, белковый и углеводный обмены, в своей активной форме — пропио-
нил-КоА — карбоксилируется при участии пропионил-КоА-карбоксилазы
в метилмалонил-КоА [114, 115]
СНз
CHSCH,CO — SKoA + N5-CO2 — Биотин HOOCCHCO — SKoA + Биотин
Далее метилмалонил-КоА при каталитическом участии кофермента ви-
тамина В12, входящего в состав метилмалонил-КоА-мутазы, изомеризует-
ся в сукцинил-КоА
СН,
I
HOOCCHCO - SKoA«> НООССН2СН2СО -SKoA
В результате такого превращения пропионовой кислоты в янтарную кис-
лоту происходит ее включение в окислительный цикл трикарбоновых кис-
лот (см. с. 320).
Ng-Карбоксибиотин участвует в обмене аминокислот. Так, L-лейцин,
(CH3)2CHCH2CH(NH2)COOH, расщепляется до ацетоуксусной кислоты и
ацетил-КоА, проходя через стадию карбоксилирования р-метилкротонил-
КоА в Р-метилглутаконил-КоА [100, 116], катализируемую метилкротонил-
КоА-карбоксилазой, содержащей биотин в форме Ы5-карбоксибиотина
[92, 117]
СНз СН,
СН3С=СНСО - SKoA + N6-COS ~ Биотин НООССН2С=СНСО - SKoA + Биотин
На одной из стадий биосинтеза аспарагиновой кислоты,
HOOCCH2CH(NH2)COOH, пировиноградная кислота в виде пирувил-КоА
карбоксилируется в щавелевоуксусную кислоту при каталитическом уча-
стии пируваткарбоксилазы [118, 119]
455
CHsCOCO -SKoA + N(5, -CO2 ~
— Биотин HOOCCH2COCO — SKoA + Биотии
Пировиноградная кислота и метил малонил-КоА подвергаются траискар-
боксилированию при каталитическом участии биотинпротеида — метилма-
лонил-КоА-транскарбоксилазы, причем двуокись углерода переносится
с остатка метилмалоновой кислоты на пировиноградную кислоту с образо-
ванием щавелевоуксусной кислоты [120]
СНз
СНзСОСООН + HOOCCHCO—SKoA z± HOOCCHSCOCOOH + CH3CH2CO~SKoA
Щавелевоуксусная кислота вовлекается в окислительный цикл трикар-
боновых кислот (см. с. 320) или переаминируется с образованием аспара-
гиновой кислоты. Биотин в составе фермента находится в виде Ы(5)-кар-
боксибиотина [121 ].
Обзоры по биохимии биотина даны в различных статьях [122—124].
СПИСОК ИСПОЛЬЗОВАННОЙ
ЛИТЕРАТУРЫ (К ГЛАВЕ X)
1. V. d u Vigneaud, К- Н о f-
m а п n, D. М е 1 v i 11 е, Р. G у-
orgy. J. Biol. Chem., 140, 643
J (1941).
»"2. V. du Vigneaud. Science, 96,
; ‘ 455 (1942).
S. H a r r i s , D. Wolf, R. Mo-
’-fz i n g o, G. A г t h, R. A n-
derson, N. Easton,
K. F о 1 k e r s. J. Am. Chem.
Soc., 67, 2096 (1945).
4. Y. N a g a s e, U. Matsumo-
to, K. Sh i m a da. Yakugaku
Zasshi, 89, 595 (1969).
5. V. d u V i g n e a u d, К. H о f-
m a n n, D. M e 1 w i 1 1 e,
J. Rachel e. J. Biol. Chem.,
140, 763 (1941).
6. R. Ghosh, J.McOmie,
J. W i 1 s о n. J. Chem. Soc.,
1945, 705.
7. G. Brown, V. du Vigneaud.
J. Biol. Chem., 141, 85 (1941);
146, 351 (1942).
8. F. Kogi, L. P о n s. Z. physiol.
Chem., 269, 61 (1941).
9. R. Peck, D. Wolf, K. Fol-
k e r s. J. Am. Chem. Soc., 74,
1999 (1952).
10. D. Wolf, J. Valiant,
K. F о 1 k e r s. J. Am. Chem.
Soc., 73, 4142 (1951).
11. M. Goldberg, L. S t e r n-
b a c h. Пат. США 2489237; С. A„
45, 187 (1951).
12. L. W r i g h t, E. C r e s s о n,
J. V a 1 i a n t, D. Wolf,
K. F о 1 k e r s. J. Am. Chem.
Soc., 76, 4163 (1954).
13. D. Melville et al. J. Biol.
Chem., 208, 495, 503 (1954).
14. H. R u i s, D. M с С о r m i c k,
L. Wright. J. Org. Chem. 32,
2010 (1967); Methods Enzymol., 18,
386 (1970).
15. R. L e t t, A. M ar q ue t. C. r.
Acad. Sci., 268, 2348 (1969); Tetra-
hedron lett., 1971, 2851, 2855.
16. L. C h e n e у, J. R i e n i n g.
J. Am. Chem. Soc., 66, 1040 (1944);
67, 731 (1945).
17. F. Kogi. Ber., 68A, 16 (1935). f -
18. F. Ko g 1, В. T 6 n n is. Z. phy-
siol. Chem., 242, 43 (1936).
19. D. M e 1 v i 1 1 e, К. H of ma п n.^f
V. duVigneaud. Science, 94,
308 (1941); J. Biol. Chem., 141,
207 (1941).
20. D. M e 1 w i 1 1 e, К. H о f m a n n,“f •
V. duVigneaud. J. Biol,
Chem., 145, 101 (1942).
21. K. Hofmann, D. Melville,
V. du Vigneaud. J. Am.
Chem. Soc., 63, 3237 (1941).
22. H. H о f m a n n, D. M e 1 v i 11 e,
V. du V i g n e a u d. J. Biol.
Chem., 144, 513 (1942).
23. V. d u V i g n e a u d, К. H о f-
m a n п, В. M e 1 w i 1 1 e. J. Am.
Chem. Soc., 64, 188 (1942).
24. К . H о f m a n n, G. К i 1 m e r,
D. M e 1 v i 1 1 e, V. du Vi-
gneaud, H. Darby. J. Biol.
Chem., 145, 503 (1942).
25. G. Kilmer, M. Arms-
trong, G. В г о w n, V. du
Vigneaud. J. Biol. Chem.,
145, 495 (1942).
26. F. Kogi, T. de Man. Z. phy-
siol. Chem., 269, 81 (1941).
27. J. В о u g a u 1 t, E. C a t t e-
lain, P. C h a b r i e r. Bull,
soc. chim., France, 7, 781 (1940).
28. V. du Vigneaud, D. Mel-
ville, K. F о 1 k e r s, D. W о 1 f,
R. Mozingo, J. Kereszte-
s y, S. H a r r i s. J. Biol. Chem.,
146, 475 (1942).
29. D. M e 1 v i 1 1 e. J. Am. Chem.
Soc., 66, 1422 (1944).
30. D. M e 1 v i 1 1 e, A. Moyer,
К. H о f m a n n, V. du Vi-
gneaud. J. Biol.Chem., 146,487
(1942).
456
4
i
j
i
.•к
81. S. H a r r i s, * R. M ozi n go,
D. Wolf, A. Wilson, K. Fol-
ic e r s. J. Am. Chem. Soc., 67,
2102 (1945).
32. В. В a к e r, W. McEwen,
W. К i п 1 e y. J. Org. Chem., 12,
322 (1947).
33. F. Kogi. Naturwissensch., 25,
465 (1937).
34. P. G у б r g y, R. Kuh п, E. L e-
derer. J. Biol. Chem., 131, 745
(1939).
35. P. Gyorgy, C. Ros e,
K. Hoffmann, D. M e 1 v i 1 1 e,
V. du Vigneaud. Science, 92,
609 (1940).
36. D. Melville, K. Hofmann,
E. H a g u e, V. du Vig-
neaud. J. Biol. Chem., 142,
615 (1942).
37. S. H a r r i s, N. E a s t о n,
D. H e у 1, A. W i 1 son, К. Fol-
k e r s. J. Am. Chem. Soc., 66,
1757 (1944).
38. S. H a r r i s, D. W о 1 f, R. Mo-
zing o, K. F о 1 к e r s, Science,
97, 447 (1943).
39. С. Д. M и x н о, В. M. Б e p e-
зовский, H. А. Преобра-
жен с к и й. ЖВХО им. Менделее-
ва, 8, 357 (1963).
40. В. М. Березовский,
С. Д. М и х н о, Н. С. К у л а ч-
к и н а, В. В. Ж у к,
Н. А. Преображенский.
ЖОХ, 33, 2888 (1963).
41. С. Д. М и х н о, В. М. Б е ре-
зо в с к и й. Хим. гетер, соед.,
1968, 639
42. С. Д. М и х и о, Н. С. К у л а ч-
к и н а, В. М. Березовс-
кий. Хим. гетер, соед., 1966, 179.
43. D. W о 1 f, R. М о z i n g о,
S. Harris, A. Anderson,
К. F о 1 k е г s. J. Am. Chem. Soc.,
67, 2100 (1945).
44. F. К б g 1, Е. Т е и Н а Ш.
Z. physiol. Chem., 279, 141
(1943); Naturwissensch., 31, 208
(1943).
45. С. Д. М и х и о, В. М. Б е р е-
зовский, Н. А. Преобра-
жен с к и й. ЖОХ, 32, 2829
(1962).
46. С. Д. М и х н о, И. А. Со л у-
н и н а, В. А. Д е в я т н и н,
В. М. Березовский. ЖАХ,
22, 1419 (1967).
47. A. G г u s s n е г, J. В о и г-
q и i п, О. S с h n i d е г. Helv.
Chim. Acta, 28, 517 (1945).
48. О. S с h n i d е г, J. В о и r-
quin, A. Griissner. Helv.
Chim. Acta, 28, 510 (1945).
49. H. S c h m i d. Helv. Chim. Acta,
27, 128 (1944).
50. A. G г й s s n e r, J. В о и r-
q и i n, 0. S c h n i d e r. Helv.
Chim. Acta, 29, 770 (1946).
51. B. Baker, M. Q и e r r y,
W. M с E w e n, E. В e r n s t e i n,
S. S a f i r, L. D о r f m a n.
Y. Subbarow. J. Org. Chem.,
12, 186 (1947).
52. В. В a k e r, M. Q и e г г y,
S. S a f i r, W. M с E v e n,
E. В e r n s t e i n. J. Org. Chem.,
12, 174 (1947).
53. B. Baker, M. Querry,
S. S a f i r, E. В e r n s t e i n. J.
Org. Chem., 12, 138 (1947).
54. В. В a k e r, M. Querry,
E. В e r n s t e i n, S. S a f i r,
Y. S и b b a г о w. J. Org. Chem.,
12, 167 (1947).
55. G. В г о w n, M. A r m s t г о n g,
A. M о у e r, W. A n s 1 о w,
B. Baker, M. Querry,
E. В e r n s t e i n, S. S a f i r.
J. Org. Chem., 12, 160 (1947).
56. G. В г о w n, B. Baker,
E. Bernstein, S. S a f i r.
J. Org. Chem., 12, 155 (1947).
57. C. G г о b, H. S p r e c h e r.
Helv. Chim. Acta, 35, 885 (1952).
58. M. G о 1 d b e г g, L. S t er n-
b a c h. Пат. США 2489232—
2489236, 2489238; С. A., 45, 184—
187 (1951).
59. I. I s a k а, К. К и b о, M. Т а-
k ash i ma, M. Murukami.
Yakugaku Zasshi, 88, 1062 (1968).
60. M. G e r e c k e, J. Zimmer-
mann, W. Aschwanden.
Helv. chim. acta, 53, 991 (1970).
61. I. I s a k а, К. К и b о, M. T а-
kashima, М. Murukami.
Yakugaku Zasshi, 88, 1068 (1968).
62. I. I s a k а, К. К и b о, M. T a-
kashima, M. Murukami.
Yakugaku Zasshi, 88, 964 (1968).
63. M. Murakami, К. К и b o,
I. I s a k а. Яп. пат. 31669,
37776 (1970); С. A., 74, 100018,
100055 (1971).
64. В. Филлипов, М. Ильина.
ДАН СССР, 95, 1267 (1954).
65. V. du Vigneaud, D. Mel-
ville, P. G у 6 r g у, C. R о s e.
Science, 92, 62 (1940).
66. R. Eakin, E. Snell,
R. W i 1 1 i a m s. J. Biol. Chem.
136, 801 (1940); 140, 535 (1941).
67. F. К 6 g 1 , A. HaagenSmit.
Z. physiol Chem., 243, 209 (1936).
68. А. В и t e n a n d t. Ber., 75A, 1 86
(1942).
69. A. J a n s e n, P. S t о k e s. J.
Chem. Soc., 1962, 4909; 1964, 1530.
70. S. S a f i г, E. В e r n s t e i n,
В. В a k e r, . W. McEwen,
Y. Subbarow, J. Org. Chem.,
12, 475 (1947).
71. K. Hof in a n n. J. Am. Chem.
Soc., 67. 421, 738, 1165, 1459 (1945).
72. C. G г о b, F. R e b e r. Helv.
Chim. Acta, 33, 1776 (1950).
73. К. H о f m a n n. J. Am. Chem.
Soc., 66, 157 (1944).
74. J. E n g 1 i s h, R. Clapp,
Q. Cole, J. Halverstodt,
J. L a m p e n, R. R о b b i n. J.
Am. Chem. Soc., 67, 295, 2263
(1945).
457
75. D. М е 1 v i 11 е, К. D i t f m e r,
G. Brown, V. du Vi-
gne a u d Science, 98, 497
(1943).
76. G. E m e r s о n. J. Biol. Chem.,
157, 127 (1945).
77. J. Wo od, V. du V j g n e a u d.
J. Am. Chem. Soc., 67, 210 (1945).
78. R. Dushinsky, L. D о 1 a n.
J. Am. Chem. Soc., 67, 2079 (1945).
79. В. M. Родионов. Успехи хи-
мии, 20, 273 (1951).
80. M. Н. Al е й се л ь. Биохимия, 15,
361 (1949).
81. В. М. Родионов. Изв. АН
СССР, ОХН, 1945, 233.
82. В. М. Р од я он ов, В. К. 3 в о-
р ы к и н а. Изв. АН СССР, ОХН,
1943, 216.
83. J. М u 1 1 е г, J. S u Ь а г о w. J.
Bacterio!., 34, 153 (1937).
84. Т. W i п п i с к, К. Hol-
man п, F. Р i 1 g г i m, А. А х el-
r od. J. Biol. Chem., 161, 405
(1945).
85. V. du V i g n e a u d. Chem. Eng.
News, 23, 620 (1945).
86. К. H о f m a n n, A. В r i d g-
water, A. Axelrod. J. Am.
Chem. Soc., 71, 1253 (1949); пат.
США 2506594, 2520404; Zbl., 1951,
I, 1638, 1915.
87. R. D u s c h i n s к y, S. Ru-
bin. J. Am. Chem. Soc., 70,
2546 (1948).
88. G. F i e 1 d, W. Z a 1 1 y,
L. S t e r n b a c h. J. Am. Chem.
Soc., 92, 3520 (1970); пат. ФРГ
2008626; С. A. 73, 131047 (1970).
89. D. Al a r t i n, L. H a n к a.
L. R e i n e к e. Tetrahedron Lett,
1971, 3791.
90. J. К n a p p e. Deut. Med. Wochen-
schrift, 85, 1892 (1960).
91. А. А. Познанская,
В. 3. Горкин. Вопр. мед. хим.,
8, 115 (1962).
92. J. К п а р р е, Е. Ringel-
m a n n, F. L у n е n. Biochem.
Z„ 335, 168 (1961).
93. L. W r i g h t, E. C r e s s о n,
H. S k e g g s, T. W о о d,
R. Pec k, D. Wolf, K. Fol-
k e r s. J. Am. Chem. Soc., 74, 1996
(1952).
94. B. F i e s t о n e, S. К о s e r.
J. Bacteriol., 79, 674 (1960).
95. A. G i 1 g e n, F. L e u t h a r t.
Helv. chim. Acta, 45, 1833
(1962).
96. M. L a n e, F. L у n e n. Proc.
Natl. Acad. Sci. U. S., 49, 379
(1963).
97. F. Lyne n. Proc. Nat. Acad.
Sci. U. S., 49, 384 (1963).
98. J. К n a p p e, B. Wenger,
V. Vie ga n d. Biochem. Z.,
337, 232 (1963).
99. D. W о 1 f, I. V a 1 i a n t,
R. P e c k, K. F о 1 k e r s. J.
Am. Chem. Soc., 74, 2002 (1952).
100. F. L у n e n, J. К n a p p e,
E. Lorch. 5-й Межд. биохим.
конгресс, симпозиум IV, тетрадь 8,
Москва, 1961 г.
101. Н. L а г d у, J. Adler. J.
Biol, chem., 219, 933 (1956).
102. В. Bachhawat, AL Coon.
J. Biol. Chem., 231, 625 (1958).
103. K. F 1 e t c h e r, N. Al у a n t.
Nature, 188, 585 (1960). =' <
104. S. W a k i 1, E. T i t c h e n e r,
D. G i b s о n. Biochim. biophys.
Acta, 29, 225 (1958).
105. M. С a p 1 о w. J. Am. Chem. Soc.,
87, 5774 (1965).
106. M. С a p 1 о w, M. Y a g e r.
J. Am. Chem. Soc., 89, 4513
(1967).
107. D. G i b s о n, E. T i t c he-,
n e r, S. W a k i 1. J. Am. Chem..
Soc., 80, 2908 (1958). "«-j
108. S. W a k i 1. J. Am. Chem.
Soc., 80, 6465 (1958).
109. F. L у n e n, M. T a d a. Angew. лг
Chem., 73, 513 (1961).
ПО. M. W a i t e, S. W a k i I. X
Biol. Chem., 238, 77, 81J2
(1963). ?'
111. M. Vallotton, F. Le n- - Л
t h a r d t. Helv. chim. Acta, 47, ?
311 (1964). '
112. F. Ly nen. Angew. Chem., 77,
929 (1965).
113. M. L a n e, D. Halen zr-
D. К о s о w, С. H e g r e. J.’^S
Biol. Chem., 235, 3082 (1960).
114. E. S t a d t m а п, P. О v e<S>
rath, H. E g ge r e r, F. L у nen./
Biochem. Biophys. Res. Com-";?
muns, 2, 1 (1960).
115. D. H a 1 e n z, M. L a n e. J.
Biol. Chem., 235, 878 (1960).
116. F. L у n e n, J. К n a p p e,
E. L о г c h, G. Jutting,
E. Ringelmann. Angew.
Chem., 71, 481 (1959).
117. J. К n a p p e, K. Bieder-
b i c k, W. Bruemmer. Angew.
Chem., 74, 432 (1962).
118. M. U t t e r, D. К e e c h. J.
BioL chem., 235, PC17 (I960).
119. W. S e u b e г t, U. R em-
ber g e r. Biochem. Z., 334, 401
(1961).
120. R. S w i c k, H. W о о d. Proc.
NatL Acad. Sci. U. S., 46, 28
(1960).
121. H. W о о d, H. Lochmu 1-
ler, C. Riepertinger,
F. L у n e n. Biochem. Z., 337,
247 (1963).
122. С. E. Эпельбаум. Успехи
современной биологии, 26, 819
(1948).
123. Edith J u-H w a-C h u. Che-
mistry and Industry, 1948, 115.
124. H. L i c h s t e i n. Vitamines
and Hormones, vol. 9, 27; New
York, 1951; J. Bacteriol, 60, 485
(1950).
458
' ГЛАВА XI
ПТЕРИНОВЫЕ ВИТАМИНЫ И КОФЕРМЕНТЫ
ПТЕРОИЛ-Е-ГЛУТАМИНОВАЯ КИСЛОТА (ФОЛИЕВАЯ КИСЛОТА) И ЕЕ
ПОЛИПЕПТИДЫ
Птероил-£(+)-глутаминовая кислота —4'-N- [(2-амино-4-окси-6-птери-
дил)метил ]аминобензоил-£(+)-глутаминовая кислота, витамин Вс (I) —
впервые выделенная из листьев шпината и обычно называемая фолиевой
кислотой [1]
CHgCOOH
относится к производным конденсированных пиримидо[4,5-&]пиразиновых
циклических систем, которые носят наименование «птеридинов»1, и их 2-ами-
но-4-оксиптеридина, известного под названием «птерина», для которого
установлена лактамная таутомерная форма [3].
Из различных природных источников выделены полипептиды фолиевой
кислоты; птероил-£(+)-т,т-триглутаминовая кислота (ферментативный
L-casei-фактор) (I I) [4—6 ] — из культуры Corynebacteria — и птероил-£(-[-)-
гептаглутаминовая кислота, витамин Вс-конъюгат, витамин М (III) [7] —
из дрожжед
соон
«I
CHjCOHNCH
СН,
СН2СООН
СООН
*1
HNCH
в I
СН,
CHjCOOH
П~т е р о и л-£(+)-г лутаминовая кислота (I) представляет
собой тонкие бледно-желтые заостренной формы листочки, не имеющие
определенной температуры плавления (разложение около 250° С), вследст-
вие гигроскопичности желтеющие на воздухе. Птероил-£-глутаминовая
кислота воздушной сушки содержит кристаллизационную воду в количест-
ве, несколько меньшем, чем это соответствует ее дигидрату (т. е. меньше 8%).
Она не изменяется при сушке в вакууме до 140° С [8], но выше 145° С в
высоком вакууме теряет кристаллизационную воду. Птероил-£-глутамино-
вая кислота имеет в 0,1 н. NaOH спектр поглощения с максимумами абсорб-
ции при 255, 282, 365 нм; ЕрсМ 565, 350 и 195 соответственно [91. Максиму-
мы 255 и 365 нм относятся к птеридиновой части молекулы, а максимум
282 нм — к n-аминобензойной кислоте; £-глутаминовая кислота почти не
1 Терминология и нумерация атомов углерода птеридиновых соединений дана
в опубликованной работе [2J.
459
оказывает влияния на спектр поглощения фолиевой кислоты. Спектроско-
пические исследования фолиевой кислоты (фосфоресценция и парамагнитные
свойства) позволяют предположить некопланарность фрагментов ее мо-
лекулы [101. Удельное вращение птероил-£-глутаминовой кислоты fal^o
4-16° (7,6 г в 1 л 0,1 н. NaOH) [111.
Птероил-£-глутаминовая кислота очень мало растворима в воде: 10 мг
в 1 л (0,001 %) [12] или, по другим данным, 1 мг при 0° С и около 2 мг при
30° С в 1л [13]. Лучше она растворяется в кипящей воде — 500 мг в 1 л
(0,05%) [12], а также в ледяной уксусной кислоте, феноле, в метиловом
спирте, очень плохо — в этиловом и бутиловом спиртах и совсем нераство-
рима в ацетоне, эфире, хлороформе и углеводородах.
Присутствие электролитов, таких кай хлористый натрий и хлористый
кальций, повышает растворимость птероил-£-глутаминовой кислоты в вод7
ном растворе: при pH 2,8 растворимость в 1 мл — 3 мг при 80° С и 0,1 мг
при 5° С.
Концентрированная соляная кислота легко растворяет птероил-£-глу-
таминовую кислоту, которая почти полностью выделяется при разбавлении
водой. Она адсорбируется активированным углем, различными активными
глинами, углекислым барием; вымывание из адсорбентов производится раз-
бавленным раствором едкого натра или 3—5%-ным водным аммиаком.
В чистом виде птероил-£-глутаминовая кислота не флуоресцирует, для
растворов ее нечистых образцов характерна сине-зеленая флуоресценция,
зависящая от примеси простых птеринов.
ХИМИЧЕСКИЕ СВОЙСТВА ПТЕРОИЛ-£-ГЛУТАМИНОВОЙ КИСЛОТЫ
Птероил-£-глутаминовая кислота (I) в водных растворах щелочей или
карбонатов щелочных металлов образует одно-, двух- и трехосновные соли,
которые хорошо растворимы в воде. Растворимость динатриевой соли сос-
тавляет около 1,5% при 0° С; аммонийная соль хорошо растворима в водных
спиртах. Серебряная, цинковая, кадмиевая соли и соли тяжелых металлов
птероилглутаминовой кислоты практически нерастворимы в воде. Птеро-
ил-£-глутаминовая кислота из водных растворов солей осаждается при
pH 3—7.
Фолиевая кислота (I) с катионами Си++, Со++и Fe++ образует нераствори-
мые хелатные комплексы зеленого, желтого и красного цвета, соответствен-
но— соединение IV [14].
14
HOCXCH/THXHNHOCQH^HNCH/N
СООН |
О—Me—О
.CHjNHQH^COHNCHCHjCHjCOOH
СООН
Водные растворы птероил-£-глутаминовой кислоты на свету (особенно
ультрафиолетовом с X 365 нм) быстро разлагаются с образованием п-амино-
бензоилглутаминовой кислоты (VI) и 6-формилптерина, окисляющегося в
присутствии кислорода воздуха в 6-птеринкарбоновую кислоту (V); час-
тичное разложение наблюдается и для твердого продукта. В водных щелоч-
ных растворах, особенно быстро при pH 9,2, птероил-£-глутаминовая кис-
лота в аэробных условиях расщепляется с образованием тех же соедине-
ний (V и VI) [15, 16], однако в щелочных растворах в отсутствие кислорода
460
воздуха она устойчива. Фолиевая кислота легко расщепляется гидролити-
чески при нагревании с минеральными кислотами с образованием 6-метил-
птерина (VII) [17]. В качестве стабилизатора птероил-L-глутаминовой кис-
лоты в водных растворах применяется n-аминобензойная кислота или, еще
лучше, этилендиаминотетрауксусная кислота [18].
^оон HfA/V4*
HjN -е Vcohnch X Л J
ClipyZOOH HJN Xn X
Полипептиды фолиевой кислоты в щелочных растворах в отсутствие
кислорода воздуха отщепляют L-глутаминовую кислоту и превращаются
в фолиевую кислоту [19]. Незамещенный птеридин, лежащий в основе мо-
лекулы фолиевой кислоты, нестоек к сильным окислителям и расщепляется
с образованием производных гуанидина. При нагревании с серной кислотой
расщепление протекает с образованием пиразинового ядра, причем гомологи
птеридина расщепляются в более мягких условиях.
Окисление хлором приводит к расщеплению птероил-А-глутаминовой
кислоты на 6-птеринкарбоновую кислоту (V) и 4-амино-3,5-дихлорбензоил-
- L-глутаминовую кислоту [20].
В мягких условиях (в формамиде) хлор (или бром) замещает 3'- или 3',
б'-положения ароматического цикла без расщепления молекулы птероил-
-L-глутаминовой кислоты [21, 22]. При действии азотной кислоты нитру-
ется ароматический цикл птероил-Л-глутаминовой кислоты с образованием
соответствующих 3',5'-динитропроизводных [22].
Аминогруппы, замещающие положение 2 или 4 в пиримидиновой части
молекулы птеридина, неравноценны. 4-Аминогруппа по своим реакциям по-
добна ароматической аминогруппе и легко удаляется; 2-аминогруппа проч-
но связана с ядром и в обычных условиях при действии азотистой кислоты
не дезаминируется. Так, 1 моля нитрита натрия в холодном растворе соля-
ной кислоты достаточно, чтобы нитрозировать птероил-L-глутаминовую кис-
лоту (I) с образованием lO-N-нитрозоптероил-L-глутаминовой кислоты
(VIII), выделяемой в виде белого вещества [23]. Избыток азотистой кислоты
в более жестких условиях в смеси с уксусной и минеральной кислотами одно-
временно приводит и к дезаминированию 2-аминогруппы — соединение IX.
Однако почти полное дезаминирование наблюдается только в 4 н. сер-
ной кислоте в смеси с уксусной кислотой при 50° С [24].
При действии фенола и соляной кислоты 10-М-нитрозо-Д-птероилглутами-
новая кислота снова превращается в птероил-L-глутаминовую кислоту (I)
[23]. Другие заместители в положении 4 ведут себя аналогично заместите-
лям ароматического ядра, и гидроксильная группа при действии пятихло-
ристого фосфора замещается хлором, который легко гидролизуется щелочью
или элиминируется йодистоводородной кислотой.
Химические связи в положениях 6 и 7 пиразиновой части молекулы пте-
ридина неравноценны. Связь углерод — водороде положении6 более проч-
на, чем связь углерод —кислород или углерод — углерод. Обратное явле-
ние наблюдается для положения 7. Так, при электровосстановлении у лей-
коптерина элиминируется только 6-гидроксил [25]. При декарбоксилиро-
461
вании удаляется только карбоксильная группа, находящаяся в положе-
нии 6, и, например, 6-окси-7-птеринкарбоновая и 2,4-диокси-7-птеринкар-
боновая кислоты в обычных условиях не могут быть декарбоксилированы
[26, 27].
Птероил-L-глутаминовая кислота (I) при действии 80—98%-ной муравьи-
ной кислоты образует lO-N-формилптероил-Л-глутаминовую кислоту (X)
CI^CHjCOOH о сно сн2сн2соон
CryiNC6H4COHNCH CH^QH/lOHbCH
СООН---* L 4 J COOH
, H,N N N
[28], которая под влиянием едкого натра при комнатной температуре отще-
пляет муравьиную кислоту и вновь превращается в исходное соединение I
Характерным свойством птероил-L-глутаминовой кислоты, как и других
сложных птеринов, является ее способность к восстановительно-окислитель-
ным превращениям, связанным с передачей водорода в восстановительно-
окислительных ферментных системах [30]. Она легко восстанавливается в
дигидровитамин с присоединением двух атомов водорода. Образующееся
лейкосоединение в отличие от витамина, окрашенного в желтый цвет, бес-
цветно. Присоединение водорода к замещенному птерину I с образованием
лейкосоединения происходит по С(7>—С(в> двойной связи пиразинового кольца
[9]. При осторожном гидрировании в уксусной кислоте образуется 7,8-ди-
гидро- (XI) и 5,6,7,8-тетрагидроптероил-Л-глутаминовая кислота (XII)
R=CeH4COHNCHCH2CH2COOH
соон
эти соединения неустойчивы и легко дегидрируются даже кислородом
воздуха в птероил-£-глутамнновую кислоту (I).
Стремление к ароматизации пиразинового цикла столь велико, что ди-
гидроптеридины, содержащие в положении 6 или 7 оксиметильные, амино-
метильные или галогенометильные заместители, в отсутствие кислорода
воздуха отщепляют воду, аммиак или галогеноводород и перегруппировы-
ваются в птеридины. •
Птероил-L-глутаминовая кислота (I) при восстановлении в минерально-
кислой среде претерпевает расщепление на 6-метилптерин (VII) и п-амино-
бензойную кислоту.
Свойства простых птеринов рассмотрены в обзорах 12, 32—34].
Для аналитического определения птероил-/.-глутаминовой кислоты, ее
аналогов и производных служит химический метод [35], основанный на опре-
делении ароматического амина, образующегося наряду с 6-метилптерином
в результате количественного восстановительного расщепления, проводи-
мого цинковой пылью с 0,5 н. соляной кислотой. Рекомендуется прибавле-
ние небольшого количества (0,005%) желатина для стабилизации освобо-
ждающегося амина. n-Аминобензойная кислота определяется путем ее ди-
азотирования, азосочетания с Ы-(а -нафтил)этилендиамином и колориметри-
рования полученного красителя, имеющего характерный Хмакс 550 нм [36].
На измерении флуоресценции продуктов окисления птероил-/.-глутами-
новой кислоты основан метод обнаружения малых количеств витамина в био-
логических материалах [37—39].
462
ПРИРОДНЫЕ ВЕЩЕСТВА ПТЕРИНОВОЙ СТРУКТУРЫ
Простейшие соединения птеринового ряда впервые были обнаружены
в чешуйках крыльев бабочек Гопкинсом [40 ] в 1890 г. в виде белого, желтого
и красного пигментов. Так как природные птерины с трудом подвергаются
очистке, не плавятся при высокой температуре и при определении элемент-
ного состава сжигаются с трудом [32], то для установления их строения
потребовалось в течение последующих 50 лет провести большое количество
исследований, многие из которых, к сожалению, приводили к ошибочным
выводам. Только начиная с 1940—1941 гг., после того как были получены
чистые соединения и анализу подверглись вещества, высушенные при
170° С в вакууме, был установлен истинный состав ксантоптерина и дру-
гих простейших природных птеринов [41, 42].
Из природных источников, главным образом из чешуек крыльев бабочек,
микроорганизмов и др., выделены производные птерина: ксантоптерин
(XIII), нзоксантоптерин (XIV), лейкоптерин (XV), биоптерин (XVI), ихтио-
птерин (XVII), эритроптерин (XVIII), экаптерин (XIX), лепидоптерин (XX),
сепиаптерин (XXI), птероиновая кислота (XXII), ризоптерин (XXIII), пте-
рородин (XXIV) и др.
Ксантоптерин, 2-амино-4,6-диоксиптеридин (XIII), — желтый
пигмент крыльев бабочек—лимонниц; он также находится среди конечных
продуктов обмена веществ животных, но не обнаружен в растениях [43 ].
Ксантоптерин (XIII) представляет собой порошок желтого цвета, не пла-
вящийся до 350° С. Он очень плохо растворим в воде (1 : 40 000) и в органи-
ческих растворителях. Для его водных растворов характерна флуоресцен-
ция. Ксантоптерин обладает амфотерными свойствами и ведет себя как двух-
основная кислота и однокислотное основание [44], но свойства его более
основны, чем изоксантоптерина (XIV) и лейкоптерина (XV) [45]. Строение
ксантоптерина установлено на основании реакций его расщепления [40, 41,
46, 47] и превращений в другие соединения. Он находится не в окси-, а
в оксоформе, и его структура отвечает 2-амино-4,6-диоксо-3,4,5,6-тетрагид-
роптеридину (XIII).
Ксантоптерин синтезирован из 2,4,5-триамино-6-оксипиримидина и мо-
нохлоруксусной кислоты [48], дихлоруксусной кислоты [42] или полу-
ацеталя глиоксиловой кислоты [49], а также из 2-амино-4-хлор-6-оксипири-
мндина, хлористого фенилдиазония и эфира аминоуксусной кислоты [50].
Нзоксантоптерин, 7-оксиптерин, 2-амино-4,7-диоксиптеридин
(XIV), и лейкоптерин, 6,7-диоксиптерин, 2-амино-4,6,7-триоксипте-
рин (XV), — бесцветные пигменты чешуек крыльев бабочек-капустниц.
Оба этих соединения находятся в оксоформе. На основании данных УФ-спек-
тров поглощения структура лейкоптерина отвечает 2-амино-4,6,7-триоксо-
гексагидроптеридину [51 ]. Строение лейкоптерина (XV) было окончательно
установлено Пуррманом [42] в результате синтеза из 2,4,5-триамино-6-окси-
пиримидина и щавелевой кислоты.
Биоптерин, 6-А-эритро-Г,2'-пропилптерин (XVI) [52, 53], вы-
делен из человеческой мочи [54], из бабочек Drosophila melanogaster [55],
Ephestia kuhniella [56] и микроорганизмов Crithidia fasciculata [54].
463
о
R
XXVIU
о
HN
tyr
ГТК"!
s''ch(o^ci{3oh
xxix
OH OH OH
R- CHj-(t—c—CHjOH
Анн
Синтез биоптерина (XVI) осуществлен из 2,4,5-триамино-6-оксипирими-
дина и рамнотетрозы [57] или 5-дезокси-£-арабинозы [58], а также другими
методами.
И х т и о п т е р и н, 6-(а,р-диоксипропил)изоксантоптерин (XVII)
[59], — вещество с фиолетово-синей флуоресценцией; выделено из кожи рыб
[60]. ।
Эритроптерин (XVIII) — оранжево-красный пигмент бабочек
Catopsilia argante [61 ].
Эритроптерин — ростовой фактор для некоторых микроорганизмов
[62]; он выделен из туберкулезных бактерий [63]; труднорастворим в воде, 1
обладает более основными свойствами, чем ксантоптерин. При восстановле-
нии эритроптерин переходит в бесцветное лейкосоединение [44]. Строение
464
эритроптерина на основании спектрофотометрических данных отвечает
структуре XVIII [64] (ср. [62]).
Эритроптерин (XVIII) синтезирован из 2,4,5-триамино-6-оксипиримиди-
на и ацетощавелевой кислоты [62] или из ксантоптерина (XIII) и пирови-
ноградной кислоты [65,661.
Из эритроптерина (XVIII) путем его восстановления боргидридом нат-
рия и последующего окисления получен экаптерин (XIX), а при действии
аммиака—лепидоптерин (XX) [66].
Экаптерин (XIX) выделен из головок мучной моли Ephestia kiih-
niella [56].
Лепидоптерин (XX), кетимин эритроптерина [67], выделен
также из Ephestia kuhniella [56].
Сепиаптерин (XXI) — желтый флуоресцирующий птерин; вы-
делен из Drosophila melanogaster [68].
Птероиловая кислота, 4'-N- [(2-амино-4-окси-6-птеридил)-
метил]аминобензойная кислота (XXII), усиливает рост микроорганизмов
Streptococcus faecal is R.
Ризоптер ин, lO-N-формилптероиновая кислота (XXIII), выделен
из культуральной жидкости грибка Rhizopus nigricans [69], который являет-
ся продуцентом фумаровой кислоты из глюкозы.
Птерородин (XXIV) выделен из крыльев бабочек Appias пего
[70].
6-(1 ',2',3',4',5'-Пентаоксипентил-2'-р-глюкуронид)птерин (XXV) вы-
делен из Mycobacterium smegmatis [71 ].
6-А миноптерин, 2,6-диамино-4-оксиптеридин (XXVI) выделен
из Drosophila melanogaster, культуры Anacystis nidulans и др. [72]. Выде-
лены также дрозоптерин [73] и другие птерины.
У рот ион (XXIX) — оранжево-красные кристаллы; la Id —12°;
выделен в 1943 г. из человеческой мочи [74]; уточненная структура уста-
новлена в 1967 г. [75].
Различные природные производные 5,6,7,8-тетрагидроптероил-Л-глу-
таминовой кислоты, принимающие участие в качестве кофакторов в биохи-
мических превращениях, рассматриваются в разделе «Птериновые кофер-
менты».
Помимо производных птерина, в природных источниках обнаружены
различные другие птеридины. Так, из дрожжей Eremothecium ashbyii
выделены 6-метил-(1'-О-рибитил)-2,4,7-триоксиптеридин (XXVII) и 6,7-ди-
метил-8-(1'-О-рибитил)-2,4-диоксиптеридин (XXVIII) —• биологический
предшественник рибофлавина [76, 77].
СТРОЕНИЕ ВИТАМИНОВ ГРУППЫ ФОЛИЕВОЙ КИСЛОТЫ И НЕКОТОРЫХ
ПТЕРИНОВ
Строение птероил-£(+)-глутаминовой кислоты (печеночного L.
casei-фактора, витамина Вс)
При окислении нефлуоресцирующей птероил-£-глутаминовой кислоты
(I) [78] или ее диметилового эфира хлорноватистой кислотой образуется
кристаллическое флуоресцирующее вещество; при дальнейшем окислении
получается гуанидин [79]. Такое же флуоресцирующее вещество получено
при расщеплении «ферментативного L. casei-фактора». Так, при щелочном
расщеплении без доступа воздуха ферментативный L. casei-фактор (II)
дает L-глутаминовую кислоту и птероил-£-глутаминовую кислоту (I) (схе-
ма 104). При дальнейшем гидролизе из нее в присутствии воздуха образу-
ется два вещества в равномолекулярных количествах: ароматический амин
(XXXIII) и двухосновная кислота, оказавшаяся 6-птерин карбоновой кисло-
той (V) с рДа 3,9 и 7,7; она обладает сильной флуоресценцией и показывает
наличие пиримидинового ядра с аминогруппой в положении 2, что доказы-
465
I
I'
I
I
вается образованием гуанидина при расщеплении этого вещества хлорной
водой и последующем гидролизе [19]. Проба Вебера [80] на гуанидин давала
положительную реакцию, проба Сулливана [81 ], позволяющая определять
только гуанидин, но не метилгуанидин, также давала положительную реак-
цию [19], в то время как проба Сакагуши [82] на метилгуанидин, но не сво-
бодный гуанидин давала отрицательную реакцию.
Из двух групп кислотного характера в этом соединении V наличие одной
в виде карбоксильной группы доказывалось выделением при декарбоксили-
ровании двуокиси углерода. Полученное при этом флуоресцирующее веще-
ство, содержащее вторую группу кислотного характера (>С=СЬ±>С—ОН)
с рКо 8,0, оказалось идентичным 2-амино-4-окси-3,4-дигидроптеридину
(XXXVI), синтетически полученному из 2,4,5-триамино-6-окси-1,6-дигид-
ропиримидина (XXXV) и глиоксаля [83] (схема 104). Этим синтезом
установлена природа цикла и положение 2-амино- и 4-оксигрупп; показано,
уго карбоксильная группа находится в положении 6 пиразинового цикла
При расщеплении L. casei-фактора (I) в кислой среде в отсутствие воз-
духа наблюдалось образование другого продукта расщепления — 6-метил-
птерина (VII) [19] (схема 104); строение этого соединения доказано рас-
щеплением его в свою очередь при действии раствора едкого натра при
170° С, приводящего к расщеплению пиримидинового цикла с образованием
2-амино-3-карбокси-5-метилпиразина (XXXI), который после декарбокси-
лирования дает 2-амино-5-метилпиразин (XXXII) [83]. Строение соедине-
ния VII было доказано также по его идентичности с продуктом де-
карбоксилирования 6-птеринуксусной кислоты [19].
Схема 104
Установление строения птероил-£-глутами новой кислоты реакциями расщепления
[o](NaOH), Ю00
W H2SO4
Ayh0
XXX
HOOCyVCH3 NHa
XXXI
__HN
HjN
COOH
о
-CO2
, I COOH
HjN-\ y-COHNCH
СИ,
CHjCOOH
H2o
54)
I
COOH
HjNCH
b
CHjCOOH
XXX1Y
HjN
XXXIII
COOH
о
HjN N N
NHa
NH»
XXXY
CHO
CHO
Образование 6-метилптерина (VII) одновременно с 6-карбоксиптерином
(V) также наблюдалось при сульфитном расщеплении ферментативного
466
L. casei-фактора (II) [41 и при стоянии со щелочью в отсутствие воздуха
птериновой фракции, полученной при кислом анаэробном расщеплении
этого вещества.
Для объяснения образования 6-метилптерина [11] предложен следующий
механизм реакции. Первоначально из соединения I в результате перемеще-
ния атомов водорода в пиразиновый цикл образуется ди гидрооснование Шиф-
фа (XXXVII), которое затем гидролизуется в 6-дигидроптеринальдегид
(XXXVIII); последнее соединение в результате реакции Канниццаро пе-
реходит в дигидроптеринкарбоновую кислоту (XXXIX) и дигидроптерин-
карбннол (XL). Перегруппировка этого соединения с внутримолекулярным
отщеплением молекулы воды приводит к получению 6-метилптерина (VII),
а образующаяся при этой дисмутации дигидроптеринкарбоновая кислота
(XXXIX) кислородом воздуха дегидрируется в 6-карбоксиптерин (V).
КИСЛОТЫ
6-Дигидроптеринальдегид при конденсации с п-аминобензоил-А-глутами-
новой кислотой может превращаться в птероил-L-глутаминовую кислоту [841.
Помимо 6-птерин кар боновой кислоты (V), при окислении птероил-А-глу-
таминовой кислоты хлорноватистой кислотой образуется вещество, при рас-
щеплении которого получается 3,5-дихлор-4-аминобензойная кислота и
DL-пирролидонкарбоновая кислота, являющаяся у-лактамом глутаминовой
кислоты, следовательно, амином, входящим в молекулу птероил-£-глутами-
новой кислоты, является п-аминобензоил-£-глутаминовая кислота (VI,
схема 104). Прямое доказательство этого было получено при обнаружении
соединения VI в продуктах аэробного щелочного расщепления фермента-
тивного L. casei-фактора. Соединение VI гидролизовалось при нагревании
с серной кислотой; из раствора экстракцией этилацетатом была выделена
n-аминобензойная кислота (XXXIII) [19], идентифицированная по отсутст-
вию депрессии температуры плавления пробы смешения с известным ве-
ществом и микробиологически с Acetobacter suboxydans. п-Аминобензой-
ная кислота (XXXIII) образуется и при сульфитном расщеплении фермен-
тативного L. casei-фактора [4]. Второй атом азота был определен как об-
разующий пептидную связь азот а-аминокислоты, которая была идентифи-
цирована как £(+)-глутаминовая кислота (XXXIV) (см. схему 104).
Доказательство положения связи птеридинового ядра с ароматической
частью молекулы было получено на основании следующих данных. При
восстановлении происходило расщепление исходной молекулы витамина на
ароматическую и птериновую части; последняя представляет собой 6-метил-
птерин (VII). Образование первичного ароматического амина и птерина при
гидролизе в щелочной и кислой среде при сульфитном расщеплении и при
гидрировании делают вероятным связь этих соединений по ароматичес-
кой аминогруппе. Птериновая часть присоединяется по шестому углеродному
атому, так как при расщеплении образуются 6-метилптерин (VII), 6-карбок-
сиптерин (V) или промежуточный 6-формилптерин. Поэтому атом углерода,
467
соединяющий птеридиновое и ароматическое ядра, представлен в виде ме-
тиленовой группы, что находится в соответствии с поглощением кислорода
при щелочном расщеплении и с поглощением водорода при восстановитель-
ном образовании 6-метилптерина.
Строение птероил-А(-[-)-глутаминовой кислоты (I) доказано ее синтезом
в 1945 г. [85, 86] из 2,4,5-триамино-6-оксипиримидина (XXXV), л-амино-
бензоилглутаминовой кислоты (VI) и а, 0-дибромпропионового альдегида
как непосредственной конденсацией этих соединений в ацетатном буфере в
присутствии кислорода воздуха [85], так и путем предварительного полу-
чения промежуточной четвертичной аммониевой соли пиридина с а, р-ди-
бромпропионовым альдегидом [86].
Строение птероил-£(+)-7,т-триглутаминовой кислоты (ферментативного
L. casei-фактора)
Структура этого соединения II была установлена путем расщепления
в кислой и щелочной среде и выделения ряда продуктов, показавших спектры
поглощения, сходные со спектрами ряда известных птеринбв. Ферментатив-
ный L. casei-фактор (II) при щелочном расщеплении без доступа воздуха
первоначально дает рацемическую птероил-ОА-глутаминовую кислоту («пе-
ченочный L. casei-фактор») и 2 моля дикарбоновой а-аминокислоты (глу-
таминовой), выделяемой в виде бариевой соли или этилового эфира. При
дальнейшем расщеплении в присутствии воздуха были получены п-амино-
бензойная кислота (ХХХШ) и 6-птеринкарбоновая кислота (V) [19, 83,
86]. Кроме того, в результате гидролиза L-пирролидонкарбоновой кислоты,
образующейся при нагревании ферментативного L. casei-фактора при pH 4,
была получена £(+)-глутаминовая кислота (XXXIV) [4].
Таким образом, в молекулу ферментативного L. casei-фактора входят пте-
риновая часть (см. выше), л-аминобензойная кислота и три молекулы
£(4-)-глутаминовой кислоты, соединенные в виде пептида.
Окончательное строение ферментативного L. casei-фактора как птероил-
у-глутамил-у-глутамилглутаминовой кислоты (II) (у,у-трипептида) было
СООН
VcOHNCH СООН
J • I
CH,CH,COHNCH соон
CH,CH,COHNCH
И СН,СН,СООН
установлено в результате синтеза всех пяти возможных изомеров и
испытания их на микробиологическую активность [87—91 ]. Строение изо-
мерных трипептидов отвечает следующим формулам:
соон 1 соон соон
CO-HNCH Г 1 R-HNCH 1 1 CO.-HNCH
CO-HNCH 1 1 сн, сн2 соон 1 л 1 R-HNCH СН, J 1 *
R-HNCH СН, । । * сн2 СН, CO-HNCH 1 * f г сн, сн2 соон
СН, СН2 СООН CO-HNCH СН, СН, СООН СН, сн, СООН СН, соон cL,cC -Трипептид СООН •у,сС-Трипептид CH2CHjCOOH CO-HNCH СН. CO-HNCH 1 1 соон сн, сн, соон об,у -Трипептид
R-HNCH СООН JI N CHjHN-/~VcO
*= 1 Т У
СН, СООН H2N">|^>|
CO-HNCH
CHjCHjCOOH
сС,с£-Диглутаминовая кислота
468
Строение ризоптерина (фактора Streptococcus lactis R.)
Потенциометрическим титрованием в ризоптерине (XXIII) установлено
присутствие карбоксильной и слабой кислой группы [29]. Он образует "моно-
ацетильное, метоксиацетильное, фен ила цетильное и бензоильное производ-
ные. При действии щелочных растворов образуется динатриевая соль дез-
формилризоптерина с формулой C14H10N6O3Na2, являющаяся солью птерои-
новой кислоты, апоризоптерина (XXII)
Микробиологическая активность при этом снижается до 10% первоначаль-
ной [92].
Наличие 2-аминопиримидинового ядра в апоризоптерине (XXII) дока-
зывалось получением оксалилгуанидина при окислении хлорноватокислым
калием в присутствии соляной кислоты [29], а также подобием спектров
поглощения с птероил-L-глутаминовой кислотой, отличающихся только
интенсивностью [92].
Ризоптерин содержит одну первичную аминогруппу в пиримидино-
вом ядре, что определялось по реакции с нитритом натрия и соляной
кислотой. При окислении апоризоптерина наряду с оксалилгуанидином
образуется n-аминобензойная кислота, которая выделена из продуктов
щелочного гидролиза в виде n-ацетаминобензойной кислоты [93].
При образовании птероиновой кислоты (XXII) из ризоптерина (XXIII)
одновременно образуется муравьиная кислота [29]. Формильная группа
связана с атомом азота остатка n-аминобензойной кислоты; это вытекает
из тех соображений, что ризоптерин содержит только одну группу, обра-
зующую ацильное производное, а птероиновая кислота присоединяет две
ацильные группы, т. е. ацилированию подвергается аминогруппа птериди-
нового ядра и вторичная аминогруппа фенильного остатка [29].
Помимо формильной группы, птеринового производного и п-аминобен-
зойной кислоты молекула ризоптерина содержит еще один атом углерода,
который является связующим звеном между птериновой и ароматической
частями молекулы. Окончательным подтверждением строения ризоптерина
послужил его синтез нз птериновой и муравьиной кислот.
СИНТЕЗ ПТЕРОИЛ-ЬГЛУТАМИНОВОЙ КИСЛОТЫ
В соответствии со строением птероил-£-глутаминовой кислоты (II)
ее синтез осуществляется или путем непосредственного соединения отдельных
звеньев в одну стадию: пиримидинового производного, трехуглеродного ком-
понента и п-аминобензоил-£-глутаминовой кислоты по схеме А-фБф-В
(метод 1), или постепенным наращиванием молекулы на основе п-ампнобен-
зоил-£-глутаминовойкислоты, предварительно соединяемой с трехуглерод-
ным компонентом, а только затем — с пиримидиновым производным по схе-
469
ме А+БВ (метод 2), или, наконец, путем наращивания молекулы на основе
пиримидинового производного, предварительно конденсируемого с трех-
углеродным компонентом в птериновое производное с активной углеродной
группой в положении 6, затем соединяемое с п- аминобензоил-Б-глутами-
новой кислотой по схеме АБ-J-B (метод 3).
1. Синтез фолиевой кислоты построением ее молекулы трехкомпонентной
конденсацией
По одному из методов синтез птероил-L-глутаминовой кислоты (I) про-
водят одностадийной конденсацией (схема 105) 2,4,5-триамино-6-оксипирими-
дина (XXXV), 2,3-дибромпропионового альдегида (XLI) и п-аминобен-
зоил-А(4 )-глутампновой кислоты (VI) [15, 94].
Схема 105
Синтез птероил-Б-глутамкиокой кислоты из 2,4,5-триамино-б-оксипиримндина,
п-амииобемзоил-Б-глутамииовой кислоты и 2,3-дибромпропиоиового альдегида
/CHjBr
ВгСН
ОНС Б
XU
соон
COHNCH -----------—
I
снг
снасоон
Он соон
UM X^N CHjHNCeH^OHNCH
III" &
H>N N N xLU CHjCOOH
О соон
им X/ N _CHjHNC6H4COHNCH
- L IL J ch2
HjN N N ! CH2CO0H
Реакция идет через промежуточное образование 5,6-дигидроптероил-
L-глутаминовой кислоты (XLII) неоднозначно, сопровождается побочным
образованием 6-метилптерина (VII) и 6-оксиметилптерина (XLV) [95] и,
по-видимому, протекает следующим образом [95]: первоначально в буфер-
ном растворе при pH 4 2,3-дибромпропионовый альдегид (XLI) конден-
сируется с 2,4,5-триамино-6-оксипиримидином (XXXV) и образуется
5,6-дигидро-6-бромметил птерин (XLIII), который в условиях реакции пре-
терпевает ряд последующих превращений: а) конденсацию с п-аминобен-
зоил-Б-глутаминовой кислотой (VI) в 5,6-дигидроптериол-Б-глутамино-
вую кислоту (XLII); б) дегидробромирование и последующую изомери-
зацию семициклической двойной связи таутомерной формы 6-метилпте-
рина, приводящие к 6-метилптерину (VII); в) гидролиз в соединение
(XLIV) и последующее дегидрирование, дающие 6-оксиметилптерин (XLV)
[95].
470
Для дегидрирования промежуточной 5,6-дигидроптероил-А-глутами-
новой кислоты (XLII) в фолиевую кислоту (I) применяют йод [94, 96],
двухромовокислый натрий или калий [89], уксуснокислую ртуть [97],
или, что менее совершенно, кислород воздуха. Общий выход фолиевой
кислоты с чистотой около 30% составляет не выше 25% [941.
Синтезом птероил-А-глутаминовой кислоты-9-14С с 14С-2,3-дибром-
пропионовым альдегидом, содержащим меченый атом в группе СН2 Вг,
доказано, что метиленовый мостик птероил-А-глутаминовой кислоты об-
разуется за счет СН2Вг-группы 2,3-дибромпропионового альдегида [98],
а не из альдегидной группы. Была синтезирована также птероил-А-глу-
таминовая кислота с меченым атомом углерода (2-14С) в пиримидиновом
адре [99].
При проведении синтеза одностадийной конденсацией к водному раст-
вору смеси н-аминобензоил-А-глутаминовой кислоты (VI) и 2,4,5-три-
амино-6-оксипиримидина (XXXV) постепенно прибавляют спиртовой раствор
2,3-дибромпропионового альдегида (XLI). Если же прибавлять п-ами-
нобензоил-А-глутаминовую кислоту к смеси остальных реагентов, то вы-
ход уменьшается [15]. Выход несколько увеличивается, если предваритель-
но 2,4,5-триамино-6-оксипиримидин и спиртовой раствор 2,3-дибромпропио-
нового альдегида кипятить в водном растворе уксуснокислого натрия, а
затем продолжить кипячение с n-аминобензоил-А-глутаминовой кислотой
[100]. Оптический антипод (птероил-£(—)-глутаминовая кислота) был по-
лучен из тех же компонентов [101 ] в токе азота с небольшим выходом [102].
Конденсацию проводят также с восстановлением, однако выделение осуще-
ствляется в присутствии кислорода воздуха.
Интересно остановиться на некоторых особенностях рассматриваемого
синтеза. В 2,4,5-триамино-6-оксипиримидине (XXXV) 5-аминогруппа пири-
мидинового ядра неравноценна 4-аминогруппе. Так, ацилирование протекает
предпочтительно в положении 5 [48,103] вследствие большей реакционной
способности сильноосновной 5-аминогруппы по сравнению с 4-аминогруп-
пой; в химии пиримидина для такого характера ацилирования неизвестно ни-
каких исключений [26, 103]. Однако, когда в реакцию с 2,4,5-триамино-6-
-оксипиримидином вводятся соединения с двумя или несколькими группами
или атомами, активными к конденсации с аминогруппами, то наблюдается
двойственное течение реакции, зависящее от реакционной способности
этих групп или атомов и от характера среды.
По-видимому, бром при атоме углерода положения 2 молекулы 2,3-ди-
бромпропионового альдегида (XLI) не настолько активнее альдегидной
группы этого соединения в условиях реакции, чтобы преимущественно кон-
денсироваться с 5-аминогруппой пиримидинового ядра и привести к высо-
кому выходу птероил-А-глутаминовой кислоты (I). Несомненно, что конден-
сация 5-аминогруппы 2,4,5-триамино-6-оксипиримидина протекает и с аль-
дегидной группой, а это приводит к образованию 7-птероилглутаминовой
кислоты (XLVI). Ее строение доказано синтезом из 7-бромметилптерина и
n-аминобензоил-А-глутаминовой кислоты [104].
соон
।
XLYI
CH2CHSCOOH
Однако 7-птероилглутаминовая кислота (XLVI) и ее дегидроформа ла-
бильны как в кристаллическом виде, так и в растворах, кислых и особенно
щелочных и легко подвергаются расщеплению. В нестойкости 7-птероилглу-
таминовой кислоты находится причина ее отсутствия в качестве примеси
471
в птероил-L-глутаминовой кислоте [104]. Спектр поглощения 7-птероилглу-
таминовой кислоты идентичен со спектром птероил-£-глутаминовой кисло-
ты (I).
2,3-Дибромпропионовый альдегид (XLI) применяют в виде его бисульфит-
ного производного [105]; он может быть заменен его ацеталем [106], 3-эт-
окси-2-бром(хлор)пропионовымальдегидом [1071,2 : З-оксидо-1-пропионил-
дихлоридом [108] и толуолсульфокислым эфиром глицеринового альдеги-
да; реакцию проводят в присутствии йодидов [109].
Чтобы исключить образование промежуточной дигидроформы птерина
(XLII), получающейся при конденсации пиримидинового ядра с дигалоген-
альдегидами, применяют 2,2,3-тригалогенпропионовый альдегид и 1,1,3-
тригалогенацетон [ПО, 111], конденсация с которыми приводит к непо-
средственному образованию птеридинового цикла.
При конденсации 2,4,5-триамино-6-оксипиримидина (XXXV), п-амино-
бензоил-£-глутаминовой кислоты (VI) и 1,1,3-трнхлорацетона (XLVII)
в водном растворе при pH 4 птероил-L-глутаминовая кислота образуется
с выходом всего около 1,4% [8]. j
Прибавление бисульфита натрия предохраняет трихлорацетон от разложе-
ния, в результате чего общий выход фолиевой кислоты с чистотой 60% повы-
шается до 37%; бисульфиту натрия приписываются не только антиокисли- '
тельные функции, но и каталитические [8]. Этот синтез имеет преимущества
перед многими другими.
О СООН о соон
zCHjCI H2NC6H4COHNCH HN></M<%/CHiiHNC6H4COHNCH
.T cq x о -t _____ ПП т I |
11 + f *' CH2 11 J CH2
H2N N NHj CHO, CHXOOH H.N N N C^COOH
XXXV XLVII VI I
Применение в качестве трехуглеродного компонента тетрагалогензаме-
щенных производных: 1,3-дибром-1,1-дихлорацетона [112], 1,1-дибром-
3,3-дихлор-и 1-бром-1,3,3-трихлорацетона [113] — для конденсации с сое-
динениями XXXV и VI в присутствии бисульфита натрия приводит к не-
большому повышению выхода фолиевой кислоты (с последними двумя сое-
динениями).
Если заменить трихлорацетон 1,1-дихлор-З-бромацетоном, то выход пте-
роил-А-глутаминовой кислоты снижается до 15% [8,114], а с 1,1,3-трибром-
ацетоном при конденсации в водной среде в присутствии гликоля выход пте-
роил-£-глутаминовой кислоты составляет только 6—14 % [ 111 ], в спиртовой
среде выход снижается еще больше. Как показано синтезом с 1,1,3-три-
бромацетоном-3-14С [Вг14СН2СОСНВг2], метиленовая группа птероил-L-глу-
таминовой кислоты на 37% образуется за счет СН2Вг-группы и на 63% за
счет СНВг2-группы трибромацетона [115].
Аналогичным образом из 2,4,5-триамино-6-оксипиримидина (XXXV),
п-аминобензойной кислоты и 1,1,3-трибромацетона получается птеронновая
кислота с выходом 11% [111]; применение 1,1-дихлор-З-бромацетона повы-
шает ее выход до 17%.
Если производить конденсацию не с тригалогенацетоном, а с 2,2,3-три-
бромпропионовым альдегидом кипячением в спирто-водном растворе уксус-
нокислого натрия, то выход птероил-А-глутаминовой кислоты остается низ-
ким и не превышает 9% [111]. Применение в этой реакции малых количеств
гидросульфита связано с защитой галогенальдегида от окисления [116].
Показано, что для синтеза птероил-Д-глутаминовой кислоты можно ис-
пользовать 1,1,3-тригалоген-З-ацплоксиацетон (выход около 7%) [117],
1,1-дигалоген-2,3-эпоксипропан(выходменее 1 %) [118] и 1,2-дихлор-З-ими-
нопропан [119]. Для синтеза применяют также З-хлор-2-нитрозопропионовый
альдегид, получаемый непосредственно в реакционной среде из акролеина
472
и хлористого нитрозила, образующегося при прибавлении в реакционную
6 среду амилнитрита и соляной кислоты [120], и диоксиакролеин—НОСН=
i =С(ОН)СНО [121].
Так как получаемая по методам 1, 2 и 3 сырая птероил-L-глутаминовая
кислота содержит примесь родственных птеринов, вопросу ее очистки уде-
I лено значительное внимание. Основной принцип очистки основан на полу-
> чении солей птериновых соединений с основаниями при pH выше 12, пре-
1 имущественном осаждении примесей при pH выше 10,2 и осаждении обога-
щенной птероил-А-глутаминовой кислоты при pH 3—4. Применяют очень
разбавленные растворы (1 : 1000—10 000). Очистку повторяют многократно,
з. затем производят обработку активированным углем и перекристаллизацию
из воды при pH 3 [15]. Известны методы очистки через натриевые, магние-
1 вые, бариевые, цинковые и другие соли [87, 88, 122—130].
В некоторых методах для очистки применяют переосаждение из концен-
трированной соляной кислоты с последующим разбавлением водой [94, 131],
а также адсорбцию на кислой окиси алюминия и выделение раствором фос-
> фата при pH 6 [11].
f Птероил-А-глутаминовая кислота также очищается через ее lO-N-фор-
S мильное производное (X) с последующим гидролизом [132—134].
i 2. Синтез фолиевой кислоты построением ее молекулы на ароматическом
< цикле
|
Следующая группа синтезов птероил-£-глутаминовой кислоты (I) осно-
вана на конденсации 2,4,5-триамино-6-оксипиримидина (XXXV) с уже
готовой частью молекулы птероил-£-глутаминовой кислоты, содержащей
трехуглеродную систему с двумя функциональными группами или атомами,
[ способными реагировать с аминами (схема 106). В качестве такого соедине-
ния применена Н-[п-(2,3-диоксиен-2-пропилиденамино) бензоил l-L-глутами-
новая кислота (XLIX) [135], получаемая из п-аминобензоил-L-глутаминовой
I кислоты (VI) и редуктона (XLVIII).
Схема 106
Синтез птероил-А-глутамииовой кислоты из 2.4,5-триамино-6-оксипиримидииа,
л-амииобеизоил-А-глутаминовой кислоты и редуктона
HN
ноч/сно
С
п
носн
D
XLVIH
о
ДкНг
H2N N Д NH,
XXXV
С
И
НОСН
YI
соон
СН3СООН
БВ
XUX
СН.
но ch=nc6h4cohnch
СН„СООН H2N
СООН
н
N ch=nqh4cohnch
уС.. । Фолиевая
СН2 *"кислота
" хххи. '
Реакция протекает через дигидрооснование Шиффа (XXXVII). Общий
выход птероил^-глутаминовой кислоты по этому методу составляет 10—15%.
Первичная аминогруппа л-аминобензоил^-глутаминовой кислоты (VI)
в условиях синтеза птероил-L-глутаминовой кислоты легко подвергается
окислению, что приводит к понижению выхода. С целью ее защиты поль-
зуются n-толуолсульфокислотой. Применяя при алкилировании п-аминобен-
зоил^-глутаминовой кислоты, защищенной тозильной группой, замещенные
пропиленоксиды, получают соединения, которые при конденсации с
2,4,5-триамино-6-оксипиримидином дают фолиевую кислоту (I) [136—140].
473
Для этого из ацеталя акролеина при действии хлорноватистой кислоты с
последующим элиминированием хлористого водорода твердым едким нат-
ром с выходом 60% получают ацеталь 2 : 3-оксидопропионового альдегида
(LI), который применяют для алкилирования диэтил-(М-тозил-п-аминобен-
зоил)глутамата (L) в присутствии щелочного катализатора (схема 107).
Образующийся диэтил-(П-тозил-П-а-окси-р, р -диэтоксипропил-n- аминобен-
зоил)глутамат (L11) при окислении хромовым ангидридом дает кетон (LIII),
который при конденсации с 2,4,5-триамино-6-оксипиримидином (XXXV)
в присутствии уксуснокислого натрия образует диэтиловый эфир N-10-то-
зил птероил-L-гл утамиповой кислоты (LIV, схема 107). После детозилиро-
вания 30%-ным бромистым водородом в уксусной кислоте в присутствии фе-
нола (без фенола птероил-А-глутаминовая кислота неустойчива к 30 %-ному
бромистому водороду [136, 137 ]) с последующим гидролизом эфирных групп
получают птероил-L-глутаминовую кислоту (I) с выходом 40—50%, исходя
из соединения L [137].
Схема 107
Синтез птероил-А-глутамииовой кислоты из 2,4,5-триамиио-6-оксипиримидииа,
диацеталя 2 : 3-оксидопропиоиового альдегида и замещенного
п-аминобензоил-А-глутаминового эфира
CHjCOOR
О
СНлСНСН(ОИ)г
U
III
COOR
COHNCH
СН,
CHjCOOR
-
H2N"T< NM2
XXXY A
Ts ._. COOR
1 /7А 1
zCH,N-< 7-COHNCH •
=C CH2
CH(or)2 БВ CH,CO
1Ш
Ts COOR l) 3096-ный НВг, o COOH
N CHNC^COHNCH 2>SoJJ°H HINTS' %^HNWOHNCH
Д CH2 —------------------------- A A J <4
N uv CH2COOR HjN N N j снсоон
Если в реакции применить соединения со свободной альдегидной груп-
пой, а не защищенной в виде ацеталя, то выход птероил-Л-глутаминовой
кислоты (I) уменьшается до 15% [137].
При использовании для синтеза пропиленоксидов:
OCH2CHCH2R,
где R=C1, ОСОСН3, ОСН3, ОСНО, выходы получаются меньшие, чем с аце-
талем 2 : 3-оксидопропионового альдегида (LI) [136].
По такой же схеме получают и птероиновую кислоту путем замены про-
изводных п-аминобензоил-А-глутампновой кислоты производными п-амино-
бензойной кислоты. Птероиновая кислота может быть превращена в ее хлор-
ангидрид, а затем в птероил-L-глутаминовую кислоту конденсацией с эфи-
ром L-глутаминовой кислоты с последующим гидролизом [141].
Для синтеза фолиевой кислоты (I) предложен диэтил-(Ы-тознл-Ы-а-
-бром-р -оксипропил-п-аминобензоил)глутамат [ 142 ].
К этому же методу синтеза птероил-£-глутаминовой кислоты (I) из ком-
понентов А-|-БВ относится синтез, в котором исходят из 2-амино(дезокси)-
474
-5,5-дибромбарбитуровой кислоты (LV), конденсируемой с п- [N-(2,3-ди-
аминопропил) ]аминобензоил-L-глутаминовой кислотой (LVI) [143].
соон
CHjHNQH/ZOHNCH
* HjNCH СН,
HjNCHj CHjCOOH
о соон
n^X^N^CHjHNCsH/rOHNCH
” A A J см,
h,n n n сн,соон
3. Синтез фолиевой кислоты построением ее молекулы иа пиримидиновом
цикле
Известен ряд синтезов птероил-L-глутаминовой кислоты, в которых исхо-
дят из замещенных птеринов, имеющих в положении 6 углеродсодержащие
функциональные группы или атомы, способные конденсироваться с амино-
группой второго компонента, такие, как оксиметильная, галогенметильная,
альдегидная и др.
Для постепенного построения молекулы птероил-L-глутаминовой кис-
лоты первоначально из 2,3-дибромпропионового альдегида и пиридина по-
лучают соль четвертичного аммониевого основания (LVII), которую конден-
сируют с 2,4,5-триамино-6-оксипиримидином (XXXV) в промежуточное
дигидропроизводное (LVIII), окисляющееся в процессе реакции в полностью
ароматизированное соединение LIX (схема 108) [144]. Реакцию проводят в
среде ацетона с прибавлением йода, растворенного в водном растворе йоди-
стого калия [145]. 5-Аминогруппа 2,4,5-триамино-6-оксипиримидина
(XXXV) взаимодействует с бромом в а-положении четвертичного аммоние-
вого основания (LVII) с образованием четвертичной аммониевой соли 6-ме-
тилдигидроптерина (LVIII).
Для получения птероил-£-глутаминовой кислоты четвертичнуюсоль (LIX)
конденсируют с п-аминобензоил-£-глутаминовой кислотой (VI) в растворе
метилата натрия в этиленгликоле [ 144 ]. Однако этот метод дает относитель-
но малый выход (4—10%) птероил-L-глутаминовой кислоты (I) с 25%-ной
чистотой.
Схема 108
Синтез птероил-£-глутаминовой кислоты из 2,4,5-триамино-б-оксипиримидииа,
п-аминобензоил-Л-глутаминовой кислоты и 2,3-дибромпропионового альдегида,
применяемого в виде соли четвертичного аммониевого основания
Соединение LIX при восстановлении цинковой пылью в щелочной среде
с последующим точным окислением марганцовокислым калием образовавше-
гося тетра гидросоединения дает с хорошим выходом 6-метилптерин (VII)
1145—147].
475
Из других замещенных птеринов применяют 6-галогенметилптерин, ко-
торый конденсируют с п-аминобензоил-А-глутаминовой кислотой или ее
эфиром в птероил-£-глутаминовую кислоту в присутствии муравьиной кис-
лоты [147—149]. 6-Галогенметилптерин получают или действием хлористого
тионила на 6-птеринкарбинол (XLV) [148, 149] или галогенированием 6-ме-
тилптерина (VII) [147].
Если применить для конденсации с п-аминобензоил-Е-глутаминовой
кислотой (VI) 2-меркапто-4-окси-6-хлорметилптеридин (LX) и полученное
соединение LXI ацетилировать йодистым метилом в 2-алкилмеркаптопро-
изводное (LXII), то после насыщения аммиаком получается также птероил-
L-глутаминовая кислота (I) [150].
R*=C^H4COHNCHCH2CH2COOH
Для конденсации с п-аминобензоил-Е-глутаминовой кислотой или ее
динатриевой солью применяют также непосредственно 6-птеринкарбинол
(XLV) [151—153]. Катализаторами этой реакции являются уксусная кислота,
метилат натрия и безводный хлористый цинк [152]. Конденсация протекает
также в муравьиной кислоте при одновременном гидрировании с палладие-
вым катализатором [153].
Можно 6-формилптерин (XXX) конденсировать также с п-аминобензо-
ил-£-глутаминовой кислотой (VI) при одновременном каталитическом гид-
рировании в присутствии платинового или палладиевого катализатора [11,
154, 155] по схеме 109.
Схема 109
Синтез птероил-А-глутаминовой кислоты из 6-формилптерииа и п-амииобензоил-А-
глут аминов ой кислоты
СООН
HNC^COHNCH
СН,
СН.СООН
VI 2
в
соон
N CH=NC6H4COHNCH
J "
N ,Х11| СНСООН
H,N
СООН
N CHjHNCeH/TOHNCH
АН сн,
н н Х1| СН2СООН
О соон
Д.И ^CH3HNC6H/:OHNCH
- A I J
H,N N N [ СН2СООН
Первоначально образующееся Шиффово основание птероил- L-глутами-
новой кислоты (LXIII) гидрируется, причем происходит также гидрирова-
ние пиразинового цикла; промежуточное соединение XII дегидрируют
йодом или другими окислителями в птероил-Е-глутаминовую кислоту (I)
с общим выходом около 20% [11, 154]. Конденсация проводится в присут-
ствии бикарбоната натрия, ацетата натрия или муравьиной кислоты при
50° С [11, 154, 155]. В этом синтезе промежуточное шиффово основание
(LXIII) устойчиво только в кислой среде (в отличие от обыкновенных осно-
ваний Шиффа); в условиях реакции птероил-Л-глутаминовая кислота под-
вергается расщеплению. На примере синтеза птероиновой кислоты установ-
лено [156], что выход фолиевой кислоты может быть значительно повышен,
476
если взять большой избыток ароматического компонента и образующееся
шиффово основание подвергнуть восстановлению боргидридом натрия.
Схема 110
Синтез птероил-£-глутами новой кислоты из 2-М-ацетнл-6-формилптерина и
л-аминобеизоил-£-глутаминовой кислоты
О
X ,NHj
HN у *
\ NHj
XXXV
о
HjN
HN
(с,Н^о)2СНСНВгСНВгОС2Н5-
LXV
cr%|NaHC°3
CHBrCH(OC2Hs)2
СНО
LXVI
NaHCO,
(CH.CO).о
(C2HS6)2CI ICI l=CHOC2Hs
LXIV
HjO,
63%
85%
CH.COHN
LXVil COOH
л I
o HNQHjCOHNCH
JL^N.CHO CHjCHjCOOH
^OHN N’
CHj
N
LXIX
О
HN
HjN N N
N
LXVIII
Ч^снЬс,|кйон
95%
COOH
,CH=NCeH4COHNCH
CHjCOOH
CjHySH
54%
CHjCOHN' X xn
p
HN
COHN N N
4
CHj
o COOH
A.N. .CH,HNCbH4COHNCH
CHjCOOH
COOH
HCOOH |
56% ’
CHjCOHN
LXX о
НС
CHNCsH/TOHNCH
<fn2
CH2COOH
LXXI
86%
0,lH.NaOH
Фолиевая
кислота
I
1з
В другом синтезе [157] аминогруппу 6-формилптерина защищают ацети-
лированием (схема ПО). Первоначально 1,3,3-триэтоксипропен LXIV) бро-
нируют в инертном растворителе в бромоэфир (LXV), затем подвергают это
соединение частичному гидролизу в водном растворе бикарбоната натрия
в 2-бром-3,3-диэтоксипропиоальдегид (LXVI), который конденсируют с
2, 4,5-триамино-6-оксипиримидином (XXXV). Образующийся промежуточ-
ный дигидроптеридин окисляется перекисью водорода в 2-амино-4-окси-6-ди-
этоксиметилптерин (LXVII). После ацетилирования этого соединения и по-
следующего гидролиза ацетальной группы соединения LXVIII с муравьиной
кислотой получают 2-М-ацетил-6-формилптерин (LXIX) [157, 158]. Кон-
денсация птеринальдегида (LXIX) с п-аминобензонл-£-глутаминовой кис-
лотой (VI) в присутствии n-тнокрезола или других веществ, содержащих
тиольную группу, как восстановителей промежуточно образующегося азо-
метина приводит к получению 2-М-ацетилптероил-£-глутаминовой кислоты
(LXX); в том случае, если в качестве восстанавливающего агента применяет-
ся муравьиная кислота, образуется 10-М-формил-2-М-ацетилптероил-£-глу-
таминовая кислота (LXXI) [157, 159]. Оба эти вещества в результате гид-
ролиза образуют 98—100%-ную птероил-£-глутаминовую кислоту (I),
не требующую сложной очистки. Общий выход фолиевой кислоты из соедине-
ния VI составляет 35,9% при применении п-тиокрезола и 31% — муравьи-
ной кислоты. В аналогичной реакции для синтеза птероиновой кислоты
в качестве восстановителя предложен диметиламиноборан (CH3)2HN->-BH3,
что несколько повышает выход [160].
477
Для конденсации с п-амннобензоил-£-глутаминовой кислотой в среде пи-
ридина или диоксана в присутствии n-тиокрезола или других меркаптосое-
динений с успехом можно применять неацилированный 6-формилптерин
(XXX); реакция приводит непосредственно к получению фолиевой кислоты
(I) 1159].
Для конденсации с п-аминобензоил-А-глутаминовой кислотой предло-
жена также 6-птерин-а-бромуксусная кислота; реакция протекает с отщеп-
лением углекислоты [13, 161].
Вызывает интерес рассмотрение реакций образования замещенных пте-
ринов, необходимых для осуществления синтезов по методу 3.
6-Птеринкарбинол (XLV) получают конденсацией 2,4,5-триамино-6-окси-
пиримидина (XXXV) с диоксиацетоном (LXXII) в присутствии гидразин-
гидрата и уксусной кислоты [148, 162] через промежуточный 7,8-дигидро-
-6-птеринкарбинол (LXXIII).
о
A NHj
HLjN NHa
XXXV
ZCH,OH
+ со
СН2ОН
LXXII
В этом случае, как и во многих других, более реакционноспособная 5-ами-
ногруппа пиримидинового ядра преимущественно конденсируется с карбо-
нильной группой. Однако если конденсацию проводить в отсутствие окисли-
телей, то 6-оксиметилптерин (XLV) получить не удается — образуется 6-ме-
тилптерин (VII). Эта реакция носит общий характер.
, Конденсация диаминопиримидинов с алифатическими производными с
целью получения дигидроптеридинов, в положении 6 или 7 которых ожи-
даются заместители типа СН2ОН, CH2NH2, CH2NHC6H4COOH [11, 163, 164]
и СН2На1 [164], в отсутствие кислорода воздуха приводит к полностью аро-
матизированным птеридинам с отщеплением элементов воды, аммиака,
n-аминобензойной кислоты [11,163, 164] или галогеноводорода [164]. Так,
при конденсации 2,4,5-триамино-6-оксипиримидина (XXXV) с различными
соединениями, содержащими три атома углерода, первоначально образует-
ся оксиметил- (XLIV и LXXV) или галогенометилдигидроптерины (LXXIV
и LXXVI), которые затем превращаются преимущественно в полностью аро-
матизированные метилптерины (VII и LXXVII) с незамещенной метильной
группой. Реакция проводится в отсутствие легко окисляющих или дегид-
рирующих агентов и протекает с потерей воды или галогеноводорода, при-
чем получающийся промежуточный метиленптерин перегруппировывается
в метилптерин [164].
HOHjCCOCHjOH
ОНССНВгСН2Вг
ОНССНС1СН„С1
онссн(он)сн2он
ВгН2ССОСН„ОН
ВгН2ССОСН2Вг
С1Н2ССОСН2С1
О
CH2
о
HN^
HSN N IN'
YU
H (Hat)
Л CH,OH
H -
HjN'V "H
XLIV (LXXIY)
LXXY1I
H.N IN IN CH2OH
н (Hal)
LXXY (LXXYl)
H2N IN N CH3
В зависимости от характера применяемых трехуглеродных компонен-
тов конденсация протекает в двух направлениях — с образованием 6- или
7-производных птерина. Конденсация соединения XXXV с 2,3-дибромпропио-
новым альдегидом приводит к получению 6-метилптерина (VII) с 90%-ным
выходом [84]; так же протекает реакция с диоксиацетоном [149]. При
478
изменении условий реакции . наблюдается образование 7-метилптерина
(LXXVII) [15, 149, 165].
Компонентами, содержащими три атома углерода и применяемыми в
конденсации, могут быть 1,3-дибромацетон [84], глицериновый альдегид
[166], 2,3-дихлорпропионовый альдегид, производные а-бромтетроновой
кислоты (ОНСН2СОСНВгСООН), лактон которой при гидролизе в концен-
трированной соляной кислоте, по-видимому, отщепляет углекислоту с обра-
зованием 1-бром-З-оксиацетона, вступающего затем в реакцию [164].
6-Метилптерин (VII) необходим для получения 6-галогенометилптерина.
Для его синтеза может служить и конденсация 2,4,5-триамино-б-оксипири-
мидина с ацетолом (СН3СОСН2ОН) в присутствии гидразина [165] или
с метилглиоксалем в водном растворе в присутствии сульфита и бисульфита
натрия при pH 7 [167]. Если метилглиоксаль заменить 1,1-дихлорацетоном,
то в присутствии уксуснокислого натрия образуется 6-метилптерин [168],
нос 1,1-дибромацетоном образуется 7-метилптерин [84].
Для получения 6-формилптерина (XXX) исходным продуктом может
служить 6-полиоксиалкилптерин (LXXVIII, п — 3), подвергаемый мягкому
окислению; для окисления применяют тетраацетат свинца, Pb3O4 HNO3
и ШО4 [11, 154, 155, 169]. Строение 6-птеринальдегида доказывалось по-
лучением из него 6-птеринкарбоновой кислоты при окислении щелочным
раствором марганцовокислого калия [170, 171 ], а также превращением при
действии щелочи по реакции Канниццаро в 6-птеринкарбоновую кислоту
(V) и 6-оксиметилптерин (XLV) [169, 172].
6-Формил птерин (XXX) можно также получить из 6-мети л птерина (VII)
действием на него бромом в 48%-ной бромистоводородной кислоте при 150° С
с последующим гидролизом образующегося дибромметилпроизводного
(LXXIX) [147, 172.1
XXX ¥
| | Реакция Канниццаро I
LXXIX XLV
Реакции получения 6-полиоксиалкилптеринов представляют значитель-
ный интерес. Так, при конденсации озонированных углеводов — глюкозо-
на (LXXX) с 2,4,5-триамино-6-оксипиримидином (XXXV) при pH 5—9
с хорошим выходом образуется 6-тетраоксибутилптерин (LXXVIII, п = 3)
о
х(снопХсн2он
I II + VC
Н2ЬГ bl NH2 OHC
XXXV LXXX
Однако в сильнокислой среде в условиях блокирования 5-аминогруппы пи-
римидинового ядра наблюдается образование 7-тетраоксибутилптерина
(LXXXIII, п = 3) [169].
479
NH,
NH,
XXXY
CH,он снуэн
^нон)" нн^Ч^”01^
co -----» 7 |T J
CH,OH H,N'''%T Nil
LXXXI
CHO
CHOH
(снон)п
CH,OH
LXXXII
LXXVIII
LXXXIII СНг°Н
о
CH,OH
2,4,5-Триамино-6-оксипиримидин (XXXV) способен конденсировать-
ся с альдозами (LXXXII) и кетозами (LXXXI), причем альдозы образуют
преимущественно 7-полиоксиалкилпроизводные (LXXXIII), а кетозы —
6-производные птеридинов (LXXVIII) [166].
При конденсации с альдозами, помимо образования 7-птеринзамещен-
ных (LXXXIII), образующихся в присутствии гидразина, получаются и
6-полиоксиалкилпроизводные птеринов (LXXVIII) [173]. Эта реакция
подробно изучена [11, 166, 169, 170, 171, 173].
Конденсация 2,4,5-триамино-6-оксипиримидина (XXXV) с фруктозой
или сорбозой проводится в разбавленных кислых растворах, содержащих
уксусную кислоту или ее соль в присутствии гидразина [155, 174]. В усло-
виях реакции гидразин является окислителем и, вероятно, переходит в
аммиак, в то время как в щелочном растворе гидразин—восстановитель
[173]. Лучший выход получается при pH ниже 7: выход птерина из фруктозы
и сорбозы составляет 55—60% [173]. Борная кислота является катализа-
тором этой реакции [171, 174], но она не необходима [173]. Конденсация
приводит главным образом к получению 6-тетраоксиалкилптерина, что
доказано путем его окисления в 6-карбоксиптерин (V) [171].
6-Формилптерин получен из а-аминоциануксусного эфира, диизонитро-
зоацетона и гуанидина [175].
4. Получение некоторых исходных продуктов, необходимых для синтеза
птериновых витаминов
2,4,5-Триамино-6-оксипиримидин (XXXV) является важнейшим ключе-
вым исходным веществом для синтеза витаминов группы фолиевой кислоты
и многочисленных других птеринов. Это соединение получают по методу Тра-
убе [103] путем конденсации гидрохлорида гуанидина с циануксусным эфи-
ром в спиртовом растворе этилата натрия через промежуточную ацикличе-
скую форму LXXXIV в 2,4-диамино-6-оксипиримидин (LXXXV) [103, 176]
с последующим нитрозированием в положение 5 и восстановлением полу-
ченного нитрозосоединения (LXXXVI) сернистым аммонием в 2,4,5-триами-
но-6-оксипиримидин (XXXV, схема 111).
Значительно экономичнее использовать для конденсации азотнокислую
соль гуанидина и проводить реакцию с алкоголятом натрия без выделения
соединений LXXXIV [177] и LXXXV [178], что приводит к увеличению вы-
хода 2,4-диамино-5-нитрозо-6-оксипиримидина (LXXXV1). Так как это ве-
щество в слабощелочной среде ведет себя подобно о-изонитрозофенолам и,
по-видимому, способно реагировать в форме хинонимина О=С—C=NOH,
то восстановление нитрозосоединения оказалось возможным легко провести
каталитическим путем со скелетным никелевым катализатором с выходом
80% в щелочной [177, 178] или нейтральной среде [177] или осуществить
с 50%-ным сплавом никеля с алюминием в растворе едкого натра [179].
480
Схема 111
Синтез 2.4.5-триамино-6-оксипиримидина
NH,
С +
HN4 XNH,
LXXXV
' QHgONa HN- "CH,
Vi ь> I I
I CN
CN HN- NH,
LXXXIV
О
LXXXVI
88%
XXXV
Представляет интерес непосредственное получение 2,4-диамино-5-нитро-
зо-6-оксипиримидина (LXXXVI), минуя соединение LXXXV, осуществляе-
мое путем конденсации гуанидина с оксииминоциануксусным эфиром
NCC(=NOH)COOC2H5 [180—1821.
Для получения п-аминобензоил-£(+)-глутаминовой кислоты (VI) с
т. пл. 171—172° С, [clJd +16,76°, входящей в структуру фолиевой кислоты,
£(+)-глутаминовую кислоту (XXXIV) по методу Фишера [183] конденси-
руют с n-нитробензоилхлоридом (LXXXVI I) в среде водного раствора бикар-
боната натрия в и-нитробензоил-£-глутаминовую кислоту (LXXXVIII), кото-
рую освобождают от примеси n-нитробензойной кислоты экстракцией эфи-
ром и затем подвергают восстановлению (схема 112).
Схема 112
Синтез л-аминобензоил-£-глутамииовой кислоты
соон
СООН
сум
LXXXV1I
COCI + HjNCH сум
। / тТО
СН2СН,СООН
XXXIV
COHNCH
90%
сцсцсоон
LXXXVIH
VI
соон
COHNCH
СН,СН,СООН
Конденсацию L-глутаминовой кислоты с n-нитробензоилхлоридом про-
водят в воднобензольной среде с прибавлением для нейтрализации Mg(OH)2,
Са(ОН)2 [184] или едкого натра [183, 185—187]; применяются и другие
органические растворители, например хлороформ.
Если восстановление соединения LXXXVIII проводить сернистым аммо-
нием, то аминосоединение VI выделяют в виде медной соли, которую затем
подвергают обработке сероводородом [188]. Однако значительно лучше про-
текает каталитическое восстановление со скелетным никелевым катализа-
тором [102], при этом сразу получается чистый водный раствор и-амино-
бензоил-А-глутаминовой кислоты (VI).
/г-Аминобензойная кислота (XXXIII) выделена в виде бензоильного
производного из дрожжей после их кислотного или щелочного гидролиза
в жестких условиях [189]. Для синтетического получения /г-аминобензойной
кислоты (схема 113) практически может быть использовано несколько ме-
тодов, в которых исходят из /г-нитротолуола.
По одному из методов n-нитротолуол (LXXXIX) восстанавливают чу-
гунными стружками в присутствии соляной кислоты в и-толуидин [1901,
который ацетилируют в ацетотолуидид (ХС) действием 80 %-ной уксусной
кислоты; при нагревании до 140° С отгоняется водная уксусная кислота,
16—69 481
затем продукт реакции выливают в воду. Соединение ХС окисляют
марганцовокислым калием [191] в присутствии сернокислого магния при
80° С и n-ацетиламинобензойную кислоту (XCI) выделяют из раствора соля-
ной кислотой [192], в результате ее деацетилирования кипячением с раз-
бавленной соляной кислотой образуется n-аминобензойная кислота (X X X111)
[1911 с общим выходом поэтому четырехстадийному методу около 56%.
Схема ИЗ
Синтез л-амииобензойной кислоты
По другому варианту n-нитротолуол первоначально окисляют двухромо-
вокислым натрием (или калием) в присутствии серной кислоты в водном
растворе в n-нитробензойную кислоту (ХСП), которую отделяют и очи-
щают через растворимую в воде натриевую соль [190, 192]. Нитробензой-
ную кислоту (ХСП) можно предварительно этерифицировать, затем восста-
новить в анестезин (ХСШ) и гидролизовать в n-аминобензойную кислоту
(ХХХШ). Однако проще n-нитробензойную кислоту непосредственно вос-
становить в n-аминобензойную кислоту с выходом 78—80%, что может
быть произведено сернистым аммонием [193], оловом и соляной кислотой
[194], чугунными стружками в присутствии уксусной кислоты или серно-
кислым железом и аммиаком [195], гидросульфитом натрия [196] или лучше
каталитическим гидрированием с платиновым катализатором [197] или со
скелетным никелевым катализатором в водно-спиртовой среде под давле-
нием. n-Нитробензойная кислота практически нерастворима в воде и спирте,
что препятствует гидрированию, но при повышенной температуре (60—70° С)
гидрирование протекает с высоким выходом. Общий выход п-аминобензой-
ной кислоты из соединения LXXXIX через n-нитробензойную кислоту
(ХСП) составляет около 64%.
Электролитическое восстановление п-нитробензойной кислоты (ХСП)
может быть осуществлено при перемешивании ее суспензии в 2—14%-ной
соляной кислоте на оловянном, свинцовом или амальгамированном цин-
ковом катоде в приборе с диафрагмой при температуре 603 С с выходом
95—98% в растворе [198]. Применяется плотность тока от 2 до 14 А/дм2,
причем на свинцовом катоде выход возрастает с 83 до 95% при увеличении
плотности тока с 2 до 12 А/дм2. Губчатый слой на поверхности катодов из
свинца, меди или графита влияет на процесс, по-видимому, каталитически.
Для получения и-аминобензойной кислоты в бесцветных моноклиниче-
ских призмах с т. пл. 186—187° С [194] вещество растворяют в растворе со-
ды, осветляют активным углем в атмосфере азота при 80° С, обрабатывают
гидросульфитом и вновь углем и затем осаждают соляной кислотой до
pH 3,5—4,0 [199]. Она мало растворима в воде и очень хорошо — в спирте.
В 100 мл растворяется n-аминобензойной кислоты: в 90%-ном спирте при
9,6° С —11,3 г, в эфире при 5,8° С —8,2 г, в уксусной кислоте при 12,5° С —
482
8,1 г, в этилацетате при 9,5° С—7,0 г, в воде при 12,8° С — 0,34 г, в серо-
углероде при 9,5° С—0,01 г; в петролейном эфире кислота нерастворима.
Спектр поглощения ультрафиолетового света n-аминобензойной кислоты
имеет максимумы: в 95%-ном спирте — 220 нм (s 8,22-103) и 288 нм (s
17,4-103) [200]; в водном растворе — 266,271 и 284 нм (е 14,0-103) [201].
СИНТЕЗ ПТЕРОИЛПОЛИГЛУТАМИНОВЫХ кислот
В получении птероил-L-ди- и птероил-Л-триглутаминовых кислот слож-
ность представляет синтез пептидной части молекулы определенного стро-
ения, что связано с необходимостью блокирования тех амино- и карбоксиль-
ных групп L-глутаминовой кислоты, которые не должны вводиться в по-
следующие реакции. В остальном, т. е. в дальнейшем полном построении мо-
лекулы, синтез осуществляется по методам, описанным выше для получения
птероил-L-глутаминовой кислоты. n-Аминобензоилди- или /г-аминобензоил-
трипептиды конденсируют с 2,3-дибромпропионовым альдегидом и 2,4,5-три-
амино-6-оксипиримидином; образующееся промежуточное дигидросоедине-
ние окисляют двухромовокислым натрием [15]. В процессе очистки эфирная
группа сложного птерина гидролизуется и образуется птероил--рглутамил-
7-глутамилглутаминовая кислота (II) или другие птероилполиглутаминовые
кислоты [87—91 ]. Для получения пептидов применяется два основных
метода: синтез через ангидриды [91, 202] и синтез через азиды [89].
Получение пептидов L-r лутаминовой кислоты
через ангидриды
Для получения птероил^-триглутаминовой кислоты (II) через ангидри-
ды [91 ] ключевым промежуточным продуктом является у-этиловый эфир
карбобензилокси-а-глутамилглутами новой кислоты (XCVI), получаемый из
карбобензилоксиглутаминового ангидрида (XCIV) и -у-моноглутаминового
эфира (XCV). 1
СН,СО
XCIV
—oc2hs
XCVll
соон
HjN^H -----
сн2
СЦСОООД;
XCV
I—OC,H$
н-Г-осгн5
—OCjHj
XCVI
co-
-HNCH
I
= CH,
CH,
co-
Полученный моноэтиловый эфир карбобензилоксидипептида (XCVI)
этерифицируют до триэтилового эфира и после декарбобензилоксилирова-
ния методом каталитического восстановления получают дипептид, триэти-
ловый эфира- (XCVI1) [91 ] или -у-глутамилглутаминовой кислоты (XCVIII)
[202], который вновь конденсируют с карбобензилоксиглутаминовым ангид-
ридом, и затем после повторения указанных превращений получают три-
пептид определенного строения. Конденсация трипептида с л-нитробензоил-
хлоридом с последующим восстановлением полученного нитросоединения
дает n-аминобензоилтрипептид, применяв мый для получения птероил-L-TpH-
глутаминовой кислоты (II) [91, 202].
Получение пептидов L-rлутаминовой кислоты
через азиды
Рядом исследований показано [203, 204], что азиды кислот легко реа-
гируют с эфирами аминокислот с образованием пептидной связи. Эта реак-
ция положена в основу синтезов пептидов L-глутаминовой кислоты через
16*
483
азиды. Конденсацией у-моноэтилового эфира L-глутаминовой кислоты
(XCIX) с п-нитробензоилхлоридом получают у-этиловый эфир п-нитробен-
зоил-Л-глутаминовой кислоты (С), который в результате взаимодействия
с гидразин-гидратом переводят в гидразид кислоты (CI) [89]. При действии
азотистой кислоты (см. схему 114) из гидразида образуется у-азид п-нитро-
бензоил-L-глутаминовой кислоты (СП).
Азид (СП) реагирует с у-этиловым эфиром L-глутаминовой кислоты или
с L-глутаминовой кислотой в водном растворе бикарбоната натрия, образуя
л-нитробензоилднпептид в виде моноэфира (СШ) [89].
n-Нитробензоилтрипептид получается по двум вариантам — или в виде
триэтилового эфира (CIV) при взаимодействии у-азида п-нитробензоил-L-
-глутамнновой кислоты (СП) с триэтиловым эфиром у-глутамилглутамино-
вой кислоты, или в виде моноэтилового эфира (CVI1) путем дальнейшего по-
строения молекулы, исходя из у-моноэтилового эфира п-нитробензоил-у-глу-
тамилглутаминовой кислоты (СШ). С этой целью из соединения СШ после-
довательно получают динатриевую соль, гидразид ди натриевой соли и у-азид
n-нитробензоил-у-глутамилглутаминовой кислоты (CVI), который затем
обрабатывают у-этиловым эфиром L-глутаминовой кислоты и получают
п-нитробензоилтрипептид в виде моноэтилового эфира (СУП) [89]. Для
синтеза птероилтрипептида применяют п-аминобензоил-у-глутамил-у-глу-
тамилглутаминовую кислоту в виде моно- (CVIII) или триэтилового эфира
(CV), получаемых восстановлением цинковой пылью соединений CVII
и CIV соответственно.
Схема 114
Синтез у.у-трипептида л-амииобеизоилглутамииовой кислоты
БИОЛОГИЧЕСКОЕ ЗНАЧЕНИЕ ВИТАМИНОВ ГРУППЫ ФОЛИЕВОЙ
КИСЛОТЫ. ЗАВИСИМОСТЬ МЕЖДУ СТРОЕНИЕМ И БИОЛОГИЧЕСКОЙ
АКТИВНОСТЬЮ СОЕДИНЕНИЙ ПТЕРИНОВОГО РЯДА. АНТИВИТАМИНЫ
Первоначально противоанемический птернновый фактор был выделен
в 1941 г. из листьев шпината [1, 205]. В последующие годы была произ-
ведена идентификация многих соединений, обладающих той же биологи-
ческой активностью. ,
Птероил-А-глугампновая кислота, фолиевая кислота (VI) оказывает
активное противоанемическое действие и является витамином кровообра-
зованияТХЗна^ылечивает от «тропической» макроцитарной" анемии и спру
484
[206], против которой витамин В12 неактивен. Птероил-L-глутаминовая
кислота применяется в медицине как терапевтическое средство при мегало-
бластных -Злокачественных анемиях(перпициозной анемии), связанных с
наругиением функций ^кроветворен и я? При этом заболевании происходит
задержка в созревании мегалобластов (лейкоцитов) в костном мозгу и связан-
ное с этим постепенное уменьшение количества форменных элементов крови.
Фолиевая, кислота применяется jipu лечении «лучевых болезней», свя-
занных с облучением рентгеновскими и другими-проникающими лучами.
В виде коферментов фолиевая кислота осуществляет каталитические функ-
ции реакций переноса «одноуглеродных фрагментов» при синтезе пуринов,’
тимина, метионина и других метаболитов в процессах обмена веществ [207]
(см. с. 497).
Птероил-Л-глутаминовая кислота — активный ростовой фактор для мик-
роорганизмов, особенно для Lactobacillus casei и Streptococcus faecalis R.
[15, 83, 85, 86, 97]. Образование пептидных связей с новыми молекулами
L-глутаминовой кислоты дает птероил-Л-триглутаминовую и птероил-
L-гептагл утами новую кислоты [15, 87—91, 208]. Эти соединения как
ростовые факторы малоактивны для микроорганизмов. Так, активность
птероил-Т-триглутаминовой кислоты — ферментативного L. casei-факто-
ра (II) для L. casei равна 64%, а для S. faecalis — всего 2,4% активности
фолиевой кислоты [497]. ’
Птероил-Т-гептаглутаминовая кислота, витамин Вс-конъюгат (III) как.
ростовой фактор обладает 1 % активности фолиевой кислоты для L. casei.
и S. faecalis R. Птероил-Т-триглутаминовая кислота (II) и птероил-L-ren-
таглутаминовая кислота (III) при действии фермента конъюгазы отщепляют
две или шесть молекул L-глутаминовой кислоты и переходят в свободную
форму — птероил-Т-глутаминовую кислоту (I), активную для микроорга-
низмов.
Природная птероил^-триглутаминовая кислота представляет собой
7,7-трипептид. Синтетически полученный 7-дипептид, птероил-7-глутамил-
глутаминовая кислота обладает 63 %-ной активностью для L. casei и
70%-ной для S. faecalis R. [91]. Другие синтетические ди-и трипеп-.
тиды с иным расположением пептидных связей показывают значительно
меньшую активность, чем птероил-7-глутамил-7-глутаминовая кислота
[91, 202].
На зависимости роста микроорганизмов от количества фолиевой кислоты
в питательной среде основан ее анализ микробиологическим методом [209 ].
Птероил-Т-глутаминовая кислота в естественных продуктах находится
главным образом в связанном состоянии в виде полипептидов и освобождает-
ся при действии ферментов; она широко распространена в растительном
мире. В наиболее значительных количествах фолиевая кислота находится
в свежих зеленых овощах, цветной капусте, печени, почках; в меньших ко-
личествах — в мясе и хлебе; в малых количествах — в корнеплодах, молоке
и рисе.
Детальный обзор биологических свойств, методов выделения из природ-
ных веществ и распространения птериновых витаминов дан в опубликован-
ных работах [210—212].
Потребность в фолиевой кислоте для человека составляет 0,1—0,5 мг
ежедневно. Лечебная доза фолиевой кислоты — 5—10 мг ежедневно в те-
чение 10—20 дней. Токсичность птероил-L-глутаминовой кислоты при
введении внутривенно составляет 410 мг/кг для кролика и 120 мг/кг для
морской свинки.
Молекула фолиевой кислоты (I) и ее производных, осуществляющих фун-
кции кофакторов в процессах метаболизма, таких, как 5,6,7,8-тетрагидро-
птероил^-глутаминовая кислота, 5-1Ч-формил-5,6,7,8-тетрагидроптероил-
L-глутаминовая кислота (фолиновая кислота) и др. (см. раздел «Птери-
новые коферменты»), в основной своей части высокоспецифична. Так,
для проявления витаминных свойств обязательна птериновая структура,
485
т. е. наличие 2-амино-4-оксиптеридина в молекуле. Среди птеринов, как ука-
зано выше (см. с. 465), имеется много других природных представителей,
например, птероиновая кислота (XXII) активна как ростовой фактор для
микроорганизмов Streptococcus faecalis R., но неактивна для L. casei и
для цыплят (86, 106], ее эфир менее активен, а амид совсем неактивен 1106],
ризоптерин—lO-N-формилптероиновая кислота (XXIII) — ростовой фактор
для S. faecalis R. и др.
По своему строению птеридины близки к флавинам — соединениям изо-
аллоксазинового ряда, содержащим также пиримидопиразиновую цикли-
ческую систему, конденсированную с бензольным кольцом, главнейший
представитель которых — рибофлавин, витамин В2 (CIX).
сн/снон)3сцрн
CIX
Характерно, что у птериновых витаминов и кофакторов фолиевой кис-
лоты п-аминобензоил-£-глутаминовая кислота присоединена через метиле-
новый мостик в положение 6 птеринового цикла; 7-птероил-£(ф-)-глутами-
новая кислота (XLVI) биологической активностью как ростовой фактор |
или как антивитамин не обладает [104].
Модификация молекулы птероил-£-глутаминовой кислоты (I) возможна
только при формилировании или нитрозировании аминогруппы в положе-
нии 10 или метилировании и формилировании по циклическому атому азо-
та положения 5.10-Ь1-Нитрозоптероил-£-глутаминовая кислота (IX) по био-
логическому действию равнозначна птероил-£-глутаминовой кислоте. Вве-
дение других заместителей в различные положения молекулы птероил-£-глу- j
таминовой кислоты (I) приводит к аналогам, почти лишенным витаминных
свойств или обладающих антагонистическими свойствами, выраженными
в различной степени.
Аналог птероил-£-глутаминовой кислоты, содержащий о-аминобензой-
ную кислоту, сохраняет активность только как ростовой фактор [100].
Аналог, содержащий остаток аминоадипиновой кислоты, обладает 5—6%-ной
активностью для L. casei и S. faecalis R. [213]; аналог с п-аминотолуиловой
кислотой обладает антагонистическими свойствами [214]. Замена /.-глута-
миновой кислоты другими аминокислотами почти полностью лишает аналог
витаминных свойств [215 ], а в отдельных случаях аналоги приобретают анта-
гонистические свойства. lO-N-n-Нитробензолсульфопроизводное птероил-
-£-глутаминовой кислоты еще сохраняет 3—8%-ную активность при испы-
тании на S. faecalis R. [216]. Аналог, содержащий остаток аспарагиновой
кислоты НООССН (NH2)CH2COOH, является антагонистом для различных
видов микроорганизмов [122].
Замещение атома водорода у первичной аминогруппы положения 2 бен-
зоильной или ацетильной группами, а также обработка птероил-£-глутами-
новой кислоты йодистым метилом обычно лишают соединение витаминной
активности. При защите 2-аминогруппы ризоптерина различными ацильны-
ми остатками: СНО, СОСН3, СОСН2ОСН3 и СОСН2СеН5 образующиеся сое-
динения сохраняют ростовые свойства для микроорганизмов [217]. 2-Дез-
аминоптероил-£-глутаминовая кислота биологически неактивна [218].
Замещение аминогруппы в положении 2 на гидроксильную группу вызывает
появление антибактериальных свойств [219].
Замещение атома водорода вторичной аминогруппы (в положении 10)
метильной группой изменяет витаминные свойства птероилглутаминовой
кислоты на антивитаминные [123]. 10-Ы-Метилптероил-£-(-]-)-глутами-
новая кислота является простейшим членом такого гомологического ряда
486
и наиболее активным антагонистом [2201. Антивитаминная активность
установлена также для производных 10-N-CH2COCfiH5 и для производных
lO-N-птероиновой кислоты с заместителями; СН3, С2Н5, СН2С6Н5,
СН2СООН, СН2СОСвН5 [123] и др.
Антагонистом для S. faecalis R. и некоторых животных является и
аналог, в положение 9 которого введена метильная группа [221, 222].
9-Метилптероил-£(-Ь)-глутаминовая кислота может быть превращена в
Ю-М-нитрозо-Э-метилптероилглутаминовую кислоту ]223]. Антивитамином
является и 9-метилптероил-0-(—)-гл утами нова я кислота [222].
Как антивитамины описаны также 9,10-N-диметилптероил-L-глутаминовая
и 9,10-Ы-диметилптероиновая кислоты [221 ], а также 7-метил- и 7,10-М-ди-
метилптероил-£-глутаминовые кислоты [220, 224 ].
4-О-Алкилптероил-А-глутаминовая кислота легко подвергается гидро-
лизу в птероил-£-глутаминовую кислоту [225].
Замена 4-оксигруппы 4-аминогруппой в птероил-А-глутаминовой кис-
лоте приводит к получению соединений с сильными антагонистическими
свойствами [226]. К таким соединениям относится 4-N-[(2,4-диамино-б-пте-:
ридил) метил ]аминобензоил-£-глутаминовая кислота, 4-амино(дезокси)-пте-
роил-А-глутаминовая кислота, «аминоптерин» (СХ); это один из сильных
антивитаминов для крыс, цыплят и для S. faecalis R. Антивитаминными
свойствами обладает и аналог, в котором одновременно находится и 4-ами-
ногруппа, и lO-N-метильная группа, — «аметоптерин», «метотрексат» (CXI)
[96, 124]. Оба эти соединения высокотоксичны.
NH, соон
N N^CHjHNQH^COHNCH
11 10 X
N ™ сх СНгСООН
СН, СООН
CH,NCgH4COHNCH
сн2
СХ! СН2СООН
Соединение СХ получают из 2,4,5,6-тетраминопиримидина, л-аминобензоил-
-L-глутаминовой кислоты и 2,3-дибромпропионового альдегида [96,227,228]
или 1,1,3-трибром(хлор)ацетона [229]. «Аметоптерин» синтезирован из
2,4,5,6-тетрааминопиримидина, n-метиламинобензоил-А-глутаминовой кис-
лоты и 1,1,3-трихлорацетона [230].
Интересно, что при расщеплении 4-амино(дезокси)-10-М-метилптероил-£-
-глутаминовой кислоты (CXI) 4-аминогруппа легко гидролизуется и полу-
чается не 2,4-диаминозамещенный птеридин, а 2-амино-4-оксипроизводное
[96]. Осторожный гидролиз 4-аминогруппы создает возможность превраще-
ния «аминоптерина» в птероил-£-глутаминовую кислоту — посредством гид-
ролиза в щелочной среде без доступа воздуха [165, 227]: так же ведут се-
бя и 4-алкиламинопроизводные [124].
Антивитамином является и 4-амино(дезокси)-9, lO-N-диметилптероил-А-
глутаминовая кислота [231], а также 3-фтор- и 3,5-дифторпроизводные
метотре ксата [232].
Как аминоптерин (СХ) и аметоптерин (CXI), так и многие другие антиви-
тамины фолиевой кислоты по своей функции выступают как ингибиторы ди-
и тетрагидрофолатредуктазы, осуществляющей восстановление фолиевой
кислоты в кофакторы: 7.8- ди- и 5,6,7,8-тетрагидрофолиевую кислоту (см.
ниже). В результате становится невозможным биосинтез пуриновых и пи-
римидиновых нуклеиновых кислот в процессах метаболизма [233]. Анти-
витамины конкурентно тормозят действие фолиевой кислоты, в то же время
их токсичность снимается одновременным применением фолиновой кислоты
(лейковорина).
Аминоптерин (СХ) и аметоптерин (CXI) обладают способностью тормо-
зить развитие злокачественных опухолей и применяются при лечении острой
лейкемии у детей [234, 235] в дозировках 0,25—1 и 2,5—7,5 мг в день соот-
ветственно. Аметоптерин (CXI) используется также при лечении хориокар-
487
1 i&tfv
циномы. Эти соединения оказывают иммунодепрессивное действие; аме-
топтерин применяется для преодоления тканевой несовместимости при пере-
садке органов и тканей [236].
4-Аминогруппе следует приписать наиболее сильную антивитаминную
активность по сравнению с другими заместителями.
Введение в положение 4 птеридинового цикла фолиевой кислоты не сво-
бодной, а замещенной аминогруппы, такой, как метиламино-, диметиламино-
или 1-пиперидилгруппы, дает соединения, менее токсичные, но также и бо-
лее слабые как антагонисты, чем «аминоптерин» [124].
Из других аналогов с антагонистическими свойствами следует указать
на 7-оксиптероилглутаминовую кислоту [237] и 4-амино(дезокси)-7-окси-
птероилглутаминовую кислоту [238]. Испытаны аналоги птероиновой кис-
лоты с N-метилантраниловой кислотой, с М-(4-карбоксифенил)глицином,
n-фенациламино-А-глутаминовой кислотой [106], с 4-амино-3,5-дихлорбен-
зоил-А-глутаминовой кислотой [21 ], хлор- и бромзаместителями в других -
положениях ароматического цикла фолиевой кислоты [21 ]. Аналоги пте-
роил-А-глутаминовой кислоты и аминоптерина с различными галогенозаме-
стителями в бензольном ядре не обладают антагонистической активностью,
большей, чем у 4-амино(дезокси)-3',5'-дихлорптероил-А-глутаминовой кис- J
лоты [22]. J
Увеличение метиленового мостика между птеридиновой и ароматичес-
кой частями молекулы до двух и трех звеньев приводит к гомо- и бнс-гомофо-
лиевой кислоте с антагонистическими свойствами [239—241 ].
Аналог фолиевой кислоты с сульфамидной группой (СХП) [242]
*
° /—\ соон ]
hnAn CH^N-T Vso,hnch j
A A J ” ~ сн» i
HjN N N CXH СЦСООН
обладает свойством понижать кровяное давление.
К антагонистам относится и аналог птероил-А-глутаминовой кислоты,
в котором атом азота в положении 10 заменен кислородом [243].
Антивитаминами оказались и многочисленные птеридиновые соединения,
среди них 2,4-диамино-6-оксиметил-7,8-дигидроптеридин, ингибирующий
биосинтез фолиевой кислоты и применяемый в виде различных лекарственных
форм [244], птеридины с меркаптогруппой в положении 2 общей формулы
[25, 245—247]
R’
R'-OH. NH,; R" R=OH.CH, и др.
различные производные 2,4-диамино-6,7-дифенилптеридина [248] и другие
птеридины.
Помимо птериновых антивитаминов синтезирована и изучена на инги-
бирующую способность очень большая группа пиримидиновых и других
аналогов фолиевой кислоты.
Отдельные фрагменты молекулы фолиевой кислоты, не содержащие пте-
ридинового цикла, проявляют определенную ростовую активность, напри-
мер по отношению к микроорганизмам. Так, п-аминобензоил-А-(+)-глута-
миновая кислота (VI) обладает высокой ростовой активностью на культурах
Streptobacterium plantarum [196], однако она проявляет малую активность
на многих других микроорганизмах.
488
Большее биологическое значение имеет n-аминобензойная кислота
(XXXIII)
XXXII1
которая была отнесена к ростовым факторам в 1940 г. после того, как Вудс
и Фильдс [249] установили, что она необходима для развития многих мик-
роорганизмов и что действие сульфаниламидных препаратов подавляется
малыми количествами n-аминобензойной кислоты, ингибиторами которой
они являются [250].
n-Аминобензойная кислота необходима для биосинтеза витаминов группы
фолиевой кислоты, в состав молекулы которых она входит.
n-Аминобензойная кислота (витамин Нх) необходима для нормальной
пигментации волос у млекопитающих [251 ]; с недостаточностью п-аминобен-
зойной кислоты связано поседение шерсти у крыс [197]. Однако применение
n-аминобензойной кислоты для предохранения людей от поседения волос не
нашло достаточного подтверждения. Возможно, что она необходима для
нормальной лактации [252] и влияет на функцию щитовидной железы.
n-Аминобензойная кислота имеет огромное значение в развитии однокле-
точных организмов [253, 254], в частности для ацетобутиловых и многих
других микроорганизмов [189]. Так как n-аминобензойная кислота широко
распространена в природе и проявляет свою активность в чрезвычайно ма-
лых количествах (разведение 1 : 1011 еще достаточно для стимулирования
роста микроорганизмов), то наряду с витамином В12 и биотином она является
наиболее активным ростовым фактором микроорганизмов из всех до сих пор
известных. Однако она не усиливает роста сыпнотифозных бактерий и
применяется в лечебных мероприятиях против этого заболевания.
n-Аминобензойная кислота является ростовым фактором высших рас-
тений [255] и водорослей [256].
В состав многих природных продуктов n-аминобензойная кислота вхо-
дит как в связанном, так и в свободном состоянии. В дрожжах она содер-
жится в виде полипептида, состоящего из 10—12 молекул L-глутаминовой
кислоты, одной молекулы аминокислоты неустановленного строения и
одной молекулы n-аминобензойной кислоты, связанной с остальной частью
молекулы по карбоксильной группе [257]. На сложные соединения п-ами-
нобензойной кислоты приходится в дрожжах 40%, а в печени 80% общего ее
содержания.
n-Аминобензойная кислота не токсична в больших дозах: токсичность
проявляется для мышей при дозе 2,85 г/кг, для собак 1,3 г/кг [258].
Антагонистами n-аминобензойной кислоты являются сульфаниламидные
препараты, содержащие характерную группу: —SO2NH—. Простейший
сульфаниламидный препарат — n-аминобензолсульфамид, белый стрепто-
цид, сульфаниламид (СХШ)
схш
отличается от n-аминобензойной кислоты (XXXIII) тем, что карбоксильная
группа n-аминобензойной кислоты заменена в нем сульфамидной группой.
Химиотерапевтическое значение производных сульфаниламида в 1935 г.
открыл Домак [259] на красном стрептоциде. С того времени синтезировано
более 10 000 сульфаниламидных соединений, из которых многие оказались
ценными лечебными средствами против стрептококковых, менингококковых,
гонококковых (ангина, крупозное воспаление легких, менингит, гоноррея)
и других инфекционных заболеваний [260].
489
Как антиростовые факторы сульфаниламидные препараты действуют
бактериостатически; При их воздействии процессы метаболизма бактерий
снижаются, жизненные функции ослабляются, прекращается или заме-
дляется размножение бактерий, что облегчает макроорганизму борьбу
с ними. Особенно сильно на повышение бактериостатической способности
сульфаниламидных препаратов влияет повышение температуры: так, на-
пример, повышение температуры с 37 до 38° С увеличивает эффект в 100
раз. Чтобы подавить ростовое действие одной части и-амииобензойной кис-
лоты, необходимо применить очень большие количества ингибиторов: 36 ч.
норсульфазола, 100 ч. сульгина или сульфидина, 1500 ч. белого стрепто-
цида и т. д. [261 ].
ПТЕРИНОВЫЕ КОФЕРМЕНТЫ
В живой клетке фолиевая кислота находится в виде кофермента —
5,6,7,8-тетрагидрофолиевой кислоты и ее активированных форм — ацилиро-
ванных или алкилированных по циклическому атому азота положения 5
или по вторичной аминогруппе положения 10. Из природных источников
выделены: 5,6,7,8-тетрагидрофолиевая кислота, промежуточный продукт
частичного восстановления — 7,8-дигидрофолиевая кислота, 5-М-формил-,
5-М-оксиметил-, 5-М-формимино-, lO-N-формил-, циклические 5, lO-N-мети-
лен- и 5,10-Ы-метенил-5,6,7,8-тетрагидрофолиевые кислоты и, кроме того,
5-М-метил-5,6,7,8-тетрагидрофолиевая кислота. Из различных активирован-
ных форм кофермента 5-М-формил-5,6,7,8-тетрагидрофолиевая кислота ста-
бильна и устойчива к окислителям [262]. Остальные соединения неустойчивы
и легко подвергаются воздействию окислителей, в том числе кислорода воз-
духа, превращаясь при этом с отщеплением одноуглеродных групп в фолие-
вую кислоту.
5,6,7,8-Тетрагидрофолиевая кислота и ее активированные формы в сое-
динении со специфическими белками образуют птеринопротеидные фермент-
ные системы, в виде которых и осуществляют свои биокаталитические функ-
ции по транспорту одноуглеродных фрагментов между различными субст-
ратами.
7,8-Д игидрофолиевая кислота, 7,8-дигидроптероил-
А(+)-глутаминовая кислота —ДГФК (XI) [9],—бесцветное микрокрис-
таллическое вещество
Спектр поглощения в 0,1 н. NaOH имеет лмакс 284 нм по сравнению с Хмаис
5,6,7,8-тетрагидрофолиевой кислоты, смещенный в ультрафиолетовую
область. В нейтральных или щелочных растворах ДГФК окисляется кисло-
родом воздуха с расщеплением на 6-птеринкарбоновую кислоту и п-амино-
бензоил-£-глутаминовую кислоту.
5,6,7,8-Т етрагидрофолиевая кислота, 5,6,7,8-тетрагид-
роптероил-£(4~)-глутаминовая кислота, 4'-N- [(2-амино-4-окси-5,6,7,8-тет-
рагидро-6-птеридил)метил ]аминобензоил-Л(+)-глутаминовая кислота —
ТГФК (XII)
,, ._. соон
,, ___CH.HN —\ VCOHNCH
H,N N N H
н хп
490
представляет собой бесцветный порошок, плохо растворимый в воде и хо-
рошо— в щелочных растворах. Спектр поглощения в 0,1 н. NaOH имеет
\,акс 290 нм. Для ТГФК характерна интенсивная флуоресценция с ).макс
360 нм.
Природная ТГФК представляет собой оптически активную форму по
двум асимметрическим центрам — она является С*6-/,/.-5,6,7,8-тетрагидро-
фолисвой кислотой с [a ]J —16,9° (0,1 н. NaOH) (263]. ТГФК синтетическая
представляет собой рацемическую смесь двух стереоизомеров, так как при
каталитическом восстановлении фолиевой кислоты атом углерода положе-
ния 6 становится асимметрическим [264].
ТГФК (XII) на воздухе последовательно быстро окисляется в 7,8-дигид-
рофолиевую кислоту(XI) и затем в фолиевую кислоту (I). В качестве стаби-
лизатора ТГФК используют L-аскорбиновую кислоту и меркаптоэтанол.
В кислой среде ТГФК более устойчива, чем в нейтральной.
5-П-Ф о р м и л-5,6,7,8-т етрагидрофолиевая кислота,
5-Н-формил-5,6,7,8-тетрагидроптероил-£(+)-глутаминовая кислота, 4'-N-
[(2-амино-4-окси-5-Ы-формил-5,6,7,8-тетрагидро-6-птеридил)метил ]амино-
бензоил-Г(+)-глутаминовая кислота, фолиновая кислота, цитроворум-
фактор, лейковорин — 5-И-формил-ТГФК (CXIV)
CHjHN
CXIV
СООН
COHNCH
CHjCHjCOOH
представляет собой белое кристаллическое вещество, разлагающееся при
240—250° С без плавления; кристаллизуется в листочках с тремя молекула-
ми воды. Фолиновая кислота малорастворима в воде. Она неустойчива в раз-
бавленных кислых растворах, но стабильна в щелочных, даже при 100° С.
Стабильность молекулы связана с наличием формильной группы при цик-
лическом атоме азота положения 5. Фолиновая кислота флуоресценцией не
обладает.
Природная С*6-/,£-фолиновая кислота вращает плоскость поляризации
влево; [а] о —15,1° (Н2О); спектр поглощения в 0,1 н. NaOH имеет Хмакс
при 282 нм; Е\^ 545; Хмин 242 нм [265, 266].
Синтетическая фолиновая кислота является рацемическим соединением по
оптически активному центру асимметрии у С(6) птеридинового цикла, дающе-
го два диастереомера (второй асимметрический атом находится в остатке
глутаминовой кислоты, представляющей собой L-конфигурацию); С* б-d/,
L-форма имеет (а Ь—2,82° (Н2О[265]. С* 6-/,L-Фолиновая кислота получена
из рацемата кристаллизацией кальциевых солей.
Биологическая активность С* 6-/,/.-фолиновой кислоты в 2 раза больше
активности С* G-dl,/.-формы. С* б-d,/.-форма с [a]D-|-28,3o (Н2О) обладает
низкой биологической активностью [265]; оптическая активность во всех
случаях определялась на кальциевых солях.
Фолиновая кислота при потенциометрическом титровании характеризу-
ется как трехосновная кислота с рК„ 3,1, 4,8 и 10,4; образует двух- и трех-
основные соли щелочноземельных металлов [264].
Фолиновая кислота (CXIV) при нагревании с соляной кислотой (pH 1,3)
легко подвергается циклизации с образованием нового имидазольного
цикла по циклическому атому азота положения 5 и атому азота положе-
ния 10 с образованием четвертичной соли изофолиновой кислоты (CXV)
[267 ] (для хлорида т. пл. 250° С с разл., в 0,1 н. НС1 Хмакс 355 нм), которая
в щелочной среде переходит в 10-П-формил-5,6,7,8-тетрагидроптероил-Б-глу-
таминовую кислоту (CXVI)
491
соон
CHjHNCjH^COHNCH
СН3
CXIV ^соон
H,N
сн соон
N ^CHj-NCgH^COHNCH
N Н
и CXV
сн2 -
CHjCOOH
о ,, сно соон
„ Н t |
HINX,N CH^Ce^COHNCH
L J CH,
N Й CXVI снсоон
Фолиновая кислота при нагревании при pH 10—12 стабильна в отличие
от 10-Ы-формилитероил-£-глутаминовой кислоты (CXVI) (268].
Формильная группа у S-N-формил-ТГФК более прочно связана с ато-
мом азота, чем у lO-N-формил- и 5,10-М-метенил-5,6,7,8-тетрагидрофолиевой
кислоты.
При осторожном дегидрировании фолиновой кислоты (CXIV) происхо-
дит изомеризация формильной группы с образованием lO-N-формилптеро-
ил-А-глутаминовой кислоты (X, с. 00).
В реакциях переноса одноуглеродных групп фолиновую кислоту (CXIV)
можно рассматривать как одно из конечных стабильных звеньев превращений
биокатализатора.
Фолиновая кислота аналогично фолиевой кислоте образует полипепти-
ды. Она является ростовым фактором для Leuconostoc citrovorum [269, 270],
а также более активным биологическим фактором, чем фолиевая кислота,
для подавления антивитаминных свойств метилптероил-£(+)-глутамино-
вой кислоты [269] и «аминоптерина» [227].
Фолиновая кислота имеет терапевтическое применение одновременно с
противораковыми препаратами птеринового характера; она совместное окси-
кобаламином — витамином В12а (гепафактор, ПНР) применяется при забо-
леваниях печени (которые сопровождаются нарушением метаболитического
процесса превращения фолиевой кислоты в фолиновую кислоту) для вос-
становления ее функции, при гипохромной анемии, белковой недоста-
точности и др.
5-М-Ф ормимино-5, 6, 7, 8-тетрагидрофолиевая кис-
лота — б-Ы-формимино-ТГФК (CXVII)
соон
।
COHNCH
CHjCHjCOOH
имеет Хмакс 285 нм при pH 7, легко гидролизуется с выделением аммиака,
в кислом растворе превращается в 5,10-М-метенил-ТГФК (CXV).
IO-N-Ф орми л-5, 6, 7, 8-тетрагидрофолиевая кисло-
та — lO-N-формил-ТГФ К (CXVI)
о сно.—. соон
VCKN -V _ V COHNCH
A- Jk J X' CHjCHjCOOH
HjN N N
H CXVI
желтоватые кристаллы, плохо растворимые в воде; Хмажс в 0,1 н. NaOH
253 нм. Для энзиматически активной С* 6-/,£-10-Ы-формил-ТГФК [a ]d
—42° [271 ], для химически полученной С* 6-<1/,£-10-М-формил-ТГФК
[а ]d —9° [271 ]. Она легко окисляется кислородом воздуха в lO-N-формил-
492
фолиевую кислоту (X), при действии щелочей при нагревании легко претер-
певает перегруппировку формильной группы из N-10- в N-5-положение пи-
разинового цикла с образованием фолиновой кислоты (CXIV).
5,10-N-M е т и л е н-5,6,7,8-т етра ги д рофо л иев а я кисло-
та— 5,IO-N-метилен-ТГФК (CXVIII)
HJW
СООН
COHNCH
CHjCHsCOOH
N
H CXVIII
представляет собой «активный формальдегид» [272]. Она имеет несколько
большую стабильность, чем другие активные формы ТГФК, хотя фолиновая
кислота более устойчива. 5,10-М-Метилен-ТГФК выделена из природных
источников и синтезирована в виде солей с Хмакс 285 нм при pH 7—11 [273,
2741. Природная активная диастереомерная форма С* 6-<1,£-5,10-П-мети-
лен-ТГФК характеризуется [ci]d+I56 ±9° [275].
5,10-N-M етен и л-5,6,7,8-т етрагидрофолиевая кисло-
та, изофолиновая кислота—5,10-М-метенил-ТГФК (CXV)
имеет спектр поглощения в 0,1 н. НС1 с Хмакс 355 нм [267], обладает интен-
сивной флуоресценцией с Хмакс 470 нм. У энзиматически активной С* 6-d,
£-5, lO-N-метенил-ТГФК [a Id+68° [271].
5-N-M е т и л-5,6,7,8-т етрагидрофолиевая кислота,
«префолиевая кислота»—S-N-метил-ТГФК (CXIX) [276—279]
CXIX
соон
COHNCH
CHjCHjCOOH
с ^макс 290 нм при pH 7 образуется при восстановлении 5,10-Н-метилен-ТГФК
(CXVIII) боргидридом натрия и другими путями.
Так же, как и фолиновая кислота, другие коферментные формы 5,6,7,8-те-
трагидрофолиевой кислоты образуют полиглутаматы, которые, возможно,
и являются основными активными коферментными формами в живой клетке
[31 ].
СТРОЕНИЕ ФОЛИНОВОЙ КИСЛОТЫ
5-П-Формил-5,6,7,8-тетрагидроптероил-£-глута.миновая кислота, фоли-
новая кислота (CXIV) [264, 269, 270, 280—282], имеет в своей структуре
5,6,7,8-тетрагпдроптеридиновый цикл, что следует из подобия ее спектра
поглощения спектру ТГФК (XII), а также из химических реакций [93, 262,
267]. Фолиновая кислота при окислении дает гуанидин, щавелевую кислоту
и хлоранил, что указывает на наличие как пиримидинового ядра, так и за-
мещенного бензольного ядра [93].
Фолиевая кислота (I) может быть восстановлена в 5,6,7,8-тетрагидропте-
роил-Г-глутаминовую кислоту (XII) и формилированием последней переве-
493
дена в фолиновую кислоту (CXIV) по схеме 115. При формилировании в при-
сутствии уксусного ангидрида как из фолиевой кислоты (I), так и из фоли-
новой кислоты (CXIV) образуется lO-N-формилфолиновая кислота (СХХ)
с двумя формильными группами в положениях N-5 и N-10 [931; эта кислота
при действии щелочей переходит в фолиновую кислоту с отщеплением одной
формильной группы.
Схема 115
Установление строения фолиновон кислоты реакциями ее получения и превращения
О
HJN N N
N C^HN-C^-
СООН
I
COHNCH
сн2
СЦСООН
H2N
соон
N CHjHN-CeHfCOHNCH НСООН
Тн ™
й нхп
сна
СН2СООН
HN
СООН
О НС=О I
A A CH3HN-C6HfCOHNCH .
j d
CHjCOOH
I JI Ьн
HJN N N Н
Н CX1Y
НСООН I
NaOH
I h2n
О НС=О НС=О
N СН^-СвНг'
$
N »
н СХХ
СООН
COHNCH
I
СН,
СН/ХЮН
О
I
СИНТЕЗ 5,6,7,8-ТЕТРАГИДРОФОЛИЕВОЙ КИСЛОТЫ И ЕЕ
АКТИВИРОВАННЫХ ФОРМ — IO-N-ФОРМИЛ-ТГФК, 5-М-Ф0РМИЛ-ТГФК
И ДРУГИХ
7,8-Дигидрофолиевую кислоту (XI) синтезируют при осторожном ката-
литическом гидрировании фолиевой кислоты (I) с Pt- или Pd-катализатором
в уксуснокислом растворе [9, 31 ].
5,6,7,8-Тетрагидрофолиевую кислоту (XII) получают гидрированием фо-
лиевой кислоты (I) на Pt- или Pd-катализаторе в растворе уксусной кислоты
(9, 31, 264] или на Pt- и Rh-катализаторе в трифторуксусной кислоте [283],
а также восстановлением гидросульфитом натрия [284, 285] с выходом 68%
при 75° С [286] или боргидридом натрия [287].
Кристаллическая фолиновая кислота, 5-формил-ТГФК (CXIV), получает-
ся из фолиевой кислоты (I) в результате ее формилирования 80—100%-ной
муравьиной кислотой при 0—50° С первоначально в lO-N-формилпте-
роил-L-глутаминовую кислоту (X), восстановления электрохимическим [288]
или каталитическим путем с платиновым катализатором в среде уксусной
или муравьиной кислоты в 10-П-формил-5,6,7,8-тетрагидроптероил-£-глу-
таминовую кислоту (CXVI) [264, 268, 280, 289—291 ] и нагревания получен-
ного продукта в щелочном растворе при pH 10—12 и 90—100° С [264,268,
281] (схема 116). При этом происходит миграция формильной группы из
N-10- в более стабильное N-5-положение. Чтобы избежать влияния кислоро-
да, восстановление и нагревание проводят в присутствии L-аскорбиновой
кислоты [281, 291 ]. Общий выход (в растворе) составляет 40—50%. Фолино-
вая кислота очищается хроматографической адсорбцией ее нейтральной каль-
циевой или бариевой соли.
Схема 116
Синтез фолииовой кислоты
о СООН О СНО соон
НСООН HNAYr\CH’ NC6H4C°HNCH [н]
Л 1 J СН2 ~ ди сн,
HJN N N | CHjCOOH HjN N N x CH2COOH
_ , CHO COOH
О H i I
A U V* -------------------------
N pi CHjCOOH HjN
CHO coon
N CH,HNC6H<COHNCH
J
N CHjCOOH
H CXIV ’
494
lO-N-Формилптероил-А-глутаминовая кислота (X) получается в ваде
желтого порошка и в результате прямого синтеза путем нагревания 6-пте-
ринальдегида, п-аминобензоил-£-глутаминовой кислоты и муравьиной
кислоты 1132, 292, 293].
10-М-Формил-5, 6, 7,8-тетрагидрофолиевая кислота (CXVI) полу-
чается также формилированием 5, 6, 7, 8-тетрагидрофолиевой кислоты
(XII).
5, lO-N-Метилен-б, 6, 7, 8-тетрагидрофолиевая кислота (CXVIII) син-
тезирована из ТГФК (XII) и формальдегида (2 моля) при pH 4—5 и при
комнатной температуре [272—274].
ВЗАИМОПРЕВРАЩЕНИЯ ТГФК И АКТИВНЫХ ФОРМ КОФЕРМЕНТА
Фолиевая кислота (I) ферментативно превращается в свой кофермент —
5, 6, 7, 8-тетрагидрофолиевую кислоту, ТГФК (XII) — в одну или две стадии
(схема 117). Первоначально при каталитическом восстановлении НАДФ-Н
при участии дигидрофолатдегидрогеназы (1)* фолиевая кислота переходит
« 7, 8-дигидрофолиевую кислоту, ДГФК (XI), а затем с НАДФ-Н при дей-
ствии тетрагидрофолатдегидрогеназы (2) ДГФКвосстанавливается в ТГФК
(XII) [294]. Химическое восстановление гидросульфитом натрия также пре-
вращает фолиевую кислоту в 7,8-дигидрофолиевую кислоту (XI).
При взаимодействии с муравьиной кислотой и АТФ по вторичной ами-
ногруппе положения 10 ТГФК (XII) происходит формилирование. АТФ
является источником энергии для осуществления этой реакции, при этом
он превращается в АМФ и ортофосфорную кислоту. Реакция формилиро-
вания катализируется формилтетрагидрофолатсинтетазой (3). Образовав-
шаяся IO-N-формил-ТГФК (CXVI) может обратно деформилироваться в
ТГФК (XII) при каталитическом участии формилтетрагидрофолатдеформи-
лазы (4). Химическим путем формилирование ТГФК (XII) может быть
осуществлено в lO-N-формил-ТГФК (CXVI) при действии муравьиной кис-
лоты и дегидратирующих агентов в сильнокислой среде.
lO-N-Формил-ТГФК вступает в ферментативные реакции формилирова-
ния субстрата или подвергается дальнейшим взаимопревращениям в дру-
гие активные формы кофермента, которые в свою очередь участвуют в
различных реакциях обмена одноуглеродных групп.
lO-N-Формил-ТГФК (CXVI) в результате восстановления превращает-
ся в циклическую 5, IO-N-метилен-ТГФК (CXVIII). Эта активная форма
восстанавливается далее в 5-М-метил-ТГФК (CXIX) НАД-Н при участии
5, lO-N-метилентетрагидрофолатредуктазы (5).
5, lO-N-Метилен-ТГФК (CXVIII) может быть получена химически из
протонированной по N<5> ТГФК при pH 4,45 через промежуточно обра-
зующуюся протонированную по N(5) lO-N-оксиметил-ТГФК (CXXI) [295].
В циклическую 5, IO-N-метилен-ТГФК (CXVIII) могут превращаться как
S-N-формил-ТГФК (CXIV), так и IO-N-формил-ТГФК (CXVI) химическим
путем [78, 93]. Малоактивная S-N-формил-ТГФК (фолиновая кислота)
(CXIV) может включаться в систему активных трансформаций кофермент-
ных форм ТГФК путем или непосредственного ферментативного превра-
щения в IO-N-формил-ТГФК (CXVI) [296], или превращения в это соеди-
нение через циклическую 5, IO-N-метилен-ТГФК (CXVIII) [297].
5, lO-N-Метилен-ТГФК (CXVIII) в результате окисления НАДФ при
участии метилентетрагидрофолатдегидрогеназы (6) превращается в 5, 10-
-N-метенил-ТГФК (CXV). Это соединение подвергается гидролитическому
превращению при участии воды и метенилтетрагидрофолатциклогидролазы
(7) в 10-формил-ТГФК (CXVI). Реакция гидролиза 5, lO-N-метенил-ТГФК
в IO-N-формил-ТГФК легко осуществляется и химическим путем. 5, 10-N-Me-
• На схеме арабские цифры обозначают ферментные системы.
495
тенил-ТГФК также химически восстанавливается боргидридом натрия в
5, lO-N-метилен-ТГФК (CXVIII) (2951.
5, IO-N-Метенил-ТГФК (CXV) гидролитически превращается в 5-М-фор-
мил-ТГФК (CXIV), представляющую собой устойчивую к окислению актив-
ную форму кофермента. Выделяющийся при реакции протон Н+ переходит
в растворитель.
5, IO-N-Метенил-ТГФК (CXV) ферментативно при участии формимино-
тетрагидрофолатциклодезаминазы (8) образуется из 5-М-формимино-ТГФК
(CXVII); реакция протекает с отщеплением молекулы аммиака, протон
берется из растворителя.
Источником получения 5-М-формимино-ТГФК (CXVII) является ТГФК
(XII), которая участвует в реакциях превращений М-формимино-А-глута-
миновой кислоты в L-глутаминовую кислоту и N-формиминоглицина в гли-
цин, катализируемых глутаматформиминотрансферазой (9) и глицинфор-
миминотрансферазой (10).
Реакция взаимодействия ТГФК (XII) с формальдегидом в виде СН2(ОН),
идет по циклическому атому азота положения 5 (а не по вторичной амино-
группе положения 10, как при ацилировании муравьиной кислотой) и при-
водит к получению 5-М-оксиметил-ТГФК (CXXI). Эта реакция может про-
текать и неферментативно в нейтральной среде [273].
Схема 117
Взаимопревращения ТГФК и активных форм кофермента
CH=NH
HjN
о
№ .’’CHjNHR
н
in^,ch2nhr
h2n in n'
XII
(-HCOOli) и HCOOH; АТФ ч
HjO ||(-АМФ;-Н3РО^
О ®Н
N -yW^R H/)(-H4)
N-CHjNHR
Н
CXVII
Ф+н-
СНО
N
н
CXY
н,о
СН.
CH^NR
HN
H2N tn n
H
CXYI
1
+2Н , +2е
HjN
сно
I
n а<ж
CXVHI CXIY
R-остаток вензоилглутаминовой кислоты
н
CXXI
1+2Н* +2е
(-М
сн3
CHJNHR . НАД-Н+ н м CHj-lNR
' ' * (2Н.2е)(~НАД+) ® НЫ
H,N
HjN
СХ1Х
N'
Н
В результате реакции ферментативного восстановления 5-М-оксиме-
тил-ТГФК (CXXI) переходит в 5-М-метил-ТГФК (CXIX).
Следует отметить, что в реакциях оксиметилирования субстратов 5-N-ok-
симетил-ТГФК (CXXI) не принимает непосредственного участия; предва-
496
рительно это соединение должно дегидратироваться в циклическую
5, lO-N-метилен-ТГФК (CXVIII). Только 5,10-!Ч-метилен-ТГФК (CXVIII)
является активной формой кофермента, осуществляющей передачу окси-
метильной группы на субстрат.
Реакции взаимных превращений различных активных форм кофермен-
та фолиевой кислоты предшествуют, сопровождают или заключают реак-
ции субстратов, связанные с транспортом формильной, оксиметильной
или метильной групп.
Необходимо отметить, что многое в реакциях превращений «активного
формальдегида» и его участии в ферментативных реакциях субстратов не-
ясно и требует дальнейших уточнений. Проблема «активного формальде-
гида» изучалась на упрощенных моделях активных форм кофермента 1298].
БИОХИМИЧЕСКИЕ ФУНКЦИИ ПТЕРИНОВЫХ КОФЕРМЕНТОВ
Птериновый кофермент — 5, 6, 7, 8-тетрагидрофолиевая кислота и ее ак-
тивные формы — катализирует реакции, приводящие к образованию или
разрыву связей: С—С, С-—N, С—S, С—О, С—Н, N—Н.
К основным реакциям обмена веществ, протекающим в животном орга-
низме или микроорганизмах, связанным с синтезом и расщеплением не-
которых а-аминокислот, пиримидинов, пуринов, нуклеиновых кислот и
других соединений, катализируемых птериновым коферментом и его актив-
ными формами в содержащих их ферментных системах, относятся три типа
обратимого переноса «одноуглеродных групп» — формильной и формиминой
(—СНО и —CH=NH), оксиметильной (—СН2ОН) и метильной (—СН3) [299].
Эти реакции следующие:
формил и рование L-глутаминовой кислоты, глицинамидриботида, 5-ами-
но-4-имидазолилкарбоксамидриботида и формиминолирование L-глутами-
новой кислоты и глицина [300, 301];
оксиметилирование глицина в L-серин [302, 303];
метилирование (оксиметилирование и последующее восстановление)
гомоцистеина в метионин, урацила в тимин, цитозина в 5-метилцитозин,
аминоэтилового спирта в холин и др.
Поставщиками одноуглеродных групп могут быть некоторые вещества
за счет своих отдельных атомов углерода, как это доказано путем приме-
нения меченых атомов. К ним относятся: муравьиная кислота [302], уксус-
ная кислота [51, глицин [302], серин (₽-оксиаланин), холин, метионин [304],
пировиноградная кислота и др.
В реакциях формилирования (формиминолирования) участвуют 5-М-фор-
мил-, 5-1Ч-формимино-, IO-N-формил- и 5,10-Ы-метенил-ТГФК.
Важное значение на промежуточных стадиях синтеза пуриновых осно-
ваний имеет введение одного из углеродных звеньев, связанных с азотом.
Это относится к атомам углерода положений 2 и 8 синтезируемого пури-
нового цикла.
Необратимое формилирование 5-фосфорибозилглицинамида протекает
при участии 5, lO-N-метенил-ТГФК фосфорибозилглицинамид-формилтранс-
феразы в присутствии воды. В результате реакции получается 5-фосфори-
бозил-N-формил глицинамид, образуется ТГФК и выделяется протон,
который переходит в раствор [305].
он он
? н4гзЬн
(НО),РОН2С О ЫН
СО
5,10—N—Метенил -
-ТГФК
ОН ОН
Н Н
,с о nh+-n-ch-ch2-n-+h*
сб 8 ' -°
ГИЛ ТГФК
497
Формилирование П-5'-фосфорибозил-5-амино-4-карбоксамидимидазола
при действии lO-N-формил-ТГФК фосфорибозиламиноимидазолкарбоксамид-
формилтрансферазы в присутствии ионов К+ приводит к N-S'-фосфорибозил-
-5-формамино-4-карбоксамидимидазолу и ТГФК (305].
он он
Н2М
н2к
СО N
СНО
-N-CH-CHj-N---
S I ю
lO-N-Формил-
-ТГФК
Последующее замыкание полученного пуринового предшественника
в цикл с образованием гипоксантина приводит к получению инозиновой
кислоты [305], которая в организме обычно образуется при дезаминирова-
нии аденозинфосфатов.
Обратимую реакцию формилирования L-глутаминовой кислоты (XXXIV)
в М-формил-£-глутаминовую кислоту осуществляет 5-Ы-формил-ТГФК (фо-
линовая кислота) формилглутамат-формилтрансферазы [306, 307].
соон сно соон
1 | Н I
HSNCH 4---N —СН—СН2—N— OHCHNCH +
I 6 I 10 I
CH2CHSCOOH 5-Формил-ТГФК СН2СН2СООН
•X- Н Н
•> + —N-СН-СН,—N—
5 | 10
ТГФК
В реакциях ферментативного расщепления гистидина промежуточно
образующаяся N-формимино-М-глутаминовая кислота (СХХП) при ката-
литическом участии ТГФК глутаматформиминотрансферазы деацилируется
в L-глутаминовую кислоту (XXXIV), причем получается 5-Ь1-формимино-
СООН
HN=HCHNCH Н Н
I + —N—СН—СН2—N—
СН2 5 | 10
I
СН2СООН
СХХП ТГФК
СООН
H,NCH CH=NH
—► I I I H
— + — N—CH—CH2—N —
। 2 5 | 10
CH2COOH
XXXIV 5-М-Формимино-ТГФК
Один из продуктов пуринового обмена, например продукт расщепления
ксантина некоторыми микроорганизмами — N-формиминоглицин, перехо-
дит в глицин при каталитическом участии ТГФК глицинформиминотранс-
феразы, образующей при этом 5-М-формимино-ТГФК [3081
Н Н
HN=HCHNCH2COOH+—N—СН—CHS—N— “Г
5 | 10
ТГФК
498
CH=NH
I H
H2NCH2COOH + —N—CH—CH->—N—
5 | 10
5-М-Формимино-ТГФК
В реакциях оксиметиллрованпя в качестве активного оксиметилирую-
щего агента выступает только 5,10-М-метилен-ТГФК.
Обратимая реакция трансоксиметилирования осуществляется между
двумя аминокислотами — серином и глицином при участии ТГФК и 5, 10-
N-метилен-ТГФК и сопровождается выделением или поглощением одной
молекулы воды [31, 309]. Реакция катализируется серинальдолазой (серин-
оксиметилтрансферазой), содержащей в качестве простетической группы
пиридоксальфосфат и ионы Мп++ [3101
/ СН2 х
СН2ОН НН / \
I 4-------N—СН—СН2— N H2NCH2COOH+—N-СН—СН2—N— + Н2О
H2NCHCOOH 5 I 10 5 | 10
ТГФК 5,10-1\'-Метилен-ТГФК
Синтез серина из глицина, вероятно, идет через промежуточную форму
шиффова основания пиридоксальфосфата с глицином
носн2снсоон
N
11 О
СН у ,
снг НО T .CI^OPtOH^
+ —N—СН—CH^-N— + HjO— I I]
S I Ю
1 • CH3 N
5,Ю—N—Метилен-ТГФК
+ — N-CH-CHj-N—
S । z to
ТГФК
Первичным акцептором оксиметильной группы является промежуточ-
ная форма шиффова основания, причем присоединение оксиметильной груп-
пы идет по ц-углеродному атому остатка глицина.
Осуществлено неэнзиматическое гидроксилирование фенилаланина в
тирозин
CeH5CH2CH (NH2) СООН -> НОС6Н4СН2СН (NH2) соон
кислородом воздуха в водном растворе в присутствии 5, 6, 7, 8-тетрагидро-
птерина, Ре++илиГ"е+++ и этилендиаминтетрауксусной кислоты в. фосфатном
буфере при pH 6,9 [311].
В реакциях метилирования, по-видимому, первоначально происходит
оксиметилирование субстрата с последующим его восстановлением.
Введение метильной группы в положение 5 урацильного основания с
образованием производного тимина в реакции превращения 2-дезоксиури-
дин-5'-фосфата через первоначальное оксиметилирование урацилового осно-
вания с последующим восстановлением оксиметильной группы промежу-
точно образовавшейся формы в метильную группу тимидин-5'-фосфата, одного
из компонентов дезоксирибонуклеиновых кислот, протекает при ката-
литическом участии 5,10-М-метилен-ТГФК птеринопротеидов и витами-
на В12 [312, 313].
499
Реакция восстановления, возможно, осуществляется ТГФК, которая
передает два протона и два электрона акцептору и сама при этом окисляется
в ДГФК 1313, 314]. ДГФК вновь может регенерироваться в ТГФК при фер-
ментативном восстановлении тетрагидрофолатдегидрогеназой [314].
Метилирование цитозинового основания в положение 5 ведет к обра-
зованию 5-метилцитозина в реакции превращения компонента нуклеино-
вых кислот цитидин-5'-фосфата, приводящего к 5-метилцитидин-5'-фосфату;
реакция катализируется активной формой ТГФК [313].
он он
Птеринопрст:иды при участии 5-Ь1-метил-5,6,7,8-тетрагидрофолиевой
кислоты и кофермента витамина В12 (выполняющего функцию переноса
метильной группы) осуществляют реакцию метилирования гомоцистеина,
возможно, через промежуточный S-оксиметилгомоцистеин в метионин
[309, 315]
COOH COOH COOH
H2NCH -i ► H2NCH -> H2NCH
CH2CH2SH CH2CH2SCH2OH CH2CH2SOHs
Имеются данные в пользу того, что эта реакция идет через S-аденозил-
аминокислотные промежуточные производные. 5-Ы-Метил-5,6,7,8-тетра-
гид роптерои л-L-тр и гл утами новая кислота также используется в качестве
донора метильной группы.
Синтез холина из аминоэтилового спирта осуществляется путем мети-
лирования при участии активных форм ТГФК [316]
H2NCII2CH2OH -> (CH,),NCH2CH2OH
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ (К ГЛАВЕ XI)
1. М. Mi t с he 1 1, Е. S ne 11,
R. W i 1 1 i a m s. J. Am. Chem.
Soc., 63, 2284 (1941).
2. В. M. Березовский. Успе-
хи химии, 22, 191 (1953).
3. W. P f 1 e i d e г e г, E. L i e-
d e k, R. Lohrmann, T. Ru-
kwie d. Chem. Ber., 93, 2015
(1960).
4. В. H u t c h i n g s, E. S t о k-
st a d, J. M о \v a t, J. В о о t h e,
C. Waller, R. Angier,
J. Se mb, Y. Subbar ow. J.
Am. Chem. Soc., 70, 10 (1948).
5. J. Buchanan, I.Sonne,
A. D e 1 1 u v e. J. Biol. Chem.,
166, 395 (1946).
6. В. H u t c h i n g s, E. S t о k-
s t a d, N. Bohonos, N. Slo-
bodkin. Science, 99, 371 (1944).
7. S. В i n k 1 e у, O. Bird,
E. Bloom, R. Brown,
D. С a 1 k i n s. С. С о m p b e 1 1,
A. Emmett, J. J. Pfiffner.
Science, 100, 36 (1944).
8. S. U у e o, S. M i z u k a m i,
T. К u b о t a, S. T a k a g i. J.
Am. Chem. Soc., 72, 5339 (1950).
9. B. O’Del 1, J. Vanden-
b e 1 t, E. В 1 oo m, J. P f i f-
f r. e r. J. Am. Chem. Soc., 69, 250
(1947).
10. С. 'C a i 1 1 y, S. F 1 a v i a n,
P. D о u z о u. Comp, rend, C263,
272 (1966).
11. F. W e у g a n d, A. Wacker,
V. S c h m i e d-K о w a r z i k.
Chem. Ber., 82, 25 (1949).
12. J. P f i f f n e r, S. В i n k 1 e y.
E. В 1 о о m, В. O’D ell. J.
Am. Chem. Soc., 69, 1476 (1947).
13. R. T s c h e s c h e, R. P e t e r-
s e n, F. Korte. Chem. Ber., 84,
579 (1951).
14. A. Albert. Biochem. J., 9, 47
(1950).
500
15. C. W a 11 е г. В' H u t c h i n gs,
J.Mowat, E. Stokstad,
J. Boothe, R. Angier,
J.Semb, Y. Sub bar ow,
D. С о s u 1 i c h, M. Fa hren-
b a c h, M. H u H q u i s t,
E. Kuh, E. N о e t h e y,
D. Seeger, J.Sickels,
J Smit h. J. Am. Chem. Soc., 70,
19(1948).
16. В. К о f t, M. S e v a g. J. Am.
Chem. Soc., 71, 3245 (1949).
17. E. D a n i e 1, О. К 1 i n e. J.
Biol. Chem., 170, 739 (1947).
18. J. '.Veidenheimer, J. C a r-
s t e n s e n. Канад, пат. 511524;
РЖХ, 1958, 19008.
19. E. S t о к s t a d, B. Hut-
chings, J. Mow at, J. Boothe,
C. Waller, R. Angier,
J.Semb, Y. Sub bar ow.
J. Am. Chem. Soc., 70, 5, (1948).
20. E. W i t t 1 e, B. O’D e 1 1,
J. V a n d e n b e 1 t. J. P f i f-
f n e r. J. Am. Chem. Soc., 69,
1786 (1947).
21. D. С о s u 1 i c h, D. S e e g e r,
M. Fahrenbach, B. Roth,
J. Mowat, J. Smith, M.Hult-
q u i s t. J. Am. Chem. Soc., 73,
2554 (1951).
22. D. С о s u 1 i c h, D. S e e g e r,
M. Fahrenbach, K. Col-
li n s, B. R о t h, E. H u 1 t-
quist, J. Smith. J. Am.
Chem. Soc., 75, 4675 (1953).
23. D. С о s u 1 i c h, J. S m i t h.
J. Am. Chem. Soc., 71, 3574 (1949).
24. R. A n g i e r, J. В о о t h e,
J.Mowat, C. Waller,
J. Se m b. J. Am. Chem. Soc.,
74, 408 (1952).
25. H. W i e 1 a n d, R. L i e b i g.
Lieb. Ann., 555, 46, 150 (1944).
26. R. P u r r m a n n. Lieb. Ann., 548,
284 (1941).
27. С. C a i n, M. M a 1 I e t t e,
E. T а у 1 о r. J. Am. Chem. Soc.,
70, 3026 (1948).
28. M. G or d on, J. R a v e 1,
R. E a к i n, W. S h i v e. J.
Am. Chem. Soc., 70, 878 (1948).
29. D.W о 1 f, R. Anderson,
E. К a c z к a, S. Harris,
G. A r t h, P. Southwick,
R. M о z i n g о, K. F о 1 к e r s.
J. Am. Chem. Soc. 69, 2753 (1947).
30. M. P о 1 о n о v s к i, R. Bu n-
sel, M. P e s s о n. Helv. Chim.
Acta, 29, 1328 (1946).
31. R. Bia к I e v. Biochem. J., 65,
331, 342 (1957).'
32. M. Gate s. Chem. Revs., 41, 63
(1947).
33. H. С. В у л ь ф с о и. Успехи хи-
мии, 17, 249 (1948).
34. R. L. Blakley. The Biochemi-
stry of folic acid and related pteri-
dines, Amsterdam—London, 1969.
35. В. H u t c h i n g s, E.Stok-
s t a d, J. В о о t h e, J.Mowat,
C. Waller, R. Angier,
J.Semb, Y.Subbarow.
J. Biol. Chem., 168, 705 (1947).
36. A. Bratton, E. M a r s c hall
J. Biol. Chem., 128, 537 (1939).
37. E. Becker. Z. physiol. Chem.,
246, 177 (1937).
38. H. Wieland, R. Liebig.
Lieb. Ann., 555, 146 (1944).
39. H. А. Андреева, В. H. Б у-
ки и. ДАН СССР, 64, 95 (1949).
40. Т. Hopkins. Nature, 45, 197
(1891); 45, 581 (1892).
41. Н. W i е 1 а п d, R. Purr-
m а п п. Lieb. Ann., 544, 163 (1940).
42. R. Purrman п. Lieb. Ann., 544,
182; 546, 98 (1940).
43. W. К о s h a r a. Z. phvsiol. Chem.,
240, 127 (1936); 277, ' 159 (1943).
44. C. Schop p, E. В ec ke r.
Lieb. Ann., 507, 266 (1933).
45. H. W i e 1 a n d, R. P и r r-
m a n n. Lieb. Ann., 539, 179 (1939).
46. C. S c h б p p, A. К 6 t t 1 e r.
Lieb. Ann., 539, 128 (1939).
47. C. S c h б p p, E. В e с к e r,
R. Reichert. Lieb. Ann., 539,
156 (1939).
48. G. Hutchings, G. Elion.
J. Am. Chem. Soc., 71, 467 (1949).
49. F. Kort e. Chem. Ber., 87, 1062
(1954).
50. W. В о о n, T. L e i g h. J. Chem.
Soc., 1951, 1497.
51. W. P f e i d e г e r, M. R и k-
w i e d. Chem. Ber., 94, 118 (1961).
52. E. Patterson, R. Milst-
r e v, E. S t о к s t a d. J. Am.
Chem. Soc., 78, 5868 (1956).
53. R. G r e e n, H. R e m b о 1 d.
Chem. Ber., 99, 2162 (1966).
54. E. P a t t e r s о n, H.Bro-
q и i s t, A. Albrecht, M.
Saltza, E. Stokstad. J.Am.
Chem. Soc., 77, 3167 (1955).
55. H. Forrest, H. Mitchell.
J. Am. Chem. Soc., 77, 4865 (1955).
56. M. V i s с о n t i n i, H. S t i e r-
1 i n. Helv. chim. Acta, 44, 1783
(1961).
57. R. Tschesche, B. Hess,
I. Ziegler, К. M a c h-
le i d t. Lieb. Ann., 658, 193 (1962).
58. M. V i sc о n t i n i, R. P г о -
v e n z a 1 e. Helv. Chim. Acta, 52,
1225 (1969).
59. R. H й t t e 1, D. S c h r e c k.
Chem. Ber., 93, 2439 (1960).
60. R. H й t t e 1, G. S p r e n g-
1 i n g. Lieb. Ann., 554, 69 (1943).
61. C. S c h б p p, E. В e с к e r.
Lieb. Ann., 524, 49, 55 (1936).
62. R. Tschesche, F. Korte.
Chem. Ber., 84, 77 (1951).
63. M. С г о w e, A. Walker.
Science, 110, 166 (1949).
64. W. Pfleiderer. Angew. Chem.,
73, 581 (1961); Chem. Ber., 95, 2195
(1962).
65. C. S n о p f, K. G a n s h i r t.
Angew. Chem., 74, 153 (1962).
66. M. V i s с о n t i n i, H. S ii e r-
501
. 1 i n. Helv. chim. Acta, 46, 51
(1963).
67. M. V i s с о n t i-n i, ,H. S t i e r-
1 i n. Helv. Chim. Acta, 45, 2479
(1962).
68. H. Forrest, H. M i t c h e 1 1.
J. Am. Chem. Soc., 76, 5656, 5658
(1954).
69. J. Kerestesy et al. Science,
97, 465 (1943).
70. W. P f I e i de r e r. Z. Natur-
forsch., 18b, 420 (1963).
71. M. G о t о, К. К о b a у a s h i,
H. S a t o, F. К or t e. Lieb.
Ann., 689, 221 (1965).
72. С. В a a 1 e n, H. F о r r e s t.
J. Am. Chem. Soc., 81, 1770 (1959).
73. M. V i sc о n t i n i, E. Mo h 1-
m a n n. Helv. Chim. Acta, 42,
1679 (1959).
74. W. К о s c h a r a. Z. physiol. Chem.,
277, 284 (1943).
75. M. G о t o, A. S а к u г a i,
К. О h t a, H. Y a m a к a m i.
Tetrahedron Lett., 1967, 4507.
76. M. McNutt. J. Am. Chem.
Soc., 82, 217 (1960).
77. W. P f 1 e i d e r e r, G. Nubel.
Chem. Ber., 93, 1406 (1960).
78. M. M a 1 1 e t t e, E. T а у 1 о r,
С. C a i n. J. Am. Chem. Soc., 69,
1814 (1947).
79. E. W i t t 1 e, B. O’D e 1 1,
J. Vandenbelt, J. P f i f-
iner. J. Am. Chem. Soc., 69,
1768 (1947).
80. J. E. A n d e s, V. С. M у e r s.
J. Biol. Chem., 118, 137 (1937).
81. O. Sullivan. Proc. Soc. Exp.
Biol. Med., 33, 106 (1935).
82. J. D u b n о f f. J. Biol. Chem.,
141, 711 (1941).
83. J. Mowa t, J. Boothe,
В. H u t c h i n g s, E. S t о k-
s t a d, R. An gier, J. Se m b,
D. С о s u 1 i c h, Y. S u b b a-
г о w. J. Am. Chem. Soc., 70, 14
(1948).
84. F. W e у g a n d, V. S c h m i e d-
Kowarzik, A. Wacker,
W. Ruff. Chem. Ber., 83, 460
(1950).
85. R. Angier et. al. Science, 102,
227 (1945).
86. R. Angier, J. Boothe,
B. Hutchings, J. Mowa t,
J. S e m b, E. S t о к s t a d,
Y. Subbarow, C. W a 1 1 e r,
D. С о s u 1 i c h, M. F a h г e n-
bach, M. Hultquist,
E. Kuh, E. N о r t h e y, D.
Seeger, J. Sickels, J.
Smith. Science, 103, 667 (1946).
87, J. Al о w a t, В. H u t c h i n g s,
R. A n g i e r, E. S t о к s t a d,
J. Boothe, C. Waller,
J. Se mb, Y. Subbarow.
J. Am. Chem. Soc., 70, 1096(1948).
88, J. В oot he, J. M о w a t
B. Hutchings, R. Angier,
C. W a 1 1 e r, E. Stokstad,
J. Se m b, A. Gazzo la,
Y. Subbar ow. J. Am. Chem.
Soc., 70, 1099 (1948).
89. J. Boothe, J. Semb, C. Wa 1-
1 e r, R. A n g i e r, J. Al о w a t,
В. H u t c h i n g s, E. S t о k-
s t a d, Y. S u b b а г о w. J. Am
Chem. Soc., 71, 2304 (1949).
90. J. M о w a t, A. G a z z о 1 a,
B. Hutchings, J. Boothe,
C. W a 1 1 e r, R. A n g i e r,
J. S e ra b, Y. Subbarow.
J. Am. Chem. Soc., 71, 2308 (1949).
91. J. Semb, J. Boothe,
R. A n g i e r, C. W a 1 1 e r,
J.Mowa t, B. Hutchings,
Y. S u b b a г о w. J. Am. Chem.
Soc., 71, 2310 (1949).
92. E. R i с к e s, N. Tre n ner,
J.Conn, J. Keresztesy. J.
Am. Chem. Soc., 69, 2751 (1947).
93. A. P о h 1 a n d, E. Flynn,
R. J о n e s, W. S h i v e. J. Am.
Chem. Soc., 73, 3247 (1951).
94. В. M. Березовский,
Л. И. Стрельчунас,
M. Я. Каган. ЖОХ, 27, 1717
ne Q957>-
95. В. М. Березовский,
А. М. Ю р к е в и ч, И. К- Кри-
во ш е и н а. ЖОХ, 31, 2782 (1961).
96. D. S е е g е г, D. С о s u 1 i с h,
J. S m i t h, M. H и 1 t q и i s t.
J. Am. Chem. Soc., 71, 1753 (1949).
97. E. U у e o, S. M i z и k a m i.
Japan J. Pharmacy and Chem., 21,
237 (1949).
98. F. Weygand, G. Schaefer.
Chem. Ber., 85, 307 (1952).
99. F. W e у g a n d, H. M a n n,
H. Simon. Chem. Ber., 85,
463 (1952).
100. C. W a I ler, J. M о w a t.
Пат. США 2500296; С. A., 44, 5401
(1950).
101. P. L i t t 1 e, A. S a m p a t h,
V. P a g a n e 1 1 i, E. Locke,
Y. Subbarow. Trans. N. Y.
Acad. Sci., 10, 91 (1948).
102. В. К и p с а н о в а, А. Тру-
фа н о в. Биохимия, 14, 413
(1949).
103. W. Т г а и b е. Вег., 33, 1371,
3035 (1900).
104. С. W а 1 1 е г, М. F г а п к е п-
b а с h, J. В о о t h е, R. А п-
g i е г, В. Н и t с h i n g s, J. At o-
w a t, J. P о 1 e t t o, J. S e m b.
J. Am. Chem. Soc., 74, 5405 (1952).
. О к i I i n s h i c h i, Ai о r i t a
H о s h i г а. Яп. пат. 1083 (1953);
С. A., 48, 2124 (1954).
106. Англ. пат. 638415; пат. США
2512572; С. А., 44, 9488, 9989
(1950).
107. J. G е г о с i. Пат. США 2501168;
С. А., 44, 5924 (1950).
108. М. Н и 1 q и i s t, Р. Dreis-
b а с h. Англ. пат. 657830; С. А.,
46, 9622 (1952).
109. Пат. США 2520882; Off. Gaz.
U. S. Pat. Office, 637, 1570 (1950).
НО. М. Н и 1 t q и i s t, P, Drei-
502
sb a ch. Пат. США 2443165;
С. A., 42, 7944 (1948).
ill. F. Weygand, V. Schmied-
K о w a г z i k. Chem. Ber.,
82, 333 (1949).
112. S. К a w a n i s h i, К. M a t-
s u i. Яп. пат. 3327—3329 (1959);
С. A., 54, 14283 (1960).
113. S. Kawanis i. Яп. пат. 3329
(1959); РЖХ, 1960, 93548; англ. пат.
823827; С. А., 54, 5713 (1960).
114. F. К i п g, Р. S р е п s 1 е у. J.
Chem. Soc., 1952, 2144.
115. F. W е у g a n d, О. Swobo-
d a Chem. Ber., 89, 18 (1956).
116 J. В о о t h e. Пат. CHIA 2444002;
C. A., 43, 1812 (1949).
117. M. H u 1 t q u i s t, D. S e e-
ger. Пат. США 2472481; С. A.,
43, 6672 (1949).
118. M. Hultquist, P. Dreis-
b a c h. Пат. США 2472482; С. A.,
43, 3672 (1949).
119. S. Mar uy a m а. Яп. пат. 7978
(1958); С. A., 54, 4634 (1960).
120. D. Cosu 1 i ch. Пат. США
2444005; С. A., 42, 6862 (1948).
121. Англ. пат. 640092; С. А., 45, 2513
(1951).
122. В. Н u t с h i n g s, J. M о w a t,
J.Oleson, E. Stokstad,
J.Boothe, C. Waller,
R. Angier, J. Semb, Y. Sub-
barow. J. Biol. Chem. 170,
323 (1947).
123. D. Cosulich, J. Smith.
J. Am. Chem. Soc., 70, 1922 (1948).
124. B. Roth, J. S mi th, M. Hult-
quist. J. Am. Chem. Soc., 72,
1914 (1950).
125. B. Hutchings. Пат. CHIA
2470490, 2470491; C. A., 43, 6672
(1949).
126. M. О e s t e r 1 i n. Пат. ФРГ
931285; РЖХ, 1957, 9684.
127. Англ. пат. 713853; J. Appl.
Chem., 5, i 616 (1955).
128. Англ. пат. 680662; С. А., 48,
747 (1954).
129. А. Н a n ze. Пат. США 2662081;
С. А., 48, 4185 (1954).
130. Е. К u h. J. Smith. Пат.
США 2474184; С. А., 43, 7052
(1949).
131. С. Waller. Пат. США 2474022;
С. А., 43, 7974 (1949).
132. Швейц, пат. 263281; 268328; С. А.,
44, 4047 (1950); 45, 5193 (1951).
133. Н. Lindlar. Пат. США
2634271; С. А., 48, 5888 (1954).
134. Франц, пат. 987800; С. А., 50,
10802 (1956).
135. R. Angier, Е. Stokstad,
J. Mowa t, В. Hutchings,
J. Booth, C. Waller,
J. Semb, Y. Subbarow,
D. Cosulich, M. Fahren-
bach, M. H u 1 t q u i s,
E. Kuh, E. N о r t h e y,
D. Seeger, J. S i с к e 1 s,
J. Smith. J. Am. Chem.
Soc., 70, 25 (1948).
136. D. W e i s b 1 a t t, В. M a g e r-
1 e i n, A. H a n z e, D. M y-
e r s, S. Rolf son. J. Am.
Chem. Soc., 75, 3625 (1953).
137. D. W e i s b 1 a t t, В. M a g e r-
1 e i n, D. My e г s , A. H a n-
z e, E. F a i r b u r n, S. R о 1 f-
s о n. J. Am. Chem. Soc 75,
5893 (1953).
138. D. W e i s b 1 a t, E. F a i n-
b u г n. Пат. США 2650241;
РЖХ, 1954, 42271.
139. D. Weisblat, В. Mage r-
1 e i n. Пат. США 2631149; С. A.,
48, 1446 (1954).
140. D. Weisblat. Пат. ФРГ
913175; Zbl., 1955, 3205.
141. Англ. пат. 653068; С. А., 46,
3092 (1952).
142. Мекс. пат. 58886; С. А., 54,
24826 (1960).
143. Е. Н а е h п е г, Н. N a f z i-
g е г, Н. L u d e s. Klin. Wo-
chenschr., 29, 571 (1951).
144. M. H u 1 t q u i s t, E. К u h,
D. С о s u 1 i c h, M. F a h-
renbach, E. Northey,
D. Seeger, J.Sickels,
J. S m i t h, R. A n g i e r,
J. Boothe, H. Hutchings,
J. M о w a t, J. Semb,
E. Stokstad, Y. Subba-
row, C. W a 1 1 e r. J. Am.
Chem. Soc., 70, 23 (1948).
145. M. Hultquist. Пат. США
2466897; С. A., 43, 6245 (1949).
146. J. Boothe. Пат. США 2454751;
С. A., 43, 1813 (1949).
147. J. Boothe, C. Waller,
E. Stokstad, B. Hut-
chings, J. Mowa t,
R. Angier, J. Semb,
Y. S u b b a г о w, D. С о s u-
lich, M. Fahrenbach,
M. H u 1 t q u i s t, E. К u h,
E. Northey, D. Seeger,
J.Sickels, J. Smith. J.
Am. Chem. Soc., 70, 27 (1948).
148. Англ. пат. 624394 , 629440; С. A.,
44, 2574, 7356 (1950).
149. H. F о г г e s t, J. Walker.
J. Chem. Soc., 1949, 2077.
150. Пат. США 2561658; С. A., 46,
1052 (1952).
151. Англ. пат. 656403; С. А., 46,
7595 Л 9559
152. J. Semb. Пат. США 2491285;
С. А., 44, 3040 (1950).
153. Н. S р i е g е 1 b е г g. Пат. США
2487393; С. А., 44, 7356 (1950).
154. Швейц, пат. 256346; С. А., 44,
3040 (1950).
155. Англ. пат. 626171; 628305; С. А.,
44, 3536, 4046 (1950).
156. М. Viscontini, J. В i е г i.
Helv. Chim. Acta, 54, 2291 (1971).
157. M. S 1 e t z i n g e r, D. R e i n-
h о 1 d, J. G г i e г, M. В e a-
c h e m, M. T i s h 1 e r. J. Am.
Chem. Soc., 77, 6365 (1955); пат.
США 2740784; С. A., 50, 15601
(1956).
503
158. M. Sletzinger, M. T i s h-
1 e г. Пат. США 2786056, 2791586,
2816109, 2821527, 2821528; С. A.,
52, 9228, :9229, 10221, 11972 (1958).
159. N. Sletzinger, D. R e i n-
hol d. Франц, пат. 1113380;
РЖХ, 1958, 40732; пат. США
2816110; РЖХ, 1960, 6197.
160. L. Р 1 a n t е. J. Org. Chem., 36,
860 (1971).
161. R. Tschesche, F. Korte.
Пат. ФРГ 869961; С. A„ 52, 16386
(1958).
162. Швейц, пат. 253838; С. А., 44,
3040 (1950).
163. F. W е у g a n d, A. Wacker,
V. Schmied-Kowarzik.
Experientia, 4, 427 (1948).
164. R. A n g i е г, C. Wai let,
J. Boothe, J.Mowat,
J. S e m b, В. H u t c h i n g s,
F. S t о к s t a d, Y. S u b b a-
r о w. J. Am. Chem. Soc., 70,
3029 (1948).
165. P. Karrer, R. Schwyzer.
Helv. Chim. Acta, 33, 39 (1950).
166. P. Karrer, R. Schwyzer,
В. E r de n, A. S i e g w a r t.
Helv. Chim. Acta, 30, 1031, 1034
(1947).
167. J. S e m b. Пат. США 2477426;
С. A., 44, 1146 (1950).
168. F. King, P. S p e n s 1 e y. Na-
ture, 164, 574 (1949).
169. H. Petering, D. Weis-
b 1 a t. J. Am. Chem. Soc., 69,
2566 (1947).
170. P. Karrer, R. Schwyzer.
Helv. Chim. Acta, 31, 777, 782
(1948).
171. H. F or re st, J. W a 1 ke r.
J. Chem. Soc., 1949, 79, 83; Na-
ture, 161, 308 (1948).
172. C. W a 1 1 e r, A. Gol d man,
R. A n g i e r, J. В о о t h e,
В. H u t c h i n g s, J.Mowat,
J. S e m b. J. Am. Chem. Soc.,
72, 4630 (1950).
173. H. P e t e r i n g, J, S c h m i d t.
J. Am. Chem. Soc., 71, 3977 (1949).
174. Швейц, пат. 254801; С. A., 44, 669
(1950).
175. E. T а у 1 о г, К. Ее n ar d.
Lieb. Ann., 726, 100 (1969).
176. Синтезы органических препара-
тов, 4, 119; ИЛ, 1953.
177. В. М. Б е р е з о в с к и й,
Л. И. Стрельчунас. Тру-
ды ВНИ витаминного института,
5, 28, М., Пищепромиздат, 1954.
178. В. М. Березовский, М. Я.
Каган, Л. И. Стрельчу-
нас. Авт. свид. СССР 103777;
Бюлл. изобрет., 1956, № 7, 10.
179. N. R о у, N. К u п d u. Nature,
188, 581 (1960).
180. Ph. Landauer, Н. Ry-
d о п. J. Chem. Soc., 1953, 3721.
181. Герм. пат. 206453; С. А., 3,
1816 (1909).
182. А. А 1 b е г t, Н. W о о d. Na-
ture 172, 118 (1953); J. Appl. Chem.,
3, 521 (1953).
183. E. Fischer. Ber., 32, 2464
(1899).
184. S h . Y о s h i d а, К. I m a k i,
S. A k a g i. C. A., 44, 3442
(1950); Яп. пат. 989 (1953); С. A .
48, 2089 (1954).
185. H. Winter. J. Am. Chem. Soc ,
62, 3266 (1940).
186. M. Ste i ger. J. Org. Chem., 9,
396 (1944).
187. F. К i n g, P. S p e n s 1 e y,
R. Nimmo-Smith. Nature,
162, 153 (1940); С. A., 42, 8800
(1948).
188. I. Vander Scheek,
K. L a n d s t e i n. J. Immuno-
logy, 29, 373 (1953).
189. S. Rubbo, J. Gillespie.
Nature, 146, 838 (1940).
190. В. M. Родионов, Б. M. Бого-
слов с к и й, A. M. Ф e д о p o-
в а. Лабор. руководство по хи-
мии промежуточных продуктов и
красителей, М., Госхимиздат, 1948.
191. A. Hofmann. Вег., 9, 1299
(1876).
192. А. М. Б е р к е и г е й м. Химия
и технол. синтетич. лекарственных
средств, стр. 349, 350 и 352,
ОНТИ, 1935.
193. G. Fischer. Lieb. Ann., 127,
137 (1863).
194. J. W i 1 b г a n d, F.Beil-
stein. Lieb. Ann., 128, 257,
262 (1863).
195. W. L e w i s, H. C h e e t h a m.
Lieb. Ann., 43, 2117 (1921).
196. J. H i r a t а. Яп. пат. 109708;
С. A., 29, 4776 (1935).
197. R. Adams, F. Cohen,
O. R e e s. J. Am. Chem. Soc.,
49, 1093 (1927).
198. H. А. И з г a p ы ш e в, M. Я.
Ф и о ш и и. ДАН СССР, 90, 189
(1953).
199. A. L у d i п g. Пат. США 2735865;
С. А., 50, 10776 (1956).
200. W. К u m 1 е г. J. Am. Chem. Soc.,
68, 1184 (1946).
201. L. Doub, 1. Vandenbelt.
J. Am. Chem. Soc., 69, 2114 (1947).
202. J. Boothe, J.Mowat,
B. Hutchings, R. Angier,
C. W a 1 1 e r, E. S t о k-
s t a d, J. S e m b, A. G a z-
zola, Y. Subbarow. Trans.
N. Y. Acad. Sci., 10, 70 (1948).
203. J. Fruton, M. Bergman.
J. Biol. Chem., 127, 637 (1939).
204. A. A. P 1 e n t 1, J. H. P a g e.
J. Biol. Chem., 1963, 59 (1946).
205. H. M i t c he 1 1, E. S n e 1 1,
R. W i 1 1 i a m s. J. Am. Chem.
Soc., 66, 267 (1944).
206. W. Darby, E. Jones,
H. J о h n s о n. J. Am. Med.
Assoc., 130, 780 (1946).
207. J. Guest, S. Friedman,
M. D i 1 w о r t h, D. W о о d s.
504
Ann. N. Y. Acad. Sci., 112, 774
(1964).
208. J. P f i f f n e r, D. С a 1 к i n s,
E. Bloom, B. O’D e 1 1. J. Am.
Chem. Soc., 68, 1392 (1946).
209. Г. И. Першин, Л. Щ e p 6 a-
к о в а. Биохимия, 16, 171 (1951).
210. J. P f i f f n e г, A. Hogan.
Vitamins and Hormones, vol. 4, 1;
New York, 1946.
211. В. H u t c h i n g s, J.Mowat.
Vitamins and Hormones, vol. 6, 1;
New York, 1948.
212. H. А. Андреева. Витамины
группы фолиевой кислоты, изд.
АН СССР, М., 1963.
213. В. Кирсанова, А. Тру-
фа н о в. Биохимия, 15, 243 (1950).
214. Р. Р а 1. Sci. Culture 19, 160 (1953);
J. Indian. Chem. Soc., 31, 673
(1955); C. A., 50, 1035 (1956).
215. W. Wright, D. С о s u 1 i c h,
M. Fahrenbach, C. Wal-
ler, J. Smith, M. Hult-
quist. J. Am. Chem. Soc., 71,
3014 (1949).
216. B. Magerlein, D. Weis-
b 1 a t. J. Am. Chem. Soc., 76,
1702 (1954).
217. D. W о 1 f, K. F о 1 к e r s. Пат.
США 2515483; С. A., 44, 8963
(1950).
218. D. В г о w n. J. Chem. Soc., 1953,
1644.
219. G. M a r t i n, H. U r i s t. Пат.
США 2478873; С. A., 43, 9389
(1949).
220. J. Boot he, J. Mowat,
C. W a 1 1 e r, R. Angier,
J. S e m b, A. G a z z о 1 a.
J. Am. Chem. Soc., 74, 5407 (1952).
221. M. Hultquist, J. Smith,
D. S e e g e r, D. С о s u 1 i c h,
E. Kuh. J. Am. Chem. Soc., 71,
619 (1949).
222. G. M a r t i n, L. T о 1 m a n,
J. Moss. Archiv. Biochem.,
12, 318 (1947).
223. H. S a u b e r 1 i c h, С. В a u-
m a n n. J. Biol. Chem., 176, 165
(1948).
224. R.Tschesche, F. К or-
t e. Пат. ФРГ 887812; С. A., 52,
16385 (1958).
225. В. R oth, J. S m i t h, M. Hu-
ltquist. J. Am. Chem. Soc.,
73, 2864, 2869 (1951).
226. D. Seeger, J. Smith,
M. H u 1 t q u i s t. J. Am. Chem.
Soc., 69, 2567 (1947).
227. J. S m i t h. Пат. США 2525150;
С. A., 45. 3429 (1951).
228. Датск. пат. 76602; РЖХ, 1956,
7996.
229. В. М. Березовский, Г. Д.
Глебова, Е. М. Б и р и н-
берг, Л. В. Казанская.
Хим. фарм. ж., 2, № 12, 15 (1968).
230. В. М. Березовский, Г. Д.
Глебова, Е. М. Б и р и н-
берг, Л. В. Казанская.
Хим. фарм. ж., 1968, № 12, 15.
231. С. Waller, В. Hutchings,
J. Mowat, Е. Stokstad,
J. Boothe, R. Angier,
J. S e m b, Y. S u b b а г о w,
D. Cosulich, At. Fahren-
bach, M. Hultquist,
E. Kuh, E. N о r t h e y,
D. Seeger, J. S i e k e 1 s,
J. S m i t h. J. Am. Chem. Soc.,
70, 19 (1948).
232. A. T о m c uf c i k, D. S e e-
ger. J. Org. Chem., 26, 3351
(1961).
233. J. T о t t e г, A. В e s t. Arch.
Bioph. Biophys., 54, 318 (1955).
234. E. F a г b e r, L. D i a m о n d,
R. Mercer, R. Sylvester,
J. Wolff. New England J.
Med., 238, 787 (1948).
235. J. Smith, N. Jersey.
J. Pharm., 22, 15 (1949).
236. Г. H. Першин. ЖВХО им.
Д. И. M e н д e л e e в a, 15, 216
(1970).
237. R.Tschesche, Z. Zakoze-
w s k у, F. Korte. Chem. Ber.,
86, 450 (1953).
238. Пат. ФРГ 921698; С. A,, 52,
13808 (1958).
239. L. Goodman, J. DeGraw,
R. К i s 1 i n k, M. F r i e d-
kin, E. Paslore, E. Craw-
ford, L. P 1 a n t e, Aly
Ab-Nakas, J. Morning-
star, G. К w о k, L. W i 1-
son, E. Donovan, J. Rat-
z a n. J. Am. Chem. Soc., 86,
308 (1964).
240. J. DeGraw, J.Marsch,
E. A c t о n, О. С r e w s,
С. M о s h e r. J. Org. Chem., 30,
3404 (1965).
241. С. M о s h e r, E. A c t о n,
О. С г e w s, L. G о о d m a n.
J. Org. Chem., 32, 1452 (1967).
242. M. Viscontini, J. Meier.
Helv. Chim. Acta, 32, 877 (1949).
243. E. Fairburn, B. Mager-
lein, L. Stubberfield,
E. Starert, D. Weisr
b 1 a t. J. Am. Chem. Soc., 76,
676 (1954).
244. G. Elion. Пат. США 3242178;
РЖХ, 1967, 7H356.
245. M. Polonovski, V i е lie-
fosse, M. Pesson. Bull,
soc. chim. France, (5), 12, 78
(1945).
246. G. E 1 i о n, G. H u t c h i n g s.
J. Am. Chem. Soc., 69, 2553 (1947).
247. E. G a 1. J. Am. Chem. Soc., 72,
3532, 5315 (1950).
248. E. T а у 1 о r. J. Am. Chem. Soc.,
74, 1648 (1952).
249. D. Woods. P. F i e 1 d s. Chem.
Ind., 59, 133 (1940).
250. P. Fields. Lancet, 1940, 944,
955.
251. S. Ansbacher. Science, 93,
163 (1941).
252. B. Sure. Science, 94, 167 (1941).
505
253. R. Ku h п, К. Sc h warz, Ber.,
74, 1617 (1941).
254. H. Д. Иерусалимский.
Азотное и витаминное питание мик-
робов, Изд. АН СССР, 1949.
255. F. R i b е г i о. J. Biol. Chem.,
152, 665 (1944).
256. S. W i e d 1 i n g s. Science, 94,
389 (1941).
257. S. Ratner, M. Blanchard,
D. G г e e n. J. Biol. Chem., 164,
691 (1946).
258. C. Scott, E. Robbins.
Proc. Soc. Exptl. Biol. Med., 49,
184 (1942).
259. G. D om a gk. Klin. Wochen-
schr., 15, 1576 (1936).
260. О. Ю. Магидсон, Успехи
химии, 15, 101 (1946).
261. D. Woods. J. Exp. Med., 75,
369, 383 (1942).
262. W. Allen, R. Posternak,
W. S e a m о n. J. Am. Chem.
Soc., 74, 3264 (1952).
263. С. M a t h e w s, F. H u e n n e-
k e n s. J. Biol. Chem., 235, 3304
(1960).
264. B. R о t h, M. H u 1 f q u i st,
M. F a h r e n b a c h, D.Co-
sulich, H. Broquist,
J. Brockman, J. Smith,
R. Parker, E. Stock-
s t a d, T. J u k e s. J. Am. Chem.
. Soc., 74, 3247 (1952).
265. D. C osu I i c h, J. S m i t h,
H. В r oq u i st. J. Am. Chem.
Soc., 74, 4215 (1952).
266. D. С о s u 1 i c h. Пат. США
2688018; С. A., 49, 11727 (1955);
аигл. пат. 735112; С. А., 50, 7884
(1956).
267. М. М а у, Т. Bardos, F. Ваг-
ger, М. Lansford, J. В а -
v е 1, G. Sutherland,
V. S h i v е. J. Am. Chem. Soc.,
73, 3067 (1951).
268. D. С о s u 1 i c h, B. R о t h,
J. S m i t h, M. Hultquist,
R. P a г k e r. J. Am. Chem. Soc.,
74, 3252 (1952).
269. T. Bond, T. В a r d о s,
M. S i b 1 e y, W. S h i v e. J. Am.
Chem. Soc., 71, 3852 (1949).
270. J. Brockman n, B. Roth,
H. В г о q u i s t, M.Hult-
q u i s t, J. S m i t h, M. F a h-
renbach, D. Cosulich,
R. Parker, E. Stokstad,
T. J u k e s. J. Am. Chem. Soc.,
72, 4325 (1950).
271. P. Ho, L. J о n e s. Biochim.
Biophys. Acta, 148, 622 (1967).
272. M. О s b о г n, P. T a 1 b e r t,
F. Huennekens. J. Am.
Chem. Soc., 82, 4961 (1960).
273. R. Blakley. Biochem. J., 72,
707 (1959).
274. M. О s b о г n, P. T a 1 b e r t,
F. H u j ne kensa. J. Am.
Chem. Soc., 82, 4921 (1960).
275. R. Blakley. J. Biol. Chem.,
238, 2113 (1963).
276. A. Larrabee, S. Rosen-
thal, R. C a n t h о n, J. В a-
c h a n a n. J. Am. Chem. Soc -
83, 4094 (1961).
277. J. C h a n a r i n, J. P e r r y.
Biochem. J., 105, 633 (1967).
278. V. Gupta, F. Huenne-
kens. Arch. Biochem. Biophys
120, 712 (1967).
279. K. Donaldson, J. Keres-
z t e s y. Biochem. Biophys. Res.
Communs, 5, 286, 289 (1960).
280. W. S h i v e, T. Bardos,
T. В о n d, L. Rogers. J.
Am. Chem. Soc., 72, 2817 (1950)
281. E. Flynn, T. Bond, T. Bar-
dos, W. S h i v e. J. Am.
Chem. Soc., 73, 1979 (1951).
282. D. С о s u 1 i c h, B. R о t h,
J. Smith, M. Hult-
q u i s t, R. Parker. J. Am.
Chem. Soc., 73, 5006 (1951).
283. А. В о b s t, M. V i s с о n t i-
n i. Helv. Chim. Acta, 49, 875
(1966).
284. R. Blakley. Nature, 188, 231
(1960).
285. D. L e о d i s. Anal. Biochem.,
26, 459 (1968).
286. M. S i 1 v e r m a n n, J.No-
r о n h a. Biochem. Biophys. Res.
Communs, 4, 180 (1961).
287. В. H i 1 1 с о a t, R. В 1 a k-
1 e y. Biochem. Biophys. Res. Com-
muns, 4, 180 (1961).
288. J. Brockman, B. Roth.
Пат. ФРГ 941848; РЖХ, 1958.
33591
289. E. К a c z k a, K. F о 1 k e r s.
Пат. США 2632759; РЖХ, 1954,
42272.
290. Швейц, пат. 294897; Zbl., 1954,
6290; Англ. пат. 733062; С. А., 50,
7884 (1956).
291. W. S h i v е. Пат. США 2741608;
РЖХ, 1958, 33592.
292. Швейц, пат. 259123; Англ. пат.
631516; С. А., 44, 3040, 5400
(1950).
293. Н. L i п d 1 а г, Н. К 1 а е п.
Пат. США 2520156; С. А., 45, 3423
(1951).
294. S. Z a k г z е w s к i, С. N i-
с h о 1. Biochem. Biophys Acta,
27, 425 (1958).
295. M. О s b о г п, P. T a 1 b e r t,
F. Huennekens. J. Am.
Chem. Soc., 82, 492 (1960).
296. J. К а у, M. О s b о r n, Y. H a-
t i f, F. H u e n n e к e n s. J.
Biol. Chem., 235, 195 (1960).
297. J. Peters, D. Green-
berg. J. Am. Chem. Soc., 80,
2719 (1958).
298. M. V i s с о n t i n i, J. В i e r i,
Helv. Chim. Acta, 55, 21 (1972).
299. T. J u к e s. Intern. Z. Vitamin-
forsch, 23, 356 (1952).
300. M. S c h u 1 m a n, J. В uch a-
n a n. J. Biol. Chem., 196, 513
(1952).
506
301. G. Gree nb e r g. J. Am. Chem.
Soc., 76, 1458 (1954).
302. T. Winnick, J. Morin g-
Glesson, D. Greenberg.
J. Biol. Chem., 175, 127 (1948).
303. В. H о 1 I a n d, W. M e i n к e.
J. Biol. Chem., 178, (1949).
304. W. S a к a ni i. Federation Proc.,
8, 246 (1949).
305. S. H a г t m a n, J. В u c h a-
n a n. J. Biol. Chem., 234, 1812
(1959).
306. A. M i 1 1 e r, H. W a e 1 s c h.
J. Biol. Chem., 228, 365 (1957).
307. H. T a b о r, L. W у n g a a r-
den. Federation Proc., 18, 336
(1959); J. Biol. Chem., 234, 1830
(1959).
308. J. Rabinowitz, W. P r i-
c e r. J. Am. Chem. Soc., 78,
5702 (1956).
309. D. Brown, O. Silva,
R. G a r d i n e r, M. Silver-
man. J. Biol. Chem., 235, 2058
(1960).
310. B. Bright, M. Anderson.
J. Biol. Chem., 230, 1, 271 (1958).
311. A. Bobs t, M. Viscont i-
n i. Helv. Chim. Acta, 49, 884
312. ^.Huennekens, M. Os-
born. Advances in Enzymol.,
21, 369 (1959).
313. B. McDougall, R. В 1 a k-
1 e v. J. Biol. Chem., 236, 832
(1961).
314. D. Greenberg, G. Humph-
rey s. Feredation Proc., 17,
234 (1958); J. Biol. Chem., 236,
2217 (1961).
315. A. P e г a u 1 t, В. P u 1 1-
m a n. Bioch. Biophys. Acta, 44,
251 (1960).
316. K. G i b s о n, J. W i 1 s о n,
A. Ude nfr iend.. J. Biol.
Chem., 236, 673 (1961).
ГЛАВА XII
ФЛАВИНОВЫЕ ВИТАМИНЫ И КОФЕРМЕНТЫ
РИБОФЛАВИН
7, 8-Диметил-10-№(Г-Д-рибитил)изоаллоксазин (I), рибофлавин1 — ви-
тамин В2 11—3]—относится к флавинам — желтым красящим вещест-
вам, в основе строения которых лежит система изоаллоксазина, являющаяся
пиримидо[4,5-/1хиноксалиновой конденсированной системой из срощенных
ароматического, пиразинового и пиримидинового циклов или 6eH3o[g]nTe-
ридиновой системой из ароматического и птеридинового циклов2
СН2ОН
НОСН
I
НОСН
Рибофлавин (I) представляет собой желто-оранжевого цвета иглы, со-
бранные в друзы, горького вкуса ст. пл. 282° С (с разл.). Для кристаллов
1 7,8-Диметилизоаллоксазины обозначаются термином «флавины». Тривиальные
названия конкретных соединений включают приставку, характеризующую замести-
тель в положении 10, причем пентитные и гекситные заместители обозначаются по
названию соответствующего им сахара, например, 7,8-диметил-10-Ы-(1'-£>-дульцитил)-
изоаллоксазии называется О-галактофлавином.
2 Нумерация цикла приводится в соответствии с номенклатурой JUPAC [4].
В ряде источников применяется иная нумерация атомов азота и углерода молекулы
изоаллоксазина: 10, 9 для атомов азота пиразинового цикла (вместо 5, 10) и 5, 6, 7, 8
для атомов углерода ароматического цикла (вместо 6, 7, 8, 9).
507
рибофлавина характерен полиморфизм: он может кристаллизоваться в виде
пластинок с т. пл. 290° С 15]. Рибофлавин мало растворим в воде — 0,012%
при 27,5° С, 0,019% при 40° С и 0,230% при 100° С; еще меньше — в мети-
ловом, этиловом (0,0045%), н-бутиловом и амиловом спиртах, в феноле,
пиридине, амилацетате и циклогексане; нерастворим в ацетоне, эфире,
бензоле, хлороформе и бензине.
В соляной кислоте растворимость рибофлавина при 15° С составляет
(в %): 0,026 в 3%-ной, 0,06 в 6%-ной, 0,22 в 9%-ной, 0,61 в 12%-ной,
1,0 в 15%-ной, 2,9 в 18%-ной, 4,3 в 21%-ной, 6,7 в 24%-ной и 18 в 30%-ной
[6]. Рибофлавин умеренно растворим в ледяной уксусной и муравьиной
кислотах (до 1%).
Концентрированные водные растворы натриевой соли п-аминобензой-
ной кислоты (до 30%) применяются для приготовления препаратов рибо-
флавина; в них можно растворить до 4% рибофлавина [7]. Водный раствор
никотинамида в концентрации 5—50% растворяет от 0,1 до 2,5% рибофла-
вина [81. Многие другие вещества в водном растворе создают среду, в кото-
рой растворяется значительно большее количество рибофлавина, чем в
воде [8].
Из кислых водных растворов рибофлавин осаждается фосфорноволь-
фрамовой кислотой, сернокислой ртутью и частично уксуснокислым свин-
цом; из нейтральных растворов — азотнокислым серебром. Из таких же
растворов рибофлавин хорошо адсорбируется фуллеровой землей, нори-
том, асканитом, а из нейтральных — франконитом. Из адсорбентов он
вымывается водно-пиридиново-метанольной смесью [9,10].
Насыщенный водный раствор рибофлавина имеет pH около 6,0. Он обла-
дает зеленовато-желтой окраской и характерной для флавинов интенсив-
ной желто-зеленой флуоресценцией; в кислой и щелочной среде окраска
ослабевает. Спектр флуоресценции имеет полосу в области зеленого и
желтого между 515 и 615 нм [11] с максимумом около 530 нм (в воде) [12].
и наибольшей интенсивностью при pH 6—8 [13] и зависит от концентрации
раствора [14]. Флуоресцирующая способность рибофлавина положена в;
основу аналитического его определения в природных продуктах.
Рибофлавин имеет спектр поглощения с максимумами (в воде): 445 нм
(в видимой области), 374 , 268 , 223 нм (в ультрафиолетовой области) [15];
е 12,3-103, 10,8-103 31,4-103 и 30,1-103 соответственно [16].
Константы диссоциации рибофлавина составляют: Ко 6,3-10~12 и
Kfc 0,5-10“5; изоэлектрическая точка находится при pH 6 [17].
В слабощелочном растворе рибофлавин показываем левое вращение
[ц]о —Н4° (0,125% в 0,1 н. NaOH) [14,18]; удельное вращение зависит от
концентрации раствора и при 0,06% составляет [ц]®’ —70°; в нейтраль-
ных и кислых растворах оптического вращения не наблюдается [19].
Помимо рибофлавина, в тканях организма, например в сетчатке глаза,
находятся продукты его частичного расщепления — 7, 8, 10-триметилизоал-
локсазин, люмифлавин (II) и 7, 8-диметилаллоксазин, люмихром (III)
сн.
Люмифлавин (II), имеющий т. пл. 330° С, в растворах обладает желтой
окраской и флуоресценцией, подобной окраске и флуоресценции рибофла-
вина, однако более интенсивной [20]; кривая абсорбции и максимумы по-
глощения также подобны, но в отличие от рибофлавина люмифлавин раст-
508
ворим в хлороформе. Он обладает окислительно-восстановительными свой-
ствами, но биологически неактивен [21, 221.
Люмихром (III) —лимонно-желтоватое вещество — в щелочных раст-
ворах обладает интенсивной желтой окраской с желто-зеленой флуорес-
ценцией; его нейтральные, водные и хлороформенные растворы имеют не-
бесно-голубую флуоресценцию [201. Максимумы поглощения люмихрома:
в воде — 356, 262 и 220 нм; в спирте — 387, 339, 260, 221 нм.
ХИМИЧЕСКИЕ СВОЙСТВА РИБОФЛАВИНА И ДРУГИХ АЛЛО-
И ИЗОАЛЛО КС АЗИНОВ
Гидроксильные группы рибитильной цепи рибофлавина легко образуют
простые и сложные эфиры. Первоначально этерификации подвергается
5'-первичная гидроксильная группа, а затем вторичные гидроксилы в по-
ложениях 2', 3' и 4'. При действии на рибофлавин трифенилхлорметана
в среде пиридина тритилируется первичная гидроксильная группа и обра-
зуется 5'-тритилрибофлавин [231. Уксусный ангидрид или хлористый аце-
тил ацетилируют рибофлавин по гидроксилам рибитильной цепи с обра-
зованием 2', 3', 4', 5'-тетраацетилрибофлавина (IV) с т. пл. 242—244° С,
лучше растворимого в воде, чем рибофлавин [22, 24, 25]. При действии бу-
тилового эфира в 60%-ном водном растворе хлорной кислоты или бутирил-
хлорида [26] на рибофлавин образуется 2', 3', 4', 5'-тетрабутилрибофлавин
(т. пл. 145—147° С). Подобным образом получаются 2', 3', 4', 5'-тетрапаль-
митилрибофлавин [26,27] с т. пл. 78,5° С, 2', 3', 4', 5'-тетраникотинилрибо-
флавин [28] и другие эфиры.
При действии на рибофлавин различных фосфорилирующих агентов,
в качестве которых применяют хлорокись фосфора, монохлорангидрид
ортофосфорной кислоты, хлорангидриды диалкилзамещенных ортофосфор-
ной кислоты, реагирует первичная гидроксильная группа и образуется ри-
бофлавин-5'-фосфат (V) [29—37], а также другие эфиры — ди- и поли-5'-
фосфаты рибофлавина.
снрсосц,
снэсоосн
сн3соосн
СН3СООСН
сн,
он
СН,О-Р-ОН
I 2 II
носн о
носн
носн
Рибофлавин-5'-фосфат, флавинмононуклеотид, является коферментом и
в соединении со специфическим белком в виде соответствующих ферментов
катализирует окислительно-восстановительные реакции обмена веществ
(см. флавиновые коферменты).
Рибофлавин реагирует с ацетоном, образуя по гидроксильным группам
соответствующие моно- и диизопропилиденовые производные [38].
При взаимодействии рибофлавина с концентрированной серной кисло-
той [39] или с хлорсульфоновой кислотой в хлороформе получаются моно-,
ди-, три- и тетрасульфокислые эфиры рибофлавина [40, 41], не обладающие
витаминной активностью [39].
Рибофлавин, подвергнутый обработке соляной кислотой, испытывает
дегидратацию и дает 2', 5'-ангидрорибофлавин (VI) [42, 43], а при действии
n-толуолсульфохлорида образует 4', 5'-ангидрорибофлавин (VII). Он спо-
собен образовывать глюкозиды пираноидной формы (VIII), выделенные
из экстрактов печени крыс [44] или из культуральных сред [45, 46].
509
ОН ОН о
Г I
сн2-с-с—с—сн,
I Н Н «. S’
С борной кислотой рибофлавин образует хорошо растворимый в воде
комплекс 147] состава Q7Hj9O6N4Na-Na2B4O7-10Н2О [391; комплексообра-
зование, возможно, идет по 2', З'-гидроксильным группам. С большим избыт-
ком борной кислоты в образование комплекса вовлекаются все гидроксиль-
ные группы [48].
С солями некоторых металлов рибофлавин образует нерастворимые,
интенсивно окрашенные хелатные, или «клешневидные», комплексы, по-ви-
димому, по атому азота положения 5 и кислороду карбонильной группы
положения 4; точное строение комплексов не установлено. В качестве ме-
таллов в таких комплексах участвуют: Ag* [49], Hg*, Cu+, Fe**, Co**, Ni**,
Cu** Zn**, Mn** [50—54]. Хелатные комплексы с одновалентными металлами
более устойчивы, чем с двухвалентными. Следует отметить, что биокаталити-
ческая активность многих флавиновых ферментов связана с содержащимися
в них ионами металлов, такими, как железо, молибден, медь или марганец,
[50, 51, 55].
Рибофлавин и другие алло- и изоаллоксазины стабильны в водных
кислых растворах. Характерной особенностью соединений алло- и изоалло-
ксазинового ряда является неустойчивость гетероциклического ядра мо-
лекулы в щелочных растворах, особенно при нагревании. Рибофлавин
полностью расщепляется в 1%-ном водном растворе едкого натра за 24 ч
при комнатной температуре. Расщепление рибофлавина (I) в щелочной сре-
де (1 н. едкий натр, нагревание на кипящей бане в течение нескольких ми-
нут) протекает с образованием мочевины и 6, 7-диметил-1-К-(Г-£)-рибитил)-
-2-oKco-l, 2-дигидрохиноксалин-З-карбоновой кислоты (IX) — т. пл. 183—
183,5° С (с разл.) [56, 57].
CHJCHOHJjCHjOH
IX
4- HjNCONHj
Подобным образом происходит расщепление люмифлавина (II) при на-
гревании с баритовой водой в 1, 6, 7-триметил-2-оксо-1, 2-дигидрохинокса-
лин-3-карбоновую кислоту [22] и люмихрома (III)—при нагревании в раст-
воре едкого натра в 6, 7-диметил-2-оксихиноксалин-3-карбоновую кисло-
ту [58].
Расщепление пиримидинового цикла аллоксазинов водным аммиаком
происходит иначе, чем едким натром или баритовой водой, — из алло-
ксазина получается 2-аминохиноксалин-З-карбоновая кислота, декарбокси-
лирующаяся в 2-аминохиноксалин [58, 59].
Алло- и изоаллоксазины различаются по положению системы двойных
связей (—Ч=С10!1—C4a=N5—или—Nlc==C10a—С4а—N5—), характеризуются
II I I
510
значительным различием химических, физико-химических и биологических
свойств.
Осуществлен переход от изоаллоксазинов к аллоксазинам путем отще-
пления боковой цепи при 10-N. Так, при нагревании люмифлавина (II)
в уксусной кислоте, содержащей гидроксиламин, в течение 120 ч получен
люмихром (III) 160]. Обратный переход — получение флавинов путем алки-
лирования люмихрома — не известен.
Пиролитическое расщепление рибофлавина при температуре выше его
температуры плавления приводит к образованию люмифлавина (II) и люми-
хрома (III) [61].
В присутствии света, особенно прямого солнечного [5,19] или ультрафио-
летового, рибофлавин в кислых растворах подвергается расщеплению; фото-
лиз протекает сильнее в слабых концентрациях. Скорость фотореакции
увеличивается с повышением температуры и pH и с увеличением интенсив-
ности облучения (особенно при 365—590 нм) [5, 62, 63].
В нейтральных или кислых растворах происходит фотолитическоедез-
алкилирование рибофлавина (I) в положении 10-N, приводящее к образо-
ванию люмихрома (III). Фотолиз рибофлавина в щелочных растворах со-
провождается расщеплением боковой рибитильной цепи с образованием
люмифлавина (II) и только частично люмихрома (III) [19, 21, 22]
При фотолизе рибофлавина (I) в щелочных растворах с одновременным
окислением перекисью водорода происходит расщепление С(2-) — С(3 ) связи
боковой цепи в положении 10 с образованием около 7% 7,8-ди метил-10-кар-
боксиметилизоаллоксазина (X) [64] наряду с соединениями II и III.
Первичным продуктом фоторасщепления рибофлавина, по-видимому,
является 10а-формиллюмифлавин (XI) [65]
Неустойчивая к свету рибитильная группа рибофлавина стабилизируется
пропусканием кислорода в щелочной раствор [63, 66, 67]. Однако в водном
растворе в отсутствие кислорода рибофлавин на свету не испытывает фото-
расщепления, а восстанавливается в 1,5-дигидрорибофлавин (XII), причем
донором протонов служит вода [68].
В отличие от рибофлавина 1,5-дигидрорибофлавин (XII) устойчив к фо-
толизу, следовательно, для фотореакции необходимо наличие в молекуле
сопряженной системы—Nj=C—C=N5—[69]. Эта же система обусловливает
и флуоресценцию изоаллоксазинов [70]. Люмифлавин (II) не расщепляется
при 20° С, однако в горячем нейтральном или слабокислом растворе он
511
фотолитически дезалкилируется в люмихром (III) 160]. Это характерная ре-
акция превращения изоаллоксазинов в аллоксазины.
Свет не является специфическим катализатором реакции расщепления
изоаллоксазинов в аллоксазины. Собственно расщепление катализируется
ионами металлов [66, 67, 71], такими, как Fe++, Sn++, Со++, в некоторых слу-
чаях основаниями [72] и реагентами на карбонильную группу [60], четвер-
тичными аминами и тпоэфирами [73]. Фотолиз тормозится третичными
ами нами.
Показательно, что светочувствительность флавинов, приводящая к рас-
щеплению до люмихрома (III), обусловливается наличием в боковой цепи
свободной гидроксильной группы в 2'-положении [74]. 2', 3', 4', б'-Тетра-
ацетилрибофлавин (IV), у которого в этом положении отсутствует свобод-
ная гидроксильная группа, при облучении не разрушается. Светочувстви-
тельностью обладают 10-(2'-оксиэтил)- (XIII) и 10-(2', З'-диоксипропил)
изоаллоксазин (XV), имеющие свободную гидроксильную группу в
2'-положении; они расщепляются до аллоксазина (XIV).
Скорость фотолиза связана с характером гидроксильных групп в поло-
жении 2'; для первичной, вторичной и эфирной групп она находится в
соотношении 100 : 15 : 5 соответственно [75].
Смещение гидроксильной группы из 2'- в З'-положение приводит к силь-
ному ослаблению светочувствительности, что наблюдается у 10-(3'-окси-
пропил)изоаллоксазина. Расщепление флавинов зависит от рода облучения,
и некоторые соединения, не содержащие в 2'-положении гидроксильной
группы, хотя и более устойчивы к свету, чем рибофлавин, но в нейтральных
растворах также переходят в аллоксазин, например 7,8-диметил-10-М-(£>-2'-
-дезоксирибитил)изоаллоксазин [76]. При расщеплении флавингликозидов
под влиянием света также образуется люмихром (III) [24].
Реакция фоторасщепления флавинов изучалась в зависимости от длины
и степени гидроксилирования боковой цепи при С(ю) [77]. Из реакционной
смеси были выделены различные промежуточные продукты фотолитиче-
ского расщепления изоаллоксазинов [63]. Предложен механизм фотолиза
[75, 78, 79].
Рибофлавин (I) устойчив к окислителям, даже таким сильным, как азот-
ная кислота, перекись водорода и бром, но окисляется хромовой кислотой
[13] и марганцовокислым калием. При окислении тетраацетатом свинца
или йодной кислотой расщепляется боковая цепь между соседними гидрок-
сильными группами и образуется 7,8-диметил-10-формилметилизоаллокса-
зин (XI) [80, 81]; это же соединение получается и при фотолизе.
Люмифлавин в кипящей уксусной кислоте окисляется избытком азо-
тистой кислоты по метильной группе положения 8 с образованием 8-кар-
боксинорлюмифлавина [82]. Это соединение малоустойчиво и в щелочном
растворе расщепляется до 8-карбоксинорлюмихрома [82].
Другие изоаллоксазины также устойчивы к перекиси водорода. В отли-
чие от них аллоксазины окисляются перекисью водорода при нагревании
в муравьиной кислоте, однако с образованием только 10-моно-П-окисей
[83—85]. При таком окислении, например, люмихрома (III) и 8-хлоралло-
ксазина были получены IO-N-окись люмихрома (XVI) и IO-N-окись 8тхло-
раллоксазина (XVII) [85].
512
Однако, помимо lO-N-окисей аллоксазинов, к существованию способны
и синтетически полученные S-N-окиси аллоксазинов 186]. В то же время
5,10-ди-М-окиси аллоксазинов имеют неравноценные по стабильности
N-окисные группы; так, при нагревании, например, 5,10-ди-М-окиси 8-нор-
люмихрома (XVIII) в муравьиной кислоте происходит отщепление кислоро-
да у атома азота в положении 5 и образование lO-N-окиси 8-норлюмихрома
(XIX) [87].
XVIH XIX
Примечательно, что недавно синтетически получены и N-окиси изо-
аллоксазинов, например, 5-М-окись рибофлавина .(с. 523), неизвестная в
в природе.
Все естественные и многочисленные синтетические флавины обладают
характерными окислительно-восстановительными свойствами благодаря на-
личию в их молекуле окислительно-восстановительной системы
I 2 [Н] : |
N=C—C=N— —N—С=С—N—
| — 2[H] H |- - H <“
Они способны восстанавливаться химически или каталитически в бес-
цветные дигидросоединения, которые при окислении вновь превращаются
в окисленную форму. Хромофором, обусловливающим окрашенность со-
единений изоаллоксазинового ряда, является азометиновая группировка
>C=N—.
Рибофлавин (I-) легко восстанавливается водородом каталитически, элек-
тролитически, гидросульфитом натрия, цинком в присутствии кислоты и
другими восстановителями в 1,5-дигидрорибофлавин, лейкорибофлавин
(XII) с присоединением двух протоков и двух электронов; при этом окраска
раствора сильно ослабляется, а флуоресценция исчезает [10]. 1,5-Дигидро-
рибофлавин легко реокисляется в рибофлавин уже при воздействии кис-
лорода воздуха.
При восстановлении рибофлавина (I) образуются промежуточные со-
единения с интенсивной окраской, являющиеся комплексами рибофлавина
или его дигидроформы с семихиноидными радикалами (радикалами частично
восстановленной формы рибофлавина) [88]. Семихиноидный радикал —
моногидрорибофлавин (XX), отдавая один электрон, превращается в катион
флавина или, присоединяя электрон и протон, —в 1,5-дигидрорибофлавин
(XII) (схема 118).
На свойстве рибофлавина образовывать частично восстановленные про-
межуточные соединения, обладающие меньшей растворимостью в воде, чем
рибофлавин (растворимость в 100 мл: рибофлавин — 12 мг, вердофлавин —
5 мг, хлорофлавин — 2 мг, родофлавин—1мг и 1,5-дигидрорибофлавин
0,8 мг [5]), основан метод его выделения из разбавленных растворов или
культуральных жидкостей после микробиологического синтеза.
17-69
513
Флавины находятся на воздухе в устойчивой окисленной форме (ФлН),
поскольку их окислительно-восстановительный потенциал (например, для
рибофлавина он составляет—0,21 В) значительно ниже потенциала атмосфер-
ного кислорода (+0,82 В) 1891. На схеме 119 представлена окислительно-
Схема 118
Промежуточные соединения, образующиеся при восстановлении рибофлавина
ДигпдрориБофлаеин (бесцветный)
XU
ононон
I I I
Rib=CHjCHCHCHCHjOH
Схема 119
Окислительно-восстановительная система флавинов в зависимости от pH
ОНОНОН
I I I
RIb=CH3CHCHCHCH3OH
514
восстановительная система флавина для его окисленной формы, флавосе-
михинона и дигидрофлавина в зависимости от pH 190].
Протонирование флавинов при рН< 1 идет по атому азота положения 1
(ФлН2+) [91] однако имеются данные, что протон в сильнокислой среде при-
соединяется к кислороду карбонильной группы положения 2 [92].
Восстановительное ацилирование алло- и изоаллоксазинов с помощью
цинка, уксусного ангидрида и уксусной кислоты дает стабильные к окис-
лению S-N-ацил-1,5-дигидропроизводные, например ацилирование люми-
флавина (II) приводит к получению 5-М-ацетил-1,5-дигидролюмифлавина
(XXI) 160, 93]. Ацетильная группа при N(5> тормозит реокисление, однако
при 50° С в разбавленной минеральной кислоте при доступе кислорода
воздуха она гидролизуется [93]. Восстановительное ацилирование рибо-
флавина (I) цинковой пылью и смесью уксусного ангидрида с уксусной кис-
лотой в присутствии серной кислоты дает 2',3',4',5',5-пентаацетил-1,5-ди-
гидрорибофлавин (XXII) [60].
XXI
XXII
При действии хлоругольного эфира и щелочного раствора гидросульфита
натрия ацильной группой замещается только положение 5 [43].
Вообще из четырех атомов азота молекулы алло- и изоаллоксазинов аци-
лированию подвергается только атом азота положения 5 и только у 5,10-ди-
гидроаллоксазинов или 1,5-дигидроизоаллоксазинов.
Подобным образом получают 5-М-формил-1,5-дигидролюмифлавин [601
и 2-тио-5-ацетил-1,5-дигидролюмифлавин [93].
Фотолиз 2',3',4',5',5-пентаацетил-1,5-дигидрорибофлавина приводит к
отщеплению 5-Н-ацетильной группы [42].
Для 1,5-дигидрофлавинов характерна непланарная структура молекулы,
она сложена по оси 5-N—10-N, и, как следует из рентгеноструктурного ана-
лиза, плоскости безольного и пиримидинового циклов расположены под
углом 140—160° [91, 94].
При исчерпывающем восстановлении водородом под давлением с
платиновым катализатором у флавинов восстанавливается ароматическое
ядро, причем образуются бесцветные октагидросоединения, имеющие в
водном растворе желто-зеленую флуоресценцию (при облучении ультрафио-
летовым светом), изменяющуюся при прибавлении щелочи в фиолетово-го-
лубую флуоресценцию [47].
Наблюдается различие химического поведения алло- и изоаллоксазинов
и их 1,5-дигидропроизводных в реакции алкилирования. Алкилирование
йодистым алкилом, диазометаном или диалкилсульфатом приводит к N-ал-
килпроизводным, причем аллоксазины алкилируются по N(।> и No,, но не по
атому азота положения 10 или 5 160]. Изоаллоксазины алкилируются по
атому азота положения 3. Из люмихрома (III) при алкилировании диазоме-
таном или смесью избытка диметилсульфата и диметилформамида в присут-
ствии углекислого калия при 120—150° С с большим выходом образуется
1,3-диметиллюмихром (XXIII) [95], а из рибофлавина (I) при действии
диметилсульфата в присутствии едкого натра — З-Ы-метилрибофлавин
(XXIX, R = СН2(СНОН)3СН2ОН); люмифлавин (II)соответственно обра-
зует З-М-метиллюмифлавин (XXIX, R = СН3) [96] (схема 120). Изоаллокса-
зины noN(i) не алкилируются.
17*
515
• Если аллоксазины метилируются при более низкой температуре, то
возникает главным образом смесь 1- или З-М-монометилпроизводных [60].
Следует отметить, что реакция алкилирования рибофлавина и люмифлавина
диалкилсульфатами сопровождается окислительной димеризацией (см. ни-
же). •
Восстановительное ацилирование 1,3-диметиллюмихрома (XXIII) дает
1,3-диметил-5-ацетил-1,5-дигидролюмихром (XXIV), а его метилирование
идет в положение 10 и приводит к получению 1,3, 10-триметил-5-ацетил-1,5-
-дигидролюмифлавина (XXV, R — СН3). В результате деацилирования при
кислотном гидролизе образуется 1,5-дигидропроизводное (XXVI), которое
окисляется азотистой кислотой в четвертичную флавиниевую соль 1,3,7,8,-
10-пентаметилизоаллоксазина (XXVII, R — СН3) [60,97]. 1,5-Дигидро-
производное (XXVI) стабильно к окислению; оно может быть подвергнуто
метилированию в 1,3,5,7,8,10-гексаметил-1,5-дигидроизоаллоксазин (X XVI11,
R = СН3) [60].
Схема 120
' О- и N-Алкилироваиие алло- и изоаллоксазинов
R(l)=C^dlOH)3CHOH И R(1I)=CH3
В реакциях алкилирования или хелатообразования изоаллоксазинов
наличие незамещенной циклической иминогруппы обеспечивает возмож-
ность таутомерии с карбонилом положения 4 (или 2). Поэтому в отличие от
соответствующих аллоксазинов алкилирование 5-М-ацетил-1,5-дигидроизо-
аллоксазинов (XXX) приводит главным образом к получению О(2), ОИ)-диал-
килзамещенных [60, 97] и только в некоторой степени — 3-О,4-1Ч-диалкил-
производных [60]. Так, алкилирование 5-Кт-ацетил-1,5-дигидролюмифлави-
на (XXX, R = СН3) диметилсульфатом в диметилформамиде в присутствии
516
углекислого калия протекает не по азоту, а по кислороду (лактимная тауто-
мерная форма) с образованием 2-О,4-О-диметил-5-ацетил-й,5-дигидролюми-
флавина (XXXI, R — СН3) [60] (схема 120). После осторожного дезацили-
рования и окисления образуется четвертичная соль (XXXII), которая мо-
жет быть дезалкилирована в люмифлавин (II).
N-Окиси аллоксазинов также, как это характерно для изоаллоксазинов,
не алкилируются по азоту положения 1 198].
Алло- и изоаллоксазины могут быть превращены в свои 2- или 4-тиопро-
изводные. При действии пятисернистого фосфора на 2,3,4,5-тетраацетилрибо-
флавин образуется 4-тиопроизводное, которое в результате гидролиза прев-
ращается в 4-тиорибофлавин [99].
Замещение атома кислорода оксогруппы в положении 4 изоаллоксази-
новой молекулы на серу возможно, по-видимому, только для дигидроформы
[93]. При действии на люмифлавин (II) пятисернистого фосфора в пиридине
образуется нефлуоресцирующий 4-тиолюмифлавин (ХХХШ), причем пер-
воначально люмифлавин восстанавливается сульфидом в 1,5-дигидролюми-
флавин, и только затем происходит реакция замещения с последующим окис-
лением кислородом воздуха промежуточного 4-тио-1,5-дигидролюмифлавина
[93].
2-Оксогруппа устойчива к действию пятисернистого фосфора.
Атом серы в тиопроизводных легко замещается кислородом с образова-
нием алло- и изоаллоксазинов. Так, 2-тиорибофлавин, 2-тио-2'-дезоксирибо>-
флавин, 2-тиолюмифлавин легко окисляются при действии разбавленной пе-
рекиси водорода или кислорода воздуха в рибофлавин [100], 2'-дезоксирибц-
флавин[101] и люмифлавин [102] соответственно. А при окислении перекисью
водорода в метиловом спирте из 2-тиолюмифлавина образуется 2-О-метил-
люмифлавин [103]. Это соединение в водном растворе при pH 2 или pH
13 гидролизуется в люмифлавин [103].
Целый ряд реакций нуклеофильного замещения атома серы тионовой
группы может быть осуществлен, исходя из 2-тиорибофлавина (XXXIV)
или 2-тиолюмифлавина [93, 104]. Так, при действии на 2-тиорибофлавин
(XXXIV) йодистого метила в безводных условиях при комнатной тем-
пературе получается 2-метилтиорибофлавин (XXXVI) [105]. Это соединение
с аминами, например с морфолином, анилином или этаноламином, дает
2-морфолино-2-дезоксирибофлавин, 2-фенилимино- й 2-Р-оксиэтилиминори-
бофлавин [105, 106]. 2-Тиорибофлавин (XXXIV) реагирует с гидразином,
образуя 2-гидразонрибофлавин (XXXV) [105].
Rib-риБитил
Подобные реакции осуществлены с 2* и с 4-тиолюмифлавином [105].
Все полученные соединения при окислении перекисью водорода образуют ри-
бофлавин (I). *
2-Тио-5-ацетил-1,5-дигидролюмифлавин (XXXVII) при обессеривании
по Мозинго с помощью никеля Ренея превращается в 2-дезоксо-7,8,10-трц-
517
XXXVII
XXXVIII
метил-5-ацетил-1,5-дигидроизоаллоксазин (XXXVIII), который легко оки-
сляется кислородом воздуха в люмифлавин (II) [93].
Бромирование 2',3',4', 5'-тетраацетилрибофлавина N-бромсукцинимидом
в трихлоруксусной кислоте в присутствии каталитических количеств пере-
киси бензоила идет в положениеЭ [107] с образованием соединения XXXIX
Br CH^CHOA^CHjOAc
Л
NH
сн,
XXXIX °
сн,
Аллоксазин нитруется дымящий азотной кислотой в смеси с концен-
трированной серной кислотой в положения 7 и 9 с образованием мононит-
роаллоксазинов, а в более жестких условиях — в присутствии олеума при
70° С реакция приводит к получению 7,9-динитроаллоксазина [108].
Нитрование люмифлавина, люмихрома и N-окиси люмихрома дымящей
азотной кислотой в смеси с концентрированной серной кислотой приводит
к образованию соответствующих 6,9-динитросоединений [109].
В результате восстановления нитросоединений образуются соответст-
вующие аминоалло- и аминоизоаллоксазины. Аминогруппа этих соедине-
ний может быть ацилирована. 8-Аминонорлюмифлавин ацетилируют через
1,5-дигидроформу с последующим деацетилированием образовавшегося сое-
динения по N(5) [104]. Слабоосновная аминогруппа 8-аминонорлюмихрома
реагирует с уксусным ангидридом при длительном нагревании в среде пи-
ридина при 60° С, образуется 8-К[-ацетиламинонорлюмихром; подобным
образом получаются e-N-бензоил- и S-N-тозиламинонорлюмихром [110].
8-Аминоноррибофлавин (XL), 8-аминонорлюмифлавин и 8-аминонорлю-
михром диазотируются обычным образом. Диазопроизводные этих соедине-
ний при гидролитическом разложении их солей образуют соответствующие
8-оксиноррибофлавин (XLI) 8-оксинорлюмифлавин и 8-оксинорлюмихром
r-ch/choh^ch.oh
Осуществлено превращение 8-аминоизоаллоксазинов в 8-галогеноизоал-
локсазины через хлориды или бромиды диазосоединений по реакции Занд-
мейера; в результате реакции были получены 8-хлор-, 8-бромнорлюмифла-
вин [104, 112], а из соединения XL—8-хлорноррибофлавин (XLII) [1121,
один из сильных антагонистов рибофлавина
518
CH/CHOH^CHjOU
ch,(chohXch,oh
XLII
9-Аминолюмнфлавин (он не флуоресцирует) может быть дезаминирован
при действии азотистой кислоты с последующим разложением полученного
диазосоединения в люмифлавин [1131.
8-Аминонорлюмихром и 8-оксинорлюмихром вступают в реакцию азосо-
четания с диазотированными ароматическими аминами в среде водного
диметилформамида или диметилсульфоксида с образованием 9-арилазо-
8-аминонорлюмихрома и 9-арилазо-8-оксинорлюмихрома [114].
8-Аминоаллоксазины и 8-аминоизоаллоксазины по-различному ведут
себя в реакции присоединения окиси этилена. К 8-аминонорлюмифлавину
присоединение окиси этилена происходит по циклической иминогруппе
положения 3 — соединение XLIII, в то время как присоединение к 8-ами-
нонорлюмихрому идет одновременно и по N^j, и по аминогруппе положения
8 с образованием соединения XLIV [115].
В реакцию с нуклеофильными агентами вступает 8-хлорнорлюмифла-
вин — при взаимодействии с этаноламином или метилатом натрия он об-
разует 8-оксиметиламино- и 8-метоксинорлюмифлавин соответственно [82].
Так же реагирует 8-хлоризоаллоксазин с циклическими аминами (пипери-
дином, морфолином) при нагревании в растворе диметилформамида [116].
Интересно отметить, что галоген аллоксазинов к нуклеофильному заме-
щению в этих условиях нереакционноспособен.
2-0,3-N- и 2-О,4-О-Диалкилфлавиниевые соли реагируют с нуклеофиль-
ными агентами, например четвертичная соль 2,4-диэтокси-7, 8, 10-триме-
тилизоаллоксазина (типа XXXII, схема 120) при действии аммиака об-
разует люмилфлавин-2,4-диимин [97].
По своей реакционной способности метильные группы положений 7 и
8 молекулы рибофлавина или люмихрома различны и характеризуются
рядом особенностей. Так, метильная группа положения 8 рибофлавина
подвергается селективному бромированию избытком элементарного брома
в смеси пиридина и диоксана.
2', 3', 4', 5'-Тетраацетилрибофлавин (IV) превращается в 8а-дибром-
-2', 3', 4', 5'-тетраацетилрибофлавин (XLV) 1117], а затем в результате
последующего гидролиза нагреванием с соляной кислотой — в 8-формилнор-
рибофлавин (XLVI) [117].
Ас=СОСН,
сн/снонУнрн
519
При непродолжительном действии эквимолекулярного количества брома
в диоксане в присутствии катализатора — перекиси бензоила — 2', 3', 4',
5'-тетраацетилрибофлавин (IV) превращается в 8а-монобром-2', 3', 4',
5'-тетраацетилрибофлавип (XLVII) [118, 1191.
Метильная группа положения 8 изоаллоксазина имеет подвижные атомы
водорода, вследствие чего обладает функциональными свойствами. Так,
она вступает в реакцию с альдегидами.
Рибофлавин (I) вступает с п-ди метилами нобензальдегидом в реакцию
Кневенагеля с образованием соответствующего стильбена — темно-си-
него 8а-п-диметиламинобензальрибофлавнна (XLVIII); подобным обра-
зом получается соответствующее производное люмифлавина [120].
CH^HOH^CHjOH СН/СНОН^СНрН
(CH^NC^CH^H^^x^N^N^O
XX XX * —- ™ХДДЛ
ch/y^n к • • сн, v к if
о о
I • XLVIII
Следует отметить, что рибофлавин (I) и люмифлавин (II) в безводных
условиях реакции алкилирования — в среде, содержащей диметилсуль-
фат, диметилформамид и углекислый калий,— ведут себя аномально:
наряду с метилированием по иминогруппе положения 3 (соединение XXIX)
происходит катализируемая основаниями необратимая реакция окисли-
тельной димёризации по метильной группе положения 8 [82] с образованием
N(з)-метилбензохиноидного бис-флавина
R(ll)=CH3 и R(l)= ЩЬ(рибитил)
Окислительная димеризация рибофлавина (I) и люмифлавина (II) наблю-
дается при взаимодействии этих флавинов с углекислым калием в горячем
диметилформамиде в атмосфере азота [821; в результате реакции получается
бензохиноидный бис-рибофлавин (L, R = ,Rib) или бензохиноидный бис-
люмифлавин (L, R = СН3) темно-красного цвета.
Ml
L
р(||)=СН,и R^Xch/cHOH^CHjOH
В сильно дегидратирующих условиях фосфорилирования рибофлавина
(I) (ортофосфорная кислота и фосфорный ангидрид) частично также
имеет место окислительная димеризация по метильной группе положения
(520
8 с образованием бензохиноидного бас-рибофлавин-5'-дифосфата [121].
Реакционная способность алло- и изоаллоксазинов детально рассмот-
рена и обсуждена [122].
СТРОЕНИЕ РИБОФЛАВИНА
Строение рибофлавина на основании изучения продуктов его расщепле-
ния и полного синтеза установили Каррер с сотр. [1, 2] и Кун с сотр. [3]
в 1935 г. При облучении нейтральных или кислых растворов рибофлавина
происходит полное отщепление бокового пентитного остатка в виде гли-
колевого и глицеринового альдегидов [123, 124] и образуется 7, 8-диметил-
аллоксазин, люмихром (III) [20] (схема 121).
Схема 121
Установление строения рибофлавина реакциями расщепления
Как известно, при фотолизе щелочных растворов рибофлавина (I) рас-
щепление идет частично до люмихрома (III), но главным образом отщепление
боковой цепи происходит между C(ij и С(2) с образованием 7, 8, 10-триме-
тилизоаллоксазина, люмифлавина (II) [19, 21, 22]. Его строение было ус-
тановлено следующим образом. При нагревании люмифлавина с баритовой
водой образуется мочевина [125], чем доказывается [126] присутствие в его
молекуле группировки >N—СО—N<- Вторым продуктом разложения
является 4, 6, 7-триметил-3-оксо-3,4-дигидрохиноксалин-2-карбоновая ки-
слота (LI) [22], переходящая при декарбоксилировании в хиноксалиновое
производное (LII); из этого соединения при кипячении с раствором щелочи
получен известный 4,5-диметил-М-метил-о-фенилендиамин (ЫП) [96].
Строение люмифлавина (II) было подтверждено его синтезом из о-кси-
лола [127] через ряд стадий превращаемого в 4,5-диметил-М-метил-о-фе-
нилендиамин (ЫП), который конденсировался с аллоксаном по методу
Кулинга [128] (см. с. 524).
Строение люмихрома (III) также подтверждено его синтезом [20].
При облучении рибофлавина (I), помимо люмифлавина (II), отщепляет-
ся остаток (С4Н8О4), имеющий углеводную природу. Так как рибофлавин
образует тетраацетат [22], а люмифлавин не ацетилируется, то, следователь-
но, четыре гидроксильные группы находятся в углеводном остатке. Обра-
зование формальдегида при окислении рибофлавина тетрацетатом свинца
указывает на наличие одной первичной спиртовой группы [21]. Получение
моно- и дпизопропилиденовых производных рибофлавина при взаимодей-
ствии с ацетоном говорит о попарном пространственном расположении гид-
роксильных групп [38].
На основании выяснения строения продуктов расщепления рибофлави-
на— люмихрома (III) и люмифлавина (II) — и углеводной природы остат-
ка в положении 10 формула рибофлавина была установлена как 7,8 -диме-
521:
тил-10-тетраоксипентилизоаллоксазина. Неясным оставался вопрос об али-
фатической боковой цепи из пяти углеродных атомов, содержащей четыре
гидроксильные группы, и их пространственном расположении. Строение
рибофлавина окончательно было установлено только его синтезом, для
чего потребовалось искусственное получение большого количества флави-
нов, имеющих в положении 10 остатки различных спиртов. Этот синтез
осуществили в 1935 г. Каррер, Шопп, Бенц, Эйлер, Мальмберг, Беккер и
Фрей 11, 21 и одновременно Кун, Рейнемунд, Вейганд и Штробеле 13].
Полиоксиалкильной цепью, замещающей положение 10 в молекуле рибо-
флавина, оказался остаток D-рибита.
ВЫДЕЛЕНИЕ РИБОФЛАВИНА
Впервые пигмент, содержащий флавин, выделен в 1879 г. из сыворотки
коровьего молока [129]. Затем из молока был выделен желтый водораство-
римый фактор [130, 131], названный лактохромом [131], он же был выделен
из одуванчика, солода [132], яичного желтка [133], печени [133, 134]; из
свиного сердца выделено желтое вещество — цитофлав (оказавшееся
фосфорнокислым эфиром рибофлавина) [135]. Варбург и Христиан [22]
в 1932 г. выделили из дрожжей желтый окислительный фермент, в котором
ими был открыт рибофлавин (лактофлавин), являющийся составной частью
этого фермента.
В кристаллическом виде рибофлавин получили почти одновременно
Эллингер и Кошара [9] из молочной сыворотки (лактофлавин) и Кун, Дьор-
дьи и Вагнер-Яурегг [10] — из сыворотки и из яичного белка (овофлавин).
Для получения 1 г рибофлавина необходимо было переработать около
5000 л молочной сыворотки [88].
ПОЛУЧЕНИЕ РИБОФЛАВИНА МИКРОБИОЛОГИЧЕСКИМ ПУТЕМ
В природе рибофлавин синтезируется растительной клеткой, дрожжами,
мицеллярными грибами и бактериями. Мицеллярный гриб, например
Eremothecium ashbyii, может производить его в больших количествах
[136, 137]. В высушенных маслянокислых бактериях его содержится 136 мкг
в 1 г 122]. Для некоторых микроорганизмов рибофлавин на свету имеет
бактериостатическую активность [1381.
Продуцент рибофлавина Eremothecium ashbyii относится к одним из
эффективных [136, 139]. При культивировании на твердых средах этот гриб
образует до 2% рибофлавина (20 мг в 1г) [140], на жидких средах образо-
вание рибофлавина достигает 2,5 мг в 1 мл среды [141]. Ashbua gossypii
продуцирует до 5,0 мг в 1 мл среды. Промышленное значение имеет куль-
тура Sarcina lutea.
Из культуральной жидкости рибофлавин выделяют при pH 4,5—7 в
труднорастворимой восстановленной семихиноидной форме с выходом око-
ло 90% [142]. Для осаждения рибофлавина в качестве восстановителя при-
меняют гидросульфит [143]. Осадок рибофлавина в виде примеси содержит
рибофлавин-5'-фосфат, из которого витамин В2 получается в результате
гидролиза. Биосинтез рибофлавина микробиологическим путем по своей
эффективности уступает химическому синтезу.
СИНТЕЗ РИБОФЛАВИНА, ЛЮМИФЛАВИНА, ЛЮМИХРОМА И НЕКОТОРЫХ
РОДСТВЕННЫХ СОЕДИНЕНИЙ
Молекула рибофлавина и других биологически активных алло- и изоал-
локсазинов построена из конденсированных ароматического, пиразинового
и пиримидинового циклов и алифатических заместителей, вследствие чего
важнейшие синтезы этих соединений осуществляются тремя основными ме-
тодами: конденсацией ароматического и пиримидинового компонентов с
522
одновременным созданием срощенного пиразинового цикла (синтезы 1—5),
построением молекулы на птеридиновом цикле с созданием конденсирован-
ного ароматического кольца (синтез 6) и построением молекулы на хинокса-
линовом цикле с достройкой пиримидинового цикла (синтез 7). Метильная
группа или полиоксиалкильная цепь в положение 10 молекулы изоаллок-
сазинов вводится предварительно с ароматическим или гетероциклическим
компонентом.
I. Синтез рибофлавина конденсацией ароматического моноамина
и виолуровой кислоты
В этом синтезе один атом азота, необходимый для образования изоаллок-
сазинового цикла, вводится с ароматическим компонентом; второй же атом
азота вводится не с ароматическим ядром, а с пиримидиновым. Поэтому для
конденсации непосредственно с 3,4-диметилфенил-Р-рибитиламином (LIV)
применяют виолуровую кислоту (LV)
сн2он
носн
носн
носн
।
CHj
L1Y
ноу\о
HON=C\ JNH
СО
LY
Для получения рибофлавина (I) конденсацию соединений LIV и LV
проводят в спиртовой, диоксановой [144] или водной среде, а также в среде
органических кислот и фенола при температуре до 100° С [145]. Выходы
рибофлавина по этому методу небольшие вследствие малой реакционной
способности ароматического моноамина к орто-замещению нитрозосоеди-
нениями (виолуровая кислота).
2. Синтез рибофлавина и люмифлавина из ароматического моноамина
и 6-хлорурацила
Значительно более интересно протекает синтез рибофлавина через
5-N-OKHCb рибофлавина [145а]. При нагревании 3,4-диметилфенил-О-риби-
тиламина (LIV) с 6-хлорурацилом до 160° С в течение 5 мин с выходом
90 % образуется сиропообразный 6-(3,4-диметилфенил-Р-рибитиламино)
урацил (А), нитрозирование которого в уксусной кислоте избытком нитри-
та натрия в течение 2 ч с последующим частичным выпариванием и раз-
бавлением водой приводит к получению S-N-окиси рибофлавина (Б) с вы-
ходом 85%.
Восстановление S-N-окиси рибофлавина гидросульфитом натрия в воде
дает рибофлавин (I) почти с количественным выходом.
523
3? СинтёЗ- рибофлавина, Люмифлавина, люмихрома и их 2-тиопроизводных
конденсацией ароматических о-диаминов с аллоксаном
Важнейший метод синтеза алло- и изоаллоксазинов заключается в кон-
денсации ароматических о-диаминов с а-дикарбонильными соединениями
пиримидинового ряда, в качестве которых применяют производные барби-
туровой кислоты, имеющие активные к конденсации с аминами группы
или атомы. Такими соединениями являются: аллоксан (LVI) [1—31, 5,5-
дихлорбарбитуровая [81, 146] и монохлорбарбитуровая кислоты и аллок-
сантин [147].
Аллоксан, 5-оксобарбитуровая кислота (LVI), представляет собой цик-
лический трикетон [148] с кислыми свойствами, он неустойчив г щелочам—
баритовая вода превращает его в ациклическую аллоксановую кислоту [149],
которая с о-диаминами образует хиноксалиновые производные.
' Для получения ароматического компонента соединяют ароматический
амин с алифатической частью молекулы. Полученный вторичный ароматичен
ский алкиламин — ароматический компонент — конденсируют с пирими-
диновым компонентом, содержащим функциональные группы или атомы,
способные взаимодействовать с диаминами, в аллоксазин или изоаллокса-
зин необходимого строения.
Для синтеза 7, 8, 10-триметилизоаллоксазина—люмифлавина (II)
гидрохлорид 4,5-диметил-М-метил-о-фенилендиамина (LIII) конденсируют
с аллоксаном (LVI) — в воде при 50—60°С с выходом 75% [127, 150]
Если реакцию проводить с большим избытком аллоксана в безводном
спирте при 15—20° С, то преимущественно образуется уреид хинокса-
линкарбоновой кислоты (LVII) [151]. Побочная реакция образования про-
изводного хиноксалина облегчается при недостаточной кислотности среды.
Синтез рибофлавина впервые осуществлен конденсацией 3,4-диметил-
6-аминофенил-Д-рибитиламина (LVIII) с аллоксаном (LVI) [1—31; однако
реакция в присутствии соляной кислоты в отличие от синтеза 10-алкилза-
мещенных изоаллоксазинов идет с низким выходом — всего 5—10% [151].
Следует отметить, что при конденсации 3,4-диметил-6-аминофенил-Д-ри-
битиламина (LVIII) с аллоксаном (LVI) в среде уксусной кислоты, помимо
реакции, идущей с выделением двух молекул воды и приводящей к рибо-
флавину (I) с выходом 5—10%, протекает вторая реакция с выделением
только одной молекулы воды [38] и образованием производного хинокса-
лина — уреида 6,7-диметил-1-И-(1'-0-рибитил)-2-оксо-1,2-дигидрохинонса-
лин-3-карбоновой кислоты (LIX) 156, 152]
LVIII
сн2он
носн
носн
носн
сн2
СН3^\^ NH
Т 1Г
СИ, NHj
524
Как оказалось, прибавление борной кислоты приводит-к повышению
выхода флавинов; в результате ее применения как катализатора, ускоряю-
щего первую реакцию, выход рибофлавина увеличен до 70—90% 11531.
Каталитическая активность борной кислоты не зависит от ее комплексо-
образования с гидроксильными группами (что обычно характерно для этого
соединения), так как присутствие этой кислоты в реакционной среде также
увеличивает выход изоаллоксазина, не содержащего гидроксильных групп
в боковой цепи при 10-N [Г54]. В качестве катализатора реакции конден-
сации может быть использован пироборацетат 1(СНзСОО)2В]2О, получаемый
из борной кислоты и уксусного ангидрида [154].
Конденсацию о-диаминов с аллоксаном (аллоксан-гидратом) осущест-
вляют в суспензии или в растворе воды, спирта, ледяной уксусной кислоты,
водном или спиртовом растворе соляной кислоты. Для синтеза рибофла-
вина [3, 1551, 6-метилрибофлавина [70], и других флавинов применяют
смесь борной кислоты и ледяной уксусной кислоты. Конденсацию обычно
^проводят при 100°С или выше (при температуре кипения среды) [156].
• Хотя из первично-вторичных ароматических о-диамйнов и аллоксана
" возможно образование двух изомерных изоаллоксазинов, однако вслед-
,х ствие преимущественной реакции первичной аминогруппы, с 5-оксогруппой
аллоксана конденсация протекает однозначно с образованием изоаллок-
>’ сазинового соединения со строго определенной структурой.
Подобным образом синтезирован 2-дезоксиробофлавин [157]. Конден-
сация 4,5-диметил-о-фенилендиамина и аллоксана в описанных выше ус-
ловиях приводит к получению люмихрома (III) [95].
Следует отметить, что природа N-заместителя и заместителей аромати-
ческого цикла оказывает влияние на ход реакции и выход флавинов.
Для конденсации с аллоксаном применяют производные о-фенйленди-
амина, в котором аминогруппа защищена ацетильной группой. При
конденсации 3,4-диметил-6-ацетиламинофенил-М-£)-рибиталамина (LX) с
аллоксаном рибофлавин получен с малым выходом, так как основная реак-
ция проходит с отщеплением элементов воды от исходного диамина и
циклизацией в производное 2, 5, 6-триметил бензимидазол a (LXI) [1581.
CHjtHOH^CHOH
CH3x^x,NH CH3,
/Ц.Jk —HjO*
CH, NHCOCH, ’ CH,
LX
сн/снон)3сн2он
|| ?с-сн,
N
LXI
Помимо аллоксана в реакцию с о-диаминами вступает продукт его вос-
становления — аллоксантин. Однако в этом случае конденсация проте-
кает несколько иначе, чем с аллоксаном,— один из гетероциклов аллоксан-
тина (LXII) отщепляется в виде диалуровой кислоты (LXIII), которая в
свою очередь медленно окисляется кислородом воздуха в аллоксантин.
Рибофлавин получают в результате конденсации 3,4-диметил-6-амино-
фенил-О-рибитиламина (LVIII) и смеси аллоксантина (LXII) с аллоксаном
1159, 160], или одного аллоксантина, или диалуровой кислоты (LXIII)
в присутствии воздуха в спиртовом солянокислом растворе [147, 161]
CH.CIIOH;3CHjOH
NH
H
-N.
oc co
или Щ I 1 ———-Рибофлавин
^C. NH
HO co 1
LXIII
OH HC> -N.
OCX ^C^ C co
II II
HN ,C--------C NH
CO OH HO co
LXII
CH,.
ch; v nh2
LVIII
Для синтеза люмифлавина (II) использована изодиалуровая кислота
(LXIV); ее конденсация с 4,5-диметил-М-метил-о-фенилендиамином (LIII)
в ледяной уксусной кислоте протекает с трудом, через промежуточный
525
7, 8, 10-триметил-3,4-дигидроизоаллоксазин (LXV), который в кислой
среде кислородом воздуха окисляется в люмифлавин (II) [93, 1621
В качестве пиримидинового компонента для конденсации с ароматичес-
кими о-диаминами можно применить 5-моно- и 5,5-дигалогенбарбитуровые
кислоты, которые по своему строению являются триоксогексагидропири-
мидинами [1631. Атомы галогена в 5,5-дигалогенбарбитуровой кислоте не-
равноценны и различаются по своей реакционной способности [164, 165].
Конденсация 3,4-диметил-6-аминофенил-£)-рибитиламина (LVIII) с 5,5-
дихлорбарбитуровой кислотой (LXVI) приводит к образованию рибофла-
вина (I) с выходом 85% [56, 146].
ivm
сЩснон^снрн
CUI 1
.NH
Cl co
LXVI
В циклизации co вторичной аминогруппой принимает участие 4-ок-
согруппа галогензамещенной барбитуровой кислоты, чем определяется
однозначность реакции. Конденсация осуществляется в пиридине при
100° С. Возможно применение в качестве среды уксусной кислоты в при-
сутствии ацетата или бората натрия, однако выход в этих условиях умень-
шается. Из соответствующих диаминов с 5,5-дихлорбарбитуровой кислотой
в пиридине люмифлавин получен с выходом 78%, а люмихром — 90% [56].
Для конденсации с о-диаминами можно использовать 5,5-дибромбарби-
туровую кислоту, но выход получается более низкий [56, 166].
5-Моногалогенбарбитуровая кислота в реакции конденсации с аромати-
ческими о-диаминами образует промежуточные дигидроизоаллоксазины,
которые подвергаются дегидрированию в присутствии кислорода воздуха в
изоаллоксазины. Выходы с этим пиримидиновым компонентом получаются
ниже, чем с 5,5-дигалогенбарбитуровыми кислотами. Так, с 5-монохлорбарби-
туровой кислотой люмифлавин получен с 70%-ным выходом [561, а с 5-моно-
бромбарбитуровой кислотой выход рибофлавина был значительно ниже [1661-
Конденсацией первично-вторичного ароматического о-диамина с 2-ими-
но-5,5-дибромбарбитуровой кислотой в щелочной среде получен 2-люми-
флавинимин [1131.
Если применить для конденсации с первичными ароматическими
о-диаминами виолуровую кислоту, то образуются соединения аллоксазино-
вого ряда [167]. Так, из 4,5-диметил-о-фенилендиамина (LXVII) и виолу-
ровой кислоты (LV) в смеси спирта и уксусной кислоты в присутствии
борной кислоты как катализатора реакции образуется люмихром (III)
LXVIII
526
В отсутствие борной кислоты или в водном спирте [168] наблюдается
образование уреида 6,7-диметил-2-оксихиноксалин-3-карбоновой кислоты
(LXVIII). Пиразиновый цикл соединений образуется из атомов азота
о-фенилендиамина, что доказано применением виолуровой кислоты, меченной
UN по изонитрозогруппе положения 5; атом азота изонитрозогруппы от-
щепляется в виде гидроксиламина [167].
Подобным методом из первично-вторичных ароматических о-диаминов и
виолуровой кислоты синтезированы люмифлавин и рибофлавин [169].
При конденсации в разбавленной соляной кислоте выход люмихрома
достигает 70% [167]. Из 4-хлор-о-фенилендиамина и виолуровой кислоты
синтезирован 8-хлораллоксазин [170], а из 4,5-диметил-о-фенилендиамина
и 2-тиовиолуровой кислоты в спиртовом растворе хлористого водорода
получен 2-тиолюмихром (выход 58%) [167].
Следует отметить, что виолуровая кислота используется для конден-
сации с ароматическими м-диаминами, однако в этом случае образуются
только 8-аминозамещенные алло- и изоаллоксазины [104, 171, 172]. Этим
методом синтезированы 8-аминонор- и 8-рибитиламиноноррибофлавин [173].
Очистка флавинов производится перекристаллизацией из низших али-
фатических спиртов, пиридина, фенола, диметилформамида, N-метилацет-
амида, растворением в щелочи и осаждением кислотой.
4. Синтез рибофлавина и некоторых других алло- и изоаллоксазинов
конденсацией ароматических орто-аминоазосоед и нений с барбитуровой
кислотой
Применение для синтеза в качестве пиримидинового компонента бар-
битуровой кислоты (LXX) и для конденсации с ней вторичных орто-ами-
ноазосоединений ароматического ряда, содержащих два атома азота в ор-
то-положении, необходимых для построения срощенного пиразинового
цикла, например 3,4-диметилфенил-6-фенилазо-М-Ь-рибитиламина (LXIX),
приводит к получению рибофлавина (I) с выходом 71% [25, 174, 175]. Об-
разование рибофлавина основано на реакции восстановительной цикли-
зации, протекающей по активной метиленовой группе барбитуровой кислоты
и атому азота азогруппы ароматического компонента. Одновременно про-
текает отщепление ароматического амина, например в рассматриваемой
реакции — анилина
сн2он
носн
носн
носн
сн2
CHj^^rx^NH HO^_«bk_n
XI + J./h
СН, N=NC6HS СО
LXIX LXX
СН2ОН
носн
носн
носн
I
Следует отметить, что соединение LXIX является промежуточным про-
дуктом по одной из схем получения 3,4-диметил-6-аминофенил-П-£)-ри-
битиламина (LVIII), который, как описано ранее, используется для син-
теза рибофлавина.
В качестве среды для конденсации применяют диоксан [156, 174—177],
этиловый спирт [178], н-бутиловый спирт [102, 178—183], w-амиловый спирт
[183], этилацетат [184], н-бутилацетат [100, 101], метилэтоксиацетат [185],
этиленгликоль [183], бутиловый эфир [186] и др.
527
Реакция конденсации 3,4-диметилфеиил-6-арилазо-М-©-рибитиламина с
барбитуровой кислотой катализируется слабыми органическими кислотами
с константой диссоциации К от 1,2-Ю-5 до 6,89-10-5: уксусной кислотой
[1731, бензойной, пара-аминобензойной, фенилуксусной, никотиновой, ян-
тарной и др. [177]. Выходы рибофлавина составляют 62—71%. В отсут-
ствие катализатора выход достигает всего 21% [177].
Вместе с уксусной кислотой в реакционной среде используют SnCl2
или А1С13 [186, 187], однако эти соединения не имеют явно выраженного
каталитического эффекта.
3,4-Диметилфенил-6-арилазо-^'-0-рибитиламин (LXIX) в своей моле-
куле, помимо остатка анилина, в качеств диазосоставляющей может содер-
жать остатки о- и n-нитроанилина, п-толуидина [173], п-аминобенсойной
кислоты [53]. Однако выход рибофлавина находится в зависимости от при-
роды арилазогруппы в положении 6. Азосоединения, содержащие в виде
азогруппы остатки о- и п-хлоранилина, n-метоксианилина, 2,4-диметилани-
лина, дают значительно пониженный выход рибофлавина [173]. По некото-
рым данным [188], при использовании 3,4-диметилфенил-6-(о-дифенила-
зо)-М-О-рибитиламина выход рибофлавина достигает 80%, а азосоединение
с п,п-дифенильным остатком дает выход всего 14% [173]. При конденсации
барбитуровой кислоты с о-аминоазосоединениями симметричного строения
выход алло- и изоаллоксазинов уменьшается в 3—10 раз [189, 190].
Лучший выход рибофлавина наблюдается при соотношении барбиту-
ровой кислоты и аминоазосоединения 1,6 : 1 (в молях) или более [177] при
конденсации в н-бутиловом спирте и уксусной кислоте. С другой стороны,
имеется замечание, что в среде диоксана и уксусной кислоты избыток бар-
битуровой кислоты не имеет существенного влияния на выход [156].
Важное значение для скорости реакции взаимодействия ароматических
орто-аминоазосоединений с барбитуровой кислотой имеет температурный
фактор. Реакция проводится при 78—120° в зависимости от применяемого
растворителя и других условий. Так, при получении рибофлавина в среде
этилового или н-бутилового спирта и уксусной кислоты при 115° С (с эти-
ловым спиртом под давлением) конденсация продолжается 4—5 ч. При
применении этилового спирта происходит частичная его этерификация ук-
сусной кислотой в этилацетат.
- При получении рибофлавина минеральные кислоты ингибируют реак-
цию конденсации компонентов и одновременно способствуют расщеплению-
аминоазосоединения.
Изомерный азокраситель, 3,4-диметилфенил-2-фенилазо-М-О-рибитил-
амин (изоазорибитиламин), образующийся в виде примеси при получении
3,4-диметилфенил-6-фенилазо-М-£)-рибитиламина (LXIX), в тех же усло-
виях в конденсацию с барбитуровой кислотой не вступает [173] и поэтому
не может служить источником загрязнения рибофлавина (I) изомерным
6,7-диметил-10-М-(Г-£)-рибитил)изоаллоксазином.
Неспособность изоазорибитиламина к конденсации с барбитуровой кис-
лотой, возможно, может найти объяснение в наличии пространственных
затруднений, вызываемых орто-положением метильной группы к объемис-
той азогруппе, «зажатой» с другой стороны орто-расположенной вторичной
аминогруппой. Это обстоятельство согласуется с отсутствием реакционной
способности у 3,4,5-триметилфенил-2-фенилазо-М-£)-рибиталамина к цикли-
ческой конденсации с барбитуровой кислотой [176]. Следует отметить,
что ароматические орто-аминоазосоединения, имеющие в качестве заме-
стителя атом хлора в пара-положении к аминогруппе (.мета-положении,
к азогруппе), также не реагируют с барбитуровой кислотой в среде дио-
ксана и уксусной кислоты [156].
Рассматриваемый метод синтеза рибофлавина прост в осуществлении,
так как барбитуровая кислота доступнее аллоксана или 5,5-дихлорбарби-
туровой кислоты, а вторичные орто-аминоазосоединения легче получить,
чем первично-вторичные о-фенилендиамины.
528
На схеме 12 представлен полный синтез рибофлавина, имеющий тех-
ническое значение; в схему включены исходные и промежуточные продукты
синтеза.
Аналогично описанной реакции из 3,4-диметилфенил-6-(п-толилазо)-
-N-D-рибитиламина и барбитуровой кислоты-2-14С получен рибофлавин-
2-14С [1911, а из соответствующих орто-аминоазосоединений и барбитуровой
кислоты — 2',3',4',5'-тетраацетилрибофлавин [173], 5'-дезоксирибофлавин
[192], 2'-дезоксирибофлавин (выход 65,4%) [101], природный лимифлавин
[102, 193]. Конденсацией 3,4-ди.метилфенил-6-фенилазо-П-О-рибитиламина
(LXIX) с 2-тиобарбитуровой кислотой (LXXI) нагреванием в среде н-бутил-
ацетата и уксусной кислоты получен 2-тиорибофлавин (XXXIV) с выхо-
дом 60,4 %[100]
ch/chohYchoh сн/аю^сн2он
H><L oL/NH
CH, • N=NCeHs CO CH, N H
LXIX LXXI XXXIV °
Подобным путем синтезирован 2-тио-2'-дезоксиробофлавин с выходом 69,6%
11011. Отмечалось что 3,4-диметилфенил-6-(п-толилазо)-М-О-рибитиламин
при нагревании в среде диоксана и уксусной кислоты в реакцию с 2-тио-
барбитуровой кислотой не вступает [194].
Конденсацией барбитуровой кислоты с 3,4-диметилфенил-6-(3',4'-ди-
метилфенилазо)анилином при каталитическом участии уксусной или щаг
велевой кислоты синтезирован природный люмихром (выход 84%) [182],
а при конденсации с 2-тиобарбитуровой кислотой — 2-тиолюмихром (выход
79,7%) [100].
Для очистки рибофлавина применяют перекристаллизацию из воды,
слабой соляной или азотной кислоты, 5%-ной уксусной кислоты [195],
переосаждение из щелочных растворов — при этом возможно расщепление
рибофлавина. Лучшим является способ [174, 196], по которому раствор
рибофлавина в 24—36%-ной соляной кислоте фильтруют от примесей, под-
вергают действию окислителей, например перекиси водорода, а затем веще-
ство осаждают избытком воды при нагревании. По другому варианту ра-
створ рибофлавина в соляной кислоте нейтрализуют водным раствором ед-
кого натра и нагревают с водным анилином [197].
5. Синтез рибофлавина и некоторых других алло- и изоаллоксазинов
конденсацией орто-бензохинонов с диаминоурацилами
Построение молекулы алло- и изоаллоксазинов можно осуществить из
пиримидиновых о-диаминов путем их конденсации с ароматическими
а-дикарбонильными соединениями. Так, при взаимодействии 5-амино-4-
D-рибитиламиноурацила (LXXIII) с димеризующимся во время реакции
4,5-диметил-о-бензохиноном (LXXII) при нагревании в уксусной кислоте
получен рибофлавин (I) с выходом 29% [198, 199]
сн2(снон),сн2он
HNX
с со
II I
• /С. дмн
h2n со
LXXIII
CH^CHOI^CHjOH
Конденсация проводилась при нагревании в спирте или водном растйоре
в присутствии щелочи или уксусной кислоты [200]. Подобным образбм,
исходя из 4,5-диметил-о-бензохинона (LXXII) и соответствующих о-ди-
529
аминопиримидинов, был получен люмифлавин (выход 40%) [198] и люми-
хром (выход 68%) [200].
Следует отметить, что о-бензохинон в реакцию с 4,5-диаминопирими-
дином не вступает [201].
6. Синтез рибофлавина и некоторых других алло- и изоаллоксазинов
из люмазинов
Рассматриваемый метод синтеза заключается в построении молекулы
алло- и изоаллоксазинов на птеридиновом цикле с достройкой на нем аро-
матического цикла.
Для получения рибофлавина (I) 6,7-диметил-8-М-Д-рибитил-2,4-диок-
соптеридин—6,7-диметил-8-рибитиллюмазин (LXXIV) конденсируют с диа-
цетилом при 130° С [202]
CH^CHOH^CHjQH
СН,
сн,
LXXIV
CH^IIOI^CHjOH
I
При аналогичной конденсации из 6,7,8-триметиллюмазина получают
люмифлавин [2021.
л. 6,7-Диметил-8-рибитиллюмазин (LXXIV) наряду с другим веществами
находится в клетках Eremothecium ashbyii [203]; это соединение, получен-
ное синтетически, при обработке ферментсодержащими экстрактами из
ч микробных клеток Ashbya gossypii образует рибофлавин [202, 204], однако
такое направление биохимической реакции в живой клетке не является ос-
новным [205]. Соединение LXXIV синтезировано из 5-амино-4-О-рибитил-
аминоурацила (LXXIII) и диацетила [202]
сн-со
I
.со
сн/
CH^CHOH^CHjOH
♦ HI
сн,(сно^,сн,он
CH,^,N 14^,0
LXXIII
CH, N V
о
LXXIV
Использование для конденсации с 5-амино-4-£)-рибитиламиноурацилом
(LXXIII) димерного альдоля диацетила — 5-ацетил-2-окси-2,5-диметил-
-3-оксотетрагидрофурана (LXXV) — с последующей циклизацией под вли-
янием 0,1 н. раствора едкого натра приводит к образованию смеси рибо-
флавина (I) и 6,7-диметил-8-рибитиллюмазина (LXXV) [205]
сн,.
сн.со'
^о
' С-он
чо/хсн3
LXXV
сн,—со
сн,сон со
сн,со сн,
сн^снон)3сн,он
мЦЖ сн,.
Ц-Асб™
LXXIII
сн2(снон),сн2он
+ Соединение
LXXIV
Если исходить из 4,5-диаминоурацила и димерного альдоля диацетила,
то получается люмихром, а из 5-амино-4-метиламиноурацила и димера диа-
цетила — люмифлавин [206].
530
7. Синтез алло- и изоаллоксазинов построением их молекулы
на хиноксалиновом цикле
Хиноксалины путем достраивания на их молекуле пиримидинового
цикла превращают в аллоксазины или изоаллоксазины. 2-Рибофлавин-
имин (LXXVII) получен конденсацией N-рибитилпроизводного 6,7-диме-
тил-2-оксо-1,2-дигидрохиноксалин-3-карбоновой кислоты (LXXVI) с гуа-
нидином в темноте в атмосфере азота [72, 104].
LXXVI
NH,
Т Т Т
CH3'X^^N COCH,
Амид-2-аминохиноксалин-З-карбоновой кислоты при взаимодействии с
хлоругольным эфиром образует соответствующий уретан, который в при-
сутствии этилата натрия циклизуется в аллоксазин [207, 108]. Это же сое-
динение получают из диамида хиноксалин-2,3-дикарбоновой кислоты по
реакции Гофмана при действии 2 молей гипобромида натрия с последующей
обработкой щелочью [209].
8. Получение исходных веществ, необходимых для синтеза рибофлавина
Синтез 3,4-д им ети л фени л - D - р и б и т и л а м и н а и его про-
изводных
V
Для синтеза рибофлавина ключевыми исходными соединениями явля-
ются 3,4-диметилфенил-£)-рибитиламин (LIV) и его замещенные в положе-
нии 6 aMHHO-(LVIII) или арилазо- (LXIX) производные. Многочисленные
синтезы этих веществ могут быть сведены в несколько типовых схем, охва-
тывающих также основные пути получения различных синтетических фла-
винов.
I) Синтез из ароматических моноаминов н
D - р и б о з ы.
Важнейшее практическое значение для получения 3,4-диметилфенил-
D-рибитиламина (LIV) и его производных имеет способ (схема 122), по ко-
торому 3,4-диметиламинобензол, о-4-ксилидин (LXXIX) конденсируют с
.D-рибозой (LXXVIII) и образующийся N-рибозид (LXXX и LXXXI) вос-
станавливают в 3,4-диметилфенил-Р-рибитиламин (LIV) [210, 211].
Конденсация ариламина с углеводом в ариламиногексозид подробно
исследована Сорокиным [212]. Было показано [213], что при конденсации
ароматических аминов с альдозами образуются не шиффовы основания,
как ранее полагали, а N-гликозиды.
Конденсация D-рибозы с ароматическим амином протекает с образова-
нием двух форм N-гликозида: в среде безводного спирта при кратковре-
менном кипячении образуется 3,4-диметиланилин-М-а-Р-рибофуранозид
(LXXX), а в условиях комнатной температуры в слабокислой водно-спир-
товой среде [214] или в слабокислом водном растворе [215]—3,4-диметил-
анилин-М-а-Р-рибопиранозид (LXXXI). Пиранозиды легко переходят в
фуранозиды при нагревании в спирте. N-D-Рибопиранозид (LXXXI) об-
ладает способностью образовывать труднорастворимые комплексы с соля-
ми щелочных металлов [214, 216], например с сульфатом натрия. Инте-
ресно отметить, что способность к образованию комплексов имеют только
производные углеводов с quc-гидроксилами в положении 2 и 3, т. е. D-ри-
боза, D-ликсоза и D-манноза. D-Ксилоза, D-арабиноза и D-галактоза
комплексов не образуют [214].
531
I-'.’ . Схема 122 .. 1 . .
Синтез рибофлавина из D-рибозы, 3,4-диметиламинобензола, мочевины и малонового
эфира
сцон
чЛ~~°хн
Rh
но он
он
Р-Риьоза
LXXVIII
СН3.
СН,
IH,
СН,'
H Н
hJ----1сн,он
NH ---------
СН,ОН
носн
s’
4*
72%
LXXX
н н
82%
сн,
НОСН
НО^Н
си,
NH
г'
а'
сн,
IH
ОН
н
1;4-Диметиламиновензол
; * LXX1X
сн^
LXXX1
Ц'З;.
CH,.
СНГ.
сн,
3,4-Диметилфенил-
D - риьитиламин
’ LIV
85%
СООС3Н5
СН,
COOC,HS
Малоновый
Эфир
H,N'
Мочевина
•N—N
3,4-Диметилфенил-в-фенилаэо-
-D-рибитиламин
LXIX
он он он
СН,-С—с—с—сн,он,
Л н н н
со
iC /NH
СО
Барбитуровая
кислота
NH
ОС
СН,
сн,.
сн,-
сн,он
носн
носн
О
Рибофлавин
I
f
LXX
Для конденсации с ариламином (LXXIX) можно применять не труд-
нодоступную в кристаллическом виде D-рибозу (LXXVIII), а ее растворы,
не содержащие эпимерных примесей 1217]; конденсацию проводят в при-
сутствии борной кислоты, и образовавшийся борнокислый комплекс восста-
навливают в 3,4-Диметилфенил-Л-рибитиламин (LIV) обычным образом.
D-Рибозу (LXXVIII) в растворах, получаемых от восстановления чистого
кристаллического D-рибоно-у-лактона амальгамой натрия, также ус-
пешно конденсируют с ариламином (LXXIX) и без выделения продукт
конденсации непосредственно гидрируют в 3,4-диметилфенил-О-рибитил-
амин (LIV) [218].
Важное практическое значение имеет применение для получения 3,4-
диметиланилин-Г\тО-рибопиранозида (LXXXI) неочищенных растворов-D-
рибозы (LXXVIII), содержащих в качестве примеси эпимерную D-араби-
нозу. Из раствора такой смеси после конденсации D-рибозы с 3,4-диме-
тиламинобензолом (LXXIX) выделяют комплекс 3,4-диметиланилин-М-
D-рибопиранозида (LXXXI) с сульфатом натрия [219, 220] без примеси
эпимера. Этот метод получения 3,4-диметиланилин-М-О-рибопиранозида
дает возможность применить более простой синтез D-рибозы из D-глюкозы
через D-арабоновую кислоту, минуя D-рибонат кадмия или кристалличес-
кий D-рибоно-у-лактон (см. с. 542). Изучено влияние различных факторов
(pH, температуры, концентрации альдозы и спирта) на выход 3,4-диметил-
анилин-М-£>-рибопиранозида [220].
При каталитическом восстановлении в присутствии скелетного никеле-
вого катализатора обе формы N-рибозида (LXXX и LXXXI) дают один и
532
.тот же 3,4-диметилфенил-О-рибитиламин (LIV). Изучено влияние среды «
температуры на выход соединения LI V 1220].
Для последующей конденсации с пиримидиновым компонентом в рибо-
флавин в положение 6 молекулы 3,4-диметилфенил-О-рибитиламина (LIV)
необходимо ввести еще один атом азота в орто-положение к существующему
(методы синтеза рибофлавина 3 и 4). Для этого используется активность
атома водорода в положении 6 ароматического цикла соединения LIV к
сочетанию с солями диазония, которая усилена n-ориентирующим влиянием
метильной группы положения 3 и значительно превосходит подвижность
атома водорода в положении 2. Однако азосочетание всегда идет немного
и в положение 2; на количество образующихся азоизомеров оказывает
влияние входящая азогруппа. При азосочетании с солями диазония 3,4-
диметилфенил-6-фенилазо-ЬМ)-рибитиламин (LXIX, Аг = С6Н5) образует-
ся с выходом около 92%, а изомерный 3,4-диметилфенил-2-фенилазо-Ы-
D-рибитиламин (LXXXIII, Аг —СеН5)— только с выходом 6% (1731. Если
применить в качестве диазосоставляющей n-нитроанилин, то побочный
3,4-диметилфенил-2-п-нитрофенилазо-М-Д-рибитиламин (LXXXIII, Аг ==
= C6H4NO2) получается с выходом до 15%. Азосочетание проводят прй
pH 3—4 [221]. При pH 5 образуется не азосоединение LXIX, а диазоими-
носоединение (LXXXII) [176], которое не способно к конденсации в рибо-
флавин (схема 123)
Схема 123
Синтез производных 3,4-диметилфенйл-£>-рибитиламина
СН2ОН
носн
I
СН2ОН
носн
носн
носн
I
сн,
LXIX
CHjOH
носн
I
носн
сн2он
носн
I
носн
носн
I
CHjOH
носн
I
сн,он
носн
I
носн
LXXXIV
LXXXII LXXXIII
При азосочетании в муравьиной кислоте обычно образуется азосоеди-
нение, в то время как диазоаминосоединение (триазен) получается в аце-
татном буфере [102].
Обращает на себя внимание то обстоятельство, что скорость реакции
азосочетания находится в зависимости от расположения гидроксильных
групп алифатического заместителя у вторичной аминогруппы цикла. Пока-
зано [221], что при одном и том же значении pH среды — pH 4 —
стереоизомерные арилглюкамины (производные рибозы — LIV, араби-
нозы — LXXV, ксилозы — LXXXVI и ликсозы — LXXXVII) имеют раз-
533
личную реакционную способность при азосочетании с солями фенилдиа-
зония или л-нитрофенилдиазония, которая находится в связи с изменением
скорости реакции замещения в ядро из-за пространственных затруднений;
зто приводит к образованию различных конечных продуктов: азосоединений
и диазоиминосоединений.
S’ СН.ОН I СН2ОН 1 * енрн СН,ОН
4* носн неон носн HOCH 1
S’ носн носн неон неон
2’ носн неон 1 носн неон 1
I’ сн2 сн, сн, 1 * сн, СН.^
RNH RJ4H RNH RNH R= JI J
UV LXXXV LXXXVI LXXXVI! CHj^^
Пространственные затруднения образования азосоединений вызывают-
ся транс-расположением 2'- и З'-гидроксильных групп, например, у 3,4-
диметилфенил-Д-арабитиламина (LXXXV) и 3,4-диметилфенил-Ь-ксили-
тиламина (LXXXVI) и не наблюдаются при рядовом цис-расположении
гидроксильных групп при 2' и 3' атомах углерода полиоксиалкилзамес-
тителя вторичной аминогруппы, в том числе и 3,4-диметилфенил-О-ри-
битиламина (LIV) и 3,4-диметилфенил-Д-ликситиламина (LXXXVII).
3,4-Диметилфенил-б-фенилозо-О-рибитиламин (LXIX) применяют не-
посредственно для конденсации во флавин (синтез 4) или превращают в
3,4-диметил-6-аминофенил-£>-рибитиламин (LVIII) [210, 222J,. который при-
меняют также для получения рибофлавина (синтез 3). Восстановление
азосоединений в о-фенилендиамины осуществляют или каталитическим
путем над никелем (70, 102, 176, 177, 182,223], платиной, палладием [147,
155], или гидросульфитом натрия [223, 224], хлористым оловом и соляной
кислотой [225], цинком и уксусной кислотой [223].
При использовании рассматриваемого метода получения ароматичес-
кого компонента LXIX выход рибофлавина составляет 38% на кристалли-
ческую D-рибозу.
При восстановлении изоазорибитиламина (LXXXIII) образуется изо-
мерный 3,4-диметил-2-аминофенил-Л-рибитиламин (LXXXIV), который
способен к конденсации с аллоксаном в изорибофлавин (схема 122).
Для синтеза 3,4-диметил-6-аминофенил-О-рибитиламина используют ви-
доизмененный метод получения [226], по которому первоначально исходят из
3,4-диметиламинобензола (LXXIX), а образовавшийся 6-фенилазоксилидин
(LXXXVIII) конденсируют с D-рибозой (LXXVIII) в присутствии бор-
ной кислоты в 3,4-диметиланилин-6-фенилазо-Г4-Д-рибозид (LXXXIX);
в этом D-рибозиде одновременно восстанавливают азогруппу и оксидную
связь боковой цепи в соединение LVIII 1195].
LXXIX
LXXXY1H
Nu Л-рилэа
1 LXXV"1
N=NAr
снЬюфщон
NH
NH,
LXXXIX LVIII
Борнокислый комплекс 3,4-диметиланилин-6-фенилазо-М-Б-рибозида
(LXXXIX) можно непосредственно конденсировать с аллоксаном при од-
новременном каталитическом гидрировании в рибофлавин в среде метило-
вого спирта [195].
Синтез некоторых первичных и вторичных орто-аминоазосоединений
бензольного ряда осуществлен методом перегруппировки триазенов [101,
227, 2281.
534
2)Синтез из ароматических моноаминов и
D-арабинозы с использованием перегруппи-
ровки Амадори
Известен метод получения 3,4-диметилфенил-П-рибитиламина, в кото-
ром исходят не из труднодоступной D-рибозы, а из более легко получае-
мой D-арабинозы [229], отличающейся от D-рибозы противоположной кон-
фигурацией гидроксильной группы положения 2. В основу этого метода
положена перегруппировка Амадори [230], представляющая собой изоме-
ризацию ариламиногексозидов (пентозидов) в арилизогексозамины [231].
При нагревании ариламиногексозидов без растворителя или при дли-
тельном кипячении в спиртовом растворе [232] или в присутствии минераль-
ных и органических кислот происходит изомеризация в производные 1-
дезокси-1-аминокетоз; образующийся арилизогексозид в ряде случаев был
выделен в кристаллическом виде [231]. Такая изомеризация для производ-
ных пентоз протекает труднее [126, 229].
3, 4-Диметиламинобензол (LXXIX) конденсируют с D-арабинозой в 3,4-
Диметиланилин-П-арабинозид (XCI), который одновременно изомеризуют
в 3,4-диметилфенил-П-изоарабинозамин (ХСП) в присутствии бензойной
кислоты (схема 124) [229].
Схема 124
Синтез ЗЛ-диметилфенил-Р-рибитиламина из 3,4-диметнламинобензола и О-арабииозы
он н
н J---ксн/эн
|(н нср| -
нотгн -
ХС +
СИ, - ^NH,
снЛ -
иоах сн,
СН, NH,
LY1II
сн,^ - ,мн
сн,"
сн(снон),снсн,он
СИ, - NH
CH,CO(cHOt^CH,OH
NH
XCI
NH [н] СН,
СИ, N=NAr
хеш
сн,
UY
СЦ,(сНОН\сНрН
NH
аДснонУнрн
СН, NH
СН, - N=NAr
LXIX
При каталитическом восстановлении изоарабинозамина (ХСП) можно
было бы ожидать образования двух эпимеров: 3,4-диметилфенил-П-риби-
тиламина (LIV) и 3,4-диметилфенил-П-арабитиламина (LXXXV, с. 534),
по пространственному положению гидроксилов у С(2) и С(3) отвечающего
исходному арабинозиду (XCI). Получение того или иного эпимера зависит
от условий: при гидрировании с платиновым катализатором в органически
кислой среде образуется арабитиламин (LXXXV), а в щелочной среде —
рибитиламин (LIV); в присутствии минеральной кислоты восстанавливает-
ся ароматический цикл, а карбонильная группа остается неизменной. Вы-
ход 3,4-диметилфенил-Й-рибитиламина по этому методу составляет около
13%. Гидрирование в присутствии никелевого катализатора приводит к
получению смеси изомеров [233].
Видоизменением приведенного выше метода является получение изоара-
бинозамина в виде его азосоединения (ХСШ) [225]), которое затем восста-
навливается в смесь 3,4-диметил-6-аминофенил-Д-рибитиламина (LVIII)
и 3,4-диметил-6-аминофенил-П-арабитиламина (схема 124).
В результате конденсации D-арабинозы с 3,4-диметиламинобензолом,
перегруппировки образующегося арабинозида (XCI) в изоарабинозамин
(ХСП), восстановления его в рибитиламин (LIV) с последующим превра-
щением в азосоединение(LXIX), которое затем конденсировалось с барби-
туровой кислотой, рибофлавин синтезирован с общим выходом 7,5% на
D-арабинозу [175].
535
3) С и н т е з из ароматических моноаминов и /
ацетильных производных D-р нбоз ы
Конденсацию 3,4-диметиламинобензола (LXXIX) проводят с тетра-
ацетил-П-рибононитрилом (XCIV) и продукт конденсации каталитическим
гидрированием превращают в 2' ,3',4',5'-тетраацетил-3,4-диметилфенил- £
D-рибитиламин (XCVI, схема 125) (146, 234]. Предпочтительнее конденси- $
ровать с 3,4-диметиламинобензолом (LXXIX) тетраацетил-П-рибозу (XCV)
и продукт конденсации восстанавливать в 2',3',4',5'-тетраацетил-3,4-ди- 1
метилфенил-П-рибитиламин (XCVI) (235]. Его или деацетилируют в риби-
тиламин (LIV), или непосредственно сочетают с солями диазония в ’
2' ,3' ,4',5'-тетраацетил-3,4-диметилфенил-6-арилазо-£)-рибитиламин (XCVI I) 1
1173, 236], который далее применяют для конденсации с барбитуровой кис- 5
лотой в 2',3',4',5'-тетраацетилрибофлавин.
Существенное упрощение этого синтеза заключается в проведении кон-
денсации 3,4-диметиламинобензола (LXXIX) и тетраацетил-П-рибозы
(XCV) одновременно с восстановлением, с последующим гидролизом, что
приводит непосредственно к получению 3,4-диметилфенил-П-рибитилами-
на (LIV) без выделения промежуточных соединений [237].
Схема 125
Синтез 3,4-диметилфеннл-О-рибитнламина из 3.4-днметиламинобензола и ацетильных
производных D-рибозы
CHjfCHOA^jCHjOAc
СН, . / . NC(CHOAc)3CH2OAc -
у |] + XCIV
OHC(CI IOAcJjCHjQAc
LXXIX XCV
CH^CHOhXcHjOH
CH.
—~ 1T
CIL,-"^/
LIV
XCVI
I СН,(СНОАс)/:Н,ОАс
CH3
CH/^^N'-NAr
xcvu
Ac=COCH3
4) С и н т e з из ароматических моноаминов и
D-рибоно-у-лактона
Заслуживает внимания метод, в котором конденсацию 3,4-диметил-
аминобензола (LXXIX) проводят с D-рибоно-у-лактоном (XCVIII) при од-
новременном каталитическом восстановлении с платиновым катализатором
при 150 ат и 70—80° С в течение 30 ч и получают 3,4-диметилфенил-£)-ри-
битиламин (LIV) с выходом до 50% (схема 126); эта реакция протекает
также с 4-нитро-о-ксилолом, но с малым выходом [238].
Можно полагать, что альдонолактон (XCVIII) в этой реакции предва-
рительно восстанавливается в D-рибозу (LXXVIII), которая конденси-
руется с 3,4-диметиламинобензолом (LXXIX) в N-гликозид (LXXX),
далее восстанавливаемый в 3,4-диметилфенил-Р-рибитиламин (LIV). Это
следует из аналогичной реакции восстановительного алкилирования аро-
матических аминов эфирами альдоновых кислот, например при взаимодей-
ствии метилового эфира D-арабоновой кислоты и 3,4-диметиламинобензола
[2391. Процесс сопровождается одновременным образованием анилида
(XCIX), который, однако, в ариглюкамин (LIV) не восстанавливается.
По другому варианту (схема 126) D-рибоно-у-лактон (XCVIII) конден-
сируют с 3,4-диметил аминобензол ом (LXXIX) [234, 240] в анилид (XCIX),
ацетилируют, превращают в имидохлорцд, восстанавливают . в
536
2',3',4'>5,-тетраацетил-3,4-диметилфенил^-рибитиламин (XCVII), а затем
гидролизуют в 3,4-диметилфенил-О-рибитиламин (LIV).
Схема 126
Синтез 3.4-диметилфеиил-В-рибитиламииа из 3.4-диметиламинобензола и D-рибоко-
•у-лактона
[онс(снон)эсн2он]
LXXVIII
ОС(СНО11)2СНС112ОН
-------1 XCVIII
со(снон)2сн2он
NH
сн,
Сн* | XCIX
CO^HOAc^CHjOAc
ас(сНОАс)зСН,ОАс
I---О----)
сн(снон)2снсн2он
[н]
CH^CHOH^CHjOH
Ac=COCHj
CH/cHOAc^CHjOAc
5) Синтез ' из ароматических диаминов и D-
. ,£ рибозы
Производные о-фенилендиамина, в которых одна аминогруппа блоки-
рована в виде этоксикарбонильной группы — фенилуретаны (С) 11, 2, 221,
имеют то преимущество, что этоксикарбонильная группа в последующем
легко подвергается гидролизу с едким натром [157] или гидроокисью бария
[31. Следует отметить, что если аминогруппу защитить ацетильной группой,
то при гидролизе происходит образование 2-метилбензимидазола. Соеди-
нение С конденсируют с D-рибозой (LXXVIII) в N-рибозид (CI), затем вос-
станавливают в соединение СП и гидролизуют в 3,4-диметил-6-аминофе-
нил-И-рибитиламин (LVIII) [241]. Выход рибофлавина при использовании
этого варианта составляет 15% на кристаллическую D-рибозу.
н н
id-----Lch/jh
но тэ н
LXXVIII
CH(CHOh)jCHCH2OH
СН,.^. NH
CH/^^NHCOOCjHs
CI
CH^CHOHJjCHjOH
CH’ ПЙ-
Un
CHj NHCOOCjHs
CH2(CHOH)3CH2OII
CH,.-. NH
LVIII
6) Синтез из о-нитрозамещенных ароматических
соединений и D-рибозы
Производное о-динитро-(СП1) или о-хлорнитробензола (CIV) кон-
денсируют при повышенной температуре с D-рибозамином (CV) (получае-
мым из D-рибозы восстановительной конденсацией с аммиаком с выходом
80%) в 1,2-диметил-4-нитро-5-(1-О-рибитиламино)бензол (CVI), выход сос-
тавляет 40%. Восстановлением соединения CVI получают 3,4-диметйл-
537
6-аминофенил-П-рибитил амин (LVIII) [56, 74, 242]. Выход рибофлавина,
синтезируемого этим путем, составляет около 20%, считая на кристалличес-
кую D-рибозу (схема 127).
По другому варианту, исходя из 3,4-диметил-о-нитроанилина (CVII)
[3, 243], путем его конденсации cD-рибозой (LXXVIII) получают о-нитро-
анилин-М-П-рибофуранозид (CVIII). Хотя реакционная способность ами-
ногруппы к конденсации с сахарами понижается введением нитрогруппы
в ароматический цикл, однако разбавленная серная кислота в безводном
спирте является хорошим катализатором реакции [214, 244]. Хлористый
аммоний или метиламин также катализируют реакцию [243, 245].
о-Нитроанилин-М-П-рибозид (CVIII) каталитическим гидрированием
превращают в соединение LVIII. Восстановление протекает значительно
лучше в щелочной среде с Pd/BaSO4 под давлением в присутствии солей
борной кислоты в качестве буфера [243]. Использование гидразин-гидрата
для восстановления нитрогруппы дает хорошие результаты [246]. Выход
рибофлавина по этому методу синтеза около 16%, считая на D-рибозу.
Схема 127
Синтез 3(4-диметилфенил-Р-рибитиламина из производных иитро-о-ксилола
и D-рибозы или рибозами на
cm(civ)
.(Ct)
+ hnch3(choh)1ch1oh
CV
?CHr
Г CH,
H H
.NH, H J------kCH,OH
NO, НО О н
evil LXXVIII
^н,(снон),сн,он
CHj'^^'NO,
CVI I
<^HO^CHCH,OH
CHf^So,
CVIII
Офюн\рн/5Н
CH,v^4TiH
- YT
CH3 "/NH2
LVIII
С
Синтез 3,4 - диметиламинобензола (o-4 - ксилидина)
В качестве ароматического амина, необходимого для синтеза рибофла-
вина, важнейшее значение имеет 3,4-диметиламинобензол (о-4-ксилидин);
для получения его имеется несколько путей, в которых исходят из о-кси-
лола. Технический ксилол, состоящий из смеси о, ль и n-изомеров, имеющих
близкие температуры кипения, содержит всего 6—8%- о-ксилола [247].
Для его выделения в чистом виде применяют фракционированную перегон-
ку технического ксилола и последующее вымораживание.
о-Ксилол может быть превращен в 3,4-диметиламинобензол (LXXIX)
следующими методами (схема 128). При действии ацетилхлорида на о-кси-
лол в присутствии хлористого алюминия ’ получают 3,4-диметилацетил-
ацетофенон (CIX) [248], оксим которого (СХ) через стадию бекмановской
перегруппировки превращают в о-4 ксилидин (LXXIX) [249]. Метод нитро-
вания о-ксилола и последующего восстановления 3,4-диметилнитробен-
зола (СХШ) в о-4-ксилидин (LXXX) дает выход несколько выше 30%
[250J; нитрование сопровождается образованием изомерного 2,3-диметил-
нитробензола (СХП) с выходом свыше 50% [251].
Важное техническое значение имеет метод бромирования о-ксилола в
3,4-диметилбромбензол (CXI) с последующим его амонолизом 28%-ным вод-
ным аммиаком под давлением при 195° С в присутствии однохлористой и
металлической меди [252]. Продолжительность реакции бромирования при
30—40° С составляет 1—2 ч; выход соединения CXI около 90% [2531.
Интересен также метод, в котором исходят из n-нитротолуола, подвер-
гаемого хлорметилированию и последующему восстановлению в о-4-кси-
538
Схема 128
Синтез 3.4-дкметиламинобензола (о-4-ксилидина)
лидин (LXXIX). Хлорметилирование осуществляют сшил1-дих лор метило-
вым эфиром в присутствии в качестве конденсирующих агентов низкопро-
центного олеума [226, 254] или хлорсульфоновой кислоты [254, 2551; выход
2-хлорметил-4-нитротолуола (CXIV) почти количественный. С избытком
дихлорметилового эфира 2-хлорметил-4-нитротолуол образует 2,6-бнс-хлор-
метил-4-нитротолуол. 2-Хлорметил-4-нитротолуол (CXIV) подвергают ка-
талитическому гидрированию, которое проводят с платиновым катализа-
тором в уксуснокислой среде [2261 или с никелевым катализатором в спирто-
во-щелочной среде (содержание NaOH 0,6%) [225], или соединение CXIV
восстанавливают оловом и соляной кислотой [256], электролитическим пу-
тем [257], на свинцовом или цинковом катоде в среде 85%-ного спирта в
присутствии серной [257] или соляной [258] кислоты (выход 77—79%).
Синтез D-p и б о з ы
Необходимая для синтеза ароматического компонента рибофлавина
(3,4-диметилфенил-7)-рибитиламина и его производных) D-рибоза может
быть получена из продуктов гидролиза дрожжевых нуклеиновых кислот,
таких, как гуанозин (CXVI) и аденозин (CXVII) [259, 260].
CXVII
Источником нуклеиновых кислот служат сульфитные щелока, а также
дрожжи или их отходы от производства эргостерина для витамина D2,
из которых выделяется около 2—3% нуклеиновых кислот.
Однако значительно больший практический интерес представляют син-
тетические пути получения D-рибозы. Для синтеза ароматического ком-
понента рибофлавина применяют и промежуточные продукты получения
D-рибозы (LXXVIII), такие, как D-рибоно-у-лактон (XCVIII), тетрааце-
тил-D-рибоза (XCV) и др. Основным исходным сырьем для синтеза этих
539
соединений служит D-глюкоза, получаемая гидролизом кукурузного крах-
мала, или сахароза, после инвертирования дающая смесь D-глюкозы и
D-фруктозы.
1) Синтез D-рибозы через D-эритрозу
D-Рибозу получают из D-эритрозы наращиванием одного атома угле-
рода циангидриновым синтезом. При действии цианистого калия на D-
эритрозу (CXVIII) образуются нитрилы D-рибоновой и D-арабоновой кис-
лот, которые в результате гидролитического отщепления аммиака превра-
щаются в смесь D-рибоно-у-лактона и D-арабоно-у-лактона (XCVIII и
CX.IX)- Восстановление смеси лактонов амальгамой натрия приводит к
получению
ходом 25%
D-рибозы (LXXVIII) и D-арабинозы (ХС) с общим вы-
смеси
[261].
сно
I
НСОН
неон
сн2он
CXVIII
Альдозы разделяются и
HCN
NH3
со
неон I
I о
НСОН j
НС 1
СН2ОН
XCVIII
со
носн
।
неон
I
нс-----
СН2ОН
CXIX
I СН2ОН
О , н J—— О н
I—RLH^I
HO^f он
он
LXXVIII
сн2он
н о н
|ч^он^
но^г он
н
ХС
отделяются от лактонов на
амберлите: IR-120.
2)Синтез D-рибозы через производные
D-ксилозы
5-Бензоил-1,2-о-метилэтилиден-а-0-ксилофуранозу (СХХ), синтезиро-
ванную из D-ксилозы, окисляют в растворе хлороформ-бензола ди(трет-
бутоксид)хромом в 5-бензоил-1,2-0-метилэтилиден-а-0-эритропентафурано-
зидулозу-3 (CXXI), которую гидрируют на платиновом катализаторе в 5-
бензоил-1,2,-О-метилэтилиден-а-О-рибофуранозйд (СХХП), а из него ги-
дролизом с едким натром получают D-рибозу (LXXVIН) с выходом около
20% [2621
CHjOCOQHs
н]——он
- к* н
НО Of |-|
CXXII
3) Синтез D-рибозы через 1,2; 5,6 - д и - О - и з о п р о -
пилиден-3-мезил(тозил)-О-глюкозу
В этом синтезе D-глюкозу (СХХIII) первоначально при взаимодействии
с ацетоном в кислой среде превращают в 1,2; 5,6-ди-О-изопропилиден-а-
D-глюкофуранозу (СХХIV) с выходом 65%, при действии на которую метан-
сульфохлорида в пиридине с выходом 88%получают З-О-мезильное про-
изводное (CXXV); его гидролиз в 30%-ной серной кислоте приводит к
образованию З-О-мезилглюкозы (CXXVI) с выходом 80—90% [263, 264].
Тозильное производное получается со значительно меньшим выходом.
>хн,
(снэ)асх I
QCH
НО/1----Он
О'~Х(СН3)Э
CXXIV
(сНзХ I
осн
(CH,)2so2oJ—Он
К? н —
н т?
°~~с(снД,
CXXV
540
CXXVI
CHjOH
OHCOCH
HOCH
HCOSOjCH,
CHO
CXXVII
CH,OH r
н>—qh
кн и n
HO^|< OH
OH
LXXVIII
З-О-Мезилглюкоза (CXXVI) подвергается окислению перйодной кисло-
той, в результате чего образуется 4-О-формил-2-мезил-£>-арабиноза
(CXXVII), инверсия и гидролиз ацильной группы которой с помощью ед-
кого натра приводят к получению D-рибозы (XXVIII) с выходом 60% с
примесью 23% D-арабинозы. D-Рибозу выделяют в виде п-толилсульфонил-
гидразона, который расщепляют с помощью бензальдегида и серной кис-
лоты (выход 70%). [263, 264]. Общий выход кристаллической D-риббзы
составляет 19%.
4) С и н т е^з D-рибозы через D - а р а би н ал ь '
Основным промежуточным продуктом в синтезе D-рибозы через D-apa-
биналь [241, 265, 266] служит D-арабиноза (ХС), получаемая из D-глюкозы
(СХХШ) путем окисления последней бромной водой или электролитичес-
ким путем в D-глюконовую кислоту (CXXVIII), выделяемую в виде D-
глюконата кальция, который последующим окислением перекисью водорода
в присутствии солей трехвалентного железа (метод Руффа [267]) превра-
щают в D-арабинозу {ХС). Для реакции применяют 0,11—0,18 моля уксус-
нокислого железа (или его смеси с сернокислым железом) [268, 269]. Ус-
тойчивые выходы (44%) могут быть получены, если применять всего 0,028
моля уксуснокислого железа и проводить реакцию при 50—70° С [270].
D-Арабинозу (ХС) при действии уксусного ангидрида и бромистого во-
дорода переводят в триацетилбром-О-арабинозу (СХХГХ), затем обработ-
кой цинком превращают в диацетиларабиналь (СХХХ), деацетилируют его
окисью бария в арабиналь (CXXXI) и после окисления пербензойной кис-
лотой получают D-рибозу (LXXVIII) вместе с примесью (около 20%) D-
арабинозы. D-Арабинозу (ХС) можно окислить бромной водой в D-apa-
_боновую кислоту и превратить в D-арабоно-у-лактон (CXIX), который
можно использовать в синтезе D-рибозы в результате эпимеризации через
D-рибопо-у-лактон (см. ниже) [271—273].
Общий выход D-рибозы на D-арабинозу составляет 9—10,6%, а на
D-глюкозу — около 3,2%.
СООН^Са) )___________
неон неон
I I I
носн н,ог носн о
неон 44% неон
неон нс---
। ।
сн3он СН3ОН
CXXVIII ХС
HCBrj
AcOCH О
- I
HCOAc
I
HC-----
CHaOAc
CXXIX
- 1 1
HCOAc
HC------’
CH3OAc
CXXX
Ac-COCHj
87%|Вг3+Н3С
*„О
С^----1
НОСН I
I о
неон |
нс----1
снгон
LXXVIII
HC
HC 1
I о
неон |
HC----'
CXXXI
CXIX
541
5)Синтез D-рибозы через т ет р а а ц е т и л - D -
’рибоновую кислоту
Принципиальная схема синтеза известна из патентной литературы 1274].
В результате детальной разработки получаемая в этом синтезе тетрааце-
тил-П-рибоза (XCV) приобрела значение для получения рибофлавина.
D-Рибозу получают, исходя из D-рибоно-у-лактона (XCVIII) (синтез
см. ниже) через D-рибонат кадмия (СХХХП), тетраацетил-П-рибоновую
кислоту (CXXXIII), ее хлорангидрид и тетраацетил-а/-О-рибозу (XCV).
D-Рибонат кадмия (СХХХП) подвергают непосредственному ацетилиро-
ванию уксусным ангидридом в присутствии хлористого водорода [2751.
После образования хлорангидрида тетраацетил-О-рибоновой кислоты пос-
ледний переводится каталитическим гидрированием с палладиевым ката-
лизатором на активированном угле в кипящем ксилольном растворе в
тетраацетил-а/-О-рибозу (XCV) [274], а затем гидролизом — в D-рибозу
(LXXVIII); общий выход на D-глюкозу составляет около 8,4%.
CHjOH COOCd 1 ц соон 1 нс=о 1 енрн
н J—о неон НСОАс НСОАс н j—— о н
'н н Ь 1 =О—гНСОН (СН,СО)аО нЛ» *”М -НСОАс °И . - К»
но 'а НСОН 70% неодс 5096 НСОАс «** но он
он ’ Л СН2ОН CHjOAc CHjOAc он
XCVII1 СХХХП СХХХШ XCY LXXY1H
Ac=COCHj
6) Синтез D-рибозы
через D - ри б о и о - у - л а ктон
По этому синтезу, первоначально предложенному Фишером [271] для
L-рибозы, D-арабинозу (ХС) через D-арабоновую кислоту и ее лактон,
эпимеризацию в D-рибоновую кислоту и ее лактон с последующим разделе-
нием посредством их кадмиевых солей превращают в D-рибоно-у-лактон
(XCVIII) [271, 272, 276]; его восстанавливают амальгамой натрия в D-ри-
бозу, очищаемую затем через бромфенилгидразон [272, 276]; выход на D-
арабинозу составил 17% [272]. Рибоновую кислоту можно выделить в виде
ртутной [272] или аммонийной соли [277].
Однако значительно лучше D-арабоновую кислоту получать из D-глю-
козы не через D-арабинозу, а непосредственным окислением D-глюкозы
[278—2801.
При изучении кинетики расщепительного окисления D-глюкозы кис-
лородом в щелочных растворах в D-арабоновую кислоту показано, что ре-
акция интенсивно протекает при 40—45° С с выходом 70% [280]. Более
медленно, но с несколько большим выходом реакция идет при 35° С. При
температуре выше 45° С реакция идет по иному направлению, минуя обра-
зование D-арабоновой кислоты. Вероятный механизм этой реакции сос-
тоит в том, что под влиянием едкого кали (или едкого натра) D-глюкоза
(СХХIII) таутомерно превращается в ендиольную форму, и затем окисле-
нию молекулярным кислородом одновременно подвергаются первый и вто-
рой атомы углерода с разрывом этиленовой связи и образованием муравьи-
ной и D-арабоновой кислот [280]. D-Арабоновую кислоту выделяют в виде
калиевой соли (CXXXV, К) и затем обменной реакцией с хлористым каль-
цием переводят в более трудно растворимую кальциевую соль (CXXXV,
Са)
542
E -Phsoho- у-лактон
XCVIII
•Р-Ривоза
LXXVIII
Для синтеза с успехом используется сахароза; предварительно под
влиянием очень небольших количеств соляной кислоты в водном растворе
она инвертируется в D-глюкозу (CXXIII) и D-фруктозу (CXXXIV) — эта
смесь и подвергается окислению кислородом в щелочном растворе в обычных
условиях, только при несколько пониженной температуре; выход D-ара-
боната калия тот же.
Реакцию окисления осуществляют при высокой дисперсности кислорода
с самовсасывающей мешалкой без давления или под давлением 3—13 атм
(0,3—1,3 МПа), что значительно ускоряет процесс. Так как альдозы и
кетозы в щелочной среде изомеризуются в многочисленные продукты, в
том числе и с укороченной углеродной цепью, то целесообразно раствор ще-
лочи прибавлять постепенно. Метиленовый голубой [281] и индиго [282]
оказывают каталитическое влияние на реакцию окисления D-глюкозы и
D-фруктозы — выход D-арабоновой кислоты повышается на 8—12%. Воз-
можно и непосредственное выделение D-арабоната кальция из реакцион-
ного раствора, минуя выделение D-арабоната калия, после нейтрализации
соляной кислотой до pH 8,0.
Общий выход D-арабоната кальция (CXXXV, Са) из D-глюкозы (СХХШ)
и D-фруктозы (CXXXIV) достигает 80%.
Предложен способ противоточного окисления D-глюкозы кислородом
в колоннах тарельчатого типа или с насадкой [283].
Окислением кислородом 18%-ного водного раствора D-глюкозы при
постепенном прибавлении раствора едкого натра с последующей нейтра-
лизацией и взаимодействием с хлористым кальцием непосредственно по-
лучают D-арабонат кальция (CXXXV) [284] с выходом свыше 55%.
Классическая реакция эпимеризации обычно проводится в водных ра-
створах свободной альдоновой кислоты, получаемой из ее кальциевой
соли осаждением иона кальция щавелевой или серной кислотой, при 135° С
в течение 3 ч или при 100° С в течение 48 ч в присутствии пиридина [217].
Эпимеризацию осуществляют также в глицерине [273]. Показано [237, 285],
что хорошие результаты дает эпимеризация кальциевых солей альдоновых
кислот в присутствии органических гетероциклических оснований или
окислов щелочноземельных металлов; в этом случае процесс связан только
с однократной обработкой растворов щавелевой или серной кислотой (вмес-
то двукратной). Арабонат кальция подвергают эпимеризации также в вод-
ном аммиаке в течение 4 ч при 140° С под давлением [277].
Из растворов технической рибоновой кислоты D-рибоно-у-лактон
(XCVIII) может быть выделен в кристаллическом виде (т. пл. 77—79° С)
путем прибавления бутилового спирта и отгонки части растворителя [286].
Очистку рибоновой кислоты осуществляют на катионите сульфостироль-
ного типа [287].
Для восстановления лактонов альдоновых кислот в соответствующие
монозы известно применение различных гидридов металлов, на-
543
пример боргидрида натрия [288], однако выход D-рибозы из D-рибоно-у-
лактона не превышает 30% (в водном растворе).
Восстановление С-рибоно-у-лактона (XCVIII) в D-рибозу (LXXVIII)
осуществляется единственно реальным путем: с помощью амальгамы нат-
рия, получаемой или химическим способом, или в процессе электролиза,
или на амальгамированном катоде.
При восстановлении П-рибоно-^-лактона высокопроцентной (2,5—3%)
твердой амальгамой натрия для получения лучших результатов необхо-
димо поддерживать реакцию среды в пределах pH 3—4 [289] и применять
большой избыток амальгамы, вследствие чего в первое время, когда ско-
рость реакции особенно большая, трудно удержать в нужных пределах
кислотность среды; в нейтральной же и щелочной среде восстановление
протекает нес образованием полиоксиальдегида, а дальше — с образованием
соответствующего многоатомного спирта — рибита. Низкая температура
реакции (0, +2° С) позволяет уменьшить скорость побочной реакции об-
разования рибита и повысить выход D-рибозы до 75%. Изучена зависи-
мость выхода D-рибозы (LXXVIII) от температуры, pH, концентрации
D-рибоно-т-лактона (XCVIII) в растворе, содержания натрия в амальга-
ме [290].
Восстановление D-рибоно-т-лактона (XCVIII) и других альдонолакто-
нов осуществлено [291, 292] электролитическим путем на ртутном катоде
с диафрагмой В качестве катода могут применяться амальгамированные
катоды из различных металлов, однако восстановление на неамальгами-
рованных катодах не идет. Электролитическое восстановление может быть
проведено с применением в растворе католита борной кислоты, а также
сернокислого натрия и фосфорной кислоты [293] или ионов аммония
[294].
Хорошие результаты дает совмещенный способ электролитического
получения амальгамы с содержанием натрия 1—1,5% и последующего
восстановления П-рибоно-у-лактона в одном приборе при низких темпера^
турах (0, +5° С); выход D-рибозы составляет 60—70% [290].
Предложен способ получения D-рибозы (LXXVIII) и других моноз
Посредством электровосстановления лактона альдоновой кислоты [98],
в частности низкопроцентной амальгамой (ниже 0,1 %) щелочных металлов,
при непрерывной циркуляции реагента и ртути в процессе ее регенерации
[291, 295]. Такие условия дают возможность осуществить реакцию восста-
новления в мягких, строго регулируемых условиях.
На основе проведенных выше исследований разработан технологичес-
кий способ получения D-рибозы восстановлением D-рибоно-у-лактона не-
прерывно получаемой электролитическим путем жидкой амальгамой на-
трия (>0,3%), смешиваемой в проточных условиях с раствором лактона,
подксиленным соляной кислотой [296, 297].
D-Рибоза может быть выделена из раствора через анилин-М-П-рибо-
зид: горячий водный раствор его пропускают через амберлит 1R-120 и
после концентрирования получают кристаллическую D-рибозу 1298].
Так как выделение рибозы в твердом виде для синтеза рибофлавина не
вызывается необходимостью, а при получении D-рибозы (LXXVIII) с при-
месью (25—30%) эпимерной D-арабинозы (ХС) эффективная очистка от
эпимерных примесей достигается на последующих стадиях синтеза рибо-
флавина, а именно при получении 3,4-диметиланплин-М-£>-рибоппранозида
(LXXXI) в виде комплекса с сульфатом натрия [220], то весь цикл син-
теза D-рибозы можно упростить и сократить. Таким образом, синтез зак-
лючается в эпимеризации арабоната кальция (CXXXV, Са), выделении
свободной рибоновой кислоты с примесью арабоновой, лактонизации BD-ри-
боно-у-лактон (XCVIII) с примесью 0-арабоно-?-лактона и восстановле-
нии лактона в D-рибозу (LXXVIII) с примесью D-арабинозы [219]. Общий
выход D-рибозы, используемой в дальнейшем синтезе рибофлавина, свыше
25% (считая на D-глюкозу или на смесь D-глюкозы и D-фруктозы. •
544
7) Ферментативный синтез D-рибозы
Ферменты хлебопекарных дрожжей осуществляют биосинтез D-рибозы
(LXXVIII) альдольной конденсацией из D-глицеринового альдегида и из-
бытка оксиппровиноградной кислоты с выходом 88% за 2 ч на соответствую-
щих питательных средах [299].
Синтез аллоксана и барбитуровой кислоты
В качестве пиримидинового компонента для синтеза рибофлавина при-
меняется аллоксан или барбитуровая кислота. Аллоксан (LVI, см. с. 524)
можно получить из 5,5-дихлорбарбитуровой кислоты, из барбитуровой
кислоты через бензальбарбитуровую кислоту и из мочевой кислоты через
аллоксантин [300].
Синтез барбитуровой кислоты (LXX) осуществлен конденсацией амида
малоновой кислоты и эфира угольной кислоты — (С2Н5О)2СО — в жидком
аммиаке в присутствии едких щелочей с выходом 56%; т. пл. 243—244° С
(с разл.) [301]. Из-за недостаточно высоких выходов этот метод не имеет
технического значения.
Для получения барбитуровой кислоты (LXX) исходят из диэтилового
эфира малоновой кислоты и мочевины (формулы см. на с. 532), которые кон-
денсируют под влиянием спиртового раствора этилата натрия при равно-
молекулярных соотношениях реагентов [302—305]. Однако лучший выход
(80%) барбитуровой кислоты получен при проведении реакции с 1,25 мо-
ля этилата натрия [144]. Первоначально образующийся натриймалоновый
эфир в своей таутомерной форме ацилирует одну аминогруппу мочевины в
уреид эфира малоновой кислоты; ацилирование второй аминогруппы с за-
мыканием цикла может проходить в более мягких условиях, в присутствии
поташа или аммиака.
Виолуровую кислоту (LV) получают нитрозированием барбитуровой
кислоты нитритом натрия с высоким выходом.
БИОЛОГИЧЕСКОЕ ЗНАЧЕНИЕ РИБОФЛАВИНА. ЗАВИСИМОСТЬ МЕЖДУ
СТРОЕНИЕМ И БИОЛОГИЧЕСКОЙ АКТИВНОСТЬЮ. АНТИВИТАМИНЫ
Значение рибофлавина для животного организма заключается в том,
что в процессах метаболизма он в составе флавиновых ферментов осуще-
ствляет реакции дегидрирования. Биокаталитическое дегидрирование фла-
виновыми ферментами является только звеном в цепи окислительно-восста-
новительных процессов, в которой и восстановленная форма ферментов
претерпевает окисление (восстановление дегидрирующих свойств).
Рибофлавин широко распространен в природе — в микроорганизмах,
в растительных и животных клетках, однако животные организмы не спо-
собны к самостоятельному биосинтезу рибофлавина и получают его или
с пищей, или в результате деятельности микрофлоры желудочно-кишечного
тракта. В виде соединения с фосфорной кислотой (рибофлавин-5'-фосфат,
ФМН), или соединения ФМН с АМФ (флавинадениндинуклеотид, ФАД),
или 8 а-производных ФАД, ковалентно связанных с белком, которые в
свою очередь связываются с апоферментами и обычно с металлами
(флавиновые ферменты), рибофлавин находится в различных органах и
тканях [308], почти во всех аэробных клетках. В свободном виде рибофлавин
найден в молоке, моче и пигментном слое сетчатки глаз [309, 310]. В сет-
чатке глаз рыбы рибофлавина содержится до 20 мкг в 1 г свежей ткани
[19]. Однако в животных тканях рибофлавин находится преимущественно
в виде ФМН, ФАД и 8 а-производных ФАД. В тканях человеческого орга-
низма содержится около 85% ФАД, от 10 до 15% ФМН и только несколько
процентов рибофлавина.
Специфическое влияние рибофлавина на организм человека было обна-
18-69 545
ружено в ряде случаев. Арибофлавиноз выражается в особом кожном за-
болевании в области рта, а в тяжелых случаях происходит поражение глаз.
Витамин В2 принимает важнейшее участие в механизме зрения. Благо-
даря своей светочувствительности он превращает действующие на него фио-
летовые и синие лучи в более длинноволновые (свет зеленой флуоресценции),
к которым глаз обладает большой чувствительностью [306], т. е. выполняет
как бы роль сенсибилизатора в зрении, производя батохромный эффект.
Некоторые глазные заболевания успешно излечиваются рибофлавином.
При патологических процессах, связанных с нарушением обмена веществ,
например в опухолях при раковых заболеваниях [307], обнаружено увели-
ченное содержание рибофлавина по сравнению с содержанием его в окру-
жающих тканях.
Физиологическая потребность человека в рибофлавине составляет
2—2,5 мг в день. Следует отметить, что в обычном пищевом рационе чело-
века содержание витамина В2 недостаточно.
Наряду с тиамином и никотиновой кислотой рибофлавин применяется
для витаминизации сортов белого хлеба и других изделий из пшеничной
муки.
Специальные добавки рибофлавина оказывают стимулирующее действие
на птиц и крыс: при его недостатке происходит задержка роста молодых
животных, а у крыс наблюдается общее заболевание кожи, так называе-
мый «неспецифический» дерматит (при одновременном недостатке биотина).
Рибофлавин занимает важное место в животноводстве; эффективное
бройлерное производство цыплят и высокая яйценоскость кур невозможны
без специальных добавок в корм витамина В2.
Рибофлавин применяется для витаминизации комбикормов — 5—8 г/т.
При поступлении внутрь в слизистой оболочке кишечника рибофлавин
под действием флавокиназы в своей большей части фосфорилируется в ФМН,
а затем в печени в присутствии АТФ и ионов магния превращается в ФАД
[311]. Рибофлавин (излишек) выводится из организма с большей скоростью
[312]. а-Оксиэтилфлавин — метаболит рибофлавина — выделен из козьего
молока [313], а также из мочи овец и коз при кормлении их большими коли-
чествами рибофлавина [313].
В строении молекулы рибофлавина можно обнаружить сродство ее от-
дельных фрагментов со структурными частями различных важнейших
биологически активных соединений (314). Так, с витаминами группы
фолиевой кислоты (см. с. 459) рибофлавин объединяет общность конден-
сированных пиримидинового и пиразинового циклов [315]. С тиамином
(см. с. 376) рибофлавин объединяет общность пиримидинового цикла (в
молекуле рибофлавина он срощен с хиноксалиновым циклом). Нуклеиновые
кислоты включают в свою молекулу D-рибозу, а рибофлавин — ее вос-
становленную форму — D-рибит. о-Ди метил бен зол с двумя атомами азота
в орто-положении входит как в молекулу рибофлавина, так и в молекулу
цианокобаламина, витамина В12 (см. с. 586).
По своей структуре рибофлавин высокоспецифичен; уже небольшие
изменения в его молекуле приводят к потере витаминной активности или
к появлению антивитаминных свойств. Среди многочисленных искусствен-
ных флавинов и других изоаллоксазинов лишь немногие обладают частич-
ной витаминной активностью рибофлавина (табл. 21).
Следует отметить, что при замене радикала D-рибитила L-арабнтилом
хотя и сохраняется V3 активности, но этот флавин вызывает лишь прохо-
дящее стимулирование роста крыс, приводя затем к смерти животных [2,
322]. При замещении другими радикалами пяти- и шестиатомных спиртов
активность пропадает полностью или, как для радикала L-ксилитила,
сохраняется лишь в незначительной степени. Замещение одной метильной
группы в положении 7 или 8 одной этильной группой сохраняет только 50
или 38% соответственно активности рибофлавина, однако наблюдается
уменьшение воспроизводимости животных [323]. Замена обеих метильных
546
Таблица 21
Витаминная активность флавинов
Название флавинов Витаминная ак- тивность по срав- нению с рибофла- вином, взятым за 1 T. пл., °C Литера- турная ссылка
7,8-Диметил-10-(Г-О-рибитил) изоаллоксазии (ри- бофлавин) 2-Тио (дезоксо)рибофлавин 7-Метил-Ю-(Г-О-рибитил) изоаллоксазии 8-Метил-Ю-(Г-О-рибитил) изоаллоксазии 7-Этил-8-метил-10-(1'-О-рибитил) изоаллоксазии 7,8- Д иметил-10-( Г - L-a рабитил)изоа ллоксазин 7,8-Тетраметилен-10-( Г - Ь-арабитил)изоаллоксазин 7,8-Т риметилен-10-( Г-О-арабитил)изоа ллоксазин 7,8-Диметил-10-(1'-Д-ксилитил)изоаллоксазин 7,8-Диметил-10-(Г-О-ликситил)изоа ллоксазин (£)-ликсофлавин) 1 Около 1 0,5 0,5 0,5 0.33 0,25 0,25 Слабая Очень слабая 282 282 285—286 238—240 298 285—286 300 280—282 [1-3] [100 [316 [157 [224 [158, 317] [318] [318] [319] [320]
Примечание. Из сердечной мышцы человека был выделен флавин, которому
приписывалось строение L-ликсофлавина и высокая биологическая активность [320].
Это соединение было синтезировано различными путями [179, 180 , 320], однако дан-
ные о содержании L-ликсофлавина в сердечной мышце и его витаминной активности
ие подтвердились [321].
групп тетраметиленовыми или триметиленовыми группами сохраняет
витаминные свойства только на 1/4. Удаление одной метильной группы из
положения 7 или 8 сохраняет только ’/2 активности. 2-Тиорибофлавин
по своей витаминной активности на крысах и цыплятах почти равноценен
рибофлавину [100], в то время как 4-тиорибофлавин имеет всего 10% ак-
тивности рибофлавина [324]. Все же другие заместители в положении 7 и
8 или же в 6 и 9, а также различные заместители в положении 10 приводят
к устранению всякого коферментного действия и витаминной активности
флавина.
При испытании на Lactobacillus casei аналоги рибофлавина с одним за-
местителем в бензольном кольце обладали ростовой активностью: 7-этил —
3% [325], 7-метил — 3,6% и 8-метил — 21% [326] от активности рибофла-
вина. Аналог с незамещенным 7- и 8-положением обладает высокой токсич-
ностью [327].
Высокая специфичность молекулы рибофлавина подчеркивается еще
тем, что витаминная активность сохраняется только при наличии свобод-
ной иминогруппы в положении 3, ее метилирование уничтожает витаминные
свойства [326]. Это, возможно, связано со способностью иминогруппы фос-
форного эфира рибофлавина участвовать в образовании комплексов в
активном центре фермента [328]. Остаток пентита также должен иметь в
положении 2 гидроксильную группу, расположенную так, как это изобра-
жается для D-рибозы, L-ликсозы и L-арабинозы. Однако ацетилирование
гидроксильных групп, которое могло бы уничтожить всякое коферментное
действие, не снижает активности рибофлавина, по-видимому, вследствие
легкой способности 2',3',4',5'-тетраацетилрибофлавина к гидролизу в ор-
ганизме. Рибофлавин-5'-фосфорный эфир обладает активностью свободного
рибофлавина. Рибофлавин-5'-пирофосфорный эфир имеет 33% активности
витамина В2 [329].
Рибофлавин — важнейшее ростовое вещество для многих микроорга-
низмов. Некоторые аналоги рибофлавина, наоборот, являются антаго-
нистами его свойств, бактериальными ингибиторами; к ним относятся
люмифлавин [330], арабофлавин [331], галактофлавин [227, 332], изорибофла-
вин, 6,7-диметил-10-М-(1'-О-рибитил)изоаллоксазин [333] (витаминный
индекс 200; в дозе 2 мг в день на крысу он совершенно подавляет
витаминную активность 10 мкг рибофлавина [333]). При замене метильной
18*
547
группы в 7- или 8-положении молекулы рибофлавина атомом хлора обра-
зуются антивитамины 1334—336], например 8-хлор-7-метил-Ю-М-(Г-О-риби-
тил )изоаллоксази и.
а^снонУнрн
hoch^choi <\сн2. сн/а о ^снрн
При испытании на Lactobacillus casei его индекс ингибирования
составил 85 1112]. К сильным антивитаминам относятся б'-дезоксирибо-
флавин [337] и рибофлавин-5'-сульфат [40]. Однако наиболее сильное
ингибирование вызывает введение в изоаллоксазиновую молекулу амино-
группы [115, 172, 338]: 8-амино- и 8-рибитиламиноноррибофлавин имеют
индекс ингибирования 32 и 9 соответственно [112].
Соединения, в которых сохранены функциональные группы, характер-
ные для рибофлавина, но внесены глубокие изменения в изоаллоксазино-
вое ядро путем замены двух атомов азота на внециклические, например
2,4-диамино-7,8-диметил-10-(Г-П-рибитил)дигидрофеназин, также явля-
ются бактериальными ингибиторами [339].
Многие антималярийные препараты подавляют витаминные свойства
рибофлавина; это обстоятельство вызвало многочисленные исследования
по изысканию среди соединений аллоксазинового ряда таких производных,
которые обладали бы лечебными свойствами против малярии [340—343].
Некоторые изоаллоксазиновые производные оказались активными про-
тив развития искусственно вызванных злокачественных опухолей у мышей.
К таким флавинам относится 7-хлор-10-(Г-П-сорбитил)изоаллоксазин н
другие 7-моно- и 7,8-дихлорпроизводные [344,345], а также 2-тиолюмифла-
вин и 2-люмифлавинимин [721.
Среди изоаллоксазинов обнаружены соединения с сильным противовос-
палительным эффектом или жаропонижающим действием [346].
Так как флавины обладают типичной способностью к флуоресценции
и некоторые из них — способностью к свечению в ультрафиолете, то пред-
ставляет значительный интерес применение их в качестве люминесцентных
соединений.
ФЛАВИНОВЫЕ КОФЕРМЕНТЫ
Свою биологическую функцию в живом организме рибофлавин осуще-
ствляет в виде флавиновых нуклеотидных коферментов— фосфорного эфира
рибофлавина (ФМН) и производного этого эфира — соединения с адени-
ловой кислотой, P-N(9) -аденин-0-рибофуранозил-5'-фосфатом—флавин-
адениндинуклеогида (ФАД); кроме того, 8а-производные рибофлавина обра-
зуют коферменты: 8а-гистидил-ФАД и 8а-цистеинил-ФАД. В свою очередь
флавиннуклеотиды являются простетической активной группой флавиновых
ферментов и осуществляют окислительную биокаталитическую функцию
только в соединении с апоферментом — белковой составной частью моле-
кулы ферментов.
Рибофлавин-5'-фосфорный эфир, рибофлавин-5'-
фосфат, флавинмононуклеотид1— РМФ, ФМН (V) — кристаллическое веще-
ство желто-оранжевого цвета с т. пл. 215° С, [alo + 44,5° (Н2О).
1 Термин «флавинмоноиуклеотид» — не точный: рибофлавин-5'-фосфат представ-
ляет собой нуклеотид с D-рибозой, восстановленной в D-рибит.
548
он
I
S’ CHjO-P-OH
< носн о
I
? HOCH
I
2- HOCH
I
CH2
ФМН образует фосфорноэфирную связь по первичной гидроксильной
группе полиоксиалифатической цепи (23, 347, 3481. Он хорошо раство-
рим в воде, почти нерастворим в спирте, нерастворим в эфире, хлороформе
и многих других органических растворителях.
Спектр поглощения рибофлавин-5'-фосфата — Хмакс (£) в воде: 445
(12,5-103), 373 (10,4-10s), 266 (31,8-103) нм. ФМН обладает желто-зеленой
флуоресценцией в ультрафиолетовом свете с Хмакс 525 нм. Он неустойчив
в водных растворах на свету, в кислых растворах в темноте относительно
стабилен, в щелочных растворах довольно быстро разлагается с расщепле-
нием пиримидинового цикла. ФМН начинает подвергаться гидролитичес-
кому расщеплению в водных растворах в темноте при температуре выше
80° С; при 100° С за 48 ч среди продуктов распада обнаруживается около
70% рибофлавина и около 15% люмифлавина [349]. Для стабилизации
ФМН в водных растворах используют этилендиаминотетрауксусную кис-
лоту [350]. РМФ является двухосновной кислотой с двумя точками пере-
хода — при pH 4,5 и 8,5. Он образует хорошо растворимую в воде натрие-
вую соль, которая растворима примерно в 200 раз больше, чем рибофлавин.
Бариевая соль нерастворима в воде.
Флавинадениндинуклеотид, Р,-(рибофлавин-5')-Р2-(аде-
нозин-5')пирофосфат — ФАД (CXXXVI)—состоит из молекулы рибо-
флавина, этерифицированной по первичной гидроксильной группе остатком
фосфорной кислоты и соединенной фосфоангидридной связью с аденозин-
5'-фосфатом (АМФ)
ФАД представляет собой’вещество желто-оранжевого цвета; хорошо рас-
творим в воде, почти не растворим в спирте.
ФАД имеет подобный рибофлавину спектр поглощения с Хмакс (е) в
воде: 450 (11,3- 10s), 375 (9,3-103), 263 (38,0-103) нм. В водном растворе
ФАД при возбуждении ультрафиолетовым светом флуоресцирует (желто-
зеленая флуоресценция) с Хмакс 520—530 нм, причем интенсивность флу-
оресценции составляет 18—20% таковой для рибофлавина.
В отличие от рибофлавина ФАД в водных растворах устойчивее к фо-
толизу примерно более чем в 20 раз, однако в водных растворах при нагре-
вании он быстро дезактивируется; при 100° С за 4 ч активность ФАД умень-
549
шается на 30%. Для стабилизации ФАД к растворам прибавляют этиленди-
аминотетрауксусную кислоту [351]. В щелочных растворах он быстро
расщепляется. ФАД является слабой, двухосновной кислотой.
8а- (3-N-L-F и стид и л) флавин аденинди нуклеотид,
Р1-[8а-(М-А-гистидил)рибофлавин-5']-Р2-(аденозин-5')пирофосфат —8а-гисти-
дил-ФАД (СХ XXVII) [352, 353]
8а-Гистидил-ФАД — недиссоциирующий кофактор сукцинатдегидро-
геназы; в индивидуальном виде не выделен. Флавин этого кофактора по
8-СН2-группе ковалентно связан с атомом азота L-гистидина. Имеет по-
добный ФАД спектр поглощения, однако второй максимум (375 нм) имеет
гипсохромный сдвиг на 20—25 нм; в ультрафиолетовом свете флуоресци-
рует желто-зеленой флуоресценцией, характерной для ФАД.
8а-(S- Цисте и нил)флавин аденинди нуклеотид,
Р1-[8а-(8-цистеинил)рибофлавин-5']-Р2-(аденозин-5')-пнрофосфат — 8а-цис-
теинил-ФАД (CXXXVIII) [354—356]
8а-Цистеинил-ФАД — недиссоциирующий кофактор моноаминоокси-
дазы, флавин которой ковалентно связан по 8-СН2-группе с атомом серы
аминокислоты, в индивидуальном виде не выделен. Спектр поглощения
8а-цистеинил-ФАД подобен спектру 8а-гистидил-ФАД, так же как и его жел-
то-зеленая флуоресценция, которая имеет низкий выход по всей области pH.
Флавиновые коферменты — ФМН, ФАД и 8а-замещенные ФАД — легко
присоединяют водород (протон и электрон) по положениям 1-N и 5-N
изоаллоксазинового цикла, отщепляя его, как правило, от восстановленных
форм никотинамидных коферментов (НАД-Н и НАДФ-Н) или в некоторых
случаях] непосредственно от субстрата — донора водорода. При восстано-
влении флавиновых коферментов образуются промежуточные окрашенные
семихиноидные формы и затем, с присоединением всего двух электронов и
двух протонов, — бесцветные дигидросоединения: дигидро-ФМН и диги-
дро-ФАД (ФМН-Н2 и ФАД-Нг), обратимо окисляющиеся кислородом
550
воздуха или цитохромами а, Ь, с в организме [357] вновь в исходные оки-
сленные коферментные формы (ФМН и ФАД); то же самое относится к
8а-гистидил-ФАД и 8а-цистеинил-ФАД.
Ц— остато* кофермента
Флавины имеют сравнительно высокое сродство к электрону. Поэтому,
пс-видимому, одно- и двухэлектронное восстановление ФМН и ФАД про-
текает не путем первоначального присоединения протона к изоаллоксази-
новой молекуле по типу А или Б
и затем присоединения электрона с образованием
нейтрального свобод-
ного флавинового радикала ФлН2 (ФлН — флавин: рибофлавин, ФМН,
ФАД и замещенные ФАД), а в результате присоединения электрона с обра-
зованием анионрадикала ФлН- и последующей протонизации в флавин-
радикал ФлН2
е" . Н+ . е- Н+
ФлН----сФлН-----> ФлН2---->-ФлН2---► ФлН3.
Присоединение второго электрона приводит к образованию флавинового
аниона ФлН2-, который со вторым протоном дает дигидрофлавиновое сое-
динение ФлН3. Дигидроизоаллоксазины легко обменивают свой протон с
протоном окружающей среды. Следует отметить, что чрезвычайно высокая
активность флавиновых ферментов как биокатализаторов, по-видимому, и
связана с тем, что флавиновые ферменты участвуют в переносе электрона в
цепи тканевого дыхания в виде промежуточного флавосемихиноидного
соединения свободного флавин-радикала хлорофлавина ФлН2.
Показательно, что флавиновые ферменты имеют значительно более вы-
сокий окислительно-восстановительный потенциал (например, старый жел-
тый фермент — 0,12 В), т. е. обладают большей способностью к восста-
новлению, чем рибофлавин (—0,21 В) или ФМН и ФАД (от —0,187 до
—0,191 В при 20° С и —0,219 В при 30° С при pH 7) [358].
ФАД и ФМН образуют эквимолекулярные хелатные комплексы с ме-
таллами: Са, Mg, Ba, Cd, Mn++, Со++; Ni++ [359]. При действии азотистой
кислоты происходит дезаминирование ФАД (в структуре аденина) с обра-
зованием 6-окси(дезамино)-ФАД. Воздействие света в щелочной среде
приводит к расщеплению ФАД с образованием люмифлавина.
При действии мягких дегидратирующих агентов, например N, N'-
дициклогексилкарбодиимида в водном пиридине РМФ превращается в
рибофлавин-4',5'-циклофосфат (CXXXIX) с количественным выходом [360].
ОН ОН О о
1111
СН —СН—С Н —С H-CHj
CXXXIX
551
Рибофлавин-4',5'-циклофосфат при кислом гидролизе преимущественно
дает РМФ (V).
РМФ легко ацилируется; при действии соответствующих ацилирующих
агентов получены 2',3',4'-триацетил- и 2',3',4'-трибензоилрибофлавин-
5'-фосфат [361]. Если РМФ обработать трифторуксусным ангидридом, то
он превращается в 2',3'-дифторацетилрибофлавин-4',5'-циклофосфат [362],
который при действии спирта и аммиака гидролизуется в рибофлавин-
4',5'-циклофосфат [362, 363].
Рибофлавин-2',3',4'-трифосфат нестабилен; при его образовании путем
гидролиза 5'-тритилрибофлавин-2',3',4'-три(|3-циан)этилфосфата (перво-
начально в 10%-ной уксусной кислоте и затем в аммиаке) единственным
продуктом реакции является рибофлавин-5'-фосфат (V), что связано с
миграцией фосфорных групп от вторичных гидроксилов к первичной гидрок-
сильной группе положения 5' [364].
Рибофлавин-5'-фосфоамиды легко гидролизуются в РМФ [365].
СТРОЕНИЕ И ВЫДЕЛЕНИЕ ФЛАВИНОВЫХ КОФЕРМЕНТОВ
А. Сент-Дьерди и Банга в 1932 г. [135] впервые выделили из сердечной
мышцы желтый флавиновый кофермент (ФМН), названный ими «цитофла-
вом»; этот кофермент принимает участие в биохимическом окислении глю-
козо-6-фосфата. Варбург и Христиан в 1932 г. [19] открыли и изолировали
из дрожжей первый флавопротеид, так называемый «старый желтый фер-
мент». Теорелл в 1934—1935 гг. [366] выделил простетическую группу —
ФМН — как кофактор «старого желтого фермента» (дегидрогеназы восста-
новленного НАД) и установил, что в этом веществе содержится 1 моль
фосфата.
Желтый окислительный фермент Теорелл разложил на его составные
части, из которых фермент впервые в истории биохимии был вновь синте-
зирован [366]. В белковой части желтого окислительного фермента уста-
новлено присутствие следующих аминокислот: 33 мол. аргинина, 13 мол.
гистидина, 12 мол. аспарагиновой кислоты, 34 мол. глутаминовой кисло-
ты, 66 мол. лизина, 40 мол. пролина, 30 мол. тирозина, 17 мол. триптофа-
на, 24 мол. фенилаланина и 1—2 мол. цистина [367].
Положение фосфорного остатка в молекуле ФМН было определено ме-
тодом перйодатного окисления [347] и подтверждено синтезом флавинмоно-
нуклеотида, осуществленным в результате прямого фосфорилирования
рибофлавина хлорокисью фосфора в пиридине [29]. Однако окончательно
структура ФМН была доказана последовательным синтезом из рибофла-
вина в результате предварительной защиты первичной гидроксильной
группы рибитильной цепи трифенилметильной (тритильной) группой с
последующим ацетилированием вторичных гидроксильных групп с обра-
зованием ^'.З'Л'-триацетил-б'-тритилрибофлавина, избирательного снятия
тритильной защиты и последующего фосфорилирования хлорокисью фос-
фора в пиридине, что привело к образованию 2',3',4'-триацетилрибофла-
вин-5'-фосфата; это соединение мягким щелочным гидролизом было превра-
щено в рибофлавин-5'-фосфат [368].
(С6Н5)аСС1 (СНЭСО)2О
RCHj (СНОН),СН2ОН---------> RQi2(CHOH)sCH2(3C (С(;НЬ)2 ------*
Частичный
гидролиз
RCH2 (CHOCOCH3)sCH2OC (С8Нь)а-------►
POCIs Гидролиз
-> RCH3(CHOCOCH3)3CH2OH------> RCH2 (СНОСОСН3)3СН3О po2h2---------♦
-> RCH2(CHOH)3CH2OPO3Ht,
R = 7,8-диметилизоаллоксазин.
552
Позднее, в 1938 г. [369] Варбург и Христиан выделили из почек и пече-
ни овцы и лошади и дрожжей флавинадениндинуклеотид как кофактор окси-
дазы D-аминокислот. Структура ФАД была установлена на основании
идентификации продуктов его расщепления, которыми оказались ФМН
и АМФ [370, 371]. Позднее строение ФАД было доказано синтезом из се-
ребряной соли рибофлавин-5'-фосфата и 2',3'-изопропилиденаденозин-5'-
бензилхлорфосфата [372].
Было известно, что простетнческая группа сукцинатдегидрогеназы не
идентична ФМН или ФАД и в отличие от них ковалентно связана с одной
из аминокислот белка [373]. Последующими исследованиями установлено,
что этот кофермент имеет структуру 8а-(М-£-гистидил)-ФАД. При воздей-
ствии протеолитическими ферментами из сукцинатидегидрогеназы было
выделено пептидное производное — СД-ФАД [373]. Затем в результате
кислотного гидролиза из этого соединения получено 6—7-аминокислот —
аланин, серин, глутаминовая кислота, валин, треонин и, кроме того, СД-
рибофлавин, содержащий еще один аминокислотный остаток [373]. Были
основания считать, что аминокислота связана с 8-СН3-группой рибофлавин!
При дальнейшем жестком кислотном гидролизе выделен гистидин, связь
которого с флавином осуществлялась по положению З-N. Таким образом
СД-рибофлавин представляет собой 8а-(3-1Ч-А-гистидил)рибофлавин [353].
Это соединение было получено встречным синтезом в результате нагревания
8а-бром-2',3',4',5'-тетраадетилрибофлавина с бензоилгистидином в безвод-
ном диметилформамиде с последующим гидролизом 6 н. соляной кисло-
той [352].
Структура 8а-(8-цистеинил)-ФАД установлена на основании следующих
исследований. Недиссоциирующий кофактор моноаминооксидазы с пептид-
ной связью при ступенчатом гидролизе дал рибофлавин-олигопептид,
содержащий цистеин, серин, тирозин и 2 мол. глицина, а затем 8а-(5-ци-
стеинил) рибофлавин [354, 355]. Это соединение было синтезировано при
взаимодействии 8а-бром-2',3',4',5'-тетраацегилрибофлавина с гидрохлори-
дом цистеина с последующим деацилированием [356].
Следует отметить, что, помимо ФМН, ФАД и двух производных ФАД,
выделенных из животных тканей, дрожжей и ферментов, из культуральной
жидкости Rhizopus oryzae выделена ФМН-О-глюкоза [375].
СИНТЕЗ ФЛАВИНОВЫХ КОФЕРМЕНТОВ
1. Синтез ФМН
Рибофлавин-5'-фосфорный эфир, ФМН (V), получают путем непосред-
ственной этерификации рибофлавина (I) хлорокисью фосфора [29], однако
при этом получается трудно разделимая смесь различных продуктов, в
том числе состоящая из рибофлавин-5'-фосфорного эфира (V) и цикличес-
кого рибофлавпн-4',5'-фосфата (CXXXIX) [30].
Более четкие результаты получаются, если предварительно защитить
вторичные гидроксильные группы рибофлавина, а затем провести этерифи-
кацию хлорокисью фосфора по первичному гидроксилу [368] (см. выше).
Для фосфорилирования рибофлавина предложена также пирофосфор-
ная кислота [31] и смесь фенола, хлороформа и фосфорного ангидрида
[32].
При фосфорилировании рибофлавина метафосфорной кислотой обра-
зуется смесь высших эфиров; в результате их гидролиза получают рибо-
флавинполифосфаты, которые могут быть отделены от рибофлавин-5'-
фосфорного эфира на основе их лучшей растворимости в воде [33, 376, 377].
Для непосредственного фосфорилирования рибофлавина лучшие ре-
зультаты дает применение гидратированной хлорокиси фосфора (хлоран-
553
гидридов ортофосфор ной кислоты) различного состава 134—36]. При фос-
форилировании рибофлавина диметилхлорфосфорной кислотой первоначаль-
но получается промежуточный рибофлавин-5'-диметилфосфорный эфир
(CXL), который гидролизуется с отщеплением двух молекул метилового
спирта при действии разбавленной соляной кислоты [37, 378] в рибофла-
вин-5'-фосфорный эфир (V)
сн/снон),ск,о-Р^
\ он
Офю^снр-Р^
Исследована реакция этерификации рибофлавина различными фосфо-
рилирующими агентами, при этом оказалось, что реакция во всех случаях
проходит неоднозначно, и по мере ее протекания наблюдается постепенное
наращивание фосфатных групп, что приводит к образованию, помимо мо-
нофосфата, ди- и полифосфорных эфиров рибофлавина [37].
Рибофлавин-5'-фосфорный эфир синтезирован из фосфорных эфиров
3,4-диметилфенил-Ы-Б-рибитиламина и 3,4-диметилфенил-6-фенилазо-Ы-
D-рибитил амина [379].
Для очистки монофосфорного эфира применяют превращение эфиров
в соли морфолина или диэтаноламина, так как эти соли рибофлавин-5'-
фосфорного эфира легко отделяются от других фосфорных эфиров путем
их фракционной кристаллизации [34, 380]. Полная очистка растворов
ФМН от рибофлавина достигается на катионите КУ-2 (Н+); выход состав-
ляет 85% [381].
Карбодиимидным методом путем взаимодействия ФМН с 85%-ной орто-
фосфорной кислотой в присутствии N.N'-дициклогексилкарбодиимида был
получен рибофлавин-5'-дифосфат [382].
2-Тио-РМФ (CXL1) синтезирован из монофосфата 3,4-диметилфенил-
6-фенилазо N-D-рибитиламина- и 2-тиобарбатуровой кислоты [383] или зна-
чительно эффективнее — непосредственным фосфорилированием 2-тиори-
бофлавина гидратированной хлорокисью фосфора [384].
CH^CHOHjjCHjOH
XXXIV
сн/сно^снр-р^
оМХЛ
О
CXU
2. Синтез ФАД
В синтезе флавинадениндпнуклеотида (ФАД), являющегося несиммет-
ричным Р1,Р2-диэфиром пирофосфорной кислоты, пирофосфатная связь
создается двумя основными путями: 1) конденсацией двух различных
нуклеозидмонофосфатов — аденозин-5'-фосфата и рибофлавин-5'-фосфата;
2) конденсацией нуклеозиддифосфата с нефосфорилированным нуклеози-
дом — аденозин-5'-дифосфата и рибофлавина или рибофлавин-5'-дифос-
фата и аденозина.
Создание пирофосфатной связи конденсацией двух нуклеозидмонофос-
фатов осуществляется четырьмя основными методами: 1) хлорфосфатным
методом, который заключается в конденсации галогенфосфатов или гало-
геннуклеотидов, имеющих защищенные гидроксильные группы, с солями
фосфатов или нуклеотидов с последующим удалением защитных групп;
554
2) карбодиимидным методом, состоящим в непосредственной конденсации
двух монофосфатов нуклеозидов, содержащих незащищенные гидроксилы,
под влиянием карбодиимидов (М.М'-дициклогексилкарбодиимида, ди-л-
толилкарбодиимида и др.); 3) фосфоамидным методом, основанным на кон-
денсации фосфоамидного производного нуклеотида, имеющего незащищенные
гидроксилы, с другим незащищенным мононуклеотидом; 4) анионооб-
менным методом, который состоит в направленном нуклеофильном заме-
щении одного из двух компонентов нуклеотидангидрида третьим компо-
нентом.
Хлорфосфатный метод основан на реакции обмена пирофосфатов или
смешанных ангидридов фосфорных кислот или других кислот с фосфат-
ными анионами.ч При фосфорилировании хлорфосфатным методом исполь-
зуются полностью или частично этерифицированные фосфаты. Полностью
этерифицированные пирофосфаты лабильны и способны легко вступать в
реакцию обмена с другими анионами, вследствие чего они являются более
активными фосфорилирующими агентами, чем частично этерифицирован-
ные фосфаты. Однако благодаря легкости, с которой они вступают во вза-
имодействие с другими анионами, образуются очень сложные и трудно
разделимые смеси.
Первый успешный синтез ФАД (CXXXVI) осуществили Кристи, Кен-
нер и Тодд в 1954 г. [372] хлорфосфатным методом конденсации серебряной
соли рибофлавин-5'-фосфата (CXLII) и 2'3'-О-изопропилиденаденозин-5'
бензил хлорфосфата (CXLIII) [385, 386] в присутствии триэтиламина. В ка-
честве растворителя был выбран фенол, так как ФМН хорошо в нем раст-
ворим; в этой среде довольно легко идет образование пирофосфатной связи,
фенолы способны монодебензилировать образующиеся бензилпирофосфаты.
Изопропилиденовая защитная группировка в последующем удалялась
кислотной обработкой. Однако выход ФАД составлял всего 6%, что было
частично связано с расщеплением образовавшейся пирофосфатной связи
при удалении защитных групп -
он он
Карбодиимидным методом ФАД (CXXXVI) был получен из ФМН (V)
и АМФ (CXLIV) в водном пиридине в присутствии ди-п-толилкарбодиими-
да [387]. Реакцию проводят в темноте в течение 24 ч, после чего образовав-
шуюся ди-п-толилмочевину отфильтровывают и кофермент выделяют из
реакционной смеси хроматографией на бумаге с последующей очисткой
через ураниловую соль [388]. Выход составил 4%, содержание 90%.
555
ои |
ho-p-ohsc 'o’
II
О
OH OH
*
CXXXV!
CXLIV
Низкий выход ФАД, синтезируемого карбодиимидным методом, свя-
зан с тем, что основная реакция направляется в сторону образования цик-
лического рибофлавин-4',5'-фосфата (CXXXIX), что находится в соответ-
ствии с известными данными о направлении реакции с N,N'-дициклогек-
силкарбодиимидом, часто приводящей к циклофосфатным нуклеозидам
[389, 390]. Так, при взаимодействии ФМН с АМФ (CXLIV) в присутствии
N.N'-дициклогексилкарбодиимида единственным продуктом реакции ока-
зался циклический рибофлавин-4',5'-фосфат [30, 391].
Выход ФАД можно несколько увеличить (до 11,6%), если уменьшить
избыток ди-п-толилкарбодиимида и проводить реакцию при 50—55° С
[392]. Таким путем был получен 2-дезоксифлавинадениндинуклеотид [393].
Карбодиимидным методом синтезирован Р1-(2',3',4'-триацетилрибо-
флавин-5')-Р2-тиаминпирофосфат [394].
Используемый в синтезе ФАД аденозин-5'-фосфоамид (CXLV) получают
из АМФ (CXLIV) и аммиака в присутствии избытка N.N'-дициклогексил-
карбодиимида в трет-бутаноле и формамиде при 80° С [395, 396] и выделяют
в виде кристаллической дициклогексилгуанидиниевой соли с выходом 85—
92%. Синтез ФАД (CXXXVI) осуществляют из этой соли (CXLV) и избыт-
ка пиридиниевой соли ФМН (V) при комнатной температуре в течение 96 ч
[397].
он он он он
til 1
СН£-СН-СН—CH-CHjO-P-OH
I о
— ФАД
CXXXVI
Для разделения продуктов реакции успешно использована диэтилами-
ноэтил целлюлоза (в хлоридной форме) [397], на которой ФАД был выде-
лен в виде литиевой соли с выходом 40% и содержанием 90%.
Фосфоамидным методом, конденсацией пиридиниевой соли ФМН с фос-
фоморфолидными производными соответствующих нуклеозидов синтезиро-
ваны аналоги ФАД, которые вместо аденина содержат: тимин, цитозин,
ксантин, гуанин, никотинамид и их дезокси производные [398, 399]. Синте-
зированы также 2',3',4'-три-0-ацил-[400], 7,8-дихлор-2'-дезокси-[401],
М(3)-метил-ФАД [401, 402] и производные, имеющие вместо рибитильной цепи
радикалы: D-эритро-, 4'-оксибутил-, 5'-оксипентил-[401].
Так как триалкиламмониевые соли способствуют увеличению раство-
римости фосфорных эфиров в безводных растворителях, то для синтеза ФАД
можно использовать алкиламмониевые соли мононуклеотидов. В качестве
нуклеозидфосфоамида преимущество имеет применение нуклеозидфосфо-
морфолидов. Аденозин-5'-фосфоморфолид получают в виде кристаллической
4-морфолин-М,М'-дициклогексилкарбоксиамидиниевой соли в результате
медленного приливания раствора М,М'-дициклогексилкарбодиимида в
/пре/п-бутаноле в кипящий раствор АМФ (CXLIV) и морфолина fe водном
556
трет-бутаноле. Из 4-морфолин-Н,Н'-дициклогексилкарбоксиамидиниевой
соли АМФ (CXLVII) и триэтил-, три-н-октил- или цетилтриметиламмоние-
вой соли РМФ (CXLVI) в растворе диметилформамида или его смеси с пи-
ридином при 40—50° С в течение нескольких часов ФАД (CXXXVI) полу-
чают с выходом 40—50% [399—403].
сн3
сн.
CXLVI
он он он о
Т । Т и
СНгСН-СН-СН-СН/Э-Р-ОН
ст
«{•(СН,),
Скн33
С фосфоморфолидами соответствующих нуклеозидов были получены
различные аналоги ФАД — флавинуридиндинуклеотид, флавингуанозин-
динуклеотид [404], флавин-8-бромадениндинуклеотид [405] и др.
Для синтеза ФАД были использованы также пиперидатные производ-
ные АМФ [406]. Кроме того, был получен аденозин-5'-фосфоимидазолид
(АМФ-И) [407]. При конденсации АМФ-И с РМФ (V) образуется ФАД с
выходом 67% [398]. Получен также флавинбензимидазолдинуклеотид [408];
он не связывается с активным центром оксидазы D-аминокислот.
Фосфоамидный метод образования пирофосфатной связи эффективнее
некоторых других методов, так как конденсация двух нуклеотидных мо-
лекул проводится без предварительной защиты гидроксильных групп.
Ценность метода заключается в возможности использования водной среды,
что важно в тех случаях, когда присутствие уже небольшого количества
воды увеличивает растворимость реагирующих компонентов.
ФАД (CXXXVI) синтезирован анионообменным методом с выходом 70%
в результате взаимодействия Р1-дифенил-Р2-(аденозин-5')пирофосфата с ани-
оном — этерифицированным рибофлавин-5'-фосфатом [409].
Для синтеза ФАД из ФМН (в виде натриевой соли) и АМФ в качестве
конденсирующего средства был применен трифторуксусный ангидрид [410].
Реакция протекает в результате атаки трифторуксусного ангидрида фос-
фатным анионом с образованием промежуточного ангидрида, который,
взаимодействуя со вторым фосфатным анионом, образует Р‘,Р2-(динуклео-
зид-5')пирофосфат. Этим методом можно получать значительные количе-
ства ФАД, однако выход вещества невысокий (~10%). Из ФМН и соот-
ветствующих нуклеозид-5'-фосфатов с применением трифторуксусного ан-
гидрида были получены аналоги ФАД, содержащие вместо аденина урацил,
цитозин, гуанин [410] и никотинамид [410, 411].
Применение смеси хлорокиси фосфора и фосфорного ангидрида для
конденсации двух соответствующих незащищенных мононуклеотидов
дает возможность получить ФАД [412]. Реакцию проводят в феноле, в
котором хорошо растворяются реагирующие вещества [413]. В этом син-
тезе, помимо ФМН и АМФ„ применяют рибофлавин-5'-дифосфат и адено-
зин, рибофлавин и АДФ, а также другие исходные вещества.
ФАД получают также из ^иридиниевых солей ФМН и АМФ в о-хлор-
феноле в присутствии трихлорацетонитрила [414].
Особо следует отметить синтез аналогов ФАД с моно-, три- и тетра-фос-
фатными группами, связывающими флавиновые и пуриновые частицы моле-
кулы — Р1-(рибофлавин-5')-Р,-(аденозин-5')монофосфат, Р1-(рибофлавин-5')-
Р3-(аденозин-5')трифосфата и Р1 -(рибофлавин-5')-Р4-(аденозин-5')тетрафос-
фата (CXLVIII) [415, 416]
557
fl”1
П —3
П—4
а также Р1-(рибофлавин-5')-Р2-(адснозин-2'(3')фосфо-5')пирофосфат
(ФАДФ) — аналог ФАД с дополнительной фосфатной группой в аденози-
новой части молекулы 1416]. Некоторые из этих соединений при испытании
на оксидазе О-аминокислот обнаруживают частичную коферментную актив-
ность, характерную для ФАД.
Выделение коферментов как из природных источников, так и из син-
тетических продуктов представляет собой сложную и трудоемкую работу.
у; Выделение и очистка ФАД может быть достигнута на ионообменной смоле
Н IRC-50.
С лучшими результатами была использована распределительная хрома-
тография в системе фенол—бутанол—вода [417], хроматография на бу-
: . маге [387], колоночный электрофорез [418], разделение на полисахарид-
:?,ном геле сефадексе [403] и хроматография на порошке целлюлозы [419].
Для идентификации флавиновых соединений использовалась хроматогра-
Дфия на бумаге в различных системах растворителей [420].
3. Ферментативный синтез ФАД
A- С большим успехом ФАД получают микробиологическим методом. Так,
для биосинтеза ФАД широкое распространение находит культура
-Eremothecium ashbyii [421, 422]; в культуральной жидкости содержа-
Д ние ФАД составляет 60—120 мкг/л и в сухом мицелии 20—40 мг/г [45].
Г Использование для образования ФАД бактерий Sarcina lutea имеет техни-
ческое значение. Для выделения и концентрирования ФАД из продуктов
биосинтеза используют ионообменные смолы, сефадекс или применяют
метод его восстановления гидросульфитом натрия в труднорастворимые
семихиноидные формы [423].
Биосинтез рибофлавин-5'-фосфата в организме осуществляется из ри-
бофлавина и АТФ при каталитическом участии рибофлавинкиназы (фла-
вокиназы):
Рибофлавин + АТФ —► ФМН + АДФ
Рибофлавинфосфотрансфераза катализирует реакцию фосфорилиро-
вания рибофлавина фосфатной группой О-глюкозо-1-фосфата:
ЬТлкжозо-1 -фосфат + Рибофлавин D-Глюкоза -f- ФМН.
Биосинтез ФАД осуществляется ФМН-аденинтрансферазой из ФМН и
АТФ с выделением неорганического пирофосфата
ФМН 4- АТФ -> ФАД + Н4Р2О7.
Биосинтез ФАД происходит главным образом в цитоплазме, около 60%
клеточного ФАД содержится в митохондриях [410].
БИОХИМИЧЕСКИЕ ФУНКЦИИ ФЛАВИНОВЫХ КОФЕРМЕНТОВ
Флавиновые коферменты в составе флавопротеидов (ферменты класса
оксидоредуктаз) катализируют окислительно-восстановительные реак-
ции различных соединений в процессах обмена веществ животного орга-
низма с образованием химических связей: С—С, С=О, N—Н, S—Н и
расщеплением связей: С—N, С—О, N—О, S—S.
Флавопротеиды принимают участие в окислении спиртов в карбоновые
кислоты, альдегидов и кетонов также в кислоты и двуокись углерода,
насыщенных углерод — углеродных связей в двойные, аминосоединений
в иминосоединения, N-замещенных дигидропиридинов в четвертичные пи-
ридиниевые производные и др.
558
Известно несколько типов реакций переноса водорода от субстрата (до-
нора) на акцептор, осуществляемых флавиновыми коферментами флавопро-
теидных систем:
1. Первичное дегидрирование субстрата при участии НАД(Ф) с обра-
зованием восстановленных форм НАД(Ф), отщепление от них водорода
(протона и электронов) ФМН и ФАД, передача от восстановленных флави-
нов протона через среду, а электронов по системе цитохромов цепи дыха-
ния на молекулярный кислород.
2. Дегидрирование субстрата непосредственно флавиновыми кофер-
ментами без участия НАД(Ф) с передачей водорода (протона через среду,
электронов по системе цитохромов цепи дыхания) на молекулярный кис-
лород.
3. Окисление субстрата, катализируемое ФМН и ФАД без участия
НАД(Ф), с передачей водорода непосредственно на молекулярный кислород
(минуя систему цитохромов).
4. Окисление восстановленного НАД при участии ФАД с передачей
водорода (электрона и протона) непосредственно на перекись водорода.
Участие флавопротеидовв тканевом дыхании
и окислительном фосфорилировании (первый
тип реакции переноса водорода)
Завершающим этапом биологического окисления является тканевое
дыхание, в результате которого происходит перенос водорода (протонов
и электронов) от субстрата (НАД-Н или сукцината) на молекулярный кис-
’йлород. Этот процесс осуществляется при каталитическом участии системы
« коферментов, входящих в электроно-транспортную дыхательную цепь ми-
тохондрий животных тканей, последовательно осуществляющих реакции
окислительно-восстановительных превращений.
Никотинамидные ферменты дегидрируют субстрат, причем НАД+ пе-
реходит в восстановленную форму — НАД-Н — и одновременно в буфер-
ную среду митохондрий переходит протон. Протоны и электроны акцепти-
руются ФАД и передаются на убихинон (кофермент Q) [424] и далее на
систему цитохромов. Эту реакцию осуществляет флавопротеид — дегидро-
геназа восстановленного НАД (цитохром-с-редуктаза), выделенная из сер-
дечной мышцы 1425], печени [426]. В состав фермента, помимо ФАД, вхо-
дит четыре атома негеминового железа на моль флавина; молекулярная
масса 78 000. Из системы ферментов цепи дыхания выделен флавопротеид
с простетической группой ФМН и двумя атомами железа на моль флавина
1427].
Дегидрогеназа восстановленного НАДФ из дрожжей [366, 428, 429]
содержит ФМН. Она имеет молекулярную массу около 75 000 [366] и
катализирует окисление в течение минуты при 38° С 8500 молекул восста-
новленной формы никотинамидадениндинуклеотидфосфата (НАДФ-Н) в
никотинамидадениндинуклеотидфосфат (НАДФ+, НАДФ).
Система цитохромов представляет собой ферментативную цепь пере-
носа электронов на молекулярный кислород. Она состоит из цитохромов
с1г с и а. Система переноса электронов бактерий имеет другой набор цито-
хромов: alt а2, blf с2, с3, ск.
Цитохромы — гемопротеиды, в качестве простетической группы со-
держащие порфириновые системы. Цитохромы группы с имеют гем железо-
порфирина
559
Цитохром ct (из митохондрий) — гемопротеид с молекулярной массой 40 000—
содержит четыре геминовые группы на моль фермента. Цитохром с раство-
рим в воде, имеет молекулярную массу 13 000. Цитохром а входит в состав
цитохромоксидазы, катализирующей завершающую фазу реакции пере-
носа электронов от восстановленного цитохрома с (окисляя его железо в
Fe+++) на молекулярный кислород, используя при этом протон воды и
образуя воду. Цитохром а представляет собой железоцитопорфирин. Ци-
тохромоксидаза содержит два атома меди на моль фермента; молекулярная
масса 70 000.
При окислении янтарной кислоты сукцинатдегидрогеназой восстанов-
ленный флавиновый кофермент передает электроны первоначально на ци-
тохром Ь, а затем на цитохром сг и другие звенья цепи дыхания. Цитохром
Ь содержит две винильные группы и является железопротопорфирином IX.
Возможность протекания реакции по цепи дыхания при участии фер-
ментов находится в соответствии с их способностью последовательного
восстановления и обратимого окисления и последовательным изменением
величин их окислительно-восстановительных потенциалов (Ёо)-
Название системы В
Водородный электрод (Н2 Z+ 2Н+ + 2е) —0,42
НАД. НАДФ —0,28
Рибофлавин —0,21
ФМН. ФАД —0,18
Флавопротеид (старый желтый фермент) —0,12
Цитохром b (переносящий электрон при окислении ян- —0,05
тарной кислоты)
Убихинон 4-0.12
Цитохром с +0,26
Цитохром а +0,29
оа +0.82
Перенос электронов по цепи дыхания от НАД-Н на1 убихинон с регене-
рацией НАД осуществляет флавопротеид дегидрогеназа восстановленного
560
НАД — цитохром-с-редуктаза. Дегидрирование восстановленного НАДФ
осуществляется другим, специфичным к НАДФ-Н ферментом.
Биологическое окисление и транспорт электронов по цепи дыхания
тесно связаны с окислительным фосфорилированием, являющимся главным
источником накопления свободной энергии в клетках в легко используемой
форме — в виде богатых энергией фосфорных соединений, главным образом
в АТФ. В окислительном цикле трикарбоновых кислот на каждую молеку-
лу уксусной кислоты, окисленной до двуокиси углерода, образуется 8 про-
тонов и 8 электронов, которые транспортируются по цепи дыхания и вос-
станавливают молекулярный кислород в воду. Отщепление атомов водорода
происходит на следующих этапах цикла трикарбоновых кислот:
^ис-аконитовая кислота —> изо-лимонная кислота;
а-кетоглутаровая кислота —> сукцинил-КоА;
L-яблочная кислота —> щавелевоуксусная кислота;
янтарная кислота —> фумаровая кислота.
В первых трех реакциях акцептором протона и электрона является
НАД(Ф), передающий их ФАД, в четвертой реакции — 8а-гистидил-ФАД.
Одновременно с окислением уксусной кислоты и переносом водорода вдоль
цепи дыхания происходит фосфорилирование АДФ в АТФ [430], причем на
один атом восстанавливаемого кислорода образуется 3 моля АТФ, вероят-
но, на следующих этапах:
НАД -> ФАД;
цитохром b (cj) —> цитохром с;
цитохром а —> молекулярный кислород.
Таким образом, при расщеплении 1 моля уксусной кислоты [высвобо-
ждаемая энергия аккумулируется в 12 молях макроэргического АТФ.
Для одного из этапов (НАД -> ФАД) области цепи дыхания предло-
жено следующее гипотетическое объяснение сопряженной реакции окисли-
тельного фосфорилирования с образованием АТФ [431, 432].
R—остаток остальной части молекулы ' кофермента;
Ad-остаток аденозина
561
Участие фл а в опротеидо в в непосредственном
окислении жирных кислот (второй тип
реакции переноса водорода — без участияНАД)
Окислительное (3-расщепление жирных кислот начинается с активации
жирной кислоты в результате ее реакции с аденозин-5'-трифосфатом (АТФ)
и коферментом А (см. с. 91). При этом образуется ацил-КоА с макроэргичес-
кой связью и одновременно АТФ превращается в аденозин-5'-фосфат и
неорганический пирофосфат. Следующая стадия расщепления жирных
кислот — дегидрирование активированной формы жирной кислоты до
а, ^-ненасыщенной кислоты в связанной с коферментом А форме
ФАД
СН3 (СИ2)ПСН2СН2СО - SKoA------► CHS (СН2)„СН=СНСО ~ SKoA.
-фад-н2
катализируют металлофлавопротеиды — ацил-КоА-дегидрогеназа и бути-
рил-КоА-дегидрогеназа (на окончательной фазе (3-расщепления), содержащие
ФАД в качестве простатической группы, но отличающиеся по природе
металла. Ацил-КоА-дегидрогеназа содержит железо, не связанное с гемом
1433]; бутирил-КоА-дегидрогеназа содержит медь (2 атома на моль флавина),
катализирует дегидрирование ацил-КоА-производных с короткой (С4—
С8) углеродной цепью (55, 433]. Из бутирил-КоА при дегидрировании об-
разуется кротонил-КоА
ФАД
СН,СН2СН2СО ~ SKoA---------► СН,СН=СНСО ~ SKoA.
—ФАД-Н2
Участие фл а в опротеидо в в окислительном
цикле трикарбоновых кислот (второй тип
реакции переноса водорода —без участия НАД)
Превращения в цикле трикарбоновых кислот (см. с. 320) — главный
источник энергии в организме. Флавопротеидные коферменты катализи-
руют в цикле трикарбоновых кислот две реакции: непосредственное дегид-
рирование янтарной кислоты в фумаровую кислоту без участия в этой
реакции никотинамидных протеидов и окислительное декарбоксилирова-
ние а-кетоглутаровой кислоты с образованием сукцинил-КоА (см. с. 91). По-
мимо этого, они переносят водород от НАД-Н и НАДФ-Н, принимающих
участие в цикле трикарбоновых кислот, на другие ферменты тканевого
дыхания.
Дегидрирование янтарной кислоты в фумаровую кислоту катализирует
сукцинатдегидрогеназа, простатической группой ее служит 8а-гистидил-
ФАД (СХ XXVII), который пептидной связью соединен с апоферментом
[352, 353, 373].
НООССН2СН2СООН + 8а-Гистидил-ФАД —>
-> НООССН=СНСООН 4- 8а-Гистнднл-ФАД-Н2.
Далее восстановленный флавиновый кофермент сукцинатдегидрогеназы
передает водород (протон и электрон) на систему цитохромов по ферментной
цепи дыхания
8а-Гистидил-ФАД-Н2 + 2 Цитохром с (Fe+++) —►
-> 8я-Гистидил-ФАД + 2 Цитохром с (Fe++) -f-2H+.
В состав фермента сукцинатдегидрогеназы входит негеминовое железо
(4 атома на моль флавина), по-видимому, связанное с SH-группами апофер-
мента.
Окислительное декарбоксилирование а-кетоглутаровой кислоты в сук-
цинил-КоА в окислительном цикле трикарбоновых кислот осуществляет
562
комплекс ферментов с простатическими группами: липоевой кислоты, КоА,
НАД, ТДФ, ФАД. Флавопротеид липоамиддегидрогеназа [4341 ката-
лизирует окисление восстановленной липоевой кислоты (ее амида), обра-
зующейся в этой сложной реакции наряду с сукцинил-КоА, и перенос
водорода на НАД
СН2СН,СН (CH2)«CONH2 + ФАД уГ СН2СН2СН (CH,)4CONHa + ФАД-Н»
>1 <! J
SH SH S S
ФАД-Н2+НАД+ ФАД + НАД-Н+Н+.
Липоамиддегидрогеназа найдена во всех животных тканях, растениях
и бактериях [4351. Дегидрирование флавиновыми ферментами может
происходить и в присутствии кислорода воздуха.
Участие флавопротеидов в аэробных
реакциях (третий тип реакции переноса
водорода)
•Многие флавопротеидные ферменты — оксидазы — обладают способ-
ностью к самоокислению, т. е. к непосредственной передаче водорода на
молекулярный кислород. Подобных флавопротеидных оксидаз известно 14.
Окисление гликолевой кислоты в глиокси-
ловую кислоту. Реакцию катализирует гликолатоксидаза из
табачных листьев, содержащая ФМН [436]. Гликолатоксидаза входит в
состав ферментных систем, переносящих водород в растениях:
ФМН
НООССН2ОН ф О2-> НОООСНО 4-Н2О2
Окислительное декарбоксилирование молоч-
ной кислоты в уксусную кислоту. Катализатором
реакции служит лактатоксидаза, содержащая ФМН [4371. Фермент выпол-
няет двойную функцию — катализирует окисление и декарбоксилирова-
ние Л-молочной кислоты с образованием уксусной кислоты и двуокиси
углерода. Возможно, промежуточным продуктом окисления является пи-
ровиноградная кислота
1 ФМН
CHjCH (ОН)СООН + 1 у Os-♦ [СН3СОСООН] -> СН3СООН + со2 + Н2О2.
Окисление p-D-глюкозы в D - г л ю к о н о - б-л а к-
т о н. Эту реакцию катализирует глюкозооксидаза (нотатин, пенициллин
В), содержащая ФАД (2 моля на моль фермента) [438]. Молекулярная масса
фермента 154 000; он содержится в плесневых грибах Penicillium notatum
и др. [439, 440]. Механизм ферментного окисления изучен с помощью Н^Э,
меченной 18О [441], при этом оказалось, что кислород, выделяющийся при
разложении перекиси водорода под действием каталазы, не содержит 18О,
т. е. глюкозооксидаза катализирует перенос водорода от глюкозы в газо-
вой фазе, акцептором водорода служит газообразный кислород. По ради-
кальному механизму в первой ступени реакции |3-£)-глюкопираноза (а)
теряет протон и электрон с образованием свободного радикала (б), который,
теряя второй электрон, образует оксониевый ион (в). Прототропная реак-
ция между оксониевым ионом и перекисным анионом завершает окисление
с образованием £)-глюконо-б -лактона (г).
563
Механизм бактериостатического действия нотатина (глюкозооксидазы),
вероятно, заключается в образовании Н2О2 при окислении глюкозы.
Окисление альдегидов в карбоновые кис-
лоты. Реакция протекает при участии альдегидоксидазы; фермент из
печени свиньи содержит ФАД, молибден, железопротопорфирин [4421:
ФАД
RCHO + О, + Н2О----♦ RCOOH 4- Н2Оа.
Окислительные превращения пуринов. Реакцию
окисления пуринов (а также птеринов и многих альдегидов) катализирует
ксантиноксидаза, содержащая ФАД — две молекулы — и молибден — два
атома на моль фермента. Этим ферментом, например, ксантин окисляется
в мочевую кислоту [443] — конечный продукт пуринового обмена
Ог; НдОСФАД)
-нА
Акцепторами водорода, помимо кислорода, может быть метиленовый
голубой, хиноны, цитохром с. Ксантиноксидаза (из молока) имеет молеку-
лярную массу около 290 000.
Окислительное декарбоксилирование пиро-
в иноградной кислоты. Реакцию катализирует пируваток-
сидаза с простатической группой ФАД и Мп в присутствии тиаминдифос-
фата и ортофосфорной кислоты. В первой стадии процесса под действием
фермента образуются ацетилфосфат и перекись водорода, которая затем
неферментно окисляет пировиноградную кислоту в уксусную
ФАД, ТДФ
CHjCOCOOH 4- О2 + Н3Р04--------» СН3СООРО3Н2 4- СО2 4- Н2О2,
СН3СОСООН 4- Н2О2 -> СН3СООН 4- СО2 + Н,О.
Ацетилфосфат под влиянием фермента фосфотрансацетилазы, содержащей
кофермент А, превращается в ацетилкофермент А и неорганический фос-
фат
СН3СООРО3Н2 4- HSKoA -> CH3COSKoA 4- H3P04.
Окислительное расщепление щавелевой кис-
лоты. Реакция протекает при участии оксалатоксидазы, содержащей ри-
бофлавин или 'ФМН:
НООССООН 4- о2 -* 2СО2 4- Н2О2
Окисление 4,5- дигидрооротовой кислоты в о ро-
товую кислоту. Реакция окисления протекает под влиянием ди-
гидрооротатдегвдрогеназы, содержащей ФМН, ФАД и негеминовое желе-
зо [444]:
ФМН, ФАД
564
о
II
-»HN |
I | +HA
о H COOH
При восстановлении дигидрооротатдегидрогеназы НАД-Н наблюдается
образование семихиноидного радикала [445].
Оротовая кислота является предшественником пиримидиновых осно-
ваний в процессе биосинтеза нуклеиновых кислот.
Окислительное деметилирование N-метил-
L-аминокислот в L-аминокислоты. Специфическая ок-
сидаза N-метиламинокислот реагирует при наличии свободной a-карбок-
сильной группы субстрата. Этот флавопротеид, содержащий ФАД, не дей-
ствует на N-диметиламинокислоты. Реакция сопровождается выделением
перекиси водорода и формальдегида
RCHCOOH О, (ФАД) RCHCOOH н,о RCHCOOH RCHCOOH
।-----------> | -----> | .------> I
HNCHS --н2о, N=CHt NHCH2OH - hcho NH2
Окислительное дезаминирование а-амино-
кислот в a - к е т о к и с л о т ы и моноаминов в аль-
дегиды. Окислительное дезаминирование a-аминокислот катализируют
флавопротеиды—оксидазы L-аминокислот, катализирующие окисление при-
родных аминокислот L-ряда, и оксидазы D-аминокислот, действующие на
неприродные аминокислоты D-ряда. Ферменты высокоспецифичны по от-
ношению к стереохимической конфигурации аминокислот и малоспеци-
фичны по отношению к боковой цепи, а-Аминокислота при каталитичес-
ком действии флавопротеида дегидрогенизируется до иминокислоты, об-'../
разующийся дигидро-ФАД передает водород на молекулярный кислород, V
а иминокислота неферментно превращается в а-кетокислоту [369] 4$
RCHCOOH 4- ФАД-----> RCCOOH + ФАД-Н^
I II
NH2 NH
ФАД-Н2 + О,----* ФАД + Н2О2
RCCCOH + Н2О------ RCOCOOH + NH3
Оксидаза D-аминокислот с простетической группой ФАД [369] окисляет
в соответствующие иминокислоты DL-лейцин, D- и DL-метионин, D- и
DL-аланин, DL-a-аминомасляную кислоту. Оксидаза L-аминокислот, со-
держащая ФАД, дезаминирует L-аргининв a-кето-б-гуанидовалериановую
кислоту, L-орнитин в a-кето-б-аминовалериановую кислоту, L-лизин в
1-кето-е-аминокапроновую кислоту.
ФАД-содержащие ферменты катализируют дезаминирование свыше
13 различных аминокислот, среди них, кроме упомянутых ранее, L-фе-
нилаланин, L-цитруллин, DL-валин и др.
Выделена оксидаза L-аминокислот, в состав которой входит ФМН [446].
Специфическая D-аспартатоксидаза катализирует окислительное деза-
минирование D-аспарагиновой кислоты в щавелевоуксусную кислоту.
Природа флавиновой простетической группы фермента не установлена.
Дезаминирование моноаминов катализирует купропротеидмоноамино-
оксидаза, содержащая, как недавно установлено, в качестве простетической
группы 8а-Цистеинил-ФАД [354—356]. Так, образующийся в результате
565
декарбоксилирования тирозина тирамин дезаминируется в соответствую-
щий альдегид.
СУ, к исленйе пиридоксин -5а- фосфата и пири-
докса мин -5а- фосфата в пиридоксаль -5а- фо с-
ф а т. Реакцию катализирует пиридоксаминфосфатоксидаза с простетичес-
кой группой ФМН [447]. При окислении пиридоксамин-5а-фосфата в
пиридоксаль-5а-фосфат выделяется аммиак и перекись водорода
CH2NH2
СНО
Этот же фермент подвергает окислению и пиридоксин в пиридоксаль.
Флавопротеиды катализируют процессы фиксации атмосферного азота
микроорганизмами (азотобактер и др.) первоначально в аммиак или обра-
тимые реакции восстановления нитратов через нитриты в аммиак с после-
дующим его использованием для обменных реакций
Восстановление нитратов и нитритов катализируют три нитратредук-
тазы, содержащие ФАД и молибден, и нитритредуктаза с ФАД и марганцем.
Донором водорода являются восстановленные формы НАД или НАДФ.
НАД-Н + Н+ + ФАД ФАД-Н, + НАД*,
ФАД-Н2 4-’HNO3 ФАД + HNO, + HSO.
ЗФАД-Н2 + HNO2 ЗФАД + NH*OH 4- HSO.
Реакции окисления азота в окисьГазота и окиси азота в нитрит катализи-
руют флавопротеиды — редуктаза окиси а§ота, содержащая ФАД, медь
и железо, и нитритредуктаза соответственно.
Участие флавопротеидов в восстановлении
перекиси водорода (четвертый тип реакции
переноса водорода)
Перекись водорода, возникающая в аэробных реакциях окисления
субстрата, используется в некотором количестве для катализируемого НАД-
пероксидазой, содержащей ФАД, окисления восстановленной формы НАД
в окисленный, НАД
НАД-Н +-Н+ + ФАД -> НАД* + ФАД-Н,,
ФАД-Н2 + Н;О2 -> ФАД + 2Н2О
Б66
В этой реакции перекись водорода восстанавливается в воду.
Перекись водорода, образующаяся в аэробных реакциях, если она не
используется для других обменных процессов, полностью выводится из
окислительно-восстановительного цикла разложением на молекулярный
кислород и воду при участии гемопротеидного фермента каталазы
2Н£О, -> 2Н2О -+ О2.
Связь кофермента с апопротеином в флавопротеидных ферментах дос-
таточно прочная, недиссоциирующая. Флавопротеиды расщепляются с
различной степенью трудности с отделением апофермента и образованием
рибофлавина или его коферментов. Однако простетическая группа в D-
аспартатоксидазе отделяется уже в процессе очистки. В оксидазе /.-ами-
нокислот простетическая группа более прочно связана с апоферментом и
диализ, а также повторное осаждение недостаточны для разрыва этой связи.
Оксидаза £)-аминокислот и дегидрогеназа восстановленного НАД (цито-
хром-с-редуктаза) диализируются на холоду разбавленной минеральной
кислотой с выделением протеина.
Возможно [448], что в связывании кофермента с белком принимает
участие фосфатная группа как дианион и первичные аминогруппы протеи-
на — а- и е-аминогруппы лизина, а с другой стороны — имино- или окси-
группы изоаллоксазина и тирозиновый остаток протеина
сн,
сн,'
ОН ОН ОН О
CHj-CH—СН—СН—СН,О—Р-О"
N 'О О"
хл_______________
N С
со...
HjNCHCHjCHjCHjCHjOi,
+ или
HjNC^CHaCHjCHjCHCO.^
-О СНаСН^Ь...
Н 4=7 NH,
О--
—но
снхнсо...
NH,
Связь флавина с протеином не во всех ферментах осуществляется по
иминогруппе положения 3. В связывании флавина с белком в оксидазе D-
аминокислот и в липоамиддегидрогеназе [449] участвует рибитильная и аде-
ниловая части ФАД; иминогруппа, по-видимому, остается свободной, так
как эти ферменты обладают способностью к флуоресценции.
В сукцинатдегидрогеназе между простетической группой и белком
имеется пептидная связь (3731; то же относится к моноаминооксидазе,
саркозинокспдазе и 6-оксиникотиноксидазе.
В процессе выполнения биокаталитической функции кофермент в фла-
вопротеидном комплексе не может отщепляться и переходить на другой
апофермент.
Особую группу флавиновых ферментов составляют металлофлавопро-
теиды, содержащие наряду с флавиновой частью атомы металлов — Fe,
Со, Ni, Мп, Zn, Си [54, 450].
Применение флавиновых коферментов
В медицине ФМН применяется в виде инъекций [451] при кожных за-
болеваниях (различных дерматозах, нейродерматитах), при заболеваниях
глаз (поражениях роговицы и др.). ФАД, флавитан, применяется для ле-
чения кожных заболеваний — себоррейного дерматита, акнэ («вульгарной»,
«розовой»), лицевой рассеянной фолликулярной волчанки, при заболева-
ниях глаз, при тяжелых формах эндогенного арибофлавиноза.
667
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ (К ГЛАВЕ XII)
I. Р. Karrer, ’ К. S с h б р р,
F. Benz. Helv. Chim. Acta, 18,
426 (1935).
2. H. Euler, P. Karrer,
M. Mai mberg, K. Schopp,
F. Benz, B. Becker,
P. F r e i. Helv. Chim. Acta, 18,
522 (1935).
3. R. Kuhn, K. Reinemund,
F. We v ga n d, P. S t г о b e-
1 e. Ber., 68, 1756 (1935).
4. Номенклатура IUPAC, J. Biol.
Chem., 241, 2987 (1966).
5. S h. Shimizu. J. Ferment. Tech-\
nol. Japan, 28, 139, 320, 399
(1950); C. A., 47, 1755 (1953).
6. Sh. Shimizu. J. Ferment.
Technol. Japan, 28, 135 (1950);
C. A., 47, 12435 (1953).
7. A. L e a 1, A. A n dr a de,
M. Lourdes-Alves. Rev. Por.
Farm., 2, 126 (1952).
8. D. F г о s t. J. Am. Chem. Soc.,
69, 1064 (1947).
9. P. E 1 I i n g e r, W. К о sc h a-
r a. Ber., 66, 315, 808, 1411 (1933).
10. R. Kuhn, P. Gyorgy,
T. Wagner-Jauregg. Ber.,
66, 317, 576, 1034 (1933).
11. H. В i e г г у, B. Gouzon.
Comp. rend. soc. biol., 119, 101
(1935).
12. J. Koziol. Photochem. Photo-
biol., 5, 41 (1966).
13. L. Booher. J. Am. Med. Ass.,
110, 1105 (1938).
14. P. Karrer, H. Fritzsche.
Helv. Chim. Acta, 18, 911, 1026
(1935).
15. R. К u h n, H. R u d y. Ber.,
68, 169, 300 (1935).
16. D. Adamson. Analyst, 73, 442
(1948).
17. R. К u h n, G. M о r u z z i. Ber.,
67, 888 (1934).
18. P. Karrer, T. К о b n e r,
F. Zehender. Helv. Chim.
Acta, 19, 261 (1936).
19. A. W a r b u r g, W. Chri-
st i a n. Biochem. Z., 254, 438
(1932); 266, 377 (1933).
20. P. К a r r e r, H. S a 1 о m о n,
K. S c h б p p, E. S c h 1 i t t-
ler, H. Fritzsche. Helv.
Chim. Acta, 17, 1010 (1934).
21. R. К u h n, H. R u d у, T. W a-
gner-J auregg. Ber., 66, 1950
(1933).
22. B. Kuhn, T. Wagner-Jau-
regg. Ber., 66, 1577 (1933).
23. R. К u h n, H. Rudy, F. Wey-
g a n d. Ber., 69, 1543 (1936); пат.
США 2111491; C. A., 31, 3768 (1937).
24. R. Kuhn, R. Strobe le. Ber.,
70, 747 (1937).
25. M. T i s h 1 e r, J. W e 1 I m a n.
Пат. США 2370093; С. A., 39, 4098
(1945).
26. К. Y a g i. Яп. пат. 5841 (1963);
С. A., 59,” 6208 (1963); Яп. пат.
26416 (1964); РЖХ, 1967, 17Н389;
Англ. пат. 943078; С. А., 60, 5523
(1964); Бельг, пат. 620984; С А ,
58, 13733 (1963).
27. К. Y a g i, Y. О k u d а,
A. D m i t г о v s k i i. J. Bio-
chem. (Tokyo), 46, 621 (1960); C. A.,
65, 6482 (1961).
28. А. К о t a k u, K. Y a g i. Bi-
tamin, 36, 212 (1967); C. A., 67,
106381 (1967).
29. R. К u h n, H. R u d y. Ber.,
68, 383 (1935).
30. H. F о г r e s t, A. T о d d. J.
Chem. Soc., 1950, 3295.
31. P. В г e i v о g e 1. Пат. США
2535385; С. A., 45, 3428 (1951).
32. A. M о r r i s о n, F. A t h е г-
t о п. Англ. пат. 687980; С. А., -
48, 4598 (1954); пат. США 2677680;./
РЖХ, 1956, 30473; швейц, пат.
297192; РЖХ, 1956, 55934.
33. М. V i s с о п t i п i, С. Е b n б-
t h е г, Р. К а г г е г. Helv.
Chim. Acta, 35, 457 (1952).
34. L. F 1 е к s е г, W. F a r k a s.
Пат. США 2610179; С. A., 47, 8781,
(1953).
35. T. Sato, J.Yoshimura,
T. T a k a о k a. Proc. Japan Acad.,
29, 260 (1953); C. A., 49, 1063 (1955).
36. T. S a t о, H. Yoshimura.
Яп. пат. 3675 (1956); С. A., 51,
13943 (1957).
37. В. M. Березовский, Р. В.
Артемкина, Е. Д. Хому-
това. ЖОХ, 34, 2791 (1964).
38. R. Kuhn, Н. Rudy, F. W е у -
g a n d. Вег., 68, 625 (1935); герм,
пат. 632131; С. А., 30, 6897 (1936).
39. G. Stone. Science, 111, 283
(1950).
40. М. Т a k a h a s h i, К. J a g i,
F. E g a m i. J. Chem. Soc. Japan,
78, 1287 (1957); РЖХ, 1958, 39789.
41. F. Egami, M. Takahashi.
Яп. пат. 2624 (1959); С. A., 54,
13151 (1960).
42. J. В a d d i 1 e у, J. В u c h a-
n a n, В. С a r s s. J. Chem. Soc.,
1957, 4058.
43. V. Sumi. Yakugaku Zasshi, 81,
647, 652 (1961); C. A., 55, 24760,
24761 (1961).
44. L. W h i try. Biochem. J., 50,
433 (1952).
45. H. К a t a g i r i, H. Y a m a-
d а. Яп. пат. 6192 (1963); С. A.,
59, 6960 (1963).
46. Y. S u z u k i, K. Uc h i d a. J.
Vitaminol., 11, 313 (1965); J. Agric.
Chem. Soc. (Japan), 42, 233 (1968).
47. P. К a r r e r. R. О s twald, Rec.
trav. chim., 57, 500 (1938).
48. D. F г о s t. J. Biol. Chem., 145,
693 (1942).
49. G. W e b e r. Biochem. J., 47,
114 (1950).
50. W. Foye, W. Lange. J.
Am. Chem. Soc., 76, 2199 (1954).
568
51. A. Albert. Biochem. J., 47,
27 (1950).
52. T. H a г к i n s, H. F r e i s e r.
J. Phys. Chem., 63, 309 (1949).
53. P. В a m b e r g, P. H e m m e-
r i c h. Helv. Chim. Acta, 44,
1001 (1961).
54. P. H e m m e г i c h, S. F a I-
1 a b. Helv. Chem. Acta, 41, 498
(1958).
55. D. G r e e n, S. M i i, H. M a h-
1 e r, B. Bock. J. Biol. Chem.,
206, 1 (1954).
56. M. T i s h 1 e r, J. Wei 1 ma n,
K. La den b ur g. J. Am. Chem.
Soc., 67, 2165 (1945).
57. A. S u r r e y, F. N a c h о d.
J. Am. Chem. Soc., 73, 2336 (1951).
58. J. W e i j 1 a r d, M. T i s h-
1 e r, A. E r i с к s о n. J. Am.
Chem. Soc., 66, 1957 (1944).
59. F. W о 1 f, R, Be u tel,
J. Stevens. J. Am. Chem.
Soc., 70, 2572 (1948).
60. P. H e m m e r i c h, В. P r i j s,
H. Erlenmeyer. Helv. Chim.
Acta, 43, 372 (1960).
61. К. H о t t о, M. T e r a o. Bi-
tamin, 17, 542 (1959); C. A., 59,
9015 (1963).
62. K. Y a g i. Igaku to Seibut Su-
gaku, 28, 54 (1953); C. A., 48, 2141
(1954).
63. T. W a d a, Y. S а к u r a i e t al.
J. Japan. Soc. Food. Nutrition, 5,
44, 97, 208 (1952—1953); 6, 3
(1953—1954); C. A., 50, 16899, 16900
(1956).
64. C. F u к a m a c h i, Y. S a к u-
r a i. Vitamins, 7, 939 (1954);
C. A., 51, 18026 (1957); Vitaminolo-
gy, 1, 217 (1955); C. A., 49, 14769
(1955).
65. E. S m i t h, D. Metrler. J.
Am. Chem. Soc., 85, 3285 (1963).
66. J. Merkel, W. Nickerson.
Biochem. Biophys. Acta, 14, 303 (1954).
67. A. S w a 1 1 о w. Nature, 176, 793
(1955).
68. W. N i с к e r s о n, G. S t r au s s.
J. Am. Chem. Soc., 82, 5007(1960).
69. С. O’M о 1 1 e у, C. S i e v e r t.
Ind. Eng. Chem., 34, 1117 (1942).
70. В. M. Березовский, E. П.
Родионова. ЖОХ, 28, 1046
(1958).
71. К. Sakai. Nagoya J. Med. Sci.,
18, 222 (1956); C. A., 50, 11828
(1956).
72. H. E r 1 e n m e у e r, S. Fa I-
1 a b, P. H e m m e г i c h. Пат.
ФРГ 1031310; С. A., 54, 1459 (1960).
73. Vernon. Bioch. Biophys. Acta,
36, 177 (1959).
74. P. К a r r e r, H. S a 1 о m о n,
K. Schopp, E. Schlit-
tler. Helv. Chim. Acta, 17, 1165
(1934).
75. M. H a 1 v e r. J. Am. Chem. Soc.,
73. 4870 (1951).
76. P. К a r r e r, H. M e e r w e i n.
Helv. Chim. Acta, 18, 1126 (1935).
77. C h u n g Shu Yang,
D. Me С о r m i c k. J. Am.
Chem. Soc., 87, 576 (1965).
78. B. Holmstrom. Arkiv. Ke-
mi, 22, 329 (1964).
79. B. Holmstrom, G. Oster.
J. Am. Chem. Soc., 83, 1867 (1961).
80. H. F a 1 1, H. P e t e r i n g. J.
Am. Chem. Soc., 78, 377 (1956); пат.
США 2825729; С. A., 52, 18477
(1958).
81. Англ. пат. 721426; С. А., 50, 2687
(1956).
82. Р. Н е m m е г i с h, В. Р г i j s,
Н. Erlenmeyer. Helv.
Chim. Acta, 42, 2164 (1959).
83. H. Petering. Пат. США
2973359; РЖХ, 1962, ЗЛ313.
84. H. Petering, G. Giessen.
J. Pharm. Sci., 52, 1192 (1963).
85. В. M. Березовский, Ж. И.
Аксельрод. ДАН СССР, 168,
577 (1966).
86. Н. Gol d пег, G. D i е t z,
Е. Carstens. Lieb. Ann.,
694, 142 (1966).
87. В. M. Березовский, Ж. И.
Аксельрод, Н. Д. Гри-
гор ь е в а, В. 3. М е л ь н и-
к о в, Н. И. К и р и л л о в а.
ДАН СССР, 198, 829 (1971).
88. R. Kuhn, R. Strobel е. Вег.,
70, 753 (1937).
89. W. С 1 а г k. Oxydation-reduc-
tion potentials of organic systems,
1960, p. 441.
90. A. Ehrenberg, P. Hemme-
ric h. В книге Biological Oxida-
tions, ed. by T. Singer, N. Y.,1968,
стр. 239.
91. P. H e m m e r i c h, С. V e e-
ger, H. Wood. Angew. Chem.
(Int. Ed.), 77, 699 (1965).
92. T. П. Ф e т и с о в a, A. В o-
лодарский, В. 3. Мель-
ник о в, С. Д. Д ы м о в а,
Ж. К. Т о р о с я н, И. М. Кус-
та н ов и ч, В. М. Березов-
с к и й. ЖОХ, 41, 2305 (1971).
93. Р. Н е m m е г i с h, Н. Erlen-
meyer. Helv. Chim. Acta, 40,
180 (1957).
94. R. N о г r e s t a m, P. К i r-
kegaard, B. Stlusland,
L. T о r b j о r n s s о n. J. Chem.
Soc. «D», 1969, 1250.
95. R. К u h n, H. R u d y. Ber.,
67, 1826 (1934).
96. R. К u h п, H. R u d y. Ber.,
67, 892, 1125, 1298, 1770, 1936
(1934).
97. K. D u d I e у, P. H e m m e-
r i c h. Helv. Chim. Acta, 50, 355
(1967).
98. В. M. Березовский, Ж. И.
Аксельрод. ДАН СССР, 171,
1101 (1966).
99. J. Sumi. Vitamin, 13, 204 (1957);
С. А., 54, 4608.
100. В. М. Б е р е з о в с К и й, Л. М.
Мельникова. ЖОХ, 31, 3827
569
(1961); авт. свид. СССР 137922;
Бюлл. изобрет. 1961, № 9, 25.
101. В. М. Б е р е з о в с к и й, Т. В.
Еременко. ЖОХ, 31, 3831 (1961).
102. Р. Н е in m е г i с h, S. F а 1-
1 a b, Н. Erlenmeyer. Helv.
Chim. Acta, 39, 1242 (1956).
103. F. Мй 1 I er, W. Wa 1 ker,
P. H e m m e r i c h. Helv. Chim.
Acta, 49, 2365 (1966).
104. P. H e m m e r i c h, В. P r i j s,
H. Erlenmeyer. Helv. chim.
Acta, 42, 1604 (1959).
105. P. M й 1 1 e r, P. H e m m e-
r i c h. Helv. Chim. Acta, 49,
2352 (1966).
106. F. Muller, P. Hemme-
ric h, H. E r 1 e n m e у e r.
Experientia, 18, 498 (1962).
107. D. McCormick. J. Heter.
Chem., 4, 629 (1967).
108. Ж. И. Аксельрод, В. M.
Березовский. ЖОХ, 39,
1630 (1969).
109. В. M. Березовский, Ж. И.
Аксельрод. ДАН СССР, 180,
607 (1968).
110. В. М. Березовский, Ж. И.
Аксельрод. ЖОХ, 38, 2440
(1968).
111. Н. А. Полякова, Л. С.
Тульчинская, Л. Г. 3 а-
песочная, В. М. Березов-
ский, ЖОХ, 42, 1135 (1972).
112. Л. С. Тульчинская,
Т. А. Ж и л и и а, В. Д. К л е-
банова, Л. М. Солодки-
н а, В. А. Миронов,
В. М. Березовский. ЖОХ,
42, 1135 (1972).
113. Р. Н е m m е г i с h. Helv. Chim.
Acta, 41, 514 (1958).
114. Л. С. Тульчинская,
Н. А. П о л я к о в а, В. М.
Березовский, ЖОХ, 40,
1859 (1970).
115. В. М. Березовский,
Н. А. П о л я к о в а, Л. С.
Тульчинская. Хнм. гете-
рой. соед., 1967, 729.
116. 3. В. П у ш к а р е в а, Л. Ф.
Петрова, В. Ф. Грязев.
Хим. гетерой, соед., 1965, 438.
117. D. McCormick. J. Heter.
Chem., 7, 447 (1970).
118. S. G h i s I a, U. Hart maun,
P. H e m m e r i c h. Angew.
Chem., 82, 669 (1970).
119. W. W a 1 k e г, T. Singer,
S. G h i s 1 a, P. H e m m e-
r i c h. Eur. J. Biochem., 26,
279 (1972).
120. P. Hemmeric h. Helv. Chim.
Acta, 43, 1942 (1960).
121. В. M. Березовский, P. B.
А д а н я e в a, H. Д. Г p и-
г о p ь e в а. ЖОХ, 38, 1704
(1968).
122. В. M. Б e p e з о в с к и й, Л. С.
Тульчинская, Н. А. По-
ля к о в а. Усп. химии, 41,
1277 (1972).
123. А. К о с е n t. Chem. listy, 47,
195 (1953); РЖХ, 1953, 3300.
124. М. Н а 1 w е г. J. Am. Chem. Soc.,
73, 4870 (1951).
125. A. W а г b u г g, W. Chri-
st i a n. Biochem. Z„ 263, 228
(1933)
126. F. We у ga n d. Ber., 72, 1664
(1939).
127. R. Kuhn, K. R e i n e m u n d,
F. W e v g a n d. Ber., 67, 1460,
1032 <19341
128. O. Kuh 1’i n g. Ber. 24, 2363
(1891); 27, 2116 (1894); 28, 1968
(1895).
129. А. В 1 у t h. J. Chem. Soc., 35,
530 (1879).
130. T. Osborne, L. Mendel.
J. Biol. Chem., 15, 311 (1913).
131. В. В 1 e у e r, O. Kallmann.
Biochem. Z., 155, 54 (1925).
132. P. К a r r e r, K. S c h opp.
Helv. Chim. Acta, 17, 771, 1013
(1934).
133. E. M с С о 1 I u m, M. D a v i s.
J. Biol. Chem., 20, 641 (1915); 21,
179 (1915).
134. P. К a r r e r, H. Salmon,
K. S c h 6 p p. Helv. Chim. Acta,
17, 419 (1934).
135. J. Banga, A. Szen t-G у о r g i,
Biochem. Z„ 246, 203 (1932).
136. Moore, В e c z e. J. Bacte-
rid., 54, 40 (1947).
137. Э. M. Д и к а н с к а я. Микро-
биология, 19 (1950); 22, 256 (1953).
138. M. H. M e й с e л ь, Э. M. Д и-
канская. ДАН СССР, 85, 1377
(1952).
139. М. Г. Голышева. ДАН СССР,
73, 585 (1950).
140. R. Т a k a t a. J. Japan Bio-
chem. Soc., 20, 130 (1948); С. А.,
44, 8063 (1950).
141. J. Hastings. Chem. and Ind
(London), 1957, 274.
142. R. T а к a t a, S. U m e m o-
t o. J. Ferment. Technol. Japan,
26, 333 (1948).
143. R. Hickey. Arch. Biochem, 11,
259 (1946).
144. В. M. Березовский, Тру-
ды ВНИ витаминного института,
4', 13, Пищепромиздат, 1953.
145. W. F а г k a s, L. F 1 е х s е г.
Пат. США 2472007; С. А., 43,
7053 (1949).
145а. F. J о n е d а, М. I с h i b а.
Chem. Pharm. Bull., 20, 1832
(1972).
146. M. T i s h 1 e r, J. Wei 1 m an.
Пат. США 2261608; С. A., 36,
1050 (1942); пат. США 2342438;
С. А., 38, 4624 (1944).
147. F. В е г g е 1, А. С о h е п,
J. Haworth. J. Chem. Soc.,
1945, 165; пат. США 2374661; С. А.,
40, 614 (1946).
148. О. К й h 1 i п g, О. К a s е-
1 i t z. Ber., 39, 1314 (1906).
149. О. H i n s b e r g. Ber. 18, 1228
(1885).
570
150. R. Ku h n, * F. We у ga n d.
Ber., 67, 1409 (1934).
151. H. P e t e r i n g, G. Giessen.
J. Org. Chem., 26, 2818 (1961);
пат. США 2993898; С. A., 56,
3490 (1962).
152. F. King, J. С 1 а г к - L e -
w is. J. Chem. Soc., 1959, 3379.
153. R. Kuhn, F. Wey ga nd.
Ber., 68, 1282 (1935).
154. R. Kuhn, F. Weygand.
Ber., 68, 1289 (1935).
155. S. Ikuma, M. Takemura,
S. К u b о t a, S. К a m a t a,
H. К am a m u r a, H. I t a g a -
к i, S. H a b u. Ann. Rept.
Tamamine Lab., 3, 6 (1951); C. A.,
49, 340 (1955).
156. E. H a 1 e y, J. L a m b о о у.
J. Am. Chem. Soc., 76, 2926, 5093
(1954).
157. P. К a r r e r, H. S a 1 о m о n,
K. S c h 6 p p, F. В e n z. Helv.
Chim. Acta, 18, 1143 (1935).
158. P. К a r r e r, K. S c h 6 p p,
F. В e n z, К. P f a e h 1 e r.
Helv. Chim. Acta, 18, 69 (1935);
Ber., 68, 216 (1935).
159. L. F 1 e x s e r, W. F a г к a s.
Пат. США 2456395; С. A., 43,
1813 (1949).
160. Англ. пат. 628410; С. А., 44,
4935 (1950).
161. F. Ber gel, A. Cohen,
J. Wynne. Англ. пат. 550836;
С. А., 38, 1752 (1944).
162. Р. В a m b е г g, Р. Н е m m е-
rich, Н. Erlenmeyer.
Helv. Chim. Acta, 43, 395 (1960).
163. H. G r a h a m, A. Macle t h,
W. О r r. J. Chem. Soc., 1927, 740.
164. А. В a e у e r. Lieb. Ann., 130,
130 (1864k
165. J. Backes, R. West,
M. W h i t e 1 e y. J. Chem. Soc.,
119, 359 (1921).
166. T. Matsukawa, K. Shi-
ra к a w а. Яп. пат. 155208,
157988 (1941); J. Pharm. Soc. Ja-
pan, 69, 458 (1949); C. A., 44,
3504 (1950).
167. В. M. Березовский, Г. Д.
Гл е б о в а. ДАН СССР, 146,
355 (1962).
168. В. М. Б е р е з о в с к и й, Г. Д.
Глебова. ДАН СССР, 143,
1341 (1962).
169. В. М. Березовский,
Г. Д. Глебова. ЖОХ, 34, 1014
(1964).
170. В. М. Березовский, Г. Д.
Глебова. Хим. гетероц. соед.
1965, 121.
171. К. Ganapati. Indian. Chem.
Soc., 15, 17 (1938).
172. S. N i s h i d a. Bull. Inst. Phys.
Chem. Research (Tokyo), 22, 872
(1943); C. A., 43, 7938 (1949).
173. В. M. Б e p e з о в с к и й, Л. С.
Тульчинская, Н. А. По-
ля к о в а. ЖОХ, 35, 673 (1965).
174. М. Т i s h 1 е г, К. Р f i s t e r,
R. Babson, K. Lade.n
burg, A. F 1 e m i n g. J. Am.
Chem. Soc., 69, 1487 (1947).
175. В. Березовский, В. Ку p -
д юк о в а, Н. П р е о б ра-
же и с к и й. ЖПХ, 22, 527 (1949).
176. В. М. Березовский. Тру-
ды ВНИ витаминного института, 5,
12; Пищепромиздат, 1954.
177. В. М. Б е р е з о в с к и й, Л. С.
Ту л ь ч и н с к а я, Т. В. Ер е-
м е н к о, Е. П. Р о д и о н о-
в а, М. А. Барска я. ЖОХ,
31, 3689 (1961).
178. В. М. Березовский, Е. П.
Родионова. Авт. св. СССР
93306; Бюлл. изобрет., 1952,
№ 2-3, 7.
179. В. М. Бер езов с к и й, Е. П.
Родионова, Л. И. Стре-
льну н а с. ЖОХ, 24, 628 (1954).
180. D. Н е i 1, Е. С h a s е, F. К о-
n i n s z у, К. F о 1 k е г s. J. Ат.
Chem. Soc., 73 3826 (1951).
181. Shun к, Lavigne, К. F о I-
к е г s. J. Am. Chem. Soc., 77,
2210 (1955).
182. В. М. Б е р е з о в с к и й, Л. С.
Тульчинская, ЖОХ, 31,
2779 (1961); авт. свид. СССР
139321; Бюлл. изобрет. (1961),
№ 13, 22.
183. Т. Н о s h i п о, Т. Sato,
М. Kato. Яп. пат. 3575 (1957);
С. А., 52, 7364 (1958).
184. С. Howe. Пат. США 2807611
(1957); С. А., 52, 663 (1958).
185. К. Р а 1 ze 1 t, J. С t у г t п i к,
J. О р a v s к у. Чехосл. пат.
131622; С. А., 72, 111780 (1970).
186. Т. Н о s h i п о, Т. Sato,
М. Kato. Яп. пат. 3575 (1957);
С. А., 52, 7364 (1958).
187. Т. N a i t о, Т. Y о s h i к а-
w а. Яп. пат. 7737 (1963); С. А.,
59, 11646 (1963).
188. С. Howe. Пат. США 2807611;
РЖХ, 1959, 72361.
189. Л. С. Тульчинская, Н. Б.
К а р л с т э д т, Н. А. П о л я-
к о в а, В. М. Березов-
ский. ЖОХ, 38, 2457 (1968).
190. Л. С. Т у л ь ч и н с к а я, Н. Б.
К а р л с т э д т, В. At. Б е ре-
зо в с к и й. Хнм. гетероц. соед.
1968, 538.
191. Е. Н а 1 е v, J. L a m b о о у.
J. Am. Chem. Soc., 76, 2926 (1954).
192. К. Felkers, С. S h u n k.
Пат. США 2847413; С.A., 53, 3252
(1959).
193. Н. В i n е г t. J. Biol. Chem., 225,
465 (1957); Biochem. Biophys. Acta,
20, 588 (1956).
194. E. Holey, P. Thesis. Uni-
versity of Rochester, Rochester,
New York, 1954, p. 99.
195. T. Sato. Яп. пат. 3328 (1954);
С. A., 50, 1094 (1956).
196. R. Pasternack, E. Brown,
Пат. США 2324800; С. A., 38,
221 (1944).
671
197. К. Tarukawa, К. Ue no.
Яп. пат. 19377 (1961); С. А.. 57,
8667 (1962).
198. R. Cresswell, Т. N е i 1-
s е п, Н. Wood. J. Chem.
Soc., 1961, 476.
199. Т. N е i s 1 е n, H. W oo d.
J. Chem. Soc., 1962, 44.
200. T. Bardos, D. Olsen,
T. E n к о j i. J. Am. Chem. Soc.,
79, 4704 (1957). Пат. США 3057865;
С. A., 58. 4582 (1963).
201. R. Kuhn, А. С о о k. Ber., 70,
761 (1937).
202. T. К u w a d a, T. M a s u d a,
T. К i s h i, N. A s a i. Chem.
Pharm. Bull. (Tokyo), 6, 361, 366
(1958); C. A., 52, 16358 (1958).
203. T. M a s u d a. Chem. Pharm.
Bull. (Tokyo), 4, 375 (1956); 5, 28
(1957); C. A., 50, 14859 (1956); 51,
9931 C1Q57I
204. G. Ma le y, G. P 1 a u t. J. Am.
Chem. Soc., 81, 2025 (1959).
205. R. C r e s we I 1, H. Wood.
Proc. Chem. Soc. 1959, 387; J.
Chem. Soc., 1960, 4768.
206. A. Birch, C. Moye. J.
Chem. Soc., 1957, 412; 1958, 2622.
207. H. В r e d e r e c k, W. P f 1 ei-
der e r. Chem. Ber., 87, 1119
(1954).
208. A. Gowenlock, G. N e w -
h о 1 d, F. S p r i n g. J. Chem.
Soc., 1948, 517.
209. R. В a x t e r, F. S p r i n g.
Nature, 154, 462 (1944).
210. P. Karrer, H. Meer wei n.
Helv. Chim. Acta, 18, 1130 (1935),
19, 264 (1936).
211. P. Salzberg. Пат. США
2193433; С. A., 34, 4742 (1940).
212. Б. С о p о к и н. J. prakt. Chem.,
(2), 37, 281 (1888).
213. J. I г с i n, R. G i b u о u г.
J. Chem. Soc., 93, 94, 1429 (1908),
95, 1545 (1909); 97, 1449 (1910);
99, 161 (1911).
214. L. Berger, J. Lee. J. Org.
Chem., 11, 75, 84 (1946).
215. Англ. пат. 599013; С. A., 42, 6847
(1948).
216. J. Lee, U. Solmssen,
L. Berger. Пат. США 2384105;
С. A., 40, 600 (1946).
217. H. Spieg'elberg. Пат. США
2429244; С. А., 42, 1605 (1948).
218. L. F 1 е х s е г, W. S с h п у d е г.
Пат. США 2477560; С. А., 44,
169 (1950).
219. В. М. Березовский. Авт.
св. СССР 97366; Бюлл, изобрет.,
1954, № 3, 15.
220. В. М. Березовский, Е. П.
Родионова. Труды ВНИ
витаминного института, т. 6, 5
(1959).
221. В. М. Березовский, Е. П.
Родионова. ДАН СССР, 87,
585 (1952).
222. Р. Karrer. Герм. пат. 677515
(1939); С. А., 33, 9325 (1939).
223. J. К a m 1 е t. Пат. США 2406774;
С. А., 41, 780 (1947).
224. Р. К а г г е г, Т. Q u i b е 1 1.
Helv. Chim. Acta, 19, 1034 (1936).
225. W. R о s s, J. Chem. Soc., 1948,
219.
226. K. Sahas hi, H.Acamat-
s и, H. Ge nd a. Bull. Inst.
Phys. Chem. Research (Tokyo), 24
72 (1948); C. A., 42, 5458 (1948).
227. В. M. Березовский, T. В.
Еременко. ЖОХ, 32, 4056
(1962).
228. В. M. Б ер ез овс к и й, Л. С.
Тульчинская. ЖОХ, 31,
2774, 3614 (1961); 32, 853 (1962).
229. F. W е у g a n d. Вег., 73, 1252,
1259 (1940).
230. М. A ma d or i. Atti R. Acad.
Lincei (Roma), (6), 2, 337 (1925);
9, 68, 226 (1929); 13, 72, 195 (1931).
231. R. Kuhn, F. W e у g a n d.
Ber., 70, 769 (1937); 71, 1535 (1938).
232. R. К u h n, A. D a n s i. Ber.,
69, 1745 (1936).
233. В. А. Конькова. ЖОХ, 22,
1896 (1952).
234. M. Tishler, N. Wendler,
K- Ladenburg, J. Well-
man. J. Am. Chem. Soc., 66, 1328
(1944).
235. R. P a s t e г п a c k, E. Brown.
Пат. США 2250999; С. A., 35,
7120 (1941).
236. M. T i s h 1 e г, J. Wellman.
Пат. США 2370093; С. A., 39,
4098 (1945).
237. В. M. Б e p e з о в с к и й, Е. П.
Родионова. ЖОХ, сб. II,
939 (1953).
238. L. Jampolsky, Н. Wuest.
J. Am. Chem. Soc., 68, 1777 (1946).
239. В. M. Б e p e з о в с к и й, В. А.
Курдюкова. ДАН СССР, 76,
839 (1951).
240. F. В е г g е 1, A. Cohen,
J. Н a w о г t h. J. Chem. Soc.,
1945, 165; англ. пат. 550169;
С. А., 38, 1247 (1944).
241. Р. К а г г е г, В. В е с к е г,
F. В е n z, Р. F г е i, Н. S а 1 о-
mon, К. Schopp. Helv.
Chim. Acta, 18, 1435 (1935).
242. R. К u h n, F. W e у g a n d.
Ber., 68, 1001 (1935).
243. R. Kuhn, R.Strobel e. Ber., 70,
773 (1937); герм. пат. 664048, 664439,
С. A., 33, 644 (1939); герм. пат.
624148; Zbl., 1937, II, 4421; герм,
пат. 679000; Zbl., 1940, 1, 760.
244. J. Lambooy. J. Am. Chem.
Soc., 72, 5225 (1950); 80, 110 (1958).
245. A. Surrev. Пат^ США 2351721;
С. A., 38, 5228 (1944).
246. 3. В. Пушкарева, В. Н.
Конюхов, Г. С. С а к о-
в в ч. Хим. гетерой, соед., 1965,
604.
247. Н. К и ж н е р. ЖРХО, 57, 7
(1925); ЖОХ, 6, 748 (1936).
248. А. С 1 a u s. J. pr. Chem., (2
41. 409 (1890).
572
249. В. М. Березовский, В. А.
Курдюкова, Н. А. П р е-
ображенский. ЖПХ, 22,
533 (1949).
250. Р. Karrer. Helv. Chim. Acta,
18, 1435, 1446 (1935).
251. К. К о b e, Р h. Р г i t с h е t t.
Ind. Eng. Chem., 44, 1398 (1952).
252. В. Визанский, С. Ансба-
хе p. В сборнике Синтезы орга-
нических препаратов, 4, 93, 170;
М„ ИЛ, 1953.
253. А. А. Шерешевский,
В. М. Березовский. Хим.
фарм. ж., 1969, № 10, 52.
254. В. М. Березовский, В. А.
Курдюкова, Н. А. Пре-
ображенский. Авт. св. СССР
81929; Бюлл. изобрет., 1950, № 5,
15.
255. В. М. Березовский, В. А.
Курдюкова, Н. А. Пре-
ображенский. ЖОХ, 21,
1163 (1951).
256. Н. S t е р h е n, W. S h о г t,
G. G 1 a d d i n g. J. Chem. Soc.,
117, 510 (1920).
257. В. M. Березовский, В. С.
В a p к о в. ЖОХ, 23, 100 (1953).
258. В. М. Березовский, В. С.
Вар ков. Труды ВНИ витамин-
ного института, 4, 87; Пищепром-
издат (1953).
259. Р. L е v е n е. J. Biol. Chem., 108,
41Q Л 9451
260. Н. В г е d e г е с k, М. К б t h-
n i g, E. Berger. Ber., 73,
956 (1940).
261. K. Uehara, T. Mizogu-
chi. J. Vitaminol., 11, 159
(1965).
262. К. О k a, H. W a d a. J. Pharm.
Soc. Japan, 83, 890 (1962); яп. пат.
15107 (1964); С. A„ 61, 16142
(1964).
263. D. Smith. Chemistry and Indu-
stry, 1955, 92.
264. H. S t r oh, D. D a r g e 1,
R. H 6 u s s 1 e r. J. prakt. chem.,
23, 309 (1964).
265. M. G e h r k e, F. A i c h n e r.
Ber., 60, 918 (1927).
266. W. A u s t i n, F. H u m о 1-
1 e r. J. Am. Chem. Soc., 56, 1152
(1934).
267. O. Ruff. Ber., 31, 1573 (1898); 32,
553 (1899); 35, 2360 (1902).
268. R. Hockett, C. Hudson.
J. Am. Chem. Soc., 56, 1632 (1934).
269. J. J о n e s, P. Ke n t, M. S t a-
c e y. J. Chem. Soc., 1947, 1341.
270. В. M. Березовский, В. A.
Курдюкова. ЖПХ, 22, 1116
(1949).
271. E. F i s c h e г, О. P i I о t у.
Ber., 24, 4214 (1891).
272. M. Steiger. Helv. Chim. Acta,
19, 189 (1936).
273, H. S t о p s a с k. Пат. ГДР 21547,
РЖХ, 1962, 7Л78; пат. ФРГ
1148991, С. А., 59, 14101 (1963).
274. R. Pasternack, Е. Brown.
Пат. США 2237263; С. А„ 35, 4394
(1941).
275. К. Ladenburg, М. Т i s h-
ler, J. Wellman, R. Bab-
son. J. Am. Chem. Soc., 66, 1217
(1944).
276. M. T i s h 1 e г. Пат. США 2409455,
2417143; С. A., 41, 989, 3824 (1947).
277. T. N a i t о, T. Y о s h i k a w a,
S. Kubota. Яп. пат. 8211 (1964);
С. A., 61, 12074 (1964).
278. О. S p e n g 1 e r, A.Pfannen-
s t i e 1. Z. Wirtschaftsgr. Zu-
ckerind., 85, 546 (1935).
279. H. Isbell. Bur. Stand., 29, 227
(1942).
280. В. M. Березовский. ЖОХ,
сб. II, 944 (1953).
281. H. Schmidt. Ам. пат. 2587906;
С. А., 46, 9123 (1952).
282. X. А к а б а, М. Ц у р у г а. Яп.
пат. 16528, РЖХ, 1968, 11Н328.
283. А. А. Ш е р е ш е в с к и й, Э. М.
Г усейнов. Хим.-фарм. ж.,
1969, № 9, 45.
284. D. А п d о, S. К i п о u d i. Яп.
пат. 11442 (1968); С. А., 70, 4546
(1969).
285. В. М. Бер е з овс к и й, Л. В.
Коновалова. Авт. свид.
СССР 76376; Бюлл. изобрет., 1949,
№ 9, 17.
286. L. Sternbach. Пат. США
2438881, 2438883; С. А., 42, 5048
(1947).
287. Z. Per i па, Z. V о к а с. Че-
хосл. пат. 91755; РЖХ, 1961,
5Л326.
288. М. Wolfrom, Н. Wood.
J. Am. Chem. Soc., 73, 2933 (1951).
289. N. S p e r b e r, H. Z a u g g,
W. S a n d s t г о m. J. Am. Chem.
Soc., 69, 915 (1947).
290. В. M. Березовский, Ю. П.
Соболев. Труды ВНИ вита-
минного института, Т. 6, 34 (1959).
291. В. М. Березовский. ЖОХ,
25, 789 (1955).
292. В. М. Березовский, В. С.
В а р к о в. Авт. свид. СССР
86536; Бюлл. изобрет., 1950, № 10,
15.
293. Т. S a t о, К. S a t о. Яп. пат.
3277 (1957); С. А., 52, 9228 (1958).
294. S. Sugasawa, М. М a t s
m о t о. Яп. пат. 613 (1964); С. А
60, 12096 (1964).
295. В. М. Березовский, В. С.
В а р к о в. Авт. свид. СССР
93682; Бюлл. изобрет., 1952, № 5,
31.
296. С. Ф. С у з д а л ь ц е в а,
В. Н. Красюк. Авт. свид.
СССР 194080; Бюлл. изобрет., 1967,
Ks 44, 20.
297. Е. И. Григорашвили,
Н. С. 3 о л о т а р е в, Г. И. 3 а-
р е ц к и й. Хим.-фарм. ж., 1971,
№ 3, 45
298. S. I t о, S. Asa k a, Y. К а-
w a m и г а. Яп. пат. 955 (1957);
С. А., 52. 4684 (1958).
573
299. К. Uehara, Т. М a t su к а-
w а. Яп. пат. 2279 (1959); С. А.,
54, 13016 (1960).
300. Синтезы органических препара-
тов, 3, 9, 12, 14; М„ ИЛ, 1952.
301. К. S h i m о, S. W a k a m a t-
s u. J. Soc. Org. Smth. Chem. (Ja-
pan), 17, 232 (1959); РЖХ, 1960,
38735.
302. A. Michael. J. prakt. Chem.
(2), 35, 456 (1887).
303. S. G a b r i e 1, J. С о 1 m a n.
Ber., 37, 3657 (1904).
304. Синтезы органических препара-
тов, 2, 79; M., ИЛ, 1949.
305. J. Tafel, A. Weinschenk.
Ber., 33, 3383 (1900).
306. Н. Euler. Rapp, et discuss. 6-me
Conseil de Chimie, Paris, 1938.
307. H. L e e m a n n. Klin. Wochen-
schr., 21, 60 (1942).
308. H. E u 1 e r, E. A d 1 e r. Sv.
Vet. Akad. Arkhiv. Kemi, B. 11,
28 (1934).
309. H. E u 1 e r, E. A d 1 e r. Z.
phys. Chem., 223, 105; 228, 1
X. (1934).
4 310. O. Brunner, E. Baroni.
yf' Monatsh., 68, 264 (1936).
M 311. K. Yagi, J. Biochem., 41, 575
(1954).
312. Chung Shu Yang, D.
McCormick. J. Nutr., 93,
445 (1967).
313. E. Owen. Proc. 1st. Inter. Congr.
Food Sci. Technol., 1962, III, 669;
Biochem. J., 84, 96 (1962).
314. В. M. Березовский, Успе-
хи химии, 18, 737 (1949).
315. M. P о 1 о n о v s k i, R. В u s-
n e 1, M. P e s s о n. Helv. Chim.
Acta, 29, 1328 (1946).
316. P. К a r r e r, F. S t г о n g.
Helv. Chim. Acta, 18, 1343 (1935).
317. R. К u h n, F. Weygand.
Ber., 68, 166 (1935).
318. R. Kuhn, H. V e t t e r,
H. R z e p p a. Ber., 70, 1302
(1937).
319. P. К ar r e r, H. S a 1 о m о n,
K. S c h 6 p p, F. В e n z, B. Be-
cker. Helv. Chim. Acta, 18, 908
(1935).
320. E. Pellajes, H. Garza.
Arch. inst. cardiol. Mex. 19, 735
(1949); C. A., 44, 5368 (1950).
321. J. Baddiley, J. Bucha-
nan, В. C a r s s. J. Chem. Soc.,
1957, 4058.
322. H. E u 1 e r, P. К a r r e r,
M. M a 1 m b e r g. Helv. Chim.
Acta, 18, 1336 (1935).
323. J. L a in b о о у. Biochim. Bio-
phys. Acta, 29, 221 (1958); J. Nutr.
75, 116 (1961).
324. J. Sumi. Bitamin, 20, 532 (1960);
C. A., 61, 16351 (1964).
325. H. A p о s h i a n, J. Lam-
b о о y. J. Am. Chem. Soc., 76,
1307 (1954).
326. E. S n e 1 1, F. S t г о n g. Enzy-
mologia, 6, 186 (1939).
327. R. Kuhn, P. Boulanger
Z. physiol. Chem., 241, 233 (1936).
328. R. К u h n, H. R u d y. Ber.,
69, 2557 (1936).
329. G. Serchi, G. Alberta z-
z i. Chimia, 29, 54 (1953).
330. H. S a r e t t. J. Biol. Chem., 162,
87 (1946).
331. H. F u 1 e r, P. Karrer. Helv.
Chim. Acta, 29, 353 (1946).
332. G. E m e r s о n, E. W u r t z,
O. J о h n s о n. J. Biol. Chem.,
160, 165 (1945).
333. G. E m e r s о n, M. T i s h-
1 e r. Proc. Soc. Exper. Biol.
Med., 55, 184 (1944).
334. R. К u h n, F. We у ga nd,
E. Moller. Ber., 76, 1044 (1943).
335. E. Haley, J.Lambooy.
J. Am. Chem. Soc., 76, 5093 (1954).
336. R. В a r I о w. J. Chem. Soc., 1951,
2225
337. K. F о 1 k e r s, C. S h u n k.
Пат. США 2847413; С. A., 53,
3252 (1959).
338. C. A r s e n i s, D. McCor-
mick. J. Biol. Chem., 239, 3093
(1964).
339. D. W о о 1 1 e y. J. Biol. Chem.,
154, 31; 152, 225 (1944).
340. R. Adams, C. Weisel,
H. M о s h e r. J. Am. Chem.
Soc., 68, 883 (1946).
341. F. К i n g, H. A c h e s о n,
A. York e-L о n g. J. Chem. Soc.,
1948, 1926.
342. H. В u r k e t t. J. Am. Chem. Soc.,
69, 2555 (1947).
343. H. H i p p c h e n. Chem. Ber.,
80, 263 (1947).
344. F. Holly, E. Peel, J. C a-
h i 1 1, F. К о n i u s z у, K. F o-
I k e r s. J. Am. Chem. Soc., 74,
4047 (1952).
345. C. S h u n k, F. К о n i u s z y,
K. F о 1 k e r s. J. Am. Chem. Soc.,
74, 4251 (1952).
346. T. Wagner-Jauregg,
О. В ii c h, J. M о 1 n a r. Che-
motherapia, 2, 96 (1961).
347. P. Karrer, P. Frei, H. Meer-
wein. Helv. Chim. Acta, 20,
79 (1937).
348. R. К u h n, H. R u d v. Ber.,
69, 1974 (1936).
349. В. M. Березовский, P. В.
Артемкина, M. А. Ч e p-
н о в а. ЖОХ, 35, 677 (1965).
350. Ю. К о ё с и, М. И в а к и. Яп.
пат. 12288 (1962); РЖХ, 1963,
17Н174.
351. Т. Комацу. Яп. пат. 1740
(1962); РЖХ, 1963, 14Н227.
352. S. G h i s 1 a, U. Hartmann,
P. Hemmerich. Angew. Chem.,
82, 669 (1970).
353. W. W a 1 k e r, T. S i n g e r. J.
Biol. Chem., 245, 4224 (1970).
354. W. Walker, E. Kearney,
R. S e n g, T. S i n g e r. Bio-
chem. Biophys. Res. Commun., 44,
287 (1971).
574
355. E. Kearney, J. S а 1 а с h,
W. W а 1 ке г, R. S е n g,
T. Singer. Biochem. Biophys.
Res. Commun., 42, 490 (1971).
356. S. G h i s I a, P. H e m m e-
r i c h. FEBS Letters, 16,229 (1971).
357. D. G r e e n, J. D e w a n. Bio-
chem. J. 32, 626 (1938).
358. R. К u h n, P. В ou I a n ger.
Ber., 69, 1557 (1936).
359. T. Kobayashi. Bitamin, 20,
371 (1960).
360. T. Tanaka. Яп. пат. 9973 (1958),
С. A., 54, 5714 (1960); Yakugaku
Zasshi, 78, 627 (1958); C. A., 52,
18460 (1958).
361. E. Д. X о м у т о в a, T. A.
Шапиро, M. В. M e з e н ц e-
в а, В. M. Березовский.
ЖОХ, сборник «Синтез природных
соед. их аналог, и фрагм.», 1965,
241.
362. Н. F о г г е s t, Н. Ma s on,
A. T о d d. J. Chem. Soc., 1952,
2530.
363. S. Shimizu. Vitamins (Kyo-
to) 17, 48 (1959); C. A., 58, 4641
(1963).
364. E. Д. X о м у т о в a, T. А. Ш a-
n и p о, В. M. Березов-
ск и й. ЖОХ, 36. 1749 (1966).
365. Р. В. Артем кии а, В. М. Б е-
резовский. ЖОХ, 36, 823
(1966).
366. Н. Т h е о г е 1 1. Biochem. Z.,
272, 155 (1934); 275, 37, 344 (1934);
278, 263, 279 (1935).
367. R. К u hn, Р. D е s п u е 1 1 е.
Вег., 70, 1907 (1937).
368. R. Kuhn, Н. Rudy, F. Wey-
g а п d. Ber., 69, 1549 (1936).
369. О. Warburg, W. Chri-
stian. Naturwissensch, 26, 235
(1938); Biochem. Z„ 295, 261
(1938); 298, 150 (1938).
370. E. Abraham. Biochem. J., 33,
543 (1939).
371. P. Karrer, H. Frank.
Helv. Chim. Acta, 23, 948 (1940).
372. S. C h r i s ti e, G. Kenner,
A. Todd. Nature. 170, 924
(1952); J. Chem. Soc., 1954, 46.
373. E. Kearney. J. Biol. Chem.,
235, 865 (1960).
374. P. H e m m e r i c h, A. Eh-
renberg. W. Walker,
L. Erihsson, J.Salach,
P. В a d e r. T. Si nger.
FEBS Letters, 3, 37 (1969).
375. S. Tachibana. J. Vitaminol.,
7, 274 (1961).
376. V. C a n a 1 i, G. C a s c i о t-
t i. Bull. Soc. ital. bioL, 27,
1478 (1951); C. A., 47, 11208 (1953).
377. Англ. пат. 712387; С. A., 49,
11725 (1955); швейц, пат. 299878;
РЖХ, 1956, 52327; инд. пат.
48340; РЖХ, 1955, 47366; австр.
пат. 157818; РЖХ, 1956, 17358.
378. L. F 1 е х s е г, W. Farkas.
Пат. США 2610178; С. А., 47,
8781 (1953); англ. пат. 690463;
РЖХ, 1956, 55932.
379. L. F 1 е х s е г, W. F а г k a s.
Пат. США 2610176; С. А., 47,
8782 (1953); Англ. пат. 694470;
С. А., 48, 10787 (1954); франц,
пат. 1043090, РЖХ, 1956, 30475.
380. L. F 1 е х s е г, W. F а г к a s.
Chem. Eng. News, 29, 3997 (1951).
381. P. В. А д а н я e в а, Л. Я. Де-
ни с о в a, А. Н. Б о р о в и-
к о в а, В. М. Березов-
ский. Хим. фарм. ж., 1, 16
(1957).
382. С. К i 1 g о и г, F. Н е u n п е-
k е п. J. Am. Chem. Soc., 79, 2256
(1957).
383. W. F б г у, Р. Н е т т е г i с h.
Helv. Chim. acta, 50, 1766 (1967).
384. В. M. Б e p e з о в с к и й, Л. М.
Мельникова. ЖОХ. 43, 921
(1973).
385. G. К е n п е г, А. Т о d d,
F. W е у т о n t h. J. Chem. Soc.,
1952, 3675.
386. N. С о r b y, G. Ke n ner,
A. T о d d. J. Chem. Soc., 1952,
3669.
387. F. H e u n n e k e n s, G. К i 1-
g о u r. J. Am. Chem. Soc., 77,
6716 (1955).
388. F. Heunnekens, S. Feb
ton. Methodes in Enzymology,
III, New York, 1957.
389. C. Dekker, H. Khorana.
J. Am. Chem. Soc., 76, 3522 (1954).
390. G. T e n e г, H. К h or a na.
J. Am. Chem. Soc., 77, 5349 (1955).
391. M. Smith, J. Moffatt,
H. К h о r a n a. J. Am. Chem.
Soc., 80, 6204 (1958).
392. E. Д. X о м у т о в a, T. А. Ша-
ли p о, В. M. Березов-
ск u й. ЖОХ, сб. Биол. акт.
соед., 1965, 230.
393. В. M. Березовский, Т. В.
Еременко, ЖОХ, сб. Биол.
акт. соед., 1965, 234.
394. М. Susai, Т. Takamatsu.
Яп. пат. 3180 (1966); С. А., 64,
19750 (1966).
395. Н. Khorana. J. Am. Chem.
Soc., 79, 4240 (1957).
396. R. Chambers, J. Mof-
fatt. J. Am. Chem. Soc., 79,
3752 (1958).
397. J. Moffatt, H. Khorana.
J. Am. Chem. Soc., 79, 3755 (1958).
398. F. Cramer, H. Neunhoef-
f e r, K. Sc he it, G. S c hn e i -
d e r, J. T e n n i g k e i t.Angew.
Chem., 74, 387 (1962).
399. D. McCormick, В. C h a s-
s y, J. T r i b r i s. Biochim.
Biophys. Acta, 89, 447 (1964).
400. T. Kuroda. Bitamin, 29, 437
(1964).
401. B. Ch a ssy, D. McCor-
mick. Biochim. Biophys. Acta,
110, 91 (1965).
402. W. F б г у, P. H e m m e r i c h.
Helv. Chim. Acta, 50, 1766 (1967).
57Б
403. Е. Д. Хомутова, Т. А. Ша-
пиро, М. В. Мезенцева,
В. М. Березовский. Хим.
фарм. ж., 1967, № 1, 11.
404. В. М. Березовский, Р. В.
Аданяева. ЖОХ, 37, 2639
(1967).
405. D. McCormick. J. Med.
Chem., 12, 333 (1969).
406. Т. Kuroda. Яп. пат. 5842
(1963); С. А., 60, 652 (1964).
407. L. G о 1 d m а п, J. М а г s i-
с о, G. A n d er so n. J. Ат.
Chem. Soc., 82, 2969 (1960); пат.
США 2951838; С. А., 55, 4536
(1961).
408. Р. Z u m р е, С. Woenckhaus.
Z. Naturforsch., В21, 1149 (1966).
409. A. Michelson, Chem. and
Ind., 1960, 1267; Biochim. Biophys.
Acta, 91, 1 (1964).
410. C. D e L u c a, N. К a p 1 a n,
J. Biol. Chem., 223, 569 (1956);
Biochim. Biophys. Acta, 30, 6
(1958).
411. Л. M. Мельникова, В. М.
Березовский. ЖОХ, 40, 918
(1970).
412. К. S u z u k i, Н. I i t о. Яп.
пат. 8971 (1958); С. А., 54, 5716
(1У60).
413. К. М а к i п о, Т. К и г о d а,
К. О h i к е. Kobayashi Jikei-
kai Med. J., 6, 58 (1959); C. A.,
55, 11426 (1961).
414. M. S и s a i, T. T a к a m a t-
s и. Яп. пат. 3708 (1966); С. A.,
65, 793 (1966).
415. E. Д. X о м у т о в a, Т. A. Illja-
п и р о, В. М. Б е р е з о в-
ский. ЖОХ, 40. 470 (1970).
416. Т. А. Ш а п и р о, Е. Д. Хо-
мутова, В. М. Б е р е з о в-
ский. ЖОХ, 42,1634 (1972).
417. Е. D i m а п t, D. S а п a d 1,
F. Heunnekens. J. Am. Chem.
Soc., 74, 5440 (1952).
418. N. S i 1 i p r a n d i, P.Cer-
1 e t t i. Arch. Biochem. bio-
phys., 76, 214 (1958).
419. L. Whitby. Biochem. J., 54,
437 (1953); Biochim. Biophys. Acta,
15, 148 (1954).
420. G. Kilgour, S. Felton
F. Heunnekens. J. Am.
Chem. Soc., 79, 2254 (1957).
421. K. Suzuki. Яп. пат. 7430,
7431 (1959); С. A., 54, 2672 (1960).
422. H. Kobayashi, K.Tsu-
k i h a r a. S. О k a m о t о. Яп.
пат. 6650 (1958); С. A., 54, 1571
(1960).
423. T. Masud а. Яп. пат. 9594
(1956); пат. ФРГ 1114198; С. А.,
52, 15847 (1958).
424. Y. Н a t е f i, А. Н a a v i k,
D. Griffiths. Biochim. Bio-
phys. Res. Commons., 4, 441, 447
(1961).
425. C. F г i e d e n. Biochim. Bio-
phys. Acta, 24, 241 (1957).
426. P. Strittmatter, S. V e-
1 i c k. J. Biol. Chem., 228, 785
(1957).
427. T. К i n g, R. H о war d. J.
Biol. Chem., 237, 1686 (1962).
428. F. S t r a u b, H. С о r r a n,
D. Green. Biochem., J., 33,
7Q4 119ЧСП
429. H. Euler, К. H a sse. Na-
turwissensch., 26, 187 (1938).
430. A. L e h n i n g e r. J. Biol.
Chem., 190, 345 (1951); Rev. Mod.
Phys., 31, 136 (1959).
431. B. G r a b e. Biochim. Biophys.
Acta, 30, 560 (1958).
432. L. L i n d be r g, B. G r a b e,
H. L 6 w, P. S i e k v i t z,
L. E r n s t e r. Acta chem. scand.,
598 (1958).
433. J. Hauge, F. Crane,
H. В e i n e r t. J. Biol. Chem.,
219, 727 (1956).
434. V. Massey. Biochim. Biophys.
Acta, 30, 205 (1958); 32, 286 (1959);
37, 314 (1960); 38, 447 (1960).
435. E. Lockhart. Biochem. J., 33,
613 (1939).
436. E. Richardson, N. Tol-
bert. J. Biol. Chem., 236, 1280
(1961).
437. A. Shoswell. Biochem. J., 79,
22 (1961).
438. W. F r a n k e, M. D e f f n e r.
Lieb. Ann., 532, 1 (1937); 541, 117
(1939).
439. E. К e i 1 i n, E. H о r t r e e.
Nature, 1946, 157, 801; Biochem.
- J., 42, 221, 230 (1948).
440. R. С e c i 1, A. О g s t о n. Bio-
chem. J. 42, 229 (1948).
441. R. Bentley, A. Neuber-
ger. Biochem. J., 42, 221 (1948);
45, 584 (1949).
442. A. Gordon, D. Green,
V. Subrahmanyan. Bio-,
chem. J., 24, 764 (1940).
443. H. С о r r a n, D. G r e e n. Bio-
chem. J., 32, 2231 (1938).
444. S. U d a k а, В. V e n n e s-
1 a n d. J. Biol. Chem., 237,
2018 (1962).
445. G. G i b s о n, V. M a s s e y,
N. Atherton. Biochem. J., 85,
369 (1962).
446. A. Kornberg, J. Pricer.
J. Biol. Chem., 182, 763 (1950);
186, 557 (1950).
447. H. W a d a, E. S n e 1 I. J.
Biol. Chem., 236, 2089 (1961).
448. H. T h e о r e 1 1, A. N у g a-
a r d. Acta chem. scand., 8, 877,
1649 (1954); 9, 1587 (1955).
449. E. W a 1 a a s, O. W a 1 a a s.
Acta chem. scand., 10, 122 (1956).
450. H. Mahler. J. Am. Chem. Soc.,
75, 3288 (1953); J. Biol. Chem.,
206, 13 (1953); Advanc. Enzymol.,
17, 233 (1956).
451. L. F I e x s e r, W. F a r k a s.
Food Manufacture, 27, 57 (1952).
576
ГЛАВА XIII
КОРРИНОВЫЕ ВИТАМИНЫ И КОФЕРМЕНТЫ
ЦИАНОКОБАЛАМИН, ОКСИКОБАЛАМИН
Кобаламины, витамины группы В12, относятся к производным внутрен-
него кобальтового комплекса нуклеотида бензимидазола и макроцикличес-
кой корриновой системы. Они включают в свою молекулу 3-фосфорный
эфир l-a-D-рибофуранозил-б, 6-диметилбензимидазола (а-рибазола), свя-
занный с 0^-1-аминопропанолом-2*, образующим пептидную связь с про-
пионовой кислотой положения 17 корриновой системы’, все 3-положения
гидрированных пиррольных циклов которой замещены метильными, а
также семью карбоксиметильными и карбоксиэтильными заместителями,
представленными в виде амидов кислот. Атом кобальта ковалентно и ко-
ординационно связан в виде «клешнеобразного», или «хелатного», комплек-
са с циангруппой и с атомами азота гидрированных пиррольных колец
и бензимидазола.
Примечательно необычное присутствие в молекуле витамина В12
а-анамерной конфигурации N-гликозидной связи между D-рибозой и 5,6-
диметилбензимидазолом, в то время как все нуклеозиды природных пури-
нов и пиримидинов имеют 3-конфигурацию [2]. Это связано с тем, что в
хелатном комплексе координационная связь атома кобальта стереохими-
чески образуется только с атомом азота рибофуранозилдиметилбензими-
дазола а-анамерной конфигурации [3].
Макроциклическая планарная корриновая система содержит четыре
азотсодержащих пятичленных цикла, соединенных по а,а-положениям
колец А—В, В—С и C—D тремя лгезо-углеродными атомами и одной непос-
редственной а, а-связью между кольцами А и D. Эта система имеет шесть
сопряженных двойных связей (4, 5] и девять асимметрических атомов угле-
рода, из которых шесть находятся в группе колец А—D.
Ранее макроциклические пиррольные структуры были известны в жиз-
ненноважных природных системах: порфиринах, например в гемине (же-
лезный комплекс), лежащем в основе красящего вещества крови — гемо-
глобина, и цитохромных коферментах, а также в дигидропорфиринах или
хлоринах, например в веществе зеленых листьев — хлорофилле (магние-
вый комплекс). Однако в этих системах четыре пиррольных кольца соеди-
нены по а, а-положениям четырьмя лезо-углеродными атомами.
Цианокобаламин, а-(5,6-диметилбензимидазолил)кобамид циа-
нид—витамин В12 (I) — кристаллизуется в рубиново-красных иглах или приз-
мах орторомбической системы (из водного ацетона); он содержит14—17% во-
ды, которая удаляется в вакууме при 100° С. Безводный витамин гигро-
скопичен (поглощает около 12% влаги). Он достаточно стабилен.
Цианокобаламин не имеет четко выраженной температуры плавления:
при температуре выше 200° С он начинает постепенно темнеть и разлагаться,
но не плавится до 320° С [6]. Цианокобаламин относительно легко раство-
ряется в воде (1,25% при 25° С), образуя растворы с нейтральной реакцией,
а также в метиловом и этиловом спиртах [7], в уксусной кислоте, феноле,
диметилформамиде, диметилсульфоксиде, нерастворим во многих неполяр-
ных органических растворителях, например в хлороформе, ацетоне, бен-
золе, пиридине и эфире. Водные растворы наиболее устойчивы при pH 4—7
[8]. Удельное вращение [аШ33 — 59 ± 9°[4] и [а]^ —НО ± 10°
(1,98 мг в 0,4 мл воды) [9J.
* Индекс g, так же как и s, указывает на связь с глицериновым альдегидом или
серином, используемыми как стандарты конфигурации [Ц.
1 О нумерации атомов коррииового цикла см. с. 581.
Vs 20-69
577
(i)H2NOCCH2CH2.
(fl)HjNOCCHa.
CH-
CH/
ц CH, CH .
^A/JL>C11,CONH,(c)
H |e XrCHsCHjCONH,^)
N CNN^/'h
(я) H2NOCCH2<
сн.
Спектр поглощения цианокобаламина в ультрафиолетовом и видимом
свете, измеренный в водном растворе или в растворе метилового спирта,
имеет максимумы: 278, 361, 525 и 550 нм (Е}с°и 115, 204, 57 и 63 соответ-
ственно) [7, 9, 101. Максимум поглощения при 278 нм, по-видимому, от-
носится к бензимидазол ьной части молекулы. В других полосах спектра
наблюдается некоторое подобие спектру поглощения порфирина 111].
При спектрофотометрическом анализе цианокобаламина соотношение аб-
сорбции должно быть при длинах волн (нм): 361/278 как 1,70—1,90 и 361/550
как 3,15—3,40. Спектр поглощения цианокобаламина измерен в инфракрас-
ной области [12, 13].
Цианокобаламин из растворов в метиловом спирте осаждается ацетоном;
из водного раствора извлекается фенол-хлороформной смесью, а из раство-
ра, насыщенного сернокислым аммонием, — «-бутиловым спиртом. Циано-
кобаламин легко адсорбируется из водного раствора активированным углем
и вымывается 65%-ным спиртом, водным раствором фенола или водно-пи-
ридиновым раствором; он адсорбируется из водного раствора или раствора
в метаноле окисью алюминия.
Вследствие большой молекулярной массы цианокобаламина — 1355,5
[14] определить его элементарный состав представляло большие трудности;
предлагались различные эмпирические формулы [7, 15]. Цианокобаламин
имеет элементарный состав C63H88N14O)4PCo [16]. Молекулярная масса
его определялась по повышению температуры кипения [7] и рентгенострук-
турным анализом [14].
Для цианокобаламина (I) на основании рентгеноструктурного анализа,
данных химического расщепления и частичного синтеза в 1955 г. предло-
жена структурная формула [16—19], впоследствии уточненная, в пользу
которой приводится и ряд новых данных [4, 51; эта формула подтверждена
полным синтезом.
Хелатный кобальтовый комплекс цианокобаламина (витамина В12) по
своему характеру практически нейтрален (очень слабо основен). Цианоко-
баламин диамагнитен, кобальт в нем трехвалентен и обладает координа-
ционным числом 6 [20 — 23]. Это подтверждается данными полярографи-
ческого восстановления [24, 25]. Три координационные связи кобальта
насыщаются тремя парами электронов атомов азота частично гидрированных
пиррольных циклов, а четвертая координационная связь — атомом азота
положения 3 диметилбензимидазола. Кобальт ковалентно связан с пирро-
лидиновым кольцом [26] и с циангруппой. Кобаламиновые комплексы очень
прочны: атом кобальта нельзя удалить из них без распада всей молекулы.
578
Фосфорный остаток нуклеотидного лиганда циано кобаламина этерифициро-
ван [27], однако не полностью, так как третий гидроксил в виде аниона фос-
форной группы компенсирован положительным зарядом атома кобальта.
Таким образом, витамин В52 представляет собой внутреннюю соль.
По отношению к макроциклической планарной корриновой системе
с атомом кобальта в центре аксиальными лигандами являются CN“ и 5,6-
диметилбензимидазол. Бензимидазольный цикл расположен почти перпен-
дикулярно корриновому циклу (под углом 80°) и рибофуранозному циклу,
который почти параллелен с коррином. Рибофуранозный цикл имеет кон-
формацию конверта, и С(2) выходит из плоскости четырех других атомов.
Следует отметить, что пирролиновые кольца корриновой системы не имеют
полной планарности, а сопряженная система почти полностью планарна.
Кольца А, В, С и D корринового цикла цианокобаламина в качестве
^-заместителей имеют те же остатки уксусной и пропионовой кислот, кото-
рые расположены при аналогичных ₽-атомах углерода уропорфирина III, кро-
ме C(i2), где вместо карбоксиметильного остатка имеется метильная группа.
Важно отметить, что все четыре пропионамидные группы и бензимидазол-
нуклеотид находятся по одну сторону планарной корриновой части моле-
кулы цианокобаламина, все три ацетамидные группы расположены на про-
тивоположной стороне. Образующийся из цианокобаламина в кислой среде
темно-красного цвета биологически почти неактивный циано-13-эпикобал-
амин (неовитамин В12) в кольце С коррина имеет пропионамидную груп-
пу в пространственно противоположной конфигурации [28].
С\
N—£Н3
// С
'СПз
Н CH2CH..CONH2(e)
Цианокобаламин, однако, нельзя рассматривать в качестве природной
формы. Скорее всего [29], витамин В12 в цианоформе образуется при выде-
лении из природных источников с применением CN“-ионов, так как без
цианистых соединений был выделен только оксикобаламин (в щелочной
среде) или аквокобаламин (в кислой среде) [29, 30].
Оксикобаламин1, а-(5,6-диметилбензимидазолил)оксикоб-
амид (II),— витамин В12а из печени; идентичный ему витамин В12Ь [26, 32]
выделен из продуктов жизнедеятельности актиномицетов; ему идентичен и
витамин B12d, [33, 34].
Оксикобаламин [II] содержит координационно связанную оксигруппу
(вместо циангруппы цианокобаламина). Он кристаллизуется в фиолетово-
красных иглах или пластинках орторомбической системы (из водного аце-
тона) [32]; окраска его кристаллов более темная, чем у цианокобаламина.
При температуре около 200° С кристаллы оксикобаламина начинают
темнеть, но плавятся при температуре около 300° С.
Оксикобаламин легче растворим в воде, чем цианокобаламин, вслед-
ствие чего цианокобаламин может быть очищен от него перекристаллиза-
цией из воды. В смеси бензиловый спирт — вода (1 : 1) эти витамины имеют
следующий коэффициент распределения2: 0,78 для цианокобаламина и
1 В соответствии с номенклатурой Интернационального союза по чистой и при-
кладной химии (ШРАС) [31] лнганд, связанный с металлом, называют по системе,
применяемой в номенклатуре комплексных соединений неорганической химии, с со-
единительной гласной «о», например «цианокобаламнн», а не «циаикобаламин» и т. д.
Однако в тексте используется термин «оксикобаламин», так как применение в русском
языке наименования «оксокобаламин» придает соединению иное смысловое содер-
жание.
3 Коэффициент распределения — это отношение растворимости витамина в орга-
ническом растворителе к растворимости его в воде.
»/s20* 579
HjNOCCHjCH,
Н2МОССН2ч/7
X Л
си, CH3
CHjCONH,
•N OH N^/ H
В \-CH,CH^ONHi
Н^ОССН/
HNO€CH2CH2 CHa j C^CHjCONH,
N
OH
CHj
o,
0,055 для оксикобаламина (т. e. последний находится почти целиком в воде)
[35].
Удельное вращение окси кобаламина [а]^ от — 15 до — 20° (Н2О);
максимумы поглощения по сравнению с максимумами цианокобаламина сме-
щены в коротковолновую область [26,32,361:270—277 нм (E/tt 137), 352 нм
(Е{'ем 150), 500 и 530 нм (EJ’cm 56); их точное положение зависит от кон-
центрации водородных ионов.
Водный раствор оксикобаламина ведет себя как слабое основание,
имеющее pH около 9. Оксикобаламин (а) в водном растворе в зависимости
от pH может находиться в форме аквокобаламина (б)
Н+
RCo—[(RCo—ОН) Н]+.
а б
Аквокобаламин образует с некоторыми анионами ионные соединения
[26, 37], которые можно рассматривать как соединения, образованные по
типу солей. Аквокобаламин менее устойчив, чем цианокобаламин, осо-
бенно в щелочном растворе.
Помимо оксикобаламина, в животных тканях находится кофермент-
кобаламин [38], в виде которого витамин В12 преимущественно и содержит-
ся в организме.
5'-Д езоксиаденозил-кобаламин, кофермент-кобаламин—
кофермент витамина В12, содержащий вместо циангруппы цианокобаламина
5-дезоксиаденозильный лиганд, — впервые выделен из Clostridium teta-
nomorphum и Bacterium propionicum shermanii [39] (формулу и свойства
см. на с. 607). В виде этого кофермента витамин В12 проявляет свое
биокаталитическое действие и осуществляет метаболические функции.
5-Дезоксиаденозильный лиганд кофермента менее прочно связан с
атомом кобальта, чем нуклеотидный диметилбензимидазольный лиганд
цианокобаламина; он отщепляется при более мягком кислотном гидролизе.
ПРОДУКТЫ ЧАСТИЧНОГО РАСЩЕПЛЕНИЯ2ЦИАНОКОБАЛАМИНА
Цианокобаламин при воздействии различных химических реагентов
частично расщепляется с образованием более простых корриновых произ-
водных — карбоновых кислот и их амидов, причем кобальт у этих соеди-
580
нений остается треквалентным. Для этих структур типа III—VIII
(jjROCCHjCH,.
(a) RQCCH.y'pj
сн/
сн;
(fJROCCH,
(/) r'occh2ch3
сн,
CH,COR(c)
- - СН,СН,СОК (г/)
сн,
сн,
Нз CH,CH,COR(»)
Ш-VIII
(обозначения R и R* см. в тексте)
применяется следующая терминология1:
кобириновая кислота (III; R=R'=OH, гептакарбоновая кислота);-
кобировая кислота, abcdeg-гексамид кобириновой кислоты, фактор 1
Vla (IV; R—NH2, R'—ОН, монокарбоновая кислота);
кобиновая кислота, /-2'-оксипропиламид кобириновой кислоты (V;/;
R=OH, R'=HNCH2CH(OH)CH3, гексакарбоновая кислота);
кобинамид, abcdeg-гексамид кобиновой кислот, /-2-оксипропиламид коби- л
новой кислоты, фактор В, кобаламин без нуклеотида (VI; R=NH2, R'=
HN СН2СН(ОН)СН3);
кобамовая кислота, f-D-рибофуранозил-З'-фосфокобиновая кислота (VII;
R=OH, a R' =
CH2HN—
I
СНСН,
I
о
гексакарбоновая кислота);
кобамид, abcdeg-гексамид кобамовой кислоты, кобаламин без 5,6-диме-
тилбензимидазола (VIII).
Многие из продуктов частичного расщепления витамина В12 выделены
из природных источников. К ним относятся:
фактор Vla — кобировая кислота (IV), выделенная в виде цианида акво-
кобировой кислоты из активного ила полей орошения [40] и др. Синтети-
чески она получается из кобинампда (VI) или цианокобаламина [41]. При
ферментации с Bacterium propionicum shermanii кобировая кислота прев-
ращается в кобинамид (фактор В, VI) и витамин В12 [42];
фактор V(ib) — пентамид кобиновой кислоты (содержит остаток аминопро-
панола), монокарбоновая кислота, наряду с другими моно-, ди- и трикарбо-
новыми кислотами выделенный из ферментных растворов В. propionicum
shermanii [42];
1 Карбоксильные группы или их модификации обозначаются буквами от а до g.
19-69
581
фактор Ib—кобамид дицианид (VIII) (вместо 5,6-диметилбензимида-
зольного лиганда содержит циан-ион) [43J;
фактор В — кобинамид дицианид (VI) 127], образующийся при фермен-
тации В, propionicum shermanii, — монокислотное основание. Синтети-
чески получается из цианокобаламина расщеплением с помощью гидроокиси
церия [44]. Спектр поглощения (в воде): Хмакс 277, 367, 540, 580 нм 145];
фактор В-фосфат—кобинамидфосфат ди цианид, выделенный из продуктов
гидролиза природного Р'-^уанозин-бЭ-Р'^-кобинамидпирофосфата [461;
цианокобаламин-5'-фосфат—кобамид, содержащий 5,6-ди мети л бензимид-
азол с дополнительной фосфатной группой при первичной гидроксильной
группе положения 5', выделен из ферментной жидкости В. propionicum
shermanii [47].
ХИМИЧЕСКИЕ СВОЙСТВА КОБАЛАМИНОВ
При нагревании кристаллического цианокобаламина при 100° С про-
исходит очень медленное разложение. Нагревание его водных растворов
в пределах pH 4—7 практически не вызывает разложения; в очень малых
концентрациях (0,2—10 мг в 1 мл) при pH 2 происходит медленная потеря
активности, а в щелочной среде, при pH 9,— быстрое расщепление (около
90% за сутки). Уже при хроматографировании на бумаге наблюдается час-
тичное отщепление циангруппы с образованием пятна оксикобаламина.
Растворы цианокобаламина неустойчивы к свету [8, 91; фотолиз сопровож-
дается отщеплением циан-иона.
При мягком гидролизе цианокобаламина разбавленной соляной или
фосфорной кислотой различной концентрации происходит постепенное
частичное дезаминирование и образование моно-, ди-, три-, тетра-, пента-,
гекса- и гептакарбоновых кислот с сохранением нуклеотида [48]. Кратко-
временный гидролиз концентрированной соляной или хлорной кислотой
при 65° С в контролируемых условиях приводит к отщеплению нуклеотида
(образование кобинамида, фактора В, VI), а затем аминопропанола (образо-
вание кобировой кислоты, фактора Vla, IV) [27, 49], причем все другие
амидные группировки остаются неизменными. Продолжительный гидролиз
с соляной кислотой средней крепости приводит к образованию безнуклео-
тидных моно—гексакарбоновых кислот (кобировой кислоты и др.) (соответ-
ственно гекса—моноамидов) и в конечном итоге к кобириновой кислоте, геп-
такарбоновой кислоте (III).
Жесткий щелочной гидролиз цианокобаламина сопровождается отщеп-
лением 5,6-диметилбензимидазола и образованием дилактампента- и мо-
нолактамгексакарбоновых кислот, содержащих весь координационносвя-
занный кобальт [19, 50].
При окислении перекисью водорода в кислом растворе получаются
продукты, обладающие антагонистическими свойствами для Lactobacillus
leichmannii [51] в отличие от ростового действия для Euglena gracilis [52].
При окислении витамина В12 марганцовокислым калием отщепляется си-
нильная кислота.
Хлор обесцвечивает раствор цианокобаламина, однако другие галогены
(Br, J) не дают такой реакции [53]. Промежуточный продукт хлорирования
содержит 3,8% хлора и может быть выделен в виде фиолетового кристалли-
ческого вещества [53].
При действии одним эквивалентом хлора или хлорамина Т на витамин
В12 (I) при pH 4 происходит окислительная циклизация (соединение IX) с
последующей реакцией замещения и образованием стабильного цикличес-
кого лактона по кольцу В — абск^-пентамид-с-лактона а-(5,6-диметил-
бензпмидазолил)-8-оксикобамовой кислоты (X) [5] и лишь последующее
хлорирование приводит к замещению в положении 10 с образованием лак-
тона 10-хлорвитамина В12 (XI) [5, 53].
582
При непродолжительном (10 мин) окислении кислородом воздуха вита?
мина В12 (I) в горячем 0,1 н. щелочном растворе образуется дегидровитамйЦ
В12 (XII) [5] — afafeg-пентамид-с-лактам а-(5,6-диметилбензимидазолил)-
8-аминокобамовой кислоты
Образование лактама происходит по положению 7,8 кольца В; он имеет
цис-конфигурацию. В мягких условиях хлорирования дегидровитамина
В12 происходит замещение атомом хлора в положение 10 с образованием
10-хлордегидровитамина В12 — ahd^-пентамид-с-лактама а-(5,6-диметил-
бензимидазолил)г8-амино-10-хлоркобамовой кислоты (XIII).
При каталитическом гидрировании с платиновым катализатором циано-
кобаламин присоединяет 5 атомов водорода и отщепляет метиламин, причем
в остающейся части молекулы трехвалентный кобальт переходит в двух'?
валентный (32, 54] с образованием витамина B12r (XIV)
„ 1H]/Pt [Н]
RCo[III]—CN --------RCo [II]----> RCo[III] H t—>RCo[I] H+.
-ch3nh2
Цианокобаламин Витамин Bllr Витамин B125
I XIV XV
В качестве продуктов восстановления циано кобаламина (I) известны
два вещества — витамины В12. (XIV) и Вш (XV). Первый получают при
восстановлении витамина В12 цинком в растворе хлористого аммония [55,
56], едкого натра или уксусной кислоты либо хлористым оловом [57] без
доступа воздуха [29]. Так как витамин В12г диамагнитен, возможно, что он
содержит одновалентный кобальт; это подтверждается некоторыми титро-
метрическими данными [57]. По другим титрометрическим и полярографи-
ческим данным [25, 58, 59], кобальт в этом соединении двухвалентен, о
чем свидетельствует также спектр электронного парамагнитного резонанса
[60]. Витамин В12г, по-видимому, является продуктом одноэлектронного
восстановления цианокобаламина и содержит двухвалентный кобальт
[61]; он выделен в твердом состоянии [62].
При более длительном восстановлении, а также при использовании
более сильных восстановителей, таких, как ацетат хрома при pH 9,5 [24,
19* 583
56] или боргидрид натрия, получают продукт двухэлектронного восстанов-
ления цианокобаламина — витамин В12? (XV) (24, 56, 63, 64—66]. В при-
веденном выше уравнении структура витамина BI2v (XV) представлена в
виде двух форм, причем равновесие, по-видимому, в значительной степени
сдвинуто в сторону одновалентного кобальта [67].
Витамин B12s устойчив в водной среде в отсутствие кислорода
воздуха. На воздухе он очень быстро переходит в окси(акво)кобаламин
(32, 54]. Атом кобальта при восстановлении приобретает нуклеофильный
характер и реагируете соединениями, содержащими электрофильный центр.
При обработке витамина В12л бромцианом образуется витамин В12 [68].
При разложении цианокобаламина в щелочном растворе образуются зеле-
новато-коричневые продукты, имеющие спектр поглощения, аналогичный
спектрам витаминов В12г и B12i, представляющие собой восстановленные
соединения кобальта [69].
Водный раствор витамина В12г имеет спектр поглощения с максимумами
при 312,5, 405 и 473 нм [56, 58], а раствор витамина B12s — при 385, 460,
554, 680 и 800 нм [57, 58]
При гидрировании в нормальной соляной кислоте при 100° С витамин
В12 испытывает более глубокие изменения необратимого характера [53].
Цианокобаламин с синильной кислотой в щелочном растворе образует
продукт присоединения, обладающий пурпурной окраской,— дицианокоба-
ламин (32, 33] (см. с. 592). В результате замещения циангруппы основаниями
цианокобаламин способен образовывать комплексы с аминами и аминокис-
лотами, так называемые кобалихромы [37].
Цианокобаламин дает различные производные при замещении циангруп-
пы другими лигандами, например ОН', NO", SO", Cl", Вт", SCN~ и пр.,
образуя оксикобаламин, нитритокобаламин, сульфатокобаламин, хлоро-
кобаламин, бромокобаламин, тиоцианатокобаламин и т. п. Все эти произ-
водные в присутствии циан-ионов вновь превращаются в цианокобаламин
(57]. Механизм реакции замещения подробно исследован [26, 37, 70].
Цианокобаламин (I) в водном растворе при мягком гидролизе [71] или
фотолитически [72] превращается в оксикобаламин (II). Оксикобаламин
образуется из цианокобаламина при его каталитическом гидрировании с
платиновым катализатором через промежуточно образующийся витамин В12г
с последующим окислением кислородом воздуха; при этом первоначально
образовавшаяся светло-коричневая окраска переходит в фиолетово-крас-
ную [26, 32, 55, 73—77]. Он же получается при восстановлении цианокоба-
ламина гидросульфитом натрия [55], метабисульфитом натрия в уксусной
кислоте [78], цинковой пылью в растворе хлористого аммония [55, 79] с
последующим воздействием кислорода воздуха
[HJ; [О]
RCo [III]—CN -----* RCo [III]— ОН.
HCN
При действии на оксикобаламин (витамины В12а, В12Ь и B12d) синильной
кислотой происходит его превращение в цианокобаламин (I) 113, 26, 55,
80].
Если при этом применить синильную кислоту с изотопным атомом угле-
рода 14С, то образуется 14СЫ-кобаламин. Витамин В12 с меченым атомом
60Со получается биосинтезом при прибавлении радиоактивного кобальта
в культуральную жидкость Streptomyces griseus [81, 82].
Цианокобаламин подвергается алкилированию диметилсульфатом, при-
чем метилирование идет по N(3) бензимидазольного цикла с разрывом связи
N(3)—Со и образованием четвертичного 3,5,6-триметилбензимидазолийко-
бамида дицианида [83].
584
Кобинамид, фактор В (VI) в условиях кислого катализа —при обработке
смесью метилового спирта, фтористого водорода и хлористого цинка — ис-
пытывает N, О-ацильную перегруппировку с образованием р-аминоизопро-
пилового эфира кобировой кислоты (XVI); продукт его ацилирования по
аминогруппе гидролизуется пиперидиновой водой в кобировую кислоту
(IV). В присутствии основания происходит обратная О, N-ацильная пере-
группировка эфира (XVI) в кобинамид (VI) [41].
QCONHCH2CHOH QCOOCHCH2NH2
СН3 СНз
VI XVI
Q — остаток кобировой кислоты.
Для определения витамина Bj2 известны физические и химические мето-
ды, основанные на спектроскопическом и колориметрическом определении
цианокобаламина, на расщеплении его молекулы и определении циан-
иона и 5,6-диметилбензимидазола или на определении по дицианидному
комплексу [84—86].
СТРОЕНИЕ ЦИАНОКОБАЛАМИНА
В кристаллическом виде цианокобаламин впервые выделили из печени
в 1948 г. почти одновременно Рикес и Фолькерс с сотр. [6] и Смит [87].
При установлении строения цианокобаламина химическим путем изучена
только меньшая часть молекулы; строение остальной части молекулы опре-
делено рентгеноструктурным методом анализа.
В молекуле цианокобаламина содержится около 4,5% кобальта, т. е.
один атом, что доказано спектроскопически [6, 87]. Присутствие в цианоко-
баламине циан-иона установлено на основании образования и выделения
цианистого водорода при нагревании витамина в растворе соляной или ща-
велевой кислоты [13]. Выделения синильной кислоты не происходит, если
гидролиз производить в растворе серной кислоты, имеющей малую склон-
ность к образованию координационной связи.
При кислотном гидролитическом расщеплении витамина BJ2 при 100° С
в 20 %-ной соляной кислоте в запаянной трубке, помимо отщепления синиль-
ной кислоты, выделяется одна молекула фосфорной кислоты [11], шесть
молекул аммиака [11, 27, 88] и вещество, обнаруженное в виде пятнана
бумажной хроматограмме, дающее положительную реакцию с нингидрином
[11, 88, 89].
Большие трудности представила идентификация этого так называемого
«нингидрин-реагирующего» вещества, отделяемого хроматографией на бу-
маге от других продуктов кислотного гидролиза витамина В12. Этот алифа-
тический осколок не являлся аминокислотой, и его нельзя было отделить
от искусственной смеси с 2-амннопропанолом-1. Однако хроматографией
на бумаге его нельзя было отделить и от 1-аминопропанола-2 [88]. Так как
2-аминопропанол-1 при окислении дает а-аланин, а «нингидрин-реагирую-
щее» вещество аланина не дает, то вопрос об их идентичности отпал.
«Нингидрин-реагирующее» вещество оказалось £>?-1-ампнопропанолом-2
(XVII), что было установлено структурным анализом и синтезом (схема
129) [90]. Из кислотного гидролизата цианокобаламина Dg.-1-аминопропа-
нол-2 (XVII) [90, 91] был выделен в виде дибензоата с т. пл. 73—74° С и
[a]D —72 ± 1°. Строение свободного амина доказано реакцией его расщеп-
ления йодной кислотой до ацетальдегида и формальдегида. Установлено
[88], что в цианокобаламине присутствует только одна молекула
аминопропанола-2.
Другим осколком молекулы цианокобаламина в результате жесткого
кислотного гидролиза в 6 н. соляной кислоте при 150° С в течение 20 ч [92]
585
оказался 5,6-диметилбензимидазол (XVIII) с т. пл. 205—206° С [92—94],
идентифицированный по спектру поглощения [93] и по его пикрату (с т.
пл. 273—275° С). Строение 5,6-диметилбензимидазола доказано его расщеп-
лением бензоилхлоридом в 4,5-диметил-1,2-дибензоиламинобензол (XX)
и синтезом из 4,5-диметил-1,2-диаминобензола (XIX) и муравьиной кис-
лоты [92] (схема 129).
Схема 129
Продукты расщепления цианокобаламина
При несколько более мягком кислотном гидролизе цианокобаламина —
в 6 н. соляной кислоте при 120° С в течение 8 ч— выделено новое вещество
(XXI) [95]. С целью установления его структуры методом хроматографии
на бумаге было охарактеризовано ультрафиолетовым спектром 22 заме-
щенных бензимидазола, специально синтезированных в качестве возмож-
ных производных 5,6-диметилбензимидазола, и изучено их поведение. Этими
методами выяснено, что новое вещество представляет собой l-a-D-рибофура-
нозил-5,6-диметилбензимидазол, а-рибазол (XXI); по пикрату (т. пл. 212—
224° С; [а]р + 9,1°) оно идентично с синтетическим [94 , 961.
а-Конфигурация гликозидного атома углерода рибазола (XXI) [97]
установлена на основе сравнения оптической активности а- и Р-рибазолов
[98]. Доказательство присутствия рибозного остатка фураноидной струк-
туры получено на основании окисления пикрата а-рибазола (XXI) йодной
кислотой, которая реагировала (1 моль) с образованием диальдегида пикрата
(XXIII) с т. пл. 180—185° С [97]. Для окисления соединения пираноидной
структуры потребовалось бы 2 моля йодной кислоты. При окислении синте-
тического 1-р-Д-рибофуранозил-5,6-диметилбензимидазола (Р-рибазола) в
виде его пикрата получен диальдегид пикрат (XXIV) с т. пл. 141—144° С,
отличающийся от соответствующего производного а-рибазола.
hCHOOHCcHjOh
R ХУ IН
XXIII
КСНООНССЦОН
XXIV
586
Гликозидная С—N-связь а-рибазола (XXI) очень прочна и не гидроли-
зуется при кипячении с соляной кислотой при 100° С [99]. Ее расщепление
происходит только при 120° С в отличие от легко гидролизующихся пури-
новых нуклеозидов и от пиримидовых нуклеозидов, которые разбавлен-
ными кислотами не гидролизуются [97], а более концентрированными кис-
лотами расщепляются (в углеводной части молекулы).
При еще более мягком кислотном гидролизе цианокобаламина — в
20%-ной соляной кислоте при 100е С 11001 или в н. соляной кислоте в тече-
ние часа [101] — выделен фосфорный эфир а-рибазола (XXII) [100]. Так
как этот фосфорный эфир а-рибазола, имеющий т. пл. 240—241е С (с разл.),
не обнаруживает а-гликольной группировки, способной расщепляться с
йодной кислотой, то, по-видимому, эфир образован по 2- или З'-гидроксиль-
ным группам [1001. При кислотном или щелочном гидролизе фосфорный
остаток может мигрировать, вероятно, между положениями 2' и 3', и
поэтому в полученном соединении эфирная связь не обязательно должна
быть образована с тем же гидроксилом, что и в витамине В12 1102,
103].
В дальнейшем было установлено, что а-рибазолфосфат (XXII) является
З'-фосфатом [104], доказательством чего служит более низкое значение
Rf его пятна на бумажной хроматограмме, что аналогично более низ-
кому значению Rf для аденозин-З'-фосфата по сравнению с аденозин-2'-
фосфатом; окончательное доказательство строения соединения XXII как
З'-фосфата получено рентгеноструктурным анализом [105].
При кратковременном нагревании с концёнтрированной соляной кислотой
при 60° С цианокобаламин (I) отщепляет 5,6-диметилбензимидазолнуклео-
тид (XXII) и образует фактор В, кобинамид (VI), сохраняющий все амидные
группировки неизменными.
HjNOCCHjCHs.
НзЮССНз
СН/ Y-N
СНэ'
СНз сНз
CHjCONH,
—СЦСНзСОМНз
Тб
CN
"7\
г в
Со
ю,
Н2ЬЮССНг-Ф
HNOCCH2CH3
СН2
СН3СНОН
•СИ,
"СНз
Нз CHaCHjCONH,
см N'
. С 12)
Ди/
СН3
VI
В отличие от нейтрального цианокобаламина кобинамид электрофоре-
тически ведет себя как однокислотное основание [27].
Цианокобаламин имеет полиамидный характер, что установлено по вы-
делению аммиака (6 молей) при кислом гидролизе [11, 27, 88] и данным ин-
фракрасного спектра. При анализе продуктов гидролиза цианокобаламина
в кислых, нейтральных и щелочных растворах эффективно применен метод
электрофореза и хроматографического разделения. Электрофорез на бу-
маге при pH 6,5 и 10 позволил разделить продукты расщепления на отдель-
ные соединения по их ионным зарядам. Ступенчатый гидролиз в холодной
разбавленной соляной кислоте показывает присутствие трех амидных групп,
относящихся, по-видимому, к боковым цепям пропионовых кислот. Полу-
чены три одно-, три двух-, одна трех- и одна четырехосновная кислоты,
содержащие нуклеотидную часть молекулы витамина; эти кислоты были
превращены с хлоругольным эфиром в смешанные ангидриды и затем
с аммиаком в цианокобаламин [27].
В условиях щелочного гидролиза, протекающего с отщеплением 5,6-
диметилбензимидазолнуклеотида, обнаружено шесть первичных амидных
587
групп, из которых три менее лабильны к щелочам и кислотам; они, по-ви-
димому, относятся к боковым цепям уксусной кислоты. Установлено при-
сутствие одной вторичной амидной группы, образуемой аминопропанолом,
H2NCH2CH(CH3)OH [271.
При гидролизе цианокобаламина, помимо идентифицированных выше
продуктов расщепления, остается красный аморфный остаток, который
содержит весь координационно связанный кобальт. В результате кислого
гидролиза витамина горячим раствором соляной кислоты средней крепости
образуется красный аморфный остаток, представляющий собой смесь кислот,
содержащих от одной до семи карбоксильных групп [27], которые могут быть
разделены электрофорезом. Гептакарбоновая кислота, не содержащая 5,6-
диметилбензимидазолиуклеотида и Dg-l-аминопропанола-2, представляет
собой кобириновую кислоту (III, схема 130), выделенную в виде дихло-
рида. То обстоятельство, что отщепление Dg-]-аминопропанола-2 проис-
ходит после отщепления нуклеотида, а также отсутствие основных групп
в красном кобальтсодержащем продукте гидролиза дает основание заклю-
чить, что аминопропиловый спирт этерифицирован фосфорной кислотой и
соединен амидной связью с остальной молекулой [27].
Таким образом, на основании химических данных и физических методов
исследования можно считать доказанным, что в молекулу цианокобала-
мина включаются фосфорная кислота, D-рибоза и 5,6-диметилбензимид-
азол (XVIII), образующие фосфорный эфир 1-а-£)-рибофуранозил-5,6-
диметилбензимидазола (XXII); этот нуклеотид связан фосфорноэфирной
связью с 1-аминопропанолом-2 (XVII), который в свою очередь связан амид-
ной связью с карбоксильной группой остальной части молекулы, содержа-
щей кобальтовый комплекс, образующий координационную связь с цикли-
ческим атомом азота бензимидазола и с нитрильной группой.
При дальнейших исследованиях кобальтсодержащего комплекса из
гидролизата цианокобаламина, освобожденного от Dg-l-аминопропанола-2,
нуклеозида бензимидазола и фосфорной кислоты, после окисления 5%-ным
раствором марганцовокислого калия было получено девять различных ор-
ганических кислот. Из них шесть идентифицированы как уксусная, щаве-
левая, янтарная, метил янтарная, диметилмалоновая и 2,2-диметил-З-кар-
боксиадипиновая (XXV) кислоты (схема 130) [53, 106]; эти кислоты могут
образоваться в результате окислительного расщепления циклической сис-
темы гептакарбоновой кислоты (III).
Нагревание цианокобаламина с едким натром приводит к его расщепле-
нию, причем в летучих продуктах обнаруживаются пиррольные соедине-
ния (образование красной окраски с соляной кислотой и п-диметиламино-
бензальдегидом), что дало основание сделать заключение о присутствии в
молекуле витамина макроциклической пиррольной структуры, возможно,
типа порфирина или пигмента желчи [7, 72].
При окислительном расщеплении цианокобаламина (I) бихроматом нат-
рия в уксуснокислом растворе выделен амид 3,3-диметил-2,5-диоксопирро-
лидин-4-пропионовой кислоты (XXVI) [1071 (отвечающий кольцу С молекулы
цианокобаламина), а при гидролитическом расщеплении и окислении би-
хроматом натрия — 3,3-диметил-2,5-диоксопирролидин-4-пропионовая кис-
лота (XXVII) [108] с т. пл. 151—152° С и лактон ее 4-оксипроизводного
(XXVIII) с т. пл. 143—144° С, по-видимому, образующийся из кислоты
при ее окислении [108] (схема 130). При гидролизе амида (XXVI) с 6 н.
соляной кислотой в течение 20 ч получается 2,2-диметил-З-карбоксиадипи-
новая кислота (XXIX) с т. пл. 152—153° С [107, 108], при сравнении иден-
тичная с синтетической [108], которая может быть обратно превращена в
амид (XXVI) при действии аммиака на ее хлорангидрид [107]. Эта же 2,2-
диметил-3-карбоксиадипиновая кислота (XXIX) образуется не только при
окислении цианокобаламина марганцовокислым калием [106], но и при
гидролизе соединения XXVII, в которое аммонийная соль кислоты (XXIX)
может быть обратно превращена при пиролизе [108].
588
Схема 130
Реакции расщепления цнанокобаламина и синтез замещенного дноксопнрролидина
Цианоио-
каламии*
I
HOOCCHi
НООССН2СН
HOOGCH,
СН3'
сн;
HOOCCHjCH, СНа СН,СН,СООН
СНз СН
СН,СООН
CHjCHsCOOlI
сн3
снэ
СНз СН,
CHj-l——[-CHjCHjCONH, сн,—I—
N^O
н
XXVI
N О
н
XXVII
’ NH;
пиролиз
СН.
СН3СН
COOR
XXX
сн3ссюн
—ноос-соон
— НООССН2СН3СООН
— НООССЦСНСООН
сн3
HOOCCC-GOOH
СН, СН,
--------•- НООС-С —(JHCHjCHjCOOH
СН3СН3СООН
XXV
СН.О
СН/НСРОН сн,-]——_
сг^^о
н ц
f-7 XXVIII
Пиролиз
CHj-C ---СНСН,СН2СООН
СООН СООН
XXIX
+CtCOCH2CH2COOR-
XXXI
СНз
CHjCCOCHjCHjCOOR
COOR
XXXII
<НС°°Н
СТ > CHjCOOH
-^СНСООН
CHf
СН3С-----с—сн,
соон соон
XXXIV
(Гидролиз и
лактонизация
сн3 он
С Н 3С— CCHjCHjCOOR
COORXCN
хххш
о
Лактон 3,3-диметил-2,5-диоксо-4-оксипирролидин-4-пропионовой кис-
лоты (XXVIII) при действии едкого кали расщепляется с образованием
янтарной и изомасляной кислот; при перегонке лактон дает реакцию на
пиррол 1108]. Для получения лактона (XXVIII) исходят из этилового эфира
изомасляной кислоты (XXX) и хлорангидрида моноянтарного эфира
(XXXI), которые под влиянием этилата натрия конденсируют в соединение
XXXII, циангидриновым синтезом превращаемое в нитрил (ХХХШ):
после его гидролиза и лактонизации получают лактон дикарбоновой кис-
лоты (XXXIV), пиролиз аммонийной соли которого и приводит к получе-
нию лактона (XXVIII) [108].
Щелочной гидролиз цианокобаламина характеризуется многими осо-
бенностями по сравнению с кислотным. При энергичном щелочном гидро-
лизе кобальтсодержащего комплекса ионогенносвязанный кобальт отще-
пить не удалось [109]; это дает основание полагать, что атом кобальта в
молекуле витамина связан координационно с атомом азота в положении 3
молекулы бензимидазола [ПО].
Из гидролизата цианокобаламина, полученного в результате гидролиза
с 30%-ной щелочью при температуре 150° С в течение часа, электрофорезом
на бумаге были выделены безнуклеотидные пяти- и шестиосновные карбо-
новые кислоты. Что особенно ценно, была получена кристаллическая опти-
чески активная кобальтсодержащая лактамгексакарбоновая кислота
(XXXV) [19], которая очищена на ионообменных смолах [111]. В кристал-
лическом виде кобальтсодержащая лактамгексакарбоновая кислота была
589
получена в хлоридцианидной форме. Спектр поглощения ультрафиолетового
и видимого света для нее характеризуется теми же максимумами, что и для
цианокобаламина, кроме отсутствия максимума при 278 нм, характерного
для ди метил бенз и мидазол а.
Показательно, что конечным продуктом щелочного гидролиза являет-
ся не гептакарбоновая кислота (как при кислом гидролизе), а лактамгек-
сакарбоновая кислота (XXXV), так как одна алифатическая цепь с карбок-
сильной группой замыкается в устойчивое к щелочам лактамное кольцо.
Пентакарбоновая кислота, образующаяся при щелочном гидролизе [191,
по-видимому, представляет собой а-моноамид лактамгексакарбоновой
кислоты (см. формулу XXXV) или соответствующий лактам этой кисло-
ты, связанный амьдной группой в положении 3 кольца А (XXXVI) [501.
Ходжкин, Пикуорт, Робертсон с сотр. [171 провели исчерпывающий рент-
геноструктурный анализ лактамгексакарбоновой кислоты (XXXV). Ха-
рактерной особенностью этого соединения оказалась кольцевая система
вокруг атома кобальта, состоящая из частично гидрированных четырех
5 пиррольных колец, соединенных в макроцикл. Эта система имеет сходство
с порфином, однако в ней отсутствует один лезо-углеродный атом, и поэтому
г два гетероцикла соединены непосредственно прямой Са—Са-связью.
^ Боковые цепи, содержащие два остатка уксусной и четыре остатка пропио-
' новой кислот, расположены в ^-положениях пятичленных гетероциклов,
что также характерно для уропорфирина III.
Атом кобальта связан с циангруппой и атомом хлора, которые распо-
ложены перпендикулярно основной плоскости молекулы. Он также связан
с четырьмя атомами азота гетероциклических колец, находящихся в одной
плоскости с ним. Все внутренние атомы большого кольца лежат в той же
плоскости, что характерно для наличия сопряженной системы двойных
связей. Все ₽, 0'-заместители гетероциклов не планарны с плоскостью коль-
ца; это указывает на то, что ₽, Р'-углеродные атомы пиррольного кольца
гидрированы [1011. Молекула лактамгексакарбоновой кислоты характери-
зуется наличием десяти центров асимметрии.
Новый гетероцикл, образующийся за счет срощенного лактамного коль-
ца с кольцом В лактамгексакарбоновой кислоты (XXXV), по-видимому,
образуется из промежуточного продукта с гидроксильной группой (кото-
рую несет атом углерода положения 8) с последующим замыканием цикла
через амидный остаток и гидроксильную группу [16, 191. Промежуточный
продукт возникает в результате окисления кислородом воздуха, сопровож-
дающего щелочной гидролиз цианокобаламина. Интересно отметить, что
реакция окисления катализируется кобальтом расщепленной часта моле-
кулы [501.
Образование лактамного кольца, по-видимому, первоначально про-
исходит при воздействии щелочи в присутствии кислорода воздуха на циа-
нокобаламин. Получающийся дегидровитамин В12 с лактамным кольцом,
образованным по атомам углерода положения 7 и 8, только затем гидроли-
тически расщепляется в лактамгексакарбоновую кислоту (XXXV) [51.
590
По форме макроцикла цианокобаламина и лактамгексакарбоновой
кислоты, имеющих планарную структуру [4, 17], можно сделать заклю-
чение, что хромофорная система, обусловливающая красную окраску этих
соединений, состоит из полиеновой сопряженной системы [16—18]. Рас-
положение системы двойных связей установлено на основании исследований
по хлорированию витамина Bj2 N-хлорамидами кислот [53, 89], витамина В12,
дегидровитамина В12 и лактамгексакарбоновой кислоты — хлор-
амином Т, а также на основании данных спектрофотометрических опреде-
лений [5]. Установлено, что сопряженная система циано кобаламина и лак-
тамгексакарбоновой кислоты состоит из шести двойных связей [4, 5].
В последнем соединении сопряженная система в зависимости от обстоятельств
может занимать различное положение.
Таким образом, по данным рентгеноструктурных исследований и хими-
ческому поведению была установлена полная структура и пространствен-
ная формула лактамгексакарбоновой кислоты (XXXV), содержащая час-
тично гидрированную тетрапиррольную циклическую систему и координа-
ционно связанный кобальт [4, 5].
На основании рентгеноструктурных кристаллографических исследо-
ваний сначала цианокобаламина, а затем лактамгексакарбоновой кислоты,
проводившихся в течение 7 лет Ходжкин-Кроуфут с сотр. в Оксфорде [4,
14, 17, 105, 112—114], и материалов по химическому изучению витами-
на В12 Ходжкин, Тодд и Джонсон с сотр. установили структурную формулу
цианокобаламина (I), приведенную выше [4, 5, 17—19, 101]. Эту струк-
туру окончательно подтвердили полным синтезом витамина В12 в 1972 г.
Вудвард с сотр. и Эшенмозер с сотр. (см. ниже).
ПРОИЗВОДНЫЕ ЦИАНОКОБАЛАМИНА С ДРУГИМИ ЛИГАНДАМИ
(АНИОНАМИ)
Помимо цианокобаламина и окси кобаламина, имеющих в качестве
аниона циан- или оксигруппы, из природных источников или синтетически
получены кобаламины с другими анионами1. Такие кобаламины представ-
ляют собой соединения красного цвета; они проявляют биологическую ак-
тивность, подобную активности витамина В12 на животных [32], по-види-
мому, вследствие их легкой способности превращаться в витамин В12 в
процессе обмена веществ.
Нитритокобаламин, а-(5,6-диметилбензимидазолил)нитрито-
кобамид, или витамин В12с, является комплексным кобальтовым произ1-
водным, содержащим координационно связанную нитритогруппу
(ONO-) [26, 115]. Он представляет собой фиолетово-красные крис-
таллы. Молекула витамина В12с значительно более окислена, чем цианоко-
баламина [100]. При кислотном гидролитическом расщеплении он дает тот
же 5,6-диметилбензимидазол, что и цианокобаламин.
Удельное вращение нптритокобаламина [сИмзв —50° (Н2О); спектр его
поглощения смещен в коротковолновую область к 412 нм по сравнению со
спектрами оксикобаламина и цианокобаламина.
Витамин В12с выделен [33] из культуральной жидкости Streptomyces
griseus наряду с витамином В12Ь (оксикобаламином). Он получен также из
окси кобаламина (витамина В12Ь) обработкой азотистой кислотой [26,115,116].
С у л ь ф а т о к о б а л а м и н, а-(5,6-диметилбензимидазолил) суль-
фатокобамид — содержит SO3-rpynny (вместо CN-группы). Он получается
при действии сернистой кислоты на цианокобалампн [26, 115]; серусодер-
жащая группа в сульфатокобаламине по своим свойствам не является
сульфатной, сульфитной или сульфидной [117].
1 Наименования производных витамина В12 получаются в результате присоедине-
ния к основной части молекулы витамина (катиону) без аксиально связанной с ней
циангруппы (аниона), называемой «кобаламином», названия другого лиганда, вводи-
мого в молекулу [26].
591
Хлорокобаламин получается при обработке раствора вита-
мина В12 соляной кислотой [26, 1151.
Бромокобаламин получается из цианокобаламина и бромисто-
водородной кислоты [115].
Т иоцианатокобаламин получается из цианокобаламина
при действии SCN-ионов [1181, причем роданистая группа в нем более
прочно связана [1171, чем у других производных.
Дицианокобаламин [(RCo—CN)CN]_— продукт присоеди-
нения синильной кислоты к цианокобаламину, образуется при действии
циан-иона на цианокобаламин при pH выше 10; реакция характеризуется
изменением окраски раствора до яркого темно-красного цвета [1101. При
этой обратимой реакции происходит образование связи комплекса с новым
циан-ионом [80], причем в растворах оснований циан-ион вытесняет нукле-
отидный лиганд [37]. Соединение неустойчиво в слабокислой среде.
К молекуле цианокобаламина возможно присоединение двух циан-
ионов [117]. Цианокобаламин образует хорошо кристаллизующийся гекса-
перхлорат с хлорной кислотой [15].
Спектры поглощения производных цианокобаламина [26, 37, 1191
(в воде) представлены в табл. 22.
Таблица 22
Спектры поглощения производных цнанокобаламина
Производные цнанокобаламина ^макс* ®“ Л % BlcJ,, соответственно
Нитритокобаламин (витамин в12с) Сульфатокобаламин Хлорокобаламин Бромокобаламин Днцианокобаламин Продукт реакции витамина В12 и H2S 255—275 , 354 , 412 , 530 272—275 , 352 , 520—530 274—275 , 352—353 , 525—530 274—275 , 352 , 520—530 279, 368, 540, 582 273—275, 352 , 525—530 187, 185—,75 135, 163, 55 142, 174, 59 140, 168, 59 140, 170, 58
ПСЕВДОВИТАМИНЫ Bi2
В природных источниках, помимо витамина В12, встречаются родствен-
ные ему соединения, так называемые псевдовитамины В12, которые проду-
цируются в анаэробных условиях микроорганизмами, обитающими в ки-
шечнике жвачных и других животных (собственно псевдовитамин В12),
или микроорганизмами гнилостного шлама канализационных отбросов
(многие другие псевдовитамины). Псевдовитамины В12 являются ростовыми
факторами для некоторых микроорганизмов (Lactobacillus lactis и leich-
mannii), но они не обладают витаминной активностью для животных и не
излечивают у них пернициозной анемии [1201.
Псевдовитамины В12 имеют ту же основную структурную часть моле-
кулы, что и цианокобаламин, но в отличие от него в качестве аксиального
лиганда содержат не 5,6-дпметилбензимидазолнуклеозид (XXI), а дру-
гие основания. Такими основаниями являются следующие: a-D-рнбофура-
нозил-5-метилбензимидазол (XXXVII) и a-D-рибофуранозилбензимидазол
(XXXVIII), обнаруженные в псевдовитаминах Bj2 гнилостного шлама ка-
нализационных отбросов [121] (они были получены также биосинтетически);
5-оксибензимидазол (XL) в виде а-£)-рибофуранозил-5-оксибензимидазо-
ла (XXXIX), находящегося в структуре фактора III, также выделенного
из гнилостного шлама [87, 122—124]; пуриновая система аденина (XLII)
в виде 7а-аденозина (XLI), собственно псевдовитамина В12 [125—127];
система 2-метиладенина (XLV), неизвестного до этого в природе, в виде
7а-2-метиладенозина, фактора A (XLIV7) [128—130]; продукты их дез-
аминирования — гипоксантин (XLIII) и 2-метилгипоксантин (XLVI) в
592
HOCHs о н
XXI
нощ. о н
XXXVII
> НОСН2 о н
XXXVIII
XLIV
XLVU
XLVIII
593
виде соответствующих 7а-нуклеозидов — фактора G и фактора Н [131—
132];система 2-метилмеркаптоаденина (XLVIII) в виде 7а-2-метилмеркап-
тоаденозина (XLVII) [133], а также система гуанина (L) в виде 7а-гуано-
зина (XLIX) [134]. Кроме того, продукт ферментации Nacardia—фак-
тор С [46] — содержит гуанин в виде нуклеотида, обычного для всех
природных нуклеотидов (кроме производных кобамидов) 9|3-строения с допол-
нительной фосфатной группой (LI), однако вследствие стерических затруд-
нений гуаниновое основание по 7-N не образует координационной связи
с кобальтом. Фактор С представляет собой 9₽-гуанозинил-5'-пирофосфо-
кобинамид дицианид [46, 135].
При кислотном деструктивном гидролизе с нормальной соляной кис-
лотой при 100° С псевдовитамин В12 дает аммиак, фосфорную кислоту,
D-1-аминопропанол-2, аденин (XLII) и гипоксантин (XLIII) [125, 136].
В молекулу псевдовитампна В12 и псевдовитамина B12d, помимо D-1-ами-
нопропанола-2 и фосфорной кислоты, входит также D-рибоза [120, 128].
В более мягких условиях гидролиза получается фосфорная кислота, си-
нильная кислота, фактор В (кобинамид) и 7а-£>-рибофуранозиладенин
(XLI) [137].
Фактор В, кобинамид (VI), представляющий собой красного цвета ко-
бальтовый комплекс основной структурной части молекулы витамина Ви
и псевдовитаминов [27], может подвергаться взаимопревращениям с нукле-
отидными остатками по следующей схеме [119]:
5,6-диметилбен-
зимидазол
нуклеотид
Циаиокобаламин г
I
Фактор В
(кобинамид)
VI
Адениннуклеотид
.... Псевдовитамин В1а
t *~ Псевдовитамин Bi2d,
2-Метиладенин- Фактор А
нуклеотид
Различные псевдовитамины В12, выделенные из природных источников, представлены в табл. 23. Таблица 23 Псевдовитамины В12
Рациональное наименование Различные названия с установ- ленной нл:( предполагаемой идентичностью Спектр поглощения нм
а-Бензимидазолилкобамид Цианид а-(5-Метилбеизимидазолил) - кобамид цианид а-(5-Оксибензимидазолил) кобамид цианид 7а-Аденилкобамид цианид Фактор III Псевдовитамин Вх2, : псевдовитамнн В12ь, цнан-,3-кобаламии, циан-7-коба ламин 295, 361, 518, 550 278 , 361, 518 , 548—550 (Е}%, 130, 204 , 545 , 58 Соответственно)
7а-(2-Метиладенил)кобамид цианид 7а-Г ипоксаитилкобамид цианид 7а-(2-Мети лг ипоксантил) ко- бамид цианид 7а-(2-Метилмеркаптоаденил)- кобамид цианид 7а-Гуанозилкобамид цианид 98-Гуанозил-5-пирофосфоко- бииампд дицианид Фактор А псевдовитамин Bi2d, псевдовитамин Bi2f, псевдовитамин Bi2m, циан-«-кобаламин Фактор G (дезаминированный псевдо- витамин Bi2) Фактор Н (дезаминирован- ный псевдовитамин B12d) В12-фактор (?) Фактор С 278, 359, 358, 276, 361, 518, 516, 540 517, 540 367, 540 548-550 580
594
Обычно псевдовитамины извлекаются из природных источников в виде
смесей, разделение которых представляет значительные трудности. Для
разделения смесей, кажущихся химически однородными, применяется
электрофорез 1138].
Псевдовитамин В12 выделен из кишечника жвачных в кристаллическом
виде 1120]. В сухих фекалиях жвачных его содержание составляет 100—
300 мкг/кг [136].
Витамин В12т (идентичный псевдовитамину B12d) выделен из свиного
экскрета.
В гнилостном шламе канализационных отбросов от общей суммы псев-
довитаминов В12 и цианокобаламина последнего содержится около 30%;
псевдовитамины разделены на многочисленные индивидуальные формы
[40].
При испытании на Lactobacillus leichmanii ростовая активность псев-
довитаминов В12 и B12d составляет соответственно 70 и 20% активности
цианокобаламина [128].
а-(5-Оксибензимидазолил)кобамид цианид (фактор III) при действии со-
ответствующих реагентов образует эфиры по фенольной гидроксильной
группе, такие, как бензиловый, 2-оксиэтиловый, 2,4-динитрофениловый
и др. [139].
ПОЛУЧЕНИЕ ЦИАНО КОБАЛАМИНА
Майнот и Марфи в 1926 г. [140] установили, что в печени и ее экстрак-
тах содержится вещество, которое излечивает тяжелое заболевание — зло-
качественное малокровие, называемое пернициозной (адиссон—бирмеров-
ской) анемией. Однако это активное вещество, оказавшееся витамином
В12, было выделено только в 1948 г. [6, 87].
Выделение витамина В12 связано с большой сложностью из-за его мало-
го содержания в природных продуктах. Из 1 т свежей говяжьей печени мож-
но получить около 28 мг чистого цианокобаламина НО, 141] (при его со-
держании 1 : 1 000 000) путем водно-спиртовой экстракции, освобождения
от белков, адсорбции, хроматографического разделения и с помощью дру-
гих тонких процессов. Из печени, помимо цианокобаламина [9, 10, 89,
1411, выделен и оксикобаламин (В12ь) [9, 142].
Оксикобаламин выделяют из печени свиней или крупного рогатого ско-
та экстракцией спиртом. Из 1 т измельченной печени получают 1 г оксико-
баламина [143].
Из культуральной жидкости и мицелия Streptomyces griseur получены
цианокобаламин [6, 20], оксикобаламин (В12а Ь) [20, 32, 142] (B12d) [33]
и нитритокобаламин (В12с) [33].
Однако единственным продуцентом витамина В12 в природе являются
микроорганизмы, находящиеся в кишечнике животных, в почве, в навозе,
в гнилостном шламе канализационных отбросов и других природных сре-
дах, которые благоприятны для интенсивного развития микроорганизмов.
Для получения витамина В12 известны методы, использующие ил (шлам)
сточных вод, подвергнувшийся аэробным бактериальным процессам [40];
однако наряду с цианокобаламином из шлама извлекаются в значительном
количестве неактивные псевдовитамины В12 и другие, еще не изученные
продукты, очистка от которых чрезвычайно затруднительна.
Для извлечения витамина В12 и псевдовитаминов В12 из гнилостного
шлама первоначально производят горячую экстракцию с последующей
фильтрацией, затем адсорбцию и вымывание в водный раствор (при этом
получаются концентраты 1/1000 первоначального объема) [40]. Из водного
концентрата В12-факторы извлекают фенолом в неполярных растворителях
и затем вновь переводят в водный раствор, получая при этом рубиново-
красные концентраты. После осаждения В12-факторов n-хлорфенолом на
595
кизельгур производят хроматографическое разделение их раствора в н-бу-
тиловом спирте на колонке с целлюлозным порошком и затем выделение
и кристаллизацию отдельных В12-факторов [40] (см. также [144]). Методы
получения цианокобаламина из гнилостного шлама канализационных от-
бросов технически разработаны; из 1 т сухого ила можно получить около
1 г витамина В12 1145, 146]. Предложен непрерывный процесс обработки
шлама с целью извлечения из него витамина BJ2 [147].
Для технического получения витамина В12 используют производство
антибиотиков. Цианокобаламин наряду со стрептомицином продуцируется
актиномицетами и выделяется из мицелия и культуральной жидкости Strep-
tomyces griseus [6, 148] или Streptomyces olivaceus [149]. Так, по одному
из методов после удаления антибиотиков из ферментной жидкости витамин
адсорбируют активированным углем (или ионообменными смолами), из ко-
торого вымывают водным бутиловым спиртом или разбавленной щелочью,
а затем выделяют и очищают с применением органических растворителей
и различных адсорбентов. Ферментативный метод совместного получения
цианокобаламина и антибиотиков с применением актиномицетов имеет не-
большое распространение.
Однако существенные преимущества имеет направленная ферментация
с применением специальных бактерий, которым свойственно накапливать
значительные количества цианокобаламина. Для этой цели применяют
Bacillus megatherium [150], а также Bacterium propionicum [1511, раз-
вивающиеся в анаэробных условиях. В процессе ферментации происходит
образование витамина В12, главным образом в форме его кофермента —
б'-дезоксиаденозилкобаламина [152] (до 80%), который при последующей
обработке раствором цианистого калия превращается в устойчивый циано-
кобаламин [77 ]. Неустойчивый к нагреванию оксикобаламин и некоторые
другие кобамиды в этих условиях также переходят в цианокобаламин.
Содержащийся в ферментном растворе цианокобаламин очищается от
примесей с применением органических растворителей путем разделитель-
ного распределения м^жду двух жидких фаз, например между бензиловым
спиртом и водой [153], фенолом или о-крезолом и водой [154] и другими
операциями [9, 89, 141 ], затем подвергается хроматографированию на оки-
си алюминия и кристаллизации из ацетона или горячей воды. Для очистки
применяют адсорбцию и элюцию на ионообменных смолах [155, 156].
Для получения высокоочищенных препаратов витамина Вг2 используют
выделение из водных растворов в виде кристаллического комплекса циано-
кобаламина с фенолом [157, 158] или резорцином [158] с последующим
разложением. Описаны многочисленные патенты по выделению и очистке
цианокобаламина [159—163].
При культивировании с Bacterium propionicum выход цианокобаламина
составляет 1—3 мг на 1 л питательной среды. При одновременном культи-
вировании с молочнокислыми бактериями выход повышается до 4,3 мг на
1 л культуральной среды. Продуцирование витамина В12 стимулируется
прибавлением солей кобальта [164, 165] и осколка молекулы витамина
В12 — 5,6-диметилбензимидазола.
Кормовой препарат витамина В12 получают микробиологическим синте-
зом с Bacterium propionicum shermannii на углеводных средах — от-
ходах свеклосахарного производства (мелассе). В сухой биомассе содержа-
ние витамина В12 составляет 350—400 мг в 1 кг.
При ферментации с термофильными метанообразующими бактериями
при 55—57° С (при этой температуре подавляется развитие всей посторон-
ней микрофлоры) используют в качестве среды барду ацетоново-бутиловых
и спиртовых заводов, работающих на зерне и мелассе [166]; для повышения
выхода в среду прибавляют метанол [167]. Полученная сухая биомасса
содержит 16—25 мг витамина В12 в 1 кг и биологически неактивные псевдо-
витамины В12, около половины количества от всех кобамидных соедине-
ний.
596
Для аналитического определения цианокобаламина, помимо химиче-
ских, применяется микробиологический метод с культурой Lactobacillus
lactis 1168] и др.
СИНТЕЗ ЦИАНОКОБАЛАМИНА И ФРАГМЕНТОВ ЕГО МОЛЕКУЛЫ
В 1972 г. Р. Вудвард (США) с сотр. и Н. Эшенмозер (Швейцария) с
сотр. [177] осуществили полный синтез цианокобаламина. Синтезированы
отдельные фрагменты его молекулы, такие, как Dg-l-аминопропанол-2,
а-рибазол и его фосфат; проведены исследования по стереонаправлен-
ному синтезу макроциклической корриновой системы со всеми заместите-
лями, определяющими структуру кобириновой кислоты, осуществлен
частичный синтез кобинамида и витамина В12, исходя из природной кобиро
вой кислоты. Синтезированы также различные цианокобаламины по месту
нуклеотидного лиганда.
Синтез Dg-l- аминопропанола-2
Синтез £>^-1-аминопропанола-2 (XVII) осуществлен, исходя из Dg-Mo-
лочной кислоты (LII), путем переведения ее в эфир, затем в амид и восста-
новления алюмогидридом лития [901. Из более легко доступного рацеми-
ческого аминопропилового спирта D-антипод был получен в результате
асимметрического расщепления его n-нитробензоата с Ц+)-винной кислотой
[169].
ОН ОН ОН он
| I | (Hl/LiAlHi 1
СН3СНС00Н -> CHSCHCOOC2H6 -* CHsCHCONH2--------► CHjCHCH2NH2
LII XVII
Синтез а-p и б а з о л a
При конденсации 5-трифенилметил-Д-рибофуранозы (LIV) с 2-нитро-
4,5-диметиламинобензолом (LIII) путем кипячения в бензоле с уксусной
кислотой как катализатором получен 2-нитро-4,5-диметил-М-(5'-трифенил-
метил-Р-рибофуранозил)анилин (LV), который каталитическим гидрирова-
нием с палладиевым катализатором в метиловом спирте со строго дозиро-
ванным количеством водорода превращен в 4,5-диметил-о-фенилендиамин-
N-рибофуранозид (LVI). При действии на последний гидрохлоридом
изопропилформиминоэфира (схема 131) через промежуточное соединение по-
лучают 5'-О-трифенилметилпроизводное а-рибазола (LVII), которое после-
дующим кислотным гидролизом трифенилметильной группы дает N-(a-D-pn-
бофуранозил)-5,6-диметилбензимидазол, а-рибазол (XXI), в виде кристал-
лического пикрата. Пикрат разлагается в водной суспензии при pH 2,
пикриновая кислота извлекается хлороформом и после нейтрализации
и сгущения водного раствора выделяется а-рибазол (XXI) с выходом 6%
от соединения LV [98].
Более просто [170] а-рибазол синтезирован из 2,3,5-три-О-бензил-1-О-
и-нитробензоил(или п-фенилазобензонл)-Р-0-рибофуранозы (LVIII) превра-
щением в 2,3,5-три-О-бензил-О-рибофуранозилхлорид (LIX), его конден-
сацией с избытком 5,6-диметилбензимидазола в гликозид — М-(2,3,5-три-
0-бензил-а-0-рибофуранозил)-5,6-диметилбензимидазол ,(LX) с последую-
щим каталитическим дебензилированием
R-O/iCJ^CO; Bz - CeHjCHj
597
Для получения анамерного 0-рибазола, М-ф-О-рибофуранозил)-
5,6-диметилбензимидазола соединение LV ацетилируют уксусным ангид-
ридом в пиридине в 2-нитро-4,5-диметил-М-(2',3'-диацетил б'-трифенилметил-
£>-рибофуранозил)анилин, затем восстанавливают и конденсируют с гидро-
хлоридом этилформиминоэфира через промежуточное соединение в
бензимидазольное производное, которое гидролизуют в 0-рибазол (LXI) [98].
Соединение LIX получают также конденсацией 5,6-диметилбензимида-
зола с 1-бром-2,3,5-трибензил-£>-рибозой [171] или 1-хлор-2,3,5-триацетил-
D-рибофуранозой [2]. Подобными синтетическими методами получены N-
гликозиды бензимидазола с D-глюкозой, D-маннозой, D-галактозой, D-ара-
бинозой и др. [99]. Изучены различные методы замыкания имидазольного
кольца [172].
Синтез а - ри б азолфосфата
а-Рибазол-2'-фосфат или а-рибазол-З'-фосфат (XXII) получен фосфорили-
рованием б'-трифенилметил-а-рибазола (LVII) хлорангидридом дифенил-
фосфорной кислоты, (С6Н5О)2РОС1. Из этого соединения гидролизом уда-
лялись трифенилметильная и фенильная группы, затем а-рибазолфосфат
(XXII) выделялся в виде свинцовой соли, которая разлагалась сероводоро-
дом. Вещество было получено с т. пл. 235—236° С [102] и отвечало З'-фос-
фату а-рибазола [104].
598
Для фосфорилирования применяется и хлорангидрид дибензилфосфорной
кислоты, (СВН5СН2О)2РОС1. Взаимодействие 5'-тритил-а-рибазола (LVII)
с [3-цианэтилфосфатом в пиридине в присутствии М,М'-дициклогексилка-
рбодиимида с последующим детритилированием и гидролизом также приво-
дит ка-рибазолфосфату (XXII), обычно в виде 2',3'-циклофосфата (173].
Стереонаправленный синтез структурной части
молекулы витамина В12 — кобировой кислоты
Корриновый цикл витамина В12 содержит девять асимметрических цент-
ров, шесть из которых находятся в системе колец А—D. Для синтеза кор-
ринового цикла витамина В12 со всеми заместителями в правильной абсо-
лютной ориентации Вудвард и Эшенмозер получили соединения, пред-
ставляющие собой кольца В, С и систему колец А—D в оптически активной
форме и соответствующей специфической абсолютной конфигурации (174].
Кольца A—D получают [174], исходя из 6-метокси-2,3-диметил-индола
(LXII) [175]. Первый асимметрический центр создается при реакции маг-
ниевого производного LXII с пропаргилбромидом. Пропаргилиндоленин
(LXIII) при действии фтористого бора и окиси ртути в метаноле циклизует-
ся стереоспецифическим образом, давая трициклический кетон (LXIV);
при этом создается второй асимметрический центр. Трициклический кетон
был разделен на оптические изомеры при кристаллизации диастереоизо-
мерных мочевин, образующихся при реакции рацемического кетона (LXIV)
с оптически активным а-фенилэтилизоцианатом.
При конденсации правовращающего трициклического кетона (LXIV)
с левовращающей кислотой (LXV) образуется амид (LXVI), который при
обработке третичным бутилатом калия дает пентациклпческий лактам
(LXVII), содержащий два новых асимметрических центра. Для защиты
активных кетонной и амидной групп соединение LXVII переводят в кеталь
метоксиенамина (LXVIII), который при действии лития в жидком аммиаке
превращается в дигидропроизводное (LXIX). Кислотная обработка соеди-
нения LXIX приводит к пентацикленону (LXX), при этом образуется шес-
той, последний асимметрический центр, необходимый для построения ко-
лец А—D. Для введения в молекулу пентацикленона (LXX) второго атома
азота снимают кетальную защиту и получают оксим, который при обработ-
ке озоном, а затем HJO3 и диазометаном превращают в соединение (LXXI).
Это вещество переводят в кетоэфир (LXXII). Бекмановская перегруппи-
ровка последнего приводит к соединению LXXIII, которое содержит два
пятичленных азотистых цикла, представляющих кольца A—D.
599
В результате реакции соединения LXXIII с тиофенолом в присутствии
хлористого водорода образуется меркаптопроизводное (LXXIV), озонолиз
которого и последующая обработка жидким аммиаком приводит к полу-
чению соединения LXXV, имеющего в кольце D /-пропионамидную и аль-
дегидную группы. После восстановления боргидридом натрия получается
спиртовая группа, ее мезилируют ангидридом метансульфокислоты, затем
мезильную группу замещают бромом; одновременно при мезилировании
амидная группа дегидратируется в нитрильную. Получают соединение
LXXVI, которое представляй собой левую половину молекулы витамина
В,2 с дифференцированной /-пропиононитрильной цепочкой в кольце D [177].
LXXIY
CH^CHXOOCHj
CH,OOCHSC /<tjo
Н,С*
H3c
NH
H3C CHjCHaCONHj
LXXV
CH3CIT,COOCHj
CHjOOCH2C
n3C
H3C CHjCHjCN
LXXVI
Синтез кольца В [174 j начинают конденсацией [J-метил-р-ацетилакри-
ловой кислоты с бутадиеном, приводящей к получению циклогексенкарбо-
новой кислоты (LXXVII), которая была разделена на оптические изомеры
кристаллизацией ее солей с оптически активным а-фенилэтиламином. При
окислении правовращающей кислоты (LXXVII) была получена дилактоно-
600
вая кислота (LXXVIII). Удлинение ацетатной цепи приводит к кислоте
(LXXIX), которая после обработки аммиаком превращается в лактам
(LXXX), т. е. в кольцо В. Абсолютная конфигурация соединения LXXX
установлена превращением его в сукцинимид, продукт озонирования геп-
таметилкобирината.
Синтез кольца С [174] осуществляют, исходя из правовращающего кам-
ферхинона (LXXXI), который рядом реакций превращают в амид (LXXXII),
при этом образуется асимметрический центр требуемой абсолютной
конфигурации. Обработка этого амида озоном после ряда превра-
щений приводит к образованию озонида (LXXXIII), которой восстанав-
ливают в соединение LXXXIV, а последнее в кислой среде дает соединение
LXXXV. Наконец, пиролиз соединения LXXXV приводит к получению
лактама (LXXXVI), который представляет собой кольцо С.
Соединение колец В и С [174] осуществляют путем введения мостиково-
го атома серы [176]. Предварительно лактам (LXXX) (кольцо В) превраща-
ют в тиолактам (LXXXVII). Обработка этого тиолактама и енамида
(LXXXVI) (кольцо С) перекисью бензоила приводит к образованию S-мос-
тикового производного (LXXXVIII), которое при нагревании с (С2Н5О)3Р
превращается в соединение LXXXIX, являющееся системой колец В—С
со всеми заместителями в правильной абсолютной конфигурации.
21—69
601
Схема 132
Синтез кобировой кислоты
602
Для осуществления конденсации в коррин (174, 177} (схема 132) соеди-
нение LXXXIX превращают в тиолактам (ХС), далее в присутствии трет’
тичного бутилата калия он соединяется с бромидом (LXXVI), при этом
образуется очень лабильный тиоэфир (XCI). Это соединение превращается
в изомер (ХСП), десульфирующийся при обработке анэтилфосфином и
дающий линейную структуру колец ADCB XCIII.
Окончательное завершение циклизации линейной структуры
ADCB в коррин Вудвард и Эшенмозер осуществили через мостиковый
атом серы (177, 178], для чего с помощью пятисернистбго фосфора из
соединения XCIII получают тиолактам-тиолактонное производное (XCIV),
тиольную группу кольца А метилируют триметилоксонийфторборатом в
S-метил производное (XCIV). В это соединение вводят атом кобальта и при
действии диметиламина раскрывают тиолактамный цикл, при этом образу-
ется диметиламидное производное дицианида кобальтового комплекса (XCV);
дальнейшее нагревание с диазобициклононаном в диметилацетамиде при-
водит к получению циклического корринового производного (XCVI). Амид-
ную группу кольца В удаляют, образуется лактонное кольцо; в мезополо-
жения 5 и 15 полученного соединения вводят замещенную метильную
группу при действии хлорбензилового эфира в сульфолане и тиофенола —
образуется 5,15-бис-фенилтиометильное производное (XCVII). Десульфиро-
вание этого соединения с Ni Ренея происходит одновременно с раскрытием
лактонного кольца цикла В и после этерификации карбоксильной группы
с образованием f-нитрила гексаметилкобириновой кислоты (XCVIII)
[177].
/-Нитрильную группу кольца D соединения XCVIII гидролизуют в
амидную и осуществляют разделение стереоизомеров по положению 13, на-
личие таких стереоизомеров отмечалось на всех стадиях соединения колец
AD и ВС. Затем f-амидную группу соответствующего стереоизомера при дей-
ствии N?O4 превращают в /-карбоксильную группу, амидируют жидким
аммиаком карбоксиэфирные группы в амидные и получают кобировую
кислоту (IV), идентичную с выделенной при расщеплении природного ци-
анокобаламина (177].
Синтез кобинамида, его фосфорного эфира и
цианокобалам ин а
Исходя из кобировой кислоты, фактора Vla (IV), полученной из при-
родных источников или продуктов кислотного расщепления витамина В12,
осуществлен синтез кобинамида (VI) (179, 180], его фосфорного эфира (С)
и витамина В12 (I) П81 ].
Кобировая кислота, abcdeg-гексамид кобириновой кислоты (IV), имею-
щая свободную /-карбоксильную группу, вводится в реакцию с хлоруголь-
ным эфиром в безводном диметилформамиде в присутствии триэтиламина
и превращается в смешанный ангидрид кобировой кислоты (XCIX) [179,
180]. При действии на смешанный ангидрид кобировой кислоты (ХС1Х)
Dg-l-аминопропанолом-2 образуется кобинамид, фактор В (VI), с выходом
90% [179, 180]. Его можно получить и непосредственно из кобировой кис-
лоты (IV) и 0„-1-аминопропанола-2 карбодиимидным методом с выходом
22,5% [179]. Из кобинамида (VI) [182] реакцией с р-цианэтилфосфатом
в безводном диметилформамиде и пиридине в присутствии N, N'-дицикло-
гексилкарбодиимида с последующим гидролизом синтезирован кобннамид-
фосфат, фактор В-фосфат (С), с выходом 78% 1183, 184].
Необходимый для синтеза витамина В12 фосфодиэфир, а-рибазолфосфо-
аминопропанол (CI), получают конденсацией З'-фосфо-а-рибазола (XXII)
с карбобензокси-О^.-!-аминопропанолом-2 с последующим гидрированием.
Можно использовать 2,'3'-циклофосфат а-рибазола с последующим разде-
лением образовавшихся 2'- и З'-изомеров [181]. При конденсации смеша н-
21
603
QCOIINCHXHCHj
А-₽е°
I он
с он
QCOOH ,,,a,Nav:,
Коа<хвая\ о ЛН/ Кобинамид
кислота \'о ^2^/ VI
iv \0ч
QCOOCOOCJIj
XCIX
4 K6hnch,ch(oh)ch,
*)[n]/Pd
CHjOH
QCOOCOOQH^ Цианоко-
XCIX саламин
Н
РС°
ioh
о
I
ьучсн2снсн3
d
Q-остаток кОБТфоаой
кислоты
; ного ангидрида кобировой кислоты (XCIX) с а-рибазолфосфоаминопропа-
“ йолом (CI) в Среде диметилформамида при 0° С получается кристаллический
Цианокобаламин (I) с выходом около 60% [181 ].
Подобными методами были синтезированы 2'- и 5'-фосфоаналоги вита-
$,Мина В12 [181, 182, 1841.
' Моно-, ди- и трикарбоновые кислоты, содержащие нуклеотидную часть
Молекулы, получаемые из кислых гидролизатов витамина В12 [27] методом
смешанных ангидридов с последующим амидированием аммиаком, вновь
ресинтезируются в цианокобаламин.
Были синтезированы аналоги витамина В12: Р1-(аденозин-5')-Р2-кобин-
амидпирофосфат [183], Р1-(гуанозин-5,)-Р2-кобинамидпирофосфат [135],
сЬ(5-метоксибензимидазолил)кобамид цианид, 7а-(2-метиладенил)кобамид
Цианид [18], аналоге 2-метил-2-аминопропанолом [185, 186] и др.
Синтезированы различные производные продуктов расщепления циано-
йобаламина. Из кобировой кислоты (IV), хлоругольного эфира и а-амино-
кислот, таких, как серин, треонин и др., и их метиловых эфиров получены
соответствующие пептиды кобировой кислоты [187, 1881.
Обзор по химии корриновых систем дан в литературе [189].
БИОЛОГИЧЕСКОЕ ЗНАЧЕНИЕ ВИТАМИНА В12. ЗАВИСИМОСТЬ МЕЖДУ
СТРОЕНИЕМ И БИОЛОГИЧЕСКОЙ АКТИВНОСТЬЮ
Витамин В12 (цианокобаламин и др.) связан с осуществлением биокаталити-
ческих реакций, обеспечивающих кроветворную функцию организма, он
способствует нормализации функции печени, благоприятно влияет на ре-
генерацию нервных волокон. Витамин В12 применяют для лечения перни-
циозной и некоторых других видов анемии. Он активирует созревание фор-
менных элементов крови. Для излечения пернициозной(аддисон—бирмеров-
Ской) анемии достаточно нескольких инъекций цианокобаламина по 10 мкг
С трехдневными промежутками (витамин задерживается в организме 72 ч);
при этом в огромном количестве происходит выработка новых красных кро-
вяных телец. При лечении макроцитарной анемии фолиевая кислота может
быть только частично заменена витамином В12. Этот витамин не излечивает
лейкемии. Цианокобаламин применяют для лечения полиневрита, тройнич-
ного нерва и других невралгических заболеваний, а также спру, острых
хронических гепатитов и циррозов печени [191 ], остеоартритов, аллерги-
ческих и некоторых других заболеваний. Витамин В12 способствует усилен-
ному кровеобразованию после лучевого воздействия расщепляющимися ве-
ществами [189, 190]. Комбинированный препарат из оксикобаламина и фо-
лиевой кислоты обладает выраженной противоанемической активностью
604
11921, a npenapat из оксикобаламина и 5-формил-5,6,-7,8-тетрагидрофолие-
вой кислоты (фолинсвой кислоты), так называемый гепа-фактор, активен
для восстановления нормальной функции печени и нарушенных процессов
метаболизма. Цианокобаламин и оксикобаламин активны в чрезвычайно
малых количествах.
Витамин В12 необходим для нормального роста и функционирования
организма человека и животных; совместно с холином и метионином он
обладает эффективным липотропным действием (предотвращает отложение
жира в печени).
Цианокобаламин связан с обменом жиров, поддерживая в восстановлен-
ном состоянии сульфгидрильную группу коэнзима А, осуществляющего
в обмене веществ реакции ацетилирования [50, 189]. Биокаталитическое
действие цианокобаламин проявляет в качестве кофермента или комплекса
с белком, в виде которого он находится в печени [193].
Усвоению витамина В12 при его приеме внутрь с пищей животного про-
исхождения или в виде препаратов способствует так называемый «внутрен-
ний фактор», выделяемый желудком [194]. Он представляет собой гомо-
генный мукопротеид с молекулярной массой около 20 000 [165]. «Внут-
ренний фактор» может быть получен из высушенной ткани слизистой
свиного желудка [195]. Причина заболевания пернициозной анемией — не-
достаточность в выделении «внутреннего фактора», следствием чего является
неудовлетворительная способность организма к всасыванию цианокобал-1
амина.
У человека синтез витамина В12 симбионтами, обитающими в пищевая
рительном тракте, очень незначителен, поэтому в организм человека он
должен поступать с пищей [197]. Авитаминоз В12 наблюдается у полных
вегетарианцев, которые совершенно не употребляют животной пищи. По-',
требность в витамине В12 для человека составляет около 5 мкг в суткщ, ч
Недостаток витамина В12 у животных характеризуется потерей аппети-
та, замедлением роста, огрублением волосяного покрова, нарушением ко-
ординации движений, некрозами печени; у домашних птиц происходит
неправильное эмбриональное развитие яиц, уменьшается выводимость,;
увеличивается смертность цыплят.
Анемия у крупного рогатого скота и.овец излечивается при применении-
солей кобальта [196], который, по-видимому, используется микроорганиз-
мами, синтезирующими витамин В12.
Цианокобаламин, как правило, не встречается в продуктах раститель-
ного происхождения, за исключением некоторых видов водорослей (напри-
мер, альга) и бобовых растений, у которых он образуется на корневой си-
стеме в результате деятельности клубеньковых бактерий.
Источником витамина В12 для животных служит микрофлора желудка
и кишечника жвачных или продукты животного происхождения.
Витамин В12 содержится в печени, рыбной муке, яйцах, молоке, соевых
бобах, в мицелии и культуральной среде актиномицетов Streptomyces
griseus и aureofaciens различных видов Pennicillium, в микроорганизмах
Mycobacterium smegmatis, Lactobacillus arabinosus, Bacylus subtilis [6]
и др. Кроме того, витамин Bj2 находится в свином, коровьем и курином по-
мете, в гнилостном шламе канализационных отбросов, а также в большом-
количестве в желудочном содержимом жвачных животных [196].
В животных продуктах витамин Bj2 содержится в виде белкового комп-.;
лекса, из которого выделяется в свободном виде нагреванием или фермен-
тативным путем. f
Витамин В12 в количестве 100 мг/1 кг не токсичен для морских свинок
и крыс.
В основе биологической активности витамина В12 лежит его корриновая
макроциклическая система с соответствующими заместителями, находящая-
ся в координировании с атомом кобальта. Этот кобальтовый комплекс, i
включающий нуклеотид 5,6-диметилбензимидазола, обладает очень высокой >
605
витаминной активйбстью. Производные цианокобаламина, у которых вмес-
то циангруппы находятся в качестве лигандов такие группы и атомы,
как ОН, ONO, CI, Br, HSO4 и др., обладают почти такой же витаминной
активностью, как и цианокобаламин.
При замене 5,6-диметилбензимидазола другими производными имидазо-
ла аналоги витамина В,2 сохраняют частичную биологическую активность
на цыплятах (при введении per os): с 5 (или 6)-метилбензимидазолом —
35% [40], с 5-оксибензимидазолом (фактор III) — 10—30% [131], с бензи-
мидазолом— 23% [40, 198], с 5,6-динитробензимидазолом — 75% [198],
с 2,3-нафтимидазолом — 85% [199]. При замене метильных групп 5,6-ди-
метилбензимидазола на атомы хлора молекула аналога частично сохраняет
биологические свойства против злокачественной анемии [50].
Следует отметить, что орто-положение диметильных групп в конденси-
рованном бензольном ядре, по-видимому, имеет существенное физиологи-
ческое значение, так как opmo-диметильная группировка находится также
в конденсированном бензольном цикле изоаллоксазинового ядра молекулы
витамина В2 (рибофлавин), хотя среди других природных веществ она не
обнаружена.
Наоборот, система адениловой кислоты в виде нуклеотида аденина или
более сложных аденозинди- и аденозинтрифосфорных эфиров (АМФ, АДФ,
АТФ) широко распространена в природе, так как она входит не только
в коферменты никотинамида, пантотеновой кислоты и рибофлавина (нико-
тинамидадениндинуклеотид, кофермент А, флавинадениндинуклеотид), но
и в нуклеопротеиды клеточных ядер. Однако в составе псевдовитаминов В1а
эта система не обеспечивает необходимых биокаталитических функций.
Псевдовитамины В12, вместо 5,6-диметилбензимидазола содержащие аденин,
2-метиладенин, 2-метилмеркаптоаденин, гипоксантин, 2-метилгипоксантин
и др., полностью утрачивают витаминную активность для животных, одна-
ко для некоторых микроорганизмов сохраняется частичная ростовая актив-
ность (например, для Lactobacillus lactisи Lactobacillus leihmannii) [40, 125,
128]. Аналог витамина В12 с фосфорной группой положения 2' рибозы
(вместо 3') почти не обладает активностью [181].
Дезаминирование карбоксамидных групп положений а—е и d цианоко-
баламина приводит к неактивным или антагонистическим аналогам по от-
ношению к Esherishea coli [197]. Изменение пространственной конфигура-
ции пропионамидной группы положения 13 корринового макроцикла на
противоположную — неовитамин В12 — уменьшает ростовую активность
на Е. coli до 10%. [28].
- Удаление, метильного заместителя из положения 2 группы 1-аминопро-
панола-2 цианокобаламина приводит к аналогу с сильным конкурентным
ингибиторным действием [200], так же как замещение метильной группы
на объемистые заместители или введение их в положение 1. Для аналога,
содержащего 2-аминопропанол-1 вместо 1-аминопропанола-2, также харак-
терны антагонистические свойства [185, 186].
Биологической активностью витамина В12 обладают некоторые осколки
его молекулы. а-Рпбазол (XXI) и его анамер —Р-рибазол (LIX) обладают
1/400 активности витамина при испытании на крысах [201 ]; такую же ак-
тивность имеет и фосфорный эфир а-рибазола [100]. 5,6-Диметилбензими-
дазол (XVIII), 5-монометилбензимидазол, 1,2-диамино-4,5-диметилбензол
(XIX) и £)-рибитил-5,6-диметилбензимидазол также активны для крыс
(202], но в меньшей степени; их активность составляет около 1/1000 актив-
ности витамина В12 [2031.
КОРРИНОВЫЕ КОФЕРМЕНТЫ
Кофермент-кобаламин — а-(5,6-диметилбензимидазолил)-Со-5'-дезокси-
аденозилкобамид, 5'-дезоксиаденозилкобаламин (СП),—обнаружен у микро-
организмов [39], а также в печени человека, овцы, кролика, цыпленка
606
[38]. 5'-Дезоксиаденозилкобаламин представляет собой коррин, имеющий;
шесть сопряженных двойных связей в цикле и трехвалентный атом кобаль-:
та с координационным числом 6. Атом кобальта связан с атомом углерода
положения 5'-дезоксиаденозильного лиганда (вместо циангруппы цианоко-
баламина) и с атомом азота пирролидинового цикла, имеет три координа-
ционные связи с тремя атомами азота частично гидрированных пиррольных
циклов и одну — с атомом азота беизимидазольного цикла; положительный
заряд атома кобальта компенсирован отрицательным зарядом атома кисло-
рода в фосфорном остатке нуклеотидного лиганда.
Кофермент витамина В]2 кристаллизуется в виде игл или плоских ром-
бических кристаллов красного цвета. В кристаллическом виде кофермент-
кобаламин относительно устойчив -— сохраняет активность при хранении
в течение нескольких месяцев при —10° С или нескольких дней при 20—
25° С. Его растворы наиболее стабильны при pH 6,0—7,0. Кофермент хо-
рошо растворим (как и его аналоги) в воде, этаноле и феноле, нерастворим
в ацетоне, эфире, дихлорэтане, диоксане и других неполярных растворите-
лях. В кислом растворе он электрофоретически подвижен.
Для спектра поглощения кофермента витамина В12 характерен ярко вы-
раженный максимум при 260 нм и плечо при 375 нм; лмакс 260, 315, 340, 375,
522 нм [204]. Спектры аналогов кофермента различаются лишь максиму-
мами в длинноволновой части в зависимости от наличия других лигандов.
Кофермент-кобаламин и его аналоги отличаются от витамина В12 повы-
шенной чувствительностью к действию света и цианистых соединений. . ;
сн
Кофермент витамина В12 легко подвергается фотолизу. Фотолитическое
анаэробное расщепление является гомолитической реакцией, что следует
на основании изучения продуктов реакции [68]. Сначала при гомолитиче-
ском разложении образуется 5'-дезоксиаденозильный радикал (СП!) и ви-
тамин Bi2r(XIV). Первый связывается с С(8> аденинового основания, в ре-
зультате чего образуется 5',8-циклический аденозин - (CIV) [205, 206].
В присутствии воздуха 5'-дезоксиаденозильный радикал, окисляясь, пре-
вращается в аденозин-5'-карбоновую кислоту (CV) или в аденозин-5'-альде-
607
гид (CV, СНО). Витамин В12г (XIV) в свою очередь при окислении кисло-
родом воздуха чрезвычайно легко превращается в аквокобаламин [207].
RCo[ll]-
Витамин BUr
XIV
|[О]
[(rco—oh)h]+
Аквокобаламин
и ОН он
NHj
Аденозин-5'-карбоновая кислота гидролизуется в аденин и D-рибоно-
вую кислоту [208].
Фотолитическое расщепление уридинового аналога приводит к аквоко-
баламину даже при полном отсутствии кислорода [68].
Если фотолиз протекает в присутствии акцепторов радикалов, таких,
как соединения с тиольными группами, то присоединение радикала к ак-
цептору происходит раньше, чем описанные выше превращения [209, 2101.
Так, фотолиз метил кобаламина в присутствии цистеина или гомоцистеина
дает соответственно S-метил цистеин или метионин. Аналогичная реакция
кофермента витамина В12 (СП) в присутствии гомоцистеина приводит к
S-аденозилгомоцистеину (CVI).
RCoOlO-C^Ad+HOOCCHOVHSH^^
Кофермент-поьаламин
СП
он он
Перенести аденозильную, группу на метионин оказалось невозможным
[211].
Связь Со—С в алкилкобаламинах, например в 0-цианэтилкобаламине,
может быть при действии щелочи обратимо расщеплена с образованием
витамина B12s (XV) (с. 583) и акрил нитрила, причем оба электрона оста-
ются на кобальте [64].
Кофермент витамина В12 (СП) легко подвергается действию CN-ионов,
причем расщепляется Со—С-связь с СН2-группой аденозина и возникает
608
новая Со—С-связь с CN-группой. Эта реакция осуществляется по гетеро-
литическому механизму [212 ] (в отличие от фотолиза, проходящего по го-
молитическому механизму) с образованием аденина (XLII), дицианокобала-
мина и 2,3,4-триоксигекс-5-енонитрила (CVII)
Интересно отметить, что кофермент витамина В12 и его аналоги с нук-
леозидным лигандом реагируют с синильной кислотой гораздо быстрее,
чем с цианистым калием, возможно, потому, что начальное протонирование
нуклеотида облегчает реакцию замещения [68], которая проходит даже в
темноте.
В отличие от витамина В12, который в щелочных растворах при дейст-
вии кислорода воздуха превращается в дегидровитамин В12 (XII), 60%
кофермент-кобаламина не изменяется и лишь 40% превращается в оксико-
баламин, который затем быстро окисляется в дегидрооксикобаламин
(CVIII) [213].
R= остальная часть молекулы
кобаламинов или ИХ производил
Имеются различия между витамином В12 и его коферментом в протека-
нии реакций циклизации ацетамидного заместителя кольца В и хлориро-
вания атома углерода в положении 10. Цианокобаламин при действии
хлорамина Т первоначально образует лактон, затем протекает реакция
хлорирования. При действии первого эквивалента хлорамина Т (в 0,002544
растворе соляной кислоты) на метилкобаламин (CIX) первоначально про-
исходит замещение хлором по углероду положения 10, а затем избыток
хлорирующего агента приводит к образованию монохлорлактона метил-
кобаламина (СХ), обнаруживаемого по характерному поглощению при
1778 см"1 [681.
При обработке полученного соединения CN-ионами на свету образуется
лактон 10-хлорцианокобаламина (CXI).
Метилкобаламин легче бромируется, чем цианокобаламин. Так, при
бромировании N-бромсукцинимидом в ледяной уксусной кислоте образует-
ся 88% 10-бромметилкобаламина и 12% метилкобаламинлактона, в то вре-
мя как в 0,5 М растворе уксусной кислоты получается 50% 10-бромметил-
кобаламина и 50% метилкобаламинлактона [214].
609
СТРУКТУРА КОФЕРМЕНТ-КОБАЛАМИНА
Структура 5'-дезоксиаденозилкобаламина (СП) была установлена на ос-
новании кристаллографического и химического исследования. Сначала ато-
му кобальта в этом соединении приписывалось двухвалентное состояние
[212]. Однако на основании электронного парамагнитного резонанса и из-
мерения магнитной восприимчивости установлено, что кофермент витамина
В12 имеет в своей молекуле диамагнитный трехвалентный кобальт [57, 60,
215 J. Это подтверждено также данными рентгеноструктурного анализа [63,
1521 и частичным химическим синтезом кофермент-кобаламина [63].
По данным синтеза метилкобаламина с применением трития установлено,
что сопряженная система корринового кольца кофермент-кобаламина иден-
тична корриновой системе цианокобаламина [216].
Кофермент-кобаламин имеет металл-алкильную связь [152]. Он содер-
жит 5'-дезоксиаденозильный нуклеотид с обычной природной 0-гликозид-
ной связью [208].
Кофермент-кобаламин в отличие от цианокобаламина относительно лег-
ко расщепляется минеральными кислотами. При действии на кофермент-ко-
баламин и его аналоги разбавленных кислот отщепляется только нуклео-
зидный лиганд. а-Гликозидная связь D-рибозы с 5,6:диметилбензимидазо-
лом в нуклеотидной части молекулы, по-видимому, очень прочна. Так, при
действии на кофермент витамина В12 (СП) 0,1 н. раствором соляной кисло-
ты (100°С, 90 мин) образуется аденин (XLII), аквокобаламин (II) и корри-
ноза—П-зршпро-2,3-диоксипент-4-еналь (СХП) [204, 206, 217].
н
N ьг
^Co-CH2Ad ----— С Т N + [(RCo~Oh)h]++ сн^снсн(он)сн(он)сно
Кофермент-кобаламин
СП XUI ЬЦ II CXII
Структура корринозы была подтверждена синтезом эритро-1,2,3-три-
оксипент-4-ена — продукта восстановления корринозы боргидридом натрия
[206].
Следует отметить, что у аденилкобамидкофермента — соединения, отли-
чающегося от кофермента витамина В12 тем, что нуклеотидным лигандом
в нем является аденин, связанный с D-рибозой р-гликозидной связью, —
в описанных выше условиях разрываются гликозидные связи обоих лиган-
дов, причем образуются аденин (2 моля) и корриноза. Нуклеотидный ли-
ганд с р-гликозидной связью легко гидролизуется уже .0,07 н. раствором
соляной кислоты (85° С, 10 мин), а гликозидная связь б'-дезоксиаденозиль-
ного лиганда и его связь с кобальтом оказываются стабильными.
ПОЛУЧЕНИЕ КОФЕРМЕНТ-КОБАЛАМИНА
Кофермент-кобаламин, —5'-дезоксиаденозилкобаламин (СП) — впер-
вые обнаружили в бактериальных клетках Clostridium tetanomorphum Бар-
кер с сотр. [39] в 1958 г., затем он был выделен из ферментных растворов
этого микроорганизма и Propionibacterium Shermanii в кристаллическом
виде [204, 218].
Однако первой коферментной формой, обнаруженной у микроорганиз-
мов, был 7а-аденил-5'-дезоксиаденозилкобамид [391 — аналог кофермент-
кобаламина, не обладающий биологической активностью. Это вещество
окрашено в желтый цвет в широком диапазоне концентраций водородных
ионов [30].
При ферментации с Propionibacterium Shermanii в анаэробной фазе
развития микроорганизмов образуются большие количества кобинамида,
фактора В (VI), — безнуклеотидного витамина В12 и кром.е того 5'-де-
GW
зоксиаденозилкобйнамид (безнуклеотидный кофермент-кобаламин), ко-
торый в течение аэробной фазы ферментации присоединяет нуклеотидный
лиганд (5,6-диметилбензимидазолнуклеотид) и превращается в кофермент-
кобаламин (СП) [219]. При ферментации с Propionibacterium fleudenreihii
кофермента образуется от 8 [219] до 19 мг/л [220], а с Propionibacterium
Shermanii — 23 мг/л [220]. В полностью анаэробном процессе ферментации
с Bacterium butyricuni rettgeri получается 5 мг/л кофермент-кобаламина
[221].
Безнуклеотидный кофермент-кобинамид выделен из Propionibacterium
Shermanii в присутствии солей кобальта, но без добавления гетероцикли-
ческих оснований [222, 223]. Его можно получить также химическим путем,
действуя гидроокисью трехвалентного церия на кофермент витамина В12
[224]. Обнаружена также кофермент-кобировая кислота [225].
Ферментативным методом были получены различные кофермент-коба-
миды — соединения, родственные коферменту витамина В12, у которых
нуклеотидным лигандом являются производные бензимидазола или пури-
на: бензимидазолил- [226], аденил- [227], 5(6)-метилбензимидазолил-, 5(6)-
нитробензимидазолил- [227 ], 5-аминобензимидазолил-, 6-аминобензимида-
золил- [227, 228], 5,6-дихлорбензимидазолил- [229], 2,6-диаминопуринил-
[227] и др.
Бензимидазолилкобамид-кофермент был получен в кристаллическом
виде; он выделен из пропионовокислых бактерий [226] и Clostridium teta-
nomorphum в присутствии бензимидазола, и также выделен из печени овцы,
кролика, кур и человека [38].
Соединения этого типа могут быть получены из клеток Propionibacte-
rium arabinosus, выращенных на среде с добавкой соответствующих гетеро-
циклических оснований [227].
ЧАСТИЧНЫЙ СИНТЕЗ КОФЕРМЕНТ-КОБАЛАМИНА
Кофермент-кобаламин (СП) и его нуклеозидные аналоги могут быть по-
лучены путем частичного синтеза. Одним из компонентов такого синтеза
служит витамин B12s (XV), другим — подходящим образом защищенный
нуклеозид. Витамин B12s для синтеза кофермента получают не из циано-
кобаламина, а из оксикобаламина, чтобы устранить присутствие циан-ионов.
Частичный синтез самого кофермент-кобаламина (СП) осуществляют
следующими путями (схема 133).
Аденозин (СХШ) превращают в 2',3'-О-изопропилиденаденозин (CXIV),
который реакцией с n-толуолсульфохлоридом в пиридине переводят в 5'-то-
зил-2', З'-О-изопропилиденаденозин (CXV), конденсацией последнего с ви-
тамином B12s (XV) получают 2', З'-О-изопропилиденовое производное ко-
фермента витамина В12, которое в результате гидролиза 0,1 н. раствором
соляной кислоты при 90° С в течение 15 мин превращается в кофермент-ко-
баламин (СП) [63, 230—233] с выходом 54%.
По другому методу аденозин (СХШ) действием трифенилхлорметана
легко превращают сначала в б'-тритил-, а затем в 5'-тритил-2',З'-диацетил-
аденозин, из которого тритильную группу легко удаляют разбавленными
кислотами. Полученный 2',3'-диацетиладенозин (CXVI) тозилируют, 5'-то-
зил-2',3'-диацетиладенозин (CXVII) конденсируют с витамином B12s (XV),
промежуточное соединение подвергают деацетилированию в мягких усло-
виях с помощью органического основания и с хорошим выходом получают
кофермент витамина В12 (СП) [231].
Аденозин (СХШ) может быть подвергнут тозилированию непосред-
ственно в 5'-О-тозиладенозин (CXVIII), при его взаимодействии с ви-
тамином B12s (XV) с последующей очисткой получается чистый кофермент-
кобаламин с выходом 68% (СП). Соединение CXVIII может быть получено
из аденозина (СХШ) через 2',3'-п-диметиламинобензилиденаденозин [234,
235].
вп
Конденсацию обоих компонентов в синтезе кофермента осуществляют
в темноте, обычно в водно-метанольном растворе в атмосфере азота, с по-
следующей очисткой на сефадексе G-15, диэтиламиноэтил- или карбоксиме-
тилцеллюлозе, конечное вещество экстрагируют смесью фенола и четырех-
хлористого углерода или хлороформа [235, 236].
Кофермент-кобаламин может быть получен из трифенилфосфинового
или резорцинового комплекса цианокобаламина и 5'-О-тозиладенозина
[235].
Синтезированы многочисленные аналоги кофермент-кобаламина, содер-
жащие в качестве нуклеозидного лиганда 5'-дезоксицитидил-, 5'-дезокси-
гуанозил-, 5'-дезокситимидил-, 5'-дезоксиуридил-, 1-1Ч-метил-5'-дезоксиаде-
нозил- и другие нуклеозидные заместители, а также ацилированные произ-
водные [69, 230, 231, 237—241].
Соединения типа кофермент-кобаламина, у которых вместо нуклеозид-
ного лиганда имеются алкил-, алкен-, ацил- или S—Co-связанные группы,
получают из витамина B12s (XV), действуя на него различными реаген-
тами, имеющими сродство к электрону. Обычно это сильные алкилирующие
и ацилирующие вещества. Так, для синтеза метилкобаламина можно
использовать диазометан, диметилсульфат [63, 242] или йодистый метил,
причем последний применяют как в щелочной среде, так и в уксусной
кислоте [231 ]. Конденсацию с витамином В12з проводят в атмосфере инерт-
Схема 133
Синтез кофермент-кобаламина
' CH, CH,
о о
о
носн, °
CXIV
NH,
он он
о
носн, °
СХШ NH,
сн, сн,
о о
о
БОСН/ЧТ N
OH OH
О
TsOCH,xr -
CXV
L_
NH,
CXVIII NH,
RCo [I] H +
Витамин Bt2s
XV
ОН он
- СН NH.
R—остальная часть
молекулы витамина Вв
ного газа в темноте или при красном свете, об окончании реакции судят по
изменению цвета раствора от серо-зеленого до красного. Подобными метода-
ми получены аналоги кофермент-кобаламина с различными лигандами [243].
612:
БИОХИМИЧЕСКИЕ ФУНКЦИИ КОРРИНОВОГО КОФЕРМЕНТА
Кофермент осуществляет некоторые ферментативные реакции изомери-
зации и диспропорционирования [39, 218, 244, 245], рассматриваемые
ниже.
1. Изомеризация L-глутаминовой кислоты в L-mpeo-$ -метиласпарагино-
вую кислоту при участии метиласпартатмутазы (глутаматизомеразы)
NH, NH„
I I ’
НООССНСН2СН2СООН НООССНСНСООН
сн3
2. Изомеризация метилмалонилкофермента А в сукцинил кофермент А
под влиянием метил малонил-КоА-мутазы [246, 247 J
СН3
I
HOOCCHCO ~ SKoA Z± НООССН2СН2СО - SKoA
Реакция изомеризации протекает, по-видимому, межмолекулярно, в ре-
зультате взаимного обмена карбонилтиоэфирных групп двух молекул ме-
тилмалонил-КоА, как это следует из опытов с 13С меченными соединениями
1248, 249].
СООН
I
о сн
KoAS-^/XcH,
+ ‘--------------*
CHS С—SKoA
\ZU
сн о
о
KoAS-ч!
сн2 +
Ан2
«I
соон
соон
I
сн2
С—SKoA
*соон
3. Внутримолекулярные окислительно-восстановительные превращения
1,2-пропандиола в пропионовый альдегид при участии пропандиолдегидра-
тазы [245]
ОН он
I 1
CHSCH—СН2 CH3CH2CHO + H2O
Подобная реакция при участии глицеролдегидратазы наблюдается при
превращении глицерина в р-оксипропионовый альдегид
НОСН2 СН (ОН) СН2 ОН —НОСН., СН2 CHO+HgO.
Активность глицеролдегидратазы ингибируется аналогами кофермент-ко-
баламина, например 5'-дезокси-М(6)-п-нитробензиладенозилкобаламином
[241 ].
Кроме того, кофермент-кобаламин, по-видимому, участвует в реакции
расщепления лизина в масляную и уксусную кислоты [250, 251 ]
СН2СН2СН2СН2СНСООН + 2НгО -> СН3СН2СН2СООН + СН3СООН + 2NHj
NH, NH2
и реакции превращения рибозида в дезоксирибозид [252]. Отмечено, что
кофермент витамина В12 стимулирует включение лейцина-14С в молекулу
белка [253].
При пернициозной анемии гематологические эффекты кофермента вита-
мина В12 и самого витамина В12 эквивалентны [254].
613
Помимо кофермент-кобаламина, биохимическую активность проявляют
и его аналоги, полученные из природных источников. Так, метилкобаламин
является донором метильной группы в ферментативном синтезе метионина
из гомоцистеина 169, 210]
HSCH2CH2CH (NH2) СООН -> CH3SCH2CH2CH (NHS) соон
Метилирование сульфгидрильной группы гомоцистеина осуществляется
по сложному, до конца не выясненному механизму, в котором принимает
участие 5-ЬТметил-5,6,7,8-тетрагидрофолиевая кислота (5-М-метил-ТГФК).
Возможно, что метилкобаламин является переносчиком метильной группы
от 5-М-метил-ТГФК к гомоцистеину {210]. Метилирование гомоцистеина
метилкобаламином было осуществлено также in vitro (180].
Следует отметить, что метаболитические функции фолиевой кислоты
и витамина В12 еще не разграничены. Производные витамина В12 кофер-
ментного типа и фолиевая кислота оказывают, видимо, совместно катали-
тическое действие на обмен одноуглеродных соединений, важнейшими реак-
циями которого являются синтезы пуриновых и пиримидиновых оснований,
входящих в структуру РНК и ДНК 1255—261 ]. Многие кобамидные про-
изводные кофермента витамина В12, содержащие измененный нуклеотид-
ный лиганд, активны в тесте глутаматизомеразы [262], а аденилкобамид-
кофермент и бензимидазолилкобамид-кофермент участвуют также в изо-
меризации сукцинил-КоА в метилмалонил-КоА и в окислении гликолей
до соответствующих дезоксиальдегидов [245, 246, 250, 262, 263].
Однако in vivo у животных и человека только кофермент-кобаламин
выполняет жизненно важные биохимические функции; замена 5'-дезокси-
аденозильной группы на другие лиганды (в том числе алкильные группы)
ведет к потере активности или вызывает антагонистическое действие [63,
237].
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ (К ГЛАВЕ XIII)
1. Н. V i с к е г у. J. Biol. Chem.,
169, 237 (1947).
2. A. J о h n s о п, G. М i 1 1 е г,
J. М i 1 1 s, А. Т о d d. J. Chem.
Soc., 1953, 3061.
3. D. Hodgkin. Biochem. Soc.
Symp. (Cambridge, Engl.), № 13,
28 (1955).
4. D. H о d g к i n, J. К a m p e r,
M. M а с к a y, J. P i ck wor t h,
K. Trueblood, J. White.
Nature, 178, 64 (1956).
5. R. Bonnett, J. Cannon,
V. Clark, A. J о h n s о n,
L. Parker, E. L. Smith,
A. Todd. J. Chem. Soc., 1957,
1158.
6. E. R i c ke s, N. В r i n k,
F. К о n i u s z у, T. W о о d,
К. Folkers. Science, 107,
396; 108, 634 (1948).
7. N. Brink, D. Wolf,
E. К a c z к a, E. Riekes,
F. К о n i u s к у, T. W о о d,
К. F о 1 к e r s. J. Am. Chem.
Soc., 71, 1854 (1949).
8. F. H a r t 1 e у, P. S t г о s s,
R. Stuckey. J. Pharm. Phar-
macol., 2, 648 (1950).
9. K. F a n t e s, J. Page,
F. Parker, E. S m i t h.
Proc. Royal. Soc. London, 136B,
592 (1949).
10. H. W i j m e n g a, J. Le n s,
A. Middelbeek. Chem. Week-
blad, 45, 342 (1949).
11. B. Ellis, V. Petrov,
G. Snook. J. Pharm. Pharma-
col., 1, 287, 735, 950 (1949).
12. R. Barer, А. Cole.H. Tomp-
son. Nature, 163, 198 (1949).
- 13. N. В r i n k, F. К u e h 1, K. F о 1-
k e r s. Science, 112, 354 (1950).
14. D. H о d g к i n, M. P о r t er,
R. Spiller. Proc. Roy. Soc.
(London), B136, 609 (1949).
15. J. A 1 i c i n o. J. Am. Chem. Soc.,
73, 4051 (1951).
16. A. T о d d, A. J о h n s о n.
Angew. Chem. 67, 428 (1955).
17. D. Hodgkin, J. Pick-
worth, J. Robertson,
K. Trueblood, R. Pro-
se n, J. White. Nature, 176,
325 (1955).
18. R. В о п n e t t, J. С a n n о n,
A. Johnson, J. Suther-
land, A. T о d d, E. L.
Sm i t h, Nature, 176, 328 (1955).
19. R. Bonnett, J. Cannon,
A. Johnson, A. T о d d.
J. Chem. Soc., 1957, 1148.
20. W. J a к s о n, G. W h i t f i e 1 d,
W. De Vries, H. N e 1 s о n,
J. E v a n s. J. Am. Chem. Soc.,
73, 335 (1951).
614
21. F. G r fl n, R. e n a s se. Expe-
rienzia, 6, 263 (1950).
22. J. W a 1 1 m a n n, B. Cun n i n-
g h a m, M. С a 1 v i n. Science,
113, 55 (1951).
23. H. Diehl, R.Wander-
haar, R. Seabock. J. Am.
Chem. Soc., 72, 5312 (1950).
24. R. В о о s, J. C a r r, J. Conn,
Science, 117, 603 (1953).
25. B. J a s e 1 s к i s, H. D i e h 1.
J. Am. Chem. Soc., 76, 4345 (1954).
26. E. К a c z к a, D. Wolf,
F. Kuehl, K. F о 1 к e r s.
Science, 112, 354 (1950); J. Am.
Chem. Soc., 73, 3569 (1951).
27. J. A r in i t a g e, J. Cannon,
A. Johnson, L. Parker,
E. L. S m i t h, W. St a If or d,
A. Todd. J. Chem. Soc., 1953, 3849.
28. R. Bonnett, J. Godfrey,
V. Ma t h. J. Chem. Soc., ,,C”,
1971, 3736:
J29. K. Bernhauer, O. Muller,
F. Wagner. Angew. Chem., 75,
1145 (1963).
30. R. Bonnett. Chem. Rev., 63,
573 (1963).
31. Номенклатура IUPAC, J. Am.
Chem. Soc., 82, 5575 (1960).
32- E. Kaczka, R. Denkewal-
t e г, A. H о 1 1 a n d, K. F о 1-
k e r s. J. Am. Chem. Soc., 73, 335
(1951).
33. W. A n s I о w, S. В a 1 1,
W. E m e г у, K. F a n t e s,
E. L. Smith, ’A. Walker.
Chemistry and Industry, 1950, 574.
34. E. L. Smit h. Nature, 169, 60
(1952).
35. J. H e a t h с о t e. J. Pharm.
Pharmacol., 4, 641 (1952).
36. J. R о 1 о v i c h, G. Fabr i-
z i o, L. C a s i 1 I i. Farmaco Ed.
prat., 17, 553 (1962).
37. G. С о о 1 e у, В. E I 1 i s, V. P e- < «
t г о v, G. В e a v a n, E. H o-; -
1 i d а у, E. J о h n s о n. J, '£«“
Pharm. Pharmacol., 3, 271, 607 Й
(1951).
38. J. Toohey, H. Barker. J.
Biol. Chem., 236, 560 (1961).
39. H. В a г к e r, H. W e i s s -
bach, R. S m у t h. Proc. Natl.
Acad. Sci. U. S„ 44, 1093 (1958).
40. K. Bernhauer, W. Fried-
rich. Angew. Chem., 66, 776 (1954).
41. G. Muller, О. M ii 1 1 e r.
Angew. Chem., 78, 143 (1966); Z.
Naturforsch, 21b, 1159 (1966).
42. K. Bernhauer, E. Becher,
G. G г о s s, G. W i 1 h a r m.
Biochem. Z„ 322, 562 (1960).
43. H. D e 1 1- w e g, К. В e r n h a-
u e r. Arch. Biochem. Biophys., 69,
74 (1957).
44. K. Bernhauer, W. F r i d-
r ich; Пат. ФРГ 1032256; C.A., 54,
22693 (1960).
45. К. В e r n h a u e r, P. R e n z,
F. Wagner. Biochem. Z„ 335,
443 (1962).
46. R. В о r c h i e 1 1 i, G. В о r e t-
t 1, A. Di Mor co, P. J ul j.
t a, A. M i g 1 i a с c i, A. M i n-
ghetti, C. S p a 1 1 a. Biochem.
J., 74, 382 (I960).
47. P. Benz, Angew. Chem., 79, 311
(1967).
48. К. В e r n h a u e r, F. W a g-
n e r, H. Beisbar t b,
P. R i e t z, H. V о g e 1 m a n n.
Biochem. Z., 334, 289 (1966).
49. R. Bonnet, J. Godfrey,
D. Redman. Chem. Com-
muns, 1965, 466; J. Chem. Soc,,
1969, 11363.
50. E. L. Smith. Chemistry and
Industry, 1957, 572.
51. J. Beiler, J. Moss, G. Mar-
t i n. Science, 114, 122 (1951).
52. G. V i 1 1 e 1 a, L. A b r e u. Science,
115, 205 (1952).
53. H. Schmid, A. Ebnot her,
P. Karrer. Helv. Chim. Acta,
36, 65 (1953).
54. J. E 1 1 i n g b о e, J. M о r r i-
s о n, H. D i e h 1. Jowa State
Coll. J. Sci., 30, 263 (1955); РЖХ,
1957, 66237.
55. O. Schindler. Helv. Chim.
Acta, 34, 101, 1356 (1951).
56. G. Beavan, E. Johnson.
Nature, 176, 1264 (1955).
57. H. H i 1 1, G. P r a t t, R. W i 1-
1 i a m s. J. Theor. Biol., 3, 423
(1962).
58. H. Diehl, R. M u r i e. Iowa
State Coll. J. Sci., 26, 555 (1952).
59. H. H i 11, G. P r a t t, R. W i 1-
1 i a m s. Chem. and Ind, 1964, 197.
60. H. H о g e n к a m p, H. В a r-
k e r, H. M a s о n. Arch. Bio-
chem., 100, 353 (1963).
61. T. К a t o, S. S h i m i z u,
S. Fukui. Bitamin, 31, 175
(1965).
62. R.Iamodo, Sh. Shimi-
zu, S. F u к u i. Ach. Biochem.
Biophys., 117, 675 (1966).
63. К. В e r n h a u e г, О. M fl 1-
1 e r, G. M fl 1 1 e r. Biochem.
Z_, 336, 102 (1962).
64. R. В a r n e t t, H. H о g e n-
k a m p, R. A b e 1 e s. J. Biol.
Chem., 241, 1483 (1966). . .
65. О. M fl I i e r, G. M fl 1 1 e r. Bio-
chem. Z., 336, 299 (1962).
66. E. L. S m i t h, L. Me r v у n.
Biochem. J„ 86, 2P (1963).
67. J. С о 1 1 a t, J. Abbot. J.
Am. Chem. Soc., 86, 2308 (1964).
68. D. D о 1 p h i n, A. J о h n s о n,
R. Rodrigo. Ann. N. Y. Acad.
Sci., 112, 590 (1964).
69. O. Muller, K. Bernhauer.
Ann. N. Y. Acad. Sci., 112, 575
(1964).
70. J. Conn, T. Wartman n.
Science, 115, 72 (1952).
71. J. Brockman n, J. Pierce,
E. S t о к s t a d, H.Bro-
q u i s t, T. J u к e s.- J; Am.
Chem. Soc., 72, 1042 (1950).
615
72. W. Veer, J. Edelhausen,
H. Wijmenga, J. Lens.
Bioch. Biophys. Acta, 6, 225 (1950).
73. J. В oige, R. Cote. Франц,
пат. 1346256; РЖХ, 1965, 20H214.
74. J. Bayer. Pharmazie, 19 , 602(1964).
75. S. P i e r r e 1. Бельг, пат. 618755;
С. A., 58, 9108 (1963).
76. H. Hogenkamp, J. Rush.
Biochem. Prep., 12, 121 (1968).
77. E. К a c z к a, D. W о 1 f, F. Ku-
ehl, K. F о 1 к e г s. J. Am.
Chem. Soc., 71, 1514 (1949).
78. E. L. Smith. Англ. пат. 974284;
РЖХ, 1966, 23H457.
79. G. F a b r i z i о, I. R о 1 o-
v i c h. Англ. пат. 1012360; РЖХ,
1967, 14H360.
80. H. W i j me n ga, W. V e e r,
J. Lens. Bioch. Biophys. Acta,
6, 229 (1950).
81. A. C h a i e t. Ch. Rosen-
blum, D. Woodbury. Sci-
ence, 111, 601 (1950).
82. C h. Rosenblum, D. Woo d-
b u r y. Science, 113, 215 (1951).
83. W. F r i e d r i c h, K.Bern-
h a u e r. Chem. Ber, 89, 2030
(1956).
84. G. Boxer, J. Rickards.
Arch, bioch., 29, 75 (1950); 30, 372,
382, 392 (1951); Arch, bioch. bio-
phys., 39, 281 (1952).
85. G. Rudkin, R. Taylor.
Anal. Chem., 24, 1155 (1952).
.86. В. H. Б у к и и, Л. Я. А р е ш-
к и н а, Л. С. К у ц е в а. Био-
химия, 19, 713 (1954).
87. Е. L. Smith. Nature, 161, 638;
162, 144 (1948).
88. Е. Chargaff, С. Levine,
С. G г е е n, J. К г е a m. Ехре-
• rienzia, 6, 229 (1950).
89. В. Е 1 1 i s, V. Р е t г о V,
G. S п о о к. J. Pharm. Pharma-
col., 1, 60 (1949).
90. D. W о 1 f, W. J о n e s, J. Va-
lia n t, K. F о 1 к e r s. J. Am.
Chem. Soc., 72, 2820 (1950).
91. G. С о о 1 e у, M. D avies,
В. E 1 1 i s, V. P e t г о v,
В. S t u r g e о n. J. Pharm.
Pharmacol., 5, 257 (1953).
92. N. В г i n к, K. F о 1 к e r s. J.
Am. Chem. Soc., 71, 2951, 4442
(1949).
93. E. Holiday, V. P e t г о w. J.
Pharm. Pharmacol., 1, 734 (1949).
94. G. В e a v a n, E. H о 1 i d a y,
E. J о h n s о n, В. E 1 1 i s,
P. Mama I is, V. P e t г о w,
B. Sturgeon. J. Pharm. Phar-
macol., 1, 957 (1949).
95. G. С о о 1 e у, В. E 1 1 i s, P. Ma-
rn a 1 i s, V. P e t г о v, B. S t u r-
ge о n. J. Pharm. Pharmacol.,
2, 579 (1950).
96. N. В г i n k, F. Holley,
C. S h u n к, E. P e 1 1, J. C a-
h i 1 1, K. F о 1 к e г s. J. Am.
Chem. Soc., 72, 1866 (1950).
97. N. Brink, K. F о 1 к e r s. J.
Am. Chem. Soc., 74, 2856 (1952).
98. F. Holly, C. S h u n k.
E. P e e 1, J. C a h i 1 1, J. L a-
vigne, K. Folkers. J. Am.
Chem. Soc., 74, 4521 (1952).
99. P. M a m a 1 i s, V. P e t г о w,
B. Petrow, B. Sturgeon.
J. Pharmacol., 2, 491, 503, 512
(1950).
100. J. Buchanan, A. John-
son, I. M i 1 1 s, A. T о d d.
Chemistry and Industry, 1950, 426;
J. Chem. Soc., 1950, 2845.
101. D. H о d g к i n, A John-
son, A. T о d d. Recent work on
naturally occurring nitrogen hete-
rocyclic compounds, Spec. Publ.
№ 3, p. 109; London, 1955.
102. E. К a c z к a, D. H e у 1,
W. J о n e s, K. F о 1 к e r s. J.
Am. Chem. Soc., 74, 5549 (1952).
103. R. Bonnet, J. Buchanan,-
A. J о h n s о n, A. T о d d. J.
Chem. Soc., 1957, 1168.
104. E. К a c z к a, K. F о 1 ke r s.
J. Am. Chem. Soc., 75, 6317 (1953).
105. N. Brink, D. Hodgkin,
J. Lindsey, J. Pick»
worth, J. Robertson,
J. White. Nature, 174, 1169
(1954).
106. C. Garbers, H. Schmid,
P. Karrer. Helv. Chim. Acta,
38, 1490 (1955).
107. F. Kuehl, C. S h u n k,
M. M о or e, K. F о 1 к e r s.
J. Am. Chem. Soc., 77, 4418 (1955).
108. F. К u e h 1, C. S h u n k,
K. F о 1 к e r s. J. Am. Chem. Soc.,
77, 251 (1955); Angew. Chem., 67,
658 (1955).
109. O. S c h i n d 1 e r. Z. Vitamin-,
Hormon-, und Fermentforschung,
5, 66 (1952).
110. G. В e a v a n, E. H о 1 i d a y,
E. J о h n s о n, В. E 1 1 i s,
V. P e t г о w. J. Pharm. Phar-
macol., 2, 733, 944 (1950).
111. J. С a n n о n, A. J о h n s о n,
A. Todd. Nature, 174, 1168
(1954).
112. D. Hodgkin, J. Kamper,
J. Lindsey, M. MacKay,
J. Pickworth, J. Robert-
son, C. Shoemaker,
J. White, R. P г о s e n,
K. Trueblood, Proc. Roy.
Soc. (London), A242, 228 (1957).
113. D. H о d g к i n, J. L i n d s e y,
M. MacKay, K. True-
blood. Proc. Roy. Soc. (London),
A266, 475 (1962).
114. D. H о d g к i n, J. L i n d s e y,
R. Sparkes, K. True-
bio о d, J. W h i t e. Proc. Roy.
Soc. (London), A 266, 494 (1962).
115. E. К a c z к a, D. Wolf,
K. Folkers Пат. США
2738302; С. A., 50, 11621 (1956).
116. E. L. S m i t h et. al. Bioch. J.,
48, 1 (1951).
616
117. E. L. S m i t h," E. В a 1 1,
D. Ireland. Biochem. J., 52,
395 (1952).
118. R. В u h s, E. Newstea d,
N. T r e n n e r. Science, 113, 625
(1951).
119. K. F о 1 к e r s, D. Wolf,
Vitamins and Hormones, vol. 12,
1; New York, 1954.
120. J. Pf i I f ner, D. С a 1 к i n s,
R. P e t e r s e n, О. В i r d,
V. M c G 1 о h о w, R. S t i p e k.
Abstr. Amer. Chem. Soc. Meeting,
Sept. 1951, 22c, 120th.
121. W. Friedrich, K. Bern-
h a u e r. Chem. Ber., 91, 1665,
2061 (1958).
122. F. R о b i n s о n, J. M i 1 1 e r,
J. M с P h e r s о n, K. F о 1-
k e r s. J. Am. Chem. Soc., 77,
, 5192 (1955).
й. 123. W. F r i e dr i ch, К. В e r n-
h a u e r. Z. Naturforsch. 9b, 685
.124. C. Shunk, F. R о b i n s о n,
J. McPherson, M. Gas-
ser, K. F о 1 к e r s. J. Am.
Chem. Soc., 78, 3228 (1956).
125. H. D i о n, D. С a 1 к i n s,
J. P f i f f ne r. J. Am. Chem.
> Soc., 74, 1108 (1952).
126. W. F r i e d r i c h, K.Bern-
h a u e r. Chem, Ber., 89, 2507
(1956).
127. J. M on t gome r-y, H. Tho-
mas. J. Am. Chem. Soc., 85,
2673 (1963); 87, 5442 (1965).
128. H. D i о n, D. C a 1 к i n s,
J. Pfiffner. J. Am. Chem. Soc.,
76, 948 (1954).
129. W. F r i e d r i c h, K.Bern-
hauer. Chem. Ber., 90, 465
(1957).
130. W. F r i e d r i c h, H. Hei n-
r i c h. Biochem. Z., 333, 550
(1960).
131. F. Brown, J. Cain*
D. G r a n t, L. P a г к e r,
E. L. Smit h. Biochem. J., 59,
82 (1955).
132. J. Pfiffner, H. Dion,
D. Calkins. Federation Proc.,
13, 274 (1954).
133. W. F r i e d r 1 c h, K. Bern-
hauer. Angew. Chem., 69,
478 (1957); Chem. Ber., 90, 1966
(1957).
134. W. F r ie dr ic h, K.Bern-
h a u e r. Angew. Chem., 71, 311,
(1959); Z. physiol. Chem., 317,
116 (1959).
135. К. В e r n h a u e r, F. W a g-
n e r. Biochem. Z., 335, 453 (1962).
136. J. F о r d, J. P о r t e r. Brit.
J. Nutrition, 7, 326 (1953).
137. W. F r i e d r ic h, K.Bern-
h a u e r. Angew. Chem., 68, 580
(1956).
138. E. Holdsworth, J. Ford,
S. К о n, J. Porter. Nature,
171, 148, 150. (1953).
139. G. Gross, W. Friedrich,
K. Bernhauer. Chem. Ber.,
90, 1202 (1957).
140. R. Minot, W. Murphy.
J. Am. Med. Assoc., 87, 470 (1926).
141. O. S c h i n d 1 e r, T. R e i c fa-
st e i n. Helv. Chim. Acta, 35,
307 (1952).
142. J. Pierce, A. Page,
E. Stockstad, T. Jukes.
J. Am. Chem. Soc., 71, 2615,
2952 (1949).
143. R. Cote. Англ. пат. 954965;
РЖХ, 1966, 2H329.
144. К. Bernhauer, W. Fried-
rich. Пат. ФРГ 922126,
932982, 941150, 944216; РЖХ, 1957,
5936, 42616, 64612, 55698; пат. ФРГ
1037653; РЖХ, 1961, 7Л381.
145. W. S t е v е n s, В. W о 1 п а к,
R. Zinn. Chem. Eng. Progr., 51,
163 (1955).
.146. T а к а т a. Vitamins, 8, 369 (1955);
РЖХ, 1957, 58785.
147. В. Wol nar, R. Z i n n. Пат.
США 2833692; РЖХ, 1960, 70673.
148. К. Bernhauer, W. Fried-
rich. Пат. ФРГ 936287; РЖХ,
1957, 5937.
149. G. Ledingham. Ann. Rev. Mi-
crobiol., 7, 433 (1953).
150. J. Garibaldi, K. Ijihi,
N. S n e 1 1, J. Lewis. Ind.
Eng. Chem., 45, 838 (1953).
151. A. Leviton, R.Hargro-
v e. Ind. Eng.' Chem., 44, 2651
(1952).
152. P. Le n he r t, D.Crow-
f о о t - H о d g k-1 n. Nature, 192,
937 (1961).
153. N. В r i n k, F. W о 1 f. Пат.
США 2607717; С. A., 46, 11594
(1952).
154. N. В r i n к, T. Woo d. Am.
пат. 2609325; C. A., 47, 833 (1953).
155. H. В u n g а у, M. Mar sk,
R. P e t e r s о n. J. Biochem. Mi-
crobiol. Technol. Eng., 2, 419 (1960).
156. Г. В. Самсонов, Г. 3.
Елькин, Л. В. Д митр и-
е н к о и др. Авт. свид. СССР
173884; РЖХ, 1966, 18Н332.
157. G. N о m i п е, L. Р е п a s s е,
Р. Barthelemy. Франц,
пат. 1216221, 75254; РЖХ, 1961,
7Л379; 1962, 18Л259.
158. А. М. Ю р к е в и ч, И. П. Р у-
дакова, Т.Н. Поспело-
в а. ЖОХ, 36, 850 (1966).
159. Пат. США 2683680, 2695862,
2709669, 2731389, 2787578. 2793161,
2899360; С. А., 49, 6550. (1955);
РЖХ, 1957, 5933, 5935, 61877;
С. А., 51, 142 0, 13325 (1957);
РЖХ, 1960, 70672.
160. Аигл. пат. 675247, 692803,
708038, 718454, 721414, 765537;
С. А., 46, 10555 (1952); 48, 14130
(1954); 49, 4723 (1955); РЖХ,
1957, 5934, 42617; С. А., 51, 14210
(1957).
161. Канад, пат. 509642, 510309,
617
510310, 513750, 513776; РЖХ, 1957,
24589, 20821, 20822, 52572, 52573.
162. Пат. ФРГ 920933, 930651, 932981,
933052, 937372, 940369, 940423,
940489, 944216: РЖХ, 1956, 26961,
79420; РЖХ, 1957, 39093, 35708,
20823, 52575, 32351, 52574, 55698.
163. Д. Г. Колесников, Ю. В.
Шостенко, И. С. Ситом,
Н. Г. Божко. Авт, свид. СССР
105644; Бюлл. изобрет., 1957, № 3,
19.
164. V. К ос her, Е. S о г k i п.
Helv. Chim. Acta, 35, 1741 (1952).
165. A. Latner, R. Merrill s,
L. R a i n e. Lancet. 1, 497
(1954); Biochem. J., 57, XIX (1954).
166. В. H. Б у к и и, И. С. Л а г о т-
к и и, Э. Д. Михлин, В. Я.
Быховский, Е. С. Па и ц-
х а в а, И. И. П е л е в и и,
И. М. 3 а р и ц к и й. Авт. свид.
СССР 140539; РЖХ, 1962, ЦЛ285.
167. В. Н. Б у к и и, В. Я- Б ы х о в-
с к и й, Е. С. П а н ц х а в а,
Л. Н. К о н д а к о в а, И. С.
Лаготкин. Авт. свид. СССР
175182; РЖХ, 1966, 18НЗЗЗ.
168. М. S h о г b. Science, 107, 397
(1948).
169. R. Clark; W. J о nes,
W. R a i c h, K. F о I k e r s. J.
Am. Chem. Soc.,. 76, 3995 (1954).
170. J. Stevens, R. Ness,
H. F 1 e t c h e r. J. Org. Chem.,
33, 1806 (1968).
171. F. W e у g a n d, F. W i r t h.
Chem. Ber., 85, 1000 (1952).
172. D. H e у 1, E. Chase,
C. S h u n k, M. Moore,
G. Emerson, K. Folkers.
J. Am. Chem. Soc., 76, 1355 (1954).
173. W. F r i e d r i c h. Z. Naturforsch.,
18b, 455 (1963).
174. R. B. Woodward. Pure and
Appl. Chem., 17, 519 (1968).
175. R. B. Woodward. Angew.
Chem., 75, 871 (1963).
176. J. Y a m a d a, D. M i.l j k o-
v i с, P. W e h r I i, B. Gol-
din g, P. L б 1 i g e r, R. Kee-
s e, К. M й 1 le r, H. E s c hen-
mo s e r. Angew. Chem., 81, 301
(1969).
177. R. B. Woodward. Pure and
Appl. Chem., 25, 283 (1971); 33,
145 (1973).
178. A. Eschenmoser. Quart.
F- Rev. 24, 366 (1970).
179. K. Bernhauer, F. W a g-
n e r, P. Z e 1 1 e r. Helv. chim.
Acta, 43, 696 (1960).
180. K. Bernhauer, F. W a g-
i пег, H. Dellweg, P.Zel-
1 e r. Helv. chim. Acta, 43, 700
(1960).
181. W. F r i e d r i c h, G. G г о s s,
K. Bernhauer, P. Zeller.
Helv. chim. Acta, 43, 704 (1960).
182. F. W a g п ед. Biochem. Z„ 336,
99 (1962).
183. G. T e n e r. J. Am. Chem. Soc.,
83, 159 (1961).
184. F. Wagner. Biochem, Z., 336
99 fl9621
185. К. В e r n h a u e r, F. W a g-
n e r. Z. physiol. Chem., 322, 184
(1960); Biochem. Z„ 335, 325 (1962).
186. H. H e i n r i c h, W. Fr ied-
r i c h, P. R i e d e 1. Biochem.
Z„ 334, 284 (1961).
187. K. Bernhayer, F. W a g-
n e r. Z. physiol. Chem., 322, 194
(1960).
188. К. В e r n h а у e r, H. D e 1 1-
w e g. Z. physiol, chem., 322, 190
(1960).
189. T. А. Меле нтье в a, H. Д.
Пекель, В. М. Березов-
ский. Успехи химии, 38, 2016
(1969).
190. Е. L. S m i t h. Nutr. abstr.
reviews, 20, 795 (1951); Биохи-
мия и физиол. витаминов, 6, М.,
ИЛ, 1953.
191. G. N о г m a n, L. Р о n a s s е,
Р. В а г t he 1 е m у. Пат. США
3268409; С. А., 65, 15167 (1966).
192. L. Rolland. Фраиц. пат.
1477М (1962); РЖХ, 1964, 7Н214.
193. Е. Cohn, R. G г е е n. Scien-
ce, 109, 443 (1949).
194. Е. Stockstad, Т. Jukes,
J. Р i е г с е, А. Р a g е. Fran-
klin. J. Biol. Chem., 180, 647
(1949).
195. Е. Meulengracht. Brit.
Med. J., 1954, 838.
196. В. В. Ковалевский. При-
рода, 1954, № 4, 11.
197. E. Л. Смит, Витамин Вц, М.,
1962.
.198. J. Paweikiewicz, К, N о-
wakowska. Acta Biochim. Ро-
lon., 2, 313 (1955).
199. Н. D е 1 1 w е g, Е. В е с h е г,
К. Bernhauer. Biochem.,
Z., 328, 96 (1956).
200. К. Bernhauer. Швейц, пат.
385871; РЖХ, 1967, 12Н443.
201. G. Е m е г s о п, F. Н о 1 1 у,
С. S h u п k, N. Brink,
К. F о 1 к е 1 s. J. Am. Chem.
Soc., 73, 1068 (1951).
202. G. E m e r so n, N. В r i n k,
F. H о 1 1 y, F. К о n i и s z y,
D; Hey 1, K. F о I ker s. J.
Am. Chem. Soc., 72, 3084 (1950).
203. K- Folkers. Intern. Z. Vi-
taminforsch., 23, 429 (1952).
204. H. В a г к e r, R. S m у t h,
H. We issbach, J. Too-
hey, J. Ladd, B. Volcan i,
J. Biol. Chem., 235, 480 (1960).
205. К. В e r n h а и e г, О. M й 1-
1 e r. Biochem. Z„ 334, 199 (1961).
206. A. J о h n s о n, N. S h a w. J.
Chem. Soc., 1962, 4608.
207. R. Brady, H. Barker. Bio-
chem. Biophys. Res. Communs, 4,
373 (1961).
208. G. Moss, C. Reese,
K. S h о f i e I d, . R. S h a p i-
r о, A. T od d. J. Chem. Soc.,
1963, 1149.
618
209. A. Johnson,' N. S h a w,
F. Wagner. Biochim. Biophys.
Acta, 72, 107 (1963).
210. J. G u e s t, S. F r i e d m a n,
D Woods, E. L. S m i t h.
Nature, 195, 340 (1962).
211. S. S h a p i r o, F. S c h 1 e n k.
Advanc. EnzymoL, 22, 243 (1960).
212. A. J о h n s о n, N. S h a w.
Proc. Chem. Soc. (London), 1960,
420.
213. F. Wagner, K. Bernhau-
er. Ann. N.Y. Acad. Sci., 112,
580 (1964).
214. F. Wagner. Proc. roy. Soc.
(London), Ser. A, 228, 344 (1965).
215. T. Kato, S. Shimizu,
S. Fukui. J. Chem. Soc. Ja-
pan, 66, 1047 (1963).
216. F. W a g n e r, P. R e n z. Tet-
rahedron Letters, 1963, 259.
217. A. J о h n s о n, N. S h a w. Proc.
Chem. Soc., 1961, 447. _‘r •;
J./218. H. Weissbach, J. To o-LA .
hey, H. В a г к e r. Proc. '
Natl. Acad. Sci. U.S., 45, 52!^-'
(1959). ,
:: .219. P. R i 1 e у, P. J а с к s о n,
D. Ross, P. Savage.
Soc. Chem. Ind. (London), Mono-
graph., 12, 127 (1961).
,220. J. Speedie, G. Hull, пат. США
2951017; С. A., 54, 25561 (1960).
221. D. P e г 1 m a n, J. S e m a r.
BiotechnoL Bioeng., 5, 21 (1963).
222. К. В e г n h a u e r, P. G a i-
s e г, О. M u 1 1 e r, O. W a g-
ne r. Biochem. Z„ 333, 106 (I960).'
223. J. Pawelkiewicz, В. В ar-
iosi n s к i, W. Walerych.
Bull. Acad. Pol. Sci., Ser. Sci.
biol, 8, 123 (1960).
224. К. В e r n h a u e г, О. M ii 1-
1 e r. Biochem. Z., 335, 44 (1961).
225. A. M i g 1 i a с c i, A. R u s c o-
n i. Biochim. Biophys. Acta, 50,
370 (1961).
226. J. T о о h e у, H. В a г к e r.
Proc. NatL Acad. Sci. U. S., 45,
521 (1959).
227. J. T о о h e y, D. P e r 1 m a n,
H. В a г к e r. J. Biol. Chem., 236,
2119 (1961).
228. K. Bernhauer, G. Reber.
Biochem. Z„ 335, 463 (1962).
229. M. К a m p e r, D. H о d g к i n.
Nature, 176, 551 (1955).
230. E. L. S ni i t h, L. M e r v у n,
A. J о h n s о n, N. S h a w. Na-
ture, 194, 1175 (1962).
231. E. L. Smi t h, L. Me r v у n,
R. Muggleton et al. Ann.
N. Y. Acad. Sci., 112, 565 (1964).
232. E. L. S m i t h, L. M e r v у n,
V. Clark. Англ. пат. 963373;
РЖХ, 1966, 22H293.
233. H. H о ge n к a m p, W. P a i-
1 e s. Biochem. Prep., 12, 124
(1968).
234. R. S c h m i d t, F.Huen-
n e к e n s. Arch. Biochem. Bio-
phys., 118, 253 (1967).
235. A. M. Ю p к e в и ч, И. П. P y-
д а к о в а, Т. А. Поспело-
в а. ЖОХ, 39, 425 (1969).
236. Е. L. S m i t h, L. Mer vy n,
V. Clark. Англ. пат. 963373;
РЖХ, 1966, 22H293.
237. P. О v e r a t h, E. S t a d t-
m a n, G. Ke Her man,
F. L у n e n. Biochem. Z-„ 336,
77 (1962).
238. H. H о g e n к a m p, T. О i к a-
m a. J. Biol. Chem., 239, 1911
(1964).
239. B. Z a g a 1 a k, L. Pawel-
kiewicz. Acta biochem. pol.,
12, 103 (1965).
240. И. П. P у д а к о в a, T. А. По-
спел о в a, А. М. Ю р к е-
в и ч. ЖОХ, 40, 2493 (1970).
241. В. А. Я к о в л е в, А. А. П о з-
н а н с к а я, Н. К. Просве-
те в а, И. П. Р у д а к о в а,
Т. А. Поспелова, А. М.
Юр ке в и ч. ДАН СССР, 197,
230 (1971).
242. R. Barnett, Н. Hogen-
к a m р, R. A b е 1 е s. J. Biol.
Chem., 241, 1483 (1966).
243. В. М. Б е р е з о в с к и й, Т. В.
Еременко. Хим.-фарм. ж.,
1967, № 7, 8.
244. Н. Eg ger е г, Р. Overath,
F. Lynen, Е. Stadtman.
J. Am. Chem. Soc., 82, 2643 (1960).
245. R. A b e 1 e s, H. Lee. J. Biol;
Chem., 236, 2347 (1961).
246. J. S t e r n, G. F г i e d m a n.
Biochem. Biophys. Res. Communs,
2, 82 (1960).
247. H. Wood, R. Ke 1 ler me-
yer, R. Stjernholm,
S. Allen. Ann. N. Y. Acad. Sci.,
112, 661 (1964).
248. E. Phares, M. Long,
S. Carson. Biochem. Biophys.
Res. Communs, 8, 142 (1962); Ann.
N. Y. Acad. Sci., 112, 680 (1964).
249. R. Kellermeyer, H. Wood.
Biochemistry, 1, 1124 (1962).
250. S. G u r n a n i, S. M i s t r y,
B. J о h n s о n. Biochim. Bio-
phys. Acta, 38, 187 (1960).
251. T. Stadman. J. Biol. Chem.,
238, 2766 (1963); Ann. N. Y. Acad.
Sci., 112, 728 (1964).
252. R. В 1 a к I e у. H. В a г к e r.
Biochem. Biophys. Res. Communs,
10, 391 (1964).
253. J. Z a 1 t a, F. M e у e r. Expe-
rientia, 21, 519 (1965).
254. H. Uchi no, J. J a g i г i,
T. Yoshino et al. J. Vita-
minol., 9, 251 (1963).
255. J. G u e s t, G. W о о d s. В ки.:
Vitamin B12 und Intrinsic Factor,
Hamburg, 1961, 686.
256. H. A r n s t e i n, A. W t i t e.
В кн.: Vitamin Bj2 und Intrinsic
Factor, Hamburg, 1961, 211.
257. R. D e W a g n e, A. N i col.
J. Biol. Chem., 237, 2278 (1962).
258. S. Takeyam a, J. Bucha-
nan. J. Biochem., 49, 576 (1961).
259. S. Та к e у a m a, F. H a t c h.
619
J. Buchanan. J. Biol. Chem.,
236, 1102 (1961).
В. H. Букин, ДАН СССР
148, 707 (1963).
260. Л. Я. A p e ш к и н а, Л. С. 262. Н. W е i s s b a c h, H. В a r-
Куцева, E. П. Скоробо-
гат о в a. ДАН СССР, 169, 227
(1966).
к е г. Труды 5-го Междунар. био-
хим. конгресса М., 1962, симп. 4,
281.
261. Л. Я. Арешки на, Н. А.
Андреева, Л. С. Куцева,
263. R. Stjernholm, Н. С.
Wood. Proc. Natl. Acad. Sci.
U. S., 47, 303 (1961),
ГЛАВА XIV
НЕКОТОРЫЕ ПРИРОДНЫЕ БИОЛОГИЧЕСКИ АКТИВНЫЕ ВЕЩЕСТВА
Кратко рассматриваемые в этой главе некоторые важные природные био-
логически активные вещества по тем или иным причинам ранее относили
к витаминам, однако на самом деле к витаминам они причислены быть не
могут.
' Наряду с незаменимыми аминокислотами незаменимые ненасыщенные
высшие жирные кислоты (витамин F) и аминоспирт холин участвуют в по-
строении различных тканей.
Другая группа соединений — пангамовая кислота, карнитин, S-метил-
метионин (последние два соединения в этой главе не рассматриваются)
и некоторые другие — относятся к многочисленным известным к настоя-
щему времени метаболитам.
Биофлаваноиды являются фармакодинамическими веществами. Ни у че-
ловека, ни у животных отсутствие биофлаваноидов не вызывает специфи-
ческой недостаточности, и им не свойственны какие-либо явно выражен-
ные биокаталитические функции.
пара-Аминобензойная кислота (витамин Нх) (рассматривается в гл. XI)
и мезо-инозит представляют собой ростовые вещества микроорганизмов.
ЛИНОЛЕВАЯ, ЛИНОЛЕНОВАЯ И АРАХИДОНОВАЯ КИСЛОТЫ
К незаменимым насыщенным высшим жирным кислотам (витамин F)
[1] относятся три кислоты, содержащие 18 или 20 атомов углерода и от
двух до четырех несопряженных двойных связей с полной z/пс-конфигура-
цией [2, 3]:
линолевая кислота (С18) — 9, 12-ди-1/ис-октадекадиеновая кислота (I)
Н Н НН
линоленовая кислота (С18) — 9,12,15-три-г/ыс-октадекатриеновая ки-
слота (II)
НН НН НН
С~Сч уС—Сч ,с=с>
СН3СН*
(СНг)7 соон
арахидоновая кислота (С^) —5,8,11,14-тетра-цис-эшсозатетраеновая ки-
слота (III)
620
Линолевая кислота (I) имеет т.кип. 149,5°С при 1 мм,
182,4°С при 4 мм; т.пл. —5° [51, +11° [41; d? 0,903; 1,4697, п*
1,4588; йодное число 181,0; бромное число 100; максимум поглощения
Хиакс 190 нм [6]. Производные линолевой кислоты: тетрабромид—т.пл.
114—115° С; этиловый эфир—т.кип. 173,5° С при 0,5 мм; метиловый
эфир — т.кип. 145—150°С при 0,05—0,1 мм [7].
Линоленовая кислота (II) имеет т.кип. 157—158° С при
0,001—0,002 мм, 184° С при 4 мм [8], 230—232° С при 17 мм; т.пл. — 11° С;
0,914; пд 1,4780, п'о 1,4678; йодное число 273,5; максимум поглоще-
ния 195 и 175 нм. Производное линоленовой кислоты: гексабромид —
т.пл. 180—181° С [9].
Арахидоновая кислота (III) имеет т.кип. 160—165° С при
1 мм; т.пл. — 49,5° С; йодное число 333,5; максимум поглощения Хмажс
294 нм. Производные арахидоновой кислоты: октабромид [101; метиловый
эфир —т.кип. 177—178° С при 0,35 мм; октабромид метилового эфира —
т.пл. 227—230° С [11].
Для незаменимых жирных кислот (не синтезируемых животным орга-
низмом) характерно специфическое биологическое действие, вследствие
чего их относили к витаминам под названием витамин F [4, 5]; однако они
не являются витаминами [12].
Впервые потребность животных (крыс) в незаменимых жирных кисло-
тах обнаружили Г. и М. Бурр в 1929 г. [1 ].
Незаменимые жирные- кислоты — линолевая, линоленовая и арахидо-
новая, по-видимому, осуществляют в животном организме функции окис-
ления насыщенных жирных кислот, участвуя тем самым в процессе усвое-
ния жиров [13] и в жировом обмене кожных покровов [14].
Следует отметить, что транс-изомеры незаменимых жирных кислот и не-
насыщенные кислоты с сопряженными двойными связями, например 9,11-
октадекадиеновая кислота, физиологической активностью не обладают.
По своей активности арахидоновая кислота превышает активность ли-
нолевой и линоленовой кислот приблизительно в 10 раз.
Линолевая и линоленовая кислоты содержатся в различных раститель-
ных продуктах в виде их триглицеридов, т. е. в виде ненасыщенных жиров,
и в некоторых животных продуктах в виде фосфолипидов. Арахидоновая
кислота встречается только в животных жирах.
Линолевая кислота находится также в масле зародышей зерен ржи
и других злаковых.
Жир человека содержит 8,2—11 % линолевой кислоты и 0,3—1 % ара-
хидоновой кислоты по отношению ко всем жирным кислотам [16].
Полноценная пища должна иметь в своем составе 0,1% арахидоновой
кислоты или 1% линолевой и линоленовой кислот [15].
Вопросы химии, выделения и синтеза незаменимых ненасыщенных выс-
ших жирных кислот рассмотрены в опубликованных работах [17, 18].
БИОФЛАВАНОИДЫ
Среди многочисленных широко распространенных в растениях дубиль-
ных и красящих веществ, в молекулу которых включается ядро хромана
(бензо-у-дигидропирана), имеется группа соединений, производных флава-
на (2-фенилхромана), обладающих способностью уменьшать проницаемость
кровеносных капиллярных сосудов. Некоторые исследователи относили эти
621
биологически активные вещества к витаминам группы Р (permeability —
проницаемость) [19, 201. Однако биологическая активность их носит фар-
макодинамический характер [19] и они не являются витаминами. Предста-
вители веществ, уменьшающих проницаемость кровеносных капилляров,
встречаются среди следующих соединений:
катехинов, 3-оксипроизводных флавана (2-фенилхромана, IV), с двумя
асимметрическими центрами, представляющих собой бесцветные кристал-
лические вещества, обладающие свойствами дубильных веществ;
флаванонов, производных 4-оксофлавана (4-оксо-2-фенилхромана, V), с
одним асимметрическим центром,
красящих веществ плодов и ягод;
флавонов, производных 2,3-дегидрофлавана (2-фенилбензо-у-пирона,
2-фен ил хромона, VI,) — соединений, включающих в свою молекулу ядро
7-пирона, желтых красящих веществ цветов, корней и стеблей растений.
Биологически активные вещества, снижающие проницаемость капилля-
ров, характеризуются обязательным присутствием двух гидроксильных
групп в положениях 5 и 7 ядра бензопирана, а также заместителями в по-
ложениях 3',4' бензольного ядра.
В природе катехины, флаваноны и флавоны представлены или в свобод-
ном виде, в форме так называемых агликонов, или в виде гликозидов, обра-
зующихся по гидроксильной группе в положении 3, 5 или 7.
Катехины имеют два асимметрических атома углерода и представ-
лены в двух эпимерных формах, каждая из которых существует в виде ра-
цемата и двух оптических антиподов: d-, I- и dl-катехинов и их диастерео-
изомеров — d-, I- и dZ-эпикатехинов. В природе встречаются, по-видимому,
только d-, /-катехин и /-эпикатехин.
d-Катехин — б/-3,5,7,3'',4'-пентаокси-2-фенилхроман (VII) [211 —
представляет собой бесцветные иглы с т.пл. 174—175° С; [а 1578 + 16,9°
и [а ]р +18,7° (в ацетоне), [а 1о + 3,7° и [а ]578 ± 0° (в спирте). Из воды
он кристаллизуется с 4 молекулами воды; плохо растворим в холодной
воде и эфире, легко растворим в горячей воде и спирте, нерастворим в бен-
золе и хлороформе. d-Катехин встречается в гамбире, экстракте из малай-
ской манны, Uncaria gambir, бобах какао и др.
/-Эпи катехин — /-3,5,7,3',4'-пентаокси-2-фенилхроман (VIII) —
диастереоизомер d-катехина, представляет собой бесцветные призмы с т.пл.
237—239° С; [а $8 — 68° и [а Id — 69° (7% в спирте); [а ]578 — 60° (4% в
ацетоне). Из водных растворов он кристаллизуется с 4 молекулами воды;
плохо растворим в холодной воде и эфире, легко растворим в спирте и аце-
тоне. /-Эпикатехин встречается в древесине индийской Acacia catechu [221,
в чайном танине, в орехах Gila и др.
Галлокатехин — 3,5,7,3',4',5'-гексаокси-2-фенилхроман [231 —
содержится в чайном катехине (выделен из зеленого чая) (24, 25 ].
622
Флаваноны представлены в виде тетраоксипроизводных 4-оксо-2-
-фен ил хромана, их метиловых эфиров и гликозидов, находящихся в цве-
тах и плодах, по-видимому, в связанном состоянии с протеинами; они вхо-
дят также в состав лигнина.
Эриодиктиол — 5,7,3',4'-тетраоксифлаванон (IX) — представ-
ляет собой бесцветные листочки с т.пл. 267° С. Он умеренно растворим в
воде, плохо — в спирте и очень мало — в других органических раствори-
телях.
Эр и од иктин (X) по своему строению является 7-рамноглюкози-
дом эриодиктиола.
Гесперетин — 4'-метиловый эфир эриодиктиола, 5,7,3'-триокси-
4'-метоксифлаванон (XI) — кристаллизуется в листочках желтого цвета
с т. пл. 224—226° С. Он очень трудно растворьм в воде, бензоле, эфире;
растворим в спирте. Встречается в плодах цитрусовых [26] — апельсинах,
лимонах и др. — главным образом в виде гликозида гесперидина.
Гесперидии — 7-рамноглюкрзид гесперетина [271, 7-рамноглюко-
зидо-5,7,3'-триокси-4'-метоксифлаванон (XII) — представляет собой бес-
цветные иглы с т.пл. 252° С (по другим данным 261—262 и 266° С), трудно
растворим в воде, однако в два раза более активен, чем его агликон геспере-
тин. Гесперидии содержится в апельсинах [26], лимонах, черной смо-'
родине, шиповнике; содержание его в сухой кожуре мандарина составляет
5,68%, в сухом апельсине 3,07—8,2% [19]. Смесь флаванонов (глав-
ным образом гесперидина) и флавонов, выделяемых из лимонного сока,
называется цитрином [28].
Рамноглюкозидный остаток (С]2Н21Ов), состоящий из дисахарида ру-
тинозы, при гидролизе распадающейся на L-рамнозу и D-глюкозу, может
быть представлен следующей структурной формулой:
jS-l-Z-рамнозидо -6-Z7-глюкозид
1^—остаток агликона
К флавонам относятся хризин, апигенин, лютеолин, дисомерин,
скутеллареин, трицин, а к флавонолам (3-оксифлавонам) — кверцетин, га-
лантин, кемферол, физетин и многие другие. Наиболее важные биологиче-
ски активные флавоны следующие.
Кверцетин (Ci5H10O7 • ЗН2О) — 3,5,7,3',4'-пентаоксифлавон
(XIII) — представляет собой лимонно-желтые иглы с т. пл. 316—317° С
623
[29]; он является флавонолом лутеолина [30]. Кверцетин нерастворим в
холодной воде, очень плохо растворим в горячей воде; хорошо растворим
в уксусной кислоте и спирте, а также в разбавленных щелочах с образова-
нием интенсивной желтой окраски. С минеральными кислотами кверцетин
дает хорошо кристаллизующиеся соединения. Кверцетин известен с 1854 г.
[31 ]. Он встречается (как в свободном виде, так и в виде гликозида) во
многих растениях: в чае, хмеле, в листьях табака, в цветах (желтый лев-
кой, мать-мачеха, красная роза и др.), в конском каштане, сумахе, оболоч-
ках луковиц, в виноградной лозе, дубе Quercus tinctoria и др.
Рутин (С27Н3()О16-ЗН2О) —3-рутинозид кверцетина.З-рамногликозил-
3,5,7,3',4'-пентаоксифлавон (XIV) — представляет собой светло-желтые иг-
лыст. пл. 180—190° С [32 ] (разлагается при 214—215°С); [а ]д +12,19° (1 г
в 100 мл С2Н5ОН), —38,83° (1 г в 100 мл пиридина). Спектр поглощения:
Хыакс 257 и 362 нм (£1’См 32,5). Рутин очень мало растворим в холодной
воде (0,013%); лучше — в горячей (0,5 г в 100 мл при 100° С), в метиловом
спирте (5,5 г в 100 мл), ацетоне (0,4%); легко растворим в пиридине и раст-
ворах щелочей, нерастворим в эфире, бензоле, хлороформе. Рутин впервые
выделен в 1860 г. из зеленой гречихи [33], в которой его содержание дохо-
дит до 6% (на сухую массу) [34]. Он встречается в листьях табака, в цве-
точных почках и цветах (бузины, томатов и др.). Содержание рутина в цве-
точных почках Sophora japonia достигает 25% и выше.
Кверцетин Рутин
XIII XIV
Прочность капиллярных сосудов у человека, обезьяны и морской свин-
ки снижается при недостатке веществ, относимых к биофлаваноидам.
Впервые активность флавановых производных (эриодиктиол, геспери-
дии) как веществ, снижающих проницаемость капилляров, установлена
в 1936 г. [35].
Биологической активностью разнообразного характера обладают много-
численные представители ряда флавана: катехины, флаваноны и флавоны
[36], а также кумарин и его производные, такие, как 6,7-диоксикума-
рин [37 ].
По биологической активности 100 единиц витамина Р отвечают 1 г гес-
перидина или 0,15 г цитрина (смесь флаванонов и флавонов из лимонного
сока), а также 1,05 г рутина или 2—4 г гесперетина.
Вопросы синтеза биофлавоноидов детально рассмотрены в опубликован-
ной работе [19].
МЕЗО-ИНОЗИТ
Инозит — гексаоксициклогексан — представляет собой шестиатомный
циклогексановый спирт [38].
По своей пространственной структуре инозит может существовать в виде
восьми цис-, пгранс-стереоизомеров, семь из которых представляют собой
оптически неактивные мезо-формы, а один существует в виде двух оптиче-
ских антиподов и одного рацемата.
Из стереоизомеров инозита к веществам с физиологической активностью
относится только оптически недеятельный циклогексан, (1,2,3,5-^ис)гексол
(XV), обычно называемый мезо-инозитом, t-инозитом или мио-инозитом
(по-гречески <миос» — мышцы)
н он
XV
624
Конформационная формула мезо-инозита может быть представлена в
виде «кресла» с единственной северо-аксиальной гидроксильной группой
в положении 2, характеризующейся своим особо выступающим положением
в молекуле (еаееее) 1391.
связи:
са — северо-аксиальные,
юа — южно-аксиальные,
е — экваториальные.
Остальные гидроксильные группы (соединенные пунктирными линиями)
относятся к экваториальным группам.
У мезо(мио)-инозита аксиальная гидроксильная группа положения 2
и три экваториальные гидроксильные группы положений 1, 3, 5 располо-
жены по одну сторону кольца и обозначаются так же, как цис-гидроксилы.
В природных продуктах обнаружены мезо(мио)-инозит (XV), L- и .D-ино-
зиты и сциллит. Остальные формы в природе не найдены и, кроме полного
цис-инозита, получены только синтетически [40].
мезо-Инозит широко распространен в природе; он входит в состав жи-
вотных и растительных тканей, находится в мышцах [41], веществе мозга
(в виде кефалин-фосфолипида) и других органах. В растениях он встреча-
ется или в свободном виде, или в виде гексафосфорного эфира, фитина,
впервые найденного [42] в зернах злаков.
мезо-Инозит кристаллизуется в бесцветных моноклинических призмах
(из воды) при температуре ниже 50° С с 2 молекулами кристаллизационной
воды; на воздухе выветривается. Он перегоняется в вакууме, хорошо раст-
ворим в воде (при 12° С в 10 ч., при 23,6° С в 5,7 ч.), хуже — в разбавлен-
ной уксусной кислоте, почти нерастворим в безводном спирте, нерастворим
в эфире; обладает сладким вкусом.
жезо-Инозит обладает физиологической активностью для мышей в ка-
честве фактора против алопеции [44 ] (своеобразного выпадения шерсти —
плешивости). Он является ростовым фактором для некоторых культурных
рас дрожжей; однако дикие расы дрожжей и многие виды бактерий в полу-
чении инозита не нуждаются, так как способны его синтезировать.
Фитин обнаружен в растениях в виде кальциевой соли или смеси каль-
циевой и магниевой солей [43]. Источником для технического получения
фитина служит кукуруза.
Фитин (фосфорнокислый эфир мезо-инозита) обладает активностью для
мышей, но неактивен для микроорганизмов, по-видимому, вследствие их
неспособности расщеплять связанную форму инозита. Алкильные эфиры
мезо-инозита активностью для микроорганизмов также не обладают [43].
Причина благоприятного действия инозита в различных случаях состоит
в том, что, являясь ростовым фактором для некоторых микроорганизмов
кишечной флоры, он стимулирует микробиологический синтез недостающих
витаминов (например, биотина) [45].
холин
Холин (XVI) представляет собой метилированное производное р-амиио-
этилового спирта, коламина, и является четвертичным аммониевым основа-
нием, гидратом окиси триметил-р-оксиэтиламмония
СНЯ\ ОН-
сн,—й— сн,—сн,он
сн/
625
Холин (XVI)—вещество, которое входит в клеточные структуры как
составная часть двух фосфолипидов — лецитина и сфингомиелина — и од-
новременно является веществом, поставляющим метильные группы в реак-
циях метилирования различных соединений в организме. Для нормальной
жизнедеятельности организма необходимо поступление холина из внешней
среды. Холин не является витамином, его следует отнести к незаменимым
аминоспиртам; он используется как пластическое вещество для построения
клеток.
Холин представляет собой бесцветные, с запахом триметиламина, силь-
но гигроскопичные кристаллы, легко превращающиеся в вязкую жидкость.
Он легко растворим в воде и в безводном этиловом спирте.
Холин необходим для всех животных. В частности, в пищевой рацион
цыплят и индюшат нужно добавлять холин до содержания 0,15—0,20%.
Холин принимает участие в биосинтезе незаменимой аминокислоты —ме-
тионина—и некоторых других аминокислот в организме. Однако при недо-
статке холина в реакциях переноса метильной группы он частично может
быть заменен метионином [47 ], бетаином или бетаин-альдегидом.
Физиологическая активность и пищевое значение холина рассмотрены
в опубликованной работе [481, химические свойства и синтезы холина опи-
саны [17J.
ПАНГАМОВАЯ КИСЛОТА
Пангамовая кислота — витамин В15 — представляет собой 6-О-диме-.
тилглициновый эфир О-глюконовой кислоты (XVII)
СООН
I
неон
I
носн
I
неон
I
неон
CH2OCOCH2N(CH3)2
XVII
Это соединение было названо витамином В15 [49 J. Пангамовая кислота об-
ладает физиологической активностью. Она была выделена из водного экс-
тракта абрикосовых косточек, из ростков риса, пивных дрожжей, печени
быка и лошади [49, 50 J.
Кальциевая соль пангамовой кислоты кристаллизуется с одной моле-
кулой воды, хорошо растворима в воде. Описан синтез пангамовой кислоты
[511.
Физиологическое действие пангамовой кислоты заключается в активи-
ровании кислородного обмена в клетках различных тканей организма и в ее
способности быть донором метильных групп в реакциях метилирования.
Пангамовая кислота обладает выраженным липотропным действием и при-
меняется при различных заболеваниях печени [52 J.
СПИСОК ИСПОЛЬЗОВАННОЙ
ЛИТЕРАТУРЫ (К ГЛАВЕ XIV)
.1 . G. В и г г, М. В и г г. J. Biol.
Chem., 82, 345 (1929); 86, 587
(1930).
2. Т. Hilditch, Jasperson.
J. Soc. Chem. Ind., 58, 233 (1939).
3. A. M c-K ay, A. В a de r. J.
Org. Chem., 13, 75 (1948).
4. The Vitamins, Ed. W. Ser-
bell, R. Harris, vol. 11, New
York, 1954.
5. H. Vogel. Chemie und Technik
der Vitamine, vol. 1. Stuttgart,
1950.
6. J. R ussoH, J. P 1 a f t„
H.Klevens, G. Burr. J.
Am. Chem. Soc.; 67, 673 (1945).
7. C. Swift, W. Rose, G.Ja-
'm i e son. Oil and Soap, 20. 24t>
(1943).
626
'8 . J. Ma t tews, ‘ W. В rode,
J. В r own. J. Am. Chem. Soc.,
63, 1064 (1941).
9. G. S h i п о w a г a, J. В r own.
J. Am. Chem. Soc., 60, 2734 (1938).
10. J. Smedley-Mac-Lean,
L. Nunn. Biochem. J., 35, 983
(1941).
11. W. Ault, I. Brown. J. Biol.
Chem., 107, 615 (1934).
12. E. Schaunstein. Oster. Chem.
Z., 52, 28 (1951).
13. R. Sinclair. J. Biol. Chem.,
174, 343, 355 (1948).
14. F. G r a n d e 1. Fette und Seifen,
45, 94 (1938); 46, 150 (1939).
15. H. Aviles. Bui. extr. de inf.
del centro de investig. de plantes
у animales medicinales, 1953.
16. D. Cr a mer, I. В г о w n. J.
Biol. Chem., 151, 427 (1943).
17. В. M. Березовский. Хи-
мия витаминов, M., Пищепром-
издат, 1959.
18. „Vitamine”. Под ред. J. Fragner,
Jena, 1964—1965.
19. Н. Scarborough, A. Ba-
ch а г а с h. Vitamins and Hor-
mones, vol. 7, 1 (1949).
20. А. Л. К у p с а н о в, В. H. Б y-
кин, К. Л. Поволоцкая,
М. Н. Запрометов. Биохи-
мия, 15, 337 (1950).
21. Т. М a s о n. J. Soc. Chem. Ind.,
1928, 269.
22. W. Hutchings, T. Whee-
ler. J. Chem. Soc., 1939, 91.
23. С. M e n t z e r, D. P i 1 1 о n.
Bull. soc. chim. France, 1953, 538.
24. W. D e i j s. Rec. trav. chim., 58,
805 (1939).
25. M. H. Запрометов, ДАН
СССР, 87, 649 (1952); Биохимия, 17,
97 (1952).
26. G. Zemple n, R. Bognar.
. Ber., 75, 1043 (1942); 76, 452, 773
(1943).
27. Y. A sa h i n a, J. S h i п о d a,
M. Inubuse. J. Pharm. Soc.
Japan, 48, 29 (1928).
28. L. A r m e n t a n o. Z. Klin. Med.,
1936, 129, 685; Z. ges. exptl. Med.,
97, 630 (1936).
29. N. Waliaschko. Arch. Pharm.,
242, 232 (1904); 246, 249 (1908).
30. S. Kostanecki, V. Lam-
pe, I. T a m b о r. Ber., 37, 1402
(1904).
31. L. R i g a u d. Ann. Chem. Pharm.,
90, 283 (1854).
32. C. S a n d о, H. В a r t 1 e 11. J.
Biol. Chem., 41, 495 (1920).
33. F. S c h u n k. Mem. lit. phil. Soc.
Manchester, 15, 122 (I860).
34. I. N a g h s k i, W. P or ter,
J. С о z c h. J. Am. Chem. Soc., 69,
572 (1947).
35. L. Armentano, A. Bent-
sat h, T. В er e s, I. R u s z-
n у a k. Deut. med. Woschr., 62,
1326 (1936).
36. J. Lavollay, J. Neumann.
Compt. rend., 212, 251 (1941).
37. M. Javillier, J.Lavol-
' lay. Hel’’. Chim. Acta, 29, 1283
(1946).
38. L. M a q u e n n e. Lieb. Ann.,
12, 80 (1887).
39. T. P о s t e г n a k, D. Rey-
mond. Helv. Chim. Acta, 36, 260
(1953).
40. T. P о s t e r n a c k. Helv. Chim.
Acta, 19, 1333 (1936).
41. D. Scherer, Lieb. Ann., 73, 322
(1850).
42. В. И. Палладии. Z. Biol., 31,
199 (1895).
43. D. Woolley. J. Biol. Chem.,
136, 113 (1940); 139, 29 (1941);
J. Nutrition, 21, Suppl., 17
(1941).
44. D. Wooley. Science, 92, 384
(1940).
45. К. M. Л e у т с к и й. Витамины
(комплекс В2), изд. Львовского
ун-та, Львов, 1949.
46. F. В е i 1 s t ei п. Handbuch der
Organ. Chemie, 4, 277 (425)
(720).
47. V. D u V i gn e a u d, M. С о h n,
J. C h a n d 1 e r, J. S c h e п c k,
S. S i m m о n d s. J. Biol. Chem.,
140, 625 (1941).
48. Л. А. Ч e p к e с. Холин, как пи-
щевой фактор и патология холи-
нового обмена, М„ изд. АМН, 1953.
49. Т. Т о m i j a m a, Y. Y о п е.
Proc. Japan Acad., 29, 178
(1953).
50. E. К r e b s, E. К r e b s, J r.,
H. Beard, R. M a 1 i n, A. Har-
ris, С. В a r 1 e t t. Internal.
Rec. Med., 164, 19 (1951).
51. A. M. Ю p к e в н ч, С. Г. В e-
p e н н к и н a, M. С. Д о л г и x,
Н. А. Преображенский.
ЖОХ, 37, 1267 (1967).
52. В. Н. Буки и. Пангамат каль-
ция (витамин В1Б). М., нзд-во «Нау-
ка», 1968.
ОГЛАВЛЕНИЕ
ПРЕДИСЛОВИЕ 3
ВВЕДЕНИЕ 5
Классификация витаминов 6
Провитамины 10
Биокаталитическне функции витаминов 12
Антивитамины 15
Определение витаминов 17
Потребность в витаминах 17
Список использованной литературы (к введению) 17
ЧАСТЬ ПЕРВАЯ. ВИТАМИНЫ АЛИФАТИЧЕСКОГО РЯДА
Глава I. Витамины — производные ненасыщенных полиокси-7-лактонов 19
L-аскорбиновая кислота 19
Химические свойства L-аскорбиновой кислоты 21
Строение L-аскорбнновой кислоты 26
Аскорбиген 30
Синтез L-аскорбиновой кислоты 30
1. Синтез L-аскорбиновой кислоты конденсацией бензоинового типа 31
2. Синтез L-аскорбиновой кислоты конденсацией эфиров замещенных
а-оксикислот 31
3. Синтез L-аскорбиновой кислоты ци ан гидр н новым (озои-цианидным)
методом 32
4. Синтез L-аскорбиновой кислоты изомеризацией и лактонизацией 2-
или 3-кетогексоновых кислот 33
Получение препаратов L-аскорбииовой кислоты 47
Биологическое значение L-аскорбнновой кислоты. Зависимость между
строением и биологической активностью 47
Список использованной литературы (к главе I) 51
Глава II. Витамины и коферменты — производные ^-аминокислот 57
Пантотеновая кислота 57
Химические свойства пантотеновой кислоты 57
Строение пантотеновой кислоты 58
Синтез пантотеновой кислоты 60
Конденсация пантолактона с ^-аланином 60
Синтез пантолактона 63
Синтез Р-аланина 65
Биологическое значение пантотеновой кислоты. Зависимость между
строением н биологической активностью. Антивитамины 69
Кофермент А и другие коферменты пантотеновой кислоты 72
Строение кофермента А 74
Выделение кофермента А 75-
Синтез пантетина н пантетеина 76-
Синтез кофермента А 80
Биохимические функции кофермента А 85-
Список использованной литературы (к главе II) 92
ЧАСТЬ ВТОРАЯ. ВИТАМИНЫ АЛИЦИКЛИЧЕСКОГО РЯДА
Глава III. Циклогексанол-этиленгидриндановые витамины их провитамины 97
Кальциферолы 97
Химические свойства кальциферолов 100
Строение эргокальциферола и других витаминов 104
Выделение кальциферолов (витаминов группы D) 109
Фотосинтез кальциферолов из провитаминов 109
Фотонзомернзация эргостерина, 7-дегидрохолестерина и других прови-
таминов в эргокальциферол, холекальциферол и другие витамины 109
628
Механизм реакции фотоизомеризации стериновых провитаминов в каль-
циферолы 112
Выделение кристаллических кальциферолов из продуктов фотоизомери-
зацни 115
Строение продуктов фотоизомеризацнн стериновых провитаминов и каль-
циферолов и нх физико-химическая характеристика 115
Синтез кальциферолов и их изомеров 120
Биологическое значение кальциферолов. Зависимость между строением
и биологической активностью кальциферолов 123
Провитамины кальциферолов (провитамины D) 126
Физические н химические свойства провитаминов кальциферолов 127
Выделение и синтез провитаминов кальциферолов 130
Список использованной литературы (к главе III) 134
Глава IV. Циклогексенилизопреиоидиые витамины и провитамины. 140
Ретинол, дегидроретинол 140
Химические свойства ретинолов 145
Строение ретинолов 153
Ретинол 153
Дегидроретинол 155
Выделение ретинола (витамина А) из природных источников 156
Синтез ретинолов 157
1. Синтез исходных веществ для получения ретинола —цитраля, псев-
доионоиа и Р-нонона 157
2. Синтез ретинола нз замещенного циклогексанона 161
3. Синтез ретинола нз цитраля 162
4. Синтез ретинола из 3-ноиона через углеводороды С1в и С1в или соеди-
нением с цепочкой С, 163
5. Синтез ретинола из р-ионоиа через кетон С18 или альдегид Си 166
6. Синтез ретинола из р-ионона через альдегид С14 176
7. Направленный стернческий синтез геометрических изомеров рети-
нола 180
8. Синтез дегидроретинола 182
Биологическое значение ретинолов. Зависимость между строением и
А-витаминной активностью 183
Провитамины ретинола н дегидроретинола 187
Строение провитаминов 198
Выделение каротиноидных провитаминов 200
Получение p-каротина ферментативным путем 201
Синтез каротиноидных провитаминов 201
Список использованной литературы (к главе IV) 214
ЧАСТЬ ТРЕТЬЯ. ВИТАМИНЫ АРОМАТИЧЕСКОГО РЯДА
Глава V. Нафтохиноиовые витамины и провитамины 223
Филлохинон, менахиноны 223
Химические свойства нафтохиноновых витаминов 225
Строение нафтохиноновых витаминов 229
Филлохинон 229
Менахиноны 230
Вещества, родственные нафтохиноновым витаминам 231
Синтез нафтохиноновых витаминов 235
Синтез филлохинона 235
Синтез менахнионов 237
Получение исходных веществ для синтеза нафтохийоновых витаминов 237
Биологическое значение нафтохиноновых витаминов. Зависимость между
строением н биологической активностью, антивитамины 239
Провитамины 245
Синтез провитаминов 246
Список использованной литературы (к главе V) 249
ЧАСТЬ ЧЕТВЕРТАЯ. ВИТАМИНЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
Глава VI. Хромановые витамины 253
Токоферолы 253
Химические свойства токоферолов 255
Строение токоферолов 262
а-Токоферол — 5,7,8-триметнлтокол 262
f-Токоферол — 5,8-днметилтокол 263
7-Токоферол — 7,8-диметилтокол 264
Ь-Токоферол — 8-метнлтокол 264
Сг-Токоферол — 5,7-диметилтокол 264
629
е-Токоферол — ₽-токотриеиол 265
Выделение токоферолов 265
Природные вещества, родственные токоферолам 266
Синтез токоферолов 269
Синтез а-токоферола 270
Полусинтез а-токоферола нз f- и В-токоферолов 274
Синтез других токоферолов 275
Получение исходных веществ для синтеза токоферолов — фитола, изо-
фитола и триметил-п-гидрохинона 278
Биологическое значение токоферолов. Зависимость между строением и
биологической активностью. 284
Список использованной литературы (к главе VI) 288
Глава VII. Витамины и коферменты — производные пиридинкарбоиовых кис-
лот 293
Никотинамид и никотиновая кислота 293
Химические свойства никотиновой кислоты и никотинамида 294
Строение никотиновой кислоты 297
Синтез никотиновой кислоты и никотинамида 297
1. Синтез никотиновой кислоты из никотина н анабазина 298
2. Синтез никотиновой кислоты из пиридина 299
3. Синтез никотиновой кислоты из р-алкилзамещеиных пиридина 299
4. Синтез никотиновой кислоты из хинолина 304
5. Биосинтез никотиновой кислоты из незаменимых аминокислот 306
6. Синтез никотинамида 307
Биологическое значение никотиновой кислоты и никотинамида. Зависи-
мость между строением и биологической активностью. Антивитамины 307
Никотинамидные коферменты 309
Строение никотинамидных коферментов 312
Выделение никотинамидных коферментов - 313
Синтез никотинамидных коферментов 314
Биохимические функции никотинамидных коферментов 316
Список использованной литературы (к главе VII) 324
Глава VIII. Окснметилпиридииовые витамины и коферменты 330
Пиридоксин, пиридоксаль, пиридоксамин 330
Химические свойства оксиметилпиридиновых витаминов 331
Строение пиридоксина, пиридоксаля и пиридоксамина 336
Синтез пиридоксина, пиридоксаля и пиридоксамина 338
1. Синтез пиридоксина через производные иитрнла никотиновой кислоты 338
2. Синтез пиридоксина через производные азида никотиновой кислоты 342
3. Синтез пиридоксина через производные моио- и динитрила цинхоме-
роновой кислоты 343
4. Синтез пиридоксина через производные эфира цинхомероновой кис-
лоты 344
5. Синтез пиридоксина через производные хинолина и изохинолина 346
6. Синтез пиридоксина через производные фурана 347
7. Синтез пиридоксина через конденсированные пиримидо-фурановые
системы 349
8. Диеновый синтез пиридоксина из замещенных оксазолов и тиазолов. 350
9. Синтез пиридоксамина и пиридоксаля , 354
Биологическое значение оксиметилпириднновых витаминов. Зависимость
между строением и биологической активностью. Антивитамины 355
Пиридоксалевые коферменты ' 358
Строение пиридоксаль-5а-фосфата 359
Синтез пиридоксаль-5а-фосфата 359
Биохимические функции пиридоксалевых коферментов 361
Переаминированне 362
Рацемизация 364
р-Замещение 365
Дегидратирование, дегидроксилирование и детиолирование 366
Декарбоксилирование 36/
а, Р-Расщепленне 369
Гидролитическое расщепление 370
Список использованной литературы (к главе VIII) 371
Глава IX. Пиримидилметилтиазолиевые витамины коферменты 376
Тиамнн 376
Химические свойства тиамииа 377
Реакции и производные тиольной формы тиамииа 380
Строение тиамина 390
Синтез тиамина 392
630
1. Синтез тиамина конденсацией пиримидинового и тиазолового компо-
нентов 392
• 2. Синтез тиамииа построением его молекулы на пиримидиновом цикле 394
3. Синтез гомотнамнна построением его молекулы на тиазоловом цикле 397
4. Синтез важнейших структурных частей молекулы тиамина 398
Биологическое значение тиамина. Зависимость между строением и био-
логической активностью. Антивитамины 413
Тиаминовые коферменты 417
Химические свойства фосфорных эфиров тиамина , 418
Синтез тиаминдифосфорного эфира и его производных 419
Биохимические функции тиаминдифосфата 421
Список использованной литературы (к главе IX) 425
Глава X. Гексагидроимидазолотиеновые витамины и коферменты 433
Биотин 433
Химические свойства биотина 434
Строение биотина 435
Стереоизомерия биотина 437
Выделение биотипа 439
Синтез биотина и его стереоизомеров 439
1. Синтез биотина, аллобиотина и эпналлобиотина из L-цистеина через
3-оксотиофан без заместителя в положении 2 439
2. Синтез биотииа через 3-оксотиофан с «-метоксибутильным заместите-
лем в положении 2 441
, 3, Синтез биотина, эпибиотина и эпиаллобиотииа через /пра«с-3,4-ди-
карбокси-2-(8-карбоксибутил)тиофаи 443
4. Синтез эпибиотина и эпиаллобиотина через 3-иитротиофан ’ 445
5. Синтез биотина через мезо-а,₽-дибеизиламинояитариую кислоту 445
Биологическое значение биотина. Зависимость между строением и био-
логической активностью 449
Биотиновые коферменты 452
Строение и синтез кофермента биотина 452
Синтез биоцитина 453
Биохимические функции кофермента биотииа 453
Список использованной литературы (к главе X) 4 456
Глава XI. Птерниовые витамины и коферменты 459
Птероил-£-глутаминовая кислота (фолиевая кислота) и ее полипептиды 459
Химические свойства птероил-Б-глутаминовой кислоты 460
Природные вещества птериновой структуры - 463
Строение витаминов группы фолиевой кислоты и некоторых птеринов 465
Строение птеронл-Б(ф)-глутаминовой кислоты (печеночного L. casei-
фактора, витамина Вс) 465
Строение птероил-Б(-|-)-7.7-триглутамнновой кислоты (ферментативного
L. casei-фактора) 468
Строение ризоптерина (фактора Streptococcus lactis R.) 469
Синтез птероил-Б-глутаминовой кислоты • 469
1. Синтез фолиевой кислоты построением ее молекулы трехкомпоиент-
ной конденсацией 470
2. Синтез фолиевой кислоты построением ее молекулы на ароматиче-
ском цикле ' 473
3. Синтез фолиевой кислоты построением ее молекулы на пиримидино-
вом цикле 475
4. Получение некоторых исходных продуктов, необходимых для синте-
за птериновых витаминов 480
Синтез птероилполнглутамнновых кислот 483
Биологическое значение витаминов группы фолиевой кислоты. Зависи-
мость между строением и биологической активностью соединений птери-
нового ряда. Антивитамины 484
Птерниовые коферменты 490
Строение фолиновой кислоты 493
Синтез 5,6,7,8-тетрагидрофолиевой кислоты и ее активированных форм —
lO-N-формнл-ТГФК, S-N-формил-ТГФК и других 494
Взаимопревращения ТГФК и активных форм кофермента 495
Биохимические функции птернновых коферментов 497
Список использованной литературы (к главе XI) 500
Глава XII. Флавиновые витамины и коферменты 507
Рибофлавин 507
Химические свойства рибофлавина и других алло- и изоалло-
ксазииов 509
631
Строение рибофлавина 521
Выделение рибофлавина 522
Получение рибофлавина микробиологическим путем 522
Синтез рибофлавина, люмифлавина, люмихрома и некоторых родствен-
ных соединений 522
1. Синтез рибофлавина конденсацией ароматического моноамина и вио-
луровой кислоты 523
2. Синтез рибофлавина и люмифлавина из ароматического моноамина и
6-хлорурапила 523
3. Синтез рибофлавина, люмифлавина, люмихрома н их 2-тиопронзвод-
иых конденсацией ароматических о-диамннов с аллоксаном 524
4. Синтез рибофлавина и некоторых других алло- и изоаллоксазинов
конденсацией ароматических орто-аминоазосоединений с барбитуровой
кислотой 527
5. Синтез рибофлавина и некоторых других алло- и изоаллоксазинов
конденсацией орто-бензохинонов с диаминоурацилами 529
6. Синтез рибофлавина и некоторых других алло- и изоаллоксазинов
из люмазинов 530
7. Синтез алло- и изоаллоксазинов построением их молекулы на хино-
ксалиновом цикле 531
8. Получение исходных веществ, необходимых для синтеза рибофлавина 531
Биологическое значение рибофлавина. Зависимость между строением
и биологической активностью. Антивитамины 545
Флавиновые коферменты 548
Строение и выделение флавиновых коферментов 552
Синтез флавиновых коферментов 553
1. Синтез ФМН 553
2. Синтез ФАД 554
3. Ферментативный синтез ФАД ‘ 558
Биохимические функции флавиновых коферментов 558
Список использованной литературы (к главе XII) 568
Глава XIII. Коррииовые витамины и коферменты 577
Цианокобаламин, оксикобаламин 577
Продукты частичного расщепления циаиокобаламина 580
Химические свойства кобаламинов 582
Строение цианокобаламина 585
Производные цианокобаламина с другими лигандами (анионами) 591
Псевдовитамины Big 592
Получение цианокобаламииа . 595
Синтез цианокобаламина и фрагментов его молекулы 597
Биологическое значение витамина Bit. Зависимость между строением
и биологической активностью 604
Коррииовые коферменты 606
Структура кофермент-кобаламина 610
Получение кофермент-кобаламина 610
Частичный синтез кофермент-кобаламина 611
Биохимические функции корринового кофермента 613
Список использованной литературы (к главе XIII) 614
Глава XIV. Некоторые природные биологически активные вещества 620
Линолевая, линоленовая и арахидоновая кислоты 620
Биофлаваноиды 621
жезо-Инознт ' 624
Холин 625
Пангамовая кислота 626
Список использованной литературы (к главе XIV) 626