/




























































































































































































































































































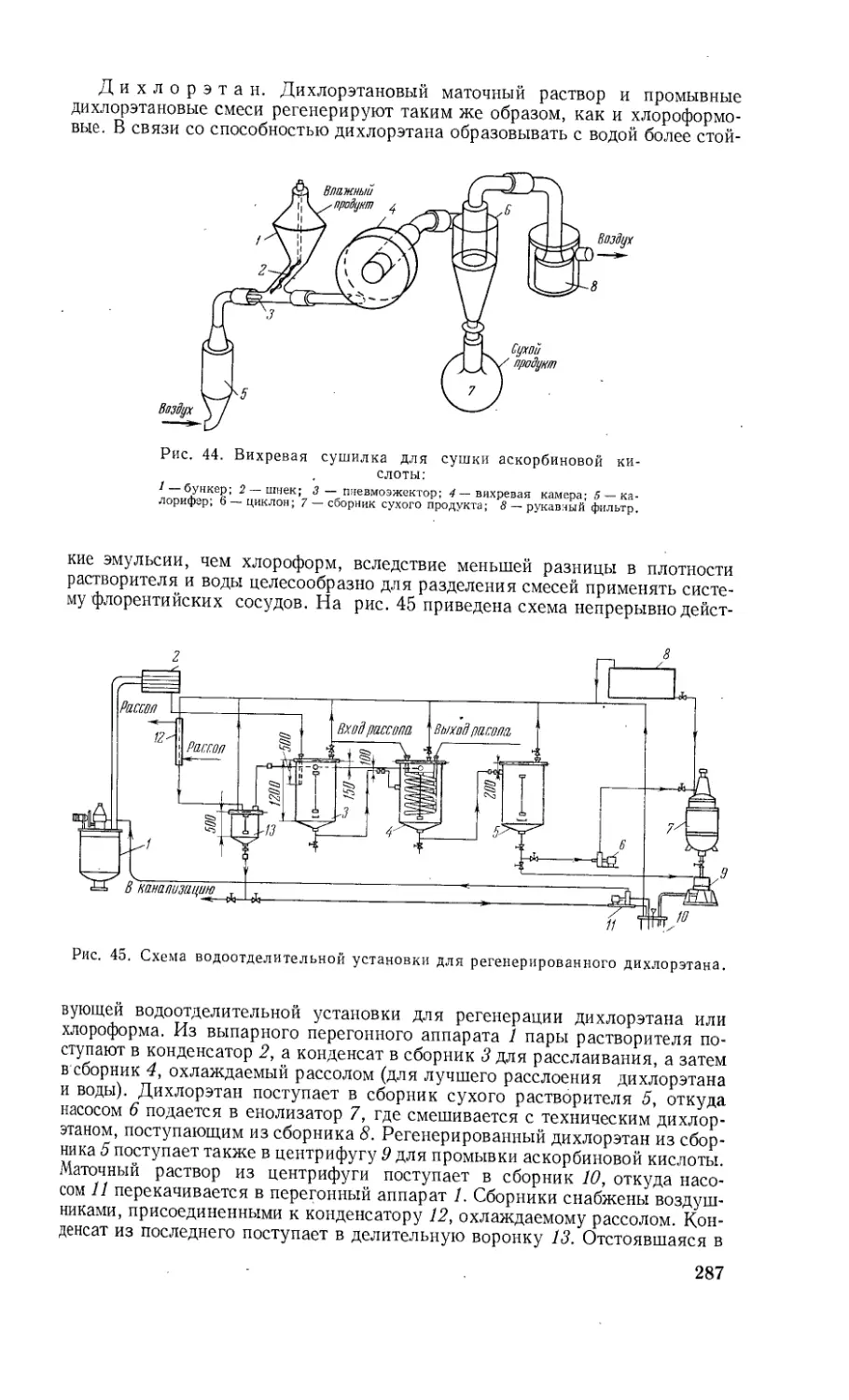



























































































































































Автор: Шнайдман Л.О.
Теги: материальные основы жизни биохимия молекулярная биология биофизика биология фармакология витамины
Год: 1973




Текст
Л. О. Шна-йама/н,
произвол гео
ВИТАМИНОВ
Л.О.Шний^ман
Доктор технических наук
лауреат Государственной премии СССР
ПРОИЗВОДСТВО
ВИТАМИНОВ
ИЗДАНИЕ ВТОРОЕ, ПЕРЕРАБОТАННОЕ И ДОПОЛНЕННОЕ
Москва «Пищевая промышленность» 19 73
УДК 577. 16. 002. 2
Производство витаминов. Шнайдман Л. О., 1973.
В книге изложены основные принципы правильного построения техноло-
гии производства. Рассмотрены по опубликованным литературным данным ме-
тоды синтеза витаминов: А, Вх, В2, В3, В6, Вх5, РР, фолиевой и ли-
поевой кислот, С, D2, D3 и К.
Материал, относящийся к синтезу витаминов, изложен в следующем по-
рядке: значение и применение, физико-химические свойства, методы синтеза
и выбор рационального метода для производства, технологическая схема в ап-
паратурном оформлении, технологические показатели. Описаны основные ап-
параты преимущественно непрерывного действия, рекомендуемые для приме-
нения в витаминной промышленности.
Описана технология производства витаминных препаратов: из плодов ши-
повника; из плодов облепихи; Р-витаминного сырья (чайного листа, черно-
плодной рябины, черной смородины, гречихи, бадана, отходов цитрусовых
и столовой свеклы); из моркови и тыквы; из печени рыб (витамин А) и из дрож-
жей (эргостерин и D2).
Приведен состав биологически активных веществ, содержащихся как в при-
родном сырье, так и в витаминных препаратах. Эти вещества экспериментально
изучены при помощи современных химических, физических и физико-химиче-
ских методов — хроматографии, спектроскопии и др.
Кратко рассмотрены природные пищевые биологически активные краси-
тели.
Таблиц 47, иллюстраций 101, список литературы — 1411 названий.
Рецензент проф. доктор техн, наук Б. Г. Савинов.
Издательство «Пищевая промышленность», 1973 г.
3177—042
42—73
044(01)—73
ПРЕДИСЛОВИЕ
В Директивах XXIV съезда КПСС по пятилетнему
плану развития народного хозяйства СССР на 1971 —
1975 гг. предусмотрено повысить качество, расширить
ассортимент и улучшить питательную ценность и вку-
совые достоинства продуктов питания; значительно
увеличить выпуск витаминов и антибиотиков; шире
использовать отходы пищевой промышленности для
кормовых целей.
Рост производства витаминов происходит и будет
происходить в основном в результате расширения вы-
пуска синтетических витаминов. Однако будущие от-
крытия биологически активных вешеств всецело зависят
от химических и биологических исследований в области
природного сырья.
Пищевые продукты — основной источник витаминов
для населения. Поэтому исследование биологически
активных веществ сельскохозяйственного сырья — основ-
ного сырья пищевой промышленности — представляет
важную для народного хозяйства задачу, направленную
на повышение питательной ценности продуктов питания.
Комплексное (химическое, биологическое и техноло-
гическое) изучение природного сырья и пищевых про-
дуктов должно привести к обогащению их биологически
активными веществами. Таким образом, будет создана
еще одна возможность для дальнейшего повышения
качества пищевых продуктов.
Важно также обогащение витаминами рационов сель-
скохозяйственных животных.
Интерес представляют следующие данные: свекольной
ботвы в стране получают примерно 30—40 млн. т в год'.
В ней содержится приблизительно 8 тыс. т аскорбино-
вой кислоты, 900 т витаминов группы В и 2 тыс. т
каротина, т. е. почти столько же, сколько этих синте-
тических препаратов производится во всем мире. Отсюда
видно, какое большое значение имеет использование
отходов пищевой промышленности в животноводстве.
Наука о витаминах и техника производства их интен-
сивно развиваются. За период, прошедший со времени
первого издания книги «Производство витаминов», осу-
ществлен в промышленных масштабах синтез витаминов:
В3, В15, D3, Е, РР, липоевой кислоты. Разработан
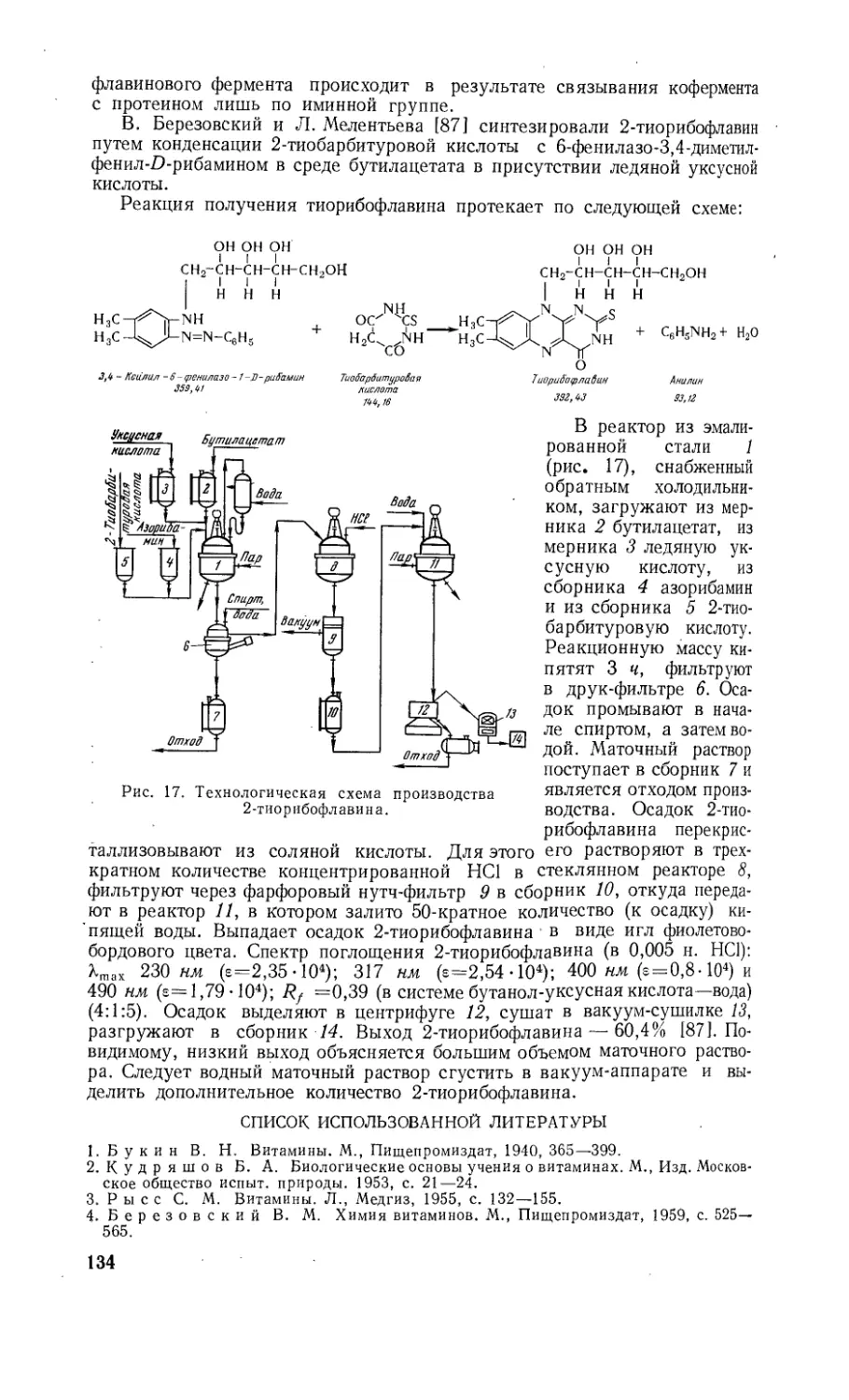
синтез аналогов, гомологов и коферментов: тиаминпро-
3
пилдисульфида, пиридоксаля, тиорибофлавина, амида
никотиновой кислоты, аскорбината натрия и др.
В последние годы в науке и технике производства
витаминов из природного сырья.развилось новое научное
направление, известное под названием «Идентификация
биологически активных веществ природного сырья с це-
лью их комплексного промышленного использования».
При комплексном использовании предусматривается по-
лучение ряда витаминных препаратов из одного вида
сырья. Разработана технология получения витаминных
препаратов из новых видов природного сырья: черно-
плодной рябины, столовой свеклы, бадана и др.
Учитывая большой удельный вес синтетических ви-
таминов в промышленности, в первой части книги изла-
гается технология синтеза витаминов, во второй —
производство витаминных препаратов из природного
сырья.
Рассмотрены основные требования к технологичес-
кой аппаратуре для производства витаминов, описаны
конструкции аппаратов, в основном непрерывно дейст-
вующих.
Второе издание книги отличается от первого увели-
чением числа глав по синтезу витаминов и технологии
производства витаминов из природного сырья, а также
обновлением материала в переиздаваемых главах.
Большая помощь при подготовке рукописи была ока-
зана рецензентом проф. Б. Г. Савиновым, которому
автор выражает искреннюю благодарность.
Часть I. ПРОИЗВОДСТВО СИНТЕТИЧЕСКИХ ВИТАМИНОВ
Глава 1. ОСНОВНЫЕ ПРИНЦИПЫ ПРАВИЛЬНОГО ПОСТРОЕНИЯ
ТЕХНОЛОГИИ ПРОИЗВОДСТВА
Основными показателями эффективности производства являются: высо-
кое качество целевого продукта, низкая себестоимость его и высокая про-
изводительность труда.
Высокое качество целевого продукта достигается, при соблюдении сле-
дующих правил.
1. Технический продукт, поступающий на рафинирование (перекристал-
лизация или ректификация) должен обладать высокой чистотой, так как
превратить загрязненный и интенсивно окрашенный технический продукт
в рафинированный при однократной кристаллизации или одной перегонке
невозможно. Это обусловлено тем, что при кристаллизации на поверхности
граней растущего кристалла адсорбируются посторонние вещества, содер-
жащиеся в неочищенном растворе; при перегонке вещества пары уносят
с собой частицы перегоняемого вещества. Кроме того, с загрязняющими
веществами могут образовываться азеотропные смеси, следовательно, чем
ниже качество технического продукта, тем больше посторонних веществ
поступит в готовый продукт.
2. Качество технического продукта зависит от качества предшествующих
полупродуктов. Отсюда следует, что полупродукты, поступающие на после-
дующие стадии производства, должны удовлетворять предъявляемым к
ним требованиям по чистоте и цветности. Как правило, чем выше качество
компонентов, участвующих в реакциях, тем полнее и эффективнее проте-
кают реакции, тем больше выход продуктов и тем выше их качество, С этой
точки зрения, наиболее оптимальными являются полупродукты, выделяе-
мые в твердсм виде и подвергнутые перекристаллизации.
3. Для достижения высокого эффекта очистки необходимо при рафини-
ровании применять два процесса — адсорбционный (осуществляемый при
помощи активированного угля, а в' отдельных случаях ионообменным спо-
собом) и кристаллизационный. Пределы рационального сочетания адсорбци-
онного и кристаллизационного процессов определяются исходя из следую-
щего положения [1 ]: в тех случаях, когда задачей очистительного процесса
является глубокое разделение веществ в низкокачественных продуктах,
основная роль очистки принадлежит перекристаллизационному процессу.
Очистка же растворов высокого качества и низкой цветности может быть
произведена одним только адсорбционным процессом. Например, для
превращения технической аскорбиновой кислоты с чистотой 97,6—98%
и цветностью — 20—40 ед. в продукт фармакопейного качества необходимы
оба очистительных процесса (адсорбция и кристаллизация). Если качество
аскорбиновой кислоты 11 высокое (чистота 98,0—98,5 % и цветность 5—7 ед.),
то для получения фармакопейного продукта из аскорбиновой кислоты II
можно применять лишь один адсорбционный процесс.
Из указанного положения следует, что адсорбционный процесс может
также дать существенный эффект при сочетании его с ректификацией мно-
гокомпонентных смесей. Такое сочетание, т. е. очистка последних перед
ректификацией активированным углем, в практике, обычно, не применяется.
По мнению автора, такое сочетание было бы весьма эффективным.
4. Цветность (оптическая плотность) продукта является одним из самых
чувствительных индикаторов качества протекания процессов во всех слу-
чаях, когда классическими химическими или физико-химическими метода-
5
ми невозможно определить отклонение содержания вещества (в %) от
нормы.
Растворы, из которых кристаллизуются витаминные препараты фарма-
копейного качества, должны быть практически бесцветными. Автором раз-
работана методика определения цветности растворов [2]: цветность (цв)
определяется по формуле ( в ед. ВНИВИ):
где Pi — оптическая плотность испытуемого раствора (светофильтр 400 нм);
Р2 — оптическая плотность эталонного раствора № 5 по Госфармакопее [3];
D — навеска вещества, а;
V — общая масса (навеска + растворитель), а.
Примечание. Для получения эталонного раствора приготовляют два раст-
вора: Раствор А —0,1 а растертого бихромата калия, высушенного при 100—105° С
до постоянной массы, помещают в мерную колбу вместимостью 500 мл и доводят во-
дой до метки.
Раствор Б— 2,975 а растертого хлорида кобальта (СоСБ-бНгО) помещают в
мерную колбу вместимостью 500 мл, прибавляют 1,5 мл соляной кислоты и до-
водят содержимое колбы водой до метки.
Для определения цветности применяют основной раствор, получаемый
смешением растворов А и Б в соотношении 70:30. Для получения эталона
цветности № 5, принятого за единицу ВНИВИ, основной раствор разбав-
ляют водой в соотношении 1:4 (на 1 мл основного раствора 4 мл воды).
Для неокрашенных препаратов фармакопейного качества допускается
цветность не более 2—3 ед.
5. На стадии кристаллизации, как и на протяжении всего технологичес-
кого процесса, необходимо направлять на совместную переработку лишь
те продукты, которые обладают одинаковой чистотой и цветностью. На ста-
дии кристаллизации, как правило, все полупродукты получают следующее
направление — твердые — на одну ступень выше, а жидкие — на одну
ступень ниже'. Например, кристаллы тиамина II поступают для переработ-
ки на первую кристаллизацию ( на одну ступень выше), а маточный раст-
вор I, получаемый на первой кристаллизации, направляют на вторую
кристаллизацию (на одну ступень ниже). Многолетняя практика показала,
что нарушение этого правила (например, направление на первую кристалли-
зацию аскорбиновой кислоты III вместо переработки ее по правилу на
второй кристаллизации) приводит к понижению качества целевого продук-
та и к повышению потерь.
В связи с этим применяемые довольно широко в химической технологии
так называемые рециклы, т. е. возврат в реакционный аппарат истощенной
реакционной массы (за счет частичного извлечения из нее целевого продук-
та реакции), нельзя признать рациональными. Например: при окислении
2-метил-5-этилпиридина азотной кислотой под давлением [4 ] в непрерывном
процессе по методу Б. Уставщикова и других из окисленной реакционной
массы частично выделяют изоцинхомероновую кислоту, а маточный раствор
обедненного состава возвращают (рецикл) в реактор-окислитель. Этим
непрерывно ухудшают качество реакционной массы, а следовательно, и
выделяющейся изоцинхомероновой кислоты. С точки зрения сформулиро-
ванного автором положения, маточный раствор, содержащий изоцинхомеро-
новую и частично никотиновую кислоты, должен быть сгущен под вакуумом
и подвергнут кристаллизации с выделением указанных кислот и перера-
боткой полученного маточного раствора II путем выделения целевого про-
дукта через медные соли.
.6. Важное значение имеет процесс промывки кристаллов в центрифуге.
Этот процесс позволяет очистить поверхность кристаллов от маточных
загрязнений и ускорить процесс сушки кристаллов. В связи с этим оконча-
тельно промывать кристаллы следует спиртом в центрифуге. Если первая
промывка осуществляется водой, как, например, для аскорбиновой кислоты,
6
то спирт повышает эффективность процесса, так как он обладает более
универсальной растворяющей способностью. Кроме того, спиртовая обо-
лочка кристаллов способствует ускорению процесса и смягчению режима
сушки, что снижает потери вещества в этом процессе.
7. Сохранение качества витаминных препаратов при длительном хране-
нии имеет первостепенное значение. Для предотвращения разрушения
витаминных препаратов фармакопейного качества в процессе длительного
хранения необходимо обеспечить низкую влажность продукта и герметич-
ность тары. В качестве поглотителя влаги внутри тары следует применять
силикагель. Для упаковки порошкообразных препаратов целесообразно
применять: а) двойной полиэтиленовый мешочек, б) жестяную тару, про-
паянную по шву корпуса, с крышками, уплотненными каучуковыми про-
кладками, и в) силикагелевый поглотитель влаги, содержащейся в воздухе,
заключенном в таре. В этих условиях можно гарантировать сохранность
препарата в течение 2—3 лет. Для сохранения качества жидких препаратов
важное значение имеет: дезаэрация продукта инертным газом, заполнение
тары до пробки с минимальным зазором (недолив 0,0007) и полная герме-
тизация тары [5].
Совершенно отдельно стоит вопрос о применении стабилизаторов — ан-
тиокислителей в виде токоферолов, гидрохинона, эфиров галловой кислоты,
сантохина и др. К сожалению, эти вопросы недостаточно изучены.
Уровень и степень совершенства технологии производства имеет решаю-
щее влияние на себестоимость витаминов. В калькуляции себестоимости син-
тетических витаминов затраты на сырье и материалы составляют 50—60%.
Следовательно, низкая себестоимость витаминов может быть достигнута
при небольших затратах на сырье. Это в свою очередь зависит от ряда фак-
торов:
выбора рационального метода синтеза;
использования дешевого недефицитного сырья;
получения высоких выходов полупродуктов и целевого продукта;
полной регенерации летучих органических растворителей;
комплексного использования сырья и отходов производства.
Можно привести ряд примеров, обосновывающих влияние указанных
факторов на себестоимость витаминов.
1. Например, из анализа двух методов синтеза никотиновой кислоты
видно, что при синтезе ее из ₽-пиколина с окислением его перманганатом
калия затраты на сырье в 3 раза выше, чем при синтезе из хинолина или
из 2-метил-5-этилпиридина с применением в качестве окислителя азотной
кислоты. Использование в качестве растворителя хлороформа вместо ди-
хлорэтана в производстве тиамина или применение в качестве окислителя
перманганата калия вместо гипохлорита натрия в производстве аскорбино-
вой кислоты резко повышают затраты на сырье.
2. Можно привести пример, показывающий влияние локального хими-
ческого метода на перспективу синтеза в целом. Прогрессивный синтез
амида никотиновой кислоты парофазным окислительным аммонолизом |3-пи-
колина сделался неэффективным из-за применения перекиси водорода в
щелочной среде для гидратации нитрила. При этом расход перекиси водо-
рода составил 10,5 и этилацетата 16,8 кг на 1 кг амида. При этих усло-
виях метод неперспективен из-за высокой стоимости химикалиев. Однако
проведение метода гидратации нитрила с применением ионитов в ОН-форме
сделало окислительный аммонолиз весьма эффективным [6,7].
3. Выход полупродуктов и целевого продукта влияет в прямой про-
порции на расход химикалиев. Пример можно привести из синтеза фолие-
вой кислоты. При замене в реакции трехкомпонентной конденсации 2,3-ди-
бромпропионового альдегида трихлорацетоном удалось более чем вдвое
повысить выход фолиевой кислоты и соответственно снизить затраты на
сырье.
7
4. Регенерация летучих органических растворителей способствует су-
щественному снижению затрат на сырье. В виде примера можно привести
расход ацетона в синтезе аскорбиновой кислоты. В производство вводят
20 кг ацетона на 1 кг аскорбиновой кислоты, а расход ацетона вследствие
эффективной регенерации его составляет 2 кг, т. е. 90% вводимого ацетона
регенерируется.
При высокой летучести органических растворителей необходимо обеспе-
чить надлежащую герметичность аппаратов и трубопроводов и соответ-
ствующие улавливающие и конденсирующие устройства.
5. Комплексное использование сырья является одним из показателей
степени совершенства технологии производства. Это подтверждается сле-
дующими примерами. Ранее из 1 т плодов шиповника получали 2920 кг
сиропа с витамином С. Жом, получаемый в качестве отхода, не использовал-
ся. При организации комплексной переработки плодов заводы стали полу-
чать дополнительно: концентрат витамина Р — 63,5 кг, масляный препарат
каротолин — 30 кг, масло семян шиповника — 22,8 кг. Наиболее полное
использование сырья способствовало значительному повышению эффектив-
ности производства.
Весьма существенным является также использование отходов. Так, в
производстве аскорбиновой кислоты на стадии ацетонирования в качестве
отходов образуется осадок десятиводного сульфата натрия (Na2SO4- 10Н2О)
в количестве 2,5 т на 1 т аскорбиновой кислоты. Разработан метод обез-
воживания гидрата. Выход безводного сульфата натрия составляет 95%
от теоретического [8] и по своему качеству соответствует требованиям
ГОСТа на безводный сульфат натрия. Разработан также метод использова-
ния калийного отхода в этом же производстве на стадии окисления диаце-
тонсорбозы с получением концентрата с содержанием 33% К2О [8]. Ис-
пользование отходов производства не только снижает затраты на сырье,
но и облегчает задачу очистки сточных вод.
6. В связи с большой лабильностью витаминных препаратов и их полу-
продуктов важное значение для повышения эффективности технологических
процессов имеют следующие факторы: скорость процессов термической
обработки, дезаэрация лабильных реакционных масс, величина объема
верстата в цехе, предотвращение побочных реакций.
Скорость процессов термической обработки продуктов реакций при
наиболее низкой допустимой температуре имеет важное значение для пре-
дотвращения процессов разрушения лабильных веществ. Выпаривание
растворов следует производить в пленочных аппаратах, высушивание раст-
воров — в распылительных или вакуум-вальцовых сушилках, а высушива-
ние осадков — в вихревых или с ожиженным слоем. Следует учитывать
что скорость тепловых процессов имеет решающее значение для сохранения
качества веществ. Поэтому при решении вопроса, какому из двух факто-
ров — скорости процесса или уровню температуры — отдать предпочтение,
необходимо положительно ответить в пользу первого. В этой области боль-
шую роль играет теплоноситель и размер поверхности нагрева. Оптималь-
ным теплоносителем является водяной пар, обладающий высоким теплосо-
держанием (640 кал! кг). Однако он может быть использован в сравнительно
узком диапазоне температур —до 150—180° С, что соответствует давлению
5—10 кгс!см~.
При необходимости подогрева реакционных масс до более высокой
температуры (200—250° С) целесообразно применять паровые нагреватель-
ные установки высококипящих органических жидкостей, как, например,
дифенильных смесей, дитолилметана, нефтяных масел и др. Непосредствен-
ный электрический обогрев поверхности реактора неприемлем из-за воз-
можных местных перегревов реакционной массы. Применение горячей воды
в качестве теплоносителя при температуре кипения раствора 50—70° С
в большинстве случаев неприемлемо, так как это приводит к значительному
удлинению процесса выпаривания с вытекающими отсюда последствиями
8
разрушения лабильных веществ. Мнение некоторых химиков о необходи-
мости применения в качестве теплоносителя горячей воды для сгущения
растворов лабильных веществ, как правило, не является обоснованным.
В этом вопросе главную роль играет скорость процесса. Конечно, темпера-
турный фактор также имеет первостепенное значение. Поэтому оптималь-
ными условиями для процессов с тепловой обработкой реакционных масс
является большая скорость при низкой температуре. Последняя достигает-
ся при применении сравнительно глубокого вакуума при выпаривании и
сушке. При выпаривании растворов или при их сушке на вальцовых
сушилках остаточное давление в аппаратах не должно превышать
70—80 мм рт. ст. Однако в практике часто эти тепловые процессы осуще-
ствляют при остаточном давлении 150—250 мм рт. ст., что приводит к
снижению скорости процесса и ухудшению качества реакционной массы.
, Скорость протекания реакции зависит также от эффективности хладаген-
та и поверхности охлаждения. При экзотермических реакциях с высоким
тепловым эффектом скорость реакций будет зависеть от интенсивности
охлаждающих устройств.
В связи с изложенным выше при проектировании витаминных заводов
следует особое внимание уделять вопросам обеспечения процессов достаточ-
ной глубиной вакуума и холодом.
Дезаэрация лабильных реакционных масс в ряде случаев предотвра-
щает окислительные процессы и способствует повышению эффективности
технологических процессов. Дезаэрацию осуществляют орошением реакци-
онной массы углекислотой или азотом. Этот процесс особенно полезен на
последней стадии синтеза лабильных витаминов А, В, С, D, и D3, Е и каро-
тина. Известно, что основным фактором, влияющим на распад витамина А,
каротина, аскорбиновой кислоты, тиамина, витамина Е является кислород
воздуха. Он неблагоприятно влияет также на процесс облучения в ультра-
фиолетовом свете раствора эргостерина и 7-дегидрохолестерина. В связи
с этим применение инертных газов на процессах выпаривания, облучения,
а также при расфасовке витаминов в тару, в особенности в ампулы, являет-
ся целесообразным.
Величина верстата цеха, т. е. сумма объемов аппаратов, сборников,
мерников, в которых осуществляют реакции или хранят химикалии и
промежуточные продукты, влияет на технологические результаты произ-
водства. Чем меньше верстат цеха, тем меньше потери от разложения проме-
жуточных продуктов. Поэтому необходимо лимитировать объем верстата
каждого цеха. При необходимости хранения лабильных полупродуктов,
как, например, маточных растворов на стадии кристаллизации аскорбино-
вой кислоты, необходимо сборники хранения обеспечить охлаждающими
рубашками для поддержания нулевой температуры. Как правило, следует
считать что чем меньше объем верстата материалов и полупродуктов, тем
эффективнее технология производства.
Немаловажную роль в технологии производства играют вопросы кор-
розии. Известно, что металлы в ряде процессов служат катализаторами
различных реакций. Поэтому содержание их в реакционной массе может
способствовать течению побочных реакций, увеличивать цветность ее и
снижать качество целевого полупродукта. Содержание металлов в готовой
продукции может обусловить нестабильность ее при хранении. Поэтому
для процессов, осуществляемых при участии агрессивных реагентов (кислот,
щелочей, галогенов) необходимо обосновывать род конструктивных мате-
риалов аппаратуры. Для большинства процессов, применяемых в производ-
стве витаминов, пригодны аппараты, изготовленные либо из эмалированной
стали, либо из нержавеющей стали марки IXI8H9T. Однако наряду с этим
в синтезе витаминов имеются процессы, для которых указанные материалы
непригодны, например, процессы окисления хинолина или 2-метил-5-этил-
пиридина азотной кислотой под давлением при температуре выше 170° С.
Для этих условий реакции необходимы реакторы из тантала. При сниже-
9
нии степени агрессивности среды реакций можно применять реакторы,
изготовленные из титана. Процессам коррозии в производстве витаминов
следует уделить серьезное внимание при конкретном изучении их для
каждой данной реакции.
Производительность труда также зависит от уровня и степени совершен-
ства технологии производства. Наилучшие условия могут быть обеспечены
при непрерывном осуществлении процессов и их автоматизации.
Исследования в области непрерывных процессов в производстве аскор-
биновой кислоты [8,9] показали, что все периодические процессы, действую-
щие в производстве, могут быть заменены непрерывными процессами.
Это позволит резко сократить количество аппаратов, уменьшить площадь
цеха и примерно в 5 раз повысить производительность труда.
список использованной литературы
1. Шнайдман Л. О. Производство сахара-рафинада. М., Снабтехиздат, 1935, т. I,
с. 27.
2. Шнайдман Л. О. О снижении потерь и повышении качества аскорбиновой
кислоты. — «Труды ВНИВИ», 1953, № 4, с. 54—62.
3. Государственная фармакопея СССР, IX, 1946, с. 606.
4. Уставщиков Б. Ф., Титова Т. С., Дегтярев Е. В., Фарбе-
ров М. И. Технический синтез никотиновой кислоты окислением 2-метил-5-этил-
пиридина разбавленной азотной кислотой. «ЖПХ», 1966, т. 39, с. 1388—1393, с ил.
5. Ш н а й д м а н Л. О., Дульчина Б. М., Павлова А. М. Влияние
различных физико-химических факторов на устойчивость каротина в растворах.
«Труды ВНИВИ», 1954, № 5, с. 51—64, с ил.
6. Жданович Е. С., Чекмарева И. Б., Преображенский Н. А.
Получение нитрила и амида никотиновой кислоты. ЖОХ, 1961, 31, 10, с. 3272—3274
с ил.
7. Чекмарева И. Б., Жданович Е. С., Сазонова Т., Преобра-
женский Н. А. Авт. свидет. № 164601, 1963; Бюлл. изобрет. 1964, № 16, с. 11.
8. Ш н а й Д м а н Л. О. Исследования в области химии и технологии производства
витаминов, доклад до кт. дисс., М., 1967, с. 18.
9. Ш н а й д м а н Л. О. Перспективы усовершенствования синтеза аскорбиновой кис-
лоты. — Сб. «Витамины». М., ГОСИНТИ, 1959, №5, с. 5—22. Перспективы усовер-
шенствования производства витаминов на Уфимском витаминном заводе.—Сб. «Вита-
мины», Уфа, Башизд., 1959, с. 41—54.
Глава 2. ПРОИЗВОДСТВО СИНТЕТИЧЕСКОГО ВИТАМИНА А
(РЕТИНОЛА) [1, 2, 3, 4, 5, 6]
Витамин А (ретинол) носит название антиксерофтальмического, антиин-
фекционного витамина, фактора роста, витамина, предохраняющего эпите-
лий, второго витамина размножения. При недостатке витамина А эпителий
слизистых оболочек превращается в роговидный эпителий. Это вызывает
поражение глаз, известное под названием ксерофтальмии (греч. xeros —
сухой, ophthalmos— глаз). Поэтому витамин А назван антиксерофталь-
мическим. При этом снижается способность глаза приспосабливаться к
слабому освещению (I стадия — гемералопия — куриная слепота; II ста-
дия — ксерофтальмия; III стадия — кератомаляция и полная потеря зре-
ния; греч. keros — рог, malacia — размягчение).
Ороговение эпителия кожи приводит к его повреждению, что облегчает
внедрение инфекции [4]. Снижение барьерной функции кожи к инфекции
ведет к возникновению дерматитов, а сухость слизистых дыхательных путей
способствует заболеванию бронхитом, катаром дыхательных путей. По при-
чине ороговения слизистых оболочек могут возникать и другие заболевания
(стоматит, воспаление почечных лоханок и мочевого пузыря, гастрит, ко-
лит).
Лечебное применение витамина А вытекает из указанных функций его.
Суточная потребность человека в витамине А равна 1,5—2 мг [5].
10
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА [7, 8, 9, 10]
Витамин Аг Структурная формула витамина А была установлена в
1931—1933 гг. [И, 12] в следующем виде:
сЦз ^СН3 СН3 сн3
Н2С< 6^.C,-J3H=сн-с = сн- сн= сн—с=сн-сн,он
I ’.II ¥ 2 3 4 5 в 7 8 9 2
H2.c<3.2q-CH3
'₽Н2
¥ * .
битаитн А,
Эмпирическая формула витамина At — С20Н30О. Спиртовая функция
витамина А4 установлена путем окисления его в альдегид (ретиналь). При
каталитическом гидрировании витамин At образует пергидровитамин А4
с эмпирической формулой С20Н40О, что указывает на наличие пяти двойных
связей. При окислении озоном из одной молекулы витамина А4 получают
одну молекулу героновой кислоты, что обусловлено содержанием в молеку-
ле одного кольца (3-ионона.
При окислении витамина Ai хромовой кислотой образуются три моле-
кулы уксусной кислоты, что свидетельствует о наличии в молекуле трех
групп СН3—С=; при окислении перманганатом калия в щелочной среде
образуются две молекулы уксусной кислоты, что указывает на то, что одна
метильная группа (в кольце) отличается своим положением от других двух
(в боровой цепи).
Наконец, был синтезирован пергидровитамин [11] и показано, что
он идентичен соединению, получаемому при каталитическом гидрировании
витамина АР
} Стереоизомеры витамина Ар Витамин Ai может сущест-
вовать в следующих стереоизомерных формах [7]:
сн3 Н,С у у Су3
>< >4 >4 >4 /Сн,он
L 1 .1 I I ’I
н н н
2,3,4,5-Тетра-иранс-витамин Ai, или витамин A, k„fH5°H = 325 нм; Е\%м = 1830,
температура плавления 64° С.
2,4,5-Три-транс-З-цис-витамин А
^.паГ,0Н =323 нм, гщс-пик=260 нм,
(25% биологической активности витамина А),
температура плавления 81,5—82,5° С.
„ г Н- сн3 н СН3Н
I I II
сн,он
(ill
Н Н • Н
сн3
1:1
2,3,4-Три-траис-5-чис-витамин А или неовитамин А (75% биологической актив-
ности), Z^2a^5°H=328 «л; Е}%м =1690, температура плавления 58—59° С.
2,4-Ди-транс-3,5-ди-({ис-витамин А (25% биологической активности), ^тах°Н =
=324 нм, г{ис-пик=260 нм, температура плавления 81,5—82,5° С.
СН3 Н,С Н СНз н н н
УХ ХСН2ОН
кЛсНз” Н СН3
2,3,5-Три-транс-4-чис-витамин А (3% биологической активности), ~
= 312 нм, температура плавления эфира р-фенилазобензоата — 99—100° С [12].
гн н г Н СН3 Н Н Н
•Ч^З гьу III | |
Х/С=С\ X /А
у у у у >у хн2он
н /Н сн3
3,4,5-Три-отранс-2-^ис-витамин А (25% биологической активности), л^2^4<)н=
= 322 нм [12].
Природный витамин А (ретинол), полученный из жира печени морских
рыб, содержит полный игрпнс-изомер в количестве 65 и 35% неовитамина
А, [13].
На рис. 1 изображена кривая поглощения ультрафиолетового света [7]
раствором витамина Aj в
Рис. 1. Спектр поглощения
витаминов At и А2 в УФ-свете.
этаноле, XmS6°H=325 нм, Е}°^=1830. Вита-
мин А дает с раствором треххлористой
сурьмы в хлороформе интенсивное синее
окрашивание с максимумом поглощения
620 нм. Окраска быстро меняется, и через
несколько минут максимум снижается до
580 нм. Неорганические галоидные соеди-
нения (AsCl3, SnCl4H др.), а также кислые
земли дают интенсивную окраску с вита-
мином А.
Витамин А хорошо адсорбируется ней-
тральными или щелочными адсорбентами
(окись алюминия, окись магния и др.), что
позволяет выделить его из смеси при помо-
щи хроматографирования. Кислые адсор-
бенты его разрушают. В ультрафиолетовом
свете витамин А флуоресцирует [11]. Как
первичный спирт витамин А образует про-
стые и сложные эфиры. Из простых эфи-
ров следует отметить метиловый с температурой плавления 34—35° С
и фенильный с температурой плавления 90—92° С. Оба эфира являются
твердыми веществами.
12
Витамин А быстро разрушается под действием кислорода воздуха, окис-
лительных агентов и ультрафиолетового света. Кислород воздуха при тем-
пературе 120° С полностью разрушает витамин А в течение 4 ч. В отсутст-
вии кислорода витамин А термоустойчив и не разрушается при температуре
120° С. Озон, перекись водорода так же, как и перекиси, образующиеся
в маслах при прогоркании, разрушают этот витамин.
Исследования показали, что основным фактором, влияющим на стой-
кость витамина А в концентратах рыбьего жира, является кислород возду-
ха. Кислотность концентрата по своему разрушающему действию занимает
второе место. Щелочи и олово не оказывают какого-либо влияния на про-
цесс распада витамина А [13а].
К’ Для повышения устойчивости витамина А в его масляных растворах
рационально добавить специальные вещества, поглощающие кислород воз-
духа. К этим веществам, названным антиоксидантами, относятся: гидро-
хинон, лецитин, токоферол, масла, богатые токоферолом (соевое масло),
сантоквин (1,2-дегидро-6-этокси-2,2,4-триметилхинолин), сложные эфиры
галловой кислоты (пропилгаллат, додецилгаллат и др.). Минеральные
кислоты инактивируют витамин А.
Ангидровитамин At получают из витамина At в присутствии
кислот (небольших количеств НС1) по схеме [11]:
СНз СН3 СН3 СН3
<4—СН=СН-С=СН-СН=СН-С=СН-СН2ОН н+
I Л” WT
6 Витамин Л
СЖ СН3 сн3 сн3
г><Ч=сн-сн=с-сн=сн-сн=с-сн=сн2
3 ЛнгидроВшпамин Л,
Ангидровитамин Aj— оранжево-желтые кристаллы, имеют температу-
ру плавления 76—77° С и максимум поглощения в ультрафиолетовом свете
351, 371, 392 нм, Ei°m — 2540, 3680, 3200. По другим источникам [8] в
спирте — 350, 368, 389 нм.
Ангидровитамин Aj в малых концентрациях содержится в печеночных
жирах рыб и других естественных источниках витамина А.
Витамин А-альдегид (ретинен, ретиналь) явля-
ется пигментом зрительного пурпура. Он был выделен Вальдом [14] и ис-
следован Мортоном [15]. Путем окисления витамина А перманганатом ка-
лия [16] или еще лучше двуокисью марганца [17] получают витамин А-аль-
дегид-2,3,4,5-тетра транс (ретиналь), температура плавления 61-—62° С;
7тах=368 нм, Е\%я = 1050 (в этиловом спирте) [18]; по другим источникам
[17] Кщах = 369 нм, E\°jM = 1685 — в петролейном эфире; или [8] темпера-
тура плавления 57 и 65° С (диморфная форма) Хтах = 381 нм — в спирте.
Витамин А2(С2,Н27ОН). В 1937 г. во ВНИВИ Е. Ледерером и В. Розано-
вой [19, 20] было показано, что в неомыляемой фракции жира печени прес-
новодных рыб содержится вещество, обладающее А-витаминной активностью
и имеющее максимум поглощения в спирте 351—352 нм, вместо 325 нм —
для витамина А. Это же вещество с треххлористой сурьмой (реакция Карр-
Прайса) имеет максимум 690 вместо 620 нм (см. рис. 1). Этому веществу
авторами присвоено название витамина А2 [21]. Позднее витамин А2 был
выделен хроматографически в виде желтого масла, а также кристаллов
фенилазобензоата с температурой плавления 76—77° С; Хтах = 341нл;
№ = 1190 [22].
13
Рядом исследователей [23, 24, 25] была установлена структурная фор-
мула витамина Л2 (дегидроретинол)
СНз СН3 СН3 сн3
— сн=сн-с=сн-сн=сн-с=сн-сн2он
^^СНз
Витамин Аг (дегидроретинол)
На рис. 1 изображена кривая поглощения витамина А2 в ультрафиоле-
товом свете. Максимум поглощения для витамина Л2 установлен в 351 —
352 нм, ЕЦм = 1460. Биологическая активность витамина Л2 составляет
40% от биологической активности витамина Аь Витамин А2 в кристалли-
ческом виде не получен.
Дегидроретиналь. При окислении витамина А2 образуется
дегидроретиналь (С20Н26О). Температура плавления дегидроретиналя 77—
78° С; Хтах5°н = 400 нм, £1^= 1380 [26]. Биологическая активность де-
гидроретиналя около 40% от биологической активности витамина At.
Ангидровитамин А2 — температура плавления 89,5° С, Хтах—
—352; 370 нм\ £1^=2040; 2980 — спирт [22].
Витамин А3 (С40Н66). В 1966 г. Л. Шнайдман, М. Ушакова, А. Ефимов и
И. Кущинская показали, что каротиноид, выделенный из мякоти плодов
шиповника и томатов, предупреждает развитие А-авитаминоза и излечива-
ет А-авитаминоз у лабораторных животных, содержащихся на А-авитами-
нозной диете. Биологическую активность ликопина указанные исследовате-
ли проверяли в двух направлениях.
1. Опыты предупреждения А-авитаминоза ликопином [27] проводились
на белых крысах-самцах массой 40—44 г. Животных содержали на А-ави-
таминозной диете. Препараты вводили внутрь в двух дозах (6 и 12 мкг).
Контролем служили животные, получавшие только А-авитаминозную диету
(отрицательный контроль), а также группы животных, получавшие витамин
А-ацетат и синтетический |3-каротин. Тестом, характеризующим активность
препаратов, служил привес животных. Опыт йродолжался 12 недель. При-
вес животных, содержащихся на А-авитаминозной диете, приведен в табл. 1.
Таблица 1
Препарат
Доза,
мкг
Привес
животных
% к
г контро-
лю
Препарат
Доза,
мкг
Привес
животных
% К
г контро-
лю
Ликопин, выделенный из
шиповника.............
То же.................
₽-Каротин.............
6
12
6
23,8
26,9
24,5
145,1
164,0
149,4
р-каротин............
Витамин А-ацетат . .
То же . .'...........
Контроль отрицатель-
ный .................
12
6
12
28,1
24,7
27,8
171,3
150,6
170,0
16,4 100,0
Статистически достоверные результаты опыта показали, что каротиноид
ликопин предохраняет от А-авитаминоза животных, содержащихся на ави-
таминозной диете.
2. Опыты излечения А-авитаминозных животных ликопином [27]. Ли-
копин был выделен из томатной пасты в кристаллическом виде. Идентич-
ность установлена хроматографически и спектром поглощения в видимой
области света Хтах—445; 470; 502 нм (в петролейном эфире). Кристаллы ли-
копина растворяли в подсолнечном масле. Опыт проводили на белых кры-
14
сах-самцах массой 55—65 г в течение 15 недель. В первом периоде опыта
животные содержались 6 недель на А-авитаминозной диете. Во втором
периоде животные (по 6 в каждой группе) подвергались лечению в течение
9 недель. Животные были разделены на четыре группы: первая группа
получила витамин А-ацетат в дозе 50 мкг-, вторая — |3-каротин — 185 мкг-,
третья — ликопин — 185 мкг в сутки (все препараты были растворены в
подсолнечном масле). Четвертая группа получила подсолнечное масло без
препаратов (препараты давали внутрь через зонд). Результаты лечения
А-авитаминозных животных различными препаратами [27] приведены в
табл. 2. •
Таблица 2
Препарат Масса жи- вотного до лечения, г Результаты наблюде- ний через 7 недель Состояние животных после лечения
привес, г погибло из 6 животных
А-ацетат ....... 101,7 72,5 0 Состояние хорошее без
Р-каротин 99,8 86,0 1 признаков А-авитаминоза То же
Ликопин 95,4 51,4 1 »
Контроль отрицатель- ный 90,6 3,8 6 Все погибли
Результаты данного опыта, полученные аналогично при двукратном
повторении, свидетельствуют об успешном лечении ликопином А-авитами-
нозных заболеваний животных. Во всех случаях в печени излеченных жи-
вотных найден ликопин, а витамин А не обнаружен.
Рис. 3. ИК-спектр поглощения ликопина.
Рис. 2. Спектр по-
глощения витами-
на А3 (ликопина)
в видимой области
света.
Таким образом, экспериментально установлено, что ликопин обладает
А-витаминным действием. Автор полагает, что ликопин может быть отнесен
к витаминам группы А в качестве витамина А3.
Существовавшие до настоящего времени мнения, что ликопин, как каро-
тиноид, в структуре которого отсутствует |3-ионон, не обладает А-витамин-
ной активностью, не подтвердилось [27а].
Витамин А3 (ликопин) имеет максимумы поглощения в видимой области
спектра в петролейном эфире Хтах—443; 471; 503 [28]; 446; 470; 500 нм
[29]. Rf при тонкослойной хроматографии (на окиси алюминия и подвиж-
ном растворителе — петролейный эфир: бензол: метанол 60:10:1)—0,21.
На рис. 2 изображен спектр поглощения ликопина в видимой области
света, на рис. 3 — спектр поглощения в инфракрасной области света; тем-
15
пература плавления ликопина 174—175° С [30]. Ликопин легко растворим
в бензоле и в сероуглероде, плохо — в кипящем спирте.
Структурная форма ликопина (С40Нв6)
сн3 сн
сн3 сн3
I сн3 сн3 сн3 сн3
сн=сн-с=сн-сн=сн-с=сн-сн=сн-сн=с-сн=сн-сн=с-сн=сн
3
СН3
Витамин
Н3с
Физико-химические константы витаминов группы А и некоторых их
производных приведены в табл. 3 [7, 9].
Таблица 3
Витамины группы А и некото- рые их производные [7, 9] Молеку- лярная масса Температура плавления °C, или Абсорбционный максимум в этиловом спирте, нм pi% ^1см Биологическая активность 1 мг, и. е.
Витамин А (алкоголь) . . Сложные эфиры: 286,4 64° 325 1830 3330
ацетат* 328,5 57—58° 326 1550 2910
бутират 256,5 1,5986 325—328 1345 2200
бензоат 390,5 1,6305 325-328 1240 1800
пальмитат 524,8 28-29° 325—328 975 1600
стеарат Простые эфиры: 552,9 1,5548 325-328 940 1500
метиловый 300,5 34-35° 326** 1660 3500
фениловый . . . . . Витамин А-альдегид (ре- 362,5' 90—92° 327 1460 100
тиналь) 284,4 61—62° 368-369,5 (петролей- ный эфир) 1050-1685 3000
Витамин Ai-кислота . . Витамин А2 (дегидрорети- 272,4 179—181,5 347-353 1460-1600 1320
нол) Витамин Аа-альдегид (де- 284,4 351-352; 287—288 1460-1454; 820—771 1320
гидроретиналь) 282,4 77—78 400 1380-1350 1320
Витамин А3-(ликопин) . 536,9 174-175 443; 471; 503 — —
* Витамин А-ацетат витамина А в и. е. * * В изопропиловом с служит эталоном для пирте. установления биологической активности
МЕТОДЫ СИНТЕЗА ВИТАМИНА Ах И ВЫБОР 4
РАЦИОНАЛЬНОГО МЕТОДА ДЛЯ ПРОИЗВОДСТВА [7, 31, 32]
В течение короткого периода (1947—1950 гг.) было опубликовано боль-
шое число весьма важных работ по синтезу витамина А [33—42]. Одновре-
менно в различных странах был осуществлен синтез витамина А: в Швей-
царии [43], в США [44], в Голландии [45], в ФРГ [39].
Разработаны следующие три основные схемы синтеза витамина А: 1) из
Р-ионона через кетон С18 с использованием реакции Реформатского; 2) из
2,6,6-триметилциклогексанона; 3) из |3-ионона через альдегид С14 с приме-
нением реакции Дарзана.
СИНТЕЗ ВИТАМИНА А ИЗ ₽-ИОНОНА ЧЕРЕЗ КЕТОН С18
Первые попытки синтезировать витамин А из Р-ионона с применением
реакции Реформатского были сделаны Куном и Моррисом в 1937 г. [46].
Однако недостаточная очистка промежуточных продуктов от различных 16
16
Примесей и изомерных соединений обусловили получение препарата с весь-
ма низким содержанием витамина А (7,5%). В связи с этим другие исследо-
ватели не воспроизводили этот синтез [35, 47].
Более поздние исследователи [42] объясняли неудачу синтеза Куна
загрязнением этилового эфира |3-ионолиденацетата |3- и ^-изомерами (см.
стр. 18).
Большое значение для этого синтеза имело открытие свойств алюмогид-
рида лития, который избирательно восстанавливает карбоксильную группу
в спиртовую, не затрагивая двойные углерод-углеродные связи [35, 48].
Кетон С18 был впервые синтезирован из |3-ионона с эфирами рбром-
кротоновой кислоты в присутствии цинка (реакция Реформатского) с полу-
чением транс-|3-ионолиденкротоновой кислоты. При воздействии на по-
следнюю метиллития [45] получили кетон С18 по следующей схеме:
сн3
СН=СН-С=О+ВгСН2СН=СНСООС2Н5
Zn + бензол
а _ ионон Зтилобый эфир -у - бром крота -
“ нобой кислоты
сн3
сн=сн-с=сн-сн=сн-соон
сн3
СН=СН-С=СН-СН=СНСООС2Нз
Зтилобый эсрир-^З-ионолиденкротонабой л и ело ты
кон
CH3Li
Трине -J3 - ионолиденкротоноёая кислоте
сн3 сн3л,
I 3 I JOLi
сн=сн-с=сн-сн=сн-с^
OLi
Н2О
сн3 СН3
сн=сн-с=сн-сн=сн-с=о
Кетон С}8
Повторение с кетоном С18 реакции Реформатского, т. е. конденсация его
с эфиром бромуксусной кислоты, дает витамин А-кислоту. С другой стороны,
конденсация кетона С18 с броммагнийэтоксиацетиленом, частичное гидри-
рование и омыление получающегося винилового эфира приводит к получе-
нию витамина А-альдегида, который при восстановлении с алюмогидридом
лития дает витамин А.
сн3 сн3
СН=СН-С=СН~СН=СН-С=О + MgBrOC-OC2H5
Кетон Ctg
сн3 сн3
СН=СН-С=СН-СН=СН-С-С=С-ОС2Н5 H/Pd
он ’
сн3 сн3
СН=СН-С=СН-СН=СН-С-СН=СН-ОС2Н5
он
сн3 сн3 н
сн=сн-с=сн-сн=сн-с=сн-с-о
Витамин Л ~ альдегид
LiA1H4
сн3 сн3
сн=сн-с=сн-сн=сн-с=сн-сн2он
Витамин А
17
Однако при этом синтезе получили вещество, загрязненное различными
изомерами, о чем свидетельствовал абсорбционный спектр в ультрафиоле-
товом свете (максимум при 315—320 вместо 325 нм для витамина А), причем
выход вещества был крайне низок (около 5% на |3-ионон).
Для повышения выхода были исследованы другие варианты этого син-
теза [49—55], в частности для удлинения углеродной цепи (3-ионона в реак-
ции Реформатского вместо эфиров рбромкротоновой кислоты использовали
этиловый эфир бромуксусной кислоты (ВгСН2СООС2НБ) или броммагний-
этоксиацетилена (BrMgC = СОС2НБ). Однако и эти варианты синтеза ке-
тона С18 не дали заметного повышения выхода витамина А.
Впоследствии причина низкого выхода витамина А при синтезе его из
Р-ионона и эфира бромуксусной кислоты в присутствии цинка (реакция
Реформатского) или броммагнийэтоксиацетилена была выяснена в 1952 г.
голландскими исследователями [56]. Аналогичные работы в 1953—1955 гг.
проводились в СССР [54, 55, 57, 58]. Было показано, что дегидратация
промежуточного оксиэфира сопровождаётся следующей аллильной пере-
группировкой в циклогексеновое кольцо:
сн3
сн=сн-с-сн2-соос2н5_Ш
он
сн3
=сн-сн=с-сн2-соос2н5
сн3
= CII-CH=C-CH2-COOC2HS
он
Полученное соединение проявляет способность к другим перегруппиров-
кам [59], которые в дальнейшем синтезе обусловливают образование изо-
мерных веществ, не обладающих биологической активностью. Таким обра-
зом, метод синтеза витамина А из |3-ионона через кетон С18 не получил прак-
тического применения, вследствие низкого выхода витамина.
СИНТЕЗ ВИТАМИНА А ЧЕРЕЗ 2,6-6-ТРИМЕТИЛЦИКЛОГЕКСАНОН
Дефицитность цитраля, из которого синтезируют |3-ионон, побудила ис-
следователей искать новые пути синтеза витамина А без применения (3-ионо-
на. Такой метод был разработан, исходя из триметилциклогексанона, кото-
рый при действии ацетилида натрия в жидком аммиаке дает 2,6,6-триметил-
1-этинилциклогексанол-1 с хорошим выходом [60—62]. Последний конден-
сируют по реакции Гриньяра с кетоном С9 в гликоль С20, который в кислой
среде претерпевает аллильную перегруппировку. При восстановлении гли-
коля посредством алюмогидрида лития [63] и ацетилировании получают
моноацетат, дегидратация которого при помощи р-толуолсульфокислоты
дает продукт, содержащий 50% витамина А-ацетата, ангидровитамин А и
другие вещества. Синтез осуществлен по следующей схеме:
Триметилциклогексанон
NaC=CH
сн3 сн3
о=с-сн=сн-сн=с-сн=сн2
2C2H5MgBr
2,6,6-?риметил-1-этинил- Лотон Cs
циклогексанол -1
18
СН3 СН3 СИ3 СН3
></С=С-С-СН=СН-СН=С-СН=СН2 ...
г г\ । Н
-------Н J?OH он -------
СН3
° Гликоль Ого
СН3 СН3 СН3 СН3
><,СеС-С=СН-СН=СН~С=СН-СН2ОН
Аллильная \ уС LlAlHj.
и > ОН г
перегруппировка Селективное
С Н з Л- Г'' Г ацетилирование
СН3 СНз ^Из VH3
'Х\хСН=СН-С=СН-СН=СН-С=СН-СН2-ОСОСНз
..... Г Р-Толуол-
*[ 1 ОН Китами^
слота
Моноацетат
Витамин А может быть получен в чистом виде-после соответствующей
хроматографической очистки.
Этот метод синтеза представляет лишь теоретический интерес, так как
получение кетона С9 и триметилциклогексанона требует нескольких стадий
синтеза с низкими выходами полупродуктов, и в связи с этим получается
очень низкий выход витамина А.
СИНТЕЗ ВИТАМИНА А ИЗ ₽-ИОНОНА ЧЕРЕЗ АЛЬДЕГИД С14
Как было указано, удлинение цепи Р-ионона сопровождается образова-
нием значительного количества изомерных веществ, что влечет за собой
снижение выхода биологически активного вещества. Впоследствии была
установлена возможность удлинения боковой цепи Р-ионона без образова-
ния побочных соединений путем глицидного синтеза Р-ионона с эфирами
хлоруксусной кислоты в альдегид С14. Впервые альдегид С14 был получен
в 1937 г. [641 из Р-ионона с применением реакции Дарзана.
Синтез витамина А через альдегид С14 был разработан рядом исследова-
телей [65,66,43]. Большое число исследований посвящено изучению альде-
гида С14 [67—71].
Синтез состоит из следующих основных стадий: 1) синтез Р-ионона;
2) синтез альдегида С14; 3) синтез ацетиленового карбинола; 4) синтез
витамина А или его ацетата.
Синтезр-ионона. Исходным сырьем для синтеза витамина А через альде-
гид С14 является цитраль, получаемый из лемонграссового или кориандро-
вого масла или синтетическим путем [71а].
При конденсации цитраля и ацетона в присутствии щелочи получается
псевдоионон. Последний под влиянием минеральных кислот (концентри-
рованная H2SO4) циклизуется с изомеризацией в р-ионон по следующей
схеме:
Цитраль
СН3СОСН3
NaOH
Псебдоионон
Синтез альдегида С14. р-Ионон конденсируется с хлоруксусным эфиром
под влиянием метилата натрия или изобутилата калия по реакции Дарзана
19
[64, 66] в альдегид С14. Реакция проходит через промежуточное неустой-
чивое соединение — глицидный эфир [43], который после омыления и де-
карбоксилирования на холоду дает альдегид С14 [72] согласно следующей
схеме:
С1СН2СООСНз >
CH3ONa LJ-
сн3
СН=СН-С-СН-СООСН3 ОН'
о
Метилобый хлорукусный Глицидный эфир
зцзир и метилат натрия
fl - Ионон
Альдегид
Выход альдегида С14 по этой схеме составляет 80% от теоретического1.
Синтез ацетиленового карбинола. Следующим промежуточным продук-
СН3
I
том синтеза является «цепочка» — ацетиленовый карбинол НС=С—С=
=СН—СН2ОН, который синтезируют путем конденсации метилвинилке-
тона и литийацетилида в жидком аммиаке в присутствии ацетилена (под
давлением). Что касается метилвинилкетона, то синтез осуществляют пу-
тем конденсации ацетона и муравьиного альдегида в присутствии щелочи
с последующей дегидратацией фосфорной кислотой. Метилвинилкетон
может быть также получен из винилацетилена при действии слабой серной
кислоты в присутствии сернокислой ртути. Синтез ацетиленового карбинола
идет по следующей схеме:
fl* NaOH HSPO4
СН3—СО—СН3 + НС ------------> СН3СОСН2СН2ОН------> СН3СОСН=СН2 —
хн
V Метилвинилкетон
сн3 сн3
сн=сы | HCI |
------> НСнС—С—СН=СН2------------> СНг С—С=СН—СН2ОН.
i Аллильная _
' перегруппи- Первичный ацетиленовый
ОН ровка карбинол
Другой вариант этого синтеза протекает по такой схеме:
Н2О (НС = С)2 Са
HC.-.C—СН=СН2-----------> Н3С—СО—СН=СН2------------>
H.SO. • HgS04
Винилацетилен
сн3
НСееС-СН=СН2 —►
' ОН
Третичный ацетиленовый
карбинол
сн3
НС = С—С=СН—СН2ОН.
Первичный ацетиленовый
карбинол
1 По данным советских исследователей (Г. Самохвалов, Л. Жукова и Н. Преоб- •
раженский, ЖОХ, 1956, 26, 3105), альдегид С14 имеет следующее строение:
СН3 н .
СНз- сн=с-с
%
20
Синтез витамина А-ацетата. Имеются три варианта получения витамина
А-ацетата из альдегида С14 путем конденсации его: 1) с третичным ацети-
леновым карбинолом [73]
СН3
HCsC—СН=СН2;
I
ОН
си3
2) с первичным ацетиленовым карбинолом HCz^C—С=СН—СН2ОН
[74—76] и 3) с литийацетилидом, а затем с 4-ацетоксибутанон-2
(СН 3СОСН2СН2ОСОСН 3)‘.
Из указанных трех вариантов наилучшие результаты [7 ] получены при
использовании второго варианта конденсации альдегида С14 с первичным
ацетиленовым карбинолом по реакции Гриньяра. При этом образуется аце-
тиленовый гликоль С20 (температура плавления 59° С) с высоким выходом.
Его подвергают в присутствии -частично дезактивированного палладиевого
катализатора [77] селективной гидрогенезации (с количественным выхо-
дом). При этом ацетиленовая связь гликоля С20 гидрогенизуется до этиле-
новой, другие непредельные связи в молекуле не затрагиваются. Гликоль
далее частично ацилируют в присутствии пиридина при 0° С и получают
гликоль-моноацетат (температура плавления 74° С). Затем последний под-
вергают дегидратации. Для дегидратации применяют различные ме-
тоды: 1) йод в петролейном эфире [751; 2) хлорокись фосфора в присутст-
вии пиридина [75]; 3) пиридингидробромид в ледяной уксусной кислоте
[73]. Наилучшие результаты (выход 45%) дает хлорокись фосфора в при-
сутствии пиридина.
Синтез витамина А-ацетата из альдегида С14 протекает по следующей
схеме:
сн3 н
сн=сн-сн-с=о
Альдегид
сн3
+ нс=с=сн-сн2он
Первичный ацетиленовый
карбинол
2C2HsMgBr
СНо СН3 сн3 сн3
С|Г-СН=СН-СН-СН-С=С-С=СН-СН2ОН
<АСНз ОН
Ацетиленовый гликоль Сго
И
Pd
СНо СН3 сн3 сн3
pV-CH=CH-CH-CH-CH=CH-CH=CH-CH2OH
кАСНз он
Гликоль С 20
СН3СОС1
Пиридин
СН, СН3 СН3 сн3
C>Sr-CH=CH-C-CH-CH=CH-C=CH-CH2OCOCH3 Дегидратация
н ОН Пири3ин + РОС13’
Гликоль-моноацетат
СНо СН3 СН3 сн3
. Plj—сн=сн—С= СН-СН=СН-С=СН-СН2ОСОСН3
kx"zK'CH3
Витамин А-ацетат
21
Общий выход витамина А по этой схеме, считая на р-ионон, составляет
25—45 % [7], т. е. наиболее высокий из рассматриваемых трех схем синте-
за. Поэтому схема синтеза витамина А через альдегид С14 из р-ионона нашла
широкое практическое применение и должна быть выбрана как наиболее
эффективная.
Для осуществления синтеза витамина А из альдегида С14 необходим
р-ионон, который до последнего времени получают из природного альде-
гида-цитраля. Последний выделяют из лемонграссового или кориандрового
масла путем окисления содержащегося в них линалоола. Однако ресурсы
природного цитраля ограничены и для промышленного производства ви-
тамина А необходим синтетический цитраль [78]. Одним из перспективных
методов синтеза цитраля является получение его из изопрена через гера-
нилхлорид [79, 80]. Для этвго на изопрен действуют хлористым водородом
и полученную смесь а, а, и 7, 7-диметилаллилгалогенидов совместно с изо-
преном подвергают теломеризации в присутствии SnCl4 в геранилхлорид
(совместно с терпенилхлоридом). Последний выделяют в виде уротропино-
вой соли четвертичного основания и нагреванием с формальдегидом превра-
щают в цитраль.
Изопрен Геранилхлорид
Цитраль
Цитраль может быть синтезирован и другим методом, в котором исход-
ным сырьем является ацетон и ацетилен. Ацетон конденсируют с ацетиле-
нидом натрия в диметилэтинилкарбинол с выходом в 80%, который гид-
рируют в присутствии частично отравленного палладиевого катализатора
на углекислом кальции в диметилвинилкарбинол по схеме [81]:
Ацетон
NaC = CH
Ацетиленид
наФрия
сн
Диметилзтинил- Диметилбинил-
карбцнол карбинол
Диметилвинилкарбинол далее превращают в 2-метилгептен-2-он-6 (ме-
тилгептенон). Это может быть осуществлено различными методами:
1) на диметилвинилкарбинол действуют бромистым водородом и, не вы-
деляя получаемого а.а-диметилаллилбромида, конденсируют с натрийаце-
тоуксусным эфиром; после омыления получают 2-метилгептен-2-он-6 с
выходом 75% по схеме [82]:
НВг
СН2Вг
COOR
I
NaCHCOCH3
Диметилбинил- cC,ct - Диметилал-
карбинал лилбромид
Натрийацетоуксусный метилгептенин
Эфир
2) метилгептенон может быть также получен, с выходом 60—70% из
диметилвинилкарбинола этерификацией ацетоуксусным эфиром с последую-
22
щим пиролизом при температуре 160— 165° С под давлением по схеме
182]:
СШСН3
|| он
сн2
COOR
I
СН2СОСН3
Диметилбинил-
карбинол
или
2. О=С5 ,ДС=СН2
^CfT2
ГЛ-1 I
сн3 сЩУ I
. f !_о____jCH2
сн2 .
сн3
С^
1. Ацетоуксусный Эфир Ацетоацетат
2. Дикетен диметилбинилкарбинола
Пиролиз
160-165 °C
Метилгелтенон
Промежуточным продуктом, который не выделяется, но может быть
выделен [82], является ацетоацетат (ацетоуксусный эфир диметилвинил-
карбинола). Это же соединение может быть получено методом этерификации
диметилвинилкарбинола дикетеном [83, 84] в присутствии металлического
натрия с выходом 60% или в присутствии пиридина с выходом 92% [82].
Превращение метилгептенона в псевдоионом может быть осуществлено
через 3-7-диметилоктаен-6-ин-ол-3 (дегидролиноол). Последний получают
конденсацией метилгептенона по методу Назарова [84] с ацетиленом в
среде эфира под влиянием едкого кали при температуре 0—20° С при давле-
нии 5—10 кгс/см2 с выходом 92% [85] по схеме:
Метилгелтенон
нс^сн
кон
Дегидролиналоол
Дегидролиналоол можно превратить в псевдоионон следующими мето-
дами:
1) ацетоацилируют дикетеном [83, 86] и полученный ацетоацетат подвер-
гают пиролизу. При этом в результате внутримолекулярной перегруппиров-
ки (с отщеплением СО2) образуется псевдоионон с выходом 50—55 %. Вмес-
то дикетена для ацилирования может быть применен ацетоуксусный эфир
[87] с тем же выходом по схеме:
/О.
1. О = сД ДС=СН2
хсн2
Ацвтоацетат
Дегидролиналоол
COOR
СН2СОСН3
/. Дикетен
2. Ацетоуксусный зсрир
СНз,СН3 СН3
no-iao’o^ р||—СН=СН—СО
СН3
Псевдоионон
2) дегидролиналоол лучше превратить в псевдоионон по методу Излера
[88] без применения дикетена или ацетоуксусного эфира, а ацетилировани-
23
ем дегидролиналоола, изомеризацией полученного ацетата, присоединени-
ем уксусной кислоты с последующим гидролизом в присутствии ацетона
в псевдоионон с выходом 80%.
Дегидролиналоол
сн3 сн3 сн3 сн3 . СЩ СПз сн3
(СН3)2СО ^снососнз (сн3)2со Ж'|-сн=сн-со
ДгСН3 сн3
ососн3 сн3
Псебдоионон
. Из анализа приведенных различных методов синтеза псевдоионона
следует, что наиболее перспективным является метод получения его из изо-
прена через геранилхлорид (метод Лээтса). Этот синтез состоит из неболь-
шого числа стадий и не требует дефицитного сырья. Однако этот метод
технологически еще не отработан. По-видимому, наиболее отработанным
для промышленного применения является синтез псевдоионона из ацетона
и ацетилена через диметилэтинилкарбинол, диметилвинилкарбинол, метил-
гептенон и дегидролиналоол.
технологическая схема производства синтетического витамина а
Технологическая схема, разработанная в основном Н. Преображенским,
Г. Самохваловым и их учениками [89], состоит из изложенных ниже стадий
синтеза, включающих получение псевдоионона, ₽-ионона, альдегида С14
и витамина А (ацетата и пальмитата).
СИНТЕЗ ПСЕВДОИОНОНА
Синтез псевдоионона (рис. 4) осуществляют по следующей химической
схеме [81, 82, 85, 88]:
сн3
сн3-с=о
Ацетон
НС = СН I н, ।
——^-с-осн —Са^ сн3-с-сн=сн2
он он
А цетилен Диметилэтинилкарбинол Диметилвинилкарбинол
Диметилэтинилкарбинол [81]. Синтез его осуществляют по следующей
химической реакции:
О СН3
II КОН |
СН3—С—СН3 + HCsCH ---------> СН3—С— С~СН.
о!н
Ацетон 58,08 Ацетилен Диметилэтинилкарбинол
26,04 84,11
24
Рис. 4. Технологическая схема
производства синтетического псевдоионона.
В реактор 1 из нержавеющей стали, снабженный мешалкой и обратным
холодильником, загружают из мерника 2 дибутилформаль, а затем едкий
калий (технический 85—88%), нагревают до 130—135° С и при интенсивном
перемешивании суспендируют расплавленный едкий калий в течение 30 —
40 мин. Затем охлаждают до ПО—115° Сив течение 2 ч из баллона 3 насы-
щают ацетиленом, после чего охлаждают до минус 8—10° С в реакторе 4
и вводят ацетон из мерника 5 в течение 3 ч, перемешивают 3—4 ч при не-
большом токе ацетилена. После добавления ледяной воды из мерника 6
(четырехкратное к массе ацетона) и перемешивания 15—20 мин разделяют
слои в делительной воронке 7. Водный слой экстрагируют эфиром, сливае-
мым в экстрактор 9 из мерника 8. Экстракт объединяют в реакторе 10 с ди-
бутилформальным раствором, нейтрализуют углекислотой из баллона 11
и сушат сульфатом натрия. Разгонку ведут в перегонном аппарате с
колонкой 12: первая фракция (36—50° С) — эфир используют для экст-
ракции; вторая фракция (50-—100° С); третья фракция 103—105° С содер-
жит диметилэтинилкарбинол 97%. Выход 75—76%. Ее собирают в прием-
нике 13.
Диметилэтинилкарбинол представляет собой бесцветную жидкость,
температура кипения 101—102° С при давлении 760 . мм рт. ст.-,
С6Н8О, молекулярная масса 84,11; п® = 1,4208. Хорошо растворим в орга-
нических растворителях, смешивается с водой.
Диметилвинилкарбинол. Это соединение получают из диметилэтинил-
карбинола селективным гидрированием тройной связи до двойной в присут-
ствии палладиевого катализатора по следующей химической схеме [81]:
СНз СН3
I [HJ I
сн3—с—с=сн----------> сн3—с—сн=сн2.
I Pd/CaCO3 I
ОН ОН
Диметилэтинилкарбинол Диметилвинилкарбинол
84,И 86.13
Гидрирование ведут в автоклаве 14, снабженном мешалкой и рубашкой,
обогреваемой водой, куда через мерник 15 загружают диметилэтинил-
карбинол; катализатор (палладированный СаСО3) добавляют в количестве
1 % к массе карбинола. Температура гидрирования 23—25° С, давление во-
дорода 3—3,5 кгс/см2. Автоклав предварительно продувают азотом, подавае-
мым из сети, либо из баллона 16. Водород подается из баллона 17. Контроль
ведут на наличие тройной связи аммиачным раствором азотнокислого се-
ребра. По окончании гидрирования фильтруют реакционную массу на нутч-
фильтре 18 и фильтрат собирают в приемнике 19. Выход около 95%, содер-
жание вещества 83—85%. !
Катализатор — палладированный мел получают следующим образом.
В смесителе 20 приготовляют водный раствор хлористого кальция, а в сме-
сителе 21 — водный раствор углекислого натрия. Оба раствора сливают в
реактор 22. Осадок мела отфильтровывают на нутч-фильтре 23 и промы-
вают водой. Осадок мела смешивают с водой в реакторе 24. В реактор 25
из мерника 26 сливают разбавленную соляную кислоту (8%-ную) и в ней
растворяют хлористый палладий. Раствор сливают в реактор 24 и при тем-
пературе 80-—90° С перемешивают, фильтруют на нутч-фильтре 26. Осадок
после промывки (до отсутствия ионов хлора) высушивают в вакуум-сушилке
27 и хранят в сборнике 28.
Диметилвинилкарбинол — бесцветная жидкость, температура кипения
95—96° С при давлении 760 мм рт. ст.; С5Н10О, молекулярная масса 86,13;
По - 1,4140; <*4° = 0,8618.
26
Метилгелтенон (6-метилгептен-5-он-2). Получают его конденсацией ди-
метилвинилкарбинола и ацетоуксусного эфира при температуре 160—165° С
по следующей химической схеме [821:
СН3 COOC2IIS
СН3-С-СН=СН2 + СН2СОСН3
160 -165 °C
Диметилдинилкарбанол
86,13
Ацетоуксусный
эсрир
130, 74
сн3
сн3-с-сн=сн2
ОСОСН2СОСН3
Ацетоицетат диметилдинил -
ларбинола
170,20
Метилеептенон
126,13
В реактор 29 из нержавеющей стали, снабженный колонкой с дефлегма-
тором и конденсатором, из мерника загружают вазелиновое масло (высо-
кокипящий разбавитель) и при температуре 210° С (в масле) загружают
диметилвинилкарбинол и ацетоуксусный эфир так, чтобы температура ре-
акционной массы была не ниже 160—165° С. Затем нагревание продолжают
при температуре 160—180° С 3 ч до прекращения выделения газа (СО2).
В сборник после конденсатора собирают отгон (спирт с примесью ацетона).
Кубовый остаток разгоняют при остаточном давлении 5—6 мм рт. ст. в
вакуум-перегонном аппарате 30. Готовый продукт поступает в приемник.
Выход 60% [82].
Метилгелтенон — бесцветная жидкость, температура кипения 52—53° С
при остаточном давлении 5 мм pm. cm. CgH14O, молекулярная масса 126,19;
По = 1,4404; dВ * * * * * * * * * * * 20=0,8616, хорошо перегоняется с водяным паром; Хтах =
= 243 нм (в спирте), Igs =2,54.
Дегидролиналоол (3,7-диметилоктаен-6-ин-Гол-3). Дегидролиналоол
синтезируют по следующей химической реакции [85]:
НС^СН
Ацетилен
26,09
СНя сн3
КОН с=сн
СН3
Дегидролиналоол
152,23
В реактор из эмалированной стали 31, снабженный мешалкой, барботе-
ром для подвода ацетилена загружают толуол из мерника 32 и порошко-
образное едкое кали, нагревают до 80° С и из баллона 33 пропускают аце-
тилен при перемешивании в течение 2 ч. После прекращения нагревания
уменьшают ток ацетилена, охлаждают рассолом до —12—10° С и постепен-
но в течение 3 ч приливают метилгелтенон из мерника 34. Затем добавляют
воды и после перемешивания разделяют слои в делительной воронке 35.
Толуольный раствор переводят в реактор 36, в котором нейтрализуют угле-
кислотой. В перегонном аппарате 37 отгоняют толуол, а затем при остаточ-
ном давлении 12—14 мм рт. ст. собирают фракцию, кипящую при темпера-
туре 89—91 ° С. Выход 76—80 %.
Дегидроналоол — бесцветная жидкость, температура кипения 78—80°С
при остаточном давлении 8 мм рт. ст.-, С10Н1вО, молекулярная масса
152,23; «0 = 1,4632. Хорошо растворим в органических растворителях,
плохо — в воде.
Псевдоионон. Псевдоионон получают из дегидролиналоола путем аци-
лирования его, изомеризации ацетата, омыленйя его и конденсации с аце-
27
тоном в присутствии едкого натра. Синтез протекает по следующей схеме
[881:
Дееидролиналоол
+ (сн3со)2о
Ацетат дегидролиналоола
199,26
+ СН3СООН
Ацетат дегидролиналоола
Уксусный ангидрид
102, 09
Уксусная кислота
60, 05
В реактор из нержавеющей стали 38 загружают из мерника 39 дегидро-
линалоол, из мерника 40уксусный ангидрид и из мерника 41 каталитическое
количество фосфорной кислоты, перемешивают (температура не выше 50° С)
и выдерживают 14—15 ч при температуре 18° С. Затем вводят в реактор из
баллона 42 азот, нагревают реакционную массу до 90° С и добавляют
каталитическое количество карбоната серебра, продолжая перемешивание
1,5 ч при температуре 90° С. Далее реакционную массу охлаж-
дают до 20° С и передают под давлением в реактор 43, в который
из мерника 44 загружают 20%-ный водный раствор хлористого натрия.
После перемешивания разделяют слои в делительной воронке 45. В ней
же промывают верхний слой раствором хлористого натрия до нейтральной
реакции. Затем верхний слой переводят в реактор 46 и вводят в него из
мерника 47 ацетон и из мерника 48 8%-ный водный раствор едкого натра,
нагревают до 40° С и перемешивают 2,5—3 ч. Реакционную массу при темпе-
ратуре 20° С нейтрализуют уксусной кислотой из мерника 49. В делитель-
ной воронке 50 разделяют слои: нижний слой поступает в сборник 51,
откуда далее направляют на регенерацию. Верхний слой промывают в ко-
лонке 52 раствором хлористого натрия. Промытый слой (технический псев-
доионон) передают в сборник 53 и далее в вакуум-перегонный аппарат 54,
снабженный колонкой, дефлегматором и конденсатором. Перегонку ведут
при остаточном давлении 6—7 мм рт. ст., отбирают фракцию, кипящую
при 131—135° С в сборник 55. Выход 54—55%.
Псевдоионон — желтоватая маслянистая жидкость, хорошо растворима
в органических растворителях, плохо — в воде, температура кипения при
остаточном давлении 5 мм рт. ст.— 120° С; С13Н£0О, молекулярная мас-
са 192,29; Hd = 1,5300, df = 0,8954; Xmax = 291 нм, E\°jM = 1205; содержание
не ниже 95%.
СИНТЕЗ р-ИОНОНА
Р-Ионон [90, 91] получают процессом циклизации псевдоионона под
влиянием смеси концентрированной серной кислоты и ледяной уксусной
кислоты в среде толуола по химической схеме:
СН1 сн3 сн3
кгсн-сн=сн-со
г г
\^СН3
. Исебдоионин
192,29
H2SO4
сн3 сн3 сн3
сн=сн-со
\ЧЧСНз
- Ионон
192,29
28
Рис. 5. Технологическая схема производства синтетического витамина А.
В реактор (рис. 5) 1 из сборника 2 загружают псевдоионон и из сборника
3 толуол и перемешиванием получают толуольный раствор псевдоионона
(плотность 890—900 кг/м3), подаваемый насосом 4 в мерник 5. В реактор
из эмалированной стали 6 сливают концентрированную серную кислоту
из мерника 7, которую в реакторе 6 охлаждают до 0°, а затем медленно за-
гружают из мерника 8 ледяную уксусную так, чтобы температура не подни-
малась выше 15° С. Смесь кислот насосом 8 подают в мерник 9. В аппарат
для циклизации 10 из нержавеющей стали, снабженный мешалкой и рубаш-
кой, подают из мерника 9 смесь кислот, а из мерника 5 толуольный раствор
псевдоионона. Реакция протекает при температуре минус 7—10° С в тече-
ние 1 ч. Для нейтрализации реакционной массы применяют 18—20%-ный
раствор углекислого натрия. В реактор 11 загружают углекислый натрий,
из мерника 12 воду и при перемешивании насыщенный раствор насосом 13
подают в мерник 14. Из аппарата циклизации 10 нейтрализованная реакци-
онная масса поступает в делительную воронку 15, где промывается раство-
ром карбоната натрия и далее поступает в сборник 16 и в перегонный аппа-
рат 17. В нем отгоня ют толуол в сборники 18 и 19 при остаточном давлении
20 мм рт. ст. Остаток перегоняют при остаточном давлении 1 мм рт. ст.
в перегонном аппарате 20 и собирают в приемнике. Выход 75%.
|3-Ионон — желтоватая маслянистая жидкость, температура кипения
118—120° С при остаточном давлении 5 мм рт. ст. и 132° С при остаточном
давлении 12 мм рт. ст., С13Н2оО, молекулярная масса 192,29; Пд =1,5210;
хорошо растворим в органических растворителях, плохо в воде; Хтах=
= 296 нм, £^=557.
СИНТЕЗ АЛЬДЕГИДА См [4(2', 6', 6'-ТРИМЕТИЛЦИКЛОГЕКСЕН-1'-ИЛ)-2-
МЕТИЛБУТЕН-З-АЛЬ-1]
Синтез альдегида С14 осуществляют по реакции Дарзана путем конден-
сации |3-ионона с метиловым или этиловым эфиром монохлоруксусной кис-
лоты в присутствии метилата натрия1. Реакции протекают по следующей
схеме:
J3 - Ионон
192,29
ОС2Н5 СН, СН3 сн3
С1СН2-С=О г^ху1— сн=сн-с-сн-соос2н3
122^6 L L V
CH3ONa \ СН3
Этиловый з<рир /лоруксус-
ной кислоты и метилат Глицидный Эфир
натрия 278, 73
54.03
+ NaCl + СН3ОН
з СН3
CH=CH«-C-CH-COOC2H
v
сн3
+ NaOH
з СН3
CH=CH—С—СН—COONa + Н2О
V
СН3
CH, CHJ сн3
CH=CH—C—CH—COONa
11
Натриевая соль глицидной кислоты
272,2
Альдегид -J5 -С
206, 32
+ NaHCO3
бикарбонат натрия
83,9
S
Реакция конденсации. В реактор 21, снабженный охлаждающей рубаш-
кой и мешалкой, загружают |3-ионон из сборника 22 и в течение 2—3 ч при-
1 Применение в качестве конденсирующего агента метилата натрия связано с
технологическими затруднениями. В дальнейшем он был заменен изобутилатом калия
(Миропольская М., Егорова В., Валашек И. и Самохвалов Г. Авторское свидетельство.
№ 202 102, 1963; Бюллетень изобретений и открытий при Совете Министров СССР,
1967, № 14).
30
ливают из мерника 23 этиловый эфир хлоруксусной кислоты, а из сборника
24 сухой метилат натрия. Температуру при этом поддерживают минус
5—7° С. В результате реакции конденсации получается глицидный эфир,
который из раствора не выделяют.
Омыление. Глицидный эфир омыляют раствором едкого натра в водном
метаноле, который добавляют из смесителя 25 в тот же реактор в течение
1,5—2 ч при температуре 18—20° С. В результате омыления получают
натриевую соль глицидного эфира.
Декарбоксилирование. В реактор 21 добавляют воду и дихлорэтан, пере-
мешивают, а затем направляют реакционную массу в делительную ворон-
ку 26. Нижний дихлорэтановый слой отделяют в воронке и в смесителе 27
промывают водным раствором поваренной соли, приготовленном в смеси-
теле 28. Нижний слой спускают в смеситель 29, затем добавляют в этот
смеситель сульфат натрия и перемешивают. Сухой экстракт переводят в
вакуум-перегонный аппарат 30, отгоняют дихлорэтан, а затем под глубо-
ким вакуумом (0,1 мм рт. ст. при температуре около 100° С) отгоняют аль-
дегид С14. При необходимости альдегид подвергают ректификации при оста-
точном давлении 0,3—0,5 мм рт. ст.
Альдегид С14 — светло-желтая маслянистая жидкость с температурой
кипения 103—106° С при остаточном давлении 0,2 мм рт. ст., хорошо рас-
творим в органических растворителях, плохо—в воде. При хранении
неустойчив. Формула С14Н22О, молекулярная масса 206,14, пд = 1,5111.
СИНТЕЗ ПЕРВИЧНОГО АЦЕТИЛЕНОВОГО КАРБИНОЛА (3-МЕТИЛ-ПЕНТА-
ЕН-2-ИН-4-ОЛ-1)
Получение первичного ацетиленового карбинола состоит из следующих
стадий.
1. Получение ацетиленида • кальция
Са + 2СН=СН-------------> (НС = С)2Са + Н2.
NH3 (жидкий)
40,08* Ацетилен Ацетиленид кальция
. 2-26,04 90,10
2. Конденсация метилвинилкетона с ацетиленидом кальция
ОН
2NH4C1 |
(HCsC)2Ca + 2 /О=С—СН=СН2\----------> 2НС-С—С—СН=СН2 + СаС12 + 2NH3
I nh3 |
\ СН3 ) СН3
Ацетиленид Метилвинилкетон 3-Метил-3-окси-пента-ен-1-ин-4
кальция 2-70,09 2-96,12
3. Аллильная перегруппировка и получение первичного ацетиленового
карбинола
ОН
I
нс=с-с—сн=сн2-----------> нс = с—с=сн—СН2ОН
| H2SO4 |
сн3 сн3
3-метил-3-окси-пента-ен-1-ин-4 3-метил-пента-ен-2-ин-4-ол-1
96,12 96,12
Получение ацетиленида кальция. Из баллона 31 в реактор 32 загружают
жидкий аммиак, охлаждаемый до минус 40° С, туда же порциями добавля-
ют кальций в виде стружки и в течение 30 мин растворяют его.
В реактор 33 при охлаждении до минус 40° С загружают другую часть
аммиака. Затем пускают из баллона 34 в реактор 32 ацетилен, аммиачный
раствор кальция передают под давлением в реактор 33 в течение 0,5—1 ч,
получают ацетиленид кальция (исчезновение синего окрашивания).
31
Конденсация ацетиленида кальция с метилвинилкетоном. В реактор 33
из смесителя 36 загружают метилвинилкетон. Реакционную массу переме-
шивают 1 ч в токе ацетилена. Затем прекращают подачу ацетилена и вводят
в аппарат порошкообразный хлористый аммоний, прекращают охлаждение,
слегка подогревают реакционную массу водой через змеевик, расположен-
ный внутри реактора 33, для испарения аммиака. Последний сгущают в
холодильнике, и жидкий аммиак спускают в охлаждаемый сборник 37.
Остаток отгоняют из реактора 33 водяным паром. Дистиллят поступает в
сборник 38, а оттуда в экстрактор 39. Туда же добавляют хлористый натрий
и экстрагируют ацетиленовый карбинол петролейным эфиром. Экстракт
собирают в приемнике 40. Процесс экстракции третичного карбинола может
быть заменен повторной перегонкой при температуре 60—65° С (остаточное
давление 60 мм рт. ст.).
Третичный ацетиленовый карбинол — бесцветная жидкость с темпера-
турой кипения 72—76°С(при остаточном давлении 100 мм рт. ст.), 60—65°С
(при остаточном давлении 60 мм рт. cm.), n7!j--= 1,4460; формула С0Н8О,
молекулярная масса 96,12. Хорошо растворим в воде и в органических рас-
творителях. При хранении темнеет. Хранить следует в атмосфере инертно-
го газа.
Изомеризация третичного ацетиленового карбинола. В реактор 41 за-
гружают раствор третичного карбинола в петролейном эфире, добавляют
из мерника 42 разбавленной (20%-ной) серной кислоты и перемешивают
реакционную массу в присутствии азота при температуре 60—65° С в те-
чение 1 ч.
Реакционную массу спускают в делительную воронку 43, отделяют
верхний эфирный слой, который направляют в смеситель 44 для промывки
5%-ным раствором бикарбоната натрия. Нижний воднощелочной слой спус-
кают, а эфирный экстракт подсушивают прокаленным сернокислым натри-
ем. Сухой экстракт поступает в сборник 45, а оттуда в вакуум-перегонный
аппарат 46, где вначале отгоняют эфир, который возвращают для исполь-
зования в мерник 35, а остаток в вакуум-перегонном аппарате дистиллиру-
ют (при температуре 70—75° С и остаточном давлении при 15 мм рт. ст.).
Первичный ацетиленовый карбинол поступает в сборник 47.
Первичный ацетиленовый карбинол представляет собой бесцветную
жидкость с температурой кипения 73—75° С (при остаточном давлении
20 мм рт. ст.), по = 1Д834, химическая формула С6Н8О, молекулярная
масса 96,12. Хорошо растворим в органических растворителях, слабо —
в воде. Следует хранить в герметической таре, заполненной азотом.
СИНТЕЗ АЦЕТИЛЕНОВОГО ГЛИКОЛЯ С20 [9(2', 6', б'-ТРИМЕТИЛЦИКЛО-
ГЕКСЕН-1'-ИЛ)-3,7-ДИМЕТИЛ-НОН-2,7-ДИ-ЕН-4-ИН-1,6-ДИОЛ]
Синтез ацетиленового гликоля С20 состоит из следующих стадий:
1) магнийбромэтил
Вг
Mg + СН3СН2Вг —> MgZ
С2Н5
Магний Бромистый Магнийбромэтил
24,32 этил 133,30
108,99
2) магнийорганический комплекс первичного ацетиленового карбинола
Rr СН3 СН3
/Вг I I
2Mg + СН = С—С=СН=СН2ОН —с MgBrCsC—С=СН—CH2OMgBr + 2С2Н6.
С2Н5
Магнийбромэтил 3-метил-пеита-ен-2-ии-4-ол-1 Магнийорганический комплекс Этан
2 • 133,30 96,12 302,59 2 • 30,07
32
Конденсация альдегида С14 и магнийорганического комплекса «цепочки»
СН3 СН3 СН3 0
сн2-сн=с-с^
н
СН3
Альдегид
206,32
сн3
+ MgBr C=C-C=CH-CH2OMg-Br
Магнийорганический комплекс
302, 59
СНо СН3 СНз СНз
Г|Г- CH2-CH=C-CH-C=C-C=CH-CH2OMgB
xjk СН3 OMgBr
Магнийорганический комплекс ацетиленового гликоля С^о
508, 92
СНзСИз СН3 СН3
CH2-CH=C-CH-C=C-C=CH-CH20MgBr+2H2O + 2NH4Ct
СН3 0MgBr
, Хлористый
Магнийорганический комплекс гликоля Оро
2'18,02
аммоний
2'53,50
3 сн3 сн3
сн2—сн=с—сн—с=с—с=сн—СН2ОН
9 8 7 6| 5432 1
+ 2Mg-BrCl + 2NH4OH
Ацетиленовый гликоль Срр ..
302,46
Гидрат окиси
Маенийхлордромид аммония
2'139,70 2'53,50
Получение магнийбромэтила. В реактор 48, снабженный мешалкой,
обратным холодильником, змеевиком для подогрева и рубашкой для охлаж-
дения загружают металлический магний и абсолютный эфир. В смесителе
49 приготовляют раствор бромистого этила в эфире. Этот раствор медленно
приливают в реактор 48 и подогревают массу до кипения и полного раство-
рения магния и охлаждают до 15—20° С. Выход реактива Гриньяра 90%
(на бромистый этил).
Получение магнийорганического комплекса «цепочки». При переме-
шивании в присутствии азота медленно приливают из смесителя 50 в реак-
тор 48 раствор первичного ацетиленового карбинола в эфире. Нагревают
до кипения и получения отрицательной реакции на присутствие свободного
этилмагнийбромида (с кетоном Михлера).
Получение магнийорганического комплекса ацетиленового гликоля С20.
Массу в реакторе 48 охлаждают до 0°, из смесителя 51 приливают раствор
альдегида С14 в абсолютном эфире, подогревают 20—30 мин до кипения
и отрицательной реакции на присутствие альдегида С14.
Разложение комплекса. Из реактора 48 реакционную массу спускают
в реактор 52, в котором содержится охлажденная до 0° смесь воды и хлорис-
того аммония, и 10%-ной серной кислотой доводят до pH 8,0—8,5. Эфир-
ный слой отделяют в делительной воронке 53 и направляют в сборник 54
и далее в перегонный аппарат 55. Отгоняют эфир, а остаток растворяют в
петролейном эфире (50° С). Раствор промывают водным метиловым спир-
том (1:2) в делительной воронке 56. Эфирный слой отделяют и подсушивают
сернокислым натрием в смесителе 57. В вакуум-перегонном аппарате 58
при температуре 30—40° С отгоняют часть петролейного эфира до получе-
ния 20% сухого вещества. Сгущенный раствор спускают в кристаллизатор
59 и кристаллизуют при температуре минус 10° С. Кристаллы технического
гликоля отфильтровывают на друк-фильтре 60. Кристаллы перекристалли-
зовывают из петролейного эфира в реакторе 61, кристаллизаторе 62, друк-
фильтре 63. Маточный раствор 1 сгущают. Затем из него выкристаллизовы-
вают дополнительное количество гликоля, направляемого на перекристал-
2-522 33
лизацию совместно с техническим гликолем. Маточный раствор является
отходом производства.
Ацетиленовый гликоль С20 представляет собой бесцветные кристаллы
с температурой плавления 54—56° С; формула СвН30О2, молекулярная мас-
са 302,46. Хорошо растворим в органических растворителях, плохо — в
воде. Хранить следует в герметическом сборнике в атмосфере азота.
СИНТЕЗ ГИДРИРОВАННОГО АЦЕТИЛЕНОВОГО ГЛИКОЛЯ С20 [9(2', 6', 6'-
ТРИМЕТИЛЦИКЛОГЕКСЕН-1'-ИЛ)-3,7-ДИМЕТИЛ-НОН-2,4,7-ТРИЕН-1,6-ДИОЛ]
Получение гидрированного ацетиленового гликоля протекает по следую-
щей реакции:
СНз СНз СНз СН3 СНз СН3 снз СН3
Ply—сн2—сн=с—сн~с=с—с—сн—сн2он + н2 _ <Ч-сн2-сн=с-сн-сн=сн-с=сн-сн2он
ОН Pd/CaCO3 он
СН3 . v СНз
Ацетиленовый гликоль Сго Гидрированный ацетиленовый гликоль С го
302, 44 304, 46
В горизонтальный аппарат (типа автоклава) 64, снабженный мешалкой,
загружают кристаллы ацетиленового гликоля С2о, из мерника 65 добавляют
циклогексан и при перемешивании растворяют кристаллы. Затем загружа-
ют катализатор, представляющий собой палладированный карбонат каль-
ция, отравленный чистым хинолином. Добавляют 2% катализатора
к массе кристаллов гликоля и 3% хинолина из мерника 66. Воздух из ап-
парата вытесняют азотом. Гидрирование ведут под атмосферным давлением
при температуре 25—30° С до поглощения 1 моля водорода на 1 моль гли-
коля С20. Затем спускают реакционную массу в друк-фильтр 67, отфильтро-
вывают катализатор, а фильтрат направляют в вакуум-перегонный аппа-
рат 68, где отгоняют циклогексан при температуре 20—30° С. Остаток рас-
творяют в петролейном эфире при температуре 50° С, спускают в кристалли-
затор 69, где в течение 8 ч кристаллизуют при минус 15—20° С. Затем крис-
таллы отфильтровывают от маточника в друк-фильтре 70 и высушивают в
вакуум-сушилке 71. Маточный раствор 1 сгущают и выкристаллизовывают
гликоль С20, который поступает на первую кристаллизацию.
Гидрированный гликоль С2о представляет собой бесцветные кристаллы
с температурой плавления 70—72° С; формула С20Н32О2, молекулярная
масса 304,46. Хорошо растворим в органических растворителях, плохо —
в воде. Продукт нестойкий, следует хранить в герметизированном сборнике
в среде азота.
Приготовление катализатора. Палладированный карбонат кальция, от-
равленный хинолином, получают следующим образом. Путем растворе-
ния мела в соляной кислоте и действия на полученный раствор хлористого
кальция углекислым аммонием получают свежеосажденный карбонат каль-
ция. Последний отфильтровывают, смешивают с водой и обрабатывают го-
рячим раствором хлористого палладия (86° С) в соляной кислоте. Затем
горячую смесь обрабатывают водородом до прекращения поглощения по-
следнего. Катализатор отфильтровывают, промывают водой, а затем разме-
шивают, добавляют хинолин и нагревают 40 мин на кипящей водяной
бане. Катализатор отфильтровывают, промывают дистиллированной во-
дой и высушивают под вакуумом.
Катализатор представляет собой темно-серый порошок, содержащий
5% палладия.
34
СИНТЕЗ ВИТАМИНА А-АЦЕТАТА И ПАЛЬМИТАТА
Получение витамина А-ацетата протекает по следующим стадиям.
Частично ацетилированный гликоль С2о
сн, сн3 сн, сн,
сн,-сн=с-сн-сн=сн-с=сн-сн2он
L Jk он
с н
3 Гидрированный гликоль Сро
304, 46
+ (сн3со)2о
Уксусный ангидрид
102, 09
Аллильная
перегруппировка
C5H5N
3 СНз СНз
сн2-сн-с=сн-сн=сн^с=сн-сн2ососн3
он
Ьцетилиройллный ллиполъ Брл^9-(21,6',В'-1лриые1лилципло^
гексен- Г-ил 1-3,7 - диметил-1 -ацетокси-нон-2,-4,6-триен
346,49]
Витамин А-ацетат (отщепление гидроксильной группы)
СНз,СН3 СН3 сн3
<S1— СН,-СН-С=СН-СН=СН-С=СН-СН2ОСОСН3
I + НВг ------*"
<^-сн3 он
Частично ацетилированный гликоль Орд
346,49 80,92
СН3 СН3 СН, СН3
><f 1“ ।
J СН2-СН-С=СН-СН=СН-С=СН-СН2ОСОСН3 + NaHCo3____________>-
ЪП3 409,40 84,02
СН3СН3 сн3 сн3
СН=СН-С=СН-СН=СН-С=СН-СН2ОСОСНЭ ±,.тгг
U I + NaBr +H2O + CC
^^'СНз
Витамин 4 -ацетат
328,48
Получение ацетилированного гликоля С20. В реактор 72, снабженный
охлаждающей рубашкой и мешалкой, загружают гидрированный гликоль
С20, дихлорэтан и пиридин (из мерников 73 к 74). В реакционную массу при
перемешивании добавляют из смесителя 75 раствор уксусного ангидрида
в дихлорэтане и перемешивают 2 ч при температуре 25—30° С. Затем добав-
ляют воду и направляют реакционную массу в промывные колонки 76, 77
для промывки 5%-ным раствором серной кислоты и водой.
Отщепление гидроксильной группы и получение витамина А-ацетата.
Дихлорэтановый раствор пропускают через колонку 78 с сульфатом натрия
для подсушки. Далее его направляют в реактор 79, снабженный рубашкой
и мешалкой, куда из мерника 80 приливают 48%-ный раствор бромистово-
дородной кислоты в уксусной кислоте при 0°, приливают воду, перемеши-
вают 10 мин и через стеклянный фонарь на спускном штуцере отделяют ор-
ганический слой. Последний спускают в реактор 81, куда из смесителя 82
направляют водный раствор бикарбоната натрия, ц-токоферол и перемеши-
вают 3 ч. Органический слой отделяют через стеклянный фонарь реактора
81 и направляют в смеситель 83, куда добавляют сернокислый натрий для
подсушки.
Раствор сливают в сборник 84, а затем в вакуум-перегонном аппарате 85
отгоняют под вакууме м дихлорэтан при температуре 30° С. В остатке по-
лучают технический продукт — витамин А-ацетат в виде оранжевого
масла.
2*
35
Кристаллизация. Масло растворяют в этиловом спирте, подаваемом из
мерника 86 в вакуум-перегонный аппарат 85, спускают в кристаллизатор 87
и кристаллизуют 12 ч при температуре минус 5° С. Выпавшие кристаллы
отфуговывают в центрифуге 88. Маточник направляют в сборник 89, сгу-
щают в вакуум-аппарате 90, кристаллизуют в кристаллизаторе 91 и фугуют
в центрифуге 92. Кристаллы ацетата II направляют на первую кристалли-
зацию. Переработка маточного раствора на ацетат II требует дополнитель-
ного изучения.
Витамин А-ацетат представляет собой светло-желтые кристаллы с тем-
пературой плавления 55—57° С; формула С22Н32О2, молекулярная масса
328,48. Хорошо растворим в органических растворителях, растительном
масле, плохо — в воде.
Витамин А-ацетат неустойчив при хранении, поэтому кристаллы расфа-
совывают в ампулы, запаянные в присутствии азота. При выпуске в виде
растворов в рафинированном растительном масле следует добавлять стаби-
лизаторы (токоферол, додецилгаллат, гидрохинон, бутилоксианизол, про-
пилгаллат 0,05%). Витамины А хорошо сохраняются в растворе пищевого
нерафинированного соевого масла при наполнении флакона до пробки (с
учетом коэффициента расширения масла 0,0007).
Синтез пальмитата витамина А. Эфиры ретинола обладают большей
стойкостью, чем свободный витамин А-спирт.
В этом отношении витамин А-пальмитат является более стойким, нежели
витамин А-ацетат. Пальмитиновый эфир витамина А может быть получен
путем переэтерификации ацетата витамина А метиловым эфиром пальмити-
новой кислоты [90].
СНзСНз сн3 сн3
Г[|— сн=сн-с=сн-сн=сн-с=сн-сн2ососн3
<'-А'СН3
витамин А -ацетат
328, 4 <9
+ C1SH31COOCH3
. Метиловый эаоир
пальмитиновой кислоты
270, 44
CH3ONa
СН3 СН3
CH=CH-C=CH-CH = CH-C = CH-CH2OCOC1SH31
витамин А - пальмитат
52 к, <94
+ СН3СООСН3
Метилацетат
7й,08
ТЕХНОЛОГИЧЕСКИЕ ПОКАЗАТЕЛИ
РАСХОД ХИМИКАЛИЕВ, КГ НА 1 КГ
100%-НОГО ПОЛУПРОДУКТА
Синтез псевдоионона
Диметилэтинилкарбинол
Ацетон....................... 0,9
Ацетилен . . ................. 1,1
Кали едкое (техническое) .... 2,0
Дибутилформаль ............... 2,0
Эфир..................... 1,0
Углекислота.............. 0,7
Сульфат натрия ............... 0,5
Выход (на ацетон), %.....75,0
Димергилвинилкарбинол
Диметилэтинилкарбинол .... 1,0
Водород, .и3................. 0,2
Палладий хлористый .......... 0,005
Выход (на диметилэтинилкарби-
нол), % ...................95,0
36
Метилгептенон
Диметилвинилкарбинол ........... 0,9
Ацетоуксусный эфир.............. 1,7
Вазелиновое масло............... 0.6
Выход (на диметилвинилкарби-
нол), % ...................... 60,0
Дегидролиналоол
Метилгелтенон .................. 1,0
Кали едкое ..................... 1,7
Толуол.......................... 1,0
Выход (на метилгептенон), % . . 78,0
Псевдоионон
Дигидролиналоол ................ 1,2
Уксусный ангидрид .............. 1,1
Ортофосфорная кислота .... 0,01
Уксусная кислота............... 3,5
Карбонат серебра .............. 0,004
Ацетон......................... 15,0
Натр едкий..................... 0,6
Натрий хлористый................ 7,6
Выход (на дегидролиналоол), % 55,0
Синтез витамина А из ^-ионона
$-Ионон
Псевдоионон ...................... 1,3
Толуол............................ 0,4
Серная кислота ................... 9,0
Уксусная кислота.................. 2,0
Углекислый натрий................. 4,0
Альдегид Си
Р-Ионон . . . '................ 1,51
Этиловый эфир хлоруксусной ки-
слоты .......................... 1,25
Метилат натрия....................0,60
Едкий натр........................0,70
Метиловый спирт с учетом регене-
рации ...........................0,5
Дихлорэтан с учетом регенерации . 1,0
Натрий сульфат.................0,3
Выход альдегида Сц с температу-
рой кипения 101 — 102С (при ос-
таточном давлении 0,15 мм рт.
ст.) (на р-ионон), %.............60,0
Первичный ацетиленовый карбинол
Метилвинилкетон................. 1,81
Кальций металлический............1,03
Ацетилен ’.......................2,70
Аммиак жидкий с учетом регенера-
ции .............................3,36
Эфир этиловый абсолютный с уче-
том регенерации................. 1,20
Эфир петролейный 80—100°С с уче-
том регенерации................ 1,2
Натрий хлористый ................0,66
Серная кислота (плотность
1840 кг/м3)....................2,20
Бикарбонат натрия ...............0,13
Натрий сульфат...................0,40
Выход третичного ацетиленового
карбинола, % от теоретического
(на метилвинилкетон).............52,0
Выход первичного карбинола (на
метилвинилкетон), %..............40,0
Ацетиленовый гликоль С2о
Альдегид Ci4.................. 1,0
Первичный ацетиленовый карбинол 0,50
Магний металлический ...... 0,25
Бромистый этил.................. 1,10
Эфир абсолютный с учетом регене-
рации ...........................3,40
Аммоний хлористый............... 1,20
Метиловый спирт................. 1,00
Натрий сульфат ..................0,50
Петролейный эфир с учетом реге-
нерации .........................3,00
Выход ацетиленового гликоля С20
(на альдегид С14), %.............70,0
Гидрированный ацетиленовый гликоль С20
Ацетиленовый гликоль С20 .... 1,4
Катализатор (хлористый палладий) 0,05
Циклогексан с учетом регенерации 0,5
Хинолин .......................0,05
Петролейный эфир (60°) с учетом
регенерации.................... 1,0
Водород, .it3....................0,04
Выход (на ацетиленовый гли-
коль), % .................70,0
Витамин А-ацетат
Гидрированный ацетиленовый гли-
коль С20....................... 1,7
Уксусный ангидрид...........0,5
Пиридин.................... 1,5
Дихлорэтан с учетом регенерации . 2,0
Бромистоводородная кислота .... 1,5
Бикарбонат натрия.......... 1,0
Этиловый спирт с учетом регенера-
ции ........................... 1,5
Натрий сульфат....................0,5
Выход (на гидрированный ацетилено-
вый гликоль С20), %..........60,0
РАСХОД ПОЛУПРОДУКТОВ, КГ
на синтез 1 кг псевдоионона
Диметилэтинилкарбинол......... 1,08
Диметилвинилкарбинол.......... 1,08
Метилгептенон ................ 1,2
Дегидролиналоол............... 1,2
на синтез 1 кг витамина
A-а ц е т а т а
Псевдоионона...................4,67
р-Ионон........................3,59
Метилвинилкетон................2,15
Первичный ацетиленовый карбинол 1,19
Альдегид С14...................2,38
Ацетиленовый гликоль С20 .... 2,38
Гидрированный ацетиленовый гли-
коль С20 ................... 1,7
РАСХОД ХИМИКАЛИЕВ, КГ
на синтез 1 кг псевдоионона
Апетилен...................... 1,19
Ацетон........................ 15,97
Ацетоуксусный эфир............ 2,04
Вазелиновое масло........... 0,72.
Водород, м3 .................. 0,22
Дибутилформаль ............... 2,16
Кали едкое.................... 2,04
Карбонат серебра ............ 0,004
Натр едкий . ................. 0,60
Натрий хлористый.............. 7,60
Натрий сульфат................ 0,54
Ортофосфорная кислота .... 0,01
Палладий хлористый ........... 0,05
Толуол........................ 1,20
Углекислота .................. 0,76
Уксусный ангидрид............. 1,10
Уксусная кислота . .......... 3,50
Эфир этиловый................. 1,08
на синтез 1 кг витамина.
А-ацетата из псевдоионона
Ацётилен ...................... 3,21
Аммиак жидкий с учетом регене-
рации ..................... ... 4,00
Аммоний хлористый .............. 2,86
Бикарбонат натрия .............. 1,15
Бромистоводородная кислота . . 1,50
Бромистый этил..................2,62
Водород, м3 .....................0,07
Дихлорэтан с учетом регенерации 4,38
Кальций металлический........... 1,23
Кислота серная (плотность
37
1840 кг/л3)..................34,93
Кислота уксусная ............... 7,18
Магний металлический.............0,60
Метилат натрия ................. 1,43
Метиловый спирт..................3,57
Метилвинилкетон..................2,15
Натр едкий...................... 1,67
Натрий хлористый................2,62
Натрийсульфат ................. 2,80
Натрий углекислый..............18,68
Палладий хлористый...............0,09
Пиридин ........................ 1,50
Псевдоионон ....................4,67
Спирт этиловый с учетом регене-
рации ......................... 1,44
Толуол......................... 1,50
Уксусный ангидрид...............0,50
Этиловый эфир хлоруксусной ки-
слоты ..........................2,98
Эфир петролейный (80—ЮО'С) с
учетом регенерации ........... 10,27
Эфир этиловый абсолютный с уче-
том регенерации.................9,52
Циклогексан.......................0,85
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. Букин В. Н. Витамины. М., Пищепромиздат, 1940. с. 16—108.
2. Ефремов В. В. Применение витаминов в медицине. М., Медгиз, 1946, с. 80.
3. К У Д Р я ш о в Б. А. Витамины, их физиологическое и биохимическое значение.
Московское общество испытателей природы, 1953, с. 41—43, 68—72.
4. Б р е м е н е р С. М. Витамины, М., Медгиз, 1959, с. 7.
5. Ш и л о в П. И., Яковлев Т.Н. Основы клинической витаминологии.М.,
«Медицина», 1946, с. ПО.
6. Ш н а й д м а н Л. О. Значение витаминов в'питании человека. — В сб. «Труды
Всесоюзного совещания по витаминам», Уфа, Башиздат, 1963, с. 11—25.
7. S е b г е 1 1 W., Harris R. The Vitamins, New York Academic Press, 1967, 1—303.
8. Rosenberg H. Cemistry a. Physiology of the Vitamins, Interscience Publishers,
N. Y., 1942, 57—96.
9 Березовский В. M. Химия витаминов. М., Пищепромиздат, 1959, с. 176—
252.
10. Беэр А. А., Рубцов И. А. Синтез витаминов, М., Пищепромиздат, 1956,
с. 36.
11. Karrer Р., Morf R., Schopp К-, Helv. Chim. Acta, 1931, 14, 1036, 1431;
1933, 16, 557, 625.
12. Heilbron I., Heslop R., Morton R., Webster E., ReaJ.,
Drummond J., Biochem, J., 1932, 26, 1178.
13. R о b e s о n C„ Baxter J., J. Am. Chem. Soc., 1947, 69, 136.
13a. Шнайдман Л. О., Павлова A. M. Устойчивость концентрата витамина
А из рыбьего жира. —«Труды ВНИВИ», М., Пищепромиздат, 1959, № 6, с. 116—118.
14. Wald G., J. Gen. Physiol., 1935, 19, 351.
15. М’о г t о n R. Nature, 1944, 153, 69.
16. M о r t о n R., Goodwin T, Nature, 1944, 153, 405.
17. В a 1 1 S., Goodwin T., Morton R. Biochem. J. 1948, 42, 516.
18. H u n t e r R., Hawkins E. J. Chem. Soc., 1944, 411.
19. Ледерер Э. А. иРозанова В. А. Исследования по витамину А в рыбьих
жирах. — «Биохимия», 1937, т. 2, 293—303 с ил.
20. G i 1 1 a m A., Heilbron I., J о n е s Е., Lederer Е., Rosano-
v a W. Biochem J., 1938, 32, 405.
21. Lederer Е., Rosanova W., С i 1 1 a m А., Н е i 1b г о n I. Nature,
1937, 140, 233.
22. S с h n a t z E., Science, 1948, 108, 417.
23. G г а у E., Cawley I. J. Biol. Chem., 1939, 131, 317; 1940, 134, 397.
24. Farrar K., Hamlet I. и др., J. Chem. Soc., 1952, 2657.
25. M о r t о n R., Salah M., Stubbs A. Nature, 1947, 159, 744.
26. Salah M., Morton R. Biochem. J., 1948, 43, 56.
27. Шнайдман Л. О., Ушакова M. Т., Ефимов А. 3., К у щ и н с-
к и й И. Н. А-витаминная активность ликопина плодов шиповника. — «Меди-
цинская промышленность СССР», 1966, № 1, с. 15—17. То же. ВИамш Ai каротини,
Матер1али конференцп Чсрн1вцц 1969, 108. То же. Биологическая активность
ликопина. — В сб. «Труды по витаминам из природного сырья», Уфа, Башиздат,
1971, с. 209—214.
27а Euler В., Euler Н., Karrer Р. Helv. Chim. Acta, 1929, 12, 281.
28. С u г 1 A. Food Res., 1960, 25, 190, 670.
29. Кущи иска я И. Н„ Шнайдман Л. О. Идентификация каротиноидов,
содержащихся в сухих плодах шиповника. — «Медицинская промышленность
СССР», 1964, № 4, с. 38—40.
30. Ш н а й д м а н Л. О. Исследования в области химии и технологии производства
витаминов. Докт. диссертация, М., 1967. То же. Бельштейн, т. 30, 1938, с. 81.
31. Сам о хвалов Г. И. Промышленный синтез витамина А (Обзор литературы).
— В сб.: «Пищевая промышленность за рубежом. Витамины», М., Пищепромиздат,
1957, № 3, с. 4—25.
31а. Самохвалов Г. И. Исследования по химии полиеновых соединений. Авто-
реф. докт. дисс., М„ 1960.
38
32. О г о s h n i к W., Encyclopedia Chem. Technology, USA, 1955, 14, 791.
33. С а в и н о в Б. Г., Третьякова Г. С, О синтезе соединений, обладающих
А-витаминной активностью. —«Успехи химии», 1951, т. 20, с. 336—359.
34. Embree N., Ann. Rev. Biochem., 1947, XVI, 323.
35. M i 1 a s N. Vitamins, a. Hormones, 1947, 5, 1.
36. Johnson A., Science Prog., 1948, 36, 496.
37. H e i 1 b г о n I. J. Chem. Soc., 1948, 386.
38. К a r r e r P. Osterr. Chem. Ztg., 1948, 49, 215.
39. Inhoffen H., В о h 1 m a n F. Fortschr. Chem. Forsch., 1949, 1, 175.
40. Hunter R., Research (London), 1950, 3, 453.
41. Isler O., Chimia (Швейцария), 1950, 4, 103.
42. В a x t e r J., Fortschr, chem. org. Naturstoffe, 1952, 9, 41.
43. Isler O., Huber W., К о e f 1 e r M., Ronco A. Experientia, 1946, 2, 31;
Helv. Chim. Acta. 1947, 30, 1911.
44. M i 1 a s N., Science, 1946, 103, 581.
45. D. Van Dorp., J. Arens, Rec. trav. chim., 1946, 65, 338; Nature, 1947, 160, 189.
46. К u h n R., Mor r is C., Ber., 1937, 70, 853.
47. Вульфсон H. С. К вопросу о синтезе витамина А. — В сб. «Труды конферен-
ции по витаминам», 1940, с. 132—134.
48. М i 1 a s N., Harrington Т., J. Am. Chem. Soc., 1947, 69, 2247; 1948; 70,
4275.
49. М i 1 a s N. Пат. США No 2424994, 5/VIII, 1947.
50. Н е i 1 b г о п I., Jones Е. O’Sullivan D., J. Chem. Soc., 1946, 886.
51. К а г г е г Р., J u с k е г Е., Schick Е. Helv. Chim. Acta., 1946, 29, 704.
52. Inhoffen H., В о hl ma n n F., Bohlmann.M., Ann., 1950, 568, 47.
53. H и i s m a n H. Rec. trav. Chim., 1950, 69, 851.
54. С а м о x в а л о в Г. И., Миропольская М. А., Вакулова Л. А.,
Преображенский Н. А. Синтетические реакции в области полиеновых
соединений с помощью металлоорганических производных алкоксивинилацетиле-
нов. — «Труды ВНИВИ», 1953, № 4, с. 10—12, То же. 1954, № 5, с. 5—10.
55. Самохвалов Г. И., Миропольская М. А., Вакулова Л. А.
и др. Анионтропные и прототропные перегруппировки при синтезе полиеновых сое-
динений. — ДАН СССР, 1954, т. 99, с. 273—276 с ил.
56. Н u i s m a n Н., Smit А., V г о m е г S., Fischer L., Rec. trav. chim.,
1952, 71, 899.
57. Словохотова H. А., Самохвалов Г. И., Куницкая Г. М. и
др. Применение инфракрасных спектров поглощения к исследованию промежуточ-
ных продуктов синтеза витамина А и каротина. — ЖОХ, 1954, т. 24, с. 2222—30.
58. Миропольская М. А. Исследования в области полиеновых соединений..
Канд, дисс., М., 1955.
59. О г о s h п i k W., К а г mas Р., Mebane A. J. Am. Chem. Soc., 1952, 74,
295; 1953, 75, 1050.
60. Attenburrow J., Cameron A., Chapman J., Evans R.,
Hems B., Jansen А. и др. J. Chem. Soc., 1952, 1094.
61. Cheeseman G. J. Chem. Soc., 1949, 2031.
62. M i 1 a s N., MacDonald N. и др. J. Am. Chem. Soc., 1948, 70, 1829.
63. C h a n 1 e у J., S о b о t к a H. J. Am. Chem. Soc., 1949, 71, 4140.
64. I s h i к a w a S., Matsuur a T. Sci Repts, Tokyo Bunrika Daigaka, 1937, ЗА,
173.
65. M i 1 a s N., Пат. США 2369156, 13/11, 1945; Science, 1946, 103, 581.
66. Heilbron I., Johnson A., Jones E., Spinks A. J. Chem. Soc.
1942, 727.
67. С у m e r m a n J., Heilbron I., J о n e s E., Lacey R., J. Chem. Soc.,
1946, 500.
68. Milas N., Lee S. и др. J. Am. Chem. Soc., 1948, 70, 1584.
69. Cheesma n G., Heilbron I., Jones E. и др., J. Chem. Soc., 1949, 1516.
70. Inhoffen H., Bohlmann F., Linhof G. Ann., 1950, 570, 73.
71. N a v e s J. K-> Barbier H., A r d i z i о P., Bull. Soc. Chim. France, 1950,
1188.
71a. Артемьев В. И., Богородская Л. П., Самохвалов Г. И. и
др. Способ получения цитраля. Авт. свидет., № 268404, Бюлл. изобрет., 1970, № 14,
с. 20.
72. L i n d 1 а г Н. Пат. США No 2451740, 7/VI 1947; Off. gaz. 1948, 615, No3, 717.
73. Milas N. Пат. США № 2577538, 4/XII, 1951; Англ. пат. 664815, 16/1, 1952;
Швейц, пат. № 288645, П/Ш, 1953.
74. Isler О., Ronco A., G u е х W. и др., Helv. Chim. Acta, 1949, 32, 489.
75. I s 1 е г О. Пат. США, No 2451739, 1948; Chem. Eng. News, 1951, 29 , 3962.
76. М i 1 a s N., Пат. США, No 2567572, 28/XI 1947; Off. gaz., 1951, 650, No2, 505. •
77. L i n d 1 a r H., Helv. Chim. Acta, 1952, 35, 446.
78. Б e p e з о в с к и й В. M. Современные задачи в области химии и технологии ви-
таминов и перспективы развития их производства. — «Медицинская промышлен-
ность СССР», 1960, № 5, с. 7—23.
39
79. Л э э т с К- В., Шумейко А. К., Роз ено ер А. А. и др. Новый синтез
цитраля из изопрена. — ЖОХ, 1957, т. 27, с. 1510—1512.
80. Л э э т с К- В.. Авт. свидет., № 105428, 1955; Бюлл. изобрет., 1957, № 3, с. 17.
81.Назаров И. Н., Ракчеева В. Н. и др. Производные ацетилена. — «Из-
вестия АН СССР, ОХН», 1946, с. 305—311.
82. Н а з а р о в И. Н., Яновская Л. А., Гусев Б. П. и др. Синтез метил-
гептенона и метилгептадиенона. —ДАН СССР, 1957, 114, с. 331—34.
83 Kimmel W. Пат. США № 2638484, 1954; С. А. 1954, 48, 2763; Kimmel W.,
Sax N., Пат. США № 2661368, 1955; С. А., 1955, 49, № 3, 1784.
84. Н а з а р о в И. Н., Ко т л я р е в с к и й. И. Л. , Рябченко В. Ф. Кон-
денсация альдегидов и кетонов с ацетиленом под давлением. — ЖОХ, 1953, 23, с.
1900—1904. То же. «Известия АН СССР, ОХН», 1956, с. 60.
85. Н а з а р о в И. Н., Гусев Б. П., Макин С. М. и др. Конденсация аце-
тилена с метилгептеноном и его аналогами. — «ДАН СССР», 1957, 114, с. 796—802.
86. Самохвалов Г. И., Миропольская М. А., Преображенс-
кий Н. А. Новый метод синтеза полиеновых кетонов с сопряженными двойными
связями. — «ДАН СССР», 1956, 107, с. 103—104. То же. Авт. свидет. № 103776,
1955; Бюлл. изобрет.; 1956, № 7, с. 10.
87. Н а з а р о в И. Н., Яновская Л. А., Гусев Б. П. и др. Синтез гера-
нилацетона, 3-метилгеранилацетона, псевдоионона и псевдоирона. — «ДАН СССР»,
1957, 114, с. 1029—1032.
88. Isler О., Helv. Chim. Acta., 1956, 39, 249; Chimia, 1958, 12, 326.
88a. Hoffman W., Pasedach H., Fischer R., Пат. ФРГ, Nol, 793,
445; Auszuge, 1971, 27/V, No22, 1393.
89. Преображенский H. А., Самохвалов Г. И., Миропольс-
кая М. А. Технологическая схема производства синтетического витамина А.
В кн.: Шнайдман Л. О. Производство витаминов, 1958, с. 393—402.
90. S t i е g W., Nilsen A. (Pfiser), Пат. США, 2971966, 30/VIII, 1957; Off. gaz.,
1961, 763, No2, 469.
91. Франц, пат. № 1523361, 25/Ш 1968; РЖХим, 1970, Н 526 П.
Глава 3. ПРОИЗВОДСТВО СИНТЕТИЧЕСКОГО ^-КАРОТИНА
Еще в 1919 г. Стинбок [1] обратил внимание на то, что растительный
пигмент каротин подобно витамину А стимулирует рост животных и излечи-
вает ксерофтальмию крыс. Эйлер, Каррер и другие подтвердили А-витамин-
ное действие каротина [2, 3, 4]. Наконец, Муур в своих исследованиях [5]
показал, что при введении в А-авитаминозную диету каротина в печени под-
опытных крыс откладывается витамин А. Таким образом была установлена
генетическая связь между витамином А и каротином, т. е. было показано,
что каротин является провитамином А. Витамин А встречается лишь в про-
дуктах животного происхождения и источником его является каротин,
поступающий в организм с растительной пищей. Каротин, подвергаясь
ферментативному гидролизу, расщепляется в печени и дает витамин А [6].
Существенное значение для всасывания каротина через стенки кишечника
имеет наличие в диете жира.
Ограниченные ресурсы витамина А и богатство флоры каротиноидными
пигментами способствовало широкому исследованию последних. Это сти-
мулировалось также установленным значением каротина в борьбе с авита-
минозами и гиповитаминозами как у людей, так и у животных. На ряде
примеров показано, что применение каротиновых препаратов в животно-
водстве и птицеводстве позволяет получить значительный экономический
эффект [7]. Кроме того, каротиноиды повышают товарное качество мяса,
в особенности птичьих тушек. Помимо носителя А-витаминных свойств,
|3-каротин является одним из наиболее ценных красителей жиров [8] и в
частности маргарина. Важное значение |3-каротина в народном хозяйстве
предопределяет перспективу его производства как из растительного сырья,
так и синтетическим методом.
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Каротин принадлежит к классу полиеновых углеводов, характеризуе-
мых наличием длинной алифатической цепи с системой конъюгированных
(чередующихся) двойных связей. В каротине полиеновая цепь состоит
40
из четырех изопреновых (2-метил-2-бутен) остатков, ограниченных с одно-
го или с обоих концов р-иононовым кольцом (триметилциклогексен).
Строение молекулы каротина было доказано при помощи реакции окис-
ления. Применяя для окисления каротина хромовый ангидрид, получаем
шесть молекул уксусной кислоты, что свидетельствует о содержании в мо-
лекуле шести метильных групп* 1 [9].
При окислении каротина перманганатом калия получают четыре моле-
кулы уксусной кислоты. Это послужило доказательством того, что две ме-
тильные группы отличаются своим положением от четырех других [10].
При окислении каротина озоном [11] была получена героновая кислота
в количестве в 2 раза большем, чем при окислении р-ионона, это указывает
на содержание в молекуле каротина двух колец р-ионона. При окислении
последнего получают героновую, диметилглутаровую, диметилянтарную
и диметилмалоновую кислоту, что видно из следующей схемы:
Р - Ионон
Героновая кислота Диметилелутаровая Диметилянтарная
кислота кислота
Диметилмалоновая
кислота
Те же кислоты образуются при окислении каротина озоном и перманга-
натом.
При каталитическом гидрировании p-каротин присоединяет 11 молекул
Нг, что указывает на наличие в молекуле каротина одиннадцати двойных
связей [12].
Таким образом было установлено, что молекула каротина содержит
два кольца р-ионона, четыре метильные группы в алифатической цепи и
одиннадцать двойных связей. Эти данные и ряд других позволили П. Карре-
ру в 1930 г. установить следующую структурную формулу р-каротина:
СН3.СН3 сн3 СН3 ' снз СНз СНд сн3
Г|Г- СН=СН-С=СН-СН=СН-С=СН-СН=СН-СН=С-СН=СН-СН=С-СН=СН—Т|<''1
СН3 каротин Н3С
Однако вскоре было доказано, что кристаллический каротин, получае-
мый из растений, содержит три изомера каротина, а, р, у, различающиеся
лишь структурой колец, находящихся на концах алифатической цепи.
У p-каротина оба кольца имеют структуру р-ионона, поэтому его называют
также Р, P-каротин. У а-каротина одно кольцо р-ионона, а другое а-ионона,
поэтому ц-каротин называют также а, p-каротин. У у-каротина, с одной
стороны алифатической цепи находится кольцо р-ионона, ас другой— не-
замкнутое кольцо, названное псевдоиононом. Строение ^-каротина установ-
лено: а) реакцией окисления, при которой получается, кроме уже извест-
ных продуктов, еще ацетон; б) реакцией гидрогенизации, при которой к
молекуле у-каротина присоединяется не 11, а 12 молекул водорода Н2.
СН3
1 При окислении хромовой кислотой системы =СН—С= и более
СН3
типа —Н2С—С—, получают количественно уксусную кислоту.
насыщенной,
41
Природные изомеры каротина. Структурная формула а-каротина при-
ведена ниже.
3
сн3
СН3
СНз
СН3
СН=СН-С=СН-СН=СН-С=СН-СН=СН-СН=С-СН=СН-СН=С-СН=СН
сн3
of - Каротин
СНз
а-Каротин (С40Н5в, молекулярная масса 536,85) кристаллизуется из
петролейного эфира [13] в виде призм фиолетового цвета с точкой плавле-
ния 187—-188° С. Он оптически активен [а1Ь8+385°С и имеет спектр по-
глощения в петролейном эфире при 478 и 447,5, в гексане 446 и 478 нж;
Е\см при 446 нл1=2700. На рис. 6 изображена кривая поглощения в ультра-
фиолетовом и видимом свете для раствора а-каротина в гексане.
15
500 Л,нм
300 WO
а
Рис. 6. Кривые поглощения изомеров каротина в гексане в видимой об-
ласти света:
а — а-каротин; б — {3-каротин; в — у-каротин
Витаминная активность а-каротина вдвое меньше (3-каротина. В морко-
ви а-каротина содержится до 15% от всего количества изомеров каротина.
|3-каротин (С4оН6в, молекулярная масса 536,85) из раствора в бензине
кристаллизуется в форме гексагональных призм оранжево-красного цвета
с фиолетовым отливом. Температура плавления кристаллов 181—182° С
[13, 14].
|3-Каротин оптически недеятелен. Спектр поглощения в сероуглероде
имеет три максимума при длине волны 520, 485 и 450 нм и в гексане при
длине волны 450, 480 нм\ Е^м при 450 нж=2400. На рис. 6 изображена
кривая поглощения в ультрафиолетовом и видимом свете для растворов
(3-каротина в гексане. (3-Каротин слабо растворим в бензине и петролейном
эфире (около 1,0 а в 1,5 л), лучше растворяется в хлороформе, бензоле и
маслах.
(3-Каротин почти нерастворим в холодном этиловом и метиловом алко-
голе, несколько лучше — в кипящем алкоголе. В моркови (3-каротина
содержится около 85% от общего количества изомеров каротина [15].
Структурную формулу ^-каротина см. на стр. 41.
Физиологическая витаминная активность у-каротина составляет 27%
активности (3-каротина
СН3 _СН3
сн=сн-с=сн-сн=сн-с=сн-сн=сн-сн=с-сн=сн-сн=с-сн=сн
СН3
СН3
СНз
СН3
СН3
у - Каротин
Н3С
42
7-Каротин (С40Н5в, молекулярная масса 536,85) кристаллизуется из рас-
творов в бензине в виде темно-красных призм с точкой плавления 178° С.
Спектр поглощения у-каротина в сероуглероде 533, 496 и 463 нм\ Ё\с°м при
463 нм = 2760; 7-каротин оптически инактивен. В моркови 7-каротина
содержится около 0,1% от общего содержания изомеров каротина.
а,- |3-, у-Каротины являются наиболее распространенными природными
изомерами каротина.
Хроматографический метод тонкого разделения пигментов по Цвету
позволил обнаружить новые природные изомеры каротина. Так, А. Винтер-
штейн [16] выделил из плодов гонокариума четвертый изомер каротина,
названный 3-каротин.
Г. Фрепс и А. Кеммерер [17] выделили из желтой кукурузы пятый
изомер к-каротин. В последние годы обнаружены еще два изомера каро-
тина, z-каротин, найденный в моркови, и s-каротин— в диатомовых водо-
рослях [15]. Однако 3, z-, и s-каротины встречаются в природе в очень
небольших количествах.
Изомеризация каротина. Природные изомеры каротина -а- 0- и 7-об-
ладают способностью изомеризоваться. Изомеризация заключается как в
перемещении двойной связи в циклах, так и в ^цс-транс-превращениях
в алифатической цепи молекулы и обусловливает изменение спектральной
характеристики и провитаминной активности вещества. Изомеризация про-
исходит в момент растворения пигмента, т. е. контакта его с растворителем,
зависит от времени и температуры [18] и стремится к равновесному значе-
нию. Установлено, что процесс изомеризации является обратимым процес-
сом [19]. г{цс-Изомеры получают обычно действием света, тепла, йода, кис-
лот и катализаторов [14].
В 1942 г. впервые было обнаружено, что неизомеризованные (3-, у-каро-
тины обладают большей интенсивностью поглощения в видимой части спект-
ра, чем полученные из них изомеры [20].
Л. Цехмейстер и А. Польгар [20 ] широко исследовали вопрос об изоме-
ризации а- и (3-каротина под влиянием нагревания растворов, при обра-
ботке йодом, соляной и йодистоводородной кислотами, а также при плавле-
нии кристаллического продукта.
При хроматографировании были выделены следующие изомеры а- и
(3-каротина, отличающиеся один от другого максимумами поглощения в пет-
ролейноэфирном растворе, перечисленные в порядке адсорбции изомеров
по высоте колонки (табл. 4).
Таблица 4
Изомеры (3-каротина Максимум пог- лощения, нм Изомеры а-каротнна Максимум пог- лощения, нм
Нео-^-каротин U 481,0; 450,0 Нео-а-каротин U 471,5:441,5
Heo-fi-каротин V 472,5; 441,5 Нео-а-каротин V 465,5:437,0
₽-каротин 486,0; 454,0 Нео-а-каротин W 470,5;441,0
Нео-З-каротин А 469,0; 437,5 Нео-а-каротин X 463,5;435,О
Нео-З-каротин В 475,5; 444,5 Нео-а-каротин Y 467,5;437,0
Нео-^-каротин С 465,5; 433,0 Нео-а-каротин 477,0;446,5
НеоА -каротин D 474,5; 441,5 Нео-а-каротин А 468,5;439,0
Нео-З-каротин Е 477,5; 445,0 Нео-а-каротин В 466,5;437,0
Без названия 468,5; 437,0 Нео-а-каротин С 472,5:442,5
» » 473,5; 443,0 Нео-а-каротин D 46О,О;432,О
476,5; 445,5 Нео а-каротин Е 461,5;433,5
471,0; 440,0
» » 473,0; 444,0
Рядом исследований установлена [21] взаимосвязь между спектральной
характеристикой и стереопревращениями в молекулах каротиновых пиг-
ментов. Отклонение от полной транс-формы вызывает понижение интен-
43
сивности поглощения в видимой части спектра и некоторый сдвиг макси-
мума в сторону более коротких волн.
Кроме того, установлено, что в результате изомеризации на кривой по-
глощения появляется новый максимум в ультрафиолетовой области (320—
380 нм). Новый максимум назван цис-пиком. Расстояние между гщс-пиком
и первым максимумом (при наибольшей длине волны) неизомеризованного
исходного вещества в растворе гексана оказалось величиной постоянной
(142+2 нм).
Все описанные выше а- и |3-изомеры каротина, по-видимому, являются
пространственными изомерами.
Известно, что наибольшая интенсивность окраски полиенов должна
соответствовать максимально выпрямленной плоскостной структуре цепи,
т. е. полной транс-форме.
Полная транс-форма для |3-каротина может быть представлена в сле-
дующем виде:
СН3 Н CH, Н СН3 Н Н Н Н Ни
। 6 i । * । । 6 । > 1 I । п:
снзн н
tfi - Каротин [полная транс -форма.)
Если расстояния между атомами в полиеновой цепи молекулы |3-каро-
тина обозначить окружностями, радиусы которых выдержаны в относитель-
ном масштабе, как показано для полной транс-формы молекулы |3-каротина,
то видно, что двойные связи 3, 5, 6, 7 и 9 являются стереохимически актив-
ными. Около этих связей возможны г{цс-повороты без возникновения про-
странственных затруднений [21]. У двойных связей-2, 4, 8, 10 такие цис-по-
вороты молекулы затруднены, так как при этом водород и метильная группа
сближались бы на значительно меньшее расстояние, чем это допускают
размеры атомов. В связи с этим до последнего времени считали, что цис-
поворот возможен только по типу II и затруднен по типу I [14] из-за про-
странственных затруднений
Это обстоятельство ограничивало ожидаемое число изомеров в молекуле
|3-каротина с 272 до 20 и в молекуле а-каротина с 512 до 32 [18]. В табл. 5
приведены некоторые цис-транс-стереоизомеры |3-каротина, существование
которых считали возможным [23].
44
Таблица 5
Г Номер изомера Конфигурация Номер изомера Конфигурация
I Полный транс XI 3,5,6-три-^ис
II 3-цис XII 3,5,7-три-цис
III 5-цис XIII 3,5,9-три-^ис
IV S-цис XIV 3,6,7-три-^ис
V 3,5-цн-цис XV 3,6,9-три-^ис
VI 3,6-ди-^ис XVI 5,6,7-трп-цис
VII 3,7-ди-цис XVII 3,5,6,7-тетра-^ис
VIII 3,9-цн-цис XVIII 3,5,6,9-тетра-^ис
IX 5,6-дп-цис XIX 3,5,7,9-тетра-^ис
X 5,7-цн-цис XX 3,5,6,7,9-пента-^ис
Однако недавно было доказано, что пространственно затрудненные
^uc-конфигурации в изопреноидных полиенах не только возможны, но до-
вольно стойки [22, 23, 24]. Они получаются синтетическим путем. П. Кар-
рер [25] таким путем получил 4,8-ди-г{Мс-|3-каротин, а Г. Ингофен [26] —
центральный 6-цис-р-каротин. В табл. 6 приведены данные об А-провита-
минной активности пространственных изомеров каротина [14]:
Таблица 6
Пространственные изомеры каротина Конфигурация А-провитаминная активность, % к полному транс-3’ каротину
а-каротин Полный транс 53
Нео-а-каротин-U 3-или 9-моно-^ис 13
Нео-а-каротин-В 3,6-(или 7)-ди-(<ис 16
р-каротин Полный транс 100
Нео-^-каротин-U 3-ысто-цис 38
Нео-^-каротин-В 3,6-(или 7)-ди-^ис 53
6-цис-р-каротин 6-моно-цис 50
4,8-цис-З-каротин 4,8-ди-^ис 20
7-каротин Полный транс 27
Нео-7-каротин 3-моно-цис 19
Про-'!-каротин Поли-(пента)-£{ис 44
Криптоксантин Полный транс 57
Нео-криптоксантин-U 3- моно-цис 27
Нео-криптоксантин-А S-тлот-цис 42
Каротиноиды. В растительных материалах изомеры каротина сопро-
вождают родственные им изопреноидные полиены, которые имеют общее
название каротиноиды. Значение последних заключается в провитаминной
активности отдельных представителей этого класса веществ, а также в их
красящей способности (пищевые красители).
Из каротиноидов углеводородного характера известны:
а) ликопин, содержащийся в томатах, плодах шиповника, облепихе и
других фруктах и ягодах. Ликопин отличается от p-каротина тем, что в его
молекуле |3-иононовые циклы заменены псевдоиононовыми (см. витамин А3
стр. 15);
б) биксин (Bixa orellana) — каротиноид, содержащийся в кожуре се-
мян индийского кустарника oriean tree. Биксин представляет собой пиг-
мент кислотного характера. В воде не растворяется, трудно растворяется
в холодных органических растворителях. Кристаллизуется в форме темно-
красных лепестков. Температура плавления выше 300° С. Спектр погло-
щения биксина в хлороформе — 509; 474,5; 442 нм. Химическая формула
45
Ci
Структура молекулы каротиноидов
1
Таблица 7
Название Основные физико-химические константы [15, 29, 30]
темпера- тура плавления, СС количе- ство двойных связей оптическое вращение максимум поглощения, нм
cs2 СНС13 петролей- иый эфир
2 3 4 5 6 7 8
Фитоин С^оНба — 9 — — —
Фитофлюин СщНвг — 10 \ — — 332 347 366
z-Каротин С^оНво — 11 — — — 378 400 425
Нейроспорин 12 — — —
Ликопин С4оН66 175 13 0 548 507,8 477 517 480 453 506 475,5 447
у-Каротин C4oHse 178 12 0 533 496 463 508,5 475 446 495 462 431
S-Каротин C^gHje 181—182 11 — 520 485 450 493 463 436 483,5 452 426
м
S-Каротин 12 526 503 490
C.joHse 490 470 458
457 440 420 (в гек-
а-Каротин 187—188 11 4-385 500 485 сане) 478
QioHse 477 454 447,5
Криптоксантин 169 И 0 519 497 485,5
483 463 452
452 433 424
Рубиксантин ено 160 12 0 533 509 495,5
494 474 463
461 439 432
Ликоксантин СаоНзвО 168 13 — 546 506 504 473
472 — 444
Зеаксантин 205 11 —40 517 495 483,5
С4оН5602 50 482 462 451,5
450 429 423
Ксантофилл 193 11 4-145 508 487 477,5
С4оН5602 475 456 447,5
445 428 420
Ксантофилл- 192 10 — 501,5 482 471
эпоксид 472 453 442
С4оН56Оз в бензоле
Флавоксантин 184 10 4-190 479 459 450
QloHseOg 449 430 421
Родоксантин 219 12 564 546 521
С4оН5в02 525 510 487
491 482 456
QO
Структура молекулы каротиноидов
Продолжение табл. 7
Название Основные физико-химические константы [15, 29, 30]
темпера- тура плавления, °C количе- ство двойных связей оптическое вращение максимум поглощения, н.и
cs2 СНС13 петролей- ный эфир
2 3 4 5 6 7 8
Антераксантин 205 10 — 510 490,5 —
С4оН5603 478 460,5 —
Фукоксантин 159,5— 11 +72,5 510 492 -
с40н56о6 160,5 477 457 —
445 — —
Виолаксантин 200 9 +35° 501 482 472
^40^56^4 470 451,5 443
440 424 417,5
Тараксантин 185—186 — — 501 — 472
С4оН66С)4 469 — 443
441 — —
Ауроксантин 203 9 0 454 — —
С4оН5604 423 — —
Миксоксантин 168—169 12 488 473 465
С40Н54О
Миксоксанто- 182 10 —255 526 518 503
филл 489 484 471
С4оН5607 458 454 445
в пири- в этаноле
дине
сн=сн — (к)— сн=сн
сн —сн ~ (к) — сн= сн
н3соос-сн=сн — (к) — сн=сн —соон
ноос— (к) —соон
о
Астацин 240—243 11 Около Около
С40Н43О4 510 500
в пири-
дине
Капсантин 176 10 +36° 542 520 505
С4оН58Оз 503 486 475
в бензоле
Капсорубин 201 9 — 541,5 520 506
С4оН6004 503 486 474
468 455 444
в бензоле
Биксин Выше 300 9 527,5 509
С25Б130О4 492 474,5 —
457,5 442 —
Кроцетин 285 7 — 482 463 450,5
С20Н24О4 453 434,5 424,5
426 — -—
Азафрин 212—214 7 — 483 472 454
СгтНзаС^ 452 440 429
Афанин 176 12 0 533,5 504 494
С40Н54О 494 474 460
Целаксантин 209-210 13 0 562 520,5 520
С4оН560 521 488 486,5
487 455 456
в этаноле
Афаницин 190 23 .— 533 504 494
СзоЦцвОз 195 494 474 462
Эхиненон 178—179 11 (520)
С4оН580 488 .— —
(450)
СП
о
Структура молекулы каротиноидов
Продолжение табл. 7
Название Основные физико-химические константы [15, 29, 30]
темпера- тура плавления, °C количе- ство двойных связей оптическое вращение [«Го максимум поглощения, нм
cs2 СНС1, петролей- ный эфир
2 3 4 5 6 7 8
Торулародин СзтЩзОа 201—203 12 0 582 541 502 554 515 (483) 537 501 (467)
Торулин С40Н54 185 13 f — 565 525 491 539 501 469 520 486 456
Ликофилл CjoHjeOa 179 13 . — 546 506 472 521 487 456 в бензоле в этаноле 504 473 444
/
метилового эфира С25Н30О4, молекулярная масса 394,515. Структурная
формула:
СН3 СН3 СН3
Н3СООС-СН=СН—с=сн—сн=сн—с=сн—сн=сн—сн=с—сн=сн—сн=
сн3
=с—сн=сн—соон
Биксин (С25Н:,О4)
Отдельную группу каротиноидов представляют собой ненасыщенные
спирты — производные каротина—ксантофиллы. К ним относятся крипток-
сантин — пигмент шиповника, кукурузы, рябины [27, 28], имеющий био-
логическую активность в полной транс-форме — 57% от р-каротина
сн3 сн3 сн3 сн3
<|Г~СН=СН-С=СН-СН=СН-С-СН-СН=СН-СН=С- сн=сн-сн=с-сн = сн
3 _ Криптоксантин ^3*^
Второй представитель этой группы рубиксантин (пигмент шиповника)
подобно криптоксантину имеет один гидроксилированный цикл р-ионона
[28], но другой цикл у него раскрыт (псевдоионон)
Й'з СН3 СН3 ( СН3 QU
сн=сн-с=сн-сн=сн-с=сн-сн=сн-сн=с-сн=сн-сн=с-сн=сн
а - P</iuttcanmuH ^3*-
Третий представитель этой группы — зеаксантин (пигмент яичного
желтка, облепихи) с двумя гидроксилированными р-ионовыми циклами
[15]
сн=сн-с=сн-сн=сн-с=сн-сн=сн-сн=с-сн=сн-сн=с-сн=сн
сн3 . н3с
Зеаксантин (HS6 02 )
Изомером зеаксантина является ксантофилл желтых листьев — лютеин,
у которого один гидроксилированный цикл Р-ионона, а второй — а-ионона.
К группе ксантофиллов относятся также флавоксантин (пигмент лютика,
тыквы) с эмпирической формулой С40Н56Оа, изомеры — виолаксантин (пиг-
мент анютиных глазок и тыквы) и тараксантин С4оН5б04(пигмент одуван-
чика).
Следующая группа каротиноидов принадлежит к оксисоединениям. Из
них наиболее изучены родоксантин (С40Н66О2), афанин (С40Н54О) — моно-
кетопроизводное p-каротина и миксоксантин (С4оН540) — монокетопроиз-
водное у-каротина. К монокетонам относится также каротиноид животного
происхождения эхиненон. В табл. 7 дан перечень каротиноидов и приведе-
ны их физико-химические константы [15, 29, 30], где
СН3 СН3 СН3 СН3
II II
к=—с=сн—сн=сн—с=сн—сн=сн—сн=с—сн=сн—сн=с—.
Влияние различных факторов на устойчивость каротина. Вопросу устой-
чивости каротина и его препаратов посвящено большое число исследований,
из которых вытекает: каротин как в кристаллическом состоянии, так и в
51
растворах весьма лабилен [15, 31, 32, 33], процессы распада интенсифици-
руются с повышением температуры [34]; на интенсивность процесса распа-
да каротина влияет свет, тяжелые металлы, в особенности медь [15, 31, 32].
Каротин лабилен в кислой среде, но сравнительно стоек в щелочной.
На величину потерь каротина при хранении его растворов оказывает влия-
ние вид растворителя и концентрация каротина в растворах [15 , 35, 36, 37].
Применение антиокислителей в виде а-токоферола, гидрохинона, сантохи-
на (1,2-дигидро-6-этокси-2,2,4-триметилхинолин) оказывает положительный
эффект [38, 39].
Исследования показали, что основным фактором, влияющим на распад
каротина, является кислород воздуха. В исследованном диапазоне темпе-
ратур (до 100° С) в отсутствие кислорода распада каротина не происходит
[40, 41]. На распад каротина оказывает влияние растворенный в жидкой
фазе воздух. В масляном концентрате, содержащем 1,5 мг!г каротина, рас-
творенное в масле (подсолнечном) количество воздуха способно окислить
26% каротина.
Судя по экспериментальным данным, свет не является самостоятельно
действующим фактором распада каротина, а играет роль катализатора [41].
Так как потери каротина при хранении в герметически укупоренной
таре зависят от количества воздуха, содержащегося в флаконе (в раство-
ренном и нерастворенном виде), то, следовательно, чем выше концентра-
ция каротина в растворе, тем ниже относительные потери каротина. С дру-
гой стороны, влияние рода растворителя (при отсутствии в нем окислитель-
ных агентов) будет определяться способностью его к растворению воздуха.
Для снижения потерь каротина в производстве и при хранении препа-
ратов необходимо:
на всех этапах производства по возможности устранять аэрацию полу-
продуктов;
при расфасовке каротина максимально заполнять тару концентратом,
оставив недолив в соответствии с коэффициентом расширения масла (0,0007);
перед укупоркой тары производить дезаэрацию концентратов инертным
газом в течение 15 мин при комнатной температуре [42].
МЕТОДЫ СИНТЕЗА р-КАРОТИНА И ВЫБОР РАЦИОНАЛЬНОГО МЕТОДА
ДЛЯ ПРОИЗВОДСТВА [43, 44, 45]
Из сравнения структурных химических формул |3-каротина и витамина
А видно их химическое родство. Две молекулы витамина А, соединяясь
посредством двойной связи, дают молекулу p-каротина. В связи с этим ста-
ло очевидно, что для синтеза p-каротина следует исходить из р-ионона или
альдегида С14.
Одной из задач в этой области — выбор фрагментов для построения мо-
лекулы p-каротина. Решение этой задачи может быть облегчено путем кон-
денсации трех фрагментов, из которых два крайних являются одноимен-
ными. В общем виде этот синтез может быть выражен следующей общей
формулой: Сх+Су+Сх = С40. Теоретически изучали следующие варианты:
1. С19+С2+С19=С4о. Здесь две молекулы р-С19-альдегида конденсиро-
вали с ацетилендимагнийбромидом по реакции Гриньяра [46]. Этот вари-
ант был Излером и др. [47] усовершенствован в заводской практике. Полу-
ченный при конденсации диол переводили в 15,15'-дегидро-р-каротин
путем аллильной перегруппировки и одновременной дегидратации. Частич-
ное гидрирование в присутствии катализатора Линдлара (палладиевый де-
зактивированный) дало 15,15'-1{цс-р-каротин, который был изомеризован
в полной шра«с-[3-каротин в высококипящем петролейном эфире. Участ-
вующий в синтезе альдегид С19 [9,13-диметил-7-(1,1,5-триметилциклогек-
сен-5-ил-6)-октатриен-8,10,12-аль-14] был получен Ингоффеном из альде-
гида С14 [48,49]—полупродукта синтеза витамина А, синтезированного из
Р-ионона глицидным синтезом [50]. Альдегид С14 при конденсации с орто-
52
муравьиным эфиром при участии катализатора (п-толуолсульфокислота)
дает а-, ^-ненасыщенный ацеталь альдегида С14, который при повторной кон-
денсации с виниловым эфиром (катализатор ZnCl2) превращается в эфиро-
ацеталь. Последний гидролизуют кипячением с уксусной или фосфорной
кислотой и получают альдегид С16 (кристаллический продукт с температу-
рой плавления 78—79° С). Все-эти превращения (ацетализирование, конден-
сация и гидролиз) осуществляют в одну стадию с выходом в 65% [47, 51].
Из альдегида С16 таким же методом конденсацией с пропениловым эфиром
(вместо винилового) получают альдегид С19 с выходом около 70%. Весь
синтез осуществляют по следующей схеме:
Альдегид
CH(OC2HS)3
ACH3C6H4SO3H
Ацеталь альдегида CJlt
СН2= CHOR
Д ZnCt2
з сн3 OR
СН2-СН = С-СН-СН2-СН
OR OR
CH3COOH
СН, СН3 СН3
рЧ—CH2-CH=C-CH=CH-CHO CH(OC2Hs)3
ЧЧ<сн3
Альдегид Cjg
Зрироацеталь
Ацеталь альдегида Сц
СН3СН3 СН3 СН3
сн2—сн=с—сн =сн -сн=с-сно
\ X
'/т:н3
Альдегид Ctg
CH3-CH=CHOR
сн3 сн3
СН3 CH3/OR
сн2-сн=с-сн=сн-сн-с~сн сн3соон
I \ —-—-
OR OR
СН3
Зфироацеталь альдегида. Ctg
+ Вг М (г CsC MgBr + альдегид C/g
Ацетилендимагнийдромид
сн
сн3
сн3 сн3
-сн = сн-сн=сн-сн=с-сн-с=с-сн-с=сн-сн=сн-с=сн-сн2-
он
он
Диол
Н3с
сн3 сн
ifQ
сн3
сн3
сн3
13 СН;
сн=сн-с=сн-сн=сн-с=сн-с = с - сн=с-сн=сн-сн=с-сн=сн
сн
75,15'-Дегидро - fl-каротин
Н3С
сн
сн.
сн.
СН.
сн=сн-с=сн-сн=сн-с=сн-сн=сн-сн=с-сн=сн-сн=с-сн=сн
СН3
fl - каротин
н-
Общий выход p-каротина из альдегида С14 составляет около 25%. По
этой химической схеме синтезированы некоторые каротиноиды, как напри-
мер зеаксантин и криптоксантин [52].
2. С16+С8+С6=С40. По этой схеме Р-каротин был синтезирован в 1950 г.
Каррером [53], Ингоффеном [54] и Майласом [55]. Синтез Р-каротина по
этому варианту был осуществлен, исходя из ацетиленового карбинола С16,
который получали конденсацией р-ионона и пропаргил бромида (ВгСН2=
=СН). Для этого конденсировали две молекулы ацетиленового карбинола
С16 по реакции Гриньяра с ненасыщенным дикетоном С8 (октен-4-дион-2,7).
Получаемый при этом С40 — тетрол ацетиленового ряда подвергали
частичному каталитическому гидрированию на дезактивированном палла-
53
диевом катализаторе в полиеновый тетрол С40 и дегидратации п-толуол-
сульфокислотой в Р-каротин. Недостатком метода является получение про-
межуточных продуктов с ОН в аллильном положении к двойной связи в
цикле р-ионона. Это влечет за собой ретроионилиденовую перегруппировку,
резко снижающую выход p-каротина (около 3%) [53]. Несколько лучшие
результаты могут быть получены, если синтез Р-каротина осуществить из
альдегида С14 и ацетиленового комплекса Иоцича (BrMgC=CMgBr) через
ацетиленовый карбинол С16, так как в данном случае гидроксил в нем не
находится в аллильном положении к двойной связи в цикле. Этот синтез
осуществлен по следующей схеме:,.
СН3СН3 СН3
<|1-СН2—СН=6-СН-С = СН
I 11 он
^ СН,
Карбинол С!В
BriMgC=CMgB
СНз сн3
+ ос-сн2- СН=СН-СН2СО
+ Карбинол C/g
Дикетон Сд
СНз СНд pit рц
3 СН3 СН3 сн3
P|1—СН2—СН=С-СН-С^С-С-СН2-СН=СН-СН2 —C-CsC-
I 1L он он он
сн3
СНз
сн-с=сн-сн2
он
н3с
3
Диацетиленобый тетрол
Гидрирование и дегидратация
- /(apoтин
3. С18+С4+С18=С40. Для синтеза использован кетон С18, причем две'
молекулы кетона были соединены молекулой димагнийбром диацетилена
(BrMgC— С—C=CMgBr) [56] в диацетиленовый диол, который частичным
гидрированием и дегидратацией при действии фосфордийодида превращают
в Р-каротин [57]. Дийодид, получающийся в ходе реакции, неустойчив и
распадается с выделением молекулы йода [58]. Синтез осуществлен по схе-
ме:
СНзДНз сн3 сн,
Plj—сн=сн—с=сн—сн=сн—с=о
СНз Кетон С;р
BrMgC=C—С=СМдгВг + Кетон
СНзСНз СН3 СНз СНз СН3 СНзСНз
P~'>-CH = CH-C=CH-CH = CH-C-CSC-C=C-C-CH=CH-CH = C-CH=CH—Г|>
11 OH OH JI J
сн3 н3с
Диацетиленобый диол
Pd | частичное гидриробание
Полиенобьш диол
J3 - Каротин
Выход p-каротина на последней стадии при обработке Р214 оказался
очень низким (около 1%).
4. С14+С12+С14 = С4о. Известны еще ряд схем синтеза Р-каротина,
как, например, C16+C10+Ci6 [59, 60]; C14+C12+Ci4 [61, 62]; Ci3+C14+Ci3
[59]; С10+С20+С10 [59, 63]; 025+С18 [64]; С21+С19 [65, 66]; С20+С20 [66,
59, 67, 68]. Из приведенных схем интерес представляет схема С14+С12+
+С14, сущность которой заключается в конденсации под влиянием хлорис-
того цинка двух молекул а, P-ненасыщенного ацеталя альдегида Ci4 с од-
ной молекулой эфира диенолина С12. Образующийся при этом диэфир ди-
54
кеталя Р-С40 омылением и отщеплением спирта превращают в дикетон Р-С40
и далее в диол (3-С40, 15,15-дегидро-Р-каротин и в р-каротин [51 ].
Этот вариант синтеза является двухкомпонентным и в нем довольно
сложен синтез промежуточной цепочки — эфира диенолина С12 из тигли-
нового альдегида, являющегося дефицитным. Таким образом, из рассмат-
риваемых четырех вариантов синтеза Р-каротина наиболее эффективным
является первый вариант, обозначенный формулой С1в+С2+С19=С40. Судя
по литературным данным, этот вариант синтеза нашел практическое приме-
нение за рубежом.
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА СИНТЕТИЧЕСКОГО р-КАРОТИНА
Технология производства базируется на однокомпонентном методе син-
теза, разработанном Ингоффеном [46, 49] и усовершенствованном Изле-
ром [47] с их соавторами. Этот метод нашел свое дальнейшее развитие в
исследованиях Н. Преображенского, Г. Самохвалова и Л. Вакуловой [70,
71, 72, 73]. Метод заключается в конденсации двух молекул альдегида С19
с молекулой ацетилена по реакции Гриньяра. Технология включает сле-
дующие стадии синтеза: синтез Р-С16-альдегида из Р-С14-альдегида; синтез
₽-С)9-альдегида из Р-С16-альдегида; синтез 15,15-дегидро-Р-каротина изС19-
альдегида и ацетилена; синтез транс-Р-каротина из 15,15-цис-Р-каротина.
СИНТЕЗ р-С1в-АЛЬДЕГИДА’ [9-МЕТИЛ-6-(1,1,5-ТРИМЕТИЛЦИКЛОГЕКСЕН-5
ИЛ)-ГЕКСАДИЕН-8, 10-АЛЬ-12]
Химические реакции получения альдегида С16 заключаются в ацетали-
зировании альдегида-С 14, конденсации полученного ацеталя с виниловым
эфиром в присутствии хлористого цинка и омыления алкоксиацеталя аль-
дегида С16. Химизм реакций синтеза альдегида С16 основан на склонности
виниловых эфиров присоединяться к ацеталям а, Р-непредельных карбо-
нильных соединений [51, 74], причем одна алкооксигруппа [OR] переме-
щается от ацеталя к двойной связи винилового эфира. Остаток ацеталя
присоединяется к Р-углеродному атому винилового эфира. Конденсация
и перегруппировка протекает по схеме:
CH3 OR
R-CH=C-CH + ch2=ch-or—►r-ch=c-ch-ch2-ch
J3 cl ' '
СН3
OR
OR
OR
OR
По этой схеме протекает следующий синтез.
Схема реакций синтеза Р-С16-альдегида.
Реакция ацетализирования
СН3 СН3 СН3
Г|Т-СН2— сн=с-сно
-альдегид
206, 32
СИ, сн3 CH3oc2Hs
+ нс(ос2н5)3 —^Г>сн2-сн=с-сн
ос2н5
Ортомурабьиный э/pup Диэтилацеталь ~ альдегида
148,2 280,44
55
Реакция конденсации
сн3 ос2н5
сн2-сн=с-сн
I
oc2Hs
280, «4
CH2=CH-OC2HSUI^—
Зтилбинилобый Эфир
72,10
СН3 ' OC2HS
сн2-сн=с-сн-сн2-сн
ОС2Н5 OC2HS
Зтоксиацеталь fl-Ctf - альдегида
380,34
Реакция омыления
сн, сн3 сн3 ос2н5
СН2—СН=С-СН_СН2—СН
^<:н2 °с2н5 oc2Hs
380,34
СН3СН3 сн3
СНдСООН <5гсн2-сн=с-сн=сн-сно „„ „ „
III + зс^н^он
J3~ C;g - альдегид
232,35
Для успешного протекания реакций ацетализирования и конденсации
важно, чтобы влажность реагентов была минимальной (в %): абсолютного
спирта — 0,15; этилвинилового эфира — 0,2; ортомуравьиного эфира —
0,09; альдегида-С14 — 0,04. Хлористый цинк предварительно должен быть
сплавлен и высушен в вакуум-эксикаторе над концентрированной серной
кислотой.
Ацетализирование. В реактор 1 (рис. 7) из нержавеющей стали, снабжен-
ный мешалкой, обратным холодильником и барботером для азота, загру-
жают через мерник 2 альдегид-С14 («д = 1,510), из мерника 3 ортомуравьи-
ный эфир (температура кипения 144—145° С, плотность 897 кг/л3), из мер-
ника 4 — раствор паратолуолсульфокислоты в абсолютном этаноле. Реак-
цию ведут в присутствии азота, вводимого в реактор из баллона 5. Переме-
шивают в течение 20—24 ч при комнатной температуре. Затем в реакционную
массу вводят из мерника 6 лигроин и нейтрализуют 2,5%-ным раствором
бикарбоната натрия, загружаемым из мерника 7. После этого отделяют ор-
ганический слой в делительной воронке 8 и после просушки поташом на-
правляют в сборник 9, а из него в перегонный аппарат 10, где при темпера-
туре около 50° С и остаточном давлении 3—5 мм рт. ст. отгоняют раствори-
тель. Технический продукт содержит около 95% ацеталя, Пд =1,4820.
Выход ацеталя из Р-С14-альдегида составляет около 75 %г. На выход ацета-
ля из альдегида-С 16 значительно влияет чистота альдегида-С14. Диэтил-
ацеталь Р-С14-альдегида С18Н32О2 представляет собой маслянистую жид-
кость желтого цвета с температурой кипения 87—96° С при остаточном
давлении 0,2 мм рт. cm.-, d20 =0,9279; п1 2® =1,4773.
Конденсация с этилвиниловым эфиром. В реактор 11, снабженный хо-
лодильником, загружают из мерника 12 диэтилацеталь-Р-С ^-альдеги-
да, затем из мерника 13 медленно добавляют при температуре 35 —40° С
этилвиниловый эфир (температура кипения 35° С, остаточная влажность
не выше 0,2%), а из мерника 14 — 10%-ный раствор сплавленного хлорис-
того цинка в ледяной уксусной кислоте. Реакцию проводят в присутствии
азота, вводимого из баллона 15, при температуре 35—40° С в течение 1 ч.
В результате реакции образуется этоксиацеталь Р-С 16-альдегида (см. хи-
мическую схему), представляющий собой (перегонка при остаточном
давлении 0,02 мм рт. ст.) вязкое светло-желтое масло, d23 = 0,9315,
«20=1,4727. Выход 66—70% [70].
1 Имеются данные [70], что при ацетализировании 3-С14-альдегида при помощи
тетраэтоксисилана — этилового эфира ортокремневой кислоты Si(OC2H5)4 — в при-
сутствии катализатора ортофосфорной кислоты выход диэтилацеталя можно увели-
чить До 93%.
56
Омыление этоксиацеталя. В процессе омыления ацетальной группы
происходит также отщепление молекулы спирта. В реактор 11 из мерника
16 добавляют смесь ледяной уксусной кислоты, ацетата натрия, воды и
гидрохинона (небольшое количество). Реакционную массу медленно нагре-
вают до 90—95° С и перемешивают 3 ч. Затем раствор (темно-вишневого
цвета) переводят в реактор-охладитель 17. Охлаждают до 0°, выкристалли-
зовывают технический ₽-С 16-альдегид и отфуговывают его в центрифуге 18.
Перекристаллизация технического альдегида С16. Процессы ведут в
этаноле в реакторе 19 по двухступенчатой схеме. После обработки активи-
рованным углем раствор фильтруют через нутч-фильтр 20. Кристаллизуют
в кристаллизаторе 21, отфуговывают кристаллы в центрифуге 22. Маточ-
ный раствор I поступает в сборник 23. Сгущение его производят в вакуум-
аппарате 24 и далее кристаллизуют в кристаллизаторе 25. Кристаллы вто-
рой кристаллизации отфуговывают в центрифуге 26, а маточный раствор II
направляют в сборник 27. Он является отходом производства. Кристаллы
второй кристаллизации поступают на перекристаллизацию в реактор 19
совместно с техническим продуктом.
Альдегид-|3-С16 (С16Н24О) представляет собой светло-желтые кристаллы
с температурой плавления 77—78° С, хорошо растворим в органических
растворителях, плохо в воде; Хгаах—276—280 нм (в спирте), Е\ °/°м—1347.
СИНТЕЗ 3-С1в-АЛЬДЕГИДА [9,13-ДИМЕТИЛ-7-(1,1,5-ТРИМЕТИЛЦИКЛОГЕК-
СЕН-5-ИЛ)-ОКТАТРИЕН-8,10,12-АЛЬ-14]
Химические реакции получения альдегида-С )9 заключаются в ацетали-
зировании альдегида С16, конденсации полученного ацеталя с виниловым
эфиром в присутствии хлористого цинка и омыления алкоксиацеталя аль-
дегида С19 по следующей химической схеме.
Схема реакций синтеза р-С19-альдегида
Реакция ацетализирования
СН3СН3 сн3
<$гсня-сн=с-сн=сн-сно . > н3ро4.
+Hcl°W3 с^5он‘
СН.з
-альдегид Ортомурабьиный эфир
232,35 М8,2
СНз/СНз СН3 ОС2Н5
_<>S-CH2-CH=:C-CH=CH-CH
\^СН3 ОС2Н5
Дизтилацеталь Д~С1в -альдегида
306,47
НСООС2Н5
Реакция конденсации
СНз/СНз СН3 ОС2Н5
<ф-СН2-СН=С-СН=СН-СН +
11 » 1
ОС2Н5
сн3
сн=сн-ос2н5
306, hl
Зтилпропенилодый эфир
86,13
57
СН3 СНз СНз СН3 ОС2Н5
ZnC12 |^ХЧ-СН2-СН=С-СН=СН-СН-СН-СН
СН3СООН <^ЖСНз ОС2Н5 OC2HS
Этоксиацеталь fl-Ojg- альдегида
392, 60
Реакция омыления
СНзСН3 СН3 СН3 ОС2Н5
г^>т-сн2-сн=с-сн=сн-сн-сн-сн СН3СООН
\^СН3 ОС2Н3 6с2Н5 СНзСООКа, Н2О
392,60
СЬ{з/СНз СНз СНз
____ГП-СН2-СН=С-СН=СН-СН=С-СНО
I || 3C>2*i5'^*^
^^^СНз
fl -Cjg- аль де г ид
272,^1
Для успешного протекания указанных реакций необходимы те же усло-
вия обезвоживания химических реагентов, как и в синтезе Р-С 16-альдегида.
Ацетализирование. Процессы проводят так же, как и для синтеза 0-С16-
альдегида и в аналогичной аппаратуре. К ней относятся реактор 28 и сбор-
ники: для альдегида-С 16 29, ортомуравьиного эфира 3, катализатора 4,
лигроина 6, нейтрализующего раствора бикарбоната натрия 7. Азот в реак-
тор подается из баллона 30. Разделение слоев осуществляют в делительной
воронке 31 и после просушки органического слоя поташом направляют
его в сборник 32 и далее в перегонный аппарат 33, где отгоняют раствори-
тель и не вошедший в реактор ортомуравьиный эфир (при температуре
50—55° С и остаточном давлении 2—3 мм рт. ст.). Получают технический
диэтилацеталь Р-С16-альдегида с содержанием основного вещества 95—97%,
п2°= 1,5026—1,5070; маслянистая жидкость, температура кипения около
145° С при остаточном давлении 0,05 мм рт. ст. Выход 75—80% (в пере-
счете на альдегид [3-С1в).
Конденсация с этилпропениловым эфиром. Процесс осуществляют в
реакторе 34, в который загружают диэтилацеталь альдегида-С 16 из мерни-
ка 35, а из мерника 14 раствор (10%) сплавленного хлористого цинка в ле-
дяной уксусной кислоте. Масса принимает темно-вишневый цвет. Затем при
температуре 25—30° С из мерника 36 медленно добавляют этилпропени-
ловый эфир (температура кипения 69—71° С, остаточная влага не выше
0,15%). Масса постепенно окрашивается в желтый цвет. Реакция протекает
в' присутствии азота, вводимого из баллона 37, при перемешивании.
Омыление этоксиацеталя. В реактор 34 из мерника 16 добавляют смесь
ледяной уксусной кислоты, ацетата натрия, воды и гидрохинона. Реакци-
онную массу медленно нагревают до 90—95° С и перемешивают 3 ч. Затем
раствор темно-вишневого цвета направляют в реактор-охладитель 38,
охлаждают до минус 5—7° С и кристаллизуют. Кристаллы технического про-
дукта отфуговывают в центрифуге 39. Получаютжжелтые кристаллы с содер-
жанием основного вещества около 95%. Маточный раствор направляют в
сборник 40; он является отходом.
Перекристаллизация технического альдегида С19. Процессы ведут в эта-
ноле по схеме перекристаллизации альдегида С16 в следующей аппаратуре:
для первого продукта — реактор-растворитель 41, нутч-фильтр 42,
кристаллизатор 43, центрифуга 44;
58
для маточного раствора I — сборник 45\
для второго продукта — вакуум-аппарат 46, кристаллизатор 47, цент-
рифуга 48, сборник маточного раствора II-—отхода производства — 49.
Выход альдегида на диэтилацеталь составляет 55—57% (от теоретиче-
ского). Альдегид |3-С)9 представляет собой ярко-желтые ромбические крис-
таллы с температурой плавления 63—65° С; хорошо растворим в органиче-
ских растворителях, плохо — в воде; Хтах=325 нм (в спирте); = 1825.
СИНТЕЗ 15, 15'-ДЕГИДРОф-КАРОТИНА
Вещество получают конденсацией альдегида С19 с ацетиленовым комп-
лексом Иоцича с последующей дегидратацией образующегося диола С40.
Реакцию конденсации начинают с приготовления реактива Гриньяра
(C2H8MgBr), который с ацетиленом в среде сухого эфира дает комплекс
Иоцича по схеме:
2C2H5MgBr + СНгСН —> BrMgC CMgBr + 2С2Н6.
2-133,3 26,04 232.49 2-30,07
Димагнийбромацетилеп
Ацетилен пропускают при температуре 18—20° С до полного исчезнове-
ния магний бромэтила, что контролируется реакцией с кетоном Михлера
(наличие вызывает изумрудно-зеленое окрашивание). Дегидратацию диола
С4о осуществляют в среде сухого серного эфира спиртовым раствором хло-
ристого водорода в присутствии азота. Реакции протекают по следующей
схеме:
Схема реакций синтеза 15,15'-дегидро-0-каротина
Конденсация
3 сн3 сн3
СН2-СН = С-СН = СН-СН=С-СНО* ВгМдС = СМдВ
сн,
yd - Cfff -Альдегид
272,47
сн3 сн3 сн3 сн3
-сн=с-сн=сн-сн=с-сн-с=с-сн-с=сн-сн=сн-с=сн-сн
OMgBr ОМдВг
Маенийорг&нический комплекс
Диолин - С40
Дегидратация
3 сн3 сн3 сн3 сн3
сн=сн-с=сн-сн=сн-с=сн-с=с-сн=с-сн=сн-сн=с-сн=сн
15,15' - Дегидро -fl - каротин
534. 83
Конденсация. В стальной эмалированный реактор 50, снабженный ме-
шалкой и обратным холодильником, предварительно тщательно высушен-
ный, загружают через люк магниевую стружку из сборника 51, сухой сер-
ный эфир (влажность не выше 0,1 %) из мерника 52 и медленно из мерника
53 приливают в течение 1 ч раствор сухого бромистого этила в сухом эфире.
Затем в течение 1 ч нагревают реакционную массу до кипения и перемешива-
ют до полного растворения магния. Затем охлаждают массу до 15—18° С
и в течение 5—6 ч пропускают из баллона 54 предварительно осушенный
через вымораживатель 55 ацетилен до получения отрицательной пробы с
кетоном Михлера. Затем реакционную массу охлаждают до 10—12° С и
59
из мерника 56 медленно добавляют раствор альдегида-С )9 в сухом эфире
так, чтобы температура не превышала 12—13° С. Раствор окрашивается в
ярко-оранжевый цвет. Реакция при перемешивании протекает в присутст-
вии азота в течение 1,5—2 ч с повышением в конце процесса температуры
до 20—25° С. Полноту реакции конденсации определяют по исчезновению
альдегида-С 19 (реактив Легаля). После этого реакционную массу сливают
в реактор-охладитель 57 с ледяной водой, куда из мерника 58 залит хлорис-
тый аммоний. Массу сливают в делительную воронку 59. Органический
слой промывают водой, просушивают сульфатом натрия из сборника 60
и направляют через сборник 61 в вакуум-аппарат 62. Растворитель удаля-
ют в вакууме в токе азота при температуре не выше 30° С и получают |3-С4о-
диолин в виде твердого желтого осадка.
Дегидратация. Процесс осуществляют при помощи хлористого водорода.
Для этого из мерника 63 сливают в вакуум-аппарат 62 хлористый метилен,
растворяют диолин-С4о и переводят раствор в реактор 64, снабженный ме-
шалкой и рассольным охлаждением. Массу охлаждают до минус 15—18° С,
а затем из мерника 65 постепенно добавляют 8%-ный раствор сухого НС1
в абсолютном спирте с таким расчетом, чтобы температура реакционной
массы не превышала к концу процесса+3,+5° С. Затем в делительной
воронке 66 отделяют органический слой, промывают его насыщенным раство-
ром бикарбоната из мерника 67 и направляют в сборник 68 и далее в вакуум-
аппарат 69, где под вакуумом в токе азота при температуре 30—35° С от-
гоняют хлористый метилен. Кристаллизующуюся массу направляют в крис-
таллизатор 70, где при температуре —2, —3° С в течение 8—10 ч в присутст-
вии азота выпадают кристаллы 15,15'-дегидро-р-каротина. Последние от-
фуговывают в центрифуге 71, промывают этиловым спиртом. Выход около
50%. Маточный раствор поступает в сборник 72 и является отходом произ-
водства. Вопрос о выделении вещества из маточного раствора еще недоста-
точно изучен. 15,15'-дегидро-|3-каротин представляет собой кристаллы крас-
ного цвета с металлическим блеском; температура плавления 153—154° С;
хорошо растворим в органических неполярных растворителях, плохо —
в воде; Хтах=454 и 430 нм\ £["^=1568 и 1873. Выход 48—50% [70].
СИНТЕЗ ТРАНСф-КАРОТИНА
Синтез осуществляют путем гидрогенизации 15,15'-дегидро-|3-каротина
в растворе толуола на частично отравленном палладиевом катализаторе
[75] с целью превращения ацетиленовой связи до этиленовой и получения
[бДб'-^ис-Р-каротина. Изомеризация в среде петролейного эфира превра-
щает последний в транс-|3-каротин. Для успешного проведения реакции
гидрирования необходимо применять тщательно очищенный толуол с при-
менением палладиевого катализатора на меле, приготовляемого как указа-
но на стр. 34. Реакции протекают по следующей схеме:
60
15,15'-Моно-дис-р-каротин. В реактор 73 из эмалированной стали
загружают через люк 15,15'-дегидро-|3-каротин, а из мерника 74 толуол
и при нагревании до 35—40° С и перемешивании растворяют кристаллы.
Затем добавляют палладиевый катализатор, нанесенный на мел. Аппарат
дважды продувают азотом из баллона 75, а затем водородом из баллона 76,
после чего при температуре 20° С и избыточном давлении до 0,5 кгс!см2
при перемешивании осуществляют процесс гидрогенизации. Реакцию конт-
ролируют по количеству поглощенного водорода. Далее реакционную мас-
су фильтруют через нутч-фильтр 77 и сборник 78, откуда фильтрат направ-
ляют в перегонный аппарат 79 для отгонки толуола при вакууме (остаточ-
ное давление 8—10 мм рт. ст.) в токе азота. Кубовый остаток сливают в
кристаллизатор 80, где при минус 5—8° С выкристаллизовывают 15,15'-мо-
HO-Zjuc-p-каротин. Кристаллы выделяют при помощи центрифуги 81; ма-
точный раствор поступает в сборник 82 и является отходом производства.
Катализатор с нутч-фильтра 77 направляют на регенерацию. Выход цис-
фкаротина составляет 90—95% [70], темно-вишневые кристаллы; темпе-
ратура плавления 148—150° С; Хтах=338 (цыс-пик), 450, 480 нм (в гексане);
£i % =1040, 1765, 1430.
Транс-f)-Каротин. В эмалированный реактор 83, снабженный мешалкой
и обратным холодильником, загружают цис- |3-каротин, из мерника 84 петро-
лейныйэфир (80—90° С), нагревают массу до кипения и продолжают переме-
шивать в течение 10—12 ч (изомеризация). Затем сливают в кристаллизатор
85, охлаждают до 0 — минус 2° С и кристаллизуют в течение 6 ч. Крис-
таллы выделяют в центрифуге 86, а маточный раствор I направляют в сбор-
ник 87 и после сгущения в вакуум-аппарате 88, кристаллизации в кристалли-
заторе 89, выделения кристаллов в центрифуге 90 получают дополнительное
количество кристаллов транс-Р-каротина II, которые поступают для
перекристаллизации в кристаллизатор 85. Маточный раствор II является
отходом производства.
Перекристаллизация технического транс-р-каротина. Перекристаллиза-
цию ведут из петролейного эфира по двухступенчатой схеме: для первой сту-
пени — реактор-растворитель 91, нутч-фильтр 92, кристаллизатор 93, цент-
рифуга 94, сборник маточного раствора I 95; для второй ступени — вакуум-
аппарат 96, кристаллизатор 97, центрифуга 98, сборник маточного раствора
11 99. .Кристаллы |3-каротина II поступают на перекристаллизацию
совместно с техническим |3-каротином в реактор-растворитель 91.
ТЕХНОЛОГИЧЕСКИЕ ПОКАЗАТЕЛИ
РАСХОД ХИМИКАЛИЕВ, КГ НА 1 КГ
100%-НОГО ПОЛУПРОДУКТА
$-С16-Альдегид
^-Сц-альдегид.................... 1,3
о-Муравьиный эфир.................. 1,0
Этилвиниловый эфир.................0,5
Спирт этиловый абс...............0,3
Уксусная кислота ледяная .... 4,2
Ацетат натрия ...................0,6
Эфир серный ..................... 0,8
Сода двууглекислая...............0,2
Калий углекислый безводный . . 0,3
Цинк хлористый безводный ... 0,01
Паратолуолсульфокислота .... 0,01
J-Cig- А льдегид
P-C1J-альдегид....................2,1
о-Муравьиный эфир.................. 1,8
Эфир этилпропениловый...........0,9
Эфир серный.................... 1,8
Уксусная кислота ледяная .... 7,2
Спирт этиловый абс.............0,4
Калий углекислый безводный . . 0,4
Сода двууглекислая............. 0,15
паратолуолсульфокислота .... 0,01
Ацетат натрия ..................0,07
15,15'-Дегидрокаротин
₽-С19- Альдегид................2,6
Эфир серный....................7,0
Ацетилен.......................0,3
Бромистый этил................. 1,6
Магний металлический............0,34
Аммоний хлористый..............4,5
Сульфат натрия ................ 1,0
Этанол абсолютный..............2,9
Хлористый водород (газ).........0,35
Метилен хлористый..............4,0
Сода двууглекислая............. 0,6
61
Т ранс-$-каротин
15,15'-Дегидро-^-каротин............ 1,4
Толуол............................12,0
Катализатор Pd-CaCO3................0,3
Эфир петролейный....................5,0
РАСХОД ПОЛУПРОДУКТОВ НА
СИНТЕЗ 1 КГ ТРАНСА-КАРОТИНА,
КГ
15,15'-Дегидро-р-каротин .... 1,40
₽-Ci9-Альдегид...................3,64
₽-Ci6-Альдегид..................7,64
р-Сц- Альдегид..................9,93
РАСХОД ХИМИКАЛИЕВ НА СИНТЕЗ
1 КГ ТРАНС-р-КАРОТИНА, КГ
Аммоний хлористый................ 6,3
Альдегид-С14.................... 9,9
Ацетат натрия .................... 4,9
Ацетилен..................... 4,2
Водород хлористый (газ)...... 0,4
Калий углекислый............. 3,8
Кислота паратолуолсульфуровая . 0,1
Кислота уксусная.............58,3
Метилен хлористый............ 5,6
Магний металлический......... 0,5
Натрий сернокислый безводный . . 1,4
Спирт этиловый абс........... 7,9
Сода двууглекислая........... 2,8
Цинк хлористый безводный ... 0,1
Эфир ортомуравьиный.............14,2
Эфир серный.....................22,5
Эфир этилвиниловый ............. 3,8
Эфир этилпропениловый........... 3,3
Этил бромистый.................. 2,2
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. S t е е n b о с k I., J. Biol. Chem., 1919, 40, 511; 1920, 41, 81; 41, 149; 42, 131; 1921
46, 32; 1922, 51, 63.
2. Euler В., Euler Н., Hellstrom Н. Biochem Z., 1928, 203, 370.
3. Euler H„ Karrer F., Demole V., Walker D. Helv. Chim. Acta,
1930, 13, 1078.
4. Euler H., Karrer P., Hellstrom H., R ibgom M. Helv. Chim.
Acta, 1931, 14, 839.
5. M о о r e J., В i о c h e m. J., 1929, 23, 803; 23, 1267; 1930, 24, 692; 1931, 25 , 275.
6. В у л ь ф с о н Н. С. Химическая природа витамина А. — «Успехи химии», 1944,
т. 13, вып. 6, с. 437.
7. Кудрнцкая С. Е. Потребность в А-витаминоактивных препаратах для живот-
новодства УССР и пути ее удовлетворения. Автореф. дисс., Киев, 1965.
8. Schuchardt W. Fette und Seifen, 1951, 53, 689.
9. Kuhn R., Winterstein А., К a r 1 о w i t z L., Helv. Chim. Acta, 1929,
12, 66, 907.
10. К a r r er P. и др., Helv. Chim. Actq., 1929, 12, 1142; 1930, 13, 87; 1930, 13, 268.
11. Karrer P., Helf enstein A., Wehrli H., Winterstein A.,
Helv. Chim. Acta, 1930, 13, 1084.
12. Smith I., J. Biol., Chem., 1931, 90, 597.
13. S e b r e 1 W., Harris R., The Vitamins, N. Y., Academ. Press., 1954, 3—26.
14. О г о s h n i k W., Encyclopedia of Chem. Technology, U. S., 1955, 14, 791.
15. С а в и н о в Б. Г. Каротин. Киев, Изд-во АН УССР, 1948, 230 с.
16. Winterstein A., Z. Physiol. Chem., 1933, 219, 249.
17. F г а р s G., Kemmerer А. и др. Ind. Eng. Chem., 1941, 13, 806; 1943, 15,
714.
18. Zechmeister L., Tuzson P., Biochem. J., 1938, 32, 1305; Ber.,
1939, 72, 1350.
19. Garter G., Gillam A., Biochem J., 1939, 33, 1325.
20. P о 1 g a r A., Zechmeister L.,J. Am. Chem. Soc., 1942, 64, 1856; 1944, 66,
137.
21. С а в и н о в Б. Г., Гринберг Ф. Л. Изомеризация каротина. — «Успехи
химии», 1948, т. 17, вып. I, с. 74—84.
22. F о у I., М о г g а г iedge К-, Annal. Chem., 1948, 20, 304.
23. О г о s h n i k W., К a r m a s G., Mebane A., J. Am. Chem. Soc., 1952,
74, 295.
24. О г о s h n i k W., Mebane A., J. Am. Chem. Soc., 1954, 76, 5719.
25. Garbers C., Eugster C., Karrer P. Helv. Chim. Acta., 1952, 35, 1850.
26. Inhoffen H., S i e m e r H., Fortschr. Chem. Qrg. N.aturstoffe, 1952, 9, 1.
27. Савннов Б. Г. и Проценко Л. Д. — «Украинский химический журнал»,
1944, т. 20, вып. 4, с. 399.
28. К У Ш и н с к а я И. Н., Шнайдман Л. О. Идентификация каротиноидов,
содержащихся в сухих плодах шиповника. — «Медицинская промышленность
СССР», 1964, № 4, с. 38—40.
29. К а г г е г Р., J u с k е г Е. Carotinoide, Basel, 1948.
30. Проценко Л. Д. Исследование химической природы пигментов некоторых
новых каротиноносителей. Автореф. канд. дисс., Киев, 1953.
31. Кирсанова В. А. К вопросу об окислении каротина. — «Биохимия», 1938,
т. 3, вып. 2, с. 191—201.
32. Савинов Б. Г., С в и щ у к А. А. Каталитическое влияние металлов на
62
окисление каротина. —«Украинский химический-журнал», 1950, т. 16, вып. 1,
с. 57—63, с ил.
33. Варламов Г.— «Консервная н овощесушильная промышленность», 1957, 6, 31.
34. Михайловннна А. А. Исследования некоторых превращений каротина и
его устойчивости. —Автореф. канд. дисс., Киев, 1952.
35. Г е н и н а Р. Б. Материалы к стандарту на каротин.— В сб.: «Витамины в тео-
рии и практике», М., Пищепромиздат, 1941, т. 3, с. 124—129.
36. В a u ш а п п С., J. Biol. Chem., 1933, 101, 547; 103, 339; 1934, 105, 167; 107, 705.
37. Euler Н. Helv. Chim. Acta, 1929, 12, 278; 1930, 13, 1078, 1932, 15, 469; 1934,
17, 24; 1938, 21, 21.
38. T h о m p s о n С. Пат. США, 2. 562. 970, 7. VIII. 1951.
39. C i e g 1 e r A., Nelson V., Hall H., J. Agr. food, Chem., 1961, 9, 447.
40. Шнайдман Л. О., Д ул ь чин а Б. M., Павлова А. М. Влияние
различных физико-химических факторов на устойчивость каротина в растворах.—
«Труды ВНИВИ», 1954, № 5, с. 51—64 с ил.
41. Ш н а й д м а н Л. О. Исследование в области химии и технологии производства
витаминов. Доклад докт. дисс., М., 1967, с. 46.
42. Ш н а й д м а н Л. О. Усовершенствование технологии производства каротина из
моркови и тыквы. — В сб.: «Технология и применение витамина А и каротина»,
Пищепромиздат, 1956, с. 30—38.
43. I s 1 е г О., S с h u d е 1 Р., Hoffmann-La Roche, Advances in Organic
Chemistry, 1963, 4, 115—224.
44. С а м о x в а л о в Г. И. Синтез fi-каротина. — В сб.: «Витамины. Пищевая
промышленность за рубежом», М., Пищепромиздат, 1957, № 3, с. 26—48.
45. Schuchard W., О 1 е a g i n е u х, 10, № 4, 1955. — В сб.: «Витамины.
Пищевая промышленность за рубежом», 1956, № 1, с. 108.
46. I п h о f f е n Н., Р о m m е г Н., В о h 1 m a n n F., Ann., 1948, 561, 26; 1949,
565, 45, 1950, 569, 74; 1950, 570, 54.
L e i b n e r G., Ann., 1952, 575, 105.
S i e m e r H., M 6 h 1 e K- Ann., 1954, 585, 126.
Пищепромиздат, 1959, с. 231.
Lindlar Н., Montavon М., R й е g g R., R и s е г G.,
Helv. Chim. Acta., 1956, 139, 456; 1956, 39, 2041; 1957, 40, 456.
, Eugster C. Helv. Chim. Acta., 1950, 33, 1172.
, _ . L i _1F., Bartram K-, Pommer
47. I s 1 e r O., Lindlar H., Montavon M., Riiegg R., Zeller P.,
Helv. Chim. Acta, 1956, 39, 249.
48. InhoffenH
49. I n h о f f e n 11 ,
50. I s h i k a w a S., M a t s u u r a T., Zbl., 1937, 11, 3452.
51. Б e p e з о в с к и й В. M. Химия витаминов. М. “
52. I s 1 ег О., .............
Zeller Р.,
53. К а г г е г Р.,_„___
54. Inhoffen Н., Bohlmann
Ztg., 1950, 74, 285.
55. М i 1 a s N.,
4844.
56. Inhoffen H., В
57. Kuhn R.,
58. С а м о x в а л о в Г. И. Синтез р-каротина. — В сб.: «Витамины. Пищевая про-
Н. Chem.
1950, 72,
с I., F 1 е s D., J. Am. Chem. Soc.,
s
D a v i
Р., В е 1 i
F., A 1 d а у н др., Ann., 1951, 573,
K-, 'Ber., 1938, 71, 1889.
1.
h 1 m a n n
n f e 1 s
о
e
Wall
мышленность за рубежом», 1957, № 3, с. 26—48.
59. Р о m m е г Н. Angew. Chem. 1960, 72, 11.
60. Р о m m е г Н. Sarnecki W. Пат. ФРГ, 1. 068, 709, 1959; С. А., 1961, 55
134731.
61. Isler О., Chopard L., Montavon М., R ii е g g R., Zeller P.,
Helv., Chim., Acta., 1957, 40, 1256.
62. I s 1 e r O., Montavon M., R ii egg R., Zeller P., Ann., 1957, 603,
129.
63. I s 1 e r O., Gutmann H., Lindlar H., Montavon M., R ii e g g
R., Zeller P., Helv. Chim. Acta, 1956, 39, 463.
64. Riiegg R., Schwieter U., Ruser G., Schudel P., Isler O.,
Helv. Chim. Acta., 1961, 44, 985.
65. Inhoffen H., Sch w i e t e r U., Chichester C., Mackinney G.
J. Am. Chem. Soc., 1955, 77, 1053.
66. Isler O., Lindlar H., Montavon M., R i e g g R., Zeller P.,
Helv. Chim. Acta., 1956, 39, 249.
67. R о b e s о п С. Пат. США № 2 932 674, 1960; С. A., 1960, 54, 24852 д.
68. S t e г п M., Пат. США № 2945069, 1960; С. А., 1961, 55, 608е.
69. I s 1 е г О., Montavon М., R ii е g g R., Р. Zeller, Ann. 1957, 603,
129.
70. В а к у л о в а Л. А. Синтетические исследования в области каротиноидных сое-
динений. Автореф. кандид. дисс., М., 1959.
71. Самохвалов Г. И., Миропольская М. А., Вакулова Л. А.,
Преображенский Н. А. Полный синтез псевдоионона, иононов, геранио-
ла и нерола. — «ДАН СССР», 1952, 84, с. 1179—1182 с ил.
72. Самохвалов Г. И., Миропольская М. А., Вакулова Л. А.,
Преображенский Н. А. Полный синтез псевдоинона. — ЖОХ, 1955,
25, с. 545-550.
73. Самохвалов Г. И., Давыдова Л. П., Захаркин Л. И. и др.
63
Синтетические исследования в области полиеновых соединений. Новый синтез
ретиналя. — ЖОХ, 1960, 30, с. 1823—1828 с ил.
74. Н о a g 1 i n R., N i г s h D., J. Am. Chem. Soc., 1949, 71, 3468.
75. L i n d 1 a r H. Helv. Chim. Acta., 1952, 53, 446.
Глава 4. ПРОИЗВОДСТВО СИНТЕТИЧЕСКОГО ВИТАМИНА В3
(ТИАМИНА)
В 1882 г. было замечено [1 ], что возникновение распространенной в то
время среди моряков болезни бери-бери связано с рисовым питанием. Одна-
ко большинство врачей склонялось к инфекционному происхождению бо-
лезни.
Голландский врач Эйкман [2] в 1890—1897 гг. занимался изучением
бери-бери; он впервые вызвал это заболевание у кур, питавшихся очищен-
ным рисом.
Вначале Эйкман связывал эту болезнь с токсичностью полированного
риса. Лишь в 1906 г. он согласился с мнением своего сотрудника Гринса [3],
считавшего, что причина возникновения бери-бери состоит в неполноцен-
ном питании.
Препарат, излечивающий полиневрит, был выделен Функом [4] лишь
в 1911—1912 гг. из дрожжей и рисовых отрубей.
В 1916 г. Мак-Коллум и Кеннеди [5] предложили назвать вещество,
излечивающее полиневрит, витамином В п
Виндаус выделил витамин Bi в чистом виде [6] и в 1932 г. установил
его эмпирическую формулу C12H18ON4SC12-H2O. Витамин Bd имеет важное
значение для животного организма. Он входит в состав фермента карбокси-
лазы, катализирующего реакции декарбоксилирования пировиноградной
кислоты и других ц,-кето кислот. При недостатке тиамина в организме про-
исходит накопление пировиноградной кислоты — продукта обмена угле-
водов, что нарушает нормальную функцию нервной системы и вызывает
заболевание полиневритом (бери-бери). Тиамин излечивает эту болезнь.
Кроме того, дифосфат тиамина входит в состав многих других ферментов
в качестве кофермента, связанного с апоферментом — белком. Сюда отно-
сятся и ферменты, катализирующие реакции обмена углеводов типа альдоль-
ных конденсаций и др. Витамин Bd связан также с функцией органов кро-
ветворения, участвует в обмене воды, углеводов, жиров и минеральных со-
лей [7, 8, 9, 10]. Витамином В4 богаты дрожжи (пивные и пекарские) и
злаки, не очищенные от отрубей. Ржаной, а также пшеничный цельный
хлеб, крупы (в особенности гречневая) являются для человека основным
источником витамина В(,
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ВИТАМИНА Bj
После того как Виндаус [6] в 1932 г. установил эмпирическую фор-
мулу витамина В 1( а Виллиаме открыл реакцию его сульфитного расщеп-
ления:
C12H18ON4SC12 + Na2SO3 —> C6H9O3N3S + C6H9ONS + 2NaCl,
стало успешно развиваться изучение химического строения молекулы вита-
мина Bj.
При сульфитном расщеплении витамина В t образуется аминосульфо-
новая кислота, она растворима в органических растворителях и слабо рас-
творима в воде
СН
N^xC-CH2-SO3H
Н3С-С^ „C-NH,
d IN 2
Лиримидиламиносулыроновая
кислота
64
При действии соляной кислоты пиримидиламиносульфоновая кислота
превращается в оксисульфоновую кислоту, что указывает на наличие пер-
вичной аминогруппы. При нагревании с водой под давлением (до 200° С)
получается серная кислота, а при плавлении со щелочами — сернистая
кислота, что свидетельствует о наличии сульфогруппы [11]. Ультрафиоле-
товый спектр поглощения аминосульфоновой кислоты показал характер-
ные данные для 4-аминопиримидина [12, 13, 14].
При окислении витамина В ^сульфата марганцовокислым барием или
при действии жидким аммиаком получено соединение C6HJoN4 [15], кото-
рое при синтезе идентифицировано как 2-метил-4-амино-5-аминометилпи-
римидин.
Строение второго продукта сульфитного расщепления, легко раствори-
мого в воде основания
N___С- СН3
II II
нс с-сн2сн2он
4SZ
4-метил-5-оксиэтилтиазола, было установлено в 1935 г. [16]. При окислении
последнего азотной кислотой так же, как и при окислении витамина Въ
образуется вещество, сходное с синтетической 4-метил-тиазол-5-карбокси-
линовой кислотой
N___С-СН3
II II
нс с-соон
Изучая тиазоловый компонент молекулы витамина В ь Кларк и Вилли-
аме пришли к заключению, что витамин В 4 является солью четвертичного
основания (четвертичный азот). В 1935 г. Виллиаме предложил следующую
структурную формулу витамина В4 [17]:
CI-
CH / сн,+
N^C2 N3—4С-СН3
I и5 IId 4||
Н3С-С4з'х-С4' НСЪхС-СНоСНоОН
N NH2-HC1 S
Витамин Bj ,
Витамин Bt—(С12Н 17ON4S)+C1_• НС1 • 0,5Н2О, его химическое название
4-метил-5-|3-оксиэтил-М-[[21-метил-41-аминспирРмидил(5/)]-метрл)-тиазслил-
хлоридгидрохлорид.
Независимо от Виллиамса такую же структуру витамина В 4 предложи-
ли Греве [18], Андерзаг и Вестфаль [19].
Тиамингидрохлорид, молекулярная масса 337,27, твердое кристалли-
ческое вещество, из алкоголя кристаллизуется в виде бесцветных игл мо-
ноклинической системы с температурой плавления 248—250° и 233—244° С
(диморфизм) с разложением [20]. Тиамингидробромид, молекулярная мас-
са 436,21, белый кристаллический порошок желтоватого оттенка, со слабым
характерным запахом, горького вкуса, с температурой плавления 220° [16],
229—231° [11], 227—231 ° С [21]. Витамин В t с минеральными кислотами
образует соли, водные растворы которых имеют кислую реакцию, напри-
мер pH 0,12%-ного раствора тиамингидробромида равен 3,0. Соли тиамина
3-522
65
растворителях. При температуре
Рис. 8.
тамина
Спектр поглощения ви-
Bj-гидрохлорида в УФ-
свете
имеют следующие температуры плавления: сульфат 203°, тетрасульфат
276—278°, нитрат 164—165° [22], пикрат 208°, хлораурат 189°С.
Витамин Bt хорошо растворим в воде и метиловом спирте, трудно рас-
творим в этиловом спирте и нерастворим в эфире и других неполярных
“ 25°С 1 г тиамингидрохлорида растворим
в воде — в 1 мл, в 95%-ном спирте —
в 100 мл, в абсолютном алкоголе — в
315 мл, в глицерине — в 18 мл\ в водном
85 %-ном спирте при кипении в соотно-
шении 1:6.
Витамин В г гидрохлорид ‘имеет
спектр поглощения в ультрафиолетовом
свете с максимумами, зависящими от
pH: 235 и 267 нм [20, 23] при pH 7 и выше
(рис. 8); 245—247 нм при pH 5.5 и
менее [13]. Таким образом, абсорбцион-
ный спектр поглощения витамина В 4 яв-
ляется функцией концентрации водород-
ных ионов [24]. Витамин В 4 стабилен
в кислой среде. Чистый витамин В гги-
дрохлорид в водном растворе при pH
120° С без разложения, а в более сла-
3,5 выдерживает нагревание при
бых кислотах разрушается. Крепкие минеральные кислоты разрушают мо-
лекулу с выделением аммиака [21]. В присутствии сульфита молекула ви-
тамина В4 расщепляется на свои составные компоненты [25]:
сг
СН2>--трСНз Na2SO3 CH2SO3H + N —pCHs
н,сЛ A <JLCH сн он H3c4 Anh2 < Лсн2сн2он
3 N NH2-HC1^^ 22 N S
Витамин В; Пиримидиновый компонент Тиазоловый компонент
Расщепление витамина В 4 происходит также в нейтральных растворах
ацетата натрия и нитрата бария. В щелочных растворах витамин В быстро
разрушается. При простом титровании щелочью на 1 моль витамина расхо-
дуется 3 моля щелочи, что связано с разрушением тиазолового цикла. Од-
нако при немедленном обратном титровании кислотой витамин полностью
восстанавл ивается.
В щелочной среде в присутствии красной кровяной соли витамин Bj
легко превращается в тио хром [26]
С1-
СИ2\ '
n--------гснз NaOH ---Гснз
НГ1 L AcH2CH2OH K3[Fe(CN)6fH3cA A A. AcH2CH2OH
IN 11П2’Н^1 О iN П 5
Витамин В/ Тио хром
Витамин В4 очень чувствителен к окислителям и восстановителям.
Окисление перманганатом калия или азотной кислотой приводит к глубо-
кому разрушению молекулы. При гидрогенизации витамина В 4 с плати-
новой чернью поглощаются два атома водорода [27 [. При восстановлении
витамина В 4 гипосульфитом в присутствии бикарбоната натрия присоеди-
няются два атома водорода к тиазоловой части молекулы.
К производным тиамина относятся различные соли, эфиры, а к произ-
водным тиола тиамина — различные дисульфиды. Из солей тиамина,
кроме бромида и хлорида, следует отметить тиаминмононитрат [28] —бе-
лые кристаллы с температурой плавления 196—200° С (с разложением).
Его растворимость в воде: при температуре 25° С — 1 г в 37 мл, при темпе-
66
ратуре 100° С— 1 г в 3,4 мл. Водные растворы тиаминмононитрата нейт-
ральны. Стабильность их значительно превышает стабильность тиаминга-
логенидов и поэтому они применяются для витаминизации хлеба, а также
в клинической практике [29]. Синтезирован также сульфаттиамин [30],
соли с различными органическими кислотами и др. [10].
Из эфиров тиамина важное значение имеют фосфорные эфиры (тиамин-
моно- и дифосфат), которые получают обработкой тиамина хлорокисью фос-
фора [31 ] или смесью пирофосфорной кислоты и фосфорного ангидрида [32]
с последующим хроматографическим разделением продуктов на колонках.
Пирофосфорный эфир тиамина известен под названием кокарбоксилазы.
Его структурная формула:
Н3С
N NH2HC1
Кокарбоксилаза
|| ||
СН2СН2О-Р-О-Р-ОН
он он
Описан другой метод получения фосфатов тиамина [33], который за-
ключается в фосфорилировании 4-метил-5-|3-оксиэтилтиазола, а затем полу-
ченные моно- или дифосфорные эфиры тиазола конденсируют с 2-метил-4-
амино-5-бромметилпиримидином. При этом получают высокий выход
эфиров. К производным тиольной формы тиамина относятся тиаминпропилди-
сульфид и тиаминтетрагидрофурфурилдисульфид и др. [34]. Из многих
дисульфидных производных тиольной формы тиамина особое внимание за-
служивает тиаминпропилдисульфид, который обладает активностью вита-
мина В1, но быстрее и полнее усваивается организмом и не разрушается
тиаминазой [35, 36].
МЕТОДЫ СИНТЕЗА ВИТАМИНА В3 И ВЫБОР РАЦИОНАЛЬНОГО МЕТОДА
ДЛЯ ПРОИЗВОДСТВА [10, 11, 37, 38, 39]
Существуют следующие три направления синтеза витамина В4.
1. Раздельный синтез пиримидинового и тиазолового компонентов с
последующей их конденсацией по схеме:
h3c-U.nJLNH2.hc1
Пиримидиновый компонент
, сн2 -
сн2сн2он н3с-ЧкХ Лсн,сн,он
2 N NH2-HC1 s
Тиазоловый компонент витамин в)
с2.. Постепенное построение тиазолового цикла на азоте пиримидинового
цикла через тиоформильное производное, которое конденсируется с а-гало-
идированным кетосоединением соответствующего строения по следующей
схеме:
„ Г1 сн2. +
N^fr-CH2NH2 N^>t-CH2N=CH-SH . COH=CC1-R2 N |f ’ll—jTCH3
H3c\^NH2-HC1 H3c-U J’-NHfHCl H3C-kN>NlfeHa4s>CH2CH2OH
Пиримидиновый компонент Тиорормильное производное св - Галоидированное витамин в;
кетосоединение
Этот вариант синтеза успешно осуществлен в Японии [40, 41, 42, 43]
путем конденсации 2-метил-4-амино-5-аминометилпиримидина (диамина)
с сероуглеродом и у-хлор-у-ацетопропилацетатсм в присутствии аммиака
3* 67
с образованием у-ацето-а-ацетоксипропил-N- [(2'-метил-4'-аминопирими-
дил-5'-метил)-дитиокарбомата 1.
Последний при нагревании с НС1 дает тиотиамин по следующей схеме:
Н3С
3 N
со-сн, сн2.
CH,NH, । NH3 1NH СО-СН, HC1
nh2 + CS2 + C1-CH-CH2-CH2OR —NH2 I I H_ ’ -
R=-H; -COCH3 N s S
2 - Метил - 4-амино -5 - Сероуглерод у-Хлор-у-ацетолропил- Тиотиан
аминометилпиримидин ацетат
СГ(Вг)
СН2Х , , ___ ^•СН2^ +
_ N |< 7—С~СНз Н2о2; НС1 (НВг) Ni[ ^N—с-сн3
Н3С-^ >NH2 / Лсн2сн2он NaHCO3 НС А Дч , СН ,_С-СН2СН2ОН
N S S N NH2-HCl(HBrPs
Тиотиамин
Тиаминхлорид (бромид)
3. Постепенное построение пиримидинового
схеме синтеза И. Г. Фарбениндустри гомолога
цикла на тиазоловом по
витамина В 1; а именно:
ROOC СН2Вг
СН
I
CN
Вг"
снз ROOC CH2-N
сн2сн2он—- II
ь I
СН3
СН2СН2ОН
NH
Н3С-С
nh2
Эфир оС-циан-уд-
бромпропионовой кислоты
Тиазоловый компонент
CN
3<рир тиазола -of-циан -fl • бромпропионовой кислоты Ацетамидин
Окситиамин
сн3
СН2СН2ОН
N NH2-HC1 S
Тиаминхларид
Этот вариант синтеза витамина В 4 развития не получил в виду его слож-
ности. Практическое применение нашли первый и второй методы синтеза.
В связи с этим вопрос о преимуществе первого или второго метода синтеза
тиамина должен быть обсужден после детального рассмотрения технологи-
ческих схем производства и технико-экономических показателей обоих
методов. Необходимо, однако, учесть, что в связи со сложностью синтеза
тиамина в каждом методе, в особенности в первом, возник ряд химических
и технологических вариантов на отдельных стадиях синтеза. Чтобы пра-
вильно сформулировать химию и технологию данного метода синтеза тиа-
мина, необходимо рассмотреть эти варианты.
Итак, по первому методу, предусматривающему раздельный синтез пи-
римидинового и тиазолового компонентов, необходимо:
1. Выбрать вариант синтеза пиримидинового компонента и при этом
решить следующие вопросы:
II
а) определить химическую структуру трехуглеродной цепочки С—R2,
I
R3
которая при конденсации с ацетамидином должна дать пиримидиновый
компонент;
б) найти наиболее эффективный вариант получения ацетамидина;
в) установить тип галоида (хлор, бром) для галогенирования пиримиди-
нового компонента.
68
2. Выбрать вариант синтеза тиазолового компонента и при этом решить
следующие вопросы:
а) синтез из производных тиазола, замещенных в положении 2, или
синтез на основе тиоформамида;
б) определить вариант получения у-хлор-у-ацетопропилового спирта:
из сильвана или из у-хлор-у-ацетобутиролактона.
3. Выбрать вариант конденсации пиримидинового и тиазолового компо-
нентов с получением тиамина галогенида и при этом рассмотреть возможные
способы конденсации компонентов, получения целевого продукта и
непосредственно синтеза тиаминхлорида:
конденсация компонентов сплавлением их или в среде органического
растворителя;
получение целевого продукта в виде тиаминхлорида или тиаминбромида;
непосредственный синтез тиаминхлорида путем конденсации 2-метил-
4-амино-5-хлорметилпиримидина с 4-метил-5-|3-оксиэтилтиазолом или син-
тез его путем обменных реакций, в том числе ионообменным методом.
По второму методу синтеза тиамина, предусматривающему построение
его молекулы на пиримидиновом цикле, необходимо:
1. Выбрать вариант синтеза тиамина и рассмотреть пути получения его
через тиоформаминометилпиримидин или через тиотиамин.’
2. Обсудить вопрос о типе целевого продукта (тиаминбромид, хлорид,
нитрат и др.).
МЕТОД СИНТЕЗА ТИАМИНА, ОСУЩЕСТВЛЯЕМЫЙ КОНДЕНСАЦИЕЙ
ПИРИМИДИНОВОГО И ТИАЗОЛОВОГО КОМПОНЕНТОВ
Выбор метода синтеза пиримидинового компонента. Ряд исследователей
установили [44, 45], что синтез пиримидиновых производных, алкилиро-
ванных в положении 2, достигается довольно легко при конденсации ами-
динов с [i-альдегидо- и |3-кетокислотами или Р-дикетонами в присутствии
щелочи.
Применение в синтезе ацетамидина обеспечивает получение метильной
группы в положении 2. Подбирая для конденсации с ацетамидином вто-
рой компонент этой реакции, можно получать производные пиримидина,
замещенные в положениях 4 и 5
Ацетамидин образует часть молекулы пиримидинового компонента (I)
N7
Н3С—С
(I)
СН
С—СН2С1
С—NH2-HC1
R1
II
с—r2
(III)
Другая часть молекулы (II) должна представлять собой трехуглеродную
цепочку типа (III), где Rt представляет собой оксиметиленовую (=CHOR)
или аминометильную группу (=CHNH2), a R3 — нитрильную (—CN) или
карбоксильную группу (—COOR). Что касается заместителя R2, то наибо-
лее эффективной является алкоксиметильная группа (—СН2ОС2Н5), кото-
рая при действии галоидоводородной кислоты образует необходимую галои-
дометильную группу (СН2 Гал).
По-видимому, наилучшим вариантом синтеза пиримидинового компонен-
та будет при трехуглеродной цепочке с R3—CN, так как при конденсации
с ацетамидином получим в этих условиях пиримидиновый компонент с
группой NH2 в положении 4.
Из сопоставления приведенных ниже методов синтеза пиримидинового
компонента видно, что сложность его зависит от применяемых заместите-
лей R ь R2 и R3, поэтому правильный выбор их очень важен.
69
Метод Виллиаме а, Клайна и Финкельштейна
[20 ] заключается в конденсации ацетамидина гидрохлорида с натрийформил-
|3-этоксиэтилпропионовым эфиром и получении 2-метил-4-гидрокси-5-это-
ксиметилпиримидина по следующей схеме:
NH2
н3с-с
NH
Ацетамидин
I
NaOCH
С—С Н2О С2Н5 г н £ С
СООС2Н5
Натрийформил -fl-эток -
сиэтилпропионобый эфир
сн2ос2н5
он
2 - Метил - 4 - гидрокси - '
5 - этоксиметилпиримидин
Соединение II авторы получали по схеме:
нсоос2н5 +
Этилформиат
СН2-СН2ОС2Н5
СООС2Н5
fl - Этоксиэтилпропи-
оновый эфир
NaOCH
с-сн2ос2н5
СООС2Н5
Натрийформил -fl-этокси-
этилпропионовый эфир
Метод Греве [46] заключается в
этоксиметилмалоновым нитрилом по схеме:
конденсации ацетамидина с
NH2
н3с—с
NH
Ацетамидин
снос2н5
%
C-CN -
I
CN
Этоксиметиленма -
лонобый динитрил
Nf^siT-CN
~H3C-U3*Lnh2
3 N
2 - Метил - 4 - амино-
5 - цианпиримидин
Метод Андер
денсации ацетамидина
зага и Вестфаля [471 заключается в кон-
с формилянтарным эфиром по схеме:
NH2
Н3с-сх
NH
Ацетамидин
СООС2Н5
C-CH2COOC2HS
II 2 5
снон
Формилянтарный эфир
СН2СООС2Н5
он
2 - Метил -4- оксипиримидил -
5 -этоксиуксусный эфир
Метод Челинцева и Беневоленской [48] заключает-
ся в конденсации ацетамидина с а-ацетоксиметилен-[3-этоксипропионитри-
лом по схеме:
NH2 СНОСОСН3
I
Н3С —С + С-СН2ОС2Щ
I 5
NH (2]\|
Ацетамидин сС -Ацетоксиметилен-
J3 -зтоксипропионитрил
Nf^sjT СН2ОС2Н5
НзС-Цз^МНг
2 - Метил -k- амино -
5 - этоксиметилпиримидин
70
Чтобы оценить эффективность четырех указанных методов синтеза
аминопиримидиновой части молекулы витамина В 4 рассмотрим количество
стадий синтеза, необходимых для преобразований полученного промежуточ-,
ного пиримидинового соединения в компонент структуры 2-метил-4-амино-
5-бромметилгидробромид.
По методу Виллиамса
1. Зн3с- —ЧХ N -CH2OC2HS -он РОС13 -3 Н3С N' CH2OC2HS LCl + Р(ОН)3
2. Н3С- “4s N -CH2OC2HS -Cl 2NH3 н3с- N' I; |-СН2ОС2Н5 lnh2 + NH4C1
3. н3с- О: -CH2OC2H3 -NH2 2HBr н3с N- рСН2Вг L-NH2HBr + с2н5он
В этом синтезе переход от оксигруппы в положении 4 к аминогруппе
осуществляется через хлорпроизводное. Преимущество способа состоит
в простоте превращения радикала в положении 5 в галоидметиленовый
радикал.
По методу Греве N^At-CN +Н2 1. h3cAnJJ-nh2 I N^]-CH2NH2 + NaNO2 + НС 2. H3cAnAnH2 11 N<Ai-CH2OH + 2HBr 3. h3cAnAnh2 111 , Г^АрСНгМНг н3с—k Anh2 3 N 2 Заметил -b~ амино- 5-амцнометилпиримидин ' 11 1 r N^CH2OH H3CAnAnh2 2-Метил -k~ амино- 5-оксиметил^иримидин 111 г ЬИАг-СН2Вг H3C-kNANH2-HBr Z - Метил - 4 -амино ~5- бромметилпиримидингидробромид
По этому методу образование аминогруппы в положении 4 происходит
в процессе циклизации и ее не надо вводить в ядро посредством двух стадий.
Однако для преобразования циангруппы в положении 5 в бромметильную
требуются три стадии: 1) каталитическое гидрирование; 2) обработка азо-
тистой кислотой соединения II и 3) обработка бромистым водородом соеди-
нения Ill.
71
По методу Андерзага и Вестфаля
1. CH2COOC2HS
Н3С он
IN
2 - Метил - 4 - оксипиримидин-5 -
зтоуксусный зрир
РОС1г СН2СООС2Н3 +
н3с4 Л-ci
3 N
2-Метил-4 -хлорлиримидил-
5-зтоуксусный зрир
Р(он)3
N<^^|r-CH2COOC2H5 3lNH3 _
Н3С -U 3- С1
3 N
cX^nhCONH*C2H5°h + nh4c1
П ON-1 I 1 ПО
3 N
2 - Метил -4- аминопиримидил -
5 -ацетамид
n^>-ch2conh2
h3c-U Anh2
3 IN 2
NaOCl
H3C
^-аС1 + СО2
N^^^i-CHaNHa
HjC-kj >-NH2
IN
lNalNO2 + HCl
IN
2 - Метил - 4 - амино -5- амино -
метилпиримидин
СН2ОН .
Н3С 1NH2 + N2 + н2° + INaGl
3 N
5. ГМ<г?Х|Г-СН2ОН
>-nh2
3 N 2
2HBr
2 - Метил -4- амино -5-
оксиметилпиримидин
N^YCH2Br
H3(A Лмн2.нвг 2
3 IN 2
2 - Метил - 4 -амино -5- бром *
метилпиримидингидробромид
По методу Челинцева и Беневоленской
. N^]-CH2OC2H5
h3cAnAnh2
2HBr
N^>i-CH2Br +
Н3С^>^Н2-НВг +
2 -Метил-4- амино -5-
бромметилпиримидинеидробромид
C2HSOH
Таким образом, для преобразования промежуточного пиримидинового
соединения в 2-метил-4-амино-5-бромметилпиримидингидробромид раз-
ными методами требуется следующее количество стадий: методом Вилли-
амса — 3; Греве — 3; Андерзага и Вестфаля—5; Челинцева и Беневолен-
ской — 1. Кроме того, по методу Виллиамса весьма низок выход 2-метил-
4-окси-5-этоксиметилпиримидина (продукта циклизации); по методу Греве
встречаются большие трудности при каталитическом гидрировании циано-
группы в соединении I. Метод Андерзага и Вестфаля весьма сложен и мно-
гостадиен. Следовательно, наиболее эффективным методом синтеза пири-
мидинового компонента является метод Челинцева и Беневоленской [48].
' Остановившись на варианте метода Челинцева и Беневоленской, необхо-
димо отметить, что авторы, применяя для конденсации с ацетамидином
а-ацетоксиметилен-|3-этоксипропионитрил, предложили синтез последнего
из этиленциангидрина путем алкилирования его эфиром муравьиной кис-
лоты в присутствии этилата натрия, свободного от спирта по схеме:
, C2H5ONa
HOCH2CH2CN + НСООС2Н5----------> C2H5OCH2CH2CN —>
Эгилезциангидрин Эгилфэрмиат 65% З-Этоксипропиояитрил
НСООС2Нб СН3СОС1
-----C2H5OCH2CCN > C2H5OCH2CCN
C2H6ONa ||-II
CHONa СНОСОСНз
40% Натрэаолят-а- Хлорис- а-Ацетоксиметилеч-
оксиметилеч-3-эгоксипро- тый 3-этоксипропиолитрил
пионитрил ацетил
72
Выход натрэнолята можно значительно увеличить, если конденсацию
вести в среде акрилонитрила [49]. Последний связывает спирт, выделяю-
щийся в реакции, и образует с ним |3-этоксипропионитрил. Вследствие
этого равновесие в реакции смещается в сторону образования натрэнолята.
Затем было показано [50], что |3-этоксипропионитрил с лучшим выходом
(95%) можно получить при цианэтилировании этилового спирта акрило-
нитрилом в присутствии щелочи:
СН2=СН—CN + С2Н5ОН------------> C2H6OCH2CH2CN
NaOH
Акрилонитрил {З-Этоксипропионитрил
В дальнейшем синтез пиримидинового цикла был усовершенствован
Т. Фодором с сотрудниками [51 ] в связи с применением ими для конденса-
ции с ацетамидином простых эфиров а-оксиметилен-|3-этоксипропионит-
рила вместо сложных. При использовании сложного эфира в реакции кон-
денсации выделяется уксусная кислота, которая связывает одну молекулу
ацетамидина, вызывая непроизводительный расход его. Применяя простой
эфир, как, например, метоксиметилен-[3-этоксипропионитрил, образование
пиримидинового цикла не сопровождается выделением кислоты, а следова-
тельно, для реакции конденсации достаточно брать один моль ацетамидина
на один моль эфира:
NH, СН-ОСОСН,
I \\ 3
н3с-с + с-сн2ос2н5
3 У. ! 2 2 5
NH CN
А цетамидин Метоксиметилен -fl-
зтокси пропионитрил
кон
H3cA>NH2
2-Метил - 4-амино-5-*
этоксиметилпиримидин
Простой эфир метоксиметилен-|3-этоксипропионитрил может быть полу-
чен действием на натрэнолят диметилсульфатом (CH3)2SO4 или монохлор-
метиловым эфиром С1СН2ОСН3.
Далее возникает вопрос о путях синтеза ацетамидина — первого компо-
нента пиримидинового цикла. Для получения ацетамидина необходим аце-
тонитрил, который превращают в ацетамидин путем этерификации его спир-
том с получением ацетоиминоэфира и его аммонолиза. Несмотря на наличие
ряда лабораторных способов получения ацетонитрила, промышленный
способ синтеза не был достаточно разработан [38]. Способ получения
ацетонитрила метилированием цианистого калия применим- лишь в лабо-
раторных условиях [52]. Методы получения ацетонитрила из ацетамида
путем дегидратации его фосфорным ангидридом [53, 54], хлорокисью фосфо-
ра [55] или толуолсульфохлоридом [56] весьма сложны и дороги.
Наиболее перспективным промышленным методом получения ацетонит-
рила является непрерывный каталитический метод. При получении аце-
тонитрила этим методом пары уксусной кислоты взаимодействуют с парами
аммиака при температуре 350° С над катализаторами — окисью алюминия
или двуокисью тория [57], силикагелем [58], смесью фосфорного и кварце-
вого катализатора [101 ]. Реакция протекает по схеме:
Катализатор
СНзСООН + NH3----------> CH3CN + 2Н2О.
350°
Ацетонитрил
В настоящее время этот метод широко применяется в промышленности.
Наконец, необходимо решить вопрос о типе галоида, применяемого для
галогенирования аминопиримидина.. С точки зрения технологической эф-
фективности получение бромпроизводных пиримидина и их конденсация с ти-
азоловым компонентом протекает более успешно и с более высоким выходом,
чем хлорпроизводных. Если целевым продуктом синтеза является тиамин-
бромид, то понятно, что галогенизация аминопиримидина осуществляется
73
бромистым водородом. Если же методом двухкомпонентной конденсации
необходимо получить в качестве товарного продукта — тиаминхлорид,
то логичным является получение хлорпроизводного пиримидина; послед-
ний при конденсации с тиазоловым компонентом образует тиаминхлорид.
Необходимо учесть, что хлор более дешев и менее дефицитен, чем бром,
и технология его применения в газообразном виде более совершенна по срав-
нению с применением брома в жидком виде в стеклянной таре. Из изложен-
ного следует, что наиболее эффективным методом синтеза пиримидинового
компонента явлется метод Челинцева—Беневоленской. Этот метод усовер-
шенствован путем замены исходного сырья — этиленциангидрина на акри-
лонитрил; применяемый для конденсации в 2-метил-4-амино-5-этоксиметил-
пиримидин ацетамидин рационально получать непрерывным каталитическим
способом из аммиака и уксусной кислоты; галогенирование аминопиримидина
должно производиться хлором, если целевым продуктом является тиамин-
хлорид. Принципиальная схема синтеза пиримидинового компонента может
быть изображена следующей схемой:
ch2=ch-cn
нсоос2н5
C2H5ONa
CHONa
%
C2H5OCH-C
CN
(CH3)2SO4
СНОСНз
------ с2н5осн2-хс
CN
NH2
H3C-C
d \\ „ .
______NH X-CH2OC2H5_____ n-^vch2ci
Н3сД> JLnH2 HC1 H3C-^n>NH2-HC1
Выбор метода синтеза тиазолового компонента. Варианты синтеза 4-ме-
тил-5-оксиэтилтиазола можно обобщить по двум основным группам [57].
Первая группа объединяет варианты, созданные на применении тиофор-
мамида. Эти синтезы приводят к построению тиазолового цикла с необхо-
димыми заместителями в одну стадию и базируются на общем методе получе-
ния производных тиазола Ганча, заключающемся во взаимодействии тиа-
мидов кислот с а-галоидированными кетонами по схеме [58, 59]
NH
II
н3с-сх
SH
О=С —R
+ /н2
С1
N----C-R
II II
Н3С-Сх с
SX
К данной группе относятся синтез через сложные эфиры -у-галогено-
рацетопропилового спирта, как, например, при конденсации у-хлор-р-аце-
топропилацетата с тиоформамидом получают ацетилированный тиазол,
который при действии щелочи гидролизуют в 4-метил-5-|3-оксиэтилтиазол
по схеме [60, 61, 62, 63]:
NH N — с-сн3
и и и
СН3-СО-СН-СН2-СН2ОСОСН3+ СН ------------*~НСХ С-СН2СН2ОСОСН3
С1 4SH S
/.лорацетопропипаиетат Тиодзормамид 4 - Метил -5- ацетоксизтилтиазол
NaOH
N3—-7С-СН,
II3 4||
НС\ Лс-снгсн2°н
• S
4 - метил-5-Ji-оксизтилтиазол
Использование в реакции галоидирования рацетопропилового спирта
вместо ацетата резко снижает выход галоидопроизводного. Причиной этого
может явиться способность у-ацетопропилового спирта к таутомерным пре-
вращениям [64], а также, как показано Я. Слободиным и Е. Гельме
[65], при бромировании свободного рацетопропилового спирта происходит
74
частичная изомеризация первичного у-ацетопропилового спирта во вторич-
ный — пентанол-2-он-4. Поэтому было предложено галоидировать не у-аце-
топропиловый спирт, а его эфир —ацетат. Однако, как показано В. Турси-
нымидр. [66, 67], при хлорировании у-ацетопропил ацетата, кроме у-хлор-
у-ацетопропилацетата, образуется а, у-дихлор-у-ацетопропилацетат, трудно
разделяемый при перегонке в вакууме. При хлорировании же у-ацето-
у-бутиролактона получают лишь у-хлор-у-ацетопропилацетат с выходом
60%. Этими же авторами было показано, что при получении тиотиона вы-
ход его увеличивается на 13,3%, если применен для конденсации у-хлор-
у-ацетопропилацетат, полученный хлорированием ацетобутиролактона,
вместо ацетопропилацетата. С этой точки зрения представляет интерес
предложенный Бухманом [68] метод получения ацетобутиролактона и при-
менение его для получения у-хлорацетопропилового спирта и далее для кон-
денсации с тиоформамидом с получением 4-метил-5-|3-оксиэтилтиазола
по схеме:
снэ
со
I +
сн2
COOR.
СН, СН, СНз и,,
I 3 I 3 I 3 СН
ch2xq t со ____со __________со XSH
сн2 сн-сн2 С1-С— СН2 С1-СН
СО СН2 СО ZCH2 СН2СН2ОН
о
Сложный эфир Окись у - Ацетобд -
ацетоуксусной этилена тиролактон
у-Хлор - у-ацето-
пролилоВый спирт
N —С-СН,
II II
СН С~СН2СН2ОН
xs
4 -Метил-5-у-оксиэтилтиазол
Дальнейшее упрощение метода дал Венц [69], который показал, что
у-хлор-у-ацетобутиролактон можно непосредственно конденсировать с тио-
формамидом в тиазоловый компонент по схеме:
NH
и
СН
\н
Тиоформамид
СН3
СО
I
+ С1-С — сн2 ------------
со сн2
'хо/
у-Хлор-у-ацетобутиролактон
N---С-СН,
II II
нс с-сн2сн2он
х8
4- Метил-5-ft-
- оксиэтилтиазол
Упрощение метода позволило исключить процессы омыления с после-
дующим декарбоксилированием у-хлор-у-ацетобутиролактона для получе-
ния у-хлор-у-ацетопропилового спирта, а также ацетилирование спиртовой
группы. Этот вариант метода также создает наиболее благоприятные усло-
вия для реакции хлорирования, а именно: в ацетопропиловом спирте атомом
галоида могут замещаться два атома водорода, находящихся у углеродов,
соседних с карбонильной группой. Между тем в у-ацетобутиролактоне эта
возможность исключена, так как лишь один водород сильно активирован
двумя соседними карбонильными группами. Это подтверждено работой
В. Турсина и сотрудниками [66, 67], где показано важное значение этого
фактора на следующей стадии синтеза.
Дальнейшее усовершенствование внесли Громатка, Жданович, Лис-
нянский [70], которые непосредственно конденсировали формамид с у-хлор-
у-ацетипропиловым спиртом или ацетатом в присутствии пятисернистого
фосфора n<j схеме:
75
nh2 ch3
НС + P c + co ______________
II p2b5 + !
О Cl-CH
I
CH2CH2OH
Нормами В у -Хлор -у - ацето пропи-
ловый спирт
N----С-СН3
II II
нс с-сн2сн2он
sz
4 - Метил -5 -J3- оксиэтил -
тиазол
Ко второй группе вариантов относятся синтезы тиазола через его про-
изводные, замещенные в положении 2, например, пуРем конденсации
а-галоидированных альдегидов или кетонов с роданидами металлов по
схеме:
СН3
С = О + NCSMe —Ju f-CH3
I , HO-C XC-R
C1-CH-R S к
В приведенной схеме R = — СН2СН2ОСОСН3 или — СН2ОСОС2Н6.
Такой синтез тиазола был предложен Андерзагом и Вестфалем [71].
Он заключается в следующем. у-Ацетопропилацетат бромируется и конден-
сируется с роданистым барием, образуя у-тиоциано-у-ацетопропилаце-
тат. Последний в кислой среде перегруппировывается в 2-гидрокси-4-метил-
5-ацетоксиэтилтиазол. Гидроксил в положении 2 при помощи хлорокиси
фосфора превращается в хлор, а хлорид действием цинка и уксусной кисло-
ты превращается в 4-метил-5-ацетоксиэтилтиазол, который при омылении
дает 4-метил-5-|3-оксиэтилтиазол по следующей схеме:
сн3. сн3 сн3
СО + Вг СО BafCNSj N СО СН3СООН
СН2 Вг-СН *~С СН-СН'2СН2ОСОСН3
СН3СН2ОСОСН3 СН2СН2ОСОСН3 * 4sz
у - Ацето пропил ацетат
у- Тиоциано -у- ацетопропилацетат
N.—;С—СН3
II3 4||
НО-С2 (5.С-СН2СН2ОСОСН3
S
2-Гидракси-4 -метил-5-ацетоксиэтилтиазол
РОС1
N—С-СН3
С1-С .с-сн2сн2ососн3
4S
2-Хлор-метил-5-ацетоксиэтилтиазол
N—С-СН,
_______ II II
НС уС—СН2СН2ОСОСН3
У
4 - Метил -5 - ацетоксиэтилтиазол
izr^L-i N С СН*
конг II II
НС С-СН2СН2ОН
sz .
4 - Метил -5 -J3- оксиэтилтиазол
Метод Андерзага и Вестфаля является многостадийным и, кроме того,
стадия циклизации и образования 2-гидрокси-4-метил-5-ацетоксиэтилтиа-
зола дает малый выход продукта (8-ж-10%). Всесоюзный научно-исследова-
тельский химико-фармацевтический институт (ВНИХФИ) разработал ана-
логичную схему синтеза тиазолового компонента:
76
N---C-CH3
nh2csnh4 +
CH3COCHCH2CH2OCOCH3
Дитиокарбамат
аммония
Н2О2
С1
Метил -с£ - хлор - у-ацетоксипропилкетон
N— С-СН3
II II
НС1,ВаС12 нс с-сн2сн2ососн3
4 -Метил -5-fl-au,emo-
ксизтилтиазол
Окисление
NaOH
hs-c с-сн2сн2ососн3
S
2 - Меркапто -4- метил -
5 - ацетоксиэтилтиазол
N----С-СН3
II 3
нс сн2сн2он
А
4 - Метил - 5 -fl-оксиэтил тиазол
Недостаток вариантов второй группы синтеза тиазола заключается в
том, что при конденсации исходные компоненты дают тиазоловое соедине-
ние, замещенное в положении 2 функциональной группой (ОН, HS). Элими-
нирование этой группы значительно усложняет синтез. Наиболее короткий
путь синтеза тиазолового компонента — получение при конденсации про-
дукта, незамещенного в положении 2, что достигается при применении
для конденсации тиоформамида.
Таким образом, исследования Бухмана, Венца, Турсина, применивших
для конденсации с тиоформамидом у-хлор-у-ацетобутиролактон, а также
Громатки и Лиснянского, исключивших процесс выделения нестойкого
тиоформамида как самостоятельного полупродукта, привели к следующей
химической схеме получения 4-метил-5-|3-оксиэтилтиазола [66—70]:
сн3
со
I ।
NH С1-С--СН, N—С-СН3
II . I I II II NaOH
сн —со сн2—*-нс С-СН2СН2ОСОСН3----*-
Ан
Тиосрормамид
у -Хлор -у-ацетобутиро-
лактон
4 -Метил-5-ДЗ-аиртоксизтилтиазол
N—С-СН3
Il II
нс с-сн2сн2он
4- Метил-5-^'0ксизтилтиазол.
Вместо у-хлор-у-ацетобутиролактона может быть применен 2-метил-
2-алкокси-З-хлортетрагидрофуран [71а]. Предложено также применение
для конденсации с тиоформамидом 2-метил-2,3-дихлортетрагидрофурана
[94]. Вследствие термолабильности тиоформамида авторы применили рас-
твор тиоформамида в дихлорэтане, в котором он довольно стоек.
Синтез 4-метил-5-|3-оксиэтилтиазола осуществлен в присутствии муравьи-
ной кислоты и формиата натрия с выходом 68,1%. Температура кипения
120—122° С (при остаточном давлении 3 мм рт. ст, ); = 1,5483; темпе-
ратура плавления пикрата— 164° С; УФ-спектр—Хтах=250 нм; ИК-спектр:
1540, 1410, 1380, 1050, 920, 855, 780 см-1; содержание — 95—97%.
Выбор метода конденсации пиримидинового и тиазолового компонентов,
целевой продукт и технология его получения. Заключительной стадией
двухкомпонентного синтеза является конденсация 2-метил-4-амино-5-хлор
(бром) метилпиримидина (I) и 4-метил-5-|3-оксиэтилтиазола (II) по схеме:
N^j]-CH2Cl(Br)
н3сА Anh2
3 N
”ТСНз
А- сн2сн2он
и
_ /-сн2\сС
N [Г CH3
h3cA A , A Ach2ch2oh
3 N NH2-HCl(HBr) S
111
Конденсацию можно осуществить сплавлением обоих компонентов при
температуре 100—120° С [54]. Однако несколько лучшие результаты дости-
гаются при проведении этого процесса в среде органических растворителей,
как, например, бутанола [20], ацетонитрила [72], бромоформа [73],
77
|3-этоксипропионитрила [74] и керосина (Волкова, Лейбина). Однако указан-
ные растворители также недостаточно эффективны для процесса конденса-
ции: в среде бутанола возможна побочная реакция между галогенметилпи-
римидиновым компонентом и спиртом с образованием алкооксиметиламино-
пиримидина; керосин трудно регенерировать; остальные указанные раство-
рители дефицитны и дороги. По-видимому, более рациональным являются
такие растворители, как толуол или ксилол.
Далее возникает вопрос о целевом продукте синтеза — тиаминбромид
или тиаминхлорид (III). Как известно, в СССР широко развито производст-
во тиаминбромида, а за рубежом производится исключительно тиаминхло-
рид. К сожалению, в медицинской литературе мы не встречаем какие-либо
экспериментальные данные, которые свидетельствовали бы о преимущест-
вах одной формы тиамина над другой. Предполагая тождественную физио-
логическую ценность обеих форм тиамина, необходимо все же отметить
следующие технологические и качественные преимущества тиаминхлорида
перед тиаминбромидом:
в связи с большей реакционной способностью бромпроизводных нежели
хлорпроизводных тиаминхлорид должен быть более стойким при хранении
нежели тиаминбромид;
по биологической активности объем продукции тиаминбромида в соот-
ветствии с большим молекулярным весом в 1,3 раза выше, чем тиаминхло-
рида; это влечет за собой увеличение объема тары, затрат на расфасовку,
упаковку и на транспорт;
бром по сравнению с хлором является дефицитным и более дорогим про-
дуктом;
технологическое применение брома в связи с расфасовкой его в мелкую
стеклянную тару значительно сложнее, нежели применение газообразного
хлора;
природной формой тиамина все же является не бромидная, а хлоридная
и надо полагать, что для профилактических целей систематическое
применение бромистой соли тиамина и накопление брома в организме вряд
ли целесообразно; в то же время соли хлора естественны для организма.
Исходя из изложенного, следует признать, что наиболее эффективной
формой целевого продукта синтеза является тиаминхлорид. Это, конечно,
не исключает производство также некоторого количества тиаминбромида,
который, по-видимому, для лечебных целей в некоторых случаях может
более успешно использоваться, чем тиаминхлорид. Что же касается тиамин-
мононитрата, то его применение для витаминизации хлеба оправдано более
высокой стабильностью его при хлебопечении, а также важной физиологи-
ческой ролью азотистых соединений в организме.
Остановившись на тиаминхлориде, как на более эффективной форме
тиамина, следует обсудить вопрос о наиболее целесообразных методах его
получения при синтезе тиамина способом двухкомпонентной конденсации.
Казалось бы, что самым эффективным должен быть метод непосредствен-
ной конденсации 2-метил-4-амино-5-хлорметилпиримидина с 4-метил-5ф-ок-
сиэтилтиазолом [57, 75, 76]. Однако в литературе появились указания [77],
что в этих условиях выход тиаминхлорида в реакции конденсации снижается
на 30% по сравнению с выходом тиаминбромида; известно также, что конден-
сация бромпроизводных протекает более успешно, чем хлорпроизводных
[64]. При этом было высказано мнение [77], что низкий выход тиамина
при конденсации хлорпроизводных обусловлен тем, что часть 4-метил-5ф-ок-
сиэтилтиазола расходуется на связывание хлористого водорода, которого в
хлораминпиримидине содержится больше Одного моля. По-видимому, вслед-
ствие причин, указанных выше, исследователи считали более перспектив-
ным получение тиаминхлорида через тиаминбромид с применением обмен-
ных реакций, как, например, при действии -на тиаминбромид хлористым
серебром [19]; при действии хлора в присутствии органических адсорбен-
тов брома (пиридина, фенола) [78]; при действии хлористого кальция в
78
среде метилового спирта [79]; обработкой тиаминбромида безводным хло-
ристым водородом в растворе низшего алифатического спирта [80].
Более сложным и громоздким является метод получения тиаминхлори-
да из тиаминбромида через промежуточное соединение—тиаминроданат [81 ].
В водный раствор тиаминбромида загружают роданистый аммоний и
при температуре 60° С водным аммиаком доводят pH до 9,0, кристалли-
зуют и высушивают. Полученный тиаминроданат растворяют в 7%-ном НО,
очищают активированным углем и пропускают через колонку со специально
подготовленным анионитом. В колонке происходит ионный обмен CNS на
С1 и в элюате получают водный раствор тиаминхлорида, который выделяют
очисткой углем, сгущением в вакууме и кристаллизацией. Преимущества
данного метода заключаются в его высоком эффекте очистки тиамина от
посторонних примесей в результате выделения его через промежуточное со-
единение тиаминроданат. В практике нашел применение способ превраще-
ния тиаминбромида в тиаминхлорид через тиаминроданат и ионный обмен,
хотя более технологичным является метод превращения бромида в хлорид
реакцией обмена в среде метилового спирта или фенола, насыщенных хло-
ристым водородом. Однако вопрос о способе осуществления реакции обмена
брома на хлор в бромаминопиримидине требует дополнительных технико-
экономических исследований. Во всяком случае решение этого вопроса в
пользу метода через тиаминроданат является преждевременным. Все же с
точки зрения рациональной технологии производства получение тиамин-
хлорида методом двухкомпонентной конденсации будет в перспективе
решаться в пользу непосредственной конденсации хлоргидрата 2-метил-
4-амино-5-хлорметилпиримидина с 4-метил-5-|3-оксиэтилтиазолом [75, 76].
Переходим к рассмотрению второго конкурирующего метода синтеза
тиамина.
МЕТОД СИНТЕЗА, ПРЕДУСМАТРИВАЮЩИЙ ПОСТРОЕНИЕ МОЛЕКУЛЫ
ТИАМИНА НА ПИРИМИДИНОВОМ ЦИКЛЕ
Этот метод синтеза исходит из основного полупродукта 2-метил-4-ами-
но-5-аминометилпиримидина (диамин). Однако последующие стадии синте-
за, хотя имеют различные варианты, но главных направлений синтеза су-
ществуют два: через 2-метил-4-амино-5-тиоформаминометилпиримидин (I)
Н3С
CH2xnh
НС^
N NH2
1
и через (2-метил-4-амино-5-пиримидилметил)-дитиокарбамат аммония (II)
N^XxCH2xNH
Нзс4 ILnh2,cx/nh4
S S
11
Рассмотрим оба направления синтеза.
Соединение I получают по двум вариантам:
непосредственно путем конденсации диамина с дитиоформиатом калия в
водном растворе [82—84]:
. H<i __ jn:CH2>H
v н3с—Anh2 СН
s N Ws
79
через формильное производное диамина с последующей обработкой его
пятисернистым фосфором [85—87].
ch2nh2
h3c-^n^nh2
Соединение I при взаимодействии с эфиром у-хлор-у-ацетопропилового спир-
та в присутствии минеральной кислоты превращают в эфир тиамина; по-
следний гидролизом переводят в тиамин по следующей схеме:
СН.
N^iT NH
НзсЧ^Н2 сн
N
S
СН3
со
+ I ---
С1-СН
I
ch2-ch2or
сн3 „ ______
ch2ch2or
СН2
н\ "N—|Т-СН3 .
н3с >-nh2 >-сн2сн2он
3 N S
Необходимо однако отметить, что данное направление синтеза тиамина
является мало перспективным из-за низкого выхода тиамина на стадии кон-
денсации, что, по-видимому, связано с лабильностью соединения I, вслед-
ствие легкого отщепления от него тиоформильной группы.
Более перспективным является второе направление синтеза через соеди-
нение II. Этот метод технически разработан и освоен в промышленном мас-
штабе в Японии [43, 88—90] и в некоторых других странах. Сущность ме-
тода заключается в воздействии на диамин аммиаком и сероуглеродом и
образовании (2-метил-4-амино-5-аминометилпиримидил)-дитиокарбамат ам-
мония; последний при конденсации с "[-хлор-рацетопропилацетатом и при
нагревании с соляной кислотой превращается в дихлорид меркаптотиамин,
а при обработке его щелочью получают тиотиамин. Последний окислением
перекисью водорода в среде минеральной кислоты превращают в соответ-
ствующую соль тиамина [911 по следующей схеме:
о=с-сн3
NYt~ch2NH2 CS2+NH3, Г4^г/СН2~'^|н C1-CH~CH2-CH2OR.
Н,сЛ^ Л-nh, ^н3с-к ^-nh2 C zNH4 R=COCH3; H
3 N 3 N S* S
Z-Метил - 4-амино-5-амино- 2-Метил-Ь-амино-5-аминометилпири- у-Хлор-у-ацетопролил-
мвтилпиримидин (диамин) мидин-дитиокардамат аммония ацетат
С1"
________ NH ОС-СН3 НС1. N if N п-СН3 _
НзС-к т^с^ /СН-сн2— сн2од н3с-к Л Лсн2-сн,он
IN 11 ling iic*i о
of-Ацетопси -у - ацетопропил-N- Дихлоридмврпаптотиамин
—(2 -метил - it-амино -5-аминометилпиримидин) -
дитиопардамат
N-(2 -метил -4- амино-5-аминометилпиримидин) - Тиаминхлорид
4 - метил -5-ft- оксизтил -2 - тиотиазолон
(тиотиамин)
80
Таким образом, второй метод синтеза, предусматривающий построение
молекулы тиамина на пиримидиновом цикле, оказался маловариантным и
получил промышленное применение в некоторых странах. Он осуществля-
ется по схеме, изложенной выше.
Далее рассмотрим технологические схемы производства тиаминхлорида
по обоим методам и их наилучшим, по нашему мнению, вариантам.
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА ТИАМИНХЛОРИДА МЕТОДОМ
КОНДЕНСАЦИИ ПИРИМИДИНОВОГО И ТИАЗОЛОВОГО ЦИКЛОВ
(ВАРИАНТ I)
Пиримидиновый цикл. Пиримидиновый цикл включает синтез следую-
щих продуктов:
Р-этоксипропионитрила из акрилнитрила, спирта и едкого натра;
этилформиата из этилового спирта и муравьиной кислоты;
а-метоксиметилен-р-этоксипропионитрила из акрилнитрила, диметилсу-
льфата и этилформиата;
ацетонитрила из уксусной кислоты и аммиака в присутствии катализа-
тора;
ацетоиминоэфира из ацетонитрила и хлористого водорода;
хлористоводородной соли ацетамидина (хлоргидратацетамидина) из
ацетоиминоэфира и спиртового раствора аммиака;
2-метил-4-амино-5-этоксиметилпиримидина (аминопиримидина) из хлор-
гидрата ацетамидина и а-метоксиметилен-р-этоксипропионитрила;
хлоргидрата 2-метил-4-амино-5-хлорметилпиримидина (хлораминопи-
римидина) из аминопиримидина и хлорводорода.
Тиазоловый цикл получения тиаминхлорида состоит из нескольких ста-
дий синтеза:
"[-ацетобутиролактона;
хлорацетопропил ацетата из "[-ацетобутиролактона и хлора;
формамида из этилформиата и аммиака;
4-метил-5-р-оксиэтилтиазола из формамида, пятисернистого фосфора
(тиоформамида) и хлорацетопропилацетата.
Тиаминхлорид получают путем конденсации хлораминопиримидина и
4-метил-5-|3-оксиэтилтиазола.
Р-ЭТОКСИПРОПИОНИТРИЛ [92]
Реакция получения
NaOH
СН2=СН—CN + С2Н6ОН-------> C2H6OCH2CH2CN
Акрилонитрил Спирт (З-Этоксипропионитрил
53,05 46,07 99,13
Акрилонитрил загружают в реактор 1 (рис. 9), снабженный обратным
холодильником из нержавеющей стали, туда же вводят из мерника 2 этило-
вый спирт, а из сборника 3 порошкообразный едкий натр. Реакцию прово-
дят при температуре 50° С. Затем массу нейтрализуют уксусной кислотой
до pH 6,0—6,5 и перегоняют в аппарате 4 с холодильником 5. В приемник 6
собирают фракцию, кипящую при температуре 170—173° С. Остальные
фракции поступают в сборник 7, откуда их вторично направляют на раз-
гонку. Процесс получения p-этоксипропионитрила усовершенствован путем
непрерывного егб осуществления в тонкопленочном реакторе [94, 95].
Реакцию цианэтилирования осуществляют в пленке в присутствии 0,5—•
1,0% этилата натрия при температуре 45—50° С. Достигнут выход в 95%
(в пересчете на акрилонитрил). Реактор снабжен многорядной лопастной
мешалкой и нагревательной рубашкой для воды.
Р-Этоксипропионитрил, молекулярная масса 99,13, представляет собой
бесцветную жидкость со специфическим запахом, температура кипения
81
170—173° С (при остаточном давлении 13 мм рт. ст. температура кипения
63° С).
ЭТИЛ ФОРМИАТ (ЭТИЛОВЫЙ ЭФИР МУРАВЬИНОЙ КИСЛОТЫ)
Реакция получения
H2SO4
нсоон + СаН5ОН-------> НСООС2Н5 + Н2О.
Муравьиная 46,07 Этилформиат
кислота 74,08
4ф03
В ректификационный (из эмалированной стали) аппарат 8 с мешалкой,
снабженный дефлегматором 9 и конденсатором 10 загружают-из мерника 11
муравьиную кислоту, из мерника 12 — этиловый спирт и из мерника 13 —
серную кислоту. Смесь нагревают до кипения при перемешивании и при
включенном на полный возврат флегмы дефлегматоре кипятят 40—45 мин.
Затем включают конденсатор и отгоняют в сборник 14 при температу-
ре 54—56° С этилформиат. Разработан также непрерывный процесс произ-
водства этилформиата с применением силикагелевого катализатора [93].
Этилформиат, молекулярная масса 74,08, летучая бесцветная жидкость
с эфирным запахом, температура кипения 54,5° С, плотность 907,8 кг/м3.
НАТРИЕВАЯ СОЛЬ я-ОКСИМЕТИЛЕНф-ЭТОКСИПРОПИОНИТРИЛА [48]
(НАТРЭНОЛЯТ)
Реакция получения
Na
С2н6—О—СН2— СН2—CN + НСООС2Н5 —>- С2Н5ОСН2—С—CN + С2Н5ОН
II
CHONa
(З-Эгоксипропионитрил Этилформиат Натрэнолят-а-оксиметилен-
99,12 74,08 (3-этоксипропионитрил 149,12
Этилформиат конденсируют с |3-этоксипрбпионитрилом по типу сложно-
эфирных конденсаций Клайзена в присутствии металлического натрия.
В реактор из нержавеющей стали 15 приливают из мерника 17 толуол
и добавляют из сборника 16 нарезанный кусочками металлический натрий.
Содержимое реактора охлаждают до 15° С, а затем постепенно (в течение
3 ч) добавляют смесь p-этоксипропионитрила и чистого этилформиата. По-
следнюю готовят в реакторе 18, куда подают fl-этоксипропионитрил из мер-
ника 19, а этилформиат —• из мерника 20. Реакционную массу перемешива-
ют 2—3 ч при температуре 20—22° С (подогревают теплым керосином),
а затем выдерживают 10—12 ч. Потом массу фильтруют1 на нутч-фильтре 21.
Осадок промывают сухим толуолом из мерника 17 и высушивают в ва-
куум-сушилке 22. Продукт поступает в сборник 23. Хранение его не допус-
кается. Натрэнолят, молекулярная масса 149,12, порошок желтоватого
цвета, хорошо растворим в воде, хуже в метиловом и этиловом спиртах,
не растворяется в органических растворителях — бензоле, толуоле и др.
я-АЦЕТОКСИМЕТИЛЕНф-ЭТОКСИПРОПИОНИТРИЛ (АЦИЛЭНОЛЯТ)
*
Реакция получения
C2H6OCH2CCN + С1СОСН3 —► C2H5OCH2CCN + NaCl
II II
CHONa СНОСОСН3
Натрэнолят а-окси- Хлористый а-Ацетоксиметилеи-(3-этоксипро-
метилен-р-этокси- ацетил пионитрил
пропионитрил 78,50 169,18
149,12
1 Существует мнение о нецелесообразности выделения натрэнолята из реакцион-
ной массы при ацилировании или метилировании его.
82
В реактор 24 загружают порошкообразный натрэнолят из сборника 23
и толуол из мерника 25, перемешивают и постепенно добавляют из мерни-
ка 26 хлористый ацетил. После охлаждения массу фильтруют через нутч-
фильтр 27 для отделения хлористого натрия, который промывают толуо-
лом. Фильтрат направляют в вакуум-перегонный аппарат 28. Сначала от-
гоняют толуол в сборник 29, затем ацилэнолят в сборник 30 при остаточном
давлении 22 мм рт. ст. и температуре 122—144° С.
Ацилэнолят представляет собой бесцветную жидкость, трудно раство-
римую в воде, смешивающуюся с обычными органическими растворителями,
плотность d15= 1069 кг!м3, п^= 1,460; при хранении заметно не изменяется.
Для синтеза пиримидинового компонента вместо сложного эфира а-аце-
токсиметилен-|3-этоксипропионитрила могут быть применены с успехом
простые эфиры а-оксиметилен-|3-этоксипропионитрила [51], т. е. оксимети-
леновая группа в Na-эноляте может быть защищена, помимо ацетильного
остатка, другими группами: метоксиметиленовой — действием хлорметило-
вого эфира, или алкильной [96] —действием эфиров серной кислоты (ди-
метилсерной кислоты)
' С1СН2ОСН3---► c2h5och2-c-cn
C2HSO-CH2- C-CN СНОСН2ОСН3
II +
CHONa
(CH3)2SO4 ---------------*~с2н5осн2—c—cn
CHOCH-j
При применении простого эфира уменьшается расход ацетамидина, так
как при конденсации последнего с простым эфиром не образуется кислота,
а потому можно применять ацетамидин в эквимолекулярном количестве.
Для усовершенствования синтеза пиримидинового цикла проводились ис-
следования [96] в условиях осуществления реакций получения в одном
аппарате натриевой солиа-оксиметилен-[3-этоксипропионитрила, затем ц-ме-
токсиметилен-|3-этоксипропионитрила и, наконец, 2-метил-4-амино-5-эток-
симетилпиримвдина (без выделения промежуточных продуктов). Устано-
влено влияние на выход аминопиримидина следующих факторов:
скорости всех реакций и непрерывности процесса формилирования |3-эток-
сипропионитрила;
температуры формилирования (не выше 26° С), метилирования ц-эноля-
та при pH 5,5—6,0 (около 40° С) и конденсации метильного производного
с ацетамидином (55—65° С) [104];
интенсивности перемешивания реакционной массы.
АЦЕТОНИТРИЛ
Способ получения ацетонитрила метилированием цианистого калия [52]
применим лишь в лабораторных условиях [38]. Получение ацетонитрила
из ацетамида путем дегидратации его фосфорным ангидридом [53, 54],
хлорокисью фосфора [97] или толуолсульфохлоридом [98] сложен и дорог.
Наиболее перспективным промышленным методом получения ацетонит-
рила является каталитический метод, при котором пары уксусной кислоты
взаимодействуют с парами аммиака при температуре 350—500° С над ката-
лизаторами — окисью алюминия, двуокисью тория [99], силикагелем [100],
фосфорной кислотой на силикагеле [101, 102].
83
Реакция получения
СН3СООН + NH3 —с CH3COONH4
Уксуснокислый
аммоний
Каталитическая
CH3COONH4------------> CH3CN + 2Н3О;
Дегидратация Ацетонитрил
СН3СООН + NH3 —> CH3CN + 2Н3О
60,05 17,03 41,05
Из мерника 31 уксусная кислота поступает в контактную печь, имею-
щую три секции: испаритель уксусной кислоты 32, аммиачную печь 33 и
каталитическую печь 34. Последнюю загружают кольцами Рашига и фос-
форным катализатором. В испарителе поддерживают температуру 150° С.
Здесь происходит процесс превращения жидкой уксусной кислоты в пар.
В аммиачной печи температуру повышают до 500° С, а в каталитической
печи поддерживают 300—350° С. Аммиак из баллонов 35 поступает в печь
и подогревается. Таким образом, в каталитическую секцию печи поступают
пары уксусной кислоты и газообразный аммиак. Продукты реакции посту-
пают в холодильник 36, охлаждаемый рассолом, конденсируются и посту-
пают в сборник 37; конденсат поступает в смеситель 38 для нейтрализации
соляной или уксусной кислотой. Нейтрализованную массу переводят в
делительную воронку 39, нижний слой сливают, а верхний подсушивают в
воронке с поташом. Высушенный ацетонитрил поступает в сборник 40.
Ацетонитрил, молекулярная масса 41,05, представляет собой бес-
цветную жидкость с температурой кипения 81,5° С при давлении
760 мм рт. cm., df=0,7828, «^=1,3442. Ацетонитрил хорошо растворяет
многие неорганические соли, а также лаки, смолы, жиры, масла. Ацетони-
трил хорошо растворяется в воде и в спирте.
ХЛОРГИДРАТ ЭТИЛОВОГО АЦЕТОИМИНОЭФИРА [44, 103]
Реакция получения
^NH • НС1
CH3CN + С2Н6ОН + НС1 —» СН3С
ХОС2Н5
Ацетонитрил Хлоргидрат ацетоиминоэфира
41,05 123,59
При производстве хлоргидрата ацетоиминоэфира процесс ведут непре-
рывно (И. Лиснянский).
В колонку 41 из эмалированной стали, снабженную рубашкой для ох-
лаждения, помещают насадку из колец Рашига, из мерников 42 и 43 непре-
рывно подают ацетонитрил и абсолютный этиловый спирт. Снизу из генера-
тора непрерывно поступает газообразный хлористый водород. При выходе
из колонки газ поглощается водой в ловушке 44. Насыщение хлористым
водородом ведется до концентрации НО 37—40% * при температуре 14—
18° С. Реакционная масса поступает в кристаллизатор 45, где она кристал-
лизуется при температуре минус 2—3°С. Газообразный хлористый водород
получают либо контактным методом из элементов, либо в генераторе 46,
куда вводят серную кислоту из мерника 47 и соляную кислоту из мерни-
* По данным А. Шерешевского и И. Лиснянского, при содержании общего хлора
в смеси, выходящей из колонны, более 37% ацетоиминный эфир не кристаллизуется.
84
ка 48\ газ пропускают через предохранительную туриллу 49. Из кристал-
лизатора 45 масса поступает на друк-фильтр 50, на котором отфильтровы-
вают ацетоиминоэфир, промывают и размалывают в дробилке 51.
Хлоргидрат этилового ацетоиминоэфира, молекулярная масса 123,59,
представляет собой белое кристаллическое вещество, хорошо растворимое
в воде, метиловом и этиловом спирте. Кристаллы гигроскопичны и на воз-
духе расплываются.
Ацетоиминоэфир легко разлагается в присутствии влаги, спирта, а также
при хранении, давая различные побочные продукты.
ХЛОРГИДРАТ АЦЕТАМИДИНА
Реакция получения
NH • НС1 NH • НС1
СНз—С -j- NH3 * > СН3—С -f- С2Н5ОН
I I
ОС2Н5 nh2
Хлоргидрат ацетон- Хлоргидрат ацетамидина
миноэфира 94.56
123,59
Хлоргидрат ацетамидина получают при взаимодействии хлоргидрата
ацетоиминоэфира и 10%-ного спиртового раствора аммиака при температуре
10°С. Для этого в реактор 52 загружают из мерника 53 абсолютный спирт,
а из баллона 54 подают аммиак. Насыщение спирта аммиаком ведут до
10%-ной концентрации. Затем спиртовый раствор аммиака спускают в ре-
актор 55, охлаждают рассолом до 10°С и постепенно добавляют в него ацетои-
миноэфир. Реакционную массу перемешивают 6 ч и на друк-фильтре 56,
снабженном паровой рубашкой, отфильтровывают осадок хлористого ам-
мония. Этот осадок тщательно промывают абсолютным спиртом при нагре-
вании и перемешивании. Промывка производится 3—4 раза для полного
извлечения хлоргидрата ацетамидина. Фильтрат и спиртовые промой по-
ступают в сборник 57, а затем в вакуум-аппарат 58 для отгонки спирта,
который собирают в приемнике 59, откуда его направляют на повторное
использование в мерник 53. Упаренную массу направляют в кристалли-
затор 60, где в течение 4 ч кристаллизуют при 0°. Кристаллы отфильтровы-
вают в центрифуге 61, промывают спиртом и высушивают в вакуум-сушил-
ке 62 при температуре 30—40° С. Маточный раствор из центрифуги
поступает в сборник 63, а далее в вакуум-аппарат 64, где его под вакуумом
упаривают, затем кристаллизуют в кристаллизаторе 65 и фильтруют в
центрифуге 66. Кристаллы ацетамидина II после растворения поступают
на повторную перекристаллизацию в вакуум-аппарат 58. Маточный
раствор II поступает в сборник 67 и является отходом производства.
Хлоргидрат ацетамидина, молекулярная масса 94,56, представляет со-
бой белое кристаллическое гигроскопическое вещество с температурой
плавления 166—167° С, легко растворяющееся в воде и спирте.
2-МЕТИЛ-4-АМИНО-5-ЭТОКСИМЕТИЛПИРИМИДИН (АМИНОПИРИМИДИН)
Реакция получения
Аминопиримидин получают конденсацией а-ацетоксиметилен-|3-этокси-
пропионитрила с ацетамидин-основанием. Ацетамидин-основание полу-
чают при действии на солянокислый ацетамидин этилата натрия. Реакции
протекают по следующим уравнениям:
85
^NH-HCl
1. сн3-с^
ЧШ
Хлоргидрат ацетамидина
9k,56
,NH
+ C2HsONa, CH3-C^ + NaCl + C2HSOH
Ацетамидин - основание
58,08
NH
и
2. H3C-C +
nh2
А цетам ид ин - основание
58,08
сн-ососн3
4C-CH2-OC2HS
CN
а-Ацетоксиметилен -
J3 - зтоксипропионитрил
169,18
sj]-CH2OC2Hg
H3C-C4.34c-NH2
N
2 - Метил - 4 - амино - 5-
этоксиметилпиримидин
167,20
В реактор 68 загружают солянокислый ацетамидин, из сборника 63
прибавляют абсолютный спирт в соотношении 1:2,5, массу нагревают до
полного растворения ацетамидина. Затем приливают из сборника 70 спир-
товой раствор этилата натрия из расчета 0,6 кг на 1 кг ацетамидина. Массу
перемешивают 10—15 мин, охлаждают до 35° С и фильтруют через друк-
фильтр 71 для отделения осадка поваренной соли. Осадок промывают спир-
том, а фильтрат (ацетамидин-основание) поступает в сборник 72.
В реакционный аппарат 73, снабженный холодильником, из сборника 30
вводят ацилэнолят, а из мерника 74 — толуол и перемешивают. К сме-
си приливают из сборника 72 ацетамидин-основание, перемешивают в те-
чение 1 ч и выдерживают 20 ч. Затем отгоняют растворитель в сборник 75
при остаточном давлении 100—150 мм рт. ст. Спирто-толуольную смесь
разделяют в делительной воронке 76, толуол поступает в сборник 77,
спирт — в сборник 78, откуда они далее идут на регенерацию.
Кубовый остаток растворяют в 10 %-ной щелочи в реакторе 79 и нагре-
вают 2,5 ч при температуре 100° С. Затем масса поступает в кристаллизатор
30, где после охлаждения до 20° С из мерника 81 вводят 50%-ную щелочь
и высаливают аминопиримидин, в виде маслянистой жидкости, которая
быстро кристаллизуется. Массу фугуют в центрифуге 82. Щелочной маточ-
ник является отходом производства. Кристаллы аминопиримидина посту-
пают на перекристаллизацию в растворитель 83, куда вводят из мерника 74
толуол. Раствор фильтруют через друк-фильтр 84, кристаллизуют в кристал-
лизаторе 85, фильтруют через друк-фильтр 86. Кристаллы аминопиримиди-
на I направляют на хлоргидрирование. Маточный раствор из сборника
поступает на выпаривание в вакуум-аппарат 87. Сгущенный раствор крис-
таллизуют в аппарате 88, фильтруют в центрифуге 89. Кристаллы амино-
пиримидина II возвращают на перекристаллизацию в растворитель 83.
Маточный раствор II поступает из центрифуги в сборник 90 и является от-
ходом производства.
Аминопиримидин, молекулярная масса 167,08, представляет собой бес-
цветные игольчатые кристаллы с температурой плавления 89—90° С, рас-
творимые в воде и в органических растворителях.
Низкий выход 2-метил-4-амино-5-этоксиметилпиримидина (около 30%
на |3-этоксипропионитрил), по-видимому, вызывается тем, что оксиметиле-
новые производные существуют в цис-н транс-изомерных формах
ROCH2—С—CN
II
Н—С—OR
ROCH2—С—CN
и II
RO—С—Н
цис транс
Экспериментально установлено [51 ], что только цис-форма способна
образовывать пиримидиновое ядро. По данным Н. Золотарева и др. [104],-
достигнуто повышение выхода аминопиримидина до 44,2% за счет усовер-
шенствования технологии.
.86
ХЛОРГИДРАТ 2-МЕТИЛ-4-АМИНО-5-ХЛОРМЕТИЛ-ПИРИМИДИНА
(ХЛОРАМИНОПИРИМИДИН)
При действии хлористого водорода на аминопиримидин в среде абсолют-
ного этилового спирта происходит замена этоксигруппы в положении 5 на
хлор с образованием хлоргидрата. Реакция протекает по следующему урав-
нению:
N<^-CH2OC2H5
Н3сЛ'/-М12
з N 2
2 - Метил - k - амино -
5 - зтоксиметилпиримидин
167,20
+ 2НС1
n^>-ch2ci
h3cA-nAnh2- hci
Хлорзидрат 2 - метил -к- амино -
5-хлорметилпиримидин
19k, 07
В реактор 91 загружают из мерника 92 абсолютный спирт и в течение
10—12 ч пропускают в реактор сухой хлористый водород, получаемый на
специальной установке контактным методом из элементов (хлора и водоро-
да). Насыщение хлорводородом продолжают, пока концентрация НС1 в
спирте достигает 24—25%. Затем из сборника 93 загружают в реактор 91
аминопиримидин и продолжают пропускать НО. Температуру реакцион-
ной массы медленно повышают до 65—70° С при непрерывном и энергичном
насыщении НС1 в течение 16—20 ч. Насыщение прекращают по анализу
контрольной пробы кристаллов (температура плавления около 200° С, либо
процент содержание хлора — около 36%). Реакционную массу охлаждают
до 5—10° С и после выдержки в течение 5—6 ч фильтруют через друк-
фидьтр 94. Кристаллы промывают спиртом, тщательно отжимают и во влаж-
ном виде направляют на следующую стадию в сборник 95. Маточный раствор
направляют в сборник 96, мерник 97 и далее в реактор 98 для насыщения
НС1 и в друк-фильтр 99. Кристаллы хлораминопиримидина II используют-
ся на следующей стадии совместно с кристаллами I. Маточный раствор в
сборнике 100 является отходом производства.
Хлоргидрат 2-метил-4-амино-5-хлорметилпиримидин, молекулярная
масса 194,07, представляет собой кристаллы белого цвета, температура
плавления около 200° С, хорошо растворимы в воде, плохо в органических
растворителях. Содержание хлораминопиримидина—96—97%, общее со-
держание хлора — 36,0—36,5%.
7-АЦЕТО-7-БУТИРОЛАКТОН
При действии окиси этилена на ацетоуксусный эфир в среде метанола в
присутствии метилата натрия образуется у-ацето-у-бутиролактон. Реакция
протекает по следующему уравнению:
СН3
I
с=о
I
сн2
I
СООС2Н5
сн2—сн2
о
Ацетоуксусный Окись
эфир этилена
130,14 44,05
сн3
CH3ONa С=О
----> I
НС—со
I
сн2
I
сн2—о
7 - Ацето - 7 -бутиро лактон
128,12
В реактор из нержавеющей стали 101 загружают из мерника 102 мета-
нол, охлаждают его до нуля и пропускают из баллона 103 окись этилена;
затем в реактор заливают из мерника 104 ацетоуксусный эфир, из мерника
102 небольшое количество метанола и из мерника 105 метилат натрия при
87
температуре не выше 25° С, перемешивают с выдержкой 10—12 ч. После
этого реакционную массу нейтрализуют соляной кислотой из мерника 106
до pH 6,0—6,5; осадок хлористого натрия отфильтровывают на нутч-фильт-
ре 107, а фильтрат упаривают в вакуум-аппарате 108. К кубовому остатку
добавляют воды и ацетобутиролактон экстрагируют хлороформом на экст-
ракционной установке непрерывного действия [Ю5], состоящей из экст-
рактора 109, напорных сборников для кубового остатка ПО и для хлоро-
форма 111, пленочного аппарата 112 для отгонки растворителя, сборника
технического ацетобутиролактона 113. После дистилляции последнего в
вакуумперегонном аппарате 114 отбирают в сборник 115 фракцию с темпе-
ратурой кипения 92—94° С при остаточном давлении 1 мм рт. ст.; =
= 1,4590; d2°= 1,1860. Выход 70% (на ацетоуксусный эфир).
7-Х Л ОР-Т-АЦЕТОПРОПИЛ АЦЕТ АТ [66]
При действии хлора на у-ацето-у-бутиролактон с последующим декарбо-
ксилированием и ацетилированием получают у-хлор-у-ацетопропилацетат.
Реакция протекает по следующему уравнению:
СН3 СН3
I I
с=о с=о
I СЬ. CaCO. I
НС—со —
сн2
I
сн2—о
•[-Ацето - т-бутиро
лактон
128,12
> С1—С----СО
СН2
СН2—О
Т-Хлор-у-ацето-
бутиролактон
162.57
сн.соон
(СН3СО0)2О
СНз
I
с=о
С1—сн
сн2
I
СН2ОСОСН3
7-Хлор-7-ацетопропилацетат
178,62
В реактор 116 загружают из мерника 118 у-ацето-у-бутиролактон, из
мерника 117 дихлорэтан, а затем мел, добавляют воды и в течение 3 ч при
перемешивании при температуре 12—15° С пропускают хлор. Затем в де-
лительной воронке 119 разделяют слои; водный слой промывают дихлор-
этаном; органический слой направляют в сборник 120 и далее в перегон-
ный аппарат 121 для отгонки дихлорэтана, а остаток перегоняют в вакууме
(4^-5 мм) и направляют в сборник 122. Выход у-хлор-у-ацетобутиролактона
составляет 92%; температура кипения 100—102° С при остаточном давле-
нии 4 мм рт. cm.; 1,4709; 1,186; содержание хлора 22%. Затем
у-хлор-у-ацетобутиролактон направляют из сборника 122 в реактор 123
из эмалированной стали, добавляют из мерника 124 уксусной кислоты, из
мерника 125 небольшое количество соляной кислоты, нагревают до 90° С
и перемешивают 4 ч. После этого реакционную массу охлаждают до 50—
60° С, приливают уксусного ангидрида из мерника 126 и перемешивают 4 ч
при температуре 90° Сив этом же реакторе, снабженном прямым и обрат-
ным холодильником, отгоняют вначале уксусную кислоту (в слабом вакуу-
ме) в сборник 127, а затем при остаточном давлении 5 мм рт. ст. дистил-
лируют у-хлор-у-ацетопропилацетат в сборник 128. Выход — 71 %
(в пересчете на бутиролактон); температура кипения 105—107° С при
остаточном давлении 5 лии рт. cm.; n^=l,4469; d2°— 1,165.
ФОРМАМИД (АМИД МУРАВЬИНОЙ КИСЛОТЫ)
Известны различные методы синтеза формамида — взаимодействием ам-
миака с окисью углерода [106, 107, 108], аммиака с муравьиной кислотой
[109] и ее сложными эфирами [ПО—112]. Наиболее эффективным следует
считать непрерывный процесс аммонолиза газообразным аммиаком этило-
88
вого эфира муравьиной кислоты [113] (этилформиата). Реакция протекает
по следующему уравнению:
НСООС2Н6 + NH3 —► НС + С2Н6ОН.
xnh2
Этилформиат Аммиак Формамид Спирт
74,08 17,03 45,04 46,07
Экспериментально установлено [113], что' наиболее селективно процесс
протекает в пленке в насадочной колонке при добавлении каталитиче-
ских количеств воды (около 2%); молярное соотношение этилформиата и
аммиака 1:1,1; оптимальная температура реакции — минус 6—8° С.
В колонный реактор 129 из мерника 130 подается этилформиат, а из
баллона 131 газообразный аммиак противотоком снизу. Продукт реакции
собирают в приемнике 132, откуда поступает в вакуум-перегонный аппарат
133 для отгонки легколетучих веществ, направляемых в сборник 134, а
кубовый остаток, содержащий 95% формамида, сливают в сборник 135.
Формамид представляет собой бесцветную жидкость с температурой
кипения 114° С при остаточном давлении 18 мм рт. ст.-, плотность
1133,4 кг/м3; «д = 1,4472. Растворяется в воде.
4-МЕТИЛ-5-Р-ОКСИЭТИЛТИАЗОЛ (ТИАЗОЛОВЫЙ КОМПОНЕНТ)
Процессы получения тиазолового компонента заключаются в двух са-
мостоятельных реакциях: 1) образования тиоформамида из формамида и
пятисернистого фосфора и 2) конденсации тиоформамида с у-хлор-у-ацето-
лропилацетатом с смылением получаемого промежуточного продукта.
ГЧН2
1. 5 НС. +
V
Формамид
5-45,04
nh2
P2S5------
Пятисернистый $
tpocqjop Тиощормамид
222,29 5-61,1
nh2
НС +
%
Тиосрормамид
61,1
сн3
СО -------'
I
С1-СНСН2СН2ОСОСН3
Хларацетопропилацетат
178,63
N----С-СН3
II II
- нс с-сн2сн2ососн3
xs
2- Метил -5-ацетоксиэтилтиазол
HC1
N— С-СН,
Il II
нсч ХС-СН2СН2ОН+ СН3СООН
4 -Метил -5 - J3- оксизтилтиазол
143,21
Эти две реакции могут быть осуществлены либо раздельно, либо совмест-
но без выделения тиоформамида. Последнее обосновывалось лабильностью
тиоформамида. Однако имеются также данные [113], что тиоформамид в
органическом растворителе достаточно долго сохраняется. Имеются также
экспериментальные данные [113], указывающие на лучшие результаты омы-
ления 4-метил-5-ацетоксиэтил тиазола в кислой среде (pH 1—2), чем в ще-
лочной (pH 8,5—9,0). Экспериментально также показано [114, 115], что
хлороформ, дихлорметилен и дихлорэтан, как имеющие наибольшие коэф-
фициенты распределения, являются наиболее эффективными растворителя-
ми, причем экстрагирующая способность убывает в ряду: дихлорметилен,
хлороформ, дихлорэтан. Бензол, толуол и четыреххлористый углерод
непригодны в качестве экстрагентов. Эти данные учтены в технологической
схеме.
89
В реактор 136 загружают формамид из мерника 137 и хлористый мети-
лен из мерника 138 и при размешивании постепенно добавляют мелко
измельченный пятисернистый фосфор; температуру поддерживают 17—
18° С. Затем температуру повышают до 35° С и продолжают размешивание
24 ч. Затем экстракт сливают и производят вторичную экстракцию. Экстракт
собирают в смесителе 139 и высушивают прокаленным сульфатом натрия.
Сухой экстракт направляют в вакуум-перегонный аппарат 140, отгоняют
хлористый метилен, кубовый остаток растворяют в абсолютном спирте и
направляют в сборник 141.
В реактор 142 загружают из мерника 143 хлорацетопропилацетат и по-
степенно добавляют раствор тисформамида в абсолютном спирте, наблю-
дая за тем, чтобы температура реакционной массы не поднималась выше
60° С. По окончании реакции массу выдерживают 12 ч при температуре
15—20° С. Затем ее нагревают до 80° С в течение 2 ч, разбавляют холодной
водой, из мерника добавляют соляной кислоты и фильтруют через нутч-
фильтр 144. Осадок промывают, а фильтрат и промывные воды направляют
в сборник, а отсюда в реактор для омыления 145, куда подают из мерника
соляной кислоты до pH 1,0—2,0. Для омыления смесь кипятят 3 ч,
охлаждают и фильтруют через друк-фильтр 146 для выделения осадка.
Последний промывают хлороформом, который подается из мерника 147.
Осадок идет в отход. Фильтрат поступает в сборник 148, а из него в экстрак-
тор 149 для экстракции хлороформом. Водный слой спускают в канализа-
цию, а хлороформенный осушают в смесителе 150 поташом. Экстракт
отфильтровывают от поташа на друк-фильтре 151 и направляют в
вакуум-перегонный аппарат 152, отгоняют хлороформ (без вакуума) и отгон
направляют в приемник 153, затем включают вакуум и отгоняют тиазол при
остаточном давлении 3—7 мм рт. ст. в приемник 154.
4-Метил-5-|3-этоксиэтилтиазол представляет собой густую маслянистую
бесцветную жидкость с характерным запахом; температура кипения 135° С
при остаточном давлении 7 мм рт. ст и 123—124° С при остаточном давле-
нии 3 мм рт. ст. Он растворяется в спирте, хлороформе, дихлорэтане, эфи-
ре и воде; плотность d^°=1840 ка/ж3; показатель преломления = 1,544;
УФ-спектр Хтах 250 нм\ £j%M—285; содержание основного вещества 94—
95%.
СИНТЕЗ 7-ХЛОР-7-АЦЕТОПРОПИЛАЦЕТАТА ИЗ СИЛЬВАНА [118]
Выше было указано (стр. 75, 91) на преимущества использования для
данной цели у-ацетобутиролактона, Однако среди некоторых витаминологов
существует мнение о целесообразности получения хлорацетопропилацетата
из сильвана через ацетопропиловый спирт и ацетата его. Сторонники этого
направления исходят из соображений, что стоимость фурфурола — сырья
для получения сильвана, ниже стоимости ацетоуксусного эфира — сырья
для получения у-ацетобутиролактона. Рассмотрим вкратце этот путь син-
теза.
Сильван из фурфурола [116, 116а, 118]. Синтез протекает по следующей
схеме:
НС___СН
II' И
нс с-с<
чо н
2Н2 .
Катализатор
НС____СН
. II II
нс с-сн3
чо
^УРРИрМ
36,08
2 - Метилфуран
(сильван)
82,10
90
Процесс гидрогенизации осуществляют в каталитическом реакторе, запол-
ненном медно-алюминиевым катализатором в парогазовой фазе при темпе-
ратуре 200—250° С и давлении в 5 кгс/см2. Продукты реакции конденси-
руются в холодильнике; их разгоняют в ректификационной колонне.
Фракцию с температурой 56—62° С направляют для получения ацетопро-
пилового спирта. Выход сильвана 70—80%. Ранее применяли сильван,
выпускаемый лесохимической промышленностью. [116а].
Ацетопропиловый спирт (АПС). у-Ацетопропиловый спирт из сильвана
получают по методу К. Топчиева и Л. Павлова [117] путем гидрирования
2-метилфурана [118]. Реакция протекает двухступенчато по схеме:
2^ Метилфуран
82,10
2~Метил-Ь,5-
дигидроруран
[ Кон —► СН3СОСН2СН2СН2ОН
O'THj
Циклическая форма 'у-Ацетолролиловый слиргп
АПС ’ 102, 13
Гидрирование сильвана осуществляют в автоклаве при избыточном давле-
нии 2—2,5 кгс!см2 при температуре 55—60° С на палладиевом катализаторе
(хлористый палладий) в водной среде. Реакционную массу нейтрализуют
кальцинированной содой до слабощелочной реакции (pH 8,0—9,0), от-
фильтровывают катализатор и фильтрат разгоняют, отбирают фракцию с
температурой кипения 100—106° С при остаточном давлении 15 мм рт. ст.
Выход ацетопропилового спирта составляет 54% [118] (на сильван). Ацето-
пропиловый спирт — бесцветная жидкость с характерным запахом. Раство-
рим в воде (1:14), также в спирте и эфире; d20= 1,00197; «^=1,4350;
температура кипения 209° С (760 мм рт. ст.), 100—106°С (15 мм рт. ст.).
Однако для синтеза 4-метил-5-|3-оксиэтилтиазола нецелесообразно примене-
ние у-галоген-у-ацетопропилового спирта, так как он подвержен таутомерным
превращениям. Кроме того, при галогенировании возможна изомеризация
его во вторичный спирт СН3С(ХН2СН(ОН)СН3; после галогенирования и
конденсации с тиоформамидом дает изомерный 4-метил-5-а-оксиэтилтиазол.
В связи с этим для галогенирования в практике стали применять уксусный
эфир у-ацетопропилового спирта. Однако и в данном случае, как это показа-
но В. Турсиным [66, 67], при хлорировании, кроме монохлорпроизводного,
образуется дихлорпроизводное ацетопропилацетата, что способствует сни-
жению выхода тиазолового ксмпонента. По этой причине в схеме синтеза
тиазола нами ацетопропиловый спирт заменен ацетобутиролактоном.
ВИТАМИН Вх — КОНДЕНСАЦИЯ ПИРИМИДИНОВОГО И ТИАЗОЛОВОГО
КОМПОНЕНТОВ
Химическая реакция получения витамина В! (тиамина) заключается в
конденсации 2-метил-4-амино-5-хлорметилпиримидингидрохлорида (пи-
римидинового компонента) и 4-метил-5-|3-оксиэтилтиазола. (тиазолового
компонента), согласно следующему уравнению:
сн
'С-СН2С1
нс-сК Л]\1Н2-НС1
N
2 - Метил амино -5-
хлорметилпиримидингидрохлорид
Z44, 07
СН СН2Х +
N—C-CHj -С" 2ХЬ1 —СН-СН3
нс. Jc-ch2ch2oh нсД,СЧ LI Асн2сн2он-о,5н2о
S г'’ -НС1
4- Метил-5 -оксизтилтиазол 2
1Р3.21 Витамин В, (тиаминхлорид)
331, 21
Процесс конденсации осуществляют в реакторе 155 из эмалированной ста-
ли, снабженном обратным холодильником. Загружают в него из мерника
156 толуол, из мерника 157 тиазол и после тщательного перемешивания
91
постепенно из сборника 158 добавляют хлораминопиримидин и при темпера-
туре ПО—115° С перемешивают 5 ч. После этого реакционную массу
охлаждают, фильтруют через центрифугу 158 а, промывают спиртом из
мерника 159. Затем осадок направляют в реактор 160, загружают спирт и ки-
пятят 2—3 ч. Затем охлаждают массу до 20° С и отфуговывают осадок техни-
ческого тиаминхлорида в центрифуге 161. Последний поступает в сборник
162 и далее его направляют на перекристаллизацию. Фильтрат из сборни-
ков 163 и 163а передается для регенерации спирта, тиазола и толуола.
ПЕРЕКРИСТАЛЛИЗАЦИЯ ВИТАМИНА Вх
Первая кристаллизация. Технический витамин Вг растворяют в реакто-
ре 164 в 80%-ном спирте, подаваемом из мерника 165. Затем вводят активи-
рованный уголь (10% к массе тиамина) и перемешивают 15 мин при темпе-
ратуре 70° С, фильтруют через нутч-фильтр 166, кристаллизуют в кристал-
лизаторе 167 при 0° 8 ч и фугуют в центрифуге 168. Кристаллы промывают
спиртом. Кристаллы тиамина I подвергают вторичной перекристаллиза-
ции, а маточный раствор I направляют в сборник 169, а оттуда на перера-
ботку на четвертую кристаллизацию.
Вторая кристаллизация. На вторую кристаллизацию поступают: крис-
таллы тиамина I, центрифугальные промой после отгонки спирта и крис-
таллы тиамина II. Указанные полупродукты растворяют в 80 %-ном спирте,
добавляют 2—5% к массе витамина активированного угля и в течение 15 мин
перемешивают при температуре 80° С в реакторе 170. Затем раствор фильт-
руют через нутч-фильтр 171, кристаллизуют в аппарате 172, фугуют крис-
таллы в центрифуге 173, промывают их абсолютным спиртом и высушива-
ют в вакуум-сушилке 174.
Сухие кристаллы расфасовывают. Маточный раствор II поступает в
сборник 175 и оттуда его направляют на третью кристаллизацию.
Третья кристаллизация. На третью кристаллизацию поступают: маточ-
ный раствор II, угольные промой II, центрифугальные промой III и крис-
таллы тиамина IV. Указанные полупродукты растворяют в воде в реакторе
176, добавляют 5% к массе содержащегося в полупродуктах витамина Bt
активированного угля, фильтруют через нутч-фильтр 177 и упаривают в
вакуум-аппарате 178, массу спускают в кристаллизатор 179, кристаллизуют
12 ч при 0°, фугуют в центрифуге 180. Кристаллы после промывки 96%-ным
спиртом направляют в реактор 170, а маточный раствор III поступает в
сборник, откуда далее его направляют на IV кристаллизацию.
Четвертая кристаллизация. На четвертую кристаллизацию поступают:
маточный раствор III, центрифугальные промой IV и маточный раствор
первой кристаллизации.
Эти полупродукты растворяют в реакторе 181 в воде, обрабатывают ак-
тивированным углем, фильтруют через нутч-фильтр 182, упаривают в ва-
куум-аппарате 183, кристаллизуют в кристаллизаторе 184 при температуре
минус 5—10° С 3—5 суток и фугуют в центрифуге 185. Кристаллы тиамина
IV поступают на перекристаллизацию в реактор 176, а маточный раствор —
в сборник 186, откуда его направляют либо в канализацию, либо на пятую
кристаллизацию, если чистота его выше 40—45%.
ТЕХНОЛОГИЧЕСКИЕ ПОКАЗАТЕЛИ
РАСХОД ХИМИКАЛИЕВ, КГ НА 1 кг
100%-НОГО ПОЛУПРОДУКТА ПО
МЕТОДУ ДВУХКОМПОНЕНТНОЙ
КОНДЕНСАЦИИ (ПЕРВЫЙ МЕТОД)
^-Этоксипропионитрил
Акрилонитрил .................. 0,6
Спирт этиловый .................0,5
Едкий натр....................0,01
Уксусная кислота..............0,01
Выход, % к теоретическому (на
акрилонитрил) ................95,0
Этилформиат
Муравьиная кислота.............0,9
Спирт этиловый.................0,9
92
Серная кислота.....................0,2
Выход, % к теоретическому (на
муравьиную кислоту)...............80,0
Метанол .........................6,0
Выход, % к теоретическому (на
ацетоуксусный эфир) ........... 70,0
Пагпрэнолят-а-оксиметилен-$-этокси-
пропионитрил
р-Этоксипропионитрил............... 0,9
Этилформиат........................ 0,7
Этилат натрия ..................... 0,6
Толуол ............................ 1,0
Выход, % к теоретическому (на
этилформиат) у.....................75,0
а-Ацгтоксиметилен-^-этоксипропионит-
рил
Натрэнолят......................... 1,4
Толуол ............................ 1,4
Хлористый ацетил....................0,8
Выход, % к теоретическому (на
натрэнолят) .......................65,0
Ацетонитрил
Аммиак .......................... 0,6
Уксусная кислота ................ 2,1
Поташ............................ 2,0
Выход, % к теоретическому (на
уксусную кислоту)............... 70,0
Хлоргидрат этилового ацетсиминоэфира
Ацетонитрил..................... 0,36
Абсолютный спирт................ 0,40
Хлористый водород.............. 0,4
Эфир этиловый.................. 0,10
Выход, % к теоретическому (на
ацетонитрил).....................90,0
Хлоргидрат ацетамидина
Ацетоиминоэфир................... 1,7
Спирт абсолютный ................ 0,2
Аммиак........................... 0,3
Выход, % к теоретическому (на
ацетоиминоэфир) .................80,0
2-Метил-4-амино-5-этоксиметилпирими-
дин (аминопиримидин)
Ацилэнолят........................ 1,7
Абсолютный спирт ................. 1,0
Толуол ........................... 5,2
Ацетамидинхлоргидрат.............. 1,0
Едкое кали........................ 5,0
Этилат натрия (сухой) ............ 1,2
Выход, % к теоретическому (на
ацилэнолят) ......................60,0
2-Метил-4-амино-5-хлорметилпирими-
дин-гидрохлорид (хлораминопиримидин)
Аминопиримидин .................. 1,0
Хлористый водород................ 1,0
Спирт абсолютный ................ 1,5
Выход, % к теоретическому (на
аминопиримидин)..............85,0
-[ Ацето-ц-бутиролак/пон
Ацетоуксусный эфир ..............1,47
Окись этилена .................1,5
Натрий металлический ............0,3
Хлороформ......................1,0
7 - Хлор-у -ацетопропилацетат.
у-Ацето-у-бутиролактон.........1,0
Дихлорэтан.....................0,5
Уксусная кислота...............0,8
Уксусный ангидрид..............0,8
Мел............................0,08
Хлор.............................0,4
Выход, % от теоретического (на
лактон) ........................71,0
Формамид (амид муравьиной кис-
лоты )
Этилформиат.................... 2,0
Аммиак 25%-ный раствор .... 1,9
Выход, % к теоретическому (на
этилформиат)...................80,0
4 Метил -5-$- оксиэтилтиазол ( тиазо-
ловый компонент)
Пятисернистый фосфор............1,4
Хлористый метилен ..............1,0
Формамид........................1,8
Хлорацетопропилацетат ..........3,0
Хлороформ ......................0,6
Поташ ........................ 0,7
Соляная кислота ............... 1,0
Спирт абс...................... 1,0
Выход, % к теоретическому (на
хлорацетопропилацетат) .... 50,0
Витамин Bj медицинский
Хлораминопиримидин ............ 1,0
Тиазоловый компонент............1,0
Толуол .........................0,5
Ацетон на промывку..............0,5
Спирт этиловый..................0,5
Активированный уголь............0,2
Выход, % к теоретическому (на
хлораминопиримидин) .......... 65,0
Выход медицинского витамина Bi
из технического, % .........88,0
РАСХОД ПОЛУПРОДУКТОВ НА
СИНТЕЗ 1 КГ МЕДИЦИНСКОГО
ВИТАМИНА Вт, КГ
Технический витамин Bi........ 1,14
Хлораминопиримидин ........... 1,0
Тиазол........................ 1,0
Хлорацетопропилацетат ........ 3,0
Формамид ..................... 1,8
Этилформиат...................5,27
Ацетобутиролактон.............3,0
Аминопиримидин ............... 1,0
Ацилэнолят................ . 1,7
Хлоргидрат ацетамидина 1,0
Хлоргидрат ацетоиминоэфира ... 1,7
Ацетонитрил .................. 0,61
Натрэнолят....................2,38
fi-ЭтоксипрОпионитрил.........2,14
Акрилонитрил.................. 1,28
93
РАСХОД ХИМИКАЛИЕВ НА
ПРОИЗВОДСТВО 1 кг
МЕДИЦИНСКОГО ТИАМИНХЛОРИДА
(С УЧЕТОМ РЕГЕНЕРАЦИИ
РАСТВОРИТЕЛЕЙ ПО ПЕРВОМУ
ВАРИАНТУ), КГ
Акрилонитрил .................. 1,28
Аммиак (жидкий)................ 1,52
Ацетон......................... 0,50
Ацетил хлористый................1,36
Водород, _и:| ..................0,50
Дихлорэтан .................... 1,50
Кислота муравьиная..............4,74
Кислота серная..................0,95
Кислота соляная ............... 1,00
Кислота уксусная .............. 3,71
Кали едкое......................5,00
Метилен хлористый.............. 1,00
Мел ............................0,24
Метанол ......................18,-0
Натр едкий......................0,02
Натрий металлический ...........0,90
Окись этилена ..................4,50
Поташ...........................1,92
Спирт этиловый..................6,31
Спирт абсолютный................4,38
Толуол........................10,46
Уголь активированный............0,20
Уксусный ангидрид...............2,40
Фосфор пятисернистый ...........1,40
Хлороформ.......................3,60
Хлор............................2,60
Этилат натрия .................2,63
Эфир ацетоуксусный .............4,40
Эфир этиловый...................0,17
Итого. . . . 87,19
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА ТИАМИНХЛОРИДА МЕТОДОМ
ПОСТРОЕНИЯ МОЛЕКУЛЫ НА ПИРИМИДИНОВОМ ЦИКЛЕ (ВАРИАНТ 11)
Метод включает следующие стадии синтеза:
1. (З-Метоксипропионитрила из акрилонитрила и метилового спирта;
2. а-Диметоксиметил-|3-метоксипропионитрила (ацеталя) из метоксипро-
пионитрила, этилформиата, металлического натрия и диметилсульфата;
3. Этилформиата из муравьиной кислоты и этилового спирта;
4. Ацетонитрила из уксусной кислоты и аммиака в присутствии катали-
затора;
5. Ацетоиминоэфира из ацетонитрила и хлористого водорода;
6. Хлоргидрата ацетамидина из ацетоиминоэфира и аммиака;
7. 2-Метил-4-амино-5-ацетамидометилпиримидина из ацеталя и ацета-
мидина;
8. у-Ацетобутиролактона из ацетоуксусного эфира, окиси этилена и
метилата натрия;
9. 7-Хлор-у-ацетопропилацетата из у-ацетобутиролактона, хлора и ук-
сусного ангидрида;
10. 4-Метил-5-|3-оксиэтил-М-(2'-метил-4'-аминопиримидил-5/-метил)-2-
тиотиозолона (тиотиамин) из 2-метил-4-амино-5-ацетамидометилпиримиди-
на, аммиака и сероуглерода;
11. Тиаминхлорида хлоргидрата из тиотиамина окислением его пере-
кисью водорода с перекристаллизацией получаемого технического тиамин-
хлорида.
Из указанных 11 стадий 7 (№ 1, 3—9) являются общими для данного
метода синтеза и Метода синтеза при помощи двухкомпонентной конденсации.
Рассмотрим специфичные для данного метода четыре стадии синтеза.
ПОЛУЧЕНИЕ а-ДИМЕТОКСИМЕТИЛ-[3-МЕТОКСИПРОПИОНИТРИЛА
[121, 122, 123]
Процесс осуществляют путем формилирования |3-метоксипропионитрила
с последующим метилированием диметилсульфатом по следующей схеме:
94
CH3O-CH2-CH2-CN + HCOOC2HS + Na-
jO - Метоксипропионитрил Этитрормиат
85,11 n,OS 23,00
сн3о—сн2—c-cn
d 2 n
NaO-CH
Na-соль cL-оксиметилен-
Д - метоксипропионитрил
735,10
(CH3O)2SO2
Диметилсерная
кислота
126,13
CH3OCH2- C~CN
3 2 II
UHCOCH3
изомер - >
NaOH
ch3och2-c-cn
II
Н3СОСН
Траноизомер
сн3он ch3och2-ch-cn
7Л5% н-с(осн3)2
-Диметоксиметил -J3-
метоксипропионитрил (ацеталь)
159, 18
В реактор 1 (рис. 10) из нержавеющей стали, снабженный обратным
холодильником, из мерника 2 загружают безводный толуол, а затем ме-
таллический натрий, нагревают до 105° С, перемешивают 10 мин. После
измельчения натрия в мелкие гранулы из мерника 3 приливают сухой ме-
тиловый спирт в течение 30 мин, а затем перемешивают 1 ч при температу-
ре 100—105° С для превращения натрия в метилат натрия. Затем массу
охлаждают до 0° и из мерника 4 приливают смесь этилформиата и |3-мето-
ксипропионитрила в течение 30 мин и перемешивают 4 ч, повышая темпера-
туру постепенно до 18—20° С. После 10—12-часовой выдержки температуру
4-Метил-5-^-онсиэтил- ьНД-метил- ^-аминопиримидил-^метил)*
- г-тиотиозолон (тиотиамин)
ТиаминхлориВ хлоргидрат
(витамин 81)
Рис. 10. Технологическая схема производства тиаминхлорида (вариант II).
95
повышают до 25—30°С и перемешивают 2 ч, затем охлаждают до 0°, прили-
вают из мерника 5 диметилсульфат, поддерживая при перемешивании тем-
пературу 45—50° С в течение 3—4 ч.
Натриевую соль диметилсерной кислоты отфильтровывают в нутч-фильт-
ре 6. Фильтрат поступает в сборник 7 и далее в вакуум-перегонный аппа-
рат 8. Собирают в приемник 9 фракцию, кипящую при температуре 90—
95° С (при остаточном давлении 2 мм рт. ст.), представляющую собой
смесь цис- и транс-изомеров а-алкоксиметилен-|3-метоксипропионитрил
[119, 121]. В реакции присоединения спирта к этим изомерам с целью
получения диметоксиметилен-|3-метоксипропионитрила в качестве катали-
затора вместо едкого натра применен анионит АВ-17 в ОН-форме. Для этой
цели в реактор 10 из мерника 3 загружают метанол, смесь изомеров и анио-
нит АВ-17 перемешивают 3 ч при температуре 25—30° С, отфильтровывают
анионит в нутч-фильтре 11, фильтрат поступает в сборник 12, вакуум-пере-
гонный аппарат 13, где перегоняют при температуре 75—80° С (при остаточ-
ном давлении 2 мм рт. ст.). Фракцию направляют в сборник 14. Выход
73,8%. о,-Диметоксиметил-|3-метоксипропионитрил — бесцветная жидкость,
молекулярная масса 159,18, растворяется в органических растворителях;
температура кипения 85—86° С (при остаточном давлении 2 мм рт. ст.)-,
1,0140; 4Э= 1,4270 [121].
ПОЛУЧЕНИЕ 2-МЕТИЛ-4-АМИНО-5-АЦЕТАМИДОМЕТИЛПИРИМИДИНА
Получение основано на реакции взаимодействия указанного ацеталя с осно-
ванием ацетамидина. При этом образуется в качестве промежуточного
соединения 2,7-диметил-5,6-дигидропиримидо-4,5-пиримидин. Реакция про-
текает по следующей схеме:
NH
2 Н
Ацетамидин
58,09
(сн3о)2-сн
НС-СН2ОСН3
CN
2,7 -Ди метил -5,6-
дигидропиримидо -
4,5-пиримидин
оС ~ Диметоксиметил-
-J3 - метокси пропионитрил
159,18
N<?>t-CH2-NH-COCH3
h3cAn^nh2
2-Метил -4- амино - 5-ацетамидометилпиримидин
180,22
В реактор 15 из нержавеющей стали, снабженный обратным холодиль-
ником, загружают солянокислый ацетамидин и из мерника 16 метанольный
раствор метилата натрия, перемешивают 30 мин при температуре 20°С.
Из мерника 17 вводят в реактор а-диметоксиметил-|3-метоксипропионитрил,
перемешивают 3 ч при кипении, затем охлаждают и фильтруют через нутч?
фильтр 18 и сборник 19, откуда направляют в вакуум-перегонный аппарат
20 для отгонки метанола. Затем добавляют воды, едкого натра, нагревают
3 ч при температуре 85—90° С и сливают в кристаллизатор 21, где при 0°
выкристаллизовывают продукт, отфуговывают его в центрифуге 22, промы-
вают охлажденной водой и высушивают в вакуум-сушилке 23 и хранят в
сборнике 24. Маточный раствор направляют в сборник 25 и он является от-
ходом производства. В случае необходимости ацетамидо пиримидин пере-
кристаллизовывают из воды. Выход 51,5% (на ацеталь). Ацетамидопири-
мидин— молекулярная масса 180,22, бесцветные кристаллы, температура
плавления 203—204° С, растворимы в воде,'нерастворимы в органических
растворителях.
96
ПОЛУЧЕНИЕ 4-МЕТИЛ-5ф-ОКСИЭТИЛ-Ы-(2'-МЕТИЛ-4'-АМИНОПИРИМИДИЛ-
5'-МЕТИЛ)-2ТИОТИОЗОЛОНА (ТИОТИАМИНА)
Получение основано на реакции омыления ацетамидопиримидина в
2-метил-4-амино-5-аминометилпиримидин (диамин), который с сероуглеро-
дом и у-хлор-у-ацетопропилацетатом в присутствии аммиака образует а-аце-
тил-у-ацетоксипропил-Ы-(2/-метил-4л- аминопиримидил-5'-метил) - дитиокар-
бомат, превращающийся при нагревании с соляной кислотой в тиотиамин.
Реакции протекают по схеме:
„ —н cXXnh'™1
П3С—INH2 н3с—1NH2
Ацетамидааиримидин
180,22
CH2~NH2
nh2
+ NH3
Диамин
40,01 138,18
сн3
с=о
I
+ С1-СН + cs2
сн2
СН2ОСОСН3
+ CH3COONa;
8г, оь
Ди амин у-Хлор--у- ацетопропилацетат
138,18
17,03 178,57 76,15
С н
— 1i)^Y н п°~СНз НС1
Н,сЛ> ,Ль1Н2 .с. сн-сн,-сн2ососн3
N V 2
Дихлорид'мвркаптотиа мин
Дитиокардамат
NaOH
—п-СИ3
Д-сн2-сн2он
Тиатиамин
'296,41 -
В реактор 26 из эмалированной стали загружают из сборника 24 ацето-
амидопиримидин и из сборника 27 раствор NaOH (15%), нагревают 4 ч
при температуре 105—110° С. Полученный водный раствор диамина нейтра-
лизуют до слабощелочной реакции соляной кислотой из мерника 28. Ре-
акционную массу охлаждают в реакторе 29 до 18—20° С, приливают
25%-ный водный раствор аммиака из мерника 30, спиртовый раствор у-хлор-
у-ацетопропилацетат из мерника 31 и сероуглерод из мерника 32. Реакци-
онную массу перемешивают 1 ч при температуре 35° С до появления крис-
таллов дитиокарбомата. Затем из мерника 28 приливают соляную кислоту
до кислой реакции, нагревают 10 мин при температуре 70° С для превра-
щения дитиокарбомата в дихлорид меркаптотиамин, переводят в реактор-
охладитель 33, где реакционную массу охлаждают до 10° С и из мерника 34
приливают 42%-ной щелочи до слабощелочной реакции и выделяют тио-
тиамин. Осадок отфильтровывают на нутч-фильтре 35 и промывают водой.
Маточный раствор и промывные воды поступают в сборник и являются от-
ходом. Влажный тиотиамин направляют в сборник 36 и далее на перекрис-
таллизацию. Для этого технический продукт растворяют в реакторе 37 в
70%-ном спирте и при температуре 70° С вводят 3—5% активированного
угля. Спирт приливают из мерника 38. Массу фильтруют через нутч-фильтр
39, кристаллизуют в кристаллизаторе 40, фугуют в центрифуге 41 и выгру-
жают в сборник 42, откуда в сыром виде направляют на следующую стадию.
Маточный раствор поступает в сборник 43, а из него в вакуум-аппарат 44,
где его сгущают и сливают в кристаллизатор 45, центрифугу 46. Кристаллы
тиотиамина II во влажном виде направляют для перекристаллизации сов-
местно с техническим продуктом в реактор 37. Маточный раствор II поступа-
етвсборник47 и является отходом производства. Выход тиотиамина 61,8%'
4—522 ду
Тиотиамин представляет собой бесцветные кристаллы с температурой плав-
ления 235—238° С, растворим в водных растворах спиртов и трудно раство-
рим в воде и ацетоне. Нерастворим в бензоле, хлороформе. Спектр погло-
щения в УФ-245 и 322 нм (в 5% растворе НС1).
ПОЛУЧЕНИЕ ТИАМИНХЛОРИДА ХЛОРГИДРАТА (ВИТАМИН BJ
Витамин Bt получают окислением тиотиамина перекисью водорода,
обработкой соляной кислотой или азотной кислотой [139] и перекристалли-
зацией технического продукта. Реакции протекают по следующей схеме:
/СН2х
|ГСНз
Тиотиамин
236, 41
Н2О2
на
Н3с
N Ь1Н2-НС1 S
сн3
сн2-сн2он
Тиаминхлорид гидрохлорид
337,27
В реактор из эмалированной стали 48 из сборника 42 загружают тиоти-
амин, добавляют воды и осторожно приливают 28%-ный раствор перекиси
водорода и при температуре 30° С перемешивают 1 ч. Затем из мерника 28
при температуре 20° С добавляют концентрированную соляную кислоту.
Реакционную массу перемешивают 30 мин и из мерника 49 сливают насы-
щенный раствор хлористого бария, а затем активированного угля. Уголь
и выделившийся осадок сернокислого бария отфильтровывают в нутч-фильт-
ре 50 и далее через сборник 51 фильтрат засасывают в вакуум-аппарат 52
и сгущают до содержания сухих веществ 70% и сливают в кристаллизатор
53, где выделяют его спиртом, добавляемым из мерника 54. Кристаллы тех-
нического тиамина отфуговывают в центрифуге 55. Кристаллы поступают
в сборник 56 и далее на перекристаллизацию, а маточный раствор поступает
в сборник 57, а затем на переработку совместно со вторым маточным раство-
ром, получаемым при четырехступенчатой перекристаллизации технического
тиаминхлорида. Перекристаллизацию ведут по схеме, изложенной на
стр. 92. Выход технического продукта составляет 74%, а медицинского —
69% (в пересчете на тиотиамин). Чистота технического тиамина 95—96%,
а медицинского — 98%.
ТЕХНОЛОГИЧЕСКИЕ ПОКАЗАТЕЛИ
РАСХОД ХИМИКАЛИЕВ, КГ НА 1 КГ
100%-НОГО ПОЛУПРОДУКТА по
МЕТОДУ ПОСТРОЕНИЯ МОЛЕКУЛЫ
ТИАМИНА НА ПИРИМИДИНОВОМ
ЦИКЛЕ (ВТОРОЙ МЕТОД)
Р-Метоксипропионитрил
Акрилонитрил ................ 0,7
Спирт метиловый.............. 0,5
Метилат натрия................ 0,03
Уксусная кислота.............. 0,01
Выход, % к теоретическому (на
на акрилонитрил) .............85,0
Этилформиат
Диметилсульфат ...............1,44
Спирт метиловый...............0,55
Толуол........................1,60
Анионит АВ-17.................0,05
Натр едкий....................0,10
Ацетонитрил
Аммиак .................... 0,6
Уксусная кислота............. 2,1
Поташ......................... 2,0
Выход, % к теоретическому (на
уксусную кислоту) ............70,0
Хлоргидрат этилового ацетоиминоэфира
Муравьиная кислота............... 0,9
Спирт этиловый................... 0,9
Серная кислота .................. 0,2
Выход, % к теоретическому (на
муравьиную кислоту)...............80,0
Диметоксиметилен-^-метоксипропио-
нитрил (ацеталь)
Р-Метоксипропионитрил ..........0,9
Этилформиат.....................0,9
Натрий металлический.............0,27
Ацетонитрил ...................0,36
Спирт абсолютный...............0,40
Водород хлористый..............0,40
Эфир этиловый..................0,10
Выход, % к теоретическому (на
ацетонитрил)...................90,0
Хлоргидрат ацетамидина
Ацетоиминоэфир................. 1,7
Спирт абс...................... 0,2
Аммиак ........................ 0,3
98
Выход, % к теоретическому (на
ацетоиминоэфир) .................80,0
2-Метил-4-амино-5-ацетамидометил-
пиримидин
Барий хлористый................... 0,7
Спирт этиловый.................... 2,0
Выход, % от теоретического (на
тиотиамин).........................69,0
Ацеталь........................... 1,80
Ацетамидин хлоргидрат............. 2,40
Натрий металлический.............. 0,53
Спирт метиловый................... 5,00
Натр едкий........................ 0,04
Выход, % от теоретического (на
ацеталь) .........................51,5
\-Ацето-^-бутиролактон
Ацетоуксусный эфир ............... 1,47
Окись этилена ................. 1,5
Натрий металлический...............0,3
Хлороформ......................... 1,0
Метанол............................6,0
Выход, % к теоретическому (на
ацетоуксусный эфир) ..............70,0
1-Хлор-~(-ацетопропи лацетат
f-Ацето--f-бутиролактон .... 1,0
Дихлорэтан...................... 0,5
Уксусная кислота................ 0,8
Уксусный ангидрид .............. 0,8
Мел............................. 0,08
Хлор.............................. 0,4
Выход, % к теоретическому (на
лактон).........................71,0
Тиотиамин.
Ацетамидопиримидин ................ 1,0
Натр едкий......................... 0,5
Аммиак ............................ 0,1
Сероуглерод ....................... 0,5
Спирт.............................. 4,0
р-Хлор-у-хлорацетопропилацетат . 1,0
Кислота соляная.................... 2,0
Уголь активированный ............. 0,1
Выход, % от теоретического (на
ацетамидопиримидин)................61,8
Витамин медицинский
Тиотиамин.......................... 1,3
Перекись водорода (28%) .... 0,6
РАСХОД ХИМИКАЛИЕВ НА
ПРОИЗВОДСТВО 1 КГ МЕДИЦИН-
СКОГО ТИАМИНХЛОРИДА
(С УЧЕТОМ РЕГЕНЕРАЦИИ
РАСТВОРИТЕЛЕЙ) ПО ВТОРОМУ
ВАРИАНТУ, КГ
Акрилонитрил....................1,48
Аммиак .........................2,22
Анионит АВ-17...................0,12
Барий хлористый.................0,70
Водород хлористый................2,12
Диметилсульфат .................3,37
Дихлорэтан......................0,65
Кислота муравьиная............. 1,90
Кислота серная..................0,42
Кислота соляная ................2,60
Кислота уксусная .............. 5,09
Мел............................ 1,10
Натр едкий..................... 1,40
Натрий металлический .......... 1,71
Окись этилена ................. 1,95
Перекись водорода................0,60
Поташ.......................... 3,84
Сероуглерод .....................0,65
Спирт метиловый................16,65
Спирт этиловый..................9,10
Спирт этиловый абс..............2,74
Толуол.............•............3,74
Уголь активированный...........0,13
Уксусный ангидрид.............. 1,04
Хлор...........................0,52
Хлороформ...................... 1 ,30
Эфир этиловый..................0,53
Эфир ацетоуксусный.............2,60
Итого
70,27
Сравнительная технико-эконсмическая эффективность обоих вариантов
синтеза тиаминхлорида характеризуется данными, приведенными в табл. 8.
Таблица 8
Варианты Количество стадий Расход хими- ка лиев, кг/кг Относительная стоимость сырья, % Выхот тиамин- хлорида из акрилонитрила, % от теоре- тического Примечание
Первый 12 87 135 13,5 Получение аминопи- римидина из р-это- ксипропионитрила по- считано за одну ста- дию
Второй 10 70 100 13,8 Получение ацетами- допиримидина из р- метоксипропионитрила посчитано за одну стадию
Прим второй — че 4* е ч а н и е. ' ?ез тиотиами Лервый вари н. энт — метод с двухкомпоь гентной конденсацией, 99
Таким образом, с технико-экономической точки зрения более эффекти-
вен метод синтеза тиаминхлорида через тиотиамин. Однако учитывая, что
вариант I освоен в многотоннажном производстве, а вариант II находится
лишь в стадии освоения (хотя в Венгрии и Японии применяется ряд лет)
следует признать целесообразным продолжить испытание обоих методов и
впредь для окончательного решения вопроса выбора метода в будущем.
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА ТИАМИНПРОПИЛДИСУЛЬФИДА
(ХЛОРИСТОВОДОРОДНАЯ СОЛЬ) [129]
В связи с недостаточной стабильностью тиамина и небольшой продол-
жительностью его пребывания в организме проводятся исследования но-
вых устойчивых форм тиамина [124, 125]. Из дисульфидных производных
тиольной формы тиамина наиболее физиологически активным оказался
тиаминпропилдисульфид [126]. Испытание тиаминпропилдисульфида в кли-
никах показало, что этот препарат может применяться как лечебный в
комплексе с другими средствами при послеоперационных нервных нару-
шениях и нейрохирургических вмешательствах; сахарном диабете с нали-
чием астенического состояния или полиневрита и др. Синтез тиаминпро-
пилдисульфида описан в литературе [127, 128]. Синтез его основан на ре-
акции между тиольной формой тиамина и пропилтиосульфатом натрия
[129].
Реакция получения
Тиаминхлорид хлоргидрат
337, 27
NaOH , N^yCH2-N-C-CH3 CH3CH2CH2S-SO3Na
H3C-k AnH2 НСО С-СН2СН2ОН ‘И"™ ”
' N SNa
Натриедан саль тиольной формы тиамина
304,36
n^z^n-c-ch,
HjC-k^JL-NHs НСО с-сн2сн2он
S-S-CH2CH2CH3
Тиаминпропилдисуль/рид
356, 52
СН
2"n— с-сн3 .
НэсА А - НСО С-СН2СН2ОН
N NH2-HC1 / 2 2
о о V.Г12'-•*;
Тиаминпропилдисулырид гидрохлорид
392,99
В реактор 1 (рис. 11) из нержавеющей стали загружают тиаминхлорид,
растворяют его в воде (3:2), охлаждают раствор до 5° С, а затем постепенно
приливают из мерника 2 15%-ный раствор едкого натра до pH 9. Из сбор-
Рис. 11. Технологическая схема производства тиаминпропилди-
сульфида.
100
ника 3 вводят пропилтиосульфат натрия (соль Бунте). Реакционную массу
перемешивают 3 ч при .температуре 18—20° С, отфильтровывают осадок
тиаминпропилдисульфида через нутч-фильтр 4 и промывают водой. Маточ-
ный раствор поступает в сборник 5 и является отходом. Выход техниче-
ского тиаминпропилдисульфида составляет 89,7% (на тиаминхлорид) со-
держание вещества 97%. Технический продукт перекристаллизовывают из
водного спирта (50%). Для этого из нутч-фильтра 4 технический продукт
загружают в реактор из нержавеющей стали 6, из мерника 7 вводят водный
спирт. Растворение ведут при температуре 70° С, добавляют активирован-
ный уголь (3—5% к массе тиамина), фильтруют через нутч-фильтр 8, фильт-
рат направляют в кристаллизатор 9, где кристаллизация ведется 7—8 ч
при температуре 0 плюс 2° С, после чего кристаллы фугуют в центрифуге
10, промывают водой и высушивают в вакуум-сушилке 11. Маточный раст-
вор I поступает в сборник 12, а оттуда в вакуум-аппарат 13, из которого
после сгущения направляют для кристаллизации в кристаллизатор 14 и
для фуговки в центрифугу 15. Промытые кристаллы поступают на пере-
кристаллизацию в реактор 6. Маточный раствор 11 поступает в сборник
16 и является отходом производства. Выход медицинского продукта состав-
ляет 85—87%, содержание вещества 98,5—99,0%, температура плавления
128—129° С. Тиаминпропилдисульфид — белые кристаллы, плохо раство-
римые в воде, хорошо — в органических растворителях, обладают чесноч-
ным запахом. Воднорастворимая хлористоводородная соль может быть по-
лучена путем обработки концентрированной соляной кислотой спиртового
раствора тиаминпропилдисульфида и выделения хлористоводородной соли
ацетоном. Выход около 85%. Формула C15H24N4O2S2-HCI, температура
плавления 159—160° С. Таким же образом'может быть получена бромисто-
водородная соль (C15H24N4O2S2-НВг, температура плавления 140—141° С)
и азотнокислая соль (C16H24N4O2S2-HNO3, температура плавления 106—
110°С) [129]. Соль Бунте синтезируют из бромистого пропила и тиосуль-
фата натрия в спирто-водной среде по схеме:
СН3СН2СН2Вг + Na2S2O3 • 5Н2О —CH3CH2CH2S—SO3Na + NaBr.
Бромистый про- Тиосульфат натрия Пропилтиосульфат натрия Натрий бромистый
пил 123,01 248,19 178,21 102,91
Соль Бунте — белые кристаллы, обладающие специфическим запахом, со-
держание пропилтиосульфата натрия около 55%.
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА КО КАРБОКСИЛАЗЫ —
ДИФОСФОРНОГО ЭФИРА ТИАМИНА [130, 131]
Кокарбоксилаза была впервые выделена из дрожжей в 1937 г. [132,
133]. Синтез ее был осуществлен из тиамина фосфорилированием различ-
ными агентами: хлорокисью фосфора [134], смесью пирофосфата натрия
и ортофосфорной кислоты [135], пирофосфорной кислотой [136], фосфор-
ным ангидридом [137] и др. Указанные агенты в реакции фосфорилирова-
ния не позволяют получить индивидуальное вещество, а приводят к обра-
зованию смеси эфиров моно- ди- и трифосфорных (ТМФ, ТДФ, ТТФ), из
которых кокарбоксилаза выделяется с трудом. Исследования в области
фосфорилирования тиамина, проведенные В. Березовским и В. Колтуно-
вой) [130, 131], показали, что наилучшие результаты достигаются при
продолжительности реакции 30 мин. и использовании в качестве агента
смеси фосфорного ангидрида и ортофосфорной кислоты, обезвоженной при
температуре 125° С в течение 6 ч при вакууме 10—15 мм рт. ст. (содержа-
ние кокарбоксилазы в продукте реакции 35,7—41,0%); и что в промышлен-
ных условиях чистый продукт можно выделить методом хроматографии;
[138, 139]. Разделение смеси фосфорных эфиров тиамина авторы [130,
131 ] осуществили в виде внутренних солей «бетаинового» типа так назы-
ваемых гидратных форм. Для этой цели анионитом АН-25 в ОН-форме
101
из смеси эфиров была удалена солеобразующая и свободная фосфорная
кислота (pH увеличился с 2 до 5). Затем на катионите КБ-1 в Н-форме
была разделена смесь эфиров. При элюировании водой кокарбоксилаза
выделяется к концу процесса. Из концентрированного элюата кокарбокси-
лаза выделяется спиртом.
Реакция получения
сн3
сн2сн2он
Тиаминхлорид
Н3с
Уч1»
Тиаминмонофосфорный эфир
J II
СН2СН2О-Р-О -Н3РО4
он
(ТМЯ>) 442,34
СН3 О о
СН2СН2-О-Р-0-Р-0 -Н3Р04
он он
Тиаминдифоофорный Эфир
СН3 ООО
3 II II II
СН2СН2-О-Р-О-Р-О-Р-О -Н3РО4
он он он
ТиаминтрифОСфОрнь/й Эфир
(ТТФ) 602,31
Фосфорилирование тиамина (рис. 12). В реактор из эмалированной .
стали 1, снабженный ловушкой для улавливания HCI, загружают обезво-
женную (в вакууме 10—20 мм рт. ст. при температуре 125° С) 98%-ную
ортофосфорную кислоту, постепенно фосфорный ангидрид и медленно во
избежание сильного вспенивания тиаминхлорид-гидрохлорид; перемеши-
вают 30—60 мин. Затем добавляют ледяной воды и перемешивают реакци-
онную массу до полного растворения; ее переводят в реактор 2, куда зара-
нее наливают из мерника 3 безводный ацетон при температуре 10° С. После
перемешивания массу оставляют в аппарате на 24 ч при температуре 10° С
для осаждения фосфорных эфиров. Водно-ацетоновый слой декантируют
в сборник 4, откуда направляют на регенерацию в сборник-мерник 5 и
далее в ректификационную колонну 6. Фосфорилированный тиамин оста-
ется в реакторе 2.
Очистка фосфорных эфиров. В реактор 2 заливают воду и растворяют
фосфорилированный тиамин, состав которого примерно следующий (в %)
ТМФ — 37,7; ТДФ — 35,7; ТТФ —22,10, тиамин — 4,6. Раствор подают
в напорный сборник 7, а затем на колонну 8, наполненную анионитом
АН-25 в ОН-форме. Фосфорные эфиры элюируют дистиллированной водой
из мерника 9. Элюат собирают в приемнике 10, а последние промывные
с очень низким содержанием эфиров направляют в сборник 11 и далее в
отход. Элюат передают через мерник 12 в реактор-смеситель 13, куда вводят
активированный уголь, фильтруют через нутч-фильтр 14‘, фильтрат посту-
пает в сборник 15.
Разделение фосфорных эфиров тиамина. Эту операцию осуществляют
при помощи катионитной хроматографии. Очищенный раствор фосфорных
эфиров через мерник 16 пропускают через колонну 17, заполненную катио-
нитом КБ-1 в Н-форме. Элюируют ТТФ и ТДФ водой, а ТМФ — 5%-ным
раствором ортофосфорной кислоты. Элюаты направляют в сборники соот-
ветственно 18, 19 и 20. Состав фракций контролируют хроматографией
на бумаге.
Выделение фосфорных эфиров в кристаллическом виде. Фосфорные эфи-
ры выделяют путем очистки углем, сгущения и кристаллизации. Для этого
элюаты переводят в мерники 21, 22 и 23. Затем их направляют в реактор-
смеситель 24, где обрабатывают углем при температуре 60—65° С 15—
20 мин. Затем фильтруют через нутч-фильтр 25 и сливают в сборники 26,
102
27 и 28. Далее фильтраты сгущают в вакуум-аппарате 29 и кристаллизуют
добавлением из мерника 30 спирта в кристаллизаторе 31, выделяют ТДФ
и ТТФ. Кристаллы отфуговывают в центрифуге 32 и сушат в вакуум-су-
шилке 33 при температуре 18—20° С, ТМФ-фосфат выделяют аналогичным
образом при помощи ацетона.
ТМФ-фосфат, ТДФ и ТТФ представляют собой белый кристаллический
порошок. При бумажной хроматографии в системе растворителей: н-про-
Фосфорилирование тиамина
Ацетон
Рчистна фотрорнь/х лрироб
Рис. 12. Технологическая схема производства кокарбоксилазы.
пиоловый спирт — вода — 0,1 ж фосфатный буфер pH 5; 3:1:1 характерны
следующие R^: ТМФ — 0,46; ТДФ — 0,3; ТТФ — 0,22.
Для превращения в ТДФ подвергают ТТФ гидролитическому расщеп-
лению соляной кислотой и направляют раствор в реактор 2, где он с основ-,
ным раствором фосфорных эфиров проходит стадии очистки, разделения
и выделения.
ТМФ в гидратной форме получают из ТМФ-фосфата пропусканием его
через анионит в ОН-форме с последующим выделением из безводного аце-
тона или спирта.
Для превращения в ТДФ возможно ТМФ-фосфат направлять для фос-
форилирования в реактор 1 совместно с тиаминхлоридом. Выход кокарбо,-
ксилазы составляет около 30% от теоретического (на тиаминхлорид).
103
ТДФ-хлоргидрат получают из ТДФ-тетрагидрата путем обработки по-
следнего 5%-ным раствором соляной кислоты с последующим выделением
безводным спиртом — белый мелкокристаллический порошок [131].
Для получения устойчивой кокарбоксилазы для инъекций даны реко-
мендации в патентной литературе:
1) добавление Ь-ацетил-Ё-цистеина в количестве не ингибирующем ле-
карственное действие (японский патент № 1295, 21/1 1969 г.);
2) добавление физиологически неактивного количества дисульфида ди-
фениленамина или алкиламина (японский пат. № 27326, 25/XI 1968 г.).
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. Т а к а к i К- Prevention of какке in Japanese, navy 1885; Lancet, 1887, 11, 189.
2. E i j к m a n C. Virchow’ s Archiv, 1897, 149, 187; Gen. Tijdschr. V. Nederl. Indie,
1890, 30, 295; 1896, 36, 214; Arch. Hyg., 1906, 58, 150.
3. G r i j n s G. Gen Tijdschr. V. Nederl. Indie, 1901, 41, 3.
4. F u n к C. Brit. Med. J., 1912, 2700, 788; J. Physiol., 1912, 44, 50; 45, 75; 45 , 489.
5. Me Collum E., Kennedy C. J. Biol. Chem., 1916, 24, 491.
6. Wind aus A., Tschesche R., Ruhkopf H., L a q u e r F.,
Schultz F., Z. Physiol., Chem., 1932, 204, 123.
7, Шилов П. И., Яковлев T. Н. Основы клинической витаминологии. М.,
«Медицина», 1964, с. 52.
8. Диксон М., Уэбб Э. Ферменты. М., ИЛ, 1960, 814 с.
9. Abderhalden R. Ztschr. fiir Vitamin, Hormon und Fermentforsch, 1962,
12, 2/3, 83.
10. Ю p к e в и ч A. M. Успехи химии тиамина. — «Успехи химии», 1964, т. 33, вып.
4, с. 418.
11. Rosenberg Н. Chemistry and Physiology of the Vitamins, 1942, 99.
12. Williams R., J. Am. Chem. Soc., 1935, 57, 229.
13. Smakula A. Z. physiol. Chem., 1934, 230, 231.
14. Heyroth F., Loofbonrow J. Bull. Basic. Sci. Research., 1932, 4, 35.
15. W i n d a u s A., Tschesche R., G r e w e R. Z. physiol. Chem., 1935, 237,
98.
16. С 1 a r k e H., Gurin S. J. Am. Chem. Soc., 1935, 57, 1876.
17. W i 1 1 i a m s R., В uchman E., R u c h 1 e A. J. Am. Chem. Soc., 1935, 57,
1093; Williams R., J. Am. Chem. Soc., 1935, 57, 229.
18. G r e w e R. Nature, 1936, 24, 657; Z. physiol. Chem. 1936, 242, 89.
19. Andersag H., Westphal K- Ber., 1937, 70, 2035.
20. С 1 i n e I., Williams R., Finkelstein J. J. Am. Chem., Soc., 1937, 59, 1052;
Williams R., Cline I., Ibid., 1937, 59, 216.
21. V о g e 1 H. Chemie und Technik der Vitamine, 1943.
22. Kinnersley H., O'Brien I. Peters R. Bioch. J., 1935, 29, 701.
23. Wintersteiner O., Williams R., RuchleA. J. Am. Chem. Soc.,
1935, 57, 517.
24. Holiday E., Biochem J., 1935, 29, 718.
25. Williams R., Waterman R., Keresztesy J., Buchman E.,
J. Am. Chem. Soc., 1935, 57, 536.
26. В a r g e r G., В e r g e 1 F., Todd A. Nature, 1935, 136, 25; Ber., 1935, 68,
2257
27. L i p p m a n n F., Nature, 1936, 138, 1097.
28. P i e r s о n E., Encyclopedia of Chemical Technology, N. Y., 1955, 14, 38.
29. T a k a m i z a w a A., N a k a j i m a S., Cato H., C. A., 1959, 53, 16139.
30. Японский пат. № 6726, 1959; С. A., 1960, 54, 15405.
31. Stern К., Hofer J., Science, 1937, 85, 483.
32. S e r c h i G. Chimica, 1954, 30, 39.
33. Leichssenring G., Schmidt I. Ber., 1962, 95, 767.
34. Ohara M., Vitamins, 1963, 27, 393.
35. M a t s u k a w a T., J u r u g i S., Oka I., Ann. N. Y., Acad, of Sci., 1962, 98,
430.
36. Кругликов а-Л ь в о в а Р. П. Новый витаминный препарат тиаминпро-
пилсульфид и другие дисульфидные производные тиамина. — «Хим.-фарм. ж.»,
1970, № 11, с. 59—60.
37. Т р а в и н А. И. Синтез витамина Вг —ЖПХ, 1943, 16, с. 105—117.
38. Л и с и я н с к и й И. М. Витамин В, — тиамин. — В сб. «Витамины. Пищевая
промышленность за рубежом», 1956, Xs I, с. 127—137. Тоже. Синтез тиазолового
компонента молекулы витамина В1; 1957, Xs 3, с. 49—56.
39. Березовский В. М. Современные задачи в области химии и технологии
104
витаминов и перспективы развития их производства. — «Успехи химии», I960,
т g с 7__23.
40. М а х i bn Е. Пат. США № 2799676, 1957; С. А., 1958, 52, 1284.
41. I о s h i d a S., Y s h i z u k a W., К a t a о k a M. Япон. пат. № 10232, 1956;
С. A., 1958, 52, 15599.
42. I о s h i d a S., J s h i z u k a W. Япон. пат. 8285, 1956; С. A., 1958, 52, 11950.
43. U п о k i К. J. Pharmac. Soc. Japan., 1954, 74, 4; «РЖХим», 1956, 7, 19379.
44. P i п n e r A., Ber., 1884, 17, 2519; 1885, 18, 759; 1889, 22, 1615; 1893, 26, 2124;
1907, 42, 3020.
45. Gabriel S., Ber., 1904, 37, 3639.
46. G r e w e R., Z. physiol. Chem., 1936, 242, 89.
47. Anders ag H., Westphal К., Герм. пат. 685032, 11/XII, 1939; Англ. пат.
471416/30, VIII 1937; Франц, пат. 816432, 7/VIII 1937; Chem. Zbl. 1938, I, 937,
3800.
48. Челинцев Г. В. и Беневоленская 3. В. Новый способ получения
витамина Bt —ЖОХ, 1944, т. 14, с. 1142—1147. То же. О некоторых ацидэнолах.
1947, т. 17, с. 273—277.
49. Р у б ц о в И. А., Балякина М. В., Жданович Е. С., Преобра-
женский Н. А. О взаимодействии акрилонитрила с эфирами муравьиной и
щавелевой кислот. — «Труды ВНИВИ», 1953, № 4, с. 23—25.
50. Преображенский Н. А., Рубцов И. А., Лиснянский И. М.
Авт. свидет., № 93885, 1948. Бюлл. изобрет., 1952; № 6, с. 9.
51. Фодор Г., Герч А., Киш И., Колонии Я., В е й н Я-, Ковач Э.
Видоизмененный синтез 2-метил-4-амино-5-этоксиметилпиримидина. ЖОХ, 1951,
т. 21, с. 1897—1902.
52. Walden Р., Вег., 1907,40,3215.
53. В и с h m а п Е., Hoffman A. Lieb. Ann., 1936, 100, 130.
54. Сборник технологических инструкций по производству витаминов. М., Пищепром-
издат, 1943, с. 111.
55. В г a u п I., Вег., 1934, 67, 1769.
56. В о л к о в а А. ЖРХО, 1871, № 3, с. 239.
57. Б е р е з о в с к и й В. М. Химия витаминов. М., Пищепромиздат, 1959, с. 420.
58. Н а п t z s с h . А. Ann., 1889, 250, 262.
59. Н а п t z s c'h A., Popp G., Lurcher H. Ann., 1889, 250, 257.
60. Barger G., В er gel F., Todd A., Nature, 1935, 136, 259; Ber., 1935, 68,
2257.
61. Todd A., Bergel F., Karimullah. Ber., 1936, 69, 217.
62. Todd A., Bergel F., Karimullah, Keller R. J. Chem. Soc.,
1937, 361.
63. T о d d A., Bergel F., Jacob A. J. Chem. Soc., 1936, 1555.
64. Б e э p А. А., Рубцов И. А. Синтез витаминов. M., Пищепромиздат, 1956,
с. 231" 217
65. Слободин Я. М. , Гельме Е. Э., — «ДАН СССР», 1943, т. 39, с. 152.
66. Т у р с и н В. М., Канина Т. И., Шмуйлович Л. М. Хлорирование
5-ацетоксипептанона-2—ЖОрХ. В сб. «Проблемы получения полупродуктов
промышл. орг. синтеза», 1967, с. 49.
67. Т у р с и н В. М., Канина Т. И. Продукты хлорирования 3-ацетбпропил-
ацетата и факторы, влияющие на их состав. — ЖПХ, 1970, т. 43, с. 377—380 с ил.
68. В u с h m а п Е. J. Am. Chem. Soc., 1936, 58, 1803.
69. Wenz А., Герм. пат. № 664789, 5/IX 1938.
70. G г о m a t к а О., Пат. США № 2160867, 6/VI 1939; Герм. пат. № 670131, 12/1
1939; Жданович Е. С., Лиснянский И. М. Авт. свидет., № 99150, 1954; Бюлл. изо-
брет., 1954, № 10, с. 13.
71. Andersag Н., Westphal К-, Вег., 1937, 70, 2035.
71а. Londergan Т. J. Am. Chem. Soc., 1953, 75, 4456.
72. Baumgart en Р., D о г п о w А. Вег., 1940, 73, 44.
73. Магидсон О. Ю., Травин А. И. Авт. свидет., № 59808, 1939; Бюлл.
изобрет., 1941, № 4, с. 19.
74. Р у б ц о в И. А., Балякина М. В., Жданович Е. С. и др. Авт. сви-
дет. № 99479, 1954; Бюлл. изобрет., 1954, № 12, с. 13.
75. S е b г е 1 1 W., Harris S. The Vitamins, N. Y., 1954, 3, 411.
76. Будешинский 3., Копецкий Я. Chem. Listy, 1954, 1364.
77. Рубцов И. А., Балякина М. В., Жданович Е. С., Преобра-
женский Н. А. Синтез хлоргидрата хлорида тиамина (витамин Вх). — «Тру-
ды ВНИВИ», М., Пищепромиздат, 1954, № 5, с. 10—12.
78. С а г n a h а п В., Пат. США 2678312; С. А., 1955, 49, 6323; Аигл. пат. № 717445;
РЖХим., 1956, 69768.
79. Англ. пат. 768773; С. А., 1957, 51, 15590.
80. W. Stieg. Пат. США 2833769, 21/VI 1955; Offic. gaz., 1958, 730 № 1, с. 213.
81. Пат. ГДР, 11163, 1956; 34854, 1964.
82. I m a i Т., Ma kino К-, Z. physiol. Chem. 1938, 252, 76.
83. Т о d d A., Bergel F., J. Chem. Soc., 1937, 364.
105
84. М. К 1 i п g е n f u s s, Франц, пат. 831110, 1938; Zbl., 1939, I, 729.
85. Andersag H., Westphal К. Герм. пат. 671787, Zbl. 1938, 1, 3800.
86. Matsukawa T., J. Pharm. Soc. Japan, 1942, 62, 417; C. A. 1951 45, 4723.
87. M a t s u к a w a T.( О lh t a M. Пат. США 2184720 , 26/XII, 1939; Zbl 1939,1,
3930.
88. Matsukawa T., H. H i r a n o. J. Pharm. Soc., Japan., 1953, 73 4 375;
«РЖХим», 1955, 52070.
89. I о s h i d a S., U n о к i К- J. Pharm. Soc., Japan, 1953, 73, 6, 631; «РЖХим»,
1956, 68485.
90. Matsukawa T., В i t a m i n , 1953, 6, 3, 73; 6, 631; РЖХим, 1954, 39571,
91. T у p с и н В. M., Чеботарева Л. Г., Белоусова Л. Н. и др. По-
лучение витамина Bv ЖПХ, 1961, т. 34, с. 229—232. То же. Авт. свидет., № 118502,
1958; Бюлл. изобрет., 1959, № 6, с. 10.
92. Kolsch, J. Am. Chem. Soc., 1943, 65, 457.
93. Лиснянский И. М., Золотарев Н. С., С и р о т е н к о В. И. Не-
прерывный способ получения этилового эфира муравьиной кислоты. — «Хим,-
фарм. ж.», 1969, 6, с. 49—50.
94. Шапиро Б. И. Исследования в области промышленного синтеза витамина Bb
Автореф. кандид. дисс., М., 1968.
95. Лиснянский И. М., Корнилов А. А., Шапиро Б. И. и др.
Авт. свидет., № 190890, 1955; Бюлл. изобрет., 1967, 3, с. 24.
96. Шнайдман Л. О., Дульчина Б. М. Ускоренный метод получения а-
метоксиметилен-р-этоксипропионитрила. — В сб.: «Витаминная промышленность»,
М., Пищепромиздат, 1958, Ns 5, с. 13—17. То же. 1960, № 6, 32—37.
97. Braun I. Вег., 1934, 67, 1769.
98. В о л к о в а А., ЖРХО, 1871, 3, 239.
99. Epps D., Reid Е. J. Am. Chem. Soc., 1916, 38, 2128.
100. Mitchell J., Reid E. J. Am. Chem. Soc., 1936, 53, 321.
101. Лиснянский И. M., Жданович Е. С. Авт. свидет., № 100776,
2/XI 1954; Бюлл. изобрет.; 1955, № 6, с. 9.
102. Ж Д а н о в и ч Е. С. Исследования в области синтеза и разработка методов про-
изводства витаминов группы В. Докл. докторск. дисс., М., 1965, с. 49.
103. Knorr Р„ Вег., 1917, 50, 229.
104. 3 о л о т а р е в Н. С., Е н ю т и н Ю. В., Ржевский Н. К. и др.
О некоторых усовершенствованиях технологии получения 2-метил-4-амино-5-это-
ксиметилпиримидина. — «Хим.-фарм. ж.», 1971, № 8, с. 45—47.
105. Бурова Л. Е., Плановский А. Н., Белая Е. И. Исследование
экстракции а-окси-р, p-диметил-у-бутиролактона непрерывным методом. — «Меди-
цинская промышленность СССР», 1966, 2, с. 47—50.
106. Драгунов С., Шилина А. Авторское свидетельство СССР, № 55842,
1938; Бюлл. изобрет., 1939; Ns 10, с. 11.
107. Смейкал К-, Кетнинг М. Авт. свидет., № 110576 1956; Бюлл. изо-
брет., 1958, № 1, с. 29.
108. Банковски О., Кетнинг М., К л е м т Г. Авт. свидет., № 113530,
1956; Бюлл. изобрет., 1958, Ns 6, с. 23.
109. Braun A., J. Am. Chem. Soc., 1918, 793.
НО. Brill J., Plummer R. Пат. США. 2092723, 1937.
111. Klempt W., Герм. пат. 658158, 1934.
112. T a n n er W., Пат. США., 2106579, 1938.
113. Лиснянский И. М., К и п е р Н. Я., Ш а п и р а Б. И. Непрерывный
способ получения формамида. — «Хим.-фарм. ж.», 1968, 5, с. 47—48 с ил.
114. Бурова Л. Е. —«Медицинская промышленность СССР», 1964, 12, 27.
115. Б у р о в а Л. Е., Плановский А. Н. О предельной скорости движения
дисперсий фазы в колонном экстракторе с механическим перемешиванием. — «Ме-
дицинская промышленность СССР», 1966, 7, с. 35—39.
116. Григорашвили Е. И., Золотарев Н. С., Буймов А. А. и
др. Непрерывный способ получения 2-метилфурана методом каталитического гид-
рирования фурфурола. — «Хим.-фарм. ж.», 1969, 6, с. 50—52 с ил.
116а. НатрадзеА. Г., Коган С. , Новикова К. Е. — «Медицинская
промышленность СССР», 1950, 3, 15.
117. Топчиев К. С., Павлов Л. Н. Способ получения ацетопропилового
спирта из метилфурана. Авт. свидет. № 48114, 1937; Топчиев К. С. О механизме
образования у-ацетопропилового спирта при гидрировании — гидратации а-ме-
тилфурана. — «ДАН СССР», 1938, 19, с. 497—98.
118. 3 о л о т а р е в Н.' С., Л а т в и с П. П., Буймов А. А. и др. Исследо-
вание процесса получения у-ацетопропилового спирта из фурфурола. — «Хим,-
фарм. ж.», 1972, 3, с. 52—56 с ил.
119. Swadish S., Smith S., Dunlop A., J. org. Chem., 1949, 14, 692; 1951,
16, 476.
120. С о p e 1 i n H. Пат. США 2682546, 20/II 1952; Offic. gaz., 1954, 683, № 5, c. 1149.
121 .JT у p с и н В. M., Чеботарева Л. Г., Г p о й з и к Т. Ю. Применение
106
ионообменной смолы как катализатор в синтезе 2-диметоксиметил-З-метоксипро-
пионитрила. — ЖПХ, 1965, т. 38, с. 443—445.
122. Т о m i t а М., U у е о S., Takamizawa A., Moeda R., J. P h a r m.
Soc. Japan, 1954, 74, 742.
123. Takamizawa A. J. Pharm. Soc. Japan, 1954, 74, 748; 1958, 78, 682.
124. Matsukawa T., Vitamins (Japan), 1954, 7, 815.
125. Matsukawa T. J. Vitaminology (Japan), 1954, 1, 13.
126. Кругликова-Львова P., Лобанова M. Материалы VI научной
сессии ГГМИ и Всесоюзного симпозиума по тиамину. Минск, 1966, с. 414; То же.—
Хим. -фарм. ж., Xs 11, с. 59.
127. Matsukawa Т., Jurugi S., Oka I. Ann. of the N. Y. Acad, of Sci.,
1962, 98, 430.
128. Matsukawa T., J. Pharm. Soc., Japan, 1953, 73, 497, 216; РДХим, 1955,
52069, 52071,
129. T у p с и н В. M., Иванова E. А. Тиаминпропилдисульфид.— ЖОрХ, 1965,
т. 1, с. 1151—53.
130. Березовский В. М., Колтунова В. И., Шлимович Е. А.,
Девятнин В. А. Синтез монофосфорного эфира тиамиифосфата.—ЖОХ, 1962,
т. 32, с. 3890—92. То же. Колтунова В. И. Синтез фосфорных эфиров тиамина и их
производных. Автореф. канд. дисс. М., 1969.
131. Березовский В. М., Колтунова В. И., П е к е л ь Н. Д.,
Шлимович Е. А. Синтез кокарбоксилазы— ЖОХ, 1963, т. 33, с. 49—55 сил.
132. Auhagen Е., Z. physiol. Chem., 1932, 209, 20.
133. Lohman К., Schuster Р., Biochem Z., 1937, 294, 188.
134. Stern К- J- Hofer, Science, 1937, 85, 483.
135. Tauber H., J. Am. Chem. Soc., 1938, 60, 730.
136. Wei j lard J., Tauber H. J. Am. Chem. Soc., 1938, 60, 2269.
137. К i h a c h r i о Uchara. Япон. пат. 10872, 1960; С. A., 1961, 55, 9, 8774.
138. Tanaka T., J. Pharm. Soc. Japan, 1956, 76, 1314; C. A., 1957, 51, 3607.
139. Турсин В. M., Чеботарева Л. Т., ХФЖ, 1971, Xs 5, с. 27.
Глава 5. ЕРОИЗВОДСТВОГСИНТЕТИЧЕСКОГО РИБОФЛАВИНА
(ВИТАМИНА В2)
Витамин В2 (рибофлавин) входит в состав ряда ферментных систем,
регулирующих окислительные процессы в клетках. Рибофлавиновые кофер-
менты катализируют дегидрирование в животном организме а-аминокис-
лот, альдегидов, моносахаров и др.
Рибофлавин участвует в синтезе белков и жиров, в клеточном дыхании,
оказывает регулирующее влияние на состояние центральной нервной си-
стемы, воздействует на процессы обмена в роговице и сетчатке глаз (на
функцию зрения). Оказывает регулирующее влияние на кровеносную си-
стему, на функцию печени, кожи и слизистых оболочек рта [1, 2, 3].
Специфическое влияние недостатка рибофлавина на человеческий ор-
ганизм выражается в авитаминозном заболевании, известном под названием
арибофлавиноз. Он выражается в поражении слизистой оболочки рта и
глаз [5]. Рибофлавин влияет на рост птиц [4]; при его недостатке проис-
ходит задержка роста.
Открытие и выделение витамина В2. После того как в 1881 г. Н. И. Лу-
нин доказал неполноценность искусственного молока и содержание в на-
туральном молоке, кроме белков, жиров, углеводов и минеральных со-
лей, жизненно необходимых дополнительных веществ, было установлено
[6], что в натуральном молоке содержится воднорастворимое вещество,
способствующее росту животных. В 1925 г. из молока был выделен желтый
пигмент, названный лактохром [7]. В 1932 г. из дрожжей был выделен
и изучен «желтый дыхательный фермент Варбурга» [8]. Было установлено,
что он построен из двух частей—белка и желтого красителя. Последний был
впоследствии идентифицирован как витамин В2 и был назван лактофлави-
ном (флавин молока). Когда выяснилось, что в состав витамина В2 входит
рибоза, то тогда ему было присвоено название рибофлавина [9]. Одно-
временно было установлено, что в свободном виде рибофлавин встречается
лишь в молоке, моче и в сетчатке глаз. Во всех других естественных источ-
никах он находится в виде моно- или динуклеотида (кофермента). Выде-
107
ление рибофлавина из природных источников осуществляется методом ад-
сорбции'на фуллеровой земле [10], либо асканите (тальк, окись алюминия,
мел, каолин и кизельгур не адсорбируют витамина В2). Элюирование про-
изводится либо ацетоном, либо смесью пиридина, метанола и воды. После
очистки и удаления растворителя рибофлавин выкристаллизовывают из
воды, либо из водного спирта, либо из разбавленной уксусной кислоты
[11].
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ВИТАМИНА В2 (РИБОФЛАВИНА)
Витамин В2-6,7-диметил-9-(£)-1-рибитил)-изоаллоксазин имеет следую-
щую структурную формулу:
ОН ОН он
I I I
сн2—сн—сн-сн-сн2он
1| 2’ 3' 4’ 5'
,, „ ^сн N N
Н3С-С^8'-С-'9ХС^ЛСО
I' II f I 2I
H3C-C^5^Cxio<c^4jNH
СН N СО
РибогрлаВин
37В, 36
Его эмпирическая формула C17H2aN4Oe, молекулярная масса 376,36.
Он кристаллизуется в виде оранжево-желтых игл с температурой плавле-
ния 282° С с разложением (темнеет при температуре 240° С). Рибофлавин
нерастворим в жирах и в растворителях жиров,
слабо растворим в воде. При температуре 27,5° С
насыщенный водный раствор содержит 0,012%
рибофлавина, при 40° — 0,01944%, а при 100°—
0,23% [11, 12]. В щелочной среде рибофлавин
лучше растворим, нежели в нейтральной. Слабо
растворим в нормальном бутиловом, амиловом,
этиловом (0,0045% при 27,5°) и метиловом
спиртах. В нейтральных водных растворах он
имеет зеленовато-желтую окраску с интенсивной
желто-зеленой флуоресценцией, которая умень-
шается вплоть до исчезновения как в кислотной,
Рис. 13. Спектр поглоще- так и в щелочной среде (максимум при pH 6—
ния рибофлавина в УФ- 7). Интенсивность флуоресценции зависит от кон-
лучах- центрации пигмента [13] и достигает максимума
при концентрации 0,003%. Изоэлектрическая
точка витамина В2 находится при pH 6,0.
Спектр поглощения рибофлавина (в воде) имеет один максимум в
видимой части света при длине волны 445 нм и три максимума в ультрафио-
летовой части спектра при длине волны 372; 269; 225 нм (рис. 13). Е%ы ра-
вен для 7.225=750; Х2в9= 830; Х372= 275; X44s = 310 [14, 15]. Растворимость
рибофлавина в соляной кислоте при температуре 15° С составляет [161:
Концентрация НС1, % .......... 3 16 19 112 115 118
Растворимость, %................ 0,026 |0,060 |0,220 |0,610 |1,000 |2,900
Рибофлавин умеренно растворим в ледяной уксусной и муравьиной
кислотах [4]: Он имеет удельное вращение [я]^= —114° в 0,1 н. растворе
NaOH. Максимум спектра флуоресценции наблюдается при 565 нм [11].
Рибофлавин в щелочной среде переходит в люмифлавин (6,7,9-триметил-
изоаллоксазин), растворяющийся в хлороформе и не обладающий биологи-
ческими свойствами витамина В2. При облучении солнечным светом в нейт-
108
ральной и кислой среде образуется биологически неактивный люмихром
(6,7-диметилаллоксазин), обладающий сильной голубой флуоресценцией
[17].
Люмифлабин
Установлено [13], что прямой солнечный свет быстро разрушает рибо-
флавин при любом pH среды. Наиболее губительное действие на рибофла-
вин оказывают лучи света с длиной волны 440 нм.
Рибофлавин в больших концентрациях и в растворах с низким значе-
нием pH более устойчив к действию света, чем при низких концентрациях
в щелочных условиях. Аскорбиновая кислота, чайный танин способны за-
медлять разрушение рибофлавина. Рибофлавин устойчив к кислотам, бро-
му и к таким окислителям, как перекись водорода и концентрированная
азотная кислота. Хромовая кислота окисляет его до аммиака, углекислоты
и азота [6]. Рибофлавин в естественных источниках сырья связан в значи-
тельной части с белком; связь эта расщепляется протеолитическими фер-
ментами [18].
Флавиновые коферменты. В природных источниках рибофлавин находит-
ся в связанном состоянии, входя в состав желтого окислительного фермен-
та, цитохромредуктазы, диафораз, оксидаз, объединяемых в группу флави-
новых ферментов [8]. Рибофлавин входит в молекулу фермента в виде
соединения с фосфорной, адениловой кислотами в виде кофермента. По-
следний приобретает ферментные свойства только в сочетании с протеи-
ном (апоферментом). Наиболее простой флавиновый кофермент — рибофла-
вин-5'-фосфорный эфир или рибофлавинмононуклеотид, который образует
эфирную связь по первичной гидроксильной группе рибозного остатка
[19, 20]. Строение рибофлавина-5-фосфата доказано синтезом путем фос-
форилирования рибофлавина метафосфорной кислотой при температуре
50—60° С [21, 22], либо пирофосфорной кислотой [23]:
ОН ОН ОН о
1’ 2’1 3'1 4'1 5’ и
СН2-СН-СН-СН-СН2О-Р-ОН
о
Рибофлавин - 5'~ фосфорный эфир (РМФ)
456,36
Его эмпирическая формула C17H21N4O9P; температура плавления 215° С;
[а]о=+44,5° (в соляной кислоте). В воде растворим в 200 раз более, чем
рибофлавин. Его спектр поглощения (в воде) имеет максимумы при длине
волны 442 ; 372 ; 268; 222 нм. Применяется для внутривенной инъекции
в медицинской практике [24]. Синтез РМФ см. стр. 132. Из наиболее слож-
ных флавиновых коферментов следует отметить рибофлавинадениннуклео-
тид, представляющий собой соединение пирофосфатной связью фосфорного
эфира рибофлавина и фосфорного эфира аденин-И-рибозида (адениловой
кислоты). Он входит в состав желтого окислительного фермента, диафора-
зы II. Строение кофермента доказано синтезом его из аденозин-5-фосфата
109
и рибофлавин-5'-фосфорного эфира [25] и выражается следующей струк-
турной формулой:
ОН
он
СН2О-Р-О-Р-О-СН2
но-с-н о
МН
но-с-н
но-с-н
н
он
он
о
Рибофлабинадениннуклеотид
(Ф/\Д)
ФАД может быть синтезирован из морфолидата аденозин-б'-монофос-
фата и триэтиламмониевой или децилтриметиламмониевой соли рибофла-
вин-5'-монофосфата (В. Березовский, Е. Хомутова).
Флавиновые ферменты катализируют реакции дегидрирования различ-
ных соединений в животном организме, как, например: а-аминокислот
в а,-кето кислоты [8], альдегидов — в карбоновые кислоты [26], глюкозы —
в глюконовую кислоту [27]. Флавиновые ферменты обладают высоким
окислительно-восстановительным потенциалом благодаря их способности
присоединять электроны с превращением в восстановленную форму фла-
вина. Присоединенные электроны флавины передают соответствующим
акцепторам, окисляясь в исходную форму. Из флавиновых коферментов
широкое применение имеет рибофлавин-5'-фосфорный (рибофлавинмонону-
клеотид) и его натриевая соль.
МЕТОДЫ СИНТЕЗА РИБОФЛАВИНА И ВЫБОР РАЦИОНАЛЬНОГО МЕТОДА
ДЛЯ ПРОИЗВОДСТВА
Молекула рибофлавина состоит из трех компонентов: ароматического,
алифатического и пиримидинового.
В качестве ароматического компонента применяют 3,4-диметиламино-
бензол (0-4-ксилидин), алифатического-£>-рибозу, а пиримидинового — ал-
локсан или барбитуровую кислоту
h3c-t<Ynh2
н3с-Ч>
Н Н Н
I I I
носн2-с-с-с-с^
2 I I I хн
ононон
О-4-- Ксилидин D - Ри доза
N
нос* 4=0.
O=CVxNH
V.
II
о
Аллоксан
NH
О=С* 4=0
H2CxcxNH
I)
о
барбитуродая кислота
Существующие методы синтеза рибофлавина в основном сводятся к по-
лучению 3,4-диметил-6-аминофенил-£>-рибамина и конденсации его с алло-
ксаном [28, 29] или'с дихлорбарбитуровой кислотой [30], или к конденса-
ции 3,4-ксилил-6-фенилазо-1-£>-рибамина с барбитуровой кислотой [31]
по следующей схеме:
110
сн2он
но-с-н
I
но-с-н
I
но-с-н
СН2ОН
но-с-н
I
но-с-н
I
но-с-н
I
J, 4 -Диметил -6- а мини -
фенил-1 -Д-рибамин
сн2
о о
Аллоксан Рибофлавин
сн2он
I
но-с-н
I
но-с-н
но-с-н
ЗД-Ксилил -6-фенилазо- барбитуробая
1 -Л-рибамин кислата
Реакцию конденсации проводят в кислой среде в присутствии борной
кислоты [32, 33], что значительно повышает выход (до 90%). В противном
случае выход резко снижается вследствие образования побочного продук-
та. Катализаторами этой реакции являются также H2S и SnCl2 [34].
Все синтезы рибофлавина в основном различаются методами получения
диметиламинорибамина.
1. Конденсация о-нитроксилидина с D-рибозой и каталитическое вос-
становление образовавшегося рибозида в диамин [35] по следующей схеме
(метод Куна):
Н3С-1^ |1-NH2 . HoC-r^>[-NH-CH-fcHOH\-CHo +Н2] Ндс%-nh-ch2’(CHOH)3CH2OH
НзС-Чх М°2 HjC-Ux^JJ-NOa Н3С-^/НЧН2
О - Нитраксилидин
6-Нитро -3,k - диметилфенил -Л-рибозид 3,Ь-Диметил - б-аминорзенил-П-рибамин (диамин)
Выход рибофлавина по этому методу составляет 16% (на D-рибозу).
2. о-Нитрохлорксилол конденсируют с аминосахарами и продукт реак-
ции восстанавливают водородом в диамин по следующей схеме (метод Кар-
рера):
.HaC-f^Vci
'H3cA^^NO2
+ H2NCH2-(CHOH)xCH2OH
О-Нитррцларксилол
___^HjC-r^jpNH-CHa-fcHOH^CHaOH
н3сД>^Лмо2
27 - Рибозид
+ Н2?
____Н3С-f^i]- nh-ch2 -(choh)xch2oh
Н3С—
Диамин
Этот метод дает удовлетворительный выход, если аминосахар содержит
менее четырех гидроксилов. При х>3 выход диамина очень низок [36—38].
3. о-Динитроксилол конденсируют с D-рибозоамином в водном спирто-
вом растворе и каталитически восстанавливают в диамин по следующей
схеме [39]:
H3C7irNO2+ Н INCH (СИОН-) СН пи _Н3С^ИН-СН2-(СНОН)3СН2ОН _+Н;
h3c-4>No2+ H2NCH2^HOH^H2OH—h3CJ<JLNo2
О -Динитроксилол
6-нитро-3,Ь -диметилфенил - И -рибамин
Н 3С—TN н-сн2-Сс нон)3- СН2ОН
Н3С-^Ч>^-^Н2
3,Р -Диметил -6-аминофенил-В-рибамин
Выход рибофлавина по этому методу составляет 4,5%, считая на рибозу
111
4. Конденсируют о-3,4-ксилидин с D-рибозой в спиртовом растворе
(40—42]. Образуемый при этом о-3,4-ксилидин-М-Д-рибозид без выделе-
ния из реакционной массы восстанавливают каталитически водородом [43]
в 3,4-диметилфенил-Р-рибамин. Вторая аминогруппа вводится в послед-
ний путем сочетания с фенилдиазонием (или его производными). Выход
азосоединения составляет 92% от теоретического [44]; восстановление его
в диамин может быть осуществлено с выходом 85% [20]. Синтез осуществ-
ляют по схеме Каррера
Н3С|Г-NНз +D-рибоза _ н3С|Т-n н- с н - (с нон)3-с н 2 +н2]
H3c-4jJ H3C-4J
О -3,4-Ксилидин о -3,4 -Ксилидин -N -I-рибозид
___ н 5 с-N Н-С Н2-£ Н он)3- с Н2ОН_________Ji jC-r^jr-N н-сн2- £ н он)3-сн2он + Н2
Н3С~Ч^ + C6HSN=NC1 Н3С A^J-N=N-C6H5 ’
0-3,4 - Ксилил - 27 - рибамин о -Ксилил -6- фенилазо-1-В -рибамин
Н 3C Н-С н 2 - (с н он)3 - с Н2ОН
3.4 - Диметил - 6 - аминофенил -Б-рибамин
Выход рибофлавина по этому методу довольно высок и составляет 38%
к теоретическому (на рибозу).
5. Для конденсации с D-рибозой применяют 3,4-диметил-1,6-фенилен-
диамин, в котором одна аминогруппа находится в виде уретана [28]. Син-
тез идет по следующей схеме:
ИзС-г^^ГМЩ,
НзС-Ц^Д-мнсооСзН.;
I---о-----,
^п-рибит НзС-р^р-NН-CН-(СНОН)3-СН2
НзС-’^Ч/ NH-COOC2H5
+ н2
____H3C-p^>r-NH-CH2-(CHOH)3-CH2OH_____^HjCqi^^NH-C^-CCHOHlg-C^OH
H3C'*^4/"NH^COOC2>I5 ОшаиДр Нзс
В данном методе аминогруппу защищают при помощи карбоэтокси-
группы
Н3С-^Ч-ЫО2 СзЩОН H.C-p^p-NOj +Н2, ЩС-г^р-ЦН,
H3C-U4^LNH2 СОС12 НзС-Ч^Д-мнсООСзНг ’"h3C-4^A-NHCOOC2Hs
Выход рибофлавина составляет 14—15% по рибозе [11].
Во всех описанных методах синтеза диметиламинофенилрибамина в ка-
честве алифатического компонента использовали D-рибозу. Оригинальным
методом синтеза Диамина является конденсация о-4-ксилидина с D-араби-
нозой, которая более доступна, чем D-рибоза [45]. При наличии неболь-
ших количеств бензойной кислоты образуемый D-арабинозид после нагре-
вания до 75° С претерпевает перегруппировку Амадори и переходит в
D-изоарабинозамин. После каталитической гидрогенизации в щелочном
растворе D-изоарабинозамин образует из двух один эпимер •— 3,4-диметил-
фенил-Д-рибамин, выход его составляет 13% к теоретическому (на D-ара-
бинозу) [11]. Далее путем сочетания последнего с фенилдиазонием полу-
чают диамин. Синтез протекает по следующей схеме (метод Вейганда):
112
О ОН Н Н ОНН Н
~ II I I I , III
Н3С-Г YNH2 + C -С — С-С-СН2ОН______ HjC-r^S-NH-CH-C-C-C-CHsOH_________
H3c~kJ н н он он кт H3c-^J I Н ОН I
О - 3,4 - Ксилидин ...О ।
В-Арабинозид
бензойная
кислота
В - Арабинозамин
н н н
ын—сн2—с—с—с—сн2он
он он он
н н
I I
со-с-С-СН2ОН
ОНОН
Изомеризация
3,h - Диметилфвнил -В - рибамин
+ н2
Опись платины 6 щелочном
растворе
н н н
I I 1
Н 3С-н-сн2- с-с-с-сн2он
h3c_L iLn=n-c6h5ohOh oh
3,4-Ксилил-6 -фвнилазо- 1-В-рибамин
Таким образом, в основу промышленного синтеза рибофлавина могут
быть взяты три схемы: Куна [35], Каррера [40] и Вейганда [45].
ОСНОВНЫЕ ПОЛУПРОДУКТЫ ДЛЯ СИНТЕЗА РИБОФЛАВИНА
D-Арабиноза. Для синтеза диамина можно исходить из D-арабинозы и в
последующем продукт конденсации ее с ксилидином подвергнуть изо-
меризации. Для получения D-арабинозы существует ряд методов [45—
49]. Интерес представляет метод, основанный на окислении D-глюконата
кальция перекисью водорода в присутствии небольших количеств уксусно-
кислого железа в качестве катализатора [50].
D-Рибоза. D-Рибоза может быть получена из дрожжей путем гидроли-
за содержащихся в них нуклеиновых кислот [51 ]. Из 2 кг дрожжей можно
получить 1—2 г чистой рибозы [52]. Синтетическим методом D-рибозу
получают из глюкозы путем окисления ее в щелочной среде кислородом
воздуха в арабоновую кислоту, эпимеризации последней, получения рибо-
нолактона и восстановления последнего амальгамой натрия в D-рибозу
[37, 53, 54].
Один из наиболее эффективных методов получения D-рибозы заклю-
чается в следующем [53]. Глюкозу окисляют в щелочном растворе при тем-
пературе 40—48° С в избытке мелкораспыленного воздуха под давлением.
Полученный D-арабонат калия переводят в арабонат кальция обменной
реакцией с хлористым кальцием. Арабонат кальция подвергают эпимери-
зации в присутствии пиридина или гидрата окиси кальция в рибонат каль-
ция. Последний в результате лактонизации под вакуумом при нагревании
переходит в D-рибоно-у-лактон, который восстанавливают амальгамой
натрия в D-рибозу. Реакцию получения D-рибозы можно изобразить сле-
дующей схемой:
СООН соон С 1 н-с=о
но-с-н н-с-он н-с-он 1 н-с-он
1 1 1 о 1
н-с-он н-с-он Н-С-ОН 1 н-с-он
н-с-он н-с-он Н-С 1 н-с-он 1
СН2ОН СН2ОН сн2он СН2ОН
В - АрабоноЗая В - РибоноЗая Рибонолактон В- Рибоза
кислота кислота
Переход от рибоновой кислоты к D-рибозе для получения 3,4-ксилил-
D-рибамина может быть осуществлен и другими методами, среди которых
113
можно отметить следующие: 1) рибоновую кислоту превращают в хлор-
О
ангидрид — ROCH2(CHOR)3C\ , который восстанавливают в альдо-
С1
О
зу — ROCH2(CHOR)3C\ , а затем в П-рибозу [55]; 2) рибонолактон кон-
Н
денсируют с о-4-ксилидином с получением анилида, превращаемого в хлор-
амин. Последний восстанавливают в 3,4-ксилил-П-рибамин [56]; 3) рибо-
О
нолактон превращают в D-рибонамид — ROCH2(CHOR)3C^ ; последний
nh2
превращают в рибонитрил, который конденсируют с о-4-ксилидином [30,
57].
Однако из описанных выше методов в практике нашел применение ме-
тод получения рибозы путем восстановления D-рибонолактона амаль-
гамой натрия.
0-4-Ксилидин (3,4-диметиламинобензол). Получение о-4-ксилидина из
технического ксилидина, который в свою очередь синтезируют из техниче-
ского ксилола, является неэффективным из-за низкого содержания в этих
продуктах чистого вещества [58, 59]. Поэтому о-4-ксилидин необходимо
синтезировать в чистом виде.
Существует несколько методов его синтеза [27]:
1) из чистого о-ксилола I нитрованием получают смесь изомеров, из
которой выделяют 3,4-диметилнитробензол II, который восстанавливают
в ксилидин IV [60]; при этом получают также изомер V [61];
2) из чистого о-ксилола бромированием на холоду [62, 63] получают
3,4-диметилбромбензол III.
Аминирование последнего водным аммиаком под давлением около
75 кгс/см2 при температуре 195° С в присутствии однохлористой и метал-
лической меди дает ксилидин с выходом 66% [63]. Общий выход о-4-кси-
лидина составляет 62%-;
3) из р-нитротолуола обработкой дихлорметиловым эфиром (В. Бере-
зовский) в присутствии хлорсульфоновой или разбавленной серной кисло-
ты получают с хорошим выходом (около 95%) 2-хлорметил-4-нитротолуол,
который после каталитического восстановления с катализатором платино-
вым в уксуснокислой [64] или никелевым в спирто-щелочной среде [651
или оловом и НС1 [66], или электролитического восстановления на свин-
цовом катоде даёт о-4-ксилидин с выходом в последнем случае 74—78%
к теоретическому. Процесс протекает по следующей схеме [53]:
114
Выход о-4-ксилидина по этому методу равен 50% (на р-нитротолуол).
Из анализа эффективности методов синтеза о-4-ксилидина следует, что
в промышленных условиях могут быть применены два метода синтеза:
из о-ксилола бромированием на холоду с последующим аминированием
получаемого бромксилола;
из р-нитротолуола обработкой дихлорметиловым эфиром с последую-
щим электролитическим восстановлением получаемого 2-хлорметил-4-нит-
ротолуола.
Первый метод более эффективен, так как он предусматривает две стадии
вместо трех (учитывая стадию синтеза дихлорметилового эфира из пара-
форма).
Пиримидиновый компонент. В качестве пиримидинового компонента
для синтеза витамина В2 можно применять аллоксан или барбитуровую
кислоту. Хотя при конденсации диамина с аллоксаном можно получить
несколько лучший выход нежели с барбитуровой кислотой [33 ], но все же
при выборе пиримидинового компонента следует отдать предпочтение бар-
битуровой кислоте, так как при синтезе аллоксана исходят из барбитуро-
вой или дихлорбарбитуровой кислоты или из мочевой кислоты [64].
Барбитуровую кислоту синтезируют из диэтилового эфира малоновой
кислоты и мочевины под влиянием этилата натрия.
Конденсацию проводят в абсолютном спирте при нагревании [64] по
схеме [53], изложенной на стр. 111.
сС„,, сс„ „ z , ONa Nil ONa NH NH
I OC2H5. C2H5ONa И OC2H5 CO<NH2)2_ ''O' kCO _____________________________ CO HC1_°C
<rH2 -С2Н5ОНГ СИ -C2H,OH HC4 NH2 - C2H5OH* HCx /NH Н2Щ /NH
COOC2H5 COOC2H5 COOC2H5 сб co
Диэтилмалонобый Натрий малонобый • Уреид малонобого ' барбитуробая
Эфир Эфир aipupa кислота
Этот синтез осуществляют с выходом 80% к теоретическому.
Для данного синтеза может быть применен этилат натрия, полученный
из спирта, едкого натра и бензола азеотропным способом.
Из описанных выше методов синтеза компонентов молекулы рибофлавина
вытекает возможность промышленного применения следующих методов:
о-4-Ксилидин (из р-нитротолуола) конденсируют с £>-рибонолактоном
(из D-глюкозы) в о-3,4-ксилил-О-рибамин. Последний путем сочетания
с фенилдиазонием образует 3,4-ксилйл-6-фенилазо-£)-рибамин, который
при конденсации с барбитуровой кислотой дает рибофлавин;
о-4-Ксилидин (из бромксилола) конденсируют с £>-арабинозой (из глю-
коната кальция) в о-3,4-ксилил-1)-арабинозид, который изомеризуют под
влиянием бензойной кислоты в о-3,4-ксилилизоарабинозамин. Последний
при каталитическом гидрировании с платиновым катализатором в щелоч-
ной среде превращают в один из изомеров о-3,4-ксилил-0-рибамин. Соче-
тают последний с фенилдиазонием и конденсируют полученный о-3,4-кси-
лил-6-фенилазо-О-рибамин с барбитуровой кислотой в рибофлавин;
наконец, эффективным синтезом может быть комбинированный метод,
а именно: о-3,4-ксилидин (из бромксилола) конденсируют с восстановлен-
ным D-рибонолактоном, а полученный рибамин сочетают с фенилдиазонием
и конденсируют с барбитуровой кислотой.
115
Ч-Срои-а-ксилм о-З-ИсилиЗин
Я- Арабиноза.
Рис. 14. Технологическая схема производства синтетического рибофлавина (вариант I).
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА СИНТЕТИЧЕСКОГО
РИБОФЛАВИНА (ВАРИАНТ I)
Технология базируется на следующих методах синтеза отдельных ком-
понентов молекулы рибофлавина (рис. 14):
синтез о-4-ксилидина из о-ксилола по методу Визанского и Ансбахера
[62, 631;
синтез о-3,4-ксилил-6-фенилазо-1-£>-рибамина из D-арабинозы и о-4-кси-
лидина по методу Вейганда [45];
синтез рибофлавина путем конденсации о-3,4-ксилил-6-фенилазо-1-0-
рибамина с барбитуровой кислотой по методу Березовского [4, 68, 72].
Синтез о-4-ксилидина. При синтезе о-4-ксилидина вначале получают
4-бром-о-ксилол из о-ксилола, а затем о-4-ксилидин из 4-бром-о-ксилола.
4-Бром-о-ксилол. Химическая реакция получения 4-бром-о-ксилола
заключается во взаимодействии о-ксилола и брома в присутствии катали-
затора согласно следующему химическому уравнению:
Н-.С-Т^ Г Вг
Н,сХД
О-Ксилол
106, 16
+ вг2_£1
^2.
бром
2. 79,92
4 - бром -О-ксилол
185, 07
В реактор 1 (рис. 14) из эмалированной стали, снабженный обратным
холодильником, сливают из сборника 2 о-ксилол, добавляют желез-
ные опилки и кристаллический йод (катализатор). Массу охлаждают
до 0°. Затем из мерника 3 добавляют в течение 3 ч бром из расчета 1,15 кг
на 1 кг о-ксилола. Температуру поддерживают минус 5° С. При повышении
температуры до плюс 10° С увеличивается количество образующегося ди-
бромксилола1. Реакционную массу после прибавления брома выдержива-
ют при температуре минус 5° С от 6 до 8 ч. Выделяющийся бромистый во-
дород поглощают ловушкой 4. После этого реакционную массу промывают
в делительной воронке 6 водой (в количестве 100%), 3%-ным раствором
едкого натра (в количестве 200%) из мерника 5 и снова водой (100% к мас-
се ксилола). Промытую массу направляют в сборник, оттуда в вакуум-
перегонный аппарат 7 для перегонки с водяным паром. Дистиллят соби-
рают в приемниках 8. В делительной воронке 9 препарат отделяют от воды
и подсушивают хлористым кальцием, после чего вторично перегоняют под
вакуумом (14—15 остаточное давление мм рт. ст.) и собирают фракцию
с температурой кипения 92—94° С (п^= 1,5558) в сборник 10. Выход
4-бром-о-ксилола составляет 85% (на бром). Имеются указания [63],
что можно повысить чистоту препарата (212—215° С при давлении
760 мм рт. ст.) путем сульфирования его, получения бариевой соли суль-
фокислоты, перекристаллизации последней и получения 4-бром-о-ксилола
путем гидролиза бариевой соли в кислой среде.
4-бром-о-ксилол — жидкость, молекулярная масса 185,07, температура
кипения 214,5° С, cf2°=l,369: n2j=l,556 [65].
О-4-Ксилидин. Химическая реакция получения о-4-ксилидина заклю-
чается во взаимодействии 4-бром-о-ксилола и аммиака в присутствии ка-
тализатора согласно следующему уравнению:
4- Ьром -О ксилол
185, 08
СиС1
“ей
НзС^^Г^Но
О - 6 - Ксилидин
121,18
+ NH4Br
1 По последним данным (А. Шерешевский, В. Березовский, ХФЖ, 1969 г., № 10),
с возрастанием температуры реакции до 40° С продолжительность реакции сокраща-
ется с 8 ч до 30 мин, а количество моно- и дибромксилола остаются в тех же пределах.
117
В стальной автоклав высокого давления 11 сливают 4-бром-о-ксилол
из мерника 12, помещают 7% медной проволоки и 6% однохлористой меди
к массе бромксилола. Из мерника 13 прибавляют водный аммиак (28-%ный)
в количестве 270%. Повышают температуру реакционной массы до 195°С
и поддерживают ее на этом уровне при помощи терморегулятора в течение
12—14 ч (повышение температуры приводит к снижению выхода). Давление
при этом повышается до 50—70 кгс/см2*. Затем автоклав охлаждают, сни-
жают давление, реакционную массу сливают в делительную воронку 14
и отделяют верхний слой органического вещества, направляя его в сборник
15 и далее в перегонный аппарат 16, куда добавляют 40%-ный раствор
NaOH (на 100 кг бромксилола 20 л). Перегонку ведут водяным паром.
Дистиллят поступает в приемник 17 и далее в кристаллизатор 18, где его
охлаждают до 0+5°. Кристаллы амина отделяют в центрифуге 19, про-
мывают водой (20° С), а затем подвергают перекристаллизации. Маточный
раствор является отходом, а промывные воды собирают в приемнике 20.
Технический ксилидин растворяют в спирте в аппарате 21, обрабатывают
активированным углем при температуре 80—85° С, фильтруют через нутч-
фильтр 22, кристаллизуют при 0 — минус 2° С в кристаллизаторе 23 и фу-
гуют в центрифуге 24. Кристаллы о-4-ксилидина поступают в сушку 25,
а маточный раствор в сборник 26 и далее в аппарат 27 для обработки уг-
лем и фильтрации в нутч-фильтре 28. Фильтрат упаривают в вакуум-ап-
парате 29, кристаллизуют в аппарате 30 и фугуют в центрифуге 31. Крис-
таллы ксилидина 11 поступают на первую кристаллизацию в аппарат-раст-
воритель 21. Маточный раствор является отходом. Выход о-4-ксилидина
составляет около 66% к теоретическому [64]. о-4-Ксилидин, молекулярная
масса 121,18; кристаллизуется в виде бесцветных игл с температурой плав-
ления 49° С; температура застывания 48,7°; температура кипения 105° С
при остаточном давлении 11 мм рт. ст.-, 125° С при остаточном давлении
30 мм рт. ст.-, 192,5—193,5° С при давлении 760 мм рт. ст.-, =
= 1,5582; «20=0,992. Растворимость в воде при температуре 22° С —
0,38%, хорошо растворим в спирте, бензоле и других органических раст-
ворителях [53, 64, 65].
Синтез D-арабинозы. Получают О-арабинозу из D-глюконата каль-
ция окислением его перекисью водорода в присутствии солей железа
согласно следующему уравнению [46, 48, 49, 66, 67]:
СН2ОН . сн2он Н-С-ОН Н-С-ОН Н-С-СШ + Fe2(SO4)3-9H2O H-C-QH +Н2О но—с—н 2 2 ВаСсн3соо)2-н2о’но-с-н ” Н-С-ОН с=о с^° с с'о'& Глюконат кальция 275, 75 СН2ОН Н-С-ОН 1 Н-С-ОН Ho-cLh +СО2+Са(ОН)2 1 с=о 1 н В -Арабиноза 750, 73
В реакторе 32 растворяют D-глюконат кальция (10%-ный раствор),
прибавляют 1,75% к массе глюконата сернокислого железа Fe2(SO4)3-9H20
в виде 10%-ного водного раствора из мерника 33 и 2,5% уксуснокислого
бария (СН3СОЭ)2Ва • Н3О в виде 15 %-ного водного раствора из мерника 34.
Массу подогревают до 60° С и из мерника 35 начинают постепенно при пе-
ремешивании приливать 30%-ный раствор перекиси водорода 1 л на 1 кг
D-глюконата кальция. Перекись водорода предварительно разбавляют в
* Уровень давления зависит от величины заполнения автоклава, рекомендуемый
в пределах 50—60% (не более).
118
шесть раз водой. Через несколько минут начинает выделяться углекислота.
После прибавления всей перекиси водорода и прекращения выделения СО2
реакционную массу фильтруют через нутч-фильтр 36 в сборник 37.
Фильтрат обрабатывают активированным углем в количестве 3% к мас-
се D-глюконата в смесителе 38, фильтруют через нутч-фильтр 39. Очищен-
ный раствор поступает в сборник 40, а затем в вакуум-аппарат 41, где его
упаривают, а затем спускают в кристаллизатор 42. Кристаллизация про-
должается 10—12 ч при температуре 5—10° С. Затем массу фугуют в цент-
рифугах 43, кристаллы промывают метиловым спиртом и сушат в сушил-
ке 44. Маточный раствор поступает из центрифуги в приемник 45, откуда он
насосом подается в аппарат-смеситель 46 для обработки активированным
углем. Далее массу .фильтруют через нутч-фильтр 47. Фильтрат поступает
в сборник 48 и далее в вакуум-аппарат 49, где его упаривают и затем спус-
кают из вакуум-аппарата в кристаллизатор 50, где он 24 ч кристаллизу-
ется при температуре 10° С, затем его фугуют в центрифуге 51. Кристаллы
D-арабинозы II поступают на перекристаллизацию в смеситель 38. Маточ-
ный раствор из центрифуги поступает в сборник 52 и является отходом про-
изводства. Если кристаллизация продукта будет неудовлетворительной, то
следует полуспущенный фильтрат первой кристаллизации обработать
смесью метилового спирта и ацетона.
Установлено [67], что на выход D-арабинозы существенно влияет ко-
личество катализатора (0,028 моля катализатора на моль глюконата), в
меньшей степени выход D-арабинозы зависит от температуры. Оптималь-
ная температура 50—70° С. Количество окислителя должно соответство-
вать 2 молям активного кислорода на 1 моль глюконата. Выход D-араби-
нозы 44%. D-Арабиноза, молекулярная масса 150,13, температура плавле-
ния 155,5—156,5° С, fa]2j=108° (в воде).
3,4-Ксилил-О-рибамин. Далее проводят конденсацию D-арабинозы с
о-4-ксилидином в 3, 4-ксилил-М^-арабинозид, который под влия-
нием бензойной кислоты изомеризуется в изоарабинозамин, с восстановле-
нием последнего в присутствии платинового катализатора в щелочной сре-
де в 3, 4-ксилил^-рибамин согласно следующему уравнению:
(7-4 -Ксилидин
121,18
СН2ОН
I
н-с-он
I
н-с-он
I
с6н5соон
СН2
3,4 - Ксилил - D-
изоарабинозамин
сн2он
н-с-он
I
н-с-он
I
но-с-н
I
с=о
I
н
D - Арабиноза
150,13
J,4 -Ксилил-
-D-арабинозид
СН2ОН
I
н-с-он
I
•н-с-он
I
н-с-он
Р1
NaOH
сн2
3,4 - К силил - D -рибамин
255, 31
119
В реактор 53, снабженный паровой рубашкой, загружают D-арабинозу,
о-4-ксилидин, бензойную кислоту и воду в отношении 10:8:0,5:3. Интенсив-
но перемешивают и в течение 6 мин нагревают до 90—95° С. Затем из мер-
ника 54 вводят спирт в количестве 2 л на 1 кг D-арабинозы, быстро раство-
ряют реакционную массу и рассолом охлаждают до комнатной темпера-
туры.
Затем в реактор вводят восстановленную окись платины в количестве
5% к массе D-арабинозы и 0,4 л 8%-ного NaOH на 1 кг D-арабинозы и при
перемешивании пропускают водород в течение 1,5—2 ч. Количество погло-
щенного водорода составляет около 60 л на 1 кг D-арабинозы. По оконча-
нии реакции массу фильтруют через нутч-фильтр 55, отделяют катализа-
тор, а фильтрат направляют в перегонный аппарат 56 для отгонки спирта.
Отгонку проводят водяным паром. Смолообразную коричневую массу экст-
рагируют водой в перегонном аппарате, спускают экстракт в смеситель 57,
обрабатывают активированным углем и фильтруют через нутч-фильтр 58.
Фильтрат поступает в сборник 59, затем в вакуум-перегонный аппарат 60
для упаривания. Сгущенный раствор поступает в кристаллизатор 61.
После кристаллизации в течение 12 ч при 0° масса поступает в центрифугу
62. Полученный рибамин-сырец перекристаллизовывают из спирта по нор-
мальной трехступенчатой схеме.
3,4-Ксилил-1)-рибамин C13H31O4N, молекулярная масса 255,31, крис-
таллизуется в виде бесцветных блестящих пластинок с температурой плав-
ления 144° С, 1а]д=—29° (при концентрации 5%), хорошо растворим
в.горячей воде, слабо в холодной. Хорошо растворим в горячем этиловом
и w-бутиловом спирте, плохо в холодном.
При исследованиях [53] установлено, что на реакцию перегруппировки
оказывает влияние искусственное охлаждение реакционной массы и пере-
мешивание.
Выход 3,4-ксилил-П-рибамина составляет 13% (на D-арабинозу).
3,4-Ксилил-6-фенилазо- l-D-рибамин получают сочетанием 3,4-ксилил-Д-
рибамина с раствором хлористоводородного фенилдиазония при pH 3,2—
4,0; пэстоянную концентрацию водородных ионов поддерживают добавле-
нием раствора уксуснокислого натрия [53, 67]. В результате изучения
(В. Березовский) механизма реакции азосочетания для стереоизомерных
арилгликаминов [72] определены оптимальные условия pH (3,2—4,0) для
данной реакции, обеспечивающие высокий выход (выше 90%) 3,4-ксилил-
6-фенилазо-1-О-рибамина. В этих условиях при азосочетании с 3,4-кси-
лил-О-арабамином образуется триазен —
Н Н Н
н Зс N-С Н2-С - С - С - С н2он
I Ан Ан Ан
n=n-c6h5
Триазен в конденсацию с барбитуровой кислотой не вступает и рибо-
флавина не образует. Использование результатов этих исследований обес-
печивает получение чистого рибофлавина без примеси арабофлавина. Реак-
ции протекают по следующей схеме:
н н н
h3c-y^-nh-ch2-c- с— с-сн2он
нзс~Ц>^ он он он
3,4 - Л силил -В- рибамин
255. 31
C6H5-N=N-C1
Хлористоводородный
Фвнилдиазоний
CH3COONaK
Уксуснокислый
натрий
III
HjC-^^jy-NH-CHa-C- С- С-СН20Н
h3C-^JJ~N=N-C6H5 он ОН ОН
3,4 - Ксилил- 6-фенилаза-1~й-рибамин
359,41
н н н
120
Хлористоводородный фенилдиазоний получают из анилина диазотиро-
ванием его нитритом натрия в солянокислой среде, согласно уравнению
C6H6NH2+ NaNO2 + 2НС1 —> С6Н6—N=N—Cl + NaCl + 2H2O.
93,12 69,01 2-36,47 140,88
В реактор 62а загружают анилин из мерника 63, соляную кислоту
(плотность 1190 кг/м3) из мерника 64 и воду из мерника 65 в количестве:
НС1 — 2,3 л, воды — 1,7 л на 1 кг анилина.
Смесь при перемешивании охлаждают до —7° С прибавляют 3 кг льда
и вводят раствор нитрита натрия в количестве 0,8 кг в 1,3 л воды.
Температуру поддерживают на уровне 3° С, реакция должна быть кис-
лая на конго. Раствор направляют в сборник 66, откуда расходуют на азо-
сочетание.
Для получения азорибамина в реакторе 67 растворяют рибамин в воде,
подкисленной соляной кислотой (1 кг рибамина, 7 л воды и 0,7 л НС1 плот-
ностью 1190кг/м3). Из сборника 66 приливают при размешивании холодный
раствор хлористого фенилдиазония в течение 2—3 мин. При этом темпера-
тура повышается до 16—18° С. Затем в течение 30 мин перемешивания
из мерника 68 добавляют 28%-ный раствор уксуснокислого натрия из рас-
чета 16 л на 1 кг рибамина [31 ], температуру поддерживают на уровне 20—
24° С, а pH 3—4. Выделяется красный осадок. При более высоком pH (4—5)
и при избытке уксуснокислого натрия образуется диазоиминосоединение.
Реакционную массу отфильтровывают на нутч-фильтре 69 и промывают
водой. Осадок поступает в сборник 70, а промывные воды в канализацию.
Полученный технический азорибамин подвергают перекристаллизации.
Технический продукт содержит изомер 3,4-диметилфенил-2-фенилазо-1-
О-рибамин [53].
Технический продукт растворяют в спирте при температуре 80—85° С
в реакторе 71, фильтруют через нутч-фильтр 72 и кристаллизуют в аппара-
те 73 при 0°. Чистый азорибамин I выделяют в центрифуге 74, промывают
горячим спиртом. Маточный раствор I и промой поступают на упаривание
в вакуум-аппарат 75 и далее на кристаллизацию в кристаллизатор 76 и
центрифугу 77. Кристаллы азорибамина II поступают на перекристаллиза-
цию в реактор 71, а маточный раствор в зависимости от степени истощения
либо направляют на кристаллизацию, либо в отход. Азорибамин I высуши-
вают в вакуум-сушилке 78. 3,4-Ксилил-6-фенилазо-1-£)-рибитиламин пред-
ставляет собой кристаллы в виде игл оранжево-красного цвета с темпера-
турой плавления 174—175° С.
Кристаллы нерастворимы в воде, слабо растворимы в спирте на холоду
и лучше при нагревании. Выход азорибамина 90% от теоретического [53].
Синтез рибофлавина [68]. Барбитуровая кислота. Вначале получают бар-
битуровую кислоту, для этого проводят конденсацию этилового эфира
малоновой кислоты и мочевины в присутствии этилата натрия [53, 64].
Полагают [53], что получающийся натриймалоновый эфир в таутомерной
форме ацилирует одну аминогруппу мочевины с образованием уреида эфи-
ра малоновой кислоты. Ацилирование второй аминогруппы с замыканием
цикла проходит в присутствии щелочных агентов. Реакция протекает по
-следующей схеме:
С< С С ONa NH
I OC2HS + c2HsONa II OC2HS 4-coCNH2)2 , учсо - C2HSOH
к3 -C2H5OH I -C2HSOH нсх Лн2
СООС2Н5 COOC2HS COOC2HS
Дизти левый эфир
пало новой колоты
160,17
Натренолят ма- Мочевина
лонового эдоира 60,06
Урсид малонового
здэира
ONa NH NH
Сх ''СО НС1, ОСХ ''СО
нщ Ан *н2с. Ан
со со
Барбитуровая
кислота
128,09
121
В реактор 79, снабженный обратным холодильником, загружают этилат
натрия из мерника 80, нагревают до 45—50° С и при перемешивании при-
бавляют из мерника 81 малоновый эфир, а затем сухую мочевину: на 1 кг
малонового эфира 3,5 л этилата натрия (плотность 880 кг/м3) и 0,4 кг мо-
чевины. Смесь нагревают до кипения и выдерживают при перемешивании
при температуре кипения 7 ч. Затем добавляют в реактор 6 л горячей воды
(50° С) и из мерника 82 около 0,6 л на 1 кг малонового эфира соляной кис-
лоты (реакция кислая). При этом осадок натриевой соли барбитуровой
кислоты растворяется. Раствор фильтруют через нутч-фильтр 83. Фильтрат
переводят в кристаллизатор 84, в котором массу охлаждают до 0° и отфу-
говывают в центрифуге 85, промывают ледяной водой и высушивают в> ап-
парате 86.
Выход 75% к теоретическому. Маточный раствор, содержащий спирт,
поступает в сборник 87 и далее в перегонный аппарат 88 для отгонки спир-
та. Последний поступает в сборник 89.
Барбитуровая кислота C4H4N2O3, молекулярная масса 128,09, пред-
ставляет собой бесцветные кристаллы, трудно растворимые в холодной
воде, лучше в горячей.
Рибофлавин. Далее проводят конденсацию 3,4-ксилил-6-фенилазо-1-
D-рибамина с барбитуровой кислотой в присутствии уксусной кислоты
и спирта [68] при нагревании до кипения согласно следующему уравне-
нию:
сн2он
н-с-он
।
н-с-он
I
н-с-он
I
сн2
НзСп#:?хггNH
Н3С-Ц^Д_ N=N-C6HS
3,4 - Ксилил -6 ~ ере ни л азо -1-D-
рибамин
359,41
NH
ОС' ^О
Н2Щ XNH
сб
барбитуровая
кислота
128,09
СН2ОН
Н-С-ОН
I
н-с-он
I
н-с-он
I
о
Рибофлавин
376,36
C6HSNH2 + H2O
Анилин
93,12
В реактор 90, снабженный обратным холодильником, загружают 1 кг
азорибамина, 0,6 кг барбитуровой кислоты, 8 л этилового спирта и 1,4 л
ледяной уксусной кислоты и при температуре кипения и размешивании
нагревают 15 ч. Из раствора постепенно выделяется рибофлавин. Реакцион-
ную массу охлаждают до 10° С и фугуют на центрифуге 91, промывают
спиртом, горячей водой и осадок — технический рибофлавин — подверга-
ют перекристаллизации. По Японскому патенту [68а] конденсацию азори-
бамина и барбитуровой кислоты ведут в трехкомпонентной смеси из уксус-
ной кислоты, нитробензола и бензола и получают витамин В2 высокой
чистоты.
Технический рибофлавин растворяют в 0,1 н. соляной кислоте в реак-
торе 92 из расчета 1 кг рибофлавина в 100 кг разбавленной кислоты при
температуре 85° С. Раствор фильтруют через нутч-фильтр 93 и кристалли-
зуют в аппарате 94 при охлаждении до 0 минус 2° С в течение 8 ч. Массу
фугуют в центрифуге 95, кристаллы промывают холодным спиртом и вы-
сушивают в вакуум-сушилке 96. Маточный раствор из центрифуги посту-
пает в сборник 97, а из него в вакуум-аппарат 98, где его сгущают в 15—
20 раз, спускают в кристаллизатор 99, где кристаллизуют 12 ч с постепен-
ным доведением температуры до 0°. Затем массу фугуют в центрифуге 100.
Кристаллы рибофлавина II (второй кристаллизации) поступают на пере-
кристаллизацию совместно с техническим рибофлавином в реактор 92.
Маточный раствор II поступает в сборник 101, а оттуда на третью кристал-
122
лизацию — в вакуум-аппарат 102, кристаллизатор 103, центрифугу 104.
Кристаллы рибофлавина III (третьей кристаллизации) поступают на пе-
рекристаллизацию совместно с маточным раствором I в сборник 97, а ма-
точный раствор III направляют в канализацию через сборник 105.
Выход рибофлавина на стадии конденсации 67%, а на перекристалли-
зации— 93%. Свойства рибофлавина (см. стр. 108).
ТЕХНОЛОГИЧЕСКИЕ ПОКАЗАТЕЛИ
РАСХОД ХИМИК АЛИЕВ, КГ НА 1 КГ
ЮО-%-НОГО ПОЛУПРОДУКТА
(ВАРИАНТ I)
4-Бром-о-ксилол
Выход, % к теоретическому (на ма-
о-Ксилол........................0,70
Бром............................ 1,00
Йод.............................0,01
Натр едкий твердый.............0, ГО
Кальций хлористый...............0,10
Выход, % к теоретическому
(на бром)........................85,0
о-4-Ксилидин
лоновый эфир) ..................75,0
Рибофлавин
Азорибамин ......................1,5
Барбитуровая кислота ........... 0,7
Кислота уксусная.................2,0
Спирт ...........................0,8
Кислота соляная .................3,5
Выход, % к теоретическому (на
азорибамин).....................67,0
[53]
Выход рибофлавина при перекри-
сталлизации, % ..............93,0
4-Бром-о-ксилол.........' . . . . 2,30
Аммиак 99%-ный....................0,50
Однохлористая медь................0,15
Медная проволока..................0,16
Натр едкий (100%-ный).............1,00
Спирт этиловый ...................0,30
Уголь активированный..............0,06
Выход, % к теоретическому (на
4-бром-о-ксилол) ................66,0
РАСХОД ПОЛУПРОДУКТОВ, КГ
НА СИНТЕЗ 1 КГ РИБОФЛАВИНА
ПО ВАРИАНТУ I ЧЕРЕЗ
D-АРАБИНОЗУ
D-Арабиноза
D-Глюконат кальция .............3,3
Перекись водорода ..............1,3
Сернокислое железо................0,06
Уксуснокислый барий.............0,09
Активированный уголь.............0,10
Метиловый спирт...................0,20
Ацетон..........................0,15
Выход, % к теоретическому (на
глюконат кальция) .............. 44,0
Азорибамин ...................... 1,5
Барбитуровая кислота .......... 0,7
Рибамин........................ 1,2
D-Арабиноза ..................5,4
о-Ксилидин.......................4,3
4-Бром-о-ксилол .................9,9
РАСХОД ХИМИКАЛИЕВ, КГ
НА ПРОИЗВОДСТВО
1 КГ РИБОФЛАВИНА С УЧЕТОМ
РЕГЕНЕРАЦИИ РАСТВОРИТЕЛЕЙ
ПО ВАРИАНТУ 1
(ЧЕРЕЗ D-АРАБИНОЗУ)
3,4-Ксилил-О-рибамин (рибамин)
D-Арабиноза .....................4,5
о-4-Ксилидин.....................3,6
Бензойная кислота................0,2
Едкий натр.......................0,2
Водород, м3 .....................0,27
Окись платины....................0,02
Уголь активированный.............0,05
Выход, % к теоретическому (на
D-арабинозу) .................... 13,0
.3,4-Ксилил-6-фенилазо-1 -D-рибамин
(азорибамин)
Рибамин ..........................0,80
Анилин............................0,30
Натрий нитрит.....................0,25
Кислота соляная ................. 1,00
Спирт ............................0,80
Выход, % к теоретическому (на
рибамин).........................90,0
Барбитуровая кислота
Эфир малоновый .................1,6
Мочевина........................0,6
Кислота соляная ................1,2
Спирт абсолютный ...............1,0
Натрий металлический............0,23
о-Ксилол.........................6,9
Бром.............................9,9
Йод .............................0,1
Натр едкий (100%-ный)...........4,3
Кальций хлористый плавленый . . 1,0
Аммиак 99%-ный..................2,2
Медь однохлористая..............0,7
Медная проволока................0,7
Спирт этиловый...................4,0
Уголь активированный ...........0,9
D-Глюконат кальция ..............17,8
Перекись водорода ............. 7,0
Железо сернокислое...............0,3
Барий уксуснокислый.............0,5
Спирт метиловый..................10,8
Ацетон....................... 0,8
Кислота бензойная ........... 0,3
Водород, м3 ................. 0,3
Окись платины................. 0,002
Анилин....................... 0,5
Натрий нитрит................ 0,4
Натрий уксуснокислый .... 5,9
Кислота соляная ............. 3,0
Эфир малоновый .............. 1,1
Мочевина..................... 0,4
Натрий металлический......... 0,1
Кислота уксусная ледяная ... 2,0
Итого 81,902
123
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА СИНТЕТИЧЕСКОГО
РИБОФЛАВИНА (ВАРИАНТ II)
Вариант II синтеза рибофлавина отличается от варианта I методом
синтеза 3,4-ксилил-П-рибамина. Вариант II синтеза показан на следую-
щей схеме [531
н,с
н,с
Вг
О-ксилол
н,с
н,с
4 - бром - 0 -к си ЛОЛ
П
NH.
н,с
nh2
Н3С
О-U- Ксилидин
Ш
он
сн2он
н 1—— о н
1/н \J 1
Хон н/|
НО )-----К он
н он
В - Глюкоза
COOK
но-с-н
+ 02,
кон
н-с-он
н-с-он
сн2он
Арабонат палия
IV
COO (I Са)
но-с-н
Н-С-ОН -
| ризация
Н-С-ОН I
н-с-он ?
+н2
н-с-
н-с-он I
I о
Н-С-ОН I.
н-с-он
н-с
н-с
СН2ОН
D-Арабонат кальция
V
сн2он
В-Рибоно -у - лактон
VI
сн2он
В-Рибоза
чи
В
Н2С
Н-С-ОН
Н3С
Н3С
NH
сн2он
(снон)
сн2
C6HSN= NCt H3C
Н3с
сн2он
(снон)3
сн2
NH
н2
н-с-он 0
н-с-он I
сн
Н3с
Н3С
NH
3,4 -Ксилил-В-рибамин
а
3,1* - Ксилил -б- фенилазо -
-1 -В -рибамин
н-с-он
I
н-с-он
I
н-с-он
3,4- Ксилидин -N-D -рибо -
лиранозид
УШ
NH
00х со
нЛс^н .
барбитуробая кислотЦ
XI
Стадии получения 4-бром-о-ксилола и о-4-ксилидина аналогичны для
обоих вариантов синтеза рибофлавина. Здесь рассмотрим отличительные
стадии второго варианта синтеза. К ним относятся стадии, предусматрива-
ющие окисление D-глюкозы в D-арабоновую кислоту V [69—711, которую
подвергают эпимеризации в D-рибоновую кислоту с превращением ее в
D-рибоно-у-лактон VI; последний амальгамой натрия восстанавливают в
D-рибозу VII. При конденсации D-рибозы с о-4-ксилидином III получают
3,4-ксилидин-М-П-рибопиранозид VIII, который водород восстанавливает
в 3,4-ксилил-О-рпбамин IX. Последний при сочетании с хлористоводород-
ным фенилдиазонием дает азорибамин X. Ниже приводится технологиче-
ская схема (рис. 15) с описанием этих стадий.
D-Арабонат кальция получают в результате деструктивного окисления
глюкозы кислородом воздуха в щелочном растворе с получением D-арабо-
ната калия, превращаемого обменной реакцией с хлористым кальцием
в D-арабонат кальция. Такое превращение необходимо для снижения тру-
доемкости процесса удаления катиона соли альдоновой кислоты (Са вместо
124
Рис. 15. Технологическая схема производства синтетического рибофлавина (вариант II).
К). Была изучена [53, 72] кинетика реакции деструктивного окисления
D-глюкозы кислородом воздуха в щелочном растворе. Показано, что об-
разование D-арабинозы происходит только в узких пределах температуры
(40—50° С). Реакция определена как реакция первого порядка, определе-
ны константы скорости при температуре 40 и 50° С (К4О=0,01161 и К60=
=0,03742, энергия активации £=23 500 кал). При недостатке кислорода
под влиянием щелочи глюкоза частично изомеризуется в фруктозу, ман-
нозу и другие продукты [53].
Реакция получения D-арабоната кальция протекает по следующей схеме;
2
Н-С-------
I
н-с-он
I
но-с-н о
I
н-с-он
I
Н-С-------
I
СН2ОН
Л - Глюкоза
COOK
I
но-с-н
I
+ О2 + 2КОН---*- H-C-OH +НСООК+2Н2О
Н-С-ОН
I
СН2ОН
D-Арабонат калия
204,22
г. 56, ю
180,16
COOK
I
но-с-н
I
н-с-он
I
н-с-он
I
сн2он
+ СаС12 + 5Н2О-----►
СОО-
I
но-с-н
I
н-с-он
I
н-с-он
I
сн2он
z- + +
Са
• 5Н2О +2КС1
ш, 44
Л-Арабонат кальция
460,40
149,11
В реакторе 1 (рис. 15) приготовляют 15%-ный раствор глюкозы и через
мерник 2 загружают в автоклав 3, через мерник 4 подают раствор КОН
(15%) и из баллона 5 подают кислород. Температуру в автоклаве поддер-
живают 40—45° С, а давление кислорода 10—12 кгс/см2. По окончании
реакции (3—3,5 ч) массу остаточным давлением кислорода передают в реак-
тор 6 для обработки активированным углем, фильтруют через нутч-фильтр
7 в сборник 8; фильтрат направляют в вакуум-аппарат 9. В нем при ва-
кууме 600—650 мм рт. ст. сгущают до получения подвижной кристалли-
ческой массы, которую сливают в кристаллизатор 10, где при 0° кристал-
лизуют 6—8 ч. Кристаллы арабоната калия выделяют в центрифуге 1Г,
маточный раствор направляют в сборник 12 и далее в вакуум-аппарат 13
для сгущения, в кристаллизатор 14, центрифугу 15. В ней отфуговывают
кристаллы арабоната калия II, которые направляют для перекристалли-
зации в реактор 6; маточный раствор из центрифуги 15 поступает в сбор-
ник 16 и является отходом производства.
D-Арабзнат калия (С5Н9ОвК, молекулярная масса 204,22). Он пред-
ставляет собой бесцветные кристаллы, хорошо растворимые в воде, нераст-
воримые в метиловом и этиловом спирте. Выход 70%. Кристаллы арабо-
ната калия растворяют в воде в реакторе 17 (насыщенный раствор при
температуре 87—90° С), куда из мерника 18 загружают водный раствор
хлористого кальция (30—35%), добавляют активированный уголь и фильтру-
ют через нутч-фильтр 19, сборник 20. Из последнего фильтрат переводят
в кристаллизатор 21, где ведут кристаллизацию 7—8 ч при 0°. Затем крис-
таллы арабоната кальция отфуговывают в центрифуге 22 и высушивают
в сушилке 23. Маточный раствор из сборника 24 засасывают в вакуум-ап-
126
парат 25, сгущают, кристаллизуют в аппарате 26 и фугуют в центрифуге
27. Кристаллы направляют на перекристаллизацию в реактор 17, а маточ-
ный раствор в сборнике 28 является отходом производства. Выход — 90%
(на глюкозу).
D-Арабонат кальция (C10Hi8O12Ca-5H2O, молекулярная масса 460,40)
представляет собой бесцветные кристаллы, плохо растворимые в холодной
воде (1 часть в 78 частях воды при температуре 12°С), хорошо растворимые
в горячей воде [53].
D-Рибоно-у-лактон. Путем эпимеризации арабоната кальция в присутст-
вии гидрата окиси кальция при температуре 135—137° С получают D-ри-
боновую кислоту. Последнюю при нагревании в вакууме при температуре
100° С подвергают лактонизации с превращением ее в D-рибоно-у-лактон.
Реакции протекают по следующей схеме:
о
СОО+а) СОО(2Са) соон
но-с-н Н-С-ОН н-с-он н-с-он 1
1 , Са(ОН% Н-С-ОН • 22Н2О —-— 1 (СООН)2 • • н-с-он — 2H2O 1 —н-с-он 100\ н-с-он ?
1 1 -Н2о 1
н-с-он [ н-с-он н-с-он 2 н-с 1
СН2ОН сн2он сн2он сн2он
б-Ьрабомт кальция Я-Рибонат кальция 2? -Рибоновая кислота 11 - Рибоно-у-лактан
гзо,2 195,18 166,13 М8,И
В реактор из эмалированной стали 29 загружают из вакуум-сушилки
23 D-арабонат кальция, растворяют в воде, добавляют около 1% окиси
кальция и нагревают при 135—137° С (давление 3,2—3,3 кгс/см2) в течение
5—6 ч. Осадок извести отфильтровывают при температуре 75—80° С на
нутч-фильтре 30, сливают в кристаллизатор 31, где при 0° выкристаллизо-
вывают не подвергшийся эпимеризации D-арабонат кальция, который от-
фуговывают в центрифуге 32 и направляют обратно на эпимеризацию в
реактор+9. Фильтрат поступает в сборник 33, а из него в реактор-осади-
тель 34, куда подают раствор щавелевой кислоты из мерника 35 с целью
осаждения иона кальция. Раствор фильтруют через нутч-фильтр 36. Оса-
док щавелевокислый кальций из сборника <37 либо 'идет на регенерацию,
либо является отходом производства. Фильтрат из сборника 38 поступает
в смеситель 39, где обрабатывают активированным углем, фильтруют че-
рез нутч-фильтр 40 в сборник 41 и сгущают в вакуум-аппарате 42 с сохра-
нением подвижности сиропа при температуре 20° С. В конце выпаривания
температуру повышают до 100° С в течение 2 ч с целью лактонизации D-ри-
боновой кислоты. Сироп содержит значительную примесь D-арабонолак-
тона. Выход — 62% от теоретического.
Кристаллический D-рибоно-у-лактон (С5Н8О5, молекулярная масса
148,11) имеет температуру плавления 78—80° С, [а,1д = + 18,4 (концентра-
ция 5%), растворим в воде и в спирте, трудно растворим в этилацетате
[53].
D-Рибоза [72, 73]. D-рибоно-у-лактон восстанавливают в D-рибозу амаль-
гамой натрия. Амальгаму получают электролизом раствора едкого натра
на ртутном катоде. Процесс восстановления протекает по следующей схеме:
с------- н-с=о
I I
н-с-он н-с-он
н-с-он 9 +2Н++2е—* н-с-он
н-с----------------------1 н-с-он
I I
сн2он СН2ОН
0,8,11 2,01В 150,13
127
Исследовалась реакция электролитического восстановления альдоно-
лактонов амальгамой натрия [72]. Показано влияние температуры, интен-
сивности перемешивания и pH раствора альдонолактонов на выход альдоз.
В частности, при pH выше 7,0 процесс восстановления идет до образования
спирта; при pH ниже 3,0 возможен непроизводительный расход тока.
С. Суздальцева и В. Красюк, Е. Григорашвили, Н. Золотарев, Г. За-
рецкий [73] предложили непрерывный процесс восстановления D-рибоно-
у-лактона в D-рибозу, который осуществляют на установке, состоящей
из следующих аппаратов [73]: электролизера с ртутным катодом и нике-
левым или стальным никелированным анодом для получения амальгамы
натрия электролизом раствора едкого натра, реактора для восстановления,
разделителя фаз (газ, раствор, амальгама), холодильника для поддержа-
ния температуры реакционного раствора и вспомогательных аппаратов.
Установка работает следующим образом (рис. 15). В электролизер 43 через
промыватель 44 поступает отработанная амальгама с содержанием натрия
2,3—2,5 г-экв/л. Сюда же направляют 25%-ный раствор едкого натра
из мерника 45. Из электролизера амальгама с содержанием натрия в 2,5—
2,72 г-экв/л проходит промыватель 46, где отмывается водой от щелочи.
Далее амальгама поступает в реактор-восстановитель 47. Сюда же по-
дают очищенный подкисленный соляной кислотой раствор D-рибоно-у-лак-
тона из мерника 48 и через холодильник 49; в этом реакторе при темпера-
туре 10—12° С и pH 3,0—4,0 протекает процесс восстановления. Отсюда
реакционная масса поступает в разделитель 50, где происходит разделение
фаз; отработанную амальгаму направляют в промыватель 44 (для отмывки
водой от остатков рибозы и лактона) и далее в электролизер 43. Восстанов-
ленный раствор из разделителя 50 проходит контрольный отстойник 51
и поступает в сборник восстановленного лактона 52. Выход — 60%. Авто-
ры приводят следующие данные испытания опытной установки [73]. Ре-
зультаты испытания установки по непрерывному восстановлению D-рибо-
но-у-лактона электролизной амальгамой натрия приведены в табл. 9.
Таблица 9
Номер опыта Концентрация Содержание в про- дукте, г/л Выход ри- бозы, %
амальгамы, г-экв/л рибонолакто- на в исход- ном раство- ре, г/л КИСЛОТЫ в исходном растворе, г-экв/л
при входе в реактор при выходе из реактора рибоно- лактона рибозы
1 3,5 3,2 120 4,0 15 75 60
2 3,4 3,2 180 5,6 19 108 60
3 4,2 4,0 180 5,6 17 132 73
Опубликованы данные по расчету скрубберных разлагателей амальга-
мы и определению в них количества ртути [74]. D-Рибоза кристаллическая,
С5Н!0О5, молекулярная масса 150,13, имеет температуру плавления 89° С,
[аЬ=—23,7°. На следующую стадию D-рибоза поступает в водном раство-
ре с содержанием около 100 г/л с примесью лактона — 15—25 г/л, плот-
ность 1190—1210 кг/м3, pH — 3,5—4,0. Выход D-рибозы 60%.
Очистка ртути. После 6—7 суток работы ртуть загрязняется различными
посторонними веществами и эффективность электролитического процесса
резко снижается. В связи с этим возникает необходимость в очистке рту-
ти, заключающейся в обработке ее азотной кислотой, щелочью, промывке
водой и вакуум-дистилляцией (остаточное давление 1—2 мм рт. ст., 190-
200° С). Очищенную ртуть направляют на электролиз. Опубликованы ре-
зультаты прямого электровосстановления D-рибоно-у-лактона на ртутном
катоде вместо восстановления амальгамой натрия с выходом 80% [74а].
Этот метод представляет интерес.
3,4-Ксилидин-Л,-/)-рибопиранозид. Получают путем конденсации о-4-кси-
лидина с D-рибозой в спиртовой среде и выделения в кристаллическом виде
128
комплексной соли с сернокислым натрием [72]. Эта стадия является очень
важной, так как здесь происходит очистка D-рибопиранозида от примеси
£>-арабопиранозида, который не образует нерастворимой комплексной соли
с сернокислым натрием.
Реакция протекает по следующей схеме:
н2с
- н-с-он
Z7-4— Ксилидин
121,18
/ОН н-с-он о
н-с-----1 н-с-он
1 I
н-с-он I н-с-----
Н-С-°Н I tNa2SO4rH3C-^YNH .
Н-с----1 NaOH НзС-Ц^
сн2он
D- Рида за
150,13
3,4 -Ксилидин -N-D -рибопиранозид
(комплексная соль)
253,29 142, Ов
В реактор из нержавеющей стали 53 загружают из сборника 52 раствор
рибозы, безводного сульфата натрия, из мерника 54 раствором NaOH до-
водят pH до 5,0, из мерника 55 загружают спирт и из мерника 56 спир-
товый раствор о-4-ксилидина. При перемешивании при температуре 28—
30° С выкристаллизовывается комплексная соль, которую отфуговывают
в центрифуге 53a и высушивают в вакуум-сушилке 57 при температуре
20—25° С. Маточный раствор поступает в сборник 58 и направляется для
регенерации спирта. Выход — 65%. 3,4-Ксилидин-Ы-П-рибопиранозид
(C13H18O4N, молекулярная масса 253, 29) комплексная соль, плохо раст-
ворима в воде, нерастворима в спирте. Нестойка при нагревании.
3,4-Ксилил-/)-рибамин. Получают путем гидрогенизации на скелетно-
никелевом катализаторе 3,4-ксилидин-Ы-Р-рибопиранозида в спиртовой
среде. Реакция протекает по следующей схеме:
Н2С------
н-с-он
I
н-с-он о
I
н-с-он
I
н-с-------
I
H3C-r^>j-NH + н2
H3cAJJ Ni
Ридопиранозид
253,29
сн2он
н-с-он
I
н-с-он
I
н-с-он
I
сн2
I
Н3С—NH
H3C-1^JJ
Рибамин '
255,31
В автоклав из нержавеющей стали 59 загружают 3, 4-ксилидин-Ы-7)-пи-
ранозид (комплексную соль), спирт из мерника 60 и скелетный никелевый
катализатор. Автоклав продувают азотом из баллона 61, водородом из бал-
лона 62, затем в течение 3—4 ч при избыточном давлении 30—40 кгс!см2
и температуре 50—60° С проводят гидрирование. Отфильтровывают ката-
лизатор на нутч-фильтре 63. Катализатор хранят под водой и направляют
на регенерацию. Фильтрат из сборника 64 направляют в кристаллизатор 65,
при 0° кристаллизуют 6—8 ч и отфуговывают кристаллы рибамина в
центрифуге 66, в вакуум-сушилке 67 их высушивают и направляют в
сборник. Маточный раствор из сборника 68 направляют в реактор-сме-
ситель 69, где обрабатывают активированным углем (10% к массе сухих
веществ), фильтруют через нутч-фильтр 70, в сборник 71. Фильтрат
5-522 1 29
засасывают в вакуум-аппарат 72, добавляют щелочи для разрушения
рибо но ксилидина, сопутствующего рибамин [53] и освобождения ксили-
дина, который отгоняется в сборник 73. Спирт дистиллируют в сбор-
ник 74. Кубовый остаток сливают в кристаллизатор 75, кристаллизуют
12 ч при температуре +5° С, фугуют в центрифуге 76 и высушивают в ва-
куум-сушилке 67. Выход рибамина 79,0%.
3, 4-Ксилил-й-рибамин (C13H21O4N, молекулярная масса 255,31) —бес-
цветные кристаллы, температура плавления 143—144° С, Ictlo —29° (вода
5%) [53], хорошо растворим в воде, спирте, в кислотах; плохо растворим
в щелочах. Стоек к нагреванию.
3,4-Ксилил-6-фенилазо-1-£>-рибамин (см. рис. 120).
Барбитуровая кислота (см. стр. 121).
Рибофлавин (см. стр. 122).
ТЕХНОЛОГИЧЕСКИЕ ПОКАЗАТЕЛИ
РАСХОД ХИМИКАЛИЕВ, КГ НА 1 КГ
100%-ГО ПОЛУПРОДУКТА
(вариант II)
4-Бром-о-ксилол
о-Ксилол........................0,70
Бром............................ 1,00
Йод ............................0,01
Едкий натр твердый..............0,10
Хлористый кальций плавленый . . 0,1
Выход, % к теоретическому (на
бром)............................85,0
о-4-Ксилидин
4-Бром-о-ксилол.................2,3
Аммиак 99%-ный..................0,15
Медь однохлористая..............0,15
Медная проволока................0,16
Натр едкий (100%-ный)........... 1,0
Спирт этиловый..................0,30
Уголь активированный ...........0,06
Выход, % к теоретическому (на
4-бром-о-ксилол) .................66
D-Арабонат кальция
Глюкоза ........................ 1,30
Кали едкое......................0,80
Кальций хлористый плавленый . . 0,80
Кислород, м3....................0,50
Уголь активированный............0,05
Выход, % к теоретическому (на
глюкозу)........................63,0
D-Рибоно-'^лактон
Арабопат кальция ...............2,50
Щавелевая кислота...............0,60
Гидрат окиси кальция ........... 0,10
Уголь активированный............0,05
Выход, % от теоретического (на
арабонат кальция) ............. 62,0
D-Рибоза
Рибонолактон .................. 1,7
Ртуть металлическая ............ 0,003
Кислота азотная................0,15
Натр едкий.....................2,50
Выход, % от теоретического (на
рибонолактон) ................. 60,0
3, А-Ксилидин-и-О-рибопиранозид
.D-Рибоза...........................0,9
’о-4-Ксилидин.......................0,7
Натр едкий.........................0,2
Сульфат натрия.................... 1,0
Спирт .............................5,0
Выход, % от теоретического (на
D-рибозу).........................65,0
3 А-Ксилил-О-рибамин
Рибопиранозид ................1,27
Спирт ........................2,50
Натр едкий....................0,05
Водород, м3 .......... 0,6
Выход, % от теоретического (на
рибопиранозид)..............79,0
3,4-Ксилил-6-фенилазо-1 -D-рибамин
(Азорибамин)
Рибамин ......................0,80
Анилин........................0,30
Натрий нитрит ................0,25
Кислота соляная ..............1,00
Спирт ........................0,80
Выход, % к теоретическому
(на рибамин)................90,0
Барбитуровая кислота
Эфир малоновый ...............1,60
Мочевина......................0,60
Кислота соляная ..............1,20
Спирт абсолютный .............1,00
Натрий металлический..........0,23
Выход, % к теоретическому
(на малоновый эфир).........75,0
Рибофлавин
Азорибамин....................1,50
Барбитуровая кислота..........0,70
Кислота уксусная..............2,00
Спирт ........................0,80
Кислота соляная ..............3,50
Выход медицинского В2, % к тео-
ретическому (на азорибамин) . . 67,0
Выход (в %) рибофлавина при пе-
рекристаллизации ...........93,0
РАСХОД ПОЛУПРОДУКТОВ НА
СИНТЕЗ РИБОФЛАВИНА
(по варианту II), кг
Азорибамин......................1,50
Барбитуровая кислота .......... 0,70
Рибамин ........................1,20
Рибопиранозид ..................1,52
D-Рибоза (100%-ная) ............1,35
D-рибонолактон..................2,30
D-Арабонат кальция..............5,75
3,4-К.силидин................. 1,06
4-Бромксилол....................2,44
130
РАСХОД ХИМИК А ЛИ ЕВ, КГ
НА ПРОИЗВОДСТВО 1 КГ РИБОФЛА-
ВИНА ПО ВАРИАНТУ II
ЧЕРЕЗ Б-РИБОЗУ
Аммиак 99%-ный....................0,53
Анилин............................0,45
Бром..............................2,44
Глюкоза.......................... 7,48
Йод...............................0,03
Кали едкое .......................4,60
Кальций гидроокись .............. 0,23
Кальций хлористый плавленый . . 4,84
Катализатор никелевый.............0,40
Кислота азотная ..................0,20
Кислота соляная ..................5,84
Кислота уксусная..................2,00
Кислота щавелевая................ 1,38
Ксилол.......................... 1,7!
Медь однохлористая...............0,16
Мочевина........................0,42
Натр едкий 100%-ный 5,04
Натрий металлический.............0,23
Натрий нитрит...................0,38
Натрий сульфат безводный .... 1,52
Натрий уксуснокислый ............1,30
Ртуть металлическая.............0,15
Спирт .........................12,62
Спирт абсолютный ................0,74
Уголь активированный............0,47
Эфир малоновый ..................1,12
Итого . . 56,28
Сравнительная технико-экономическая эффективность обоих методов
синтеза рибофлавина приведена в табл. 10.
Таблица 10
Наименование метода Количество стадий Расход основных химикали- ев, к,г/кг Относи- тельная стоимость основного сырья Выход рибофла- вина, %
Синтез через Б-арабинозу (вариант 1) Синтез через Б-рибозу 7 81,9 248 3,4
(вариант 11) 10 56,3 100 4,5
Из сопоставления двух вариантов синтеза рибофлавина видно, что ва-
риант II, который 15 лет назад [75] уступал варианту I как по числу ста-
дий (12 вместо 7), так и по расходу химикалиев (160,3 вместо 76,5), в на-
стоящее время по эффективности значительно превзошел вариант I как
по расходу химикалиев (56,3 вместо 81,9), так и по стоимости сырья
(100% вместо 248) и по выходу (4,5 в пересчете на D-глюкозу, вместо
3,4% — на глюконат кальция). Объясняется это значительным усовершен-
ствованием как самого синтеза, так и технологии производства (получение
о-4-ксилидина из о-ксилола вместо параформа, непрерывный процесс вос-
становления О-рибоно-у-лактона амальгамой натрия и др.). Это способст-
вовало резкому снижению расхода химикалиев и увеличению выхода рибо-
флавина по варианту II.
Между тем синтез рибофлавина по варианту I в производстве не осуще-
ствлялся и в лабораторных условиях усовершенствованию не подвергался.
Следует также подчеркнуть, что очень высокая стоимость сырья по вариан-
ту 1 вызвана высоким расходом D-глюконата кальция (17,8 кг) и высокой
его стоимостью около 60% стоимости всего сырья.
Из изложенного приходим к выводу, что в данных условиях наиболее
эффективным вариантом синтеза рибофлавина является вариант II (через
0-рибозу). Однако если учесть возможное снижение стоимости глюконата
кальция и усовершенствование технологии синтеза по варианту I, то этот
вариант получит более высокую оценку.
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА КОФЕРМЕНТА РИБОФЛАВИН-
МОНОНУКЛЕОТИДА И ЕГО НАТРИЕВОЙ СОЛИ [76 -79]
Рибофлавин-б'-фосфат был впервые синтезирован Куном [79а] при фос-
форилировании рибофлавина хлорокисью фосфора с предварительной за»
5* 131
щитой вторичных гидроксильных групп. В дальнейшем было показано,
что фосфорилирование может быть осуществлено без их защиты. В этих
условиях фосфорилирование проводилось орто-, мета- и пирофосфорными
кислотами [801, хлорокисью фосфора [81 ], окси- и алкоксипроизводными
хлорокиси фосфора [82].
В. Березовский и Р. Артемкина разработали метод, по которому рибо-
флавин-5'-фосфат получают фосфорилированием рибофлавина диметиловым
эфиром хлорида ортофосфорной кислоты с последующим гидролизом диме-
тилового эфира рибофлавина-5'-фосфата. Натриевую соль рибофлавина-5'-
фосфата синтезируют путем взаимодействия рибофлавин-5'-фосфата со ще-
лочью.
Реакции протекают по следующей схеме:
н3с
ЩС
он он он
। । ।
СН2-СН-СН-СН-СН2ОН
Рибофлавин Диметилобый эфир
376,36 хлорида ортофосфор -
ной кислоты
744,50
ОН он он ' осн3
сн2-с - С -С-СН2О-Р-ОСН3
I н н н о
Диметилобый зфир ридофлабина ~5'~ фосфата
484, чО
онон он он ононон он
III I III I
СН2-С-С-С-СН2О-Р-ОН СН2—С —С —С—СН2О-Р~ОМа
I । 1 । A I I I I II
IHHH о IHHH О
Рибофлавин-в-фосфорный эфор Натриевая соль рибофлавин-5- фосфата
456, 35 476,34
Диметиловый эфир хлорида ортофосфорной кислоты получают по реак-
ции
С1
I
С1-Р-С1
Хлороокись
фосфора
153,3k
осн,
I 3
+ 2.СН3ОН------*-СН3О-Р-С1 + 2НС1
II
О
2-32,0k Диметилабый эфир 2'36,k$
хлорида ортофосфор-
ной кислоты
/44,50
В реактор из эмалированной стали 1 (рис. 16), снабженный обратным
графитовым холодильником, загружают из мерника 2 хлорокись фосфора
и из мерника 3 медленно приливают абсолютный метиловый спирт. Реак-
ционную массу выдерживают при температуре 16—24° С в течение 10—
12 ч. Хлористый водород, выделяющийся при перемешивании, поглоща-
ется щелочными ловушками 4. Затем в реактор 5 сливают фосфорилирую-
щую смесь из реактора 1 и из сборника 6 загружают рибофлавин. Реакцию
фосфорилирования ведут 1 ч при температуре 40—45° С. Затем из мерника/
медленно добавляют воду для гидролиза фосфорилирующего агента.
К концу реакции температуру повышают до 75—80° С. Затем охлаждают
до 20° С и осаждают рибофлавин-5-фосфат спиртом из мерника 8, вы-
держивают массу при 0° 6—8 ч, фильтруют через центрифугу 9. Кристаллы
направляют в сборник 10 и далее для получения натриевой соли. Маточ-
ный раствор поступает в сборник 11 и является отходом. Выход рибофла-
вина-5'-фосфата составляет 87—90%. Кристаллы имеют желтый цвет, тем-
пература плавления 183—184° С (с разложением). Имеется указание 176],
132
Рис. 16. Технологическая схема производства рибофлавина-5-фосфата натрия.
что в этих условиях синтеза рибофлавин-5'-фосфат загрязнен некоторыми
количествами дифосфата и рибофлавина. Влажные кристаллы рибофлавин-
5-фосфата загружают в реактор из нержавеющей стали 12, сливают в него
воду из мерника 13 и после размешивания из мерника 14 медленно слива-
ют раствор едкого натра до pH 5,0—6,0. Затем раствор сгущают примерно
вдвое в вакуум-аппарате 15, фильтруют через нутч-фильтр 16 в сборник
17. Фильтрат засасывают в реактор 18, куда сливают из мерника 8 спирт
и кристаллизуют 6—8 ч при 0°.
Кристаллы отфуговывают в центрифуге 19 и высушивают в вакуум-су-
шилке 20. Маточный раствор I направляют в сборник 21, откуда его заса-
сывают в вакуум-аппарат 22, где его сгущают при вакууме 650—
700 мм рт. ст. и сливают в кристаллизатор 23. Кристаллизацию ведут
10—12 ч при 0+5° С, после чего в центрифуге 24 отфуговывают кристаллы
рибофлавин-5'-фосфата II и направляют их в реактор 18 для перекристал-
лизации. Маточный раствор II поступает в сборник 25 и оттуда либо по-
ступает на третью кристаллизацию через те же аппараты 22—25, либо явля-
ется отходом производства. Выход натриевой соли 55—60% от теоретиче-
ского. Имеется также вариант [79] очистки рибофлавин-5'-фосфата от при-
месей (рибофлавина) путем пропускания раствора с pH около 6,0 через
катионит КУ-2 в Н+-форме. Натриевая соль рибофлавин-5-фосфата
(C17H2oN409PNa -2Н2О, молекулярная масса 514,39) представляет собой
желтые кристаллы. Растворимость соли — около 6 г в 100 мл воды при
температуре 25° С. Хорошо растворима в щелочах и нерастворима в орга-
нических растворителях. Содержание РО)” не менее 0,5%, вкус горький,
спектр поглощения натриевой соли рибофлавин-5-фосфата в видимой об-
ласти 445 нм, е — 11 200 (в воде); в ультрафиолетовом светеХтах—372 ; 266;
222 нм; е — соответственно 9600; 29 200; 27 800. Препарат применяется
в виде инъекционных растворов при арибофлавинозе, при дерматозах и
глазных заболеваниях.
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА 2-ТИОРИБОФЛАВИНА [87]
Известно [83—86 ], что аналоги рибофлавина либо вовсе лишены витамин-
ной активности, либо обладают значительно сниженной активностью. Напри-
мер, при метилировании в молекуле рибофлавина иминогруппы в положении 3
витаминные свойства аналога уничтожаются. Полагают [85], что образование
133
флавинового фермента происходит в результате связывания кофермента
с протеином лишь по иминной группе.
В. Березовский и Л. Мелентьева [87] синтезировали 2-тиорибофлавин
путем конденсации 2-тиобарбитуровой кислоты с 6-фенилазо-3,4-диметил-
фенил-О-рибамином в среде бутилацетата в присутствии ледяной уксусной
кислоты.
Реакция получения тиорибофлавина протекает по следующей схеме:
н3с
н3с
он он он
। । ।
сн2-сн-сн-сн-сн2ои
I 1 1 1
н н н
1 NH
ri]-NH ОЩ ks Н3'
M=N-CeHs Н2Щ ДмН *~Н3
он он он
сн2-сн-сн-сн-сн2он
I н н н
1иориборлабин
392,1(3
н20
Анилин
93,12
Тиобарбитуровая
кислота
744,16
3,4 - Ксилил - 6- (ренилазо - 7 -77 -рибамин
359,1(1
Рис. 17. Технологическая схема производства
2-тиорибофлавин а.
В реактор из эмали-
рованной стали 1
(рис. 17), снабженный
обратным холодильни-
ком, загружают из мер-
ника 2 бутилацетат, из
мерника 3 ледяную ук-
сусную кислоту, из
сборника 4 азорибамин
и из сборника 5 2-тио-
барбитуровую кислоту.
Реакционную массу ки-
пятят 3 ч, фильтруют
в друк-фильтре 6. Оса-
док промывают в нача-
ле спиртом, а затем во-
дой. Маточный раствор
поступает в сборник 7 и
является отходом произ-
водства. Осадок 2-тио-
рибофлавина перекрис-
таллизовывают из соляной кислоты. Для этого его растворяют в трех-
кратном количестве концентрированной НС1 в стеклянном реакторе 8,
фильтруют через фарфоровый нутч-фильтр 9 в сборник 10, откуда переда-
ют в реактор 11, в котором залито 50-кратное количество (к осадку) ки-
пящей воды. Выпадает осадок 2-тиорибофлавина в виде игл фиолетово-
бордового цвета. Спектр поглощения 2-тиорибофлавина (в 0,005 н. НС1):
Хтах 230 нм (е=2,35 • 104); 317 нм (е=2,54 • 104); 400 нм (s=0,8-104) и
490 нм (е= 1,79 • 104); Rf =0,39 (в системе бутанол-уксусная кислота—вода)
(4:1:5). Осадок выделяют в центрифуге 12, сушат в вакуум-сушилке 13,
разгружают в сборник 14. Выход 2-тиорибофлавина — 60,4% [87]. По-
видимому, низкий выход объясняется большим объемом маточного раство-
ра. Следует водный маточный раствор сгустить в вакуум-аппарате и вы-
делить дополнительное количество 2-тиорибофлавина.
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. Б у к и н В. Н. Витамины. М., Пищепромиздат, 1940, 365—399.
2. К У Д р я ш о в Б. А. Биологические основы учения о витаминах. М., Изд. Москов-
ское общество испыт. природы. 1953, с. 21—24.
3. Р ы с с С. М. Витамины. Л., Медгиз, 1955, с. 132—155.
4. Березовский В. М. Химия витаминов. М., Пищепромиздат, 1959, с. 525—
565.
134
5. Ш и л о в П. И., Яковлев Т. Н. Основы клинической витаминологии. ИзД.
«Медицина», 1964, с. 59—64.
6. Osborne Т., М. endel L., J. Biol., Chem., 1913, 15, 311.
7. Bleyer В., Kallmann О., Biochem Z., 1925, 155, 54.
8. W a r b u r g O., Christian W., Biochem. Z., 1932, 254, 438; 1933, 257, 492
1933, 266, 377.
9. Б e э p А. А., Рубцов И. А. Синтез витаминов. M., Пищепромиздат, 1956
с. 187—207.
9а. П о в о л о ц к а я К- Л. О некоторых коферментных функциях рибофлавина. —
В сб.: «Витамины», изд. АН УССР, Киев, 1956, № 2, 134—143 с ил.
10. Salmon W., Gn errant N., Haus I. J. Biol. Chem., 1928, 80, 9,1.
11. Rosenberg H. Chemistry a. Physiology of the Vitamins, 1942, 153—195.
12. S i t a G. Encyclopedia of Chemical Technology, N. J., 1955, 11, 749.
13. К a r r e r P., Fritzsche H. Helv. Chimica Acta, 1935, 18, 911.
14. К u h n R., Rudy H., Ber., 1935, 68, 169.
15. A d a m s о n D., Analyst., 1948, 73, 442.
16. Shoishi Shimizu. Ferment. Technology, 1950, 28, 135; C. A., 1953, 47, 12435.
17. К a r r e r P., Solomon H., Schopp K., Schlittler E., F r i t z-
s c h e H. Helv. Chim. Acta, 1934, 17, 1010.
18. Поволоцкая К. Л., Зайцева Н. И. Разрушение рибофлавина под
влиянием видимого света. — «Труды ВНИВИ», 1954, № 5, 145—151 с ил. То же.
ДАН СССР, 1951, т. 77, с. 317.
19. К а г г е г Р., Frei Р., М е е г w 1 i п Н. Helv. Chim. Acta., 1937, 20, 79.
20. К u h n R., Rudy H. Ber., 1936, 69, 1974.
21. Karrer P„ Helv. Chim. Acta, 1952, 35, 457.
22. Англ. пат. 712387; С. A., 1955, 49, 11725.
23. Breivogel P. Пат. США. 2535385; С. A., 1951, 45, 3428.
24. F 1 e x s e r L., Farkas W., Food Manufacture, 1952, 27, 57.
25. Huennekens F., Kilgour G., J. Am. Chem. Soc., 1955, 77, 6716.
26. Gordon A., Green D. Subrahmanyan V. Biochem., J. 1940, 34, 764.
27. F r a n k e W., D e f f n e r M. Ann., 1937, 532, 1; 1939, 541, 117.
28. К a r r e r P., Schopp К., В e n z F. Helv. Chim. Acta., 1935, 18, 426;
Euler H., Karrer P. Helv. Chim. Acta, 1935, 18, 522.
29. К u h n R., Reinemund K-, Weygand F., Strobel e R. Ber/,
1935, 68, 1765.
30. T i s h 1 e r M., Wellman J. Пат. США 2261608, 21/Ш 1940; Off. gaz. 1941,
532, № 1, 160.
31. T i s h 1 о r M., Pfister K., Babson R., Ladenburg K-, Fle-
ming A. J. Am. Chem. Soc., 1947; 69, 1487. .
32. К u h n R. Angew. Chem., 1936, 49, 6.
33. Kuhn R., Weygand F. Ber., 1935, 68, 1282; 1938, 71, 1535.
34. Hoffmann — La Roche. Англ. пат. 628410, 1949; С. A., 1950, 44, 4935.
35. Kuhn R., Strobel e R. Ber., 1937, 70, 773.
36. Karrer P., Salomon H., Schopp K-, Schlitter E., Helv.-.
Chim. Acta., 1934, 17, 1165.
37. S eb r ell W., Harris S. The Vitamins N. Y., 1954,300—394.
38. Karrer P., Schlittler E., Pfaehler K-, Benz F. Helv. Chim.
Acta, 1934, 17, 1516.
39. К u h n R., Weygand F., Ber., 1935, 68, 1001.
40. Karrer P., Becker B., BenzF., Frei P., Salomon H., Schopp K-
Helv. Chim. Acta., 1935, 18, 1435.
41. W i s a n s k у W. Ansbacher S. J. Am. Chem. Soc., 1941, 63, 2532.
42. Kuhn R., Birkhofer L., Ber., 1938,71,621.
43. Hoffmann-— La Roche. Пат. США 2477560, 1949; С. A., 1951, 44, 169,
44. К a г г e г Р., М е е г w е i n Н. Helv. Chim. Acta, 1935, 18, ИЗО; 1936, 19, 264.
45. W е у g a n d F. Ber., 1940, 73, 1259.
46. Ruff 0., Ber., 1898, 31, 1573; 1899, 32, 553; 1902, 35, 2360.
47. С 1 a r k E. J. Biol. Chem., 1917, 31, 605.
48. J о n e s J., Kent P., Stacey M. J. Chem. Soc., 1947, 1341.
49. Hockett R., Hudson C. J. Chem. Soc., 1934, 56, 1632.
50. Березовский В. M., Курдюкова В. А. Получение D-арабинозы.—-
ЖПХ, 1949, т. 22, с. 1116—1117.
51. Bredereck Н. Вег., 1938, 71, 408; Bredereck Н., К б t h n i g M.,
Berger E., Ber., 1940, 73, 956.
52. S i t a G. Encyclopedia of Chemical Technology, N.—Y., 1955, 1.
53. Б e p e з о в с к и й В. M. Химические исследования в области витамина В2.-—
«Труды ВНИВИ», М., Пищепромиздат, 1953, № 4, с. 13—19. То же. 1954. № 5.,
с. 12-17.
54. S с h m i d t H. Пат. США 2587906, 17/IX 1949; Offic. gaz. 1952, 656, № 1 148
55. T i s h 1 e r M. Пат. США 2428437, 26/VII 1945; Offic. gaz. 1947, 603 № 1 86
56. T i s hl er M. Пат. США 2420210, 12/IX 1943; Offic. gaz. 1947, 598, № 1’ 132.
57. T i s h 1 e r M., Wellmann J. Пат. США 2342438, 18/VI 1941; 2358191
24/V 1941; Offic. gaz., 1944, 559, № 4, 652; 1944, 566, № 1, 310.
135
58. К и ж н е р. Н. Сравнительные исследования состава каменноугольного и нефтяного
ксилола. —ЖОХ, 1936, 6, 748—756.
59. Спрысков А. А. Выделение м-4 и п-ксилидинов из технической смеси изоме-
ров,— ЖПХ, 1948, 21, 157—163 с ил.
60. Karrer Р. Helv. Chim. Acta, 1935, 18, 1435, 1446.
61. Kobe К. Ph. Pritchett Ph. Ind. Eng. Chem., 1952, 44, 1398.
62. Ghigi E„ Ber., 1938, 71, 686.
63. Виз а некий В., Ан'сбахер С. Синтезы органических препаратов, М., ИЛ,
1949, 2, 79; 1953, 4, 93—170.
64. Синтезы органических препаратов, М., ИЛ, 1953, 3, 9, 12, 14.
65. Техническая энциклопедия, физические, химические и технические величины, 1927, 1,
269, 339.
66. Clark Е. J. Biol. Chem., 1917, 31, 605.
67. Березовский В. М., Родионова Е. П. О пространственных затруднениях
при образовании стериоизомерных N-полиоксиалкилзамещенных 0-аминоазокрасите-
лей. —ДАН СССР, 1952, т. 87, 585.
68. Березовский В. М. и Родионова Е. П. Авт. свидет. № 93306, 12/Х 1950;
Бюлл. изобрет. 1952, № 2—3, с. 7.
68а. Фирма Танабэ сэйяку, Японский пат. 10151, 1968.
69. S р е п g 1 е г О., Pfannenstiel A. Z. Wirtschaftsgr. Zuckerind., 1935, 85, 546.
70. Isbell Н., J. of Research of the National Bureau of Standards, 1942, 29, 227.
71. Б e p ез о в с к и й В. М. Исследования в области окисления альдоз. — ЖОХ, 1953,
И, 944.
72. Березовский В. М. Исследование в области синтеза конденсированных диази-
новых систем. Автореф. докт. дисс., М., 1962.
73. Суздальцева С. Ф. и Красюк В. П. Авт. свидет. Ns 194080, 1965;
Бюлл. изобрет., 1967, № 8, с. 20. То же. Григорашвили Е. И., Зо-
лотарев Н. С., Зарецкий Г. И. Непрерывная схема получения D-ри-
бозы. — «Хим.-фарм. ж.», 1971, № 3, с. 45—47 с ил.
74. Кубасов В. Л., Рыбкин В. И. ЖПХ, 1970, 43, № 2, 322; 327.
74а. Авруцкая И., Ф и о ш и н М., Громова Е. В.—ЖПХ, 1970, 43, № 5,
1084.
75. Ш н а й д м а и Л. О. Производство витаминов. Пищепромиздат, 1958, 332.
76. Березовский В. М., Артемкина Р., Хомутова Е. Д.—ЖОХ, 1964,
34, 2791.
77. Артемкина Р., Березовский В. — ЖОХ, 1966, 36, 823.
78. Б ерезовский В. М., Артемкина Р., Чернова М.— ЖОХ, 1965,
35, 677.
79. Аданяева Р., Денисова Л., Боровикова А., Березов-
ский В. — ХФЖ, 1967, 1, 16.
79а. Kuhn R., Rudy Н., Вег., 1935, 68, 383.
80. V i s к о n t i n i M., Eb not her C., Karrer P. Helv. Chim. Acta, 1952,
35, 457; Англ. пат. 712387; РЖХим, 1956, 30474; С. А., 1955, 49, 11725.
81. Forrest Н., Todd A., J. Chem. Soc., 1950, 3295.
82. Пат. США 2604470; С. А.„ 1953, 47, 3354; Пат. США 2610177; С. А., 1953, 47, 8781.
83. А р о s h i а n Н., J. Lambooy, J. Am. Chem. Soc., 1954, 76, 1307.
84. S n e 1 1 E., Strong F. Enzimologia, 1939, 6, 186.
85. К u h n R., Rudy H., Ber., 1936, 19, 2557.
86. К u h n R., Boulanger P. Z. physiol. Chem., 1936, 241, 233.
87. Березовский В., Мелентьева Л. — ЖОХ, 1961, 31, 3827.
Глава 6. ПРОИЗВОДСТВО СИНТЕТИЧЕСКОГО ПАНТОТЕНАТА
КАЛЬЦИЯ (ВИТАМИНА В3)
Пантотеновая кислота (витамин В3) открыта Р. Вильямсом в 1933 г.
11]. Она была им охарактеризована как стимулятор роста дрожжей. На-
звание свое витамин В3 получил от греческого термина «вездесущий», так
как пантотеновая кислота была обнаружена почти во всех растительных
и животных тканях. Хорошими источниками витамина В3 являются дрож-
• жи, отруби риса, печень [2]. В печени содержание пантотеновой кислоты
составляет 10 мг в 1 кг. В 1939 г. было установлено, что цыплячий фактор
и пантотеновая кислота идентичны [3, 4]. Вскоре было установлено, что
все животные, в особенности молодые, нуждаются в витамине В3 [5]. Не-
достаток пантотеновой кислоты вызывает преждевременное поседение во-
лос у крыс и серебристо-черных лисиц [6]. Характерным признаком В3-
авитаминоза у животных является потеря шерстного покрова [7] и де-
136
пигментация его. При недостатке пантотеновой кислоты цыплята заболе-
вают пеллагрой [81; вследствие этого пантотеновая кислота вначале была
названа цыплячьим фактором [9]. Установлено, что добавление в р щион
домашней птицы этого фактора приводит к значительному увеличению их
массы [10] и к повышению яйценоскости [9]. Потребность в витамине В3
животных колеблется в пределах 0,1—2,5 мг на 1 кг массы [11].
В 1945—1947 гг. Липманом с сотрудниками был открыт коэнзим А (от
слова ацетилирование), участвующий в ацетилировании холина в ацетил-
холин и в других реакциях ацетилирования, причем было доказано, что
этот коэнзим содержит пантотеновую кислоту [12]. Дальнейшее изучение
показало, что в состав молекулы коэнзима А входят: монофосфорный эфир
пантотеновой кислоты, адениннуклеотид и 2-меркаптоэтиламин [13, 14].
Кофермент А также участвует в окислительном распаде жирных кислот
и играет большую роль в образовании фосфолипидов [15].
Пантотеновая кислота благоприятно влияет на водный обмен, на усвое-
ние глюкозы. Имеется также указание на ее защитные действия при радио-
активном облучении. Таким образом, пантотеновая кислота имеет широкие
перспективы применения в профилактической и клинической практике,
а также в сельском хозяйстве. Суточная потребность человека в пантоте-
новой кислоте составляет 5—12 мг [15]. При конденсации пантотеновой
кислоты с p-меркаптоэтиламином образуется пантотетин, который в 100
раз активнее пантотеновой кислоты [14, 16].
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ВИТАМИНА В3
Пантотеновая кислота представляет собой производное |3-аланина и пан-
толактона (а,-окси-|3,|3-диметил--[-бутиролактона). Химическая формула
пантотеновой кислоты [1]:
СН3
I *
НОСН2—С—СН(ОН)—СО—NH—сн2—сн2—соон
7 ₽ | а
сн3
а, у, Диоксид, р«диметилбутирил-М-амид-₽'-аминопропионовой
кислоты 219,23
Ее эмпирическая формула С9Н17О5 N. Пантотеновая кислота имеет
один 'асимметрический атом углерода (обозначен звездочкой), вследствие
чего она имеет два оптических антипода D(+) и L(—) и рацемат. Биологи-
ческой активностью обладает правовращающая /)(+)-пантотеновая кисло-
та, левовращающая L(—) — биологически неактивна [17, 18, 19]. Раце-
мическая пантотеновая кислота обладает 50% активности правовращаю-
щей пантотеновой кислоты.
7)(+)-Пантотеновая кислота — маслянистое вещество, растворимое в
воде, спирте, уксусной кислоте, нерастворима в хлороформе, бензоле [20].
Ее удобнее употреблять в виде кристаллических солей натрия и кальция.
D-Пантотенат кальция имеет температуру плавления 193,5—195° С [21];
[ц]д+24,3 (вода) [19]; хорошо растворим в воде, нерастворим в органи-
ческих растворителях, мало в спирте. Кристаллы обладают гигроскопич-
ностью.
D-Пантотенат натрия представляет собой сильно гигроскопические иглы
с температурой плавления 121—122° С (с разложением); [aPg+27 (вода)
[22].
Пантотеновая кислота была идентифицирована следующими реакциями
[16, 22]: этерификация ее метиловым спиртом или диазометаном дает слож-
ный эфир, который омыляется с освобождением пантотеновой кислоты [23,
24].
сн3он
C8H17O6N----> (C3H16O3N)COOCH3.
137
Образование монометилового эфира указывает на то, что пантотеновая кис-
лота одноосновна. При ее ацилировании образуется' диацильное производ-
ное, что свидетельствует о наличии в молекуле двух гидроксильных групп:
сн coci /СООН
(C8H16O3N)COOH —----> (CsHuON)
Х(ОСОСН3)2
При щелочном гидролизе пантотеновая кислота расщепляется на р-аланин
и а, 7-диокси-р, |3-диметилмасляную кислоту по схеме:
н2о /СООН
(OH)2(C8HuON) СООН —-> NH2CH2CH2COOH + (С5Н9)— он
\он
Диоксикислота была выделена в виде кристаллического оксилактона
после нагревания подкисленного раствора [3] и его экстракции [25, 26].
Строение оксилактона, как а-окси-р, р-диметил-у-бутиролактон, было до-
казано превращением его в а, а-диметил-р-оксипропионовую кислоту.
Таким образом, к 1940 г. было окончательно установлено строение пан-
тотеновой кислоты. В соответствии со своей химической структурой панто-
теновая кислота может образовать простые и сложные эфиры по окси-
и карбоксильным группам, хлор ангидриды, амиды и другие соединения
[27]. С холином образует комплекс, обладающий биологическими свойст-
вами обоих витаминов [28]. Устойчива к кислороду воздуха [22]. Наибо-
лее важное биокаталитическое действие пантотеновая кислота проявляет в
составе коферментных и ферментных систем (реакции ацетилирования хо-
лина, уксусной кислоты, аминов, спиртов) [29, 30, 31]. Простейшим био-
логически активным коферментом является пантетеин [14], который пред-
ставляет собой продукт конденсации пантотеновой кислоты и 2-меркапто-
этиламина H2NCH2CH2SH и имеет следующую химическую структуру
HOCH2C(CH3)2CH(OH)CONHCH2CH2CONHCH2CH2SH.
Пантетеин
Пантетеин является ростовым фактором молочнокислых бактерий (La-
ctobacillus bulgaricus) [32].
Самым ответственным и наиболее сложным по химической структуре
биологически активным производным пантотеновой кислоты является ко-
фермент А, катализирующий различные реакции переноса и присоедине-
ния ацильных остатков в процессах жирового и углеводного обмена. Ак-
тивной группой кофермента, осуществляющей эти реакции, является суль-
фогидрильная группа 2-меркаптоэтиламина. Строение кофермента А было
изучено реакциями его гидролитического расщепления [14, 33]; на осно-
вании полученных данных установлена следующая химическая формула:
Г
ОН
РО3Н2
н о
н
о
О
NH3
сн2-о-Р-он
Н
Тршросфону-
клвозидаденин
I ^о
о-PC
I он
о
Н2С
1
Н3С-С-СН3
но-сн
о=с
ch2-coHnh-ch2-ch2-sh
СН2
XNHZ
Пантотеновая пиелита
| 2 - Меркаптоэтиламин
Кофермент А
138
Из химической формулы видно, что в коферменте А пантотеновая кисло-
та карбоксильной группой связана с 2-меркаптоэтиламином, а у-оксигруп-
пой с трифосфонуклеозид-аденином.
МЕТОДЫ СИНТЕЗА ПАНТОТЕНОВОЙ КИСЛОТЫ И ВЫБОР РАЦИОНАЛЬНОГО
МЕТОДА ДЛЯ ПРОИЗВОДСТВА
Как указано было выше (стр. 137), наивысшей биологической актив-
ностью (100%) обладает правовращающая 0(+)-пантотеновая кислота; ле-
вовращающая инактивна, а рацемическая имеет лишь 50% активности.
Обычно для лечебных или профилактических целей применяют оптически
активную £>(+), а для животноводства рацемическую пантотеновую кис-
лоту. В связи с этим целесообразно рассмотреть вопрос о выборе схем син-
теза обоих препаратов.
После того как Вильямс в 1938 г. выделил пантотеновую кислоту в
кристаллическом виде [34], им же в 1940 г. было установлено ее строение
как аланида, а, у-диокси-Р, (3-диметил масляной кислоты и осуществлен син-
тез [24, 26, 35]. В основу синтеза принята реакция конденсации двух ком-
понентов — эфиров или солей р-аланина и алифатической диоксикисло-
ты-а, у-диокси-р, p-диметил-у-бутиролактона (пантолектона). В полученном
продукте реакции р-аланин связан с безазотистой частью молекулы пеп-
тидной связью. Синтез пантотеновой кислоты был одновременно осуществ-
лен различными исследователями по этой же реакции [17, 19, 36].
Таким образом, синтез пантотеновой кислоты сводится к следующим
стадиям: а) синтезу р-аланина, б) синтезу пантолактона и в) конденсации
этих веществ. Особо стоит вопрос о синтезе О(—)-пантолактона, необходи-
мого для синтеза 0(+)-пантотеновой кислоты. Рассмотрим известные ва-
рианты указанных стадий синтеза пантотеновой кислоты.
СИНТЕЗ ₽-АЛАНИНА ф-АМИНОПРОПИОНОВОЙ КИСЛОТЫ)
Существует несколько методов синтеза р-аланина, различающихся по
реакции образования аминогруппы, а именно:
1. Из имида янтарной кислоты (сукцинимида) по реакции Гофмана [22,
37, 38] при взаимодействии с гипохлоритом или гипобромитом натрия или
калия в присутствии щелочи по схеме:
сн2-сох СН2—СООН w пГ1 СН2—СООН
|\ г^и 1 JN aOCl I
NH—> | ----> |
СН2—со/ сн2—conh2 Na0H сн2—nh2
Сукцинимид р-Аланин
Выход Р-аланина, получаемого по этому методу, низкий и составляет
41-45% [22].
2. Из циануксусного эфира путем каталитического гидрирования и омы-
ления его гидратом окиси бария [38, 39, 40] по схеме:
СООС2Н5 + н2 СООСН2Н5 Ва<он)2 СН2СООН
| ----------> | ---------------> |
СН2—CN сн2—сн2—nh2 сн2—nh2
Циануксусный Этиловый эфир р-амино- . р-Аланин
эфир пропионовой кислоты
Выход р-аланина достигает 72% [22], но циануксусный эфир дорог.
3. Из галогенопропионовой кислоты аммонолизом [41, 42] по схеме:
СН2—СООН nh, СН2—СООН
| ---> | +НС1
СН2С1 сн2—nh2
Р-Хлорпропионовая р-Аланин
кислота
р-Аланин получают в смеси с иминодипропионовой кислотой, из кото-
рой его выделяют с выходом в 50%. Метод дает недостаточный выход и ус-
ложнен как выделением р-аланина из реакционной массы, так и синтезом
139
хлорпропионовой кислоты (из акрилонитрила и соляной кислоты).
4. Из акрилонитрила аммонолизом [43, 44, 45] с последующим гид-
ролизом p-аминопропионитрила соляной кислотой или щелочью [46—48]
по следующей схеме:
СН2=СН—CN + NH3 H2N-CH2—СН2—CN —> H2N—СН2—СН2—СООН.
Акрилонитрил (З-Аминопропиочигрил [3-Аланин
Однако реакция цианэтилирования аммиака идет не однозначно с обра-
зованием смеси первичного, вторичного и третичного аминов [43, 44, 45,
50]: H2NCH2CH2CN; HN(CH2CH2CN)2; N(CH2CH2CN)3, что снижает выход
р-аланина. Для сдвига равновесной системы в сторону образования пер-
вичного амина была изучена зависимость выхода р-аланина от концентра-
ции раствора аммиака, относительного количества акрилонитрила и тем-
пературы проведения процесса [50 и 50а]. По данным Е. Жданович и
Е. Вялой, р-аланин с максимальным выходом получают при температуре
154—158° С и избыточном давлении 26—32 кгскм2 в 10%-ном растворе
аммиака при соотношении 10%-ного раствора аммиака к акрилонитрилу
18,5:1 и углекислого аммония к акрилонитрилу 3,7:1 [50]. Однако прямой
выход р-аланина составляет 40—44% [50а]. В последнее время разработан
метод превращения смеси вторичного и третичного аминов в р-аланин.
При этом выход повышается до 65—67%. Метод этот представляет интерес,
но нуждается в практической отработке.
5. Из акрилонитрила аминированием фталимидом [40, 51, 52] в при-
сутствии катализатора с получением в качестве промежуточного продукта
фталимидопропионитрила по следующей схеме:
^^СО /\/С°
ch2=ch-cn + Г V NH - » \-ch2-ch2-cn-
' I iclvJri XX .хХ. /
co co
Акрилонитрил Фталимид Фталимидпропионитрил
В качестве катализаторов известны: этилат натрия [22], триметилфенил-
аммоний [51], спиртовой 1%-ный раствор едкого натра [52]. При исполь-
зовании последнего выход полученного хлоргидрат р-аланина составил
77,8% [52]. Метод представляет существенный практический интерес.
Выделение свободного р-аланина или его соли. Метод выделения зависит
от того, в виде какой соли получают р-аланин при гидролизе. Если этот
процесс осуществляют соляной кислотой, то получают хлоргидрат р-ала-
нина. Для выделения из этой соли свободного р-аланина предложены раз-
личные реагенты: гидрат окиси лития [40, 51 ], окись свинца, а затем об-
работка водородом [40]. Эти реагенты либо дороги, либо требуют сложной
обработки во вредных условиях. Наиболее эффективным является метод
ионообмена на катионите КУ-2 [52] с элюированием 2%-ным раствором
аммиака с последующим выпариванием и кристаллизацией. Выход 92,0%.
Возможен и другой вариант — омыление серной кислотой с последую-
щим выделением ее окисью кальция в виде гипса, а р-аланина в виде каль-
циевой соли по следующей схеме:
2
СО
N-CH2-CH2-CN + 8Н2О + 2H2SO4
СО
у?- Фталимидлролионитрил
СООН
СООН
' Фталебая кислота .
ЗСаО
2
+ h2so4(h2n-ch2- сн2- СООН)2р+ (NH4)2SO4j
Сулырозидрат fl- аланина
(H2N-CH2-сн2-СОО)2Са + 2CaSO4 + ЗН2О + 2NH3
Кальциевая соль fl-аланина
140
При изучении режима омыления установлена оптимальная концентра-
ция серной кислоты в 25%, а pH при нейтрализации кислоты •— 6,8—7,0,
а для получения кальциевой соли около 8,0. Недостатком метода гидроли-
за серной кислотой с выделением ее в виде гипса и с получением кальцие-
вой соли р-аланина является трудность получения последней в чистом
виде, что отрицательно влияет на качество патентоната кальция.
Из анализа изложенных методов следует отдать предпочтение двум ме-
тодам синтеза р-аланина: 1) аммонолиз акрилонитрила под давлением и
2) аминированием акрилонитрила фталимидом с выделением свободного
Р-аланина на ионитах или в виде кальциевой соли.
СИНТЕЗ а-ОКСИ-8, р-ДИМЕТИЛ-7-БУТИРОЛАКТОНА (ПАНТОЛАКТОНА)
Вторым компонентом молекулы пантотеновой кислоты является а-окси-
Р, р-диметил-у-бутиролактон или (другое название) — а, у-диокси-р, р-диме-
тилмасляная кислота. Синтез пантолактона осуществлен, исходя из изома-
сляного альдегида, общим методом получения а-оксикислот — циангидрин-
ным синтезом по следующей схеме [19, 36, 531:
СН3 СН3
I К2СО3 I NaCN(KCN)
нсно + НС—СНО-------------> НОСН2—С—СНО --------:----->
I (NaaCO3) | СаС12
сн3 сн3
Формальдегид Изомасляный (З-Окси-а, а-диметил-
альдегид пропионовый альдегид
сн3 сн3
I H2so4 I
—> НОСН2—С— СН—CN-------------> НОСН2—С—сн—соон -
II II
сн3 он сн3 он
Нитрил а, 7-диокси-З . (3-ДИ- а, у-Диокси-р, (З’ДИметилмасляиая
метилмасляной кислоты кислота
СН3
I
—> СН2—С—сн—со
I I
СНз ОН
-----о-----
Пантолактон
Химизм процессов заключается в альдольной конденсации изомасляно-
го альдегида и формальдегида в присутствии поташа или соды в р-окси-а,
а-ДИметилпропионовый альдегид; последний конденсируют с синильной кис-
лотой или цианистым калием в присутствии хлористого кальция [53] или
с цианистым натрием [37] и получают а, у-диокси-р, р-диметилмасляную
кислоту и при ее лактонизации — рацемический пантолактон. Дальней-
шее усовершенствование синтеза пантолактона привело к упрощению тех-
нологического процесса в результате замены цианирующего агента — циа-
нистого калия — ацетонциангидрином и других технологических усовер-
шенствований. В результате четырехстадийный синтез пантолактона стали
осуществлять в одну стадию [54]. Таким образом, исходным сырьем для
получения пантолактона является изомасляный альдегид, который может
быть получен различными методами синтеза [22, 55]:
1) гидратацией хлористого диметилэтилена с последующим омылением
и дегидратацией:
СН3 . СН3 С1 СН3 ОН 1СН3
I +Н2О | | II |
С=СНС1---> СН—СН —► СН—СН-----> СН—СНО
I 1111 -н‘° I
сн3 сн3 он сн3 он сн3
141
2) гидролизом с одновременной дегидратацией эфиров:
СН3 СН3
I H2so4 I
С(ОН)—СН2ОС2Н5-------> СН—сно
I 190° |
сн3 сн3
3) окислением изобутилового спирта хромовой смесью:
СН3 СН3
I К2СГ2О7 |
СН—СН2ОН--------> сн—сно
I I
сн3 сн3
Первые два метода для промышленного применения не приемлемы’из-за
дефицитности сырья. Что касается третьего метода, то недостатком его
является побочная реакция, обусловливающая дальнейшее окисление аль-
дегида в изомасляную кислоту и этерификация ее изобутиловым спиртом,
не вошедшим в реакцию. В результате этой реакции образуется в значи-
тельном количестве изобутиловый эфир масляной кислоты, что значительно
снижает выход изомасляного альдегида (около 40%). Кроме того, бихро-
мат калия также дорог и дефицитен [55]. Наилучшую перспективу для
промышленного применения имеет метод синтеза изомасляного альдегида
путем каталитического дегидрирования изобутилового спирта кислородом
воздуха на медном или серебряном катализаторе при температуре 230—
300° С с выходом 80—90% [56]. В дальнейшем было показано [55], что
серебряный катализатор, нанесенный на пемзу, при температуре 500—
600° С более эффективен по сравнению с медным. По-видимому, вопрос
о выборе катализатора для данного процесса должен быть дополнительно
изучен. Имеются также указания [16], что в качестве промышленного ме-
тода может быть осуществлен оксосинтез:
Катализатор
СН3—СН=СН2 + со + Н2О------------> сн3
Пропилен 1н-сно
I
СН3
Изомасляный альдегид
Из изложенного можно сделать заключение, что наиболее рациональ-
ным методом синтеза пантолактона является одностадийный метод, пред-
ложенный Е. Жданович и Е. Вялой, заключающийся в альдольной кон-
денсации изомасляного альдегида и формальдегида с цианированием аце-
тонциангидрином и дальнейшим омылением и лактонизацией [54]. Этим
методом получают рацемический D, L-пантолактон. Для синтеза оптически
активной £)(+)-пантотеновой кислоты считают более целесообразным кон-
денсировать левовращающий О(—)-пантолактон с |3-аланином, чем рас-
щепление на свои антиподы D, Е-пантотеновой кислоты. Для получения
£>(—)-пантолактона необходимо пантолактон-рацемат разложить на опти-
ческие антиподы. Для этого на рацемат действуют каким-либо оптически
деятельным органическим основанием-алколоидом, например, хинином [16,
19, 36, 57] бруцином [58] или оптически деятельными синтетическими ами-
нами, как, например, ц-фенилэтиламином [59]. Если право- и левовращаю-
щие пантолактоны обладают одинаковыми свойствами, за исключением
вращения плоскости поляризации и кристаллизации в энантиаморфных
формах, т. е. с различной пространственной ориентировкой атомов, то
полученные соли с алколоидами вследствие вхождения в их молекулы но-
вого асимметрического углеродного атома обладают различными свойства-
ми, как, например, растворимостью. Поэтому мы получаем возможность
их разделить дробной кристаллизацией. Разделив эти соли и разложив
их кислотой, мы получаем стереизомеры в чистом виде. Таким путем уда-
142
ется из пантолактона рацемата выделять D(—)-пантолактон. Предложен
также способ расщепления рацемического пантолактона при помощи
/.(+)-трео-1(и-нитрофенил)-2-амино-1,3-пропандиола [50, 60, 61]. Поэто-
му методу выход комплексной соли О(+)-лактона и Е(+)-треоамина со-
ставляет 70,9%, а выход О(—)-пантолактона из комплексной соли состав-
ляет 62,9% [60]. После выделения D(—)-пантолактона остающаяся L(+)-
форма пантолактона в виде натриевой соли подвергается при 130—150° С
рацемизации едким натром и после перегонки в глубоком вакууме (5—
10 мм рт. ст.) получают пантолактон-рацемат с выходом около 90%. По-
лученный D, Ё-пантолактон-рацемат можно снова подвергнуть расщепле-
нию на оптические антиподы и т. д.,. пока весь рацемат не будет превращен
в D (—)-форму.
СИНТЕЗ П(+)-ПАНТОТЕНОВОЙ КИСЛОТЫ ИЛИ ЕЕ РАЦЕМАТА
Последней стадией синтеза является конденсация О(—) или D, L-пан-
толактона с |3-аланином. Этот процесс осуществляют различными путями:
а) при нагревании до 70° С D(—)-пантолактона с этиловым или мети-
ловым эфиром |3-аланина [24, 36, 35] с последующим омылением с выхо-
дом О(+)-пантотеновой кислоты 50% по схеме:
СН3
_Н,С-С----СНОН
I I
Н2С со
о
СН3
н3с-с-СНОН HqSOa
+ h2nch2ch2cooc2h5—*- I 1хон ' 2 4»
Н2С ^^NHCH^COOC^.
J) (-) - пантопактон Этилобый эфир
fl-аланина
Эфир D (+) -пантотенобой кислоты
О(+)-пантотеновая кислота;
б) лучшие результаты получают при конденсации пантолактона с нат-
риевой или кальциевой солью |3-аланина в среде безводного спирта [37,
62 , 63];
в) при конденсации сухой натриевой соли |3-аланина с £>-(—)-пантолак-
тоном при температуре 100—105° С получают пантотенат натрия с высоким
выходом (92%) [64]. Имеются указания [22], что при использовании в
реакции конденсации свободного |3-аланина выход пантотеновой кислоты
весьма низок;
г) однако выход значительно повышается, если конденсацию вести в
среде вторичных или третичных аминов в присутствии окиси кальция или
этилата натрия [65]. Е. Жданович указывает [50], что кальциевая соль
пантотеновой кислоты была получена при конденсации |3-аланина и О,
/.-пантолактона в среде метилового спирта в присутствии диэтиламина
с обработкой реакционной массы окисью кальция с выходом в 90,2%.
Из всего изложенного можно прийти к технологической схеме произ-
водства Р(+)-пантотеновой кислоты или ее рацемата, заключающейся в
следующих стадиях синтеза:
получение (3-аланина аммонолизом акрилонитрила в одну стадию;
одностадийный синтез О, Е-пантолактона путем альдольной конденса-
ции изомасляного альдегида и формальдегида и цианирования |3-окси-а,
а-диметилпропионового альдегида ацетонциангидрином и последующего
омыления и лактонизации;
расщепление О, Е-пантолактона-рацемата и выделение О(—)-пантолак-
тона с помощью А(+)-треоамина;
конденсация D(—)-пантолактона и |3-аланина в среде метилового спир-
та в присутствии диэтиламина.
143
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА £>(+)-ПАНТОТЕНАТА КАЛЬЦИЯ
И ЕГО РАЦЕМАТА
В основу технологической схемы положены известные в литературе
данные [19, 36, 43—46, 49, 53, 65], а также опубликованные исследования
Е. Жданович, Н. Преображенского и их учеников [50, 50а, 52 , 54 , 55, 60,
61].
СИНТЕЗ Р-АЛАНИНА
Синтез р-аланина осуществляют аммонолизом акрилонитрила. При этом
образуется смесь первичного, вторичного и третичного аминов [43—45,
50] по следующей схеме:
+ 2ЩО
. H2NCH2CH2CN-----> H2NCH2CH2COOH + NH3
X I 89,09
/ -J- 4H2O z
6H2C=CH—CN + 3NH3 > HN(CH2CH2CN)2----------> HN(CH2CH2COOH)2 + 2NH3
6H2O
4N(CH2CH2CN)3-----> N(CH2CH2COOH), 4- 3NH3
Акрилонитрил 17,03
53,06
Для сдвига равновесия реакции в сторону образования Р-аланина сле-
дует обеспечить большой избыток аммиака и высокую температуру [44,
66]. По данным Е. Жданович [50], требуется температура реакции 154—
158° С (избыточное давление 26—32 /егс/с.и2), соотношение 10%-ного раст-
вора аммиака к акрилонитрилу 18,5:1 и углекислого аммония к акрило-
нитрилу 3,7:1. На основании этих данных технологический процесс заклю-
чается в следующем: в горизонтальный автоклав 1 (рис. 18) с вращающейся
мешалкой и паровой рубашкой загружают из мерника 2 водный раствор
(10—15%) аммиака и из сборника 3 двууглекислого аммония и из мерника
4 акрилонитрил. Нагревают реакционную массу до 154—158° С, при этом
избыточное давление повышается до 30—40 кгс/см2. Не допускается за-
грузка более 0,4 объема автоклава. Из автоклава реакционную массу вы-
гружают в перегонный аппарат 5, где отгоняют водный раствор аммиака.
Кубовый остаток сливают в реактор 6, разбавляют водой и очищают акти-
вированным углем при температуре 40—50° С; уголь отфильтровывают на
нутч-фильтре 7, фильтрат направляют в сборник 8, а затем в вакуум-ап-
парат 9 для сгущения. Сгущенный раствор сливают в кристаллизатор 10,
где выделяют р-аланин добавлением из мерника 11 этилового абсолютиро-
ванного спирта при температуре 0+5° С. Затем осадок фугуют в центри-
фуге 12. Кристаллы сушат в вакуум-сушилке 13 и направляют в сборник
14. Маточный раствор поступает в сборник 15, откуда засасывают в ваку-
ум-аппарат 16, сгущают, сливают в кристаллизатор 17, где спиртом выде-
ляют дополнительное количество р-аланина, который отфуговывают в цент-
рифуге 18. Кристаллы р-аланина II для переосаждения направляют в реак-
тор-кристаллизатор 10. Маточный раствор II из центрифуги 18 собирают
в приемнике 19, он является либо отходом производства, либо его направ-
ляют на переработку в р-аланин. Выход р-аланина — прямой 40—50%,
а при регенерации р-аланина из вторичного и третичного аминов выход
может быть увеличен до 65—70%. P-Аланин (Р-аминопропионовая кислота)
C3H7O2N представляет собой бесцветные кристаллы с температурой 199—
200° С [52], молекулярная масса 89,09, хорошо растворим в воде, труднее
в метиловом, этиловом и изопропиловом спиртах; нерастворим в эфире
и ацетоне.
СИНТЕЗ а-ОКСИф, Р-ДИМЕТИЛ-7-БУТИРОЛАКТОНА (ПАНТОЛАКТОНА)
[54, 60]
Для синтеза О(+)-пантотеновой кислоты необходимо синтезировать
О(—)-пантолактон, который может быть получен из D, L-пантолактона
(рацемата). Следовательно, вначале рассмотрим синтез пантолактона-ра-
144
Рис. 18. Технологическая схема производства £)(+)-пантотената кальция.
цемата. Известный синтез пантолактона из изомасляного альдегида [19,
36, 53] был в последнее время усовершенствован [50, 50а]. Изучены усло-
вия конденсации изомасляного альдегида с формальдегидом. Показано,
что для стабильного и максимального выхода а, а-диметил-|3-оксипропионо-
вого альдегида необходимо поддерживать pH среды 7,5—8,0, что позволяет
без выделения подвергнуть последний цианированию. Для цианирования
ядовитые цианистые соли заменены ацетонциангидрином; реакция протека-
ет в течение 5 ч\ омыление нитрила и лактонизация проводятся при тем-
пературе 65—70° С в течение 4 ч 18%?ной соляной кислотой.
В результате проведенных исследований ранее известный четырехста-
дийный синтез пантолактона осуществлен в одну стадию по следующей
схеме:
сн3
сн-сно
I
сн3
+ нсно---►
pH 8,0
СН3
носн2-с-сно
сн3
Н3С. /ОН
Н3С^ CN
Изомасляный
альдегид
72,09-
Формальдегид
30,03
СНз
НО-СН2-С -CH-CN
с£,с£- ди мети л -J3- ом си ~
пропионобый альдегид
102, 07
/i ц етонциангидрин
85,10
нсг
сн3 он
сн3
НОС Но- с -сн-соон
I I
сн3 он.
Нитрил -сС, у- диаке и - fl, диме -
тилмасляной кислоты
128, 74
г (сн3)2-с-сн(он)
Н2Сх хСО
V
об-Окси -Д,Д - ди мет ил ~у-бутиролактон
130,74 •
of, у -Диокси -Д,Д~ ди мет ил-
масляная кислота
135, 13
В реактор 20 из эмалированной стали загружают из мерника 21 фор-
мальдегид (35%-ный), из мерника 22 изомасляный альдегид и из мерника
23 водный раствор соды (20%-ный). Реакционную массу нагревают до
70° С и перемешивают 40 мин, охлаждают до 20° С, подкисляют из мерника
24 18%-ной соляной кислотой до pH 8,0. Затем из мерника 25 загружают
ацетонциангидрин. Реакцию цианирования ведут 4 ч при 18—22° С. Далее
из мерника 24 приливают в реактор соляную кислоту и при температуре
70—80° С проводят процесс омыления натрила в течение 3 ч. После этого
реакционную массу нейтрализуют углекислым натрием до pH 5,0. Из ре-
акционной массы пантолактон экстрагируют метиленхлоридом в колон-
ном экстракторе с механическим перемешиванием [67 ]. Для этого реак-
ционную массу из реактора 20 засасывают в напорный сборник 26, откуда
она поступает в экстракционную колонну 27 снизу, а метиленхлорид из
сборника 28 поступает в колонну сверху. Экстракт пантолактона из колон-
ны идет в сборник 29 и далее направляется в напорный сборник 30 и в пле-
ночный испаритель непрерывного действия 31, где отгоняется метиленхло-
рид. Концентрат пантолактона непрерывно поступает в сборник 32, откуда
его передают в вакуум-перегонный аппарат 33, где отбирают фракцию
при температуре 112—114° С (остаточном давлении 8 мм рт. ст.), которую
направляют в сборник 34. Выход продукта 61,6%. Результаты исследова-
ния процесса экстракции пантолактона непрерывным методом [67] уста-
новлены следующие оптимальные условия: а) объемное отношение раство-
рителя к водному раствору пантолактона — для дихлорэтана 2,5:1, а для
метиленхлорида — 2:1; б) нагрузка по обеим фазам — 30—35 м3/м2 в 1 ч;
в) окружная скорость мешалки 0,65—0,70 м/сек.
146
а-0кси-|3, p-диметил-у-бутиролактон представляет собой бесцветное ве-
щество аморфного строения, температура плавления 74,6—75,4° С [54];
хорошо растворим в воде и в органических растворителях.
СИНТЕЗ D(—)-а-ОКСИф, р-ДИМЕТИЛ-7-БУТИРОЛАКТОНА (ОПТИЧЕСКИ
АКТИВНОГО ЛЕВОВРАЩАЮЩЕГО) [50, 60, 61]
£>(—)-пантолактон получают расщеплением рацемата при помощи L(+)-
трео-1-(п-нитрофенил)-2-амино-1,3-пропандиола — Ц+)-треоамина
(C9H12N2O4) [60 ], представляющего отход производства антибиотика левоми-
цетина. Метод заключается в конденсации Ь(+)-треоамина с рацематом
пантолактона с разложением диастереомерных солей серной кислотой.
Предложены два варианта получения комплексной соли: 1) непосредствен-
ная конденсация пантолактона-рацемата с Ь(+)-треоамина и 2) конденса-
ция натриевой соли рацемата с сернокислой солью Ь(+)-треоамина.
Реакция протекает по следующей схеме:
СНз
2 HOH2C-C-CHOH-COONa + (o2N~С6Н4- CHOH-CH-CH2OH)2H2SO4 + H2SO4
сн3 nh2
Пантолактон натрия L (н)-треоамин (C9Hf2N20^)
2'153, /4 ’ 2-212,2
СНз Na2SO4 СН3
hoch2-c-choh-cooh-c9h12n2o4 носн2-с-снон-соон- c9h12n2o4
СН3 СН3
леЗая tpopwi правая срорма
<5 о
i/) СУ)
3D (+) т пантолактон
130, 14
494, 43
D (-)- пантолактон
130,14
По первому варианту выход комплексной соли составляет 53,7%, а по вто-
рому— 78,81%. Следовательно, оптимальным является второй вариант
[50]. Разложение диастереомерных солей проводят серной кислотой с
целью получения сернокислой соли Ь(+)-треоамина, которую вновь ис-
пользуют для расщепления пантолактона-рацемата.
Сернокислая соль Л(ф-)-треоамина. В реактор из эмалированной стали
35 загружают из мерника 36 серную кислоту (7—8%-ную) и при темпера-
туре 40—45° С прибавляют Ь(+)-треоамина (температура плавления
159,5—160,5° С), [oJd +28°. Реакционную массу обрабатывают активиро-
ванным углем (1,5—2,0%), фильтруют через нутч-фильтр 37 в кристалли-
затор 38. Кристаллы фугуют в центрифуге 39, сушат в вакуум-сушилке 40
и направляют в сборник 41. Маточный раствор из центрифуги через сборник
42 засасывают в вакуум-аппарат 43, сгущают, кристаллизуют в кристалли-
заторе 44 при температуре 2—4° С, фугуют в центрифуге 45 и направляют
на перекристаллизацию в реактор 35. Маточный раствор поступает в сбор-
147
ник 46 и является отходом производства. Выход 93,4%; температура плав-
ления 231—231,5° С (разложение); [а1о = +24,20 (с—5%, вода).
Комплексная соль £)(-[-)-пантолактона и Ь(4-)-треоамина. В реактор
47, снабженный прямым холодильником, загружают из сборника 34 пан-
толактон-рацемат, растворяют его в воде, добавляют из мерника 48
42%-ный раствор едкого натра. Раствор перемешивают 1 ч при температуре
70—73° С (pH 7,0). Затем из сборника 41 прибавляют сернокислую соль
Ь(+)-треоамина, нагревают реакционную массу 3 ч и раствор выпаривают
до состояния густой, но подвижной кассы, приливают из мерника 49 спирт
из расчета получения пересыщенного раствора и перемешивают. Выпав-
ший в осадок сернокислый натрий отфильтровывают на нутч-фильтре 50.
Фильтрат сливают в кристаллизатор 51, где выкристаллизовывают ком-
плексную соль правовращающего пантолактона и Ь(+)-треоамина при тем-
пературе 2—4° С. В центрифуге 52 отфуговывают кристаллы, высушивают
в вакуум-сушилке 53 и собирают в приемнике 55. Маточный раствор посту-
пает в сборник 54. Выход 82,2%, температура плавления 135,5—136,3°С
(из спирта); [а]о = +24,5° (с=2%, вода).
D(—)-пантолактон. В реактор из эмалированной стали 56 растворяют
полученную соль Л(4-)-треоамина и £>(+)-пантолактона в воде, добавляют
из мерника 57 серной кислоты (30%-ную), перемешивают 2 ч при темпера-
туре 85° С, а затем раствор подают в сборник 58 и далее на экстракцию
в колонный экстрактор непрерывного действия 59 снизу [67]; раствори-
тель (метиленхлорид) из сборника 60 поступает в колонну сверху. Экстракт
пантолактона из колонны непрерывно выходит, через сборник 61 его на-
правляют в пленочный испаритель 62, где отгоняют растворитель до
объема. Концентрат через сборник 63 подают в кристаллизатор 64, откуда
после кристаллизации в течение 20 ч при температуре 3° С направляют
в центрифугу 65, далее в сушку 66 и сборник 67. Выход 25% (на рацемат).
Температура плавления 90—91° С; [а]р=+50,25° (с=2%, вода). Маточ-
ный раствор направляют в сборник 68.
Регенерация А(+)-треоамина. Водный слой из экстрактора 59 (сверху)
поступает в кристаллизатор 64, где по охлаждении до 3° С в течение 3 ч
выкристаллизовывают Т(+)-треоамин. Выход 81%, температура плавления
231—233°С; [а]р =+21° (с=5%, вода).
7.(-|-)-пантолактон. Маточный раствор, получаемый после выделения
комплексной соли правого пантолактона, из сборника 54 поступает в ва-
куум-аппарат для отгонки спирта 69, сливают в реактор 70 и добавляют
серной кислоты из мерника 57, нагревают при 85° С 2 ч, а затем раствор
подают в сборник 71, далее в колонный экстрактор 72, куда непрерывно
подают метиленхлорид из мерника 73. Экстракт Л(+)-пантолактона снизу
колонны выходит в сборник 74 и далее в пленочный испаритель 75, снаб-
женный конденсатором 76 и приемником 77, а концентрат поступает в крис-
таллизатор 78, центрифугу 79 на сушку 80 и в сборник 81. Выход 95%,
температура плавления 90—9ГС (из бензола); [а]20=+50,3° (с—2%,
вода).
Рацемизация А(-]-)-пантолактона. В реактор из нержавеющей стали 82
загружают L (+)-пантолактон из сборника 81, добавляют из мерника 82
42%-ный раствор едкого натра. Смесь нагревают 8 ч при 130—150° С, сли-
вают в сборник 83, засасывают в вакуум-аппарат 84 для разгонки в ваку-
уме (5 мм рт. ст). Выход 92,4%, [а]д=0°. Рацемат из сборника 85 воз-
вращают на расщепление в реактор 47.
/)(+)-Пантотенат кальция. Получают путем реакции конденсации
П(—)-пантолактона с (3-аланином в присутствии диэтиламина [50] с по-
следующей обработкой образовавшейся пантотеновой кислоты на ионите
КУ-2 и нейтрализацией углекислым кальцием. Реакции протекают по сле-
дующей схеме:
148
сн3
н3с-с—сн(он)
2 , । । _ + 2NH2CH2CH2COOH
HgC . .с — о
о
Л (-) - пантолактон р-алаиин
130, 10 99,03
NH(C2HS)2_
Катионит K6~Z
Диэтиламин
73, №
. , , \ СаСО3
—►гноен2с(сн3)2сн(он)conhcн2сн2соон mgg*
D(+)-лантотенобая кислота
---►[онс н2с(сн3)2с HOHCON НС Н2С Н2СОО]3Са
]](+)- пантотенат кальция
476,53
Полагают, что диэтиламин образует с пантотеновой кислотой соль, ко-
торая при обработке ионитом КУ-2 разлагается. В реактор из нержавею-
щей стали 86 загружают из мерника 87 спирт, из сборника 67 D)—)-пан-
толактон, из сборника /4-|3-аланин и из мерника 88 диэтиламин, подогре-
вают до 70—75° С при перемешивании. После растворения компонентов
добавляют активированного угля, перемешивают 10—15 мин и фильтруют
через нутч-фильтр 89 в сборник 90. Затем фильтрат засасывают в вакуум-
аппарат 91, отгоняют спирт, добавляют воды и сливают раствор в сборник
92, а оттуда подают в напорный мерник 93 и далее на ионитную колонну
94, сборник очищенного раствора 95. Отсюда раствор передают в реактор-
нейтрализатор 96, куда добавляют мел для нейтрализации пантотеновой
кислоты. Избыточный мел отфильтровывают на нутч- фильтре 97 в сборник
98. Фильтрат засасывают в вакуум-аппарат 99, сгущают при вакууме (10—
15 мм рт. ст.) до содержания сухих веществ 90—92%, сливают в кристал-
лизатор 100, куда вводят абсолютированный спирт из мерника 101. Крис-
таллы отфуговывают в центрифуге 102, сушат в вакуум-сушилке 103 и на-
правляют в приемник 104. Маточный раствор из сборника после центрифу-
ги 105 подвергается переработке по схеме: вакуум-аппарат 106, кристалли-
затор 107, центрифуга 108. Кристаллы второго продукта направляют для
перекристаллизации в вакуум-аппарат 99, маточный раствор II поступает
в сборник 109 и является отходом производства. Пантотенат-рацемат
можно получать таким же методом.
Выход £>(+)-пантотената кальция составляет 90,29-6 [50]. Считают, что
выход пантотената кальция рацемата аналогичен. Выход 1)(+)-пантотената
кальция при перекристаллизации 80%. О(+)-пантотенат кальция при не-
обходимости перекристаллизовывают из спирта с обработкой активиро-
ванным углем или очищают ионообменным способом (катионит КУ-23)
с выходом 88% [68].
Синтез /?(+)-пантетеина [69, 70]. Как уже было указано (стр. 138)
пантетеин является простейшим коферментом, являющимся ростовым фак-
тором молочнокислых бактерий. Синтез пантетеина заключается в конден-
сации £)(+)-пантотената кальция и 2-мер.каптоэтиламина по следующей
химической схеме:
(HOCH2C(CH3)2CHOHCONHCH2CH2COO)2Ca + СООН + 2 (C2H5)3N —>
СООН
D (+) Пачтотенат кальция Щавелевая кислота Триэтиламин
[HOCH2C(CH3)2CHOHCONHCH2CH2COO-NH+(C2H5)3]2 —>
- С1СООС2Н5 + (H2NCH2CH2SH)3
Этиловый эфир 2-Меркаптоэгиламин
хлоругольчой
кислоты
[HOCH2C(CH3)2CHOHCOMHCH2CH2CONHCH2CH2SH]2
D (4-)-ПдН етси.ч
149
Ниже описан лабораторный метод получения пантетеина [69]. К раство-
ру 10 г £>(+)-пантотената кальция в 25 мл воды приливают 10 мл три-
этиламина и 25 мл 7,6%-ного раствора щавелевой кислоты. Осадок от-
фильтровывают. Фильтрат упаривают. Маслянистый остаток сушат до по-
стоянной массы в вакууме (10 мм рт. ст.) и растворяют (12,5 г) в 50 мл
диметилфорамида. В охлажденный раствор до минус 5—10°С приливают
4,1 г этилового эфира хлоругольной кислоты, перемешивают и затем при-
ливают 25 мл раствора цистамина (меркаптоэтиламин) в метаноле. Вы-
павший осадок отделяют, а растворитель выпаривают. Маслообразное ве-
щество растворяют в 400 мл воды и пропускают через колонки со смолой
КУ-2 (Н+-форма) и Вофатид LD — 50(ОН~-форма). Колонки промывают
водой. Элюат упаривают досуха. Выход 4,81 г (43,6%); [а]д + 16,6° (с=3%,
вода).
Описаны методы синтеза S-ацильных производных (S-пальмитоил-,
S-стеароил-, S-бензил-) £)-пантетеина [71 ].
ТЕХНОЛОГИЧЕСКИЕ ПОКАЗАТЕЛИ
РАСХОД ХИМИКАЛИЕВ, КГ НА 1 КГ
100%-НОГО ПОЛУПРОДУКТА
В СИНТЕЗЕ D(+)-nAHTOTEHATA
• КАЛЬЦИЯ
^-Аланин
Акрилонитрил................... 1,2
Аммиак жидкий..................2,2
Аммоний углекислый ...... 2,2
Уголь активированный ...........0,05
Спирт этиловый.................0,5
Вмход, % от теоретического
(на акрилонитрил)..............60,0
а-Окси-'fi, ^-диметил-'рбутиролактон
(пантолактон)
Альдегид изомасляный............0,9
Ацетоциангидрин................ 1,0
Формальдегид 35%-ный........... 1,1
Метиленхлорид...................2,5
Сода кальцинированная ......... 0,6
Кислота соляная ............... 1,0
Выход, % от теоретического (на
изомасляный альдегид)..........61,6
D (—уа-Окси-1), ’удиметил-у
бутиролактон
Сернокислая соль
Е(+)-т реоамина
А(+)-Треоамин...................0,88
Кислота серная (1,83)...........0,20
Уголь активированный ..........0,05
Выход, % от теоретического (на
треоамин)......................93,4
D (—УПантолактон
Пантолактон (рацемат) .'........4,8
Натр едкий (42%-ный) ...........3,0
Сульфат треоамина (с учетом воз-
врата) .........................2,0
Кислота серная (1,83).......... 1,4
Метиленхлорид...................2,8
Спирт ..........................2,8
Выход, % от теоретического (на пан-
толактон рацемат)..............25,0
£>(+)-Пантотенат кальция
(технический)
£>(—)-Пантолактон ..............0,60
З-Аланин........................0,42
Диэтиламин......................0,40
Спирт ..........................0,50
Ионит КУ-2 .....................0,15
Мел ............................0,28
Выход, % от теоретического (на
£>(—)-Пантолактон)..............90,2
О(~у)-Пантотенат кальция (меди-
цинский)
£>(-(-)-Пантотенат кальция техни-
ческий .......................1,25
Спирт ......................0,50
Уголь активированный ...........0,05
Выход, % .....................80,0
РАСХОД ХИМИКАЛИЕВ, КГ
НА ПРОИЗВОДСТВО 1 КГ
£>(+)-ПАНТОТЕНАТА КАЛЬЦИЯ
Альдегид изомасляный...........3,24
Акрилонитрил ..................0,62
Аммиак жидкий...................1,14
Аммоний углекислый .............1,14
Ацетонциангидрин...............3,60
Диэтиламин......................0,50
Ионит КУ-2'....................0,19
Мел ...........................0,35
Метиленхлорид.................11,10
Кислота соленая ...............3,60
Кислота серная..................1,31
Натр едкий (42%-ный) ..........2,25
Спирт ..........................3,46
Уголь активированный............0,12
Формальдегид.............-. . .• 3,96
Ь(+)-Треоамин...................0,66
РАСХОД ПОЛУПРОДУКТОВ, КГ
НА 1 КГ £>(+)-ПАНТОТЕНАТА
КАЛЬЦИЯ
^-Алании........................0,52
£>(—)-Пантолактон ..............0,75
Пантолактон-рацемат .............3,6
Технический О(+)-пантотенат каль-
ция ............................1,25
Сульфаттреоамин................1,5
150
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. W i 1 1 i a m s R., Lyman C., Goodyear G., T r u e s d a i 1 T.,
Holiday D., J. Am. Chem. Soc., 1933, 55, 2912.
2. J u n k e s J., Biolog. Chem., 1937, 117, 11.
3. Wooley D., Waisman H., E 1 v e h j e m C., J. Am. Chem. Soc., 1939,
61, 977; J. Biol. Chem., 1939, 129, 673.
4. J u k e s T., J. Am. Chem. Soc., 1939, 61, 975.
5. Morga n A., Simms H. J. Nutrition, 1940, 19, 233.
6. Веселкина В. Успехи современной биологии. 1947, 24, 33.
7. Mickelson О., Waisman Н., Е 1 v е h j е m С., J. Biol. Chem., 1938,
124 313.
8. Е 1 v е h j е m С., Koehn С., J. Biol. Chem., 1935, 108, 709.
9. S n e 1 1 E. и др., J. Nutrition, 1940, 21, 201.
10. S n e 1 1 E. и др., J. Biol. Chem., 1941, 133, 552.
11. S t e r n J., C a m p i 1 1 о A., Coon M. J. Am. Chem., Soc., 1953, 75, 1517, 2277.
12. Lipmann F., Kaplan N., N о v e 1 1 i G. и др. J. Biol. Chem., 1947,
167, 869.
13. В e i n e r t H., К о r f f R., J. Am. Chem. Soc., 1953, 74, 854.
14. В a d d i 1 e у J., T h a i n E., J. Chem. Soc., 1952, 800; 1951, 3421; 1952, 3783.
15. Ill и л о в П. И, Я к о в л e в Т. Н. Основы клинической витаминологии. М.,
«Медицина», 1964, 88.
16. Б е э р А. А., Рубцов И. А. Синтез витаминов. М., Пищепромиздат, 1956,
17. Kuhn R., Wieland Т„ Вег., 1940, 73, 971, 1134.
18. Griissner A., G a t z i—F i c h t e r M., Reichstein T., Helv. Chim.
Acta, 1940, 23, 1276.
19. S t i 1 1 e r E., Harris S., Finkelstein J., Keresztesy J.,
F a 1 k e r s K-, J. Am. Chem. Soc., 1940, 62, 1785.
20. W о о 1 1 e у D., Waisman H., Mickelson О., E 1 v e h j e m C.,
J. Biol. Chem., 1938, 125, 715.
21. Kogan F., Heinzelman R., W e i s b 1 a t D., Greiner W. J. Am.
Chem. Soc., 1957, 79, 3545.
22. Б e p e з о в с к и й В. M. Химия витаминов. М., Пищепромиздат, 1959, с. 91.
23. Williams R., Weinstock Н., Rohr mann Е., Truesdail J.,
Mitchell H., Meyer C. J. Am. Chem. Soc., 1939, 61, 454.
24. W i 1 1 i a m s R., M i t c h e 1 1 H., Weinstock H., Snell E. J. Am.
Chem. Soc., 1940, 62, 1784.
25. W о о 1 1 e у D. Science, 1940, 91, 245.
26. W i 1 1 i a m s R., Major R. Science, 1940, 91, 246.
27. H a r r i s S., F о 1 k e r s К. Пат. США 2557284; С. A., 1952, 46, 3071.
28. F r e e d M. Пат. США 2653968; С. A., 1954, 48, 8817.
29. Kaplan N., S о о d a k M., Federation. Proc.., 1949, 8, 211.
30. Браунштейн A. E., Ефимочкина E. Ф. Влияние недостаточности
пантотеновой кислоты на синтез гиппуровой кислоты в организме крысы. — «ДАН
СССР», 1950, т. 71, с. 347—353.
31. L у п е n F., Reichert Е., Angew. Chemie, 1951, 63, 47.
32. S п е 1 1 F., Brown G., Peters V., Craig J., W i t t 1 e E.,
Moore J., Me G1 о hon V., Bird O., J. Am. Soc., 1950, 72, 5349.
33. H о a d 1 a n d M., N о v e 1 1 i G. J. Biol. Chem., 1954, 207, 767.
34. W i 1 1 i a m s R. J. Am. Chem. Soc., 1939, 61, 454.
35. W i 1 1 i a m s R., Science, 1939, 89, 486.
36. Reichstein T., Griissner A. Helv. Chim. Acta, 1940, 23, 650.
37. Б e эр А. А., Преображенский H. А. Химия пантотеновой кислоты
и ее синтез. — В сб.: «Новое в науке и технике витаминов», М., Пищепромиздат,
1946, № 1, с. 43—47.
38. К л а р к X., Б э р Л. Синтезы органических препаратов. М., ИЛ, 1949, 2, с. 20
39. W е у g а п d F., Вег., 1941, 74, 256. '
40. Р о д и о н о в В. М., Ярцева Н. Г. Простой лабораторный способ приготов-
ления р-аланина. — «Известия АН СССР», 1948, № 2, с. 251—255.
41. Н е i п t z W. Ann. 1870, 156, 36.
42. Mulder Е., Вег., 1876, 9, 1903.
43. В а у е г О. Angew. Chem., 1949, 61, 229.
44. Т е р е н т ь е в А. П., Чурсина К. И., Кост А. Н. О реакции акрило-
нитрила с аммиаком и получении триметилендиамина. —ЖОХ, 1950, т. 20,
с. 1073—1078.
45. К о с т А. Н. —«Ученые записки МГУ, серия хим.», 1950, VI, 131, 37.
46. Ф о р д т Д., Б э к С. Синтез органических препаратов. ИЛ, 1553, 4, 14, 46.
47. F о г d I., J. Am. Chem. Soc., 1945, 67, 876.
48. С a г 1 s о n G., Hotchkiss С., Пат. США, 2335997; С. А., 1944, 38, 2972 с.
49. В и с S., Ford J.,’ W i s е Е., J. Am. Chem. Soc., 1945, 67, 92.
50. Ж Д а н о в и ч Е. С. Исследования в области синтеза и разработка методов про-
изводства витаминов группы В. Доклад докт. дисс., М., 1965, 36 с.
151
50а. Б я л а я Е. И., ДД а нови ч Е. С., Евдокимова Г. С. и др. Изуче-
ние прямого аммонолиза акрилонитрила. — «Хим.-фарм. ж», 1969, № 12, с. 40—
41 с ил.
51. G а 1 a t A. J. Am. Chem. Soc., 1945, 67, 1414.
52. Ж Д а и о в и ч Е. С., Бялая Е. И., Крылова В. Н., Преобра-
женский Н. А. и др. Исследования в области синтеза пантотеина. — ЖОХ,
1961, т. 31, с. 446. То же — «Труды ВНИВИ», М., Пищепромиздат, 1961, № 7,
с. 16—17.
53. Carter Н., Ney L., J. Am. Chem., Soc., 1941, 63, 312.
54. Жданович E. С., Бялая Е. И., Крылова В. Н. и др. Авт. свидет.,
№ 150525, 1962; Бюлл. изобрет., 1962, № 19.
55. Жданович Е. С., Бялая Е. И., Преображенский Н. А. Син-
тез пантотеновой кислоты. — «Труды ВНИВИ», М., Пищепромиздат, 1959, № 6,
с. 14—17.
56. Губен И. Методы органической химии. Госхимиздат, 1941, 2, с. 40.
57. Theilacker W., Winker Н. Вег., 1954, 87, 690.
58. В е u t е 1 R., Т i s h 1 е г М. J. Am. Chem. Soc., 1946, 68, 1463.
59. Ozegowski W., H a e r i n g H. Pharmazie, 1957, 12, 254.
60. Ж Д а н о в и ч E. С., Козлова Г. С., Крылова В. Н., Преобра-
женский Н. А. Авт. свидет., № 147182, 1961; Бюлл. изобрет., 1962,
№ 10, с. 24; То же. Пат. ГДР 16982, 1959; С. А.; 1960, 54, 19494 f.
61. Жданович Е. С., Крылова В. Н., Козлова Г. С., Преобра-
женский Н. А. Синтез оптически активных а-окси-3,В-диметил-т-бутиролак-
тонов. — ЖОрХ, 1967, т. 3, с. 826—829.
62. Р i с k е 1 F., Weinstock Н., Пат. США 2442143; С. А., 1948 , 42 , 5174.
63. В а й д л и х Г. Успехи химии. 1941, т. 40, с. 804.
64. В а b с о с к S. Jukes Т. J. Am. Chem. Soc., 1940, 62, 1628.
65. W i 1 s о п E., W e i 1 a r d J., T i s h 1 e г M. Пат. США 2496363, 27/VIII
1948; Off. gaz. 1950, 631, 17/11; To же, J. Am. Chem. Soc. 1954, 76, 5177.
66. F о r d J., В u c S., Greiner J. J. Am. Chem. Soc., 1947, 69, 844.
67. Бурова Л. E., Плановский A. H., Бялая E. И. Исследование
экстракции а-окси-Э,р-диметил-бутиролактона непрерывным методом. —«Меди-
цинская промышленность СССР», 1966, № 2, с. 47—50 с ил.
68. Ж данович Е. С., Козлова Г. С., Кибалова Н. Ю. Выделение
и очистка £)-пантотеновой кислоты ионообменным методом. — «Хим.-фарм. ж»,
1970, № 2, с. 27—29, с ил.
69. Жданович Е. С., Копелевич В. М., Преображенс-
кий Н. А. Синтез £>-(+)-пантетина. —ЖОХ, 1967, т. 37, с. 361—363.
70. W i е 1 а п d Т., В о k е 1 m а п п Е. Naturwiss., 1951, 38, 384.
71. Копелевич В. М., Евдокимова Г. С., Рязанцева А. Ф.,
Жданович Е. С. Синтез S-ацильных производных £>-пантетеина — «Хим.-
фарм. ж.», 1970, № 11, с. 28—30.
Глава 7. ПРОИЗВОДСТВО СИНТЕТИЧЕСКОГО ПИРИДОКСИНА
(ВИТАМИНА Вв)
Экспериментальный Вв-авитаминоз был впервые изучен в 1934 г. [1].
При отсутствии этого витамина у крыс возникает пеллагра, выражающая-
ся в появлении симметричного дерматита с поражением конечностей, кон-
чиков ушей и носа [2]. У собак и свиней В6-а'витаминоз протекает без кож-
ных явлений и преимущественно характеризуется нервными симптомами
[3].
В6-авитаминоз у человека наблюдать не удалось [2], однако признано,,
что ряд болезненных явлений вызван недостатком витамина Вв. Установ-
лено [2], что витамин В6 оказывает регулирующее влияние на нервную
систему. Недостаток витамина Вв вызывает своеобразные изменения кожи,
а также слизистой оболочки полости рта и языка.
Известно, что В8 участвует в аминокислотном обмене и прежде всего
в обмене триптофана, влияет на эритропоэз, лейкопоэз и обмен железа.
Он показан при лечении следующих заболеваний [2]: прогрессивная мы-
шечная дистрофия, миастения, себоррея, рвота и тошнота у беременных
женщин, лучевая болезнь, кишечные инфекции, спру, пеллагра и др.
В 1938 г. из рисовых отрубей был выделен, кристаллический продукт
[4], который в 1939 г. идентифицировали как витамин Вя [51, в этом же
году Гаррис и Фолькерс разработали его синтез [6]. По предложению
152
Гиорги витамин В6 в соответствии с его химической структурой был на-
зван пиридоксином. Позднее было установлено, что пиридоксин в живот-
ных тканях и дрожжах содержится в весьма активной форме; повышение
его активности обусловлено превращением пиридоксина в пиридоксамин
и пиридоксаль [7, 8, 9, 10]. На долю пиридоксина приходится 20%, а пи-
ридоксаля и пиридоксамина — 80% от общего содержания витаминов груп-
пы В6. Витамин В6 в виде кофермента пиридоксаль-фосфорного эфира
(кодекарбоксилазы) входит в состав различных ферментов аминокислотно-
го обмена: декарбоксилаз, аминофераз и др. Разнообразные биохимические
функции витаминов группы Вв нашли широкое освещение в литературе
[11—16]. Ряд работ посвящен содержанию пиридоксина в пищевых про-
дуктах [17—20].
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ВИТАМИНА Вв
К витаминам группы В6 принадлежат три вещества, которые в организ-
ме могут превращаться одно в другое по следующей схеме:
Пиридоксин
(2-Метил-3-окси-ь,5 -
диоксиметиллиридин)
СН2ОН
СН2ОН ~2К» н
i2H. н
сно
сн2он
+ nh3-h2o
+ 2Н
Пиридоксамин
(2-Метил-З-окси-к-амино-
метил-5-оксиметилпиридин)
Пиридоксаль
(2-Метил - 3-окси - 4- формил -
5-оксиметилпиридин )
Пиридоксин гидрохлорид C8HtlNO3HCl, молекулярная масса 205,64,
представляет собой кристаллы белого цвета, имеющие форму пластинок
с температурой плавления 204—208° С [21], а по другому источнику [22]
с температурой 205—212° С с
разложением. Вещество оптиче-
ски неактивно [23], легко су-
блимируется без разложения.
Гидрохлорид хорошо раство-
рим в воде, но плохо в спирте и
ацетоне [4]. В 100 мл воды при
комнатной температуре раство-
ряется 20 г, а в 100 мл спирта—
1,1 г; pH 3,0 (в водном раство-
ре). На свету пиридоксин бы-
стро разлагается в щелочных и
дейтральных растворах [3]. В
'0,1 н. растворе НС1 он сравни-
тельно устойчив.
Таутомерные свойства пири-
доксина подтверждаются измен- рис jg Спектр поглощения витамина В6
чивостью его абсорбционного УФ-свете.
спектра в ультрафиолетовом
свете при разных pH [23]: при
pH 2,0 кривая имеет максимум при Х=292,5 нж; при pH 4,0 значение
Е повышается и появляется новый максимум при 327,5 нж; при pH 6,75
Ез27,5 повышается, максимум при 292,5 нм исчезает и появляется макси-
мум при 256 нм. При повышении pH до 10,2 для обоих максимумов увели-
чивается E, а максимумы сдвигаются в сторону более коротких волн [21 ].
На рис. 19 показан абсорбционный спектр пиридоксина для pH 4,0 и 6,75.
Пиридоксин-основание, C8HnNO3, получают в виде бесцветных приз-
матических кристаллов с температурой плавления 160° С, хорошо раство-
римых в воде, спирте и ацетоне, плохо растворимых в эфире и хлорофор-
153
ме, легко сублимируются. Они обладают слегка горьким вкусом. Пири-
доксаль кристаллизуется в виде оксима с температурой плавления 225—
226° Сив виде хлоргидрата с температурой плавления 165° С (с разложе-
нием). Растворимость хлоргидрата пиридоксаля 1 г в 2 мл воды; 1,7 г в
100 мл 95%-ного спирта; pH 1%-ного водного раствора — 2,65.
Пиридоксамин-основание — бесцветные кристаллы с температурой
плавления 193—193,5° С, пиридоксамин хлоргидрат — температура плав-
ления 226—227° С (с разложением). Растворимость хлоргидрата пиридок-
самина: 1 г в 2 мл воды; 0,65 г в 100 мл 95%-ного спирта; рН1 %-ного вод-
ного раствора 2,4 [24].
В химическом отношении пиридоксин проявляет свойства стабильного
азотистого основания. Из растворов он осаждается фосфорновольфрамовой
кислотой; солями тяжелых металлов (свинца, ртути, серебра, платины),
его осадить нельзя. Минеральные кислоты, нагревание или охлаждение
на витамин В6 не оказывают влияния [23]. Не действует на него также
жидкость Фелинга [21]. С хлорным железом пиридоксин подобно фенолам
дает красновато-коричневую окраску. Изучением химической структуры
пиридоксина занимались в 1938—1939 гг. различные исследователи: Стил-
лер, Керештези, Стивенс и Гаррис [5, 23, 25] в США; Кун, Вендт и Вест-
фаль [26—29] в Германии; Итиба и Мити [30] в Японии.
Для установления структуры важное значение имело получение при
окислении пиридоксина 2-метил-3-метокси-4,5-пиридин-карбоновой кисло-
ты, из которой в дальнейшем был синтезирован пиридоксин
Пиридинкарбоновую кислоту превращали в 2-метил-З-мето кси-4,5-ди-
цианпиримидин, из последнего путем каталитической гидрогенизации по-
лучали 2-метил-3-метокси-4,5-диаминометилпиридин. При действии нанего
азотистой кислоты аминометильные группы превращали в оксиметильные.
Реакции протекают по следующей схеме [29]:
СООН
2-Метил -3-метон си-
4,5 -пиридин - нардонодая
пиелита
2 -Метил -3- метонси-
4,5 - дицианпиридин
2 - Метил - 3- метопси -
4,5- диаминометилпиридин
ГЧН3
РОСГ
(~н2о)
HNO<
СНоОН
2 - Метил -3 -метопси-
4,5- дионсиметилпиридин
2 -Метил - 3 - окси-
4,5-диоксиметилпиридин
Впоследствии [31] синтез пиридоксина из пиридиндикарбоновой кисло-
ты был осуществлен в две стадии. Проводилось восстановление эфира пи-
ридинкарбоновой кислоты литийалюмгидридом до 2-метил-3-метокси-4,5-
диоксиметилпиридина и омыление последнего в пиридоксин. Реакции про-
текают по следующей схеме:
154
СООС2Н5
Н3СО-|^\-СООС2Н5
н3с—U ф
N
Диэтилобый эфир 2 - мети л -
3-метокси - 4,5-пиридинкар-
бонобои кислоты
СН2ОН
[Н] Н3СО-г^^СН2ОН Н2О
L1AIH4 H3c-U JJ НС1Г
N
2 - Метил -3- метокси-
4,5- диоксиметилпиридин
СН2ОН
Пиридоксин
Строение пиридоксаля
ния из метилового эфира
и пиридоксамина доказано реакцией их получе-
пиридоксина по следующей схеме:
НО
Н3С
Метилобый эфир
пиридоксина
NH3
Пиридоксамин
СН2ОН
Пиридоксин
-2[Н]
СНО
Пиридоксальные коферменты. Несмотря на разнообразие химических
реакций превращений аминокислот, катализируемых витаминами группы
В, установлено, что во всех случаях участвует один и тот же кофермент—
пиридоксаль-5-фосфат (кодекарбоксилаза), имеющий следующее строение:
N
СНО
НО СН2ОРО3Н2
Н3С
По-видимоМу, специфичность каждой реакции зависит от вида протеина,
связанного с кодекарбоксилазой [11]. Строение пиридоксальфосфата до-
казано синтезом его из пиридоксамина или из пиродоксина реакциями
фосфорилирования и окисления [32, 33].
МЕТОДЫ СИНТЕЗА ВИТАМИНА В6 И ВЫБОР РАЦИОНАЛЬНОГО МЕТОДА
ДЛЯ ПРОИЗВОДСТВА [21, 24, 34—38]
Химическая структура молекулы пиридоксина открывает перспективу
многих путей синтеза ее. Казалось, наиболее эффективным должен быть
путь синтеза через производное пиридина, как, например, 2-метил-5-этил-
пиридин или Р-пиколин. Однако введение заместителей в пиридиновый
цикл (кроме ^-положения) является весьма сложным и малодоступным.
Следовательно, остаются два варианта возможного осуществления синте-
за пиридоксина: 1) применение таких производных пиридина или хиноли-
на, которые уже содержали бы заместители в требуемых положениях (2; 3;
4 и 5), либо 2) синтез из алифатических фрагментов пиридинового цикла
с функциональными группами в соответствующих положениях. К первому
варианту относится синтез пиридоксина через производные хинолина или
изохинолина, а ко второму варианту — синтез пиридоксина: а) через про-
изводные динитрила цинхомероновой кислоты и б) через производные нит-
рила никотиновой кислоты.
155
СИНТЕЗ ПИРИДОКСИНА ЧЕРЕЗ ПРОИЗВОДНЫЕ ХИНОЛИНА И
ИЗОХИНОЛИНА [29, 30]
Этот синтез основан на деструкции сложных молекул — производных
хинолина и изохинолина — и на образовании в качестве промежуточного
продукта синтеза цинхомероновой кислоты. Для синтеза используются
производные хинолина, содержащие в пиридиновом кольце соответствую-
щие заместители: в положении 2 метильную, а в положении 3 метоксигруп-
пу. Для предотвращения разрушения пиридинового кольца, ослабленного
указанными заместителями, в бензольное кольцо хинолинового или изо-
хинолинового производного вводят аминогруппу, это достигают нитрова-
нием (обработкой азотной кислотой) и последующим восстановлением
водородом в присутствии палладиевого катализатора. Затем полученный про-
дукт окисляют перманганатом калия в щелочной среде, при этом образу-
ется а, (3-, у-пиридинтрикарбоновая кислота или для б) 2-метил-З-мето-
ксицинхомероновая кислота. У первой при температуре 180° С отщепля-
ют СО2 и тоже получают 2-метил-З-метоксицинхомероновую кислоту,
которую этерифицируют, затем восстанавливают литийалюмогидридом в
2-метил-3-метокси-4,5-диоксиметилпиридин и омыляют. Реакция происхо-
дит по следующей схеме (а — для хинолинового производного, б — для
изохинолинового производного):
Пиридоксин
2~Метил -3-метокси -
4,5- диоксиметилпиридин
2 - Метил -3- метоксицин -
хомероновая кислота
ДизтилоВый. эфир 2-метил-
З-метоксицинхомеронобой кислоту
Недостатком этого метода является
производных.
трудность получения хинолиновых
МЕТОДЫ СИНТЕЗА ПИРИДОКСИНА, ОСНОВАННЫЕ НА ПОСТРОЕНИИ
ПИРИДИНОВОГО ЦИКЛА ИЗ ДВУХ АЛИФАТИЧЕСКИХ ФРАГМЕНТОВ
Молекулу пиридоксина можно рассматривать, как состоящую из двух
структур [37]:
СН2ОН
|НО-С7^ ^с-СН2ОН
Н3С-С^
N
Второй и четвертый углеродные атомы структуры I должны иметь функ-
циональные группы, которые при взаимодействии со структурой II обеспе-
чат образование пиридинового цикла. Углерод в положении 3 может не
иметь функциональной группы, так как введение ее в |3-положении доступ-
156
но. Таким образом, структура I может быть использована в виде оксиаце-
тилацетона или амида ацетопировиноградной кислоты:
СН2ОН
н2с;4^о
Н3С-С2
ХО
Оксиацетипацетон
СО1\Н2
н2с;Ьо
н3с-ск.
хо
Амид ацетопиродиноградной
кислоты
Структура II должна иметь в положении 6 аминогруппу, которая с
С=О в положении 2 структуры I создает углерод-азотную связь. В поло-
жении 5 углерод должен иметь электроотрицательный заместитель, кото-
рый увеличил бы подвижность его водородных атомов, как, например,
карбоксильную или еще лучше нитрильную группу. Последняя легко мо-
жет быть превращена в оксиметильную. Таким образом, структура II мо-
жет быть представлена в виде:
,СН2— CN
• 1
JC\
nh2 хо
Цианацетамид
Из изложенного выше вытекают два варианта синтеза.
Вариант синтеза пиридоксина через производные динитрила цинхомеро-
новой кислоты [35, 36, 39, 40]. Исходным сырьем для синтеза служат амид
ацетопировиноградной кислоты и цианацетамид. При конденсации указан-
ных веществ в присутствии диэтиламина [39, 40] образуется 2-метил-4-кар-
боамид-5-циан-6-пиридон с выходом 73%. При дегидратации пиридона
хлорокисью фосфора в среде пиридина образуется 2-Метил-4,5-дициан-6-
пиридон (выход 92%). При нитровании последнего крепкой азотной кисло-
той в присутствии уксусного ангидрида получают 2-метил-3-нитро-4,5-
дициан-6-пиридон (выход 68%), который при нагревании с пятихлористым
фосфором дает хлор производное (выход 96%). Последнее при восстановле-
нии в присутствии катализатора (палладия) на угле под давлением дает
2-метил-3-амино-4,5-диаминометилпиридин (выход 41%). Далее при диазо-
тировании нитритом натрия в солянокислом растворе получают пиридок-
син (выход 45%). Реакция протекает по следующей схеме:
сн2-см
nh2 хо
РОС13
-Н2О
2-Метил-4-кардомид-
HNO3
2-Метил - 4,5-дии.иан-
Цианацетамид
CN
5-циа.н-В-пиридон 6-лиридон
conh2
н2Ч 'о
н3с-с^.
о
Амид ацетолиробино-
градной кислоты
CN
2 - Метил- J- нитро-
4,5-дициан - В-пиридон
PC1S O2N-44-CN [н]
Н,С “Ч С1 Рсруж®
.N
2-Метал-д-нитро-4,5-
дициан -6-клорпиридин
ch2nh2
H2N-p|r СН2Ь1Н2 NaNO2; НС1 НО
п3сЛ ') "н3с
N 3
Пиридоксин
СН2ОН
СН2ОН )
N
2-Метил - 3- амино-4,5-
диаминометиллиридин
Этот метод дает вполне удовлетворительный выход пиридоксина, но
внедрение его затруднено дефицитностью исходного сырья — амида ацето-
пировиноградной кислоты.
157
Вариант синтеза пиридоксина через нитрил никотиновой кислоты. Ниже
приведена схема синтеза, разработанного Гаррисом и Фолькерсомв перво-
начальном варианте и с введенными в последующем изменениями.
Схема синтеза пиридоксина по Гаррису и
Фолькерсу
СН2ОС2Н5
Н2С хо
I
НзС-С.
о
Этоксиацетилацетон
I
СН2ОС2Н5
2-Метил-^ -этоксимегпил-
5-циан-6-пиридон
ш
CH2OC2HS
V
и
Pd
ch2-cn
^с.
NH2 о
Циан ацетамид
o2n
н3с
СН2ОС2Н
NH
С2Н5ОН
Пиперидин
2-Метил -3-нитро - 4 -зто-
ксиметил-5-циан-6-пиридон
IV
CH2OC2HS
С6Н5С1
PC1S
h2n
Н3С
O2N CN
Н3С Cl
N
Z -Метил -3- нитро -4-
зтоксиметил -5-циан-
6-хлорпиридин
V
H2N —у >т- CN
н3с Д-ci
N
Z-Метил-З- амино-4-
этоксиметил - 5-циан-
6-хлорпиридин * ___
VI
Ml
Pd 1
NH
Z -Метил -3-амино -k-
этоксиметил -5 - циан -
6-пиридон
а
СН2ОС2Н5
СНоОН MONO
N
CH2OC2HS
CH2NH2
НС1
h2n
H3C
2 ’
2 - Метил -3 - амино-4 - окси -
метил -5-аминометилпири-
дин
Villa
H2SC>4
h2n
н3с
3 N
но
Н3С.
2-Метил-3-окси-Ц-этокси- 2-Метил-З-амино-Ь-зтокси-
метил -5-метоксипиридин метил - 5~аминометилпиридин
Z - Метил -3 -окси - Р, 5-
бромметилпиридин гидродром ид
IX
Пиридоксин гидрохлорид
X
Исходным сырьем для данного метода синтеза служит этоксиацетилаце-
тон и цианацетамид, которые при конденсации в присутствии пиперидина
дают 2-метил-4-этоксиметил-5-циан-6-пиридон (выход 81 %). Последний под-
вергают нитрованию дымящей азотной кислотой в среде уксусного ангид-
рида при температуре 40—45° С, в результате чего получают 2-метил-З-
нитро-4-этоксиметил-5-циан-6-пиридон (выход 70%). Полученный пиридон
обрабатывают пятихлористым фосфором при нагревании в хлорбензоле
и получают 2-метил-3-нитро-4-этоксиметил-5-циан-6-хлорпиридин (выход
31 %). Далее гидрогенизацией под давлением в присутствии платинового
или палладиевого катализатора нитрогруппу переводят в аминогруппу,
158
при этом получают 2-метил-3-амино-4-этоксиметил-5-циан-6-хлорпиридин
(выход 76%). Для восстановления в последнем нитрильной группы в ами-
нометильную и одновременном элиминировании атома хлора осуществляют
гидрогенизацию хлорпиридина в присутствии палладия на костяном угле.
В результате получают 2-метил-3-амино-4-этоксиметил-5-аминометилпири-
дин (выход 55%). Для замещения аминогруппы на оксигруппу производят
диазотирование аминометилпиридина и разложение соли диазония в горя-
чем растворе смеси серной кислоты и нитрита натрия. В результате этой
реакции получают 2-метил-3-окси-4-этоксиметил-5-метоксипиридин (этило-
вый эфир пиридоксина); (выход —около 30%). При обработке этилового
эфира пиридоксина 48 %-ной бромистоводородной кислоты получают 2-ме-
тил-З-окси-4,5-дибромометил гидробромид (выход 66%). Его омыляют пу-
тем кипячения в водном растворе, удаляют ионы брома хлористым сереб-
ром и получают пиридоксин (выход 75%).
Этот первоначальный вариант синтеза был авторами затем несколько
изменен. Например, при переходе от нитропиридона (IV) к хлораминопи-
ридину (VI) вначале получали аминопиридон (Va), а затем хлорпроизвод-
ное (VI). Для перехода от эфира (VII) к пиридоксину вначале проводили
омыление эфира в (Villa), а затем диазотирование. Для перехода от эфира
пиридоксина (VIII) к пиридоксину (X) проводили непосредственный гид-
ролиз эфира 2,5 н. соляной кислотой под давлением при 160° С [41] с
исключением стадии бромирования. Были внесены и другие изменения
[42, 43].
Этот метод синтеза благодаря доступности исходного сырья нашел ши-
рокое применение в заводской практике.
При дальнейшей разработке данного метода было показано, что наибо-
лее высокий выход пиридоксина получают при использовании для синтеза
метоксиацетилацетона вместо этоксиацетилацетона [44—52]. Метоксиаце-
тилацетон может быть получен из монохлоруксусной кислоты, которая
метилированием и последующей этерификацией превращается в метиловый
эфир метоксиуксусной кислоты [53]. Последний путем сложноэфирной кон-
денсации с ацетоном образует метоксиацетилацетон по следующей схеме:
CH3ONa ' СН3ОН
С1СН2СООН-------> СН3ОСН2СООН-------> СН3ОСН2СООСН3 —►
Монохлоруксусная Метиловый эфир Метиловый эфир
кислота уксусной кислоты метоуксусной
кислоты
сн3сосн3
------СН3ОСН2СОСН2СОСН3
Метоксиацетилацетон
Для дальнейшего упрощения синтеза пиридоксина было предложено
[54 и 54а] сочетать реакции получения цианацетамида, метоксиацетилаце-
тона и пиридона в одной стадии путем взаимодействия метилового эфира
метоксиуксусной кислоты и ацетона в присутствии метилата натрия. За-
тем, не выделяя натрийметоксиацетилацетона, в реактор вводят цианук-
сусный эфир и аммиак, при этом получается цианацетамид, который также
не выделяют. При добавлении серной кислоты происходит конденсация
метоксиацетилацетона и цианацетамида с образованием пиридона.
Однако нагромождение трех реакций в одной стадии вряд ли будет це-
лесообразным Выход пиридона в этих условиях низок (39—55% к теоре-
тическому). При отсутствии каких-либо особых причин, как, например,
нестойкость или сильная ядовитость промежуточных продуктов реакции,
сочетание большого числа химических реагентов в одном аппарате следует
считать неэффективным. Поэтому в рекомендуемой схеме синтеза принято
выделение цианацетамида и метоксиацетилацетона как самостоятельных
промежуточных продуктов.
Новый синтез пиридоксина. Разработан Гаррисом и др. [65] на основе
нового метода синтеза пиридиновых соединений Кондратьевой и Хуан Чжи
159
Хен (из производных оксазола и диенофилов) [66]. Синтез Гарриса заклю-
чается в конденсации 4-метил-5-этоксиоксазола с диэтиловым эфиром ма-
леиновой кислоты или с динитрилом фумаровой кислоты, или с 2,5-дигид-
рофураном.
Реакции протекают по следующей схеме:
соос2н5 сн2он
"ТТосн *
\ П3С-Ч> / П3С-1^. >
О ММ
Ь-Метил-5-зтоксиоксазол Диэтилобый эфир малеиновой кислоты Дизтилобый зрир-2-метил- Пиридоксин
-3-окси-4,5 -дикарбоксипиридин
4-Метил-5-этоксиоксазол синтезирован из этил-£), Л-аланина гидрохло-
рида; при воздействии на последний муравьиной кислоты получен этил-
N-формил, D, L-аланин с выходом 78% (температура кипения 100°С,
CeHnNO3). Это соединение циклизуют с фосфорным ангидридом в среде
хлороформа (5 ч) по методу Каррера [67], нейтрализуют едким кали, ди-
стиллируют и получают 4-метил-5-этоксиоксазол с выходом 60 % (темпера-
тура кипения 78—80° С, при остаточном давлении 50 мм рт. ст.) ?.тах =
=229 нм', е=4440 (метанол), CeH9NO2. Оксазол смешивают с двумя моля-
ми диэтилового эфира малеиновой кислоты, нагревают при 110° С в тече-
ние 2 ч и после обработки аддукта соляной кислотой получают диэтиловый
эфир 2-метил-3-окси-4,5-дикарбоксипиридина с выходом 85%,
температура плавления 140—144° С (лит. 144—145° С) [68]. Этот диэфир
восстанавливают литийалюмогидридом (LiAlH4) в пиридоксин. Авторами
также предложен вариант конденсации оксазола с динитрилом фумаровой
кислоты (NCHC=CHCN) в среде метанола в течение 5 ч. Аддукт обрабаты-
вают соляной кислотой и получают кристаллы 2-метил-3-окси-4,5-дициан-
пиридин, температура плавления 189—191° С, Хгаах=235; 360 «ж; е=12 900;
9300, (pH 7,0, фосфатный буфер), C8H5N3O. Дицианпиридин превращают
в пиридоксин гидрогенизацией на палладиевом катализаторе с получением
2-метил-3-окси-4,5-диаминометилпиридина (выход 70%, температура плав-
ления 290° С, лит. 296° С) и диазотированием (выход 79%). Этот метод син-
теза усовершенствован Г. Кондратьевой; однако отсутствие публикаций
по технологии метода не позволяет отдать ему предпочтение перед другими
методами.
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА ПИРИДОКСИНА (ВИТАМИНА Вв)
Технология производства пиридоксина состоит из отдельных стадий
[35, 53—57] (рис. 20) получения следующих продуктов:
метилового эфира метоксиуксусной кислоты из монохлоруксусной кис-
лоты;
цианацетамида из этилового эфира циануксусной кислоты и аммиака;
натрийметоксиацетилацетона из метилового эфира метоксиуксусной кис-
лоты;
2-метил-4-метоксиметил-5-циан-6-пиридона (пиридона) из метилового
эфира метоксиуксусной кислоты и цианацетамида;
2-метил-3-нитро-4-метоксиметил-5-циан-6-пиридона (нитропиридона) из
пиридона;
получение ' 2-метил-3-нитро-4-метоксиметил-5-циан-6-хлорпиридина
(нитрохлорпиридина) из нитропиридона;
получение 2-метил-3-окси-4-метоксиметил-5-метоксипиридина (метилово-
го эфира пиридоксина) из нитрохлорпиридина;
получение 2-метил-3-окси-4,5-диоксиметилпиридина (хлоргидрата пи-
ридоксина) из метилового эфира пиридоксина.
160
Рис, 20. Технологическая
схема производства синтетического пиридоксина.
6-522
Синтез метилового эфира метоксиуксусной кислоты. Эфир получают
из монохлоруксусной кислоты при действии на нее метилата натрия в сре-
де метилового спирта и присутствии серной кислоты согласно следующему
химическому уравнению:
НщО,
С1СН2СООН + СН3ОН + CH3ONa----------Н3СОСН2СООСН3 + NaCl + Н2О
Монохлоруксусная 32,04 54,03 Метиловый эфир метокси- 58,46
кислота 94,50 уксусной кислоты 104,10
В реактор 1 из нержавеющей стали, снабженный прямым и обратным
холодильником, загружают из мерника 2 раствор метилата натрия в аб-
солютированном метиловом спирте, приготовленный в отдельном аппарате
3 в специальных безопасных условиях (на схеме не показан). Из мерника 4
постепенно приливают раствор монохлоруксусной кислоты в метиловом
спирте. Затем осторожно из мерника 5 приливают серную кислоту (плот-
ность 1840 кг!см3}. Реакционную массу кипятят 2 ч и отгоняют метиловый
спирт в сборник 6. Далее массу охлаждают до 20° С, нейтрализуют
NaHCO3 до pH 7,0 и фильтруют через друк-фильтр 7. Осадок промывают
хлороформом из мерника 8. Фильтрат и промывные фракции хлороформа
поступают в сборник 9, а из него в экстрактор 10 для обработки хлорофор-
мом. Хлороформовый экстракт поступает в делительную воронку 11, а
затем в сборник 12 и на сушку в смеситель 13, куда вводят сульфат натрия.
После перемешивания экстракт поступает в сборник 14, а из него в ваку-
ум-перегонный аппарат 15 для отгонки хлороформа без вакуума. Затем
при вакууме (остаточное давление 50 мм рт. ст.} разгоняют кубовый оста-
ток. Отбирают фракцию при температуре 56—-58° С. Последняя поступает
в сборник 16. Метиловый эфир метоксиуксусной кислоты, С4Н8О3, моле-
кулярная масса 104,10, температура кипения при 50 мм рт. ст. 56—58° С,
Пд = 1,399—1,401. Выход 70%.
Синтез цианацетамица. Циан ацетамид получают при взаимодействии
аммиака и этилового эфира циануксусной кислоты по следующему уравне-
нию:
NH3 + CNCH2COOC2H5 —> cnch2conh2 + С2Н5ОН
Аммиак Этиловый циачуксус- Циачацетамид Спирт
15,03 ный эфир 113,11 84,08 46,07
В реактор 17 из нержавеющей стали, снабженный мешалкой, загружа-
ют из мерника 18 расчетное количество водного 25%-ного аммиака, а из
мерника 19 соответствующее количество этилового циануксусного эфира.
После перемешивания реакционную массу охлаждают до 0°. При этом
выпадает осадок цианацетамида, затем массу фильтруют через друк-фильтр
20, осадок промывают спиртом из мерника 21, охлажденным до 0° в труб-
чатом холодильнике 22. Осадок собирают в приемнике 23, откуда его на-
правляют на синтез пиридона. Промывной спирт поступает в сборник 24,
а из него поступает на регенерацию.
Цианацетамид — белый кристаллический порошок, растворимый в воде
и в спирте, температура плавления 118—120° С. Выход 86% [58].
Синтез натрийметоксиацетилацетона. Натрийметоксиацетилацетон по-
лучают путем взаимодействия метилового эфира метоксиуксусной кислоты
и ацетона в среде метилата натрия согласно следующему химическому урав-
— нению:-
CH,ONa
Н3СОСН2СООСН3 + СН3СОСН3----------н3с—сн—сн2—со—сн2осн3 + СН3ОН
I
ONa
Метиловый эфир Ацетон Натриевая соль метоксиацетилацетона Метиловый
метоксиуксусной 58,08 152,13 спирт
кислоты 32,04
101,12
162
В реактор 25, снабженный обратным холодильником, из мерника 26
загружают раствор метилата натрия в метиловом спирте.
При температуре 55° С из мерника 27 постепенно добавляют смесь ме-
тилового эфира метоксиуксусной кислоты и ацетона, перемешивают 3 ч
при температуре 58—60° С. Затем реакционную массу охлаждают до 20° С,
при этом выделяется кристаллический осадок натриевой соли метоксиаце-
тилацетона, который отфильтровывают на друк-фильтре 28. Метиловый
эфир метоксиуксусной кислоты и ацетона смешивают в реакторе 29, куда
вводят из мерника 30 метиловый эфир метоксиуксусной кислоты, а из мер-
ника 31 ацетон.
Натриевую соль промывают на друк-фильтре метиловым спиртом, после
отжима ее направляют в сборник 32, а из него на следующую стадию.
Фильтрат поступает в сборник 33, а из него в перегонный аппарат 34 для
отгонки метилового спирта. Выход 80%. Натриевая соль метоксиацетил-
ацетона, C6H9O3Na, молекулярная масса 152,13; температура кипения
42—44°С (при остаточном давлении 3 мм рт. ст.) «0 = 1,461, d4°=l,0499
[55].
Синтез 2-метил-4-метоксиметил-5-циан-6-пиридона (пиридона). Пири-
дон получают путем взаимодействия натриевой соли метоксиацетилацетона
и. цианацетамида в присутствии серной кислоты согласно следующему хи-
мическому уравнению:
СН2ОСН3
Н2С О С Но— CN
I +21
н3с—СН /С.
ONa NHo O
+ Na2SO4
H2so4,9
”н3С
NH
Натрийметоксиаце -
тилацетон
15Z, 13
Цианацетамид
80,08
2 -Метил-0 -метоксиметил-
5-ц и ан - В- пир ид ан
178, 19
В эмалированный реактор 35, снабженный мешалкой и обратным хо-
лодильником, загружают натриевую соль метоксиацетилацетона, из сбор-
ника 32, вводят воду, массу подогревают до 50° С и перемешивают до пол-
ного растворения осадка. Затем из приемника 23 загружают цианацетамид,
из мерника 36 постепенно добавляют серную кислоту до кислой реакции
(на конго) при температуре не выше 50° С, при этом выделяется осадок
2-метил-4-метоксиметил-5-циан-6-пиридона. Реакционную массу охлажда-
ют при перемешивании, осадок отфильтровывают на центрифуге или нутч-
фильтре 37, промывают ледяной водой до нейтральной реакции, высуши-
вают в вакуум-сушилке 38 до влажности 0,5% и хранят в герметичном
сборнике 39. Выход 78% [25].
2-Метил-4-метоксиметил-5-циан-6-пиридон, С9Н10C2N2, молекулярная
масса 178,19, белый кристаллический порошок с температурой плавления
230-232° С.
В литературе [54, 55] приведены данные по получению 2-метил-4-мето-
ксиметил-5-циан-6-пиридона без выделения промежуточных полупродуктов
при оптимальных условиях, разработанных для получения натриевой соли
метоксиацетилацетона. Имеется в виду получение натриевой соли метокси-
ацетилацетона и без выделения подвергнуть ее иминированию (действием
аммиака), а затем конденсации с циануксусным эфиром и циклизации.
Таким образом, четыре химических реакции осуществляют без выделения
промежуточных продуктов. Реакции протекают по следующей схеме:
ONa
Na |
СН3ОСН2СООСН3 + СН3СОСН3 —> СН2ОСН3—СН—СН2—С—СН3 —>
II
О
Метиловый эфир мето- Натриевая соль метоксиа-
ксиуксусной кислоты цетилацетона
6*
163
ONa
NH3 |
—> CH2OCH3—CH—CH2—C—CH3
Имин натриевой соли метоксиацетил-
ацетояа
CH2CN—COOC2H5
Этиловый цианук-
сусный эфир
СН2ОСН
С “ONa
НС
H3c-^N
ch2cn
со
CH2OCH3
H2SO4 CN
Пиридон
Выход пиридона достигает 56% при условиях конденсации метилового
эфира метоксиуксусной кислоты с ацетоном при температуре 45—55° С
и применении 5,3-кратного количества бензола (мнение автора см. стр. 159).
Синтез 2-метил-3-нитро-4-метоксиметил-5-циан-6-пиридона (нитропи-
ридона). Для превращения пиридона в пиридоксин необходимо:
ввести гидроксил в положение 3, что достигается путем нитрования,
восстановления группы NO2 в аминогруппу NH2, а последней в ОН путем
диазотирования;
превратить циангруппу в оксиметильную СН2ОН, что достигается вос-
становлением CN до аминометильной группы с переводом последней в окси-
метильную диазотированием;
восстановить кислород в 6-ом положении в водород, что обеспечивается
получением хлорида и восстановлением его;
омылить метоксиметильную группу в положении 4 с превращением ее
в оксиметильную.
Из всех указанных реакций в первую очередь должно быть проведено
нитрование для того, чтобы не потребовалась защита функциональных
групп пиридоксина от окисляющего действия азотной кислоты. В соответ-
ствии с указанным выше пятой стадией синтеза должно быть получение
нитропиридона путем нитрования пиридона согласно следующему химиче-
скому уравнению:
СН2ОСН3
NH
HNO3
(СН3СО)2О
СН2ОСН3
2-Метил~3-нитро-^~мето-
ксимегпил-5~циан-6-лиридон
223, 19
2-Метил-4 -ме/лохсиметил-
5 - циан ~6- пиридон
178, 19
В реактор 40 из эмалированной стали заливают из мерника 41 уксусный
ангидрид и засыпают расчетное количество пиридона и мочевины. В реак-
торе 42 из эмалированной стали приготовляют нитрующую смесь из азот-
ной кислоты (плотность 1400 кг/ж3) и уксусного ангидрида. Смесь посту-
пает в мерник 43, а из него медленно (в течение 40—45 мин) в реактор 40.
Температуру реакционной массы поддерживают не выше 45° С, используя
охлаждение водой. Затем реакционную массу спускают в реактор 44, в
котором находится вода, охлажденная до 0°. При смешении с водой выпада-
ют кристаллы нитропиридона, которые отфильтровывают на друк-фильтре
45, промывают ледяной водой. Осадок высушивают в сушилке 46. Сухие
кристаллы поступают в сборник 47. Лучший выход получают при
выдержке (кристаллизации) осадка при 0° около 10 ч. Выход 70%.
Нитропиридон, C9H9O4N3, представляет собой белые кристаллы, тем-
пература плавления 203—204° С, молекулярная масса 223,19.
164
При проектировании и осуществлении процесса нитрования должны
быть предусмотрены все условия для безопасной работы, как, например:
противовзрывные мембраны в реакторе 40 и 42, интенсивное перемешива-
ние реакционной массы, автоматическая регулировка температуры (не вы-
ше 45° С) скорости подачи нитрующей смеси, улавливание окислов азота
из аппаратов.
Синтез 2-метил-3-нитро-4-метоксиметил-5-циан-6-хлорпиридина (нит-
рохлорпиридина) [57, 59—61]. Нитрохлорпиридин получают из нитропи-
ридона путем обработки последнего пятихлористым фосфором в среде хлор-
бензола.
Реакция протекает по следующему уравнению:
СН2ОСН3
2-Метил-3-нитро
метоксиметил ~5-циан-
6-пир идон
223,19
PClg
С6Н5С1
СН2ОСН3
2 - Метил -3- нитро -4-
метоксиметил - 5-циан-6-
хлорпиридин
241, 54
В реактор 48, снабженный обратным холодильником и с отводной тру-
бой с ловушкой для выделяющегося НС1, загружают сухой хлорбензол
из мерника 49, сухой нитропиридон из сборника 47 и пятихлористый фос-
фор. Массу нагревают постепенно до 110—115° С и перемешивают 2,5 ч.
После охлаждения до 20° С раствор фильтруют через друк-фильтр 50.
Фильтрат поступает в сборник 51 и далее в вакуум-перегонный аппарат
52 для отгонки хлорбензола под вакуумом (остаточное давление 5—
10 мм рт. ст.). Кубовый остаток спускают в кристаллизатор 53, добавля-
ют 96%-ный спирт и кристаллизуют 8 ч при 0°. Кристаллы выделяют в
центрифуге 54, промывают спиртом из сборника 55 и высушивают в ваку-
ум-сушилке 56.
Маточный раствор I поступает в сборник 57, его обрабатывают углем
в смесителе 58, фильтруют, через нутч-фильтр 59 и кристаллизуют при 0°
8 ч в кристаллизаторе 60. При этом выкристаллизовывают непрореагиро-
вавший нитропиридон. Кристаллы последнего выделяют в центрифуге 61,
откуда они поступают на повторную переработку. Маточный раствор II
поступает в сборник 62 и далее в смеситель 63, где его обрабатывают ак-
тивированным углем (2% к массе сухих веществ), затем фильтруют в
нутч-фильтре 64 и направляют в сборник 65. Затем из них отгоняют спирт,,
упаривают в вакуум-аппарате 66, кристаллизуют в кристаллизаторе 67
при 0° 8 ч. Кристаллы выделяют в центрифуге 68, откуда они поступают
в смеситель 69 и далее на перекристаллизацию. Маточный раствор III
либо направляют в сборник 70 и далее на следующую кристаллизацию,
а если он истощен, то направляют как отход. Выход 66% [57].
Нитрохлоропиридин, C9H8O3N3C1, молекулярная масса
241,64, порошок с температурой плавления 75—76° С.
Были также исследованы [57 ] методы хлорирования пиридона хлор-
окисью фосфора в присутствии пиридина (выход 65%), хинолина (выход
56,1%), диэтиламина (выход 52,3%). При применении тионилхлорида в
среде хлористого метилена и катализатора диметилформамида выход 92%.
Таким образом, на стадии хлорирования пиридона пятихлористый фосфор
может быть заменен хлорокисью фосфора или тианилхлоридом [54].
Синтез 2-метил-3-окси-4-метоксиметил-5-оксиметилпиридина (метило-
вого эфира пиридоксина). По методу Гарриса и Фолькерса [9] восстановле-
ние группы NO2, CN и Cl проводили в две стадии: вначале в присутствии
платинового катализатора восстанавливали нитрогруппу в аминогруппу,
165
а затем, применяя палладиевый катализатор, восстанавливали нитриль-
ную группу и элиминировали хлор в положении 6 [62].
Впоследствии было показано, что все три группы можно восстановить
одновременно как в присутствии палладиевого, так и никелевого катали-
затора [57, 61—64].
Превращение же аминогруппы и аминометильной группы в оксигруппы
путем диазотирования оказалось эффективным без выделения диамина, при
прибавлении к реакционной массе нитрита натрия после подкисления со-
ляной кислотой. Реакция протекает по следующей схеме:
O2N
н3с
СН2ОСН3
-f^SrCN [Н]
-Ч-Лс! Ni(Pd)’
N
СН2ОСН3
CH2NH2 NaNO2
HCl
СН2ОСН3
2-Memuo-3-oKCU-k -*
метоксиметил -5-
оксиметилпиридин
163,20
2 -Метил - 3-нитро -
4 -метоксиметил -5-
циан-6-хлорлиридин
2k1, 6k
2-Метил-З-амино-k-мето-
ксиметил-5-аминометил-
пиридин
131,2k
Гидрирование хлорида осуществляют ступенчато [61 ]: вначале восста-
навливают нитрогруппу, а затем нитрильную с элиминированием хлора.
Процесс осуществляют в автоклаве из нержавеющей стали, снабженном
метательным прибором, в спиртовом растворе. Для этого в реактор 71
загружают из мерника 72 метиловый спирт, из сборника 56а хлорид, ни-
келевый катализатор1 из сборника 73. После перемешивания раствор спус-
кают в автоклав 74, куда подают водород из баллона 75 под избыточным
давлением 50 кгс/см2. После окончания восстановления нитрогруппы, что
продолжается около часа, из мерника 76 загружают 25%-ный раствор
аммиака, добавляют катализатор и продолжают гидрирование еще около
1,5 ч при избыточном давлении водорода 50 кгОсм2. Хорошие результаты
при гидрировании получают при тщательной промывке катализатора с по-
следующим активированием его водородом [56]. По окончании процесса
реакционную массу при помощи остаточного давления водорода в авто-
клаве подают в друк-фильтр 77 для отделения катализатора, направляемо-
го на регенерацию.
Фильтрат поступает в мерник 78, а из него в реактор 79 на диазотирова-
ние. В последний добавляют соляную кислоту, при температуре 80° С
и перемешивании из реактора 80 — раствор нитрита натрия.
Процесс диазотирования осуществляют при температуре 80° С [54] и
конец реакции определяют по исчезновению окрашивания йодокрахмаль-
ной бумаги. Затем реакционную массу спускают в сборник 81, откуда ва-
куумом засасывают в перегонный вакуум-аппарат 82, снабженный мешал-
кой, и выпаривают досуха. Сухой остаток экстрагируют при температуре
70—75° С абсолютным спиртом из мерника 83. Экстракт сливают в смеси-
тель 84, где его обрабатывают активированным углем и фильтруют через
друк-фильтр 85. Фильтрат поступает в сборник и далее в перегонный ап-
парат 86 для отгонки спирта. Сгущенный концентрат поступает на кристал-
лизацию при 0° в кристаллизатор 87, а затем в центрифугу 88. Кристаллы
эфира высушивают в сушилке 89. Маточный раствор I из центрифуги по-
ступает в сборник 90, а далее в смеситель 91, где его 15 мин обрабатывают
активированным углем при температуре 60° С и фильтруют через нутч-
1 Никелевый катализатор, как более дешевый, здесь применен на основании ли-
тературных данных [56]. Некоторые исследователи считают более рациональным при-
менение палладиевого катализатора [54, 55, 62], причем наилучшие результаты по-
лучены при использовании 1,5% к массе хлорида палладия, 12% активированного
угля в разбавленной соляной кислоте.
166
фильтр 92. Фильтрат через сборник направляют в перегонный аппарат
93, где его сгущают, а затем спускают в кристаллизатор 94. Кристаллы
отфуговывают в центрифуге 95. Кристаллы второго продукта поступают
на перекристаллизацию в смеситель 84. Маточный раствор II собирают
в приемнике 96. При истощении его направляют в отход; в противном слу-
чае подвергают III кристаллизации по такой же схеме.
Метиловый эфир пиридоксина, C9H13O3N, молекулярная масса 183,20,
температура плавления 173—175° С.
Синтез хлоргидрата 2-метил-3-окси-4,5-диоксиметилпиридина (хлор-
гидрата пиридоксина). Хлоргидрат пиридоксина получают путем гидролиза
эфира пиридоксина соляной кислотой при температуре 145—147° С соглас-
но следующему уравнению:
сн2осн3
СН2ОН 2НС1
но
Н3С
N
2-Метил-3-окси-Ь-меток-
симетил -5- оксиметиллиридин
183,2
СН2ОН
2-Метил- 3-окси -Ь,5- ди окси -
метилпиридин (пиридоксин хлоргидрат)
205,65 -
В автоклав из эмалированной стали 97, снабженный мешалкой, загружают
из реактора 98 водный раствор эфира пиридоксина, добавляют из мерника
соляную кислоту (плотность 1190 кг/ж3), раствор нагревают до 145—147° С
(избыточное давление 5—6 кгс/сж2). При этой температуре реакционную
массу выдерживают 6 ч. Затем раствор охлаждают, спускают в сборник 99,
откуда переводят в смеситель 100. В последнем его 10 мин обрабатывают
активированным углем при 75° С, фильтруют через нутч-фильтр 101.
Фильтрат поступает в сборник 102 и далее в вакуум-перегонный аппарат
103. Концентрат спускают в кристаллизатор 104, где при 0° кристаллизу-
ют 8 ч. Затем массу фугуют в центрифуге 105. Технический продукт посту-
пает на перекристаллизацию, а маточный раствор из сборника 106 направ-
ляют на очистку активированным углем в смеситель 107. После фильтрации
в нутч-фильтре 108 раствор сгущают в вакуум-аппарате 109, кристал-
лизуют в кристаллизаторе 110, фугуют в центрифуге 111. Кристаллы вто-
рого продукта поступают на перекристаллизацию в смеситель 100, где их
растворяют и обрабатывают углем.
Маточный раствор II из центрифуги 111 является отходом.
Перекристаллизация технического пиридоксина. Перекристаллизацию
технического пиридоксина ведут в 75 %-ном спирте. Отношение пиридоксина
к спирту 1:5 [61 ].
В реактор 112 из мерника 113 наливают 75%-ный спирт и загружают
технический пиридоксин из сборника 114. Подогревают раствор до 75° С
и добавляют активированный уголь (2% к массе пиридоксина). Перемеши-
вание ведут в течение 10 мин.
Раствор фильтруют через нутч-фильтр 115\ фильтрат поступает в сбор-
ник 116 и далее для упаривания в вакуум-аппарат 117. Сгущенную
массу спускают в кристаллизатор 118, где он кристаллизуется 8 ч при 0°,
а затем его отфуговывают на центрифуге 119, промывают 96%-ным спиртом
и высушивают в вакуум-сушилке 120. Маточный раствор I направляют
в сборник 121 и далее в смеситель 122, куда вводят 2% угля к массе пири-
доксина; полученную массу фильтруют через нутч-фильтр 123. Фильтрат
из сборника 124 поступает для сгущения в вакуум-аппарат 125, затем для
кристаллизации при 0° в кристаллизатор 126. После этого массу фугуют
в центрифуге 127. Получают пиридоксин II, направляемый для перекрис-
167
таллизации в реактор 112. Маточный раствор II направляют в сборник
128, а в зависимости от концентрации в нем пиридоксина его либо направ-
ляют на третью кристаллизацию, либо в отход.
Для контроля качества хлоргидрата пиридоксина и определения при-
месей его метилового эфира были сняты спектры в инфракрасной-об-
ласти на спектрофотометре ИКС-11 [54]. В спектре (рис. 21) хлор-
гидрата пиридоксина имеются полосы поглощения при 1215 и 1100 см"1
(деформационные колебания ОН фенольного и спиртового гидроксилов).
В спектре метилового эфира пиридоксина установлена дополнительно по-
лоса в 1050 см"1, характерная для колебаний СО эфирной связи. Разра-
ботан также метод анализа при помощи восходящей распределительной
Рис. 21. ИК-спектры пропускания, хлоргидрата: 4-ме-
тилового эфира пиридоксина (/) и хлоргидрата пири-
доксина (//).
хроматографии на бумаге (система растворителей м-бутанол—уксусная кис-
лота—вода=4:1:5). Метод позволяет определить примесь эфира пиридо-
ксина.
КОФЕРМЕНТЫ И ПРОИЗВОДНЫЕ ПИРИДОКСИНА
Известно [71—73], что фосфорные эфиры пиридоксина, пиридоксаля
и пиридоксамина в качестве коферментов входят в состав различных фер-
ментов, катализирующих белковый обмен в организме. В этом обмене
особо важную роль играет пиридоксаль-5-фосфат. Известно также (см.
стр. 153), что в животных тканях и дрожжах преимущественно содержатся
пиридоксаль и пиридоксамин. В связи с этим синтез указанных веществ
и их коферментов представляет большой интерес.
Хлоргидрат пиридоксаля может быть получен из хлоргидрата пири-
доксина окислением его перманганатом калия в щелочной среде и выде-
лением его солянокислым гидроксиламином в виде оксима [74]. В качестве
окислителя применяли также двуххромовокислый калий [75], дву-
окись марганца [76, 54]; в последнем случае выход оксима составил
59%.
Оксим пиридоксаля может быть превращен либо в пиридоксаль через
хлоргидрат моноэтилацеталя пиридоксаля путем обработки нитритом нат-
рия и соляной кислотой, либо в пиридоксамин восстановлением водородом
на палладиевом катализаторе [77] по следующей схеме [54].
168
CH2OH
HO CH2OH KMnO4 HO
H3C
.N-HCl
CH=NOH
CH2OH NaNO2
HC1; C2H5OH
N
NH2OH H3C
H5C2O-CHO
~ho-AA-ch2
N-HCl
Хлоргидрат пиридоксина
205, 65
Оксимпиридоксаля
782,78
Моноэтилацеталь
пиридоксаля
231,67
|HCl
СНО
[H]|Pd
ch2nh2
Дихлоргидрат
пиридоксамина
ZU 7, 7Z
CH2OH
Хлоргидрат
пиридоксаля
203,63
но
H3C
N-HCl
Пиридоксамин дихлоргидрат (C8H12O2N2.2HC1 молекулярная масса
241,12). Получают [77] пиридоксаминдихлоргидрат по следующей методике.
В реактор из эмалированной стали загружают воду, активированный уголь
и раствор хлористого палладия в концентрированной соляной кислоте
и пропускают водород до восстановления хлористого палладия. Затем в
реактор загружают раствор оксима пиридоксаля в воде и продолжают
восстановление водородом оксима до пиридоксамина. Количество участ-
вующего катализатора в пересчете на палладий—3% к массе оксима. Затем
отфильтровывают катализатор; фильтрат сгущают в вакууме и кристалли-
зуют. Выход пиридоксамина 98,2%, температура плавления 198—200° С.
Оксим пиридоксаля получают из хлоргидрата пиридоксина окислением
двуокисью марганца при участии серной кислоты [54, 77] при температуре
60—70° С и pH 6,0 [24]. В этой реакции образуется пиридоксаль, который
выделяют в виде оксима добавлением солянокислого гидроксиламина в
среде уксуснокислого натрия. Реакцию ведут при нагревании (85—90° С).
Затем по охлаждении выделяются кристаллы оксима, которые перекристал-
лизовывают из спирта. Выход 59,0% [77]. Температура плавления 225—
226° С.
Пиридоксаль хлоргидрат (C8H9O3N-HC1, молекулярная масса 203,62).
Получают пиридоксаль хлоргидрат из оксима пиридоксаля путем обработ-
ки его при температуре 80—85° С нитритом натрия в солянокислой среде,
упаривания и экстракции спиртом; после фильтрации и отгонки' спирта
выделяют моноэтилацеталь пиридоксаля (С10Н13O3N-НС1, температура
плавления 142—143° С). Последний растворяют в воде, омыляют соляной
кислотой, сгущают и выделяют хлоргидрат пиридоксаля. Выход 80%, тем-
пература плавления 173—174° С.
Пиридоксаль-5-фосфат (кодекарбоксилаза). Синтез кодекарбоксилазы
был осуществлен в 1951 г. [78—80] из пиридоксамина путем фосфорили-
рования его и окисления аминометильной группы в альдегидную. Фосфо-
рилирование осуществляли хлорокисью фосфора [11] или смесью фосфор-
ного ангидрида и ортофосфорной кислоты [78, 81], или метафосфорной
кислотой [10]. Реакции протекают по следующей схеме:
но
Н3С
ch2nh2
СН2ОН
N-2НС1
Н3РО4 4- Р2О5
Пиридоксамин
2Н1, 12
сн2мн2 о
НО СН2О-р-ОН МпО2
НдС пи
N • 2Н2О UH
Пиридоксамин -5-ipoctpam
270,21
о
но сн2о-р-он
или НС Ди
CH3COCOONa + 3 n-h2O
+ (CH3COO)2CU Пиридоксаль -5- рзосроат
257,16
СНО
169
Пи р И ДО КС амин-5-фосфат (C8H]0NO6 -Н2О, молекулярная
масса 251,16) получают из хлоргидрата пиридоксамина путем обработки
его фосфорилирующей смесью, состоящей из ортофосфорной кислоты и пя-
тиокиси фосфора. Реакцию проводят при нагревании (65—70° С). Затем
раствор обрабатывают активированным углем, либо катионитом, фильтру-
ют, упаривают при остаточном давлении 10—20 мм рт. ст. и кристалли-
зуют при 0°. Получают белые кристаллы с температурой плавления 230—
232° С, хорошо растворимые в воде. C8H13N2O6P-2Н2О, молекулярная
масса 270,21. Выход 45—50%. Превращение пиридоксамина-5-фосфата в
пиридоксаль-5-фосфат может быть осуществлено либо осторожным окисле-
нием двуокисью марганца [78, 81, 82], либо действием пировиноградной
или глиоксалевой кислоты в присутствии медных солей [83, 84]. В послед-
нем случае загружают в реактор пиридоксамин-5-фосфат, воду, пировино-
градокислый натрий, ацетат меди, перемешивают при слабом нагревании,
обрабатывают углем, либо катионитом (для удаления катионов), сгущают
и выделяют осаждением ацетоном. Получение в свободном виде затрудне-
но, значительно легче выделение его в виде оксима с температурой плав-
ления 229—230° С [24].
Производные пиридоксина [85]. Известна важная роль пиридоксина в
обмене непредельных жирных кислот [86], как, например, превращение
линолевой кислоты в арахидоновую. Известно также, что жирнокислотные
эфиры пиридоксина (например, пальмитат) обладают повышенной стой-
костью [87]. Синтезирован тристеарат пиридоксина (2-метил-З-стеароило-
кси-4,5-дистеароил-окси-метилпиридин) этерификацией хлоргидрата пири-
доксина хлорангидридом стеариновой кислоты в среде пиридина по сле-
дующей схеме [85]
НО
Н3С
сн2он
сн2он
N-HC1
Хлоргидрат пиридоксина
205, 65
СН2ОСОС17Н35
+ зс н coci -CsHs1\ н3кс17соо^усняосос17нж
17 35 Н3с-С>
J N
Клирингидрид стгаринобои Гристеарат пиридоксина
3-302,317 968,53
В реактор загружают суспензию хлоргидрата пиридоксина в пириди-
не и медленно приливают хлорангидрид стеариновой кислоты (температу-
ра кипения 180—183° С при остаточном давлении 3 мм рт. ст.), переме.
Рис. 22. УФ-спектры поглоще-
ния эфиров жирных кислот
пиридоксина:
1 — тристеарин пиридоксин; 2 — три-
пальмитат пиридоксина; 3 —трилино-
леат пиридоксина.
Рис. 23. ИК-спектры пропускания эфиров жир-
ных кислот и пиридоксина:
а — хлоргидрат пиридоксина; б — трипальмитат пиридокси-
на; в — тристеарат пиридоксина; г — трилинолеат пиридок-
сина.
170
шивают, отфильтровывают в друк-фильтре, промывают спиртом и пере-
кристаллизовывают из гексана. Выход 75,3% [85]. Кристаллы имеют тем-
пературу плавления 77—78,3° С; Хтах=271 нм, е=4,28-103, C62H113OeN.
Тристеарат пиридоксина может быть получен из трилинолеата пири-
доксина [88] путем гидрирования последнего в гексане при участии ката-
лизатора палладиевой черни при температуре 20° С. Катализатор отфильт-
ровывают, фильтрат сгущают и при 0° кристаллизуют. Выход 94,8%.
Синтезированы тем же методом (ацилированием хлоргидрата пиридокси-
на хлорангидридом соответствующих жирных кислот) пальмитат пиридо-
ксина с выходом 75,4% (температура плавления 73,5—74,2° С; Zmax =
=272 нм.-, е=3,88-103, C6eH101O6N) и линолеат пиридоксина с выходом
41,1% (d4°=0,9529; Пд = 1,4851, C62H101ObN). На рис. 22 и 23 даны спект-
ры в УФ-свете и ИК-лучах для жирнокислотных эфиров пиридоксина.
В реакции этерификации, кроме пиридина и гексана, применены раз-
личные дизамещенные амиды, как, например, диметилформамид и др. [89].
> В Японии получены производные пиридоксина, обладающие пролонги-
рованной активностью [90] или являющиеся источником аминокислот, как,
например, производные пиридоксина и гомоцистеина [91].
ТЕХНОЛОГИЧЕСКИЕ ПОКАЗАТЕЛИ
РАСХОД МАТЕРИАЛОВ, КГ, \\Х\КГ
100%-НОГО ПОЛУПРОДУКТА
Метиловый эфир метоксиуксусной
кислоты
Монохлоруксусная кислота .... 1,3
Натрий металлический.............0,5
Метиловый спирт................. 1,2
Серная кислота ................. 1,8
Бикарбонат натрия ............. 1,1
Сульфат натрия.................. 1,0
Хлороформ....................... 1,2
Выход, % к теоретическому (на
монохлоруксусную кислоту) . . . 70,0
Цианацетамид
Этиловый циануксусный эфир . . 1,5
Аммиак 25%-ный, плотность
900 кг/м3....................... 1,9
Спирт этиловый...................0,2
Выход, % к теоретическому (на
этиловый циануксусный эфир) . . 86,0
Натрийметоксиацетилацетон
Метиловый эфир метоксиуксусной
кислоты .......................0,9
Ацетон..........................0,5
Натрий металлический.............0,25
Метиловый спирт абсолютный . . 0,5
Выход, % к теоретическому (на ме-
тиловый эфир метоксиуксусной кис-
лоты) ..........................80,0
2-Метил-4-Метоксиметил-5-цаан-6-пири-
дон (пиридон)
Натрийметоксиацетилацетон ... 1,,1
Цианацетамид ....................0,6
Серная кислота ................. 0,4
Выход, % к теоретическому (на
цианацетамид) ..................78,0
2-Метил-3-нитро-4-метоксиметил-
5-циан-6-пиридон (нитропиридон)
Пиридон......................... 1,15
Азотная кислота, плотность
1400 кг/м? ......................0,8
Уксусный ангидрид..............3,2
Мочевина.......................0,05
Выход, % к теоретическому
(на пиридон) ...................70,0
2-Метил-3-нитро-4-метоксиметил-
5-циан-6-хлорпиридин
Нитропиридон.................. 1,5
Пятихлористый фосфор.......... 1,5
Хлорбензол.....................0,8
Спирт 96%-ный..................0,5
Уголь активированный ..........0,1
Выход, % к теоретическому
(на нитропиридон)..............66,0
2-Метил-3-окси-4-метоксиметил-
5 - оксиметилпиридин
(метиловый эфир пиридоксина)
Нитрохлорпиридин ............. 1,9
Метиловый спирт............... 1,0
Аммиак 25%-ный................ 1,1
Никелевый катализатор..........0,5
Спирт 96%-ный................. 1,0
Уголь активированный...........0,1
Нитрит натрия................. 1,0
Выход, % к теоретическому (на
нитрохлорпиридин) .............68,0
Хлоргидрат-2-метил-3-окси-4,5-
диоксиметилпиридин
(пиридоксин гидрохлорид)
Метиловый эфир пиридоксина . . 1,2
Соляная кислота, плотность
1190 кг/м3 .....................0,2
Спирт ...........................0,8
Активированный уголь...........0,1
Выход, % к теоретическому (на
эфир)..........................85,0
Выход пиридоксина гидрохлорида
при перекристаллизации, % ... 92,0
РАСХОД ПОЛУПРОДУКТОВ, КГ
НА СИНТЕЗ 1 КГ ПИРИДОКСИН
ГИДРОХЛОРИДА МЕДИЦИНСКОГО
Пиридоксин гидрохлорид техниче-
ский .......................... 1,1
Метиловый эфир пиридоксина . . 1,2
Нитрохлорпиридин ............. 2,3
Нитропиридон ................. 3,5
Пиридон.................... . . 4,0
Натрийметоксиацетилацетон ... 4,4
171
Метиловый эфир метоуксусной кис-
лоты ............................4,0
Цианацетамид....................2,4
РАСХОД ХИМИКАЛИЕВ, КГ
НА СИНТЕЗ 1 КГ ПИРИДОКСИНА
ГИДРОХЛОРИДА МЕДИЦИНСКОГО
Аммиак 25%-ный ..................5,9
Ангидрид уксусный...............11,2
Ацетон...........................2,2
Катализатор никелевый ...........0,6
Кислота азотная, плотность
1400 кг/м3 . . ..................2,8
Кислота соляная, плотность
1190 кг/м3 ......................0,2
Кислота серная, плотность
1860 кг/м3......................8,8
Кислота монохлоруксусная . . . 5,2
Мочевина...................., 0,2
Натрий бикарбонат ..............4,4
Натрий металлический............3,1
Натрий сульфат..................4,0
Нитрит натрия...................1,2
Спирт метиловый..............8,2
Спирт этиловый...............3,6
Фосфор пятихлористый ...........3,5
Уголь активированный.........0,4
Хлорбензол...................1,8
Хлороформ....................4,8
Этиловый циануксусный эфир . . 3,6
Итого . . 75,7 кг
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. G у б г g у Р., Nature, 1934, 133, 498; Biochem. J., 1935, 29, 741, 760, 767.
2. Р ы с с С. М. Витамины. Медгиз, 1955, с. 156.
3. К у д р я ш о в Б. А. Витамины, их физическое и биохимическое значение. М.,
Изд. Московское Общество испытателей природы, 1953, с. 25.
4. Keresztesy I., Stevens I., Proc. Soc. exp. Biol. Med., 1938, 38, 64.
5. S t i 1 1 e r E., Keresztesy I., S t e v e n s I., J. Am. Chem. Soc., 1939, 61,
1237.
6. Harris S., Folkers K-, Science, 1939, 89, 347.
7. S n e 1 1 E., G u i r a r d B., W i 1 1 i a m s R., J. Biol. Chem., 1942, 143, 159.
8. К u h n R., W e n d t C., Ber., 1938, 61, 1534.
9. H a r r i s S., H e у 1 D., Folkers K., J. Biol. Chem., 1944, 154, 315; J. Am.
Chem. Soc., 1944, 66, 2088.
10. Snell E., J. Am. Chem. Soc., 1944, 66, 2082; J. Biol. Chem., 1944, 154, 313.
11. Браунштейн A. E. Путь превращения триптофана в организме животных
и функции витамина В6 в этих’превращениях —«ДАН СССР», 1949, 65, с. 715—718.
12. Браунштейн А. Е., Горяченкова Е. Биохимия. 1949, т. 14, с. 1631.
13. Горяченкова Е. Новые данные о роли витамина В6 в обмене веществ. —
В сб.« Материалы VI сессии ВНИИ Витаминологии», 1967, с. 14 —15.
14. Shejderman S., Carretero R., Jacobs K-, J. Clin. Nutrit, 1953,
1, 200.
15. M о 1 1 e r E. Angew. Chem., 1949, 53, 204.
16. Knox W., Mehler A., J. Biol. Chem., 1950, 187, 419, 431.
17. H о d s о n A., J. Nutrit, 1944, 27, 415.
18. T e p 1 у L., Strong F., Elvehjem C., J. Nutrit; 1942, 24, 167.
19. Williams R., Chel del in V., Mitchell H., Univ, of Техас Pub.,
1942, 4237, 97.
20. Henderson L., Waisman H., Elvehjem C. J. Nutrit., 1941, 21,
589.
21. Sebrell W., Harris R. The Witamins, N. Y., 1954, 3, 219.
22. H a r r i s S., S i t a G., Stecher P. Encyclopedia of Chemical Technology,
1953 11 293.
23. Keresztesy I., Stevens I. J. Am. Chem. Soc., 1938, 60, 1267.
24. Б e p e з о в с к и й В. M. Химия витаминов. М., Пищепромиздат, 1959, с. 367.
25. Н а г г i s S., Stiller Е., Folkers К- J- Am. Chem. Soc., 1939, 61, 1242.
26. К u h n R., Wendt G. Ber., 1939, 72, 305.
27. Kuhn R., And ersag H., Westphal K-, Wendt G., Ber., 1939
72, 1309.
28. К u h n R., Wendt G., Westphal K., Ber., 1939, 72, 310.
29. Kuhn R., Westphal K-, Wendt G., Westphal O., Naturwissen-
schaften, 1939, 27, 469.
30. I t i b a A., M i t i K-, Sci Papers Inst. Phys. Chem. Research (Tokyo), 1938, 35,
73; 1939, 36, 1; 1939, 36, 173.
31. J ones R., Kornfeld E. J. Am. Chem. Soc., 1951, 78, 107.
32. Baddiley J., Mathias A., J. Chem. Soc., 1952, 2583.
33. H e у 1 D., L u z E., Harris S., Folkers K. J. Am. Chem. Soc., 1951,
73, 3436.
34. R о s e n b e r g H. Chemistry and Physiology of the Vitamins, 1942, 195.
35. Преображенский H. А., Генкин Э. И. Химия органических и ле-
карственных веществ. М., Госхимиздат, 1953, с. 141.
36. Балякина М. В., Преображенский Н. А., Ждано-
вич Е. С. К вопросу о синтезе витамина В6. — В сб.: «Витамины», Киев, Изд.
АН УССР, 1958; № 3, с. 17—31.
172
37. Б е э р А. А., Рубцов И. А. Синтез витаминов. М., Пищепромиздат, 1956,
с. 139.
38. Ш и а й д м а н Л. О. Производство витаминов. М., Пищепромиздат, 1958,
с. 356.
39. М о w a t J., Pilgrim F., Carlson G., J. Am. Chem. Soc., 1943, 65, 954.
40. Англ. пат. 567611; С. A., 1947, 41, 2755.
41. H a r r i s S. Пат. США 2399347, 1946; С. А., 1946, 40, 418.
42. S t i 1 1 е г Е. Пат. США 2372690; С. А., 1945, 39, 4199.
43. Н а г г i s S. Пат. США 2266754 23/XII 1941; С. А. 1942, 36, 2378.
44. М о г i i S., Ма к i no К. Enzimologia, 1939, 7, 385.
45. H a r r i s S., J. Am. Chem. Soc., 1940, 62, 3203.
46. К e r e s z t e s у I., S t e v e n s I., Пат. США 2280831, 1942.
47. Bruce W., Coover H., J. Am. Chem. Soc., 1944, 66, 2092.
48. Harris S. Англ. пат. 593796; С. A., 1948, 42, 1710.
49. H a r r i s S. Пат. США 2422627, 1947; С. А., 1947, 41, 5554.
50. Hoffman — La Roche. Англ. пат. 570365, 1946; С. А., 1946, 40, 5210.
51. Hoffman — La Roche. Герм. пат. 732238; С. А., 1944, 38, 1251.
52. Б е р е з о в с к и й В. М. Химия витаминов группы В6. — «Успехи химии»,
1952, т. 21, с. 40—68.
53. Мих ай л о в Г. С., Конькова В. А., Басова А. К. К получению
этилового эфира этоксиуксусной кислоты. — ЖПХ, 1952, т. 25, с. 1329—1330.
54. Ж Д а н о в и ч Е. С. Исследования в области синтеза и разработка методов произ-
водства витаминов группы В. Докл. докт. дисс. М., 1965.
54а. Михайлов Г. С., Балякина М. В., Басова А. К., Ждано-
вич Е. С. К методике синтеза метилового эфира метоксиуксусной кислоты. —
«Медицинская промышленность СССР», 1962, № 12, с. 24—27.
55. Балякина М. В., Жданович Е. С., Преображенский Н. А.
Исследования в области синтеза витаминов группы Вв. — «Труды ВНИВИ», М.,
Пищепромиздат, 1961, т. № 7, с. 8—16 с ил.
56. Михайлова М. К., Шпунтова М. Т. Совершенствование метода полу-
чения активного скелетного катализатора. —В сб.: «Из опыта витаминных заводов
Москвы и Ленинграда», М., Пищепромиздат, 1956, с. 8—9.
57. Балякина М. В., Жданович Е. С., Преображенский Н. А.
Синтез 2-метил-3-нитро-4-метоксиметил-5-циан-6-хлорпиридина. — ЖПХ, 1962,
т. 35, 1864; То же. ЖОХ, 1961, т. 31, с. 542; То же. — ЖОХ, 1961, т. 31, с. 2983.
58. Синтезы органических препаратов. М., ИЛ, 1949, 1, 498.
59. Н а г г i s S., F о I к е г s К., J. Am. Chem. Soc., 1939, 61, 1245.
60. Н а г г i s S., F о 1 к е г s К., Пат. США 2766250, 1956; РЖХим, 1958, т. II,
54940.
61. Б а с о в а А. К-, Михайлов Г. С., Васильева Е. Я- и др. — В сб.:
«Материалы о творческом содружестве», Лениздат, 1954, 29.
62. Ко н ь к о в а В. А.., Петрова Л. А. Выбор катализатора при получении
промежуточных продуктов синтеза пиридоксина. — «Труды ВНИВИ», М., Пище-
промиздат, 1959, № 6, с. 10.—14.
63. S m i t h H., В e d о 1 t W., Fusee J., J. Am. Chem. Soc. 1949, 71, 3709.
64. T u c k e r S., J. Chem. Education, 1950, 27, 449.
65. H a r r i s E., Firestone R., Pfister K., Boettcher R.,
Cross F., J. Org. Chem., 1962, 2705.
66. Кондратьева Г. Я-, Хуан Чжи Хен. Реакция алкил-и алкоксиок-
сазолов с имидом малеиновой кислоты. — «ДАН СССР», 1961, т. 141, вып. 3, с. 628—
631.
67. К а г г е г Р., М у a m i с h i Е., Storm Н., Widmer R., Helv. Chim.
Acta 1925 8 205.
68. Cohen A., Hughes E., Англ. пат. 625997, 1949.
69. Cohen A., Haworth J., Hughes E., J. Chem. Soc., 1952, 4374.
70. M a k i n о К., M о r i i S., Chang F., T a g a m i I., Bull. Chem. Soc.,
Japan, 1944, 19, 1.
71. Б p а у н ш т e й н A. E., Шемякин M. M. Теория процессов аминокислот-
ного обмена, катализируемых пиридоксалевыми энзимами. — «ДАН СССР», 1952,
т. 85, с. 1115—1118.
72. Б р а у н ш т е й н А. Е. Успехи современной биологии. 1953, 35, 27.
73. Б р а у н ш т е й н А. Е. The Enzymes, 1960, 2, 113.
74. Н а г г i s S., Н е у I D., F о I k е г s К., J. Am. Chem. Soc., 1944, 66, 2088;
J. Biol. Chem., 1944, 154, 315; Пат. США 2497730, С. А., 1950, 44, 5923.
75. Snel 1 Е. Англ. пат. 603289; 603290; С. А., 1949, 43, 725.
76. Не у 1 D., J. Am. Chem. Soc., 1948, 70, 3434.
77. Ж Д а н о в и ч Е. С., Балякина М. В., Преображенский Н. А.,
Авт. свидет., № 133885, 1960, Бюлл. изобрет., 1960, № 23, с. 17.
78. W i 1 s о п A., Harris S., J. Am. Chem. Soc., 1951, 73, 4693.
79. V i s с о n t i n i M., E bnother C., Karrer P., Helv. Chim. Acta, 1951,
34, 1834.
173
80. V i s с о n t i n i M., E bno t her C., Karrer P., Helv. Chim. Acta, 1951,
34 2198" 2199.
81. Peterson E., Sober H., Meister A., J. Am. Chem. Soc., 1952, 74, 570.
82. H a r r i s S., Wilson А., Пат. США 2660061; С. A., 1954, 48, 12811.
83. L о n g R. Англ. пат. 749800; С. А., 1957, 51, 1297.
84. К у р о д а Т. Витамин (Япония), 1963, 28, 3, 211.
85. Б а л я к и н а М. В., Жданович Е. С., Земскова А. Г. и др. Син-
тез производных пиридоксина, содержащих остатки кислот алифатического ряда.—
ЖОХ, 1962, т. 32, с. 1172.
86. М с Henry Е., Gavin G., J. Biol. Chem., 1941, 138, 471.
87. R о с h е g g i а п i G., Prakt. Chem., 1960, 11, 162.
88. S a k u r a g i T.,. Kummerow F. J. Am. Chem. Soc., 1956, 78, 839.
89. К о б о я с и К. Пат. ФРГ 1296140, 1969; Ausziige, 1969, № 20.
90. О к у м а р а К. Фирма «Танабэ». Япон. пат. 0271, 1968; С. А., 1968, 69, № 15,
59107 t.
91. Фирма «Танабэ». Япон. пат. 9833, 1967; Витамин, 1967, 36, 2, 195.
Глава 8. ПРОИЗВОДСТВО СИНТЕТИЧЕСКОЙ ПАНГАМОВОЙ
КИСЛОТЫ (ВИТАМИНА В1в) [1]
Термин, витамин В16—был предложен в 1950 г. японским исследова-
телем Томиямой [2] для названия воднорастворимого вещества, выделен-
ного им из печени быка-и отличающегося от витамина В12. Кребс с сотруд-
никами [3] в 1951 г. обнаружил в водном экстракте ядер абрикосовых
косточек кислоту, которая затем была выделена йз отрубей риса, пивных
дрожжей, бычьей крови и печени лошади. Кислота эта была им названа
пангамовой из-за ее распространения в семенах различных растений (греч.
«пан»—всюду, «гами» — семя). В 1955 г. Кребс получил патент на откры-
тие пангамовой кислоты и метод синтеза ее [4]. Затем была установлена
идентичность пангамовой кислоты и витамина В16. До настоящего времени
отсутствуют какие-либо работы, подтверждающие витаминные свойства
пангамовой кислоты. До сих пор неизвестно является ли она веществом
эндогенного происхождения или же результатом поступления в организм
с пищей.
Клинические исследования, проведенные рядом авторов как в СССР,
так и за рубежом, выявили следующие лечебные свойства пангамовой кис-
лоты [5]:
липотропное действие, обнаруженное при лечении гепатита и цирроза
печени [6—8] и при циррозе сердечной мышцы [9];
активирование кислородного обмена в клетках тканей, выявленное при
лечении гипоксических и аноксических состояний, ревмокардита, инфарк-
та миокарда, стенокардии и др. [3, 10—12]. При лечении отмечено [5]
уменьшение или полное прекращение стенокардических болей, уменьше-
ние одышки, нормализации ритма сердца, нормализации сна и улучшение
аппетита;
детоксицирующее действие [5] при острых и хронических отравлениях
алкоголем и другими наркотиками [7, 8, 10], при отравлениях хлористыми
углеводородами, как, например, дихлорэтаном [13], антибиотиками тетра-
циклинового ряда [14].
Можно предположить (И. Гаркина), что кажущаяся универсальность
действия пангамовой кислоты обусловлена ее метилирующей способностью
и высокой активностью в окислительных процессах, протекающих в клет-
ках и тканях организма.
Кребс допускает возможность коферментной роли пангамовой кислоты
[3, 7]. Соловьева и Гаркина [15] полагают, что указанные свойства пан-
гамовой кислоты в какой-то степени связаны со способностью ее подвер-
гаться окислительному деметилированию.
Согласно инструкции, утвержденной фармакологическим комитетом
[16], пангамат кальция способен улучшать липидный обмен, повышать
174
усвоение кислорода тканями, устранять явления гипоксии, обладает де-
токсирующим действием. Он показан к применению при атеросклерозе,
эмфиземе легких, хронических гепатитах, кожно-венерических заболева-
ниях, при хронической алкогольной интоксикации и при применении кор-
тикостероидов и сульфамидов.
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ВИТАМИНА В15
Пангамовая кислота является эфиром D-глюконовой кислоты и диме-
тилглицина и выражается следующей формулой:
СООН
I
н—С—ОН
I
но—с—н
н—с—он
н—с—он
I
CH2OCOCH2N(CH3)2
6-о-Диметилглицил-2?-глюконовая
кислота
173,26
Из-за лабильности сложноэфирной связи получение пангамовой кисло-
ты в чистом виде представляет большие трудности. В связи с этим зарубеж-
ные фирмы создали ряд препаратов аналогов и производных пангамовой
кислоты, как, например, пангаметин, биопангамин и др. Исследование
этих препаратов [17] показало, что в их состав входят в различных соот-
ношениях дихлорацетат диизопропиламмония, глюконат кальция или нат-
рия и глицин [18]. У нас выпускают препарат пангамат кальция, имею-
щий следующую формулу: [СОО~—СНОН—СНОН—СНОН—
CHOHCH2OCCX2H2N(CH3)2]2Caa+-Н2О, молекулярная масса 618,60. Он пред-
ставляет собой белый аморфный порошок, легко растворимый в воде (1:1),
нерастворим в спирте и других органических растворителях. Температура
плавления 132—140° С (с разложением).
Изучение физико-химических свойств пангамовой кислоты крайне за-
труднено из-за отсутствия индивидуального вещества. Опубликованный
в 1955 г. [4] Кребсом синтез пангамовой кислоты не был воспроизведен
в Японии. Пангамовая кислота в чистом виде не была выделена и охарак-
теризована [19]. В связи с этим возникла необходимость в получении ин-
дивидуальных веществ, как, например, пангамолактона и амида пангамо-
вой кислоты. Последние, синтезированные Юркевичем и Вереникиной
[20], позволили изучить свойства и гидролитическую устойчивость панга-
мовой кислоты и ее амида [17, 21]. Гидролитическая устойчивость изучена
методом перйодатного окисления [17]. О степени расщепления 6-0-ациль-
ной связи судили по количеству формальдегида, выделяющегося при окис-
лении гидролизованного сахара. Исследования показали, что степень гид-
ролиза пангамовой кислоты и ее производных находится в зависимости
от pH среды и температуры. В кислых средах они более стойки, нежели
в щелочных. При кипячении и pH 10—12 распад происходит через 3—
5 мин, а при pH 2,0 и температуре 20° С пангамолактон за 5 ч гидролизу-
ется на 22%, после чего распад прекращается. Амид пангамовой кислоты
в этих условиях гидролизуется вдвое быстрее.
Хлоргидрат-6-о-диметилглюцил-1,4-£>-глюконолактона (пангамовой
кислоты), C10H18NO7Cl, температура плавления 69—73; [а]д+32,3о (с=
= 1,18%, метанол) [20].
175
МЕТОДЫ СИНТЕЗА ВИТАМИНА В15 И ВЫБОР РАЦИОНАЛЬНОГО МЕТОДА
ДЛЯ ПРОИЗВОДСТВА [1, 5, 20, 21]
Кребс в своем патенте предложил два варианта синтеза витамина В15:
I) путем взаимодействия D-глюконолактона с монохлоруксусной кисло-
той (или с ее хлорангидридом) получают глюконохлорацетат, который с
диметиламином дает пангамовую кислоту и 2) путем этерификации глю-
коновой кислоты диметилглицином. Реакции протекают по следующей
схеме:
I
н-с-он
но-с-н
I
. н-с-он
I ' -
н-с-----
I
сн2он
Лактон глтконобой
кислоты
т^он
н-с-он
но-с-н
н-с-он
н-с-он
I
сн2он
Глюконобая кислота
с<
I он
н-с-он
О+ СН2С1-С^„Л—►но-с-н
Un |
н-с-он
+ hn(ch3)2
, Монохлоруксусная
кислота
н-с-он _
I др -
СН2ОС-СН2С1^
Хлррацетат гл/оконобой ।
+ hoooch2-n(ch3j2
Диметиламиноуксусная кислота
(Диметилглицин)
кислота
Диметиламин
V^OH
-Н2О; НС1
35-00°
н-с-он
но-с-н
I
н-с-он
I
н-с-он „
1 х
сн2оссн2м(сн3)2г
6 -О-Диметилглицил-D -
глюконобая кислота
(Пангамобая кислота)
При воспроизводстве обоих вариантов выход пангамовой кислоты по
первому варианту составил 56%, а по второму — 25% (на глюконовую
кислоту) [5]. Этерификация глюконовой кислоты осуществляется в при-
сутствии соляной кислоты, которая в первом варианте выделяется в реак-
ции метилирования, а во втором — добавляется. Затем соляная кислота
удаляется путем диализа. Диализат нейтрализуется едким натром до pH
7,0—7,5 и высушивается при низкой температуре (лиолифилизация). По-
лучают натриевую соль пангамовой кислоты, представляющую собой бе-
лый порошок, нерастворимый в органических растворителях, весьма ги-
гроскопичный, с температурой плавления 186° С. Недостатками метода
являются: а) большая продолжительность реакции этерификации с
применением газообразного хлористого водорода в качестве катализатора;
б) применение процесса диализа для удаления соляной кислоты и других
примесей через специальную пленку и большой продолжительности (7—
8 ч) значительно усложняет технологию производства и повышает потери
целевого продукта; в) усложняет технологию также применение лиофиль-
ной сушки; г) высокая гигроскопичность пангамата натрия.
Метод Кребса усовершенствован Букиным, Гаркиной и Сошниковым
в направлении устранения указанных недостатков и замены целевого про-
дукта на пангамат кальция. Проверка последнего в 20 клиниках на тысяче
больных подтвердила действенность его лечебных свойств [1].
В 1968 г. Сошников и Петунина разработали микробиологический метод
получения глюконата кальция с применением штамма Gluconobacter Su-
boxydans [23]. Другие исследователи для этой цели предложили штамм
Acetobacter Suboxydans [23а].
176
В 1967 г. Юркевич, Вереникина, Долгих и Преображенский разработа-
ли карбодиамидный синтез [24—27] лактона пангамовой кислоты [20].
Этот метод был успешно использован для получения сложных эфиров аци-
ламинокислот и глюкозы [28]. Указанными авторами были изучены усло-
вия образования эфиров ди метил глицин а под действием дициклогексил-
карбодиимида [28, 29] и показано, что хлоргидрат диметилглицина I в
спиртовом растворе с дициклогексилкарбодиимидом II образует хлоргид-
рат этилового эфира диметилглицина III и дициклогексил мочевину IV:
_ + с2н6он
CeHuN=C=NCeHu + C1(CH3)2NHCH2COOH--------->
п i
—> С1](СН3)2ЙНСН2СООС2Н5)] + CeHnNHCNHCjHn
II
О
III IV
По этой схеме авторами [20] был получен лактон пангамовой кислоты
путем конденсации а-Й-глюконолактона с хлоргидратом диметилглицина
в присутствии дициклогексилкарбодиимида в пиридине^по схеме:
СН2ОН
НО——Н
/°\ ' + c8h..n=c=nc6h
\ОН h>=O-i-C1(CH3)2NHCH2COOH -----------—
CeHsN
СН2ОССН2ТЧН(СН3)2С1
о
=о
н он н он
у - D - Глюконолампан КлорзиЗра/п Зиметилзлииила Дициклогексилкарбодиимид б-О-Диметилзлицил-О-
/4 139,59 206,32 глюконолактон
299,71
CeHnNHCNHCeHn
О
Дициклогексилмочевина
22^,21
Хлоргидрат пангамолактона получают после перекристаллизации из
спирта в виде аморфного порошка довольно стойкого при хранении. Между
тем при получении по методу Кребса натриевой соли пангамовой кислоты
при подщелачивании происходит разрушение эфирной связи.
Таким образом, в настоящее время известны два метода синтеза вита-
мина В15: метод Кребса, усовершенствованный Букиным, Гаркиной и Сош-
никовым, и карбодиимидный синтез, разработанный Юркевичем и Вере-
никиной.
В виде примера приводим опубликованный в литературе [20] карбо-
диимидный синтез пангамата кальция через хлоргидрат лактона панга-
мовой кислоты.
технологическая схема производства синтетического пангамата
КАЛЬЦИЯ
Технология производства витамина В16 состоит из следующих стадий:
синтез лактона ZJ-глюконовой кислоты из глюконата кальция [30];
синтез диметилглицина солянокислого из диметиламина и хлоруксус-
ной кислоты [31];
синтез пангамата кальция из D-глюконолактона и диметилглицина кар-
бодиимидным методом [20, 21].
Лактон /Углюконовой кислоты. Получают путем взаимодействия глюко-
ната кальция и щавелевой кислоты и дегидратацией полученной глюконо-
вой кислоты1. Реакция протекает по следующей схеме:
1 D-Глюконовая кислота может быть получена из D-глюконата кальция по ме-
тоду Сошникова, Сюхиной и Петуниной ионообменом при помощи катионита КУ-2
с выходом около 90% [31а].
177
1.
coo-
н-с-он
но-с-н
н-с-он
СООН
Са • Н2О + | •
СООН - 2Н2О
СООН
н-с-он
но-с-н
н-с-он
СаС2О4- ЗН2О
н-с-он
н-с-он
СН2ОН
J 2
Л- Глюконат кальция
448,39
СООН
Щавелевая кислота
126,07
с*°-
СН2ОН
27- Глюкановая кислота
2'196,16
Щавелевокислый,
кальций
128,1
н-с-он
2. НО-С-Н
-н2о
Н-С-ОН I
I о
НО-С-Н 1
н-с-он
н-с
н-с-он
СН2ОН
J3 - Глюкановая кислота
196, 16
Н-С-ОН
СН2ОН
И - Глюконолактон
178,14
В реактор 1 (рис. 24) из эмалированной стали загружают глюконат
кальция, воду, подогревают паром до 70—75° С и при перемешивании раст-
воряют кристаллы. Затем добавляют щавелевую кислоту и осаждают ща-
велевокислый кальций, который отфильтровывают через нутч-фильтр 2.
Фильтрат из сборника 3 переводят в реактор 4 из эмалированной стали,
добавляют активированный уголь, перемешивают 15 мин при температуре
Рис. 24. Технологическая схема производства синтетического пангамата кальция.
178
60—65° С, фильтруют через нутч-фильтр 5 в сборник 6. Фильтрат засасы-
вают в вакуум-аппарат 7, упаривают до густого сиропа, а затем при ва-
кууме 10—15 мм рт. ст. и нагревании лактонизируют глюконовую кис-
лоту. Затем из мерника 8 загружают в вакуум-аппарат спирт и при нагре-
вании растворяют полузатвердевшую массу; на нутч-фильтре 9 отфильтро-
вывают нерастворившуюся часть. Фильтрат поступает в сборник 10 и да-
лее в вакуум-аппарат 11 для отгонки спирта. Сгущенную массу лактона
сливают в кристаллизатор 12. Из кристаллизатора массу направляют в
центрифугу 13, где осадок промывают спиртом или отгоном спирта. Массу
лактона разгружают в открытый сборник 14, а маточный раствор направ-
ляют в сборник 15. Вопрос о технологии переработки маточного раствора
подлежит изучению. Выход D-глюконолактона 80% (на глюконат кальций).
Диметилглицин хлоргидрат. Может быть получен различными методами:
а) конденсацией глицина с формальдегидом в солянокислой среде [32];
б) взаимодействием натриевой соли хлоруксусной кислоты с диметилами-
ном [33]; в) непосредственной реакцией диметиламина (при значительном
избытке) с монохлоруксусной кислотой [31 ]. Наиболее дешевым по затра-
там на сырье является последний метод. Синтез по этому методу протекает
по следующей схеме:
(CH3)2NH + С1СН2СООН —> (CH3)2NCH2COOH • НС1.
Диметиламин Монохлоруксусная Диметилглицин хлоргидрат
45,08 кислота 94,50 139,59
В реактор 16 из эмалированной стали из мерника 17 загружают водный
раствор диметиламина и при охлаждении до 0° медленно добавляют из мер-
ника 18 водный раствор монохлоруксусной кислоты. Затем реакционную
массу сливают в реактор 19, снабженный прямым холодильником и прием-
ником. В нем реакционную массу подогревают при 60—65° С и выдержи-
вают при этой температуре до завершения реакции. Затем при вакууме
раствор сгущают, сливают в кристаллизатор 20. Из последнего сгущенный
раствор направляют в центрифугу 21, отфуговывают кристаллы, промы-
вают их спиртом из мерника 22 и сушат в вакуум-сушилке 23. Маточный
раствор поступает в сборник 24 \ использование его требует изучения. Су-
хой диметилглицин собирают в приемнике 25. Диметилглицин солянокис-
лый представляет собой бесцветные кристаллы, температура плавления
189° С [32]. Выход 88% (на монохлоруксусную кислоту).
Пангамат кальция. Получают путем конденсации у, D-глюконолактона
с хлоргидратом диметилглицина в растворе пиридина в присутствии ди-
циклогексилкарбодиимида с получением лактона пангамовой кислоты и
с нейтрализацией его окисью кальция. Схему реакции получения лактона
пангамовой кислоты см. стр. 178.
/)-Пангамолактон. В реактор 26 из эмалированной стали загружают
из мерника 27 пиридин, из сборника 14 глюконолактон, из сборника 25
хлоргидрат диметилглицина и перемешивают до полного растворения ин-
гридиентов. Затем добавляют дициклогексилкарбодиимид и продолжают
перемешивание несколько часов. В результате реакции выпадает осадок
дициклогексилмочевины, который отфильтровывают на нутч-фильтре 28.
Фильтрат поступает в сборник 29 и далее в вакуум-аппарат 30 для упари-
вания. Сгущенный раствор сливают в реактор 31, куда добавляют ацетон
из мерника 32 и выделяют осадок лактона пангамовой кислоты. Осадок
отфильтровывают на друк-фильтре 33. Фильтрат из сборника 34 идет на
регенерацию ацетона, а осадок направляют в реактор 35 для перекристал-
лизации из спирта, поступающего из мерника 36. По растворению осадка
при температуре 60—65° С в реактор вводят активированный уголь, пере-
мешивают 15 мин и фильтруют через нутч-фильтр 37. Фильтрат поступает
непосредственно в кристаллизатор 38, где при —5° С выкристаллизовыва-
ют лактон пангамовой кислоты. Затем в центрифуге 39 отфуговывают крис-
таллы и высушивают в вакуум-сушилке 40 при глубоком вакууме (темпе-
ратура 20—30° С). Выход 78%; температура плавления 69—73° С; [а]д =
179
=+32,3° (с= 1,18, метанол). ИК-спектр 1750 см-1, характеризующий слож-
ноэфирную связь лактона (рис. 25). Маточный раствор поступает в сборник
41. Вопрос об использовании его требует дополнительного изучения.' Как
указано было выше, гидролитическая устойчивость лактона значительно
выше, чем у производных или солей пангамовой кислоты. В виде примера
можно привести кривые гидролиза лактона и амида пангамовой кислоты
при pH 5,0 и 7,0 и температуре 22° С (рис. 26). Возможно, что в связи с
/00
-1---1---1----1-----1—।—।-------1_____।______।____I—j
3600 2800 /800/750/000 /000 /200 /000 800См!
Рис. 25. ИК-спектр хлоргидрата 6-О-диметилглицил, 1,4-0-
глюконолактона.
Рис. 26. Кривые гидролиза хлоргидрата:
I—3 -О-диметилглицил 1,4-2?-глюконолактона; 2 —6,9-диметилглицил-1,4-D-
глюкояоамида.
этим встанет вопрос о производстве пангамолактона вместо кальциевой
соли. О степени расщепления 6-о-ацильной связи судили по количеству
выделившегося формальдегида. При pH 5,0 гидролиз лактона проходит
медленно (в сутки выделилось лишь 0,192 моля формальдегида), а амид
разрушился почти на 50% (выделилось 0,422 моля формальдегида).
Кальциевая соль пангамовой кислоты. Ее получают путем нейтрализации
водного раствора D-пангамолактона. Реакция протекает по схеме:
CH2OCCH2NH(CH3)2C1"
но—н о
2 (ОН 'рЬ=О + 2СаО
н он
Хлоргидрат 6-0- диметил-
глицил -D-глюконолактон
2. 299,71
СОО- Са + +
н-с-он но-с-н н-с-он + СаС12
1 н-с-он 1 CH2OCCH21N(CH3)2 о 2-Н2О
Лангамат кальция
618,60
180
Для получения пангамата кальция кристаллы лактона пангамовой кис-
лоты из центрифуги 39 (см. рис. 24) переводят в реактор 41а, растворяют
в воде и нейтрализуют 10%-ным водным раствором гидрата окиси кальция,
приготовленным в смесителе 42, до pH 7,0. Сюда же добавляют активиро-
ванный уголь, перемешивают 5 мин при температуре 20° С, фильтруют
через нутч-фильтр 43 в сборник 44. Сгущение фильтрата производят в ва-
куум-аппарате 45 до сиропообразного состояния. Массу сливают в крис-
таллизатор 46, добавляют спирт из мерника 36 и при охлаждении до 0°
в течение 5—6 ч выделяют пангамат кальция. Из кристаллизатора 46 массу
сливают в центрифугу 47, отфуговывают кристаллы, промывают их спир-
том и высушивают в вакуум-сушилке 48. Маточный раствор через сборник
49 направляют в вакуум-аппарат 50 для отгонки спирта. Вопрос об исполь-
зовании кубового остатка для дополнительного извлечения пангамата каль-
ция требует своего изучения. В процессе превращения пангамолактона
в пангамат кальция при pH 7,0 значительная часть препарата будет гид-
ролизована (см. рис. 26).
ТЕХНОЛОГИЧЕСКИЕ ПОКАЗАТЕЛИ
РАСХОД ХИМИКАЛИЕВ, КГ
НА 1 КГ 100%-НОГО ПОЛУПРОДУКТА
б) Кальциевая соль пангамовой кислоты
Лактон-О-глюконовой кислоты
Глюконат кальция................ 1,5
Щавелевая кислота ..............0,4
Спирт этиловый (с регенерацией) . 0,3
Уголь активированный............0,05
Выход D-глюконолактона (на глю-
конат кальция), %...............80
D-Пангамолактон............... 1,3
Окись кальция ................0,4
Спирт (с регенерацией) .......0,5
Активированный уголь..........0,05
Выход пангамата кальция (на пан-
гамолактон), %.................75
РАСХОД ПОЛУПРОДУКТОВ, кг
Диме/пилглицин хлоргидрат
НА СИНТЕЗ 1 КГ МЕДИЦИНСКОГО
ПАНГАМАТА КАЛЬЦИЯ
Диметиламин....................0,7
Монохлоруксусная кислота .... 0,8
Спирт .........................0,2
Выход хлоргидрата диметилгли-
цина (на хлоруксусную), % ... 88
D-Пангамолактон................. 1,3
Хлоргидрат диметилглицина ... 0,8
f-Лактон-О-глюконовой кислоты . . 1,1
Пангамат кальция
а) D-Пангамолактон
f-Лактон О-глюконовой кислоты . 0,8
РАСХОД ХИМИКАЛИЕВ, КГ
НА ПРОИЗВОДСТВО 1 КГ
МЕДИЦИНСКОГО ПАНГАМАТА
КАЛЬЦИЯ (С УЧЕТОМ
РЕГЕНЕРАЦИИ РАСТВОРИТЕЛЕЙ)
Хлоргидрат диметилглицина ... 0,6
Дициклогексилкарбо диимид ... 0,9
Пиридин (с регенерацией) .... 10,0
Ацетон (с регенерацией).........3,0
Спирт (с регенерацией) .........0,2
Активированный уголь............0,05
Выход Й-пангамолактона (на D-глю-
конолактон), %...................78
Ацетон..................... 3,9
Диметиламин................ 0,6
Дициклогексилкарбодиимид ... 1,2
Кислота щавелевая.......... 0,5
Кислота монохлоруксусная .... 0,7
Окись кальция .................... 0,4
Пиридин ....................... 13,0
Спирт .......................... 1,3
Уголь активированный....... 0,2
Производные пангамовой кислоты. В медицинской практике часто под
названием витамина В15 применяются синтетические аналоги пангамовой
кислоты. Здесь рассмотрим методы синтеза амида пангамовой кислоты,
эфира диизопропилглицина и фармакологического аналога пангамовой кис-
лоты— дихлорацетата диизопропиламмония. В патенте Кребса [4] имеет-
ся указание на получение производных пангамовой кислоты, содержащих
8 и даже 12 метильных групп. Однако их синтез сложен и выход крайне
низок. Кроме того, в литературе отсутствуют какие-либо сведения об их
биологической активности.
Хлоргидрат 6-о-диметилглицил-О-глюконо-
а м и д получают путем конденсации D- глюко но амида и хлоргидрата ди-
метилглицина в среде пиридина при участии дициклогексилкарбодиимида.
181
Технология примерно такая же, как и для хлоргидрата П-пангамолактона
[20]. Реакция протекает по следующей схеме:
О ОН Н ОН ОН
|| | | | | _ + C.HnN=C=NC,Hn
H2N—С—С—С—С—С—СН2ОН + C1(CH3)2NHCH2COOH------------------
|||| - Пиридин
н онн н
D-Глюконоамид Хлоргидрат диметилглицина
о
с—nh2
I
н—с—он
I
—> НО—С—Н + С6Нц NHCNHCoHu
I II
н—С—ОН о
I
н—С—ОН Дициклогексилмочевииа
СН2ОССН2ЙН(СН3)2С1-
D-Пангамолактонамид
Температура плавления 92—95° С (с разложением); выход 62,6% [20].
Хлоргидрат 6-о-диизопропилглицил-О-глюко-
нолактон получают путем конденсации D-глюконолактона и хлор-
гидрата диизопропилглицина в среде пиридина при участии дициклогек-
силкарбодиимида по той же схеме, как D-пангамолактона с той лишь раз-
ницей, что вместо диметилглицина применяют диизопропилглицин; темпе-
ратура плавления 55—60° С. Выход 45% [20].
Дихлорацетат диизопропиламмония является ак-
тивным фармакологическим аналогом пангамовой кислоты. Изучены фар-
макологические и физиологические действия препарата [34]. Показано,
что препарат снижает жировую инфильтрацию печени лабораторных жи-
вотных в остром опыте (10—20%) или при безбелковой диете (11,8—48,8%).
В области синтеза препарата имеются в литературе скудные данные [35,
36]. У нас синтез препарата разработан в 1965 г. [34]. Синтез дихлораце-
тата диизопропиламмония состоит из трех стадий. Первая стадия заключа-
ется в конденсации изопропиламина I и ацетона II и получают диметил-
изопропилкетимин III, температура кипения 93,5° С; й?4°=0,7480; выход
85,6%
СН3 СН3 СНз СНз
I III
CHNH2 + СО —> СН—N=C
I III
сн3 сн3 сн3 сн3
I II III
Вторая стадия предусматривает гидрирование диметилизопропилкети-
мина на никелевом катализаторе с получением диизопропиламина IV; тем-
пература кипения 83,9° С; dl° = 0,7163; Пд— 1,3518; выход 80%
СН3 СН3
СН—N=C
I I
СНз СНз
щ
+ н2
Ni
“ СНз '
I
СН—
_СНз
IV
NH
2
182
Третья стадия заключается в получении дихлорацетата диизопропил-
аммония VI путем конденсации диизопропиламина с дихлоруксусной кис-
лотой V; температура плавления 119—120° С; выход 70%.
СН—
I
_сн3 _2
NH + С12НССООН
NHCOOHCHC1
IV
VI
Судя по литературным данным [34], производные пангамовой кислоты
при соответствующей биологической и клинической апробации могут в
перспективе найти применение как аналоги витамина В15.
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. В сб.: «Витамин В15». (Пангамовая кислота), М., «Наука», 1965, 280 с.; Б у к и н В.
Пангамат кальция, М., «Наука», 1963.
2. Т о m i j a m а Т., V о n е V., Proc. Japan Acad., 1953, 29, 178; С. А., 1954, 48,
5961.
3. К г е b s Е. Sr., Krebs Е. Jr., Beard Н., Malin R., Harris A.,
Barlett C., Internal. Record. Med., 1951, 164, 18.
4. Krebs E. Sr., Krebs E. Jr. Пат. США 2710876, 1955; C. A., 1955, 50,
5733 c.
5. Гаркина И. Пангамовая кислота и ее производные. — В сб. : «Витамин В16»,
М., «Наука», 1965, с. 5—19.
6. V a i 1 a t i G., I n v е г n i z z i G., Minerva medica, 1957, 48, 3431.
7. Krebs E., Johnson V., J. Appl. Nutr., 1955, 8, 360.
8. Стрельчук И. В. О лечении хронического алкоголизма и других наркоманий
витамином В15 — В сб.: «Витамин В16», М., «Наука», 1965, с. 104—109.
9. Leone М., Longareiti A*., L u с с h е 1 i i Р. Minerva med., 1957, 48,
3438.
10. С и g a d t 1 а Е., Dispense Е. Minerva med., 1957, 48, 3428.
11. I d z и m i а , Vitamins (Japan), 1959, 16, 279.
12. Marschall F., Adamson R., Long J. Proc. Soc., Exper. Biol, and
Med., 1961,. 107, 420.
13. А л п а т о в И. M., Гайда макин Н. А., Удалов Ю. Ф. Опыт ис-
пользования пангамовой кислоты при экспериментальном отравлении дихлорэта-
ном. — В сб.: «Витамин В16». М., «Наука», 1965, с. 91—93.
14. Б р а у д е А. И., Шевнюк Л. А., У р янская В. И. Эксперименталь-
ное изучение некоторых проявлений биологического действия пангамата кальция.
Там же, 1965, 38—43 с ил.
15. С о л о в ь е в а Н. В., Гаркина И. Н. Окислительное деметилирование
пангамовой кислоты. — Там же, 1965, 28—33.
16. Инструкция по применению кальция пангамата. Фармакологический комитет Ми-
нистерства здравоохранения СССР, 21 июля, 1965.
17. Ю р к е в и ч А. М., Вереникина С. Г., Преображенский Н. А.
Изучение гидролитической устойчивости пангамовой кислоты. — «Хим.-фарм. ж.»,
1967, № 12, с. 26—30 с ил.
18. С a s и В., D a n s i А. и др., Bull. chim. farmaceut, 1958, 97, 3; С. А., 1958,
52, 9521.
19. Ковахара К-, Кобаяси X. Япон. пат. 4765, 1962;
20. Ю р к е в и ч А. М., Вереникина С. Г., Долгих М. 'С. Лактон
6-о-диметилглицил-О-глюконовой кислоты и его производные.—ЖОХ, 1967, т. 37,
с. 1267—1272 с ил.
21. Вереникина С. Синтетические исследования в области пангамовой кислоты
и ее производных, автореф. канд. диссерт., М., 1967.
22. G u t г i е R., Methods in Carbohydrate Chemistry, N. Y. 1962, I.
23. С о ш н и к о в Д. Я-, П е т у н и н а А. Г., Малышева Е. А. Получение
глюконата кальция микробиологическим методом — «Прикл. биохим. и микробиол.»,
1970, т. 6, вып. 1, с. 83—85.
23а. Разумовская 3. Г., Щелкунова С. А., Г р и д ю ш к о Е. С.,
Сергеева Т. Н., Кантор Б. Б., Васильева О. А. Авт. свидет.,
№ 275031, 1968; Бюлл. изобрет., 1970, № 22, с. 12.
24. Smith М., Moffatt I., Khorana Н., J. Am. Chem. Soc., 1958, 80,
6204.
25. Khorana H., J. Am. Chem. Soc., 1954, 76, 35171.
26. Chambers R., Moffatt I., Khorana H., J. Am. Chem. Soc., 1957,
79, 4240.
183
27. Braunuger H., Delzer W., Pharmazie, 1965, 20, 279.
28. Кочетков H. K-, Деревицкая В. А., Лихошерстов Л. M.
и др. Синтез 6-о-х-глицилглюкозы и 6-о-(й,£-аланил)-глюкозы—ЖОХ, 1962
т. 32, 1159.
29. Кочетков Н. К-, Деревицкая В. А., Молодцов Н. В. Синтез
N-ацильных производных аминосахаров. —ЖОХ, 1962, т. 32, 2500.
30. Грабилина Г. О., Уткин Л. М. Об ацетоипроизводных D-глюконовой
кислоты и ее -(-лактона. —ЖОХ, 1960, т. 30, с. 3126—3128.
31. С а 1 с о t t W., S р i е g 1 е г L., Tinker J. Пат. США 2203009, 26/VIII 1937-
Off. gaz. 1940, 515, № 1, 95.
31 а. С о ш н и к о в Д. Я., С ю х и н а В. И. Пет у н и н а А. Г. Выделение
глюконовой кислоты из глюконата кальция ионообменным методом. — «Прикл.
биохим. и микробиол.»., 1968, т. 4, вып. 4, с. 440—443 с ил.
32. С 1 а г k е Н. и др. J. Am. Chem. Soc., 1933, 55, 4571.
33. М i с h а е 1 i s L., Schubert M., J. Biol. Chem., 1936, 115, 221.
34. Кругликова-Львова P. П., Рекунова В. H., Ушако-
ва М. Т., Ю р к е в и ч А. М. Синтез дихлорацетата диизопропиламмония и
изучение его фармакологических свойств.—«Медицинская промышленность СССР»,
1965, № 12, с. 19.
35. Пат. ФРГ 1105877, 1959.
36. Italosever, Англ. пат. 862248, 30/VII 1959; Abridgments of specifications , 1961, 62.
Глава 9. ПРОИЗВОДСТВО СИНТЕТИЧЕСКОЙ
НИКОТИНОВОЙ КИСЛОТЫ (ВИТАМИНА РР)
В 1912—1914 гг. Функ [1, 2] и Сузуки [3] в поисках витамина против
полиневрита выделили никотиновую кислоту из дрожжей и рисовых от-
рубей. Однако указанные исследователи не придали ей значения как ви-
тамину. В течение 1917—1930 гг. проводились исследования по экспери-
ментальному созданию у лабораторных животных болезни «черный язык»,
идентичной по происхождению пеллагры у человека (pelle agra — грубая
кожа, итальянск.). Изыскивались диеты, вызывающие это заболевание
и излечивающие его [4—8]. В 1935 г. было установлено, что никотинамид
входит в состав окислительных ферментов дегидрогеназ [9—11]; этим была
подчеркнута важная физиологическая роль никотиновой кислоты. Нако-
нец, в 1937 г. были опубликованы экспериментальные данные [12] об ус-
пешном применении никотинамида для лечения пеллагры человека. С этого
времени стали придавать никотиновой кислоте и ее амиду значение вита-
мина РР (Pellagra preventive). В настоящее время считается твердо уста-
новленным, что витамин РР играет важную роль в жизнедеятельности
живого организма. Он входит в виде амида никотиновой кислоты в состав
двух коферментов (козимазы и кодегидразы). Специфическое действие ви-
тамина выражается в лечении и предохранении организма от пеллагры.
Это заболевание возникает при длительном потреблении продуктов, не
содержащих достаточного количества никотиновой кислоты или провита-
мина РР — аминокислоты триптофана. Антагонистом никотиновой кисло-
ты (антивитамин РР) является |3-индол-3-уксусная кислота.
Кроме антипеллагрического действия, никотиновая кислота принимает
участие в углеводном, белковом, холестериновом и основном обмене ве-
ществ [14—17]. Никотиновая кислота обладает инсулиноподобным дейст-
вием — регулирует содержание сахара в крови. Имеются данные о том,
что никотиновая кислота снижает избыточное содержание пировиноградной
кислоты в крови и повышает содержание холестерина в ней. Никотиновая
кислота повышает кислотность желудочного сока; в ряде случаев в послед-
нем появляется свободная соляная кислота, когда обычные физические
раздражители (бульон, капустный отвар) этого не обеспечивают.
Никотиновая кислота оказывает благоприятное влияние на функцио-
нальную деятельность печени, в частности на ее антитоксическую и пиг-
ментную функции [17]. Под влиянием никотиновой кислоты наступает
сосудистая реакция (чувство жара, покраснение, ощущение покалывания,
зуда, появление мурашек), которая длится 5—20 рин и бесследно исчеза-
•184
ет. Экспериментально установлено, что большие дозы никотиновой кисло-
ты сужают венечные сосуды сердца, а средние дозы (50—100 мг) расширя-
ют их или вовсе не оказывают влияния на просвет сосудов [17]. Никоти-
новая кислота не токсична и в лечебных дозах (20—50 мг) хорошо перено-
сится.
Среди производных никотиновой кислоты важное физиологическое зна-
чение имеет амид никотиновой кислоты. Наиболее богаты никотиновой
кислотой дрожжи, пшеничные и рисовые отруби, грибы, печень. Значение
витамина РР для животноводства возросло с расширением использования
Кукурузы, содержащей недостаточное количество никотиновой кислоты и
аминокислоты триптофан. Обогащение кукурузных рационов никотиновой
кислотой способствует лучшему усвоению кормов и повышению на 15—
18% привесов [18].
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ВИТАМИНА РР
Никотиновая, или |3-пиридинкарбоновая, кислота представляет собой
белые кристаллы с температурой плавления 235,5—236,6 [20]. Возгоня-
ется при 210—225° С без разложения. Эмпирическая формула C6H5O2N,
молекулярная масса 123,11, структурная формула:
СН
НС^ с—соон
I II
нс^ ^сн
N
Растворимость в воде при температуре 25° С—1 : 60, в спирте—1 : 80.
Никотиновая кислота и ее производные легко растворимы в кипящей воде,
кипящем спирте (табл. 11) и водных щелочных растворах; в эфире нико-
тиновая кислота практически не растворяется.
Таблица 11
Температу- ра, °C Растворимость, % [20,21]
никотиновой кислоты никотината натрия никотиновой кислоты гидро- хлорида
вода спирт вода вода
0 0,86 0,57 9,5 1,73
15 1,30 0,92 23,11 8,50
38 2,47 2,10 31,16 (30°) 14,23
61 4,06 4,20 39,11 17,01
78 6,00 7,06 43,20 19,50
100 9,76 — 49,78 21,48
В сухом чистом виде никотиновая кислота негигроскопична и весьма
стабильна. Растворы ее могут быть автоклавированы 20 мин при 120° С
без разложения. Она также стабильна при нагревании в 1—2 н. раство-
рах минеральных кислот и щелочей [20]. Изоэлектрическая точка лежит
при pH 4,2—4,3.
Никотиновая кислота обладает амфотерным характером: растворяется
в кислотах с образованием четвертичных солей, как, например, C6H-O2N-
НС1, и в щелочах с образованием нормальных солей, как, например,
никотината натрия CeH4O2NNa, никотината меди (CeH4O2N)2Cu, нераст-
воримого в воде, никотината кальция (CeH4O2N)2Ca, трудно растворимого
в воде. Некоторые свойства никотиновой кислоты и ее производных при-
ведены в табл. 12.
185
Таблица 12
Название и формула Физико-химические свойства никотиновой кислоты и ее производных [20]
молеку- лярная масса форма кристаллов температу- ра плав- ления, сс абсорбци- онный максимум в ультра- фиолето- вом свете, нм растворимость
Никотиновая кислота C5H4NCOOH 123,11 Иглы (из спир- та) 235,5— —236,6 261,5; 385 [24] Хорошо в горячей воде и спирте, плохо в эфире
Никотинамид C5H4NCONH2 122,12 Иглы (из бен- зина) 133; 129-131 261,5 То же
№-Метил никотинамид C5H4NCH3C1CONH2 172,62 Листочки (из абсолютного алкоголя) 230,5; 233—234; 240 264,5 Растворим в воде и спирте, плохо в горячем абсолют- ном алкоголе '
Никотиновая кисло- та — гидрохлорид C5H4NHC1-COOH 159,58 Призмы (из воды) 274-275; 272 Растворима в во- де и спирте, пло- хо в эфире и бен- золе
Хинолиновая кислота C5H3N(COOH)2 167,12 Кристаллы мо- ноклинической системы (из воды) 232—237 Растворима в го- рячей воде, хоро- шо в эфире, сла- бо в спирте, не- растворима в бен- золе
Амид никотиновой кислоты представляет собой гигроскопичный бес-
цветный кристаллический порошок с температурой плавления 129—13ГС.
2615
240
250 260 270 НМ
Рис. 27. УФ-спектр поглощения никоти-
новой кислоты при различном pH.
Легко растворим в воде (1:1), в
спирте (1:1,5); нерастворим в
эфире, бензоле; из водных рас-
творов экстрагируется хлорофор-
мом, этилацетатом и бутило-
вым спиртом 122]. Никотинамид
сублимируется в вакууме при
температуре 150—160° С и при
остаточном давлении 0,0005 мм
рт. ст. [23]. Стоек к нагрева-
нию, чувствителен к щелочам.
На рис. 27 изображены кри-
вые поглощения водного рас-
твора никотиновой кислоты в
воде при различном значении
pH в ультрафиолете при
261,5 нм [24, 25].
Изомерные кислоты: пиколи-
новая (а-пиридинкарбоновая) и
изоникотиновая (у-пиридинкар-
боновая) по своим свойствам
значительно отличаются от ни-
котиковой кислоты. Раствори-
мость в воде резко возрастает
от у- к а-положению. Например,
растворимость в воде изонико-
тиновой кислоты в 100 раз меньше, чем никотиновой кислоты. При вы-
сокой температуре пиридинкарбоновые кислоты декарбоксилируются,
причем легче других декарбоксилируется пиколиновая кислота, а труд-
нее других никотиновая кислота
186
СООН
Пиколиновая
кислота •
Изопиколиновая ’
кислота
Такая же последовательность наблюдается при декарбоксилировании
пиридин-ди и трикарбоновых кислот, что видно из следующей схемы:
СООН
г^'С-СООН
k JJ-COOH
N
cLtJ3,y- Пиридин-
кардонобая кислота
СООН
^уг-СООН_______
'n
Ц инхомеронобая
Никотиновая Хинолиновая
кислота кислота
СООН
СООН
кислота
Указанное свойство используется при синтезе никотиновой кислоты
из более сложных гетероциклических соединений, как, например, из хи-
ьолина, 2-метил-5-этилпиридина.
МЕТОДЫ СИНТЕЗА НИКОТИНОВОЙ КИСЛОТЫ, ЕЕ АМИДА И
ВЫБОР РАЦИОНАЛЬНОГО МЕТОДА ДЛЯ ПРОИЗВОДСТВА
Никотиновая кислота. Для синтеза витамина РР (р-пиридинкарбоновой
кислоты) могут быть использованы как пиридин, так и его производные
(замещенные в p-положении). Последние могут быть получены либо из при-
родного сырья, как, например, никотин-основание из отходов табачного
листа, анабазин — (5-(а-пиперидил)-пиридин из растения Anabasis aphylla,
Р-пиколин и хинолин из каменноугольной смолы, либо синтетическим пу-
тем, как, например, 2-метил-5-этилпиридин. Рассмотрим основные источ-
ники сырья и методы синтеза никотиновой кислоты, имеющие промышлен-
ное применение, и выберем те из них, которые представляются наиболее
эффективными.
СИНТЕЗ НИКОТИНОВОЙ КИСЛОТЫ ИЗ ПИРИДИНА
Пиридин является одним из продуктов перегонки каменноугольной смо-
лы и легко получается в чистом виде.
Никотиновая кислота может быть синтезирована из пиридина двумя
способами:
1) сульфирование пиридина олеумом и получение 3-пиридинсульфоно-
вой кислоты, сплавление ее калиевой или натриевой соли синильной кис-
лоты и получение цианпиридина, последующий гидролиз его едким
датром
.Пиридин
HgSO4; 230°
Олеум
7О°/а
N
3 -Лиридинсулыро -
новая кислота
350° NaOH
KCN* U S 30°/o *
40°/o N
Цианпиридин
СООН
N
Никотиновая
кислота
2) более рациональным методом является синтез из пиридина через
бромпроизводное (бромирование при 300° С) с последующим замещением
атома брома на нитрильную группу при действии цианистой меди и гид-
ролизом едким натром. Синтез осуществляют в несколько стадий [26—
187
31]: на стадии I выход от теоретического составляет 30%; на стадии II —
50%; на стадии III — 90%.
ЗВг2
Cu(CN)2
ЗНВг
Пиридин
3 - Цианпиридин
СиВг2
Цианистая
медь
coonh4
NaOH
COONa НС1
Никотинат
Никотинат Никотинобая
аммония
натрия кислота
Недостаток метода — низкий выход никотиновой кислоты (13,5%), вы-
сокий расход брома и применение токсической цианистой меди. Пири-
дин является недефицитным продуктом коксохимической промышленности.
Однако трудность введения заместителей в пиридиновое ядро, а также
указанные выше недостатки умаляют промышленное значение пиридина
как источника сырья для производства никотиновой кислоты. Наиболее
перспективными являются |3-алкилзамещенные пиридина, а также хино-
лин.
СИНТЕЗ никотиновой кислоты ИЗ ПРОИЗВОДНЫХ ПИРИДИНА
Синтез никотиновой
может быть получена из
кислоты из р-пиколина. Никотиновая кислота
р-пиколина с наименьшим расходом окислителя:
Н2О
С этой точки зрения р-пиколин является самым эффективным сырьем.
Окисление его в никотиновую кислоту может быть осуществлено различ-
ными окислителями [32—34]: перманганатом в щелочной среде с выходом
60—77% [35—38]; хромовокислым калием (хромпиком) при 200—250° С
под давлением с выходом 83% [39]; азотной кислотой в присутствии сер-
ной с выходом 55—58% [40]; азотной кислотой при 200° С [40]; 30%-ной
азотной кислотой при избыточном давлении 15—20 кгс/сж2 при температу-
ре 130—150° С [41 ];'серной кислотой в присутствии селена при темпера-
туре 300° С [42].
В 1944—1948 гг. появились сообщения о синтезе никотиновой кислоты
из р-пиколина парофазным процессом — окислением кислородом воздуха
в присутствии катализатора с выходом 17% [44, 45]; в присутствии аммиа-
ка в результате реакции получают нитрил никотиновой кислоты с выходом
66% [46]. Нитрилы пиридиновых кислот получают пропусканием смеси
паров пиколина, аммиака и воздуха при 300—350° С над пятьюокисью
ванадия, нанесенного на активированной окиси алюминия [47, 47а].
С выходом 65% получен никотиннитрил над гранулированным ванадатом
олова при температуре 370° С [48, 49]. Лучшие результаты (выход нитри-
ла 93,6%) получены при использовании катализатора ванадата алюминия.
Омыление нитрила осуществляют в водно-аммиачном растворе в присут-
188
ствии едкого кали и 10%-ного раствора перекиси водорода в течение 2 ч
при температуре 42° С. Выход никотинамида (на р-пиколин) 64,9% [50],
по другим данным [51], выход 50—52%. В 1946—1961 гг. появилось не-
большое число исследований [36, 52—55] по окислению р-пиколина элект-
ролитическим путем.. Процесс проводят в электролизере с керамиковой
диафрагмой с применением анолита из р-пиколина в 30%-ной серной кис-
лоте при температуре 40—45° С и плотности тока 5 а/дцм2. Анолит нейтра-
лизуют щелочью, упаривают и при pH 3,4 выделяют никотиновую кислоту
с выходом 60%. Из всех изложенных выше методов превращения р-пико-
лина в.никотиновую кислоту наиболее перспективным является парофаз-
ный аммонолиз его с получением никотинамида. Этот метод не требует де-
фицитного и дорогостоящего перманганата калия, агрессивных окислите-
лей (азотная и серная кислоты). Процесс можно осуществлять непрерывно
и в менее опасных условиях труда. Важным является также получение
целевого продукта в виде амида никотиновой кислоты. Химизм процесса:
СН3 [o];nh3
N
J3-Пиколин
93,12
Аммонийная соль
никотиновой кислоты
-2Н2О
conh2
Нитрил никоти- Никотинамид
новой кислоты 122,13
ЮН, 11
Казалось, что в условиях высокой эффективности р-пиколина как сырья
для производства витамина РР следовало на нем базировать промышлен-
ное производство. К сожалению, источники сырья для его получения (пи-
колиновая фракция каменноугольной смолы) весьма ограничены. Кроме
того, р-пиколин (температура кипения 143° С) в них содержится вместе
с ^-пиколином (144° С) и 2,6-лутидином (142° С) в соотношении (приблизи-
тельно) 3:2:5. Для очистки р-пиколина применяют различные химические
реакции, в которые вступают примеси, а р-пиколин не вступает. К этим
реакциям относятся: конденсация с формальдегидом [56, 57], с фталевым
ангидридом [56, 58, 59], с фталевым и уксусным ангидридом [60], с моче-
виной [61 ], с бензойной кислотой [62], с муравьиной кислотой [63]. При-
меняется также метод очистки пиколиновой смеси от 2,6-лутидина путем
связывания р-пиколина с хлористым цинком в комплексную соль с после-
дующим разложением ее щелочью по следующей схеме:
J3 - Пиколин
Комплексная соль
2,6-Лутидин в этих условиях комплексной соли не образует.
Разработан также метод фракционной жидкой экстракции 2,6-лутиди-
на, р-пиколина и у-пиколина, позволяющий получать р-пиколин 95%-ной
чистоты [64]. Очистка р-пиколина от сопровождающих его других пико-
линов стала необходимой, так как в противном случае при окислении по-
лучали бы смесь кислот — пиколиновой из ct-пиколина и 2,6-лутидина,
никотиновой из р-пиколина, изоникотиновой из '(•-пиколина. Однако чис-
тый р-пиколин может быть получен синтетическим путем из акролеина
и аммиака согласно следующей схеме:
нс>СН2 сн=сн2
I ?
НС;. ^СН
ХО +' ск
NH3
Акролеин + аммиак
А1РО4
J3 - Пиколин
189
Конденсация проводится в присутствии фосфорнокислого алюминия [39,
65]. Этот синтез, раньше имевший лишь теоретическое значение, в настоя-
щее время в связи с открытием возможности получения акролеина из про-
дуктов переработки нефти приобретает перспективу промышленного ис-
пользования.
Синтез никотиновой кислоты из 2-метил-5-этилпиридина (МЭП) [66].
Бесцветная жидкость с резким неприятным запахом; температура кипения
178,3° С; эмпирическая формула C8IInN; молекулярная масса 121,18;
= 0,9208; = 1,4970; растворимость в воде при температуре 20° С—
1,22%; растворяется в спирте, эфире, бензоле, хлороформе, дихлорэтане.
Концентрация МЭП определяется инфракрасной спектроскопией. Синтез
МЭП осуществляют по методу Чичибабина конденсацией аммиака с ацет-
альдегидом в паровой фазе в присутствии катализатора [67]. Позднее было
установлено, что хороший выход МЭП получают при использовании для
реакции конденсации паральдегида и аммиака в водном растворе в присутст-
вии уксуснокислого аммония при температуре 200—250° С (под давлением)
[68, 69]. Этот синтез также разработан в непрерывном процессе при темпе-
ратуре 250—265° С, избыточном давлении 80—120 кгс/см2 [70]. Химическая
схема получения никотиновой кислоты из МЭП:
2 -Метил - 5- зтилпиридин
СООН
Пиридин -2,5-дикардонобая
кислота'
(изоцинхомеронобая)
-СО2
Никотиновая
кислота
В пиридинкарбоновых кислотах вследствие пониженной электронной
плотности в положениях 2 и 6 пиридинового цикла карбоксильные группы
элиминируются значительно легче, чем в положении 5.
Окисление МЭП может быть осуществлено в жидкой или паровой фазе,
различными окислителями при нормальном или повышенном давлении пери-
одическим или непрерывным процессом. Никотиновая кислота может быть
получена из МЭП в одну стадию без выделения изоцинхомероновой кислоты
или в 2 стадии с выделением последней с последующим декарбоксилиро-
ванием.
Рассмотрим различные способы окисления МЭП в зависимости от исполь-
зуемого окислителя.
Перманганат калия. [71] Японские авторы указывают, что
максимальный выход имели при использовании 8 молей перманганата ка-
лия на 1 моль МЭП. Выход в 76,1 % был получен при окислении 12,1г МЭП в
400 мл воды, добавлении 126,4 г перманганата калия при температуре 90—
95° С. Вследствие дефицитности окислителя метод не имеет перспектив для
промышленного применения. Химическая схема:
+ 6К,МпО4
коос
4КОН + 6МпО2 + СО2 + 2Н2О
Серная кислота. Окисление МЭП серной кислотой осуществля-
ют в присутствии селенового катализатора при температуре 300—350° С [72].
На 1 моль МЭП берут 21 моль серной кислоты (плотность 1840 кг/м3) и 5—8%
к массе МЭП селена. Конечным продуктом реакции является изоцинхо-
мероновая кислота с выходом 62—82%. Имеется указание о возможности
окисления МЭП двуокисью марганца и серной кислотой с селеновым ката-
190
лизатором [73], а также действием хлора, серной кислоты с селеновым ка-
тализатором [74]. Химическая схема:
,, Se r^VCOOH
н с Jj + 10H2SO4 ----------- J + 12H2O+ 9SO2+2CO2
3 N "n-H2SO4
Серная кислота является дешевым окислителем. Однако жесткие усло-
вия процесса окисления (300—350° С), выделение больших объемов сернис-
того газа, высокая агрессивность среды создают большие трудности в ап-
паратурном оформлении процессов. Применение токсичного селена связа-
но со специальной очисткой от него никотиновой кислоты. В связи с этим
данный метод впредь до появления соответствующих антикоррозий-
ных материалов не имеет перспектив промышленного применения.
Азотная кислота. Проведено наибольшее число исследований
и в ряде случаев достигнуты практические результаты при использовании в
качестве окислителя МЭП азотной кислоты, являющейся самым дешевым
окислителем. Работы проводились в направлении получения никотиновой
кислоты: а) процесс осуществлялся в одну или две стадии, б) при периоди-
ческом или непрерывном осуществлении реакции окисления, в) при исполь-
зовании в качестве окислителя свободной азотной кислоты или гидролизу-
ющихся нитратов; г) при применении концентрированной или разбавленной
азотной кислоты.
Известно [66, 75], что в США при промышленном осуществлении окисле-
ния МЭП азотной кислотой процесс ведут при температуре 185° С и избыточ-
ном давлении 20 кгс!см2 и 3,56 частей азотной кислоты на 1 часть МЭП. Ре-
акция идет в направлении получения изоцинхомероновой кислоты; послед-
няя во второй ступени реактора декарбоксилируется при температуре 195° С
в никотиновую кислоту. Выход никотиновой кислоты по обоим процессам
составляет 70%. Аналогичный процесс описан в непрерывном потоке [76].
По ряду исследований и патентов процесс осуществляют непрерывно двух-
ступенчато, с получением на первой ступени изоцинхомероновой кислоты.
Так, она получается при окислении МЭП 20%-ной азотной кислотой при тем-
пературе 175 ° С в течение 1 ч [77]. При окислении МЭП 40—70%-ной азотной
кислотой при температуре 170—200° С и избыточном давлении 40 кгс/см1 в
течение 30 мин получают изоцинхомероновую кислоту с выходом 60,5—72,7%
[78]; при окислении МЭП 25%-ной HNO3 и при температуре 185°Св автокла-
ве под давлением выход был 80% [78а ]. Представляют интерес способы окис-
ления МЭП при атмосферном давлении в периодическом процессе. Так описан
процесс, в котором раствор МЭП в 60—65%-ной азотной кислоте приливают
к концентрированной серной кислоте, содержащей в качестве катализатора
ванадат аммония [79]. Описан также метод окисления МЭП 57%-ной азот-
ной кислотой в присутствии серной кислоты (1:1 объем) и участии 1 % ката-
лизатора ванадата аммония при температуре 205—210° С в течение 4 ч с
выходом 58—60% [80, 81 ]. В сообщении Уставщикова, Фарберова и др. [82]
приведены данные по окислению МЭП разбавленной (10%-ной) азотной кис-
лотой под давлением в ампулах из нержавеющей стали (1Х18Н9Т) при тем-
пературе 190° С. Получена изоцинхомероновая кислота в смеси с никотино-
вой кислотой с суммарным выходом в 92%.
Описан также непрерывный одностадийный процесс получения никоти-
новой кислоты из МЭП, при котором раствор МЭП в HNO3 в соотношении
1: 6 прокачивают через реактор, нагретый до 253° С под избыточным давле-
нием около 50 кгс!см2. Выход никотиновой кислоты 64,2% [83].
При исследовании одностадийного процесса окисления МЭП азотной кис-
лотой были получены (Лобеев, Шнайдман) следующие оптимальные данные:
молярное соотношение МЭП и HNO3 (1 : 6), температура 230° С, избыточное
191
давление 35—40 кгс-см-\ состав исходной смеси (в %): МЭП — 9,5; HN03
(100%-ная) — 29,5, вода — 61,0. Выход на прореагировавший МЭП
61,0%.
Известен также метод окисления паров МЭП парами азотной кислоты
[84] при температуре 210—270° С и атмосферном давлении. На 1 часть (ве-
совую) МЭП применяют 20 частей 40%-ной азотной кислоты и 0,06 части
борной кислоты. При температуре 230° С выход никотиновой кислоты сос-
тавил 51%.
Для окисления МЭП вместо свободной азотной кислоты могут быть при-
менены гидролизующиеся нитраты железа, алюминия или меди. Окисление
МЭП азотнокислой медью изучали японские авторы [71, 85]. Реакцию про-
водили в автоклаве из нержавеющей стали при температуре 210° С в течение
3 ч. Азотнокислую медь применяли в виде 31%-ного раствора в количестве
110% от теоретического. Выход изоцинхомероновой кислоты составил
92,8%.
Наши исследования [80, 81] показали положительные результаты при
окислении МЭП нитратом меди при температуре 210° С, загрузке МЭП 63 г,
Cu(NO3)2. ЗН2О — 421 г, продолжительности реакции 4 ч, избыточном
давлении 70 кгс/см2. Реакционную массу обрабатывали едким натром при
кипячении, окись меди отфильтровывали, а из фильтрата при pH 1,5—2,
выделяли изоцинхомероновую кислоту с выходом 80,9—84,1%.
При окислении МЭП азотной кислотой в присутствии азотнокислой меди
получен выход изоцинхомероновой кислоты в 92,7% [87]. Разработан метод
окисления МЭП кислородом воздуха в присутствии нитрата меди при повы-
шенной температуре и избыточном давлении 17—25 кгс/см2 [88]. Этому же
вопросу посвящен ряд других работ [89—92].
Несмотря на наличие большого, числа исследований и вариантов техно-
логических схем в области применения азотной кислоты для окисленияМЭП,
выбор наиболее эффективного варианта затруднен из-за сильно коррозиру-
ющих сред и жестких режимов, требующих особо стойких к коррозии мате-
риалов. Действительно, наиболее эффективным вариантом был бы непрерыв-
ный одностадийный процесс при содержании азотной кислоты в реакцион-
ной массе 29,5%, МЭП 9,5%, при температуре 230° С и избыточном давлении
30—40 кгс!см2. Однако для таких жестких условий требуется реактор-окис-
литель из тантала, дорогого и дефицитного материала. Применение же дру-
гих материалов (нержавеющая или эмалированная сталь, титан) для данно-
го режима, а также для других вариантов окисления МЭП азотной кислотой
под давлением угрожает взрывами и несчастными случаями. Окисление МЭП
нитратом меди возможно в автоклаве из нержавеющей стали, но здесь воз-
никают трудности с обеспечением безопасной работы из-за возможных
резких скачков давления, вызываемых обильным выделением газов (на
1 моль МЭП выделяется 5 молей газов — 4NO + СО) в начальный период
реакции. Кроме того, нитрат меди дефицитен и дорог; разложение медной
соли для выделения изоцинхомероновой кислоты — процесс недостаточно
технологичный. Следовательно, в условиях ненадежности конструкционных
материалов для реакторов-окислителей приемлемым вариантом является
окисление МЭПа при атмосферном давлении и периодическим процессом.
Этот вариант предусматривает [80] окисление МЭП 57%-ной азотной
кислотой в присутствии серной кислоты (1:1 объем) и 1% катализатора
ванадата аммония при температуре 205° С в течение 4 ч с выходом изо-
цинхомероновой кислоты 68%. Реакцию окисления возможно было бы
проводить в реакторе из нержавеющей стали 1Х18Н9Т или в эмалиро-
ванном аппарате при условии систематического контроля коррозийного
процесса.
Для вариантов окисления МЭП азотной кислотой или азотной кислотой
в присутствии серной кислоты и катализатора ванадата аммония реакции
протекают по следующим схемам:
192
2-Метил-5-
этилпиридин
121,18
+ 6HNO3-----
СООН
Изоцинхомероновая
кислота
157, 77
СО2 + 6NO+ 6Н2О
r^yc2Hs nh4vo3 ,
Н,сЛ> 2 6НЬЮ3
n-h2so4
2~ Метил-5-
зтилсулыратпиридин
218, 26
\ |ГСООН + СО2 + 61NO + H2SO4
"n
№7,11
Реакция декарбоксилирования изоцинхомероновой кислоты протекает
по схеме:
ноос
Изоцинхомероновая кислота
167, 11
200-205°
+ со2
Никотиновая кислота
123,-11
Парофазный окислительный аммонолиз. Начиная
с 1952 г. в печати стали появляться отдельные работы, посвященные парофаз-
ному окислению МЭИ кислородом воздуха в присутствии аммиака и ката-
лизатора с получением нитрила никотиновой кислоты [93] по схеме:
2-Метил '5~ этилпиридин
эИ + NH3 + 2СО2 + 5Н2О
'n
Нитрил никотиновой
кислоты
(3-цианпиридин)
При окислении МЭП при температуре 320—350° С с ванадиевым катали-
затором, нанесенным на окись алюминия, получен никотиннитрил с выходом
37,5%. Аналогичные результаты по выходу 3-цианпиридина получены на
ванадиймолибденовом катализаторе при температуре 440—450° С [95]. На
этом же катализаторе этим же автором были окислены 2,3-лутидин и 2,5-
лутидин (лутидиновая фракция, сопровождающая МЭП при его синтезе) с
выходами соответственно 72,2 и 54,3%. Описан [94] также метод окисления
алкилпиридинов и МЭПа кислородом воздуха с добавлением паров воды над
V2O6—SiO2 катализатором с выходом никотиновой кислоты при температуре
260° С в количестве 85%. Однако указанный выход пока экспериментально
не подтвержден. Более подробно об окислительном аммонолизе см. [96].
З-Цианпиридин может быть различными способами превращен в никоти-
новую кислоту или в никотинамид, например, при кипячении в течение 8—
10 вводного раствора 3-цианпиридина с гидроокисью бария получают никоти-
новую кислоту с хорошим выходом [97]. Для получения никотинамида
водный раствор 3-цианпиридина нагревают в присутствии аммиака [98]
или других слабых оснований, например Na2CO3, NaHCO3, Na2B4O7 [99],
СаСО3 [100]. Почти количественный выход амида получают при обработке
нитрила в слабощелочном растворе при температуре 40—45° С разбавленным
раствором перекиси водрода [97, 101, 102 ]. Известен также метод гидролиза
7-522
193
3-цианпиридина в амид никотиновой кислоты обработкой в водном растворе
в течение 2 ч при температуре 60—65° С анионитом в ОН-форме с выходом
87,5% [95].
Метод синтеза никотиновой кислоты и ее амида парофазным окислитель-
ным аммонолизом весьма перспективен. Однако технология этого метода
находится в начальной стадии развития и пока еще не обеспечивает при-
емлемый выход целевого продукта.
Другие окислители также изучались в жидкофазном процес-
се. Сюда относятся: нитрозилхлорид [103], гипохлорит натрия [104], хлор
в водном растворе [105, 106], перекись водорода [107 ] и др. Однако указан-
ные окислители практического применения не нашли. Что касается окисле-
ния МЭП озоном, а также электрохимическим методом [55], то такие иссле-
дования широко ведутся преимущественно с хинолином. По этим вопросам
опубликован ряд работ (стр. 197). Начаты также исследования и в области
МЭП.
Декарбоксилирование изоцинхомероновой (кислоты. Большинство ме-
тодов окисления МЭП предусматривает раздельное проведение процессов
окисления и декарбоксилирования. Это позволяет избежать интенсивной
коррозии аппаратуры при более высокой температуре окисления, необходи-
мой для реакции декарбоксилирования. Последнюю можно проводить в
различных средах, как, например, в минеральном масле при температуре
216° С, в тетралине, дифениле [108, 109], хинолине. Более рационально де-
карбоксилирование проводить в водном растворе под давлением при темпе-
ратуре 190—200° С [71, 77, 108, ПО]. Например, при соотношении изоцин-
хомероновой кислоты и воды от 1 : 2 до 1 : 10, температуре 170—190° С и
продолжительности процесса 2 ч выход никотиновой кислоты составил
90,7— 95,6%, при температуре выше 200° С выход снижается, вследствие
частичного превращения изоцинхомероновой кислоты в пиридин.
Известен метод декарбоксилирования твердых гетероциклических ди-
карбоновых кислот при нагревании в замкнутой системе при температуре
205° С (для изоцинхомероновой кислоты) [111]. По-видимому, более эффек-
тивен процесс декарбоксилирования с одновременной очисткой получаемой
никотиновой кислоты методом сублимации изоцинхомероновой кислоты (Ло-
беев).
Процесс очистки никотиновой кислоты методом сублимации в среде воз-
духа, азота или углекислоты при температуре 160—250° С описан как для
лабораторных [112], так и для промышленных условий [113, 114]. Наши ис-
следования [86] показали, что для декарбоксилирования изоцинхомероновой
кислоты в водном растворе оптимальными условиями являются: соотно-
шение изоцинхомероновой кислоты и воды — 1,0 : 3,5, температура среды в
автоклаве 200—205° С, продолжительность процесса 1,5 ч, выход 89%.
Очистку технического продукта производили двухступенчатой кристал-
лизацией из воды (1 : 15), растворяли при температуре 95° С, обрабатывали
активированным углем (5%), охлаждали до 0° и выделяли кристаллы I.
Маточный раствор, обрабатывали углем, сгущали под вакуумом, кристалли-
зовали при 0° и выделяли никотиновую кислоту IL Выход медицинской ни-
котиновой кислоты из технической составил 89%.
Синтез никотиновой кислоты из хинолина. Светло-желтая жидкость
(на воздухе темнеет), температура кипения 238,5° С, C9H7N, молекулярная
масса 129,15, df= 1,0929 — 1,0933; температура плавления пикрата201,8—
202,3° С [114], содержится в фракции хинолиновых оснований каменно-
угольной смолы коксобензольной промышленности. Смола из кузнецких
углей содержит 3,3%, а из донецких углей 1,2% хинолиновых оснований
[115]. В указанной фракции, кроме хинолина, содержатся его изомер изо-'
хинолин (температура кипения 243,5° С) и аналог хинальдин (а-метилхино-
лин, температура кипения 247,6° С). Путем двухкратной ректификации вы-
деляют технический хинолин (95%-ный), выкипающий в интервале 2°С
194
(237,5—239,5). Превращение хинолина в никотиновую кислоту происходит
по следующей общей химической схеме:
Хинолин
J СООН
'a k J-LCOOH
N
Хинолиноёая кислота
-СО2
N
Никотинобая кислота
Бензольное кольцо хинолина менее устойчиво к окислителям, чем гетеро-
циклическое и в результате окисления образуется хинолиновая кислота;
в последней а-карбоксильная группа менее устойчива, чем |3- и при нагре-
вании легче отщепляется с образованием никотиновой кислоты.
Хинолин может быть окислен различными окислителями в жидкой или
паровой фазе периодически или непрерывным процессом в одну или две
стадии. Рассмотрим методы окисления хинолина в зависимости от рода окис-
лителя.
Серная кислота. В основу реакции окисления хинолина поло-
жен метод Вудварда [116], заключающийся в окислении хинолина серной
кислотой при температуре 300° С в присутствии катализатора — металличе-
ского селена. Выход около 40%. Эта реакция протекает по следующей
схеме:
N
Хинолин
СООН
+ 10H2SO,
N
Хинолиновая кислота
СООН -со2
СООН
N
Н X'HSO4
Никотиновая
кислота - сульфат
В качестве катализатора применяют также двуокись селена [117, 118J,
галогенные соединения или йод [119]. При окислении хинолина серной кис-
лотой в присутствии селена применялась и более высокая температура, при
этом никотиновая кислота очищалась либо через медную соль, либо ионита-
ми (амберлитом) [120].
Недостатком метода окисления хинолина серной кислотой является:
высокая температура, сложное аппаратурное оформление процесса, обиль-
ное выделение сернистого газа и токсичность катализатора — селена.
Азотная кислота. При окислении хинолина чистой азотной кис-
лотой под давлением при температуре 150—180° С выход никотиновой кис-
лоты низок (около 30%) [41]. С повышением температуры до 200—250° С
выход увеличивается [121]. Однако большинство исследований проведено
при окислении хинолина азотной кислотой в присутствии серной кислоты и
катализаторов при различных температурах: селен при 260—270° С [122];
селен, ртуть, медь при 305—310° С (выход 88%) [123]; йод (выход 86—90%)
[124]; соляная кислота при 215—225° С (выход 73%) [125]; молибденово-
кислый аммоний при 245—250° С (выход 69,2%) [126]. Наиболее подробно
разработан периодический процесс получения никотиновой кислоты из хи-
нолина в присутствии серной кислоты (1 : 1,5 объем) окислением азотной
кислотой при участии катализатора пятиокиси ванадия (1%) при температу-
ре 220—230° С. Выход технического продукта 50,8%, а медицинского 45,2%
[81, 127, 128]. Реакция протекает по следующей схеме:
7*
195
2 jT J} + 18HNO3 + 2H2SO4 —°Ч 2 ^|]-С°°Н+ 9N'O2 + 9NO+6CO2 + 11H2O
^N^
H HSO4
Хинолин Никотиновая
2.129,16 18.63,02 2.98,08 кислота - сулырат
2. 221, 18
+ 3NaOH
221, 18 3. 40,03
10H2O COONa
----*-*- IH + Na2SO4 . 10H2O + 3H2O
N
Натриевая соль •
никотиновой кислоты
145, 09
+ NaCl
Никотиновая кислота
145,09 36,47
58,45
Недостатком метода является неудовлетворительное использование объ-
ема реактора-окислителя (25-4-30%), большое количество отходов в виде суль-
фата натрия, периодический процесс. Метод проверен в полузаводских ус-
ловиях.
Парофазный окислительный аммонолиз хинолина. Этот процесс изучали
Жданович, Чекмарева, Суворов [129,130] на плавленом смешанном ката-
лизаторе из окислов ванадия и олова при температуре 420—450° С. Они
показали, что основными продуктами окислительных превращений чистого
хинолина в присутствии аммиака являются никотинонитрил, бензонитрил,
пиридин и бензол. Оптимальный выход никотинонитрила 60,9% был получен
на катализаторе V2O6/SnO2 при соотношении, равном 1 : 1,5, и температуре
450° С и девятикратном избытке аммиака. В этих же условиях из техничес-
кого хинолина (67% хинолина и 25% изохинолина, температура кипения
237—239° С) выход составил 21,7%. Выход газообразных продуктов реакции,
по данным авторов, зависит от количества аммиака и температуры. Из-
быток аммиака подавляет процессы окисления пиридинового цикла; удли-
нение времени контакта повышает в газообразных продуктах содержание
окислов углерода.
Для анализа продуктов парофазного окислительного аммонолиза хино-
лина разработан метод их газо-жидкостной хроматографии [131—133]. Не-
который интерес представляет каталитическое парофазное окисление хино-
лина кислородом воздуха. Имеются указания [134] на достижение выхода
никотиновой кислоты, равного 75%, при окислении хинолина кислородом
воздуха на смешанном катализаторе [Sn(VO3)4 : SnO2 = 1 : 3] и при темпе-
ратуре 400° С. Другие исследователи [135] отмечают, что при применении
этого катализатора вообще не удавалось получить никотиновую кислоту.
Е. Жданович [130] указывает, что при окислении хинолина кислородом воз-
духа при температуре 420° С катализатор пятиокись ванадия непригоден
(сгорание хинолина). При смешанных катализаторах (V2O6 : SnO2 = 1 : 1,5)
выход никотиновой кислоты достигал 20%. Однако при подаче воды в систе-
му (0,42 кг на 1 кг катализатора) выход возрастал до 70—72%. Вторым важ-
ным фактором является концентрация кислорода. При увеличении подачи
воздуха с 4 до 18 молей кислорода на 1 моль хинолина выход никотиновой
кислоты возрастал с 26,8 до 72,4 %. Необходимо отметить, что парафазный
каталитический процесс окисления хинолина кислородом воздуха без аммо-
нолиза или с его применением имеет в будущем перспективу промышленного
использования. Для этого метода не требуются агрессивные среды. Менее
жесткие антикоррозийные требования предъявляются к аппаратуре, отсут-
ствует угроза взрывов реакционной массы, процесс осуществляется непре-
196
рывным потоком. Однако внедрение данного метода тормозится из-за отсутст-
вия опытно-промышленных исследований.
Озон. В последние годы (1960—1968 гг). в печати появились патенты
и отдельные работы по окислению хинолина озоном в среде азотной и дру-
гих кислот с декарбоксилированием получаемой хинолиновой кислоты.
Описано [136] окислительное расщепление хинолина озоном в среде мине-
ральных кислот(HNO3, H2SO4, Н3РО4) или в среде уксусной в присутствии
азотной кислоты с выходом хинолиновой кислоты 90%; в частности, 10 мл
хинолина в 276 мл 60% -ной НNO3 нагревали до 40° С, а затем пропускали
Од—О2 (8% Од) и кипятили 2 ч. После отгонки азотной кислоты хинолиновую
кислоту выкристаллизовывали из воды. Выход хинолиновой кислоты
составлял 90%. Никотиновая кислота получена путем сублимации хиноли-
новой кислоты. Аналогичное исследование проведено Медниковым, Дзба-
новским, Воронковой и др. Оно заключалось в озонировании хинолина в
56%-ной HNO3 при температуре 70—75° С, кипячении 2 ч, отгонке HNO3,
декарбоксилировании при температуре 200—210° С и перекристаллизации.
Выход никотиновой кислоты достигал 64%.
Механизм озонолиза хинолина мало изучен. Предполагают, что в раство-
рах хлороформа и ледяной уксусной кислоты реакция в основном идет по
связям 5—6 и 7—8 с образованием устойчивого диозонида, превращаемого
в хинолиновую кислоту под влиянием окислителя, например азотной кисло-
ты, по схеме:
2[Оз] ;
Диозонид хинолина
и
с-н
с-н
II
о
Пиридин -2,3-диальдегад-
нитрат
^Х-СООН
L- Д-соон
N
OjN'" ХН
Нитрат хинолинодои:
кислоты
Диозонид и диальдегид хинолина не выделены и их существование в сво-
бодном виде неизвестно. Юркина, Русьянова и др. изучали механизм озо-
нолиза хинолина в различных растворителях (хлороформ, метанол, уксус-
ная кислота безводная и с добавлением воды) [137—140]. Ими было показа-
но, что наилучшим растворителем является 90%-ная уксусная кислота;
что гидролиз диозонида в воде происходит очень быстро; в растворе обнару-
жены пиридин-2,3-диальдегид, глиоксаль, щавелевая кислота и смолистые
продукты. Превращение диальдегида в никотиновую кислоту удалось авто-
рам достичь действием на него атомарным кислородом, получаемым при тер-
мическом разложении озона. Для этого раствор диозонида в 90%-ной ук-
сусной кислоте нагревали до 106° С и пропускали через него озон в присутст-
вии катализатора — ацетата кобальта. Таким образом, авторы разработали
одностадийный процесс превращения хинолина в никотиновую кислоту с
применением одного окислителя озона. Процесс проводится в два периода,
отличающиеся только температурными условиями. Оптимальными режима-
ми являются: для первого периода— температура 20—25° С, концентрация
уксусной кислоты — 90%, хинолина 100 г/л, расход озона 3 моля на 1 моль
хинолина; для второго периода — содержание воды 10%, количество ацета-
та кобальта 0,5—1,0% к массе хинолина, температура 106° С, расход озона
1 моль на 1 моль хинолина. Выход медицинской никотиновой кислоты сос-
тавляет 80%.
197
Расход химикалиев, кг на 1 кг никотиновой кислоты
Хинолин технический......................... 1,43
Уксусная кислота .............' .......... 1,25
Кислород, .и3................................2,40
Ацетат кобальта ............................ 0,02
Уголь активированный ........................0,06
Расход электроэнергии составляет 45,2 кет -ч на 1 кг никотиновой кис-
лоты.
Из приведенных данных следует, что озонолиз является весьма перспек-
тивным методом окисления хинолина. Однако для практического приме-
нения этого метода необходимы экспериментальные данные о степени взры-
ваемости промежуточных продуктов озонолиза хинолина, так как известно,
что органические озониды обладают взрывчатыми свойствами. Этот вопрос
можно будет решить положительно лишь после длительной эксплуатации по-
лузаводской установки с применением предупредительных устройств для
безопасной работы.
Другие окислители хинолина также исследовались, как например, пере-
кись водорода в присутствии уксуснокислой меди с выходом медной соли
хинолиновой кислоты 90% [141 ]; двуокисью марганца в концентрированной
серной кислоте при температуре 175° С [142]; смесь серной и азотной кислот
при температуре 300° С в присутствии катализатора окиси ртути с выходом
никотиновой кислоты через медную соль 88% [123].
По электрохимическому окислению хинолина имеются лишь отдельные
работы. В среде 50—70%-ной серной кислоты хинолин окисляется на аноде
из платины или окиси свинца при температуре 70° С с применением катали-
заторов V2O6, SeO2 и др. Выход хинолиновой кислоты достигал 77% [36].
Другие исследователи [52, 143 ] на свинцовом аноде при температуре 30—40°С
получили выход хинолиновой кислоты, равный 60—62%.
Указанные окислители и методы их применения не нашли практического
использования. Интерес представляет метод окисления хинолина кислоро-
дом воздуха в водно-щелочной среде под давлением 80 кгс/см2, с выходом в
54% [143а].
Синтез никотиновой кислоты из никотин-основания (пиридинметил-
пирролидина). Температура кипения 246° С; d^0 == 1,0099, алкалоид расте-
ния Nicotiana tobacum получают из отходов производства табака. Никоти-
новую кислоту получают окислением: азотной кислотой [143, 144], перман-
ганатом калия [145]; серной кислотой в присутствии селена [146], двуокисью
марганца в среде серной кислоты [147] гипохлоритом натрия [13]. Более
подробно см. [148, 149]. В настоящее время никотин-основание вследствие
Дефицитности и высокой стоимости потерял свое значение как сырье для про-
изводства никотиновой кислоты. По этой же причине и алколоид анабазин,
получаемой из растения Anabasis aphylla (пиридин-3-пиперидин, температура
кипения 276° С; d%° 1,0455) не нашел промышленного применения [148, 149].
Синтез никотиновой кислоты из 2,5-лутидина. В. Трубников показал
[95], что при парофазном окислительном аммонолизе лутидина (смеси ал-
килпроизводных пиридина) получен выход нитрила, близкий к выходам,
получаемым при раздельном окислительном аммонолизе. Отсюда автор
предложил лутидиновые фракции использовать как источник сырья.
ВЫБОР РАЦИОНАЛЬНОЙ ТЕХНОЛОГИЧЕСКОЙ СХЕМЫ
ПРОИЗВОДСТВА никотиновой кислоты
Выбор рационального метода производства связан с выбором вида сырья.
Само собой разумеется, что с технологической стороны наиболее эффектив-
ным видом сырья является |3-пиколин, так как переработка его на никотино-
вую кислоту или ее амид вполне осуществима по наиболее прогрессивному
методу — парофазному окислительному аммонолизу (стр. 188). Единствен-
198
ный недостаток — небольшие ресурсы природного |3-пиколина, а синтез его
не организован. Следовательно, при освоении синтеза дешевого |3-пиколина
вопрос о синтезе никотиновой кислоты и ее амида был бы решен однозначно
методом парофазного окислительного аммонолиза. Однако при дефицитнос-
ти р-пиколина приходится использовать менее эффективное с технологичес-
кой стороны, но более дешевое сырье — 2-метил-5-этилпиридин и хинолин;
из них наиболее перспективным является МЭП, как продукт химического
синтеза, требующий менее жесткого режима окисления. Выше (стр. 190) опи-
сан ряд методов окисления МЭП, из которых наиболее прогрессивным был бы
парофазный окислительный аммонолиз. Однако по этому методу имеются
пока лишь4 поисковые исследования недостаточно результативные. Наибо-
лее дешевым окислителем МЭП является азотная кислота. Из изложенного
(стр. 191) видно, что оптимальным вариантом для данного окисления явля-
ется одностадийный процесс под давлением при температуре 230° С с приме-
нением реактора-окислителя из тантала. Применение нержавеющей стали,
титана, эмалированной стали даже для менее жестких условий окисления
азотной кислотой под давлением (температура 175—180° С, концентрирован-
ная HNO3—10—20%) не является надежным. При отсутствии танталовых
реакторов окисление МЭП азотной кислотой в аппаратах из нержавеющей
стали или кислотоупорной эмали следует производить периодическим про-
цессом при атмосферном давлении, т. е. в условиях, позволяющих осущест-
влять систематический контроль за состоянием внутренней поверхности
реактора-окислителя.
Что касается хинолина, то при наличии ряда прогрессивных методов
окисления (парофазный окислительный аммонолиз, озонолиз, электрохими-
ческое окисление) ни один из них не разработан на уровне полузаводских
установок. С положительными результатами проверен в полузаводских усло-
виях периодический процесс окисления хинолина азотной кислотой в при-
сутствии серной кислоты с участием катализатора пятиокиси ванадия при
атмосферном давлении. Ниже описана технология производства никотино-
вой кислоты по методам наиболее изученным и опубликованным.
АМИД никотиновой кислоты
Из производных никотиновой кислоты наиболее важное физиологическое
значение имеет амид никотиновой кислоты. Он входит в состав молекул ко-
ферментов кодегидразы I (козимазы) и кодегидразы II (кофермент Варбурга).
Механизм действия указанных коферментов заключается в окислительно-
восстановительных реакциях.
Известны следующие методы синтеза никотинамида:
1. Амидирование эфиров никотиновой кислоты, заключающееся в этери-
фикации никотиновой кислоты и обработке полученного эфира аммиаком по
схеме:
СООН
N
с2н5он
н2$о4
Имеются различные модификации указанных реакций: а) этерификацию
никотиновой кислоты ведут различными спиртами: изопропиловым [150],
метиловым [151], этиловым, пропиловым, изопропиловым, бутиловым,
изобутиловым [152], при этом было показано, что с увеличением
молекулярной массы спирта выход эфира уменьшается, особенно для спир-
тов изостроения. В этой же зависимости изменяется выход никотинамида при
обработке эфира 30%-ным водным аммиаком: для метилового — 63%, эти-
лового— 43%, пропилового — 39,8%, изопропилового — 28,3%; б) реак-
цию амидирования ведут в растворе [150, 151, 153] или в отсутствии раст-
199
ворителя под давлением [154]; в) наилучшие результаты были получены при
молярных соотношениях никотиновой кислоты метилового спирта и серной
кислоты 0,81 : 6,1 : 1,53. Представляет интерес непрерывный процесс ами-
дирования эфира никотиновой кислоты при температуре 200—285°С и давле-
нии 49—98 кгс!см? [155а].
2. Непосредственное амидирование никотиновой кислоты газообразным
аммиаком в течение 2,5 ч при температуре 220—230° С при избыточном дав-
лении 27—28 кгс/см- с выходом 84% [71, 155]; менее эффективно амидирова-
ние при 180—185° С в течение 30 ч с выходом 78,8% [156].
3. Гидролиз нитрила, получаемого при парофазном окислительном аммо-
нолизе fl-пиколина или хинолина. Метод заключается в омылении 3-цианпи-
ридина в водно-щелочном растворе [157]. Никотинамид можно также полу-
чить при действии на 3-цианпиридин перекисью водорода в щелочной среде
[158]. Выход амида из нитрила, равный 64%, получен при нагревании вод-
но-аммиачного раствора нитрила до 42° С в течение 2 ч при участии КОН и
перекиси водорода [50, 51 ]. Наиболее прогрессивным является метод гид-
ратации нитрила при помощи ионитов в ОН-форме по следующей схеме:
„ Анионит
+ Н2О --------
conh2
К водному раствору нитрила добавляют в качестве катализатора ионо-
обменную смолу АВ-17, кипятят 70 мин, фильтруют и после выпаривания
выделяют амид никотиновой кислоты (выход 85,2%) [159]. Разработан так-
же ионообменный метод выделения никотинамида из технического продук-
та, полученного прямой амидизацией никотиновой кислоты [160, 161].
Метод заключается в следующем: в водный 10%-ный раствор, содержащий
(на сухое вещество в %): никотинамида 93,0, никотиновой кислоты 4,0 и
никотината аммония 3,0, загружают смесь смол КБ-1 в Н-форме и АВ-17
в ОН-форме в соотношении 1 : 5 по объему; после 2 ч контакта смолу
отфильтровывают, а фильтрат упаривают. Выход 95—97%.
Из рассмотрения методов, описанных выше, можно сделать вывод, что
наиболее целесообразным является непосредственное получение амида нико-
тиновой кислоты из fl-пиколина парофазным окислительным аммонолизом
с гидратацией 3-цианпиридина ионитом АВ-17 в ОН-форме (стадия выделе-
ния никотиновой кислоты отпадает). При превращении никотиновой кисло-
ты в никотинамид наиболее эффективным является метод непосредственного
амидирования никотиновой кислоты под давлением при температуре 220—
230° С. Этот метод дает высокий выход целевого продукта (84%) при односта-
дийном процессе.
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА НИКОТИНОВОЙ КИСЛОТЫ И ЕЕ
АМИДА ИЗ р-ПИКОЛИНА ПАРОФАЗНЫМ ОКИСЛИТЕЛЬНЫМ АММОНОЛИЗОМ
[47а, 50, 51]
Химическая схема процессов описана на стр. 189. Исследования [50] по-
казали, что лучшим катализатором для парофазного окислительного аммо-
нолиза является пятиокись ванадия, осажденная на окиси алюминия с до-
бавлением сернокислого калия, оптимальная температура окисления 300—
320° С, оптимальная нагрузка fl-пиколина на 1 л катализатора в 1 ч состав-
ляет 50 г; температура испарения fl-пиколина 35° С; количество fl-пиколина,
испаряемого 1 л воздуха — 0,03—0,05 г. Метод может быть рекомендован к
внедрению только по получении данных о взрывобезопасности при исполь-
зовании смесей паров fl-пиколина, аммиака и воздуха, а также о конструкции
контактного реактора. Технологическая схема предусматривает три стадии
200
синтеза: а) нитрила никотиновой кислоты; б) никотиновой кислоты и в) ни-
котинамида.
Нитрил никотиновой кислоты. В реактор-испаритель 1 колонного типа,
снабженный рубашкой для обогрева водой и каплеотбойником, непрерывно
из мерника 2 загружают снизу fl-пиколин. Сюда же подают воздух через
скруббер 3, влагоотделитель 4, компрессор 5, ватный фильтр 6 и счетчик 7.
В испарителе поддерживают температуру 35° С. Воздух с парами fl-пиколина
из испарителя 1 поступает в горизонтальный трубчатый перегреватель 8, куда
также поступают пары аммиака из баллона 9 через счетчик 10. Благодаря
электрообогреву смесь паров и воздуха нагревается в аппарате до 280—
Рис. 28. Технологическая схема производства никотиновой кислоты и ее амида из
P-пиколина парофазным окислительным аммонолизом.
300° С и с этой температурой поступает в вертикальный реактор-контактор
11, изготовленный из нержавеющей стали и снабженный электрообогревом.
В контактор загружают из сборника 12 ванадиевый катализатор, на окиси
алюминия, причем снизу и сверху слоя катализатора помещают насадку из
колец Рашига. В контакторе поддерживают температуру 300—320° С. Ре-
акционные пары и газы далее поступают в охладитель 13, представляющий
собой реактор с рубашкой, охлаждаемый водой. Отсюда конденсат продук-
тов реакции непрерывно стекает в приемник 14, частично наполненный во-
дой. Несконденсированные пары и газы из приемника 14 направляются в
ловушку 15, охлажденную рассолом, а из нее в ловушку 16, заполненную
водой. Получают водно-аммиачный раствор нитрила никотиновой кислоты
и около 19% непрореагировавшего fl-пиколина. Выход на прореагировав-
ший р-пиколин 93,6% [50], температура плавления 56—57° С (из воды).
Водно-аммиачный раствор нитрила из приемника 14 переводят в сборник 17,
откуда далее направляют на получение либо никотиновой кислоты, либо
амида.
Никотиновая кислота. Из сборника 17 водно-аммиачный раствор пере-
дают в реактор 18, снабженный обратным холодильником и водяной ловуш-
кой для поглощения выделяемого аммиака, загружают гидрат окиси бария
201
и кипятят до прекращения выделения аммиака. Затем из мерника 19 слива-
ют в реактор 18 разбавленную (1 : 1) серную кислоту и нейтрализуют ре-
акционную массу до pH 2—3. Осадок отфильтровывают на нутч-фильтре 20
в сборник 21. Фильтрат упаривают в вакуум-аппарате 22, сливают в крис-
таллизатор 23, кристаллизуют при минус 1—5° С; кристаллы отфуговыва-
ют в центрифуге 24 и высушивают в вакуум-сушилке 25, откуда они посту-
пают в сборник. При необходимости никотиновую кислоту перекристалли-
зовывают по трехступенчатой схеме (стр. 206). Выход — 50%.
Амид никотиновой кислоты. Его можно получать непосредственно
из водно-аммиачного раствора. Для этого его направляют из сборника 17
в реактор 26, снабженный обратным холодильником, куда добавляют в ка-
честве катализатора сильноосновную ионообменную смолу АВ-17 в ОН-
форме, кипятят 70 мин. Затем на нутч-фильтре 27 отфильтровывают смолу,
а фильтрат направляют в сборник 28 и далее в вакуум-аппарат 29, где упа-
ривают до сиропообразной консистенции, сливают в кристаллизатор 30
и кристаллизуют при 0°. Кристаллы отфуговывают в центрифуге 31, высу-
шивают в вакуум-сушилке 32, откуда через сборник 33 направляют на пере-
кристаллизацию по трехступенчатой схеме (стр. 206). Выход на нитрил сос-
тавляет 75%; на пиколин — 64,9% [50].
Подготовка анионита АВ-17 в ОН-форме [159—160]. Воздушно-сухой
анионит загружают в смеситель 34, заливают пятикратным количеством во-
ды, перемешивают 2—3 ч и выдерживают 8—10 ч при температуре 20° С,
сливают воду, промывают 2%-ным раствором соляной кислоты из мерни-
ка 35 до исчезновения ионов железа. После промывки водой смолу обрабаты-
вают 5%-ным водным раствором едкого натра из мерника 36 для превраще-
ния ее в ОН-форму; сливают щелочь и смолу промывают водой до нейтраль-
ной реакции по фенолфталеину. Затем смолу разгружают в сборник 37,
откуда переводят в реактор 26.
Приготовление катализатора для окислительного аммонолиза. [47а 50].
В реакторе 38, снабженном рубашкой, конденсатором и приемником, в 200 л
воды растворяют 7,5 кг метаванадатаммония и 4 кг сернокислого калия,
нагревают до 80—85° С при перемешивании. В горячий раствор засыпают
7,5 кг окиси алюминия (размер частиц 2—3 мм в поперечнике) и упаривают
до подвижной густой консистенции. Затем массу сливают в шнековую су-
шилку 39, высушивают при температуре 110° С и хранят в приемнике 12.
Перед употреблением каталйзатор активируют в реакторе-контакторе И
воздухом при температуре 400° С в течение 3—4 ч.
Необходимо подчеркнуть, что метод парофазного окислительного аммо-
нолиза, несмотря на его прогрессивность и современное аппаратурное
оформление, в первом своем варианте (с применением перекиси водорода и
этилацетата для получения никотинамида из нитрила) оказался бы нерента-
бельным по себестоимости.
Это лишний раз показывает, что несмотря на всю важность прогрессив-
ного аппаратурного оформления, решающее значение имеет рациональная
технологическая разработка процессов. Замена метода превращения нитри-
ла никотиновой кислоты в никотинамид перекисью водорода в щелочной сре-
де методом каталитической гидратации нитрила анионитом в ОН-форме обес-
печила этому методу большую эффективность и перспективу.
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА НИКОТИНОВОЙ КИСЛОТЫ ИЗ
2-МЕТИЛ-5-ЭТИЛПИРИДИНА ОКИСЛЕНИЕМ АЗОТНОЙ КИСЛОТОЙ ПОД ДАВ-
ЛЕНИЕМ
Как выше было указано (стр. 191), одностадийный синтез никотиновой
кислоты из МЭПа при температуре 230° С, давлении 35—40 кгс/см2, и содер-
жании HNO3 в реакционной массе 10% был бы наиболее эффективным, так
как при этой температуре одновременно протекал бы процесс декарбокси-
лирования; тепловой эффект более экономичен, так как при окислении вы-
202
деляется около 200 ккал/(г-моль), а при декарбоксилировании поглощает-
ся около 100 ккал!(г• моль); сокращается продолжительность реакции до
15 мин вместо 1 ч при температуре 180—185° С. Однако столь жесткий ре-
жим окисления может быть обеспечен лишь реактором-окислителем, изго-
товленным из тантала. При отсутствии тантала, по данным Уставщикова,
Фарберова и др. [82], можно использовать нержавеющую сталь марки
1Х18Н9Т для реактора-окислителя при концентрации азотной кислоты 10%;
температуре 190° С и избыточном давлении 18—20 кгс/см2. Однако опыт ис-
пользования стали указанной марки в этих условиях недостаточный, поэто-
му для положительного решения вопроса требуются дополнительные испы-
тания.
Технологическая схема, описываемая ниже, в основном базируется на
этих данных [82].
Химическая схема, следовательно, предусматривает две стадии синтеза:
Первая стадия протекает с выделением тепла 200 ккал/(г моль), вто-
рая— с поглощением тепла 100 ккал/(г-моль).
Условия процесса: концентрация азотной кислоты—10%; молярное
отношение МЭП : HNO3— 1 : 7; температура реакции 180—185° С, избы-
точное давление 18—20 кгс/см2, продолжительность 1 ч.
Окисление МЭП. В реактор-смеситель 1 (рис. 29) сливают из мерника 2
азотную кислоту (плотность 1350 кг/м3), из мерника 3 — воду, а из мерни-
ка 4 — МЭП в указанных выше соотношениях. Смесь фильтруют через ком-
муникационный фильтр 5 в сборник 6. Из последнего дозировочным насосом
7 раствор нагнетают в трубчатый подогреватель 8, а из последнего в реак-
тор-автоклав 9 трубчатой конструкции со специальной регулировкой темпе-
ратуры реакции. Продукты реакции поступают в холодильник-газосепара-
тор 10, охлаждаемый водой; после дросселировки давления до атмосферного
давления и охлаждения до 80—85° С они направляются в вакуум-аппарат 11
для сгущения в 3—4 раза. Из мерника 12 добавляют в вакуум-аппарат ед-
кий натр для доведения pH до 2,0. Массу сливают в кристаллизатор 13 и
при температуре минус 2° С выкристаллизовывают изоцинхомероновую и
частично никотиновую кислоту.
Выделяемые из сепаратора 10 газообразные продукты (NO, NO2 и СО2)
поступают в колонный поглотитель 14, орошаемый водой. Из кристаллиза-
тора массу передают в центрифугу 15 для выделения кристаллов.
Маточный раствор через/сборник 16 переводят в реактор-нейтрализатор
17. После нейтрализации маточного раствора щелочью до pH 8,0—9,0 на-
правляют в вакуум-аппарат 18, снабженный насадочной колонной, и отгоня-
ют не прореагированный МЭП, который направляют в мерник 19 и частич-
но (с учетом влажности) добавляют в реактор-смеситель 1. Кубовый остаток,
содержащий изоцинхомероновую и никотиновую кислоты направляют для
регенерации указанных кислот через их медные соли. Кристаллы, получен-
ные в центрифуге 15, направляют на декарбоксилирование.
Декарбоксилирование [80]. Кристаллы изоцинхомероновой с примесью
никотиновой кислоты растворяют в реакторе 20 в воде (1 : 3,5), передают в
автоклав 21 (наполнение 0,5) и нагревают при перемешивании в течение
1,5 ч до 200—205° С. Избыточное давление в автоклаве должно автомати-
чески регулироваться и поддерживаться в пределах 30—40 кгс/см2. По
окончании реакции давление в автоклаве дросселируют до атмосферного
203
через поглотительную ловушку со щелочью 22; массу разбавляют водой
(1: 0,5), сливают в реактор-смеситель 23, куда загружают активированный
уголь (2—3% к массе сухих веществ), нагревают до 65—70° С, перемешива-
ют 10—15 мин и сливают на нутч-фильтр 24. Фильтрат поступает в кристал-
лизатор 25, центрифугу 26; кристаллы направляют на перекристаллизацию
по трехступенчатой схеме (стр. 206). Маточный раствор из сборника 27 за-
сасывают в вакуум-аппарат 28, упаривают в 3—4 раза, сливают в кристал-
лизатор 29 (0°) и отфуговывают кристаллы в центрифуге 30. Кристаллы
никотиновой кислоты II направляют на перекристаллизацию совместное
Окисление МОЛ
Рис. 29. Технологическая схема производства никотиновой кислоты из
2-метил-5-этилпиридина окислением азотной кислотой под давлением.
техническим продуктом. Маточный раствор II из центрифуги 30 стекает в
сборник 31 и является отходом производства.
Переработка кубового остатка. [80]. Остаток, получаемый в вакуум-
аппарате 18 после отгонки, перерабатывают — осаждают изоцинхомероно-
вую и никотиновую кислоты через их медные соли. Для этого кубовый оста-
ток через сборник 32 переводят в реактор 33, куда вводят сульфат меди и
при перемешивании осаждают медные соли. Последние отфильтровывают
на нутч-фильтре 34; фильтрат поступает в сборник 35 и является отходом.
Осадок медных солей переводят в реактор 36, в который вводят едкий натр
из мерника 37, кипятят 30 мин и отфильтровывают выделяющуюся окись
меди на нутч-фильтре 38. Фильтрат через сборник 39 направляют в реактор
40, куда вводят из мерника 41 соляную кислоту и доводят pH до 2,0. Выде-
ляющуюся изоцинхомероновую кислоту отфуговывают в центрифуге 42
и направляют ее на декарбоксилирование в реактор 20. Маточный раствор
удаляют как отход через сборник 43. Общий выход никотиновой кислоты
204
50—60% (на МЭП). Установлена следующая химическая структура молеку-
лы медной соли изоцинхомероновой кислоты [81 ]:
О-----ОС
СО------1
Разложение медной соли изоцинхомероновой кислоты происходит по
следующей схеме:
(C7O4H4n)2Cu + 4NaOH-► ^>-COONa
NaOOC—+CuO+3H2Q
N
COOlNa conu
МаООсЛ- J *2HCl HOOcX J ' + 2NaCl
IN N
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА НИКОТИНОВОЙ КИСЛОТЫ ИЗ
2-МЕТИЛ-5-ЭТИЛПИРИДИНА ОКИСЛЕНИЕМ АЗОТНОЙ КИСЛОТОЙ В ПРИСУТ-
СТВИИ СЕРНОЙ ПРИ АТМОСФЕРНОМ ДАВЛЕНИИ [81, 86]
В условиях ненадежности конструкционных материалов для работы в
среде азотной кислоты под давлением удовлетворительным вариантом мож-
но считать периодический процесс окисления МЭП при атмосферном дав-
лении.
Химическая схема процесса см. стр. 193.
При изучении процесса окисления МЭП азотной кислотой в присутствии
серной и ванадиевого катализатора установлено [80], что наилучший выход
изоцинхомероновой кислоты достигается при температуре окисления 205—
210° С и применении азотной кислоты с концентрацией 57%. Ниже описана
лабораторная методика осуществления процессов по окислению МЭП и вы-
делению изоцинхомероновой кислоты [80].
К 15 мл концентрированной серной кислоты и 0,16 г ванадата аммония в
течение 4 ч при температуре 205—210° С приливают раствор 16,5 г МЭП в
210 мл 57 %-ной азотной кислоты. Затем в течение 2 ч при той же темпера-
туре добавляют еще 60—80 мл азотной кислоты. После 10-минутной выдержки
реакционную смесь охлаждают до 130° С, после чего ее выливают в 185 мл
холодной воды и подщелачивают едким натром до pH 2,0. При этом выпадает
белый осадок изоцинхомероновой кислоты, который отфильтровывают и вы-
сушивают. Выход составляет 58—60%, в том числе около 3% в виде мед-
ной соли.
Изоцинхомероновая (пиридин-2,5-дикарбоновая) кислота C7H5O4N, моле-
кулярная масса 167,11, кристаллы бесцветные, температура плавления
254° С (с разложением); трудно растворима в воде, спирте и эфире, растворя-
ется в кислотах с образованием четвертичных солей. Соли Na, К и NH4 хо-
рошо растворимы в воде; медная соль практически нерастворима.
Декарбоксилирование изоцинхомероновой кислоты см. стр. 203. Пере-
кристаллизация технической никотиновой кислоты см. стр. 206.
технологическая схема производства никотиновой кислоты из
ХИНОЛИНА ОКИСЛЕНИЕМ АЗОТНОЙ КИСЛОТОЙ В ПРИСУТСТВИИ СЕРНОЙ
КИСЛОТЫ и ВАНАДИЕВОГО КАТАЛИЗАТОРА [127, 128]
Химическая схема процесса изложена на стр. 196. Для разработки тех-
нологии процессов получения никотиновой кислоты из хинолина проводи-
лось изучение влияния количества добавляемой серной кислоты, рода ката-
205
лизатора, концентрации азотной кислоты на выход никотиновой кислоты,
а также технологии перекристаллизации технического продукта. В резуль-
тате было установлено: а) оптимальное количество серной кислоты, вводи-
мое в реакцию в объемном соотношении хинолин—серная кислота (1 : 1,5);
б) наилучшие результаты были получены при использовании в качестве ка-
тализатора порошкообразной пятиокиси ванадия в количестве 1% к массе
хинолина; в) концентрация азотной кислоты в 57% (плотность 1350 кг/м3)
признана оптимальной.
Ниже описана технологическая схема производства, в основу которой
положены указанные результаты [127].
В реактор 1 (рис. 30), снабженный рубашкой для масла или дитолилме-
тана, мешалкой с частотой вращения 200 об/мин и прямым холодильником
2, загружают 150 л серной кислоты плотностью 1840 кг/м? из мерника 3 и
100 л хинолина (температура кипения 236—238° С) из мерника 4\ затем до-
бавляют 1 кг пятиокиси ванадия, подогревают реакционную массу до 220—
230° С и начинают постепенно приливать азотную кислоту (регенерирован-
ную) плотность 1220 кг/м3 из мерника 5, а затем плотностью 1350 кг/м3 из
мерника 6 в соотношении 1 : 1, в пересчете на 100%-ную азотную кислоту
630 кг. Реакция окисления продолжается 12 ч при температуре 220—230° С,
после чего реакционную смесь выдерживают в течение 1 ч при той же темпе-
ратуре.
Реакционную массу разбавляют 250 л воды из мерника / и спускают в ре-
актор 8, где разбавленную реакционную смесь нейтрализуют 33 %-ным раст-
вором едкого натра из мерника 9 до pH 7,5—7,8 и охлаждают до 0° в
течение 3 ч для выделения сульфата натрия. Сульфат натрия отфильтровы-
вают в центрифуге 10, промывают ледяной водой и направляют в вальцовую
сушилку 11', безводный сухой сульфат расфасовывают в мешки 12. Фильт-
рат вместе с промывными водами направляют в сборник 13, откуда насосом
14 подают в реактор 15, где обрабатывают углем и фильтруют через нутч-
фильтр 16. Фильтрат поступает в сборник 17 и затем в вакуум-аппарат 18
для сгущения до содержания сухих веществ 44—46%.
Сгущенную массу спускают в кристаллизатор 19, охлаждают до 0° в
течение 2 ч для выделения остаточного сульфата. Выделившийся сульфат
отфильтровывают в центрифуге 20. Осадок после промывки ледяной водой,
направляют в сборник 21 и далее в сушилку 11.
Фильтрат из центрифуги 20 вместе с промывными водами поступает в
сборник 22, где его подогревают до 70° С. Далее фильтрат направляют в
реактор 24 для выделения технической никотиновой кислоты. Сюда подают
из мерника 25 соляную кислоту до pH 3,2—3,4, охлаждают до 0°, выдержи-
вают 8 ч, отфильтровывают техническую никотиновую кислоту в центрифу-
ге 26, промывают ледяной водой и направляют в сушилку 27. Маточный
раствор поступает в сборник 28, затем в реактор 29 для обработки углем;
уголь отфильтровывают в нутч-фильтр 30. Маточный раствор сгущают в
вакуум-аппарате 31, кристаллизуют в кристаллизаторе 32, отфильтровыва-
ют никотиновую кислоту в центрифуге 33 и промывают ледяной водой. Ни-
котиновую кислоту направляют в сушилку 27, а маточный раствор в сборник
28 а.
Перекристаллизация технической никотиновой кислоты. Первая кристал-
лизация. В реактор-смеситель 34 загружают никотиновую кислоту и дис-
тиллированную воду в отношении 1 : 15, подогревают до 95° С, добавляют
5% к массе кислоты активированного угля, перемешивают в течение 20мин.
Раствор сливают в нутч-фильтр 35, обогреваемый паром. Фильтрат направ-
ляют в кристаллизатор 36. В кристаллизаторе раствор кристаллизуют 8 ч
(4 ч без охлаждения и 4 ч при 0°), затем фугуют в центрифуге 37, осадок про-
мывают ледяной водой, спиртом и высушивают в вакуум-сушилке 38.
Вторая кристаллизация. Маточный раствор I из центрифуги 37 направ-
ляют в сборник 39, откуда насосом подают в' реактор 40, где обрабатывают
при температуре 75° С активированным углем (3% к массе содержащейся
206
hpowMcinSo технической никотиюбой кишты
Регенерации
В сборник 58
Рис. 30. Технологическая схема производства никотиновой кислоты из хинолина окислением азотной кислотой при атмосферном давлении.
в растворе никотиновой кислоты), а затем фильтруют через нутч-фильтр 41.
Фильтрат поступает в сборник 42 и далее в вакуум-аппарат 43 для сгу-
щения; сгущенный раствор кристаллизуют в кристаллизаторе 44 в течение
8 ч при температуре 20°С и 8 ч при 0°, а затем отфильтровывают никотиновую
кислоту в центрифуге 45. Никотиновую кислоту II направляют в реактор-
растворитель 34 для перекристаллизации.
Третья кристаллизация. Маточный раствор II собирают в приемнике 46г
обрабатывают в реакторе 47 активированным углем при температуре 60° С,
фильтруют через нутч-фильтр 48. Фильтрат направляют в сборник 49, сгу-
щают в вакуум-аппарате 50. Сгущенный раствор кристаллизуют в кристалли-
заторе 51 в течение 30 ч при температуре 20° С и 18 ч при 5° С, а затем фугу-
ют в центрифуге 52. Маточный раствор III поступает в сборник 53 и являет-
ся отходом производства. Кристаллы никотиновой кислоты третьей кристал-
лизации направляют в реактор-растворитель 40 для перекристаллизации.
Регенерация азотной кислоты. При окислении пары азотной кислоты
конденсируются в холодильнике 2 и собираются в приемнике 54, откуда
азотную кислоту плотностью 1220 кг!мъ возвращают на окисление в мерник 5.
Окислы азота поглощаются в колонке 55, наполненной кольцами Рашига
и орошаемой водой из мерника 56. Разбавленная азотная кислота, получае-
мая при поглощении NO2 водой, насосом 57 перекачивается в мерник 56
и систематически циркулирует до получения кислоты плотностью,
1100 кг!мй, после чего ее направляют в сборник 58, откуда подают на разгон-
ку в перегонный аппарат 59. При аппарате имеются три сборника, в которые-
собирают азотную кислоту трех сортов.
ТЕХНОЛОГИЧЕСКИЕ ПОКАЗАТЕЛИ
РАСХОД ХИМИКАЛИЕВ, КГ НА 1 КГ НИКОТИ-
НОВОЙ КИСЛОТЫ (НИКОТИНАМИДА) И ВЫХОД
ПОЛУПРОДУКТОВ, % ОТ ТЕОРЕТИЧЕСКОГО
Синтез из пиридина методом цианирования
Пиридин (с регенерацией) ................. 1,7
Бром (с регенерацией)......................4,3
Поташ .................................... 2,2
Натр едкий ............................... 1,7
Медь сернокислая ......................... 3,0
Медь цианистая............................ 1,2
Кислота соляная . ........................ 3,2
Итого . . . 17,3 кг
Выход (в %): З-Бромпиридина...............30,0
З-Цианпиридина..................50,0
Никотиновой кислоты (на пири-
дин) .......................... 13,5
Синтез из ^-пиколина парофазным окислительным аммонолизом
а) никотиновая кислота
^-Пиколин................... 1,5
Аммиак........................5,6
Гидрат окиси бария восьмивод-
ный .........................12,0
Кислота серная ............. 4,0
Уголь активированный . . . .0,15
Ванадат аммония..............0,02
Калий сернокислый .... 0,01
Окись алюминия.........0,02
Итого .... 23,3 кг
Выход (в %): нитрила никоти-
новой кислоты (на р-пиколин) . 65,0
Никотиновой кислоты в пере-
счете
на нитрил..................77,0
на р-пиколин .............50,0
208
б) никотинамид с гидратацией нитрила
ионообменной смолой
3-Пиколин .................1,5
Аммиак.................5,6
Ионообменная смола АВ-17 . 2,0
Кислота соляная ...........0,3
Натр едкий.............0,3
Уголь активированный . . 0,15
Спирт этиловый..........0,5
Ванадат аммония.........0,02
Калий сернокислый .... 0,01
Окись алюминия ............0,02
Выход (в %):
Нитрил никотиновой кислоты . 65,0
Амид никотиновой кислоты
технический (иа р-пиколин) . . 57,0
Амид никотиновой кислоты ме-
дицинский ....................52,0
Итого . . . 10,4 кг
Синтез из 2-метил-5-этилпиридина
а) окислением 10%-ной азотной кислотой под давлением
2-Метил-5-этилпиридин . . 2,0 Выход (%):
Азотная кислота 57%-ная . 8,0 Изоцинхомероновая кислота
Натр едкий 42%-ный . . . 0,2 (на МЭП) 65,0'
Соляная кислота 0,3 Никотиновая техническая кис-
Сульфат меди 0,05 лота (на изоцинхомероновую) . 87,0
Уголь активированный . . . 0,15 Никотиновая медицинская в
Спирт этиловый 0,5 пересчете
на техническую .......89,0
Итого ... 11,2 кг на МЭП ......................50,3
б) окислением азотной кислотой
ванадиевого катализатора при атмосферном
в присутствии серной и
давлении
2-Метил-5-этилпиридин .... 2,0
Азотная кислота ............ 25,0
Серная кислота 94%-ная ... 3,0
Соляная кислота ............. 0,3
Натр едкий (42%-ный) .... 0,2
Спирт этиловый...............0,2
Сульфат меди .................0,05
Ванадат аммония...............0,02
Уголь активированный .... 0,15
Выход (в %):
Изоцинхомероновой кисло-
ты (на МЭП)................68,0
Технический никотиновой
кислоты (на изоцинхомеро-
новую кислоту).............87,0
Медицинской никотиновой
кислоты в пересчете
Итого . . . 30,92 кг
на техническую кисло-
ту ....................89,0
на МЭП ................52,6
Синтез из хинолина
а) окислением азотной кислотой в присутствии серной и
ванадиевого катализатора при атмосферном давлении
Хинолин технический . . . 2,6 Выход (в %):
Азотная кислота 56%-ная . 13,8 Технической никотиновой кис-
Серная кислота техническая лоты (на хинолин).............50,8
96%-ная ................6,6 Медицинской никотиновой кис-
Натр едкий 42%-ный . . . 17,0 лоты (на хинолин).............45,2
Соляная кислота 31%-ная . 1,0
Пятиокись ванадия 98%-ая 0,02
Уголь активированный . . 0,15
Итого. . . . 41,17 кг
б) окислением озоном
Хинолин технический ... 1,43
Уксусная кислота .......... 1,25
Кислород, м3..........2,40
Ацетат кобальта ............0,02
Уголь активированный . . 0,15
Итого... 5,25 кг
Расход электроэнергии составляет 45,2 квт ч.
Выход медицинской никотиновой кислоты (на хинолин), % ..... 80
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. F и п к С., J. Physiol., 1913, 46, 173; 1913, 1, 814.
2. D г u m m о п d J., F и п к С., Biochem., 1914, 8,594.
3. S и z и к i U., Sh a m i m и г а Т., О к a d е S., Biochem. Z., 1912, 43, 89, 99,.
209
4. Chittenden R., Underhill F., Am. J. Physiol., 1917, 44, 13.
5. G о 1 d b e r g e r J., L i 1 1 i e R. U. S. Pub. Health Service Pub. Health Repts
1926, 41, 1025.
6. Rosenberg H., Chemistry and Physiology of the Vitamins, N. Y., 1942.
7. Aykroyd W., R о s с о 1 M. Biochem. J. 1929, 23, 483.
8. Norris L., Ringrose A. Science, 1930, 71, 643.
9. Warburg О., C hr i st io n W. Biochem. Z., 1935, 275, 46.
10. Warburg O., Christion W., G r i e s e A. Biochem. Z., 1935, 279, 143.
11. Euler H., Albers H., Schlenk F., Z. Physiol. Chem., 1935, 237, 1.
12. F о u t s P., Helmer O., L e p к о v s к у S., Jukes T., Proc. Soc., Exptl.
Biol. Med., 1937, 37, 405.
13. Преображенский H. А., Г e н к и н Э. И. Химия органических и ле-
карственных веществ. М., Госхимиздат, 1953, с. 126.
14. Р ы с с С. М. Витамины. Л., Медгиз, 1955.
15. X а е с С. Влияние никотиновой кислоты на содержание сахара, гликогена и мо-
лочной кислоты крови. Автореф. кандид. дисс., Горький, 1956.
16. Ш и л о в П. И., Яковлев Т. Н. Основы клинической витаминологии. М.,
«Медицина», 1964, с. 65, 179.
17. Главвитаминпром (редакц. А. Гессена). Никотиновая кислота и ее практическое
применение. Л., 1953.
18. G о г d i n g R., F 1 e x s e r L. J. Am. Pharm. Assoc., 1940, 29, 230.
19. Б у к и н В. H. Значение витаминов в народном хозяйстве. — В сб.: «Труды Все-
российского совещания по витаминам», Уфа, Башиздат, 1963, с. 1—10.
20. S е b г е 1 1 W., Harris R. The Vitamins N. Y., 1954, 2, 1452.
21. Слободин Я. M., Гольдман М. М. Растворимость никотиновой и изо-
никотииовой кислот. —ЖПХ, 1948, т. 21, с. 859—861.
22. К а г г е г Р., Keller Н., Helv. Chim. Acta, 1939, 22, 1292.
23. Kuhn R., Vetter H. Ber., 1935, 68, 2374.
24. H ii n e c k e H. Ber., 1927, 60, 1451.
25. Hugnes E., lellinek H., Ambrose B. J. Phys., Colloid. Chem., 1949,
53, 414.
26. Fischer O., Ber., 1882, 15, 63.
27. M с E 1 v a i n S., G о e s e M., J. Am. Chem. Soc., 1941, 63, 2283; 1943, 65, 2233.
28. Ч у м а к о в Ю. И. Методы получения химических реактивов и препаратов,
ИРЕА, М, 1963, 7, 86.
29. К г о s М., Пат. ФРГ 828246, 1953, С. А., 1956 50, 3504.
30. S hive W., Glenn Р. Пат. США 2409806, 1946; С. А., 1947, 41, 2087.
31. Чумаков Ю. И. Пиридиновые основания. «Техника», Киев, 1965, с. 42.
32. Wei del Н., Вег., 1879, 12, 1992, 2004.
33. Ost Н., J. prakt. Chem. (2) 1883, 27, 286.
34. S е у f f е г t h Е., J. prakt. Chem. (2), 1886, 34, 258.
35. W о о d w a r d C., Badgett C., Willman J. Ind. Eng. Chem., 1944, 36,
540.
36. ; К u 1 k a M., J. Am. Chem. Soc., 1946, 58, 2472.
37ДР убцовМ. В., Яхонтов Л. Н., Яценко С. В. Получение никотиновой
кислоты из ^-пиколина—отхода от производства фтивазида.— ЖПХ, 1957, т. 30,
с. 315—318.
38. В 1 а с k е G., Depp Е., Corson В. J. org. Chem., 1949, 14, 114.
394O'g i 1 v i 1 J., S w e e t А. Пат. США 2415147; С- A., 1947, 41, 2754.
40. C i s 1 a к F., Wheeler W. Пат. США 2396457, 1945; С. A., 1946, 40, 3142.
41. P 1 a z e к E., Kozdrojowna H. Roczniki Chem., 1951, 25, 509; C. A., 1954,
48, 5, 5863.
42. Woodward E., Badgett C. Kaufman J. Ind. Eng., Chem., 1944,
36, 544.
43. C i s 1 a к F., Wheeler W. Пат. США 2522163, 23/Ш 1942; Off. gaz 1950, 638,
№ 2, 522.
44. C i s 1 a к F., Wheeler W. Пат. США 2437938, 1948; С. A., 42, 4204.
45. L e’w i s R., Brown O. Ind. Eng. Chem. 1944, 36, 890.
46. M a" у urnik G., Moschetto А. и др., Ind. Eng. Chem., 1952, 44, 1630.
47. H a d 1 e у D., Wood В. Аигл . пат. 777746, 1957; РЖХ, 1959, 39758.
47a. Жданович E. С., Галкин А. Ф., Чекмарева И. Б. и др. Полу-
чение пиридиикарбоновых кислот.— ЖПХ, 1959, т. 32, с. 2821—2822. То же. С. А.,
1960, 54, 9920.
48. Р а ф и к о в С. Р., Суворов Б. В., Жубанов Б. А. и др. Синтез ни-
котиновой кислоты и ее амида через никотиноиитрил.—«ДАН СССР», 1959, т. 126,
1286—1288 с ил.
49. С у в о р о в Б. В., Рафиков С. Р., Жубанов Б. А., X м у р а М. И.
Авт. свидет., № 119878, 1958; Бюлл. изобрет., 1959, № 10, с. 15. То же. Авт. сви-
дет. № 123155, 1959; Бюлл. изобрет., 1959, № 20, с. 17.
50. Жданович Е. С., Чекмарева И. Б., Преображенский Н. А.
Получение нитрила и амида никотиновой кислоты. —ЖОХ, 1961, т 30, вып. 9,
с. 3272—3274 с ил.
210
51. Пат. ГДР 828246, 1953; С. А., 1956, 50, 39504.
52. Б о р х и Л. Д., Хомяков В. Г. Авт. свидет., № 187024, 1965, Бюлл. изо-
брет. 1966, 20, с. 38.
53. X о м я к о в В. Г., Кругликов С. Авт. свидет. № 122147, 1959; Бюлл.
изобрет., 1959, № 17, с. 14.
54. Кругликов С., Хомяков В. Г. — «Труды Московского химико-техно-
логического института им. Менделеева», 1961, 32, с. 194.
55. Кругликов С., Хомяков В. Г., Бахчисарайцьяи Н. Авт.
свидет., № 140062, 1961; Бюлл. изобрет., 1961, № 15, с. 19..
56. П о л я к о в а И. М. Получение р-пиколина из легких пиридиновых основа-
ний. — ЖПХ, 1947, т. 20, с. 845—853 с ил.
57. Магидсон О. Ю. Достижения и перспективы химического синтеза витами-
нов. — В сб.: «Труды Всесоюзной конференции по витаминам», Изд-во АН СССР,
1940, с. 106—115.
58. С i s 1 a k F. Пат. США 2272159; С. А., 1942, 36, 3514.
59. С i s 1 а к F., Wheeler W., Пат. США 2430804; С. А., 1948, 42, 1322.
60. R i с t h о f G. Пат. США 2295606; С. А., 1948, 42, 1131.
61. J 6 п е s J. Аигл. пат. 598036; С. А., 1948, 42, 4614.
62. S 1 a g е К-, В о w m а п R. Пат. США 2452191; С. А., 1949, 43, 3043.
63. В о w m а п R. Пат. США 2459359; С. А., 1949, 43, 3043.
64. Karr A., Scheibel Е. Ind. Eng. Chem., 1954, 46, 1583.
65. S t i t z F., Osterr. Chem., Ztg., 1942, 45, 159.
66. С и л и и г M. И., Шиайдмаи Л. О. Синтез никотиновой кислоты из 2-ме-
тил-5-этилпиридииа. — В сб.: «Витамины (по зарубежным материалам)», М., Гос-
инти, 1960, № 6, 3—15.
67. Ч и ч и б а б и н А. Е., М о ш к и н П. А., Тяжелова Л. С. ЖРФХО,
1920, 54, 413.
68. Frank R., BlegenJ. и др. у. Am. Chem. Soc., 1946, 68, 1368.
69. N а г a s a k i Н., S u d s и k i Н. Repts. Govt. Chem. Industr. Res. Inst. Tokyo,
1957, 52, 1, 8; РЖХим, 1958, 29709.
70. D и n n J. Англ. пат. 706816, 1954; 742268, 1955, J. Appl. Chem. 1954, 4, 635;
C. A., 1956, 50, 16874; РЖХим; 1957, 5787.
71. Aral K., Osyka, K- Tanabe, F e r a m e t о , I m i к i d s a к i,
J. Chem. Soc. Japan, 1954, 57, 495; РЖХим, 1955, 45897.
72. J a r d a n T., Ind. Eng. Chem., 1952, 44, 332.
73. S e b r e 1 1 W., Harris R., The Vitamins, New York, 1954, 2, 479.
74. M u e 1 1 e r M. Пат. США 2793213, 1957; РЖХим, 1959, 50793.
75. F e u b e r W., Laboratoriums — Praxis, 1957, 9, 64.
76. Aries R., Sachs А., Пат. США 2708196 (1955); РЖХим, 1957,17063.
77. A r i e s R. Пат. США 2702802, 27/ХП 1951; Off. gaz. 1955, 691, № 4, 564.
78. Burrows L., Herring H. Пат. США 2524957, 15/V 1947; Off. gaz. 1950,
639, № 2, 404.
78a. Stager R. Швейц, пат. 336066, 1959, С. A., 1960, 54, 19719.
79. Швейц, пат. 297015, 1954.
80. Ш и а й д м а и Л. О., С и л и н г М. И. Синтез изоцинхомероновой и никоти-
новой кислот из 2-метил-5-этилпиридииа. — «Труды ВНИВИ», М., Пищепромиз-
дат, 1961, № 7, с. 18—26 с ил.
81. Ш н а й д м а н Л. О. Исследования в области химии и технологии производства
витаминов. Доклад, докт. дисс., М., 1967, с. 30.
82. Уставщиков Б. Ф., Титова Т. С., Дегтярев Е. В., Ф а р б е-
р о в М. И. Технический синтез никотиновой кислоты окислением 2-метил-5-
этилпиридина разбавленной азотной кислотой. —ЖПХ, 1966, т. 39. с. 1388—1393
с ил.
83. 11 1 i с h G. Пат. США 2905608, 1959; С. А., 1960, 54, 2369.
84. Lonza Electric. Chem. Works Ltd., Англ. пат. 757958, 24 VIII/1954; Abridgments,
1956.
85. T a n a b e К., A r a i К. Япон. пат. 2730, 1953; С. A., 1955, 49, 2523.
86. Ш н а й д м а и Л. О., С и л и н г М. И. Синтез изоцинхомероновой кислоты
окислением 2-метил-5-этилпиридииа азотной кислотой при атмосферном давлении.—
«Труды ВНИВИ», М., Пищепромиздат, 1961, № 8, с. 5—11 с ил.
87. Уставщиков Б. Ф., Фарберов М. И., КутьинА. М. и др. Уче-
ные записки Ярославского технологического института, 1960, . 5, 71.
88. Б а р а и о в а Т. И., К У т ь и и А. М., Фарберов М. И., Уставщи-
ков Б. Ф. Авт. свидет., № 161755, 1963; Бюлл. изобрет., 1964, № 8, с. 20.
89. Hiroshi Sob u е, Akiza, F о m i t а и др. Япон. пат. 13081, 1959; С. А.,
1964, 60, 570.
90. S с h w а г z е W., Пат. ФРГ, 1010524, 1957; С. А., 1959, 53, 18970.
91. Kato Т., Tsunoda V., К о g у о К agaki Lasshi, 1960, 63, 1278;
С. А., 1962, 56, 10091.
92. А г a i A., Inst. Phys. Res., 1957, 7, 116; РЖХим, 1958, 77665.
93. М а у г n i k G., Moschetto A., Bloch H., J. S c u d i, Ind. Eng. Chem.,
1952, 44, 1630.
211
• 94. W e t t s t e i n W. Пат. США 2845428, 1958; С. A., 1959, 53, 3250.
• 95. Трубников В. И. Исследования в области парофазного окислительного ам-
монолиза производных пиридина. Автореф. каидид. дисс., М., 1969; Трубни-
ков В. И., Жданович Е. С. и др. Фонды ВИНИТИ, 1969, 452—469.
96. Суворов Б. В., Рафиков С. Р., Кагарлицкий А. Д. Окисли-
тельный аммонолиз органических соединений. — «Успехи химии», 1965, т. 34, 9,
с. 1526—1549 с ил.
• 97. Krohs W. Пат. ФРГ, 828246, 1953.
98. R о s е п b е г g И. Пат. США 2446957, 1948; С. А., 1949, 43, 694.
•99. D u е s е 1 В., Friedmann Н. Пат. США 2471518, 1949; С. А., 1949, 43, 7513.
100. Gasson Е., Hadley D. Англ. пат. 777517, 1956; С. А. 1958, 52, 7361,
101. J. de Kamp. М. Т i s h 1 е г. Пат. США 2435809; С. А., 1948, 42, 2732.
102. Couch J., Krewson Ch.. Пат. США 2453496, Off. gaz., 1948, 616, № 2,
499, 19/X 1943; С. A., 1949, 43, 1811.
103. Martin А. Пат. США 2694070; 1954, С. A. 1956, 50, 4237.
104. С о r b e 1 1 i n i A., Paoli E. Итал. пат. 629859, 1961; С. A., 1962, 57, 15080.
105. H 6 f 1 i ng W., E i 1 h а п e r D., Reckling G., Andreas F., Швейц,
пат. 411879, 30/1 1963; Pat.— Liste, 1966, № 8, 418.
106. Garcia E., Greco C., Hunsberger I., J. Am. Chem. Soc., 1960, 82,
4430.
107. Hawkinson A., Elston А. Пат. США 2371691, 1945; С. A., 1945, 39,
4336.
108. Benner R. Пат. США 2829144, 14/IV 1955; Off. gaz. 1958, 729, № 1, 168.
109. Roche Prod. Ltd. Англ. пат. 714664, 1954; С. A., 1951, 45, 9054.
ПО. T а и a b е К-, А г a i К- Япои. пат. 2734, 1953; С. А., 1955, 49, 2523.
111. Н а р р f Н., Krieger A., Helv. Chim. Acta, 1961, 44, 1058.
112. В г о с h t а I. Пат. США 2916494, 15/IV 1958; Off. gaz. 1959, 749, № 2, 476.
ИЗ. Англ. пат. 855819, 1960; С. А., 1961, 55, 12430.
114. Р е г г е г R., Moser W. Швейц, пат. 352332, 4/Ш 1957; Pat.—List. 1961, X» 1,
123.
.115. П о т а ш и и к о в М. М. Исследования в области выделения и использования
соединений каменноугольной смолы. Автореф. докт. дисс., Нижиий Тагил, 1962.
116. Woodward С., Badgett С., Willman I. Ind. Eng. Chem. 1944, 36,
540.
117. M u e 1 1 e г M. Пат. США 2436660, 1948; С. A., 1948, 42, 4203.
.118. P о r t e r F., Bumpus M., Cosby J. Пат. США 2513251, С. A., 1950, 44,
8380.
119. T e e t e r s W. Пат. США 2476004, 1949; С. A., 1949, 43, 8402.
120. Англ. пат. 718007, 30/X 1952; J. Appl. Chem., 1955, 5, 782.
121. M u e 1 1 e г M. Пат. США 2449906, 21/XI 1944; Offic. gaz. 1948, 614, № 3, 768.
122. L a r r i s о n M. Пат. США 2475969, 1949; С. A., 1950, 44, 2038.
.123. Saha S., J. Indian. Chem. Soc., 1956, 2, 95; C. A., 1958, 52, 18407.
124. Лин Си-Цзи, ж. Хуасюэ Шицзе, 1955, 6, 271; РЖХим, 1958, 2,
5560.
425. Жданович Е. С., Чекмарева И. Б., Кап л ин а Л. И., Пре-
ображенский Н. А. Авт. свидет., № 148411, 1961; Бюлл. изобрет., 1962,
№ 13, с. 16.
126. Чекмарева И. Б., Жданович Е. С., Преображен-
ский Н. А. Получение p-пиридинкарбоновой (никотиновой) кислоты. —«ЖПХ»,
1965, т. 38, с. 220—221.
127. Шнайдман Л. О., Кондрашова А. .А., БалаценкоС. В. Син-
тез никотиновой кислоты из хинолина. — «Труды ВНИВИ», М., Пищепромиздат,
1961, № 7, с. 27—36 с ил.
.128. Кондрашова А. А., Шнайдман Л. О., Б а л а ц е и к о С. В.,
Жданович Е. С. Авт. свидет. № 132225, 1960; Бюлл. изобрет., 1960, № 19,
с. 22.
.129. Трубников В. И., Петров В. В., Жданович Е. С., П р е о б-
жеиский Н. А. Синтез никотиновой кислоты и ее амида методом парофаз-
ного окислительного аммонолиза хинолина. — ХФЖ, 1969, 9, с. 49—52 с ил.;
Суворов Б. В., Рафиков С. Р., КагалицкийА. Д. и др. Авт.
свидет., № 235764; Бюлл. изобрет., 1969, № 6, с. 27.
130. Жданович Е. С. Исследование в области синтеза и разрабоки методов про-
изводства витаминов группы В. Доклад, докт. дисс., М., 1965.
131. Чекмарева И. Б., Трубников В. И., Жданов и'ч Е. С. Ана-
лиз продуктов парофазного окислительного аммонолиза. — В сб.: «Вестник тех-
нической и экономической информации», НИИТЭХИМ, М., 1965, № 5, с. 5.
.132. Чекмарева И. Б., Трубников В. И., Жданович Е. С. и др.—
«Вестник технической и экономической информации», НИИТЭХИМ, М., 1964,
№ 10, с. 26.
.133. Т р у б и и к о в В. И., Малахов'а Л. М., Жданович Е. С., Пре-
ображенский Н. А. Газо-жидкостная хроматография карбоновых кислот
пиридинового ряда и их амидов. — «Хим.-фарм. ж.», 1967, № 12, с. 14—18 с ил.
212
То же, 1968, № 1, с. 31.
134. V о m a t s u F., (Япония), С. A., 1960, 54, 2911.
135. Юркииа Л. П., Русьяиова Н. Д., Малышева Н. — «Сб. статей
ВУХИН», Свердловск, 1967, № 4, с. 259.
136. Sturr ock М., Cline. Е., Robinson К., Zercher К. Пат. США.
2964529, 1960; С. А., 1961, 55, 11443.
137. Юркина Л. П. Получение никотиновой кислоты озонолизом хинолина.
Свердловск, Автореф. кандид. дисс., 1968.
138. Юркииа Л. П., Русьянова Н. Д., Малышева Н. В. — В сб.:
«Химические продукты коксования углей», ВУХИН, Свердловск, 1967, № 4,
с. 251.
139. Русьяиова Н. Д., Малышева Н. В., Юркииа Л. П. Там же,
1967, 4, 231.
140. Р у с ь я н о в а Н. Д., Малышева Н. В., Юркина Л. П., Конд-
ратов В. К- Авт. свидет., № 191562, 1965; Бюлл. изобрет., 1967, № 4, с. 35.
141. Hawkinson A., Elston А. Пат. США 2347410, 27/XI 1941; Off. gaz.
1944, 501, № 3, 608.
142. Kamp V., Sletzinger. Пат. США 2392437; С. А., 1946, 40, 2473.
143. Мак-Э льве н. — В сб.: «Синтезы органических препаратов», М., ИЛ., 1949,
1, 228.
143а. Говорова Л. М., Проскуряков В. А., Чистяков А. Н.
Окисление хинолина кислородом воздуха в водно-щелочной среде. — ЖПХ, 1970,
т. 43, вып. 10, с. 2356—2360 с ил.
144. Ш м у к А. А. Получение никотиновой кислоты (витамина РР). — ЖПХ, 1947,
т. 20, с. 245—250 с ил.
145. L a i bl i n R., Вег 1877,10,2136.
146. Е 1 v a i n М., G о s е М. J. Am. Chem. Soc., 1941, 63, 2283.
147. В а с'ю и и и а Н. А,, Б е э р А. А., Преображенский Н. А. — ЖПХ,
1943, 16, 206.
148. Шнайдмаи Л. О. Производство витаминов. М., Пищепромиздат, 1958,
с. 336.
149. Березовский В. М. Химия витаминов М., Пищепромиздат, 1959, с. 343.
150. Lustig О. Пат. США 2752355, 1956, РЖХим. 1958, 33593.
151. Чекмарева И. Б., ЖдаиовичЕ. С., Новопокровская Т. С.,
Преображенский Н. А. Получение амида р-пиридинкарбоновой (ни-
котиновой) кислоты. —ЖПХ, 1962, т. 35, с. 1157—1159.
152. Чекмарева И. Б. Жданович Е. С., Преображенский Н. А.
Исследования в области получения амида никотиновой кислоты. — «Медицинская
промышленность СССР», 1965, № 7, с. 11.
153. F г е i f е 1 d е г М., I 1 1 i с h G., Robinson R. Пат. США 2923715, 1960;
С. А., 1960, 54, 10247.
154. Англ пат. 822579, 1959; С. А., 1960, 54, 4632.
155. Т г и с h a n A., Davidson I. Пат. США 2993051, 1961; С. А., 1961, 55;
23942.
155а. Michael W., фирма Abbot, пат. ФРГ, 1290938, 1969; Ausziige, 1969, № 12.
156. Жданович Е. С., Чекмарева И. Б., Новопокровс-
кая Т. С. и др. Получение амида р-пиридинкарбоновой кислоты. —ЖОХ,
1962, т. 32, с. 2828.
157. Dues el В., Friedman Н. Пат. США 2471518, 28/VIII 1942; Off. gaz.
1949, 622, № 5, 1409.
158. Couch J., Krecvson С. Пат. США 2453496, 1948; С. A., 1949, 43, 1811.
159. Чекмарева И. Б., Жданович Е. С., Сазонова Г., Преоб-
раженский Н. А. Авт. свидет., № 164601, 1963; Бюлл. изобрет., 1964,
№ 16, с. 11.
160. Ч е к м а р е в а И. Б., Жданович Е. С., Л у щи к Н. А., Преоб-
раженский Н. А. Выделение амида никотиновой кислоты методом ион-
ного обмена. —ЖОргХ, 1965, т. 1, с. 375.
161. Ч е к м а р е в а И. Б., Жданович Е. С., Паньшина Т. А., Пре-
ображенский Н. А. Авт. свидет. № 170509, 1963; Бюлл. изобрет., 1965,
№ 9, с. 24.
Глава 10. ПРОИЗВОДСТВО СИНТЕТИЧЕСКОЙ ФОЛИЕВОЙ КИСЛОТЫ
[1, 2, 3]
Фолиевая кислота является витамином, стимулирующим и регулирую-
щим процессы кроветворения. Она обладает антианемическими свойствами.
Антианемические препараты, которые преимущественно приготовлялись из
дрожжей и печени, носили различные названия: фактор Билля [4], витамин
213
М [5], фактор U [6], витамин Вс[7], L. casei — фактор [8]. Название «фо-
лиевая кислота» (лат. folium — лист) было дано в 1941 г. препара-
ту, полученному из шпината [9], который стимулировал рост Streptococcus
faecalis R. и L. casei.
В последнее время установлено, что антианемическими свойствами об-
ладают ряд химических соединений птеридинового ряда, к которому отно-
сится фолиевая (птероиглутаминовая) кислота. К этому же ряду относится
фолиновая кислота, которая по активности примерно в 100 раз сильнее фо-
лиевой кислоты.
Фолиевая и фолиновая кислоты излечивают макроцитарную (тропичес-
кую) анемию и спру [101. Помимо анти анемического действия они катали-
зируют синтез белков (аминокислот), участвуют в обмене холина, применя-
ются при лечении злокачественной (пернициозной) анемии, а также при за-
болеваниях крови, вызванных радиоактивным облучением.
В процессе эндогенного образования холина и метионина активное учас-
тие принимает фолиевая кислота. Этим самым фолиевая кислота способст-
вует предупреждению жировой инфильтрации печени при нарушении жиро-
вого обмена. Фолиевая кислота понижает потребность организма в холине
[10]. ’Суточная потребность человека в фолиевой кислоте составляет
0,3—0,5 мг [11]. Лечебная доза 5—10 мг [11].
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ФОЛИЕВОЙ КИСЛОТЫ
Фолиевая кислота состоит из следующих компонентов:
пиримидиновой части А(2,4,5-триамино-6-оксипиримидин);
трехуглеродного компонента Б;
р-аминобензойной кислоты В;
L-глутаминовой кислоты Г.
Структурная формула N[р- [(2-амино-4-окси-6-птеридилметил)-амино]-
бензоил)-глутаминовая кислота [12] (фолиевая) имеет следующее строение:
Эмпирическая формула — C19H19OeN7, молекулярная масса 441,40.
Фолиевая кислота из воды кристаллизуется в виде желтых листочков.
При нагревании кристаллы не плавятся, но темнеют и при температуре
250° С обугливаются [13]. Растворимость в воде при 0° — 1 лгг/100 мл-, при
температуре 100° С—50 мг/100 мл [14]. Концентрированная соляная кисло-
та легко растворяет фолиевую кислоту, которая при разбавлении водой поч-
ти полностью выделяется. Растворимость двунатриевой соли при температу-
ре 100° С — 1,5 г/100 мл. Цинковые, свинцовые и серебряные соли фолие-
вой кислоты в воде нерастворимы [13, 14]. Соли щелочных металлов хорошо
растворимы в воде, из растворов они могут быть осаждены спиртом.
Фолиевая кислота практически нерастворима в органических раствори-
телях и слабо растворима в уксусной кислоте. Удельное вращение [ц]о° =
= +16° в 0,1 н. растворе NaOH при концентрации 0,76 г/100 мл [15]. При
высушивании под атмосферным давлением и при умеренной температуре
кристаллы содержат две молекулы воды. Кристаллизационная вода удаля-
ется при температуре 145° С под глубоким вакуумом, при этом вещество ста-
новится гигроскопичным.
Фолиевая кислота имеет характерный абсорбционный спектр в ультра-
фиолетовом свете [12, 13, 16, 17], показанный на рис. 31. Кривая имеет
максимум при длине волн 256, 282 и 365 нм и коэффициент экстинкции
214
Ekl = 585; 570; 206 в 0,1 н. растворе NaOH; при pH 11,0 максимумы по-
глощения остаются теми же, несколько увеличивается Ей-}, — соответствен-
но 603; 600; 213. Фолиевая кислота хорошо адсорбируется активированным
углем, фуллеровой землей и другими адсорбентами [18].
Фолиевая кислота в щелочном рас-
творе в присутствии катализатора оки-
си платины присоединяет одну моле-
кулу водорода, образуя бесцветное
дигидропроизводное с абсорбционным
максимум 284 нм. В ледяной уксус-
ной кислоте в этих же условиях обра-
зуется тетрагидроформа. Дигидрофор-
ма на воздухе окисляется в фолиевую
кислоту [19].
При действии кислорода в щелоч-
ной среде на фолиевую кислоту про-
исходит ее расщепление с образова-
нием 6-птеринкарбоновой кислоты
[17]. Расщепление происходит также
под влиянием ультрафиолетового све-
та, причем сначала образуется 6-птеринальдегид, а затем 6-птеринкарбоно-
вая кислота [2].
Рис. 31. УФ-спектр поглощения фо-
лиевой кислоты.
МЕТОДЫ СИНТЕЗА ФОЛИЕВОЙ КИСЛОТЫ И ВЫБОР РАЦИОНАЛЬНОГО
МЕТОДА ДЛЯ ПРОИЗВОДСТВА [1,2,3]
Существуют три основных направления синтеза фолиевой кислоты [1].
1. Одностадийная конденсация 2,4,5-триамино-6-оксипиримидина с
трехуглеродным компонентом и с р-аминобензоил-А-глутаминовой кислотой.
2. Построение путем наращивания на птерин активной углеродной группы
в положении 6 и конденсации полученного соединения с р-аминобензо-
илглутаминовой кислотой.
3. Конденсация 2,4,5-триамино-6-оксипиридина с готовой боковой цепью
фолиевой кислоты.
Одностадийная конденсация трех компонентов по методу Валлера [12,
17, 20]. Метод заключается в одновременной конденсации трех компонен-
тов — 2,4,5-триамино-6-оксипиримидина-2,3-дибромпропионового альдеги-
да и р-аминобензоилглутаминовой кислоты по следующей схеме:
2,4,5 - Триамино-
6 ~ оксипиримидин
СН2Вг
НСВг
+ I +
,с=о
н'
2,3- Диброипролио-
ноВый апьЗвеиЗ
О
C-NH-CH-сн2- сн2- СООН
соон
р - Амино&нзоилзлутаииноЗая-
кислота.
II
C-NH-CH-CHa- СН2-СООН
СООН
ДивидроптвроилелитаминоЗал кислота
н о
CH2-N-Z Vc-NH-CH-CHa-’CHa-COOH
СООН
Ртероилглутаминобая (фолиебая) кислвта
Предполагается следующий механизм реакции: в буферном растворе при
pH 4 дибромпропионовый альдегид переходит в а-бромакролеин (СН2=
21S
СВгСНО); взаимодействуя с группой NH2, содержащейся в р-аминобензоил-
глутаминовой кислоте, образует соединение CeH4NHCH2CHBrCHO. Это
соединение конденсируется с 2,4,5-триамино-6-оксипиримидином, образу-
ет пиразиновый цикл и переходит в дигидроптероилглутаминовую кислоту
[21]. Для дегидрирования последней применяют йод [22], двухромовокис-
лый калий [23], уксуснокислую ртуть [24] или кислород воздуха.
Метиленовый мостик СН2 в молекуле фолиевой кислоты образуется за
счет группы СН2Вг в дибромпропионовом альдегиде [21 ]. Экспериментально
установлено, что при одностадийной конденсации лучшие результаты полу-
чают, если дибромпропионовый альдегид в спиртовом растворе прибавлять
к водному раствору двух других компонентов [17].
В качестве трехуглеродного компонента вместо дибромпропионового
альдегида были использованы соединения: производные акролеина типа
НО—СН=СОН—СНО [2, 25, 26] и типа СН2=СГал.—СНО [27], З-этокси-2-
Гал-пропионовый альдегид С2Н5О—СН2СНГал.—СНО [28], производные
ацетона Гал—СН2—СОСНГал2 [29, 30, 31] и др.
Указанные выше компоненты для одностадийной конденсации получают
следующим путем:
а) 2,4,5-триамино-6-оксипиримидин — по методу Траубе [32, 33] путем
конденсации гуанидинхлорида с циануксусным эфиром в спиртовом раст-
воре этилата натрия, получая 2,4-диамино-6-оксипиримидин. Последний
нитрозируют в положении 5 и путем каталитического восстановления нитро-
зосоединение превращают в 2,4,5-триамино-6-оксипиримидин.
Вместо гуанидинхлорида можно использовать азотнокислую соль гуа-
нидина [34, 35];
б) р-аминобензоил-Г-глутаминовую кислоту получают путем конденса-
ции £(+)-глутаминовой кислоты с р-нитробензолхлоридом в среде водного
раствора бикарбоната натрия [36]. Образующуюся р-нитробензоилглутами-
новую кислоту каталитическим гидрированием восстанавливают в р-амино-
бензоилглутаминовую кислоту. По методу ВНИХФИ конденсацию Ц+)-глу-
таминовой кислоты с р-нитробензоилхлоридом осуществляют в водно-бен-
зольной среде или в среде хлороформа с прибавлением для нейтрализации
соды, MgO, СаО [37] или NaOH [36, 38—40];
в) 2,3-дибромпропионовый альдегид получают бромированием акролеи-
на в среде четыреххлористого углерода или хлороформа с последующей
перегонкой в вакууме.
Описанный метод синтеза позволяет получить выход фолиевой кислоты —
30% от теоретического, считая на р-аминобензоил-£-глутаминовую кислоту.
Для предотвращения образования в качестве промежуточного продукта
при конденсации 2,4,5-триамино-6-оксипиримидина с 2,3-дибромпропионо-
вым альдегидом дигидроптероилглутаминовой кислоты более ра-
циональным является применение в качестве трехуглеродного компонента
1,1,3-тригалогенацетон. Применение, например, трихлор ацетона приводит
к непосредственному образованию птеридинового ядра [29,30]. Однако при
этом необходимо добавление бисульфита натрия, который, с одной стороны,
предохраняет трихлорацетон от окисления, а с другой, служит катализато-
ром реакции, обеспечивая выход фолиевой кислоты в 37% [41, 42].
Фолиевая кислота, получаемая при одностадийной конденсации, за-
грязнена примесями различных птеридинов и содержит 40—60% чистого
вещества.
Для очистки продукта применяют несколько способов.
1. Растворяют технический продукт в разбавленной щелочи и, добавляя
хлористый барий, при pH 7,0 осаждают различные примеси. Фильтрат
трехкратно экстрагируют девятью объемами бутанола. Водный раствор кон-
центрируют под вакуумом, подкисляют до pH 3,0 и охлаждают до 0°. Вы-
кристаллизовавшийся продукт растворяют в разбавленной щелочи. Полу-
ченный раствор обрабатывают костяным углем, подкисляют до pH 3,0 и из
него после кристаллизации получают чистую фолиевую кислоту [1].
216
2. Для очистки применяют переосажДение из концентрированной соля-
ной кислоты с последующим разбавлением водой [17, 20, 36, 43].
3. Растворяют технический продукте 1 н. растворе NaOH и при pH 10,5—
11,2 вводят хлористый цинк, гидрат окиси кальция и перекись водорода.
Осадок отфильтровывают и выделяют фолиевую кислоту избытком горячей
1%-ной уксусной кислоты [44].
4. Получают соли птериновых соединений при действии оснований при pH
выше 12,0; примеси осаждают при pH выше 10,2, а фолиевую кислоту при
pH 3—4. Очистку производят в очень разбавленных растворах (1 : 10000)
и повторяют многократно. Производят также обработку продукта углем и
перекристаллизацию его из воды при pH 3,0 [17].
Для очистки кислоту переводят в натриевые, бариевые, цинковые, маг-
ниевые и другие соли [45—49].
Конденсация предварительно синтезированного птерина с р-аминобен-
зоил-L-глутаминовой кислотой. Метод заключается в конденсации 2,3-ди-
бромпропионового альдегида. 2,4-5-триамино-6-оксипиримидина и пиридина
в присутствии йодистого калия [50]. При этом получают N-[(2-амино-4-
окси-6-птеридил)-метил]-пиридинбромид. Последний конденсируют с р-
аминобензоилглутаминовой кислотой в этиленгликоле с образованием пте-
роилглутаминовой кислоты по следующей схеме:
h2n
h2n
. он
2,4, 5 - Триамино -S-
он
П-](2-амино-А—окси-6-птеридил}-метил} -
_ сно
+ Вг-СН2—СНВг +
Пиридин 2,3- ДибромпропионоВый
альдегид
оксипиримидин
пиридинбромид
р-Аминобензоилглутаминобая кислота
СООН
Фолиебая кислота
Для конденсации с р-аминобензоил-Z.-глутаминовой кислотой могут
быть использованы и другие производные птерина, имеющие в положении 6
вместо метилпиридина следующие функциональные группы: — СН2С1 [51,
52];—СНО [53, 54]; — СН2ОН+нитробензоилглутаминовая кислота при
каталитической гидрогенизации [55]; —СН2ОН [56];—СН2Вг [57]; —
-ВгСН-СООН [58] и др.
Другие варианты этого синтеза приводятся в ряде патентов [59—66].
Как видно из изложенного, этот метод синтеза сложнее, чем метод Вал-
лера, но двухстадийная конденсация компонентов, возможно, обеспечит
более высокий выход фолиевой кислоты.
Конденсация 2,4,5-триамино-6-оксипиримидина с готовой боковой
цепью молекулы фолиевой кислоты. Этот метод заключается в предвари-
тельном синтезе боковой цепи молекулы фолиевой кислоты и последующей
конденсации ее с 1,4,5-триамино-6-оксипиримидином. К соединениям, ко-
торые могут быть использованы в качестве боковой цепи, относятся такие,
которые способны образовать пиразиновый цикл при взаимодействии с со-
седними аминогруппами 2,4,5-триамино-6-оксипиримидина, как, например:
217
но он о н н
1 1 11 1 1
нс=с-hc=n—v у-с—n-c-cooh ;
сн2
сн2-соон
Вг Вт ОН Н
। 1 /Г~и 1 ।
сн2- ch-h2c-n-c Vc-n-c-cooh
со сн2
сн3 сн2-соон
N [р-(2,3-диокси -2 - ен-пропилиденамино) - бензоил] -
глутамат [25]
N - ацетил -N -(2,3 - дибромпрооил) -у - аминобензоил-
глитаминобая кислота
Мы видим, что и этот метод связан с двухстадийной конденсацией и яв-
ляется более сложным нежели метод Валлера.
Пока в заводской практике применяют метод Валлера. Выход фолиевой
кислоты, получаемый этим способом, недостаточный. В будущем, возможно,
что предпочтение будет отдано методам двухступенчатой конденсации.
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА СИНТЕТИЧЕСКОЙ ФОЛИЕВОЙ
КИСЛОТЫ
Технологическая схема состоит из следующих основных стадий:
1. Синтез р-аминобензоилглутаминовой кислоты путем конденсации
L-глутаминовой кислоты с р-нитробензоилхлоридом и каталитического
гидрирования получаемой р-нитробензоилглутаминовой кислоты. Эта ста-
дия синтеза включает получение следующих промежуточных продуктов:
L-глутаминовой кислоты;
р-нитробензоилхлорида;
р-нитробензоилглутаминовой кислоты;
р-аминобензоилглутаминовой кислоты.
2. Синтез 2,3-дибромпропионового альдегида из глицерина. Эта стадия
включает получение следующих промежуточных продуктов:
акролеина;
2,3-дибромпропионового альдегида.
3. Синтез 2,4,5-триамино-6-оксипиримидина сульфата путем конденса-
ции гуанидина с циануксусным эфиром. Эта стадия включает получение сле-
дующих промежуточных продуктов:
2,4-диамино-6-оксипиримидина;
2,4-диамино-5-нитрозо-6-оксипиримидина;
2,4,5-триамино-6-оксипиримидина сульфата.
4. Конденсация р-аминобензоилглутаминовой кислоты с 2,4,5-триамино-6-
оксипиримидином с 2,3-дибромпропионовым альдегидом и получение фолие-
вой кислоты.
ПОЛУЧЕНИЕ Р-АМИНОБЕНЗОИЛГЛУТАМИНОВОЙ КИСЛОТЫ
Получение £(4~)-глутаминовой кислоты из глутена. 72%-ную пшенич-
ную муку замешивают с водой в густое тесто в тестомесильной машине 1
(рис. 32). Затем в эту же тестомесильную машину подводят водопроводную
воду и вымывают крахмал. Конец отмывания крахмала определяют по окон-
чании окрашивания отжима при пробе с йодом. Клейковину выгружают в
сборник 2, откуда подают в вальцовую сушилку <3. Высушенную клейковину
измельчают в молотковой дробилке 4 в тонкую муку и хранят в бункере 5.
В реакторе 6 из эмалированной стали, снабженном обратным холодиль-
ником 7 из эмалированных труб, клейковинную муку обрабатывают соляной
кислотой (плотность 1190 кг/м3), подаваемой из мерника 8. После этого в
рубашку аппарата пускают холодную воду и охлаждают реакционную смесь
до комнатной температуры. Затем добавляют в охлажденный раствор акти-
вированный уголь, смесь тщательно перемешивают в течение 15 мин, а затем
фильтруют через слой асбеста на нутч-фильтре 9. Уголь промывают соляной
кислотой, фильтрат и промывные фракции собирают в приемнике 10, отку-
да они поступают на упаривание в вакуум-аппарат 11. Из вакуум-аппарата
218
Рис. 32. Технологическая схема производства синтетической фолиевой кислоты.
концентрат I поступает в кристаллизатор 12,. охлаждаемый хладагентом до
температуры минус 2—3° С. Выделившиеся кристаллы гидрохлорида I
глутаминовой кислоты отфуговывают на центрифуге 13 из эмалированной
стали. Маточный раствор I собирают в приемнике 14, откуда его засасывают
в вакуум-аппарат 15, где упаривают до уменьшения объема на 50%. Из ва-
куум-аппарата концентрат II спускают в кристаллизатор 16, откуда он
поступает на центрифугу 17, где отделяют гидрохлорид II от маточного
раствора II, являющегося отходом производства. Гидрохлорид I подвергают
перекристаллизации из водного раствора, для чего его растворяют в смеси-
теле 18, упаривают в вакуум-аппарате 19, кристаллизуют в кристаллизато-
ре 20. Кристаллы отфуговывают на центрифуге 21 и высушивают в вакуум-
сушилке 22; из последней выходит готовый полупродукт,, который хранят
в сборнике 23.
В смесителе 24 растворяют в маточном растворе 1а гидрохлорид II, затем
раствор засасывают в вакуум-аппарат 25, далее кристаллизуют в кристал-
лизаторе 26, кристаллы отделяют на центрифуге 27; маточный раствор Па
направляют для переработки в сборник 14.
При небольшом объеме производства обычно предусматривают одну ли-
нию перекристаллизации для готового продукта (гидрохлорида /.-глутами-
новой кислоты), а другую линию (вакуум-аппарат — кристаллизатор —
центрифуга) для промежуточных продуктов.
Вопрос о коррозиии аппаратуры данной технологической схемы не изу-
чен.
Получение глутаминовой кислоты из мелассы1 [70, 72]. Более ра-
циональным является метод М. Аймухамедовой получения глутаминовой кис-
лоты из мелассы по комплексной схеме. Сущность метода заклю-
чается в адсорбции из раствора мелассы глутаминовой кислоты анионитом
ЭДЭ-10П, десорбции ее при регенерации анионита 1 н. раствором NaOH
или аммиаком, выпаривании раствора и кристаллизации. На получение 1 кг
глутаминовой кислоты расходуют (в кг): мелассы 16,5, анионита — 0,003,
едкого натра — 2,6, уголь активированный — 0,4.
Получение р-нитробензоилхлорида. Химическая реакция получения:
2o2n V соон + pci5 —► 2O2n -4 у СОС1 + РОСТ3 + Н2О
р-Нитробензойная кислота Пятихлорис-
2 • 767, 72 тый ipocpop
208,31
р - Нитробекзоилхлорид Хлорокись
2 785,57 фосфора.
753,39
Паранитробензоилхлорид получают в эмалированном реакторе-дистил
ляторе 28, снабженном прямым и обратным эмалированным холодильником
29 с обогревом дитолилметаном (т. кип. 300° С) и двумя приемниками 30
и 31. В дистиллятор вводят пятихлористый фосфор и сухую паранитробен-
зойную кислоту, взятые в отношении 132 : 100, и медленно нагревают до
100° С в течение 35—40 мин при постоянном перемешивании до образования
гомогенной массы.
Затем массу подвергают фракционированной перегонке. Вначале отго-
няют при нормальном давлении хлорокись фосфора при температуре 200—
250° С в приемнике 30. Затем включают вакуум и при остаточном давлении
20—22 мм рт. ст. и температуре 163—166° С отгоняют паранитробензоил-
хлорид в приемник 31, откуда в жидком виде либо в растворе хлороформа
передают в реактор 33. Выход хлорида составляет 85% от паранитробензой-
ной кислоты.
Паранитробензоилхлорид, C7H4O3NC1, молекулярная масса 185,57, крис-
1 По-видимому, еще более эффективным является микробиологический метод по-
лучения L-глутаминовой кислоты с применением Brevibacterium или micobacterium
(См. японские патенты № 673, № 1834, 1835, 1972, Derwent Т02; ТОЗ, 1972 г.).
220
таллизуется в иглах светло-желтого цвета, температура плавления 73° С,
температура кипения при остаточном давлении 20 мм рт. ст. 155° С, хоро-
шо растворим в органических растворителях.
ПОЛУЧЕНИЕ Р-НИТРОБЕНЗОИЛГЛУТАМИНОВОЙ КИСЛОТЫ
Химическая реакция получения:
СООН
o2n
h2nch
СОС1 + I
COONa
СН2
2NaHCO3 O2N
CO-NH-CH
o2n
p - Нитродензоил-
xл о pud
CO-NH-CH
сн2соон
L - Глутаминовая
кислота
COONa
2HC1
CH2
СН2
CH2COONa
Натриевая саль
нитрадензоилглутаминабай кислоты
СООН
o2n
CO-NH-CH
+ 2NaCl
CH2.
CH2COONa
CH2COOH
p - Нитрадезоилглутаминобая кислота
L-Глутаминовая кислота из сборника 52 поступает в охлажденный реак-
тор 33, где смешивается с четырехкратным количеством бикарбоната нат-
рия, подаваемого из приемника 34, и двадцатипятикратным количеством
воды из сборника 35. Затем сюда вводят паранитробензоилхлорид и продол-
жают перемешивание при охлаждении реактора рассолом. Затем непрореа-
гированный паранитробензоилхлорид отсасывают на нутч-фильтре 36 и
дважды промывают водой. Фильтрат и промывные воды направляют в эма-
лированный реактор 37 при помощи насоса 38, где их подвергают нейтрали-
зации и подкислению соляной кислотой до фиолетового окрашивания капли
конго-красного от капли раствора (pH около 3,0). При этом выпадает оса-
док нитробензойной кислоты, получаемой при частичном разложении р-
нитробензоилхлорида. Осадок отфильтровывают на нутч-фильтре 39 и про-
мывают водой. Фильтрат и промывные воды упаривают в вакуум-аппарате
40 и кристаллизуют в кристаллизаторе 41 при охлаждении до минус 2° С.
Кристаллы отделяют на центрифуге 42.
Нитробензоил глутаминовую кислоту перекристаллизовывают из горя-
чей воды с переработкой маточного раствора. Для этого кислоту растворяют
в аппарате 43 при нагревании и затем охлаждают в кристаллизаторе 44.
Кристаллы отделяют в центрифуге 45. Маточный раствор I собирают в при-
емнике 46, упаривают в вакуум-аппарате 47, кристаллизуют в кристаллиза-
торе 48 и фугуют в центрифуге 49. Кристаллы первого продукта высушива-
ют в вакуум-сушилке 50. Маточный раствор II поступает в сборник 51,
откуда удаляется как отход производства. Кристаллы второго продукта
перекристаллизовывают совместно с технической паранитробензоилглу-
таминовой кислотой, для чего направляют в реактор 43.
р-Нитробензоилглутаминовая кислота, C12H12O7N2, молекулярная масса
296,23, представляет собой бесцветные пластинки с температурой плавления
114—116° С; [а]д =+16,02° (2%-ный раствор в 2н. растворе NaOH).
В воде и спирте растворима, нерастворима в эфире.
Получение р-аминобензоилглутаминовой кислоты. Химическая реакция
получения:
221
СООН
O2N-< у—CO-NH-CH +
\=/ ।
СН2
I
сн2сооп
р - НитровензоилглутамитИая кислота
236,23
СООН
ЗН2 J^H2N4^2y>--CO-NH-CH + 2Н2О
сн2
I
СН2СООН
р - Аминобензоилглутаминобая кислота.
266,25
р-Нитробен'зоилглутаминовую кислоту растворяют в воде при температу-
ре 35° С в аппарате-растворителе 52, откуда раствор после фильтрации на
нутч-фильтре 53 поступает в приемник 54 перед автоклавом 55. Гидрирова-
ние осуществляют в автоклаве при избыточном давлении водорода 25—
30 кгс/см2 при 50° С в присутствии алюминиево-никелевого катализатора.
Катализатора добавляют 10% к массе кислоты.
По окончании процесса гидрирования раствор направляют в приемник
56, откуда он поступает на нутч-фильтр 57 для отделения катализатора. Из
нутч-фильтра фильтрат поступает в сборник 58, а из него в вакуум-аппарат
59, где его упаривают в 8 раз. Концентрат спускают в кристаллизатор 60,
а из него в центрифугу 61.
Кристаллы р-аминобензоилглутаминовой кислоты при необходимости
сушат в вакуум-сушилке 50. Маточный раствор упаривают и повторно крис-
таллизуют, получают иглы, температура плавления 166—167° С; [ц]2^ =
= +27,4° С (2%-ный раствор в 2 н. растворе едкого натра). Растворима в
воде, плохо растворима в органических растворителях.
СИНТЕЗ 2,3-ДИБРОМИРОНИОНОВОГО АЛЬДЕГИДА
Получение акролеина. Химическая реакция получения:
СН2
СН2ОН ||
I рьо СН
СНОН + KHSO4 —> | + PbSO4 + КОН + 2Н2О
| С=О
СН2ОН |
н
Глицерин Акролеин
92,08 56,06
Процессы осуществляются в герметической аппаратуре.
В реактор-дистиллятор 62, снабженный масляным подогревом и холо-
дильником, загружают глицерин и высушивают его при температуре 170° С.
Затем вводят в реактор кислый сернокислый калий и првышают температу-
ру в реакторе до 200° С. При этой температуре начинает перегоняться акро-
леин. Перегонка продолжается 2 ч. Дистиллят собирают в реактор 63, где
обрабатывают свинцовым глетом (РЬО) для освобождения дистиллята от
SO3 с образованием труднорастворимого осадка PbSO4. Затем дистиллят в
этом же аппарате, снабженном холодильником, перегоняют. Дистиллят со-
бирают в реакторе 64, куда для стабилизации добавляют гидрохинон и хло-
ристый кальций, фильтруют через нутч-фильтр 65 и вторично перегоняют в
перегонном аппарате 66, дистиллят собирают в приемнике. Выход 46%.
Акролеин, С3Н4О, молекулярная масса 56,06, представляет собой бесцвет-
ную жидкость резкого запаха с температурой кипения 52,5° С, плотностью
845 кг!м\ хорошо растворим в воде. На свету быстро полимеризуется. Ак-
ролеин токсичен и вызывает слезоточивость.
222
Получение 2,3-дибромпропионового альдегида. Химическая реакция по-
лучения:
сн2
Вг—СН2
сн
с=о
Br—CH
Br2 |
C=O
н н
Акролеин 2-79,92 2, З-Дибромпропноновый
56,62 альдегид 215,90
Акролеин легко полимеризуется, поэтому необходимо его защищать от
света, хранить на холоду, а главное — немедленно подвергать бромирова-
нию. Процесс бромирования осуществляют в вытяжном шкафу.
В эмалированном реакторе 67 приготовляют раствор брома в четырех-
хлористом углероде.
Под реактором 67 помещается реактор для бромирования 68, в который
загружают акролеин и пятикратное количество свежеперегнанного четырех-
хлористого углерода. В реактор вводят рассол в качестве хладагента и
охлаждают раствор акролеина до минус 2—3° С. Бромирование осущест-
вляют при плюс 5° С и постоянном перемешивании с добавлением неболь-
шими порциями брома (из реактора 67) до ^прекращающегося оранжево-
красного окрашивания от избытка брома. Затем в дистилляторе 69 отгоня-
ют четыреххлористый углерод в приемник 70, а в приемник 71 отгоняют
2,3-дибромпропионовый альдегид. При этом собирают фракцию, кипящую
при температуре 90° С и остаточном давлении 24 мм рт. ст. Выход 85%
(на акролеин).
2,3-Дибромпропионовый альдегид, С3Н4Вг2О, молекулярная масса 215,90,
светло-желтая жидкость, d15 = 2,198. При хранении полимеризуется в
кристаллический продукт при температуре кипения 79—84° С при остаточном
давлении 5—6 мм рт. ст.
В качестве трехуглеродного компонента более рационально применять
1,1,3-трихлорацетон [73, 75]. Его получают непосредственным хлорирова-
нием ацетона сначала при температуре 35—40°С (до образования 1,1-ди-
хлорацетона), а затем при температуре 85—90° С для получения 1,1,3-три-
хлорацетона. Затем отгоняют моно- и дихлорацетон, а из кубового остатка
извлекают водой трихлорацетон. Выход 40%; температура кипения 172° С;
= 1,4917 [74]. Л. Бугрова, Г. Руднев и др. [76] усовершенствовали
синтез трихлорацетона путем применения в качестве катализаторов заме-
щенных аминов.
СИНТЕЗ 2,4,5-ТРИАМИНО-б-ГИДРОКСИПИРИМИДИНА
Получение 2,4-диамино-6-оксипиримидина-сульфата. Химическая реак-
ция получения:
nh2 СООС2Н5
I 2 I
hn=c-nh2- НС1 + сн2
CN
Циануксусный
зтрир
113,11
Гуанцдинхлорид
95,59
со
HN "'СН2 NaOH
HN=CX CN
nh2
1Z6,1Z
C2H5ONa
ОН
Алкоголят
натрия
68,06
.со
HN ХСН2
I I +2C2HsOH+NaCl
HN=C^ CN
nh2
Цианаиртил-
гуанидин
126,12
он
N^ СН
I II
h2n-c^_ ^.c-nh2
N
2,4 -Диамина -6-
оксипиримибин
118,12
H2SO4
H2o'
nh,-|h2so44h2o
2,9 -Диамино-6-окси-
пиримидин - сульфат
176,17
223
Открытая изомерная форма цианацетилгуанидин, образующаяся при
действии NaOH, превращается в циклическую пиримидиновую форму; при
действии серной кислоты получают 2,4-диамино-6-оксипиримидин-сульфат.
В реактор 72 с обратным холодильником загружают порошок гуанидин-
хлорида, абсолютный спирт и алкоголят натрия, взятые соответственно от-
ношению 1:5:3. Реактор охлаждают рассолом. Выделившийся осадок пова-
ренной соли отфильтровывают на нутч-фильтре 75, а фильтрат насосом
перекачивают в реактор 74, куда постепенно приливают циануксусноэтило-
вый эфир. Реакционную массу подогревают до кипения и кипятят 15 лшн
при перемешивании, затем ее оставляют на 8 ч при комнатной температуре.
Реакционную массу фильтруют через друк-фильтр 75. Осадок переносят в
реактор 76 и кипятят в течение 10 мин с 6%-ным NaOH. Затем ее охлаждают
и вводят в реактор 76 50%-ную H2SO4 (до фиолетового оттенка конго-крас-
ного) и осаждают диаминоксипиримидинсульфат. Его отфильтровывают на
нутч-фильтре 77 и направляют для сушки в сушильный аппарат 78. Фильт-
рат из друк-фильтра 75 и нутч-фильтра 77 засасывают в вакуум-аппарат 79,
где растворитель отгоняют досуха. Затем вводят в вакуум-аппарат 6%-ный
NaOH, кипятят 10 мин, затем щелочной раствор переводят в реактор 80 из
нержавеющей-стали, охлаждают массу рассолом и осаждают диаминооксипи-
римидинсульфат 50%-ной H2SO4. Выделившиеся кристаллы отфильтровы-
вают на нутч-фильтре 81 и сушат в сушилке 78.
2,4-Диамино-6-оксипиримидин-сульфат (C4H6ON4- 0,5H2S04-0,5H20), мо-
лекулярная масса 176,17, бесцветные кристаллы, плохо растворяются в хо-
лодной и лучше в горячей воде.
Получение 2,4-диамино-5-нитрозо-6-оксипиримидина. Химическая ре-
акция получения:
он он
н2т^1н2 • °’5Н*° + NaN°2---*нД^Кн2 + 0'3Na2S°4 +
N 2 N 2
2,U -Диамино-З-оксипиримидин- 2,4 -Диамино-5-нигпрозо-
сулырзт 6-оксипиримидин
176,17 155,12
Диаминооксипиримидин-сульфат растворяют в горячей воде в реакторе
82, отфильтровывают от нерастворимого осадка на нутч-фильтре 83. Затем
фильтрат поступает в реактор 84 для нитрозирования, где охлаждается до
0°. В реактор вводят 60%-ный раствор нитрита натрия и 20%-ный раствор
H2SO4; при перемешивании тотчас выделяется розово-красный осадок нитро-
зосоединения. Реакционную массу охлаждают до 0° и фильтруют через нутч-
фильтр 85. Осадок сушат в сушилке 86. Выход 95%.
2,4-Диамино-5-нитрозо-6-оксипиримидин, C4H5O2N5, молекулярная мас-
са 155,12, розово-красные игольчатые кристаллы. Плохо растворим в воде,
спирте и разбавленных кислотах, лучше в разбавленных щелочах.
Получение 2,4,5-триамино-6-оксипиримидинсульфата. Химическая ре-
акция получения:
ОН
N |р№ *2Н2; H2SO4
H2N-U Д-1МН2 Ni "
2 N
2И-Диамино-5-нитрозо-
6 - онсипиримидин
155,12
ОН
2,4,5-Гриамино-6- оксипиримидин -
сульфат
257,23
н2о
Восстановление нитрозосоединения осуществляют водородом в автокла-
ве в присутствии никелевого катализатора. Для этой цели в автоклав 87
224
загружают нитрозосоединение, никелевый катализатор и 4%-ный раствор
едкого натра из мерника 88. Гидрирование ведут при перемешивании при
температуре 85—90° С и давлении водорода 20—25 кгскм2. После окончания
гидрирования раствор спускают в сборник. Затем на нутч-фильтре 89 от-
фильтровывают катализатор, а фильтрат направляют в аппарат-смеситель
90, в котором находится 50%-ный раствор серной кислоты, спускаемый в
него из мерника 91. Выделяемый осадок триаминооксипиримидинсульфата.
отфильтровывают в нутч-фильтре 92, промывают водой и высушивают в ва-
куум-сушилке 93, откуда он поступает в приемник 94. 2,4,5-Триамино-6-
оксипиримидинсульфат, C4H,ONS • H2SO4 • Н2О, молекулярная Масса
257,23, легко растворим в воде, окисляется на воздухе.
С целью упрощения процесса получения 2,4-диамино-5-нитрозо-6-оксипи-
римидина предложено конденсацию и нитрозирование проводить в одну ста-
дию [75]. Для этого гуанидин нитрат переводят в гуанидин-основание в сре-
де этилата натрия при температуре 68—72° С в течение 2,5 ч Затем добавля-
ют циануксусный эфир, перемешивают 1,5 ч и отгоняют растворитель; оса-
док растворяют в воде и фильтруют. После этого проводят нитрозирование
добавлением нитрита натрия и соляной кислоты до полного осаждения 2,4-
диамино-5-нитрозо-6-оксипиримидина. Осадок фильтруют и высушивают.
Выход 76,5—-79,0% (на гуанидин нитрат).
ПОЛУЧЕНИЕ ФОЛИЕВОЙ КИСЛОТЫ
Химическая реакция получения:
h2n
соон
Вт-СН2
Вг—СН
I +
с=о
I
н
он
H2SO4 -Н2О
р-Аминобензоипгпутаминобая кислота
266,25
2,3 ~ Ди бром про- 2,4,5- Три а ми но -
пионобый альде- 6-оксипиримидин-сульфат
ФолиеВая кислота
447, 40
Конденсация трех компонентов. (Метод А. Труфанова и В. Кирсановой).
В реактор 95 вводят р-аминобензоилглутаминовую кислоту и 2,4,5-триами-
но-6-оксипиримидин-сульфат, добавляют воду (примерно двадцатипяти-
кратное количество к общей массе реагентов), нагревают до 50 °C и фильтру-
ют через нутч-фильтр 96. Фильтрат направляют в сборник 97, а затем в
реактор 98, охлаждают водой до 25° С, добавляют ацетатного буфера, чтобы
pH был равен 4,0 (1 г-моль уксусной кислоты и 1 г-моль уксуснокислого
натрия) и 20%-ного едкого натра для нейтрализации H2SO4. Затем вво-
дят в реактор азот и при постоянном перемешивании небольшими
порциями приливают 2,3-дибромпропионовый альдегид, растворенный
в этиловом спирте в реакторе 99. По мере хода реакции выделяется НВг,
который изменяет оптимальный pH. Поэтому непрерывно добавляют в ре-
актор 20%-ный NaOH, следя за реакцией по индикатору конго-красному.
Дибромпропионовый альдегид добавляют в течение 2,5 ч, затем массу пере-
мешивают еще 60 мин без пропускания азота при охлаждении. После это-
го реакционную массу направляют в нутч-фильтр 100, где осадок техничес-
кой фолиевой кислоты отсасываютдосуха, промывают на фильтре водой и
спиртом, а затем направляют в вакуум-сушилку 101.
8-522
225
Маточный раствор упаривают в вакуум-аппарате 102 до 50%-ного объе-
ма, кристаллизуют на холоду при 0° в кристаллизаторе 103 и отфуговывают
в центрифуге 104. Промытый осадок направляют в вакуум-сушилку 101.
Конденсация компонентов фолиевой кислоты может быть также осущест-
влена по методу О. Магидсона и К. Чхиквадзе следующим образом. Вначале
приготовляют водный раствор р-аминобензоилглутаминовой кислоты. Затем
в этот раствор вводят йодистый калий и 2,4,5-триамино-6-оксипиримидин;
pH раствора едким натром доводят до 4,0. Наконец, после нагревания раст-
вора до 40° С в него наливают раствор 2,3-дибромпропионового альдегида и
йодистого калия в изопропиловом спирте и одновременно 1 н. раствор едко-
го натра. По окончании реакции массу охлаждают до 8—10° С и подкисляют
соляной кислотой, до pH 3,0. Выделяющийся осадок фолиевой кислоты отфу-
говывают в центрифуге. Чистота технической кислоты 50—60%. По методу
Березовского и др. в реакцию конденсации применяют бариевую соль р-
аминобензоилглутаминовой кислоты и в качестве трехуглеродного компо-
нента применяют 1,1,3-трихлорацетон [771. При этом в реакцию конденса-
ции вводят бисульфит натрия, который предохраняет промежуточные сое-
динения от окисления и, по-видимому, служит катализатором реакции.
Очистка фолиевой кислоты. Техническую фолиевую кислоту растворяют
в 0,1 н. растворе NaOH в реакторе 105 при температуре 40° С. Раствор филь-
труют через нутч-фильтр 106. Фильтрат поступает в смеситель 107, где
его обрабатывают активированным углем в количестве 3—5% к массе техни-
ческой фолиевой кислоты. Затем отфильтровывают осадок угля в нутч-фильт-
ре 108, промывают разбавленным раствором щелочи. Фильтрат насосом 109
передают в реактор 110, куда вводят НС1 до фиолетового оттенка капли кон-
го-красного; массу охлаждают до минус 3° С и кристаллизуют. Затем крис-
таллы отфуговывают в центрифуге 111, промывают осадок водой и спиртом.
Получают получистую фолиевую кислоту.
Маточный раствор I упаривают в вакумм-аппарате 112 др 50%-ного объе-
ма, кристаллизуют в кристаллизаторе 113, кристаллы отделяют на центри-
фуге 114. Они поступают в переработку совместно с технической фолиевой
кислотой. Маточный раствор II является отходом.
Получистую фолиевую кислоту растворяют в реакторе 115 в 0,1 н. раст-
воре NaOH, обрабатывают активированным углем (2—3% к массе кислоты),
фильтруют через нутч-фильтр 116, фильтрат направляют в реактор 117,
куда вводят НО до pH 2,5—3,0, охлаждают до минус 3° С, осадок отделяют
в центрифуге 118, промывают водой и спиртом, высушивают в вакуум-су-
шилке, 119. Маточный раствор II поступает на переработку совместно с тех-
нической фолиевой кислотой.
Кроме указанного метода, применяют очистку фолиевой кислоты путем
ее растворения в крепкой соляной кислоте и осаждения при большом раз-
бавлении солянокислого раствора водой. Как щелочной, так и кислотный
метод очистки не дают хороших результатов по качеству и выходу фолие-
вой кислоты. Качество фолиевой кислоты можно будет значительно улуч-
шить, если удастся процесс осаждения заменить процессом кристаллизации,
например, путем растворения технического продукта в 0,1 н. НС1 при тем-
пературе 75—80° С и выкристаллизовывании ее при минус 2—3° С.Однако
этот вопрос требует особого изучения.
ТЕХНОЛОГИЧЕСКИЕ ПОКАЗАТЕЛИ
РАСХОД МАТЕРИАЛОВ, КГ НА 1 КГ
ПОЛУПРОДУКТА
L-Глутаминовая кислота
Мука пшеничная .................34,6
Кислота соляная (плотность
1190 кг! м2).....................17,3
Уголь активированный ............0,46
Спирт с учетом регенерации . . 0,68
Выход L-глутаминовой кислоты,
% к муке ........................2,6
р-Нитробензоилхлорид
р-Нитробензойная кислота .... 1,18
Пятихлористый фосфор...........1,46
226
Выход, % от теоретического (на
р-нитробензойную кислоту) . . . 76,5
р-Нитробензоилглутаминовая кислота
I.-Глутаминовая кислота ........ 0,67
Бикарбонат натрия................2,67
р-Нитробензоилхлорид............ 1,33
Кислота соляная................. 1,74
Выход, % от теоретического (на L-
глутаминовую кислоту) ..........93,0
р-Аминобензоилглутаминовая кислота
р-Нитробензоилглутаминовая кис-
лота . ........................ 1,67
Водород, л3......................0,40
Сплав (Ni + Al) ...............0,2
Выход, % от теоретического (на
р-нитробеизоилглутаминовую кис-
лоту) ..........................66,7
Акролеин
Глицерин безводный...............3,8
Калий кислый сернокислый ... 6,5
Бикарбонат натрия ...............0,05
Гидрохинон.......................0,01
Выход, % от теоретического (на
глицерин).......................43,0
2,3-Дибромпропионовый альдегид
Акролеин.........................0,36
Бром ........................... 1,0
Углерод четыреххлористый с уче-
там регенерации .................0,3
Выход % от теоретического (на
акролеин/.......................85,0
2-4-Диамино 6-оксипиримидинсульфат
Гуанидинхлорид...................0,6
Цианоуксусный эфир ..............0,7
Спирт............................ 1,0
Натрий металлический ............0,2
Едкий натр 50%-ный...............0,4
Кислота серная...................1,2
Выход, % от теоретического (на
гуанидинхлорид) ................88,0
2-4-Диамино-5-нитрозо-6-оксипиримидин
Диаминоксипиримидинсульфат . . 1,20
Нитрит натрия....................0,45
Выход, % от теоретического (на
диаминооксипиримидин)...........95,0
2,4,5-Триамино-6-оксипиримидинсульфат
Диаминонитрозооксипиримидин . . 0,95
Сплав (Al+Ni)....................0,40
Едкий натр.......................0,50
Кислота серная...................1,3
Водород, м3 .....................1,0
Выход, % от теоретического (на
риаминооксипиримид ..............75,0
ФОЛИЕВАЯ КИСЛОТА
р-Аминобензоилглутаминовая кис-
лота ............................2,0
2,3-Дибромпропионовый альдегид 1,7
2,4,5-Триамино-6-оксипиримидин . 1,7
Соляная кислота ................ 0,5
Едкий натр технический..........1,6
Спирт ...........................3,0
Активированный уголь ............0,2
Уксусная кислота ледяная ... 0,3
Натрий уксуснокислый 28%-ный . 1,0
Ацетон...........................0,5
Выход, % от теоретического (на
р-аминобензоилглутаминовую кис-
лоту) ..........................30,0
Расход полупродуктов, кг на 1 кг чис-
той фолиевой кислоты
L-Глутаминовая кислота ........ 2,24
р-Нитробензоилхлорид.............4,45
р-Нитробензоилглутаминовая кис-
лота ............................3,34
р-Аминобензоилглутаминовая кис-
лота ............................2,0
Акролеин.......................0,61
2,3-Дибромпропионовый альдегид 1,7
2,4-Диамино-6-оксипиримидинсуль-
фат............................. 1,94
2,4-Диамино-5-нитрозо-6-оксипири-
мидин .......................... 1,62
2,45- Триамино-6-оксипиримидин -
сульфат ........................ 1,7
Расход химикалиев, кг на 1 кг фолие-
вой кислоты
Кислота соляная (плотность
1190 кг/л3).................45,1
Едкий натр технический......2,9
Спирт с учетом регенерации . . . 6,5
Сплав (Al-f-Ni)............. 1,1
Водород, м3 .....................2,5
Нитрит натрия ................. 0,7
Гуанидинхлорид........... . . . 1,2
Циануксусный эфир........... 1,4
Натрий металлический’.......0,4
Бром ............................1,7
Углерод четыреххлористый с уче-
том регенерации .................0,5
Глицерин....................2,3
Уголь активированный........ 1,2
Кислота уксусная ледяная .... 0,3
Натрий уксуснокислый 28%-ный . 1,0
Ацетон ..........................0,5
Кислота серная контактная ... 4,6
Калий кислый сернокислый . . -. 4,0
Бикарбонат натрия ...............9,0
Гидрохинон ......................0,02
р-Нитробензойная кислота .... 5,3
Фосфор пятихлористый.......6,5
Мука пшеничная.............77,5
Итог о . .176,2
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. S е Ь г е 1 1 W., Harris R., The Vitamins, 1954, III, 87—217.
2. Березовский В. Успехи химии, 1953, XXII, 2, с. 191—232.
3. Hultquist М., Jukes Т., Encyclopedia of the Chemical Technology, 1955, 6,
778.
4. W i 1 1 s L., С о n t a b a M., Lond, Brit. Med., J. 1931, 1, 1059.
8* 227
5. Day P., Langston W., Darby W. Proc. Soc. Exptl. Biol. Med., 1938, 38,
860.
6. Stocstad E., Manning P., J. Biol. Chem., 1938, 125, 687.
7. H о g a n A., Parrott E., J. Biol. Chem., 1938, 125, 687.
8. S t о c s t a d E„ J . Biol., Chem., 1943, 149, 573.
9. Mitchell H., Snell E., Williams R., J. Am. Chem. Soc., 1941, 63,
2284.
10. HI и л о в П. И., Яковлев Т. Н. Основы клинической витаминологии. М.,
«Медицина», 1964, с. 83.
И. Р ы с с С. М. Витамины. М., Медгиз, 1955, с. 165
12. Angier R. и др., Science, 1945, 102, 227; 1946, 103, 667.
13. Pfiffner I., Binkley S. и др., J. Am. Chem. Soc., 1947, 69, 1476.
14. S t о c s t a d E. и др., J. Am. Chem. Soc., 1948, 70, 3.
15. W e у g a n d F., Wacker' A., Schmied — Kowarzik V., Ber., 1949,
82, 25.
16. В 1 о о m E. и др., Science, 1944, 100, 295.
17. W a 1 1 e г С. и др. J. Am. Chem. Soc., 1948, 70, 19. Пат. США 2474022, 15/X
1946; Off. gaz. 1949, 623, № 3, 886.
18. T p у ф а н о в А. В. Витамины и антивитамины. M., Пищепромиздат, 1950,
с. 47.
19. О' Dell, Vandenbeld I., Bloom E., Pfiffner J. J., Am. Chem.
Soc., 1947, 69, 250.
20. Березовский В. M., Стрельчунас Л. И., Коган М. Я. Син-
тез птероил-Б-(-|-)-глутаминовой кислоты. — ЖОХ, 1957, т. 27, с. 1717—1722.
21. Weygand F., Schaefer G., Ber., 1952, 85, 307.
22. Seeger D., Cosulich D., Smith, J. Hultquist M., J. Am. Chem. Soc.,
1949, 71, 1753.
23. В о о t h e J. и др., J. Am. Chem. Soc., 1949, 71, 2304.
24. Uy eo S., lap. I., Pharmacy and Chem., 1949, 21, 237.
25. Angier R., Stocstad E. и др., J. Am. Chem., Soc., 1948, 70, 25.
26. Angier R. Пат. США № 2442836, 29/XI 1945; № 2442837,. 5/II 1946; Off.
gaz 1948, 611, № 2, 432, № 2466670, 29/XI 1945; Off. gaz. 1949, 621, № 2, 438.
27. Doub L., В a mb as L. Пат. США № 2476360, 14/XI 1946; Off. gaz. 1949, 624,
№ 3, 780.
28. G e г о c i J. Пат. США № 2501168, 28/VI 1947; Off gaz. 1950, 632, № 3, 797.
29. Weygand F., Schmied — Kowarzik V., Chem. Ber., 1949, 82, 833.
30. H u 1 t q u i s t M., Dreisbach P. Пат. США № 2443165, 4/X 1946; off. gaz.
1948, 611, № 2, 513.
31. U у e о S., M i z u k a m i S., Kubota T., T a k a g i S. J. Am. Chem.
Soc., 1950, 72, 5339.
32. T r a u b e W., Be r., 1900, 33, 1371, 3035.
33. Синтезы органических препаратов. M., ИЛ., 1953, 4, с. 149.
34. Березовский В. М., Стрельчунас Л. — «Труды ВНИВИ», М.,
Пищепромиздат, 1954, № 5, с. 28.
35. Березовский В. М., Каган М. Я., Стрельчунас Л. И. Авт.
свидет., № 103777, 1956; Бюлл. изобрет., 1956, № 7, с. 10.
36. F i s с h е г Е., Вег., 1899, 32, 2464.
37. J о s h i d a S h. I m a k i K., A k a g i S., C. A., 1950, 44, 3442. Япон. пат.
989, 1953; С. A., 1954, 48, 2089.
38. W i n t e r H., J. Am. Chem. Soc., 1940, 62, 3266.
39. Steiger M., J. Org. Chem., 1944, 9, 396.
40. К i n g F., S p e n s 1 e у P., N i m m о - Smith R., Nature, 1940, 162, 153;
C. A., 1948, 42, 8800.
41. U у e о S., M i z u k a m i S., Kubota T., T a k a g i S., J. Am. Chem.,
Soc., 1950, 72, 5339.
42. Б e p e з о в с к и й В. М. Химия витаминов. М., Пищепромиздат, 1959, с. 474.
43. Березовский В. М., Коган М. Я-, Стрельчунас Л. И. Авт.
свидет., № 104992, 1956; Бк?лл. изобрет., 1957, № 1, с. 13.
44. Березовский В. М., Юркевич А. М., Родионова Е. П. и
др. Авт. свидет., № 145567, 1961; Бюлл. изобрет., 1962, № 6, с. 23.
45. Boothe J., Mowat J. и др., J. Am. Chem. Soc., 1948, 70, 1099.
46. Mowat J., Hutchings В. и др., J. Am. Chem., Soc., 1948, 70, 1096. '
47. К u h E„ Smith J., U. S. Pat. № 2474184; C. A., 1949, 43, 7052.
48. H u t c h i n g s B., U. S. Pat. № 2470490; № 2470491; C. A., 1949, 43, 6672.
49. С о s u 1 i c h D., S m i t h J. J., Am. Chem. Soc., 1948, 70, 1922.
50. H u 1 t q u i s t M., Kuh E. и др., J. Am. Chem. Soc., 1949, 70, 23.
51. Roche Products, Brit. Pat. № 624394, 7/VI, 1949.
52. В о о t h e J., U. S., Pat. № 2547519, 3/IV, 1951.
53. Roche Products, Brit. Pat., № 631516, 3/XI, 1949.
54. L i n d 1 a r a H., К 1 a e n i H., U. S. Pat. № 2520156, 29/VIII, 1950.
55. S p i e g e 1 b e г g H., U. S. Pat. № 2487393, 8/XI, 1949.
56. S e m b J., U. S„ Pat. № 2491285, 13/XII, 1949.
228
57. В о о t h e J., W а 1 1 е г С. и др., J. Am. Chem. Soc., 1948, 70, 27.
58. Т s с h е s с h е В., Korte F., Р etersen R., Chem. Ber., 1951, 84, 579.
59. В о о t h е J. Пат. США № 2547519—520, 27-го VII 1946; Off. gaz. 1951, 645, 1,
220; № 2444002, 18/VII, 1946; Off. gaz. 1948, 611, 2, 513.
60. Co s u 1 1 c h В. Пат. США № 2444005, 31/XII, 1946; Off. gaz., 1948, 611, № 4,
991; № 2537006, 2/VII, 1949; Off. gaz., 1951, 642, 2, 442.
61. A n g i e г R. Пат. США. № 2442836, 20/XI, 1945; № 2442837, 5/II, 1946, Off. gaz.
1948, 611, 2, 432.
62. W a 1 1 e г С. Пат. США № 2442867, 12/III 1946; Off. gaz., 1948, 611, 2, 440.
63. H u 1 t q u i s t M., Dreisbach P. Пат. США № 2443165, 4/X 1946; Off. gaz.,
1948, 611, 2, 513.
64. W a 1 1 e г С., M о w a t J. Пат. США № 2500296, 23/II 1945; Off. gaz. 1950, 632,
2, 452.
65. S e e g e г D. Пат. США № 2570392, 23/Ш 1950; Off. gaz., 1951, 651. № 2, 426.
66. S e e g e г D. Пат. США № 2568597, 22/VIII 1947; Off. gaz., 1951, 650, № 3, 870.
67. Karrer P., Schwyzer R., Helv. Chim., Acta., 1948, 31, 777.
68. Roche Products Lim. Англ, пат.:
№ 624394, 23/IV 1947: опубл.7/VI 1949;
№ 626171, 23/IV 1947; » 11/VII 1949;
№ 628305, 13/VI 1947; » 25/VIII 1949;
№ 630751, 18/VII 1947; » 20/X 1949;
№ 714664, 1/X 1952; » 1/IX 1954;
69. В о о t h e J. Пат. США № 2472462; С. A., 2949, 43, 6673.
70. Аймухамедова M. Б. Получение глутаминовой кислоты, бетаина и их
практически важных производных ионообменным способом. Автореф. докт. дисс.,
М., 1965.
71. Аймухамедова М. Б. Значение и методы получения глутаминовой кисло-
ты, бетаина и их производных. Фрунзе, Изд-во АН Киргизской ССР, 1962.
72. Аймухамедова М. Б. Авт. свидет., № 162293, I960; Бюлл. изобрет.,
1964, № 9, с. 69.
73. Березовский В. М., Глебова Г. Д., Биринберг Е. М., К а-
занская Л. В. Синтез 4-амино(дезокси)-10-метилптероил-Б-глутаминовой
кислоты. — «Хим-фарм. ж.», 1968, № 12, с. 12—18 с ил.
74. Polaczkowa W., R о с z n i k i Chemi., 1956, 30, 119.
75. Березовский В. М. Каган М. Я-, Стрельчунас Л. И. Авт. •
свидет., № 103777, 1956, Бюлл. изобрет., 1956, № 7, с. 10.
76. Б у г р о в а Л. В., Руднев Г. К-, Радченко В. И. Способ получения
1,1,3-трихлорацетона. Авт. свидет., № 264385; Бюлл. изобрет., 1970, № 9, с. 27.
77. Березовский В. М., Биринберг Е. М., Глебова Г. Д. Авт.
свидет., № 245115; Бюлл. изобрет.; 1969, № 19, с. 32.
Глава 11. ПРОИЗВОДСТВО СИНТЕТИЧЕСКОЙ ЛИПОЕВОЙ КИСЛОТЫ
Липоевая (6,8-дитиоктановая) кислота является простетической группой
фермента декарбоксилирования пировиноградной кислоты и других а-ке-
токислот [1—31. Она впервые была выделена в 1951 г. в кристаллическом ви-
де из печени [41 и синтезирована в 1953 г. [5].
Липоевая кислота имеет перспективу широкого применения в лечебной
практике [6, 7]. Исходя из механизма действия, ее применяют при заболе-
ваниях печени (гепатит, цирроз). Заслуживает внимание факт нормализации
холестеринового обмена при действии липоевой кислоты [81. Замечено [7],
что желтуха при остром гепатите проходит быстрее. Липоевая кислота пре-
пятствует ожирению печени. Имеются указания на ее мочегонное действие
[6,7]; обезвреживает яды при интоксикациях [9]. Является фактором роста
цыплят [10]. Установлено, что липоевая кислота в растениях принимает
участие в фотосинтезе — в превращении световой энергии в химическую
[11, 12].
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ЛИПОЕВОЙ КИСЛОТЫ
Липоевая кислота имеет следующую химическую структуру:
НООС— (СН2)4— СН—СН2—СН2: C8HUO2S2; молекулярная масса 206,33.
229
Имеет два оптических антипода [13]
(+)-форма
(~) -форма
Кристаллы бледно-желтого цвета; температура плавления 60,5—61,5° С
[14]; 57—60° С [15]; температура плавления (+)—формы 45—48,5° С;
(:—) — формы 45—47° С [15]; (ф-) — кислота [а])3 ф- 104° (с = 0,88, бен-
зол); (—)—кислота [а]^ =—113° (с = 1,88, бензол). Для D, Л-формы Хтах =
= 332; 280 нм\ £)'“м = 142 (в этиловом спирте — для Хтах = 332) ; темпе-
ратура плавления 60—61° С (из циклогексана); ИК-спектр — 1700 см-1',
температура кипения 155—160° С [16].
Липоевая кислота хорошо растворима в органических растворителях,
нерастворима в воде. Считают, что липоевая кислота существует в форме
а и |3 [17, 18]. Однако этот вопрос дотстаточно не изучен.
МЕТОДЫ СИНТЕЗА ЛИПОЕВОЙ КИСЛОТЫ И ВЫБОР РАЦИОНАЛЬНОГО
МЕТОДА ДЛЯ ПРОИЗВОДСТВА [19]
Начиная с 1953 г., когда впервые была ситнезирована липоевая кислота
[5], до настоящего времени разработано более десятка методов синтеза ее.
Большинство этих методов предусматривает в качестве исходного сырья
адипиновую кислоту. Рассмотрим некоторые из наиболее перспективных
методов.
Синтез по методу Баллока и Хенда [20]. При синтезе липоевой кислоты
этим методом исходят из хлорангидрида этилового эфира адипиновой кис-
лоты [1], превращаемого реакцией Фриделя-Крафтса при действии этилена
(в присутствии безводного хлористого алюминия) в этокси-6-кето-7-октено-
ат (II). При гидрировании последнего под давлением в присутствии серово-
дорода получают эфир дигидролипоевой кислоты (III), который окисляют в
липоевую кислоту (IV), в присутствии Fe3\ Сероводород образуется в ав-
токлаве при гидрировании из добавляемой элементарной серы и водорода в
присутствии катализатора полисульфида кобальта.
Реакции протекают по следующей схеме:
сн2=сн2
С2Н5ООС—(СН2)4—СОС1-------> С2Н5ООС—(СН2)4—СО—СН2—СН2С1 —>
А1С!3
I
CH3COONa CoSn[H]
------> С2Н5-ООС—(СН2)4-С0—сн=сн2 —>
S
II
[01
—> С2Н5ООС—(СН2)4—СН—СН2—СН2-------> НООС— (СН2)4—сн—сн2-сн2
I I Fe3+ I I
SH—— SH S-------S
III IV
Выход липоевой кислоты составляет 29% (на II).
Недостатком метода является высокое давление в автоклаве при гидриро-
вании (150 кгс/см2), высокая температура (150° С) в среде уксусной кислоты
и выделение сероводорода.
Синтез, предложенный Шмидтом и Графеном [21]. При синтезе липоевой
кислоты этим методом также исходят из хлорангидрида моноэтилового эфи-
ра адипиновой кислоты (I), который конденсируют с ацетиленом (по Фриде-
230
лю-Крафтсу) в хлорвинилкетон (II). Последний без выделения при взаимо-
действии со спиртовой щелочью образует ацеталькетон (III), который вна-
чале гидрируют в присутствии частично «отравленного» никелевого катали-
затора и третичного амина в оксиацеталь (IV); затем с активным катализа-
тором Ренея при давлении 150 кгс/см2 и температуре 160—180° С восстанав-
ливают оксиацеталь (IV) в 6-окси-8-метоксиоктановую кислоту (V). При
ацетилировании (V) уксусным ангидридом в пиридине получают эфир 6-
ацето-8-метоксиоктановой кислоты (VI), который действием тиомочевины и
йодистоводородной кислоты превращают в дигидролипоевую кислоту (VII).
Последнюю окисляют кислородом в присутствии Fe3+ в липоевую кислоту
(VIII).
Реакции протекают по следующей схеме:
СИ СИ сн,он
C1COR------> [CI—СН=СН—СО—R] ——> (СН8О)2—CH—СН2—СО—R —»•
NaOH
I II III
[Н] [Н
—»• (СН8О)2—СН—СН2—СН—R —> СН8О—СН2—СН2—СН—R —»•
NI I Ni I
160—180° ОН ОН
IV V
nh2
(СН3СО 2о nh2
------> СН8О—СН2—СН2—СН—R-----------> СН2—СН2—СН—R
I HI I I
ОСОСНз SH------SH
VI . VII
[О]
----> СН2—СН2—СН—R', где R= —(СН2)4—СООС2Н5;
Fe3+ ] |
S--------S R'= —(СН2)4—СООН
VIII
При проверке метода (В. Турсин) выход полупродуктов на отдельных
стадиях оказался нестабильным. Перегонка промежуточных продуктов воз-
можна при низком остаточном давлении (0,1—0,3 мм рт. ст.), гидрирование
осуществляется при давлении 150 кгс/см2.
Синтез пе методу Аскера [22]. Здесь также исходят из хлорангидрида
моноэтилового эфира адипиновой кислоты (I), который конденсируют с
этиленом по реакции Фиделя-Крафтса в хлорвинилкетон (II). Последний
без выделения восстанавливают натрийборгидридом в этокси-6-окси-8-
хлороктаноат (III), который обрабатывают тионилхлоридом и превращают в
этокси-6,8-дихлороктаноат (IV). Далее взаимодействием этого дихлорида с
дисульфидом натрия получают липоевую кислоту (V). По данным автора,
выход липоевой кислоты составляет 68%. Синтез осуществляют по следую-
щей химической схеме:
сн2=сн2
С2Н5ООС— (СНа)4—COCI------> [С2Н5ООС—(СН2)4—СО—СН2—СНа—С1] -+
AIC1,
I II
NaBH4 SOCI2
---> С2Н5ООС—(СН2)4—сн—сн2 ---->
ХЗгБ S
—> С2Н5ООС—(СН2)4—СН—СН2—СН2-> НООС—(СН2)4—сн—сн2—сн2
II II
Cl Cl S-----S
IV V
231
В. Турсин считает этот метод наиболее приемлемым в связи со сравни-
тельно высокими и стабильными выходами полупродуктов, а также доступ-
ностью аппаратурного оформления. Этот метод усовершенствован В. Турси-
ным и Л. Чеботаревой [19].
ТЕХНОЛОГИЧЕСКАЯ СХЕМА СИНТЕЗА ЛИПОЕВОЙ КИСЛОТЫ [16, 19]
Синтез липоевой кислоты состоит из следующих стадий:
синтеза хлорангидрида моноэтилового эфира адипиновой кислоты из
моноэтилового эфира адипиновой кислоты действием тионилхлорида;
синтеза этилового эфира 6-окси-8-хлороктаноата из хлорангидрида
моноэтилового эфира адипиновой кислоты действием этилена в присутствии
хлористого алюминия и восстановлением боргидридом натрия полученного
хлорвинилкетона;
синтеза этилового эфира 6,8-дихлороктановой кислоты из этокси-6-
окси-8-хлороктаноата действием тионилхлорида;
синтеза липоевой кислоты из этилового эфира 6,8-дихлороктановой кис-
лоты и дисульфида натрия.
Хлорангидрицмоноэтилового эфира адипиновой кислоты получают по
следующей химической схеме:
С2Н5ООС—(СН2)4—СООН + SOC12 —> С2Н5ООС—(СН2)4—СОС1.
Моноэтиловый эфир адипиио- 118,98 Хлорангидрид моноЭтилового
вой кислоты 174,19 эфира адипиновой кислоты
192,64
В реактор из эмалированной стали 1 (рис. 33), снабженный прямым хо-
лодильником, приемником и щелочной ловушкой для улавливания НС1 и
SO2, загружают из сборника 2 моноэтиловый эфир адипиновой кислоты
(МЭАК) и из мерника 3 тионилхлорид. Реакционную массу перемешивают
30 мин и отгоняют в вакууме избыток тионилхлорида в сборник 4. Выход
79,3% [19]. Продукт сливают в обогреваемый сборник 7.
Хлорангидрид моноэтилового эфира адипиновой кислоты С8Н130зС1,
молекулярная масса 192,64, бесцветная жидкость с резким запахом, темпе-
ратура кипения 96—98° С (остаточное давление 3 мм рт. cm.); df 1,006;
tv® — 1,4428; хорошо растворим в органических растворителях. Моноэти-
ловый эфир адипиновой кислоты С8Н14О4, молекулярная масса 174,19,
имеет температуру плавления 28—29° С, температура кипения 148—152° С
(4 мм рт. cm.); d30 = 1,0805; п3® — 1,4337.
Этиловый эфир 6-окси-8-хлороктаноата (хлороспирт) получают по
реакции Фриделя-Крафтса из хлорангидрида моноэтилового эфира адипи-
новой кислоты и этилена с последующим гидрированием натрийборгидридом
кетогруппы этокси-6-кето-8-хлороктаноата (хлоркетона) по следующей схе:
ме:
сн2=сн2
QH5OOC—(СН2)4—СОС1--------> С2Н5ОО— (СН2)4—СО—СН2—СН2-С1 —>
AICI3
Хлорангидрид мояоэгилового -28,05 Эгокси-6-кето-8-хлороктаюата 220,71
эфира адипиновой кислоты
192,64
NaBH4
----> С2Н5ООС—(СН2)4—сн-сн2—сн2
I I
.37,85 ОН CI
Этокси-З-окси-З-хлорокЫнОаГ
222,72
В реактор из эмалированной стали 5 загружают Хлористый алюминий и
из мерника 6 дихлорэтан [23], перемешивают, а затем при охлаждении из
мерника 7 медленно добавляют хлорангидрид МЭАК. Затем при температу-
ре 25° С из баллона 8 через барботер пропускают этилен. Затем реакцион-
ную массу направляют в реактор 9 со льдом, образованным из воды при ох-
лаждении рассолом, и дихлорэтаном, сливаемым из мерника 10, эКстрагиру-
232
ют вещество. Массу сливают в делительную воронку И, где разделяют слои.
Органический слой, а также экстракт от промывки водного слоя дихлорэта-
ном переводят в сборник 12, а из него в вакуум-аппарат 13, где отгоняют
дихлорэтан в вакууме. Кубовый остаток растворяют в спирте, сливаемом в
вакуум-аппарат из мерника 14. Массу сливают в реактор 15, охлаждаемый
рассолом, куда постепенно загружают при перемешивании и охлаждении из-
мельченный боргидрид натрия. Затем из мерника 16 сливают аммиачную
воду, перемешивают и массу направляют в делительную воронку 17, в кото-
Рис. 33. Технологическая схема производства синтетической липоевой кислоты.
рой вещество извлекают дихлорэтаном (из мерника 10). Экстракт промывают
5%-ным раствором соляной кислоты из мерника 78, водный слой является
отходом, а органический передают в сборник 18а и далее в вакуум-аппарат
19, где отгоняют растворитель, а затем в глубоком вакууме (остаточное дав-
ление 0,06 мм рт. ст.) собирают фракцию, кипящую при температуре 112—
120° С. Выход 63,2%; = 1,4560; df = 1,0826; С10Н18О3С1, молекуляр-
ная масса 222,72. Фракцию направляют в мерник 20.
Этиловый эфир 6,8-дихлороктановой кислоты [19] (дихлорид) получают
из этокси-6-окси-8-хлороктаноата действием тионилхлорида по следующей
химической схеме:
soci2
С2Н6ООС—(СН2)4—СН—СН2—СН2------------> С2Н5ООС— (СН2)4—СН—СН2—СН2
ОН С1 С1 С1
Эгокси-6-окси-8-хлороктаноат Этокси-6.8-дихлороктаиоат
222,72 118,98 241,17
В реактор 27-, снабженный прямым и обратным холодильником и ловуш-
кой со щелочью, загружают из мерника 20 хлороспирт, из мерника 3
тионилхлорид, из мерника 22 бензол, а из мерника 23 каталитическое коли-
233
чество пиридина. Массу кипятят 1 ч, затем сливают в делительную воронку
24; бензольный слой направляют через сборник 25 в вакуум-аппарат 26,
где отгоняют бензол и при глубоком вакууме (остаточное давление 0,5 мм
рт. ст.) и температуре 104—108° С отбирают фракцию дихлорида и направ-
ляют ее в мерник 27. Дихлорид — бесцветная жидкость, температура кипе-
ния 104—108° С (остаточное давление 0,5 мм рт. cm.); d?° = 1,1026; п2° =
= 1,4620; Ci0H18CI2O2, молекулярная масса 241,17. Выход 88,6%.
Липоевая кислота. Ее получают по химической схеме:
NagS S
С2Н5ООС—(СН2)4—СН—СН2—СН2---------> НООС—(СН2)4—СН—СН2-СН2
II II
Cl Cl S-------S
Этокси-6,8-дихлороктаноат Липоевая кислота
241,17 78,05; 32,07 2М6,33
В реактор 28 из эмалированной стали, снабженной прямым и обратным
холодильником, загружают из мерника 29 спирт, затем сернистый натр и
серу и в токе азота, подаваемого из баллона 30, нагревают до кипения для
растворения осадка. Далее из мерника 27 медленно сливают дихлорид. Пос-
ле прибавления едкого натра (для омыления эфира) из мерника 31 и нагре-
вания в течение 10 ч при кипении спирт отгоняют, массу охлаждают водой
(через рубашку), подкисляют соляной кислотой из мерника 32 до pH 2,0,
сливают в делительную воронку 33, где вещество извлекают бензолом, пода-
ваемым из мерника 22. Органический слой направляют в сборник 34, а из
него в вакуум-аппарат 35, соединенный с холодильником 36, и охлаждае-
мым водой с температурой 30—35° С и приемником 37, обогреваемым водой
с температурой 55—60° С для предотвращения кристаллизации липоевой
кислоты в трубках холодильника и в приемнике. Вначале отгоняют бензол,
а затем при глубоком вакууме (остаточное давление 0,05 мм рт. ст.) и при
температуре 155—160° С отбирают фракцию липоевой кислоты и направляют
ее для перекристаллизации в обогреваемый водой мерник 38. Выход 68,4%
[19]. Для получения медицинской липоевой кислоты ее перекристаллизо-
вывают из циклогексана по двухступенчатой схеме с выходом около 80%
(на техническую кислоту).
ПРОИЗВОДНЫЕ ЛИПОЕВОЙ кислоты
Заслуживают внимания разработанные В. Турсиным и Л. Чеботаревой
методы синтеза следующих производных липоевой кислоты.
Амид липоевой кислоты [19] получают в среде тетрагидрофурана пои
участии триэтиламина пропусканием через реакционную массу аммиака
с выходом 73,4%; C8H15NOS2, молекулярная масса 205,34; температура
плавления 129,5—130° С.
В Японии запатентован метод получения амида липоевой кислоты [29]
взаимодействием эфира липоевой кислоты с аммиаком под давлением в при-
сутствии галоида аммония.
Натриевую соль липоевой кислоты [ 19] получают взаимодействием рас-
твора липоевой кислоты в метиловом спирте с раствором щелочи в метаноле с
выходом 90,4%. Аналогичным путем получены калиевые и кальциевые соли
липоевой кислоты.
D, Е-Липоил-Е-фенилаланин; -Е-валин; -Е-метионин [24]. Эти же со-
единения описаны в литературе [25]. Комплексы липоевой кислоты с амино-
кислотами, по-видимому, играют важную роль в обмене веществ [2, 26, 27 ].
Синтез заключается в конденсации липоевой кислоты с соответствующей
аминокислотой при участии изобутилхлоругольного эфира.
Комплекс имеет следующий химический состав:
СН2—СН2—СН—(СН2)4—СО—NH—R,
I I
S-------S
где R—остаток аминокислоты.
234
В связи с трудностью кристаллизации соединения выделяются в виде
бензгидриламмониевых солей [24].
Тиамин-липоат — производные обоих витаминов синтезированы ука-
занными авторами [28] в виде двух соединений: тиамин-8-(этил-6-ацетилди-
гидротиооктаноат-дисульфида (I) и дитиамин-6,8-(этилдигидротиоктаноата)-
дисульфида (II).
Н3С
Г)1 — С-СН;
нсо
NH2-HC1
''ТМ-
I
НСО
ЫН2-НС1
с—сн3
’ll J
С~СН2~СН2ОН
S-S-CH2-CH2-
I с-сн2- СН2ОН
S~S~CH2
\н„
N S^S-CH-fc^X-COOCaHs
H3C-J^ % нсо с-сн2—CH2OH
СН-(СН2)4~СООС2Н5 /N—С-СН3
SCOCH3 СН2
11
Исследование биологической активности указанных производных пред-
ставляет большой интерес.
ТЕХНОЛОГИЧЕСКИЕ ПОКАЗАТЕЛИ
РАСХОД ХИМИКАЛИЕВ, КГ НА 1 КГ
100%-НОГО ПОЛУПРОДУКТА
Хлорангидрид МЭАК
Моноэтиловый эфир адипиновой
кислоты (МЭАК)................1,14
Тионилхлорид...................0,78
Натр едкий (твердый)...........0,05
Выход (на МЭАК)...............79,3%
Этиловый эфир б-окси-8-хлорок.таноата
Хлорангидрид МЭАК............1,37
Этилен.........................0,40
Боргидрид натрия...............0,27
Дихлорэтан (с регенерацией) . 1,40
Алюминий хл’ористый............2,00
Аммиачная вода 25%-ная ... 1,0
Кислота соляная................0,10
Выход (на хлорангидрид МЭАК) 63,2%
Этиловый эфир 6, 8-дихлорок.тановой
кислоты
Оксихлороктаноат ..............1,05
Тионилхлорид .................. 0,56
Бензол (с регенерацией)........0,30
Натр едкий 4 ..................0,10
Пиридин........................0,002
Выход (на оксихлороктаноат) . . 88,6%
ЛИПОЕВАЯ КИСЛОТА
Дихлороктаноат (дихлорид) ... 2,14
Натрий сернистый 19..............2,30
Серный цвет ....................0,37
Натр едкий (твердый)............0,50
Спирт (с регенерацией)..........3,10
Кислота соляная.................3,20
Бензол (с регенерацией).........2,20
Расход полупродуктов, кг на 1 кг
липоевой кислоты
Этиловый эфир 6, 8-дихлорокта-
ноата...........................2,14
Этиловый эфир 6-окси-8-хлорок-
таноата.........................2,25
Хлорангидрид моноэтилового эфи-
ра адипиновой кислоты ......... 3,08
Моноэтиловый эфир адипиновой
кислоты.........................3,51
Расход химикалиев, кг на производство
1 кг медицинской липоевой кислоты
Алюминий хлористый..........4,50
Аммиачная вода 25%-ная .... 2,24
Бензол (с регенерацией) .... 2,84
Боргидрид натрия ............. 0,61
Дихлорэтан (с регенерацией) . . 3,15
Кислота соляная .............. 3,43
Натр едкий (твердый) .........0,91
Натрий сернистый............2,30
Пиридин....................0,005
Серный цвет.................0,37
Спирт....................• . . 3,10
Тионилхлорид ................. 3,60
Этилен ........................0,90
Эфир моноэтиловый адипиновой
кислоты.........................3,51
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
ЕЗбарский Б. И., Иванов И. И., Мордашов С. Р. Биологическая
химия. М., Медгиз, 1965, 520с.
2. М э с с и В., Гибсон К- 5-й Международный биохимический Конгресс, Сим-
позиум, V, 1961, 5, 21.
3. D a i g о К., Brady W., Reed L., J. Am. Chem. Soc., 1962, 84, 652.
4. R e e d L., Advances in Enzymology, 1957, 18, 319.
235
5. Hornberger C., Heitmiiller R., Gunsalus J., Schnakenberg G.,
Reed L., J. Am. Chem. Soc.', 1953, 75, 1273.
6. R a u s c h F., Atti del Simposio Internazionale Su 1’acido Tioctio, 1955, 177.
7. Colarusso А. Там же, 1955, 197 (Международный Симпозиум по липоевой кис-
лоте).
8, R a u s с h F., Klin. Wschr., 1956, 34, 737.
9. Cutolo, Experimentia, 1956, 12, 214.
10. В u s k B., Williams R., Archiv. Biochem et Biophys. Acta, 1957, 23, 34.
11. Calvin M., В a r 1 t г о p J., J. Am. Chem. Soc., 1956, 78, 4120.
12. В a s s h a m I., J. Am. Chem. Soc., 1956, 78, 4120.
13. Mislow K-> M e 1 u c h W., J. Am. Chem. Soc., 1956, 78, 2341.
14. A d a m s P., J. Am. Chem. Soc., 1955, 77, 5357.
15. W a 1 t о п E., Wagner А. и др., J. Am. Chem. Soc., 1955, 77, 5144.
16. Чеботарева Л. Разработка синтеза липоевой кислоты и исследования в об-
ласти ее производных. Автореферат кандидатской диссертации. М., 1965.
17. Reed L., Science, 1951, 114, 93.
18; Р a t t е г s о п Е., J. Am. Chem. Soc., 1951, 73, 5919.
19. Т у р с и н В. М., Чеботарева Л. Г., Филонова Л. М., П о п о-
в а С. М. и др. Синтез рацемической липоевой кислоты и ее производных. —
ЖОХ, 1964, т. 34, с. 3662—3664.
20. В u 1 1 о с k М., Hand J., Chem. Eng. News., 1955, 33, 1526.
21. Schmidt U., G r a f e п P., Ber., 1959, 92, 5, 1177.
22. A s к e r D., W а у n e W., J. Am. Chem. Soc., 1957, 79, 6483.
23. R e e d L., Ching -i-Niu, J. Am. Chem. Soc., 1955, 77, 416.
24. Ч e б о т a p e в а Л. Г., T у p с и н В. M., Лукьянова Л. 9- и ДР- Син-
тез бензгидриламмониевых солей D, А,а-липоил-А-фенилаланина, L-метилонина,
-А-валина. — ЖОХ, 1964, т. 34, с. 3665—3667.
25. D a i g о К. и др., J. Am. Chem. Soc., 1962, 84, 666, 662.
26. М a s s е у V., Biochim Biophys Acta, 1960, 38, 447.
27. N a w H. и др., J. Am. Chem. Soc., 1960, 82, 896.
28. Ч е б о т а р е в а Л. Г., Т у р с и н В. М., Садкова Л. С. и др. Синтез
коферментов, содержащих тиамин и липоевую кислоту. —ЖОХ, «Биологически
активные соединения», 1965, с. 221—224.
29. Фирма Фудзисава якухин когё. Япон. пат. № 15942, 1967.
Глава 12. ПРОИЗВОДСТВО СИНТЕТИЧЕСКОЙ
АСКОРБИНОВОЙ КИСЛОТЫ
Цинга была подробно описана в 16 в., но до 1912 г. не знали, что причи-
ной ее возникновения является недостаток витамина, которому Друммонд
[1 ] в 1920 г. дал название витамина С. До 1927 г. природа витамина С не
была установлена, несмотря на то что Бессонов уже в 1922 г. получил пре-
парат витамина С из капусты, а Зильва усиленно работал над выделением
витамина С из лимонного сока [2, 3].
В 1928 г. Сцент-Гиорги [4] впервые выделил из коры надпочечников
вещество с резко выраженными восстанавливающими свойствами и молеку-
лярной массой 176. Полагая, что по химической структуре это вещество
является кетогексоновой кислотой, он назвал его гексуроновой кислотой,
переименованной в 1933 г. в аскорбиновую кислоту [5].
Вскоре была установлена структура аскорбиновой кислоты [6, 7] и в
1933 г. осуществлен ее синтез Рейхштейном в Швейцарии [8, 9] и Гевортом
в Англии [10, 11, 12].
Аскорбиновая кислота принимает активное участие в окислительно-вос-
становительных процессах в организме и входит в состав ряда сложных
ферментов, обусловливающих процессы клеточного дыхания [13]. Витамин
С участвует в процессах углеводного и белкового обмена; повышает сопро-
тивляемость организма к инфекционным заболеваниям; регулирует холесте-
риновый обмен; участвует в нормальном функционировании желудка, ки-
шечника и поджелудочной железы; совместно с витамином Р обеспечивает
нормальную эластичность стенок кровеносных капилляров; стимулирует
образование протромбина; обезвреживает действие ряда лекарственных ве-
ществ (мышьяковая группа) и промышленных ядов (свинец). Аскорбиновая
236
кислота применяется при лечении цинги и является единственным средством,
излечивающим ее. Кроме того, аскорбиновую кислоту с успехом используют
при инфекционных заболеваниях, крупозной пневмонии, ревматизме, ту-
беркулезе, дифтерии, поражении сердечной мышцы, ахилии, язвенной бо-
лезни, при гепатитах и циррозах печени, базедовой и бронзовой болезнях,
при полиомиелите, шоковом состоянии и ряде других болезней [14].
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА АСКОРБИНОВОЙ КИСЛОТЫ
Витамин С — аскорбиновая кислота С6Н8О6, молекулярная масса 176,12,
представляет собой твердое вещество белого цвета. Оно кристаллизуется из
пересыщенных растворов в виде кристаллов моноклинической системы с
температурой плавления 192° С (с разложением).
ОН ОН
I I
н он с = с
III I
но-с-с-нс. .с=о
нА °
(у-Лактон -2,3-дезидро-Ь -гулонобой.
кислоты)
Аскорбиновая кислота оптически деятельна; ее удельное вращение в вод-
ном растворе [ojg1 = +23°, а в метаноле tcclj3 = +48° (при концентра-
ции с =0,85%). Удельное вращение, таким образом, зависит от вида
растворителя и, кроме того, от концентрации (с) вещества в растворе. Так,
например, для водного раствора аскорбиновой кислоты различных концен-
траций [15] удельное вращение характеризуется следующими данными:
С= 1%, [а$ = +24°,
С= 1,6%, [а]2“ = +23°,
с= 2,2%, = + 22,4°,
с = 14,0%, [а]“= +20,0°.
Натриевая соль аскорбиновой кислоты имеет [а д° = +105° [15]. Ас-
корбиновая кислота является одноосновной кислотой с
константами диссоциаций рК}=4,17, и рК2= 11,57 [12].
Аскорбиновая кислота в водном растворе имеет ти-
пичный спектр поглощения ультрафиолетовых лучей с
максимумом при 265 нм [16] при 1§£“°ль = 3,98 и неболь-
шую полосу между 350 —400 нм [17] при 1§£)'°ль =1,0.
На рис. 34 показан спектр поглощения ультрафиолето-
вого света для водного раствора аскорбиновой кислоты,
стабилизованной KCN [17 ] в эквимолекулярном коли-
честве. Окислительно-восстановительный потенциал
аскорбиновой кислоты Д’ равен при pH 4,0 и температуре
35° С +0,166 в. Децинормальный раствор аскорбиновой
кислоты в воде имеет pH 2,2. Аскорбиновая кислота хо-
рошо растворяется в воде. Растворимость ее в спиртах
зависит от числа атомов углерода в их молекуле. В мети-
ловом спирте она растворяется хорошо, в этиловом —
труднее, а в амиловом спирте трудно. В эфире, бензине,
бензоле, хлороформе, дихлорэтане и других неполярных
Рис. 34. УФ-спектр
поглощения аскор-
биновой кислоты.
237
растворителях аскорбиновая кислота практически нерастворима. Трудно
растворима в ацетоне. Растворимость аскорбиновой кислоты в воде и 96%-
ном этиловом спирте приведена в табл. 13 [18].
Таблица 13
Температура, Растворимость аскорбиновой кислоты, % Температура, °C Растворимость аскорбиновой кислоты, %
в воде в спирте в воде в спирте
0 13,59 3,33 45 35,46
5 15,57 — 50 38,24 8,27
10 17,79 — 55 40,64 —
15 19,85 — 60 42,34 10,65
20 22,42 4,61 70 46,57 —
25 24,48 — 78 17,76
30 27,10 . 5,50 80 50,47
35 29,97 —. 100 57,51
40 30,75 6,62 — — —
Соли аскорбиновой кислоты — аскорбинаты Na, Са , Fe и NH4 — раст-
воримы в воде [19—28]. Основная свинцовая соль нерастворима в воде и
спирте. Нейтральная свинцовая соль растворима в воде, но нерастворима
в алкоголе. Аскорбиновая кислота легко диффундирует через полупрони-
цаемые перегородки, сильно адсорбируется активированным углем и окис-
ляется.
Аскорбиновая кислота обладает сильной восстановительной способно-
стью. Раствор Фелинга, азотнокислое серебро и перманганат калия вос-
станавливаются при комнатной температуре; йод в кислом растворе обес-
цвечивается; красящие вещества восстанавливаются в свои лейкооснования.
На этом свойстве основаны методы ее количественного определения с 2,6-
дихлорфенолиндофенолом, йодом и йодноватокислым калием.
Большая восстановительная способность аскорбиновой кислоты обус-
ловливает ее неустойчивость к окислителям. Сухая чистая кристаллическая
аскорбиновая кислота устойчива по отношению к кислороду воздуха. В вод-
ных растворах в присутствии воздуха и особенно в щелочной или кислой сре-
де она быстро окисляется. Окисление аскорбиновой кислоты усиливается
при каталитическом действии тяжелых металлов, в особенности меди, а
также ферментативных систем [29, 30 ] рибофлавина и при действии ультра-
фиолетового света.
В. Вадова экспериментально установила влияние тяжелых металлов на
снижение устойчивости аскорбиновой кислоты в водных растворах [21 ]:
наиболее сильное разрушающее действие на аскорбиновую кислоту ока-
зывают медь и железо. Если металлы расположить по степени убывающего
действия их на окисление аскорбиновой кислоты, то получим ряд: медь, же-
лезо, алюминий, олово, свинец, никель, серебро и нержавеющая сталь;
интенсивность разрушающего действия тяжелых металлов увеличивается
с понижением концентрации растворов аскорбиновой кислоты:
растворы солей меди и железа с концентрацией около 10 мг!л уже су-
щественно влияют на стойкость аскорбиновой кислоты;
Химизм реакций окисления и распада аскорбиновой кислоты. Химиче-
ские процессы окисления и распада аскорбиновой кислоты еще не вполне
изучены. До последнего времени считали [31, 32], что эти процессы проте-
кают по следующим стадиям:
процесс окисления, при котором аскорбиновая кислота превращается в
дегидроаскорбиновую кислоту. Этот процесс является обратимым. При дей-
ствии сероводорода дегидроаскорбиновая кислота восстанавливается в ас-
корбиновую;
238
предполагают, что происходит разрыв лактонного кольца с превращени-
ем дегидроаскорбиновой кислоты в 2,3-дикето-/.-гулоновую кислоту. Этот
процесс необратим и протекает без участия кислорода (не является окисли-
тельным);
окисление 2,3-дикето-Б-гулоновой кислоты с разрывом цепи между ке-
тонными группировками с образованием щавелевой и L-треоновой кислот.
Таким образом, химизм распада аскорбиновой кислоты представляли в
следующем виде:
,О Х> СООН 1
X—1 X—1 соон соон
с-он 1 с=о 1 с=о Щавелевая
II о । о 1 кислота
С—ОН I ——- С=О 1 с=о соон
н-с 1 н-с 1 [ 1 н-с-он н-с-он
но-с-н но-с-н 1 1 но-с-н но-с-н
сн2он СН2ОН СН2ОН СН2ОН
L-А скорби новая Дегидроаскорбинобая 2,3-Дикето- L - Треоновая
кислота кислота Ь^гулонобая кислота кислота
Действительно, при окислении аскорбиновой кислоты йодноватокислым
натрием в щелочном растворе образуются щавелевая и Б-треоновая кислоты.
В щелочной среде возможно дальнейшее окисление Б-треоновой кислоты в
L-винную кислоту:
СООН
Н—С—ОН
НО—С—н
I
СН2ОН
L-Трэояовая кислота
СООН
I
Н—С—ОН
I
НО—С—Н
СООН
L-Вииная кислота
До последнего времени считали, что окисление аскорбиновой кислоты
протекает по указанной выше схеме в щелочной среде, а водные растворы ас-
корбиновой кислоты при pH < 7,0 устойчивы. Более того, прибавление
кислоты в раствор рассматривалось как фактор стабилизации аскорбино-
вой кислоты.
Однако последующие работы [33] показали, что при длительном хране-
нии или нагревании водных растворов аскорбиновой кислоты в них обна-
руживается фурфурол, а не щавелевая кислота. Кроме того, при распаде
аскорбиновой кислоты в водных растворах обнаружено нарушение эквимо-
лекулярного соотношения между израсходованным кислородом и окислен-
ной аскорбиновой кислотой. Например, в ампулированном водном растворе
содержалось 100 мг аскорбиновой кислоты при объеме воздуха в ампуле
0,5 мл. При хранении было разрушено 82 мг аскорбиновой кислоты, на что
требуется 7,5 мг кислорода. В ампуле же содержалось около 0,15 мг кисло-
рода. Возникает вопрос, откуда поступило 7,35 мг кислорода?
С другой стороны, в ампульном растворе обнаружен фурфурол, который
мог образоваться лишь в результате внутреннего процесса восстановления,
сопровождаемого выделением двух атомов водорода, необходимых для пре-
вращения 2,3-дикето-Б-гулоновой кислоты после ее декарбоксилирования в
фурфурол. Из этого примера видно, что в процессе распада аскорбиновой
кислоты участвует окислительно-восстановительный процесс,.причем можно
предполагать, что переносчиком водорода является аскорбиновая кислота.
239
В результате некоторых исследований [331 высказано предположение о
следующем химизме распада аскорбиновой кислоты в водных растворах при
pH < 7,0:
Л /О
С------п С—-п СООР
c-ОН I С=О I оо
II о г 1 I О I
c-ОН I [о] С=О I +Н2О с=о -со2
н-с------' н-с----------1 н-с-он "
но-с-н но-с-н но-с-н
сн2он СН2ОН СН2ОН
L - Аскорбиновая
кислота
Дегидроаскорбиновая
кислота
II
2,3 - диквто -L- гцлоно-
вая кислота
III
___________I
с=о
--► Н-С-ОН
I
но-с-н
I
СН2ОН
н-с=о
।
н-с-он
I
н-с-он
I
но-с-н
I
СН2ОН
-зн2о
н-с=о
I
с—
II
н-с
I
с-н
II
н-с—
Ксилозон
IV
L - Ксилоза
V
Ч’ИР'РУРОЛ
VI
н
Аскорбиновая кислота (I) под влиянием кислорода воздуха окисляется в
дегидроаскорбиновуюкислоту (II), которая при гидролизе[34] при рН<7,0
дает 2,3-дикето-А-гулоновую кислоту (III). Последняя под влиянием Н+-
ионов декарбоксилируется и превращается в ксилозон (IV) — весьма реак-
ционноспособное соединение, восстанавливаемое аскорбиновой кислотой в
L-ксилозу (V). Последняя циклизуется в фурфурол (VI). Сама же аскорби-
новая кислота,отдавая два атома водорода соединению (IV), окисляется в
дегидроаскорбиновую кислоту и далее в 2,3-дикето-А-гулоновую кислоту,
которая декарбоксилируясь, превращается в ксилозон и т. д.
Таким образом, можно предположить, что в данном случае происходит
цепная реакция окисления аскорбиновой кислоты. Для возбуждения цеп-
ной реакции достаточно наличия следов кислорода, а самый процесс ката-
лизируется наличием водородных ионов. Реакция развития или роста цепи
не требует кислорода извне. Отсюда становится понятным, почему в приве-
денном выше примере окисление значительного количества аскорбиновой
кислоты происходит при минимальных количествах кислорода. Автором
также установлено [33], что интенсивность распада аскорбиновой кислоты
значительно возрастает при понижении pH водного раствора аскорбиновой
кислоты (в диапазоне pH < 7,0), повышении температуры и увеличении ко-
личества кислорода, вовлекаемого в реакцию.
Приведенная концепция автора для химизма распада аскорбиновой кис-
лоты в водных растворах при pH < 7,0 требует дополнительного исследова-
ния. Что касается направления процессов распада аскорбиновой кислоты в
водных щелочных растворах, то этот вопрос также недостаточно изучен.
Следует отметить также, что в связи с опубликованием указанных выше
предположений о направлении распада аскорбиновой кислоты в водных раст-
ворах при pH < 7,0 высказано мнение о гидролитическом расщеплении ас-
корбиновой кислоты [35] по следующей схеме:
240
+ н2о
I
н-с-----
но-с-н
I
СН2ОН
L -Аскорбиновая
кислота
соон
I
с=о
НО-С-Н -СО2
н-с-он
I
но-с-н
I
сн2он
Z-Kemo-L-
гулоновая кислота
н
I
с=о
но-с-ГнП -н2о
н-с фон] ’
но-с-н
।
СН2ОН
L-Ксилоза
но-сн-сн о
-I II II
----*-сн2 с — с
1 г—J—I 1
оу он, н
Гнофсн—сн о
I II II
нфсн с - с
1—1 \ / I
-Н2о
сн—СН о
II II- II
•Рургрурм
Дигидрсоксиеруррурол
По этой схеме предполагается, что аскорбиновая кислота в растворе на-
ходится в равновесии с каким-то количеством 2-кето-А-гулоновой кислоты.
Последняя при определенных условиях декарбоксилируется с выделением
С02 и образованием А-ксилозы, которая, как пентоза, подвергается дегид-
ратации с образованием фурфурола.
Однако такая схема гидролитического распада аскорбиновой кислоты не
подтверждается экспериментально. В водном растворе чистой аскорбиновой
кислоты при длительном хранении обнаруживается дегидроаскорбиновая
кислота [33], образование которой не предусматривает данная схема. Пере-
ход же от аскорбиновой кислоты к дегидроаскорбиновой кислоте возможен
лишь в результате окислительного процесса.
Кроме того, установлено [36], что интенсивность распада аскорбиновой
кислоты в ампулированных водных растворах находится в прямой зависи-
мости от количества воздуха, находящегося в ампуле. Так, например, при
нагревании водного раствора, содержащего 352 мг % аскорбиновой
кислоты, в ампулах в течение 3 ч при температуре 100° С было раз-
рушено следующее количество аскорбиновой кислоты в зависимости от
объема воздуха (табл. 14). Если бы процесс распада аскор- Таблица 14
бйНОВОЙ КИСЛОТЫ НОСИЛ Характер ГИ- Количество дролитического расщепления, т. е. юоФФаскорФ- протекал без участия кислорода, то новой кислоты, Потери аскор- биновой кис- лоты, % Цветность раствора, ед. ВНИВИ
этот процесс. 183 0 В литературе [37] имеются также 40^6 данные по изучению распада аскорби- 28,2 новой кислоты в инертной атмосфере. 2>° Изучалась кинетика термолитическо- го распада; установлена константа скорости реакцт X 10 4, продолжительность периода полураспада 44 ч г и термический коэффициент 1,98 (реакция 1-го поряд гают, что механизм распада аскорбиновой кислоты в 30,1 25,0 17,6 5,1 ги распада 1ри темпера ка). Авторь инертной с 3,0 1,3 1,0 0,5 К = 157х туре 100° С I предпола- эеде проис-
ходит по схеме:
с —
н
I
с=о
с-он
II
с-он
I
н-с—
но-с-н
I
СН2ОН
С —
I
с=о
I о
____Н-с-он I -2НгО
н-с-----1
I
но-с-н
I
СН2ОИ
соон-
I
с=о
+Н2О I--c-Н -со2_
I Н-С-ОП_^2°
О I
с-н
II
с-н
241
Однако здесь же авторы отмечают: 1) что в опытах явление окисления в
начальном периоде (6 ч) наблюдалось (повышение К. почти вдвое) из-за не-
избежных следов кислорода; 2) что искусственный раствор L-ксилозы в анало-
гичных условиях (100°С) приводит к образованию незначительных количеств
ФУРФУРола (следов) только через 6 ч и при дальнейшем нагревании; 3) ссы-
лаясь на литературные данные [38], авторы указывают, что дегидроаскор-
биновая кислота в этих же условиях не приводит к образованию фурфурола
и что для фурановой циклизации необходимо присутствие восстановленной
аскорбиновой кислоты. Полагаем, что и в данном случае не имеем доказа-
тельства о протекании реакции распада аскорбиновой кислоты в отсутствие
кислорода, так как, по нашим воззрениям, достаточны следы кислорода, что-
бы реакция стала цепной.
Другие исследователи [39—45 ] также пытались определять скорость рас-
пада аскорбиновой кислоты в аэробных и анаэробных условиях при различ-
ных условиях проведения реакций. Однако при опытах, проведенных в ана-
эробных условиях, они также не учитывали следов кислорода, которые ос-
таются в растворе и в среде инертного газа [36]. В связи с этим эксперимен-
таторы отмечали так называемый неокислительный распад аскорбиновой
кислоты, хотя в значительно меньшей степени, чем в присутствии кислорода
воздуха [45]. По мнению автора, это вызвано вовлечением в цепную реак-
цию малых количеств кислорода.
Дегидроаскорбиновая кислота [35, 46], безводная, кристаллы бесцветные
с температурой плавления 225° С (с разложением); растворима в нейтраль-
ных органических растворителях; в воде растворима хорошо при температу-
ре60°С; сероводородом восстанавливается в Б-аскорбиновую кислоту; [а]д=
= 4-50°, 1%-ный водный раствор (по другим данным [47] + 55°); после
2 ч + 44°; после трех дней 4-16°; после 6 дней 0°.'Водный 1 %-ный раствор
имеет pH 3,37, а после 6 дней 2,08; 2,4-динитрофенилозазон имеет темпера-
туру плавления 282° С.
Безводную дегидроаскорбиновую кислоту получают [34] путем встряхи-
вания 8,8 г аскорбиновой кислоты с 12,4 г ресублимированными йодом в 75 мл
метанола (влажность 0,5%). Добавляют 30—35 г карбоната свинца, отфильт-
ровывают осадок свинцовых солей. Следы свинца удаляют из фильтрата
сероводородом, фильтруют через кизельгур. Фильтрат упаривают при ваку-
уме (температура 30—40° С). После охлаждения получаемую стекловидную
массу взбалтывают с 30 мл абсолютного спирта и кристаллизуют в течение
двух дней при 0°. После удаления спирта промывкой и высушиванием
получают 2,0 г дегидроаскорбиновой кислоты.
Изомеры и аналоги L-аскорбиновой кислоты. Аскорбиновая кислота
имеет два асимметрических атома углерода в положениях 4 и 5 и образует
четыре оптических изомера и два рацемата. Оптические изомеры представ-
лены следующими формулами:
ОН ОН он он
сн2он сн2он
Z - Аскорбиновая D - Аскорбиновая
сн2он сн2он
L - Изоаскорбиновая D -Изоасксрбиновая
кислота кислота
(L - Арабоаскорбиновая) (П-Арабоаскорбиновая)
кислота кислота
Примечание. Температура плавления /.-аскорбиновой кислоты 192° С,
[а]д =-[- 23°(НаО); температура плавления D-аскорбиновой кислоты 192° С, [а]д=
= —23°(НаО); температура плавления L-изоаскорбиновой кислоты (Т-арабоаскорби-
новой) 174° С, [а]д =+17°; температура плавления О-изоаскорбиновой кислоты
(О-арабоаскорбиновой) 174°, [а]д = —17°.
242
Витаминная активность изомеров и аналогов Л-аскорбиновой кислоты
обусловлена наличием систем А, а,|3-бутенолида, причем 7-лактонное кольцо
должно быть образовано по гидроксильной группе положения 4£)-конфигу-
рации. В противном случае (по A-конфигурации) соединения неактивны
(например, А-арабоаскорбиновая кислота).
Положение гидроксила в положении 5 должно соответствовать /.-конфи-
гурации; в противном случае активность уменьшается в 20 раз, как это наб-
людается у D-арабоаскорбиновой кислоты [46—51 ]. Элиминирование груп-
пы ОН в положении 6, как, например, у 6-дезокси-А-аскорбиновой кислоты,
снижает активность в 3 раза [52]. Также снижается активность при увели-
чении в молекуле числа углеродных атомов, как, например, в 5 раз для
L-рамноаскорбиновой кислоты, в 40 раз для глюко-и в 60 раз для А-галак-
тоаскорбиновой кислоты.
МЕТОДЫ СИНТЕЗА АСКОРБИНОВОЙ КИСЛОТЫ И ВЫБОР РАЦИОНАЛЬНОГО
МЕТОДА ДЛЯ ПРОИЗВОДСТВА
Синтез аскорбиновой кислоты принципиально может быть осуществлен
следующими четырьмя методами [12, 21, 35, 53, 54].
изомеризацией и лактонизацией 2-кетогексоновых кислот;
из пентоз через озазон и нитрил (озонпианидный метод);
конденсацией бензоинового типа двух альдегидов с низкой молекуляр-
ной массой под каталитическим влиянием KCN с образованием кетоспирта;
конденсацией эфиров а-оксикислот.
Анализируя указанные методы (подробно см. [35, 53, 54]), необходимо
отметить следующее:
озон-цианидный метод синтеза через 3-кетогексоновые кислоты не может
найти практического применения из-за отсутствия промышленных ресурсов
ликсозы. Синтез /,-ликсозы из D-галактозы или из D-глюкозы очень сложен;
метод бензоиновой конденсации двух альдегидов, как, например, этил-
глиоксилата и L-треозы, не перспективен, как из-за дефицитности сырья,
так из-за неоднозначности реакции конденсации альдегидов (альдольная
конденсация, образование бензоинов из одного итого же альдегида). Это
приводит к низкому выходу целевого продукта;
метод конденсации эфиров а-оксикислот также неэффективен из-за
сложности синтеза этих эфиров и из-за низкого выхода аскорбиновой кисло-
ты.
Промышленное применение нашел метод синтеза /.-аскорбиновой кисло-
ты из 2-кето-/.-гексоновой кислоты по методу Рейхштейна [55]. Это обус-
ловлено тем, что основное сырье (D-глюкоза) и вспомогательные химикалии,
применяемые для синтеза, используются в пищевой и химической промыш-
ленности. Технология синтеза сравнительно несложная и состоит лишь из
пяти стадий.
Некоторый интерес представляет аналогичный синтез /.-аскорбиновой
кислоты из галактуроновой кислоты [56]. Сущность метода заключается в
обработке пектиновых веществ ферментом пектиназой; в результате этого
получают галактуроновую кислоту, которую выкристаллизовывают в виде
кальциевой соли. Последнюю водородом в присутствии катализатора вос-
станавливают в /.-галактоновую кислоту, которую окисляют в 2-кето-/.-
галактоновую кислоту, лактонизируют и енолизируют в /.-аскорбиновую
кислоту (более подробно [21 ]). Этот метод мог бы найти применение при усо-
вершенствовании первой стадии— получение Са-соли галактуроновой кис-
лоты из свекловичного жома, выход которой менее 1 % к массе сухого жома.
Интерес также представляет микробиологический метод получения /.-ас-
корбиновой кислоты из D-глюкозы, включающий пять стадий, в том числе
три стадии химических [57, 58]. Метод заключается в окислении D-глюко-
зы уксуснокислыми бактериями Acetobacter Suboxydans в Са-5-Kero-D-
243
глюконат, который водородом в присутствии никелевого катализатора
восстанавливают и в связи с возникновением нового асимметрического атома
углерода образуются два оптических изомера — Ca-D-глюконат (50%) +
+ Ca-L-идонат (50%). После эпимеризации полученную смесь сбраживают
культурой Pseudomonas chromospirans, в результате этого получают Са-2-
кето-Й-глюконат (50%) + Са-2-кето-Т.-идонат (50%). Последнюю лактониза-
цией и енолизацией превращают в L-аскорбиновую кислоту (Г. Фербер).
Этот метод привлекает внимание ограниченным применением химических
реагентов и почти полным исключением органических растворителей. Од-
нако, как известно, процессы биохимического окисления трудно управляе-
мы; выход получаемых в этих процессах полупродуктов низок в связи с об-
разованием оптических антиподов в виде производных глюконовой и идоно-
вой кислот.
Широко внедренный в промышленности синтез L-аскорбиновой кислоты
из D-глюкозы по методу Reichsteina, усовершенствованному в СССР, сос-
тоит из следующих пяти стадий:
получение D-сорбита из D-глюкозы методом каталитического восстанов-
ления последней водородом при давлении 80—100 кгс/см2 и температуре
135—140° С;
получение /.-сорбозы из D-сорбита путем биохимического глубинного
окисления последнего уксуснокислыми бактериями;
получение диацетон-/.-сорбозы из /.-сорбозы путем обработки последней
ацетоном и в присутствии катализатора серной кислоты, отгонки ацетона и
выделения диацетонсорбозы щелочью;
окисление диацетон-/.-сорбозы в гидрат диацетон-2-кето-/.-гулоновую
кислоту перманганатом калия или гипохлоритом в щелочной среде с выде-
лением гидрата диацетон-2-кето-/.-гулоновой кислоты соляной кислотой;
енолизация и лактонизация гидрата диацетон-2-кето-Т.-гулоновой кис-
лоты в аскорбиновую кислоту в среде хлороформа или дихлорэтана и в
присутствии катализатора хлористого водорода, а также перекристаллиза-
ция технической аскорбиновой кислоты в чистый продукт.
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА
ПРОИЗВОДСТВО D-СОРБИТА ИЗ О-ГЛЮКОЗЫ
В производстве синтетической аскорбиновой кислоты D-сорбит является
первым промежуточным продуктом синтеза. Сорбит представляет собой шес-
тиатомный спирт, изомер маннита, талита, идита и дульцита. Он представ-
ляет собой белый кристаллический порошок, легко растворимый в воде.
Кристаллизуется с 0,5 молекулы воды. Химическая формула сорбита
С6Н8(ОН)(<-О,5Н2О, молекулярная масса безводного вещества 182,17. В 96%-
ном спирте трудно растворим, а в абсолютном алкоголе почти нерастворим.
Температура плавления безводного вещества 110° С. Удельное вращение
[59] водного раствора [ц]^’ = —1,7°. Показатель преломления водного
раствора определяется по формуле [53]:
[«]*> = 1,33298 + 0,00014327 5,
где В—концентрация сорбита, г/л.
Плотность (при температуре 20° С) и содержание сухих веществ в вод-
ных растворах сорбита по показаниям сахарной шкалы рефрактометра при-
ведены в табл. 15 [53].
244
Таблица 15
Показания са- харной шкалы рефрактометра при 20сС Плотность, кг/м* Содержание сухих ве- ществ в водном рас- творе сорбита Показания сахарной шка- лы рефракто- метра при 20°С Плот- ность, кг/м2 Содержание сухих веществ в водном рас- творе сорбита
% г в 100 мл % г в 100 мл
5 1017 5,0 5,1 35 1138 35,2 40,1
10 1035 10,0 10,4 40 1159 40,6 47,1
15 1055 15,0 15,8 45 1182 46,0 54,4
20 1074 20,1 21,6 50 1206 51,4 62,0
25 1094 25,1 27,5 55 1231 56,7 69,8
30 1115 30,2 33,7
Сырьем для производства D-сорбита в настоящее время служит D-глюко-
за, кторая является сравнительно дорогим видом сырья. А. Баландин,
Н. Васюнина, С. Чепыго и Г. Барышева [60, 611 разработали метод полу-
чения D-сорбита из непищевого растительного сырья (хлопковый линт,
сульфитная целлюлоза) путем гидролитического гидрирования последних.
Процесс представляет собой совмещение двух каталитических реакций —
гидролиза полисахаридов с образованием моноз и гидрирования последних
в многоатомные спирты. Гидролитическое гидрирование протекает в присут-
ствии двух катализаторов: гидролизующего катализатора — фосфорной
кислоты (0,7%) и гидрирующего катализатора — рутения на угле или сили-
кагеле (0,5%) при температуре 170—180° С и давлении 80—90 кгс/см\ Этот
метод является весьма перспективным, но в связи с дороговизной катализа-
торов требует тщательной технологической отработки.
Конечно, для данной цели может быть также использована сахароза при
раздельном процессе гидролиза и гидрирования с применением более деше-
вого катализатора (например, никелевого). Однако этот вопрос требует
специального исследования. В настоящее время на витаминных заводах
D-сорбит получают восстановлением D-глюкозы путем гидрирования по-
следней в присутствии катализатора.
Реакция восстановления глюкозы протекает по следующей схеме;
СН2ОН СН2ОН
ОН—С—Н НО— С—Н
I I
НО—С—Н [Н] НО—С—н
I —>1 I
н—с—он н—с—он
I I
но—с—н но—с—н
н—с=о СН2ОН
D-Глюкоза D-Сорбит
183,16 182,17 '
Каталитическое гидрирование глюкозы. Процесс восстановления D-глю-
козы в D-сорбит может быть осуществлен двумя способами:
каталитической гидрогенизацией;
электролизом глюкозного раствора.
Каталитическая гидрогенизация обеспечивает получение высококачест-
венного сорбита и высокий выход его. Однако сам процесс гидрогенизации
требует сложной аппаратуры, работающей под давлением в 100 кгс!см\
Изготовление катализатора и его регенерация осуществляются в специаль-
ной аппаратуре.
Электролитическое восстановление D-глюкозы в D-сорбит осуществля-
ется при комнатной температуре и не нуждается в применении дорогостоя-
щего катализатора — в этом его преимущество. Однако в процессе электро-
литического восстановления получается раствор сорбита, загрязненный изо-
245
мером маннитом. Разделение этих изомеров представляет большие труднос-
ти. Поэтому в настоящее время на витаминных заводах применяют метод ка-
талитической гидрогенизации глюкозы под давлением в присутствии ката-
лизатора.
Установлено, что для этого процесса имеет значение вид катализатора,
способ его приготовления и регенерации и чистота водорода. Следы окиси
углерода, сероводорода и других сернистых соединений, фосфористых и
мышьяковистых соединений, хлористого водорода действуют как каталити-
ческие яды.
Для гидрогенизации необходимо использовать водород, полученный
электролитическим путем. В качестве катализатора применяют скелетный
никель, получаемый выщелачиванием алюминия щелочью из сплава, содер-
жащего 67—70% алюминия и 30—33% никеля. Кроме никеля, для гидроге-
низации могут применяться катализаторы из других металлов: медь, вос-
становленная из окиси меди при температуре 200° С; железо и кобальт, вос-
становленные из соответствующих окисей при температуре 400—500° С.
Железный и кобальтовый катализаторы приготовляют на трегерах, так как
индивидуальные катализаторы легко спекаются при высокой температуре.
Приготовление и регенерация скелетного никелевого катализатора.
Для приготовления алюминиево-никелевого сплава применяют никель сор-
тов Н4 и Н2 и алюминий марки А, сорт I, содержащий 99,5% А1. Болванки
сплава могут дробиться в дробилках типа Блека либо на строгальных стан-
ках до величины зерен 3—5 мм. Крупные зерна отсеивают ситом с диаметром
ячеек 3 мм, а пыль шелковым ситом.
Из алюминиево-никелевого сплава изготовляют катализатор путем раст-
ворения части алюминия в щелочи. Реакция протекает по уравнению:
2А1 + 2NaOH + 2Н2О —> 2NaA102 + ЗН2 или 2А1 + 2NaOH + 6Н2О -+
—>• 2Na [Al (ОН)41 + ЗН2.
Вследствие растворения и удаления почти 50% алюминия оставшийся
никель приобретает развитую контактную поверхность (скелетную струк-
туру), обусловливающую его высокую активность. Процесс обработки
сплава щелочью известен под названием «выщелачивание сплава». Этот про-
цесс осуществляют в аппарате-выщелачивателе. Он представляет собой же-
лезный котел, снабженный мешалкой и водяной рубашкой. Загрузку сплава
осуществляют через бункер, установленный над люком. В аппарат через
штуцер трубопроводом подводят 25 %-ный раствор едкого натра из расчета
8—10 л щелочи на' 1 кг сплава. При размешивании прибавляют небольшими
порциями измельченный сплав. Охлаждением водой температуру реакцион-
ной массы поддерживают не выше 70° С. Тотчас же начинается выделение
водорода. Его отводят через трубопровод и гравийный предохранительный
фильтр в атмосферу. При засыпке сплава непрерывно продувают вы-
щелачиватель азотом (1—2 Л13/ч).
После прекращения выделения водорода, что служит признаком оконча-
ния реакции растворения алюминия, массу охлаждают до 50—60° С путем
подачи холодной воды. Осадку катализатора дают отстояться в течение
2—3 ч. После продувки азотом отстоявшийся раствор алюмината натрия
декантируют, а катализатор фильтруют через нутч-фильтр для отделения
осадка катализатора от остатка раствора алюмината натрия.
По окончании фильтрации осадок (черного цвета) промывают горячей
водой (желательно конденсатом водяного пара) до нейтральной реакции на
фенолфталеин. Промытый катализатор немедленно снимают с фильтра и
помещают в закрытый приемник, где он хранится под слоем воды или спир-
та для предотвращения его соприкосновения с воздухом.
Отработанный катализатор регенерирует путем обработки его 5%-ным
раствором NaOH при кипении и промывкой дистиллированной водой.
246
Регенерированный катализатор добавляют в двойном количестве вместо
свежего. Активность катализатора определяют по его пирофорности — вос-
пламенению фильтрованной бумаги после высыхания нанесенного на нее
катализатора (свежего) и по степени разогрева при высушивании на фильт-
ровальной бумаге (отработанного).
Истираемость катализатора может быть определена по опубликованной
методике [621.
В последние годы проведены некоторые исследования, имеющие отноше-
ние к технологии приготовления никелевого катализатора.
Исследовано влияние способа приготовления на свойства скелетного ни-
келевого катализатора [63]. Показано, что температура и продолжитель-
ность выщелачивания алюминия, а также температура использования ка-
тализатора существенно влияют на распределение водорода по поверхнос-
ти катализатора.
Изучен процесс гидрирования глюкозы на никелевом катализаторе с до-
бавкой от 0,01 до 0,1 % платины или родия [64]. Добавка платины или родия
существенно не влияет на активность катализатора. Активность и стабиль-
ность катализатора повышается при введении в качестве промотора марган-
ца в количестве 5—10% [64].
В качестве промотора для скелетно-никелевого катализатора, применяе-
мого для гидрогенизации масел, был предложен палладий в количестве 0,1 %
[64].
Интерес представляет работа по выявлению способа хранения никелевого
катализатора, который не снижал бы его активность [65]. Свежеприготов-
ленный катализатор представляет собой нестабильную систему. Она дости-
гает равновесия через 10—30 дней. Для поддержания высокой активности
катализатора рекомендуется хранить его при низкой температуре и в среде
полярного растворителя. Так, при хранении образца в 96%-ном спирте при
температуре —78° С активность его через 450 дней сохранилась полно-
стью.
При выщелачивании сплава и фильтрации необходимо соблюдать сле-
дующие правила:
измельченный сплав следует вводить в котел-выщелачиватель небольши-
ми порциями во избежание бурного выделения водорода и выбрасывания
щелочи и катализатора в вытяжную трубу;
едкий натр следует применять в количестве не более 10 л на 1 кг сплава,
так как избыток щелочи приводит к полному растворению алюминия и зна-
чительному уменьшению механической прочности катализатора;
при фильтрации необходимо следить за тем, чтобы катализатор находил-
ся под слоем жидкости и не соприкасался с воздухом во избежание «отрав-
ления» катализатора;
процесс выщелачивания алюминия щелочью следует вести в присутствии
азота;
после слива реакционной массы на нутч-фильтр следует при помощи
душевого устройства смыть водой с крышки и внутренней поверхности реак-
тора частицы катализатора.
Приготовление раствора глюкозы. Кристаллическая глюкоза С6Н12О6х
х Н2О (ГОСТ 975—63) представляет собой белый кристаллический
порошок. Кристаллы должны проходить через сито с диаметром отверстий
1,5 мм. Для гидрогенизации применяют 50—55%-ный водный раствор глю-
козы, который приготовляют в специальном реакторе-растворителе, снаб-
женном паровой рубашкой и мешалкой. В котле подогревают воду до 70—
75°С, а затем прибавляют глюкозу из расчета 122 кг гидратной глюкозы на
100 л воды. Затем в котел добавляют активированный уголь из расчета 0,5 кг
на 100 кг глюкозы, продолжают перемешивание в течение 5—10 мин при тем-
пературе 75° С и фильтруют через нутч-фильтр. В очищенный углем раствор
добавляют известковой воды до pH 8,0—8,1 [70 а] и в таком виде раствор на-
правляют для гидрогенизации. Известковую воду получают путем настаива-
247
ния извести (строительная, гидратная, ГОСТ 9179—59) в воде (1 : 20). От-
стой воды должен содержать 1,5—1,7 г!л извести.
Процесс гидрогенизации [66—68 а]. Гидрогенизацию глюкозы осу-
ществляют либо автоклавным периодическим способом, либо в непрерывно
действующих установках. Автоклавное гидрирование производят в горизон-
тальных автоклавах высокого давления. .
Автоклавный процесс гидрогенизации осуществляют следующим образом.
Автоклав промывают горячей водой, масленки заполняют смазочным мас-
лом, закрывают спускной вентиль и засасывают в автоклав раствор глюкозы
и отдельно катализатор (его вводят 5% к массе товарной глюкозы). Затем
для удаления воздуха автоклав продувают два раза азотом и один раз водо-
родом. Когда давление водорода в автоклаве снизится до 5 кгс!см2, закрыва-
ют продувной кран, осторожно открывают водородный вентиль и доводят
давление в автоклаве до 80 кгс!см2.
Затем пускают пар в рубашку, открывают вентиль для конденсата на
конденсационный горшок, открывают воду на сальник и приводят в движе-
ние мешалку. Температуру раствора в автоклаве повышают до 135—140° С.
Для поддержания в автоклаве постоянного давления водорода (около
80 кгс/см2) необходимо непрерывно подавать водород в автоклав. После окон-
чания гидрирования, определяемого прекращением падения давления водо-
рода в автоклаве в течение 20 мин, выключают подачу пара в рубашку, за-
тем пускают в нее холодную воду, охлаждают раствор сорбита до 75—80° С,
выпускают водород через продувной трубопровод в следующий готовый к
пуску автоклав, а под конец процесса — в атмосферу. Когда давление в ав-
токлаве снизится до 5—7 кгс!см2, осторожно открывают продуктовый спуск-
ной кран. Под давлением раствор сорбита совместно с катализатором посту-
пает в сборник, а из сборника для отделения катализатора и тщательной про-
мывки его горячей водой масса поступает в нутч-фильтр. Промытый катали-
затор поступает на регенерацию.
Имеются данные, что американская фирма «Мерк», несмотря на большие
промышленные масштабы производства D-сорбита, применяет периодичес-
кий процесс гидрогенизации глюкозы в автоклавах при температуре 150° Си
избыточном давлении 70 кгс!см2 [68 а].
Американская фирма «Атлас Пудер Ко» [69] осуществляет непрерывный
процесс гидрогенизации глюкозы со суспензированным катализатором Ре-
ней-никель, который отфильтровывают на фильтр-прессах.
В Венгрии на одном из заводов производят сорбит по методу Института
высокого давления [70] непрерывным процессом. Гидрогенизацию D-глю-
козы осуществляют при избыточном давлении 200 кгс!см2 и температуре 180—
200° С синтез-газом в присутствии суспензированного катализатора Реней-
никель.
В ГДР на заводе «Гидрирверк» гидрирование глюкозного раствора про-
изводится непрерывным процессом при температуре 120—140° С и избыточ-
ном давлении 201—240 кгс!см2 при участии стационарного медно-никелевого
катализатора. По-видимому, при непрерывном процессе более эффективным
является применение суспензированного катализатора, так как при этом
достигается лучшее использование объема автоклава и повышение контакт-
ной поверхности катализатора нежели при стационарном катализаторе.
Последний обычно применяют либо в виде крупных зерен (5—8 мм), либо в
виде таблеток, которые занимают значительную часть объема автоклава и
обладают малой контактной поверхностью.
При осуществлении технологического процесса гидрогенизации Д-глю-
козы необходимо учитывать побочные реакции, протекающие в этом про-
цессе.
Замечено, что при смешении щелочного раствора D-глюкозы (I) с катали-
затором после некоторого стояния смеси происходит быстрое снижение pH
раствора, по-видимому, в результате энергичного окисления D-глюкозы в
D-глюконовую кислоту (II) кислородом воздуха в присутствии катализатора.
248
Установлено также, что в процессе гидрогенизации наблюдается сниже-
ние pH вследствие возможного протекания реакции Канниццаро с образо-
ванием D-сорбита (III) и D-глюконовой кислоты (II).
Известно также, что D-глюкоза в щелочной среде может подвергаться
енолизации и изомеризации в D-фруктозу (IV) и /)-маннозу(У). D-фруктоза
вследствие образования при восстановлении кетогруппы нового асимметри-
ческого атома углерода при гидрогенизации превращается в D-сорбит (III)
и D-маннит (VI). Последние исследования показали [706], что в процессе
гидрогенолиза глюкозы, кроме D-сорбита, образуются: этиленгликоль,
глицерин, эритрит, ксилит и пропиленгликоль.
Ниже приведены схемы указанных реакций. .
СООН
• .1
н-с-он
I
но-с-н
н-с-он
I
н-с-он
I
сн2он
с=о
но-с-н
н-с-он
СН2ОН
н-с=о н-с-он
I II
н-с-он с-он
I I .
но-с-н но-с-н
н-с-он-* н-с-он
н-с-он н-с-он
I I
сн2он сн2он
I
н-с-он
, сн2он
< IV
н-с=о
I ,
но-с-н
I
но-с-н
' н-с-он
I
н-с-он
I
сн2он
сн2он
н-с-он
но-с-н
н-с-он
н-с-он
СН2ОН
111
СН2ОН
но-с-н
I
но-с-н
н-с-он
н-с-он
I
сн2он
.VI
соон
I
н-с-он
I
но-с-н
н-с-он
I
н-с-он
I
сн2он
СН2ОН
н-с-он
I
но-с-н
I
н-с-он
I
н-с-он
I
сн2он
II
Для того чтобы свести к минимуму отрицательный эффект побочных ре-
акций, необходимо:
в периодическом процессе не допускать хранения щелочного раствора
Д-глюкозы с катализатором. Рационально катализатор вводить отдельно в
автоклав в присутствии азота перед началом продувки;
реакцию гидрирования рационально вести при pH, близком к нейтраль-
ному (7,3—7,5), так как в щелочной среде при температуре 135—140° С
D-глюкоза будет подвергаться распаду. Однако при этом надо учесть неко-
торое снижение pH при смешивании катализатора с раствором D-глюкозы в
автоклаве. Поэтому pH раствора добавлением известковой воды следует до-
вести до 8,0;
подщелачивание раствора D-глюкозы следует производить известковой
-водой (отстоем); едкий натр или кальцинированная сода интенсифицирует
249
распад глюкозы и, кроме того, являются сильными патокообразователями и
снижают выход кристаллической L-сорбозы [70а];
вода для растворения глюкозы не должна быть жесткой, лучше дистил-
лированная;
глюкозный раствор должен быть бесцветным и не содержать посторонних
солей, в особенности сернистых. Реакция раствора должна быть слабоще-
лочной (pH 8,0);
водород не должен быть загрязнен побочными газами (SO2, СО, СО2 и
др.), так как последние отравляют катализатор. Рекомендуется применять
электролитический водород;
катализатор должет быть тщательно подготовлен и промыт. Величина
зерен катализатора должна быть 1—2 мм\
автоклав следует загружать глюкозным раствором на 0,6—0,7 емкости.
Повышенная загрузка автоклава приводит к удлинению процесса, а пони-
женная — к увеличению потери водорода;
следует осуществлять глубокую гидрогенизацию глюкозного раствора.
Остаточная глюкоза не должна превышать 0,1%;
Непрерывный процесс гидрогенизации позволяет применять автомати-
ческий контроль и регулирование его, что обеспечивает более высокое ка-
чество продукта и увеличение производительности труда. Поэтому при боль-
шой мощности сорбитного цеха целесообразно процесс гидрирования осу-
ществлять непрерывно.
Очистка сорбитного раствора от тяжелых металлов1. Метод осаж-
дения. Получаемый раствор D-сорбита содержит примеси солей тяжелых
металлов (железа, меди, никеля) и алюминия. Эти примеси оказывают отри-
цательное влияние на последующий процесс окисления сорбита в сорбозу.
Проведенные анализы показали, что в исследованном сорбитном растворе
(из автоклава) содержалось (в%): железа 0,041, алюминия 0,0163, никеля
0,0036 [21].
Для удаления солей тяжелых металлов раствор сорбита подвергают об-
работке двухзамещенным фосфорнокислым натрием (Na2HPO4) и мелом
(СаСО3) [71 ]. Химизм процесса очистки заключается в осаждении тяжелых
металлов в виде фосфатов и карбонатов. Кроме того, получаемые аморфные
осадки адсорбируют из раствора какое-то количество примесей и красящих
веществ, очищая растворы.
Процесс очистки ведут в реакторе, снабженном паровой рубашкой и ме-
шательным прибором. Предварительно раствор сорбита разбавляют водой
или промывными водами до содержания 20—25% сорбита и затем при пере-
мешивании добавляют 1,5—2,0% Na2HPO4 и 2—3% мела к массе сорбита в
растворе. Смесь нагревают до 85—90° С, выдерживают при этой температу-
ре 30—60 мин. Отфильтрованная проба должна показывать с диметилглиок-
симом отрицательную реакцию на никель. Раствор фильтруют через нутч-
фильтр или фильтр-пресс с применением асбестовой или угольной подушки.
Осадок промывают на фильтре, а промывные воды направляют на разбав-
ление технического сорбита перед чисткой. По окончании фильтрации раст-
вор сорбита подвергают анализу на содержание сорбита, глюкозы и тяжелых
металлов.
Очистка сорбита ионообменными смолами [72].
Процесс очистки сорбита двузамещенным фосфатом натрия и мелом является
трудоемким, продолжительным и требует большого расхода химических реа-
гентов. В последние годы широкое применение в технике очистки растворов
нашли ионообменные смолы.
Очистка основана на способности этих смол заменять ионы тяжелых ме-
таллов раствора сорбита ионами, входящими в состав смолы. Для этого
1 Существует мнение (Н. Золотарев, 3. Тхоревская и др. ХФЖ, 1970, № 6, 28),
что при содержании никеля менее 0,06 г!л и при добавлении NH4NO3 1—2 г!л можно
исключить процесс очистки. Автор считает это недоказанным.
250
25—30%-ный раствор сорбита вначале обрабатывают катионитом КУ 2
для обмена катионитов никеля и железа на ионы водорода; при этом значи-
тельно снижается pH раствора за счет остающихся в растворе анионов. Для
повышения pH до 4,0—4,6 раствор обрабатывают слабо основным анионитом
ЭДЭ-ЮП, который обменивают анионы раствора на анион СО3, содержащий-
ся в смоле. Процесс очистки осуществляют в непрерывно действующих ко-
лоннах.
Регенерация смол. Когда обменная способность ионитов истощается и
наступает проскок никеля, очистку раствора прекращают и колонну пере-
водят на регенерацию. Для этого смолу в колонне промывают дистиллиро-
ванной либо обессоленной водой (10—12% к объему очищенного раствора).
Регенерацию смолы КУ-2 осуществляют 5 %-ным раствором соляной кис-
лоты до исчезновения иона никеля в промывной жидкости. Затем промывают
смолу водой до нейтральной реакции промывных вод (по метилоранжу). В
процессе регенерации смолу в колонне взрыхляют 1—2 раза путем подачи
насосом воды обратным током (снизу вверх). Один объем набухшего катио-
нита может очистить до 15—20 объемов раствора D-сорбита при содержании
никеля 150—200 мг!л.
Смолу ЭДЭ-ЮП вначале регенерируют 5%-ным раствором соляной кис-
лоты, затем после промывки водой 5%-ным раствором кальцинированной со-
ды с последующей отмывкой щелочи водой.
Получение кристаллического сорбита. Очищенный раствор сорбита вы-
паривают в вакуум-аппарате при вакууме не ниже 650 мм рт. ст. до содер-
жания 95% сухих веществ. Сгущенный сорбит растворяют в 2—3-кратном
количестве 96%-ного этилового спирта при температуре 78° С, азатем про-
водят кристаллизацию при интенсивном перемешивании и постепенном ох-
лаждении до температуры 18—20° С. Затем полученную массу фугуют через
центрифугу. Кристаллы сорбита промывают спиртом и высушивают при тем-
пературе 35—40° С.
Из маточного раствора выделяют кристаллы сорбита II.- По изложенному
методу получают чистый медицинский сорбит [53, 731.
Наши заводы выпускают сгущенный технический сорбит. Для этого
раствор очищенного сорбита, отфильтрованный от катализатора, выпари-
вают под вакуумом до содержания сухих веществ около 95% и разливают в
кюветы, которые предварительно смазывают в нагретом виде маслом какао.
Для пищевых целей выпускают плитки массой 100 и 200 г, а для технических
целей — массой до 10 кг..
Технические условия на пищевой и техничес-
кий сорбит.
Внешний вид—твердая стекловидная масса с не-
значительным зеленоватым оттенком
Содержание влаги, % , не более . . 5
Содержание сорбита (на сухое ве-
щество), %...................... 99,4
Содержание золы, %, не более . . 0,5
Присутствие тяжелых металлов ... Не допускается
Запах карамели....................Не допускается
Технические .условия на жидкий сорбиточи-
щен и ы й
Раствор очищенного сорбита должен быть про-
зрачен и бесцветен. Допускается слабый оттенок
желтизны.
Содержание сорбита в растворе, %,
не менее......................... 25
Содержание редуцирующих веществ, %
к массе сухих веществ, не более . 0,1
Тяжелые металлы ..................Не допускаются
'Применение D-сорбита. Кроме синтеза аскорбиновой кислоты, D-cop-
бит нашел широкое использование в медицине как сахар для диабетиков и
для лечебных целей — для больных холециститом, запорами кишечного
251
Рис. 35. Технологическая схема производства D-сорбита непрерывным процессом гидрогенизации и ионообменной очистки
тракта, в дерматологии [75], в офтальмологии [76], как кондиционер влаги
в косметической промышленности [77], в кондитерской промышленности
для сохранения мягкости и свежести кондитерских и хлебных изделий, в
кожевенной, клеевой, текстильной, табачной и других отраслях для стабили-
зации влаги в продукции и изделиях.
На рис. 35 изображена технологическая схема производства D-сорбита с
применением непрерывного процесса гидрогенизации D-глюкозы и ионооб-
менной очистки сорбитного раствора. Элеватором / глюкозу загружают через
бункер 2 в реактор смеситель 3, в котором приготовляют 30%-ный водный
раствор. Добавляют 0,5% к массе глюкозы активированного угля и после
перемешивания в течение 5—10 мин при температуре 75° С фильтруют
через нутч-фильтр 4 в сборник 5, откуда насосом 6 перекачивают в смеси-
тель 7 (небольшого объема). Туда же непрерывно подают настой известковой
воды из мерника-смесителя 8 и катализатор Реней-никель. Раствор глюкозы
насосом высокого давления 9 подают в тройник смешения 10. Сюда же ком-
прессором И нагнетают водород под давлением 80—100 кгс/слР и
суспензию направляют в подогреватель 12, где температуру газо-жидкост-
ной смеси повышают до 135—140° С. Далее суспензия непрерывно поступа-
ет последовательно в три реактора 13, проходит холодильник 14, где
охлаждается до 30—40° С, сепаратор 15, каплеотделитель 16. Гидрированный
раствор направляют в сборник 17 и далее на очистку ионитами. Водород из
каплеотделителя 16 многоступенчатым компрессором 18 подают в тройник
смешения 10. Убыль водорода в системе компенсируют нагнетанием свежего
водорода компрессором 11 из газгольдера 19. Для безопасной работы сис-
темы должны быть предусмотрены необходимые предохранительные клапаны
и аварийные вентили для сброса водорода из системы через вытяжную трубу
с предохранительной свечой в атмосферу. Раствор сорбита из сборника 17
насосом 20 передают в смеситель 21, в котором раствор водой или промывны-
ми водами, получаемыми при отмывке смол от сорбита, разбавляют до нуж-
ного содержания сухих веществ, фильтруют через нутч-фильтр 22, сливают
в сборник 23 и далее насосом 24 нагнетают в колонну с катионитом КУ-2,
а из нее в колонну с анионитом, где pH раствора повышается до 4,0—4,5.
Из колонн 25—26 очищенный раствор направляют в сборник 27 и далее на
окисление.
Приготовление катализатора. В реакторе 28 приготовляют 25-ный рас-
твор едкого натра, фильтруют через нутч-фильтр 29 в сборник 30, откуда
насосом 31 подают в напорный мерник 32, а из него в реактор-выщелачива-
тель 33. Туда же подают мелкими порциями измельченный сплав никеля и
алюминия. По окончании реакции массу сливают в нутч-фильтр 34. Фильт-
рат-раствор алюмината натрия направляют в сборник 35, а катализатор—
в сборник 36, где его хранят под водой.
Производство водорода. Водород для гидрогенизации D-глюкозы по-
лучают электролизом воды, так как водород, полученный химическим мето-
дом, может быть загрязненным примесями газов (СО, СО2, N, Н2), отрица-
тельно влияющих на работу никелевого катализатора.
При разложении 1 кг воды электрическим методом выделяется 1,244 м3
водорода и 0,622 м3 кислорода согласно уравнению Н2О -> Н2 + 0,5О2.
Количество электроэнергии, расходуемой на разложение воды в п часов,
определяется из формулы
Г = 1пЕ,
где I — сила тока,
Е — разность потенциалов на клеммах электродов.
Согласно закону Фарадея при электролизе для выделения эквивалента
вещества необходимо затратить 96500 кулонов или ампер-секунд электри-
чества. Следовательно, для выделения 1 г-моля водорода (2 г-экв) потребу-
ется 96500 -2 = 193 000 а • сек или = 53,611 а ч.
253
При этом будет получено 22,414 л водорода при 0° и давлении
760 мм рт. ст., или 22293 = 24,056 л при температуре 20° С.
Следовательно, на выработку 1 м3 водорода при температуре 20° С
(и одновременно 0,5 м3 кислорода) необходимо затратить —24 5’ — =
= 2225 а ч электричества.
Теоретически для процесса электролиза воды требуется разность потен-
циалов на электродах 1,23 в. Однако практически эта разность значительно
больше и достигает от 2 до 4 в. Расход электроэнергии на выработку 1 м3
водорода определяется по формуле
2225 Е
N =--------кет ч,
1000
где Е — напряжение тока.
N колеблется от 4,5 до 6 кет ч. Для снижения расхода электроэнергии
применяют для электролиза не чистую воду (она имеет низкую электропро-
водность), а 25%-ный раствор едкого натра.
Soda из ШопроМа,
Рис. 36. Схема водородно-кислородной станции Уралхиммаша.
Установка для производства эл е к т р о л ия ес к о го
водорода. На рис. 36 изображена схема водородно-кислородной стан-
ции производительностью 50 м3 водорода в час. Генератор 1 (или выпрями-
тель тока) снабжает электролизер 2 постоянным током, подводимым к кон-
цевым плитам электролизера. Электролит подается через фильтр 3. После
заполнения электролитом электролизер продувают азотом. Водород и кис-
лород, образующиеся в ячейках, отводятся по соответствующим трубкам в
водородный и кислородный каналы вместе с циркулирующим электролитом,
который затем отделяется в разделительных колонках 4 и возвращается в
электролизер через фильтр 3. Водород и кислород после промывки в аппара-
тах 5 направляется через регуляторы давления 6, в ресиверы для кислорода 1
и для водорода 8. Электролит поступает в электролизер через питатель 9.
Насос 10 из бака 11 подает в питатель щелочь. Из ресивера (или газгольде-
ра) 8 водород поступает в трехступенчатый компрессор, где после каждой
ступени охлаждается в холодильниках змеевикового типа. Водород, сжатый
до избыточного давления 150 кгс/см3, подают для очистки в водомаслоотде-
литель и далее на рампу, снабженную 6—10 баллонами. С рампы через водо-
родную гребенку водород под избыточным давлением 120—130 кгс!см‘2' по-
дают на гидрирование. В системе всасывания компрессора должно быть
избыточное давление для предотвращения попадания воздуха и образования
гремучей смеси.
254
При эксплуатации электролизерной установки необходимо соблюдать сле-
дующие правила:
концентрация NaOH в электролите должна поддерживаться в пределах
200—250 г/л;
электролит не должен содержать карбонатов более 100 мг!л\ железа
3 жг/л; иона хлора 800 мг!л\
температура электролита должна быть 80 + 5° С;
чистота получаемых газов должна быть не ниже: водорода 99%, кисло-
рода 98%.
Использование отходов. При производстве D-сорбита в виде отхода
производства в процессе обработки алюминиево-никелевого катализатора
щелочью и регенерации катализатора получают алюминат натрия в коли-
честве около 0,6 кг на 1 кг сорбита. В строительной технике эффективно при-
меняют алюминат натрия при производстве работ по заделке фильтрующих
трещин, щелей и каверн в бетонных и железобетонных сооружениях, а так-
же по устройству водонепроницаемых цементных штукатурок при капилляр-
ной фильтрации [74]. Алюмйнат натрия в виде 2—5%-ного раствора добав-
ляют в воду для приготовления растворов бетона. Применение алюмината
натрия значительно повышает сопротивляемость свежих смесей вследствие
быстрого схватывания, повышенной потребности в воде, повышенной стой-
кости против размыва водой, отсутствия расслоения и водоотделения.
Указанные свойства алюминат сообщает свежим смесям вследствие ускоре-
ния процесса образования гидроалюмината кальция (ЗСаО • А12О3 • пН2О),
обусловливающего твердость бетона.
Следует отметить, что для строительной техники пригоден алюминат нат-
рия, не загрязненный органическими веществами.
ПРОИЗВОДСТВО L-СОРБОЗЫ ИЗ О-СОРБИТА1
А-Сорбоза является кетогексозой, молекулярная масса 180,16, имеет
следующую структурную формулу:
СН2ОН
но-с-----
I
но-с-н
I
н-с-он
I
но-с-н
I
н2с----
А-Сорбоза представляет бесцветные кристаллы сладкого вкуса ромбичес-
кой формы с температурой плавления 165° С. Удельное вращение в воде
[а]^° ~ минус 42,9—43,2°; по другим данным [78] — минус 43,65°; не об-
ладает мутаротацией. Сорбоза хорошо растворима в воде, плохо растворима
в спирте и нерастворима в этиловом эфире; в кристаллическом виде имеет
[i-форму пиранозы.
В табл. 16 приведены показатели растворимости сорбозы при различной
температуре. В табл. 17 — показатели преломления и плотности водных
растворов сорбозы при температуре 20° С.
1 В Чехословакии опубликован патент на способ получения L-сорбозы из карто-
фельного крахмала путем гидролиза его HC1 и окончательного гидролиза глюкоами-
лазой с последующей гидрогенизацией и ферментацией обычным методом [77а].
255
Таблица 16
Темпера- тура, °C Раствори- мость сор- бозы в воде, % Темпера- тура, °C Раствори- мость сор- бозы в спирте, %
0 37,8 20 0,5
20 44,9 40 0,96
40 51,1 60 1,85
60 57,7 75 2,9
80 64,4
100 71,8
Таблица 17
Процент сорбозы Показа- тель пре- ломления Плотность раствора, Процент сорбозы Показа- тель пре- ломления Плотность ; раствора, кг,'м.г
1,007 1,3350 1003 30,787 1,3835 изо
6,026 1,3423 1022 35,858 1,3917 1154
10,990 1,3500 1042 38,846 1,3987 1173
16,229 1,3583 1064 45,189 1,4103 1203
21,049 1,3658 1085 48,030 1,4199 1225
25,734 1,3739 1110
Технические условия на сорбозу
Внешний вид-—светло-желтые или почти бес-
цветные кристаллы
Влажность, %, не более ................ 0,2
Содержание сорбозы, %.................. 97—98,8
Стойкость водных растворов /,-сорбозы к нагреванию. Исследования
стойкости водных растворов сорбозы показали [79], что при нагревании их
происходит разрушение /.-сорбозы, характеризуемое изменением цвета,
количества редуцирующих веществ и максимумов поглощения в ультра-
фиолетовом свете. Интенсивность этого процесса зависит от значения pH
раствора, от продолжительности нагревания раствора и от температуры.
Судя по спектру поглощения, в ультрафиолетовом свете распад /.-сорбозы в
кислой среде идет в сторону образования оксиметилфурфурола и далее му-
равьиной и левулиновой кислот [80]:
СН2ОН
I
но—с-----
I
но—с—н
I
н—с—он
но—с—н
u I
Н2С------
£-Сорбоза
Н—С-0
с-
с-н
н—с
СН2ОН
Оксиметилфурфурол
НСООН Муравьиная
кислота
СООН
I
сн2
I
сн2
I
со
СН3
Левулиновая
кислота
с
о
Наименьшие потери редуцирующих веществ наблюдаются при pH 3,0,
и в этих условиях /.-сорбоза.является наиболее стойкой.
В отношении механизма процессов распада /.-сорбозы в водных раство-
рах имеются следующие предположения [79]:
1. При нагревании щелочных водных растворов сорбозы происходит рас-
пад последней с образованием кислот. Последние понижают pH раствора:
при pH < 7,0 начинается процесс дегидратации/.-сорбозы с образованием
оксиметилфурфурола, который подвергается распаду с образованием мура-
вьиной и левулиновой кислот. При pH < 3,0 наступает процесс реверсии ок-
симетилфурфурола. При этом как процесс реверсии, так и процесс образо-
вания оксиметилфурфурола ускоряются, что влечет за собой повышение
цветности раствора и повышение интенсивности поглощения в ультрафиоле-
товой части спектра.
2. При нагревании кислых водных растворов /.-сорбозы процесс распада
идет в сторону образования оксиметилфурфурола, затем муравьиной и леву-
линовой кислот; при pH ниже 3,0 наступает процесс реверсии, который идет
параллельно с процессом дегидратации и образования оксиметилфурфурола.
Сущность процесса бактериального окисления />-сорбита в £-сорбозу.
Сорбоза является вторым промежуточным продуктом в процессе синтеза
256
аскорбиновой кислоты. Реакция окисления протекает по следующей схеме:
СН2ОН СН2ОН
НО—С— Н НО—С---------;
НО—С—н но—с—н |
I I о
Н—С—ОН [О] н—с—он
но—с—н но-с—н
I I
СН2ОН Н2С--------
D-Сорбит /.-Сорбоза
182,17 183,13
Процесс окисления О-сорбита в /.-сорбозу осуществляется биохимичес-
ким методом и является результатом жизнедеятельности аэробных, кето-
генных, уксуснокислых бактерий, культивируемых на питательной среде,
состоящей из О-сорбита и дрожжевого автолизата или экстракта.
Изучено окислительное действие следующих микроорганизмов: Aceto-
bacter xylinum, Ac. xylinoides, Ac. suboxydans, Ac. mesoxydans, Ac. melano-
genum.
Установлено [81], что наиболее эффективными биохимическими окисли-
телями D-сорбита являются Ac. melanogenum и Ac. suboxydans.
Необходимо отметить, что механизм окислительного действия уксусно-
кислых бактерий еще недостаточно изучен [82—84].
Факторы, влияющие на процесс окисления D-сорбита в L-сорбозу.
К основным факторам, влияющим на этот процесс, относятся:
1. Качество питательной среды, зависящее от степени очистки раствора
D-сорбита от тяжелых металлов, от содержания моносахаридов и изомерных
гекситов и от содержания биологически активных веществ. Известно, что
никель при концентрации 5 мг/л, железо 150 мг!л и алюминий 500 мг!л угне-
тают процесс окисления.
При недостаточно четком проведении процесса Гидрогенизации возмож-
но содержание в сорбите повышенного количества D-глюкозы, которая при
окислении дает D-глюконовую кислоту, а последняя может окисляться в
З-кето-D-глюконовую кислоту. Из маннита при окислении может образо-
ваться фруктоза, а последняя в кислой среде подвергается дегидратации в
оксиметилфурфурол и далее в триоксимасляную и гликолевую кислоты.
Важное значение имеет введение в питательную среду биологически ак-
тивных веществ, без которых жизнедеятельность уксуснокислых бактерий
тормозится и процесс окисления D-сорбита в /.-сорбозу прекращается.
2. Процесс окисления D-сорбита в L -сорбозу является аэробным, по-
этому интенсивность его зависит от количества и качества воздуха, подавае-
мого для аэрации питательной среды. Практикой установлено, что на 1 л
питательной среды в 1 мин требуется 2—3 л воздуха.
3. Наконец, одним из наиболее ответственных требований является гер-
метичность аппаратуры и недопустимость заражения среды посторонней
микрофлорой, для чего необходимо:
ст ерилизовать масло, добавляемое в ферментаторы, в специальных
бачках и стерильно подавать его в аппараты;
по стоянно поддерживать стерильность трубопроводов для подачи посев-
ного материала из инокуляторов и питательной среды из стерилизаторов,
а также пробных кранов путем создания в трубопроводах и арматуре паро-
вой завесы;
устранять возможность попадания окисленного раствора сорбита в рабо-
тающие ферментаторы при разгрузке путем отключения действующих фер-
ментаторов от общего разгрузочного трубопровода.
Процесс биохимического окисления D-сорбита в L-сорбозу. Техноло-
гический процесс окисления D-сорбита в L-сорбозу состоит из следующих
стадий [53, 86, 87]:
9-522
257
приготовление и поддержание посевной культуры;
приготовление рабочей культуры;
проведение процесса биохимического окисления в ферментаторах;
очистка окисленного раствора и перекристаллизация /.-сорбозы.
Для получения высококачественной питательной среды необходимо, что-
бы она была биологически активной, т. е. содержала в своем составе, кроме
углеводов, азота и фосфора, биологические катализаторы (витамины и фер-
менты) и ряд минеральных солей. При отсутствии биокатализаторов и мине-
ральных солей рост и размножение уксуснокислых бактерий невозможен.
Вот почему для проведения биохимического окисления D-сорбита в /.-сор-
бозу необходимо к раствору очищенного сорбита добавлять биохимически
активный субстрат в виде дрожжевого автолизата богатого витаминами, фер-
ментами и минеральными солями. Дрожжевой экстракт может быть заменен
экстрактом или концентратом, полученным из кукурузы или из барды вино-
куренных заводов. Однако необходимо отметить, что ни один из этих экст-
рактов по своей биологической активности не может конкурировать с экст-
рактом из дрожжей.
В последнее время вместо автолизата дрожжей на заводах стали приме-
нять концентрат витаминов группы В, получаемый при производстве вита-
мина D2 из пекарских дрожжей. Спиртовой экстракт дрожжей уваривают в
вакуум-аппарате примерно до следующей концентрации (состав в %): су-
хие вещества 45,5; зола 19,0; азот 6,0; углеводы в пересчете на глюкозу 13,0.
Содержание в концентрате витаминов группы В (мг/г) составляет [85]:
Bi
в2
рр
0,04 В6.......................0,10
0,163 Холин..................7,1
2,0 Пантотеновая кислота . 0,35
Целесообразность замены автолизата концентратом витаминов группы В
сомнительна, так как биологическая активность концентрата значительно
ниже активности автолизата дрожжей.
Приготовление и поддержание посевной культуры. Для культивирова-
ния уксуснокислых бактерий применяют кристаллический чистый сорбит и
автолизат пекарских дрожжей; pH среды должен быть 4,8—5,5. Регулиру-
ется pH добавлением уксусной кислоты.
Состав среды (в % сухих веществ): сорбит 12,0—14,0, автолизат 0,15.
Вместо автолизата можно использовать и препарат В-комплекс в коли-
честве 1,5% объемных.
Среду разливают в пробирки (по 10 мл), закрываемые ватно-марлевыми
пробками. Затем пробирки стерилизуют в автоклаве 30 мин при избыточном
давлении пара 1,0 кгс/см2, охлаждают и помещают в термостат при темпера-
туре 32—34° С на 48 ч для проверки стерильности среды. Последнюю хранят
при комнатной температуре не более 1 месяца. Посев культуры производят
в 2—4 пробирки. Для посева выбирают пробирки, в которых культура хоро-
шо развита, активна и проверена на чистоту. Засеянные пробирки выдержи-
вают в термостатной камере при температуре 32—34° С в течение 24—48 ч
для проверки стерильности, а затем их хранят при комнатной температуре.
В производстве постоянно должны храниться не менее 2—4 пробирок со
средой. Культуры пересевают на новую жидкую среду каждые 7—10 дней,
а на твердую среду — 1 раз в 30 дней.
Твердую питательную среду готовят также, как и жидкую, но с прибав-
лением 2—3% агара.
После стерилизации в течение 30 мин при избыточном давлении 1 кгс/см2
агар скашивают или оставляют столбиками в пробирках. Косой агар засе-
вают культурой, которую проверяют на чистоту путем микроскопирования и
на активность — путем определения содержания редуцирующих веществ.
Активность уксуснокислых бактерий устанавливают следующим обра-
зом. В две конические колбы с питательной средой, указанной выше, добав-
ляют 5% односуточной культуры. Культивирование производят в неподвиж-
258
ном слое (1 см) в течение 24—36 ч в термостате при температуре 32—34° С.
Через 24 ч в первой и через 36 ч во второй колбе определяют содержание
редуцирующих веществ. Культуру считают активной, если через 24 ч она
дает 80—85% окисления D-сорбита в /.-сорбозу, а через 36 ч — 95—98%.
Приготовление рабочей культуры. Питательной средой для рабочей
культуры служит та же среда, что и для чистой культуры, только вместо
кристаллического сорбита применяют очищенный раствор сорбита. Среду
разливают в литровые колбы (по 500 мл} и стерилизуют в автоклаве 30 мин
при избыточном давлении 1 кгс/см2. Засев питательной среды в колбах про-
изводят односуточной культурой, выращенной в пробирках на жидкой пи-
тательной среде. Для посева отбирают пробирки, гарантирующие нормаль-
ный рост культуры, чистоту ее и активность. Из каждой пробирки произво-
дится посев в одну колбу. Засеянные колбы выдерживают в термостате 24 ч.
После этого культуру из литровых колб переводят в 20-литровые бутыли
(из расчета одной колбы на одну бутыль), содержащие 5 л питательной сре-
ды. Бутыли выдерживают в термостате в горизонтальном положении в свя-
зи с тем, что рост культуры должен проводиться в тонком слое. Из бутылей
культуру переводят в аппараты-инокуляторы, а из последних в посевные
ферментаторы.
Посевной материал выращивают глубинным способом в специальных ап-
паратах-инокуляторах и посевных ферментаторах. Аппараты предваритель-
но стерилизуют острым паром в течение 20 мин, затем при помощи вакуума
засасывают питательную среду следующего состава: 10%-ный раствор очи-
щенного сорбита и 0,2% (на сухое вещество) автолизата дрожжей к массе
сорбита или 1% (к объему среды) концентрата витаминов группы В.
В питательную среду добавляют серную кислоту и доводят pH до 4,5—
5,5. Затем раствор стерилизуют при температуре 120° С в течение 45 мин.
По окончании стерилизации раствор охлаждают до 35° С, засасывают в ино-
кулятор рабочую культуру в количестве 10% по объему к производственной
среде и начинают продувку воздухом. Глубинную культуру выращивают
при температуре 30—32° С в течение 12—24 ч, после чего ее стерильно пере-
водят в посевные ферментаторы из расчета 5—10% к объему среды. Культу-
ру, задаваемую из инокулятора или посевного ферментатора, проверяют
на чистоту и процент окисления, который не должен быть ниже 30% для пер-
вого и 50% для второго.
Проведение процесса биохимического окисления в ферментаторах
Для эффективности проведения процесса глубинного окисления очень
важно, чтобы сжатый воздух, нагнетаемый компрессором, был подвергнут
тщательной очистке от бактериальных загрязнений. Технологическая схема
окисления D-сорбита в /.-сорбозу приведена на рис. 37. Компрессор 1 на-
гнетает воздух в ресивер 2. Воздух проходит маслоотделитель 3, представ-
ляющий собой вертикальный стальной цилиндр высотой 2,0—2,5 м, запол-
ненный кольцами Рашига. Проходя через извилистые каналы, образованные
кольцами Рашига, воздух освобождается от капелек масла, уносимых из
компрессора. Затем сжатый воздух проходит через скруббер 4, имеющий
такую же конструкцию, как и маслоотделитель (высотой 2,5 Л4).
В скруббер сверху поступает вода, которая встречает на своем пути воз-
дух и, промывая его, стекает вниз и уходит в канализацию, а воздух уходит
через верх скруббера в влагоотделитель 5. Последний имеет высоту около
1,2 м и также заполнен кольцами Рашига. Затем воздух поступает в ватно-
угольный фильтр 6, представляющий собой стальной цилиндр, снабженный
паровой рубашкой и двумя решетками, расположенными над днищем и под
крышкой. На дно фильтра помещают слой стеклянной ваты толщиной 10—
15 см, затем слой гранулированного активированного угля толщиной 3—4 см,
затем снова слой стеклянной ваты толщиной 10—45 см и, наконец,
слой хлопчатобумажной ваты — 2—5 см. Зарядку фильтра производят раз
9*
259
в месяц. Из угольного филь-
тра воздух поступает в ватный
фильтр 7, который имеется у
каждого инокулятора и фер-
ментатора.
Фильтры стерилизуют че-
рез паровую рубашку острым
паром (избыточное давление
1,2—2 кгс/сдД) в течение 2—3 ч
перед пуском в работу. После
стерилизации фильтры просу-
шивают воздухом в течение
1—2 ч с подогревом фильтра
паром через паровую руба-
шку.
Окисление проводят глу-
бинным способом в специаль-
ных аппаратах-ферментато-
рах, представляющих верти-
кальные реакторы из эмали-
рованной стали1, снабженные
рубашками для обогрева го-
рячей водой, метательными
приборами, и приспособлени-
ями для распыления воздуха
в виде барботеров или кера-
миковых свечей с регулирую-
щими устройствами. Произ-
водственную питательную
среду готовят в стерилизаторе
8, куда поступает очищенный
раствор сорбита с содержа-
нием около 20% сухих ве-
ществ и 0,2% автолизата (на
сухое вещество). Добавлени-
ем серной кислоты pH среды
доводят до 4,5—5,5. Среду
стерилизуют 30 мин при тем-
пературе 120° С и перемеши-
вании. В процессе стерилиза-
ции через 15 мин через ни-
жний штуцер сливают 5—6 л
среды и продолжают стерили-
зацию еще 15 мин. После это-
го среду переводят в стериль-
ный ферментатор 9, где ее
охлаждают до 32—34° С. Про-
цесс стерилизации может быть
осуществлен в стерилизаторе
непрерывного действия ост-
рым паром с последующим
охлаждением в трубчатом ап-
парате непрерывного действия.
1 Имеются лабораторные
опытные данные [87а], что ма-
териалы фторопласта и полипро-
пилена не влияют отрицательно
на процесс окисления.
260
Перед пуском в работу ферментатор стерилизуют острым паром в тече-
ние 1 ч при избыточном давлении 1,5—2,0 кгс/см2.
В случае заражения установки посторонней микрофлорой стерилизация
паром не дает необходимого эффекта. В этом случае следует всю аппаратуру
и коммуникацию обработать формалином. Для этого герметически закрыва-
ют стерилизаторы, ферментаторы, фильтры и коммуникацию и избыточным
давлением сжатого воздуха до 3 кгс/см2 проверяют герметичность всей сис-
темы. Затем давление воздуха снижают, в ферментатор загружают 2 л 38 % -
ного формалина и пускают в ферментатор пар до возгонки формалина (тем-
пература 140—142° С) и заполнения им всей системы. В таком состоянии
аппаратура находится 3—4 ч. Затем в ферментатор подают острый пар
(избыточное давление 3 кгс/см2) для испарения и удаления паров формалина.
После этого установка считается стерильной.
Процесс окисления проводят следующим образом. В охлажденную пита-
тельную среду переводят стерильно из инокуляторов или посевных фермен-
таторов 10 рабочую культуру из расчета 8—10% к объему среды, после чего
в ферментатор подают для аэрации культуральной жидкости воздух, нагне-
таемый компрессором через систему аппаратов для очистки. Распыление
воздуха осуществляют через барботеры, либо через керамиковые свечи1.
Для гашения пены, образующейся в ферментаторе, используют воздух, рас-
пыляемый в верхней части ферментатора через перфорированные трубки из
нержавеющей стали по всему сечению ферментатора.
Процесс окисления осуществляют при температуре 32—34° С и избыточ-
ном давлении воздуха в ферментаторах 0,2—0,5 кгс/см2. Расход воздуха ко-
леблется в пределах 2—3 л/мин. на 1 л среды.
Для контроля процесса окисления из ферментатора отбирают пробы че-
рез 12—15 ч от начала процесса, а затем каждые 2 ч в зависимости от хода
процесса; глубина окисления должна быть 95—98%.
При нормальном течении процесса окисление заканчивается через 18—
24 ч и раствор сливают в сборник 9а.
Очистка окисленного раствора сорбита. Окисленный раствор сорбита
содержит 20—25% сухих ьеществ. Между тем для последующего процесса—
ацетонирования — L-сорбоза применяется в виде тонкоизмельченного по-
рошка с влажностью около 0,2%. В таком виде L-сорбозу получают после
сгущения окисленного раствора D-сорбита и кристаллизации L-сорбозы.
Известно, что эффективность процессов выделения кристаллических веществ
из растворов зависит от чистоты последних. Чем меньше посторонних при-
месей, в особенности коллоидальных и красящих веществ, тем быстрее и
полнее идет процесс кристаллизации и тем ниже потери вещества в так
называемой патоке (последний продукт при многократной кристаллизации
отходов).
Окисленный раствор D-сорбита содержит большое количество коллои-
дальных веществ в виде бактериальных клеток, а поэтому хотя бы частичное
удаление этих веществ из раствора повышает выход и качество кристалличес-
кой сорбозы. Очистку раствора целесообразно осуществлять при помощи
активированного угля. Для этого раствор из сборника 9а направляют в ап-
парат-смеситель 11, куда вводят активированный уголь в количестве 1%
к массе сухих веществ раствора, нагревают до 70° С при перемешивании 5—
10 мин, а затем насосом нагнетают раствор в фильтр-пресс 12, откуда посту-
пает в сборник фильтрованного раствора; фильтр-пресс промывают горячей
водой. Промывные воды используют на второй кристаллизации сор-
бозы.
Перекристаллизация L-сорбозы. Сорбоза является нестойким продуктом
и при длительном нагревании подвергается распаду. Поэтому для повыше-
1 Керамиковые свечи более эффективны, так как кроме функции тонкого распы-
ления воздуха они служат фильтром для микробов.
261
ния выхода L-сорбозы в кристаллическом виде необходимо быстро прово-
дить процессы выпаривания и кристаллизации растворов L-сорбозы и все
процессы осуществлять по возможности при низкой температуре (по так на-
зываемому скоростному холодному методу).
Из сборника фильтрованный раствор засасывают в вакуум-аппарат 13,
в котором он упаривается под вакуумом 675—700 мм рт. ст. до содержа-
ния 90% сухих веществ. Процесс упаривания должен протекать быстро (не
более 2—Зч), для чего требуется, чтобы вакуум-аппарат имел развитую
трубчатую поверхность нагрева и чтобы избыточное давление греющего па-
ра было не ниже 3—4 кгс/см2. При большом объеме производства (более
25 кг кристаллической сорбозы в 1 ч) рационально для снижения расхода
пара применять выпарные установки многократного выпаривания.
Сваренный утфель первой кристаллизации спускают из вакуум-аппарата
13 в кристаллизатор 14, где его постепенно охлаждают до 18° С в течение
9 ч. Из кристаллизатора утфель поступает в центрифугу 15, отфуговывают
маточный раствор, а кристаллы промывают ледяной водой и высушивают
в барабанной сушилке 16; маточный раствор I поступает в сборник 17.
На первой кристаллизации получают до 70—75% сорбозы от содержания
ее в утфеле; 25—30% ее остается в маточном растворе. Маточный раствор
обязательно перерабатывают, чтобы извлечь из него кристаллическую сор-
бозу.
Маточный раствор I из сборника 17 и угольные промывные воды, полу-
ченные при очистке окисленного раствора сорбита, обрабатывают активиро-
ванным углем в количестве 2% к массе сорбозы в аппарате-смесителе 18 при
температуре 65° С в течение 10 мин. Затем фильтруют через фильтр-пресс 19.
Фильтрат поступает в сборник 20. Угольную лепешку промывают горя-
чей водой и полученные промывные воды II поступают в сборник 25 маточ-
ного раствора II. Профильтрованный маточный раствор I упаривают в ва-
куум-аппарате 21 до содержания в нем сухих веществ 85—86%. Утфель
спускают в предварительно подогретый кристаллизатор 22, где в течение
36 ч при работающей мешалке он охлаждается до 26—27° С, затем в рубаш-
ку кристаллизатора пускают холодную воду и охлаждают в течение 24 ч
до 18—20° С. Полученные кристаллы отфуговывают в центрифуге 23, про-
мывают ледяной водой, высушивают в сушилке 16 и измельчают в микро-
мельнице 24. Полученную сорбозу II также направляют на ацетониро-
вание.
Маточный раствор II поступает из центрифуги 23 в сборник 25, где он
смешивается с угольными промывными водами II и поступает на очистку
углем в аппарат-смеситель 26, куда вводят 2 % (по массе сорбозы) активиро-
ванного угля и обрабатывают 10 мин при температуре 65° С при перемеши-
вании. Затем массу фильтруют через фильтр-пресс 27. Фильтрованный ма-
точный раствор поступает в сборник 28 и далее на упаривание в вакуум-
аппарат 29, где упаривается до 75—80% сухих веществ.
Утфель кристаллизуют 72—120 ч при температуре 20° С в кристаллиза-
торе 30 и фугуют в центрифуге 31. Полученную сорбозу направляют на пе-
реработку совместно с маточным раствором I. Маточный раствор III (сор-
бозная патока) является отходом производства, его направляют в сбор-
ник 32.
Получение дрожжевого автолизата. Сухие дрожжи заливают пятикрат-
ным по массе количеством воды, нагревают в смесителе 33 при перемешива-
нии до температуры 40—45° С и выдерживают при этой температуре в те-
чение 2 ч. Затем дрожжевую массу охлаждают, декантируют и фугуют в се-
параторе или центрифуге отстойного типа 34. Остаток дрожжей размешивают
с трехкратным количеством воды при температуре 55—60° С; в течение
30—40 мин и повторно фугуют. Полученные экстракты поступают в сбор-
ник 35 и представляют дрожжевые автолизаты с содержанием 4—6% сухих
веществ. При этом методе извлекается из дрожжей 63,4—76,1% азотистых
веществ, 66% тиамина и 80% рибофлавина [88].
262
Непрерывный, способ получения А-сорбозы из D-сорбита
Способ состоит из двух стадий: а) непрерывного культивирования уксус-
нокислых бактерий при биохимическом окислении D-сорбита в проточных
средах и б) непрерывного выделения кристаллической L-сорбозы из окислен-
ного раствора. Некоторые теоретические основы непрерывного культивиро-
вания микроорганизмов изложены в работах различных исследователей
[89—92]. Изучены также пути интенсификации процесса за счет улучшения
условий аэрирования среды [93, 94]. Существенные исследования в области
биохимического окисления D-сорбита в L-сорбозу были проведены в тарель-
чатом колонном ферментаторе непрерывного действия [95, 96] и была пока-
зана эффективность его работы. При сравнительно низком расходе воздуха
Рис. 38. Технологическая схема производства L-сорбозы из D-сорбита непрерывным
процессом.
(1—2 л!л в минуту) продолжительность окисления 11—18%-ного раствора
сорбита составила около 15 ч. Теоретически обоснована и экспериментально
подтверждена возможность увеличения производительности ферментатора
при работе с рециркуляцией культуральной жидкости путем частичного ее
возврата с тарелок верхней зоны в нижнюю зону аппарата [96].
Экспериментально также установлена [97] возможность замены процес-
сов многоступенчатой кристаллизации L-сорбозы процессами предваритель-
ной очистки окисленного раствора сорбита при pH 3,0 активированным уг-
лем (3—5% к массе сорбозы) или ионообменными смолами [98] и обезвожи-
ванием его в распылительной сушилке. Ниже описана технологическая схе-
ма производства L-сорбозы из D-сорбита непрерывным процессом (рис. 38)
[53, 97]. Питательную среду из сборника 1 непрерывно насосом подают в
стерилизатор 2 и далее в сборник-выдерживатель 5, охладитель 4 и сборник
охлажденной среды 5. В этот сборник непрерывно стерильно поступает ра-
бочая культура. Из сборника 5 питательная среда непрерывно поступает в
ферментатор 6. Параллельно со средой в ферментатор снизу подают сжатый
воздух. Для гашения пены ферментатор сверху снабжен пеногасителем 7 и
брызгоуловителем 8. Воздух из колонны выходит через фильтр 9, а окислен-
ный раствор поступает в сборник 10. Температуру среды в колонне поддер-
живают водяным обогревом через секционные рубашки. Давление воздуха
регулируется прибором 11, а рециркуляция питательной среды1— регулято-
1 В дальнейшем было показано (В. Зарак, Л. Шнайдман, Авторское свидетель-
ство СССР № 257 721, 1967; Бюлл. изобр. № 36, 1969, 20/XI), что наиболее эффек-
тивно процесс протекает при трехступенчатой схеме — при непрерывном поступле-
нии в ферментатор свежей посевной культуры, непрерывной ферментации и непре-
рывном дозревании, т. е. установка должна состоять из трех непрерывно действую-
щих аппаратов — инокулятора, ферментатора и дозревателя.
263
ром 12. Окисленный раствор непрерывно из сборника 10 поступает в сбор-
ник 13 и далее в сепаратор 14 для очистки от белковых частиц. Далее рас-
твор направляют через сборник 15 в колонну 16 с катионитом КБ-2, а затем
в колонну 17 с анионитом ЭДЭ-ЮП. Очищенный раствор из сборника 18
насосом высокого давления 19 подают в распылительную сушилку 20. Суш-
ка протекает при температуре 70° С и порошок сорбозы непрерывно посту-
пает на досушку до влажности 0,1—0,2 % в шнековую сушилку 21 и далее
в сборник 22. Отсюда сорбозу направляют для ацетонирования.
ПРОИЗВОДСТВО ДИАЦЕТОН-Б-СОРБОЗЫ ИЗ Б-СОРБОЗЫ
Диацетон-Z,-сорбоза (2 : 3,4 : б-О-диизопропилиден-а-Б-сорбофураноза)
С12Н2оОв, молекулярная масса 260,28, кристаллическое вещество с темпера-
турой плавления 78° С хорошо растворимо в воде, бензине, эфире, бензоле,
Рис. 39. ИК-спектр диацетон-Б-сорбозы.
хлороформе, дихлорэтане и других растворителях. Растворимость в петро-
лейном эфире (в %): при температуре 40° С — 15,3; 20° С — 3,4; при 0° —
,1,3. На рис. 39 дан ПК — спектр диацетон-Ь-сорбозы, в котором обнаруже-
ны полосы поглощения с частотой (в см"1) : 3550 (наличие гидроксилов),
1370 [С(СН3)2], 1180—1200 (СО); —18,1° (с = 1,38%, ацетон).
Исследования в области устойчивости водных и ацетоновых растворов
диацетон-Z-сорбозы при различных pH и нагревании при 98—100° С пока-
зали:
1) при pH ниже 7,0 диацетонсорбоза в водных растворах крайне нестой-
ка, причем в числе продуктов распада имеется оксиметилфурфурол (макси-
мум поглощения в ультрафиолетовом свете при 280 и 230 нм). Еще до нагре-
вания при pH 0,5 вся диацетонсорбоза исчезает. В щелочной среде диаце-
тонсорбоза сравнительно устойчива, но длительное нагревание также
приводит ее к частичному разрушению. При pH 11,8 образуется небольшое
количество оксиметилфурфурола.
Наличие оксиметилфурфурола показывает, что распад диацетонсорбо-
зы не ограничивается гидролизом ее до сорбозы, а идет дальше;
2) в растворах ацетона при температуре 50° С и pH 7,0 и выше 7,0 диаце-
тонсорбоза сравнительно устойчива. В кислой среде процесс распада идет
интенсивно.
Механизм реакции ацетонирования. Рядом экспериментов, проведен-
ных различными исследователями [55, 99], установлено, что при ацетониро-
вании Z-сорбозы вначале образуется моноацетонсорбоза, которая в зависи-
мости от температурных условий переходит в диацетонсорбозу.
При этом было обнаружено образование двух форм моноацетонсорбозы:
одна из них имеет температуру плавления 93° С и [o.Jd= +7° (с — 1,42%,
вода) [100], а другая — температуру плавления 142—143° С и [а]д =
= —85° (с — 1,5%, вода) [99].
Однако механизм реакции ацетонирования вследствие своей сложности
недостаточно изучен. Прежде всего неизвестно, в какой форме находится
в реакционной массе Z-сорбоза из тех четырех возможных форм — a-и fl-
264
сорбофураноз и а- и |3-сорбопираноз [101 ]. Предполагают, что моноацето-
нированию подвергаются те формы L-сорбозы, которые имеют гидроксиль-
ную группу, расположенную рядом с полуацетальным гидроксилом по той
же стороне цикла (Z,—а-сорбофураноза и L—а-сорбопираноза):
ОН Н
Z, -Сорборураноза
ОН Н
Z. сС-Сорбопираноза
Далее при моноацетонировании затрагивается кетогруппа в положении 2
(в форме гидроксила циклической структуры сорбозы). Это доказано тем,
что моноацетонсорбоза не дает характерную для кетоз реакцию Фелинга.
Затем установлено [100, 102], что моноацетонирование происходит по двум
соседним гидроксилам (либо 1,2, либо 2,3), что обусловлено образованием
при этом Долее стабильного пятичленного цикла (включающего остаток аце-
тона), нежели шестичленного при ацетонировании несоседних гидроксилов.
Остается решить вопрос, по каким гидроксилам идет моноацетонирова-
ние (1,2 либо 2,3) и какова форма Z-сорбозы (фураноидная или пираноидная).
Существовало мнение [35, 101 ], что этот процесс идет главным образом
по гидроксилам 2,3 с образованием 2,3-о-моноизопропилиден-а-А-сорбофу-
ранозы (IV); 1,2-о-моноизопропилиден-а-Л-сорбопираноза (II) образуется
с малым выходом и что последняя неспособна к дальнейшему ацетонирова-
нию [101 ]. При этом предполагалось [35], что Z-сорбоза I имеет пираноид-
ную структуру, которая при ацетонировании изомеризуется в фураноидную.
Отсюда механизм реакции ацетонировании представляли в следующем
виде [53]:
cP - L -а^-ИзопропилиНенсор-
Иопираноза, т пл. М3 °
II
Однако позднейшие исследования, проведенные японскими авторами
[103, 104], не подтвердили указанный механизм реакции ацетонировании.
Для исследования ими были выделены следующие чистые вещества, кроме
L-сорбозы (I): a-L-(l : 2)-изопропилиденсорбопираноза (//);a-L-(2 : 3)-(4 : 6)-
диизопропилиденсорбофураноза (///); a-L-(2 : 3)-изопропилиденсорбофу-
раноза (/У).
Проводя ацетонирование указанных веществ, авторы исследовали кине-
тику следующих реакций: II -> III; III -> IV; IV -> III. Для определения
265
скорости реакций был применен метод К- Фишера титрования воды, образу-
ющейся в реакции [105]. Продукты реакций были идентифицированы газо-
жидкостной хроматографией (200° С, газ водород, скорость ЮОлм/лшн;
время удержания (в мин)-. II — 13,25; III — 16,5; IV — 20,5). Применена
также хроматография на бумаге (растворитель — бутанол-уксусная кис-
лота-вода 4:1:5, проявитель — бензидиновый раствор [1061; Rf: I—
0,25, II— 0,65, III — 0,88, IV — 0,68).
Значительный интерес представляют полученные авторами результаты
гидролиза соединения II (0,1 г) серной кислотой (0,038 г) в среде диоксана
(1,86 г) при температуре 30° С 1 ч. Хроматографией газо-жидкостной и на
бумаге были идентифицированы I, II, III и IV. Образование III из II в от-
сутствие ацетона, по мнению авторов, возможно в результате гидролиза ди-
мера (П)2. На основании своих кинетических исследований Токияма, Гонда
и Гоки предложили следующий механизм реакции ацетонирования вещест-
ва II:
II---> [П]2----> Ш IV
Остается неясным механизм реакции на первой стадии ацетонирования,
исходя из L-сорбозы, хотя в последующей работе [107] имеются указания,
что в начале процесса ацетонирования образуются лишь II и III, а IV явля-
ется продуктом гидролиза III.
Однако в наших исследованиях [107а] уже через 15 мин от начала аце-
тонирования обнаруживают (в % от теоретического) II — 8,4; III—28,3;
IV— 11,2. Следует полагать, что механизм реакции ацетонирования дол-
жен учитывать существование L-сорбозы в двух формах — фураноидной I
и пираноидной 1а и отсюда 2 самостоятельных направления реакции:
1) I IV —> III
2) la II—[И]2
III IV
+
I
сн3/сн3
х
сн3 сн3
с'
ОН II
Об-L-Сорбопираноза
он н
сб-L -(1 -2)-изопропилиден-
сорболираноза
ОН н
Димер
ОН н
of -L - Сорбопираноза
la II И
1а
Влияние различных факторов на процесс ацетонирования. Реакция аце-
тонирования L-сорбозы является обратимой и направление ее зависит от
различных условий.
Установлено влияние следующих факторов [21, 53, 108]:
1. При уменьшении температуры состояние равновесия между a-L-
(2 : 3), (4 : 6)-диацетонсорбозой и другими ацетоновыми производными сме-
266
щается в сторону образования диацетонсорбозы; при повышении температу-
ры состояние равновесия смещается в обратном направлении. В связи с этим
реакция ацетонирования должна проводиться при возможно допустимо низ-
кой температуре (12—14° С) с последующим охлаждением до —10° С; по
возможности должна быть снижена температура нейтрализации (доО+5°С).
2. Не менее важным фактором является присутствие или возникновение
в процессе реакции воды, так как известно, что ацетоновые производные
А-сорбозы нестойки и в присутствии воды быстро омыляются. Источниками
воды могут быть как влажность химикалиев, поступающих в реакцию (L-
сорбоза, ацетон и серная кислота), так и реакция конденсации ацетона. По-
следняя протекает по следующей схеме:
н3с—с—сн3 н3с—сон—сн3 н3с—с-сн3
II 1 II
о сн2 сн
+
СН2Н > — —> + Н2О
1 со со со
сн3 сн3 сн3
Ацетон Кетон Окись мезитила
Спектрофотометрические ис-
следования [109] динамики ре-
акции уплотнения ацетона под
влиянием серной кислоты при
температуре 26—28° С показали
прямую зависимость между про-
должительностью реакции аце-
тонирования и количеством об-
разующейся окиси мезитила. Это
видно из рис. 40 по изменению
величины показателя экстинк-
ции Е при лтах = 332 нм.
Для максимального сниже-
ния количества воды, поступаю-
щей в реакцию с химикалиями,
необходимо направлять на аце-
тонирование сорбозу с влажно-
стью не более 0,1 %, ацетон вла-
жностью 0,1—0,2%* и серную
кислоту в виде олеума с содер-
жанием 18% свободного SO3 и
плотности 1890 кг/м3.
Рис. 40. Влияние продолжительности (в
мин) ацетонирования на накопление оки-
си мезитила:
1 — 20; 2—40; 3—60; 4—80; 5—120;
3. Реакция уплотнения протекает не только в присутствии кислоты, нэ
и в щелочной среде по следующей схеме [110]:
сь [3' сн3 сн3 сн3 сн3 сн3
'С^ОН хок ХСОН Хс/ II
+ [КОН] + сн2 — I —> СН + [Н2О] 1
сн3 сн3 СН2Н со со
1 \ /ОН с=о + КОН —> С( 1 с=о сн3
1 1ХОК; [
сн3 сн3 сн3 сн3
Окись мезитила
* Интерес представляет метод обезвоживания ацетона (до. содержания воды
0,005%) при помощи цеолитов (И. Сосонкин и др. ЖПХ, 1971, 44, № 4, 950).
267
Катализатором этой реакции является щелочь. В связи с этим необходи-
мо строго следить за реакцией массы после нейтрализации, не допуская вы-
сокой щелочности (реакция должна быть слабощелочной по фенолфталеину).
В случае повышенной щелочности при отгонке ацетона при повышении
температуры будет происходить конденсация ацетона с образованием окиси
мезитила. Это ухудшает качество регенерируемого ацетона1.
4. Важное значение имеет количество серной кислоты, добавляемой с
целью связывания выделяющейся при ацетонировании воды.
Теоретическое количество серной кислоты на 1 кг сорбозы может быть
подсчитано [53] и составляет около 0,65 л на 1 кг сорбозы.
Увеличение количества серной кислоты обусловит интенсификацию про-
цесса конденсации ацетона, а уменьшение ее количества будет способство-
вать гидролизу части диацетон-L-сорбозы несвязанной частью воды. В за-
водской практике применяют 0,5 л олеума на 1 кг сорбозы.
5. Для величины выхода диацетонсорбозы имеет значение порядок сме-
шения компонентов [21,102], т. е. прибавление раствора щелочного реагента
к кислому ацетоновому раствору (прямая нейтрализация) или, наоборот,
прибавление кислого ацетонового раствора к раствору щелочи (обратная
нейтрализация)
При прямой .нейтрализации ацетонового раствора диацетонсорбозы вод-
ной щелочью возможен частичный гидролиз диацетонсорбозы вследствие
сохранения в течение всего процесса нейтрализации кислой среды, появле-
ния в ацетоновом растворе воды и местного повышения температуры
в результате выделяющейся теплоты нейтрализации.
При обратной нейтрализации ацетоновый раствор сразу попадает в ще-
лочную среду, где диацетонсорбоза сравнительно стойка.
Экспериментально установлено [102], что при прямой нейтрализации
уже при 0° гидролизуется в моноацетонсорбозу около 20% диацетонсорбозы.
При обратной нейтрализации при 0° гидролиз незначителен (1,6%). Поэтому
в целях увеличения выхода диацетонсорбозы следует применять обратную
нейтрализацию ацетонового раствора.
6. На выход диацстон-L-сорбозы существенно влияют температура нейт-
рализации и интенсивность перемешивания реакционной массы в ацетона-
торе и нейтрализаторе. Результаты наших исследований (при участии А.Фон-
даренко) показали:
а) при понижении температуры нейтрализации от +5, +7 ° С до 0° выход
диацетон-Z,-сорбозы повышается на 4—5%;
б) при увеличении интенсивности перемешивания реакционной массы в
ацетонаторе выход. диацетон-L-сорбозы составил (в % от теоретического):
Частота вращения мешалки, об/мин 100 200 400 750
Выход, % ...................... 62,7 70,8 77,6 81,0
Периодический процесс производства диацетон L-сорбозы
Синтез диацетон-L-сорбозы состоит из нескольких стадий: подготовки
сырья, процессов ацетонировании L-сорбозы, нейтрализации, выделения
диацетон-L-сорбозы и доацетонирования моноацетон-L-сорбозы (рис. 41).
Сырье. Качество поступающих в реакцию химических реактивов влияет
на качество и выход диацетонсорбозы. Поэтому необходимо особое внимание
уделять вопросам их приемки и хранения, а также подготовки'для произ-
водства. При большом объеме производства требуется организация специ-
ального отделения по подготовке реактивов и их стандартизации. Качество
сырья должно соответствовать ГОСТам (ацетон — 2768—60; олеум — 4204—
65; натр едкий — 2263—59).
1 Окись мезитила, С6Н10О, температура кипения 130—131° С, с^°=0,86532,
Пд=1,44478 [111].
268
Процесс ацетонирования. Процесс взаимодействия L-сорбозы с ацетоном
в присутствии серной кислоты называется ацетонированием и осуществля-
ется в специальном аппарате — ацетонаторе.
Ацетонатор представляет собой реактор из нержавеющей стали или из
стали, покрытой кислотоупорной эмалью, снабженный рубашкой для ох-
лаждения, метательным прибором (частота вращения 200 об/мин), гильзой
с термометром, люком для загрузки сорбозы, штуцером для подачи олеума
из мерника, штуцером для подачи ацетона из общего мерника и штуцером
воздушным.
Рис. 41. Технологическая схема производства диацетон-Д-сорбозы периодическим
процессом.
Каждый ацетонатор имеет свой мерник олеума во избежание усложнения
коммуникации для распределения кислоты. Наиболее эффективным является
метод ацетонирования с применением олеума при температуре 12—15° С
[53]. По этому методу в реактор-ацетонатор 1 вводят из охлаждаемого мер-
ника 2 ацетон температурой минус 5° С и постепенно в течение 30 мин из
мерника 3 приливают олеум1 из расчета 0,5 л на 1 кг сорбозы (кислотность—
80 г/л H2SO4).' Концентрация свободного SO3 в олеуме около 18%. Затем
быстро добавляют в ацетон всю порцию сорбозы, поддерживают температуру
массы 12—15° С и перемешивают до полного растворения диацетон-сорбозы.
Остаточная сорбоза не должна превышать 2 г/л. Далее массу спускают в
охладитель 4, где охлаждают до минус 10—12° С и выдерживают 1 ч [112].
1 Опубликован патент [112а] на метод ацетонирования без олеума при помощи
ионообменных смол (Амберлит 15) при 16-кратном количестве ацетона при темпера-
туре 35° С. Вода удерживается специальным ионитом типа молекулярного сита. Вы-
ход 88%; чистота -96% .
269
Нейтрализация. Для процесса нейтрализации реакционной массы при-
меняют такой же реактор, как и для ацетонирования, но на 25—30% боль-
шего объема. Скорость вращения мешалки также больше (200—250 об/мин).
Нейтрализатор изготовляют из нержавеющей стали, так как эмаль слабо
сопротивляется щелочам.
В нейтрализатор 5 загружают 18—20%-ный раствор едкого натра из
мерника 6 в количестве, необходимом для нейтрализации всей серной кис-
лоты и создания слегка щелочной среды (слаборозовое окрашивание фенол-
фталеиновой бумажки) и охлаждают до минус 1—2° С. Затем охлажденный
ацетоновый раствор постепенно приливают в энергично перемешиваемый
охлажденный раствор едкого натра с таким расчетом, чтобы продолжитель-
ность нейтрализации не превышала 30—60 мин и температура не поднима-
лась выше 0+2°.
Для нейтрализации не допускается применение щелочного маточного
раствора, получаемого при выделении диацетон-Ь-сорбозы, так как это про-
тиворечит основным принципам технологии (стр. 6). По окончании процес-
са нейтрализации раствор, отстоявшийся от осадка, должен иметь слабоще-
лочную реакцию на фенолфталеин (конечная щелочность 0,5—1,0 г/л). Не-
обходимо иметь в виду, что кислая реакция среды обусловливает распад
диацетонсорбозы во время выпаривания ацетона. Сильнощелочная среда вы-
зывает конденсацию ацетона в окись мезитила и ухудшает качество регене-
рированного ацетона. Нейтрализованный раствор далее направляется на
выделение диацетонсорбозы.
Выделение диацетон-Л-сорбозы. Оно может быть осуществлено лишь
после изоляции осадка десятиводного гидрата сульфата натрия и отгонки
ацетона. Здесь возникает вопрос, какая из этих операций должна предшест-
вовать. Казалось, что в первую очередь реакционная масса должна быть ос-
вобождена от осадка сульфата, из фильтрата должен быть отогнан ацетон,
а затем из кубового остатка выделена диацетонсорбоза. Первоначально этот
путь и был признан в производстве [21 ]. Затем в порядке рационализации
процесса отделения осадка сульфата натрия был предложен метод деканта-
ции [113], основанный на различии в удельных весах ацетонового раствора
(0,9) и десятиводного гидрата Na2SO4 (1,46) и на низкой температуре плавле-
ния последнего (32,4°С). Однако практика показала, что при декантации
верхнего ацетонового слоя трудно добиться четкого разделения ингредиен-
тов. В результате этого возникли большие потери диацетон-сорбозы в ниж-
нем слое (расплавленном сульфате). Кроме того, при этом методе несколько
ухудшается качество диацетонсорбозы, вследствие перехода в нее красящих
веществ, адсорбируемых осадком сульфата и десорбируемых при расплавле-
нии кристаллов [53]. Тогда возник вариант предварительной отгонки из
реакционной массы ацетона с последующим расслаиванием кубового остат-
ка. Этот метод нашел применение в производстве в связи с одновременным
внедрением процесса доацетонирования моноацетонсорбозы (Л. Петрова и
Е. Ярош). Сущность метода заключается в следующем. После отгонки аце-
тона и охлаждения до 40—42° С происходит расслаивание массы: наверху
диацетонсорбоза (плотность 1170—1200 кг/м3), а внизу насыщенный
раствор сульфата натрия (плотность 1250—1260 кг/м3). Диацетонсорбозу
направляют на отгонку окиси мезитила и на очистку переосаждением ще-
лочью, а раствор сульфата натрия, содержащий моноацетонсорбозу, на до-
ацетонирование. Последний охлаждают до 20—25° С, нейтрализуют серной
кислотой до содержания едкого натра 1—2 г/л. Затем при температуре 40—
45° С ацетоном экстрагируют моноацетонсорбозу и остаточную диацетонсор-
бозу. Затем реакционную массу расслаивают и верхний слой (ацетоновый
экстракт) направляют в вакуум-выпар нои аппарат, отгоняют ацетон и ох-
лаждают до 18—20° С, нейтрализуют до остаточного содержания NaOH 1—
2 г/л и отфильтровывают выделившиеся кристаллы. Фильтрат сгущают при
вакууме 600—650 мм рт. ст. и подвергают ацетонированию в обычных ус-
270
ловиях с применением пятикратного весового количества ацетона к массе
моноацетонового сиропа.
При этом варианте прямой выход диацетон-L-сорбозы составляет 72—
73%, а при доацетонировании получают дополнительно 7—8%, а всего 79—
81 % от теоретического.
Необходимо однако подчеркнуть, что этот вариант в технологическом от-
ношении не является оптимальным и имеет следующие недостатки:
усложняется и ухудшается процесс отгонки ацетона в связи с загрязне-
нием поверхности нагрева вакуум-аппарата осадком сульфата;
понижается качество диацетонсорбозы из-за десорбции красящих ве-
ществ при расплавлении десятиводного сульфата;
вследствие трудности четкого разделения ингридиентов часть диацетон-
сорбозы остается в расплаве сульфата, которая в последующих процессах
разрушается;
процессы доацетонирования являются очень громоздкими; для них тре-
буется большое количество аппаратов.
Учитывая изложенное, следует считать более рациональным, чтобы отде-
ление осадка сульфата натрия предшествовало отгонке ацетона.
В связи с этим отпадает необходимость в процессе доацетонирования,
так как показано [53, 114], что после выделения диацетонсорбозы в маточ-
ном растворе остается незначительное количество ацетоновых производных,
способных превращаться в диацетонсорбозу. Далее возникает вопрос о ме-
тодевыделения диацетонсорбозы из кубового остатка после отгонки ацетона.
Прежде всего необходимо отогнать водяным паром окись мезитила для сни-
жения расхода окислителя. Для этого вводят в вакуум-аппарат воду и при
вакууме 600—700 мм рт. ст. и температуре 100° С отгоняют окись мезитила.
Затем из кубового остатка следует выделить диацетонсорбозу. Существуют
два метода: экстракция органическими растворителями и высаливание рас-
твором едкого натра.
Для экстракции диацетонсорбозы предлагались различные растворители:
эфир [55], дихлорэтан [115], бензин [116], хлороформ (А. Басова, Г. Ми-
хайлов, Л. Петрова), бензол [117]. Однако исследования [53, 97] показа-
ли, что экстракционный метод выделения диацетонсорбозы не позволяет
заметно повысить выход целевого продукта и вместе с тем усложняет и удо-
рожает производство, а также резко ухудшает условия труда.
Поэтому практическое применение имеет у нас метод выделения диацетон-
сорбозы щелочью. Таким образом, после нейтрализации ацетонового раство-
ра реакционную массу сливают в охлаждаемый сборник 7 (см. рис. 41) и
далее в центрифугу 8 взрывобезопасного типа из нержавеющей стали. В ней
отфуговывают кристаллы сульфата натрия, которые промывают ацетоном.
Фильтрат вместе с промывным ацетоном поступает в сборник 9 и далее в
выпарной вакуум-аппарат 10, где отгоняют ацетон, а затем из мерника Па
вводят воду в количестве 200—300 % к массе сорбозы и отгоняют при темпе-
ратуре 100° С окись мезитила. Объем кубового остатка должен составить
2,5—3,0 л (плотность 1120—ИЗО кг/м*} на 1 кг сорбозы. Сгущенный раствор
сливают в кристаллизатор 11, куда добавляют щелочной маточник II в ко-
личестве 1 : 1 (по объему) из мерника 12 и выделяют диацетонсорбозу при
температуре 50° С.
В кристаллизаторе массу выдерживают 2—4 ч в зависимости от степени
охлаждения, не давая массе затвердеть. Отслоившийся маточный раствор I
спускают через нижний штуцер с разделительным стеклом в сборник 13,
снабженный рубашкой для подогрева и охлаждения. Путем подогрева ма-
точника I до 70° С и последующего охлаждения до 20° С можно достигнуть
выделения в твердом виде на поверхности маточника всей остаточной диаце-
тонсорбозы, которую направляют на очистку. Оставшуюся в кристаллизато-
ре 11 диацетонсорбозу растворяют в горячей воде в соотношении 1:1, затем из
мерника 14 подают в кристаллизатор чистую натриевую щелочь (42—44 % -
ную) из расчета 0,5 л на 1 л раствора при температуре 50° С. Массу,не давая ей
271
затвердеть, выдерживают до 2 ч пока полностью не расслоится диацетонсор-
боза и маточник II. Последний спускают в сборник 15, откуда его насосом 16
перекачивают в сборник 12. Жидкую диацетонсорбозу спускают в аппарат-
смеситель 17, где ее растворяют в воде (5 частей воды на 1 часть диацетон-
сорбозы) до содержания 15—18% сухих веществ. Водный раствор диацетон-
сорбозы далее направляют на окисление.
Ацетон, полученный в вакуум-перегонном аппарате 10, содержит до 15%
влаги. Для ректификации его направляют через сборник 18 в ректификаци-
онную колонну 19. Сухой ацетон собирают в приемниках 20 и возвращают его
в сборник 2. Осадок десятиводного сульфата натрия из центрифуги 8 на-
правляют в плавильный аппарат 21, откуда засасывают в перегонный аппа-
рат 22, отгоняют ацетон, а расплавленные кристаллы сульфата передают в
вальцовую сушилку 23. Обезвоженный продукт содержит 98% Na2SO4 и по
своему составу соответствует ГОСТу 6318—52 на безводный сульфат натрия
I сорта. Выход 41,7% к десятиводному гидрату или 95% от теоретического.
Непрерывный процесс производства диацетон-Л-сорбозы [97, 118—120]
На основании различных опубликованных исследований [97, 118—120]
разработана следующая технологическая схема непрерывного процесса аце-
тонирования (рис. 42). В смеситель 1 через дозаторы 2, 3 непрерывно посту-
Рис. 42. Т ехнологическая схема непрерывного производства диацетон-Ц-сорбозы.
пают ацетон и олеум, где при охлаждении (—5 Ч-------7° С) смешиваются.
Смесь поступает в ацетонатор 4, куда непрерывно поступает сорбоза из до-
затора 5. Ацетонатор снабжен скоростной мешалкой (частота вращения
250—350 об/мин}. Затем раствор через холодильник 6 идет в аппарат-вы-
держиватель 7, где при минус 10—15° С выдерживается в течение 1 ч. Реак-
ция нейтрализации осуществляется в аппарате со скоростной мешалкой 8,
272
куда непрерывно поступает из мерника 9 щелочь, охлажденная в холодиль-
нике 10. Нейтрализованный раствор далее направляется в аппарат с мешал-
кой 11, а из него в центрифугу непрерывного действия 12. Фильтрат после
отделения десятиводного гидрата передается в сборник 13. В пленочном ис-
парителе 14 отгоняется ацетон. К кубовсму остатку в смесителе 15 добавля-
ется вода, а в пленочном испарителе 16 под вакуумом отгоняется окись ме-
зитила. В смесителе 17 сироп монодиацетонсорбозы смешивается с 42 %-ной
щелочью, а в расслоительной колонке 18 раствор диацетонсорбозы плот-
ностью 1110 кг/м? при температуре 60° С отделяется от щелочного маточно-
го раствора (плотность 1270 кг/м‘д). Диацетонсорбоза непрерывно поступа-
ет в сборник 19, откуда далее на окисление. Щелочной маточный раствор,
направляется в сборник 20.
Полузаводские испытания подтвердили результаты лабораторных иссле-
дований по выходу диацетон-L-сорбозы (72—73%) [118].
ПРОИЗВОДСТВО ГИДРАТА ДИАЦЕТОН-2-КЕТО-£-ГУЛОНОВОЙ КИСЛОТЫ
Проведенные в последние годы исследования показали, что L-сорбозу
можно непосредственно (без применения защиты ацетоновыми группами)
окислить в 2-кето-£-гулоновую кислоту. Для этого могут быть применены
следующие методы: а) химический — азотной кислотой [121, 122, 123],
гипохлоритом натрия [124]; кислородом в присутствии платинового или пал-
ладиевого катализатора в нейтральной или щелочной среде [125, 126, 127];
б) электролитический [128]; в) биохимический — при помощи микроорга-
низмов класса Pseudomonas в аэробных условиях (например, Р. viridiflava,
или Р. aerogunosa, или Р. mildewagi при температуре 25—40°С .[129, 130].
Реакция окисления L-сорбозы в 2-кетоД-гулоновую кислоту протекает
по следующей
СН2ОН
I
НО-С------
НО-С-Н
I <
Н-С—ОН
НО-С—н
Н2С-----
Д'СорЗоза
Н—с=о
I
но—с----.
I
но—с—н |
I С
н—с—он
но—с—н
I
Н2С----
Д-Гулозон
СООН
I
но—с----,
НО—С—Н I
I С
н—с—он
I
но—с—н
Н2С-----
соон
I
с=о
но—с—н
I
н—с—он
I
но—с—н
I
Н2СОН
2-Кето-Д-гуло-
новая кислота
Такое направление процесса окисления обосновывается большой реак-
ционной способностью первичной спиртовой группы, вследствие активиро-
вания ее соседней карбонильной группой С=О.
Однако не исключено и другое направление реакции, обусловленное
большой активностью менее гидрогенизированной вторичной спиртовой
группы (СНОН) по соседству с карбонилом. Тогда в результате реакции
будут получены гликолевая и эритритовые кислоты [53]. Реакция может
идти еще в третьем направлении с образованием 2-кето-Т-гулосахарн6й кис-
лоты [35].
При непосредственном окислении L-сорбозы возникают значительные
трудности, связанные с регулированием процесса окисления, а также с вы-
делением 2-кето-Ь-гулоновой кислоты, в связи с чем этот метод пока не по-
лучил промышленного применения. Однако прогрессивность данного мето-
да и связанные с ними перспективы изъятия из производства огнеопасного
ацетона заслуживают проведения углубленных исследований в этой области
с целью скорейшего освоения процесса.
Диацетон-2-кетоЩ-гулоновая кислота (гидрат) — представляет четвер-
тый промежуточный продукт синтеза аскорбиновой кислоты из D-глюкозы.
273
Формула безводного вещества С12О7Н18, молекулярная масса 274,26, гид-
рата С12О8Н20, молекулярная масса 292,28; он представляет собой бесцвет-
ные кристаллы хорошо растворимые в спирте, эфире и трудно растворимы в
ледяной воде (в 1 л растворяется около 1 г гидрата). Температура плавле-
ния 98—99° С.
При кипячении с водой гидрат легко омыляется и переходит в 2-кето-Ь-
гулоновую кислоту, которая выделяется в кристаллическом виде путем вы-
паривания раствора под вакуумом до консистенции густого сиропа, из ко-
торого она быстро кристаллизуется. Процесс гидролиза также легко проте-
кает в кислой среде.
Химизм процесса окисления диацетон-А-сорбозы в диацетон-2-кето-Т-
гулоновую кислоту. Для окисления диацетон-Т-сорбозы предложены различ-
ные окислители: перманганат калия [55], гипохлорит натрия [131, 132] в
присутствии катализатора, манганат калия [133], электролиз [134] в при-
сутствии небольших количеств КМпО4 (около 25%). Практическое приме-
нение получили два окислителя: КМпО4 и NaClO.
При окислении диацетонсорбозы перманганатом калия процесс осущест-
вляется в две стадии согласно следующим химическим уравнениям:
।— -----^сн2он
I н3сх .о-с
о ।
н3с" хо-с-н
з н-с-о сн,
। \ /
-----------с-н с
н2с-о \н3
Диацетин -L- сорбоза
3'260,28
4KMnO4 + 3NaOH
_______ COONa
Н,С„ хо-с
_СХ I
Н;С о-с-н
н-с-о
I \
+ 4МпОо +
СН3
4КОН + 4Н20
4'158,03 + 3-40,01
-----------с-н с
I / \
н2с-о сн3
Натриебая соль диацетон-2-кето-
L - гулонобой кислоты
3-236,25
О-'8 6,93 + it-56,1
При действии НС1 на холоду (2° С) нейтрализуют щелочь и выделяют ди-
ацетон-2-кето-Ь-гулоновую кислоту в виде гидрата
________^COONa
I н3сх хо-с
о юб I
нс хо—с—н
3 Н-С-О\ /СНз
---------с-н с
I / \
н2с-о сн3
Натриебая соль диацетон -2 .- кето -
L - гулонобой кислоты
3'296,25
4КОН + 7НС1
4'58,1 <-7-38,47
_________^соон • Н2О
I н3сх .о-с
о X ।
Н{С о-с-н
Н-С-О сн3
----------с-н с
I / \
н2с-о сн3
Гидрат диацетон-2- кета-
L -гулонобой кислоты
4KCl + 3NaC1 + H2O
3-292, 28
Ц-Щ,55+3-58,U5
Процесс окисления диацетон-7,-сорбозы перманганатом калия в щелоч-
ной среде основан на следующей реакции:
2КМпО4 + Н2О —> 2МпО2 + 2КОН + 30.
Эта реакция в условиях щелочной среды протекает стадийно. Вначале
образуется манганат калия с выделением двух атомов кислорода по следую-
щему уравнению:
4КМпО4 + 4К0Н —> 4К2МпО4 + 2Н2О + 02.
Образованный К2МпО4 далее также действует как окислитель, выделяя
один атом кислорода, по следующему уравнению:
К2МпО4 + Н20 —> MnO2 + 2К0Н + О.
Если исходить из уравнения окисления диацетонсорбозы, то расход ед-
кого натра на 1 кг диацетонсорбозы составит 0,154 кг, а концентрация щело-
274
чи в реакционной среде 2%. Однако исследования показали [135], что вы-
сокая конечная щелочность обусловливает излишний расход перманганата
калия, так как часть перманганата по окончании процесса окисления и при
наличии свободной щелочи раскисляется до манганата калия. Количество
потерянного перманганата зависит от величины конечной щелочности реак-
ционной массы. Установлено, что наилучшие результаты достигаются при
конечной щелочности среды 0,3—0,5%, что соответствует введению на окис-
ление около 0,10 кг щелочи на 1 кг диацетонсорбозы [135].
При окислении диацетонсорбозы гипохлоритом натрия или кальция в
присутствии катализатора NiSO4 процессы также осуществляют в две ста-
дии. Вначале окисляют диацетонсорбозу и получают натриевую соль диаце-
тон-2-кето-Ь-гулоновой кислоты, а затем соляной кислотой выделяют ее
гидрат [131, 132, 136, 137].
Реакция окисления идет по следующей схеме:
-------сн2он
н3сХ(,хо-с
H3CZ ХО-С-Н
/СНз
•--------с-н с
Н2С-О/ \нз
Диацетон - L -сорбоза
260,28
+ 2NaClO + NaOH
NiSO4
_________ COON а
I Н3Сх ^о-с
О Cf I
H3CZ о-с-н
„ । +2NaCl+2H2O
Н~С~О /СН3
•-----------
н2с-ох \н3
натриевая соль диацетон-2- кето-
L - гулонобой кислоты
236.25
Механизм окислительного процесса, осуществляемого при помощи ги-
похлорита в присутствии сернокислого никеля, заключается в следующем.
Реакция окисления диацетонсорбозы гипохлоритом (при несоблюдении
определенных условий) протекает бурно с быстрым повышением темпера-
туры до 100—102° С.
Это явление может быть объяснено при рассмотрении механизма реак-
ции окисления диацетонсорбозы гипохлоритом в присутствии сернокислого
никеля или смешанного катализатора (соли никеля и кобальта) [138 В.
В щелочной среде никель осаждается в виде гидрата закиси, который •
гипохлоритом окисляется до окиси никеля со следующим термохимическим
эффектом (Q):
NiSO4 + 2NaOH —> Ni (ОН)2 + Na2SO4
213,6— 15,1 2(102—10,1) 129,8 332,2—0,3; Q4 = — 79,4 ккал;
2Ni (OH)2 + NaClO —> Ni2O3 + 2H2O + NaCl
2-129,8 83,4 196,7 2-68,4 98,3— 1,2; Q2 = — 87,6 ккал.
Далее окись никеля действует как катализатор; выделяя кислород'из
гипохлорита натрия, окись никеля превращается в закись, которая гипо-
хлоритом окисляется в окись никеля и т. д.
NaClO + Ni2O3 —> 2NiO + NaCl + O2
83,4 196,7 2-58,4 98,3— 1,2 60,5; Q3 = + 5,7 ккал.
Наконец, четвертая фаза — окисление выделяющимся кислородом ди-
ацетонсорбозы сопровождается обильным выделением тепла (по расчету
288 ккал!(г моль).
1 В Японском патенте [138] рекомендуется применение смешанного катализато-
ра, состоящего из 70% никеля и 30% кобальта. В этих условиях скорость реакции
увеличивается в 1,8 раза. На 500 кг диацетонсорбозы вводят 5 кг хлорида никеля
и 2 кг хлорида кобальта. Реакцию ведут при температуре 60° С. Выход гидрата ди-
ацетон-2-кето-£-гулоновой кислоты составляет 525 кг.
275
Для иницирования процесса окисления требуется подогрев реакцион-
ной массы, так как и Q2 имеют отрицательное значение. Затем обильное
выделение тепла при непосредственном процессе окисления вовлекает в ре-
акцию все большие количества катализатора, который еще более ускоряет
реакцию окисления ит.д. Реакция окисления все ускоряется, а вследствие
этого увеличивается количество выделяемого тепла и быстро повышается тем-
пература (температурная вспышка). Это подтверждается следующим термо-
химическим расчетом при следующих параметрах процесса окисления; объ-
ем реактора-окислителя 3000 л. Загружают водный раствор диацетон-Z,-
сорбозы (ДАС) 900 л с содержанием 20%; d = 1,07. Расход гипохлорита нат-
рия на окисление при расходе активного хлора в 0,9 кг на 1 кг ДАС или
1300 л с содержанием 133 г/л активного хлора. Содержание ДАС в растворе
900 • 1,07 • 0,2 = 192,6 кг. При термическом эффекте + 288 ккал/(г моль)
при окислении 192,6 кг ДАС будет выделено тепла:
1000-192,6
260,28
где 260,28 — молекулярная масса ДАС.
Из этого количества тепла на подогрев реакционной массы от 65 до 85° С
расходуется тепла:
Qi = (900- 1,07 + 1300- 1,2) 0,9(85 — 65) =45414 ккал,
где 1300 — количество гипохлорита натрия, л\
0,9 — теплоемкость массы.
При охлаждении реакционной массы рассолом температурой —10° С
при F = 3 м2 и коэффициенте теплопередачи в 5 ккал/мин получим, что в 1 ч
может быть локализовано следующее количество тепла:
Q2 = 3 5 • 60 [85 — (—10)] = 85500 ккал.
Следовательно, продолжительность реакции, допускаемая поверхностью
охлаждения, составляет:
213109 — 45414
-------------2 ч.
85500
При отсутствии охлаждения и мгновенном протекании реакции выделяе-
мое тепло может повысить температуру на
213109
= 93°С,
0,9(900 1,07 + 1300 1,2)
что обусловливает бурное вскипание реакционной массы и ее переброс. Од-
нако продолжительность реакции можно регулировать путем непрерывного
и постепенного ввода в реакцию катализатора и гипохлорита; в крайнем слу-
чае,’ необходимо ступенчатое добавление катализатора (в четыре приема).
Что касается моноацетонсорбозы, то она, имея четыре незащищенных
углеродных атома, подвергается под влйянием гипохлорита натрия гидроли-
зу и расщепительному окислению с образованием натриевой соли щавелевой,
уксусной кислот и хлороформа по следующему уравнению:
_________СН2ОН
I н3с. п-с
о с' I
н3с о-с-н , ,
I + 12NaOCl + 4NaOH---------»-3(COONa)2+ СН3СООМа+ СНС13 + 9МаС1 + 8Н2О
н-с-он
I
----------с-н
I
СН2ОН
Моноацетон -L - сорбоза
276
Таким образом, загрязнение диацетон-А-сорбозы моноацетон-Е-сорбозой
приводит к значительному увеличению расхода окислителя (в 6 раз) и
щелочи (в 4 раза). Это влечет за собой увеличение количества NaCl и оксала-
та натрия, ухудшающих качество гидрата диацетон-2-кето-Е-гулоновой кис-
лоты.
Влияние различных факторов на процесс окисления. На выход и качест-
во гидрата оказывают влияние при окислении перманганатом калия [53]:
концентрация диацетон-Е-сорбозы в растворе. В сильно разбавленных
растворах увеличиваются потери гидрата в маточном растворе после осаж-
дения гидрата соляной кислотой. Оптимальная концентрация диацетон-L-
сорбозы составляет 17—18%;
качество диацетон-L-сорбозы, поступающей на окисление. При большом
загрязнении ее моноацетонсорбозой увеличивается зольность гидрата, что
обусловливает увеличение количества промывных вод, а следовательно,
потери гидрата;
контроль полноты реакции окисления и соблюдения технологического
режима процесса. При недоокислении части диацетонсорбозы не только сни-
жается выход гидрата, но и ухудшается его качество. Имеются предположе-
ния, что так называемый «несохнущий» гидрат получается при наличии в
растворе значительного количества недоокисленной диацетон-L-сорбозы;
pH среды при выделении гидрата диацетон-2-кето-Е-гулоновой кисло-
ты НС1. Полнота осаждения достигается при pH 1,5—2,0. При pH ниже 1,5,
кроме снижения выхода гидрата, возможно образование «несохнущего»
гидрата.
При применении в качестве окислителя гипохлорита на выход гидрата и
на его качество влияют:
концентрация щелочи в гипохлорите, которая должна поддерживаться
на уровне 70 г1л при концентрации активного хлора 150—170 г!л\
количество гипохлорита не должно превышать теоретического, так как
большой избыток его приводит к загрязнению гидрата поваренной солью,
что обусловливает увеличение количества промывных вод, а следовательно,
и потерь гидрата в них. Кроме того, это способствует выделению свободно-
го хлора при разложении натриевой соли диацетон-2-кето-Е-гулоновой
кислоты;
температура окисления должна быть около 80° С;
концентрация диацетонсорбозы в реакционной массе 18—20%. При
более высокой концентрации реакция окисления протекает слишком энер-
гично с потерями гипохлорита.
На расход окислителя большое влияние оказывает чистота диацетонсор-
бозы. Увеличение содержания моноацетонсорбозы вызывает резкое увели-
чение количества окислителя. Поэтому рекомендуется диацетон-L-сорбозу
подвергать очистке путем двойного переосаждения (стр. 271).
На выход гидрата диацетон-2-кето-Ё-гулоновой кислоты влияет pH реак-
ционной массы. По-видимому, нельзя допускать местного понижения pH
(менее 7,5—8,0) [138а].
К вопросу о выборе окислителя. Известно, что перманганат калия явля-
ется окислителем очень эффективным и удобным для применения. Однако
вследствие дороговизны и дефицитности КМпО4 не имеет перспектив для
широкого применения в производстве аскорбиновой кислоты.
Наиболее перспективным окислителем диацетон-Е-сорбозы является
гипохлорит натрия или хлор как дешевый и широко распространенный хи-
микалий. Его применение лишь тормозится по санитарным причинам на за-
водах, расположенных в крупных городах. В этих условиях в перспективе
могут быть использованы два других метода окисления диацетон-Е-сорбозы:
1) электрохимический или 2) каталитический кислородом воздуха. Электро-
химический метод применяется с использованием галогенидов никеля или
их смеси с кобальтом [139], солей марганца или бромидов в качестве пере-
носчиков кислорода. В частности, этот метод применяется для электрохими-
277
ческого окисления D-глюкозы в D-глюконовую кислоту с получением в при-
сутствии бромистого калия (натрия) и карбоната кальция глюконата каль-
ция [139а]. Здесь окисление осуществляет гипобромид калия (натрия),
образуемый при электролизе щелочного раствора бромида. Выделяясь на
аноде, бром реагирует с NaOH и образует гипобромит натрия по схеме:
2NaOH + Вг2 —> NaOBr + NaBr + Н2О.
Проверен каталитический метод окисления диацетон-Z,-сорбозы кисло-
родом воздуха в присутствии палладиевого катализатора [140—143].
Рис. 43. Технологическая схема окисления диацетон-Д-сорбозы перманганатом
калия.
Разработан метод приготовления палладиевого катализатора на угле
путем постепенного добавления формальдегида к щелочной водной суспен-
зии хлористого палладия [141 ]. Исследован вопрос о многократном исполь-
зовании отработанного катализатора. Активность катализатора зависит от
качества формальдегида, и этот вопрос требует дополнительного изучения.
Синтез диацетон-2-кето-/,гулоновой кислоты (ДК.ГК.) включает разделы,
касающиеся качества сырья, процесса окисления диацетон-Z,-сорбозы и вы-
деления гидрата ДКГК-
Сырье. Концентрация водного раствора диацетон-Z,-сорбозы должна
быть 18—20% при окислении гипохлоритом или перманганатом калия. По-
следний должен содержать КМпО4 не ниже 96%. Хлор для приготовления
гипохлорита натрия должен содержать С1 не менее 99,5%, а влаги не более
0,06%.
Окисление диацетон-Z,сорбозы марганцовокислым калием (рис. 43). Го-
товят раствор диацетон-Z,-сорбозы, содержащий 18% сухих веществ. Для
растворения используют промывные воды, получаемые при фильтрации и
промывке осадка перекиси марганца. На 1 кг технической диацетонсорбозы
вводят 5,0—5,5 л промывных вод. Затем вводят водную щелочь из расчета
1,2—1,5 л 42—44% NaOH на 10 кг перманганата калия. Процесс окисления
осуществляют в реакторе-окислителе, изготовляемом из нержавеющей ста-
ли, снабженном мешалкой (с частотой вращения 180—20.0 об/жнн) и рас-
сольным охлаждением.
После проверки щелочности (12—14 г/л) раствор нагревают до 34° С и
начинают подачу перманганата калия в сухом виде через специальный доза-
тор, устроенный по принципу трясучки, приводимой в движение от вала ме-
шательного прибора.
Одновременно в рубашку пускают рассол и поддерживают температуру
34—36° С в течение всего процесса окисления; продолжительность добавле-
ния перманганата не должна превышать 2 ч. В конце процесса окисления
щелочность раствора снижают до 0,3—0,5%.
278
К концу процесса окисления наносят каплю раствора на фильтровальную
бумагу. Фиолетовый цвет указывает на наличие свободного перманганата
калия, а зеленый цвет — на присутствие манганата, являющегося первым
промежуточным продуктом раскисления КМпО4. При отсутствии зеленого
кольца необходимо в реакционную массу добавить щелочь, а при отсутствии
фиолетового КМпО4. После окончания добавления перманганата реакцион-
ную массу выдерживают 40 мин при температуре 34—36° С. Если по истече-
нии этого времени взятая проба на фильтровальной бумаге дает зелено-
фиолетовый ореол, а анализ показывает содержание остаточной диацетон-
сорбозы в реакционной массе не выше 1 г в 1 л , то процесс окисления следу-
ет считать законченным. После этого реакционную массу подогревают до
50—55° С при перемешивании. При этом раствор обесцвечивается. Из реак-
тора 1 (см. рис. 43) масса поступает в нутч-фильтр 2 для отделения осадка
перекиси марганца. Осадок промывают горячей водой (70—75° С) до исчез-
новения в промывных водах натриевой соли диацетон-2-кето-Е-гулоновой
кислоты. Первые порции промывных вод присоединяют к основному раство-
ру, а остальные промывные воды используют для растворения диацетон-
сорбозы.
При спуске реакционной массы в нутч-фильтр во избежание попадания на
центрифужную ткань кристалликов перманганата и ее разрушения часть
реакционной массы (20—30 л) спускают из нижнего штуцера и возвращают в
окислитель. Зарядка нутч-фильтра фильтровальной тканью производится
следующим образом.
На решетке нутч-фильтра помещают салфетку из бязи, поверх нее два
слоя фильтровальной бумаги, затем салфетку, наконец, мешок из бельтин-
га, который надевают на всю внутреннюю поверхность приемной части
фильтра. Целесообразно применять контрольный фильтр для предотвраще-
ния попадания осадка в фильтрат МпО2.
Фильтрат из нутч-фильтра поступает в сборник 3, промывные воды в сбор-
ник 4, а промытый осадок направляют в сушилку 5. Высушенный осадок
перекиси марганца измельчают, упаковывают в ящики и отправляют по-
требителю. Фильтрованный раствор из сборников 3 и 4 насосом 6 перекачи-
вают в реактор-гидратор из эмалированной стали 7, охлаждают до 0 плюс 2° С
и при перемешивании нейтрализуют разбавленной вдвое соляной кислотой
или смесью серной и соляной кислот (плотность 1150—1200 кг/м3), получае-
мой в виде отхода производства при насыщении спирта хлористым водоро-
дом (на стадии енолизации). Кислоту прибавляют тонкой струей до кислой
реакции (pH 1,5—2,0 — фиолетовый цвет бумаги конго), при которой выде-
ляется кристаллический гидрат диацетон-2-кето-А-гулоновой кислоты. Пол-
ноту осаждения определяют анализом. Затем массу фильтруют через цент-
рифугу 8 и цромывают ледяной дистиллированной водой до исчезновения в
промывных водах ионов СЕ и SO4".
Промывные воды собирают в приемнике 9 и используют для растворения
диацетонсорбозы. Отжатые кристаллы гидрата (влажность 8—10%) посту-
пают в воздушную сушилку барабанного типа 10, где при температуре 45—
50° С кристаллы высушивают до содержания влаги не более 7—8% (включая
кристаллизационную воду).
Окисление диацетон-Е-сорбозы гипохлоритом натрия. На основании
результатов исследований [136, 137] можно рекомендовать следующую тех-
нологию процесса окисления диацетон-Е-сорбозы гипохлоритом натрия.
В смеситель загружают водный раствор диацетон-Е-сорбозы с содержа-
нием 18—20% сухих веществ. Массу размешивают и нагревают до 60—50° С.
К раствору добавляют в виде водного раствора непрерывно в течение всего
процесса сернокислый никель из расчета 1,0—1,5% к массе диацетонсорбо-
зы, либо в виде соли порциями в четыре ступени через каждые 30 мин. В ре-
актор вводят непрерывно раствор гипохлорита натрия (из расчета полного
расхода 0,9—1 кг активного хлора на 1 кг чистой диацетонсорбозы). Темпе-
ратуру поддерживают на уровне 75—80° С путем нагревания. При высокой
279
концентрации диацетонсорбозы (21—25%) и при определенном соотношении
действующих масс температура может быстро повыситься до 90—100° С
(температурная вспышка) и обусловить выброс реакционной массы. Для
предотвращения температурной вспышки и рекомендуется непрерывно
постепенно вводить катализатор, либо ступенчато, либо концентрацию ак-
тивного хлора в гипохлорите снизить до 100—120 г/л. В этих условиях ре-
акция окисления протекает без бурного выделения кислорода, что обеспе-
чивает лучшее его использование.
Реакция также протекает спокойно при концентрации диацетонсорбозы
в 15%. Затем массу фильтруют через нутч-фильтры. Выделение гидрата ди-
ацетон-2-кето-Г-гулоновой кислоты производится при помощи НС1. Не до-
пускается применять для этой цели отход — смесь серной и соляной кислот,
так как это обусловит высокую зольность гидрата ДКГК вследствие более
низкой растворимости сульфата натрия при 0° по сравнению с NaCl.
Получение водного раствора гипохлорита натрия из хлора и щелочи.
Процесс протекает по следующему уравнению:
2NaOH + С12 NaClO + NaCl + Н2О.
В аппарат загружают водный раствор NaOH с плотностью 1230 кг/м3
(30%), охлаждают до 5° С и при этой температуре при перемешивании и ох-
лаждении начинают пропускать в раствор хлор1. Скорость пропускания
хлора регулируют таким образом, чтобы температура реакционной массы
не поднималась выше 5° С. Раствор считают готовым при содержании в нем
150—170 г/л активного хлора. Раствор фильтруют через нутч-фильтр (через
стеклянное полотно). Плотность раствора 1100—1200 кг/м3. Раствор должен
быть прозрачным и иметь светло-зеленый цвет. Содержание свободной щело-
чи около 50—70 г/л. На производство 1 кг раствора гипохлорита натрия с
концентрацией активного хлора 120 г/л и щелочи 70 г/л требуется едкого
натра 0,08 кг-, хлора 0,22 кг.
Получение водного раствора гипохлорита натрия из гипохлорита каль-
ция. Гипохлорит кальция Са(С1О)2 вырабатывают в виде стабильных основ-
ных щелочных солей Са(СЮ)2 • 2Са(ОН)2 или ЗСа(С1О)2 • 2Са(ОН)2. Содер-
жит до 75% активного хлора. Для приготовления водного раствора гипохло-
рита натрия в реактор из эмалированной стали объемом в 5 м3 загружают
2500 л воды, 450—500 л 42%-ного едкого натра и 900 кг гипохлорита каль-
ция. После перемешивания в течение 1 ч и отстоя выделяющегося в осадок
гидрата окиси кальция жидкость декантируют и подают в мерник для окис-
ления. Получают 2200 л раствора гипохлорита натрия с содержанием в нем
активного хлора 140—150 г/л и щелочи около 60 г/л. Выход активного хло-
ра по гипохлориту кальция составляет 85—90%. Реакция получения гипо-
хлорита натрия из гипохлорита кальция протекает по следующему урав-
нению:
Са (ОС1)а + 2NaOH —* Са (ОН)2 + 2NaOCl.
Так как гидрат окиси кальция выделяется в осадок, то водный раствор
NaOCl, полученный из гипохлорита кальция менее загрязнен побочными
солями, чем NaOCl, полученный из хлора и щелочи.
Использование отходов. Получение калийного концентрата [97]. Калий-
ный отход получают на стадии окисления ди ацетон-L-сорбозы пермангана-
том калия в количестве 12 м3 на 1 т аскорбиновой кислоты. Отход представ-
ляет собой слабоокрашенную прозрачную жидкость, плотность
1070 кг/м3, pH 2,0, содержание сухих веществ 7,5%. После сгущения в пере-
гонном аппарате втрое раствор обезвоживают на вальцовой сушилке. Полу-
ченный калийный концентрат представляет собой темно-желтый порошок,
1 В журнале «Химическая промышленность» [144] приведены данные по транс-
портировке хлора в жидком и газообразном виде по трубопроводам и передвижной
таре.
280
растворимый в воде (при температуре 20° С— 18%). Состав (в %): Кг$О4 —
61,1; Na2SO4— 17,4; NaCl — 5,9; летучие вещества— 10,9, влажность —
5,0. Содержание КгО — 33%. Калийный концентрат после проверки в поле-
вых условиях может быть использован как калийное удобрение.
ПРОИЗВОДСТВО Д-АСКОРБИНОВОЙ КИСЛОТЫ ИЗ ГИДРАТА ДИАЦЕТОН-
2-КЕТО-Д-ГУЛОНОВОЙ кислоты
Химизм процессов лактонизации и енолизации. Превращение гидрата
диацетон-2-кето-А-гулоновой кислоты в Д-аскор би новую кислоту в среде ин-
дифферентного растворителя происходит в две основные стадии.
Первая стадия заключается в образовании в присутствии этилового спир-
та и хлористого водорода этилового эфира 2-кето-А-гулоновой кислоты [145]
по схеме:
________\COOH • н2о
I н3с. дэ-с
о Ж 1
н3с о-с-н
I
И-С-О^ /СН3
C2HSOH
HC1
I / \
с-о СН3
Гидрат диацетон-2 ~кето-
-L-гулонобой кислоты
|'ОС2Н5
с=о
I
но-с-н
I +
н-с-он
1
но-с-н
I
сн2он
2СН3СОСН3
Этилодый эфир 2-кето-
-L-гулонобай кислоты
Вторая стадия заключается в лактонизации и енолизации этилового
эфира 2-кето-А-гулоновой кислоты с образованием аскорбиновой кислоты
по схеме:
| ХОС2Н5
С=О
| на
НО—С—Н ---------->
Н—С—ОН
НО—С—н
I
СН2ОН
Этиловый эфир 2-кето-
L-гулоновой кислоты
cZ?_____
I
с—он
II О
с—ОН I + С2Н5ОН
н-с_______I
I
НО—С—н
СН2ОН
L-Аскороиновая кислота
Как первая, так и вторая стадии состоят из двух химических процессов,
а именно: первая стадия —• из этерификации и гидролиза, вторая — из лак-
тонизации и енолизации. Однако последовательность протекания этих про-
цессов до настоящего времени не изучена. Считают, что в первой стадии про-
цесса образования эфира превалирует процесс этерификации, а в конце —
процесс гидролиза [146]. Это обусловлено тем, что для процесса гидролиза
требуется две молекулы воды. Но в системе имеется лишь одна молекула
кристаллизационной воды и очень небольшое количество воды, внесенной с
реагентами.
Следовательно, в начале процесса в реакционной массе не содержится
необходимого количества воды для обеспечения гидролиза; имеющаяся во-
да удерживается газообразным хлористым водородом, растворенным в спир-
те. Для этерификации же в начале процесса имеются соответствующие ус-
ловия, так как выделяемая при этой реакции вода удерживается хлористым
водородом. По мере снижения концентрации хлористого водорода в резуль-
281
тате улетучивания и адсорбции его осадком эфира вода освобождается, что
обеспечивает процесс гидролиза (отщепление ацетоновых групп).
По этой гипотезе процессы первой стадии должны протекать по следую-
щей схеме:
соон • н.
12о
ct°
I Н,С О-С
О X I
н3с чо-с—н
н-с-о
,СН3
с2н5он
HC1
I Н3С. .о—с
о I
Н3С о-с-н
•
^"ОСгНз
с=о
с-н с
н-с-о
сн.
но-с-н z ч
I +2(СН3)9СО
н-с-он
с-н с
но-с-н
н2с—о сн3
Гидрат диацетан-2,-кето-
-L-гулонедой кислоты
Н2С—О СН;
Этилодый з<рир диацетон-
2-кето-1~гулонодой кислоты
СН2ОН
Этилодый з<рир-2-кето~
-L - гулок одой кислоты-
Другие считают [147], что на первой стадии вначале протекает процесс
гидролиза, а затем идет процесс этерификации. Однако если гидролиз гидра-
та протекает вначале, то откуда появляется вторая молекула воды, необхо-
димая для гидролиза гидрата?
Теоретические предпосылки позволяют высказать следующие сообра-
жения о механизме реакций, протекающих при енолизации.
1. Спиртовой раствор хлористого водорода связывает всю воду, находя-
щуюся в реакционной массе. Благодаря этому создаются благоприятные ус-
ловия для процесса этерификации. Образующийся эфир диацетон-2-кето-£-
гулоновой кислоты выделяется в осадок в виде аморфного вещества, адсор-
бируя значительное количество хлористого водорода.
2. Освобожденная при этерификации вода обеспечивает гидролиз с от-
щеплением ацетоновых групп. В результате этого образуется этиловый эфир
2-кето-Б-гулоновой кислоты, который является конечным продуктом I ста-
дии енолизации.
3. Этиловый эфир подвергается лактонизации с отщеплением спирта,
образованием лактона 2-кето-Б-гулоновой кислоты и последующей еноли-
зацией последнего и превращением в Б-аскорбиновую кислоту по следующей
схеме:
с\0
|ХОС2Н5
с=о
но—с—н
I
н—с—он
но—с—н
СН2ОН
Этиловый эфир 2-К5ТО-
L-гулоновой кислоты
с—-------. ------
I I
С=О I с—он I
I о II о
но—с—н -------------> с—он
I I
Н—С--------- Н—С---------
I I
но—с—н он—с—н
сн2он сн2он
Лактон 2-кето- L-Аскорбиновая кислота
L-гулоновой кислоты
Влияние различных факторов на процессы, протекающие на стадии ено-
лизации в среде хлороформа. Состав растворителя. При применении реге-
нерированного хлороформа, содержащего около 8—10% спирта и 15—20%
ацетона, необходимо знать влияние каждого ингредиента на эффективность
процесса. Этиловый спирт участвует в реакции этерификации, необходимое
количество его на 1 кг гидрата может быть подсчитано по формуле:
1 • 0,93 • 46,07 п
=0,156 кг,
274,26
где 0,93 — содержание безводной диацетон-2-кето-£-гулоновой кислоты в 1 кг
гидрата (заводского продукта);
282
46,07 — молекулярная масса этилового спирта;
274,26 — молекулярная масса безводной диацетон-2-кето-Ь-гулоновой кислоты;
0,156 кг этилового спирта соответствует 0,2 л абсолютного спирта или 0, 22 л
96°-ного.
Опыты показали [146], что как при большом избытке, так и при недо-
статке спирта в среде енолизации снижается выход аскорбиновой кислоты.
Отмечено также, что при избытке спирта наряду со снижением выхода ас-
корбиновой кислоты повышается ее качество, а при недостатке спирта или
при попадании воды в хлороформ аскорбиновая кислота получается в виде
гранул и плохо промывается. Добавление больших количеств ацетона (20%)
ухудшает процесс енолизации и снижает выход.
Концентрация хлористого водорода. Установлено, что при концентра-
ции НО более 50—60 г!кг выход аскорбиновой кислоты снижается. Низок
выход аскорбиновой кислоты при концентрации НС1 28 г!кг. Наилучшие ре-
зультаты получены при концентрации 47—50 г/кг НО [146].
Количество индифферентного растворителя. Уменьшение количества хло-
роформа с 200 до 150 и даже до 100% к массе гидрата приводит не к сниже-
нию, а к увеличению выхода аскорбиновой кислоты [146]. При снижении
вдвое количества растворителя — хлороформа — и при сохранении абсолют-
ного количества спирта, ацетона и хлористого водорода концентрация по-
следних в среде увеличивается в два раза, что положительно влияет на про-
цесс енолизации.
Ступенчатость процессов енолизации. В связи со стадийностью процес-
сов енолизации (этерификация, гидролиз, лактонизация и енолизации) воз-
никла мысль о проведении ступенчатой енолизации, которая заключается в
следующем [146]:
проводят процесс этерификации с добавлением части хлористого водоро-
да в герметизированном аппарате (I ступень), чтобы сохранить весь хлорис-
тый водород для удержания максимального количества воды, выделяемой
при этерификации;
проводят процессы лактонизации и енолизации: добавляют вторую часть
хлористого водорода (II ступень) и включают «воздушку» для удаления час-
ти хлористого водорода из раствора.
Опыты показали, что ступенчатая енолизация позволяет получить более
высокий выход аскорбиновой кислоты, чем обычная.
Режим процесса выделения и промывки аскорбиновой кислоты. Иссле-
дования показали:
замена нутч-фильтра центрифугой способствует повышению качества
аскорбиновой кислоты, понижению потерь и сокращению расхода раствори-
теля на промывку (с 1500 до 700—800% к массе аскорбиновой кислоты);
температура хлороформа в пределах 20—40° С не оказывает какого-либо
существенного влияния на потери аскорбиновой кислоты в процессе промыв-
ки хлороформом;
на результаты процесса большое влияние оказывает материал енолизато-
ра. Енолизатор должен быть изготовлен из эмалированной стали. Обнажение
стали хотя бы на небольших участках корпуса енолизатора приводит к быст-
рому разрушению эмали. Соляная кислота растворяет железо; последнее
ускоряет разрушение аскорбиновой кислоты, загрязняет ее;
качество поступающих на енолизацию гидрата ДКГК и химикалиев.
Качество гидрата диацетон-2-кето-Е-гулоновой кислоты оказывает большое
влияние на результаты процесса енолизации. Зольность гидрата не должна
превышать 0,1—0,2%, а содержание безводного вещества составлять
91,5—92,5%;
хлороформ должен быть тщательно отделен от воды и поступать на ено-
лизацию в сухом виде. Попадание воды с хлороформом в результате небреж-
ности при его регенерации может привести к порче целых партий аскорбино-
вой кислоты.
283
Влияние различных факторов на процессы, протекающие на стадии ено-
лизации в среде дихлорэтана [21,148]:
а) при проведении процесса енолизации в среде дихлорэтана действуют
те же факторы, что и при енолизации в среде хлороформа. В частности, весь-
ма важно, чтобы количество спирта в среде составляло 0,22 л на 1 кг гид-
рата;
б) при использовании регенерированного дихлорэтана с плотностью
1080—1150 кг/м? необходимо следить за тем, чтобы плотность смеси техни-
ческого и регенерированного дихлорэтана была в пределах 1150—1170.
Повышение плотности указывает на недостаток спирта в растворителе, что
может обусловить образование трудно промывающихся гранул аскорбино-
вой кислоты;
в) в связи с более высокой температурой кипения азеотропных смесей
дихлорэтана (70,1—70,6° С), чем хлороформа (59,4—59,9° С), представля-
ется возможность значительно ускорить процесс енолизации в среде дихлор-
этана при атмосферном давлении и довести продолжительность енолизации
до 13 ч [149]. Однако при этом ухудшается качество и снижается выход ас-
корбиновой кислоты.
Необходимо поэтому подчеркнуть, что скоростной метод енолизации в
среде дихлорэтана требует дополнительных исследований;
г) в литературе имеются данные о деструкции фурфурола в присутствии
кислот, причиной которой является молекулярно растворенный кислород
[148а]. По-видимому, это явление может иметь место на стадии енолизации,
а следовательно, применение СО2 или азота на этой стадии должно дать
положительный эффект.
Влияние различных факторов на процессы перекристаллизации аскорби-
новой кислоты [150].1 1. Повышение температуры водных растворов аскор-
биновой кислоты способствует интенсификации процесса ее распада. В свя-
зи с этим необходимо понизить температуру: всех жидких продуктов (маточ-
ников и промывных вод) при хранении до 0°; утфеля при фуговке до 0 ми-
нус 2° С; массы при упаривании в утфель до 50—55° С.
2. С возрастанием количества добавляемого активированного угля ад-
сорбционный эффект увеличивается, но наряду с этим повышаются потери
аскорбиновой кислоты. При добавлении 5% активированного угля к массе
аскорбиновой кислоты потери ее составляют 6,6—8,7%, а при снижении ко-
личества угля до 1,5—2,0% потери уменьшаются до 2% [150].
3. Самые низкие потери аскорбиновой кислоты получены при продолжи-
тельности контакта угля с водным раствором аскорбиновой кислоты 2 мин.
4. Окислительное свойство активированного угля может быть устранено
путем специальной обработки его (восстановления) по следующему методу
(В.. Букин). Уголь размешивают с десятикратным количеством холодной
(15—17°С) известковой воды. В смесь вводят 100—150 г глюкозы на 1 кг
угля, подогревают до кипения и кипятят в течение 3 мин. Затем уголь от-
фильтровывают и промывают горячей водой до исчезновения щелочной реак-
ции по фенолфталеину. Отфильтрованный уголь помещают в герметизиро-
ванный сосуд, наполненный СО2. Применение «восстановленного» угля для
очистки аскорбиновой кислоты увеличивает ее выход на стадии перекристал-
лизации на 1,5—2,0% к массе введенной технической аскорбиновой кислоты
[1501.
5. Продолжительное хранение полупродуктов перекристаллизации при-
водит к большим потерям аскорбиновой кислоты. Следовательно, при пере-
кристаллизации необходимо устранить задержку полупродуктов на завод-
ском верстате.
1 В связи с обескислораживанием воды при замерзании (И. И. Баденко,
А. Н. Горшков, ЖПХ, 1971, 44, 2, 433) автор полагает, что замораживание раство-
ров аскорбиновой кислоты с быстрым оттаиванием перед термической обработкой,
приведет к снижению потерь и увеличению выхода аскорбиновой кислоты.
284
6. Во всех процессах перекристаллизации необходимо строго соблюдать
четкость в разделении продуктов по их качеству, т. е. направлять на совмест-
ную переработку лишь те продукты, которые имеют одинаковую доброка-
чественность и цветность.
7. Для обеспечения сохранности аскорбиновой кислоты необходимо исклю-
чить факторы, способствующие распаду ее. К ним относятся влага, кислород
воздуха, тяжелые металлы (катализаторы).
Влажность медицинской аскорбиновой кислоты не должна превышать
0,1%. Для предотвращения отсыревания аскорбиновой кислоты рекоменду-
ется при расфасовке в крупной таре снабдить ее бязевыми пакетами с сили-
кагелью. Для исключения притока кислорода воздуха тара, в которую рас-
фасовывают аскорбиновую кислоту, должна быть так же герметичной, как
для консервов.
Для доведения до минимума содержания железа в продукте рационально
вводить на первой кристаллизации трилон-Б (динатриевая соль этиленди-
аминтетрауксусной кислоты) в количестве 40 г на 100 кг технической аскор-
биновой кислоты. Содержание железа в техническом продукте составляет
(2—5)10-3%, а в медицинском — (3—15)10-6%.
Исходя из указанных выше положений, можно следующим образом сфор-
мулировать основные требования, предъявляемые к процессам перекристал-
лизации аскорбиновой кислоты:
необходимо максимально ускорить все процессы очистки и перекристал-
лизации аскорбиновой кислоты (скоростной метод);
необходимо понизить температуру на всех стадиях производства и хра-
нить промежуточные продукты при температуре около 0° (холодный метод);
необходимо строго ограничить количество применяемого для очистки
активированного угля и до применения подвергать его «восстановлению»;
чистота технической аскорбиновой кислоты, поступающей на перекрис-
таллизацию, должна быть не ниже 98,0%, а цветность не выше 20 ед.ВНИВИ.
Получение технической аскорбиновой кислоты и ее перекристаллизация
Сырье и химические реактивы. Эффективность процесса енолизации гид-
рата диацетон-2-кето-1-гулоновой кислоты (ДКГК) во многом зависит от
качества сырья и в первую очередь гидрата ДКГК и органического раство-
рителя. Гидрат должен содержать 91,5—92,5% вещества и не более 0,1—
0,2% золы. Хлороформ (дихлорэтан) должны быть освобождены от воды.
Плотность (в кг/м3) растворителей, вводимых в реакцию, для хлороформа
составляет 1290—1350 кг/м3, а для дихлорэтана— 1150—1170 кг!м\
Спиртовой раствор хлороводорода получают либо периодическим про-
цессом путем взаимодействия концентрированных серной и соляной кислот
[53], либо синтезом из элементов. В последнем случае хлористый водород
получают в результате взаимодействия хлора с водородом в реакционном
аппарате (печь) при температуре 600—700° С. Водород и хлор из ресиверов
через ротаметры подаются в печь. Перед включением систему продувают
вначале азотом 10—15 мин, а затем водородом. Включают запальник (элект-
роспираль) и подают в печь хлор. Для полноты реакции необходим неболь-
шой избыток водорода 1,1 : 1,0. При установившейся реакции выключают
запальник, так как реакция идет автотермично вследствие выделения боль-
шого количества тепла (22,1 ккалЦг • моль). Из реакционного аппарата
газообразный хлороводород охлаждают и направляют в абсорбционную ко-
лонную установку барботажно-пенного типа или в батарею стеклянных
бутылей, помещенную в баке с проточной холодной водой. Насыщение спир-
та ведут до содержания НС1 250—300 г!л. На 1 кг газообразного хлористого
водорода расходуют 0,98 кг жидкого хлора и 0,39—0,4 м3 водорода.
Енолизация в среде хлороформа. Процесс енолизации осуществляют в
аппарате-енолизаторе. В реактор загружают смесь технического и регене-
рированного хлороформа в таком соотношении, чтобы плотность смеси была
285
равна 1290—1350 кг/м3 из расчета 1,5 л хлороформа на 1 кг гидрата диаце-
тон-2-кето-А-гулоновой кислоты. Последний загружают при непрерывном
перемешивании. Затем в реакционную массу добавляют спиртовой раствор
хлористого водорода из расчета 50 г хлористого водорода на 1 кг гидрата.
Необходимо следить за тем, чтобы содержание спирта в реакционной массе
составляло 0,22 л на 1 кг гидрата. Реакционную смесь нагревают в течение
2 ч*до 58—60° С и при этой температуре и непрерывном помешивании (час-
тота вращения мешалки 50 об/мин) выдерживают в течение 48 ч. После это-
го процесс енолизации и лактонизации считается законченным, реакционную
массу охлаждают до 15° С и выгружают.
Для стабилизации процесса енолизации и снижения расхода хлороформа
целесообразно проводить ступенчатую енолизацию в малом объеме хлоро-
форма [150]. Для этой цели гидрат диацетон-2-кето-Ь-гулоновой кислоты
растворяют в 1,0—1,1 л хлороформа, в начале процесса вводят 40 г хлорис-
того водорода, а через 24 ч после начала енолизации — 15 г. Енолизатор
должен быть герметизирован для сохранения хлористого водорода. Перед
спуском реакционной массы рекомендуется добавлять в енолизатор регене-
рированный хлороформ в количестве около 0,5 л на 1 кг гидрата. Необхо-
димо также предусмотреть в енолизаторе спускной штуцер с диаметром не
менее 100 мм, а в качестве запорного клапана кран вместо вентиля. Следует
также подвести сжатый инертный газ к енолизатору для продувки спускной
коммуникации.
Енолизация в среде дихлорэтана. В реактор загружают смесь техниче-
ского и регенерированного дихлорэтана плотностью 1150—1170 кг/м9 (плот-
ность регенерированного дихлорэтана 1080—1150 кг/м3). Общее количество
спирта должно быть 0,22 л на 1 кг гидрата. Дихлорэтана вводят 1,5 л на 1 кг
гидрата. Спиртового хлористого водорода добавляют из расчета 45—50 г
НС1 на 1 кг гидрата ступенчато: 75% при загрузке и 25% через 24 ч. Реак-
цию ведут при температуре 58—60° С в течение 48 ч.
Выделение технической аскорбиновой кислоты. Аскорбиновая кислота
из реакционной массы должна быть выделена центрифугированием. Аскор-
биновую кислоту отфуговывают в центрифуге от маточного раствора, затем
хорошо отжимают.
Промывку аскорбиновой кислоты осуществляют легкой фракцией реге-
нерированного растворителя (для хлороформа плотность 1200—1220 кг/л3,
для дихлорэтана 1080—1100 кг/м3). Промывка производится 3 раза (по 6—7 л)
на центрифугу типа ТВ-600. Необходимо тщательно следить за качеством
промывки аскорбиновой кислоты.
При необходимости аскорбиновую кислоту промывают спиртом из рас-
чета 0,1 л на 1 кг аскорбиновой кислоты. В качестве фильтрующего материа-
ла применяют мешок из бельтинга.
Сушка аскорбиновой кислоты. Промытую аскорбиновую кислоту выгру-
жают, измельчают на кусочки и сушат в вихревой сушилке (рис. 44) при тем-
пературе нагнетаемого воздуха 90—95° С (см. стр. 351).
Регенерация растворителя. Хлороформ. Хлороформовый маточ-
ный раствор и промывные хлороформовые смеси нейтрализуют до слабоще-
лочной реакции (по фенолфталеину) и разгоняют по фракциям: отгон с плот-
ностью 1290—1350 кг/м3 используется для енолизации в смеси с техническим
хлороформом, а фракции с меньшей плотностью применяют для промывки
аскорбиновой кислоты. При необходимости фракции с пониженной плот-
ностью отмываются от спирта и ацетона водой до плотности их
1290—1350 кг/м3.
Разгонка хлороформа производится в перегонном аппарате из нержавею-
щей стали, снабженном поверхностным конденсатором.
* Существует мнение, что при более продолжительном нагреве реакционной мас-
сы (до 8 ч) достигаются лучшие результаты.
286
Дихлорэтан. Дихлорэтановый маточный раствор и промывные
дихлорэтановые смеси регенерируют таким же образом, как и хлороформо-
вые. В связи со способностью дихлорэтана образовывать с водой более стой-
Рис. 44. Вихревая сушилка для сушки аскорбиновой ки-
слоты:
1 — бункер; 2 — шнек; 3 — пневмоэжектор; 4—вихревая камера; 5 — ка-
лорифер; 6 — циклон; 7 — сборник сухого продукта; 8 — рукавный фильтр.
кие эмульсии, чем хлороформ, вследствие меньшей разницы в плотности
растворителя и воды целесообразно для разделения смесей применять систе-
му флорентийских сосудов. На рис. 45 приведена схема непрерывно дейст-
Рис. 45. Схема водоотделительной установки для регенерированного дихлорэтана.
вующей водоотделительной установки для регенерации дихлорэтана или
хлороформа. Из выпарного перегонного аппарата 1 пары растворителя по-
ступают в конденсатор 2, а конденсат в сборник 3 для расслаивания, а затем
в сборник 4, охлаждаемый рассолом (для лучшего расслоения дихлорэтана
и воды). Дихлорэтан поступает в сборник сухого растворителя 5, откуда
насосом 6 подается в енолизатор 7, где смешивается с техническим дихлор-
этаном, поступающим из сборника 8. Регенерированный дихлорэтан из сбор-
ника 5 поступает также в центрифугу 9 для промывки аскорбиновой кислоты.
Маточный раствор из центрифуги поступает в сборник 10, откуда насо-
сом 11 перекачивается в перегонный аппарат 1. Сборники снабжены воздуш-
никами, присоединенными к конденсатору 12, охлаждаемому рассолом. Кон-
денсат из последнего поступает в делительную воронку 13. Отстоявшаяся в
287
флорентийских сосудах вода непрерывно выводится сверху в контрольный
сборник.
Для обеспечения низкого расхода растворителя необходимо обеспечить
полную герметизацию всех сборников, коммуникации и фильтрующих ап-
паратов. Уплотняющие прокладки должны быть изготовлены из клингери-
та или паранита, прогретого в олифе и смазанного свинцовым суриком на
олифе или на свинцовых белилах. Для сальников применяется прографи-
ченная пеньковая набивка.
Другие перспективные методы енолизации диацетон-2-кето-Ь-гулоновой
кислоты.
1. Предложено смесь расплавленного гидрата диацетон-2-кето-А-гуло-
новой кислоты в присутствии каталитического количества концентрирован-
ной соляной кислоты нагревать при температуре 70—75° С с одновременной
отгонкой отщепляющегося ацетона [150а]. Это позволяет исключить из про-
изводства токсические органические растворители и сократить продолжи-
тельность процесса.
2. Процесс енолизации проводят в присутствии ионообменных смол.
Вначале осуществляют этерификацию диацетон-2-кето-А-гулоновой кислоты
в спиртовом растворе в присутствии катионита в Н-форме. Реакционную
массу без выделения эфира подвергают енолизации в присутствии анионо-
обменных смол при температуре около 60° С. Аскорбиновая кислота адсор-
бируется на смоле и выделяется обычным методом1. Приводится один из
следующих примеров [1506]. 30 г диацетон-2-кето-Ь-гуло новой кислоты
растворяют в 200 мл метанола, добавляют 8 г Амберлита 200 (Н-формы) и
при перемешивании нагревают 1 ч с обратным холодильником. После от-
гонки растворителя (постепенно в течение 4 ч) удаляют смолу, вводят анио-
нит Амберлит 1RA-410 (100 мл) и проводят реакцию енолизации. Аскорби-
новую кислоту элюируют из смолы 4%-ным водным раствором едкого натра.
Получают 15,9 г аскорбиновой кислоты. Выход составляет 88%.
Перекристаллизация аскорбиновой кислоты. В результате лабораторных
исследований доказано, что аскорбиновая кислота в водных растворах при
pH ниже 7 распадается по следующей схеме: аскорбиновая кислота -» де-
гидроаскорбиновая кислота -> дикето-А-гулоновая кислота -> фурфурол +
+ СО2 -> продукты конденсации фурфурола ->смола (см. стр. 240). Эта реак-
ция протекает при pH <7,0, и при этом чем ниже pH, тем интенсивнее идет
процесс распада.
Распад аскорбиновой кислоты ускоряется при повышении температуры,
увеличении времени воздействия температуры и увеличении количества ак-
тивированного угля.
Исходя из этих теоретических соображений, выявляется необходимость:
ускорения всех процессов очистки и перекристаллизации аскорбиновой
кислоты (скоростной процесс);
понижения температуры на всех стадиях производства медицинской ас-
корбиновой кислоты и хранения всех промежуточных продуктов при темпе-
ратуре около 0° (холодный процесс);
строгого ограничения количества применяемого для очистки активиро-
ванного угля.
Первая кристаллизация. Техническая аскорбиновая кис-
лота, поступающая на перекристаллизацию, должна иметь чистоту не ниже
98,0% и цветность не выше 20 ед. ВНИВИ.
В реактор-смеситель 1 (рис. 46) подают воду из сборника 2, исходя из
расхода 1,1 л воды на 1,0 кг аскорбиновой кислоты, и подогревают ее до
85—90° С. Затем в горячую воду подают техническую аскорбиновую кисло-
ту, которую быстро растворяют при температуре 70—75° С. Далее в смеси-
1 По английскому патенту № 1 222 322, 1971 (Derwent, 1971, December) процесс
осуществляют одним катионитом «Ксеролит 225», нагревают 7 ч, фильтруют, кон-
центрируют и выделяют аскорбиновую кислоту кристаллизацией.
288
тель вводят «восстановленный» активированный уголь из расчета 1,5—2,5%
к массе технической аскорбиновой кислоты и раствор с углем выдерживают
2 мин, потом быстро фильтруют (7—8 мин) через нутч-фильтр 5. Фильтро-
ванный раствор направляют в кристаллизатор 4, где он кристаллизуется
6—8 ч. Конечная температура кристаллизации 0 минус 2° С. Затем утфель
подают в центрифугу 5, где аскорбиновую кислоту отфуговывают от маточ-
ного раствора, промывают сначала ледяной водой из расчета 6—10 л на
100 кг технической аскорбиновой кислоты, затем 96 %-ным спиртом из рас-
чета 6—10 л на 100 кг кислоты.
Промытую кислоту под вакуумом сушат в сушилке 6. Выход перекрис-
таллизованной аскорбиновой кислоты I составляет 65—70% к массе введен-
ной кислоты.
Техническая acmf-
бинобая киспота
Рис. 46. Технологическая схема многоступенчатой кристаллиза-
ции аскорбиновой кислоты.
Вторая кристаллизация. Маточный раствор, поступающий
в сборник 7 из центрифуги, насосом 8 направляют в аппарат-смеситель 9,
где при температуре 50° С его обрабатывают в течение 2 мин восстановлен-
ным активированным углем в количестве 1,5—2,5% к массе введенной
аскорбиновой кислоты.
В этот же смеситель направляют угольные промывные воды из сборни-
ка 10, в который их подают из нутч-фильтра 3 насосом 8. Из смесителя 9
маточный раствор поступает в нутч-фильтр 11, затем в сборник 12, откуда
его засасывают в вакуум-аппарат 13, в котором выпаривание воды ведется
под вакуумом 675—700 мм рт. ст. до содержания 80—82% сухих веществ1.
Утфель спускают в кристаллизатор 14, где он кристаллизуется в тече-
ние 12 ч при температуре около 0°. Затем утфель поступает в центрифугу
15, в которой его после отфуговки маточника промывают ледяной водой и
50 %-ным спиртом. Кристаллы аскорбиновой кислоты II направляют для
перекристаллизации в аппарат-смеситель 1 и выпускают ее в качестве
готового продукта.
Третья кристаллизация. Маточный раствор II из центри-
фуги 15 направляют в сборник 16, из которого насосом 17 его подают в ап-
парат-смеситель 18. Туда же поступают угольные промывные II из сбор-
ника 19, подаваемые в него из нутч-фильтра 11 насосом 17.
1 Для снижения потерь аскорбиновой кислоты весьма важно применение пленоч-
ного выпарного аппарата.
Ю-522 2 89
Раствор при температуре 50° С обрабатывают в течение 2 мин активиро-
ванным углем в количестве 1,5% к массе введенной аскорбиновой кислоты.
Затем маточный раствор II поступает в нутч-фильтр 20, из него в сборник 21,
вакуум-аппарат 22, кристаллизатор 23, центрифугу 24. Продолжительность
кристаллизации 24 ч. Кристаллы аскорбиновой кислоты III после промывки
ледяной водой вместе с маточным раствором I направляют на перекристал-
лизацию в аппарат-смеситель 9.
Четвертая кристаллизация. Маточный раствор III из
центрифуги 24 поступает в сборник 25. В этот же сборник поступают уголь-
ные промывные воды III из нутч-фильтра 20. Далее раствор идет в вакуум-
аппарат 26, гке. его упаривают до содержания 70% сухих веществ. Затем
утфель спускают в кристаллизатор 27. Процесс кристаллизации продолжа-
ется 48—72 ч. Перед фуговкой при необходимости утфель раскачивают эти-
ловым спиртом или маточным раствором III для облегчения фуговки утфеля
в центрифуге 28.
Кристаллы аскорбиновой кислоты IV направляют для перекристаллщ
зации в аппарат-смеситель 18. Маточный раствор IV, поступающий в сбор-
ник 29 из центрифуги 28, является отходом производства.
Выход 100%-ной аскорбиновой кислоты при перекристаллизации тех-
нической аскорбиновой кислоты составляет 92—94%.
ПРОИЗВОДНЫЕ АСКОРБИНОВОЙ КИСЛОТЫ
Аскорбиновая кислота как одноосновная кислота способна с металлами
давать соли по типу С6Н7О6Ме, а как спирт дает сложные эфиры с кислота-
ми. Из солей аскорбиновой кислоты ниже рассмотрены аскорбинаты Na, Са
и Fe, а из эфиров — пальмитат аскорбиновой кислоты.
Аскорбинат натрия C6H7O6Na, молекулярная масса 198,11; [а]о =4-105
(вода); хорошо растворим в воде, нерастворим в безводном спирте, эфире
и других органических растворителях. Аскорбинат натрия получают путем
нейтрализации насыщенного водного раствора аскорбиновой кислоты би-
карбонатом натрия при температуре 55—70° С и осаждения полученной
соли из водного раствора спиртом [35] или путем добавления горячего спир-
тового раствора NaOH к спиртовому раствору аскорбиновой кислоты и по-
следующей кристаллизации [21]. Аскорбинат натрия применяют вместо
аскорбиновой кислоты для приготовления инъекционных растворов [151],
а также для витаминизации пищевых продуктов.
Аскорбинат кальция (С6Н7О6)2Са .2Н2О, кристаллы, легко растворимы в
воде, трудно в спирте, нерастворимы в эфире. Получают добавлением из-
бытка мела к водному раствору аскорбиновой кислоты и осаждением аскор-
бинатаспиртом [19, 20]; [ci]d=+96 (вода).
Аскорбинат железа C6H8O6.FeO [20, 21, 152—154] получают путем до-
бавления к водному раствору аскорбиновой кислоты избытка FeCO3. Из
фильтрата спиртом осаждают аскорбинат в виде темно-фиолетового порошка
с содержанием железа от 18 до 22% [27]. При получении другим мето-
дом [154] в качестве сырья применяют аскорбиновую кислоту и сернокис-
лое закисное железо, а осаждение ведут ацетоном.
Пальмитат аскорбиновой кислоты получают путем взаимодействия
ОН ОН
ОН С=С
I I I
СН3 (СН2)14 со—ОН2С—с—сн с=о
I
н
ее с хлорангидридом пальмитиновой кислоты [155]. Получаемые бесцвет-
ные кристаллы нерастворимы в воде, хорошо растворимы в жирах и органи-
290
ческих растворителях. Применяется для витаминизации, а также для ста-
билизации жиров как антиоксидант. По японскому патенту [156] для эте-
рификации применяют 5,6-изопропилиден-аскорбиновую кислоту и после
гидролиза получают 3-алифатический ацил аскорбиновой кислоты.
ТЕХНОЛОГИЧЕСКИЕ ПОКАЗАТЕЛИ
ПАРАМЕТРЫ И РАСХОД ОСНОВНЫХ
МАТЕРИАЛОВ НА СИНТЕЗ
L-АСКОРБИНОВОЙ КИСЛОТЫ ИЗ
О-ГЛЮКОЗЫ
Стадия получения D-сорбита
Содержание сухих веществ в
растворе глюкозы, % .... 50,0
Температура гидрирования, °C 135—140
Избыточное давление водорода
в автоклаве, кгс/см2 ....... 80
Количество добавляемого ката-
лизатора, % к глюкозе ... 5,0
Количество веществ, добавля-
емых для очистки раствора, %
к массе сорбита в растворе
Na2HPO4 . ...........1,5—2,0
СаСОз...................2,0—3,0
Температура раствора при хи-
мической очистке, СС .... 85—90
Для ионообменной очистки при-
меняют катионит КУ-2 и ани-
онит ЭДЭ-1ОП
Выход сорбита, %
к теоретическому .... 97,5
к глюкозе................. 98,5
Расход на 1 кг сорбита, %
глюкозы, кг................. 1,02
водорода, м3.............0,2—0,21
сплава, кг........... 0,02
мела, кг............ 0,025
фосфорнокислого натра, кг 0,025
щелочи (NaOH) 92%-ной, кг 0,03
уголь активированный, кг 0,01
Стадия получения L-сорбозы
Содержание сухих веществ в
окисленном растворе, % . . 20—22
Продолжительность окисле-
ния, ч.................... 18—24
Глубина окисления (доброка-
чественность) раствора, % 96—98
Выход сорбозы, %:
к теоретическому ... 90,0
к сорбиту ............... 89,0
Чистота сорбозы, % ... 97,0—98,8
Расход дрожжей сухих, кг 0,02
Расход концентрата В комп-
лекса в сухом веществе на
1 кг сорбозы.............. 0,02
Расход серной кислоты, кг 0,02
для ацетонатора .... 200—250
для нейтрализатора . . 250—300
Температура выделения ди-
ацетонсорбозы щелочью, °C 45—50
Выход диацетонсорбозы, %
к теоретическому.......... 80—87
Выход диацетонсорбозы, к
массе 100%-ной сорбозы . . 115—125
Выход щелочного маточника,
% к массе 100%-ной сорбо-
зы ....................... 300,0
Расход на 1 кг 100%-ной
диацетонсорбозы, кг
100%-ной сорбозы . . . 0,88
ацетона .................. 0,78
едкого натра 100%-ного 2,0
олеума........... 1,10
Стадия получения диацетон-2-кето -
L-гулоновой кислоты
Температура окисления, °C
перманганатом калия . . . 34—35
гипохлоритом натрия . . . 75—80
Температура раскисления пер-
манганата калия, °C..........50—55
Содержание сухих веществ, %
при окислении КМпО4 . . 18—20
при окислении гидрохлори-
том .........................18—20
Выход диацетон-2-кето-Т-гуло-
новой кислоты, %
к теоретическому .... 96,8
к диацетон-7,-сорбозе ... 102,0
Выход сухой перекиси марган-
ца, % к диацетонсорбозе . . 45
Расход кг на 1 кг гидрата диа-
цетон-2-кето-£-гулоновой кис-
лоты
при окислении пермангана-
том калия
диацетон-L -сорбозы ... 0,98
перманганата калия . . . 0,9—1,0
едкого натра 100%-ный . 0,09
при окислении гипохлоритом
натрия
гипохлорита натрия в пе-
реводе на хлор, кг ... . 1,2—1,3
сернокислого никеля ... 0,02
едкого натра 92%-кого . . 1,2—1,5
соляной кислоты.......... 0,5
Стадия получения диацетон-L-сорбозы
Стадия получения аскорбиновой кислоты
Температура растворения
L-сорбозы (применение оле-
ума), СС.................... 12—14
Температура выдержки аце-
тонового раствора, °C . . . —7—10
Температура нейтрализа-
ции, °C ....................0, -)-5
Частота вращения мешал-
ки, об/мин
Температура на стадии ено-
лизации, °C .............. 58—60
Температура массы енолиза-
ции перед фуговкой, °C . . 5—15
Температура растворения
технической аскорбиновой
кислоты и обработки активи-
рованным углем, °C ... 70—75
Температура кристаллизации
10*
291
утфеля I, °C.............. —2—3
Продолжительность
енолизации, ч .... 48
растворения технической
аскорбиновой кислоты,
обработки углем и филь-
трации, мин........... 30—40
кристаллизации утфеля
I, ч.................. 6—8
Количество хлороформа или
дихлорэтана, загружаемого
на енолизации, л на 1 кг
гидрата .................. 1,5—2,0
Необходимое количество
спирта в реакционной среде,
л на 1 кг гидрата......... 0,22
Выход технической аскорби-
новой кислоты, %
к теоретическому ... 89,4
к гидрату (по массе) . . 57,5
Выход медицинской аскорби-
новой кислоты от техничес-
кой, %.................... 92—94,0
Расход, кг на 1 кг медицин-
ской аскорбиновой .кислоты
гидрата .............. 1,83
технической аскорбино-
вой кислоты ..........1,07—1,09
хлороформа или дихлор-
этана ................ 1,7—1,9
спирта этилового 96% -
ного ...................... 0,5
активированного угля . 0,03
РАСХОД ВСПОМОГАТЕЛЬНЫХ
МАТЕРИАЛОВ, КГ НА 1 КГ
МЕДИЦИНСКОЙ АСКОРБИНОВОЙ
КИСЛОТЫ
Масло растительное .... 0,02
Бумага фильтройальная . . 0,05
Бумага подпергаментная . . 0,03
Вата........................ 0,002
Фильтрующие ткани
бязь и миткаль .... 0,17
бельтинг.............. 0,08
диагональ................. 0,10
гризбон............... 0,08
марля ................ 0,05
Объемные массы, кг/л
Кристаллическая глюкоза
сухая ....................0,77—0,82
Кристаллическая сорбоза су-
хая ...................... 0,74
Гидрат диацетон-2-кето-Т-
гулоновой кислоты сухой . 0,47
Техническая аскорбиновая
кислота
сухая . ................. 0,57
сырая ................... 0,50
Медицинская аскорбиновая
кислота сухая ............... 0,75
Перманганата калия в по-
рошке .................... 1,26—1,36
Плотность растворов, кг/м3
Глюкоза (50%-ная) .... 1200
Окисленный раствор сорбита 1054
Маточный раствор сорбозы
первой кристаллизации . . 1324
Маточный раствор аскорби-
новой кислоты
первой кристаллизации . . 1080
второй кристаллизации . . 1320
Монодиацетоновый раствор
после ацетонирования . . . 893
Водный раствор диацетон-
сорбозы для окисления пер-
манганатом калия......... 1036
Водный раствор натриевой
соли гидрата диацетон-2-
кето-Д-гулоновой кислоты
для выделения гидрата . . 1084
Водный раствор технической
аскорбиновой кислоты для
перекристаллизации . . . 1252
Скорость фильтрации веществ через
различные ткани
Фильтрующая Скорость
ткань фильтрации
л/м2 в час
Диагональ
для фильтрования глюкозного
раствора..................... 500
Бязь суровая
для отделения осадков
никелевого катализатора . . . 350
мела и дифосфата ............ 200
Шинельное сукно + бязь суровая
для фильтрования окисленно-
го раствора сорбита.......... 100
Бязь суровая или шелковое си-
то № 21
для фуговки сорбозы .... 50
Бязь и диагональ или один слой
бельтинга
для отделения осадка МпО2 . 50
Бязь и диагональ или один слой
бельтинга
для фильтрования гипохлори-
та .......................... 400
Бельтинг + бумага + бязь
для отделения осадка гидрата
ДКГК на нутч-фильтре . . . 100
Бельтинг
для отделения технической ас-
корбиновой кислоты на центри-
фуге ......................... 20
Бязь + бумага + бязь
для раствора технической ас-
корбиновой кислоты с углем 200
Гризбон или отбельная бязь
для отделения медицинской ас-
корбиновой кислоты на центри- ,
фуге.......................... 15
292
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. Drummond J., Biochem. J., 1920, 14, 660. '
2. Бессонов Н. Bull. Soc. Sci Hyg. aliment., 1923, 11, 4. Compt. rend. Ac. Sci.,
1925, 180, 970.
3. Z i 1 v a S., Biochem J., 1925, 19, 589; 1930, 24, 1687.
4. A. Szent-Gyorgyi, Biochem. J., 1928, 22, 1387.
5. A. Szent-Gyorgyi. Hawort W., Nature, 1933, 131, 23.
6. Herbert R., Percival E. и др. J. Soc. Chem. Ind., 1933, 52, 221, 481.
7. Micheel F., Kraft K-, Z. physiol. Chem., 1933, 222, 295.
8. R e i c h s t e i n T., Nature, 1933, 132, 280.
9. Reichstein T., Griissner A., Oppenauer R., Helv. Chim. Acta,
1933, 16, 561, 1019.
10. A u 1 t R., Baird D., Carrington H., Hawort W., Herbert!?.,
Hirst E., Percival E., Smith F., Stacey M., J. Chem. Soc., 1933, 1419.
11. Baird D., Hawort W., Herbert R., Hirst E., Smith F., Stacey M., J. Chem. Soc., 1934,
62. .
12. Rosenberg H., Chemistry and Physiology of the Vitamins, N. J., 1942, 289.
13. Г e с с e н А. Аскорбиновая кислота и ее практическое применение. Л., 1953, с. 9.
14. Ши л о в П. И., Яковлев Т. Н. Основы клинической витаминологии. М.,
«Медицина», 1964, с. 26.
15. Vogel Н., Chemie und Technik der Vitamine, 1943, 175.
16. Bowden F., Snow C., Nature, 1932, 129, 720.
17. Mohler H., Lohr H., Helv. Chim. Acta, 1938, 21, 485.
18. С л о б о д и н Я. M., Басова А. К., Гельме E. Э. Растворимость ас-
корбиновой кислоты и промежуточных продуктов ее синтеза.— ЖПХ, 1946, т. 19,
с. 170—171 с ил.
19. Девятнин В. А. Фармация, 1940, с. 11.
20. Д е в я т н и н В. А. Витамины. Пищепромиздат, 1948, с. 157; 151.
21. Ш н а й д м а н Л. О. Производство синтетической аскорбиновой кислоты. М., Пи-
щепромиздат, 1948, 228с.
22. R u s k i п S. Пат. США № 2596103; С. А., 1953, 47, 275.
23. A. Szent-Gyorgyi, Z. Physiol., Chem., 1934, 225, 168.
24. Р i j о n M., Science, 1937, 86, 80.
25. К а н ч у x Ш. Ф., Рубцов И. А. Авт. свидет., № 101626, 1955; Бюлл. изо-
брет., 1955, № 10, с. 12.
26. В а й с м а н С. Б., Кручакова Ф. А. К вопросу о методике получения
препарата железоаскорбинового комплекса. — В сб.: «Витамины», АН УССР,
1953, т. 1, с. 158—165.
27. Д е в я т н и н В. А., Н о с и к о в а В. М. Аскорбинат железа. — «Вопросы
питания», 1940, №> 1, с. 50—53.
28. Maurer, Sciedt, Biochem, Ztschr., 1936, 285, 67.
29. Э н г e л ь г a p д т В. А., Букин В. Н. О ферментативном окислении аскор-
биновой кислоты. — В сб.: «Проблема витаминов», 1937, № 2, с. 255—269 с ил.
30. Ш м и д т А. А. Аскорбиновая кислота, ее природа и значение в животном орга-
низме. Л., Пищепромиздат, 1941, 134 с.
31. Herbert R., Hirst Е., Percival Е., Reynolds R., Smith F.,
J. Chem. Soc., 1933, 1270.
32. Б у к и н В. H. Витамины. М., Пищепромиздат, 1940, 472 с.
33. Ш н а й д м а н Л. О. О химизме распада аскорбиновой кислоты. — «Труды
ВНИВИ», 1954, № 4, с. 47—53.
34. Kenyon J., Munro N., J. Chem. Soc., 1948, 158.
35. Б e p e з о в с к и й В. M. Химия витаминов. М., Пищепромиздат, 1959, с. 40.
36. Ш н а й д м а н Л. О., К а р п о в М. Ф. Усовершенствование технологии про-
изводства жидкого концентрата витамина С из шиповника. —В сб.: «Передовой
опыт производства концентратов витамина С из плодов шиповника», М., Пищепром-
издат, 1955, с. 20—26.
37. С i е г A., N о f г е С., D г е v о п В., L е f i е г A., Bull. Soc. Chim. France,
1959, 1.74; —Сб. «Витамины», ГОСИНТИ, М., 1960, 6, 26.
38. N о f г е С., С i е г A., D г е v о п В. Compt. Rend., 1958, 246, 1611.
39. F i s с h е г - J е п s е п Е., Dansk Tidsskr. Farm., 1955, 29, 125.
40. F i п h о 1 t P., J. of Pharm. Sci., 1963, 52, 1948; 1965, 54, 124, 181.
41. N e r 1 о H., Acta Polon. Pharm., 1952, 8, 289.
42. D u d z i k Z., Acta Polon Pharm., 1956, 13, 225.
43. Zwolinska Z., Farm. Pol., 1962, 18, 548.
44. G a r r e t E., J. Pharm. Sci., 1962, 51, 811.
45. Krowczy nski L., Kr asowska H., M a k о s z A., Acta Polonia]
Pharmaceutica, 1966, 6, 493.
46. H i r s t E., Woodward C., Biochem. J., 1944, 38, 10.
47. О h 1 e H., Erlbach H., Carls H., Ber., 1934, 67, 324.
48. Reichstein T., Griisner A., Oppenauer R. Heiv. Chim. Acta,
1934, 17, 510.
49. M a u г e г K-, S c h i e d t B., Ber., 1933, 66, 1054; 1934, 67, 1239.
293
50. D e m о 1 е V., Biochem J., 1934, -28, 770.
51. Z i 1 v a S., Biochem. J., 1935, 29, 1612.
52. M u 1 1 e г H., Reichstein T., Helv. Chim. Acta., 1938, 21, 273.
53. Шнайдман Л. О. Производство витаминов. М., Пищепромиздат, 1958, с. 219.
54. S е Ь г е 1 1 W., Harris R. The Vitamins. N. J., 1954, 177.
55. Reichstein T., Griissner A., Heiv. Chim. Acta., 1934, 17, 311; 1938,
21, 254.
56. I s b e 1 1 H., J. Research National Bureau Standards, 1944, 33, 45.
57. G г а у В. Пат. США, 2421611, 4/V 1945; 2421612 4/V 1945; Off. gaz., 1947, 599,
№ 1, 107.
58. I a m a z a к i M., M i к i T., J. Ferment. Technol., 1953, 31, 39; lamazaki, M.,
J. Agr., Chem. Soc., Japan., 1953, 27, 462, 633, 1954, 28, 748, 890.
59. P ю м и л e p А. Сахарное производство. Петроград, Научн. хим.-техн. изд.
ВСНХ, 1924, с. 98.
60. Ч е п и г о С. В., Баландин А. А., Васюнина Н. А. и др. Получе-
ние многоатомных спиртов путем каталитического превращения полисахаридов
растительных материалов. — «Химическая наука и промышленность», 1957, 2,
с. 416—424 с ил.
61. Б а л а н д и н А. А., Васюнина Н. А., Барышева Г. С., Ч е п ii-
го С. В. Катализаторы гидрирования полисахаридов. —«Известия АН СССР,
ОХН», 1957, № 3, с. 392. То же. Гидролитическое гидрирование целлюлозы. —
«ДАН СССР», 1959, т. 128, с. 941.
62. М у х л е н о в И. П., Добкина Е. И., Д е р ю ж к и н а В. И. и др.
Определение истираемости катализаторов. —«Кинетика и катализ», 1970, т. 11,
3, с. 771—74 с ил.
63. Я кубено к Э. Ф. , Подвязкин Ю. А., Юкельсон И. И. Влия-
ние способа приготовления на свойства скелетного никелевого катализатора. —
ЖПХ, 1969, т. 42, вып. II, с. 2605—2608 с ил.
64. Б и ж а н о в Ф. Б., Сокольский Д., В., Малкина И. Я- и др. Гид-
рирование глюкозы на скелетно-никелевых катализаторах. —«Кинетика и ката-
лиз», 1969, т. 10, вып. 4, с. 801. То же. Авт. свидет., № 237111, 1966; Бюлл. изоб-
рет., № 8, с. 14; То же Авт. свид. № 242147; Бюлл. изобрет., 1969, №15, с. 21.
65. Zapletal V., Soukup I., Chem. Ing. Techn., 1969, 41, 1/2, 78.
66. Борисоглебский С. Д. Метод получения пищевого кристаллического
сорбита из £)-глюкозы. — ЖПХ, 1940, т. 13, вып. 4, с. 571—574.
67. Tettamanzi А., А г п а 1 d i N., С. А., 1944, 38, 3841.
68. К а г b i п о s V., В а 1 1 u п A., J. Am. Soc., 1953, 75, 4510.
68а. Fedor W., Millet I., А с с о 1 a A., Sorbitol, Ind. Eng. Chem., 1960,
32, 282.
69. Atlas Puder C , Anon: «Sorbitol and derivates», Chemical Eng., 1952, 208.
70. Хайдеггер Эрне. Промышленное значение использования сорбита. —
«Журнал венгерских химиков», 1961, 8.
70а . Харин С. Е., Палаш И. П. Роль сахаратов натрия и кальция в мелассо-
образовании. —.«Известия ВУЗов. Пищевая технология», 1969, № 1, с. 125—128.
706. jP ождественская И. Д., Фадеева Т. Н., Титова Н. Б., Ш и-
л е й к о Л. В. О некоторых вопросах механизма реакции гидрогенолиза глюко-
зы. — «Кинетика и катализ», 1970, т. 11, вып. 3, с. 696—703 с ил.
71. Поспеев А. Д., Чебан Е. А. Авт. свидет., № 64925, 1943; Бюлл. изобрет;
1945, № 6, с. 12.
72. М о и с е е в А. С. Очистка сорбита ионообменными смолами. •— В сб. «Витамин-
ная промышленность», М., Пищепромиздат, 1955, № 1, с. 3—8.
73. Поспеев А. Д. — «Медицинская промышленность», 1964, 7, с. 39.
74. М и т г о р ц Л. Б., Яку б И. А. Опыт борьбы с фильтрацией сооружений.—
«Строительная промышленность», 1954, № 1.
75. Kwoczek I., Noers-Messmer W., Z. fiir Haut und geschlechten —
krankemieiten, 1952, 12, 9.
76. В e 1 1 о m s I. и др., Arch. Ophtalm, 1938, 20, 1036; C. A., 1939, 33, 22108.
77. I a n i s t у n N., Parfumerie und Kosmetik, 1950, 3, 31.
77a. Kulhanek N., Tadra M., M a п c f e 1 d V., nam. ЧССР 124995, 1967;
C. A. 1968, 58388 g.
78. И д e л ь с о н Б. И. Разработка поляриметрических методов анализа. Л., ЦБТИ,
1958, 4 с.
79. Ш н а й д м а н Л. О. О стойкости водных растворов L-сорбозы к нагреванию. —
«Труды ВНИВИ»,М., Пищепромиздат, 1954, Ns 5, с. 46—50.
80. Ч и ч и б а б и н А. Е. Основные начала органической химии. М., Госхимиздат,
1957, I, с. 558.
81. Креслинг Е. Окисление сорбита при помощи микроорганизмов. — «Микро-
биология», 1937, т. 6, вып. 7, 898—901.
82. Wieland, Ergebnisse d. Physiologie, 1922, 20, 477.
83. Митюшова H. М. О газообмене уксуснокислых бактерий при окислении сор-
бита в сорбозу. —«Микробиология», 1954, т. 23, вып. 4, с. 400—409 с ил.
84. К р е с л и н г Е. — «Микробиология», 1942, т. 11, 4, с. 115.
294
85. Ждан-Пушкин С. М. Динамика размножения Acetobacter Suboxydans и
окисление им сорбита в средах с комплексом витаминов В. — «Микробиология»,
1955, 24,"5, 545—49 с ил.
86. М и х л и н Э. Д., Голышева М. Г., К е п п е н В. А. Влияние условий
аэрации на развитие уксуснокислых кетогенных микроорганизмов.— «Микробио-
логия», 1952, 21, вып. 5, с. 521—27 с ил.
87. W е 1 1 s Р., Stubbs I., L о с k w о о d L., Roe Е., Ind. Eng. Chem. 1937,
29, 1385; 1939, 31, 1518.
87a. Косте л ьм ан В. Н., Наумов Е. Г., Новикова Ф. Б. О воз-
можности применения фторопластов при биохимическом процессе окисления сор-
бита в сорбозу. — «Прикладная биохимия и микробиология», 1970, т. 6, вып. 5,
с. 621—623 с ил.
88. С м и р н о в а А. Э. Дрожжевой автолизат как источник азотистого и витаминно-
го питания микроорганизмов. — «Труды ВНИВИ», М., Пищепромиздат, 1954,№ 4,
с. 65—73 с ил.
89. Continuous cultivation of microorganisms, Simposium, Praha, 1958.
90. Иерусалимсклй H. Д. Теория и практика проточных культур микроор-
ганизмов. — «Микробиология», 1959, т. 28, вып. 1, с. 152—55.
91. Monod J., Ann. Inst. Pasteur, 1950, 79, 4.
92. N о v i c k A., Szilard Z., Science, 1950, 112, 715.
93. M и хл и н Э. Д., Голышева М. Г. Влияние метода холодной стерилизации
питательной среды на биохимическую активность бактерий. — «Труды ВНИВИ»,
1953, № 4, с. 83—87,
94. М и х л и н Э. Д., Голышева М. Г. и др. Окисление сорбита в сорбозу в
системе жидкость •— газ. — «Труды ВНИБИ», 1954, № 5, с. 66—73 с ил.
95. К о г а н М. И., Буйко Д., Лебедев В. Г., Савостьянов Г. И.,
Чесночков Е. А. Авт. свидет., № 102532, 1954; Бюлл. изобрет., 1956, № 2,
10.
96. К о г а н М. И., Белякова М. С., Савостьянов Г. И. и др. Био-
химическое окисление D-сорбита в L-сорбозу в тарельчатом колонном фермента-
торе непрерывного действия. —«Труды ВНИВИ», М., Пищепромиздат, 1961, № 8,
с. 22—35.
97. Ш н а й д м а н Л. О. Исследование в области химии и технологии производства
витаминов. Доклад, докт. дисс., М., 1967, с. 18.
98. Ш н а й д м а н Л. О. Перспективы усовершенствования синтеза аскорбиновой
кислоты. — В сб.: «Витамины», М., ГОСИНТИ, 1959, № 5, с. 5—22.
99. Ohle Н., Вег., 1938, 73,3. 562.
100. Темникова Т. И., Склярова В. В. Определение моно- и диацетон-
сорбозы при совместном присутствии и получение моноацетонсорбозы. —ЖПХ,
1954, т. 21, с. 1131—32.
101. Б е э р А. А., Рубцов И. А. Синтез витаминов. М., Пищепромиздат, 1956,
с. 5.
102. Березовский В. М., Стрельчунас Л. Н. Исследование в области
ацетонирования гексоз. — ЖОХ, «Сб. статей по общей химии», 1953, № 1, с. 453.
103. Tokuyama К., Honda Е., Hoki N., J. Org. Chem., 1964, 29, 1, 133.
104. Tokuyama K-, Honda E., Bull. Chemie. Soc. Japan, 1964, 37, 4, 591;
Tokashi Madea, Yoshiyki Miichi and Kanji Tokuyama. Bull, Chem. Soc. Japan,
1969, 42, 2648.
105. Fischer K., Z. angew. Chem., 1935, 48, 394.
106. Horrock R,. Nature, 1949, 164, 144.
107. Patil J., Rose J., .Indian Chem. Soc., 1966, 43, No3, 161.
107a. Шнайдман Л. О., Фондаренко А. В. О механизме реакции ацето-
нирования L-сорбозы. — «Хим.-фарм. ж.», 1971, № 1, с. 55—59.
108. Кристалинская Р. Г, Разработка метода получения диацетонсорбозы с
максимальным выходом продукта. — В сб.: «Витамины в теории и практике», Л.,
Пищепромиздат, 1941, 3, с. 78—87.
109. Шнайдман Л. О., Дульчина Б. М., Мавричева О. А., III е-
вырева О. Н. Ускорение процесса окисления диацетон-Г-сорбозы в диацетон-
2-кето-Б-гулоновую кислоту перманганатом калия. — В сб.: «Витаминная про-
мышленность», М., Пищепромиздат, 1955, Ns 1, с. 26—27.
ПО. Фаворский А. Е. Курс органической химии. Л., КУБУЧ, 1930, с. 227.
111. Словарь органических соединений. М., ИЛ, 1949, 1, с. 593.
112. Краснокутский Г. И. Способ быстрого охлаждения монодиацетонового
раствора. — В сб.: «Витаминная промышленность», М., Пищепромиздат, 1955, № 1,
с. 19.
112а. Фирма «Merck», пат. ФРГ, № 1176506, 1966; Auszuge, 1970, № 9.
113. Буль П. Е., Болгова А. Н., Ярош Е. П. Авт. свидет., № 93300,
1950; Бюлл. изобрет., 1952, № 2—3, с. 7.
114. Шнайдман Л. О. Рациональная технологическая схема многоступенчатой
очистки и перекристаллизации аскорбиновой кислоты. — Тезисы докладов на со-
вещании по техническому усовершенствованию производства аскорбиновой кисло-
ты, Главвитаминпром, ВНИВИ, М., 1954, с. 21—24 с ил. Там же. Некоторые
295
вопросы химии и технологии в производстве синтетической аскорбиновой кислоты,
с. 3—7.
115. С т р у к о в И. Г., Копылова Н. А., Фармация, 1947, 3, с. 8.
116. Т у р с и н В. М., Критский А. Г., Авт. свидет., Ns 65364, 1944, Бюлл.
изобрет., 1945, Ns 9—10, с. 10.
117. Березовский В. М., Цимар кина Г. Е., СтрельчунасЛ. Н.
О выделении и ацетонировании (3-Б-2,3-моноацетонсорбофуранозы. — «Труды
ВНИВИ», М., Пищепромиздат, 1954, Ns 5, с. 21—25 с ил.
118. Шнайдман Л. О., Хохлов И. М., Дульчина Б. М., С о л я н-
к и н а Л. И. Непрерывный процесс ацетонирования L-сорбозы. — В сб.: «Ви-
таминная промышленность», М., ГОСИНТИ, 1960, № 6, с. 12—21 с ил.
119. Ш н а й д м а н Л. О. Перспективы усовершенствования синтеза аскорбиновой
кислоты. —-В сб.: «Витамины», М., ГОСИНТИ, 1959, 5, с. 5—22 с ил.
120. Шнайдман Л. О. Перспективы усовершенствования производства витами-
нов на Уфимском витаминном заводе. — В сб.: «Витамины», Уфа, Башиздат, 1959,
с. 41—54.
121. О v е г h о f f J., Н и у s е г W. Пат. США 2467442, 6/IV, 1946; Off. gaz., 1949,
621, 3, 779.
122. Haworth W., Nature, 1934, 134, 724; Англ. пат. 443901, 2/Ш 1936; пат. США
9П7Ч907 9/ТТТ 1937
123. Голл. пат. № 59301, 1947; С. А., 1947, 41, 5895.
124. Goldschmidt S. Голл. пат. № 57143, 1946.
125. Trenner N. Пат. США 2483251, 1/V 1942; Off. gaz., 1949 626, No 4, 1121.
126. Damler О., Heyns К- Пат. США 2189778, 13/11, 1940; 2190377, 13/11, 1940.
127. Heyns К., Ann 1947, 558, 177.
128. S m i d t h F. Датск. пат. Ns 68836; C. A., 1949, 43, 8919.
129. Huang H. Пат. США 3043749, 1962; С. A., 1963, 59, 5739.
130. Фирма «Такэда Якухин Когё», Японск. пат. № 159, 28/IX 1962; 160, 1/Х 1962;
Токкё Кохо, 1966, 1, сб. 305.
131. Weijlard, J. Am. Chem. Soc., 1945, 67, 1031.
132. Беер А. А., Преображенский H. А. Окисление диацетон-Д-сорбозы
гипохлоритом. —ЖПХ, 1946, т. 19, с. 1121—24 с ил.
133. Т у р с и н В. М., Русакова М. Окисление диацетон-Д-сорбозы ман-
ганат калия. — ЖПХ, 1945, т. 18, вып. 9—10, с. 564. То же. Новое в науке и тех-
нике витаминов. М., Пищепромиздат, 1946, I.
134. A. Verhegden. Пат. США, 2559034, 13/VIII, 1946; Off. gaz., 1951, 648, 1,
163.
135. Шнайдман Л. О., Авт. свидет., № 104879, 1956; Бюлл. изобрет., 1957,
№ 1, с. 12. То же. Снижение расхода перманганата калия при окислении диацетон-
сорбозы в диацетон-2-кето-Б-гулоновую кислоту. — В сб.: «Витаминная промыш-
ленность», 1957, № 4, с. 26—29.
136. Рубцов И. А,, Балякина М. В., Грызлова Л. Г. и др. Усовер-
шенствование процесса окисления диацетон-Д-сорбозы гипохлоритом натрия. —
«Труды ВНИВИ», М„ Пищепромиздат, 1954, № 5, с. 17; То же. В сб.: «Витамин-
ная промышленность», М., Пищепромиздат, 1955, Ns 1, с. 23—24.
137. Рубцов И. А. Исследования в области производственных методов синтеза ви-
таминов А, Вь С. Доклад, докт. дисс., М., 1967, с. 37.
138. X а с и и Э. Японск. пат. Ns 26693, 14/Ш 1963; Токкё Кохо, 1965, II сборн.,
1606.
138а. Никольский Б. П., Крунчак В. Г. и др. Спектрофотометрическое
исследование устойчивости растворов гипохлорита и хлорноватистой кислоты. —
«ДАН СССР», 1970, 191, вып. 6, 1324—26 с ил.
139. Авруцкая А. А., Ф и о ш и н М. Я., БорисовА. И. Авт. свидет.,
Ns 255235, 1968; Бюлл. изобрет., 1969, № 33, с. 20.
139а. Майофис Л. С. Химия и технология химико-фармацевтических препаратов.
М., Медицина, 1964, с. 165.
140. Шнайдман Л. О., Кущинская И. Н. Каталитическое окисление ди-
ацетон-Д-сорбозы в диацетон-2-кето-Д-гулоновую кислоту кислородом воздуха. —
«Труды ВНИВИ», М„ Пищепромиздат, 1951, № 8, с. 13—21.
141. Ш н а й д м а н Л. О., Рюмина, И. В., Васильева Г. А. Усовершен-
ствование процесса каталитического окисления диацетон-Д-сорбозы кислородом
воздуха. — В сб.: «Витаминная промышленность», М., ГОСИНТИ, 1961, Ns 7,
с. 26—31, То же. Авт. свидет., Ns 137913, 1960; Бюлл. изобрет., 1961, Ns 9.
.142. Шнайдман Л. О., К. е й м а х Р. Я-, Определение конца реакции окисле-
ния диацетон-Д-сорбозы в диатетон-2-кето-Д-гулоновую кислоту. — В сб.: «Ви-
таминная промышленность», М., Пищепромиздат, 1958, № 5, с. 57—63.
143. Н е у г s К.., Paulsen Н., Angew. Chemie., 1957, 18—19, 600.
144. Бережковский М. И., Свистулев В. М. Транспорт жидкого хло-
ра. — «Химическая промышленность», 1970, № 10, с. 776—781 с ил.
145. Березовский В. М., Стрельчунас Л. И. Механизм превращения
диацетон-2-кето-Д-гулоновой кислоты в L-аскорбиновую кислоту. — ЖОХ, 1950,
т. 20, вып. II, с. 2072—75 с ил.
296
146. Шнайдман Л. О. Техническое усовершенствование процессов лактониза-
ции и енолизации диацетон-2-кето-Д-гулоновой кислоты. — «Труды ВНИВИ»,
М., Пищепромиздат, 1954, № 5, с. 32—41.
147. Векслер В. И., Ш а л т ы к о Г. Е. Исследование реакции превращения
гидрата диацетон-2-кето-Д-гулоновой кислоты в 4-аскорбиновую кислоту. —
ЖПХ, 1955, т. 28, вып. 7, с. 761—765.
148. Струков И. Г., Копылова Н. А. Енолизации диацетон-2-кето-£-
гулоновой кислоты в среде дихлорэтана. — В сб.: «Витаминная промышленность»,
М., Пищепромиздат, 1956, № 2, с. 20—26.
148а. Кульневич В. Г., Абрамян С. В. Влияние кислорода на разложе-
ние фурфурола в водных растворах. — «Известия вузов. Химия и химическая
технология», 1969, т. 12, вып. 4, с. 423—436 с ил.
149. Березовский В. М., Стрельчунас Л. И. Лактонизация и еноли-
зация 2-кето-Т-гулоновой кислоты и ее производных. —ЖПХ, 1949, т. 21, вып.
10, с. 1113—15 с ил.
150. Шнайдман Л. О. Снижение потерь и повышения качества аскорбиновой кис-
лоты при ее перекристаллизации. — «Труды ВНИВИ», 1953, № 4, с. 54—62. То
же. 1959, 6, 62. То же. — В сб.:«Витаминная промышленность», М., Пищепромиз-
дат., 1955, 1, 28.
150а. Шнайдман Л. О., Дульчина Б. М., Авт. свидет., № 141988, 1959;
Бюлл. изобрет., 1965, № 4, с. 116.
1506. Сугимото К- и др. Японск. пат. № 27054, 26/II 1965.
151. Ш н а й д м а н Л. О. Авт. свидет., № 95074, 1952; Бюлл. изобрет., 1953, № 2,
с. 9.
152. A. Szent—Gyorgyi, Z. Physiol. Chem., 1934, 225, 168.
153. Гольдштейн Б. И., Волькензон Д. В., — «Биохимия», 1940, т. 5,
602 с.
154. Вайсман С. Б., Кручакова Ф. А. К вопросу о методике получения
препарата железоаскорбинового комплекса. — В сб.: «Витамины», Изд-во
АН УССР, 1953, № 1, с. 158—165.
155. Swern D., J. Am. Chem., 1949, 71, 3256.
156. Фирма «Такэда Якухин Когё». Японск. пат. № 9550, 1968.
Глава 13. ПРОИЗВОДСТВО ВИТАМИНОВ D2 и D3
Возникновение детской болезни рахита связано с недостатком в орга-
низме витамина D. Последний катализирует реакции обмена кальция в
детском организме и отложения фосфорнокислого кальция в костных тка-
нях. Для лечения рахита издавна особой популярностью пользовался ры-
бий жир. С другой стороны, в начале XX в. исследователи различных стран
установили важное значение в профилактике и лечении рахита солнечного
света [1, 21. Вопрос о благотворном влиянии на больных рахитом, с одной
стороны, рыбьего жира, с другой, солнечного света долгое время казался
непонятным. Лишь в 1924 г. было установлено [3, 4], что путем облучения
ультрафиолетом можно некоторым пищевым продуктам придать антирахи-
тическую активность. Вскоре было установлено, что при облучении акти-
вируется жировая фракция продукта и конкретно вещество неомыляемой
фракции, принадлежащее к высокомолекулярным спиртам — стеролам.
В 1927 г. Виндаус [5] выделил из дрожжей, а Розенгейм [6] из спорыньи
чистые препараты эргостерина, которые облучением ультрафиолетом пре-
вращали в витамин D2. Однако было затем показано, что витамин D2 мало-
активен для птиц. При УФ-облучении 7-дегидрохолестерина был получен
витамин D3 более активный для птиц, чем витамин Г)2 17]. В 1935 г.
Виндаусу с сотрудниками удалось синтезировать 7-дегидрохолестерин [8],
а в 1936 г. из рыбьего жира тунца и палтуса были выделены кристаллы
витамина D3, идентичные с витамином D3, полученным при УФ-облучении
7-дегидрохолестерина [9].
Таким образом, к 1937 г. было установлено существование двух прови-
таминов — эргостерина и 7-дегидрохолестерина, которые при УФ-облу-
чении дают соответственно витамины D3 и D3.
Исследования последних лет показывают [101, что синтетические вита-
мины холекальцеферол и эргокальциферол, введенные в животный орга-
низм, функционируют в нем не в виде неизменных кальциферолов, а в виде
297
других химических структур. Более того, В. Вендт и 3. Даценко показа-
ли, что при облучении животного ультрафиолетовым светом ни его кожа,
ни печень не содержат витамина D3; что в неомыляемой фракции кожи крыс,
облученных in vivo содержится кроме 7-дегидрохолестерина, пять неиз-
вестных веществ стероидной природы, не являющихся витамином D2 или
D3 [11]. В свете этих исследований (о наличии in vivo каких-то других
биологически активных форм витамина D) вопрос о преимуществах холекаль-
циферола над эргокальциферолом становится бездоказательным, так как
ссылка сторонников D3 на специфическое действие холекальциферола in
vivo не подтверждается.
Необходимо также подчеркнуть значение витаминов D2 и D3 в животно-
водстве. Активность этих витаминов для крупного рогатого скота овец и
свиней почти одинакова. Для птицы активность витамина D2 составляет
активности витамина D3 [12].
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ВИТАМИНОВ D
Витамин D2 — эргокальциферол (феро —греч. — несущий), С28Н44О,
молекулярная масса 396,63, температура плавления 115—117° С [13];
120—121° С [14]; бесцветные кристаллы; [а]^ = + 106° в спирте; +82,6°—
в ацетоне; +91,2° — в эфире; Хтах = 265 нм-, E™°™ = 19400 (в спирте); Ejсм =
= 490 [14, 15]. Витамин D2 получают из эргостерина (провитамина D2) в
результате его облучения УФ.
Витамин D2 нерастворим в воде, хорошо растворим в ацетоне, хлоро-
форме, этаноле, в растительных жирах. Активность витамина D2—40 000
и. е. в 1 мг. С раствором треххлористой сурьмы в хлороформе витамин D2
дает характерное розовое окрашиваемое с >-Шах=500 нм.
При хранении кристаллы эргокальциферола постепенно разрушаются
под влиянием кислорода воздуха, влаги и света; под вакуумом в ампулах
из оранжевого стекла на холоду в течение 9 месяцев не было заметно при-
знаков разложения [15]. Масляные эмульсии и водно-коллоидные растворы
эргокальциферола нестойки [16]. В нейтральной и щелочной среде витамин
D2 стоек к нагреванию; в кислой среде разрушается [17]; при омылении
жиров не разрушается.-Перекись водорода, сернистый ангидрид, формаль-
дегид разрушают витамин D2 [18]. Сложные эфиры эргокальциферола не
обладают антирахитической активностью.
Изомеры эргокальциферола — пирокальциферол .(температура плавле-
ния 94° С) и изопирокальциферол (температура плавления 112—115° С)
образуются при нагревании витамина D2 до 160—190° С. Оба изомера не-
активны и нетоксичны. Эмпирическая формула как и для эргостерина
С28Н44О.
Витамин D3—холекальциферол С27Н44О, молекулярная масса 384,62, тем-
пература плавления 82—86° С [17,19]; [а]о =+84,8° в ацетоне [201; + 111,2°
в спирте [21 ]; ?.тах =265 нм; £^"=18200 в спирте или гексане. Активность
витамина D3 40000 и. е. в 1 мг. Витамин D3 получают из 7-дегидрохолесте-
рина путем его облучения УФ-светом.
Холекальциферол дигитонином не осаждается, цветные реакции те же,
что и для витамина D2.
Кристаллический холекальциферол более стабилен, чем витамин D2.
При хранении в негерметичной таре быстро наступает разложение препа-
рата; в герметичной таре и при хранении на холоду в течение года незамет-
но его разрушение [19, 20].
МЕХАНИЗМ РЕАКЦИЙ ФОТОХИМИЧЕСКИХ ПРЕВРАЩЕНИЙ ЭРГОСТЕРИНА
И 7-ДЕГИДРОХОЛЕСТЕРИНА В ВИТАМИНЫ D2 И D3
При облучении растворов эргостерина происходит сложная фотохими-
ческая реакция с образованием ряда фотодериватов. Изучение этой реакции
показало, что образование фотодериватов происходит по определенной
298
закономерности, установленной Виндаусом в 1932 г. [22], а именно: внача-
ле образуется люмистерин, затем п ротах истер ин, тахистерин, прекальцифе-
рол, кальциферол (витамин D2) и, наконец, супрастерин I и II и токсисте-
рин (см. схему)
эргостерин —» люмистерин —>• протахистерин —> тахистерин —>- прекальциферол —>-
витамин D2
{токсистерин
супрастерин II
супрастерин I
До сих пор еще не наблюдали обратного процесса — получения эргосте-
рина из фотодериватов. Не выяснено также, образуются ли три последних
продукта — супрастерины и токсистерин — непосредственно из витамина
D2 или в данном случае наблюдается последовательный переход одного
фотодеривата в другой.
Приводим структурные формулы и краткую физико-химическую харак-
теристику фотодериватов, получаемых при облучении эргостерина
Эргостерин, С28Н44О • Н2О. Температура плавления 166°С
[а]д = 132° в хлороформе, абсорбционный максимум 260, 270,
282 и 293,5 нм.
Люмистерин, С28Н44О. Температура плавления 118°С, [а]д =
= 192° в ацетоне, абсорбционный максимум 265 и 279 нм [19],
молекулярный коэффициент экстинкции при 279 нм — 8500.
Протахистерин не выделен.
Тахистерин, С28Н44О. Температура плавления эфира 3,5-дини-
тро-4-метилбензоата 154—155°С, [«]д =— 70° в бензоле, аб-
сорбционный максимум 268, 280 и 294 нм, молекулярный коэф-
фициент экстинкции при 280 нм 24000.
Прекальциферол, С28Н44О. Температура плавления 3,5-дини-
тробензоата 103—104°С; [а]д эфира = + 45° в хлороформе и
+ 30° в бензоле.
Витамин D2 (кальциферол) (С28Н44О). Температура плавления
115—117°С [21]; [а]д = + 106° в спирте; ЛШах=265 нм;
£МОПЬ = 19400.
Токсистерин, С28Н44О.
Температура плавления
около 50°С; [а]д = —16°
в хлороформе; абсорбци-
онный максимум 248 нм;
молекулярный коэффи-
циент экстинкции 18300
[И]
Супрастерин I, С28Н44О.
Температура плавления
104°С; [а]д = —76° в
хлороформе; абсорбцион-
ный максимум 250 нм.
Супрастерин II, С28Н44О.
Температура плавления
110°С; [а]д =—63° в хло-
роформе; абсорбционный
максимум 250 нм
На рис. 47 даны спектры в УФ-свете промежуточных продуктов облуче-
ния эргостерина.
299
В указанную выше схему фотолиза Виндауса были внесены существен-
ные изменения Л. Веллюзом в 1949 г. в связи с открытием прекальциферола
и новыми данными по структуре тахистерина и эргокальциферола [23, 24].
Им было показано, что при облучении УФ-светом чистого прекальциферола
в эфирном растворе образуется смесь из прекальциферола (40%), тахисте-
Рис. 47, УФ-спектры промежуточных продуктов облучения эргосте-
рина.
рина (30%), эргостерина (3%) и люмистерина (3%)/ На основе этих работ
и других ([25, 26] была предложена новая модель фотолиза 7-дегидрохоле-
стерина (или эргостерина):
7 - Дегидро холестерин
Люмистерин 3
300
ВЛИЯНИЕ РАЗЛИЧНЫХ ФАКТОРОВ НА ЭФФЕКТИВНОСТЬ ФОТОРЕАКЦИИ
ПРЕВРАЩЕНИЯ ПРОВИТАМИНОВ D В ВИТАМИНЫ D
Источники лучистой энергии. Для правильного выбора источника лу-
чистой энергии прежде всего необходимо выяснить, какая область спектра
является наиболее эффективной для процесса фотолиза. По этому вопросу
имеется ряд исследований.
Еще в 1927 г. было установлено [27], что эффективность процесса фото-
лиза при 313 нм очень низка. Позднее экспериментально было показано
[28], что при 302,5 нм фотохимическая реакция для эргостерина мало актив-
на; для 7-дегидрохолестерина > длины волн 245,3; 253,7; 265,2; 280,4 и
302,5 нм дают одинаковый эффект на 1 квант энергии.
При 313 нм не наблюдалось активации провитамина.
. Линия же в 296,7 нм дает несколько больший эффект, чем другие. Эти
же авторы еще ранее показали на крысах, что при 296,7 нм фотолиз дает
наибольшую эффективность.
Между тем другие исследователи [29] также на крысах показали, что
линия 280,4 нм дает наилучшие результаты.
Г. Розенберг [17] утверждает, что наибольший выход витамина D при
наименьшем количестве побочных продуктов получают при длине волны об-
лучающего света 275—300 нм. В. Вендт, исходя из спектров светопоглоще-
ния фотодериватов, предложил ступенчатый процесс облучения 7-дегидро-
холестерина лампами с различными спектрами излучения: первая стадия
облучения осуществляется люминесцентными эритемными лампами с излу-
чением в области 280—340 нм и с максимумом при 310—312 нм. Затем не
выключая эритемных ламп, зажигают бактерицидные лампы ([253,7 нм].
На этой стадии происходит превращение накопившегося люмистерина в
тахистерин. Последний под влиянием эритемных ламп (/.гаах=280 нм)
превращается в витамин D3 [30].
Данные различных исследователей об оптимальной области спектра,
хотя не вполне сходятся, но очень близки. По-видимому, область спектра
275—300 нм можно принять как оптимальную для процесса фотолиза эрго-
стерина и 7-дегидрохолестерина.
Казалось бы, что при помощи специальных светофильтров можно изо-
лировать область спектра 275—300 нм и таким образом добиться наивыс-
шего эффекта фотохимической реакции. В этом направлении были проведе-
ны многие исследования и изучены светофильтры, поглощающие область
спектра с длиной волны короче 275 нм и больше 313 нм. Для этой цели
применяли стекла специального состава, селективно пропускающие свет
[31, 32], или различные растворы органических и неорганических соеди-
нений, как, например, бензол [33, 34], ксилол, дифенил [34], уксуснокис-
лый свинец [35], нитрит калия [33], сероуглерод [36]. Для поглощения
области спектра короче 275 нм применяют 5%-ный раствор бензола в спир-
те, ксилола, дифенила в бензоле (концентрация 0,005%), 5%-ный раствор
уксуснокислого свинца, а для поглощения света с длиной волны больше
313 нм — четыреххлористый углерод [17].
Необходимо, однако, отметить, что светофильтры не нашли широкого
производственного применения. Кривые абсорбционных максимумов фото-
дериватов, получаемых при фотолизе (см. рис. 47), показывают насколько
близки они между собой, а следовательно, и абсорбция света происходит
в трудно разграничиваемых пределах [19].
Влияние растворителя на эффективность фотолиза. Принципиально
эргостерин может активироваться при облучении его в твердом, жидком
и парообразном состоянии. При облучении его в твердом состоянии продук-
ты облучения на поверхности вещества действуют как светофильтр и защи-
щают лежащие ниже слои от проникновения ультрафиолетового света.
Практически эффективность процесса облучения эргостерина в твердом
виде не превышает 10% эффективности облучения в растворах. Облучение
301
эргостерина в парообразном виде еще более низко, так как точка плавления
(165° С) близка к температуре изомеризации кальциферола (160—190° С)
в пирокальциферол и изопирокальциферол, а следовательно, часть обра-
зуемого кальциферола изомеризуется. В связи с этим в практике эрго-
стерин обычно облучают в растворенном состоянии.
Процесс превращения эргостерина в эргокальциферол протекает лишь
при условии поглощения раствором лучистой энергии, поэтому весьма
важно при облучении раствора создать условия надлежащего контакта
между потоком ультрафиолетовых лучей и частицами растворенного ве-
щества. Необходимо, чтобы растворитель был проницаем для ультрафиоле-
товых лучей. Если растворитель поглощает эти лучи, то контакт между
частицами эргостерина и ультрафиолетовыми лучами установится лишь на
Рис. 48. Изменение антирахитиче-
ской активности растворов эргосте-
рина:
поверхности раствора в молекуляр-
ном слое. В этом случае раствори-
тель будет играть роль внутреннего
светофильтра. Поэтому выбор раство-
рителя имеет весьма важное значение
для эффективности фотохимической
реакции при облучении эргостерина.
Об этом свидетельствуют данные,
приведенные на рис. 48, по антирахи-
тической активности растворов эрго-
стерина в эфире, циклогексане и спир-
те. Из рис. 48 видно, что максимум
активности достигается значительно
медленнее (5—6 раз) для эфирного
раствора, чем для спиртового; вели-
чина максимума активности для эфира
почти в 3 раза больше, нежели для
1 — в эфире; 2 —в циклогексане; 3 —в спирте. СПИрТЯ.
В настоящее время серный эфир
общепризнан наилучшим растворителем эргостерина при его облучении.
Более медленный (5—6 раз) процесс активации эргостерина в среде эфира
позволяет избегать его переоблучение.
Влияние кислорода. По этому вопросу в литературе имеются разноре-
чивые данные. В одних работах [17, 19, 37] подчеркивается вредное влия-
ние кислорода воздуха на процесс фотолиза эргостерина; при этом указы-
вается, что фотодериваты более чувствительны к окислению кислородом,
чем провитамины и витамины D; что присутствие окисленных веществ мало
отражается на выход витамина, но очень затрудняет его последующее вы-
деление в кристаллическом виде.
В других работах [38, 39] указывается, что при удалении кислорода в
процессе облучения не замечено увеличение антирахитической активности
препарата.
Разноречивость данных, вероятно, обусловлена наличием растворенно-
го воздуха в растворителе, которое не всегда учитывается исследователями.
Лишь полная деаэрация раствора может выявить влияние кислорода воз-
духа на эффективность процесса фотолиза.
Вне сомнения, что проведение процесса фотолиза в присутствии воздуха
значительно ухудшает его качество и деаэрация раствора в процессе облу-
чения должна считаться обязательной.
Влияние температуры. Большинство исследователей отмечает незначи-
тельное влияние температуры на эффективность процесса фотолиза [40].
Исследователи [40] при облучении эргостерина при температуре между
—195 и +78° С наблюдали умеренный температурный эффект. Имеется так-
же указание [40], что температура не влияет на процесс активации эрго-
стерина.
302
Одновременно установлено [19], что активация провитамина протекает
более интенсивно в кипящих растворах.
Объясняется это тем, что повышенная температура влияет на смещение
равновесия реакции превращения прекальциферола в кальциферол вправо.
Кроме того, при кипении наблюдается интенсивное перемешивание раство-
ра, а следовательно, более тесный контакт частиц раствора со световым
потоком.
Из изложенного следует, что с наибольшим эффектом процесс фотолиза
может быть осуществлен в состоянии кипения раствора. Такое состояние
раствора является также рациональным с точки зрения максимального уда-
ления растворенного воздуха.
Влияние концентрации эргостерина. Имеются данные [39], что при облу-
чении эргостерина в спиртовом растворе при концентрации 0,3% провита-
мина достигается выход кальциферола на 5,2—6,3% больший, чем при
концентр ации 0,4%.
Надо полагать, что фактор концентрации провитамина в облучаемом
растворе влияет на производительность облучающего аппарата.
Для каждого аппарата опытным путем должна быть установлена опти-
мальная концентрация провитамина в облучаемом растворе.
Влияние чистоты эргостерина. Проведенные исследования [42] показали
что фотохимическая реакция облучения эргостерина идет тем быстрее, чем
выше температура плавления эргостерина.
При облучении эргостерина с пониженной температурой плавления
следует удлинить время облучения.
Из изложенных кратких сведений, полученных при изучении химии
витаминов группы D, а также влияния различных факторов на фотолиз про-
витаминов D, можно сделать следующие основные выводы:
1. Экспериментально установлено, что наиболее эффективное превра-
щение эргостерина в кальциферол происходит под влиянием ультрафиоле-
тового света с длиной волны 275—300 нм. Область с длиной волны 218—
280 нм разрушает кальциферол и превращает его в токсистерины и супра-
стер ины.
2. Можно рекомендовать следующие относительно наиболее эффектив-
ные условия для проведения фотолиза провитаминов D в витамин D;
растворитель для получения кристаллического витамина — эфир; для
масляных растворов витамина—либо эфир, либо спирт;
ведение процесса фотолиза в деаэрированном растворе в присутствии
инертного газа:
температура фотолиза — температура кипения растворителя.
Вопросы применения светофильтров, оптимальной интенсивности лу-
чистой энергии, концентрации провитамина в растворе требуют дополни-
тельного изучения.
ВЫБОР РАЦИОНАЛЬНОГО МЕТОДА ПРОИЗВОДСТВА ЭРГОКАЛЬЦИФЕРОЛА
(D2) И ХОЛЕКАЛЬЦИФЕРОЛА (D3)
Полный химический синтез кальциферолов или их провитаминов пред-
ставляет пока что теоретический интерес и не имеет практического значе-
ния вследствие сложности синтеза пергидроциклопентанофенантренового
скелета молекулы стерина. Несравненно проще пользоваться в качестве ис-
ходного сырья для получения кальциферолов стероидными структурами,
синтезируемыми в природе. В этой области особый интерес представляет
эргостерин — провитамин D2, синтезируемый пекарскими дрожжами. Тех-
нология извлечения эргостерина из дрожжей подробно описана (стр. 423),
[18, 43]. Превращение эргостерина в эргокальциферол осуществляется
фотосинтезом.
Провитамином холекальциферола является 7-дегидрохолестерин, ко-
торый более скупо синтезируется в природе беспозвоночными животными
303
(моллюсками) [43, 44]. Более широко распространен холестерин. Его осо-
бенно много в органах и тканях высших животных (мозг, кожное сало,
почки, печень).
В промышленности холестерин получают из мозга крупного рогатого
скота или из шерстяного овечьего жира •—ланолина. Холестерин по своей
химической структуре отличается от 7-дегидрохолестерина только отсутст-
вием второй двойной связи между седьмым и восьмым атомами углерода в
цикле В стериновой молекулы.
Впервые синтез 7-дегидрохолестерина был осуществлен Виндаусом с
сотрудниками по схеме [45]:
Однако, по данным ряда авторов [46—49], этот метод синтеза не нашел
применения из-за низкого выхода 7-дегидрохолестерина (8—12%). В связи
с этим метод подвергли совершенствованию в первую очередь на стадиях
получения 7-оксихолестерина и превращения дибензоата в монобензоат
[50—53].
Более эффективным оказался другой метод образования в кольце В вто-
рой двойной связи при помощи последовательного бромирования и дегидро-
бромирования стерина в положении С7. Этот метод базируется на результа-
тах исследований Циглера о возможности избирательного замещения во-
дородного атома стерина, находящегося в аллильном положении к двойной
связи, бромом при применении N-бромсукцинимида в качестве бронирую-
щего агента [54—58]. Данный метод был тщательно проверен и изучен
В. Вендтом [44]. Общий выход 7-дегидрохолестерина составил 26—28% (на
исходный бензоат холестерина). Из анализа литературных данных можно
придти к следующему заключению:
исходным сырьем для фотосинтеза эргокальциферола должен быть эрго-
стерин, а для холекальциферола 7-дегидрохолестерин;
эргостерин получают известными методами [18, 43] из дрожжей, а 7-де-
гидрохолестерин — из холестерина методом бромирования и дегидробро-
мирования;
фотопревращение эргостерина и 7-дегидрохолестерина в эргокальцифе-
рол и холекальциферол протекает наиболее эффективно в серном эфире.
Второе место занимает этиловый спирт.
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА КРИСТАЛЛИЧЕСКОГО ЭРГО-
КАЛЬЦИФЕРОЛА — ВИТАМИНА D2
Схема предусматривает следующие основные стадии [18] (рис. 49):
освобождение фотосмолы от эргостерина;
получение 3,5-динитробензоата витамина D2;
выделение свободного витамина D2.
304
Освобождение фотосмо-
лы от эргостерина. При
облучении спиртового рас-
твора эргостерина, отгонки
растворителя и выделения
непрореагировавшего эрго-
стерина получают фотосмо-
лу. Она представляет собой
вязкую жидкость, хорошо
растворимую в органичес-
ких растворителях. Ее при-
мерный состав: эргокаль-
циферол—55—60 %, люми-
стерин 15—20%, тахисте-
рин—10—12%, эргосте-
рин — 10—13%. Облучен-
ный раствор эргостерина
в серном эфире из сборника
1 (рис. 49) поступает в пе-
регонный аппарат 2, где
отгоняют эфир и в 100 раз
концентрируют раствор.
Затем при температуре
минус 10° С выкристалли-
зовывают непрореагиро-
вавший эргостерин в кри-
сталлизаторе 3. Кристаллы
эргостерина отделяют на
центрифуге 4. Маточный
раствор, из которого уда-
лен эргостерин, поступает
в сборник 5, откуда далее
его направляют в неболь-
шой эмалированный ваку-
ум-аппарат 6, где при ос-
таточном давлении не
выше 50 мм рт. ст. отго-
няют остаток эфира.
Дальнейшую очистку
смолы осуществляют путем
ее растворения в этом же
аппарате в смеси метило-
вого спирта и сухого сер-
ного эфира (2:1).
Далее отгоняют 50 %
растворителя и массу на-
правляют в кристаллиза-
тор 7 для дополнительного
выделения стеринов. По-
следние отфильтровывают
во взрывобезопасной цент-
рифуге или в друк-фильтре
8. Маточный раствор по-
ступает в сборник 9, откуда
далее идет в вакуум-пер е-
гонный аппарат 10 для
отгонки остатков раствори-
телей. Очищенной фотосмо-
Рис. 49. Технологическая схема производства кристаллического эргокальциферола — витамина
305
лы получают 90% к массе эргостерина, загруженного на облучение. Ее со-
бирают в приемнике И.
Получение 3,5-динитробензоата витамина D2. 3,5-Динитробензоат
эргокальциферола получают путем взаимодействия фотосмолы, содержа-
щей около 60% эргокальциферола, с 3,5-динитробензилхлоридом в рас-
творе пиридина. Реакция этерификации протекает по следующей схеме:
Эргокальциферол
(6 фотосмоле)
396, 63
3,5- динитробензоат
зргока ль и, и. (рерола.
590,74
Динитробензоил -
хлорид
230,57
Очищенную фотосмолу из приемника 11 направл яют в реактор-раство-
ритель 12, изготовленный из эмалированной стали, где ее растворяют в су-
хом перегнанном пиридине (1 кг в 2,5 л), подаваемом из сборника 13. К по-
лученному раствору в присутствии СО2 добавляют порошкообразный или
расплавленный свежеперегнанный 3,5-динитробензоилхлорид из сборни-
ка 14 из расчета 0,8 кг на 1 кг фотосмолы. Для предотвращения перегревания
(выше 60° С) реактор охлаждают водой, массу перемешивают 3—4 ч и остав-
ляют на 8—10 ч.
Затем включают вакуум и при остаточном давлении 5О.и.м рт. ст. отго-
няют 50% пиридина. Затем добавляют теплую воду (40—50° С) в шести-
или семикратном количестве к массе смолы, и после энергичного переме-
шивания воду декантируют. Промывку ведут до полного удаления пириди-
на.
Для удаления избыточной динитробензойной кислоты массу промывают
(с декантацией) метиловым спиртом (2,5 л на 1 кг смолы) из сборника 15
при температуре кипения массы. Оставшуюся нерастворенной смолистую
массу обрабатывают при нагревании до кипения ацетоном, поступающим из
сборника 16, до ее полного растворения. Туда же в реактор /2вводятак-
тивированный уголь в количестве 2% к массе смолы. Затем раствор спуска-
ют в нутч-фильтр 17 и далее в кристаллизатор 18. Раствор кристаллизуют
при минус 10° С. Через 12 ч отфуговывают кристаллы динитробензоата на
взрывобезопасной центрифуге 19.
Для получения рафинированного продукта кристаллы 3,5-динитробен-
зоата растворяют в ацетоне (1 кг в 15 л) в смесителе 20 при температуре
кипения, куда вводят активированный уголь (около 2% к массе кристал-
лов), отфильтровывают раствор от угля в нутч-фильтре 21, кристаллизуют
в кристаллизаторе 22 и отфуговывают кристаллы в центрифуге 23. Кристал-
лы высушивают в аппарате 24 под вакуумом. Маточник из сборника 25 на-
правляют для переработки в реактор 12.
Основной маточник из сборника 26 обрабатывают в смесителе 27 активи-
рованным углем, фильтруют в нутч-фильтре 28. Фильтрат поступает в
сборник 29, откуда далее его направляют в вакуум-аппарат 30 для сгуще-
ния, кристаллизуют при минус 8—5° С в кристаллизаторе 31 и фугуют в
центрифуге 32.
Полученные кристаллы 3,5-динитробензоата направляют для перекрис-
таллизации в реактор 12, а маточный раствор II поступает в сборник 33 и
является отходом производства.
306
Выход 3,5-динитробензоата составляет около 40 % к массе очищенной
фотосмолы. Его температура плавления^ 145—147° С [а]2°=+79,5о (аце-
тон); +55° (бензол).
Выделение свободного витамина D2. Эргокальциферол получают пу-
тем омыления 3,5-динитробензоата эргокальциферола спиртовой щело-
чью по следующей химической схеме:
(NO2)2C6H3COO
Эрга киль цирерол р о бензойной кислоты
3,5 - Динитро - бензоат В2
590,7k 395,63 23k,21
Для получения чистого эргокальциферола проводят омыление эфира
динитробензоата в реакторе 34, снабженным обратным холодильником,
при помощи 5%-ного раствора едкого кали в метиловом спирте, приготов-
ленного в реакторе 35. Желтый цвет динитробензоата исчезает и начина-
ется осаждение фиолетового осадка калиевой соли 3,5-динитробензойной
кислоты. Раствор фильтруют в горячем виде через друк-фильтр 36 при по-
мощи инертного газа.
Фильтрат поступает в кристаллизатор 37, куда добавляют на 1 кг ди-
нитробензоата 0,6 л горячей воды.
Затем раствор постепенно охлаждают и снижают температуру до минус
2—3° С. Полученные кристаллы отделяют на центрифуге 38, промывают
небольшим количеством водного 10—50 %-ного метилового спирта и дистил-
лированной водой. Промытые кристаллы эргокальциферола высушивают в
сушилке 39.
Выход витамина D2 80% к теоретическому, считая на динитробензоат.
Маточный раствор эргокальциферола перекристаллизовывают по той же
схеме, как и динитробензоат.
Кристаллы витамина D2 должны быть белого цвета с температурой плав-
ления 113—114° С [(Hd =31° (ацетон). Если кристаллы витамина D2
не соответствуют указанным нормам, то их перекристаллизовывают из ме-
тилового спирта.
В производстве кристаллического витамина D2 необходимо особое вни-
мание уделить аппаратуре, которая должна быть изготовлена из эмалиро-
ванной стали. Процессы должны протекать в присутствии инертного газа в
тех случаях, когда в них участвует облученный эргостерин или кристалли-
ческий эргокальциферол.
Динитробензоилхлорид. C7H3O5N2C1 получают путем химического взаи-
модействия динитробензойной кислоты с тионилхлоридом по уравнению
МО2
СООН + SOC12—*-
NO2
hcl+sq2
Динил/робензойная Гионилхлорид Динитробензоилхлорид
кислота 83,53 230,57
212. 09
Загружают динитробензойной кислоты 1,0 кг, тионилхлорида (плот-
ность 1680 кг/.и3) 3,0 кг.
307
Выход динитробензоилхлорида с температурой плавления 66—67° С
составляет 0,9—1,0 кг, или 85—90% к теоретическому.
Процесс осуществляют в реакторе из эмалированной стали с прямым
и обратным эмалированным холодильником. Смесь указанных химикалиев
нагревают при перемешивании при температуре 100° С до полного раство-
рения динитробензойной кислоты. Затем включают вакуум и через прямой
холодильник отгоняют тионилхлорид. Затем остаток переводят в вакуум-
аппарат (холодильник обогревается горячей водой) и при остаточном давле-
нии 2—3 мм рт. ст. отгоняют при температуре 160—170° С динитробензо-
илхлорид (светло-желтого цвета, полностью застывает). Кристаллы долж-
ны растворяться в бензоле без остатка.
Для реакции применяют динитробензоилхлорид в измельченном виде.
Его следует защищать от влаги воздуха.
Из калиевой соли динитробензойной кислоты, полученной при омыле-
нии динитробензоата, регенерируют 3,5-динитробензойную кислоту. Для
этого калиевую соль суспензируют с водой и обрабатывают разведенной
серной кислотой. При этом выделяется техническая динитробензойная кис-
лота, которая перекристаллизовывается из спирта.
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА ХОЛЕКАЛЬЦИФЕРОЛА—
ВИТАМИНА D3
Схема включает следующие стадии синтеза [48]: бензоата холестерина,
7-бромхолестеринбензоата, 7-дегидрохолестеринбензоата, холекальцифе-
рола и получение витаминно-белкового комплекса «Видеин-3».
' Бензоат холестерина получают взаимодействием холесте-
рина в растворе пиридина с хлористым бензоилом по схеме:
Холестерин Хлористый бензоил бензоат холестерин 36,47
386,6В 740,57 490, 7и
В реактор 1 (рис. 50) загружают холестерин и из мерника 2 — пиридин,
перемешивают до полного растворения холестерина. Затем из мерника 3
постепенно прибавляют хлористый бензоил, поддерживая температуру
реакционной массы (охлаждение водой в рубашке) 45—50° С. Затем осадок
бензоата отфильтровывают в центрифуге 4, промывают тщательно водой,
затем метанолом из мерника 5. Промытый осадок разгружают в кюветы и
высушивают в вакуум-сушилке 6 при 55—60° С. Метанольные промывные
из центрифуги направляют в сборник 7 и далее на регенерацию. 7-Бензоат-
холестерина имеет температуру плавления 148° С, содержание 98—99%.
Выход 90%.
7-Бромхолестеринбензоат получают бромированием бензоатхолестерина
N-бромсукциимидом [48] или 1,3-дибром-5,5-диметилгидантоином [50]
по следующей схеме:
Бензаатхолестерин
490, 76
7-Брашолестерин - бензоат
569, 65
308
бензоат холестерин
7Бромхолестериндснзоат
7 Бромхояестеринбензоат
Спиртовый
раствор ВО.
'‘‘Аснордина
да из поз.65
Па
Вакуум
НСВ
Вада
Вихру.
50
7- Дееидрохолестерин
Метиловый Толуол
спирт
Отход
Вода
Пар
65
Виде ин - В
Спиртовый раст-
вор ВиВехола
створ казеина
^UJJiH.pac
•оредаОН
Спиро
Горя-
чая
Вода
Вануу
Уксусная
кислота
ИГподготовление
масляного Ви деин а
раствора О3
75
7В
Витамин В3
Рис. 50. Технологическая схема производства холикальциферола—витамина D3,
По литературным данным, на выход 7-бромпроизводного влияют каче-
ство бромирующего агента, природа растворителя и температура. Свет ка-
тализирует процесс бромирования. По данным В. Вендта, процесс бромиро-
вания N-бромсукцинимидом осуществляют следующим образом [48].
. В реактор 8, снабженный обратным холодильником, загружают 10,0 кг
бензоата холестерина, а из мерника 9 четыреххлористого углерода (35 л),
массу нагревают при температуре 60—65° С до полного растворения бен-
зоата и сливают в друк-фильтр 10. Фильтрат поступает в сборник И, а из
него в аппарат-броматор 12. Последний представляет собой стеклянный
реактор, снабженный обратным холодильником, мешалкой и змеевиком для
нагревания и охлаждения реакционной массы. С двух сторон броматора
на. расстоянии 200 мм от боковой поверхности его смонтированы ртутно-
кварцевые лампы типа ПРК-2. Одновременно в реактор 13 загружают 5 кг
N-бромсукцинимида и из мерника 9 четыреххлористого углерода 25—30 л и
перемешивают. Когда раствор бензоата в броматоре доведен до кипения,
включают лампы; при облучении из реактора 13 вводят в броматор 12 бро-
мирующий агент и продолжают перемешивание при кипении 10—15 мин.
Затем подачей холодной воды в змеевик броматора охлаждают реакцион-
ную массу до 20—25° С и сливают ее на друк-фильтр 14. Фильтрат посту-
пает в сборник 15, а из него в вакуум-аппарат 16 для отгонки растворите-
ля. Отогнанный четыреххлористый углерод из сборника 17 направляют
для повторного использования в мерник 9, а кубовый остаток растворяют
в ксилоле, подаваемом в вакуум-аппарат 16 из мерника 18. Ксилольный
раствор 7-бромхолестеринбензоата фильтруют через нутч-фильтр 19 в сбор-
ник 20, откуда насосом 21 подают в мерник 22 на дегидробромирование.
Выход 7-бромхолестеринбензоата составляет 40% (на бензоат).
7-Д егидрохолестеринбензоат получают из 7-бромхо-
лестеринбензоата отщеплением из него молекулы бромистого водорода на-
греванием в ксилольном растворе с двууглекислым натрием последующей
химической схеме:
Для реакции отщепления бромистого водорода от 7-бромхолестеринбен-
зоата были предложены различные соединения, связывающие НВг: диме-
тиланилин [60], коллидин [61], хинальдин [63], окись кальция [63], би-
карбонаты и карбонаты различных щелочноземельных металлов.
Имеются указания о влиянии на этот процесс температуры, природы
растворителя, наличия влаги [48], а также о получении наилучших резуль-
татов при проведении реакции в ксилоле при температуре 135—140° С [57].
В. Вендт рекомендует следующий режим процесса дегидробромирова-
ния [48]. Раствор бромида в ксилоле (35—40%) из мерника 22 направляют
в реактор-дегидроброматор 23, снабженный прямым холодильником и при-
емниками. Туда же загружают из бункера 24 сухой тонко измельченный
порошок бикарбоната натрия и из баллона 25 подают азот. Реакцию ведут
при температуре 135—140° С (температура теплоносителя — 160—170° С).
Реакцию ведут до полного удаления воды из массы, которая вместе с
ксилолом отгоняется в сборники 26. Затем Ксилольный раствор 7-дегидро-
холестеринбензоата фильтруют через нутч-фильтр 27 в сборник 28. Из по-
следнего фильтрат засасывают в вакуум-аппарат 29, отгоняют часть раство-
рителя и сливают в кристаллизатор 30, куда добавляют при температуре
25—30° С ацетон из мерника 31 и кристаллизуют при —10° С в течение
10—12 ч. Выделившиеся кристаллы отфуговывают во взрывобезопасной
310
центрифуге 32, промывают ацетоном из мерника 31, разгружают в кюветы
и сушат в вакуум-сушилке 32 при температуре 25—30° С. Маточный раствор
поступает в сборник 33. Вопрос о технологии извлечения из него оставше-
гося в растворе продукта подлежит изучению.
Выход 7-дегидрохолестеринбензоата составляет 50%.
7-Д егидрохолестерин получают путем омыления 7-дегидро-
холестеринбензоата спиртовой щелочью по следующей химической схеме:
Процесс омыления эфиров стеринов обычно ведут спиртовым раствором
едкого кали [64]. Для предотвращения окисления 7-дегидрохолестерина
кислородом воздуха, В. Вендт рекомендует вводить в реакционную массу
небольшое количество аскорбината натрия или пирогаллола.
В реактор 34, снабженный обратным холодильником, загружают из
вакуум-сушилки 35 кристаллы 7-дегидрохолестеринбензоата, а из мерника
36 спиртовый раствор едкого кали и аскорбинат натрия. Затем из баллона
37 вводят азот и нагревают реакционную массу при кипении в течение 1 ч..
Затем нейтрализуют соляной кислотой из мерника 38, перемешивают и
фильтруют на нутч-фильтре 39. После промывки водой до нейтральной
реакции осадок высушивают в вакуум-сушилке 40. Технический продукт
подвергают перекристаллизации из смеси метилового спирта и толуола
(1:1) в реакторе 41. Метиловый спирт сливают из мерника 42, а толуол —
из мерника 43. Растворение ведут при нагревании до 60—70° С, фильтруют
через нутч-фильтр 44 в кристаллизатор 45. Кристаллизацию ведут в тече-
ние 10—12 ч при —10—15° С. Кристаллы I отфуговывают в центрифуге 46,
а маточный раствор поступает в сборник 47 и далее на переработку путем
сгущения в вакуум-аппарате 48, кристаллизации в кристаллизаторе 49,
фуговки в центрифуге 50. Кристаллы II поступают на переработку совмест-
но с техническим продуктом в реактор 41, а маточный раствор — в сборник
51 и является отходом. Кристаллы I из центрифуги 46 направляют в ва-
куум-сушилку 52 и далее в сборник 53. Общий выход провитамина состав-
ляет 85% (на 7-дегидрохолестеринбензоат), температура плавления 149—
150° С; [ц]д=—120° (хлороформ); С27Н44ОН, молекулярная масса 384,64;
легко растворим в эфире, трудно в метиловом спирте. £Т°л|Ь= 10920 при'
^тах=281,5 НМ.
Холекальциферол получают путем облучения спиртовых
растворов 7-дегидрохолестерина ультрафиолетом.
Фото реакция протекает по следующей химической схеме:
7-Дегидрохолестерин
384, 64
А7ле паль ц шрерол
334,64
311
На основании исследований В. Вендта применяют двухступенчатый
процесс облучения [30] с применением эритемных и бактерицидных ламп.
Процесс осуществляют следующим образом.
В реактор-смеситель 54 загружают 7-дегидрохолестерин, из мерника 55
спирт и при перемешивании приготовляют при температуре 40—50° С рас-
твор, содержащий 0,1—0,2% провитамина. Раствор фильтруют через нутч-
фильтр 56 в сборник 57, откуда поступает в мерник 58. Из последнего
раствор 7-дегидрохолестерина поступает й облучательные аппараты 59, снаб-
женные люминесцентными эритемными (Хтах=310—312 нм) и бактерицид-
ными (Хтах =253,7 нм) лампами. Вначале из баллона 60 подают азот, за-
тем включают эритемные лампы. Через 60— 100 мин (пока 35% провита-
мина не превратится в люмистерин) [30] включают бактерицидные лампы,
излучение которых превращает люмистерин в тахистерин. Последний под
влиянием излучения эритемных ламп превращается в провитамин и вита-
мин D3. Общая продолжительность процесса облучения в аппаратах пе-
риодического действия составляет 2,0—2,5 ч. Из облучательного аппарата
облученный раствор направляют в сборник 61, а из него в вакуум-аппарат
62 для отгонки спирта и сгущения (в 8—10 раз); далее сгущенный раствор
поступает в кристаллизатор 63, где при температуре — 10—-15° С выкри-
сталлизовывают непрореагировавший 7-дегидрохолестерин, который от-
фильтровывают на нучт-фильтре 64, осадок промывают спиртом из сборни-
ка 65 и направляют в реактор 41 для перекристаллизации. Фильтрованный
облученный раствор из нутч-фильтра 64 засасывают в вакуум-аппарат 66
для дополнительной отгонки спирта. Затем полученный концентрат фильт-
руют через нутч-фильтр 67 в сборник 68, откуда далее его направляют
либо для приготовления масляного препарата, либо для получения виде-
ина-3. Выход холекальциферола составляет 55,7% [30].
В и де и н - 3 является искусственным комплексом витамина D3 с ка-
зеином [65]. Применяется для профилактики и лечения рахита у детей, а
также для витаминизации продуктов детского питания. Препарат обладает
большей антирахитической активностью по сравнению с другими препа-
ратами витамина D [66—69]. Метод получения видеина основан на совме-
щении процесса осаждения белка в момент изоэлектрического его состоя-
ния с процессом образования коллоидного раствора витамина D3, происхо-
дящего в результате замены растворителя. Витамин D3 применяется в виде
кристаллического комплекса его с холестерином, названного авторами ви-
дехолом.
Получение видеина предусматривает следующие процессы: приготовле-
ние раствора препарата П3-видехола; приготовление раствора казеина;
осаждение комплекса витамина D3 с казеином.
В реактор 69 наливают из мерника 70 раствор казеина в 0,05 н. растворе
едкого натра. Затем тонкой сильной струей азота (баллон 71) из мерника 72
вводят при энергичном перемешивании спиртовый раствор видехола. Пред-
варительно в мерник 72 добавляют из мерника 73 уксусную кислоту в ко-
личестве, необходимом для нейтрализации щелочи и достижения изоэлект-
рической точки казеинового раствора [70]. Процесс осаждения проводят в
течение 1,5—2 мин. После отстаивания осадок выделяют в центрифуге 74,
высушивают в сушилке 75 при температуре 55—60° С и измельчают в мик-
ромельнице 76. В 1 г препарата содержится 200 тыс. и. е. витамина D3.
ТЕХНОЛОГИЧЕСКИЕ ПОКАЗАТЕЛИ
ПРОИЗВОДСТВО КРИСТАЛЛИЧЕСКО-
ГО ЭРГОКАЛЬЦИФЕРОЛА
(ВИТАМИНА D2)
Расход материалов, кг на 1 кг полу-
продукта
3,5-Ди.нитробензои.лхлорид
Динит робензойная кислота .... 1,1
Тионилхлорид ................. 3,3
312
Выход (на динитробензойную кисло-
ту), % .........................85
3,5-Динитробензватэргокальциферола
Фотосмола (или эргостерин 2,8) . 2,5
3,5-Динитробензоилхлорид ... 2,0
Пиридин (с регенерацией) .... 1,5
Метиловый спирт (с регенерацией) 1,5
Ацетон (с регенерацией) .... 3,0
Активированный уголь ...........0,10
Выход (на фотосмолу), % .... 40
Эргокальциферол
3,5-Динитробензоатэргокальцифе-
рола ............................ 1,9
Кали едкое (100%)...............0,5
Спирт метиловый (с регенерацией) 1,0
Выход (на динитробензоат эрго-
кальциферола) , % ...............80
Расход химикалиев и полупродуктов,
кг на производство I кг кристалличес-
кого эргокальциферола
Химикалии
Ацетон..........................5,70
Динитробензойная кислота .... 4,20
Кали едкое .....................0,50
Пиридин.........................2,85
Тионилхлорид ................. 12,54
Уголь активированный ..........0,19
Эргостерин.....................5,3
Спирт метиловый . . .'........3,85
Полупродукты
3,5-Динитробензоилхлорид .... 3,8
3,5-Динитробензоатэргокальцифе-
рола.............................1,9
Эргостерин .....................5,3
7-Дегидрохолестеринбензоат
7-Бромхолестеринбензоат .... 2,4
Ксилол (с регенерацией) .... 1,6
Бикарбонат натрия...............5,0
Ацетон (с регенерацией) .... 3,0
Выход (на 7-бромхолестеринбензо-
ат), %...........................50
7-Де гид рохолесте рин
7-Дегидрохолестеринбензоат . . 1,5
Кали едкое........................0,7
Спирт метиловый (с регенерацией) 2,0
Кислота соляная...................0,3
Аскорбинат натрия ............... 0,02
Толуол (с регенерацией) .... 1,5
Выход (на 7-дегидрохолестерин-
бензоат), %......................85,0
Холекальциферол (на 100%-ный)
7-Дегидрохолестерин.............2,0
Спирт этиловый ........ 120
Выход (на 7-дегидрохолестерин), % 55,7
Расход химикалиев и полупродуктов,
кг на производство 1 кг холекальци-
ферола (в виде спиртового концентрата
с содержанием 400 тыс. и. е. в 1 мл)
ПРОИЗВОДСТВО ХОЛЕКАЛЬЦИФЕРО-
ЛА (ВИТАМИНА Д3)
Бензоат холестерина
Холестерин.....................0,9
Пиридин (с регенерацией) .... 0,5
Хлористый бензоил..............0,6
Спирт метиловый................0,2
Выход (на холестерин), % ... 90
7-Бромхолестерин5ензоат
Бензоат холестерина ...........2,0
Четыреххлористый углерод (с ре-
генерацией) ...................4,0
N-Бромсукцинимид...............1,0
Выход (на бензоат холестерина),% 40
Химикалии
Ацетон.........................15,0
Аскорбинат натрия .............. 0,04
Бензоилхлорид...................8,60
Бикарбонат натрия ............ 15,00
N-Бромсукцинимид .............. 7,20
Кали едкое......................1,40
Кислота соляная .................0,60
Ксилол ..........................4,80
Метанол ........................6,90
Пиридин..........................7,20
Толуол ................... . . 3,00
Спирт . . -...................120,0
Четыреххлористый углерод . . . 28,8
Холестерин.....................13,0
Полупродукты
Бензоат холестерина .......... 14,4
7-Бромхолестеринбензоат..........7,2
7-Дегидрохолестеринбензоат ... 3,0
7-Дегидрохолестерин..............2,0
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. Huldschinsky К-, Deut. med. Wochschr., 1919, 45, 712.
2. H e s s A., Gutman M., J. Am. Med. Assoc., 1922, 78, 29.
3. Steenbock H., Science, 1924, 60, 224.
4. H e s s A., Weinstock M., J. Biol., Chem., 1924, 62, 301.
5. Windaus A., Hess A., Nachr. Ges. Wiss. Gottingen, Math, physik. klasse,
1927, 3, 185.
6. Rosenheim O., Wester T., Biochem J., 1927, 21, 389.
/.Waddell J., J. Biolg. Chem., 1934, 105, 711.
8. W i n d a u s A., Bock F., Z. physiol. Chem., 1937, 245, 168.
9. Brockmann H., Z. physiol. Chem., 1936, 241, 104; 1937, 245, 96.
10. В e н д т В. П. Некоторые данные об обменно-активных формах витамина D. —
В сб.: «Материалы совещания по химии и биохимии витаминов D и их применение
в медицине и животноводстве», Киев, «Наукова думка», 1968, с. 9—10.
И. Даценко 3. Исследования антирахитической активности продуктов фотопре-
вращения стеринов кожи животных. Автореф. кандид. дисс., Киев, 1968.
12. Рекомендации по применению витаминных концентратов в животноводстве. М.,
«Колос», 1965, 5.
13. Rosenberg Н., Chemistry and Physiology of the Vitamins, N. J., 1942, 341.
14. R e n a u H., Hagemann G., Helv. Chim. Acta, 1946, 29, 1366.
15. Crews S., Smith E., Analyst, 1939, 64, 568.
16. S h a 1 1 i n g D., Proc. Soc. Exp. Biol. Med., 1946, 35, 660.
17. Rosenberg H., Encyclopedia of Chemical Technology., 1955, 14, 828.
18. Ш н а й д м а н Л. О. Производство витаминов. M., Пищепромиздат, 1958, с. 174.
19. S е b г е 1 1 W., Harris R., The Vitamins, Acad. Press II, 1954, 132.
313
20. H u b е г a W., Barlow О., J. Biol. Chem., 1943, 149, 125.
21. U. S. P. XV, 1955.
22. Windaus A., Werder F., Lflttringhaus A., Ann., 1932, 499, 188.
23. V e 1 1 u z L., Petit A., Michel G., R onssau G., Compt. rend., 1948,
226, 1287.
24. V e 1 1 u z L., Petit A., A m i a r d G., Bull. Soc. Chim. France, 1948, 15, 1115,
1949, 16, 501. ' '
25. V e 1 1 u z L., A m i a r d G., G о f f i n e t B., Compt. rend., 1955, 240, 2326,
26. V e 1 1 u z L., Gaston A., G о f f i n e t B., Bull. Soc. Chim. France, 1955,
1341.
27. H e s s A., Anderson W., J. Am. Med. Assoc., 1927, 89, 1222.
28. Bunker J., Harris R., Mosher L., J. Am. Chem. Soc., 1940, 62, 1760.
29. К n u d s e n A., Benford T., J. Biol. Chem., 1938, 124, 287.
30. В e н д т В. П. О применении новых источников ультрафиолетового излучения
для фотохимического превращения 7-дегидрохолестерина в витамин D3.— В сб:.
«Витамины», Киев, Изд-во АН УССР, 1958, 3, с. 44—49.
31. Н е i 1 b о г п I., Kamm Е., Morton R., Nature., 1927, 120, 617.
32. W i n d a u s A., Dent. med. Wochschr., 1931, 57, 678.
33. Reenrink E„ Wiyk V., Biochem. J., 1929,23, 1294.
34. I. G. Farbenindustrie, O. Linsert. Герм. пат. 565900, 7/XII, 1932.
35. S p e r t i G., Norris R., Withrow R., Schneider H. Пат.
США 1982029, 27/XI 1934.
36. P h i 1 i p s- N. Англ. пат. 385626, 20/XII 1932.
37. В i 1 1 s C., Honeywell E., Cox W., J. Biol. Chem., 1931, 92, 601.
38. Beard H., et ol., J. Biol. Chem., 1932, 96, 37.
39. M а н к о С,- C. — В кн.: Л. О. Шнайдман. Производство витаминов. М.,
Пищепромиздат, 1958, с. 187.
40. W е b s t е г Т., В о n г d i 1 1 о п R., Biochem., J., 1928, 22, 1223.
41. D a s 1 е г W., Summaries of Doctoral. Univ. Wisconsin, 1938, 3, 219.
42. Вадимов В. M. Биологический и спектрографический методы исследования
препаратов витамина'D3, М., Пищепромиздат, 1946, 63с. с ил.
43. Шнайдман Л. О. Производство витаминов из растительного и животного
сырья. М., Пищепромиздат, 1950, 323с.
44. В е н д т В. П. Беспозвоночные как источник витаминов группы D. — В сб.:
«Витамины», Киев, Изд-во АН УССР, 1953, № 1, с. 106—124 с ил.
45. W i n d a u s A., D е р р г е М., Wunderlich W., Ann. Chem., 1937, 532,
2, 118.
46. Van-der-Vliet, Chem. Weekblad, 1948, 44, 48, 692.
47. Гольцева P. Ученые записки Московского педиатрического института им.
Ленина, 1948, 53, 6, 9.
48. В е н д т В. П., Белявская В. В., Лопотько Н. В. Синтез 7-дегид-
рохолестерина. — В сб.: «Витамины», Киев, Изд-во АН УССР, 1958, № 3, с. 32—
43 с ил.
49. Я х и м о в и ч Р. И., Гируштин Г. Г., Яворский Я- 3. Пути син-
теза провитамина D3. — В сб.: «Материалы совещания по химии и биохимии' вита-
минов D и их применение в медицине и животноводстве», Киев, «Наукова думка»,
1968, с. 42—43.
50. W i п d a u s A., Schenck F. Пат. США 2098984, 16/XI 1937.
51. F i е s е г L. и др. J. Am. Chem. Soc., 1949, 76, 6, 2226.
52. Haslewood G., J. Chem. Soc., 1938, 224; Biochem. J., 1939, 33, 454.
53. Wintersteiner O., Ruigh W., J. Am. Chem. Soc., 1942, 64, 1177, 2453.
Пат. США 2411177; С. A., 1947, 41, 1398.
54. Ф и з e p Л., Ф и з e p М. Химия природных соединений фенантренового ряда.
М., Госхимиздат, 1953.
55. Buisman J., Stevens W., Van-der-Vliet J., Rec. Trav. Chim.,
1947, 66, 1—2, 83.
56. В i d e A., Henbest H. и др., J. Chem., Soc., 1948, 1783.
57. В e r n s t e i n S. и др. J. Org. Chem., 1949, 14, 3, 433.
58. Schaltegger H., Helv. Chim. Acta., 1950, 33, 2101.
59. Гируштин Г. Г., Яхимович Р. И. Исследование процесса бромиро-
вания эфиров холестерина. — В сб.: «Материалы совещания химии и биохимии ви-
таминов D и их применение в медицине и животноводстве», Киев, «Наукова думка»,
1968, 12.
60. W а п d е г А. Швейц, пат. 278937; С. А., 1953, 47, 4923.
61. S t г a t i п g J. и др. Nutr. Abstr., 1950, 3, 581.
62. Пат. США 2542241; 2546787; С. А., 1951, 45, 7606.
63. S о b е 1 V. и др., J. Am. Chem. Soc., 1949, 71, 1887.
64. Vogel Н., Knobloch Н., Chemie und Technik Vitam. Stuttgart, 1950.
65. T p о ф и м о в а С. И. Получение медицинского препарата видеина-3 в полупро-
изводственных условиях. — В сб.: «Материалы совещания химии, биохимии вита-
минов D и их применение в медицине и животноводстве», Киев, «Наукова думка»,
1968, 39.
314
66. Говсеева Н. Там же, 1968, 13.
67. К У Ц е н о к Я. Там же, 1968, 18.
68. Лукьянова Е., Вендт В., Хохол И., Навроцкая Г. Там же,
1968, 25.
69. Ш к н р я к 3. Там же, 1968, стр. 40.
70. В е н д т В. П., Дрокова И. Г. Искусственные комплексы белков с витами-
нами A, D2, D3 и Е, их получение и некоторые свойства. — В сб.: «Витамины», Ки-
ев, Изд-во АН УССР, 1956, № 2, с. 25—29 с ил.
Глава 14. ПРОИЗВОДСТВО СИНТЕТИЧЕСКОГО ВИТАМИНА Е
(а-ТОКОФЕРИЛАЦЕТАТА)
Еще в 1920 г. было замечено [1] бесплодие крыс, содержавшихся на
одном цельном молоке. Работы Иванса и Шура [2, 3] в этом направлении
привели к мысли о существовании витамина воспроизводства, предохра-
няющего мужские и женские особи от бесплодия и нарушений функции
размножения. Этот биологический фактор был назван витамином Е. Первые
высокоактивные концентраты витамина Е были получены из пшеничных
зародышей еще в 1925 г. [4]. В 1939 г. В. Девятнин и В. Иосикова [5] раз-
работали схему получения высокоактивных концентратов витамина Е из
пшеничных зародышей экстракцией органическим растворителем. В 1936 г.
Иване и Эмерсон выделили в виде кристаллических веществ эфиры а- и
[3-токоферолов [6]. В 1937—1938 гг. Фернгольц изучил химическую струк-
туру а-токоферола [7]. В 1938 г. Каррер с сотрудниками синтезировали
ОД-а-токоферол [8].
Физиологическое действие витамина Е [91 обусловлено его участием в
окислительно-восстановительных реакциях организма. Это действие, по-
видимому, сводится к задержке процессов окисления и интенсификации
обмена кислорода в тканях. а-Токоферол является антикоагулянтом и пре-
дотвращает коагуляцию крови в сосудах; предохраняет витамин А и нена-
сыщенные жирные кислоты от окисления.
Наиболее чувствительны к недостатку витамина Е половые органы, при
этом у самок это влечет за собой нарушение процесса беременности [10],
а у самцов — нарушение сперматогенеза и способности сперматозоидов к
оплодотворению. В связи с этим витамин Е называют антистерильным вита-
мином. Имеются указания на успешное применение витамина Е при лече-
нии серьезных сердечных заболеваний [11], при сосудистых расстройствах
(хронические язвы, тромбофлебиты, ранние гангрены конечностей, ожоги)
[И—14]; при лечении туберкулеза, воспалении мочевого пузыря и др. [15].
При хроническом Е-авитаминозе развивается мышечная дистрофия. Весьма
важна роль витамина Е в животноводстве [16]. На симпозиуме по витамину
Е в Японии [16а] были приведены данные о взаимосвязи витамина Е и
старения организма, а также взаимосвязи с заболеванием печени.
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ВИТАМИНА Е
Токоферолы (греч. «потомство несу») являются производными хромана
(бензо-у-дигидропирана). По данным Фернгольца [7], изучившего строе-
ние молекулы витамина Е, а также Каррера, синтезировавшего его, в осно-
ве токоферолов лежит ядро хромана, в котором водороды при С в положе-
нии 2 замещены метильной группой и фитольным радикалом, а в положе-
нии 6 гидроксильной группой
Токол
315
В зависимости от степени метилирования бензольного цикла различают
7 изомерных форм токоферола, а именно:
5,7,8- триметилтокол 5,8- диметилтокол
у - токоферол t, - токоферол
7,8- диметилтокол 5,7- диметилтокол
Все указанные изомеры выделены из природных источников [17].
Изомеры токоферола представляют собой вязкие масла, не кристаллизующие-
Рис. 51. УФ-спектры а- и [3-токо-
феролов:
1 — а-токоферол; 2 — (3-токоферол.
ся. Лишь эфиры токоферолов (алло-
фанаты, бензоаты, пальмитаты) полу-
чают в кристаллическом виде. Токо-
феролы неустойчивы к окислителям:
в особенности легко окисляются! б- и
у-изомеры, в связи с чем и являются
хорошими антиоксидантами. В отсут-
ствие кислорода токоферолы весьма
стабильны даже при температуре 200°
С, а также к действию минеральных
кислот при температуре 100° С.
С едкими щелочами реагируют медлен-
но и при омылении растительных
объектов не разрушаются [18]. Рас-
творимы в жирах и органических
растворителях.
Наиболее физиологически актив-
ной является природная правовра-
щающая форма. Синтетический
D,L-а-токоферол обладает70—75% ее
активности; |3-токоферол— 40—50%,
у-токоферол— 8% [19].
а-Токоферол, С29Н50О2, молекуляр-
ная масса 430,69; плотность 953 кг!м9\
для Д,7.-а-токоферола: d|0—0,947—0,958; п®— 1,5030—1,5070, темпе-
ратура кийения (при остаточном давлении 0,01 мм рт. ст.) 225—230° С,
йодное число 143; Хтах— 292—295 нм, Ei/м— 71—77 (рис. 51).
Ацетат П,Л-а-токоферола, С31Н62О3, температура кипения 205—208° С
при остаточном давлении 0,3 мм рт. cm.: di — 0,954—0,966; Пд 1,4958—
1,4972; Хтах =285 нм-, Е\ — 42,5—43,5; вязкое масло.
316
Аллофанат (эфир циановой кислоты — HNCO) температура плавления
172° С; 2,4-динитробензоат, температура плавления 63° С.
В молекуле токоферолов имеются три асимметрических атома углерода
в положениях 2,4' и 8'. Следовательно, для каждого токоферола возможно
восемь оптических изомеров и четыре рацемата.
ВЫБОР РАЦИОНАЛЬНОГО МЕТОДА СИНТЕЗА ДЛЯ ПРОИЗВОДСТВА
В связи с тем что из семи известных изомерных форм а-токоферол обла-
дает наивысшей активностью, вопрос о его синтезе стал первоочередным.
Химическое строение молекулы а-токоферола предопределило и путь син-
теза его из триметилгидрохинона и фитола. Действительно, в 1938 г. почти
одновременно был синтезирован а-токоферол различными исследователями
методом конденсации триметилгидрохинона с фитолом [221 или с фитил-
бромидом [8] или с фитадиеном [23].
Рассмотрим наиболее эффективные пути синтеза триметилгидрохинона,
фитола, а также технологию их конденсации и очистки целевого продукта.
Синтез триметилгидрохинона, 2,3,5-триметилгидрохинон может быть
получен:
а) из псевдокумидина Д,2,4-триметил-5-аминобензол) путем ацилиро-
вания, нитрования, омыления, диазотирования, восстановления динитро-
производного до n-диаминопсевдокумола, окисления до псевдокумохинона
и восстановления последнего [24];
б)1 проще получение триметилгидрохинона из псевдокумола методом
бромирования (или сульфирования), нитрования, восстановления, окисли-
тельного замещения аминогрупп на оксогруппы и их восстановления по
следующей химической схеме [24—26] :
л - Диаминопсевдокумол Псевдокумсхинон
Триметил -п-гидрохинон
г) еще проще получение триметилгидрохинона из нитромезителена [27]
путем избирательного восстановления в мезитилгидроксиламин и превра-
щения его в триметилгидрохинон по реакции Бамбергера — перегруппиров-
ка хинолов в соответствующие гидрохиноны [28] по следующей химиче-
ской схеме:
Нитромезителен
Мезитилгидроксиламин С Н3
Триметилгидрохинон
1 По японскому, патенту [24а] триметилгидрохинон получают из 3,6-диметил-
4-нитрофенола через 3,6-диметил-2-хлорметил-4-нитрофенол, восстанавливаемый в
2,3,6-триметил-4-аминофенол, окисляемый FeCl3 в 2,3,6-триметилгидрохинон.
317
Из изложенного видно, что наиболее простым и эффективным по выхо-
дам полупродуктов является метод синтеза триметилгидрохинона из нитро-
мезителена или из мезитилгидроксиламина [27].
Синтез фитола. Фитол, второй компонент молекулы токоферолов, может
быть получен различными методами. Однако все они являются весьма слож-
ными. Впервые фитол, был синтезирован Фишером и Левенбергом в 1929 г.
[29] из псевдоионона через тетрагидрофарнезол, кетон С18Нз4О, кетон
С]8Н36О, С2о — ацетиленкарбинол и С2о — этиленкарбинол. Синтез вклю-
чает 11 стадий и вследствие своей сложности не мог найти практического
применения. В 1939 г. фитол был синтезирован Каррером и Ранжье [30]
также из псевдоионона через гексагидрофарнезол и кетон С18Н36О с полу-
чением бромпроизводного фитола — фитилбромида. Этот метод состоит из
10 стадий и так же весьма сложен.
Вслед за этим в 1943 г. Смитз [30а] показал возможность замены в син-
тезе а-токоферола фитола на изофитол, что упрощает синтез фитольного
компонента. По методу Смитза при наличии псевдоионона, бромистого-
3-этоксипропила и 4-хлор-2-бутанона может быть осуществлен синтез хлор-
гидрата изофитола в четыре стадии по следующей химической схеме
(см. ниже). Выход хлоргидрата изофитола составляет 10% (на псевдоионон)
[21]. Необходимо, однако, отметить, что для синтеза указанных трех про-
межуточных продуктов потребуется дополнительно ряд стадий, что в ко-
нечном итоге также усложняет метод Смитза. По-видимому, преимущест-
вом будут обладать те методы синтеза изофитола, в которых многократно
повторяются однотипные реакции. К ним относятся метод Сарычевой [31],
Излера [32] (синтез изофитола из псевдоионона) и метод Маурита и др.
[33] (синтез изофитола из ацетона и ацетилена).
° + BrCH2-CH2- CH2OC2Hg-—।
Псейдоионон Бромистый 3'-этоксипролил
Л ос2н5 >-
Вг
,oc2hs г
[Н] izs’
Ni нош/ш*
II — I I + СН3СОСН2СН2С1-----------►
Зтилодый э/pup Р,3р2~триметилтрид5лаи 3,8р2-Триметил-тридраан- 1-дром З-Хлор-З-бутанан
НгС1
он
Хлоргидрат иза/ритола
Псевдоионон в 6 стадий наращивают до изофитола [31, 32] при много-
кратном повторении однотипных реакций конденсации кетонов с ацетиле-
ном, частичном гидрировании тройных связей ацетиленовых спиртов до
двойных, кроме того, применяется конденсация третичного ненасыщенного
спирта с дикетеном или ацетоуксусным эфиром с последующей перегруппи-
ровкой [34].
Синтез осуществляют по следующей химической схеме:
318
В большинстве описанных в литературе методов исходным сырьем для
'синтеза фитола или изофитола применяют псевдоионон или цитраль или
линалоол. М. Маурит и др. в 1961 г. опубликовали синтез изофитола, ис-
ходя из ацетона и ацетилена [33]. В основу синтеза положены две последо-
вательно повторяющиеся реакции наращивания цепи: 1) на два атома уг-
лерода — путем конденсации кетонов с ацетиленом в среде ксилола, толуо-
ла и др. в присутствии едкого калия; 2) на три атома углерода — путем
получения ненасыщенных кетонов и с их конденсацией с ацетоном. Синтез
осуществляют по следующей химической схеме:
сн3 сн, сн, г , сн, сн,
он
ЗгМетилбутин- 2 - Метилеептадиен-2,и - 2-Мети л гептанон-6
з-ол-г -он—6
сн3 сн3 сн3 СН, СН, г . ,
—— сн3~сн_(сн2)3—с-с=сн СН3-СН-(СН2)3-С=СН-СН=СН-СО
он
2,6 - Диистилолтин - 2,6 -Диме/пилундекадиен -
-7-ол-В -6,8-он-10
СН3 СН3 нг=г СН3 СН3
—►сн3-(сн-сн2-сн2-сн2)2-со - сн3(сн-сн2-сн2-сн2)2-с-с=сн "Н^°5'Нз>
он
2,6-Диметилундвканон -10 2,6,10 ~ Тримвтилдодеиан -
-Ц-ОЛ-10
—►СН3(СН-СН2-СН2-СН2)2-С‘=СН-СН=СН-СО-И^-Ч:Н3(СН-СН2-СН2-СН2)3-СО
2,6, 10 - Гримв/пилпентаДекадивн - 2,6, /О - ГримвтилрентаОвнанон -14
~10Д2-он-/4
СН3 СН3 г . СН3 сн3
'—►сн3(сн-сн2-сн2-сн2)3-с-свсн зд^ » сн3(сн-сн2-сн2-сн2)3-с-сн=сн2
он ' ОН
2,6, Ю, 14 - Тетраметилгенсадецин- 2,6,10,74 -Гетраметил8ексадецен-15-ол-14
-15-ол~14 % (Изофитол)
Из изложенного видно, что наиболее простым и удобным является син-
тез изофитола из псевдоионона, а еще лучше из линаллоола по методу Са-
рычевой, Преображенского и др. [31 ]. При дефицитности псевдоионона
синтез изофитола из ацетона и ацетилена по методу Маурит, Преображен-
ского и др. может оказаться эффективным, несмотря на его сравнительную-
сложность.
Фитол из растений. Распространен в природе как компонент молекулы
хлорофилла. Последний сосредоточен в пластидах растительной клетки,
в которых содержание его достигает 7% [35,36]. Для получения фитола
следует применять листья растений с повышенным содержанием хлорофил-
ла. К таким растениям относятся крапива [21], мята [37—39], отходы
чайных листьев [40]. Кроме того, предложен метод получения фитола из
отходов шелковичного червя [Букин, Великославинская). Химическая
схема получения фитола из хлорофилла может быть представлена в следую-
щем виде:
/
СООСН3 [н+] СООСН3 кон
[MgN4C32H30O] -----> [N4C32H32O] ----->
Хлорофилл соос20н39 соос20н39
* [N4C31H33] (СООК)2 + С29н39он + СН3ОН
Фитол
319
Разработаны различные методы выделения фитола из растительных
объектов. Все они сводятся к процессам экстракции хлорофилла, включая
образующийся в процессе сушки феофитин, органическим растворите-
лем, отгонка последнего, омыление кубового остатка и дистилляция неомы-
ляемой части в глубоком вакууме. В качестве растворителя предложен
ацетон и исходное сырье — листья крапивы [41], изопропиловый спирт и
отходы перечной мяты [37—39]. Выход фитола из растительных объектов
колеблется в пределах 0,11—0,30% к массе сухого сырья. Фитол,
СгоНддОН, молекулярная масса 296,52; dl°— 0,852; температура кипения
150—151° С (остаточное давление 0,06 мм рт. ст.), i?d =1,4637; ?.тах =
=212 нм (спирт); хорошо растворяется в органических растворителях.
Технология конденсации триметилгидрохинона с фитолом. Конденса-
ция триметилгидрохипопа и изофитола изучалась различными исследова-
телями. По Карреру [8] реакцию конденсации триметилгидрохинона с фи-
тилбромидом проводят в среде безводного бензина в токе азота в присутст-
вии катализатора хлористого цинка. По Швейцарским патентам фирмы
«Ля-Рош» конденсацию проводят в присутствии этилата натрия, либо NaOH,
КОН. Берджел [22] считает рациональным проводить конденсацию триме-
тилгидрохинона не с фитилбромидом, а с фитолом в присутствии в качестве
конденсирующего агента муравьиной кислоты. Смитз [23] применял смесь
2,6,10- Триметилпентаденанон-1Ц
Рис. 52. Технологическая схема производства
320
муравьиной и уксусной кислоты (5:2). Маурит, Преображенский и др. [33]
предлагают сочетать реакцию конденсации с реакцией этерификации. Для
этого они рекомендуют конденсацию компонентов проводить в среде уксус-
ной кислоты в присутствии эфира трехфтористого бора и хлористого цинка.
В результате получают ацетат токоферола с выходом в 69,5%. По-видимому,
последний метод является наиболее эффективным.
Из анализа изложенного выше можно сделать вывод о целесообразности
построения схемы синтеза токоферилацетата в следующем виде;
синтез 2,3,5-триметилгидрохинона из мезитил гидроксиламина по
методу Крафта и Цыгановой [27];
синтез изофитола по методу Сарычевой, Преображенского и др. [31 ].
Конденсацию компонентов необходимо сочетать с этерификацией а-токо-
ферола по методу Маурит, Преображенского и др. [33].
Необходимо также подчеркнуть, что при небольшом объеме производ-
ства а-токоферилацетата может оказаться более эффективным применение
фитола из растений, чем изофитола синтетического.
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА СИНТЕТИЧЕСКОГО а-ТОКОФЕ-
РИЛАЦЕТАТА
Синтез включает получение 2,3,5-триметилгидрохинона и изофитола и
их конденсацию. Метод содержит следующие стадии (рис. 52):
2,6,10- ТриметиМехаЗиен-?,6-ИН-11-ол-ю(Зегидронероли&ол) 2,6,10-Триметилпетадекагг1етраен-!,Б,11},12-ОН-1Ч
2,6,10,1^- Тетраметилгексабеиин-15-ОЛ-К,
(ацетиленобый третичный карбинол Сго)
Мн поз 33
синтетического а-токоферилацетата.
11^522
321
синтез 2,3,5-триметилгидрохинона (I);
синтез геранилацетона (II) из линалоола;
синтез дегидронеролидола (III) из геранилацетона;
синтез 2,6,10-триметилпентадекатетраен-2,6,10,12-он-14 (IV) из де-
гидронеролидола;
синтез 2,6,10-триметилпентадеканона-14 (V) из (IV);
синтез 2,6.10,14-тетраметилгексадецин-15-ол-14 (VI) из (V);
синтез 2,6,10,14-тетраметилгексадецин-15-ол-14 (VII) из (VI);
синтез изофитола, 2,6,10,14-тетраметилгексадецен-15-ол-14 из VII.
Синтез 2,3,5- триметилгидрохинона осуществляют по
методу Крафта [27) (см. стр. 317). Синтез изофитола осуществляют по ме-
тоду Сарычевой и др. [31 ].
Синтез 2,6- диметилундекадиен -2,6-он-10 (геранил-
ацетона) (II) осуществляют по химической схеме:
СН3 СН3
I I
сн3—с=сн—-сн2—сн2—с—сн=сн2
I
он
2,6- Диметилоктадиен-2,7-ол-6,
линалоол (I) 154,24
СН3СОСН2СООС2Н5
———-------------->
Азот, 160—170°
Ацетоуксусный эфир
130,14
сн3 сн3
I I
сн3—с=сн—сн2—сн2-с—сн=сн2 ----------->
I 55,6%
ОСОСН2СОСН3
Ацетоацетат линалоола 238,32
сн3 сн3
сн3—с=сн—сн2—сн2—с=сн—сн2—сн2—сосн3
2,6-Диметилундекадиеи-2,6-он-10, геранилацетон (II)
194.31
t В реактор 12 загружают 0,194 г-моль линалоола из мерника 13 и
0,68 г-моль ацетоуксусного эфира из мерника 14, нагревают в токе азота
(из баллона 15) при температуре 160—170° С (около 50 ч). Затем от реакци-
онной массы отгоняют непрореагировавший ацетоуксусный эфир в сбор-
ник. В вакуум-перегонном аппарате 16 и при остаточном давлении
7 мм рт. ст. и температуре 118—120° С отгоняют геранилацетон (II). Вы-
ход 55,6%; температура кипения 118—120° С (при остаточном давлении
7 мм рт. cm.), dl°=0,8801; Пд =1,4689, С13Н22О, молекулярная масса
194,31.
Синтез 2,6,10- триметилдодекадие н-2,6-ин-11-ол-Ю,
дегидронеролидола (III) осуществляют по химической схеме:
СН3 СН3
I I сн=сн
СН3—С=СН—сн2—сн2—с=сн—сн2—сн2—сосн3---------->
Na
Геранилацетон
194,31
сн3 сн3 сн3
1'1 I
—>• сн3—с=сн—сн2—сн2—с=сн—сн2—сн2—с—с=сн
он
2, 6, 10’Тримзтилдодекадчгн-2,6-ин-11-ол-10
Дэгидронеролидол (III) 220,34
В реактор 17 из нержавеющей стали, снабженный мешалкой, барботе-
ром и рубашкой, холодильником и приемниками, из баллона 18 загружают
жидкий безводный аммиак и пропускают быстрый ток сухого ацетилена
из баллона 19. Затем при перемешивании вводят металлический натрий
322
(1 г-атом) с такой скоростью, чтобы не возникало стойкого посинения реак-
ционной массы. Далее при уменьшенной скорости пропускания ацети-
лена в течение 40 мин из мерника 20 приливают геранилацетон (II) в ко-
личестве 0,52 г-моль, перемешивают 2 ч и выдерживают 16 ч при температуре
20° С. В реакторе 17 образуется густая масса желтого цвета, которую охлаж-
дают до —5° С и разлагают хлористым аммонием (порошком), подкисляют
50%-ной серной кислотой из мерника 22 до pH 6,5. Массу переводят в де-
лительную воронку 23, где происходит расслаивание; верхний органиче-
ский слой сливают в смеситель 24, где подсушивают его сернокислым нат-
рием, сливают в сборник 25 и отгоняют в вакуум-перегонном аппарате 26
аммиак, а затем при температуре 107—110° С (при остаточном давлении
2,5 мм рт. ст.) — полупродукт. Выход 70,9%. Дегидронеролидол,
С15н24о, молекулярная масса 220,34; di°=0,8894; Пд = 1,4790 (Обратить
внимание на технику безопасности).
Синтез 2,6,10- триметилпентадекатетрае н-2,6,10,
12-он-14 (IV)-k е т о н С18 осуществляют по химической схеме:
СН3 СН3
I | сн3сосн2соос2н5
СН3—(С=СН—СН2СН2)2—С—С-СН ----------------------->
| 165—170°
ОН
Дегидроиеролидол (III) Ацетоуксусный эфир
220,34 130,14
। 3 /СНз
—»• СН3—(С=СН СН2 СН2)2—С—С-СН —>
N)COCH2COCH3
Ацетоацетат геранилацетона
СНз
, I
СН3—(С=СНСН2СН2)3 СОСНз
2, 6, 10-Триметилпентадекатетраен-2, 6, 10, 12-он-Н, кетон CI8 (IV)
260,40
Из мерника 27 загружают в реактор 28 из нержавеющей стали, снабжен-
ный холодильником и приемником дегидронеролидол (III) 0,317 г-моль,
а из мерника 14 свежеперегнанный ацетоуксусный эфир 1,192 г-моль и на-
гревают в токе азота из баллона 29 при температуре 165—170° С в течение
55 ч. При этом выделяется углекислый газ. Затем отгоняют непрореагиро-
вавший ацетоуксусный эфир. Кубовый остаток сливают в вакуум-перегон-
ный аппарат 30, в котором при температуре 110—111° С (при остаточном
давлении 0,3 мм рт. ст.), 125—127° С (при остаточном давлении
0,6 лии рт. ст.) отгоняют кетон С18. Выход 54,3%; С18Н28О; молекулярная
масса 260,40; Д;о=О,9114; пЬ°= 1,4951.
Синтез 2,6,10- триметилпентадеканон-14 (V) осуще-
ствляют по химической схеме:
СНз г СН3 -1
I [Н] I
СНз—(С=СНСН2СН2)з СОСНз-------> СН3—(СН—СН2 СН2 СН2)з СН—СНз
Ni I
он
Кетон СХ8 2,6,10-Триметилпеитадсканол-14
260,40 спирт С18
СН3
Ni; СН3СОСН3; H2SO4 |
---------------> СНз—(СНСН2СН2СН2)з СОСНз
К2СГ2О7
2,6,10-Триметилпеитадеканон-14 (V)
268,47
В автоклав 31 из мерника 32 загружают кетон С18 (IV), из мерника 33
спирт и скелетный никелевый катализатор (7—10%) и из баллона 34 проду-
П* 323
вают азот, а затем пропускают водород из баллона 35 (избыточное давление
90 кгс/см2) в течение 2 ч. Затем отфильтровывают катализатор на нутч-
фильтре 36 и через сборник 37 фильтрат поступает в перегонный аппарат 38
и отгоняют спирт. Кубовый остаток сливают в реактор 39, где спирт С18
окисляют хромпиком в присутствии ледяной уксусной кислоты, подавае-
мой из мерника 40, и азота из баллона 34. Затем приливают серную кислоту
из мерника 22. Для завершения реакции массу нагревают в течение 50 мин
до 80—85° С. Далее добавляют из мерника 41 дихлорэтан, перемешивают
и сливают в делительную воронку 42. Нижний слой отделяют в сборник 43,
в вакуум-перегонном аппарате 44 вначале отгоняют дихлорэтан, затем при
температуре 110—113° С (остаточном давлении 0,2 мм рт. ст.) гидриро-
ванный кетон С ]8. Выход 69,3%; С18Н36О, молекулярная масса 268,47;
d2°=0,8740; п2д= 1,4652. •
Синтез 2,6,10,14- тетраметилгексадеци н-15-ол-14,
ацетиленовый третичный карбинола С2о осуществляют
по следующей химической схеме:
СН3 СН3 СН3
I СН=СН | I
СН3—(СНСН2СН2СН2)3СОСН3----------> СН3— (СНСН2СН2СН2)3—с—с-сн
Na; NH3 [
он
2,6,10’Триметилпентадеканой'1‘1 2.6,10, 14-Тетрамзтилгексадецин-15-ол-14,
268,47 третичный ацетиленовый карбинол С20 (VI)
294,50
В реактор 45 из нержавеющей стали, снабженный мешалкой, рубашкой
для рассола, барботером, холодильником и приемниками, загружают из
баллона 46 аммиак, пропускают ток ацетилена из баллона 47 и при
перемешивании добавляют металлический натрий (0,08 г-атом). Затем в
течение 40 мин из мерника 48 приливают 2,6,10-триметилпентадеканон-14
(0,037 г-мол). Массу перемешивают 1 ч и выдерживают 15 ч при температуре
20° С> Затем из сборника 21 добавляют хлористый аммоний (кристалличе-
ский) для разложения реакционной массы и подкисляют серной кислотой
из мерника 22 до pH 6,5. Реакционную массу сливают в делительную во-
ронку 49. Верхний органический слой сливают в сборник 50, откуда его за-
сасывают в вакуум-перегонный аппарат 51. Вначале отгоняют аммиак. Затем
при температуре при 119—121° С (остаточном давлении 0,2 мм рт. ст)
отгоняют карбинол С2о. Выход 53,6%; С2оН380, молекулярная масса 294,50;
d4°=0,8798; «д = 1,4698.
Синтез 2,6,10,14-т етраметилгексадеце н-15-ол-14,
изофитола осуществляют по следующей химической схеме:
СН3 СН3
I I
СН3—(СНСН2 СН2 СН2)3—с—с = сн
I
он
[Н]
------------>
Pd, хинолин
I 3 /СН3
СН3—(СНСН2СН2СН2)3 С—СН=СН2
ХОН
Третичный ацетиленовый карбинол С20
294,5
Изофитол (VII)
296,52
В автоклав 52 загружают 2,6,10,14-тетраметилгексадецин-15-ол-14 и
спирт из мерника 33, продувают азотом из баллона 53 и гидрируют водоро-
дом из баллона 54 в присутствии катализатора Линдлера [42] (палладиевый
катализатор, отравленный хинолином). Катализатор отфильтровывают на
нутч-фильтре 55. Фильтрат через сборник 56 засасывают в вакуум-перегон-
ный аппарат 57, отгоняют спирт; затем перегоняют изофитол при темпера-
туре 114—115° С (остаточном давлении 0,4 мм рт. ст.) или 170—175° С
(остаточное давление 5 мм рт. ст.) в сборник 58. Выход 84,4%. Изофитол,
С20Н40О, молекулярная масса 296,52, бесцветная жидкость, Пл — 1,4718;
Й4°=0,8680.
324
Синтез ацетата D,L-a-4 окоферола осуществляют путем
конденсации фитилхлорида с 2,3,5-триметилгидрохиноном в среде уксус-
ной кислоты в присутствии эфирата трехфтористого бора и хлористого цин-
ка по следующей химической схеме:
СН3 СН3
I I НС1
СН3—(СНСН2СН2СН2)3—с—сн=сн2------>
он
Изофитол
296,52 35,47
сн3 сн3
I I
—»• СН3—(СНСН2 СН2 СН2)3—С=СН—СН2 С1 +
Фитилхлорид
315,00
СН3СОО
Н3С
сн3
СН3
(сн2-сн2-сн2-сн)3сн3
2Н3
D, L — сб—токоферилацетатп
(сн3со)2о
Эфирам
BF3 a ZflCl2
472, 42
юг, оз
В реактор 59 из эмалированной стали из мерника 60 загружают изофи-
тол, а из мерника 61 концентрированную соляную кислоту и перемешивают
при температуре 20° С. Затем добавляют воду, перемешивают, сливают в
делительную воронку 62\ верхний слой направляют в аппарат-смеситель
63, где фитилхлорид промывают до нейтральной реакции и сливают в сбор-
ник 64, а из последнего в мерник 65.
В реактор 66 из эмалированной стали, снабженный рубашкой для обо-
грева паром и охлаждения водой, холодильником и приемниками, загру-
жают из мерника 67 уксусную кислоту, из мерника 65 фитилхлорид,
из вакуум-сушилки 11 триметилгидрохинон и катализатор хлористый цинк
и эфират трехфтористого бора, нагревают в присутствии азота (из баллона
68) в течение 3—4 ч при температуре 120—122° С. После охлаждения водой
сливают в делительную воронку 69, где промывают водой, отделяют
верхний слой, который направляют в реактор 66, добавляют из мерника 70
уксусного ангидрида и при температуре 120° С нагревают 2 ч с отгонкой
образующейся уксусной кислоты. Кубовый остаток разгоняют в сборник
71 и отбирают фракцию при температуре 205—208° С (остаточном давлении
0,3 мм рт. ст.). Выход 69,5% (на изофитол).
Ацетат витамина Е — густое, вязкое масло светло-желтого цвета, рас-
творимое в органических растворителях. С31Н52О3, молекулярная мас-
са 472,42; dlo=O,9773; Пд = 1,5021; Е}”^=43,35 при Хтах=285 нм.
325
ТЕХНОЛОГИЧЕСКИЕ ПОКАЗАТЕЛИ
РАСХОД ХИМИКАЛИЕВ, КГ НА 1 КГ
100 %-НОГО ПОЛУПРОДУКТА
2,3,5-Триметилгидрохинон
Мезитилгидроксиламин ............1,75
Сода кальцинированная .......... 5,0
Серная кислота ................. 17,0
Уголь активированный............0,12
Натр едкий ..................... 1,0
Бисульфит натрия.................0,2
Вазелиновое масло .............. 8,3
Выход, % (на мезитилгидрокси-
ламин)..........................60,0
2,6-Диметилундекадиен-2,6-он-10, гера-
нилацетон
Линалоол ....................... 1,4
Ацетоуксусный эфир .............2,0
Выход (на линалоол), % ... . 55,6
2,6,10-Триметилдодекадиен-2,6-ин-11 -
ол-10, дегидронеролидол
Натрий сернокислый . , ... 2,0
Выход, % (на гидрированный ке-
тон Си) .......................53,6
Изофитол
Третичный ацетиленовый карбинол 1,2
Спирт (с регенерацией)..........2,0
Палладий хлористый . . . . • . 0,003
Хинолин.....................0,003
Водород .......................0,005
D, А-а-ТОКОФЕРИЛАЦЕТАТ
Изофитол ......................0,90
Триметилгидрохинон ............ 0,46
Кислота соляная ............... 2,00
Кислота уксусная (с регенерацией) 0,50
Цинк хлористый ................0,05
Эфират трехфтористого бора . . . 0,03
Ангидрид уксусный............0,70
Выход, % (на изофитол) . . . .69,50
Геранилацетон ................. 1,25
Аммиак жидкий .................7,5
Ацетилен.......................0,5
Натрий металлический...........0,3
Натрий сернокислый ............2,0
Кислота серная.................5,0
Выход, % (на геранилацетон) . . 70,9
2,6-10-Т риметилпентадекатетраен-
2,6,10,12-он-14 (ненасыщенный кетон Ci8)
Расход полупродуктов, кг на 1 кг D,L-
а-токоферилацетата
Триметилгидрохинон ............ 0,46
Третичный ацетиленовый карбинол 1,08
Гидрированный кетон Ci8 .... 1,84
Ненасыщенный кетон С18 .... 4,78
Дегидронеролидол................7,65
Геранилацетон ................. 9,56
Дегидронеролидол ...............1,6
Ацетоуксусный эфир (с регенера-
цией) ......................... 1,2
Расход химикалиев, кг на синтез 1 кг
Выход, % (на дегидронеролидол) 54,3
2,6,10-Триметилпентадеканон-14 (ги-
дрированный кетон С18)
Ненасыщенный кетон Ci8 .... 2,6
Никелевыц.катализатор скелетный 0,3
Спирт (с регенерацией)...........0,6
Хромпик.......................... 1,1
Кислота уксусная ледяная .... 3,0
Кислота серная (плотность
1840 кг/м3)..................... 1,0
Натрий сернокислый ..............2,0
Водород ..........................0,03
Выход, % (на ненасыщенный ке-
тон С18) .......................69,3
2,6,10,14-Тетраметилгексадецин-
15-0Л-14 (Ацетиленовый третичный кар-
бинол)
Гидрированный кетон Ci8 .... 1,7
Натрий металлический............0,3
Ацетилен .......................0,3
Аммиак жидкий (с частичной ре-
генерацией) .....................7,0
Кислота серная (плотность
1840 кг/м3)..................... 1,0
D, А-а-токоферилацетата
Аммиак жидкий с частичной реге-
нерацией .......................13,3
Ангидрид уксусный...............0,7
Ацетилен .......................8,3
Водород ........................0,054
Катализатор никелевый..........0,55
Кислота соляная................2,0
Кислота уксусная...............6,02
Кислота серная.................49,60
Масло вазелиновое...............4,0
Мезитилгидроксиламин ...........0,8
Натр едкий .....................0,5
Натрий сернокислый ............21,14
Натрий металлический...........2,52
Натрий бисульфат...............0,10
Палладий хлористый ............0,003
Спирт ..........................2,9
Сода кальцинированная .......... 2,5
Уголь активированный............0,06
Двухромовокислый калий . . . .2,02
Хинолин ........................0,003
Эфир ацетоуксусный ..............5,74
Эфират трехфтористого бора . . . 0,03
Цинк хлористый ..................0,05
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. М a t i 1 1 Н., Conklin R., J. Biol. Chem., 1920, 44, 137.
2 Evans H., Bishop K-, Science, 1922, 56, 650.
3. Sure B„ J. Biol. Chem., 1923, 58, 681; 1925, 63, 211.
4. Evans H., Burr G., Science, 1925, 61, 519.
5. Д e в я т н и н В. А., И о с и к о в а В. М. Пищевая промышленность СССР,
о, с. а. . « , _
6 Evans Н., Emerson О., Emerson G., J. Biol. Chem., 1936, 113, 319.
7. Fernholz E. , J. Am. Chem. Soc., 1937, 59, 154; 1938, 60, 700.
8 Karrer P, Fritzsche H., Ringier B., Salomon H., Helv. Chim.
Acta, 1938, 21, 520, 820; Nature, 1937, 141, 1057.
9. Ш и л о в П. И. , Яковлев Т.Н. Основы клинической витаминологии.
М., «Медицина», 1964, с. 124.
10. Д е р а н к о в а Е. Акушерство и гинекология. М., Медгиз, 1954, 2.
326
11. Weitzel G., Schon H., Gey F., В u d e с к e F., Hoppe S. Z.
physiol. Chem., 1956, 304, 297.
12. Mason K., The Vitamins N. J., 1954, 652.
13. Frey I., Strahlentherapie, 1954, 95, 3, 440.
14. H ii t t e r E., Dtsch. Gesundheitswesen 1954, 9, 9, 227.
15. Д p о к о в a H. Г. Применение витамина Е в медицине. — В сб.: «Витамины.
Пищевая промышленность за рубежом», М. Пищепромиздат, 1958, № 4, с 115—
121.
16. Рекомендации по применению витаминных концентратов в животноводстве. М.,
«Колос», 1965, с. 6.
16а. Japan Med. gaz., 1968, 5, 6, 4; Ind. Journ. of Pharmacy, 1968, 30, 10, 236.
17. Б e p e з о в с к и й В. М. Химия витаминов. М., Пищепромиздат, 1959, с. 295.
18. Rosenberg Н., Chemistry and Physiology of the Vitamins, N. J., 1942, 435.
19. J о f f e M., Harris P., J. Am. Chem. Soc., 1943, 65, 925.
20. С а в и н о в Б. Г., Гринберг Ф. Л. Синтез витамина Е. — В сб.: «Вита-
мины. Пищевая промышленность за рубежом», М., Пищепромиздат, 1958, № 4,
с. 4—16.
21. С а в и н о в Б. Г., Гринберг Ф. Л., С в и щ у к А. А. Получение синте-
тического и природного фитола. — Там же, 1958, № 4, с. 17—35.
22. В е г g е 1 F., Jacobs A., Todd A., Work Т., Nature , 1938, 142, 36.
23. Smith L., U n g п a d е H., Prichard W., Science, 1938, 88, 37.
24. Smith L., J. Am. Chdm. Soc., 1934, 56, 472.
24a. Японский пат. № 10852, 1967; Витамин, 1967, 36, 2, 196.
25. Meyer R., Meyer W., Ber., dtsch.Chem. Ges., 1948, 51, 1571.
26. Г p и н б e p г Ф. Л. , Св и щу к А. А. Восстановление 5-бром-3,6-динитро-
псевдокумола в 3,6-диаминопсевдокумол. — «Украинский химический журнал»,
1957, 23, т. 79.
27. К р а ф т М. Я., Цыганова А., М. Авторское свидетельство, № 122152, 1958;
, Бюллетень изобретений, 1959, 17, 16.
28. В a m b е г g е г Е., Rising А., Вег., 1900, 33, 3623—3636; 1926, 59, 418.
29. Fischer F., Lowenberg К., Ann., 1929, 475, 183.
30. К а г г е г Р., R i n g i е г В., Helv. Chim. Acta., 1939, 22, 610.
30а. Smith L., S р г u п g J., J. Am. Chem. Soc., 1943, 65, 1276.
31. Сарычева И. К., Воробьева Г. А., Кузнецова Н. А. и др.
Новый синтез 2,6,10,14-тетраметилгексадецен-15-ола-14, изофитола. —ЖОХ,
1958, т. 28, с. 647—51.
32. Isler О., Angew. Chem., 1959, 71, 7.
33. М а у р и т М. Е., Смирнова Г. В., Парфенов Э. А., Сарыче-
ва И. К., Преображенский Н. А. Полный синтез витамина Е, а-токо-
ферола и его производных. — «ДАН СССР», 1961, т. 140, № 6, с. 1330—1333.
34. Б е р е з о в с к и й В. М. Современные задачи в области химии и технологии ви-
таминов и перспективы развития их производства. — «Медицинская промышлен-
ность СССР», 1960, № 5, с. 7—23.
35. Г о д н е в Т. Н., Осипова О. П. О природе связи хлорофилла и белка в
хлоропластах. — «ДАН СССР», 1947, т. 57, с. 161—64.
36. Euler Н., Ztschr., physiol. Chem., 1929, 188, 177.
37. М и х л и н Э. Д., Шахова М. Ф., Лукьянова Л. В. Препарат фито-
ла из отходов мяты перечной. — «Труды ВНИВИ», М., Пищепромиздат, 1961, № 8,
с. 57—65 с ил.
38. Ш а х о в а М. Ф., Ш найдман Л. О. Содержание биологически активных
веществ в производственных отходах мяты перечной. — «Растительные ресурсы»,
1968, 4, 1, с. 53—62 с ил.
39. К а л у г и н П. И., Шахова М. Ф. Авт. свидет., № 136855, 1959; Бюлл.
изобрет., 1961, № 6, с. 42.
40. А г а п о в а Е. В. Усовершенствование технологии переработки чайного листа
на витамины группы Р. — В сб.: «Витаминная промышленность», М., Пищепромиз-
дат, 1957, № 4, с. 45—53.
41. С в и щ у к А. А., Савинов Б. Г. Получение фитола из хлорофилла расте-
ний. — «Украинский химический журнал», 1956, т. 22, с. 518—522.
42. Lindlar Н., Helv. Chem. Acta., 1952, 35, 446.
Глава 15. ПРОИЗВОДСТВО СИНТЕТИЧЕСКИХ ВИТАМИНОВ ГРУППЫ К
Авитаминоз К был открыт в 1929 г. Г. Дамом [1]. При изучении холе-
стеринового обмена у цыплят Дам отметил внутренние кровоизлияния,
несмотря на обеспечение их рациона всеми известными тогда витаминами.
Эти наблюдения в последующем [2, 3] привели к открытию антигеморраги-
ческого фактора. Название витамина К было предложено Дамом в 1935 г.
327
[4] для обозначения жирорастворимого фактора, необходимого для сверты-
вания крови у цыплят (Koagulations vitamin). Механизм физиологического
действия витамина К еще недостаточно изучен [5]. Считают [6—81, что
витамины группы К играют важную роль в клеточном метаболизме, участ-
вуя в транспорте электронов и окислительном фосфорилировании. Отсутст-
вие витаминов К приводит к явлениям геморрагического синдрома — нару-
шению синтеза протромбина в печени. Б. Кудряшов [9, 10] показал, что
при участии витаминов К синтезируется новый белковый компонент крови
тромботропин. Из протромбина и тромботропина при участии ионов каль-
ция и под влиянием фермента тромбокиназы образуется фермент тромбин.
Последний свертывает кровь путем превращения растворимого белка фиб-
риногена в нерастворимый протеин фибрин, образующий тромб.
Таким образом, механизм процесса свертывания крови можно предста-
вить в следующем виде: 1) протромбин-Ьтромботропин4-Са+++тромбоки-
наза -> тромбин; 2) тромбин-(-фибриноген -> фибрин.
Недостаток витамина К в рационе понижает содержание протромбина
в крови, что увеличивает геморрагию. Аналогичное явление имеет место
в связи с понижением усвояемости витамина К при недостатке желчи в ки-
шечнике, например при закупорке желчных протоков. Известно, что вита-
мин К обладает кровеостанавливающим действием также при кровоточи-
вости, которая не обусловливается пониженным содержанием протромбина
[11], что связывают с положительным влиянием витамина К на сосудистый
эндотелий.
Установлены следующие показания для применения витамина К с ле-
чебной целью [12]: кровотечения различного характера, геморрагические
диатезы, механическая желтуха, болезнь Боткина (эпидемический гепатит),
циррозы печени, язвенная болезнь, хронические поражения кишечника.
Витамин "К также является стимулятором мышечной деятельности, спо-
собствует усилению регенерации тканей, ускоряет заживление ран, обла-
дает болеутоляющим действием и повышает сопротивляемость организма
к инфекции [13, 14].
В 1939 г. Дам, Каррер и др. выделили из люцерны витамин Ki в чистом
виде [15], а Дойзи с сотрудниками выделили из гнилой рыбной муки ви-
тамин Кг [16]. В этом же году витамин Ki был одновременно синтезирован
Алмквистом [17], Дойзи [18] и Физером [19].
Ансбахер и Фернгольц в 1939 г. открыли К-витаминную активность
2-метил-1,4-нафтохинона [20], получившего название витамина К3.
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ВИТАМИНОВ ГРУППЫ К
Установлено, что существуют два природных вещества, обладающих
активностью витамина К — вещество, содержащееся в зеленых листьях
растений (Ki), и вещество, содержащееся в микроорганизмах (Кг)- Соответ-
ствующие исследования показали, что антигеморрагической активностью
обладают многие производные нафтохинона, причем наибольшей актив-
ностью обладает 2-метил-1,4-нафтохинон [21], названный витамином Кз-
С другой стороны, в литературе имеются новейшие данные о том, что природ-
ные витамины Ki и Кг обладают значительно большей биологической ак-
тивностью, чем витамин Кз [22—24].
Витамин Ki (2-метил-3-фитил-1,4-нафтохинон), фитонадион, филлохи-
нон) имеет следующую химическую структуру:
О
СН, СН, СН, СН,
Г Т Т 3 I z > 1 z ч I z X I
С Н2С Н= С (С Н2)3С н (С Н2)3С Н (с Н2)3С НС н3
о
328
Витамин Кг (2-метил-3-дифарнезил-1,4-нафтохинон), фарнохинон имеет
следующую структуру (в формуле п=2; 3; 5).
О
г и СН3 СН3
|Г 2|рСН3 । 3 ! з
-^Д^Л-СН2СН=С СН2СН2СН=С/пСН3
о
Витамин К3 (2-метил-1,4-нафтохинон) имеет следующую структуру:
Витамин Ki, С31Н46О2, молекулярная масса 450,68, представляет собой
подвижное желтое масло; нерастворим в воде, плохо растворим в метиловом
и этиловом спирте, хорошо — в петролейном эфире и других органических
растворителях. Устойчив к действию воздуха и влаги. Витамин можно
перегнать без разложения при остаточном давлении <^0,001 мм рт. ст. Он
чувствителен к ультрафиолетовому свету, а волны длиной 400—800 нм не
оказывают какого-либо влияния [25]. Разбавленные кислоты не действуют
на витамин, но щелочи его быстро разрушают.
Удельное вращение [а]д—0,4° (в бензоле).
Витамин Ki дает спектр поглощения с максимумами при 243—244 нм
(1g е = 4,25—4,27), 248—249 нм (1g г = 4,28), 261—263 нм (1g е = 4,24),
270 нм (1g s = 4,24) и 325—328 нм (1g е = 3,49—3,59) [4].
При’остаточном давлении Ю-4—Ю-5 мм рт. ст. витамин Ki перегоняют
при температуре 120—140° С [26]. Витамин Кг, С41Н56О2, молекулярная
масса 580,86, представляет собой бледно-желтое твердое кристаллическое
вещество, температура плавления 53,5—54,5° С, Хтах (гексан) = 243 нм
(1g е = 4,20), 249 нм (1g е = 4,24), 260 нм (1g е = 4,18), 270 нм (1g е =
= 4,18), 325 нм (1g е = 3,45) [27, 28], температура кипения 200° С
(2-Ю-4 мм рт. ст. с разложением).
По многим свойствам витамин Кг сходен с витамином Ki, он представ-
ляет собой более ненасыщенное соединение, так как в положении 3 он вмес-
то фитила имеет дифарнезил [29]. В растворе этилата натрия витамин Кг
дает интенсивное синее окрашивание.
Витамин К3, менадион, метинон, CnHgOa, молекулярная масса 172,17,
твердое кристаллическое вещество, с температурой плавления 105—106° С;
растворим в бензоле, эфире и несколько менее в ледяной уксусной кислоте.
Его растворимость в спирте 17 мг/мл, а в кунжутном масле 13 мг/мл [4].
Слабо растворим в воде, чувствителен к нагреванию. Под действием света
превращается в бесцветное вещество (димер).
329
Xmax (гексан)=244 нм (1g е = 4,3), 253 нм (1g е = 4,29—4,31), 263—264 нм
(1g е = 4,22) и 334 нм (1g е = 3,4—3,5) [27]. Щелочи и кислоты разруша-
ют витамин Кз- В водных растворах витамин Кз легко реагирует с бисуль-
фитом-натрия и дает два ряда изомерных продуктов присоединения [4]:
Натриевая соль 2-метил-
-нафтолинон -2-сульфо-
кислоты
он
он
Натриевая соль 2-метил-!,4-
на/ртигидрохинон -3-сульфо-
кислоты
Соединение I обладает высокой ан-
тигеморрагической активностью [30],
а II —слабой [31]. На рис. 53 даны
кривые поглощения ультрафиолетово-
го света витаминами Ki, Кг и Кз-
Рис. 53. УФ-спектры витаминов К1( К2 и К3.
МЕТОДЫ СИНТЕЗА ВИТАМИНОВ ГРУППЫ К И ВЫБОР РАЦИОНАЛЬНОГО
МЕТОДА ДЛЯ ПРОИЗВОДСТВА
Витамин Ki- В соответствии со структурой витамина Щ синтез его был
первоначально осуществлен [17] путем конденсации фитилбромида и 2-ме-
тил-1,4-нафтохинона в присутствии ледяной уксусной кислоты и цинковой
пыли. Были предложены и другие варианты синтеза (32—34). Из всех ме-
тодов более эффективным [4] оказался метод Физера [19]. Он заключается
в конденсации в течение 36 ч при температуре 75° С фитола с 2-метил-1,4-
нафтогидрохиноном в растворе диоксана в присутствии щавелевой кисло-
ты1.
В результате реакции получают дигидровитамин Ki- Непрореагировав-
ший 2-метил-1,4-нафтогидрохинон извлекают раствором разбавленной ще-
лочи.
Дигидровитамин Ki очищают и окисляют до витамина Ki- Выход вита-
мина составляет 29% к теоретическому (на фитол). Синтез осуществляют
по следующей схеме:
1 По японскому патенту [19а] конденсацию фитола и 2-метил-1,4-нафтогидро-
хинона проводят в присутствии пирофосфорной кислоты.
330
он
Z - Метил - 7,4 - нсирто-
гидрохинон
сн3 сн3 сн3 сн3
Lrl3 I 1,1 I
+ носн2сн=с(сн2)3сн(сн2)3сн(сн2)3снсн3
Фитол
Ag2O .
MgSO4
он
Далее было установлено, что эфиры фтористого бора [35, 36] и кислый
сернокислый калий [35] как катализаторы более эффективны, чем щавеле-
вая кислота. Установлено также, что вместо фитола можно применять его
простые и сложные эфиры [19, 33, 35, 36], а также изофитол или его произ-
водные [19].
Витамин К2. До последнего времени витамин К2 считался второстепен-
ным витамином группы К [4, 37]. Однако исследования биологов послед-
них лет [23, 24] показали, что биологически активной формой в организме
животных является витамин Кг, в который как будто превращаются все
другие витамины группы К. Вслед за этим начались у нас интенсивные ис-
следования по созданию новых и усовершенствованию существующих схем
синтеза изопреноидных спиртов, необходимых для получения витамина Кг,
а также схем синтеза самого витамина [38—42]. Синтез витамина Кг/15 и
К2/20 был осуществлен Э. Козловым [39] по следующей химической схеме:
СН3 СН3 СН3 СН3
I I сн=сн | I
СН3 (С=СНСН2СН2)2—С=О-----------------> СН3—(С=СНСН2СН2)2—С—с = сн
(СН3)2 снсщок |
ОН
транс-Герачилац&гон(1) Ацзтнлен и изобутилат Дегндронеролидол (II)
калия
сн3 сн3
[Н] | | а) РВг3
-------> СН3—(С=СНСН2СН2)2—С—сн=сн2------------------>
Pd/CaCO3 I б) CHsCOCH2COOR
в) NaOH
, Неролидол
(HI)
сн3 сн3
—>• СН3—(С=СНСН2СН2)3-С=О
СН=СН
(СН3)2СНСН2ОК
Фарнезилацетон (кетон С18)
IV
сн3 сн3 сн3 сн3
I I СН] I I
СН3—(С=СНСН2СН2)3—С—С СН----------СН3-(С=СНСН2СН2)3—с—сн=сн2
I Pd/СаСОз j
он он
Геранилдегидролиналоол
V
mpawc-Гераниллиналоол
VI
трпнс-Геранилацетон I конденсируют с ацетиленом в присутствии изо-
бутилата калия в среде толуола. Полученный дегидронеролидол II восста-
навливают в неролидол III, который путем последовательных реакций бро-
мирования, конденсации с натрийацетоуксусным эфиром и омыления пере-
331
водят в фарнезилацетон IV (кетон С18). Далее повторением реакций этини-
лирования фарнезилацетона и восстановления полученного геранилдегид-
ролиналоола V синтезирован транс-гераниллиналоол VI.
Второй компонент молекулы витамина К2-1-ацилокси-2-метилнафтогид-
рохинон синтезируют по следующей химической схеме:
Мепшлнафтохинон
VII
Гиврохинон
VIII
Метилнафтохинон восстанавливают в гидрохинон VIII; затем послед-
ний ацилируют ангидридом или хлорангидридом кислоты и избирательно
омыляют в среде диоксана в присутствии аммиака (выход 92%).
Конденсацию 1-ацилокси-2-метилнафтогидрохинона IX с неролидолом
III (для К2/15) или с гераниллиналоолом VI (для К2/20) осуществляют в при-
сутствии катализаторов хлористого цинка или эфирата трехфтористого
бора с выходом 33—39%. Полученные 1-ацилоксиэфиры омыляют и окис-
ляют окисью серебра и получают витамины К2/15 и Кг/го- Указанные реак-
ции осуществляют Xi о следующей химической схеме:
OR
он
7-Аи,илокси-2-
метилнафтогидрохинон
OR
СНз + СН.
СН3 СН3
I Н BF3; ZnCl2
i2= сн-с-(сн2сн2сн=с)п-сн3 ^2Hg)"2'o"
он «:2з
Неролидол -
гераниллиналоол
ОН
СН3 СН3 NaOH
СН=С-(СН2СН2СН=с)п-СН3 , Ag2O
/ - Ацилоксиэфир
Витамин К3 был синтезирован из 2-метилнафталина при окислении его
ангидридом хромовой кислоты в уксуснокислом растворе со следующими
выходами (на 2-метилнафталин): 25—40% [43], 37—39% [44], 38—42%
[45], 45% [46], 50—60% [47]. Для окисления 2-метилнафталина могут
быть применены другие окислители [4]: перекись водорода в уксуснокис-
лой среде (выход 30%), окись хрома, бихромат натрия, серная кислота
(выход 35%).
Для синтеза витамина Кз могут быть использованы различные произ-
водные 2-метилнафталина, содержащие в положении 1 или 4 алкоксильную
группу, аминогруппу или группу ОН.
332
Промышленный синтез витамина Кз из 2-метилнафталина идет в одну
стадию по следующей схеме [51]:
2 -.Метилнарталин
142, 19
О -
Ма2Сг2О7 —СНо / \ _
—J-кД » [ 1 Г 3 + Na2SO4 + Cr2(SO4)3 + 5Н2О
о
2В2, 02 2-Memuo-1,U-BaipnioxuHOH
4. 08,08 172, 77
Исходное сырье — 2-метилнафталин получают путем перегонки камен-
ноугольной смолы.
Другой путь промышленного синтеза витамина Кз — конденсация то-
лухинона с бутадиеном [48] и окисление продукта конденсации хром-
пиком с выходом около 30% на ортотолуидин. Последний является исход-
ным сырьем для получения толухинона путем окисления МпО2.
Синтез осуществляют по следующей схеме
/СИ,
НС^
Толухинон 2 - Метил -1,4- нарто-
гидрохинон
НС.
сн2
бутадиен
Окисление промежуточного продукта.
2-Метил - 1,4 -нартохинон
Этот метод синтеза витамина Кз имеет лишь одно преимущество — до-
ступность сырья (бутадиен получают на заводах синтетического каучука),
но сам синтез довольно сложен, и выход метинона на о-толуидин практиче-
ски не превышает 30—35%.
Анализируя приведенные материалы по синтезу витаминов группы К
необходимо отметить следующее.
1. Витамин Кз (метинон) по своей биологической активности не уступа-
ет витаминам Ki и К2, а синтез его значительно проще, так как исключа-
ется громоздкий технологический процесс получения фитола или гера-
ниллиналоола — компонентов молекулы витамина К i и К2-
2. Более эффективным методом синтеза витамина К3 является получе-
ние его в одну стадию из 2-метилнафталина с выходом 40—50% вместо
трех стадий синтеза при получении витамина Кз из о-толуидина с выходом
30—35%.
Плохая растворимость метинона в воде заставила ряд исследователей
(А. Палладии, А. Шмук, А. Гусева) искать пути превращения метинона в
333
водорастворимый витамин Кз путем введения в его молекулу сульфогруппы
[51, 52]. Это достигалось действием на метинон сульфита или бисульфита
щелочного металла. Реакция получения эфира сернистой кислоты проте-
кает по следующей схеме [531:
ONa
I
S=O
I
НО
Викасол
276,25
2-М&тил - 1,4-на(рглохином бисульфит* натрия
172,17 104,08
Получаемое бисульфитное производное витамин Кз названо викасолом.
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА ВИТАМИНА К3 (МЕТИНОНА)
И ВИ КАСОЛА [54, 55]
Синтез метинона и викасола состоит из следующих стадий.
I. Окисление 2-метилнафталина в растворе уксусной кислоты двух-
хромовокислым натрием и серной кислотой в метинон с очисткой послед-
него перекристаллизацией.
2. Получение викасола из метинона и раствора бисульфита натрия.
СИНТЕЗ 2-МЕТИЛ-1,4-НАФТОХИНОНА (МЕТИНОНА)
Химическое уравнение реакции см. на стр. 333.
В реактор 1 (рис. 54) загружают двухромовокислый натрий и воду до
получения раствора с плотностью 1500 кг/л3. Затем из мерника 2 вводят
Рис. 54. Технологическая схема производства витамина К3.
334
серную кислоту плотностью 1840 кг!м9 и перемешивают при охлаж-
дении окислительной смеси до 0° . Далее приготовляют раствор 2-метил-
нафталина в ледяной уксусной кислоте, для чего в реактор 3 загружают
2-метилнафталин и из мерника 4 вводят в реактор 3 при температуре 20° С
ледяную уксусную кислоту (на 1 кг 2-метилнафталина 5 л кислоты). В окис-
лительную смесь в реакторе 1 в течение 2—3 ч приливают постепенно при
перемешивании раствор 2-метилнафталина.
Реакционную массу нагревают до 60° С и выдерживают при перемеши-
вании 15 ч. Затем ее спускают в смеситель 5, в который предварительно
наливают холодную воду (1:1). Массу размешивают и спускают на нутч-
фильтр 6. Осадок промывают водой до получения неокрашенного фильтра-
та. На фильтре находится технический метинон.
Первая кристаллизация. Очистку и перекристаллизацию метинона ведут
из спиртового раствора. Для этого сырой осадок помещают в реактор 7 и
при температуре 75° С растворяют в спирте (1:5), который добавляют из
мерника 8. Туда же вносят 2% (к массе сухого вещества) активированного
угля, фильтруют через нутч-фильтр 9 и кристаллизуют в кристаллизаторе
10. Кристаллы отделяют на центрифуге 11, промывают спиртом и высуши-
вают в вакуум-сушилке 12.
Вторая кристаллизация. Маточный раствор I из сборника 13 поступает
в смеситель 14, где его обрабатывают активированным углем. Фильтруют
через нутч-фильтр 15, направляют в сборник 16, затем упаривают в вакуум-
аппарате 17 и кристаллизуют в кристаллизаторе 18. Полученные кристал-
лы метинона отделяют на центрифуге 19. Кристаллы поступают на пере-
кристаллизацию в реактор 7. Маточный раствор II поступает в сборник 20,
при полном истощении он является отходом производства, в противном
случае его направляют на третью кристаллизацию.
СИНТЕЗ ВИКАСОЛА ИЗ МЕТИНОНА И БИСУЛЬФИТА НАТРИЯ
Викасол является монобисульфитным производным витамина Кз и полу-
чается при взаимодействии 2-метил-1,4-нафтохинона с водным раствором
бисульфита натрия. Уравнение химической реакции см. на стр. 334.
Процесс осуществляют следующим образом.
Метинон после сушки" измельчают в дробилке 21, откуда он поступает
в сборник 22. Затем измельченный метинон направляют в тестомесилку 23,
где его растирают с небольшим количеством воды в кашицеобразную массу.
Сюда же добавляют 40%-ный водный раствор бисульфита натрия. На одну
часть метинона берут 2,5 части раствора бисульфита. Смесь слегка разогре-
вается и масса густеет от выделяющихся кристаллов викасола. Кашице-
образную массу высушивают на вакуум-вальцовой сушилке 24. Сухой про-
дукт измельчают в микромельнице 25 и направляют в расфасовку.
Викасол, CuHjOeNaS, молекулярная масса 276,25, кристаллизуется в
виде бесцветных или слегка окрашенных в желтый цвет шелковистых лис-
точков, легко растворяющихся в воде. Вещество не гигроскопично и хорошо
сохраняется в обычных условиях. При действии щелочей викасол разлага-
ется с выделением 2-метил-1,4-нафтохинона.
ТЕХНОЛОГИЧЕСКИЕ ПОКАЗАТЕЛИ
РАСХОД СЫРЬЯ И МАТЕРИАЛОВ, КГ
НА 1 КГ 100%-НОГО ПОЛУПРОДУКТА
2-Метил-1,4-нафтохинон (метинон)
2-Метилнафталин..................1,7
Двухромовокислый натрий ... 3,3
Серная кислота ................ 5,0
Ледяная уксусная кислота с уче-
том регенерации................. 1,0
Спирт 96%-ный....................1,0
Выход, % от теоретического
(на 2-метилнафталин) ...........50,0
Викасол
Метинон ......................... 0,7
Бисульфит натрия . ........... 0,7
Выход, % от теоретического
(на метинон) ....................90,0
335
РАСХОД СЫРЬЯ И МАТЕРИАЛОВ, КГ
НА СИНТЕЗ 1 КГ ВИКАСОЛА
С УЧЕТОМ РЕГЕНЕРАЦИИ
2-Метилнафталин................ 1,2
Двухромовокислый натрий ... 2,3
Серная кислота ................. 3,5
Ледяная уксусная кислота .... 0,7
Спирт 96%-ный................... 0,7
Бисульфит натрия ............... 0,7
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. D a m Н., Biochem. Z., 1929, 215, 475; 1930, 220, 158.
2. D a m Н., Nature, 1934, 133, 909.
3. Schonheyder F., Nature, 1935, 35, 653; Biochem., J., 1936, 38, 890.
4. Hirschmann R., Encyclopedia of Chemical Technology, New Jork, 1955, 14, 858.
5. P ы с с С. M. Витамины. Л., Медгиз, 1955, с. 272.
6. Ciba Foundation Symposium on Quinones in Electron transport, London, 1961.
7. Asano A., Brodie A., Biochem., Biophys. Res. Commun., 1963, 16, 423.
8. Dallam R., J. Hamilton, Arch. Biochem., 1964, 105, 630.
9. К у д p я ш о в Б. А. Витамины, их физиологическое и биохимическое значение.
Моск, общество испытат. природы, 1953, с. 49.
10. К У Д р я ш о в Б. А. — «ДАН СССР», 1948, 60, с. 1469.
11. Палладии А. В. — «ДАН СССР», 1943, 41, 85, 258.
12. Витамин К и его практическое применение (ред. А. Гессен), Главвитаминпром,
Л., 1953, с. 28.
13. Ober doer st er F., H. H о r n., Zschr. ges. Hyg., 1956, 5.
14. HI и л о в П. И., Яковлев Т. Н. Основы клинической витаминологии. М.,
«Медицина», 1964,“с. 130.
15. D a m Н., Karrer Р. и др., Helv. Chirm Acta., 1939, 22, 310.
16. D о i s у E. и др. J. Am. Chem. Soc., 1939, 61, 1295.
17. A 1 m q u i s t H. , К 1 о s e А. Там же, 1939, 61, 2557.
18. D о i s у E., и др. Там же, 1939, 61, 2558.
19. F i е s е г L. Там же, 1939, 61, 2559.
19а. Фирма «Ниссей сэйфун». Японск. пат. 20702, 1967; Витамин, 1968, 37, 1, 132.
20. Ansbacher S. Fernholz Е. J. Am. Chem. Soc., 1939, 61, 1924.
21. В a c h e r F., Boley A., S h о n k C., Anal. Chem., 1954, 26, 1146.
22. I s 1 e г О. и др., Hoppe — Seylers Z. physiol. Chem., 1953, 295, 290.
23. M a г t i n s C., Angew. Chem., 1961, 73, 597.
24. Idem., Schweiz, med. Wschr., 1963, 93, 1264.
25. Ewing D., Tomkins F., Kamm O., J. Biol. Chem., 1943, 147, 233.
26. Sebrell W., Harris R., The Vitamins, Academic Press, N. J., 1954, 2, 388.
27. E w i n g D., Vandenbelt I., Kamm O., J. Biol. Chem., 1939, 131, 345.
28. Morton R., E a r 1 a m W., J. Chem. Soc., 1941, 159.
29. В i n k 1 e у S., M c k e e R. и др., J. Biol. Chem., 1940, 133, 721.
30. Carmack M., Moore M., Balis M., J. Am. Chem. Soc., 1950, 72, 844
31. Baker B., Davies T. и др., J. Am. Chem. Soc., 1942, 64, 1096.
32. McCorquodale D., Cheney L. и др., J. Biol. Chem., 1939, 131, 357.
33. I s 1 e г О. Пат. CHIA 2325681, 3/VIII, 1943; C. A., 1944, 38, 1077.
34. В i n k 1 e у S., D о i s у E. и др. , J. Am. Chem. Soc., 1939, 61, 2558.
35. Hirschmann R., Miller R., Wendler N., J. Am. Chem. Soc., 1954,
76, 4592.
36. I s 1 e г О., D о e b e 1 K-, Helv. Chim. Acta., 1954, 37, 225.
37. Г и p ш м а н P. Ф. Витамин К- — В сб.: «Витамины. Пищевая промышленность
за рубежом», М., Пищепромиздат, 1956, № 2, с. 98—124.
38. К о з л о в Э. И., Я нотовски й М. Ц., Самохвалов Г. И. Син-
тез трансгераниллинолоола. — ЖОХ, 1964, т. 34, с. 2748—51 с ил.
39. Козлов Э. И. Синтез витаминов Кг/15, Кг/го и их 1-ацилоксиэфиров. — «Ме-
дицинская промышленность СССР», 1965, № 4, с. 16—21 с ил.
40. К о з л о в Э. И., Самохвалов Г. И. Синтез нафтохроманолов витаминов
Кг/го, Кг/15 и их производных. — ЖОХ, 1966, т. 36, с. 2120—24 с ил.
41. К о з л о в Э. И., Обольникова Е. А., Самохвалов Г. И. Окис-
ление 6-хроманолов витаминов Кг/15 и Кг/го кислородом воздуха. —ЖОХ, 1967,
т. 37, с. 544—46 с ил.
42. Янотовский М., Козлов Э., Обольникова Е., Волкова О.,
Самохвалов Г., «ДАН СССР», 1968, 179, 3,733.
43. S m i t h L., Webster I., J. Am. Chem. Soc., 1937, 59, 662.
44. P r i c e R., С a r 1 s о n G. Пат. США, 2331725, 12/X, 1943.
45. Fieser L„ J. Biol. Chem., 1940, 133, 391.
46. V e 1 d s t z a H., W i a r d i P., Res. trav. chim. , 1943, 62, 75.
47. Японский пат; № 162647, 8/Ш, 1944; С. А., 1948, 42, 4201.
48. F i е s е г L., С h а п g F., J. Am. Chem. Soc., 1942/64, 2043.
49. F i e s e r L., Oxford A., J. Am. Chem. Soc., 1942, 64, 2060.
50. F i e s e r L., T i s h 1 e r M., Wendler N., J. Am. Chem. Soc.,1940, 62, 2861.
51. П а л л а д и н А. В., Ш м у к A. A. — В сб.: «Технологические инструкции по
производству витаминов», М., Пищепромиздат, 1943, с. 107.
336
52. Ш м у к А. А., Гусева А. Авторское свидетельство, № 63828, 1942; Бюлл.
изобрет., 1944, № 6, с. 11.
53. Ш е м я к и н М. М. и др. — ЖОХ, 1943, т. 13, 327; 1946, т. 16, с. 2033.
54. Союзвитаминпром. Синтез витамина К. — В сб.: Технологические инструкции по
производству витаминов, 1943, М., Пищепромиздат.
55. Девятнин В. А. Витамины. М., Пищепромиздат, 1948, 'с. 246.
Глава 16. ТЕХНОЛОГИЧЕСКАЯ АППАРАТУРА
ДЛЯ ПРОИЗВОДСТВА ВИТАМИНОВ
К принципиальным основам рациональной технологии относится- ка-
чество технологической аппаратуры, к которой должны быть предъявлены
следующие требования:
1. Прогрессивность конструкции, под которой подразумевается обеспе-
чение непрерывности процессов, эффективное проведение их с точки зрения
полноты протекания реакций с большой скоростью, с минимальными затра-
тами труда, тепла и с минимальными потерями вещества. Сюда относятся,
например, следующие аппараты непрерывного действия: колонные реакторы,
барабанные вакуум-фильтры, пленочные реакторы и испарители, ректи-
фикационные, диффузионные, сушильные аппараты и др.
2. Конструкция аппарата должна удовлетворять требованиям техно-
логического процесса в части герметичности, размера поверхности нагре-
вания или охлаждения, удобства опорожнения и заполнения. Например,
при отсутствии достаточно развитой поверхности нагрева в выпарных аппа-
ратах замедляется процесс выпаривания, что приводит к разрушению ве-
щества, находящегося под влиянием высокой температуры. Потери веществ
вследствие разрушения могут происходить при негерметичности аппаратов
и трубопроводов, работающих под вакуумом. Большое значение для повы-
шения эффективности производства имеет также развитость поверхностей
охлаждения.
Необходимо иметь в виду, что в технологии производства витаминов
решающее значение имеет возможность обеспечения процессов глубоким
вакуумом и большой скоростью выпаривания и охлаждения реакционных
масс.
Поэтому при проектировании заводов следует обеспечивать процессы
достаточными по производительности вакуум-насосами, холодильными уста-
новками и соответствующими скоростными аппаратами, преимущественно
пленочного и вихревого режимов.
3. Не менее важно качество материала, из которого изготовлены техно-
логические аппараты. Материал должен быть подобран таким образом, что-
бы предотвратить переход в реакционную среду тяжелых металлов (железо,
медь, никель и др.).
В практике известны многочисленные случаи порчи продукции и полу-
фабрикатов в связи с коррозией металла аппаратов. Кроме того, интенсив-
ная коррозия стенок аппарата может привести к их прободению. Отсюда
возникает опасность взрывов и пожаров, влекущих за собой несчастные
случаи с людьми.
4. Большим тормозом в работе технологической аппаратуры является
недостаточный диаметр спускных штуцеров, в особенности при гетероген-
ной реакционной массе, либо при кристаллизующихся густых растворах.
Применение в этих случаях спускных штуцеров диаметром менее 75—
100 мм недопустимо. При передаче пересыщенных растворов по трубопро-
водам последние должны быть снабжены обогреваемыми водой или паром
спутниками.
5. Охрана труда и техники безопасности диктует необходимость обеспе-
чения технологических аппаратов в особенности работающих под давле-
нием, а также аппаратов, работающих с пылевидными продуктами предо-
337
хранительными устройствами и приборами, контролирующими параметры
процесса, гарантирующими безопасность работы.
Автор не ставит своей целью в данном труде рассмотрение и расчеты всей
аппаратуры, применяемой в производстве, так как эти вопросы достаточно
подробно рассматриваются в специальных руководствах, как, например,
в книге «Процессы и аппараты химической и нефтехимической технологии»
А. Плановского и П. Николаева. Гостоптехизат, 1960, а также И. Ле-
бедева, изд-во «Химия», 1971.
Автор имеет в виду фиксировать внимание читателя на специфическую
аппаратуру, применяемую в витаминной промышленности, а также на не-
которые прогрессивные конструкции аппаратов, как работающих на заводах,
так и рекомендуемых к внедрению.
Аппаратуру, применяемую для производства витаминов, подразделяют
на две группы:
аппараты, используемые в химическом синтезе и предназначенные для
проведения реакций выделения вещества путем осаждения, фильтрации,
либо экстракции, выпаривания маточных растворов, фуговки кристалли-
ческих масс, сушки кристаллов, обработки отходов;
аппараты, применяемые в производстве витаминов из природного сырья,
включая аппараты диффузионные, прессовые, выпарные, сушильные и др.
В этой главе описаны аппараты преимущественно непрерывного дейст-
вия, изготовляемые на машиностроительных заводах СССР.
К ним относятся реакторы, барабанные вакуум-фильтры, установки
для выпаривания растворов, кристаллизации и фуговки витаминных про-
дуктов, ректификационные аппараты, сушильные, диффузионные, моеч-
ные и др. Перечисленные аппараты работают на отдельных витаминных за-
водах.
Диффузионный аппарат системы Гузенко, пленочные выпарные аппара-
ты, вихревые сушилки, пресс Яна изготовляются в механических мастер-
ских витаминных заводов [1, 2, 5, 6, 6а, 9—12, 14—18].
ч—•— Рис. 55. Реактор-ацетонатор.
Рис. 56. Реакционный аппарат непрерывного
действия для вязких масс.
338
Кроме того, приведены некоторые аппараты, изготовляемые зарубежны-
ми фирмами. Эти аппараты представляют интерес для внедрения в витамин-
ной промышленности.
АППАРАТЫ НЕПРЕРЫВНОГО ДЕЙСТВИЯ ДЛЯ ХИМИЧЕСКОГО СИНТЕЗА
ВИТАМИНОВ
Реактор-ацетонатор (рис. 55) [1]. Ацетонатор — аппарат, изготовленный
из нержавеющей стали. Для охлаждения раствора реактор снабжен рубаш-
кой. Крышка 1 ацетонатора соединяется с аппаратом при помощи фланцев
2 через прокладку 3. Днище аппарата 4 приваривается к наружному ци-
линдру и снабжено отверстием для спуска промоев. Наружный цилиндр 5
приваривается к фланцу 6, к которому в свою очередь приваривается внут-
ренний цилиндр 7. Высота его меньше высоты наружного цилиндра. Смесь
олеума и, ацетона загружают через штуцер 8, а сорбозу через штуцер 9.
Непрерывный выход реакционной массы из ацетонатора осуществляется
через коллектор 10, соединяющий два штуцера.
Продукт перемешивается пропеллерно-лопастной мешалкой 11. Вал
мешалки с частотой вращения 235 об/мин получает движение от электро-
двигателя.
Реакционный аппарат непрерывного дей-
ствия для вязких масс (рис. 56). Смешивание
компонентов происходит в смесителе 1, отку-
да масса поступает в реактор шнекового
типа 2, обогреваемого паром или охлажда-
емого рассолом. Реакция протекает по мере
продвижения по шнеку 2. Густая вязкая
масса направляется в бункер 3. Аппарат мо-
жет быть присоединен к холодильнику-кон-
денсатору, и тогда в процессе реакции может
отгоняться растворитель. Массу компонентов
можно нагревать или охлаждать через полый
вал шнека.
Реактор пленочного типа. На рис. 57
изображен реактор пленочного типа. Он
представляет собой цилиндрический сосуд 1,
внутри которого вращается рамная мешалка
2 с подвижными прилегающими к боковой
стенке скребками 3. Компоненты реакции
подают при помощи дозировочных насосов в
верхний расширительный сосуд, откуда они,
распределяясь по стенкам, стекают в виде
тонкой пленки, в которой интенсивно Проте-
кают реакции. Стенки аппарата обогрева-
ются паром, горячей водой или высококипя-
щей жидкостью [2].
Аппараты пленочного типа применяются
в качестве испарителей-концентраторов.
Техническая характеристика испарителей-кон-
центраторов, выпускаемых фирмой «Sames-
penther» (ФРГ) [2], приведена в табл. 18.
Опубликованы обзоры: 1) о реакторах
типа барботажной колонны, приведены ре-
комендации по выбору, автоматизации и
оптимизации реакторов [3]; 2) о реакторах
для проведения реакций в тонкой пленке [4].
Рис. 57. Реактор пленочного
типа.
339
Таблица 18
Показатели Испарители-концентраторы марок
Т-100 Т-300 Т-600
Поверхность нагрева, м? . . .* . 0,314 2,0 8,8
Внутренний диаметр, мм .... 100 300 600
Полезная высота, мм 1100 2400 5000
Общая высота, мм 2200 4300 8000
Частота вращения вала, об/мин . 350 150 60
Расход энергии, кет Рабочее избыточное давление в ру- 0,37 1,47 2,57
башке, кгс/см2' 30 30 10
Барабанный вакуум-фильтр непрерывного действия (ГОСТ 5748—51)
[51. Б 5-1,75/0,9; Б 10-2,6/1,3; Б 10-1,75/1,8; Б 20-2,6/2,6; Б 15-3,0/1,1 (пер-
вая цифра показывает фильтрующую поверхность (в м2), вторая — диа-
метр барабана, третья —длину барабана (в м). Фильтр (рис. 58) состоит
Рис. 58. Барабанный вакуум-фильтр непрерывного действия.
из горизонтального ячейкового вращающегося барабана 1, частично погру-
женного в корыто 2 с фильтруемой жидкостью. Барабан покрыт перфо-
рированным ситом 3, поверх которого проволокой закрепляют фильтроваль-
ную ткань. Барабан опирается на подшипники 4, установленные на корыте,
цапфами с ячейковыми каналами, которые служат продолжением ячеек,
барабана. При помощи распределительной головки 5, прижатой к торцу,
ячейки сообщаются с вакуумом или сжатым воздухом в зависимости от по-
ложения ячейки на окружности вращения. В зоне фильтрации, просушки
и промывки ячейки соединены с вакуумом, в зоне съема осадка — с сжатым
воздухом, промывные ячейки соединены с вакуумом, в зоне съема осадка
с сжатым воздухом.
В корыте фильтра установлена мешалка 6 для предотвращения осажде-
ния твердых частиц. Привод барабана осуществлен от двигателя 7 через
редуктор-вариатор 8.
Мешалка приводится в движение от двигателя 9 через червяк и систему
зубчатых колес. Барабан снабжен ножом 10, валиком 11 для съема- осадка
(приводной обрезиненный валик, прижимаемый к поверхности барабана),
приспособлением для промывки осадка 12, приспособлением для заглажи-
вания трещин 13. Последняя состоит из бесконечной тканевой ленты, рас-
тянутой на четырех роликах 14 и обтягивающей поверхность осадка в верх-
ней части барабана.
Габариты аппарата Б 5-1,75/0,9 (в мм): длина 2570, ширина 2680, вы-
сота 2310, диаметр барабана 1750, масса 4800 кг.
340
Мощность двигателя для привода барабана 1 кет, для привода мешалки
1 кет, частота вращения двигателя 930 мин, частота вращения барабана
от 0,13 до 1,5 об!мин.
Рис. 59. Друк-вакуум-фильтр фирмы BHS (ФРГ).
Барабанный друк-вакуум-фильтр. На выставке «Ахема» (ФРГ) был экс-
друк-вакуум-фильтр • новой конструкции
понирован барабанный
«BHS» („Bauerische Berg”, „Hiitten
und Salzwerke”) [2]. Вместо ко-
рыта установлен фильтрующий ба-
рабан 1 с кожухом 2 (рис. 59).
Суспензия и промывная жидкость
подаются через соответствующие
штуцеры. Полое пространство ме-
жду внутренней стороной корпуса
и поверхностью кожуха фильтро-
вального барабана разделено эле-
ментами 3 на камеры 41г 4Z, 43
и 4±. В камере 4t происходит раз-
деление осадка и жидкости, в ка-
мере 42—промывка, в камере 43 —
сушильное отсасывание, в камере
4^— разгрузка осадка. Каждая
ячейка фильтра, образованная
планками 5, снабжена отводной
трубкой 6, присоединяемой к ре-
гулирующему устройству 7, позво-
ляющему вести разделение маточ-
ных и промывных жидкостей. Кор-
пус барабана для герметичности
снабжен сальниками с обеих сто-
рон. Барабан установлен на под-
шипниках, образованных боковыми
крышками 9. Привод состоит из
механизма регулирования числа
оборотов 10 и редуктора 11. Филь-
трация может производиться либо
под избыточным давлением, либо
под вакуумом.
фирмы
Рис. 60. Установка для выпаривания,
кристаллизации и фуговки.
341
Установка для выпаривания растворов, кристаллизации и фуговки
(рис. 60). Эти процессы широко применяются в синтезе витаминов.
Рекомендуется показанная на рис. 60 компоновка аппаратов [5]. Установка
состоит из вакуум-аппарата 1, кристаллизатора 2, центрифуги 3. Вакуум-
аппарат снабжен ребристой поверхностью нагрева 4, барометрическим
конденсатором 5. Вакуум создает вакуум-насос 6. Упаренный раствор спус-
кают из вакуум-аппарата 1 в кристаллизатор 2, снабженный метатель-
ным прибором. Сгущенный раствор из кристаллизатора поступает в центри-
фугу 3. Кристаллическую массу из центрифуги разгружают на трясучку 7.
Маточные растворы отводят из центрифуги в специальные приемники.
Центрифуги. Для выделения кристаллических веществ в технологии
производства почти всех витаминов широко применяются центрифуги пе-
риодического действия. Однако более перспективными являются центрифу-
ги непрерывного действия. В табл. 19 приведены типы центрифуг по ката-
логу-справочнику Главхиммаша [6].
Таблица 19
Марка центрифуги Техническая характеристика
ТВ-600 Трехколонная с верхней ручной выгрузкой, фильтрующая, периоди- ческого действия, диаметр барабана, 600 мм, мощность 2,8 кет, п = 1420 об/мин, габариты 1450 X 1080 X 820 мм
ПС-1200 Подвесная, саморазгружающаяся, периодического действия, диаметр барабана 1200 мм, мощность 40 кет, я = 1000 об/мин, габариты 2200x2100x3500 мм
НОГШ-230 Непрерывного действия, отстойная, горизонтальная со шнековой вы- грузкой осадка, со сплошным барабаном, наибольший диаметр—230 мм, мощность 2—3 кет, частота вращения барабана 1600 об/мин, габа- риты 1275 х 925 х 430 мм
НОГШ-600 Непрерывного действия, отстойная, горизонтальная с шнековой вы- грузкой осадка, со сплошным барабаном, наибольший диаметр 600 мм, мощность 40 кет, частота вращения барабана 1400 об/мин, габари- ты 2815 X 1 100 X 1200 м.ц
На рис. 61 изображена принципиальная схема шнековой центрифуги.
Суспензия поступает через полый горизонтальный вал конусного бараба-
на 2, установленного в кожухе 1. Выгрузка осадка осуществляется шне-
ком 3. Барабан приводится во вращение шкивом 4, а шнек — шкивом 5.
Направление жидкости и осадка видно на рисунке.
Центрифуга с пульсирующим толкателем. Среди непрерывно действую-
щих центрифуг наиболее перспективными являются центрифуги с пульси-
рующим толкателем [2]. Схема такой центрифуги изображена на рис. 62.
Подача суспензии осуществляется по трубе 1 и через распределительную
воронку 2 равномерно растекается по стенке барабана 3. Жидкость прохо-
дит через отверстия в барабане, а осадок накапливается на внутренней
поверхности его. Толкателем 5 (поршень 4 гидравлического устройства)
осадок выталкивается вперед к разгрузочному устройству. Промывка осад-
ка происходит через разбрызгивательную трубу 6.
На Международной выставке «Химия в промышленности, строительстве
и сельском хозяйстве» в 1965 г. [7 ] в Москве было экспонировано 17 фир-
мами 36 типов центрифуг, из которых для витаминной промышленности
особый интерес представляют центрифуги во взрывобезопасном исполне-
нии:
фирмы «Эшер Висс» (ФРГ) центрифуга непрерывно действующая, че-
тырехкаскадная, пульсирующая, обогреваемая или охлаждаемая, фактор
разделения 1008, диаметр ротора 250/450 мм;
фирмы «Краусс-Маффей» (ФРГ) — трехколонная центрифуга с верх-
342
ней и нижней выгрузкой из нержавеющей стали; фактор разделения 476,
диаметр ротора 700—1450 мм, масса загрузки 150 кг;
Рис. 61Принципиальная схема шнековой
центрифуги непрерывного действия.
Рис. 62. Центрифуга с пульсирую-
щим толкателем.
фирмы «Робатель и Мюлатье» (Франция) — непрерывно действующая
горизонтальная осадительная со шнековой выгрузкой осадка; оригинально
сконструированы двухступенчатый планетарный редуктор с механизмом
блокировки и герметичные сальниковые уплотнения; фактор разделения
Рнс. 63. Вакуум-выпарная установка системы «Виганд».
1100, диаметр ротора 160—800 мм, частота вращения 1600 об/мин, произ-
водительность до 10 т/ч.
Вакуум-выпарные аппараты. Интерес представляют пленочные аппара-
ты и многокорпусные аппараты непрерывного действия с выносной поверх-
ностью нагрева. К последним следует отнести вакуум-выпарную установку
системы «Виганд» (рис. 63). Установка состоит из трех выпарных корпусов
I, II и III, трубчатого поверхностного конденсатора IV, насоса для откач-
ки концентрата V и мокровоздушного вакуум-насоса VI. Выпарные корпу-
са 7 и /7 состоят из подогревателя и испарителя. Установка работает сле-
дующим образом. Разбавленный раствор из сборника 1 через систему по-
плавковых клапанов 2 непрерывно поступает в подогреватель 3 корпуса 7,
343
где вскипает и далее направляется в испаритель 4 и по трубе 5 частично воз-
вращается в подогреватель 3. Часть сока по трубе 7 переходит в следующий
корпус, затем через подогреватель 6 поступает в испаритель 8, оттуда по
трубе 9 частично возвращается в подогреватель 6, а частично через откры-
тый вентиль 11 переходит в корпус III, а из последнего откачивается насо-
Рис. 64. Тонкопленочный
роторный испаритель
Т-160 «Самбай».
сом V в сборник 10. Корпус I обогревается острым
паром, корпус II — соковым паром корпуса I, а
корпус///—соковым паром корпуса //. Конден-
сат корпуса / переходит в корпус //, из него в кор-
пус ///, а из последнего поступает в конденсатор,
откуда откачивается мокровоздушным насосом VI.
Переход сока из корпуса в корпус регулируется
вентилем //, а конденсат — мембранами 12.
Техническая характеристика: избыточное дав-
ление греющего пара —3 кгс/сж2; расход острого
пара на 1 кг выпаренной воды — 0,4 кг-, габари-
ты— 5,5x3,3x3,0 м.
В последних моделях аппарата «Виганд» приме-
няется термокомпрессия сокового пара при помощи
пароструйного компрессора, что еще более снижает
расход пара на корпус I.
При высокой теплотехнической эффективности
недостатком аппарата является сравнительно боль-
шая продолжительность процесса выпаривания.
Тонкопленочный испаритель. Для выпаривания
лабильных веществ весьма эффективными являются
тонкопленочный испаритель. На Международной
выставке в Москве [7] экспонировался тонкопле-
ночный роторный испаритель Т- 160 «Самбай» фирмы
SMS (ФРГ).
Техническая характеристика испарителя Т-160
«Самбай» (рис. 64): поверхность нагрева 0,75 м2;
диаметр аппарата 160 мм\ общая высота 3420 мм.
Скорость вращения ротора при испарении 220 и
при сушке 400 об/мш-с, наибольший вакуум
2 мм рт. ст. Аппарат изготовляется фирмой по
американской лицензии. Выпарные аппараты ро-
торные пленочного типа отечественного производ-
ства имеют внутренний диаметр 160 и 300 мм, а по-
верхность нагрева — 0,8 и 2,0 м2.
МАССООБМЕННЫЕ ПРОЦЕССЫ И АППАРАТЫ
В синтезе витаминов широко применяются аппараты для массообменных
процессов, к которым относятся аппараты для адсорбции, экстракции и
ректификации.
Аппараты для адсорбции. Они предназначены для очистки растворов
активированными углями; применяются в технологии производства всех
синтетических витаминов. Процесс обычно осуществляют в реакторах из
нержавеющей или эмалированной стали, снабженных обогреваемой рубаш-
кой и мешалкой. Применяются мешалки различных типов: лопастные,
якорные, рамные, пропеллерные (с диффузором), турбинные [8]. Для ад-
сорбционных процессов (обработка углями) конструкция мешалок не имеет
существенного значения. Обычно применяют якорную мешалку при часто-
те вращения 50—60 об/мин. Для реакционных аппаратов интенсивность
перемешивания реакционной массы имеет важное значение. Например, в
производстве аскорбиновой кислоты эффективность процессов ацетониро-
вания L-сорбозы, нейтрализации монодиацетонового раствора и окисления
344
диацетонсорбозы зависит от конструкции мешалок и их окружной скорости.
Интенсивное перемешивание достигается при комбинированной пропеллер-
но-лопастной мешалке и частоте вращения 200—250 об/мин. Рекомендации
по выбору типов мешалок и номограмма для расчета мощности приведена
в обзоре .[8а ].
Аппараты для ионообменного процесса (рис. 65). Аппарат применен для
выделения глюконовой кислоты из глюконата кальция при помощи катио-
нита КУ-2 в производстве витамина В15 [9]. Аппарат состоит из сборника
глюконата кальция 1, ионообменных колонн 2, сборника соляной кислоты
3 и сборника дистиллированной воды 4. Через колонки пропускают 15%-ный
Рис. 65. Укрупненная ионитная установка по полу-
чению глюконовой кислоты на смоле КУ-2.
раствор глюконата кальция при температуре 60—70° С со скоростью 15 л/ч.
Регенерацию смолы осуществляют разбавленным раствором соляной кис-
лоты.
Интерес представляет комплектная ионообменная установка (габариты
1400 X 700 X 1600 мм) типа Дестионекс, Хемокомплекс (Венгрия) произво-
дительностью 500 л/ч. Установка предназначена для очистки жесткой
воды. Она состоит из двух колонок анионовых и двух катионовых.
Снабжена двумя сборниками для 10%-ной соляной кислоты и 40%-ного
раствора едкого натра, используемых для регенерации ионитов. Установка
снабжена контрольно-измерительными приборами.
Экстракционные аппараты непрерывного действия. В синтезе витаминов
они применяются недостаточно широко. Наиболее эффективными экстрак-
торами являются колонные смесительно-отстойные аппараты [8]. Переме-
шивание жидких компонентов осуществляется турбинными или пропеллер-
ными мешалками. Расслаивание проводится в зонах аппарата, заполняе-
мых для успокоения потоков либо насадочными телами, либо статорными
кольцами. Экстрактор такого типа применен в синтезе витамина В3 для экс-
тракции D (—) — пантолактона метиленхлоридом из водного раствора
комплексной соли. На рис. 66 показана конструкция крупнолабораторного
колонного смесительно-отстойного экстрактора [10]. На рис. 67 изображена
схема непрерывно действующей экстракционной установки промышлен-
345
ного типа [8] для извлечения растворенных веществ органическими раство-
рителями. Легкая жидкость из сборника 1 насосом 2 нагнетается в напор-
ный сборник 3 и через ротаметр 4 поступает в нижнюю часть экстрактора.
Таким же путем тяжелая жидкость из сборника 5 насосом 6 подается в на-
порный сборник 7 и через ротаметр 8 направляется в верхнюю часть колон-
ны 9. Контактное взаимодействие компонентов осуществляется перемеши-
ванием. В зонах с насадочными телами происходит расслаивание жидкос-
тей. Легкая жидкость поступает в сборник 11, а тяжелая в сборник 10.
Ректификационные аппараты. Эти аппара-
ты широко применяются в синтезё витаминов
в основном для регенерации растворителей.
На крупных предприятиях применяются рек-
тификационные аппараты непрерывного дей-
ствия, как, например, для регенерации аце-
тона в производстве L-аскорбиновой кислоты.
Рис. 66. Колонный экстрактор
смесительно-отстойного типа:
1 — колонна; 2 — статор юе кольцо;
3 — мгшатгльный элемент.
Рис. 67. Схема непрерывно действующей экстрак-
ционной установки промышленного типа.
На! рис. 68 приведена схема ректификационной установки непрерыв-
ного действия для разделения смеси на два компонента [8]. Раствор
поступает в подогреватель 1, где температура его повышается за
счет пропускания через подогреватель кубового остатка. Нагретый раствор
направляют в ректификационную колонну 2 на одну из верхних тарелок,
где смешивается с флегмой, поступающей из дефлегматора 3. Ректифика-
ция осуществляется за счет тепла, сообщаемого раствору в кипятильнике 4.
В колонну возвращают лишь часть флегмы; остальная часть проходит че-
рез холодильник 5 и поступает в сборник 7. Кубовая жидкость непрерывно
из нижней части колонны отводится в сборник 6. Для ректификации высо-
ко кипящих лабильных веществ необходимы аппараты, работающие при
глубоком вакууме. На рис. 69 показана модель такого аппарата, разрабо-
танная К. Новиковой и Ю. Шведовым [11]. Колонна 1 с насадкой из колец
Рашига внизу заканчивается кубом 2. Пары из колонны поступают в дефлег-
матор 3. Часть флегмы возвращается в колонну на орошение при помощи
автоматически регулируемой электромагнитной направляющей воронки 4.
Из куба через U-образную трубку с холодильником типа «труба в трубе»
кубовый остаток непрерывно откачивается насосом. Вакуум создается на-
сосом ВН-2 через сборник дистиллята. Для измерения перепада давления
между верхом и низом колонн установлен дифференциальный манометр 7
с холодильником 8.
346
Технический продукт подается через дозатор 5 и подогреватель 6. При
ректификации в колонне технического (3-альдегида С14 получен чистый
альдегид при следующих параметрах: температура в кубе 145—150° С;
остаточное давление (мм рт. ст.) 6-Ю-1—8-Ю"1; флегмовое число 10.
Рис. 68. Схема ректификационной уста-
новки непрерывного действия.
Рис. 69. Вакуумная ректификационная
колонна непрерывного действия.
СУШИЛЬНЫЕ АППАРАТЫ
Сушильные аппараты широко используются в технологии синтеза всех
витаминов. Здесь приведены наиболее перспективные сушильные аппара-
ты непрерывного действия.
Барабанные сушилки. Применяются для сушки сорбозы и гидрата ди-
ацетон-2-кето-£-гулоновой кислоты. На рис. 70 изображена барабанная
Рис. 70. Барабанная сушилка.
сушилка [8].'Аппарат состоит из барабана 1, установленного под неболь-
шим углом к горизонту. Барабан снабжен бандажами 2, катящимися по
опорным роликам 3 и фиксируемыми опорными роликами 4. Барабан приво-
дится во вращение зубчатой передачей 5. Влажный материал поступает
через бункер 6. На внутренней поверхности барабана монтируются направ-
ляющие лопатки, которые при вращении барабана распыляют сыпучий
347
материал и передвигают его навстречу теплому воздуху к бункеру 7. Теп-
лый воздух поступает через колорифер 8, нагнетается вентилятором 9 и
1 выводится в атмосферу через трубу 10.
Вакуум-вальцовая сушилка (рис. 71). Принцип действия этой сушилки
заключается в следующем [5]. Два полых, тщательно отшлифованных ци-
линдра 1 медленно вращаются в противоположные стороны. Цилиндры обо-
греваются острым паром (давление 5 кгс/сж2) через трубопровод 2. Влажный
Рис. 71. Вакуум-вальцовая сушилка.
или жидкий продукт поступает сверху через распределительное устройст-
во в полость между цилиндрами, образуя на них пленку. В течение одного
оборота цилиндров вода из пленки испаряется, а сухая пленка, полученная
на поверхности цилиндра, снимается скребком и сбрасывается в шнек 3,
расположенный под барабанами. Шнеки транспортируют сухой продукт в
камеры 4. Вся установка герметизирована специальным кожухом 5. Сушил-
ка для разгрузки не останавливается. Для этого только отключают камеры
вентилями 6. Цилиндры и шнеки приводятся в движение трансмиссионным
приводом 7. Вакуум в сушилке создается специальным вакуум-насосом.
Пары конденсируются в поверхностном конденсаторе. Техническая харак-
теристика вакуум-вальцовой сушилки приведена в табл. 20 [5].
Таблица 20
Модель вакуум- вальцовой сушилки Поверхность нагрева вальцов, м2 Испарительная способность 1 м2 , кг/ч воды Расход мощности, кет Габариты, м
длина ширина высота
II 5,1 35—65 3,0—4,5 4,15 1,70 1,7
III 6,9 35—65 3,7—5,2 4,60 1,70 1,7
IV 9,6 35—65 5,2—6,6 5,53 2,40 1,9
V 12,4 35—65 6,0—7,4 6,08 2,40 1,9
VI 14,7 , 35—65 7,4—9,0 6,48 2,40 1,9
Сушилка с центробежным распылением (рис. 72). Распыление жидкости
в этой сушилке происходит под действием центробежной силы быстровра-
щающегося диска или колокола. Сушилка работает следующим образом.
Раствор, подогретый до 80—85° С, из сборника 1 под напором поступает в
распылительную шайбу 2, вращающуюся с большой скоростью (125—
160 м!сек, при частоте вращения 10—12 тыс. об!мин) внутри башни 3. Жид-
кость распыляется, образуя туман. Капли тумана интенсивно перемеши-
ваются с подогретым воздухом, подаваемым в башню через калорифер 4.
Отработанный воздух отсасывается'вентилятором 5 через рукавные филыг-
348
ры 6, улавливающие пыль. Высушенные частицы оседают на пол камеры,
откуда снимаются скребком 7 и шнеком 8 удаляются из башни.
Рис. 72. Сушилка с центробежным распылением.
Техническая характеристика [5]
Производительность, кг/ч испаряемой влаги . . . . 100, 200, 500
и 2000
Габариты (длина X ширина х высота) для произво-
дительности 2000 кг/ч, м........................ 15 х Ю х Ю
Частота вращения шайбы, тыс. об/мин ........... 10—12
Сушилка с форсуночным распылением (рис. 73) [5]. Она состоит из
башни 1, в центре которой установлена распылительная форсунка 2. Вы-
сушиваемый раствор подают в форсунку насосом под избыточным давле-
нием 100 кгс/см2. Раствор выбрасывается из форсунки в виде мельчайших
капель, образуя туман. Вентилятор 3 нагнетает воздух через калорифер 4.
Порошок удаляют из сушилки конвейером 5 при помощи специального
скребка. Отработанный воздух вентилятором 6 отсасывается из башни че-
рез фильтр Бетта 7 и выбрасывается наружу через выхлопную трубу 8.
Техническая характеристика: испарительная способность влаги кг/ч 100;
температура воздуха 125°; поверхность нагрева калорифера в м2 — 6,0:
скорость движения ленты транспортера 0,2 м1мин\ число рукавов в фильт-
ре Бетта —44. Габариты сушилки: 12,5x7x8,02 м. Интерес представляет
применение на Уфимском заводе в распылительной башне щеточного меха-
низма для непрерывной чистки сферической поверхности башни.
Сушилки со взвешенным слоем (псевдокипящим). Материал, подлежа-
щий сушке, загружают в конический сосуд 1 (рис. 74) с перфорированным
днищем на тележке 2 и устанавливают в герметично закрывающуюся каме-
ру 3. Сосуд прижимают при помощи специального рычажного механизма
к горизонтальной перегородке 4, имеющей отверстие, соответствующее
диаметру сосуда. Над перегородкой установлен рукавный фильтр для предот-
вращения уноса высушиваемого продукта. В верхней части камеры смонти-
рован вентилятор с калорифером, просасывающий воздух через камеру.
Для снятия зарядов статического электричества служат заземленные ме-
таллические стержни, установленные в слое псевдоожиженного материала.
349
Продолжительность сушки колеблется от нескольких минут до 2 ч в зави-
симости от начальной влажности материала и его специфических свойств.
В Швейцарии такие сушилки выпускают 2 фирмы («Аэроматик» и «Ро-
берт Мюнстер»). Производительность сушилок марок ST-2 и ST-200 колеб-
<W
Рис. 737 Сушилка с форсуночным распылением:
а — вид спереди; б — план.
лется от 2—4 до 200—400 кг единовременной загрузки при наибольшей
температуре воздуха соответственно 200 и 110° С, расход воздуха 2 и
200 мЧмин. Выпускаются также сушилки с загрузкой 5,30, 60 и 100 кг.
Вакуум-фильтр ленточный непрерывного действия (рис. 75) [14]. Он
предназначен для фильтрации различных жидких суспензий; состоит из
станины 1 и двух барабанов 2 с натянутой на них рифленой лентой 3, по-
крытой фильтротканью. Для вращения барабана имеется привод. Фильт-
руемая суспензия чефез лоток 4 поступает на движущуюся ленту. Фильт-
рат и промывная жидкость отсасываются через вакуум-камеру 5 в ловушку
6, а осадок снимается ножом 7 и сбрасывается вниз. Фильтр изготовляют
в углеродистом и нержавеющем исполнении.
350
Техническая характеристика
Поверхность фильтрации, м- 1,6
Температура, °C
фильтрации..................... 40
промывки................... 60
Длина ленты, мм
рабочая...................... 3200
общая................... 10560
Ширина ленты, мм .... 650
Скорость движения ленты,
м/мин....................0,75—11,8
Мощность привода, кет, . . 1,7
Габаритные размеры, мм
длина....................... 5200
ширина.................. 1868
высота............... 1740
Общая масса, кг............. 5140
Рис. 74. Сушилка
со взвешенным слоем.
Рис. 75. Вакуум-фильтр ленточный непрерывного действия,
ти
Сушилка с вихревым режимом [12]. На рис. 44 (см. стр. 287) дана схема
вихревой сушилки, в которой обработка материала производится методом
вихревого пневмотранспорта. Отличительная особенность этого типа су-
шилок заключается в следующем. При сушке в шкафах процесс состоит из
двух периодов: 1) испарение поверхностной влаги при постоянной и зна-
чительной скорости и 2) испарение внутренней влаги, которое лимитиру-
ется диффузией влаги из внутренних слоев к поверхности при падающей
скорости.
При сушке материала в вихревом режиме конвективный обогрев продук-
та в шкафах заменяется конвекционным, что резко ускоряет процесс.
Вихревая сушилка работает следующим образом. Влажный продукт из
бункера 1 шнеком 2 и пневмоэжектором 3 направляется в вихревую камеру
4, куда подается воздух, нагретый до 80—100° С в калорифере 5. В камере
и трубопроводе быстро протекает процесс сушки. Высушенный продукт и
воздух далее поступают в циклон 6; продукт в нем выделяется и поступает
в сборник 7, а воздух через рукавный фильтр <5 уходит в атмосферу. При
351
испытании данной установки Соколовским и Хлуденевым получены сле-
дующие результаты (пример для аскорбиновой кислоты) [12]: температура
начальная 90° С, в камере 55° С; время пребывания продукта в камере
45 сек, влажность высушенной аскорбиновой кислоты — 0,4%, удельная
производительность 3650 кг/ч/м?.
Опубликован обзор по сушке материалов в аппаратах с псевдоожижен-
ным слоем и во взвешенном состоянии [13]. Авторы отмечают, что сушилки
данных типов просты в конструктивном отношении, работают с высокой
интенсивностью, компактны и допускают полную автоматизацию про-
цесса.
АППАРАТЫ НЕПРЕРЫВНОГО ДЕЙСТВИЯ ДЛЯ ПРОИЗВОДСТВА ВИТАМИННЫХ
КОНЦЕНТРАТОВ ИЗ ПРИРОДНОГО СЫРЬЯ
К этому разделу относятся аппараты, служащие для специфических
процессов, связанных с извлечением биологически активных веществ из
природного сырья и превращением их в витаминные концентраты. Из мно-
Рис. 76. Кулачная мойка Добровольского.
гочисленных аппаратов, применяемых в данной отрасли производства,
рассмотрим лишь основные.
Кулачная мойка Добровольского [5]. Она применяется в производстве
концентратов каротина из моркови. Мойка (рис. 76) представляет собой
горизонтальный корытообразный аппарат 1, состоящий из трех отделений.
Первое отделение (I) моечное снабжено горизонтальным вращающимся ва-
лом 2 с винтообразно расположенными кулачками 3 для перемешивания
352
и передвижения корней. Корыто имеет ложное дырчатое дно 4. Земля про-
ходит через отверстия в ложном дне и удаляется через спускные трубы 5
с шиберами. Второе отделение (II) железной перегородкой 6 изолировано
от третьего отделения (III). Третье отделение перегородкой 7, не доходящей
до дна, разделено на две части. Над вторым и третьим отделениями прохо-
дит вал <5 со специальными черпаками 9, служащими для перебрасывания
моркови из одного отделения в другое.
Второе и третье отделения снабжены люками 10 для улавливания и уда-
ления камней. Частота вращения вала с кулачками 15 об/мин, а вала с чер-
паками — 10 об/мин. Морковь поступает в первое отделение мойки, откуда
вдов
Рис. 77. Диффузор Гузенко.
передвигается кулачками через отверстия в перегородке во второе отделе-
ние. Из второго отделения морковь черпаками 9 перебрасывается в третье
отделение, а камни оседают и удаляются через люк 10. Из третьего отделе-
ния морковь черпаками 9 выбрасывается в элеватор, подающий ее в тероч-
ную машину. Кулачную мойку следует установить на кирпичном фунда-
менте высотой 1—1,5 м над уровнем пола для удобства очистки ее от грязи.
Пол под мойкой цементируют с уклоном 5 мм на 1 м для быстрого удаления
сточных вод.
Диффузор Гузенко. Применяется для извлечения экстрактивных ве-
ществ из плодов шиповника [5]. Диффузор (рис. 77) является аппаратом
непрерывного действия. Он состоит из двух1 расположенных один над дру-
гим барабанов 1, 2. Нижний барабан опирается двумя бандажами 3 на че-
тыре ролика 4. Головная часть барабана опирается на подшипники непо-
движной сокоотборной коробки 5, а хвостовая — на корпус шнека 6, выгру-
жающего жом из диффузора. Верхний барабан1 — ошпариватель длиной 2 м
своими концами сочленяется с неподвижными камерами для загрузки сырья
7 и для отбора сока 8. Вращение барабана осуществляется зубчатой переда-
чей 9. Верхний и нижний барабаны по своей длине разделены поперечными
алюминиевыми дисками на отдельные секции. Каждый из дисков имеет по
3 эллипсоидальных выреза аъ а2, а3, к которым прикреплены ковши спе-
циальной конструкции из штампованных сит. Примыкая один к другому,
эти диски образуют в барабане ряд камер, сообщающихся между собой
указанными эксцентрическими вырезами в перегородках. Когда ковши
1 При переработке сухих плодов верхний барабан не нужен; он необходим при
переработке сырых плодов.
12—522
353
находятся в верхнем положении, то через эти вырезы измельченное сырье
сбрасывается в следующую камеру, а когда ковши находятся в нижнем
положении, через них проходит сок. Питание аппарата осуществляется
через бункер 10 с дозатором И. Вода поступает через нижний вал шнека 12.
Диффузор приводится в движение от двигателя через червячный редуктор
и систему зубчатых колес 13. Техническая характеристика: производи-
Рис.‘ 78. Пресс Яна.
, • /
тельность 5 т сухого шиповника в сутки; частота вращения барабанов
0,5 об/мин-, диаметр барабанов 1,0 л; габариты 11,21x2,05x3,39 м.
Пресс Яна [5]. Предназначен для отжатия сока из морковной мезги в
производстве каротина из моркови. Пресс (рис. 78) имеет следующее устрой-
ство: два полых барабана 1 покрыты ситами с диаметром отверстий 0,5—
0,7 мм. Барабаны подобно вальцам вращаются в противоположные сторо-
ны. Расстояние между барабанами регулируется винтом с маховиком 2.
Над барабанами устанавливают бункер 3, внутри которого расположен
снабженный лопатками направляющий прибор 4, разбивающий комья
мезги, равномерно распределяя ее в бункере. Под барабанами смонтирова-
на камера 5, закрываемая крышкой 6. Масса крышки и жома уравновеши-
ваются грузом 7, расположенным на рычаге 8.
Из терки, которую обычно устанавливают над прессом, мезга поступает
в бункер 3, а из него попадает в пространство между двумя барабанами,
где подвергается прессованию. Через отверстия в сите сок проходит внутрь
354
барабанов, откуда и удаляется через торцовую часть пресса. Затем мезга
попадает в камеру допрессовывания, мезги 5, где дополнительно выделяет-
ся некоторое количество сока. Частота вращения барабанов пресса 4 об/мин.
Расход мощности 2,5—3 кет.
Часовая производительность пресса до 1500 кг моркови или тыквы.
Выход сока после первого прессования составляет 70% к массе сырья.
Шнековый пресс непрерывного действия. Применяется для отжатия
воды из жома шиповника перед процессом сушки. Пресс изготовляется из
стали и имеет следующую характеристику: производительность по сырью
5000 кг/ч\ мощность двигателя 10 квт\ выход сока 80—85%; марка пресса
ПНД-5. На рис. 79 дан внешний вид пресса [15].
Выпарной аппарат системы «Виганд» (стр. 343). Используется для
сгущения диффузионного сока в водный концентрат.
Сушилка с форсуночным распылением (стр. 350). Применяется для вы-
сушивания водного концентрата в порошок с витамином С или с витамином Р.
Рис. 80. Паровая конвейерная сушилка непрерывного действия.
12*
355
Экстракционный аппарат непрерывного действия. Используется для
экстракции каротина из сухого белкового лепестка тыквы дихлорэтаном
(стр. 346).
Паровая конвейерная сушилка непрерывного действия ПКС-20. (рис. 80).
Сушилка представляет собой [16] железный шкаф, внутри которого один
над другим установлены четыре конвейера с сетчатыми лентами из нержа-
веющей стали 1. Рабочая поверхность каждой ленты 5 м2. Паровые калори-
феры 2 установлены под рабочей поверхностью ленты. Общая поверхность
Рис. 81. Центрифужный молекулярно-дистиляционный аппарат НИИХИММАШа.
калориферов 248,4 м2. Подогретый воздух поступает под ленты. Отрабо-
танный воздух отсасывается через вытяжную трубу 3. Применяется для
сушки жома. Шрот на сушильную ленту подается элеватором 4. Скорости
движения лент на четырех транспортерах имеют отношение 1:0,66; 0,5:0,4.
Продолжительность сушки составляет около 140 мин. На первых трех лен-
тах установлены ворошители продукта 5.
Центрифужный молекулярно-дистилляционный аппарат НИИХИММАШа
[17]. Используется для дистилляции витаминов А из рыбьего жира и
витамина Е из растительных масел. Устройство аппарата показано на
рис. 81. Ротор 1 аппарата изготовлен из алюминия и имеет по верхнему
краю диаметр 0,4 м. Дистиллируемая жидкость подается на дно ротора
через трубу 2, распределительную тарелку 3 и трубки 4. Под действием
центробежной силы жидкость поднимается по стенкам ротора весьма
тонкой пленкой (доли мм) и сбрасывается в алюминиевый приемный
желоб 5. Стенки ротора обогреваются электронагревателем 6. Остаток
жидкости из желоба 5' через карман 7 выводится из аппарата.
Внутри ротора помещены два конденсатора 8 и 9. Конденсатор 8, распо-
ложенный против поверхности испарения, состоит из полых элементов.
356
Внутри элементного конденсатора расположен змеевиковый конденсатор 9.
Температура поверхности первого конденсатора более высокая нежели
второго. Благодаря этому на элементном конденсаторе конденсируются
пары более тяжелой фракции. Пары наиболее летучих веществ проходят
через зазоры между элементами и конденсируются на втором конденсаторе.
Расстояние между элементным конденсатором и внутренней поверх-
ностью ротора составляет 15 мм. Между конденсатором и ротором установ-
лена съемная сетка 10, которая играет роль дефлегматора: наиболее тяже-
лые компоненты паров дистиллята конденсируются на сетке и стекают
обратно на дно ротора в виде флегмы. Под конденсаторами расположены под-
доны И и 12 для сбора дистиллята. Из поддонов дистиллят через специаль-
Рис. 82. Аппарат непрерывного действия для облу-
чения растворов эргостерина.
ные приспособления сбрасывается и по трубкам 13 и 14 выводится из ап-
парата.
Корпус аппарата 15 снабжен крышкой 16, на которой расположены два
смотровых люка 17. Под крышкой расположен отражательный козырек 18.
Вал 19 проходит через стакан 20 со специальным уплотняющим устрой-
ством. Откачка неконденсирующихся газов из аппарата производится че-
рез боковой штуцер 21.
Техническая характеристика аппарата
Поверхность испарителя, ж2.......... 0,3
Мощность электронагревателя, кет . . 3,5
Максимальная температура дистилля-
ции, °C ............................ 270
Частота вращения, об/мин ........... до 2000
Рабочий вакуум в аппарате, мм рт. ст. до 1-Ю-3
Габаритные размеры, мм
высота........................... 1365
максимальный диаметр по фланцу . 740
Масса аппарата, кг .................... 240
Расход мощности на вращение ротора
при 2000 об/мин, кет ......... 0,8
Аппарат непрерывного действия для облучения растворов эргостерина
показан на рис. 82 и применяется в производстве витамина D2. Аппарат
состоит из двух кольцеобразных сосудов из кварцевого стекла, установлен-
ных один в другой. Каждый сосуд представляет собой двухстенный цилиндр,
полый внутри. Наружный сосуд 1 служит для пропускания раствора эрго-
стерина (вход в штуцер 2 и выход в штуцер 3). Внутри сосуда 1 расположен
сосуд 4 для водяного охлаждения (вход в штуцер 5 и выход из штуцера 6).
Во внутренней полости сосуда 4 устанавливают ртутно-кварцевые лам-
пы/. Ультрафиолетовые лучи, проходя через кварцевый сосуд 4 с холодной
водой, охлаждаются, а затем поступают в сосуд облучения 1. Установка
из трех последовательно включенных аппаратов имеет производительность
10 л/ч [5].
357
НОВЫЕ МАТЕРИАЛЫ ДЛЯ ИЗГОТОВЛЕНИЯ АППАРАТУРЫ, РАБОТАЮЩЕЙ
С АГРЕССИВНЫМИ СРЕДАМИ [2]
К таким материалам следует отнести титан, тантал, цирконий.
Титан. Плотность титана 4500 кг/м3, коэффициент линейного расши-
рения в интервале 20 — 100°С —9,0 -10-6; удельная теплоемкость
0,13 кал!(г-°C); температура плавления 1725° С, может применяться при
температуре не выше 350° С. Титан устойчив по отношению к агрессивным
средам при следующих условиях (табл. 21). Титан применяют для футеров-
, Таблица 21
Среда Концент- рация, % Темпера- тура, °C Среда Концент- рация, % Темпера- тура, °C
Муравьиная кислота . 90 100 Щавелевая кислота 10 35
Хлор с содержанием Фосфорная кислота . 30 35
воды более 0,013% —— 75 Азотная кислота . . 70 100
Уксусная кислота . . Любая Кипение Соляная кислота . . 1,0 70
Серная кислота . . . 5 35
ки стальной аппаратуры с применением специального метода точечной
сварки.
Тантал чрезвычайно устойчив к хлору и к кислотам азотной, соляной,
серной и фосфорной при температуре до 250° С. Тантал не взаимодействует
с 98%-ной серной кислотой при температуре до 150° С. При температуре
175° С скорость коррозии достигает 0,0025 мм, а при 200° С — 0,038 мм в
год. Тантал обладает хорошими физико-механическими свойствами: темпе-
ратура плавления 3000° С ; плотность 16600 кг/м3-, коэффициент линейного
расширения 6,58 • 10~6; удельная теплоемкость 0,036 кал/(г-°С). При рабо-
те при температуре свыше 300° С тантал становится хрупким, что ограни-
чивает его применение. Тантал также не применим для растворов плавико-
вой кислоты и горячих крепких щелоков.
Цирконий. В последнее время за рубежом начали изготовлять стальные
аппараты, футерованные цирконием (в виде тонкой фольги), имеющим сле-
дующие физико-механические свойства:- температура плавления 1845° С;
плотность 6500 кг/м3-, коэффициент линейного расширения — 6,58-10"6;
удельная теплоемкость 0,068 кал/(г-°С).
Цирконий применяют главным образом для аппаратов, в которых че-
редуются реакции со щелочами и кислотами. Очень стоек по отношению
к уксусной, щавелевой и фосфорной кислотам. Нестоек к азотной и соляной
кислотам.
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
I. Шнайдман Л. О., Хохлов И . М., Дульчина Б. М., Сол ян-
кина Л. Н. Непрерывный процесс ацетонирования L-сорбозы.—Веб.: «Вита-
минная промышленность». М., ГОСИНТИ, 1960, 6, 12.
2. Н и к о л а е в В., У д ы м а П. Новое оборудование для заводов химической
промышленности. М., Машгиз, 1960, 50. То же. ЦИНТИхимнефтемаш. Зарубежное
химическое машиностроение на Международной выставке в Москве. М., 1966.
3. К о 1 b е 1 Н., Hammer Н., L а п g е m а п п Н., Chem. Ztg. Chem. Apparat,
1968, 92, 581.
4. Mutzenberg A., Giger A., Chem. Techn., 1969, 21, 1, 52.
5. Шнайдман Л. О. Производство витаминов. M., Пищепромиздат, 1958, 414 с.
6. Каталог-справочник. Промышленные центрифуги. М., ЦИНТИхимнефтемаш,
1966.
6а. Каталог «Колонные аппараты», ЦИНТИхимнефтемаш, 1966.
7. Зарубежное химическое машиностроение на Международной выставке в Москве. М.,
ЦИНТИхимнефтемаш, 1966.
8. Л е б е д е в И. И. Процессы и аппараты химической и нефтехимической техноло-
гии (учебник). М., «Химия», 1971, 840 с.
358
8a. Fritz Peter, Chem. Ztg. Chem. Apparat, 1969, 93, 2, 71.
9. Сошников Д. Я-, С ю x и н а В. И., Петунина А. Г. Выделение
глюконовой кислоты из глюконата кальция ионообменным методом. — «Приклад-
ная биохимия и микробиология», 1968, т. 4, вып. 4, с. 440—443 с ил.
10. Б у р о в а Л. Е., Плановский А. Н. О предельной скорости движения
дисперсной фазы в колонном экстракторе с механическим перемешиванием. — «Ме-
дицинская промышленность СССР», 1966, № 7, с. 35—38 с ил.
11. И о ви ко в а К- Е., Шведов Ю. П., Метельников В. А., Конд-
ратьева И. М. Получение р-альдегида С14 методом непрерывной ректифика-
ции под вакуумом. —«Хим.-фарм. ж.»., 1971, № 2, с. 49—52 с ил.
12. Соколовский А. А., Хлуденев И. К-, Кондратьева Н. М.,
Кондакова Т. С. Сушилки с вихревым режимом. — «Хим.-фарм. ж.»., 1970,
№ 10, с. 43—46 с ил. То же. Кочетов Л. М., Сажин Б. С. Вихревые
камеры для сушки волокнистых материалов. — «Химическая промышленность»,
1971, № 6, с. 474 с ил.
13. Г е л ь п е р и н Н. И., Филорикьян Д. Ф. Сушка материалов в аппара-
тах с псевдоожиженным слоем и во взвешенном состоянии. — «Хим.-фарм. ж.».,
1969, № 2, с. 37—46 с ил.
14. Каталог химического оборудования, М., «Машиностроение», 1964, 38.
15. Пресс ПНД-5, ВДНХ, Киев, Гостехиздат, 1959.
16. Ш е л а м о в а А. С., Бабак А. М. Сушка яблок на паровых конвейерных
сушилках ПКС-20. М., Пищепромиздат, 1955.
17. М а т р о з о в' В. М. Молекулярно-дистилляционный аппарат центрифужного
типа. — В сб.: «Исследования сублимационных и дистилляционных аппаратов и
гидродинамики мешалок». М., Машгиз., 1954, 16, с. 63.
18. Каталог «Сушильные аппараты», ЦИНТИхимнефтемаш, 1965.
Часть П. ПРОИЗВОДСТВО ВИТАМИННЫХ ПРЕПАРАТОВ
ИЗ ПРИРОДНОГО СЫРЬЯ
Роль научных исследований в биологической оценке природных пи-
щевых продуктов и в развитии витаминологической науки. Еще в 1881 г.
Н. И. Лунин показал, что подопытные мыши при их кормлении искус-
ственным молоком гибли, а молоком матери нормально развивались. Со-
поставление биологической ценности искусственного и природного молока
привело II. И. Лунина к мысли о содержании в естественной пище в малых
количествах каких-то дополнительных пищевых ингредиентов. Исследо-
вания голландского врача Эйкмана показали, что куры заболевали по-
линевритом при их кормлении белым рисом и излечивались при введении
в рацион неочищенного риса. Отсюда исследователь пришел к заключению,
что в рисовых отрубях имеется какой-то дополнительный фактор питания,
отсутствие которого вызывает полиневрит. Исследование отрубей риса при-
вело к открытию витамина Вх.
Исследования норвежских ученых Гольста и Фролиха и английско-
го ученого Гопкинса показали, что причиной возникновения цинги
является недостаток в пище свежих овощей и фруктов. Эти исследования,
а также изучение антицинготных свойств хвои, капусты, цитрусовых при-
вели к открытию витамина С.
Изучение действия печеночного трескового жира, а также облученного
эргостерина дрожжей на заболевание рахитом и ксерофтальмией привело
к открытию ростового фактора витамина А и антирахитического витамина D.
В 1919 г. Стинбок обратил внимание на то, что растительный пиг-
мент каротин, выделенный из моркови, подобно витамину А стимулирует
рост животных.
В 1937 г. Е. Ледерер и В. Розанова, изучая состав печеночного жира
пресноводных рыб, открыли витамин А2.
В 1966 г. Л. Шнайдман, М. Ушакова, А. Ефимов и И. Кущинская, изу-
чая каротиноиды плодов шиповника и томатов, открыли А-витаминное
'действие ликопина (витамин А3). Витамин В15 был выделен Кребсом
из ядер абрикосовых косточек. Дам, Каррер и др. открыли витамин
Кг в результате исследования люцерны.
Эти весьма краткие данные из истории витаминологии приведены для
того, чтобы показать, что открытия в будущем новых биологически актив-
ных веществ будут всецело зависеть от размаха химических, биологических
и агротехнических исследований природного сырья. Прогресс витами-
нологической науки будет в основном обусловлен этими исследова-
ниями.
Глава 17. ПЛОДЫ ШИПОВНИКА И КОМПЛЕКСНАЯ ПЕРЕРАБОТКА
ИХ (R3SAE)
Лечебные свойства шиповника известны давно. Об этом свидетельствуют
рецепты лечебников XVII в. Например, о масле шиповника в них указыва-
ется: «Аще кто внутрь то масло приемлет или же шею помазует, пользу
творит горлу, кои хрипит. То масло, аще мешаем с молоком козьим и прием-
360
лем, тогда стружение в мйхире (мочевом пузыре) уймет и болезнь пузырную
и печенную выведет и залегание водяное отворит». Однако применение
плодов шиповника для лекарственных целей было крайне ограниченным.
Переработка шиповника на концентраты витамина С началась после того,
как в СССР Всесоюзным институтом растениеводства была установлена вы-
сокая антицинготная активность шиповника.
Ботаническая характеристика и географическое распространение. Ши-
повник принадлежит к семейству розовоцветных и представляет собой
многолетний кустарник высотой от 0,5 до 6 м. Ветки и молодые побеги
шиповника густо усажены шипами;, листья сложные, эллиптической или
яйцевидной формы (с пильчатыми зубчиками по краям). Цветы розовые
или белые. Плоды шиповника — ложные ягоды — имеют шаровидную,
•бутыльчатую или грушевидную форму и содержат большое количество се-
мян. Цвет зрелых плодов — желтовато-красный или темно-красный. Мя-
коть плода внутри покрыта волосками. Шиповник растет по склонам овра-
гов, гор и в поймах рек, образуя заросли по лесным опушкам. Плоды созре-
вают в августе-сентябре.
Наиболее богаты витамином С плоды шиповника, произрастающие в
северной и средней полосе европейской части СССР, в Киргизской ССР и
на Дальнем Востоке. Плоды шиповника, распространенного в южных райо-
нах (шиповник собачий), содержит меньшее количество витамина С (0,1—
0,2% на сухую массу) при почти одинаковом содержании витамина Р с
Розой коричной. Самыми распространенными видами шиповника являются
Роза коричная и Роза иглистая. Роза коричная с крупными розовыми или
темно-красными пяти лепестковыми цветами у основания черешка листьев
имеет два изогнутых шипа. Плоды Розы коричной окрашены в оранжево-
красный цвет, снабжены пятидольной чашечкой, не опадающей после со-
зревания плодов. Роза иглистая в отличие от Розы коричной имеет на ство-
ле и ветках тонкие прямые шипы. Плоды оранжево-красные, но не блестя-
щие, а матовые.
Роза коричная произрастает в Рязанской, Горьковской, Пензенской,
Куйбышевской, Архангельской, Вологодской областях, в Марийской, Чу-
вашской,' Мордовской, Татарской, Башкирской автономных республиках
и Красноярском крае. Роза иглистая произрастает главным образом в
Сибири и на Алтае.
На основании исследований Воронцовской центральной биологической
станции Всесоюзного научно-исследовательского витаминного института
(ВНИВИ) могут быть рекомендованы для введения в культуру следующие
виды шиповника: Роза коричная, Роза иглистая, Роза морщинистая, Роза
сизая, Роза яблочная, Роза Федченко, Роза Уэбба, Роза Альберта, Роза
даурская как высоковитаминные, урожайные, зимостойкие, засухоустой-
чивые, стойкие против грибных болезней и вредителей [2—7].
Сырьевые запасы шиповника. СССР богат зарослями дикорастущего
шиповника. Общие ежегодные запасы шиповника превышают 56 тыс. т
сырых плодов. Около 80% всех выявленных запасов шиповника находится
в РСФСР. Шиповником богаты районы Центральной черноземной полосы,
Поволжья и Урала, Сибирь, Дальний Восток, Средняя Азия и Казах-
стан.
Объем заготовок плодов шиповника колеблется в пределах 11—21% от
его валового урожая. Лучше других используются ресурсы шиповника
Волжского бассейна (около 60%).
Основное количество сырья поступает из пойм рек Волги, Камы, Оки,
Белой, Вятки, Ветлуги и Суры. Ведущими районами по заготовке плодов
шиповника являются: Татарская АССР, Башкирская АССР, Чувашская
АССР, Рязанская, Горьковская, Куйбышевская и Ульяновская области.
В одних этих районах ежегодно может быть заготовлено 3—4 тыс. т
плодов шиповника.
361
ХИМИЧЕСКИЙ И ВИТАМИННЫЙ СОСТАВ ПЛОДОВ ШИПОВНИКА
Химический состав плодов шиповника видов Р. коричная и Р. иглистая
исследовала Вадова [8], а видов Р. даурская и Р. собачья — Сабуров [9].
В сухой мякоти плодов Р. коричной (R. Cinnamomea) найдено [8] (в %):
общего сахара 23,93; инвертного сахара 18,56; клетчатки сырой 12,52; пекти-
новых веществ 14,1; золы сырой 6,4. Общая кислотность составляет 2,84 %.
В золе отмечено высокое содержание солей калия, магния и фосфора.
410 ЧОО 490 нм
IX
Рис. 83. Спектры погло-
щения каротиноидов ши-
повника в видимой
области света:
1—фитофлюин; 11— полицис
ликопин a; 111— криптоксан-
тин; IV — а-каротин; V .— по-
лицис ликопин b; VI — ру-
биксантин; VII — {3-каротин;
V111 — полицис ликопин с;
/X — тараксантин; X — Z-ка-
ротин; X/ —ликопин.
Плоды шиповника богаты витаминами. Среднее содержание аскорбино-
вой кислоты в сухих плодах шиповника, поступающих на витаминные
заводы, составляет 1200—1500 мг% и первоначально плоды оценивали ис-
ключительно по содержанию в них аскорбиновой кислоты. Однако
дальнейшие исследования показали, что плоды шиповника содержат
и другие витамины. Так, изучен состав каротиноидов плодов вида
R. Cinnamomea [10]. Методами хроматографии и спектрофотометрии опре-
делен следующий состав каротиноидов (в % к общему содержанию):
1) группа каротинов С4оН58 (а-, |3-, z-каротин) — 5,5; 2) группа ликопина
С4оН5в (полицис-лакопин-а, полицис-ликопин-Ь и ликопин) — 53,2 и 3)кис-
лородсодержащие каротиноиды (криптоксантин, С40Н56О; рубиксантин,
С4оН560; тараксантин, С40Н5вО4) —41,3. На рис. 83 даны спектры погло-
щения этих каротиноидов в видимой области света. Биологические испыта-
ния групп каротиноидов на лабораторных животных показали, что А-ви-
таминной активностью обладают каротины и ликопины. Кислород-
содержащие каротиноиды обнаружили весьма слабую биологическую ак-
тивность [12].
В результате изучения в плодах шиповника состава флавоноидных ве-
ществ (витамины группы Р) идентифицированы [11, 13]: кверцетин,
С)5Н10О7; кемпферол, С15Н9О6; изокверцитрин, С21Н20О12; тилирозид-кемп-
362
ферол-7-р-кумароил-3-|3-глюкозид, С3оН2в013. На рис. 84 даны УФ-спект-
ры поглощения указанных флавоноидов шиповника.
Общее содержание витамина Р составило в свежих плодах (в % на су-
хую массу): для Розы коричной 4,0, для Розы морщинистой — 2,13.
Идентифицированы катехины: (—) — эпигаллокатехин, (—) — галлока-
техин, (—)-эпигаллокатехингаллат и (—) — эпикатехингаллат. Общее
содержание дубильных веществ, определенное методом Левенталя, составило
Рис. 84. УФ-спектры поглощения флавоноидов шиповника:
а — кверцетин; б — кемпферол; в — изокворцитрин; г — тилирозид.
в сухих плодах 4,4%. Из антоциановых веществ идентифицирован циани-
дин, а общее их содержание— 45 мг%. Изучен состав токоферолов в масле
семян шиповника и установлено, что их общее содержание составляет
170 мг%. Идентифицированы а- и |3-токоферолы.
ИССЛЕДОВАНИЯ ТЕХНОЛОГИИ КОМПЛЕКСНОЙ ПЕРЕРАБОТКИ ПЛОДОВ
ШИПОВНИКА НА ВИТАМИННЫЕ ПРЕПАРАТЫ
Идентификация биологически активных веществ показала, что, кроме
аскорбиновой кислоты, в плодах шиповника содержатся флавоновые,
катехиновые, антоциановые вещества (витамин Р), каротиноиды и токофе-
ролы [Ю, 13 , 14]. Задачей исследований в области технологии
явилась разработка промышленных методов комплексного использо-
вания этих биологически активных веществ. При этом был изучен ряд тех-
нологических вопросов:
исследованы различные схемы подготовки плодов шиповника для диффу-
зионного процесса и показано, что для этой цели целесообразно исполь-
зовать плоды шиповника без отделения семян, так как при сепарации семян
от мякоти потери витамина С составляют 7,3% [15];
разработана технология получения препарата витамина Р из жома ши-
повника. Производственные испытания, проведенные совместно с Щел-
ковским витаминным заводом (Е. М. Потак), подтвердили наши ла-
бораторные исследования о возможности замены при экстракции флаво-
ноидов спирта водой. В обоих случаях выход витамина Р составил
около 50%, а содержание его в порошке 20—22% [16];
показано, что предварительная горячая водная экстракция Р-витамин-
ных веществ из жома шиповника способствует в дальнейшем более полно-
му извлечению из него каротиноидов [16];
исследована технология экстракции каротиноидов из мякоти плодов
шиповника и показано, что применение органического растворителя вместо
растительных масел позволяет увеличить выход пигмента с 72,75—78,75
до 92,2%. Из растительных масел наивысший эффект дает соевое масло —
78,75% вместо 72,75% для подсолнечного масла [171;
363
изучена сохранность каротиноидного пигмента в сухом жоме шиповника
и в растворе растительных масел и установлена удовлетворительная ста-
бильность каротиноидов в растительных маслах, в особенности в соевом.
При хранении в течение 133 дней при температуре 20° С потери каротинои-
дов составили: для подсолнечного масла — 9,4%, для кукурузного — 7,7%,
для соевого — 2,64%. В сухом жоме каротиноидный пигмент нестоек:
за 40 дней хранения при 20° С потери его составили в неразмолотом жоме —
30,0%, а в размолотом — 48,6% [18]. Высокая эффективность соевого мае-
Сироп с витаминомС
расфасовку
Линия получения концентрата с витамином С
Линия получения концентрата с
витамином Р
{Порошкообразный
/концентрат с вита-
пином С на тавлети-
Жам 7 с семенами Р°^ани-е
1
j СО {Порошкообразный
Ь | ^концентрат с
витаминомР на
Рис. 85. Технологическая схема комплексной промышленной переработ-
ки плодов шиповника.
ла обусловлена содержанием в нем сильных антиоксидантов — у- и 8-то-
коферолов.
На основании результатов лабораторных, полузаводских и заводских
исследований автором при участии И. Н. Кущинской, Д. Я. Сошникова,
М. Е. Воликова и П.Н. Трофимовой разработана технология промышлен-
ного комплексного использования плодов шиповника для производства
витаминных препаратов [16—24]. Технологическая схема комплексной
промышленной переработки плодов шиповника приведена на рис. 85.
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА ВИТАМИННЫХ ПРЕПАРАТОВ
Концентрат с витамином С. Сухие плоды шиповника элеватором 1
(рис. 85) подают в диффузор Гузенко 2 (температура 70—75° С). Диффу-
зионный сок направляют через фильтр-пресс 3 в трехкорпусный выпарной
аппарат 4, где сок сгущают до плотности 60% и перекачивают далее,
либо в распылительную сушилку 5 для получения порошкообразного кон-
центрата с витамином С, идущего на таблетирование, либо для приготовле-
ния сиропа с витаминами С и Р.
Концентрат с витамином группы Р. Сырой жом I направляют во второй
диффузор Гузенко 2, где процесс диффузии протекает при повышенной тем-
пературе (98—99° С). Диффузионный сок через фильтр-пресс 3 посту-
пает в выпарной аппарат 4 и оттуда в вакуум-вальцовую сушилку 6. Сухой
порошок поступает на таблетирование.
Каротиноидный препарат. Сырой жом II направляют в барабанную су-
шилку 7 и далее в сепаратор 8 для отделения семян. Сухой жом подают в
непрерывно-действующий экстракционный аппарат колонного типа 13.
Экстракцию ведут дихлорэтаном, либо хлористым метиленом. После отгон-
ки растворителя в вакуум-аппарате 16 получают каротиноидный пигмент
в виде пасты. Если экстракцию пигмента ведут маслом, то получают масля-
ный препарат — каротолин [23].
364
Масло семян (концентрат витамина Е). Семена после сепаратора 8 на-
правляют в дробилку 9 и в экстрактор 10 и экстракцию ведут органическим
растворителем. Последний удаляют в вакуум-аппарате 12. По данной тех-
нологической схеме из 1 т сухих плодов шиповника получают (в кг):
Сироп с витамином С...................... 2920,0
Р-витаминный препарат...................... 63,5
Каротиноидный пигмент в виде пасты .... 27,4
Масляный препарат (каротолнн).............. 30,0
Масло семян шиповника...................... 22,8
Все новые препараты проверены биологически, клинически и Фарма-
кологическим комитетом Министерства здравоохранения СССР, разрешены
к, выпуску для использования в профилактических и лечебных целях.
Технология комплексной переработки плодов шиповника внедрена на
Уфимском, Йошкар-Олинском витаминных заводах.
ТЕХНОЛОГИЧЕСКИЕ ПОКАЗАТЕЛИ
Подробное описание технологических процесов производства и аппара-
туры дано в литературе [4, 5, 25]. Основные технологические показатели
приведены в табл. 22.
Таблица 22
Показатели Получение диффузноичого сока из шиповника (плоды)
свежие сушеные
" целые | МЯКОТЬ
Характеристика сырья, содержание аскорбиновой кислоты, мг% 800—1400 1200—1500 2000
влажность, % 60—65 11—15 5—8
содержание семян, % 34—38 45—50 —
Диффузионный процесс Диффузия батарейная продолжительность оборота диффу- зионной батареи, мин 120 50 60
наивысшая температура на диффузо- ре, °C 75 75 . 75
нагрузка сырья на 100 л емкости диффузора, кг 40 30 25
откачка диффузионного сока, % . . 150 300 350
выход жома, % выход диффузионной воды, % . . . 160—170 215—250 250
100 100 100
содержание сухих веществ в диффу- зионном соке, % содержание''аскорбиновой кислоты, % в жоме 8 6 8
0,02 0,02 0,02
в воде 0,01 0,01 0,01
Диффузия непрерывная откачка сока, % 150 200 200
выход жома, % 150 170 200
содержЛие сухого вещества в диф- фузионном соке, % потери аскорбиновой кислоты в жо- ме, % — 6—8 —
1,7—2,3 —
неопределенные потери аскорбиновой кислоты, % — 0,2—0,3 —
Состав сырого жома шиповника (из целых плодов), %
Вода ...................... 68—73
Клетчатка.................. 17—15
Пектиновые вещества........2,5—2,0
Зола сырая ............2,5—2,0
Общие сахара...............2,5—2,0
Органические кислоты . . . 1,0—0,5
Белковые вещества .... 3,0—2,5
Жир....................... 2,8—2,4
Аскорбиновая кислота . . . 0,05—0,02
365
Очистка диффузионного сока шиповника энзимами. Производство энзи-
матического препарата аспергиллюс нигер1. Питательная среда: морковь
65%; отруби 35%. Смесь варят в воде (50% к массе смеси), охлаждают до
30° С. Грибницу выращивают в кюветах при температуре 30—32° С в те-
чение 48 ч., Затем ее сушат при температуре 40—45° С, измельчают и про-
сеивают через сито № 16.
Процесс ферментации. Ферментация мезги имеет преимущество перед
ферментацией диффузионного сока. При ферментации мезги происходит
более полное извлечение аскорбиновой кислоты и других экстрактивных
веществ. Расход препарата фермента 2—3% к массе шиповника. Продолжи-
тельность процесса ферментации 8—12 ч, температура 43—45° С.
Процесс упаривания диффузионного сока из плодов шиповника. Темпе-
ратурный режим в трехкорпусной выпарной установке (в °C): в первом
корпусе 90, во втором 75, в третьем 55—60. Потери аскорбиновой кислоты
в процессе выпаривания составляют 2—3%, содержание сухих веществ
в водном концентрате 50—55%.
Химический состав водного концентрата (в % на сухое вещество)
Зола сырая ........... 6,2—7,7 Фосфор (Р2О5) ...... 0,7—0,8
Клетчатка............ 1,4—4,7 Белковые вещества .... 4,5—6,9
Пектиновые вещества . . 7,6—9,4 Аскорбиновая кислота . . . 3,0—5,0
Общий сахар .......... 27,2—32,2
Производство жидких концентратов витамина С
[4, 22, 25] (табл. 23)
Таблица 23
Вид концентрата Расход спир- та, л на 1 л концентрата Потери витами- на С при пере- работке водного концентрата, %
Спиртоочищенный . . . 0,25 10,0
Спиртованный 0,20 1,0
Спирто-сахарный .... Витаминизированный си- 0,15 1,0
роп — 2,0
Витаминизированный сахарный сироп1. При содержании аскорбиновой
кислоты в 1л сиропа 5 г, сухих веществ 73% й плотности 1340 кгДи3
расход сахара в зависимости от содержания в диффузионном соке аскор-
биновой кислоты и сухих веществ можно определить по формуле
Сх = 730
1,34 —
5000 Вх
100 А
= 978,2
50 Вх
А
где Сх — расход сахара, кг/1000 л;
А — содержание аскорбиновой кислоты в 1 л диффузионного сока, г;
Вх — содержание сухих веществ в диффузионном соке.
Сушка водного концентрата
Продолжительность сушки на вальцах, сек.......... 8—10
Продолжительность сушки при распылении, сек . . . 0,02—0,03
Потери аскорбиновой кислоты при сушке, % ... . 10—12
Производство порошка шиповника размолом [4]
Выход плодовой мякоти (кожуры) из целых пло-
дов, %........................................... 45—50
Выход зерна из целых плодов, %................... 40—45
Потери массы при фракционировании, %............. 1—1,5
Неопределенные потери аскорбиновой кислоты, % . . 5,0
Потери аскорбгновой кислоты при сушке кожуры
с 11—12 до 5—8% содержания влаги, % ........ 7—8
1 Подробное описание см. [4].
366
Оптимальная влажность плодов шиповника,
поступающих на сепарацию, % ..................... 11 —14
кожуры, поступающей на размол, % ........ 5,0
Предельная величина частиц порошка, мм......... 0,4
Оптимальный размер частиц (сход с сита № 43 и про-
ход через сито № 23), мм ...................... 0,2—0,35
Таблетирование порошка
Влажность порошка не должна превышать, %........... 6
Допускается мучнистой фракции (0,16 мм), % ..... 20—25
Производство пюре и пасты из плодов шиповника.
черной смородины и других ягод
Выход пюре, %
из плодов шиповника...............................55—60
из черной смородины ............................82—85
Отношение пюре к сахару................................ 1:1
Температура розлива шоре и пасты, °C ...............90—95
Выход аскорбиновой кислоты в пюре и пасте к массе вве-
денной с сырьем, % ...............................85,0
Потери аскорбиновой кислоты при хранении в течение
6 месяцев при комнатной температуре, %.............. 18,0
Примерные нормы расхода сырья и материалов на производство кон-
центратов витамина С из плодов шиповника приведены в табл. 24 и 25.
Таблица 24
Наименование сырья Нормы расхода
на 1000 л сиропа с витамином С на 1 tn порошка с витамином С на 1 tn таблеток с витами- ном С
без добав- ления ягод- ных экст- рактов с добав- лением ягодных экстрактов из концен- трата пло- дов ши- повника из плодов- шиповни- ка
Плоды шиповника сухие, кг1 .... 476 488 462 367 4280 1874 4032
Черная смородина свежая в кг . . . — 73,7 — — —
Сахар, кг 812 838 — — —
Тальк, кг — — — — 50
1 Расход сырья (в кг): для батарейной диффузии — 476; для мацерационного спо-
соба— 488; для диффузии Гузенко — 462.
Таблица 25
Сырье и материалы
Расход, кг
Производство концентрата ви-
таминов группы Р
Жом шиповника (сырой) .
Сухой концентрат витами-
нов группы Р (22%-ный) .
Тальк ...................
Сахар ...................
Производство каротиноидного
пигмента (пищевого красителя)
Шрот II (сухой)..........
Масло растительное соевое
Дихлорэтан...............
Спирт ...................
Производство масла из семян
шиповника
Семена шиповника ....
Дихлорэтан...............
Спирт......................
На 1 т таблеток массой 0,5 г
каждая и содержанием витами-
на Р 30 мг
16282
273
50
677
На 1 т пищевого красителя в
виде пасты и масляного раст-
вора
1000 9710
1070 500
— 971
— 50
На 1 т масляного концентрата
15380
1600
50
367
368
Нормы качества готовой продукции, получаемой из плодов шиповника
Таблица 26
Наименование препаратов гсст, МРТУ Общая характеристика 9 Содержание витаминов, мг в 1 мл, не менее Содержание сахара, %, не менее । Содержание сухих ве- ществ, %, не 1 менее Влажность, %, ие более Зола, раство- римая в 10%- , ной НС1, %, i не более Плотность, кг/м8 Показатель преломления
с Р
Сироп с витамином С из шиповника без добавле- ния ягодных экстрактов ГОСТ 8460—57 Однородная консистенция, сладкого вкуса, краснова- то-коричневого цвета 5 — 62,5 72,0 — о,1 — —
Тоже, с добавлением ягодных экстрактов ГОСТ 8460—57 То же, с добавлением ягодного экстракта 4 —- 62,5 72,0 — 0,1 — —
Витаминный сироп пло- дов шиповника МРТУ—42 Яг 3018—62 Густоватая жидкость, ко- ричневого цвета, со свое- образным запахом и кис- ловато-сладким вкусом 27—33 13,5—16,5 — ’ — — — 1354—1368 1,468—1,474
Порошок витамина С из концентрата плодов ши- повника ГОСТ 8460—57 Порошок без комков и по- сторонних примесей 22 — — 93,0 — 0,15 — —
Таблетки с витаминами Р и С из плодов шипов- ника ВТУ-Ф № 2591 Серого или светло-серого цвета с мраморной поверх- ностью 45—55 в 1 Т< 22,5—27,5 1блетке — — 10,0 — — —
Каротолин М РТУ-42 3409—66 Оранжевого цвета в тон- ком слое со специфическим запахом и вкусом Кислотное число не бс Содержание каротинои не менее 120 мг % лее 3,5 дов в пе зесчете на р-кар отин 925 1,477—1,480
Масло шиповника МРТУ-42 № 3304—65 Маслянистая жидкость бу- рого цвета с зеленым оттен- ком и горьковатым вкусом Кислотное число не более 5,5 Содержание токоферолов не менее 40 ротиноидов не менее 55,0 мг % мг % и ка- 940 1,480—1,490
Выход готовых продуктов И ОТХОДЫ из 1 т целых плодов
шиповника, кг
Мякоть.....................
Сироп с витамином С, л . . .
Сырой жом..................
Сухой концентрат витамина Р
Флавоны, содержащиеся в кон-
центрате ..................
Жом II.....................
Краситель в виде пасты (1,2%
пигмента).............• . .
Содержание пигмента . . .
550
2163
2150
63,5
13,3
266,0
27,4
0,328
Шрот (при экстракции дихлор-
этаном) ....................253
Шрот масляный...............288
Масло из семян.............. 22,8
с содержанием
каротина ............... 0,0062
пигмента .................. 0,0353
витамина Е.............. 0,057
Шрот от семян...............322
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. Р о ж к о в М. И. Шиповник — витаминная культура. М., Пищепромиздат, 1948г
с. 8.
2. В а д о в а В. А. Биохимия шиповника. — «Биохимия культурных растений», 1949г
т. 7, с. 531.
3. В а с и л ь е в В. П. Шиповник в Марийской и Чувашской автономных респуб-
ликах. Чебоксары, Марийск, Госиздат, 1941.
4. Ш н а й д м а н Л. О. Производство концентратов витамина С из растительного-
сырья. М., Пищепромиздат, 1944, 121с.
5. Шнайдман Л. О. Производство витаминов из животного и растительного сырья.
М., Пищепромиздат, 1950. с. 29—79.
6. И г н а т ь е в Б. Д. Шиповник и его использование. Новосибирск, Изд-во АН
СССР, 1946, 320с.
7. Флора СССР. Изд-во АН СССР, 1942, 10.
8. В а д о в а В. А., Меньшиков В. Н., Я нишевская М. В. Об измен-
чивости химического состава шиповника. — В сб.: Витамины в теории и практике.
Л., Пищепромиздат, 1941, № 3, с. 165—169.
9. С а б у р о в Н. В., Г р ж и в о В. С. — «Труды ЦЕНИБИ Наркомпищепрома»,
1936, 1—4, с. 136.
10. К У Щ и и с к а я И. Н„ Шнайдман Л. О. Идентификация каротиноидов,
содержащихся в сухих плодах шиповника. — «Медицинская промышленность
СССР», 1964, № 4, с. 38—40. То же. Материалы совещания по витаминам из природ-
ного сырья, Куйбышев, ЦБТИ, 1964, с. 141—144.
11. Шнайдман Л. О. Исследования в области химии и технологии производст-
ва витаминов. Доклад, докт. дисс., М., 1967.
12. Ш н а й д м а н Л. О., Ушакова М. Т., Е ф и м о в А. 3., К У Щ и н -
ска я И. Н. А-витаминная активность ликопина плодов шиповника.—«Медицин-
ская промышленность СССР», 1966, № 1, с. 15—17.
13. Ш н а й д м а н Л. О., К у щ и н с к а я И. Н. Идентификация флавоновых и
катехиновых веществ плодов шиповника. — «Медицинская промышленность СССР»,
1965, № 2, с. 14—17. .
14. Ш н а й д м а н Л. О., Кущинская И. Н. Идентификация токоферолов-
масла семян шиповника. —«Медицинская промышленность СССР», 1964, № 5,
с. 44—46.
15. К у Щ и н с к а я И. Н., Шнайдман Л. О. Усовершенствование техноло-
гии переработки плодов шиповника на концентраты витамина С. — В сб.: «Вита-
минная промышленность», М., Пищепромиздат, 1957, № 4, с. 37—38.
16. К У Щ и н с к а я И. Н., Шнайдман Л. О. Комплексное использование
плодов шиповника в производстве витаминов. — В сб.: «Витаминная промышлен-
ность», М., Пищепромиздат, 1958, № 5, с. 28—47 с ил.
17. Шнайдман Л. О., Кущинская И. Н. Комплексная переработка пло-
дов шиповника на препараты витаминов. — «Труды ВНИВИ», М., Пищепромиздат,
1961, № 8, с. 66—71.
18. К у Щ и н с к а я И. Н., Шнайдман Л. О. Пути увеличения выхода каро-
тиноидного пигмента при переработке жома шиповника. — В сб.: «Витаминная
промышленность», М., Пищепромиздат, 1957, № 4, с. 38—40.
19. Ш найдман Л. О. Передовой опыт производства концентратов витамина С
из плодов шиповника. М., Пищепромиздат, 1955, 26с.
20. Ш н а й д м а н Л. О., К У щ и н с к а я И. Н., Сошников Д. Я-, Тро-
фимова П. Н. Полузаводские опыты комплексной переработки плодов шипов-
ника на препараты витаминов. — В сб.: «Материалы совещания по витаминам из
природного сырья», Куйбышев, ЦБТИ, 1964, с. 138—140.
21. Шнайдман Л. О., Кущинская И. Н. Авт. свидет., № 118265, 1958;
Бюлл. изобрет., 1959, № 4, с. 13.
22. Ш н а й д м а н Л. О., Кущинская И. Н. Авт. свидет., № 146740, 1958;
Бюлл. изобрет., 1962, № 9.
369
23. Ш н а й д м а н Л. О., Кущи некая И. Н., Ушакова М.Т., Сошни-
ков Д. Я., Воликов М. Е., Трофимова П. Н. Авт. свидет.,
№ 180304, 1962; Бюлл. изобрет., 1966, № 7.
24. Кущи н ская И. Н. Исследование биологически активных веществ плодов
гшиповника и технология их комплексной переработки на витаминные препараты.
Автореф. кандид. дисс., М., 1965.
25. Ш н а й д м а н Л. О. Производство витаминов. М., Пищепромиздат, 1958,
с. 14—38.
Г/а'ва 18. ПЛОДЫ ОБЛЕПИХИ (Н1РРЭРНАЁ RHAMNOIDES)
Облепиха представляет собой ветвистый колючий кустарник высотой
до 3,5—5 м. Ягоды облепихи созревают в конце августа и настолько обиль-
ны, что как бы облепляют ветки (отсюда и название облепихи). Сбор ягод
обычно происходит при наступлении заморозков. Ягоды имеют приятный
кислый вкус со специфическим ароматом. Мощные заросли облепихи име-
ются в Восточной Сибири, к западу от Байкала, на Алтае, а также в Сред-
ней Азии и на Кавказе [1]. Обширные массивы облепихи расположены в
пойме реки Катунь [2, 3]. Селекцией различных форм облепихи и агротех-
никой ее выращивания занимается Алтайская плодово-ягодная опытная
станция. Среди наиболее урожайных форм, ею выведенных, является «Но-
вость Алтая» (4,3% масла и 2,4 мг% каротина). Урожайность 10—
15 ml га. Имеются данные, что в Грузинской ССР встречаются виды облепи-
хи, содержащей до 13,6% масла и до 5,3 мг% (3-каротина.
ХИМИЧЕСКИЙ СОСТАВ ПЛОДОВ ОБЛЕПИХИ
В табл. 27 химический состав облепихи (в %) приведен по данным ряда
авторов.
Таблица 27
Состав плодов оэлепихи По А. Афа- насьевой [4] По Ж. Га- тину [3] По Д. Обо- ' довской [6] По дачным Бурятской АССР
Вода 82,45 83,2—85,8
Общий сахар В том числе: 2,36 2,5—6,6 — 3,5—7,1
инвертный 2,36 2,1—4,6 — —
общая кислотность . . 2,30 1,2—2,5 — 1,5—1,8
Семена — 4,0—11,0 — 5,5—12,7
Масло — 2,8—8,0 3,5—8,0 2,7—6,1
По исследованиям В. Ручкина, сок из мороженых ягод облепихи Крас-
ноярского края имеет следующий состав [5]:
Химический состав сока мороженой облепихи, %
Вода...................................... 91,53
Сухие вещества.............................. 8,47
В том числе:
инвертный сахар........................ 3,36
кислота яблочная........................ 2,49
азотистые вещества ..................... 0,98
зола общая.............................. 0,65
Плотность сока, кг!м3...................1038
Химический состав семян облепихи, %
Вода................................... . 6,45
Азотистые вещества..........................24,38
Жир ...................................... 12,13
Пентозаны................................. 14,90
Дубильные вещества......................... 2,37
Зола....................................... 2,11
Клетчатка и пр..............................30,66
370
Содержание семян в плодах облепихи составляет 16% (к массе плодов).
Витаминный состав Алтайской свежей облепихи, мг% [7]
Каротин............................... до 10,9
Витамин Bj.........................0,016—0,039
Фолиевая кислота....................... 0,79
Аскорбиновая кислота ................. до 272,8
Витамин В2 ........................ 0,03—0,056
Витамин Е ............................ до 8,0
Содержание витаминов в масле плодов Алтайской облепихи, мг%
Каротин................................... 55
Каротиноиды .............................200
Витамин Е ...............................до 160
В табл. 28 приведены физико-химические показатели масла из семян
и мякоти облепихи [7].
Таблица 28
Физико-химические показатели Масло Физико-химические - показатели Масло
плодо- вой мякоти семян плодо- вой мякоти семян
Плотность при 20°С, кг/ м3 Кислотное число .... Число омыления .... Йодное число Родановое число .... Общее содержание жир- ных кислот, % .... Содержание летучих жир- ных кислот, % .... 913,7 6,2 200 74,9 61,5 95,3 0,69 959,0 9,5 199,0 155,0 95,7 95,6 Неомыляемые вещества, % Содержание каротина, мг % Содержание витамина Е, мг % Сумма каротиноидов, мг % Содержание кислот, % насыщенных олеиновой линолевой линоленовой 3,23 35 160 250 32,8 50,6 15,6 1,85 3,02 105,0 13,38 16,35 47,61 18,35
По данным В. Кириллова, Д. Ободовской и Н. Чернобаевой, в масле
облепихи содержатся следующие жирные кислоты (табл. 29).
Как видно из табл. 28, масло семян содержит факторы витамина F — линолевую (47,6%) и лино- леновую кислоты (18,35%), что подтверждает биологическую цен- жирные кисло™ ность масла. Масло плодовой мя- коти облепихи состоит преимущест- Т а б j Содержа п кислот облеш- из плодо- вой мякоти I и ц а 29 ie жирных з масле хи, % из семяи облепихи
венно из глицеридов олеиновой КИСЛОТЫ. Е. Шишкина [10] изуча- Насыщенные ла содержание масла в плодах “Линолевая облепихи различного географичес- линоленовая . . . кого происхождения и установила, олеиновая .... что по этому показателю отличается Кантунская облепиха, содержащая масло (в среднем за 1963—1965 гг.) 25,1% и Тянь-Шане 24,01 3,73 77,79 кая — 2 8,98 38,28 4,01 61,73 2,5% К
массе целых сухих плодов. В этой же работе имеется указание о различ-
ном содержании витаминов в масле семян и мякоти плодов. Так, в первом
каротина 3 мг% и токоферолов 105—247 мг%, тогда как в масле из
плодовой мякоти соответственно 27—62 и 100 — 160 мг%.
Наиболее полно с привлечением современных физико-химических ме-
тодов было изучено содержание биологически активных веществ масла в
1966—1969 гг. [8, 9, 10].
371
ФИЗИКО-ХИМИЧЕСКИЕ ПОКАЗАТЕЛИ ПРЕССОВОГО МАСЛА ИЗ МЯКОТИ
ОБЛЕПИХИ
Плотность, кг!ж3...............921,0
........................... 1,4698
Кислотное число............ 4—10
Йодное число .............. 83
Число омыления ............ 195
Эфирное число.............. 185
Родановое число ........... 62,5
Число Рейхерта-Мейсля .... 1,61
Неомыляемые вещества, % . . 3,2
Жирные кислоты, %.............. 77,0
Свободные жирные кислоты . . 5,05
Число нейтрализации смеси жир-
ных кислот, % ................ 198,70
Глицерин, %.................. 8,4
Из биологически активных веществ прессового масла мякоти определе-
ны следующие.
Каротиноиды [9]. Общее содержание 259 мг%, в том числе (в мг%):
каротины (Р+у)—51,8; ликопин — 82,1; зеаксантин — 37,3%; неиденти-
фицированные — 87,8.
В табл. 30 приведены данные полученной хроматограммы в тонком слое
окиси алюминия каротиноидов масла облепихи. Идентификация дополни-
тельно проведена по Rz в сопоставлении с метчиками: для ликопина, р-каро-
тина и а-каротина. Метчики были получены для ликопина из томатов; для.
а-каротина — из моркови, для Р-каротина— синтетический.
Таблица 30
Номер Окраска зон Хшах в ВИДИМОЙ области света, нм Идентифицировано
пигмента ЗОНЫ
и 8 Бесцветная 0,99 330; 345; 366 Неидентифицировано
10 7 Желтая 0,98 420; 445; 475 а-Каротин
9 з 6 Ярко-оранжевая 0,92 425; 450; 475 р-Каротин
7 5 Оранжево-желтая 0,63 440; 465 Полицис, ликопин b
6 4 Розовая 0,46 440; 460; 495 Полицис, ликопин с
5 3 Ярко-розовая 0,32 445; 470; 500 Ликопин
4 476; 445; 420 Ксантофилл
3 2 Желтая 0,17 430; 455 Не идентифицирован
2 400; 430; (455) Не идентифицирован
1 1 Оранжево-желтая 0,07 415; 445; 470 Тараксантин
Токоферолы [8]. Общее содержание составляет 0,2%. Идентификация
изомеров токоферолов проведена как тонкослойной, так и газо-жидкост-
ной хроматографией. В табл. 31 дана хроматограмма в тонком слое силика-
Рис. 86. Спектр поглощения
токоферолов масла облепихи в
УФ-свете и их газо-жидкост-
ная хроматограмма:
а—УФ-токоферолов /—а. 2—у. 5—8;
б — газо-жидкостная хроматограмма
токоферолов: 1 — а-токоферола (сви-
детель) , 2 — соевого масла, 5— масла
облепихи.
372
гель — гипс (91,5:8,5) в растворителе хлороформ — циклогексан (2:1). Иден-
тификация проведена по максимумам поглощения в УФ-свете, а также по Ry
в сопоставлении с метчиками (синтетический а-токоферол и токоферолы
соевого масла).
На рис. 86 дан спектр поглощения токоферолов масла облепихи в УФ
(спирт) и газо-жидкостная^ хроматограмма (температура колонки 230° С,
газ-носитель —• аргон, скорость газа 40 мл!мин, время удерживания для
токоферолов в мин: а — 13; у — 8: 8 — 4). Содержание отдельных изоме-
ров (в % к общему количеству составляет): а-токоферол—51; у-токофе-
рол — 12; 8-токоферол — 37.
Жирные кислоты [9]. Общее содержание составляет 77%. Идентифика-
ция их произведена газо-жидкостной хроматографией метиловых эфиров.
Последние получают этерификацией жирных кислот диазометаном [9, 11].
Условия хроматографирования: температура 160° С, газ-носитель — ар-
гон; скорость газа 60 мл!мин. Время удерживания в мин: Ci4 — 6; С15—12;
С16 - 14; С18 - 28; Cj8 - 32; Q8 - 37; - 48.
Таблица 31
Номер пятна на хромато- грамме Rf Максиму- мы погло- щения в УФ-све- те, нм Идентифика- ция
1 0,42 290 а-Токоферол
2 0,26 299 -(-Токоферол
3 0,08 297 S-Токоферол
На рис. 87 показана газо-жидкост-
ная хроматограмма метиловых эфиров
жирных кислот по сравнению с метчи-
ками (штриховая линия). Установлено
следующее содержание отдельных жир-
ных кислот в масле облепихи (в % к
общему содержанию). Миристиновая
(С14) — 0,3; пальмитиновая (С16) — 26,2;
стеариновая (С18) — 3,2; пальмитинолеи-
новая (С' ie) — 45,6; олеиновая (С'18) —
9,4; линолевая (С"18) — 10,8; линолено-
вая (С"'18) — 4,5.
Фосфолипиды. Они входят в состав
Рис. 87. Газо-жидкостная хрома-
тограмма метиловых эфиров жир-
ных кислот облепихового масла.
всех важных органов животного орга-
низма: (мозг, печень, почки, сердце, легкие). Фосфолипиды играют
важную биологическую роль. Они участвуют в белковом обмене; облада-
ют тромбопластической активностью, участвуют в процессе свертывания
крови. Применяются при лечении атеросклероза [13]. По химическому
строению фосфолипиды являются сложными эфирами многоатомных спир-
тов (глицерина, сфингозина) и жирных кислот. К ним относятся:
CH2OOCR СН2 OOCR1
RCOO—СН О RCOO—СН О
I II + I II +
СН2О—Р—ОСН2 СН2 N (СН3)3 СН2О—Р осн2 сн2 nh2
о- . о-
Фосфатидилхолии (лецитии) « Фосфатидилэтаноламии
(кефалин)
373
CH2 OOCR1
RCOO CH О
I II
CH2O—P—О—инозит
ОН
Фосфатидилинозит (липозитол)
Общее содержание фосфолипидов в масле облепихи, определенное по
количеству фосфора [14], составляет 1%. Идентификация их произведена
хроматографией в тонком слое силикагеля-гипса. Газо-жидкостной хрома-
тографией исследованы жирнокислотный состав фосфолипидов.
Для тонкослойной хроматографии применяют подвижный растворитель
хлороформ-метанол-вода (65:25:4) или хлороформ-метанол-уксусная кисло-
та-вода (25:15:4:2); проявляют раствором родамина или йодом. Для иден-
тификации аминофосфатидов (кефалина) хроматограмму опрыскивают раст-
вором нингидрина; для холинсодержащих фосфолипидов (лецитина) приме-
няют реактив Драгендорфа [ 15 ]. Идентифицированы следующие фосфолипиды
[9]: лецитин (Rf — 0,6) и кефалин (Rf —0,32). В качестве метчиков при-
меняли синтетический лецитин и фосфолипиды желтка яйца.
Газо-жидкостной хроматографией идентифицированы метиловые эфиры
жирных кислот фосфолипидов. Последние получены этерификацией фосфо-
липидов метанолом в присутствии H2SO4 при температуре 80° С в течение
3 ч. Идентифицированы следующие жирные кислоты в их процентном соот-
ношении: миристиновая С14 — 0,8, пальмитиновая С1в—37, пальмитин-
олеиновая С'1в—39,5, Стеариновая С18 — 3,3, олеиновая С'18 — 9,7, лино-
левая С" 18 — 8,8, линоленовая С"' 18— 0,9.
Стерины. Идентифицированы методами тонкослойной и газо-жидкост-
ной хроматографии. Общее содержание стеринов в масле, определенное
колориметрическим методом, составило 1,4%. Из стеринов идентифициро-
ван [3-ситостерин. 2 других стерина не идентифицированы [9].
Витамин Идентифицирован хроматографией в тонком слое алюми-
ния III степени активности. Подвижный растворитель — петролейный
эфир (70—100° С) — серный эфир (19:1); проявитель — пары йода или
0,001 %-ный раствор родамина;’метчик синтетический витамин Ki- Содер-
жание витамина Ki определено спектрофотометрически; 7.1Г1ах = 249 нм;
Е\°м = 435. Установлено высокое содержание витамина Ki в масле —
0,2% [9].
БИОЛОГИЧЕСКИ АКТИВНЫЕ ВЕЩЕСТВА СОКА ОБЛЕПИХИ
Сок облепихи недостаточно исследован. Химические и биологические
исследования, проведенные автором и Н. Шугам [16], приведены ниже.
Состав сока облепихи, по данным исследований, следующий (в %).
Сухие вещества..........7,0
Зела....................0,4
Редуцирующие сахара . . 1,2
Белковые вещества ... 2,7
Клетчатка ............0,6
Кислотность .......... 1,6
Плотность, кгIм3 ... 1037
Качественным эмиссионным анализом установлено содержание в соке
следующих микроэлементов: кальций, натрий, медь, магний, алюминий,
кремний и титан. В наибольшем количестве содержатся магний, а затем
кальций. Хроматографией на бумаге исследован аминокислотный состав
сока [17]. Состав свободных аминокислот сока (мг%): цистеин — 520,
а-аланин — 80, фенилаланин — 72, лейцин — 96. Из них фенилаланин и
лейцин являются незаменимыми аминокислотами [16].
В соке облепихи содержится значительное количество бетаина (0,7%),
определенного по методу Бенина и Шнайдера [18]. Содержание холина не-
значительно (0,03%) [19]. Эти вещества, как известно, обладают липотроп-
374
ным действием. Биологические испытания действительно показали, что сок
облепихи уменьшает жировую инфильтрацию печени, вызванную введени-
ем лабораторным животным четыреххлористого углерода [16].
Значительный интерес предста'вляют исследования биологической ак-
тивности масла и сока облепихи. Было установлено [20], что облепиховое
масло ускоряет процесс заживления химических ожоговых травм (на 40—
60%) и что стерины являются одним из наиболее активных компонентов
неомыляемой фракции масла. Дальнейшие исследования (Н. Шугам,
Л. Шнайдман) касались вопроса выяснения влияния отдельных компонен-
тов масла на скорость заживления химических ожогов. Были получены
биологически активные компоненты облепихового масла, которые в отдель-
ности смешивали с вазелиновым маслом в концентрации, соответствующей
их содержанию в масле. Ими смазывали пораженную поверхность кожи,
вызванную химическим ожогом (втиранием 0,02 мл 20%-ного раствора КОН
в течение 30 сек) и определяли скорость заживления ожогов (в день в ел2).
Последняя составила для: неомыленной части масла в целом — 9,1; то же
без стеринов — 7,9; стеринов — 9,0; жирных кислот — 7,3; фосфолипи-
дов — 6,6; вазелинового масла — 6,3; контроля —• 5,9. Эксперимент про-
водили над 119 лабораторными белыми крысами массой 200+ 10 г. Получен-
ные результаты были статистически достоверны. Таким образом, было по-
казано, что для лечения ожогов преимущественное значение имеют стери-
ны, что открывает путь применения стериновых препаратов в медицине.
Биологические исследования сока облепихи впервые проводились в
1969 г. [16] в связи с высоким содержанием в нем бетаина-липотропного
фактора. Жировую инфильтрацию печени вызывали однократным внутри-
брюшинным введением четыреххлористого углерода (1 мл!кг массы). Сок
или кристаллический бетаин вводили из расчета 45—50 мг в день.
Степень жировой инфильтрации печени определяли по содержанию в
ней жира, холестерина и снижению жирового коэффициента. Результаты
действия сока облепихи при жировой инфильтрации печени приведены в
табл. 32.
Таблица 32
Группа
животных
Условия опыта
Количе-
ство
животных
Содержание в печени, %
жира
холесте-
рина
леци-
тина
^Кировой
коэффициент
лецитин
холестерин
1
2
3
4
5
Контроль положительный
Кршроль отрицательный (СС14)
Сок (pH 3,3) + СС14
Сок (pH 5,2) + СС14
Бетаин -f- СС14
38
45
42
14
10
11,0
27,4
21,3
19,7
20,2
0,78
1,52
1,01
0,95
1,08
4,45
4,47
4,47
4,16
4,60
5,7-
2,89
4,40
3,87
3,80
Результаты эксперимента указывают,
ну обладают липотропным действием.
что сок облепихи, подобно бетаи-
ТЕХНОЛОГИЯ КОМПЛЕКСНОГО ИСПОЛЬЗОВАНИЯ ПЛОДОВ ОБЛЕПИХИ
[7, 21, 22]
Предусмотрены следующие стадии переработки плодов облепихи: 1) по-
лучение сока; 2) извлечение масла из мякоти плодов; 3) экстракция масла
из семян; 4) получение препарата витаминов группы Р.
Получение сока. В связи с тем что данные о биологической активности
сока облепихи отсутствовали, не было достаточного внимания к его исполь-
зованию: частично его направляют для переработки в винодельческую про-
мышленность, частично перерабатывают на сироп, содержащий 50—55%
сухих веществ; имеется также заводской опыт выпуска стерилизованного
375
сока с сахаром, разбавленного в 2—3 раза водой для снижения кислотнос-
ти. Когда был изучен состав сока облепихи и показано его липотропное
действие, встал вопрос о рациональной технологии его получения. Задача
снижения кислотности сока была решена путем обработки его анионитом
ЭДЭ-10П в ОН-форме. В результате взаимодействия сока в течение 60 мин
с анионитом в количестве 5% к массе сока pH сока повышался с 3,3 до 5,2.
Липотропное действие сока от этой обработки не изменилось (стр. 375).
При отжатии сока из мякоти в него переходит небольшое количество масла
(от 1,5 до 7 кг из 1 т плодов, или около 10% к общему содержанию масла
в плодах [7]). Отделение масла осуществляют либо отстаиванием и декан-
тацией, либо сепарацией. Таким образом, технология получения сока в
основном сводится к отжатию его из плодов, выделению сепарацией масла,
купажу с сахаром, расфасовке и стерилизации.
Получение масла из мякоти плодов. Процесс сводится к сушке жома
(жмыха), измельчению и извлечению из него масла. Для этой цели жмых
измельчают в дробилке и подвергают сушке на паровой конвейерной сушил-
ке типа ПКС-10 при 75° в течение 1—1,5 ч до влажности 6—7%. Выход
сухого жмыха составляет 7,5—9,0% к массе свежего сырья. Состав сухого
жмыха (в %): масла в плодовой мякоти — 15—27, каротина — 12—16 мг%,
семян — 45—55%, влажность 4,0—7,0. Процесс экстракции масла из жмы-
ха осуществляют в настоящее время по методу В. Казанцева и А. Охина
в батарее из 22 диффузоров подсолнечным или кунжутным маслом при 50—
65° С. Полный оборот батареи 24 ч. Отбор масла из головного диффузора
происходит каждые 1,0—1,5 ч. Из хвостового диффузора соответственно вы-
гружают жмых с масличностью 45—50%. В специальном шнековом прессе
(экспеллере) отжимают масло из жмыха. Недостатками данного метода диф-
фузии являются: потери каротина достигают 20—22%, получаемое масло
содержит 15—20% подсолнечного, высокое кислотное число масла, дости-
гающее 10,0—15,0. В связи с этим возник вопрос о применении органическо-
го растворителя для экстракции липидов облепихи. В результате прове-
денных исследований процесса экстракций с различными растворителями
(петролейный эфир, дихлорэтан, бензол и хлористый метилен) наиболее
эффективным является хлористый метилен (дихлорметан, СН2С12). Послед-
ний имеет низкую температуру кипения (41—42°), плотность при 20° С
1336 кг/м3, малотоксичен. При экстракции этим растворителем может
быть получен высокий выход масла (95%) и каротина (97%) [21]. По-види-
мому, Экстракция масла из жмыха хлористым метиленом будет наиболее
эффективна. Необходимо лишь отработать вопрос полного удаления раст-
ворителя из готового продукта.
Экстракция масла из семян и получение препарата витамина группы Р
до настоящего времени не разработаны. Получение препарата витаминов
группы Р в достаточной мере не изучено. Известно, что выход витамина Р
в полу заводских опытах составил 79%, а содержание витамина Р в порошке
17—19,76%, выход препарата — порошка составил из 1 т плодов 50 кг
при содержании 17,0%.
технологическая схема комплексной переработки плодов облепихи
(рис. 88).
Получение сока. Ягоды облепихи на инспекционном конвейере 1 подвер-
гаются проверке. При этом удаляют гнилые и пораженные ягоды, а также
посторонние примеси. На конвейере ягоды проходят паровую бланширов-
ку 2. Затем ягоды элеватором «Гусиная шея» 3 через сборник 4 подают в
дробилку 5 и вальцовый пресс Яна1 6 для отжима сока. Сок поступает на
сито-трясучку 7 (предотвращающее попадание в сок выжимок), а затем в
сборник сока 8. Насосом 9 сок подают в напорный сборник 10, а из него в
1 Работа пресса Яна при переработке плодов облепихи нуждается в практиче-
ской проверке.
376
сепаратор И для выделения масла. Сок из сепаратора поступает в сборник
12, далее насосом 13 подается в смеситель 14, где смешивается с анионитом
ЭДЭ-10П в количестве 5% к массе сока, насосом 15 подают в фильтр-пресс 16.
Из фильтр-пресса сок поступает в сборник 17 и далее на линию розлива
сока, состоящую из разливного автомата 18, укупорочного автомата 19,
автомата для инспекции 20 системы Бартеньева производительностью 3 тыс.
бутылок в 1 ч и автомата для этикетировки 21. Мойка бутылок производит-
ся в бутылкомоечном автомате 22 при температуре воды 25—30° С и щелоч-
Рис. 88. Технологическая схема комплексной переработки плодов облепихи.
ности раствора 1—2%. Анионит с фильтр-пресса 16 направляют на регене-
рацию.
Масло из мякоти облепихи из сепаратора 11 поступает в сборник 23 и
нутч-фильтр 24, затем в сборник 25. Из последнего масла разливают в фла-
коны через наполнитель 26. Укупорку и этикетировку флаконов осуществ-
ляют на конвейере 27.
Сушка жома. Жом после пресса Яна 6 подается шнеком в сборник 28,
затем насосом или элеватором — в напорный сборник, вакуум-вальцовую
сушилку 29 для высушивания до содержания 90% сухих веществ.
Сухой жом поступает в дробилку 30 и после раздробления комьев посту-
пает в сборник 31 и сепаратор 32 (типа зернового). В сепараторе семена от-
деляются от мякоти. Семена поступают в сборник 33, а мякоть — в сбор-
ник 34.
Получение масла из мякоти плодов. Мякоть плодов поступает в дробил-
ку 35 для измельчения в порошок. Последний направляют в сборник 36 и
далее на экстракцию в экстракционный аппарат 37, снабженный холодиль-
ником-конденсатором 38, испарителем 39 и сборником 40. Экстракцию ве-
дут четырех-пятикратным количеством хлористого метилена при темпера-
туре около 40° С. В испарителе отгоняют ’растворитель, а затем масло из
испарителя переводят в вакуум-аппарат 41 для отгонки в среде углекисло-
ты остатков хлористого метилена при добавлении небольшого количества
воды (под вакуумом в 650—700 мм рт. ст.). Из вакуум-аппарата масло
спускают в сборник 42, а из него направляют на расфасовку.
Получение масла из семян облепихи. Масло из семян выделяют отдельно
из-за его специфических лечебных свойств. Семена измельчают в дробилке
377
43 в порошок, который направляют в сборник 44 и далее в экстракционный
аппарат 45, снабженный холодильником 46, испарителем 47, сборником 48.
Экстракцию ведут хлористым метиленом, направляемым из сборника 49.
Остатки растворителя из экстракта отгоняют в вакуум-аппарате 50 в при-
сутствии воды под вакуумом, масло спускают в сборник 51 и направляют
его на расфасовку.
Производство концентрата витамина Р. Из шрота мякоти плодов в экс-
тракторе 37 отгоняют растворитель. Сухой шрот направляют в сборник 52,
а из него на экстракцию 60%-ным спиртом, добавляемым в пяти-шестикрат-
ном количестве к массе сухого шрота, в экстрактор 53, снабженный холо-
дильником 54, испарителем 55 и приемником 56. В испарителе отгоняют
спирт, а концентрат насосом 57 подают в сборник 58, а из него на вакуум-
вальцовую сушилку 59. Сухой концентрат измельчают в дробилке 60 в
порошок, который поступает в сборник 61. Из последнего концентрат ви-
тамина Р поступает либо на расфасовку в виде порошка, либо на таблетиро-
вание с наполнителем — сахаром.
Использование шрота. Из шрота плодовой мякоти и семян отгоняют
растворитель в экстракторах 53 и 45. Шрот выгружают в приемники 62, 63,
подсушивают в сушилке 64 и направляют в сборник 65 для расфасовки.
ТЕХНОЛОГИЧЕСКИЕ ПОКАЗАТЕЛИ [7, 9, 22]
Сырье (плоды облепихи), сухие целые плоды
Сухие вещества, 14,2—16,8 Витамин Е, мг % . 40—50
Масло, % ... . 20—25 Витамин С, мг % . 350—700
Каротин, мг % . . 13—33 Семена, % .... 35—50
Технология производства
Получение сока
Сок, % к свежему сырью............................65—70
Жом » » » » ..........................30—35
Расход плодов на 1000 л сока, кг .'............. 1428
Состав сока, %
Сухие вещества Белковые вещества Зола pH Бетаин
7,0 2,7 0,4 5,0 0,5
Состав жома сухого [7]
Сухие вещества, % Семена, % Каротин, мг % Масло мякоти, %
93—96 45—55 12—16 15—27
Показатели масла мякоти
913,7—921,0
1,4698
3,2—6,2
75—83
Плотность, кг{м3
п20
nD............
Кислотное число
Йодное число
Жирные кислоты, % 77,0
Получение масла экстракцией:
Каротиноиды, мг % .
Токоферолы, % . . .
Фосфолипиды, % . .
Витамины KL, % . .
Стерины, % . . . .
240—260
160—200
1,0
0,2
1,4
а) хлористым метиленом [21]
выход, %
масла................................................95,1
каротина.............................................96,0
витамина P-в шроте..................................91,6
б) подсолнечным маслом
выход, %
масла облепихи .................................. 80—85
каротина .................................... 77,9—88,0
витамина Р в шроте.............................. 95,7
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. П а в л о в Н. В. Дикие, полезные и технические растения СССР. М., изд. Госпла-
на, 1942.
2. Евтушенко А. Облепиха как высокоценная витаминная культура. Автореф.
канд. дисс., М„ 1950.
378
3. Г а т и н ж. И. Облепиха. М., Сельхозиздат, 1963.
4. Афанасьева Л. И., Горшкова А. И. — «Труды НИИплодо-овощного и
Энохимического институтах, 1931, № 2, с. НО.
5. Р у ч к и н В. Н. — В кн.: Ф. В. Церевитинов. Химия свежих плодов и
овощей. М., Пищепромиздат, 1938, с. 431.
6. Ободовская Д. А. Облепиха как сырье для витаминной промышленности.
М., Пищепромиздат, 1957, 24с.
7. Ободовская Д. А., Девятнин В. А. Бюллетень технической инфор-
мации, Пищепромиздат, 1952, 6.
8. Шнайдман Л. О., Ш у г а м Н. А. Исследование и идентификация токофе-
ролов в плодах и масле облепихи. — «Изв. вузов. Пищевая технология», 1966, № 2,
с. 39—41. с ил.
9. Ш у г а м Н. А., Шнайдман Л. О. Жирные кислоты и фосфолипиды в мас-
ле облепихи. — «Изв. вузов. Пищевая технология», 1967, 1, с. 52—54 с ил.
Ш у г а м Н. А. Исследования биологической активности веществ облепихи. Ав-
тореф. дисс., М., 1969.
10. Ш и ш к и н а Е. Е. Содержание масла в плодах облепихи различного географи-
ческого происхождения. — «Труды Всесоюзного семинара по БАВ плодов и ягод»,
Свердловск, .1968, с. 314—318.
11. Б л этт А. Синтез органических препаратов. ИЛ, М., 1949, 2.
12. Данилевский В. Лецитин, Харьков, 1938.
13. Мясников А. Л. Атеросклероз. М., Медгиз, 1960.
14. 3 и н о в ь е в А. А. Химия жиров. М., Пищепромиздат, 1952.
15. А х р е м А. А., Кузнецова А. И. Тонкослойная хроматография. М., «На-
ука», 1965, с. 168.
16. Ш н а й д м а н Л. О., Ш у г а м Н. А., Ушакова М. Т. Изучение состава
и биологических свойств сока облепихи. — «Прикладная биохимия и микробиоло-
гия», 1969, т. 5, № 3, с. 371—373.
17. П а с х и н а Т. С. Количественное выделение аминокислот при помощи хрома-
тографии на бумаге. —.В сб.: «Современные методы в биохимии», М., «Медицина»,
1964, № 1, с. 162.
18. Б е н и н С. Г., Шнайдер Е. Е. Бетаин и его определение в продуктах са-
харного производства. —«Сахарная промышленность», 1951, 11, с. 44.
.19 . Трусов В. И. Метод определения холина в биологическом материале. — «Био-
химия», 1950, т. 15, № 6, 495с.
20. Ч укаева В. Н., Г у р в и ч А. И., У ш а к о в а М. Т. и др. К изучению
биологической активности масла облепихи и его компонентов. — В сб.: «Материалы
совещания по витаминам из природного сырья», Куйбышев, ЦБТИ, 1964, с. 172—177.
21. Шнайдман Л. О., Ч укаева В. Н., Клименцов Н. И. и др. Ком-
плексная переработка плодов облепихи. Там же, 1964, с. 145—151.
22. Ш н а й д м а н Л. О. Производство витаминов. М., Пищепромиздат, 1958, с. 135.
Глава 19. ПРИРОДНЫЕ ПРОМЫШЛЕННЫЕ ИСТОЧНИКИ
Р-ВИТАМИННОГО СЫРЬЯ И ПРОИЗВОДСТВО ИЗ НИХ
ВИТАМИННЫХ ПРЕПАРАТОВ [1—3]
В 1936 г. А. Сцент-Гиорги и Л. Арментано с сотрудниками [4, 5] уста-
новили, что венгерский красный перец и лимонный сок содержат вещество,
способное увеличить прочность и снижать проницаемость капиллярных кро-
веносных сосудов. В дальнейшем было установлено, что активное вещество
состояло из флавонов и флавоновых глюкозидов. Это вещество было названо
витамином Р — витамином проницаемости (первая буква англ, слова
permeability). Кристаллическая флавоновая фракция, полученная из ли-
монов, названа цитрином. При анализе цитрина было установлено, что он
в основном состоит из флавоидных глюкозидов-гесперидина и эриодиктио-
ла. Этим двум глюкозидам вначале и приписывалось капилляроукрепляю-
щее действие. В дальнейшем выяснилось, что подобным же действием облада-
ет ряд других веществ, как, например, рутин, кверцетин, маклурин, эску-
лин (—)-эпикатехин и другие катехины.
К витамину Р относятся три группы веществ: флавоны (рутин, кверце-
тин, гесперидии, эриодиктиол, тилирозид, кемпферол и др.), антоцианы
(цианидин, пеларгонидин и дельфинидин) и L-катехины: (—)-эпикатехин,
(—)-галлокатехин, (—)-эпикатехингаллат и (—)-галлокатехингаллат и
др. В природе эти группы взаимопревращаемы, причем флавонолы через
379
лейкоантоцианидины могут превращаться либо в антоцианидины, либо в
катехины [6]. По-видимому, процесс превращения протекает непрерывно,
поэтому в растительном мире эти группы сопутствуют одна другой. Напри-
мер, в плодах шиповника [7] наряду с преобладанием веществ группы фла-
вонов (рутин, кверцетин, изокверцетин, тилирозид) содержатся катехины
(L (—)-эпикатехин, (—)-галлокатехингаллат и др.) и антоцианы (циани-
дин). В листьях чая (отходах) содержатся преимущественно катехины, но
наряду с ними флавоны (рутин, кверцетин около 2% к массе листьев)-и ан-
тоцианы [8]. По-видимому, в любом растительном материале вещества этих
групп содержатся в том или ином соотношении.
Почти все исследователи [9—11 ] утверждают, что витамины группы Р
способствуют накоплению и лучшему использованию в организме аскорби-
новой кислоты. По мнению Е. Шамрая [12], полифенолы (витамины груп-
пы Р) способствуют переходу аскорбиновой кислоты в дегидроаскорбиновую
кислоту, которая является транспортной формой первой. Проходя через мем-
браны внутрь клеток тканей, дегидроаскорбиновая кислота восстанавливает-
ся там в аскорбиновую кислоту, способствуя ее накоплению. Общеизвестно
специфическое действие витамина Р, выражающееся в укреплении стенок
кровеносных капилляров [13, 14], в подавлении гиперфункции щитовид-
ной железы [13, 15]. Имеются также соображения, что эти витамины обра-
зуют с протеином ферментный комплекс, являющийся переносчиком водо-
рода в организме [16, 17]. Указанное свидетельствует о важном значении
витаминов группы Р для человека.
Предполагают, что вещества, обладающие действием витамина Р, попа-
дая в организм, включаются в сложную систему реакций окисления — вос-
становления [13]; обладают диуретическим действием и токсичностью по
отношению к микроорганизмам [13а].
Предполагают также [18] о существовании какой-то связи между кан-
церогенными заболеваниями и витамином Р. Эти предположения базируют
на работах доктора А. Ференци (Венгрия) о лечении рака соком столовой
свеклы и окрашенным виноградным вином (антоцианы) [19, 20], а также на
работе О. Кабиева и др. о противоопухолевой активности лейкоантициани-
динов и катехинов [21 ] и некоторых других [22, 23].
Автор считает также целесообразным сделать вывод, который по сущест-
ву вытекает из работы Н. Березовской [15], хотя он ею и не отмечен — о
двух различных функциях витамина Р: каталитической и ингибирующей.
С одной стороны, в этой работе доказана способность биофлавоноидов уси-
ливать тканевое окисление, а с другой — подавление ферментативного пре-
вращения тирозина в печени, а также подавления активности щитовидной
железы, подавления системы гиалуроновая кислота — гиалуронидаза. Та-
ким образом, витамины группы Р обладают замечательной способностью
катализировать одни реакции и ингибировать другие. Если эту закономер-
ность обобщить, то станут несколько более понятными условия, при кото-
рых в микроскопической клетке протекают параллельно и одновременно
сотни сложнейших химических реакций, не тормозя одна другую.
ХИМИЯ ВИТАМИНА Р
Вещества, обладающие Р-витаминной активностью, имеют много общего
в химической структуре. Скелет у них состоит из бензольного кольца, кон-
денсированного с у-пироновым или у-пирановым кольцом.
Особенностью некоторых из этих соединений является разрыв (пироно-
вого кольца между кислородом и соседним углеродом с образованием хал-
кона) вещества, обладающего сильно восстановительными свойствами. Су-
ществует мнение [24], что витаминной активностью обладают только хал-
коны, а не циклические соединения.
Все вещества, обладающие Р-витаминной активностью, имеют химиче-
скую структуру — 2-фенил-1,4-бензопирон (фенил-хромон), либо 2-фенил-
380
1,4-бензопиран. Соединения, имеющие у-пироновое кольцо, представляют
группу флавонов — желтых пигментов растений. Соединения, имеющие пи-
рановое кольцо, представляют группу катехинов и антоцианов
О
2 - Фенил - бензо -у- пирон
2 - Фенил - бензо-у-пиран
В основе строения всех Р-витаминных веществ (флавоноидов) лежит
структура Св—С3—С6 углеродных единиц. Различаются они по степени окис-
ленности пропановой цепочки С3, связывающей оба ароматические кольца.
Ниже^приведена структура для различных групп этого класса соединений:
он о
(Лейкоцианидин)
(Дельфинидин)
Подавляющее большинство встречающихся в природе флавоноидов со-
держат гидроксильные группы в положениях [5, 7, 3', 4'] (эридиктиол,
цианидин, кверцетин), а также в положениях 5, 7, 3', 4', 5' (дельфинидин,
петунидин, мальвидин). Встречаются также флавоноиды с алкилирован-
ными гидроксильными группами.
Гликозиды флавоноидов содержат остаток сахара в определенном поло-
жении. Например: антоцианы — в 3-м или в 3-м и 5-м (для дигликозидов);
флавонолы — в 3-м; флавоны — в 7-м положении. С агликонами чаще всего
сочетаются сахара: D-глюкоза, Г-рамноза, D-галактоза и рутиноза (|3-1-Г-
рамнозидо-6-D-гликозид).
Флавоноиды —• пигменты растений — представляют собой кристалли-
ческие вещества желтого или оранжевого цвета с высокой точкой плавления
(например, для эриодиктиола 267° С, для гесперидина 261° С, для рутина
192°С), трудно растворимы в воде. Лучше других растворяется рутин [25].
Некоторые из них хорошо растворимы в щелочах (эриодиктиол), другие —
в горячей уксусной кислоте (гесперидии). Катехины — конденсированные
дубильные вещества. Они содержатся в зеленых листьях чая (не прошедших
ферментацию) и в черном чае. Препарат танина из зеленых чайных листьев
представляет собой бесцветный аморфный порошок, легко растворимый в
воде и спирте. Аморфный танин из черного чая окрашен в красновато-бу-
рый цвет. Из зеленых чайных листьев был также выделен L(—)-эпикатехин
в виде бесцветных кристаллов с температурой плавления 235—237° С [9].
381
Танин из листьев Цейлонского чая [26] содержит (—)-эпикатехина 6,5%,
(±)-галлокатехина 24,2%, (—)-эпикатехингаллата §,0%, (—)-галлокате-
хингаллата 49,0%.
Из перечисленных выше Р-витаминных веществ промышленное приме-
нение получили: рутин — глюкозид флавонола, смесь катехинов — препа-
рат чайного танина, препарат антоцианидина из черноплодной рябины,
смесь флавонов и катехинов — препарат шиповника; перспективу промыш-
ленного применения имеют: гесперидии из отходов цитрусовых и смесь ка-
техинов из бадана (Bergenia crassifolia)1.
Рутин является спутником аскорбиновой кислоты в растениях. Он пред-
ставляет собой кристаллы игольчатой формы бледно-желтого цвета. При
гидролизе разбавленными кислотами рутин дает кверцетин, глюкозу и рам-
нозу. Кривая поглощения спиртового раствора рутина в ультрафиолетовом
свете имеет два максимума при длине волны 362,7 и 257,7 нм, Е\см для
^257,7=345; для Х362,7=2б5. УФ-спектры поглощения других флавоноидов
см. стр. 363.
СЫРЬЕ И ТЕХНОЛОГИЯ ПРОИЗВОДСТВА Р-ВИТАМИННЫХ ПРЕПАРАТОВ
Исследования автора [18] показали, что в продуктах широкого потреб-
ления (хлеб, картофель, капуста) содержание Р-витаминных веществ
крайне мало (около 5 мг%), что может обусловить Р-витаминную недоста-
точность в весенне-зимний период у отдельных групп населения. В связи с
этим в литературе имеется указание [1] о необходимости доведения произ-
водства витамина Р в ближайшие годы до 500 т в год. В связи со слож-
ностью и дороговизной синтетических методов производства флавоноидов
практика пошла по пути получения их из растительного сырья. При выборе
перспективного сырья необходимо руководствоваться биологической цен-
ностью получаемых препаратов и доступностью сырья. В табл. 33 приве-
дены сравнительные результаты биологических исследований Р-витамин-
ных препаратов, полученных из различных видов сырья [27].
Из данных, приведенных, в табл. 33, видно, что все указанные препараты
обладали Р-витаминной активностью, при этом более высокой — препара-
ты из черной смородины и черноплодной рябины, а менее — препарат из
шрота облепихи. Следовательно, к перспективным видам промышленного
Р-витаминного сырья следует отнести: черную смородину, черноплодную
рябину, отходы чайных листьев, отходы сокового производства цитрусовых
и жом шиповника. Эти виды сырья в той или иной мере уже проверены в
производстве. Положительные результаты при биологической проверке на
биоактивность дали препараты из бадана (Bergenia crassifolia), из горной
эфедры (Ephedra equisatina) [27, 28] и из корня кермека (Statice gmelini
willd) [29]. Значительный интерес в качестве сырья для производства Р-ви-
таминных препаратов представляют отходы сокового и винодельческого
производства. Показано [30], что только на предприятиях Краснодарского
края может быть получено препаратов витамина Р (100%-ного вещества
в т) из отходов: винограда 130, яблок и слив 24 и абрикосов 5.
Особо стоит вопрос о сырье для производства рутина. Применяемые в
настоящее время для этой цели бутоны софоры японской не перспективны
из-за дефицитности. В литературе имеются данные о применении зеленой
массы гречихи [31—33] для производства рутина. По-видимому, на этот
1 Сырьем для промышленного получения препаратов витамина Р служат: для
производства препарата чайного танина — отходы чайного листа; для рутина — се-
мена софоры японской; для антоцианидина — черноплодная рябина; для смеси ка-
техинов — бадан; для гесперидина—отходы цитрусовых (сокового производства); для
смеси флавонов и катехинов — жом плодов шиповника.
382
Таблица 33
Р-витамииные препараты Изменение капилляр- ной реакции у живот- ных, получавших Р-ви- таминные препараты Р-витаминные препараты Изменение капилляр- ной реакции у живот- ных, получавших Р- витаминные препараты
время появления- петехий, сек количе- ство жи- вотных с положи- тельной реакцией, % время поивления петехий, сек количе- ство жи- вотных с положи- тельной реакцией, %
Опыт № 1 Опыт № 3
Паста Гесперидии из отходов
из черной сморо- производств а ма нд а р и-
дины 42,78 82,6 нового сока 21,8 77,8
из рябины черно- Катехины из чая .... 23,5 75,7
ПЛОДНОЙ .... 38,60 75,0 Контроль (отрицатель-
Катехины из листьев ный) —18,1 38,2
чая 32,64 84,0
Порошок из шрота Опыт № 4
облепихи . . • ... 12,00 66,7 Концентрат
Контроль (отрица- витамина Р из жома
тельный) —2,00 42,9 шиповника .... пищевого красителя 21,5 16,6 80,9
Опыт № 2 из винограда . . . 73,6
Лейкодельфинидин из горной эфедры . . Катехины 29,7 70,8 Катехины из листьев чая Контроль (отрицатель- ный) 20,1 —5,2 79,1 53,3
из корня керме- ка 21,6 79,2
из чая 23,5 68,4
Контроль (отрица- тельный) —4,5 35,3
вид сырья придется ориентировать производство в случае недостатка или
отсутствия софоры японской.
Отдельно также стоит вопрос об использовании столовой свеклы (Beta
vulgaris hortensis) в связи с исследованиями Ференци [19, 20].
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА ВИТАМИНА Р ИЗ ОТДЕЛЬНЫХ
ВИДОВ СЫРЬЯ
ВИТАМИН Р ИЗ ЛИСТЬЕВ ЧАЯ [1, 34—37]
Чай [8] — растение относится к семейству чайных (Theaceae). Влаж-
ность листьев колеблется от 70 до 78%; экстрактивные вещества зеленого
листа чая составляют 42—45/6. Чайный танин представляет собой смесь
нескольких катехинов и их галловых эфиров: (—)-эпикатехин, (—)-эпигал-
локатехин, (—)-эпикатехингаллат, (±)-галлокатехин, (±)-катехин, (—)-
эпигаллокатехингаллат и (—)-галлокатехингаллат. Общее содержание та-
нидов в зеленом листе составляет 22—24% к массе сухих веществ.
• В чайном листе содержатся различные флавоновые глюкозиды: рутин
(1%), кверцитрин (около 1%), дающий при гидролизе кверцетин (флаво-
нол с Р-витаминными свойствами); глюкозиды из группы антоцианов, игра-
ющие важную роль в качестве пигментов листьев, цветов и плодов. Счита-
ют, что от количества флавонов и антоцианов зависит степень окраски и
вкусовые достоинства чая. Чайное растение вырабатывает также алколои-
ды — кофеин, теофиллин, теобромин; пигменты — каротин, ксантофилл и
хлорофилл; эфирные масла, стерины и другие соединения. Из алкалоидов
чая наиболее важным является кофеин, содержание которого колеблется
в пределах 1,8—2,8% и хлорофилл (0,8%) на сухое вещество.
383
Содержание пектиновых веществ в чайном листе 6—7%, золы — 4—5%,
Для производства чайного танина используют отходы чая (сухие грубые
листья), в которых содержание сухих веществ равно 93%, витамина Р —
15—16% и кофеина—1,5—2,0% на сухое вещество.
Экстракция кофеина. Чайный лист слегка измельчается в дробилке 1
(рис. 89) и поступает на предварительную экстракцию дихлорэтаном в
экстрактор 2. .На предварительной экстракции из чайного листа извлека-
ются кофеин, смолы и пигменты. Этот процесс называется экстракцией ко-
феина. На 1 кг листа вводят в экстрактор 2,0—2,5 л дихлорэтана. Продол-
жительность экстракции 40—60 мин при температуре 45—50° С. Затем:
Рис. 89. Технологическая схема производства витамина Р из чайного листа.
экстрактор разгружают в друк-фильтр 3, снабженный паровой рубашкой
и соединенный с поверхностным конденсатором 4 и приемником дихлорэта-
на 5. По окончании фильтрации отгоняют из шрота растворитель путем пус-
ка пара в рубашку друк-фильтра.
Экстракция витамина Р. Сухой шрот из друк-фильтра 3 поступает в
сборник 7, а из него в экстракционную батарею 8, состоящую из 6—8 аппа-
ратов, для экстракции чайного танина спиртом при температуре 50° С. Пе-
ред разгрузкой экстрактора из шрота отгоняют спирт. Для обеспечения
отгонки растворителя экстракторы батареи соединены через конденсатор 9
со сборником 6 и снабжены паровыми рубашками.
Сгущение экстракта и получение препарата витамина Р. Спиртовой
экстракт витамина Р поступает в сборник 10, а из него в вакуум-перегон-
ный аппарат И для отгонки растворителя до содержания 40% сухих ве-
ществ. Концентрат поступает в сборник 12, а из него в вакуум-вальцовую
сушилку 13. Полученную сухую пленку измельчают в дробилке 14, просеи-
вают через бурат 15 и порошок направляют в расфасовку.
Спирт, отгоняемый при сушке, собирают в приемник 16, откуда он по-
ступает в сборник спирта 17. В этот же сборник поступает отгон спир-
та из вакуум-перегонного аппарата И.
384
Получение технического кофеина. Экстракт кофеина из друк-фильтра <3
направляют в сборник 18, откуда он поступает в вакуум-аппарат 19 для
сгущения до содержания в нем 80% сухих веществ1.
Сгущенный раствор сливают в кристаллизатор 20, где он кристаллизу-
ется 6—8 ч при 0°. Затем его фугуют в центрифуге 21, промывают спиртом
и высушивают в вакуум-сушилке 22.
Маточный раствор I из центрифуги поступает в сборник 23, откуда да-
лее направляется в вакуум-аппарат 24 для сгущения до содержания в нем
85% сухих веществ.
Сгущенный раствор II направляют в кристаллизатор 25, где он кристал-
лизуется 12 ч, затем поступает в центрифугу 26. Кристаллы кофеина II рас-
творяют в дихлорэтане в аппарате 27 и раствор направляют на первую крис-
таллизацию в сборник 18-, маточный раствор II является отходом производ-
ства.
Регенерация растворителей. Спирт из сборника 17 поступает в ректифи-
кационную колонну 28, где подвергается ректификации. Спирт-ректифи-
кат направляют в сборник ректификата 29. Дихлорэтан из вакуум-аппара-
та 19 поступает в систему флорентийских сосудов 30. Сухой дихлорэтан
поступает в общий сборник дихлорэтана 31.
В литературе имеется указание [38], что из маточного раствора II мож-
но выделить фитол. Однако примененная технология выделения весьма
сложна и связана с использованием еще двух органических растворителей
(ацетона и четыреххлористого углерода), что едва ли оправдает затраты по
сравнению с более простой технологией получения фитола из отходов мяты
одним растворителем (изопропиловым спиртом) [39, 40].
ВИТАМИН Р ИЗ ЖОМА ШИПОВНИКА (ROSAE)
Жом шиповника является отходом производства концентратов витами-
на С из плодов шиповника. Сырого жома на заводах получают в количестве
250% к массе сухих плодов шиповника. Состав сырого жома шиповника
(в %): вода 68—73; клетчатка 17—15; пектиновые вещества 2,5—2,0; зола
сырая 2,5—2,0; общий сахар 2,5—2,0; органические кислоты 1,0—1,5; бел-
ковые вещества 3,0—2,5; жир 2,8—2,4.
Из плодов шиповника после извлечения диффузионным способом аскор-
биновой кислоты получают жом, содержащий 0,9—1,5% витаминов груп-
пы Р (на сухое вещество).
Последние экстрагируют путем повторной диффузии водой при темпера-
туре 100° С в течение 1,5—2,0 ч. Получаемый диффузионный сок упаривают
в трехкорпусном выпарном аппарате под вакуумом в сироп, содержащий
30—40% сухих веществ. Сироп высушивают в вакуум-вальцовой или рас-
пылительной сушилке. Сухую пленку измельчают в порошок — концент-
рат, содержащий 20—22% витаминов группы Р (подробно см. стр. 364).
ВИТАМИН Р ИЗ ЧЕРНОПЛОДНОЙ РЯБИНЫ (FRUCTUS SORBUS ARONICAL)
[18, 41, 42]
Для расширения ассортимента Р-витаминных препаратов и антоциано-
вых красителей исследованы биологически активные вещества черноплод-
ной рябины. Для этого были проведены следующие исследования:
1 Экспериментально показано [38], что сгущенный экстракт кофеина сильно
загрязнен. Для очистки необходимо его растворить в воде и из раствора экстрагиро-
вать органическим растворителем (ДХЭ) — отогнать растворитель и далее, как ука-
зано ниже.
1/2 14—522
385
1. Разработана методика определения антоцианов фотоколориметриче-
ским методом по калибровочной кривой (рис. 90), построенной по выделен-
ному из черноплодной рябины цианидину [43].
2. В плодах методом распределительной хроматографии на бумаге иден-
тифицированы флавоноиды: рутин, кверцетин и геспередин.
Методом тонкослойной хроматографии на окиси алюминия в петролей-
ном эфире идентифицированы: а-каротин (>.„,„ =420; 445; 473 нм\ 7^=0 93):
р-Каротии (420; 450; 480 нм) Rf......... 0,88
Ксантофил-эпоксид (445; 475 нм) Rf .. . 0,42
Ксантофил (420; 447; 475 нм) Rf......... 0,24
Тараксантин (420; 442; 470 нм) Rf ... 0,16
Методом тонкослойной хроматогра-
фии на силикагеле [18] идентифициро-
ваны лецитин и кефалин.
3. Разработана технология комплек-
сного использования черноплодной ря-
бины [42], предусматривающая следую-
щие стадии: получение сока, получение
пищевого красителя, получение препа-
рата витамина Р.
Получение сока. Подготовляют пло-
ды (инспекция, мойка, измельчение в
вальцовой дробилке), прессуют массу в
прессах (типа Яна, шнековых, пакпрес-
сах), фильтруют, купажируют сок с са-
Рис. 90. Калибровочная кривая
для количественного определения
цианидина.
харом, расфасовывают в бутылки или банки и стерилизуют.
Получение пищевого красителя. Жом дважды экстрагируют водой
(1:4, 1:3) при температуре 90—95° С, 40 мин-, pH 5, сгущают в вакууме до
60% сухих веществ. Краситель расфасовывают в банки в горячем виде
(80° С) и герметически закрывают.
Получение препарата витамина Р. Жом II с влажностью 40% подверга-
ют сушке до остаточной влаги 7—8%, измельчают и таблетируют.
Из 1 т сырья получают (в кг) [18]:
Натуральный сок (содержание витамина Р
3—3,5%)............................... 654
Пищевой витаминный краситель (содержа-
ние цианидина 3,4%, витамина Р 6,6%) . 59,0
Препарат витамина Р (порошок, содержа-
ние витамина Р — 21,3%) ..............97,0
Разработана также технология получения высокоактивного порошко-
образного красителя путем экстракции жома I спиртом (60%-ным) при
pH 2,0, сгущения экстракта под вакуумом до 75% сухих веществ, осажде-
ния двукратным количеством 25%-ной серной кислоты при температуре
85° С и перекристаллизации осадка из спирта, содержащего 1% НО. Кра-
ситель содержит 54,1% цианидина. Выход его составляет 20 кг из 1 т пло-
дов.
ВИТАМИН Р ИЗ ЧЕРНОЙ СМОРОДИНЫ (RIBES NIGRUM) [1, 44—47]i
Ягоды черной смородины богаты витаминами: аскорбиновая кислота —
0,1—0,3%, витамин Р—1,2—1,5%, каротин — 0,7 мг%, витамин Вх —
0,06 мг%. Сок содержит большое количество пектиновых веществ.
Технология комплексного использования черной смородины предусмат-
ривает следующие две стадии производства: получение ягодного пюре, по-
лучение антоцианового препарата или пищевого красителя.
1 В работе, кроме автора, участвовали В. Афанасьева и О. Хорина.
386
Получение ягодного пюре [44]. Ягоды подвергают инспекции, мойке в
душевых мойках, бланшируют в воде при температуре 90—100° С от 3 до
8 мин, протирают через протирочные машины с ситами из нержавеющей
стали с отверстиями диаметром в 2 мм. Затем массу протирают через фини-
шер с диаметром отверстий 0,75—0,8 мм. Пюре расфасовывают в стеклян-
ные банки, герметически укупоривают и стерилизуют при температуре
100°С 30 мин. Содержание сухих веществ — 9,4—9,5%, антоцианов 1,15—
1,20%.
Для детского питания пюре перед расфасовкой смешивают с сахарным
сиропом (20 кг сиропа на 80 кг пюре) [47]. Содержание сухих веществ в
пюре с сахаром должно быть не менее 22%, антоциановых веществ — 1,0—
1,1%. Выход красящих веществ составляет 74—79%.
Получение антоцианового препарата или пищевого красителя. Отходы
черной смородины после протирочной машины содержат 3,3—3,9% крася-
щих веществ в виде глюкозидов цианидина и дельфинидина. Препарат по-
лучают экстракцией водой при температуре 90—95° С. Экстракт упаривают
в вакууме до содержания сухих веществ 35—40% и антоцианов 2,4—2,7%.
Выход красящих веществ составляет около 20% от введенных с ягодами.
Жом II высушивают и используют в качестве корма скота. Его выход со-
ставляет— 2,4—2,7% к массе свежих ягод. Содержание антоцианов в
нем — 0,85%, витамина Р — 3,4—3,7%.
Несмотря на то что черная смородина является весьма распространен-
ной и старинной культурой, состав биологически активных веществ ее ягод
современными методами не изучен.
ВИТАМИН Р ИЗ ЗЕЛЕНОЙ МАССЫ ГРЕЧИХИ (FAGOPYRUM SAGITATUM)
Гречиха из семейства гречишных имеет слегка мясистые сердцевидные
листья. Цветы собраны в кисти, образуя щитовидную метелку. Стебли име-
ют красную, а цветы — белую или розовую окраску. Высота растения
30—60 см. Содержание рутина в гречихе сильно колеблется. Отдельные ви-
ды гречихи содержат небольшое количество рутина (меньше 1%), а другие—
2% и более. Содержание рутина в отдельных сортах гречихи составляет
[48]:
Сорт гречихи Содержание рутина, % на сухое веще- ство листьев и цветов Сорт гречихи Содержание рутина, % на сухое веще- ство листьев и цветов
Богатырь . . . 1,7 Большевик .... 2,8
Ольгинская . . 1,57 Епифанская мест-
Тереховская . . 1,4 ная 2,0
Красноуфимская 1,73 Литовская . . . . 0,42
Из исследованных образцов гречихи наиболее богаты рутином Епифан-
ская местная (2%) и Большевик (2,8%).
Содержание рутина в стеблях незначительно (около 0,4%). Отношение
массы стеблей к массе листьев 1:1. Содержание сухих веществ .в зеленой
массе составляет около 10%.
Максимальное содержание рутина бывает в ранней стадии цветения (на
40—50-й[день с момента посева).
Урожайность зеленой массы гречихи составляет 20 т, а при двух укосах
в течение лета — 40 т с 1 га.
Производство кристаллического рутина из зеленой массы гречихи осу-
ществляется в различных странах по различным технологическим схемам
[31—33], но сущность их одна и та же. Все они сводятся к экстракции ру-
тина спиртом или водой, отгонке спирта и повторной очистке полученного
концентрата индифферентным растворителем (дихлорэтан, хлороформ) и
кристаллизации.
х/214* 387
Во ВНИВИ (В. Кириллов и Шахова) разработана схема производства
рутина из зеленой массы гречихи [49], заключающаяся в следующих стади-
ях [1]:
получение из зеленой массы гречихи сухой муки для экстракции;
экстракция рутина из муки 70%-ным спиртом при температуре 80° С
и получение сырого продукта;
очистка сырого рутина дихлорэтаном;
кристаллизация и перекристаллизация рутина.
Более подробно см. [1].
Получение из зеленой массы гречихи сухого материала для экстракции
[!].', При переработке зеленой массы растений значительные трудности
Рис. 91. Технологическая схема переработки зеленой массы гречихина
сухую муку.
представляют сушка и разделение стеблей и листьев. Необходимость в этих
процессах возникает потому, что стебли бедны витаминами, в том числе и
витамином Р (в 5—6 раз). Поэтому отделение стебля от листьев повышает
эффективность последующих процессов.
Во ВНИВИ разработана и в полузаводских условиях проверена сле-
дующая схема переработки зеленой массы люцерны на муку. Она может
быть применена для переработки зеленой массы гречихи.
Зеленая масса гречихи (рис. 91) с сырьевой площадки 1 поступает на
инспекционный конвейер 2 для удаления посторонних примесей и далее
проходит паровой бланширователь 3, где она подвергается ошпариванию.
Бланшированная масса дробится на табакорезальной машине 4 на крошку
типа табачной. Крошка элеватором 5 передается в протирочную машину 6.
Вытирки (жом) далее шнеком 7 транспортируются во вторую протирочную
машину 8, куда добавляют 100—150% воды к массе жома. Протертая мас-
са-пюре из протирочной машины 6 поступает в сборник 9 и далее через
сборник 10 насосом 12 в приемник пюре 13 и в вакуум-вальцовую сушилку
14. Сухое пюре собирают в приемнике 15 и затем при необходимости разма-
лывают в дробилке 16. Мука поступает в сборник 17. Жом II из протироч-
ной машины S-попадает на конвейер 11 и удаляется как отход производства.
При данном методе обработки зеленой массы обеспечивается наилучшая
сохранность содержащихся в ней витаминов (Р, Е и каротина) и высокое
качество материала для экстракции.
ВИТАМИН Р ИЗ БАДАНА (BERGENIA CRASSIFOLIA) [50]
Бадан широко распространен в Западной и Восточной Сибири. Вслед-
ствие высокого содержания дубильных веществ в листьях и корневище
(до 23—24% на сухое вещество) он обратил на себя внимание как сырье
388
для получения дубильных экстрактов. Кроме того, в 1929 г. Чичибабин,
Кирсанов и др. открыли высокое содержание в нем кристаллического
вещества, названного бергенином. Вещество получено при гидролизе та-
нидов в количестве 22% к массе их и было идентифицировано как произ-
водное изокумарина [51 ] со следующим, строением:
СНзО-^^^^р-СНОН-СНОЧ-СНОН-СНгОН
3-[тетраокси (1, 2, 3, 4-бутил)]-6-окси-7-метокси-изокумарии
Связь бергенина и танидов не установлена. Безводный бергенин (С14Н16О9)
плавится при температуре 233° С, а водный (С14Н18О]0) имеет две точки
плавления 138° и 233° С; [а,]о — 37,3° (спирт) и — 47,4° (вода); легко рас-
творим в щелочах и трудно в воде и спирте.
В листьях бадана, кроме дубильных веществ (около 23% на сухое ве-
щество) содержится арбутин, представляющий собой гликозид гидрохинона
В 1935 г. Якимовым и др. была разработана технология получения из
листьев бадана танина и гидрохинона с выходами 10% и 3,8—4,5% к массе
сухого листа соответственно [52].
Водные экстракты бадана применялись в народной медицине от поно-
сов и лихорадки; кроме того, последние исследования показали эффектив-
ность лечения экстрактами бадана эрозии шейки матки, дизентерии и дру-
гих кишечных заболеваний [53, 54].
Наши исследования [50] показали, что в корневище бадана содержится
9,8—14,3% дубильных веществ, 0,4—0,5% бетаина, 0,1% холина. Каро-
тиноиды и токоферолы не обнаружены. Методом распределительной хрома-
тографии на бумаге с предварительной очисткой через колонку с полиамид-
ной смолой удалось идентифицировать качественно: (—)-эпикатехин,
(—)-эпикатехингаллат и (—)-эпигаллокатехин.
Технология получения Р-витаминных препаратов заключается в экст-
ракции измельченного корневища бадана водой, либо 60%-ным спиртом.
Экстракты сгущают в вакууме и высушивают. Получаемый после измельче-
ния порошок содержит 30—35% дубильных веществ (растворителем может
быть вода и спирт). Однако выход дубильных веществ при водной экстрак-
ции (55—60%) значительно ниже, чем при экстракции 60%-ным спиртом
(80—85%). Препараты витамина Р из бадана, полученные водной и спирто-
вой экстракцией, были биологически исследованы на токсичность и Р-ви-
таминную активность методом капиллярной реакции [55]. Показана
высокая Р-витаминная активность препаратов бадана, выражающаяся в уве-
личении более чем вдвое (по сравнению с исходным) резистентности капил-
ляров у 100% опытных животных, содержавшихся на Р-авитаминной диете
и получавших по 10 мг препарата в день. Результаты опытов были стати-
стически достоверны. Препараты бадана оказались более активными, чем
препараты катехинов чая. Время появления петехий до опыта в течение
26 сек. увеличивалось до 55,7 сек при 100% животных с положительной реак-
цией для препаратов бадана и соответственно 50,1 сек и 88% для катехи-
нов чая.
Из изложенного видно, что бадан является весьма перспективным про-
мышленным сырьем для производства Р-витаминных препаратов1.
1 В исследованиях бадана, кроме автора, принимали участие М. Ушакова,
А. Ефимов, И. Кущинская, М. Мительман, С. Власов и- М. Стратийчук.
13-522 389
ВИТАМИН Р ИЗ ОТХОДОВ ЦИТРУСОВОГО СОКОВОГО ПРОИЗВОДСТВА
Цитрусовые плоды имеют разнообразное применение в пищевой про-
мышленности. Из Них приготовляют соки, компоты, джем, цукаты, а из
отходов (кожуры) — пектин. В производстве в основном применяют апельси-
ны и мандарины.
Химический состав кавказских апельсинов и мандаринов (по Ф. Цере-
витинову и В. Реутову) [56, 57]
Апельсины Мандарины
Масса 1 плода, а................. 157—161 43—51,6
Кожица » , %................... 16,7—17,1 18—23,8
Мякоть » , %................. 82,1—82,9 68,5—79,2
Семена » , %................. О—1,2 0—7,7
Состав мякоти (в %)
Сухие вещества .................. 13,87—16,67 15,04—19,40
Лимонная кислота................. 0,65—0,71 0,31—0,84
Инвертный сахар ................. 3,63—4,82 2,56—5,74
Общий сахар ................. 6,01—7,99 3,42—11,70
Кожица апельсинов и мандаринов содержит большое количество эфир-
ного масла 1,2—2,1 и 1,86—2,5% от массы кожицы (соответственно).
В белом слое кожицы (albedo) содержится значительное количество про-
топектина (50% на сухое вещество).
Технология получения витамина Р (гесперидина) из отходов производ-
ства мандаринового сока [58, 59]. Отходы производства цитрусового сока
(кожура и мезга) после отжима и размельчения подвергают водно-щелочной
экстракции (обработка окисью кальция) при pH 10,4—10,8. При этом фла-
воновый комплекс переходит в экстракт. Последний фильтруют и из фильт-
рата выделяют гесперидии добавлением соляной кислоты до pH 4,0—4,4.
Осадок гесперидина отфильтровывают, сушат и измельчают. Порошок
должен содержать не менее 90% гесперидина. Гесперидии представляет
собой белый кристаллический порошок с легким специфическим запахом
со слабогорьковатым вкусом, легко растворим в пиридине и в растворах
щелочей (pH 11 и выше). Растворим в 50%-ном этаноле, в кипящей ук-
сусной кислоте. Нерастворим в воде и в органических растворителях.
Отходы производства мандаринового сока составляют 60% к общей
массе сырья. Они содержат гесперидина 1,2—1,5%, эфирного масла 1,2%
и пектина 1,5%. Выход препарата гесперидина составляет 0,8% к массе
сырья [58].
ВИТАМИН Р ИЗ СТОЛОВОЙ СВЕКЛЫ (BETA VULGARIS HORTENSIS L.)
Столовая свекла по литературным данным [60—62] содержит следую-
щие витамины (в мг%): С — 4,0—22,0; Р — 15—40; Вх — 0,01—0,05; В2—
0,023—0,07; РР — 0,56—0,65; В3 — 0,10—0,11; В6 — 0,013; фолиевая кис-
лота — 0,045. Зола содержит (в % от массы золы) [63]: КгО — 33,4—46,6;
Na2O — 8,4—18,3; СаО — 6,5—6,7; MgO — 2,5—5,1; Р2О5 — 14,0—19,4;
С1 — 0,6—1,3; SiO2 — 3,1—7,4; SO3—10,8—15,6. Благодаря высокому
содержанию солей калия столовая свекла обладает диуретическим действи-
ем. Свекла является одним из лучших продуктов, где кальций и фосфор
находятся в наиболее благоприятном для усвоения сочетании.
Венгерский доктор А. Ференци в клинических условиях выявил проти-
вораковое действие столовой свеклы [19, 20].
Красящие вещества столовой свеклы. Они отличаются от других анто-
циановых растительных пигментов по своим физико-химическим свойствам.
Основной пигмент столовой свеклы — бетанин — относится к особой груп-
пе азотсодержащих красящих веществ — бетацианам. Он впервые был вы-
390
делен и охарактеризован Р. Вильштеттером и Г. Шуделем, как азотсодер-
жащий антоциан, в 1918 г.
Пигменты столовой свеклы изучены мало. Они отличаются от типичных
антоцианов наличием в молекуле 6% азота, устойчивостью к HNO3, хоро-
шей растворимостью в воде и нерастворимостью в органических раствори-
телях, кроме метилового и этилового спирта.
В отличие от типичных антоцианов не удается получить аглюконов пиг-
ментов столовой свеклы путем обычного кислотного гидролиза, так как
они полностью разрушаются.
Пигменты столовой свеклы представляют собой малоустойчивые соеди-
нения, которые легко окисляются на воздухе и при нагревании. Доволь-
но трудно получить их в чистом виде. Поэтому до сих пор нет точных дан-
ных о химической природе пигментов столовой свеклы.
Эмпирическая формула основного пигмента — бетанина установлена
Н. Wyler [64] в виде C25H2g_30O13N2, молекулярная масса — 564—566.
Хроматографически было установлено, что бетанин распадается на бета-
нидин и глюкозу. Позднее (в 1965 г.) [65] была предложена следующая
химическая формула для бетанидина:
20
,СНЭ СН2 ю с-соон
НО-СД4'сн 3 Ун-COO-
I 6 Н ,1+ I ,3 II 19
Н3СО-С^7/СН — N=CH-CH=C,1 Х-Н-СООН
сн8 11 12 сн217
18
Химическое название бетанидина: 5-окси-6-метокси-2-карбокси-индол-
М-[1-этил-(пиридил-15,17-дикарбоновая кислота)] -1,12-диен.
Бетанин прочно адсорбируется на анионите, слабо на катионите. Он
легко превращается в желтый пигмент, который под действием кислорода
может снова превратиться в красно-фиолетовый пигмент. Наибольшая ус-
тойчивость пигментов столовой свеклы наблюдается в растворе с pH 4,0;
при pH 9,2 окраска пигмента изменяется от красной через фиолетовую к
желтой (при pH 10,5).
Наши исследования при участии В. Афанасьевой и О. Хориной показа-
ли следующий химический состав столовой свеклы сорта Бордо и ее сока
(табл. 34).
Таблица 34
Показатели Содержание, %
свекла СОК
Сухие вещества 23,5 15,3
Красящие вещества (по циани-
дину) 1,3 0,75
Сумма сахаров 12,9 12,2
В том числе:
редуцирующие 0,7 0,8
пектиновые вещества . . 1,75 0,5
зола 1,05 —
клетчатка . 1,15 —
Общая кислотность (на яблоч-
ную кислоту) 0,57 2,44
Сырой белок (Nx6,25) . . . . 1,3 —
Холин 0,034 0,002
Бетаин — 0,437
Фосфолипиды 0,015 —
13'
391
Рис. 92. Хроматографическое разделе-
ние красящих веществ столовой свек-
лы и их идентификация:
/—желтый пигмент; 2 и 2а—красно-фиолетовый.
Из биологически активных веществ столовой свеклы интерес представ-
ляют красящие вещества (бетанин), бетаин (липотропный фактор) и элемен-
ты золы.
Исследование красящих веществ столовой свеклы производилось мето-
дом тонкослойной хроматографии и спектрофотометрии. Из большого чис-
ла испытанных адсорбентов четкое разделение пигментов в тонком слое
дал тальк, при растворителе — вода или вода — метанол (3:1). Полученные
пятна пигментов элюировали водой и элюаты были подвергнуты спектро-
фотометрированию в видимой области света (рис. 92). Фиолетовые пигмен-
ты № 1 и 2 идентифицированы по максимуму поглощения 537 нм (Е}%—•
1100) как бетанин. Желтый пигмент
№ 2 а имеет максимум поглощения
480 нм и не идентифицирован.
Наличие трех флавоноидов в
соке столовой свеклы установлено
хроматографией на бумаге с при-
менением растворителя 60%-ной
уксусной кислоты и проявителя —
хлористого алюминия. Идентифи-
цирован (по метчику) кверцетин,-
Два других флавоноида не иденти-
фицированы.
Исследовались также фосфоли-
пиды свеклы и установлено их со-
держание около 0,15% к массе
свежей свеклы. Методом хромато-
графического разделения в тонком
слое силикагеля идентифицированы
кефалин и лецитин. Начаты техно-
логические исследования по полу-
чению биологически активного красителя и препарата из столовой свеклы.
БИОЛОГИЧЕСКИ АКТИВНЫЕ ВЕЩЕСТВА РЯБИНЫ ОБЫКНОВЕННОЙ
(SORBUS AUCUPARIA) [66, 67, 67а]
Имеются указания о содержании в плодах рябины каротиноидов [68,
69], £>-сорбита [70, 71], парасорбиновой кислоты [72]. Исследования, про-
веденные автором (совместно с И. Кущинской и М. Мительман), показали,
что в сухих плодах рябины содержатся флавоноиды, антоциановые вещест-
ва, каротиноиды и др. (табл. 35).
Таблица 35
Биологически активные вещества
Содержа-
ние,
мг %
Биологически активные
вещества
Содержа-
ние,
мг %
Аскорбиновая кислота..........
Витамины группы
Р (кверцетин, изокверцетин,
рутин) ...................
Антоциановые вещества (по циа-
иидину).......................
Дубильные вещества............
Каротиноиды..................
Фосфолипиды (кефалин, лецитин)
Токоферолы....................
Рибофлавин ...................
11,0
2600
795
610
27,1
70,4
2,41
0,8
Сорбит ......................
Парасорбиновая кислота . . .
Пектиновые вещества ....
Кислотность (в пересчете на
яблочную кислоту)............
15—20
6,86
2,0
5,95
392
Для определения общего содержания и идентификации отдельных ве-
ществ были использованы следующие методы: для флавоноидов — метод
Лоренца-Арнольди, усовершенствованный Вадовой [7]; для идентифика-
ции применен метод хроматографии на бумаге в 60%-ной уксусной кислоте
с использованием проявителя 1%-ного спиртового раствора хлористого
алюминия. Антоциановые вещества количественно определены по калибро-
вочной кривой цианидина [43] и идентифицированы хроматографией на
бумаге (одно пятно как цианидин; Хтах =555 нм; второе пятно не иденти-
фицировано). Каротиноиды идентифицированы при помощи тонкослойной
хроматографии на окиси алюминия [74] в виде p-каротина, р-каротин —
моноэпоксида и криптоксантина по величинам Ry и максимумам поглоще-
ния (450 нм в петролейном эфире и 460 нм в хлороформе).
Фосфолипиды выделяли методом Фольча [75], идентифицировали хро-
матографией в тонком слое силикагеля с содой (1:2) с проявлением пятен
парами йода или раствором родамина [76].
Парасорбиновую кислоту определяли путем превращения ее в сорбино-
вую кислоту щелочным гидролизом [72, 77].
Разработана технология получения двух концентратов из рябины —
рябиновой пасты и рябинового порошка. Также показана целесообразность
получения сиропа с витамином С из смеси шиповника и рябины в составе
(70—80:30—20).
Биологическими исследованиями на лабораторных животных установлено,
что препараты рябины (паста и порошок) приводят к снижению количества
жира в печени и холестерина в крови; не оказывают существенного влияния
на углеводный и белковый обмен [67а].
ТЕХНОЛОГИЧЕСКИЕ ПОКАЗАТЕЛИ
ПРОИЗВОДСТВО ПРЕПАРАТА ВИТАМИНА Р
(КАТЕХИНЫ) ИЗ ОТХОДОВ ЧАЙНОГО ЛИСТА
Сухие грубые листья чая (отходы), %
Сухие вещества................... 93,0—94,0
Катехины и флавоновые глюкозиды 15,0—16,0
Кофеин........................... 1,5—2,0
Технология производства
Экстракция кофеина дихлорэтаном (2—2,5 л на
1 кг листьев) при температуре,°C ........... 45—50
Экстракция витамина Р спиртом при температуре,
°C •.......................................... 65—70
Откачка на мерник, %............................ 200
Сухие вещества в экстракте, % .............2,8—3,0
Отгонка спирта при остаточном давлении,
мм рт. ст................... 40
Температура отгонки, °C .................... 28—30
Потери катехинов [38], %
при выпаривании............................ 3—5
при сушке на вакуум-вальцах ............ 1,2—2,0
Содержание катехинов в препарате, %......... 80—91
в том числе [38];
(—)-эпигаллокатехин....................... 19,5
(+)-галлокатехин ........................... 2,5
(—)-эпикатехин и (+)-катехин................ 6,2
(—)-эпигаллокатехингаллат ................. 49,0
(—)-эпикатехингаллат ...................... 10,8
Выход катехинов, % . . ................. 77,0—79,0
Расход чайного листа, кг на 1 кг 100%-ного
препарата...................................6,6—7,8
100%-ного спирта, л..................... 12—14
100%-ного дихлорэтана, кг............... 15—20
393
ПРОИЗВОДСТВО ПРЕПАРАТОВ ВИТА-
МИНА Р ИЗ ЖОМА ШИПОВНИКА
(см. стр. 367—368)
ПРОИЗВОДСТВО РУТИНА ИЗ ЗЕЛЕ-
НОЙ МАССЫ ГРЕЧИХИ
Сухая зеленая масса гречихи, %
Влажность.................10,0—12,0
Рутин ...................2,8
Технология производства
Выход муки, % к сырью . 7,0—8,0
Насыпная масса муки,
кг!м3.................... 340
Экстракция 70%-ным спир-
том при температуре,°C . . 80
Выход сырого рутина, %,к
массе сырья.......... 15,0
Выход сырого очищенного
рутина, %............ 9—10
Выход готового препарата
рутина, % к массе сырья . 1,5—2,0
Расход сырья, кг на 1 кг рутина
Сухая зелень гречихи ... 75,0
Спирт ...................... 10,0
Дихлорэтан .............. 1,0
Выход витамина Р .... 10,3—14,2
Выход красящих веществ . 12,4—12,8
Сырой жом................ 44,7—45,7
Препарат витамина Р, %
Сухие вещества........... 94,5—96,7
Витамин Р................19,7—23,3
Антоцианы................9,0—10,0
Выход препарата, % к сы-
рью ........................9,0—10,3
Выход витамина Р, % к со-
держанию в сырье .... 66,7—67,8
Выход антоцианов, % к со-
держанию в сырье .... 57,6—59,5
Выход препаратов, кг из 1 т плодов
Натуральный сок.......... 654
Пищевой краситель .... 59
Препарат витамина Р . . . 97
ПРОИЗВОДСТВО ПРЕПАРАТА ВИТА-
МИНА Р ИЗ ЧЕРНОЙ СМОРОДИНЫ
Плоды, %
Сухие вещества ..... 13—15,5
Витамин Р...............0,4—0,5
Антоцианы............... 1,2—1,5
ВитаминС . ...........0,1—0,3
ПРОИЗВОДСТВО ПРЕПАРАТА ВИТА.
МИНА Р ИЗ ЧЕРНОПЛОДНОЙ
РЯБИНЫ
Плоды, %
Сухие вещества....... 23,0—25,0
Витамин Р..............3,0—4,0
Антоцианы.............. 1,5—2,0
Технология производства
Натуральный сок, %
Сухие вещества .........19,0—20,5
Витамин Р...............3,0—3,5
Антоцианы...............0,4—0,5
Выход сока, % к сырью . 65,0—66,0
Выход витамина Р в соке,
% к содержанию в сырье . 10,0—13,0
Выжимки................. 33,6—34,6
Пищевой краситель, %
Сухие вещества.......... 68,0—75,0
Витамин Р...............6,0—7,0
Антоцианы (красящие ве-
щества) ................ 3,3—3,8
Технология производства
Пюре без сахара, %
Сухие вещества...........9,4—9,5
Витамин Р................0,2—0,25
Антоцианы................1,15—1,20
Выход
витамина Р........... 46—50
красящих веществ . . 73,8—79,1
Выход пюре (к сырью) . . 86,4—88,2
Пищевой краситель, %
Сухие вещества .......... 35—40
Красящие вещества .... 2,4—2,7
Выход красителя, % к сы-
рью ..................... 8,2—9,1
Выход сухого жома, % к
сырью ...................2,4—2,7
ПРОИЗВОДСТВО ВИТАМИНА Р ИЗ
БАДАНА (табл. 36).
Выход красителя, % сырью 5,3—6,1
Сырье, %
Дубильные вещества . . . 9,8—14,3
Бергенин ............... 2,2—3,1
Бетаин..................0,4—0,5
Холин ..................0,1—0,15
Технология производства (табл. 36)
Таблица 36
Способ экстракции Содержание дубиль- ных веществ, % Выход, %
в сырье в сухом препарате дубиль- ных ве- ществ препарата к сырью
Водой .... 60%-ным спир- 14,3 30,2 56,9 26,9
ТОМ 14,3 32,2 81,6 35,1
394
ПРОИЗВОДСТВО ВИТАМИНА Р ИЗ ОТХОДОВ ЦИТРУСОВОГО сокового
ПРОИЗВОДСТВА
Отходы производства мандаринового
сока, %
Гесперидии........................... 1,2—1,5
Эфирное масло........................ 1,2—1,3
Пектиновые вещества.................. 1,5—1,6
Выход отходов к сырью................ 60—65
Технология производства
pH при водно-щелочной экстракции . . 10,4—10,8
pH при выделении гесперидина .... 4,0—4,4
Содержание гесперидина в препарате, % 90,0—91,0
Выход препарата, % к массе отходов . 0,8—0,9
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. Ш н а й д м а н Л. О. Производство витаминов. М., Пищепромиздат, 1958, с. 39.
То же. Ш н а й д м а н Л. О., Сошников Д. Я-, Солодухин А. И. —
«Изв. вузов. Пищевая технология», 1965, № 3, с. 19—21.
2. Запрометов М. Н. Биохимия катехинов. М., «Наука», 1964, с. 295.
3. Чумбалов Т. К- и др. Сб. работ кафедры органической химии по изучению
химического состава растительного сырья, Казгосполитиздат, 1962, 136с.
4. A. Szent-Gyorgyi, S. Rusznyak, Nature, 1936, 138, 3479.
5. Armentano L., Z. Ges. exptl. Med., 1936, 97, 630; Z. Klin. Med., 1936, 129, 685.
6. lochi C., Kulkarni A., J. Sci. Ind. Res., 1957, 16, 307.
7. HI н а й д м а.н Л. О., Кущи некая И. H. Идентификация флавоновых и
катехиновых веществ плодов шиповника. — «Медицинская промышленность СССР»,
1965, № 2, с. ,14—17.
8. В о р о н ц o’bj В. Биохимия чая. М., Пищепромиздат, 1946.
9. Курсанов А. Л., Букин В. Н., Поволоцкая К- Л., Запроме-
тов М. Н. Биологическое действие чайного танина. —«Биохимия», 1950, т. 15,
вып. 4, с. 337—345.
10. Н и к о л а е в Р. П., Поволоцкая К- Л., Водолазская Н. А.
Биологическая ценность различных концентратов и препаратов витамина С. —
«Биохимия», 1953, т. 18, вып. 2, с. 169—174.
11. Cotereak Н., G а b е и др., Nature, 1948, 161, 557.
12. 'Ш а м р а й Е. Ф. О механизме биологического действия витамина Р. — В сб.:
«Материалы VI научной„сессии ВНИИВитаминологии», М., 1967, с. 67—68.
13. К У Р с а н о в А. Л., ""Запрометов М. Н., Ерофеева Н. Н. Вита-
минная активность катехинов чайного листа. — «Биохимия», 1952, т. 17. вып. 6,
с. 729—733.
13а. S е s h a d г i Т., Ann. Rev. Biochem., California, 1951, 20, 487.
14. P а г г о t J., Onicka A., Prac. Inst. Med., Chicago, 1946, 16, 94.
15. Березовская H. H. Материалы физиологического действия флавоноидных
соединений и их связи с аскорбиновой кислотой в организме животных. Автореф.
до кт. дисс., М., 1968.
16. Ш е м я к и н М. М. Успехи химии витаминов и их инактиваторов за последние
годы. — «Успехи химии», 1947, т. 16, вып. 3, с. 279—314.
17. Л е о н т ь е в И. Успехи современной биологии. 1945, т. 19, с. 134.
18. Ш н а й д м а н Л. О. Исследования в области химии и технологии производст-
ва витаминов. Доклад, докт. дисс., М., 1967, 45.
19. F е г е п с z i A., Zschr. inn. Med., 1959, 8, 408.
20. F е г е n с z i A., Zschr. inn. Med., 1961, 10, 437.
21. К а б и e в О. К., Верменычев С. М., Чумбалов Т. К. О противо-
опухолевой активности лейкоантоцианидинов и катехинов. — В сб.: «Вопросы он-
кологии и радиологии», Алма-Ата, «Наука», 1965, с. 236.
22. Е в с е е н к о Л. С. Клинические аспекты применения малотоксичных феноль-
ных соединений. Тезисы симпозиума по фенольным соединениям, АН СССР, М.,
1966, с. 49.
23. Б о г д а н о в Г. Н., Бурлакова Е. Б. и др. Противоопухолевые и радио-
защитные свойства фенольных сред. Там же, с. 48.
24. С к а р б о р о Г., Бахарах А. А. Витамин Р. — В сб.: «Биохимия и фи-
зиология витаминов», 1950, № 2, с. 7—57.
25. Р ы с с С. М. Витамины. Медгиз. 1955, с. 259.
26. 3 а п р о м е т о в М. Н. Биохимия чайного производства. М., «Наука», 1950,
6, с. 110.
27. У ш а к о в а М. Т., Ефимов А. 3., Анисимова А. Д. и др. Сравни-
тельная биологическая оценка новых Р-витаминных препаратов. — В сб.: «Мате-
риалы совещания по витаминам из природного сырья», Куйбышев, ЦБТИ, 1964,
с. 178—182.
28. Т а р а с к и н а К- В., Чумбалов Т. К-, У ш а к о в а М. Т., Аниси-
мова А. Д. Получение лейкоэфдина и эфдина из горной эфедры и изучение их
395
• Р-витамннной активности. — «Медицинская промышленность СССР», 1966, № 4,
с. 27—29.
29. Ч у м б а л о в Т. К-, Киль Т. А. Сб. работ кафедры органической химии по
изучению химического состава растительного сырья, Казгосполитиздат, 1962, с. 6;
там же, с. 21—36.
30. Шнайдман Л. О., Афанасьева В. С., Солодухин А. И., Па-
панов В. А. Использование отходов сокового и винодельческого производства
для получения витамина Р и каротина. — «Консервная и овощесушильная промыш-
ленность», 1965, № 12, с. 31—33.
31. И а г г у М., Пат. США 2626869, 12/1, 1950; Offic. gaz., 1953, 666, № 4, 1060.
32. С о n с h J., N a g h s k i J., Porter W. Пат. США 2520127, 31/X 1947; Off.
gaz., 1950, 637, 5, 1375.
33. К г e w s о n C., Couch J., J. Amer., Pharm. Assoc., 1950, 3, 163.
34. А г а п о в a E. В. Усовершенствование технологии переработки чайного листа
на витамины группы Р. — В сб.: «Витаминная промышленность», Пищепромиздат,
1957, № 4, с. 45—53' с ил.
35. А г а п о в а Е. В. Технология производства витамина Р из листьев чая. — В сб.:
«Витаминные ресурсы», Изд-во АН СССР, 1959, № 4, с. 213—224 с ил.
36. Запрометов М. Н., Агапова Е. В. — «Медицинская промышлен-
ность», 1964, № 10, с. 46.
37. К У р с а н о в А. Л., Запрометов М. Н., Агапова Е. В., Бере-
зовский В. М. и др. Авт. свидет., № 117043, 1958; Бюлл. изобрет., 1959, № 1,
с. 18.
38. Агапова Е. В. Витамин Р из листьев чая. Автореф. дисс. М., 1965.
39. К а л у г и н П. И., Шахова М. Ф. Авт. свидет., № 136855, 1959; Бюлл.
изобрет., 1961, № 6, с. 42. .
40. М и х л н н Э. Д., Шахова М. Ф., Лукьянова Л. В. Препарат фи-
тола из отходов мяты перечной. —«Труды ВНИВИ», М., Пищепромиздат, 1961,
№ 8, с. 57—65.
41. Ш н а й д м а н Л. О. Народнохозяйственное значение витаминов и перспективы
расширения их производства. — В сб.: «Материалы совещания по витаминам из
природного сырья», Куйбышев, 1964, 17—30.
42. Шнайдман Л. О., Афанасьева В. С., X о р н н а О. П. Исследо-
вания в области получения биологически активных красителей. Там же, с. 156 —
163.
43. Шнайдман Л. О., Афанасьева В. С. Методика определения анто-
циановых веществ. Рефераты докладов IX Менделеевского съезда, М., «Наука»,
1965, № 8, с. 79—80.
44. Сборник технологических инструкций по производству консервов, М. Пищепромиз-
дат, 1960, № 2, с. 104.
45. Степанова Е. М. Черная смородина — витаминная культура, М., Пищепром-
издат, 1950.
46. Сборник технологических инструкций по производству витаминов, М., Пищепром-
издат, 1943, с. И.
47. МРТУ 18/8—65. Консервы. Пюре плодовое и ягодное, Сб. межреспубликанских
технических условий, Изд. стандартов, 1965.
48. Кириллов В. Г., Шахова М. Ф. Рутин, его производство и лечебно-
профилактическое применение.— В сб.: «Витамины. Пищевая промышленность за
рубежом», М., Пищепромиздат, 1955, № 1, с. 76—88.
49. К и р и л л о в В. Г., Шахова М. Ф., Соловьев Б. М. Авт. свидет.,
№. 103713, 1955; Бюлл. изобрет., 1956, № 6, с. 13.
50. Атлас лекарственных растений СССР, 1962, с. 60. То же. Шнайд-
ман Л. О., Ефимов А. 3., Кущинская И. Н., С т р а т и ft-
чу к М. А. и др. Биологически активные вещества Bergenia crassifolia fritsch и
их промышленное использование. «Растительные ресурсы», 1970, № 6, с. 415—419.
51. Чичибабин А. Е., Кирсанов А. В., Королев А. И., Ворож-
цов Н. М. Недубильные вещества экстракта корневища бадана,— «Известия
АН СССР», 1929, № 4, с. 323.
52. Я к и м о в П. А., К о з л о в и ч Н. Б., К P у с с е р О. В. Техническое
растение бадан. —ЖПХ, 1935, т. 8, вып. 4, с. 654.
53. Гефштадт Н. Ф. К фармакологии бадана. — В сб.: Лекарственные растения
Сибири, их лечебные препараты и применение, Томск, 1953, № 4, с. 148.
54. Г л е з и н В. М. Новое лекарственное средство —бадан. «Медицинская про-
мышленность СССР», 1950, № 6, с. 36.
55. Е р о ф е е в а Н. Н. Биологическая методика и результаты испытания актив-
ности веществ, обладающих действием витамина Р. — В сб.: «Витаминные ресурсы
и их использование», М., АН СССР, 1959, № 4, с. 171—178 с ил.
56. Ц е р е в и т и н о в Ф. В. Химия свежих плодов и овощей. Сельхозгиз, М.,
1933, с. 565.
57. Цер евитинов Ф. В., Реутов В. — «Пищевая промышленность», 1924,
с. 7—8.
58. Романенко Е. В. — «Консервная и овощесушильная промышленность»,
1964, № 1, с. 19.
396
59. Р о м а н е н к о Е. В. Метод определения гесперидина в препарате витамина Р.
— Прикл. биохим. и микробиол., 1966, т. 2, вып..З, с. 308—312 с ил.
60. Бабичев И. Биохимия столовой кормовой свеклы. — В кн.: Биохимия овощ-
ных культур, М„ Сельхозиздат, 1961, с. 400.
61. В а д о в а В. А., Т о п е х а Е. Ф. Содержание витамина Р в плодах и ово-
щах. — «Труды ВНИВИ», М., Пищепромиздат, 1953, № 4, с. 192 —195.
62. Дев ятнин В. А. Витамины. М., Пищепромиздат, 1948, с. 262.
63. Церевитинов Ф. В. Химия и товароведение свежих плодов и овощей,
М., Госторгиздат, 1949, с. 360.
64. W у 1 е г И. и др., Helv. Chim. Acta., 1959, 42, 1696.
65. W i 1 с о k M., Wyler H., M о 1 г у Т., D г е i d i n g A., Helv. Chim.
Acta., 1965, 48, 24, 252.
66. Ш м у к А. А., Замыслов А. Ягоды рябины как сырье для получения ка-
ротина и каротиновых препаратов. Доклады ВАСХНИЛ, 1945, № 4—5, с.7.
67. П р о ц е н к о Л. Д. Исследования химической природы некоторых каротино-
носителей. Авторефер. кандид. дисс., Киев, 1953.
67а. Шнайдман Л. О., Кущинская И. И., Ефимов А. 3. и др. Био-
логически активные вещества плодов рябины обыкновенной и перспективы их про-
мышленного использования. — «Растительные ресурсы», 1971, т. 7, вып. I, с. 68—
71.
68. Розенблюм Ю., Левина Л., — «Медицинская промышленность СССР»,
1948, 1, 126.
69. Niirnberger Н., Pharmazie, 1964, 19, 10, 677.
70. И в а н о в Н. Н. Методы физиологии и биологии растений. 1946, с. 81.
71. Б а л а н д и н А. А., Васютина Н. А., Ч е п и г о С. В., Барыше-
ва Г. С. ДАН, 1959, 128, 5, 941.
72.. Н andschack W., Flora, 1963, 153, 4, 514; Е. Letzig, Nahrung, 1964, 8, 1, 49.
73. Девятнин В. А. Методы химического анализа в производстве витаминов.
«Медицина», 1964, с. 291.
74. Кущинская И., Шнайдман Л. Идентификация каротиноидов, содер-
жащихся в сухих плодах шиповника вида R. Cinnamomea. — «Медицинская про-
мышленность», 1964, № 4, с. 38—40.
75. F о 1 с h J., Z е е п i s М., Stanley Q., J. Biol. Chem., 1957, 226, 491.
76. Ш у г а м Н. А., Шнайдман Л. О. Жирные кислоты и фосфолипиды в мас-
ле облепихи. — «Изв. вузов. Пищевая технология», 1967, № 1, с. 52.
77. Овчарова Г. Сорбиновая кислота — консервант пищевых продуктов.М.,
ЦИНТИ, Пищепромиздат, 1964.
Глава 20. МОРКОВЬ (DAUCUS CAROTA) И ТЫКВА (CUCURBITА) —
ПРОМЫШЛЕННОЕ СЫРЬЕ ДЛЯ ПРОИЗВОДСТВА КОНЦЕНТРАТОВ
КАРОТИНА
МОРКОВЬ [1]
Одним из видов сырья для промышленного производства каротина слу-
жит морковь, принадлежащая к группе овощей-корнеплодов. Морковь
(Daucus carota) — двухлетнее растение из семейства зонтичных. В первом
году она дает корнеплод и листья, а во втором году — цветоносные стебли
и семена. Для производства каротина используются лишь корнеплоды
моркови. В моркови содержится 10—15 мг% каротина.
По форме морковь бывает следующих видов: каротель, полудлинная и
длинная. Средний химический состав моркови приведен в табл. 37.
Таблица 37
Состав Количе- ство, % Состав Количе- ство, %
Влага Сухие вещества В том числе: экстрактивные .... водорастворимые . . . Общий сахар Азот 85,5 14,5 9,1 5,4 7,0 0,9 Клетчатка Зола Жир Крахмал Пектиновые вещества . . . Пентозаны 1,6 1,0 0,3 0,2—0,9 0,6—2,9 1,0—1,2
397
Примерный состав золы моркови, %
К2О Na2O CaO MgO Fe2O3 Р2О5 SO3 SiO2 Cl
37 21 И 4 1 13 6 2 5
Наши исследования (совместно с М. Ф. Шаховой) методами хроматогра-
фии и спектрофотометрии выявили в моркови следующие биологически
активные вещества: каротиноиды — а- и p-каротин, ликопин, полицис-
ликопин, флавоксантин и тараксантин; аминокислоты — лизин, орнитин,
гистидин, цистеин, аспарагин, серин, треонин, пролин, метионин, тирозин,
лейцин; витамины группы В (холин, бетаин).
Необходимо подчеркнуть, что морковь, кроме каротиноидов, содержит
важный набор незаменимых аминокислот (лизин, метионин, лейцин, трео-
нин), а также бетаин (300 мг%), холин (11 мг%). Последние обладают ли-
потропным действием и участвуют в биосинтезе метионина, фосфолипидов
и в кроветворении.
1. Наиболее урожайными сортами моркови являются Московская позд-
няя, Шантанэ и Лосиноостровская-13. Урожайность этих сортов моркови
колеблется от 16—18 т 1га (в Краснодарском крае) [2] до 79,0 т/га в Челя-
бинской области [-1].
Наилучшими сортами моркови признаны Нантская-14 (ранний сорт),
Шантанэ № 2461 и Лосиноостровская-13 [2].
2. Указанные сорта моркови наиболее богаты каротином, содержание
которого колеблется от 11 до 23 мг% на сырую массу. Содержание каротина
сильно варьирует в пределах одного и того же сорта в зависимости от поч-
венных и климатических условий, а также от продолжительности вегета-
ционного периода.
Хранение моркови. Так как каротиновый цех начинает обычно работать
одновременно с началом копки моркови, то часть сырья с поля поступает
непосредственно на сырьевую площадку для переработки, а другая часть
закладывается на длительное хранение в бурты. При поступлении моркови
с поля в переработку с нее срезают ботву и зачищают головку и стеблевую
часть. При направлении моркови в овощехранилище или в бурты необхо-
димо тщательно ее проверять, не оставлять на корнеплодах ростовой поч-
ки и не допускать увеличенного среза головки. Не принимается на хране-
ние морковь гнилая, мороженая или сильно загрязненная.
На сырьевую площадку морковь должна поступать в ящиках или кор-
зинах, содержащих по 15—20 кг корнеплодов. Ящики укладывают пра-
вильными рядами в штабеля высотой в 2—2,5 м. На каждом штабеле выве-
шивают паспорт с указанием сорта, массы, наименования поставщика и
даты приемки.
Часть моркови, предназначенная для длительного хранения, поступает
на буртовую площадку для укладки в бурты. Буртовая площадка должна
быть соответствующим образом подготовлена. Для этого нивелируют учас-
ток, устраивают дренаж его для удаления поверхностных вод и предотвра-
щения скопления на нем атмосферных осадков. Необходимо также преду-
смотреть устройство подъездных путей и ветрозащиты.
Морковь укладывают в бурты (рис. 93) с углублением 1 (котлован) в
почву на 20 см. Расстояние между соседними буртами 3,5 м, в проезжей
части 7 м. Размеры бурта: длина 30 м, ширина 1,5 м, высота 1 м. Перед
укладкой моркови дно котлована обрабатывают известью, просушивают и
рыхлят. Уложенный первый ряд моркови засыпают сухим песком или зем-
лей, затем укладывают второй ряд, который также закрывают песком и
землей и продолжают так до вершины бурта. Последний ряд корней закры-
вают слоем песка или земли 2 толщиной 15 см.
При укладке моркови необходимо правильно оформлять бурт в виде
треугольника со срезанной вершиной. После укладки бурт укрывают слоем
земли толщиной 10—15 см у гребня и 15—20 см у основания, а затем соло-
менными матами. Бурты окапывают на расстоянии 0,5 м от края канавкой 3
398
шириной 0,5 и глубиной 0,4 м. Между расположенными попарно буртами
проводят канавку, которую выводят в сбросную канаву для отвода воды с
территории участка.
Находящаяся в буртах морковь при дыхании выделяет углекислоту,
воду и тепло, для удаления которых целесообразно устроить вентиляцию.
Для этого вдоль всего бурта внизу укладывают горизонтальную деревян-
ную трубу 4 с отверстиями. К этой трубе через 5—6 м присоединяют верти-
/ 4
Рис. 93. Бурт моркови.
кальные трубы 5 с заслонками для регулирования тока воздуха. Ежеднев-
но утром и вечером проверяют температуру внутри бурта специальными
буртовыми термометрами.
ТЫКВА
Ботанические свойства кормовой тыквы [3]. Тыква принадлежит к одно-
летним ползучим растениям семейства тыквенных. Тыква имеет мощную
корневую систему и хорошо развитые стелющиеся стебли. Листья крупные,
их площадь для одного растения достигает 30 .и2. Цветки у тыквы раздель-
нополые, венчик пятираздельный, лепестки сросшиеся желтого и оран-
жевого цвета. Опыление тыквы происходит при помощи насекомых и ветра.
Кормовая тыква вызревает не только в южных районах, но и в средней
полосе СССР. Плоды тыквы разнообразны по форме и величине и содержат
разное количество каротина, каротиноидов, сахара и сухих веществ.
Виды тыквы. Широкое распространение имеют следующие три вида
тыквы.
1. Крупноплодная кормовая (Cucurbita maxima Duch). К этому виду
относятся следующие сорта: Серая волжская, Стофунтовая, Бирючекутс-
кая 630, Белая медовая, Столовая зимняя.
2. Обыкновенная столовая (Cucurbita pepo). К этому виду относятся
кустовые тыквы — кабачки и патиссоны.
3. Мускатная (Cucurbita moschata Duch). К этому виду относятся сто-
ловые и кормовые сорта: Перехватка, Бирючекутская, 627, Витаминная.
Тыква содержит (в %): кожицы 17,0, мякоти 73 и семян 10. По другим
данным [1], выход семян колеблется от 1,3 до 5% от массы плода. Состав
семян тыквы (в %): воды 6,31; жира 38,4; азотистых веществ 27,47; сахара,
крахмала и пентозанов 11,02; клетчатки 14,84; золы 1,99. Физические свой-
ства тыквенного масла: плотность 921,7 кг/л3; кислотное число 0,675; чис-
ло омыления 196,3; йодное число 108,7; неомыляемые вещества 1,13.
Состав жирных кислот (в %): олеиновая кислота 25,0; линоленовая кис-
лота 45,0; пальмитиновая и стеариновая кислоты около 30,0.
Л. Кревченко на Бирючекутской селекционной опытной станции (Рос-
товская область) вырастил сорт тыквы под названием БК-627, богатый каро-
тином. Впоследствии в связи с дальнейшей селекционной работой этот сорт
тыквы назван Витаминная. Содержание каротина в ней составляет (в мг%):
8—16 на сырую массу или 120—240 на сухую массу. Отдельные экземпляры
этого сорта тыквы содержат около 37 мг% каротина на сырую массу или
399
около 500 мг% на сухую [4]. Масло семян тыквы содержит
тамина Е [5].
Химический состав тыквы Витаминная (без кожуры)
78 мг% ви-
Сахар общий, % ... 5,9
Сахароза, % .........1,0
Белковые вещества, % 1,5
Жир, % ................0,1
Сухие вещества, % . . 8,0
Каротин, мг % .... 16,0
Каротин + каротинои-
ды, мг% .............' . 20,0
Пигмент тыквы состоит на 70% из а- и p-изомеров каротина и на 30%
из каротиноидов (виолаксантин, флавоксантин и ксантофилл) [6]. По
исследованиям автора из каротиноидов идентифицированы: ликопин, фла-
воксантин и тараксантин.
Накопление каротина в тыкве происходит в последние дни вегетацион-
ного периода. За последние 20 дней из 150 дней вегетационного периода в
тыкве накапливается каротина в 20 раз больше, чем за весь предшествую-
щий период [4].
Сравнительная характеристика производственных показателей для мор-
кови и тыквы, по данным Краснодарского опорного пункта ВНИВИ, при-
ведена в табл. 38 [7].
Таблица 38
Овощи - корнеплоды Урожай с 1 га, ц Содержание каротина на сырую массу, мг % Выход каротина с 1 га, кг
Морковь Шантанэ 171 12,45 2,13
Тыква Витаминная 342 17,65 6,04
По данным В. Папанова [8], содержание каротина (мг%) в тыкве 15—
16 вместо 10—12 в моркови и далее соответственно: урожайность 20—30 и
10—12 ml га, трудоемкость в человеко-днях на 1 т сырья 1,6 и 5,5; выход
каротина с 1 га (в кг) 6,02 и 2,13; себестоимость 1 т сырья (в руб.) 15—20 и
25—31. Приведенные показатели свидетельствуют о большой перспектив-
ности тыквы как сырья для производства каротина.
Хранение тыквы. Плоды тыквы, не имеющие повреждений при хране-
нии, хорошо сохраняются на стеллажах при температуре 1—3° С и отно-
сительной влажности воздуха 60—70%. Частое и резкое изменение темпе-
ратуры воздуха отрицательно влияет на стойкость плодов.
При хранении неповрежденных плодов тыквы содержание каротина в
них не уменьшается, а даже несколько увеличивается, в среднем на 3—4%
в течение 120 дней хранения, что, по-видимому, обусловлено освобождени-
ем связанного с белком каротина в результате ферментативных процессов.
Силосование тыквы. Неустойчивость плодов тыквы при хранении пре-
пятствует ее широкому промышленному использованию. В связи с этим
проводились исследования по силосованию плодов тыквы [4, 9]. Плоды
тыквы измельчали на терочной машине и загружали в стеклянные бутылки.
Через четыре дня pH силоса снижался с 6,9 до 4,1, содержание молочной
кислоты достигало 0,988%, а уксусной кислоты — 0,334%. Спирта в сило-
се не найдено. Силос имел запах квашеных бахчевых овощей, приятно-кис-
лый вкус и оранжевый цвет.
При хранении силоса до трех месяцев содержание каротина увеличилось
на 16,4% к каротину, введенному с сырьем, а после шести месяцев хране-
ния потери каротина составили 12,9%. Первоначальное увеличение содер-
жания каротина при хранении и последующее снижение его по сравнению
с первоначальным, по-видимому, обусловлено освобождением связанной с
белком части каротина в результате ферментативных процессов при сило-
совании. При этом вначале наблюдается накопление каротина, а затем,
естественно, каротин подвергается окислению, вследствие чего содержание
400
его в силосе начинает снижаться. При силосовании тыквы в производствен-
ных условиях (18,5 т) и хранении силоса в течение 138 дней отмечено уве-
личение содержания каротина на 29,3%.
Для силосования тыквы требуется специальное оборудование и камеры
для хранения и созревания силоса. Оборудование обеспечивает мойку пло-
дов, удаление плодоножек, семян, измельчение мякоти, транспортирова-
ние измельченной мякоти (мезги) в отсеки хранилища и из него — в произ-
водство.
Хранилище — полузаглубленное, камеры сооружаются с усиленной
гидроизоляцией. На стены камер наносят кислотостойкое покрытие.
По данным В. Папанова [8], при силосовании тыквы никаких добавок
в измельченную массу тыквы не делают — процесс идет естественным пу-
тем. Брожение длится несколько десятков дней в зависимости от качествен-
ного состава мякоти тыквы (содержания сахаров), степени ее измельчения
и температуры среды. Через 30—40 дней с момента загрузки силосная масса
становится пригодной для дальнейшей переработки.
Силосованная тыква при хорошей герметизации хранилища, предотвра-
щающей проникновение воздуха, хорошо хранится. В порядке эксперимен-
та на Краснодарском комбинате было произведено хранение силосованной
тыквы в течение 2 лет. Силосная масса и каротин в этой массе сохранились
почти полностью.
По окончании процесса молочнокислого брожения в силосных ячейках
происходит довольно четкое разделение всей массы на две фазы — жид-
кую, находящуюся в нижнем слое, являющуюся используемым отходом,
и плотную массу, находящуюся в верхнем слое, используемую для получе-
ния каротина. Содержание последнего в массе составляет 250—350 мг!кг.
Рациональная организация производства каротина из моркови и тыквы.
Основным видом сырья для производства каротина до последнего времени
была морковь. Значительным достижением явилось открытие нового вида
сырья — витаминной тыквы. Силосование тыквы оказалось эффективным
методом длительного ее сохранения (200—250 суток).
Учитывая, что морковь созревает в августе, а тыква в октябре, целесооб-
разно в течение августа — ноября перерабатывать морковь, а с декабря
до июля тыкву.
При выборе технологических схем для производства каротина из морко-
ви и тыквы можно руководствоваться следующими соображениями:
1. При переработке моркови целесообразно применять коагуляцион-
ный метод, заключающийся в получении сока и коагуляции содержащихся
в нем белков (стр. 402). Выход каротина при этом методе составляет 66%
к введенному с сырьем. Для увеличения выхода каротина можно жом пос-
ле сушки подвергать экстракции органическими растворителями (дихлор-
этан, дихлорметилен). При этом выход каротина повышается до 78%.
2. Для переработки силосованной тыквы можно рекомендовать техноло-
гическую схему, заключающуюся в сушке силосованной массы и экстрак-
ции из нее каротина соевым маслом или органическим растворителем.
При выборе органического растворителя необходимо учесть, что наи-
лучшие результаты достигаются при использовании дихлорметилена, по-
зволяющего процессы экстракции и отгонки растворителя вести при низ-
кой температуре.
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА МАСЛЯНОГО КОНЦЕНТРАТА
КАРОТИНА ИЗ МОРКОВИ
Подготовка сырья. Морковь из бурта 1 (рис. 94) подвозят автомашиной
2 в бункер 3, откуда гидравлическим транспортером 4 и шнеком 5 морковь
подают в кулачную мойку 6, где ее промывают. Мойка должна быть обеспе-
чена чистой и проточной водой. Ежесменно необходимо мойку очищать
от грязи и камней. Далее элеватором 7 морковь подается на автоматические
401
весы 8 типа Хронос, затем на контрольный конвейер 9 для инспекции
и удаления посторонних примесей.
Получение сока. Для получения максимального выхода сока из сырья
необходимо разорвать клетки. Для этой цели применяют пильные терки.
На степень измельчения сырья влияют следующие факторы: качество пилок,
удельное число зубьев и размер выступа зуба над поверхностью барабана.
На производительность терки и на степень измельчения влияет окружная
скорость барабана. Установлено, что наилучшие результаты достигаются
Рис. 94. Технологическая схема производства масляного концентрата каротина из
моркови.
при 8 зубьях на 1 см длины полотна пилки, при высоте зуба 1—1,5|лл, при
окружной скорости барабана 50 м/сек. Необходимо следить за равномерным
поступлением моркови в терку, а также за правильным положением прижи-
мов терки и состоянием пилок. При наборе пил следует пользоваться шаб-
лоном.
Первое прессование мезги. Из терки 10 мезга поступает в бункер над
прессом Яна И. Необходимо следить за равномерным питанием пресса
мезгой, не допуская перелива ее, поддерживать установленное расстояние
между прессующими валками, следить за стоком опрессованного сока и не
допускать скопления отпрессованной мезги. Для устранения попадания мез-
ги в сок необходимо наблюдать за тщательной пригонкой сит; диаметр их
ячеек не должен превышать 0,6—0,7 мм. Сита должны быть из нержавею-
щей стали. В результате первого прессования получают сок I, загрязнен-
ный мелкими частицами клетчатки. При попадании в коагулят они увели-
чивают его количество и ухудшают последующие процессы.
Фильтрация. Для отделения от сока указанных примесей применяют
сотрясательное сито — трясучку 12. Сито по длине имеет уклон не менее
25 мм на 1 м.
Сито получает от привода 300—400 колебаний в 1 мин, размах качаний
7—9 мм. Под ситом устанавливают желоб-поддон из алюминия или нержа-
веющей стали. Сок из пресса Яна поступает на сотрясательное сито, прохо-
дит через сито в желоб и далее направляется вниз в сборник фильтрован-
ного сока 13, откуда насосом 14 выкачивают сок I в сборник 15.
Второе прессование. После первого прессования в мезге остается около
30—40% каротина, введенного с морковью. Поэтому полученный жом I
402
подвергают перемешиванию с водой в специальном смесителе 16, располо-
женном под прессом Яна. Воды добавляют 60% к массе жома.
Увлажненный жом насосом 14 перекачивают во второй агрегат терку-
пресс 10' и 11', где происходит повторная перетирка жома и его прессова-
ние. В результате получают сок 11 и жом II. Сок II подвергают фильтрации
на сотрясательном сите 12'. Фильтрованный сок поступает в сборник со-
ка 16'.
Третье прессование. Практически установлено, что двойная обработка
сырья на агрегате терке-прессе не обеспечивает полного извлечения каро-
тина из сырья. Потери каротина в жоме при этом составляют около 20%
к введенному каротину. Применение третьего цикла обработки путем промыв-
ки жома II и третьего прессования его увеличивает выход каротина на
4—5%. В связи с этим в технологической схеме предусмотрено направле-
ние жома II насосом 14" в терку 10" и пресс 11" для отжатия сока III. По-
следний насосом 14" перекачивается в сборник 15' сока II и III.
Получение сухого белкового коагулята. Метод основан на коагуляции
растительных белковых веществ, содержащихся в соке и адсорбции каро-
тина выделяемым белковым осадком [10]. Для этой цели насосом 14 сок I,
II и III подают в трубчатые подогреватели 17, где его нагревают до 70—
75° С. После нагрева сок направляют в отстойник 19, а затем часть чистого
сока спускают в сборник 39, откуда его направляют на выпарку, а часть
сока с белковым коагулятом насосом 14 перекачивают в фильтр-пресс 20.
Фильтрация. Для фильтрации применяют фильтр-прессы закрытого
типа, рамы и плиты которых изготовлены из нержавеющей стали. После
выделения белка сок должен содержать каротина не более 1,0 мг%. Сок
поступает в сборник 21, откуда он далее поступает в выпарной агрегат.
Белковый осадок после разгрузки фильтр-пресса поступает в вакуум-вальцо-
вую сушилку 22. Задержка белкового осадка во влажном виде не допуска-
ется.
Сушка коагулята. Влажный коагулят, содержащий 15—20% сухих ве-
ществ, поступает в вакуум-вальцовую сушилку 22, где процесс сушки про-
текает под вакуумом на барабанах в течение 15—20 сек. Не допускается
хранение высушенного коагулята в вакуум-шкафе сушилки более 2 ч. Су-
хой коагулят должен содержать не более 15% влаги.
Экстракция каротина маслом. На эффективность экстракции каротина
влияет дисперсность коагулята: чем мельче частицы белкового осадка, тем
больше их суммарная поверхность и тем скорее протекает процесс экстрак-
ции.
Сухая пленка, полученная из вакуум-вальцовой сушилки, легко измель-
чается. Для измельчения сухого коагулята применяют диабазовые или гра-
нитные вальцы 23. Каротин извлекают из белкового коагулята маслом в
экстракционной батарее, состоящей из 8—10 диффузоров 24 с калоризатора-
ми при них. Батарея работает по принципу противотока [11]. Измельчен-
ный белковый коагулят загружают в диффузоры. Температура в диффузо-
рах должна быть 70° С.
Выход каротина в масляных мисцеллах составляет около 94%, потери
в шроте 5% и неопределенные потери около 1% [12].
Более высокие потери каротина получаются при использовании менее
эффективного метода настаивания белкового коагулята маслом в экстрак-
торах с механическими мешалками. При этом методе применяют трехкрат-
ное настаивание при температуре 60—65° С. Первое настаивание произво-
дят двойным к массе коагулята количеством мисцеллы II, содержащей 1,2—
1,5 мг/мл каротина; в результате настаивания в течение 40 мин получают
мисцеллу I с содержанием каротина 2 мг/мл. Мисцеллу I выделяют либо
фильтрацией на фильтр-прессе, либо в центрифуге. Шрот I подвергают
настаиванию двойным количеством мисцеллы III, содержащей 0,8 мг/мл
каротина, в течение 40 мин при температуре 65—70° С.
Третье настаивание производят рафинированным маслом.
403
Выход каротина в мисцеллах при методе настаивания составляет 88—
92% к введенному с белковым коагулятом.
Масляную мисцеллу, получаемую диффузионным методом, в сборнике 25
фильтруют через фильтр-пресс 26.
При разгрузке диффузора шрот вместе с маслом спускают на центрифу-
гу 27 для отделения масла, возвращаемого в сборник масла 28. Шрот из
центрифуги направляют для отжатия масла в гидравлический пресс 29.
Отжатое масло поступает из пресса в сборник 30, откуда его насосом перека-
чивают в общий сборник масла 28.
Купажирование мисцелл. Для получения масляного концентрата, со-
держащего 2 мг!мл каротина, производят купаж мисцелл с повышенной и
пониженной концентрацией каротина (сборники 31 и 32) в аппарате-смеси-
теле 33, откуда насосом перекачивают концентрат в сборник 34.
Использование отходов производства [13, 14]. В масляный концентрат
каротина переходит лишь незначительное количество жирорастворимых ве-
ществ (десятые доли процента сырья). По существу вся морковь переходит
в полупродукты, являющиеся отходами производства. К ним относятся
жом, сок и масляный шрот. Жом представляет собой ценный корм, содер-
жащий основные питательные вещества моркови и около 10 мг% каротина.
Однако при большой производительности завода жом не всегда может быть
использован для скармливания животным в день его получения. Поэтому
возникает необходимость силосования или сушки жома. В последнем слу-
чае жом насосом подается в сборник 35, а из него в вальцовую сушилку 36.
Сухой жом смешивают с патокой в шнековом смесителе 37 и брикетируют
в прессе 38.
Сок I содержит около 6—8%, а сок II и III —2—3% экстрактивных
веществ. Последние преимущественно состоят из сахаров и элементов золы.
Морковный сок можно использовать по четырем направлениям: 1) упари-
вание в морковную или тыквенную патоку; 2) переработка на дрожжи;
3) переработка на спирт и 4) использование в качестве питательной среды
для биосинтеза антибиотиков и витаминов В12 и В2.
В первом случае сок после отстойника 19, фильтр-пресса 20 и сбор-
ника 21 насосом 14 подают в сборник 39, а из него в трехкорпусный вы-
парной аппарат 40, где его упаривают в патоку, содержащую 70% сухих
веществ. Патоку из третьего корпуса выпарного аппарата выкачивают в
сборник 41, из которого разливают в бочки 42. Патока в бочках может
быть направлена на спиртовые заводы для переработки на спирт. Из 1 т
сахара, содержащегося в патоке, можно получить 62 дал спирта [13].
Шрот является ценным питательным продуктом для животных и может
быть использован для витаминизации кормов. Содержание каротина в шро-
те составляет 10—15 мг%.
ТЕХНОЛОГИЧЕСКАЯ схема производства концентрата каротина из
СИЛОСОВАННОЙ ТЫКВЫ [8, 15]
Технологическая схема производства концентрата каротина из силосо-
ванной тыквы, проверенная на Краснодарском витаминном комбинате [15],
заключается в следующем (рис. 95).
Силосованную массу из хранилища подают насосом через сборник в
протирочный агрегат типа «Дуплекс» 1, снабженный ситами с диаметром
отверстий 2 мм (в верхней протирочной машине) и 1 мм (в нижней). Про-
тертую массу направляют в сборник 2, а из него в нейтрализатор 3 для нейтра-
лизации при помощи NaOH до pH 7,0 и далее на фильтр-пресс 4. Отфильтро-
ванную силосованную массу подают в вакуум-вальцовую сушилку 5, где вы-
сушивают массу до влажности 10—15%. Сухие лепестки тыквы направляют
в экстрактор 6 непрерывного действия НД-200 или НД-500 (типа Гильдебран-
дта). Экстрактор состоит из выгрузочной 6 и экстракционной 7 колонн, снаб-
404
женных шнеками для передвижения лепестка внутри колонн и из колон-
ны 6 в колонну 7 шнеком 8. Движение лепестка и растворителя (хлористый
метилен) происходит по принципу противотока. Свежий растворитель подают
насосом 9 из сборника 10 через водоотделитель 11 и нагреватель 12 в экстрак-
ционную колонну 7. Каротинсодержащую мисцеллу, выходящую из колон-
ны 6 насосом 13 нагнетают в фильтр-пресс 14. Отфильтрованная мисцелла
поступает в сборник 15, а из него в вакуум-дистиллятор 16, снабженный
конденсатором 17. Окончательное сгущение мисцеллы производят в дистил-
ляторе с меньшим объемом 18 (с конденсатором 19). Мисцеллу засасывают
из сборника 20. Концентрация каротина в мисцелле после экстрактора 200—
250; после дистиллятора I — 1500—2000, а после дистиллятора II — 30—
Рис. 95. Технологическая схема производства концентрата каротина из силосованной
тыквы.
40 тыс. мг/кг. Для удаления остатков растворителя добавляют в дистилля-
тор 18 к концу выпаривания из сборника 21 воды в количестве 5—6% к
массе концентрата. Готовый концентрат направляют на расфасовку.
Шрот из экстракционной колонны поступает в шнековой испаритель
22, соединенный с конденсатором 23. После удаления растворителя шрот
направляют в шнек 24 и используется как корм. Растворитель из конденса-
тора 23 поступает в флорентийский сосуд 25, а из последнего — в сборник 10.
В этот же сборник через приемники 26 поступает конденсат хлористого
метилена из вакуум-дистилляторов 16 и 18.
Пары растворителя из воздуха, выходящего из флорентийских сосудов,
мисцеллосборников и вакуум-дистилляторов, улавливаются при помощи
дефлегматорной установки 27. Воздух с парами растворителя поступает в
конденсатор 27, а из него последовательно в две колонны 27, заполненные
кольцами Рашига, орошаемые водой, и удаляется через вытяжную трубу
28. Орошающая дефлегматоры жидкость проходит через флорентийский
сосуд для удаления воды и сбора растворителя. Экстракторы НД-200 и
НД-500 характеризуются высокими показателями в работе. Получаемый
концентрат содержит 30—40 мг/г р-каротина.
Отходы и их использование. Отходами производства при переработке
силосованной тыквы являются: 1) сок силосный, получаемый после выде-
ления твердой фазы в протирочном агрегате I в количестве 58—60% к мас-
405
се свежей тыквы и 2) шрот, получаемый на стадии экстракции после шнеко-
вого испарителя 22 в количестве 2,1—2,5% к массе сырья. Наши исследо-
вания (совместно с М. Шаховой, М. Ушаковой и А. Ефимовым) силосного
сока показали: высокое содержание в нем белковых веществ (26% на сухое
вещество), минеральных веществ (20%) (в том числе Р2О5 — 1,5—2,6%
и СаО — 0,9%), бетаина (7%). В состав золы, исследованной при помощи
эмиссионного анализа, входят следующие элементы: Na, Са, Си, Mg, Al.
Ni, Cr, Si, Fe и Ti.
В соке, сгущенном до 48% сухих веществ, методом хроматографического
разделения на бумаге идентифицированы следующие аминокислоты: цис-
теин, гистидин, аргинин, серин, глутаминовая кислота, пролин, (3-фенил-
а-аланин, триптофан, лизин, тирозин.
Биологические испытания на лабораторных животных показали, что
силосный сок обладает ростстимулирующим действием, выражающимся
в повышении привеса крыс на 23,7% и цыплят на 7,3% по сравнению с конт-
ролем. Препарат не обладает токсичностью. В связи с этим сгущенный сок
является эффективной биологически активной добавкой к кормам для жи-
вотноводства и птицеводства.
ПРОИЗВОДСТВО КРИСТАЛЛИЧЕСКОГО Р-КАРОТИНА ИЗ. МОРКОВИ [1, 14, 16]
Исходным сырьем для получения кристаллического p-каротина являет-
ся морковь, содержащая среди каротиноидов 85—90% Р-каротина. Наобо-
рот, в тыкве содержание p-каротина составляет лишь 60—70% [17]. Про-
изводство кристаллического каротина включает следующие стадии: 1) экс-
тракция каротина из сухого коагулята белков органическим растворителем;
2) омыление концентрата; 3) экстракция каротина из омыленной массы и
4) кристаллизация каротина.
Экстракция каротина. Большинство исследователей [14, 16, 18] схо-
дятся на применении в качестве органического растворителя для экстрак-
ции Р-каротина хлорированных углеводородов (в основном дихлорэтан).
Существует мнение (А. Вечер [14]) о целесообразности предварительной
экстракции белкового коагулята спиртом для удаления стеринов, фосфа-
тидов, свободных жирных кислот и других веществ. Однако дополнитель-
ная экстракция спиртом сильно осложнит технологию производства, поэто-
му необходимость этого процесса нуждается в технико-экономическом обос-
новании. Экстракцию осуществляют дихлорэтаном в экстракторах непре-
рывного действия (при крупном производстве) или в аппаратах типа Сокс-
лета при небольших масштабах производства. Дихлорэтана в реактор 1
(рис. 96) загружают 400% к массе сухого коагулята. Экстракцию ведут в
течение 1—1,5 ч. Содержание каротина в шроте не должно превышать 5%
к введенному каротину с белковым коагулятом. Затем в испарителе 2 в
присутствии СО2 отгоняют дихлорэтан (температура не должна быть выше
50° С).
Омыление концентрата. Омыление производят 10%-ным раствором ед-
кого кали, которого добавляют около 10% к массе концентрата. Процесс
проводят в реакторе 3 с обратным холодильником в течение 20 мин при
50° С. При омылении образуется осадок, содержащий до 80% каротина и
жидкое мыло. Осадок отфильтровывают на нутч-фильтре 4 и промывают
спиртом от мыла и красящих веществ.
Б. Савинов и А. Свищук [18] указывают на образование нерасслаиваю-
щихся эмульсий при омылении липоидных экстрактов в хлорированных
углеводородах. Это явление ими было успешно устранено совмещением
стадии омыления со стадией экстракции (подробно см. [18]).
Экстракция каротина из омыленной массы. Каротин экстрагируют ди-
хлорэтаном в количестве, необходимом для растворения каротина при
406
комнатной температуре, исходя из того, что в 100 мл дихлорэтана (ДХЭ)
растворяется при температуре 25° С 1,16 г каротина [16].
Экстракцию ведут при комнатной температуре в реакционном аппарате
5, снабженном обратным холодильником и мешалкой. Затем массу фильт-
руют на нутч-фильтре 6, промывают осадок чистым ДХЭ. Экстракт с про-
мывным ДХЭ сгущают в вакуум-перегонном аппарате 7 до получения пере-
сыщенного раствора.
Первая кристаллизация. Пересыщенный раствор спускают в кристалли-
затор 8, где в течение 8 ч идет процесс кристаллизации вначале при комнат-
ной температуре, а зат,ем через 4 ч при охлаждении, к концу процесса тем-
пературу доводят до 5° С. Для увеличения выхода каротина на первой крис-
Рис. 96. Технологическая схема производства кристаллического каротина.
таллизации в пересыщенный раствор вводят этиловый спирт в отношении
1:2. Затем отфуговывают в центрифуге 9 выделившиеся кристаллы, промы-
вают их спиртом и высушивают в вакуум-сушилке 10. Маточный раствор
I поступает в сборник 11.
Вторая кристаллизация. Маточный раствор I перерабатывают совместно
с промывными и мыльной массой. Для этого мыльную массу экстрагируют
два раза ДХЭ в нутч-фильтре 4, а экстракт промывают водой в смесителе 12.
Экстракт и маточник I направляют в сборник 13, откуда они поступают в
вакуум-аппарат 14 для упаривания в концентрат II. Последний поступает
в кристаллизатор 15, где кристаллизуется 24 ч. Фуговку производят при
температуре 5° С в центрифуге 16. Кристаллы каротина II промывают спир-
том и направляют на переработку совместно с экстрактом омыленной мас-
сы (до первой кристаллизации). Маточный раствор II поступает в сборник
17.
Третья кристаллизация. Маточный раствор II совместно с промывными
второй кристаллизации упаривают в вакуум-аппарате 18, кристаллизуют
72 ч в кристаллизаторе 19, фугуют в центрифуге 20. Кристаллы промыва-
ют спиртом. Получают кристаллы каротина III, направляемые на переработ-
ку в маточный раствор I и в виде отхода маточный раствор II — в сборник
21.
Нормы качества готовой продукции. Кристаллический каротин должен
быть однородным, мелкокристаллическим сухим порошком без слежав-
407
шихся комков лилово-красноватого цвета с металлическим блеском. Точ-
ка плавления каротина должна быть не ниже 160° С. Содержание (3-кароти-
на в кристаллах не менее 90%.
Вопросы усовершенствования технологии производства каротина из мор-
кови. Интересные исследования в этой области были проведены Б. Савино-
вым и его учениками (ИОХ АН УССР). Исходя из факта локализации каро-
тина на хромопластах, им было предложено заменить процесс прессования
мезги моркови процессом вымывания пластид из клеток интенсивным пере-
мешиванием мезги с водой в суспензионном экстракторе [19]. Им же был
разработан метод получения масляных концентратов каротина из влажного
белкового коагулята путем применения центробежного смесителя [20].
Разработан метод получения каротина из моркови и тыквы методом термиче-
ской коагуляции белков в клетке [21, 22], изучены вопросы экстракции
каротина в многочленной батарее [23]. К сожалению, эти методы пока
не нашли применения. Важное значение в области усовершенствования
химии и технологии каротина имела книга Б. Савинова «Каротин» (изд.
АН СССР, 1948 г.).
ТЕХНОЛОГИЧЕСКИЕ ПОКАЗАТЕЛИ
ПРОИЗВОДСТВО КОНЦЕНТРАТОВ КАРОТИНА ИЗ МОРКОВИ
Показатели качества и выходов полупродуктов в производстве масля-
ных концентратов из моркови приведены в табл. 39.
Таблица 39
Полупродукты Сухие вещества, % Содержа- ние каро- тина, мг% Выход по- лупродук- тов, % к сырью । Полупродукты Сухие вещества, % П) X X- f3 и и £ Выход по- лупродук- тов, % к сырью
Мезга 15,0 12 97 Коагулят ....
Сок сырой . . . 20,0 160 6
I 12,5 12 64 сухой . . . 80 640 1,5
II 4,0 6,0 25 Шрот 95—96,0 10—15 1,8
III 2,0 3,0 20 1 Содержание каро-
Жом тина в масля-
I 21,0 12 33 ном концентра-
II 20 8,5 28 те — 200 66
III 23 8 22
Производство кристаллического каротина
Мисцеллы каротина, % к сухому коагу-
ляту ................................. 200
Сгущенной мисцеллы, % к сухому коа-
гуляту ............................... 30—40
Выход кристаллического каротина, %
к введенному с морковью............... 55—60
Показатели качества кристаллического каротина
содержание каротина, %, не менее 90,0
температура плавления, °C, не ниже 160
выход кристаллического каротина из
1 т моркови, г........................ 72
Выход и потери каротина, полученные при производственных испыта-
ниях сокового коагуляционного метода на Краснодарском витаминном
заводе (в % к каротину, введенному с сырьем), приведены в табл. 40.
408
Таблица 40
Показатели Морковь Тыква
Содержание каротина, % 78,5 67,5
в сыром белковом коагуляте . .
в сухом белковом коагуляте . . 76,9 65,9
в готовой продукции 66,0 55,0
Потери каротина, % 16,2
в жоме 27,2
неопределенные на стадиях коа- 5,3 5,3
гуляции и фильтрации
при сушке коагулята 1,6 1 >6
при экстракции каротина маслом 8,9 8,9
при купаже и фильтрации мисцелл 2,0 2,0
Итого потерь 34,0 45,0
ПРОИЗВОДСТВО КОНЦЕНТРАТОВ КАРОТИНА
ИЗ СИЛОСОВАННОЙ ТЫКВЫ [8]
Свежая тыква
Содержание каротина, жг/кг .........72,1—165
Влажность, % ........................91,4—93,2
Объемная масса, кг/л................ 0,45
Силосованная тыква
Содержание каротина в 'массе, мг)кг . . 292—334,7
Влажность массы, %..................93,6—91,6
Кислотность массы, pH ..............3,8—4,0
Объемная масса сухого силоса (после
сушки и измельчения), кг/л.......... 0,12
Отходы
Таблица 41
Наименование
Сок силосный . .
Шрот..............
Выход, % К сырью Химический состав, сырую массу % на Содержание каро- тина, Л42% Плотность, к.г/м? Использование
сухие ве- щества белок общий сахар зола общая
58 5 1,0 о,1 1,03 1,о 1010 Добавка к корму для животновод- ства
2,1 94,5 11,0 Нет 6,2 4—6 — Корм ных для живот-
Дихлорэтановые мисцеллы
1. После экстрактора НД-500
содержание каротина, .иг/кг . . .
плотность, кг/м?...................
2. После 1-й дистилляции
содержание каротина, мг/кг .
плотность, кг/м3 ..................
3. После 2-й дистилляции
содержание каротина, мг/кг . . .
плотность, кг/м3...................
250—450
1250—1260
1100—1900
1200—1180
20—30 тыс.
1100
15—522
409
Выход полупродуктов (табл. 42)
Таблица 42
Наименование Сухие вещества, % Каротин, мг % Выход, % к сырью
Силосованная протертая мезга Сырой силос после фильтр -прес- 7—8 29,0—35,0 32
са 19—21 115-120 8—9
Сухой силос (лепесток) .... Выход каротина, % к введен- 80—83 400—500 2,1
ному с сырьем — — 75,0
Сок силосованной тыквы . . . 5,0 1,0 58,25
Шрот (после сушки) 94,5 6,0 2,1
Потери каротина на отдельных стадиях производства
(в % к введенному с сырьем)
При силосовании (хранение 6 месяцев) ... 4,0
С соком..................................... 0,5
При сушке силоса........................... 13,0
При экстракции (в процессе и в шроте) . . 3,0
При дистилляции............................... 4,5
Общие потери............ 25,0%
Выход каротина от введенного с сырьем 75,0%
Удельные и объемные массы сырья и полупродуктов
Сырье Объемная масса, л/кг Сырье Объемная масса, л /кг
Морковь 0,67 Коагулят и сырой силос
Тыква 0,45 морковный коагулят 0,94
Мезга тыквенный коагулят 0,94
морковная 0,65 сырой тыквенный силос . . 0,79
тыквенная 0,61 Сухие коагулят, мезга и силос
тыквенная силосованная пос- морковный коагулят 0,15
ле перетира 0,93 тыквенный силос 0,12
Сок жом свежей тыквы 0,20
морковный I 1,03 Мисцеллы и концентрат
морковный II 1,02 мисцеллы дихлорэтана .... 1,26
морковный III 1,01 масляный концентрат .... 0,92
тыквенный I 1,02 Шрот
силосованной тыквы . . . 1,01 масляный морковный 0,40
Жом дихлорэтановый тыквенный
морковный III 0,41 после пропарки 0,22
тыквенный I 0,680 после сушки 0,13
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
ПЦеревитинов Ф. В. Химия свежих плодов и овощей. М., Пищепромиздат,
1938, с. 431.
2. Ш и б р я Г. И. Достижения в области селекции и агротехники витаминных куль-
тур. — В сб.: «Производство н использование витаминов, антибиотиков и биологи-
чески активных веществ», Краснодар, 1965, с. 116—139.
3. Р о ж к о в М. И.,. Кириллов Г. Н., Смирнов Н. Е. Тыква — вита-
минная культура. М., Пищепромиздат, 1954, 23с.
4. Белоножко Ф. И. Новый вид сырья и способы его переработки на препараты
каротина. Автореф. кандид. дисс. М., 1955.
5. 3 а х а р о в а М. П. Содержание витамина Е в некоторых продуктах. — «Тру-
ды ВНИВИ», М., Пищепромиздат, 1954, № 5, с. 178—180.
6. П р о ц е н к о Л. Д. Исследование химической природы пигментов некоторых ка-
ротиноносителей. Автореф. кандид. дисс., Киев, 1953.
7. Ш и б р я Г. И. В кн.: Л. О. Шнайдман. Производство витаминов. М., Пище-
промиздат, 1958, с. 134.
8. Папанов В.’ А. Промышленный метод переработки тыквы на препараты каро-
410
тина. — В сб.: «Труды по витаминам из природного сырья», Уфа, Башиздат, 1971,
с. 173—179.
9. Николаев Р. П., Белоножко Ф. И., Бабичева О. И., Пав-
лова А. М. Силосованная тыква как сырье для производства каротина. —«Тру-
ды ВНИВИ», М., Пищепромиздат, 1954, № 4, с. 106—111.
10. В е ч е р А. С. -Плодоовощная промышленность, 1935, 2. То же. Биохимия,
1936, 1, 567. То же. Пластиды моркови как коллоидные системы и носители витами-
нов. 1947, т. 12, вып. 3, с. 196—200.
11. Шнайдман Л. О. Производство концентратов витамина С из растительного
сырья. М., Пищепромиздат, 1944, с. 25.
12. Ш н а й д м а н Л. О., Дульчина Б. М. Извлечение каротина из сухого
белкового коагулята растительным маслом в экстракционной батарее. — «Труды
ВНИВИ», М., Пищепромиздат, 1959, № 6, с. 108—110 с ил.
13. Ш н а й д м а н Л. О. Производство витаминов из животного и растительного
сырья, М., Пищепромиздат, 1950, с. 134.
14. В е ч е р А. С. Получение кристаллического каротина и каротинового масла из
каротиновых концентратов. — Веб.: «Технология и применение витамина А в каро-
тина», М., Пищепромиздат, 1956, с. 15—25.
15. Шнайдман Л. О., Папанов В. А., Солодухин А. И., Дуль-
чина Б. М. Получение высокоактивных концентратов из силосованной тыквы
непрерывной экстракцией. — В сб.: «Витаминная промышленность», М.,
ГОСИНТИ, 1961, № 7, с. 41—44 сил. Авт. свидет., № 128566 1959; Бюлл. изоб-
рел, 1960, № 10.
16. С а в н н о в Б. Г. Каротин. Киев, изд-во АН УССР, 1948, 230с.
17. С а в и н о в Б. Г. Каротиноидный состав основных витаминных культур и хро-
матография каротиноидов. — В сб.: Технология и применение витамина А и каро-
тина. М., Пищепромиздат, 1956, с. 46—53.
18. С в и щ у к А. А., Савинов Б. Г. Получение кристаллического каротина
методом фракционирования компонентов белкового коагулята. Там же, с. 25—28.
19. С а в и н о в Б. Г., С в и щ у к А. А. Авт. свидет., № 88517, 1949; Бюлл. изо-
брет., 1951, № 1, с. 28; № 89121, 1949; Бюлл. изобрет., 1951, № 3, с. 38.
20. Савинов Б., Вернер Д., С в н щ у к А., Авт. свидет., № 88021, 1949;
Бюлл. изобрет.; 1951, Xs 1, с. 15.
21. Шнайдман Л. О., Кириллов В. Г., Павлова А. М., Кан-
ч у х Ш. Ф. Авт. свидет., №99082, 1954; Бюлл. изобрет., 1954, № 10, с. 13.
22. Шнайдман Л. О. Усовершенствование технологии производства каротина
нз моркови и тыквы.— Веб.: «Технология и применение витамина А и каротина»,
М., Пищепромиздат, 1956, с. 30—38 с ил.
23. Ш н а й д м а н Л. О., Дульчина Б. М. Уточнение некоторых вопросов
новой технологии производства каротина нз моркови и тыквы. — «Труды ВНИВИ»,
М.., Пищепромиздат, 1959, № 6, с. 110—115.
Глава 21. КОНЦЕНТРАТЫ ВИТАМИНА А ИЗ ПЕЧЕНИ РЫБ
Промышленным сырьем для производства концентратов витамина А
служит рыбная печень. По содержанию витамина А печень рыб занимает
первое место среди известных в природе источников этого витамина. При-
чины накопления витамина А в печени некоторых рыб не изучены. Пола-
гают, что первоисточником витамина А в печени рыб является каротин,
содержащийся в зеленых морских водорослях и в фитопланктоне. Послед-
ние поедаются мелкими рыбами и морскими животными, поступающими
в свою очередь в пищу более крупным рыбам. Эта гипотеза подтверждается
закономерностью, наблюдаемой в колебаниях концентрации витамина А
в печени рыб в зависимости от количества фитопланктона в море.
Содержание витамина А в печени рыб колеблется в широких пределах
не только в зависимости от вида рыб. Даже для одного и того же вида рыб
концентрация витамина А в печени меняется от условий ее существования,
причем значительно увеличивается с возрастом рыбы. Например, у зрелой
трески активность печеночного жира в 10 раз выше, чем у молодой трески.
Температура среды обитания рыб также влияет на процесс накопления
витамина А.
На содержание витамина А в печени рыб влияют также и другие факто-
ры: особенности питания, пол, место улова и др.
Ряд исследователей изучали содержание витамина А в печени различ-
ных рыб основных промысловых бассейнов: Балтийского, Азово-Черномор-
15!
411
ского [1], Дальнего Востока [2], Волго-Каспийского [3], Черного моря
14, 511.
Для выбора рационального вида промышленного сырья для производ-
ства концентрата витамина А необходимо в первую очередь учитывать
удельный расход рыбы, который зависит от трех показателей: 1) массы
печени (в % к массе рыбы), 2) процента содержания жира в печени и
3) концентрации витамина А в печеночном жире. Зная эти три показателя,
можно определить эффективность переработки рыбы того или иного
вида.
Удельный расход рыбного сырья при производстве концентратов вита-
мина А [2, 6, 7, 8] приведен в табл. 43.
Таблица 43
Название Масса печени, % к массе рыбы Содержание жира в печени, % Содержание витамина А в 1 г жира, и. е. Расход сырья на 1 трил- лион и. е. витамина А, т
печени рыбы
Полярная акула (Squalus acanthias) 10 50 12000 167 1670
Кит тихоокеанский (ка- шалот, финвал) (Physalus antiquorum) . 4 3,5 300000 100 2500
Минтай (Theragra chalco- gramma) . 4 22 10000 455 11375
Треска тихоокеанская (Gadus morhua macroce- phalus) 6 50 2000 1000 16670
Палтус (Hippoglossus stenolepis) 2 20 50000 100 5000
Частиковые 1,5 4 3000 8325 555000
Осетровые(Acipenser giil- denstadti) 1,5 15 1500 4450 296700
Лососевые (нерка) (Опсо- rhynehus nerka) .... 2,1 4,0 . 106000 235,9 11233
Примечание. По данным 3. Виноградовой [4] в печеночном жире черноморского
осетра содержится 32900 и. е., а по данным В. Розановой [6], 1254—3245 и. е. вита-
мина А в 1 а жира.
Зная эти три показателя, можно определить эффективность использо-
вания того или иного вида рыбы для производства концентратов витами-
на А.
Из данных табл. 43 видно, что частиковая рыба не может служить сырь-
ем для широкого промышленного производства концентратов витамина А.
Рациональными видами сырья являются следующие: акула, кит, минтай,
палтус и треска тихоокеанская. Наиболее эффективным сырьем, как видно
из этих же данных, является печень акулы и кита.
В оценке осетровых рыб с точки зрения содержания витамина А в пе-
ченочном жире мнения исследователей расходятся. Этот вопрос требует
дальнейшего изучения.
ТЕХНОЛОГИЧЕСКАЯ схема производства концентрата витамина а
[8, 9, 10]
Результаты испытания различных методов извлечения витаминного жи-
ра из печени акулы приведены в табл. 44 [9].
1 Подробно см. Л.'Шнайдман. Производство витаминов. М., Пищепромиздат,
1958, с. 89.
412
Результаты исследова-
Таблица 44
ний показывают, что наи-
больший выход витамина
А (без применения орга-
нического растворителя)
дает метод мягкого двух-
Способ обработки печени акулы
ступенчатого гидрол иза.
Это обусловлено тем, что
витамин А в печени нахо-
дится в виде протеидных
адсорбционных соединений
[10]. Связь витамина А с
белком является довольно
непрочной и легко разру-
шается при слабощелочном
гидролизе.
Далее было установлено
[9], что наиболее эффек-
Вытапливание жира паром . .
Сухое жиротопление под ваку-
умом ........................
Экстракция жира из высушенной
печени органическим раствори-
телем .......................
То же, после частичного гидро-
лиза ........................
Мягкий двухступенчатый гид-
ролиз .......................
56
56
56
56
56
66 42
71 51
98 80
94 94
89 95
тивным является двухступенчатый температурный режим гидролиза: на-
гревание до определенной температуры с добавлением 50% щелочи, затем
повышение температуры и добавление остального количества щелочи. На-
конец, показано, что при обработке тощей печени (например, китовой)
достигается надлежащее извлечение витамина А, если к печени перед
гидролизом добавлять жир в количестве, равном массе печени [9].
S с
ПРОИЗВОДСТВО ВИТАМИННОГО ЖИРА ИЗ ПЕЧЕНИ РЫБ
Хранение сырья. Витаминный жир обычно получают из свежей печени,
так как это производство организуют на рыбных промыслах или в близ-
лежащих к ним пунктах. Если печень поступает в засоленном виде, то хра-
Сырьевая
площадка
сушильное"^ gg
^ZZZZZZnZZZZ2ZZ7HZZZZZ^ZZZZZZZZZZZZZZZZZnZZZZZP,
z?zzzzzzzzzz.
Экспедиция Склад готовой продукции
Аммиачно-
компрессорная
У установка й & 12 /4
bzzzz^zzzzzzazzzuzz2zz2zzz^^^^z^zuzip^^zzz^2i2z^zzzzzzzzii2zzzzuzzzuzuzzzzzzuzzzzzz2zz^
Рис. 97. План цеха по производству А-витамннного жира из печени рыб.
некие ее в течение непродолжительного времени может осуществляться
в обычных условиях. Если же печень доставляют в свежем виде, то ее не-
обходимо хранить при пониженной температуре в холодильной камере.
Замораживание облегчает процесс гидролиза печени. Из холодильной ка-
меры 1 (рис. 97) печень подается в камеру размораживания 2, откуда от-
таивая поступает в производство на конвейер 3.
Инспекция и мойка. На конвейере проверяют качество печени, удаляют
посторонние примеси, срезают пленки или оставшиеся частицы внутренно-
стей рыбы и на конце конвейера в душевой мойке 4 промывают печень хо-
лодной водой.
Измельчение печени. От степени измельчения печени зависит эффектив-
ность выделения жира. Наиболее простая машина для измельчения — вол-
чок (мясорубка) 5. Маложирная печень после измельчения поступает в
смеситель, где перемешивается с жиром.
Гидролиз печени. Для разрушения связи между белковыми веществами
и витамином А печень подвергают двухступенчатому гидролизу при pH
9—10 и температуре 82—85° С. Режим гидролиза зависит от качества пе-
413
чени и в каждом отдельном случае устанавливается лабораторией. При
гидролизе добавляют 40% воды и 3% сухой щелочи к массе печени в виде
25%-ного раствора едкого натра. Процесс осуществляют следующим об-
разом: измельченная печень шестеренчатым насосом 6 перекачивается в
гидролизаторы 7, представляющие собой котлы из нержавеющей или эма-
лированной стали с мешалками и паровой рубашкой. После перекачки
печени в гидролизаторы добавляют воду, подогревают массу двухступен-
чато до 80° С, а затем также двухступенчато вводят раствор NaOH до по-
лучения pH 9—10. В течение 30 мин процесс гидролиза печени протекает
до конца, и белковые вещества полностью растворяются.
Сепарация жира. Из гидролизатора масса поступает в сепаратор 8. Гид-
ролизат, имеющий большую плотность, отводится в нижнюю трубу. Жир
с меньшей плотностью поднимается вверх и отводится в верхнюю трубу.
Жир, отделенный от гидролизата, поступает в отстойник 9, гидролизат •—
в отстойник 10, откуда насосом 11 его перекачивают в гидролизатор 7
и сепаратор 8 для повторного извлечения жира.
Повторный процесс извлечения заключается в добавлении рыбьего жира
низкой активности и вторичном сепарировании жира. Первичный жир
из отстойника 9 насосом 12 перекачивают в промывной котел 13, снабжен-
ный мешалкой и паровой рубашкой, где жир подогревают до температуры
80° С и промывают водой. Расход воды равен почти десятикратному коли-
честву по отношению к общей массе гидролизата. Промытый жир с водой
поступает в промывные сепараторы 14, где жир отделяется от воды. По-
следняя уходит в канализацию, и жир поступает в сборник 15, откуда на-
сосом 16 перекачивается в резервуары 17. Рационально после промывки
жир просушить под вакуумом для удаления остатков влаги и коагуляции
белковой взвеси, профильтровать и охладить.
Белковая масса после повторного сепарирования поступает в сборник
18, откуда насосом 19 перекачивается в распылительную или вальцовую
сушилку 20, где высушивается до содержания влаги 5%.
Полученный порошок упаковывают в мешки или ящики и в зависимо-
сти от качества используют как белковый корм или для удобрения. Если
витаминный жир предназначен для хранения, его расфасовывают в желез-
ные бочки, которые заполняют инертным газом (азотом или углекислотой)
и герметически укупоривают.
Проведенные исследования показали [9], что витамин А в рыбьих жи-
рах, полученных по методу мягкого щелочного гидролиза, сравнительно
устойчив вследствие сохранения в этих условиях переработки натураль-
ных антиоксидантов и эфирной формы витамина А.
ПРОИЗВОДСТВО КОНЦЕНТРАТА ВИТАМИНА А ИЗ РЫБЬЕГО ЖИРА
Технология производства заключается в омылении жира спиртовой
щелочью, двойной экстракции витамина А из мыльного клея хлористым
метиленом, охлаждении и фильтрации с целью выделения мыла, промывке
экстракта водой, отгонке растворителя, выделении стеринов и стабилиза-
ции концентрата антиоксидантами (подробно см. [81).
ОСНОВНЫЕ УСЛОВИЯ ПРОИЗВОДСТВА КОНЦЕНТРАТОВ ВИТАМИНА А ИЗ
ПЕЧЕНИ РЫБ
Витамин А, как один из наиболее лабильных витаминов, подвергается
быстрому разрушению под действием воздуха, окислительных агентов и
света. Химические процессы распада витамина А усиливаются в присут-
ствии тяжелых металлов, в особенности меди, олова, свинца. В связи с
неустойчивостью витамина А необходимо в производстве строго соблюдать
установленный технологический режим и следующие основные правила.
414
1. Соленую и даже консервированную печень необходимо хранить на
холоду при температуре не выше минус 1° С. При наличии морозильных
камер. рационально печень хранить в замороженном виде.
2. От степени измельчения печени зависят выход жира, а следователь-
но, и потери витамина А в белковых отходах; для полного извлечения жира
требуется тонкое измельчение печени.
3. Щелочной гидролиз печени служит для разрушения химической свя-
зи между белком и витамином А, и, следовательно, потери витамина А
в белковых отходах будут увеличены, если процесс гидролиза будет непол-
ным; технологический дэежим гидролиза должен быть установлен для каж-
дой партии печени.
4. Витамин А в получаемом рыбьем жире в присутствии воздуха не-
стоек, поэтому требуется:
хранить рыбий жир в алюминиевых бочках; заполненных инертным
газом и герметически укупоренных;
добавлять к рыбьему жиру (5% к массе его) масло пшеничных зароды-
шей или соевое, так как содержащийся в них токоферол (антиокислитель)
и играет роль стабилизатора витамина А; положительные результаты дает
также подсолнечное нерафинированное масло, добавленное в значительно
большем количестве, чем пшеничное масло. Необходимо специально изу-
чить и установить оптимальное количество растительного масла, обеспе-
чивающее стабильность рыбьего жира при его хранении. Стабилизация
витамина А может быть достигнута прибавлением разрешенных синтети-
ческих антиоксидантов [11] в количестве 0,04—0,05% (додецилгаллат,
пропилгаллат, сантохин и др.);
температура хранения рыбьего жира не должна превышать 0°;
5. Для правильного осуществления процессов омыления рыбьего жира
и экстракции из него витамина А хлористым метиленом необходимо:
строго контролировать количество щелочи, вводимой в реактор для
омыления жира, имея в виду, что как избыток щелочи, так и недостаток
ее приводит к отрицательным последствиям;
экстракцию витамина А вести при температуре 25—30° С в течение
90 мин-,
экстракт необходимо промывать кипяченой водой, освобожденной от
кислорода воздуха.
6. Для обеспечения высокого качества и стабильности концентрата ви-
тамина А необходимо:
выделить из охлажденного (после отгонки растворителя) концентрата
остатки мыла;
выделить из концентрата стерины путем охлаждения его до минус 10° С,
выстаивания в течение 3—5 ч, фуговки его в центрифуге или фильтрации;
стабилизовать концентрат путем добавления натуральных масел (пше-
ничное, соевое), хранить его на холоду в темном месте в герметически уку-
поренных бочках, заполненных инертным газом.
7. Все процессы в производстве витамина А из печени рыб должны
протекать с максимальной быстротой в-эмалированной, алюминиевой или
из нержавеющей стали аппаратуре.
8. Во всех аппаратах, в которых осуществляют процессы переработки
печени на концентраты витамина А, воздух вытесняют путем заполнения
аппаратов инертным газом.
ПРОИЗВОДСТВО КОНЦЕНТРАТОВ ВИТАМИНОВ А И Е МЕТОДОМ МОЛЕКУЛЯР-
НОЙ дистилляции
Технология производства концентратов витаминов А и Е методом омы-
ления и экстракции органическим растворителем страдает крупными не-
достатками: большой расход химикатов для омыления жира и экстракции
витаминов из омыленной массы; большие потери исходного сырья, так как
415
жир в процессе омыления превращается в мыло; большие потери витами-
нов, вызванные потерей в процессе омыления естественных антиоксидантов
в виде эфиров.
Разработанный в 1937—1946 гг. [12—18] промышленный метод моле-
кулярной дистилляции жиров, основанный на способности витамина А
дистиллироваться при температуре 110—250° С, а витамина Е — при тем-
пературе 140—200° С при остаточном давлении 0,001 мм рт. ст., лишен
указанных недостатков.
ФИЗИЧЕСКИЕ ОСНОВЫ ПРОЦЕССА МОЛЕКУЛЯРНОЙ ДИСТИЛЛЯЦИИ
[17—21]
Молекулярная дистилляция отличается от обычной дистилляции сле-
дующими признаками:
обычная дистилляция жидкостей осуществляется при температуре ки-
пения, а молекулярная дистилляция — путем испарения со свободной по-
верхности, что позволяет вести процесс на 100° ниже температуры кипе-
ния;
процесс испарения при молекулярной дистилляции осуществляется под
весьма высоким вакуумом. При этом увеличивается средняя длина свобод-
ного пробега молекул, т. е. того пути, который проходят молекулы, не
сталкиваясь одна с другой. В этих условиях, молекулы, испаряющиеся
с поверхности, не встречая на своем пути сопротивления частиц окружаю-
щего газа, свободно достигают конденсатора.
При обычной дистилляции наблюдается реконденсация значительной
части молекул на испаряющей поверхности, чего при молекулярной дис-
стилляции не бывает.
При вакууме 1 • 10"4 мм рт. ст. средний свободный пробег молекул
достигает десятков и даже сотен миллиметров. Если конденсатор распо-
ложить от испарителя на расстоянии меньшем, чем длина свободного про-
бега молекул, то последние будут беспрепятственно переходить с поверх-
ности испарителя к поверхности конденсатора. Скорость этого перехода
(скорость дистилляции) будет определяться скоростью теплового движения
самих молекул.
Средний свободный пробег молекул может быть определен, исходя из
кинетической теории газов по следующей формуле:
1
X = —г-----, (1)
К2 ппсР
где п — число молекул в единице объема газа;
d. — диаметр молекулы.
Для определения средней величины свободного пробега удобно исполь-
зовать связь между X и вязкостью газа т]. Путем соответствующих преоб-
разований [19] формула (1) принимает вид
X = 5,737 . 1/_£-сл1, (2)
р, Г М '
где т] —вязкость, пз (1 из=0,1 н-сек!м?)',
р—давление, мм рт. ст.-,
М — молекулярная масса;
Т — абсолютная температура.
Из уравнения (2) следует, что величина среднего свободного пробега
молекул обратно пропорциональна давлению газа.
Ниже приведены величины X в зависимости от р [19, 21]:
р X, см р см
760 ........... 6,21-10-е 1-10-* . . . .47,2
1.............42,7-10“3 1 10-е .... 470
1-10-2 . . . 4,72-10'1 1-10-’ .... 47000
416
Таким образом, при р=1-10"4 величина X достигает почти 0,5 м. Сле-
довательно, с уменьшением давления воздуха или газов в сосуде, в котором
происходит дистилляция, можно добиться увеличения свободного пробега
молекул до расстояния, равного или большего, чем расстояние между
испаряющей и конденсирующей поверхностями. В этих условиях молеку-
лы пара после отрыва от поверхности испарения свободно достигают кон-
денсатора. Создание таких условий является основой молекулярной ди-
стилляции.
3. При обычной дистилляции существует равновесие между паром и ки-
пящей жидкостью, определяющее разделяющую способность процесса —
степень повышения концентрации более летучего компонента в паре по
сравнению с его концентрацией в жидкости.
Степень разделения при молекулярной дистилляции не связана с рав-
новесием между паром и жидкостью, так как в данном случае имеем сво-
бодный поток пара от испарителя к конденсатору. Следовательно, в данном
случае степень разделения будет зависеть от соотношения скоростей теп-
лового движения молекул отдельных компонентов.
Скорость молекулярной дистилляции может быть выражена следующи-
ми формулами [20, 21]:
Р
V = 5,833 • 10-2 • —- моль/(см2 • сек), (3)
У МТ
0 = 5,833 10"2р с/(см2 • сек), (4)
где V — объем пара, проходящего через сечение 1 см2;
G — масса пара, проходящего через сечение 1 см2;
р —давление насыщенного пара при температуре Т, мм рт. ст.;
М — молекулярная масса;
Т — абсолютная температура.
Для идеальной бинарной смеси коэффициент разделения (коэффициент
относительной летучести) в случае обычной равновесной дистилляции ра-
вен
Pi
Gt « “
р р2
Для молекулярной дистилляции коэффициент разделения равен
Таким образом, ат больше в раз.
Следовательно, при Р i=P2 коэффициент 0-^=1, а ат=11/ т. е. при
г Mi
одинаковой упругости паров компонентов их разделение обычной равно-
весной дистилляцией невозможно. При молекулярной дистилляции ком-
поненты можно разделить с различными молекулярными массами. Если же
Mi=M2, то а=1; таким образом, при молекулярной дистилляции разде-
ление компонентов невозможно, если молекулярные массы и упругость
паров их одинаковы.
Практически скорость дистилляции составляет 70—90% от вычислен-
ной по формуле (4). Скорость дистилляции уменьшается вследствие столк-
новения дистиллируемых молекул между собой, а также с молекулами
остаточного газа (воздуха).
4. В процессе молекулярной дистилляции наблюдаются следующие ста-
дии:
диффузия молекул более летучих компонентов из глубинных слоев жид-
кости к ее поверхности;
417
испарение молекул;
переход молекул с поверхности испарения на поверхность конденсации;
конденсация молекул на поверхности конденсации.
Особенности молекулярной дистилляции определяют область ее приме-
нения — для разделения смесей жидких органических веществ с высокой
молекулярной массой, разлагающихся при температуре кипения. Нецеле-
сообразно применять молекулярную дистилляцию в тех случаях, когда
процесс разделения компонентов может быть осуществлен при обычной
дистилляции.
ТРЕБОВАНИЯ, ПРЕДЪЯВЛЯЕМЫЕ К ПРОЦЕССУ И АППАРАТУРЕ ПРИ
МОЛЕКУЛЯРНОЙ ДИСТИЛЛЯЦИИ
Для эффективного осуществления процесса молекулярной дистилляции
необходимо следующее:
максимальная скорость процесса. Это обеспечивает минимальную про-
должительность воздействия высокой температуры дистилляции на лабиль-
ное вещество;
глубокий вакуум (1-Ю-4 мм рт. ст.). Он достигается применением
мощных вакуумных насосов и трубопроводов с малым сопротивлением;
форма и положение поверхности должны обеспечивать беспрепятствен-
ный отвод неконденсирующихся газов, выделяющихся в процессе дистил-
ляций. Расстояние поверхности конденсации от поверхности испарения
должно быть равно или меньше длины свободного пробега молекул;
предварительная дегазация жидкости (вне дистилляционного аппарата)
для освобождения ее от большого объема газов. В противном случае
трудно будет поддерживать глубокий вакуум в аппарате. Кроме того,
без дегазации жидкость подверглась бы бурному вспениванию и на кон-
денсатор попадали бы не пары дистилляции, а пена. Дегазация должна
производиться в отдельных аппаратах, называемых дегазаторами;
толщина слоя дистиллируемой жидкости должна быть небольшой, что-
бы обеспечить интенсивное ее перемешивание. Чем меньше толщина слоя
жидкости и чем лучше она перемешивается, тем быстрее выравнивается
концентрация дистиллируемой жидкости в различных ее слоях.
Подробные исследования процесса фракционированной молекулярной
дистилляции рыбьего жира проводил К- Хикман [13—16].
Как известно, в печеночном жире витамин А находится в двух формах:
1) в виде алкоголя с максимумом дистилляции при температуре 123° С
и 2) в виде сложных эфиров с максимумом дистилляции при температуре
190—230° С, причем эфирная фракция количественно преобладает над ал-
когольной.
Кроме указанных модификаций, идет фракционирование и других ком-
понентов жира, а именно:
Интервал Дистиллируемые продукты
температур, °C
80—100 Дистиллируется эфирное масло
. 100—170 Дистиллируются жирные кислоты
100—160 Дистиллируется витамин А (алкоголь)
160—255 Дистиллируется витамин А (сложные
эфиры)
170—250 Дистиллируется токоферол и эфиры
стеринов
На рис. 98 приведены кривые фракционной дистилляции печеночного
жира [161. В начале процесса дистилляции рыбьего жира удаляются эфир-
ные масла, обладающие резким запахом и вкусом (кривые 1, 2), а также
свободные жирные кислоты (кривая 3).
Масса этой фракции составляет около 3% к массе дистиллируемого
жира. Следующая фракция дистиллята содержит стерины и витамин D
418
(кривая 4), токоферол и его эфиры (кривая 5), обе формы витамина А (кри-
вая 6), глицериды (кривая 7). Выход основной фракции концентрата —
витамин А — составляет 5% к массе первоначального жира. Недистил-
лированный остаток представляет собой рыбий жир, освобожденный от
пахучих веществ, свободных жирных кислот и витаминов. Из этих кривых
видно, что для промышленной дистилляции витамина А должен быть при-
менен температурный интервал 160—255° С.
М. Коган [22] изучал процесс дистилляции рыбьего жира на молеку-
лярном перегонном аппарате типа «падающей пленки», причем им были
Рис. 98. Кривые фракционной дистилля-
ции печеночного жира.
получены концентраты витамина
А, обогащенные в 14—42 раза,
с максимальной активностью до
1 млн. и. е. в 1 г. Выход вита-
мина А в интервале температур
160—270° С составил 83%. Дав-
ление в перегонном аппарате
должно поддерживаться 0,001—
0,0001 мм рт. ст. [8].
М. Коган и А. Азарова изу-
чали в аппарате типа «падаю-
щей пленки» процесс дистилля-
ции некоторых растительных
масел [23]. При дистилляции
хлопкового и соевого масла при
температуре 140—200° С среднее
увеличение концентрации токоферолов во фракциях составляет 30—40
крат по отношению к исходной. В отдельных фракциях хлопкового масла
содержалось до 6,8%, а соевого — 8,5% токоферолов. В наиболее актив-
ных фракциях, весовое количество которых не превышало 1,2%, выход
токоферолов составлял более 50% от исходного.
Степень обогащения токоферолами наиболее активных фракций дистил-
лятов масел пшеничных и кукурузных зародышей не превышала 5—7 крат
при низком выходе токоферолов (30—34%).
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА КОНЦЕНТРАТОВ ВИТАМИНА
А ИЗ РЫБЬИХ ЖИРОВ МОЛЕКУЛЯРНОЙ ДИСТИЛЛЯЦИЕЙ
На рис. 99 дана принципиальная схема однокубовой установки [22].
Дистиллируемый жир из резервуара 1 через монжю 2 передавливается
инертным газом в мерник 3, откуда поступает в дегазатор 4, снабженный
ловушкой 5. Предварительная дегазация проводится при вакууме (оста-
точное давление 0,1—0,05 мм рт. ст.). Из дегазатора 4 жир, пройдя по-
догреватель 6, поступает в дегазатор 7 высокого вакуума, представляющий
собой колонку, заполненную кольцами Рашига, в которой поддерживает-
ся остаточное давление в пределах 0,001—0,004 мм рт. ст. Дегазирован-
ный жир из сборника 8 насосом 9 подают через трубчатый теплообменник
10 в дистиллятор центрифужного типа 11. Фракции дистиллятов собира-
ются в приемники 14, 15, 16, 17, а недистиллированный остаток поступает
в приемник 12, откуда он может быть возвращен на дистилляцию или
через сборник 13 выведен из системы. Установка оборудована самостоя-
тельными вакуумными системами: для дегазации и дистилляции 18, 19, 20;
21 — сборник сжатого инертного газа; 22 — пульт управления.
Сырье. Сырьем для производства концентратов витамина А служит
жир печени рыб и морского зверя, содержащий не менее 10 тыс. и. е. вита-
мина А в 1 г. На заводе рыбий жир следует хранить при температуре
0—5° С.
Подготовка сырья. Жир предварительно подсушивают под вакуумом
(не менее 600 мм рт. ст.) при температуре 125—135° С в течение 2 ч.
419
В процессе подогревания жира происходит коагуляция белковых и сли-
зистых веществ. Высушенный жир охлаждают до 80° С, фильтруют на нутч-
фильтре и далее охлаждают до 30° С.
Дегазация жира. Для удаления воздуха, растворенного в жире, воды и
легколетучих веществ жир подвергают дегазации в две ступени: 1) при
вакууме 10~2—10-1 мм рт. ст. и температуре 18—20° С и 2) при вакууме
10~3—10“2 мм рт. ст. и температуре 60—70° С.
При дегазации необходимо контролировать вакуум, температуру по-
догрева, скорость подачи жира и охлаждение ловушек.
Рис. 99. Принципиальная схема однокубовой установки для получения концентра-
та витамина А из рыбьих жиров молекулярной дистилляцией.
Подогрев жира перед дистилляцией. Жир в электроподогревателе
нагревают до температуры на 60° более низкой, чем температура дистил-
ляции. Электронагреватель включают при работающем питательном на-
сосе для предотвращения образования нагара на трубках подогревателя.
Жир в подогреватель следует подавать лишь при вращающемся роторе ди-
стилляционного аппарата.
Дистилляция жира. Технологический режим работы дистиллятора за-
ключается в поддержании температуры дистилляции, постоянной скорости
подачи жира, вакуума и постоянного числа оборотов ротора.
ТЕХНОЛОГИЧЕСКИЕ ПОКАЗАТЕЛИ
Сырье (см. стр. 412)
Производство витаминного жира из печени рыб
Гидролиз печени
Температура, °C..................82—85
pH ...............................9—10
Количество добавляемой щелочи,
% к массе печени.................... 3
Количество воды ........ десятикратное
к массе гид-
ролизата
Производство концентрата витамина А
Омыление жира
Температура, °C ................45—50
Сепарация жира и промывка
водой
Температура воды, °C ....... 80
Сушка белковой массы
Влажность высушенной белковой
массы, %.................... .5
из рыбьего жира методом омыления
Охлаждение экстракта
Температура охлаждения, °C . . минус
1-2
420
Количество спирта, % к массе
жира................................ 10
Экстракци я (периодическая)
Температура, °C .................... 50
Продолжительность, мин.......... 90
Отношение массы жира к массе
растворителя ..................... 1:4
Производство концентратов витамина А
Сырье
Содержание витамина А в рыбьем жире, тыс.
не менее
Температура хранения жира, °C
Подготовка сырья
Температура подсушивания, °C
Вакуум, мм рт. ст., не менее
Температура фильтрации, °C
Дегазация жира
Вакуум, мм рт. ст
Температура, °C
Отгонка хлористого метилена
Присутствие СО2 или азота... обязательно
Вакуум, мм рт. ст. . . . 700—720
Выделение стеринов
Температура охлаждения, °C.... минус 5
Продолжительность, ч . . . 3—5
методом молекулярной дистилляции
и е.,
10
0—5
125—135
600
80
II ступень
10-3—Ю-2
60—70
I ступень
0-е—ю-i
18—20
Дистилляция жира (см. стр. 419)
Очистка концентрата витамина А
Этерификация
Количество метилового спирта, объем к концентрату . . . ........двукратный
Количество твердой щелочи, % к массе концентрата................ 1
Температура, °C................................................. 40
Продолжительность, мин.......................................... 75—90
Подготовка к дистилляции
Отгонка избытка метилового спирта ..............................в токе СОа
Температура воды при промывке, °C............................... 75
Температура подсушки в вакууме, °C.............................. 50
Дистилляция
Вакуум, мм рт. ст...............................................2—4-10~3
Температура дистилляции витамина А, °C.......................... 170—230
Температура дистилляции витамина Е, °С ...................... 140—200
Содержание витамина А в концентрате, тыс. и. е./г, не менее . ... . 300
Содержание витамина Е в концентрате из хлопкового масла, %...... 5—6
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. Переплетчик Р. Р. Содержание витамина А в печени и других внутренних
органах промысловых рыб и дельфинов Балтийского и Азово-Черноморского бас*
сейнов. — В сб.: «Витаминные ресурсы и их использование», М., Изд-во АН СССР,
1951, № 1, с. 139—181.
2. Кизеветтер И. В., Лаговская Е. А. Содержание витамина А в рыбах
Дальнего Востока. Там же, с. 71—138.
3. К о л ч е в В. В. Содержание витамина А в основных промысловых рыбах и тюле-
не Волго-Каспийского бассейна. Там же, с. 182—206.
4. Виноградова 3. А. Сравнительная характеристика содержания витамина А
в печени рыб Черного моря. — В сб.: «Витамины», Киев, Изд. АН УССР, 1953, № 1,
с. 82—99 с ил. То же. Витамин А в печени рыб Черного моря, Изд-во АН УССР, 1957,
167с.
5. Кузнецова Л. Н. Витамин А в печени Черноморских скатов.— В сб.: «Ви-
тамины», Киев, Изд-во АН УССР, 1953, № 1, с. 100—105 с ил.
6. Р о з а н о в а В. А. Получение концентрата витамина А из печени рыб. — В сб.:
«Витамины в теории и практике», Л., Пищепромиздат, 1941, с. 105—113. То же. Со-
держание витамина А в различных рыбьих жирах. Там же, с 130—139.
7. Б у к и н В. Н„ Скоробогатова Е. П. — «Рыбное хозяйство», 1946,
№ 2—3, с. 39; То же. Печень китов как сырье для получения витамина А. — В сб.:
«Витаминные ресурсы и их использование», М., Йзд-во АН СССР, 1951, № 1, с. 207—
215.
8. Ш н а й д м а н Л. О. Производство витаминов. Пищепромиздат, 1950, с. 149;
1958, с. 93.
9. Лагунов Л. Л., Букин В. Н., Березин Н. Т., Прозорс-
к а я М. К- Гидролитический метод производства витаминных рыбьих жиров. —
В сб.: «Витаминные ресурсы и их использование», М., Изд-во АН СССР, 1951, 1, 22.
10. Б у к и н В. Н., О р е ш к и н а Л. Я- Протеидные соединения провитаминов А
и D. — В сб.: «Витаминные ресурсы», М., «Наука», 1951, № 1, с. 7—21.
421
11. Николаев Р. П. Применение антиоксидантов для стабилизации витаминных
препаратов и пищевых продуктов. — В сб.: «Витамины. Пищевая промышленность
за рубежом», М., Пищепромиздат, 1957, Ns 3, с. 103—107.
12. Hickman К., Ind. Eng. Chem., 1937, 29, 968, 1107; 1947, 39, 686.
13. Hickman К., E m b г e e N., там же, 1940, 40, 135.
14. Hickman К., Mess G., там же, 1946, 38, 28.
15. H i c k m ап К., F г e v о у D., там же, 1952, 44, 1903.
16. Н i с k m а п К. Пат. США 1925559, 5/IX 1933; 2113302 , 5/IV 1938; 2124879,
26/VII 1938; 2117802, 17/V 1938; 2117803, 17/V 1938; 2126466, 9/VIII 1938; 2126467,
9/VIII 1938; 2147894, 14/11 1938; 2150683, 14/III 1939; 2150684, 14/III 1939; 2150685,
14/III 1939; 2180356, 21/IX 1939; 2199994, 7/V 1940; 2210927, 13/VIII 1940; 2229173,
21/1 1941; 2249524, 5/VII 1941.
17. Ко г а н M. И. Получение концентратов витамина А из рыбьего жира методом
молекулярной дистилляции. — В сб.: «Новое в науке и технике витаминов», М.,
Пищепромиздат, 1946, Ns 1, с. 86—95 с ил.
18. Квятковский А., Коган М. И. Молекулярная дистилляция и ее при-
менение в США. — В сб.: «Бюллетень технической информации», М., Пищепромиз-
дат, 1945, с. 28—37 с ил.
19. Жаворонков Н., Майер А. Методы и процессы химической технологии,
М., Изд-во АН СССР, 1955, 1, с. 5.
20. М а т р о з о в В. М. Молекулярно-дистилляционный аппарат центрифужного
типа. —В сб.: «Исследования сублимационных и дистилляционных аппаратов и
гидродинамики мешалок», М., Машгиз, 1954, № 16, с. 63.
21. М а т р о з о в В. М. Аппаратура для молекулярной дистилляции. Машгиз. 1954.
22. Коган М. И. Промышленное производство концентратов витамина А методом
молекулярной дистилляции. — В сб.: Технология и применение витамина А и ка-
ротина, М., Пищепромиздат, 1956, с. 58—69 с ил.
23. Ко г а н М. И., Азарова А. X. Выделение и исследование обогащенных
токоферолом фракций растительных масел, полученных молекулярной дистилля-
цией. — «Труды ВНИВИ», М., Пищепромиздат, 1959, № 6, с. 64—74.
Глава 22. ПРОИЗВОДСТВО ЭРГОСТЕРИНА ИЗ ДРОЖЖЕЙ И
КОНЦЕНТРАТОВ ВИТАМИНА D2 ДЛЯ НУЖД ЖИВОТНОВОДСТВА
СЫРЬЕ ДЛЯ ПРОИЗВОДСТВА ВИТАМИНА D2 [1]
Естественные источники витамина D2 весьма ограничены.
Известно, что витамин D встречается лишь в некоторых животных
и рыбных продуктах (рыбьем жире, печени рыб, сливочном масле, молоке,
икре), а также в яичном желтке. Растения, как правило, не содержат ви-
таминов группы D, однако среди стеринов растительных жиров обнаружен
провитамин D2 — эргостерин.
Витамином D богаты лишь печеночные жиры рыб. Некоторые из них,
например печеночный жир тунца, содержит в 1 гот 40 до 60 тыс. и. е. ви-
тамина D2.
В молоке, сливочном масЛе и яйцах содержание витамина D2 ограниче-
но. Например, в 100 а молока содержится всего около 1 и. е. витамина D2,
в сливочном масле — 40—320 и. е. (в зависимости от времени года и ка-
чества кормов).
Незначительное содержание витамина D2 в естественных ресурсах за-
ставило научную мысль искать природные источники провитамина D2 —
эргостерина.
Дрожжи. В 30-х годах XX в. рядом исследователей было установлено,
что хлебопекарные (пекарские) дрожжи являются богатым источником эр-
гостерина. Позднее было установлено, что некоторые микроорганизмы мо-
гут служить источником витамина D.
Примерный химический состав дрожжей приведен в табл. 45.
Кроме указанных витаминов, в дрожжах содержатся и другие витами-
ны группы В (В3, В6, Н, Вс, холин, инозит, р-аминобензойная кислота
и др.). В получаемых с дрожжевых заводов пекарских дрожжах содержит-
ся эргостерина 0,18—0,25 к массе прессованных дрожжей [3].
По данным Ф. Трайниной [4], содержание эргостерина в дрожжах,
культивируемых при аэрации на зерновом сусле или выращиваемых на
422
Таблица 45
Содержание в дрожжах [2]
Примерный химический состав
пивных пекарских
Сырой протеин (Nx6,25) ....;.
Жир................................
Углеводы ..........................
Зола...............................
Прочие ............................
Витамин Bj (тиамин)................
Витамин В2 (рибофлавин)............
Витамин РР (никотиновая кислота) .
Провитамин D2 (эргостерин) . . . .
В % на сухое вещество
54 45
10 14
8 , 8
26 31
В мг % на сухое вещество
60—200 20—66
25—30 36—40
400—500 280—400
200 1000—2000
2 2
мелассе при соответствующем увеличении количества посевных дрожжей,
может быть увеличено до 2%, а по другим данным [5] до 2,3—3,0% к су-
хому веществу дрожжей. Таким образом, пекарские дрожжи являются
сырьем, весьма важным и имеющим промышленное значение.
Мицелий пенициллина. Ранее считали, что наиболее эффективным сырь-
ем для производства эргостерина являются мицелий плесневых грибов
Aspergillus niger и Penicillium. Мицелий последнего, по данным К- Пово-
лоцкой и Е. Скоробогатовой [6], содержит (на сухое вещество): стеринов
1,2—1,4%, тиамина 26—37 мг и рибофлавина 12—26 мг в 1 кг. Мицелий
пенициллиновой промышленности — отход производства — содержал
14,5% сухих веществ и 0,5% стеринов на сухое вещество. В отходах мице-
лия гриба содержится эргостерина в 2 раза меньше, чем в дрожжах, но
стоимость мицелия в 20 раз ниже стоимости дрожжей. Казалось, что этот
вид сырья для производства эргостерина является весьма перспективным.
Однако впоследствии оказалось, что в связи с изменением технологии про-
изводства резко снизилось содержание в мицелии стеринов. Впрочем, этот
вопрос требует дополнительного изучения.
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА ЭРГОСТЕРИНА И КОНЦЕНТРАТОВ
ВИТАМИНА D2 ИЗ ДРОЖЖЕЙ
Витаминный состав дрожжей богат и разнообразен. В них содержится
14 витаминов уже исследованных в той или иной мере. За исключением
провитамина эргостерина, растворяющегося в жирах, все остальные ви-
тамины, содержащиеся в дрожжах, растворяются в воде. В дрожжах со-
держатся также фосфолипиды [7, 8], ферменты и нуклеиновые кислоты.
Кроме того, дрожжи содержат в своем составе 48—50% белков, которые
по аминокислотному составу приближаются к животным белкам и поэтому
обладают значительной питательной ценностью.
Следовательно, рациональная технологическая схема переработки дрож-
жей должна базироваться на процессах, обеспечивающих полное извлече-
ние витаминов, содержащихся в дрожжах, и сохранение дрожжевого белка.
Однако извлечение из дрожжей эргостерина и водорастворимых вита-
минов связано с большими трудностями, так как витамины прочно связаны
с белковой частью дрожжевой клетки. Следовательно, для освобождения
витаминов необходимо разрушить белковый комплекс клетки, что может
быть достигнуто различными методами.
А. Труфанов и В. Кирсанова [9] экспериментально установили, что
часть эргостерина в дрожжах так прочно связана с клеткой, что даже в
результате 2-часового гидролиза щелочью при температуре 100° С он не
извлекается из дрожжей.
423
Витамины группы В также находятся в связанном состоянии. Так,
например, витамин Bi входит в состав кофермента кокарбоксилазы; ви-
тамин В2 является составной частью желтого фермента.
А. Труфанов [10] считает, что процесс освобождения витаминов Bj
и В2 наиболее полно протекает при энзиматическом расщеплении дрож-
жей. Установлено, что при автолизе дрожжей выход эргостерина увели-
чивается до 30%, а выход тиамина за 6—10 ч автолиза увеличивается в не-
сколько раз.
К- Поволоцкая и Е. Скоробогатова [6] считают, что в условиях авто-
лиза прекращается рост и размножение клеток и наступает процесс их
старения с накоплением стеринов. Экспериментально [11] удалось полу-
чить большое накопление стеринов в дрожжах на плотных питательных
средах при задержке их размножения и роста.
Гидролиз белка может быть также осуществлен путем нагревания и ки-
пячения дрожжей, разведенных и подкисленных молочной или соляной
кислотой до pH 5 (М. Мейсель, Ф. Трайнина), под небольшим давлением
в автоклаве. При этом методе возможно комплексное использование дрож-
жей с получением концентрата витаминов группы В, кристаллического
эргостерина и белкового кормового средства.
По методу ферментативного гидролиза хлебопекарные дрожжи подвер-
гают автолизу при температуре 45° С. В процессе автолиза фермент, со-
держащийся в дрожжах, разрушает белковые вещества протоплазмы,
освобождая витамины, входящие в их состав.
Автолизированные или гидролизированные дрожжи обрабатывают спир-
том для извлечения витаминов группы В и для обеспечения надлежащей
фильтруемости дрожжевой массы. Спиртом или другим растворителем экст-
рагируют стерины, а дрожжевой шрот после отгонки спирта используется
как кормовой продукт.
Дрожжевую массу для обезвоживания дрожжевых клеток и коагуля-
ции белка обрабатывают спиртом. Дрожжевые клетки становятся плотны-
ми, легко оседают на дно сосуда и хорошо фильтруются.
Из изложенного вытекает, что для максимального извлечения витами-
нов и использования ценных питательных свойств дрожжевого шрота тех-
нологическую схему целесообразно строить следующим образом.
1. Для освобождения витаминов, прочно связанных с белково-фермен-
тативными комплексами, и увеличения таким образом их выхода, надо
подвергать дрожжи гидролизу или автолизу.
2. Для обеспечения надлежащей консистенции целесообразно автоли-
зированную или гидролизированную дрожжевую массу подвергать обработке
спиртом с экстракцией водорастворимых витаминов группы В.
3. Для извлечения стеринов и фосфолипидов высушенный дрожжевой
шрот следует подвергать повторной обработке спиртом или другим рас-
творителем.
4. Полученный после экстракции стеринов белково-дрожжевой шрот
после освобождения от спирта должен быть использован для пищевых или
кормовых целей, а также для получения нуклеиновых кислот. На рис. 100
изображена технологическая схема по комплексной переработке дрожжей
в объеме проверенных методов [1].
ПРОИЗВОДСТВО КОНЦЕНТРАТА ВИТАМИНОВ ГРУППЫ В [1]
Хлебопекарные дрожжи или мицелий пенициллина измельчают в шне-
ковом прессе 1 и направляют в автоклав 2, где дрожжевая масса подверга-
ется кислотному гидролизу при температуре 110° С в течение 20—30 мин.
К 100 кг дрожжей добавляют 20 л воды и 10 мл соляной кислоты (плотность
1190 кг/м9). Кислотный гидролиз увеличивает выход эргостерина за счет
освобождения эргостерина, связанного с белком. Из автоклава дрожжи
поступают в коагулятор 3, снабженный обратным холодильником 4, куда
424
Спирт
Шелпчь
Зшир
Вода.
flap
Мицелий пеници -
л ин а или дрожжи
Получение концентрата вита-
минов группы В Получение эреостврина-сырца
Вода
А Спирт -
=1 бензол
< _ Вода
Перекристаллизация обличение зргостерина и получение концентрата
эргостерина-сырца________£-------2-----_------, дат о мина И,
Вода
Вода
ЗВ\-
Вода
28
Пар
Пар.
'ода
7/
Пар
33 А°л°Я
ZI
Лар'
Спирт на ректификацию
Отход-мыло
Рис. 100. Технологическая схема комплексной переработки дрожжей на эргостерин и концентраты витамина D2.
ia pactpa-
сооку
-г-, , л о -• белкодаямука
। | iPacyawo/ta г~—z—
Z5
Колод
Вода
(Г7ПД
Лар
Отход и.
(L
7 но
вводят на каждые 100 кг дрожжей 85 л спирта с доведением крепости рас-
твора до 50°. В коагуляторе массу выдерживают 40—50 мин при интенсив-
ном перемешивании и температуре 75—78° С. При этом происходит
коагуляция белковых веществ, облегчающая процесс фильтрации, и экстрак-
ции водорастворимых витаминов. После этого массу переводят в аппарат-
охладитель 5, где ее охлаждают до 10° С и затем фильтруют через бара-
банный вакуум-фильтр 6. Фильтрат поступает в сборник 7, а из него в
вакуум-перегонный аппарат 8 с поверхностным конденсатором 9. Конден-
сат спирта направляют в сборник спирта-дистиллята 10, а из него на рек-
тификацию. После отгонки спирта и части воды в вакуум-перегонном ап-
парате остается жидкий концентрат витаминов группы В с содержанием
50% сухих веществ. Концентрат направляют в сборник 11, затем в раз-
ливочный прибор 12 и на расфасовочном конвейере 13 разливают в фла-
коны.
Белковый остаток с вакуум-фильтра 6 поступает в дистилляционный
аппарат 14 для отгонки спирта.
После отгонки спирта белковый осадок поступает в бункер 15, а из него
в вальцовую сушилку 16, где дрожжи высушивают — содержание влаги в
дрожжах не должно превышать 2%.
Полученную лепешку измельчают в дробилке 17 в муку, которая по-
ступает на дальнейшую переработку.
ПРОИЗВОДСТВО КОНЦЕНТРАТА ВИТАМИНА D2 [1]
Получение эргостерина-сырца. В экстракторе 18 с обратным холодиль-
ником дрожжевую муку подвергают обработке трехкратным количеством
(к массе дрожжевой муки) спирта-ректификата при температуре 78° С.
Затем дрожжи отфильтровывают в друк-фильтре 19. Первый экстракт по-
ступает в сборник 20, а осадок — во второй экстрактор 21, где его снова
обрабатывают трехкратным количеством спирта. Белковый осадок отфильт-
ровывают на друк-фильтре 22. Второй экстракт также поступает в сборник
20. Из последнего — оба экстракта переходят в вакуум-перегонный аппа-
рат 23, где спирт отгоняют под вакуумом.
Спирт после отгонки направляют в сборник 24, откуда он поступает
на ректификацию. Фильтрат упаривают до содержания 70% сухих веществ.
Из 100 кг сухих дрожжей получают 20—25 кг липидного концентрата.
В вакуум-аппарат вводят 45%-ный раствор едкого натра в количестве
6 кг на 100 кг дрожжевой муки и омыляют жиры. Омыленный раствор эр-
гостерина подвергают кристаллизации при 0° в аппарате-кристаллизаторе
25. Выделяющиеся кристаллы эргостерина-сырца отфильтровывают на
нутч-фильтре 26 и направляют на перекристаллизацию. Межкристальный
спиртовой раствор поступает в вакуум-перегонный аппарат 27 для отгонки
спирта. Остающийся в перегонном аппарате щелочной раствор является
отходом производства. Спирт-отгон из конденсатора 28 поступает в сбор-
ник 29, а отсюда направляется на ректификацию. Белковую массу из друк-
фильтра 22 направляют в дистиллятор 30, где ее разбавляют водой в ко-
личестве, равном удвоенной массе осадка. Затем отгоняют спирт и направ-
ляют его в сборник 31. Белковая масса из дистиллятора после нейтрали-
зации поступает в бункер 32, затем в вальцовую сушилку 33, дробилку
34 и далее на расфасовку.
Перекристаллизация эргостерина-сырца. Эргостерин-сырец подвергают
аффинации в аппарате 35 спиртом крепостью 65% (весовых) в количестве
10 кг на 1 кг сырца. Аффинированную массу направляют в друк-фильтр
36. Отфильтрованные аффинированные кристаллы эргостерина направля-
ют на перекристаллизацию в аппарат 37 в спирто-бензольном растворе,
состоящем из 80 частей спирта и 20 частей бензола. Маточный раствор,
получающийся при аффинации, поступает в сборник 38, а затем в перегон-
ный аппарат 39 для отгонки спирта. Из перегонного аппарата спирт-отгон
426
попадает в сборник 40, а из последнего — в сборник 65а и в ректифи-
кационную колонку 64. Раствор эргостерина в спирто-бензольной смеси
из аппарата 37 направляют в кристаллизатор 41 и нутч-фильтр 42. Крис-
таллы эргостерина I после промывания высушиваются в вакуум-сушилке
43 (температура плавления должна быть не ниже 160° С). Маточник крис-
таллизации I поступает в сборник 44, а затем в вакуум-перегонный аппа-
рат 45 для отгонки спирто-бензольного растворителя, который собирают
в приемнике 46.
Сгущенный маточник подвергают кристаллизации в аппарате 47. Крис-
таллы эргостерина II отделяют от маточника II на нутч-фильтре 48.
Эргостерин II направляют на аффинацию совместно с эргостерином-
сырцом. Маточник II из нутч-фильтра 48 поступает в сборник 49, а из него
в вакуум-аппарат 50. Отгонный спирт направляют в сборник 51, откуда
он через сборник 46а поступает на повторное использование.
Облучение эргостерина и получение концентрата витамина D2. Ряд ис-
следований показывает, что в эфирном растворе процесс активации эрго-
стерина протекает не так интенсивно, как в спирте. Если в последнем мак-
симум активации наступает через 30—40 мин, то в среде эфира через 150—
200 мин, что позволяет избежать переоблучения раствора. Это особенно
важно для производства кристаллического эргокальциферола, так как про-
дукты переоблучения эргостерина удерживают эргокальциферол в рас-
творе [5, 12], препятствуя его кристаллизации. Сухой эргостерин I подвер-
гают облучению в растворе серного эфира. Предполагают, что эргостерин
вначале изомеризуется в прекальциферол, который при нагревании пере-
ходит в эргокальциферол и частично в люмистерин (см. стр. 300). В реак-
тор 52 из сушилки поступает эргостерин, а из сборника 53 — эфир. По-
лученный раствор фильтруют через друк-фильтр 54, фильтрат через мер-
ник 55 подают в сборник 56, откуда он непрерывно течет через облучающие
аппараты 57. Облученный раствор переходит в сборник 58, затем в пере-
гонный аппарат 59 для отгонки эфира и выделения непрореагировавшего
эргостерина, который отфильтровывают в друк-фильтре 60 и передают на
перекристаллизацию. Этот эргостерин должен быть перекристаллизован
вследствие низкой чистоты (80—90%). Фильтрат стандартизуют по актив-
ности маслом в смесителе 61, фильтруют через нутч-фильтр 62 в сборник
63, откуда направляют на расфасовку.
Эргостерин (провитамин D2). Эмпирическая формула эргостерина
С28Н44О. Молекулярная масса гидрата C2gH44O-H2O— 414,648, а безвод-
ного — 396,632. Кислород в молекулу эргостерина входит в виде гидро-
ксильной группы, следовательно, эргостерин является высокомолекуляр-
ным спиртом, который с кислотами может образовать эфиры.
Большое число работ было проведено для определения структурной
формулы эргостерина. Работами Виндауса и его сотрудников установлена
структурная формула эргостерина:
Таким образом, молекула эргостерина состоит из четырех колец. В пер-
вом кольце имеется спиртовая группа, во втором — две конъюгированные
двойные связи; к четвертому кольцу присоединена боковая цепь из 9 уг-
427
леродных атомов. Являясь ненасыщенным соединением (3 двойные связи),
эргостерин легко окисляется и темнеет в присутствии света и кислорода
воздуха даже при обычной температуре.
Форма кристаллов эргостерина зависит от рода растворителя: из 95%-но-
го алкоголя он кристаллизуется в виде бесцветных листочков, из хлоро-
форма, эфира, ацетона — в виде красивых игл. Крупные кристаллы могут
быть получены при кристаллизации из смеси эфир-ацетон в отношении 1:3
[13].
Присутствие небольшого количества воды в смесях бензин-спирт или
этилацетат-спирт способствует росту кристаллов эргостерина [14].
Температура плавления эргостерина зависит от степени гидратации
кристаллов. Тщательно перекристаллизованное вещество, содержащее 1,5
молекулы кристаллизационной воды, имеет
температуру плавления 166°С [15].
Получение обезвоженных препаратов
весьма затруднено. Их температура плав-
ления колеблется от 168 до 183° С. Рас-
творы эргостерина дают разные углы вра-
щения плоскости поляризации в зависи-
мости от растворителя [aJz> = 132° — в
2 %-ном растворе хлороформа; 94° — в
эфире; 125° — в бензоле; 93° — в абсолют-
ном этиловом спирте; 92° — в ацетоне. При
глубоком вакууме (остаточное давление
0,4 мм рт. ст.) эргостерин перегоняется
без разложения при температуре 250° С.
Окислители (галоиды, КМпО4 и др.) раз-
рушают эргостерин. В воде эргостерин не-
растворим, в органических растворителях
(хлороформе, дихлорэтане, эфире) раство-
Рис. 101. Спектр поглощения
эргостерина в УФ-свете.
ряется хорошо.
Эргостерин обладает способностью сильно поглощать ультрафиолетовые
лучи с абсорбционным максимумом при длине волны 263, 271, 282 и
293,5 нм в этиловом спирте (рис. 101). Молекулярный коэффициент эк-
стинкции при 282 нм=11 500, теплота сгорания 1 а — 9,950 кал, плот-
ность 1040 кг!м3 [12].
Эргостерин легко окисляется, в особенности в растворе под влиянием
тепла и света, при этом кристаллы желтеют и температура их плавления
снижается. Отмечено [16], что обезвоженные кристаллы быстрее окисля-
ются, чем гидратные. Эргостерин рекомендуется сохранять в условиях
вакуума или в инертном газе. Экспериментально установлено, что эрго-
стерин можно полностью сохранить в герметически укупоренной таре,
заполненной инертным газом, при хранении на холоде (0°) и в тем-
ноте.
Эргостерин нерастворим в воде, но присутствие 0,2% воды в этилаце-'
тате увеличивает его растворимость. Слабые коллоидные растворы эрго-
стерина могут быть приготовлены при добавлении ацетона, при применении
ультразвука или при помощи других диспергирующих факторов. Наи-
лучшие коллоидные растворы могут быть получены при растворении нат-
риевых, калиевых и аммониевых солей сложных эфиров эргостерина, как,
например, моноэргостеринового эфира фталевой, янтарной кислот [17, 18].
Устойчивость эргостерина в различных растворителях различна. В спирте
эргостерин может сохраняться несколько недель в темноте. Пиридин раз-
рушает эргостерин [16]. В дихлорэтане эргостерин нестоек вследствие при-
сутствия НС1.
В табл. 46 приведена растворимость эргостерина.
428
Таблица 46
Растворитель Темпера- тура, °C Растворимость эргостерина [12], частей раствори- теля на 1 часть эргостерина Растворитель Темпера- тура, °C Растворимость эргостерина [12], частей раствори- теля на 1 часть эргостерина
Ацетон 20 200 Бензин .... 16 94
» 56 32 Хлороформ . . 18 50
Спирт 95%-ный 15 526 Эфир серный . 20 50
То же 78 36 То же .... 35 28
ПОЛУЧЕНИЕ КОНЦЕНТРАТА ВИТАМИНА D2 В ВИДЕ СУХИХ ОБЛУЧЕННЫХ
ДРОЖЖЕЙ [19]
Способ заключается в следующем. Сырье, содержащее эргостерин (на-
пример, дрожжи), размешивают с водой до легкотекучей консистенции
смеси (содержание дрожжей в водной суспензии 5%). После этого смесь
подвергают облучению ультрафиолетовым светом в любом центробежном
пленочном аппарате или аппарате, работающем на принципе падающей
пленки. Облученные дрожжи высушивают на вакуум-вальцовой сушилке.
При данном методе значительно повышается витаминная активность дрож-
жей 10—15 тыс. и. е. в 1 а вместо 1,0—1,5 тыс. и. е. в 1 г, получаемых при
облучении сухих измельченных дрожжей на конвейере [19—22].
Это обусловлено, по-видимому, двумя причинами: набуханием дрожже-
вых клеток в водной суспензии, способствующим утончению клеточной
оболочки и облегчающим проникновение ультрафиолетовых лучей внутрь
клетки; достаточной дисперсностью дрожжевой массы в водной среде,
облегчающей контакт ее с ультрафиолетовым светом.
Идентификация фосфолипидов и выделение их из хлебопекарских дрож-
жей. Совместно с Н. Богословским и Е. Кузнецовой исследовали фосфо-
липиды хлебопекарских дрожжей. Фосфолипиды выделяли осаждением
ацетоном из раствора липидного остатка дрожжей в хлороформе [8]. Вы-
ход фосфолипидов составляет 3,3% от липидного остатка. Для идентифи-
кации фосфолипиды хроматографически разделяют в тонком слое кремне-
вой кислоты [23]. Идентификацию производили по характерным цветным
реакциям с молибденовой синью [24] и 0,5%-ным раствором нингидрина
в 95%-ном изопропиловом спирте.
Количественное содержание отдельных фракций фосфолипидов опреде-
ляли по фосфору (метод Беверидже [25]). Идентифицированы следующие
фосфолипиды, в % от общего содержания: фосфатидилэтаноламин — 41,0,
фосфатидилинозит — 15,0, фосфатидилхолин — 22,0.
ТЕХНОЛОГИЧЕСКИЕ ПОКАЗАТЕЛИ
ПОЛУЧЕНИЕ КОНЦЕНТРАТА ВИТАМИНОВ ГРУППЫ В
Температура автоклавирования, °C ............................. ПО—115
Продолжительность автоклавирования, мин .............' . . 10—20
Количество добавляемого спирта, л на 1 кг дрожжей ............ 0,85
Крепость спиртового раствора в коагуляторе, %................. 50,0
Продолжительность процесса коагуляции, мин.................... 45
Температура охлаждения дрожжевой массы, °C.................... 15—20
Содержание сухих веществ в скоагулироваиной массе, % . . . . 13—15
Концентрат витаминов группы В, % на сухое вещество
Сухие вещества.................................................. 50,0
Общий азот....................................................... 6,4
Углеводы (считая на глюкозу).................................... 13,2
Зола ............................................................ 19,4
В том числе:
К2О........................... 11,0 MgO..........................0,6
Р2О5 ........................... 6,2 СаО 0,1
Fe2O3........................... 1,5
429
Витаминный состав, мг/г [26]
Bi ........................
В2.........................
Холин......................
0,04
0,17
7,1
Никотиновая кислота..........
В6 ..........................
В3...........................
2,0
0,1
0,4
ПОЛУЧЕНИЕ ЭРГОСТЕРИНА-СЫРЦА
Содержание стеринов в оухой дрожжевой муке, %.................0,8—1,0
Выход дрожжевой муки, % от массы прессованных дрожжей . . 18—20
Количество вводимого спирта на каждую экстракцию (I и II), %
к массе дрожжевой муки ....................................... 300
Количество липидного остатка, % от массы дрожжевой муки . . 20—25
Содержание эргостерина в липидном остатке, % .................3,3—3,5
Режим омыления липидного остатка
Количество 45% спиртовой щелочи, кг на 100 кг дрожжевой муки 6
Продолжительность омыления при кипении, ч .................... 4
Выход товарного эргостерина из 1 т прессованных дрожжей,г . . 1400—1500
Выход эргостерина, % к введенному............................. 60—68
Качество эргостерина:
температура плавления, °C ............................... 160—162
содержание летучих веществ, %, не более ................. 3,0
Облучение эргостерина
Концентрация эргостерина в растворе, %
спиртовом....................................................0,3—0,4
эфирном .................................................... 1,0
Продолжительность облучения в спиртовом растворе, мин .... 30—40
Температура, °C .............................................. 50
Отгонка спирта до объема концентрата, % ...................... 15—20
Температура выкристаллизовывания эргостерина, °C ................минус 5—10
Количество выделяемого непрореагировавшего эргостерина, % к
введенному.................................................... 40—50
Выход витамина D2 из 1 т прессованных дрожжей, млрд. и. е. . 33—35
Выход эргостерина из 1 т прессованных дрожжей, кг............. 1,10—1,29
Расход сырья и материалов на переработку 1 т прессованных
дрожжей
спирт-ректификат, л..................................... 125—150
бензол, л................................................ 5—6
кали едкое, кг........................................... 7—8
Расход эргостерина, г на 1 млрд и. е.
витамина D2 ............................................ 35—39
Состав фотосмолы, %
эргокальциферола......................................... 55—65
люмистерина ............................................. 15—20
тахистерина.............................................. 10—13
эргостерина................................................... 10
Растворимость эргостерина при различной температуре приведена в
табл. 47 [3].
Таблица 47
Растворитель Растворимость эргостерина, г на 100 г растворителя при температуре. GC
0 10 20 50 70
Дихлорэтан 0,04 0,083 0,308 2,570 9,040
Бензол — 0,260 0,521 4,010 16,160
Спирт 0,02 0,077 0,110 0,740 0,790
Спирто-бензольная смесь (80% спирта и 20% бензола) 0,05 0,170 0,300 2,280 5,180
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. Шнайдман Л. О. Производство витаминов. М., Пищепромиздат, 1958, с. 174.
2. Г и в а р т о в с к и й Р. В., П л е в а к о Е. А. Технология дрожжевого про-
изводства. М., Пищепромиздат, 1943, 253с.
3. Татьянин А. Р. Производство витамина D, М., Пищепромиздат, 1943, 62с.
4. Т р а й н и н а Ф. Л. Накопление эргостерола в расах дрожжей при различных
условиях их культивирования. — В сб.: «Витамины в теории и практике», Л., Пи-
щепромиздат, 1937, т. 2, вып. 1, с. 53—62.
430
5. Rosenberg H., Encyclopedia of Chemical Technology U. S., 1955, 14, 828.
6. Поволоцкая К- Л., Скоробогатова Е. П. Витаминные и фермен-
тативные свойства мицелия Penicillium. — «Биохимия», 1947, т. 12, вып. 3, с. 268—
275 с ил.
7. Богословский Н. А. Синтетические исследования в области фосфатидов.
Автореф. кандид. дисс., М., 1962.
8. Folich J., Lees М. и др. J. Biol. Chem., 1957, 226, 497.
9. Труфанов А. В., Кирсанова В. А. Содержание эргостерина в дрожжах
при автолизе.— «Биохимия», 1939, т. 4, вып. 4, с. 377—380. То же, 1940, т. 5, с. 234.
10. Т р у ф а н о в А. В. Одновременное получение из дрожжей эргостерина, рибо-
флавина и аневрина. — В сб.: «Труды Всесоюзной конференции по витаминам»,
М., Пищепромиздат, 1940, с. 141—143.
11. Проскуряков Н. И., Попова Е. М., Осипов Ф. М. К определе-
нию и содержанию эргостерина в различных дрожжах. — «Биохимия», 1938,
т. 3, вып. 3, с. 397—400.
12. S е Ь г е 1 1 W., Harris R., The Vitamins, Acad. Press., 1954, 2, 132.
13. В i 1 1 s С., Пат. США 1775548, 9/IX 1930.
14. Carlson W., Пат США 2167272, 25/VII, 1939.
15. R о s e n b e r g R., Chemistry and Physiology of the Vitamins, 1942, 57.
16. Collow R., Biochem. J., 1931, 25, 79.
17. Emerson H., Heyl F., J. Am. Chem. Soc., 1930, 52, 2015.
18. Hoff ma nn - La Roche, Немецкий пат. 495450, 30/VI, 1927.
19. К а н ч у x Ш. Ф. Авт. свидет., № 85886, 26/IX 1949; Бюлл. изобрет., 1950,
№ 8, с. 50.
20. Шнайдман Л. О. Производство витаминов из животного и растительного
сырья. М., Пищепромиздат, 1950.
21. В ад и м о в' В. Электрификация сельского хозяйства. 1931, № 5, 6, 9, 11; 1932,
№ 8; 1933, № 4.
22. Ма ц ко С. Н. — «Бродильная промышленность», 1932, 10.
23. Skipski V., Peterson R., Barclay M., Biochem. J, 1964, 90, 374.
24. D i t m e г J., Lester R., J. Lipid Res., 1964, 5, 126.
25. Beveridge, Johnson, Canad., J. Res. E., 1949, 27, 159.
26. Ж д а и- П у ш к и н С. — «Микробиология», 1955, т. 24, 5, с. 545.
Глава 23. ПРИРОДНЫЕ ПИЩЕВЫЕ БИОЛОГИЧЕСКИ
АКТИВНЫЕ КРАСИТЕЛИ
Для окраски пищевых продуктов и фармацевтических препаратов ра-
нее применялись преимущественно синтетические красители: красный —
амарант, синий — индигокармин и желтый — нафтол, тартразин. В послед-
нее время Министерство здравоохранения СССР начало ограничивать при-
менение для указанных целей синтетических красителей, так как они в
большинстве случаев не безвредны. В связи с этим встал вопрос о замене
синтетических красителей природными.
КЛАССИФИКАЦИЯ ПРИРОДНЫХ КРАСЯЩИХ ВЕЩЕСТВ
Наиболее приемлемой является классификация по химической струк-
туре [1, 2, 3]. По этому принципу красящие вещества делятся на три груп-
пы {1 ]:
красящие вещества алифатического ряда, к которым относятся кароти-
ноиды и ряд антибиотиков, как, например, пентамицин;
красящие вещества ароматического ряда, к которым относятся халко-
ны, бензохиноны и нафтохиноны;
красящие вещества гетероциклического ряда и флавоноиды, антоцианы
и другие, например производные индигоиндола, производные пирролахло-
рофилла, витамин В12.
Имеется также классификация красящих веществ по биологической ак-
тивности, а именно: красители каротиноидные, обладающие А-провитамин-
ной активностью; красители биофлавоноидные, обладающие Р-витаминной
активностью; красители хлорофилловые, биологическая активность кото-
рых известна, но вполне еще не изучена.
431
ПИЩЕВЫЕ БИОЛОГИЧЕСКИ АКТИВНЫЕ ПРИРОДНЫЕ КРАСИТЕЛИ, ИЗУЧЕН-
НЫЕ И ОСВОЕННЫЕ В ПРОИЗВОДСТВЕ
Полный ассортимент синтетических красителей изложен в каталоге
английской, фирмы [4].
Освоенные в производстве природные красители делятся на две группы:
жирорастворимые и водорастворимые. К первым относится каротиноидный
краситель, вырабатываемый из моркови, тыквы, томатов и из семян тро-
пического кустарника Bixa orellana. К водорастворимым относятся анто-
циановые красители, полученные из отходов красных сортов винограда
(Саперави, Каберне, Изабелла и др.), из черноплодной рябины, столовой
свеклы.-
Жирорастворимые красители. Морковь (Daucus carota) и тыква (Cucur-
bita) служат сырьем для производства каротинового препарата-красителя.
Он применяется в медицине для лечебных целей, а также в животноводстве.
В маргариновой промышленности он применяется для подкраски марга-
рина.
Нашими исследованиями (совместно с М. Шаховой) выявлены:
в моркови: каротиноиды (10—15 мг%), в том числе — а- и 0-каротин,
ликопин, флавоксантин и тараксантин; аминокислоты — лизин, орнитин,
гистидин, цистеин, серин, аспарагин, треонин, пролин, метионин, тиро-
зин, лейцин; витамины —бетаин (300 мг%), холин (11 мг%);
в тыкве витаминной: каротиноиды (ликопин, флавоксантин, виола-
ксантин и тараксантин) [4—6].
Каротиновый краситель из моркови получают путем отжатия сока мор-
кови, коагуляции в нем белковых веществ, адсорбирующих весь каротин;
коагулят отфильтровывают, высушивают, измельчают и экстрагируют ка-
ротин растительным маслом [5]. Каротиновый краситель из тыквы полу-
чают путем силосования ее, протирки силосной массы, нейтрализации, ко-
агуляции белков, высушивания, экстракции органическим растворителем,
отгонки растворителя и растворения концентрата в растительном масле.
Содержание каротина в масляном растворе 2 мг!мл.
Томаты служат сырьем для производства ликопинового красителя,
который получил широкое применение и как краситель для маргарина
(совместно с каротином), и как ростовой фактор в животноводстве. Тех-
нология производства красителя заключается в экстракции ликопина из
томатной массы растительным маслом при температуре 80—85° С при ва-
кууме 600—650 мм рт. ст.
При исследовании образцов красителя было установлено следующее
содержание (в мг%): витамина Е — 89,0; каротиноидов — 66,0; 0-кароти-
на — 6,5—8,0. Состав каротиноидов (в %): а- и 0-каротин — 6,5; лико-
пин— 61,8; кислородсодержащие каротиноиды — 31,7.
Семена аннато — кустарника Bixa orellana содержат два кра-
сящих вещества:
каротиноид биксин (С25Н30О4, монометиловый эфир дикарбоновой кис-
лоты), листочки темно-красного цвета с металлическим блеском, темпера-
тура плавления 191,5° С, нерастворим в воде;
орелин малоизученный, хорошо растворимый в воде краситель желтого
цвета.
Краситель аннато получают экстракцией растительным маслом при тем-
пературе 90° С. При исследовании красителя аннато установлено, что в
нем содержится (в мг%): витамин Е— 150, каротиноиды — 1,9, стерины
200—300.
Водорастворимые красители (красно-фиолетовые). К этой группе отно-
сятся красители, получаемые из черноплодной рябины, столовой свеклы,
о которых выше приведены некоторые данные. Интерес представляет кра-
ситель, получаемый из выжимок винограда красных сортов и содержащий
432
в основном антоциан винограда — энин [6]. Последний представляет собой
моногликозид энидина, являющийся диметиловым эфиром дельфинидина
Технология получения энокрасителя по методу Леонова и Руднева [7 ]
заключается в экстракции выжимок винограда 1%-ным раствором соляной
кислоты в соотношении 1:1 при Температуре 65—70° С, фильтрации и сгу-
щении в вакуум-выпарной установке до содержания 35% сухих веществ
или красящих веществ — 60—62 г/л; pH — 2,0—3,0. Энокраситель — ин-
дикаторный краситель, меняющий цвет в зависимости от pH от красного
(при pH 1—2), фиолетового (pH 4,0—5,0) до синего (при pH 6,0—7,0) [8].
В настоящее время основной задачей в области пищевых природных
красителей является изучение и освоение в производстве стабильных хло-
рофилловых красителей, а также красителей из столовой свеклы и чер-
ноплодной рябины.
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. Вульфсон Н. С. — «Успехи химии», 1963, 32, 6.
2. П е т р о в В. —«Труды Ботанического института Азербайджанского филиала
АН СССР», Баку, 1939, 6.
3. Лазурьевский Г., Гранитов И. — «Труды Института химии АН Уз-
бекской ССР». Ташкент, 1948, 1.
4. Каталог I. С. I. Technical Information Soc. Imperial Chemical Industries, 1960, 542.
5. Шнайдман Л. О. Производство витаминов. М., Пищепромиздат, 1958, 121.
6. Майоров Б., Бегунова Р. Использование отходов виноделия для про-
изводства естественных красителей. ЦИНТИпищепром, М., 1962.
7. Руднев Н., Леонов Б. Натуральный пищевой краситель из виноградной
выжимки. М., ЦИНТИпищепром, 1961, 2.
8. Солодухин А. И. Производство и использование витаминов, антибиотиков и
биологически активных веществ. Краснодариздат, 1965, 145.
ОГЛАВЛЕНИЕ
Предисловие 3
ЧАСТЬ I. ПРОИЗВОДСТВО СИНТЕТИЧЕСКИХ ВИТАМИНОВ
• Глава 1. Основные принципы правильного построения технологии производства 5
Список использованной литературы 10
Глава 2. Производство синтетического витамина А (ретинола) 10
Физико-химические свойства 11
Методы синтеза витамина Ах и выбор рационального метода для про-
изводства 16
Технологическая схема производства синтетического витамина А 24
Технологические показатели 36
Список использованной литературы 38
Глава 3. Производство синтетического ^-каротина 40
Физико-химические свойства 40
Методы синтеза, ^-каротина и выбор рационального метода для произ-
водства 52
Технологическая схема производства синтетического Р-каротина 55
Технологические показатели 61
Список использованной литературы 62
Глава 4. Производство синтетического витамина Вх (тиамина) 64
Физико-химические свойства витамина Вх 64
Методы синтеза витамина Вх и выбор рационального метода для произ-
водства 67
Технологическая схема производства тиаминхлорида методом конденса-
ции пиримидинового и тиазолового циклов (вариант I) 81
Технологические показатели 92
Технологическая схема производства тиаминхлорида методом построения
молекулы на пиримидиновом цикле (вариант II) 94
Технологические показатели 98
Технологическая схема производства тиаминпропилдисульфида 100
Технологическая схема производства кокарбоксилазы—дифосфор-
ного эфира тиамина 101
Список использованной литературы 104
\у Глава 5. Производство синтетического рибофлавина (витамина В2) 107
Физико-химические свойства витамина В2 108
Методы синтеза рибофлавина и выбор, рационального метода для
производства ПО
Технологическая схема производства синтетического рибофлавина
(вариант I) 117
Технологические показатели 123
Технологическая схема производства синтетического рибофлавина
(вариант II) 124
Технологические показатели 130
Технологическая схема производства кофермента рибофлавинмоно-
нуклеотида и его натриевой соли 131
434
Технологическая схема производства 2-тиорибофлавина 133
Список использованной литературы 134
Глава 6. Производство синтетического пантотената кальция (витамина В3) 136
Физико-химические свойства витамина В3 137
Методы синтеза пантотеновой кислоты и выбор рационального метода
для производства 139
Технологическая схема производства £>(-/-(-пантотената кальция и
его рацемата 144
Технологические показатели 150
Список использованной литературы 151
'Глава 7. Производство синтетического пиридоксина (витамина В6) 152
Физико-химические свойства витамина В6 153
Методы синтеза витамина В6 и выбор рационального метода для про-
изводства 155
Технологическая схема производства пиридоксина (витамина В6) 160
Коферменты и производные пиридоксина 168
Технологические показатели 171
Список использованной литературы 172
Глава 8. Производство синтетической пангамовой кислоты (витамина В15) 174
Физико-химические свойства витамина В15 175
Методы синтеза витамина В15 и выбор рационального метода для
производства 176
Технологическая схема производства синтетического пангамата каль-
ция 177
Технологические показатели 181
Список использованной литературы 183
Глава 9. Производство синтетической никотиновой кислоты (витамина РР) 184
Физико-химические свойства витамина РР 185
Методы синтеза никотиновой кислоты, ее амида и выбор рационального
метода для производства 187
Выбор рациональной технологической схемы производства никотиновой
кислоты 198
Технологическая схема производства никотиновой кислоты и ее ами-
да из fl-пиколина парофазным окислительным аммонолизом 200
Технологическая схема производства никотиновой кислоты из 2-ме-
тил-5-этилпиридина окислением азотной кислотой под давлением 202
Технологическая схема производства никотиновой кислоты из 2-ме-
тил-5-этилпиридина окислением азотной кислотой в присутствии сер-
ной при атмосферном давлении 205
Технологическая схема производства никотиновой кислоты из хино-
лина окислением азотной кислотой в присутствии серной кислоты
и ванадиевого катализатора 205
Технологические показатели 208
Список использованной литературы 209
Глава 10. Производство синтетической фолиевой кислоты 213
Физико-химические свойства фолиевой кислоты 214
Методы синтеза фолиевой кислоты и выбор рационального метода
для производства 215
Технологическая схема производства синтетической фолиевой кис-
лоты 218
Технологические показатели 226
Список использованной литературы 227
Глава 11. Производство синтетической липоевой кислоты 229
Физико-химические свойства липоевой кислоты 229
435
Методы синтеза липоевой кислоты и выбор рационального метода
для производства 230
Технологическая схема синтеза липоевой кислоты 232
Производные липоевой кислоты 234
Технологические показатели 235
Список использованной литературы 235
Глава 12. Производство синтетической аскорбиновой кислоты 236
Физико-химические свойства аскорбиновой кислоты 237
Методы синтеза аскорбиновой кислоты и выбор рационального мето-
да для производства 243
Технологическая схема производства 244
Производство О-сорбита из D-глюкозы 244
Производство L-сорбозы из D-сорбита 255
Производство диацетон-Д-сорбозы из Д-сорбозы 264
Производство гидрата диацетон-2-кето-Д-гулоновой кислоты 273
Производство Д-аскорбиновой кислоты из гидрата диацетон-2
кето-Д-гулоновой кислоты 281
Производные аскорбиновой кислоты 290
Технологические показатели 291
Список использованной литературы 293
Глава 13. Производство витаминов D2 и D3 297
Физико-химические свойства витаминов D 298
Выбор рационального метода производства эргокальциферола (D2)
и холекальциферола (D3) 303
Технологическая схема производства кристаллического эргокальци.
ферола (витамина D2) 304
Технологическая схема производства холекальциферола — витамин
(D3) 308
Технологические показатели 312
Список использованной литературы 313
Глава 14. Производство синтетического витамина Е (a-то кофер ил ацетата) 315
Физико-химические свойства витамина Е 315
Выбор рационального метода синтеза для производства 317
Технологическая схема производства синтетического а-токоферил-
ацетата 321
Технологические показатели 326
Список использованной литературы 326
Глава 15. Производство синтетических витаминов группы К 327
Физико-химические свойства витаминов группы К 328
Методы синтеза витаминов группы К и выбор рационального метода
для производства 330
Технологическая схема производства витамина К3 (метинона) и ви-
касола 334
Технологические показатели 335
Список использованной литературы 336
Глава 16. Технологическая аппаратура для производства витаминов 337
Аппараты непрерывного действия для химического синтеза витами-
нов 339
Аппараты непрерывного действия для производства витаминных кон-
центратов из природного сырья 352
Новые материалы для изготовления аппаратуры, работающей с аг-
рессивными средами 358
Список использованной литературы 358
436
ЧАСТЬ II. ПРОИЗВОДСТВО ВИТАМИННЫХ ПРЕПАРАТОВ ИЗ ПРИ-
РОДНОГО СЫРЬЯ
Глава 17. Плоды шиповника и комплексная переработка их 360
Химический и витаминный состав плодов шиповника 362
Исследования технологии комплексной переработки плодов шипов-
ника на витаминные препараты 363
Технологическая схема производства витаминных препаратов 364
Технологические показатели 365
Список использованной литературы 369
Глава 18. Плоды облепихи 370
Химический состав плодов облепихи 370
Физико-химические показатели прессового масла из мякоти обле-
пихи 372
Биологически активные вещества сока облепихи 374
Технология комплексного использования плодов облепихи 375
Технологическая схема комплексной переработки плодов облепихи 376
Технологические показатели 378
Список использованной литературы 378
Глава 19. Природные промышленные источники Р-витаминного сырья и про-
изводство из них витаминных препаратов 379
Химия витамина Р 380
Сырье, и технология производства Р-витаминных препаратов 382
Технологическая схема производства витамина Р из отдельных
видов сырья 383
Витамин Р из листьев чая 383
Витамин Р из жома шиповника 385 ,
Витамин Р из черноплодной рябины 385
Витамин Р из черной смородины 386
Витамин Р из зеленой массы гречихи 387
Витамин Р из бадана 388
Витамин Р из отходов цитрусового сокового производства 390
Витамин Р из столовой свеклы 390
Биологически активные вещества рябины обыкновенной 392
Технологические показатели 393
Список использованной литературы 395
Глава 20. Морковь и тыква — промышленное сырье для производства кон-
центратов каротина 397
Морковь 397
Тыква 399
Технологическая схема производства масляного концентрата кароти-
на из моркови 401
Технологическая схема производства концентрата каротина из сило-
сованной тыквы 404
Производство кристаллического p-каротина из моркови 406
Технологические показатели 408
Список использованной литературы 410
Глава 21. Концентраты витамина А из печени рыб 411
Технологическая схема производства концентрата витамина А 412
Производство концентратов витаминов А и Е методом молекулярной
дистилляции 415
Технологические показатели 420
Список использованной литературы 421
437
Глава 22. Производство эргостерина из дрожжей и концентратов витамина D2
для нужд животноводства 422
Сырье для производства витамина D2 422
Технологическая схема производства эргостерина и концентратов ви-
тамина D2 и дрожжей 423
Производство концентрата витаминов группы В 424
Производство концентрата витамина D2 426
Получение концентрата витамина D2 в виде сухих облученных дрож-
жей 429
Технологические показатели 429
Список использованной литературы 430
Глава 23. Природные пищевые биологически активные красители 431
Классификация природных красящих веществ 431
Пищевые биологически активные природные красители, изученные
и освоенные в производстве 432
Список использованной литературы 433
ЛЕВ ОСИПОВИЧ ШНАЙДМАН
ПРОИЗВОДСТВО ВИТАМИНОВ
Редактор И. И. Морозова
Художник Г. Р. Левин
Худож. редактор С. Р. Н а к
Технический редактор Т. С. Пронченкова
Корректор 3. В. Коршунова
Т-01875. Сдано в набор 26/VII 1972 г. Подписано
к печати 18/IV 1973 г. Формат 70X 1081/16. Бумага
книжно-журнальная. Печ. л. 27,5+0,25 вклейка=
27,75. Усл. п. л. 38-85. Уч.-изд. л. 40,11. Тираж
4000 экз. Цена 2 р. 34 к. Заказ 522.
Издательство «Пищевая промышленность»
113035 Москва М-35 1-й-Кадашевский, 12
Ярославский полиграфкомбинат «Союзполиграф-
прома» при Государственном комитете Совета Ми-
нистров СССР по делам издательств, полиграфии
и книжной торговли. Ярославль, ул. Свободы, 97.
Издательство «Пищевая промышленность»
В 1973 Г. ВЫЙДЕТ В СВЕТ И ПОСТУПИТ
В ПРОДАЖУ КНИГА
Березовского В. М. Химия витаминов. Изд. 2-е доп.,
перераб. 50 л., 3000 экз., 5 р. 25 к.,
1-е издание вышло в 1959 г. За период, прошедший
со времени выпуска 1-го издания книги, в области хи-
мии витаминов появилось большое количество иссле-
дований, посвященных уточнению структуры витами-
нов, новым методам их синтеза, изучению их физиче-
ских, химических и биологических свойств, открыты
новые коферменты.
Книга представляет собой монографию, в которой
рассматриваются вопросы химии витаминов в ее со-
временном состоянии.
Книга рассчитана на научных сотрудников и ин-
женеров, работающих в области химии природных ор-
ганических соединений пищевой химии, в фармацев-
тической, витаминной промышленности, на аспирантов
и студентов вузов. Она представит интерес и для спе-
циалистов смежных областей.
Заказы на книгу (без денежных переводов) следу-
ет направлять по адресу:
113035, Москва, М-35
1-й Кадашевский пер., дом 12.
Отдел распространения издательства
Ji - альдегид - C№
Ji-альдегид Ca
IS,>5“-Дегидро - fi- каротин
Транс - Ji- нар a тин
Петро /синий
Толуол
doom
4
дщир Альдегид Хлористый м
серный fytyOr cfg NH^CSметилен НСд
Альдегид 0-муравьи-
~С^ ны Зфцр
ы
Аигроин Ufa HOOj
2Д
вакуум
Pd/СаСО,
Азот
Пар
35
вода
вода.
Азо
Ла
Вануум
РасеарУ ZS
I SO Отход
Отход
45
Отход
Отход
Отход
Г - , Ацетат Ма
"г зтилви- Ацеталь Н20
CHiCOOH
Рассол
1 [на расточение
J гг № мерное 5G
] л Jmuonpone-
Ацгл, чль
S2
Рис. 7. Технологическая схема пронзводства^сиитетического ^-каротина.
J/pup
№
CH-CH
Вода
NoHCO.
\Pd/CaCOj на реге
1J нерацию
дтанол Рассм
Уголь
Лар
Уголь
вода
вода
peep не -
рацию
Толуол
Рассол
Рассол
Потролейный
otpup
2S
Зак. 522
Рассол
Спирт
Натрэнолят -ел-охсиметилен^
~fi~ это ней пропионитрил
Ацилзнялят
NaOH
2—
Голуол
/7~Л
fi~3moxcunponuoHumpun
ЬН'ОИ
Хлористый
ацетил
29
2В
34 =,
Ацетонитрил
Уксусная
\кислота
J 32
33
~/Амниан
нес
40
38
39
Хлоргидрат ацетоининоэ/рира
42 Адсрлют-H^SOf
ыислир
47
Абсолютный
епирт
Зтилат
натрия
Аминепиримидин
Толиол ИОН 10% НОН 50%
Неточна/
'Отход
На ^егенераии/А
отход
88
Вода
30
81
12 13 Ла
10
Азо/П
Зтилформиат
НСООН СгН^ОН Н^ЗОи
Хлоргидрат ацетамидина
мое
152
Лотам.
XzCO.i
62
Рассол
753 154
вакуум
Тиорормамид
у - Хлор -р - ацето пропилацетат
АнтиОироВан-
ный уоаль
- Ацето -у - Зутиролантон
Ацетоунсус-
Летакол нь/и эмир Нстилат
Хлороформ
Ю1
97
Отход
4 -Нетил- 5-в-оксазтилтиамл
Хлоооформ нее
Тиам*
Формами^
Зтилформиат
Хлорметилея
1311 '
Хлораминопиримидин
Абсолютный
спирт
32
33
97
38
ЗУ
п 700 -
Отход
HCZ
дитанин Bj медицинский
Спирт 80Х
ЖГ-
164-
ЛЯ
1S9
734
136^гу
горя-^
ч ая Вода
НагЗОч
"осячая Хода
Вода
116
Дихлорзтан\
113
121
дихлорэтан Унсуснь/й
»---£— Уксусная НСВ ангидрид
X. наслал ‘ *
124-
117
129
С& 121^
Чодй
122.
/о/.
123727 128
Витамин St технически#
Толуол
Спирт
Na регенерацию
Спирт
^ктиойроВанный уголь
[1 АнтиОиробанный —
170\
уголь
178
I
'183
Лар
167
tea
дата'
174 175
Рнс. 9. Технологическая схема производства тиаминхлорида (вариант 1).
•^172
°асеол
173
177
,179
icea.
134
'Рассол
рирп
180
185
,186
3 реантор
170
Omxtfi