/


























































































































































































































































































































































































































































































































































































































































Автор: Фрешни Р.Я.
Теги: биологическая техника, экспериментальные методы и оборудование в целом общая биология биология
ISBN: 978-5-94774-596-2
Год: 2010




Текст
R. Jan Freshney
CULTURE
OF ANIMAL CELLS
A MANUAL OF BASIC TECHNIQUE
Fifth Edition
RJan Freshney
Cancer Research UK Centre for Oncology and Applied Pharmacology
Cancer Research UK Beatson Laboratories
University of Glasgow
©WILEY-LI SS
A JOHN WILEY & SONS, INC., PUBLICATION
Р. Я.Фрешни
КУЛЬТУРА
животных
КЛЕТОК
ПРАКТИЧЕСКОЕ РУКОВОДСТВО
Перевод с 5-го английского издания
д-ра биол. наук Ю. Н. Хомякова,
канд. мед. наук Т. И. Хомяковой
Москва
БИНОМ. Лаборатория знаний
2010
УДК 57.08
ББК 28.03
Ф87
Фрешни Р. Я.
Ф87 Культура животных клеток : практическое руководство /
Р. Я. Фрешни ; пер. 5-го англ. изд. — М. : БИНОМ. Лаборатория
знаний, 2010. — 691 с. : ил., [24] с. цв. вкл.
ISBN 978-5-94774-596-2
Учебное издание, написанное ведущим специалистом в данной области,
содержит наиболее полное описание теоретических основ и практических
приемов работы с культурами животных клеток, а также необходимого оборудо¬
вания, включая лабораторный дизайн. В достаточном объеме освещены вопросы
техники безопасности. Подробно обсуждаются методика подготовки сред, приемы
работы с первичной культурой и клеточными линиями. Описано специальное
оборудование, в том числе для манипуляций с культурами животных клеток.
Книга прекрасно иллюстрирована и удобна для использования как практическое
руководство в лаборатории.
Для студентов-биологов, биотехнологов, медиков, а также исследователей,
специалистов биофармацевтических центров и сотрудников диагностических
лабораторий.
УДК 57.08
ББК 28.03
По вопросам приобретения обращаться:
«БИНОМ. Лаборатория знаний»
Телефон: (499)157-5272
e-mail: binom@Lbz.ru, http://www.Lbz.ru
Copyright © 2005 by John Wiley & Sons. Inc.
Все права защищены. Перевод
с английского языка по договору.
ISBN 978-5-94774 596~2 © БИНОМ. Лаборатория знании, 2010
Оглавление
Список рисунков 15
Список цветных вкладок 18
Предисловие 19
Список сокращений 21
ГЛАВА 1. ВВЕДЕНИЕ 25
1.1. История вопроса 25
1.2. Преимущества метода культуры ткани 30
1.2.1. Контроль окружения 30
1.2.2. Характеристика и однородность
образца 31
1.2.3. Экономичность, эффективность
и автоматизация процесса 31
1.2.4. Моделирование in vitro условий
in vivo 31
1.3. Ограничения метода культуры ткани 31
1.3.1. Наличие специальных навыков 31
1.3.2. Затраты 32
1.3.3. Дифференцировка и селекция 32
1.3.4. Происхождение клеток 32
1.3.5. Нестабильность 32
1.4. Основные отличия культуры in vitro 32
1.5. Типы культуры ткани 33
ГЛАВА 2. ТРЕНИРОВОЧНЫЕ
ПРОГРАММЫ 36
2.1. Цели и задачи главы 36
2.2. Упражнения для начинающих
(основной курс) 36
Упражнение 1. Асептический метод I:
Пипетирование и перенос жидкостей 38
Упражнение 38
Упражнение 2. Введение в технику ведения
культуры ткани 39
Упражнение 39
Упражнение 3. Асептический метод II:
Приготовление питательной среды 40
Упражнение 41
Упражнение 4. Смена среды в монослойной
культуре 41
Упражнение 41
Упражнение 5. Мытье и стерилизация
стеклянной посуды 42
Упражнение 42
Упражнение 6. Подготовка и стерилизация воды 42
Упражнение 42
Упражнение 7. Подготовка и стерилизация
фосфатно-буферного раствора для среды
Дюльбекко (D-PBS) без Са?*и Mg2* 43
Упражнение 43
Упражнение 8. Приготовление стандартных
растворов для контроля pH 43
Упражнение 43
Упражнение 9. Приготовление основной
питательной среды из порошка
и стерилизация методом фильтрования 44
Упражнение 44
Упражнение 10. Приготовление полной среды
из концентрата 10х 44
Упражнение ‘45
Упражнение 11. Приготовление полной среды
из порошка 45
Упражнение 12. Подсчет клеток в гемоцитометре
и электронном счетчике 45
Упражнение 46
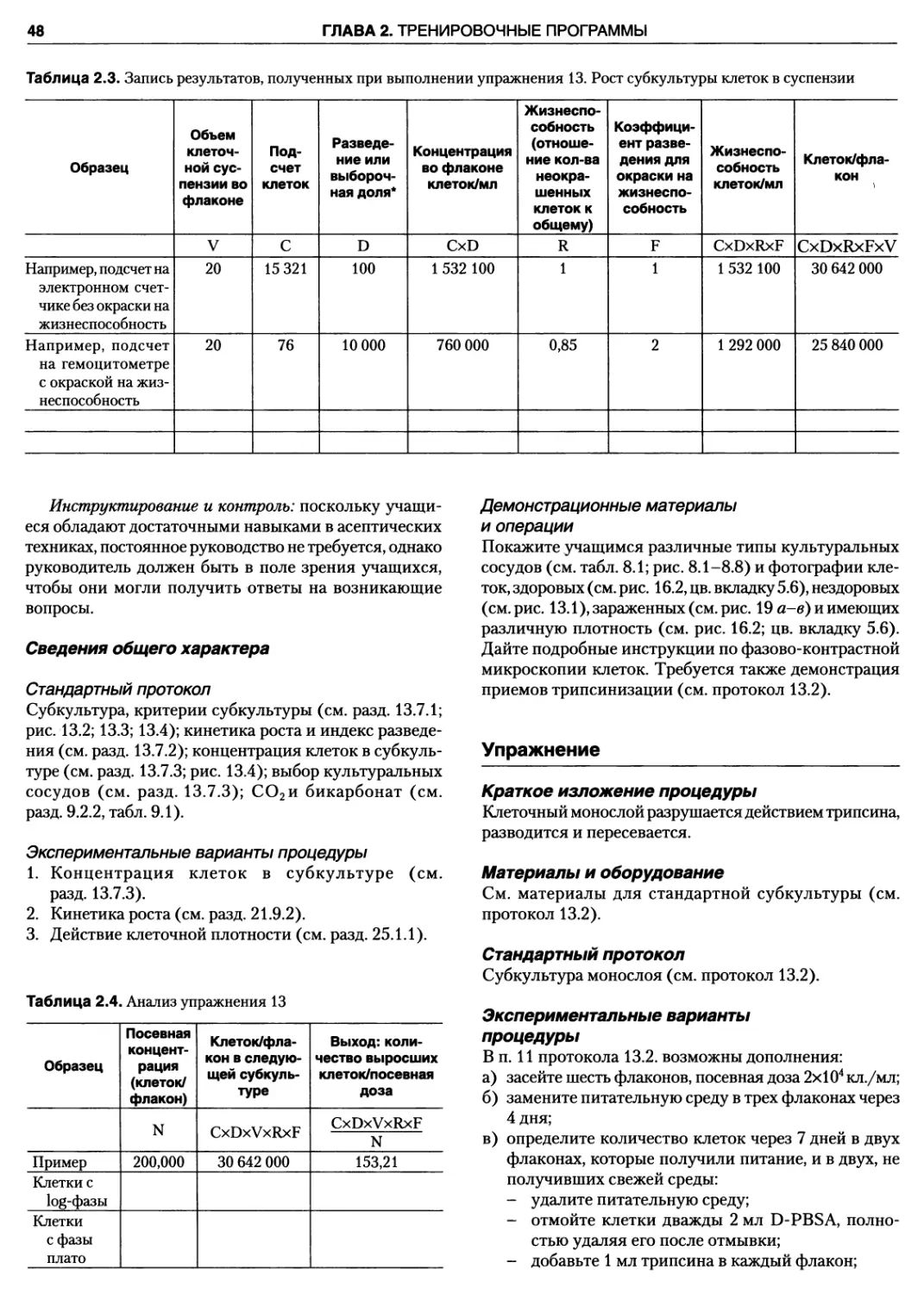
Упражнение 13. Рост субкультуры клеток
в суспензии 46
Упражнение 47
Упражнение 14. Рост субкультуры постоянной
клеточной линии в монослое 47
Упражнение 48
Упражнение 15. Окрашивание монослойной
клеточной культуры по Гимза 49
Упражнение 50
Упражнение 16. Построение и анализ
кривой роста 50
Упражнение 50
2.3. Упражнения повышенной сложности 51
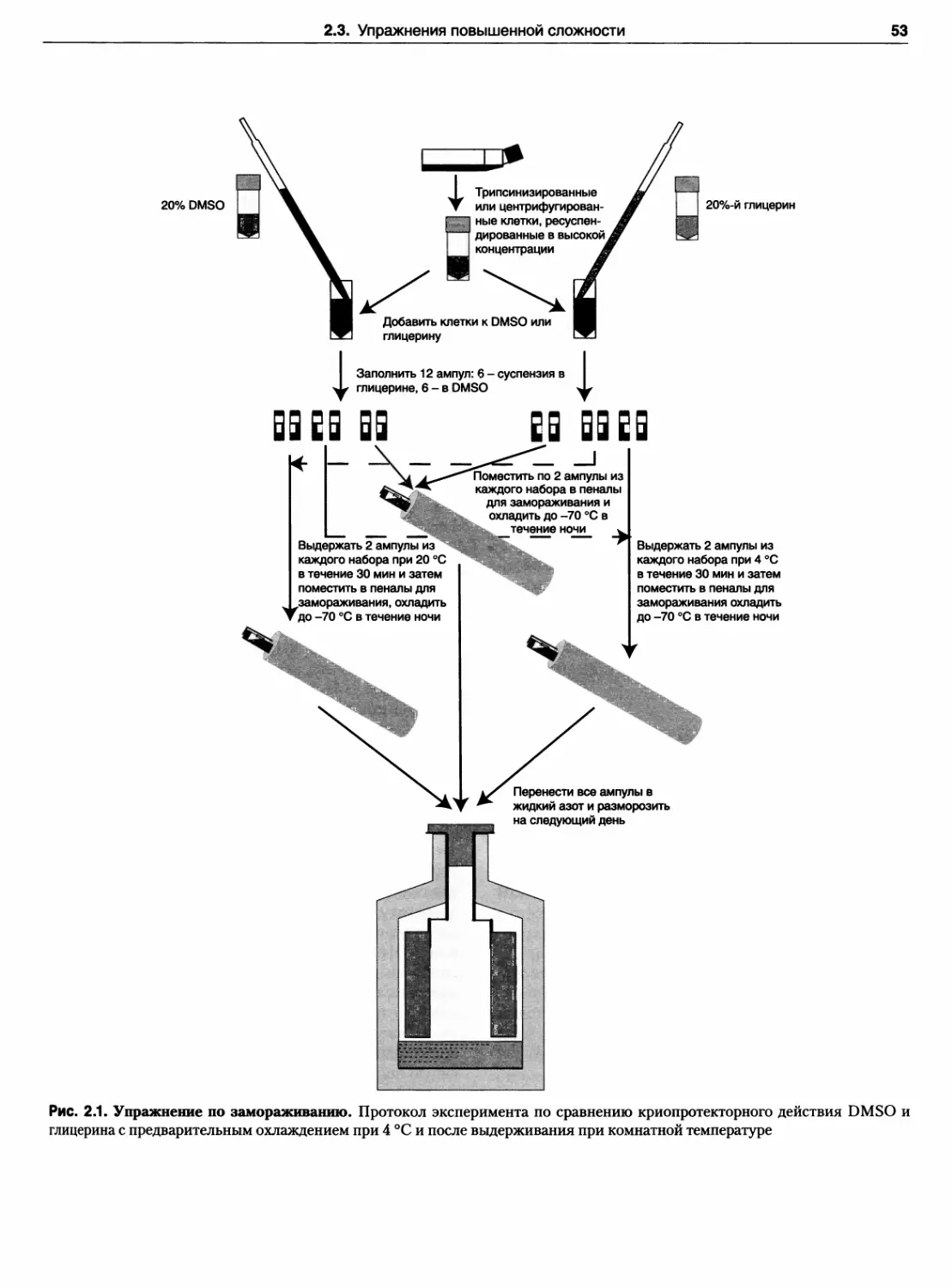
Упражнение 17. Криоконсервация
культивируемых клеток 51
Упражнение 52
Упражнение 18. Обнаружение микоплазм 52
Упражнение 54
Упражнение 19. Характеристика клеточных
линий 54
Упражнение 55
Упражнение 20. Первичная культура 55
Упражнение 56
Упражнение 21. Клонирование в монослое 56
Упражнение 57
2.4. Специализированные упражнения 57
ГЛАВА 3. БИОЛОГИЯ КУЛЬТИВИРУЕМЫХ
КЛЕТОК 58
3.1. Влияние окружающей среды
на культуру клеток 58
3.2. Клеточная адгезия 58
3.2.1. Молекулы клеточной адгезии 58
3.2.2. Межклеточные контакты 59
3.2.3. Внеклеточный матрикс 60
3.2.4. Цитоскелет 61
3.2.5. Клеточная подвижность 61
3.3. Клеточная пролиферация 62
3.3.1. Клеточный цикл 62
3.3.2. Контроль клеточной пролиферации... 62
3.4. Дифференцировка 63
6
Оглавление
3.4.1. Поддержание и индукция
дифференцировки 63
3.4.2. Дедифференцировка 64
3.5. Передача клеточных сигналов 66
3.6. Энергетический метаболизм 67
3.7. Получение первичной культуры 67
3.8. Эволюция клеточных линий 68
3.8.1. Старение 68
3.9. Возникновение постоянных
клеточных линий 68
3.10. Происхождение культивируемых клеток... 70
ГЛАВА 4. СТРУКТУРА И ПЛАНИРОВАНИЕ
ЛАБОРАТОРНЫХ ПОМЕЩЕНИЙ 72
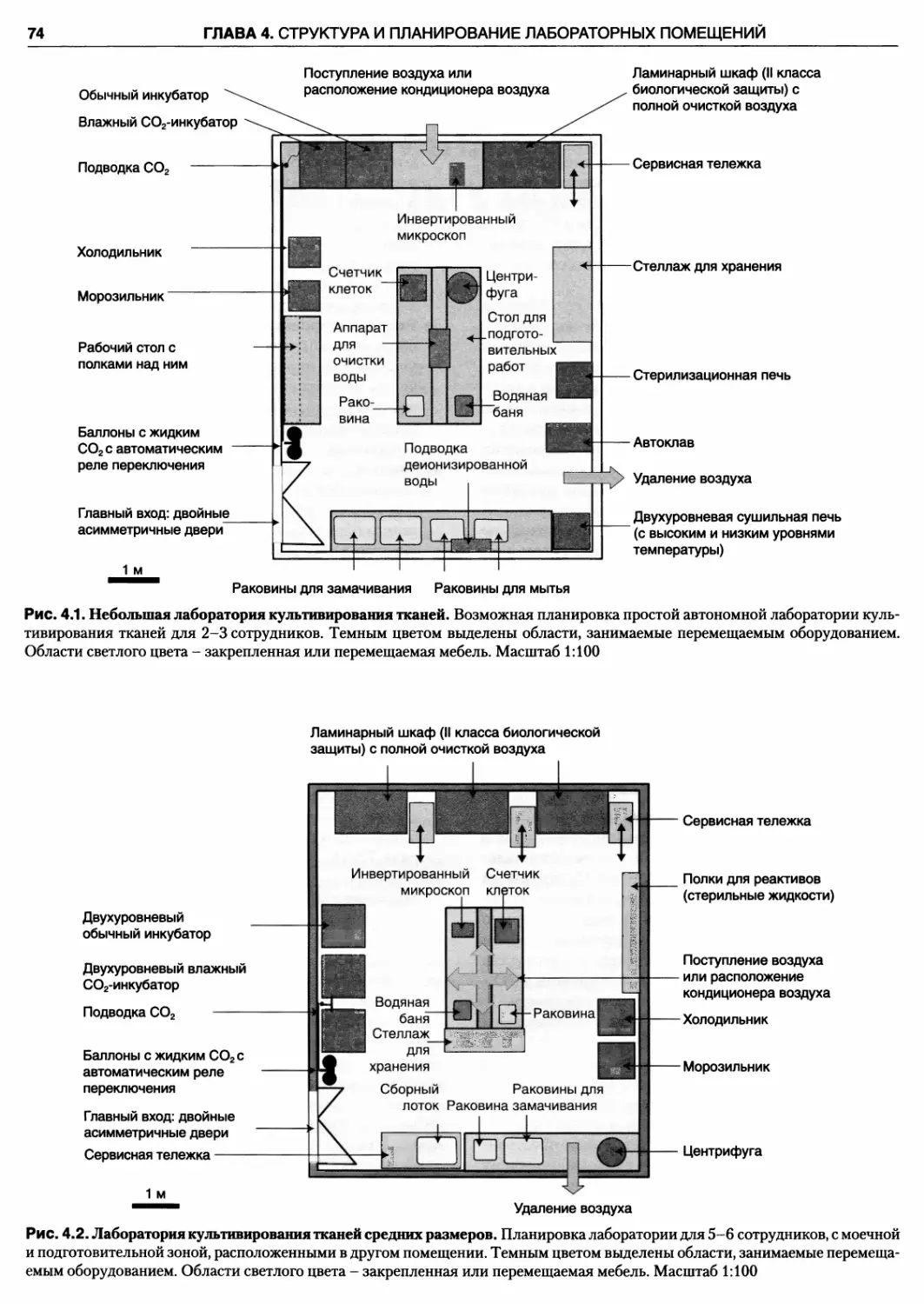
4.1. Планирование 72
4.2. Обслуживающие системы
и вспомогательные службы 76
4.3. Планирование асептических комнат
или блоков 76
4.3.1. Помещение для стерильных
манипуляций 77
4.3.2. Ламинарное оборудование 77
4.3.3. Карантин и изоляция 78
4.3.4. Помещения для сервисных служб 78
4.4. Инкубация 78
4.4.1. Инкубаторы 78
4.4.2. Термальная комната 78
4.5. Помещения для подготовительных работ... 81
4.5.1. Приготовление сред 81
4.5.2. Помещение для мытья посуды 81
4.5.3. Хранение 82
ГЛАВА 5. ОБОРУДОВАНИЕ 85
5.1. Потребности лаборатории культур тканей .. 85
5.2. Асептическая зона 85
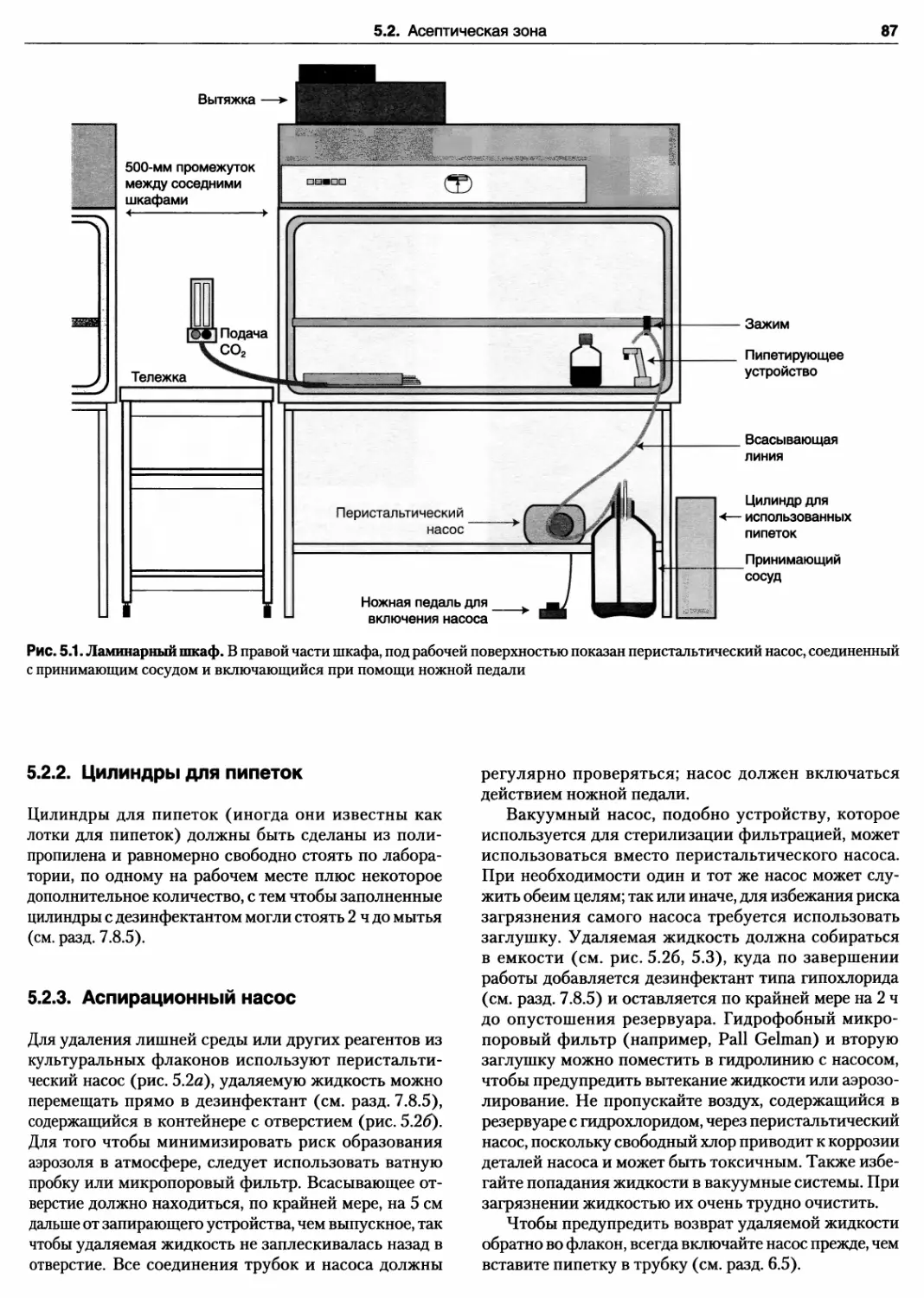
5.2.1. Ламинарные шкафы 85
5.2.2. Цилиндры для пипеток 87
5.2.3. Аспирационный насос 87
5.2.4. Сервисные тележки 88
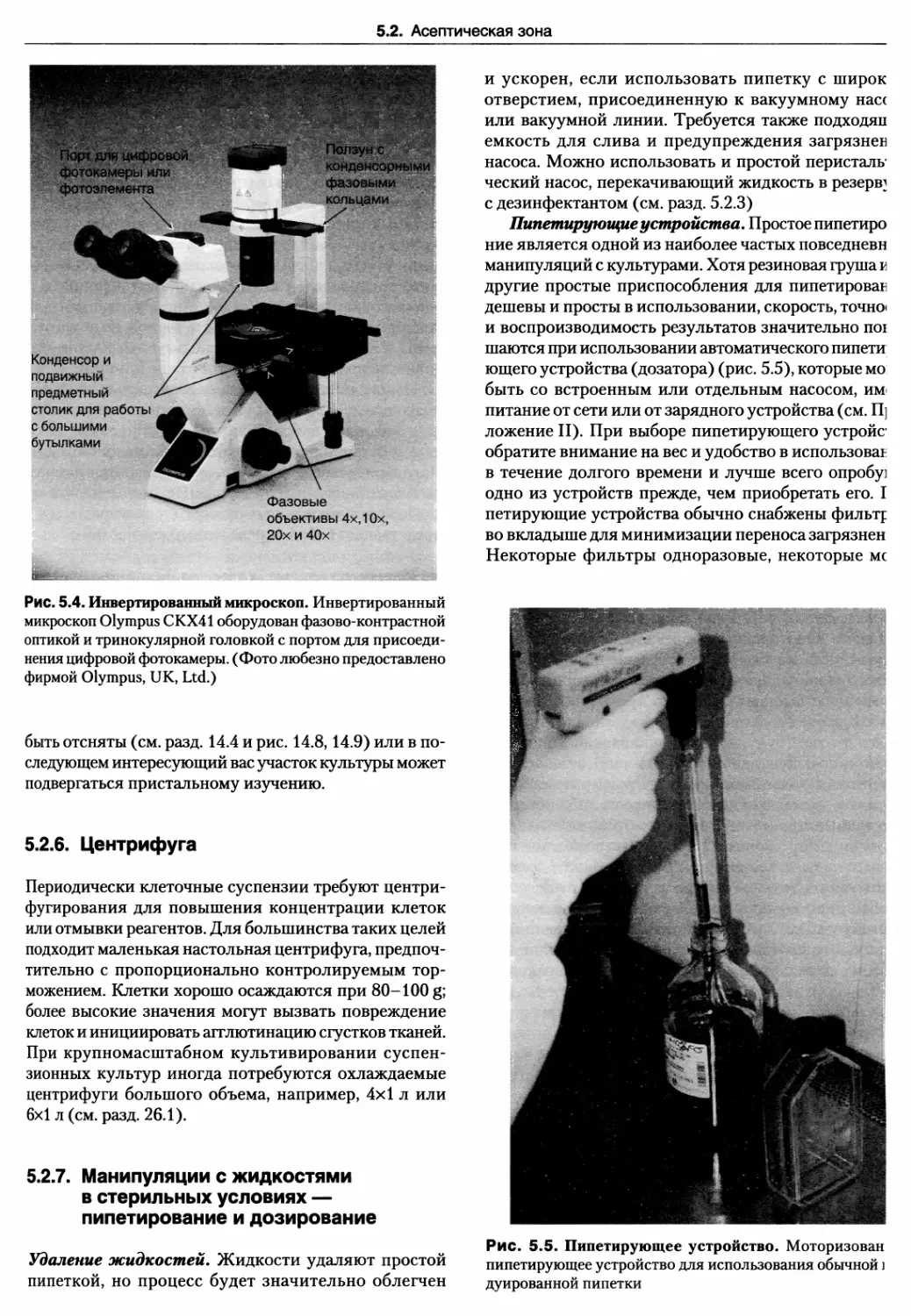
5.2.5. Инвертированный микроскоп 88
5.2.6. Центрифуга 89
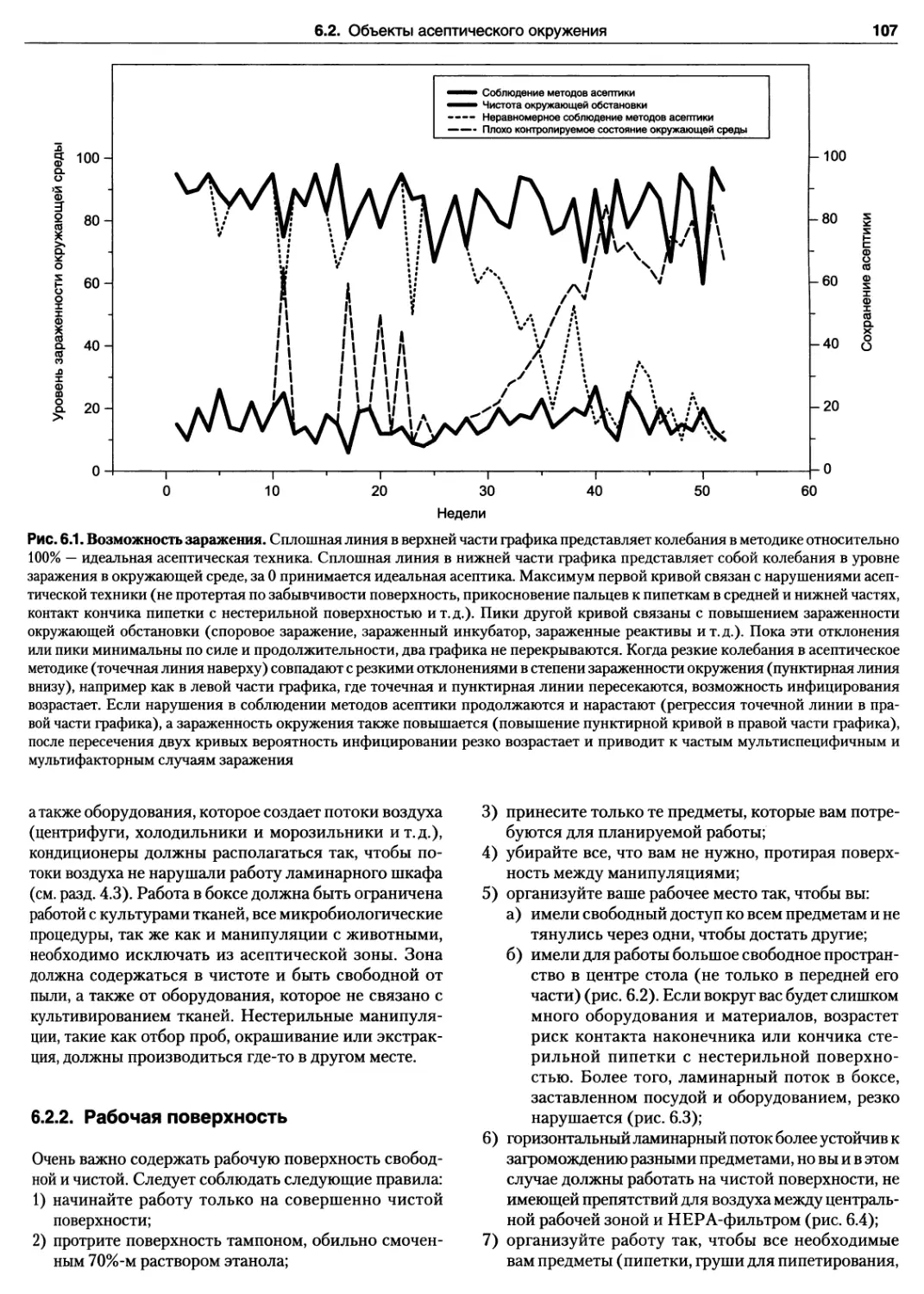
5.2.7. Манипуляции с жидкостями
в стерильных условиях —
пипетирование и дозирование 89
5.2.8. Счетчик клеток 92
5.2.9. CCD-камера и монитор 93
5.2.10. Препаровальная лупа 93
5.3. Инкубация 94
5.3.1. Инкубатор 94
5.3.2. Влажный С02-инкубатор 94
5.3.3. Регистратор температуры 95
5.3.4. Роллерные штативы 96
5.3.5. Магнитная мешалка 96
5.4. Подготовительные работы
и стерилизация 96
5.4.1. Мытье посуды 96
5.4.2. Очиститель воды 96
5.4.3. Стерилизация и сушильные шкафы ... 97
5.4.4. Паровой стерилизатор (автоклав) 98
5.4.5. Весы 98
5.4.6. рН-Метр 99
5.4.7. Магнитная мешалка с подогревом 99
5.4.8. Автоматический диспенсер 100
5.4.9. Измеритель электропроводности
(кондуктометр) 100
5.4.10. Осмометр 100
5.4.11. Моечная машина для стеклянной
посуды 100
5.5. Хранение 101
5.5.1. Холодильники и морозильники 101
5.5.2. Контейнеры для криоконсервации 102
5.5.3. Морозильники с управляемым
процессом замораживания 102
5.6. Лабораторные приборы 102
5.6.1. Компьютеры и сети 102
5.6.2. Прямой микроскоп 102
5.6.3. Низкотемпературный морозильник ... 102
5.6.4. Конфокальный микроскоп 103
5.6.5. Термоциклер 103
5.7. Специальное оборудование 103
5.7.1. Установка для микроинъекций 103
5.7.2. Счетчик колоний 103
5.7.3. Центрифужный элютриатор 104
5.7.4. Проточный цитометр 104
5.8. Расходные материалы 104
5.8.1. Пипетки 104
5.8.2. Гемоцитометр 104
5.8.3. Культуральные сосуды 105
5.8.4. Стерильные контейнеры 105
5.8.5. Шприцы и иглы 105
5.8.6. Стерилизационные фильтры 105
5.8.7. Бумажные полотенца и салфетки 105
5.8.8. Дезинфектанты 105
ГЛАВА 6. МЕТОДЫ АСЕПТИКИ 106
6.1. Цели асептики 106
6.1.1. Поддержание стерильности 106
6.2. Объекты асептического окружения 106
6.2.1. Стерильная зона 106
6.2.2. Рабочая поверхность 107
6.2.3. Личная гигиена 108
6.2.4. Реагенты и среды 109
6.2.5. Культуры 109
6.3. Стерилизующие манипуляции 109
6.3.1. Протирание поверхностей 109
6.3.2. Закрывание емкостей 110
6.3.3. Работа с горелкой 110
6.3.4. Манипуляции с бутылками
и колбами 110
6.3.5. Пипетирование 110
6.3.6. Переливание 112
6.4. Ламинарный поток 112
6.5. Стандартная процедура 113
6.5.1. Чашки Петри и многолуночные
планшеты 113
6.6. Приборы и оборудование 114
6.6.1. Инкубаторы 114
Протокол 6.1. Асептические методы работы
в вертикальном ламинарном потоке 115
Протокол 6.2. Работа на открытой поверхности... 116
Протокол 63. Манипуляции с чашками
и планшетами 118
6.6.2. Коробки для влажного инкубатора 119
6.6.3. Культивирование в атмосфере С02 ... 119
ГЛАВА 7. БЕЗОПАСНОСТЬ, БИОЭТИКА
И ВАЛИДАЦИЯ 120
7.1. Лабораторная безопасность 120
7.2. Оценка риска 120
Оглавление
7
7.3. Стандартные операционные процедуры 120
7.4. Контроль безопасности 120
7.5. Общая безопасность 122
7.5.1. Оператор 122
7.5.2. Оборудование 122
7.5.3. Стеклянные и острые предметы 123
7.5.4. Химическая токсичность 124
7.5.5. Газы 125
7.5.6. Жидкий азот 126
7.5.7. Ожоги 126
7.6. Пожар 126
7.7. Ионизирующее излучение 126
7.7.1. Попадание в организм 127
7.7.2. Утилизация радиоактивных отходов .. 127
7.7.3. Излучение от меченых реагентов 127
7.7.4. Излучение от высокоэнергетичных
источников 127
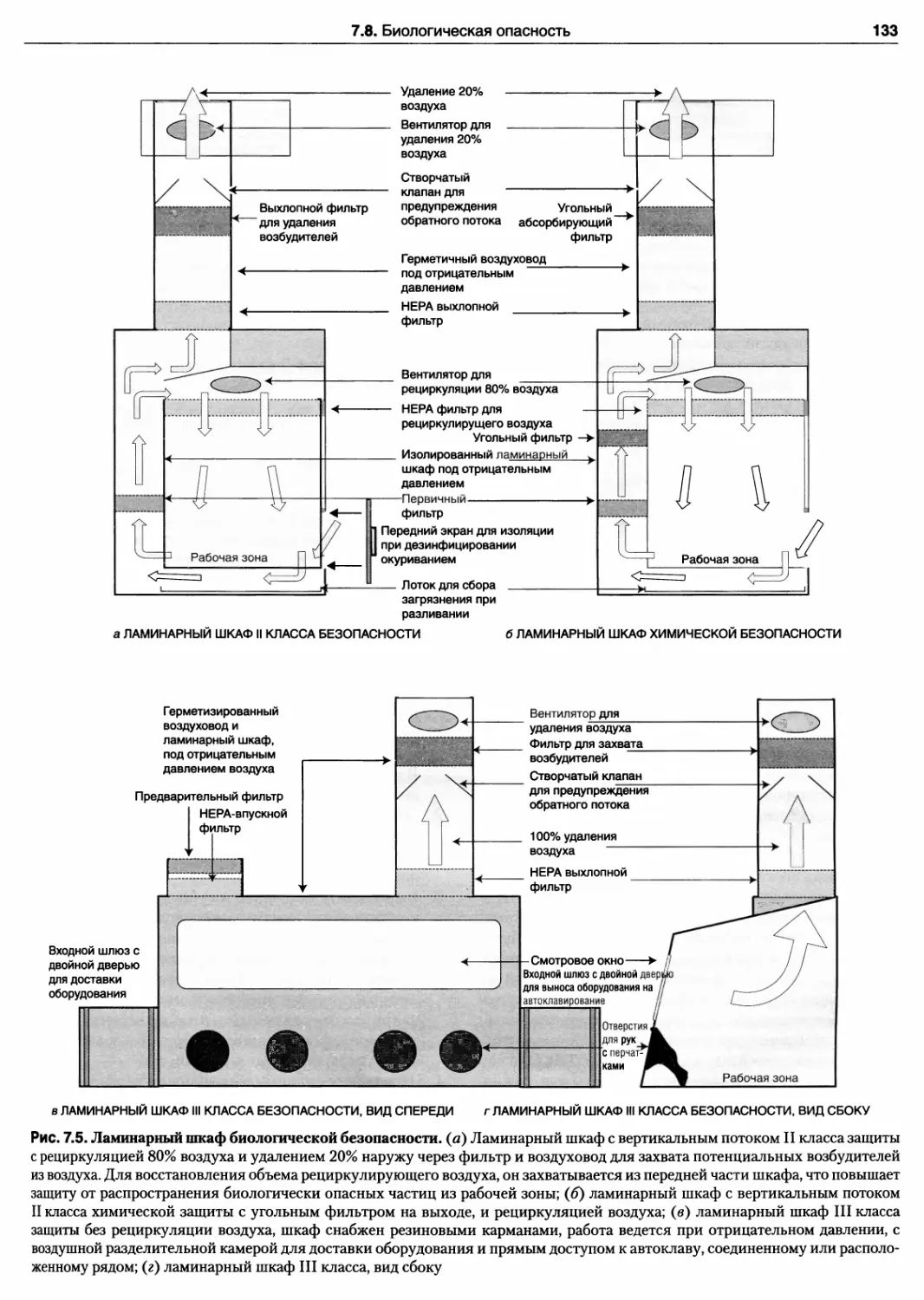
7.8. Биологическая опасность 129
7.8.1. Уровни биологической защиты 129
7.8.2. Ламинарные шкафы
микробиологической защиты 129
7.8.3. Человеческий биопсийный материал.. 131
7.8.4. Манипуляции с генетическим
материалом 135
7.8.5. Уничтожение биологически
опасных отходов 136
7.8.6. Дезинфекция окуриванием 136
7.9. Биоэтика 136
7.9.1. Ткани животных 136
7.9.2. Ткани человека 136
7.10. Валидация 137
7.10.1. Подтверждение аутентичности 138
7.10.2. Происхождение 138
7.10.3. Контаминация 138
ГЛАВА 8. ПОСУДА И СУБСТРАТЫ
ДЛЯ КУЛЬТИВИРОВАНИЯ КЛЕТОК 139
8.1. Субстрат 139
8.1.1. Прикрепление и рост клеток 139
8.1.2. Материалы 139
8.2. Выбор сосудов для культивирования
клеток 139
8.2.1. Клеточный выход в культуральном
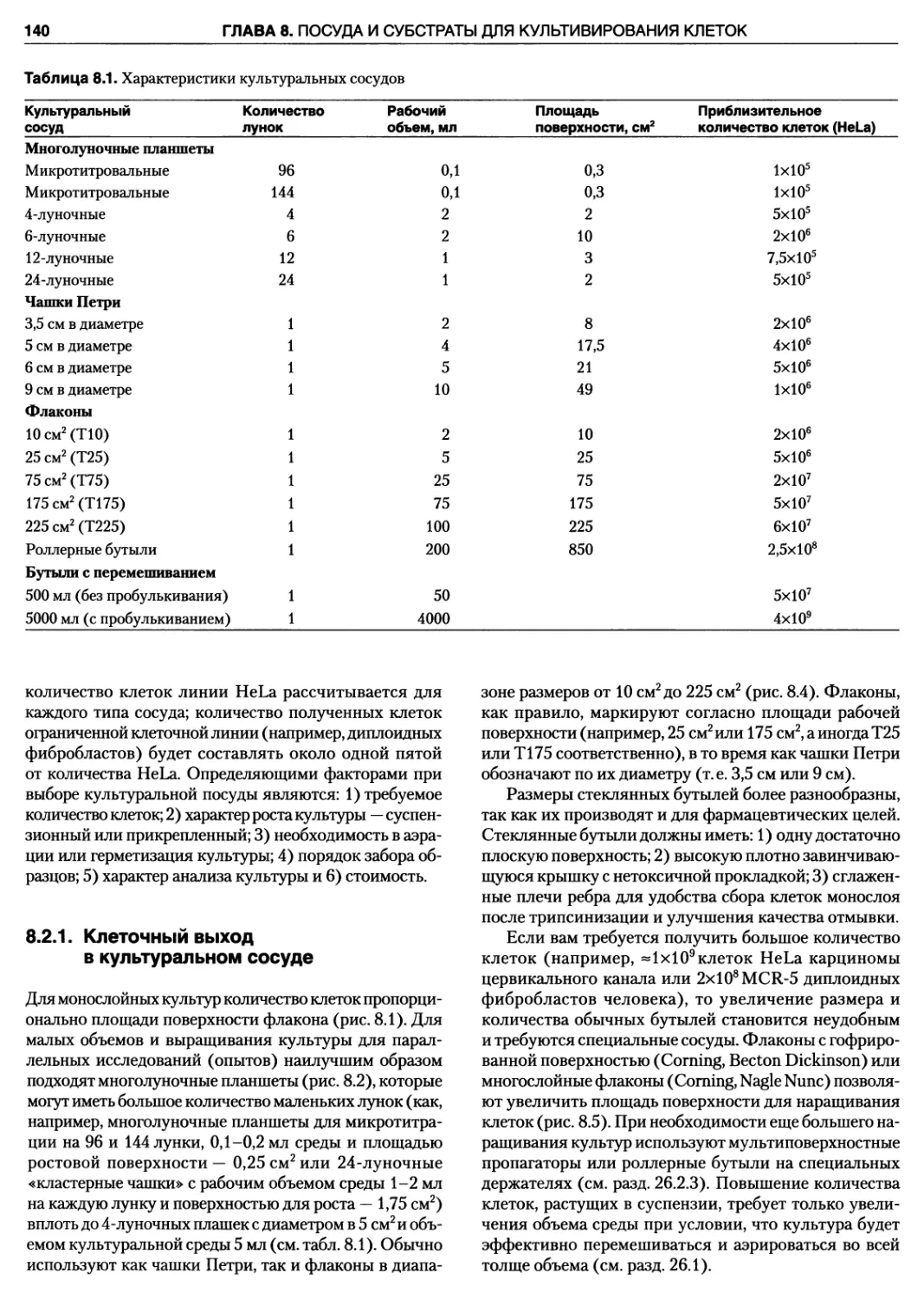
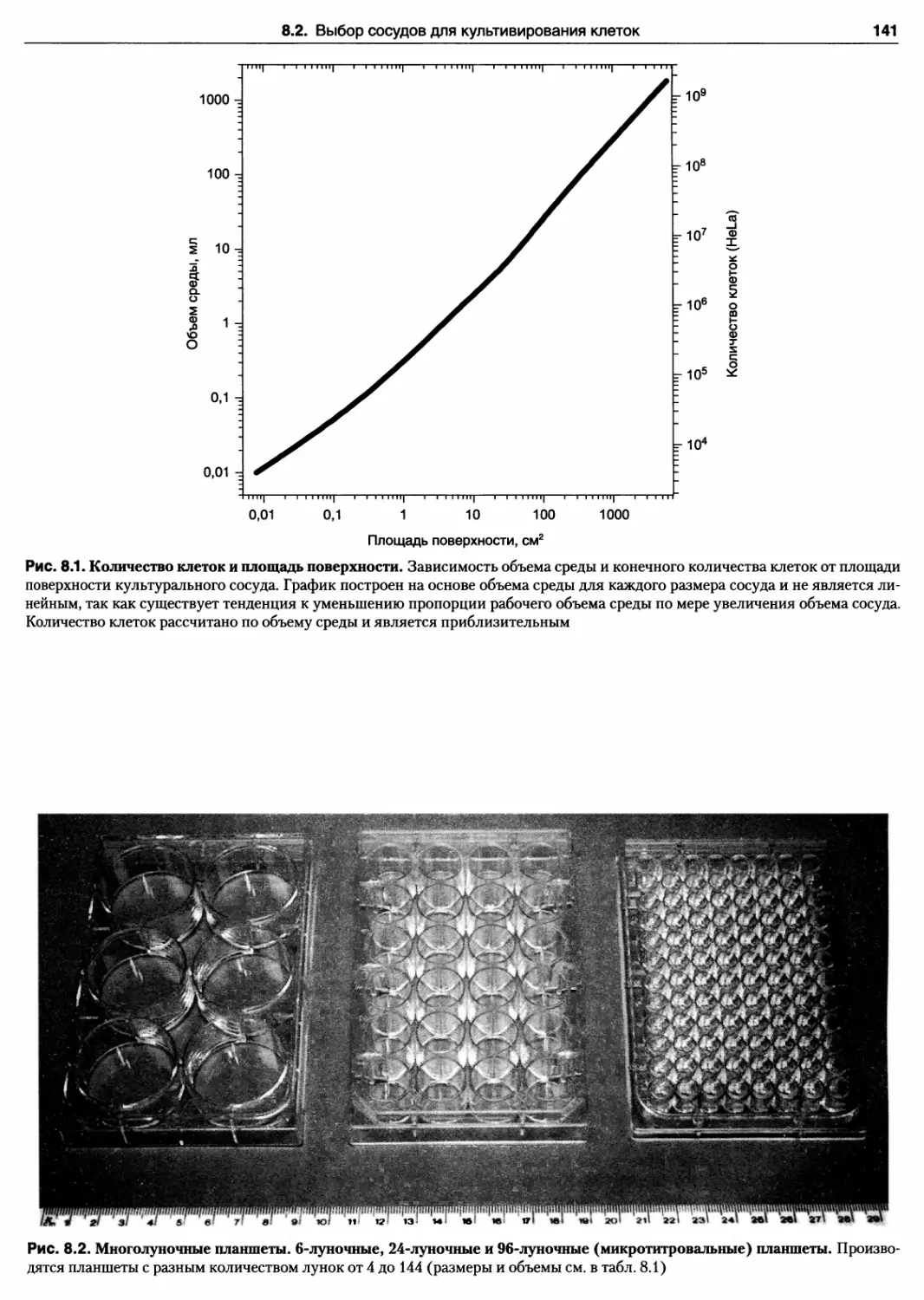
сосуде 140
8.2.2. Суспензионная культура 142
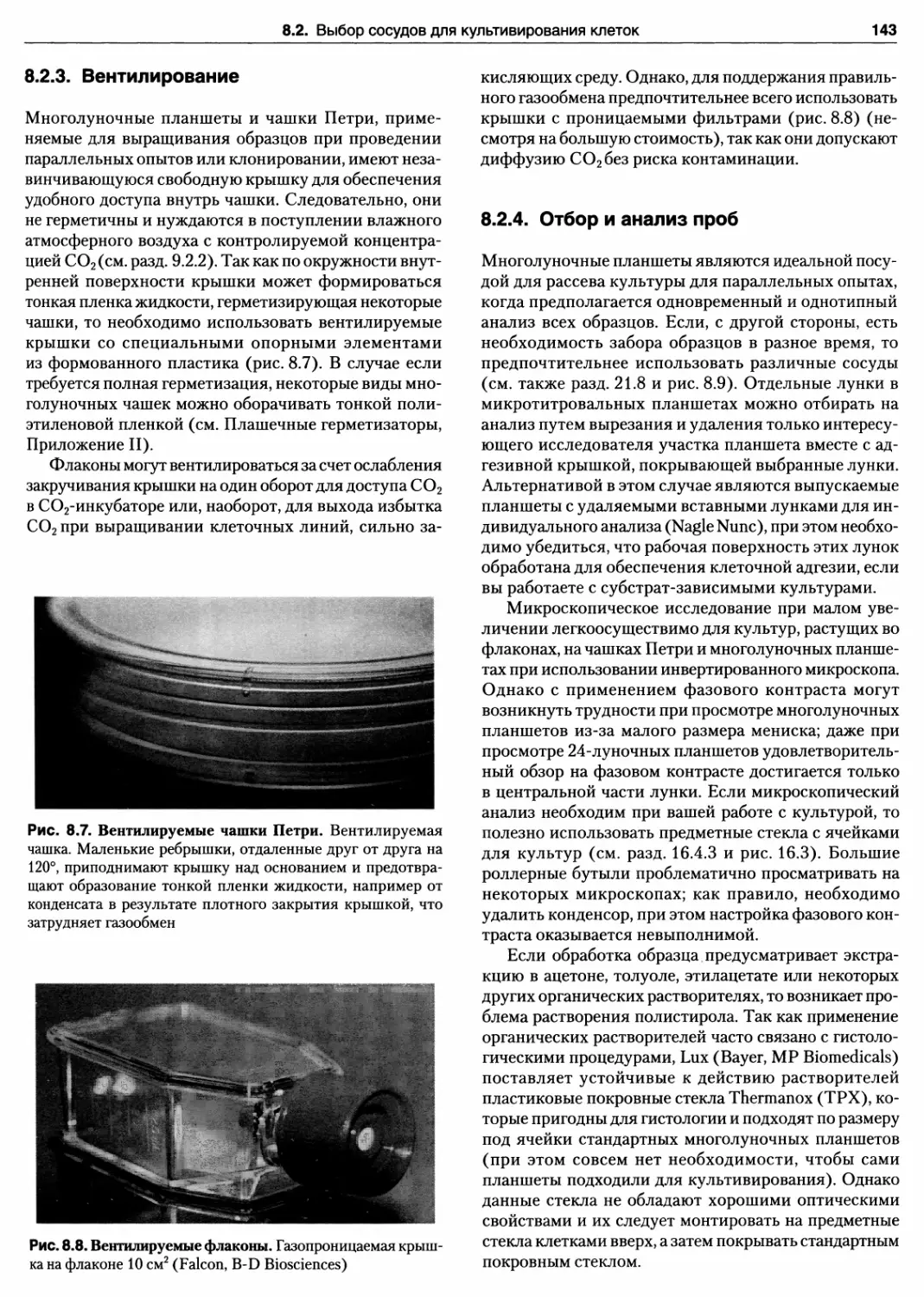
8.2.3. Вентилирование 143
8.2.4. Отбор и анализ проб 143
8.2.5. Неравномерный рост 144
8.2.6. Стоимость 144
8.3. Специализированные системы 145
8.3.1. Проницаемые подложки 145
8.4. Обработка поверхности 145
8.4.1. Покрытие матриксом 145
Протокол 8.1. Приготовление ВКМ 146
8.4.2. Фидерные слои 146
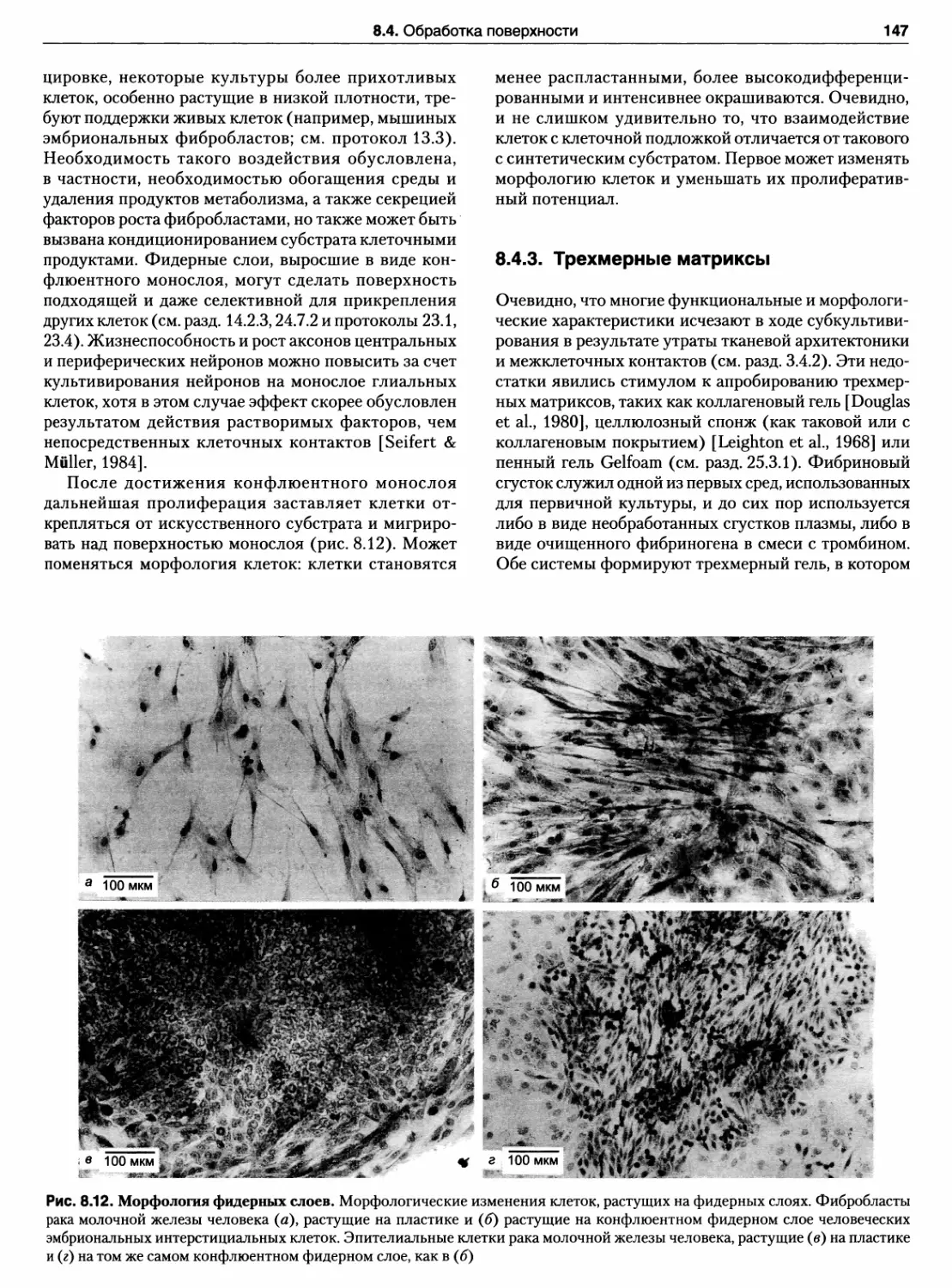
8.4.3. Трехмерные матриксы 147
8.4.4. Металлические субстраты 148
8.4.5. Неадгезивные субстраты 148
ГЛАВА 9. СРЕДЫ ОПРЕДЕЛЕННОГО
ХИМИЧЕСКОГО СОСТАВА
И ДОБАВКИ К СРЕДАМ 149
9.1. Составление сред 149
9.2. Физико-химические свойства 149
9.2.1. pH 149
Протокол 9.1. Приготовление стандартов PH 150
9.2.2. С02 и бикарбонат натрия 150
9.2.3. Буферизация 151
9.2.4. Кислород 152
9.2.5. Осмотическое давление 152
9.2.6. Температура 153
9.2.7. Вязкость 154
9.2.8. Поверхностное натяжение
и пенообразование 154
9.3. Сбалансированные солевые растворы 154
9.4. Полные питательные среды 154
9.4.1. Аминокислоты 155
9.4.2. Витамины 155
9.4.3. Соли 155
9.4.4. Глюкоза 155
9.4.5. Органические добавки 158
9.4.6. Гормоны и факторы роста 158
9.4.7. Антибиотики 158
9.5. Сыворотка 159
9.5.1. Белки 159
9.5.2. Факторы роста 160
9.5.4. Питательные вещества и метаболиты.. 160
9.5.5. Липиды 160
9.5.6. Неорганические элементы 160
9.5.7. Ингибиторы 160
9.6. Выбор среды и сыворотки 160
9.6.1. Резервирование партии сыворотки 161
9.6.2. Тестирование сыворотки 162
9.6.3. Тепловая инактивация 163
9.7. Другие добавки 163
9.7.1. Гидролизат аминокислот 163
9.7.2. Эмбриональный экстракт 163
9.7.3. Кондиционированная среда 163
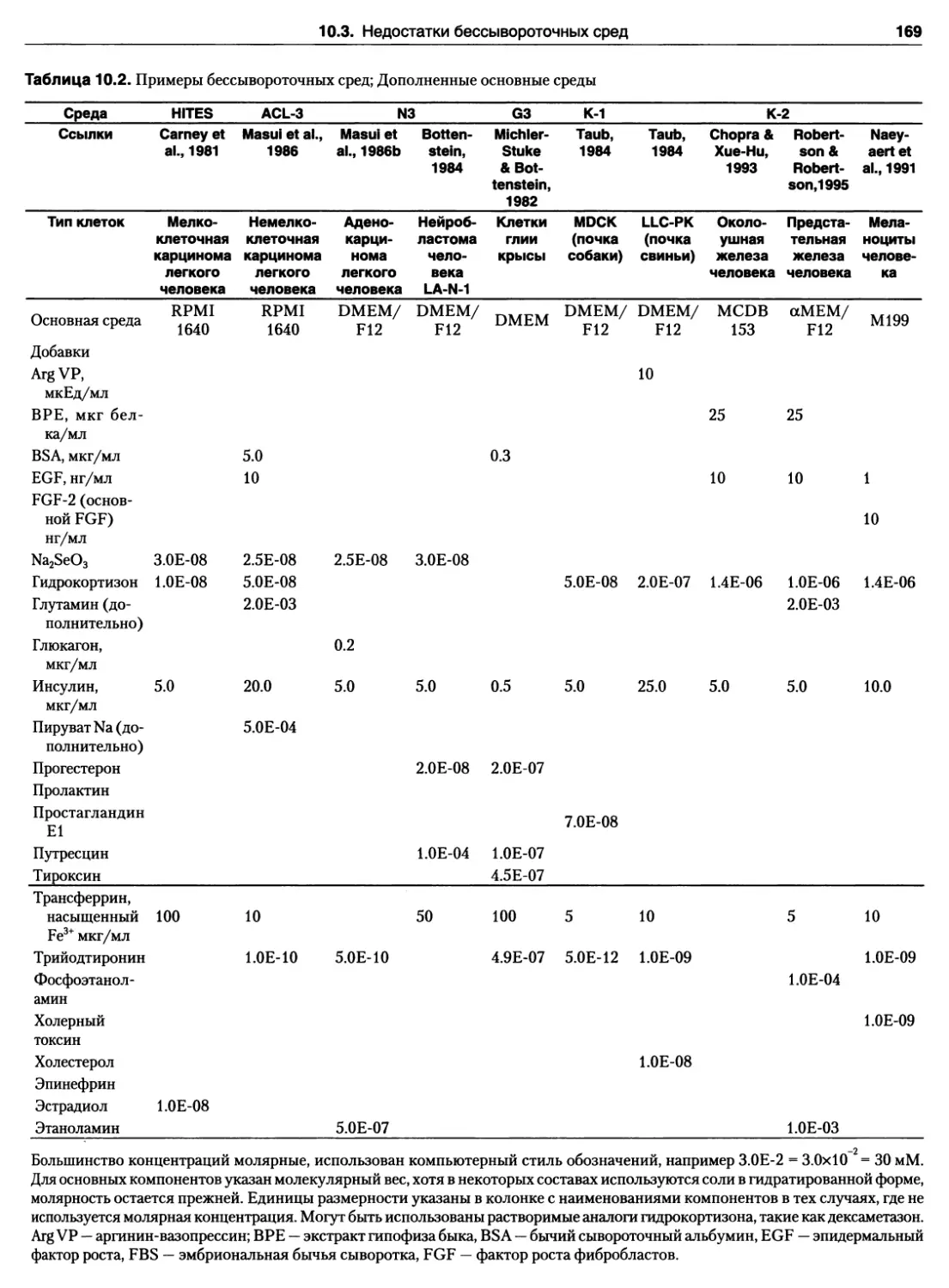
ГЛАВА 10. БЕССЫВОРОТОЧНЫЕ СРЕДЫ . . . 164
10.1. Недостатки сыворотки 164
10.2. Преимущества бессывороточных сред 168
10.2.1. Селективные среды 168
10.2.2. Регуляция пролиферации
и дифференцировки 168
10.3. Недостатки бессывороточных сред 168
10.4. Замена сыворотки 170
10.4.1. Компоненты для бессывороточного
культивирования 170
10.4.2. Гормоны 170
10.4.3. Факторы роста 171
10.4.4. Питательные вещества сыворотки ... 171
10.4.5. Белки и полиамины 171
10.4.6. Матрикс 171
10.5. Выбор бессывороточной среды 171
10.5.1. Имеющиеся в продаже
бессывороточные среды 176
10.5.2. Заменители сыворотки 176
10.5.3. Адаптация клеток
к бессывороточной среде 176
10.6. Разработка бессывороточной среды 178
10.7. Приготовление бессывороточной среды 179
10.8. Среды, не содержащие белков 179
10.9. Заключение 179
ГЛАВА 11. ПОДГОТОВИТЕЛЬНЫЕ
РАБОТЫ И СТЕРИЛИЗАЦИЯ 180
11.1. Приготовление реактивов
и материалов 180
8
Оглавление
11.2. Стерилизация оборудования
и жидкостей 180
11.3. Оборудование 180
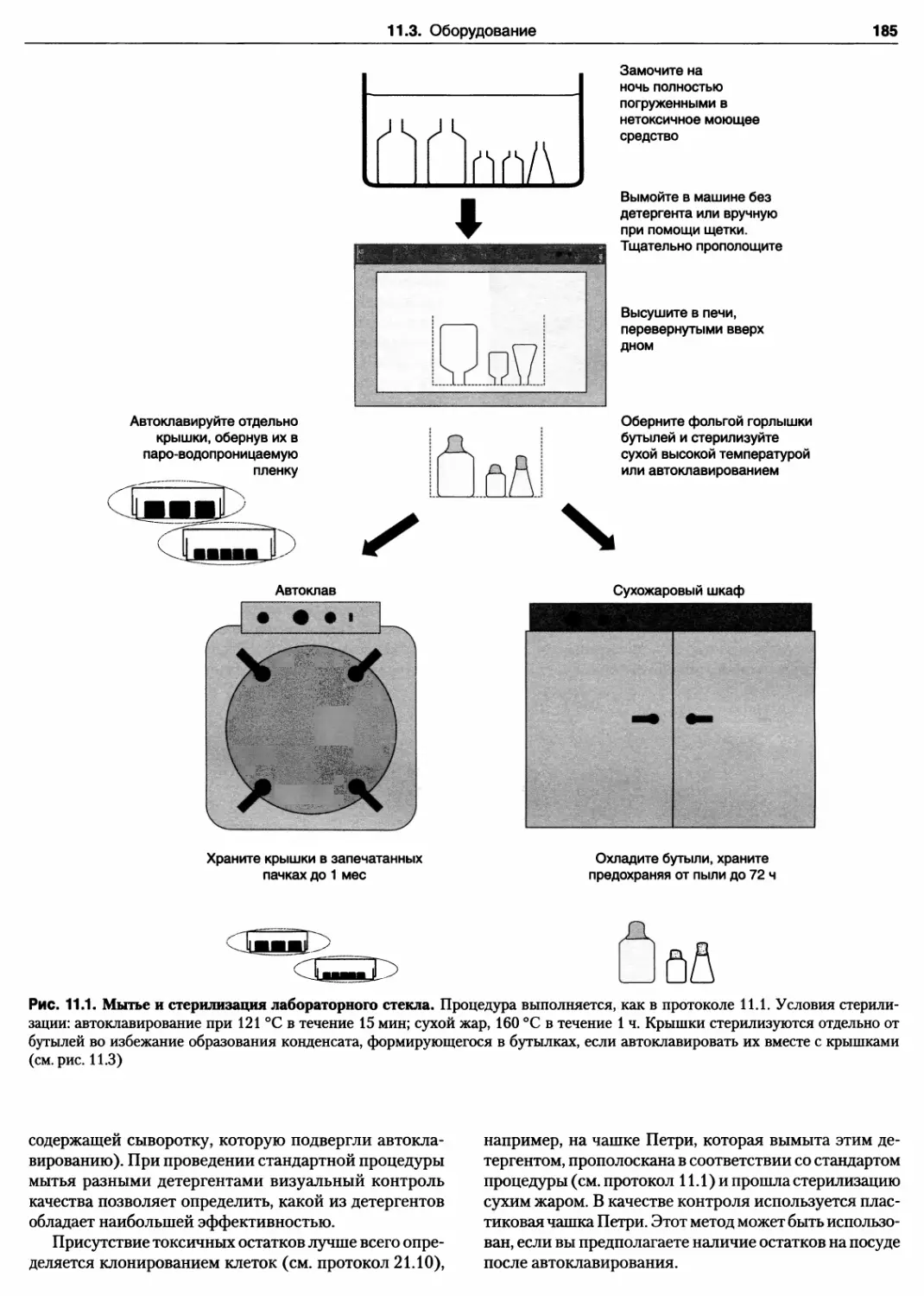
11.3.1. Стеклянная посуда 180
Протокол 11.1. Подготовка и стерилизация
изделий из стекла 182
11.3.2. Стеклянные пипетки 183
11.3.3. Завинчивающиеся крышки 183
11.3.4. Выбор детергента 184
11.3.5. Прочее оборудование 186
Протокол 11.2. Подготовка и стерилизация
стеклянных пипеток 187
Протокол 113. Подготовка и стерилизация
завинчивающихся крышек 188
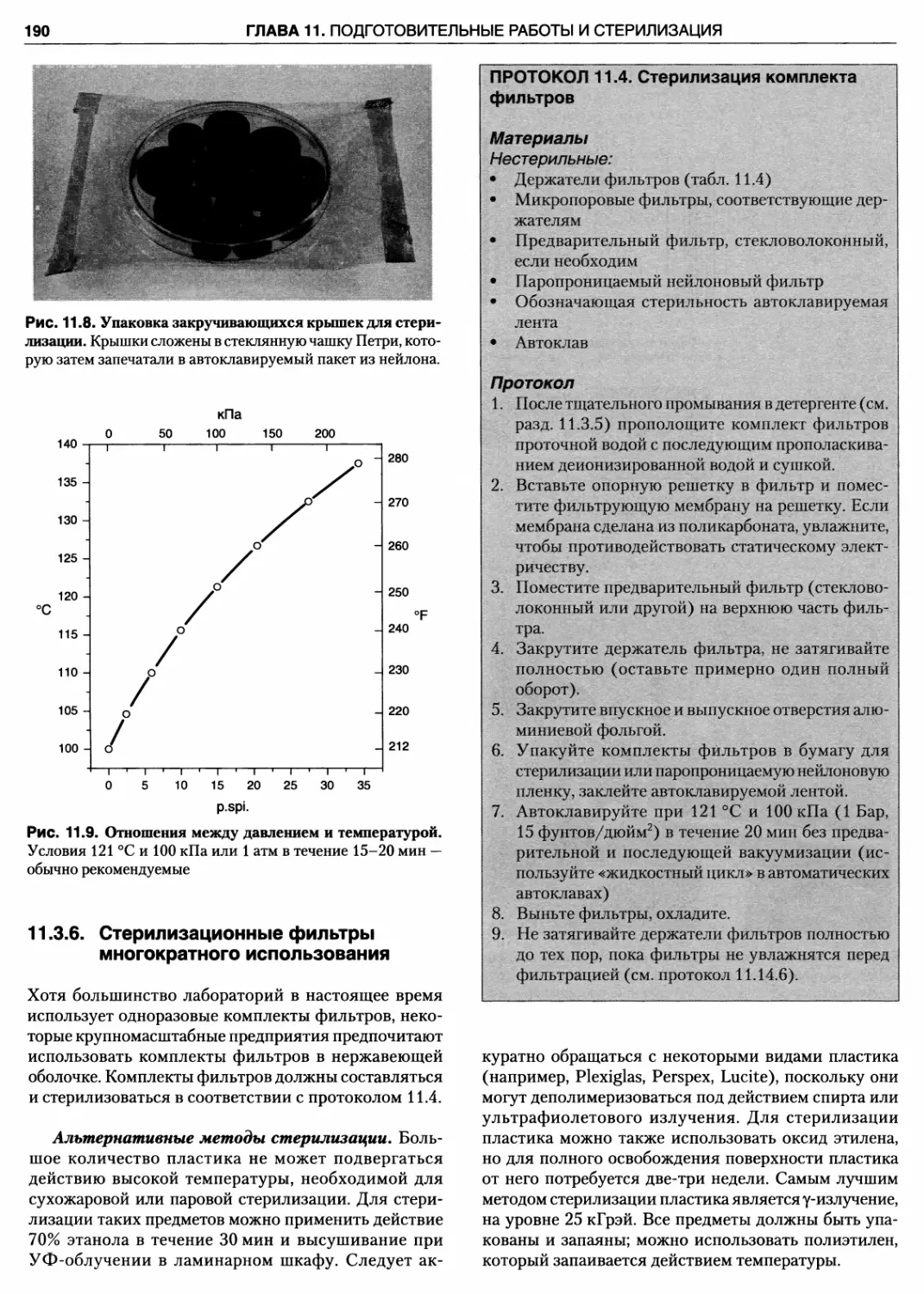
11.3.6. Стерилизационные фильтры
многократного использования 190
Протокол 11.4. Стерилизация комплекта
фильтров 190
11.4. Реагенты и среды 191
11.4.1. Вода 191
Протокол 11.5. Приготовление и стерилизация
сверхчистой воды (UPW) 192
11.4.2. Сбалансированный солевой
раствор (BSS) 193
Протокол 11.6. Приготовление и стерилизация
D-PBSA 193
11.4.3. Приготовление и стерилизация
среды 194
Протокол 11.7. Приготовление среды
из концентрата с кратностью 1 х 194
Протокол 11.8. Приготовление среды
из концентрата с кратностью 10 х 196
11.4.4. Порошкообразные среды 197
11.4.5. Специализированная среда 198
Протокол 11.9. Приготовление среды из порошка ... 198
11.5. Стерилизация среды 199
11.5.1. Автоклавируемые среды 199
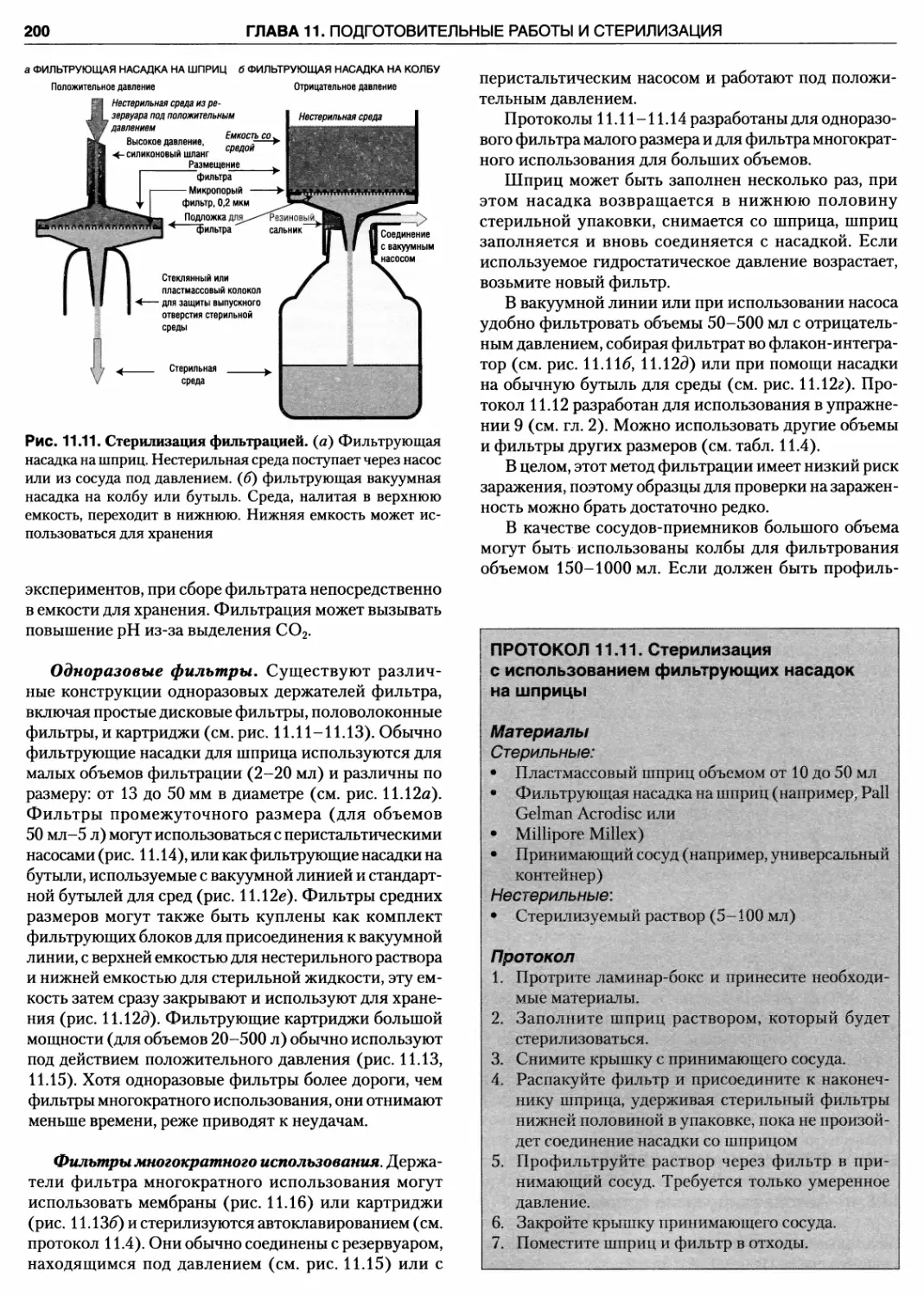
11.5.2. Стерилизация методом фильтрации .. 199
Протокол 11.10. Приготовление
специализированной среды 199
Протокол 11.11. Стерилизация с использованием
фильтрующих насадок на шприцы 200
Протокол 11.12. Стерилизация с использованием
фильтрующей вакуумной насадки на колбу 202
Протокол 11.13. Стерилизация с использованием
малого встроенного фильтра 202
11.5.3. Сыворотка 203
Протокол 11.14. Стерилизация с использованием
большого встроенного фильтра 205
Протокол 11.15. Сбор и стерилизация сывороток... 206
Протокол 11. 16. Диализ сыворотки 207
11.5.4. Приготовление и стерилизация
других реагентов 208
11.6. Контроль, проверка качества
и хранение сред 208
11.6.1. Контроль качества 208
11.6.2. Проверка стерильности 209
11.6.3. Проверка культуральных свойств ... 209
11.6.4. Хранение 210
ГЛАВА 12. ПЕРВИЧНАЯ КУЛЬТУРА 211
12.1. Типы первичных культур клеток 211
12.2. Выделение образцов ткани 211
Протокол 12.1. Выделение мышиного эмбриона 212
12.2.1. Мышиный эмбрион 212
12.2.2. Куриный эмбрион 215
Протокол 12.2. Извлечение куриных эмбрионов 216
12.2.3. Биопсийный материал человека 216
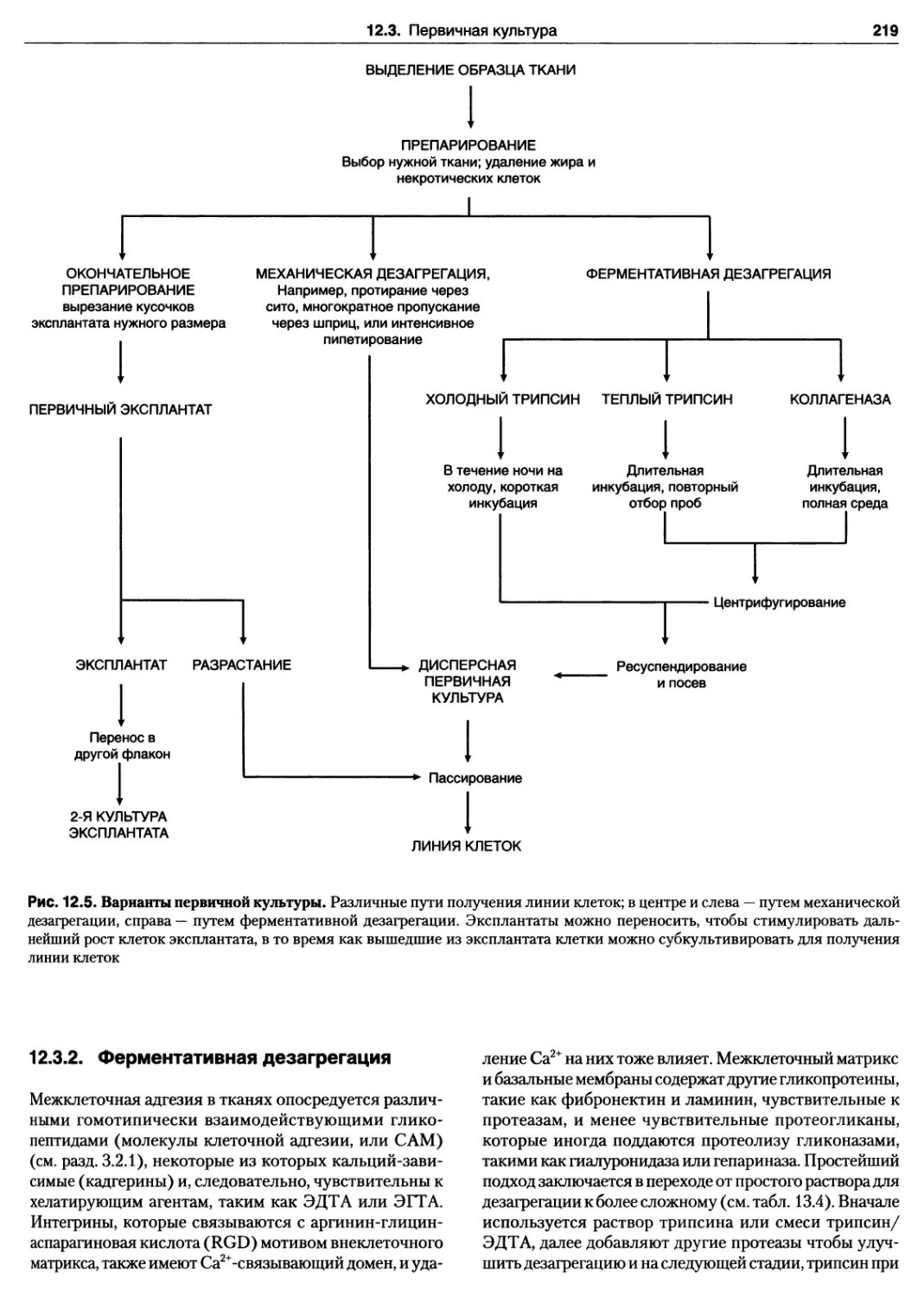
12.3. Первичная культура 217
Протокол 12.3. Биопсийный материал человека 218
12.3.1. Первичный эксплантат 218
12.3.2. Ферментативная дезагрегация 219
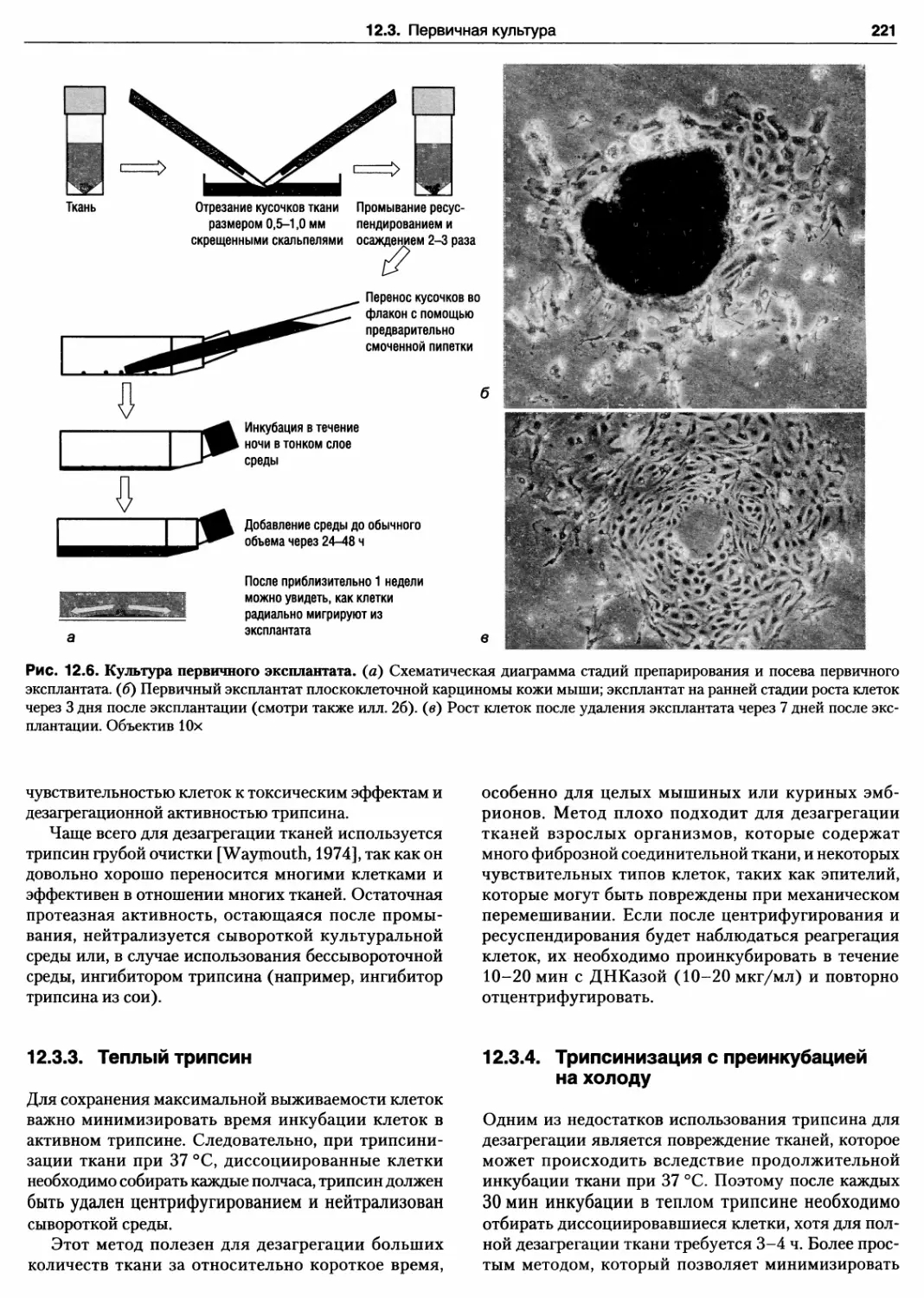
Протокол 12.4. Первичные эксплантаты 220
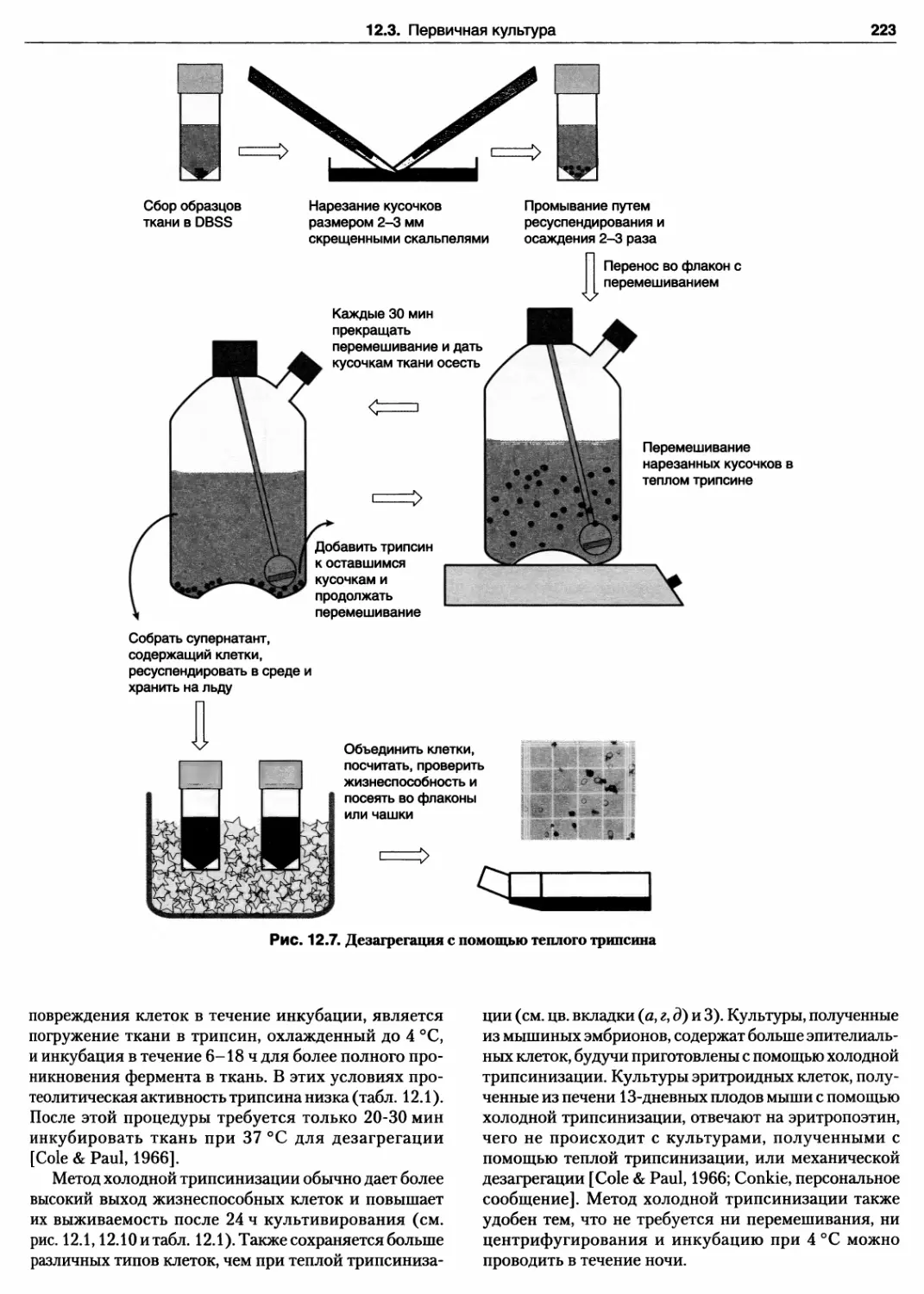
12.3.3. Теплый трипсин 221
12.3.4. Трипсинизация с преинкубацией
на холоду 221
Протокол 12.5. Дезагрегация ткани теплым
трипсином 222
12.3.5. Рудиментарные органы куриного
эмбриона 224
12.3.6. Дезагрегация с использованием
других ферментов 224
Протокол 12.6. Дезагрегация ткани холодным
трипсином 225
Протокол 12.7. Рудиментарные органы
куриного эмбриона 226
12.3.7. Коллагеназа 226
12.3.8. Механическая дезагрегация 227
Протокол 12.8. Дезагрегация ткани
с помощью коллагеназы 230
12.3.9. Разделение жизнеспособных
и нежизнеспособных клеток 230
12.3.10. Первичная культура, краткие
выводы 231
12.3.11. Первичная документация 231
Протокол 12.9. Механическая дезагрегация
путем просеивания 233
Протокол 12.10. Обогащение жизнеспособных
клеток 234
ГЛАВА 13. СУБКУЛЬТУРА
И КЛЕТОЧНЫЕ ЛИНИИ 235
13.1. Субкультивирование 235
13.2. Терминология 235
13.3. Возраст культуры 235
13.4. Маркировка клеточной культуры 240
13.5. Выбор клеточной линии 240
13.6. Обычный порядок поддержания
культуры 241
13.6.1. Необходимость исследования
клеточной морфологии 241
13.6.2. Замена среды 242
13.7. Субкультура 243
Протокол 13.1. Смена среды в монослойной
культуре 243
13.7.1. Критерии субкультивирования 244
Протокол 13.2. Субкультивирование монослойной
культуры клеток 246
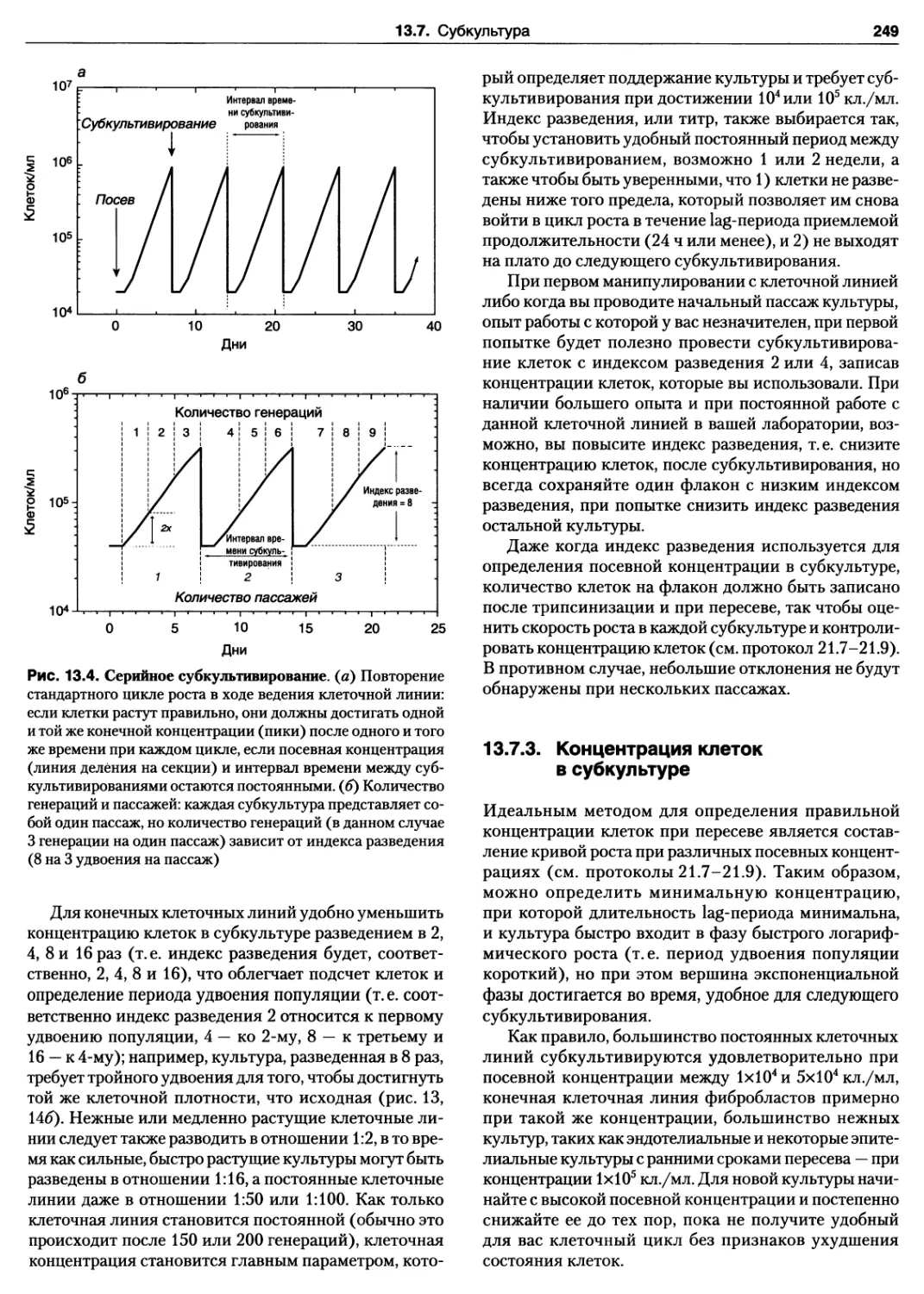
13.7.2. Цикл роста и индекс разведения 248
13.7.3. Концентрация клеток в субкультуре.. 249
13.7.4. Культивирование в суспензии 250
13.7.5. Субкультура клеток, растущих
в суспензии 250
Протокол 13.3. Субкультивирование суспензионной
культуры клеток 251
13.7.6. Стандартизация условий
культивирования 252
13.7.7. Использование антибиотиков 252
13.7.8. Ведение документации 253
Оглавление
9
ГЛАВА 14. КЛОНИРОВАНИЕ
И СЕЛЕКЦИЯ 255
14.1. Клонирование клеток 255
Протокол 14.1. Клонирование с разведением 256
14.2. Стимуляция эффективности посева 257
14.2.1. Условия, которые улучшают
клональный рост 258
Протокол 14.2. Приготовление кондиционированной
среды 260
Протокол 14.3. Приготовление фидерных слоев 260
14.2.2. Кондиционированные среды 260
14.2.3. Фидерные слои 261
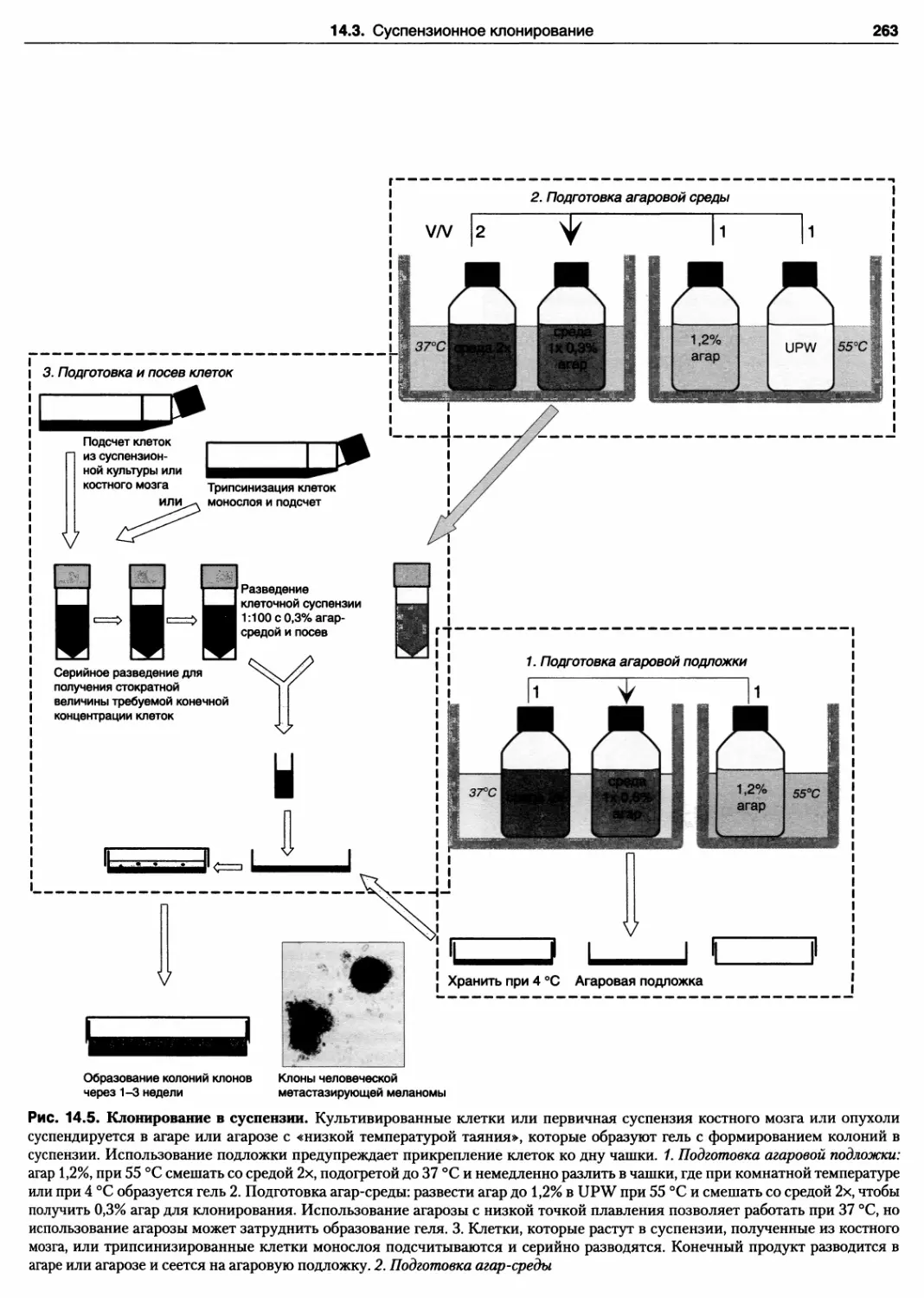
14.3. Суспензионное влонирование 261
Протокол 14.4. Клонирование в агаре 262
Протокол 14.5. Клонирование в метилцеллюлозе 264
14.4. Выделение клонов 265
Протокол 14.6. Выделение клонов
с использованием колец для клонирования 266
Протокол 14.7. Выделение клеточных колоний
путем облучения 266
14.4.1. Другие методы выделения
монослойных клонов 267
Протокол 14.8. Выделение суспензионных клонов 268
14.4.2. Суспензионные клоны 268
14.5. Получение репликативных колоний 268
14.6. Селективные ингибиторы 268
Протокол 14.9. Метотрексат-резистентность
и DHFR-амплификация 270
14.7. Выделение генетических вариантов 270
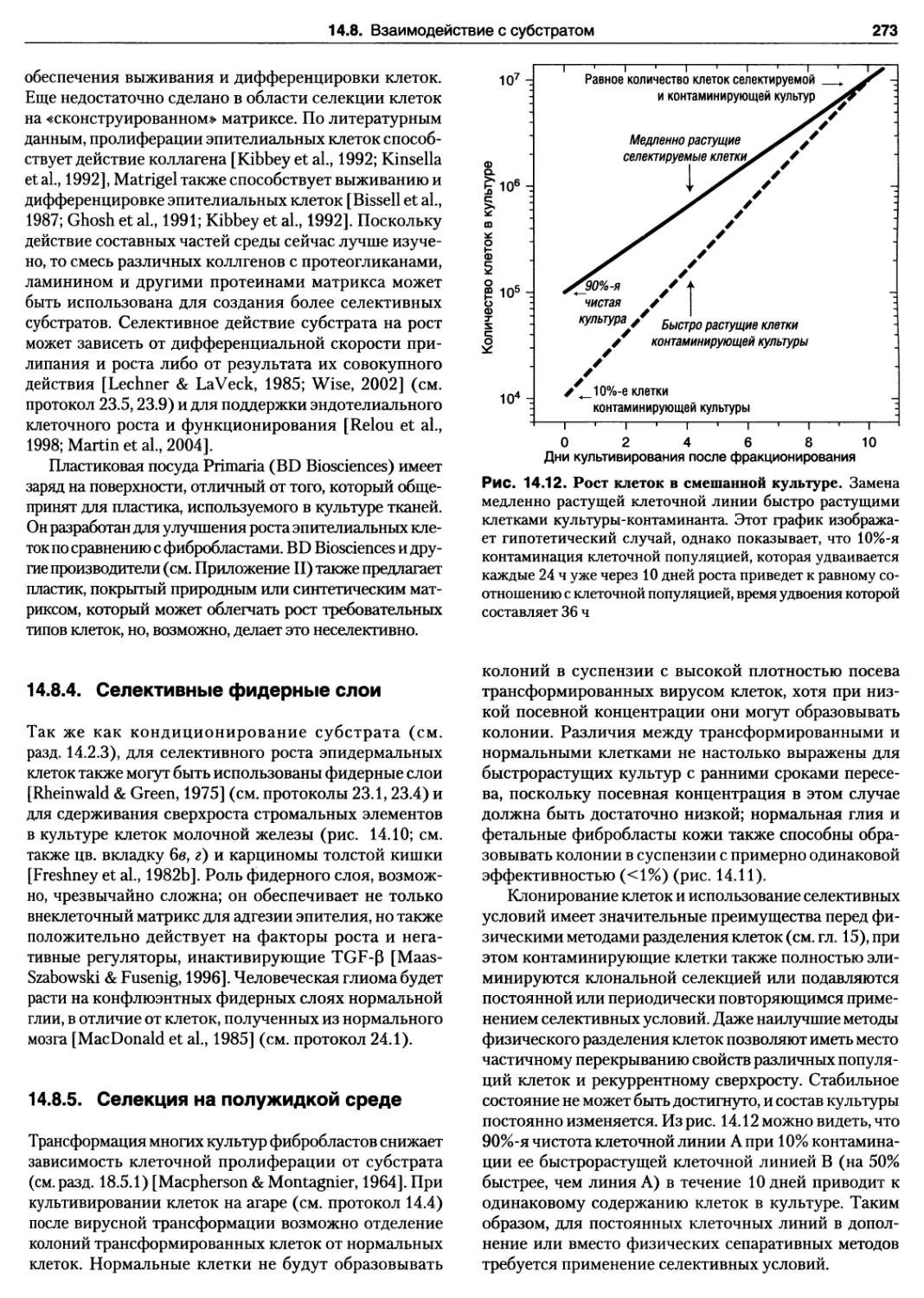
14.8. Взаимодействие с субстратом 272
14.8.1. Селективная адгезия 272
14.8.2. Селективное открепление 272
14.8.3. Природа субстрата 272
14.8.4. Селективные фидерные слои 273
14.8.5. Селекция на полужидкой среде 273
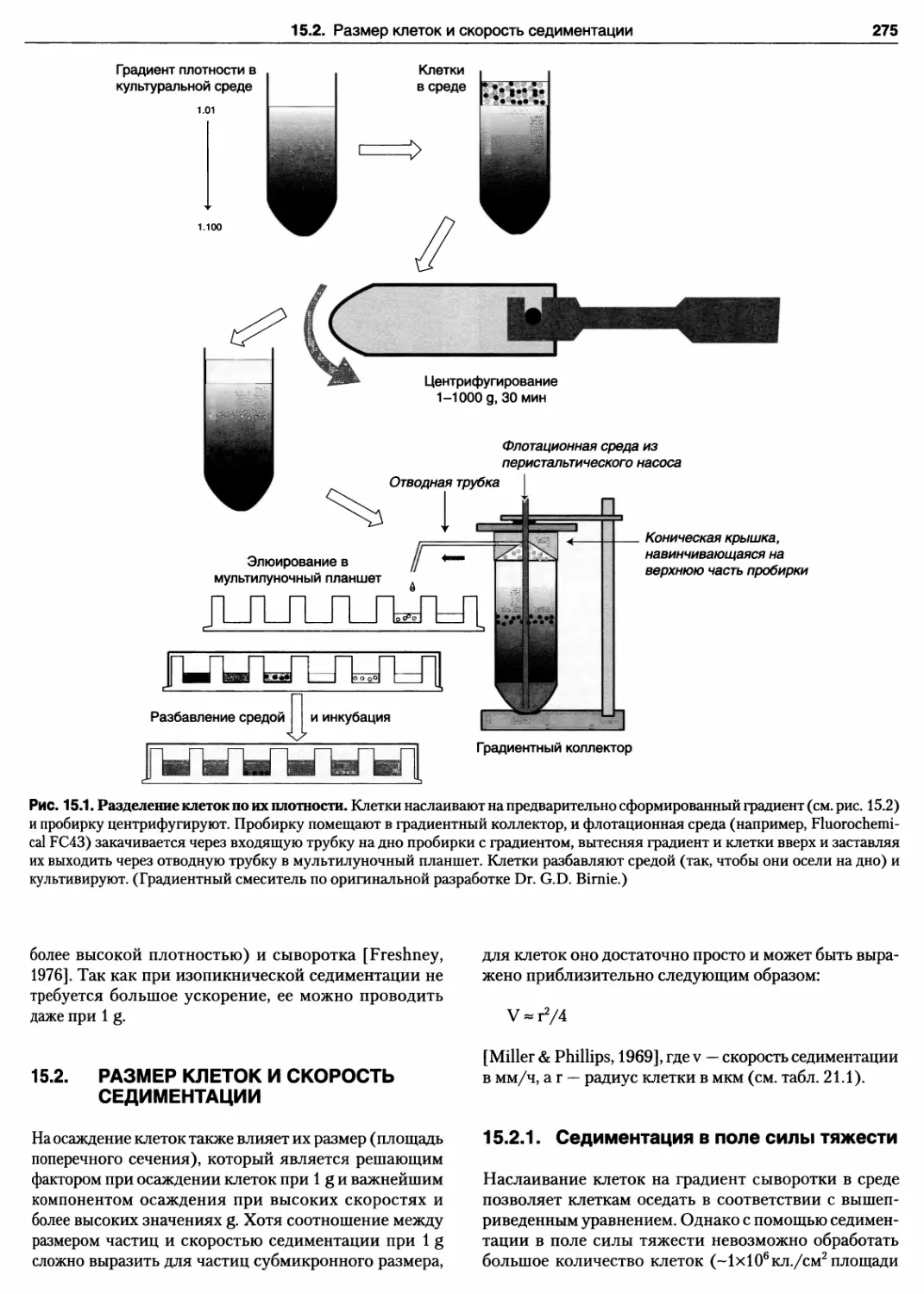
ГЛАВА 15. РАЗДЕЛЕНИЕ КЛЕТОК 274
15.1. Плотность клеток и изопикническая
седиментация 274
Варианты 274
15.2. Размер клеток и скорость седиментации ... 275
15.2.1. Седиментация в поле
силы тяжести 275
15.2.2. Элюатриационное
центрифугирование 276
Протокол 15.1. Разделение клеток
центрифугированием в градиенте плотности ... 276
15.3. Методы, основанные на применении
антител 277
15.3.1. Иммунный пэннинг 278
15.3.2. Магнитный сортинг 278
Протокол 15.2. Магнитно-активированный
клеточный сортинг (MACS) 279
15.4. Флюоресцентно-активируемый
клеточный сортинг 281
15.5. Другие методы 283
15.6. Советы начинающему 283
ГЛАВА 16. ХАРАКТЕРИСТИКА КЛЕТОК 285
16.1. Необходимость характеристики 285
16.2. Ведение документации
и происхождение клеток 285
16.3. Подтверждение аутентичности 285
16.3.1. Видовая идентификация 286
16.3.2. Маркеры дифференцировки
или маркеры ткани 286
16.3.3. Уникальные маркеры 288
16.3.4. Трансформация 288
16.4. Морфология клеток 288
16.4.1. Микроскопия 289
Протокол 16.1. Использование инвертированного
микроскопа 289
Протокол 16.2. Окрашивание по Гимза 290
16.4.2. Окрашивание 290
16.4.3. Культуральные сосуды
для цитологии: монослойные
культуры 291
Протокол 16.3. Окрашивание кристаллвиолетом ... 291
Протокол 16.4. Подготовка суспензионной
культуры клеток для цитологических
исследований методом
цитоцентрифунгирования 292
16.4.4. Приготовление суспензионной
культуры для цитологии 292
Протокол 16.5. Фильтрование суспензии клеток
для цитологического исследования 298
16.4.5. Микрофотография 298
Протокол 16.6. Цифровая фотография
на микроскопе 300
16.5. Хромосомный состав 300
Протокол 16.7. Приготовление хромосомного
препарата 301
Варианты 302
16.5.1. Дифференциальное окрашивание
хромосом 302
16.5.2. Хромосомный анализ 303
16.5.3. Окрашивание хромосом 304
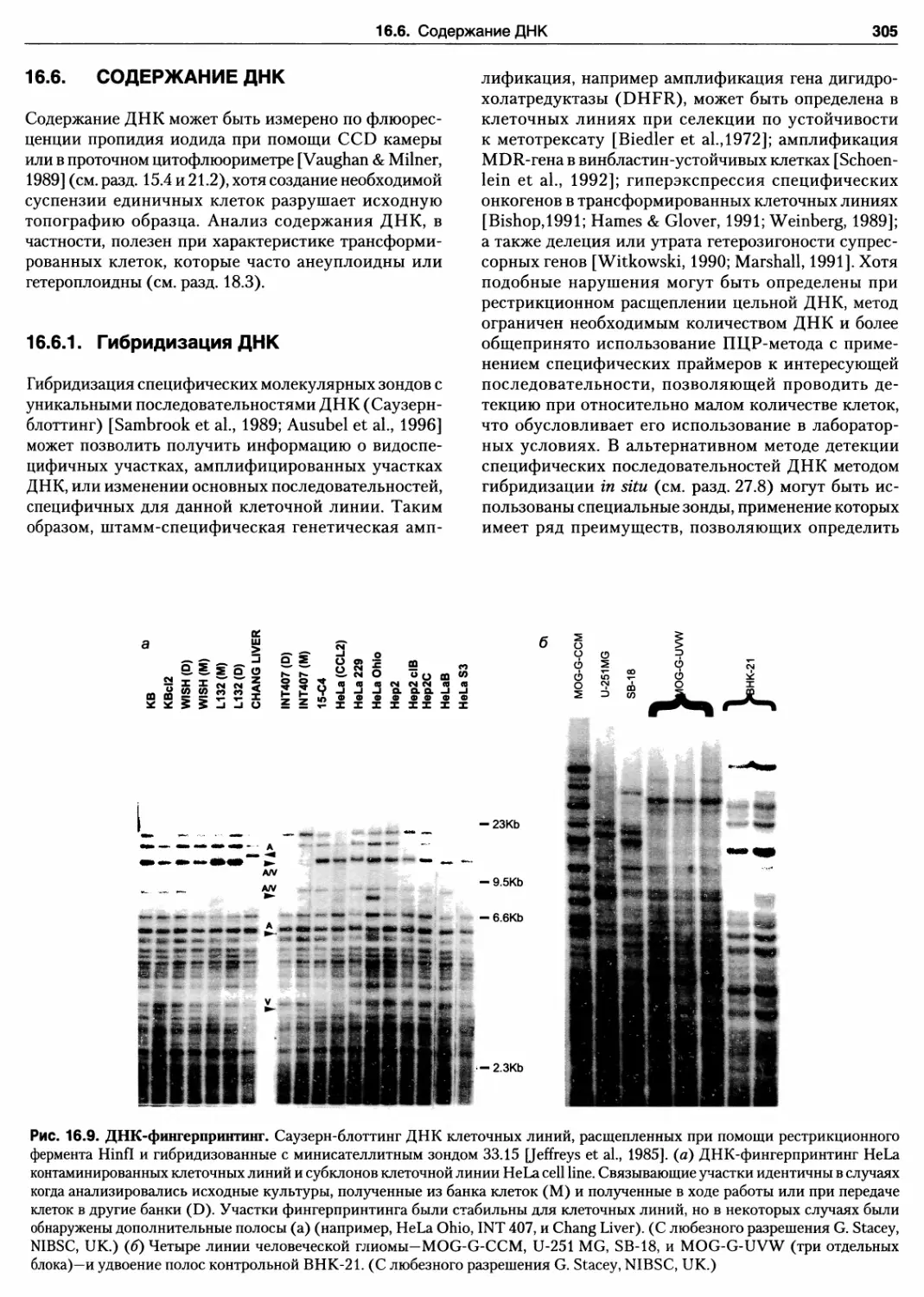
16.6. Содержание ДНК 305
16.6.1. Гибридизация ДНК 305
Протокол 16.8. Мультилокусный фингерпринтинг
ДНК клеточных линий 306
16.6.2. Фингерпринтинг ДНК 306
Протокол 16.9. STR-анализ профиля ДНК
клеточных линий 310
16.6.3. Анализ профиля ДНК 310
16.7. РНК и экспрессия белка 312
16.8. Активность ферментов 313
16.8.1. Изоферменты 313
16.8.2. Изоферментный электрофорез
при помощи Authentikit 314
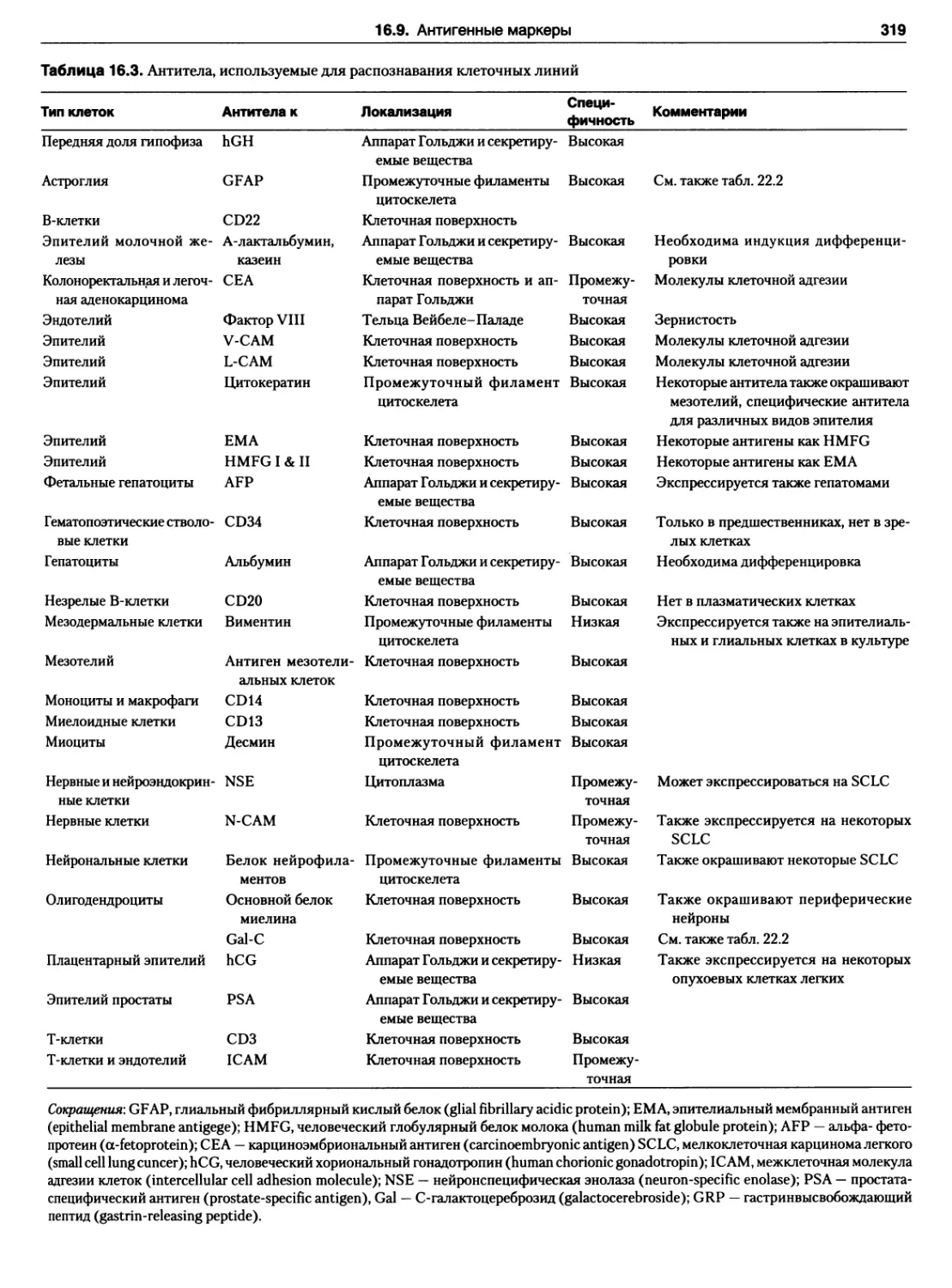
16.9. Антигенные маркеры 315
Протокол 16.10. Изоферментный анализ 316
Протокол 16.11. Непрямая иммунофлюоресценция.. 318
16.9.1. Иммунное окрашивание 318
Варианты 318
16.9.2. Иммунный анализ 320
16.10. Дифференцировка 320
ГЛАВА 17. ДИФФЕРЕНЦИРОВКА 321
17.1. Экспрессия фенотипа in vivo 321
17.1.1. Дедифференцировка 321
17.1.2. Линейная селекция 321
17.2. Стадии дифференцировки 321
17.3. Пролиферация и дифференцировка 322
17.4. Коммитирование и линии
дифференцировки 322
17.5. Пластичность стволовых клеток 324
17.6. Маркеры дифференцировки 324
17.7. Индукция дифференцировки 325
10
Оглавление
17.7.1. Межклеточные взаимодействия 325
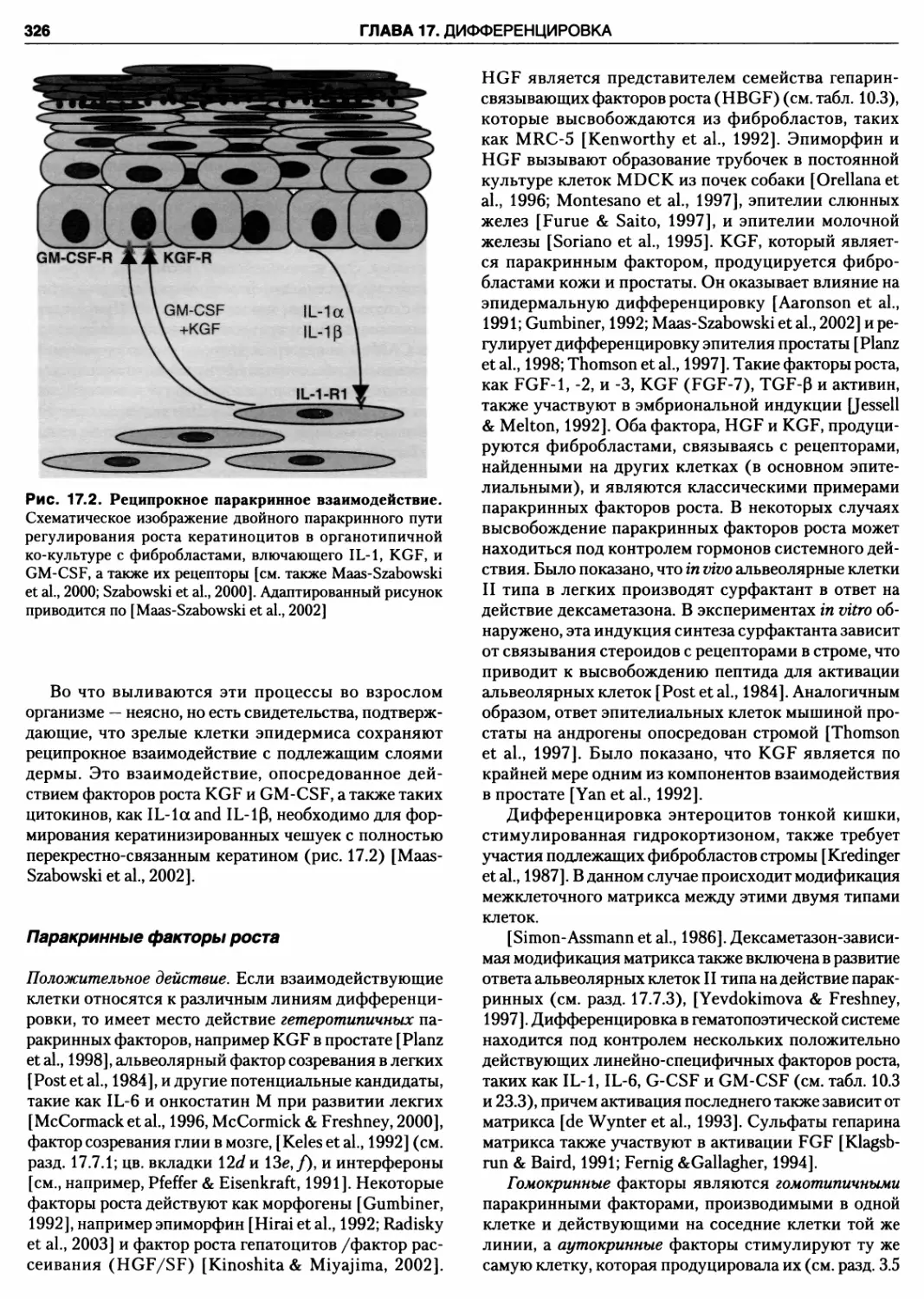
Паракринные факторы роста 326
17.7.2. Системные или экзогенные факторы.. 327
17.7.3. Взаимодействия клетки с матриксом.. 327
17.7.4. Полярность и форма клетки 329
17.7.5. Давление кислорода 329
17.8. Дифференцировка и злокачественность 330
17.9. Практические аспекты 330
ГЛАВА 18. ТРАНСФОРМАЦИЯ
И ИММОРТАЛИЗАЦИЯ 332
18.1. Роль в характеристике клеточных
линий 332
18.2. Что такое трансформация? 332
18.3. Генетическая нестабильность 332
18.3.1. Хромосомные аберрации 334
18.3.2. Изменение содержания ДНК 335
18.4. Иммортализация 335
18.4.1. Контроль физиологического
старения 336
18.4.2. Иммортализация с использованием
вирусных генов 336
18.4.3. Иммортализация человеческих
фибробластов 337
Протокол 18.1. Иммортализация фибробластов 338
18.4.4. Теломераза-индуцированная
иммортализация 340
18.4.5. Трансгенные мыши 341
Протокол 18.2. Иммортализация мезенхимальных
стволовых клеток человека с помощью
теломеразы 341
18.5. Аберрантный контроль роста 343
18.5.1. Независимость от прикрепления
к субстрату 343
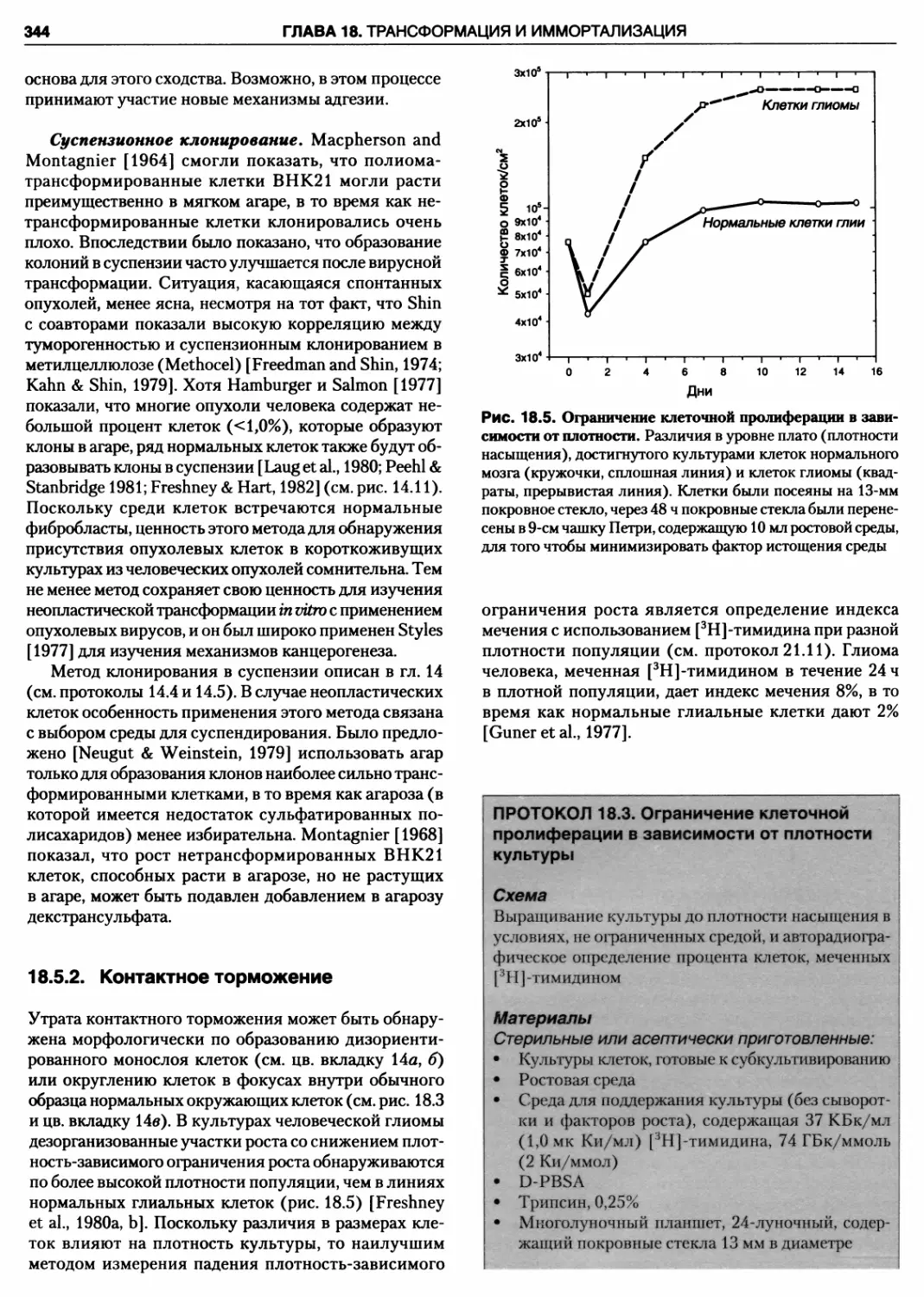
18.5.2. Контактное торможение 344
Протокол 18.3. Ограничение клеточной
пролиферации в зависимости от плотности
культуры 344
18.5.3. Зависимость от сыворотки 345
18.5.4. Онкогены 346
18.6. Туморогенность 346
18.6.1. Малигнизация 346
18.6.2. Опухолевая трансплантация 347
18.6.3. Инвазивность 347
18.6.4. Ангиогенез 348
18.6.5. Активаторы плазминогена 349
ГЛАВА 19. КОНТАМИНАЦИЯ 350
19.1. Источники контаминации 350
19.1.1. Основные приемы стерильной
работы 350
19.1.2. Состояние внешней среды 350
19.1.3. Использование и поддержание
в рабочем состоянии ламинарного
шкафа 350
19.1.4. Инкубаторы с увлажнением 353
Протокол 19.1. Обработка инкубаторов 353
19.1.5. Холодильные камеры 354
19.1.6. Стерильные материалы 354
19.1.7. Привезенные клеточные линии
и биопсийные образцы 354
19.1.8. Карантин 354
19.2. Виды микробной контаминации 354
19.3. Контроль контаминации 355
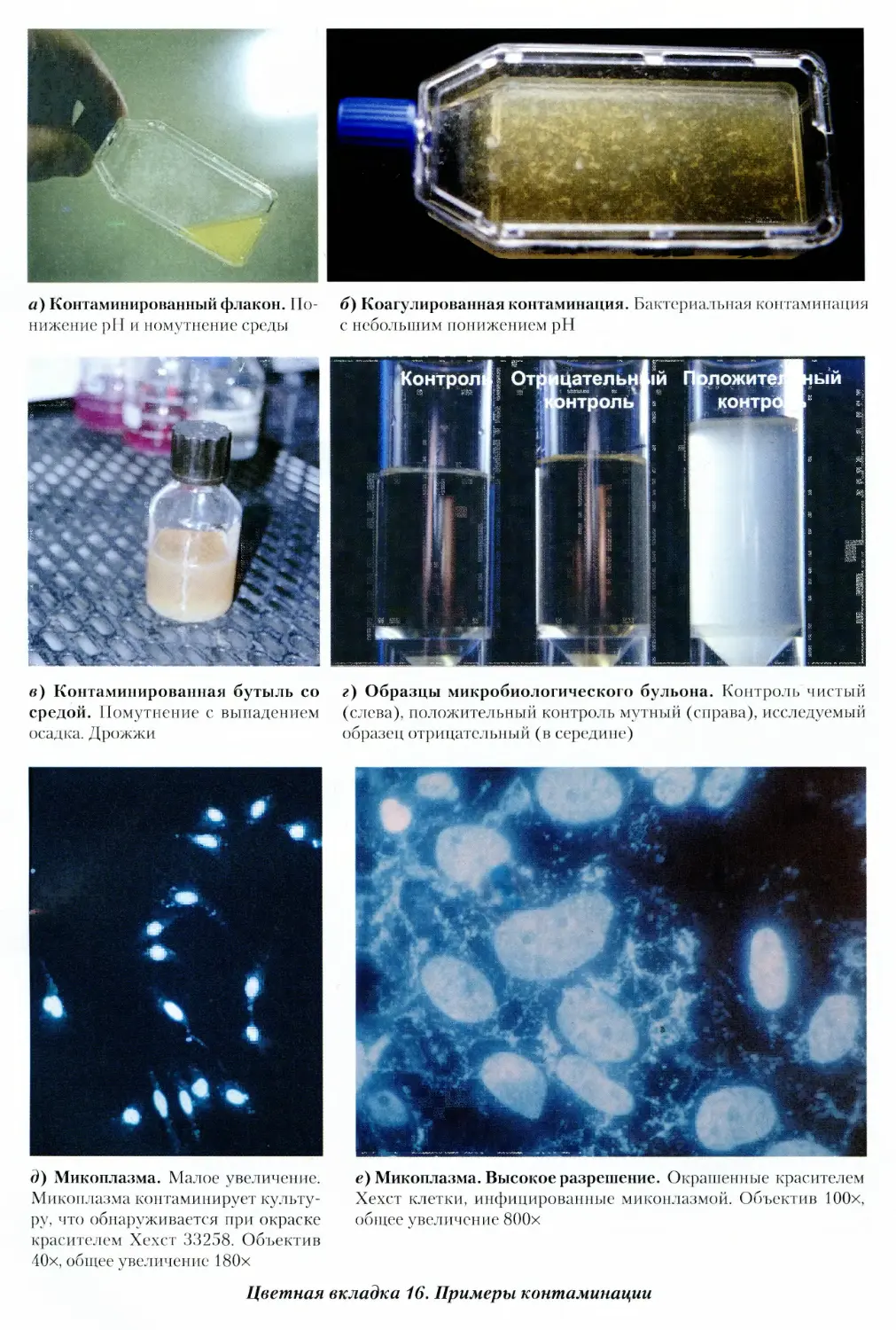
19.3.1. Визуально определяемая
микробная контаминация 355
19.3.2. Микоплазма 357
Протокол 19.2. Выявление микоплазмы методом
флюоресценции 357
19.3.3. Окрашивание микоплазмы
флюоресцентными красителями 358
19.3.4. Использование ПЦР для детекции
микоплазм 358
Протокол 19.3. Выявление микоплазмы
с помощью ПЦР 359
Интерпретация результатов 360
19.3.5. Альтернативные методы детекции
микоплазмы 361
19.3.6. Вирусная контаминация 361
19.4. Устранение контаминации 361
19.4.1. Бактерии, грибы и дрожжи 361
19.4.2. Устранение микоплазмы 362
19.4.3. Устранение вирусной контаминации.. 362
19.4.4. Персистирующая контаминация 362
Протокол 19.4. Ликвидация микробной
контаминации 362
19.5. Перекрестная контаминация 363
19.6. Заключение 363
ГЛАВА 20. КРИОКОНСЕРВАЦИЯ 365
20.1. Предпосылки метода замораживания 365
20.2. Получение клеточных линий
для криоконсервации 365
20.3. Принципы криоконсервации 365
20.3.1. Теоретическое обоснование
замораживания клеток 365
20.3.2. Концентрация клеток 366
20.3.3. Среда для замораживания 366
20.3.4. Скорость охлаждения 367
20.3.5. Морозильные камеры 369
Протокол 20.1. Замораживание клеток 372
20.3.6. Протоколирование работы
морозильника 373
Протокол 20.2. Размораживание замороженных
клеток 374
20.3.7. Размораживание хранившихся
ампул 374
20.4. Планирование и контроль
замораживания культур клеток 377
20.4.1. Контроль за состоянием запасов
клеток в морозильнике 377
20.4.2. Периодическая замена
культивируемых клеток 377
20.5. Банки клеток 378
20.6. Транспортировка клеточных культур 379
20.6.1. Замороженная ампула 379
20.6.2. Живые культуры 379
ГЛАВА 21. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ ... 380
21.1. Подсчет клеток 380
21.1.1. Гемоцитометр 380
Протокол 21.1. Подсчет клеток при помощи
гемоцитометра 380
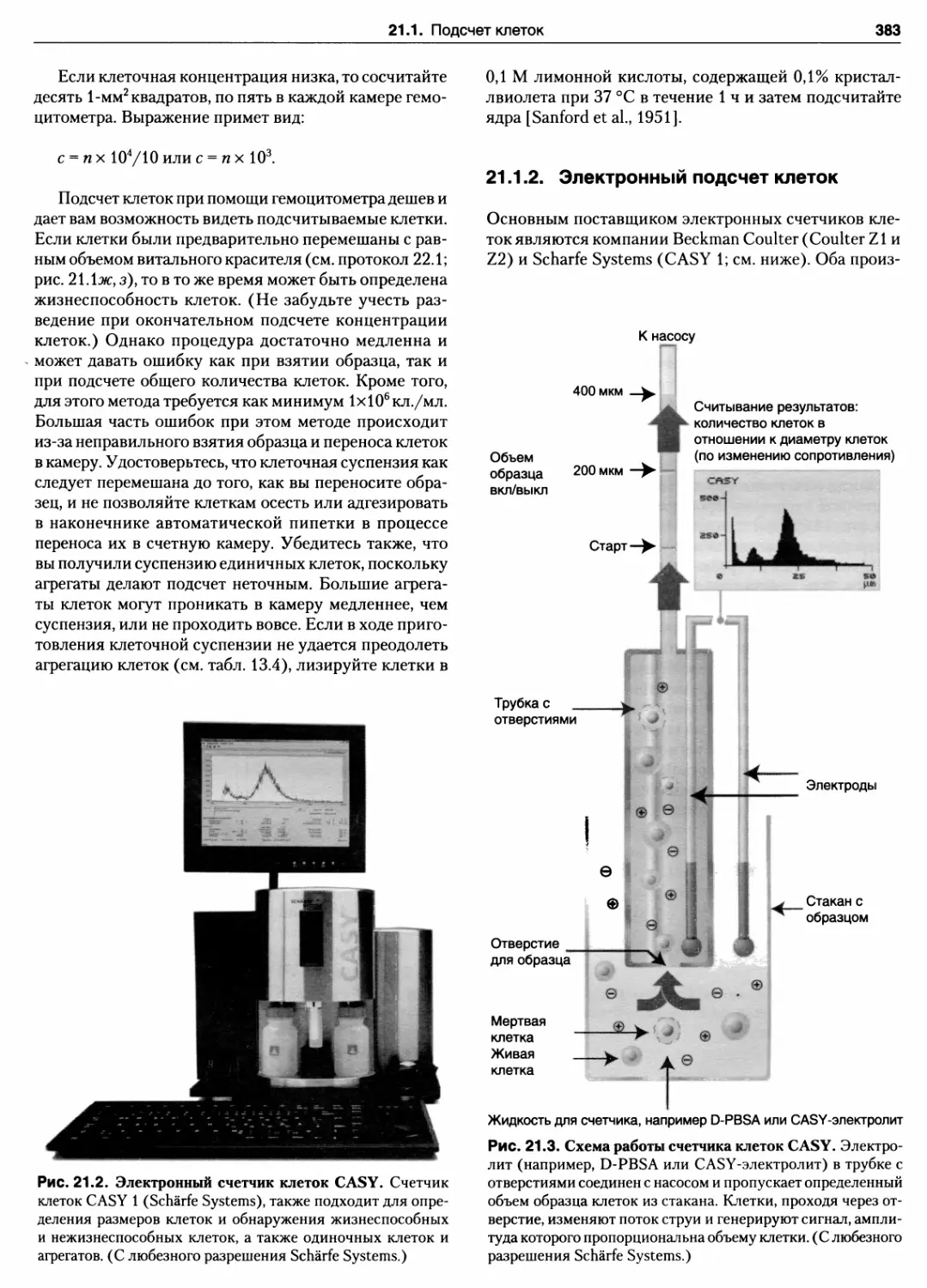
21.1.2. Электронный подсчет клеток 383
Протокол 21.2. Электронный подсчет клеток
на основании электрического сопротивления 384
21.1.3. Окрашенные монослои 386
21.2. Вес клеток 386
Оглавление
11
21.3. Содержание ДНК 386
21.4. Белок 386
21.4.1. Солюбилизация образца 387
21.5. Скорость синтеза 387
21.5.1. Синтез ДНК 387
Протокол 213. Оценка содержания ДНК
с использованием красителя HOECHST33258 ... 387
Протокол 21.4. Оценка содержания белка
методом Брэдфорда 387
Протокол 21.5. Оценка уровня синтеза ДНК
методом включения Н3-тимидиновой метки 388
21.5.2. Синтез белка 389
Протокол 21.6. Синтез белка 389
21.6. Приготовление образцов
для ферментного и иммуноферментного
анализа 390
21.7. Цитометрия 390
21.7.1. Мечение in situ 390
21.7.2. Проточная цитометрия 390
21.8. Увеличение количества образцов
для статистического анализа 390
21.8.1. Получение данных 391
21.8.2. Анализ данных 391
21.9. Клеточная пролиферация 391
21.9.1. Планирование эксперимента 391
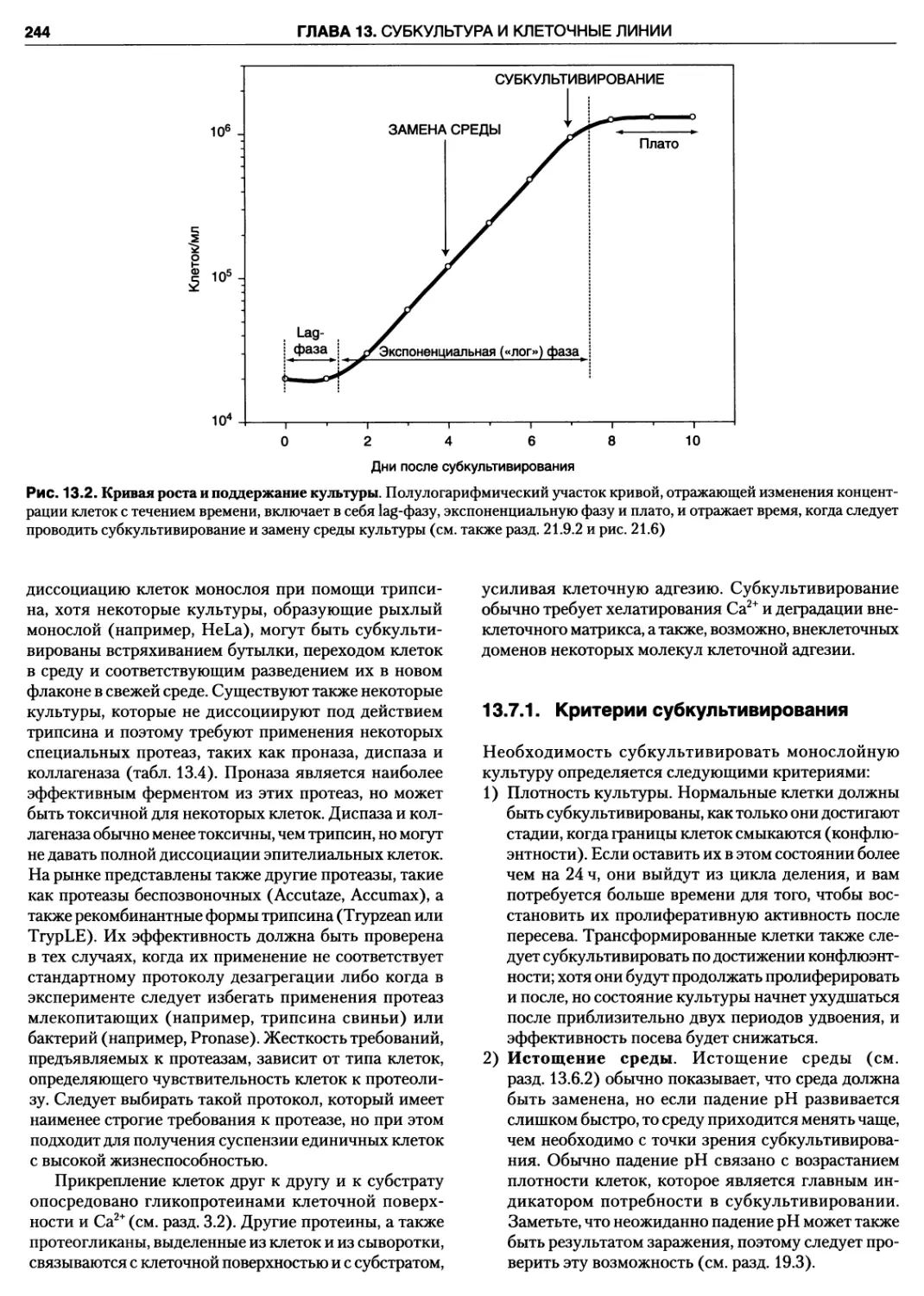
21.9.2. Цикл роста 392
Протокол 21.7. Кривая роста монослоя
во флаконах 393
Протокол 21.8. Кривая роста монослоя
в многолуночном планшете 394
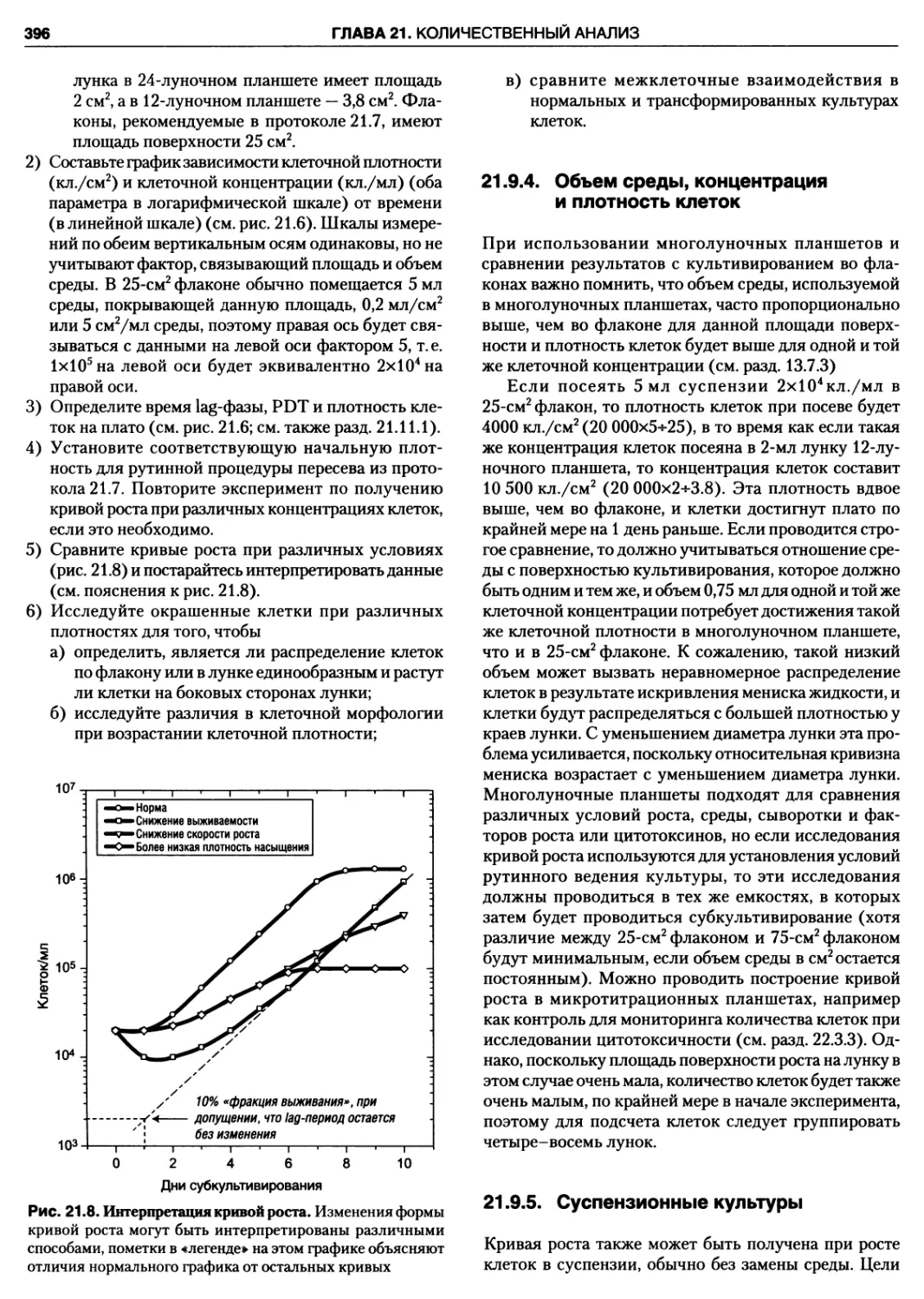
21.9.3. Анализ кривой роста монослоя 395
21.9.4. Объем среды, концентрация
и плотность клеток 396
21.9.5. Суспензионные культуры 396
21.9.6. Фазы цикла роста 397
Протокол 21.9. Кривая роста суспензионной
культуры 397
21.9.7. Данные, получаемые при исследовании
кривой роста 399
21.10. Эффективность культивирования 399
Протокол 21.10. Определение эффективности
посева на чашках Петри 400
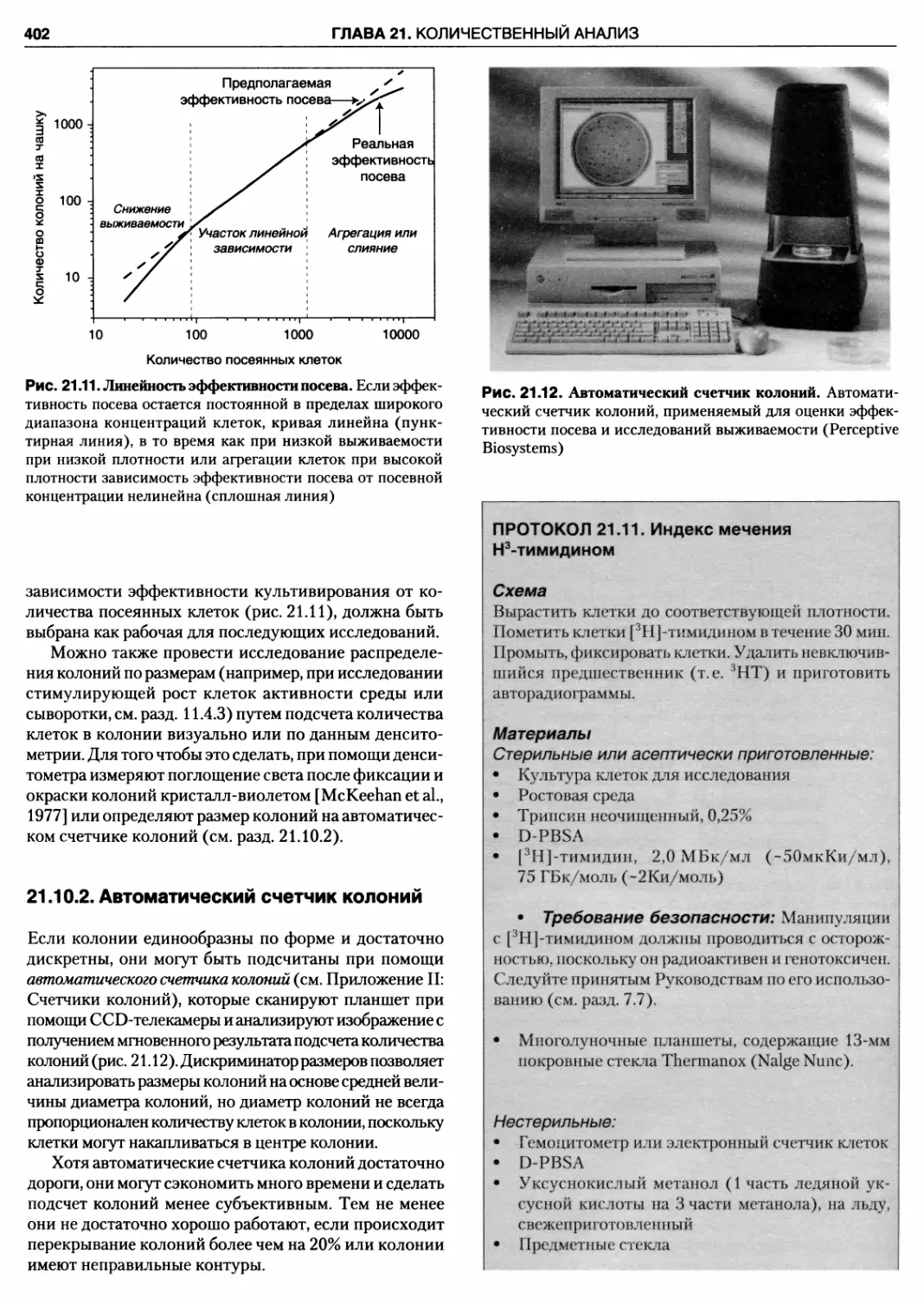
21.10.1. Анализ формирования колоний 401
21.10.2. Автоматический счетчик колоний .. 402
Протокол 21.11. Индекс мечения Н3-тимидином 402
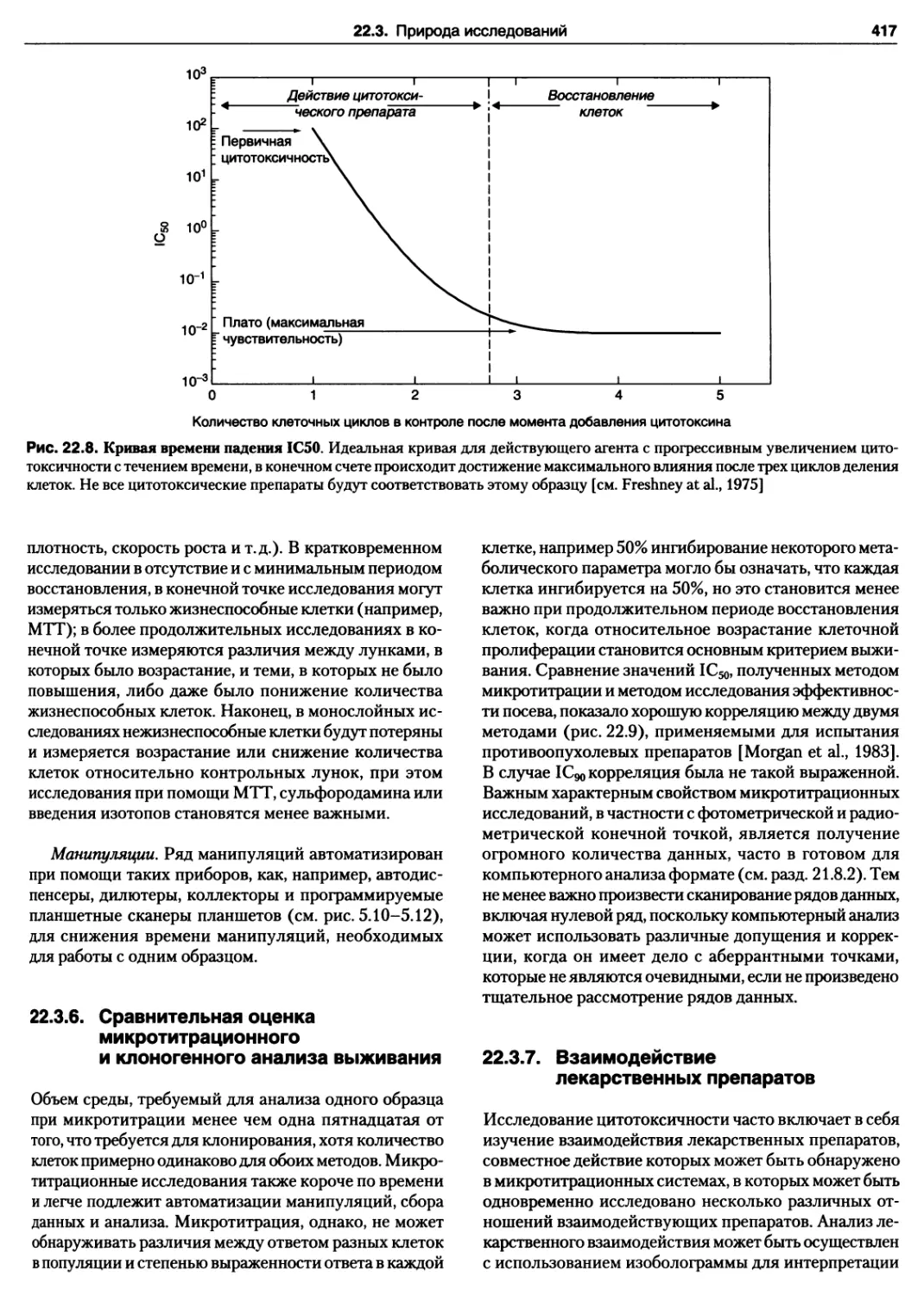
21.11. Индекс мечения 403
21.11.1. Фракция пролиферирующих
клеток 404
21.11.2. Митотический индекс 404
21.11.3. Индекс пролиферации 404
Протокол 21.12. Определение фракции
пролиферирующих клеток 404
21.12. Время клеточного цикла 405
21.13. Клеточная миграция 405
ГЛАВА 22. ЦИТОТОКСИЧНОСТЬ 406
22.1. Жизнеспособность, токсичность
и выживаемость 406
22.2. Ограничения in vitro 407
22.3. Природа исследований 407
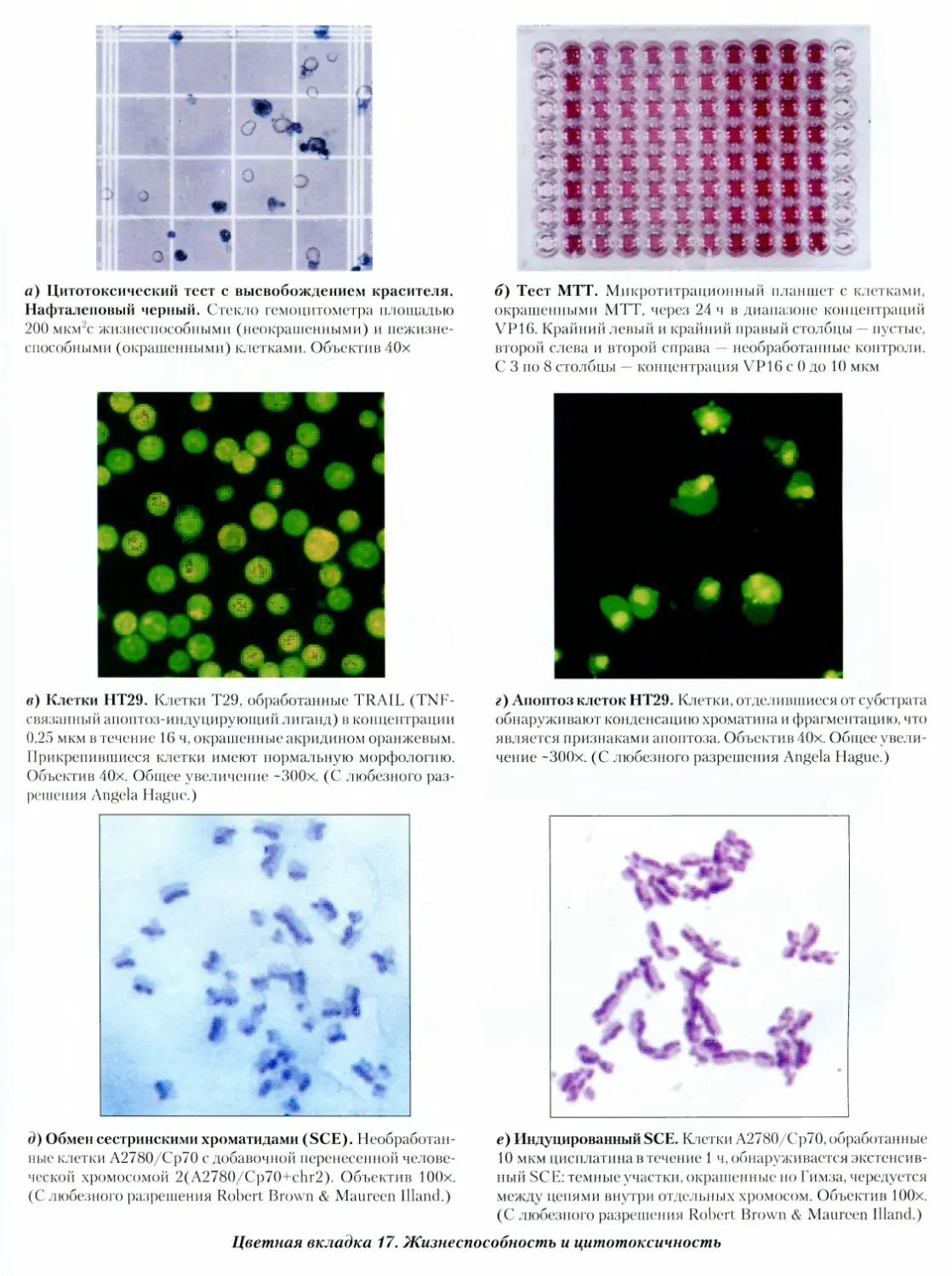
22.3.1. Жизнеспособность 407
Протокол 22.1. Оценка жизнеспособности
методом отторжения красителя 408
Протокол 22.2. Оценка жизнеспособности
методом поглощения красителя 408
22.3.2. Выживаемость 409
Протокол 22.3. Клоногенный анализ
прикрепившихся клеток 410
22.3.3. Анализ, основанный на клеточной
пролиферации 413
22.3.4. Метаболический анализ
цитотоксичности :.. 413
22.3.5. Микротитрационный анализ 413
Протокол 22.4. Оценка цитотоксичности
при помощи МТТ-теста 414
22.3.6. Сравнительная оценка
микротитрационного и клоногенного
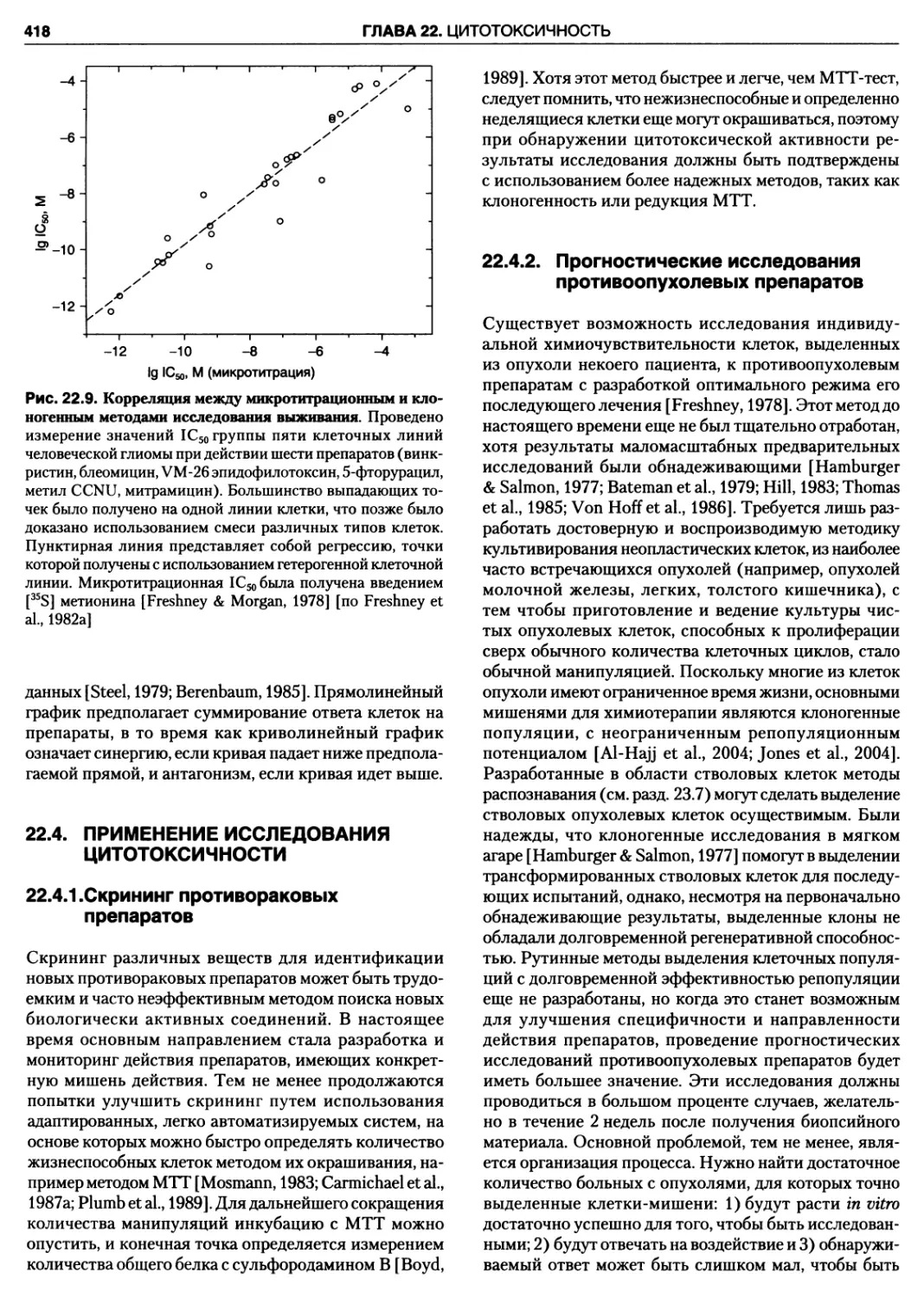
анализа выживания 417
22.3.7. Взаимодействие лекарственных
препаратов 417
22.4. Применение исследования
цитотоксичности 418
22.4.1. Скрининг противораковых
препаратов 418
22.4.2. Прогностические исследования
противоопухолевых препаратов 418
22.4.3. Фармацевтическое тестирование 419
22.5. Трансформация и мутагенез 419
22.5.1. Анализ мутагенеза методом обмена
сестринских хроматид 419
Протокол 22.5. Обмен сестринских хроматид 419
22.5.2. Канцерогенность 421
22.6. Воспаление 422
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ
ТИПОВ КЛЕТОК 423
23.1. Клеточные культуры
специализированных
клеток 423
23.2. Эпителиальные клетки 423
23.2.1. Эпидермис 424
Протокол 23.1. Эпидермальные кератиноциты 425
23.2.2. Роговица 428
Протокол 23.2. Эпителиальные клетки роговицы— 429
Протокол 233. Эпителий молочной железы 430
23.2.3. Молочная железа 430
23.2.4. Шейка матки 431
Протокол 23.4. Цервикальный эпителий 431
23.2.5. Желудочно-кишечный тракт 433
Протокол 23.5. Выделение и культивирование
клеток крипт толстого кишечника 434
23.2.6. Печень 435
Протокол 23.6. Выделение гепатоцитов крысы 436
23.2.7. Поджелудочная железа 437
Протокол 23.7. Поджелудочный эпителий 437
23.2.8. Почка 438
Протокол 23.8. Эпителий почки 439
Протокол 23.9. Бронхиальный и трахеальный
эпителий 440
23.2.9. Бронхиальный и трахеальный
эпителий 440
23.2.10. Эпителий ротовой полости 441
Протокол 23.10. Кератиноциты ротовой
полости 441
Протокол 23.11. Эпителий простаты 442
23.2.11. Простата 442
23.3. Мезенхимные клетки 444
23.3.1. Соединительная ткань 444
23.3.2. Жировая ткань 444
Протокол 23.12. Первичная культура
адипоцитов 445
12
Оглавление
Протокол 23.13. Выделение и культивирование
гладкомышечных клеток 446
23.3.3. Мышцы 446
Протокол 23.14. Культура миобластов
взрослой мышцы взрослого человека 447
Протокол 23.15. Культура отдельных
мышечных волокон скелетных мышц 449
Протокол 23.16. Хондроциты на альгинатных
бусинах 450
23.3.4. Хрящ 450
23.3.5. Кость 453
Протокол 23.17. Остеобласты 453
23.3.6. Эндотелий 454
Протокол 23.18. Выделение и культивирование
клеток сосудистого эндотелия 455
23.4. Нейроэктодермальные клетки 458
23.4.1. Нейроны 458
Протокол 23.19. Гранулярные клетки мозжечка 459
23.4.2. Глиальные клетки 460
Протокол 23.20. Ольфакторные капсульные
клетки (ОЕС) 461
23.4.3. Эндокринные клетки 463
23.4.4. Меланоциты 463
Протокол 23.21. Меланоциты 464
23.4.5. Подтверждение идентичности
меланоцитов 465
23.5. Гематопоэтические клетки 465
23.5.1. Долгоживущие культуры клеток
костного мозга 467
Протокол 23.22. Долгоживущая культура
гематопоэтических клеток костного мозга 467
Протокол 23.23. Анализ колониеобразования
культуры гематопоэтических клеток 468
23.5.2. Анализ колониеобразования
культуры гематопоэтических клеток.. 468
23.6. Гонады 469
23.7. Стволовые клетки 470
23.7.1. Эмбриональные стволовые клетки... 470
23.7.2. Мультипотентные стволовые
клетки взрослого организма 470
23.7.3. Источники стволовых клеток
и работа с ними 471
ГЛАВА 24. КУЛЬТУРА ОПУХОЛЕВЫХ
КЛЕТОК 472
24.1. Проблемы культивирования
опухолевых клеток 472
24.2. Взятие образцов 473
24.3. Дезагрегация 473
24.4. Первичная культура 474
24.5. Характеристика 474
24.6. Размножение клеточных линий 475
24.6.1. Постоянные клеточные линии 476
24.7. Селективная культура опухолевых
клеток 476
24.7.1. Селективные среды 476
24.7.2. Конфлюэнтные фидерные слои 476
Протокол 24.1. Выращивание клеток
на конфлюэнтном фидерном слое 477
24.7.3. Суспензионное клонирование 478
24.7.4. Гистотипическая культура 478
24.7.5. Ксенотрансплантат 479
24.7.6. Характеризация культур
опухолевых клеток 479
24.7.7. Сохранение тканей замораживанием.. 479
Протокол 24.2. Замораживание биопсии 479
24.8. Специфические опухолевые культуры 480
24.8.1. Молочная железа 480
24.8.2. Легкое 480
24.8.3. Желудок 481
24.8.4. Толстый кишечник 481
Протокол 24.3. Культура колоректальных
опухолей 482
24.8.5. Поджелудочная железа 483
24.8.6. Яичники 484
24.8.7. Простата 484
24.8.8. Мочевой пузырь 484
24.8.9. Кожа 484
24.8.10. Шейка матки 485
24.8.11. Глиома 485
24.8.12. Нейробластома 485
24.8.13. Сперматоцитома 486
24.8.14. Лимфома и лейкемия 486
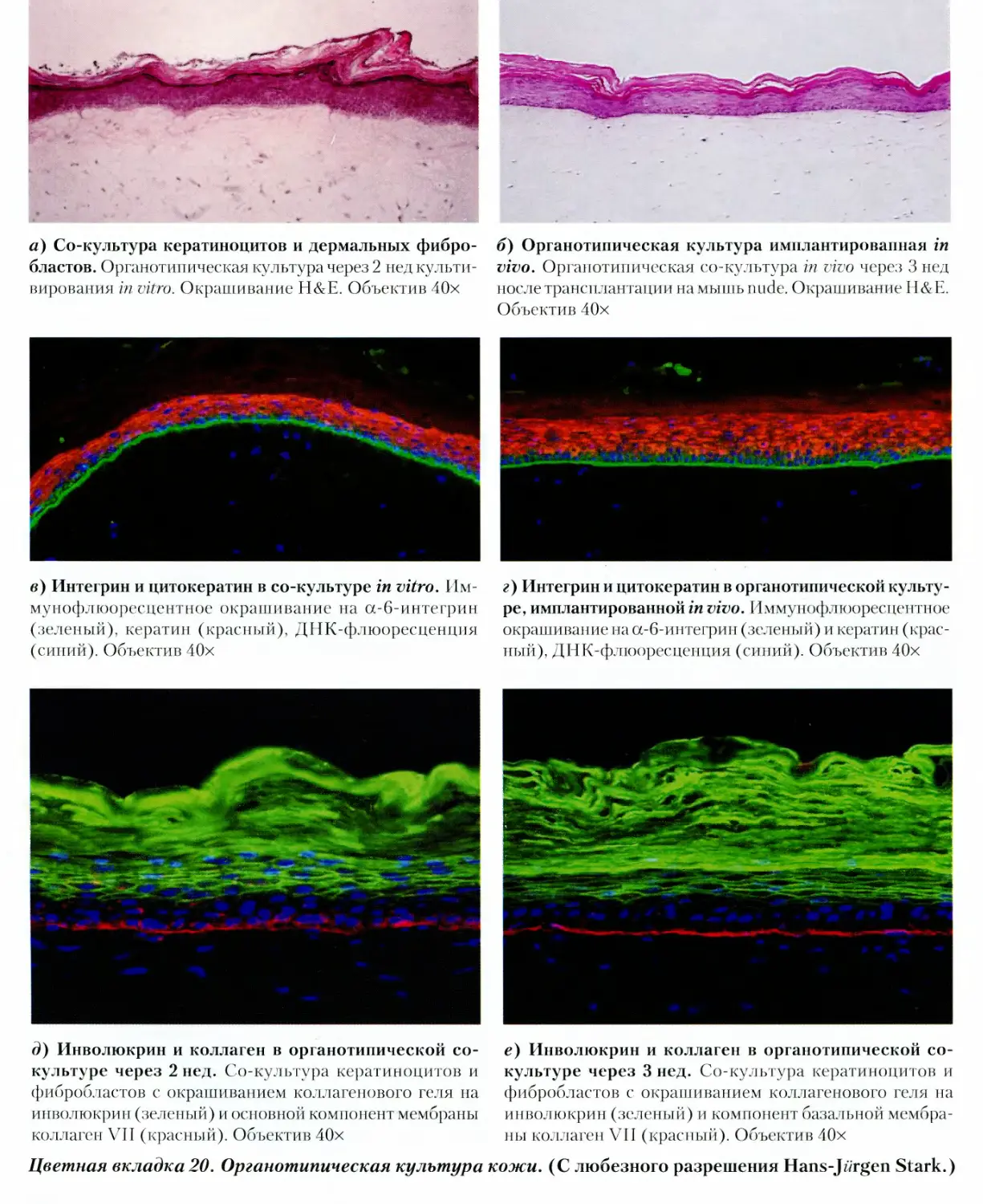
ГЛАВА 25. ОРГАНОТИПИЧЕСКАЯ
КУЛЬТУРА 487
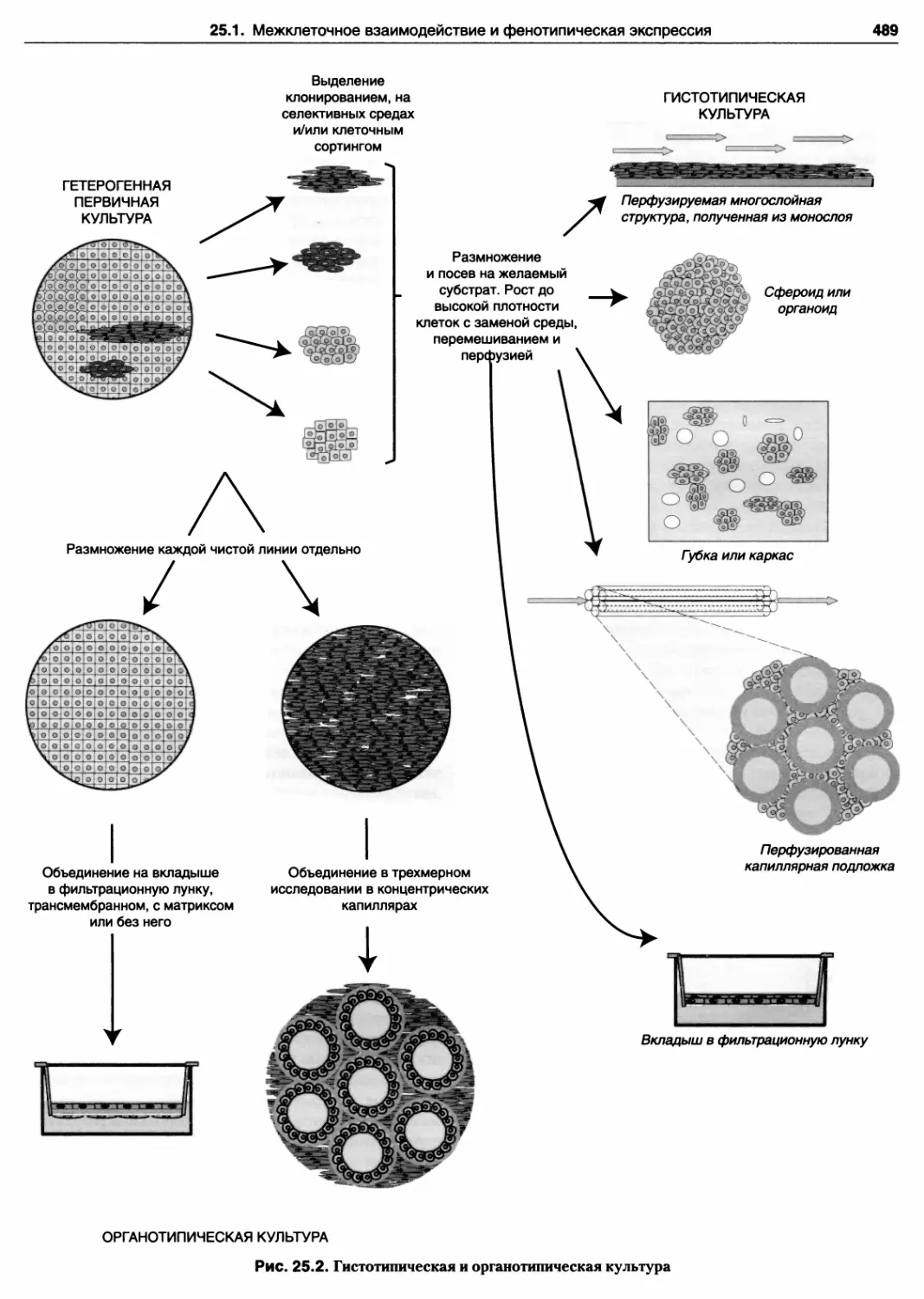
25.1. Межклеточное взаимодействие
и фенотипическая экспрессия 487
25.1.1. Роль клеточной плотности 487
25.1.2. Реципрокные взаимодействия 487
25.1.3. Выбор модели 488
25.2. Органная культура 490
25.2.1. Обмен питательных веществ
и газов 490
25.2.2. Структурное единство 490
25.2.3. Рост и дифференцировка 490
25.2.4. Ограничения метода органной
культуры 491
25.2.5. Типы органных культур 491
Протокол 25.1. Органная культура 491
25.3. Гистотипическая культура 492
25.3.1. Метод геля и губки 492
25.3.2. Пустотелые волокна 493
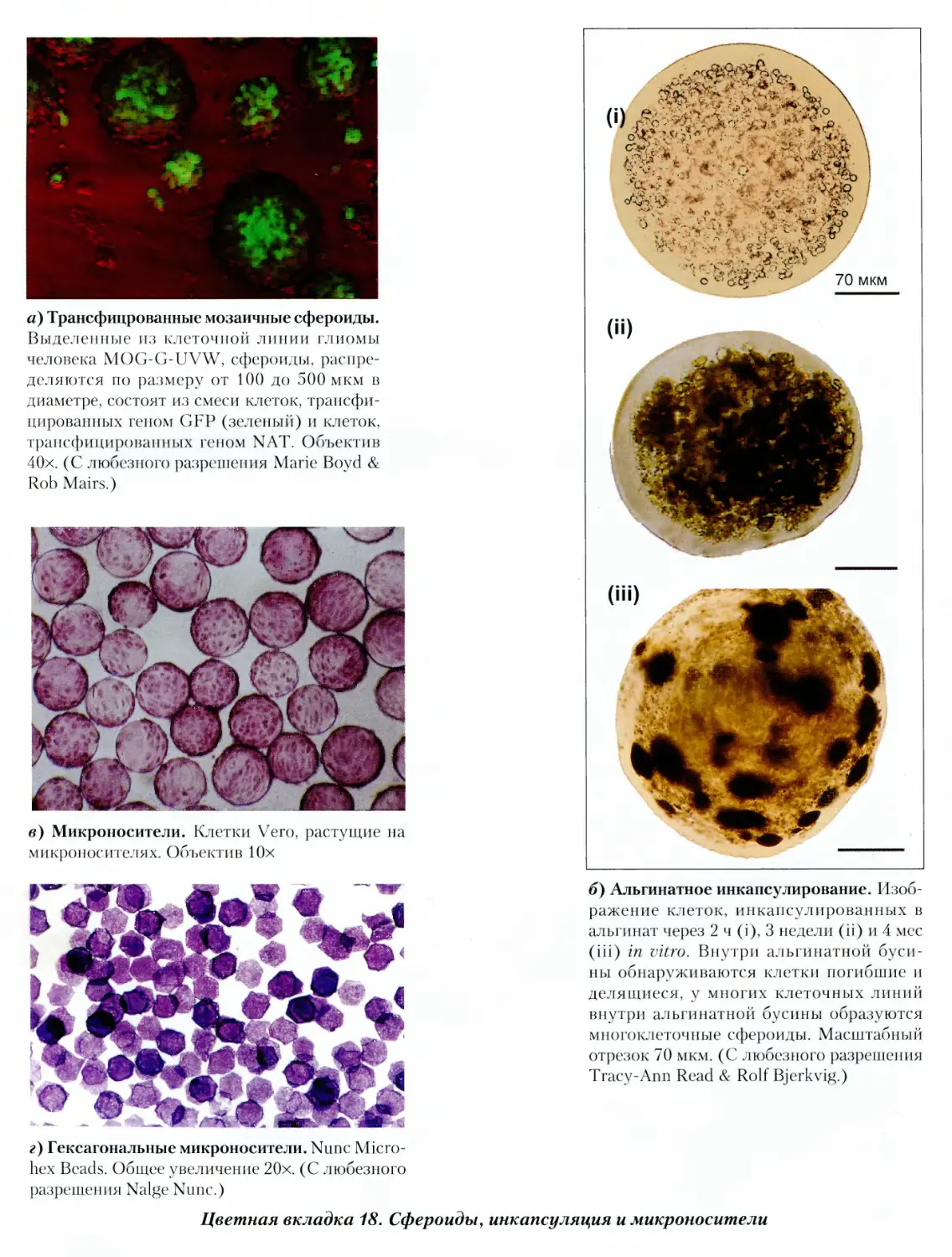
25.3.3. Сфероиды 493
Протокол 25.2. Сфероиды 494
25.3.4. Вращающиеся камерные системы ... 496
25.3.5. Иммобилизация живых клеток
в альгинате 496
Протокол 25.3. Альгинатное капсулирование 497
Протокол 25.4. Вкладыши для фильтрационных
лунок 498
25.3.6. Вкладыши для фильтрационных
лунок 498
25.3.7. Культуры нейрональных агрегатов .. 500
25.3.8. Тканевой эквивалент и тканевая
инженерия 501
Протокол 25.5. Нейрональные агрегаты 501
25.4. Создание трехмерных изображений
клеток (З-В-реконструкция) 504
ГЛАВА 26. КРУПНОМАСШТАБНОЕ
ПРОИЗВОДСТВО КЛЕТОК 505
26.1. Крупномасштабное производство
суспензионных культур 505
Протокол 26.1. Перемешивание 4-литровой
емкости с суспензионной культурой 506
26.1.1. Постоянная культура 507
26.1.2. Масштаб и комплексность 508
26.1.3. Перемешивание и аэрация 508
Оглавление
13
26.2. Крупномасштабное производство
монослойных культур 510
26.2.1. Многослойные пропагаторы
(культиваторы) 510
Протокол 26.2. Система NUNC CELL FACTORY 511
26.2.2. Многоцелевые диски, спирали
и пробирки 512
26.2.3. Роллерные культуры 512
Протокол 26.3. Роллерная бутылочная культура ... 514
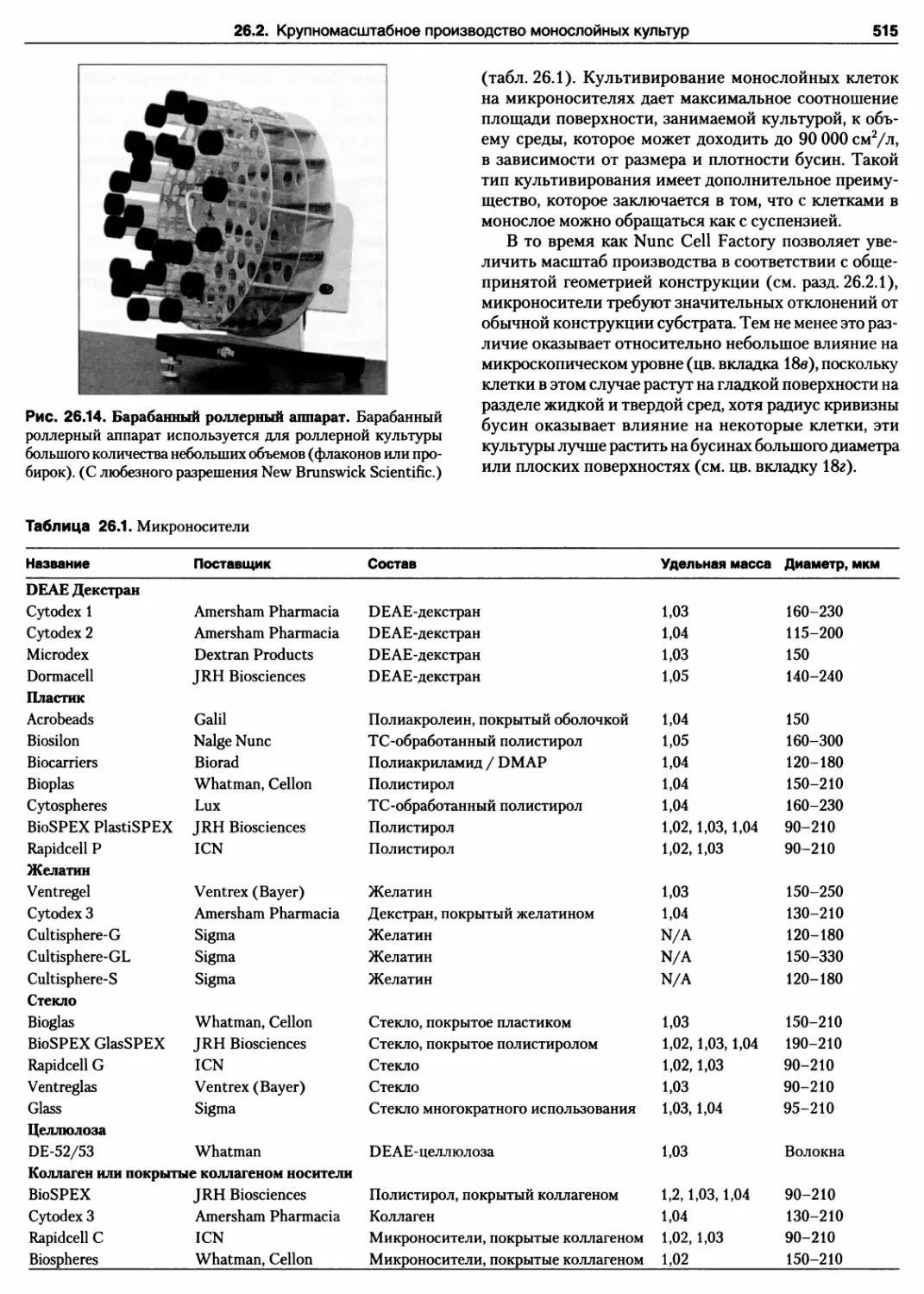
26.2.4. Микроносители 514
Протокол 26.4. Микроносители 516
26.2.5. Макроносители 516
26.2.6. Перфузионные монослойные
культуры 516
26.3. Контроль процесса 518
ГЛАВА 27. СПЕЦИАЛЬНЫЕ МЕТОДЫ 520
27.1. Методы выделения и исследования
лимфоцитов 520
Протокол 27.1. Приготовление лимфоцитов 520
27.1.1. Бласттрансформация 521
27.2. Авторадиография 521
Протокол 27.2. ФГА-стимуляция лимфоцитов 521
Протокол 27.3. Микрорадиография 522
27.3. Съемка в режиме замедленного
времени 526
Протокол 27.4. Видеозапись в замедленном
режиме времени 526
27.4. Конфокальная микроскопия 528
27.5. Синхронизация клеток 528
27.5.1. Разделение клеток 528
27.5.2. Блокирование клеточного цикла 529
27.6. Культура амниоцитов 529
Протокол 27.5. Культура амниоцитов 529
27.7. Культуры клеток пойкилотермных
животных 534
27.7.1. Клетки рыб 535
Протокол 27.6. Культуры клеток эмбриона
полосатого данио 536
27.7.2. Клетки насекомых 538
Протокол 27.7. Культуры клеток насекомых 538
27.8. Молекулярная гибридизация in situ 539
27.8.1. Анализ экспрессии генов РНК
методом гибридизации in situ 539
Протокол 27.8. Авторадиографическая
гибридизация in situ 539
27.8.2. Флюоресцентная гибридизация
in situ (FISH) при анализе генов
и хромосом 543
Протокол 27.9. FISH с использованием
однокопийных геномных зондов
и хромосомных красителей 543
27.9. Слияние соматических клеток 545
Протокол 27.10. Гибридизация клеток 545
27.9.1. Ядерный перенос 547
27.10. Продукция моноклональных антител 547
Протокол 27.11. Продукция моноклональных
антител 548
27.11. Перенос ДНК 551
27.11.1. Копреципитация с фосфатом
кальция 551
27.11.2. Липофекция 551
Протокол 27.12. Транзиторная трансфекция
методом липофекции 552
27.11.3. Электропорация 553
Протокол 27.13. Устойчивая трансфекция
методом электропорации 553
27.11.4. Другие методы переноса ДНК 554
27.11.5. Репортерные гены 555
Протокол 27.14. Окраска in situ
на $-галактозидазу 555
Протокол 27.15. Определение активности
хлорамфениколацетилтрансферазы
(CATASSAY) 556
ГЛАВА 28. РЕШЕНИЕ ПРОБЛЕМ 557
28.1. Медленный рост клеток 557
28.1.1. Проблемы, ограниченные
вашими собственными
исходными культурами 557
28.1.2. Более общие проблемы,
возникающие и у других
сотрудников 557
28.1.3. Не было ли у вас в лаборатории
каких-либо изменений? 558
28.2. Среда 558
28.2.1. Выбор среды 559
28.2.2. Нестабильные реактивы 559
28.3. Чистота составляющих частей 560
28.3.1. Правильно ли работает система
очистки воды? 560
28.3.2. Бикарбонат 560
28.3.3. Антибиотики 560
28.3.4. Сыворотка 560
28.4. Пластиковая посуда 560
28.5. Стеклянная посуда 561
28.5.1. Мытье посуды 561
28.6. Микробиологическая контаминация 561
28.6.1. Контаминация, ограниченная
одним исследователем 561
28.6.2. Распространенная контаминация 562
28.6.3. Идентификация контаминации 563
28.6.4. Деконтаминация 563
28.7. Химическая контаминация 563
28.7.1. Стеклянная посуда 563
28.7.2. Пипетки 563
28.7.3. Очистка воды 564
28.7.4. Другие реактивы 564
28.7.5. Порошки и аэрозоли 564
28.8. Первичная культура 564
28.8.1. Низкий выход клеток
в первичной культуре 564
28.8.2. Неверно выбраны клетки 565
28.8.3. Контаминация 565
28.9. Дифференцировка 565
28.9.1. Клетки не дифференцированы 565
28.9.2. Снижение образования продукта — 565
28.10. Смена среды 565
28.10.1. Обычные монослои 565
28.10.2. Клоны 565
28.11. Субкультура 566
28.11.1. Фазы клеточного цикла
при субкультуре 566
28.11.2. Старение 566
28.11.3. Среда 566
28.11.4. Неравномерный рост 566
28.12. Клонирование 566
28.12.1. Низкая эффективность посева 566
28.12.2. Диффузные колонии 567
28.12.3. Слишком много колоний на чашку.. 567
14
Оглавление
28.12.4. Неслучайное распределение 567
28.12.5. Неприкрепляющиеся клетки 567
28.13. Прекрестная контаминация 567
28.13.1. Симптомы перекрестной
контаминации 567
28.13.2. Предупреждение перекрестной
контаминации 568
28.13.3. Элиминация перекрестной
контаминации 568
28.14. Криоконсервация 568
28.14.1. Плохое восстановление 568
28.14.2. Изменение внешнего вида культуры
после криоконсервации 568
28.14.3. Контаминация 569
28.14.4. Утрата рабочей культуры 569
28.15. Грануляция клеток 569
28.16. Подсчет клеток 569
28.16.1. Гемоцитометр 569
28.16.2. Электронный счетчик клеток
посредством измерения
сопротивления при пропускании
через дроссель 569
28.17. Жизнеспособность 570
28.17.1. Морфологические признаки 570
28.17.2. Определение жизнеспособности 570
28.17.3. Цитотоксичность 570
ГЛАВА 29. ЗАКЛЮЧЕНИЕ 571
Приложение I. ПРИГОТОВЛЕНИЕ
РЕАКТИВОВ 572
Приложение II. ИСТОЧНИКИ
ОБОРУДОВАНИЯ И МАТЕРИАЛОВ 579
Приложение III. ПРОИЗВОДИТЕЛИ
ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ 601
Приложение IV. СЛОВАРЬ
УПОТРЕБЛЯЕМЫХ ТЕРМИНОВ
[по Schaeffer, 1990] 633
Приложение V. ОСНОВНАЯ ЛИТЕРАТУРА .. 640
Список литературы 641
Предметный указатель 689
Список рисунков
1.1.
Рост количества публикаций по культуре тканей
6.1.
Возможность заражения
1.2.
Области применения культуры тканей
6.2.
Возможное расположение оборудования в рабо¬
1.3.
Типы клеточных культур
чей зоне ламинарного шкафа
2.1.
Упражнение по замораживанию
6.3.
Плохо организованное расположение предметов
3.1.
Клеточная адгезия
в рабочей зоне
3.2.
Межклеточные контакты
6.4.
Планировка рабочей зоны в ламинарном шкафу
3.3.
Рост клеток А549 на матригеле
с горизонтальным воздушным потоком
3.4.
Клеточный цикл
6.5.
Планировка рабочей зоны на открытом столе
3.5.
Регуляции клеточного цикла
6.6.
Способ удерживания груши и крышки от фла¬
3.6.
Дифференцировка из стволовых клеток
кона в руке
3.7.
Клеточное взаимодействие и передача сигнала
6.7.
Воздушные потоки в ламинарном шкафу
3.8.
Развитие клеточной линии
6.8.
Стакан для слива
3.9.
Количество хромосом в клетках конечной и пос¬
6.9.
Правильное введение пипетки в пипетирующее
тоянной клеточных линий
устройство
4.1.
Небольшая лаборатория культивирования тканей
6.10.
Чашки Петри в коробке
4.2.
Лаборатория культивирования тканей средних
6.11.
Заполнение флакона газом
размеров
7.1.
Переполненные цилиндры для использованных
4.3.
Лаборатория культивирования тканей с приле¬
пипеток
жащими препаративными комнатами
7.2.
Правильное введение пипетки в пипетирующее
4.4.
Большая лаборатория культивирования тканей
устройство
4.5.
Термальная комната
7.3.
Хомут для баллона
4.6.
Раковина для мойки и моечная машина для пи¬
7.4.
Колба для спиртовой стерилизации инструментов
петок
7.5.
Ламинарный шкаф биологической безопасности
4.7.
Хранилище для жидкого азота и морозильное
8.1.
Количество клеток и площадь поверхности
оборудование
8.2.
Многолуночные плашки
5.1.
Ламинарный шкаф
8.3.
Чашки Петри
5.2.
Удаление среды с помощью насоса
8.4.
Пластиковые флаконы
5.3.
Вакуумный насос Integra
8.5.
Многопластинчатый флакон
5.4.
Инвертированный микроскоп
8.6.
Маленькие флаконы с перемешиванием
5.5.
Пипетирующее устройство
8.7.
Вентилируемые чашки Петри
5.6.
Автоматическая пипетка (пипеттор)
8.8.
Вентилируемые флаконы
5.7.
Градуированный бутылочный диспенсер
8.9.
Пузырьки и флаконы с закручивающимися
5.8.
Пипетка с многократно дозируемым объемом
крышками
5.9.
Автоматический диспенсер
8.10.
Случайный рост
5.10.
Многоканальная пипетка
8.11.
Культивирование на полых волокнах
5.11.
Автоматическое заполняющее устройство
8.12.
Морфология фидерных слоев
5.12.
Планшетный сканер
9.1.
Осмометр
5.13.
Устройство для переноса
11.1.
Мытье и стерилизация лабораторного стекла
5.14.
Замкнутая телевизионная система
11.2.
Механические щетки для мытья бутылок
5.15.
С02-инкубатор
11.3.
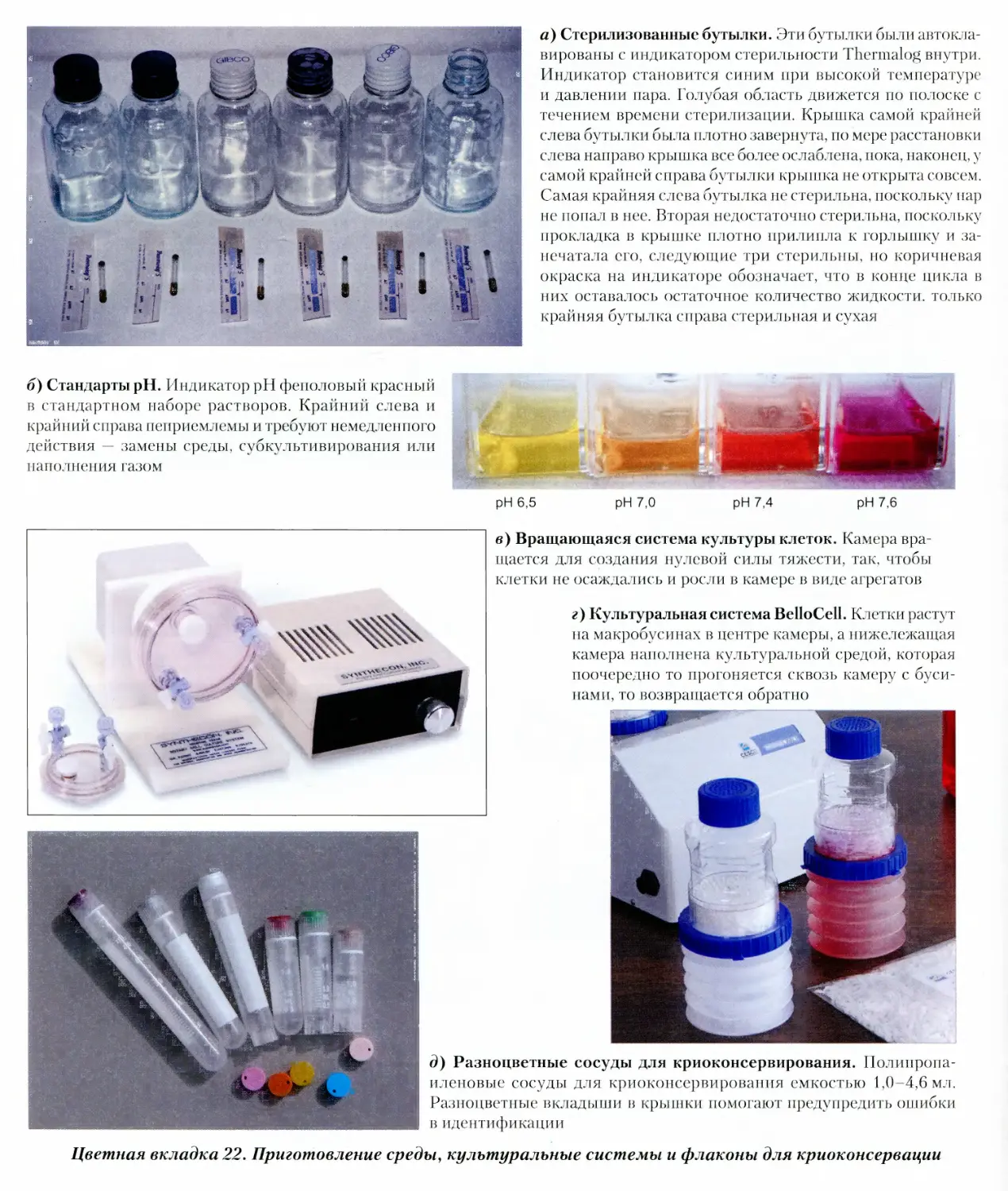
Стерилизованные бутылки
5.16.
Устройство С02-инкубатора
11.4.
Сифонное моющее устройство для пипеток
5.17.
Очиститель воды
11.5.
Мытье и стерилизация пипеток
5.18.
Настольный автоклав
11.6.
Полуавтоматическое устройство для затыкания
5.19.
Напольный автоклав
пипеток ватными пробками (Bellco)
5.20.
Диспенсер, соединяющийся с бутылкой
11.7.
Стерилизационный шкаф
5.21.
Моечная машина для стекла
11.8.
Упаковка закручивающихся крышек для стери¬
5.22.
Микроманипуляторы и подогреваемый столик
лизации
16
Список рисунков
11.9. Отношения между давлением и температурой
11.10. Очистка воды
11.11. Стерилизация фильтрацией
11.12. Одноразовые стерилизационные фильтры
11.13. Фильтрационная установка большой мощности
11.14. Фильтрация при помощи перистальтического
насоса
11.15. Крупномасштабная стерилизация фильтрацией
с использованием большого встроенного комп¬
лекта фильтров
11.16. Фильтры многократного использования
11.17. Предварительный фильтр
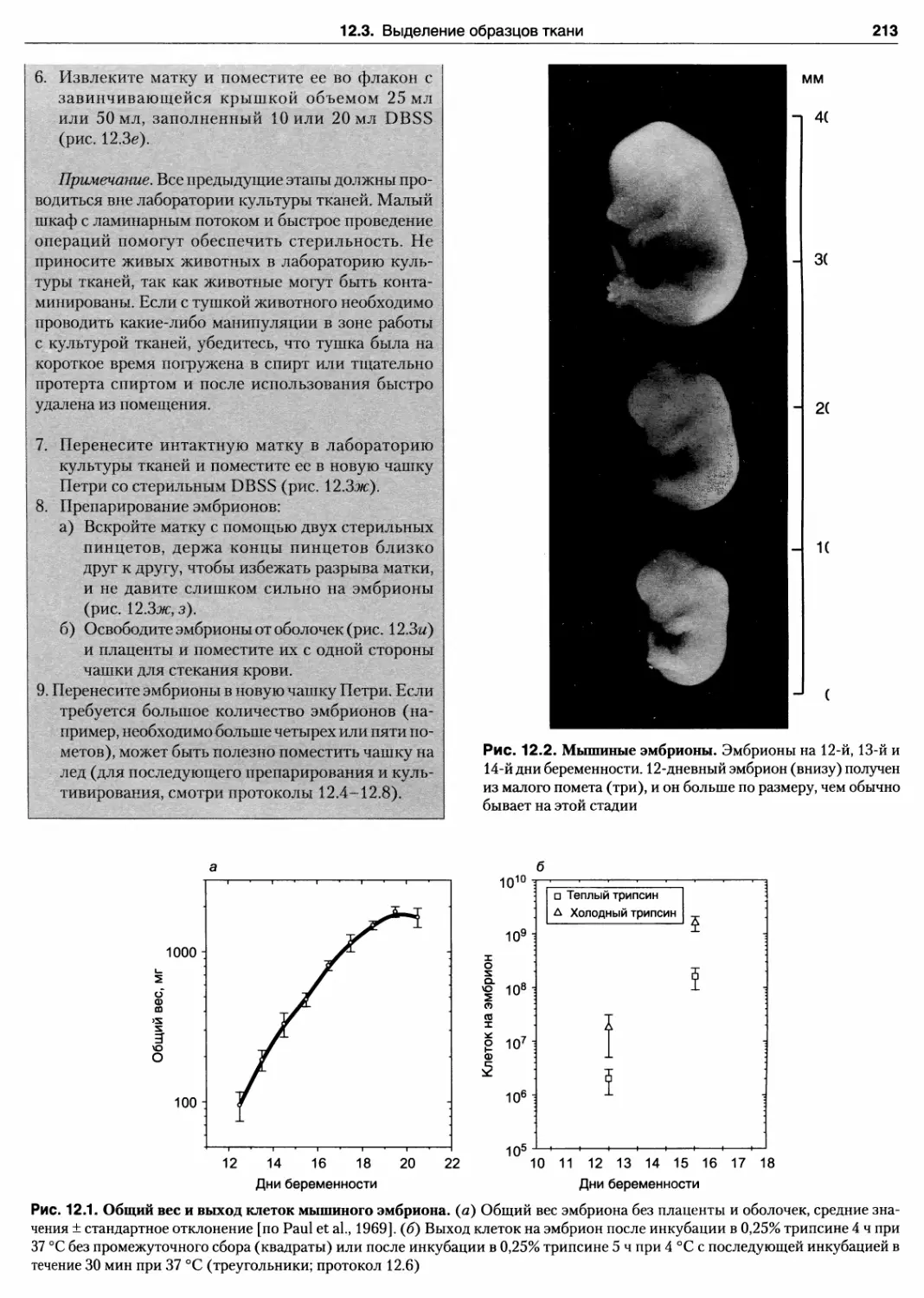
12.1. Общий вес и выход клеток мышиного эмбриона
12.2. Мышиные эмбрионы
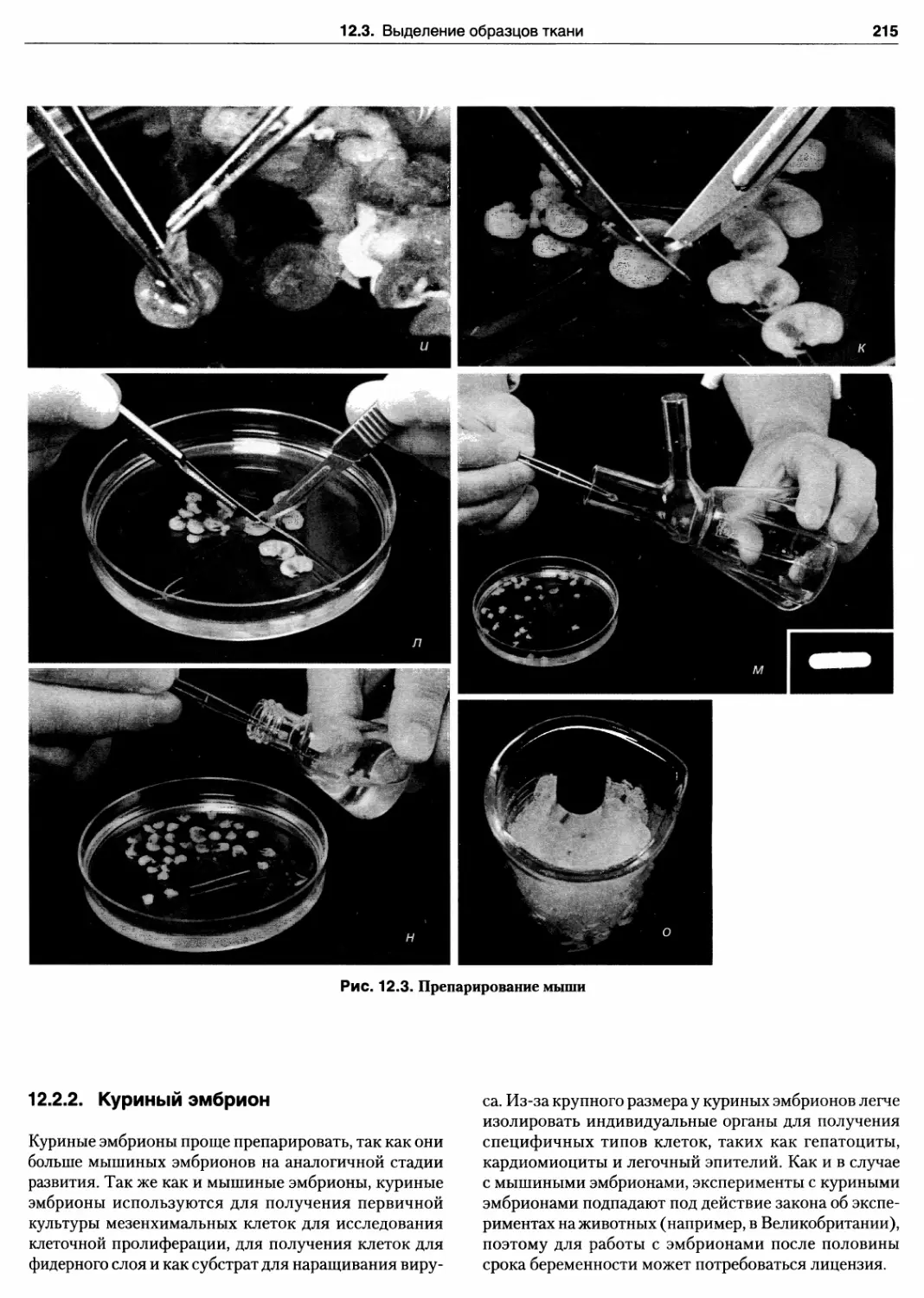
12.3. Препарирование мыши
12.4. Извлечение куриного эмбриона из яйца
12.5. Варианты первичной культуры
12.6. Культура первичного эксплантата
12.7. Дезагрегация с помощью теплого трипсина
12.8. Клеточное сито
12.9. Дезагрегация в холодном трипсине
12.10. Теплая и холодная трипсинизация
12.11. Препарирование куриного эмбриона
12.12. Дезагрегация ткани с помощью коллагеназы
12.13. Механическая дезагрегация
13.1. Плохое состояние культуры
13.2. Кривая роста и поддержание культуры
13.3. Субкультивирование монослоя
13.4. Серийное субкультивирование
13.5. Перемешиваемая культура
13.6. Параллельные культуры и антибиотики
14.1. Изменение числа клеток при клонировании
14.2. Клонирование с разведением
14.3. Клонирование в планшетах для микротитрации
14.4. Фидерные слои
14.5. Клонирование в суспензии
14.6. Клонирование в метилцеллюлозе
14.7. Кольца для клонирования
14.8. Выделение монослойного клона
14.9. Выделение суспензионных клонов
14.10. Селективные фидерные слои
14.11. Рост клеток меланомы, фибробластов и глии в
суспензии
14.12. Рост клеток в смешанной культуре
15.1. Разделение клеток по их плотности
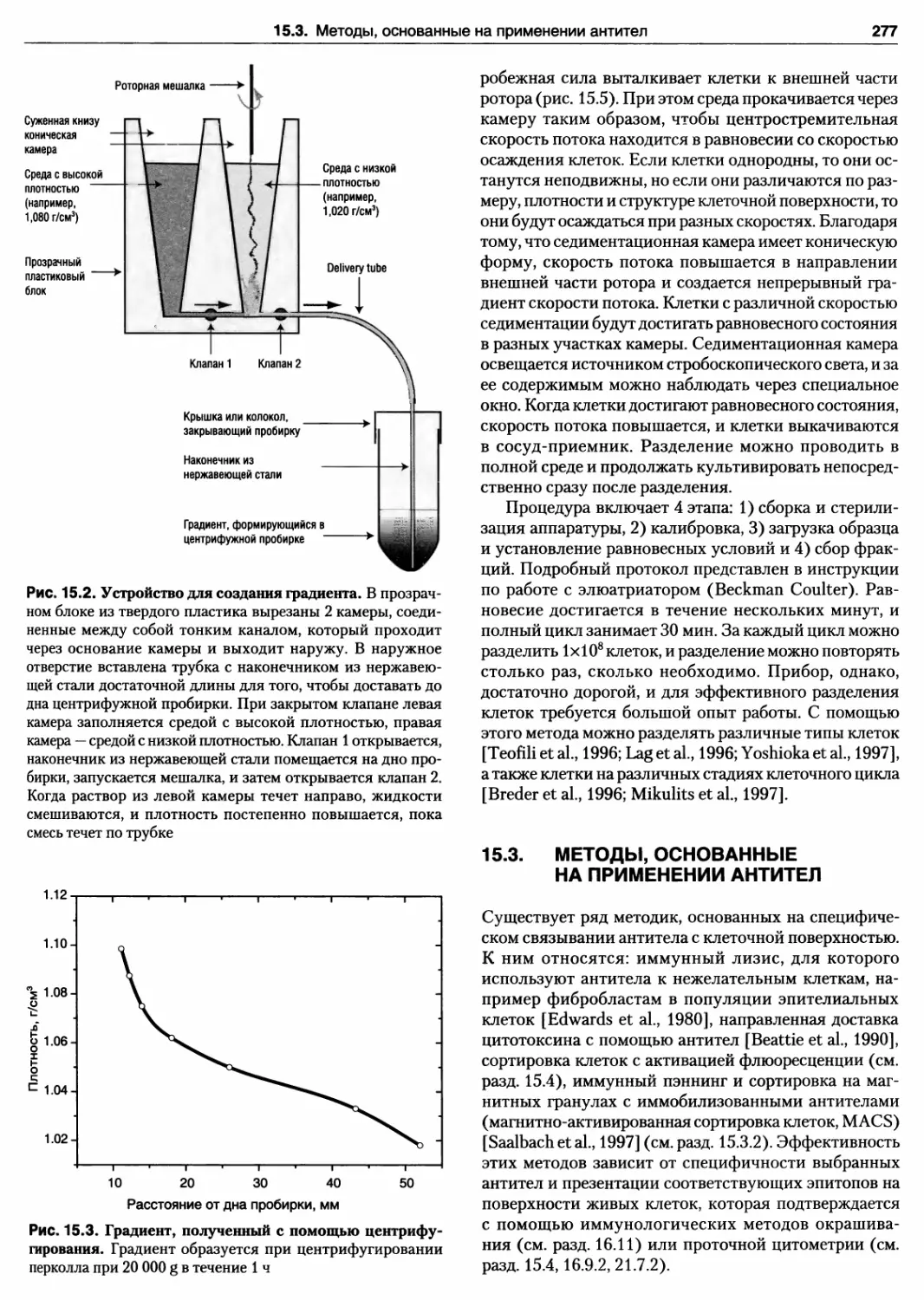
15.2. Устройство для создания градиента
15.3. Градиент, полученный с помощью центрифуги¬
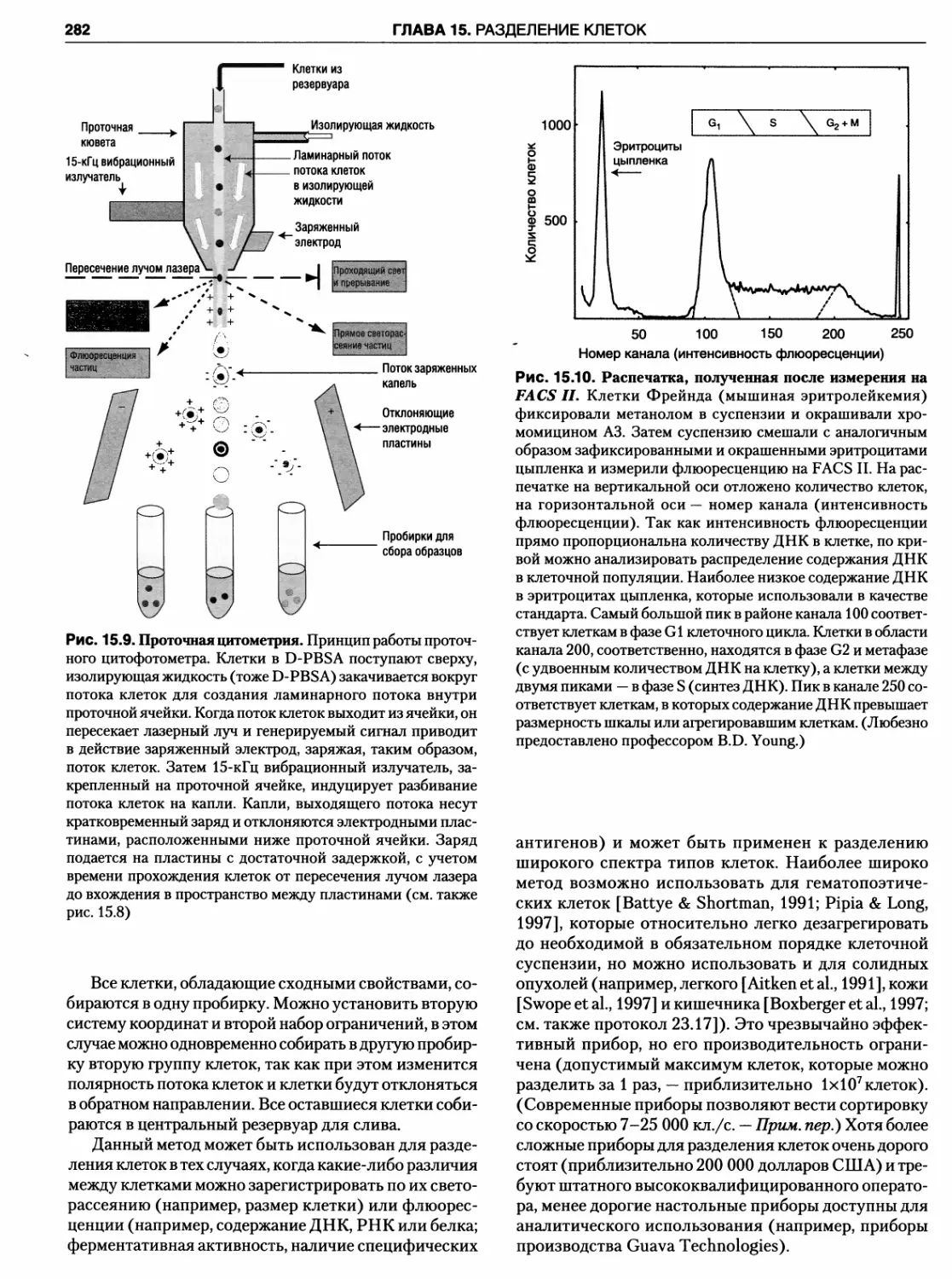
рования
15.4. Центрифужный элютриаторный ротор (Beckman)
15.5. Разделительная камера элюатриаторнного ротора
15.6. Магнитный сортинг
15.7. Магнитный сортинг клеток (MACS® Technology)
15.8. Флюоресцентно-активируемый сортер клеток
(FACS)
15.9. Проточная цитометрия
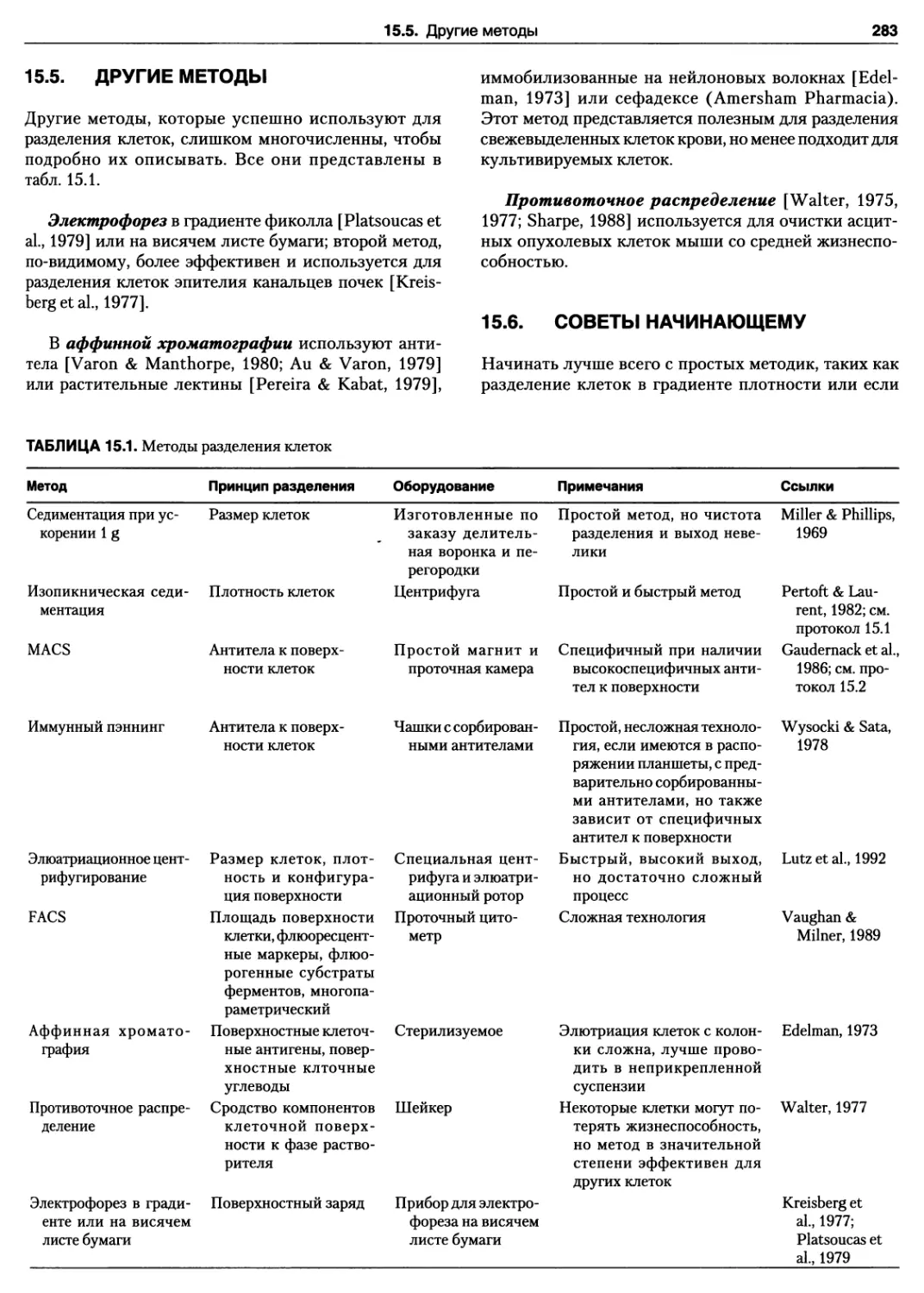
15.10. Распечатка, полученная после измерения на
FACS II
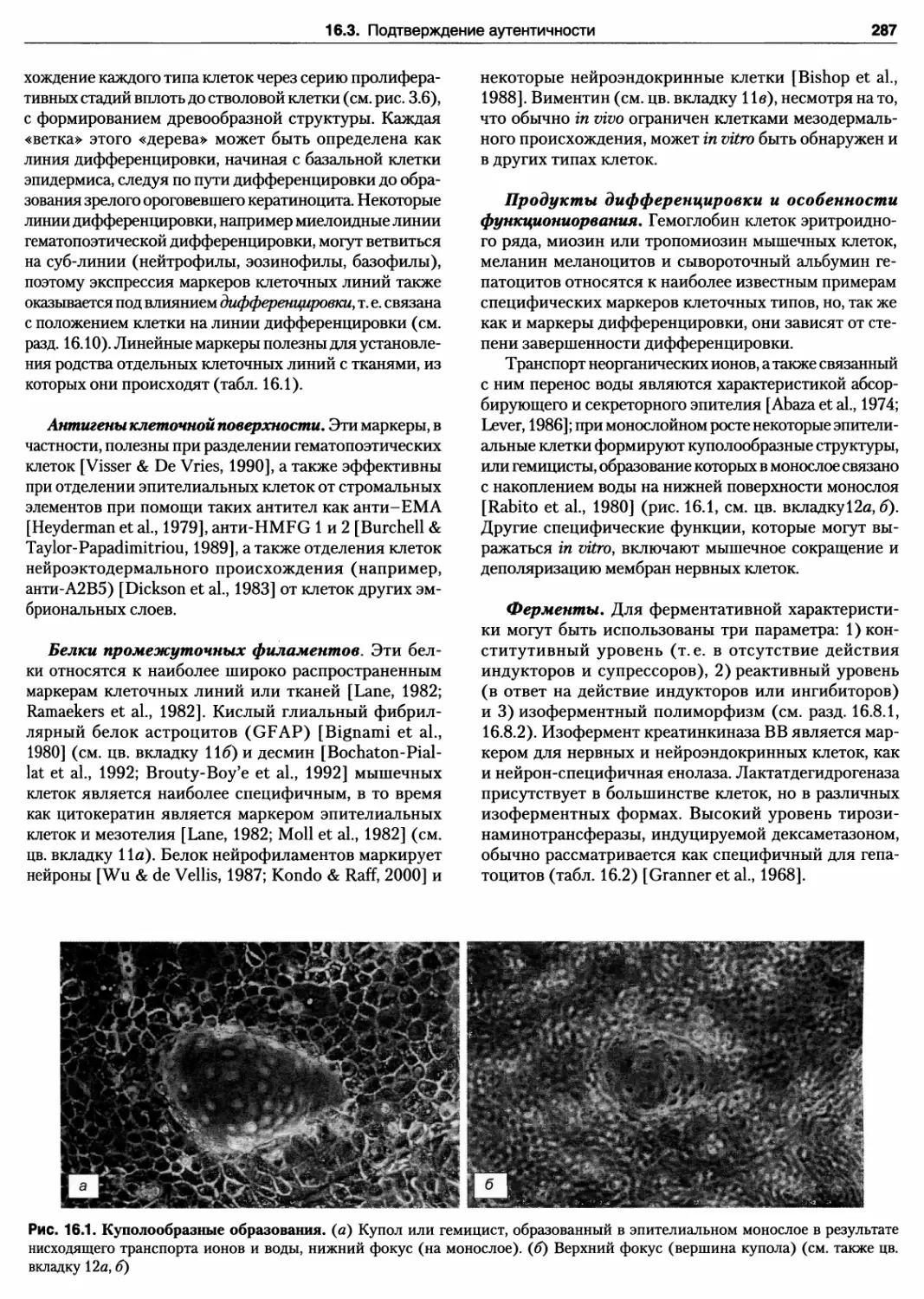
16.1. Куполообразные образования
16.2. Примеры морфологии клеток в культуре
16.3. Культуральные сосуды для цитологии
16.4. Цитоцентрифуга
16.5. Цитологический фильтр
16.6. Приготовление препарата хромосом
16.7. Окрашивание хромосом
16.8. Препарат кариотип
16.9. ДНК-фингерпринтинг
16.10. Сиквенирование ДНК
16.11. Изучение профиля ДНК
16.12. Изоферментный электрофорез
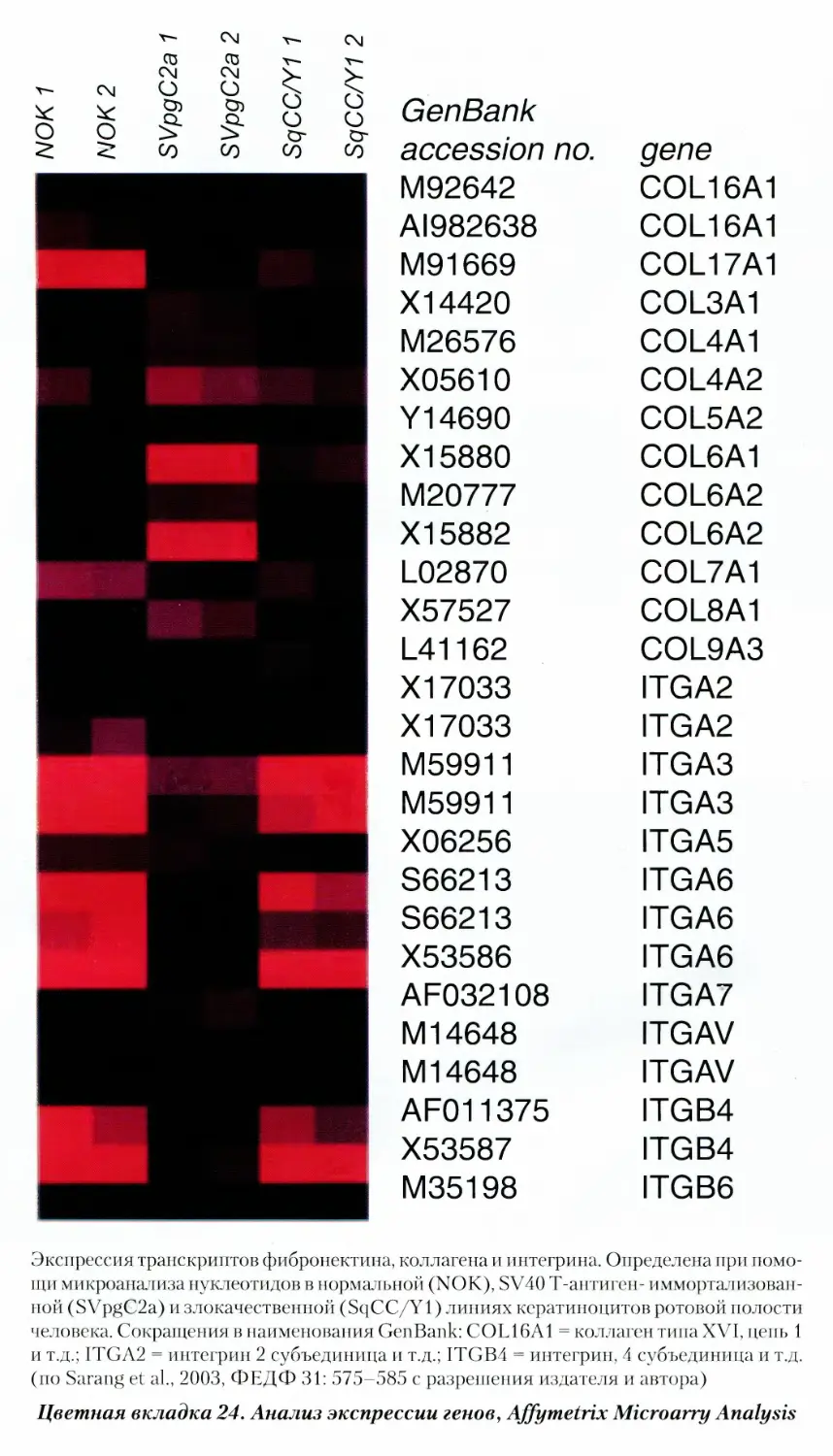
16.13. Прибор для иммуноанализа Agilent
17.1. Регуляция дифференцировки
17.2. Реципрокное паракринное взаимодействие
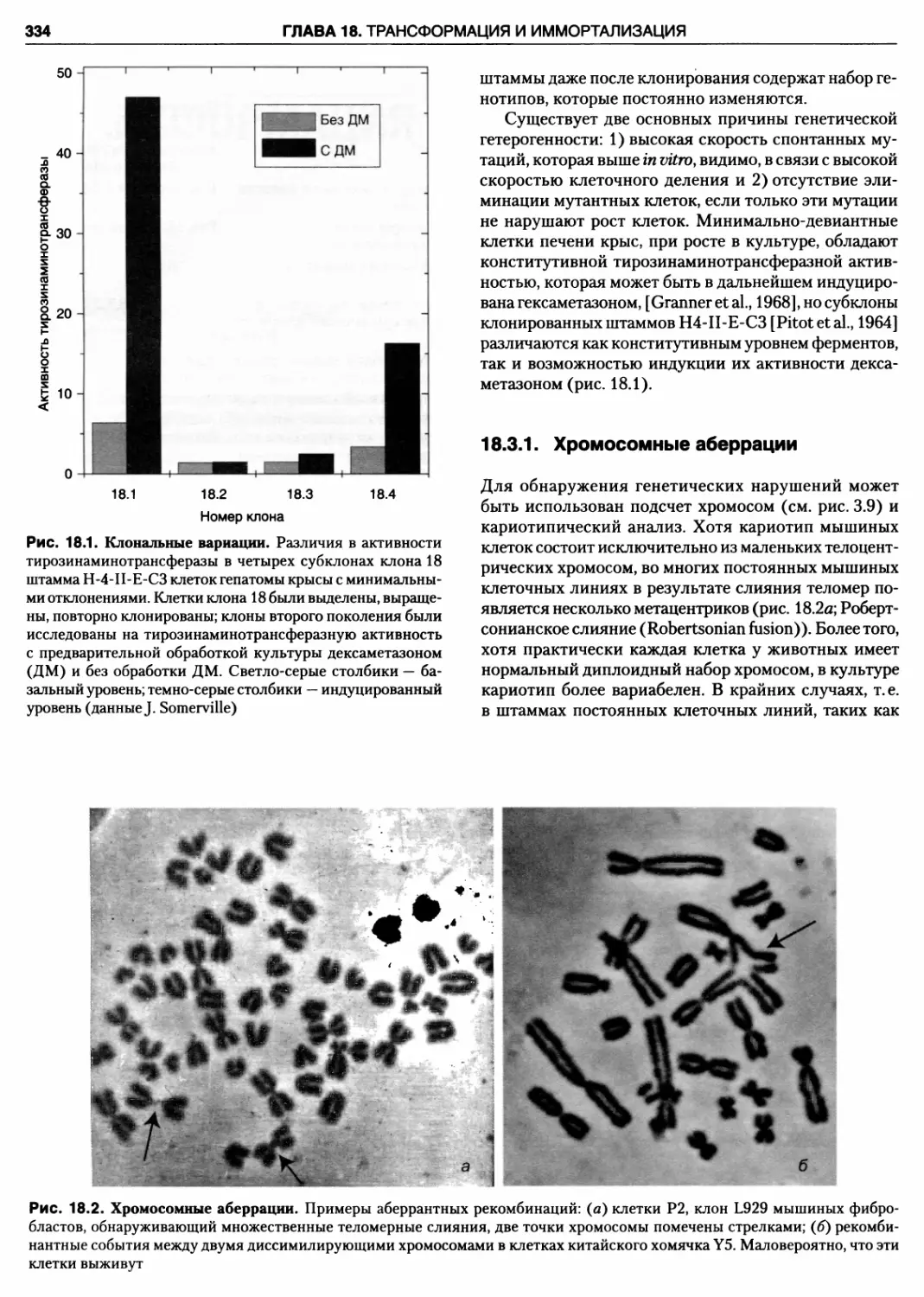
18.1. Клональные вариации
18.2. Хромосомные аберрации
18.3. Фокусы трансформации
18.4. Совокупное количество периодов удвоения
популяции (PD) иммортализованных клеток
hTERT по сравнению со стареющими контроль¬
ными клетками
18.5. Ограничение клеточной пролиферации
18.6. Эксперимент с использованием сердца цыпленка
18.7. Инвазия в фильтрующей лунке
18.8. Активатор плазминогена (РА)
19.1. Типы контаминации
19.2. Детекция микоплазмы с помощью ПЦР
20.1. Кривая замораживания
20.2. Ампулы на стержне
20.3. Пробка для узкогорлого морозильника
20.4. Охлаждающее устройство Nalge Nunc Cooler
20.5. Программируемый морозильник
20.6. Морозильники с жидким азотом
20.7. Конструкция азотного морозильника
20.8. Замораживание клеток
20.9. Размораживание клеток
20.10. Серийное возобновление культуры
20.11. Контейнер для транспортировки клеток
21.1. Использование гемоцитометра
21.2. Электронный счетчик клеток С AS Y
21.3. Схема работы счетчика клеток С AS Y
21.4. Аналоговая распечатка результатов подсчета
клеток на электронном счетчике СASY
21.5. Электронный счетчик клеток Beckman Coulter
Vi-CELL
21.6. Кривая роста
21.7. Внешний вид многолуночных планшетов
21.8. Интерпретация кривой роста
21.10. Разведение клеток для клонирования
21.11. Линейность эффективности посева
21.12. Автоматический счетчик колоний
21.13. Индекс мечения
21.14. Направление сканирования препаратов или
чашек
21.15. Фракции роста
22.1. Клоногенные исследования монослойных клеток
22.2. Кривая выживания
22.3. Интерпретация кривых выживания
22.4. Влияние фидерного слоя на долю выживания
клеток
Список рисунков
17
22.5. Микротитрационное исследование
22.6. Кривая ингибирования
22.7. Продолжительность исследования
22.8. Кривая падения IC50
22.9. Корреляция между микротитрационным и кло¬
ногенным методами исследования выживания
22.10. Органотипическое исследование
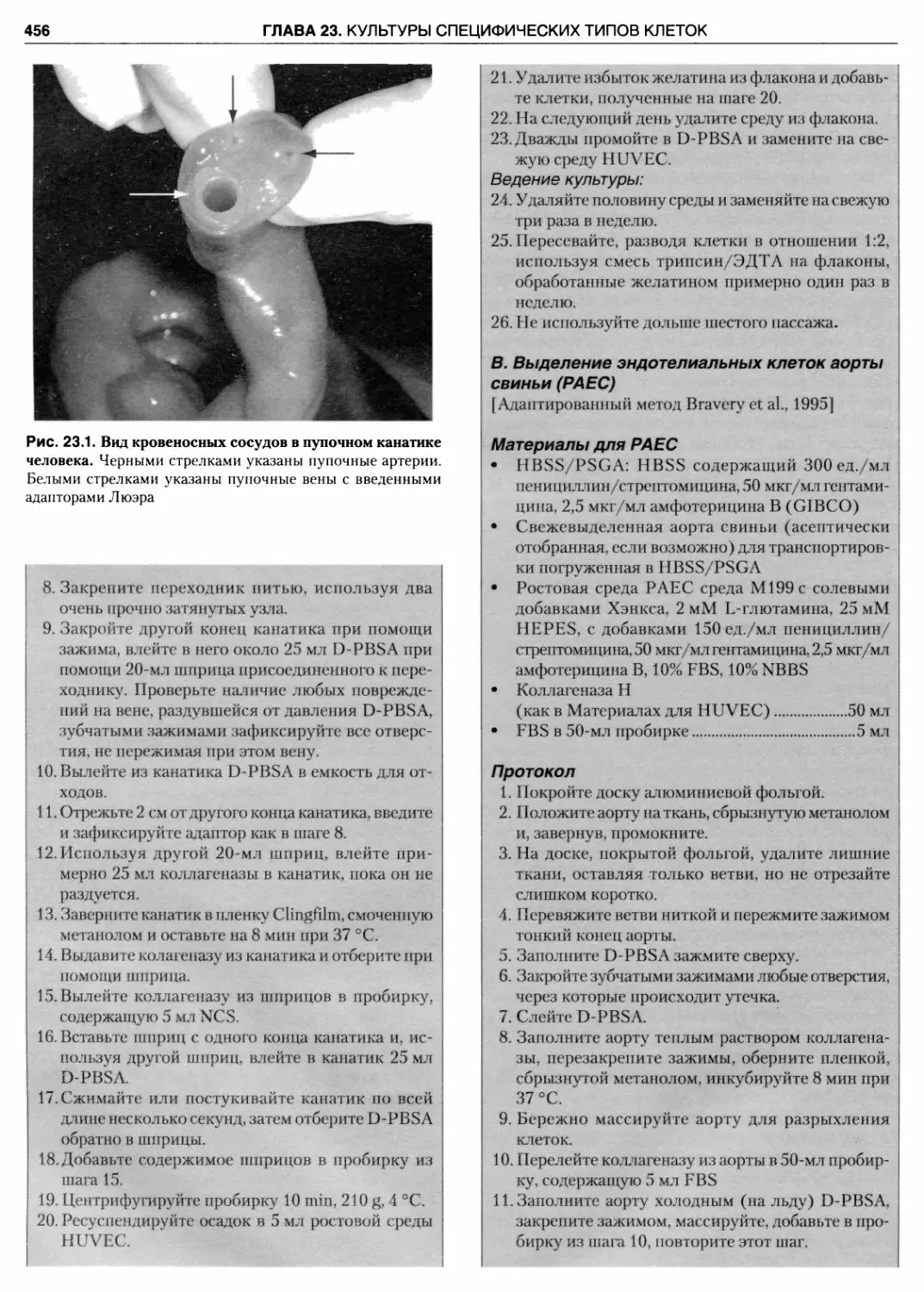
23.1. Вид кровеносных сосудов в пупочном канатике
человека
23.2. Клетки сосудистого эндотелия в культуре
23.3. Иссечение обонятельной луковицы
23.4. Культура меланоцитов
24.1. Конфлюэнтные фидерные слои
24.2. Клеточные линии карциномы желудка
24.3. Клеточные линии рака поджелудочной железы
24.4. Культуры глиомы человека
25.1. Влияние клеточной плотности на экспрессию
GFAP в клетках С6
25.2. Гистотипическая и органотипическая культура
25.3. Органная культура
25.4. Разделение клеток в сфероидах
25.5. Вращающаяся камерная система
25.6. Вращающаяся система для культуры клеток
Synthecon Rotatory Cell Culture System
25.7. Вкладыши в фильтровальный колодец
25.8. Вкладыши Transwells
25.9. Каркасы и матрицы
25.10. MRI-мониторинг клеток в 3D конструктах
25.11. MRI конструкта хряща
26.1. Большой сосуд для перемешивания
26.2. Перемешиваемая культура
26.3. Биостат
26.4. Ферментер с барботированием
26.5. Аэратор культуры BelloCell
26.6. Перфузия на полых волокнах
26.7. Система Membroferm
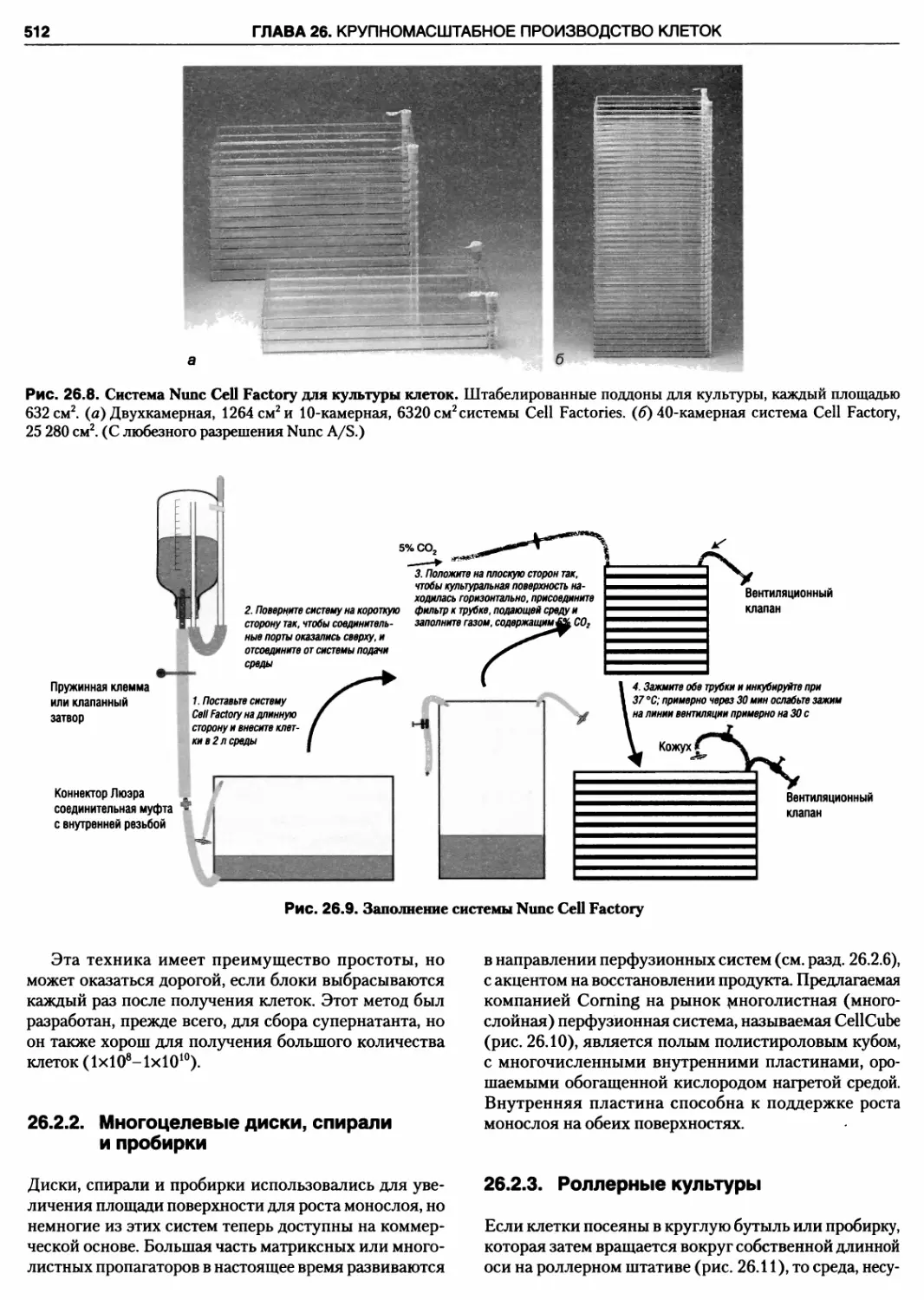
26.8. Система Nunc Cell Factory для культуры кле
ток
26.9. Заполнение системы Nunc Cell Factory
26.10. Система для культуры клеток Corning Cell
Cube
26.11. Бутыли для ролерных культур на стеллажах
26.12. Схема работы роллерной системы культуры
26.13. Примеры бутылей для роллерных культур
26.14. Барабанный роллерный аппарат
26.15. Реакторы с неподвижным слоем
26.16. Реакторы с псевдоожиженным слоем
26.17. Системы контроля биореактора
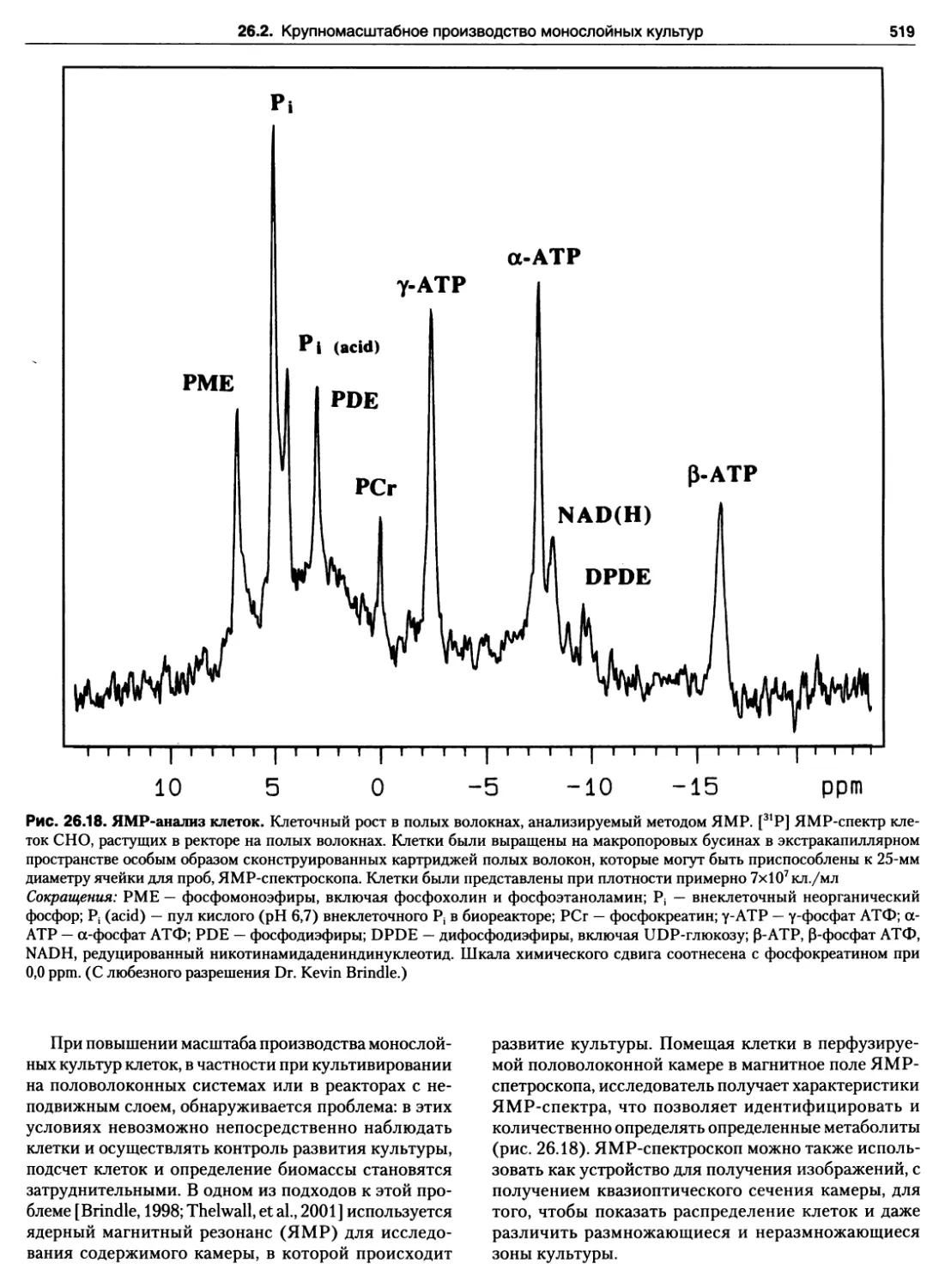
26.18. ЯМР-анализ клеток
27.1. Микроавторадиография
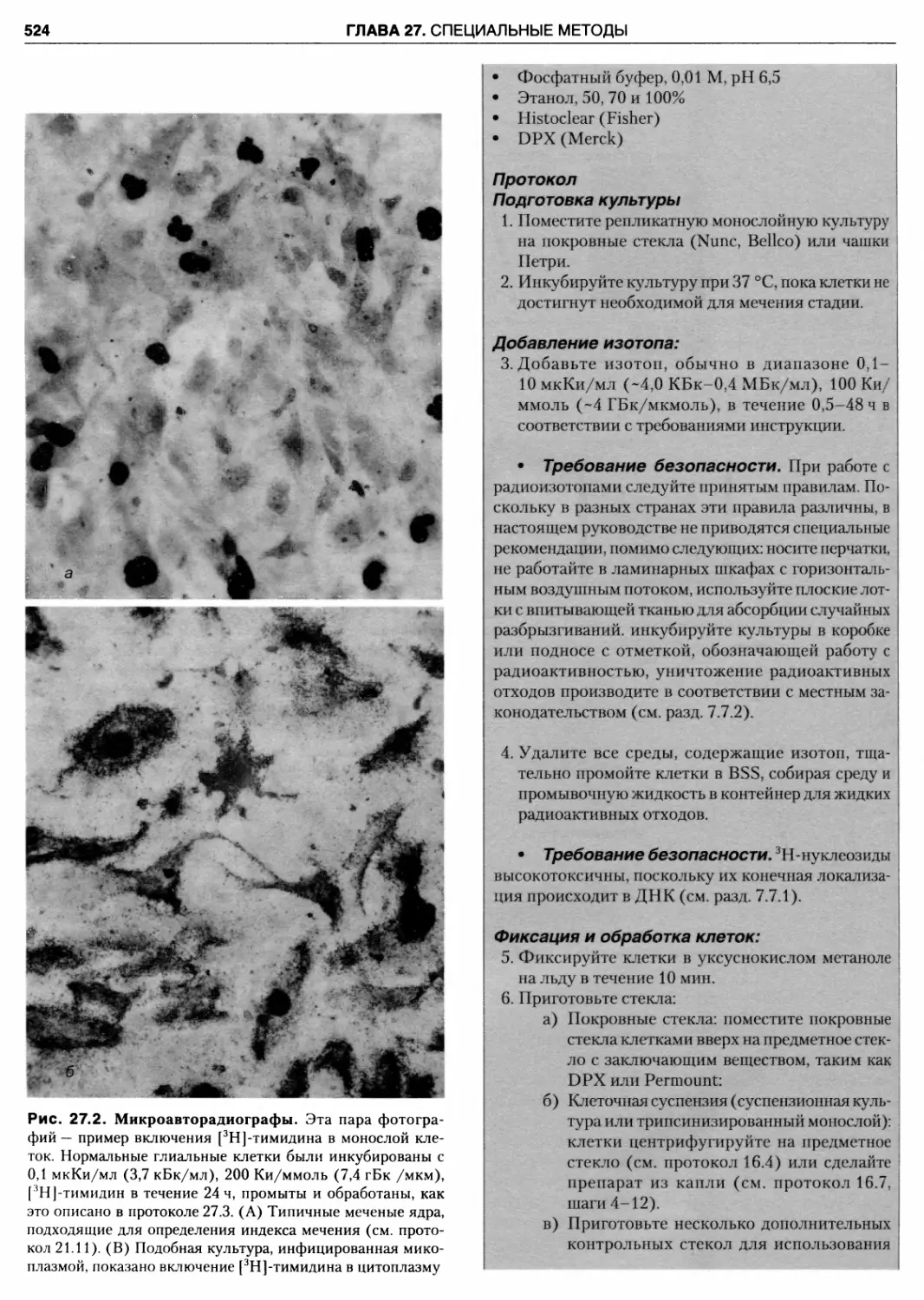
27.2. Микроавторадиографы
27.3. Гибридизация соматических клеток
27.4. Получение гибридом
27.5. Перенос ДНК
Список цветных вкладок
1. Первичная культура, человеческая
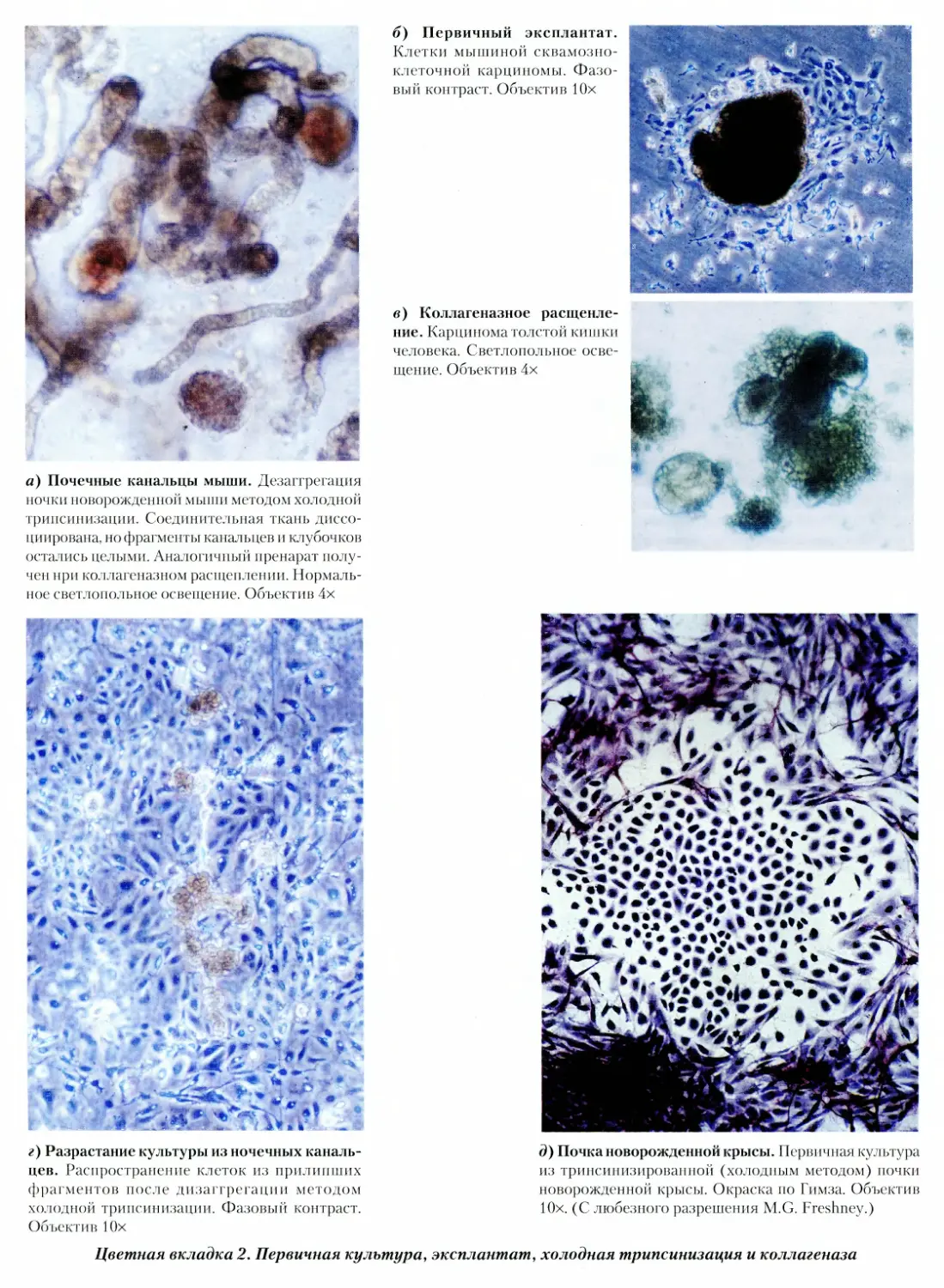
2. Первичная культура, эксплантат, холодная трип-
синизация и коллагеназа
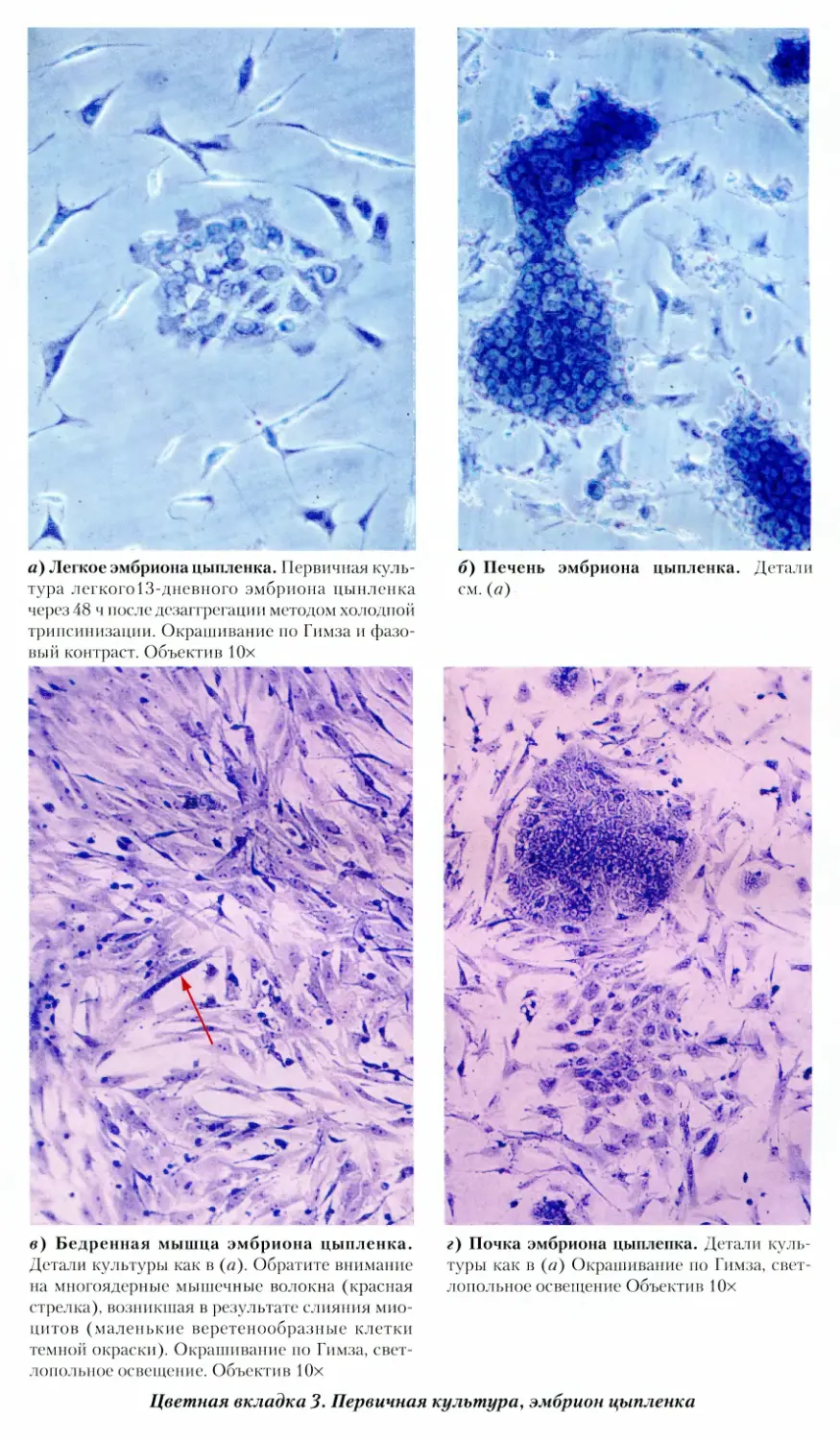
3. Первичная культура, эмбрион цыпленка
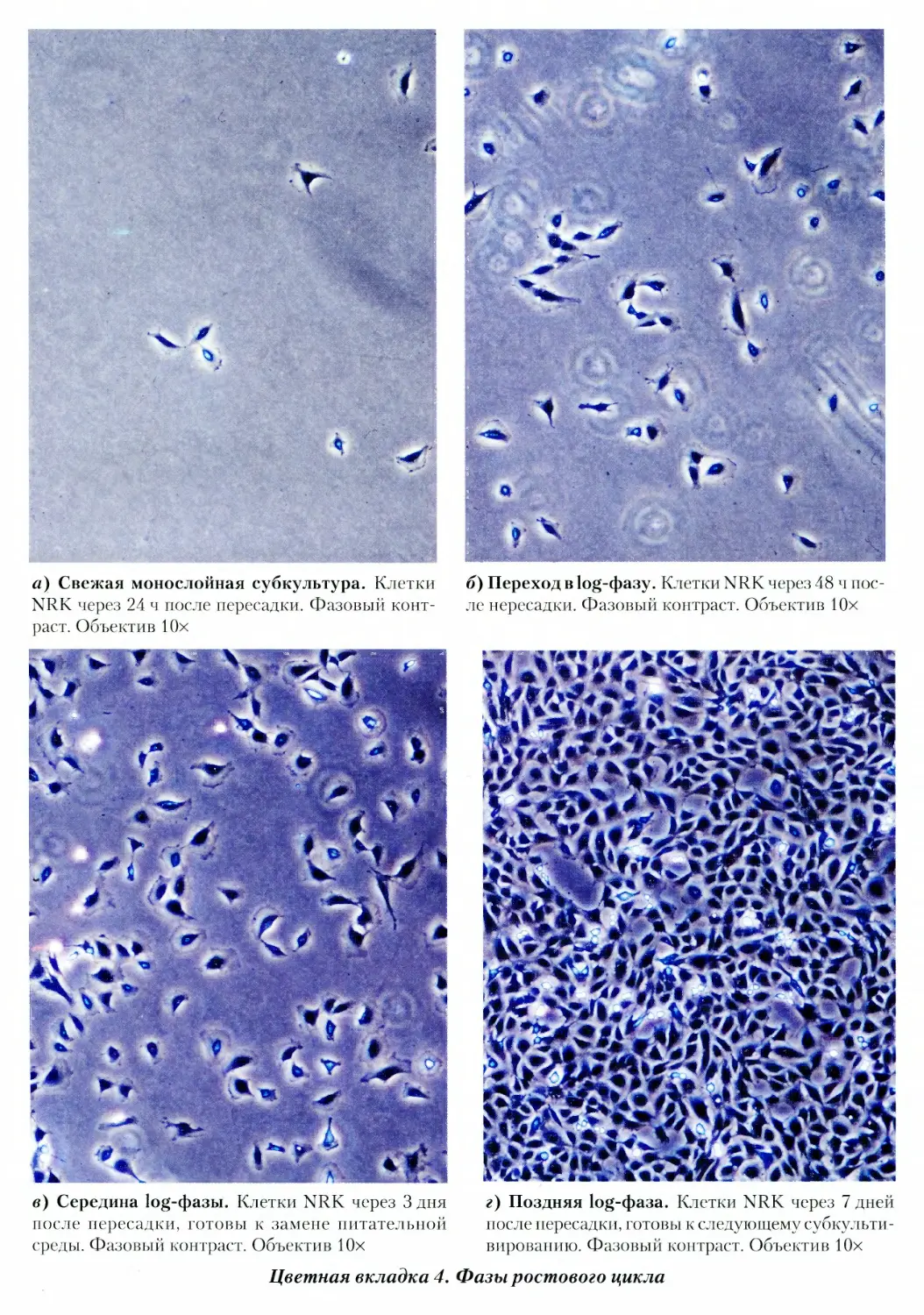
4. Фазы ростового цикла
5. Субкультивирование методом трипсинизации
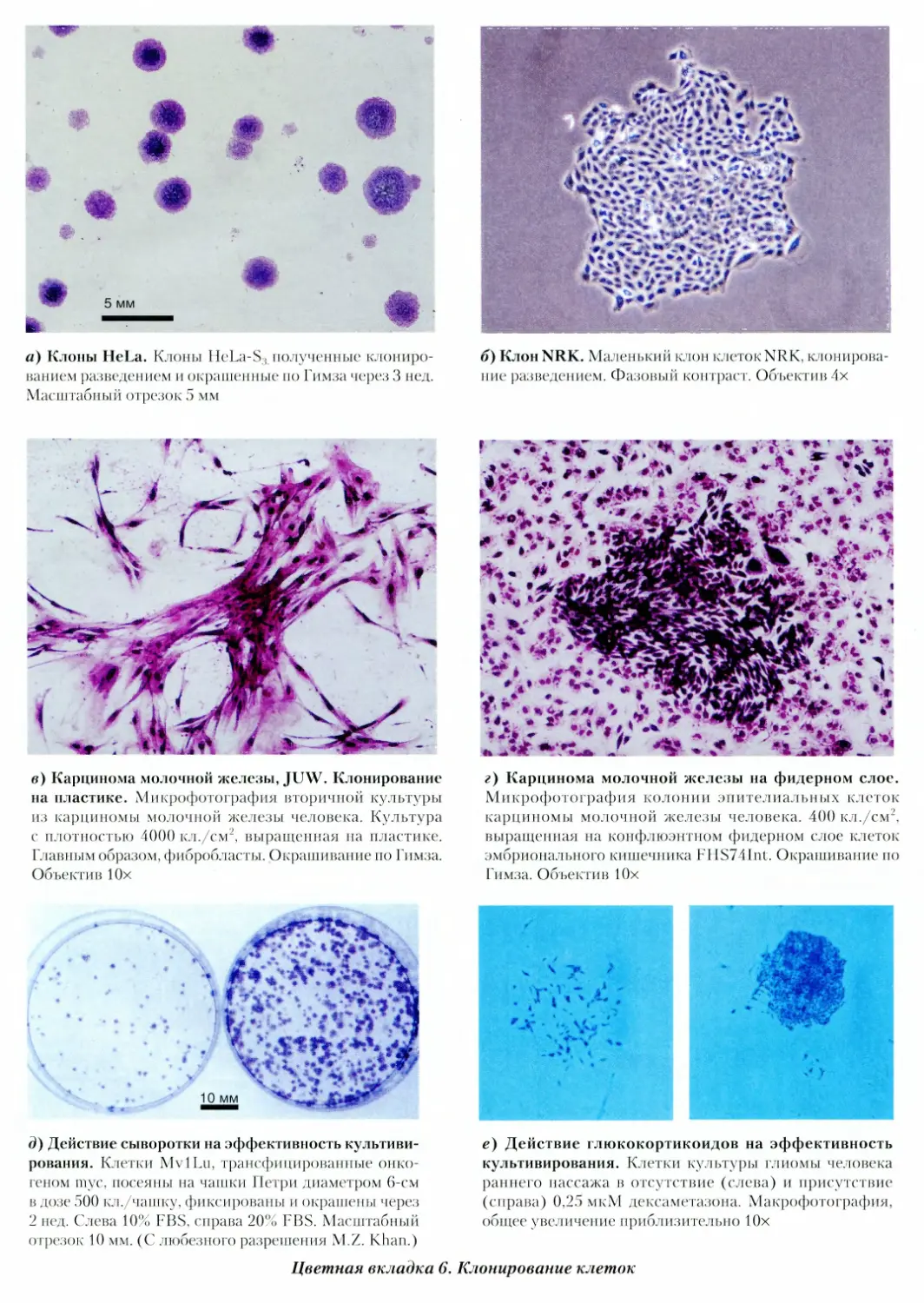
6. Клонирование клеток
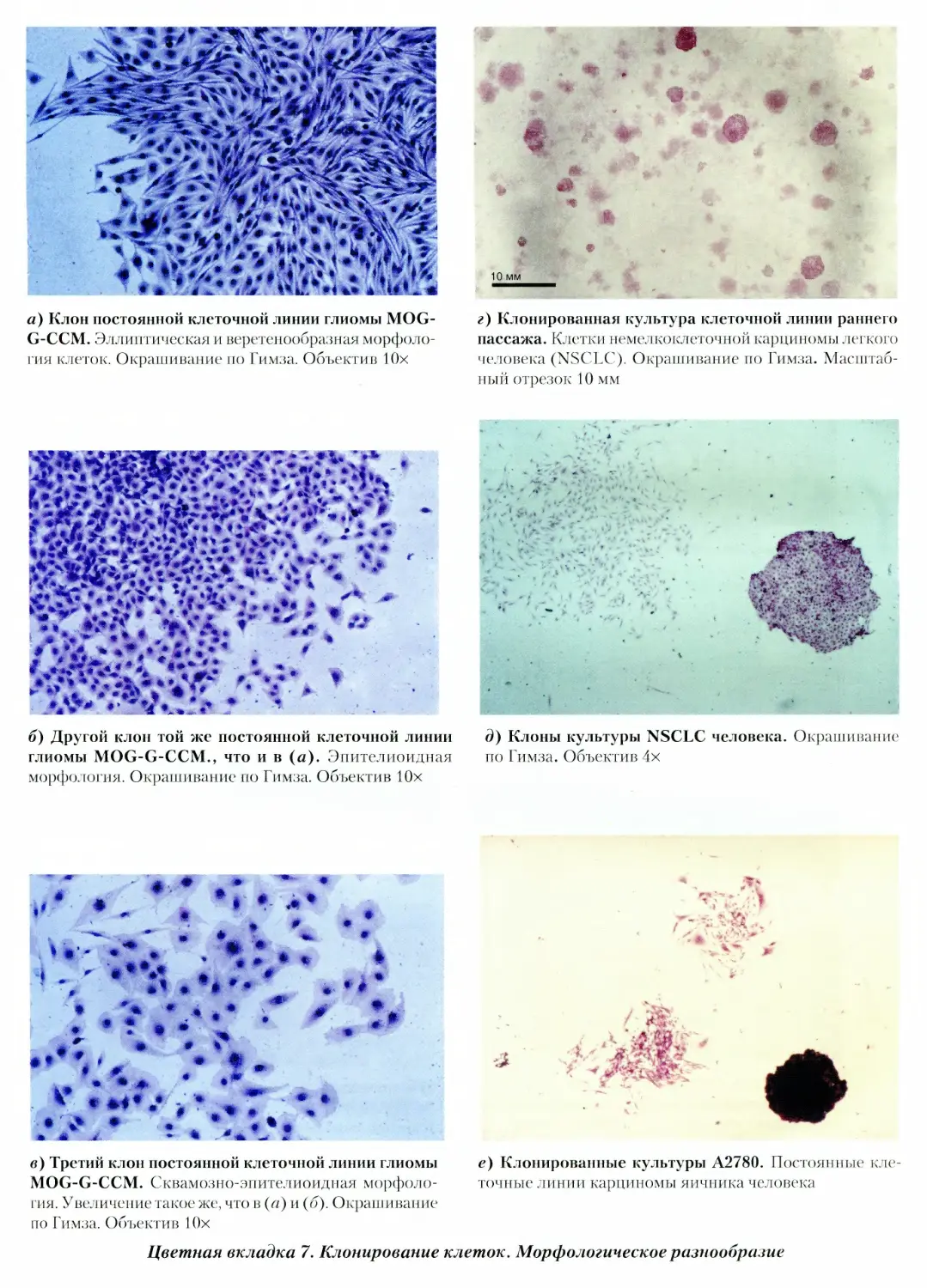
7. Клонирование клеток. Морфологическое разно¬
образие
8. Конечные клеточные линии
9. Постоянные клеточные линии из опухоли человека
10. Постоянные клеточные линии из нормальных
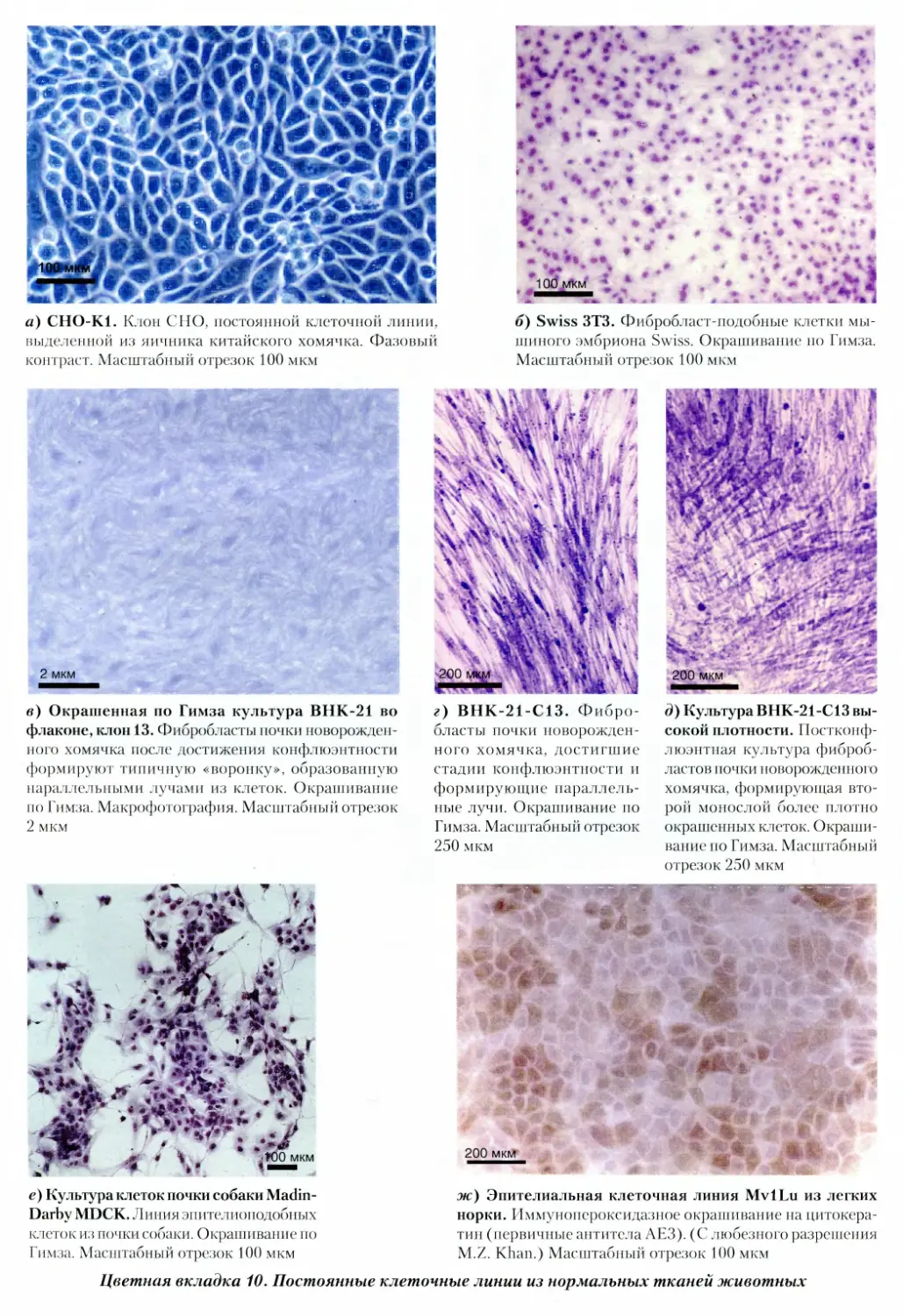
тканей животных
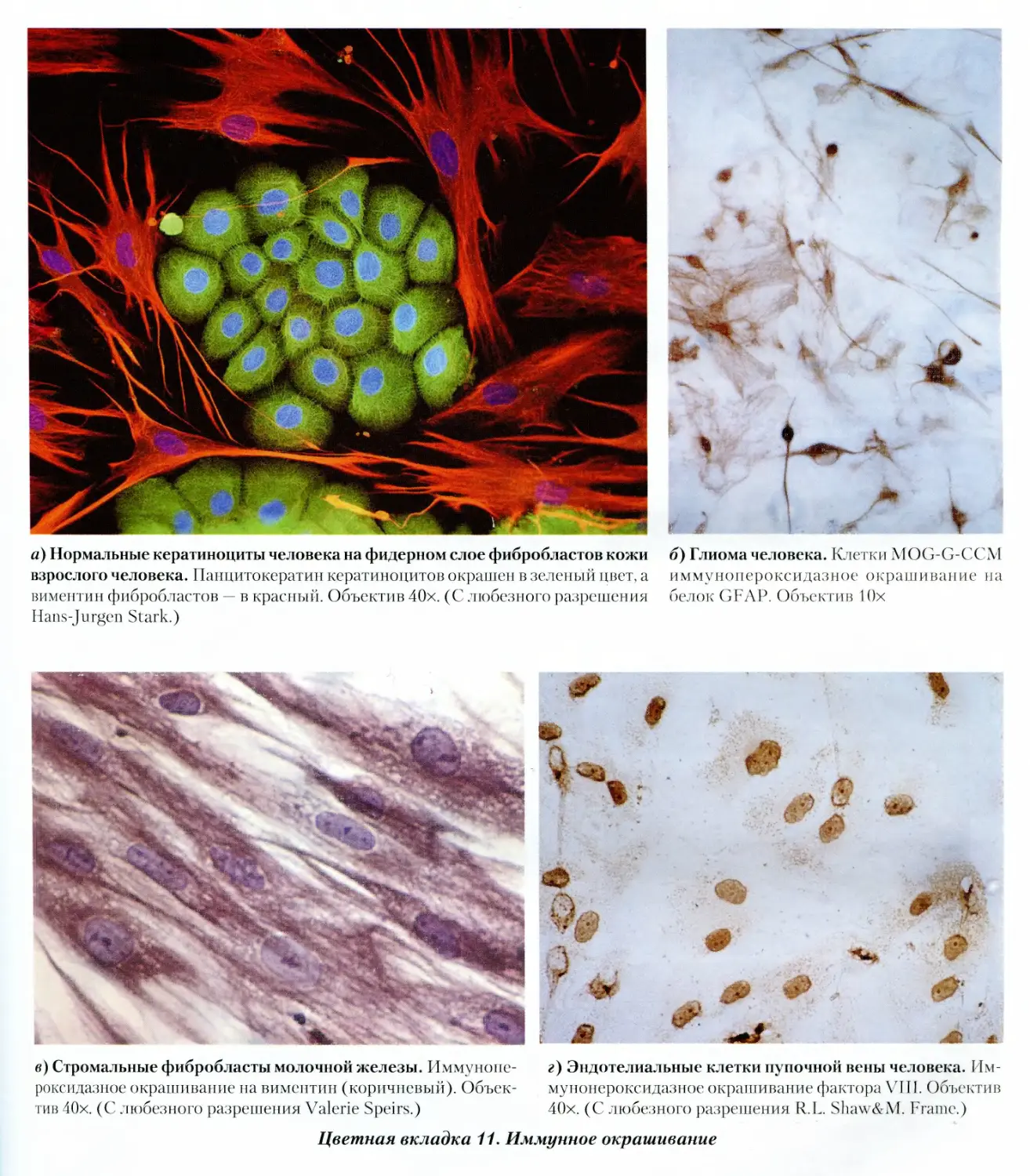
11. Иммунное окрашивание
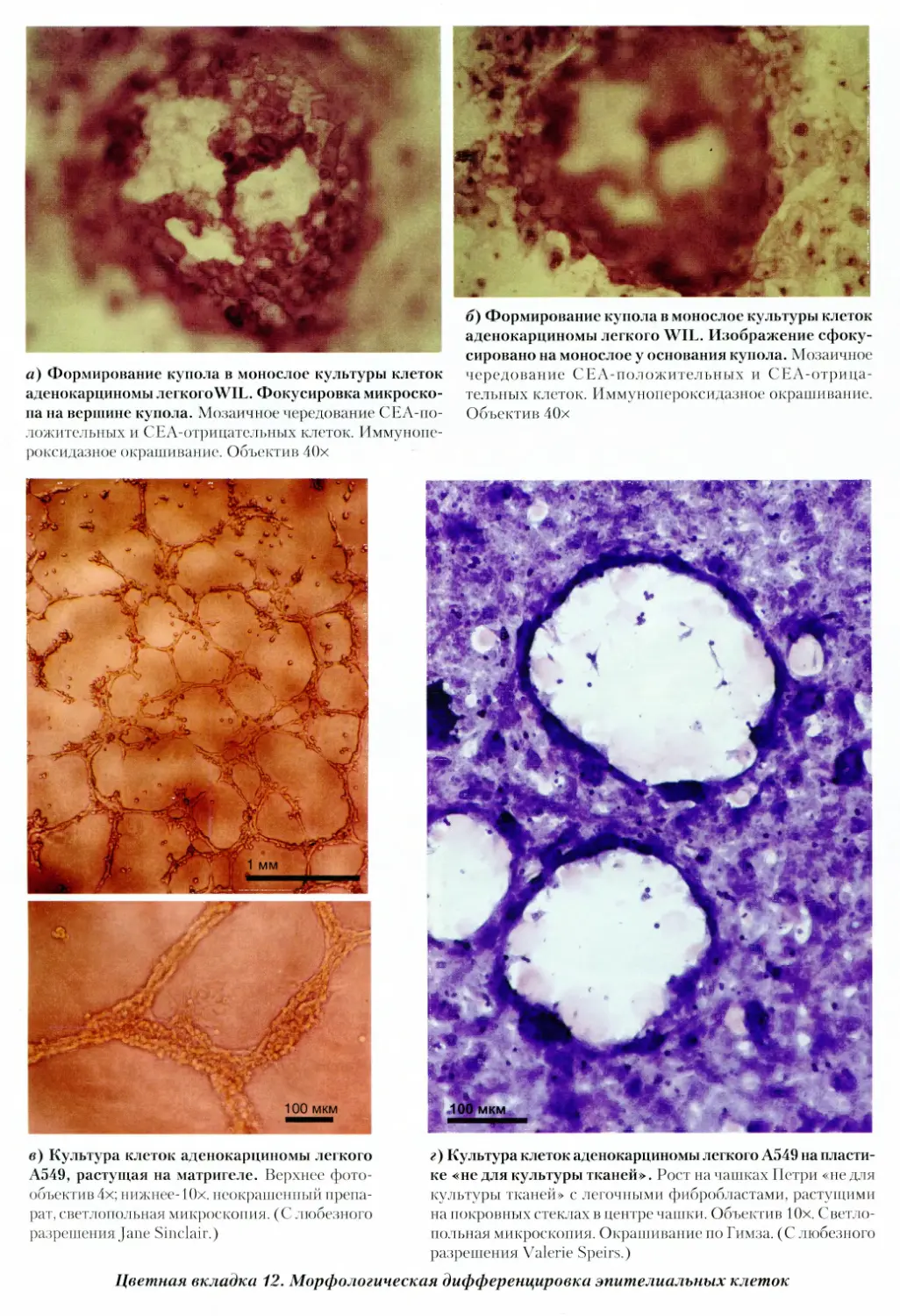
12. Морфологическая дифференцировка эпителиаль¬
ных клеток
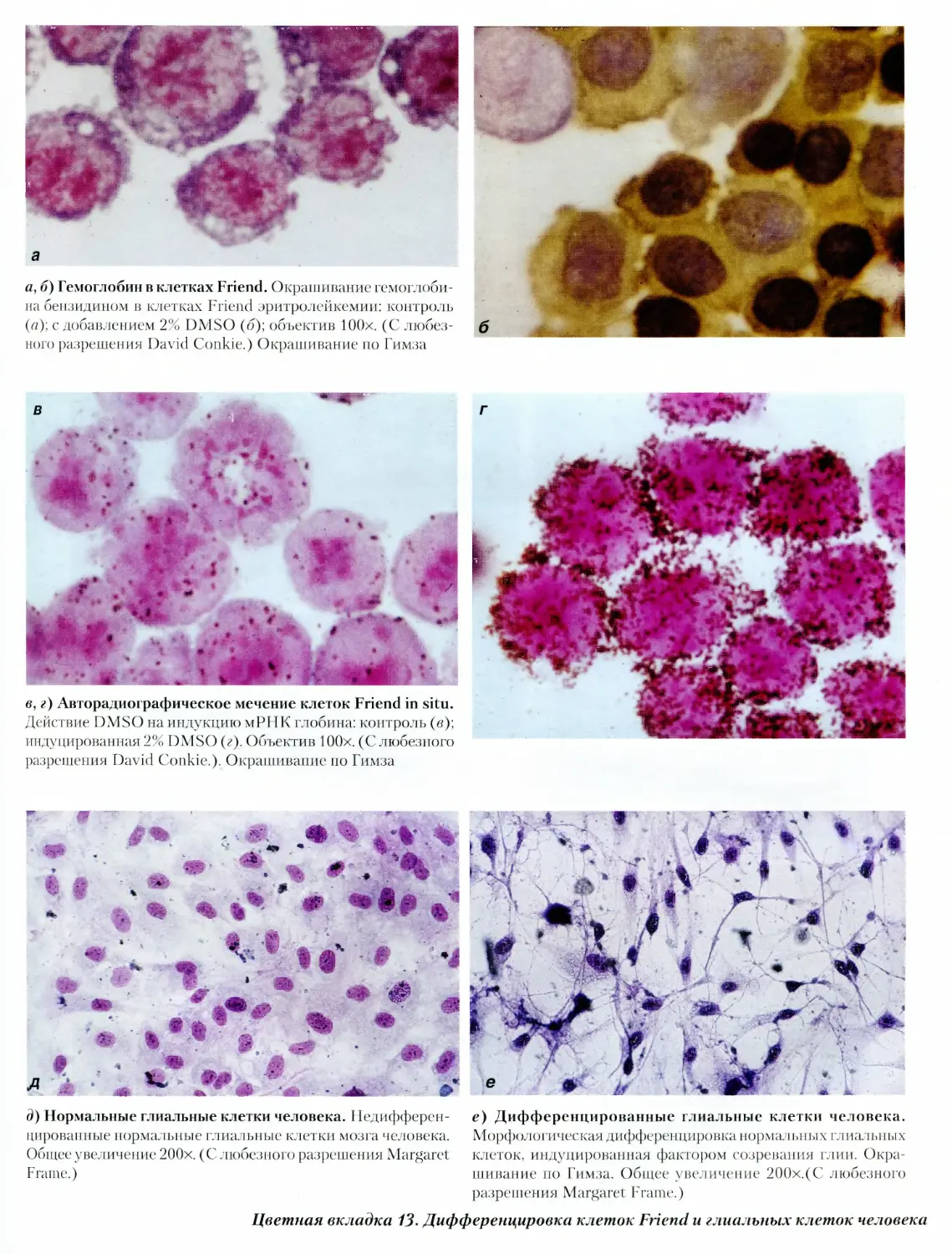
13. Дифференцировка клеток Friend и глиальных
клеток человека
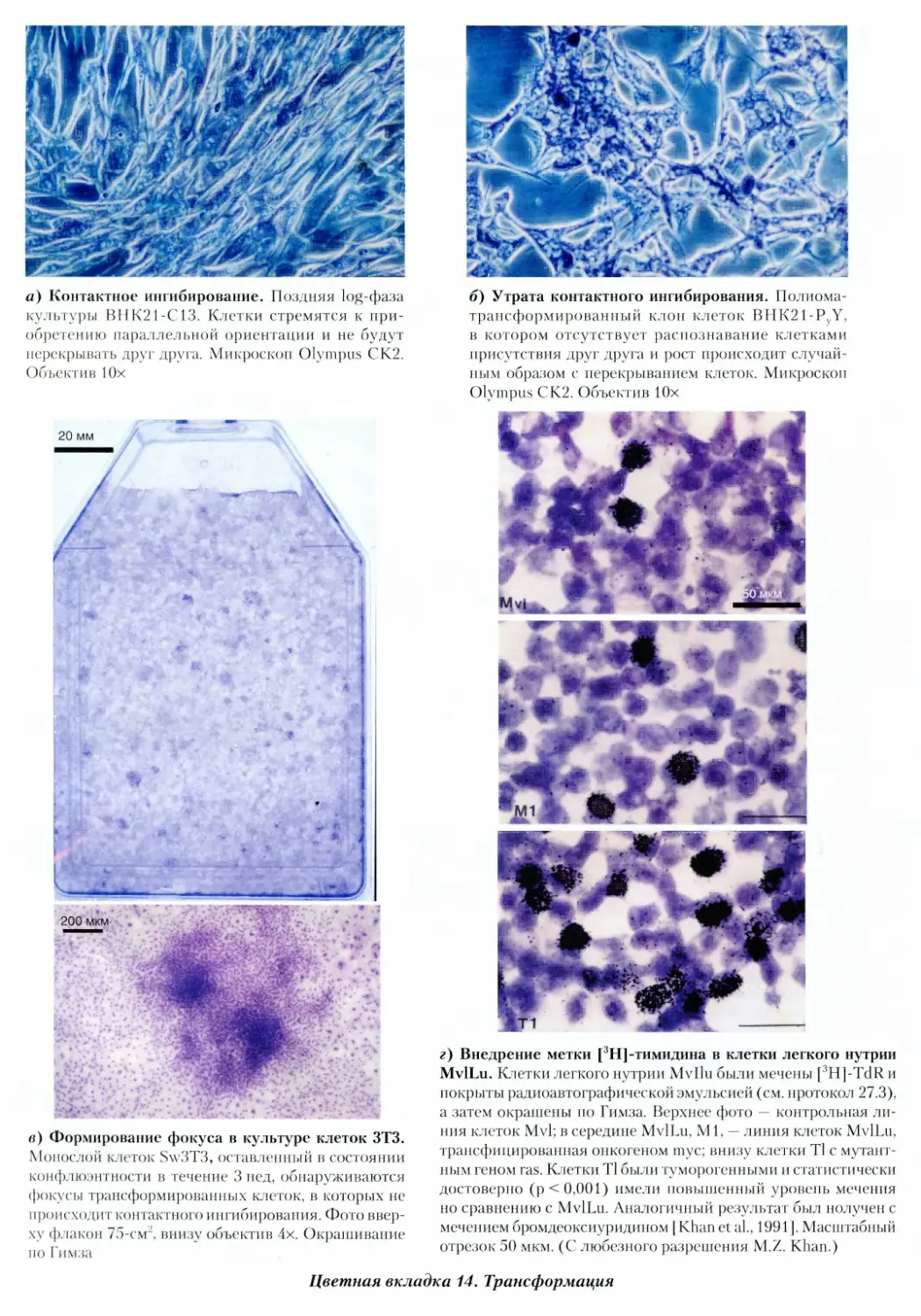
14. Трансформация
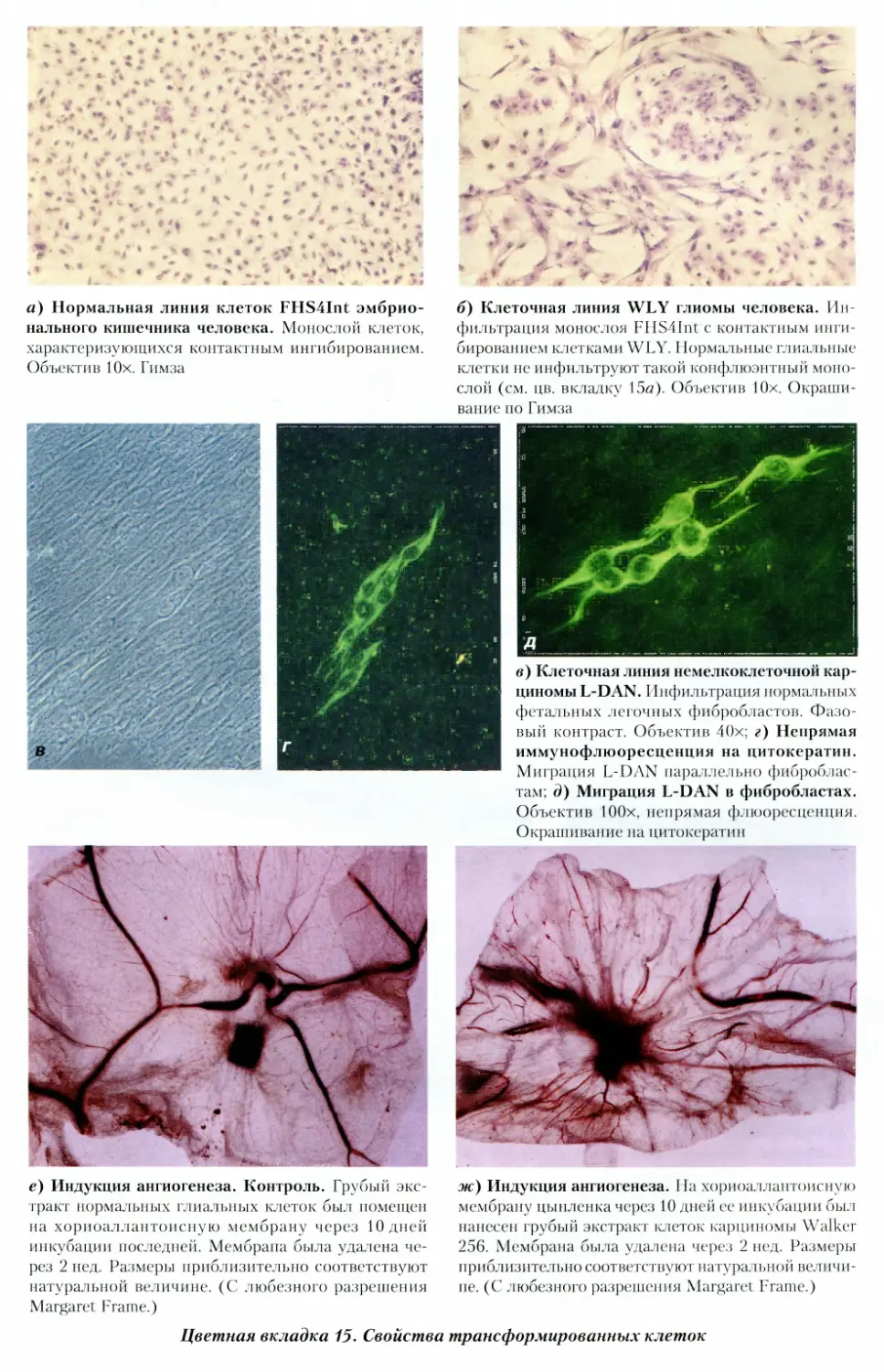
15. Свойства трансформированных клеток
16. Примеры контаминации
17. Жизнеспособность и цитотоксичность
18. Сфероиды, инкапсуляция и микроносители
19. Органотипическая культура в фильтрующей лунке
20. Органотипическая культура кожи
21. Токсичность в органотипической модели in vitro
22. Приготовление среды, культуральные системы и
флаконы для криоконсервации
23. Магнитноактивированный клеточный сортинг
24. Микроматричный анализ экспрессии генов (Af-
fymetrix UK)
Благодарю за помощь и поддержку на протяжении
многих лет моих друзей и коллег. Без них не было бы
этой книги.
Предисловие
Настоящее издание книги «Культура животных кле¬
ток» по структуре сходно с предыдущим изданием, но
содержит некоторые значительные изменения. Введена
новая глава «Тренировочные программы», которая по
своему построению, так же как сноски и ссылки, должна
облегчить использование этой книги преподавателями
в качестве учебного пособия. Содержание данной гла¬
вы предполагает овладение новыми практическими
навыками учащимися или техническим персоналом,
поэтому включает в некоторых протоколах как экс¬
периментальные, так и расчетные элементы. Все это
сделает процесс обучения интереснее, информативнее
и несколько разнообразнее.
Приведенные в книге ссылки тщательно выверены
и усовершенствованы. Отдельные разделы теперь
имеют нумерацию, соответствующую нумерации главы.
Биномиальная ссылка, например 4.1, означает гл. 4, раз¬
дел 1. Триномиальная ссылка 14.6.2 отсылает к гл. 14,
разделу 6, подразделу 2. Поэтому первая цифра ссылки,
независимо от того, относится она к тексту, таблице или
иллюстрации, будет всегда означать соответствующую
главу. Такая структура книги, в частности, вызвана
необходимостью облегчить создание гиперссылок в
будущей электронной версии книги, которая должна
появиться на веб-сайте издательства Wiley (www.wiley.
com) вскоре после выхода печатного издания.
Количество цветных вкладок увеличено вдвое. На
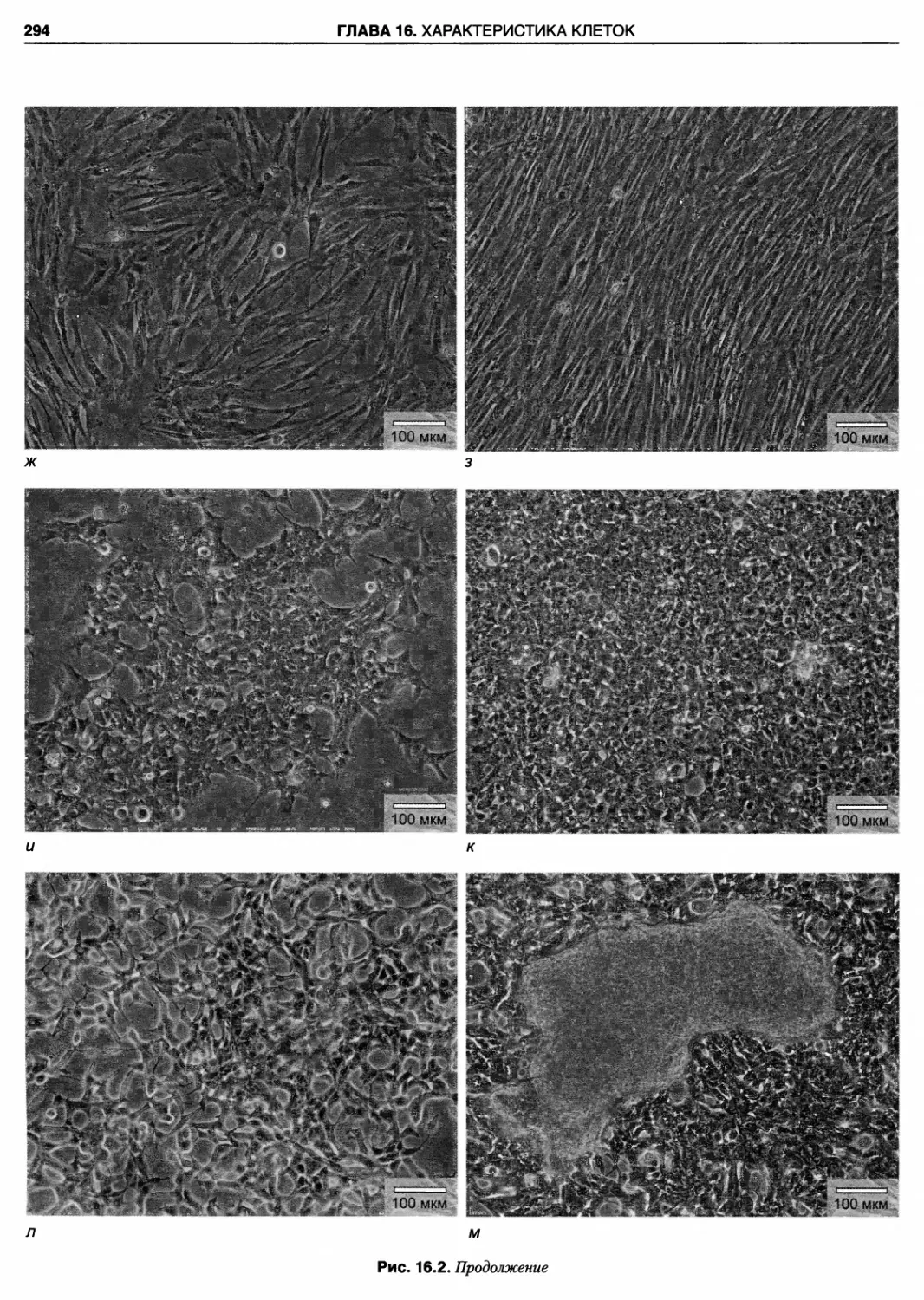
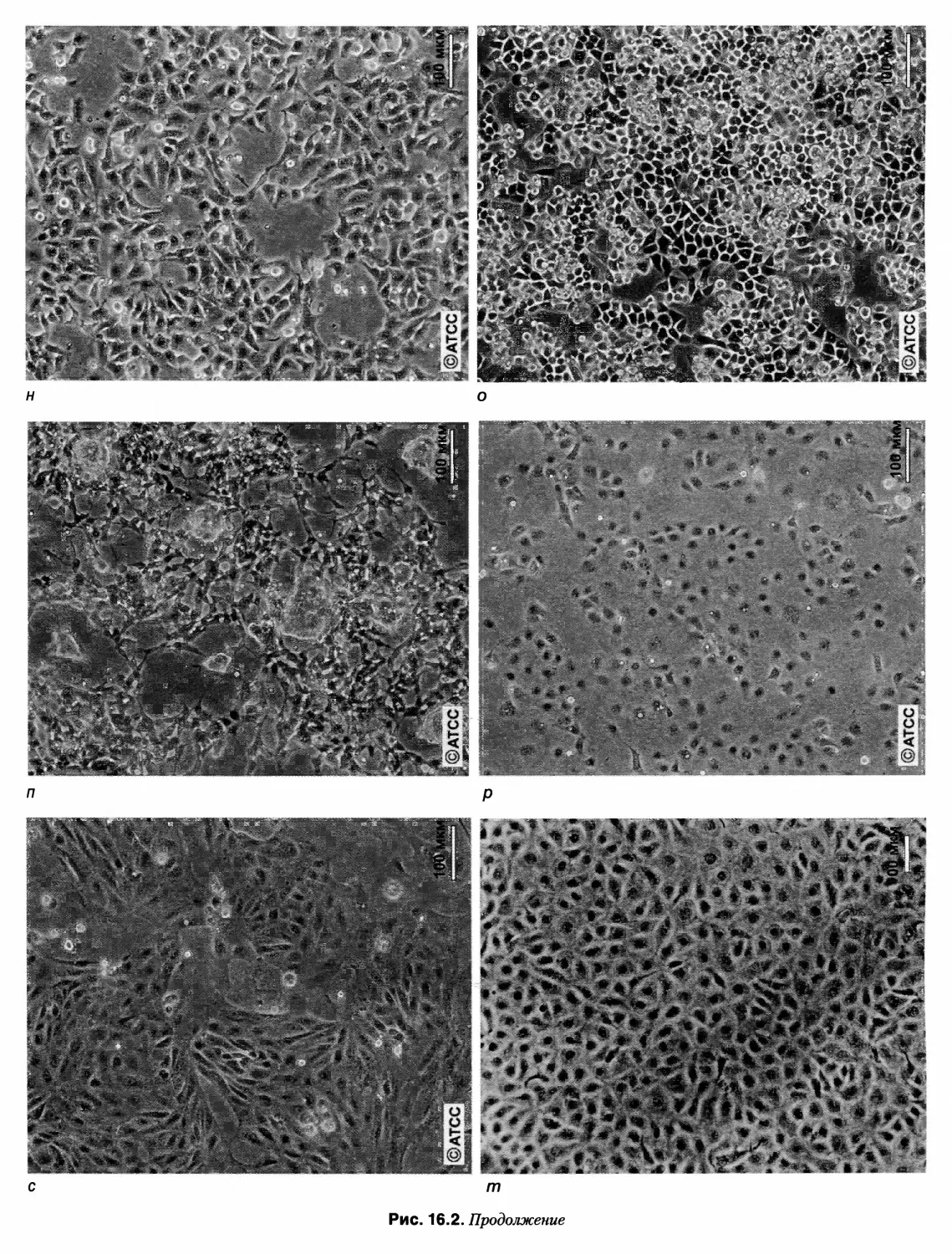
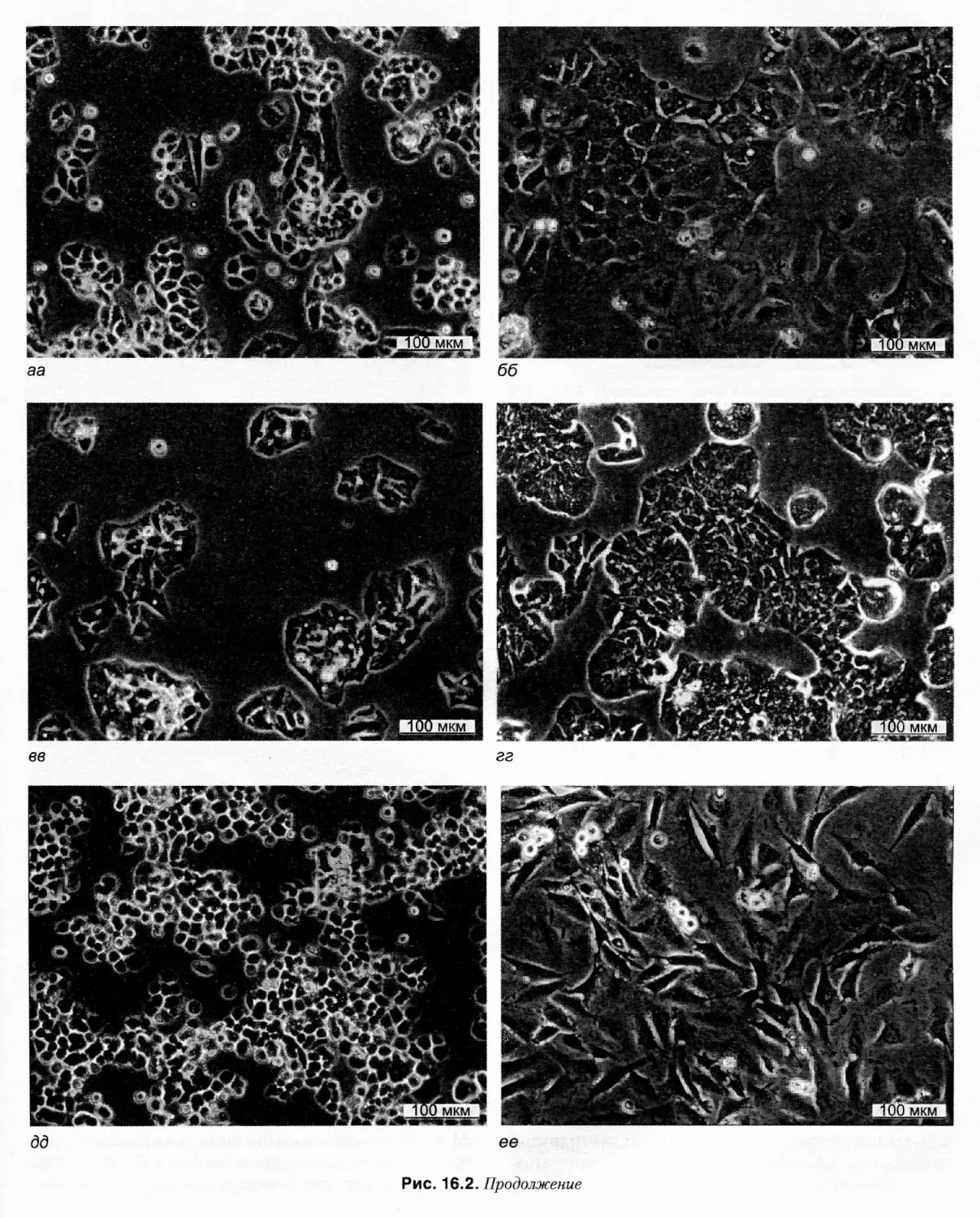
рис. 16.2 представлены изображения примерно сорока
различных клеточных линий, а также первичных куль¬
тур, оборудования и стадий рабочего процесса. Я глубо¬
ко благодарен Ивонне Рейд и Грэгу Сайкису из АТСС,
Питеру Трэвису из ЕСАСС и многим другим за любез¬
но предоставленные новые иллюстрации. Надеюсь, что
это воодушевит читателей, позволит им взглянуть на
клетки, с которыми они работают, более внимательно и
разовьет чуткость к любым изменениям, которые могут
возникнуть в ходе повседневной работы.
В целом я сохранил акцент, который был в предыду¬
щих редакциях, и сосредоточил внимание на основных
методах работы с культурами животных клеток, опи¬
сав особые приемы работы со специализированными
культурами.
Эти методы приведены подробно, с использованием
пошаговых протоколов, которые должны дать полную
информацию о порядке проведения всей процедуры
без обращения к другим литературным источникам.
Каждому протоколу в книге предшествует вводный
материал, который дает теоретическое обоснование и
дополнительную информацию для проведения альтер¬
нативных процедур и применения данного протокола.
Некоторые основные понятия биологии объясняются
в тексте, но подразумевается, что читатель обладает
начальными знаниями в анатомии, гистологии, био¬
химии, клеточной и молекулярной биологии. Книга
предназначена для тех, кто не имеет навыков или имеет
незначительный опыт в работе с культурами тканей,
включая лаборантов-исследователей, студентов стар¬
ших курсов, дипломников, аспирантов и клиницистов,
имеющих интерес к научно-исследовательской работе.
Кажется нелогичным особо выделять молекулярные
методы, тем более что точной границы между молеку¬
лярными и клеточными методами не существует.
Молекулярные методы, описанные в книге, имеют
прямое отношение к культурам клеток. Не было даже
попытки представить их все, поскольку такие методы
доступно изложены в других работах [Samrook et al.,
1989, Ausubel et al., 2002 и др.]. Тема повышения выхода
клеточной массы близка области биотехнологии, и глава,
посвященная этой проблеме, дает описание некоторых
приемов и техник повышения количества получаемых
клеток. Однако эта глава не рассматривает процессы
производства биофармацевтической продукции.
Список оборудования и оснащения обновлен и ко
времени публикации книги, надеюсь, еще будет соот¬
ветствовать современному уровню. Однако меняются
названия компаний, происходит слияние фирм, исчеза¬
ют некоторые виды оборудования, поэтому достаточно
трудно сохранять постоянство в этом отношении. Тем
не менее я надеюсь, что в будущем этот аспект будет
учитываться более эффективно на веб-сайте.
В тексте используются сокращения, которые при¬
водятся отдельным списком после предисловия. Об¬
щеприняты такие условные обозначения, как D-PBSA
(фосфатно-солевой буфер Дюльбекко без Са2+ и Mg2+),
UPW (сверхчистая вода, независимо от способа ее
получения). Там, где это возможно, используются
молярные концентрации. Реальным весом (в граммах)
обычно пренебрегают, используя в списке питатель¬
ных сред молярность, поскольку предполагается, что
очень небольшое число людей попытается составлять
собственные питательные среды из отдельных компо¬
нентов. Но возможно, что при необходимости сравнить
компоненты будет полезным использовать молярные
эквиваленты.
20
Предисловие
Протоколы выделены в тексте рамкой и особым фо¬
ном. Реагенты, специфические для данного отдельного
протокола, указаны в материалах соответствующего
протоколу раздела, прописи для общеупотребительных
реагентов, таких как буферный раствор Хэнкса или
трипсин, приводятся в Приложении II. Приложения
также содержат подробные описания компаний, произ¬
водящих реагенты, оборудование и другие ресурсы.
Как обычно, я должен выразить свою благодарность
авторам, которые участвовали в разработке протоко¬
лов, а также помогали мне советами в тех областях, в
которых мои знания несовершенны. Это Роберт Ау¬
эрбах, Боб Браун, Кеннет Кельман, Ричард Хэм, Роб
Хэй, Стэн Кей, Николь Кейт, Уолли Мак Киэн, Рона
Мак Кай, Стефан Мерри, Джейн Плам, Питер Вогхэм,
Поль Уокман, Роландж Графстрем и Джон Поль. Я
был счастлив работать вместе с клиницистами Дэви¬
дом И. Грэмом и Дэвидом Дж. Т. Томасом и Джоном
Максвеллом Андерсеном.
На ранних этапах работы над книгой я получил
большую пользу от бесед с Доном Дугласом, Питером
дель Веккьо, Сергеем Федоровым и Майком Гэбрид-
жем, Дженом Лундином, Джоном Райаном, Джимом
Смитом и Чэрити Уэймаут. Я бесконечно благодарен
Полю Чэппелю, который первым убедил меня в необ¬
ходимости написать руководство по основным мето¬
дам работы с культурами тканей и который недавно
предложил перевести этот текст в мультимедийный
формат и опубликовать в издательстве Wiley-Liss.
Многие из иллюстраций этой книги были выполнены
Джейн Джиллис и Мариной ла Дюк, хотя часть из них
была заменена в связи с требованиями электронной
публикации. Я очень признателен руководству Бри¬
танских лабораторий Битсона по исследованию рака
за разрешение фотографировать их оборудование
и использовать фотографии для иллюстрирования
этой книги. Некоторые из данных, представленных
в книге, были получены теми, кто работал со мной в
течение многих лет, это: Шейла Браун, Ян Каннигэм,
Лин Эванс, Маргарет Фрэйм, Элайн Харт, Кэрол Мак
Кормик, Джон МакЛин, Элистар МакНэб, Диана Мор¬
ган, Элисон Мюррей, Элисон Мэкки, Ирэн Оспрей,
Мохаммед Зарин Хан и Наташа Евдокимова.
Мне посчастливилось получить отличные сове¬
ты и поддержку от сотрудников издательства John
Wiley&Sons. С искренней благодарностью я бы хотел
упомянуть и всех тех, кто побеспокоился и прислал нам
письма с своими советами и конструктивной критикой
по поводу предыдущих редакций издания. Приятно и
радостно слышать высказывания о том, что наша книга
полезна, но еще более важно услышать от читателей
критику по поводу недостатков книги, которые я попы¬
таюсь учесть в дальнейшем. Можно лишь надеяться, что
те из вас, кто использует кни1у в работе, сохранят в душе
то же волнение, которое я ощущал, размышляя о буду¬
щих перспективах, открывающихся в этой области.
Я бы хотел поблагодарить мою дочь Джилиан и
сына Норманна за помощь, которую они много лет
назад оказывали мне в подготовке первого издания,
за их постоянные советы и поддержку. Кроме того, я
благодарен моей жене Мэри за те часы, которые она
провела за составлением, правкой и другой работой. Без
ее помощи и поддержки первоначальный текст никогда
не был бы написан, и я не завершил бы подготовку это¬
го переиздания к оговоренным срокам и не достиг бы
необходимого уровня технической точности, которая
является основным требованием хорошего руководства
по культурам тканей.
Ян Фрешни
Список сокращений
AFP Альфа-фетопротеин
Arg VP Аргинин-вазопрессин
AST Аспартатаминотрансфераза
АТСС Американская коллекция культур клеток
BMP Костный морфогенетический белок
Ьр Нуклеотидная пара (нп)
ВРЕ Экстракт гипофиза быка
bGH Гормон роста быка
BrdU Бромдезоксиуридин
BSA Бычий сывороточный альбумин
BSL Уровень биобезопасности
BSS Сбалансированный солевой раствор
BudR Бромдеоксиуридин
САМ Хориоаллантоисная мембрана
сАМР Циклический аденозинмонофосфат
CAMs Молекулы межклеточной адгезии
CCD Устройство с зарядовой связью
CCTV Встроенное телевизионное устройство
СЕ Эффективность клонирования
СЕА Карциноэмбриональный антиген
CMF Солевой раствор без кальция и магния
CMRL Лаборатория Медицинских исследований
Коннаута
СРМ Число импульсов в минуту
DAG Диацилглицерин
D-BPSB Раствор Са2+ и Mg2+для добавления в
D-PBSA
DDAB Диметилдиоктадециламмония бромид
DEPC Диэтилпирокарбонат
DHFR Дигидрофолатредуктаза
DIC Дифференциальный интерференционный
контраст
DMEM Модификация Дюльбекко среды Игла
DMSO Диметилсульфоксид
DOSPA 2,3-диолеилокси-М[2(сперминкарбокса-
мид)этил ] -К,М-диметил-1 -пропанамин
трифторацетат
DOPE Диолеилфосфатидилэтаноламин
DOTMA М-[1-(2,Здиолеилокси)-пропил]-М,К,К-
триметиламмония хлорид
D-PBS Фосфатно-буферный солевой раствор
Дюльбекко
DPDE Дифосфодиэфиры
DT Время удвоения популяции
DTT Дитиотреитол
EBSS Сбалансированный солевой раствор
Эрла
a-fetoprotein
Arginin-Vasopressin
Aspartate aminotransferase
American Type Culture Collection
Bone morphogenetic protein
Base pairs
Bovine pituitare extract
Bovine growth hormone
Bromodeoxyuridin
Bovine Serum Albumine
BioSafety Level
Balanced salt solution
Bromodeoxyuridine
Chorioallantoic membrane
Cyclic adenosinmonophosphate
Cell adhesion molecules
Charge-coupled device
Closet-circuit television
Cloning efficiency
Carcinoembryonic antigen
Calciun- and magnesium -free saline
Connaught Medical Research Laboratory
Counts per minute
Diacylglicerol
Dimethyl dioctadecylammonium bromide
Diethyl pyrocarbonate
Dihydrofolate reductase
Differential interference contrast
Dulbecco’s modification of Eagle’s medium
Dimetilsulphoxid
2,3-dioleyloxy-N[2(sperminecarboxamido)
ethyl] -N,N-dimethyl-1 -trifluoroacetate
Dioleoylphosphatidylethanolamine
N-[l-(2,3 dioleyloxy)-propyl]-N,N,N-tri-
methylammonium chloride
Dulbecco’s phosphate-buffered saline
Diphosphodiesters
Doubling time population
Dithiothreitol
Earle’s balanced salt solution
22
Список сокращений
EBV
Вирус Эпштейна-Барра
Epstein-Barr virus
ЕСАСС
Европейская коллекция животных куль¬
тур клеток
European collection of Animal cell cultures
ECGF
Фактор роста эндотелиальных клеток
Endothelial cell growth factor
ECM
Внеклеточный матрикс
Extracellular matrix
EDTA
Этилендиаминтетрауксусная кислота
Etylendiamintetraacetat
Diaminoethane tetraacetic acid
EGF
Эпидермальный фактор роста
Epidermal grouth factor
EGTA
Этиленгликоль-бис-(Р-аминоэтилэфир)-
Ethylene glycol-bis(P-aminoethylether)-
4п-тетрауксусная кислота
N,n,n,n-tetraacetic acid
EM
Электронный микроскоп
Electrone microscope
EMA
Эпителиальный мембранный антиген
Epithelial membrane antigene
FBS
Эмбриональная бычья сыворотка
Fetal bovine serum
FCS
Эмбриональная сыворотка теленка
Fetal calf serum
FGF
Фактор роста фибробластов
Fibroblasts grouth factor
G6PD
Глюкоза-6-фосфат-дегидрогеназа
Glucose-6-phosphate dehydrogenase
Gal-С
Г алактоцереброзид
Galactocerebroside
GFAP
Глиальный фибриллярный кислый белок
Glial fibrillary acidic protein
GLP
Хорошая лабораторная практика
Good laboratory practice
GMP
Хорошая производственная практика
Good manufacturing practice
GPDH
Глиальный фибриллярный кислотный
Glial fibrillary acidic
протеин
Protein
GRP
Гастрин-высвобождающий пептид
Gastrin-releasing peptide
H&E
Г ематокси л ин/эозин
Hematoxlin&eosin
HAT
Гипоксантин, аминоптерин, тимидин
Hypoxantine, aminopterin, thymidine
HBGF
Гепарин-связывающий фактор роста
Heparin binding grouth factor
HBS
Hepes-забуференный солевой раствор
Hepes- buffered saline
HBSS
Сбалансированный солевой раствор
Хэнкса
Hanks’ balansed salt solution
HC
Гидрокортизон
Hydrocortisone
hCG,
Человеческий хориональный гонадотропин
Human chorionic gonadotropin
HEPA
Высокоэффективный дисперсный воз¬
душный поток
High-efficiency particulate air
HES
Гидроксилэтиловый крахмал
Hydroxyethylstarch
HGPRT
Гипоксантин гуанозин фосфорибозил
Hypoxantinguanosine phosphoribosyl trans¬
трансфераза
ferase
Hgh
Человеческий гормон роста
Human growth hormone
HITES
Гидрокортизон, инсулин, трансферрин,
Hydrocortisone, insuline, transferrin, estra¬
эстрадиол и селен
diol and selenium
Hmfg
Глобулярный белок молока человека
Human milk fat globule protein
HPV
Вирус папилломы человека
Human papilloma virus
Hspgs
Г епарансульфатпротеогл иканы
Heparan sulfate proteoglycans
Ht
Г ипоксантин /тимидин
Hypoxantine/timidine
ICAM
Межклеточная молекула адгезии клеток
Intercellular cell adhesion molecule
ISO
Международная организация стандар¬
тизации
International standardization organization
Its
Инсулин, трансферрин, селен
Insulin, transferrin, selen
IUdr
Иод-дезоксиуридин
Iododeoxiuridin
Kbm
Основная среда для кератиноцитов
Keratinocyte basal medium
Kdm
Селективная среда для выделения кера¬
тиноцитов
Keratinocyte-defined medium
Kgm
Среда для роста кератиноцитов
Keratinocyte growth medium
LI
Индекс мечения
Labeling index
M199
Среда 199
Medium 199
Maa
Микроматричный анализ
Microarray analysis,
MACs
Искусственные хромосомы млекопита¬
ющих
Mammalian artificial chromosomes
MACS
Магнитно-активируемый клеточный
сортинг
Magnetic-activated cell sorting
Список сокращений
23
Md
Малатдегидрогеназа
Malate dehydrogenase
MEM
Минимальная питательная среда игла
Eagle’s minimal essential medium
MPI
Манноза-6-фосфат-изомераза
Mannose-6-phosphate isomerase
MSC
Микробиологические боксы биологичес¬
кой безопасности
Microbiological safety cabinet
NBCS
Сыворотка новорожденного теленка
Newborn calf serum
NCI
Государственный институт рака
National cancer institute
NP
Нуклеозидфосфорилаза
Nucleoside phosphorylase
NSE
Нейрон-специфическая энолаза
Neuron-specific enolase
O.D.
Оптическая плотность
Optical density
PA
Активатор плазминогена
Plasminogen activator
PBS
Фосфатно-буферный раствор
Phosphate-buffered saline
PBSA
Фосфатно-буферный раствор — а
Phosphate-buffered saline-a
PBSB
Фосфатно-буферный раствор — в
Phosphate-buffered saline-b
PCA
Перхлорная кислота
Perchloric acid
PCR
Полимеразная цепная реакция
Polymerase chain reaction
PCR
Фосфокреатин
Phosphocreatine
PDE
Фосфодиэфиры
Phosphodiesters
PDGF
Тромбоцитарный фактор роста
Platelet-derived growth factor
PDT
Время удвоения популяции
Population doubling time
Pe
Эффективность культивирования
Plate efficiency
PE
Фосфатно-буферный раствор - а/эдта
Pbsa/edta
PEG
Полиэтиленгликоль
Polyethyleneglycol
Pepb
Пептидаза b
Peptidase b
PGA
Полигликолевая кислота
Polyglycolic acid
Pla
Полимолочная кислота
Polylactic acid
Pma
Форбол-миристиновокислая соль уксус¬
ной кислоты
Phorbol myristate acetate
Pme
Фосфомоноэфиры
Phosphomonoesters
Ptfe
Политетрафторэтилен
Polytetrafluorethylene
Pvp
Поливинилпирролидон
Polyvinylpyrrolidone
PHA
Фитогемагглютинин
Phytohemagglutinin
PWM
Митоген лаконоса
Pokeweed mitogen
Rt-pcr
ПЦР, основанная на использовании обрат¬
ной транскриптазы
Reverse transcriptase per
Sees
Обмен сестринских хроматид
Sister chromatide exchange
Scid
Тяжелый комбинированный иммуноде¬
фицит
Severe combine immunodeficite syndrome
Sclc
Мелкоклеточная карцинома легких
Small cell lung cuncer
Sd
Плотность насыщения
Saturation density
SDS
Додецилсульфат натрия
Sodium dodecyl sulfate
Se
Эффективность посева
Seeding efficiency
Sit
Селен, инсулин, трансферрин
Selen, insulin, transferrin
Skdm
Селективная среда с добавками для выде¬
ления кератиноцитов
Supplemented kdm
Sis
Лаурилсульфат натрия
Sodium lauryl sulfate
Ssc
Цитрат натрия/хлорид натрия
Sodium citrate/sodium chloride
Sv40
Вирус обезьяны 40
Simian virus 40
Sv40lt
Ген sv40 для большого т-антигена
Sv40 gene for large t-antigene
Teb
Буфер трис/эдта
Tris/edta buffer
Teer
Трансэпителиальное электрическое со¬
противление
Transepithelial electrical resistance
Tgf
Трансформирующий фактор роста
Transforming growth factor
Tk
Т имидинкиназа
Thymidine kinase
Toe
Общий органический углерод
Total organic carbone
UPW
Сверхчистая вода
Ultra pure water
VEGF
Фактор роста сосудистого эндотелия
Vascular endotelial growth factor
Vntrs
Вариабельное число тандемных повторов
Variable number tandem repeats
Yacs
Искусственные хромосомы дрожжей
Yeast artificial chromosomes
24
Список сокращений
соп
Стандартная операционная процедура
МЦ
Метилцеллюлоза
ТХУ
Трихлоруксусная кислота
ФГА
Фитогемагглютинин
Pi
Внеклеточный неорганический фосфор
УФ
Ультрафиолет
Птфэ
Политетрафторэтилен
ССР
Сбалансированный солевой раствор
мкм
Микрометр
мл
Миллилитр
мм
Миллиметр
кгр
Килогрэй
кПа
Килопаскаль
Глава 1
ВВЕДЕНИЕ
I
1.1. ИСТОРИЯ ВОПРОСА
Техника культуры ткани была впервые задумана
и разработана в начале двадцатого века [Harrison,
1907; Carel, 1912] (табл. 1.1) для изучения свойств
животных клеток, свободных от системных влияний,
возникающих in vivo как при нормальном гомеостазе,
так и в условиях стресса, вызванного экспериментом.
Как видно из названия, эта технология первоначально
разрабатывалась для недезагрегированных фрагментов
тканей; рост культуры был ограничен возможностью
миграции клеток из кусочка ткани и нерегулярностью
митозов в первичном разрастании. Культивирование
клеток из такого первичного эксплантата тканей доми¬
нировало в данной области более 50 лет [Fischer, 1925;
Parker, 1961]. Поэтому неудивительно, что исходный
термин «культура ткани» продолжает использоваться,
несмотря на то что бурное развитие в этой области в
течение второй половины XX в. (рис. 1.1) было связа¬
но с возможностью использования диспергированных
клеточных культур.
Впервые возможность дезагрегации клеток экс¬
плантата с последующим выращиванием на чашках
диспергированных клеток была показана Роусом [Rous
and Jones, 1916], хотя в то время чаще всего пересев
культуры для создания клеточного штамма осущест¬
вляли путем оперативного отделения ее фрагмента.
Первым клонированным клеточным штаммом был
L929, выделенный из мышиных L-клеток методом
капиллярного клонирования [Sanford et al., 1948].
В 1950-х годах для анализа вирусных бляшек на
монослое клеточных культур Дюльбекко [Dulbecco,
1952] описал метод получения монослойных культур с
использованием трипсина, который стал затем широко
применяться в практике культивирования. Возмож¬
ность создания клеточной суспензии из единичной
клетки, полученной путем предварительной трипси-
низации эксплантата, способствовала дальнейшему
развитию клонирования клеточных линий. Первая по¬
стоянно пересеваемая клеточная линия человеческого
происхождения HeLa была получена Геем [Gey at al.,
1952]. Впоследствии она была клонирована Паком
[Puck and Marcus, 1955] с использованием облученно¬
го рентгеном фидерного слоя клеток. Культура тканей
стала в то время шире использоваться благодаря появ¬
лению антибиотиков, которые позволили разработать
долгоживущие клеточные линии. Тогда же появились
первые предупреждения о риске существования скры¬
той или антибиотико-резистентной контаминации
долгоживущей культуры [Parker, 1961]. Пятидесятые
годы были также годами разработки и создания многих
питательных сред с определенным составом [Morgan
et al., 1950; Parker et al., 1954; Eagle, 1955, 1959; Way-
mouth, 1959], которые в конечном итоге легли в основу
разработки бессывороточных сред [Наш, 1963, 1965]
(см. разд. 10.6).
В тексте этой книги термин культура ткани исполь¬
зуется как обобщенный термин, обозначающий как
органные, так и клеточные культуры. Термин органная
культура подразумевает трехмерную культуру неде-
загрегированной ткани, сохраняющей полностью или
частично все гистологические особенности этой ткани
in vivo. Термин клеточная культура имеет отношение
к культуре, полученной от диспергированной исход¬
ной ткани из первичной культуры или из клеточного
штамма путем ферментативной, механической или
химической дезагрегации. Термин гистотипическая
культура подразумевает, что клетки реагрегировали
или наросли с формированием трехмерной структуры,
соответствующей таковой в ткани. Гистотипическая
культура формируется путем культивирования при вы¬
сокой плотности клеток в ячейке фильтра, вторичном
росте монослоя во флаконе или на чашке, в суспензии
в агаре, в условиях реального или смоделированного
40 ООО
35 ООО
§ 30 000
3
i 25 000
0
= 20 000
g 15 000
Т
1 10 000
*
5000
о
/* 1960 1970 1980 1990 2000
Количество сообщений Год публикации
по [Fisher, 1925]
Рис. 1.1. Рост количества публикаций по культуре ткани.
Количество сообщений в PubMed начиная с 1965 г. Коли¬
чество публикаций до 1960 г. приводится по библиографии
Fisher [1925]
26
ГЛАВА 1. ВВЕДЕНИЕ
Таблица 1.1. Ключевые события в развитии методов культивирования тканей и клеток
Год/годы Событие
Ссылка
1907 Рост in vitro нервного волокна эмбриона лягушки
1912 Эксплантаты соединительной ткани цыпленка, сокращение сердеч¬
ной мышцы в течение 2-3 месяцев
1916 Трипсинизация и субкультура эксплантата
1920-30-е Субкультуры клеточных линий фибробластов
1925-26 Дифференцировка в органной культуре in vitro
1940-е Внедрение использования антибиотиков в культуре ткани
1943 Создание линии L-клеток мышиных фибробластов;
первая постоянная клеточная линия
1948 Клонирование L-клеток
1949 Выращивание вирусов в клеточной культуре
1952 Использование трипсина для воспроизведения культуры; метод
вирусных бляшек
1952-1955 Создание первой линии клеток человека HeLa из цервикальной
карциномы
1952 Трансплантация ядра
1954 Контактное торможение клеточной подвижности в культуре
фибробластов
Противополиомиелитная вакцина Солка, полученная в культуре
клеток почек обезьян
1955 Клонирование клеток HeLa на гомологичном фидерном слое
Разработка питательных сред. Необходимость сывороточных фак¬
торов роста в питательных средах
Конец Понимание важности инфицирования микоплазмами (PPLO)
1950-х
1961 Объяснение конечности времени жизни нормальных клеток челове¬
ка. Клеточное слияние — гибридизация соматических клеток
1962 Создание и трансформация линии ВНК21. Поддержание диффе¬
ренцировки (опухоли гипофиза и надпочечника)
1963 ЗТЗ клетки и спонтанная трансформация
1964 Плюрипотентность эмбриональных клеток
Селекция трансформированных клеток в агаре
1964-1969 Бешенство. Противокраснушная вакцина в культуре фибробластов
легкого человека WI-38
1965 Клонирование клеток китайского хомяка в бессывороточной
среде
Г етерокарион-гибрид: человек-мышь
1966 Фактор роста нервов
Дифференцировка гепатомы крысы
1967 Эпидермальный фактор роста
Перекрестная контаминация клетками HeLa
Ограничение клеточной пролиферации суспензии плотности
Клетки линии лимфобластомы
1968 Поддержание уровня дифференцировки в культуре нормаль¬
ных миобластов. Клеточная пролиферация, независимая от
подложки
1969 Формирование колоний гематопоэтических клеток
1970-е Изобретение ламинарных шкафов
1973 Перенос ДНК, фосфат кальция
1975 Фактор роста фибробластов. Гибридомы. Моноклональные анти¬
тела
1976 Тотипотентные эмбриональные стволовые клетки. Добавление
факторов роста к бессывороточной питательной среде
Harrison, 1907
Carrel, 1912; Burrows, 1912
Rous & Jones, 1916
Carrel & Ebeling, 1923
Strangeways & Fell, 1925,1926
Keilova, 1948; Cruikshank & Lowbury, 1952
Earle et al., 1943
Sanford et al., 1948
Enders et al., 1949
Dulbecco, 1952
Dulbecco, 1952
Gey et al., 1952
Cm. Briggs & King, 1960
Abercrombie & Heaysman, 1954
cm. Griffiths, 1991
Puck & Marcus, 1955; Eagle, 1955,1959; San¬
ford et al., 1955
Harris, 1959; Coriell et al., 1958; Rothblat &
Morton, 1959; Nelson, 1960
Hayflick & Moorhead, 1961; Sorieul &
Ephrussi, 1961
Macpherson & Stoker, 1962; Buonassisi et al.,
1962; Yasamura et al., 1966; Sato & Yas-
umura, 1966
Todaro & Green, 1963
Kleinsmith & Pierce, 1964; Macpherson &
Montagnier, 1964
Wiktor et al., 1964; Andzaparidze, 1968
Ham, 1965; Harris & Watkins, 1965
Levi-Montalcini, 1966; Thompson et al.,
1966
Hoober & Cohen, 1967; Gartler, 1967; Stoker
& Rubin, 1967; Moore et al., 1967; Gerper et
al., 1969; Miller etal., 1971
Yaffe, 1968; Stoker et al., 1968
Metcalf, 1969; see alsoMetcalf, 1990
see Kruse et al., 1991; Collins & Kennedy,
1999
Graham & Van der Eb, 1973
Gospodarowicz et al., 1975
Kohler & Milstein, 1975; Illmensee & Mintz,
1976; Hayashi & Sato, 1976
1.1. История вопроса
27
Таблица 1.1. Ключевые события в развитии методов культивирования тканей и клеток (окончание)
Год/годы Событие
Ссылка
1977 Подтверждение перекрестной контаминации многих линий клеток
клетками HeLa
ЗТЗ фидерный слой и культура клеток кожи
1978 MCDB-селективная бессывороточная среда
Взаимодействие с матриксом
Форма клетки и контроль роста
1980-е Регуляция экспрессии генов. Онкогенез, злокачественность и
трансформация
1980 Матрикс саркомы EHS (Позже Matrigel™)
1983 Регуляция клеточного цикла. Иммортализация культуры с исполь¬
зованием SV40
1980-1987 Разработка многих специализированных клеточных линий
1983 Реконструированная культура клеток кожи
1984 Продукция рекомбинантного тканетипичного активатора плазми-
ногена в клетках млекопитающих
1990-е Культура трансфицированных клеток в промышленном масштабе
для нужд биофарамацевтики
1991 Культура мезенхимальных стволовых клеток взрослого человека
1998 Выращенный в культуре хрящ
1998 Культура эмбриональных стволовых клеток человека
Nelson-Rees & Flandermeyer, 1977; Rheinwald
& Green, 1975
Ham & McKeehan, 1978; Gospodarowicz et
al., 1978b; Reid & Rojkind, 1979; Folkman
& Moscona, 1978
e.g., Darnell, 1982; cm. Weinberg, 1989
Hassell et al., 1980
Evans et al., 1983; см.также Nurse, 1990;
Huschtscha & Holliday, 1983
Peehl & Ham, 1980; Hammond et al., 1984;
Knedler & Ham, 1987
Bell et al., 1983
Collen et al., 1984
Butler, 1991
Caplan, 1991
Aigner et al., 1998
Thomson et al., 1998
отсутствия силы тяжести либо инфильтрации трех¬
мерного матрикса, такого как коллагеновый гель.
Органотипическая культура подразумевает такую же
процедуру, но с соединением клеток различных линий,
например эпидермальных кератиноцитов в комбини¬
рованной культуре с фибробластами кожи, в попытке
создать тканевой эквивалент.
В свое время Харрисон [Harrison, 1907] выбрал
лягушку как источник ткани главным образом по¬
тому, что она является холоднокровным животным,
и соответственно, культивирование не требовало
инкубации. Кроме того, поскольку регенерация тка¬
ней более распространена у низших позвоночных,
он, возможно, предположил, что рост выделенного
образца будет более успешным, чем для ткани мле¬
копитающих. Хотя его метод вызвал новую волну
интереса к культивированию тканей in vitro, лишь
некоторые ученые последовали примеру Харрисона
в выборе вида организма. Влияние медицины обусло¬
вило развитие интереса к теплокровным животным, у
которых как нормальное развитие, так и патологиче¬
ские процессы аналогичны процессам, протекающим
в организме человека. Доступность различных тканей,
многие из которых хорошо росли в культуре, сделала
излюбленным объектом для исследования эмбрион из
куриного яйца; однако развитие экспериментального
животноводства, в том числе создание инбредных
линий грызунов, вывели млекопитающих на передний
край. Хотя куриные эмбриональные ткани могли обес¬
печить разнообразные клеточные линии в первичной
культуре, ткани грызунов имели ряд преимуществ
при создании постоянных клеточных линий [Earle
et al., 1943], а также предоставили широкий спектр
перевиваемых опухолей. Создание технологии транс¬
генных мышей [Beddington, 1992; Peat et al., 1992]
при наличии хорошо изученного генома мыши дало
новый импульс выбору этого животного в качестве
излюбленной модели для исследований в области
культуры тканей.
Когда было продемонстрировано, что клетки че¬
ловеческих опухолей, такие как HeLa, также могут
давать начало постоянным клеточным линиям [Gey et
al., 1952], интерес к человеческим тканям значительно
возрос. Этому интересу позже способствовали работы
Леонарда Хейфлика по исследованию клеточного
цикла [Hayflick & Moorhead, 1961] и потребности
вирусологов и молекулярных генетиков, работающих
с человеческими клетками и тканями. Культивиро¬
вание человеческих клеток получило дальнейший
стимул тогда, когда был разработан ряд различных
бессывороточных селективных сред для специфичных
типов клеток, таких как эпидермальные кератиноциты,
клетки бронхиального эпителия, а также сосудистого
эндотелия (см. разд. 10.2.1.). Эти среды в настоящее
время доступны, хотя стоимость их остается еще доста¬
точно высокой по сравнению со стоимостью обычной
питательной среды.
В течение многих лет низшие позвоночные и
беспозвоночные не рассматривались в качестве объ¬
ектов для культивирования тканей, хотя уникальные
аспекты их развития (высокая способность к регене¬
рации амфибий, метаморфоз у насекомых) сделали их
28
ГЛАВА 1. ВВЕДЕНИЕ
привлекательными для исследований молекулярных
основ развития организма. Относительно недавно по¬
требности сельского хозяйства в области контроля над
насекомыми-вредителями простимулировали развитие
токсикологических и вирусологических исследований
на насекомых. Разработка генных технологий позволи¬
ла предположить, что создание клеточных линий насе¬
комых с бакуловирусами и другими векторами может
стать полезным для создания клеток-продуцентов в
связи со способностью включения крупных геномных
последовательностей в вирусную ДНК и снижением
риска распространения патогенных вирусов человека.
Кроме того, экономическая важность рыбоводства
и загрязнение морей и пресноводных источников
стимулировали изучение клеток и тканей рыб при
нормальном их развитии и патологических процессах.
Методы работы и приемы манипуляций с клетками не
млекопитающих организмов являются продолжением
развития методов работы, разработанных для культур
клеток млекопитающих. Кроме того, для работы с
клетками рыб и насекомых разработано ограниченное
число доступных специальных коммерческих сред (см.
разд. 27.7.1 и 27.7.2).
Типы исследований, в которых могут быть исполь¬
зованы культуры тканей, представлены на рис. 1.2:
1) исследование внутриклеточных процессов, на¬
пример репликации и транскрипции дезоксири¬
бонуклеиновой кислоты (ДНК), синтеза белка,
энергетического метаболизма, а также метаболизма
лекарственных веществ;
2) внутриклеточные потоки веществ, например РНК,
перемещение гормональных рецепторных комплек¬
сов и возникающие в результате процессы передачи
сигнала, а также мембранный транспорт;
3) взаимодействие с окружающей средой, например
питание, инфицирование, цитотоксичность, кан¬
церогенез, действие лекарственных препаратов и
лиганд-рецепторное взаимодействие;
4) межклеточное взаимодействие, например морфо¬
генез, паракринный контроль, кинетика клеточной
пролиферации, метаболическая кооперация, клеточ¬
ная адгезия и подвижность, взаимодействие с мат¬
риксом и органотипические модели для медицин¬
ского протезирования и процессов приживления;
5) генетика, включая геномный анализ в норме и пато¬
логии, генетические манипуляции, трансформация
и иммортализация;
6) образование и секреция клеточных продуктов,
биотехнология, создание биореакторов, получение
конечного продукта, технология производства и
выделения целевого продукта.
Процесс разработки клеточных культур многим
обязан двум основным отраслям медицинских иссле¬
дований: производству противовирусных вакцин и
исследованию механизмов новообразований. Стан¬
дартизация условий исследований и клеточных линий
для производства вакцин и исследования механизмов
действия вирусов, без сомнения, обеспечили стреми¬
тельное развитие современной технологии получения
культуры тканей, в частности оборудования, обес¬
печивающего выход большого количества клеток,
пригодных для биохимических исследований. Все эти
технические усовершенствования стали возможными
благодаря коммерческому производству подходящих
сред, сывороток и повышению контроля загрязнения,
путем добавки антибиотиков в среду и создания сво¬
бодного от микроорганизмов оборудования с ламинар¬
ными потоками воздуха. Эти успехи технологического
прогресса позволили внедрить культуру тканей в
разные области научных интересов.
Дополнительный вес исследованиям на культурах
тканей придают протесты различных обществ защиты
прав животных, выступающих против необоснован¬
ного использования большого количества животных
для проведения экспериментов. Общепринята идея
о необходимости использования животных в ходе
ВНУТРИКЛЕТОЧНЫЕ ПРОЦЕССЫ:
транскрипция ДНК, синтез белка,
энергетический метаболизм, метаболизм
лекарств, клеточный цикл, дифференцировка,
апоптоз
ВНУТРИКЛЕТОЧНЫЕ ПОТОКИ: процессинг
РНЦ, рецепторы гормонов, потоки
метаболитов, обмен кальция, передача
сигнала, мембранный транспорт
ФАРМАКОЛОГИЯ: действие лекарственных
препаратов, лиганд-рецепторное
взаимодействие, метаболизм лекарств,
лекарственная устойчивость
КЛЕТОЧНЫЕ ПРОДУКТЫ: протеомика,
секреция, биотехнология, конструирование
биореактора, получение конечного продукта,
направленный процессинг
МЕЖКЛЕТОЧНОЕ ВЗАИМОДЕЙСТВИЕ:
морфогенез, паракринный контроль,
кинетика клеточной пролиферации,
метаболическая кооперация, клеточная
адгезия и подвижность, взаимодействие с
матриксом, инвазия
ИММУНОЛОГИЯ: эпитопы клеточных
поверхностей, гибридомы, цитокины и
передача сигнала, воспаление
ГЕНОМИКА: генетический анализ,
трансфекция, инфекция, трансформация,
иммортализация, старение
ТКАНЕВАЯ ИНЖЕНЕРИЯ: реконструкция
тканей, матрицы и структурные каркасы,
источники стволовых клеток, разрастание
тканей, дифференцировка
ТОКСИКОЛОГИЯ: инфекция,
цитотоксичность, мутагенез, карциногенез,
раздражение, воспаление
Рис. 1.2. Области применения культуры ткани
1.1. История вопроса
29
доклинических испытаний новых фармакологиче¬
ских препаратов, однако нет морального оправдания
применению большого их количества в исследованиях
косметических препаратов и других подобных экспе¬
риментах. Таким образом, сформировано и возрастает
сообщество, в котором доминирует идея проведения
наибольшего количества исследований in vitro, хотя
эта идея связана с дополнительным подтверждением
достоверности результатов и адекватностью модели in
vitro процессам, протекающим в организме in vivo. Еще
несколько лет назад перспектива перехода большинства
исследований на модели in vitro казалась весьма отда¬
ленной. Тем не менее появление более чувствительных
методов анализа одновременно с совершенствованием
технологий и оборудования сделали работу с культура¬
ми тканей доступной широкому кругу исследователей.
Возможность исследования процессов воспаления in
vitro обеспечила беспрецедентное расширение области
применения культур тканей (см. разд. 22.4).
Помимо вирусологии и онкологии, от развития ме¬
тодов культуры тканей сильно зависят другие области
исследования. Введение в методы клеточного слияния
(гибридизации клеток, см. разд. 27.9.) и генетические
манипуляции [Maniatisetal., 1978; Sambrooketal., 1989;
Ausubel et al., 1996] определили генетический состав
соматических клеток как основной компонент генети¬
ческого анализа высших животных, включая человека.
Широкий круг методов, используемых для генетичес¬
кой рекомбинации, в настоящее время включает пере¬
нос ДНК [Ravid & Freshney, 1998], монохромосомный
перенос [Newbol & Cuthbert,1998] и ядерный перенос
[Копо, 1997], который дополнил метод соматической
гибридизации как инструмент генетического анализа и
генной манипуляции. Перенос ДНК сам по себе поро¬
дил много различных вариаций внесения ДНК в куль¬
туру клеток, включая использование фосфатно-кальцие¬
вой копреципитации, липофекции, электропорации и
ретровирусное инфицирование (см. разд. 27.11).
Особенно бурное развитие генетика человека
получила при реализации проекта Геном Человека
[Baltimore, 2001]. Данные, полученные в ходе этих
исследований, обусловили развитие методов анализа
экспрессии множественных генов [Iyer et al., 1999].
После появления моноклональных антител метод
культуры тканей внес значительный вклад в иммуно¬
логические исследования и связанные с культивиро¬
ванием клеток методы получения и анализа гематопо¬
этических клеточных линий. Глубокое проникновение
в механизм действия антител и возможность исследо¬
вания структуры эпитопа методом моноклональных
антител [Kohler & Mistein, 1975] явились, как и в
случае метода слияния соматических клеток, проло¬
гом к развитию целой новой области генетических
исследований. Возникновение этой области было
обусловлено большим количеством основополагаю¬
щих знаний о контроле транскрипции генов и новыми
технологиями. В результате из возможности внедрять
желаемый ген в прокариотическую и эукариотическую
клетку выросла отрасль промышленности с много¬
миллиардными бюджетами. Клеточные продукты,
производимые клетками с внесенным геном, такие как
гормон роста человека, инсулин, интерферон и др., в
настоящее время стали повседневным явлением. Вмес¬
те с тем отсутствие у бактерий посттранскрипционных
модификаций, таких как гликозилирование, позволяет
предположить, что клетки млекопитающих могут
обеспечить более качественный конечный продукт
[Grampp et al., 1992], особенно в свете развития тех¬
нологии получения иммортализованных клеточных
линий (см. разд. 18.4).
Другие области, связанные с развитием культуры
тканей, включают в себя изучение межклеточного
взаимодействия, внутриклеточных механизмов конт¬
роля дифференцировки и развития [Jessell and Melton,
1992; Ohmichi et al., 1998; Balkovetz & Lipschutz,
1999] и функций нейронов [Richard et al., 1998; Dunn
et al., 1998; Haynes, 1999]. Успехи неврологических
исследований еще не могут считаться значительны¬
ми, поскольку работа с пересеваемыми клеточными
линиями, полученными из нормальных клеток мозга
или нервной ткани, пересев нейронов in vitro до на¬
стоящего времени еще невозможны без использования
клеточной трансформации (см. разд. 18.4). Тем не
менее успехи работы с эмбриональными стволовыми
клетками [Thomson et al., 1998; Rathejen et al., 1998;
Wolf et al., 1998; Webber & Minger, 2004] позволяют
предположить, что этот подход может способствовать
репликации культур, которые в последующем способ¬
ны дифференцироваться в нейроны.
Технология культуры тканей также была при¬
менена во многих привычных областях медицины
и промышленности. Хромосомный анализ клеток,
полученных из амниотической жидкости пункцией
плодного пузыря беременных (см. разд. 27.6), помо¬
гает обнаружить генетические нарушения у плода.
Определение качества питьевой воды и токсического
действия фармацевтических компонентов и веществ,
загрязняющих окружающую среду, могут быть прове¬
дены на основе колониеобразующих и других методов
анализа in vitro (см. разд. 22.3.1,22.3.2,22.4). Дальней¬
шие возможные приложения технологии культуры
тканей к решению медицинских проблем связаны с
обнаруженной у культуры эпидермальных клеток
способностью формировать функционально активный
клеточный пласт [Green et al., 1979], а у клеток сосудис¬
того эндотелия — образовывать капилляры [Folkman &
Haudenschild, 1980]. Такие способности открыли новые
возможности в гомотрансплантации и восстановитель¬
ной хирургии с использованием собственных клеток
[Tuszynski et al., 1996; Gustafson et al., 1997; Limat et al.,
1996], особенно в случае обширных ожогов [Gobet et
al., 1997; Wright et al., 1998; Vunjak-Novakovic & Fresh¬
ney, 2005] (см. также разд. 25.3.8). Вместе с внесением
«нормальных» генов в генетически дефицитные клетки
стало возможным возвращать такие «исправленные»
клетки обратно в организм пациента. Показано, что
трансфецированные клетки культуры бронхиального
эпителия крыс, несущие репортерный ген Р-gal, спо-
30
ГЛАВА 1. ВВЕДЕНИЕ
собны инкорпорироваться в бронхиальную выстилку
при аэрозольном введении их в дыхательный тракт
животных [Rosenfeld et al., 1992]. Показано также, что
культивированные сателлитные клетки внедряются
в поврежденную скелетную мышцу крыс, при этом
клетки с ядром от «пересаженных» клеток обнаружи¬
ваются в зрелых синцитиальных мышечных трубочках
[Morgan et al., 1992].
Перспективные возможности имплантации нор¬
мальных клеток от здорового человека-донора или
подходящих фетальных клеток больному, а также
пересадка генетически измененных клеток от самого
пациента привели к созданию целой ветви метода
культуры тканей, которая получила название тканевая
инженерия [Atala and Lanza, 2002; Vunjak-Novakovic
and Freshney, 2005]. Тканевая инженерия включает в
себя выращивание тканевых эквивалентов с формиро¬
ванием органотипической культуры (см. разд. 25.3.8),
выделение и дифференцировку человеческих эмбри¬
ональных стволовых (ES) клеток и тотипотентных
стволовых клеток взрослого человека, таких как
мезенхимальные стволовые клетки (MSCs), перенос
гена, материаловедение, утилизацию продуктов био¬
производства и технологию трансплантации. Техниче¬
ские барьеры постоянно преодолеваются, выдвигая на
передний план вопросы биоэтики. Стало возможным
имплантирование нормальных нейронов плода паци¬
ентам с болезнью Паркинсона, но общество должно
определить законность использования фетальных ор¬
ганов и тканей для этих целей. Это касается также тех
случаев, когда культивированные собственные клетки
больного подвергаются генетической реконструкции
путем трансфекции «нормального» гена, например
внесение нормального гена продукции инсулина в
культивируемые Р-клетки островков поджелудочной
железы больных диабетом. Существуют проблемы при
трансфекции клеток другого типа, например введение
клеток-предшественников скелетных мышц [Morgan
et al., 1992], что позволяет клеткам инкорпорировать
в медленно делящиеся ткани и потенциально обеспе¬
чивать пролонгированный физиологический эффект.
Хотя этические проблемы такого типа исследований
стволовых клеток менее спорны, пока очевидны тех¬
нические ограничения этого метода.
Оплодотворение in vitro (IVF) (экстракорпораль¬
ное оплодотворение) начали разрабатывать начиная
с первых экспериментов на эмбриональной культуре
[см. Edwards, 1996], и в настоящее время оно юриди¬
чески и этически принято и широко используется во
многих странах [см. Gardner and Lane, 2003]. Однако
вызывает бурные этические дебаты формирование
in vitro гамет из герминативных стволовых клеток,
выделенных из семенников и яичников [Dennis, 2003]
или из ES-клеток. Имплантация ооцитов, полученных
культивированием клеток яичника мышиного эмбри¬
она, привела к рождению нормальных мышей [Eppig,
1996; Obata et al., 2002]. Сперматиды, выделенные из
семенников новорожденных бычков и культивиро¬
ванные вместе с клетками Сертоли [Lee et al., 2001],
так же как и сперматиды, выделенные из семенников
мышей, способны были оплодотворять яйцеклетку
с последующим рождением здоровых, фертильных
особей [Marh et al., 2003].
1.2. ПРЕИМУЩЕСТВА МЕТОДА
КУЛЬТУРЫ ТКАНИ
1.2.1. Контроль окружения
Два основных преимущества техники культуры ткани
(табл. 1.2) заключаются в возможности очень точного
контроля физико-химических свойств окружения
(pH, температуры, осмотического давления, парци¬
ального давления 02 и С02) и физиологических ус¬
ловий, которые могут поддерживаться относительно
неизменными, но не всегда могут быть определены.
Большинство клеточных линий еще пока требуют для
своего роста добавок в среду сыворотки или других
недостаточно изученных компонентов. Эти добавки
варьируют и содержат неопределенное количество
Таблица 1.2. Преимущества метода культуры ткани
Категория
Преимущества
Физико-химические
свойства окружения
Физиологическое со¬
стояние
Микроокружение
Однородность клеточ¬
ных линий
Характеристика
Сохранение
Сертификация и аккре¬
дитация
Повторяемость и вос¬
производимость ре¬
зультатов
Экономия реагентов
Контроль концентра¬
ции и времени (СхТ)
Механизация
Снижение количества
использованных жи¬
вотных
Контроль pH, температуры, ос¬
мотического давления, парци¬
ального давления растворенных
газов
Контроль гормонов и концентра¬
ций питательных веществ
Регуляция матрикса, межклеточ¬
ных взаимодействий, газовой
диффузии
Доступность селективных сред,
клонирование
Цитология и иммунное окрашива¬
ние легко осуществимо
Может храниться в жидком азоте
Происхождение, история и чистота
культуры могут быть проверены
на аутентичность и документи¬
рованы
Количественный анализ легко
осуществим
Использование меньших объемов
реагентов, прямое воздействие
на клетки, снижение стоимости
Возможность определять и конт¬
ролировать дозу, концентрацию
и время действия.
Возможность микротитрования и
роботизации
Исследование цитотоксичности,
скрининговых исследований в
фармацевтике, косметологии
и т.д.
1.2. Преимущества метода культуры ткани
31
элементов, таких как гормоны и другие регуляторные
вещества. Идентификация некоторых существенных
для клеточного роста компонентов сыворотки вместе
с более глубоким пониманием механизмов действия
факторов, регулирующих клеточную пролиферацию
(см. табл. 10.3), сделали возможным замену сыворотки
определенным набором компонентов (см. разд. 10.4).
Поскольку исследователи стремятся воспроизвести
нормальные фенотипические свойства тканей in vitro,
роль внеклеточного матрикса все более возрастает.
В настоящее время эта роль соответствует роли сы¬
воротки: матрикс часто необходим, но не всегда точно
определен, его состав может быть изменен и, со време¬
нем, когда будет доступна биохимическая идентичность
всех компонентов матрикса, состав его будет полностью
охарактеризован.
1.2.2. Характеристика
и однородность образца
Образцы тканей всегда разнородны. Повторно взятые
образцы даже из одной и той же ткани различны по
клеточному составу. Предполагается, что после одно-
го-двух пересевов культивируемые клеточные линии
однородны (или, по крайней мере, единообразны) по
составу, поскольку клетки беспорядочно перемеши¬
ваются при каждом переносе и селективное давление
условий культивирования приводит к формированию
Таблица 1.3. Ограничения метода культуры ткани
Категория
Примеры
Необходимость нали¬
чия специальных
навыков
Контроль окружаю¬
щей среды
Объем расходов и
стоимость
Генетическая неста¬
бильность
Фенотипическая
нестабильность
Идентификация ти¬
пов клеток
Стерильность манипуляций
Химические загрязнения
Микробное загрязнение
Перекрестное загрязнение
Рабочее место
Культивирование, контроль pH
Предотвращение биозагрязнения
окружающей среды и утилизация
отходов
Основное оборудование для произ¬
водства в больших масштабах
Среды, сыворотки
Одноразовое пластиковое обору¬
дование
Разнородность, изменчивость
Обратная дифференцировка
Адаптация
Избирательный чрезмерный рост
Маркеры не всегда экспрессиро¬
ваны
Сложно восстановить гистологи¬
ческую принадлежность, атипич-
ность клеток
Геометрия пространства и микроок¬
ружение изменяют цитологию
гомогенной культуры наиболее устойчивых типов кле¬
ток. Следовательно, в каждой субкультуре репликатив¬
ные образцы идентичны друг другу, и характеристика
клеточных линий может сохраняться в нескольких
поколениях и быть неизменной при замораживании
и хранении в жидком азоте. Поскольку эксперимен¬
тальные самовоспроизводящиеся клетки фактически
идентичны, необходимость вариационного статисти¬
ческого анализа уменьшается.
Возможность использования строгих тестов иденти¬
фикации клеточных линий (гл. 15) и их загрязненности
(гл. 18) означает, что сохраненные первичные культуры
и клеточные линии могут быть пригодны для будущих
исследований и коммерческого использования.
1.2.3. Экономичность, эффективность
и автоматизация процесса
В культурах тканей можно осуществить непосред¬
ственное воздействие на клетки определенных низких
концентраций реагентов. Следовательно, для достиже¬
ния аналогичного эффекта требуется использование
меньших концентраций реагентов, чем in vivo, где до
90% действующего вещества теряется при выведении и
распределении по другим тканям, которые не изучают¬
ся в данном эксперименте. Проведение скрининговых
исследований с большим количеством параметров
и использованием самовоспроизводящихся клеток
дешевле, при этом исчезает необходимость решения
юридических, моральных и этических вопросов, свя¬
занных с экспериментами на животных. Разработка
новых многолуночных планшетов и роботизация про¬
цесса также вносит значительный вклад в экономию
времени и объема работы.
1.2.4. Моделирование in vitro условий
in vivo
Перфузионные методы позволяют доставлять в
ходе эксперимента специфические компоненты, кон¬
центрация которых в ходе процесса и при различном
состоянии метаболизма клеток может регулироваться
(см. табл. 1.2). Разработка гистотипических и органо¬
типических моделей также повышает точность моде¬
лирования процессов in vivo.
1.3. ОГРАНИЧЕНИЯ МЕТОДА
КУЛЬТУРЫ ТКАНИ
1.3.1. Наличие специальных навыков
Техника культивирования клеток должна проводиться
в строго асептических условиях, поскольку животные
клетки растут намного медленнее, чем большинство
наиболее распространенных контаминантов, таких
как бактерии, плесневые грибы и дрожжи. Более того,
32
ГЛАВА 1. ВВЕДЕНИЕ
в отличие от микроорганизмов клетки многоклеточ¬
ных животных в норме изолированно не существуют
и, следовательно, не способны к жизнедеятельности
без обязательного добавления в среду компонентов,
воссоздающих условия плазмы крови или межкле¬
точной жидкости. Воспроизведение таких условий
предполагает наличие определенного уровня подготов¬
ки персонала для точного определения необходимых
условий культивирования, своевременной диагностики
возможных ошибок (табл. 1.3, см. также гл. 28) и пре¬
дотвращения перекрестной контаминации культур.
Таким образом, культура тканей не может использо¬
ваться от случая к случаю для проведения одного-двух
экспериментов.
1.3.2. Затраты
Основным ограничением культивирования тканей с
экономической точки зрения является большой объем
работы и материалов, которые необходимы для полу¬
чения относительно небольшого количества конечного
продукта. Реально максимальная партия, которую
могут нарастить небольшие лаборатории (два-три
сотрудника, занимающихся культивированием), со¬
ставляет 1-10 г клеток. Крупные лаборатории с боль¬
шими возможностями способны получить 10-100 г;
опытно-промышленные установки дают свыше 100 г
клеток в одной партии. Этот уровень превышает выход
клеток даже в крупных лабораториях, однако далек от
промышленного уровня производства, при котором
специальное оборудование позволяет получать кило¬
граммы клеток.
Стоимость производимых клеток примерно в 10 раз
выше, чем стоимость использования животных тканей.
Следовательно, если требуется большое количество
ткани (больше 10 г), причина, вынуждающая исполь¬
зовать культуру тканей, должна быть очень весома.
Для небольшого количества тканей (около 10 г) сто¬
имость получения культуры покрывается обычными
расходами, однако цена учитывает возможность
снижения стоимости анализа или подготовительных
процедур. Полумикро- или микрометоды часто можно
проводить быстрее, поскольку они требуют меньшего
общего времени работы, объема проб, времени цент¬
рифугирования и т.д. Кроме того, многие процессы
автоматизированы.
1.3.3. Дифференцировка и селекция
Когда в 1950-х годах были достигнуты первые ос¬
новные успехи воспроизводства клеточных линий,
многие исследователи отмечали утрату фенотипиче¬
ских характеристик, характерных для тех тканей, из
которых были изолированы клетки. В этом явлении
первоначально обвинили дедифференцировку — про¬
цесс, который, как предполагали, противоположен
дифференцировке. Однако позже было показано, что
такая утрата гистологических характеристик связана
с чрезмерно быстрым ростом недифференцированных
клеток того же или другого происхождения. Разработка
бессывороточных селективных сред (см. разд. 10.2.1)
позволила выделять клетки, имеющие особое проис¬
хождение, и теперь можно видеть, что при правильно
подобранных условиях многие из утраченных свойств
таких дедифференцированных клеток восстанавлива¬
ются (см. разд. 17.7).
1.3.4. Происхождение клеток
Если свойства дифференцированных клеток были
утрачены по какой-либо причине, трудно обнару¬
жить сродство клеток с функционально активными
клетками тканей, из которых они были получены.
Поэтому для характеристики и идентификации
происхождения клеток требуются их постоянные
маркеры (см. разд. 16.1). Кроме того, для экспрессии
этих маркеров могут потребоваться совершенствова¬
ние и модификация условий культивирования (см.
разд. 3.4.1, 17.7).
1.3.5. Нестабильность
Нестабильность является главной проблемой многих
постоянных клеточных линий, что связано с нестабиль¬
ностью анеуплоидного набора хромосом в животных
клетках. Даже в короткоживущих культурах ^тран¬
сформированных клеток отмечается гетерогенность
интенсивности и способности к дифференцировке,
которые могут привести к вариабельности результатов
от одного пассажа к другому (см. разд. 18.3).
1.4. ОСНОВНЫЕ ОТЛИЧИЯ КУЛЬТУРЫ
IN VITRO
Многие различия между поведением клеток в культуре
и исходной ткани in vivo проистекают из диссоциации
клеток, в свою очередь, связанной с трехмерной гео¬
метрией ткани in vivo и двухмерным распространением
клеток на субстрате in vitro. Специфические межкле¬
точные взаимодействия, характерные для гистологии
данной ткани, утрачиваются, клетки растягиваются
по площади, становясь подвижными, и во многих
случаях начинают делиться с увеличением концент¬
рации факторов роста в клеточной популяции. При
формировании клеточная линия может быть пред¬
ставлена только одним-двумя типами клеток и многие
разнообразные формы межклеточных взаимодействий
утрачиваются.
Среда культивирования также испытывает недоста¬
ток ряда системных компонентов, участвующих в регу¬
ляции гомеостаза in vivo, в том числе наиболее важных,
относящихся к нейро-гуморальной системе. Без этого
контроля клеточный метаболизм может быть более
1.5. Типы культуры ткани
33
■sLiaocju.^
Шппаппппи
■Зппоаппппо
рпппппппппо
■пппппппвппп
лпппиппипппп
аппппппппппп
□ппппппппппп
□□□□□□□□□□□□
1ППППППППППП
_1ПППОППППППП
■□□□ппппвппп
ЛППППППИППИ
■лпоопппппп
Щ2рппиаг_
постоянным in vitro, чем in vivo, но не может считаться
реально представляющим ткани, их которых эти клет¬
ки были получены. Анализ данного фактора привел к
пониманию необходимости введения в культуральную
среду различных гормонов (см. разд. 10.4.2), и, видимо,
эта тенденция будет развиваться.
Энергетический метаболизм in vitro представлен в
основном гликолизом, и, хотя цикл лимонной кислоты
также функционирует, он играет меньшую роль.
Нетрудно обнаружить другие различия между
условиями окружения клеток in vitro и in vivo (см.
разд. 22.2), и это несовпадение нередко приводит к
тому, что работы на культуре клеток часто рассматри¬
ваются скептически. Однако, хотя существование таких
различий нельзя отрицать, многие специфические
ОРГАННАЯ
КУЛЬТУРА
функции тканей и клеток можно воспроизводить в
культуре. Поэтому в той степени, в какой все описанные
ограничения позволяют рассматривать их как адекват¬
ную модель, культуры клеток являются очень ценным
инструментом для исследований.
1.5. ТИПЫ КУЛЬТУРЫ ТКАНИ
Существуют три основных метода формирования
культур [Schaeffer, 1990] (см. Приложение IV, рис. 1.3
и табл. 1.4).
1) Термин органная культура подразумевает, что в
культуре сохранены, по крайней мере частично,
структурно-функциональные характеристики
ОРГАНОТИПИЧЕСКАЯ
КУЛЬТУРА
ЭКСПЛАНТАТНАЯ ДИССОЦИИРОВАННАЯ
КУЛЬТУРА КЛЕТОЧНАЯ КУЛЬТУРА
Ткань на границе
раздела сред
жидкость-газ;
гистологическая
структура сохранена
Ткань на границе
раздела жидкость/
плотная среда,
клетки мигрируют из
эксплантата
Разрушенные ткани;
клетки формируют
монослой на границе
раздела жидкость/
плотная среда
Различные клетки
совместно культивируются
на подложке или без
нее; органотипическая
структура восстановлена
Рис. 1.3. Типы клеточных культур
Таблица 1.4. Свойства различных типов культур
Категория Органная культура Эксплантат Клеточная культура
Эмбриональные органы, фраг¬
менты взрослых тканей
Источник
Трудозатраты
Исследуемые характерис¬
тики
Гистология
Биохимическая дифферен¬
циация
Пересев
Повторный отбор проб,
воспроизводимость,
гомогенность
Количественный анализ
Высокие
Гистология
Информативна
Возможна
Невозможен
Высокая вариабельность, низ
кая воспроизводимость
Затруднен
Фрагменты тканей
Умеренные
Цитология и маркеры
Затруднена
Гетерогенна
Возможна из матричного
нароста
Высокая вариабельность,
низкая воспроизводи¬
мость
Затруднен
Измельченные ткани, первич¬
ная культура, пересеянная
клеточная линия
Низкие
Биохимические, молекулярные,
иммунологические и цитоло¬
гические исследования
Невозможна
Утрачена, но возможно восста¬
новление
Стандартная процедура
Низкая вариабельность, высо¬
кая воспроизводимость
Простой, много доступных тех-
нических средств
34
ГЛАВА 1, ВВЕДЕНИЕ
ткани in vivo. Для этого кусочки ткани культиви¬
руются на границе раздела сред жидкость-газ (на
плотике, решетке или гелевой подложке), которые
предпочтительны для фиксации сферической или
трехмерной структуры.
2) В первично эксплантируемой культуре фрагмент
ткани (эксплантат) помещается на стекло (или
пластик) в жидкую среду, где после прикрепления
активизируется миграция клеток по поверхности
плотного субстрата (см. разд. 12.3.1).
3) Термин клеточная культура предполагает, что
фрагмент ткани или полученная из первичного
эксплантата культура измельчается (механически
или ферментативно) до клеточной суспензии, ко¬
торая затем культивируется как адгезированный
монослой на плотном субстрате или как суспензия в
культуральной жидкой среде (см. разд. 12.3,13.7).
В результате сохранения клеточных взаимодей¬
ствий, изначально существовавших в ткани, которая
была использована для культивирования, органные
культуры сохраняют специфические свойства этой
ткани. Они растут не очень быстро (клеточная про¬
лиферация ограничена и снижена на периферии
эксплантата) и используются, главным образом, для
эмбриональных тканей и, следовательно, не могут
быть пересеяны. Каждый эксперимент на органной
культуре требует использования свежего эксплантата,
что требует больших трудозатрат и снижает воспро¬
изводимость результатов по сравнению с клеточной
культурой. Таким образом, количественный анализ
более сложен, и количество материала, которое может
быть выращено, ограничено размерами эксплантата
(около 1 мм3) и объемом работ, требуемых для отде¬
ления и пересадки культуры. Тем не менее органные
культуры сохраняют специфические гистологические
межклеточные взаимодействия, без которых сложно
воспроизвести характерные свойства тканей.
Из первичного эксплантата или диспергированной
клеточной суспензии можно получить клеточные
Таблица 1.5. Субкультура
Преимущества
Недостатки
Воспроизводство
Повреждение при ферментативной
или механической дезагрегации
Большое количество
Селекция клеток, приспособленных
клеток
к росту в культуре
Возможность клони¬
Чрезмерно быстрый рост неспеци¬
рования
ализированных или стромальных
клеток
Повышение однород¬
Генетическая нестабильность
ности
Характеристика реп¬
Утрата дифференцировки и свя¬
ликативных образ¬
занных с нею свойств (возможно,
цов
индуцированная)
Хранение в заморо¬
Возрастание риска неправильной
женном состоянии
идентификации или перекрестного
заражения культур
культуры. Поскольку в таких культурах часто отме¬
чается клеточная пролиферация, задача формирова¬
ния постоянных клеточных культур представляется
выполнимой. Монослой или клеточная суспензия
со значительной фракцией растущих клеток (см.
разд. 21.11.1) может быть отделен путем фермента¬
тивного воздействия или простым разведением и пе¬
ресевом (субкультивированием) во флакон со свежей
питательной средой (табл. 1.5; см. также разд. 13.1,
13.7). Эта процедура называется пассажем, а дочерняя
культура, сформированная таким образом, представ¬
ляет собой начало клеточной линии.
Формирование клеточной линии из первичной
культуры подразумевает (1) увеличение общего коли¬
чества клеток после нескольких генераций и (2) конеч¬
ное преобладание клеток или линий дифференцировки,
обладающих способностью к высокому темпу роста,
что приводит к (3) высокой степени однородности
клеточной популяции (см. табл. 1.5). Клеточную ли¬
нию можно охарактеризовать, и такие характеристики
применимы к большинству их жизненных циклов.
Получение постоянных {или «установившихся») кле¬
точных линий обычно подразумевает фенотипическое
изменение или трансформацию (см. разд. 3.8,18.2).
Сублиния, полученная при отборе клеток из куль¬
туры путем клонирования или какими-то другими
методами, называется клеточным штаммом. Подра¬
зумевается, что клеточный штамм имеет подробную
характеристику. Клеточные линии или клеточные
штаммы могут делиться и развиваться в адгезирую-
щем монослое или суспензии. Монослойная культура
означает, что при наличии необходимых условий
клетки будут прикрепляться к субстрату и в норме
будут делиться и распространяться таким способом.
Зависимость от подложки означает, что прикрепле¬
ние (и обычно степень распластанности) к субстрату
является необходимой предпосылкой для клеточной
пролиферации. Монослойная культура является ме¬
тодом культивирования, общепринятым и наиболее
распространенным для большинства нормальных
клеток, за исключением гематопоэтических. Суспензи¬
онную культуру обычно получают из клеток, которые
способны выжить и делиться без прикрепления к
субстрату (независимость от подложки); этой способ¬
ностью обладают гематопоэтические клетки, трансфор¬
мированные клеточные линии и клетки, выделенные
из злокачественных опухолей. Можно показать, что
некоторая доля клеток, способных к делению в суспен¬
зии, присутствует во многих нормальных тканях (см.
разд. 18.5.1). Принадлежность этих клеток к какому-то
типу остается неясной, но в настоящее время посту¬
лируется их сродство со стволовыми клетками или
некоммитированными предшественниками. Подра¬
зумевается, что некоторые из культивируемых клеток
представляют собой пул клеток-предшественников
из исходной ткани (см. разд. 3.10). Культивируемые
клеточные линии напоминают недифференцирован¬
ные клетки-предшественники in vivo больше, чем
полностью дифференцированные клетки, поскольку
1.5. Типы культуры ткани
35
нормальные дифференцированные клетки тканей, как
правило, не способны к делению.
Поскольку они могут размножаться как однород¬
ная клеточная суспензия или монослой, клеточные
линии имеют много преимуществ, проявляющихся
в возможности количественного анализа, точной ха¬
рактеризации и точной репликации образцов. К недо¬
статкам клеточных линий можно отнести отсутствие
или недостаточность межклеточных и клеточно-мат-
риксных взаимодействий, имеющихся у органных
культур. По этой причине многие исследователи пы¬
тались воссоздать трехмерную клеточную структуру
путем использования агрегатов в клеточной суспензии
(см. разд. 25.3.2). Такие подходы потребовали вве¬
дения новых терминов или, по крайней мере, нового
истолкования некоторых старых понятий. Термин
гистотипическая культура или гистокультура (нами
используется термин гистотипическая культура) стал
означать культуру высокой плотности, «тканеобраз¬
ную», полученную из одной клетки, в то время как ор¬
ганотипическая культура подразумевает присутствие
более чем одного типа клеток, взаимодействующих так,
как они могли бы это делать в органах, из которых они
ведут свое происхождение (либо имитируют это взаи¬
модействие). Органотипическая культура открывает
новые перспективы для изучения клеточного взаимо¬
действия внутри отдельной определенной популяции
однородных и, предположительно, генетически и
фенотипически определенных клеток.
Во многих отношениях некоторые из наиболее
многообещающих подходов к работе с культурой
ткани возникли и развились благодаря пониманию
необходимости учета межклеточного взаимодействия
в гомогенной и гетерогенной клеточной популяции в
культуре. Это понимание отмечает начало перехода от
эры фундаментальной молекулярной биологии, когда
многие регуляторные процессы были исследованы на
клеточном уровне, к эпохе клеточной или тканевой
биологии, в которой это приобретенное знание приме¬
няется к составляющей единое целое клеточной попу¬
ляции клеток и к более ясному пониманию механизмов
передачи сигналов между клетками.
Глава 2
ТРЕНИРОВОЧНЫЕ
ПРОГРАММЫ
2.1. ЦЕЛИ И ЗАДАЧИ ГЛАВЫ
Эта книга первоначально была создана как источник
информации о методах и приемах работы с культурами
тканей, предоставляющий также некоторое количество
теоретического материала и практические протоколы,
размещенные в тексте книги и разумно объясняющие
некоторые из правил проведения этих процедур.
Так или иначе, существует необходимость облег¬
чить первоначальный этап понимания и усвоения мате¬
риала начинающими работниками, а также тренировки
тех, кто уже имеет некоторые минимальные знания и
навыки работы с культурой ткани. Самостоятельный и
опытный исследователь может изучить только те части
руководства, которые имеют отношение к области его
интересов. В то же время студенту или лаборанту с
ограниченными навыками работы и знаниями рекомен¬
дуется проработать раздел тренировочных программ,
основанных на его предыдущем опыте и соответ¬
ствующий требованиям его руководителя. Эта глава
имеет целью обеспечить читателей тренировочными
программами как на начальном, так и на более высоком
уровне, и может быть использована руководителями
с возможностью модификации этих программ при
подготовке нового персонала.
Настоящие программы представлены как серия
упражнений в стандартном формате с перекрестными
ссылками на соответствующие протоколы и сноски.
Инструкции стандартных протоколов не повторяются
в упражнениях, поскольку они подробно описывают¬
ся в соответствующих главах. Однако специальные
указания позволяют модифицировать стандартные
протоколы, чтобы сделать каждое упражнение более
интересным, а полученные данные учащийся может
затем обработать и проанализировать. Большинство
упражнений предлагает использовать минимальное
количество образцов, для того чтобы сэкономить время
обучения и снизить сложность, поэтому учащийся дол¬
жен осознавать важность большего количества повто¬
рений стандартных операций и манипуляций в ходе по¬
вседневной экспериментальной практики. Упражнения
расположены в порядке усложнения с точки зрения ме¬
тодических приемов и использования оборудования: от
элементарных до более сложных. Все они представлены
в табл. 2.1. Упражнения, обязательные для выполнения,
выделены жирным шрифтом. Предполагается, что как
элементарные, так и более сложные задания послужат
хорошей основой для работы и усвоения материала,
хотя как технические возможности обучающегося, так и
время, имеющееся у него в распоряжении, могут влиять
на выбор изучаемого материала.
В тех случаях, когда выполнение упражнения тре¬
бует применения более чем одного протокола, номера
протоколов приводятся через точку с запятой. Там,
где возможен выбор, номера разделяются «или» и
инструктор (преподаватель) может решить, какой из
протоколов более уместен или лучше подходит к работе
конкретной лаборатории. Рекомендуется постараться
освоить все основные методы, описанные в упражнени¬
ях, приведенных в табл. 2.1, уделив большее внимание
наиболее существенным, выделенным жирным шриф¬
том. За инструктором остается свобода выбора заданий
в специальных разделах.
В каждом упражнении приведены дополнительные
разделы и протоколы. Они не являются необходимой
частью упражнения, но могут быть в него включены,
если это отвечает целям и интересам лаборатории или
обучающегося.
2.2. УПРАЖНЕНИЯ ДЛЯ НАЧИНАЮЩИХ
(основной курс)
Ниже приводятся упражнения, которые преподавателю
или учащемуся следует выполнить прежде всего. Боль¬
шинство из них просты и доступны для выполнения.
Протоколы и варианты этих протоколов построены так,
чтобы выполнение их становилось интересным экспе¬
риментом, и представлены подробно в текстах, на кото¬
рые есть указания в перекрестных ссылках. Количества
реагентов, указанные в разделах Материалы каждого
протокола, соответствуют описываемому методу, но
при необходимости могут быть сокращены или допол¬
нены. Упражнения, выделенные в табл. 2.1 жирным
шрифтом, представляются наиболее важными.
Обратите внимание на список необходимых мате¬
риалов и оборудования, приведенный в начале упраж¬
нений. Это позволит руководителю оценить имеющееся
у него оборудование, определить его роль и возможнос¬
ти использования, а также понять уровень подготовки
и технического обеспечения, необходимый для работы
с культурами тканей. Принципы хранения также долж¬
ны быть объяснены с тем, чтобы привлечь внимание
к особенностям упаковки и хранения образцов с раз-
2.2. Упражнения для начинающих
37
Таблица 2.1. Тренировочные программы
№ упраж- Процедура
нения
Навыки, которыми следует овладеть
Nfi протокола
8
9
10
11
12
13
14
15
16
17
18
19
20
Пипетирование и перенос жидкостей
Введение в технику ведения культуры
ткани
Асептический метод: приготовление
питательной среды
Смена среды в монослойной куль¬
туре
Мытье и стерилизация стеклянной
посуды
Подготовка и стерилизация воды
Приготовление и стерилизация фос¬
фатно-буферного раствора (PBS)
Приготовление стандартных растворов
для контроля pH
Приготовление основной питатель¬
ной среды и стерилизация методом
фильтрования
Приготовление полной среды или
концентрата основной среды 10 х
Подсчет клеток в гемоцитометре и
электронном счетчике
Основной курс
Знакомство с техникой. Точность и аккуратность мани¬
пуляций
Использование инвертированного микроскопа. Оценка
различий в морфологии клеток внутри одной линии
и межлинейные. Использование фотоаппарата и из¬
готовление референс-фотографий
Асептические манипуляции. Навыки в работе со сте¬
рильными веществами и флаконами без заражения.
Внесение добавок в питательную среду
Обследование культуры. Замена питательной среды
Знакомство со вспомогательными службами. Уяснение
необходимости чистоты и нетоксичности стеклянных
контейнеров
Понимание значения чистоты и стерильности. Приме¬
нение и ограничения. Стерилизация автоклавирова-
нием
Состав солевого раствора. Осмолярность. Забуферива-
ние и контроль pH. Стерилизация термостабильных
растворов автоклавированием
Знакомство с фенолом красным как индикатором pH
Техника фильтрации и сравнение различных методов
Асептические манипуляции. Состав среды. Контроль
pH
Количественные навыки. Подсчет клеток и оценка жиз¬
неспособности. Сравнительная оценка достоинств
двух методов
Оценка культуры. Асептические манипуляции. Подсчет
клеток и оценка жизнеспособности. Выбор концентра¬
ции для пересева
Оценка культуры. Асептические манипуляции. Методы
дезагрегации клеток, в т. ч. метод трипсинизации
Цитология клеток. Фазово-контрастная микроскопия.
Фиксация и окрашивание. Фотографирование
Репликативные культуры в многолуночных планшетах.
Подсчет клеток. Выбор концентрации для пересева
Упражнения повышенной сложности
Характеристика клеточных линий Подтверждение идентичности клеточных линий
Рост субкультуры клеток в суспензии
Рост субкультуры постоянной клеточ¬
ной линии в монослое
Окрашивание монослойной клеточной
культуры по Гимза
Построение и анализ кривой роста
Обнаружение микоплазм
Криоконсервация культивируемых
клеток
Первичная культура
Клонирование в монослое
В упражнении
16.1; 16.6
6.1; 11.7
13.1
11.1
11.5
11.6
9.1
11.11; 11.12,
11.13; 11.14
11.8; 11.9
21.1; 21.2
13.3
13.2
16.2; 16.6
21.8; 21.1,21.2
16.7 или 16.8
или 16.9 или
16.10
19.2 или 19.3
Осознание важности скрининга культур на присутствие
микоплазм. Опыт применения флюоресцентного ме¬
тода или ПЦР для постоянного скрининга клеточных
линий на предмет микоплазматического заражения
Практика замораживания клеток, подготовка клеточных 20.1; 20.2
линий, ведение записей по использованию морозиль¬
ников, контроль состояния хранящихся культур
Происхождение и разнообразие клеточных культур. Ва- 12.2; 12.6 или
рианты методологии получения первичной культуры 12.7
Техника клонирования методом серийных разведений. 14.1; 21.10; 14.6
Определение эффективности посева. Клональное
выделение
38
ГЛАВА 2. ТРЕНИРОВОЧНЫЕ ПРОГРАММЫ
Окончание табл. 2.1
№ упраж¬
нения
Процедура
Навыки, которыми следует овладеть
Ns протокола
Специализированные упражнения
21
Клонирование в суспензии
Техника клонирования методом серийных разведений
суспензионной культуры. Выделение суспензионных
клонов
14.4 или 14.5
14.8
22
Селективная среда
Демонстрация селективного роста специфических кле¬
точных типов
23.1 или 23.2
23
Разделение клеток
Выделение клеточного типа с желаемым фенотипом
одним из нескольких сепаративных методов
15.1 или 15.2
24
Приготовление фидерных слоев
Способы повышения эффективности культивирования.
Селективный эффект фидерных слоев
14.3
25
Гистотипическая культура в фильтро¬
вальном колодце с вкладышами
Знакомство с культурами с высокой плотностью. Изу¬
чение возможностей дифференцировки, транспорта
питательных веществ и инвазивных свойств
25.4
26
Исследования цитотоксичности
Знакомство с высокопроизводительными методами скри¬
нинга. Положительное и отрицательное действие
22.4
27
Исследование выживаемости (может
проводиться как компонент упраж¬
нения 20 или как отдельное упраж¬
нение)
Использование клонального роста для обнаружения
положительного и отрицательного действия на выжи¬
ваемость и пролиферативную активность клеток
22.3
делением на стерильные и нестерильные культуры
ткани. Описываются также необходимое пластиковое
оборудование, используемые реактивы с условиями
их хранения, в том числе при комнатной температуре
либо в интервале от 4 °С до -20 °С. Инструктор должен
знать положение всех образцов: предельные сроки их
хранения, какие замены производятся, кого следует
информировать при истечении сроков хранения и как
производить перемещение образцов с тем, чтобы наи¬
более старые использовались первыми.
Упражнение 1.
Асептический метод I:
Пипетирование и перенос жидкостей
Цель процедуры
Перенести жидкость быстро и аккуратно из одной
емкости в другую.
Навыки, которыми
следует овладеть
Навыки ручного пипетирования; повышение скорости,
точности и воспроизводимости пипетирования.
Инструктирование и руководство: на первом эта¬
пе — постоянные, затем оставить обучающегося пов¬
торять упражнение с подробными записями.
Время: 30 мин-1 ч.
Сведения общего характера
Манипуляции со стерильными жидкостями (см.
разд. 5.2.7); манипуляции с флаконами и бутылками
(см. разд. 6.3.4); пипетирование (см. разд. 6.3.5).
Демонстрационный материал или действия: инс¬
труктор должен показать приемы ручного пипетиро¬
вания, включая набирание жидкости в пипетку и пе¬
ренос жидкости, сопровождать процесс замечаниями,
указывающими на необходимость компромисса между
скоростью работы и аккуратностью пипетирования.
Инструктор также должен продемонстрировать уда¬
ление жидкости при помощи вакуумного насоса (если
он используется в лаборатории) и объяснить механизм
его действия и ограничения, накладываемые сообра¬
жениями безопасности.
Упражнение
Краткое изложение процедуры
Перенести жидкость пипеткой из одного сосуда в
другой.
Стандартный протокол
Асептический метод пипетирования включает в себя
работу в вертикальном ламинарном потоке (см. про¬
токол 6.1) или работу на открытом рабочем столе (см.
протокол 6.2). До начала выполнения полного прото¬
кола полезно потренироваться в простых нестерильных
манипуляциях с пипеткой, включая перенос воды из
одной емкости в другую. Это позволит более близко
познакомиться с данной задачей до начала работы в
асептических условиях.
Экспериментальные варианты
процедуры
Цель упражнения — повысить сноровку и качество
проведения манипуляции с пипетками и емкостями в
асептических условиях. Следующие дополнительные
шаги могут повысить интерес к работе и наблюдать за
тем, как обучащийся умеет:
2.2. Упражнения для начинающих
39
1. Предварительно взвесить флаконы, используемые
для приема жидкости:
а) добавить 5 мл в каждый из 5 флаконов;
б) использовать 5-мл пипетку.
2. Использовать 25-мл пипетку.
3. Записать время, которое потребовалось для завер¬
шения пипетирования.
4. Взвесить флаконы снова.
5. Инкубировать флаконы, с тем чтобы убедиться
в стерильности манипуляции и отсутствии зара¬
жения.
Данные
1. Подсчитайте средний вес жидкости во флаконе.
2. Запишите распределение данных:
а) подсчитайте процент разброса величины объема
или
б) подсчитайте величину среднего и стандартного
отклонения:
— внесите значения в таблицу в Excel в столбик;
— поместите курсор в клетку ниже столбика
цифр;
— нажмите стрелку справа от кнопки £ на
стандартной панели инструментов, выберите
опцию «средняя величина»;
— выделите столбик цифр, если он еще не
выделен, нажмите Enter. Это даст значение
средней величины ваших данных;
— поместите курсор в следующую клетку;
— нажмите стрелку справа от кнопки X на
стандартной панели инструментов, выберите
«другие функции» и затем выберите опцию
«стандартное отклонение»;
— выделите столбик цифр, если он еще не вы¬
делен, нажмите Enter.
Вы получите значение стандартного отклонения
ваших данных, которое вы затем сможете посчитать
как процент значений, чтобы оценить точность прове¬
денного пипетирования.
Анализ данных
1. Сравните результаты, полученные при помощи
разных пипеток и объясните разницу:
а) в точности,
б) во времени работы.
2. Когда будет уместнее использовать каждую из пи¬
петок?
3. Что можно принять за приемлемый уровень ошибки
в точности пипетирования?
4. Что важнее: абсолютная точность или единооб¬
разие?
Упражнение 2.
Введение в технику ведения
культуры ткани
Цель процедуры
Критический осмотр клеточных культур.
Области применения
Проверка консистенции клеточной культуры в ходе
стандартной процедуры хранения; оценка состояния
культуры до введения питательных веществ, выделения
субкультур или криоконсервации; анализ и оценка от¬
вета культуры на создание новых или эксперименталь¬
ных условий; определение явного заражения.
Навыки, которыми
следует овладеть
Более близкое знакомство с внешним видом культур
клеток разных видов и различной плотности; исполь¬
зование цифровой или пленочной фотокамеры; выяв¬
ление стерильной и зараженной, здоровой и нездоровой
культур; оценка фазы роста культуры.
Инструктирование и контроль: постоянные во
время наблюдения, периодические при работе с фото¬
графиями.
Время: 30 мин.
Сведения общего характера
Морфология, фотографирование (см. разд. 16.4.5).
Демонстрационный материал или действия: фото¬
графии, представляющие морфологию различных типов
клеток, фазово-контрастная микроскопия, фиксирован¬
ные и окрашенные клетки, иммуноокрашивание; типы
культуральных сосудов, пригодных для морфологиче¬
ских исследований, например чашки Петри (см.
рис. 8.3), пробирки с покровными стеклами, флаконы с
предметными стеклами и т.д. (см. рис. 16.3); цитоцент¬
рифуга для суспензионной культуры (см. рис. 16.4).
Безопасность: нет специальных требований к бе¬
зопасности.
Упражнение
Краткое изложение процедуры
Исследовать и фотографировать разнообразные кле¬
точные линии при различной плотности клеток.
Материалы и оборудование
• Набор плашек или чашек Петри с культурами
клеток различной плотности, предпочтительно
нормальные и трансформированные варианты од¬
ной клеточной линии (например, ЗТЗ и SV3T3 или
ВНК21-С13 и BHK21-PyY) на разных фазах роста,
включая середину log-фазы (~50%-й сливной рост с
явлениями митоза) и фазу сливного роста по всей
чашке (клетки образовали многослойную структуру
и образовали скопления, если они трансформирова¬
ны). Если возможно, исследуется также суспензи¬
онная культура с низкой и высокой концентрацией
клеток.
• Если возможно, включите в набор образцов конта-
минированные культуры (предпочтительно не на
чашках Петри во избежание риска распространения
заражения) и нездоровые культуры, например слиш¬
ком долго находящиеся без питательных веществ.
40
ГЛАВА 2. ТРЕНИРОВОЧНЫЕ ПРОГРАММЫ
• Инвертированный микроскоп с фазово-контраст¬
ными объективами 4х, 10х и 20х и конденсором.
• Автоматический фотоаппарат, предпочтительно
цифровой с монитором или пленочный фотоаппарат
с фотоокуляром.
Стандартный протокол
1. Установите микроскоп и отрегулируйте освещение
(см. протокол 16.1).
2. Принесите культуры из инкубатора. Лучше всего
за один раз брать небольшое число планшетов, чем
сразу вынимать из инкубатора на продолжительное
врем слишком много планшетов или чашек. Выбе¬
рите пару, например одни и те же клетки с низкой и
высокой плотностью или нормальные и трансформи¬
рованные варианты одного и того же вида клеток.
3. Исследуйте каждую культуру визуально, пытаясь
обнаружить мутность среды, падение pH или отде¬
лившиеся от субстрата клетки. Также постарайтесь
выявить отклонения от монослойной структуры с
формированием рельефа.
4. Исследуйте культуры при малом увеличении (объ¬
ектив 4х) инвертированного микроскопа (фазовый
контраст), обращая внимание на степень клеточной
плотности и любые признаки межклеточного взаи¬
модействия.
5. Исследуйте культуры при среднем (объектив 10х)
и большом (20х) увеличении, обращая внимание
на признаки здорового состояние культуры (см.
рис. 13.1), признаки скопления клеток в одном месте,
складки монослоя или отслоение его от субстрата.
6. Отметьте признаки контаминации культуры.
7. Поищите митотические клетки и оцените прибли¬
зительно частоту их встречаемости.
8. Сфотографируйте (см. протокол 16.6) и сделайте
записи для каждой культуры (клеточный тип, дата
последнего пассажа) и плотности клеток, а также
обо всем, что покажется вам интересным.
9. Верните культуры в инкубатор и повторите стан¬
дартную процедуру с новыми планшетами.
Служебные протоколы: окрашивание (см. про¬
токол 16.2, 16.3); цитоцентрифугирование (см. про¬
токол 16.4); непрямая иммунофлюоресценция (см.
протокол 16.11).
Экспериментальные варианты процедуры
1. Определите различия в стадии роста, клеточной
плотности и морфологии культур.
2. Оцените состояние здоровья культур.
3. Отметьте признаки контаминации.
4. Определите готовность клеток к смене культуральной
среды (см. разд. 13.6.2) или пересеву (см. разд. 13.7.1).
5. Выполните численную оценку клеточной плотности
путем вычисления площади, находящейся в поле
зрения при объективе 20х и подсчета количества
клеток в этом поле зрения. Следует использовать
самую простую цифровую камеру и монитор, в ко¬
тором экран может быть покрыт чистой пленкой и
каждую клетку можно отметить точкой маркера.
6. Попытайтесь обнаружить и сосчитать клетки в ста¬
дии митоза при этом большом увеличении.
Данные
Качественные
1. Запишите ваши наблюдения, касающиеся морфо¬
логии, формы и структурирования всех культур
клеток.
2. Отметьте все случаи заражения.
3. Подтвердите состояние здоровья культур и прочие
качественные признаки.
Количественные
1. Запишите плотность клеток (кл./см3) для каждой
культуры.
2. Запишите значение митотического индекса для
каждой культуры.
Анализ
1. Оцените различия клеточной плотности.
2. Оцените различия митотических индексов.
3. Сравните характеристики нормальных клеток и
трансформированных культур при высокой и низ¬
кой плотности культуры, попробуйте объяснить
различия.
Упражнение 3. Асептический метод II:
Приготовление питательной среды
Цель процедуры
Сохранить стерильность растворов в ходе манипуляций
с ними.
Навыки, которыми следует овладеть
Асептические манипуляции: тренировка навыков в пи-
петировании и других манипуляциях со стерильными
растворами.
Инструктирование и контроль: постоянные.
Сведения общего характера
Тренировочные объекты (см. разд. 6.1); компоненты
асептического окружения (см. разд. 6.2); манипуляции
со стерильными объектами (см. разд. 6.3); работа в
ламинарном шкафу (см. разд. 6.4); визуально опреде¬
ляемое микробное заражение (см. разд. 19.3.1).
Демонстрационные материалы или действия: не¬
обходимо показать, как нужно обрабатывать рабочие
поверхности и предметы, вносимые в ламинарный
шкаф. Объяснить принципы ламинарного потока и
фильтрации воздушных частиц. Показать учащимся,
как открывать и закрывать пробки флаконов и бутылок
и как размещать пробку на рабочей поверхности. Про¬
демонстрировать способы манипуляции с пипеткой,
включая удерживание ее в руке, забор жидкости без
соприкосновения пипетки с нестерильными объекта¬
ми, способными загрязнить переносимую жидкость,
асептический перенос пипетки в другую емкость и
2.2. Упражнения для начинающих
41
выпускание жидкости в бутыли и флаконы. Следует
сделать акцент на обработке ламинарного бокса и
уборке места под рабочей поверхностью.
Упражнение
Краткое изложение процедуры
Внести необходимые добавки и питательные элементы
к основной среде (1х).
Стандартный протокол
Приготовление питательной среды для культивирова¬
ния из основной среды (1х) (см. протокол 11.7).
Экспериментальные варианты
стандартного протокола
1. Разлить 50 мл среды в две стерильные бутылки.
2. Поместить одну из них при 4 °С.
3. Инкубировать другую бутылку в течение 1 недели
и проверить наличие признаков заражения (см.
разд. 19.3.1).
4. Использовать обе бутылки в упражнении 4.
Упражнение 4. Смена среды
в монослойной культуре
Цель процедуры
Заменить истощенную среду в монослойной культуре
на свежую.
Области применения
Используется для восполнения объема питательной
среды между пересадкой в быстро растущих культурах
или при замене одного вида среды на другой.
Навыки, которыми следует овладеть
Укрепление навыков асептических манипуляций.
Введение одного из основных принципов поддержания
культур клеток, заключающегося в замене и попол¬
нении питательной среды в ходе цикла размножения
культуры. Обучить студентов обследовать культуру,
обнаруживать признаки истощения питательной сре¬
ды, такие как клеточная плотность и/или падение pH, а
также признаки заражения. Выявление опасности пере¬
крестного заражения культур. Сравнение питательных
сред, предварительно инкубированных при комнатной
температуре и хранившихся в холодильнике.
Инструктирование и контроль: учащимся потребу¬
ется совет при интерпретации признаков истощения
питательной среды и показ приемов удаления и заме¬
щения питательной среды.
Время: 30 мин.
Сведения общего характера
Замещение питательной среды (см. разд. 13.6.2); мо¬
ниторинг заражения (см. разд. 19.4); перекрестное
заражение (см. разд. 19.5).
Демонстрационный материал или действия: уп¬
ражнение требует по крайней мере трех флаконов
постоянных клеточных линий типа HeLa или Vero,
достигших полусливного роста, с указанием даты и
посевной дозы клеток. Учащимся следует показать,
как приносить среду из холодильника, при этом сле¬
дует подчеркнуть, что для каждой клеточной линии
требуется специфическая среда (ее не следует делить
между учащимися или между клеточными линиями).
Продемонстрируйте также уборку и обработку рабо¬
чей поверхности, ламинарного шкафа, использование
инкубатора, вынимание культуры из инкубатора и
возвращение ее на прежнее место, оценку состояния
культуры «на глазок» и под микроскопом (свободу от
заражения, потребность в питательных веществах, со¬
стояние здоровья культуры). Потребуется аспиратор с
насосом для удаления питательной среды или емкость
для слива истощенной питательной среды. Необходимо
показать приемы удаления старой питательной среды
и замещения ее свежей, включая использование атмос¬
феры, содержащей 5%-й С02.
Упражнение
Краткое изложение процедуры
Истощенная среда должна быть отобрана из клеточной
культуры, отправлена в емкость для слива и замещена
свежей средой.
Стандартный протокол
Смена среды в монослойной культуре (см. прото¬
кол 13.1).
Дополнительные протоколы: приготовление полной
питательной среды (см. протоколы 11.7, 11.8 или 11.9
и упражнение 3); приготовление стандартов pH (см.
протокол 9.1); манипуляции с чашками и флаконами
(см. протокол 6.3).
Экспериментальные варианты процедуры
Флаконы с культурами, которые будут заполнены
охлажденной и предварительно инкубированной
питательными средами при выполнении этого упраж¬
нения, следует использовать позже при демонстрации
подсчета клеток (см. упражнение 12), другие такие же
флаконы с культурами должны храниться без смены
среды. Трипсинизацию всех флаконов проводят од¬
новременно.
Сведения общего характера
Полная среда (см. разд. 9.5); замещение питательной
среды (см. разд. 13.6.2).
Данные
Сравнить внешний вид и выход культуры (подсчет
клеток см. упражнение 12) из флаконов, которые полу¬
чили питательную среду, хранившуюся в холодильнике
или инкубированную при комнатной температуре, и из
флаконов, в которых не заменяли питательную среду.
42
ГЛАВА 2. ТРЕНИРОВОЧНЫЕ ПРОГРАММЫ
Стандартные действия должны записываться в
стандартные бланки для записей (см. табл. 12.5); экс¬
периментальные данные сводятся в таблицу в упраж¬
нении 12.
Упражнение 5. Мытье и стерилизация
стеклянной посуды
Цель процедуры
Вымыть и простерилизовать грязную стеклянную
посуду.
Навыки, которыми следует овладеть
Совершенствование препаративных навыков; контроль
качества обработки, проводимой вне асептической
зоны.
Инструктирование и контроль: назначенный
сотрудник из штата персонала (препараторов и лабо¬
рантов) должен продемонстрировать учащимся после¬
довательность стандартных процедур.
Время: 20-30 мин следует уделять каждому этапу
занятия, однако в целом время, посвященное этому
упражнению, должно соответствовать степени участия
учащихся в процедуре мытья и стерилизации, что оп¬
ределяется его/ее основным родом занятий и свободно
устанавливается преподавателем.
Сведения общего характера
Моечная зона (см. разд. 4.5.2); мытье (см. разд. 5.4.1);
посудомоечная машина (см. разд. 5.4.11, рис. 5.21);
стерилизатор (см. разд. 5.4.4; рис. 5.18.18); мытье и
стерилизация оборудования (см. разд. 11.3).
Демонстрационный материал или действия: уча¬
щимся следует показать все стадии приготовления,
мытья и стерилизации посуды и по возможности при¬
нять в них участие. Несколько раз посетите моечное
помещение, чтобы понаблюдать за всеми этапами.
Учащиеся должны увидеть в действии все работающее
необходимое оборудование, включая складывание в
моечную машину, контроль качества (QC) и соблю¬
дение требований техники безопасности, а также не¬
работающее оборудование, если будущие обязанности
будут включать мытье и стерилизацию.
Упражнение
Краткое изложение процедуры
Сбор, ополаскивание, замачивание, мытье и стерили¬
зация стеклянной посуды и пипеток.
Материалы и оборудование
Стандартные материалы и оборудование препаратив¬
ной зоны (см. протокол 11.1-11.3).
Стандартный протокол
1. Подготовка и стерилизация стеклянной посуды (см.
протокол 11.1).
2. Подготовка и стерилизация пипеток (см. прото¬
кол 11.2).
3. Подготовка и стерилизация заворачивающихся
крышек (см. протокол 11.23).
Дополнительные протоколы: стерилизация сложных
фильтров (см. протокол 11.4).
Данные
Учащиеся должны ближе ознакомиться с ведением
записей и пометок, таких как цифровые и графические
отметки на регистрирующем выходном устройстве
печей и автоклавов.
Упражнение 6. Подготовка
и стерилизация воды
Цель процедуры
Обеспечение постоянного снабжения чистой стериль¬
ной водой.
Навыки, которыми следует овладеть
Совершенствование и развитие препаративных навы¬
ков работы вне асептической зоны. Получение знаний
о значении обеспечения чистой стерильной водой и
знакомство с процессом ее получения.
Инструктирование и контроль: периодически.
Время: 30 мин.
Сведения общего характера
Приготовление и стерилизация сверхчистой воды
(UPW см. разд. 11.1.4; рис. 5.17; 11.10).
Демонстрационный материал или действия: под¬
готовленный специалист должен изложить принципы
работы оборудования для получения сверхчистой воды
и продемонстрировать процедуру получения UPW,
сбора, бутилирования, стерилизации и QC. Участие
учащихся определяется свободным решением этого
специалиста или преподавателя.
Упражнение
Краткое изложение процедуры
Очистить воду, разлить по бутылям и стерилизовать
автоклавированием.
Стандартный протокол
Приготовление и стерилизация сверхчистой воды (см.
протокол 11.5).
Дополнительные протоколы: приготовление стек¬
лянной посуды (см. протокол 11.1).
Данные
Получение данных
Измеритель сопротивления или проводимости на
дистилляторе, датчик общего органического углерода
(ТОС). Автоматический вывод данных из автоклава
2.2. Упражнения для начинающих
43
на печатающее устройство. Отметка о стерильности
на бутыли. Индикатор стерильности в центральной
бутыли.
Записи данных
Ознакомиться, научиться читать и делать записи в
регистрационном журнале оборудования.
Анализ данных
Обзор регистрационного журнала за периоды 1 неделя,
1 месяц и 3 месяца для определения общих тенденций
или различий в качестве воды или достижении сте¬
рильности.
Упражнение 7.
Подготовка и стерилизация
фосфатно-буферного раствора для среды
Дюльбекко (D-PBS) без Са2+и Мд2+
Цель процедуры
Приготовление изотонического солевого раствора для
использования в нормальных атмосферных условиях.
Области применения
Раствор для разведения концентратов, таких как
2,5%-й трипсин, предварительное ополаскивание при
трипсинизации, отмывающий раствор при сборе кле¬
точной массы или смене реагентов. Поскольку он не
содержит кальция, магния, бикарбоната натрия или
глюкозы, раствор не пригоден для продолжительной
инкубации клеток.
Навыки, которыми следует овладеть
Составление простого раствора солей, осмоляль-
ность. Буферные свойства раствора и доведение pH.
Стерилизация термостабильных растворов автокла-
вированием.
Инструктирование и контроль: в ходе приготовле¬
ния раствора — постоянно; на этапе QC — периодичес¬
ки. В начале и завершении процедуры стерилизации и
интерпретации данных QC — постоянно.
Время: 2 ч.
Вспомогательная информация
Сбалансированный солевой раствор (см. разд. 9.3;
табл. 9.2); использование буферных растворов (см.
разд. 9.2.3).
Демонстрируемые материалы и действия: ис¬
пользование осмометра или прибора, измеряющего
проводимость растворов. Использование автоклава
или настольного стерилизатора под наблюдением
руководителя.
Вопросы безопасности: стерилизация паром связана
с риском ожога и риском взрыва (см. разд. 7.5.2; 7.5.7).
Простые настольные стерилизаторы могут взрываться
при отсутствии пара, в связи с чем при отсутствии
автоматического температурного предохранителя
существует пожароопасность.
Упражнение
Краткое изложение процедуры
Растворение предварительно перемешанного порош¬
ка или таблетки в UPW и стерилизация автоклави-
рованием.
Стандартный протокол
Приготовление D-PBSA (см. протокол 11.6).
Данные контроля качества
Получение данных
Измерение осмоляльности или проводимости и pH
после растворения компонентов.
Запись
Сделать подробную запись в регистрационный
журнал с указанием даты и номера серии.
Упражнение 8. Приготовление стандартных
растворов для контроля pH
Цель процедуры
Приготовить серию флаконов, аналогичных ис¬
пользуемым в текущем исследовании лаборатории,
содержащих простую среду или солевые растворы с
феноловым красным. Отрегулировать pH до значений,
соответствующих диапазону pH, в норме имеющемуся
в культуре.
Области применения
Оценка pH в ходе приготовления среды и до смены
питательной среды или пересадки клеток.
Навыки, которыми следует овладеть
Более близкое знакомство с использованием феноло¬
вого красного как индикатора pH. Стерилизация при
помощи шприцевого фильтра.
Инструктирование и контроль: в начале выполне¬
ния операций — постоянно, позже — минимально.
Время: 2 ч.
Сведения общего характера
Физико-химические свойства, pH (см. разд. 9.2).
Демонстрационный материал или действия: исполь¬
зование рН-метра. Принцип действия, использование
и диапазон использования шприцевых фильтров (см.
рис. 11.12а, в).
Вопросы безопасности: до тех пор, пока для выпуск¬
ного отверстия шприца не используются иглы, проблем
безопасности нет.
Упражнение
Краткое изложение процедуры
Приготовление ряда сред с различными значениями pH.
44
ГЛАВА 2. ТРЕНИРОВОЧНЫЕ ПРОГРАММЫ
Стандартный протокол
Приготовление стандартных растворов для контроля
pH (см. протокол 9.1 и цв. вкладку 226) с использо¬
ванием 25-см2 флаконов. Стерилизация фильтрацией
с использованием шприцевых насадочных фильтров
(см. протокол 11.11).
Упражнение 9. Приготовление основной
питательной среды из порошка
и стерилизация методом фильтрования
Цель процедуры
Приготовить сложные растворы и стерилизовать тер¬
молабильные реагенты и среды.
Навыки, которыми следует овладеть
Метод фильтрации и оценка диапазона возможностей
его использования. Сравнение способов фильтрации
под действием положительного и отрицательного
давления.
Инструктирование и контроль: инструктирование
в ходе приготовления среды. Постоянно — при насадке
фильтра и при контроле качества фильтрации, перио¬
дически — в процессе фильтрации.
Время: 2 ч.
Сведения общего характера
Приготовление среды из порошка (см. протокол 11.9);
стерилизация фильтрованием (см. разд. 11.5.2; про¬
токол 11.12); альтернативные процедуры (см. прото¬
кол 11.11,11.13,11.14).
Демонстрационный материал или действия: набор
имеющихся в распоряжении одиночных и сборных
фильтров, при отсутствии набора для ознакомления
можно использовать фотографии. Сделать акцент на
размерах фильтров (площадь поверхности) и фильтру¬
емых объемах. Принципы действия и преимущества/
недостатки фильтрации под действием положительно¬
го и отрицательного давления (см. разд. 11.5.2). Проде¬
монстрировать технические приемы при манипуляции
с фильтрами, фильтрации, сборе профильтрованных
образцов и QC.
Упражнение
Краткое изложение процедуры
Растворение порошка в UPW, стерилизация филь¬
трованием, фасовка и отбор пробы для QC-стериль¬
ности.
Стандартный протокол
1. Приготовление среды из порошка (см. прото¬
кол 11.9).
2. Стерилизация 450 мл вакуумной фильтрацией (см.
протокол 11.12).
3. Стерилизация 550 мл фильтрацией при положи¬
тельном давлении (см. протокол 11.13).
Дополнительные протоколы: приготовление ком¬
мерческой среды (см. протокол 11.10); автоклавируе-
мые среды (см. разд. 11.5.1); стерилизационные фильт¬
ры многократного использования (см. разд. 11.3.6);
стерилизация фильтрованием при помощи насадочных
шприцевых фильтров (см. протокол 11.11); стерилиза¬
ция фильтрованием при помощи больших последова¬
тельно расположенных фильтров (см. протокол 11.14);
сыворотка (см. разд. 11.5.3; протокол 11.15).
Экспериментальные варианты
процедуры
Разделите растворенную среду на две части, просте-
рилизуйте 550 мл фильтрацией при положительном
давлении (см. протокол 11.13) и 450 мл фильтрацией
при отрицательном давлении (см. протокол 11.12).
Сведения общего характера
С02и бикарбонат (см. разд. 9.2.2); использование
буферных растворов (см. разд. 9.2.3); стандартные
протоколы стерилизации (см. разд. 11.5).
Данные
1. Определите и запишите значение pH до и сразу
после фильтрации.
2. Инкубируйте универсальные контейнеры или буты¬
ли со средой при 37 °С в течение недели и проверьте
наличие загрязнения.
Анализ
1. Объясните различия в значениях pH между среда¬
ми, профильтрованными с использованием поло¬
жительного и отрицательного давления.
2. Определите, восстанавливается или сохраняется
измененным значение pH.
3. В каких случаях следует предпочитать тот или иной
тип фильтрации?
4. Какие фильтры вы используете:
а) для 5 мл чистого раствора?
б) для 10 л среды?
в) для 1 л сыворотки?
Упражнение 10. Приготовление
полной среды
из концентрата 10х
Цель процедуры
Добавить нестабильные компоненты и добавки к ос¬
новной среде для получения сложной среды, необхо¬
димой для решения специфической задачи.
Области применения
Получение питательной ростовой среды, обеспечива¬
ющей пролиферацию клеток растущей культуры; под¬
держивающей среды, необходимой для поддержания
жизнеспособности клеток; или дифференциальной
среды в присутствии соответствующих индуцирующих
факторов.
2.2. Упражнения для начинающих
45
Навыки, которыми
следует овладеть
Дальнейшее приобретение опыта в асептических ма¬
нипуляциях. Улучшение понимания роли питательной
среды и добавок. Стабильность компонентов. Контроль
pH при помощи бикарбоната натрия.
Инструктирование и контроль: учащиеся, выпол¬
нившие упражнение 6, нуждаются в минимальном
руководстве, но до выполнения упражнения им требу¬
ется разъяснение необходимости некоторых добавок и
компонентов среды.
Время: 30 мин.
Сведения общего характера
Среда (см. разд. 11.4.3; 11.4.4).
Демонстрационный материал или действия: набор
стандартов pH (см. протокол 9.1). Набор емкостей,
пригодных для приготовления среды.
Безопасность: нет специальных проблем, связанных
с безопасностью, если только в среду не добавляются
токсичные вещества (например, холерный токсин
или цитотоксические препараты) или радиоактивные
компоненты.
Упражнение
Краткое изложение
процедуры
Стерильные компоненты или добавки вводятся в
предварительно стерилизованную основную среду,
чтобы приготовить полную среду, готовую к исполь¬
зованию.
Стандартный протокол
Приготовление полной среды из концентрата 10х (см.
протокол 11.8). Преподавателем могут быть выбраны
один или все пункты протокола 11.8.
Дополнительные протоколы: коммерческие среды
(см. протокол 11.10); приготовление основной среды
из порошка и стерилизация методом фильтрации (см.
протокол 11.9); приготовление стандартных растворов
для контроля pH (см. протокол 9.1).
Экспериментальные варианты
процедуры
Вентилируемые флаконы
1. Приготовьте среды в соответствии с протоко¬
лом 11.8. А без HEPES.
2. Внесите пипеткой 10 мл среды в каждый из четырех
25-см2 флаконов.
3. Добавьте 20 мкл 1М HEPES в каждый из двух фла¬
конов.
4. Герметизируйте два флакона: один — содержащий
HEPES, один — нет. Ослабьте крышки на двух
других.
5. Инкубируйте при 37 °С в течение ночи без С02-ат-
мосферы.
Данные
Запишите значения pH и внесите в таблицу условия
инкубации.
Анализ
1. Определите pH и оцените различия.
2. Какие условия подходят для низкоуглеродной пи¬
тательной среды?
3. Какое действие оказывает HEPES на стабильность
pH?
4. Когда вентиляция флакона может быть предпочти¬
тельна?
Роль концентрации бикарбоната (см. разд. 9.2.2)
1. Исключите бикарбонат из стандартной процедуры
протокола 11.86 и добавьте различное количество
бикарбоната натрия следующим образом:
а) приготовьте и пометьте 5 аликвот 10 мл безби-
карбонатной среды в 25-см2 флаконах,
б) добавьте в различные флаконы 200 мкл, 250 мкл,
300 мкл, 350 мкл, 400 мкл 7,5%-го NaHC03.
2. Оставьте крышки слабозавернутыми (только слегка
надетыми на резьбу) или используйте фильтрующие
крышки, поместите на 37 °С в С02-инкубатор.
3. Инкубируйте в течение ночи и сравните значение
pH со стандартом (см. протокол 9.1).
Данные
Запишите значения pH и внесите в таблицу в соответ¬
ствии с объемом внесенного NaHC03.
Анализ
1) Объясните, что является причиной изменения
pH?
2) Подсчитайте конечную концентрацию бикарбоната
в каждом случае и определите оптимальное для
использования количество NaHC03
3) Если ни одно из значений не оптимально, что сле¬
дует сделать для достижения правильного pH?
Упражнение 11. Приготовление
полной среды из порошка
См. протокол 11.9.
Упражнение 12. Подсчет клеток
в гемоцитометре и электронном счетчике
Данное упражнение можно использовать по-разному:
для работы как на гемоцитометре, так и на электрон¬
ном счетчике. Кроме того, можно использовать его
для сравнительного изучения двух методов подсчета
клеток и оценки их эффективности. Для экономии
времени учащихся возможна комбинация с упраж¬
нением И. Так или иначе, поскольку приобретение
первоначальных навыков в подсчете клеток происходит
достаточно медленно, рекомендуется провести подсчет
46
ГЛАВА 2. ТРЕНИРОВОЧНЫЕ ПРОГРАММЫ
клеток как отдельное самостоятельное упражнение с
использованием культур, выросших в ходе выполнения
предыдущих упражнений (например, упражнение 9),
и не использовать его как предварительный этап для
последующего упражнения. Комбинированное исполь¬
зование обоих методов подсчета будет описано ниже.
Цель процедуры
Оценить концентрацию клеток в суспензии.
Области применения
Стандартизация клеточной концентрации в повседнев¬
но используемой субкультуре; анализ количественного
роста в эксперименте и клеточной продукции в ходе
анализа кривой роста и выхода клеток.
Навыки, которыми следует овладеть
Навыки количественной оценки культуры. Подсчет кле¬
ток и оценка жизнеспособности. Оценка родственных ха¬
рактеристик гемоцитометра и электронного счетчика.
Инструктирование и контроль: требуются в ходе
подготовки и исследования образцов, а также подготов¬
ке к работе обоих устройств. Подсчет клеток в образцах
может производиться учащимися самостоятельно,
хотя им может потребоваться помощь при анализе
результатов.
Время: 45 мин.
Сведения общего характера
Подсчет клеток, гемоцитометр (см. разд. 21.1.1); элек¬
тронный счетчик (см. разд. 21.1.2).
Демонстрационный материал или действия: кле¬
точные культуры, используемые для подсчета, могут
быть получены в результате выполнения упражнения
9. Использование гемоцитометра и электронного
счетчика потребует демонстрации их работы с соот¬
ветствующей инструкцией по выполнению подсчетов
в конце работы. Следует также объяснить принципы
работы электронного счетчика.
Безопасность: при использовании человеческих
клеток манипуляции должны производиться в помеще¬
ниях II класса микробиологической биобезопасности.
Все пластиковое и стеклянное оборудование, включая
пластинки гемоцитометра и покровные стекла, после
использования необходимо поместить в дезинфициру¬
ющий раствор. Ячейки для подсчета клеток и жидкости
при подсчете на электронном счетчике также следует
обработать дезинфектантом (см. разд. 7.8.5).
Упражнение
Краткое изложение процедуры
Клеточный рост в суспензионной культуре или отде¬
ленной от монослоя трипсином суспензии подсчиты¬
вается непосредственно в оптической камере или после
разведения в D-PBS А на электронном счетчике. До под¬
счета в гемоцитометре клетки можно предварительно
окрасить для определения жизнеспособности.
Стандартный протокол
Упражнение выполняют сначала на разведенной
клеточной суспензии с использованием электронного
счетчика (см. протокол 21.2), а затем подсчет ведут на
концентрированной клеточной суспензии с помощью
гемоцитометра (см. протокол 21.1), и, используя ту же
концентрированную клеточную суспензию, проводят
оценку жизнеспособности методом исключения погиб¬
ших клеток (см. протокол 22.1).
Дополнительные протоколы: окрашивание крис-
талл-виолетом (см. протокол 16.3); содержание ДНК
(см. разд. 16.6); микротитрационное исследование (см.
разд. 22.3.5).
Экспериментальные варианты процедуры
1. Повторите подсчет 5-10 раз с использованием ге¬
моцитометра и электронного счетчика. Подсчитайте
среднее значение и стандартное отклонение (см.
упражнение 1).
2. Сравните культуры, получившие и не получившие
питание из упражнения 9.
Данные
Подсчет клеток, включая оценку жизнеспособности,
в пересчете на флакон, двумя методами.
Все детали ежедневного ведения культуры, включая
учет посевной дозы клеток на каждый флакон, должны
вноситься в регистрационный журнал (см. табл. 13.7),
экспериментальные данные заносятся в отдельную
таблицу (табл. 2.2).
Анализ
1) Подсчитайте выход жизнеспособных клеток как от¬
ношение количества клеток во флаконе к посевной
дозе.
2) Оказывает ли среда влияние на выход клеток?
Упражнение 13. Рост субкультуры
клеток в суспензии
Цель процедуры
Уменьшить концентрацию клеточной культуры про¬
порционально скорости роста, что позволит клеткам
пребывать в экспоненциальной фазе роста.
Области применения
Повседневная практика ведения культур, в т. ч. пере¬
сев неприкрепленных клеток, таких как миелома или
асцитные культуры; распространение культуры для
повышения производства клеток; отделение реплици¬
рованной культуры для экспериментальных целей.
Навыки, которыми следует овладеть
Знакомство с режимом роста клеточной культуры;
подсчет клеток и оценка жизнеспособности.
Инструктирование и контроль: требуется перво¬
начальное руководство и объяснение принципов ра¬
боты. Дальнейшие манипуляции просты и, поскольку
2.2. Упражнения для начинающих
47
Таблица 2.2. Запись результатов, полученных при выполнении упражнения 12. Подсчет клеток
Посевная
доза клеток
(кл ./флакон)
Гемоцитометрический или
электронный подсчет ко¬
личества выросших клеток
Разведение
или выбороч¬
ная доля*
Концентрация трипсини-
зированной культуры или
суспензии (клУмл)
Количество
клеток в одном
флаконе
Выход клеток:
Количество вы¬
росших клеток/
посевная доза
* Электронный счетчик разведение 50Х (например, 0,4 мл клеточной суспензии в 20 мл жидкости для разведения), для под¬
счета используется 0,5 мл, для получения значения концентрации следует умножать на фактор 100. Объем образца для камеры
гемоцитометра (усовершенствованный гемоцитометр Neubauer) обычно составляет 1 мм3, поэтому умножение на фактор 1x104
даст концентрацию кл./мл.
в результате выполнения упражнения 10 учащиеся
освоили, по крайней мере, один из методов подсчета
клеток, дальнейшее руководство не предполагается,
за исключением периодического контроля за соблюде¬
нием правил асептики.
Время: 30 мин.
Сведения общего характера
Размножение клеток в суспензии, субкультура суспен¬
зионной культуры (см. разд. 13.7.4); жизнеспособность
(см. разд. 22.3.1).
Демонстрационный материал или действия: уча¬
щимся потребуется две суспензионные культуры
разной фазы роста: одна на поздней log-фазе и одна
на плато, с указанием даты пересева и клеточной
концентрации. Следует показать, как определяют
жизнеспособность в гемоцитометре (см. разд. 21.1.1;
22.3.1; протокол 22.1). Если руководитель предпочитает
работать с перемешиваемой, а не статической культу¬
рой, до начала выполнения упражнения ознакомьте
учащихся с работой мешалки и подготовкой флаконов
с перемешиваемой культурой.
Безопасность: при использовании человеческих
клеток производите манипуляции в боксах II класса
микробиологической биобезопасности. Все пластико¬
вое и стеклянное оборудование после использования
необходимо помещать в дезинфицирующий раствор.
Упражнение
Краткое изложение процедуры
Образец клеточной суспензии отбирается из культуры,
подсчитывается количество клеток с использованием
гемоцитометра или электронного счетчика, культура
разводится в питательной среде и пересевается.
Стандартный протокол
Субкультура в суспензии (см. протокол 13.3); жизне¬
способность (см. протокол 22.1).
Дополнительные протоколы: пропорциональное
увеличение клеток в суспензии (см. протокол 26.1).
Экспериментальные варианты процедуры
Сравнительное исследование субкультур, находящихся
на log-фазе и фазе плато клеточного роста.
Теоретические предпосылки: концентрация клеток
в субкультуре (см. разд. 13.7.3).
Данные
Определите концентрацию клеток и жизнеспособность
в субкультурах, пересеянных с log-фазы и фазы плато
клеточного роста через 72 ч после пересева. Форма
записи лучше всего представлена в табл. 2.3 и может
быть переведена в электронный вид.
Анализ
1. Подсчитайте выход клеток, как описано в табл. 2.4.
2. Сравните выход из клеток, пересеянных из культур,
находящихся в разных фазах роста.
Упражнение 14. Рост субкультуры
постоянной клеточной линии в монослое
Цель процедуры
Размножение культуры путем переноса клеток в новый
культуральный сосуд. Упражнение может включать
разведение для пересева в сосуд такого же объема или
в больший сосуд, если предполагается расширение
культуры.
Навыки, которыми следует овладеть
Оценка культуры: Это упражнение требует от учащихся
умения исследовать и оценивать состояние культуры.
Следует отметить общие признаки здоровья культуры,
наличие заражения, pH среды, плотность клеток.
Асептические манипуляции: совершенствование
навыков, полученных при выполнении упражнений 5,
7 и 8.
Субкультура или пересев: это упражнение знакомит
с принципом переноса культуры из одного флакона в
другой с разведением, соответствующим предполагае¬
мой скорости роста. Следует показать учащимся техни¬
ку дезагрегирования клеточного монослоя с использо¬
ванием трипсина. Они должны знать приемы подсчета
клеток и оценки жизнеспособности, в ходе выполнения
упражнения от учащихся требуется умение определять
концентрации клеток и выбирать правильную концент¬
рацию для пересева с учетом представлений о значении
плотности клеток в культуре. Предполагается совер¬
шенствование навыков математических расчетов.
48
ГЛАВА 2. ТРЕНИРОВОЧНЫЕ ПРОГРАММЫ
Таблица 2.3. Запись результатов, полученных при выполнении упражнения 13. Рост субкультуры клеток в суспензии
Образец
Объем
клеточ¬
ной сус¬
пензии во
флаконе
Под¬
счет
клеток
Разведе¬
ние или
выбороч¬
ная доля*
Концентрация
во флаконе
клеток/мл
Жизнеспо¬
собность
(отноше¬
ние кол-ва
неокра¬
шенных
клеток к
общему)
Коэффици¬
ент разве¬
дения для
окраски на
жизнеспо¬
собность
Жизнеспо¬
собность
клеток/мл
Клеток/фла¬
кон
V
С
D
CxD
R
F
CxDxRxF
CxDxRxFxV
Например, подсчет на
электронном счет¬
чике без окраски на
жизнеспособность
20
15 321
100
1 532 100
1
1
1 532 100
30 642 000
Например, подсчет
на гемоцитометре
с окраской на жиз¬
неспособность
20
76
10 000
760 000
0,85
2
1 292 000
25 840 000
Инструктирование и контроль: поскольку учащи¬
еся обладают достаточными навыками в асептических
техниках, постоянное руководство не требуется, однако
руководитель должен быть в поле зрения учащихся,
чтобы они могли получить ответы на возникающие
вопросы.
Сведения общего характера
Стандартный протокол
Субкультура, критерии субкультуры (см. разд. 13.7.1;
рис. 13.2; 13.3; 13.4); кинетика роста и индекс разведе¬
ния (см. разд. 13.7.2); концентрация клеток в субкуль¬
туре (см. разд. 13.7.3; рис. 13.4); выбор культуральных
сосудов (см. разд. 13.7.3); С02и бикарбонат (см.
разд. 9.2.2, табл. 9.1).
Экспериментальные варианты процедуры
1. Концентрация клеток в субкультуре (см.
разд. 13.7.3).
2. Кинетика роста (см. разд. 21.9.2).
3. Действие клеточной плотности (см. разд. 25.1.1).
Таблица 2.4. Анализ упражнения 13
Образец
Посевная
концент¬
рация
(клеток/
флакон)
Клеток/фла¬
кон в следую¬
щей субкуль¬
туре
Выход: коли¬
чество выросших
клеток/посевная
доза
N
CxDxVxRxF
CxDxVxRxF
N
Пример
200,000
30 642 000
153,21
Клетки с
log-фазы
Клетки
с фазы
плато
Демонстрационные материалы
и операции
Покажите учащимся различные типы культуральных
сосудов (см. табл. 8.1; рис. 8.1-8.8) и фотографии кле¬
ток, здоровых (см. рис. 16.2, цв. вкладку 5.6), нездоровых
(см. рис. 13.1), зараженных (см. рис. 19 a-e) и имеющих
различную плотность (см. рис. 16.2; цв. вкладку 5.6).
Дайте подробные инструкции по фазово-контрастной
микроскопии клеток. Требуется также демонстрация
приемов трипсинизации (см. протокол 13.2).
Упражнение
Краткое изложение процедуры
Клеточный монослой разрушается действием трипсина,
разводится и пересевается.
Материалы и оборудование
См. материалы для стандартной субкультуры (см.
протокол 13.2).
Стандартный протокол
Субкультура монослоя (см. протокол 13.2).
Экспериментальные варианты
процедуры
В п. 11 протокола 13.2. возможны дополнения:
а) засейте шесть флаконов, посевная доза 2x104 кл./мл;
б) замените питательную среду в трех флаконах через
4 дня;
в) определите количество клеток через 7 дней в двух
флаконах, которые получили питание, и в двух, не
получивших свежей среды:
- удалите питательную среду;
- отмойте клетки дважды 2 мл D-PBSA, полно¬
стью удаляя его после отмывки;
- добавьте 1 мл трипсина в каждый флакон;
2.2. Упражнения для начинающих
49
Таблица 2.5. Запись данных упражнения 14
Образец
Объем
трипсината
Результат подсчета клеток
на гемоцитометре или электронном
счетчике
Разведение или
выборочная доля
Клеток/мл
в трипсинате
Клеток/флакон
Т
С
D
CxD
CxDxT
- инкубируйте 10 мин;
- добавьте 1 мл среды к трипсину и размешайте
клетки энергичным пипетированием;
- произведите подсчет клеток на гемоцитометре
или на электронном счетчике;
- подсчитайте количество клеток в каждом фла¬
коне, концентрацию кл./мл среды и кл./см2 во
время трипсинизации;
г) зафиксируйте и окрасьте клетки в остальных фла¬
конах (см. протокол 16.2).
Замечание: пипетки следует использовать при
подсчете клеток из изолированных образцов, а не для
выделения клеток для субкультуры, если только не
используются специальные наконечники.
Дополнительные протоколы: использование инвер¬
тированного микроскопа (см. протокол 16.1); подсчет
клеток (см. протоколы 21.1,21.2); приготовление среды
(см. протокол 11.7; 10.8; 10.9); окрашивание по Гимза
(см. протокол 16.2)
Данные
1. Подсчет клеток в начале и в одном наборе флаконов
через 1 неделю.
2. Обследование и фотографирование окрашенных
флаконов культуры, полученной при выполнении
упражнения 13. Наилучший результат можно по¬
лучить при исследовании клеточного слоя до его
высыхания.
Анализ
1. Подсчитайте кратность выхода (количество получен¬
ных клеток /количество посеянных клеток, табл. 2.6)
и объясните различия (см. разд. 18.5.2,21.9.3).
2. Требовалось ли для этих клеток промежуточное
питание?
3. Прокомментируйте различия в морфологии клеток,
получивших и не получивших питание в ходе вы¬
полнения упражнения 13.
Упражнение 15. Окрашивание
монослойной клеточной культуры
по Гимза
Цель процедуры
Окрашивание полихроматическим красителем типа
красителя Гимза, выявляющим морфологические
характеристики фиксированных клеток и позволя¬
ющий анализировать состояние и происхождение
клеток.
Области применения
Мониторинг морфологии клеток, обычно в совокуп¬
ности с фазово-контрастным исследованием, в ходе
стандартной процедуры пересева культур или в усло¬
виях эксперимента. Изготовление препаратов клеток
длительного хранения для сравнительного анализа
их морфологии. Идентификация клеточных типов,
присутствующих в первичной культуре.
Навыки, которыми следует овладеть
Следует подчеркнуть необходимость исследования
морфологии клеток в ходе ведения культуры и при¬
влечь внимание учащихся к морфологии клетки.
Таблица 2.6. Анализ данных, полученных
в упражнении
14
Образец
Посев¬
ная доза
(клетки/
флакон)
Количество
полученных
клеток/фла¬
кон1
Выход клеток
полученные
клетки /посев¬
ная доза
N
CxDxT (см.
последний
столбик
табл. 2.5)
CxDxT
N
Не получившие
питания
100 000
100 000
Получившие
питание
100 000
100 000
1 При трипсинизации жизнеспособность не учитывается,
однако при отмывке культуры до трипсинизации следует
позаботиться о том, чтобы удалить большую часть не¬
жизнеспособных клеток при манипуляциях с постоянной
клеточной линией. Тем не менее необходимость отделения
нежизнеспособных клеток не так велика, как в случае раннего
пересева или первичной культуры, когда жизнеспособность
необходимо брать в расчет (см. регистрационный лист, уп¬
ражнение 20).
50
ГЛАВА 2. ТРЕНИРОВОЧНЫЕ ПРОГРАММЫ
Инструктирование и контроль: минимальное.
Время: 30 мин.
Сведения общего характера
Морфология (см. разд. 16.4); окрашивание (см.
разд. 16.4.2).
Демонстрационный материал или действия: проде¬
монстрируйте приготовление красителя и процедуру
окрашивания. В качестве примеров используйте пред¬
варительно окрашенные препараты.
Безопасность: меры предосторожности при работе
с клетками человека аналогичны мерам, описанным в
предыдущих упражнениях.
Упражнение
Краткое изложение процедуры
Удаление среды, промывание клеточного монослоя
PBSA, фиксирование в этаноле, окрашивание краси¬
телем Гимза.
Стандартный протокол
Окрашивание красителем Гимза (см. протокол 16.2).
Дополнительные протоколы: окрашивание крис-
талл-виоллетом (см. протокол 16.3); использование
инвертированного микроскопа (см. протокол 16.1);
цифровая фотография микроскопируемого объекта
(см. протокол 16.6).
Экспериментальные варианты процедуры
1. Используйте флаконы с культурой, полученные
при выполнении упражнения 12, и сравните мор¬
фологию клеток из культур со сменой и без смены
среды.
2. Сфотографируйте культуру до (фазово-контрастная
микроскопия) и после (нормальная микроскопия в
светлом поле) окрашивания.
Упражнение 16. Построение
и анализ кривой роста
Цель процедуры
Познакомиться с образцом возобновляемого роста
субкультуры; продемонстрировать кинетику роста на
стандартно используемой субкультуре и в качестве
аналитического средства.
Области применения
Кривые роста. Исследования клеточной пролифера¬
ции; анализ цитотоксичности; исследования стимуля¬
ции роста; испытание свойств сред и сывороток.
Навыки, которыми следует овладеть
Выделение экспериментальных репликативных куль¬
тур. Подсчет клеток и жизнеспособность. Построение
и анализ кривой роста. Представление о различиях
между временем удвоения и достижением макси¬
мальной плотности популяции. Выбор пересеваемой
концентрации.
Инструктирование и контроль: если учащиеся
успешно прошли упражнение 12, трипсинизация куль¬
туры и подсчет клеток не требуют инструктирования,
но при дальнейшей работе с пересевом на чашки необ¬
ходимо некоторое руководство.
Время: 1 ч в день 0; 30 мин в день с 1 по 10 дни.
Сведения общего характера
Выбор культурального сосуда (см. разд. 8.2); мани¬
пуляции с кюветами и чашками (см. протокол 6.3);
репликация образцов (см. разд. 21.8); цикл роста
(см. разд. 21.9.2); микротитрационный анализ (см.
разд. 22.3.5).
Демонстрационный материал или действия: уча¬
щимся следует показать разнообразные многолуноч¬
ные планшеты (см. табл. 8.1 и рис. 8.2) и дать представ¬
ление об их применении. Продемонстрируйте приемы
пересева культур на чашки с применением мер, предуп¬
реждающих заражение культуры (см. разд. 6.5.1).
Безопасность: соблюдать меры предосторожности
при работе с человеческими клетками (см. упражне¬
ние 10 и разд. 7.8), особенно при работе с открытыми
чашками и кюветами в связи с повышенным риском
разбрызгивания (см. разд. 6.6.2 и рис. 6.10).
Упражнение
Краткое изложение процедуры
Существует два варианта выполнения этого упражне¬
ния: построить простую кривую роста одной клеточной
линии с использованием флаконов, как при обычной
работе с субкультурой, и использовать многолуночные
планшеты для анализа различий в росте двух клеточных
линий при различной плотности клеток или каких-либо
иных различиях в условиях культивирования. Кле¬
точный монослой трипсинизируется, подсчитывается
количество клеток, культура разводится в различных
средах и пересевается на необходимое количество куль¬
туральных флаконов или многолуночных планшетов.
Стандартный протокол
Кривая роста (см. протокол 21.7 для построения кривой
роста во флаконах и определения условий стандартной
процедуры поддержания культуры и протокол 21.8
для работы с многолуночным планшетом при анализе
кинетики роста культур различной плотности и/или
сравнения двух различных клеточных линий).
Дополнительные протоколы: манипуляции с чаш¬
ками или кюветами (см. протокол 6.3); МТТ-тест
цитотоксичности (см. протокол 22.4).
Экспериментальные варианты процедуры
Концентрация клеток при посеве
1. Посейте во флаконы постоянную клеточную линию:
быстрорастущую в дозе 2x104 кл./мл или медленно¬
2.3. Упражнения повышенной сложности
51
растущую в дозе 1х105 кл./мл. Повторите посев для
оптимизации посевной концентрации.
2. Для удобства выполнения упражнения одна кле¬
точная линия может быть посеяна в трех различных
концентрациях клеток в 12-луночный планшет
(каждая концентрация в трех рядах), одна лунка
в ряду используется для окрашивания клеток (см.
рис. 21.7а). Для исследований фазы роста для
каждого из 10 дней следует посеять отдельные
планшеты.
Различия между нормальными и трансформирован¬
ными клеточными линиями: в этом случае используйте
24-луночный планшет и посейте две клеточных линии
на каждый планшет (см. рис. 21.76).
Сведения общего характера
Цикл роста (см. разд. 21.9.2); объем, глубина и площадь
поверхности (см. разд. 13.6.2); концентрация клеток в
субкультуре (см. разд. 13.7.3); фазы роста клеток (см.
разд. 21.9.6); продукты, образующиеся в цикле роста
(см. разд. 21.9.7); контактное ингибирование (см.
разд. 18.5.2); ограничение роста клеточной популяции
плотностью клеток (см. протокол 18.3).
Данные
Подсчет клеток в ячейках каждый день.
Анализ
1. На основании количества клеток в лунке рассчитай¬
те концентрацию клеток в лунках (кл./мл и кл./см2)
и найдите для каждого дня исследования среднее
значение и величину стандартной ошибки (стандарт¬
ное отклонение, деленное на квадратный корень от
количества образцов). Постройте в логарифмичес¬
кой шкале график зависимости количества клеток
от дня после посева культуры (см. рис. 21.6).
2. Определите время lag-фазы, время удвоения массы
культуры и время достижения максимальной плот¬
ности.
3. Какая концентрация клеток оптимальна для пере¬
сева постоянно используемой субкультуры?
4. Оцените различия между нормальной и трансфор¬
мированной клеточными линиями.
2.3. УПРАЖНЕНИЯ ПОВЫШЕННОЙ
СЛОЖНОСТИ
Данные упражнения зависят от успеха в выполнении
упражнений основного курса, и не следует пытаться
выполнить их до того, как этот успех будет достигнут.
Несмотря на то что упражнения повышенной сложнос¬
ти не входят в основной курс, навыки, отрабатываемые
в ходе их выполнения, имеют широкое приложение
и потребуются каждому, кто предполагает достичь
профессионализма в работе с клеточными культура¬
ми. Однако, если время обучения ограничено, можно
отложить выполнение этих упражнений при условии,
что другие сотрудники лаборатории имеют опыт в
таких манипуляциях и вновь обученный сотрудник не
должен будет их выполнять. При этом следует учиты¬
вать, что обучение не может считаться завершенным
и соответствующим общепринятым стандартам, если
учащийся не приобрел навыки, изучаемые в упражне¬
ниях повышенной сложности.
Поскольку некоторые основные знания уже сумми¬
рованы и обобщены в ходе выполнения упражнений
основного курса, вариации стандартных протоколов
этой части не будут представлены так же детально, как
прежде. Допускается большая степень собственного
планирования эксперимента, что будет полезной и
важной частью процесса обучения.
Упражнения основного курса представлены в той
последовательности, в которой они должны выпол¬
няться, упражнения данного раздела не имеют такого
расположения и могут быть выполнены учащимися
по очереди.
Упражнение 17. Криоконсервация
культивируемых клеток
Цель процедуры
Обеспечить сохранность клеточной линии, защищен¬
ной от случайной гибели, генетической или фенотипи¬
ческой нестабильности.
Области применения
Хранение новых и постоянно пересеваемых клеточных
линий; создание и поддержание клеточного банка для
архивирования и распределения клеточных линий;
снабжение работающего клеточного банка в ходе реали¬
зации научного проекта или программы исследований;
хранение и распространение митомицин С-обработан-
ных фидерных клеток.
Навыки, которыми следует овладеть
Знакомство с процедурой замораживания и размора¬
живания клеток. Указания к возможным вариантам
улучшения процедуры для сложных клеточных ли¬
ний. Сравнение оценки жизнеспособности клеток по
высвобождению красителя с реальным выживанием
клеток.
Инструктирование и контроль: основные процеду¬
ры, такие как трипсинизация, подсчет клеток, добавка
криоконсерванта и заполнение ампул, не требуют ру¬
ководства. Оно понадобится при определении расхода
жидкого азота в системе контроля расхода материаль¬
ных средств, при замораживании и переносе ампул и
помещении их в азот, а также при размораживании и
восстановлении клеток.
Время: 1 ч в 1-й день; 15 мин во 2-й день, 30 мин в
3-й день, 1 ч в 4-й день.
Сведения общего характера
Теоретическое обоснование замораживания (см.
разд. 20.1); скорость охлаждения, аппараты для за¬
52
ГЛАВА 2. ТРЕНИРОВОЧНЫЕ ПРОГРАММЫ
мораживания, ведение записей при замораживании
(см. разд. 20.3.6.); генетическая нестабильность (см.
разд. 18.3); эволюция клеточных линий (см. разд. 3.8);
контроль физиологического старения (см. разд. 18.4.1);
периодическое замещение (см. разд. 20.4.2); банк кле¬
точных линий (см. разд. 20.5).
Демонстрационный материал или действия: уча¬
щимся следует показать типы ампул в процессе их
использования, оборудование для замораживания
(см. рис. 20.3; 20.4; 20.5) и типы морозильников (см.
разд. 20.7). Учащиеся должны ознакомиться с исполь¬
зованием и обслуживанием контрольных систем и
ведением записей по учету клеточных линий.
Безопасность: меры безопасности при работе с
человеческими клетками те же. Существует риск
отморожения, асфиксии и взрыва при погружении
ампул в жидкий азот (см. разд. 7.5.6). Настоятельно
рекомендуется, в соответствии с целью этого упраж¬
нения, не погружать ампулы в жидкий азот, а хранить
их в парах азота.
Упражнение
Краткое изложение процедуры
Клетки при высокой концентрации в среде с криокон¬
сервантом медленно охлаждаются, медленно заморажи¬
ваются и помещаются в холодильник с жидким азотом.
Затем им дают быстро оттаять и пересевают.
Сведения общего характера:
Стандартные протоколы: замораживание клеток (см.
протокол 20.1); оттаивание замороженных клеток (см.
протокол 20.2).
Дополнительные протоколы: субкультура в монос¬
лое (см. протокол 13.2); субкультура в суспензии (см.
протокол 13.3); подсчет клеток при помощи гемоцито¬
метра (см. протокол 21.1); электронный счетчик клеток
(см. протокол 20.2); оценка жизнеспособности по тесту
высвобождения красителя (см. протокол 22.1).
Экспериментальные варианты процедуры
Имеется несколько вариантов эксперимента, которые
можно использовать при выполнении упражнения:
1. Сравните свойства ДМСО и глицерина как крио¬
консервантов.
2. Сравните различные скорости замораживания.
3. Храните при комнатной температуре или при 4 °С
до замораживания.
4. Удалите криоконсервант методом замещения пи¬
тательной среды на следующий день или путем
центрифугирования после разморживания. При
выборе этого варианта процедуры затем следует
сравнить клетки суспензионной культуры (на¬
пример, лимфомы L5178Y, гибридомы или HL60)
и монослойной культуры (например, HeLa, А549,
Vero или NRK).
5. Быстро или медленно разведите в растворе после
размораживания.
Приводимая схема включает сравнение свойств
ДМСО и глицерина (1-й вариант) и сравнение резуль¬
татов хранения при комнатной температуре или при
4 °С (3-й вариант) до замораживания (рис. 2.1).
Теоретические предпосылки: криопротекторы (см.
разд. 20.3.3).
Данные
1. Ежедневные записи ведите в соответствии со стан¬
дартом регистрационных записей при заморажива¬
нии материала (см. табл. 20.2; 20.3).
2. Оцените жизнеспособность клеток в осадке в каж¬
дой ампуле после оттаивания.
3. Размороженную культуру необходимо трипсини-
зировать и провести подсчет клеток в ней в тот же
день (только в ходе упражнения; эту процедуру не
производите в ходе повседневной работы).
4. Результаты занесите в таблицу, как при выполнении
упражнения 13 и 14. Посчет жизнеспособности кле¬
ток провелите в день оттаивания для суспензионных
культур и на следующий день для монослойных
культур.
Анализ
1. Какой криоконсервант лучше для культуры, исполь¬
зованной при выполнении упражнения?
2. Согласуются ли результаты сравнения оценки жиз¬
неспособности клеток по высвобождению красителя
с реальным выживанием клеток через 24 ч?
3. Является ли повреждающим фактором промедле¬
ние при замораживании? Помогает ли выживанию
клеток охлаждение после добавление криоконсер¬
ванта?
Упражнение 18. Обнаружение микоплазм
Цель процедуры
Подтвердить, что культура свободна от микоплазмати-
ческого заражения.
Области применения
Рутинное ведение клеточных линий; контроль качества
клеточных линий перед замораживанием; проверка
поступивших на исследование клеточных линий, тка¬
ней и биопсийных образцов в ходе карантинных мер.
Навыки, которыми следует овладеть
Понимание важности отсутствия в культуре мико-
плазматического загрязнения. Приобретение опыта в
скрининге клеточных линий на предмет микоплазма-
тического заражения флюоресцентным методом или
ПЦР.
Инструктирование и контроль: пересев культуры
и инфицирование фидерного слоя не требуют руко¬
водства, нет необходимости производить настоящее
заражение культуры микоплазмами, поскольку в этом
случае все процедуры должны проводиться в условиях
строгого инструктирования и постоянного контроля
2.3. Упражнения повышенной сложности
53
20% DMSO
i
Трипсинизированные
или центрифугирован-
| ные клетки, ресуспен-
дированные в высокой
концентрации
Добавить клетки к DMSO или
глицерину
Заполнить 12 ампул: 6 - суспензия в
глицерине, 6 - в DMSO
20%-й глицерин
E3SE9 19
EQ SSEQ
Выдержать 2 ампулы из
каждого набора при 20 °С
в течение 30 мин и затем
поместить в пеналы для
I замораживания, охладить
▼ до -70 °С в течение ночи
по 2 ампулы из
каждого набора в пеналы
для замораживания и
охладить до -70 °С в
течение ночи ^
Выдержать 2 ампулы из
каждого набора при 4 °С
в течение 30 мин и затем
поместить в пеналы для
замораживания охладить
до -70 °С в течение ночи
Перенести все ампулы в
жидкий азот и разморозить
на следующий день
Рис. 2.1. Упражнение по замораживанию. Протокол эксперимента по сравнению криопротекторного действия DMSO и
глицерина с предварительным охлаждением при 4 °С и после выдерживания при комнатной температуре
54
ГЛАВА 2. ТРЕНИРОВОЧНЫЕ ПРОГРАММЫ
с соблюдением карантинных мер. Периодическое
наблюдение и руководство потребуются при окра¬
шивании образцов на микоплазмы и при экстракции
ДНК и проведении ПЦР, степень руководства зависит
от опыта учащихся в проведении этих манипуляций.
Постоянное руководство необходимо при анализе по¬
лученных результатов.
Время: 30 мин в течение 5 дней до пересева иссле¬
дуемой культуры или замены питательной среды; 1 ч в
день пересева индикаторной культуры (день 0); 30 мин
в 1-й день — перенос среды из испытываемой культуры;
2-4 ч в 5-й день для окрашивания культуры на при¬
сутствие микоплазмы или проведение ПЦР и 30 мин
в дальнейшем для оценки результатов.
Вспомогательная информация
Микоплазмы (см. разд. 19.3.2); валидация (проверка)
(см. разд. 7.10).
Сведения общего характера
Карантинные меры (см. разд. 19.1.8); использование
флюоресцентного микроскопа или ПЦР-исследова-
ние; предоставление фиксированной положительной
тест-культуры.
Безопасность: нет специальных требований к безо¬
пасности, отличных от прежде описанных для работы
с человеческими клетками (см. разд. 7.8.3). Учащиеся
должны быть предупреждены о риске для здоровья
при работе с источником УФ-света и риске, связанном
с извлечением источника света из флюоресцентного
микроскопа.
Упражнение
Краткое изложение процедуры
Исследуемая культура получает питание на среде, не
содержащей антибиотик, в течение 5 дней, образец
культуры переносится в индикаторную клеточную
линию, в которой заведомо присутствуют микоплазмы.
Присутствие микоплазм регистрируется методом ПЦР
или флюоресцентной микроскопии.
Сведения общего характера
Стандартный протокол: выявление микоплазмы
методом флюоресценции (см. протокол 19.2) или оп¬
ределение микоплазматического заражения методом
ПЦР (см. протокол 19.3).
Дополнительные протоколы: цифровая фотография
(см. разд. 16.4.5).
Экспериментальные варианты процедуры
Трудно дополнить это упражнение экспериментальны¬
ми элементами, кроме варианта выявления потенциаль¬
ного пути заражения инфицированной культуры. Ма¬
ловероятно, что в какой-либо лаборатории возникнет
желание подвергаться опасности заражения микоплаз¬
мами, если только не преследуются какие-либо особые
цели исследований, оправдывающие этот риск.
Данные
1. Результаты оцените как положительные или отри¬
цательные по сравнению с фиксированным поло¬
жительным контролем (флюоресценция) или ДНК
микоплазм (ПЦР).
2. Результаты всех исследований запишите в ре¬
гистрационный журнал или в журнал ведения
культур.
Анализ
1. В микоплазма-положительных образцах при ок¬
рашивании обнаруживаются точечные или ните¬
видные образования в цитоплазме и на периферии
клетки.
2. Электрофоретическая миграция ДНК при ПЦР мо¬
жет быть исследована в сравнении с контрольными
образцами ДНК (см. рис. 19.2).
Упражнение 19. Характеристика
клеточных линий
Наряду с детекцией микоплазматического заражения
(см. упражнение 18), умение провести характеристику
клеточной линии является одним из наиболее важных
навыков специалиста в области культивирования
клеток. Некоторые формы характеристики важны для
подтверждения идентичности используемой клеточ¬
ной линии, однако выбираемые методы характерис¬
тики определяются методологией, принятой в данной
лаборатории. При использовании в лаборатории
ДНК-фингерпринтинга или исследования профиля
ДНК эти методы могут применяться и для описания
клеточной линии, если прежде было показано, что ис¬
следуемый параметр служит характеристикой данной
линии. В противном случае для характеристики линии
потребуется более одного метода. Клеточные линии,
постоянно используемые в работе, могут иметь специ¬
фические мониторинговые характеристики, например
экспрессия отдельных рецепторов, экспрессия специ¬
фических продуктов, устойчивость к лекарственному
препарату. Возможно, для характеристики потребуется
добавить еще один параметр, например хромосомный
или изоферментный анализ. Если ДНК-фингерприн-
тинг или исследование профиля не используется в
лаборатории, то маловероятно, что кто-либо начнет раз¬
вивать эти методы только для приобретения навыков
учащимися. Скорее всего, клетки будут посланы для
идентификации в коммерческую лабораторию, име¬
ющую широкие возможности для проведения допол¬
нительных исследований, в том числе для сравнения с
референс-материалом. Однако, исходя из соображений
целесообразности, в тренировочную программу вклю¬
чено упражнение по характеристике клеточной линии,
для того чтобы убедить учащихся в важности прове¬
дения аутентификации клеточных линий, имея в виду
широкую распространенность неверно идентифициро¬
ванных клеточных линий и возможные последствия,
связанные с этим (см. разд. 16.3; 19.5; 20.2).
2.3. Упражнения повышенной сложности
55
Для проведения эксперимента в данном упраж¬
нении предлагается использовать изоферментный
электрофорез, который легко осуществим при помощи
коммерческих наборов и достаточно дешев.
Цель процедуры
Подтвердить идентичность клеточной линии.
Области применения
Проверка наличия случайного перекрестного зараже¬
ния клеточных линий; контроль качества клеточных
линий до замораживания и/или до начала реализации
проекта или программы исследований; подтверждение
идентичности поступивших клеточных линий.
Навыки, которыми следует овладеть
Подтверждение идентичности клеточной линии. Раз¬
витие понимания роли избыточного роста культуры,
неправильной идентификации и перекрестной конта¬
минации.
Инструктирование и контроль: приготовление об¬
разцов и проведение электрофореза потребуют наблю¬
дения и помощи со стороны руководителя, возможно
не постоянного.
Время: 2 ч.
Сведения общего характера
Необходимость характеристики клеточных линий
(см. разд. 16.1); морфология (см. разд. 16.4); изофер¬
менты (см. разд. 16.8.1); хромосомный состав (см.
разд. 16.5); ДНК-фингерпринтинг и исследование
профиля (см. разд. 16.6.2; 16.6.3); антигенные маркеры
(см. разд. 16.9); аутентификация (см. разд. 16.3).
Демонстрационный материал или действия: ис¬
пользование аппарата для электрофореза Authentikit™
(см. рис. 16.12); примеры ДНК-фингерпринтинга
и/или исследования профиля (см. рис. 16.9; 16.11);
примеры кариотипов (см. рис. 16.6; 16.8).
Безопасность: нет дополнительных требований
к мерам безопасности, помимо описанных выше для
работы с человеческими клетками. При использовании
ДНК-фингерпринтинга требуется соблюдение мер
осторожности при работе с радиоактивно меченными
материалами (см. разд. 7.7).
Упражнение
Краткое изложение процедуры
Подготовка экстракта клеток, проведение электро¬
фореза на агарозном геле; проведение окрашивания
хромогенными субстратами.
Стандартный протокол
Изоферментный анализ (см. протокол 16.10).
Вспомогательные протоколы: приготовление пре¬
парата хромосом (см. протокол 16.7); мультилокусный
ДНК-фингерпринтинг (см. протокол 16.8); исследо¬
вания профиля ДНК (см. протокол 16.9); непрямая
иммунофлюоресценция (см. протокол 16.11).
Экспериментальные варианты процедуры
Исследуйте шесть различных клеточных линий, вы¬
бранных из доступного лаборатории набора или из
следующего списка: HeLa; КВ или Hep-2; Vero; L929;
ЗТЗ или ЗТ6; ВНК-21-С13; СНО-К1. Большинство
клеточных линий из различных источников можно
определить с помощью четырех изоферментов: нуклео-
зидфосфорилазы, глюкозо-6-фосфат-дегидрогеназы,
лактатдегидрогеназы и малатдегидрогеназы.
Сведения общего характера
Изоферменты (см. разд. 16.8.1,16.8.2 и обзоры [Hay et
al., 2000; Stube et al., 1995]).
Данные
После окрашивания геля хромогенными субстратами
проведите фотографирование или сканирование. Гели
можно сохранить.
Анализ
1. Сравните результаты, полученные с использова¬
нием каждого фермента для разных клеточных
линий.
2. Объясните значение того, что в культурах КВ или
Нер-2 имеется тот же изофермент (глюкозо-6-фос-
фат-дегидрогеназа), что и в HeLa.
3. Как повысить разрешение между клеточными ли¬
ниями?
Упражнение 20. Первичная культура
Цель процедуры
Выделить клетки из тканей организма для создания
клеточной культуры.
Области применения
Создание клеточной культуры для производства вак¬
цин; выделение специализированных клеточных типов
для исследований; хромосомный анализ; разработка
селективных сред; создание короткоживущих клеточ¬
ных линий для тканевой инженерии.
Навыки, которыми следует овладеть
Понимание происхождения и разнообразия культи¬
вируемых клеток. Оценка различий в методологии
получения первичных культур.
Инструктирование и контроль: периодическое.
Время: 2-4 ч в день 0; 1 ч в день 1; 2 ч в день 3.
Сведения общего характера
Первичная культура (см. гл. 12).
Демонстрационный материал или действия: ткани
куриного эмбриона (или альтернативные источники
тканей).
56
ГЛАВА 2. ТРЕНИРОВОЧНЫЕ ПРОГРАММЫ
Безопасность: при использовании тканей куриного
эмбриона минимальные требования к мерам безопас¬
ности.
Упражнение
Куриный эмбрион выбран для этого упражнения по
ряду причин. Его ткани практически готовы к куль¬
тивированию с минимальной предварительной подго¬
товкой животного. Эмбрион может быть препарирован
без каких-либо ограничений, если он находится на
половинном сроке эмбрионального развития. Полный
срок составляет 21 день, потому 10-дневный эмбрион
пригоден для использования, если он достаточно мал
по размеру. Куриный эмбрион крупнее, чем мышиный,
на этой стадии развития, поэтому можно получить
больший выход клеток из целого измельченного эмб¬
риона или его изолированных органов.
Краткое изложение процедуры
Эмбрионы извлекаются из яйца и иссекаются с полу¬
чением первичного эксплантата. Выделяются дезагре¬
гированные клеточные культуры.
Стандартный протокол
1. Выделите эмбрионы из оплодотворенных яиц (см.
протокол 12.2).
2. Размельчите один эмбрион в теплом растворе трип¬
сина (см. протокол 12.5).
3. Размельчите один эмбрион таких же размеров в
холодном растворе трипсина (см. протокол 12.6).
4. Иссеките третий эмбрион и выделите отдельные
органы, например мозг, печень, сердце, кишечник,
мышечное волокно. Размельчите каждый орган
отдельно методом холодной трипсинизации (см.
протокол 12.7).
5. Соберите некоторые из оставшихся после выпол¬
нения 4-го шага тканей и получите первичные
эксплантаты (см. протокол 12.4).
Вспомогательные протоколы: дезагрегация в раст¬
воре коллагеназы (см. протокол 12.8); механическое
измельчение на ситах (см. протокол 12.9); обогащение
жизнеспособных клеток (см. протокол 12.10).
Экспериментальные варианты процедуры
1. Клетки, изолированные из различных тканей.
2. Восстановление после теплой и холодной трипси¬
низации.
Сведения общего характера
Ферментативная дезагрегация (см. разд. 12.3.2); трип-
синизация после предварительной экспозиции на
холоде (см. разд. 12.3.4).
Данные
1. Исследуйте культуры, полученные из остатков
органов, через 3-5 дней роста и отметьте морфоло¬
гические особенности и признаки сократительной
способности в клетках сердечной мышцы.
2. Подсчитайте количество клеток, полученных мето¬
дом холодной и теплой трипсинизации, и оцените
их жизнеспособность методом окрашивания.
3. Трипсинизируйте и изучите культуры, полученные
методом холодной и теплой трипсинизации, через
3 дня после посева.
4. Запишите полученные данные в регистрацион¬
ный журнал, как для первичной культуры (см.
табл. 12.2).
Анализ
1. Постарайтесь идентифицировать различные кле¬
точные типы, представленные в первичных куль¬
турах, полученных из остатков органов.
2. Как бы вы размножили эти культуры, чтобы сохра¬
нить специфические клеточные типы?
3. Произведите расчеты и занесите в таблицу общее
количество клеток на один эмбрион и на грамм, ко¬
личество жизнеспособных клеток на один эмбрион
и на один грамм.
4. Исходя из количества клеток, полученных в первой
субкультуре (3 дня после посева), высчитайте выход
клеток на эмбрион, а также в отношении к общему
количеству посеянных клеток и количеству жизне¬
способных посеянных клеток.
5. Является ли оценка жизнеспособности методом
высвобождения красителя надежным прогности¬
ческим признаком восстановления культуры?
Упражнение 21. Клонирование
в монослое
Цель процедуры
Развести клетки так, чтобы они росли как изолирован¬
ные колонии, полученные из единичных клеток.
Области применения
Выделение генотипических или фенотипических ва¬
риантов; исследование выживаемости; исследования
роста.
Навыки, которыми следует овладеть
Введение в технику клонирования клеток методом
разведения. Определение эффективности посева как
параметра роста или выживаемости клеток. Клональ¬
ное выделение определенных клеточных типов.
Инструктирование и контроль: первичное руковод¬
ство только в день 0, в последующие дни (с 10-го по
14-й) только помощь в идентификации клонов.
Время: 1 ч.
Сведения общего характера
Клонирование (см. протокол 14.1-14.4); определение
эффективности посева (см. разд. 21.10).
Демонстрационный материал или действия: пре¬
дыдущее клонирование и окрашенные культуры; ва¬
2.4. Специализированные упражнения
57
рианты клонирования, например, в чашках Петри или
на микротитрационных планшетах.
Безопасность: минимальные меры безопасности
при использовании нечеловеческих клеточных линий,
таких как СНО-К1. Использование тормозящего рост
фидерного слоя потребует привлечения внимания
учащихся к токсичности митомицина С или риска
радиоактивного излучения, в зависимости от вида
фидерных клеток.
Упражнение
Краткое изложение процедуры
Монослойная культура трипсинизируется в середине
поздней логарифмической фазы, клетки проходят
серийное разведение. Посев производится в низких
концентрациях на чашки Петри или на микротитра-
ционные планшеты.
Стандартный протокол
Для выполнения простого упражнения на клеточной
линии: использование клонирования методом разве¬
дения (см. протокол 14.1). Для усложненного вари¬
анта упражнения: использование клеточной линии с
неизвестной эффективностью посева. Определение
эффективности посева (см. протокол 21.10).
Дополнительные протоколы: приготовление конди¬
ционированной среды (см. протокол 14.2); приготовле¬
ние фидерного слоя (см. протокол 14.3); клонирование
в агаре (см. протокол 14.4); клонирование в метилцел-
люлозе (см. протокол 14.5); клоногенное исследование
(см. протокол 22.3).
Экспериментальные варианты процедуры
1. Посевные концентрации: посев 10, 20, 50, 100,
200 кл./мл (см. протокол 21.10).
2. Концентрация сыворотки: посев в различной
концентрации сыворотки от 0 до 20%. Это также
хороший способ проверки качества сыворотки (см.
протокол 14.10. Разведите клетки в бессывороточ¬
ной среде до концентрации 1000 кл./мл и разведите
200 мкл до 20 мл (1:100), что дает концентрацию
10 кл./мл в отдельном объеме, содержащем соответ¬
ствующую концентрацию сыворотки (например, 0,
0,5,0,2,1,0,2,0, 5,10,20, 50 мкг/мл).
3. Цитотоксичность: простое дополнение к упражне¬
нию состоит в добавлении цитотоксического вещества
к клеткам на 24 ч до начала клонирования (см. прото¬
кол 22.3). При использовании митомицина С полезно
предварительно подготовить фидерный слой (см.
протокол 14.3). Можно использовать экспоненци¬
альный диапазон концентраций между 1 и 50 мкг/мл,
например 0,0,1,0,2,0,5,1,0,2,0,5,10,20,50 мкг/мл.
4. Фидерный слой: повторите п. 1) в присутствии и без
фидерного слоя (см. протокол 14.3).
5. Выделение клонов клеток: выделите и сравните мор¬
фологию клонированных штаммов.
Сведения общего характера
Клонирование клеток (см. разд. 14.1); выделение кло¬
нов (см. разд. 14.6, 14.7,14.8); эффективность посева
(см. разд. 21.10); выживаемость (см. разд. 22.3.2).
Данные
1. После того как колонии на чашках станут видны
невооруженным глазом, окрасьте их кристалл-вио-
летом (см. протокол 16.3).
2. Подсчитайте количество колоний на чашках.
Анализ
1. Подсчитайте эффективность посева (отношение
количества образовавшихся колоний к количеству
посеянных клеток XI00) (см. протокол 21.10).
2. Если концентрация клеток варьирует, постройте
график зависимости количества колоний на чашке
от количества посеянных клеток (см. протокол
21.10). Должна быть линейная зависимость.
3. Если варьирует концентрация сыворотки, построй¬
те график зависимости эффективности посева от
концентрации сыворотки (см. разд. 11.6.3).
а) Почему это хороший тест для проверки качества
сыворотки?
б) Как бы вы могли сравнить различные партии
сыворотки?
4. Если использовалось цитотоксическое вещество,
вычислите отношение количества колоний на
чашку при каждой концентрации препарата к ко¬
личеству колоний на чашке в контроле. Это даст
показатель выживания, который следует построить
в логарифмических координатах против логарифма
концентрации препарата. Можно построить также в
линейных координатах при более узком диапазоне
размаха концентрации.
2.4. СПЕЦИАЛИЗИРОВАННЫЕ
УПРАЖНЕНИЯ
Допускается, что успешное выполнение специализи¬
рованных упражнений (21-27) приведет к формирова¬
нию специфических навыков и представлений и будут
выбраны соответствующие протоколы. Параметры
вариабельности определяются в соответствии с целями
обучения, поэтому эти упражнения не детализированы.
Подразумевается также, что студент/обучающийся
приобретает достаточно навыков, чтобы спланировать
и провести свой собственный эксперимент. Они вклю¬
чены в табл. 2.1, однако предполагается, что учащиеся
имеют достаточно большой интерес к работе, чтобы
расширить программу обучения и достичь более вы¬
сокого профессионального уровня.
Глава 3
БИОЛОГИЯ КУЛЬТИВИРУЕМЫХ КЛЕТОК
3.1. ВЛИЯНИЕ ОКРУЖАЮЩЕЙ СРЕДЫ
НА КУЛЬТУРУ КЛЕТОК
Достоверность и приемлемость использования культи¬
вируемых клеток в качестве модели для исследования
физиологических функций in vivo постоянно крити¬
куется. Часто клетки в культуре не проявляют того
фенотипа, который они имеют in vivo, поскольку их
микроокружение изменено. В результате недостатка
клеточной гетерогенности и трехмерной архитектуры
тканей, характерной для клеток in vivo, в культуре
редуцированы межклеточные и клеточно-матрикс-
ные взаимодействия. Отсутствуют многие гормо¬
нальные и питательные стимулы. Все это создает то
окружение, которое в большей степени благоприятно
для распространения, миграции и пролиферации
неспециализированных клеток-предшественников,
чем для проявления функциональных свойств диф¬
ференцированных клеток. Влияние окружения на
культуру осуществляется по четырем направлениям:
1) природа субстрата, на котором или в котором про¬
исходит рост культуры, — плотная, такая как пластик
или иная жесткая структура, полужидкая, например
гель (коллаген или агар), или жидкая, как в случае сус¬
пензионной культуры; 2) степень контакта с другими
клетками; 3) физико-химический и физиологический
состав среды; 4) состав газовой фазы и 5) температура
инкубации. Обеспечение соответствующего окру¬
жения, включая адгезию к субстрату, концентрация
питательных веществ, гормонов или факторов роста,
а также межклеточное взаимодействие являются
основополагающими условиями проявления специа¬
лизированных функций клеток (см. разд. 17.1, 17.7 и
Alberts et al., 2002).
3.2. КЛЕТОЧНАЯ АДГЕЗИЯ
Большинство клеток из плотных тканей растут как
прикрепленный монослой, если только не происходит
трансформации, в результате которой они могут при¬
обрести независимость от субстрата (см. разд. 18.5.1);
после дезагрегации тканей или субкультуры они нуж¬
даются в том, чтобы прикрепиться и распластаться по
субстрату до начала деления (см. разд. 13.7, 21.9.2).
Первоначально было обнаружено, что клеткам необ¬
ходимо прикрепиться и распластаться на стекле, име¬
ющем небольшой отрицательный заряд. Впоследствии
было показано, что клетки могут прикрепляться также
к пластику, такому как полистирол, предварительно
обработанному антистатиком или высокоэнергети¬
ческой ионизирующей радиацией. Теперь мы знаем,
что клеточная адгезия опосредована специфическими
поверхностно-клеточными рецепторами для молекул
во внеклеточном матриксе (см. также разд. 8.4,17.7.3),
и, по-видимому, распластыванию клетки по субстрату
предшествует секреция клетками матрикса внеклеточ¬
ных белков и протеогликанов. Матрикс адгезирует на
заряженном субстрате, и затем клетки связываются с
матриксом с использованием специфических рецеп¬
торов. Следовательно, пластик или стекло, которые
использовались для культивирования клеток, мо¬
гут обеспечивать лучшую поверхностную адгезию.
Предварительная обработка субстрата веществами,
входящими в состав клеточного матрикса, такими как
фибронектин или коллаген, либо производными, та¬
кими как желатин, может способствовать росту более
требовательных клеточных культур.
Использование фибробластподобных клеток явля¬
ется главным требованием прикрепления к субстрату и
распластывания, а также миграции отдельных клеток
при низкой плотности суспензии. Для адгезии, выжи¬
вания и роста культуры эпителиальных клеток может
также потребоваться наличие межклеточных контак¬
тов, поэтому они склонны формировать островки.
3.2.1. Молекулы клеточной адгезии
Показано, что в процессах межклеточной и клеточно-
матриксной адгезии участвуют три главных класса
трансмембранных белков (рис. 3.1). Са2+-Независи-
мые молекулы межклеточной адгезии (cell adhesion
molecules, САМ) и Са2+-зависимые кадгерины пре¬
имущественно вовлечены во взаимодействие между
гомологичными клетками. Две идентичные моле¬
кулы такого белка взаимодействуют друг с другом;
в результате этого взаимодействуют друг с другом
гомологичные молекулы располагающихся напротив
друг друга клеток [Rosenman & Galatin, 1991; Alberts
et al., 2002], вследствие чего происходит межкле¬
точное распознавание, играющее сигнальную роль
в поведении клеток [Cavallaro & Christofori, 2004].
Клеточно-субстратное взаимодействие опосредовано
3.2. Клеточная адгезия
59
CAMs
Клеточный слой
Базальная
пластинка
Соединительнотканная
строма
Интегрины Протеогликан
Рецепторы с внутриклеточным Низкоаффинные трансмембранные\
сигнальным доменом и вариабельным рецепторы без сигнального домена, 4
внеклеточным доменом, связывающим связывающие протеогликаны матрикса, '
фибронектин, витронктин, ламинин, коллаген и факторы роста
коллаген
Рис. 3.1. Клеточная адгезия. Схематическое изображение слоев эпителиальных клеток над соединительной тканью, содержащей
фиброциты и отделенной базальной мембраной. Молекулы клеточной адгезии (CAMs) и кадгерины изображены между одина¬
ковыми клетками, интегрины и протеогликаны — между слоем эпителиальных клеток и матриксом базальной пластинки
преимущественно интегринами — рецепторами для
молекул матрикса, таких как фибронектин, энтактин,
ламинин и коллаген. Интегрины связываются с ними
через специфические повторы аминокислот, обычно
содержащие последовательность аргинин-глицин-
аспарагиновая кислота (RGD) [Yamada&Geiger,
1997]. Каждый интегрин включает в себя одну а- и
одну Р-субъединицу, внеклеточные домены которых
обладают высокой степенью полиморфизма, что обус¬
ловливает значительное разнообразие интегринов.
Как интегрины, так и кадгерины взаимодействуют с
винкулином, что является звеном передачи сигнала в
клеточное ядро [Bacolitsa et al., 2004].
Третью группу молекул клеточной адгезии со¬
ставляют трансмембранные протеогликаны, также
взаимодействующие с компонентами матрикса, такими
как другие протеогликаны и коллаген, но не взаимо¬
действующие с RGD-повторами. Некоторые трансмем¬
бранные и растворимые протеогликаны действуют как
низкоаффинные рецепторы факторов роста [Subrama-
nian et al., 1977; Yevdokimova & Freshney, 1997] и могут
стабилизировать, активировать и/или перемещать фак¬
торы роста к высокоаффинным рецепторам, участвуя
в их димеризации [Schlessinger et al., 1995].
Дезагрегация ткани или отделение от субстрата мо¬
нослойной культуры под действием протеазы приведет
к расщеплению некоторых компонентов внеклеточного
матрикса и может даже способствовать деградации
некоторых внеклеточных доменов трансмембранных
белков, приводя к разобщению клеток. Эпителиальные
клетки в целом более устойчивы к дезагрегации, по¬
скольку они склонны к образованию плотных межкле¬
точных соединений (десмосом, адгезивных соединений
и плотных контактов), удерживающих их вместе, в то
время как мезенхимальные клетки больше нуждаются
в клеточно-матриксных взаимодействиях для осущест¬
вления межклеточного связывания, а поэтому легче
разобщаются. Эндотелиальные клетки могут образо¬
вывать плотные соединения в культуре, особенно при
культивировании на преформированном матриксе в
течение продолжительного времени, и диссоциация
клеток затруднена.
В любом случае, до того как начнется прикрепление
и распластывание клеток, они должны синтезировать
матричные белки либо должны иметь возможность
использования субстрата, покрытого матриксом.
3.2.2. Межклеточные контакты
В то время как некоторые молекулы клеточной адгезии
диффузно распределены на плазматической мембране,
другие организованы в межклеточные контакты. Роль
контактов различна. Механическую роль играют такие
контакты, как десмосомы и адгезивные соединения,
удерживающие вместе эпителиальные клетки, а так¬
же плотные контакты, заполняющие пространство,
например между клетками ацинуса или протока либо
между эндотелиальными клетками сосудов. Плотные
контакты позволяют ионам, питательным веществам и
небольшим сигнальным молекулам (таким как моле¬
кулы циклического АМФ, цАМФ) [см. Alberts et al.,
2002] проникать в клетки. Хотя десмосомы могут быть
распределены по всей области цитоплазматической
мембраны, имеющей контакты с другими клетками
(рис. 3.2а), они часто ассоциированы с плотными кон¬
тактами и адгезивными соединениями на апикальном
конце латеральных клеточных контактов (рис. 3.2б).
60
ГЛАВА 3. БИОЛОГИЯ КУЛЬТИВИРУЕМЫХ КЛЕТОК
Рис. 3.2. Межклеточные контакты. Электронная микрофотография культивируемых Ca-KD-клеток (ранний пассаж).
Клетки получены из вторичной аденокарциномы мозга (первичный источник неизвестен). Клетки выращены в чашке Петри
(Vivascience). (а) Десмосомы (Д) между двумя контактирующими клетками; увеличение 28.000х. (б) Плотные контакты (Т)
и комплексы контактов (JC) в области канальца; увеличение 13.500х (любезно предоставлено Каролин МакДональд)
Поскольку эпителиальные клетки дифференци¬
руются в культурах, имеющих сливной рост, они
способны формировать увеличивающееся количество
десмосом и при наличии некоторой морфологической
организации могут образовывать полные контактные
структуры. Это одна из причин, по которым эпите¬
лиальные клетки, находящиеся на стадии сливного
роста в течение слишком длительного времени, трудно
поддаются дезагрегации. Поскольку многие молекулы
адгезии в таких соединениях являются Са2+-зави-
симыми, до дезагрегации или в ее процессе вместе с
трипсином часто используются хелатирующие агенты,
такие как ЭДТА.
3.2.3. Внеклеточный матрикс
Межклеточное пространство в тканях заполнено вне¬
клеточным матриксом (extracellular matrix, ECM), со¬
став которого определяется типом клеток. Фиброциты
секретируют в матрикс коллаген I типа и фибронек¬
тин; эпителиальные клетки продуцируют ламинин.
Если контактирующие клетки относятся к различным
типам, например на границе дермы (фиброциты) и
эпидермального слоя (эпителиальные кератиноци¬
ты), оба клеточных типа вносят свой вклад в состав
ЕСМ, часто образуя базальную пластинку. Состав
ЕСМ является фактором, во многом определяющим
фенотипические проявления клеток, прилегающих
к нему, поэтому существует динамическое равнове¬
сие, при котором прилегающие к матриксу клетки
контролируют состав ЕСМ, и, в свою очередь, состав
ЕСМ регулирует фенотип этих клеток [Kleinman et al.,
2003; Zoubiane et al., 2003; Fata et al., 2004]. В связи с
этим для пролиферации мигрирующих фибробластов
необходим иной ЕСМ, чем для дифференцирующихся
эпителиальных клеток или нейронов. Культивируе¬
мые клеточные линии сами способны продуцировать
компоненты ЕСМ, однако первичная культура и
размножение некоторых специализированных кле¬
ток, а также индукция их дифференцировки могут
потребовать специальных экзогенных продуктов в
составе ЕСМ.
Внеклеточный матрикс может иметь в составе кол¬
лаген, ламинин, фибронектин, гиалуроновую кислоту,
протеогликаны и связывать факторы роста и цитокины
[Alberts et al., 1997, 2002].
Он может быть изготовлен механическим смеши¬
ванием чистых компонентов, таких как коллаген и
фибронектин. Для продукции ЕСМ используют клет¬
ки, продуцирующие ЕСМ, с последующей отмывкой
клеток-продуцентов до начала посева исследуемых
клеток (см. протокол 8.1). В культивировании часто
используется преформированный матрикс, продуци¬
рованный культурой мышиной саркомы Ehgelberth-
Holm-Swarm (ЕНС), коммерчески производимый
как матригель (Matrigel) (см. разд. 8.4.1). Матригель
часто применяется для ускорения дифференцировки
и морфогенеза в культуре, так как формирует ре¬
шеткообразную сеть с эпителиальными (см. рис. 3.3,
цв. вкладку 12в) и эндотелиальными клетками.
Во взаимодействии клеток с субстратом можно
выделить, по крайней мере, два этапа: 1) адгезию,
позволяющую осуществить прикрепление и расплас¬
тывание клеток по субстрату, необходимые для начала
клеточной пролиферации [Folkman &Moscona, 1978],
и 2) специфические взаимодействия, напоминающие
взаимодействие эпителиальной клетки с базальной
мембраной, другими компонентами ЕСМ или клет¬
ками прилегающих тканей. Эти специфические взаи¬
модействия необходимы для проявления некоторых
специфических функций клеток (см. разд. 3.4.1 и
17.7.3). Rojkind [1980], Vlodavsky et al [1980] и другие
исследователи изучали рост клеток на естественных
субстратах, аналогичных базальной мембране. В на¬
стоящее время в коммерческом варианте доступны как
3.2. Клеточная адгезия
61
Рис. 3.3. Рост клеток А549 на матригеле. Культуры клеток
аденокарциномы А549, растущие на матригеле. (а) Фо¬
тосъемка при малом увеличении, формирование решетки
через 24 ч после посева клеток в концентрации 1х105 кл./мл;
(б) фотосъемка при большом увеличении через 3 дня после
посева клеток в концентрации 1х105 кл./мл. Стрелка указы¬
вает возможные канальцевые образования (фото Jane Sinclair;
см. также цв. вкладку 12в)
естественные матрицы, так и стандартные матрицы
с определенным набором макромолекул, такие как
Matrigel, Natrigel, коллаген, ламинин и витронектин
(B-D Biosciences, Invitrogen). Эти матрицы можно
использовать для контролируемых исследований
взаимодействия с матриксом.
Использование компонентов ЕСМ может быть
чрезвычайно полезно для улучшения клеточного вы¬
живания, пролиферации, дифференцировки, однако,
если не используются рекомбинантные молекулы [см.
например, Ido et al., 2004], существует значительный
риск внедрения случайных факторов из иницииру¬
ющих животных (см. разд. 10.1). В настоящее время
доступны коммерческие препараты, представляющие
собой фрагменты фибронектина и ламинина (см. При¬
ложение II).
3.2.4. Цитоскелет
Молекулы клеточной адгезии соединяются с элемента¬
ми цитоскелета. Реципрокная передача сигнала между
поверхностью клетки и ядром обусловлена присоедине¬
нием интегринов к актиновым микрофиламентам через
связывающие белки [Fata et al., 2004]. Кадгерины также
связываются с актиновым цитоскелетом в адгезивных
соединениях, опосредуя изменения формы клеток.
Десмосомы, которые также задействуют кадгерины,
связываются с промежуточными филоментами, в дан¬
ном случае (в эпителии) с кератинами, через межкле¬
точные диски, однако еще не до конца ясно, является
эта связь чисто структурной особенностью или также
играет сигнальную роль. Промежуточные филоменты
специфичны для каждой линии клеток и могут быть ис¬
пользованы для характеристики линии (см. разд. 16.3.2;
цв. вкладку 11 a-в). Третьим компонентом цитоскелета
являются микротрубочки; их роль проявляется глав¬
ным образом в обеспечении клеточной подвижности и
внутриклеточном транспорте органелл, таких как ми¬
тохондрии и хроматиды. Кроме того, они обеспечивают
расхождение хроматид при делении клетки.
3.2.5. Клеточная подвижность
Замедленная видеозапись (см. разд. 27.3) показывает,
что культивируемые клетки способны к перемеще¬
нию по субстрату. Наибольшей подвижностью обла¬
дают фибробласты при низкой плотности культуры
(когда между клетками нет контактов), наименьшая
подвижность характерна для плотного монослоя
эпителиальных клеток. Фибробласты мигрируют как
индивидуальные клетки с видимой полярностью дви¬
жения. Ламеллиподии, образуемые за счет полимери¬
зации актина [Pollard &Borisy, 2003], вытягиваются в
направлении движения и прикрепляются к субстрату,
плазматическая мембрана на противоположной сторо¬
не клетки сокращается, заставляя клетку направленно
двигаться. Если клетка встречает другую клетку, поляр¬
ность меняется и миграция осуществляется в противо¬
положном направлении. Как показано в исследовании
с применением коллоидного золота [Scott et al., 2000],
миграция фибробластов возобновляется в случайных
направлениях до тех пор, пока клеточная плотность
культуры не достигнет фазы сливного роста. После
этого направленная миграция фибробластов прекраща¬
ется. Явление прекращения движения клеток на фазе
сливного роста культур, сопровождающееся снижением
образований морщин и складок в мембране, известно
как контактное ингибирование (см. разд. 18.5.2) и ведет,
в конечном счете, к выходу клетки из клеточного цик¬
ла. Миобласты и эндотелиальные клетки мигрируют
аналогичным образом и, как и фибробласты, способны
дифференцироваться, когда достигают сливного роста
в зависимости от микроокружения.
Эпителиальные клетки, не подвергавшиеся транс¬
формации, не совершают беспорядочного движения.
62
ГЛАВА 3. БИОЛОГИЯ КУЛЬТИВИРУЕМЫХ КЛЕТОК
При посеве в низкой концентрации клетки переме¬
щаются до осуществления контакта с другой клеткой,
после чего миграция прекращается. В конечном счете,
клетки накапливаются в клеточных островках, и затем
весь островок может проявлять признаки координиро¬
ванного движения [Casanova, 2002].
3.3. КЛЕТОЧНАЯ ПРОЛИФЕРАЦИЯ
3.3.1. Клеточный цикл
Клеточный цикл состоит из четырех фаз (рис. 3.4).
В М-фазе (Mitosis), хроматин конденсируется и обра¬
зуются хромосомы; две отдельные хроматиды, состав¬
ляющие хромосому, разделяются для того, чтобы пе¬
рейти в дочерние клетки. В Gj-фазе (Gapl) происходит
либо подготовка к синтезу ДНК и следующему циклу
клеточного деления, либо обратимый выход клетки их
клеточного цикла (выход в Go-фазу). Возможен также
необратимый переход в стадию дифференцировки.
В ходе Gt-фазы клетка частично способна контроли¬
ровать прохождение клеточного цикла в ряде точек
рестрикции, которые определяют, войдет ли клетка в
пролиферативный цикл, выйдет ли из него временно,
либо покинет его, перейдя к дифференцировке. Вслед
за Gj-фазой следует S-фаза (Synthesis ДНК), в кото¬
рой происходит репликация ДНК и которая, в свою
очередь, сменяется С2-фазой (Gap2). В ходе С2-фазы
клетка готовится вновь перейти в стадию митоза. Кон¬
трольные точки в начале фазы синтеза ДНК и С2-фазы
определяют цельность и аутентичность ДНК и могут
приостановить клеточный цикл с тем, чтобы клетка
могла репарировать повреждения ДНК или включить
механизм опоптотической гибели, если репарация не¬
возможна. Апоптоз, или программируемая клеточная
Рис. 3.4. Клеточный цикл. Клеточный цикл делится на
четыре фазы: Gl, S, G2 и М. Продвижение по циклу регули¬
руется взаимодействием циклинов с CDC-киназами и сти¬
мулируется ядерными онкогенами и цитоплазматическими
сигналами, инициируемыми взаимодействием рецепторных
киназ с лигандами. Клеточный цикл останавливается в точ¬
ках рестрикции ингибиторами клеточного цикла, такими
как Rb и р53
смерть [al-Rubeai & Singh, 1998], является физиологи¬
ческим процессом, в соответствии с которым клетка
может быть выведена из популяции. Фрагментация
ДНК, фрагментация ядра, уменьшение клеток в разме¬
ре и изменение их очертаний являются визуальными
признаками апоптотического изменения клеток (см. цв.
вкладку 17 в, г). Апоптоз можно обнаружить также по
наличию ферментов-маркеров, для детекции которых
используются наборы реактивов, такие как Apotag (Оп-
сог) или COMET- анализ [Maskell&Green, 1995].
3.3.2. Контроль клеточной пролиферации
Вход в клеточный цикл регулируется сигналами из
окружающей среды. Низкая плотность клеток приво¬
дит к тому, что клетка, не имея контактов с другими
клетками, переходит в активное состояние, способна
к размножению и поэтому входит в клеточный цикл
в присутствии митогенных факторов роста, таких
как эпидермальный фактор роста (EGF), тромбоци-
тарный фактор роста (PDGF) (см. разд. 9.5.2, 10.4.3.
и табл. 10.3), взаимодействующих с рецепторами
клеточной поверхности. Высокая плотность клеток
ингибирует пролиферацию нормальных клеток (и не
ингибирует деление трансформированных клеток) (см.
разд. 18.5.2). Ингибирование пролиферации иниции¬
руется клеточными контактами и усугубляется скопле¬
нием клеток и возникшими в результате изменениями
формы клеток, а также ограничением возможности
распространения.
Внутриклеточный контроль опосредован поло¬
жительно действующими факторами, такими как
циклины [Planas-Sliva & Weinberg, 1997; Reed, 2003]
(см. рис. 3.2), на которые влияют сигнальные каскады,
активированных фосфорилированием внутриклеточ¬
ных доменов рецепторов при их связывании с факто¬
рами роста. Негативно действующие факторы, такие
как белкир53 [Sager, 1992; Mcllwrath et al., 1994], pl6
[Russo et al., 1998] или продукты экспрессии Rb-гена
[Sager, 1992], останавливают прохождениеклеточ-
ного цикла в точках рестрикции или контрольных
точках цикла (рис. 3.5). Связь между внеклеточными
элементами контроля клеточного цикла (как положи¬
тельными, например PDGF, так и отрицательными,
например TGF-Р) и внутриклеточными эффекторами
осуществляется посредством рецепторов клеточной
мембраны и путями передачи биохимических сигна¬
лов, часто включающими фосфорилирование белка и
участие вторичных мессенджеров, таких как цАМФ,
Са2+и диацилглицерин [Alberts et al., 2002]. Имеется
множество доказательств существования этих стадий
в контроле клеточной пролиферации, полученных при
изучении онкогенов и супрессоров генной экспрессии
в опухолевых клетках. Эти исследования были пред¬
приняты с целью создания терапевтических подходов
регуляции неконтролируемого клеточного деления
при раковых заболеваниях. В ходе исследований
были получены промежуточные данные, углубившие
3.4. Дифференцировка
63
Rb
Рис. 3.5. Регуляция клеточного цикла и продвижение по
клеточному циклу. Клеточный цикл останавливается в
точках рестрикции или контрольных пунктах под действием
Rb, р53 и других ингибиторов клеточного цикла (а). При их
активации (обычно путем фосфорилирования) клеточный
цикл возобновляется (б)
понимание механизмов регуляции, происходящих в
клеточной культуре, а также природы факторов регуля¬
ции [Jenkins, 1992]. Кроме того, в ходе этих работ были
идентифицированы гены, улучшающие клеточное
деление, некоторые из которых можно использовать
для иммортализации конечных клеточных линий (см.
разд. 18.4).
3.4. ДИФФЕРЕНЦИРОВКА
Как упоминалось ранее (см. разд. 1.3.3), выражение
особенностей дифференцировки клеточной культуры
часто ограничено продвижением клеток по клеточному
циклу, необходимому для размножения клеток и расши¬
рения ростковой культуры. Условиями, необходимыми
для индукции дифференцировки, являются высокая
плотность клеток, усиление межклеточных и клеточно-
матриксных взаимодействий и присутствие различных
факторов дифференцировки (см. разд. 17.1.1; 17.7),
которые могут быть антагонистами факторов клеточ¬
ной пролиферации и наоборот. Поэтому, если есть
необходимость дифференцировки культуры, следует
различать две группы условий — одну, оптимизирую¬
щую клеточное деление, и другую, оптимизирующую
клеточную дифференцировку.
3.4.1. Поддержание и индукция
дифференцировки
Много лет назад было обнаружено, что специфические
функции клеток сохраняются дольше, когда сохранена
трехмерная структура первичной ткани, как в случае
органной культуры (см. разд. 25.2). К сожалению,
органная культура не может поддерживаться и переви¬
ваться, поэтому для каждого следующего эксперимента
требуется приготовление органной культуры de novo,
что приводит к сложностям при стандартизации и
количественном анализе данных. Восстановление трех¬
мерности пространственной структуры возможно пер¬
фузией монослойной культуры (см. разд. 25.3; 26.2.6)
или культивированием клеток на или в специальных
матрицах, таких как коллагеновый гель, целлюлоза,
желатиновая губка и др. (см. разд. 3.2.3; 8.4.1; 8.4.3;
17.7.3), в зависимости от конкретных потребностей
исследователя. Ряд коммерческих продуктов, наиболее
известным из которых является Matrigel (BD Biosci¬
ences), воспроизводит характеристики внеклеточного
матрикса, которые не определены с необходимой
точностью. Однако имеются варианты коммерчес¬
ких препаратов, из которых удалены факторы роста
(GFR-Matrigel). Эта технология имеет определенные
ограничения, но предоставляет возможность для иссле¬
дований межклеточных и клеточно-матриксных взаи¬
модействий как гомотипической, так и гетеротипиче-
ской культуры. В перспективе она также предполагает
возможность изучения тканеспецифических функций,
особенно когда взаимодействие клеток в культуре свя¬
зано с ростом культуры в фильтрационной лунке (см.
разд. 25.3.6). Для проявления фенотипических свойств
дифференцированной культуры может потребоваться
использование соответствующих селективных сред
(см. разд. 10.2.1) с соответствующими растворимыми
индукторами. Такими индукторами могут быть гид¬
рокортизон, ретиноиды или двухмерные полярные
соединения (см. разд. 17.7.1; 17.7.2). При этом обычно
не требуется добавления сыворотки.
Развитие методов моделирования нормальных
функций тканей в культуре может способствовать
исследованиям патологических изменений, таких как
демиелинизация и злокачественная инвазия. Однако с
точки зрения фундаментальных исследований клетки,
изучаемые in vitro, должны обладать всеми функциями,
характерными для исходной ткани. Проявление диф¬
ференцированного фенотипа клеток не должно быть
завершенным, поскольку появление единичного ти¬
поспецифического поверхностного антигена бывает
достаточным для того, чтобы придать клетке правиль¬
ное направление дифференцировки. Более полная
экспрессия антигенов может потребоваться для того,
чтобы поместить клетку в правильное положение в
линиях клеточной дифференцировки и использовать
64
ГЛАВА 3. БИОЛОГИЯ КУЛЬТИВИРУЕМЫХ КЛЕТОК
ее как достоверную модель для исследования свойств
клеток in vivo.
3.4.2. Дедифференцировка
Исторически в неспособности клеточных линий
проявлять фенотипические признаки, которыми они
обладают in vivot обвиняли явление дедифференциров-
ки. Согласно этой концепции, дифференцированные
клетки при их культивировании in vitro теряют свои
характерные свойства. Однако часто остается неясным,
что вызвало адаптивную и потенциально обратимую
утрату характерных для дифференцированных клеток
свойств: 1) ошибки в выделении линии клеток; 2) выде¬
ление недифференцированных клеток желаемой линии,
дающих избыточный рост по сравнению с полностью
дифференцированными клетками, обладающими по¬
ниженной способностью к делению или 3) отсутствие
соответствующих индукторов дифференцировки (гор¬
монов или цитокинов межклеточного или клеточно-
матриксного взаимодействия). В реальной практике
все эти элементы могут вносить определенный вклад
в дедифференцировку клеток. Даже при соблюдении
адекватных условий культивирования и тщательной
селекции клеток продолжительная пролиферация со¬
здает благоприятные условия для развития и деления
недифференцированных клеток-предшественников,
которые в отсутствие соответствующего окружения не
переходят в стадию дифференцировки.
Следует различать дедифференцировку, деадапта¬
цию и селекцию. Понятие «дедифференцировка» под¬
разумевает, что свойства специализированных клеток
утрачены в результате конверсии до более примитив¬
Плюрипо-
тентные
стволовые
клетки
Коммитированные
клетки-предшест¬
венники
Непролиферирую- Терминально
щие дифференци- | дифференцированные
рованные клетки | клетки
^охранение пула
тгволовых клеток
(а) НОРМАЛЬНАЯ
ДИФФЕРЕНЦИРОВКА
(6) БЛОКИРОВАННАЯ
ДИФФЕРЕНЦИРОВКА
Регуляция/адаптация
Г
Дифференцировка
Регуляция/адаптация
Амплификация
Рис. 3.6. Дифференцировка из стволовых клеток. (a) In vivo. Небольшое количество стволовых клеток дает начало пулу проли¬
ферирующих клеток-предшественников. (б) In vitro дифференцировка ограничена необходимостью пролиферации. Популяция
состоит преимущественно из клеток-предшественников, хотя в популяции могут быть также представлены стволовые клетки.
Плюрипотентные стволовые клетки (крайние слева) также могут быть выделены из некоторых тканей и культивированы, од¬
нако их отношения с тканевыми стволовыми клетками еще не ясны. Условия культивирования главным образом, определяют
селекцию пролиферирующих клеток-предшественников данной ткани или индуцируют деление частично дифференцированных
клеток, с последующим их преобразованием до состояния предшественников
3.4. Дифференцировка
65
ного фенотипа. Например, гепатоциты утратили бы
характерные ферменты (аргиназа, аминотрансферазы
и т.д.) и не могли бы накапливать гликоген или секре-
тировать сывороточные белки в результате возвращения
к состоянию клеток-предшественников [Kondo & Raff,
2004]. Деадаптация, в свою очередь, предполагает, что
синтез специфических продуктов или иные аспекты
специализированных функций клеток находятся под
регуляторным контролем гормонов, влияния межкле¬
точного и клеточно-матриксного взаимодействия и т. д.,
и может быть вновь индуцирован наличием соответству¬
ющих условий. Например, присутствие матрикса в виде
плавающего коллагенового плотика [Michalopoulos &
Pitit, 1975], Matrigel [Bissell et all., 1987] или диметил-
сульфоксида (DMSO) [Cable & Isom, 1997] позволяет
сохранить дифференцированные свойства гепатоцитов.
В настоящее время считается доказанным, что при на¬
личии специальных условий культивирования диффе¬
ренцированные фенотипические свойства клеток могут
восстанавливаться (табл. 3.1, см. также разд. 17.5).
Таблица 3.1. Клеточные линии с дифференцированными свойствами
Тип клеток
Происхождение
Клеточная
линия
N*
Вид
Маркер
Ссылка
Эндокринные
Кора надпочечни-
тгпта
Y-1
Т
Мышь
Адреналовые стероиды
Yasamura et al., 1966
Эндокринные
Опухоль гипофиза GH3
Т
Крыса
Гормон роста
Buonassisi et al., 1962
Эндокринные
Гипоталамус
С7
N
Мышь
Нейрофизин;
вазопрессин
De Vitry et al., 1974
Эндотелий
Дерма
HDMEC
Человек
Фактор VIII, CD36
Gupta et al., 1997
Эндотелий
Легочная артерия
СРАЕ
С
Корова
Фактор VIII, ангиотензин
II-конвертирующий ко-
фермент
Del Vecchio & Smith, 1981
Эндотелий
Гепатома
SK/HEP-1
т
Человек
Фактор VIII
Heffelfinger et al., 1992
Эпителий
Простата
РРЕС
N
Человек
PSA
Robertson & Robertson,
1995
Эпителий
Почка
MDCK
С
Собака
Куполообразность, Транс¬
порт
Gaush et al., 1966; Rindler
et al., 1979
Эпителий
Почка
LLC-PKI
С
Свинья
Na+ -зависимый захват глю¬
козы
Hull et al., 1976; Saier, 1984
Эпителий
Молочная железа
MCF-7
т
Человек
Куполообразность,
а-лактальбумин
Soule et al., 1973
Глия
Глиома
MOG-G-CCM
т
Человек
Глютамилсинтетаза
Balmforth et al., 1986
Глия
Глиома
С6
т
Крыса
Фибриллярный кислотный
протеин глии
Benda et al., 1968
Гепатоциты
Гепатома
Н4-11-Е-СЗ
т
Крыса
Тирозин
Pitot et al., 1964
Гепатоциты
Печень
т
Мышь
Аминотрансфераза
Yeoh et al., 1990
Кератиноциты
Эпидермис
HaCat
т
Человек
Кератинизация
Boukamp et al., 1988
Лейкемия
Селезенка
Friend
т
Мышь
Гемоглобин
Scheret al., 1971
Меланоциты
Меланома
В16
т
Мышь
Меланин
Nilos & Makarski, 1978
Миелоид
Лейкемия
К562
т
Человек
Гемоглобин
Andersson et al., 1979 a,b
Миелоид
Миелома
Various
т
Мышь
Иммуноглобулин
Horibata & Harris, 1970
Миелоид
Костный мозг
WEHI-3B D+
т
Мышь
Морфология
Nicola, 1987
Миелоид
Лейкемия
HL60
т
Человек
Фагоцитоз, восстановление
неотетразолия голубого
Olsson & Ologsson, 1981
Миоциты
Скелетные мышцы
С2
L6
С
С
Мышь
Крыса
Мышечные трубочки
Мышечные трубочки
Morgan et al., 1992; Richler
& Yaffe, 1970
Неороэндокрин-
ные клетки
Хромафинная опу¬
холь
РС12
т
Крыса
Катехоламины,
допамин,норэпинефрин
Greene & Tischler, 1976
Нейроны
Нейробластома
С1300
т
Крыса
Нейриты
Lieberman & Sachs, 1978
Пневмоциты
Карцинома легких
А549
т
Человек
Сурфактант
Giard et al., 1972
клеток Клара
II типа
NCI-H441
т
Человек
Сурфактант
Brower et al., 1986
Пневмоциты II Карцинома легких
типа
I
Мышь
Сурфактант
Wilkenheiser et al., 1991
Различные
Эмбриональная те-
ратокарцинома
F9
т
Мышь
Активатор плазминогена, ла¬
минин, коллаген IV типа
Bernstine et al., 1973
* N — нормальные; С — постоянные; I — иммортализованные; Т — трансформированные.
66
ГЛАВА 3. БИОЛОГИЯ КУЛЬТИВИРУЕМЫХ КЛЕТОК
Для того чтобы такая индукция имела место, необ¬
ходимо, чтобы в культуре имелись соответствующие
клетки. Первые попытки восстановления функции в
культурах клеток печени были неудачны, частично
в связи с избыточным ростом фибробластов соеди¬
нительной ткани или эндотелиоцитов синусоидных
кровеносных сосудов. При соблюдении правильной
техники дезагрегации ткани и правильных условий
культивирования [Guguen-Guillozo, 2002] (см. также
протокол 23.6) можно осуществить преимущественную
селекцию гепатоцитов. Таким же образом с использо¬
ванием сливного фидерного слоя [Rheinwald & Green,
1975] или селективной питательной среды [Peehl &
Ham, 1980; Tsao et al., 1982] (см. протокол 23.1) могут
быть выделены и выращены эпидермальные клетки.
Селективная среда используется также для выделения
многих других типов эпителиальных клеток [Freshney,
2002]. Эти и другие примеры [например, селектив¬
ные фидерные слои (см. протоколы 23.1, 23.4, 24.1),
использование D-валина для выделения почечного
эпителия и применение цитотоксических антител (см.
разд. 14.6) ясно доказывают, что селективное культи¬
вирование дифференцированных клеток возможно и
достижимо. Многие селективные среды были созданы,
главным образом, на основании дополненной среды
Хэма F12:DMEM или ее модификаций серии MCDB
(см. разд. 10.2.1) [Cartwright & Shah, 1994; Mather 1998],
и теперь коммерчески доступны (см. Приложение II)
часто вместе со специализированной культурой.
3.5. ПЕРЕДАЧА КЛЕТОЧНЫХ СИГНАЛОВ
Клеточное деление, миграция, дифференцировка
и апоптоз in vivo, как это обсуждалось выше (см.
разд. 3.4.1), регулируются за счет межклеточного и
клеточно-матриксного взаимодействия, гормональ¬
ных сигналов и питательных веществ. Некоторые
сигнальные цепи опосредованы молекулами клеточ¬
ной адгезии (см. разд. 3.2). Однако передача сигнала
может осуществляться также за счет растворимых
факторов, способных к диффузии. Сигнальные мо¬
лекулы, достигающие клеток из соседних тканей по
сосудистой системе, называются эндокринными; те
сигнальные молекулы (сигналы), которые диффун¬
дируют из прилегающих тканей без участия потока
крови, называются паракринными. Следует знать, что
одни и те же сигнальные молекулы синтезируются в
таких же клетках, на которые они оказывают действие.
Позже будет рассказано о гомотипической паракрин-
нойу или гомокринной, передаче сигнала (рис. 3.7).
Сигнальные молекулы, появляющиеся в клеточном
типе, отличном от реагирующих клеток, относятся к ге-
теротипической паракринной системе и в дальнейшем
будут называться просто паракринными. Клетки могут
также продуцировать свои собственные сигнальные
факторы, которые связываются с их собственными
рецепторами и это явление называется аутокринной
сигнальной системой.
Взаимодействие через
щелевые контакты
Рис. 3.7. Клеточное взаимодействие и передача сигнала.
Пути взаимодействия между клетками, (а) Факторы, влия¬
ющие на поведение клеток, включая эндокринные гормоны,
поступающие из сосудистой системы, паракринные факторы
стромы, гомокринные факторы соседних подобных клеток и
аутокринные факторы самой клетки. Матричный, раствори¬
мый и ассоциированный с клетками сульфат гепарина (HS)
может участвовать в активации, стабилизации и перемещении
паракринных факторов. (6) Единообразие ответа тканей-ми-
шеней усиливается взаимодействием через щелевые контак¬
ты, передачу сигнала и, возможно, гомокринными факторами
стимулированных клеток
Хотя in vivo все эти формы сигналов имеют место,
in vitro при нормальных условиях на основной пита¬
тельной среде реализуются только аутокринная и гомо-
кринная передачи сигнала. Неудачи в попытках пере¬
нести на чашку Петри некоторые из клеточных культур
либо низкая эффективность культивирования могут
объясняться, в частности, снижением концентрации
одного или нескольких аутокринных и гомокринных
факторов. Необходимость присутствия сигнальных
молекул является одним из разумных объяснений
Из эндокринных желез через
сосудистую систему
а
Аутокринное
действие
Действие на ту же
самую клетку
Гомокринное
(гомотипическое
паракринное)
действие
Действие на подобные
соседние клетки
Эндокринное действие
(системное действие,
например инсулина,
глюкокортикоидов)
Паракринное действие
Гетеротипическое
взаимодействие между
различными клетками
Гомокринные
диффундирующие
3.7. Получение первичной культуры
67
использования кондиционированных сред (см. про¬
токол 14.2) или фидерных слоев (см. протокол 14.3)
для повышения эффективности посева. Поскольку
интенсивность пролиферации и поддержание специ¬
ализированных клеточных линий, а также индукция
их дифференцировки могут зависеть от паракринных
и эндокринных факторов, эти факторы должны быть
идентифицированы и добавлены в дифференцирую¬
щую питательную среду (см. разд. 17.7.1, 17.7.2). Их
действие может быть достаточно сложным, и, возмож¬
но, для реализации сигнала потребуется синэргическое
действие двух или более факторов [см., например,
McCormick & Freshney, 2000]. В попытке стимулиро¬
вать межклеточное взаимодействие добавлением экзо¬
генных паракринных факторов необходимо принять во
внимание, что фенотип взаимодействующих клеток, и,
следовательно, тип сигнальных факторов, которые они
могут продуцировать, а также временные рамки, в ко¬
торые эти факторы продуцируются, изменяются в ре¬
зультате этого самого межклеточного взаимодействия.
Возможно, гетеротипическая комбинация клеток, по
крайней мере первоначально, является более простым
путем обеспечения набора сигнальных молекул в
правильном матриксном микроокружении, и анализ
этого взаимодействия может затем быть блокирован
добавлением антител либо антисенсорных РНК.
3.6. ЭНЕРГЕТИЧЕСКИЙ МЕТАБОЛИЗМ
Большинство культуральных сред содержит 4-20 мМ
глюкозы, которая, главным образом, используется как
источник углеводов для гликолиза с образованием
молочной кислоты как конечного продукта метабо¬
лизма. При нормальных условиях культивирования
(атмосферный кислород и погруженная в питатель¬
ную среду культура) имеется относительный дефицит
кислорода. В отсутствие соответствующих носителей,
таких как гемоглобин, повышение парциального дав¬
ления кислорода в атмосфере, приводит к образованию
свободных радикалов, токсичных для клеток, поэтому
концентрацию 02 обычно поддерживают на уровне
атмосферной. В результате возникают анаэробные ус¬
ловия, при которых для обеспечения энергетического
метаболизма используется гликолиз. Тем не менее
цикл лимонной кислоты (цикл Кребса) также играет
важную роль. Очевидно, ряд аминокислот, в частности
глутамин, могут быть утилизированы как источник
углевода путем окисления до глутамата при помощи
глутаминазы и поступают в цикл лимонной кислоты
через трансаминирование до 2-оксоглутарата [Reitzer
et al., 1979; Butler & Christie, 1994]. Дезаминирование
глутамина ведет к образованию аммиака, токсичного
для клеток и ограничивающего их рост. Однако при
использовании дипептидов, таких как глутамил-ала-
нин или глутамил-глицин, отмечалось минимальное
образование аммиака и более высокая стабильность
культуры, что является дополнительным преимущест¬
вом (например, Glutamax, Invitrogen).
3.7. ПОЛУЧЕНИЕ ПЕРВИЧНОЙ
КУЛЬТУРЫ
Методы получения первичной культуры подробно
будут описаны ниже (см. гл. 12). Вкратце, первичной
называется культура, полученная за счет активного рос¬
та мигрирующих клеток из фрагмента ткани или путем
ферментативной или механической дезагрегации тка¬
ней. Независимо от применяемого метода, получение
первичной культуры является первым этапом в ряде
селективных процессов (табл. 3.2) и может в конечном
итоге привести к формированию относительно едино¬
образных клеточных линий. В первичном эксплантате
(см. разд. 12.3.1) селекция осуществляется за счет
преимущественной способности клеток к миграции из
фрагмента ткани, в то время как среди диспергирован¬
ных клеток основу для формирования первичной куль¬
туры способны составить только те клетки, которые
способны как к выживанию после дезагрегации, так и
к адгезии к субстрату либо выживанию в суспензион¬
ной культуре. Если первичная культура сохраняется
несколько часов, можно переходить к следующему
шагу селекции. Количество клеток, способных к про¬
лиферации, будет увеличиваться, некоторые типы
клеток могут выживать без деления и увеличения
числа клеток, а прочие могут оказаться неспособными
к выживанию в данных условиях культивирования.
Таблица 3.2. Селекция в развитии клеточной линии
Стадия
Выделение
Первичная культура
Первая субкультура
Поддержание клеточной
линии, пассаж
Старение; трансформация
Факторы, определяющие селекцию
Первичный эксплантат
Ферментативная дезагрегация
Механическое повреждение Ферментативное повреждение
Адгезия эксплантата, избыточный рост и мигра- Клеточная адгезия и распластывание, кле-
ция, клеточная пролиферация точное деление
Чувствительность к трипсину, потребности в питательных веществах, гормональные и суб¬
стратные ограничения, пролиферативная способность
Относительная скорость роста различных клеток, селективный избыточный рост одной линии
дифференцировки. Питательные, гормональные и субстратные ограничения
Нормальные клеточные линии погибают; трансформированные клетки продолжают деление
(избыточный рост)
68
ГЛАВА 3. БИОЛОГИЯ КУЛЬТИВИРУЕМЫХ КЛЕТОК
Следовательно, относительное соотношение клеточных
типов в культуре будет постоянно изменяться. Для
монослойной культуры этот динамический процесс
будет продолжаться до тех пор, пока не будет заполнена
вся площадь субстрата. Следует понять, что первичная
культура, вполне пригодная для проведения цитогене¬
тического анализа и некоторых других целей, может не
походить для других видов исследований в результате
ее нестабильности. Как изменчивость клеточной по¬
пуляции, так и адаптивные модификации отдельных
типов клеток постоянно имеют место в ходе развития
культуры, затрудняя определение периода, в котором
культура может расцениваться как гомогенная или
стабильная.
Состояние, когда все доступные для роста области
заняты и клетки находятся в плотном контакте друг с
другом, называется стадией слитого роста, а культуру
называют конфлюэнтной. В конфлюэнтной культуре
клетки, рост которых чувствителен к контактному
ингибированию, а клеточная плотность ограничивает
клеточную пролиферацию (см. разд. 18.5.2), прекраща¬
ют деление, в то время как любые трасформированные
клетки, нечувствительные к плотности популяции,
будут продолжать избыточный рост. Поддержание
постоянно низкой плотности популяции (например,
частым субкультивированием) помогает сохранить
нормальный фенотип клеток в культуре, имеющей при
повышении плотности клеточной популяции склон¬
ность к спонтанной трансформации и избыточному
росту, например культуры мышиных фибробластов
[Todaro & Green,1963].
Некоторые аспекты специализированных функций
выражены сильнее в первичной культуре, в частности
в конфлюэнтной культуре. На этой стадии культура
обнаруживает наибольшее морфологическое сходство
с родительской тканью и сохраняет некоторое разно¬
образие клеточных типов.
3.8. ЭВОЛЮЦИЯ КЛЕТОЧНЫХ линий
После первого субкультивирования (или пассажа)
(рис. 3.8) первичная культура начинает именоваться
клеточной линией и может поддерживаться, размно¬
жаться и пассироваться несколько раз. При каждом
успешном пассаже составляющая популяции с наи¬
более высокой скоростью пролиферации начинает
доминировать и постепенно вытесняет другие ком¬
поненты популяции, обладающие меньшей пролифе-
ративной активностью. Это особенно заметно после
первого субкультивирования, при котором различия
к возможности пролиферации усиливаются различной
устойчивостью клеток к механическому воздействию
либо трипсинизации, а также переносу в другие условия
культивирования (см. разд. 13.1).
Хотя некоторые селективные и фенотипические
изменения будут продолжаться в ходе субкультивиро¬
вания после третьего пассажа клеточная линия стано¬
вится более стабильной и определяется устойчивыми к
внешним воздействиям, быстро делящимися клетками.
В присутствии сыворотки и без специфических селек¬
тивных условий часто отмечается избыточный рост
мезенхимальных клеток, имеющих в основе своего
происхождения фибробласты соединительной ткани
либо сосудистые элементы. Это явление способство¬
вало появлению очень полезных для некоторых целей
клеточных линий: легочные фибробласты человече¬
ского эмбриона WI-38 [Hayflick and Moorhead, 1961],
почечные фибробласты детенышей хомяков ВНК21
[Macpherson and Stoker, 1962], клетки COS [Gluzman,
1981], клетки CHO [Puck et al., 1958] (см. табл. 13.1),
и наиболее известные L-клетки, мышиные подкожные
фибробласты, обработанные метилхолантреном [Earle
et al., 1943, Sanford et al., 1948]. Однако в целом избы¬
точный рост является одной из сложнейших проблем
культивирования тканей, а именно: как предупре¬
дить избыточный рост при культивировании более
нежных и медленно растущих специализированных
клеток, таких как клетки печеночной паренхимы или
эпидермальные кератиноциты. Чаще всего причину
избыточного роста видят в неадекватности условий
культивирования. В области подбора селективных
сред и субстратов для поддержания многих клеточ¬
ных линий достигнуты значительные успехи (см.
разд. 10.2.1, гл. 23).
3.8.1. Старение
Нормальные клетки могут делиться ограниченное чис¬
ло раз; следовательно, клеточные линии, полученные
из нормальной ткани, погибнут после фиксирован¬
ного количества периодов удвоения популяции. Это
генетически детерминированное событие включает
участие нескольких различных генов и известно как
старение культуры. Считается, что оно определяется,
в частности, неспособностью терминальной последо¬
вательности ДНК в теломерах реплицироваться при
каждом делении клетки. В результате происходит
прогрессирующее укорочение теломеров, и в конце
концов клетка становится неспособной к дальнейшему
делению [Bodnar et al., 1998]. Исключениями из этого
правила являются половые клетки, стволовые клетки
и трансформированные клетки, которые часто экспрес¬
сируют фермент теломеразу, способный к репликации
терминальных последовательностей ДНК в теломере и
удлиняющий время жизни клеточной популяции, иног¬
да бесконечно, — в случае половых клеток и некоторых
опухолевых клеток (см. также разд. 18.4.1).
3.9. ВОЗНИКНОВЕНИЕ ПОСТОЯННЫХ
КЛЕТОЧНЫХ ЛИНИЙ
Некоторые клеточные линии могут давать начало
постоянным клеточным линиям (см. рис. 3.7). Способ¬
ность клеточной линии к постоянному росту, возможно,
отражает ее способность к генетической вариабельное-
3.9. Возникновение постоянных клеточных линий
69
Эксплантат Недели культивирования
Рис. 3.8. Развитие клеточной линии. Вертикальная ось (Y) представляет общий рост числа клеток в гипотетической клеточ¬
ной культуре (предполагается, что при пересадке уменьшения количества клеток не происходит). Общее количество клеток
(клеточный выход) представлен на этой оси в логарифмической шкале. Время культивирования показано на оси X в линей¬
ной шкале. Хотя постоянная клеточная линия изображена развивающейся в течение 14 недель, для различных клеточных
линий это время подъема различно. Соответственно, старение может начаться в разное время, для человеческих диплоидных
фибробластов старение происходит между 30-м и 60-м периодами удвоения количества клеток, или между 10-й и 20-й неде¬
лями, в зависимости от времени удвоения. Используемые термины и определения даны в словаре употребляемых терминов
(по Hayflick & Moorhead [1961].)
ти, позволяющей осуществиться дальнейшей селекции.
Генетическая вариабельность часто связана с делецией
генар55, который в норме останавливает прохождение
клеточного цикла в случае мутации ДНК, а также со
сверхэкспрессией гена теломеразы. Фибробласты
человека остаются преимущественно эуплоидными в
ходе жизненного цикла в культуре и никогда не дают
начало постоянным клеточным линиям [Hayflick and
Moorhead, 1961], в то время как мышиные фибро¬
бласты и клеточные культуры из ряда человеческих
и животных опухолей нередко в культуре становятся
анеуплоидными и дают начало постоянным клеточным
линиям. Возможно, необходимым условием является
наследственная предрасположенность большинства
из этих линий к генетической изменчивости, поэтому
неудивительно, что в них обнаруживается генетическая
нестабильность, сохраняющаяся затем в постоянных
клеточных линиях. Общим свойством многих постоян¬
ных клеточных линий человека является формирова¬
ние субтетраплоидного набора хромосом (рис. 3.9).
Изменение в культуре, которое приводит к развитию
постоянной клеточной линии, обычно называется
трансформацией in vitro (см. гл. 18) и может возникать
спонтанно или быть индуцировано химически или
вирусом (см. разд. 18.2,18.4). Термин трансформация
используется скорее приблизительно и может иметь
различный смысл при употреблении разными людьми.
В этом руководстве иммортализация означает приобре¬
тение клеточной линией способности к возникновению
неопределенного числа клеточных циклов, а трансфор¬
мация подразумевает дополнительные перестройки в
характеристиках роста (независимость от прикрепле-
70
ГЛАВА 3. БИОЛОГИЯ КУЛЬТИВИРУЕМЫХ КЛЕТОК
50- а
40
н
о
0
5
ф
СО
ф 30
Q.
6
“ 20 н
1 ,
S юн
(0
т
и
о
5
Ф
СО
т
ф
Q.
I
т
о
12
10
30 40 50 60 70
Количество хромосом
80
Рис. 3.9. Количество хромосом в клетках конечной и пос¬
тоянной клеточных линий, (а) Клеточная линия нормальных
глиальных клеток человека, (б) Постоянная клеточная линия
метастазирующей меланомы человека
ния к субстрату, утрату контактного ингибирования и
отсутствие чувствительности к плотности популяции),
которые часто, хотя и не всегда, коррелируют с онко-
генностью.
Постоянные клеточные линии обычно анеуплоидны
и часто имеют хромосомный набор между диплоидным
и тетраплоидным (см. рис. 3.9). Имеются также зна¬
чительные вариации в хромосомном наборе и составе
между клетками одной популяции (гетероплоидия)
(см. также разд. 18.3). Еще не ясно, присутствуют
клетки, которые затем дают начало постоянным клеточ¬
ным линиям, исходно в эксплантате в незначительном
количестве, либо они образуются позже как результат
трансформации одной или более клеток. На основании
анализа кинетики клеточного роста второй вариант
кажется более вероятным, поскольку постоянные
клеточные линии могут появляться достаточно позд¬
но в истории жизни клеточной культуры, намного
позже, чем они могли бы развиться при наличии и
избыточном росте предварительно существовавших
в тканях клеток. Однако остается возможность того,
что в тканевых культурах существует субпопуляция,
предрасположенная к трансформации, которая не
смешивается с остальными клетками.
Термин трансформация относится к процессу
формирования постоянной клеточной линии, в част¬
ности, в результате морфологических и кинетических
изменений клеток, но также в связи с тем, что форми¬
рование постоянной клеточной линии часто сопро¬
вождается возрастанием онкогенности. Ряд свойств
постоянных клеточных линий, таких как снижение
потребности в сыворотке, уменьшение ограничения
роста клеточной плотностью, рост на полужидкои
среде, анэуплоидия клеток (см. также табл. 18.1 и цв.
вкладку 14) и другие, связаны с малигнизационной
трансформацией (см. разд. 18.6). Похожие морфо¬
логические и функциональные изменения могут
наблюдаться в клетках, подвергшихся вирусному
или химическому воздействию. Многие нормальные
клетки (если не большинство) не могут давать начало
постоянным клеточным линиям. Классическим при¬
мером является формирование культуры фиброблас¬
тов, которые сохраняют нормальный набор хромосом
в течение жизненного цикла и затем (примерно после
50 поколений) прекращают деление, хотя остаются
жизнеспособными еще до 18 месяцев. Аналогичны¬
ми свойствами обладают глиальные клетки человека
[Ponten & Westermark, 1980] и фибробласты цыпленка
[Hay & Strehler, 1967]. С другой стороны, эпидермаль¬
ные клетки обнаруживают постепенное удлинение
клеточных циклов при усовершенствовании техники
культивирования [Rheinwald & Green, 1977; Green et
al., 1979] и, возможно, способны к формированию по¬
стоянной культуры. Постоянный рост культуры может
иметь отношение к способности тканей к регенерации
и самообновлению in vivo и успешности распростра¬
нения in vitro ее стволовых клеток (см. разд. 3.10).
Возможно образование постоянной культуры лим¬
фобластов при ее трансформации с использованием
вируса Эпштейна-Барра [Gjerset et al., 1990].
3.10. ПРОИСХОЖДЕНИЕ
КУЛЬТИВИРУЕМЫХ КЛЕТОК
Поскольку большинство исследователей работают со
стандартными условиями культивирования конечных
или постоянных пролиферирующих клеточных ли¬
ний, очень важно знать клеточный состав культуры.
Способность экспрессировать различные клеточные
маркеры под влиянием индуцирующих факторов
может означать либо то, что клетки, существующие
в данной культуре, находятся в состоянии зрелости и
требуется лишь индукция для продолжения синтеза
специализированных белков; либо то, что культура
состоит из предшественников или стволовых клеток,
которые способны к пролиферации, но остаются не¬
дифференцированными до тех пор, пока не возникнут
необходимые условия, вследствие чего все клетки или
их некоторая часть станут зрелыми и дифференци¬
руются. Полезно рассматривать клеточную культуру
как систему, находящуюся в состоянии равновесия
между мультипотентными стволовыми клетками,
недифференцированными, но уже детерминирован¬
ными клетками-предшественниками и зрелыми диф¬
ференцированными клетками (см. рис. 3.6), причем
это равновесие может быть сдвинуто в соответствии
с условиями окружения. Стандартный серийный пас¬
саж при относительно низкой клеточной плотности
стимулирует клеточную пролиферацию и сдерживает
дифференцировку, в то время как высокая концент¬
3.10. Происхождение культивируемых клеток
71
рация клеток, недостаток сыворотки и действие соот¬
ветствующих гормонов ингибируют пролиферацию и
стимулируют дифференцировку.
Источник культуры определяет наличие клеточных
компонентов, представленных в культуре. Следова¬
тельно, клеточные линии, полученные из эмбрио¬
нальных тканей, могут содержать большее количество
стволовых клеток и клеток-предшественников, а также
обладать большой способностью к самообновлению,
чем культуры, выделенные из взрослых тканей. Культу¬
ры, которые имеют высокую скорость самообновления
и регенерации in vivo (эпидермис, эпителий кишечника,
гематопоэтические клетки), могут содержать большое
число стволовых клеток, которые при соответствующих
условиях будут иметь продолжительное время жизни.
В то же время в культурах, которые обновляются ис¬
ключительно под действием стресса (фибробласты,
мышечные и глиальные клетки), могут обнаруживаться
только детерминированные клетки-предшественники
с ограниченным временем жизни.
Таким образом, идентификация культуры клеток
проводится не только по источнику in vivo (гематопо¬
этические или глиальные клетки, гепатоциты и т. п.),
но также по их положению в линии дифференциров¬
ки (стволовые клетки, предшественники или зрелые
дифференцированные клетки). Хотя предполагается,
что развитие по пути дифференцировки является
необратимым, данная концепция детерминации в
настоящее время подвергается сомнению [Kondo &
Raff, 2004; LeDouarin et al., 2004]. Возможно, некото¬
рые клетки-предшественники могут быть способны к
конверсии и реверсии до состояния стволовых клеток
и дедифференцироваться по той же или иной линии
дифференцировки.
Клеткам, происходящим из опухолевых, не требу¬
ется адгезия для пролиферации и дифференцировки.
Таким образом, клетки крысиной гепатомы могут
пролиферировать in vitro, обладая некоторыми харак¬
терными особенностями дифференцировки, однако,
чем ближе их фенотип к нормальному, тем в большей
степени индукция дифференцировки может ингибиро¬
вать пролиферацию. Отношение между положением в
линии дифференцировки и клеточной пролиферацией
может ослабляться: клетки меланомы В16 способны к
продукции большего количества пигмента при более
высокой плотности клеток и низкой скорости проли¬
ферации по сравнению с низкой плотностью и высоким
уровнем пролиферации. Несмотря на это, возможность
перехода между линиями дифференцировки еще не
установлена (см. разд. 17.4).
Глава 4
СТРУКТУРА И ПЛАНИРОВАНИЕ
ЛАБОРАТОРНЫХ ПОМЕЩЕНИЙ
4.1. ПЛАНИРОВАНИЕ
Главным требованием, которое характеризует культи¬
вирование клеток в отличие от других лабораторных
методов, является необходимость поддержания асепти¬
ческих условий. Хотя обычно экономически невыгодно
создание и поддержание большой асептической зоны,
важно, чтобы все помещения лаборатории, в которой
проводятся работы с культурами тканей, были чистыми
и не имели сквозного прохода. Появление и внедрение
ламинарных боксов (шкафов) значительно облегчило
решение этой проблемы и позволило работать с культу¬
рами клеток в неспециализированных лабораториях при
условии, что размещение оборудования соответствует
имеющимся требованиям (см. разд. 4.3.2,5.2.6,6.4).
При планировании новых помещений и приоб¬
ретении оборудования следует учитывать несколько
моментов. Будет ли строиться новое здание или уже
имеющееся помещение будет переделываться в соот¬
ветствии с новыми требованиями? В последнем случае
вы будете ограничены структурой уже имеющихся
помещений, переделывание воздуховодов и венти¬
ляционных систем может оказаться очень дорогим,
изменение структуры помещений может быть связано
с удалением несущей нагрузку стены и стать дорого¬
стоящим и сложным мероприятием. При строитель¬
стве нового здания имеется больше возможностей для
осуществления необходимой планировки и разработки
структуры лабораторных помещений, оборудование
может быть размещено более эргономично и энерго-
сберегающе, чем при необходимости распределять его
в соответствии со структурой помещений. Необходимо
учитывать следующие параметры:
1) Вентиляция
а) Равновесие давления. В идеале культуральные по¬
мещения лаборатории должны иметь повышенное
атмосферное давление относительно других зон, для
того чтобы избежать заноса зараженного воздуха
извне. Однако использование материала, выделен¬
ного из человеческих тканей, требует соблюдения
наиболее строгих требований безопасности, которые
относят лаборатории, в которых проводятся работы
по культуре тканей, к категории I (см. разд. 7.8.1).
Соответствующие требования связаны с необходи¬
мостью создания отрицательного давления в лабо¬
раторных помещениях относительно других зон.
Для того чтобы удовлетворить обоим требованиям,
предпочтительным является создание буферной
зоны с положительным давлением вне культураль¬
ных помещений, таких как подготовительная зона
или микроскопическая комната (см. рис. 4.3) или
коридор (см. рис. 4.4).
б) Ламинарные шкафы. Внимательно отнеситесь к раз¬
мещению впускных и выходных систем воздуховода.
Предпочтительно располагать ламинарный шкаф
так, чтобы поток выходящего воздуха направлялся
наружу, обеспечивая лучшую циркуляцию и уда¬
ление тепла из помещения (300-500 Вт на каждый
ламинарный шкаф). Такое расположение облегчит
проведение обеззараживания формальдегидом, если
это потребуется. Вентиляционные колпаки, обра¬
щенные во внешнюю сторону, смогут обеспечить
наилучший поток воздуха, требуемый для работы, и
необходимо лишь удостовериться, что приходящий
воздух из центральной вентиляционной системы
или локальных кондиционеров воздуха не смеши¬
вается с чистым потоком воздуха из-под колпака
ламинарный шкафа. Предпочтительно оставлять
ламинарный шкаф постоянно работающим; в слу¬
чае, если его включают только при работе, следует
выждать некоторое время для прохождения воздуха
через вентиляционный колпак.
2) Размещение
а) Количество оборудования. Количество оборудо¬
вания в помещениях культуральной лаборатории
зависит от количества работающих людей, от того,
сколько времени они будут работать еженедельно
и какой тип культуры они будут вести? С учетом
этих вопросов следует решать, сколько ламинарных
шкафов потребуется (с учетом того, смогут ли ра¬
ботники использовать бокс совместно или каждый
из них занимает ламинарный шкаф в течение всего
рабочего дня), и необходимо ли обеспечить большое
пространство для манипуляций с биореактором,
работой с животными тканями и большим количест¬
вом различных линий клеток.
Ориентировочно, при работе лаборатории, состо¬
ящей из 50 человек, выполняющих различную
работу, требуется 12 ламинарных шкафов (при их
совместном использовании).
б) Пространство. Сколько места требуется для обору¬
дования и всей лаборатории?
4.1. Планирование
73
Самая большая площадь выделяется для манипу¬
ляций с культурами, где должны быть размещены
ламинарные боксы, счетчики клеток, центрифуги,
инкубаторы, микроскопы и некоторые основные
реагенты, среды, стеклянная и пластиковая посу¬
да. Вторую по величине площадь следует отдать
помещению для мытья, препаративных работ и
стерилизации. Третью — комнате для хранения и
четвертую — для инкубации. Приблизительное со¬
отношение площади в этих помещениях составляет
соответственно 4:2:1:1.
в) Асептическая зона. Если при работе сотрудников
лаборатории будет необходимость постоянной
работы в зоне содержания животных, следует
обеспечить удобный доступ как к помещениям для
культивирования тканей, так и для комнат, в кото¬
рых осуществляются манипуляции с животными,
однако так, чтобы при этом помещения не соприка¬
сались. Окна в помещениях для культивирования
могут причинять неудобства, приводя к колебаниям
температуры в помещении, становясь причиной
ультрафиолетовой денатурации сред и инфици¬
рованию микроорганизмами, если помещения не
обеззараживаются соответствующим образом.
г) Ламинарные шкафы. Пространство между лами¬
нарными шкафами должно составлять не менее
500 мм для обеспечения свободного доступа к ним
и снижения до минимума возможности интерфе¬
ренции воздушных потоков. В это пространство
можно поместить передвижную тележку или сто¬
лик на колесиках, на которых могут располагаться
бутыли, флаконы, реактивы, а также лабораторные
журналы.
д) Инкубирование. В зависимости от того, какой тип
инкубирования используется в работе, выбираются
размеры, температура, газовая фаза и расстояние
от места расположения инкубатора до рабочего
места. Используется ли обычное инкубирование в
термостате или термальной комнате без специаль¬
ной газовой среды либо требуется С02-атмосфера
с повышенным уровнем влажности? Обычно боль¬
шое количество флаконов или флаконы большого
объема после герметизации наилучшим образом
инкубируются в термальной комнате, в то время как
открытые чашки и планшеты лучше хранить в С02-
инкубаторе с повышенным уровнем влажности.
е) Подготовительная зона. Оборудование для мытья и
стерилизации должны быть расположены: 1) близко
к асептической зоне, которую они обслуживают, и
2) вплотную к стене, выходящей наружу, для того
чтобы обеспечить лучшее удаление тепла из печей
и потоков пара из паровых вентилей автоклавов.
Обеспечьте хороший обзор и приемлемое простран¬
ство, приятное для взгляда персоналу, работающему
на стерилизационном, моющем и подготовительном
оборудовании. Обычно они выполняют свою работу
периодически, в то время как научные сотрудники
постоянно смотрят в ламинарный шкаф и не нуж¬
даются в приятной обстановке.
ж) Обслуживание асептической зоны. Выясните, бу¬
дет ли нужен лифт или достаточно эстакады или
пандуса? В последнем случае важно знать, каков
необходимый угол наклона и максимальный груз,
который можно перевозить вверх по наклону без
использования механических устройств?
з) Хранение. Каковы масштабы предполагаемого
хранения, и каких размеров пространство может по¬
требоваться для хранения одноразовой пластиковой
посуды? Каков предполагаемый объем работ с кле¬
точными линиями, обусловливающий требования
к помещению для сосудов с жидким азотом?
3) Реконструирование. Если предполагается пере¬
оборудование существовавших прежде помещений,
учитывайте определенные структурные ограни¬
чения и тщательно выбирайте местоположение,
избегая тесноты и трудновыполнимых проектов
использования комнат, в которых маневренность
ограничена.
4) Доступ. Удостоверьтесь, что обе створки дверей
открываются достаточно широко, что высота дверей
и потолков достаточна для обеспечения установки
такого оборудования, как инкубаторы, автоклавы.
При этом помните, что ламинарный шкаф требует
дополнительного пространства над верхней поверх¬
ностью для установки воздуховода. Удостоверьтесь,
что дверной проход и пространство между оборудо¬
ванием обеспечивает доступ для его технического
обслуживания.
5) Карантин. Какой карантин требуется для вашей
лаборатории? Вновь поступившие клеточные линии
и биопсийные образцы должны быть исследованы на
предмет контаминации микоплазмами до того, как
они будут переданы в те комнаты, где проводятся
работы с основными линиями. Некоторые линии
клеток человека и приматов, а также клеточные
линии, потенциально представляющие собой
биологическую опасность, требуют изоляции (см.
разд. 7.8.1).
Изучение этих вопросов позволит вам решить, каких
размеров помещения вам требуются. Каков предполага¬
емый тип устройства лаборатории: одна или две малень¬
ких комнаты (рис. 4.1, 4.2) либо комплекс помещений,
включающий моечную, стерилизационную, одну или две
асептических зоны, инкубационную комнату, темную
комнату для флюоресцентной микроскопии и микрофо¬
тографии, холодильную комнату и хранилище (рис. 4.3)?
При сравнении с традиционно расположенными в ряд
комнатами и единственным ламинарный шкафом в изо¬
лированной комнате лучшим вариантом является одно
специально спланированное для культуральных работ
помещение со смежной подготовительной зоной и ря¬
дом меньших комнат с общей подготовительной зоной.
Отдельное помещение обеспечивает лучшую защиту
от заражения, позволяет хранить клеточные ростковые
линии отдельно от реактивов и стеклянной посуды и в
любом случае потребуется при работе с клетками чело¬
века или приматов (см. разд. 7.8.1).
74
ГЛАВА 4. СТРУКТУРА И ПЛАНИРОВАНИЕ ЛАБОРАТОРНЫХ ПОМЕЩЕНИЙ
Обычный инкубатор
Влажный С02-инкубатор
Подводка С02
Холодильник
Морозильник “
Рабочий стол с
полками над ним
Баллоны с жидким
С02 с автоматическим
реле переключения
Главный вход: двойные^
асимметричные двери
1 м
Поступление воздуха или
расположение кондиционера воздуха
Инвертированный
микроскоп
Центри¬
фуга
Стол для
.подгото¬
вительных
работ
Водяная В
"баня
Подводка
деионизированной
воды
Ламинарный шкаф (II класса
биологической защиты) с
полной очисткой воздуха
Сервисная тележка
-Стеллаж для хранения
- Стерилизационная печь
Автоклав
Удаление воздуха
Двухуровневая сушильная печь
(с высоким и низким уровнями
температуры)
Раковины для замачивания Раковины для мытья
Рис. 4.1. Небольшая лаборатория культивирования тканей. Возможная планировка простой автономной лаборатории куль¬
тивирования тканей для 2-3 сотрудников. Темным цветом выделены области, занимаемые перемещаемым оборудованием.
Области светлого цвета - закрепленная или перемещаемая мебель. Масштаб 1:100
Двухуровневый
обычный инкубатор
Двухуровневый влажный
С02-инкубатор
Подводка С02
Баллоны с жидким С02с
автоматическим реле
переключения
Главный вход: двойные
асимметричные двери
Сервисная тележка
1 м
Рис. 4.2. Лаборатория культивирования тканей средних размеров. Планировка лаборатории для 5-6 сотрудников, с моечной
и подготовительной зоной, расположенными в другом помещении. Темным цветом выделены области, занимаемые перемеща¬
емым оборудованием. Области светлого цвета - закрепленная или перемещаемая мебель. Масштаб 1:100
Сервисная тележка
Полки для реактивов
(стерильные жидкости)
Поступление воздуха
или расположение
кондиционера воздуха
Холодильник
Морозильник
Центрифуга
Ламинарный шкаф (II класса биологической
защиты) с полной очисткой воздуха
Инвертированный Счетчик
микроскоп клеток
В°Дбанй |"® | \ Of Раковина
Стеллаж
для
хранения
Сборный Раковины для
лоток Раковина замачивания
Удаление воздуха
4.1. Планирование
75
Настольный
ПРЕПАРАТИВНАЯ
ЗОНА
Сервисная^
тележка
.Аппарат для
очистки воды
клеток
-Раковины
- для мытья
Стеллаж для
хранения"
Хранение
азотом
Тележка
I для сбора
грязной
посуды
Солодильни к
Морозильник
1ЕРМОСТЛ-
1ИРУЕМАЯ
<ОМНАТ/>
НА 37 °С
Стеллаж для
автоклав Стерилизационный и сушильный шкаф
I
Ламинарный шкаф биологической безопасности
(ламинар) с полной очисткой воздуха
Полки для реактивов
(стерильные жидкости)
Счетчик
Инвертированный
микроскоп
Двухуровне¬
вый инку¬
батор
Поступление
воздуха или распо<-
ложение кондицио¬
нера воздуха
Раковина -
баллонов
с жидким
-о
Баллоны с жид¬
ким С02с автос
матическим реле
переключения
С02
инкубаторы
хранения
Тележка для
сбора грязной
посуды I
Раковина Центрифуга
Холодильник
Морозильник
Подводка
со;
Раковины для Сборный
замачивания лоток
МИКРОСКО¬
ПИЧЕСКАЯ
КОМНАТА
Главный вход: двойные
асимметричные двери
Морозильная Подводка
Удаление установка С02
воздуха
Рис. 4.3. Лаборатория культивирования тканей с прилежащими препаративными комнатами. Возможная планировка лабора¬
тории культивирования тканей средних размеров (см. рис. 4.2), но с прилежащими подготовительной зоной, микроскопической
комнатой и термостатируемой комнатой на 37 °С. Масштаб 1:100
Воздухозаборник для
ламинарный шкафов
Загрязненные
предметы Инкубаторы
*
АСЕПТИЧЕСКАЯ ЗОНА
Счетчик клеток Инв. микр. Центрифуга
Мусорный
контейнер
N
МИКРО¬
СКОПИ¬
ЧЕСКАЯ
КОМНАТА
Удаление воздуха
Рис. 4.4. Большая лаборатория культивирования тканей. Предлагаемая планировка подходит для 20-30 сотрудников. Рядом
расположены моечная, стерилизационная и препаративная зоны 37 °С. Темным цветом выделены области, занимаемые обору¬
дованием. Области светлого цвета - закрепленная или перемещаемая мебель. Масштаб 1:200
76
ГЛАВА 4. СТРУКТУРА И ПЛАНИРОВАНИЕ ЛАБОРАТОРНЫХ ПОМЕЩЕНИЙ
4.2. ОБСЛУЖИВАЮЩИЕ СИСТЕМЫ
И ВСПОМОГАТЕЛЬНЫЕ СЛУЖБЫ
Комнаты должны быть оборудованы системой фильт¬
рации воздуха. Если лаборатория культивирования
тканей будет относиться к I категории безопасности
(см. разд. 7.8.1), следует обеспечить отрицательное
относительно других помещений давление воздуха.
В этом случае смежные помещения, которые ведут в
асептическую зону, необходимо рассматривать как бу¬
ферную зону и также установить систему фильтрации
воздуха, но с положительным давлением.
Оборудование в комнатах должно располагаться так,
чтобы не затруднять уборку помещений. Мебель долж¬
на прочно прикрепляться к полу или располагаться на
подставке или на ножках с тем, чтобы пространство
под ней было достаточным для уборки. Пол должен
иметь виниловое или иное пылезащитное покрытие, а
также небольшое понижение уровня по направлению
к стоку в полу, расположенному за дверью комнаты
(за пределами стерильной зоны). Это обеспечит более
свободное обращение с водой при мытье полов и, что
более важно, защитит оборудование от повреждения
при переполнении раковины или автоклава.
При возможности желательно отделить комна¬
ты, непосредственно предназначенные для работы
с культурами тканей, от препараторских, моечных
и стерилизационных помещений (см. рис. 4.3, 4.4).
Следует обеспечить необходимый водоток из препа¬
ративной/моечной зоны с небольшим наклоном пола
со стороны культуральной зоны по направлению
к моечной. Если у вас есть большая лаборатория с
отдельными помещениями для моечной и стерилиза¬
ционной, удобно разместить их на том же уровне пола,
чтобы не создавалось неудобства при перемещении
тяжестей при помощи тележки. Размещение напротив
через коридор является идеальным (см. рис. 4.4, см.
также разд. 4.3).
Постарайтесь представить себе потоки движения
людей, реактивов, тележек и т. д. и расположите обору¬
дование так, чтобы обеспечить минимум конфликтов,
легкий и близкий доступ к хранилищу, хороший доступ
к пополняемым запасам без нарушения стерильности в
асептической зоне, а также легкое удалении и уничто¬
жение испорченных и загрязненных объектов.
Требуемое обслуживание включает подключение
к источникам питания, горючим газам (бытовой ме¬
тан, пропан и т.д.), сжатому воздуху и С02и создание
вакуума. Обычно электрическое питание обеспечива¬
ется недостаточно — как в количестве электрических
розеток, так и в напряжении в сети. Определите коли¬
чество оборудования, которое должно обеспечивать¬
ся бесперебойным электроснабжением; учтите, что
число электроприборов и энергопотребление, которое
потребуется в будущем, втрое увеличится и поста¬
райтесь обеспечить необходимое электроснабжение.
Предпочтительно располагать каждый электрический
прибор рядом с розеткой или, по крайней мере, рядом
с главным распределительным щитом.
Гораздо сложнее оценить потребности в газе, по¬
скольку в данном случае необходима информация о
конкретных возможностях газоснабжения в вашем
регионе. Электричество представляет собой более чис¬
тый и легкоуправляемый источник энергии, однако газ
может быть дешевле и надежнее. Обычно потребность
в горючем газе определяют местные условия.
Если есть возможность, лучше обеспечить подводку
С02 в лабораторные помещения по трубам. Установка
такой подводки надежнее обеспечит инкубаторы и
окупит себя за счет стоимости баллонов с углекислотой
для С02-инкубируемых культур (см. разд. 5.3.2). Для
обеспечения необходимой газовой смеси (см. Прило¬
жение II) на рабочих местах могут быть установлены
счетчики газа и краны-смесители.
Сжатый воздух обычно не требуется, поскольку в
С02-инкубаторах состав газовой смеси регулируется
за счет поступления чистого С02. Однако при необ¬
ходимости должна иметься возможность получения
смеси газов на каждом рабочем месте. Сжатый воздух
также используется для удаления ватных пробок из
стеклянных пипеток до их мытья и может потребовать¬
ся для некоторых видов моечных машин (например,
Scientek 3000).
Вакуумная установка очень полезна для опорожне¬
ния культуральных флаконов, при этом для успешной
работы следует соблюдать некоторые меры предосто¬
рожности. Коллекторные сосуды должны быть обору¬
дованы промежуточными сосудами с гидрофобными
фильтрами для обеспечения защиты от попадания
жидкостей, паров или других загрязнений в систему.
Вакуумный насос также должен быть защищен от
небрежного оставления во включенном состоянии.
Обычно это достигается использованием ножной пе¬
дали, которая включает систему вакуумного отсоса и
выключается при убирании ноги с педали. Во многих
отношениях, лучше обеспечить каждое рабочее место
индивидуальным перистальтическим насосом (см.
рис. 5.1 -5.3) или установить один насос на два рабочих
места (например, Integra Vacusafe, см. рис. 5.3).
4.3. ПЛАНИРОВАНИЕ АСЕПТИЧЕСКИХ
КОМНАТ ИЛИ БЛОКОВ
В лаборатории необходимо обеспечить проведение
шести необходимых процессов: стерильных манипуля¬
ций, инкубирования, препаративных работ, мытья, сте¬
рилизации и хранения (табл. 4.1). Если используется
одна комната, то следует создать «градиент стерильнос¬
ти»; чистая зона для стерильных манипуляций должна
быть расположена на одном конце комнаты, наиболее
удаленном от дверей, а оборудование для мытья и сте¬
рилизации должно быть расположено на другом конце,
отделенное подготовительной зоной, инкубационной
зоной и хранилищем. Подготовительная зона должна
находиться рядом с моечной и стерилизационной зона¬
ми, хранилище и инкубаторы должны быть доступны
из стерильной зоны (см. рис. 4.1-4.4).
4.3. Планирование асептических комнат или блоков
77
Таблица 4.1. Помещения культуральной лаборатории
Минимальные требования
Желательное оборудование
Рекомендуемое дополнительное
оборудование
Стерильная зона, чистота, тишина, минимум пе¬
ремещения людей, нет сквозного прохода
Отдельно от помещений, в которых содержатся
животные или ведутся микробиологические
работы
Подготовительная зона
Моечная зона (не обязательно на площади куль¬
туральной лаборатории, но, по крайней мере,
по соседству с ней)
Пространство для инкубаторов
Хранилище:
Жидкости: атмосферный воздух, 4 °С, -20 °С
Стеклянная посуда (полки)
Одноразовый пластик (полки)
Небольшие предметы (ящики)
Специальное оборудование (шкафы с плавным
поворотом, стенные шкафы)
Химические реактивы (атмосферный воздух,
4 °С, -20 °С, могут располагаться рядом с
жидкостями при соблюдении условия хра¬
нения в герметичных сосудах, предупрежда¬
ющих обезвоживание)
Баллоны С02
Хранилище для жидкого азота
Морозильное оборудование
Водосток
Фильтрованный воздух (конди¬
ционер)
Система обслуживания по сосед¬
ству с культуральной зоной
Отдельная препараторская ком¬
ната
Горячая комната с регистрирующим
оборудованием
Отдельная стерилизационная
комната
Отдельное хранилище для
баллонов
Подводимый по трубам С02
Сжатый воздух
Хранилище для запасов пластика
Карантинное помещение
Изолятор (может использоваться как
карантинное помещение)
Хранилище N2 (емкость около 500 л)
и отдельное хранилище для моро¬
зильного оборудования
Микроскопическая комната
Темная комната
Вакуумная система
4.3.1. Помещение для стерильных
манипуляций
Стерильные процедуры следует проводить в спокой¬
ной части культуральной лаборатории, специально
выделенной для этих целей (не следует, например,
здесь также работать с химическими реактивами или
с другими организмами, например бактериями либо
дрожжами). Через культуральную зону не должно быть
сквозного прохода или каких-либо иных действий,
связанных с запылением или движением воздуха. Если
нет возможности использовать ламинарный шкаф,
используйте отдельную комнату или отгороженное
пространство для стерильных манипуляций. Рабочая
зона в самом простом виде представляет собой по¬
верхность, покрытую ламинатом, преимущественно
белую или нейтрально-серую для обеспечения лучшего
исследования культур, проведения анатомирования и
т. д. Такое покрытие позволяет также точнее определить
pH с помощью фенолового красного в качестве индика¬
тора. Не следует ничего хранить на поверхности этого
стола, и все полки над ним должны использоваться
только для предметов, связанных со стерильной рабо¬
той (например, для хранения стерильных пипеток и
инструментов). Рабочий стол также должен свободно
стоять (отстоять от стены) либо закрепляться у стены
уплотнительной полосой или замазкой
4.3.2. Ламинарное оборудование
Появление и введение в практику ламинарный шка¬
фов и боксов со стерильным воздухом, поступающим
на рабочую поверхность (см. разд. 5.2.1 и рис. 5.1, 6.2)
обеспечивают больший контроль стерильности при
более низкой себестоимости, чем оборудование и
поддержание стерильной комнаты. Индивидуальные
отдельно стоящие ламинарные боксы — наиболее пред¬
почтительны, поскольку они разделяют операторов и
их удобно обойти, однако можно использовать стенные
и потолочные ламинарные колпаки, объединенные в
батареи. При индивидуальных рабочих местах в сте¬
рильную область попадают только руки оператора, в то
время как во втором случае отсутствуют боксы и опера¬
торы являются частью рабочего пространства. При этом
имеется большая свобода передвижения, в том числе
с большими передвижными аппаратами (такими как
роллерные тележки с бутылями, биореакторы и т.д.),
требуется большее усилие со стороны операторов для
того, чтобы не разрушать ламинарного потока. Кроме
того, необходимо носить стерильные шапочки и накид¬
ки, чтобы избежать заражения культур.
Выбрав ламинарное оборудование, соответствующее
вашим задачам и возможностям, — свободностоящее
или настольное, — обеспечьте достаточное место для
помещения ног под ним вместе с насосами, аспиратора¬
78
ГЛАВА 4. СТРУКТУРА И ПЛАНИРОВАНИЕ ЛАБОРАТОРНЫХ ПОМЕЩЕНИЙ
ми и т. д. Свободностоящие боксы должны располагать¬
ся на блокируемых роликах, так чтобы их можно было
перемещать при необходимости. Стулья должны быть
подходящего веса, с изменяемыми высотой сиденья и
углом наклона спинки. Их конфигурация должна поз¬
волять оператору придвинуться вплотную к переднему
краю ламинарного стола для удобной работы в боксе.
Небольшая тележка, либо перемещаемый столик или
складной откидной столик (не больше 300-500 мм)
должен находиться рядом с каждым ламинарным бок¬
сом для материалов, которые могут потребоваться, но
постоянно не используются.
Расстояние между рядом стоящими боксами долж¬
но быть не менее 500 мм, а при противоположном
стоянии — 2000 мм между передними поверхностями.
Ламинарные боксы следует рассматривать как часть
конструкции здания, поскольку они будут оказывать
влияние на систему вентиляции.
4.3.3. Карантин и изоляция
Если позволяет структура помещения лаборатории,
очень ценным будет выделить отдельную комнату для
карантина и/или изоляции (см. рис. 4.4). Это отде¬
льная асептическая комната с отдельным ламинарным
колпаком (микробиологический кабинет II класса
безопасности), инкубаторами, морозильником, холо¬
дильником, центрифугами, электрической подводкой
и другими устройствами. Такая комната должна быть
отделена от остальных помещений дверью или воздуш¬
ной шлюзовой камерой и иметь отрицательное давле¬
ние относительно остальной асептической зоны. Все
манипуляции с вновь доставленными в лабораторию
клеточными линиями или биопсийным материалом
проводятся только в этом помещении, пока не будет ус¬
тановлено, что они свободны от заражения, в частности,
микоплазмами (см. разд. 19.3.2 и протоколы 19.2,19.3),
а также другими возбудителями, такими как HIV или
вирус гепатита В. Если позволяют существующие пра¬
вила внутреннего распорядка, эта же комната в другое
время может быть использована как изолятор II уровня
защиты. При использовании для более высокого уровня
защиты потребуется соответствующий кабинет, обору¬
дованный в соответствии с требованиями биологичес¬
кой опасности, либо ламинарный шкаф с отдельным
воздуховодом для выведения воздуха, оборудованным
ловушкой для возбудителей (см. разд. 7.8.2).
4.3.4. Помещения для сервисных служб
Очень удобно иметь группу помещений, в которых
будут располагаться счетчик клеток, микроскопы и т.д.
поблизости от зоны стерильных манипуляций, а так¬
же разделяющих пространство или отделяющих одну
часть комнаты от другой (см. рис. 4.1-4.4). Сервисные
помещения также включают в себя хранилища для
стерильной стеклянной посуды, одноразового пластика,
пипеток, колпачков и крышек, шприцов и т. д. в закры¬
тых ящиках внизу и открытых полках над ними. Эти
помещения могут также использоваться для другого
дополнительного оборудования, такого как маленькие
центрифуги, и следует обеспечить доступность его
использования при необходимости.
4.4. ИНКУБАЦИЯ
Требования к чистоте в зоне инкубации не такие стро¬
гие, как для зоны, в которой проводятся стерильные
манипуляции, но чистый воздух, низкий уровень фи¬
зических помех и минимальные перемещения в этой
зоне обеспечат более успешное избегание загрязнения
пылью и спорами, кроме того, здесь меньше потоки
воздуха, способные переносить их.
4.4.1. Инкубаторы
Инкубацию можно проводить в отдельных инкуба¬
торах или в термостатируемой (термальной) комнате
(рис. 4.5). Если требуется только один или два инкуба¬
тора, это экономически оправданно и выгодно с точки
зрения занимаемой площади; однако как только вам
понадобится более чем два инкубатора, их общая стои¬
мость начинает превышать стоимость оборудования
термальной комнаты, а их использование менее удобно.
Кроме того, инкубаторы теряют большое количество
тепла при открывании и медленнее восстанавливают
температуру, чем термальная комната. По приблизи¬
тельной оценке, на каждого работника потребуется
0,2 м3 (200 л) инкубируемого пространства при площа¬
ди полки 0,5 м2. Может понадобиться дополнительное
пространство для одного или более инкубаторов с конт¬
ролируемым уровнем С02 и повышенной влажностью
в атмосфере.
4.4.2. Термальная комната
Если у вас есть площадь в лабораторной зоне или
имеется смежная комната либо оборудованная и при¬
способленная для хранения холодильная камера, их
можно переоборудовать в термальную комнату (см.
рис. 4.5). Эта зона не должна иметь каких-то особых
конструктивных приспособлений, кроме теплоизоля¬
ции, предотвращающей появление холодных участков
на стенах. Для теплоизоляции поверхности обшивают
пластиковыми панелями, покрытыми ламинатом, от¬
деленными от стен слоем стекловолокна, стекловаты
или огнеупорной пеной толщиной слоя около 5 см.
Отметьте расположение крепежных элементов пане¬
лей, с тем чтобы укрепить здесь стеллажи, если они
будут использоваться. Используйте разборные полки.
Пространство между полками должно иметь высоту не
более 500-600 мм, чтобы опоры не позволяли полкам
прогибаться. Предпочтительно использование пере¬
4.4. Инкубация
79
движных полок, поскольку они позволяют произво¬
дить уборку стеллажей и комнаты. Расстояние между
полками должно составлять 200-300 мм, используйте
более широкие полки (450 мм) внизу и более узкие
(250-300 мм) выше уровня глаз. Перфорированные
полки устанавливаются на кронштейны, регулируемые
по высоте крепления. Полки должны быть плоскими и
абсолютно горизонтальными, что исключает падение.
Не стоит недооценивать пространство, которое
вам потребуется в течение всего времени существо¬
вания термальной комнаты. Оборудование несколько
большей комнаты незначительно повышает стоимость
работ. Подсчитайте стоимость работы на основе коли¬
чества полок, которое вам требуется, если вы только
начинаете работу, умножьте это количество на 5 или
10; если вы уже работаете в течение какого-то времени,
умножьте на 2 или 4.
Следует, насколько возможно, избегать деревянной
мебели, поскольку дерево может коробиться при дей¬
ствии тепла и быть источником вредителей.
В определенной части термальной комнаты должны
быть оборудованы небольшие столы и полки пред¬
почтительно из нержавеющей стали или твердого
пластика. Приспособьте эти столы для установки
инвертированного микроскопа, обследуемых фла¬
конов и тетради. Если вы предполагаете выполнять
эксперименты по синхронизации клеток или некие
стерильные манипуляции при 37 °С, также следует
обеспечить пространство для небольшого вытяжного
колпака с размерами фильтра 300x300 или 450x450 мм,
прикрепленного к стене или к подставке, расположен¬
ной выше рабочей поверхности. В этой же комнате,
помимо того, может располагаться небольшой лами¬
нарный шкаф (ширина не более 1000 мм). Двигатель
вентилятора должен быть специально приспособлен
для работы в тропических условиях и не должен по¬
стоянно работать. При постоянной работе он будет
производить тепло, и в условиях термальной комнаты
мотор перегреется и перегорит.
Как только будет оборудована термальная комната,
сотрудники других лабораторий захотят использовать
ее пространство для целей, не относящихся к куль¬
тивированию тканей, поэтому пространство рабочих
поверхностей и полок следует рассчитывать так,
чтобы обеспечить возможность инкубации пробирок,
установки шейкеров и т.д. Однако категорически
препятствуйте использованию термальной комнаты
для работы с микроорганизмами, такими как бактерии
или дрожжи.
Следует отдавать предпочтение лампам нака¬
ливания, поскольку флюоресцентные лампы могут
вызывать разрушение среды. Кроме того, некоторые
флюоресцентные трубки включаются в условиях тер¬
мальной комнаты.
Температуру в горячей комнате контролируют с
точностью до ±0,5 °С в любой момент времени, что
зависит от точности и чувствительности контроль¬
ной аппаратуры, расположения сенсоров термостата,
циркуляции воздуха в комнате, типа теплоизоляции
и выделения тепла при работе другого оборудования,
находящегося в комнате.
Нагреватели. Обогрев наилучшим образом обеспе¬
чивается путем использования теплодувных вентиля¬
торов бытового или промышленного предназначения,
в зависимости от объемов комнаты. На 20 м3 объема
комнаты потребуется нагреватель приблизительно
2-3 кВт мощности (можно использовать два нагрева¬
теля каждый мощностью 1,0-1,5 кВт), в зависимости
от типа изоляции. Вентиляторы при нагреве должны
работать постоянно, и мощность нагревательного
элемента должна регулироваться контролирующими
аппаратами.
Циркуляция воздуха. Второй вентилятор, рас¬
положенный в противоположной стороне комнаты,
должен осуществлять движение воздушного потока,
обеспечивая максимальную циркуляцию. Если комната
по размерам превышает 2x2 м, может потребоваться
специальное оборудование для распределения потоков
воздуха. Ограждение углов (см. рис. 4.5а) часто пред¬
ставляет собой наиболее простой и экономичный спо¬
соб обеспечения равномерного распределения потоков
по пространству комнаты. В длинной прямоугольной
комнате можно построить перегородки, однако поза¬
ботьтесь о том, чтобы они были теплоизолированы и
достаточны крепки, чтобы выдержать крепление на
них полок.
Термостаты. Термостаты должны быть типа «про¬
порциональный автоматический регулятор», действо¬
вать через реле, обеспечивающее поступление тепла
со скоростью, пропорциональной различию между
комнатной температурой и установленным режимом.
Когда двери открыты и температура в комнате падает,
восстановление ее должно быть быстрым; с другой
стороны, температура не должна превышать некото¬
рой установленной отметки, и чем ближе температура
комнаты к этой точки, тем меньше должно поступать
тепла.
В идеале, необходимы два отдельных нагревателя
(Н1 и Н2), каждый со своим собственным термостатом
(НТ1 и НТ2). Термостат (НТ1), установленный на
37 °С, располагают позади вентилятора (F1) напротив
нагревателя (Н1) — по диагонали комнаты. Другой
термостат (НТ2) располагается напротив нагревателя
Н2 и за вентилятором F2 и должен быть установлен на
36 °С. (см. рис. 4.5а). Два корректирующих термостата
(ST1 и ST2) обеспечивают безопасность и работают как
предохранители — один последовательно соединен с
НТ1 и установлен на 38 °С, другой находится в соеди¬
нении с основным блоком питания обоих нагревателей.
Если первый нагреватель задержится на температуре,
превышающей установленную, ST1 сработает как пре¬
дохранитель и прервет сеть и второй нагреватель (Н2)
будет обеспечивать температуру с участием регулиру¬
ющего термостата НТ2. Если второй нагреватель также
перегреется, ST2 выключит работу всей нагревающей
системы (табл. 4.2). Следует установить индикаторные
сигнальные лампы, показывающие включение ST1 и
ST2. Термостатные сенсоры для обеспечения большей
80
ГЛАВА 4. СТРУКТУРА И ПЛАНИРОВАНИЕ ЛАБОРАТОРНЫХ ПОМЕЩЕНИЙ
Тепловентилятор (Н1) Теплоизолированная стена Термостат (НТ2)
б СЕЧЕНИЕ АА
Не включая полки и
наружную оболочку
Масштаб 1:50
1 м
1 Фут
Рис. 4.5. Термальная комната. Комбинированная система отопления и предохранительное термореле (а) план, вид сверху;
(б) диагональное сечение. Стрелки показывают циркуляцию воздуха. Планировка и дизайн помещения разработаны в сотрудниче¬
стве с М. McLean (архитектурный дизайн; Boswel & Johnson) и Lindsay (инженерное решение; Kenneth Munro & Associates)
Таблица 4.2. Термостаты термальной комнаты
Термостат
37 °С
<37 °С
«37 °С
>37 °С <38 °С
>37 °С
>37 °С
>38 °С
>39 °С
НТ1
О
1
1
О
О
О
ST1
1
1
1
1
О
О
НТ2
О
О
1
О
О
О
ST2
1
1
1
1
1
О
Аварийная лампа ST1
♦
♦
Аварийная лампа ST2
♦
I = включен; О = выключен; ♦= сигнальная лампа светится.
НТ1 — регулирующий термостат для нагревателя HI; НТ2 — регулирующий термостат для нагревателя Н2; ST1 — автомати¬
ческий блокирующий термостат-предохранитель для Н1; ST2 — автоматический блокирующий термостат-предохранитель для
единого источника питания Н1 и Н2.
Вентилятор (F2)
Тепловентилятор (Н2)
Прямоугольный вентиляционный
канал, открытый у основания
Вентилятор, установленный
над вентиляционным каналом
на диагональной панели,
создающей треугольную камеру
Оборудование, установленное
на полу, например роллерная
стойка, мешалки
Микроскоп
Стол, покрытый нержавеющей
сталью или ламинатом
ПЛАН, ВИД СВЕРХУ
Регулирующий термостат (HT1)
Вентилятор (F1)
Облегченная
теплоизолирующая дверь
Отсек
4.5. Помещения для подготовительных работ
81
чувствительности должны располагаться в области
быстрых потоков воздуха, поблизости от места выхода
воздуха из второго вентилятора, обеспечивающего цир¬
куляцию. Предпочтительно использовать термисторы
или термопары, имеющие более высокую чувствитель¬
ность и высокую температурную проводимость по
сравнению с другими типами датчиков.
Перегрев. Большое внимание обычно уделяется
обеспечению достаточного количества тепла и умень¬
шению потерь энергии, при этом часто забывают о
проблеме нежелательного избыточного поступления
тепла. Оно может быть результатом: 1) возрастания
температуры в окружающей среде в лаборатории в
жаркую погоду или 2) увеличения количества тепла,
производимого работающим в горячей комнате обору¬
дованием, таким как мешалки, роллерные установки,
ламинарные мини-боксы и т. д. Постарайтесь избегать
установки теплопроизводящего оборудования в тер¬
мальной. Магнитные мешалки, работающие по прин¬
ципу магнитной индукции, производят меньше тепла,
чем механически вращаемые магниты, а двигатели,
обеспечивающие работу роллерных установок, могут
располагаться вне термальной комнаты. В тропиче¬
ских регионах или других зонах, где перегрев может
быть частой проблемой, возможно, потребуется обо¬
рудовать охлаждающие теплообменники для отвода
избытка тепла.
Доступ. При наличии необходимого указанного
выше оборудования, хорошей циркуляции воздуха и
адекватного нагрева воздушный запор не требуется.
Двери должны быть теплоизолированы (пенопластом
или стекловолокном), легкими и без труда закрываться.
Предпочтительны замозакрывающиеся двери. Полезно
также иметь шлюзную камеру, ведущую в зону культи¬
вирования тканей, с полками на обеих сторонах, чтобы
культуры могли легко передаваться в комнату. Дверь
шлюзной камеры также должна иметь изоляционное
покрытие. Расположение шлюзовой камеры над столом
позволит избежать риска появления «холодных пятен»
на полках.
Термометр. Самописец, регистрирующий колеба¬
ния температуры, необходимо установить в термальной,
чтобы регистрационная кривая была видна сотрудни¬
кам, работающим в культуральной комнате. Диаграммы
должны еженедельно сниматься с самописца. При воз¬
можности следует установить две предупреждающих
лампы: одну высокотемпературную и одну низкотемпе¬
ратурную. Лампы располагают позади самописца либо
в другом месте, доступном для наблюдения.
4.5. ПОМЕЩЕНИЯ ДЛЯ
ПОДГОТОВИТЕЛЬНЫХ РАБОТ
4.5.1. Приготовление сред
В маленькой лаборатории можно избежать потребности
в постоянном приготовлении питательных сред, если
существует испытанный источник подходящих ком¬
мерческих культуральных сред. Однако в большом
учреждении (примерно 50 человек, работающих в куль¬
тивировании тканей) может обнаружиться, что более
экономично приготовление сред непосредственно в ла¬
боратории. Меньшие лаборатории могут предпочитать
закупку готовых сред. Этим лабораториям не нужно
готовит реагенты, такие как солевые растворы, раствор
этилендиаминтетраацетата (ЭДТА), бутилировать их
и воду, упаковывать и герметизировать, подготавли¬
вать к стерилизации. В этом случае препаративная
зона должна быть чистой и тихой, но стерильность
не нужна, поскольку все покупаемые продукты будут
стерильны.
Если трудно получить приемлемые коммерческие
среды, то препаративная зона лаборатории должна
быть достаточно большой для того, чтобы расположить
там грубые и точные весы, рН-метр и, если возможно,
осмометр. Пространство рабочего стола должно быть
достаточным для разведения и перемешивания раство¬
ров, бутилирования и упаковки различных материалов.
Иногда требуются дополнительное оборудование и
полки в холодильниках. Если возможно, установите в
стерильной зоне дополнительный ламинарный шкаф
с горизонтальным потоком для фильтрации и бутили¬
рования жидкостей, а в инкубаторе обеспечьте место
для проведения контроля стерильности (например,
инкубация образцов среды в бульоне и при посеве на
чашки).
Термостабильные растворы и оборудование можно
автоклавировать или стерилизовать сухим жаром в
нестерильной части препаративной зоны. Оба пото¬
ка реагентов затем встречаются в зоне хранения (см.
рис. 4.3).
4.5.2. Помещение для мытья посуды
Лучше располагать моечное и стерилизационное поме¬
щения вне культуральной лаборатории, поскольку рас¬
пространение образующихся влажности и тепла может
быть затруднительным без превышения допустимого
уровня движения воздушных потоков. Автоклавы, печи,
дистилляторы при возможности располагают в отдель¬
ной комнате (см. рис. 4.3,4.4), с мощной системой вен¬
тиляции. Моечная зона требует много пространства для
замачивания стеклянной посуды, для расположения
моечных машин, если они вам потребуются. На рабочих
столах должно быть достаточно места для манипуля¬
ций с контейнерами с посудой, сортировки пипеток,
упаковки и герметизации оборудования, готового к
стерилизации. Кроме того, вам нужно место для мытья
и сушки пипеток. Если оборудование для стерилизации
должно располагаться на территории культуральной
лаборатории, поместите его рядом с вытяжным боксом
и подальше от стерильной зоны.
Вид моечного оборудования зависит от целей, ко¬
торые ставят перед лабораторией. Наилучшими явля¬
ются нержавеющие или полипропиленовые раковины.
Первые предпочтительнее, если вы планируете работу
82
ГЛАВА 4. СТРУКТУРА И ПЛАНИРОВАНИЕ ЛАБОРАТОРНЫХ ПОМЕЩЕНИЙ
Блок для ионизации
воды или установка для'
обратноосмотической
очистки (RO)
Деионизированная
Кран для холодной
воды со шлангом
ПОВЕРХНОСТЬ СТОЛА
Сифонная моечная
машина для пипеток
Раковина для
замачивания
900 мм
(Зфт)
Раковина
для мытья
Подача
горячей
воды
Рис. 4.6. Раковина для мойки и моечная машина для пипеток. Предполагаемый план оборудования для замачивания и мытья
с горячей, холодной и деионизированной водой. Масштаб 1:16
Подача
холодной
с изотопами, последние устойчивее к действию дезин¬
фектантов, в частности гипохлорида.
Раковины должны быть достаточно глубокими
(450 мм) для того, чтобы обеспечить работу моечной
машины и споласкивание больших предметов, и рас¬
полагаться так, чтобы не приходилось сутулиться при
работе. Ободок раковины должен находиться на высоте
около 900 мм от пола (рис. 4.6). Лучше разместить
слишком высоко, чем слишком низко, — низкорослые
работники могут всегда встать на подставку, чтобы
достать слишком высоко расположенную раковину,
но высокие люди всегда вынуждены будут наклонять¬
ся над слишком низко расположенной раковиной.
Приподнятые края раковины позволят предупредить
разбрызгивание и предохранить оператора от загрязне¬
ний и обливания, которые неизбежны при наклоне над
раковиной. Поднятые края должны проходить вокруг
раковины за кранами.
Каждая раковина, предназначенная для мытья посу¬
ды, требует четырех кранов: отдельный кран холодной
воды, смеситель с кранами горячей и холодной воды,
кран холодной воды для отводного шланга для соедине¬
ния с оборудованием для ополаскивания, а также неме¬
таллический или нержавеющий кран для деионизиро¬
ванной воды из резервуара над раковиной (см. рис. 4.6).
Избегайте централизованного обеспечения деионизиро¬
ванной водой, поскольку трубы быстро загрязняются,
зарастают водорослями и трудно очищаются.
Тележки или перемещаемые столики часто ис¬
пользуются для сбора грязной стеклянной посуды
и перераспределения свежестерилизованного обо¬
рудования и материалов. Спланируйте место для их
размещения.
4.5.3. Хранение
Обеспечьте отдельное хранение стерильных и несте¬
рильных объектов, которые должны быть снабжены
четкой маркировкой. Необходимо спланировать хра¬
нение следующих объектов:
1) Стерильные жидкости, при комнатной температу¬
ре (растворы солей, вода и т.д.), при 4 °С (среды)
и при -20 °С или -70 °С (сыворотки, трипсин,
глютамин и т.д.).
2) Стерильная и нестерильная стеклянная посуда,
включая бутылки для среды и пипетки.
3) Стерильный одноразовый пластик (например, куль¬
туральные плашки, чашки Петри, центрифужные
пробирки и флаконы, шприцы и т.д.).
4.5. Помещения для подготовительных работ
83
Дверь в коридор с отверстиями для вентиляции (дверь
открыта, когда происходит разливание азота)
Детектор кислорода
1 м (~ 3 фт)
Изолированная система подачи жидкого
азота в распределительную систему
Соединяющая дверь с отверстиями
для вентиляции (дверь открыта, когда
происходит разливание азота)
Распределительная система с гибким шлангом
Фиксатор для форсунки
Форсунка
Термоизолированный шланг
Морозильное оборудование
Аварийная сигнализация
Внешняя захлопывающаяся дверь для получения
азота и вентиляции воздуха наружу
500-литровый контейнер для хранения
жидкого азота
Рис. 4.7. Хранилище для жидкого азота и морозильное оборудование. Хранилище для жидкого азота лучше располагать рядом
с наружной стеной в помещении, оборудованном вентиляцией из здания наружу, а также дверью, обеспечивающей легкий доступ
для получения доставленного азота. Если морозильное оборудование располагается по соседству, морозильные установки могут
заполняться непосредственно путем поступления газа через гибкий шланг. В процессе заполнения двери должны оставаться
открытыми для лучшей вентиляции, настенный измеритель уровня кислорода с сигналом тревоги, детектор которого распола¬
гается на уровне пола, при обнаружении падения уровня кислорода в воздухе ниже безопасного должен подать сигнал
4) Навинчивающиеся крышки, пробки и т. д., стериль¬
ные и нестерильные.
5) Фильтры и подобные аппараты, стерильные и не¬
стерильные.
6) Перчатки, упаковочные мешки и т.д.
7) Жидкий азот для пополнения морозильных уст¬
ройств; жидкий азот может храниться двумя спо¬
собами:
а) в сосудах Дьюара (25-50 л) под рабочими сто¬
лами или
б) в больших емкостях для хранения (100-150 л)
на тележках или в хранилищах (500-1000 л),
постоянно располагающихся в комнате с соб¬
ственной вентиляционной системой или, что
предпочтительно, в отдельном охраняемом,
укрытом от непогоды строении (рис. 4.7).
Замечание. Сосуды с жидким азотом могут на¬
капливать загрязнения, поэтому следует хранить их в
чистой зоне.
• Требование безопасности. В комнате, где
хранится и разливается жидкий азот, должна быть обес¬
печена соответствующая вентиляция, предпочтительно
с сигнализатором падения содержания кислорода в
воздухе ниже допустимого уровня. Причиной этого
требования является возможность испарения азота
при манипуляциях с ним, при этом азот может заме¬
щать кислород в воздухе (1л жидкого азота —> 700 л
газообразного N2).
8) Хранение баллонов с двуокисью азота в отдельных
баллонах, при необходимости перемещаемых в по¬
мещение лаборатории.
• Требование безопасности. Баллоны должны
быть прикреплены к стене или рабочей поверхности на
стеллаже (см. рис. 7.2).
9) В культуральной лаборатории возможно центра¬
лизованное обеспечение С02 с использованием
трубопровода либо из собственного резервуара,
периодически пополняемого, из которого газ рас¬
пределяется по трубам по помещениям лаборатории.
Способ, который вы изберете, зависит от ваших
84
ГЛАВА 4. СТРУКТУРА И ПЛАНИРОВАНИЕ ЛАБОРАТОРНЫХ ПОМЕЩЕНИЙ
планов, спектра ваших задач и стоимости затрат.
При грубой оценке, работа 2-3 человек потребует
нескольких баллонов; при количестве сотрудников
10-15 человек, экономически выгоднее иметь тру¬
бопровод, соединенный со стационарно располага¬
ющимися баллонами; при количестве сотрудников
более 15 необходимо использовать систему труб,
связывающую помещения и большую емкость для
хранения С02.
Зона хранения 1 -6 должна располагаться в стериль¬
ной рабочей зоне и быть легкодоступна. Холодильники
и морозильники установите так, чтобы они были обра¬
щены к нестерильной части лаборатории, поскольку
двери и компрессорные вентиляторы создают пыль и
сквозняки и могут разносить споры грибов. Кроме того,
холодильники и морозильники требуют ухода и перио¬
дического размораживания, которые лучше проводить
отдельно от стерильной рабочей зоны.
В идеале, зона хранения должна располагаться так,
чтобы быть доступной как для изъятия, так и для заме¬
ны объектов хранения. Полезно использовать двусто¬
ронние блоки, поскольку хранящиеся предметы можно
использовать для изъятия и замены с одной стороны и
для использования в работе с другой.
• Требование безопасности. Очень важно
иметь выступающую кромку на краю с обеих сторон
полок, если они находятся на значительной высоте и на
них хранятся стеклянная посуда и реактивы. Это пре¬
дупреждает случайное выпадение предметов с полки
при их использовании и замещении.
Помните о необходимости иметь достаточно места
для хранения, поскольку это позволит вам производить
закупки большего объема, таким образом, экономя
средства, и в то же время уменьшит риск истощения
ценных запасов в тот момент, когда они не могут быть
восполнены. По грубой оценке, на каждого сотрудника
культуральной лаборатории потребуется 200 л объема
хранения при 4 °С и 100 л при -20 °С. Необходимый
объем возрастает при уменьшении количества людей.
Например, одному человеку может потребоваться
250-литровый холодильник и 150-литровый морозиль¬
ник. Конечно, эти цифры, требуемые для хранения,
приблизительны и следует сделать скидку на наличие
пространства в холодных комнатах и комнатах глубо¬
кого замораживания.
В целом, отдельные холодильники на -20 °С лучше,
чем холодильное помещение на -20 °С. Холодильни¬
ки легче мыть и эксплуатировать, они обеспечивают
лучший резерв при недостатке какой-либо единицы
хранения. Вы сами должны оценить достоинства
и недостатки холодной комнаты по сравнению с
холодильниками. Без сомнения, холодная комната
предоставляет больший объем для хранения в кубо¬
метрах, однако очень важен вопрос использования
этого пространства — насколько легко осуществлять
эксплуатацию и уборку помещения, насколько хорошо
это пространство может быть использовано индивиду¬
альными пользователями? Несколько независимых
холодильников займут больше места, чем холодная
комната эквивалентного объема, однако они могут
быть удобнее в эксплуатации и поддержании, а также
в случае поломки оборудования.
При планировании расходов стоит учитывать не¬
обходимость в дополнительном количестве морозиль¬
ников и холодильников, предоставляющих больше
возможностей как в повседневных, так и в непредска¬
зуемых обстоятельствах.
Глава 5
ОБОРУДОВАНИЕ
I
5.1. ПОТРЕБНОСТИ ЛАБОРАТОРИИ
КУЛЬТУР ТКАНЕЙ
В случае ограниченного финансирования необходимо
определить приоритетные специфические потребности
лаборатории культивирования тканей:
1) основные — оборудование, без которого вы не смо¬
жете выполнить работу с достаточной точностью и
надежностью;
2) желательные — оборудование, позволяющее осу¬
ществлять культивирование лучше, эффективнее,
быстрее или с меньшими затратами труда;
3) полезные — элементы оборудования, которые
улучшают условия работы, уменьшают усталость,
обеспечивают проведение более сложного анализа
данных или в целом делают рабочие помещения
более привлекательными (табл. 5.1). В следующих
разделах по отдельным подразделам, соответству¬
ющим предметному указателю, представлено обо¬
рудование в порядке первоочередности, как оно
видится автором, но ясно, что читатель может иметь
свое мнение о приоритетах.
Потребности в отдельных элементах оборудования
часто очень субъективны и являются результатом
личных устремлений, наличия в продаже, технических
инноваций и давления со стороны коллег. Реальные
потребности сформулировать сложнее, но они опреде¬
ляются объективно типом работы, экономией времени,
которое предоставляет это оборудование, более высо¬
ким уровнем техники в области асептики, качеством
данных, аналитических возможностей и требований к
образцам, экономией времени или трудозатрат, коли¬
чеством людей, которую будут использовать это обо¬
рудование, приемлемостью сметы расходов и размеров
потенциальной прибыли, а также специальными требо¬
ваниями, предъявляемыми вашими методиками.
5.2. АСЕПТИЧЕСКАЯ ЗОНА
5.2.1. Ламинарные шкафы
Если помещение соответствующим образом изоли¬
ровано и размещено в чистой зоне с ограниченным
доступом, то возможно выполнение асептических
работ без ламинарного потока. Однако понятно, что
для большинства лабораторий, обычно переполненных
и оживленных, использование ламинарных шкафов
является наиболее простым способом обеспечения
асептических условий (см. разд. 6.4). Обычно один
шкаф используется двумя-тремя людьми. Шкафы
с горизонтальным ламинарным потоком дешевле и
обеспечивают наилучшую защиту стерильности куль¬
туры, но в действительности они подходят только для
приготовления сред и других стерильных реагентов, а
также для культивирования клеток неприматов. Они
частично пригодны для иссечения материала неприма¬
тов при получении первичной культуры. При работе с
потенциально опасными биологическими материалами
(любые клеточные линии приматов, включая человека,
вируспродуцирующие культуры, радиоизотопы, канце¬
рогенные или токсические препараты) необходимы спе¬
циальные микробиологические шкафы II или III класса
защиты (рис. 5.1, см. также рис. 7.4). На практике
большинство лабораторий используют стандартные
микробиологические шкафы II класса защиты.
• Требование безопасности. Важно ознако¬
миться с местными и государственными требованиями
и правилами биологической опасности до установки
оборудования, поскольку требования законодательства
и рекомендации в различных регионах различаются
(см. разд. 7.8.1).
Выберите ламинарный шкаф, который: 1) достаточ¬
но большой [минимум рабочей поверхности 1200 мм
ширины х 600 мм глубины]; 2) тихий, поскольку
производящие шум ламинарные шкафы утомляют;
3) легко моется как внутри рабочей зоны, так и под
рабочей поверхностью в случае разлития жидкостей и
4) удобный для сидения рядом с ним во время работы.
Некоторые шкафы имеют неудобную систему воздухо¬
водов под рабочей поверхностью, которая не оставляет
места для колен. В других шкафах освещение или иные
приспособления над рабочей поверхностью задевают
голову, либо имеется экран, который затрудняет об¬
зор. Передний экран должен подниматься, опускаться
или полностью удаляться для обеспечения уборки и
манипуляций с громоздким оборудованием для культи¬
вирования. Помните, однако, что микробиологические
шкафы определенного уровня биозащиты не обеспе¬
чат вам как оператору необходимой защиты, если вы
удалите передний экран, кроме того, вы потеряете в
эффективности обеспечения стерильности в шкафу.
86
ГЛАВА 5. ОБОРУДОВАНИЕ
Таблица 5.1. Оборудование лаборатории культивирования тканей
Основное оборудование
Не существенное,
но желательное
Полезные дополнительные
приспособления
Ламинарный шкаф (с биозащитой для
клеток человека)
Инкубатор (влажный С02-инкубатор,
если используются открытые чашки
или флаконы)
цилиндр 5%-го С02 для создания газовой
атмосферы культивирования
баллоны с жидким С02без сифона (для
С02-инкубатора)
Весы
Стерилизатор (автоклав, термобарока¬
мера)
Холодильник
Морозильник (для хранения при
-20 °С)
Инвертированный микроскоп
Ванна или раковина для замачивания
Глубокая раковина для мытья
Цилиндры для пипеток
Моечная машина для пипеток
Дистиллятор или водоочистительная
установка
Настольная центрифуга
Морозильное оборудование, содержащее
жидкий N2 (-35 л, 1500-3000 ампул)
Сосуд Дьюара с жидким N2 (-25 л)
Аппараты для медленного заморажива¬
ния клеток (см. разд. 20.3.4)
Магнитная мешалка для суспензионных
культур
гемоцитометр
Счетчик клеток
Перистальтический насос
Дозаторы
pH-метр
Стерилизационный шкаф
Теплая комната (37 °С)
Самописцы для регистрации тем¬
пературы в стерилизаторе, авто¬
клаве и горячей комнате
Фазово-контрастный, флюорес¬
центный микроскоп
Пломбировщик пипеток
Сушка для пипеток
Автоматический диспенсер
Тележки или перемещаемые сто¬
лики
Высоко- и низкотемпературные
сушильные шкафы.
Роллерные штативы для культур
в роллер-сосудах
Трубопровод для поступления
С02 из хранилища.
Автоматический переключатель
баллонов С02
Моечная машина для стеклянной посуды
Низкотемпературный морозильник
(<-70 °С)
Измеритель проводимости
(кондуктометр)
Осмометр
Заваривающее устройство для полиэтиленовых
пакетов (для упаковки стерильного оборудо¬
вания для долговременного хранения)
Компьютер для ведения регистрационных
записей эксплуатации морозильника и база
данных по клеточным линиям
Счетчик колоний
Центрифуга большого объема (6x1 л)
Цифровая камера и монитор для инвертиро¬
ванного микроскопа
Видеооборудование для съемок в замедленном
режиме
Измеритель размеров клеток (например,
Scharfe, Coulter)
Портативный прибор для записей температу¬
ры для проверки температурного режима в
горячей комнате или инкубаторе)
Дезинтегратор/ стерилизатор пластика
Аппарат для медленного контролируемого
охлаждения клеток (для замораживания
клеток)
Клеточный сортер, активирующий флюорес¬
ценцию
Конфокальный микроскоп
Сцинтилляционный счетчик для микротитра-
ционных планшет
Элютриатор
Оператор, который будет работать в данном ламинар¬
ном шкафу, должен до его приобретения попробовать
посидеть на рабочем месте, воспроизводя обычную дея¬
тельность. Обратите внимание на следующие вопросы:
1) Можете ли вы поместить колени под рабочую по¬
верхность ламинарного шкафа, удобно ли вы при
этом сидите? Достаточно ли близко к рабочему
пространству? Располагаются ли руки, по крайней
мере наполовину, внутри ламинарного шкафа?
2) На правильном ли месте располагается подножка?
3) Можете ли вы видеть, что вы делаете, не напрягая
шею?
4) Перфорирована ли рабочая поверхность и, если да,
создаст ли это затруднения при пипетировании и
диспенсировании жидкостей (предпочтительна
сплошная рабочая поверхность с отверстиями для
вентиляции в передней и задней частях).
5) Легко ли удаляется рабочая поверхность при
уборке?
6) Если рабочая поверхность поднимается, острые
или закругленные у нее края? Нет ли опасности
порезаться при уборке?
7) Нет ли трещин на рабочей поверхности, которые
могут накапливать споры и другие загрязнения?
Некоторые шкафы имеют разделенную на секции
рабочую поверхность, что облегчает уборку, но со¬
здает капиллярные щели при нормальном рабочем
расположении секций.
8) Достаточно ли и правильно расположено освещение?
9) Разместится ли этот шкаф в лаборатории?
10) Размещенный на постоянном месте, будет ли шкаф
удобен для работы и обслуживания? (Спросите об
этом обслуживающий персонал, а не продавца!)
Между боковыми стенками шкафов должно быть
как минимум 500 мм.
11) Будет ли достаточно места над шкафом для венти¬
ляции в комнате или установления воздуховода с
выводом воздуха наружу?
12) Не будут ли воздушные потоки из других шкафов,
комнатной вентиляции, независимой системы
кондиционирования воздуха перемешиваться с
воздушными массами рабочего места данного ла¬
минарного шкафа? Не будет ли выхода воздуха или
аэрозолирования воздуха за счет турбулентных
потоков? Чтобы избежать такого явления, располо¬
жите передние поверхности противоположно сто¬
ящих шкафов как минимум на расстоянии 2000 мм
или, что предпочтительнее, 3000 мм и проведите
испытания с участием инженера с опытом работы
с микробиологическими ламинарными шкафами.
5.2. Асептическая зона
87
Зажим
Пипетирующее
устройство
Всасывающая
линия
Цилиндр для
использованных
пипеток
Принимающий
сосуд
Ножная педаль для
включения насоса
Рис. 5.1. Ламинарный шкаф. В правой части шкафа, под рабочей поверхностью показан перистальтический насос, соединенный
с принимающим сосудом и включающийся при помощи ножной педали
5.2.2. Цилиндры для пипеток
Цилиндры для пипеток (иногда они известны как
лотки для пипеток) должны быть сделаны из поли¬
пропилена и равномерно свободно стоять по лабора¬
тории, по одному на рабочем месте плюс некоторое
дополнительное количество, с тем чтобы заполненные
цилиндры с дезинфектантом могли стоять 2 ч до мытья
(см. разд. 7.8.5).
5.2.3. Аспирационный насос
Для удаления лишней среды или других реагентов из
культуральных флаконов используют перистальти¬
ческий насос (рис. 5.2я), удаляемую жидкость можно
перемещать прямо в дезинфектант (см. разд. 7.8.5),
содержащийся в контейнере с отверстием (рис. 5.2б).
Для того чтобы минимизировать риск образования
аэрозоля в атмосфере, следует использовать ватную
пробку или микропоровый фильтр. Всасывающее от¬
верстие должно находиться, по крайней мере, на 5 см
дальше от запирающего устройства, чем выпускное, так
чтобы удаляемая жидкость не заплескивалась назад в
отверстие. Все соединения трубок и насоса должны
регулярно проверяться; насос должен включаться
действием ножной педали.
Вакуумный насос, подобно устройству, которое
используется для стерилизации фильтрацией, может
использоваться вместо перистальтического насоса.
При необходимости один и тот же насос может слу¬
жить обеим целям; так или иначе, для избежания риска
загрязнения самого насоса требуется использовать
заглушку. Удаляемая жидкость должна собираться
в емкости (см. рис. 5.26, 5.3), куда по завершении
работы добавляется дезинфектант типа гипохлорида
(см. разд. 7.8.5) и оставляется по крайней мере на 2 ч
до опустошения резервуара. Гидрофобный микро¬
поровый фильтр (например, Pall Gelman) и вторую
заглушку можно поместить в гидролинию с насосом,
чтобы предупредить вытекание жидкости или аэрозо-
лирование. Не пропускайте воздух, содержащийся в
резервуаре с гидрохлоридом, через перистальтический
насос, поскольку свободный хлор приводит к коррозии
деталей насоса и может быть токсичным. Также избе¬
гайте попадания жидкости в вакуумные системы. При
загрязнении жидкостью их очень трудно очистить.
Чтобы предупредить возврат удаляемой жидкости
обратно во флакон, всегда включайте насос прежде, чем
вставите пипетку в трубку (см. разд. 6.5).
Рис. 5.2. Удаление среды с помощью насоса, (а) Пипетка,
соединенная трубкой с перистальтическим насосом, исполь¬
зуется для удаления среды из флакона; (б) перистальтический
насос во всасывающей гидролинии из ламинарного шкафа к
сосуду-приемнику для слива
5.2.4. Сервисные тележки
Различные предметы, необходимые для работы в лами¬
нарном шкафу, удобно располагать на перемещаемых
тележках. Эти тележки удобно заполняют нижнее про¬
странство между соседними шкафами и легко переме¬
щаются при обслуживании помещения и шкафов. Они
могут быть использованы обслуживающим персоналом
для перевозки материалов к шкафам и от них, а также к
другому основному оборудованию лаборатории. Боль¬
шие тележки полезны для перемещения загрязненной
стеклянной посуды и использованного оборудования
из асептической зоны в моечную. Они могут быть при¬
паркованы в удобном месте (см. рис. 4.3,4.4,5.1).
5.2.5. Инвертированный микроскоп
Будет не лишним подчеркнуть, что, несмотря на
значительный прогресс в области количественного
анализа культивированных клеток и молекулярных
зондов, остается актуальным регулярное визуальное
наблюдение за культурами клеток. Морфологические
изменения часто являются первым признаком истоще-
Рис. 5.3. Вакуумный насос Integra. Всасывающий насос и
принимающая емкость для удаления жидкости из флаконов,
предоставляется компанией Integra. (Фото любезно предо¬
ставлено Norbert Fesunig, Deutches Krebsforschungzentrum,
Гейдельберг.)
ния и старения культуры (см. разд. 13.6.1 и рис. 13.1), а
характерные черты микробиологического загрязнения
легко распознаются.
К оборудованию, необходимому для культуральной
лаборатории, относится простой инвертированный
микроскоп (рис. 5.4). Удостоверьтесь, что платфор¬
ма микроскопа достаточно велика, чтобы поместить
большой роллер-флакон между платформой и конден¬
сором (см. разд. 26.2.3) в случае, если вам потребуется
исследовать такие флаконы. На рынке доступно много
простых и недорогих инвертированных микроскопов,
особенно ценно приобрести микроскоп с фототрубкой
для цифрового фотографирования или киносъемки.
Если вы заранее знаете, что будете фотографировать
живые культуры, вам следует вложить в микроскоп
значительные средства и приобрести прибор с высоко¬
качественной оптикой и фазово-контрастным конден¬
сором и объективами с длиннофокусным расстоянием,
а также со всем необходимым для CCD-камеры и
монитора (см. рис. 5.14). Возрастающее использование
зеленого флюоресцентного белка (GFP) для мечения
живых клеток означает, что флюоресцентная оптика
также может понадобиться.
Полезной приставкой к инвертированному мик¬
роскопу может быть кольцо с маркировкой компании
Nikon. Это устройство вставляется в револьверную
головку микроскопа на место объектива и может быть
использовано для того, чтобы отмечать нижнюю часть
чашки Петри, в которой расположены интересующие
колонии или группы клеток. Затем эти колонии могут
5.2. Асептическая зона
фазовыми
кольцами
Конденсор и
подвижный
предметный
столик для работы
с большими
бутылками
Фазовые
объективы 4х, 10х,
20х и 40х
Рис. 5.4. Инвертированный микроскоп. Инвертированный
микроскоп Olympus СКХ41 оборудован фазово-контрастной
оптикой и тринокулярной головкой с портом для присоеди¬
нения цифровой фотокамеры. (Фото любезно предоставлено
фирмой Olympus, UK, Ltd.)
быть отсняты (см. разд. 14.4 и рис. 14.8,14.9) или в по¬
следующем интересующий вас участок культуры может
подвергаться пристальному изучению.
5.2.6. Центрифуга
Периодически клеточные суспензии требуют центри¬
фугирования для повышения концентрации клеток
или отмывки реагентов. Для большинства таких целей
подходит маленькая настольная центрифуга, предпоч¬
тительно с пропорционально контролируемым тор¬
можением. Клетки хорошо осаждаются при 80-100 g;
более высокие значения могут вызвать повреждение
клеток и инициировать агглютинацию сгустков тканей.
При крупномасштабном культивировании суспен¬
зионных культур иногда потребуются охлаждаемые
центрифуги большого объема, например, 4x1 л или
6x1 л (см. разд. 26.1).
5.2.7. Манипуляции с жидкостями
в стерильных условиях —
пипетирование и дозирование
Удаление жидкостей. Жидкости удаляют простой
пипеткой, но процесс будет значительно облегчен
и ускорен, если использовать пипетку с широк
отверстием, присоединенную к вакуумному насс
или вакуумной линии. Требуется также подходяп
емкость для слива и предупреждения загрязнен
насоса. Можно использовать и простой перисталь'
ческий насос, перекачивающий жидкость в резерву
с дезинфектантом (см. разд. 5.2.3)
Пипетирующиеустройства. Простое пипетиро
ние является одной из наиболее частых повседневн
манипуляций с культурами. Хотя резиновая груша и
другие простые приспособления для пипетировак
дешевы и просты в использовании, скорость, точно»
и воспроизводимость результатов значительно noi
шаются при использовании автоматического пипети
ющего устройства (дозатора) (рис. 5.5), которые мо
быть со встроенным или отдельным насосом, им<
питание от сети или от зарядного устройства (см. П]
ложение II). При выборе пипетирующего устройс
обратите внимание на вес и удобство в использовав
в течение долгого времени и лучше всего опробу]
одно из устройств прежде, чем приобретать его. I
петирующие устройства обычно снабжены фильтр
во вкладыше для минимизации переноса загрязнен
Некоторые фильтры одноразовые, некоторые мс
Рис. 5.5. Пипетирующее устройство. Моторизован
пипетирующее устройство для использования обычной ]
дуированной пипетки
90
ГЛАВА 5. ОБОРУДОВАНИЕ
но использовать повторно после стерилизации (см.
рис. 7.2 для изучения правильного метода вставления
пипеток в пипетирующее устройство).
Автоматические пипетки. Эти устройства по¬
лучили свое начало от микропипеток компании Ер-
pendorf и использовались для дозирования объемов
10-200 мкл. К настоящему времени рабочие объемы
возросли до 5 мл, поэтому термин «микропипетки»
не всегда соответствует реальности, и это устройство
называется по-разному: дозатор, автоматическая
пипетка, пипетман и т. п. (рис. 5.6). Только наконеч¬
ники этих пипеток нуждаются в стерилизации, среди
ограничений в использовании автоматических пи¬
петок — длина наконечников, определяющая выбор
используемых сосудов. Если стерильная жидкость
забирается из контейнера с помощью автоматической
пипетки, ее нестерильный корпус не должен касаться
сторон контейнера. Реагенты объемом 10-20 мл могут
быть дозированы на объемы от 5 мкл до 1 мл из уни¬
версального контейнера или на объемы 1-200 мкл из
миниатюрного флакона. Для дозирования на объемы
100 мкл-1 мл можно использовать пробирки типа Ер-
pendorf, но они требуют стерилизации.
Предполагается, что внутри автоматическая пи¬
петка стерильна либо не производит перемещения
воздуха, достаточного для заражения наконечников.
Однако в некоторых ситуациях даже минимальная
возможность заражения имеет значение. Например,
если вы осуществляете серийное субкультивирование
ростковой клеточной линии (в противоположность
кратковременному эксперименту с клетками, которые в
конечном счете будут взяты на анализ либо уничтоже¬
ны, но не предполагают их размножение), безопасность
и чистота клеточной линии становятся первостепенны¬
ми, поэтому вы должны использовать либо обычные
стеклянные пипетки с ватным фильтром, либо однора¬
зовые пипетки, которые стерильны по всей длине, что
является существенным при необходимости достичь
необходимой точки в сосуде, из которой отбираются
пробы. Если вы используете достаточно маленькие
контейнеры, то можно избежать прикосновения не¬
стерильных частей корпуса автоматической пипетки,
поэтому ее использование допустимо при условии,
Рис. 5.6. Автоматическая пипетка (пипеттор). Пипети¬
рующее устройство с изменяемым объемом, Существуют
также пипетки с фиксированным объемом. Сами пипетки
не стерилизуют, пластиковые наконечники подвергают
стерилизации
что наконечник снабжен фильтром, препятствующим
перекрестному заражению и минимизирующим микро¬
бную контаминацию. В противном случае вы рискуете
получить микробное загрязнение от нестерильных
частей автоматической пипетки или, что более значи¬
мо, перекрестное заражение из аэрозоля и жидкости,
заброшенных в ствол этой пипетки.
Обычное субкультивирование, которое должно
проводиться быстро и точно, с высокой степенью за¬
щиты от загрязнения и перекрестной контаминации,
следует проводить с использованием стеклянных или
одноразовых пластиковых пипеток. Эксперименталь¬
ная работа, которая должна быть точной, но не требует
длительного размножения используемых клеток, поз¬
воляет использовать автоматические пипетки.
Наконечники можно покупать россыпью в больших
упаковках, с тем чтобы упаковывать и стерилизовать
их в условиях лаборатории. Их можно также поку¬
пать уже стерильными и готовыми к использованию.
Предварительно упакованные наконечники намного
удобнее, но значительно дороже. Некоторые держатели
для наконечников могут повторно стерилизоваться, что
представляет собой определенный компромисс.
Диспенсеры больших объемов. Когда объем среды в
культуральных сосудах превышает 100 мл, для разлива
жидкостей можно использовать другие приспособле¬
ния. Если используется небольшое количество сосудов,
удобно применять пипетки объемом 100 мл (BD Bio-
scences) либо градуированные бутыли (рис. 5.7) или
Витая пробка
Липкая лента
Линия
аспирационного
воздуховода
Силиконовые
трубки
Зажим
Защитный колокол
для принимающего
сосуда
Рис. 5.7. Градуированный бутылочный диспенсер. Пробка
с двумя отверстиями вставлена в горлышко градуированной
бутыли, от одного отверстия отведен шланг, соединенный с
диспенсирующим колоколом, защищающий принимающий
жидкость сосуд. На шланге имеется металлический пружин¬
ный зажим, имеется также отводящая линия для равновесия
воздуха. Пробка может стерилизоваться отдельно от бутылки,
подходит к любой стандартной бутылке со средой. (Ориги¬
нальное изобретение John Paul.)
5.2, Асептическая зона
9
пакеты (Sigma). Если объемы очень большие (около
500 мл) либо требуется очень большое количество
повторного отмеривания больших объемов, может
понадобиться перистальтический насос. Простой пере¬
нос большого объема жидкости (10-10 ООО л) обычно
осуществляется приготовлением среды в закупоренном
под давлением сосуде и дальнейшим перемещением
ее в культуральные сосуды под действием положи¬
тельного давления (Alfa Laval). Можно разливать
большие объемы путем простого переливания через
горлышко сосуда, однако такого рода манипуляции
ограничиваются однократным действием с предвари¬
тельным измерением объема переливаемой жидкости
(см. разд. 6.3.6).
Многократное диспенсирование. Традиционно
диспенсер для многократного повторяющегося дис-
пенсирования представляет собой тип оборудования,
известный как шприц Корнуэла, в котором жидкость
набирается через одно отверстие, а выпускается через
другое, при этом используется простой двухходовой
клапан. Пружинный поршень шприца делает процеду¬
ру полуавтоматической и многократной. Существует
неисчислимое количество вариантов такого типа дис¬
пенсеров, многие из которых применяются в настоящее
время. Основные проблемы при их использовании
возникают из-за залипания поршня, но его можно
минимизировать, если избегать этапа сушки после
автоклавирования и прокаливания шприца со средой
или солевым раствором до и после использования.
Многократное диспенсирование небольших объемов
осуществляют пошаговым движением поршня в шпри¬
це (рис. 5.8).
Перистальтический насос также может исполь¬
зоваться для диспенсирования серийных емкостей и
имеет определенные преимущества, так как наличие у
него ножной педали позволяет оставлять руки свобод¬
ными (рис. 5.9). Использование такого оборудования
требует особенно тщательной заботы о чистоте диспен¬
сирования и предупреждения загрязнений трубок на
приемном и выпускном концах. В общем, применение
такого оборудования целесообразно, если требуется
разлить очень большое число флаконов. Автомати¬
ческое пипетирование, обеспечиваемое перистальти¬
ческим насосом, может быть контролируемо в малых
приращениях. Кроме того, необходимо автоклавиро-
вать только приносящие трубки. При разливе объемов
Рис. 5.8. Пипетка с многократно дозируемым объемом. По¬
шаговый диспенсер действует на основе движения помпового
механизма в шприце, который приводится в движение кноп¬
кой, на которую нажимают большим пальцем (Jencons)
Рис. 5.9. Автоматический диспенсер. Диспенсер Perimati
Premier, пригодный для многократного диспенсирования
разлива в диапазоне 1-1000 мл. Если оборудование испол]
зуется для стерильных операций, следует автоклавироват
только доставляющие трубки
от 10 до 100 мл точность и воспроизводимость могу
находиться на достаточно высоком уровне. Некотс
рые виды приводящих трубок можно стерилизовать
хранить в инвентаре, что позволяет быстро заменит
их при случайном загрязнении или смене клеточног
типа или разливаемого реагента.
Автоматизация. В культивировании тканей был
много попыток автоматизировать процесс разлива жи;
костей, но только некоторые системы нашли широкс
практическое применение. При стандартной систем
производства может использоваться автоматическс
снабжение растворами, однако большие затраты вр(
мени на настройку соответствующей программы и вш
сение модификаций, которые могут потребоваться пр
изменении системы, а также чрезвычайная важност
соблюдения стерильности удерживают болыпинств
лабораторий от инвестирования средств и времени в
внедрение таких систем. Появление и широкое при
менение планшета для микротитрации (см. рис. 8.2
принесло с собой автоматизацию разлива по ячейкал
учета результатов и т.д. (рис. 5.10, 5.11, 5.12). Перед*
точные устройства, использующие перфорированны
лотки, или многоканальные пипетки облегчают ш
ресев из одной плашки в другую. Существуют такж
миксеры для планшетов и держатели для центрифз
гирования плашек. Ассортимент оборудования стол
широк, что не может быть приведен здесь, рекомендуе;
ознакомиться с соответствующими торговыми катг
логами (см. Приложение II). Два вида оборудовани
заслуживают особого внимания: программируема
одноканальная или мультиканальная автоматическа
92
ГЛАВА 5, ОБОРУДОВАНИЕ
Рис. 5.10. Многоканальная пипетка. Пипетка с большим
количеством каналов может одновременно использовать
8 пластиковых наконечников. Существуют также 4- и 12-
канальные пипетки
Рис. 5.11. Автоматическое заполняющее устройство для
загрузки многолуночных планшетов. На фото показано за¬
полнение платы в нестерильных условиях, но оборудование
может быть использовано для стерильных манипуляций.
(Фото любезно предоставлено Центром исследований
для онкологии и прикладной фармакологии, Университет
Глазго.)
пипетка Rainin и оборудование для переноса среды
и реплицирования плашек Corning Coster Transtar
(см. рис. 5.13). В технику исследований при помощи
культивирования тканей сейчас вводятся роботизи¬
рованные системы (такие как Packard), что является
естественным продолжением микротитрационных
систем. Роботы полностью автоматизируют процессы,
хотя требуют перепрограммирования при изменении
условий аналитических исследований.
Выбор системы. Выбор между оборудованием с
ручным управлением и сложной автоматизированной
системой определяют пять критериев:
Рис. 5.12. Планшетный сканер. Денситометр для измерения
оптического поглощения в каждой ячейки, некоторые модели
измеряют также флюоресценцию. (Фото любезно предостав¬
лено Центром исследований для онкологии и прикладной
фармакологии, Университет Глазго.)
1) Легкость использования и эргономическая эффек¬
тивность.
2) Стоимость в соотношении с экономией времени и
возрастанием эффективности.
3) Точность и воспроизводимость в серийных и парал¬
лельных партиях.
4) Легкость стерилизации и действие ее на точность и
воспроизводимость.
5) Механическая, электрическая, химическая, биоло¬
гическая и радиологическая безопасность.
• Требование безопасности. Большинство пи-
петирующих устройств склонны выпускать жидкость с
более высокой скоростью, чем при нормальных ручных
манипуляциях, и, следовательно, больше способствуют
созданию аэрозолей. Это следует иметь в виду при
использовании потенциально опасных биологических
веществ.
5.2.8. Счетчик клеток
Счетчик клеток (см. рис. 21.2) имеет много преиму¬
ществ при одновременном ведении двух или трех
линий клеток, а также при необходимости точной
количественной оценки кинетики роста. Несколько
компаний представили на рынке различные по слож-
5.2. Асептическая зона
93
Рис. 5.13. Устройство для переноса. Transtar (Corning)
для посева культуры, переноса среды, воспроизведения
планшетов, и других аналогичных манипуляций с микро-
титрационными планшетами, требующих одновременных и
одинаковых действий со всеми 96 лунками. (Воспроизводит¬
ся с разрешения Corning.)
Рис. 5.14. Замкнутая телевизионная система. CCD-камера
присоединена к инвертированному микроскопу Zeiss Axiovert
и может использоваться для прямого распечатывания или
записи в замедленном времени при использовании видео¬
записывающего оборудования (см. разд. 27.3). Справа порт
для микроинъекций
ности модели от простых счетчиков количества частиц
до автоматизированных приборов, подсчитывающих
количество клеток и анализирующих их размеры. Для
рутинных исследований подходят представленные на
рынке счетчики Beckman Coulter и Sch rfe (см. также
разд. 21.1.2). При необходимости количественной
оценки нестерильных жидкостей для их доставки
полезны автоматические системы разлива (См. также
разд. 5.2.7). При использовании для подсчета клеток
диспенсеров, наворачивающихся на горлышко буты¬
лей, следует подождать, чтобы жидкость успокоилась
до начала подсчета, поскольку маленькие пузырьки,
появляющиеся при быстром диспенсировании, могут
учитываться счетчиком как клетки.
Анализатор размера клеток. Большинство при¬
боров для оценки размеров клеток среднего или высо¬
кого качества (Scharfe, Beckman Coulter) (см. рис. 21.2)
обеспечивают анализ размера клеток и возможность
загрузки данных в персональный компьютер, прямо
или через компьютерную сеть
5.2.9. CCD-камера и монитор
С момента появления микросхем, телевизионные
камеры и мониторы стали ценным помощником при
обсуждении культур и обучении студентов (рис. 5.14).
Выбирайте камеры с высоким разрешением и невысо¬
кой чувствительностью, поскольку чувствительность
стандартного фотоаппарата достаточно высока и
может вызвать эффект сверхсвечения. Черно-белые
камеры обычно дают лучшее разрешение и адекватны
при фазово-контрастной микроскопии живых куль¬
тур. Цветные камеры предпочтительны при изучении
фиксированных и окрашенных образцов. Если вы
предполагаете обсуждать культуры с двумя или более
ассистентами или коллегами, достаточно будет ис¬
пользовать монитор 300- или 400-мм (12-15 дюймов),
дающий хорошее разрешение, но если вы предпола¬
гаете обучать группу из 10 и более студентов, следует
приобрести 500- или 550-мм (19- или 21-дюймовый)
монитор.
CCD-видеокамера (CCD или ПСЗ — прибор с за¬
рядовой связью) с высоким разрешением может быть
полезна при записи и оцифровке изображений для по¬
следующего анализа и публикации данных, приставки
к видеооборудованию позволят снимать как в режиме
нормального времени, так и в замедленном режиме
(см. разд. 27.3).
5.2.10. Препаровальная лупа
Иссечение маленьких кусочков тканей (например,
эмбриональных органов или тканей мелких беспоз¬
воночных) потребует использования препаровальной
лупы (Nikon, Olympus, Leica), которая будет полезна
также при подсчете колоний в монослойной культуре,
94
ГЛАВА 5. ОБОРУДОВАНИЕ
а также подсчете и выборе маленьких колоний, расту¬
щих на агаре.
5.3. ИНКУБАЦИЯ
5.3.1. Инкубатор
Если нет возможности организовать термальную
комнату (см. разд. 4.4.2), может возникнуть необ¬
ходимость приобретения эквивалентного сухого
инкубатора. Даже при наличии термальной комнаты
иногда удобнее иметь дополнительный инкубатор
для трипсинизации культуры поблизости от лами¬
нарного шкафа (приблизительно 50-200 л на одного
сотрудника) с принудительной вентиляцией, темпе¬
ратурным контролем с точностью до ±0,2 °С и термо¬
статом безопасности, который прекратит нагревание,
выключив инкубатор, если температура превысит
необходимую, или, что лучше, примет на себя функ¬
цию регуляции работы инкубатора, если основной тер¬
мостат сломается. Инкубатор должен быть устойчив
к коррозии (например, из нержавеющей стали, хотя
анодированный алюминий тоже подходит для сухо¬
го инкубатора) и легок для уборки. Двойная камера
или два инкубатора, поставленные один на другой и
независимо регулируемые, — предпочтительнее, чем
один большой инкубатор, поскольку в первом случае
обеспечивается лучший температурный контроль для
большего количества культур, к тому же, если один из
инкубаторов сломается или потребует уборки, может
быть использован второй.
Многие инкубаторы имеют водяную рубашку для
равномерного распределения тепла во внутреннем
пространстве, что позволяет избежать холодовых пятен.
Эти инкубаторы дольше держат температуру при по¬
ломке нагревательной системы или выключении элект¬
роснабжения. Тем не менее новые высокоэффективные
изолирующие материалы и диффузная поверхность
нагревательных элементов дают те же преимущества,
но позволяют обойтись без водяной рубашки и делают
перемещение инкубатора более легким (при необходи¬
мости перемещения инкубатора с водяной рубашкой
необходимо слить воду).
Полки инкубатора обычно перфорированы для
улучшения циркуляции воздуха. Однако перфорация
может привести к неравномерности распределения
клеток в монослойных культурах и различию в кле¬
точной плотности в образцах, располагающихся на
разных полках. Такие различия могут возникнуть и
в результате конвекционных потоков, возникающих
над точками контактов чашек с полками по сравнению
с дырками в полках, либо могут иметь отношение к
областям местного охлаждения, возникающим при от¬
крывании дверей. Хотя при рутинных манипуляциях
обычно такие проблемы не возникают, при выполне¬
нии экспериментов, в которых однородная плотность
культуры важна, следует помещать флаконы и чашки
на изолирующий кафель или металлический поддон.
5.3.2. Влажный С02-инкубатор
Хотя культуры могут инкубироваться в закрытых
флаконах в обычном сухом инкубаторе или термальной
комнате, некоторые сосуды, например чашки Петри
или многолуночные планшеты, требуют контроли¬
руемого состава атмосферы с высокой влажностью и
более высоким уровнем С02. Самым дешевым путем
контроля газовой фазы является помещение культуры
в пластиковую коробку или камеру (Bellco, МР Bio¬
medicals), заполнение контейнера газом с необходимой
концентрацией С02 и герметизация ее. Если контейнер
не полностью заполнен чашками, поместите в него
открытые чашки Петри с водой с тем, чтобы повысить
влажность в камере.
С02-Инкубаторы (рис. 5.15) более дороги, но
простота их использования и постоянный контроль
давления С02 и температуры (анаэробные сосуды
и поглотители влаги удлиняют время нагревания)
компенсируют расходы. Контроль влажности до¬
стигается использованием увлажняющих поддонов
(рис. 5.16), а контроль давления С02 — при помощи
С02-регулирующего оборудования, которое всасывает
воздух из инкубатора в отдельную камеру, где опре¬
деляется концентрация С02, а также вводит чистый
С02 в инкубатор для пополнения дефицита. Воздух
циркулирует вокруг инкубатора путем естественной
Рис. 5.15. С02-инкубатор. С02-инкубатор (Forma) с откры¬
той дверью, позволяющей увидеть флаконы, многолуночные
планшеты и чашки Петри, помещенные в коробки (см. также
рис. 5.16)
5.3. Инкубация
95
Регулятор Термочув- I отверстие
температуры ствительныи резервный со Ддля С02 Для воды
терморегулятор 2
Pilot light key
High Heater
Регулятор
температуры
Резервный
терморегулятор
. Впускное
Детектор
I отверстие 1
для С02 , 2 Выключатель
• о
[зтЛ-
• ©1
• о!
| 36.5 |
|
Вентилятор
Рис. 5.16. Устройство С02-инкубатора. Вид спереди контрольной панели и секции внутренней камеры двух стилизованных
влажных С02-инкубаторов. (а) Инкубатор с водяной рубашкой и циркуляцией воздуха за счет вентилятора, (б) Сухостенный
инкубатор без циркуляторного вентилятора (общая схема, не относящаяся к конкретным компаниям-производителям)
конвекции или в результате использования вентиля¬
тора, который поддерживает как уровень С02, так и
температурный режим. Существует мнение, что инку¬
баторы, снабженные вентилятором, восстанавливают
уровень С02 и температуры после открывания двери
инкубатора быстрее, хотя инкубаторы с естественной
циркуляцией также могут восстанавливать режим до¬
статочно быстро, при этом ниже риск контаминации.
Кроме того, сухие, подогретые стенки инкубатора
обеспечивают меньшее заражение стенок грибами,
поскольку остаются сухими даже при относительно
высокой влажности. Некоторые типы контролирую¬
щего уровень С02 оборудования нуждаются в перио¬
дической калибровке каждые несколько месяцев, но
использование золотой проволоки или инфракрасного
детектора минимизирует дрейф нуля и многие модели
автоматически переустанавливают нулевое значение
детектора С02
Размеры инкубатора определяются потребностями
лаборатории — как количеством сотрудников, пользу¬
ющихся им, так и типами культур, с которыми они ра¬
ботают. Пять человек, пользующихся только плашками
для микротитрации, могут использовать 1000 плашек
(приблизительно 100 000 индивидуальных культур)
или проводить 10 экспериментов, каждый из которых
может потребовать инкубатор небольших размеров,
в то время как один человек, ведущий клонирование
клеток, может заполнить одну полку за один или два
эксперимента. Экономически невыгодно инкубировать
в С02-инкубаторах флаконы, особенно больших раз¬
меров. Для этих целей лучше использовать обычные
инкубаторы или термальные комнаты. Если требуется
С02, то флаконы должны быть заполнены газом из
баллонов или С02-газопровода.
Частая уборка инкубатора, в частности влажного, —
важный элемент его эксплуатации (см. разд. 19.1.4),
поэтому внутреннее пространство должно легко раз¬
бираться, не оставляя недоступных для мытья частей
и углов. Вынутые из инкубатора флаконы или чашки
либо коробки, содержащие их, при переносе в лами¬
нарный шкаф и дальнейшем использовании должны
быть обработаны тампоном с этанолом до открывания
(см. разд. 6.3.1).
5.3.3. Регистратор температуры
Регистрирующий термометр с диапазоном измере¬
ния от -50 °С и ниже до +200 °С позволит вам вести
мониторинг условий хранения в морозильной каме¬
ре, замораживания клеток, состояния инкубаторов
и стерилизационных шкафов и представляет собой
инструмент с долгосрочными проводами, покрытыми
тефлоном, и снабженный термистором или термопарой.
Печи, инкубаторы и термальные комнаты требуют по¬
стоянного контроля для осуществления стабильности
температурного режима. Регистрирующие термометры
должны беспрерывно закрепляться в термальной ком¬
нате, стерилизационных шкафах, автоклаве, а их записи
должны регулярно просматриваться для обнаружения
отклонений в работе приборов, в частности в случае
возникающих сбоев.
96
ГЛАВА 5. ОБОРУДОВАНИЕ
5.3.4. Роллерные штативы
Роллерные штативы используются в производстве
монослойных культур в расширенном масштабе (см.
разд. 26.2.3). Выбор аппарата определяется масштабом
культивирования (например, размером и количеством
роллер-бутылей). Этот масштаб может быть определен
исходя из количества клеток, которые планируется
нарастить, максимально доступной клеточной плот¬
ности, и площади рабочей поверхности бутылей (см.
табл. 8.1). Большое количество маленьких бутылей
дает наибольшую площадь поверхности, но повышает
затраты труда и интенсивность ручных манипуля¬
ций, поэтому обычно принимается компромиссное
решение — используются бутыли диаметром 125 мм
(5 дюймов) и длиной от 150-500 мм (6-20 дюймов).
Длина бутыли обусловливает конечный выход про¬
дукта, но ограничивается размерами штатива; высота
штатива будет определять количество креплений (т. е.
рядов) бутылей. Хотя дешевле купить один большой
штатив, чем несколько маленьких, последний вариант
предпочтителен, поскольку:
1) позволит установить штативы ступенчато (убеди¬
тесь, что система работает);
2) упростит расположение штативов в термальной
комнате;
3) позволит при необходимости включать изолирован¬
но один штатив.
Настольные модели Bellco или Thermo Electron
могут полностью удовлетворить запросы культи¬
вирования небольших масштабов, Belco, Integro
и New Brunswick Scientific подходят для больших
масштабов.
5.3.5. Магнитная мешалка
Культивирование тканей предъявляет определенные
специфические требования к магнитным мешалкам.
Быстрое перемешивание для растворения химических
реактивов может проводиться на любых мешалках.
При перемешивании, которое сопровождает фермен¬
тативное расщепление тканей (см. разд. 12.3.3) или
рост суспензионной культуры (см. разд. 13.7.1,13.7.5),
должны соблюдаться некоторые условия:
1) мотор не должен нагревать культуру (используйте
вращающийся индуктор или ременную передачу от
двигателя, находящегося вне мешалки);
2) скорость вращения должна контролироваться и
понижаться до 50 об./мин;
3) вращающий момент при низких оборотах должен
обеспечивать перемешивание объема до Юл
жидкости;
4) оборудование должно обеспечивать поддержание
нескольких культур одновременно;
5) каждое положение мешалки должно индивидуально
контролироваться;
6) считывание данных по частоте вращения должно
осуществляться для каждого держателя. Предпоч¬
тительно иметь специально предназначенную для
культивирования мешалку (Techno или Bellco).
5.4. ПОДГОТОВИТЕЛЬНЫЕ РАБОТЫ
И СТЕРИЛИЗАЦИЯ
5.4.1. Мытье посуды
Ванны и раковины для замачивания. Ванны и ракови¬
ны для замачивания должны быть глубокими настоль¬
ко, чтобы все ваше лабораторное стекло (кроме пипеток
и самых больших бутылей) погружалось в раствор де¬
тергента при замачивании, но не настолько глубокими,
чтобы под весом стекла разрушались мелкие предметы
на дне раковины. Примерные размеры раковины состав¬
ляют 400 мм (15 дюймов) в ширину, 600 мм (24 дюйма)
в длину и 300 мм в глубину (см. рис. 4.6).
Моечная машина для пипеток. Стеклянные пи¬
петки, пригодные для повторного употребления, легко
моются в стандартных моечных машинах сифонного
типа (см. разд. 11.3.2 и рис. 11.5). Лучше разместить
моечную машину на уровне пола, а не на уровне стола,
для того чтобы избежать неудобств при вынимании
пипеток, и соединить с оборудованием, деионизиру¬
ющим воду, и с подводкой холодной воды так, чтобы
последние несколько полосканий можно было прово¬
дить деионизованной водой. Если возможно, следует
встроить простой клапан-переключатель в систему
подводки деионизованной воды (см. рис. 4.6).
Сушка пипеток. Если в моечной машине для пи¬
петок используется корзина из нержавеющей стали,
пипетки можно прямо перенести в электрическую су¬
шилку. В другом варианте их высушивают на штативе
или в обычном сушильном шкафу.
5.4.2. Очиститель воды
Очищенная вода требуется для прополаскивания стек¬
лянной посуды, растворения порошкообразной среды
и разведения концентратов. Для первой цели обычно
достаточно деионизованной или обратноосмотически
обессоленной воды. Вторая и третья задачи требуют
сверхчистой воды (UPW), полученной в результате
трех- или четырехступенчатого процесса (рис. 5.17; см.
также рис. 11.10). Важным принципом такого процесса
является качественное отличие каждой стадии от дру¬
гих. Обратный осмос может следовать за фильтрацией
через активированный уголь, деионизацией и микро-
поровой фильтрацией (т.е. через стерилизационный
фильтр, см. рис. 11.11). Первая стадия может быть
заменена дистилляцией (с использованием элемента
в кремниевой оболочке). Обратный осмос дешевле,
если вы платите за топливо, если нет, то дистилляция
лучше и более приемлема для получения стерильного
продукта. При использовании обратного осмоса нуж¬
но выбрать такой тип картриджа, который обеспечит
необходимое значение pH; некоторые мембраны могут
5.4. Подготовительные работы и стерилизация
97
Рис. 5.17. Очиститель воды. Водопроводная вода сначала проходит через реверсивно-осмотический блок справа и затем в
емкость-хранилище слева. Затем она очищается на углеродном фильтре и проходит через деионизатор (центральный блок) и
микропоровый фильтр (Millipore Milli-Q) накапливаясь в коллекторе. (Фото любезно предоставлено Beatson Institute.)
прорываться при крайних значениях pH (уточните у
поставщика).
Деионизатор должен иметь измеритель проводи¬
мости (кондуктометр), контролирующий сточный по¬
ток, для обнаружения необходимости замены картрид¬
жа. На других картриджах необходимо помечать дату
установки и заменять в соответствии с инструкцией
производителя. Для контроля коллоидного состояния
можно использовать измеритель общего органического
углерода (TOC) (Millipore).
Очищенная вода не должна храниться, ее следует
отправлять на повторную очистку, чтобы миними¬
зировать инфицирование водорослями или другими
микроорганизмами. Все трубки и резервуары в системе
должны регулярно проверяться (каждые три месяца
или около того) на наличие водорослей, промываться
гипохлоридом и детергентом (например, Clorox или
Chloros), тщательно промываться проточной водой и
ополаскиваться дистиллированной водой перед ис¬
пользованием.
Вода является самым простым, но, возможно, наи¬
более критичным составляющим элементом всех сред
и реагентов, в частности бессывороточных сред (см.
гл. 10). Хороший контроль качества воды состоит в
исследовании эффективности посева чувствительной
клеточной линии в среде, сделанной на основе этой
воды (см. разд. 11.6.3 и протокол 21.10) через равно¬
мерные промежутки времени.
5.4.3. Стерилизация и сушильные шкафы
Хотя вся стерилизация должна проводиться в ав¬
токлаве, пипетки и другое стекло предпочтительно
стерилизовать сухим жаром, что позволяет избегать
возможности как химического загрязнения от парового
конденсата, так и коррозии пеналов для пипеток. Такая
стерилизация, однако, требует высокой температуры
(160-180 °С) и принудительно вентилируемых печей,
чтобы обеспечить прогревание всей массы загружен¬
ных материалов. Так же как в отношении автоклава,
при покупке стерилизационных шкафов не выбирайте
один большой, который может быть слишком велик
для размеров и количества используемой стеклянной
посуды. Лучше приобрести два маленьких шкафа; про¬
гревание их легче, быстрее и равномернее, крометого,
экономичнее использовать один маленький шкаф при
небольшом количестве стеклянной посуды, которую
нужно простерилизовать. Вы также в большей степени
защищены от поломок.
98
ГЛАВА 5. ОБОРУДОВАНИЕ
5.4.4. Паровой стерилизатор (автоклав)
Самый простой и дешевый способ стерилизации — бы¬
товой автоклав, который создает давление 100 кПа
(1 атм) выше атмосферного. Другой вариант — на¬
стольный автоклав (рис. 5.18), имеющий автома¬
тическое программирование и запор безопасности.
Напольная модель больших размеров с программиру¬
емым таймером, выбором режима вакуумирования до
и после стерилизации и температурным регистратором
(рис. 5.19) имеет большие возможности, обеспечивает
большую вариабельность режима, и дает перспективы
для соблюдения требования GLP (good laboratory prac¬
tice, надлежащая лабораторная практика).
«Влажный» цикл (вода, солевые растворы и т.д.)
осуществляется без удаления воздуха из камеры до
или после стерилизации. Сухие предметы (инструмен¬
ты, щетки, закручивающиеся пробки и т.д.) требуют
вакуумирования до начала стерилизации либо воздух
должен быть вытеснен в ходе стерилизации для эф¬
фективного распространения горячих потоков. Камера
должна быть также вакуумирована после стерилизации,
для удаления пара и обеспечения последующей сушки;
в противном случае все предметы останутся влажны¬
ми, появится опасность заражения из конденсата при
сушке. Для минимизации риска заражения, если у вас
нет возможности «поствакцинального» цикла, всегда
используйте деионизованную или обратноосмотически
обработанную воду для подачи в автоклав. Если вам
требуется автоклавировать большие объемы (>300 л),
предпочтительнее купить два автоклава средних раз¬
меров, поскольку в ходе рутинных манипуляций и
случайных поломок одного вы сохраните другой в ра¬
ботающем состоянии. Кроме того, оборудование сред¬
них размеров будет нагреваться и остывать быстрее и
использоваться более экономично для небольших объ¬
емов автоклавируемого материала. Оставляйте допол¬
нительное пространство вокруг автоклава для уборки
и вентиляции, обеспечивая необходимую экстракцию
воздуха для выведения тепла и пара, удостоверьтесь,
что имеется возможность вытекания конденсата.
Большинство маленьких автоклавов имеют соб¬
ственный парогенератор (калорифер); большие маши¬
ны также могут быть снабжены собственным парогене¬
ратором либо использовать подводящую пар линию.
Если в вашем распоряжении имеется паровая линия
высокого давления, то подключение к ней будет самым
простым и дешевым методом подогрева и повышения
давления в автоклаве; если нет, то лучше всего закупать
автоклав в комплекте с собственным парогенератором.
Такой автоклав будет дешевле устанавливать и легче
передвигать. При покупке больших автоклавов выбор,
вероятно, будет отсутствовать, поскольку чаще всего
предлагаются модели с отдельным генератором. В этом
случае следует планировать площадь под размещение
автоклава и генератора.
5.4.5. Весы
Хотя большинство лабораторий получают уже готовые
среды для культивирования, иногда дешевле готовить
некоторые реагенты на месте. В этом случае вам по¬
требуются весы (электронные весы с автоматическим
тарированием — наилучшие), способные взвешивать
от 10 мг до 100 г или даже до 1 кг, в зависимости от
Рис. 5.18. Настольный автоклав. Простой автоклав (Prestige Medical); слева с закрытой крышкой, справа - крышка снята.
(Фото любезно предоставлено Beatson Institute.)
5.4. Подготовительные работы и стерилизация
99
Рис. 5.19. Напольный автоклав. Лабораторный автоклав средних размеров (300 л, 10 футов3) с квадратной камерой для мак¬
симальной загрузки. Самописец на верхней консоли связан с датчиком в бутылке, располагающейся в центре загруженной
камеры. (Фото любезно предоставлено Beatson Institute.)
масштабов работы. Если вы являетесь поставщиком
услуг, то требуется готовить большое количество ре¬
агента, иногда в концентрации 10х, поэтому вес взве¬
шиваемого вещества может быть достаточно большим.
Хорошо зарекомендовало себя использование пары
весов — грубых и точных, в этом случае расходы прак¬
тически одинаковы, а точность и удобство пользования
возрастают.
5.4.6. рН-Метр
Простой рН-метр является полезным дополнением
к лабораторному оборудованию для приготовления
питательных сред и реагентов при культивировании
тканей. Хотя для мониторинга кислотности большинс¬
тва растворов достаточно фенолового красного индика¬
тора, бывают случаи, когда феноловый красный нельзя
применить (например, в приготовлении кулыур для
флюоресцентного анализа и в приготовлении основных
растворов), в этом случае потребуется рН-метр.
5.4.7. Магнитная мешалка с подогревом
В дополнение к магнитной мешалке, работающей при
комнатной температуре и используемой при трип¬
синизации культур и росте суспензионных культур,
желательно иметь магнитную мешалку с подогревом
для ускорения растворения некоторых реагентов.
Можно поместить обычную мешалку в термальной
комнате, однако при длительном нахождении солево¬
100
ГЛАВА 5. ОБОРУДОВАНИЕ
го раствора при температуре 37 °С может развиться
микробный рост, поэтому термостабильные реагенты
лучше растворять при более высокой температуре за
более короткое время.
5.4.8. Автоматический диспенсер
Для разлива объемов до 50 мл можно использовать
диспенсеры, накручивающиеся на горлышко бутыли
(см. рис. 5.20). Для больших объемов, если точность
измерения объема не критична для работы, лучше
использовать диспенсер, работающий под действием
силы тяжести. Жидкость вытекает по трубке из ре¬
зервуара, например из градуированной бутыли (см.
рис. 5.7) или пластикового пакета. Если отмеряемый
объем должен быть точным, предпочтительно исполь¬
зование перистальтического насоса (см. рис. 5.9). Точ¬
ность и объем разливаемой жидкости определяются
продолжительностью диспенсирования и диаметром
Рис. 5.20. Диспенсер, соединяющийся с бутылкой. Пру¬
жинный поршень, связанный с двусторонним клапаном, по¬
очередно откачивающим определенный объем из резервуара
и направляющим жидкость по отводной трубке в сосуд сбоку.
Устройство используется для разливания повторяющегося
объема нестерильной жидкости
трубок. Длинный цикл и тонкие подводящие трубки
могут давать большую точность, широкие трубки уско¬
ряют процесс.
5.4.9. Измеритель электропроводности
(кондуктометр)
При изготовлении растворов реагентов в лаборатории
их качество повышается (см. разд. 11.6.1). Простая
проверка концентрации ионов может проводиться
при помощи кондуктометра против известного стан¬
дарта, такого как обычный раствор поваренной соли
(0,15 М).
5.4.10. Осмометр
Одним из наиболее важных физических свойств
культуральной среды, которое к тому же трудно пред¬
сказать, является осмолярность. Проводимость опре¬
деляется концентрацией ионизированных молекул, но
в осмолярность могут также вносить вклад неионизи-
рованные молекулы. Поэтому осмометр (см. рис. 9.1)
полезен для проверки приготовленных растворов,
регулировки новых композиций питательных веществ
или для компенсации осмолярности при добавлении в
среду каких-то реагентов. Осмометры обычно работа¬
ют за счет понижения точки замерзания раствора или
среды или повышения давления пара. При покупке вы¬
бирайте прибор с маленьким объемом анализируемого
образца (<1 мл), поскольку вам может потребоваться
измерить осмолярность ценного реагента и точность
измерения (±10 моемо л/кг) будет менее важна, чем
ценность реагента.
5.4.11. Моечная машина
для стеклянной посуды
Возможно, наилучшим способом получения чистой
стеклянной посуды является наличие лаборанта, ко¬
торому вы доверяете, однако при слишком большом
количестве посуды может быть ценным приобретение
автоматической моющей машины (рис. 5.21). Некото¬
рые из тех, которые представлены на рынке, достаточно
удовлетворительны по качеству. Обратите внимание на
следующие моменты:
1) Внимательно выбирайте штативы и держатели с
индивидуальными втулками, над которыми вы мо¬
жете разместить бутыли, флаконы и т. д. Открытые
сосуды, такие как чашки Петри и химические ста¬
каны, могут быть вымыты достаточно хорошо при
помощи разбрызгивания и вращающейся формы,
но узкогорлые сосуды требуют индивидуального
высокоскоростного потока. Форсунки для потока
должны иметь прокладки или коврики в осно¬
вании для предохранения горлышка бутылки от
скалывания.
5.5. Хранение
101
Рис. 5.21. Моечная машина для стекла. Стеклянная посуда
и другие изделия помещаются на индивидуальные патрубки,
которые обеспечивают тщательное мытье и ополаскивание
(Betterbuilt). (Фото любезно предоставлено Beatson Insti¬
tute.)
2) Насос, который накачивает воду через форсунки,
должен обеспечивать достаточно высокое давление,
около 2-5 гПа, в зависимости от размеров машины.
3) Вода для мытья должна быть подогрета, по крайней
мере, до 80 °С.
4) Должно иметься приспособление для ополаски¬
вания деионизованной водой в конце цикла. Вода
должна быть подогрета до 50-60 °С; в противном
случае стекло может треснуть от резкого перепада
температуры. Вода для полоскания должна пода¬
ваться как проточная струя с удалением без повтор¬
ного использования. Если повторное использование
неизбежно, то потребуется, по меньшей мере, трое¬
кратное прополаскивание в деионизованной воде.
5) Предпочтительно удалять воду, оставшуюся от
последнего полоскания одной партии посуды, и не
оставлять ее для предварительного ополаскивания
следующей партии. Слив воды после полоскания
уменьшает риск химического загрязнения, особенно
когда машина используется для мытья химической
и радиоизотопной посуды.
6) Машина должна быть облицована нержавеющей
сталью и соединена с нержавеющими или нейло¬
новыми трубами.
7) При возможности, следует выбрать такую машину
для сушки стекла, в которой можно использовать те
же поддоны и держатели, что и в моечной машине
(см. рис. 5.21), чтобы можно было прямо перемещать
посуду, не снимая ее с держателей, с использовани¬
ем подходящих тележек или роликов.
5.5. ХРАНЕНИЕ
5.5.1. Холодильники и морозильники
Обычно бытовые холодильники и морозильники
достаточно эффективны и дешевле, чем специальное
оборудование. Существуют бытовые холодильники
без морозильной камеры («большие холодильники»)
с большим пространством и реже возникающей необ¬
ходимостью размораживания. Однако, если вам требу¬
ется хранить объем 400 л или больше (см. разд. 4.5.3),
большой больничный банк для хранения крови или
промышленный холодильник для продуктов может
оказаться лучше.
Если площадь помещения достаточно велика, а ко¬
личество людей, использующих культуру тканей, более
трех или четырех, лучше подумать о холодной комна¬
те, содержание которой по сравнению с несколькими
отдельными холодильниками экономически более
выгодно из-за большего пространства, а также легче
для доступа. Стены холодной комнаты должны быть
гладкими и легко мыться, стеллажи должны быть снаб¬
жены колесиками, чтобы можно было их перемещать
при уборке помещения. Холодная комната должна уби¬
раться регулярно, с удалением старых запасов; стены и
полки следует мыть с антисептиками для минимизации
заражения грибами.
Подобное оборудование можно использовать для
замораживания — несколько недорогих бытовых моро¬
зильников будут дешевле и эффективнее, нежели спе¬
циальный лабораторный морозильник. Большинство
реагентов для культивирования тканей хранятся при
температуре -20 °С, поэтому сверхглубокое заморажи¬
вание не требуется. Создание комнаты для глубокого
замораживания не рекомендуется — ее очень трудно и
неприятно убирать; возникает много проблем, связан¬
ных с перемещением ее содержимого при тщательной
уборке.
Хотя использование саморазмораживающихеся
морозильников нежелательно для некоторых реакти¬
вов (ферментов, антибиотиков и пр.), они достаточно
полезны для хранения большинства запасов, необходи¬
мых для культуры тканей, структура которых позволяет
выдержать колебания температуры, а природа менее
чувствительна к острому криогенному повреждению.
Теоретически сыворотки могут испортиться при ко¬
лебаниях температуры в саморазмораживающихся
морозильниках, но на практике этого не происходит.
Многие важные составляющие сыворотки — малые
протеины, полипептиды и простые органические и
неорганические компоненты — могут быть нечувстви¬
102
ГЛАВА 5. ОБОРУДОВАНИЕ
тельны к криогенному повреждению, в частности, если
растворы хранятся в объемах >100 мл.
5.5.2. Контейнеры для криоконсервации
Подробное описание контейнеров для криоконсерва¬
ции и советы по выбору этого оборудования приводятся
в гл. 20 (см. разд. 20.3.5). Коротко говоря, выбор зависит
от размеров и типа системы, которые требуются для
ваших нужд. Для маленькой лаборатории подойдет 35-л
сосуд с узким горлышком. Использование емкостей и
канистр (см. рис. 20.6а, 20.7а) или ящиков в системе
стеллажей (см. рис. 20.6г, 20.7в) должно обеспечить
хранение примерно 500-1000 ампул с культурами (см.
Приложение II). Морозильное оборудование большего
объема, которое вмещает >10 000 ампул и включает мо¬
дели со стенами, обрызганными жидким азотом, помо¬
жет снизить расходы азота и обеспечит более надежное
хранение образцов без хранения собственно жидкого
азота. При выборе хранилища с разбрызгиванием азота
по стенам важно обеспечить необходимые меры предо¬
сторожности от попадания некоторых материалов, воды
и водяных паров в перфузионную систему, поскольку
засор системы трудно, порой невозможно удалить.
Следует приобрести соответствующие сосуды для
хранения, которые позволят иметь резерв жидкого азота.
Размеры сосудов зависят от 1) размеров морозильного
оборудования; 2) частоты и возможности приобретения
жидкого азота; 3) скорости испарения жидкого азота;
для 35-л узкогорлого морозильника, использующего
5-10 л в неделю, потребуется только 25-л сосуд Дюара
при доступности регулярного его заполнения. Большие
морозильники лучше заполнять азотом по системе труб
из постоянного хранилища (например, 160-л сосуд
для хранения, соединенный с 320-л морозильником со
встроенной системой заполнения и аварийной сигнали¬
зацией, либо 500-л цистерна для большего морозильни¬
ка или нескольких меньших морозильников).
5.5.3. Морозильники с управляемым
процессом замораживания
Хотя клетки можно заморозить непосредственно поме¬
щением в изолированный шкаф при -70 °С, некоторые
клетки требуют различных скоростей охлаждения или
сложного программирования скорости охлаждения
(см. разд. 20.3.4). Программируемый морозильник
(например, Cryomed, Planer) обеспечивает управление
процессом замораживания с возможностью измене¬
ния параметров замораживания путем впрыскивания
жидкого азота в морозильную камеру под контролем
заранее установленной программы (см. рис. 20.5). Бо¬
лее дешевый вариант контроля охлаждения — клеток с
помощью систем Taylor Wharton, специальных замора¬
живающих шкафов (Nalge Nunc), а также контейнеров
из простой полистиреновой пены или пеноизоляции
труб водоотвода (см. рис. 20.2-20.4).
5.6. ЛАБОРАТОРНЫЕ ПРИБОРЫ
5.6.1. Компьютеры и сети
Большинство лабораторий оборудовано одним или бо¬
лее компьютером, которые могут быть соединены в сеть.
В зависимости от того, располагается ли компьютер или
терминал в помещении лаборатории культуры тканей,
занесение в базу данных записей по ведению клеточ¬
ных линий (см. разд. 13.7.8) первичных культур (см.
разд. 12.3.11) и протоколы экспериментов значительно
облегчают последующий поиск этой информации и ее
анализ. Данные по ведению клеточных линий лучше со¬
хранять в базе данных компьютера, что может служить
для учетного контроля замороженных в азоте культур
(см. разд. 20.4.1). В больших лабораториях контроль
запасов пластика, стекла, реактивов и питательных сред
также можно упростить использованием компьютера.
У внутренних сетей есть определенные преимущества:
поскольку индивидуальные компьютеры могут быть
дублированы централизованно на основе стандартного
программного обеспечения, постольку информация,
внесенная на любом пункте, может быть доступна всем
пользователям. Например, фотографии, сделанные
в лаборатории культивирования тканей, могут быть
сохранены на центральном сервере и извлечены из
центрального офиса или зоны ведения документации.
5.6.2. Прямой микроскоп
Наряду с инвертированным микроскопом, применяе¬
мым для хромосомного анализа, определения контами¬
нации микоплазмами и авторадиографии в лаборатории
может потребоваться прямой микроскоп. На самом деле,
инвертированный микроскоп применяется также для
оценки состояния культуры, контаминации бактериями
и грибами. Препараты авторадиографии и хромосомные
препараты можно смотреть и на прямом микроскопе. Вы¬
бирайте микроскоп с высоким разрешением (Leica, Zeiss
или Nikon), со светлопольной оптикой и увеличением
объектива до хЮО; фазово-контрастным объективом,
по крайней мере, х40, но предпочтительно хЮО и флю¬
оресцентной оптикой с подсветкой и объективами х40
и хЮО. Флюоресцентная оптика будет использоваться
для обнаружения микоплазм (см. протокол 19.2) и иссле¬
дования флюоресцентных антител. Leica предоставляет
вводно-иммерсионные объективы с увеличением 50х,
которые, в частности, полезны для изучения стандарт¬
ных препаратов микоплазм, окрашенных по Hoechst.
Автоматическая цифровая камера или CCD- обору¬
дование также полезны для фотографической формы
регистрации данных или постоянных препаратов.
5.6.3. Низкотемпературный морозильник
Большинство реактивов для ведения культивирования
могут храниться при температуре 4 °С или -20 °С, од¬
5.7. Специальное оборудование
103
нако некоторые препараты или продукты, выделенные
из культур, могут потребовать хранения при -70 °С
или -90 °С. При этой температуре замерзает вся вода
и скорость протекания большинства химических и
радиолитических реакций резко ограничивается.
Низкотемпературный морозильник -70 °С или -90 °С
также используется для замораживания клеток в
теплоизолирующем контейнере (см. протокол 20.1).
Тип морозильной камеры, располагающейся на уров¬
не груди, более экономичен и приводит к меньшему
расходу энергии, но вертикальный шкаф с выдвиж¬
ными ящиками, стоящий на полу, выглядит менее
экстравагантно и обеспечивает более легкий доступ к
хранящемуся содержимому. Если вы сделали выбор в
пользу вертикального морозильника, удостоверьтесь,
что в нем имеются 6-8 индивидуальных отделений на
объем 400 л с отдельными плотно закрывающимися
дверцами, и ожидайте, что вам придется заплатить за
него, по крайней мере, на 20% больше, чем за моро¬
зильную камеру.
Низкотемпературный морозильник производит
большое количество тепла, которое должно отводиться
для эффективной работы (или вообще работы). Такие
морозильники должны располагаться в хорошо вен¬
тилируемой или снабженной кондиционером воздуха
комнате, чтобы постоянная комнатная температура не
превышала 23 °С. Если это невозможно, вложите сред¬
ства в морозильник, приспособленный для тропических
условий, в противном случае перед вами постоянно
будет стоять проблема эксплуатации, размораживания
и более короткого срока службы со всеми соответ¬
ствующими осложнениями по перемещению ценных
запасов. Одна или две поломки вызовут расходы на
починку ($1000 и более) и потерю ценных материалов,
что вскоре сведет на нет все преимущество, которое вы
получите, покупая более дешевый морозильник.
5.6.4. Конфокальный микроскоп
Цитологические исследования флюоресценции ме¬
ченых клеток при высоком разрешении часто более
успешны, если проводятся при помощи конфокального
микроскопа. Эта техника позволяет микроскопу прово¬
дить «оптическое сечение» через образец, предоставляя
изображение в одной фокальной плоскости и избегая
интерференции, вызванной соседними клетками, рас¬
положенными в другой фокальной плоскости. Данные
накапливаются, хранятся в цифровом виде и могут
быть представлены различными способами, включая
создание образа вертикального сечения образца (т. н.
Z-сечение), что, в частности, полезно при осмотре
трехмерной культуры.
5.6.5. Термоциклер
При сертификации клеточной линии используется
большое количество дополнительного оборудования,
например для обнаружения микоплазм (см. прото¬
кол 19.3) и определения профиля ДНК (см. прото¬
кол 16.9), работа которого основана на амплификации
и обнаружении специфической последовательности
ДНК. Если вы планируете применять этот метод в
вашей лаборатории, помните, что в нем используется
полимеразная цепная реакция (ПЦР) и требуется ам-
плификатор.
5.7. СПЕЦИАЛЬНОЕ ОБОРУДОВАНИЕ
5.7.1. Установка для микроинъекций
Для введения чего-то непосредственно в клетку, напри¬
мер для ядерной трансплантации или инъекции краси¬
теля, используют микроманипуляторы (рис. 5.22).
5.7.2. Счетчик колоний
Монослойные колонии можно легко подсчитать на
глаз или при помощи препаровальной лупы и фломас¬
тера, которым отмечают колонии, но при необходи¬
мости обсчитать большое количество колоний может
помочь автоматический счетчик колоний. Самый
простой прибор включает разметочный карандаш с
электродом на конце, прикосновение которым к по¬
верхности чашки включает счетчик. Часто счетчики
снабжены мощной линзой, позволяющей помочь визу¬
ализации колоний. Дальнейшее усложнение прибора
с возрастанием стоимости приводит к электронному
счетчику с предварительно установленной програм¬
мой, которая позволяет подсчитывать колонии, ис¬
пользуя программы анализа изображения (Optomax,
Рис. 5.22. Микроманипуляторы и подогреваемый столик.
Столик с контролируемой температурой поверхности (Pel¬
tier) для видеосъемки в замедленном режиме или микрои¬
нъекций, с возможностью перфузии и отбора проб. (Фото
любезно предоставлено Beatson Institute.)
104
ГЛАВА 5. ОБОРУДОВАНИЕ
Symbiosis). Эти счетчики очень высокоскоростные и
позволяют учитывать колонии различного диаметра,
а также различать соседние слившиеся колонии (см.
разд. 21.10.2)
5.7.3. Центрифужный элютриатор
Центрифужный элютриатор представляет собой спе¬
циально адаптированную центрифугу, подходящую
для разделения клеток по размерам (см. разд. 15.2.2).
Это оборудование дорогостоящее, но эффективное, в
частности для получения большого выхода клеток.
5.7.4. Проточный цитометр
Этот прибор может анализировать клеточные популя¬
ции в соответствии с широким диапазоном параметров,
включая светорассеяние, поглощение света и флюорес¬
ценцию (см. разд. 15.4, 21.7). Многопараметрический
анализ представлен в двух- или трехмерном формате.
По типу анализа эти приборы обычно известны как
проточные цитометры (см. разд. 21.7.2, например
BD Bioscences Cytoster), но сигналы, которые они
порождают, также используют в клеточном сортере
с активированной флюоресценцией для выделения
индивидуальных клеточных популяций с высокой
степенью разрешения (например, BD Bioscences
FACStar). Стоимость проточного цитометра высока
($100 000-200 000), и наилучшие результаты могут
быть получены при помощи квалифицированного
оператора, имеющего специальные навыки.
5.8. РАСХОДНЫЕ МАТЕРИАЛЫ
Эта категория материалов включает основные пред¬
меты, такие как пипетки и контейнеры для пипеток,
культуральные флаконы, ампулы для замораживания,
центрифужные пробирки и стаканы (10-15 мл, 50 мл,
250 мл Sterlin, Nalge Nunc), одноразовые шприцы и
иглы (21-23 G для удаления жидкости из пузырька
или ампулы, 19 G для измельчения клеток), фильтры
различных размеров (см. разд. 11.5.2) для стерилиза¬
ции жидкостей, хирургические перчатки и бумажные
полотенца.
5.8.1. Пипетки
Пипетки должны быть продуваемого типа, с широким
отверстием в носике для быстрого удаления жидкости
и градуированные до носика с максимальным делением
шкалы на другом конце пипетки. Можно использовать
одноразовые пипетки, но они дороги и лучше сохранить
их в качестве запасных на период выходных дней, когда
не работает технический персонал либо на случай по¬
ломки моечной машины или стерилизатора. Пипетки
многократного использования после употребления
собираются в контейнеры для пипеток или лотки, по
одному на рабочее место. В отношении пастеровских
пипеток наилучшим образом зарекомендовали себя
одноразовые пипетки, использованные пастеровские
пипетки не следует помещать в общие цилиндры для
пипеток, используйте для этого безопасную емкость
для стеклянных отходов. Кроме того, можно пользо¬
ваться одноразовыми пластиковыми пастеровскими
пипетками (Pastettes).
Контейнеры для пипеток. Стеклянные пипетки
обычно стерилизуют в алюминиевых или никелиро¬
ванных стальных контейнерах. Квадратные в сечении
контейнеры предпочтительнее круглых, поскольку
они удобнее складируются и не покатятся по по¬
верхности рабочего стола. Существуют различные
варианты контейнеров с обложенными силиконовой
резиной крышкой и дном во избежание скола или
разбивания пипеток при манипуляциях (Thermo
Electron, Bellco).
Пластиковые либо стеклянные пипетки. Во
многих лабораториях привыкли к использованию
пластиковых пипеток, имеющих ряд преимуществ
перед стеклянными: они поступают предварительно
стерилизованными, упакованными, и не вызывают
опасности, связанной со сколом или разбиванием
стеклянных пипеток. Кроме того, их не требуется
мыть, что достаточно затруднительно, или затыкать
ватными фильтрами, что утомительно. С другой сто¬
роны, они значительно дороже, и, если они упакованы
индивидуально, их дольше вынимать из упаковки при
употреблении. Если же такие пипетки упакованы пар¬
тией, повышается риск загрязнения в случаях, когда
упаковка не подразделена на секции (см. разд. 6.3.5).
Пластиковые пипетки также увеличивают косвенные
расходы, особенно если их необходимо дезинфициро¬
вать до утилизации.
При грубой прикидке, расходы на пластиковые пи¬
петки на одного человека ежегодно составляют около
$2000. Количество людей, таким образом, определяет
выбор между стоимостью приобретения пластиковых
пипеток или оплатой труда лаборанта, моющего и
стерилизующего стеклянные пипетки. (При расчетах
помните, что на стеклянные пипетки ежегодно расхо¬
дуется $200 на человека, кроме того, требуется расход
энергии на мойку и стерилизацию.)
5.8.2. Гемоцитометр
При работе с клетками полезно иметь несколько
методов, позволяющих оценивать их количество.
Гемоцитометрические камеры наиболее дешевы и
будут полезным дополнением при оценке жизнеспо¬
собности клеток методом освобождения красителя,
(см. разд. 21.1.1, 22.3.1, рис. 21.1., цв. вкладку 17а и
протоколы 21.1, 22.1).
5.8. Расходные материалы
105
5.8.3. Культуральные сосуды
Выбор культуральных сосудов определяется: 1) требуе¬
мым выходом клеток (см. табл. 8.1); 2) типом культуры:
монослойная или суспензионная; и 3) режимом куль¬
тивирования (например, будут ли образцы отбираться
одновременно или через определенные промежутки
времени) (см. разд. 21.8). Покупая «все подряд», вы
можете выиграть в цене, но не поддавайтесь соблазну
менять продукты слишком часто, и всегда испытывайте
новые образцы культуральных сосудов, прежде чем
приобретать их партией (см. разд. 11.6.3).
Обратите особое внимание на этикетки: стерильно
или нестерильно, для культуры тканей или не для
работы с культурами тканей, качество пластика, — все
эти категории хранятся отдельно. Стеклянные бутылки
с плоской стороной (матрасы) могут использоваться
вместо пластиковых при возможности их обработки:
мытья и стерилизации. Так или иначе, более низкая цена
стеклянной посуды компенсируется превосходством
оптических характеристик, стерильностью, гарантией
качества и общим удобством использования пласти¬
ковых флаконов. Однако одноразовый пластик сейчас
составляет примерно 60% от бюджета культуральных ла¬
бораторий — даже больше, чем стоимость сывороток.
Чашки Петри менее дороги, чем флаконы, однако их
содержимое более подвержено заражению и чаще раз¬
ливается. В зависимости от вида работ и стерильности
окружения, использование чашек Петри может быть
достаточно удобно, например, при использовании в экс¬
перименте, не связанном с ведением культуры. Чашки
Петри также полезны для исследования колониеобра¬
зования, когда в конце эксперимента колонии должны
быть окрашены и подсчитаны или изолированы.
5.8.4. Стерильные контейнеры
Для иссечения тканей потребуются чашки Петри
(9 см), маленькие бутылочки (5 мл), 30-мл универ¬
сальные контейнеры или 50-мл баночки для хранения,
15- и 50-мл пробирки для центрифугирования и 1-, 2-мл
пластиковые пузырьки для консервирования (Nalge
Nunc) при замораживании в жидком азоте (см. прото¬
кол 20.1). Удобно использование пузырьков с цветовой
маркировкой (Alpha Laboratories; см. цв. вкладку 22).
5.8.5. Шприцы и иглы
Несмотря на то что при стандартных манипуляциях
частое использование игл и шприцов не рекомендуется
(в связи с опасностью травматизации, требованиями
стерильности и проблемами стресса клетки деформа¬
цией сдвига), при фильтрации в соединении с фильтру¬
ющими насадками шприцы необходимы, а иглы могут
потребоваться для экстракции реагентов (лекарствен¬
ных препаратов, антибиотиков или радиоизотопов) из
закупоренных пузырьков.
5.8.6. Стерилизационные фильтры
Хотя обычно в лаборатории культивирования тка¬
ней доступно стерилизующее оборудование, в боль¬
шинстве лабораторий используются стерилизующие
фильтры. Целесообразно иметь в запасе несколько
фильтрующих насадок различного размера, например
25-мм насадку для шприца (Pall Gelman, Millipore) и
47-мм насадку для бутылок или колбу для фильтро¬
вания (Falcon, Nalge Nunc). Хорошо иметь под рукой
небольшой набор фильтров еще больших размеров
(см. разд. 11.5.2).
5.8.7. Бумажные полотенца и салфетки
Возле каждого рабочего места или ламинарного шкафа
следует обеспечить расположение бумажных полотенец
и салфеток.
5.8.8. Дезинфектанты
Все биологические материалы после работы предпоч¬
тительно отправлять в дезинфицирующий раствор
для предупреждения роста в емкостях для утилизации
микроорганизмов, потенциально способных привести к
заражению культур. При использовании тканей чело¬
века и других приматов обязательно помещать все био¬
логические материалы в дезинфектант (см. разд. 7.8.5).
Чаще всего используются дезинфектанты на основе
хлора, которые поставляются на рынок в виде концен¬
трата (Clorox, Chloros) или таблеток (Precept).
Глава 6
МЕТОДЫ АСЕПТИКИ
6.1. ЦЕЛИ АСЕПТИКИ
Заражение микроорганизмами остается главной про¬
блемой культивирования тканей. Бактерии, микоп¬
лазмы, дрожжи и споры грибов попадают в культуру
через оператора, из воздуха, с рабочей поверхности, из
растворов и многих других источников (см. разд. 19.1 и
табл. 19.1). Асептические методы работы имеют целью
исключить заражение путем установления жестких
норм, правил и гарантий того, что каждый сотрудник,
использующий оборудование, придерживается их.
Заражение может быть незначительным и ограни¬
чиваться одной-двумя культурами, может распростра¬
ниться на несколько культур и погубить весь экспери¬
мент, либо окажется обширным и уничтожит все ваши
(и даже всей лаборатории) культуры. Катастрофу мож¬
но минимизировать, если: 1) при каждой манипуляции
с культурами тщательно проверять их визуально, при
помощи микроскопа, желательно фазово-контрастного;
2) культуры ведутся без использования антибиоти¬
ков, предпочтительно все время, но, по крайней мере,
часть времени, для установления скрытого заражения;
3) реактивы проверяются на стерильность (вами или
поставщиком) до начала их использования; 4) буты¬
ли со средой и другими реагентами не используются
несколькими людьми или для различных клеточных
линий; 5) постоянно выполняются стандартные мето¬
ды стерилизации, уровень стандартов поддерживается
на высоком уровне.
Одну из наиболее сложных проблем представ¬
ляет собой микоплазменная инфекция, невидимая
при обычной микроскопии. Если ее не обнаружить,
она может распространиться на другие культуры
по всей лаборатории. Такими образом, очень важно
дублировать визуальный контроль контаминации
проведением теста на заражение микоплазмами, осо¬
бенно если растущая культура выглядит необычно
(см. разд. 19.3.2).
6.1.1. Поддержание стерильности
Соблюдение правил и методов асептики обеспечивает
барьер между микроорганизмами окружающей среды
вне культуры и чистой, незараженной культурой во
флаконе или в чашке. Следовательно, все материалы,
которые входят в непосредственный контакт с культу¬
рой, должны быть стерильны, а все манипуляции долж¬
ны проводиться так, чтобы не было прямого контакта
между культурой и нестерильным окружением.
Установлено, что барьер стерильности не может
быть абсолютным без создания условий, которые
значительно затрудняют стандартные манипуляции.
Поскольку проверка соблюдения индивидуальных
мер предосторожности — очень обширное и затруд¬
нительное исследование, все операции адаптируются
на основе опыта и здравого смысла исследователя.
Асептические методы представляют собой комбинацию
действий, призванных снизить возможность иниции¬
рования. Корреляция между невыполнением какого-
то шага и случаями заражения не всегда абсолютна.
Оператор может отказаться от выполнения нескольких
действий, прежде чем вероятность заражения возрас¬
тет так резко, что станет почти неизбежной (рис. 6.1).
К тому же, причина заражения обычно многофакторна,
и, следовательно, не существует очевидного простого
решения проблемы. Если однажды установленные
меры предосторожности постоянно соблюдаются, то
случаи заражения имеют место значительно реже и
легче обнаруживаются.
Хотя лабораторные условия в настоящее время в
большой мере облегчают соблюдение асептики (кон¬
диционирование и фильтрация воздуха, ламинарные
шкафы и т.д.), современные лаборатории нередко
переполнены персоналом, и оборудование часто ис¬
пользуется несколькими людьми. Тем не менее при
жестком соблюдении мер предосторожности не трудно
соблюдать стерильность.
6.2. ОБЪЕКТЫ АСЕПТИЧЕСКОГО
ОКРУЖЕНИЯ
6.2.1. Стерильная зона
В отсутствие ламинарного шкафа для стерильных
работ следует использовать специальную стерильную
комнату. Если это невозможно, выделите тихий угол в
лаборатории, с минимальным перемещением людей и
отсутствием выполнения рядом какой-либо другой ра¬
боты (см. разд. 4.3.1). При наличии ламинарного шкафа
для расположения стерильной зоны следует выбрать
место, удаленное от потоков воздуха из дверей, окон и
т. д., в этой зоне не должно быть активных перемещений,
6.2. Объекты асептического окружения
107
ф
0
0S
Ф
s
т
ф
1
(0
Q.
О
О
Недели
Рис. 6.1. Возможность заражения. Сплошная линия в верхней части графика представляет колебания в методике относительно
100% — идеальная асептическая техника. Сплошная линия в нижней части графика представляет собой колебания в уровне
заражения в окружающей среде, за 0 принимается идеальная асептика. Максимум первой кривой связан с нарушениями асеп¬
тической техники (не протертая по забывчивости поверхность, прикосновение пальцев к пипеткам в средней и нижней частях,
контакт кончика пипетки с нестерильной поверхностью и т.д.). Пики другой кривой связаны с повышением зараженности
окружающей обстановки (споровое заражение, зараженный инкубатор, зараженные реактивы и т.д.). Пока эти отклонения
или пики минимальны по силе и продолжительности, два графика не перекрываются. Когда резкие колебания в асептическое
методике (точечная линия наверху) совпадают с резкими отклонениями в степени зараженности окружения (пунктирная линия
внизу), например как в левой части графика, где точечная и пунктирная линии пересекаются, возможность инфицирования
возрастает. Если нарушения в соблюдении методов асептики продолжаются и нарастают (регрессия точечной линии в пра¬
вой части графика), а зараженность окружения также повышается (повышение пунктирной кривой в правой части графика),
после пересечения двух кривых вероятность инфицировании резко возрастает и приводит к частым мультиспецифичным и
мультифакторным случаям заражения
а также оборудования, которое создает потоки воздуха
(центрифуги, холодильники и морозильники и т.д.),
кондиционеры должны располагаться так, чтобы по¬
токи воздуха не нарушали работу ламинарного шкафа
(см. разд. 4.3). Работа в боксе должна быть ограничена
работой с культурами тканей, все микробиологические
процедуры, так же как и манипуляции с животными,
необходимо исключать из асептической зоны. Зона
должна содержаться в чистоте и быть свободной от
пыли, а также от оборудования, которое не связано с
культивированием тканей. Нестерильные манипуля¬
ции, такие как отбор проб, окрашивание или экстрак¬
ция, должны производиться где-то в другом месте.
6.2.2. Рабочая поверхность
Очень важно содержать рабочую поверхность свобод¬
ной и чистой. Следует соблюдать следующие правила:
1) начинайте работу только на совершенно чистой
поверхности;
2) протрите поверхность тампоном, обильно смочен¬
ным 70%-м раствором этанола;
3) принесите только те предметы, которые вам потре¬
буются для планируемой работы;
4) убирайте все, что вам не нужно, протирая поверх¬
ность между манипуляциями;
5) организуйте ваше рабочее место так, чтобы вы:
а) имели свободный доступ ко всем предметам и не
тянулись через одни, чтобы достать другие;
б) имели для работы большое свободное простран¬
ство в центре стола (не только в передней его
части) (рис. 6.2). Если вокруг вас будет слишком
много оборудования и материалов, возрастет
риск контакта наконечника или кончика сте¬
рильной пипетки с нестерильной поверхно¬
стью. Более того, ламинарный поток в боксе,
заставленном посудой и оборудованием, резко
нарушается (рис. 6.3);
6) горизонтальный ламинарный поток более устойчив к
загромождению разными предметами, но вы и в этом
случае должны работать на чистой поверхности, не
имеющей препятствий для воздуха между централь¬
ной рабочей зоной и НЕРА-фильтром (рис. 6.4);
7) организуйте работу так, чтобы все необходимые
вам предметы (пипетки, груши для пипетирования,
108
ГЛАВА 6. МЕТОДЫ АСЕПТИКИ
Рис. 6.2. Возможное расположение оборудования в рабочей зоне ламинарного шкафа. Правильное расположение предметов
на рабочей поверхности. Для операторов-левшей расположение может быть зеркальным
Рис. 6.3. Плохо организованное расположение предметов в рабочей зоне. Ламинарный шкаф используется неправильно.
Слишком много предметов, нарушающих движение воздушных потоков, в т. ч. в передней части рабочей зоны. Нарушаются
как стерильность, так и защита оператора от биологической опасности
наконечники для автоматических пипеток и др.)
находились в поле вашего зрения и не закрывались
вашими руками при манипуляции;
8) немедленно удаляйте любые загрязнения, протирая
затем поверхность 70%-м этанолом;
9) по окончании работы уберите все, еще раз вымойте
и протрите этанолом поверхность.
6.2.3. Личная гигиена
Существуют различные мнения по поводу того, повы¬
шает или увеличивает количество микроорганизмов
на коже рук частое мытье. Независимо от результатов
этой дискуссии, мытье очищает руки и удаляет сухие
клетки кожи, которые, в противном случае, могли
6.3. Стерилизующие манируляции
109
Рис. 6.4. Планировка рабочей зоны в ламинарный шкафе с горизонтальным воздушным потоком. Для операторов-левшей
расположение может быть зеркальным
бы попасть в культуру. Мытье рук также снизит за¬
раженность слабо прикрепленными микроорганиз¬
мами, которые представляют наибольший риск для
культуры. Можно надевать хирургические перчатки
и периодически протирать их спиртом, но предпочти¬
тельной может оказаться работа без перчаток (там, где
применение перчаток не обязательно по требованиям
биобезопасности), поскольку это позволяет сохра¬
нить большую чувствительность пальцев и точность
манипуляций.
По требованиям GMP (Good Manufacturing Prac¬
tice), персоналу требуются шапочки, халаты, накидки
и хирургические маски [Food and Drug Administration,
1992; Rules and Guidance for the Pharmaceutical Manu¬
facturers and Distributes, 1997], но в них нет особой
необходимости в обычных условиях, в частности при
работе в ламинарном шкафу. Если у вас длинные во¬
лосы, завяжите их сзади. При работе в асептической
зоне на открытом столе не разговаривайте. Разговоры
допустимы при работе в ламинарном шкафу с верти¬
кальным потоком, при наличии стеклянной перегород¬
ки между вами и культурой, но и в этом случае следует
свести их до минимума. Если у вас простуда, наденьте
маску, или, еще лучше, — не проводите никаких работ
с культурами тканей, хотя бы в период выраженного
инфекционного процесса.
6.2.4. Реагенты и среды
Реактивы и среды, полученные от промышленного
производителя, должны до продажи предварительно
подвергаться строгому контролю на стерильность,
однако наружная поверхность бутылей и флаконов,
в которых они содержатся, не стерильна. Некоторые
производители обтягивают бутыли полиэтиленовой
пленкой, которая защищает от грязи и позволяет по¬
мещать их в водяную баню для размораживания или
согревания. Эту пленку следует удалить с бутыли вне
ламинарного шкафа. Бутыли после водяной бани или
холодильника, освобожденные от упаковки, должны
быть обработаны 70%-м этанолом.
6.2.5. Культуры
Культуры, полученные из других лабораторий, несут
высокий риск, поскольку они могут быть заражены как
исходно, так и при транспортировке. Импортирован¬
ные клеточные линии необходимо всегда подвергать
карантину (см. разд. 4.3.3; 19.1.8), т.е. все работы с
ними производить в отдельном от выращивания других
основных культур помещении, а также хранить без ис¬
пользования антибиотиков, пока не будет показано, что
поступившие культуры свободны от заражения. Затем
их можно присоединить к остальным культурам. Не
стоит постоянно использовать антибиотики, поскольку
они могут подавлять, но не уничтожать микроорганиз¬
мы, заразившие культуры (см. разд. 9.4.7).
6.3. СТЕРИЛИЗУЮЩИЕ МАНИПУЛЯЦИИ
6.3.1. Протирание поверхностей
Протирайте все рабочие поверхности 70%-м этиловым
спиртом до и в процессе работы, в частности после лю¬
бого разливания, а также по завершении всей работы.
Протирайте до начала работы бутылки, особенно те,
которые поступают из хранения на холоде или после
водяной бани, а также флаконы или боксы, вынутые из
инкубатора. Протирание иногда приводит к смыванию
надписей, поэтому используйте этанол-устойчивый
маркер.
110
ГЛАВА 6. МЕТОДЫ АСЕПТИКИ
6.3.2. Закрывание емкостей
Глубокие закручивающиеся крышечки предпочтитель¬
нее пробок, хотя требуется больше внимания хорошему
отмыванию детергента из внутренней поверхности
резиновых прокладок. При возможности следует ис¬
пользовать полипропиленовые крышки без прокладок.
Закручивающиеся колпачки должны быть обернуты
фольгой для предохранения горлышка бутылки от
отложения пыли, хотя использование глубоких колпач¬
ков (например, Duran) делает оборачивание фольгой
менее необходимым.
6.3.3. Работа с горелкой
При работе на открытом столе (рис. 6.5) производится
обжигание стеклянных пипеток, горлышек бутылей,
а также пробок до и после открывания емкостей; вся
работа проводится вблизи пламени, в потоках восхо¬
дящего воздуха, возникающего за счет конвекции. Не
оставляйте открытыми бутыли, если вы работаете не в
ламинарном шкафу, даже рядом с пламенем. Крышки
следует класть на чистую поверхность внутренней сто¬
роной наверх и обжигать до того, как ими снова закроют
бутыли. Колпачки можно держать в руке (рис. 6.6) во
время пипетирования, при этом отпадает необходи¬
мость класть их на стол и обжигать.
Обжигание в пламени не целесообразно, когда
вы работаете в ламинарном шкафу, поскольку это
нарушает ламинарный поток, что, в свою очередь,
нарушает стерильность культур и предохранение от
биологической опасности. Открытое пламя несет в себе
опасность пожара и может повредить НЕРА-фильтры
(high-efficiency particulate air — высокоэффективный
распыленный воздух) или расплавить некоторые внут¬
ренние пластиковые детали.
6.3.4. Манипуляции с бутылками и колбами
При работе на открытом рабочем столе не следует де¬
ржать открытые бутыли вертикально, наклоняйте их
насколько возможно без риска разливания. Штатив для
бутылей (МР Biomedical) позволяет удерживать буты¬
ли или флаконы под углом. Культуральные флаконы
должны лежать горизонтально, когда их открывают, и
затем при манипуляциях следует держать их под углом.
Когда вы работаете в ламинарном шкафу, бутыли мо¬
гут стоять вертикально открытыми, но не допускайте,
чтобы ваши руки или другие предметы двигались мимо
открытых сосудов или стерильных пипеток, а также
воздушных НЕРА-фильтров.
6.3.5. Пипетирование
Стандартные стеклянные или пластиковые пипетки
до сих пор остаются самым простым приспособлением
для манипуляций с жидкостями. Иногда используются
шприцы, но их употребление ограничено, поскольку
стандартные иглы слишком коротки для большинс¬
тва сосудов. Работа со шприцом может также создать
напряжение сдвига, повреждающее клетки, когда вы
разливаете суспензию клеток, кроме того, возрастает
риск укалывания и введения себе биологического ма¬
териала. Канюли с широким просветом имеют преиму¬
щества перед стандартными иглами, но их использо¬
вание замедляет процесс, за исключением применения
многоступенчатого диспенсера (см. рис. 5.8).
Можно выбрать пипетки удобного размера, набор
пипеток объемом 1 мл, 2 мл, 5 мл, 10 мл, и 25 мл удов¬
летворит все ваши потребности, хотя существуют и
100-мл пипетки (BD Biosciences), которые удобны при
приготовлении и разливании среды. Использование
пипеток с быстрым сливом снижает точность и акку-
тгфю
Рис. 6.5. Планировка рабочей зоны на открытом столе. Предметы расставлены в форме полумесяца вокруг чистой рабочей
поверхности в центре стола. Бунзеновская горелка расположена в центре, чтобы обеспечить обжигание предметов и создать
восходящий воздушный поток над рабочей зоной
6.3. Стерилизующие манируляции
111
Рис. 6.6. Способ удерживания груши и крышки от флакона
в руке. Крышка может быть откручена и удерживаема мизин¬
цем руки, прижимающим ее к ладони. При этом три пальца
держат грушу для пипетирования
ратность работы, но дает значительный выигрыш в ско¬
рости. Если вы используете стеклянные пипетки и вам
требуется только несколько пипеток каждого объема,
при упаковке пипеток в контейнер для стерилизации
используйте смешанный набор. Одноразовые пластико¬
вые пипетки должны иметь двойную оболочку, верхняя
из которых удаляется до помещения в ламинарный
шкаф. Неиспользованные пипетки должны храниться
в беспылевом контейнере.
Пипетирование ртом должно быть исключено,
поскольку оно является дополнительным фактором
микоплазматического заражения и может представлять
биологическую опасность для оператора (например,
при работе с вирусинфицированными клеточными
линиями, биопсийным или аутопсийным материалом
от человека, или другими объектами, обладающи¬
ми потенциальной биологической опасностью (см.
разд. 7.1.7)). Производителями предлагаются недоро¬
гие груши (рис. 6.6) и электрические пипетирующие
устройства (см. рис. 5.5); постарайтесь выбрать из этого
оборудования то, что подходит вам. Инструмент, кото¬
рый вы выберете, должен подходить к пипеткам всех
используемых размеров без применения каких-либо
усилий для вставления и удаления пипеток. Регуля¬
ция потока жидкости должна быть легкой и быстрой.
Устройство должно позволять многократно засасы¬
вать и выливать из пипетки жидкость (например, при
суспендировании) без разбрызгивания, быть легким
и удобно располагаться в руке так, чтобы длительная
работа не вызывала утомления.
Автоматическая пипетки (см. рис. 5.6), в част¬
ности, удобны для малых объемов (1 мл и меньше),
хотя большинство производителей предлагает авто¬
матические пипетки объемом до 5 мл. Поскольку за¬
труднительно достать до дна сосуда большого объема
носиком наконечника, не касаясь при этом внутренней
части его горлышка нестерильными частями корпу¬
са, автоматические пипетки следует использовать
только при работе с маленькими флаконами или с
применением промежуточных соединений с другими
емкостями, такими как универсальный контейнер.
Кроме того, при работе с большими объемами следует
использовать удлиненные наконечники. В частности,
автоматические пипетки полезны при микротитра-
ционном анализе и других манипуляциях с много¬
луночными планшетами, но не следует использовать
их при серийном ведении культуры, если только не
используются специальные наконечники с фильтра¬
ми. Многоканальные автоматические пипетки (с 4, 8
или 12 каналами) удобны для работы на микротитра-
ционных планшетах (см. рис. 5.10).
Пипетирование в культуре тканей часто представ¬
ляет собой компромисс между точностью и скоростью
выполнения работы; скорость требуется для того,
чтобы свести к минимуму повреждение клеток при
манипуляциях с ними, в т.ч. субкультивировании, а
аккуратность и точность — для воспроизводимости
результатов при стандартных условиях культиви¬
рования. Тем не менее приемлемая ошибка обычно
составляет ±5%, кроме таких условий эксперимента,
когда может потребоваться большая точность. В целом,
использование пипеток меньшего объема согласуется
с возможностью большей точности, требуемой для
получения количественных данных.
Необходимо вставлять ватную пробку в верхнюю
часть стеклянной пипетки до стерилизации, для того
чтобы сохранить стерильность пипетки в ходе исполь¬
зования. Пробка предохраняет загрязнение из груши
или пипетирующего устройства и уменьшает риск
перекрестной контаминации из содержимого пипетки.
Если ватная пробка становится влажной, замените
пипетку, поместив использованную в дезинфектант,
с последующим мытьем. Затыкание пипеток ватными
пробками перед стерилизацией — достаточно утоми¬
тельная работа, так же как удаление этих пробок перед
мытьем. Производители предлагают автоматические
устройства для затыкания пипеток ватными пробками
(см. рис. 11.6); они ускоряют и облегчают процесс.
Пластиковые пипетки поступают уже с пробками.
Однако работа с ними идет медленнее, поскольку
каждая пипетка имеет индивидуальную обертку, ко¬
торая требует удаления перед использованием и неэ¬
кономична, если несколько пипеток обернуто вместе
(часть неиспользованных пипеток всегда теряется,
поскольку нецелесообразно хранить и в следующий раз
использовать однажды открытую упаковку), а также
связана с относительно высоким риском контаминации,
поскольку процедура вынимания пипетки из обертки
более подвержена риску контаминации, чем изъятие
из контейнера. Тем не менее к достоинствам пласти¬
ковых пипеток относится то, что они более свободны
от бактериальных и химических загрязнений, чем
стеклянные, и при этом не требуют времени на мытье
и стерилизацию.
Автоматические пипетирующие устройства и
диспенсеры подробно описаны в гл. 5 (см. разд. 5.2.7).
Следует уделять большое внимание риску загрязнений
при работе с ними, но возрастание скорости работы
112
ГЛАВА 6. МЕТОДЫ АСЕПТИКИ
поможет снизить усталость и сократить время, при
котором культуральные сосуды открыты и подвержены
риску контаминации.
6.3.6. Переливание
Не переливайте жидкости из одной стерильной емкос¬
ти в другую. Переливание через горлышко допустимо,
если та бутыль, из которой выливают, используется
однократно, только чтобы перелить все ее содержи¬
мое, предварительно измерив объем, в одно приемное
устройство или сосуд. Главный риск при переливании
через горлышко заключается в появлении жидкостного
мостика между внешней стороной бутылки и внут¬
ренней ее стороной, позволяющей микроорганизмам
проникнуть внутрь бутылки в ходе хранения или
инкубации.
6.4. Ламинарный поток
Главным преимуществом работы в ламинарном шкафу
является то, что рабочая поверхность защищена от
пыли и микробного заражения постоянным стабиль¬
ным потоком фильтрованного воздуха, проходящего
над ней (рис. 6.7). Существуют два типа ламинарных
потоков: 1) горизонтальный, при котором поток воз¬
духа дует со стороны, противоположной оператору,
параллельно рабочей поверхности, и не рециркулирует
(см. рис. 6.7а); и 2) вертикальный, при котором поток
воздуха направляется сверху вниз на рабочую по¬
верхность и либо удаляется, либо рециркулирует (см.
рис. 6.76). В большинстве ламинарных шкафов 20%
воздуха удаляется, и восполняется захватом воздуха
перед поверхностью рабочего стола. Такая конфигура¬
ция создана для минимизации выхода избытка воздуха
с поверхности рабочей зоны. Горизонтальный тип
создает более стабильный поток воздуха и лучшую за¬
щиту стерильности культур и реагентов; вертикальный
тип обеспечивает лучшую защиту для оператора.
Для работы с объектами, имеющими потенциаль¬
ную биологическую опасность (культуры клеток,
выделенных из человека или приматов, вирусинфи-
цированные культуры и т.д.), следует использовать
MSC-боксы II класса биологической безопасности
(Microbiological Safety Cabinet) с вертикальным
ламинарным потоком (см. разд. 7.1.7 и рис. 1.5а). На¬
илучшую защиту от химической и радиохимической
опасности обеспечивает специальный шкаф для работ
по определению цитотоксичности, оборудованный
угольным фильтром, очищающим рециркулирующий
воздух, либо ламинарный шкаф с выведением всего
потока воздуха наружу из здания через систему вен¬
тиляции (см. разд. 7.5.4). Если оператор имеет дело с
известными возбудителями человека, приемлем бокс
для работы с возбудителями III группы патогенности
(см. разд. 7.8.2, рис. 7.5в).
Эффективность работы ламинарного шкафа зави¬
сит от минимального перепада давления, при котором
воздух проходит через фильтр. При возрастании
сопротивления перепад давления увеличивается и
поток воздуха через бокс снижается. Ниже 0,4 м/с
(80 футов/мин) стабильность потока исчезает и сте¬
рильность не может поддерживаться. Перепад давле¬
ния контролируется манометром, встроенным в бокс,
20%-е удаление воздуха
Створчатый клапан для
предупреждения обратного потока
НЕРА выхлопной фильтр
Удаляющий воздух вентилятор, 20%
Рециркулирующий вентилятор, 80%
НЕРА-фильтр для рециркулирущего воздуха
Ламинарный поток
Рециркулирующий воздух
Стеклянный экран
Захват комнатного воздуха
Удаление из рабочей зоны
Поднос для дренажа
а ГОРИЗОНТАЛЬНЫЙ ЛАМИНАРНЫЙ ПОТОК б ВЕРТИКАЛЬНЫЙ ЛАМИНАРНЫЙ ПОТОК
Рис. 6.7. Воздушные потоки в ламинарном шкафу. Стрелки указывают направление потоков, (а) Горизонтальный поток.
(б) Вертикальный поток
6.5. Стандартная процедура
113
но прямое измерение потока воздуха анемометром
предпочтительнее.
При повседневной работе проверять качество
фильтров необходимо примерно каждые 3-6 меся¬
цев. Первичный фильтр для ламинарного шкафа с
горизонтальным потоком можно снять (после выклю¬
чения вентилятора) и вымыть при помощи мыла и
воды. Встроенные первичные фильтры в ламинарных
шкафах II класса биологической опасности и боксах с
вертикальным потоком могут быть заменены только
инженером. Эти фильтры необходимо сжечь или ав-
токлавировать и заменить на новые.
Каждые 6 месяцев НЕРА-фильтр над рабочей
поверхностью должен проверяться на предмет ста¬
бильности и однородности воздушного потока, а также
дефектов и прорывов, которые определяются по мест¬
ному возрастанию воздушного потока и увеличению
подсчитанных частиц. Мониторинг, как правило,
проводят инженеры-профессионалы на контрактной
основе. В ламинарных шкафах II класса биологической
опасности также необходимо производить периодичес¬
кую замену НЕРА-фильтра при снижении качества
очищаемого воздуха. Это также должно производиться
профессиональным инженером, с соответствующей
подготовкой, включающей навыки упаковки и утили¬
зации отработанных фильтров, замены и установки
новых. Если ламинарный шкаф предназначен для
работы с объектами, имеющими потенциальную биоло¬
гическую опасность, до замены фильтра боксы должны
быть закрыты и обработаны дезинфектантом.
Регулярная проверка рабочей поверхности должна
проводиться еженедельно при любом разбрызгивании
и разливании сред и реагентов, рабочая зона немед¬
ленно вытирается и стерилизуется 5%-м феноловым
дезинфектантом или 70%-м этанолом. Иногда разлив
или разбрызгивание остаются незамеченными, поэтому
необходимо периодически проверять чистоту рабочей
поверхности. Тампоны, щетки, тканевые салфетки, пер¬
чатки, попавшие под рабочую поверхность, при уборке
могут попасть на первичный фильтр и нарушить поток
воздуха, поэтому будьте внимательны и периодически
проверяйте первичный фильтр.
Предпочтительна непрерывная работа ламинар¬
ного шкафа, поскольку она позволяет сохранять
рабочую зону чистой: любое разливание на фильтре
или на рабочей поверхности быстро высыхает в сте¬
рильном воздухе и снижается возможность роста
микроорганизмов.
Ультрафиолетовый свет также используется для
стерилизации воздуха и обработки рабочей поверх¬
ности в перерывах между работами. Эффективность
ультрафиолетовой обработки несомненна, поскольку
свет проникает в щели, куда не может проникнуть
этанол или другие дезинфицирующие агенты. Ульт¬
рафиолетовая обработка несет в себе радиационную
опасность, в частности для глаз, а также может при¬
вести к образованию трещин на пластиковых панелях
(например, Perspex) через 6 месяцев — 1 год при одно¬
временном использовании этанола.
• Требование безопасности. Если использу¬
ется ультрафиолет, следует надевать защитные очки
и закрывать все участки кожи, подвергающиеся его
воздействию.
6.5. СТАНДАРТНАЯ ПРОЦЕДУРА
Суть правил соблюдения стерильности во многом ос¬
новывается на принципах стандартной лабораторной
практики (GLP) (см. табл. 6.1). Поверхность рабочего
места должна быть ровной и чистой, на ней должны на¬
ходиться лишь те предметы, которые вам необходимы
для работы в настоящее время. Приготовьте заранее
то, что вам может понадобиться, так чтобы культуры
вынимались из инкубатора на менее продолжительный
срок, все манипуляции должны производиться так
быстро, как это возможно, без препятствий и помех.
Предметы на столе должны располагаться в зоне пря¬
мой видимости. Избегайте случайного контакта сте¬
рильных и нестерильных поверхностей. После работы
оставляйте рабочее место чистым.
В двух протоколах, которые следуют ниже, сделан
акцент на асептических методах. Приготовление среды и
другие манипуляции обсуждаются более подробно в со¬
ответствующих подразделах (см. разд. 11.4). Данный про¬
токол создан для использования в упражнении 1 (см. гл. 2,
упражнение 1), но применимым и к другим случаям.
6.5.1. Чашки Петри и многолуночные
планшеты
Возможность контаминации чашек Петри и многолу¬
ночных планшетов повышается в связи со следующими
факторами, такими как:
1) наличие большой площади поверхности, когда ем¬
кости открыта;
2) возникновение риска прикосновения к краю чашки
при манипуляциях с открытой чашкой;
3) риск переноса заражения с рабочей поверхности
на чашку через крышку, если крышку кладут вниз
краем;
4) среда, заполняющая промежуток между крышкой
и чашкой в связи с явлением капиллярности, если
чашку наклоняли или встряхнули при переноске в
инкубатор;
5) влажная атмосфера С02 инкубатора повышает риск
заражения.
Соблюдение следующих правил минимизирует
риск заражения:
1) Не оставляйте чашки открытыми на продолжи¬
тельный период, тщательно следите за открытыми
чашками и крышками.
2 ) При передвижении чашек по рабочей поверхности и
перемещении их в инкубатор или из него старайтесь
не наклонять и не трясти чашки, чтобы избежать
попадания среды на края чашки или крышки и
предотвращения заполнения промежутка между
114
ГЛАВА 6. МЕТОДЫ АСЕПТИКИ
Таблица 6.1. Основные приемы асептической техники
Объект
Правильно
Неправильно
Ламинарный шкаф
Заражение
Микоплазмы
Импорт новых клеточных
линий
Экспорт клеточных линий
Стеклянная посуда
Флаконы
Среды и реагенты
Пипетирование
Протирать до и после работы
Использовать минимум приборов, оборудова¬
ния и материалов на рабочей поверхности
Располагать материалы и оборудование по
линии прямой видимости
Работать без антибиотиков
Регулярно проверять культуру, визуально
невооруженным глазом и с помощью мик¬
роскопа
Упаковывать чашки Петри и многолуночные
планшеты в прозрачные коробки
Исследовать клетки по стандартной методике
Получать из проверенных, заслуживающих
доверие источников
Выдерживать карантин для новых линий
Проверить на микоплазмы
Подтвердить происхождение клеток
Вести записи
Проверить на микоплазмы
Подтвердить происхождение
Послать сопроводительные документы
Тройная упаковка
Хранить запасы отдельно
Наклонять флаконы при пипетировании
Заполнять газом при необходимости С02-ин-
кубирования
Быстро провентилировать при необходимости
штабелирования
Протирать бутылки до помещения их в лами¬
нарный шкаф
Открывать только в боксе
Использовать пипетки с ватными пробками
Менять пипетку, если она грязная или ватная
пробка стала влажной
Использовать пластик для агара
Выливать жидкости в стакан для отходов при
помощи воронки (рис. 6.8)
Засорять ламинарный шкаф
Оставлять ламинарный шкаф в беспорядке
Открывать зараженные флаконы в зоне работ
с культурами
Пересевать инфицированные клетки
Оставлять заражение без внимания
Пересевать зараженные клетки
Пытаться деконтаминировать («вылечить»)
культуры
Получать из источника, отдаленного от перво¬
начального производителя культуры
Посылать зараженные клеточные линии
Использовать ту же посуду для процедур, не
связанных с культивированием тканей
Открывать слишком много флаконов одновре¬
менно
Инкубировать в С02-инкубаторе, если нет га¬
зопроницаемой крышки
Укладывать в слишком высокие штабели
Использовать для нескольких клеточных ли¬
ний
Делить с другими сотрудниками
Переливать через край
Использовать одну и ту же пипетку для разных
клеточных линий
Делить с другими людьми
Создавать аэрозоль
Слишком сильно заполнять цилиндры с обезв-
реживающим раствором
крышкой и чашкой, ведущего к контаминации
вследствие явления капиллярности. Кроме того,
а) используйте вентилируемые чашки (см. рис. 8.7);
б) если среда попала в это пространство, снимите
крышку, тщательно удалите среду на наружной
поверхности края ватным тампоном, смоченным
70%-м этанолом, замените крышку на новую.
(Убедитесь, что маркировка располагается на
дне чашки!)
3) При инкубации помещайте чашки и крышки в про¬
зрачную пластиковую коробку, протрите коробку
этанолом когда вынимаете из инкубатора (см.
рис. 6.10 и разд. 6.7.1).
Для манипуляций с чашками Петри или много¬
луночными планшетами рекомендуется следующий
порядок действий (протокол 6.3).
6.6. ПРИБОРЫ И ОБОРУДОВАНИЕ
Все приборы, используемые в технике культуры ткани,
необходимо регулярно очищать для избежания накап¬
ливания пыли и предупреждения микробного роста
при случайном разбрызгивании. Заменяемые части,
такие как газовые баллоны, должны также протираться
перед внесением их в зону работы с культурами тканей.
Во время работы следует избегать перемещения при¬
боров и оборудования по асептической зоне.
6.6.1. Инкубаторы
Влажные инкубаторы являются основным источником
заражения (см. разд. 19.1.4). Их следует регулярно мыть
6.6. Приборы и оборудование
115
ПРОТОКОЛ 6.1. Асептические методы
работы в вертикальном ламинарном
потоке
Схема
Очистите и протрите рабочую зону, принесите
бутылки, пипетки и т.д. Сначала выполните подго¬
товительные действия (приготовление среды и реак¬
тивов), затем культуральную часть работы. В конце
работы очистите и вытрите рабочую поверхность
70%-м этанолом.
Материалы
Стерильные:
• Среда Игла 1х МЕМ с солями Хэнкса и НСОэ,
без антибиотиков 100 мл
• Пипетки, градуированные и заткнутые ватной
пробкой. Если используются стеклянные, то раз¬
личных размеров: 1 мл, 5 мл, 10 мл, 25 мл, в квад¬
ратном пенале, Если используется пластик, — в
индивидуальной упаковке, рассортированные по
размерам в штативе.
• Культуральные флаконы (для Упражнения 1
предварительно взвешенные), 25 см2 10
Нестерильные:
• Пипетирующее устройство или груша (см.
рис. 5.5,6.6)
• 70%-й этанол в пульверизаторе
• Не оставляющие ворса салфетки или щетки
• Абсорбирующие бумажные салфетки
• Цилиндр для пипеток, содержащий воду и дезин¬
фицирующее вещество (см. разд. 7.8.5)
• Ножницы
• Маркер с этанол-устойчивыми чернилами
• Ручка, тетрадь, протоколы и т. д.
Протокол
1. Протрите рабочую поверхность и все другие поверх¬
ности внутри ламинар-бокса, включая внутреннюю
часть переднего экрана 70%-м этанолом при помо¬
щи не оставляющих ворса салфеток или щетки.
2. Принесите среду и т. д. из холодного хранилища,
на водяной бане или иным способом разморозьте
принесенные из морозильника реактивы, про¬
трите бутылки спиртом, разместите те, что вам
потребуются сначала, в ламинарном шкафу.
3. Соберите пипетки и разместите с одной стороны
задней части рабочего места в доступном месте
(рис. 6.2).
а) Если используются стеклянные пипетки,
откройте пеналы с пипетками и поместите
крышки рядом, внутренней стороной наверх.
б) С пластиковых пипеток удалите упаковку,
рассортируйте по размерам в штативе или в
металлических банках.
4. Соберите другую стеклянную и пластиковую по¬
суду, инструменты и т. д. и поместите поблизости
(на тележке или соседнем столе).
5. Ослабьте, но не снимайте крышки всех бутылок,
которые будут использованы.
6. Снимите крышку с тех бутылок, куда вы будете
наливать жидкость, откуда будете переносить ее
пипеткой. Поместите крышки от бутылок откры¬
той стороной на самом высоком месте рабочей
поверхности в дальней части ламинар-бокса за
бутылками, так чтобы ваша рука не проходила
над ней во время манипуляций. Если вы одно¬
моментно используете только одну бутыль и
открываете только одну крышку, вы можете после
открывания бутыли удерживать крышку в руке
между мизинцем и ладонью (см. рис. 6.6) и после
пипетирования снова надевать ее на бутыль.
7. Возьмите пипетку.
а) Если пипетки стеклянные:
i) возьмите пипетку из пенала, вынимая ее
параллельно другим пипеткам в пенале и
прикасаясь к ним так мало, как это возмож¬
но, в том числе к верхним их частям (если
пипетка, которую вы достаете, коснулась
носиком верхних частей других пипеток,
еще находящихся в пенале, сбросьте ее в
цилиндр для использованных пипеток);
и) вставьте верхний конец пипетки в пипе¬
тирующее устройство, держа пипетку в
направлении от вас и закрепите ее над
градуировкой, так чтобы часть пипетки,
входящая в бутыль, не была загрязнена
(рис. 6.9).
• Требование безопасности. Если вы встав¬
ляете пипетку в грушу или пипетирующее устрой¬
ство, обратите внимание на то, чтобы не прилагать
слишком больших усилий: пипетка может сломаться
при давлении (см. разд. 7.5.3 и рис. 7.2).
б) Если пипетки пластиковые:
i) откройте упаковку в верхней части пипеток;
ii) расслоите концы упаковки, поворачивая
их снаружи внутрь;
iii) вставьте конец пипетки в грушу или пипе¬
тирующее устройство;
iv) извлеките пипетку из упаковки, не каса¬
ясь других пипеток или упаковки, а также
любых нестерильных поверхностей (см.
рис. 6.9);
v) Выбросьте обертку в мусорную корзину.
8. Пипетка в пипетирующем устройстве или груше
должна располагаться в правильном направлении
и под правильным углом в вашей руке. Просле¬
дите, чтобы кончик пипетки не касался внешней
стороны бутылки или внутренней поверхности
ламинарного бокса (см. обозначенные кружком
зоны на рис. 6.9). Всегда следите, где находится
пипетка. Выполнение этой процедуры нелегко,
когда вы овладеваете методами асептики, но это
важное требование для успешной работы и со
временем вы приобретете опыт.
16
ГЛАВА 6. МЕТОДЫ АСЕПТИКИ
9. Наклоните бутыль со средой по направлению к
пипетке так, чтобы ваша рука не проходила над
открытым горлышком и, используя 5-мл пипет¬
ку, отберите 5 мл среды и перенесите ее в 25-см3
флакон, также наклоненный.
10. Повторите еще 4 раза, заполнив 4 флакона. Когда
разливаете жидкость в несколько бутылей или
флаконов, их можно положить горизонтально
на бок. Убедитесь, что флаконы лежат надежно и
ваша рука не проходит над открытыми горлыш¬
ками (при выполнении Упражнения 1 запишите
время, которое вам потребовалось для добавления
среды в 5 флаконов).
11. Сбросьте стеклянные пипетки в цилиндр для
использованных пипеток, содержащий дезинфи¬
цирующий раствор. Пластиковые пипетки должны
быть сброшены в мешок для автоклавирования по¬
тенциально биологически опасных материалов.
12. Заверните крышки на флаконы.
13. Повторите процедуру, используя 25-мл пипетку
для переноса 5 мл в каждый из 5 флаконов (при
выполнении Упражнения 1 запишите время, ко¬
торое вам понадобилось для добавления среды в
5 флаконов).
14. Закройте крышки на бутылках со средой и фла¬
конах. Бутылки могут быть открыты, пока вы ра¬
ботаете, но всегда должны быть закрыты прежде,
чем вы покинете место у ламинарного бокса по
какой-то причине.
Замечание: В ламинарном шкафу с вертикальным
потоком не работайте непосредственно над откры¬
тым сосудом. При горизонтальном потоке не рабо¬
тайте за открытым сосудом.
15. По завершении операции плотно заверните все
крышки, уберите все растворы и материалы,
которые больше не требуются, с рабочей поверх¬
ности.
16. При выполнении Упражнения 1 взвесьте фла¬
коны, чтобы проверить точность разлива сред,
и инкубируйте при 37 °С в течение 1 недели для
проверки на возможное заражение.
Фильтровальная
воронка
Лабораторный
стакан
Рис. 6.8. Стакан для слива. Фильтровальная воронка пре-
хупреждает разбрызгивание содержимого из лабораторного
гтакана
Рис. 6.9. Правильное введение пипетки в пипетирующее
устройство. Пипетка при правильном введении захваты¬
вается как можно выше над градуировкой, конец пипетки
направлен от оператора. Обведенные зоны отмечают по¬
тенциальный риск, например случайное соприкосновение
кончика или середины пипетки с нестерильной поверхностью
бутылки или стенки шкафа
ПРОТОКОЛ 6.2. Работа на открытой
поверхности
Схема
Очистите и протрите рабочую зону, принесите
бутылки, пипетки и т.д. (см. рис. 6.5). Сначала вы¬
полните подготовительные действия. Обожгите в
пламени, если необходимо, используемые предметы,
сохраняйте рабочую поверхность свободной и чис¬
той. В конце работы очистите и вытрите рабочую
поверхность 70%-м этанолом.
Материалы
Стерильные:
• Среда Игла lxMEM с солями Хэнкса и НС03, без
антибиотиков 100 мл
• Пипетки, градуированные и заткнутые ватной
пробкой. Если используются стеклянные, то раз¬
личных размеров: 1 мл, 5 мл, 10 мл, 25 мл, в квад¬
ратном пенале, Если используется пластик, — в
индивидуальной упаковке, рассортированные по
размерам в штативе.
• Культуральные флаконы
(для Упражнения 1 предварительно взвешен¬
ные) 25см2 10
Нестерильные:
• Пипетирующее устройство или груша (см.
рис. 5.5,6.6)
• 70%-й этанол в пульверизаторе
• Не оставляющие ворса салфетки или щетки
• Абсорбирующие бумажные салфетки
• Цилиндр для пипеток, содержащий воду и дезин¬
фицирующее вещество (см. разд. 7.8.5)
• Бунзеновская горелка (или ее эквивалент) и
зажигалка
• Ножницы
• Маркер с этанол-устойчивыми чернилами
• Ручка, тетрадь, протоколы и т. д.
Протокол
1. Протрите рабочую поверхность стола 70%-м эта¬
нолом.
2. Принесите среду и т. д. из холодного хранилища,
на водяной бане или иным способом разморозьте
принесенные из морозильника реактивы, про¬
трите бутылки спиртом, разместите то, что вам
потребуется сначала, на рабочем столе, оставив
остальное поблизости в стороне.
3. Соберите пипетки и разместите с одной стороны
задней части рабочего места в доступном месте
(см. рис. 6.5).
а) Если используются стеклянные пипетки,
откройте пеналы с пипетками и поместите
крышки рядом, внутренней стороной наверх.
б) С пластиковых пипеток удалите внешнюю
упаковку, рассортируйте индивидуально упа¬
кованные пипетки по размерам в штативе или
в металлических банках.
4. Соберите другую стеклянную и пластиковую посу¬
ду, инструменты и т. д. и поместите поблизости.
5. Обожгите горлышки бутылок, быстрым движе¬
нием повернув их в пламени горелки и ослабьте
крышки.
6. Выберите пипетку.
а) Если пипетки стеклянные:
i) возьмите пипетку из пенала, поднимая ее
параллельно другим пипеткам в пенале и
прикасаясь к ним так мало, как это возмож¬
но, в частности к верхним их частям (если
пипетка, которую вы достаете, коснулась
носиком верхних частей любой из дру¬
гих пипеток, еще находящихся в пенале,
сбросьте ее);
ii) вставьте верхний конец пипетки в пипе¬
тирующее устройство, держа пипетку в
направлении от вас и закрепите ее над
градуировкой, так чтобы часть пипетки,
входящая в бутыль, не была загрязнена
(рис. 6.9).
б) Если пипетки пластиковые:
i) откройте упаковку в верхней части пи¬
петок;
ii) расслоите концы упаковки пипеток, пово¬
рачивая их снаружи внутрь;
iii) вставьте конец пипетки в грушу или пипе¬
тирующее устройство;
iv) извлеките пипетку из упаковки, не каса¬
ясь других пипеток или упаковки, а также
любых нестерильных поверхностей (см.
рис. 6.9);
v) выбросьте обертку в мусорную корзину.
7. Обожгите пипетку (только стеклянную!) в пла¬
мени горелки, проводя ее по длине через пламя и
одновременно поворачивая на 180°. Это должно
занять 2-3 с, иначе пипетка станет слишком го¬
рячей. Не стремитесь простерилизовать пипетку
таким образом; вы просто стараетесь сжечь пыль,
которая могла осесть на пипетку. Если вы прикос¬
нулись кончиком пипетки к нестерильной поверх¬
ности или могли контаминировать ее каким-то
иным образом, сбросьте ее в дезинфицирующий
раствор для повторного мытья, не пытайтесь
вновь сделать ее стерильной в пламени горелки.
8. Вставьте пипетку в пипетирующее устройство
или грушу, направляя ее от вас и закрепив ее
над градуировкой так, чтобы кончик пипетки,
который будет входить в бутылку или флакон,
не загрязнялся.
• Требование безопасности. Если вы встав¬
ляете пипетку в грушу или пипетирующее устрой¬
ство, обратите внимание на то, чтобы не прилагать
слишком больших усилий: пипетка может сломаться
при давлении (см. разд. 7.5.3 и рис. 7.2).
Не обжигайте в пламени пластиковые пипетки.
9. Пипетка в пипетирующем устройстве или груше
должна располагаться в правильном направлении
и под правильным углом в вашей руке. Проследите,
чтобы кончик пипетки не касался внешней стороны
бутылки или пенала с пипетками. Всегда осозна¬
вайте, где находится пипетка. Выполнение этой
процедуры нелегко, когда вы овладеваете методами
асептики, но это важное требование для успешной
работы и со временем вы приобретете опыт.
10. Удерживая пипетку в направлении от вас, удалите
крышку с первой бутылки, зажав ее между мизин¬
цем и ладонью (см. рис. 6.6). Если вы разливаете
жидкость в несколько бутылей или флаконов, их
можно положить горизонтально на бок. Работая
с бутылками, наклоните их так, чтобы ваша рука
не проходила над открытым горлышком. Если у
вас будут трудности с удерживанием крышки при
пипетировании, разместите ее на столе рядом с
горелкой открытой стороной вниз. Если бутылки
должны оставаться в открытом положении, их
следует наклонить так близко к горизонтальному
положению, как это возможно, на столе или опоре
для бутылок.
11. Обожгите горлышко бутылки.
Замечание. При обжигании бутылей Duran не
используйте бутыли с кольцами для разлива.
12. Наклоните бутыль со средой по направлению к
пипетке так, чтобы ваша рука не проходила над
открытым горлышком.
13. Отберите необходимое количество жидкости и
удерживайте ее в пипетке.
14. Обожгите горлышко бутылки и закройте крышку.
15. Снимите крышку с оставшейся бутылки, обож¬
гите горлышко, внесите жидкость, обожгите
горлышко, закройте крышку.
118
ГЛАВА 6. МЕТОДЫ АСЕПТИКИ
16. Плотно заверните все крышки по окончании
работы.
17. По завершении операции уберите с рабочей по-
верхности основные растворы, оставив только те
бутыли, которые потребуются.
ПРОТОКОЛ 6.3. Манипуляции с чашками
и планшетами
Материалы
Такие же, как в протоколах 6.1. и 6.2., по обстоя¬
тельствам.
Протокол
Удалите среду из чашек:
1. Сложите в штабель чашки или планшеты на од¬
ной стороне рабочей зоны.
2. Включите аспирационный насос.
3. Выберите пипетку без ватной пробки, вставьте в
аспирационную линию.
4. Поместите первую чашку или планшет в центр
рабочей зоны.
5. Удалите крышку и поместите за чашкой открытой
стороной вверх.
6. Зажмите чашку в ладони так близко к основанию,
как можно, стараясь не касаться края чашки,
и следите за тем, чтобы рука не проходила над
открытой чашкой или крышкой. При наличии
некоторой практики, возможно, вы сможете уда¬
лять жидкость из чашки, не открывая крышку
полностью. Этот метод быстрее и безопаснее, чем
описанный выше.
7. Наклоните чашку и удалите среду. Если нет ас-
пирационного насоса, перенесите среду пипеткой
и слейте в стакан с отходами через воронку (см.
рис. 6.8).
8. Закройте крышку.
9. Поставьте чашку на другую сторону рабочей зоны
отдельно от необработанных чашек, формируя
новый штабель.
10. Повторите процедуру с остальными чашками или
планшетами.
11. Сбросьте пипетку в цилиндр и выключите насос.
Добавьте среду или клетки:
1. Установите в необходимом месте нужные бутыли
и ослабьте крышку на той, которую будут исполь¬
зовать.
2. Поставьте первую чашку в центр рабочей зоны.
3. Снимите крышку с бутыли и заполнить пипетку.
4. Снимите крышку с чашки и поместите за чашкой.
5. Добавьте среду в чашку, направляя поток как
можно ближе к основанию вдоль по боковой
стороне чашки.
6. Закройте крышку.
7. Переместите чашку в ту сторону, где чашки стоя¬
ли первоначально, внимательно следя за тем,
чтобы среда не попадала на край и не заполняла
пространство между краем чашки и крышкой.
8. Повторите манипуляцию со второй чашкой и
т.д.
9. Сбросьте пипетку.
При определенной практике вы, возможно, научи¬
тесь поднимать крышку чашки и добавлять среду, не
открывая крышку полностью.
Замечание: Если вы добавляете среду в чашку
или планшет с клетками, бутылка со средой должна
быть подписана маркировкой используемой клеточ¬
ной линии и не использоваться для других линий.
Предпочтительно отлить среду в отдельную бутыль
и использовать только для этой процедуры.
через определенные промежутки времени (еженедельно
или ежемесячно, в зависимости от уровня зараженнос¬
ти атмосферы и частоты использования) с удалением
всего содержимого, включая подносы и полки, мытьем
внутренней поверхности с нетоксичным детергентом
(Decon или Roccall). Следы детергента удаляют 70%-м
этанолом, которому затем надо дать полностью испа¬
риться до установки полок и подносов.
Для предотвращения роста грибов фунгицидный
раствор (2% Roccall или 1% сульфат меди) можно по¬
местить в увлажняющий лоток на дно инкубатора, но
эффективность фунгицидного действия ограничена
поверхностями, с которыми осуществляется контакт и
такая процедура не может заменить регулярной уборки.
Некоторые инкубаторы снабжены высокотемператур¬
ным стерилизационным циклом, однако они редко
создают надолго необходимый уровень температуры.
Кроме того, такая обработка длительна, что неудобно,
поскольку на это время инкубатор не используется для
культивирования. Некоторые инкубаторы имеют мик-
ропоровый фильтр и ламинарный поток воздуха для
Рис. 6.10. Чашки Петри в коробке. Прозрачная коробка,
такая как коробка для сэндвичей или пирожных, помогает
защитить негерметично закрытые чашки или планшеты, а
также флаконы с ослабленными крышками, от заражения
во влажном инкубаторе. Этот тип контейнера может так¬
же использоваться при работе с материалом, обладающим
потенциальной биологической опасностью, для того чтобы
предупредить разлив или разбрызгивание при аварии
6.6. Приборы и оборудование
119
ингибирования циркуляции микроорганизмов (Forma,
Jencons), однако иногда лучше предпочесть небольшое
увеличение времени восстановления, отсутствие вен¬
тилятора и полагаться на конвекционную циркуляцию
воздуха (см. разд. 5.3.2).
6.6.2. Коробки для влажного инкубатора
Когда проблемы заражения во влажном инкубаторе воз¬
никают часто, можно помещать чашки Петри, планшеты
и флаконы с ослабленными крышками в пластиковые
коробки для сэндвичей (рис. 6.10). Коробки следует про¬
тереть до использования, внутри и снаружи, и высушить
в стерильном воздухе. Когда коробки будут вынуты из
инкубатора, их также обрабатывают 70%-м этанолом
до того как открыть или внести в рабочую зону. Затем
чашки аккуратно вынимают, внутренность коробки
обрабатывают перед повторным использованием.
6.6.3. Культивирование в атмосфере С02
Общепринятой практикой является ослабление кры¬
шек на флаконах при помещении их в С02-инкубатор
для проникновения газа внутрь флакона и достижения
газового равновесия, однако этот прием повышает риск
контаминации. Некоторые производители предлагают
Рис. 6.11. Заполнение флакона газом. Пипетка, вводи¬
мая во флакон, присоединена к шлангу с линией С02. Для
вытеснения воздуха из флакона используется 5% С02 без
пробулькивания среды. Буква F обозначает микропоровый
фильтр, встроенный в С02-линию
флаконы с газопроницаемыми крышечками, обеспе¬
чивающими быстрое достижение газового равновесия
в атмосфере С02без риска контаминации. Альтер¬
нативным выходом является заполнение флаконов
стерильной смесью газов (рис. 6.11) и герметичное
закрывание их. Это позволяет избежать использования
газовых инкубаторов для флаконов и обеспечивает
единообразие и быстрое достижение нужного состава
газовой среды.
Глава 7
БЕЗОПАСНОСТЬ, БИОЭТИКА
И ВАЛИДАЦИЯ
7.1. ЛАБОРАТОРНАЯ БЕЗОПАСНОСТЬ
В дополнение к ежедневным требованиям к безопаснос¬
ти работы, общим для всех рабочих мест, лаборатория
клеточных культур характеризуется рядом специфи¬
ческих рисков, связанных с культуральной работой.
Несмотря на образование и опыт большинства людей,
работающих в этих условиях, происходят различные
аварии, поскольку постоянная работа связана с обыч¬
ной, биологической и радиологической опасностью. Бо¬
лее того, отдельные лица, которые работают в подгото¬
вительной зоне, часто не имеют высшего образования,
и ответственность за их действия в лаборатории лежит
на руководителях. Важно определять потенциальную
опасность, но в то же время не преувеличивать риск.
Если нет реальной угрозы, объем предупреждающих
мер может быть сокращен, и в целом код безопасности
может быть поставлен под сомнение.
7.2. ОЦЕНКА РИСКА
Оценка риска — важный принцип, который введен
в большинство современных законов, связанных с
безопасностью. Определение природы и размеров
потенциальной опасности является только частью
процесса; способ, которым используются материалы
и оборудование, лицо, использующее их, частота ис¬
пользования, общие условия работы — все одинаково
важны при определении риска (табл. 7.1). Необхо¬
димо учитывать количество отдельных материалов,
степень и частоту контактов с опасным материалом,
процедуру манипуляций с ним, тип защитной одежды,
дополнительные опасности — такие как манипуляции
с горячими предметами, холодом, электричеством, а
также тип предварительного обучения и опыт работы
оператора — все эти элементы оказывают влияние на
риск, возникающий при проведении определенных
процедур, хотя природа опасности сама по себе остается
постоянной.
Основная проблема, постоянно выходящая на пер¬
вый план при установлении мер безопасной практики
в биомедицинских лабораториях, — непропорцио¬
нальность между чрезмерным вниманием к неясно
понимаемым, эзотерическим проблемам, связанным,
например, с генетическими манипуляциями, и недо¬
статком внимания к опасности работы с токсическими
и повреждающими химическими веществами, рас¬
творителями, потенциальными источниками пожара,
ионизирующим излучением, ударом электрическим
током и разбитой посудой. Важно, что биологическая
опасность четко подразделена на категории [Постанов¬
ление министерства здравоохранения и социального
обеспечения No. (CDC) 93-8395, 3-е издание (1993);
Комиссия по здоровью и безопасности, 1991а, 1991b,
1992, 1999а, 1999b, Caputo, 1996], при этом биологи¬
ческая опасность не переоценивается и не недооцени¬
вается. Соблюдение мер предосторожности не должно
замещать видение ежедневных проблем, связанных с
обеспечением безопасности всех видов.
7.3. СТАНДАРТНЫЕ ОПЕРАЦИОННЫЕ
ПРОЦЕДУРЫ
Опасные вещества, оборудование и условия не должны
рассматриваться отдельно от остальных составляю¬
щих элементов, следует представлять всю систему в
совокупности. Если подразумевается, что процедура
связана с неким значительным риском помимо обычно¬
го, необходимо составить стандартную операционную
процедуру (СОП), и весь персонал, работающий с
этим материалом, оборудованием и т.д. должен быть
ознакомлен с данным документом. Должны быть
определены различные этапы — покупка, хранение,
манипуляции и уничтожение, следует принять во вни¬
мание, что наличие одной или более опасностей в ходе
манипуляций умножает риск или, точнее, сложность
необходимых мер предосторожности (как, например,
проводить удаление осколков разбитой емкости, со¬
державшей линию человеческих клеток, меченных
радиоизотопом).
7.4. КОНТРОЛЬ БЕЗОПАСНОСТИ
•
Дальнейшие рекомендации не должны рассматривать¬
ся как практические инструкции по определению кода
безопасности, но, скорее, как советы, которые могут
вам помочь в составлении требований по мерам безо¬
пасности. Данная информация призвана предоставить
читателю некоторые источники и законодательные
акты, а также руководства, при помощи которых он
может составить частные нормы и правила работы в
7.4. Контроль безопасности
121
Таблица 7.1. Составляющие оценки степени риска
Продолжение табл. 7.1
Категория
Элемент, влияющий на степень риска
Категория
Элемент, влияющий на степень риска
Оператор
Опыт исследова¬
ний
Тренировка
Защитная
спецодежда
Оборудование
Возраст
Соответствие
задаче
Механическая
надежность
Электрическая
безопасность
Защитные
оболочки
Тепло
Уход
Уровень
Соответствие роду работы
Предварительная подготовка и опыт
работы
Предварительная
Соответствующая новым потребностям
Адекватная
Правильно надетая (застегивание на пу¬
говицы лабораторного халата)
Регулярная стирка
При повреждениях ремонт или замена
спецодежды
Сохранность
Соответствие законодательству
Приемлемость, объем пробы, герметиза¬
ция или изоляция
Погрузка
Крепеж
Балансировка
Соединения
Заземление
Эффект близости к воде
Аэрозоли:
Создание
Утечка из-под кожуха
Выход из рабочей зоны
Токсичные пары
Отводящая канализация:
Встроенность
Участок стока и риск с подветренной
стороны
Образование
Рассеяние
Частый
Требуется обеззараживание?
Стандартный
Требуется обеззараживание?
Захоронение
отходов
Физические риски
Интенсивный Обмораживание
холод
Электрический
шок
Огонь
Онемение
Потеря сознания
Остановка сердца
Общие меры предосторожности
Монтаж электропроводки, установка
оборудования, уход, за ним
Просачивание воды вблизи электрообо¬
рудования
Пожарные учения, процедуры и трени¬
ровки поведения при пожаре
Использование и хранение раствори¬
телей (например, не хранить в холо¬
дильниках)
Воспламеняющиеся смеси
Маркировка биологически и радиохими¬
чески опасных объектов
Химические
(включая газы
и летучие жид¬
кости)
Степень опас¬
ности
Токсичность
Используемое количество
Ядовитость
Канцерогенность
Т ератогенность
Мутагенность
Коррозионность
Раздражающие свойства
Аллергичность
Асфиксичность
Т еплообразование
Закипание
Т еплообразование
Закипание
Образование взрывчатой смеси
Отравление
Асфиксичность
Рассеивание и разбрызгивание
Реакция с водой
Реакция с раство¬
рителями
Летучесть
Образование
пыли и аэро¬
золей
Импорт, экспорт
и транспорти¬
ровка
Условия место- Доступность для необученного персо-
положения и нала
хранения Незаконный ввоз
Погодные условия, протечка воды
Надежность, устойчивость к сжатию,
поломки, утечки
Биологические опасности
Поломки и утечки
Патогенность
Вирулентность
Генетические
манипуляции
Метод изоляции
Радиоизотопы
Излучение
Изменяемость
Локализация при
приеме внутрь
Размещение
и учет
Степень
Инфекционность
Специфичность по отношению к хо¬
зяину
Заражающая доза (количество клеток)
Количество ДНК
Специфичность по отношению к хозяину
Внедрение вектора
Потеря трудоспособности
Комната
Бокс
Методы работы
Тип
Уровень энергии
Проникающая способность, защита
Взаимодействие, ионизирующее дей¬
ствие
Время полураспада
Парообразование
Распыление
Предшественники ДНК, такие как [3Н-
тимидин]
Жидкие, твердые, газообразные
Стандартные
Законодательные ограничения
122
ГЛАВА 7. БЕЗОПАСНОСТЬ, БИОЭТИКА И ВАЛИДАЦИЯ
Окончание табл. 7.1
Категория
Элемент, влияющий на степень риска
Особые обстоятельства
Беременность Иммунодефицит
Риск для плода, тератогенность
Болезни Иммунодефицит
Иммуносупрес- Иммунодефицит
сивные препа¬
раты
Раны и ссадины Возрастание риска попадания в орга¬
низм
Аллергия Порошки (например, детергенты)
Аэрозоли
Контактная (например, резиновые пер¬
чатки)
Элементы процедур
Масштаб, размах Количество используемого материала
действий Размеры оборудования и материалов
Количество персонала, необходимого
для работы
Сложность Количество этапов и стадий
Количество вариантов процедур
Взаимодействие систем и процедур
Продолжитель- Время (продолжительность) собственно
ность процедуры
Время инкубации
Время хранения
Количество лиц, Возрастание рисков?
вовлеченных в
процесс
Местоположение Секретность и доступность
конкретной лаборатории. Эти нормы и правила долж¬
ны находиться в соответствии с региональным и госу¬
дарственным законодательством и составляться при
консультации с местным комитетом по охране труда.
Настоящие рекомендации не имеют законодательной
силы и не должны рассматриваться как таковые.
Общие предписания по соблюдению мер безопас¬
ности должны быть получены из бюро безопасности
института или компании, в которой вы работаете. В до¬
полнение к ним, следует использовать рекомендации
Администрации профессиональной безопасности и
здоровья CUIA(OSHA) [www.osha-occupational-health-
and-safety.com/]. В Европе, включая Великобританию,
действует новый свод требований [в Великобритании
основанный на Руководстве по здравоохранению и
Безопасности при работе, 1999]; www.hmso.gov.uk/
si/si1999/19993242.htm (табл. 7.2). Эти требования
включают в себя все объекты общей безопасности.
Соответствующие требования и рекомендации по
биологической безопасности для США содержатся в
Руководстве «Биобезопасность в микробиологических
и биомедицинских лабораториях» [Министерства
здравоохранения и социального обеспечения США,
1993; www.orcbs.msu.edu/biological/BMBL/BMBL-
1/htm], объединенном документе, подготовленном
Центром по предупреждению и контролю болезней
в Атланте, шт. Джорджия, и Национальными инсти¬
тутами здоровья в Бетезде, шт. Мэрилэнд. На тер¬
ритории Великобритании опубликован двухтомный
сборник Информационного сообщения Комиссии
по здравоохранению и социальному обеспечению:
Безопасность работ и предупреждение инфицирова¬
ния в клинических лабораториях [Комиссия по здо¬
ровью и безопасности, 1991а] и Безопасность работ
и предупреждение инфицирования в клинических
лабораториях — типовые правила для персонала и
посетителей [Комиссия по здоровью и безопасности,
1991Ь]. Работа с генетически модифицированными
клетками проводится в соответствии с Руководством
по работе с генетически измененными микроорганиз¬
мами (ограниченного использования) (HSE, 1996), а
также распоряжениями Канцелярии Ее Высочества.
Некоторые руководства Великобритании доступны
для ознакомления по адресу www.hse.gov.uk/hthdir/
noframes/bioldex.htm/. Советы, данные в этом разделе,
носят общий характер и не должны толковаться как
удовлетворяющие законодательству.
7.5. ОБЩАЯ БЕЗОПАСНОСТЬ
В табл. 7.3 делается акцент на тех аспектах общей безо¬
пасности, которые, в частности, важны для лабораторий
культивирования тканей и должны использоваться
в совокупности с национальными и региональными
правилами и нормами безопасности.
7.5.1. Оператор
В сферу ответственности учреждения входит подго¬
товка и переподготовка работников, либо определе¬
ние степени подготовленности работника той работе,
которую ему поручают, в т. ч. знание соответствующих
лабораторных процедур. Руководитель лаборатории
должен убедиться, что новые или уже работающие со¬
трудники близко знакомы с правилами и нормами тех¬
ники безопасности. В обязанности непосредственного
руководителя каждого нижестоящего по должности
сотрудника входит проверка знаний и правильного
выполнения всех действий, а также применения ин¬
дивидуальных способов защиты, в т. ч. спец. одежды в
соответствующее время.
7.5.2. Оборудование
Руководитель подразделения должен назначить ответ¬
ственного за состояние оборудования, электрическую
безопасность и техническую надежность. Начальник
лаборатории должен быть уверен, что все оборудование
работает в соответствии со спецификацией и отвечает
требованиям техники безопасности и что все сотрудни¬
ки прошли обучение для работы с этим оборудованием.
Отдельные риски, связанные с токсичностью паров или
аэрозолированием при центрифугировании и гомоге-
7.5. Общая безопасность
123
Таблица 7.2. Руководства и нормы безопасности
Тема
США
Великобритания
Европа
Общие
Оборудова¬
ние
Химическая
безопас¬
ность
Биологиче¬
ская безо¬
пасность
Администрация профессиональной
безопасности и здоровья США
(OSHA) [www.osha-occupational-
health-and -safety.com/].
(OSHA) [www.osha-occupational-
health-and -safety.com/].
(OSHA) [www.osha-occupational-
health-and -safety.com/].
Национальный институт здоровья
[www.niehs.nih.gov/odhsb/1.
Национальный институт техни¬
ки безопасности и здоровья
(NIOSH), www.cde.gov/niosh/
homepagehtml
«Биобезопасность в микробио¬
логических и биомедицинских
лабораториях»
Министерства здравоохранения и
социального обеспечения США,
1999; [www.cdc.gov/od/ohs/bios-
fty/bmbl4/bmbl4toc/htm ]
Радиологи¬
ческая бе¬
зопасность
Регуляторная комиссия по ядерной
энергии. Медицинское, про¬
мышленное и научное исполь¬
зование ядерных материалов.
[www.nrc.gov/materials/medical.
html]
Руководстве по здравоохране¬
нию и безопасности при рабо¬
те, 1999 [www.hmso.gov.uk/si/
si1999/19993242.htm ]
Руководство по обеспечению и ис¬
пользованию рабочего оборудо¬
вания (PUWER), 1998; Уровень
безопасности, 1998, No 2306
Контроль за безопасностью ве¬
ществ для здоровья [Комиссия
по здоровью и безопасности,
HSC, 1999;www.hsegov.uk/coshh/
index/htm]
Безопасность работ и предупреж¬
дение инфицирования в клини¬
ческих лабораториях [Комиссия
по здоровью и безопасности,
1991а], а также
Безопасность работ и предупреж¬
дение инфицирования в клини¬
ческих лабораториях — типовые
правила для персонала и посети¬
телей [Комиссия по здоровью и
безопасности, 1991b]
Руководство по работе с гене¬
тически измененными микро¬
организмами (ограниченного
использования) [HSE, 1996;
www.hseygov.uk/biosafety/].
Руководство по работе с источни¬
ками ионизирующей радиации,
1999
Документы управления здравоохра¬
нения и безопасности при работе
89/391/ЕЕС
Директива по обеспечению и исполь¬
зованию рабочего оборудования
98/355/ЕЕС и 95/63/ЕС
Директива совета 98/24/ЕС от 7 ап¬
реля 1998 г. «О защите здоровья и
безопасности работников от рис¬
ков, связанных при работе с хими¬
ческим агентами» (14-я индивиду¬
альная директива в разделе Статьи
16(1) Директивы 89/391/ЕЕС).
Officional Journal, n° L131 ?
05/05/1998, p.ll
Директива EC по защите работников
от рисков, связанных при работе
с контактами с биологическими
агентами (90/679/ЕЕС).
низировании, должны быть снижены за счет дизайна
оборудования или путем установки оборудования в
изолирующие контейнеры.
Электрическая безопасность оборудования опира¬
ется в США на документы Администрации профес¬
сиональной безопасности и здоровья США (OSHA)
[;www.osha-occupational-health-and-safety.com/], в
Великобритании — на Руководство по обеспечению и
использованию рабочего оборудования [ 1998, www.hse.
gov.uk/pubns/indg291.pdf ], в странах Европейского Со¬
юза — на Директивы по обеспечению и использованию
рабочего оборудования [1995].
7.5.3. Стеклянные и острые предметы
Наиболее частыми повреждениями при проведении
работ по культуре тканей являются случайные ранения
при манипуляциях с разбитым стеклом и шприцевы-
ми иглами. В частности, опасны разбитые пипетки в
моечных цилиндрах. Разбивание и скалывание пи¬
петок происходят при вставлении с силой пипеток в
цилиндр с другими пипетками (см. рис. 7.1). Стеклян¬
ные пастеровские пипетки не должны сбрасываться в
цилиндр с обычными пипетками (см. ниже). Большую
опасность также представляют иглы, выброшенные
без соблюдения соответствующих правил вместе с
обычным мусором либо проткнувшие стенку жесткого
контейнера. Случайный укол использованной иглой,
повреждение осколком разбившегося сосуда, либо
повреждение кожи в ходе стандартных манипуляций
остаются наиболее частыми видами травм, связанных
с манипуляциями с потенциально биологически опас¬
ным материалом. Этим обусловлен также риск транс¬
плантации опухоли при уколе иглой, использованной
для работы с человеческими опухолями [Southam,
124
ГЛАВА 7. БЕЗОПАСНОСТЬ, БИОЭТИКА И ВАЛИДАЦИЯ
Рис. 7.1. Переполненные цилиндры для использованных
пипеток. В результате всовывания с усилием пипеток в уже
наполненный цилиндр, они торчат наружу; эти торчащие
пипетки не замачиваются и не дезинфицируются, а также
могут разбиться (если они стеклянные) при добавлении
новых пипеток
1958; Scanlon et al., 1965; Gugel & Sanders, 1986], хотя
сообщения о таких случаях крайне редки.
Пастеровские пипетки должны выбрасываться в
специальную корзину для острых и колющих предме¬
тов (см. приложение II и www.cdc.gov/niosh/sharpsl.
html), а не в общую мусорную корзину, поскольку
они легко разбиваются и осколки крайне опасны. При
повторном использовании их необходимо складывать
отдельно с большой аккуратностью. Производители
предлагают одноразовые пластиковые пастеровские
пипетки Kimble-Kontes; Alpha Laboratories-Pastettes,
однако они имеют более толстый кончик, чем стек¬
лянные. Избегайте использования шприцов и игл,
если только они не требуются для наполнения ампул
(используйте тупоконечные канюли) или отбора жид¬
кости из запаянных пузырьков. Пользуйтесь жесткими
пластиковыми или металлическими контейнерами
для использованных одноразовых игл. Не старайтесь
согнуть, вставить использованную иглу обратно в
упаковку или как-то иначе с ней манипулировать.
Обеспечьте отдельный ящик с жесткими стенками для
сбора отработавших колющих предметов и разбитого
стекла и не используйте его для общего мусора.
Будьте внимательны при установке пипетки в пипе¬
тирующее устройство или грушу. Выбирайте правиль¬
ный размер груши, чтобы избежать риска разбивания
пипетки и повреждения руки. Проверьте, чтобы носик
груши издавал звук, удерживайте пипетку так близко
к концу, как возможно, и, мягко надавливая, наденьте
носик, удерживая пипетку так, чтобы, случайно сло¬
мавшись, она не повредила суставов руки (рис. 7.2).
Хотя использование стеклянных пипеток в этом слу¬
чае связано с большим риском, пластиковые пипетки
тоже могут быть повреждены и сломаться, поэтому
всегда проверяйте верхнюю часть каждой пипетки до
использования.
7.5.4. Химическая
токсичность
При культивировании тканей используется отно¬
сительно мало химически опасных соединений, но
их использование должно быть согласовано с обще¬
принятыми мерами предосторожности. Обращайте
особое внимание на образование пыли и аэрозоля в
ламинарном шкафу (см. разд. 7.8.2). Детергенты, в
частности те, которые используются в моечных ма¬
шинах, обычно представляют собой каустик и могут
раздражать кожу, глаза и легкие. По возможности,
используйте жидкие детергенты в диспенсирующих
устройствах, надевайте перчатки, избегайте процедур,
сопровождающихся распылением детергента. Жидкие
детергенты в виде концентрата предпочтительны,
поскольку с ними легко обращаться и дозировать, но
они дороже. Химические дезинфектанты, такие как
гипохлорит, должны использоваться с соблюдением
всех мер предосторожности, при этом препочтительны
таблетированные формы или жидкие концентраты в
емкостях с диспенсерами. Гипохлорит-содержащие
дезинфектанты обесцвечивают одежду, вызывают
раздражение кожи, могут вызвать коррозию в местах
сварки нержавеющей стали.
При культивировании тканей используются спе¬
цифические химикаты, которые требуют особого
внимания: 1) диметилсульфоксид (ДМСО), является
мощным растворителем и проникает через кожу, по¬
этому может перенести многие вещества через кожные
покровы [Horita and Weber, 1964] и даже через неко¬
торые защитные перчатки (например, через латексную
или силиконовую резину, в меньшей степени через
нитрильную) и 2) мутагенные, канцерогенные и ци-
тотоксические вещества, работа с которыми должна
проводиться в специальных боксах. Ламинарный
шкаф II класса защиты подойдет для периодической
работы с этими веществами, но для постоянного ис¬
пользования этих соединений может потребоваться
специально разработанный бокс. Мутагены, канцеро¬
гены и другие токсичные вещества часто растворяют
в ДМСО, что повышает риск их проникновения через
кожу. Нитрильные перчатки обеспечивают лучшую
защиту, но для отдельных реагентов следует провести
7.5. Общая безопасность
125
Рис. 7.2. Правильное введение пипетки в пипетирующее устройство, (а) Неправильное положение рук. Левая рука слишком
низко удерживает пипетку, прикладывая слишком большое усилие, при котором пипетка может разбиться. Правая рука слиш¬
ком далеко захватывает устройство сверху, касаясь верхней части пипетки. Осколки при разбивании пипетки при слишком
большом усилии могут поранить суставы пальцев, (б) Правильное положение рук: левая рука в верхней части пипетки, легкий
захват, верхний конец пипетки ясно виден
испытания [см. также www.pacifica.com/NitrileGlove-
sChemicalResistance -BarrierGuide.pdf}.
Работа с химическими реактивами регулируется
документами OSHA [www.osha-gov.], а также в Вели¬
кобритании — Руководством по контролю за безопас¬
ностью веществ для здоровья [Комиссия по здоровью
и безопасности, HSC, 1999а;1999Ь]. По адресам www.
niehs.nih.gov/odhsb/nwww.cde.gov/niosh/homepage.html
доступна информация из Национального института
техники безопасности и здоровья (NIOSH) и Нацио¬
нального института здоровья (США)
7.5.5. Газы
Большинство газов, используемых в культуре тка¬
ней (С02, 02, N2) в малых количествах не оказывают
повреждающего действия, но, тем не менее, могут
быть опасны при ненадлежащем использовании. Они
должны быть заключены в баллоны под давлением,
которые затем должны соответствующим образом
храниться (рис. 7.3). Если произойдет значительная
утечка газа, то существует риск асфиксии (С02, N2) или
пожара (02). В каждом случае необходимы эвакуация
и максимальная вентиляция, при интенсивной утечке
кислорода вызовите пожарных. В комнате, в которой
хранятся запасы (С02, N2), или в комнатах, в которые
газы поступают по системе трубопровода, на уровне
пола должен быть установлен показатель содержания
кислорода.
Если используются стеклянные ампулы, они запаива¬
ются на огне газовой горелки. Наибольшая осторожность
Рис. 7.3. Хомут для баллона. Хомут прикрепляется к краю
стола или жестко закрепленной полки, предохраняя газовый
баллон при помощи тканевого ремня. Подходит к баллонам
разных размеров, может быть перемещен в другое положение
при необходимости; предлагается большинством поставщи¬
ков лабораторного оборудования
126
ГЛАВА 7. БЕЗОПАСНОСТЬ, БИОЭТИКА И ВАЛИДАЦИЯ
требуется как для работы с пламенем, так и для предуп¬
реждения непредумышленного смешивания бытового
газа и кислорода. В систему газопровода должен быть
вмонтирован однонаправленный клапан, не позволяю¬
щий кислороду проникать в газопроводные трубы.
7.5.6. Жидкий азот
С жидким азотом связаны три основных риска: об¬
морожение, асфиксия и взрыв (см. протокол 20.1).
Поскольку температура жидкого азота составляет
-196 °С, прямой контакт с жидкостью (при расплес¬
кивании и т.д.) или с чем-то другим, особенно метал¬
лическим, перед этим подверженным воздействию
азота, представляет собой серьезную опасность.
Следует надевать перчатки, достаточно толстые для
обеспечения изоляции, но достаточно мягкие и гиб¬
кие, чтобы не затруднять работы с ампулами. Когда
азот выкипает из морозильного оборудования при
рутинных манипуляциях, для удаления паров азота
достаточно работы обычной системы вентиляции;
но когда азот переливают или большое количество
образцов ставится в морозильник на замораживание,
необходимо включать дополнительную вентиляцию.
Помните, что 1 л жидкого азота образует около 700 л
газа. Датчик кислорода и сигнал тревоги должны быть
встроены (см. рис. 4.7) в вентиляционную систему, так
чтобы при изменяющемся составе воздуха и снижении
уровня кислорода звучал аварийный сигнал и вклю¬
чалась дополнительная вентиляция.
7.5.7. Ожоги
Существует три основных источника ожогов: 1) авто¬
клавы, стерилизационные печи и горячие поверхности;
2) манипуляции с предметами, только что вынутыми
из автоклавов, печей и горячих плиток и 3) открытое
пламя бунзеновской горелки при обжигании предметов
(см. табл. 7.3). Рядом со всем нагревающимся оборудо¬
ванием, в том числе горелкой, должны быть помещены
предупреждающие надписи; после автоклавирования
и стерилизации все предметы необходимо охлаждать.
Для манипуляций с горячими предметами следует
использовать теплоизолирующие перчатки.
7.6. ПОЖАР
Таблица 7.3. Общие мероприятия
Категория
Действие
Распорядитель¬
ный орган
Местный комитет
по технике бе¬
зопасности
Руководства
Стандартные дей¬
ствующие пра¬
вила (SOPs)
Защитная одежда
Контейнеры
Уровень безопас¬
ности
Тренировка
Мониторинг
Проверки
Ведение записей
Импорт и экспорт
Сортировка
мусора
Допуск
Взаимодействие с государственными и
региональными инспектирующими
органами
Назначенный представитель
Организация встреч и дискуссий
Местные и государственные
Составление местной действующей инс¬
трукции, если таковой не имеется
Определить и сделать доступными для
персонала
Обеспечение, стирка, правила ношения
Специфические нормы и правила (на¬
пример, хранение и упаковка)
Определить уровень химической, биоло¬
гической, радиологической защиты
Организация семинаров, руководство
Автоматический детектор дыма, измери¬
тель уровня кислорода
Проверка оборудования, порядка дей¬
ствий специально обученными сотруд¬
никами, назначенными для выполне¬
ния этой обязанности
Инструктор по технике безопасности и
сотрудники регулярно ведут записи
Согласно установленному распорядку,
ведение регистрационных записей
Определить порядок утилизации ко-
люще-режущего, радиоактивного
(твердые и жидкие отходы), биоло¬
гически опасного, коррозирующего,
токсичного видов мусора, а также
растворителей
Ограничить обученным персоналом
и посетителями. Исключить при¬
сутствие детей, за исключением зон
общего пользования
должен храниться в пластиковой бутыли с распыли¬
телем и не использоваться в присутствии открытого
пламени. При стерилизации инструментов в спирте с
последующим обжиганием их в пламени не возвращай¬
те инструменты обратно в спирт, пока они охвачены
огнем. Если вы используете этот метод стерилизации
инструментов, держите под рукой влажную ткань для
сбивания пламени, если спирт загорится.
Возникновение повышенной пожароопасности при
культивировании тканей связано с использованием
горелки для обжигания предметов и этилового спир¬
та для стерилизации поверхностей и инструментов.
Храните спирт в удалении от источников огня; всег¬
да следите за тем, чтобы спирт для стерилизации
инструментов содержался в минимальном объеме в
узкогорлой бутыли или колбе, которая не опрокидыва¬
ется (рис. 7.4). Спирт для дезинфекции поверхностей
7.7. ИОНИЗИРУЮЩЕЕ ИЗЛУЧЕНИЕ
С культивированием тканей связаны три основных
типа радиационной опасности: попадание внутрь орга¬
низма, излучение от меченых реагентов и излучение от
высокоэнергетичных источников. За руководством по
радиационной защите в США можно обратиться в Уп¬
равление по регуляции исследований ядерной энергии
(ядерная регуляторная комиссия США, Вашингтон,
7.7. Ионизирующее излучение
127
Рис. 7.4. Колба для спиртовой стерилизации инструментов.
Широкое основание колбы предупреждает опрокидывание,
центральная пробирка уменьшает количество необходимого
спирта, поэтому разливание, если оно происходит, минималь¬
но (оригинальная идея M.G. Freshney)
DC 20555). В Великобритании работа с радиоактивны¬
ми источниками регулируется документом «Работа с
ионизирующим излучением. Регуляция деятельности,
связанной с ионизирующим излучением [1999]».
7.7.1. Попадание в организм
Растворимые радиоактивно меченные компоненты
могут попасть в организм оператора через кожу рук
или путем аэрозолирования при пипетировании, а
также при использовании шприца. Меченные три¬
тием нуклеотиды при случайном попадании внутрь
внедряются в ДНК и могут вызвать радиоактивное
разложение ДНК из-за кратчайшего расстояния
воздействия низкоэнергетичного P-излучения от
3Н-метки. Радиоактивные изотопы йода будут кон¬
центрироваться в щитовидной железе и могут также
вызвать местное повреждение.
С изотопами работайте в боксе II класса защиты,
предохраняющего от аэрозолей, и надевайте резино¬
вые перчатки. Все предметы, с которыми вы работа¬
ете, должны располагаться на неглубоких поддонах,
выстланных фильтровальной бумагой или тканью
Benchote, впитывающими возникающие загрязнения.
Используйте наименьшее количество оборудования
(например, дозатор с одноразовыми пластиковыми
наконечниками, маленькие пробирки для образцов
и т.д.), совместимое с процедурой, для обеспечения
минимального объема при утилизации радиоактивных
отходов. Тщательно мойте руки после завершения
работы, регулярно обследуйте рабочую зону контроли¬
рующими датчиками для обнаружения радиоактивного
загрязнения.
7.7.2. Утилизация радиоактивных отходов
Процедура утилизации радиоактивных отходов опре¬
деляется местными правилами, принятыми в лабора¬
тории; совет по внедрению этих правил можно полу¬
чить из источников, приведенных выше. В целом, за
определенный период времени можно утилизировать
ограниченное количество радиоактивных отходов.
Утилизация может проводиться в особо обозначенных
устройствах. Количество выбрасываемого материала
должно быть занесено в книгу ведения радиоактив¬
ных материалов в графе «утилизация». Емкости,
используемые для утилизации, должны быть обрабо¬
таны специальными детергентами (www.oshasafety.
com/HazardLab.htm), смывные воды также должны
утилизоваться как радиоактивные отходы. Утилиза¬
ция может потребовать принятия определенных мер,
снижающих риск радиационного поражения, поэтому
те предметы, которые будут повторно использовать¬
ся, должны быть предварительно обработаны против
биологического загрязнения гипохлоритом, а затем
против радиоактивного загрязнения детергентом De-
con или аналогичным ему. Оба раствора необходимо
затем уничтожать как радиоактивные отходы.
7.7.3. Излучение от меченых реагентов
Второй тип риска связан с радиоактивным излучением
от высокоэнергетичных P-и у-источников, таких как
32Р, 1251,1311, 51 Сг. Защита обеспечивается работой за
2-мм свинцовым экраном и хранением источников в
свинцовом контейнере. При кратковременной работе
с низкими концентрациями 32Р можно использовать
экран Perspex (толщина 5 мм). Работайте на поддонах
в боксах II класса защиты (см. разд. 7.7.1).
7.7.4. Излучение от высокоэнергетичных
источников
Третий тип радиоактивной опасности связан с прибо¬
рами, использующими рентгеновское излучение, и вы-
сокоэнергетичным источником 60Со. Особой осторож¬
ности требуют также источники ультрафиолетового
излучения, которые применяются в стерилизационных
аппаратах или оборудовании для регуляции клеточной
128
ГЛАВА 7. БЕЗОПАСНОСТЬ, БИОЭТИКА И ВАЛИДАЦИЯ
Таблица 7.4. Опасные производственные факторы в лаборатории культивирования тканей
Категория
Объект
Риск
Меры предосторожности
Общие
Разбитое стекло
Пипетки
Острые инструменты
Стеклянные Пастеровские
Раны, инфекция
Раны, инфекция
Раны, инфекция
Раны, инфекция
Осторожное размещение в специальной корзине
для мусора
Проверка пипеток. Замена поврежденных. Исполь¬
зование пластиковых пипеток
Аккуратное использование, утилизация в корзине
для острых и колющих предметов
Аккуратное использование, не используйте для
Ожоги
Пожар
Радиоло¬
гическая
опасность
Биологиче¬
ская
пипетки
Иглы для шприцов
Электрические кабели
Прокладка труб
Газовые баллоны
Жидкий азот
Автоклавы, стерилизаци¬
онные печи, горячие по¬
верхности
Бунзеновская горелка,
открытое пламя, в част¬
ности в совокупности с
парами спирта;
Ручные автоклавы
Радиоизотопы в стериль¬
ном боксе
Излучение в культуре
Ввоз клеточных линий и
биопсийного материала
Генетические манипу¬
ляции
Распространение вирусов
Расположение и эксплуа¬
тация ламинарных
шкафов
Раны, инфекция
Пожар, удар током, пре¬
ткновение, падение
Утечка, затор, падение
Неустойчивость, утечка
газа
Обморожение, асфик¬
сия, взрыв
Контакт с оборудова¬
нием, манипуляции с
только что стерилизо¬
ванным материалом
Пожар, повреждение
расплавленным мате¬
риалом, риск ожога
При отсутствии воды
может загореться со¬
держимое
Излучение, разливание,
аэрозолирование,
Летучесть
Доза радиации
Инфекции
Инфекции, перенос
ДНК
Инфекции
Нарушение герметич¬
ности
потенциально биологически опасных материалов,
использование пластика
Минимизировать употребление, при возможности
отказаться от использования. Утилизация в кор¬
зине для острых и колющих предметов
Проверка надежности соединений, соединение зажи¬
мом нескольких кабелей вместе и расположение их
в безопасном месте
Проверка и регулярная замена устаревших труб
Закрепление на месте расположения, расположение
не на пути прохода персонала
Прикрепление к столу или стене, регулярная про¬
верка на предмет утечки
Ношение маски, лабораторного халата и перчаток; не
хранить ампулы в жидкой фазе, либо помещать в
оболочку при оттаивании
Предупредительные знаки, использование теплоизо¬
лирующих перчаток
Держать вне ламинарных шкафов, не помещать под
шкафы и полки, не возвращать охваченные огнем
инструменты в спирт
Устанавливать таймер и термостатический предох¬
ранитель. Удостовериться, что предохранительный
клапан в наличии и рабочем состоянии
Работать на абсорбирующем поддоне в боксе II клас¬
са химической защиты; минимизировать аэро¬
золирование
Использовать монитор дозы радиации. Носить
персональный монитор и регулярно производить
проверки
Не ввозить из зон повышенного риска, скрининг
культур на присутствие возбудителей
Следование руководствам по генетическим мани¬
пуляциям
Соблюдать требования CDC или ACDP
Работать на необходимом уровне безопасности, ми¬
нимизировать аэрозолирование
Регулярная проверка потоков в ламинарный шкафах,
перепада давления через фильтры, а также утечку
воздуха из боксов
пролиферации в фидерных слоях (см. разд. 14.2.3. и
протокол 23.1,23.4). По причине выделения высокого
уровня энергии, в частности рентгеновских лучей или
60Со, эти источники часто располагаются в специально
оборудованных строго контролируемых приспособле¬
ниях. УФ-источники могут стать причиной поврежде¬
ния кожи или глаз; они должны экранироваться для
предупреждения прямого воздействия на оператора.
Кроме того, работающему с УФ-излучением оператору
необходимо надевать специальные очки с барьерным
фильтром.
Проконсультируйтесь с местным государственным
служащим, узнайте нормы и правила работы до того,
как приступать к экспериментам с радиоизотопами.
7.8. Биологическая опасность
129
Местные правила могут иметь какие-то особен¬
ности, но большинство требует жесткого контроля
количества изотопов, которое можно использовать,
хранить и утилизовать. Эти требования следует
учитывать при планировании общего медицинского
обследования, включая планирование хранения об¬
разцов крови.
7.8. БИОЛОГИЧЕСКАЯ ОПАСНОСТЬ
Аналогично требованиям безопасности при исполь¬
зовании радиоизотопов, до начала проведения био¬
медицинских исследований требуется учесть правила
работы с биологически опасным материалом, включая
хранение образцов крови. Необходимость биологи¬
ческой защиты [см. Caputo, 1996] определяется как
источником материала, так и характером действий,
производимых над ним. Степень биологической
опасности связана также с условиями, при которых
проводится культивирование тканей. Стандартные
микробиологические приемы работы на открытой
поверхности имеют то преимущество, что эти методы
являются результатом накопления многолетнего опы¬
та работы. Проблемы появляются при введении в прак¬
тику новых методов или при увеличении количества
людей, работающих на одной площади. Использование
ламинарного шкафа с горизонтальным потоком обус¬
ловливает более высокую защиту стерильности куль¬
туры, но вероятность воздействия аэрозолированного
материала на оператора повышается. Это привело к
разработке ламинарных шкафов с вертикальным воз¬
душным экраном в передней части (см. разд. 5.2.1. и
6.4) для минимизации выхода зараженного воздуха из
объема шкафа. Такой тип ламинарного шкафа опреде¬
ляется как II класс микробиологической безопасности
(см. разд. 7.8.2).
7.8.1. Уровни биологической защиты
Национальным институтом здоровья (NIH) и Центром
по контролю и предупреждению болезней (CDC) США
выделяется четыре уровня биологической защиты
[Материалы HHS. No (CDC) 93-8395] (табл. 7.5). Они
определяют требуемые методы работы, оборудование и
меры безопасности. Европейские законы в этой области
еще окончательно не приняты, но похожи на существу¬
ющие в Великобритании Руководства, которые также
выделяют четыре уровня биологической защиты, хотя
с небольшими отличиями от классификации США
(табл. 7.6). В таблицах представлено только краткое
содержание, поэтому читатель, который предполага¬
ет работать с потенциально биологически опасными
объектами, должен обратиться к соответствующим
электронным источникам [USA: www.orcbs.msu.edu/
biological/BMBL/BMBL-l.htm;UK: www.hse.gov.uk/
biosafety/]. Руководства Европейского Союза находят¬
ся на стадии подготовки.
7.8.2. Ламинарные шкафы
микробиологической защиты
В пределах каждого уровня биологической защиты
можно выделить три уровня манипуляций, определя¬
емых типом используемого ламинарного шкафа:
1) Максимальная защита от известного возбуди¬
теля,. Изолированный бокс с фильтрацией воз¬
духа, входящего и выходящего через встроенные
фильтры-ловушки для возбудителей (бокс биоло¬
гической защиты или микробиологический бокс
III класса безопасности, рис. 7.4в). Бокс обычно
установлен в отдельной комнате с ограниченным
допуском, дезинфекцией выносимого оборудо¬
вания, твердых и жидких отходов (см. BSL4 в
табл. 7.5 и Уровень 4 в табл. 7.6), в зависимости от
природы риска.
2) Промежуточный уровень защиты от потенци¬
ального возбудителя. Вертикальный ламинарный
шкаф с передней защитой в форме воздушной
завесы и фильтрацией выходящего воздуха (бокс
биологической защиты или микробиологический
бокс II класса безопасности, рис. 1Аа) [National
Sanitation Foundation Standard 49, 1983; British
Standartisation, 1999]. Если определено наличие
возбудителя в материале, то работа должна вестись
в ламинарном шкафу, находящемся в отдельном
помещении, в соответствии со И, III или IV уровнем
защиты в зависимости от вида возбудителя. Если нет
причины предполагать инфицирование материала,
помимо случайного, то ламинарный шкаф, который
можно отнести ко II уровню защиты, допустимо
располагать в общем помещении лаборатории куль¬
тивирования тканей. При этом следует ограничить
доступ к материалу, с которым ведется работа, кон¬
тролировать утилизацию мусора, использование
защитной одежды, а также запретить употребление
пищи и напитков на территории рабочей зоны (см.
табл. 7.4; 7.5). Все боксы биологической защиты
должны быть объектом строгого контроля [Osborne
et al., 1999], с проверкой фильтров через определен¬
ные интервалы времени, соответствующей схемой
мероприятий — фумигацией боксов, заменой фильт¬
ров, безопасным уничтожением старых фильтров с
упаковкой их в двойную оболочку и последующим
сжиганием.
3) Минимальная защита. Открытая поверхность,
выполнение правил ведения микробиологических
работ. Обычно эти работы достаточно проводить в
специально выделенной части помещения, которая
может быть названа «лабораторией культивирова¬
ния тканей», но где будут соблюдаться все меры
предосторожности для работы на I уровне безо¬
пасности.
В табл. 7.7 приводится список основных мероприя¬
тий, соответствующих каждому уровню биологической
защиты. Кроме того, при организации лаборатории
следует обратиться в государственный и местный коми¬
теты по технике безопасности и использовать соответ-
130
ГЛАВА 7. БЕЗОПАСНОСТЬ, БИОЭТИКА И ВАЛИДАЦИЯ
Таблица 7,5. Уровни безопасности (BSL) в США (требуются замечания переводчика!)
BSL 1 (SMP1)
BSL 2 (SP2)
BSL 3 (SP)
BSL 4 (SP)
Допуск
Уборка
Личная
гигиена
Осколки
Допуск ограничен, когда
ведется работа
Контроль против зара¬
жения насекомыми и
грызунами
Потоки возду¬
ха и венти¬
ляция
Оборудова¬
ние
Легкость уборки, доста¬
точно пространства
между боксами, обо¬
рудованием и т. д. водо¬
упорная поверхность
рабочих столов
Ношение лабораторных
халатов, не принимать
еду и питье и т. д., не пи-
петировать ртом. Но¬
сить перчатки и защит¬
ный козырек (особенно
при наличии контак¬
тных линз). Раковина
для мытья рук
Снимать перчатки и мыть
руки, покидая поме¬
щение
Без особенностей.
Открываемые окна долж¬
ны иметь экран
Без особенностей
Без особенностей
Доступ ограничен по¬
стоянно
Оценка предраспо¬
ложенности, имму¬
низация изучение
фоновых харак¬
теристик, может
потребоваться пе¬
риодическое иссле¬
дование проб сыво¬
ротки
Аналогично BSL 1 +
регулярное обезза¬
раживание рабочих
поверхностей и обо¬
рудования
Аналогично BSL 14-
Обеспечение обезза¬
раживания и стирки
в помещении лабо¬
ратории
Оборудование для
промывки глаз
Аналогично BSL 1+
Обычное, обеззаражи¬
вание, в частности
до внесения в поме¬
щение лаборатории
или выноса его
Применение стекла
ограничено неиз¬
бежным использова¬
нием. Использован¬
ные или разбитые
предметы помеща¬
ются в контейнер,
обеззараживаются
до утилизации
Аналогично BSL 2,
+ отдельное лабораторное
помещение на два места с
самозахлопывающимися
дверьми
Когда ведется работа, двери
закрыты
Иммунизация или проверка
иммунного статуса, изуче¬
ние фоновых характерис¬
тик, периодическое иссле¬
дование проб сыворотки
Аналогично BSL 2 +
Рекомендуется работа на
абсорбирующей бумаге с
пластиковой основой. Все
поверхности в комнатах
покрыты моющимся защит¬
ным слоем
Аналогично BSL 2+ защитные
очки и маска или экран для
лица кроме BSC
Защита органов дыхания, если
образование аэрозоля не
может быть контролируемо.
Одежда со сплошной пере¬
дней частью, снимаемая при
выходе из лаборатории
Автоматический или откры¬
ваемый локтем кран на
раковине
Окна закрыты и герметизи¬
рованы
Отрицательное давление,
полная экстракция воздуха,
выхлоп воздуха наружу из
рабочей зоны или захват
воздуха
Аналогично BSL 2+ физиче¬
ские методы сдерживания,
например дополнительные
изолирующие колпаки для
центрифуг и роторов
Очищающие НЕРА-
фильтры
Вакуумные линии защище¬
ны дезинфицирующими
ловушками и НЕРА-филь-
трами
Оборудование, предупрежда¬
ющее обратный поток
Аналогично BSL 2
Аналогично BSL 3 4 только
определенные сотруд¬
ники
Охраняемые запирающиеся
двери. Раздевалка и душ.
Двери с внутренними
замками. Оборудование
и материалы вносятся
через шлюзовую камеру с
двумя дверьми (автоклав
или фумигатор)
Не вносятся никакие ма¬
териалы помимо тех, что
непосредственно необхо¬
димы для работы
Аналогично BSL 3+
герметичные соединения,
дезинфицирующие ло¬
вушки на сливных отвер¬
стиях, НЕРА-фильтры на
выпускных отверстиях.
Минимальная площадь
поверхности для пыли
Аналогично BSL 3+
Перемена одежды; одежда
автоклавируется
Душ перед уходом из лабо¬
ратории
Система вентиляции, спе¬
циально предназначен¬
ная для этого уровня,
снабженная системой
аварийной сигнализа¬
ции, без рециркуляции
воздуха
Аналогично BSL 3
Аналогично BSL 2
7.8. Биологическая опасность
131
Окончание табл. 7.5
BSL 1 (SMP1) BSL 2 (SP2) BSL 3 (SP) BSL 4 (SP)
Блок микро¬
Не требуется, миними¬
Класс II
Класс II или III, выхлоп через
Класс II или III, инди¬
биологичес¬
зировать возможность
НЕРА фильтр, для II класса
видуальный костюм с
кой защиты
аэрозолирования
возможна рециркуляция
воздуха
системой жизнеобеспе¬
чения и положительным
давлением воздуха
Дезинфекция
Любые дезинфектанты.
Аналогично BSL 1+
Аналогично BSL 2 +
Аналогично BSL 3+
Обеззараживание по¬
Специфические про¬
Разлив удаляется специально
Требуется двухсторонний
верхностей по крайней
цедуры, автокла-
подготовленными специа¬
входной автоклав, пред¬
мере один раз в день
вирование непода¬
леку
листами
почтительно из бокса III
класса защиты
Хранение и пе¬
Без особенностей
Герметичный контей¬
Аналогично BSL 2
Устойчивые к воздействию
редача мате¬
нер
материалы передаются
риала
в двойной упаковке в
небьющихся контейне¬
рах после замачивания
в дез. растворе или фу¬
мигации
Утилизация
После дезинфицирова¬
Аналогично BSL 1 +
Аналогично BSL 2 + обезза¬
Аналогично BSL 3+ обез¬
ния или обеззаражи¬
определенные меры
раживание в помещении
зараживание всех слив¬
вания автоклавирова-
обеззараживания
лаборатории
ных вод, включая душ и
нием в соседнем поме¬
туалет, а также обеззара¬
щении
живание других материа¬
лов через двусторонний
автоклав
Руководство
Без особенностей
Требуется подготови¬
Аналогично BSL 2
Аналогично BSL 3+ вы¬
по биобезо¬
тельный тренинг
сокая квалификация в
пасности и
SMP
тренинг
Аварии и раз¬
Без особенностей
Подписанная доклад¬
Аналогично BSL 2 +
Аналогично BSL 3+
ливания
ная. Возможно, пот¬
Разлив удаляется специально
Мониторинг отсутствия
ребуется медицин¬
подготовленными специа¬
персонала, забота о забо¬
ское заключение
листами
левших, карантин
Валидация
Без особенностей
Без особенностей
Без особенностей
Безопасность всех сливных
оборудова¬
жидкостей и выхлопов
ния
1 SMP — Standard Microbiological Practices, Стандартная микробиологическая практика.
2 SP — Special Practices, специальная практика. Манипуляции с агентами обладающими умеренной потенциальной опасностью.
ствующие руководства (см. разд. 7.2) для ознакомления
с требованиями действующего законодательства.
7.8.3. Человеческий биопсийный материал
Требования биологической безопасности являются
наиболее ясными, когда используется известный,
изученный возбудитель, поскольку нормы и правила
работы с каждой группой возбудителей установлены
как в США [Министерство здравоохранения и социаль¬
ного обеспечения США, 1993], так и в Великобритании
[Консультативный комитет по опасным возбудителям
(ACPD), 1995а, Ь]. Однако в двух областях исследований
существует наибольший риск, связанный с определени¬
ем природы материала. Одна из этих областей касается
новых потенциально патогенных генов и находится в
стадии развития, поскольку связана с рекомбинантными
техниками — такими как трансфекция, ретровирусное
инфицирование и межвидовая клеточная гибридиза¬
ция. Манипуляции с такими культурами в ламинарных
шкафах связаны с предполагаемым риском, который не
имеет эпидемиологического обоснования для правиль¬
ной его оценки. Трансформация вирусов, амфитропные
вирусы, трансформированные клеточные линии челове¬
ка, гибриды клеток человека и мыши, а также, например,
клеточные линии, полученные при ксенотрансплантации
у иммунодефицитных мышей, должны быть объектами,
работа с которыми ведется с большой предосторожнос¬
тью до тех пор, пока не получено достаточно данных,
подтверждающих отсутствие риска.
Другой областью риска является включение слу¬
чайных возбудителей в клеточные линии, либо в био-
псийные или аутопсийные образцы от человека или
приматов [Grizzle & Polt, 1988; Wells et al., 1989; Tedder
et al., 1995], либо в продукты животного происхождения,
132
ГЛАВА 7. БЕЗОПАСНОСТЬ, БИОЭТИКА И ВАЛИДАЦИЯ
Таблица 7.6. Уровни биологической защиты в Великобритании
УровенИ
Уровень 2
Уровень 3
Уровень 4
Допуск
При проведении работы
двери закрыты
Ограничен
Аналогично 2 +
Отдельная лаборато¬
рия с обсервацион¬
ным окном.
Когда лаборатория
не работает, двери
заперты
Аналогично 3 +
Контроль зараженности насеко¬
мыми и грызунами
Раздевалка и душ. Внутренние
запоры на дверях. Вентили¬
руемый воздушный шлюз для
оборудования. Телефон или
интерком
Площадь
Без особенностей
24 м3 на человека
Аналогично 2
Аналогично 2
Уборка
Легкость уборки, водо¬
упорная поверхность
рабочих столов
Аналогично 1 +
Обычное обеззара¬
живание рабочих
поверхностей
Аналогично 2
Аналогично 2
Личная гигиена
Носить лабораторные
Аналогично 1 + ем¬
Аналогично 2 + ноше¬
Аналогично 3 + смена одежды,
халаты (застегивают¬
кость для обез¬
ние перчаток; сни¬
автоклавирование одежды.
ся на боку или на спи¬
не), хранить, стирать
после изнашивания
правильно заменять.
Не принимать еду и пи¬
тье и т.д., не пипети-
ровать ртом
зараживания рук
рядом с выходом
мать которые при
пользовании общи¬
ми предметами, на¬
пример телефоном
Душ перед уходом.
Потоки воздуха
Предпочтительно отри¬
цательное давление
Требуется отрица¬
тельное давление
Аналогично 2
Отрицательное давление >70 Па
(7 мм Н20); воздух выпуска¬
ется через серию НЕРА-филь-
тров. Подача и экстракция
взаимосвязаны
Оборудование
Без особенностей
Без особенностей
Следует иметь все не¬
обходимое оборудо¬
вание
Необходимо иметь все необхо¬
димое оборудование
Блок микробио¬
Не требуется, миними¬
Требуется MSC или
Класс I или III
Класс III
логической за¬
щиты (MSC)
зировать возможность
аэрозолирования
изолятор
(BS5725);
класс II (BS5726)
Выхлоп через НЕРА
фильтр наружу
Дезинфекция
Любые дезинфектанты
Специфические
процедуры, ав-
токлавирование
неподалеку
Аналогично 2 +
Герметизация лабо¬
ратории для обез¬
зараживания, ав¬
то клавирование
предпочтительно в
лаборатории.
Аналогично 3
Требуется двухсторонний вход¬
ной автоклав, предпочти¬
тельно из бокса III класса
защиты
Хранение
Без особенностей
Требования безо¬
пасности при хра-
нении биологи¬
ческих объектов
Аналогично 2
Утилизация
В дезинфектант до ути¬
лизации
Аналогично 1 +
Метка на мусоре,
безопасный сбор
и утилизация
Аналогично 2
Аналогично 3+ дезинфекция
всех жидких отходов, включая
воду от душа. Двусторонний
бак для замачивания мусора,
который не может автоклави-
роваться
Аварии
Докладная
Аналогично 1
Аналогично 2
Аналогично 3+ второй сотруд¬
ник помогает в случае необ¬
ходимости
Валидация
оборудования
Без особенностей
Без особенностей
Без особенностей
Требуется
7.8. Биологическая опасность
133
С
%
Выхлопной фильтр
" для удаления
возбудителей
Удаление 20%
воздуха
Вентилятор для
удаления 20%
воздуха
Створчатый
клапан для
предупреждения Угольный _
обратного потока абсорбирующий
фильтр
Герметичный воздуховод
под отрицательным
давлением
НЕРА выхлопной
фильтр
Вентилятор для
рециркуляции 80% воздуха
НЕРА фильтр для
рециркулирущего воздуха
Угольный фильтр
Изолированный ламинарный
шкаф под отрицательным
давлением
Лервичный-
фильтр
О Передний экран для изоляции
при дезинфицировании
окуриванием
Лоток для сбора
загрязнения при
разливании
VV
а ЛАМИНАРНЫЙ ШКАФ II КЛАССА БЕЗОПАСНОСТИ
б ЛАМИНАРНЫЙ ШКАФ ХИМИЧЕСКОЙ БЕЗОПАСНОСТИ
НЕРА выхлопной
фильтр
-Смотровое окно ►
Входной шлюз с двойной двер|
для выноса оборудования на А
автоклавирование
Отверстия
для рук_^
с перчатП
ками I
Рабочая зона
Герметизированный
воздуховод и
ламинарный шкаф,
под отрицательным
давлением воздуха
Предварительный фильтр
НЕРА-впускной
фильтр
Входной шлюз с
двойной дверью
для доставки
оборудования
Вентилятор для
удаления воздуха
Фильтр для захвата
возбудителей
Створчатый клапан
для предупреждения
обратного потока
100% удаления
воздуха
в ЛАМИНАРНЫЙ ШКАФ III КЛАССА БЕЗОПАСНОСТИ, ВИД СПЕРЕДИ г ЛАМИНАРНЫЙ ШКАФ III КЛАССА БЕЗОПАСНОСТИ, ВИД СБОКУ
Рис. 7.5. Ламинарный шкаф биологической безопасности. (а) Ламинарный шкаф с вертикальным потоком II класса защиты
с рециркуляцией 80% воздуха и удалением 20% наружу через фильтр и воздуховод для захвата потенциальных возбудителей
из воздуха. Для восстановления объема рециркулирующего воздуха, он захватывается из передней части шкафа, что повышает
защиту от распространения биологически опасных частиц из рабочей зоны; (б) ламинарный шкаф с вертикальным потоком
II класса химической защиты с угольным фильтром на выходе, и рециркуляцией воздуха; (в) ламинарный шкаф III класса
защиты без рециркуляции воздуха, шкаф снабжен резиновыми карманами, работа ведется при отрицательном давлении, с
воздушной разделительной камерой для доставки оборудования и прямым доступом к автоклаву, соединенному или располо¬
женному рядом; (г) ламинарный шкаф III класса, вид сбоку
134
ГЛАВА 7. БЕЗОПАСНОСТЬ, БИОЭТИКА И ВАЛИДАЦИЯ
Таблица 7.7. Биологические процедуры и предлагаемый уровень защиты*
Процедура Уровень Рабочее пространство
защиты
Приготовление сред GLP
Первичные культуры или клеточные линии, не от 1
человека или других приматов
Первичные культуры или клеточные линии, не от 1
человека или других приматов, инфицированные
или трансфецированные
Первичные культуры или серийный пассаж клеток 2
человека или других приматов
Межвидовые гибридомы или другие рекомбинантные 2
клетки, трансфецированные клетки, человеческие
клетки, и опухолевые клетки животных
Человеческие клетки, инфицированные ретровирус- 3
ными конструктами
Вирус-продуцирующие человеческие клеточные 3
линии, клеточные линии, инфицированные амфит-
ропными вирусами
Образцы тканей и культуры, несущие известные 4
человеческие возбудители
Открытый стол со стандартной микробиологической
практикой либо горизонтальный или вертикальный
ламинарный шкаф
Открытый стол со стандартной микробиологической
практикой либо горизонтальный или вертикальный
ламинарный шкаф
Ламинарный шкаф II класса защиты
Ламинарный шкаф II класса защиты
Ламинарный шкаф II класса защиты
Ламинарный шкаф II класса защиты
Ламинарный шкаф II класса защиты
Ламинарный шкаф III класса микробиологической защиты
с перчаточными карманами, фильтрованием воздуха и
ловушкой для патогенов в системе вентиляции воздуха
* Предлагаемый уровень защиты не является законодательным основой. Ознакомьтесь с принятыми на вашей
территорией нормами и правилами техники безопасности, а также с регулирующим законодательством прежде,
чем формулировать действующее у вас Руководство.
такие как сыворотка, в частности при получении их из
тех частей света, в которых имеется высокий эпидеми¬
ческий уровень инфекционных болезней. Когда наличие
инфекции подтверждено, вид микроорганизма опреде¬
лит необходимый уровень защиты и соответствующие
меры предосторожности, но когда инфекционный агент
еще не определен, остается вероятность присутствия
в образцах возбудителей гепатита В, ВИЧ-инфекции,
туберкулеза и других еще не диагностированных ин¬
фекционных агентов. Требования конфиденциальнос¬
ти часто запрещают проводить исследования на ВИЧ
без согласия пациента, и для большинства случайных
инфекций соответствующая информация может быть
недоступна. Если возможно, то биопсийный материал
должен исследоваться на присутствие возбудителей до
начала манипуляций с ним. Полномочия на проведение
таких работ необходимо подтвердить в соответствующей
форме, подписанной работником, занимающимся куль¬
тивированием тканей (см., например, табл. 7.8). Однако
потребность получать образцы для работы как можно
быстрее приводит к тому, что культивирование начи¬
нается без предварительного тестирования на наличие
возбудителей. С такими образцами следует обращаться
со всеми необходимыми мерами предосторожности:
1. Транспортируйте образцы в двойном контейнере
(им может быть универсальный контейнер или пу¬
зырек с закручивающейся крышкой, находящийся
внутри второго сосуда с закручивающейся крышкой,
например в полипропиленовой банке). Контейнер, в
свою очередь, должен быть упакован в непрозрачный
пластиковый или водонепроницаемый бумажный
пакет, заполненный абсорбирующим материалом для
удержания внутри любых возможных протеканий.
В таком виде образец транспортируется в лаборато¬
рию специальным курьером.
2. Заносите все образцы в инвентарную книгу в графу
«поступления» и располагайте в специальном хо¬
лодильнике, с нанесенным знаком «биологическая
опасность».
3. Выполняйте иссечение и субкультивирование та¬
ких тканей и культур в ламинарном шкафу II класса
защиты, предпочтительно расположенном в комна¬
те, отдельной от общей площади лаборатории куль¬
тивирования тканей. Такие меры минимизируют
риск распространения заражения других культур,
например, микоплазмами, а также ограничат круг
лиц, контактировавших с образцом, если будет
обнаружена его инфицированность.
4. При работе с такими образцами избегайте исполь¬
зования острых и режущих инструментов (напри¬
мер, шприцов, скальпелей, стеклянных Пастеров¬
ских пипеток).
5. Помещайте все культуры в пластиковые коробки
с надписью или этикеткой, идентифицирующей
культуры как биологически опасные, с именем от¬
ветственного лица и датой.
6. Сбрасывайте все стеклянные предметы, пипетки,
инструменты и т. д. в дезинфицирующий раствор
7.8. Биологическая опасность
135
Таблица 7.8. Форма информированного согласия донора
СОГЛАСИЕ НА ИЗЪЯТИЕ ТКАНЕЙ ДЛЯ ДИАГНОСТИКИ И ИССЛЕДОВАНИЯ
Данная форма требует вашего разрешения на получение образца вашей крови или одного и более маленьких
кусочков тканей для того, чтобы использовать их в медицинских исследованиях. Этот образец, или клеточная
линия, или продукты, полученные из них, могут быть использованы рядом различных исследовательских ор¬
ганизаций, или они могут храниться в течение длительного периода, ожидая использования. Возможно также,
что образцы будут использованы какой-то коммерческой организацией для разработки новых лекарственных
препаратов. Мы бы хотели, чтобы вы были осведомленны об этом и подтвердили факт осведомленности личной
подписью. Вы также должны быть осведомлены о возможном исследовании ваших тканей на присутствие ин¬
фекционных агентов, таких как вирус СПИД или гепатит, на что вы подтверждаете согласие личной подписью
данной формы.
Я готов предоставить ткани для использования в медицинских исследованиях и разработках. Я прочел и понял
по мере своих способностей предоставленную мне информацию (если донор в слишком плохом состоянии здо¬
ровья для того, чтобы поставить подпись, форму должны подписать его близкие родственники с соблюдением
его интересов).
ФИО донора ФИО родственника
Подпись
Дата
Данный материал получит код, что гарантирует полную конфиденциальность. Ваше имя не будет известно
никому, кроме лица, непосредственно берущего образец.
Хотите ли вы получать любую информацию об этом материале, касающуюся вашего здоровья? Да
Нет
Подпись Дата
Хотите или предпочитаете ли вы, чтобы эту информацию предоставили вашему врачу? Да
Нет
Если «Да» то имя врача
Адрес врача
или мешки для автоклавирования биологически
опасных объектов.
Если соответствующие клинические диагностичес¬
кие тесты показывают, что материал не инфицирован,
стерилен и не содержит микоплазмы, он может куль¬
тивироваться, как остальные культуры. Однако если
предполагается получить более чем 1x109 клеток или
чистую ДНК, то следует посоветоваться с местным
комитетом по технике безопасности.
При обнаружении образца инфицирования он
должен быть уничтожен. Для этого его помещают в
двойной мешок для автоклавирования биологически
опасных объектов вместе со всеми реактивами, которые
использовались при работе с ним, с последующим авто-
клавированием или сжиганием. Инструменты и другие
твердые предметы помещают в контейнер с дезинфек¬
тантом, замачивают, по крайней мере, на 2 ч и затем
автоклавируют. Если есть необходимость продолжать
работу с материалом, уровень защиты должен возрасти
соответственно категории возбудителя [Отдел CDC
по здоровью и безопасности, 1999, (www.cdc.gov/od/
ohs/biosafety/bmbl4/bmbl4toc.htm); Консультативная
комиссия по Опасным Возбудителям, 2003, wwwhse.
gov.uk/aboutus/meetings/acdp/\.
7.8.4. Манипуляции с генетическим
материалом
На любые действия, связанные с изменением генети¬
ческого состава клеток или клеточной линии, с которой
вы работаете, путем переноса нуклеиновой кислоты,
136
ГЛАВА 7. БЕЗОПАСНОСТЬ, БИОЭТИКА И ВАЛИДАЦИЯ
должно быть получено разрешение местного комите¬
та по биологической безопасности. Текущие нормы
и правила могут быть получены в США из подразде¬
ления, занимающегося охраной окружающей среды
(NCI, NIH, Bldg 13, Room2W64, Bethesda, MD 20892)
(информация доступна также на сайте www.nci.nih.
gov/intra/resource/biosafe.htm, а в Великобритании — от
Комиссии по охране здоровья и безопасности (www.hse.
gov.uk/biosafety/gmo/index/htm).
7.8.5. Уничтожение биологически
опасных отходов
Материалы, потенциально биологически опасные,
должны подвергаться стерилизации до утилизации
[Национальный исследовательский совет, 1989; Кон¬
сультативный комитет по здравоохранению, 1992]. Их
можно поместить в негерметично закрытые контейнеры
и поместить в автоклав либо залить стерилизующим
веществом, например гипохлоритом. Существуют
различные дезинфицирующие растворы в виде жидких
концентратов (такие как Clorox и Chloros) или таблеток
(Precept Tablets) (см. приложение II). Рекомендуемые
концентрации зависят от местных норм и правил, но
приблизительные рекомендации обычно можно по¬
лучить из инструкции пользователя, прилагаемой к
продукту производителем препарата. Гипохлорит часто
используют в количестве 300 ppm (частей на миллион)
активного хлора, но некоторые руководства требуют
2500 ppm активного хлора (разведение Chloros 1:20),
как рекомендовано в Howie Report [1978]. Гипохло¬
рит эффективно действует и легко отмывается с тех
предметов, которые будут повторно использоваться,
но обладает высокой коррозийностью, в частности в
щелочных растворах. Он обесцвечивает одежду и даже
вызывает коррозию нержавеющей стали, поэтому при
работе с ним следует надевать перчатки и лаборатор¬
ные халаты или фартуки. Емкости для замачивания и
цилиндры должны быть сделаны из полипропилена.
7.8.6. Дезинфекция окуриванием
Некоторые виды деятельности требуют, чтобы микро¬
биологический бокс был полностью обработан после
использования. К таким видам относится работа с вы¬
сокопатогенными микроорганизмами либо обработка
бокса перед сервисным обслуживанием. Дезинфекция
окуриванием или фумигация проводится с использо¬
ванием паров формальдегида. При этом бокс должен
быть отключен от электрической сети и закрыт до
начала фумигации, которую проводят при помощи
электроподогреваемого генератора. Ламинарный шкаф
включается на короткий период для циркуляции газа
и затем оставляется на 1 ч. После этого ламинарный
шкаф включают для продувания паров на ночь, затем
открывают переднюю панель на 10 мин. Фумигация
бокса может проводиться также при помощи перекиси
водорода (Bioquell), который легче рассеивается по
завершении обработки.
7.9. БИОЭТИКА
Кроме потенциальной биологической опасности работа
с тканями человека и животных связана с рядом эти¬
ческих проблем, включая приобретение, последующие
манипуляции, а также отдаленные способы использо¬
вания материала.
7.9.1. Ткани животных
В большинстве стран, ведущих биомедицинские иссле¬
дования, вводятся в действие или будут вскоре введены
законы, регулирующие использование эксперимен¬
тальных или других животных-доноров для получения
тканей. В основном эти нормы и правила относятся к
высшим животным, поскольку предполагается, что
они имеют достаточно высокоорганизованный мозг,
чтобы чувствовать боль и дистресс, и обычно не приме¬
няются к низшим позвоночным, таким как рыбы, или
к беспозвоночным. Считается, что высшие животные
становятся разумными со второй половины эмбрио¬
нального развития, поэтому ограничения, связанные
с биоэтикой, накладываются на методы забоя или
методы обездвиживания и оперативного воздействия
в случаях, когда животное остается живым. Основные
требования направлены на минимизацию страданий
от боли и дискомфорта, причиняемых животным.
Существуют также ограничения, касающиеся содер¬
жания животных и ухода за ними как в питомнике
и виварии в условиях эксперимента, так и в ветери¬
нарной клинике в клиническом состоянии. В каждом
случае контроль осуществляется на местном уровне
комитетом по биоэтике и на уровне правительства
официально назначенным лицом. У каждой страны
свое законодательство, но при получении лицензии
для работы в любой стране первым шагом является
обращение в местный комитет по защите животных и
биоэтике (АЕС) [US: wzem.bioscience.org/guides/anim-
care/htm; UK: www.ebra.org/regulat/uk.html; www.mrc.
ac.uk/index/sitemap.htm: Использование животных в
научных исследованиях].
7.9.2. Ткани человека
Требования при работе с человеческим материалом
отличаются от правил работы с тканями животных и
более сложны. Ткани должны быть получены в усло¬
виях клиники опытным практикующим врачом, на¬
ибольшую проблему представляет юридический аспект
забора тканей и выбор целей, для которых они будут
использованы. В этом случае до начала планирования
экспериментальных работ с тканями человека также
следует прежде всего обратиться в местную комис¬
7.10. Валидация
137
сию по медицинской этике (НЕС), которая должна
решить, имеется ли достаточно причин и оправданны
ли расходы возможной прибылью. Взаимодействие с
НЕС следует осуществить на стадии планирования, по¬
скольку большинство грант-образующих организаций
до начала финансирования требуют предоставления
свидетельства этической комиссии о разрешении на
работу. Кроме того, существует вопрос собственности
на ткани и их производные — ДНК, любые клеточные
линии, полученные из них, и любые продукты или
годные для продажи технологии, которые могут быть
в конечном итоге разработаны и проданы с получением
прибыли. В связи с этим необходимо:
1) до забора тканей для исследовательских целей сверх
клинически необходимых получить информирован¬
ное согласие пациента или его родственников;
2) заполнить соответствующую письменную форму
(например, табл. 7.8), при составлении которой до¬
нор или пациент должен ясно понимать, для каких
целей будут использованы их ткани, такое согласие
должно быть письменно заверено;
3) разрешение родственников, если пациент находится
в слишком плохом состоянии, для того чтобы при¬
нимать разумное решение;
4) подготовить краткое резюме, объясняющее в тер¬
минах закона цели и задачи исследования, ход
эксперимента, ожидаемые результаты и прибыль, в
частности если они будут использованы для улуч¬
шения качества медицинской помощи;
5) гарантировать конфиденциальность происхожде¬
ния образца ткани;
6) определить собственника клеточной линии и ее
производных;
7) получить разрешение для проведения генетических
модификаций клеточных линий;
8) установить права пациента при взаимодействии с
любыми коммерческими лабораториями;
9) предоставить донору возможность определить,
должна ли генетическая информация по образцу
тканей быть передана пациенту и/или врачу;
10) согласие донора на скрининг тканей на предмет
присутствия возбудителей.
Самым простым решением будет попросить донора
или его родственников подписать отказ от права соб¬
ственности на ткани до того, как они будут извлечены;
в противном случае становится чрезвычайно затруд¬
нительным юридический аспект собственности на
клеточные линии, которые в дальнейшем могут быть
получены, как и на любые дальнейшие биофармацевти-
ческие аспекты использования этих клеточных линий,
их генов, и их продуктов. Предоставление генетичес¬
кой информации, а также свидетельства о возможной
патологической инфекции, такой как ВИЧ, является
более сложной проблемой; в случае если пациент
находится в больнице, эти данные рассматриваются
как часть диагностических исследований и предо¬
ставляются на паритетных началах лечащему врачу. В
случае когда донор не госпитализирован, вы должны
спросить его, заинтересован ли он/она ознакомиться
с данными, которые будут вами получены, а также в
соавторстве при дальнейшем их использовании. Луч¬
ше предоставить это врачу общей практики, который
лечит донора, путем подписания согласия в стандарт¬
ной форме. Такая форма может быть предоставлена
местным комитетом по биомедицинской этике, либо
составляется (см. табл. 7.8) в сотрудничестве с НЕС
и другими заинтересованными организациями, таки¬
ми как клинические лаборатории, группа поддержки
пациента, и организации, предоставляющие финанси¬
рование работ. Дальнейшая информация может быть
получена в рубрике Научно-исследовательская этика
на сайте www.bioethics.gov/topics/other_index.html, а
также в рубрике Человеческие ткани: Этические и
юридические вопросы на сайте www.nuffieldbioethics.
org/home/. Кроме того, следует обращаться в Совет по
медицинским исследованиям Великобритании и Все¬
мирную организацию здравоохранения [Рекомендации
по работе для этических комитетов, которые курируют
биомедицинские исследования]. В Великобритании
при формировании нового комитета по биомедицинс¬
кой этике либо у существующего комитета возникает
необходимость в совете и консультации, следует об¬
ращаться в Центральный офис исследовательского
этического комитета (COREC) www.corec.org.uk/.
Наиболее противоречивым аспектом процедуры
получения информированного согласия может ока¬
заться предоставление информации для донора, про¬
изводимое для того, чтобы он знал, с какой целью и как
будут использованы его ткани. Для этого требуется в
кратком виде в юридических терминах предоставить
содержание работы так, чтобы не смутить и не привести
в замешательство. Трансплантация тканей имеет ясные
цели, поэтому достаточно небольшого объяснения, но
некоторые методы и вопросы, такие как исследование
нарушений передачи клеточного сигнала в трансфор¬
мированной клетке, потребуют некоторого обобщения
и упрощения общей концепции. Часто объяснение в
простых терминах, данное устно, воспринимается легче,
чем подробное описание. Однако устное представление
тоже должно прозвучать с использованием юридичес¬
ких терминов, с акцентом на потенциальный выигрыш,
указанием этических аспектов, таких как получение
генетической информации, трансплантация клеток
другому лицу после тканевой инженерии.
7.10. ВАЛИДАЦИЯ
Правильное использование клеточных линий, в ком¬
мерческих или исследовательских целях, требует
подтверждения истинности (валидации). В промыш¬
ленных условиях в юридические обязанности произво¬
дителя входит то, что окончательный продукт должен
быть принят Федеральным управлением по контролю
качества продуктов и лекарственных средств (FDA) в
США либо Национальным институтом качества кли¬
нических исследований (NICE) в Великобритании. Од¬
нако в академических исследовательских лабораториях
138
ГЛАВА 7. БЕЗОПАСНОСТЬ, БИОЭТИКА И ВАЛИДАЦИЯ
требования могут быть менее жесткими и обязательства
по использованию клеточных линий ложатся на со¬
весть исследователя. Несмотря на это, использование
клеточных линий, не прошедших валидацию, снижает
ценность результатов и возможность повторения их
другими исследователями.
Существует три основных этапа валидации:
1) Подтверждение аутентичности: является ли клеточ¬
ная линии тем, чем она заявлена?
2) Происхождение: что произошло с клеточной линией с
тех пор, как она была выделена из первоисточника?
3) Исследование на зараженность: свободна ли клеточ¬
ная линия от заражения всеми известными формами
бактериальных возбудителей?
торая будет дополняться по мере ведения этой линии.
Это означает, что при работе необходимо регулярно
вести в правильной форме соответствующие записи
(см. разд. 12.3.11, 13.7.8, 20.3.6), в которых подробно
описаны рутинные манипуляции, важные экспери¬
ментальные наблюдения, замораживание и хранение.
Это не должно быть очень трудоемким процессом,
поскольку использование крупноформатных таблиц
и компьютерных баз данных позволяет вводить новые
данные (вновь полученные или изменившиеся) без пов¬
торного набора информации на клавиатуре. Клеточная
линия с хорошим подробным описанием происхожде¬
ния приобретает такую же ценность, как антикварная
мебель или произведение искусства.
7.10.1. Подтверждение аутентичности
Для определения специфического профиля клеточной
линии имеется несколько методов (см. разд. 16.6).
Наилучшие из них включают ДНК-фингерпринтинг
и изучение профиля ДНК, однако при этом требуется,
чтобы, во-первых, донор предоставил свою ДНК в ка¬
честве стандарта либо, во-вторых, чтобы сохранилась
ДНК ранних стадий ведения этой линии, когда ее аутен¬
тичность была несомненна. Иначе происхождение (вид
животного, тип ткани; см. разд. 16.3) при отсутствии
обоснованного сомнения может проводиться на основе
компиляции нескольких характеристик. Следует пом¬
нить, что один или более используемых критериев мо¬
гут подходить и быть специфичными для лаборатории,
в которой эта культура ведется, но недостаточными для
другой лаборатории, если происходит экспорт клеточ¬
ной линии. Важным аспектом является то, что до того,
как будут израсходованы силы, время и средства на
экспериментальную работу, должны быть предприняты
некоторые шаги для подтверждения аутентичности кле¬
точной линии, на которой предполагается проведение
экспериментов.
7.10.2. Происхождение
Частью процесса валидации является ведение под¬
робных записей, свидетельствующих о том, откуда
была выделена клеточная линия, что с ней произошло
с момента выделения, каковы условия ее культивиро¬
вания и пассирования, когда проводились проверки
на зараженность, если имели место попытки декон¬
таминации, то какие и когда, какие свойства линии
были обнаружены, какие генетические модификации
проводились, какие произошли спонтанные изменения
и т. д. Некоторые сведения о происхождении клеточной
линии, полученные из литературы или устно со слов
коллег, обусловливают выбор данной линии, однако
должна существовать независимая документация, ко¬
7.10.3. Контаминация
Как бы подробно вы ни вели записи, как бы четко ни
соблюдали методику, ценность ваших данных будет ут¬
рачена или, по крайней мере, значительно снижена, если
обнаружится, что линия заражена одним или несколь¬
кими возбудителями. В случаях, когда контаминация
налицо, клеточная линия уничтожается. Однако иногда
вопрос о чистоте линии остается неясным, если:
1) клеточные линии ведутся с использованием анти¬
биотиков (см. разд. 19.3);
2) не проводились стандартные исследования на пред¬
мет заражения микоплазмами;
3) нет стандартных методов, позволяющих обнаружить
заражение, например, некоторыми вирусами или
прионами (см. разд. 19.3.6).
Заражения можно избежать:
1) путем соблюдения правильной асептической техни¬
ки (см. гл. 6);
2) получением клеточных линий из достоверных ис¬
точников, например из банка клеток;
3) культивированием клеток в отсутствие антибио¬
тиков, хотя бы не постоянно, а лишь часть времени
(см. разд. 13.7.7);
4) регулярным скринингом на микоплазмы (окраска по
Hoechst 33258 обнаружит любые Д НК-содержащие
организмы, достаточно большие для того, чтобы
быть обнаруженными при помощи флюоресцент¬
ного микроскопа) (см. протокол 19.2); или
5) путем скрининга на большинство общеизвестных
вирусов с использованием ПЦР или коммерческих
контрактов с другими организациями.
Клеточные линии, которые прошли соответству¬
ющую валидацию, должны храниться в жидком азоте
и выдаваться конечному потребителю по требованию.
Конечный потребитель может хранить свои запасы в
ходе выполнения проекта, но, поскольку его основные
линии не прошли валидации, они не должны пере¬
даваться и новые потребители должны обращаться к
источнику, имеющему валидированные линии.
Глава 8
ПОСУДА И СУБСТРАТЫ
ДЛЯ КУЛЬТИВИРОВАНИЯ КЛЕТОК
8.1. СУБСТРАТ
8.1.1. Прикрепление и рост клеток
Большинство клеток позвоночных, культивируемых
in vitro, растут в виде монослоя на искусственном
субстрате. Следовательно, субстрат должен иметь
правильный заряд, чтобы позволить прикрепиться
клеткам или, по крайней мере, сорбировать факторы
клеточной адгезии, которые, в свою очередь, дадут
возможность клеткам прикрепиться и распластаться.
Несмотря на то что спонтанный рост в суспензии
ограничен линиями гемопоэтических клеток, асцит¬
ных опухолей грызунов и небольшим количеством
некоторых других выведенных линий, как, например,
мелкоклеточного рака легкого человека [Carney et al.,
1981], многие трансформированные клеточные линии
можно заставить расти в суспензии и не испытывать
зависимости от поверхностного заряда субстрата. Одна¬
ко большинству нормальных клеток для размножения
требуется распластывание на субстрате [Folkman &
Moscona, 1978; Ireland et al., 1989; Danen & Yamada,
2001], а неполноценное распластывание из-за слабой
адгезии или высокой плотности посева ингибирует
пролиферацию. Клетки, которым требуется прикреп¬
ление для дальнейшего роста, называются субстрат-
зависимыми; клетки, которые подвергаются процессу
трансформации, быстро становятся субстрат-незави-
симыми (см. разд. 18.5.1) и приобретают способность
расти в суспензии (см. разд. 13.7.4) в жидкой среде при
перемешивании или поддерживать суспензионный рост
в полутвердой среде, такой как агар.
8.1.2. Материалы
Стекло. Стекло было первым субстратом благодаря
своим оптическим свойствам и поверхностному за¬
ряду. Однако в настоящее время стекло заменено в
большинстве лабораторий на синтетический пластик
(обычно полистерол), который обладает большей плот¬
ностью и лучшими оптическими характеристиками.
Сейчас стекло используется редко, хотя оно является
дешевым, легко моется, не утрачивая при этом под¬
держивающих клеточный рост свойств. Обработка
сильными щелочными растворами (NaOH или едкими
детергентами) делает стекло непригодным для культи¬
вирования до тех пор, пока поверхность его не будет
нейтрализована кислотой (см. разд. 11.3.1).
Одноразовый пластик. Одноразовые стерильные
флаконы из полистирола представляют собой простой,
воспроизводимый субстрат для культивирования. Они
обычно обладают хорошими оптическими свойства¬
ми, а поверхность является достаточно плоской для
перевиваемых монослойных культур с равномерным
ростом. Промышленно произведенный полистирол
является гидрофобным субстратом, к которому клетки
не способны прикрепиться, поэтому культуральный
пластик обрабатывают коронным разрядом, газораз¬
рядной плазмой, у-радиацией или же химическими
стентами для формирования заряженной смачиваемой
поверхности. Так как качество конечного продукта мо¬
жет зависеть от производителя, образцы, полученные
из нескольких источников, необходимо тестировать
для определения скорости роста и эффективности
рапластывания клеток (см. протоколы 21.7-21.10) в
соответствующей среде, содержащей оптимальные и
полуоптимальные концентрации сыворотки или на
бессывороточной среде. (Высокие концентрации сы¬
воротки могут маскировать дефекты пластика.)
Несмотря на то что полистирол является наиболее
широко используемым и дешевым пластмассовым
субстратом, клетки также могут расти на поливинилхло¬
риде (PVC), поликарбонате, политетрафторретилене
(PTFE; Teflon), Melinex, Thermanox (ТРХ) и на ряде
других пластмасс. Для тестирования нового субстрата
необходимо вырастить на нем клетки в виде равно¬
мерного монослоя при наличии и отсутствии предва¬
рительной обработки поверхности (см. разд. 8.4.1), а
затем клонировать их (см. протокол 21.10). РТЕЕ может
использоваться как в заряженной (гидрофильной), так
и в незаряженной (гидрофобной) формах [Janssen et al.,
2003; Lehle et al., 2003]; заряженная форма может быть
использована для обычных монослойных и органотипи¬
ческих культур (Biopore, Millipore; Transwell, Coming), a
незаряженная — для макрофагов [von Briesen et al., 1990]
и некоторых трансформированных клеточных линий.
8.2. ВЫБОР СОСУДОВ ДЛЯ
КУЛЬТИВИРОВАНИЯ КЛЕТОК
Некоторые типичные сосуды для культивирования
приведены в табл. 8.1. Предполагаемое максимальное
140
ГЛАВА 8. ПОСУДА И СУБСТРАТЫ ДЛЯ КУЛЬТИВИРОВАНИЯ КЛЕТОК
Таблица 8.1. Характеристики культуральных сосудов
Культуральный Количество Рабочий Площадь Приблизительное
сосуд лунок объем, мл поверхности, см2 количество клеток (HeLa)
Многолуночные планшеты
Микротитровальные
96
0,1
0,3
1х105
Микротитровальные
144
0,1
0,3
1х105
4-луночные
4
2
2
5x105
6-луночные
6
2
10
2x106
12-лу ночные
12
1
3
7,5x105
24-луночные
Чашки Петри
24
1
2
5x105
3,5 см в диаметре
1
2
8
2x106
5 см в диаметре
1
4
17,5
4x106
6 см в диаметре
1
5
21
5x106
9 см в диаметре
Флаконы
1
10
49
1х106
10 см2 (Т10)
1
2
10
2x106
25 см2 (Т25)
1
5
25
5x106
75 см2 (Т75)
1
25
75
2x107
175 см2 (Т175)
1
75
175
5x107
225 см2 (Т225)
1
100
225
6x107
Роллерные бутыли
Бутыли с перемешиванием
1
200
850
2,5х108
500 мл (без пробулькивания)
1
50
5x107
5000 мл (с пробулькиванием)
1
4000
4x109
количество клеток линии HeLa рассчитывается для
каждого типа сосуда; количество полученных клеток
ограниченной клеточной линии (например, диплоидных
фибробластов) будет составлять около одной пятой
от количества HeLa. Определяющими факторами при
выборе культуральной посуды являются: 1) требуемое
количество клеток; 2) характер роста культуры — суспен¬
зионный или прикрепленный; 3) необходимость в аэра¬
ции или герметизация культуры; 4) порядок забора об¬
разцов; 5) характер анализа культуры и 6) стоимость.
8.2.1. Клеточный выход
в культуральном сосуде
Для монослойных культур количество клеток пропорци¬
онально площади поверхности флакона (рис. 8.1). Для
малых объемов и выращивания культуры для парал¬
лельных исследований (опытов) наилучшим образом
подходят многолуночные планшеты (рис. 8.2), которые
могут иметь большое количество маленьких лунок (как,
например, многолуночные планшеты для микротитра-
ции на 96 и 144 лунки, 0,1-0,2 мл среды и площадью
ростовой поверхности — 0,25 см2 или 24-луночные
«кластерные чашки» с рабочим объемом среды 1-2 мл
на каждую лунку и поверхностью для роста — 1,75 см2)
вплоть до 4-луночных плашек с диаметром в 5 см2 и объ¬
емом культуральной среды 5 мл (см. табл. 8.1). Обычно
используют как чашки Петри, так и флаконы в диапа¬
зоне размеров от 10 см2 до 225 см2 (рис. 8.4). Флаконы,
как правило, маркируют согласно площади рабочей
поверхности (например, 25 см2 или 175 см2, а иногда Т25
или Т175 соответственно), в то время как чашки Петри
обозначают по их диаметру (т.е. 3,5 см или 9 см).
Размеры стеклянных бутылей более разнообразны,
так как их производят и для фармацевтических целей.
Стеклянные бутыли должны иметь: 1) одну достаточно
плоскую поверхность; 2) высокую плотно завинчиваю¬
щуюся крышку с нетоксичной прокладкой; 3) сглажен¬
ные плечи ребра для удобства сбора клеток монослоя
после трипсинизации и улучшения качества отмывки.
Если вам требуется получить большое количество
клеток (например, =1x109 клеток HeLa карциномы
цервикального канала или 2xl08MCR-5 диплоидных
фибробластов человека), то увеличение размера и
количества обычных бутылей становится неудобным
и требуются специальные сосуды. Флаконы с гофриро¬
ванной поверхностью (Corning, Becton Dickinson) или
многослойные флаконы (Coming, Nagle Nunc) позволя¬
ют увеличить площадь поверхности для наращивания
клеток (рис. 8.5). При необходимости еще большего на¬
ращивания культур используют мультиповерхностные
пропагаторы или роллерные бутыли на специальных
держателях (см. разд. 26.2.3). Повышение количества
клеток, растущих в суспензии, требует только увели¬
чения объема среды при условии, что культура будет
эффективно перемешиваться и аэрироваться во всей
толще объема (см. разд. 26.1).
8.2. Выбор сосудов для культивирования клеток
141
2
0)
х
*
I
т
0
ф
т
S
1
Площадь поверхности, см2
Рис. 8.1. Количество клеток и площадь поверхности. Зависимость объема среды и конечного количества клеток от площади
поверхности культурального сосуда. График построен на основе объема среды для каждого размера сосуда и не является ли¬
нейным, так как существует тенденция к уменьшению пропорции рабочего объема среды по мере увеличения объема сосуда.
Количество клеток рассчитано по объему среды и является приблизительным
Рис. 8.2. Многолуночные планшеты. 6-луночные, 24-луночные и 96-луночные (микротитровальные) планшеты. Произво¬
дятся планшеты с разным количеством лунок от 4 до 144 (размеры и объемы см. в табл. 8.1)
2
ГЛАВА 8. ПОСУДА И СУБСТРАТЫ ДЛЯ КУЛЬТИВИРОВАНИЯ КЛЕТОК
1C. 8.3. Чашки Петри. Демонстрируемые чашки имеют
аметры 3,5 см, 5 см и 9 см. Также выпускаются квадратные
шки Петри с размерами 9x9 см. Нанесенная на дно сетка
>жет облегчить сканирование чашки, например, при пол¬
ете колоний, однако стать помехой при автоматическом
дсчете
Рис. 8.5. Многопластинчатый флакон. Nunc Triple-Flask
с тремя рабочими поверхностями по 80 см2, которые засеи¬
ваются одновременно. Несмотря на то что общая площадь
ростовой поверхности 240 см2, пространство между плас¬
тинами эквивалентно обычному флакону в 80 см2. Так как
объем газовой фазы уменьшен, такой флакон лучше всего
использовать с вентилируемой крышкой в С02-инкубаторе.
(Фотография представлена с любезного разрешения Nagle
Nunc International.)
IC. 8.4. Пластиковые флаконы. Иллюстрируемые разме-
[ — 10 и 25 см2 (Falcon, B-D Biosciences), 75 см2 (Corning)
185 см2 (Nagle Nunc) (см. табл. 8.1. для представления
змеров и объемов)
2.2. Суспензионная культура
петки, растущие в суспензии, могут культивироваться
стерильных флаконах любого вида, в плашках или
шках Петри, которые не требуют дополнительной
1работки для обеспечения клеточной адгезии. Враща-
льные бутыли используются в тех случаях, когда для
задержания клеток в суспензии требуется постоян-
ш циркуляция среды. Такие сосуды имеют разные
1змеры и изготавливаются обычно из стекла (Bellco,
echne). Перемешивание, как правило, осуществляется
агнитным стержнем, приводимым в движение внеш-
гй магнитной мешалкой (рис. 8.6; см. также рис. 12.7
25.1). Скорость вращения должна поддерживаться
13кой, -60 об./мин, во избежание действия на клетки
шреждающих центробежных сил. Обычно для мини-
Рис. 8.6. Маленькие флаконы с перемешиванием. Четыре
маленьких флакона с перемешиванием (Techne), вместимость
250 мл при объеме ростовой среды 50-100 мл на 4-местной
подставке для мешалок (Techne). Также поставляются бу¬
тыли большего объема вплоть до 10 л (см. также рис. 13.5,
26.1,26.2)
мизации центробежной силы определяющее значение
имеет форма ротора. Для постановки параллельных
опытов суспензионные культуры могут культиви¬
роваться в отдельных сосудах, либо отбор образцов
производится повторно в разное время через боковой
рукав одного и того же сосуда. Подобные сосуды могут
также применяться для поддержания долго живущей
культуры путем последовательного обновления среды
(разд. 26.1.1).
8.2. Выбор сосудов для культивирования клеток
143
8.2.3. Вентилирование
Многолуночные планшеты и чашки Петри, приме¬
няемые для выращивания образцов при проведении
параллельных опытов или клонировании, имеют неза-
винчивающуюся свободную крышку для обеспечения
удобного доступа внутрь чашки. Следовательно, они
не герметичны и нуждаются в поступлении влажного
атмосферного воздуха с контролируемой концентра¬
цией С02 (см. разд. 9.2.2). Так как по окружности внут¬
ренней поверхности крышки может формироваться
тонкая пленка жидкости, герметизирующая некоторые
чашки, то необходимо использовать вентилируемые
крышки со специальными опорными элементами
из формованного пластика (рис. 8.7). В случае если
требуется полная герметизация, некоторые виды мно¬
голуночных чашек можно оборачивать тонкой поли¬
этиленовой пленкой (см. Плашечные герметизаторы,
Приложение II).
Флаконы могут вентилироваться за счет ослабления
закручивания крышки на один оборот для доступа С02
в С02-инкубаторе или, наоборот, для выхода избытка
С02 при выращивании клеточных линий, сильно за-
Рис. 8.7. Вентилируемые чашки Петри. Вентилируемая
чашка. Маленькие ребрышки, отдаленные друг от друга на
120°, приподнимают крышку над основанием и предотвра¬
щают образование тонкой пленки жидкости, например от
конденсата в результате плотного закрытия крышкой, что
затрудняет газообмен
Рис. 8.8. Вентилируемые флаконы. Газопроницаемая крыш¬
ка на флаконе 10 см2 (Falcon, B-D Biosciences)
кисляющих среду. Однако, для поддержания правиль¬
ного газообмена предпочтительнее всего использовать
крышки с проницаемыми фильтрами (рис. 8.8) (не¬
смотря на большую стоимость), так как они допускают
диффузию С02 без риска контаминации.
8.2.4. Отбор и анализ проб
Многолуночные планшеты являются идеальной посу¬
дой для рассева культуры для параллельных опытах,
когда предполагается одновременный и однотипный
анализ всех образцов. Если, с другой стороны, есть
необходимость забора образцов в разное время, то
предпочтительнее использовать различные сосуды
(см. также разд. 21.8 и рис. 8.9). Отдельные лунки в
микротитровальных планшетах можно отбирать на
анализ путем вырезания и удаления только интересу¬
ющего исследователя участка планшета вместе с ад¬
гезивной крышкой, покрывающей выбранные лунки.
Альтернативой в этом случае являются выпускаемые
планшеты с удаляемыми вставными лунками для ин¬
дивидуального анализа (Nagle Nunc), при этом необхо¬
димо убедиться, что рабочая поверхность этих лунок
обработана для обеспечения клеточной адгезии, если
вы работаете с субстрат-зависимыми культурами.
Микроскопическое исследование при малом уве¬
личении легкоосуществимо для культур, растущих во
флаконах, на чашках Петри и многолуночных планше¬
тах при использовании инвертированного микроскопа.
Однако с применением фазового контраста могут
возникнуть трудности при просмотре многолуночных
планшетов из-за малого размера мениска; даже при
просмотре 24-луночных планшетов удовлетворитель¬
ный обзор на фазовом контрасте достигается только
в центральной части лунки. Если микроскопический
анализ необходим при вашей работе с культурой, то
полезно использовать предметные стекла с ячейками
для культур (см. разд. 16.4.3 и рис. 16.3). Большие
роллерные бутыли проблематично просматривать на
некоторых микроскопах; как правило, необходимо
удалить конденсор, при этом настройка фазового кон¬
траста оказывается невыполнимой.
Если обработка образца предусматривает экстра¬
кцию в ацетоне, толуоле, этилацетате или некоторых
других органических растворителях, то возникает про¬
блема растворения полистирола. Так как применение
органических растворителей часто связано с гистоло¬
гическими процедурами, Lux (Bayer, МР Biomedicals)
поставляет устойчивые к действию растворителей
пластиковые покровные стекла Thermanox (ТРХ), ко¬
торые пригодны для гистологии и подходят по размеру
под ячейки стандартных многолуночных планшетов
(при этом совсем нет необходимости, чтобы сами
планшеты подходили для культивирования). Однако
данные стекла не обладают хорошими оптическими
свойствами и их следует монтировать на предметные
стекла клетками вверх, а затем покрывать стандартным
покровным стеклом.
144
ГЛАВА 8. ПОСУДА И СУБСТРАТЫ ДЛЯ КУЛЬТИВИРОВАНИЯ КЛЕТОК
Рис. 8.9. Пузырьки и флаконы с закручивающимися крышками, (а) Стеклянные флаконы хорошо подходят для рассева
культуры для параллельных опытов или хранения образцов, особенно если пластик может не выдержать последующего про¬
цесса обработки образца. Закручивающиеся крышки предпочтительнее пробок из-за более низкой вероятности протекания и
лучшей защиты горлышка флакона от контаминации. (6) Сцинтилляционные пузырьки особенно необходимы для исследо¬
вания включения радиоактивных изотопов, однако их нельзя снова использовать для культивирования после того, как в них
побывала сцинтилляционная жидкость
Для таких процедур, как горячая экстракция ДНК
хлорной кислотой, требуются стеклянные сосуды.
Прямосторонние пузырьки или колбы Эрленмейера
(без краевой кромки), используемые в сочетании с
герметизирующей лентой или крышкой Oxoid, удоб¬
ны в использовании и хорошо поддерживают влаж¬
ную атмосферу с С02. Традиционные стеклянные
сцинтилляционные флаконы, или «минифлаконы»,
также являются хорошими культуральными сосу¬
дами благодаря плоскому дну. Однако даже после
однократного использования сцинтилляционной
жидкости такие флаконы становятся непригодными
для культивирования.
8.2.5. Неравномерный рост
Иногда бывает, что клетки спонтанным образом
неравномерно распределяются по рабочей поверх¬
ности сосуда. Вибрация, создаваемая открыванием
и закрыванием инкубатора, несовершенной работой
мотора или вибрацией оборудования, может вызвать
пертурбации культуральной среды, в результате чего
возникает резонанс или стоячие волны, которые, в
свою очередь, приводят к волнообразному характеру
монослоя (рис. 8.10) из-за неравномерной плотности
роста клеток. Исключение вибрации и минимизация
вторжения в инкубатор помогут избежать неправиль¬
ного роста. Помещение тяжелого груза на поддон или
бокс с платами и установка планшетов на пенопласт
может также облегчить решение данной проблемы
[Nielsen, 1989], однако в большую проблему может пре¬
вратиться мытье и стерилизация таких пенопластовых
прокладок, так как они имеют склонность к постоянной
контаминации.
Рис. 8.10. Случайный рост. Примеры гребневидного ха¬
рактера роста, видимого в монослойных культурах в чашках
и флаконах, который, возможно, возникает в результате
развития резонанса от работы мотора инкубатора или из-за
открывания и закрывания дверцы инкубатора. (С любезного
разрешения Nagle Nunc.)
8.2.6. Стоимость
Стоимость всегда нужно соотносить с прочими пре¬
имуществами; например, чашки Петри дешевле, чем
флаконы с эквивалентной площадью ростовой по¬
верхности, однако требуют поддержания влажности
и контролируемого поддува С02 и более подвержены
заражению. При этом они очень удобны для исследова¬
ния и обработки. Дешевые бутыли из Na-силикатного
стекла, хотя и не всегда обладают хорошими оптичес¬
кими свойствами, часто оказываются лучше, чем стек¬
ло высокого класса типа Ругех или оптически чистое
стекло, которое обычно содержит свинец. Наибольший
недостаток использования стекла заключается в тру¬
доемкости процесса подготовки к культивированию,
так как стеклянная посуда должна тщательно мыться
и заново стерилизоваться перед очередным исполь¬
зованием. В большинстве лабораторий в настоящее
8.4. Обработка поверхности
145
время используется пластик благодаря его удобству,
оптической чистоте и качеству.
8.3. СПЕЦИАЛИЗИРОВАННЫЕ
СИСТЕМЫ
8.3.1. Проницаемые подложки
Полупроницаемые мембраны применяются как газо¬
проницаемые субстраты, а также дают возможность про¬
никать воде и малым молекулам (<500-1000 Да). Это
свойство используется в некоторых крупномасштабных
биореакторах (см. разд. 25.3.2 и 26.2.6). Рост клеток на
водопроницаемом субстрате способствует повышенной
диффузии кислорода, С02 и питательных веществ. При¬
крепление клеток к естественному субстрату, такому
как коллаген, активирует регуляцию фенотипической
экспрессии благодаря взаимодействию интегриновых
рецепторов на поверхности клеток со специфическими
сайтами внеклеточного матрикса (см. разд. 3.2.1,3.2.3 и
17.7.3). Широкое использование приобрели натураль¬
ные гели, такие как коллаген. Культивирование клеток
на коллагеновых плотиках [Michalopoulos и Pitot, 1975;
Lillie et al., 1980] использовалось для улучшения выжи¬
ваемости эпителиальных клеток и стимуляции терми¬
нальной дифференцировки (см. разд. 17.7.3 и 25.3.8).
Вставные фильтры. Проницаемость поверхности,
к которой прикрепляются клетки, может индуциро¬
вать полярность клеток за счет симуляции базальной
мембраны. Такая полярность может оказаться жиз¬
ненно необходимой для проявления функциональных
свойств секреторного эпителия и многих других типов
клеток [Gumbiner & Simons, 1986; Chambard et al.,
1987; Artursson & Magnusson, 1990; Mullin et al., 1997].
Некоторые производители в настоящее время пос¬
тавляют проницаемые подложки в форме отдельных
фильтров, свободно вставляемых в лунки планшеты,
разных размеров, из разных материалов и с различным
диаметром пор (Costar, B-D Bioscoences, Millipore,
Nunc). Также производятся подложки, покрытые
коллагеном, ламинином или другими матриксными
материалами (например, Матригель, B-D Biosciences).
Все перечисленные виды подложек широко использу¬
ются для изучения межклеточных взаимодействий и
взаимодействий с матриксом клеток, для исследования
дифференцировки и полярности, а также трансэпите¬
лиальной проницаемости (см. разд. 25.3.6).
Полые волокна. Knazek et al. [1972] разработал
технику выращивания клеток на внешней поверх¬
ности пучков пластиковых капилляров (рис. 8.11;
разд. 23.3.2). Пористый пластик дает возможность диф¬
фундировать питательным веществам и растворенным
газам из среды, перфузируемой через просвет капилля¬
ра. Находясь на внешней стороне капиллярных трубок,
клетки растут как бы на разных уровнях по отношению
друг к другу, имитируя тканевую целостность. Полые
Рис. 8.11. Культивирование на полых волокнах. Пучок по¬
лых волокон из проницаемого пластика заключен в прозрач¬
ную пластмассовую внешнюю камеру с обеспечением доступа
к клеткам через один из двух боковых рукавов. В ходе куль¬
тивирования камера перфузируется вплоть до центра полых
капилляров через соединения, прикрепленные к обоим концам
камеры (CellMax, Spectrum Labs; см. также рис. 26.6)
волокна также используются в крупномасштабных
биореакторах (см. разд. 26.2.6).
8.4. ОБРАБОТКА ПОВЕРХНОСТИ
8.4.1. Покрытие матриксом
Кондиционирование. Клеточную адгезию и рост можно
усилить путем предварительной обработки субстрата
[Barnes et al., 1984а]. Любому профессионалу в области
культуры ткани известен тот факт, что однажды исполь¬
зованная для культуры стеклянная посуда лучше обеспе¬
чивает рост клеток, чем новая. Если это так, то, возможно,
это происходит за счет травления поверхности или
присутствия следовых остатков веществ после куль¬
тивирования. Выращивание клеток во флаконах также
улучшает поверхность для повторного посева, и конди¬
ционирование такого типа, по-видимому, происходит за
счет коллагена, фибронектина или других компонентов
матрикса [Crouch et al., 1987], продуцируемых клетками.
Субстрат также может быть кондиционирован путем
его обработки уже использованной средой от другой
культуры [Stamper et al., 1980], сывороткой, очищенным
фибронектином или коллагеном (см. протокол 23.9).
Полилизин. McKeehan и Ham [1976а] предложили
покрывать поверхность пластиковых чашек поли-
D-лизином в концентрации 1 мг/мл перед клониро¬
ванием в отсутствие сыворотки (см. разд. 14.2.1), так
что некоторые эффекты кондиционирования ростовой
поверхности могут быть связаны с ее зарядом.
Коллаген и желатин. Обработка денатурирован¬
ным коллагеном улучшает прикрепление многих типов
146
ГЛАВА 8, ПОСУДА И СУБСТРАТЫ ДЛЯ КУЛЬТИВИРОВАНИЯ КЛЕТОК
клеток, таких как эпителиальные, а неденатурирован-
ный гель может быть необходим для поддержания
дифференцированного фенотипа (см. разд. 3.2.3,17.7.3,
23.2.1). Покрытие желатином оказалось полезным при
культивировании мышечных [Richler and Yaffe, 1970]
и эндотелиальных клеток [Folkman et al., 1979] (см.
разд. 23.3.6) и необходимым для некоторых мышиных
тератом. Для создания покрытия денатурированным
коллагеном используют коллаген из сухожилий крыси¬
ных хвостов или коммерчески поставляемых аналогов
(например, Vitrogen). Раствор коллагена наливают на
поверхность планшета, удаляют его избыток и высу¬
шивают. Так как описанная процедура иногда при¬
водит к откреплению коллагенового слоя в процессе
культивирования, то Macklis et al. [1985] разработали
протокол, гарантирующий крепкое связывание колла¬
гена с субстратом за счет поперечного сшивания его с
пластиком с помощью карбодиимида. Коллаген также
может использоваться в сочетании с фибронектином
(см. протокол 23.9).
Коллаген можно применять и в форме неденатури-
рованного геля (см. разд. 17.7.3). Такой тип субстрата,
как было показано, поддерживает рост аксонов спи¬
нальных ганглиев цыпленка [Ebendal, 1976], морфоло¬
гическую дифференцировку клеток молочной железы
[Nicosia and Ottinetti, 1990; Berdichevsky et al., 1992]
и гепатоцитов [Sattler et al., 1978; Fiorino et al., 1998],
а также стимулирует проявление тканеспецифичных
свойств ряда других клеток in vitro [Maas-Szabowski
et al., 2002] (см. разд. 23.1.1). Разведение концентри¬
рованного коллагена 1:10 культуральной средой и
нейтрализация до pH 7,4 превращают коллаген в гель,
так что разведение и распределение такого раствора
должны происходить быстро. Лучше всего добавить
ростовую среду к гелю еще на 4-24 ч, чтобы достичь
равновесного состояния геля со средой перед внесени¬
ем клеток. На этой стадии также могут быть добавлены
в среду фибронектин (25-50 мкг/мл) или ламинин
(1-5 мкг/мл) или их сочетание.
Матригелъ. Коммерчески производимые вне¬
клеточные матриксы, такие как Матригель (Becton
Dickinson) из саркомы Engelbreth-Holm-Swarm (EHS),
содержат ламинин, фибронектин и протеогликаны с
превалированием ламинина (см. также разд. 17.7.3).
Другие варианты коммерческих продуктов включают
Pronectin F (Protein Polymer Technologies), лами¬
нин, фибронектин, витронектин (B-D Biosciences,
Biosource International), энтактин (US Biological),
гепаран сульфат, EHS Natrix (B-D Biosciences), ECL
(US Biological), Cell-tak (B-D Biosciences). Некоторые
из этих продуктов очищены, если не полностью хи¬
мически определены; другие же представляют собой
смесь компонентов внеклеточного матрикса, слабо
охарактеризованных и, возможно, содержащих свя¬
занные факторы роста. Если адгезия клеток является
необходимым условием их выживания, а некоторые
из субстратов неполноценны, то использование таких
матриксов приемлемо, но если проводятся механис¬
тические исследования, то матригель может быть
использован только как промежуточный этап на пути
к полностью детерминированному субстрату.
Внеклеточный матрикс. Несмотря на то что
инертное покрытие поверхности может быть вполне
достаточным, все же иногда оказывается необходимым
использование монослоя подходящего типа клеток
для формирования приемлемого матрикса с целью
поддержания некоторых специализированных клеток.
Gospodarowicz с соавторами [1980] сумели вырастить
эндотелий на внеклеточном матриксе (ВКМ) из-под
конфлюентного монослоя клеток линии ЗТЗ, удален¬
ных тритоном Х-100. Этот остаточный ВКМ также
был использован для стимуляции дифференцировки
гранулезных клеток яичника [Gospodarowicz et al.,
1980] и для исследования поведения опухолевых кле¬
ток [Vlodavsky et al., 1980].
ПРОТОКОЛ 8.1. Приготовление ВКМ
Схема
Удалить детергентом конфлюентный монослой кле¬
ток, продуцирующих матрикс, отмыть флакон или
чашку и посадить требуемые клетки на остаточный
матрикс.
Материалы
• Мышиные фибробласты, например ЗТЗ, фибро-
бласты MCR-5 человека или эндотелиальные
клетки легочной артерии теленка (или другая
подходящая клеточная линия, известная своей
способностью к наращиванию внеклеточного
матрикса)
• Стерильная вода высокой степени очистки (ВСО)
(см. часть 11.4.1)
• Тритон Х-100,1% раствор в ВСО
Протокол
1. Посадите культуру, продуцирующую матрикс, и
выращивайте ее до конфлюентного монослоя.
2. Через 3-5 дней на стадии смыкания монослоя
удалите среду и внесите в культуру равный объем
стерильного раствора Тритона Х-100 в ВСО.
3. Инкубируйте 30 мин при 37 °С.
4. Удалите раствор Тритона Х-100 и трижды
промойте остатки матрикса таким же объемом
стерильной ВСО.
5. Флаконы или чашки можно использовать сразу пос¬
ле отмывки или же хранить при 4 °С до 3 недель.
8.4.2. Фидерные слои
Несмотря на то что покрытие матриксом может
способствовать прикреплению, росту и дифферен-
8.4. Обработка поверхности
147
цировке, некоторые культуры более прихотливых
клеток, особенно растущие в низкой плотности, тре¬
буют поддержки живых клеток (например, мышиных
эмбриональных фибробластов; см. протокол 13.3).
Необходимость такого воздействия обусловлена,
в частности, необходимостью обогащения среды и
удаления продуктов метаболизма, а также секрецией
факторов роста фибробластами, но также может быть
вызвана кондиционированием субстрата клеточными
продуктами. Фидерные слои, выросшие в виде кон-
флюентного монослоя, могут сделать поверхность
подходящей и даже селективной для прикрепления
других клеток (см. разд. 14.2.3,24.7.2 и протоколы 23.1,
23.4). Жизнеспособность и рост аксонов центральных
и периферических нейронов можно повысить за счет
культивирования нейронов на монослое глиальных
клеток, хотя в этом случае эффект скорее обусловлен
результатом действия растворимых факторов, чем
непосредственных клеточных контактов [Seifert &
Muller, 1984].
После достижения конфлюентного монослоя
дальнейшая пролиферация заставляет клетки от¬
крепляться от искусственного субстрата и мигриро¬
вать над поверхностью монослоя (рис. 8.12). Может
поменяться морфология клеток: клетки становятся
менее распластанными, более высокодифференци¬
рованными и интенсивнее окрашиваются. Очевидно,
и не слишком удивительно то, что взаимодействие
клеток с клеточной подложкой отличается от такового
с синтетическим субстратом. Первое может изменять
морфологию клеток и уменьшать их пролифератив-
ный потенциал.
8.4.3. Трехмерные матриксы
Очевидно, что многие функциональные и морфологи¬
ческие характеристики исчезают в ходе субкультиви¬
рования в результате утраты тканевой архитектоники
и межклеточных контактов (см. разд. 3.4.2). Эти недо¬
статки явились стимулом к апробированию трехмер¬
ных матриксов, таких как коллагеновый гель [Douglas
et al., 1980], целлюлозный спонж (как таковой или с
коллагеновым покрытием) [Leighton et al., 1968] или
пенный гель Gelfoam (см. разд. 25.3.1). Фибриновый
сгусток служил одной из первых сред, использованных
для первичной культуры, и до сих пор используется
либо в виде необработанных сгустков плазмы, либо в
виде очищенного фибриногена в смеси с тромбином.
Обе системы формируют трехмерный гель, в котором
Рис. 8.12. Морфология фидерных слоев. Морфологические изменения клеток, растущих на фидерных слоях. Фибробласты
рака молочной железы человека (а), растущие на пластике и (б) растущие на конфлюентном фидерном слое человеческих
эмбриональных интерстициальных клеток. Эпителиальные клетки рака молочной железы человека, растущие (в) на пластике
и (г) на том же самом конфлюентном фидерном слое, как в (6)
148
ГЛАВА 8. ПОСУДА И СУБСТРАТЫ ДЛЯ КУЛЬТИВИРОВАНИЯ КЛЕТОК
клетки могут мигрировать и расти на границе твердой
поверхности и геля или непосредственно внутри геля
[Leighton, 1991].
Трехмерные матриксы широко используются в
тканевой инженерии и могут быть неорганическими,
как фосфат кальция или же органическими, как Gel-
foam (см. разд. 25.3.8). Помимо индукции клеточной
адгезии, пролиферации и дифференцировки, такие
матриксы, или каркасы, также должны быть дегради-
руемыми in vivo с последующим замещением эндоген¬
ным матриксом.
Микроносители, произведенные из полистирола
(Nugle Nunc), сефадекса, полиакриламида и коллагена
или желатина, поставляются в форме бусин и предна¬
значены для размножения субстрат зависимых клеток в
суспензии (см. разд. 26.2.4 и Приложение II). Несмотря
на трехмерность самих микроносителей, функцио¬
нально культура клеток на них представляет собой
двухмерный монослой, модифицированный радиусом
кривизны бусины. Однако некоторые микроносители
являются пористыми (см. разд. 26.2.4), в биореакто¬
рах также используются пористые микробусины (см.
разд. 26.2.5). Клетки растут в промежутках толщи мат¬
рикса в обоих типах бусин. Трехмерный рост в бусинах
из альгината происходит скорее путем инкапсуляции,
чем пенетрации матрикса (см. разд. 25.3.5), давая воз¬
можность тесному развитию межклеточного взаимо¬
действия и облегчая дифференцировку у хондроцитов
(см. протокол 23.16) и нейронов (см. протокол 25.3),
и используется для увеличения выработки антител
гибридомами [Zimmerman et al., 2003].
8.4.4. Металлические субстраты
Клетки способны расти на дисках из нержавеющей ста¬
ли [Birnie & Simons, 1967] или других металлических
поверхностях [Litwin, 1973]. Визуальный контроль
клеток, растущих на светонепроницаемом субстрате,
требует поверхностной интерференционной микро¬
скопии, за исключением случаев использования очень
тонких металлических пленок. Westermark [1978]
разработал метод выращивания фибробластов и глии
на палладии. Применив электронномикроскопическое
оборудование, используемое для напыления металлов
на образцы для создания реплик, он сформировал ос¬
тровки палладия на агарозе, которая не дает клеткам
прикрепляться в жидкой среде. Размер и форма остров¬
ков определялись рельефными участками, созданными
фототравлением, а палладий напылялся в вакуумной
камере, как это осуществляется в электронной микро¬
скопии. Так как слой палладия под клетками был очень
тонким, он оставался прозрачным и не препятствовал
визуализации.
8.4.5. Неадгезивные субстраты
Иногда прикрепление клеток является нежелательным.
Отбор колоний клеток, трансформированных вирусом,
независимых от прикрепления, может быть достигнут
путем культивирования клеток в агаре [Macpherson
& Montagnier, 1964], так как нетрансформированные
клетки спонтанно не формируют колоний в таком
матриксе. Существуют два принципа, соблюдаемых
в такой системе: 1) предотвращение прикрепления
к поверхности флакона, где должно происходить
распластывание и зависимый от прикрепления рост,
и 2) иммобилизация клеток таким образом, чтобы
дочерние клетки оставались связанными с колонией,
даже если они являются субстрат независимыми. Наи¬
более часто используются агар, агароза или Methocel
(метилцеллюлоза с вязкостью 4000 сР) (см. разд. 14.3).
Первые два представляют собой гели, а третий — золь
с высокой вязкостью. Так как Methocel является зо¬
лем, то клетки постепенно будут осаждаться в нем. По
этой причине он обычно используется с подложкой из
агара (см. протокол 14.5). Посуда, не предназначенная
для клеточных культур, может быть использована
без агара, однако и в этом случае может происходить
прикрепление и распластывание незначительного
количества клеток.
Глава 9
СРЕДЫ ОПРЕДЕЛЕННОГО ХИМИЧЕСКОГО
СОСТАВА И ДОБАВКИ К СРЕДАМ
9.1. СОСТАВЛЕНИЕ СРЕД
В ранних исследованиях для культивирования клеток
использовали природные среды на основе тканевых
экстрактов и естественных жидкостей организма,
таких как экстракт куриного эмбриона, сыворотка,
лимфа и т. д. С распространением клеточных линий
потребность в большем количестве сред стандартного
однородного качества привела к внедрению сред с
химически точным определенным составом, основан¬
ным на анализе естественных жидкостей организма и
биохимии питательных веществ. Базовая среда Игла
[Eagle, 1955] и впоследствии минимальная среда Игла
(MEM) [Eagle, 1959] получили широкое распростране¬
ние, с различными вариантами добавления сывороток
теленка, лошади или человека, белковых гидролизатов
и эмбриональных экстрактов. Так как к тому времени
уже было получено большее количество постоянных
клеточных линий (L929, HeLa и др.), стало очевидно,
что эти среды хорошо подходят для большинства кле¬
точных линий, и целью многих успешных исследова¬
ний были попытки замены сыворотки (см. разд. 10.2),
оптимизация сред для различных типов клеток (на¬
пример, для лимфобластоидных линий клеток была
разработана среда RPMI 1640), или модификация
сред для специфических условий культивирования
(например, среда Лейбовица L15 была разработана для
культивирования клеток в отсутствие С02 и NaHC03)
[Leibovitz, 1963].
Для выделения и наращивания клеток специфичес¬
кого происхождения может потребоваться селективная
бессывороточная среда (см. разд. 10.2.1), в то время как
для наращивания клеток для заражения их впослед¬
ствии вирусами или для молекулярных исследований,
не зависящих от специфичности клеток, подходят среда
Игла MEM [Eagle, 1959], среда Игла в модификации
Дюльбекко DMEM [Dulbecco & Freeman, 1959] или,
все чаще, RPMI 1640 [Moore et al., 1967], в которые
добавляется сыворотка. Однако сейчас многие про¬
мышленные технологии используют бессывороточные
среды для того, чтобы упростить процесс и снизить
риск контаминации случайными инфекционными
агентами (см. разд. 10.1). Популярным компромиссом
для многих лабораторий стала смесь сложных сред,
таких как среда Хэма Ham’s F12 [Ham, 1965] с одной
из сред, содержащей повышенные концентрации ами¬
нокислот и витаминов, таких как DMEM. Этот вариант
может шокировать сторонников использования сред с
постоянным составом, но он позволил создать полезные
многоцелевые среды, пригодные как для первичных
культур, так и для поддержания клеточных линий.
Эта глава посвящена основным принципам состав¬
ления сред. В качестве примера используются широко
применяемые среды, требующие добавления сыворот¬
ки. Глава 10 посвящена разработке и использованию
бессывороточных сред.
9.2. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
9.2.1. pH
Большинство клеток хорошо растут при pH 7,4. Хотя
оптимальные значения pH варьируют в относительно
небольшом диапазоне для разных клеточных линий,
некоторые нормальные фибробласты лучше растут при
pH 7,4-7,7, в то время как для трансформированных
клеток оптимальное значение pH находится в диапа¬
зоне 7,0-7,4 [Eagle, 1973]. По некоторым данным, эпи¬
дермальные клетки могут расти при значении pH 5,5
[Eisinger et al., 1979], однако эта точка зрения не являет¬
ся общепринятой. В особых случаях для доказательства
полезно проведение кратковременных экспериментов
по исследованию эффективности роста (см. протоко¬
лы 21.7 и 21.8), исследования эффективности посева
(см. разд. 21.10) или проведение специализированного
функционального анализа (например, см. разд. 17.7)
для определения оптимальных значений pH.
В качестве индикатора pH обычно используется фе¬
ноловый красный. При pH 7,4 он красного цвета, при pH
7,0 становится оранжевым, при pH 6,5 — желтым и ли¬
монно-желтым при более кислых значениях pH. При pH
7,6 цвет индикатора сдвигается в розовую область, при
pH 7,8 становиться фиолетовым (см. цв. вкладку 226).
Так как оценка цветов во многом субъективна, полезно
приготовить набор стандартных растворов стерильного
сбалансированного солевого раствора, содержащего
феноловый красный в необходимой концентрации, в
такой же бутыли и с таким же объемом для воздуха,
которые обычно используются для приготовления сред.
В нижеследующем примере в качестве окончательного
резервуара будет использоваться культуральный фла¬
кон площадью 25 см2, и этот пример можно использо¬
вать в сочетании с упражнением 8.
150
ГЛАВА 9. СРЕДЫ ОПРЕДЕЛЕННОГО ХИМИЧЕСКОГО СОСТАВА И ДОБАВКИ К СРЕДАМ
ПРОТОКОЛ 9.1. Приготовление
стандартов PH
Материалы
Стерильные:
• Сбалансированный солевой раствор Хэнкса
(HBSS) или среда Игла МЕМ, 10х концентрат
без бикарбоната, без глюкозы, содержащий 20 мМ
HEPES 10 мл
• Вода высшей степени очистки 90 мл
• 1нЫаОН 10 мл
• Универсальные контейнеры, 30 мл 7
• Культуральные флаконы, 25 см2 7
• Наконечники для пипеток, 100 мкл
Нестерильные:
• рН-метр
• Автоматическая пипетка, 10-100 мкл.
Протокол
1. Приготовьте раствор Хэнкса или среду с pH
6,5 или ниже и в каждый из семи подписанных
универсальных контейнеров поместите по 10 мл
раствора.
2. В течение 30 мин дайте уравновеситься воздухом.
3. С помощью 1 н NaOH доведите pH растворов в
контейнерах до 6,5, 6,8, 7,0, 7.2, 7,4, 7,6 и 7,8, из¬
меряя значения pH на рН-метре.
4. По 5 мл каждого раствора Хэнкса стерилизуйте
фильтрованием в отдельные подписанные флако¬
ны (см. протокол 11.11). Первые 5 мл используй¬
те для промывания фильтра перед сбором второй
порции в 5 мл во флакон.
5. Прочно закройте флаконы крышками.
9.2.2. С02 и бикарбонат натрия
Углекислый газ из газовой фазы растворяется в среде,
достигает равновесия с ионами НСОэ и понижает pH.
Так как растворенные С02, НСОэ и pH взаимосвяза¬
ны, трудно определить главный прямой эффект С02.
Атмосферное парциальное давление С02 будет прямо
регулировать концентрацию растворенного С02 как
функцию температуры. Эта регуляция, в свою очередь,
приводит к образованию Н2СОэ, который диссоциирует
в соответствии с реакцией
Н20 + СО, О Н2СОэ ОН+ + НСОэ- (1)
НС03“ имеет довольно низкую константу диссо¬
циации с большинством доступных катионов и, таким
образом, имеет тенденцию реассоциировать, закисляя
при этом среду. Конечный результат повышения кон¬
центрации атмосферного С02 — снижение pH, и, таким
образом, эффект повышения парциального давления
С02 нейтрализуется повышением концентрации би¬
карбоната:
NaHC03 ONa+ + HC03“ (2)
Повышенная концентрация НС03" сдвигает урав¬
нение 1) влево, пока не будет достигнуто равновесие
при pH 7.4. Если вместо NaHC03 использовать другую
щелочь (например, NaOH), окончательный результат
будет таким же:
NaOH + H2C03 ONaHCO3 + Н20
ONa+HC03" + Н20 (3)
Эквивалентные концентрации NaHC03, обычно ис¬
пользуемые при различном парциальном давлении С02,
приведены в табл. 9.1, 9.2 и 9.3. Могут использоваться
промежуточные значения С02и НС03~, при условии,
что концентрации обоих компонентов изменяются
пропорционально. Так как многие среды готовят в кис¬
лых растворах и, кроме того, в их состав может входить
буфер, трудно предсказать, сколько бикарбоната нужно
использовать, когда другие щелочи могут дать такой же
эффект, как и бикарбонат (см. уравнение (3)). Когда
новую среду готовят в первый раз, добавляют указанное
количество бикарбоната и затем достаточное количество
1н NaOH дл того, чтобы среда уравновесилась до желае¬
мого pH после инкубации в чашке Петри при 37 °С при
соответствующей концентрации С02. Если работают со
средой уже в рабочей концентрации, варьируют коли¬
чество НС03“ для того, чтобы подобрать газовую фазу
(см. табл. 9.1), и оставляют среду на ночь уравновеши¬
ваться при 37 °С. Для каждой среды есть рекомендуемые
концентрации бикарбоната и парциального давления
С02 для достижения правильных значений pH и осмо¬
тического давления, но могут быть небольшие вариации
в зависимости от метода приготовления среды.
С использованием буферов Гуда (например, HEPES,
Tricine) [Good et al., 1966] в культуре клеток, были
высказано предположение, что, так как С02 больше
не является необходимым для стабилизации pH, его
можно удалить из состава среды.
Таблица 9.1. Взаимосвязь между бикарбонатом, углекислым газом и HEPES
Вещество
Срела МЕМ Игла с солями Хэнкса
Низкая концентрация
НСОэ~ + буфер
Среда МЕМ
Игла с солями Эрла
DMEM
NaHC03
4 мМ
10 мМ
26 мМ
44 мМ
со2
Атмосферное и выделенное культурой
2%
5%
10%
HEPES* (если требуется)
10 мМ
20 мМ
50 мМ
-
9.2. Физико-химические свойства
151
Это утверждение неверно [Itagaki & Kimura, 1974],
по крайней мере для большинства типов клеток, особен¬
но при низкой их концентрации. Хотя 20 мМ HEPES
может контролировать pH в физиологическом диапазо¬
не, в отсутствие атмосферного С02 равновесие реакции
1) сдвигается влево, что в конечном итоге приводит к
элиминации растворенного С02и, далее, НС03~ из сре¬
ды. Эта цепь событий, по-видимому, ограничивает рост
клеток, хотя что именно требуется клеткам — растворен¬
ный С02, или НС03“ (или и то, и другое одновремен¬
но) — остается неясным. Рекомендуемые концентрации
С02, НС03" и HEPES указаны в табл. 9.1.
Включение в состав среды пирувата позволяет
клеткам увеличить продукцию эндогенного С02, де¬
лая их независимыми как от экзогенного С02, так и
от NaHC03. Среда Лейбовица L15 [Leibovitz, 1963]
содержит пируват натрия в высокой концентрации
(550 мг/мл) и не содержит NaHC03n не требует при¬
сутствия С02в газовой фазе. Буферизация достига¬
ется благодаря относительно высокой концентрации
аминокислот. Так как данная среда не требует С02,
L15 иногда рекомендуют использовать для транспор¬
тировки образцов тканей. (3-глицерофосфат натрия
может также использоваться в качестве буфера для
автоклавируемых сред, в которых отсутствуют С02 и
НС03" [Waymouth, 1979], и фирма Invitrogen продает
среды для культивирования в отсутствие С02. Если
отсутствие С02 важно для снижения себестоимости,
из соображений удобства или по другим причинам,
возможно, одна из этих сред заслуживает внимания, но
только после соответствующего тестирования.
Обобщая вышесказанное, клетки, культивируемые
во флаконах с доступом воздуха, необходимо инкубиро¬
вать в атмосфере С02, концентрация которого находит¬
ся в равновесии с бикарбонатом натрия, содержащимся
в среде (см. табл. 9.1, 9.2 и 9.3). Клетки в умеренно
высокой концентрации (>1x105 клеток/мл) и растущие
в закрытых флаконах не нуждаются в добавлении С02
в газовую фазу при условии, что концентрация бикар¬
боната поддерживается низкой (=4 мМ), особенно если
клетки сильно закисляют среду. Однако при низкой
концентрации клеток (например, при клонировании) и
для некоторых первичных культур необходимо добав¬
лять С02 в газовую фазу и при культивировании клеток
в закрытых флаконах. Когда требуется вентиляция
для достижения равновесия (либо для допуска С02,
либо для удаления его избытка для сильно закисля-
ющих среду клеток), необходимо оставлять крышки
флаконов приоткрытыми или использовать крышки,
пропускающие С02 (см. рис. 8.8).
9.2.3. Буферизация
Культуральная среда должна быть забуферена в двух
случаях: 1) при культивировании клеток в чашках Пет¬
ри, где образование С02 является причиной повыше¬
ния pH (см. разд. 9.2.2), и 2) при гиперпродукции С02
и молочной кислоты трансформированными клеточ¬
ными линиями, растущими в высокой концентрации,
когда pH падает. Буфер может входить в состав среды
для стабилизации pH, но 1) экзогенный С02 может,
однако, быть необходим для некоторых клеточных
линий, особенно для клеток, растущих в низкой кон¬
центрации, для предотвращения полной потери рас¬
творенного С02 и бикарбоната из среды. Несмотря на
низкую буферную емкость при физиологических pH,
бикарбонатный буфер все еще используется намного
чаще, чем другие буферы, благодаря его низкой токсич¬
ности, дешевизне и питательной пользе для культуры.
Таблица 9.2. Сбалансированные солевые растворы (BSS)
Компоненты
BSS Эрла
PBS Дюльбекко
энкса
Соли Спиннера
(как в S-MEM)
Без Са2+ и Мд2+
(D-PBSA)
о
Я
и Мд2+
BSSX
Мол. вес.
г/л
мМ
г/л
мМ
г/л
мМ
г/л
мМ
г/л
мМ
Неорганические соли
СаС12 (безводный)
111
0,02
0,18
0,2
1,80
0,14
1,3
КС1
74,55
0,4
5,37
0,2
2,68
0,2
2,68
0,4
5,4
0,40
5,37
КН2Р04
136,1
0,2
1,47
0,2
1,47
0,06
0,4
MgCl2 • 6Н20
203,3
0,1
0,5
MgS04 • 7Н20
246,5
0,2
0,81
0,98
3,98
0,1
0,4
0,20
0,81
NaCl
58,44
6,68
114,3
8
136,9
8
136,9
8
136,9
6,80
116,4
NaHC03
84,01
2,2
26,19
0,35
4,2
2,20
26,19
Na2HP04 • 7Н20
268,1
2,2
8,06
2,16
8,06
0,09
0,3
NaH2P04 • Н20
138
0,14
1,01
1,40
10,14
Общее содержание солей
147,9
149,1
154,00
149,4
158,9
Другие компоненты
D-глюкоза
180,2
1
5,55
1
5,5
1,00
5,55
Феноловый красный
354,4
0,01
0,03
0,01
0,0
0,01
0,03
Газовая фаза
5% СО,
Воздух
Воздух
5% СО,
152
ГЛАВА 9. СРЕДЫ ОПРЕДЕЛЕННОГО ХИМИЧЕСКОГО СОСТАВА И ДОБАВКИ К СРЕДАМ
HEPES является гораздо более сильным буфером в
диапазоне pH 7,2-7,6 и используется в концентрациях
10-20 мМ. Было обнаружено, что при использовании
HEPES вместе с экзогенным С02, концентрация
HEPES должна быть выше концентрации бикарбо¬
ната более чем вдвое для адекватной буферизации
(см. табл. 9.1). Вариант среды Хэма F12 (Ham's F12),
содержащей 20 мМ HEPES, 10 мМ бикарбонат и 2%
С02 успешно используется в лаборатории автора для
культивирования ряда различных клеточных линий.
Это делает возможным проведение микротитрования
и других манипуляций с многолуночными планшета¬
ми вне инкубатора без значительного повышения pH
среды и минимизирует потребность в HEPES, который
не только токсичен, но и дорогой по цене.
9.2.4. Кислород
Другим важным компонентом газовой фазы является
кислород. Несмотря на то что in vivo большинству
клеток требуется кислород для дыхания, жизнеде¬
ятельность клеток в культуре часто в большей степени
зависит от гликолиза, поэтому многие клетки, например
трансформированные, являются анаэробами. Хотя были
попытки включать в состав среды носители 02, по ана¬
логии с гемоглобином крови, эта практика не является
общепринятой. Клетки используют, главным образом,
растворенный 02, который, однако, может быть токси¬
чен, так как он повышает уровень свободных радикалов.
Обеспечение правильного парциального давления 02,
таким образом, всегда является компромиссом между
потребностями дыхания и попыткой избежать токсич¬
ности. Стратегии, включающие как повышенный, так
и пониженный уровень 02, могут использоваться при
добавлении в культуральную среду соединений-лову¬
шек для свободных радикалов, таких как глутатион,
2-меркаптоэтанол (Р-меркаптоэтанол) или дитиотре-
итол. В большинстве случаев условия подбираются
эмпирически.
Потребности культур в кислороде различаются,
главное различие лежит между органными и клеточ¬
ными культурами. Хотя атмосферное или пониженное
давление кислорода [Cooper et al., 1958; Balin et al.,
1976] для большинства клеточных культур является
предпочтительным, некоторые органные культуры,
особенно полученные из эмбрионов на поздних стади¬
ях развития, новорожденных или взрослых, требуют
содержания 02в газовой фазе до 95% [Trowell, 1959; De
Ridder & Mareel, 1978]. Требование клеток к высокому
уровню содержания 02 может являться проблемой при
его затрудненной диффузии, что, в свою очередь, свя¬
занно с геометрической формой и проницаемостью для
газов органной культуры (см. разд. 25.2.1), но может
также отражать различие между дифференцированны¬
ми и быстро пролиферирующими клетками. Наруше¬
ние диффузии кислорода может также ограничивать
применение пористых микроносителей [Preissmann et
al., 1997] (см. также разд. 26.2.4).
Большинство дисперсных клеточных культур
предпочитают низкое парциальное давление кислоро¬
да и для некоторых систем (например, для опухолевых
клеток человека в исследованиях по определению
клоногенной активности [Courtenay et al., 1978] и эм¬
бриональных фибробластов легкого человека [Balin et
al., 1976]), предпочтительней, чтобы парциальное дав¬
ление кислорода было ниже атмосферного. McKeehan
et al. [1976] предположил, что необходимость в
добавлении селена в культуральную среду связана
с токсичностью кислорода, так как селен является
кофактором синтеза глутатиона. Кислородную толе¬
рантность — так же как и селен — может обеспечить
сыворотка, таким образом контроль за парциальным
давлением 02, по-видимому, более важен для культи¬
вирования в бессывороточной среде.
Так как толщина слоя культуральной среды
может влиять на скорость диффузии кислорода в
клетки, в статической культуре рекомендуется под¬
держивать толщину слоя среды в диапазоне 2-5 мм
(0,2-0,5 мл/см2). Некоторые типы клеток, например
бронхиальный эпителий и кератиноциты, растущие
на фильтрах-вставках в лунках, по-видимому, лучше
дифференцируются на границе фаз жидкость-воздух
(см. разд. 25.3.6).
9.2.5. Осмотическое
давление
Большинство клеток в культуре проявляют довольно
широкую толерантность в отношении осмотического
давления [Waymouth, 1970]. Так как осмотичес¬
кое давление в плазме крови человека составляет
290 мосмоль/кг, резонно допустить, что этот уровень
является оптимальным для клеток человека in vitro,
хотя он может отличаться для других видов (например,
для мышей это значение составляет 310 мосмоль/кг
[Waymouth, 1970]). На практике, для большинства
клеток допустимо осмотическое давление в диапазоне
от 260 мосмоль/кг до 320 мосмоль/кг; выбранного
значения следует придерживаться, допуская разброс
±10 мосмоль/кг. Слегка гипотоническая среда может
использоваться при культивировании клеток в чашках
Петри или в открытых планшетах для компенсации
испарения во время инкубации.
Осмотическое давление обычно измеряют по
снижению температуры замерзания среды (рис. 9.1)
или по повышению давления насыщенного пара. Из¬
мерение осмотического давления является полезным
этапом по контролю качества в том случае, если вы
готовите среду самостоятельно, так как это помогает
избежать ошибок при взвешивании, разбавлении
и т.д., особенно важно контролировать осмотическое
давление, если внесены изменения в состав среды.
Добавление HEPES и веществ, которые растворены
в сильных кислотах и основаниях, и их последующая
нейтрализация могут также значительно влиять на
осмотическое давление.
9.2. Физико-химические свойства
153
эмдпк
Рис. 9.1. Осмометр. Показана передняя панель, пробирка для
образца и порт для образца осмометра Роублинга (Camlab).
Эта модель может измерять образцы объемом 50 мкл
9.2.6. Температура
Оптимальная температура для культивирования
клеток зависит от 1) температуры тела животного, из
которого клетки были получены, 2) некоторых ана¬
томических вариаций (например, температура кожи
и тестикул может быть ниже температуры покояще¬
гося тела) и 3) учета фактора безопасности, так как
могут иметь место небольшие ошибки в регулировке
инкубатора. Таким образом, в целях безопасности,
для большинства клеточных линий человека и тепло¬
кровных животных рекомендуется температура 37 °С,
которая немного ниже температуры тела, так как пе¬
регревание является более серьезной проблемой, чем
переохлаждение.
Из-за высокой температуры тела птиц, клетки птиц
необходимо культивировать при 38,5 °С для макси¬
мального роста, но они могут вполне удовлетворитель¬
но расти, хоть и несколько медленнее, при 37 °С.
Культивируемые клетки млекопитающих устой¬
чивы к значительному понижению температуры, они
выживают в течение нескольких дней при 4 °С и могут
быть заморожены и охлаждены до -196 °С (см. про¬
токол 20.1), но не выдерживают нагрева выше нормы
более чем на 2 °С (39,5 °С) более чем на несколько
часов и очень быстро погибают при температуре 40 ° С
и выше.
Необходимо обратить внимание на соответствие
температуры (в пределах ±0,5 °С), чтобы гарантиро¬
вать воспроизводимые результаты. Двери инкубатора
или термальной комнаты не должны оставаться откры¬
тыми дольше, чем необходимо, и крупные предметы
или объемы жидкости, помещенные в термальную
комнату для согревания, не должны находиться рядом
с какими-либо культурами. Распределение темпера¬
туры внутри инкубатора или термальной комнаты
должно быть также однородным (см. разд. 4.4 и 5.3);
не должно быть «холодных мест», воздух должен
свободно циркулировать. Это означает, что большое
количество флаконов не должно быть сложено стоп¬
кой, когда они в первый раз помещены в инкубатор или
термальную комнату; между флаконами должно быть
пространство, достаточное для циркуляции воздуха.
Так как газовая фаза внутри флаконов расширяется
при 37 °С, флаконы, особенно сложенные стопкой,
должны вентилироваться путем кратковременного
приоткрывания крышки (от 30 мин до 1 ч) после по¬
мещения флаконов в инкубатор.
Пойкилотермные (холоднокровные животные, ко¬
торые не способны регулировать температуру крови в
узком диапазоне) переносят изменение температуры в
широком диапазоне, между 15 °С и 26 °С. Для имита¬
ции условий in vivo (например, для клеток рыб, живу¬
щих в холодной воде) может потребоваться инкубатор,
который не только нагревает, но и охлаждает, для того
чтобы поддерживать температуру ниже температу¬
ры окружающей среды. При необходимости клетки
пойкилотермных животных можно культивировать
при комнатной температуре, но колебания темпера¬
туры окружающей среды в лабораториях делают это
нежелательным, и охлаждаемый инкубатор является
предпочтительным.
Были получены температурно-чувствительные
(ts) мутантные клеточные линии, в которых специ¬
фические гены экспрессируются при температуре
ниже стандартной [Su et al., 1991; Foster & Martin,
1992; Wyllie et al., 1992]. Эти мутанты необходимы
для проведения исследований клеточной регуляции.
Кроме того, особое значение придается узкому темпе¬
ратурному диапазону в котором можно работать, так
как две ограничивающие температуры различаются
обычно на 2-3 °С. Использование ts мутантов обычно
требует наличия инкубатора как с нагреванием, так и с
охлаждением для того, чтобы компенсировать нагрев
за счет окружающей среды.
Не считая прямого эффекта на рост клеток, темпера¬
тура может также влиять на pH из-за повышения рас¬
творимости С02 при низких температурах и, возможно,
из-за изменений степени ионизации и рКа буфера. pH
должен быть установлен для комнатной температуре на
0,2 единицы ниже, чем для 37 °С. При приготовлении
среды впервые лучшим вариантом было бы приготов¬
ление полной среды и последующее инкубирование
образца среды в течение ночи при 37 °С при правиль¬
ном давлении газа для того, чтобы проконтролировать
pH (см. разд. 9.2.2 и вкладку 226).
154
ГЛАВА 9. СРЕДЫ ОПРЕДЕЛЕННОГО ХИМИЧЕСКОГО СОСТАВА И ДОБАВКИ К СРЕДАМ
9.2.7. Вязкость
На вязкость культуральной среды влияет главным об¬
разом содержание сыворотки и, в большинстве случаев,
влияние вязкости на клеточный рост невелико. Вяз¬
кость становится важна, однако, при перемешивании
клеточной суспензии или когда клетки диссоциируют
после трипсинизации. Любые повреждения клеток,
которые происходят в этих условиях, могут быть
снижены при повышении вязкости среды с помощью
карбоксиметилцеллюлозы (КМЦ) или поливинилпир-
ролидона (ПВП) [Cherry & Papoutsakis, 1990; смотри
также Приложение I]. Это становится особенно важ¬
ным при низкой концентрации сыворотки, в отсутствие
сыворотки и для клеток, культивируемых в биореак¬
торах с перемешиванием (см. разд. 26.1), в которых
часто используется плюроник F68, хотя его эффект
по-видимому, является многофакторным.
9.2.8. Поверхностное натяжение
и пенообразование
Влияние пенообразования до сих пор не ясно, но при
пенообразовании может повышаться скорость денату¬
рации белка, может возникнуть риск контаминации,
если пена достигнет горла культурального сосуда. Пе¬
нообразование также ограничивает диффузию газов в
том случае, если пленка пены или пролившаяся часть
попадет в капиллярное пространство между крышкой
и флаконом или между крышкой и нижней частью
чашки Петри.
Пена может возникать в суспензионных культурах в
сосудах с мешалкой или в биореакторах, когда воздух с
5% С02 пропускают через среду, содержащую сыворот¬
ку. Добавление гасителя пены на основе силикона (Dow
Chemical) или плюроника F68 (Sigma) в концентрации
0,01-0,1% помогает предотвратить пенообразование в
данной ситуации, снижая поверхностное натяжение,
и может также защищать клетки от повреждающего
воздействия пузырьков. С02 не должен пропускаться
через среду при газации флакона, так как при этом
может образовываться пена и может также произойти
выброс аэрозоля из флакона.
9.3. СБАЛАНСИРОВАННЫЕ СОЛЕВЫЕ
РАСТВОРЫ
В состав сбалансированного солевого раствора (BSS)
входят неорганические соли. Кроме того, в его состав
может входить бикарбонат натрия и в некоторых слу¬
чаях глюкоза. Состав некоторых наиболее используе¬
мых сбалансированных солевых растворов представлен
в табл. 9.2. Так же при необходимости, в эти растворы
может быть добавлен буфер HEPES (5-20 мМ), при
этом удаляется эквивалентное количество NaCl для
сохранения осмотического давления. На основе BSS
готовят многие полные среды и поставщики сред, кото¬
рые продают среду Игла МЕМ на основе солей Хэнкса
[Hanks & Wallace, 1949] или Игла МЕМ на основе солей
Эрла [Earle et al., 1943] указывают, какого состава BSS
был использован при приготовлении среды. Соли Хэнк¬
са применяют при культивировании клеток в закрытых
флаконах в атмосфере воздуха, в то время как соли
Эрла применяют при высокой концентрации в среде
бикарбоната в сочетании с 5% С02в газовой фазе.
BSS также используется для разбавления концен¬
тратов аминокислот и витаминов при приготовлении
питательной среды, для изотонических промывок или
при препарировании и для кратковременной инкуба¬
ции клеток (до 4 ч, обычно с добавлением глюкозы).
Состав BSS часто модифицируют — например, исклю¬
чают глюкозу или феноловый красный из BSS Хэнкса
или исключают ионы Са2+ и Mg2+ из PBS Дюльбеко
[Dulbecco & Vogt, 1954]. PBS без Са2+ и Mg2+ известен
как раствор PBS А, и обозначение D-PBSA будет ис¬
пользоваться в данной книге чтобы указать отсутствие
этих двухвалентных катионов. Необходимо всегда
указывать модификацию при покупке BSS, а также в
публикациях и отчетах.
Выбор BSS зависит как от парциального давления
С02 (см. разд. 9.2.2 и табл. 9.1 и 9.2), так и от целей
исследователя. Если BSS используется в качестве
раствора для дезагрегации тканей или клеточного мо¬
нослоя, то Са2+ и Mg2+ обычно исключается, например,
как в солевом растворе Moscona без кальция и магния
[1952] (CMF) или в D-PBSA (см. табл. 9.2). Выбор BSS
также зависит от того, будет ли этот раствор исполь¬
зован для суспензионной культуры или монослойной
(прикрепленной) культуры клеток. S-МЕМ на основе
солевого раствора Игла в модификации Спиннера
представляет собой вариант минимальной питательной
среды Игла [1959] с пониженным содержанием Са2+
для снижения клеточной агрегации и прикрепления
клеток (см. табл. 9.2).
HBSS, EBSS и PBS как буферы на основе фосфата
имеют низкую буферную емкость при физиологичес¬
ких pH. Paul [1975] предложил BSS, забуференный
Tris, который более эффективен как буфер, но при
его применении для некоторых типов клеток требу¬
ется адаптация. Наиболее эффективными буферами
в настоящее время считаются HEPES (10-20 мМ) в
диапазоне pH 7,2-7,8 и Трицин в диапазоне pH 7,4-8,0,
но использование этих буферов в в больших масштабах
значительно удорожает исследования.
9.4. ПОЛНЫЕ ПИТАТЕЛЬНЫЕ СРЕДЫ
Термин полная питательная среда подразумевает сре¬
ду, содержащую все компоненты и добавки и подходя¬
щую для конкретного использования. Ее обычно гото¬
вят из компонентов среды определенного химического
состава, некоторые из составляющих которой, такие
как глутамин, могут быть добавлены непосредственно
перед употреблением, и различных добавок, таких как
сыворотка, факторы роста или гормоны. Состав сред
9.4. Полные питательные среды
155
определенного химического состава может быть как
относительно простым, как, например, среда Игла
MEM [Eagle, 1959], которая состоит из незаменимых
аминокислот, витаминов, солей, так и более сложным,
как в средах 199 (М199) [Morgan et al., 1950], CMRL
1066 [Parker et al., 1957], MB 752/1 [Waymouth, 1959],
RPMI 1640 [Moore et al., 1967], и F12 [Ham, 1965]
(Таблица 9.3). Кроме того, имеется большое количество
бессывороточных сред (см. табл. 10.1 и 10.2). Слож¬
ные среды содержат большое количество различных
аминокислот, в том числе заменимых аминокислот,
дополнительные витамины и часто дополнительные
метаболиты (например, нуклеозиды, компоненты цик¬
ла трикарбоновых кислот и липиды) и минеральные
компоненты. Концентрация питательных веществ,
в целом, относительно низкая в среде F12 (которая
была оптимизирована для клонирования) и высока в
среде МЕМ Игла в модификации Дюльбекко (DMEM)
[Dulbecco & Freeman, 1959; Morton, 1970], оптимизиро¬
ванной для выращивания клеток в высокой плотности
для наращивания вирусов. Barnes and Sato [1980] сме¬
шали бедную среду F12 с более богатой по питательным
веществам DMEM в соотношении 1:1 и использовали
эту смесь как основу для своих бессывороточных сред.
Хотя такой подход не всегда является целесообразным,
в данном случае эта комбинация, полученная эмпи¬
рическим путем, позволила получить базовую среду,
которая, при обогащении специальными добавками,
подходит для различных типов клеток.
9.4.1. Аминокислоты
Для культивирования клеток требуются незаменимые
аминокислоты (т.е. те, которые не синтезируются в
организме), цистин и/или цистеин, аргинин, глутамин
и тирозин. Потребность в индивидуальных аминокис¬
лотах может различаться у клеток различных типов.
Другие заменимые аминокислоты часто добавляют
для того, чтобы либо компенсировать неспособность
некоторых клеток синтезировать их, либо для компен¬
сации их утечки в среду. Концентрация аминокислот
обычно определяет максимальную плотность культуры
и при достижении равновесия влияет на выживаемость
клеток и скорость их роста. Для большинства клеток
требуется глутамин, хотя некоторые клеточные линии
могут использовать глутаминовую кислоту; есть дан¬
ные, что глутамин используется культивируемыми
клетками в качестве источника энергии и углерода
[Butler & Christie, 1994; см. также разд. 3.6]. Глутамакс
(Glutamax, Invitrogen) представляет собой дипептид
аланин-глутамин, который более стабилен по сравне¬
нию с глутамином.
9.4.2. Витамины
Среда МЕМ Игла содержит только водорастворимые
витамины, за исключением биотина (витамины группы
В, холин, фолиевую кислоту, инозитол и никотинамид,
смотри табл. 9.3); остальные необходимые компонен¬
ты клетки получают из сыворотки. Биотин входит в
состав большинства более сложных сред, в том числе и
бессывороточных; п-аминобензойная кислота (РАВА)
входит в состав М199, CMRL1066 (модифицированная
М199) и RPMI1640. Все жирорастворимые витамины
(A, D, Е и К) присутствуют только в Ml99, витамин А
входит в состав LHC-9, а витамин Е — в MCDB 110 (см.
табл. 10.1). В бессывороточных средах некоторые вита¬
мины (например, холин и никотинамид) содержатся в
повышенной концентрации. Ограничение по витами¬
нам — например, при выпадении в осадок фолиевой
кислоты из концентрированного раствора — снижает
выживаемость клеток и скорость роста, влияя на эти
параметры сильнее, чем на максимальную плотность
клеток. Аналогично тому, как это происходит с ами¬
нокислотами, потребность в витаминах определяют
эмпирически, и часто это обусловлено со свойствами
клеточной линий, используемой в исследованиях;
например в среде Фишера (Fischer) высокая концент¬
рация фолиевой кислоты потому, что клетки L5178Y,
использовавшиеся при разработке среды, являются
фолат-зависимыми [Fischer & Sartorelli, 1964].
9.4.3. Соли
Основные компоненты, обеспечивающие осмотиче¬
ское давление среды, — это соли, такие как Na+, К+,
Mg2+, Са2+, С1~, S042", Р043 и НСОэ. В большинстве
сред концентрация солей соответствует их концент¬
рации в солевых растворах Эрла (высокая концентра¬
ция бикарбоната, 5% С02 в газовой фазе) или Хэнкса
(низкая концентрация бикарбоната, газовая фаза —
воздух). Са необходим для некоторых молекул
клеточной адгезии, таких как кадгерины [Yamada &
Geiger, 1997]. Са2+ также участвует как посредник в
передаче сигналов [Alberts et al., 2002], концентрация
Са2+ в среде может влиять на процессы пролиферации
и дифференцировки (см. разд. 17.2.2, 23.2.1). Na+, К+,
и С1“ регулируют мембранный потенциал, в то вре¬
мя как S042-, Р043“ и НС03" выступают в качестве
анионов, необходимых в качестве предшественников
макромолекул, а также регуляторов внутриклеточного
заряда.
Концентрация кальция снижена в средах для сус¬
пензионных культур для того, чтобы минимизировать
клеточную агрегацию и прикрепление (см. разд. 9.3).
Концентрацию бикарбоната определяет концентра¬
ция С02 в газовой фазе (см. разд. 9.2.2); кроме того,
бикарбонат выступает в качестве не только буфера, но
и питательного компонента.
9.4.4. Глюкоза
Глюкоза входит в состав большинства сред в качестве
источника энергии. Метаболизм глюкозы происхо-
156
ГЛАВА 9. СРЕДЫ ОПРЕДЕЛЕННОГО ХИМИЧЕСКОГО СОСТАВА И ДОБАВКИ К СРЕДАМ
Таблица 9.3. Часто используемые среды
Компоненты MEM DMEM F12 {?“ЕМ/ аМЕМ М199 L15 ^Coy^S Fischer
Аминокислоты
L-аланин
L-аргинин 6,0Е-04 4,0Е-04
L-аспарагин
L-аспарагиновая
кислота
L-цистеин
L-цистин
L-глутаминовая кис¬
лота
L-глутамин
Глицин
L-гистидин
L-оксипролин
L-изолейцин
L-лейцин
L-лизин НС1
L-метионин
L-фенил аланин
L-пролин
L-серин
L-треонин
L-триптофан
L-тирозин
L-валин
Витамины
р-аминобензойная
кислота
L-аскорбиновая кис¬
лота
Биотин
кальциферол
Холин хлорид
Фолиевая кислота
Мио-инозитол
Менадион (витамин
К3)
Никотинамид
Никотиновая кис¬
лота
D-пантотенат Са
Пиридоксаль НС1
Пиридоксин НС1
Рибофлавин
Тиамин
Тиамин монофосфат
а-токоферол
Ретинол ацетат
Витамин В12
Антиоксиданты
Глутатион
Неорганические
соли
СаС12
1,8Е-03
l,8E-03
КС1
5,3E-03
5,3E-03
кн2ро4
MgCl2
Mgso4
8ДЕ-04
8ДЕ-04
NaCl
1,2E-01
1ДЕ-01
NaHC03
2,6E-02
CM
о
i
w
l,0E-04
5,0E-05
2,8E-04
2,8E-04
l,0E-03
7,0E-04
6,0E-04
3,3E-04
1ДЕ-03
l,0E-04
5,0E-05
3,3E-04
3,8E-04
l,7E-03
l,0E-04
5,0E-05
2,3E-04
2,3E-04
l,5E-04
2,0E-04
l,0E-04
5,7E-04
l,5E-03
l,0E-04
l,0E-04
8,3E-05
2ДЕ-04
l,0E-04
5,0E-05
5ДЕ-04
5ДЕ-04
l,4E-04
l,0E-03
2,5E-03
2,0E-03
6,8E-04
2ДЕ-03
l,0E-04
2,5E-04
6,7E-04
6,7E-04
l,3E-04
l,0E-04
l,5E-04
2,0E-04
9,5E-05
9,7E-05
7,6E-05
l,5E-04
3,0E-05
4,2E-04
4,0E-04
l,5E-04
3,8E-04
l,0E-04
4,5E-04
4,0E-04
4,6E-04
3,8E-04
2,0E-04
5,0E-04
4,0E-04
3,8E-04
2,2E-04
3,0E-05
l,2E-04
l,0E-04
l,0E-04
l,0E-04
3,0E-05
2,2E-04
l,9E-04
l,5E-04
9ДЕ-05
3,0E-04
l,5E-04
3,5E-04
3,5E-04
l,7E-04
l,0E-04
2,5E-04
2,4E-04
2,4E-04
2,9E-04
l,0E-04
4,5E-04
4,0E-04
2,5E-04
l,7E-04
l,0E-05
4,4E-05
4,9E-05
4,9E-05
2,5E-05
3,0E-05
2ДЕ-04
2,3E-04
2,2E-04
1ДЕ-04
l,0E-04
4,5E-04
3,9E-04
2ДЕ-04
l,7E-04
3,6E-07
7,3E-06
2,5E-04
2,8E-04
3,0E-08
l,5E-08
4ДЕ-07
4ДЕ-08
8,2E-07
l,0E-04
6,4E-05
7ДЕ-06
3,6E-06
2ДЕ-05
2,9E-06
6,0E-06
2,3E-06
2,3E-08
2,3E-06
l,0E-04
7,0E-05
1ДЕ-05
2,8E-07
l,9E-04
3,3E-07
l,7E-05
8,2E-06
2,0E-07
8,2E-06
2,0E-06
9,4E-06
4,2E-06
4,2E-08
1ДЕ-06
3,0E-07
l,0E-05
4,9E-06
l,2E-07
3,0E-07
l,5E-07
l,2E-07
4,9E-06
l,0E-07
5,8E-07
2,7E-07
2,7E-08
5,3E-07
l,0E-06
6,4E-06
3,0E-06
3,0E-08
3,0E-06
l,0E-06
5,0E-07
l,0E-06
3/7E-09
3,0E-05
3,0E-06
3,0E-04
1ДЕ-03
l,8E-03
l,8E-03
3,0E-03
4,2E-03
5,3E-04
5,3E-03
5,3E-03
1,2E-01
4,0E-04
8ДЕ-04
8ДЕ-04
4,0E-04
1,3E-01
1,2E-01
1,2E-01
1,0E-01
l,4E-02
2,9E-02
2,6E-02
2,6E-02
4,2E-03
2,8E-04
2,5E-03
1.5E-04
3,3E-04
2,9E-03
2,0E-04
7ДЕ-05
3,6E-04
3,0E-04
7,6E-05
2,3E-04
l,5E-04
4,5E-04
5,6E-07
9,9E-04
2,0E-04
5,0E-04
9,9E-05
9,9E-05
6,3E-05
4,5E-04
l,5E-04
l,0E-03
6,8E-04
2ДЕ-03
l,5E-03
l,4E-03
2,4E-03
6,7E-04
2,7E-03
l,0E-04
6,7E-04
l,0E-04
l,6E-03
l,0E-04
3,9E-04
8,3E-04
7,6E-05
l,5E-04
l,5E-04
9,5E-04
3,0E-04
5,7E-04
l,9E-04
4,6E-04
9,5E-04
3,0E-04
2,3E-04
3,8E-04
3,8E-04
5ДЕ-04
2,0E-04
2,7E-04
l,3E-03
l,0E-04
5,0E-04
l,0E-04
6,7E-04
3,4E-04
l,5E-04
7,6E-04
l,0E-04
4ДЕ-04
3,0E-04
3,5E-04
l,5E-04
4,3E-04
2,4E-04
l,9E-03
2,5E-04
l,4E-04
2,5E-04
2,5E-03
l,5E-04
3,4E-04
6,3E-04
4,9E-05
9,8E-05
l,5E-05
4,9E-05
2,0E-04
2,2E-04
l,7E-03
l,2E-04
3,3E-04
2,2E-04
2ДЕ-04
8,5E-04
l,5E-04
6,0E-04
5,6E-04
3,6E-07
7,3E-06
2,8E-07
3,2E-06
9,9E-05
4ДЕ-08
8,2E-07
4ДЕ-08
8,2E-08
2,5E-07
3,6E-06
7ДЕ-06
3,6E-05
1ДЕ-05
l,8E-03
2,3E-08
2,3E-06
2,3E-05
2,3E-05
9ДЕ-07
2,8E-07
1ДЕ-05
2,0E-04
8,3E-06
5,6E-06
6,9E-08
2,0E-07
8,2E-06
4ДЕ-06
4ДЕ-06
8,2E-06
2,0E-07
4ДЕ-06
4,2E-08
4,2E-06
8,4E-07
2ДЕ-06
4,2E-06
l,2E-07
2,5E-06
2,5E-06
l,2E-07
2,4E-06
4,9E-06
2,7E-08
l,9E-07
5,3E-07
l,3E-06
2/7E-06
CO
о
m
о
00
2,4E-06
5,9E-07
3,0E-06
3,0E-05
4,8E-06
2,3E-08
3,5E-07
1,5Е-07
l,5E-07
l,5E-06
4,5E-05
l,3E-03
l,3E-03
9,0E-04
6,2E-04
8,2E-04
5,3E-03
5,3E-03
5,3E-03
5,3E-03
2,0E-03
4,4E-04
4,4E-04
5,9E-04
l,2E-03
8ДЕ-04
l,6E-03
8ДЕ-04
4,9E-04
8ДЕ-04
1,4E-01
1,4E-01
1ДЕ-01
1,4E-01
1,0E-01
2,6E-02
l,3E-02
2,7E-02
l,0E-04
2,0E-04
2,0E-03
4,0E-03
4,0E-04
2,0E-04
2,0E-04
4,0E-04
8,0E-04
4,0E-04
8,0E-04
4,0E-04
8,0E-04
l,0E-04
2,0E-04
2,0E-04
4,0E-04
4,0E-04
4,0E-04
8,0E-04
4,9E-05
7,8E-05
2,0E-04
4,0E-04
4,0E-04
8,0E-04
7ДЕ-06 2,9Е-05
2,ЗЕ-06 9ДЕ-06
1ДЕ-05 4,0Е-05
8,2Е-06 3,3E-05
4,2Е-06 1,7Е-05
4,9Е-06 2,0Е-05
2,7Е-07 1ДЕ-06
3,0Е-06 1,2Е-05
9.4. Полные питательные среды
157
DMEM/ CMRL RPMI
F12 аМЕМ 1066 1640 Ш" L1S
Окончание табл. 9.3
McCoy’s p.. MB
5А Fl8Cher 752/1
Компоненты
MEM DMEM F12
NaH2 P04 1,0Е-03 9ДЕ-04
Na2HP04
Микроэлементы
CuS04 • 5H20
Fe(N03)3 • 9H20 2,5E-07
FeS04 ■ 7H20
ZnS04 • 7H20
Основания, нуклеозиды и т.д.
Аденин S04
Аденозин
АМФ
АТФ
Цитидин
Дезоксиаденозин
Дезоксицитидин
Дезоксигунозин
2-дезоксирибоза
НАД
ФАД
Глюкоронат Na
Гуанин
Гуанозин
Гипоксантин
5-метилдезоксицитозин
D-рибоза
Тимидин
Тимин
Т рифосфопиридиндину клео-
тид
Урацил
Уридин
УТФ
Ксантин
Компоненты энергетического
обмена
Кокарбоксилаза
Коэнзим А
D-галактоза
D-глюкоза
Ацетат натрия
Пируват натрия
Липиды и предшественники
Холестерол
Этанол (растворитель)
Линолевая кислота 3,0Е-07
Липоевая кислота
Твин 80
Другие компоненты
Пептон, мг/мл
Феноловый красный 2,7Е-05 4,0Е-05
Путресцин
Газовая фаза
С02 5% 10%
4,5Е-04
1,0Е-03 5,0Е-04
1,6Е-08 7,8Е-09
1,2Е-07
3,0Е-06 1,5Е-06
3,0Е-06 1,5Е-06
1,0Е-03 4,2Е-03 5,7Е-04
5,6Е-03 4,0Е-04 1.6Е-03 5,0Е-04 2ДЕ-03
3,7Е-05
4ДЕ-05
4,0Е-05 4,0Е-05
4,2Е-05 3,8Е-05
3,7Е-05 3,7Е-05
9,5Е-06
1,2Е-06
1,9Е-05
3,5Е-05
3,0Е-05 1,5Е-05
4ДЕ-07
3,0Е-06 1,5Е-06 4ДЕ-05 4ДЕ-05
1,ЗЕ-06
4ДЕ-05
1,8Е-06
5,4Е-05
5,8Е-07
1,8Е-05
3,7Е-06
1,6Е-06
2,2Е-06
3,3E-06
2,4Е-06
2,7Е-06
2,0Е-06
5,6Е-03 2,5Е-02
2,2Е-06
3,3E-06
5,0Е-02
1,0Е-02 1,8Е-02 5,6Е-03 5,6Е-03 1ДЕ-02 5,6Е-03 1,7Е-02 5,6Е-03 2,8Е-02
6ДЕ-04 4,5Е-04
5,0Е-03
1,0Е-03 1,0Е-03 1,0Е-03 1,0Е-03
1,5Е-07
1,0Е-06 5ДЕ-07 9,7Е-07
5,2Е-07
3,5Е-04
1,8Е-05
5,2Е-07
1,8Е-05
8,9Е-05
0,6
3,2Е-05 3,6Е-05 2,9Е-05 5,ЗЕ-05 1,ЗЕ-05 4,5Е-05 2,7Е-05 2,9Е-05 1,ЗЕ-05 2,7Е-05
1,0Е-06 5,0Е-07
2% 7% 5% 5% 5% 5% Воздух 5% 2% 5%
Все концентрации молярные, использован компьютерный стиль обозначений (например, 3,0Е-2 = 3,0x10 =30 мМ). Для неко¬
торых важных компонентов указан молекулярный вес, хотя в некоторых составах используются соли в гидратированной форме,
молярность остается прежней. Синонимы и аббревиатуры: АМФ — аденозинмонофосфат, АТФ - аденозинтрифосфат, биотин =
витамин Н; кальциферол = витамин D2; ФАД, флавинадениндинуклеотид; липоевая кислота = тиоктовая кислота; менадион = ви¬
тамин К3; мио-инозитол; = L-инозитол; никотинамид = ниацинамид; никотиновая кислота = ниацин; пиридоксин НС1 = витамин
В6; тиамин = витамин В2; а-токоферол = витамин Е; ретинол = витамин УТФ, уридинтрифосфат; витамин В12 = кобаламин.
Смотри ссылки в тексте.
158
ГЛАВА 9- СРЕДЫ ОПРЕДЕЛЕННОГО ХИМИЧЕСКОГО СОСТАВА И ДОБАВКИ К СРЕДАМ
дит главным образом путем гликолиза, из глюкозы
образуется пируват, из которого может образоваться
лактат или ацетоацетат. Пируват может окисляться в
цикле лимонной кислоты с образованием С02 и воды.
Накопление в среде молочной кислоты, особенно
характерное для эмбриональных и трансформиро¬
ванных клеток, свидетельствует о нарушениях в фун¬
кционировании цикла лимонной кислоты. Недавно
были получены данные, что большая часть углерода
в клетке образуется не из глюкозы, а из глутамина.
Это открытие может объяснить исключительно вы¬
сокие требования некоторых культур к глутамину
или глутамату.
9.4.5. Органические добавки
В состав сложных питательных сред входят разнооб¬
разные компоненты, такие как белки, пептиды, нук-
леозиды, промежуточные продукты цикла лимонной
кислоты, пируват и липиды. Было показано, что эти
компоненты необходимы для сред с пониженным
содержанием сыворотки, они помогают выжить клет¬
кам при клонировании, а также при культивировании
определенных специализированных клеток, даже в
присутствии сыворотки.
9.4.6. Гормоны и факторы роста
Гормоны и факторы роста не указываются в составах
большинства стандартных сред, хотя их часто добав¬
ляют в бессывороточные среды (см. разд. 9.5.2, 9.5.3,
10.4.2,10.4.3).
9.4.7. Антибиотики
Антибиотики исходно добавляли в культуральную
среду для того, чтобы снизить вероятность контами¬
нации. Однако использование шкафов с ламинарным
потоком совместно с применением строгих асептиче¬
ских методик сделало использование антибиотиков
необязательным. Несомненно, антибиотики имеют ряд
недостатков:
1) Они способствуют селекции микроорганизмов,
устойчивых к антибиотикам.
2) Они маскируют присутствующих в небольшом ко¬
личестве скрытых контаминантов, которые могут
активизироваться при удалении антибиотиков,
изменении условий культивирования или из них
могут развиться резистентные штаммы.
3) Они могут маскировать микоплазменную инфекцию.
4) При взаимодействии с клетками млекопитающих
они могут действовать как антиметаболиты.
5) Они позволяют неаккуратно работать в плохих
асептических условиях.
Поэтому настоятельно рекомендуется рутинное
культивирование проводить в отсутствие антибиоти¬
ков; применение антибиотиков ограничить первичны¬
ми культурами или продолжительными дорогостоя¬
щими лабораторными экспериментами. Если условия
требуют применения антибиотиков, они должны потом
быть удалены так быстро, как только возможно. Если
антибиотики используются в течение продолжительно¬
го времени, необходимо параллельно культивировать
клетки без антибиотиков (см. разд. 19.4).
Ряд антибиотиков, используемых в культуре клеток,
обладают умеренной эффективностью в отношении
бактериальных инфекций (табл. 9.4). Однако у многих
Таблица 9.4. Антибиотики, используемые при культивировании клеток
Концентрация, мкг/мл
(если не указана другая)
Антибиотик
Рабочая
Цитотоксическая
Активны в отношении:
Амфотерицин В (Фунгизон)
2,5
30
Грибы, дрожжи
Ампициллин
2,5
Грамположительные и грамотрицательные бактерии
Ципрофлоксацин
100
Микоплазма
Эритромицин
50
300
Микоплазма
Гентамицин
50
>300
Грамположительные и грамотрицательные бактерии; мико¬
плазма
Канамицин
100
10 мг/мл
Грамположительные и грамотрицательные бактерии; мико¬
плазма
MRA(ICN)
0,5
Микоплазма
Неомицин
50
3,000
Грамположительные и грамотрицательные бактерии
Нистатин
50
Грибы, дрожжи
Пенициллин-G
100 U/мл
10,000 U/мл
Грамположительные бактерии
Полимиксин В
50
1 мг/мл
Грамотрицательные бактерии
Стрептомицин сульфат
100
20 мг/мл
Грамположительные и грам-отрицательные бактерии
Тетрациклин
10
35
Грамположительные и грам-отрицательные бактерии
Тилозин
10
300
Микоплазма
По Paul, 1975,
9.5. Сыворотка
159
бактериальных штаммов развилась устойчивость к ан¬
тибиотикам, как естественным путем, так и в результате
селекции. Таким образом, контроль контаминации с
помощью антибиотиков никогда не будет абсолютным.
Особенно трудно контролировать контаминацию гри¬
бами и дрожжами, размножение этих микроорганизмов
можно сдержать, но уничтожить их удается только в
редких случаях (см. разд. 19.4).
9.5. СЫВОРОТКА
В состав сыворотки входят факторы роста, которые
способствуют клеточной пролиферации, а также фак¬
торы адгезии и вещества, обладающие антитрипсиновой
активностью, способствующие прикреплению клеток.
Сыворотка также является источником минеральных
веществ, липидов и гормонов, многие из которых могут
быть связаны с белком (табл. 9.5). Для большинства
клеточных культур используются телячья сыворотка,
эмбриональная бычья сыворотка, сыворотка взрослых
лошадей и человеческая сыворотка. Телячья (CS) и
эмбриональная бычья (FBS) сыворотки используются
наиболее широко, в особенности последняя, которую
применяют для большинства требовательных линий
клеток и для клонирования. Человеческая сыворотка
иногда используется для культивирования некоторых
линий клеток человека, но ее необходимо проверять
на вирусы, такие как ВИЧ и гепатит В. Для некоторых
исследований лошадиная сыворотка предпочтительнее
телячьей, так как она может быть получена из закрытого
донорского сердца и часто ее свойства меньше варьирует
от серии к серии. Лошадиная сыворотка также обладает
свойством меньше метаболизировать полиамины из-за
низкого уровня полиаминоксидазы; полиамины явля¬
ются митогенными факторами для некоторых типов
клеток [Hyvonen et al., 1988; Kaminska et al., 1990].
9.5.1. Белки
Хотя белки являются основным компонентом сы¬
воротки, функции многих белков in vitro не ясны;
возможно, что некоторые белки являются не только
переносчиками минеральных содинений, жирных
кислот и гормонов, но и выполняют другие функции.
К белкам с известными функциями относятся аль¬
бумин [Iscove & Melchers, 1978; Barnes & Sato, 1980],
который является важным переносчиком липидов,
минералов; глобулины [Tozer& Pirt, 1964]; фибронек-
тин (нерастворимый на холоду глобулин), который
способствует прикреплению клеток [Yamada & Geiger,
1997; Hynes, 1992], хотя, возможно, не настолько
эффективно, как фибронектин, продуцируемый клет¬
ками; и а2-макроглобулин — ингибитор трипсина [de
Vonne & Mouray, 1978]. Фетуин из эмбриональной
сыворотки способствует прикреплению клеток [Fisher
et al., 1958], а трансферрин [Guilbert & Iscove, 1976]
связывает железо, снижая его токсичность и делая
Таблица 9.5. Компоненты сыворотки
Компоненты
Диапазон концентраций8
Белки и полипептиды
Альбумин
Фетуин6
Фибронектин
Глобулины
Ингибитор протеаз: ocj-anti-
trypsin, a2-macroglobulin
Трансферрин
Факторы роста:
EGF, PDGF, IGF-I and -II,
FGF, IL-1, IL-6
Аминокислоты
Липиды
Холестерол
Жирные кислоты
Линолевая кислота
Фосфолипиды
Углеводы
Глюкоза
Гексозамин®
Молочная кислотаг
Пировиноградная кислота
Полиамины:
Путресцин, спермидин
Мочевина
Неорганические соединения:
Кальций
Хлориды
железо
калий
Phosphate
Селен
Натрий
Цинк
Гормоны:
Гидрокортизон
Инсулин
Т рийодтиронин
Тироксин
Витамины:
Витамин А
Фолат
40-80 мг/мл
20-50 мг/мл
10-20 мг/мл
1-10 мкг/мл
1-15 мг/мл
0,5-2,5 мг/мл
2-4 мг/мл
1-100 нг/мл
0,01-1,0 мкМ
2-10 мг/мл
10 мкМ
0,1-1,0 мкМ
0,01-0,1 мкМ
0,7-3,0 мг/мл
1,0-2,0 мг/мл
0,6-1,2 мг/мл
6-1,2 мг/мл
0,5-2,0 мг/мл
2-10 мкг/мл
0,1-1,0 мкМ
170-300 мкг/мл
0,14-0,16 М
4-7 мМ
100 мкМ
10-50 мкМ
5-15 мМ
2-5 мМ
0,01 мкМ
135-155 мМ
0,1-1,0 мкМ
0,1-200 нМ
10-200 нМ
1-100 нг/мл
20 нМ
100 нМ
10 нг-10 мкг/мл
10-100нг/мл
5-20 нг/мл
а Диапазон концентраций дан приблизительно и предназначен
для того, чтобы только указать порядок величины. Данные
приведены по Evans and Sanford [1978], и Cartwright and
Shah [1994].
6 Только в фетальной сыворотке.
в Больше всего в сыворотке человека.
г Больше всего в фетальной сыворотке.
его биодоступным. Другие белки, которые еще не оха¬
рактеризованы, могут использоваться для клеточного
роста и прикрепления клеток.
%6G
ГЛАВА 9. СРЕДЫ ОПРЕДЕЛЕННОГО ХИМИЧЕСКОГО СОСТАВА И ДОБАВКИ К СРЕДАМ
Белки также повышают вязкость среды, снижая
стресс от повреждений при пипетировании и пере¬
мешивании, и могут увеличивать буферную емкость
среды.
9.5.2. Факторы роста
Сыворотка, полученная естественным путем, т. е. соб¬
ранная после образования сгустка, стимулирует проли¬
ферацию клеток лучше, чем сыворотка, полученная при
физическом удалении клеток крови (например, путем
центрифугирования). Возможно, это связано с тем, что
при образовании сгустка из тромбоцитов высвобожда¬
ется тромбоцитарный фактор роста (PDGF). PDGF
[Antoniades et al., 1979; Heldin et al., 1979] — один из
представителей семейства полипептидов, обладающих
митогенной активностью и, возможно, основной фактор
роста сыворотки. Содержащийся PDGF стимулирует
рост фибробластов и глии, но другие тромбоцитарные
факторы роста, такие как TGF-|3, могут ингибировать
рост или стимулировать дифференцировку эпители¬
альных клеток [Lechner et al., 1981].
Другие факторы роста (см. табл. 10.3), такие как
факторы роста фибробластов (FGF) [Gospodarowicz,
1974], эпидермальный фактор роста (EGF) [Cohen,
1962; Carpenter & Cohen, 1977; Gospodarowicz et al.,
1978a], факторы роста эндотелия, такие как васкуляр¬
ный эндотелиальный фактор роста (VEGF) и ангиоге-
нин [Ни et al., 1997; Folkman & d’Amore, 1996; Joukov
et al., 1997; Folkman et al., 1979; Maciag et al., 1979] и
инсулиноподобные факторы роста IGF-I и IGF-II [le
Roith & Raizada, 1989], которые были выделены из тка¬
ней или секретированы в культуральную среду клетка¬
ми, значительно различаются по своей специфичности
и, возможно, присутствуют в сыворотке в небольших
количествах [Gospodarowicz & Moran, 1974]. Многие
факторы роста продаются как коммерческие препараты
(см. Приложение II) в виде рекомбинантных белков,
некоторые из них производятся в форме долгоживущих
аналогов (Sigma), которые отличаются более высокой
митогенной активностью и стабильностью.
9.5.4. Питательные вещества и метаболиты
Сыворотка может также содержать аминокислоты,
глюкозу, оксо (кето) кислоты, нуклеозиды и ряд других
питательных компонентов и промежуточных метаболи¬
тов. Эти соединения могут быть важными для простых
сред, но не так значимыми для сложных сред, особенно
для сред с высоким содержанием аминокислот и других
химически определенных добавок.
9.5.5. Липиды
Линолевая кислота, олеиновая кислота, этаноламин
и фосфоэтаноламин присутствуют в сыворотке в не¬
больших количествах, обычно в связанном с белками
(такими, как альбумин) виде.
9.5.6. Неорганические элементы
Эксперименты с заменой сыворотки [Ham & McKeehan,
1978] позволили предположить, что микроэлементы —
железо, медь и цинк — могут связываться с белками
сыворотки. McKeehan et al. [1976] продемонстриро¬
вали необходимость в селене, который, по-видимому,
помогает нейтрализовать свободные радикалы, так как
является кофактором для глутатионсинтазы.
9.5.7. Ингибиторы
Сыворотка может содержать вещества, ингибирующие
пролиферацию клеток [Harrington & Godman, 1980;
Liu et al., 1992; Varga Weisz & Barnes, 1993]. Некото¬
рые из них могут присутствовать в сыворотке из-за
ошибок при приготовлении (например, бактериальные
токсины, в случае, если сыворотка была контамини-
рована до фильтрации, или антитела, содержащиеся в
у-глобулиновой фракции, которые взаимодействуют
с эпитопами на поверхности клеток), но другие явля¬
ются отрицательными регуляторами роста, такие как
TGF-P [Massague et al., 1992]. Тепловая инактивация
удаляет из сыворотки комлемент и снижает цитотокси-
ческое действие иммуноглобулинов, не разрушая при
этом факторы роста, но может также разрушать более
лабильные компоненты сыворотки, поэтому инакти¬
вированная сыворотка может быть менее активна по
сравнению с необработанной сывороткой.
9.6. ВЫБОР СРЕДЫ И СЫВОРОТКИ
Все 12 сред, приведенных в табл. 9.3, были разработаны
для выращивания определенных клеточных линий в
определенных условиях. Многие из них были разра¬
ботаны для мышиных фибробластов L929 или клеток
карциномы шейки матки HeLa; среда Хэма F12 была
разработана для клеток яичника китайского хомячка
(СНО); однако сейчас все они имеют более широкое
применение и состав их считается классическим.
Производители свидетельствуют, что доля наиболее
популярных сред — RPMI 1640, DMEM и МЕМ со¬
ставляет 75% продаж. Доля сред другого состава редко
превышает 5% от общей суммы продаж, обычно 2-3%,
хотя доля смеси DMEM/F12 более 4%.
Минимальная питательная среда Игла (МЕМ)
была разработана путем усовершенствования Базовой
среды Игла (ВМЕ) за счет увеличения количества
и концентрации компонентов. В течение многих лет
среда Игла МЕМ использовалась чаще всех сред. Среда
МЕМ в модификации Долбекко (DMEM) была разра¬
ботана для трансформации мышиных фибробластов
и исследований, связанных с наращиванием вируса.
9.6. Выбор среды и сыворотки
161
Концентрация аминокислот в ней в 2 раза выше, чем в
МЕМ, концентрация витаминов выше в 4 раза, и двой¬
ная концентрация НСОэ (и, соответственно С02) для
лучшей буферизации. В состав аМЕМ [Stanners et al.,
1971] входят не только дополнительные аминокисло¬
ты и витамины, но и нуклеозиды и липоевая кислота;
эта среда используется для многих типов клеток,
включая гематопоэтические клетки. Среда Хэма F12
была разработана для клонирования клеток СНО при
низком содержании сыворотки, она также широко ис¬
пользуется, особенно для исследования клоногенной
активности (см. протокол 22.3.2) и первичных культур
(см. протокол 12.7).
Среды CMRL 1066, М199 и среда Вэймонта (Way-
mouth’s medium) были разработаны для культивирова¬
ния клеток L929 без добавления сыворотки, однако они
используются для многих требовательных к условиям
культур либо в чистом виде, либо в сочетании с други¬
ми средами, такими как DMEM или F12. RPMI1640 и
среда Фишера (Fischer’s medium) были разработаны
для лимфоидных клеток: среда Фишера — для лимфо-
мы L5178Y, требующей высокого содержания фолата.
RPMI 1640 особенно широко используется, часто для
прикрепляющихся клеток, хотя была исходно предна¬
значена для суспензионных культур, поэтому в ней от¬
сутствует кальций. Среда L15 была создана специально
для того, чтобы обеспечить буферизацию в отсутствие
НС03 и С02. По этой причине ее часто используют
для транспортировки клеток и в качестве среды для
первичных культур, но она используется меньше из-за
того, что в ее состав входит HEPES, что демонстрирует,
что НС03 и С02 часто необходимы для оптимального
роста клеток, невзирая на требования буферизации.
Информацию относительно выбора подходящей
среды для данного типа клеток обычно можно най¬
ти в литературе — в статьях, где описано получение
клеточной линии или сходной культуры клеток. Ин¬
формацию также можно получить от того, кто предо¬
ставляет клеточную линию. Банки клеток, такие как
АТСС (Американская коллекция клеток) и ЕСАСС
(Европейская коллекция клеток), предоставляют
информацию о средах, которые используются для
доступных в настоящее время линий клеток, и эту
информацию можно просмотреть на их веб-сайтах (см.
Приложение III, см. также табл. 9.6). В случае если
информация отсутствует, приходится либо эмпири¬
чески подбирать среду, либо провести сравнительное
тестирование нескольких сред так же, как и при выборе
сыворотки (см. разд. 9.6.2).
Многие постоянные линии клеток (например, HeLa,
L929, ВНК21), первичные культуры клеток человека,
грызунов и фибробласты птиц, и линии клеток, произо¬
шедшие от них, можно культивировать на относительно
простых средах, таких как среда Игла МЕМ, с добавле¬
нием телячьей сыворотки. Более сложные среды могут
потребоваться для специализированных клеток (см.
разд. 17.7) или когда клетки субкультивируют с низкой
плотностью посева (<1х103/мл), как при клонировании
(см. разд. 14.2). Зачастую для более требовательных
клеток, для которых необходима сложные среды,
требуется также эмбриональная бычья сыворотка, а
не телячья или лошадиная, несмотря на то что среда
исходно была разработана для культивирования клеток
в отсутствие сыворотки.
Некоторые предложения по выбору среды и не¬
сколько примеров некоторых типов клеток и исполь¬
зуемых для них сред приведены в табл. 9.6 (см. также
Mather, 1998). Если информация недоступна, за две
недели можно провести простые эксперименты по ис¬
следованию эффективности роста клеток на коммерчес¬
ких доступных средах в многолуночных планшетах (см.
протоколы 21.7-21.9). Исследование эффективности
клонообразования (клоногенной активности) (см. про¬
токол 21.10) и измерение проявления специфических
функций помогут дополнительно ограничить выбор.
Для вас может оказаться сюрпризом, если удастся
подобрать лучшие условия для культивирования
клеток, которые не согласуются с литературными
данными; воспроизвести условия, созданные в другой
лаборатории, может быть сложно из-за вариаций в
приготовлении сред или разных поставщиках, из-за
примесей, присутствующих в реактивах и воде, разницы
между сериями сыворотки. Остается надеяться, что при
снижении необходимости использования сыворотки и
повышении чистоты реактивов стандартизация среды
будет совершенствоваться.
В конце концов, вы можете найти компромисс, выби¬
рая среду или сыворотку по их цене. Автоклавируемые
среды доступны у коммерческих производителей (см.
Приложение II). Их просто готовить из порошка, и они
подходят для многих постоянных клеточных линий.
В эти среды необходимо добавлять для большинства
клеток глутамин и, обычно, сыворотку. Стоимость
сыворотки можно подсчитать, основываясь на объеме
среды, когда количество клеток не имеет значения, но
в случае, когда целью является наращивание большого
количества клеток, можно подсчитать стоимость сыво¬
ротки в расчете на клетку. Так, если культура растет до
плотности 1х106/мл в среде с сывороткой А и 2х106/мл
в среде с сывороткой Б, сыворотка Б обойдется дешевле
в 2 раза. Если необходима эмбриональная сыворотка,
попробуйте смешать ее с телячьей. Это может помочь
вам снизить концентрацию более дорогой эмбриональ¬
ной сыворотки. Если вы сможете, полностью избавьтесь
от сыворотки или снизьте ее концентрацию и исполь¬
зуйте бессывороточные составы (см. разд. 10.5)
9.6.1. Резервирование партии сыворотки
Разные партии сыворотки могут значительно разли¬
чаться по своим свойствам. Эти различия обусловлены
разными методами приготовления и стерилизации, раз¬
личным сроком и условиями хранения, а также зависят
от породы животных, у которых берут сыворотку, от их
рациона, климата, в котором животных выращивают и
от других условий окружающей среды. Важно выбрать
серию сыворотки и использовать ее как можно дольше
162
ГЛАВА 9, СРЕДЫ ОПРЕДЕЛЕННОГО ХИМИЧЕСКОГО СОСТАВА И ДОБАВКИ К СРЕДАМ
Таблица 9.6. Выбор подходящей среды
Клетки или линия клеток
Среда
Сыво¬
ротка
Клетки ЗТЗ
MEM, DMEM
CS
Эмбриональные фибро-
Eagle’s МЕМ
CS
бласты цыпленка
Яичник китайского хо¬
Eagle’s МЕМ,
CS
мячка (СНО)
Ham’s F12
Хондроциты
Ham’s F12
FB
Непрерывные клеточные
Eagle’s MEM, DMEM
CS
линии
Эндотелий
DMEM, M199, MEM
CS
Фибробласты
Eagle’s MEM
CS
Глиальные клетки
MEM, DMEM/F12
FB
Глиома
MEM, DMEM/F12
FB
Клетки HeLa
Eagle’s MEM
CS
Гематопоэтические клет¬
RPMI 1640, Fischer’s,
FB
ки
ocMEM
Диплоидные фибро¬
Eagle’s MEM
CS
бласты человека
Лейкемия человека
RPMI 1640
FB
Опухоли человека
L15, RPMI 1640,
DMEM/F12
FB
Кератиноциты
aMEM
FB
L клетки (L929, LS)
Eagle’s MEM
CS
Лимфобластоидные
RPMI 1640
FB
линии клеток (челове¬
ческие)
Эпителий молочной же¬
RPMI 1640,
FB
лезы
DMEM/F12
Эпителий почки собаки
DMEM, DMEM/F12
FB
MDCK
Меланоциты
M199
FB
Меланома
MEM, DMEM/F12
FB
Эмбриональные фибро¬
Eagle’s MEM
CS
бласты мыши
Лейкемия мыши
Fisher’s, RPMI 1640
FB, HoS
Эритролейкемия мыши
DMEM/F12,
RPMI 1640
FB, HoS
Миелома мыши
DMEM, RPMI 1640
FB
Нейробластома мыши
DMEM, DMEM/F12
FB
Нейроны
DMEM
FB
Фибробласты почки кры¬
MEM, DMEM
CS
сы NRK
Гепатома крысы с мини¬
Swim’s S77,
FB
мальным отклонением
DMEM/F12
(НТС, MDH)
Скелетные мышцы
DMEM, F12
FB, HoS
Фибробласты сирийско¬
MEM, GMEM,
CS
го хомячка (например
DMEM
ВНК21)
CS — телячья сыворотка, FB — эмбриональна бычья сыворот¬
ка; HoS — лошадиная сыворотка. SF12 — среда Хэма F12 плюс
незаменимые аминокислоты Игла и заменимые аминокис¬
лоты как в DMEM (продается как 100-кратный концентрат,
смотри Приложение II). Дополнительные рекомендации по
выбору сред можно найти у McKeehan [1977], Bames et al.
[1984(a-d)], и Mather [1998].
и заменить ее со временем на максимально сходную по
свойствам.
Стандартизировать сыворотку сложно, так как
партии могут значительно варьировать, и одна партия
может храниться от шести месяцев до года при -20 °С.
Выберите тип сыворотки, наиболее подходящей для
ваших целей, и запрашивайте партии для тестирования
у нескольких поставщиков. Большинство поставщиков
сыворотки резервируют партию, пока заказчик выберет
наиболее подходящий ему вариант (с условием, что вы¬
бор не займет более трех недель или около того). Когда
подходящая партия будет отобрана, поставщика просят
хранить необходимый объем до года и отправлять по
потребности. Других поставщиков необходимо инфор¬
мировать, что они могут вернуть зарезервированные
партии на склад.
9.6.2. Тестирование сыворотки
Качество данной сыворотки гарантируется постав¬
щиком, однако, контроль качества фирмы проводят
на одной из ряда постоянных клеточных линий. Если
ваши требования более высокие, вы должны провести
собственное тестирование. Сыворотку тестируют по
четырем основным параметрам.
Эффективность посева. При клонировании клет¬
ки растут в низкой плотности и из-за этого становятся
более чувствительными, поэтому это очень убеди¬
тельный тест. Рассейте клетки с плотностью от 10 до
100 клеток/мл и проанализируйте образование коло¬
нии в период от 10 дней до двух недель. Покрасьте и
посчитайте колонии (см. протокол 21.10) и определите
разницу в эффективности посева (отражает выживае¬
мость) и в размере колоний (отражает пролиферацию
клеток). Каждую сыворотку необходимо тестировать в
диапазоне концентраций от 2% до 20%. Такой подход
выявит, если какая-либо из сывороток будет проявлять
равную активность в более низкой концентрации, и это
поможет сэкономить деньги и использовать партию в
течение более длительного времени. Кроме того, если
в сыворотке присутствует какая-либо токсичность, при
высокой концентрации она проявится.
Кривая роста. Необходимо построить график
кривой роста клеток при их выращивании на каждой
сыворотке (см. протоколы 21.7-21.9) и определить по
кривой lag-период, время удвоения и плотность насы¬
щения (плотность клеток при выходе кривой на плато).
Длинный lag-период означает, что культура должна
адаптироваться к сыворотке; короткое время удвоения
предпочтительней, если вы хотите быстро нарастить
много клеток; высокая плотность насыщения позволит
вам вырастить больше клеток, используя данное коли¬
чество сыворотки, и поможет сэкономить средства.
Сохранение свойств культуры клеток. Ясно,
что клетки должны и с новой сывороткой сохранять
свойства и выполнять те функции, которые от них
требуются, являются ли они ьслетками-хозяевами для
заданного вируса, продуцируют ли определенный
9.7. Другие добавки
163
клеточный продукт, должны ли дифференцироваться
или проявлять специфическую чувствительность к
данному веществу.
Стерильность. Сыворотка от поставщиков с хо¬
рошей репутацией будет протестирована, и будет ука¬
зано, что в ней отсутствуют микроорганизмы. Однако
в редких случаях образцы контаминированной сыво¬
ротки проходят контроль качества. Если сыворотка
контаминирована, это выявится при тестировании на
микоплазму (см. гл. 19.3.2)
9.6.3. Тепловая инактивация
Сыворотку инактивируют путем инкубации в течение
30 мин при 56 °С. Затем ее можно разлить на порции и
хранить при -20 °С. Исходно прогревание применялось
для инактивации комплемента для использования в
иммунологических анализах, но, возможно, оно вызы¬
вает и другие эффекты, пока не описанные. Часто сы¬
воротку инактивируют потому, что это было указано в
одном из предыдущих протоколов, причем нет никаких
конкретных свидетельств, что инактивация принесет
какую-либо пользу. Утверждения, что тепловая ина¬
ктивация элиминирует микоплазму по-видимому, не¬
обоснованны, хотя тепловая обработка может снизить
титр некоторых видов микоплазмы.
9.7. ДРУГИЕ ДОБАВКИ
Кроме сыворотки по традиции в качестве добавок к
культуральным средам используют тканевые экстракты
и гидролизаты.
9.7.1. Гидролизат аминокислот
Многие добавки этого типа ведут свое происхождение
из микробиологии (автоклавируемые бульоны). Бакто-
пептон, триптоза и гидролизат лактальбумина (Difco-
B-D Biosciences) представляют собой протеолитические
гидролизаты бычьего сердца или лактальбумина и
содержат главным образом аминокислоты и малые пеп¬
тиды. Бактопептон и триптоза могут также содержать
нуклеозиды и другие термостабильные компоненты
тканей, такие как жирные кислоты и углеводы.
9.7.2. Эмбриональный экстракт
Эмбриональный экстракт представляет собой грубый
гомогенат 10-дневного эмбриона цыпленка, очищенный
центрифугированием (см. Приложение I). Coon и Cahn
[1966] фракционировали грубый экстракт и получили
фракции с высоким и низким молекулярным весом.
Низкомолекулярная фракция стимулировала проли¬
ферацию клеток, в то время как высокомолекулярная
фракция индуцировала пигментацию и дифферен-
цировку в хрящ. Хотя Coon и Cahn не охарактеризо¬
вали полностью эти фракции, более поздние данные
позволили предположить, что в низкомолекулярной
фракции, по-видимому, содержатся белковые факторы
роста, а в высокомолекулярной фракции — протеогли-
каны и другие компоненты матрикса.
Эмбриональный экстракт исходно использовали в
качестве добавки к плазме (см. разд. 3.7,12.3.1) для сти¬
муляции миграции клеток из эксплантата, и до сих пор
его используют в методиках выращивания некоторых
органных культур (см. разд. 25.2).
9.7.3. Кондиционированная
среда
Puck и Marcus [1955] обнаружили, что выживаемость
культуры клеток, растущих в низкой плотности, можно
повысить, выращивая клетки на фидерном слое (см.
разд. 14.2.3). Этот эффект, по-видимому, является
комбинацией ряда событий, включающих кондици¬
онирование субстрата и кондиционирование среды
посредством секреции в среду малых молекул мета¬
болитов и факторов роста [Takahashi & Okada, 1970].
Hauschka и Konigsberg [1966] показали, что кондици¬
онирование культуральной среды, необходимое для
роста и дифференцировки миобластов, происходит
благодаря синтезу коллагена фидерными клетками.
Использование фидерных слоев и кондиционирован¬
ной эмбриональными фибробластами или другими
клетками среды сохранилось до сих пор при культи¬
вировании трудно растущих клеток (см., например,
Stampfer et al., 1980).
На основании экспериментов по выделению актив¬
ных фракций из кондиционированной среды, были вы¬
двинуты оригинальные гипотезы, возможно, недалекие
от правильной интерпретации. Кондиционированная
среда содержит как компоненты, модифицирующие
субстрат, такие как коллаген, фибронектин и проте-
огликаны, так и факторы роста, такие как FGF, инсу¬
линоподобные факторы роста (IGF-I и — II), PDGF
и некоторые другие (см. табл. 10.3), а, кроме того, как
предполагалось ранее, промежуточные метаболиты.
Однако, при использовании кондиционированной
среды, появляются неизвестные факторы, поэтому она
должна быть удалена после того, как будут определены
ее активные компоненты.
Глава 10
БЕССЫВОРОТОЧНЫЕ СРЕДЫ
Хотя многие линии клеток все еще культивируют в
среде с добавлением сыворотки, есть много примеров
культивирования клеток в бессывороточных средах
(табл. 10.1,10.2). Необходимость 1) стандартизировать
среды для работы в разных лабораториях, 2) обеспе¬
чить специализированные среды для специфических
типов клеток и 3) удалить природные соединения, ко¬
личество и состав которых могут варьировать, привела
к разработке более сложных сред, таких как среда Ml99
[Morgan et al., 1950], CMRL 1066 [Parker et al., 1957],
NCTC109 [Evans et al., 1956], MB 572/1 [Waymouth,
1959], NCTC135 [Evans & Bryant, 1965], [Birch and Pirt,
1971] для культур фибробластов мыши L929, а также
среды F10 [Наш, 1963] и F12 [Наш, 1965] для клони¬
рования клеток яичника китайского хомячка (СНО).
Бессывороточные среды также были разработаны для
клеток карциномы шейки матки человека HeLa [Blaker
et al., 1971; Higuchi, 1977].
Для адаптации постоянных линий клеток к куль¬
тивированию в отсутствие сыворотки может потребо¬
ваться проведение селекции клеток. Тем не менее при¬
менение сред серии MCDB [Наm & McKeehan, 1978;
см. также табл. 10.1], среды на основе DMEM/F12-B
модификации Sato [Barnes & Sato, 1980] и других сред
на основе RPMI 1640 [Carney et al., 1981; Brower et al.,
1986] (см. также табл. 10.2) показало, что сыворотка мо¬
жет быть удалена из среды полностью или количество
ее может быть значительно снижено без видимой
селекции клеток в том случае, если в среду добавлены
необходимые питательные вещества и гормоны [Barnes
et al., 1984a-d; Cartwright & Shah, 1994; Mather, 1998].
Это также обеспечивает избирательные условия для
первичной культуры особых типов клеток. Среды
специального состава (например, MCDB 110 [Bettger
et al., 1981]) были разработаны для культивирования
фибробластов человека [Ham, 1984], многих нормаль¬
ных и опухолевых клеток человека и мыши [Barnes &
Sato, 1980], лимфобластоидных линий клеток [Iscove &
Melchers, 1978], а также некоторых первичных культур
[Mather & Sato, 1979а, b; Sundqvist et al., 1991; Gupta et
al., 1997; Keen & Rapson, 1995; Vonen et al., 1992] в отсут¬
ствие сыворотки. В некоторых случаях в среду добавля¬
ли необходимые белки [Tsao et al., 1982; Benders et al.,
1991]. Список типов клеток, которые выращивают на
бессывороточных средах, весьма велик, а многие среды
можно купить у фирм-производителей (см. Приложе¬
ние И). Кроме того, при производстве биофармацев-
тических препаратов, при очистке целевого продукта
необходимо удалять белки животного происхождения.
Поэтому, в целях безопасности [Merten, 1999], были
разработаны среды для постоянных клеточных линий,
таких как СНО, и гибридом [Froud, 1999; Ikonomou et
al., 2003; Shah, 1999].
10.1. НЕДОСТАТКИ СЫВОРОТКИ
Использование сыворотки в среде имеет ряд недо¬
статков.
Физиологическая вариабельность. Известны ос¬
новные компоненты сыворотки, такие как альбумин и
трансферрин, но сыворотка также содержит большое
количество минорных компонентов, которые могут
в значительной степени влиять на рост клеток (см.
табл. 9.5). Эти компоненты включают питательные
вещества (аминокислоты, нуклеозиды, сахара и т.д.),
белковые факторы роста, гормоны, неорганические со¬
единения и липиды, концентрация и действие которых
полностью не определены.
Срок хранения и постоянство состава. Состав сыво¬
ротки варьирует от партии к партии, и, в лучшем случае,
качество партии сохраняется в течение года, причем
в течение этого срока возможно ухудшение качества.
Далее сыворотку следует заменить другой партией, при
этом выбирается наиболее сходная, но она никогда не
будет идентичной сыворотке из первой партии.
Контроль качества. При использовании сыворотки
из другой партии необходимо проведение всесторон¬
него тестирования для того, чтобы убедиться, что по
своим свойствам сыворотка из новой партии макси¬
мально близка к ранее используемой. Тестирование
проводится на нескольких линиях клеток (исследуются
параметры роста, эффективность посева и специфиче¬
ские свойства клеток; см. разд. 9.6.2).
Специфичность. Если в работе используются не¬
сколько разных типов клеток, для каждого типа может
потребоваться отдельная партия сыворотки, таким
образом одновременно необходимо зарезервировать
несколько партий. При сокультивировании клеток
разных типов могут возникнуть большие проблемы.
Доступность. Время от времени поставки сыворот¬
ки прекращаются из-за засух в областях, где разводят
крупный рогатый скот, болезней животных, или по эко¬
номическим или политическим причинам. Такие про-
10.1. Недостатки сыворотки
165
Таблица 10.1. Примеры составов бессывороточных сред
MCDB
MCDB
MCDB
MCDB
MCDB
MCDB
WAJC
MCDB
Среда
110
131
170
202
302
402
404
153
Iscove’s
LHC-9
Тип клеток
Фибро-
Эндо¬
Эпите¬
Эмбрио¬
Клетки
Клетки
Эпителий
Kepa-
Клетки
Брон¬
бласты
телий
лий мо¬
нальные
CHO
3T3
предста¬
ТИНО-
лимфо¬
хиаль¬
легкого
сосудов
лочной
фибро-
тельной
ЦИТЫ
идного
ный
чело¬
чело¬
железы
бласты
железы1
проис¬
эпите¬
века
века
цыпленка
хожде¬
ния
лий
Ссылки
Bettger
Knedler
Ham¬
McKeehan
Hamil¬
Shipley
McK¬
Peehl
Iscove &
Lech-
et al.y
& Ham,
mond et
& Ham,
ton &
& Ham,
eehan et
& Ham,
Melchers,
ner &
Bettger
1987;
al., 1984
1976b
Ham,
1983
al., 1984;
1980
1978
LaVeck,
et al.,
Gupta et
1977
Chap-
1985
1981
al., 1997
roniere
& McK¬
eehan,
1986
Компоненты
Мол.
вес
Аминокислоты
L-аланин
89
1.0E-04
3.0E-05
1.0E-04
1.0E-04
1.0E-04
1.0E-04
2.8E-04
1.0E-04
L-аргинин
211
1.0E-03
3.0E-04
3.0E-04
3.0E-04
1.0E-03
3.0E-04
1.0E-03
1.0E-03
4.0E-04
2.0E-03
L-аспарагин
132
1.0E-04
1.0E-04
1.0E-03
1.0E-03
1.1E-04
1.0E-04
1.0E-04
1.0E-04
1.9E-04
1.0E-04
L-аспарагиновая кис¬
133
1.0E-04
1.0E-04
1.0E-04
1.0E-04
1.0E-04
1.0E-05
3.0E-05
3.0E-05
2.3E-04
3.0E-05
лота
L-цистеин
176
5.0E-05
2.0E-04
7.0E-05
2.0E-04
1.0E-04
2.4E-04
2.4E-04
L-цистин
240
2.0E-04
2.0E-04
4.0E-04
2.4E-04
2.9E-04
L-глутаминовая кис¬
147
1.0E-04
3.0E-05
1.0E-04
1.0E-04
1.0E-04
1.0E-05
1.0E-04
1.0E-04
5.1E-04
1.0E-04
лота
L-глутамин
146
2.5E-03
1.0E-02
2.0E-03
1.0E-03
3.0E-03
5.0E-03
6.0E-03
6.0E-03
4.0E-03
6.0E-03
Глицин
75
3.0E-04
3.0E-05
1.0E-04
1.0E-04
1.0E-04
1.0E-04
1.0E-04
1.0E-04
4.0E-04
1.0E-04
L-гистидин
210
1.0E-04
2.0E-04
1.0E-04
1.0E-04
1.0E-04
2.0E-03
8.0E-05
8.0E-05
2.0E-04
1.6E-04
L-изолейцин
131
3.0E-05
5.0E-04
1.0E-04
1.0E-04
3.0E-05
1.0E-03
1.5E-05
1.5E-05
8.0E-04
3.0E-05
L-лейцин
131
1.0E-04
1.0E-03
3.0E-04
3.0E-04
1.0E-04
2.0E-03
5.0E-04
5.0E-04
8.0E-04
1.0E-03
L-лизин НС1
183
2.0E-04
1.0E-03
2.0E-04
2.0E-04
2.0E-04
8.0E-04
1.0E-04
1.0E-04
8.0E-04
2.0E-04
L-метионин
149
3.0E-05
1.0E-04
3.0E-05
3.0E-05
3.0E-05
2.0E-04
3.0E-05
3.0E-05
2.0E-04
6.0E-05
L-фенил аланин
165
3.0E-05
2.0E-04
3.0E-05
3.0E-05
3.0E-05
3.0E-04
3.0E-05
3.0E-05
4.0E-04
6.0E-05
L-пролин
115
3.0E-04
1.0E-04
5.0E-05
5.0E-05
3.0E-04
3.0E-04
3.0E-04
3.5E-04
3.0E-04
L-серин
105
1.0E-04
3.0E-04
3.0E-04
3.0E-04
1.0E-04
1.0E-04
6.0E-04
6.0E-04
4.0E-04
1.2E-03
L-треонин
119
1.0E-04
1.0E-04
3.0E-04
3.0E-04
1.0E-04
5.0E-04
1.0E-04
1.0E-04
8.0E-04
2.0E-04
L-триптофан
204
1.0E-05
2.0E-05
3.0E-05
3.0E-05
1.0E-05
1.0E-05
1.5E-05
1.5E-05
7.8E-05
3.0E-05
L-тирозин
181
3.5E-05
1.0E-04
5.0E-05
5.0E-05
4.4E-05
2.0E-04
1.5E-05
1.5E-05
4.6E-04
3.0E-05
L-валин
117
1.0E-04
1.0E-03
3.0E-04
3.0E-04
1.0E-04
2.0E-03
3.0E-04
3.0E-04
8.0E-04
6.0E-04
Витамины
Биотин
244
3.0E-08
3.0E-08
3.0E-08
3.0E-08
3.0E-08
3.0E-08
6.0E-08
6.0E-08
5.3E-08
6.0E-08
Холин хлорид
140
1.0E-04
1.0E-04
1.0E-04
1.0E-04
1.0E-04
1.0E-04
1.0E-04
2.0E-05
2.0E-04
Фолиевая кислота
441
1.0E-04
3.0E-06
1.8E-06
1.8E-06
9.1E-06
1.8E-06
Фолиновая кислота
512
1.0E-09
1.0E-06
1.0E-08
1.0E-08
1.0E-06
Мио-Инозитол
180
1.0E-04
4.0E-05
1.0E-04
1.0E-04
1.0E-04
4.0E-05
1.0E-04
1.0E-04
4.0E-05
1.0E-04
Никотинамид
122
5.0E-05
5.0E-05
5.0E-05
5.0E-05
3.0E-07
5.0E-05
3.0E-07
3.0E-07
3.3E-05
3.0E-07
Пантотенат
238
1.0E-06
5.0E-05
1.0E-06
1.0E-06
1.0E-06
5.0E-05
1.0E-06
1.0E-06
1.7E-05
1.0E-06
Пиридоксаль НС1
204
2.0E-05
Пиридоксин НС1
206
3.0E-07
1.0E-05
3.0E-07
3.0E-07
3.0E-07
1.0E-04
3.0E-07
3.0E-07
3.0E-07
Рибофлавин
376
3.0E-07
1.0E-08
3.0E-07
3.0E-07
1.0E-07
1.0E-06
1.0E-07
1.0E-07
1.1E-06
1.0E-07
Тиамин НС1
337
1.0E-06
1.0E-05
1.0E-06
1.0E-06
1.0E-06
1.0E-04
1.0E-06
1.0E-06
1.2E-05
1.0E-06
а-токоферол
430
1.4E-07
Ретиноевая кислота
300
3.3E-07
Ретинол ацетат
329
4.2E-07
Витамин В12
1355
1.0E-07
1.0E-08
1.0E-07
1.0E-07
1.0E-07
1.0E-08
3.0E-07
3.0E-07
9.6E-09
3.0E-07
Антиоксиданты
Дитиотреитол
154
6.5E-06
Глутатион
307
6.5E-07
166
ГЛАВА 10. БЕССЫВОРОТОЧНЫЕ СРЕДЫ
Продолжение табл. 10.1
MCDB
MCDB
MCDB
MCDB
MCDB
MCDB
WAJC
MCDB
Среда
110
131
170
202
302
402
404
153
Iscove’s
LHC-9
Тип клеток
Фибро¬
Эндо¬
Эпите¬
Эмбрио¬
Клетки
Клетки
Эпителий
Kepa-
Клетки
Брон¬
бласты
телий
лий мо¬
нальные
CHO
ЗТЗ
предста¬
ТИНО-
лимфо¬
хиаль¬
легкого
сосудов
лочной
фибро¬
тельной
ЦИТЫ
идного
ный
чело¬
чело¬
железы
бласты
железы1
проис¬
эпите¬
века
века
цыпленка
хожде¬
ния
лий
Ссылки
Bettger
Knedler
Ham¬
McKeehan
Hamil¬
Shipley
McK¬
Peehl
Iscove &
Lech-
et al.y
& Ham,
mond et
& Ham,
ton &
& Ham,
eehan et
& Ham,
Melchers,
ner &
Bettger
1987;
al., 1984
1976b
Ham,
1983
al., 1984;
1980
1978
LaVeck,
et al.,
Gupta et
1977
Chap-
1985
1981
al., 1997
roniere
& McK¬
eehan,
1986
Компоненты
Мол.
вес
Неорганические соли
СаС12
147
1.0E-03
1.6E-03
2.0E-03
2.0E-03
6.0E-04
1.6E-03
1.3E-04
3.0E-05
1.5E-03
1.1E-04
КС1
75
5.0E-03
4.0E-03
3.0E-03
3.0E-03
3.0E-03
4.0E-03
1.5E-03
1.5E-03
4.4E-03
1.5E-03
KN03
160
7.5E-07
MgCl2
203
6.0E-04
Mgso4
247
1.0E-03
1.0E-02
1.5E-03
8.0E-04
8.1E-04
NaCl
58
1.1E-01
1.1E-01
1.2E-01
1.2E-01
1.2E-01
7.7E-02
NaHC03
84
1.4E-02
1.4E-02
1.4E-02
3.6E-02
na2hpo4
120
3.0E-03
5.9E-04
5.0E-04
5.0E-04
2.0E-03
1.0E-03
Микроэлементы
CuS04-5H20
160
1.0E-09
7.5E-09
1.0E-09
5.0E-09
1.0E-09
FeS04
278
5.0E-06
1.0E-06
5.0E-06
1.0E-06
5.0E-06
MnS04H20
169
1.0E-09
1.2E-09
5.0E-10
1.0E-09
1.0E-09
(NH4)6Mo7024
1236
1.0E-09
3.0E-09
1.0E-09
3.0E-09
1.0E-09
NiCl2
238
5.0E-10
4.2E-10
5.0E-12
3.0E-10
5.0E-10
H2Se03
129
3.0E-08
3.0E-08
1.0E-08
3.0E-08
1.0E-07
Na2Si03
122
5.0E-07
2.3E-05
5.0E-07
1.0E-05
5.0E-07
SnCl2
190
5.0E-10
5.0E-12
5.0E-10
nh4vo3
117
5.0E-09
5.1E-09
5.0E-09
5.0E-09
5.0E-09
ZnS04 • 7H20
288
5.0E-07
1.0E-09
1.0E-07
1.0E-06
5.0E-07
Липиды и предшественники
Холестерол
387
7.6E-06
Этаноламин
61
Линолевая кислота
280
2.0E-07
3.0E-07
Липоевая кислота
206
1.0E-08
1.0E-08
1.0E-08
1.0E-08
1.0E-06
Фосфоэтаноламин
141
Соевый лецитин,
6
мкг/мл
Соевый липид,
50
мкг/мл
Сфингомиелин,
1
мкг/мл
Гормоны и факторы роста
EGF, нг/мл
30
25
Эпинефрин
183
Г идрокортизон2
362
5.0E-07
5.0E-07
Инсулин, мкг/мл
1
10
Пролактин, мкг/мл
1
PGE!
355
2.5E-08
Трийодтиронин
673
Нуклеозиды и т. д.
Аденин S04
184
1.0E-05
1.0E-06
1.0E-06
1.0E-06
1.8E-04
Гипоксантин
136
10.1. Недостатки сыворотки
167
Окончание табл. 10.1
MCDB
MCDB
MCDB
MCDB
MCDB
MCDB
WAJC
MCDB
Среда
110
131
170
202
302
402
404
153
Iscove’s
LHC-9
Тип клеток
Фибро¬
Эндо¬
Эпите¬
Эмбрио¬
Клетки
Клетки
Эпителий
Кера¬
Клетки
Брон¬
бласты
телий
лий мо¬
нальные
СНО
3T3
предста¬
тино¬
лимфо¬
хиаль¬
легкого
сосудов
лочной
фибро¬
тельной
циты
идного
ный
чело¬
чело¬
железы
бласты
железы1
проис¬
эпите¬
века
века
цыпленка
хожде¬
ния
лий
Ссылки
Bettger
Knedler
Ham¬
McKeehan
Hamil¬
Shipley
McK¬
Peehl
lscove&
Lech-
et al.,
& Ham,
mond et
& Ham,
ton &
& Ham,
eehan et
& Ham,
Melchers,
ner &
Bettger
1987;
al., 1984
1976b
Ham,
1983
al., 1984;
1980
1978
LaVeck,
et al.,
Gupta et
1977
Chap-
1985
1981
al., 1997
roniere
& McK¬
eehan,
1986
Компоненты
Мол.
вес
Тимидин
242
3.0E-07
1.0E-07
3.0E-07
1.0E-06
3.0E-06
Компоненты энергетического обмена
D-глюкоза
180
4.0E-03
5.6E-03
8.0E-03
5.5E-03
6.0E-03
2.5E-02
Фосфоенолпируват
190
1.0E-05
Ацетат натрия ЗН20
136
1.0E
3.7E-03
3.7E-03
Пируват натрия
110
1.0E-03
1.0E-03
5.0E-04
1.1E+02
5.0E-04
Другие компоненты
Холерный токсин
2.0E-10
HEPES, натриевая
3.0E-02
3.0E-02
2.8E-02
соль
Феноловый красный
3.3E-06
3.3E-05
3.3E-06
3.3E-06
3.3E-05
3.3E-05
Путресцин 2НС1
1.0E-09
1.2E-09
1.0E-09
1.0E-06
1.0E-09
1.0E-06
Белковые добавки
ВРЕ, мкг белка/мл3
70
25
BSA, мг/мл
Диализованная FBS,
мкгР/мл
Трансферрин, Fe3+
насыщенный,
мкг/мл
Газовая фаза
С02
2%
5%
2%
5%
5%
5%
1 Смотри также полную PFMR-4A [РееЫ, 2002].
2 Можно использовать растворимые аналоги гидрокортизона, такие как дексаметазон.
3 ВРЕ можно заменить овечьим пролактином. Концентрации в основном указаны молярные, использован компьютерный стиль
обозначений, например З.ОЕ-2 = 3.0x10 = 30 мМ. Для основных компонентов указан молекулярный вес, хотя некоторые из
них присутствуют в виде солей или в гидратированной форме. Сокращения: ВРЕ — экстракт гипофиза быка, BSA — бычий
сывороточный альбумин, EGF — эпидермальный фактор роста, FBS - эмбриональная бычья сыворотка. См. ссылки в тексте.
блемы могут возникнуть в любое время, ограничивая
и количество сыворотки, и выбор среди разных партий
сыворотки. Особенно острой эта проблема становится в
условиях высокого спроса. В настоящее время спрос на
сыворотку повышенный и будет возрастать до тех пор,
пока большинство покупателей сыворотки не начнут
использовать в своей работе бессывороточные среды.
Хотя исследовательская лаборатория может резервиро¬
вать в среднем 100-200 л сыворотки в год, коммерческая
биотехнологическая лаборатория может использовать
такое количество (или больше) за неделю.
Последующая обработка продукта. Присутствие
сыворотки создает большие трудности при очистке
продукта и даже может препятствовать фармацевти¬
ческому одобрению продукта.
Контаминация. Сыворотка часто контаминиро-
вана вирусами, многие из которых хотя и не опасны
для культуры клеток, но являются дополнительным
неизвестным фактором, которые невозможно контро¬
лировать [Merten, 1999]. К счастью, усовершенство¬
вание методов стерилизации сыворотки практически
исключили риск заражения микоплазмой из сыворотки
наиболее уважаемых поставщиков, но вирусная инфек¬
ция остается проблемой, несмотря на утверждения, что
некоторые фильтры могут задерживать вирусы (Pall
Gelman). Из-за риска распространения бычьего губ¬
168
ГЛАВА 10. БЕССЫВОРОТОЧНЫЕ СРЕДЫ
чатого энцефалита среди крупного рогатого скота, для
культур клеток и сыворотки, доставленной в Соеди¬
ненные Штаты и Австралию, требуется информация
о стране происхождения и номере серии сыворотки.
Сыворотка крупного рогатого скота из Новой Зелан¬
дии, по-видимому, контаминирована эндогенными ви¬
русами в значительно меньшей степени, так как многие
вирусы крупного рогатого скота Европы и Северной
Америки в Новой Зеландии не обнаружены.
Стоимость. Стоимость часто называют одним из
недостатков применения сыворотки. Безусловно, сыво¬
ротка является самой дорогой составляющей в бутыли
со средой (она более чем в 10 раз дороже всех остальных
химических компонентов), но при замене сыворотки
компонентами определенного химического состава стои¬
мость среды может возрасти даже больше, чем в случае
применения сыворотки. Однако, так как потребность в
таких компонентах, как трансферрин, селен, инсулин
и т.д., возрастает, цена, по-видимому, будет снижаться
с увеличением рынка сбыта, и бессывороточные среды
станут относительно дешевле. Доступность рекомбинан¬
тных факторов роста вместе с увеличением рыночного
спроса может помочь снизить их стоимость.
Ингибиторы роста. Кроме стимуляторов роста сы¬
воротка содержит ингибиторы роста, и хотя стимули¬
рующий рост эффект сыворотки обычно преобладает,
конечный эффект сыворотки — это непредсказуемая
комбинация как ингибирования, так и стимуляции
роста. Хотя вещества, такие как PDGF, могут быть
митогенами для фибробластов, другие компоненты
сыворотки могут быть цитостатиками. Гидрокорти¬
зон, который присутствует в фетальной сыворотке
в концентрации приблизительно 1х10~8М, является
цитостатиком для многих типов клеток, таких как глия
[Guner et al., 1977] и легочный эпителий [McLean et al.,
1986], растущих в высокой плотности (хотя он может
быть митогеном для клеток в низкой плотности), и
TGF-P, продуцируемый тромбоцитами, является ци¬
тостатиком для многих эпителиальных клеток.
Стандартизация. Вариации в качестве сыворотки
от партии к партии затрудняют стандартизацию экспе¬
риментальных и промышленных протоколов как в раз¬
личное время, так и между разными лабораториями.
10.2. ПРЕИМУЩЕСТВА
БЕССЫВОРОТОЧНЫХ СРЕД
Все перечисленные выше проблемы могут быть разре¬
шены путем удаления сыворотки и других продуктов
животного происхождения из сыворотки. Кроме того, бес¬
сывороточные среды имеют два главных преимущества.
10.2.1. Селективные среды
Одно из главных преимуществ, которое дают бессыво¬
роточные среды, — это возможность приготовления се¬
лективных сред для индивидуальных типов клеток (см.
табл. 10.1 и 10.2). Так рост фибробластов в культурах
клеток молочной железы и кожи можно ингибировать,
используя среду MCDB 170 [Hammond et al., 1984] and
153 [Peehl and Ham, 1980], меланоциты можно культи¬
вировать в отсутствие фибробластов и кератиноцитов
[Naeyaert et al., 1991]. Подобрав правильные факторы
роста или группу факторов, можно выделить гематопо-
этические клетки определенного происхождения и даже
на одной стадии развития (см. разд. 23.4). Многие из
этих селективных сред сейчас коммерчески доступны
вместе с культурами клеток, для которых эти среды
подобраны (см. табл. 10.4).
10.2.2. Регуляция пролиферации
и дифференцировки
Бессывороточные среды позволяют не только создать
оптимальные условия для индивидуальных типов
клеток, но и дают возможность создавать условия как
для роста клеток, так и для их дифференцировки путем
изменения концентрации и типов факторов роста и
других индукторов роста и дифференцировки.
10.3. НЕДОСТАТКИ БЕССЫВОРОТОЧНЫХ
СРЕД
У бессывороточных сред имеются и недостатки:
1) Многообразие сред. Каждый тип клеток, по-ви¬
димому, требует среду определенного состава, и
культуры клеток, полученные из опухолей, могут
проявлять разные требования к составу среды,
даже если они были получены из опухолей одного
типа. Хотя эта степень специфичности может быть
преимуществом, позволяющим выделять специфи¬
ческие типы клеток, тем не менее в лабораториях
существует проблема при культивировании клеточ¬
ных линий разного происхождения.
2) Селективность. К сожалению, переход к бессыво-
роточным средам, как бы это ни было желатель¬
ным, не является таким простым, как это кажется.
Применение некоторых сред может приводить к
появлению сублиний, которые не являются типич¬
ными для основной популяции клеток. Даже для
постоянных клеточных линий может потребовать¬
ся дополнительная селекция. Клетки на разных
стадиях развития (например, стволовые клетки и
коммитированные клетки-предшественники) могут
нуждаться в средах разного состава, в особенности
факторов роста и цитокинов.
3) Чистота реагентов. Отсутствие в среде сыворотки
значительно повышает требования к чистоте реаген¬
тов и воды, а также к чистоте аппаратуры, так как
сыворотка обладает защитным и антитоксичным
действием за счет некоторых белков. Хотя исклю¬
чение этого защитного действия, без сомнения,
желательно, но достичь его при использовании
сыворотки не всегда удается.
10.3. Недостатки бессывороточных сред
169
Таблица 10.2. Примеры бессывороточных сред; Дополненные основные среды
Среда
HITES
ACL-3
N3
G3
K-1
K-2
Ссылки
Carney et
Masui et al.,
Masui et
Botten-
Michler-
Taub,
Taub,
Chopra &
Robert¬
Naey-
al., 1981
1986
al., 1986b
stein,
1984
Stuke
& Bot-
tenstein,
1982
1984
1984
Xue-Hu,
1993
son &
Robert¬
son, 1995
aertet
al., 1991
Тип клеток
Мелко¬
Немелко¬
Адено-
Нейроб-
Клетки
MDCK
LLC-PK
Около¬
Предста¬
Мела¬
клеточная
клеточная
карци¬
ластома
глии
(почка
(почка
ушная
тельная
ноциты
карцинома
карцинома
нома
чело¬
крысы
собаки)
свиньи)
железа
железа
челове¬
легкого
легкого
легкого
века
человека
человека
ка
человека
человека
человека
LA-N-1
Основная среда
RPMI
1640
RPMI
1640
DMEM/
F12
DMEM/
F12
DMEM
DMEM/ DMEM/
F12 F12
MCDB
153
ссМЕМ/
F12
M199
Добавки
Arg VP,
10
мкЕд/мл
ВРЕ, мкг бел¬
25
25
ка/мл
BSA, мкг/мл
5.0
0.3
EGF, нг/мл
10
10
10
1
FGF-2 (основ¬
ной FGF)
10
нг/мл
Na2Se03
3.0Е-08
2.5Е-08
2.5Е-08
3.0Е-08
Гидрокортизон
1.0Е-08
5.0Е-08
5.0E-08
2.0E-07
1.4E-06
1.0Е-06
1.4E-06
Глутамин (до¬
2.0Е-03
2.0Е-03
полнительно)
Глюкагон,
0.2
мкг/мл
Инсулин,
5.0
20.0
5.0
5.0
0.5
5.0
25.0
5.0
5.0
10.0
мкг/мл
ПируватЫа (до¬
5.0Е-04
полнительно)
Прогестерон
2.0Е-08
2.0Е-07
Пролактин
Простагландин
Е1
7.0E-08
Путресцин
1.0Е-04
1.0Е-07
Тироксин
4.5E-07
Трансферрин,
насыщенный
100
10
50
100
5
10
5
10
Fe3+ мкг/мл
Т рийодтиронин
1.0Е-10
5.0Е-10
4.9E-07
5.0E-12
1.0E-09
1.0E-09
Фосфоэтанол-
1.0Е-04
амин
Холерный
1.0E-09
токсин
Холестерол
1.0E-08
Эпинефрин
Эстрадиол
1.0Е-08
Этаноламин
5.0Е-07
1.0E-03
_2
Большинство концентраций молярные, использован компьютерный стиль обозначений, например З.ОЕ-2 = 3.0x10 = 30 мМ.
Для основных компонентов указан молекулярный вес, хотя в некоторых составах используются соли в гидратированной форме,
молярность остается прежней. Единицы размерности указаны в колонке с наименованиями компонентов в тех случаях, где не
используется молярная концентрация. Могут быть использованы растворимые аналоги гидрокортизона, такие как дексаметазон.
Arg VP — аргинин-вазопрессин; ВРЕ — экстракт гипофиза быка, BSA — бычий сывороточный альбумин, EGF — эпидермальный
фактор роста, FBS — эмбриональная бычья сыворотка, FGF — фактор роста фибробластов.
170
ГЛАВА 10. БЕССЫВОРОТОЧНЫЕ СРЕДЫ
4) Пролиферация клеток. На бессывороточных средах
клетки часто растут медленнее, и перевиваемые
линии клеток дают меньше поколений.
5) Доступность. Несмотря на постоянное усовершен¬
ствование, возможность использования бессыворо¬
точные среды контролируемого должным образом
качества значительно ограничена, а стоят такие
среды значительно дороже традиционных сред.
10.4. ЗАМЕНА СЫВОРОТКИ
Необходимые факторы сыворотки уже были описаны
ранее (см. гл. 9.6). К ним относятся: 1) факторы ад¬
гезии, такие как фибронектин; 2) пептиды, такие как
инсулин, PDGF и TGF-P, которые регулируют рост
и дифференцировку; 3) необходимые питательные
компоненты, такие как неорганические соединения,
витамины, жирные кислоты и их метаболиты и 4) гор¬
моны, такие как инсулин, гидрокортизон, эстрогены
и трийодтиронин, которые регулируют мембранный
транспорт, фенотипический статус и строение клеточ¬
ной поверхности. Некоторые из них входят в состав
бессывороточных сред, те же, которые не входят, иногда
требуется добавлять дополнительно (иногда требуется
оптимизация).
10.4.1. Компоненты для бессывороточного
культивирования
Факторы адгезии. В отсутствие сыворотки может
возникнуть необходимость обработать поверх¬
ность пластика фибронектином (25-50 мкг/мл)
или ламинином (1-5 мкг/мл), которые добавляют
непосредственно в среду [Barnes et al., 1984а; см.
протокол 23.9]. Было показано, что предваритель¬
ная обработка пластика поли-L-лизином (1 мг/мл)
повышает выживаемость диплоидных фибробластов
человека [McKeehan & Ham, 1976а; see also Section 8.4
and Barnes et al., 1984a].
Ингибиторы протеаз. После пересева клеток с
помощью трипсина добавление сыворотки ингибиру¬
ет протеолитическую активность остатков трипсина.
Следовательно, при использовании бессывороточных
сред, необходимо добавлять ингибиторы протеаз,
такие как ингибитор трипсина из сои или апротинин
(0,1 мг/мл, Sigma) при пересеве клеток. Кроме того,
неочищенный трипсин представляет собой сложную
смесь протеаз и для ингибирования некоторых из этих
протеаз могут потребоваться и другие ингибиторы.
Поэтому предпочтительней использовать очищенный
трипсин (например, Sigma Gr. III) и далее применять
ингибитор трипсина. Альтернативой применения
ингибиторов может служить однократная промывка
клеток и осаждение их центрифугированием для уда¬
ления трипсина.
Трипсин и другие протеазы. При трипсинизации
клеток, растущих на бессывороточной среде, может
потребоваться особая тщательность, так как клетки
становятся более уязвимыми. Возможно, придется
использовать раствор кристаллического очищенного
трипсина и охладить его до 4 °С, чтобы уменьшить
повреждения клеток [McKeehan, 1977]. Для того чтобы
избежать контакта клеток с продуктами животного
происхождения, можно использовать альтернативные
источники протеаз. Очищенный свиной трипсин мож¬
но заменить рекомбинантным трипсином (TrypLE™,
Invitrogen; TrypZean, Sigma). Также можно использо¬
вать протеазы не животного происхождения (напри¬
мер, Accutase™ or Accumax™, Sigma). Проназа, Диспаза
и коллагеназа являются бактериальными протеазами,
и для их удаления требуется центрифугирование.
Возможно, проназу можно инактивировать просто
разведением в бессывороточной среде без последую¬
щей нейтрализации [McKeehan, личное сообщение].
Проназа очень эффективна, но может проявлять ток¬
сичность для некоторых клеток; диспаза и коллагена¬
за не позволяют получить суспензию из одиночных
клеток при их использовании для рекультивирования
эпителиальных клеток.
10.4.2. Гормоны
К гормонам, используемым для замены сыворотки,
относят гормон роста (соматотропин) в концентрации
50 нг/мл, инсулин — 1-10 Ед/мл, которые повышают
эффективность посева ряда клеток различных типов,
и гидрокортизон, который улучшает эффективность
клонирования глиальных клеток и фибробластов (см.
табл. 10.1 и 10.2 и разд. 14.2.1) и, как было показано,
необходим для культивирования эпидермальных
кератиноцитов и некоторых других эпителиальных
клеток (см. разд. 23.1.1). Barnes и Sato [1980] писали,
что для клеток MDCK (почка собаки) необходимой
добавкой является трийодтиронин (Т3,10 пМ). Также
он может применяться при культивировании легочного
эпителия [Lechner & LaVeck, 1985; Masui et al., 1986b].
Было показано, что для культивирования эпителия
молочной железы необходимы различные комбинации
эстрогенов, андрогенов или прогестерона с гидрокор¬
тизоном и пролактином (концентрация около 10 нМ)
[Klevjer-Anderson & Buehring, 1980; Hammond et al.,
1984; Strange et al., 1991; Lee et al., 1996].
Было показано, что другие гормоны, действие ко¬
торых обычно не связывают с клеточной активностью,
могут быть эффективны в условиях отсутствия сыво¬
ротки, например фолликулостимулирующий гормон
(FSH) эффективен в отношении меланомы мыши В16
[Barnes and Sato, 1980]. Возможно, имеется гомология
определенных последовательностей между стимули¬
рующими рост клеток полипептидами и хорошо изу¬
ченными пептидными гормонами. Другим возможным
вариантом может быть расщепление (или протеолиз)
некоторых больших белков или полипептидов с вы¬
свобождением активных пептидов с совсем другими
функциями.
10.5. Выбор бессывороточной среды
171
10.4.3. Факторы роста
Семейство полипептидов, которые, как было обнару¬
жено, могут проявлять митогенные свойства in vitro,
довольно обширно (табл. 10.3) и включает в себя гепа-
рин-связывающие факторы роста (включая семейство
FGF), EGF, PDGF [Barnes et al., 1984а, с], IGF-I и - II
и интерлейкины [Thomson, 1991], которые проявляют
активность в диапазоне концентраций 1-10 нг/мл.
Факторы роста и цитокины обладают широкой спе¬
цифичностью, за исключением факторов, влияющих
на гематопоэтическую систему [Barnes et al.,1984d;
см. также разд. 23.4]. Фактор роста кератиноцитов
(KGF) [Aaronson et al., 1991] проявляет активность не
только в отношении эпидермальных кератиноцитов,
но также индуцирует пролиферацию и дифференци-
ровку эпителия предстательной железы [Planz et al.,
1998; Thomson et al., 1997]. Фактор роста гепатоцитов
(HGF) [Kenworthy et al., 1992] является митогеном для
гепатоцитов, а также индуцирует морфогенез почечных
канальцев. [Furue & Saito, 1997; Montesano et al., 1997;
Balkovetz & Lipschutz, 1999]. Факторы роста и цито¬
кины проявляют свою специфичность посредством
того, что их биосинтез локализован в определенных
клетках и тканях, и, таким образом, они имеют огра¬
ниченную область распространения. Большинство из
них действуют как паракринные факторы (активные в
отношении соседних клеток) и не способны к систем¬
ному распространению в крови.
Факторы роста могут действовать синергично или
аддитивно друг с другом или с другими гормонами и
паракринными факторами, такими как простагландин
F2a и гидрокортизон [Westermark & Wasteson, 1975;
Gospodarowicz, 1974]. Например, эффективность
действия интерлейкина 6(IL-6) и онкостатина М на
клетки А549 зависит от дексаметазона, синтетического
аналога гидрокортизона [McCormick et al., 1995,2000],
который индуцирует образование гепарин сульфат
протеогликана (HSPG) [Yevdokimova & Freshney,
1997]. Необходимость в гепарине или HSPG впервые
была обнаружена для FGF [Klagsbrun & Baird, 1991],
но, возможно, этот механизм является более общим.
Например, было показано, что Р-гликан учавствует в
клеточном ответе на TGF-P [Lopez-Casillas et al., 1993].
Для активации некоторых факторов роста необходим
сигнал от другого фактора роста [Phillips and Christo-
falo, 1988]; например, бомбезин не является сам по себе
митогеном для нормальных клеток, для проявления его
митогенной активности требуется одновременное или
предварительное воздействие инсулина или одного из
IGF [Aaronson et al., 1991].
10.4.4. Питательные вещества сыворотки
Железо, медь и ряд неорганических соединений входят
в состав бессывороточных сред, хотя данных о пот¬
ребности в некоторых редких соединениях пока нет.
В большинство прописей сред входит селен (Na2Se03) в
концентрации около 20 нМ. По-видимому, необходимо
включение в среды липидов или их предшественников,
таких как холин, линолевая кислота, этаноламин или
фосфоэтаноламин.
10.4.5. Белки и полиамины
Включение в состав среды белков, таких как бычий сы¬
вороточный альбумин (BSA) или тканевые экстракты,
часто способствует клеточному росту и выживаемости,
но добавляет неидентифицированные компоненты в
среду. Кроме того, при этом остается проблема случай¬
ного внесения инфекционных агентов.
К экстрактам тканей относят экстракт гипофиза
быка, используемый для культивирования керати¬
ноцитов в бессывороточной среде, но его, возможно,
можно заменить рекомбинантными факторами роста.
BSA, свободные жирные кислоты используются в кон¬
центрациях 1-10 мг/мл. Трансферрин в концентрации
около 10 нг/мл требуется как переносчик железа и так¬
же может выполнять функцию митогена. Путресцин
используется в концентрации 100 нМ.
10.4.6. Матрикс
Одним из свойств сыворотки является обеспечение
клеток белками, такими как фибронектин, которые
покрывают пластик и делают его более адгезивным (см.
разд. 10.4.1, 8.4.1). В отсутствие сыворотки пластико¬
вую подложку иногда бывает необходимо покрывать
фибронектином (см. протокол 23.9) или полилизи¬
ном [McKeehan & Ham, 1976а] (см. табл. 10.1 и 10.2 и
разд. 14.2.1).
10.5. ВЫБОР БЕССЫВОРОТОЧНОЙ
СРЕДЫ
Если причиной для использования бессывороточной
среды является задача стимулировать селективный рост
индивидуального типа клеток, то в этом случае именно
эта причина будет определять выбор среды (например,
MCDB 153 для эпидермальных кератиноцитов, LHC-9
для бронхиального эпителия, HITES для мелкокле¬
точного рака легкого, MCDB 130 для эндотелия и т.д.;
см. табл. 10.1 и 10.2). Если причина состоит попросту
в стремлении избежать использования сыворотки при
культивировании постоянных клеточных линий, таких
как клетки СНО или гибридомы, для того чтобы сни¬
зить вероятность контаминации продукта вирусными
или сывороточными белками, то в этом случае выбор
среды будет шире, и выбирать можно будет среды у
нескольких коммерческих производителей (табл. 10.4).
Если линия клеток получена от ее создателя или из
банка клеток с хорошей репутацией, поставщик обыч¬
но рекомендует подходящую среду, и единственной
причиной не использовать рекомендованную среду
172
ГЛАВА 10. БЕССЫВОРОТОЧНЫЕ СРЕДЫ
Таблица 10.3. Факторы роста и митогены
Название и синонимы
Аббревиа¬
тура
Молекулярная
масса (кДа)
Источник*
Функция
Кислый фактор роста фибробластов;
FGF-1
13 гс
Мозг быка; гипофиз
Митоген для эндотелиальных
aFGF; Гепаринсвязывающий фак¬
клеток
тор роста 1, HBGF-1; фактор роста
эндотелиальных клеток (ECGF);
фактор роста миобластов (MGF)
Активин; семейство TGF-p
гл
Гонады
Индуктор морфогенеза; стиму¬
лирует секрецию FSH
Ангиогенин
16
Фибробласты, лимфо¬
циты, эпителиальные
клетки кишечника
Ангиогенный фактор, митоген
для эндотелиальных клеток
Астроглиальный фактор 1; член се¬
AGF-1
14
Мозг
Митоген для астроглиальных
мейства кислых FGF
клеток
Астроглиальный фактор роста 2; член
AGF-2
14
Мозг
Митоген для астроглиальных
семейства основных FGF
клеток
Основной фактор роста фиброблас¬
FGF-2
13 гс
Мозг быка; гипофиз
Митоген для многих мезо-
тов; bFGF; HBGF-2; простатропин
дермальных и нейроэкто¬
дермальных клеток; ин¬
дуктор дифференцировки
адипоцитов и гранулозных
клеток яичника
Нейротрофический фактор мозга
BDNF
28
Мозг
Выживаемость нейрональных
клеток
Кахектин
TNF-oc
17
Моноциты
Катаболическое действие; ка¬
хексия; шок
Холерный токсин
XT
80-90
Холерная палочка
Митоген для некоторых нор¬
мальных эпителиальных
клеток
Цилиарный нейротрофический фак¬
CNTF
Глаз
тор; член семейства IL-6
Фактор роста эндотелия; семейство
ECGF
гл
Рекомбинантный
Митогенез эндотелия
кислого FGF
Добавки для роста эндотелия; смесь
ECGS
Гипофиз быка
Митогенез эндотелия
эндотелиальных митогенов
Эндотоксин
Бактерии
Стимулирует секрецию TNF
Эпидермальный фактор роста, уро-
EGF
6
Подчелюстные слюн¬
Активный транспорт; синтез
гастрон
ные железы мыши,
моча человека, пред¬
стательная железа
морской свинки
ДНК, РНК, белка; мито¬
ген для эпителиальных и
фибробластоидных клеток;
действует синергично с IGF-
1 и TGF-P
Эритропоэтин
EPO
34-39 гл
Околоклубочковые
клетки почки
Индукция пролиферации и
дифференцировки эритро-
идных предшественников
Фактор роста глаза 1; член семейства
EDGF-1
14
Глаз
основного FGF
Фактор роста глаза 2; член семейства
EDGF-2
14
Глаз
кислого FGF
Фактор роста фибробластов 3; про¬
FGF-3
14 гс
Опухоли молочной же¬
Митоген, индуктор морфоге¬
дукт онкогена int-2
лезы
неза, ангиогенный фактор
int-2 oncogene
Фактор роста фибробластов 4; про¬
FGF-4
14 гс
Эмбрион, опухоли
Митоген, индуктор морфоге¬
дукт онкогена hst/KS3
неза, ангиогенный фактор
oncogene
Фактор роста фибробластов 5
FGF-5
14 гс
Фибробласты, эпители¬
альные клетки, опу¬
холи F
Митоген, индуктор морфоге¬
неза, ангиогенный фактор
Фактор роста фибробластов 6; про¬
FGF-6
14 гс
Половые железы;
Митоген для фибробластов,
дукт онкогена hst-2
сердце; мышцы
индуктор морфогенеза
10.5. Выбор бессывороточной среды
173
Продолжение табл. 10.3
Название и синонимы
Аббревиа¬
тура
Молекулярная
масса (кДа)
Источник*
Функция
Фактор роста фибробластов 9
FGF-9
14 гс
Рекомбинантный
Фактор роста фибробластов 10
FGF-10
14 гс
Инвазия трофобласта; сти¬
мулирует иРА и PAI-1; ми-
тоген для альвеолярного
эпителия
Гранулоцитарный колониестиму¬
лирующий фактор; плюрипоэтин;
CFS-p
G-CSF
18-22
Стимулирует пролиферацию
предшественников грану-
лоцитов
Гранулоцитарно-макрофагальный
колониестимулирующий фактор;
CSA; человеческий CSFa
Гепаринсвязывающий EGF-подобный
фактор
factor
GM-CSF
HB-EGF
14-35 гл
Рекомбинантный
Стимулирует пролиферацию
предшественников грануло-
цитов/макрофагов
Фактор роста гепатоцитов, HBGF-8;
Фактор рассеивания
HGF
гс
Фибробласты
Стимулирует морфогенез эпи¬
телия; пролиферацию гепа¬
тоцитов
Герегулин; лиганд егЬВ2
HRG
70
Опухоли молочной же¬
лезы; рекомбинант¬
ный
Клетки молочной железы и дру¬
гие эпителиальные клетки
Фактор активации макрофагов
MAF
20-25
Активированные лим¬
фоциты
Антивирусная активность;
активация макрофагов
Ингибин; член семейства TGF-P
31 гл
Яичники
Индуктор морфогенеза; инги¬
бирует секрецию FSH
Инсулин
Инсулиноподобный фактор роста 1;
соматомедин-С; NSILA-1
Ins
IGF-1
6
Клетки Р-островков под¬
желудочной железы
Захват глюкозы и окисление;
захват аминокислот; глико-
неогенез
Опосредует действие гормона
роста на сульфатирование
хряща; инсулиноподобная
активность
Инсулиноподобный фактор роста 2;
MSA у крыс
IGF-2
7
Среда, кондициони¬
рованная клетками
BRL-3A
Опосредует эффект гормона
роста на сульфатирование
хряща; инсулиноподобная
активность
Интерферон-al; лейкоцитарный
интерферон
IFN-fll
18-20
Макрофаги
Антивирусная активность;
индуктор дифференциров¬
ки; противоопухолевая ак¬
тивность
Интерферон-а2; лейкоцитарный
интерферон
IFN-cc2
18-20
Макрофаги
Антивирусная активность;
индуктор дифференциров¬
ки; противоопухолевая ак¬
тивность
Интерферон-Р; фибробластный ин¬
терферон
IFN-pi
22-27 гл
Фибробласты
Антивирусная активность;
индуктор дифференциров¬
ки; противоопухолевая ак¬
тивность
Интерферон-Р2; фибробластный
интерферон; IL-6, BSF-2
(смотри также IL-6)
IFN-P2
22-27 гл
Активированные
Т-клетки; фибро¬
бласты; опухолевые
клетки
Индуктор дифференциров¬
ки кератиноцитов и PC 12
(смотри также IL-6)
Интерферон у ; иммунный интерфе¬
рон
IFN-y
20-25
Активированные лим¬
фоциты
Антивирусная активность; ак¬
тивация макрофагов; анти-
пролиферативное действие на
трансформированные клетки
Интерлейкин-1; фактор активации
лимфоцитов (LAF); фактор акти¬
вации В-клеток (BAF); гематопо-
этин-1
IL-1
12-18
Активированные мак¬
рофаги
Индуцирует секрецию IL-2
174
ГЛАВА 10. БЕССЫВОРОТОЧНЫЕ СРЕДЫ
Продолжение табл. 10.3
Название и синонимы
Аббревиа¬
тура
Молекулярная
масса (кДа)
Источник*
Функция
Интерлейкин-2; фактор роста Т-кле-
ток (TCGF)
IL-2
15
CD4+ve лимфоциты
(NK); мышиная
LBRM-5A4 и челове¬
ческая Jurkat FHCRC
линии клеток
Поддерживает рост активиро¬
ванных Т-клеток; стимули¬
рует клетки LAK
Интерлейкин-3; мультипотентный
колониестимулирующий фактор;
фактор роста тучных клеток
IL-3
14-28 гл
Активированные
Т-клетки; миеломо-
ноцитарная линия
клеток WEHI-3b
Поддерживает рост и диффе-
ренцировку гранулоцитов/
макрофагов
Интерлейкин-4; фактор роста В-кле-
ток; BCGF-1; BSF-1
IL-4
15-20
Активированные
CD4+ve лимфоциты
Фактор компетентности для
зрелых В-клеток; способ¬
ствует созреванию тучных
клеток (вместе с IL-3)
Интерлейкин-5; Т-клеточный заме¬
щающий фактор (TRF); фактор
дифференцировки эозинофилов
(EDF) BCGF-2
IL-5
12-18гл
Т-лимфоциты
Фактор дифференцировки
эозинофилов; фактор про¬
лиферации активированных
В-клеток
Интерлейкин-6; интерферон Р-2; фак¬
тор стимуляции В-клеток (BSF-2);
фактор стимуляции гепатоцитов;
фактор роста гибридом-плазма-
IL-6
22-27 гл
Активированные
Т-клетки
Индукция синтеза белков ост¬
рой фазы; дифференцировка
В-клеток , кератиноцитов,
РС12
цитом
Интерлейкин-7; гематопоэтический
фактор роста; лимфопоэтин 1
IL-7
15-17гл
Строма костного мозга
Фактор роста пре- и про-
В-клеток
Интерлейкин-8; моноцитарный
фактор хемотаксиса нейтрофилов
(MDNCF); фактор хемотаксиса Т-
клеток; активирующий нейтрофилы
белок (NAP-1)
IL-8
8-10 гс
Стимулированные
ЛПС моноциты; Сти¬
мулированные ФГА
лимфоциты; эндо¬
телиальные клетки;
стимулированные IL-
1 и TNF фибробласты
и кератиноциты
Фактор хемотаксиса для
нейтрофилов, базофилов и
Т-клеток
Интерлейкин-9; Р-40 человека; фак¬
тор роста Т-хелперов мыши; фактор
активации тучных клеток (MEА)
IL-9
30-40
CD4+ve Т-клетки, сти¬
мулированные анти¬
телами к CD4, ФГА
или РМА
Фактор роста Т-хелперов,
мегакариоцитов, тучных
клеток (вместе с IL-3)
Интерлейкин-10; фактор ингибирова¬
IL-10
20
Иммуносупрессор
ния синтеза цитокинов (CSIF)
Интерлейкин-11;
IL-11
21
Стимулирует пролиферацию
плазмацитомы и зависимое
от Т-клеток развитие Ig-npo-
дуцирующих В-клеток
Интерлейкин-12; фактор созревания
цитотоксических лимфоцитов
IL-12
40,35
субъединицы
Активирует рост Т-клеток и
NK; индуцирует секрецию
IFN-y
Фактор роста кератиноцитов; FGF-7
KGF
14 гс
Фибробласты
Индукция пролиферации и
дифференцировки керати¬
ноцитов и эпителия предста¬
тельной железы
Лейкемический ингибиторный фак¬
тор; HILDA; член семейства IL-6
LIF
24
Клетки SCO cells
Ингибирует дифференциров-
ку эмбриональных стволо¬
вых клеток
Липополисахарид
LPS
10
Грамположительные
бактерии
Активация лимфоцитов
Лимфотоксин
TNF-P
20-25
Лимфоциты
Цитотоксичность для опухоле¬
вых клеток
Макрофагальный белок воспале¬
ния-la
MIP-la
10
Макрофаги
Ингибитор роста гематопоэти-
ческих стволовых клеток
10.5. Выбор бессывороточной среды
175
Окончание табл. 10.3
Название и синонимы
Аббревиа¬
тура
Молекулярная
масса (кДа)
Источник*
Функция
Моноцитарно-макрофагальный ко¬
лониестимулирующий фактор;
CSF-1
M-CSF
47-74
В-и Т-клетки, моноци¬
ты, тучные клетки,
фибробласты
Индукция пролиферации и
дифференцировки предшес¬
твенников макрофагов
Мюллеров ингибирующий фактор
MIF
Тестикулы
Ингибирование развития
Мюллерова протока; инги¬
бирование пролиферации
карциномы яичника
Фактор роста нервов, Р
PNGF
27
Подчелюстные слюн¬
ные железы самцов
мышей
Трофический фактор; хемо-
таксический фактор; фак¬
тор дифференцировки; спо¬
собствует росту нейритов
нейронов периферической
нервной системы
Онкостатин М; член семейства IL-6
OSM
28
Активированные
Т-клетки и обрабо¬
танные РМА моно¬
циты
Индуктор дифференциров¬
ки (совместно с глюкокор-
тикоидами); митоген для
фибробластов
Фитогемагглютинин
ФГА
30
Красная фасоль
( Phaseolusvulgaris)
( Phaseolusvulgaris)
Активация лимфоцитов
Тромбоцитарный эндотелиальный
фактор роста; сходный с глиоста-
тином
PD-ECGF
-70
Тромбоциты крови,
фибробласты, глад¬
кая мускулатура
Стимуляция ангиогенеза; ми¬
тоген для эндотелиальных
клеток; способствует выжи¬
ванию нейронов; ингибирует
пролиферацию глии
Тромбоцитарный фактор роста
PDGF
30
Тромбоциты крови
Митоген для клеток мезо-
дермального и нейроэкто¬
дермального происхожде¬
ния; способствует зажив¬
лению ран; действует в
синергизме с EGF и IGF-1
Форбол миристат ацетат; ТРА; фор-
боловый эфир
РМА
0.617
Масло кротона
Опухолевый промотор; мито¬
ген для некоторых эпите¬
лиальных клеток и мелано-
цитов; фактор дифференци¬
ровки для HL-60 и плоского
эпителия
Митоген лаконоса
PWM
Корень фитолакки аме¬
риканской
Активация моноцитов
Фактор стволовых клеток; фактор
роста тучных клеток; стволовой
фактор; c-kit лиганд
SCF
31 g
Эндотелиальные клет¬
ки, фибробласты,
костный мозг, клет¬
ки Сертолли
Способствует первому деле¬
нию плюрипотентных гема¬
топоэтических стволовых
клеток
Трансферрин
Tfn
78
Печень
Осуществляет транспорт же¬
леза; митоген
Транформирующий фактор роста а
TGF-a
6
Индуцирует независимый от
подложки рост клеток и спо¬
собствует потере клеток кон¬
тактного ингибирования
Транформирующий фактор роста р
(шесть видов)
TGF-pi-6
23-25
Тромбоциты крови
Ингибитор пролиферации
эпителиальных клеток; ин¬
дуктор дифференцировки
сквамозных клеток
* Под источниками подразумеваются некоторые типы тканей, из которых выделяют природные факторы. Во многих случаях
природный белок заменен рекомбинантным, который имеется в продаже (смотри Приложение И). Сокращения (отличающиеся
от приведенных в колонке 2): гл — гликозилированный; гс — гепаринсвязывающий; HBGF — гепаринсвязывающий фактор
роста. С разрешения из Barnes et al. [1984а], Lange et al. [1991], Jenkins [1992], и Smith et al. [1997].
176
ГЛАВА 10. БЕССЫВОРОТОЧНЫЕ СРЕДЫ
является только невозможность ее приобретения или
ее несовместимость с другими исходными компонен¬
тами. Если возможно, лучше всего придерживаться
рекомендаций создателя культуры, так как, возможно,
это является единственным способом гарантировать
сохранение специфических свойств данной линии кле¬
ток. В табл. 10.4 суммированы данные по пригодности
и выбору бессывороточных сред, перечисленных в
табл. 10.1 и 10.2 и добавлены несколько дополнитель¬
ных советов. (См. также [Mather, 1998], [Barnes et al.,
1984a-d] и [Ham & McKeehan, 1979].)
10.5.1. Имеющиеся в продаже
бессывороточные среды
Ряд производителей (см. табл. 10.4 и Приложение II)
сейчас изготовляют бессывороточные среды. Состав
некоторых из них известен, таких как MCDB 131
(Sigma) — среда для эндотелиальных клеток, и LHC-9
(Biosource International) — для бронхиального эпите¬
лия, в то время как состав других сред запатентован
(например, среда CHO-S-SFM для клеток СНО-К1 и
Opti-MEM для гематопоэтических клеток (Invitrogen)).
Многие среды разработаны для культур гибридом, их
применяют в тех случаях, когда абсолютно необходимо
исключить присутствие в целевом продукте сывороточ¬
ных белков, но есть и другие среды, которые подходят
для других типов клеток. Хотя в литературе имеются
данные по использованию запатентованных сред для
индивидуальных типов клеток (см. табл. 10.4), состав
таких сред часто является коммерческой тайной, и
вы можете только полагаться на совет фирмы-произ¬
водителя или, что лучше, проверить несколько сред,
пассируя несколько раз на этих средах свои клетки.
Последний вариант может потребовать значительных
затрат, но будет вполне оправданным в том случае, если
вы планируете длительную работу с клетками.
Важно установить параметры контроля качества,
выполненного коммерческими производителями бес¬
сывороточных сред. В идеальном варианте среда уже
протестирована на тех клетках, которые вы хотите
культивировать (например, среда для культивирова¬
ния кератиноцитов должна быть протестирована на
кератиноцитах). Некоторые производители, такие как
Cambrex, Cascade и PromoCell, поставляют соответству¬
ющие клетки вместе со средой, прошедшей надлежащий
контроль качества, но другие поставщики могут про¬
водить контроль качества на рутинных линиях клеток,
в этом случае вы должны перед покупкой среды сами
проверить ее качество.
10.5.2. Заменители сыворотки
Коммерческими производителями разработан ряд
продуктов для замены сыворотки целиком или час¬
тично в традиционных средах. Вот некоторые из этих
продуктов: Ex-cyte (Bayer), Ventrex (JRH Biosciences),
CPSR-1,2,3 (Sigma), Nu-serum (Becton Dickinson), and
Biotain-MPS (Cambrex). Хотя состав этих заменителей
постоянен, что невозможно для обычной сыворотки,
однако все еще могут проявляться различия между
разными партиями, и состав продуктов полностью
не идентифицирован. Использование заменителей
может быть полезным при проведении специальных
экспериментов или в целях экономии, но не как заме¬
на бессывороточных сред. К добавкам определенного
химического состава относятся Nutridoma (BCL), ITS
Premix, ТСМ, TCH (ICN), SIT (Sigma), and Excell-900
(JRH Biosciences), с помощью которых можно полно¬
стью или частично заменить сыворотку. Sigma продает
серию сред MegaCell среди которых MEM, DMEM,
RPMI 1640 и МЕМ или DMEM/F12, обогащенные
факторами роста и дополнительными аминокислотами,
позволяющими снизить концентрацию сыворотки.
Выбор бессывороточной среды и заменителей сы¬
воротки сейчас настолько велик и разнообразен, что
невозможно дать индивидуальные рекомендации для
каждой индивидуальной задачи. Лучшим подходом
при выборе является изучение литературы, контакт с
производителями, получение наиболее подходящих (по
литературным данным) для ваших задач образцов их
продукции и проверка продукции в ваших собственных
тестах (например, влияние на рост, выживаемость или
специальные функции).
Гидролизаты аминокислот (см. разд. 9.7.1) исполь¬
зуются с целью снизить или полностью удалить сыво¬
ротку при наращивании культур в крупном масштабе
для биотехнологического применения. Достоинство
гидролизатов в том, что их можно стерилизовать авто-
клавированием, но состав их точно не определен.
10.5.3. Адаптация клеток
к бессывороточной среде
Многие постоянные линии клеток, такие как HeLa,
СНО-К1 или мышиные миеломы, можно адаптировать
к культивированию в отсутствие сыворотки. Процесс
адаптации включает в себя продолжительный период
отбора минорных компонентов клеточной популяции,
так как важно удостовериться, что свойства клеточной
линии не будут потеряны за время селекции.
Адаптация клеток к бессывороточной среде обычно
выполняется путем нескольких серий пассирования
с постепенным снижением концентрации сыворотки.
При достижении стабильной пролиферации клеток при
одной концентрации, клетки пассируют в среде с более
низкой концентрацией сыворотки до тех пор, пока не
восстановится стабильный рост клеток, и затем опять
понижают концентрацию сыворотки. В суспензионных
культурах контроль осуществляют путем подсчета
живых клеток и разбавляя соответственно клеточную
суспензию, при этом возможно поддерживать мини¬
мальную концентрацию клеток выше, чем при обычном
пассировании. Для монослойных культур концентра¬
цию сыворотки снижают за несколько дней до пересева
10.5. Выбор бессывороточной среды
177
Таблица 10.4. Выбор бессывороточной среды
Клетки или линия
клеток
Адипоциты
ВНК21
Бронхиальный эпителий
Куриные эмбриональные
фибробласты
Яичник китайского хомячка
(СНО)
Хондроциты
Постоянные клеточные линии
Эпителиальные клетки рого¬
вицы
COS-1,7
Эндотелий
Фибробласты
Клетки глии
Глиома
Клетки HeLa
Гематопоэтические клетки
HL-60
НТ-29
Диплоидные фибробласты
человека
Лейкоз человека и нормальные
лейкоциты
Опухоли человека
Гепатоциты, эпителий печени
Гибридомы
Клетки насекомых
Кератиноциты
Бессывороточная среда
(смотри табл. 10.1)
HyQPF СНО; HyG PF
СНО MPS; РС-1
LHC-9
MCDB 201,202
MCDB302; РС-1
Обогащенная DMEM/F12
Игла MEM, М199,
МВ752/1, CMRL 1066,
среда MCDB,
DMEM:F12 + добавки
MCDB 153
MCDB 130,131
MCDB 110,202,402
Michler-Stuke
SF12 (среда Ham’s F12 с
экстра незаменимыми и
заменимыми аминокис¬
лотами)
аМЕМ; Iscove’s
MCDB 110,202; РС-1
Iscove’s
Brower; HITES; Masui;
Bottenstein N3
Williams E, L15
Iscove’s
MCDB 153
Ссылки (смотри табл. 10.1
и 10.2)
Rodriguez et al., 2004
Pardee et al., 1984
См. протокол 22.9
McKeehan & Ham, 1976b
Hamilton & Ham, 1977
Adolphe, 1984; смотри про¬
токол 23.13
Waymouth, 1984
Смотри протокол 23.2
Doering et al., 2002
Knedler & Ham, 1987; Gup¬
ta et al., 1997; Hoheisel et
al., 1998
Bettger et al., 1981; Shipley
& Ham, 1983
Michler-Stuke &
Bottenstein, 1982
Frame et al., 1980; Freshney,
1980
Blakeret al., 1971;
Bertheussen, 1993
Stanners et al., 1971;
Iscove & Melchers, 1978
Li et al., 1997
Oh et al., 2001
Bettger et al., 1981; Ham,
1984
Breitman et al., 1984
Brower et al., 1986; Carney
et al., 1981; Masui et al.,
1986b; Bottenstein, 1984;
Chopra et al., 1996
Williams & Gunn, 1974; Mi-
taka et al., 1993]
Iscove & Melchers, 1978;
Murakami, 1984
Ikonomou et al., 2003
Peehl & Ham, 1980; Tsao et
al., 1982; Boyce & Ham,
1983; см. протокол 23.1
Коммерческие производители
(специфических сред или их
альтернативных вариантов)
PromoCell
Cambrex
Biosource; Cambrex; PromoCell
Sigma
Cambrex; Sigma; Invitrogen;
JRH Biosciences; PromoCell
PromoCell
Cambrex; Invitrogen; JRH Bio¬
sciences; ICN; Sigma
Cambrex, KGM; Cascade
Cambrex
Cambrex; Cascade; PromoCell;
Sigma
Cambrex; Cascade; PromoCell;
Cambrex; Invitrogen
Cambrex; Invitrogen
Cambrex
Sigma; Roche
Cambrex
Cambrex
Cascade; Cambrex; PromoCell
Cambrex; Hyclone; MP Bio¬
medicals Invitrogen; JRH
Biosciences; Sigma
Cambrex; Sigma
Sigma; Invitrogen; Cambrex;
JRH Biosciences; MP Bio¬
medicals; Roche; Irvine Sci¬
entific; Metachem; PromoCell
JRH Biosciences; Cell Gro
Invitrogen; Cascade; Cambrex;
PromoCell; Sigma
178
ГЛАВА 10. БЕССЫВОРОТОЧНЫЕ СРЕДЫ
Клетки или линия
клеток
L клетки (L929, LS)
Лимфобластоидные клеточные
линии человека
Эпителий молочной железы
Эпителий почки собаки MDCK
Почка свиньи LLC-PK
Меланоциты
Меланома
Мышиные эмбриональные
фибробласты; клетки ЗТЗ
Mouse leukemia
Мышиный эритролейкоз
Мышиная миелома
Мышиная нейробластома
Нейроны
Остеобласты
Предстательная железа
Почка
Гладкомышечные клетки
Скелетные миобласты
Уротелий
Подложка
Бессывороточная среда
(смотри табл. 10.1)
NCTC109; NCTC135
Iscove’s
MCDB 170
К-1; РС-1
К-2
Gilchrest
Gilchrest
MCDB 402
SF12 (среда Ham’s F12 с
экстра незаменимыми и
заменимыми аминокис¬
лотами); Iscove’s
MCDB 411; DMEM:F12/
N1
DMEM:F12/N3;
В27/ нейробаз альная
WJAC 404;
MCDB 153; KSFMc
Ссылки (смотри табл. 10.1
и 10.2)
Birch & Pirt, 1970,1971;
Higuchi, 1977
Iscove & Melchers, 1978
Hammond et al., 1984; cm.
протокол 22.3
Taub, 1984
Taub, 1984
См. протокол 22.18
См. протокол 22.18;
Halaban, 2004
Shipley & Ham, 1983; Ham,
1984
Murakami, 1984
Frame et al., 1980; Freshney,
1980; Iscove & Melchers,
1978
Murakami, 1984
Agy et al., 1981; Bottenstein,
1984
Bottenstein, 1984; Brewer,
1995
Shiga et al., 2003
См. табл. 10.11; Peehl, 2002
Goto et al., 1999
Southgate et al., 2002
Оконченние табл. 10.4
Коммерческие производители
(специфических сред или их
альтернативных вариантов)
Sigma
JRH Biosciences; CellGenix;
Amersham
Cascade; Cambrex
Cambrex
Cambrex; Cascade; PromoCell
Cambrex; Cascade; PromoCell
Biosource
MP Biomedicals; Invitrogen;
Sigma
Sigma, Invitrogen
N2, Invitrogen
OGM, Cambrex; PromoCell
Cambrex
REGM (для эпителиальных
клеток) MsGM (для мезан-
гиальных клеток), Cambrex
Cascade; Cambrex; PromoCell
PromoCell
KGM-2, Cambrex
B-D Biosciences; MP Biomedi¬
cals; Invitrogen; Biosource;
Sigma
Дополнительные рекомендации по выбору среды можно найти в работах McKeehan [1977], Barnes et al. [1984a и Mather [1998];
см. также табл. 10.1 и 10.2
и затем субкультивируют клетки в среде, содержащей
новую пониженную концентрацию сыворотки. При
адаптации клеток может возникнуть необходимость
обогатить среду известными факторами для замены
сыворотки (см. разд. 10.4,10.6).
10.6. РАЗРАБОТКА БЕССЫВОРОТОЧНОЙ
СРЕДЫ
Есть два главных подхода при разработке бессыворо-
точной среды для индивидуальной клеточной линии
или первичной культуры. Первый из них: берется из¬
вестная пропись среды для родственного типа клеток с
или без 10-20% диализованной сыворотки и вносятся
изменения в состав компонентов, индивидуально или
комбинированно, одновременно снижая концентрацию
сыворотки, до тех пор пока среда не будет оптимизи¬
рована в соответствии с вашими индивидуальными
требованиями. Этот прием был применен Наш с со¬
авторами [Наш, 1984] и обычно позволяет подобрать
оптимальные условия. Если было обнаружено, что
группа компонентов эффективна при снижении кон¬
центрации сыворотки, активные составляющие можно
идентифицировать путем систематического исключе¬
ния отдельных компонентов, и, таким образом, опти¬
мизируют концентрации необходимых добавок [Наш,
1984]. Однако этот процесс трудоемкий и занимает
10.9. Заключение
179
много времени. На каждой стадии получают кривые
клеточного роста и проводят исследования эффектив¬
ности клонообразования. Неразумно проводить, по
крайней мере, три года, разрабатывая новую среду для
нового типа клеток.
Трудоемкая природа первого подхода подводит нас
ко второму подходу: обогащение существующих сред,
таких как RPMI 1640 [Carney et al., 1981] или комби¬
нированных сред, таких как среда Ham’s F12 с DMEM
[Barnes & Sato, 1980]. При этом нет необходимости
проводить манипуляции с их составляющими для того,
чтобы уменьшить список компонентов. К последним
относятся селен, трансферрин, альбумин, инсулин,
гидрокортизон, эстрогены, трийодтиронин, этаноламин,
фосфоэтаноламин, факторы роста (EGF, FGF, PDGF,
добавки для роста эндотелия и т.д.), простагландины
(PGE1, PGF2a) и некоторые другие вещества, которые
могут иметь особую важность (см. разд. 10.4; табл. 10.3).
Как было показано, селен, трансферрин и инсулин
обычно необходимы для большинства клеток, тогда как
требования к другим компонентам могут варьировать.
10.7. ПРИГОТОВЛЕНИЕ
БЕССЫВОРОТОЧНОЙ СРЕДЫ
В настоящее время для индивидуальных типов клеток
используется ряд рецептур бессывороточных сред,
некоторые имеются в продаже (см. Приложение И)
[Cartwright & Shah, 1994; Mather, 1998; см. также
табл. 10.1 и 10.2 и гл. 23]. Процедура приготовления
бессывороточных сред сходна с приготовлением обыч¬
ных сред (см. протокол 11.9; см. также [Waymouth,
1984]). Необходимо использовать сверхчистые реаген¬
ты и воду и следует проявлять осторожность, работая
с растворами Са2+ и Fe2+ или Fe3+, чтобы избежать
выпадения осадка. Соли металлов имеют тенденцию
выпадать в осадок при щелочных pH в присутствии
фосфата, особенно если среда или солевой раствор
подвергается автоклавированию, поэтому катионы
в исходном растворе должны содержаться при низ¬
ких pH (ниже 6,5) в отсутствие фосфата. Растворы
необходимо стерилизовать путем автоклавирования
или фильтрования (см. разд. 11.3). Часто рекомен¬
дуется добавлять катионы в последнюю очередь, не¬
посредственно перед использованием среды. Другим
возможным вариантом, который часто применяется,
является приготовление серии исходных растворов
неорганических компонентов и витаминов в виде
1000-кратного концентрата, тирозина, триптофана и
фенилаланина в 0,1 Н НС1 в виде 50-кратного кон¬
центрата, незаменимых аминокислот — 100-кратного
концентрата в воде, солей — 10-кратного в воде, и
некоторые другие специальные кофакторы, липиды
и т. д. в виде 1000-кратных концентратов в соответству¬
ющих растворителях. Их комбинируют в необходимых
пропорциях и разбавляют до конечной концентрации,
после чего проверяют pH и осмотическое давление (см.
разд. 11.2.5). Факторы роста, гормоны и факторы кле¬
точной адгезии добавляют отдельно, непосредственно
перед использованием среды, так как может возникнуть
необходимость внесения изменений в соответствии с
индивидуальными условиями эксперимента.
10.8. СРЕДЫ, НЕ СОДЕРЖАЩИЕ БЕЛКОВ
Со стороны контролирующих органов усиливаются
требования удалить все продукты животного проис¬
хождения, которые контактируют с культивируемыми
клетками, при производстве биофармацевтической
продукции. Трипсин может быть заменен рекомби¬
нантным трипсином или протеазами неживотного
происхождения (см. разд. 10.4.1), а факторы роста — ре¬
комбинантными факторами роста. BSA часто может
быть заменен добавлением таких факторов, как липи¬
ды, гормоны, неорганические соединения и факторы
роста (см. разд. 10.4), которые обычно в сыворотке
связываются с BSA, так как пока не установлено, что
BSA сам играет какую-то роль. Адаптация клеток к
среде, не содержащей белков, может потребовать до¬
полнительной селекции клеток.
10.9. ЗАКЛЮЧЕНИЕ
В преимуществе использования бессывороточных
сред могут возникнуть сомнения, так как применение
сред, содержащих сыворотку, отличается простотой.
Для применения некоторых бессывороточных сред
требуются специальные методики, значительные
затраты времени, усилия и источники для приготов¬
ления новых рецептур или адаптации существующих,
многочисленность сред в случае, если необходимо
культивировать несколько типов клеток, — все это
отпугивает большинство лабораторий от использо¬
вания в своей работе бессывороточных сред. Однако
существует необходимость в создании постоянных
известных условий культивирования при исследова¬
нии регуляторных процессов, контролирующих рост и
дифференцировку. Кроме того, различные биотехноло¬
гии требуют упрощения процессов очистки продуктов.
И наконец, существует необходимость удаления всех
потенциальных источников инфекции. Все это, в ко¬
нечном счете, заставит применять бессывороточные
среды в более широком масштабе.
Глава 11
ПОДГОТОВИТЕЛЬНЫЕ РАБОТЫ
И СТЕРИЛИЗАЦИЯ
11.1. ПРИГОТОВЛЕНИЕ РЕАКТИВОВ
И МАТЕРИАЛОВ
Все растворы химических реактивов и посуда, ис¬
пользуемые при культивировании тканей, должны
храниться отдельно и предназначаться только для этих
целей. Следы тяжелых металлов или других токсичных
веществ могут быть трудноудалимы и определяться
только по постепенному ухудшению роста культуры.
Следовательно, стеклянная посуда, предназначенная
для различных целей, требует разных способов мытья.
Требования к качеству мытья посуды для культиви¬
рования тканей выше, чем в целом для лабораторных
работ. Хотя уровень загрязнения посуды при работе
может быть меньше, может потребоваться применение
специальных детергентов (см. разд. 11.3.4), следует
также избегать возможности химического загрязне¬
ния при контакте с обычной лабораторной посудой.
Практически повсеместно принятая практика исполь¬
зования одноразового пластика снимает проблему
обработки стеклянных флаконов, используемых для
культивирования животных клеток; тем не менее в
тех случаях, когда для хранения либо для культиви¬
рования используется лабораторное стекло, остается
существенной проблема химического загрязнения.
Следовательно, требованием культивирования тканей
является абсолютная чистота посуды.
11.2. СТЕРИЛИЗАЦИЯ ОБОРУДОВАНИЯ
И ЖИДКОСТЕЙ
Все оборудование и жидкости, которые контактируют с
культурами или реактивами, должны быть стерильны.
Выбор метода стерилизации зависит, главным образом,
от температурной стабильности объекта (табл. 11.1).
В целом, большинство предметов устойчивы к дей¬
ствию высокой температуры, — металлы, стекло,
термоустойчивый пластик, например политетрафтор¬
этилен (ПТФЭ), можно стерилизовать сухим жаром.
Это один из самых простых и эффективных методов
стерилизации, обеспечивающий достижение высокой
температуры во всех частях загруженной партии мате¬
риала в течение определенного времени. Стерилизация
горячим паром, или автоклавирование, также высоко¬
эффективна и может применяться для стерилизации
термостабильных жидкостей, таких как вода, растворы
солей и некоторые специально разработанные среды.
Для стерилизации термолабильных материалов может
использоваться радиоактивное излучение. Наиболее
эффективным является у-излучение, источником ко¬
торых является 60Со или 135Cs. Поскольку необходимая
эффективно действующая доза достаточно высока
(25 кГр), обычно такая обработка производится цен¬
трализованно, на оборудовании, позволяющем про¬
изводить также электронно-лучевую стерилизацию.
Для термолабильного пластика может использоваться
обработка окисью этилена, но это вещество способно
абсорбироваться на поверхности пластика и может
потребоваться несколько дней для его высвобождения
и улетучивания. Другие химические стерилизующие
соединения включают гипохлорит натрия и формаль¬
дегид, но эти вещества чаще всего используются для
обеззараживания (см. разд. 7.8.5) жидкостей и одно¬
разового пластика. Формальдегид также используется
для фумигационной обработки (см. разд. 7.8.6).
Термолабильные жидкости стерилизуются мик-
ропоровой фильтрацией (см. разд. 11.5.2), при этом
может быть использовано как положительное давление
с использованием серии насадок на шприц, располо¬
женных по размеру фильтров, — дисков диаметром
293 мм или гофрированных картриджей 1000 см2, так
и отрицательное давление с использованием флаконов
с присоединенным фильтром (см. протокол 11.12).
При наличии вакуумной линии или насоса фильт¬
рация — наиболее простой и эффективный метод
стерилизации, при котором фильтруемая жидкость
поступает в сосуды, в которых она будет храниться.
Однако фильтрация при отрицательном давлении
приводит к возрастанию pH, поскольку происходит
удаление растворенного в среде С02.
В следующих разделах описаны предпочтительные
методы стерилизации оборудования, культуральной
посуды и растворов (табл. 11.2 и 11.3).
11.3. ОБОРУДОВАНИЕ
11.3.1. Стеклянная посуда
Если для размножения клеток используются стеклян¬
ные поверхности, стекло должно не только быть чис¬
тым, но и иметь определенный заряд. Едкие щелочные
детергенты приводят поверхность стекла в состояние,
11.3. Оборудование
181
Таблица 11.1. Методы стерилизации
Метод
Условия
Материалы
Ограничения
Сухой жар
160 °С, 1ч
Термостабильные: металл, стекло,
ПТФЭ.
Возможно обугливание, например,
индикаторных этикеток и ватных
пробок
Автоклавирование
121 °С,
15-20 мин
Термостабильные жидкости: вода,
солевые растворы, автоклавируемые
среды. Умеренно-термостабильный
пластик: силикон, поликарбонат,
нейлон, полипропилен
Требуется проникновение пара ко всем
предметам, т.е. паропроницаемая
упаковка. Загрузка большого объема
требует большого времени для пол¬
ного прогревания
Излучение
у-излучение
25 кГр
Пластик, органические каркасы, тер¬
мочувствительные реактивы и фар¬
макологические препараты
Возможно химическое изменение
пластика. Макромолекулярная де¬
градация.
электронно-лучевое
25 кГр
Пластик, органические каркасы, тер¬
мочувствительные реактивы и фар¬
макологические препараты
Требуется высокоэнергетичный источ¬
ник. Не подходит для установки в
лаборатории средних размеров
микроволновое
5 мин полная
мощность
Водные растворы и гели, такие как
агар
Может использоваться только для
малых объемов, обычно для растап¬
ливания агара
коротковолновое уль¬
254 нм,
Плоские поверхности, циркулирую¬
Не достигает затененных областей.
трафиолетовое
50-100 Вт,
30 мин
щий воздух
Споры устойчивы к обработке
Химическая обработка
окись этилена
1ч
Термолабильный пластик
После обработки материалы должны
выветриваться 24-48 ч; остаются
токсичные остатки
гипохлорит
300-2500%о
30 мин
Зараженные растворы, пластик
Требуется тщательное промывание
после обработки. Могут оставаться
остатки
70% этанол
замачивание
на 1 ч
Хирургические инструменты (в ком¬
бинации с обжиганием в пламени).
Некоторые виды пластика
Не убивает споры. Риск пожара при
работе с пламенем.
Пластик Precast Регрех и Lucite может
разрушаться при действии этанола
Фильтрация
размер пор
0,1-0,2 мкм
Все водные растворы; частично
пригодна для термолабильных
реактивов и сред. Специфичные
фильтры для связывания низко¬
молекулярных белков для факто¬
ров роста и т.д.
Не пригодна для некоторых растворов,
например ДМСО. Медленно для
вязких растворов
непригодное для прикрепления клеток, и поэтому тре¬
буется последующая нейтрализация раствором НС1 или
H2S04; нейтральные детергенты не изменяют поверх¬
ности стекла и легче смываются. Наиболее эффективна
следующая процедура обработки стекла:
1) Не позволяйте грязной посуде высыхать. В воду,
заполняющую емкости для сбора грязного стекла,
должны быть добавлены стерилизующие агенты,
такие как гипохлорит натрия, для того чтобы:
• уничтожить любые источники возможной биологи¬
ческой опасности;
• предупредить размножение бактерий в воде.
2) Выбирайте детергенты, эффективные в воде вашего
региона, легко смывающиеся и нетоксичные (см.
разд. 11.3.4).
3) До высушивания посуды убедитесь, что она тща¬
тельно промыта проточной водой и ополоснута
деионизированной или дистиллированной водой.
4) Сушите посуду в перевернутом состоянии, так чтобы
остатки воды могли вытечь.
5) Стерилизацию посуды проводите сухим жаром, что
минимизирует риск переноса токсичных остатков,
возможный при стерилизации горячим паром.
Протокол проведения стерилизации разработан
таким образом, чтобы не только убить делящиеся
микроорганизмы, но и уничтожить более устойчивые
споры. Горячий пар более эффективен для этих целей,
чем сухой жар, но обработка паром связана с риском
сохранения остатков загрязнения. Сухой жар предпоч¬
тительнее, но в этом случае обработка должна продол¬
жаться в течение 1 ч при минимальной температуре
160 °С. Горячий пар (для жидкостей и скоропортящих¬
ся продуктов) следует применять при 121 °С в течение
15-20 мин (см. табл. 11.1, 11.2, 11.3). Для того чтобы
стерилизация паром была эффективнее, следует га¬
рантированно обеспечить проникновение пара ко всем
182
ГЛАВА 11. ПОДГОТОВИТЕЛЬНЫЕ РАБОТЫ И СТЕРИЛИЗАЦИЯ
Таблица 11.2. Стерилизация оборудования и
инструментов
Предметы
Стерилизация
Ампулы для замораживания,
пластик (обычно поставля¬
ется стерильным)
Ампулы для замораживания,
стекло
Закручивающиеся крышечки
Инструменты
Инструменты, имеющие стек¬
лянные части и силиконо¬
вые трубки
Магниты для магнитной ме¬
шалки
Одноразовые наконечники
для микропипеток
Плексиглас, Регрех, Lucite
Повторно используемые пи¬
петки или шприцы
Поликарбонат
Пробирки
Пробки, резиновые и сили¬
коновые
Силиконовая смазка (для
выделения клонов)
Силиконовые трубки
Стекла покровные
Стекла предметные
Стекло
Стеклянные бутылки с закру¬
чивающимися крышками
Стеклянные Пастеровские
пипетки
Стеклянные пипетки
Стеклянные шприцы
Фильтры многократного
использования
Автоклавирование1
Сухой жар2
Автоклавирование
Сухой жар
Автоклавирование
Автоклавирование
Автоклавирование; в авто-
клавируемых поддонах
или нейлоновых пакетах
70% этанол
Автоклавирование (отдельно
ПТФЭ-поршни)
Автоклавирование
Сухой жар
Автоклавирование
Автоклавирование в стеклян¬
ной чашке Петри
Автоклавирование
Сухой жар
Сухой жар
Сухой жар
Автоклавирование с ослаб¬
ленными крышками
Сухой жар
Сухой жар
Автоклавирование (отде-
льноПТФЭ-поршни )
Автоклавирование; не ис¬
пользовать предваритель¬
ное или последующее раз-
режение
1 Автоклавирование, 100 кПа (1 атм), 121 °С, 20 мин.
2 Сухой жар , 160 °С, 1 ч.
предметам, что означает, что воздух из стерилизацион¬
ной камеры должен быть полностью удален до того, как
туда поступит пар, либо воздух должен быть полностью
замещен паром, поступающим под давлением.
Помещенные в стерилизуемую посуду (или рас¬
твор), расположенный в центре стерилизационной ка¬
меры, индикаторы типа Thermalog (см. Приложение II)
и датчик температуры, связанный с самописцем, позво¬
ляют следить как за температурой, так и за влажностью
в ходе стерилизации. Для контроля процесса автокла-
вирования часто используется измерение температуры
вытекающей из автоклава воды, однако этот метод
недостаточно точно отражает температуру в камере.
ПРОТОКОЛ 11.1. Подготовка и стерилизация
изделий из стекла
Материалы
• Дезинфектанты: гипохлорит, — минимум 300 ppm
(частей на миллион) активного хлора при раство¬
рении в детергенте (например, таблетки Precept,
Clorox или Chloros
• Детергент (например, 7Х™ или Decon®)
• Ванна для замачивания
• Щетки для мытья бутылей
• Сетки-корзины из нержавеющей стали для сбора
и суппси вымытой и прополосканной посуды
• Алюминиевая фольга
• Индикаторы стерильности (Alpha Medical для су¬
хожаровой или паровой стерилизации, Thermalog
только для автоклавирования)
• Ярлык или липкая лента для обозначения сте¬
рильности (Bennett). Эти ярлыки отличаются от
используемых при автоклавировании, поскольку
температура в. сухожаровых шкафах выше.
Большинство ярлыков для автоклавирования
обугливаются и выделяют следовые количества
летучих веществ из липкого слоя, которые могут
выпасть в осадок на стенках печи или даже на
стерилизуемом стекле.
• Стерилизационная печь, снабженная вентиля¬
тором, способная давать температуру 160 °С, и,
предпочтительно, с регистратором температуры
и гибким зондом.
Протокол
Сбор и мытье стеклянной посуды (рис. 11.1)
1. Непосредственно после использования собери¬
те стеклянную посуду, поместите ее в раствор
детергента, содержащего дезинфектант. Важно,
чтобы стекло не высыхало до замачивания, иначе
отмыть посуду будет значительно труднее.
2. Замочите на ночь в ванне для замачивания.
3. Промывание:
а) На следующее утро промойте при помощи
щетки вручную или механической щеткой для
бутылей (рис. 11.2); тщательно прополощите
четырьмя полными сменами проточной воды, а
затем трижды деионизованной водой. Полезно
использовать разбрызгивающую насадку для
промывания, при ее отсутствии необходимо пол¬
ностью заполнять водой бутылки полностью их
опустошать при каждом ополаскивании.
б) Машинное промывание должно проводиться
без детергента; В этом случае можно умень¬
шить количество прополаскиваний до двух
водопроводной водой и одного деионизиро¬
ванной илиочищенной при помощи обратного
осмоса воды.
4. После тщательного ополаскивания переверните
бутыли вниз горлом, например в стальной прово¬
лочной корзине, высушите.
11.3. Оборудование
183
5. Когда посуда остынет, закройте горлышки алю¬
миниевой фольгой, поставьте на хранение
Стерилизация стеклянной посуды:
1. Приклейтёнебольшйё гаад липкой ленты
для индикации стерильности, напишите дату.
2. Поставьте посуду в стерилизационный шкаф с
вентилируемой циркуляцией воздуха, установите
температуру 160 °С.
3. Убедитесь, что температура в центре загруженной
камеры достигает 160 °С:
а) Поместите индикатор стерильности в бутыль
или типичный для этой загрузки сосуд в се¬
редине камеры.
б) При использовании регистрирующего тер¬
мометра поместите сенсорное устройство в
бутыль или типичный для этой загрузки сосуд .
в середине камеры.
в) Не забивайте камеру стерилизационной печи
слишком плотно. Оставьте место для цирку¬
ляции воздуха.
4. Закройте дверцу печи, удостоверьтесь, что темпе¬
ратура вновь составляет 160 °С, уплотните дверцу
полоской ленты с указанием времени начала
стерилизации (или установите автоматический
замок и самописец), оставьте на 1ч.
5. Через час выключите печь, позвольте ей остыть
с запертой дверью. Удобно установите автомати¬
ческий таймер так, чтобы можно было оставить
посуду стерилизоваться на ночь, с последующим
выключением и остыванием. Тогда остывшую
посуду вынимают утром. Эти меры предосторож¬
ности позволяют лучше сохранить стерильность
и минимизируют образование тейла в рабочее
время, когда жарко работать. - ; . ^ /
6. Используйте посуду в течение 24-4&Ч.: : Гл _
Регистрирующие термометры имеют то преимущест¬
во, что постоянно ведущиеся записи могут храниться
в архиве. С другой стороны, индикаторы (например,
Thermalog) обеспечивают визуальное подтверждение
необходимого режима температуры и влажности и
поэтому могут быть использованы для мониторинга
разных частей загруженной партии одновременно. Мы
рекомендуем использовать оба метода контроля.
Органические материалы должны храниться вне
жарового шкафа. Не используйте бумажную перфолен¬
ту или упаковочный материал, если вы не убеждены,
что при нагревании не произойдет выделения летучих
продуктов. Эти продукты в конечном итоге осаждаются
на внутренней поверхности стерилизационного шкафа,
что создает запах при нагревании, некоторые продукты
могут осаждаться на внутренней поверхности стекла
при стерилизации.
Кроме того, бутыли можно неплотно закрыть закру¬
чивающимися крышками и сверху накрыть фольгой
для автоклавирования. Автоклавирование проводят
в течение 20 мин при 121 °С, в режиме, включающем
создание вакуума до и после поступления пара (см.
разд. 5.4.4). Затем, когда посуда несколько остынет,
крышки следует плотно завернуть. В процессе автокла¬
вирования крышки должны быть закручены достаточно
слабо (по крайней мере один полный оборот до полного
закручивания), так, чтобы позволить пару проникнуть
в бутыли и предупредить всасывание вкладыша (если
таковой имеется) из крышки в бутыль. В противном
случае возникнет препятствие для пара, что будет
означать неполную стерилизации (рис. 11.3 и цв. вклад¬
ка 22а). К сожалению, при автоклавировании бутылей
и колб часто имеет место аэрозольное запотевание,
при конденсации пара в них образуется небольшой
остаток. Кроме того, при охлаждении колб и бутылей
существует риск попадания нестерильного воздуха и
микробного заражения до того, как они будут плотно
закрыты. Стерилизация сухим жаром предпочтитель¬
на, поскольку это позволяет охлаждать посуду в печи
(крышки автоклавируются отдельно; см. разд. 11.3.1).
11.3.2. Стеклянные пипетки
В культивировании тканей используются как стек¬
лянные, так и пластиковые пипетки. К преимущест¬
вам пластиковых пипеток относится однократность
использования, снимающая необходимость мытья
и стерилизации. Стеклянные пипетки значительно
дешевле, но требуется дополнительная работа по заты¬
канию ватных пробочек и их удалению каждый раз при
их использовании. Стеклянные пипетки необходимо
тщательно мыть в специальных моечных машинах или
устройствах для мытья пипеток, так, чтобы в них не
оставалось загрязнений, кроме того, их носики часто
забиваются.
• Требование безопасности. Стеклянные
пипетки легко повреждаются, надколотые концы
представляют собой опасность как для исследователя,
так и для обслуживающего персонала. Заменяйте или
выбрасывайте треснувшие и надколотые пипетки.
11.3.3. Завинчивающиеся крышки
Существует два основных типа крышек, которые исполь¬
зуются для стеклянных бутылей: 1) алюминиевые или
пластиковые крышки с вкладышами из синтетической
резины или силикона и 2) полипропиленовые крышки
без прокладки, которые могут повторно использоваться
(Duran); эти крышки заворачиваются на большую глу¬
бину и имеют кольцевую прокладку, обеспечивающую
надежную защиту от заражения и лучшее переливание
через край (несмотря на то что переливание не реко¬
мендуется при выполнении стерильных работ). Следует
соблюдать следующие меры предосторожности:
1) Полипропиленовые крышки прочно удерживают
содержимое только при закручивании их до упора,
при этом горлышко бутыли не должно иметь сколов
184
ГЛАВА 11. ПОДГОТОВИТЕЛЬНЫЕ РАБОТЫ И СТЕРИЛИЗАЦИЯ
Таблица 11.3. Стерилизация жидкостей
Растворенное вещество
Стерилизация
Хранение
НС1,1М
Фильтр1
Комнатная температура
HEPES
Автоклав2
Комнатная температура
NaHC03
Фильтр
Комнатная температура
NaOH, 1 М
Фильтр
Комнатная температура
Агар
Автоклав или кипячение
Комнатная температура
Аминокислоты
Фильтр
4 °С
Бакто-пептон
Автоклав
Комнатная температура
Бычий сывороточный альбумин
Фильтр (используйте стопку фильтров)
4 °С
Витамины
Фильтр
-20°
Вода
Автоклав
Комнатная температура
Гидролизат лактальбумина
Автоклав
Комнатная температура
Глицерин
Автоклав
Комнатная температура
Глюкоза, 20%
Автоклав
Комнатная температура
Глюкоза, 1-2%
Фильтр (низкие концентрации, при автоклавировании
карамелизуется)
Комнатная температура
Глютамин
Фильтр
-20°
ДМСО
Самостерилизация, диспенсирование в стерильные
пробирки
Комнатная температура, в темно¬
те, избегать контакта с резиной,
пластиком (кроме полипропи¬
лена)
Карбоксиметилцеллюлоза
Пар3,30 мин
4 °С
Коллагеназа
Фильтр
-20°
Метилцеллюлоза
Автоклав
4 °С
Пируват натрия, ЮОмМ
Фильтр
-20°
Раствор солей (без глюкозы)
Автоклав
Комнатная температура
Сыворотка
Фильтр (стопка фильтров)
-20°
Трансферрин
Фильтр
1
to
о
о
Трипсин
Фильтр
-20°
Триптоза
Автоклав
Комнатная температура
Факторы роста
Фильтр (связывающий низкомолекулярные белки)
-20°
Феноловый красный
Автоклав
Комнатная температура
ЭДТА
Автоклав
Комнатная температура
1 Фильтр, размер пор 0,2 мкм.
2 Автоклавирование, 100 кПа (1 атм), 121 °С, 20 мин.
3 Пар, 100 °С, 30 мин.
или других изъянов на верхнем краю. При их нали¬
чии замените бутыль.
2) Не оставляйте алюминиевых крышек или других
алюминиевых предметов в щелочных растворах
детергентов более, чем на 30 мин, поскольку они
подвержены коррозии.
3) Не кладите в одну емкость для замачивания стек¬
лянную посуду и крышки, поскольку алюминий
может загрязнять стекло.
4) Избегайте детергентов, которые предназначены
для машинной мойки, поскольку они слишком
щелочные.
11.3.4. Выбор детергента
Когда большая часть культуральных работ выполня¬
ется при помощи стеклянной посуды, качество (заряд,
химические загрязнения) стеклянной поверхности
становится критическим действующим фактором.
Поскольку большинство клеточных культур сейчас
ведется в одноразовом пластике, основными требова¬
ниями к чистоте стекла являются а) эффективность
детергента, удаляющего загрязнения с поверхности
стекла, и б) отсутствие токсичных остатков после
мытья, которые могли бы попасть в среду или другие
реактивы. Некоторые производители оборудования
для культуральных лабораторий предлагают ряд детер¬
гентов для ручного мытья, которые прошли испытания
на токсичность при культуральных работах (например,
7Х, MP, Biomedicals или Decon), но часто детергенты
для моечных машин поступают от производителей ма¬
шин или от общелабораторных поставщиков. Эффек¬
тивность действия детергента может быть определена
опытным путем — отмыванием сильно загрязненного
стекла (например, бутыли из под сыворотки или среды,
11.3. Оборудование
185
Автоклавируйте отдельно
крышки, обернув их в
паро-водопроницаемую
пленку
CL-^Ъ
Л|
\ы\.
*
Uo
ч\
JX
П п
а1
Замочите на
ночь полностью
погруженными в
нетоксичное моющее
средство
Вымойте в машине без
детергента или вручную
при помощи щетки.
Тщательно прополощите
Высушите в печи,
перевернутыми вверх
дном
Оберните фольгой горлышки
бутылей и стерилизуйте
сухой высокой температурой
или автоклавированием
Ч
Автоклав
Сухожаровый шкаф
Храните крышки в запечатанных
пачках до 1 мес
Охладите бутыли, храните
предохраняя от пыли до 72 ч
СЕмшйГ
О- |Р^
Рис. 11.1. Мытье и стерилизация лабораторного стекла. Процедура выполняется, как в протоколе 11.1. Условия стерили¬
зации: автоклавирование при 121 °С в течение 15 мин; сухой жар, 160 °С в течение 1 ч. Крышки стерилизуются отдельно от
бутылей во избежание образования конденсата, формирующегося в бутылках, если автоклавировать их вместе с крышками
(см. рис. 11.3)
содержащей сыворотку, которую подвергли автокла-
вированию). При проведении стандартной процедуры
мытья разными детергентами визуальный контроль
качества позволяет определить, какой из детергентов
обладает наибольшей эффективностью.
Присутствие токсичных остатков лучше всего опре¬
деляется клонированием клеток (см. протокол 21.10),
например, на чашке Петри, которая вымыта этим де¬
тергентом, прополоскана в соответствии со стандартом
процедуры (см. протокол 11.1) и прошла стерилизацию
сухим жаром. В качестве контроля используется плас¬
тиковая чашка Петри. Этот метод может быть использо¬
ван, если вы предполагаете наличие остатков на посуде
после автоклавирования.
186
ГЛАВА 11. ПОДГОТОВИТЕЛЬНЫЕ РАБОТЫ И СТЕРИЛИЗАЦИЯ
Рис. 11.2. Механические щетки для мытья бутылей. Враща¬
ющиеся щетки расположены под защитным плексигласовым
экраном. Следует тщательно следить, чтобы не прижимать
слишком плотно бутыли к щетке, чтобы бутыль не разбилась
11.3.5. Прочее оборудование
Обработка. Все новое оборудование и материалы (си¬
ликоновые трубки, держатели фильтров, инструменты
и т. д.) должны быть замочены в растворе детергента на
ночь, тщательно промыты проточной водой и прополос¬
каны в соответствии со стандартом и высушены. Все,
что может коррозировать при замачивании в щелочном
растворе детергента, — мягкая сталь, алюминий, медь,
латунь и т.д., — должно быть вымыто вручную без пред¬
варительного замачивания (или после замачивания в
течение 30 мин), ополоснуто и высушено.
После использования все предметы должны быть
промыты водопроводной водой и замочены в детер¬
генте. Оставьте их в ванне для замачивания на ночь,
утром прополощите и высушите. Еще раз напоминаем,
что предметы, подверженные коррозии, не следует
замачивать более чем на 30 мин. Алюминиевые стака¬
ны для центрифуги, как и роторы, никогда не следует
замачивать в детергенте.
Особой заботы требуют предметы, обработанные
силиконовой смазкой и силиконовыми жидкостями.
Их следует обрабатывать отдельно от остальных
предметов, при необходимости силикон удаляется
четыреххлористым углеродом. Силикон очень трудно
удаляется при случайном попадании на оборудование
и стекло.
Упаковка. В идеале, все оборудование, подвер¬
гающееся стерилизации, должно быть упаковано в
оболочку, позволяющую пару проникать внутрь, но
непроницаемую для бактерий, пыли и клещей. Службы,
обеспечивающие снабжение стерильными инструмен¬
тами клинических учреждений, предоставляют им
соответствующие пакеты, имеющие метки-индикато¬
ры, проявляющиеся после стерилизации (например,
Polysciences, Bio-Medical Products). Полупроницаемая
прозрачная нейлоновая пленка (например, Portex
Рис. 11.3. Стерилизованные бутыли. Эти бутыли были автоклавированы с индикаторами стерильности Thermalog внутри.
Thermalog становится синим при действии высокой температуры и пара. Синяя область перемещается по полосе со временем
при требуемых условиях стерилизации. Крышка на крайней левой бутыли была сильно закручена, и каждая следующая крышка
была закручена все слабее, наконец, последняя бутыль стерилизовалась без крышки.
Крайняя левая бутыль не стерильна, потому что пар не попал в нее. Вторая бутыль не стерильна также, потому что вкладыш
сместился на горлышко и запечатал ее. Следующие три бутыли полностью стерильны, но коричневая окраска на индикаторе
показывает, что в конце цикла в них имелась жидкость. Только бутыль справа стерильна и суха. Стеклянные индикаторы
(пробирки Брауна) показывают, что соответствующие бутыли были стерильны (см. также цв. вкладку 22а)
11-3. Оборудование
187
ПРОТОКОЛ 11.2. Подготовка и стерилизация
стеклянных пипеток
Материалы
• Цилиндры для пипеток (для сбора использован¬
ных пипеток)
• Дезинфектанты: гипохлорит минимум 300 ppm
активного хлора при растворе в детергенте (на¬
пример, таблетки Precept, Clorox или Chloros)
• Детергент (например, 7Х™ или Decon®)
• Сетки-корзины из нержавеющей стали (для сбора
и сушки вымытых и прополосканных пипеток)
• Пеналы для пипеток (квадратного сечения, алю¬
миниевые или из нержавеющей стали, с силико¬
новым вкладышем на обоих концах; квадратные
пеналы не скатываются с рабочей поверхности)
(Thermo Electron)
• Индикаторы стерильности
• Ярлык или липкая лента для обозначения сте¬
рильности (см. протокол 11.1). Стерилизационная
печь, снабженная вентилятором, способная нагре¬
ваться до температуры 160 °С, и предпочтительно
с регистратором температуры и гибким зондом.
Протокол
Сбор и обработка пипеток:
1. Налейте воду с детергентом и дезинфектантом в
цилиндр для пипеток.
2. Сбросьте пипетки вниз носиком в цилиндр сразу
после использования (рис. 11.5).
а) Не накапливайте грязные пипетки на рабочей
поверхности и не позволяйте использованным
пипеткам высыхать.
б) Не кладите в этот же цилиндр пипетки, ко¬
торые были использованы для шара или си¬
ликоновых соединений (водоотталкивающие
жидкости, пеногасители и т.д.). Для силико¬
новых соединений используйте одноразовые
пипетки, после агара промойте пипетки горя¬
чей проточной водой или также используйте
одноразовый пластик.
3. Замочите пипетки на ночь или как минимум на
2 ч. Если используется очень много пипеток,
заменяйте цилиндры по мере их заполнения и
замачивайте на 2 ч до передачи на мытье.
4. После замачивания удалите ватные пробки при
помощи сжатого воздуха.
5. Перенесите пипетки в моечную машину для пи¬
петок (рис. 11.4,11.5, см. также рис. 4.6) носиками
вверх.
6. Промойте проточной водой сифонным устрой¬
ством как минимум 4 ч или в автоматической мо¬
ечной машине при помощи адаптера для пипеток,
но без детергента.
7. Поверните клапан для включения в цикл де¬
ионизированной (DW) или обратно-осмотиче-
ски-очищенной (ROW) воды (см. рис. 4.6), или
дождитесь, пока не протечет последняя порция
проточной воды из крана, опустошите до конца
и заполните троекратно DW или ROW. (Ис¬
пользуйте автоматический цикл промывки DW
в моечной машине.)
8. Перенесите пипетки в сушку для пипеток или
сушильную печь и высушите в положении носи¬
ками вверх.
9. Вставьте ватные пробки (рис. 11.6).
10. Разберите по размерам и поместите на хранение,
закрытое от пыли.
Стерилизация:
1. Поместите пипетки в пеналы для пипеток (полез¬
но пометить пеналы с обоих концов ярлыками с
указанием размеров пипеток).
2. Заполните пеналы пипетками одного размера.
3. Заполните несколько пеналом наборами пипеток
объемом 1 мл, 2 мл, 10 мл и 25 мл (например, по
4 каждого объема).
4. Приклейте ярлык-индикатор стерилизации, опе¬
чатайте крышку и поставьте дату на ярлыке.
5. Стерилизуйте сухим жаром в течение 1 ч при
температуре 160 °С, используйте самое малень¬
кое количество липкой ленты или замените ее
на индикаторы температуры (Alpha medical,
Bennett), маленькие по размерам, и сделанные из
менее летучих материалов. Температура должна
измеряться в центре загруженной камеры, для
того чтобы убедиться, что эта, самая труднодо¬
ступная, часть печи прогревается достаточно для
достижения стерилизации. Оставьте место между
пеналами при загрузке печи, чтобы обеспечить
циркуляцию горячего воздуха (рис. 11.7).
6. Выньте пипетки из печи и охладите, перенесите
пеналы в лабораторию культивирования тканей.
Если вы предполагаете, что пипетки будут исполь¬
зоваться не ранее чем через 48 ч, оберните линию
соединения крышки с пеналом липкой лентой.
Plastics, Applied Scientific, KNF) продается в рулонах
или плоских упаковках разного диаметра и может ис¬
пользоваться как пакет с индикатором стерильности.
Несмотря на высокую стоимость, такая пленка может
использоваться несколько раз до того, как станет хруп¬
кой и непригодной для использования.
Пробирки и воронки должны быть завернуты при
помощи липкой ленты и бумаги или нейлоновой плен¬
ки до общей упаковки. Иголки и другие острые пред¬
меты должны быть завернуты вместе со стеклянными
тест-пробирками или другими соответствующими
предохранительными приспособлениями.
Стерилизация. Тип стерилизации зависит от вида
материала (см. табл. 11.1). Металлические предметы
лучше всего стерилизовать сухим жаром. Силиконовая
резина (которая более предпочтительна для использо¬
вания по сравнению с обычной натуральной), ПТФЭ,
поликарбонат, ацетатцеллюлозные и нитратцеллюлоз-
188
ГЛАВА 11. ПОДГОТОВИТЕЛЬНЫЕ РАБОТЫ И СТЕРИЛИЗАЦИЯ
Рис. 11.4. Сифонное моющее устройство для пипеток.
Соединяется с главной системой водоснабжения. При
медленном заполнении устанавливается принцип сифона,
промывается основная ёмкость, которая затем снова на¬
полняется. Этот процесс повторяется много раз в течение
более чем 4-6 ч. Пипетки, показанные здесь, собраны после
использования. Прежде, чем поместить корзину с пипетками
в моющее устройство, выньте ватные пробки из пипеток и
переверните их, как показано на рис. 11.5
ПРОТОКОЛ 11.3. Подготовка и стерилизация
завинчивающихся крышек
Материалы:
• Дезинфектанты: гипохлорит минимум 300 ppm
активного хлора при растворе в детергенте (на*
пример, таблетки Precept)
• Детергент (например, 7Х™ или Decon®)
• Ванны для замачивания
• Сетки-корзины из нержавеющей стали (для сбора
и сушки вымытых и прополосканных крышек)
• Индикаторы стерильности (см. также Приложе¬
ние И)
• Чашки Петри (для упаковки)
• Автоклавируемая пластиковая пленка (см.
Приложение II) или бумажные пакеты для сте¬
рилизации
• Автоклавируемая лента для обозначения сте¬
рильности
• Автоклав с регистрирующим термометром с
гибким зондом, который может быть введен в
загруженную камеру
Протокол
Сбор и мытье:
Металлические или фенолопластиковые
крышки с вкладышами:
1. Замочите минимум на 30 мин в растворе детер¬
гента.
2. Тщательно промойте в течение 2 ч (убедитесь, что
все крышки утоплены). Вкладыши должны быть
удалены и вставлены после прополаскивания,
которое можно проводить двумя способами:
а) В химическом стакане (или ковше) в про¬
точной воде, которая подводится шлангом ко
дну сосуда. Перемешивайте крышки вручную
каждые 15 мин.
б) В корзине или, лучше, в устройстве для мы¬
тья пипеток. Прополощите в автоматической
моечной машине, но не используйте в этом
случае детергент.
Полипропиленовые крышки:
Эти крышки можно мыть и прополаскивать вручную,
как описано выше (включая замачивание в растворе
детергента). Поскольку эти крышки плавают на по¬
верхности воды, следует утопить их при помощи гру¬
за при замачивании и мытье. Для автоматического
мытья после замачивания в детергенте используйте
устройство для автоматического мытья пипеток,
нормальный цикл без использования детергента.
Пробки:
Завернутые крышки предпочтительнее пробок, одна¬
ко, если последние используются, следует выбирать
силиконовые или не содержащие металл пробки из
белой резины, которые предпочтительнее обычных
черных. Мыть и стерилизовать их как крышки
(в этом случае не будет проблем с плаванием на
поверхности).
Стерилизация:
3. Поместите крышки в стеклянные чашки Петри
открытой стороной вниз.
4. Оберните чашки Петри, содержащие пробки,
плотной бумагой или паропроницаемой нейлоно¬
вой пленкой, заклейте автоклавируемой лентой
(рис. 11.8).
5. Приготовьте аналогичную упаковку с индикато¬
ром стерильности внутри и поместите в середину
загрузочной партии.
6. Автоклавируйте 20 мин при 121 °С 100 кПа
(1 атм) (рис. 11.9).
ные фильтры и т.д. следует автоклавировать в течение
20 мин при 121 °С и 100 кПа (1 Бар) при полном цикле
автоклавирования, включая вакуумирование камеры
до и после действия пара (за исключением автоклави¬
рования фильтров и комплектов фильтров, см. прото¬
кол 11.4). При использовании маленьких настольных
11.3. Оборудование
189
Замочите Удалите ватные
на ночь пробки
Промойте в сифонном Заполните и слейте Сушите в течение
устройстве: 4-6 ч деионизованную 1 ч в сушилке для
воду три раза пипеток или печи
«
Ж
Слив
Стерилизуйте сухим жаром Рассортируйте предварительно Заткните ватными J I
или автоклавированием маркированные пеналы вниз пробками
наконечниками
<=«
Охладите и храните, предохраняя от
пыли, в течение 24 ч, или герметизируйте
соединение крышки с пеналом при помощи
ленты и храните до 1 недели
Силиконовый вкладыш
'ента — индикатор
Оберните лентой соединение
крышки и корпуса пенала, с
короткой полосой ленты —
индикатора стерильности
Рис. 11.5. Мытье и стерилизация пипеток
Рис. 11.6. Полуавтоматическое устройство для затыкания
пипеток ватными пробками (Bellco). Толстый жгут из ваты
пропускается через специальную доставочную апертуру, ког¬
да соответствующая длина жгута вставлена в конец пипетки,
жгут обрезается. Для пипеток различного размера требуются
жгуты ваты различной толщины
автоклавов и термобарокамер убедитесь, что автоклав
энергично кипел 15-20 мин до герметизации, чтобы
пар заместил воздух (проследите, чтобы в этом случае
было достаточно воды до начала испарения). После
стерилизации пар выпускается. Все предметы должны
быть высушены в печи или на штативе.
• Требование безопасности. Чтобы избежать
ожога, обратите внимание на момент выпускания пара
Рис. 11.7. Стерилизационный шкаф. Пеналы с пипетками
сложены в стопки, так чтобы позволять горячему воздуху
проходить между ними. Коричневые пятна на фронтальной
поверхности шкафа являются свидетельством наличия лету¬
чих веществ в ленте-индикаторе стерильности
и вынимания горячих предметов. Надевайте изолиру¬
ющие перчатки длиной до локтя и защищайте лицо от
попадания пара при открывании крышек автоклава,
дверей и т. д. Используйте замки безопасности на
автоклавах.
uwr imp
190
ГЛАВА 11. ПОДГОТОВИТЕЛЬНЫЕ РАБОТЫ И СТЕРИЛИЗАЦИЯ
Рис. 11.8. Упаковка закручивающихся крышек для стери¬
лизации. Крышки сложены в стеклянную чашку Петри, кото¬
рую затем запечатали в автоклавируемый пакет из нейлона.
кПа
p.spi.
Рис. 11.9. Отношения между давлением и температурой.
Условия 121 °С и 100 кПа или 1 атм в течение 15-20 мин —
обычно рекомендуемые
11.3.6. Стерилизационные фильтры
многократного использования
Хотя большинство лабораторий в настоящее время
использует одноразовые комплекты фильтров, неко¬
торые крупномасштабные предприятия предпочитают
использовать комплекты фильтров в нержавеющей
оболочке. Комплекты фильтров должны составляться
и стерилизоваться в соответствии с протоколом 11.4.
Альтернативные методы стерилизации. Боль¬
шое количество пластика не может подвергаться
действию высокой температуры, необходимой для
сухожаровой или паровой стерилизации. Для стери¬
лизации таких предметов можно применить действие
70% этанола в течение 30 мин и высушивание при
УФ-облучении в ламинарном шкафу. Следует ак-
ПРОТОКОЛ 11.4. Стерилизация комплекта
фильтров
Материалы
Нестерильные:
• Держатели фильтров (табл. 11.4)
• Микропоровые фильтры, соответствующие дер¬
жателям
• Предварительный фильтр, стекловолоконный,
если необходим
• Паропроницаемый нейлоновый фильтр
• Обозначающая стерильность автоклавируемая
лента
• Автоклав
Протокол
1. После тщательного промывания в детергенте (см.
разд. 11.3.5) прополощите комплект фильтров
проточной водой с последующим прополаскива¬
нием деионизированной водой и сушкой.
2. Вставьте опорную решетку в фильтр и помес¬
тите фильтрующую мембрану на решетку. Если
мембрана сделана из поликарбоната, увлажните,
чтобы противодействовать статическому элект¬
ричеству.
3. Поместите предварительный фильтр (стеклово¬
локонный или другой) на верхнюю часть филь¬
тра.
4. Закрутите держатель фильтра, не затягивайте
полностью (оставьте примерно один полный
оборот).
5. Закрутите впускное и выпускное отверстия алю¬
миниевой фольгой.
6. Упакуйте комплекты фильтров в бумагу для
стерилизации или паропроницаемую нейлоновую
пленку, заклейте автоклавируемой лентой.
7. Автоклавируйте при 121 °С и 100 кПа (1 Бар,
15 фунтов/дюйм2) в течение 20 мин без предва¬
рительной и последующей вакуумизации (ис¬
пользуйте «жидкостный цикл» в автоматических
автоклавах)
8. Выньте фильтры, охладите.
9. Не затягивайте держатели фильтров полностью
до тех пор, пока фильтры не увлажнятся перед
фильтрацией (см. протокол 11.14.6).
куратно обращаться с некоторыми видами пластика
(например, Plexiglas, Perspex, Lucite), поскольку они
могут деполимеризоваться под действием спирта или
ультрафиолетового излучения. Для стерилизации
пластика можно также использовать оксид этилена,
но для полного освобождения поверхности пластика
от него потребуется две-три недели. Самым лучшим
методом стерилизации пластика является у-излучение,
на уровне 25 кГрэй. Все предметы должны быть упа¬
кованы и запаяны; можно использовать полиэтилен,
который запаивается действием температуры.
11.4. Реагенты и среды
191
11.4. РЕАГЕНТЫ И СРЕДЫ
Конечной целью приготовления сред и реактивов
является получение их в чистом виде, при котором
исследователь 1) избегает случайного попадания инги¬
биторов и токсичных веществ, 2) знает концентрации
и понимает функции всех составляющих элементов,
3) уменьшает риск микробного заражения.
Большинство реактивов и сред, если они термоста¬
бильны, например вода, растворы солей, гидролизаты
аминокислот, может быть стерилизовано автоклави-
рованием. Термолабильные растворы стерилизуются
фильтрацией. Растворы должны быть разлиты в
стеклянные боросиликатные или пластиковые поли-
карбонатные бутыли и плотно закрыты во избежание
испарения и химического загрязнения при автоклави-
ровании. Если используется натриевое стекло, то лучше
сохранить небольшой зазор, чтобы минимизировать
риск взрыва бутылей. Образование пара в бутыли по¬
может предупредить попадание пара из автоклава, но
уровень жидкости уменьшится. Поэтому потребуется
его восполнение добавлением стерильной сверхчистой
воды. Так же как при стерилизации оборудования, ин¬
дикаторы стерильности (например, Thermalog) и зонд
записывающего термометра должны быть расположены
в образце, помещенном в центре камеры.
Среды и реактивы, расположенные рядами в круп¬
номасштабных культуральных сосудах в промышлен¬
ных и полупромышленных биореакторах, могут быть
стерилизованы кратковременной обработкой очень
высокой температуры (Alfa-Laval). Приспосабливая
процесс к промышленному производству сред, можно
достичь высокой эффективности при значительном
снижении стоимости.
11.4.1. Вода
Вода, используемая в культивировании тканей, в
частности для приготовления бессывороточных сред
(см. также разд. 5.4.2), должна быть очень чистой. По¬
скольку снабжение водой очень различно, требуемая
степень очистки также может быть различна. Жесткая
вода может потребовать использования ионообменного
смягчения до попадания в систему очистки, мягкая
вода может непосредственно поступать из системы
водоснабжения в водоочищающую установку.
Существует четыре главных метода очистки воды:
обратный осмос, дистилляция, деионизация и угольная
фильтрация. Для получения сверхчистой воды (UPW)
первой стадии является стадия обратного осмоса, но
она может быть замещена дистилляцией (рис. 11.10;
см. также рис. 5.17). Преимуществом дистилляции
является одновременная стерилизация воды теплом,
но этот процесс экономически дороже, поскольку тре¬
бует расхода энергии и требуется регулярная очистка
бойлера. При использовании на первой стадии стек¬
лянного дистиллятора он должен контролироваться
с использованием электрического предохранителя,
нагревательные элементы должны быть выполнены из
боросиликатного стекла или покрыты кремнием. Об¬
ратный осмос зависит от целостности фильтрационных
Система водоснабжения
Первая стадия очистки
обратным осмосом или
дистилляцией
Обратная петля
Выпуск полу-
очищенной
воды
Микропоровый фильтр
Прямое использование
сверхчистой воды
Выпускное отверстие
для очищенной воды
Емкость
Угольный Высокока-
фильтр чественная
деионизация
Рис. 11.10. Очистка воды. Вода из водопровода поступает в контейнер для хранения через устройство для обратно осмотиче¬
ской очистки или стеклянный дистиллятор. Эта полуочищенная вода затем поступает обратно в емкость для хранения после
фильтрации через угольный фильтр, деионизацию и микропоровую фильтрацию. Вода необходимого качества для приго¬
товления реагентов всегда доступна для использования из емкости для хранения; вода нужного качества для приготовления
среды доступна из резервуара после микропоровой фильтрации (направо от диаграммы). Если аппарат работает непрерывно,
то вода самой высокой чистоты для приготовления среды может быть получена в первой порции в начале рабочего дня (см.
также рис. 5.17)
192
ГЛАВА 11. ПОДГОТОВИТЕЛЬНЫЕ РАБОТЫ И СТЕРИЛИЗАЦИЯ
мембран; следовательно, должен вестись мониторинг
потока. Тип картриджа, применяемого в обратно-осмо¬
тической установке, зависит от pH воды, поступающей
из системы водоснабжения (подробнее см. специфи¬
кации производителя). Если стоимость энергии для
стадий дистилляции и замены картриджей вычитается
из вашего бюджета, то обратно-осмотическая очистка
воды представляется более дешевой, но если вы не оп¬
лачиваете потребляемую электроэнергию или оплата
постоянна и не связана с реальным ее использованием,
то дистилляция дешевле.
Второй стадией является угольная фильтрация,
при которой происходит удаление органических и
неорганических коллоидных частиц. Третья стадия —
высококачественная очистка на фильтре смешанной
загрузки, при которой происходит деионизация для
удаления ионов органических веществ. Последняя
стадия — микропоровая фильтрация для удаления
любых микроорганизмов, попавших в систему, и захват
всех смол, которые могли попасть из ионообменника.
Для минимизации загрязнений при хранении вода
должна собираться после микропоровой фильтрации
непосредственно в конечные стерильные емкости.
Вода, полученная после первой стадии очистки, может
использоваться для ополаскивания при обработке по¬
суды и инструментов. Если вода постоянно (например,
в течение ночи) циркулирует из микропорового филь¬
тра в емкость, куда также поступает полуочищенная
вода из первой стадии очистки, обратно на микро-
поровую фильтрацию, то постепенно в этой емкости
происходит повышение чистоты воды («полировка»).
В такой системе вода может использоваться утром для
приготовления сред.
Качество деионизованной воды должно контролиро¬
ваться по ее проводимости (или сопротивлению) через
определенные интервалы, картридж следует заменять
при видимом возрастании проводимости. Стандарт
ISO 3696 (Международная организация стандартиза¬
ции International Standardization Organization) уста¬
новил значение сопротивления I типа воды на уровне
> 10 МОм/см (проводимость < 0,l|iS/cm) при 25 °С.
Общий органический углерод (ТОС) должен составлять
<10 частей на миллиард. Кондуктометр обычно постав¬
ляется вместе с системой очистки воды, измеритель
ТОС следует приобрести отдельно (см. Приложение II).
Дистилляция должна предшествовать деионизации,
обеспечивая поступление стерильной воды на стадию
деионизации. Стадия ультрафильтрации может нахо¬
диться между стадией деионизации и микропоровой
фильтрации для получения апирогенной воды.
Стерилизация воды достигается автоклавировани-
ем при 121 °С и 100 кПа в течение 20 мин. В емкостях
подходящего размера (например, в объеме, необхо¬
димом для разведения концентратов сред) бутыли
должны быть закрыты до автоклавирования, что тре¬
бует применения боросиликатного стекла (Ругех) или
поликарбонатного пластика (Nalge Nunc). Если бутыли
не закрыты (например, по причине применения натрие¬
вого стекла, которое может разбиться при автоклавиро-
вании), то необходимо добавить 10% дополнительного
объема воды на бутыль в расчете на испарение.
Количество, представленное в протоколе 11.5,
рассчитано на выполнение Упражнения 6 (см. гл. 2),
но может варьировать в соответствии с требованиями
лаборатории.
ПРОТОКОЛ 11.5. Приготовление
и стерилизация сверхчистой воды (UPW)
Оборудование и материалы
Нестерильные:
• Градуированные стеклянные боросиликатные
(например Ругех) или автоклавируемые плас¬
тиковые (поликарбонатные) бутыли с заворачи¬
вающимися крышками {убедитесь, чтоимеётся
достаточное свободное пространство для нослё-
дующих добавок), 500 мл **•>••••«*•»» »«>21.
• Подходящие крышки
• Индикаторные полоски для определения сте¬
рильности
• Лента для индикации стерильности --v. -
• Маркер или предварительно напечатанные яр¬
лыки
• Оборудование для очистки воды "
• Автоклав V V.
• Регистрационный журнал(длязшшс^древара^
тивных работ и стерилизации)
Стандартный протокол
1. Подпишите бутыли, о6означив дату, еодержймое
и номер партии.
2. Рано утром после ночного цикла слейте изочис-
; тителя воды в отходы около 50 мл. проверьте
проводимость (или со1фоташлеш|е) и 0бвдий ор-
ганический углерод СГОС) на соответствующих
измерительныхириборах, занесите записи в
регистрадйонный журнал.
3. Если измеренные параметры соответствуют
требованиям (сопротивление >10 МОм/см при
25 °С> ТОС <10 частей на миллиард (ррЪ), собе¬
рите воду непосредственно в емкости для хране¬
ния. “--Г
4. Наполните бутыли до определенной отметки, на¬
пример 430-450 мл, есливодабудет использовать¬
ся для разведения сред (10х) (см. протокол 11.8).
Если предполагается автоклавирование, добавьте
дополнительно 10% объема.
5. Поместите шадшаторетерильвостиводну избуты-
лей (после автоклавирования следует удалить). .
6. Закройте бутыли закручивающимися крышками.
При использовании натриевого стекла» например,
если бутыли могут лопнуть при автоклавирова-
нии, ослабьте крышки. \
7. Поставьте бутыли в автоклав вместе с бутылью,
содержащей индикатор стерильности, в центре
загрузочной партии.
11.4. Реагенты и среды
193
8. Закройте автоклав, проверьте параметры установ¬
ки: 121 °С и 100 кПа (1 Бар, 15 фунтов/дюйм2),
20 мин с отменой последующего вакуумирования.
9. Включите цикл стерилизации.
10. По завершении цикла проверьте распечатку
самописца, чтобы убедиться, что необходимые
параметры сохранялись в течение всего цикла,
занесите записи в регистрационный журнал.
11. Оставьте охладиться примерно до 50 °С.
12. Откройте автоклав, выньте бутыли. Если крыш¬
ки были ослаблены, плотно закрутите их, когда
температура бутылей опустится до комнатной.
Замечание. Запечатывание бутылей, которые были
открыты в ходе автоклавирования, еще не остывших
до комнатной температуры, может вызывать втяги¬
вание вкладышей из некоторых крышек в бутыли.
При первом открывании будет резкое втягивание
наружного воздуха в бутыль, что может вызвать за¬
ражение. До того как крышки будут плотно закрыты,
бутыли, которые были приоткрыты при автоклавиро-
вании, должны остыть до комнатной температуры в
стерильной атмосфере, например в автоклаве, или в
ламинарном шкафу с горизонтальным потоком.
Проверьте индикатор стерильности, убедитесь, что
был достигнут режим стерилизации, сделайте записи
в регистрационном журнале.
13. Поместите бутыли на кратковременное хранение
при комнатной температуре.
14. Замените запасы UPW в культуральной ком¬
нате, если это требуется, проверьте по наличию
индикаторной ленты, что бутыли прошли цикл
стерилизации.
11.4.2. Сбалансированный солевой
раствор (BSS)
Состав BSS обсуждался в предыдущих главах (см.
разд. 9.3). Формула раствора Хэнкса [Paul, 1975] вклю¬
чает в себя хлористый магний вместо некоторых других
рекомендуемых сульфатов (см. табл. 9.2); его следует
автоклавировать при pH ниже 6,5 для предупрежде¬
ния выпадения в осадок фосфатов кальция и магния
с последующей нейтрализацией pH. Аналогичным
образом, фосфатно-буферный раствор Дюльбекко
(D-PBS) готовится без кальция и магния (D-PBSA),
раствор которых готовится отдельно (D-PBSB) и до¬
бавляется при необходимости использования. DPBS
часто используется также и без добавления Са2+ и Mg2+;
в этой форме он называется (D-PBSA) либо (D-PBS)
без Са2+ и Mg2+; мы условились в этой книге называть
этот раствор «D-PBSA». Сокращение «D-PBS» так¬
же используется для того, чтобы отличить раствор
Дюльбекко от простого фосфатно-буферного раствора
(PBS), в который не входят хлорид калия и который
является изотоническим раствором хлорида натрия
в фосфатном буфере и не должен рассматриваться
как сбалансированный солевой раствор. В докладах и
публикациях всегда должно быть точно указано, какой
именно BSS был использован в работе. Большинство
BSS содержат глюкозу, которая может карамелизо¬
ваться при высокой температуре автоклавирования,
поэтому лучше не добавлять ее в ходе приготовления
и стерилизации BSS, а готовить раствор отдельно и до¬
бавлять перед использованием. Если раствор глюкозы
готовится в концентрированном виде 100х (200 г/л),
карамелизации при автоклавировании не происходит,
этот концентрат может быть затем добавляться в BSS
в объеме 5-25 мл/л с получением конечной концент¬
рации 1-5 г/л. Напротив, можно готовить полные
BSS, так же как и полные питательные среды (см.
протокол 11.8) и стерилизовать их фильтрацией (см.
протоколы 11.11-11.14). Протокол 11.6 дается для
DPBS-А, который обычно используется как раствор
для промывания и растворитель для трипсина или
EDTA, а также может быть использован для любых
BSS, в которых отсутствует глюкоза и бикарбонаты. Ко¬
личество, упомянутое в упражнении 7 (см. гл. 2), может
быть пропорционально увеличено по требованию.
ПРОТОКОЛ 11.6. Приготовление
и стерилизация D-PBSA
Схема
Растворяют порошок при постоянном перемеши¬
вании, доводят до конечного объема, проверяют
pH, проводимость, разливают на порции и авто-
клавируют.
• Порошок D-PBSA (Раствор А, с пониженным со¬
держанием Са24 и Mg2+, например Sigma D5652),
упаковка на 1 л 1
• или таблетки (Oxoid Вг 14а) 10
• Сверхчистая вода (UPW, см. разд. 11.4Л ) 1 л
• Емкости:
• Чистый стеклянный или пластиковый отсос с
пробкой на выпускном отверстии в основании,
1 л 1
• или
• Флакон или бутыль Эрленмейера, соединенный с
перистальтическим насосом, и трубками, 1 л 1
• Магнитная мешалка и покрытый политетраф¬
торэтиленом вкладыш..,. .1
• Градуированные бутыли для хранения; бороси¬
ликатное стекло (Ругех, Schott), 100 мл 10
• Кондуктометр или осмометр
• рН-метр
• Автоклавируемая лента или ярлыки-индикаторы
стерильности
• Автоклав
194
ГЛАВА 11. ПОДГОТОВИТЕЛЬНЫЕ РАБОТЫ И СТЕРИЛИЗАЦИЯ
Протокол
1. Добавьте 1 л UPW в емкость.
2. Поместите емкость на магнитную мешалку, уста¬
новите режим вращения 200 об./мин.
3. Откройте пакет с порошком D-BPSA или отсчи¬
тайте 10 таблеток, постепенно добавьте в воду
при перемешивании.
4. Перемешайте до полного растворения.
5. Проверьте pH и проводимость образца, внесите
записи в регистрационный журнал.
а) pH не должно отклоняться от установленного
более чем на 0,1 ед. pH (в различных рецепту¬
рах pH может различаться).
б) проводимость не должна отличаться от стан¬
дарта более чем на 5% от 150 |iS/cm. Можно
использовать осмолярность как альтернативу
проводимости (или в дополнение), при этом
в партии должна быть одинаковая плотность
раствора.
Замечание. Важно определять параметры плотности
раствора в качестве контроля качества, чтобы убе¬
диться, что приготовление выполнено без ошибки.
Любая коррекция, например осмолярности, должна
проводиться после полной проверки качества при¬
готовления раствора.
в) образец выбросьте в отходы, не возвращайте
обратно в основную емкость.
6. Разлейте содержимое емкости в градуированные
бутыли.
7. Закройте крышки, укупорьте. Полезно закрыть
крышки бумагой, закрепив ее эластичной лентой.
Бумага должна быть снабжена датой и пометкой,
обозначающей содержимое бутыли, используя
штамп, это позволит избежать смывания марки¬
ровки с бутыли.
8. Приклейте небольшой кусок ленты для автокла¬
вирования или ярлыка для индикации стериль¬
ности, с подписанной датой.
9. Стерилизуйте автоклавированием в течение
20 мин при 121 °С и 100 кПа в укупоренных бу¬
тылях (см. рис. 11.9).
10. Храните при комнатной температуре.
11.4.3. Приготовление
и стерилизация среды
В ходе подготовки сложных растворов следует позабо¬
титься о том, чтобы гарантировать, что все элементы
растворены и не остаются на фильтре при стерилизации
фильтрованием и что они остаются в растворе после
автоклавирования или при хранении.
Концентрированные среды часто готовят при низкой
pH (между 3,5 и 5,0), чтобы удержать все элементы в рас¬
творенном состоянии, но даже тогда может происходить
некоторое осаждение. Если все компоненты должным об¬
разом растворены, то даже при выпадении и в осадок они
обычно вновь растворяются; но если выпадение осадка
связано с разрушением некоторых компонентов среды,
то качество среды может снизиться. При образовании
осадка качество среды может быть проверено ростом
и клонированием клеток на этой среде и обеспечением
совместимости и другими соответствующими тестами
для проверки специальных функций (см. разд. 9.6.2).
Коммерческие среды предоставляются в виде
1) растворов рабочей концентрации 1х с глутамином
или без него; 2) концентраты 10х, обычно без NaHC03
и глутамина, которые поставляются как отдельные
концентраты; или 3) порошкообразные среды, с или
без NaHC03 и глутамина. Порошкообразные среды
наиболее дешевы и не намного дороже, чем составлен¬
ные в вашей собственной химический лаборатории
из отдельных компонентов, если вы учтете время на
приготовление, стерилизацию и контроль качества,
стоимость исходных материалов высокой чистоты,
стоимость дополнительных расходов, таких как энер¬
гия и заработная плата работникам. Порошкообразные
среды проходят контроль качества на производстве в
отношении обеспечения роста клеток, но, конечно, не
на предмет стерильности. Они перемешиваются очень
качественно при помощи шариковых мельниц, поэтому
теоретически одна упаковка среды может быть разде¬
лена на порции для использования за несколько раз.
Однако на практике оказывается удобнее приобретать
упаковки сред такого объема, чтобы использовать их
за один раз, поскольку в открытой упаковке качество
содержимого может испортиться, и некоторые из ком¬
понентов могут осесть на дно пакета.
Десятикратные концентраты стоят в два-три раза
дороже (в пересчете на 1 л среды рабочей концентра¬
ции) по сравнению с порошкообразными средами, од¬
нако при этом происходит экономия за счет отсутствия
стадии стерилизации. Приобретение сред рабочей
концентрации наиболее дорого (в 4-5 раз по сравнению
с концентратом 10х), но наиболее удобно, поскольку
не требует дальнейших препаративных работ, кроме
добавления сыворотки, если это необходимо. Прото¬
кол 11.7 описывает приготовление полной среды из ос¬
новного раствора 1х; упоминаемое количество связано
с выполнением упражнения 3 (см. гл. 2). Предлагаемые
объемы минимальны, хотя они могли быть предвари¬
тельно отобраны из бутылей большего объема.
ПРОТОКОЛ 11.7. Приготовление среды
из концентрата с кратностью 1 х
Схема
Проверьте рецептуру; если это полная среда, она
может использоваться непосредственно после добав¬
ления сыворотки, если это требуется (см. разд. 9.6,
13.6.2). Если состав неполный (например, при не¬
достатке глютамина), добавьте соответствующее
вещество- концентрат.
11.4, Реагенты и среды
195
Примечание. Компоненты (например, сыворотка или
антибиотики) это элемент, который добавляется к
среде и отсутствует в первоначальной рецептуре.
Необходимо описание всех компонентов, которые
вы вводите в среду, в любой публикации. Другие
компоненты (например, глютамин или NaHC03)
являются обычной составной частью рецептуры, и
они не должны описываться в публикациях, если
только вы не ввели какие-то изменения в значения
их концентраций.
Материалы
• Пипетки и т.д., как описано в Протоколе для
стерильной техники (см. протокол 6.1,6.2), плюс
дополнительное количество для обеспечения
работы предполагаемого объема:
• Основная среда, например МЕМ Эрла с солями
Хэнкса 100 мл
• Глютамин, 200 шМ (должен быть разморожен на
бане 1 мл
• Бычья сыворотка (размороженная), фетальная
или новорожденного теленка 10 мл
• Антибиотики (если требуется; не рекомендуются
для обычного использования):
• Пенициллин в ССР или D-PBSA,
10,000Ед/мл . 0,5 мл
• Стрептомицин в ССР или D-PBSA,
10 мг/мл 0,5 мл
Протокол
1. Проверьте состав среды и определите, какие ком¬
поненты требуются, например глютамин.
2. Перенесите среду вместе с необходимыми ком¬
понентами в ламинар-бокс.
3. Снимите упаковку с бутылей, если они обернуты
в полиэтилен, протрите 70%-м этанолом.
4. Снимите крышки с бутылей.
5. Перенесите соответствующий объем каждого
компонента в основной раствор для получения
раствора правильной концентрации;
Для 100 мл среды используйте следующие компо¬
ненты и количества:
Глютамин, 200 мМ.. 1 мл
Антибиотики (если используются) по 0.5 мл
Сыворотка 10 мл (для 10%)
а) Используйте различные пипетки для каждой
добавки.
б) Перемещайте каждую новую бутыль с основ¬
ной средой в противоположную сторону ла-
минар-бокса после внесения компонента так,
чтобы вы знали, что манипуляция проведена.
в) Уберите все компоненты из ламинар-бокса
после завершения изготовления среды.
6. Этот протокол предполагает использование МЕМ
с солями Хэнкса, который имеет низкую концен¬
трацию бикарбоната (4 мМ). Если используется
МЕМ с солями Эрла, или другая среда с высоким
содержанием бикарбоната (23 мМ), то необхо¬
димо добавление 5% С02 к газовой фазе. Не про¬
пускайте газ через среду, содержащую сыворотку,
так как образующиеся пузыри вызовут образова¬
ние пены, которая может выходшъ из горлышка
сосуда, что связано с риском загрязнения.
7. Вновь закройте бутыли крышками.
8. Измените этикетки, внесите записи добавленных
компонентов, даты и инициалы оператора.
9. Возвратите среду на хранение при 4 °С или не¬
посредственно используйте (см. гл. 2, упражне¬
ние 4).
10. При использовании новой среды впервые внесите
пипеткой определенное количество ее во флакон
или чашку Петри, инкубируйте, по крайней мере,
1 ч (предпочтительно в течение ночи) при ваших
стандартных условиях, чтобы гарантировать,
что устанавливается равновесие pH в нужных
пределах. Если это не так, то откорректируйте
pH среды или измените концентрацию С02 в
газовой фазе.
Примечание к протоколу 11.7. Изменение кон¬
центрации С02 в инкубаторе подействует на все
культуральные среды, находящиеся в инкубаторе.
Изменение концентрации углекислого газа должно
рассматриваться как однократное воздействие и не
должно использоваться для уменьшения колебаний pH
от партии к партии. Эти колебания лучше контролиро¬
вать добавлением стерильной кислоты или щелочи.
Приготовление среды из концентрата 10х представ¬
ляет собой компромиссное решение между экономиче¬
ским выигрышем приготовления среды из порошка (см.
протокол 11.9) и легкостью использования раствора
рабочей концентрации 1х (см. протокол 11.7). Прото¬
кол 11.8 разработан для того, чтобы быть использован¬
ным при выполнении упражнения 10 (см. гл. 2).
Если потребуется добавить какие-то иные состав¬
ляющие, растворенные в воде, то необходимо учесть
изменение объема и уменьшить соответственно объем
воды в бутыли. Если дополнительные компоненты
разведены в изотоническом растворе соли, то они могут
быть добавлены к конечному объему среды. Поскольку
сыворотка является изотоническим раствором, она
может быть также добавлена к конечному объему,
хотя это, естественно, несколько снизит концентрации
веществ в среде. Всегда контролируйте и доводите pH
до необходимого значения при 37 °С, поскольку при
повышении температуры снижается растворимость
С02, а также изменяется рКа HEPES.
Регулирование pH. Количество щелочи, необходи¬
мой для нейтрализации концентрата среды 10х (которая
сделана кислой для обеспечения хорошей растворимости
компонентов), может варьировать от партии к партии
и от одной среды к другой. На практике, титрование
среды до pH 7,4 при 37 °С может быть достаточно за¬
труднительным. Приготавливая новую среду впервые,
196
ГЛАВА 11. ПОДГОТОВИТЕЛЬНЫЕ РАБОТЫ И СТЕРИЛИЗАЦИЯ
ПРОТОКОЛ 11.8. Приготовление среды
из концентрата с кратностью 10х
Схема
Простерилизовать несколько порций деионизиро¬
ванной дистиллированной воды такого объема, что¬
бы одна аликвота использовалась в течение одной-
трех недель. Добавить концентрированную среду и
другие добавки, довести pH и использовать раствор
или поставить в холодильник на хранение.
Материалы
Пипетки и т.д. аналогично списку для стерильной
техники (см. протоколы 6.1, 6.2), плюс дополни¬
тельное количество, достаточное для обеспечения
следующих манипуляций:
А. Для запечатанной фляги культуры с воздушной
газовой фазой, низкой концентрацией НС03“, ат¬
мосферной концентрацией С02, и низкой буферной
емкостью.
Стерильные растворы для 500 мл среды:
• Предварительно определенное количество
UPW 443 мл
• Среда, концентрат 1 Ох, например, МЕМ с солями
Хэнкса 50 мл
• Глютамин (размороженный) до конечной кон¬
центрации 2 мМ, 200 тМ, 5 мл
• NaHC03, чтобы до конечной концентрации 4 мМ,
7,5% (0,89М) 5 мл
• Бычья сыворотка (размороженная), до конечной
концентрации 10%, новорожденного теленка или
фетальная 50 мл
• Антибиотики если требуется (не рекомендуются
для постоянного использования):
Пенициллин, до конечной концентрации
50 ед./мл; 10,000 ед./мл 2,5 мл
Стрептомицин до конечной концентрации
50 мкг/мл; 10 мг/мл 2,5 мл
• HEPES (если требуется), 1,0 М 5-10 мл
• NaOH, 1 М по потребности
Протокол
1. Разморозьте сыворотку и глютамин перенесите
в ламинар-бокс.
2. Перед внесением в ламинар-бокс протрите этано¬
лом все бутыли, которые были в водяной ванне
3. Добавьте вещества, как описано в разделе сте¬
рилизации растворов. Можно добавить HEPES,
чтобы увеличить буферную емкость раствора, для
некоторых клеточных линий с высокой клеточ¬
ной плотностью, если образуется много кислоты,
можно использовать вентилируемые флаконы.
4. Добавьте 1М NaOH, чтобы получить pH 7,2 при
20 °С. При инкубировании при 37 °С значение
pH среды повысится до 7,4, но первый раз, когда
используется среда данного состава, это значе¬
ние должно быть экспериментально проверено
титрованием (см. ниже, протокол 11.7, и гл. 2,
упражнение 10).
B. Для культур в открытых судах в С02 инкубаторе,
или в запечатанных флаконах, с 5% С02и высокой
концентрацией бикарбоната
Стерильные растворы на к 500 мл среды:
• Предварительно измеренное определенное коли¬
чество UPW 443 мл
• Концентрат среды 1 Ох, например, МЕМ с солями
Эрла 50 мл
• Глютамин (размороженный) до конечной кон¬
центрации 2 мМ, 200 мМ 5 мл
• NaHC03, до конечной концентрации 26 мМ; 7,5%
(0,89 М) 14,5 мл
• Бычья сыворотка (размороженная), до конечной
концентрации 10%, новорожденного теленка или
фетальная 50 мл
• Антибиотики если требуется (не рекомендуются
для постоянного использования):
Пенициллин; 10,000 ед/мл 2,5 мл
Стрептомицин 10 мг/мл 2,5 мл
• NaOH, 1 М по потребности
Протокол
1. Разморозьте сыворотку и глютамин перенесите
в ламинар-бокс.
2. Перед внесением в ламинар-бокс протрите этано¬
лом все бутыли, которые были в водяной ванне
3. Добавьте вещества, как описано в разделе сте¬
рилизации растворов. Можно добавить HEPES,
чтобы увеличить буферную емкость раствора, для
некоторых клеточных линий с высокой клеточ¬
ной плотностью, если образуется много кислоты,
можно использовать вентилируемые флаконы.
4. Добавьте 1М NaOH, чтобы получить pH 7,2 при
20 °С. При инкубировании при 37 °С значение
pH среды повысится до 7,4, но первый раз, когда
используется среда данного состава, это значение
должно быть экспериментально проверено титро¬
ванием (см. доводка pH, ниже):
а) Внесите 5 мл в каждую из 5 чашек Петри.
б) Добавьте различное количество IN NaOH, —
от 5 до50 мкл, — в каждую чашку Петри.
в) Инкубировать в атмосфере 5% С02 как мини¬
мум двух, либо, предпочтительно, в течение
ночи.
C. Для культур в открытых судах в С02 инкубаторе,
или в запечатанных флаконах, с 2% С02и высокой
концентрацией бикарбоната
Стерильные растворы на 500мл среды:
• Предварительно определенное количество
UPW 443 мл
• Среда, концентрат 10х, например, среда Хэма
F12 50 мл
11.4. Реагенты и среды
197
• Глютамин (размороженный) до конечной кон¬
центрации 2 мМ, 200 мМ * 5 мл
• NaHC03, до конечной концентрации 8 мМ, 7.5%
(0,89М) 9 мл
• HEPES, до конечной концентрации 20 мМ;
1,0 М 10 мл
• Бычья сыворотка (размороженная), до конечной
концентрации 10%, новорожденного теленка или
фетальная 50 мл
• Антибиотики, если требуется (не рекомендуются
для постоянного использования):
Пенициллин; 10,000 ед/мл .......................2,5 мл
Стрептомицин; 10 мг/мл 2,5 мл
• NaOH, 1 М по потребности
Протокол
1. Разморозьте сыворотку и глютамин перенесите
в ламинар-бокс,
2. Перед внесением в ламинар-бокс протрите этано¬
лом все бутыли, которые были в водяной ванне.
3. Добавьте вещества, как описано в разделе стери¬
лизации растворов.
4. Добавьте 1М NaOH, чтобы получить pH 7,2 при
20 °С. При инкубировании при 37 °С значение
pH среды повысится до 7,4, но первый раз, когда
используется среда данного состава, это значение
должно быть экспериментально проверено титро¬
ванием (см. шаг В4 выше и Доводка pH ниже).
добавляйте в различные порции среды определенные
количества NaHC03 и оставьте их при 37 °С в соответ¬
ствующей газовой фазе. Проверьте pH на следующее
утро и выберите правильное количество щелочи. Осталь¬
ную среду готовьте в соответствии с результатом.
Бикарбонат натрия. Концентрация бикарбоната
важна при установлении стабильного равновесия с
атмосферным С02. Независимо от количества ис¬
пользованного бикарбоната, если среда находится при
37 °С и pH 7,4, каждой концентрации С02 будет соответ¬
ствовать определенная концентрация бикарбоната (см.
разд. 9.2.2 и табл. 9.1). Некоторые среды разработаны
для использования при высоких концентрациях би¬
карбоната и повышенной концентрации С02 (напри¬
мер, среда Игла МЕМ с солями Эрла), в то время как
другие имеют низкую концентрацию бикарбоната для
использования в атмосфере атмосферного воздуха (на¬
пример, среда Игла МЕМ с солями Хэнкса, см. табл. 8.1
и 8.2). Если среда модифицируется и ее компоненты,
в том числе концентрация бикарбоната, меняются, то
важно удостовериться, что осмолярность находится в
приемлемом диапазоне. Осмолярность всегда следует
контролировать (см. разд. 9.2.5), когда производится
любое значительное изменение среды, приводящее к
отличию от первоначальной рецептуры.
Добавки к среде. Если ваш расход питательных
сред достаточно высок (> 200 л/год) и вы приобретаете
готовые среды, то будет лучше, если вы будете приобре¬
тать дополнительное количество компонентов, которые
должны быть использованы как добавки, поскольку это
будет дешевле. В частности, HEPES очень дорог, если
покупать его отдельно. Глутамин часто поставляется
отдельно от сред, поскольку он нестабилен; лучше
приобретать его отдельно и хранить в замороженном
виде. Время полужизни глутамина в среде при 4 °С
составляет около 3 недель и при 37 ° С около 1 неде¬
ли. Некоторые дипептиды глутамина повышают его
стабильность, в то время как биологическая ценность
глутамина сохраняется. К таким коммерческим препа¬
ратам глутамина относится Glutamax, производимый
компанией Invitrogen.
Выпадение осадка. Следует позаботиться о том,
чтобы все компоненты концентрата 10х находились
в растворенном виде или, по крайней мере, были тща¬
тельно ресуспендированы до разведения концентрата.
Некоторые соединения (такие как фолиевая кислота
или тирозин) могут выпадать в осадок и теряться при
разведении. Инкубация при 37 °С в течение нескольких
часов может помочь преодолеть эту проблему.
Контроль качества. Если партия среды исследо¬
вана на предмет ростовых и других свойств, и они при
этом признаны удовлетворительными, то не требуется
проводить тестирование всякий раз при использовании
среды из этой партии. Однако стерильность среды,
полученной разведением концентрата 10х, до начала
ее использования должна быть проверена инкубиро¬
ванием аликвоты полной среды при 37 °С в течение
48 ч (см. разд. 11.6.2).
11.4.4. Порошкообразные среды
Инструкция к применению порошкообразных сред
прилагается к каждой упаковке. Выберите такой раз¬
мер упаковки, который вы можете использовать за один
раз, и используйте среду в течение трех месяцев. Вы¬
бирайте рецептуру без глютамина. Если присутствуют
другие нестабильные компоненты, их также можно не
включать и добавлять позже как стерильный концент¬
рат. Хотя BSS могут быть стерилизованы автоклави-
рованием (см. протокол 11.6), при этом нежелательно
добавлять глюкозу и бикарбонат. Если BSS может быть
автоклавирован полностью, воспользуйтесь процеду¬
рой приготовления для порошкообразных сред (см.
протокол 11.9).
Для тех, кто использует меньшие количества
(< 1,0 л/нед) или несколько различных типов сред
меньших объемов, могут быть подготовлены полные
среды с глутамином, которые будут профильтрова¬
ны непосредственно в емкости для хранения: (см.
разд. 11.5.2 и протокол 11.12). При использовании
фильтрационной системы отрицательного давления
некоторое количество растворенного С02 может быть
потеряно в процессе фильтрации, и pH может повы-
198
ГЛАВА 11. ПОДГОТОВИТЕЛЬНЫЕ РАБОТЫ И СТЕРИЛИЗАЦИЯ
ПРОТОКОЛ 11.9. Приготовление среды
из порошка
Схема
Развести все содержимое пакета в нужном объеме
UPW, используя магнитную мешалку и добавляя
порошок постепенно с постоянным перемешивани¬
ем. Когда все компоненты растворятся полностью,
среда должна быть немедленно профильтрована.
Нельзя оставлять раствор нефильтрованным — в
этом случае может произойти выпадение в осадок
какого-то из компонентов среды, либо появляется
микробное загрязнение. После внесения необходи¬
мых добавок (например, глютамин, NaHC03, или
сыворотка) следует отрегулировать pH среды.
Оборудование и материалы
Стерильные:
• Градуированные бутыли для среды, 100 мл .....10
• Крышки для бутылей 10
• Универсальные контейнеры для контроля загряз¬
нения образцов 3
Нестерильные:
• Сверхчистая вода 1 л
• Порошкообразная среда, например МЕМ с соля¬
ми Эрла, без глютамина, 1-л пакет
• Градуированная бутыль или флакон Эрлен-
майера, объемом 1 л плюс дополнительный
объем 1
• Магнитная мешалка и PTFE-покрытый вкла¬
дыш, 50 мм 1
• Кондуктометр
Протокол
1. Внесите соответствующий объем сверхчистой
воды в контейнер.
2. Положите вкладыш магнитной мешалки.
3. Поставьте контейнер на магнитную мешалку,
установите режим около 200 об./мин.
4. Откройте пачку порошка и медленно добавьте
содержимое в контейнер при постоянном пере¬
мешивании.
5. Размешайте до полного растворения порошка.
6. Возьмите образец, проверьте pH и проводимость
образца и произведите записи.
а) pH должен быть в пределах 0.1 от ожидаемого
уровня для данной среды.
б) Проводимость должна быть в пределах 2% от
ожидаемой для данной среды
в) Сбросьте образец в отходы; не возвращайте
его в основную емкость со средой.
7. Стерилизуйте фильтрацией (см. разд. 11.5.2,
протоколы 11.11-11.14).
8. Закройте бутыли крышками, запечатайте.
9. Храните при 4° С.
10. Перед использованием добавьте сыворотку и
глютамин, доведите pH до 7,4.
шаться. Убедитесь, что в среде находится правильное
количество NaHC03, соответствующее данной газовой
фазе (см. разд. 9.2.2), при установлении равновесия в
инкубаторе. Это должно быть подтверждено экспери¬
ментально, когда среда используется первый раз.
Для крупномасштабного использования (> 10 л/нед)
среда может быть подготовлена в сосуде под давлением,
проверена через определенный промежуток времени
при помощи большой пипетки, чтобы определить,
является ли среда полной, с последующей стерилиза¬
цией фильтрацией при положительном давлении через
встроенный одноразовый фильтр или многоразовый
фильтр, с поступлением в принимающий сосуд (см.
разд. 11.5.2, протокол 11.14).
11.4.5. Специализированная среда
Если кто-либо намеревается использовать различные
рецептуры сред либо среду, которая должна быть
составлена в лаборатории из отдельных составляю¬
щих, то в этом случае удобно будет приготовить ряд
основных концентрированных растворов — аминокис¬
лот — 50х или 100х; витаминов — ЮООх; тирозина и
триптофана — 50х в 0,1М растворе НС1; глюкозы в кон¬
центрации 200 г/л; и однократный раствор BSS. При
необходимости смешивается требуемое количество
каждого концентрата, смесь фильтруется через сте¬
рилизационный фильтр с порами диаметром 0,2 мкм
и разводится в низко- или высокобикарбонатном BSS
(см. разд. 11.4.2, рис. 11.11, 11.12 и табл. 9.2). Пре¬
имуществом такого типа составления специализиро¬
ванных сред является возможность ее варьирования;
могут быть добавлены дополнительные питательные
вещества (оксо-кислоты, нуклеозиды, неорганические
вещества и т. д.), основная среда может быть изменена
в зависимости от требований. Однако на практике эта
система настолько трудоемка, что лишь некоторые ла¬
боратории идут на приготовление питательных сред из
отдельных компонентов, если только целью работы не
является изучение влияния постоянно меняющегося
состава питательной среды.
Надежность коммерческих сред целиком обусловле¬
на проводимым соответствующим контролем качества.
Любая среда, приготовленная из отдельных компонен¬
тов в лаборатории, также должна пройти исследования
на контроль качества (см. разд. 11.6.1).
В настоящее время существует достаточно много
надежных компаний-поставщиков стандартных ос¬
новных сред (см. Приложение II), многие из которых
могут предоставить специализированные бессыворо¬
точные композиции и среды. Важно удостовериться,
что контроль качества, проводимый поставщиком,
соответствует данной среде и тем клеткам, которые вы
предполагаете растить на ней. Вы могли бы купить сре¬
ду MCDB 153, которая исследуется на колониеобразо-
вание клеток HeLa, но этот тест не имеет отношения и
не характеризует пригодность этой культуры для куль¬
тивирования первичной культуры кератиноцитов.
11.4. Реагенты и среды
199
ПРОТОКОЛ 11.10. Приготовление
специализированной среды
Схема
Приготовьте основные концентраты сред (на осно¬
вании табл. 9.3 или 10.1), храните в замороженном
виде. Разморозьте и смешивайте при необходимости
использования.
Стерилизуйте методом фильтрации и храните,
пока не потребуется.
Материалы
• Концентрат аминокислот, 100х в воде, хранить
замороженным
• Тирозин и триптофан, 50х в 0.1 М HCL
• Витамины, ЮООх в воде
• Глюкоза, ЮОх (200 г/л в ССР)
• Растворы добавок (например, микроэлементы
и нуклеозиды, гормоны, или факторы роста,
которые должны быть добавлены только перед
использованием среды)
• Бутыли для хранения оптимального размера для
аликвоты того количества среды, которое потре¬
буется для последующего разведения (см. шаг 5
данного протокола)
Протокол
1. Разморозьте растворы, убедитесь, что все раство¬
ры полностью прозрачны.
2. Смешайте компоненты в правильных количест¬
вах:
а) концентрат аминокислот 100 мл
б) тирозин и триптофан 200 мл
в) витамины 10 мл
г) глюкоза 100 мл
3. Перемешайте, простерилизуйте фильтрацией (см.
протоколы 11.12,11.13).
4. Храните замороженным.
5. Для использования разведите 41 мл смеси кон¬
центратов с 959 мл стерильного ССР 1х.
6. Доведите pH до 7,4.
7. Храните при 4 °С.
8. Непосредственно перед использованием среды
добавьте сыворотку или другие компоненты,
типа гормонов, факторов роста и липидов. Если в
качестве микроэлементов используются металлы,
лучше добавить их также только перед использо¬
ванием среды, поскольку в концентрированных
растворах в присутствии фосфата они могут
выпадать в осадок.
11.5. СТЕРИЛИЗАЦИЯ СРЕДЫ
11.5.1. Автоклавируемые среды
Некоторые поставщики предлагают автоклавируемые
варианты среды Игла МЕМ и других сред. Автоклави-
рование намного снижает трудозатраты, экономически
дешевле и имеет более высокую надежность качества
стерильности по сравнению с фильтрацией. Процедура,
при помощи которой проводится автоклавирование,
обычно бывает описана в инструкции производителя
и аналогична описанной ранее для D-PBSA (см. про¬
токол 11.6). Кислотность среды доводят до pH 4,25 при
помощи сукцината, для того чтобы стабилизировать ви¬
тамин В при автоклавировании и после нейтрализуют.
Глутамин замещен на глутамат или глутамил, либо на
дипептиды глутамила. Можно также добавить глюкозу
после автоклавирования.
BSS автоклавируют без и глюкозы и глутамина,
а также без различных гидролизатов аминокислот,
типа триптозофосфатного бульона и других микро¬
биологических сред, которые стерилизуются также
автоклавированием.
11.5.2. Стерилизация методом фильтрации
Фильтрация через микропористые фильтры с диамет¬
ром пор от 0,1 до 0,2 мкм применяется для стерилизации
термолабильных растворов (рис. 11.11). В настоящее
время предлагаются многочисленные виды фильтров,
сделанные из различных материалов, включая поли-
эфирсульфон (PES), нейлон, поликарбонат, ацетат
целлюлозы, нитрат целлюлозы, PTFE, и керамику, и
размерами от насадки для шприца (рис. 11.12а, в) через
маленький и промежуточный встроенные фильтры
(рис. 11.126, в, ё) до фильтров в картридже (рис. 11.13).
Фильтры, связывающие белки низкой плотности, пре¬
доставляются большинством поставщиков (например,
Durapore, Millipore); обычно считается, что PES- филь¬
тры обеспечивают более высокую скорость фильтрации.
Поликарбонатные мембранные фильтры — это абсо¬
лютные фильтры с множеством отверстий одного диа¬
метра; число отверстий на единицу площади возрастает
при уменьшении диаметра отверстий, это обеспечивает
однородную скорость потока. Большинство других
фильтров имеют разнообразные структуры сит и спосо¬
бов захвата частиц на фильтре; они вообще имеют более
высокую скорость потока и уменьшенное засорение, но
могут деформироваться при высоких давлениях.
Фильтрация может быть выполнена при поло¬
жительном давлении (см. рис. 11.11а, рис. 11.126,
в, в), соединена с герметизируемым контейнером
(см. рис. 11.14) или с перистальтическим насосом
(рис. 11.15). Фильтрация под высоким давлением от
сосуда под высоким давлением быстрее, чем прокачка
при помощи перистальтического насоса, но потребует
использования сосуда-приемника, поскольку поток
не может регулироваться так легко, принимая во
внимание, что насос может быть включен и выключен
при необходимости. Высокое давление также имеет
тенденцию деформировать фильтры и не подходит
для вязких растворов типа сыворотки. Фильтрация
при отрицательном давлении часто более проста (см.
рис. 11.116, 11.12г, Э), особенно для маломасштабных
200
ГЛАВА 11. ПОДГОТОВИТЕЛЬНЫЕ РАБОТЫ И СТЕРИЛИЗАЦИЯ
перистальтическим насосом и работают под положи¬
тельным давлением.
Протоколы 11.11-11.14 разработаны для одноразо¬
вого фильтра малого размера и для фильтра многократ¬
ного использования для больших объемов.
Шприц может быть заполнен несколько раз, при
этом насадка возвращается в нижнюю половину
стерильной упаковки, снимается со шприца, шприц
заполняется и вновь соединяется с насадкой. Если
а ФИЛЬТРУЮЩАЯ НАСАДКА НА ШПРИЦ б ФИЛЬТРУЮЩАЯ НАСАДКА НА КОЛБУ
Положительное давление
Нестерильная среда из ре¬
зервуара под положительным
давлением
Высокое давление,
- силиконовый шланг
Размещение
Отрицательное давление
Емкость со
фильтра
Микропорый
фильтр, 0,2 мкм
Подложка
фильтра
Стеклянный или
пластмассовый колокол
для защиты выпускного
отверстия стерильной
среды
Стерильная
среда
Соединение
с вакуумным
Рис. 11.11. Стерилизация фильтрацией. (a) Фильтрующая
насадка на шприц. Нестерильная среда поступает через насос
или из сосуда под давлением, (б) фильтрующая вакуумная
насадка на колбу или бутыль. Среда, налитая в верхнюю
емкость, переходит в нижнюю. Нижняя емкость может ис¬
пользоваться для хранения
экспериментов, при сборе фильтрата непосредственно
в емкости для хранения. Фильтрация может вызывать
повышение pH из-за выделения С02.
Одноразовые фильтры. Существуют различ¬
ные конструкции одноразовых держателей фильтра,
включая простые дисковые фильтры, половолоконные
фильтры, и картриджи (см. рис. 11.11-11.13). Обычно
фильтрующие насадки для шприца используются для
малых объемов фильтрации (2-20 мл) и различны по
размеру: от 13 до 50 мм в диаметре (см. рис. 11.12а).
Фильтры промежуточного размера (для объемов
50 мл-5 л) могут использоваться с перистальтическими
насосами (рис. 11.14), или как фильтрующие насадки на
бутыли, используемые с вакуумной линией и стандарт¬
ной бутылей для сред (рис. 11.12е). Фильтры средних
размеров могут также быть куплены как комплект
фильтрующих блоков для присоединения к вакуумной
линии, с верхней емкостью для нестерильного раствора
и нижней емкостью для стерильной жидкости, эту ем¬
кость затем сразу закрывают и используют для хране¬
ния (рис. 11.12Э). Фильтрующие картриджи большой
мощности (для объемов 20-500 л) обычно используют
под действием положительного давления (рис. 11.13,
11.15). Хотя одноразовые фильтры более дороги, чем
фильтры многократного использования, они отнимают
меньше времени, реже приводят к неудачам.
Фильтры многократного использования. Держа¬
тели фильтра многократного использования могут
использовать мембраны (рис. 11.16) или картриджи
(рис. 11.13б) и стерилизуются автоклавированием (см.
протокол 11.4). Они обычно соединены с резервуаром,
находящимся под давлением (см. рис. 11.15) или с
используемое гидростатическое давление возрастает,
возьмите новый фильтр.
В вакуумной линии или при использовании насоса
удобно фильтровать объемы 50-500 мл с отрицатель¬
ным давлением, собирая фильтрат во флакон-интегра-
тор (см. рис. 11.116, 11.12Э) или при помощи насадки
на обычную бутыль для среды (см. рис. 11.12г). Про¬
токол 11.12 разработан для использования в упражне¬
нии 9 (см. гл. 2). Можно использовать другие объемы
и фильтры других размеров (см. табл. 11.4).
В целом, этот метод фильтрации имеет низкий риск
заражения, поэтому образцы для проверки на заражен¬
ность можно брать достаточно редко.
В качестве сосудов-приемников большого объема
могут быть использованы колбы для фильтрования
объемом 150-1000 мл. Если должен быть профиль-
ПРОТОКОЛ 11.11. Стерилизация
с использованием фильтрующих насадок
на шприцы
Материалы
Стерильные:
• Пластмассовый шприц объемом от 10 до 50 мл
• Фильтрующая насадка на шприц (например, Pall
Gelman Acrodisc или
• Millipore Millex)
• Принимающий сосуд (например, универсальный
контейнер)
Нестерильные:
• Стерилизуемый раствор (5-100 мл)
Протокол
1. Протрите ламинар-бокс и принесите необходи¬
мые материалы.
2. Заполните шприц раствором, который будет
стерилизоваться.
3. Снимите крышку с принимающего сосуда.
4. Распакуйте фильтр и присоедините к наконеч¬
нику шприца, удерживая стерильный фильтры
нижней половиной в упаковке, пока не произой¬
дет соединение насадки со шприцом
5. Профильтруйте раствор через фильтр в при¬
нимающий сосуд. Требуется только умеренное
давление.
6. Закройте крышку принимающего сосуда.
7. Поместите шприц и фильтр в отходы.
11.5. Стерилизация среды
201
Рис. 11.12. Одноразовые стерилизационные фильтры. (а) Millex 25-мм дисковая фильтрующая насадка на шприц; (6) одно¬
разовый фильтр с колоколом; (в) фильтр Sterivex с колоколом и без; (г) фильтрующая насадка на бутыль; (д) фильтрующая
крышка и емкость для хранения. (F) Большой встроенный фильтр с колоколом. (а, 6, в, г) Положительное давление, (г, д) От¬
рицательное давление (см. также рис. 11.11).
Сфотографировано с любезного разрешения Millipore (UK), Ltd.
Рис. 11.13. Фильтрационная установка большой мощности.
(а) Большие сосуды давления для фильтрации при поло¬
жительном давлении; (6) фильтры для крупномасштабной
фильтрации в виде сложенных в стопку дисков и плиссиро¬
ванного картриджа
Сфотографировано с любезного разрешения Millipore
(UK), Ltd.
трован и разлит на меньшие порции большой объем
жидкости, то лучше использовать один из нескольких
типов фильтрующих насадок на бутыли, которые могут
соединяться непосредственно со стандартной бутылью
для среды (см. рис. 11.12г). Напротив, растворы могут
быть профильтрованы под действием положительного
давления либо из сосуда под высоким давлением (см.
рис. 11.14), проходить через стерильные встроенные
мембранные фильтры или картриджи, оборудованные
раструбом для защиты принимающих сосудов от зара¬
жения (см. рис. 11.11,11.12,11.14,11.15). Протокол 11.13
разработан для фильтров малого размера и может быть
использован при выполнении упражнения 9.
Масштаб фильтрации при положительном давле¬
нии может быть увеличен путем возрастания размеров
фильтра и размеров принимающего сосуда. Перисталь¬
тический насос часто заменяют на дополнительное
положительное давление, оказываемое на резервуар-
202
ГЛАВА 11. ПОДГОТОВИТЕЛЬНЫЕ РАБОТЫ И СТЕРИЛИЗАЦИЯ
ПРОТОКОЛ 11.12. Стерилизация
с использованием фильтрующей
вакуумной насадки на колбу
Материалы
Стерильные:
• Фильтрующая насадка для объема 500 мл (см.
рис. 11.116; см. например, рис. 11.12Э) 1
• Крышка для нижерасположенной емкости (если
предполагается, что эта емкость будет использо¬
вана для хранения) 1
• Типовая пробирка или универсальный контейнер
для проверки стерильности образца 1
Нестерильные:
• Среда для стерилизации 450 мл
• Вакуумный насос или вакуумная линия
• Толстостенный соединительный шланг, соеди¬
няющий соответствующее входное отверстию
фильтрующей насадки с насосом или линией
Протокол
1. Присоедините боковую отводящую трубку фильт¬
рующей насадки к вакуумному насосу.
2. Снимите крышку с бутыли со средой и колпачок
с верхней камеры фильтрующей насадки.
3. Налейте нестерильную среду в верхнюю ем¬
кость.
4. Включите насос.
5. Снимите крышку с нижней емкости, готовой к
приему стерильной среды.
6. Когда вся жидкость переместится в нижнюю
емкость, выключите насос и отсоедините фильт¬
рующее устройство.
7. Перенесите 10 мл среды из нижней емкости в
универсальный контейнер или соответствующую
пробирку.
8. Заполните газом с 5% С02 свободное простран¬
ство в нижней емкости и универсальном контей¬
нере.
9. Закройте крышками нижнюю емкость и универ¬
сальный контейнер.
10. Снабдите емкость и универсальный контейнер
наклейками с названием среды, датой, и вашими
инициалами.
11. Храните среду при 4 °С пока она не потребуется.
12. Инкубируйте 10-мл образец в универсальном
контейнер при 37 °С в течение 1 недели для про¬
верки стерильности. Не используйте среду, пока
не доказано, что она стерильна.
приемник для среды в сосуде для высокого давления
(см. рис. 11.13). Протокол 11.14 подходит для объемов
до 50 л, но объем может быть увеличен до промышлен¬
ных масштабов путем выбора соответствующего сосуда
высокого давления и фильтра (часто с использованием
гофрированного картриджа или других фильтров боль¬
шой мощности).
ПРОТОКОЛ 11.13. Стерилизация
с использованием малого
встроенного фильтра
Материалы
Стерильные:
• Встроенный фильтр с колоколом (см. рис. 11.11а,
11.12е) 47 мм
• Градуированные бутыли для среды с горлышка¬
ми, обернутыми фольгой, стерилизованные сухим
жаром (см. протокол 11.1) 7
• Автоклавированные крышки (см. прото¬
кол 11.3) 6
• Типовая пробирка или универсальный контейнер
для проверки стерильности 1
Нестерильные:
• Среда для стерилизации (из упражнения 9 и
протокола 11.9) 550 мл
• Перистальтический насос (рис. 11.14) предпоч¬
тительно с ножной педалью 1
• Силиконовый шланг для присоединения насоса
к входному отверстию фильтра 50 см
• Зажим для фиксации фильтра 1
Протокол
1. Присоедините шланг для подачи среды через
перистальтический насос.
2. Опустите верхний конец шланга в среду, которая
будет стерилизоваться.
3. Распакуйте фильтр и соедините с выходом перис¬
тальтического насоса.
4. Закрепите фильтр в фиксаторе на подходящей
высоте, так чтобы бутыли для фильтрованной
среды с завернутым в фольгу горлышком могли
бы поместиться ниже фильтра и фольга могла
быть легко удалена перед заполнением. Если
необходимо, используйте одну из стерильных
средних бутылей при установке фильтра, но не
используйте затем эту бутыль для окончатель¬
ного собирания стерильной среды.
5. Включите насос, соберите около 20 мл в бутыль,
использованную для установки фильтра.
6. Удалите бутыль из установки, закройте ее крыш¬
кой и поставьте на этикетке № 1.
7. Удалите фольгу с первой бутыли и поместите под
колоколом фильтра.
8. Включите насос.
9. Заполните бутыль до отметки 100 мл.
10. Выключите насос.
11. Удалите бутыль, закройте крышкой, поставьте
номер и замените на новую стерильную бутыль.
12. Повторите шаги 7-11, заполняя 5 бутылей по
100 мл, соберите остаток в последней бутыли. За¬
полните газом с 5% С02 свободное пространство
в каждой бутыли.
13. Заверните в алюминиевую фольгу крышку и
горлышко каждой бутыли, чтобы предохранить
от пыли при хранении.
11.5. Стерилизация среды
203
14. Наклейте этикетки на бутыли, поставьте дату,
ваши инициалы.
15. Поместите бутыли, содержащие 100 мл среды,
для хранения на 4 °С (см. разд. 11.6.4).
16. Поместите первую и последнюю бутыли, запе¬
чатанные, на 37 °С, инкубируйте в течение 1 нед.
(см. разд. 11.6.2).
17. Храните среду при 4 °С, пока не потребуется; не
используйте среду до полного завершения иссле¬
дования стерильности. Если в испытательных
образцах обнаруживается заражение, перефиль-
труйте всю партию или выбросите как брак.
Положительное давление рекомендуется для опти¬
мального использования фильтров и позволяет избегать
понижения концентрации С02, которая обычно имеет
место при фильтрации под действием отрицательного
давления. Положительное давление может также иметь
место при использовании перистальтического насоса
(рис. 11.14), встроенного между нестерильным резер¬
вуаром и одноразовым фильтром, таким как Millipak
(Millipore) который может быть использован вместо
комплекта фильтров многоразового использования.
Подготовительные работы и стерилизация необходимы
только для принимающих бутылей, поскольку однора¬
зовые фильтры поступают стерильными (фильтруемый
поток может быть легко прерван путем выключения
перистальтического насоса).
11.5.3. Сыворотка
Подготовка сыворотки является одной из самых слож¬
ных процедур в культуре тканей в результате вариа¬
бельности качества и плотности исходного материала, а
также из-за сложностей, возникающих при фильтраци¬
онной стерилизации сыворотки, содержащей плотные
частицы и обладающей повышенной вязкостью. Более
того, сыворотка также является одним из наиболее
дорогостоящих компонентов, используемых в культу¬
ре тканей, составляющих 20-30% от общего бюджета,
если она приобретается у коммерческих поставщиков.
Приобретение стерильной сыворотки, безусловно, яв¬
ляется оптимальным с точки зрения состава и контроля
качества, однако в случае, если стерильная сыворотка
должна быть приготовлена в лаборатории, предлага¬
ется протокол 11.15. Основным принципом является
расположение нескольких фильтров по градиенту
размеров пор, при этом эти фильтры не должны быть
стерильными и служат для удаления плотных частиц до
того как сыворотка поступит при невысоком давлении
на стерилизующий фильтр с диаметром пор 0,1 мкм
(см. рис. 11.17).
Стерилизация небольшого объема сыворотки.
Если требуется небольшое количество сыворотки
(<1 л), то может быть проведена процедура, аналогич¬
ная описанной в протоколе 11.14. После сворачивания
кровяного сгустка (см. протокол 11.15; Свертывание
крови), полученное небольшое количество сыворотки
Рис. 11.14. Фильтрация при помощи перистальтического насоса. Стерилизация фильтрацией при перекачивании нестерильной
жидкости из емкости через стерилизационный фильтр (Millipak, Millipore; с любезного разрешения Millipore (UK), Ltd.)
204
ГЛАВА 11. ПОДГОТОВИТЕЛЬНЫЕ РАБОТЫ И СТЕРИЛИЗАЦИЯ
Стерильно
Пружинный зажим
Нестерильно
А
Линия давления
от насоса 15 psi,
100 кПа;
Герметизируемая
емкость
Встроенный
комплект фильтров
с держателем для
0,22-мкм фильтра
Колокол для выхода
стерильной жидкости
Сосуд-
приемник
Рис. 11.15. Крупномасштабная стерилизация фильтрацией с использованием большого встроенного комплекта фильтров.
Большой встроенный комплект фильтров, соединенный с герметизируемой емкостью (слева) и с сосудом — приемником (спра¬
ва). Стерильны только комплект фильтров и сосуд-приемник. Обычно стеклянный колокол оборачивается защитной фольгой;
на рисунке он удален с целью иллюстрации
Таблица 11.4. Размеры фильтра и объем фильтруемой
жидкости
Маркировка
или размер
фильтра
Одноразовый
(D) или много¬
разовый (R)
Приблизительный объем,
рекомендуемый
для фильтрации
Микролиты
Коллоиды
25 мм, Millex
D
1-100 mL
1-20 mL
47 мм ог
R, D
0,1-1 L
100-250 mL
Sterivex
cartridge
90 mm
R
1-10 L
0,2-2 L
Millipak-20
D
2-10 L
200 mL-2 L
Millipak-40
D
10-20 L
2-5 L
Millipak-60
D
20-30 L
5-7 L
Millipak-100
D
30-75 L
7-10 L
Millipak-200
D
75-150L
10-30 L
Millidisk
D
30-300 L
5-50 L
142 мм
R
10-50 L
1-5 L
293 мм
R
50-500 L
5-20 L
Примеры в таблице взяты из каталога Millipore. Аналогичные
продукты предлагаются компаниями Pall Gelman, Nalge Nunc,
Sartorius и рядом других.
может быть отцентрифугировано (5000-10 0000 g) и
затем профильтровано через рад одноразовых филь¬
тров (например, через 50 мм Millipore Millex или Pall
German Acrodisc) и, окончательно, через 50 мм стерили¬
зующий одноразовый фильтр с диаметром пор 0,1 мкм
(например, Millex).
Хранение. Для хранения сыворотки используйте
бутыли такого размера, чтобы использовать в течение
2-3 недель после размораживания. Замораживать сы¬
воротку следует так быстро, как это возможно, а при
случайном размораживании не следует замораживать
ее повторно, если только не предполагается продолжи¬
тельное ее хранение.
При хранении при -20 °С сыворотку лучше исполь¬
зовать в течение 6-12 месяцев после приготовления.
При -70 °С продолжительность хранения удлиняется;
но обычно создание больших запасов сыворотки ока¬
зывается непрактичным. Желательно использовать
поликарбонатные или полипропиленовые бутыли
(полипропилен высокой плотности), что снижает риск
разбивания при хранении при -70 °С. Независимо от
температуры морозильника или материала бутылей, не
11.5. Стерилизация среды
205
ПРОТОКОЛ 11.14. Стерилизация
с использованием большого
встроенного фильтра
Материалы
(См. рис. 11.13,11.15, и 10.17)
Стерильные:
• Фильтр (например, 90-мм мембранный и держа¬
тель фильтра многократного использования; см.
протокол 11.4; табл. 11.4; рис. 11.15) одноразовый
диск (например, Millipore Millex) или картридж
(например, Pall Gelman Capsule).
• Приемник для фильтрата с выпускным отверс¬
тием в основании
• Силиконовый шланг нагнетательного трубопро¬
вода от фильтра до стерильного приемника.
• Силиконовый шланг трубки со стеклянным
колоколом и пружинным зажимом на выходе
приемника (см. рис. 11.15).
• Градуированные бутыли для среды с горлышка¬
ми, обернутыми фольгой, стерилизованные сухим
жаром (см. протокол 11.1)
• Автоклавированные крышки для бутылей, (см.
протокол 11.3)
Нестерильные:
• Сосуд высокого давления, 5-50 L.
• Насос, 100 кПа (1 атм)
• Держатели зажимов и зажимы, чтобы установить
фильтр (если держатель фильтра имеет опоры) и
стеклянный колокол на выходе
• Силиконовый шланг для присоединения фильтра
к сосуду высокого давления
Протокол
1. Зафиксируйте держатель фильтра в нужном по¬
ложении.
2. Соедините выход с принимающей емкостью.
3. Расположите выход приемника на подходящей
высоте так, чтобы бутыли для фильтрованной
среды с завернутым в фольгу горлышком могли
поместиться ниже фильтра и фольга могла быть
легко удалена перед заполнением. Если необходи¬
мо, используйте одну из стерильных средних бу¬
тылей при установке фильтра, но не используйте
затем эту бутыль, для окончательного собирания
стерильной среды.
4. Соедините сосуд давления со входным отверсти¬
ем фильтра.
5. Фильтруйте среду в сосуд давления.
6. Для фильтров многократного использования
открыть ненадолго насос, так чтобы увлажнить
фильтр. Остановите насос и уплотните держатель
фильтра.
7. Включите насос, чтобы достичь 100 кПа. Когда
приемник начинает заполняться, распределите сре¬
ду по бутылям аликвотами желательного объема.
8. Закройте крышку каждой бутыли после запол¬
нения.
9. Оберните фольгой крышки.
10. Прикрепите этикетки с датой, инициалами.
11. В зависимости от числа и размера заполненных бу¬
тылей, возьмите типовые бутыли в начале, середине
и конце процесса, инкубируйте при 37 °С в течение
1 нед., чтобы проверить наличие заражения.
12. Храните среду при 4 °С, не используйте среду до
полного завершения исследования стерильности.
Рис. 11.16. Фильтры многократного использования. (a) Полипропиленовое встраиваемое приспособление Luer; (б) фильтр
дискового типа высокой мощности с держателем из нержавеющаей стали; (в) 47-мм фильтр с резервуаром; (г) встраиваемый
фильтр 90 мм со шланговыми соединениями. (С любезного разрешения Millipore, UK, Ltd.)
206
ГЛАВА 11. ПОДГОТОВИТЕЛЬНЫЕ РАБОТЫ И СТЕРИЛИЗАЦИЯ
ПРОТОКОЛ 11.15. Сбор и стерилизация
сывороток
Схема
Соберите кровь, позволив свернуться с образованием
сгустка, и отделите сыворотку.
Простерилизуйте сыворотку через фильтры с
постепенным сокращением пористости, соберите
фильтрованную сыворотку в бутыль и заморозьте.
Сбор
Могут быть приняты какие-то меры, чтобы получать
цельную кровь из скотобойни. Кровь должна быть
собрана непосредственно из кровотока тела. Кровь
может быть также получена от животных, которые
живут при надлежащем ветеринарном наблюдении.
Последний вариант, при выполнении одинаковой
последовательности действий на той же самой группе
животных, дает более воспроизводимую сыворотку,
но более низкий объем при большой затрате усилий.
Если процедура выполнена тщательно, кровь может
быть собрана асептически.
Свертывание крови
Позвольте крови свернуться, оставив ее на ночь в закры¬
той емкости при 4 °С. Эта, так называемая естественно
свернувшаяся сыворотка превосходит по качеству
сыворотку, которая физическим методом (цетрифуги-
рование и дефибринизация) отделена от клеток крови,
поскольку в ходе естественного сворачивания тромбо¬
циты выделяют в сыворотку факторы роста. Отделите
сыворотку от сгустка и центрифугируйте сыворотку в
течение 1 ч при 2,000 g. Удалите осадок.
Стерилизация
Сыворотка обычно стерилизуется фильтрацией че¬
рез стерилизующий фильтр с пористостью 0,1 мкм.
Из-за вязкости и высокого содержания микрочастиц
перед прохождением через заключительный фильтр
сыворотка должна пройти через градуируемый ряд
стекловолоконных фильтров или другой предва¬
рительный фильтр (рис. 11.17). Только последний
фильтр, например 142-К 350-мм дисковый фильтр
или его одноразовый эквивалент (например, Millipak
200), должен быть стерильным. Предварительные
фильтры могут быть сделаны из нержавеющей стали
с заменяемым картриджем, можно также применять
отдельные фильтры или их комплекты, предлагаемые
производителями (Pall Gelman). Соединенный в блок
комплект фильтров наиболее прост в употреблении,
но его труднее чистить и повторно использовать.
Для стерилизации объемов 5-20 л:
Материалы
Стерильные:
• Фильтры для стерилизации: с диаметром пор
200 мм, 0,1 мкм (например, Millipak, Millipore).
Диаметр пор 0,2 мкм достаточен для антибак¬
териальной и антигрибковой стерилизации, но
чтобы удалить микоплазмы, требуется фильтр с
размером пор 0,1 мкм.
• Стерильный приемный сосуд получения с вы¬
пускным отверстием в основании.
• Стерильные бутыль с крышками и фольгой.
• Универсальные контейнеры для проверки сте¬
рильности.
Нестерильные:
• Перистальтический насос и шланг
• Держатель зажимов и зажимы
• Нестерильные предварительные фильтры:
° Стекловолоконный одноразовый фильтр или
фильтр многократного использования 142 мм
(например, Pall Gelman)
° Одноразовый фильтр с диаметром пор 5 мкм
или фильтр многократного использования
(Например, Pall Gelman Versapore)
° Одноразовый фильтр с диаметром пор 1,2 мкм
или фильтр многократного использования
(Например, Millipore Opticap)
° Одноразовый фильтр с диаметром пор
0,45 мкм или фильтр многократного исполь¬
зования (Например, Millipore Millipak)
Примечание. Предварительные фильтры, предло¬
женные в протоколе, не обязательны для примене¬
ния. Вступить в контакт с вашим поставщиком и
запросить ряд фильтров и предварительных фильт¬
ров, которые удовлетворят вашим требованиям
при получении сыворотки. Если это однократное
действие, выбрать одноразовые фильтры. Если по¬
лучение сыворотки будет повторяться регулярно, то
экономически более выгодно приобрести держатели
фильтра многократного использования для стадий
предварительной фильтрации, с использованием од¬
норазового фильтра для стерилизации фильтрацией
для дополнительной безопасности.
Протокол
1. Вставьте соответствующие нестерильные фильт¬
ры в нестерильные держатели (если используют¬
ся держатели многократного использования).
2. Соберите один или большее количество предва¬
рительных фильтров на линии выше стерильного
одноразового или держателя фильтра много¬
кратного использования (рис. 11.17), который
содержит фильтр с диаметром пор 0,1 мкм и
связан со стерильным принимающим сосудом
через перистальтический насос.
3. Поместите всасывающий конец трубки от насоса
в емкость с сывороткой.
4 Включите насос и проверьте все фильтры много¬
кратного использования, когда они будут увлаж¬
нены. Выключите насос и затяните держатели
фильтра по мере необходимости.
11.5, Стерилизация среды
207
5. Повторно включите насос и продолжите фильтро¬
вание, контролируя наличие утечек или засоров.
Увеличение скорости потока увеличит скорость
фильтрации, но может повредить фильтры, кото¬
рые могут забиться.
6. Соберите аликвоты стерильной сыворотки в
стерильные бутыль, оставляя как минимум 20%
пустого пространства над уровнем жидкости,
чтобы учесть расширение при замораживании.
7. Отберите на контроль стерильности образцы в
начале, середине и конце процесса.
8. Закройте бутыль крышками, приклейте этикетку,
напишите номер бутыли.
9. Вновь оберните фольгой крышку и горлышко
каждой бутыли для хранения.
10. Сыворотку храните при 20 °С, пока не закончится
проверка качества (см. разд. 11.6.1).
8. Соберите сыворотку в измерительный цилиндр
и обратите внимание на собранный объем. Если
объем меньше, чем первоначальный объем сы¬
воротки, добавьте HBSS, чтобы возвратиться к
начальному объему. Если объем больше, чем на¬
чальный объем сыворотки, сделайте необходимые
поправки при добавлении сыворотки к среде в
дальнейшем.
9. Стерилизуйте сыворотку через градуируемый ряд
фильтров (см. протокол 11.15).
10. Разлейте в бутыли и заморозьте сыворотку.
следует заполнять их до верха; учитывайте расширение
жидкостей при замораживании.
Сыворотка человека. Для культуры ткани вместо
или вместе с лошадиной или бычьей кровью могут быть
использованы цельная человеческая кровь или плазма
по истечении срока их хранения в донорском банке.
Они должны быть стерильны и не требуют фильтрации.
Следует нейтрализовать антикоагулянты (гепарин или
цитрат) титрованием Са2+, оставив затем кровь на ночь
для образования сгустка, а затем отделить и заморозить
сыворотку.
• Требование безопасности. При работе с
донорской кровью следует удостовериться, что она не
заражена возбудителями гепатита, ВИЧ, туберкулеза
и другими случайными возбудителями.
Контроль качества. Используйте ту же процедуру,
что и для контроля качества среды (см. разд. 11.6).
Главной проблемой при использовании сыворотки
является возможность вирусной инфекции. Когда
на первое место по важности ставилась проблема
бактериального инфицирования, было относительно
легко решить ее методом фильтрации через фильтр
с диаметром пор 0,1 мкм. В конечном итоге, стала
стандартной процедура фильтрации через фильтр с
диаметром пор 0,45 мкм. Однако обнаружилось, что
микоплазмы способны проникать через поры диамет¬
ром 0,2 мкм, и коммерческие поставщики сыворотки
уменьшили размеры диаметра пор при фильтрации до
0,1 мкм. Уменьшение размеров привело к снижению
вероятности заражения сыворотки микоплазмами, но
вероятность вирусного заражения еще существует.
Удаление вирусов фильтрацией кажется маловероят¬
ным, но некоторые компании (например, Pall Gelmann)
заявляют, что это возможно. Получение сыворотки
из районов с низким индигенным уровнем вирусного
инфицирования и скрининг до начала работы с кровью
является лучшей альтернативой.
Диализ. На определенных стадиях культивиро¬
вания присутствие некоторых компонентов с низким
молекулярным весом (аминокислоты, глюкоза, нуклео-
зиды и т.д.) становится нежелательным. Эти вещества
могут быть удалены методом диализа.
ПРОТОКОЛ 11.16. Диализ сыворотки
Материалы
Стерильные:
• Бутыль и крышки.
• Стерилизационный фильтр, 0,1мкм.
• Предварительные фильтры: стекловолокон¬
ный 5,0 мкм, мембранные фильтры 1,2, 0,45, и
0,22 мкм (например, Durapore, Millipore; см.
протокол 11.14)
Нестерильные:
• Диализные трубки.
• Мензурка с ультрачистой водой.
• Бунзеновская горелка.
• Треножник с проволочной сеткой.
• Сыворотка для диализа.
• Раствор Хэнкса в 4° С.
• Измерительный цилиндр
Протокол
1. Прокипятите пять частей 30-ммх500-мм диа¬
лизных трубок в трех сменах дистиллированной
воды.
2. Перенесите трубки в сбалансированный раствор
солей Хэнкса (HBSS) и оставьте охлаждаться.
3. Завяжите двойные узлы на одном конце каждой
трубки.
4. До половины заполните каждую трубку сыворот¬
кой (20 мл).
5. Выдавите воздух и завяжите открытый конец
трубы, оставив место (приблизительно половины
трубки) между сывороткой и узлом.
6. Поместите трубки в 5 л HBSS, перемешиваемый
на магнитной мешалке на ночь при 4 °С.
7. Замените HBSS и повторите шаг 6 дважды.
208
ГЛАВА 11. ПОДГОТОВИТЕЛЬНЫЕ РАБОТЫ И СТЕРИЛИЗАЦИЯ
Нестерильный вход
Стекловолокно
5 мкм
1,2 мкм
мкм
Поддерживающий
экран
О-кольцо
Фиксирующая
гайка
Стерильный фильтр с
диаметром пор 0,1 или
0,22-мкм
V
Выход к сосуду-приемнику
или стерильным бутылям
Рис. 11.17. Предварительный фильтр. Предварительный фильтр для фильтрования коллоидного раствора (например, сыворот¬
ки) или раствора с высоким содержанием частиц. Несколько предварительных фильтров могут быть связаны последовательно,
и только заключительный фильтр должен быть стерильным.
(а) Схематическое изображение принципа предварительной фильтрации; среда, проходящая через фильтры с постепенно
уменьшающимся диаметром пор; (б) ряд фильтров, сложенных в нестерильном держателе фильтра многократного использо¬
вания с последующим поступлением раствора на стерильный одноразовый фильтр с диаметром пор 0,1 или 0,22-мкм; (в) один
одноразовый предварительный фильтр (нестерильный или стерильный) вставленный перед заключительным стерилизаци¬
онным фильтром 0,22 или 0,1 мкм
11.5.4. Приготовление и стерилизация
других реагентов
Индивидуальные рецепты и процедуры представлены
в Приложении I. В целом, большинство термостабиль¬
ных реактивов автоклавируются, если же они термола¬
бильны, то стерилизуются методом фильтрации (см.
табл. 11.2). Фильтры со слабыми связывающими свой¬
ствами (например, Millex-GV) могут быть использова¬
ны для стерилизации растворов белков и пептидов.
11.6. КОНТРОЛЬ, ПРОВЕРКА КАЧЕСТВА
И ХРАНЕНИЕ СРЕД
11.6.1. Контроль качества
Качество среды, приготовленной в лаборатории,
должно быть проверено до ее использования. Если
среда приобретена готовой как рабочий раствор 1х, то
ее можно использовать, надеясь на то, что контроль
качества был проведен компанией-производителем,
11.6. Контроль, проверка качества и хранение сред
209
если только у вас нет каких-либо особенных требова¬
ний к качеству и свойствам среды. При использовании
концентрата среды 10х ростовые свойства и стериль¬
ность также проверяются производителем, однако вам
следует проверить качество воды, используемой для
разведения. Учитывая, что проводимость и уровень
общего органического углерода падают в пределах
стандарта (см. разд. 11.4.1) и при добавлении воды не
происходит каких-то значительных изменений, в боль¬
шинстве лабораторий считается, что контроль качества,
проводимый компанией, достаточен.
Однако если среда готовится из порошка, а затем
стерилизуется в лаборатории, то требуется проведение
контроля качества, подтверждающее стерильность,
хотя предполагается, что при полном растворении
всех компонентов контроль ростовых качеств среды
не требуется. Среда, сделанная прямо в лаборатории
из основных компонентов, требует проведения полного
контроля качества, включая как проверку стерильнос¬
ти, так и исследование культуральных свойств.
11.6.2. Проверка стерильности
Точка образования пузырьков. По завершении фильт¬
рации при высоком давлении, когда вся жидкость про¬
шла через фильтр, поднимите давление нагнетания до
тех пор, пока не образуются пузырьки в вытекающем из
фильтра потоке. Это точка образования пузырьков, ко¬
торая соответствует давлению, более чем вдвое превы¬
шающему давление, необходимое для фильтрации (см.
инструкцию производителя). Если пузырьки появляют¬
ся на фильтре при стерилизующем давлении (100 кПа,
1 атм) или ниже, то фильтр перфорирован и должен
быть заменен. В этом случае весь собранный фильтрат
должен рассматриваться как нестерильный и повторно
фильтруется. Фильтры одноразового использования
редко не проходят тест на появление пузырьков, по¬
скольку их использование кратковременно. Фильтры
многократного использования следует подвергать этому
тесту каждый раз после завершения фильтрации.
Инкубация. Отбираются образцы в начале, середи¬
не и конце процесса фильтрации. Если бутыли, в кото¬
рые собирается фильтрат, маленькие, то можно просто
по завершении фильтрации взять для исследования
отдельные бутыли. Если бутыли большие, то для того,
чтобы не расходовать большие порции, отберите образ¬
цы в маленькие емкости через определенные интервалы
времени в ходе фильтрации. Помните, однако, что если
вы бутилируете в объемы 1000 мл, отбор 1-мл образцов
снизит чувствительность в тысячу раз. Если вас не
устраивает использование всех бутылей для хранения
такого объема, то для анализа контроля качества сле¬
дует взять, по крайней мере, 100 мл. Лучше не извлекать
образцы из отдельных бутылей, поскольку это повысит
риск контаминации бутылей для хранения и уменьшит
объем их содержимого, что приведет к последующему
изменению объема добавок в эту бутыль.
Инкубируйте образцы в соответствии с одной из
нижеописанных процедур:
1) Инкубируйте образцы среды при 37 °С в течение
1 недели. Если какой-то из образцов становится
мутным, удалите его и повторно простерилизуйте
всю партию. Если имеются признаки заражения в
других хранящихся бутылях, вся партия должна
быть выбракована.
2) Для более тщательного теста, а также в случаях,
когда фильтруемый раствор не содержит собствен¬
ных питательных веществ, возьмите образцы, как
это описано выше, и разведите одну треть каждого
объема в питательной среде (например, L-бульон,
гидролизат бычьего сердца и тиогликолевый рас¬
твор). Разделите каждый образец на две порции, и
инкубируйте при 37 °С и при 20 °С в течение 10 дней
с инокулироваными контролями. Если после инку¬
бации возникнут какие-то сомнения, перемешайте и
посейте на чашку с питательным агаром небольшое
количество и инкубируйте при 37 °С и при 20 °С.
Вторичная фильтрация по направлению струи.
Поместите разъемный 0,45мкм стерильный фильтр в
систему фильтрации после главного стерилизующего
фильтра. Любое заражение, которое может возникнуть
при повреждении первого фильтра, будет задержано на
втором. В конце процесса удалите вторичный фильтр и
поместите его на питательный агар. Если вырастут ко¬
лонии, выбросьте или профильтруйте среду повторно.
Этот метод имеет то преимущество, что позволяет вес¬
ти мониторинг всего фильтрата, а не только некоторой
его порции. Однако и в этом случае нельзя избегнуть
риска заражения при бутилировании и закрывании.
Автоклавированные растворы. Оценка стериль¬
ности автоклавированных растворов менее сущест¬
венна, поскольку считается, что при соответствующем
контроле температуры, давления и времени автокла¬
вирования при температуре стерилизации в центре
закладки происходит стерилизация всей партии (см.
разд. 11.4).
11.6.3. Проверка культуральных свойств
Среда, производимая в коммерческих масштабах,
проверяется на предмет поддержания роста одной или
нескольких клеточных линий. (Если это не делается,
поменяйте поставщика!) Тем не менее при опреде¬
ленных условиях вы можете пожелать исследовать
вашу среду на качество ростовых свойств. Это может
быть, если:
1) среда составлена в лаборатории из основных ком¬
понентов;
2) произведены любые изменения в составе среды,
например, внесены какие-то добавки;
3) среда будет использоваться для каких-то специаль¬
ных целей, которые не могут быть проверены про¬
изводителем;
210
ГЛАВА 11. ПОДГОТОВИТЕЛЬНЫЕ РАБОТЫ И СТЕРИЛИЗАЦИЯ
4) среда сделана из порошка и существует риск потери
компонентов в ходе фильтрации.
Среда может стать зараженной токсическими вещест¬
вами при фильтрации. Например, некоторые фильтры
обрабатываются детергентами, следовые количества
которых облегчают смачивание. Этот детергент может
выходить в среду при фильтрации. Такие фильтры
должны промываться проточным PBS или BSS до
использования или первая порция фильтрата должна
выбрасываться. Поликарбонатные фильтры (напри¬
мер, Nucleopore) легко смачиваются без применения
детергентов и предпочтительны для некоторых видов
работы, в частности, когда в среде низкая концентрация
сыворотки.
Существует три основных типа тестирования куль¬
туральных свойств:
1) эффективность посева;
2) изучение кривой роста при постоянной проверке
плотности;
3) изучение проявления специальных функций (напри¬
мер, дифференцировка в присутствии индуктора,
размножение вируса, образование специфического
продукта или экспрессия специфического антиге¬
на). На новой партии среды должны проводиться
все эти тесты с использованием постоянной среды,
которую вы используете в качестве контроля.
Эффективность посева. Тест на эффективность
посева (см. протокол 21.10) является наиболее чув¬
ствительный культуральным тестом, определяющим
минимальный недостаток питательных веществ или
низкую концентрацию токсинов, которые не обнару¬
живаются при более высокой клеточной плотности.
В идеале, его следует проводить при лимитированной
концентрации сыворотки, которая в противном случае
может маскировать дефицит среды. Для определения
этой лимитированной концентрации произведите
первоначальный посев при различных концентрациях
сыворотки и выберите ту концентрацию, которая соот¬
ветствует эффективности посева примерно вдвое ниже
по сравнению с обычной, но еще делает возможным
формирование колоний.
Кривая роста. Исследование роста клона не всегда
позволяет обнаружить недостаток в количестве отдель¬
ных веществ. Например, если концентрация одной или
более аминокислот низкая, это может не оказывать
влияние на рост клона, но может подействовать на
величину максимальной концентрации вырастающих
клеток.
Кривая роста (см. протоколы 21.7-21.9) позволяет
определить три параметра: ^продолжительность
lag-фазы до начала клеточной пролиферации после
субкультивирования, которая показывает возможность
адаптации клеток к условиям культивирования в дан¬
ной среде; 2) время удвоения культуры в середине фазы
экспоненциального роста, которое показывает ростовой
потенциал среды и 3) конечную плотность клеток. В тех
клеточных линиях, рост которых не чувствителен к
плотности клеток (например, постоянные клеточные
линии; см. разд. 18.5.2), конечная клеточная плотность
определяет общий возможный клеточный выход и
отражает общее содержание аминокислот и глюкозы.
Помните, что среда, которая дает половину конечной
плотности клеток, стоит вдвое дороже при пересчете
на одну полученную клетку.
Специальные функции. Если вы исследуете спе¬
циальные функции, то для испытания новой среды
следует провести стандартный тест, который вы
применяете в ходе вашей экспериментальной работы
(например, определите титр вируса в среде после опре¬
деленного срока), при этом в качестве контроля должна
быть старая среда, на которой вы уже работали.
Главной предпосылкой к проведению этого теста
является определение возможности их применения
в дальнейшем в случае истощения запасов той среды,
которую вы постоянно используете, так чтобы в работу
могли быть внесены соответствующие поправки и было
время для приготовления новой среды, если проверя¬
емая среда окажется непригодной для специальных
функций.
Ведение записей. Все качественные результаты
культуральных тестов должны записываться в регист¬
рационную книгу или компьютерную базу данных,
наряду с номером партии раствора, который подвергали
тестированию, а также именем исследователя. Лицо,
ответственное за подготовительные и стерилизацион¬
ные работы, должно следить за ведением этих записей
и определять нормы и направления, определяя необхо¬
димость дальнейших испытаний.
11.6.4. Хранение
В отношении хранения различных сред (на полках)
существуют различные мнения. По грубой оценке,
среда без глутамина может храниться, по крайней мере,
6-9 месяцев при температуре 4 °С. При добавлении
глутамина, сыворотки или антибиотиков время хране¬
ния сокращается до 2-3 недель. Следовательно, среда,
содержащая лабильные компоненты, должна быть
использована в течение 3 недель после приготовления
или храниться при -20 °С.
Некоторые формы люминесцентного света могут
вызвать разрушение рибофлавина, триптофана и ти¬
розина с образованием токсичных продуктов, главным
образом, перекисей [Wang & Nixon, 1978; Edwards et
al., 1994]. Следовательно, люминесцентные лампы не
следует использовать в холодной комнате, в которой
хранятся среды, и в термальной комнате, где куль¬
тивируются клетки. Кроме того, любой свет должен
быть выключен, если в комнатах не ведется работа.
Бутыли со средой не должны подвергаться действию
люминесцентного света более чем несколько часов.
Для долговременного хранения рекомендуется темный
морозильник.
Глава 12
ПЕРВИЧНАЯ КУЛЬТУРА
12.1. ТИПЫ ПЕРВИЧНЫХ КУЛЬТУР
КЛЕТОК
Первичной культурой называют культуру клеток на
стадии непосредственно после выделения клеток и до
первого посева. Рассмотрим 4 стадии: 1) получение
образца; 2) выделение ткани; 3) препарирование и/или
дезагрегация и 4) культура после посева в культураль¬
ный сосуд. После выделения первичная культура кле¬
ток может быть получена как путем миграции клеток
из фрагмента ткани и прикрепления их к подходящему
субстрату, так и при дезагрегации ткани (механичес¬
ким путем или с помощью ферментов) для получения
суспензии клеток, некоторые из которых, в конечном
итоге, прикрепятся к субстрату. Для большинства
нормальных ^трансформированных клеток, за исклю¬
чением гематопоэтических, необходимо прикрепление
к плоской поверхности для выживания и максимально
эффективной пролиферации. Трансформированные
клетки (см. разд. 18.5.1), в особенности индивиду¬
альные клетки из трансплантируемых опухолей жи¬
вотных, напротив, часто способны пролиферировать
в суспензии.
К ферментам, которые наиболее часто используют
для дезагрегации ткани, относятся неочищенные препа¬
раты трипсина, коллагеназа, эластаза, проназа, диспаза,
ДНКаза и гиалуронидаза, которые используют либо по
отдельности, либо в различных комбинациях, напри¬
мер эластазу с ДНКазой для выделения альвеолярных
клеток II типа [Dobbs & Gonzalez, 2002], коллагеназу
вместе с диспазой [Booth & O’Shea, 2002] и коллагеназу
вместе с гиалуронидазой [Berry & Friend, 1969; Seglen,
1975]. Другие ферменты, не животного происхожде¬
ния, такие как Trypzean (Sigma) — рекомбинантный
кукурузный трипсин, TrypLE (Invitrogen) — реком¬
бинантный трипсин, продуцируемый микробами и Ас-
cutase и Accumax (Innovative Cell Technologies), также
подходят для первичной дезагрегации. Препараты
грубой очистки часто более эффективны, чем очищен¬
ные ферментные препараты, так как первые содержат
примеси других протеаз, хотя последние обычно менее
токсичны и их действие более специфично. Трипсин
и проназа дают более полную дезагрегацию, но могут
повреждать клетки. С другой стороны, коллагеназа и
диспаза дезагрегируют клетки неполностью, но менее
токсичны. Гиалуронидаза используется в сочетании
с коллагеназой для разрушения внутриклеточного
матрикса, а ДНКаза используется для уничтожения
ДНК, высвободившейся из лизированных клеток; ДНК
снижает эффективность протеолиза и содействует
реагрегации клеток (см. табл. 13.4).
Хотя для обработки каждой ткани может потребо¬
ваться своя последовательность действий, для боль¬
шинства имеются некоторые общие требования:
1) Жир и некротические ткани лучше всего удалять во
время препарирования.
2) Ткань следует мелко нарезать острым инстру¬
ментом для того, чтобы повреждения были ми¬
нимальными.
3) Использованные для дезагрегации ферменты долж¬
ны быть впоследствии удалены с помощью мягкого
центрифугирования.
4) Концентрация клеток в первичной культуре должна
быть намного выше, чем обычно используемая при
субкультивировании, так как процент клеток, ко¬
торые выживут в первичной культуре, может быть
довольно низким.
5) Предпочтительней использовать богатые среды,
такие как среда Хэма F12, а не простые, такие как
МЕМ Игла. Если требуется сыворотка, следует
учесть, что фетальная бычья сыворотка способ¬
ствует лучшей выживаемости по сравнению с
телячьей или лошадиной. Для выделения специ¬
фических типов клеток, вероятно, потребуются
селективные среды (см. разд. 10.2.1 и гл. 23).
6) Дезагрегация эмбриональных тканей проходит
легче, чем тканей взрослого организма, выживает
больше клеток, и они быстрее начинают пролифе¬
рировать.
12.2. ВЫДЕЛЕНИЕ ОБРАЗЦОВ ТКАНИ
Перед началом работы с тканями животных или
человека вы должны быть уверены, что ваша работа
соответствует требованиям медицинской этики или
современным законодательным нормам в отношении
экспериментов с животными (см. разд. 7.9.1). Напри¬
мер, в Великобритании использование эмбрионов или
плодов после середины срока беременности регулиру¬
ется законом об экспериментах на животных 1986 года
(Animal Experiments (Scientific Procedures) Act of 1986).
Для работы с биопсийным или фетальным материалом
человека обычно требуется согласие местного комитета
212
ГЛАВА 12. ПЕРВИЧНАЯ КУЛЬТУРА
по этике и пациента и/или его или ее родственников
(см. разд. 7.9.2).
• Требование безопасности. Работа с тканями
человека должна выполняться в соответствии с Уров¬
нем защиты 2 в ламинарных шкафах II класса биоло¬
гической защиты (см. разд. 7.8.3).
Необходимо попытаться простерилизовать место
резекции 70%-м спиртом (в случае если есть вероят¬
ность загрязнения, например на коже). Асептически
вырежьте ткань и как можно скорее перенесите ее в
лабораторию культуры ткани в BSS (DBSS) или в
среде для транспортировки (см. Приложение I). Не
препарируйте животных в лаборатории культуры
тканей, так как животные могут внести бактериальную
контаминацию. Если задержка в транспортировке
ткани неизбежна, ткань можно хранить при 4 °С до
72 ч, хотя лучший результат достигается при быстрой
транспортировке.
12.2.1. Мышиный эмбрион
Эмбрионы мыши — это удобный источник клеток для
получения недифференцированных фибробластоид-
ных культур. Их часто используют в качестве фидер¬
ного слоя (см. рис. 14.2.3).
ПРОТОКОЛ 12.1. Выделение мышиного
эмбриона
Схема
Асептически удалить матку у мыши с заданным
сроком беременности и вырезать эмбрион.
Материалы
Стерильные:
• DBSS: BSS для препарирования (BSS с высокой
концентрацией антибиотиков, смотри приложе¬
ние I) в пробирке с закручивающейся пробкой
объемом 25-50 мл или в универсальном кон¬
тейнере.
• 50 мл BSS в стерильном стакане (используется
для охлаждения инструментов после стерилиза¬
ции пламенем)
• Чашки Петри диаметром 9 см
• Остроконечные пинцеты.
• Остроконечные ножницы
Нестерильные:
• Малый шкаф с ламинарным потоком
• Мышь с необходимым сроком беременности
• 70%-й спирт в промывалке
• 70%-й спирт для стерилизации инструментов
(см. рис. 7.4)
• Горелка Бунзена.
• Требование безопасности. При стери¬
лизации инструментов путем погружения их в
спирт и последующего прожигания следите за тем,
чтобы не вернуть инструменты в спирт пока они
еще горят.
Протокол
1. Индукция эстпруса. Если самцы и самки раз¬
мещены по отдельности, поместите их вместе
для спаривания. Течка у самок начнется через
3 суток. Это позволит планировать образование
эмбрионов в назначенное время. Время для
удачного спаривания можно определить, прове¬
ряя каждое утро жесткость и слизистую пробку
влагалища
2. Датировка эмбрионов. День выявления ваги¬
нальной пробки или «дата пробки» записывается
как нулевой день, и развитие эмбриона отсчиты¬
вается с этой даты. Полный срок беременности
19-21 день. Оптимальный возраст эмбриона
для получения культуры из полностью дезагре¬
гированного эмбриона — около 13 дней, когда
эмбрион уже относительно большой (рис. 12.1,
12.2), но еще содержит высокий процент не¬
дифференцированной мезенхимы, которая и
является главным источником культуры клеток.
Однако для изоляции и проведения манипуля¬
ций с эмбрионом после середины полного срока
беременности может потребоваться лицензия
(например, в Великобритании), поэтому, воз¬
можно, предпочтительней брать работать с
9-10-дневными эмбрионами. Хотя количество
ткани, полученной из этих эмбрионов, будет
существенно меньше, больший процент клеток
пойдет в рост. Большинство индивидуальных
органов, за исключением мозга и сердца, на¬
чинают формироваться примерно на 9-й день
беременности, но их трудно выделить до 11-го
дня. Препарирование индивидуальных органов
проще проводить на 13-14-й день. Большинство
органов полностью формируются к 18-му дню.
3. Забейте мышь путем дислокации шейных поз¬
вонков (процедура описана в Правилах про¬
ведения работ I Великобритании) и обильно
протрите брюшную поверхность 70%-м спиртом
(рис. 12.3а).
4. Сделайте разрез кожи с брюшной стороны
перпендикулярно медиане до диафрагмы
(рис. 12.36) и, сжав пальцами кожу с двух сто¬
рон разреза, потяните ее в противоположных
направлениях, чтобы обнажить нетронутую вен¬
тральную поверхность стенки брюшной полости
(рис. 12.3в).
5. Стерильными ножницами сделайте надрез
вдоль линии медианы, обнажая внутренности
(рис. 12.3г). На этой стадии матка с эмбрионом
хорошо видна в тазовой области брюшной полос¬
ти (рис. 12.3d).
12.3. Выделение образцов ткани
213
6. Извлеките матку и поместите ее во флакон с
завинчивающейся крышкой объемом 25 мл
или 50 мл, заполненный 10 или 20 мл DBSS
(рис. 12.3е).
Примечание. Все предыдущие этапы должны про¬
водиться вне лаборатории культуры тканей. Малый
шкаф с ламинарным потоком и быстрое проведение
операций помогут обеспечить стерильность, Не
приносите живых животных в лабораторию куль¬
туры тканей, так как животные могут быть конта-
минированы. Если с тушкой животного необходимо
проводить какие-либо манипуляции в зоне работы
с культурой тканей, убедитесь, что тушка была на
короткое время погружена в спирт или тщательно
протерта спиртом и после использования быстро
удалена из помещения.
7. Перенесите интактную матку в лабораторию
культуры тканей и поместите ее в новую чашку
Петри со стерильным DBSS (рис. 12.3ж).
8. Препарирование эмбрионов:
а) Вскройте матку с помощью двух стерильных
пинцетов, держа концы пинцетов близко
друг к другу, чтобы избежать разрыва матки,
и не давите слишком сильно на эмбрионы
(рис. 12.3 ж, з).
б) Освободите эмбрионы от оболочек (рис. 12.3и)
и плаценты и поместите их с одной стороны
чашки для стекания крови.
9. Перенесите эмбрионы в новую чашку Петри. Если
требуется большое количество эмбрионов (на¬
пример, необходимо больше четырех или пяти по¬
метов), может быть полезно поместить чашку на
лед (для последующего препарирования и куль¬
тивирования, смотри протоколы 12.4-12.8).
Рис. 12.2. Мышиные эмбрионы. Эмбрионы на 12-й, 13-й и
14-й дни беременности. 12-дневный эмбрион (внизу) получен
из малого помета (три), и он больше по размеру, чем обычно
бывает на этой стадии
Дни беременности
10Ю
109
1
о
5
О.
VO
2
о
ю8
105
107 -
106
□ Теплый трипсин
А Холодный трипсин
!
10
11 12 13 14 15 16 17 18
Дни беременности
Рис. 12.1. Общий вес и выход клеток мышиного эмбриона. (а) Общий вес эмбриона без плаценты и оболочек, средние зна¬
чения ± стандартное отклонение [по Paul et al., 1969]. (б) Выход клеток на эмбрион после инкубации в 0,25% трипсине 4 ч при
37 °С без промежуточного сбора (квадраты) или после инкубации в 0,25% трипсине 5 ч при 4 °С с последующей инкубацией в
течение 30 мин при 37 °С (треугольники; протокол 12.6)
214
ГЛАВА 12. ПЕРВИЧНАЯ КУЛЬТУРА
Рис. 12.3. Препарирование мыши. Стадии препарирования беременной мыши для забора эмбрионов (смотри протокол 12.1).
(<а) Протирание брюшной полости, (б, в) Растягивание кожи для того, чтобы обнажить стенку брюшной полости, (г) Вскрытие
брюшной полости. (Э) Выявление местонахождения матки, (ё) Извлечение матки, (ж, з) Извлечение эмбрионов из матки.
(и) Удаление оболочек, (к) Удаление головы (необязательно), (л) Измельчение эмбриона. (.м) Перенос кусочков во флакон для
трипсинизации (для тепловой трипсинизации; см. протокол 12.5). (н) Перенос кусочков в малый флакон Эрленмайера (для
холодной трипсинизации, см. протокол 12.6). (о) Помещение флакона на лед
12.3. Выделение образцов ткани
215
Рис. 12.3. Препарирование мыши
12.2.2. Куриный эмбрион
Куриные эмбрионы проще препарировать, так как они
больше мышиных эмбрионов на аналогичной стадии
развития. Так же как и мышиные эмбрионы, куриные
эмбрионы используются для получения первичной
культуры мезенхимальных клеток для исследования
клеточной пролиферации, для получения клеток для
фидерного слоя и как субстрат для наращивания виру¬
са. Из-за крупного размера у куриных эмбрионов легче
изолировать индивидуальные органы для получения
специфичных типов клеток, таких как гепатоциты,
кардиомиоциты и легочный эпителий. Как и в случае
с мышиными эмбрионами, эксперименты с куриными
эмбрионами подпадают под действие закона об экспе¬
риментах на животных (например, в Великобритании),
поэтому для работы с эмбрионами после половины
срока беременности может потребоваться лицензия.
216
ГЛАВА 12. ПЕРВИЧНАЯ КУЛЬТУРА
ПРОТОКОЛ 12.2. Извлечение куриных
эмбрионов
Схема
Извлечь из яйца эмбрион в асептических условиях
и поместить его в чашку.
Материалы
Стерильные:
• DBSS: BSS для препарирования (BSS с высокой
концентрацией антибиотиков, смотри Приложе¬
ние I) в пробирке с закручивающейся пробкой
объемом 25-50 мл или в универсальном кон¬
тейнере.
• 50 мл BSS в стерильном стакане (используется
для охлаждения инструментов после стерилиза¬
ции пламенем).
• Маленький стакан 20-50 мл или поставка для
яйца.
• Прямой и изогнутый пинцеты.
• Чашки Петри диаметром 9 см.
Нестерильные:
• Яйца с 10-дневными зародышами
• 70% спирт.
• Тампоны
• Инкубатор с увлажненной атмосферой (без до¬
полнительного со2).
Протокол:
1. Инкубируйте яйца при 38,5 °С в увлажненной
атмосфере; ежедневно переворачивайте яйца
на 180°.
2. Хотя куриные яйца насиживают от 20 до 21 дня,
продолжительность стадий развития эмбрио¬
на отличается от стадий развития мышиного
эмбриона. Для получения культуры изолиро¬
ванных клеток из целого эмбриона яйцо необ¬
ходимо инкубировать 8 дней, для выделения
рудиментарных органов — около 10-13 дней
(в Великобритании без лицензии максимально
10 дней).
3. Протрите яйцо 70%-м спиртом и поместите
его тупым концом вверх в маленький стакан
(рис. 12.4а).
4. Разбейте скорлупу в верхней части (рис. 12.4б)
и снимите скорлупу над воздушным мешком
стерильным пинцетом (рис. 12.4в).
5. Простерилизуйте пинцет (например, погружая
его в спирт и далее прожигая спирт на пинцете,
и охладив пинцет в стерильном BSS) и удалите
белую скорлуповую оболочку, чтобы обнажить
нижнюю хориоаллантоисную оболочку (ХАО)
с кровеносными сосудами (рис. 12.4г, д).
6. Прорвите ХАО стерильным изогнутым пин¬
цетом (рис. 12.4е) и вытащите эмбрион, осто¬
рожно придерживая его пинцетом ниже головы
(рис. \2Аж, з). Не сжимайте пинцет слишком
сильно, иначе порвется шея; поместите средний
палец под пинцет и используйте подушечку,
чтобы уменьшить давление указательного пальца
(рис. 12.4ж). Перенесите эмбрион в чашку Петри
диаметром 9 см с 20 мл DBSS (рис. 12.4м). (Даль¬
нейшее препарирование и культивирование
описано в протоколе 12.7.)
12.2.3. Биопсийный материал
человека
При использовании биопсийного материала челове¬
ка возникают определенные проблемы, с которыми
не сталкиваются при работе с тканями животных.
Обычно необходимо получить согласие 1) комитета
по этике больницы, 2) лечащего врача или хирурга
и 3) донора или больного или родственников боль¬
ного (см. разд. 7.9.2). Кроме того, образцы биопсии
обычно берут для диагностических целей, и, следо¬
вательно, в первую очередь учитываются интересы
патолога. Значение этого фактора снижается в случае
обширной хирургической резекции или при заборе
непатологических тканей (например, плаценты или
пуповины).
Операции часто выполняются постоянными со¬
трудниками больницы во время, не всегда подходящее
для лаборатории культуры тканей, таким образом,
должна быть лредусмотрена система временного хра¬
нения на тот период, когда вы или кто-либо из ваших
сотрудников не сможет находиться в лаборатории.
Если налажена доставка материала в вашу лаборато¬
рию, то должна быть организована система получения
образцов, записи подробностей об источнике, ткани,
патологии и т.д. (см. разд. 12.3.11) и оповещения со¬
трудника, который будет заниматься культивировани¬
ем, о прибытии образца; в противном случае ценный
материал может быть потерян или испорчен.
• Требование безопасности. При работе с
биопсийным материалом человека существует опас¬
ность инфекционного заражения сотрудников (см.
разд. 7.8.3), поэтому работать с ним необходимо в
соответствии с Уровнем защиты 2 в ламинарных
шкафах II класса биологической защиты. Все мате¬
риалы и инструменты после использования должны
быть продезинфицированы автоклавированием или
погружением в соответствующий дезинфектант (см.
разд. 7.8.5). Ткани должны быть протестированы на
наличие случайных инфекций, таких как гепатиты,
ВИЧ и туберкулез [требование Министерства здраво¬
охранения и социального обеспечения Соединенных
Штатов, 1993; Консультативный комитет по опасным
инфекциям; 1995b в Великобритании], если только
больного уже не проверяли на эти инфекции.
12.3. Первичная культура
217
Рис. 12.4. Извлечение куриного эмбриона из яйца. Стадии извлечения целого куриного эмбриона из яйца, (а) Протирание
яйца спиртом. (6) Разбивание скорлупы, (в) Удаление скорлупы, (г) Удаление скорлуповой оболочки. (д) Обнажение хорио-
аллантоисной (ХАО) оболочки с кровеносными сосудами. (е) Удаление ХАО пинцетом, (ж) Захватывание эмбриона вокруг
шеи. (з) Извлечение эмбриона из яйца, (и) Изолированный 10-дневный эмбрион в чашке Петри
12.3. ПЕРВИЧНАЯ КУЛЬТУРА
Разработано несколько методик для дезагрегации тка¬
ни при получении первичной культуры. Эти методики
можно подразделить на 1) простые механические ме¬
тоды, включающие рассечение с некоторыми видами
размягчения или без, и 2) методы с использованием
ферментативной дезагрегации (рис. 12.5). Первичные
эксплантаты применяются, когда доступны лишь
очень малые количества ткани; энзиматическая дезаг¬
регация является предпочтительной, когда имеются
в наличии большие объемы ткани. Механическую
дезагрегацию хорошо использовать, размельчая мяг¬
кие и некоторые плотные ткани, когда количество
218
ГЛАВА 12. ПЕРВИЧНАЯ КУЛЬТУРА
жизнеспособных клеток на выходе не является су¬
щественным.
12.3.1. Первичный эксплантат
Метод первичного эксплантата, разработанный Harrison
[1907], Carrel [1912] и другими учеными, был первым
методом получения культуры ткани. В оригинальном
варианте фрагмент ткани погружали в плазму крови
или лимфу, смешивали с гетерологичной сывороткой и
эмбриональным экстрактом и помещали на покровное
стекло, переворачивали и заключали в часовое стекло.
ПРОТОКОЛ 12.3. Биопсийный материал
человека
Схема
Поговорите с персоналом больницы, обеспечьте
маркированными контейнером (и) со средой и до¬
говоритесь о сборе образцов из операционной или
из кабинета патолога.
Материалы
Пробирки для образцов (15-30 мл) с герметичной
крышкой, наполовину заполненные культуральной
средой, содержащей антибиотики (смотри Приложе¬
ние I: среда для сбора образцов), на которых указаны
ваше имя, адрес и номер телефона.
Протокол
1. Подготовьте четко подписанные контейнеры
со средой для сбора образцов в приемной опера¬
ционной или лаборатории патологии.
2. Договоритесь, чтобы вам сообщили, когда мате¬
риал будет готов для сбора.
3. Заберите контейнеры после операции или найди¬
те кого-то, кто пришлет их вам сразу после сбора
и сообщит вам, когда они были отправлены.
4. Перенесите образцы в лабораторию культуры
тканей. Образцы должны быть завернуты в три
слоя (например, герметично закрытую пробирку
помещают в герметичный пакет, заполненный ад¬
сорбентом на случай протечки; пакет помещают
в упаковку с мягкой подложкой с написанным
вашим именем, адресом и номером телефона,
см. разд. 20.4). Обычно при хранении при 24 °С
клетки в образцах биопсии выживают в течение
как минимум 24 ч и даже 3 или 4 дней, хотя при
увеличении интервала времени от операции до
культивирования большая часть образцов, по-
видимому, деградирует.
5. Зарегистрируйте образец под соответствующим
номером в рукописном регистрационном жур¬
нале для последующего переноса информации в
компьютерную базу данных или занесите данные
в базу данных сразу после поступления.
6. Дезинфекция. Хотя большинство хирургических
образцов извлекают в стерильных условиях,
могут возникнуть проблемы при последующих
манипуляциях. Образцы биопсии поверхнос¬
тных тканей (например, биопсия кожи, мела¬
номы и т.д.) и желудочно-кишечного тракта
особенно подвержены контаминации, даже если
кожа предварительно продезинфицирована
перед взятием биопсии, а перед операцией же-
лудочно-кишечного тракта пациент получает
инъекцию антибиотика. Может быть полезным
проконсультироваться со специалистом по ме¬
дицинской микробиологии, чтобы определить,
какая флора представлена в данной ткани и затем
в соответствии с этим выбрать антибиотики,
которые вы будете использовать при заборе об¬
разцов и препарировании. Если хирургические
образцы достаточно большие (а именно, 200 мг
или больше), быстрое погружение (а именно,
30 с — 1 мин) в 70%-й спирт поможет снизить
поверхностную контаминацию, не повреждая
центральную часть образца ткани.
Комок плазмы удерживал ткань на месте, и эксплан¬
тат можно было рассматривать с помощью обычного
микроскопа. Гетерологичная сыворотка индуцировала
сворачивание плазмы, эмбриональный экстракт, сыво¬
ротка и плазма снабжали клетки питательными вещес¬
твами и факторами роста и стимулировали миграцию
клеток из эксплантата. Эта методика используется до
сих пор, но, по большей части, заменена упрощенным
методом, описанным в протоколе 12.4.
Этот метод особенно полезен при работе с неболь¬
шими количествами ткани, например при биопсии
кожи, когда есть риск потери клеток в ходе механичес¬
кой или ферментативной дезагрегации. Недостатки его
заключаются в низкой адгезивности некоторых тканей
и селекции клеток в процессе роста. На практике, од¬
нако, большинство клеток, особенно эмбриональных,
успешно мигрируют.
Прикрепление эксплантатов. Как прикрепление, так
и миграцию клеток можно простимулировать, помес¬
тив на поверхность эксплантата стеклянное покровное
стекло или поцарапать эксплантат пластиковой чаш¬
кой Петри перед прикреплением ткани к поверхности
флакона [Elliget & Lechner, 1992] (см. протокол 23.9).
Прикрепление также можно стимулировать или улуч¬
шить, обработав пластик полилизином или фибронек-
тином (см. протокол 8.1) или сформировав фидерный
слой (см. протокол 14.3). Исторически сложилось, что
для улучшения прикрепления использовали сгустки
плазмы. Поместите каплю плазмы на пластиковую
поверхность и заключите в ней эксплантат. Это должно
индуцировать сворачивание плазмы в течение несколь¬
ких минут, после чего можно добавить среду. В качестве
альтернативы можно использовать очищенный фибри¬
ноген и тромбин [Nicosia & Ottinetti, 1990].
12.3, Первичная культура
219
ВЫДЕЛЕНИЕ ОБРАЗЦА ТКАНИ
ПРЕПАРИРОВАНИЕ
Выбор нужной ткани; удаление жира и
некротических клеток
ОКОНЧАТЕЛЬНОЕ
ПРЕПАРИРОВАНИЕ
вырезание кусочков
эксплантата нужного размера
МЕХАНИЧЕСКАЯ ДЕЗАГРЕГАЦИЯ,
Например, протирание через
сито, многократное пропускание
через шприц, или интенсивное
пипетирование
ФЕРМЕНТАТИВНАЯ ДЕЗАГРЕГАЦИЯ
ПЕРВИЧНЫЙ ЭКСПЛАНТАТ
ЭКСПЛАНТАТ РАЗРАСТАНИЕ
ХОЛОДНЫЙ ТРИПСИН ТЕПЛЫЙ ТРИПСИН
Длительная
инкубация, повторный
В течение ночи на
холоду, короткая
КОЛЛАГЕНАЗА
Длительная
инкубация,
Перенос в
другой флакон
2-Я КУЛЬТУРА
ЭКСПЛАНТАТА
ДИСПЕРСНАЯ
ПЕРВИЧНАЯ
КУЛЬТУРА
Ресуспендирование
и посев
Пассирование
ЛИНИЯ КЛЕТОК
Рис. 12.5. Варианты первичной культуры. Различные пути получения линии клеток; в центре и слева — путем механической
дезагрегации, справа — путем ферментативной дезагрегации. Эксплантаты можно переносить, чтобы стимулировать даль¬
нейший рост клеток эксплантата, в то время как вышедшие из эксплантата клетки можно субкультивировать для получения
линии клеток
12.3.2. Ферментативная дезагрегация
Межклеточная адгезия в тканях опосредуется различ-
ными гомотипически взаимодействующими глико¬
пептидами (молекулы клеточной адгезии, или САМ)
(см. разд. 3.2.1), некоторые из которых кальций-зави-
симые (кадгерины) и, следовательно, чувствительны к
хелатирующим агентам, таким как ЭДТА или ЭГТА.
Интегрины, которые связываются с аргинин-глицин-
аспарагиновая кислота (RGD) мотивом внеклеточного
матрикса, также имеют Са2+-связывающий домен, и уда¬
ление Са2+ на них тоже влияет. Межклеточный матрикс
и базальные мембраны содержат другие гликопротеины,
такие как фибронектин и ламинин, чувствительные к
протеазам, и менее чувствительные протеогликаны,
которые иногда поддаются протеолизу гликоназами,
такими как гиалуронидаза или гепариназа. Простейший
подход заключается в переходе от простого раствора для
дезагрегации к более сложному (см. табл. 13.4). Вначале
используется раствор трипсина или смеси трипсин/
ЭДТА, далее добавляют другие протеазы чтобы улуч¬
шить дезагрегацию и на следующей стадии, трипсин при
220
ГЛАВА 12. ПЕРВИЧНАЯ КУЛЬТУРА
ПРОТОКОЛ 12.4. Первичные эксплантаты
Схема
Ткань разрезают на кусочки, промывают, помещают
на поверхность культурального флакона или чашки
Петри в маленьком объеме среды с высокой кон¬
центрацией (а именно, 40-50%) сыворотки таким
образом, чтобы поверхностное натяжение удержи¬
вало кусочки на месте, пока они не прикрепятся
спонтанно к поверхности (рис. 12.6а). После того как
это будет достигнуто, обычно следует разрастание
клеток (рис. 12.66; цв. вкладки 1а, 26)
Стерильные материалы
• Ростовая среда (например, 50:50 DMEM: F12 с
20% фетальной бычьей сыворотки.
• 100 мл DBSS.
• Чашки Петри диаметром 9 см, не для культуры
клеток.
• Пинцет.
• Скальпели.
• Пипетки на 10 мл с широким концом.
• Центрифужные пробирки на 15 или 20 мл или
универсальные контейнеры.
• Культуральные флаконы на 25 см2 или чашки
Петри для культуральных работ диаметром
5-6 см. Размер флаконов и объем ростовой сре¬
ды зависит от количества ткани: на 10 мг ткани
необходимо приблизительно пять флаконов на
25 см2.
Протокол
1. Перенесите ткань в свежий стерильный DBSS и
промойте.
2. Перенесите ткань во вторую чашку, удалите не¬
желательные ткани, такие как жир или некроти¬
ческий материал, и перенесите в третью чашку.
3. Мелко нарежьте скрещенными скальпелями (см.
рис. 12.6а, вверху) на кусочки размером около
1 кубического миллиметра.
4. С помощью пипетки (10-20 мл с широким носи¬
ком) перенесите в стерильную центрифужную
пробирку объемом 15 или 50 мл или в универ¬
сальный контейнер. (Предварительно смочите
пипетку изнутри BSS или средой, иначе кусочки
ткани будут прилипать.)
5. Дайте кусочкам осесть.
6. Промойте путем ресуспендирования кусочки в
DBSS, дайте кусочкам осесть и удалите суперна-
тант. Повторите эту операцию не менее двух раз.
7. Перенесите кусочки (не забывая смачивать пи-
петку)в культуральный флакон, приблизительно
20-30 кусочков на флакон площадью 25 см2.
8. Удалите большую часть жидкости и добавить
1 мл ростовой среды на 25 см2 ростовой поверх¬
ности.
9. Закройте флакон и поместите его в инкубатор
или термальную комнату на 37 °С на 18-24 ч.
10. Если кусочки прикрепились, далее объем среды
можно постепенно увеличивать в течение следу¬
ющих 3-5 дней до 5 мл на 25 см2 и затем менять
среду еженедельно, пока не будет наблюдаться
значительный рост клеток (см. рис. 12.66).
11. После достижения роста клеток оставшийся экс¬
плантат может быть удален с помощью скальпеля
(рис. 12.6в) и перенесен предварительно смочен¬
ной пипеткой в свежий культуральный сосуд.
(И далее повторить начиная с этапа семь.)
12. Меняйте среду в первом флаконе до тех пор, пока
растущие клетки не покроют, по крайней мере,
50% ростовой поверхности, после чего клетки
можно пересевать (см. протокол 13.2).
необходимости удаляется, для того чтобы увеличить
выживаемость клеток. Обычно повышение чистоты
ферментов позволяет лучше контролировать процесс
и, повышая специфичность, снижать токсичность, но
может также приводить к ухудшению дезагрегации.
Механическая и ферментативная дезагрегации
ткани позволяют избежать проблемы селекции кле¬
ток, возникающей при их миграции из эксплантата,
и повышают выход большего количества клеток,
которые отражают состав ткани за короткое время.
Однако, подобно тому как метод первичного эксплан¬
тата приводит к селекции клеток по их способности
мигрировать из ткани, методы диссоциации приводят
к селекции клеток по их устойчивости к протеазам и
механическому стрессу.
Эмбриональные ткани лучше поддаются дисперги¬
рованию и дают больший выход пролиферирующих
клеток, чем ткани новорожденных или взрослых. На
повышение сложности получения жизнеспособных
пролиферирующих клеток с увеличением возраста
донора влияют несколько факторов, среди которых
возникновение дифференцировки, повышение со¬
держания грубоволокнистой соединительной ткани
и внеклеточного матрикса и снижение пула недиф¬
ференцированных пролиферирующих клеток. Когда
для дезагрегации ткани требуются более жесткие про¬
цедуры (например, продолжительная трипсинизация
или интенсивное перемешивание), наиболее нежные
компоненты ткани могут разрушаться. Например, в
фиброзных опухолях очень трудно добиться полной
диссоциации клеток трипсином так, чтобы при этом
еще и остались жизнеспособные клетки карциномы.
Выбор степени чистоты трипсина всегда труден,
так как существуют две противоположные точки
зрения: 1) более чистый трипсин менее токсичен, и
результат его действия более предсказуем; 2) трипсин
грубой очистки более эффективен, возможно, из-за
присутствия в нем других протеаз. На практике могут
понадобиться предварительные эксперименты для
определения оптимальной степени чистоты, необхо¬
димой для получения жизнеспособных клеток, так
как трудно предсказать, какой будет баланс между
12.3. Первичная культура
221
Ткань
Отрезание кусочков ткани Промывание ресус-
размером 0,5-1,0 мм пендированием и
скрещенными скальпелями осаждением 2-3 раза
и
Перенос кусочков во
флакон с помощью
предварительно
смоченной пипетки
о
Инкубация в течение
ночи в тонком слое
среды
Добавление среды до обычного
объема через 24-48 ч
После приблизительно 1 недели
можно увидеть, как клетки
радиально мигрируют из
эксплантата
Рис. 12.6. Культура первичного эксплантата. (а) Схематическая диаграмма стадий препарирования и посева первичного
эксплантата, (б) Первичный эксплантат плоскоклеточной карциномы кожи мыши; эксплантат на ранней стадии роста клеток
через 3 дня после эксплантации (смотри также илл. 26). (в) Рост клеток после удаления эксплантата через 7 дней после экс¬
плантации. Объектив 10х
чувствительностью клеток к токсическим эффектам и
дезагрегационной активностью трипсина.
Чаще всего для дезагрегации тканей используется
трипсин грубой очистки [Waymouth, 1974], так как он
довольно хорошо переносится многими клетками и
эффективен в отношении многих тканей. Остаточная
протеазная активность, остающаяся после промы¬
вания, нейтрализуется сывороткой культуральной
среды или, в случае использования бессывороточной
среды, ингибитором трипсина (например, ингибитор
трипсина из сои).
особенно для целых мышиных или куриных эмб¬
рионов. Метод плохо подходит для дезагрегации
тканей взрослых организмов, которые содержат
много фиброзной соединительной ткани, и некоторых
чувствительных типов клеток, таких как эпителий,
которые могут быть повреждены при механическом
перемешивании. Если после центрифугирования и
ресуспендирования будет наблюдаться реагрегация
клеток, их необходимо проинкубировать в течение
10-20 мин с ДНКазой (10-20 мкг/мл) и повторно
отцентрифугировать.
12.3.3. Теплый трипсин
Для сохранения максимальной выживаемости клеток
важно минимизировать время инкубации клеток в
активном трипсине. Следовательно, при трипсини-
зации ткани при 37 °С, диссоциированные клетки
необходимо собирать каждые полчаса, трипсин должен
быть удален центрифугированием и нейтрализован
сывороткой среды.
Этот метод полезен для дезагрегации больших
количеств ткани за относительно короткое время,
12.3.4. Трипсинизация с преинкубацией
на холоду
Одним из недостатков использования трипсина для
дезагрегации является повреждение тканей, которое
может происходить вследствие продолжительной
инкубации ткани при 37 °С. Поэтому после каждых
30 мин инкубации в теплом трипсине необходимо
отбирать диссоциировавшиеся клетки, хотя для пол¬
ной дезагрегации ткани требуется 3-4 ч. Более прос¬
тым методом, который позволяет минимизировать
222
ГЛАВА 12. ПЕРВИЧНАЯ КУЛЬТУРА
ПРОТОКОЛ 12.5. Дезагрегация ткани теплым
трипсином
Схема
Ткань разрезают на кусочки и перемешивают в трип¬
сине в течение нескольких часов. Диссоциированные
клетки собирают каждые полчаса, центрифугиру¬
ют и объединяют в среде, содержащей сыворотку
(рис. 12.7).
Материалы
• Стерильные или приготовленные в асептических
условиях:
• Ткань, 1-5 г
• DBSS, 50 мл (см. Приложение I)
• Трипсин (грубый), 2,5% в D-PBSA или физиоло¬
гическом растворе
• D-PBSA, 200 мл
• Ростовая среда с сывороткой (например, DMEM/
F12 с 10% фетальной бычьей сыворотки)
• Чашки Петри диаметром 9 см, не для культуры
ткани
• Предварительно взвешенные флаконы
• Центрифужные пробирки на 50 мл или универ¬
сальные контейнеры, 2
• Флакон для трипсинизации: флакон Эрленмейе-
ра (предпочтительно с насечками как на рис. 12.9)
или флакон с перемешиванием (см. рис. 13.5).
• Магнит для магнитной мешалки, автоклавиро-
ванный в пробирке
• Изогнутый пинцет
• Пипетки (пастеровские на 2 и 10 мл)
Нестерильные
• Магнитная мешалка
• Гемоцитометр или счетчик клеток
Протокол
1. Перенесите ткань в чашки Петри диаметром 9 см
со свежим стерильным DBSS и промойте.
2. Перенесите ткань во вторую чашку; удалите
нежелательные ткани, такие как жир или некро¬
тический материал и перенести в третью чашку.
3. Нарежьте скрещенными скальпелями (см.
рис. 12.7) на кусочки размером около 3 кубиче¬
ских миллиметров.
4. Перенесите ткань с помощью изогнутого пинцета
в предварительно взвешенный флакон или про¬
бирку.
5. Дайте кусочкам осесть.
6. Промойте путем ресуспендирования кусочки в
DBSS, дайте кусочкам осесть и удалите супер-
натант. Повторите эту операцию не менее двух
раз.
7. Слейте жидкость из флакона или пробирки и еще
раз взвесьте.
8. Перенесите все кусочки в пустой флакон для
трипсинизации, промытые DBSS флакон или
пробирку.
9. Удалите большую часть оставшейся жидкости и
добавьте 180 мл D-PBSA
10. Добавьте 20 мл 2,5% трипсина. (На этой стадии,
если требуется, можно добавить другие фермен¬
ты, например коллагеназу, гилуаронидазу или
ДНКазу.)
11. Поместите во флакон магнит для мешалки.
12. Закройте флакон и поставьте его на магнитную
мешалку в инкубатор или в термальную комнату
на 37 °С.
13. Перемешивайте на скорости 100об./мин при
37 °С.
14. После 30 мин инкубации соберите диссоцииро¬
ванные клетки следующим образом:
а) Дайте кусочкам осесть
б) Слейте супернатант в центрифужную пробир¬
ку и поместите его на лед.
в) Добавьте свежий трипсин к оставшимся во
флаконе кусочкам и продолжайте инкуба¬
цию с перемешиванием в течение следующих
30 мин.
г) Центрифугируйте собранные на этапе 14(6)
клетки приблизительно 5 мин при 500 g.
д) Ресуспендируйте конечный осадок в 10 мл
среды с сывороткой и храните суспензию на
льду.
15. Повторяйте этап 11 до тех пор, пока не будет до¬
стигнута полная дезагрегация ткани или пока де¬
загрегация не прекратится (обычно через 3-4 ч).
16. Соберите и объедините охлажденную клеточную
суспензию, посчитайте клетки с помощью гемо¬
цитометра или электронного счетчика клеток (см.
разд. 21.1) и проверьте жизнеспособность клеток
(см. разд. 22.3.1)
17. Так как клеточная популяция может быть очень
гетерогенной, калибровка электронного счетчи¬
ка клеток может быть затруднена и потребуется
подтверждение путем подсчета клеток с помощью
гемоцитометра.
18. Удалите все оставшиеся большие агрегаты с по¬
мощью фильтрования через стерильную марлю
или специальное сито (см., например, рис. 12.8).
19. Разбавьте суспензию клеток до плотности
1х106/мл ростовой средой и посейте их в такое
количество флаконов, какое потребуется, с плот¬
ностью приблизительно 2х105кл./см2. Если про¬
цент выживания клеток неизвестен или непред¬
сказуем, подсчет клеток даст низкие значения
(например, при биопсии опухолей, когда может
быть очень высока доля некротических клеток).
В этом случае установите диапазон концентраций
от 5 до 25 мг ткани на мл.
20. Меняйте среду через регулярные интервалы
времени (2-4 дня, руководствуясь признаками
понижения pH). Проверьте, нет ли в суперна-
танте живых клеток, так как некоторые клетки
могут медленно прикрепляться или даже могут
пролиферировать в суспензии.
12.3. Первичная культура
223
Сбор образцов
ткани в DBSS
Нарезание кусочков
размером 2-3 мм
скрещенными скальпелями
Промывание путем
ресуспендирования и
осаждения 2-3 раза
Перенос во флакон с
перемешиванием
Собрать супернатант,
содержащий клетки,
ресуспендировать в среде и
хранить на льду
Рис. 12.7. Дезагрегация с помощью теплого трипсина
повреждения клеток в течение инкубации, является
погружение ткани в трипсин, охлажденный до 4 °С,
и инкубация в течение 6-18 ч для более полного про¬
никновения фермента в ткань. В этих условиях про-
теолитическая активность трипсина низка (табл. 12.1).
После этой процедуры требуется только 20-30 мин
инкубировать ткань при 37 °С для дезагрегации
[Cole & Paul, 1966].
Метод холодной трипсинизации обычно дает более
высокий выход жизнеспособных клеток и повышает
их выживаемость после 24 ч культивирования (см.
рис. 12.1,12.10 и табл. 12.1). Также сохраняется больше
различных типов клеток, чем при теплой трипсиниза¬
ции (см. цв. вкладки (а, г, д) и 3). Культуры, полученные
из мышиных эмбрионов, содержат больше эпителиаль¬
ных клеток, будучи приготовлены с помощью холодной
трипсинизации. Культуры эритроидных клеток, полу¬
ченные из печени 13-дневных плодов мыши с помощью
холодной трипсинизации, отвечают на эритропоэтин,
чего не происходит с культурами, полученными с
помощью теплой трипсинизации, или механической
дезагрегации [Cole & Paul, 1966; Conkie, персональное
сообщение]. Метод холодной трипсинизации также
удобен тем, что не требуется ни перемешивания, ни
центрифугирования и инкубацию при 4 °С можно
проводить в течение ночи.
Каждые 30 мин
прекращать
перемешивание и дать
кусочкам ткани осесть
Перемешивание
нарезанных кусочков
теплом трипсине
Добавить трипсин
к оставшимся
кусочкам и
продолжать
перемешивание
224
ГЛАВА 12. ПЕРВИЧНАЯ КУЛЬТУРА
Рис. 12.8. Клеточное сито. Одноразовые полипропиленовые
фильтр и пробирка для отделения агрегатов из суспензии
клеток первичной культуры (BD Biosciences). Можно также
использовать для дезагрегации мягких тканей (см. также
рис. 12.13)
12.3.5. Рудиментарные органы куриного
эмбриона
Метод холодной трипсинизации особенно удобен для
малых количеств ткани, таких как эмбриональные орга¬
ны. Хорошо воспроизводимые результаты получаются
при использовании протокола 12.7 при получении
культур из 10-13-дневных куриных эмбрионов, предо¬
ставляя доказательства наличия нескольких различных
типов клеток, характеризующих ткани, из которых они
были получены. Этот протокол представляет собой
хорошее упражнение для обучения.
Анализ. После 3-5 дней инкубации в культурах
сердца может наблюдаться сокращение клеток; в куль¬
турах пигментированной сетчатки появляются колонии
пигментированных клеток; в культурах скелетных
мышц — начинается образование мышечных трубочек.
Культуру можно зафиксировать и покрасить (см. про¬
токол 16.2) для дальнейшего исследования.
12.3.6. Дезагрегация с использованием
других ферментов
Дезагрегация с помощью трипсина может быть трав¬
матичной для клеток (например, для некоторых эпи¬
..
■==>
Отобрать
образцы
ткани в DBSS
Нарезать кусочки Промыть путем
размером 2-3 мм ресуспендирования и
скрещенными скальпелями осаждения 2-3 раза
Удалить трипсин
и инкубировать
20-30 мин
\7
Заменить BSS
трипсином и поместить
на ночь на лед
Диссоциировать ткань
пипетированием в
среде
Посеять во флакон
или чашку
Подсчитать клетки
и определить
жизнеспособность
Рис. 12.9. Дезагрегация в холодном трипсине. См. также
цв. вкладки 2 (а, г, д) и 3
телиальных клеток) или малоэффективной (например,
для сильно волокнистых тканей, таких как фиброзная
соединительная ткань), в таких случаях необходимо
попытаться использовать другие ферменты. Так как
внеклеточный матрикс часто содержит коллаген,
особенно в соединительной ткани и мышцах, очевид¬
ным выбором является коллагеназа [Freshney, 1972
Таблица 12.1. Количество выделенных клеток после теплой и холодной трипсинизации
После трипсинизации
После 24 ч культивирования
Продолжительность и Выделенные
температурный режим клетки/ на
трипсинизации эмбрион, х107
% выживших кле¬
ток, определяемый Общее количество
по исключению жизнеспособных
красителя (трипа- клеток х107
нового синего)
Жизнеспособные, в
% от общего коли¬
чества посеянных
клеток
Выживаемость, в
% от количества
посеянных живых
клеток
4 °С
37 °С
0ч
4 ч
1,69
86
1,45
47,2
54,9
5,5 ч
0,5 ч
3,32
60
1,99
74,5
124
24 ч
0,5 ч
3,40
75
2,55
60,3
80,2
12,3. Первичная культура
225
ПРОТОКОЛ 12.6. Дезагрегация ткани
холодным трипсином
Схема
Отрезать кусочек ткани и поместить его в охлаж¬
денный до 4 °С трипсин на 6-18 ч. После инкубации
удалить трипсин и диссоциировать клетки в теплой
среде (рис. 12.9).
Материалы
Стерильные или приготовленные в
асептических условиях:
• Ткань, 1-5 г, предварительно взвешенная
• Ростовая среда (например, DMEM/F12 с 10%
FBS)
• DBSS
• 0,25% трипсин грубой очистки в бессывороточ-
ной среде RPMI 1640 или МЕМ на солях Stirrer
(S-MEM)
• Чашки Петри диаметром 9 см, не для культуры
ткани
• Прямой и изогнутый пинцеты
• Скальпели
• Флакон Эрленмейера на 25 или 50 мл, с завин¬
чивающейся крышкой, предварительно взвешен¬
ный (или стеклянный флакон, или универсаль¬
ный контейнер)
• Культуральные флаконы площадью 25 или 75 см2
• Пипетки (пастеровские на 2 и 10 мл)
Нестерильные
• Ледяная баня
Протокол
1. Перенесите ткань в чашку Петри диаметром 9 см
со свежим стерильным DBSS и промойте.
2. Перенеситеткань во вторую чашку; удалите не¬
желательные ткани, такие как жир или некроти¬
ческий материал.
3. Перенесите в третью чашку и разрежьте скрещен¬
ными скальпелями (смотри рис. 12.9) на кусочки
размером около 3 кубических миллиметров.
Органы эмбриона, если они не превышают этот
размер, лучше брать целиком.
4. Перенесите ткань с помощью изогнутого пинцета
в предварительно взвешенный стерильный фла¬
кон объемом 15 или 50 мл.
5. Дайте кусочкам осесть.
6. Промойте путем ресуспендирования кусочков в
DBSS, дайте кусочкам осесть и удалите надоса-
дочную жидкость. Повторите эту операцию не
менее двух раз.
7. Тщательно удалите оставшуюся жидкость и еще
раз взвесьте флакон.
8. Добавьте охлажденный до 4 °С трипсин в RPMI
1640 или S-MEM (10 мл/г ткани).
9. Инкубируйте смесь 6-18 ч при 4 °С.
10. Тщательно удалите трипсин, оставив ткань толь¬
ко в следовых количествах трипсина. (На этой
стадии, если требуется, можно добавить другие
ферменты, например коллагеназу, гилуаронидазу
или ДНКазу в количестве 1-2 мл.)
11. Поместите пробирку на 37 °С на 20-30 мин.
12. Добавьте приблизительно 1 мл на каждые 100 мг
исходной ткани теплой среды и осторожно пипе-
тируйте смесь, набирая в пипетку и выливая, пока
ткань полностью не диссоциирует.
13. Если ткань не диспергирует полностью, клеточ¬
ную суспензию можно профильтровать через
стерильную марлю или сито из нержавеющей
ткани (100-200 мкм) или через одноразовый
пластиковый сетчатый фильтр (рис. 12.10), или
крупным частицам можно просто дать осесть.
Если ткани много, увеличение объема среды
для суспендирования до 20 мл на каждый грамм
ткани облегчит осаждение и последующий сбор
надосадочной жидкости. Достаточно двух-трех
минут для того, чтобы избавиться от большей
части больших кусочков.
14. С помощью гемоцитометра или электронного
счетчика клеток (см. разд. 21.1) определите
концентрацию клеток и их жизнеспособность
(смотри протокол 22.1).
15. Так как клеточная популяция может быть в
значительной степени гетерогенной, калибров¬
ка электронного счетчика клеток может быть
затруднена и потребуется подтверждение путем
подсчета клеток с помощью гемоцитометра.
16. Разбавьте суспензию клеток до плотности
1х106/мл ростовой средой и посейте их в такое
количество флаконов, какое потребуется, с плот¬
ностью приблизительно 2х105кл./см2. Если про¬
цент выживания клеток неизвестен или непред¬
сказуем, подсчет клеток даст низкие значения
(например, при биопсии опухолей, когда может
быть очень высока доля некротических клеток).
В этом случае установите диапазон концентраций
от 5 до 25 мг ткани на мл.
17 .Меняйте среду через регулярные интервалы
времени (2-4 дня, руководствуясь снижением
pH). Проверьте, нет ли в супернатанте живых
клеток, так как некоторые клетки могут медленно
прикрепляться или даже могут пролиферировать
в суспензии.
(карцинома кишечника); Speirs et al., 1996 (карцинома
молочной железы); Chen et al., 1989 (почка); Booth &
O’Shea, 2002 (кишечник); Heald et al., 1991 (клетки
островков поджелудочной железы)] (см. разд. 12.3.6,
23.2.6-23.2.8). Также, с различной степенью успеха,
используются другие бактериальные протеазы, такие
как проназа [Schaffer et al., 1997; Glavin et al., 1996] и
диспаза (Boehringer-Mannheim) [Compton et al., 1998;
Inamatsu et al., 1998]. Так как в процессах межкле¬
точной адгезии участвуют углеводы, то в сочетании
с коллагеназой используют гиалуронидазу [Berry &
226
ГЛАВА 12. ПЕРВИЧНАЯ КУЛЬТУРА
1 1 г
Преинкубация, ч при 4 °С 0 5.5 24
Инкубация, ч при 37 °С 4 0.5 0.5
Рис. 12.10. Теплая и холодная трипсинизация. Выход
жизнеспособных клеток на эмбрион при теплой и холодной
трипсинизации. Выход жизнеспособных клеток на эмбрион
повышается при холодной трипсинизации до 24 ч при 4 °С, но
восстановление роста после 24 ч культивирования выше при
менее продолжительной холодной трипсинизации (>100%,
вследствие пролиферации клеток) возможно из-за того, что
некоторые клетки, полученные с помощью холодной трип¬
синизации, не пролиферируют
Friend, 1969] и нейраминидазу. На рынке продолжают
появляться и другие протеазы (см. разд. 12.1). Так что
если трипсин, коллагеназа, диспаза, проназа, гиалуро-
нидаза и ДНКаза по отдельности или в комбинации не
эффективны, теперь есть выбор других имеющихся в
продаже протеаз.
12.3.7. Коллагеназа
Эта методика очень проста и эффективна для многих
тканей: эмбриональных, взрослых, нормальных и
опухолевых. Она крайне полезна в том случае, когда
ткань слишком волокнистая или слишком чувстви¬
тельная; в этих случаях использование трипсина ма¬
лоэффективно. Чаще используется коллагеназа грубой
очистки, возможно, частично ее действие обусловлено
контаминацией препарата другими неспецифическими
протеазами. Если неспецифическая протеолитическая
активность нежелательна, в продаже имеется коллаге¬
наза более высоких степеней очистки, но эти препара¬
ты могут быть не столь эффективны, как коллагеназа
грубой очистки.
Некоторые клетки, особенно макрофаги, могут при¬
крепляться в первом флаконе в процессе инкубации с
коллагеназой. При переносе клеток в новый флакон
после обработки коллагеназой (и последующим удале¬
нием коллагеназы) значительное количество макрофа-
ПРОТОКОЛ 12.7. Рудиментарные органы
куриного эмбриона
Схема
Извлечь отдельные органы и ткани и поместить их
(желательно целиком) в холодный трипсин на ночь.
Удалить трипсин, кратковременно инкубировать ор¬
ганы или ткань и диспергировать их в культуральной
среде. Развести и посеять в культуру.
Материалы
Стерильные:
• DBSS
• 0,25% трипсин грубой очистки (Difco 1:250 или
аналогичный) в среде RPMI 1640 или S-MEM
на льду; при использовании трипсина более
высокой степени очистки можно использовать
более низкие концентрации, например 0,05-0,1%
кристаллического трипсина производства Sigma
или Worthington Grade IV
• Культуральная среда (например, DMEM/F12 с
10% FBS), минимальный объем — 12 мл на одну
ткань
• Чашки Петри диаметром 9 см, не для культуры
ткани
• Культуральные флаконы площадью 25 см2 (2 на
1 ткань)
• Скальпели (лезвие № 11 для общих целей)
• Хирургический нож для иридэктомии для тонко¬
го препарирования
• Изогнутый и прямой тонкие пинцеты
• Пипетки (пастеровские на 2 и 10 мл)
• Пробирки, желательно стеклянные, на 10-15 мл
с закручивающимися крышками
Нестерильные:
• Куриные яйца с 10-13-дневными зародышами
(>10 дней — в Великобритании требуется ли¬
цензия)
• Ледяная баня
• Бинокулярная препаровальная лупа.
Протокол
1. Извлеките эмбрион из яйца, как описано ранее
(см. протокол 12.2) и поместите его в стерильный
DBSS.
2. Извлеките голову (рис. 12.11 а, б).
3. Извлеките глаз и аккуратно откройте его, удаляя
хрусталик, внутриглазную жидкость и стекловид¬
ное тело (рис, 12.11в, г).
4. Захватите сетчатку двумя парами тонких пин¬
цетов и осторожно отделите пигментированную
сетчатку от нейрональной сетчатки и соедини¬
тельной ткани (рис. 12.1 Id). (Для препарирова¬
ния 10-дневного эмбриона необходима препа¬
ровальная лупа. Кратковременная инкубация в
0,25% трипсине в 1 мМ ЭДТА упростит разделе¬
ние двух тканей.) Положите ткани с одного края
чашки.
12.3. Первичная культура
227
5. Проколите верхнюю часть головы кривым пин-
цетом и извлеките мозг (рис. 12. Не). Поместите
мозг рядом с сетчаткой на край чашки.
6. Разделите туловище поперечно там, где розо¬
вая печень просвечивает через кожу живота
(рис. 12.11 ж). Если сделать разрез по линии диа¬
фрагмы, то он пройдет между сердцем и печенью;
но иногда печень может находиться не в нижней
части, а в верхней части.
7. Аккуратно исследуйте изнутри разрезанную верх¬
нюю часть туловища и извлеките сердце и легкие
(рис. 12.11з; выделите органы, но не разрезайте их
до идентификации). Отделите сердце от легких и
поместите на край чашки.
8. Прозондируйте нижнюю половину и извлеките
печень со скрученным кишечником, находящимся
между долями печени (рис. 12.11м). Огдеяитепечень
от кишечника и поместите каждый на край чашки.
9. Отогните стенку тела в обратную сторону, чтобы
раскрыть внутреннюю часть задней поверхности
брюшной полости в нижней половине тела. Про¬
долговатые, состоящие из долек, почки должны
быть видны параллельно обеим сторонам средин¬
ной линии.
10. Осторожно подведите кончик скальпеля под
каждую почку и извлеките почки из брюшной
полости (рис. 12.11 и). (Для препарирования
10-дневного эмбриона потребуется препароваль¬
ная лупа.) Аккуратно вырежьте почки и помести¬
те их с одной стороны.
И. Расположите концы скальпелей вместе на сре¬
динной линии нижней части и, продвигая концы
вперед, друг за другом, выдавите спинной мозг так
же, как вы выдавливаете зубную пасту из тюбика
(рис. 12.11л). (Этот этап может быть трудновы¬
полним при работе с 10-дневными эмбрионами.)
12. Поверните вверх заднюю часть туловища эмб¬
риона и снимите кожу со спины и верхней части
ног (рис. 12.11м). Соберите и поместите эту кожу
с одной стороны.
13. Извлеките мышцу из каждого бедра и соберите
эти мышцы вместе (рис. 12.11«).
14. Перенесите все эти ткани (и любые другие, кото¬
рые вы может быть захотите) в отдельные пробир¬
ки с 0,25%-м трипсином и поместите пробирки на
лед. Убедитесь, что препараты тканей полностью
погружены в трипсин.
15. Оставьте пробирки при 4 °С на 6-18 ч.
16. Осторожно удалитетрипсинизпробирок,незатра-
гивая ткань; наклоняя и поворачивая пробирку
17. Инкубируйте ткань в следах трипсина 15-20 мин
при 37 °С.
18. Добавьте по 4 мл среды в каждый из флаконов
площадью 25 см2 (2 на каждую ткань).
19. Добавьте по 2 мл среды в пробирки с тканями и
остатками трипсина и пипетируйте для диссо¬
циации ткани, осторожно набирая в пипетку и
выливая.
20. Дайте осесть крупным кусочкам ткани.
21. Перенесите надосадочяую жидкость с помощью
пипетки в первый флакон^педомешайте и пере¬
несите 1 мл разбавленной суспензии во второй
флакон. В результате этой процедуры получится
2 флакона с различной концентрацией клеток, и
это избавляет от необходимости считать клетки.
Оптимальная концентрация клеток для исполь¬
зования в последующих экспериментах опреде¬
ляется опытным путем.
22. Смените среду, когда потребуется (например,
для мозга может быть необходимо заменить
среду через 24 ч, а для пигментированной сет¬
чатки — возможно через 5-7 дней) и проверьте
специфическую морфологию ткани и функции.
гов выводится из культуры. Если потребуется, клетки в
первом флаконе можно тоже культивировать. Мягкая
трипсинизация удалит любые прикрепляющиеся клет¬
ки, кроме макрофагов.
Метод дезагрегации тканей с помощью коллагеназы
подтвердил свою чрезвычайную полезность для полу¬
чения культур опухолей человека [Pfragner & Freshney,
2004], почки мыши, взрослого и эмбрионального мозга
человека, печени (см. протокол 23.6) легкого и многих
других тканей, в особенности эпителия [Freshney &
Freshney, 2002]. Процедура мягкая и не требует меха¬
нического перемешивания или специального оборудо¬
вания. Однако если количество ткани превышает 1 г,
процедура становится трудоемкой на стадии препа¬
рирования, а также удорожается из-за значительного
необходимого количества коллагеназы. Кроме того,
при использовании коллагеназы, из соединительной
ткани высвобождается большое количество фиброб¬
ластов, поэтому может возникнуть необходимость
в последующей селекции культуры или разделении
клеток (см. гл. 15).
Отдельные кластеры эпителиальных клеток, полу¬
ченные после дезагрегации ткани коллагеназой (см.
этап 9 протокола 12.8) и холодной трипсинизации
(см. цв. вкладки 2,3), могут быть отобраны с помощью
препаровальной лупы и перенесены в отдельные лунки
планшета для микротитрования как необработанного,
так и с облученными или обработанными митоми-
цином С клетками фидерного слоя (см. разд. 14.2.3,
23.1.1,23.1.4).
12.3.8. Механическая дезагрегация
Выход клеток из первичного эксплантата относи¬
тельно медленный процесс, который может быть в
значительной степени выборочным. Ферментативное
расщепление требует больше усилий, хотя потенциаль¬
но дает культуру, которая лучше представляет ткань.
Так как при этом имеется риск протеолитического
повреждения клеток во время обработки фермента-
228
ГЛАВА 12. ПЕРВИЧНАЯ КУЛЬТУРА
Рис. 12.11. Препарирование куриного эмбриона, (я, б) Извлечение головы, (в) Извлечение глаза, (г) Удаление хрусталика.
(Э) Отслаивание сетчатки. (е) Извлечение мозга, (ж) Разделение туловища, (з) Выделение сердца и легких из верхней половины.
(и) Выделение печени и кишечника из нижней половины, (к) Введение кончика скальпеля между левой почкой и дорзальной
стенкой тела, (л) Выдавливание спинного мозга. (м) Снятие кожи со спины и задней лапы. (н) Отделение мышцы от бедренной
кости, (о) Рудиментарные органы размещены по переферии чашки. Справа налево по часовой стрелке: мозг, сердце, легкие,
печень, мускульный желудок, почки, позвоночник, кожа и мышца
12,3. Первичная культура
229
Рис. 12.11. Препарирование куриного эмбриона
ми, многие люди выбирают альтернативный метод
механической дезагрегации. Например, сбор клеток,
которые высвободились из тщательно измельченной
ткани [Lasfargues, 1973], продавливание препарируе¬
мой ткани через серию клеточных сит с постепенным
уменьшением размера ячеек или, в другом вариан¬
те, пропускание фрагментов ткани через шприц (с
или без иглы с широким отверстием) [Zaroff et al.,
1961], или просто многократное пипетирование (см.
рис. 12.13). В результате этих процедур клеточная
суспензия получается быстрее, чем при фермента¬
тивном расщеплении, но при этом могут возникать
механические повреждения. Соскребание («стряхи¬
вание») (рис. 12.13а) и просеивание (рис. 12.136),
по-видимому, наиболее мягкий механический метод,
в то время как пипетирование (рис. 12.13г) и особенно
шприцевание (рис. 12.13в), по всей вероятности, при¬
водят к повреждениям. В протоколе 12.9 описан один
из методов механической дезагрегации, который, как
было показано, с большим успехом используется для
мягких тканей, таких как мозг.
Эта методика применима только для мягких тканей,
таких как селезенка, эмбриональная печень, взрос¬
лый мозг и некоторые мягкие опухоли человека и
животных. Даже для тканей мозга, для которых легко
достигается полная дезагрегация, выживаемость полу¬
ченных при механической дезагрегации клеток ниже,
чем при ферментативной дезагрегации, хотя затраты
времени значительно меньше. Когда количество ткани
достаточно велико, а эффективность выхода не имеет
значения, с помощью механической дезагрегации
можно выделить за короткое время столько же клеток,
230
ГЛАВА 12. ПЕРВИЧНАЯ КУЛЬТУРА
ПРОТОКОЛ 12.8. Дезагрегация ткани
с помощью коллагеназы
Схема
Поместить мелко нарезанную ткань в полную среду,
содержащую коллагеназу и инкубировать. После де¬
загрегации ткани удалить коллагеназу центрифуги¬
рованием, посеять клетки в высокой концентрации
и культивировать (рис. 12.12).
Материалы
Стерильные:
• Коллагеназа (2000 единиц/мл), Worthington
CLS или Sigma 1А
• Культуральная среда (например, DMEM/F12 с
10% FBS)
• DBSS
• Пипетки на 1 и 10 мл
• Чашки Петри диаметром 9 см, не для культуры
ткани
• Культуральные флаконы площадью 25 см2
• Центрифужные пробирки или универсальные
контейнеры объемом 15-50 мл, в зависимости от
количества ткани
• Скальпели
Нестерильные:
• Центрифуга
Протокол
1. Перенесите ткань в свежий стерильный DBSS и
промойте.
2. Перенесите ткань во вторую чашку, удалите
нежелательные ткани, такие как жир или некро¬
тический материал.
3. Перенесите в третью чашку и мелко нарежьте
скрещенными скальпелями (см. рис. 12.12) на
кусочки размером около 1 кубического милли¬
метра.
4. Перенесите ткань с помощью пипётки (10-20 мл
с широким концом) в стерильную центрифужную
пробирку объемом 15 или 50 мл или в универ¬
сальный контейнер. (Вначале смочить внутрен¬
нюю поверхность пипетки DBSS, иначе кусочки
будут прилипать.)
5. Дайте кусочкам осесть.
6. Промойте путем ресуспендирования кусочков в
DBSS, дайте кусочкам осесть и удалите надоса-
дочную жидкость. Повторите эту операцию не
менее двух раз.
7. Перенесите 20-30 кусочков в один флакон
площадью 25 см2 и 100-200 кусочков во второй
флакон.
8. Слейте DBSS и добавьте 4,5 мл ростовой среды с
сывороткой в каждый флакон.
9. Добавьте 0,5 мл коллагеназы грубой очистки
(2000 ед./мл), чтобы получить конечную концен¬
трацию коллагеназы 200 ед./мл.
10. Инкубируйте 4-48 ч при 37 °С без перемеши¬
вания. Если дезагрегация проходит медленно,
опухолевые ткани (например, фиброзные карци¬
номы молочной железы или кишечника) можно
оставить в растворе до 5 дней, хотя, если будет
наблюдаться значительное снижение pH (то есть
<рН 6,5), может понадобиться центрифугировать
ткань и ресуспендировать ее в свежей среде с
коллагеназой до окончания инкубации.
И. Проверьте эффективность дезагрегации путем
осторожного покачивания флакона, кусочки
ткани будут «размазываться^ по дну флакона
и, при осторожном пипетировании, распадать¬
ся на отдельные клетки и небольшие кластеры
(рис. 12.13).
12. В некоторых тканях (например, легком, почке и
карциномах молочной железы или кишечника)
небольшие кластеры эпителиальных клеток
могут быть устойчивы к коллагеназе. Их можно
отделить от остальных клеток, дав им осесть в
течение приблизительно 2 мин. Если потом эти
кластеры промыть DBSS путем ресуспендиро¬
вания и осаждения, ресуспендировать осадок в
среде и посеять, они могут дать начало островкам
эпителиальных клеток. Как правило, эпителиаль¬
ные клетки выживают лучше, если их диссоции¬
ровать не полностью.
13. Когда будет достигнута полная дезагрегация или
когда будут собраны клетки в надосадочной жид¬
кости (после удаления кластеров осаждением),
центрифугируйте суспензию клеток из дезагре¬
гированной ткани и всех промывочных растворов
3 мин при 50-100 g.
14. Удалите супернатант DBSS или среды, ресус-
пендируйте и объедините осадки в 5 мл среды и
посейте во флакон площадью 25 см2. Если после
обработки коллагеназой pH упадет (до pH 6,5 или
ниже в течение 48 ч), после удаления коллагеназы
разбавьте суспензию клеток в 2-3 раза средой.
15. Через 48 ч смените среду.
сколько и при ферментативном расщеплении, но для
этого понадобится гораздо больше ткани.
12.3.9. Разделение жизнеспособных
и нежизнеспособных клеток
Когда прикрепленную первичную культуру получают из
суспензии диссоциированных клеток, нежизнеспособ¬
ные клетки удаляются при первой смене среды. В случае
если клетки первичной культуры растут в суспензии,
с началом пролиферации нежизнеспособные клетки
постепенно разбавляются. Однако, если необходимо,
нежизнеспособные клетки можно удалить из суспензии
путем центрифугирования клеток на смеси фиколла
и метризоата натрия (например, Hypaque или Triosil)
12.3. Первичная культура
231
Поместить образец
ткани в DBSS
Скрещенными скальпелями нарезать
кусочки размером 2-3 мм
Промыть путем ресуспендирования
и осаждения 2-3 раза
1 Инкубировать мелко нарезан¬
ные кусочки с коллагеназой в
полной среде
Ресуспендировать клетки е
ростовой среде и посеять
во флакон
Ресуспендировать клетки е
ростовой среде и посеять
во флакон
■
щ
Собрать на-
досадочную
жидкость с
клетками и
центрифуги¬
ровать
Промыть
осадок и
дать ему
осесть
Диспергировать^
путем осторожного
пипетирования
Перенести
пробирку и
дать осесть
Рис. 12.12. Дезагрегация ткани с помощью коллагеназы. (а) Схема препарирования ткани с последующей дезагрегацией
коллагеназой. (б) Кластеры клеток, полученные из карциномы кишечника человека через 48 ч после диссоциации клеток в
коллагеназе грубой очистки (Worthington CLS grade); до удаления коллагеназы. (в) То же, что и (б), но после удаления кол¬
лагеназы, дезагрегации пипетированием и культивирования в течение 48 ч. Ясно видны круглые кластеры (указаны черными
стрелками) в (б), из которых формируются эпителиоподобные слои (указаны белыми стрелками) на фотографии (в), некото¬
рые сохраняют трехмерную структуру, некоторые распластываются, а из неравномерно оформленных кластеров образуются
фибробласты (см. также цв. вкладку 26)
[Vries et al., 1973]. Эта методика сходна с методикой
выделения лимфоцитов из периферической крови (см.
протокол 27.1). Жизнеспособные клетки собирают на
границе между средой и фиколл/метризоатом, а мерт¬
вые клетки образуют осадок на дне пробирки.
Эта методику можно модифицировать, понижая
или повышая соотношение объема фиколла к объему
суспензии клеток (например, 5 мл клеточной суспен¬
зии наслаивать на 1 мл фиколла).
12.3.10. Первичная культура,
краткие выводы
Дезагрегация ткани и получение первичной культуры
составляют первую и, возможно, наиболее существен¬
ную стадию в культивировании клеток со специфиче¬
скими функциями. Если необходимые клетки на этой
стадии потеряны, эта потеря невосполнима. Многие
клетки различных типов можно культивировать, вы¬
брав правильные методики (см. разд. 10.2.1 и гл. 23).
В общем случае трипсин является более жестким
ферментом, по сравнению с коллагеназой, но иногда он
более эффективен для получения суспензии одиночных
клеток. Использование коллагеназы не дает быстрой
диссоциации эпителиальных клеток, но это свойство
является преимуществом при отделении эпителиаль¬
ных клеток от стромальных. Механическая дезагрега¬
ция намного быстрее, чем процедура с использованием
коллагеназы, но при этом повреждается больше клеток.
Лучшим подходом будет опробовать методики, описан¬
ные в протоколах 12.4-12.9, и выбрать метод, который
лучше всего себя зарекомендует в ваших условиях.
Если не подойдет ни одна из этих методик, попробуйте
использовать дополнительные ферменты, такие как
проназа, диспаза, Аккутаза и ДНКаза, и поищите в ли¬
тературе примеры более ранних работ с теми тканями,
которые вас интересуют.
12.3.11. Первичная документация
Вне зависимости от методики, используемой для
получения культуры, важно вести точные записи о
происхождении культуры и ее источнике, включая
вид, пол и ткань, из которой она была получена, любые
значимые патологии и процедуры, использованные
для дезагрегации и получения первичной культуры
(табл. 12.2). Если в работе вы применяете нормы
правильной лабораторной практики (good labora-
232
ГЛАВА 12. ПЕРВИЧНАЯ КУЛЬТУРА
Таблица 12.2. Регистрация данных о первичной культуре
Дата Время Оператор
Регистрационные записи
Происхождение ткани
Вид
Порода или линия
Возраст
Пол
Порядковый номер или номер метки животного
Ткань
Участок
Хранение ткани/местонахождение ДНК
Патология
Агент, используемый для дезагре¬
гации
Трипсин, коллагеназа и т.д.
Концентрация
Продолжительность обработки
Растворитель
Подсчет клеток
Концентрация после ресуспендирования (Cl)
Объем (VI)
Выход (Y= Cl Ч VI)
Выход на грамм (сырой вес ткани)
Посев
Количество (N) и тип сосуда (флакон, чашка или
планшет)
Окончательная концентрация (CF)
Объем во флаконе, чашке или лунке планшета (VF)
Среда
Вид
Номер партии
Вид сыворотки и концентрация
Номер партии.
Другие добавки
Концентрация С02
Покрытие матриксом
Например, фибронектин, Matrigel, коллаген
Пассирование
Восстановление к первому пересеву, клеток/флакон
% (восстановившиеся клетки ч посеянные клетки)
Маркировка линии клеток
12,3. Первичная культура
233
ПРОТОКОЛ 12.9. Механическая
дезагрегация путем просеивания
Схема
Ткань в культуральной среде протирается через се¬
рию сит с постепенным уменьшением размера ячеек,
пока не будет получена приемлемая суспензия от¬
дельных клеток и малых агрегатов. Далее суспензию
разбавляют и сразу культивируют.
Материалы
Стерильные:
• Ростовая среда, например DMEM/F12 с 10%
FBS
• Пинцет
• Сито (рис. 12.136) или серия сит с постепен¬
ным уменьшением размера ячеек от 100 мкм
до 20 мкм, или «клеточное сито» (Falcon) (см.
рис. 12.8)
• Чашки Петри диаметром 9 см
• Скальпели
• Одноразовые пластиковые шприцы (на 2 мл или
на 5 мл)
• Культуральные флаконы
Протокол
1. После промывания и предварительного препа¬
рирования ткани (см. этапы 1 и 2 протокола 2.5)
нарежьте ткань на кусочки размером 3-5 мм в
поперечнике и поместите несколько кусочков
одновременно в сито из нержавеющей стали или
полипропилена с размером ячеек 1 мм. Сито
поместите в чашку Петри диаметром 9 см (см.
рис. 12.136) или в центрифужную пробирку (см.
рис. 12.8).
2. Протрите ткань через сито в среде, мягко надав¬
ливая поршнем от одноразового пластикового
шприца. Промойте средой из пипетки сито, чтобы
смыть клетки.
3. С помощью пипетки перенесите частично де¬
загрегированную ткань из чашки Петри в сито с
мелкими порами, возможно размером 100 мкм, и
повторите этап 2.
4. На этой стадии суспензию можно разбавить и
культивировать или ее далее можно пропустить
через сито с размером пор 20 мкм, если важно
получить суспензию одиночных клеток. Обыч¬
но при получении сильно диссоциированной
клеточной суспензии клетки подвергаются ме¬
ханическому стрессу и снижается их конечная
выживаемость.
5. Посейте клетки в культуральные флаконы с
плотностью 2х105, 1х106и 2х106кл./мл, разводя
суспензию клеток средой.
г) Ресуспендирование с помощью пипетки
Рис. 12.13. Механическая дезагрегация. (a) Соскребание
или «стряхивание». Разрезание или механическое трение по¬
верхности разреза, в результате чего высвобождаются клетки.
(6) Просеивание. Протирание ткани через сито поршнем от
шприца. (Можно использовать клеточное сито производства
Falcon; см. рис. 12.8.) (в) Шприцевание. Пропускание ткани
через шприц с полой иглой или канюлей, (г) Ресуспендиро¬
вание с помощью пипетки. Пипетирование фрагментов ткани
вперед-назад через пипетку с широким диаметром
а) Соскребание или «стряхивание»
б) Просеивание
в) Шприцевание
234
ГЛАВА 12. ПЕРВИЧНАЯ КУЛЬТУРА
ПРОТОКОЛ 12.10. Обогащение
жизнеспособных клеток
Схема
До 2х107 клеток в 9 мл среды можно наслоить на 6 мл Fi-
coll-Hypaque в центрифужном стакане на 25 мл с завин¬
чивающейся крышкой. Затем смесь центрифугируют и
из интерфазы собирают жизнеспособные клетки.
Материалы
Стерильные:
• Суспензия клеток с минимально возможным
количеством агрегатов
• Прозрачные центрифужные пробирки или уни¬
версальные контейнеры
• D-PBSA
• Ficoll Hypaque или аналогичный реактив (см.
Приложение II) (фиколл/метризоат, доведенный
до плотности 1,077 г/см3)
• Ростовая среда
• Шприц с канюлей с тупым наконечником или с
иглой с тупым концом, пастеровские пипетки или
автоматическая пипетка
Нестерильные
• Гемоцитометр или счетчик клеток
• Центрифуга
Протокол
1. Дайте осесть крупным агрегатам клеточной сус¬
пензии.
2. Наслоите 9 мл клеточной суспензии на 6 мл смеси
Ficoll-paque. Этот этап необходимо проводить,
используя широкую прозрачную центрифужную
пробирку с крышкой, такую как универсальные
контейнеры Sterilin или Nunclon или прозрачные
пластиковые пробирки на 50 мл Coming, удвоив
объемы, упомянутые выше.
3. Центрифугируйте смесь 15 мин при 400 g
4. Осторожно удалите верхний слой, не повредив
границу фаз.
5. Осторожно соберите границу фаз с помощью
шприца, пастеровской пипетки или автоматиче¬
ской пипетки.
• Требование безопасности. Если вы рабо¬
таете с материалом человека или других приматов,
не используйте острую иглу или стеклянную пасте¬
ровскую пипетку.
6. Разведите смесь средой (например, DMEM/
F12/10FB) до 20 мл.
7. Центрифугируйте смесь при 70 g 10 мин.
8. Удалите супернатант и ресуспендируйте осадок
в 5 мл ростовой среды.
9. Повторите этапы 7 и 8 для того, чтобы отмыть
клетки от фиколла.
10. Посчитайте клетки с помощью гемоцитометра
или электронного счетчика клеток.
11. Посейте в культуральный флакон(ы).
tory practice, GLP; www.oecd.org/department/0,2688,
en_2649_34381_l_l_l_l_l,00.html), то такие записи
не только желательно, но и обязательны [Food and
Drug Administration, 1992; Department of Health and
Social Security, 1986; Organisation for EconomicCo-op-
eration and Development, 2004]. Записи можно вести в
тетради или на других бланках для записи, но лучше
всего на этой стадии создать запись в компьютерной
базе данных; эта запись затем станет первым шагом в
сохранении происхождения клеточной линии. Запись в
базе данных может не пойти дальше стадии первичной
культуры, но, с другой стороны, она может быть первым
шагом в создании точных записей, характеризующих
ценную клеточную линию. Если запись станет беспо¬
лезной, ее всегда можно удалить, но если клеточная
линия приобретет значение, запись невозможно будет
создать позже с той же точностью.
Так как на более позднем этапе может возникнуть
необходимость подтвердить происхождение клеточ¬
ной линии, особенно если возникнут подозрения о
перекрестной контаминации, важно сохранить образец
ткани, крови или ДНК той же особи в момент изоляции
ткани для первичной культуры. Этот образец может в
дальнейшем использоваться как стандартный материал
для ДНК-фингерпринтинга (см. разд. 16.6.2) или для
других видов анализа ДНК (см. разд. 16.6.3).
Глава 13
СУБКУЛЬТУРА И КЛЕТОЧНЫЕ ЛИНИИ
13.1. СУБКУЛЬТИВИРОВАНИЕ
Первое субкультивирование представляет собой важ¬
ный переходный период для культуры. Необходимость
субкультивирования подразумевает, что первичная
культура разрослась до таких пределов, что заняла
весь доступный субстрат. Хотя первичная культура
может иметь различные фракции (см. разд. 21.11.1), в
зависимости от типа клеток, представленных в культу¬
ре, после первого субкультивирования фракция роста
становится достаточно высокой (80% и более). Из
очень гетерогенной первичной культуры, содержащей
много клеточных типов, представленных в исходной
ткани, возникают более гомогенные клеточные ли¬
нии. В дополнение к биологическому значению, этот
процесс имеет высокую практическую значимость,
поскольку теперь культура может поддерживаться,
размножаться, храниться, может быть охарактеризо¬
вана, и возможности культуры возрастают, включая
увеличение количества клеток и их единообразие, что
открывает широкие возможности для эксперименталь¬
ных исследований (см. табл. 1.5).
13.2. ТЕРМИНОЛОГИЯ
Как только первичная культура прошла субкульти¬
вирование (или пассаж), она именуется клеточной
линией. Этот термин подразумевает присутствие
нескольких клеточных линий как сходных, так и от¬
даленных фенотипов. Если выбрана одна клеточная
линия — методом клонирования (см. протокол 14.1),
методом физического разделения клеток (см. гл. 15)
или каким-либо иным методом селекции клеток, при
наличии некоторых специфических свойств, которые
идентифицируются у совокупности клеток в культу¬
ре, клеточная линия начинает называться клеточный
штамм (см. Приложение IV). Некоторые наиболее
используемые клеточные линии и клеточные штаммы
приведены в списке в табл. 13.1 (см. также табл. 3.1).
Если клеточная линия трансформирована in vitro, она
дает начало постоянной клеточной линии (см. разд. 3.8,
18.4), а если она клонирована и охарактеризована, то
она известна как постоянный клеточный штамм. На
этой стадии чрезвычайно важно подтвердить идентич¬
ность клеточных линий и не допускать возможности
перекрестного заражения; многие клеточные линии,
обычно используемые, на самом деле не являются тем,
чем они заявлены, поскольку перекрестно контамини-
рованы клетками HeLa или иными энергично расту¬
щими клеточными линиями (табл. 13.2). Тем не менее
постоянные клеточные линии имеют ряд преимуществ;
преимущества и недостатки конечных и постоянных
клеточных линий приведены в табл. 13.3.
Первое субкультивирование дает начало вторич¬
ной культуре, повторное — третичной и т.д., хотя на
практике эта номенклатура редко используется далее,
чем третичная культура. В работах Хейфлика [Нау-
flick & Moorhead, 1961] каждая субкультура делит
культуру пополам (т.е. индекс разведения 1:2), по¬
этому номер пассажа совпадает с номером генерации.
Тем не менее это не одно и то же. Номер пассажа это
количество раз, которое культура была пересеяна, в
то время как номер генерации — это количество удво¬
ения клеток, которому подверглась культура, при том
что количество периодов удвоения устанавливается
весьма приблизительно. Когда индекс разведения
составляет 1:2, как в экспериментах Хейфлика, номер
пассажа примерно равен номеру генерации. Тем не
менее если субкультивирование проводится при ин¬
дексе разведения выше, чем 1:2, то номер генерации,
который представляет весьма важную характеристику
возраста культуры, будет возрастать быстрее, чем
номер пассажа, основанный на количестве периодов
удвоения клеток, так что клеточная популяция, кото¬
рая произошла от предыдущего субкультивирования
(см. разд. 13.7.2). Ни одно из этих приближений не
учитывает потери клеток в результате некроза, апоп-
тоза или дифференцировки или старения культуры
с выходом из цикла деления и роста. Однако все эти
явления могут иметь место при каждом цикле роста
между субкультивированием.
13.3. ВОЗРАСТ КУЛЬТУРЫ
Клеточные линии с ограниченным временем жизни
называются конечными линиями и имеют опреде¬
ленный тип репродуктивности (см. разд. 3.8.1). Они
растут и проходят определенное конечное число де¬
лений, с формированием конечного числа клеточных
генераций, обычно от 20 до 80 периодов удвоения
клеточной популяции до начала угасания культуры.
Реальное количество удвоений зависит от вида и раз-
236
ГЛАВА 13. СУБКУЛЬТУРА И КЛЕТОЧНЫЕ ЛИНИИ
Таблица 13.1. Обычно используемые клеточные линии
Клеточная
линия
Морфология
Происхож¬
дение
Вид
Возраст
Плоидность
Характеристики
Ссылки
Конечные, из нормальных тканей
IMR-90
Фибробласты
легкое
Человек
Эмбриональный
Диплоидный
Чувствительны к
вирусным инфек¬
циям человека;
контактное ингиби¬
рование
Nichols et al., 1977
MRC-5
Фибробласты
легкое
Человек
Эмбриональный
Диплоидный
Чувствительны к
вирусным инфек¬
циям человека;
контактное ингиби¬
рование
Jacobs, 1970
MRC-9
Фибробласты
легкое
Человек
Эмбриональный
Диплоидный
Чувствительны к
вирусным инфек¬
циям человека;
контактное ингиби¬
рование
Jacobs, et al., 1979
WI-38 Фибробласты легкое
Постоянные, из нормальных тканей
Человек
Эмбриональный
Диплоидный
Чувствительны к
вирусным инфек¬
циям человека
Hayflick & Moor¬
head, 1961
293
Эпителиальные
клетки
почка
Человек
Эмбриональный
Анеуплоидный
Легко трансфециру-
ются
Graham et al.,
1977
3T3-A31
Фибробласты
Мышь
BALB/c
Эмбриональный
Анеуплоидный
Контактное ингиби¬
рование;
легко трансформи¬
руются
Aaronson & To-
daro, 1968
3T3-L1
BEAS-2B
Фибробласты
Эпителиальные
клетки
легкое
Мышь
Swiss
Человек
Эмбриональный
Взрослый
Анеуплоидный
Адипозная диффе¬
ренцировка
Green & Kehinde,
1974
Reddel et al., 1988
ВНК21-С13
Фибробласты
почка
Сирийский
хомячок
Новорожденный
Анеуплоидный
Т расформируется
вирусом полиомы
Macpherson &
Stoker, 1962
BRL3A
Эпителиальные
клетки
печень
крыса
Новорожденный
Продуцирует IGF-2
Coon, 1968
С2
С7
Фибробласт-по-
добные клетки
Эпителиоидные
клетки
скелетные
мышцы
гипоталамус
мышь
Мышь
Эмбриональный
Мышечные трубочки
Нейрофизин, ва-
зопрессин
Morgan et al.,
1992
De Vitry et al.,
1974
СНО-К1
Фибробласты
яичник
Китайский
хомячок
Взрослый
Диплоидный
Простое кариотипи-
рование
Puck et al., 1958
COS-1,
COS-7
Эпителиоидные
почка
свинья
Взрослый
Хороший реципиент
для трансфекции
ДНК
Gluzman, 1981
СРАЕ
Эндотелиальные
клетки
Эндотелий
легочной
артерии
корова
Взрослый
Диплоидный
Фактор VIII,
ангиотензин II -
конвертирующий
фермент
Del Vecchio &
Smith, 1981
НаСаТ
Эпителиальные
кератино¬
циты
Человек
Взрослый
Диплоидный
Ороговение
Boukamp et al.,
1988
L6
Фибробласт-по-
добные клетки
Скелетные
мышцы
крыса
эмбриональные
Мышечные трубочки
Richler & Yaffe,
1970
LLC-PKI
Эпителиальные
почка
свинья
Взрослый
Диплоидный
№+-зависимый за¬
хват глюкозы
Hull et al., 1976;
Saier, 1984
MDCK
Эпителиальные
почка
собака
Взрослый
Диплоидный
Куполообразность;
транспорт
Gaush et al., 1966;
Rindler et al.,
1979
NRK49F
Фибробласты
почка
Крыса
Взрослый
Анеуплоидный
Индукция роста сус¬
пензии TGF-a,P
De Larco &
Todaro, 1978
STO
Фибробласты
Мышь
Эмбриональный
Анеуплоидный
Используется как
фидерный слой
для эмбриональ¬
ных стволовых
клеток
Bernstein, 1975
13.3. Возраст культуры
237
Продолжение табл. 13.1
Клеточная Морфология
линия
Происхож- Вид
дение
Возраст
Плоидность Характеристики Ссылки
Уего
Фибробласты почка
Постоянные, из опухолевых тканей
А2780
А549
А9
В16
С1300
С6
Сасо-2
ЕВ-3
Friend
GH1, GH2,
GH3
Н4-11-Е-СЗ
HeLa
HeLa-S3
HEP-G2
HL-60
HT-29
K-562
L1210
L929
LS
MCF-7
Эпителиальные
клетки
Эпителиальные
клетки
Фибробласты
обезьяна Взрослый
Человек Взрослый
Человек
Подкожный Мышь
Фибробласт -по- Меланома Мышь
добные клетки
Нейрональные Нейроблас- Крыса
тома
Фибробласт -по- Глиома Крыса
добные клетки
Эпителий
Лимфоциты
Суспензионная
Эпителиоидные
клетки
Эпителиальные
Эпителиальные
Эпителий
Толстый ки- Человек
шечник
Перифери- Человек
ческая
кровь
Селезенка Мышь
Гипофи- Крыса
зарная
опухоль
Гепатома Крыса
Шейка мат- Человек
ки
Толстый ки- Человек
шечник
Эпителиоидные Гепатома Человек
клетки
Суспензионная Миелоид- Человек
ная лей¬
кемия
Эпителий Толстый ки- Человек
шечник
Суспензионная Миелоид- Человек
ная лей¬
кемия
Мышь
Лимфоциты
Фибробласты
Фибробласты
Мышь
Мышь
Эпителиальные Плевраль- Человек
ный вы¬
пот при
опухоли
молочной
железы
Анеуплоидный Субстрат для роста Hopps et al, 1963
и исследования
вирусов
Взрослый
Взрослый
Взрослый
Взрослый
Новорожденный
Взрослый
Юношеский
Взрослый
Взрослый
Взрослый
Взрослый
Взрослый
Взрослый
Взрослый
Взрослый
Взрослый
Взрослый
Взрослый
Взрослый
Взрослый
Анеуплоидный
Анеуплоидный
Анеуплоидный
Анеуплоидный
Анеуплоидный
Анеуплоидный
Анеуплоидный
Диплоидный
Анеуплоидный
Анеуплоидный
Анеуплоидный
Анеуплоидный
Химическая чувстви¬
тельность с нали¬
чием резистентных
вариантов
Продукция сурфак¬
танта
Получена из L929;
недостаток
HGPRT
Меланин
Аксоны
Tsuruo etal, 1986
Giard et al., 1972
Littlefield, 1964b
Nilos & Makarski,
1978
Liebermann &
Sachs, 1978
Benda et al., 1968
Глиальный фибрил¬
лярный кислотный
протеин, GPDH
Транспорт ионов и Fogh, 1977
аминокислот
Чувствительна к
вирусу Эпштейна-
Барра
Гемоглобин
Гормон роста
Анеуплоидный
Анеуплоидный
Анеуплоидный
Анеуплоидный
Тирозин
аминотрансфераза
G6PD тип А
Высокая эффек¬
тивность посева,
способна расти в
суспензии
Сохраняются неко¬
торые рибосомаль-
ные ферменты
Фагоцитоз, восста¬
новление Неотет¬
разол ия Голубого
Дифференцировка
индуцируется
NaBt
й Гемоглобин
Анеуплоидный
Анеуплоидный
Анеуплоидный
Анеуплоидный
Быстрый рост; спо¬
собность к росту в
суспензии
Клон L-клеток
Epstein & Вагг,
1964
Scheret al., 1971
Buonassisi et al,
1962; Yasamura
et al., 1966
Pitot et al., 1964
Gey et al, 1952
Puck & Marcus,
1955
Knowles et al.,
1980
Olsson & Ologs-
son, 1981
Fogh & Trempe,
1975
Andersson et al.,
1979a,b
Law etal., 1949
Рост в суспензии,
производная L929
Sanford et al.,
1948
Paul & Struthers,
личное сооб¬
щение
Рецепторы эстрогена, Soule et al., 1973
куполообразная
форма, а-лакталь-
бумин
238
ГЛАВА 13. СУБКУЛЬТУРА И КЛЕТОЧНЫЕ ЛИНИИ
Окончание табл. 13.1
Клеточная
линия
Морфология
Происхож- Вид
дение
Возраст
Плоидность Характеристики Ссылки
MCF-10
MOG-G-
ССМ
P388D1
S180
эпителиальные Фиброзно- Человек
кистозная
опухоль
молочной
железы
Эпителдиоидные Глиома Человек
Лимфоцитарные
Фибробласты
Мышь
Мышь
SK/HEP-1 Эндотелиальные Гепатома,
эндоте¬
лий
WEHI-3B D+ Суспензионная Костный
мозг
ZR-75-1 Эпителиалоьные Асцитная
жидкость
при
опухоли
молочной
железы
Мышь
Человек
Взрослый
Взрослый
Взрослый
Взрослый
Человек Взрослый
Взрослый
Взрослый
Почти дипло- Формирование купо- Soule et al., 1990
идныи
Анеуплоидный
Анеуплоидный
Анеуплоидный
Анеуплоидный
Анеуплоидный
Анеуплоидный
лообразной формы
Глутамил synthetase
Рост в суспензии
Скрининг эффектив¬
ности химиотера¬
пии рака
Фактор VIII
Продукция IL-3
Рецепторы эстрогена,
EGF-рецепторы
Balmforth et al.,
1986
Dawe & Potter,
1957; Koren et
al., 1975
Dunham & Stew¬
art, 1953
Heffelfinger et al.,
1992
Nicola, 1987
Engel et al., 1978
Таблица 13.2. Перекрестно-зараженные клеточные линии
Клеточная
Вид
Тип клеток
Контами-
Вид
Тип клеток
Источник
линия
нант
данных
207
Человек
Рге-В-лейкемия
REH
Человек
Рге-В-лейкемия
DSMZ
2474/90
Человек
Рак желудка
НТ-29
Человек
Колоноректальная рак
DSMZ
2957/90
Человек
Рак желудка
НТ-29
Человек
Колоноректальная рак
DSMZ
3051/80
Человек
Рак желудка
НТ-29
Человек
Колоноректальная рак
DSMZ
ADLC-5M2
Человек
Рак легкого
HELA/-S3
Человек
Аденорак шейки матки
DSMZ
AV3
Человек
Амниотическая жидкость
HeLa
Человек
Аденорак шейки матки
АТСС
ВСС-1/КМС Человек
Базальноклеточная рак
HELA/-S3
Человек
Аденорак шейки матки
DSMZ
ВМ-1604
Человек
Опухоль простаты
DU-145
Человек
Опухоль простаты
DSMZ
С16
Человек
Фибробласты легких эмбри¬
она клон MRC-5
HeLa
Человек
Аденорак шейки матки
ЕСАСС
CHANG liver Человек
Эпителий легких эмбриона
HeLa
Человек
Аденорак шейки матки
ATCC;JCRB
COLO-818
Человек
меланома
COLO-8OO
Человек
Меланома
DSMZ
DAMI
Человек
Мегакариотические клетки
HEL
Человек
Эритролейкемия
DSMZ
ECV304
Человек
Эндотелий
T24
Человек
Рак мочевого пузыря
АТСС
ECV304
Человек
Нормальный эндотелий
T24
Человек
Рак мочевого пузыря
DSMZ
EJ
Человек
Рак мочевого пузыря
T24
Человек
Рак мочевого пузыря
АТСС; JCRB
EPLC3-2M1
Человек
Рак легких
HELA/-S3
Человек
Аденокарцинома шейки матки
DSMZ; JCRB
EPLC-65
Человек
Рак легких
HELA/-S3
Человек
Аденокарцинома шейки матки
DSMZ
F2-4E5
Человек
Эпителий тимуса
SK-HEP-1
Человек
Гепатома
DSMZ
F2-5B6
Человек
Эпителий тимуса
SK-HEP-1
Человек
Гепатома
DSMZ
FL
Человек
Амниотическая жидкость
HeLa
Человек
Аденокарцинома шейки матки
ATCC
GHE
Человек
Астроцитома
T-24
Человек
Рак мочевого пузыря
DSMZ
Girardi Heart Свинья
Сердце взрослой свиньи
HeLa
Человек
Аденокарцинома шейки матки
ATCC
HAG
Человек
Аденоматозный зоб
T-24
Человек
Рак мочевого пузыря
DSMZ
НЕр-2
Человек
Носоглоточнеый эпителий
взрослого человека
HeLa
Человек
Аденокарцинома шейки матки
ATCC
HMV-1
Человек
Меланома
HeLa-S3
Человек
Аденокарцинома шейки матки
DSMZ
HuL-1
HeLa
Человек
Аденокарцинома шейки матки
JCRB
13.3. Возраст культуры
239
Окончание табл. 13.2
Клеточная
Вид
Тип клеток
Контами-
Вид
Тип клеток
Источник
линия
нант
данных
IMC-2
Человек
Карцинома челюсти
HeLa-S3
Человек
Аденокарцинома шейки матки
DSMZ
Intestine 407
Человек
Эпителий тонкой кишки
HeLa
Человек
Аденокарцинома шейки матки
АТСС
J-111
HeLa
Человек
Аденокарцинома шейки матки
JCRB
JOSK-I
Человек
Моноцитарная лейкемия
U-937
Человек
Гистиоцитарная лимфома
DSMZ
JOSK-K
Человек
Моноцитарная лейкемия
U-937
Человек
Гистиоцитарная лимфома
DSMZ
JOSK-M
Человек
Моноцитарная лейкемия
U-937
Человек
Гистиоцитарная лимфома
DSMZ
JOSK-S
Человек
Моноцитарная лейкемия
U-937
Человек
Гистиоцитарная лимфома
DSMZ
JTC-17
Человек
HeLa
Человек
Аденокарцинома шейки матки
Yamakage
OCRB)
КВ
Человек
Эпителий ротовой полости
HeLa
Человек
Аденокарцинома шейки матки
АТСС; JCRB
К051
K562
Человек
Миелоидная лейкемия
DSMZ; JCRB
KOSC-3
Ca9-22
JCRB
L132
Человек
Эпителий эмбриональных
легких
HeLa
Человек
Аденокарцинома шейки матки
АТСС
LR10.6
Человек
Рге-В-клеточная лейкемия
NALM-6
Человек
Рге-В-клеточная лейкемия
DSMZ
МаТи
человек
Рак молочной железы
HELA/-S3
Человек
Аденокарцинома шейки матки
DSMZ
МС-4000
Человек
Рак молочной железы
HELA/-S3
Человек
Аденокарцинома шейки матки
DSMZ
МКВ-1
Человек
Т-клеточная лейкемия
CCRF-CEM
Человек
Т-клеточная лейкемия
DSMZ
MKN28
MKN74
JCRB
MOLT-15
Человек
Т-клеточная лейкемия
CTV-1
Человек
Моноцитарная лейкемия
DSMZ
МТ-1
Человек
Рак молочной железы
HELA/-S3
Человек
Аденокарцинома шейки матки
DSMZ
NCC16
PHK16-0b
JCRB
Р1-1АЗ
Человек
Эпителий тимуса
SK-HEP-1
Человек
Гепатома
DSMZ
P1-4D6
Человек
Эпителий тимуса
SK-HEP-1
Человек
Гепатома
DSMZ
Р39 TSU
Человек
HL60
Человек
Миелоидная лейкемия
JCRB
PBEI
Человек
Пре-В-клеточная лейкемия
NALM-6
Человек
Пре-В-клеточная лейкемия
DSMZ
PSV811
Человек
Фибробласты
WI38
Человек
Фибробласты
JCRB
RAMAK-1
Человек
Синовиальная оболочка
мышц
T-24
Человек
Рак мочевого пузыря
DSMZ
SBC-2
Человек
Рак мочевого пузыря
HELA/-S3
Человек
Аденокарцинома шейки матки
DSMZ
SBC-7
Человек
Рак мочевого пузыря
HELA/-S3
Человек
Аденокарцинома шейки матки
DSMZ
SCLC-16H
Человек
SCLC
SCLC-
21/22H
Человек
SCLC
DSMZ
SCLC-24H
Человек
SCLC
SCLC-
21/22H
Человек
SCLC
DSMZ
SNB-19
U251 MG
Человек
Глиома
ATCC
SPI-801
Человек
Т-клеточная лейкемия
K-562
Человек
Миелоидная лейкемия
DSMZ
SPI-802
Человек
Т-клеточная лейкемия
K-562
Человек
Миелоидная лейкемия
DSMZ
ТК-1
Человек?
U251MG
Человек
Глиома
JCRB
ТМН-1
IHH-4
JCRB
WISH
Человек
Амниотический эпителий
HeLa
Человек
Аденокарцинома шейки матки
ATCC
новорожденного
личий в клеточных линиях, клональных вариаций и
условий культивирования, но оно обычно постоянно
для одного и того же типа клеток при одних и тех же
условиях культивирования. Таким образом, важно,
чтобы в описании клеточной линии было указано
приблизительное количество генераций или периодов
удвоения после эксплантации. Авторы подчеркивают,
что только «приблизительное», поскольку количество
генераций, которое проходит в первичной культуре,
трудно определить.
Постоянные клеточные линии (см. табл. 13.1) из¬
бегают старения, поэтому номер генерации становится
менее важным, и становится более важным номер
пассажа после посадки культуры, находившейся на
хранении (см. разд. 13.7.2). Кроме того, в результате
возрастания интенсивности пролиферации клеток и
повышения плотности культуры (см. разд. 18.5) индекс
разведения становится значительно выше (1:20-1:100)
и концентрация клеток в субкультуре становится более
критичной (см. разд. 13.7.3).
240
ГЛАВА 13. СУБКУЛЬТУРА И КЛЕТОЧНЫЕ ЛИНИИ
Таблица 13.3. Свойства конечных и постоянных клеточных линий
Свойства
Конечные
Постоянные (трансформированные)
Плоидность
Эуплоидные, диплоидные
Анеуплдоидные, гетероплоидные
Т рансформация
Нормальные
Иммортализованные, изменены контроль роста и
способность к опухолевому росту
Зависимость от подложки
Да
Нет
Контактное ингибирование
Да
Нет
Ограничение клеточного деления плотнос¬
Да
Снижена или утрачена
тью клеточной популяции
Тип роста
Монослой
Монослой или суспензия
Ведение культуры и пересев
Циклическое
Возможно устойчивое состояние
Потребность в сыворотке
Высокое
Низкое
Эффективность клонирования
Низкая
Высокая
Маркеры
Специфические тканевые
Хромосомальные, ферментативные, антигенные
Специальные функции (например, воспри¬
имчивость к вирусам, дифференцировка)
Может быть сохранена
Часто утрачена
Скорость роста
Медленная (TD 24-96 ч)
Быстрая (TD 12-24 ч)
Клеточный выход
Низкий
Высокий
Контролируемые параметры
Номер генерации, тканеспеци¬
фические маркеры
Маркеры (окраски)
13.4. МАРКИРОВКА КЛЕТОЧНОЙ
КУЛЬТУРЫ
Новым клеточным линиям должно быть дано обоз¬
начение, включающее (например, normal human brain
(NHB) (нормальный мозг человека)) номер клеточ¬
ного штамма или клеточной линии (если из одного
источника получено несколько клеточных линий;
например, NHB1, HNB2); и номер клона, если произ¬
водилось клонирование (например, NHB2-1, NHB2-2
и т.д.). Полезно вести учетный журнал или создать
компьютерную базу данных, где до инициации культу¬
ры будет регистрироваться поступление биопсийного
материала или образцов. Входящий номер в учетном
журнале или файле базы данных, возможно, связан¬
ный с буквенным идентификационным кодом, может
затем быть использован при маркировке клеточной
линии. Например, LT156 может означать материал
биопсии рака легких (lung tumor biopsy) номер 156.
Этот принцип маркировки не очень хорош из-за воз¬
можной двусмысленности толкования маркировки,
таких как одни и те же буквы для обозначения двух
различных линий клеток, и для создания автоматичес¬
ких ссылок на запись поступления культур. Правило
конфиденциальности запрещает использование ини¬
циалов донора для обозначения клеточной линии.
Для конечных клеточных линий через дробь должно
быть указано количество периодов удвоения популя¬
ции (например, NHB2/2), и это число возрастает на
один при индексе разведения 1:2 (например, NHB2/2,
NHB2/3 и т.д.) или на 2, если индекс разведения 1:4
(например, NHB2/2, NHB2/4 и т.д.) и т.д. Когда мы
имеем дело с постоянной клеточной линией, то в конце
маркировки обычно ставят «р» и далее номер пассажа
после последнего оттаивания культуры, взятой с хра¬
нения (см. разд. 20.4.2), например HeLa-S3/p4.
При упоминании клеточных линий в публикациях
и докладах полезно прибавить к маркировке клеточной
линии индикационный код той лаборатории, где была
создана данная культура (например, WI означает Wistar
Institute, NCI — National Cancer Institute, SK — Sloan-
Kettering) [Federoff, 1975]. В публикациях и докладах
при первом упоминании и в разделе «Материалы и
методы» клеточные линии должны быть даны с их пол¬
ным обозначением, а в дальнейшем может приводиться
сокращение. Существенно то, что маркировка каждой
клеточной линии уникальна. В противном случае
возникает путаница при последующих сообщениях в
литературе. Банки клеток решают эту проблему при-
даванием каждой клеточной линии входящего номера;
поэтому при упоминании клеточной линии, получен¬
ной из банка, необходимо в «Материалах и методах»
упомянуть регистрационный номер, под которым они
записаны в банке. Все знаки пунктуации также могут
быть причиной путаницы при поиске культуры в базе
данных, поэтому всегда используйте стандартную сис¬
тему знаков и не используйте апострофы и пробелы.
13.5. ВЫБОР КЛЕТОЧНОЙ ЛИНИИ
Кроме специфических функциональных требований к
культурам клеток существует ряд общих параметров, ко¬
торые следует учитывать при выборе клеточной линии.
1) Конечная или постоянная. Существует ли по¬
стоянная клеточная линия, которая имеет необ¬
ходимые функциональные свойства? Постоянную
линию клеток обычно легче поддерживать, быст¬
рее выращивать, легче клонировать, она произ¬
водит более высокий урожай клеток во флаконе
и более приспособлена к бессывороточной среде
(см. табл. 13.3).
13.6. Обычный порядок поддержания культуры
241
2) Нормальная или трансформированная. Важно, являет¬
ся линия злокачественно трансформированной или нет?
Если да, то, вероятно, следует попробовать получить
иммортализованную линию, которая не будет туморо-
генна, например линии клеток ЗТЗ или ВКК21-С13.
3) Видовая принадлежность. Важна ли видовая при¬
надлежность? Нечеловеческие линии клеток имеют
меньшее количество ограничений биобезопасности
и имеют то преимущество, что исходная ткань может
быть более доступна.
4) Ростовые характеристики. Что вам требуется в
отношении клеточного роста, выхода клеток, эф¬
фективности посева и легкости получения большого
числа клеток? Вы будете должны рассмотреть сле¬
дующие параметры:
а) время удвоения популяции (см. разд. 21.9.7);
б) плотность насыщения (на флакон; см. разд. 21.9.5);
в) эффективность посева (см. разд. 21.10);
г) фракции роста (см. разд. 21.11.1);
д) способность расти в суспензии (см. разд. 18.5.1,
табл. 13.5).
5) Эксплуатационная готовность. Если вы должны
использовать конечную линию клеток, то доста¬
точны ли доступные запасы, или вы будете должны
производить вашу собственную линию (линии)?
Если вы выбираете постоянную линию клеток, то
существует ли сертифицированный источник ис¬
ходной культуры?
6) Валидация. Насколько хорошо охарактеризована
линия (см. разд. 7.10), если она уже существует, или,
если не существует, можете ли вы определить все
необходимые характеристики (см. гл. 16)? Является
ли линия аутентичной (см. разд. 16.3)? Жизненно
важно устранить возможность взаимного загряз¬
нения линий перед осуществлением программы
работы с линией клеток, как так в литературных
источниках много сообщений о перекрестном за¬
грязнении (см. табл. 13.2).
7) Фенотипические проявления. Можно ли создать
линию, имеющую определенные фенотипические
характеристики (см. разд. 17.7)?
8) Контрольная линия клеток. Если вы используете
мутантную, трансфицированную, трансформи¬
рованную или аномальную линию клеток, то су¬
ществует ли доступный эквивалент — нормальная
клеточная линия, если это потребуется?
9) Стабильность. Насколько стабильна линия клеток
(см. разд. 18.3 и цв. вкладку 7)? Было ли проведено
клонирование? Если нет, то можете ли вы провести
клонирование и сколько времени займет процесс
клонирования для получения замороженных и го¬
товых к употреблению запасов?
13.6. ОБЫЧНЫЙ ПОРЯДОК
ПОДДЕРЖАНИЯ КУЛЬТУРЫ
Как только культура инициирована, независимо от
того, является она первичной культурой или субкуль-
Рис. 13.1. Поврежденные клетки. Вакуолизация и грануля¬
ция в клетках бронхиального эпителия (BEAS-2B) возник¬
шие, в данном случае, в результате несоответствия среды.
Цитоплазма клеток становится гранулированной, особенно
вокруг ядра, и происходит вакуолизация. Клетки на краю мо¬
нослоя могут сильно преломлять свет, если распространению
клеток что-то препятствует
турой, необходимо периодически заменять среду («пи¬
тать» культуру) с последующим субкультивированием,
если клетки делятся. В случае непролиферирующей
культуры также требуется периодическая замена пи¬
тательной среды, поскольку клетки продолжают обра¬
зовывать метаболиты, и некоторые компоненты среды
истощаются или спонтанно деградируют. Промежутки
времени между заменами среды варьируют от одной
линии к другой и зависят от темпов роста и метабо¬
лизма культуры; быстрорастущие трансформирован¬
ные линии, такие как HeLa, обычно субкультивируют
один раз в неделю, через четыре дня следует заменить
среду. Клеточные линии, которые растут медленнее, в
том числе ^трансформированные, могут потребовать
субкультивирования только каждые две, три или даже
четыре недели, а среда должна заменяться еженедель¬
но в промежутках между субкультивированиями (см.
разд. 13.6.2,13.7.1,21.9.2).
13.6.1. Необходимость исследования
клеточной морфологии
Какую бы процедуру ни производили над клетками,
чрезвычайно важно тщательно исследовать культуру
для подтверждения отсутствия признаков заражения
(см. разд. 19.3.1 и рис. 19.1). Клетки должны прове¬
ряться на наличие признаков ухудшения их состоя¬
ния, таких как грануляция вокруг ядра, вакуолизация
цитоплазмы, приобретение клетками округлой формы
с отделением их от субстрата (рис. 13.1). Такие при¬
знаки могут означать, что культура требует замены
среды, либо может означать наличие более серьезной
проблемы, например неадекватности или токсичности
среды или сыворотки, микробного заражения или ста¬
242
ГЛАВА 13- СУБКУЛЬТУРА И КЛЕТОЧНЫЕ ЛИНИИ
рения клеточной линии. Дефицит каких-то веществ в
среде может также вызывать апоптоз (см. разд. 3.3.1 и
в. вкладку 17в, г). В ходе стандартного поддержания
культуры, замены среды или периодическое субкульти¬
вирование должно иметь целью предупреждение таких
ухудшений, поскольку часто вернуть клетки в хорошее
состояние весьма затруднительно.
Близкое знакомство с морфологией клетки может
также позволить определить первые признаки пере¬
крестной контаминации или обнаружить ошибку в
идентификации культуры. Полезно иметь серию фо¬
тографий различных типов клеток, которые постоянно
используются в лаборатории (см. рис. 16.2 и цв. вклад¬
ки 9 и 10), сделанные при различной плотности клеток
(предпочтительно, известной плотности, например с
подсчетом количества клеток на см2) с пояснениями,
когда следует производить манипуляции с культурами.
В частности, это важно, когда появляется новый сотруд¬
ник, которого вводят в культуральные работы.
13.6.2. Замена среды
Существует четыре фактора, определяющих необходи¬
мость замены культуральной среды:
1) Падение pH. Следует принимать во внимание
скорость падения и абсолютное значение величи¬
ны pH. Большинство клеток перестают расти при
падении pH от 7,0 до 6,5, а также начинают терять
жизнеспособность между pH 6,5 и 6,0. Поэтому если
цвет среды меняется от красного через оранжевый
до желтого, то среду следует заменить. Постарай¬
тесь оценить скорость падения pH; если скорость
падения pH в культуре, которая растет при pH 7,0,
составляет 0,1 ед. pH в день, то задержка питания
на один-два дна по каким-то причинам не вызовет
большого повреждения клеток. В то же время при
скорости падения pH на 0,4 ед. в день необходимо
произвести замену среды в течение ближайших
24-48 ч, не оставляя культуру без питания на вы¬
ходные дни.
2) Концентрация клеток. При высокой концентрации
клеток в культуре истощение питательной среды
происходит быстрее, чем при низкой концентрации.
Этот фактор обычно (но не всегда) очевидно прояв¬
ляется в скорости падения pH.
3) Тип клеток. Нормальные клетки (например, дипло¬
идные фибробласты) обычно перестают делиться
при высокой плотности клетки (см. разд. 18.5.2) из-
за скученности клеток, истощения факторов роста и
по другим причинам. Клетки блокируются в Gj-фазе
клеточного цикла и долго сохраняются, даже будучи
оставленными в таком состоянии на две-три недели
и дольше. Однако состояние трансформированных
клеток, постоянных клеточных линий и некоторых
эмбриональных клеток быстро ухудшается при
высокой плотности клеток, если только среду не
заменяют ежедневно или не производят субкуль¬
тивирование.
4) Морфологические нарушения. Этот фактор сле¬
дует предвидеть при регулярном обследовании
и хорошем знакомстве с клеточной линией (см.
разд. 13.6.1). Если позволить этому процессу зайти
слишком далеко, то он может стать необратимым,
поскольку клетки имеют склонность входить в
апоптоз (см. разд. 3.3.1).
Объем, глубина и площадь поверхности. Обыч¬
но отношение объема среды к площади поверхности
составляет 0,2-0,5 мл/см2 (см. также разд. 21.9.3).
Верхний предел определяется диффузией газа через
слой жидкости, а оптимальное соотношение зави¬
сит от требования клеток к содержанию кислорода.
Клетки с высокой потребностью в 02 чувствуют себя
лучше в тонком слое среды (например, 2 мм), те же,
потребности которых низки, могут лучше расти в
глубоких слоях среды (например, 5 мм). Если глу¬
бина среды более чем 5 мм, газовая диффузия может
быть ограничена. В случае монослойной культуры
проблема может быть преодолена вращением бутыли
(см. разд. 26.2.3) или перфузии культуры средой и ор¬
ганизацией газообмена в промежуточном резервуаре
(см. разд. 25.3.2, 26.2.5).
Сдерживающая среда. Сдерживающая среда
может быть использована для стимуляции митозов,
которые обычно сопровождают замену среды, даже
при высокой плотности клеток, если они нежелатель¬
ны. Сдерживающая среда обычно представляет собой
обычную среду с концентрацией сыворотки, снижен¬
ной до 0,5-2% или полностью удаленной из состава
среды. В бессывороточную среду не добавляется при
этом никаких факторов роста или других митогенов.
В результате происходит ингибирование митозов у
большинства ^трансформированных клеток. Линии
трансформированных клеток не годятся для этой
процедуры, поскольку они могут успешно продолжать
делиться, либо культура испортится, поскольку транс¬
формированные клетки не блокируются в Gt-фазе
клеточного цикла, как это делают нормальные клетки
(см. разд. 3.3.1).
Сдерживающая среда используется для поддер¬
жания клеточных линий, которые имеют конечное
количество жизненных циклов, при этом не истощая
ресурс количества генераций (см. разд. 3.8.1). Редукция
сыворотки и прекращение клеточного деления также
создает благоприятные условия для проявления видо¬
измененного фенотипа некоторых линий [Maltese &
Volpe, 1979; Schousboe et al., 1979]. Среда, используемая
для сборов биопсийных образцов, может также быть
отнесена к сдерживающим средам.
Стандартный протокол смены среды. Прото¬
кол 13.1 разработан для выполнения упражнения 4,
при этом используется среда, приготовленная при
выполнении упражнения 3 (см. гл. 2). Клетки и среда в
протоколе точно определены, но могут быть изменены
в соответствии с индивидуальными потребностями.
13.7. Субкультура
243
ПРОТОКОЛ 13.1. Смена среды в монослойной
культуре
Схема
Исследовать культуру невооруженным глазом и
при помощи инвертированного микроскопа. Если
обнаружено, например, падение pH, удалить старую
среду и добавить новую среду. Возвратить культуру
в инкубатор.
Материалы
Стерильные:
• Клеточная культура А549, 4 день после посе¬
ва при концентрации 2х104кл./мл, флаконы
25 см2 4
• Питательная среда 100 мл
• Например, среда Игла MEMxl с солями Хэнкса
и 4 шМ НС03, без антибиотиков.
• Если протокол используется в качестве трени¬
ровочного упражнения, использовать две среды,
подготовленные при выполнении упражнения 3,
одна из которых хранилась при 4 °С, а другая при
37 °С в течение одной недели.
• Пипетки, градуированные и заткнутые пробками.
Если пипетки стеклянные, то в ассортименте раз¬
меров, 1 мл, 5 мл, 10 мл, 25 мл, в квадратном пе¬
нале для пипеток. Пластмассовые пипетки могут
быть индивидуально обернуты и рассортированы
по размерам на подносе.
• Пипетки без пробок для удаления среды, если
имеются перистальтический насос или вакуум¬
ная линия.
Нестерильные:
• Пипетирующее устройство или груша (см.
рис. 5.5,6.6).
• Шланг, соединяющий принимающий сосуд с ва¬
куумной линией или перистальтическим насосом
(см. рис. 5.1-5.3).
• Этанол 70% в распыляющей бутылке.
• Безворсовые салфетки или тампоны для про¬
тирки.
• Абсорбирующие бумажные полотенца.
• Цилиндр для пипеток, содержащий воду и дезин¬
фектант (см. разд. 5.8.8,7.8.5).
• Ручка-маркер с этанол-нерастворимыми черни¬
лами.
• Записная книжка, ручка, протоколы и т. д.
Протокол
1. Подготовьте ламинарный шкаф, удостоверив¬
шись, что он чист, протрите 70%-м этанолом.
2. Принесите реактивы и материалы, необходи¬
мые для процедуры, протрите бутылки 70%-м
этанолом, разместите необходимые материалы в
ламинарном шкафу (см. протокол 6.1).
3. Тщательно исследуйте культуру на признаки
контаминации или ухудшения состояния клеток
(см. рис. 13.1,19.1).
4. Проверьте предварительно описанные крите¬
рии — pH и плотность клеток или концентрацию,
и на основании ваших знаний поведения куль¬
туры решите, заменить ли среду. Если требуется
замена, действуйте следующим образом:
5. Перенесите культуру в стерильную рабочую
зону.
6. Откройте флакон.
7. Возьмите стерильную пипетку и вставьте в грушу
или пипетирующее устройство либо, используя
пипетку без ватной пробки, присоедините ее к
вакуумной линии или насосу.
8. Удалите среду, сливая ее в емкость для отходов
(см. рис. 6.8), Предпочтительно отсасывать среду
при помощи вакуумной линии, подсоединенной
к внешнему насосу (см. рис. 5.2,5.3).
9. Поменяйте пипетку.
10. Откройте бутылку со средой.
11. Возьмите новую пипетку и добавьте тот же самый
объем свежей среды, который был удален; если
важно, чтобы не было никаких задержек клеточ¬
ного роста, предварительно нагрейте среду до
37 °С. Вновь закройте бутылку.
12. Поменяйте пипетку.
13. Закройте флакон и бутылку со средой.
14. Верните культуру в инкубатор.
15. Произведите подробные записи наблюдений и
питания в регистрационном листе или журнале
лаборатории.
16. Уберите все пипетки, стеклянную посуду и т. д.
и протрите рабочую поверхность шкафа.
Замечание. Когда культура находится при низкой
плотности и медленно растет, может быть предпочти¬
тельно при выполнении шага 8 удалить только полови¬
ну среды и заместить ее на шаге 11 таким же объемом
свежей среды, какой был удален.
13.7. СУБКУЛЬТУРА
Когда клеточная линия подвергается субкультивирова¬
нию, то происходит повторный рост клеток до точки, в
которой культура становится готовой к последующему
субкультивированию. Обычно этот процесс можно
выразить в виде стандартного графика (см. рис. 13.2).
Lag-период наступает после посева, вслед за ним идет
период экспоненциального роста, называемый log-фа¬
зой. Когда плотность клеток (клеток/см2 субстрата)
достигает такого уровня, что весь доступный субстрат
оказывается занятым, или когда концентрация клеток
(клеток/мл среды) превышает возможности среды,
рост прекращается или значительно снижается (см.
рис. 16.26, г, е, ж, з, к, а также цв. вкладку 4г). Затем
следует либо заменить среду, либо культуру следует
разделить. Для адгерентных клеточных линий деление
культуры, или субкультивирование, обычно включает
в себя удаление истощенной питательной среды и
244
ГЛАВА 13. СУБКУЛЬТУРА И КЛЕТОЧНЫЕ ЛИНИИ
Дни после субкультивирования
Рис. 13.2. Кривая роста и поддержание культуры. Полулогарифмический участок кривой, отражающей изменения концент¬
рации клеток с течением времени, включает в себя lag-фазу, экспоненциальную фазу и плато, и отражает время, когда следует
проводить субкультивирование и замену среды культуры (см. также разд. 21.9.2 и рис. 21.6)
диссоциацию клеток монослоя при помощи трипси¬
на, хотя некоторые культуры, образующие рыхлый
монослой (например, HeLa), могут быть субкульти-
вированы встряхиванием бутылки, переходом клеток
в среду и соответствующим разведением их в новом
флаконе в свежей среде. Существуют также некоторые
культуры, которые не диссоциируют под действием
трипсина и поэтому требуют применения некоторых
специальных протеаз, таких как проназа, диспаза и
коллагеназа (табл. 13.4). Проназа является наиболее
эффективным ферментом из этих протеаз, но может
быть токсичной для некоторых клеток. Диспаза и кол¬
лагеназа обычно менее токсичны, чем трипсин, но могут
не давать полной диссоциации эпителиальных клеток.
На рынке представлены также другие протеазы, такие
как протеазы беспозвоночных (Accutaze, Accumax), а
также рекомбинантные формы трипсина (Trypzean или
TrypLE). Их эффективность должна быть проверена
в тех случаях, когда их применение не соответствует
стандартному протоколу дезагрегации либо когда в
эксперименте следует избегать применения протеаз
млекопитающих (например, трипсина свиньи) или
бактерий (например, Pronase). Жесткость требований,
предъявляемых к протеазам, зависит от типа клеток,
определяющего чувствительность клеток к протеоли-
зу. Следует выбирать такой протокол, который имеет
наименее строгие требования к протеазе, но при этом
подходит для получения суспензии единичных клеток
с высокой жизнеспособностью.
Прикрепление клеток друг к другу и к субстрату
опосредовано гликопротеинами клеточной поверх¬
ности и Са2+ (см. разд. 3.2). Другие протеины, а также
протеогликаны, выделенные из клеток и из сыворотки,
связываются с клеточной поверхностью и с субстратом,
усиливая клеточную адгезию. Субкультивирование
обычно требует хелатирования Са2+ и деградации вне¬
клеточного матрикса, а также, возможно, внеклеточных
доменов некоторых молекул клеточной адгезии.
13.7.1. Критерии субкультивирования
Необходимость субкультивировать монослойную
культуру определяется следующими критериями:
1) Плотность культуры. Нормальные клетки должны
быть субкультивированы, как только они достигают
стадии, когда границы клеток смыкаются (конфлю-
энтности). Если оставить их в этом состоянии более
чем на 24 ч, они выйдут из цикла деления, и вам
потребуется больше времени для того, чтобы вос¬
становить их пролиферативную активность после
пересева. Трансформированные клетки также сле¬
дует субкультивировать по достижении конфлюэнт-
ности; хотя они будут продолжать пролиферировать
и после, но состояние культуры начнет ухудшаться
после приблизительно двух периодов удвоения, и
эффективность посева будет снижаться.
2) Истощение среды. Истощение среды (см.
разд. 13.6.2) обычно показывает, что среда должна
быть заменена, но если падение pH развивается
слишком быстро, то среду приходится менять чаще,
чем необходимо с точки зрения субкультивирова¬
ния. Обычно падение pH связано с возрастанием
плотности клеток, которое является главным ин¬
дикатором потребности в субкультивировании.
Заметьте, что неожиданно падение pH может также
быть результатом заражения, поэтому следует про¬
верить эту возможность (см. разд. 19.3).
13.7. Субкультура
245
Таблица 13.4. Методы диссоциации клеток
Метод
Предваритель¬
ная обработка
Диссоциирующий агент
Среда
Области применения
Стряхивание
Нет
Мягкое механическое
стряхивание, покачи¬
вание или энергичное
пипетирование
Культуральная
среда
Митотические или другие слабо при¬
крепленные друг к другу клетки
Соскабливание
Нет
Скребок для клеток
Культуральная
среда
Клеточные линии, для которых не
могут быть использованы протеазы
(например, при анализе рецепторов
или поверхностных клеточных бел¬
ков); некоторые клетки могут быть
повреждены, редко дает однородную
клеточную суспензию
Действие трипси¬
Полное удале¬
0,01-0,5%-й р-р неочи¬
D-PBSA, CMF
Большинство постоянных клеточных
на* без других
воздействий
ние среды
щенного трипсина;
обычно 0,25%
или цитрат
линий
Предварительное
отмывание с
трипсинизацией
D-PBSA
0,25%-й р-р неочищенно¬
го трипсина
D-PBSA
Некоторые сильно адгезирующие пос¬
тоянные клеточные линии и многие
быстрорастущие часто пересеваемые
культуры
Предварительное
отмывание с
трипсинизацией
1 mM EDTA
в D-PBSA
0,25%-й р-р неочищенно¬
го трипсина
D-PBSA
Сильно адгезирующие часто пересева¬
емые культуры
Предварительное
1 mM EDTA
0,25%-й р-р неочищенно¬
D-PBSA+
Многие эпителиальные клетки, неко¬
отмывание с
трипсинизацией
в D-PBSA
го трипсина
1 mM
EDTA
торые могут быть чувствительны к
ЭДТА, в этом случае используется
EGTA
Трипсин+ колла-
1 mM EDTA
0,25%-й р-р неочищенно¬
D-PBSA+
Плотные культуры и монослои, в т.ч. с
геназа
в D-PBSA
го трипсина;
200 ед./мл неочищенной
колллагеназы
1 шМ
EDTA
фибробластами
Диспаза
Нет
0,1-1,0 мг/мл диспазы
Культуральная
среда
Удаление эпителия в монослое
(не разрушает эпителиальный слой)
Проназа
Нет
0,1-1,0 мг/мл проназы
Культуральная
среда
Образование качественной суспензии
единичных клеток, может повреж¬
дать некоторые клетки
ДНКаза
D-PBSA или
1 mM EDTA
в D-PBSA
2-10 мкг/мл кристалли¬
ческой ДНКазы
Культуральная
среда
Использование других диссоциирую¬
щих агентов, которые повреждают
клетки и способствуют высвобож¬
дению ДНК
* Пищеварительный фермент, поставляемый различными компаниями (Difco, Worthington, Roche, Sigma) в различной степени
очистки. Грубая очистка Difco trypsin, 1:250, или Worthington CLgrade collagenase, содержит другие протеазы, которые могут
быть полезны при диссоциации некоторых клеток, но также могут быть токсичны для других клеток.
Начните с грубой обработки и затем при необходимости проведите более тонкую обработку. Более тонкая обработка часто
связана с использованием более низких концентраций (мкг/мл), что связано с высокой специфичностью действия агентов (для
ферментов ед/г). Очищенный трипсин при 4 °С рекомендуется для клеток, выращенных при низкой концентрации сыворотки
или в ее отсутствие [McKeehan, 1977], и обычно считается более подходящим методом. Может быть, необходимо провести
предварительную проверку партии, аналогично проверке качества сыворотки.
3) Время, прошедшее с последнего субкультивиро¬
вания. Рутинное субкультивирование наилучшим
образом проводится по строгому расписанию так,
чтобы воспроизведение культуры было качествен¬
ным и контролируемым. Если клетки при этом не
достигают достаточно высокой плотности (т. е. не
достигают стадии конфлюэнтного монослоя) за со¬
ответствующее время, то следует повысить концен¬
трацию клеток при посеве. Если же они достигают
конфлюэнтности слишком быстро, то концентрацию
клеток при посеве следует уменьшить. Как только
установлен стандартный режим субкультивиро¬
вания, то при данной постоянной концентрации
клеток при посеве периодически повторяющийся
рост клеток должен иметь постоянную периодич¬
ность и стабильный клеточный выход. Отклонения
246
ГЛАВА 13. СУБКУЛЬТУРА И КЛЕТОЧНЫЕ ЛИНИИ
от этого стандарта будет признаком отклонения от
нормальных условий или ухудшения состояния
клеток. В идеале следует найти такую клеточную
концентрацию, которая позволит проводить суб¬
культивирование через 7 дней, со сменой среды
через 3-4 дня после субкультивирования.
4) Требования к другим процедурам. Когда клетки
требуются для каких-то конкретных целей, отличных
от стандартного режима поддержания культуры, то
также требуется субкультивирование для того, что¬
бы повысить выход культуры или изменить форму
культурального сосуда или тип среды. В идеале эта
процедура должна быть выполнена в обычное вре¬
мя субкультивирования, тогда будет известно, что
культура была выращена в стандартном режиме,
при известных условиях пересева и при ожидаемых
результатах культивирования. Тем не менее требо¬
вание в отношении получаемых клеток не всегда
совпадает с установленным режимом их ведения,
поэтому следует искать компромисс. Однако в
любом случае 1) клетки не должны быть субкуль-
тивированы в lag-периоде и 2) клетки всегда следует
отбирать в промежутке времени между серединой
log-фазы и временем, когда они входят в фазу плато
предыдущего субкультивирования (если только нет
специальных требований к получению клеток, нахо¬
дящихся на стадии плато; в этом случае потребуется
частая смена среды или постоянная перфузия).
Манипуляции с различными клеточными линия¬
ми. Манипуляции с различными клеточными линиями
должны проводиться отдельно, с различными наборами
сред и реагентов. Если они все должны осуществлять¬
ся в одно и то же время, то существует значительный
риск перекрестной контаминации. В частности, если
клетки относятся к быстрорастущим клеточным ли¬
ниям, таким как HeLa, то их культивирование должно
вестись строго отдельно от медленно растущих линий
(см. разд. 19.5).
Типичный протокол субкультивирования для кле¬
ток, растущих в монослое. Протокол 13.2 описывает
трипсинизацию монослоя (рис. 13.3, цв. вкладки 7-12)
после предварительной промывки в ЭДТА, для удале¬
ния следовых количеств среды, двухвалентных катионов
и сыворотки (если она использовалась). Эту процедуру
можно проводить и без промывки, либо только исполь¬
зуя D-PBSA в качестве промывочного раствора, добав¬
ляя 1 мМ ЭДТА в раствор трипсина, если это требуется,
в зависимости от типа клеток (см. табл. 13.4).
Количество материалов, приведенное в протоколе,
относится к упражнению 13 из гл. 2, но оно может быть
изменено по потребности.
Поскольку расширение воздуха в пластиковых фла¬
конах может вызвать набухание флаконов и нарушение
их горизонтального положения, давление воздуха во
флаконах должно быть снижено ослаблением крышек
через 30 мин после помещения флаконов в инкубатор.
Альтернативным решением этой проблемы является
сдавливание флаконов сверху и снизу при навора-
чивании крышки (будьте аккуратны, чтобы не сжать
ПРОТОКОЛ 13.2. Субкультивирование
монослойной культуры клеток
Схема
Удалить среду. Подвергнуть клетки кратковремен¬
ному действию трипсина. Инкубировать клетки. Раз¬
вести клетки в среде. Подсчитать клетки. Развести и
пересеять субкультуру.
Материалы
Стерильные:
• Клетки культуры А549, 7 дней и 14 дней пос¬
ле пересева в 2х104кл./мл, во флаконах 25см2
по 1 каждой культуры
• WI-38, MRC-5, или эквивалентная культура
нормальных диплоидных фибробластов 7 дней
и 14 дней после посева во флаконах 25см2
по 1 каждой культуры
• Ростовая питательная среда, например 1. МЕМ
с солями Игла, 23 шМ НСОэ, без антибиотиков
100 мл
• Трипсин, 0.25% в D-PBSA (см. табл. 13.4)..10 мл
• D-PBSA с 1 шМ EDTA 20 мл
• Пипетки, градуированные и заткнутые пробками.
Если пипетки стеклянные, то в ассортименте
размеров 1 мл, 5 мл, 10 мл, 25 мл, в квадратном пе¬
нале для пипеток. Пластмассовые пипетки могут
быть индивидуально обернуты и рассортированы
по размерам на подносе
• Пипетки без пробок для удаления среды, если
имеются перистальтический насос или вакуум¬
ная линия.
• Универсальные контейнеры или 50-мл центри¬
фужные пробирки 4
• Культуральные флаконы, 25 см2 8
Нестерильные:
• Пипетирующее устройство или груша (см.
рис. 5.5,6.6)
• Шланг трубки на приемник, связанный с вакуум¬
ной линией или перистальтическим насосом
• Шланг, соединяющий принимающий сосуд с ва¬
куумной линией или перистальтическим насосом
(см. рис. 5.1-5.3)
• Этанол 70%, в распыляющей бутылке
• Безворсовые салфетки или тампоны для протирки
• Абсорбирующие бумажные полотенца
• Цилиндр для пипеток, содержащий воду и дезин¬
фектант (см. разд. 5.8.8,7.8.5)
• Ручка-маркер с этанол-нерастворимыми черни¬
лами
• Записная книжка, ручка, протоколы и т. д.
• Гемоцитометр или электронная счетчик клеток
Протокол
1. Подготовьте ламинарный шкаф, принесите реак¬
тивы и материалы, необходимые для выполнения
процедуры (см. разд. 6.5).
13.7. Субкультура
247
2. Тщательно исследуйте культуру на признаки
ухудшения или заражения (см. рис. 13.1,19.1).
3. Проверьте предварительно описанные критерии
(см. разд. 13.7.1) и на основании ваших знаний
поведения культуры решите, проводить ли
субкультивирование. (Тем, кто выполняет уп¬
ражнение 13, нужно сделать записи о плотности
культуры, состоянии клеток, например наличии
митозов, многослойности, ухудшения состояния
клеток.) Если требуется субкультивирование,
действуйте следующим образом:
4. Перенесите флаконы с культурой в стерильную
рабочую зону, удалите и вылейте среду (см. про¬
токол 13.1. шаги 7-9). Произведите манипуляции
с каждой клеточной линией отдельно, повторяя
процедуру, начиная с этого шага, для каждой
клеточной линии.
5. Добавьте D-PBSA/EDTA для предварительной
промывки (0,2 мл/см2), направляя струю на сто¬
рону, противоположную прикрепленным клеткам
так, чтобы избежать смещения клеток, произ¬
ведите осторожное полоскание клеток, удалите
раствор. Этот шаг предназначен, чтобы удалить
следы сыворотки, которая ингибирует действие
трипсина и истощает двухвалентные катионы,
необходимые для прикрепления клеток.
6. Добавьте трипсин (ОД мл/см2), направляя струю
к стороне флакона, противоположной располо¬
жению клеток. Поверните флаконы, положите
их так, чтобы монослой был полностью закрыт
раствором трипсина. Оставьте флаконы в таком
положении на 15-30 с.
7. Поднимите флаконы так, чтобы удалить трипсин
с монослоя, и быстро проверьте, не отделяется ли
монослой от подложки. Использование трипсина
при 4 °С помогает предотвратить преждевремен¬
ное отделение.
8. Удалите раствор, оставив несколько капель трип¬
сина.
9. Инкубируйте флаконы в таком положении,
чтобы клетки находились горизонтально до тех
пор, пока клетки не ошарятся (рис. 13.3,13.4, цв.
вкладка 5); затем при наклоне флакона монослой
должен скользить вниз по поверхности (Это
обычно происходит после 5-15 мин инкубиро¬
вания.) Не оставляйте флаконы дольше, чем
необходимо, но, с другой стороны, не заставляйте
клетки отделяться прежде, чем они готовы это
сделать. Старайтесь, чтобы не образовывались
комки.
Примечание. В каждом случае главный дис¬
социирующий агент, трипсин или EDTA, при¬
сутствует только кратковременно, и инкубация
выполняется в его небольшом объеме, после того
как большая часть агента была удалена. Если вы
столкнетесь с трудностью в отделении и диссо¬
циации клеток, впоследствии при подготовке
суспензии единичных клеток, вы можете исполь¬
зовать альтернативные процедуры (см. табл. 13.4).
10. Добавьте среду (0,1-0,2 мл/см2) и смойте клетки
повторным пипетированием по поверхности мо¬
нослоя.
Конфлюэнтный монослой, растущий во
флаконе приблизительно через одну неделю
Инкубирование 37 °С в течение 7 дней
Через несколько часов
клетки распластались
Инкубирование при 37 °С
%
Добавлен трипсин
Клетки пересеяны в
свежий флакон
Клетки
ресуспендированы в
среде, готовы к подсчету
и пересеву
л
Трипсин удален, на поверхности моно¬
слоя остаточное количество трипсина
Инкубирование при 37 °С в течение 10 мин
Ошаривание клеток после инкубирования
Рис. 13.3. Субкультивирование монослоя. Стадии в субкультивировании и цикле роста клеток монослоя после трипсиниза¬
ции (см. также цв. вкладки 4, 5)
248
ГЛАВА 13. СУБКУЛЬТУРА И КЛЕТОЧНЫЕ ЛИНИИ
11. Наконец, пипетируйте клеточную суспензию
вверх и вниз несколько раз с таким положе¬
нием наконечника, при котором не создается
пена. Примите во внимание, что угол наклона
пипетки может изменяться от одной клеточной
линии к другой; некоторые клеточные линии
диссоциируют легко, тогда как другие требуют
энергичного пипетирования. Почти все клетки
могут претерпеть механическое повреждение,
если пипетирование ведется слишком энергично.
Первичные субкультуры и культуры с ранним
сроком пересева особенно склонны к поврежде¬
нию, частично из-за их большей недолговечности
и частично из-за их большего размера. Постоян¬
ные клеточные линии обычно более эластичны и
требуют энергичного пипетирования для полной
дизаггрегации. Обычно несколько движений
вверх-вниз достаточно для образования суспен¬
зии единичных клеток. Если этот шаг затруднен,
примените более агрессивный диссоциирующий
агент (см. табл. 13.4). Желательно получение
суспензии единичных клеток при субкультивиро¬
вании, если вы хотите обеспечить точный подсчет
клеток и стандартизовать условия клеточного
роста. Однородность и гомогенность суспен¬
зии отдельных клеток существенна также, если
проводится количественная оценка клеточного
деления или эффективности посев, а также если
предполагается изоляция клеток для последую¬
щего формирования клона.
12. Подсчитайте клетки при помощи гемоцитометра
или электронного счетчика клеток (см. разд. 21.1),
запишите полученные данные.
13. Разведите суспензию клеток до соответствующей
выбранной концентрации:
а) добавляя соответствующий объем клеточной
суспензии к предварительно отмеренному
объему среды в культуральном сосуде.
или
б) растворяя клетки в полном объеме требуемым
объемом среды в нескольких флаконах.
Процедура (а) полезна для обычного субкуль¬
тивирования, когда используется только несколько
флаконов и точное количество клеток и воспроиз¬
водимость не являются необходимыми. Процедура
(б) предпочтительна, когда производится несколько
копий, потому что общее количество манипуляций
уменьшается и концентрация клеток в каждом фла¬
коне будет идентична. Для выполнения упражне¬
ния 13 используют процедуру (б): развести клетки
из каждого флакона до концентрации 2х104 кл./в мл
в 20 мл среды и посеять по 5 мл из каждой суспензии
в два флакона.
14. Если клетки культивируются при повышенном
содержании С02 (при использовании среды
МЕМ Игла с солями Эрла и 23 mM NaHC03,
как указано в списке материалов), заполните
газом флакон, внося газовую смесь необходимого
состава (в данном случае 5% С02) из цилиндра
с предварительно образованной газовой смесью
или из газового смесителя, через фильтр, встро¬
енный в линию. Газ вносится во флакон так,
чтобы заполнить его выше среды (см. рис. 6.11),
без образования пузырей в среде, поскольку обра¬
зование пузырей может привести к денатурации
некоторых компонентов среды и увеличению
риска загрязнения. Если нормальная газовая
фаза является воздушной, как с МЕМ Игла с со¬
лями Хэнкса (см. протокол 13.1), этот шаг можно
опустить.
15. Закройте флакон, поместите в инкубатор. При¬
близительно через 1 ч проверьте pH. Если в сре¬
де с воздушной газовой фазой pH повышается,
верните флаконы в стерильную рабочую зону и
кратковременно (1-2 с) подавайте газ с повышен¬
ным содержанием (5%) С02. Поскольку каждая
культура будет вести себя аналогично в одной и
той же среде, вы, в конечном счете, будет знать,
какие клетки нуждаются в добавлении газа после
пересева. Если pH в среде повышается, когда га¬
зовая фаза содержит 5% С02 (как в этом случае),
увеличьте концентрацию С02 до 7% или 10% или
добавьте стерильный раствор 0,1 N HCL.
16. Повторите эту процедуру начиная с четвертого
шага для следующей клеточной линии.
Примечание. Процедура в шаге 15 не должна
стать долгосрочным решением проблемы снижения
высокого pH после субкультивирования. Если про¬
блема сохраняется, то уменьшите pH среды во время
ее составления, и проверьте pH среды в инкубаторе
или во флаконе, заполненном газом.
слишком сильно и не разломать флакон). При инку¬
бации происходит расширение газа, и форма флакона
восстанавливается.
13.7.2. Цикл роста и индекс разведения
Рутинное субкультивирование приводит к повторению
стандартного цикла клеточного роста (рис. 13.4а, а так¬
же рис. 13.2). Очень важно хорошо знать этот цикл для
каждого типа клеток, с которыми вы работаете, вклю¬
чая посевную дозу, продолжительность периода роста
до субкультивирования, продолжительность времени
эксперимента и соответствующее время для взятия
образцов, которые дают самую высокую плотность.
Клетки, находящиеся в различных фазах цикла роста,
ведут себя различным образом в отношении клеточ¬
ного деления, ферментативной активности, гликолиза
и дыхания, синтеза специальных продуктов и многих
других свойств (см. разд. 21.9.4, 25.1.1).
13.7, Субкультура
249
а
Дни
б
Дни
Рис. 13.4. Серийное субкультивирование, (а) Повторение
стандартного цикле роста в ходе ведения клеточной линии:
если клетки растут правильно, они должны достигать одной
и той же конечной концентрации (пики) после одного и того
же времени при каждом цикле, если посевная концентрация
(линия деления на секции) и интервал времени между суб¬
культивированиями остаются постоянными. (б) Количество
генераций и пассажей: каждая субкультура представляет со¬
бой один пассаж, но количество генераций (в данном случае
3 генерации на один пассаж) зависит от индекса разведения
(8 на 3 удвоения на пассаж)
Для конечных клеточных линий удобно уменьшить
концентрацию клеток в субкультуре разведением в 2,
4, 8 и 16 раз (т.е. индекс разведения будет, соответ¬
ственно, 2, 4, 8 и 16), что облегчает подсчет клеток и
определение периода удвоения популяции (т. е. соот¬
ветственно индекс разведения 2 относится к первому
удвоению популяции, 4 — ко 2-му, 8 — к третьему и
16 — к 4-му); например, культура, разведенная в 8 раз,
требует тройного удвоения для того, чтобы достигнуть
той же клеточной плотности, что исходная (рис. 13,
146). Нежные или медленно растущие клеточные ли¬
нии следует также разводить в отношении 1:2, в то вре¬
мя как сильные, быстро растущие культуры могут быть
разведены в отношении 1:16, а постоянные клеточные
линии даже в отношении 1:50 или 1:100. Как только
клеточная линия становится постоянной (обычно это
происходит после 150 или 200 генераций), клеточная
концентрация становится главным параметром, кото¬
рый определяет поддержание культуры и требует суб¬
культивирования при достижении 104или 105 кл./мл.
Индекс разведения, или титр, также выбирается так,
чтобы установить удобный постоянный период между
субкультивированием, возможно 1 или 2 недели, а
также чтобы быть уверенными, что 1) клетки не разве¬
дены ниже того предела, который позволяет им снова
войти в цикл роста в течение lag-периода приемлемой
продолжительности (24 ч или менее), и 2) не выходят
на плато до следующего субкультивирования.
При первом манипулировании с клеточной линией
либо когда вы проводите начальный пассаж культуры,
опыт работы с которой у вас незначителен, при первой
попытке будет полезно провести субкультивирова¬
ние клеток с индексом разведения 2 или 4, записав
концентрации клеток, которые вы использовали. При
наличии большего опыта и при постоянной работе с
данной клеточной линией в вашей лаборатории, воз¬
можно, вы повысите индекс разведения, т.е. снизите
концентрацию клеток, после субкультивирования, но
всегда сохраняйте один флакон с низким индексом
разведения, при попытке снизить индекс разведения
остальной культуры.
Даже когда индекс разведения используется для
определения посевной концентрации в субкультуре,
количество клеток на флакон должно быть записано
после трипсинизации и при пересеве, так чтобы оце¬
нить скорость роста в каждой субкультуре и контроли¬
ровать концентрацию клеток (см. протокол 21.7-21.9).
В противном случае, небольшие отклонения не будут
обнаружены при нескольких пассажах.
13.7.3. Концентрация клеток
в субкультуре
Идеальным методом для определения правильной
концентрации клеток при пересеве является состав¬
ление кривой роста при различных посевных концент¬
рациях (см. протоколы 21.7-21.9). Таким образом,
можно определить минимальную концентрацию,
при которой длительность lag-периода минимальна,
и культура быстро входит в фазу быстрого логариф¬
мического роста (т.е. период удвоения популяции
короткий), но при этом вершина экспоненциальной
фазы достигается во время, удобное для следующего
субкультивирования.
Как правило, большинство постоянных клеточных
линий субкультивируются удовлетворительно при
посевной концентрации между 1х104и 5х104 кл./мл,
конечная клеточная линия фибробластов примерно
при такой же концентрации, большинство нежных
культур, таких как эндотелиальные и некоторые эпите¬
лиальные культуры с ранними сроками пересева — при
концентрации 1х105 кл./мл. Для новой культуры начи¬
найте с высокой посевной концентрации и постепенно
снижайте ее до тех пор, пока не получите удобный
для вас клеточный цикл без признаков ухудшения
состояния клеток.
250
ГЛАВА 13. СУБКУЛЬТУРА И КЛЕТОЧНЫЕ ЛИНИИ
13.7.4. Культивирование в суспензии
Протокол 13.2. относится к субкультивированию в мо¬
нослое, поскольку большинство первичных культур и
клеточных линий растут таким образом. Тем не менее
существуют клетки, которые растут постоянно в сус¬
пензии, поскольку они неприкрепляемые (большинство
культур лейкемии или асцитных опухолей мышей), по¬
скольку они удерживаются в виде суспензии при помо¬
щи механических устройств, либо они селекционирова¬
ны как суспензионные. В этом случае их можно растить
и поддерживать как культуру бактерий или дрожжей.
Суспензионные культуры имеют ряд преимуществ
(табл. 13.5); например, не требуется воздействие трипси¬
на, поэтому субкультивирование проводится быстрее и
легче (см. также разд. 26.1). Замещение среды (питание)
в суспензионных культурах обычно не производится,
вместо этого культуру разводят и распространяют,
разводят и выбрасывают ненужную часть, или часть
клеточной суспензии изымается и остаток разводится
до соответствующей посевной концентрации. В каждом
случае цикл роста, который получается в результате,
подобен тому, что существует в монослойной культуре,
но обычно имеет более короткий lag-период.
Клетки, которые спонтанно растут в суспензии,
могут поддерживаться в обычных культуральных фла¬
конах, которые не должны обрабатываться каким-то
особенным образом (но, конечно, должны быть стериль¬
ны). Правила глубины слоя среды такие же, что и для
монослойной культуры — т. е. 2-5 мм для осуществле¬
ния газообмена. Когда объем суспензионной культуры
повышается, например при разрастании культуры, то
среда должна взбалтываться, что наилучшим обра¬
зом достигается путем использования ротационного
магнитного маятника, с помещением культурального
сосуда на магнитной мешалке (см. рис. 13.5, см. также
рис. 26.1). Роллер-флаконы, вращающиеся на подстав¬
ке, также могут быть использованы для взбалтывания
суспензионной культуры (см. табл. 8.1 и рис. 26.11).
13.7.5. Субкультура клеток,
растущих в суспензии
Протокол 13.3. описывает рутинную процедуру суб¬
культивирования суспензионной культуры в свежий
сосуд. Постоянная культура может вестись в одном и
том же сосуде, но возможность контаминации посте¬
пенно возрастает при накоплении небольших разбрыз¬
гиваний на горлышке флакона при разведении. Крите¬
рии для субкультивирования аналогичны таковым для
монослойной культуры.
1) клеточная концентрация, которая для большин¬
ства суспензионных культур не должна превышать
1х106 кл./мл.
2) pH, которая связана с клеточной концентрацией и
понижается при повышении концентрации клеток.
3) Время после последнего субкультивирования, ко¬
торое, как и для монослоя, должно соответствовать
постоянному графику.
Таблица 13.5. Сравнительная характеристика
монослойной и суспензионной культур
Монослой
Суспензия
Требования к культуре:
Регулярное техническое обслу¬
живание
Пересев с трипсинизацией
Рост ограничен площадью поверх¬
ности
Особенности культивирования:
Контактное ингибирование
Клеточное взаимодействие
Диффузионный граничный слой
Используется для:
Цитологии
Изучение митотической актив¬
ности клеток
Экстракции in situ
Применимо к:
Большинству типов клеток, вклю-
чая первичные культуры
Стационарное состоя¬
ние
Разведение
Рост ограничен объемом
(газообмен)
Гомогенная суспензия
Получение большого
количества клеток
Получение партии клеток,
находящихся в митозе
Постоянное получение
продукта
Только трансформиро¬
ванным клеткам
Суспензия
клеток
Основание с
вдавленным
центром
Контроль Включение/ Контрольная
вращения выключение лампочка
Рис. 13.5. Перемешиваемая культура. Небольшой мешалоч-
ный флакон, разработанный Techne, объемом 250-1000 мл.
Клеточная суспензия перемешивается маятником, который
вращается в кольцеобразном вдавленном пространстве в
основании сосуда
Боковое
горлышко
Флакон для
перемеши¬
вания
Намагничен¬
ный вра¬
щающийся
маятник
Индуктивно
"управляемая
магнитная
мешалка
13.7. Субкультура
251
4) Требования к полученным клеткам для экспери¬
ментальных и иных целей.
Протокол 13.3. разработан для использования в
упражнении 12 гл. 2. Клеточные линии, культуральные
сосуды и другие параметры могут быть заменены для
того, чтобы соответствовать тем линиям, с которыми
вы работаете.
Суспензионная культура имеет ряд преимуществ
(см. табл. 13.5). Во-первых, продукция и получение
конечного выхода большого количества клеток может
быть достигнута без увеличения площади поверхности
субстрата (см. разд. 26.1). Кроме того, если разведение
культуры постоянно и концентрация клеток сохраня¬
ется также постоянной, то может быть достигнуто ста-
ПРОТОКОЛ 13.3. Субкультивирование
суспензионной культуры клеток
Схема
Отобрать образец суспензии клеток, подсчитать
клетки и пересеять соответствующий объем кле¬
точной суспензии в свежую среду в новом флаконе,
восстанавливая концентрацию клеток до первона¬
чального уровня.
Материалы
Стерильные:
• Исходная культура: HL-60, L1210, или Р388,
7 дней и 10 дней после посева в концентрациях
1х104кл./мл, во флаконах объемом 25-см2
по 1 каждой
• Среда ростовая, например МЕМ с солями Спин-
нера (S-МЕМ) или RPMI1640, с 23 mM NaHC03,
сыворотка теленка 5% 200 мл
• Градуированные пипетки с ватными пробками.
Если пипетки стеклянные, то в ассортименте раз¬
меров 1 мл, 5 мл, 10 мл, 25 мл в квадратном пенале
для пипеток. Пластмассовые пипетки могут быть
индивидуально обернуты и рассортированы по
размерам на подносе
• Пипетка без пробки для отбора среды при помощи
перистальтического насоса или вакуумной линии,
если таковые имеются 1 пенал
• Универсальные контейнеры или центрифужные
пробирки объемом 50 мл.., 4
• Мешалочные флаконы объемом 500 мл с магнит¬
ным маятником-мешалкой (Techne, Bellco)...... 2
• Флаконы для культивирования 5 см2 4
Нестерильные:
• Пипетирующее устройство или груша (см.
рис. 5.5,6.6)
• Шланг, соединяющий принимающий сосуд с ва¬
куумной линией или перистальтическим насосом
(см. рис. 5.1-5.3)
• Этанол 70%, в распыляющей бутылке
• Безворсовые салфетки или тампоны для протирки
• Абсорбирующие бумажные полотенца
• Цилиндр для пипеток, содержащий воду и дезин¬
фектант (см. разд. 5.8.8,7.8.5)
• Ручка-маркер с этанол-нерастворимыми черни¬
лами
• Записная книжка, ручка, протоколы, и т. д.
• Гемоцитометр или электронный счетчик клеток
• Магнитная мешалка
Протокол
1. Подготовьте ламинарный шкаф, принесите реак¬
тивы и материалы, необходимые для выполнения
процедуры, (см. разд. 6.5).
2. Тщательно исследуйте культуру на признаки
ухудшения или заражения. По сравнению с
монослойной культурой этот шаг труднее для
суспензионной культуры, но клетки, которые
находятся в плохом состоянии, характеризуются
уменьшением объема, неправильным строением
и/или степенью детализации. Здоровые клетки
должны быть чистыми и прозрачными, с ядром,
видимым в фазовом контрасте; в статической
культуре часто образуют маленькие агрегаты.
3. Перенесите культуры в стерильную рабочую
зону, возьмите из них образцы и подсчитайте в
образцах количество клеток
4. На основании предварительно описанных кри¬
териев (см. разд. 13.7.1) и на вашем знании куль¬
туры решите, требуется ли субкультивирование.
Если требуется (для упражнения 12 потребуется
субкультура), поступите следующим образом:
5. Перемешайте клеточную суспензию и дисперги¬
руйте любые скопления клеток пипетированием
клеточной суспензии вверх и вниз.
6. Добавьте 50 мл среды в каждый мешалочный
флакон.
7. Добавьте 5 мл среды в каждый из четырех
25-см2 флаконов.
8. Добавьте достаточное количество клеток, чтобы
получить конечную концентрацию 1х105кл./мл
для медленно растущих клеток (время удвоения
36-48 ч) или 2х104кл./мл для быстрорастущей
культуры (время удвоения 12-24 ч). Для клеток,
упоминаемых в материалах, используйте конеч¬
ную концентрацию 1х104кл./мл.
9. Заполните флаконы с культурами газом с 5% С02.
10. Закройте флаконы и поместите в инкубатор.
Положите флаконы горизонтально, как в случае
монослойных культур.
11. Закройте крышками мешалочные флаконы и
поставьте на магнитные мешалки при скорости
вращения 50-100 об./мин в инкубаторе или в
культуральной комнате при 37 °С. Проследите,
чтобы двигатель мешалки не перегревал культу¬
ру. Подложите полистироловый коврик, если это
необходимо. Индукционные мешалки производят
меньшее количество тепла и не имеют движущих¬
ся частей.
252
ГЛАВА 13- СУБКУЛЬТУРА И КЛЕТОЧНЫЕ ЛИНИИ
Таблица 13.6. Запись данных. Питание
Дата Время Оператор
Дата
Клеточная линия
Обозначение
Первичная культура или субкультура
Номер генерации или пассажа
Состояние
Фаза ростового цикла
Внешний вид клеток
Плотность клеток
pH среды (примерно)
Прозрачность среды
Среда
Тип
№ партии
Тип и концентрация сыворотки
№ партии
Другие добавки
Концентрация С02
Другие параметры
бильное состояние культуры, в то время как стабильное
состояние в монослойной культуре практически недо¬
стижимо. Ведение монослойной культуры, по существу,
циклично, в результате чего темп роста и метаболизма
варьирует, в зависимости от фазы цикла роста.
13.7.6. Стандартизация условий
культивирования
Стандартизация условий культивирования — су¬
щественный фактор получения фенотипической
стабильности клеток. Хотя некоторые условия могут
меняться в зависимости от требований эксперимента,
обстоятельств и технологии, однако рутинное ведение
культуры должно придерживаться определенных стан¬
дартных условий.
Среда. Тип используемой среды оказывает влияние
на селекцию различных типов клеток и регулирует их
фенотипические признаки (см. разд. 9.6,10.2.1,10.2.2.,
17.2, 17.7). Следовательно, как только выбрана среда,
следует стандартизовать ее употребление, желательно
от одного поставщика, если среду приобретают готовой
к употреблению.
Сыворотка. Наилучшим методом элиминации
различий, связанных с сывороткой, является исполь¬
зование бессывороточных сред (см. разд. 10.2), хотя, к
сожалению, бессывороточные рецептуры разработаны
не для всех типов клеток. Кроме того, переход на бессы¬
вороточные среды потребует затрат времени и средств.
Составляющие элементы сыворотки (см. разд. 10.5.2)
могут позволить получить более высокую концентра¬
цию клеток и обычно дешевле, чем сама сыворотка или
факторы роста, используемые как добавки, но не могут
обеспечить тот контроль над физиологическим окру¬
жением, который предоставляют бессывороточные сре¬
ды. Если требуется сыворотка, выберите партию (см.
разд. 9.6.1) и используйте эту партию на всех стадиях
поддержания культуры, включая криоконсервацию
(см. разд. 20.2).
Пластик. Большинство из ведущих марок культу¬
ральных флаконов и чашек позволяют получить сход¬
ные результаты, однако могут быть незначительные
различия при использовании пластикового оборудова¬
ния различных компаний (см. разд. 8.1.2). Следователь¬
но, предпочтительно придерживаться какой-то одной
компании, а также одного типа чашек и флаконов.
Ведение клеточной линии. Клеточная линия мо¬
жет изменить свои характеристики, если ведение ее
отличается от стандартного режима (см. разд. 13.6).
Режим поддержания должен быть оптимизирован (см.
разд. 13.7) и затем оставаться постоянным в ходе всех
манипуляций с клеточной линией (см. разд. 13.7.1,
13.7.2, 13.7.3). Культуры следует также заменять раз¬
мораживанием рабочих культур (см. разд. 20.4.2)
13.7.7. Использование антибиотиков
Постоянное использование антибиотиков способствует
случайной контаминации, в частности микоплазмами,
и развитию антибиотикоустойчивых организмов (см.
разд. 9.4.7). Антибиотики также могут влиять на ис¬
следуемые клеточные процессы. Тем не менее могут
быть обстоятельства, для которых контаминация очень
распространена либо ведутся очень дорогостоящие
13.7, Субкультура
253
Таблица 13.7. Запись данных. Субкультивирование
Дата Время Оператор
Дата
Клеточная линия
Обозначение
№ пассажа или генерации
Состояние
до субкультивиро¬
вания
Фаза клеточного ростового цикла
Внешний вид клеток
Плотность клеток
pH среды (примерно)
Прозрачность
Диссоциирующий агент
Предварительная отмывка
Трипсин
EDTA
Другие
Механическое воздействие
Подсчет клеток
Концентрация после резуспендирова-
ния (Cj)
Объем (Vj)
Выход клеток
(Y=C1xV1)
Выход на флакон
Посев
Номер (N) и тип сосуда (флакон, чаш¬
ка, многолуночный планшет)
Конечная концентрация (CF)
Объем на флакон, чашку или лунку
(VF)
Коэффициент дробления
(Y:CFxVFxN) или количество засеян¬
ных флаконов / количество трипси-
низированных флаконов, если все
флаконы одного размера.
Среда /сыворотка
Тип
№ партии
Тип и концентрация сыворотки
№ партии
Другие добавки
Концентрация С02
Матричное вещество
Например, фибронектин, Matrigel,
коллаген
Другие параметры
клеточные линии, и в этих случаях могут использо¬
ваться антибиотики. В этом случае важно поддержи¬
вать несколько рабочих культур без антибиотиков
для того, чтобы выявить случайное заражение; эти
рабочие культуры могут вестись параллельно, и мож¬
но чередовать периоды введения различных культур
без антибиотиков (см. рис. 13.6), до тех пор пока не
станет возможным постоянное ведение культур без
антибиотиков. Нецелесообразно усваивать этот метод
как постоянно используемый режим, и при наличии
хронической контаминации клетки должны быть унич¬
тожены или заражение ликвидировано.
13.7.8. Ведение документации
Записывайте все детали ведения культур, включая
питание и субкультивирование (табл. 13.6, 13.7), а
также все отклонения или внесенные изменения,
которые производились в режиме поддержания кле¬
точной линии. Все эти моменты должны быть внесены
в базу данных. Такие записи требуются по правилам
GLP (Food and Drug Administration, 1992; Department
of Health and Social Security, 1986; Organisation for
Economic Co-operation and Development, 2004) для
первичных культур (см. разд. 12.3.11), но полезно
254
ГЛАВА 13, СУБКУЛЬТУРА И КЛЕТОЧНЫЕ ЛИНИИ
С антибиотиками Вез антибиотиков
Культу- | ф
ра А —— —
Культу- | р— I
раВ \
Недели 0 2^ 4 6 ^ 8 10
Используйте свободные от антибиотика культуры
для экспериментов, криоконсервации и проверки на
наличие микоплазмы. Экспериментальная культура
может содержать антибиотики до тех пор, пока из¬
вестно, что они не нарушают изучаемых процессов.
Рис. 13.6. Параллельные культуры и антибиотики. Пред¬
ложенная схема для поддержания параллельных культур
клеток с антибиотиками и без так, что каждая культура всегда
выращивается некоторое время в отсутствии антибиотиков
ввести это в лабораторную практику в отношении
всех культур в любой лаборатории. Если определены
стандарты ведения культур, то записи могут быть
ограничены пометкой «питание» или «субкульти¬
вирование» с датой и маркировкой линии. Любые
комментарии относительно визуального анализа, а
также отклонения от стандартного ведения должны
быть записаны.
Весь комплект записей составляет часть ведения
клеточной линии, и все данные, или, по крайней мере,
все основные элементы, т. е. изменение добавок к среде,
клонирование линии, трансфецирование или переход
к бессывороточной среде, должны быть внесены в базу
данных.
Глава 14
КЛОНИРОВАНИЕ И СЕЛЕКЦИЯ
В двух предыдущих главах было продемонстрировано,
что главной периодически повторяющейся проблемой
в культуре ткани является сохранение специфических
типов клеток и их специализированных свойств. Хотя
условия окружающей среды, несомненно, играют зна¬
чительную роль в поддержании дифференцированных
свойств специализированных клеток в культуре (см.
разд. 17.1,17.7), селективный избыточный рост неспе¬
циализированных клеток, а также клеток «неправиль¬
ных» линий, остается большой проблемой.
14.1. КЛОНИРОВАНИЕ КЛЕТОК
Традиционным микробиологическим подходом к ре¬
шению проблемы гетерогенности популяции является
выделение чистого клеточного штамма путем клониро¬
вания, однако хотя этот метод достаточно легко приме¬
ним для постоянных клеточных линий, его успешность
в отношении первичных культур ограничена низкой
эффективностью клонирования. Тем не менее клони¬
рование первичных культур может быть достаточно
эффективно, например для клеток Сертоли [Zwainet
al., 1991], юкстагломерулярных [Muirhead et al., 1990] и
гломерулярных [Тгоуег & Kreisberg, 1990] клеток почек,
овалоцитов из печени [Suh et al., 2003], сателлитных
клеток скелетных мышц [Zeng et al., 2002; McFarland
et al., 2003; Hashimoto, 2004], а также отдельные диф¬
ференцированные лини, полученные из популяции
зрелых стволовых клеток [Young et al., 2004].
Еще одной важной проблемой культивирования
линий, полученных из нормальных тканей, является
то, что они способны выживать только в течение не¬
скольких поколений (см. разд. 3.8, 18.4.1), и к тому
времени, когда клон может продуцировать достаточное
количество клеток, он может уже находиться на стадии
старения (рис. 14.1). Хотя клонирование постоянных
клеточных линий более успешно, чем клонирование
конечных клеточных линий, при их наращивании для
получения большого количества клеток для использо¬
вания может обнаружиться значительная гетероген¬
ность клона (см. разд. 18.3, рис. 18.1, и цв. вкладку 7).
Тем не менее клонирование может помочь снизить
гетерогенность культуры.
Клонирование также используется для оценки
выживания клеток (см. разд. 21.10, 22.3.3), для опти¬
мизации условий роста (см. разд. 10.6, 11.6.3) и для
определения химио- и радиочувствительности (см.
протокол 22.3).
Клонирование прикрепленных культур может вес¬
тись в чашках Петри, многолуночных планшетах, фла¬
конах, и в этом случае относительно легко различить
отдельные колонии. Микроманипулирование является
единственным достоверным методом для определения
чистоты клональности (т. е. происхождения клона из
единственной клетки), однако когда симметричные
колонии получаются из суспензии единичных клеток,
в частности, если формирование колоний контроли¬
руется на ранних стадиях, то существует достаточно
высокая возможность того, что колонии являются
клонами.
Клонирование также может проводиться в суспен¬
зии путем посева клеток в гель, такой как агар или
агароза, либо в вязкий раствор, например метилцел-
люлоза (МЦ) с агаровой или агарозной подложкой.
Стабильность геля или вязкость МЦ позволяет рас-
Число удвоений популяции
Рис. 14.1. Изменение числа клеток при клонировании. От¬
ношение числа клеток в клоне к числу удвоения популяции;
например, для того, чтобы произвести 106 клеток требуется
20 периодов удвоений популяции
256
ГЛАВА 14. КЛОНИРОВАНИЕ И СЕЛЕКЦИЯ
считывать на то, что дочерние клетки не отрываются от
колонии при их образовании. Даже при монослойном
клонировании некоторые клеточные линии, такие как
HeLa-S3 и СНО, слабо прикреплены и клетки могут
отсоединяться от колоний и образовывать дочерние
колонии, которые могут привести к неверной оценке
эффективности посева. Этот фактор можно миними¬
зировать путем клонирования в МЦ без подложки,
позволив клеткам оседать на пластиковую поверхность
роста. Гематопоэтические клетки обычно клонируют
в суспензии; в зависимости от используемых клеток
и факторов роста колонии продуцируют in vivo и in
vitro недифференцированные клетки с высокой эф¬
фективностью репопуляции, либо они могут дозреть
до колоний дифференцированных гематопоэтических
клеток с очень низкой эффективностью репопуляции.
Клонирование гематопоэтических клеток становится
способом исследования репродуктивного потенциала
и идентичности стволовых клеток.
В связи с трансформированным состоянием, пос¬
тоянные клеточные линии обычно имеют высокую
эффективность посева в монослое и суспензии, в то
время как нормальные клетки, которые могут иметь
умеренную эффективность клонирования в монослое,
имеют очень низкую эффективность клонирования
в суспензии из-за их потребности в прикреплении к
субстрату и распластывании до начала вхождения в
клеточный пролиферативный цикл. Это различие поз¬
воляет использовать метод клонирования в суспензии
в качестве изучения наличия и степени трансформации
клеток (см. разд. 18.5.1).
Клонирование с разведением [Puck & Marcus, 1955]
является методом, который используется наиболее
широко. Оно основано на том наблюдении, что клетки,
разведенные до определенной плотности, формируют
отдельные колонии. Количественные данные и инс¬
трукции, приведенные в протоколе 14.1, соответствуют
упражнению 20, однако могут варьировать в зависи¬
мости от индивидуальных потребностей.
ПРОТОКОЛ 14.1. Клонирование
с разведением
Схема
Посеять клетки при низкой плотности и инкубиро¬
вать до формирования колоний. Окрасить клетки
(для оценки эффективности посева и исследования
выживания; см. цв. вкладку 6а, д) или изолировать
их, и размножьте в клеточный штамм, если их ис¬
пользуют для селекции (рис. 14.2)
Материалы
Стерильные или асептически подготовленные:
• Клетки СНО с поздней bg-фазой, во флаконе
25-ст2
• Среда Хэма F12, в состоянии равновесии 5% СО2,
10% FBS 250 мл
• Трипсин, 0,25%, 1:250, или эквивалент 10 мл
• Пипетки, 1,5,10, и 25 мл по 10 пгг
каждого объема
• Чашки Петри, 6 см, 1 упаковка 20 шт
• Пробирки, или универсальные контейнеры, для
разведения 4-12 шт
• (число зависит от заключительного шага)
Нестерильные:
• Гемоцитометр или электронный счетчик клеток
Протокол
1. Трипсинизируйте клетки (см. протокол 13.2),
чтобы получить суспензию единичных клеток.
При недостаточной трипсинизации могут остать¬
ся агрегаты клеток, избыточная трипсинизация
уменьшит жизнеспособность клеток, однако
фундаментальным положением для обеспечения
клонирования является требование разведения
суспензии до единичных клеток. При проведе¬
нии клонирования новой линии клеток впервые
может быть необходимо опробовать различные
режимы трипсинизации — продолжительности
и различных рецептов (см. табл. 13.4), чтобы оп¬
ределить оптимальный режим, обеспечивающий
максимальную эффективность посева.
2. В то время как клетки трипсинизируются, прону¬
меруйте чашки (со стороны дна) и отмеряйте среду
для разведения растворения. Может потребоваться
уменьшить регулярный монослой для достижения
концентрации, подходящей для клонирования.
3. Когда клетки приняли сферическую форму и
начали отделяться, разведите монослой в 5 мл
среды, содержащей сыворотку или ингибитор
трипсина.
4. Подсчитайте клетки и разведите клеточную
суспензию до концентрации 1x10s кл./мл, затем
до концентрации, которая даст примерно 50 ко¬
лоний на чашке Петри (см. табл. 14.1).
Для СНО-К1 это составит ~ 10 кл./мл, и разве¬
дение будет состоять из следующих шагов:
а) Разведите суспензию до 1х10?кл./мл (прибли¬
зительно 1:10 или 1:20, в зависимости от числа
клеток во флаконе).
б) Разведите 200 мкл суспензии 1х105/мл до
20 мл (1:100), чтобы получить 1х103кл./мл.
в) Разведите 200 мкл суспензии 103 кл./мл до
20 мл (1:100), чтобы получить 10 кл./мл.
Если вы желаете внести изменения в условия
клонирования, например изменить диапа¬
зон концентраций сыворотки, использовать
различные сыворотки или факторы роста, то
подготовьте ряд пробирок в соответствии с ва¬
шими планами и добавьте 200 мкл суспензии
клеток 1х103кл./мл в каждую из пробирок.
При первом клонировании клеток рекомен¬
дуемый диапазон для определения эффектив¬
ности посева составляет 10,20,50,100,200 и
2000 кл./мл (см. протокол 21.10)
14.2. Стимуляция эффективности посева
257
5. Посейте на 3 чашки Петри по 5 мл каждой среды,
содержащей клетки, полученной на заключитель¬
ной стадии разведения. Является также жела¬
тельным произвести посев на чашки суспензии с
концентрацией клеток 2000 кл./мл для контроля
наличия клеток в суспензии, чтобы подтвердить
их присутствие, если не произойдет формирова¬
ние клонов при более низких концентрациях.
6. Поместите чашки в прозрачный пластмассовый
контейнер.
7. Поместите коробку во влажный СОг-инкубатор
или наполните газом запечатанный контейнер
(2-10% С02, см. разд. 9.3.2).
8. Оставьте культуры в покое в течение 1 недели.
Если колонии сформировались:
а) Для испытания эффективности посева (см.
также протокол 21.10) окрасьте и подсчитайте
колонии (см. протокол 16.3 и цв. вкладку 6).
б) Для клональной селекции изолируйте отдель¬
ную колонию (см. протокол 14.8).
Если нет видимых колоний, замените среду
и продолжите культивирование в течение
следующей недели. Смените среду еще раз
и снова культивируйте их в течение третьей
недели, если необходимо. Если колонии не
появляются к 3 неделям, то маловероятно, что
они появятся вообще.
Замена среды. Поскольку плотность клеток при
клонировании очень низкая, необходимость замены
среды культуры на чашках спорна. Замена среды,
главным образом, связана с недостатком питательных
веществ (таких как глютамин), которые нестабильны, а
также заменой факторов роста, которые разрушаются.
Тем не менее при каждой манипуляции возрастает риск
контаминации, поэтому есть смысл оставить чашки на
две недели без вмешательства и без дополнительного
внесения питательных веществ. Если необходимо оста¬
вить чашки на третью неделю, то среда (или, по крайней
мере, ее половина) должна быть заменена.
Преимущественное формирование колоний в
центре чашки может быть результатом неправильного
посева как посева клеток в центр чашки, в которой уже
содержится среда, так и образования воронки в чашке,
так что клетки собираются в центре. Однако это может
Таблица 14.1. Связь между посевной плотностью
и эффективность посева
Ожидаемая
эффектив¬
ность посева
(%)
Оптимальное количество клеток для посева
На мл
На см2
На чашку,
6 см
На чашку,
9 см
0,1
1х104
2x103
40 000
100 000
1,0
1х103
200
4000
10 000
10
100
20
400
1000
50
20
4
80
200
100
10
2
40
100
быть также связано с резонансом в инкубаторе (см.
разд. 8.2.5).
Микротитрационные планшеты. Если основной
целью клонирования является выделение колоний,
то посев на микротитрационные планшеты может
иметь некоторые преимущества (рис. 14.3). При росте
клонов выделение их достаточно легко производить,
хотя следует вести постоянный мониторинг посевов
на ранних стадиях, чтобы отметить те лунки, в которых
будут развиваться единичные клоны. Статистическая
вероятность того, что в лунке будет единичный клон,
может возрастать при снижении плотности посева до
такого уровня, чтобы предполагалось развитие клонов
только в 1 из 5 или 10 лунок на планшете; т. е. в соответ¬
ствии с табл. 14.1,100 кл./мл при 10% эффективности
посева дадут 10 колоний /мл, либо 1 колонию на 0,1 мл.
Если посевную концентрацию снижают до 10 кл./мл,
то теоретически только 1 из 10 лунок будет содержать
колонию, и возможность присутствия более, чем одной
колонии, в одной лунке очень низка.
14.2. СТИМУЛЯЦИЯ ЭФФЕКТИВНОСТИ
ПОСЕВА
Когда клетки посеяны при низкой посевной концентра¬
ции, то степень выживания снижается в большинстве
линий, за несколькими исключениями. Обычно это
не представляет большой проблемы, если речь идет о
постоянных клеточных линиях, для которых эффектив¬
ность посева редко падает ниже 10%, но для первичных
культур и конечных клеточных линий эффективность
посева может упасть достаточно низко — до 0,5-5%
или даже до нуля. Было сделано множество попыток
повышения эффективности посевов, основанных на
том предположении, что клеткам при низкой плотности
требуется большее количество питательных веществ в
связи с их потерей при утечке через мембраны. Предпо¬
лагается также, что сигнальные молекулы, выделяемые
клетками, или факторы кондиционирования, которые
присутствуют в культурах с высокой плотностью, от¬
сутствуют либо имеют слишком низкую концентрацию
в культурах с низкой плотностью. Внутриклеточные
метаболиты, выходящие из клеток через мембрану в
плотной суспензии, вскоре достигнут равновесия с
окружающей средой, но для отдельных клеток дости¬
жение этого равновесия невозможно. Этот принцип
был положен в основу разработки капиллярного метода
Sanford et al. [1948], при помощи которого был получен
клон L-клеток L929. Ограничения объема в капилляре
позволяет клеткам создать локальное микроокружение,
которое напоминает суспензию с высокой концентра¬
цией. В микрокапельной технологии, разработанной
позже, клетки были посеяны в микрокапле под жидкий
парафин, что опять же позволяло создавать достаточно
высокую концентрацию клеток, удерживать одну фор¬
мирующуюся колонию отдельно от другой и облегчая
последующее выделение отдельных клонов. При улуч-
258
ГЛАВА 14. КЛОНИРОВАНИЕ И СЕЛЕКЦИЯ
Трипсинизируйте монослой, ресуспендируйте
клетки и подсчитайте
Сделайте серию разведений от 10
до 200 кл./мл, в зависимости от
ожидаемой эффективности посева
/
Инкубируйте 1-3 недели, в
зависимости от скорости роста
Оцените для будущих разведений,
если число клеток слишком велико
Окрасьте для подсчета числа колоний
для оценки эффективности посева и
исследования выживания
I
•t
-Ш
Фазово-контрастное изображение малого
увеличения (объектив 4х) клеток клона NRK
после приблизительно 1 недели культивирования
Макрофотография с окраской по
Гимза, клетки клона HeLa-S3 после ~
2 недель культивирования
Рис. 14.2. Клонирование с помощью разведения. Клетки получены из трипсинизированной монослойной культуры. Было
подсчитано число клеток и проведено разведение, достаточное для получения изолированных колоний. Когда колонии сфор¬
мированы, они могут быть изолированы (см. рис. 14.7; 14.8). Если выделение не требуется, и клонирование выполняется для
количественного испытания (см. протоколы 21.10, 22.3), колонии фиксируются, окрашиваются и подсчитываются (см. также
цв. вкладку 6а, ё)
шении качества среды удалось повысить эффектив¬
ность посева, поэтому Puck and Marcus [1955] смогли
продемонстрировать, что клонирование клеток при
помощи простого разведения (как описано в протоко¬
ле 14.1) с использованием фидерного слоя облученных
мышиных фибробластов (см. протокол 14.3) позволяет
получить приемлемую эффективность клонирования,
хотя последующее выделение клонов требует трипси¬
низации внутри изолирующего кольца, отделяющего
каждую колонию (см. протокол 14.6).
14.2.1. Условия, которые улучшают
клональный рост
1) Среда. Выбирайте богатую среду, такую как среда
Хэма F12 или оптимизированную среду для того
типа клеток, которые вы используете (например,
MCDB 110 [Наш, 1963] для человеческих фиброб¬
ластов, среда Хэма F12 или MCDB 302 для культу¬
ры СНО [Наш, 1963; Hamilton & Ham, 1977]) (см.
разд. 9.6,10.5, табл. 10.1,10.2 и гл. 23).
14.2. Стимуляция эффективности посева
259
о
• •о
• ••
О##
• ••
• ••
• о#
• ••
о
• •
о#
• •
#о
ОФ
• •о
• ••
Посейте клетки при низкой
плотности, например
10 кл./мл в многолуночные
планшеты, 0,1 мл/лунка,
чтобы обеспечить
вероятность присутствия <
1 клетки на лунку
Отметьте, в каких лунках
имеется только одна клетка
Проверьте очаговый рост
Проверьте наличие роста по
падению pH и визуально с
помощью микроскопа
О
• ••••
• ••••
• •••©
• ••••
• ©
• •
Выберите подходящие
ячейки, в которых
предполагается
клонирование, и
трипсинизируйте
Посейте во флакон. Если
число клеток низко или
клетки медленно растут,
посейте их на дальнем краю
флакона
Трипсинизируйте начиная с дальнего края флакона и
т положите горизонтально, чтобы посеять на большей
' поверхности
Рис. 14.3. Клонирование в планшетах для микротитрации.
Клетки посеяны в достаточно низкой концентрации, чтобы
получить вероятность концентрации < 1 клетки/лунку, чтобы
в некоторых лунках была лишь 1 клетка, которая может быть
обнаружена визуально при исследовании с помощью микро¬
скопа через несколько часов после посева. За теми лунками,
которые отмечены, наблюдайте, и если обнаружится падение
pH, которое указывает на рост колонии, подтвердите ее нали¬
чие микроскопическим наблюдением. Колонию изолируют
трипсинизацией
2) Сыворотка. В тех случаях, когда требуется сыворот¬
ка, обычно лучше использовать фетальную бычью
сыворотку, а не телячью или лошадиную. Выберите
ту партию для экспериментов с клонированием, ко¬
торая при испытании дала высокую эффективность
посева.
3) Гормоны. Обнаружено, что концентрация гормонов
1хЮ“10МЕ/мл повышает эффективность посева
некоторых типов клеток [Hamilton & Ham, 1977].
Растворимый синтетический аналог гидрокорти¬
зона, дексаметазон, в концентрации 2,5х10-5М,
10 мкг/мл улучшает эффективность посева миоб-
ластов цыпленка, нормальных глиальных клеток
человека, глиомы, фибробластов и меланомы и
повышает клональный рост (размеры колоний),
если через отмыть его через пять дней после посева.
[Freshney et al., 1980а, Ь]. Обнаружено, что для эпи¬
телиальных клеток предпочтительны более низкие
концентрации (например, 1х10~7 М) (см. разд. 23.2.1,
23.2.4., 23.2.7).
4) Промежуточные метаболиты. Оксо-кислоты
(раньше известные как кето-кислоты) — напри¬
мер пируват или а-оксоглутарат (а-кетоглутарат)
[Griffiths & Pirt, 1967; McKeehan & McKeehan, 1979]
и нуклеозиды [а-МЕМ; Stanners et al., 1971] — ис¬
пользуются как добавки к среде и всегда включа¬
ются в состав богатых сред, таких как среда Хэма
F12. Пируват также добавляется в среду Игла в
модификации Дюльбекко [Dulbecco & Freeman,
1959; Morton, 1970].
5) Двуокись углерода. С02 является существенным
элементом при получении максимальной эффек¬
тивности посева для большинства клеток. Хотя
обычно используется 5% С02, для многих клеток
лучше 2% концентрация, для человеческой глии
и фибробластов возможно даже несколько лучше
пониженная концентрация С02. HEPES (20 мМ)
может использоваться с 2% С02, что защищает
клетки от колебаний pH при смене среды и в
случае нарушения работы системы подачи С02.
Использование 2%-го С02 также снижает расход
газа. Если клетки, наоборот, требуют повышенного
содержания С02, можно использовать основную
среду Игла в модификации Дюльбекко (DMEM),
которая обычно находится в равновесии с 10%-й
концентрацией С02 и часто используется для кло¬
нирования гибридом миеломы для продукции
моноклональных антител. Концентрация бикар¬
боната должна быть согласована с давлением С02,
если последняя меняется, равновесие достигается
при pH 7,4 (см. табл. 9.1).
6) Обработка субстрата. Полилизин улучшает
эффективность посева для человеческих фиброб¬
ластов при низких концентрациях сыворотки
[McKeehan & Ham, 1976а] (см. разд. 8.4):
а) Добавьте 1 мг/мл поли-Б-лизина в UPW в чашках
(~5 мл/25 см2)
б) Удалите и промойте чашки 5 мл D-PBSA на 25 см2.
Чашки могут быть сразу использованы или поме¬
щены на хранение на несколько недель до исполь¬
зования.
Фибронектин также улучшает эффективность по¬
сева многих клеток [Barnes and Sato, 1980]. Чашки
могут быть предварительно обработаны 5 мкг/мл
фибронектина, разведенного в среде.
7) Трипсин. Очищенный (дважды перекристаллизо-
ванный) трипсин, используемый в концентрации
0,05 мкг/мл, предпочтительнее, чем неочищенный
трипсин, но мнения исследователей разделяются.
260
ГЛАВА 14. КЛОНИРОВАНИЕ И СЕЛЕКЦИЯ
McKeehan [1977] отмечал повышение эффектив¬
ности посева при трипсинизации (с очищенным
трипсином) при температуре 4 °С. Введение реком¬
бинантного трипсина TrypZean™ (Sigma), TrypLE™
(GIBCO) также позволяет использовать более
высоко очищенный трипсин неживотного проис¬
хождения; Invitrogen утверждает, что TrypLE™
(GIBCO) повышает эффективность посева клеток
А549 (с сывороткой) и MDCK (без сыворотки).
14.2.2. Кондиционированные среды
Среда, которая использовалась для роста других кле¬
ток, содержит метаболиты, факторы роста и продукты
матрикса, оставшиеся от этих клеток. Такие среды
называются кондиционированными и могут повышать
эффективность посева некоторых клеток, если доба¬
вить их в обычные ростовые среды.
Замечание. Центрифугирование, замораживание и
оттаивание, а также фильтрация позволяют избежать
риска переноса любых клеток, помимо тех, которые
культивируются. Если для кондиционирования и для
клонирования используются одни и те же клетки, то эта
проблема менее важна, однако для лучшего клонирова¬
ния может быть лучше использовать различные типы
клеток или первичную культуру мышиных фиброб¬
ластов. Если клеточный штамм получен этим методом,
то идентичность клеток должна быть подтверждена
(например, см. протокол 16.10, 16.12,16.12) для того,
чтобы предупредить перекрестную контаминацию из
кондиционированной среды.
ПРОТОКОЛ 14.3. Приготовление фидерных
слоев
Схема
Посеять гомологичные или гетерологичные клетки,
например эмбриона мыши, перевести их в непроли-
феративное состояние облучением или воздействием
препарата в средней концентрации до начала клони¬
рования исследуемых клеток.
Материалы
Стерильные или асептически подготовленные:
• Вторичная культура фибробластов 13-дневного
эмбриона мыши
• (см. протоколы 12.6 и 13.2)
• Культуральная среда для клеток, которые будут
клонированы
• Основной раствор митомицина С, 100 мкг/мл, в
HBSS или бессывороточной среде
• Источник Х-лучей или ^Со, способный к из¬
лучению 30 Грэй за 30 мин или менее (вместо
митомицина С)
Протокол
1. Трипсинизируйте первичную культуру эмбрио¬
нальных фибробластов (см. протоколы 12.6
и 13.2) и пересейте клетки при концентрации
1х105кл./мл.
2. Блокируйте пролиферацию одним из следующих
четырех методов:
а) облучение
i) подвергните культуры in situ во флаконах
действию радиоактивного излучения в дозе
60 Грэй при помощи рентгена или источни¬
ка у-радиации типа ®°Со;
ii) подвергните трипсинизированную суспен¬
зию действию радиоактивного излучения
в дозе 60 Грэй при помощи рентгена или
источника у-радиации типа ^Со. Посейте
при концентрации клеток 1x105 кл./мл.
Альтернативно облученная суспензия мо¬
жет храниться при 4 °С до 5 дней.
б) обработка митомицином С
ПРОТОКОЛ 14.2. Приготовление
кондиционированной среды
Схема
Собрать среду от гомологичных клеток или клеток
другой линии, с поздней log-фазы. Профильтровать
и развести в свежей среде, как это требуется.
Материалы
• Клетки для кондиционирования: та же самая
линия клеток, другая клеточная линия (напри¬
мер, ЗТЗ клетки) или фибробласты мышиного
эмбриона (см. протоколы 12.1,12.5,12.6)
• Среды для клонирования: среда Хэма F12, с 10%
FBS или соответствующая клеткам, которые бу¬
дут клонироваться.
• Стерилизующие фильтры: 0,45 мкм или 0,22 мкм,
флаконы для фильтрования
Протокол
1. Вырастите кондиционирующие клетки до 50%
конфлюэнтности.
2. Замените среду, и инкубировать еще 48 ч.
3. Соберите среду.
4. Центрифугируйте среду при 1000 g в течение
10 мин.
5. Профильтруйте среду через 0,4 мкм стерилизу¬
ющий фильтр (может быть, сначала потребуется
очистить среду предварительной фильтрацией
через фильтры 5 мкм и 1,2 мкм; см. прото¬
кол 11.14).
6. Храните среду замороженной при -20 °С.
7. Разморозьте среду перед использованием и до¬
бавьте ее к среде для клонирования в следующей
пропорции: 1 часть кондиционирующей среды к
2 частям среды для клонирования
14.3. Суспензионное клонирование
261
3.
4.
i) добавьте митомицин С к субконфлюент-
ным клеткам в концентрации 0,25 мкг/мл,
инкубируйте при 37 °С в течение ночи
(-18 ч) [Macpherson и Bryden, 1971], заме¬
ните среду.
ii) добавьте митомицин С до конечной
концентрации 20 мкг/мл к трипсини-
зированной суспензии с концентрацией
клеток 1х107 кл./мл (2 мкг митомицина
С/106 клеток), инкубируйте 1 ч при 37 °С,
отмойте центрифугированием (4x10 мл),
чтобы удалить митомицин С,
Пересейте в 1x10^ кл./мл [Стэнли, 2002]
или храните при 4 °С до 5 дней.
После дальнейшего культивирования 24-72 ч,
трипсинизируйте клетки и пересейте или пере¬
сейте хранившиеся клетки непосредственно в
свежую среду при 5х104 кл./мл (1х104кл./см2).
Инкубируйте культуру еще 24-48 ч и затем пе¬
ресейте клетки для клонирования.
<^=
Инкубируйте 1-3 недели,в
зависимости от скорости роста
14.2.3. Фидерные слои
Облучите фидерные
клетки или подействуйте
митомицином С
Трипсинизируйте и посейте е
флаконы или чашки Петри
Инкубируйте 48 ч для
получения субконфлюэнтной
культуры
Посейте разведенную
суспензию клеток
на предварительно
образованный фидерный слой
Причина того, что некоторые клетки плохо клонируют¬
ся, связана с их неспособностью выживать при низкой
клеточной плотности. Одним из путей поддержания
клеток при клоногенной плотности, имитируя при
этом условия окружающей среды, соответствующие
высокой плотности, является метод клонирования
клеток на фидерном слое клеток с подавленной проли-
феративной активностью (рис. 14.4). Фидерные клетки
могут предоставлять питательные вещества, факторы
роста и компоненты матрикса, которые облегчают
выживание клонируемых клеток.
Фидерные клетки останутся жизнеспособными еще
в течение 3 недель, но, в конечном счете, погибнут и
не могут быть перенесены при выделении отдельной
колонии. Для повышения эффективности посева мо¬
гут быть использованы другие клеточные линии или
гомологичные клетки. Гетерологичные клетки имеют
то преимущество, что при дальнейшей необходимости
выделения клона хромосомный анализ позволяет ис¬
ключить случайную контаминацию клетками фидерно¬
го слоя. В качестве фидерного слоя обычно используют
такие культуры, как ЗТЗ, MRC-5, STO. Мышиные
эмбриональные клетки первого пассажа могут проду¬
цировать большее количество компонентов матрикса,
чем установившиеся клеточные линии, но скрининг
различных клеток — единственный способ выяснения
того, какой из типов клеток является наилучшим в
качестве фидерного слоя для конкретной культуры.
Клетки могут различаться по их чувствительности к
облучению или митомицину С, поэтому следует прово¬
дить испытания до начала попыток клонирования, для
того чтобы удостовериться, что ни одна из фидерных
клеток не выжила (см. также гл. 2, упражнение 21,
Экспериментальный вариант 3). Даже в этом случае
каждый раз при проведении клонирования в качестве
контроля желательно посеять на две или три чашки с
фидерным слоем только фидерные же клетки.
14.3. СУСПЕНЗИОННОЕ КЛОНИРОВАНИЕ
Некоторые клетки, в частности гематопоэтические
стволовые клетки, а также трансформированные
вирусами фибробласты, легко клонируются в сус¬
пензии. Для того чтобы удержать делящиеся клетки
вместе и предупредить смешивание колоний, клетки
суспендируют в агаре или метил целлюлозе (МЦ) и
сеют на агаровую подложку или в чашки, которые
не должны быть предварительно обработаны для
тканевых культур. Протокол 14.4 предоставлен Магу
Freshney, Британский центр исследований рака, Бри¬
танские лаборатории Битсона, Университет Глазго
(Cancer Research UK Beatson Institute, Garscube Estate,
Switchback Road, Bearsden, Glasgow, G61 1BD, United
Kingdom), (см. рис. 14.5. и протокол 23.23).
Колония клеток
цервикального эпителия
Дегенерация ЗТЗ клеток
фидерного слоя
Рис. 14.4. Фидерные слои. Клетки облучены и трипсиниз-
рованы (или вначале трипсинизированы и затем суспензия
облучена, или клетки обработаны митомицином С) и посеяны
при низкой плотности, чтобы повысить эффективность кло¬
нирования (фото любезно предоставлено M.G. Freshney)
262
ГЛАВА 14. КЛОНИРОВАНИЕ И СЕЛЕКЦИЯ
ПРОТОКОЛ 14.4. Клонирование в агаре
Схема
Агар является жидкостью при высоких температурах,
но переходит в гель при 37 °С. Клетки суспендируют¬
ся в теплой агаровой среде и при инкубировании пос¬
ле формирования геля образуют дискретные колонии,
которые могут быть легко изолировано (рис. 14.5).
Материалы
Стерильные:
• Высококачественный agar, Difco
• Среда в двойной концентрации (например,
F12XaMa, RPMI 1640, МЕМ Дулбекко, или
CMRL 1066). Приготовьте среду из концентрата
х10 в половине конечного объема и добавляйте
вдвое большее количество нормальной концент¬
рации сыворотки, если требуется.
• Фетальная сыворотка теленка (если требуется)
• Среда для роста, 1х, для разведения клеток
• Стерильная сверхчистая вода (UPW)
• Стерильная коническая колба
• Пипетки, включая одноразовые стерильные плас¬
тмассовые пипетки для растворов агара
• Универсальные контейнеры, флаконы-бижу или
центрифужные пробирки для разведения
• Чашки Петри, 3,5 см, сорт «не д ля культур ткани»
Нестерильный:
• Бунзеновская горелка и штатив
• Водяная баня на 55 °С
• Водяная баня на 37 °С
• Электронный счетчик клеток или гемоцитометр
• Поднос
Примечание. Перед подготовкой среды и клеток
выполните разведение клеток и маркировку чашек
Петри. Для измерения эффективности клонирова¬
ния клеточной линии клетки приготовить по три
чашки для каждого разведения клеток. Удобное
количество клеток в 3,5-см чашке: 1 ООО, 333,111, и
37 клеток — то есть серия разведений третьей части
суспензии клеток. Если в чашки должны быть до¬
бавлены любые факторы роста, гормоны или другие
добавки, они должны быть внесены в 0,6%-ю агаро¬
вую подложку.
Протокол
1. Пронумеруйте или промаркируйте чашки Петри со
стороны дна. Удобно разместить их на подносе.
2. Приготовьте 2х среду, содержащую 40% FBS, и
храните при 37 °С.
3. Взвесьте 1,2 г агара.
4. Отмерьте 100 мл стерильной UPW в стерильной
конической колбе и другие 100 мл в стерильной
бутыли. Добавьте 1,2 г агара в колбу. Закройте
флакон и кипятите раствор 2 мин. Агар может
быть стерилизован в автоклаве заранее, но если
он приготовлен заранее, то для использования
потребуется кипятить или разогреть в микровол¬
новой печи до расплавления.
5. Переместите прокипяченный агар и бутыль со
стерильной UPW в водяную баню на 55 °С.
6. Приготовьте 0,6%-ю агаровую подложку, объ¬
единяя равный объем 2х среды и 1-2% агар
(рис. 14.6). Поставьте агаровую подложку на
37 °С. Если используются любые добавки, фак¬
торы роста, гормоны и т.д., они должны быть
добавлены к подложке в этом пункте.
Примечание. Если проведена титрация факторов
роста или используется выбор различных факторов,
то следует добавлять требуемое количество в чашки
Петри до разливания подложки
7. Добавьте 1 мл 0,6%-й агаровой среды в чашки,
перемешайте, удостоверьтесь, что среда закры¬
вает дно чашки. Оставьте чашки при комнатной
температуре для охлаждения (см. рис. 14.5).
8. Приготовьте суспензию клеток и подсчитайте
клетки.
9. Приготовьте 0,3%-й агар и поместите на 37 °С.
Эта среда может быть подготовлена разведением
2х среды (при 37 °С) в 1-2% агаре (при 55 °С) и
UPW (при 55 °С) в соответствующих объемах
2:1:1 (см. рис. 14.5).
10. Приготовьте следующие разведения клеток, делая
высшую концентрацию клеток
1х105/мл
а) 1х105/мл
б) Разведите 1х105/мл в три раза так, чтобы по¬
лучить 3,3x104/мл
в) Разведите 3,3x104/мл в три раза так, чтобы
получить 1,1х104/мл
г) Разведите 1,1х104/мл в три раза так, чтобы
получить 3,7x103/мл
И. Промаркируйте четыре флакона-бижу или про¬
бирки, по одной для каждого разведения, внесите
пипеткой по 40 мкл каждого разведения клеток,
включая концентрацию 1х105/мл в соответству¬
ющую емкость. Добавьте 4 мл 0,3% агара при
37 °С в каждый объем, перемешайте, перенесите
пипеткой по 1 мл из каждого флакона на три
чашки Петри (рис. 14.5). Это даст следующие
конечные концентрации:
а) 1х103/мл/чашку
б) 30/мл/чашку
в) 10/мл/чашку
г) 7/мл/чашку
Примечание. Всегда удостоверяйтесь, что агар для
верхнего слоя имел достаточное время, чтобы охла¬
диться до 37 °С перед добавлением к нему клеток.
13. Поместите чашки Петри в чистую пластмассовую
коробку с крышкой и инкубируйте при 37 °С в
течение 10 дней.
14.3. Суспензионное клонирование
263
Разведение
клеточной суспензии
1:100 с 0,3% агар-
средой и посев
Серийное разведение для
получения стократной
величины требуемой конечной
концентрации клеток
2. Подготовка агаровой среды
3. Подготовка и посев клеток
н
1. Подготовка агаровой подложки
Подсчет клеток
из суспензион¬
ной культуры или
костного мозга
или
Трипсинизация клеток
монослоя и подсчет
Агаровая подложка
Хранить при 4 °С
Образование колоний клонов Клоны человеческой
через 1-3 недели метастазирующей меланомы
Рис. 14.5. Клонирование в суспензии. Культивированные клетки или первичная суспензия костного мозга или опухоли
суспендируется в агаре или агарозе с «низкой температурой таяния», которые образуют гель с формированием колоний в
суспензии. Использование подложки предупреждает прикрепление клеток ко дну чашки. У. Подготовка агаровой подложки:
агар 1,2%, при 55 °С смешать со средой 2х, подогретой до 37 °С и немедленно разлить в чашки, где при комнатной температуре
или при 4 °С образуется гель 2. Подготовка агар-среды: развести агар до 1,2% в UPW при 55 °С и смешать со средой 2х, чтобы
получить 0,3% агар для клонирования. Использование агарозы с низкой точкой плавления позволяет работать при 37 °С, но
использование агарозы может затруднить образование геля. 3. Клетки, которые растут в суспензии, полученные из костного
мозга, или трипсинизированные клетки монослоя подсчитываются и серийно разводятся. Конечный продукт разводится в
агаре или агарозе и сеется на агаровую подложку. 2. Подготовка агар-среды
264
ГЛАВА 14. КЛОНИРОВАНИЕ И СЕЛЕКЦИЯ
ПРОТОКОЛ 14.5. Клонирование
в метилцеллюлозе
Схема
Суспендировать клетки в среде, содержащей ме-
тилцеллюлозу (МЦ), посеять клетки в чашки,
содержащие агаровую или агарозную подложку
(рис. 14.6).
Материалы
Стерильные:
• Высококачественный агар иЛи агароза, Difco
• Среда в двойной концентрации (например, F12
Хэма, RPMI 1640, МЕМ Дулбекко, или CMRL
1066). Приготовить среду из концентрата 10х
в половине конечного объема и добавить вдвое
большее количество нормальной концентрации
сыворотки, если требуется.
• Фетальная сыворотка теленка (если требуется)
• Среда для роста, 1х, для разведения клеток
• 1,6% МЦ, 4 Pa-s (4000 сантипуаз) в UPW; хранит¬
ся на льду (см. Приложение I Methocel)
• Стерильная сверхчистая вода (UPW)
• Стерильная коническая колба
• Пипетки, включая одноразовые стерильные пласт¬
массовые пипетки для растворов агара
• Универсальные контейнеры, флаконы-бижу, или
центрифужные пробирки
• Чашки Петри, 3,5-см, сорт «не для культур
ткани»
• Шприцы для дозировки МЦ (из-за своей вязкос¬
ти МЦ прилипает к внутренней стороне пипеток,
делая разлив затруднительным и неточным)
Нестерильные:
• Бунзеновская горелка и штатив
• Водяная баня на 55 °С
• Водяная баня на 37 °С
• Электронный счетчик клеток или гемоцитометр
• Поднос
Протокол
1. Приготовьте агаровую подложку, как в протоко¬
ле 14.4, шаги 1-7.
2. Растворите МЦ до 0,8% с равным объемом 2х
среды. Хорошо перемешайте и храните на льду.
3. Трипсинизируйте клетки монослоя или соберите
клетки суспензионной культуры или костного
мозга и подсчитайте их.
4. Приготовьте следующие разведения клеток, де¬
лая высшую концентрацию клеток 1х105/мл
а) 1х105/мл
б) Разведите 1х105/мл в три раза так, чтобы по¬
лучить 3,3х104/мл
в) Разведите 3,3х104/мл в три раза так, чтобы
получить 1,1х104/мл
г) Разведите1,1х104/мл в три раза так, чтобы
получить З,7х103/мл
5. Промаркируйте четыре флакона-бижу или про¬
бирки, по одной для каждого разведения, внесите
пипеткой по 40 мкл каждого разведения клеток,
включая концентрацию 1х105/мл в соответству¬
ющую емкость. Добавьте 4 мл 0,8% МЦ в каждый
объем, хорошо перемешайте при помощи вортек-
са, если известно, что клетки хрупкие и нежные,
то перемешивать многократным осторожным
засасыванием и выдавливанием из шприца. Затем
используйте шприц, чтобы перенести из каждого
флакона в три чашки Петри (рис. 14.6). Это даст
следующие конечные концентрации:
а) 1х103/мл/чашку
б) 330/мл/чашку
в) 110/мл/чашку
г) 37/мл/чашку
6. Инкубируйте чашки во влажном инкубаторе до
формирования колоний. Поскольку колонии
формируются в промежутке между агаром и МЦ,
через 1 неделю может быть добавлена свежая
среда, 1 мл на чашку, а затем через 2 недели среда
может быть снова удалена и заменена на новую
без повреждающего действия на колонии.
Агароза, имеющая в составе меньшее количество
сульфатированных полисахаридов, может заменить
агар. Некоторые типы агарозы имеют более низкую
точку гелеобразования, что позволяет работать при
температуре 37 °С. Они загустевают при 4 °С и затем
снова переносятся на 37 °С.
В результате сложности манипуляций с растаплива¬
нием агара, содержащего клетки, а также некоторыми
примесями в агаре, некоторые лаборатории предпочи¬
тают МЦ (Methocel), который представляет собой не
гель, а вязкий раствор [Buick et al., 1979]. В подогретом
виде он имеет более высокую вязкость. Поскольку это
золь, а не гель, клетки легко могут в нем осаждаться.
Таким образом, при использовании МЦ необходимо
использовать подложку. Колонии образуются на гра¬
нице раздела между МЦ и агаровой (или агарозной)
подложкой, помещаясь в одной фокальной плоскости,
что облегчает их анализ и фотографирование.
Многие из рекомендаций, которые относятся к
компонентам сред, используемых для клонирования
на монослое, также применимы и к клонированию в
суспензии. Кроме того, иногда также используются
сульфгидрильные компоненты, такие как меркапто-
этанол (5х10-5 М), глютатион (1тМ) и а-тиоглицерин
(7,5х10“5М) [Iscove et al., 1980]. Macpherson [1973]
обнаружил, что включение декстрана DEAE оказы¬
вает положительное влияние на клонирование, что
позже было подтверждено Hamburger et al. [1978],
который также обнаружил, что макрофаги улучшают
клонирование опухолевых клеток, хотя другие авторы
показали, что макрофаги, напротив, повреждают их.
Courtenay et al. [1978] вносили красные клетки крови
14.4. Выделение клонов
265
Серийное разведение клеток до 100 ООО и 3 700 кл./мл, в
зависимости от ожидаемой эффективности посева
100,000/мл
33,000/мл
11,000/мл
3,700/мл
-J Е
1 В 0
1 1
Среда 2х
ЛЕЕЙ
. J L ■
а
1,6% раствор
метилцеллюлозы
Разведение клеток
1:100 в пробирках,
содержащих
0,8% раствор
метилцеллюлозы в
среде 1х; посев на
чашки
< Д
0,8% раствор
метилцеллюлозы в среде 1х
- 0,6% агаровая или агарозная подложка
Рис. 14.6. Клонирование в метилцеллюлозе. Серийное разведение клеток приготовлено так же, как для клонирования в агаре,
клетки разведены 1:100 в метилцеллюлозе-среде и посеяны в чашки с агаровой подложкой
крысы в среду и демонстрировали, что низкое давление
кислорода улучшает клонирование. Проблема влияния
содержания кислорода может быть связана также и с
токсичностью свободного кислорода, в присутствии
красных клеток крови он может связываться с гемог¬
лобином. Вероятно, этот процесс можно смоделировать
перфторуглеродом [Lowe et al., 1998].
Большинство различных типов клеток клонируются
в суспензии с более низкой эффективностью, чем в мо¬
нослое, некоторые клетки на два или три порядка ниже.
Однако выделение клонов из суспензии намного легче.
14.4. ВЫДЕЛЕНИЕ КЛОНОВ
Когда клонирование используется для селекции спе¬
цифических клеточных штаммов, то формируемые
колонии (см. цв. вкладку 6) должны быть изолированы
для дальнейшего размножения. Если клетки моно¬
слоя клонируются непосредственно в многолуночных
планшетах (см. протокол 14.1 — Микротитрационные
планшеты), то колонии могут быть изолированы трип-
синизацией отдельных ячеек. Так или иначе, необходи¬
мо подтвердить клональное происхождение колонии в
ходе ее формирования путем регулярного наблюдения
под микроскопом. Если клонирование проводится в
чашке Петри, то между колониями не происходит раз¬
деления. Это разделение должно быть осуществлено
удалением среды и помещением нержавеющих или
керамических колец вокруг колоний, которые предпо¬
лагается изолировать (рис. 14.7).
Могут быть применены флаконы с удаляемой верх¬
ней пленкой (Nalge Nunc), которая может отслаиваться
для того, чтобы получить клоны. В тех случаях, когда
доступен источник облучения, клоны могут быть вы¬
делены экранированием одной колонии при помощи
свинцового диска и облучением остального монослоя
в дозе 30 Грэй (см. протокол 14.7).
Рис. 14.7. Кольца для клонирования. Нержавеющие сталь¬
ные кольца, отрезанные от трубки, в 9-см стеклянной чашке
Петри. Может быть использован также фарфор (Fisher), тол¬
стостенный стекляный стакан (Scientific Laboratory Supplies)
или пластик (например, кольца, отрезанные от нейлонового,
силиконового или тефлонового толстостенного шланга).
Независимо от материала, нижний край кольца должен быть
гладким, чтобы с помощью силиконовой смазки приклеить
ко дну чашки Петри, а размер внутреннего диаметра должен
быть достаточно широк, чтобы обеспечить клонирование
одного клона без перекрывания смежного клона внешним
диаметром кольца
Если облучение и трипсинизация проводятся, когда
колония состоит примерно из 100 клеток, то повторное
клонирование трипсинизированных клеток приведет
к значительному повышению эффективности клони¬
рования. Этим методом за шесть недель может быть
осуществлено три серийных клонирования.
266
ГЛАВА 14. КЛОНИРОВАНИЕ И СЕЛЕКЦИЯ
ПРОТОКОЛ 14.6. Выделение клонов с
использованием колец для клонирования
Схема
Колонию трипсинизируют внутри фарфорового,
стеклянного, стального или PTFE кольца и перено¬
сят в одну из лунок 24- или 12-луночного планшета
либо прямо в 25-см2 флакон (рис. 14.8).
Материалы
Стерильные:
• Кольца для клонирования (см. Приложение И);
стерилизованные в чашке Петри сухим жаром
или автоклавированием
• Силиконовая смазка; стерилизованная в стек¬
лянной чашке Петри сухим жаром, 160 °С в те¬
чение 1 часа или автоклавированием при 121 °С
в течение 15 мин.
• Желтые наконечники для автоматических пи¬
петок либо Пастеровские пипетки с загнутым
концом (Ве11со#1273)
• Трипсин 0,25% в D-PBSA
• Ростовая среда
• Многолуночный планшет, 24-луночный, и/или
25-см2 флаконы
• Стерильные пинцеты
Нестерильные:
• Автоматическая пипетка, 50-100 мкл
• Фломастер или, предпочтительно, маркер для
колец Nikon или маркер для объектов, который
подходит к револьверу микроскопа вместо одного
из объективов
Протокол
1. Обследуйте клоны, отметьте те из них, которые
вы хотите выделить при помощи фломастера на
дне чашки или используйте маркер колец Nikon.
2. Удалите среду из чашки, осторожно промойте
клоны D-PBSA.
3. При помощи стерильного пинцета возьмите одно
кольцо для клонирования, погрузите в силиконо¬
вую смазку и вдавите в чашку так, чтобы смазка
распространилась вокруг основания кольца.
4. Поместите кольцо вокруг выбранной колонии.
5. Повторите шаги 4 и 5 для двух или трех колоний
в одной чашке.
6. Добавьте 0,25%-й трипсин так, чтобы заполнить
внутреннее пространство в кольцах (0,1-0,4 мл, в
зависимости от внутреннего диаметра кольца).
7. Оставьте трипсин на 20 с и затем удалите его.
8. Закройте чашку, инкубируйте в течение 15 мин
при 37 °С.
9. Добавьте 0,1-0,4 мл среды в каждое кольцо.
10. По очереди для каждого клона диспергируйте
клетки многократным пипетированием среды,
перенесите среду в 24-луночный планшет или в
25-см2 флакон, поставленный вертикально. Ис¬
пользуйте для каждого клона отдельную пипетку
или новый наконечник.
11. Промойте кольцо в 0,1-0,4 мл среды, перенесите
среду в ту же лунку планшета или флакон.
Замечание. Чашка высохнет, если оставлять ее
открытой в течение слишком долгого времени. Огра¬
ничьте число выбранных клонов или закрывайте
чашку в промежутках между манипуляциям.
12. Доведите количество среды в лунке до 1,0 мл, за¬
кройте планшет и инкубируйте его. При использо¬
вании флакона добавьте 1 мл среды в каждый фла¬
кон и инкубируйте в вертикальном положении.
13. Когда клон вырастает так, что заполняет лунку,
проведите субкультивирование в 25-см2 флакон,
инкубируйте, как обычно, в 5 мл среды. Если вы ис¬
пользуете метод культивирования в вертикально
стоящих флаконах (см. рис. 14.8), то удалите среду,
когда дно флакона будет покрыто конфлюэнтным
слоем, трипсинизируйте клетки и положите фла¬
коны плашмя. Продолжайте инкубирование.
ПРОТОКОЛ 14.7. Выделение клеточных
колоний путем облучения
Схема
Переворачивать флакон в рентгеновском аппарате
или в камере ^Со излучателя, защитив выбранные
колонии свинцом
• Требование безопасности. Рентгеновский
аппарат и ^Со излучатель должны использоваться
при соответствующем контроле соблюдения безопас¬
ности для оператора и находящихся рядом лиц (см.
разд. 7.7.4.). Свяжитесь с организацией, отвечающей
за радиологическую защиту в вашем регионе, прежде
чем приступать к экспериментам этого рода.
Материалы
Стерильные:
• Ростовая среда
• Трипсин 0,25%
• D-PBSA
Нестерильные:
• Рентгеновский аппарат или ^Со излучатель
• Кусочки свинца, вырезанные из пластины толщи¬
ной 2 мм размерами от 2 до 5 мм в диаметре
Протокол
1. Выберите желательную колонию, отметьте ее
фломастером или маркером для колец Nikon.
2. Выберите кусочек свинца соответствующего
диаметра.
3. Внесите флакон в зону облучения.
4. Переверните флакон под излучателем.
5. Закройте колонию кусочком свинца толщиной 2 мм.
6. Облучите флакон в дозе 30 Грэй.
7. Верните флакон в стерильную рабочую зону.
8. Удалите среду, трипсинизируйте клетки, оставь¬
те культуру для восстановления в том же фла¬
коне, используя облученные клетки в качестве
фидерного слоя.
14.4. Выделение клонов
267
Клетки внутри кольца
трипсинизируются и переносятся
во флакон, стоящий вертикально
Когда клетки образуют монослой
в нижней части флакона,
трипсинизируйте монослой и
положите флакон горизонтально
Удалите среду из чашки
Стерильная силиконовая смазка
Отмеченная колония
Погрузите кольца
для клонирования в
силиконовую смазку
и поместите вокруг
колонии
Стерильные кольца для клонирования
Исследуйте под микроскопом,
выберите и отметьте колонию
(колонии)
Рис. 14.8. Выделение монослойного клона. Материнские колонии исследуются под микроскопом, подходящая выбранная
колония отмечается. Среду удаляют, кольца для клонирования, погруженные в силиконовую смазку, помещаются так, чтобы
окружить каждую выбранную колонию, которая затем обрабатывается трипсином внутри кольца
14.4.1. Другие методы выделения
монослойных клонов
1) Расположите небольшие покровные стекла или
кусочки разбитого покровного стекла на дне чашки
Петри. Когда плотность клеток достигнет необхо¬
димой плотности, некоторые одиночные колонии
будут находиться на кусочках покровного стекла и
могут быть перенесены в свежую среду или в лунки
многолуночного планшета.
2) Используйте капиллярный метод Sangford и др.
[1948]. Разведенная клеточная суспензия пере¬
носится в стерильную стеклянную капиллярную
трубочку (например, 50-мкл Drummond Microcap),
позволяет колониям формироваться внутри капил¬
ляра. Затем капилляр аккуратно разбивают с обеих
сторон от колонии и переносят на свежую среду.
Этот метод использовали Echarti и Maurer [1989,
1991] для изучения клоногенности гематопоэти-
ческих клеток и опухолевых клеток, для которых
эффективность колониеобразования может быть
количественно определена путем сканирования
капилляра в денситометре.
3) Клетки могут быть клонированы в камере Opti-
Cell, которая сделана из двух расположенных друг
напротив друга поверхностей для роста на тонком
гибком пластике, который может быть отрезан при
помощи скальпеля или ножниц. Если обеспечена
стерильность наружных поверхностей, то из камеры
вырезают необходимый сегмент, содержащий коло-
268
ГЛАВА 14. КЛОНИРОВАНИЕ И СЕЛЕКЦИЯ
ПРОТОКОЛ 14.8. Выделение суспензионных
клонов
Схема
Отобрать колонию автоматической пипеткой или
пастеровской пипеткой, перенести во флакон или
лунку многолуночного планшета (рис. 14.9).
Материалы
Стерильные:
• Ростовая среда в универсальном контейнере
• Многолуночные планшеты (24 лунки)
• Культуральные флаконы, 25 см2
• Желтые наконечники для автоматической пипет¬
ки или пастеровские пипетки
Нестерильные
• Препаровальная лупа, увеличение 20-50х
• Автоматическая пипетка, 100 мкл
• Фломастер или маркер для колец Nikon
Протокол
1. Обследуйте чашки в инвертированном микро¬
скопе, отметьте колонии маркером Nikon или
фломастером.
2. Внесите пипеткой по 1 мл среды в каждую лунку
24-луночного планшета.
3. Выберите колонии, используя препаровальную
лупу.
а) Используйте отдельный наконечник или пас¬
теровскую пипетку для каждой колонии;
б) Установите на пипетке пипетируемый объем
100 мкл;
в) Наберите примерно 50 мкл среды в наконеч¬
ник, поместите кончик пипетки напротив
колонии, которую выделяете, и осторожно
отберите колонию.
4. Перенесите содержимое пипетки в 24-луночный
планшет, смойте колонию струей среды. Если
среда приготовлена с использованием метилцел-
люлозы Methocel, колония осядет, если клетки
адгерентны, то они прилипнут и начнут расти.
Клетки, обычно растущие в суспензии, осядут,
но, конечно, не прилипнут. Если среда сделана на
агаре, вам, возможно, потребуется пипетировать
колонию несколько раз в лунке, чтобы дисперги¬
ровать агар. Клоны можно также посеять прямо в
пластиковые флаконы, поставленные вертикаль¬
но (см. протокол 14.6).
нии, которые затем трипсинизируются и переносят¬
ся в многолуночные планшеты или флаконы.
14.4.2. Суспензионные клоны
Выделение колоний, выросших в суспензии, проще, но
требует применения препаровальной лупы.
14.5. ПОЛУЧЕНИЕ РЕПЛИКАТИВНЫХ
КОЛОНИЙ
Бактериальные колонии могут быть реплицированы
путем мягкого придавливания влажной подушечки к
колонии, растущей на питательном агаре, и перене¬
сением этой подушечки на другую, свежую чашку с
агаром. Предпринимались различные попытки адапта¬
ции этой методики для культур клеток, обычно путем
помещения влажного экрана или фильтра на монослой
клона на несколько дней и переноса его в новую чашку
[Hornsby et al., 1992]. Для клонов, которые образова¬
лись в многолуночных планшетах, существует ряд при¬
способлений, например Coming Transtar (см. рис. 5.13),
которые могут быть использованы для суспензионных
культур непосредственно после перемешивания или
для монослойных культур, после трипзинизации и
ресуспендирования.
14.6. СЕЛЕКТИВНЫЕ ИНГИБИТОРЫ
Манипуляция с условиями культивирования путем
использования селективных сред является стандарт¬
ным методом селекции микроорганизмов. Приложение
этого метода к клеткам животных имеет ограничения,
однако на основании метаболического сходства боль¬
шинства изолированных клеток одного животного
можно определить некоторые пищевые потребности
их культур. Проблему усложняет действие сыворотки,
которое склонно маскировать селективные свойства
селективных сред.
Большинство селективных сред, в отношении кото¬
рых обычно подтверждается успешность применения,
относятся к бессывороточным (см. разд. 10.2.1). Тем
не менее периодически демонстрируется успешность
применения ряда метаболических ингибиторов.
Gilbert and Migeon [1975, 1977] заместили L-валин в
культуральной среде D-валином и показали, что на
этой среде росли преимущественно клетки, облада¬
ющие D-аминокислотной оксидазой. Таким образом
были выделены клетки эпителия почечных канальцев
[Gross et al., 1992], эпителия молочной железы коровы
[Sordillo et al., 1988], эндотелиальные клетки кры¬
синого мозга [Abbott et al., 1992], а также культуры
Шванновских клеток [Armati & Bonner, 1990]. Однако
этот метод не слишком эффективен для выделения
человеческих фибробластов [Masson et al., 1993].
Большинство из попыток разработки селективных
условий имели целью подавление избыточного роста
фибробластов. Као and Prockop [1977] использовали
ds-OH-пролин, хотя он может оказаться токсичным
для других клеток. Fry and Bridges [1979] обнаружили,
что фенилэтилбарбитуровая кислота ингибирует из¬
быточный рост фибробластов в культуре гепатоцитов,
Braaten et al. [1974] удалось уменьшить контаминацию
фибробластами культуры неонатальных панкреати¬
ческих клеток путем воздействия на культуру этил-
меркуртиосалицилатом натрия. Фибробласты также
14,6, Селективные ингибиторы
269
Отберите среду в
наконечник пипетки
Поместите пипетку напротив
колонии и осторожно втяните
колонию в пипетку
Перенесите колонию
во флакон со средой и
диспергируйте колонию
Когда количество клеток
возрастет, положите флакон
горизонтально и увеличьте объем
среды, прикрепившиеся клетки
трипсинизируйте
Й
<=|
Рис. 14.9. Выделение суспензионных клонов. Отметьте колонию, как в случае монослоя, затем отберите при помощи пас¬
теровской пипетки (или наконечника автоматической пипетки). Перенесите колонию в культуральный флакон, разведите в
среде и инкубируйте ее. Откорректируйте среду, когда клетки начнут расти
несколько более чувствительны к действию генетицина
(G418) в дозе 100 мкг/мл [Levin et al., 1995].
Одним из наиболее перспективных методов является
разработка моноклональных антител к стромальным
клеткам карциномы молочной железы человека [Ed¬
wards et al., 1980]. Показано, что при использовании
вместе с комплементом эти антитела становятся цито-
токсичными для фибробластов из различных опухолей и
помогают очистить ряд клеточных линий злокачествен¬
ных опухолей. Клетки также могут быть избирательно
убиты при помощи конъюгатов антител с токсинами или
фармакологическими соединениями [например, Beattie
et al., 1990]. Тем не менее селективные антитела более
широко применяются при «пэннинге» или в сепаратив¬
ном методе магнитного сортинга (см. разд. 15.3.2).
Селективные среды также обычно используются
для выделения гибридных клонов в экспериментах с
гибридизацией соматических клеток. НАТ-среда (см.
Приложение I) — комбинация гипоксантина, амино-
птерина и тимидина, — позволяет проводить селекцию
гибридов, обладающих гипоксантин гуанин фосфори-
бозилтрансферазу и тимидинкиназу от родительских
клеток, дефицитных по одному или другому ферменту
(см. разд. 27.9.1) [Littlefield, 1964а].
Трансфецированные клетки также отобраны по ус¬
тойчивости к ряду лекарственных соединений, таким как
неомицин, его аналог генетицин (G418), гидромицин и
метотрексат, путем включения в генетическую конструк¬
цию для трансфекции генов устойчивости [например, пео
(аминогликозидфосфотрансфераза), hph (гидромицин В
фосфотрансфераза), или dhfr (дигидрофолат редуктаза);
см разд. 28.5]. Культивирование при правильной кон¬
центрации селективных маркеров, определенных путем
титрации против трансфецированного и нетрансфеци-
270
ГЛАВА 14. КЛОНИРОВАНИЕ И СЕЛЕКЦИЯ
рованного контроля, позволяет проводить селекцию
стабильных трансфектантов. Селекция с метотрексатом
имеет дополнительное преимущество — возрастание
концентрации метотрексата приводит к амплификации
гена dhfr, а также может приводить к ко-амплификации
других генов генетической конструкции.
Возможна также негативная селекция при помощи
Ж-гена вируса простого герпеса (HSV), который пере¬
водит ганцикловир (Ganciclovir, Syntex) в цитотокси-
ческий продукт [Jin et al., 2003]. Трансфецированные
клетки будут более чувствительными к препарату.
Когда в смеси клеток обнаруживается различная
чувствительность к факторами роста, становится
возможным стимулировать один клеточный тип со¬
ответствующим фактором роста и затем, используя
преимущества более высокой чувствительности более
быстро растущих клеток, селективно уничтожить эти
клетки облучением или действием арабинозида цито¬
зина (ara-с) (см. разд. 23.4.1). Напротив, если известен
некий ингибитор или удаление какого-то фактора роста
ингибирует рост клеток, то, выведя один тип клеток
из клеточного цикла, можно уничтожить оставшиеся
клетки облучением или ага-с.
14.7. ВЫДЕЛЕНИЕ ГЕНЕТИЧЕСКИХ
ВАРИАНТОВ
Протокол 14.9 для получения мутантных клеточных
линий, которые амплифицируют ген дигидрофолатре-
дуктазы (DHFR), разработан June Biedler (Memorial
Sloan-Kettering Cancer Center, New York, New York).
ПРОТОКОЛ 14.9. Метотрексат-
резистентность и DHFR-амплификация
Принцип
Клетки, которые подвергаются действию супенчато
возрастающих концентраций антагонистов фолие¬
вой кислоты, таких как метотрексат (МТХ), после
продолжительного периода времени приобретают
устойчивость к токсическому действию препарата
[Biedler et al., 1972]. Резистентность, возникшая в
результате амплификации гена DHFR, обычно раз¬
вивается быстрее, хотя другие механизмы, например,
изменения в транспорте антифолата и/или мутации,
действующие на структуру или аффинность фер¬
мента, могут давать частично или полностью тот же
резистентный фенотип.
Схема
Клетки подвергаются ступенчатому воздействию
МТХ по возрастанию концентраций в течение не¬
скольких недель с периодическим замещением среды
на свежую, с той же концентрацией препарата. Для
субкультивирования выбирают те флаконы, в кото¬
рых небольшой процент клеток выжил и образовал
колонии. Повторное субкультивирование таких кле¬
ток проводится при такой же или в два или в десять
раз более высокой концентрации МТХ до тех пор,
пока клетки не приобретут желаемую степень резис¬
тентности.
Материалы
Стерильные:
• Клетки китайского хомяка или быстро растущих
человеческих или мышиных клеточных линий
• Метотрексат натрия для парентерального введе¬
ния (Lederle Laboratories)
• 0,15 MNaCl
• Флаконы для культуры тканей
• Пипетки
• Культуральная среда, которая не содержит тими-
дина и гипоксантина (например, среда Игла МЕМ
с 10% фетальной сыворотки теленка)
Нестерильные
• Инвертированный микроскоп
• Морозильник с жидким азотом
Протокол
1. Клонируйте родительские клеточные линии для
получения быстро растущих, генотипически
единообразных популяций, которые будут ис¬
пользованы для селекции.
2. Разведите МТХ в стерильном 0,15 М (0,85%)
NaCl. Вещество расфасовывается для клиниче¬
ского использования в растворе с концентрацией
2,5 мг/мл.
3. Внесите 2,5х 105 клеток в серию флаконов 25-см2,
не содержащих препарата, или содержащих 0,01,
0,02, 0,05 и 0,1 мкг/мл МТХ в полной тканевой
культуральной среде. Доведите pH каждого рас¬
твора до 7,4, инкубируйте флаконы до 37 °С в
течение 5-7 дней.
4. Обследуйте культуру с помощью инвертирован¬
ного микроскопа. Замените среду на свежую, с
той же концентрацией МТХ в культурах, обнару¬
живающих клональный рост малой доли клеток
среди большого количества разросшихся, адге-
зировавших к субстрату и, возможно, погибших
клеток; повторно инкубировать эти культуры.
5. Оставьте клетки расти еще на 5-7 дней, заменяя
ростовую среду по необходимости, но сохраняя
постоянную концентрацию МТХ. Когда клеточ¬
ная плотность достигнет 2-10x106 кл./флакон,
перенесите клетки в новый флакон с той же или в
два или в десять раз более высокой концентрации
МТХ при 2,5х105кл./флакон.
6. Через 5-7 дней оцените вновь пересеянные
флаконы и культуры из предыдущих пассажей,
которые были подвергнуты действию более вы¬
соких концентраций. Замените среду и выберите
жизнеспособные клетки, как описано выше.
7. Продолжите селекцию с помощью ступенчато
повышаемых концентраций в каждой следующей
14.7. Выделение генетических вариантов
271
субкультуре, пока не будет получен желаемый
уровень резистентности: 2-3 месяца для клеток
китайского хомяка с низкой или умеренной
степенью резистентностью, повышением актив¬
ности DHFR и/или изменением транспорта;
4-6 месяца или более для более высокого уровня
резистентности и сверхпродукции фермента для
клеток хомяка, мышиных или быстро растущих
человеческих линий.
8. Периодически замораживайте образцы разви¬
вающихся линий в жидком азоте (см. прото¬
кол 20.1).
Анализ. Охарактеризуйте резистентные клетки по
уровню резистентности к препарату в исследованиях
клонального роста (см. протокол 22.3), по возрастанию
активности или количества DHFR биохимическим
или электрофоретическим методами [Albrecht et al.,
1972; Meleraet al., 1980], и/или по возрастанию mRNA
и количества копий генов редуктазы при Нозерн-,
Саузерн-, и дот-блоттинге [Scotto et al., 1986] с DHFR-
специфичными зондами для определения механизмов
резистентности.
Вариации. Может использоваться среда для клеточ¬
ной культуры, отличная от МЕМ-среды Игла; предпола¬
гается, что состав среды (например, применение фоли¬
евой кислоты) может оказывать влияние на скорость и
особенности развития МТХ-резистентности. Среды, со¬
держащие тимидин, гипоксантин и глицин (см. табл. 9.3,
10.1, 10.2), предупреждают развитие резистентности к
антифолату и поэтому их следует избегать. До селекции
клетки могут быть обработаны химическими мутагенами
[Thompson & Baker, 1973]; такая обработка может также
изменить скорость и тип селекции мутантов.
Селекцию можно также проводить с клетками, посе¬
янными на среду, содержащую препарат в 10-см чашку
для культивирования тканей при низкой плотности
(с выделением отдельных колоний при помощи колец
для клонирования; см. протокол 14.6), с использованием
единичных клеток в 96-луночном планшете или в мяг¬
ком агаре, что облегчает выделение одной из множест¬
венных клональных популяций на каждом или любом
желательном этапе в ходе развития резистентности.
Аналогичными методами клетки могут быть сдела¬
ны устойчивыми к ряду других агентов, таких как анти¬
биотики, другие антиметаболиты, токсичные металлы
и т. д. Могут иметься различия в механизмах действия
Рис. 14.10. Селективные фидерные слои. Селективное клонирование эпителия молочной железы на конфлюэнтном фидер¬
ном слое, (а) Отдельные колонии, сформированные на пластиковой чашке после посева 4000 кл./см2 (2x104 кл./мл) культуры
эпителиальной карциномы молочной железы. Маленькие, плотные колонии — эпителиальные клетки. Большие звездчатые
колонии — фибробласты. (в) Колонии клеток той же культуры, посеянные при концентрации 400 кл./см2 (2x103 кл./мл) на
конфлюэнтном фидерном слое клеток FHS74Int [Owens et al, 1974]. Эпителиальные колонии намного крупнее, чем в (а),
эффективность посева выше, нет колоний фибробластов. (в) Колонии из различных культур карциномы молочной железы,
помещенные на один и тот же фидерный слой. Отметьте различную морфологию колоний со светлым центром и кольцом в
месте контакта с фидерным слоем, (г) колонии из культуры нормального эпителия молочной железы, посеянные на фидерный
слой клеток FHI (fetal human intestine; аналог FHS74Int). В (в) и (г) присутствуют несколько маленьких колоний фибробластов
[Smith et al., 1981, A.J. Hackett, личное сообщение] (см. также цв. вкладку 6в, г)
272
ГЛАВА 14. КЛОНИРОВАНИЕ И СЕЛЕКЦИЯ
или степени токсичности агентов. Может оказаться, что
периодическое воздействие будет более эффективным,
чем постоянное, для селекции может потребоваться
более длительное время.
Клеточные линии различных видов или с более
низкой скоростью роста, такие как некоторые клеточ¬
ные линии человеческих опухолей, могут потребовать
различных (обычно более низких) начальных концен¬
траций препарата, боле длительной экспозиции при
каждой концентрации, и меньшей скорости нарастания
концентрации на каждом этапе селекции.
Солюбилизация МТХ в большей степени, чем сред¬
ство Lederle, потребует добавления эквимолярного ко¬
личества NaOH и стерилизации через 0,2 мкм фильтр.
14.8. ВЗАИМОДЕЙСТВИЕ С СУБСТРАТОМ
14.8.1. Селективная адгезия
Различные типы клеток обладают различной степенью
связывания с культуральным субстратом и прикрепля¬
ются с различной скоростью. Если первичную суспен¬
зионную культуру посеять во флакон и перенести во
второй флакон через 30 мин, а затем в третий флакон
через час и т.д. вплоть до 24 ч, то большинство при¬
крепляющихся клеток будут обнаружены в первом
флаконе, а наименее способные к прикреплению в
последнем. Макрофаги имеют тенденцию оставаться
в первом сосуде, фибробласты в следующих несколь¬
ких флаконах, эпителиальные клетки в следующих
нескольких флаконах, и, наконец, гематопоэтические
клетки в последнем флаконе.
Если в полной среде для первичной дизаггрегации
тканей используется коллагеназа (см. протокол 12.8),
большинство высвобождающихся клеток не будет при¬
крепляться в течение 48 ч, если только коллагеназа не
будет удалена из среды. Тем не менее макрофаги мигри¬
руют из фрагментов тканей и прикрепляются даже в этот
период и могут быть отдалены от других клеток путем
переноса дезагтрегата в свежий флакон через 48-72 ч
действия коллагеназы. Этот метод работает при дезагг-
регации биопсийных образцов опухолей человека.
14.8.2. Селективное открепление
Воздействие трипсина или коллагеназы на гетерогенный
монослой быстрее удалит некоторые клетки, по сравне¬
нию с остальными. Периодическая быстрая обработка
трипсином удалит фибробласты из культуры фетально¬
го кишечника человека [Owens et al., 1974] и кожи [Milo
et al., 1980]. Lasfargues [1973] обнаружил, что действие
коллагеназы на культуру клеток молочной железы в
течение нескольких дней приводит к удалению фиброб¬
ластов и сохранению эпителиальных клеток. EDTA, с
другой стороны, может приводить к высвобождению
эпителиальных клеток быстрее, чем высвобождаются
фибробласты [Paul, 1975]. Действие Dispase II (Boeh-
e 1 ММ
Рис. 14.11. Рост клеток меланомы, фибробластов и глии в
суспензии. Клетки были посеяны в количестве 5x105 клеток
на 35-мм чашку (2,5х105кл./мл) в 1,5% метилцеллюлозу на
1,25% агаровой подложке Колонии были сфотографированы
через 3 недели, а — меланома; б — нормальные фибробласты
кожи эмбриона человека; в — нормальные клетки глии взрос¬
лого человека
ringer Mannheim) приводит к селективному извлечению
эпителиальных слоев из культуры цервикальных клеток
человека, растущих на фидерных слоях клеток ЗТЗ без
разрушения самих ЗТЗ клеток (см. протокол 24.3). Эта
методика может быть эффективна при субкультивиро¬
вании эпителиальных клеток, полученных из различных
источников, для отделения их от фибробластов.
14.8.3. Природа субстрата
Хотя в настоящее время доступны различные источники
ЕСМ (см. Приложение И), остается важной проблема
14.8. Взаимодействие с субстратом
273
обеспечения выживания и дифференцировки клеток.
Еще недостаточно сделано в области селекции клеток
на «сконструированном» матриксе. По литературным
данным, пролиферации эпителиальных клеток способ¬
ствует действие коллагена [Kibbey et al., 1992; Kinsella
et al., 1992], Matrigel также способствует выживанию и
дифференцировке эпителиальных клеток [Bissell et al.,
1987; Ghosh et al., 1991; Kibbey et al., 1992]. Поскольку
действие составных частей среды сейчас лучше изуче¬
но, то смесь различных коллгенов с протеогликанами,
ламинином и другими протеинами матрикса может
быть использована для создания более селективных
субстратов. Селективное действие субстрата на рост
может зависеть от дифференциальной скорости при¬
липания и роста либо от результата их совокупного
действия [Lechner & LaVeck, 1985; Wise, 2002] (см.
протокол 23.5,23.9) и для поддержки эндотелиального
клеточного роста и функционирования [Relou et al.,
1998; Martin et al., 2004].
Пластиковая посуда Primaria (BD Biosciences) имеет
заряд на поверхности, отличный от того, который обще¬
принят для пластика, используемого в культуре тканей.
Он разработан для улучшения роста эпителиальных кле¬
ток по сравнению с фибробластами. BD Biosciences и дру¬
гие производители (см. Приложение II) также предлагает
пластик, покрытый природным или синтетическим мат¬
риксом, который может облегчать рост требовательных
типов клеток, но, возможно, делает это неселективно.
10
|7 -
ю6 -
ф
Q.
>*
!е
£
*
GQ
*
S
О
£ 105
0
ф
т
S
1
104 -
1 1 1 1 1 1 1 1 Г~
Равное количество клеток селектируемой
и контаминирующей культур
Медленно растущие
селектируемые клеткиn
культура\+ Быстро растущие клетки
контаминирующеи культуры
✓ ^Ю^о-е клетки
контаминирующей культуры
0 2 4 6 8 10
Дни культивирования после фракционирования
Рис. 14.12. Рост клеток в смешанной культуре. Замена
медленно растущей клеточной линии быстро растущими
клетками культуры-контаминанта. Этот график изобража¬
ет гипотетический случай, однако показывает, что 10%-я
контаминация клеточной популяцией, которая удваивается
каждые 24 ч уже через 10 дней роста приведет к равному со¬
отношению с клеточной популяцией, время удвоения которой
составляет 36 ч
14.8.4. Селективные фидерные слои
Так же как кондиционирование субстрата (см.
разд. 14.2.3), для селективного роста эпидермальных
клеток также могут быть использованы фидерные слои
[Rheinwald & Green, 1975] (см. протоколы 23.1,23.4) и
для сдерживания сверхроста стромальных элементов
в культуре клеток молочной железы (рис. 14.10; см.
также цв. вкладку 6в, г) и карциномы толстой кишки
[Freshney et al., 1982Ь]. Роль фидерного слоя, возмож¬
но, чрезвычайно сложна; он обеспечивает не только
внеклеточный матрикс для адгезии эпителия, но также
положительно действует на факторы роста и нега¬
тивные регуляторы, инактивирующие TGF-|3 [Maas-
Szabowski & Fusenig, 1996]. Человеческая глиома будет
расти на конфлюэнтных фидерных слоях нормальной
глии, в отличие от клеток, полученных из нормального
мозга [MacDonald et al., 1985] (см. протокол 24.1).
14.8.5. Селекция на полужидкой среде
Трансформация многих культур фибробластов снижает
зависимость клеточной пролиферации от субстрата
(см. разд. 18.5.1) [Macpherson & Montagnier, 1964]. При
культивировании клеток на агаре (см. протокол 14.4)
после вирусной трансформации возможно отделение
колоний трансформированных клеток от нормальных
клеток. Нормальные клетки не будут образовывать
колонии в суспензии с высокой плотностью посева
трансформированных вирусом клеток, хотя при низ¬
кой посевной концентрации они могут образовывать
колонии. Различия между трансформированными и
нормальными клетками не настолько выражены для
быстрорастущих культур с ранними сроками пересе¬
ва, поскольку посевная концентрация в этом случае
должна быть достаточно низкой; нормальная глия и
фетальные фибробласты кожи также способны обра¬
зовывать колонии в суспензии с примерно одинаковой
эффективностью (<1%) (рис. 14.11).
Клонирование клеток и использование селективных
условий имеет значительные преимущества перед фи¬
зическими методами разделения клеток (см. гл. 15), при
этом контаминирующие клетки также полностью эли¬
минируются клональной селекцией или подавляются
постоянной или периодически повторяющимся приме¬
нением селективных условий. Даже наилучшие методы
физического разделения клеток позволяют иметь место
частичному перекрыванию свойств различных популя¬
ций клеток и рекуррентному сверхросту. Стабильное
состояние не может быть достигнуто, и состав культуры
постоянно изменяется. Из рис. 14.12 можно видеть, что
90%-я чистота клеточной линии А при 10% контамина¬
ции ее быстрорастущей клеточной линией В (на 50%
быстрее, чем линия А) в течение 10 дней приводит к
одинаковому содержанию клеток в культуре. Таким
образом, для постоянных клеточных линий в допол¬
нение или вместо физических сепаративных методов
требуется применение селективных условий.
Глава 15
РАЗДЕЛЕНИЕ КЛЕТОК
Хотя клонирование и использование селективных ус¬
ловий культивирования являются предпочтительным
методом при получении культуры (см. разд. 10.2.1,
14.1, 14.6), но в ряде случаев клетки после посева не
дают клонов; или, по каким то причинам, невозможно
создать условия для селекции клеток. Таким образом,
может возникнуть необходимость прибегнуть к физи¬
ческим или иммунологическим методам разделения
клеток. Достоинство этих методов состоит в том, что
они дают более быстрое разделение клеток, по срав¬
нению с клонированием, хотя и с меньшей степенью
гомогенности.
Наиболее эффективные методики разделения
клеток основаны на различиях в 1) плотности клеток
(удельном весе), 2) аффинности антител к эпитопам
клеточной поверхности, 3) размеру клеток и 4) све¬
торассеянию или флюоресценции при сортировке с
помощью проточной цитометрии. Для двух первых ме¬
тодов требуется относительно несложное оборудова¬
ние, и эти методы недорогие, в то время как два вторых
метода высокотехнологичны и требуют значительных
денежных затрат. Для наиболее эффективного разде¬
ления часто используются два или более показателей
для того, чтобы получить высокий уровень чистоты
культуры [смотри, например, Calder et al., 2004; Al-
Mufti et al., 2004].
15.1. ПЛОТНОСТЬ КЛЕТОК
И ИЗОПИКНИЧЕСКАЯ
СЕДИМЕНТАЦИЯ
Разделение клеток по их плотности можно осуществить
путем центрифугирования при низких значениях g,
используя обычное оборудование [Pretlow & Pret-
low, 1989; Sharpe, 1988; Calder et al., 2004; Al-Mufti et
al., 2004]. В градиенте плотности клетки оседают до
равновесного уровня, на котором плотность среды
эквивалентна их собственной плотности (изопикни-
ческая седиментация; рис. 15.1). Среда для разделения
должна быть нетоксичной и невязкой при высоких
плотностях (1,10 г/мл) и не должна влиять на осмо¬
тическое давление раствора. Для этой цели успешно
использовали сывороточный альбумин [Turner et al.,
1967], декстран [Schulman, 1968], метризамид (Nygaard)
[Munthe-Kaas & Seglen, 1974] и перколл (Amersham
Biosciences) [Pertoft & Laurent, 1982]. Наиболее эффек¬
тивными, используемыми в настоящее время средами,
являются перколл (коллоидный кремний) и рентгено¬
контрастные йодированные соединения метризамид
и метризоат. Градиент можно создавать 1) наслаивая
перколл различной плотности пипеткой, шприцем
или с помощью насоса, 2) с помощью специального
градиентатора (рис. 15.2г) или 3) высокоскоростного
центрифугирования (рис. 15.3).
Варианты
Внесение клеток. Клетки могут быть внесены в
градиент в процессе его образования при центрифуги¬
ровании. Хотя при этом требуется только одно центри¬
фугирование, центрифугирование клеток при высоких
значениях g может повредить их. Кроме того, перколл
[Pertoft & Laurent, 1982], Isopaque [Splinteretal., 1978]
и метризамид [Freshney, 1976] могут поглощаться клет¬
ками, поэтому предпочтительней наслаивать клетки на
уже сформированный градиент.
Другие среды. Одной из наиболее распространен¬
ных сред является фиколл, потому что, как и перколл,
его можно автоклавировать. Фиколл обладает немного
более низкой вязкостью при высокой плотности, чем
перколл, что может приводить к агрегации некоторых
типов клеток. Неионное производное метризоата —
метризамид (Nycomed) — йодированное соединение,
использующееся в радиографии (Isopaque, Hypaque,
Renografin) и для выделения лимфоцитов (см. прото¬
кол 27.1), при высоких плотностях обладает меньшей
вязкостью, чем фиколл [Rickwood & Birnie, 1975], но
может использоваться для некоторых типов клеток.
Маркерные гранулы. Amersham Biosciences произ¬
водит цветные маркерные гранулы стандартной плот¬
ности, которые можно использовать для определения
плотности в различных участках градиента.
Изопикническая седиментация происходит быс¬
трее, чем скоростное осаждение с ускорением (см.
разд. 15.2), и дает более высокий выход клеток на
заданный объем градиентной среды. Идеально, когда
клетки четко различаются по плотности (>0,02 г/см3).
На плотность клеток может влиять поглощение клетка¬
ми среды, используемой при формировании градиента,
фаза клеточного цикла (в фазе плато клетки обладают
15.2, Размер клеток и скорость седиментации
275
Градиент плотности в
культуральной среде
Клетки
в среде
Центрифугирование
1-1000 д, 30 мин
Флотационная среда из
перистальтического насоса
Отводная трубка
гг
Элюирование в
мультилуночный планшет
ruijTji_ru«ryi
ы
и
О О G°
Разбавление средой
X
и инкубация
7
У*
г**
Коническая крышка,
навинчивающаяся на
верхнюю часть пробирки
Градиентный коллектор
Рис. 15.1. Разделение клеток по их плотности. Клетки наслаивают на предварительно сформированный градиент (см. рис. 15.2)
и пробирку центрифугируют. Пробирку помещают в градиентный коллектор, и флотационная среда (например, Fluorochemi-
cal FC43) закачивается через входящую трубку на дно пробирки с градиентом, вытесняя градиент и клетки вверх и заставляя
их выходить через отводную трубку в мультилуночный планшет. Клетки разбавляют средой (так, чтобы они осели на дно) и
культивируют. (Градиентный смеситель по оригинальной разработке Dr. G.D. Bimie.)
более высокой плотностью) и сыворотка [Freshney,
1976]. Так как при изопикнической седиментации не
требуется большое ускорение, ее можно проводить
даже при 1 g.
15.2. РАЗМЕР КЛЕТОК И СКОРОСТЬ
СЕДИМЕНТАЦИИ
На осаждение клеток также влияет их размер (площадь
поперечного сечения), который является решающим
фактором при осаждении клеток при 1 g и важнейшим
компонентом осаждения при высоких скоростях и
более высоких значениях g. Хотя соотношение между
размером частиц и скоростью седиментации при 1 g
сложно выразить для частиц субмикронного размера,
для клеток оно достаточно просто и может быть выра¬
жено приблизительно следующим образом:
V *1^/4
[Miller & Phillips, 1969], где v — скорость седиментации
в мм/ч, а г — радиус клетки в мкм (см. табл. 21.1).
15.2.1. Седиментация в поле силы тяжести
Наслаивание клеток на градиент сыворотки в среде
позволяет клеткам оседать в соответствии с вышеп¬
риведенным уравнением. Однако с помощью седимен¬
тации в поле силы тяжести невозможно обработать
большое количество клеток (~ 1x106 кл./см2 площади
276
ГЛАВА 15. РАЗДЕЛЕНИЕ КЛЕТОК
ПРОТОКОЛ 15.1. Разделение клеток
центрифугированием в градиенте плотности
Схема
Сформировать градиент, центрифугировать клетки
в градиенте, собрать фракции, разбавить средой и
культивировать (рис. 15.1),
Материалы
Стерильные:
• Ростовая среда
• Ростовая среда + 20%-й перколл
• Центрифужные пробирки объемом 25 мл
• D-PBSA
• 0,25%-й трипсин
• Шприц или градиентный коллектор
• Планшеты 24-луночные или для микротитро¬
вания
Нестерильные:
• Рефрактометр или ареометр
• Гемоцитометр или счетчик клеток
• Низкоскоростная центрифуга
Протокол
Приготовление градиента плотности
а) Наслаивание
i) Доведите плотность перколла до 1,10 г/см3)и
осмотическое давление до 290 мосмоль/кг.
п) Смешайте перколл и обычную среду в
различных пропорциях, чтобы получить
10 или 20 ступеней в заданном диапазоне
плотности (например, 1,020—1,100 г/см3).
iii)B центрифужной пробирке на 25 мл на¬
слаивайте одну ступень на другую (1 или
2 мл на ступень) пипеткой, шприцем или
с помощью перистальтического насоса,
начиная от раствора с самой высокой плот¬
ностью и формируя ступенчатый градиент.
Можно также (и, возможно, это будет
предпочтительнее) начинать с раствора
с самой низкой плотностью и с помощью
шприца или перистальтического насоса
подслаивать под каждый слой раствор с
постепенно увеличивающейся плотностью.
Градиент можно использовать сразу после
получения или на следующий день.
б) С помощью градиентатора: непрерывный ли¬
нейный градиент можно создать, смешивая,
например 1,020 г/мл перколла с плотностью
1,080 г/ см3 в градиентаторе (Fisons, Pharmacia,
Buchler; см. рис. 15.2).
в) Центрифугирование.
i) Поместите среду, содержащую перколл с
плотностью 1,085 г/см3, в пробирку.
ii) Центрифугируйте 1 ч при 20 000 g.
iii) В результате центрифугирования образу¬
ется сигмоидный градиент (см. рис. 15.3),
форма которого определяется начальной
концентрацией перколла, продолжитель¬
ностью центрифугирования и центро¬
бежной силой, формой пробирки и типом
ротора.
1. Трипсинизируйте клетки и ресуспендируйте их
в среде, содержащей сыворотку или ингибитор
трипсина. Убедитесь, что клетки в суспензии не
образуют агрегатов.
2. С помощью шприца, автоматической пипетки или
сужающейся пипетки наслоите до 2х107 клеток в
2 мл среды на градиент.
3. Пробирку можно оставить в штативе на 4 ч, чтобы
клетки оседали при 1 g, или можно центрифуги¬
ровать при 100-1000 g 20 мин. Если использовать
последнюю процедуру, то скорость центрифуги¬
рования увеличивать постепенно и не применять
торможение при остановке центрифуги.
4. Соберите фракции с помощью шприца или гради¬
ентного коллектора (Fisons; см. рис. 15.1). Фрак¬
ции объемом 1 мл можно собирать в 24-луночный
планшет, объемом 0,1 мл — в планшет для мик¬
ротитрования. Необходимо регулярно отбирать
образцы для подсчета клеток и для определения
плотности (р) среды с градиентом. Плотность
можно измерить рефрактометром (Hilger) или
ареометром (Рааг).
5. Добавьте в каждую лунку равный объем среды и
перемешайте, чтобы клетки осели на дно лунки.
Через 24-48 ч инкубации смените среду для уда¬
ления перколла.
поверхностью верхней части градиента) и не позволяет
хорошо разделить клетки, за исключением случаев,
когда клетки значительно различаются по размеру, а
каждая популяция гомогенна по размеру клеток. Этот
метод используется при больших различиях в размере
клеток или для отделения агрегатов от единичных
клеток, например после расщепления коллагеназой
(см. протокол 12.8).
15.2.2. Элюатриационное
центрифугирование
Большинство методик разделения клеток по их размеру
основаны или на элюатриационном центрифугиро¬
вании (которое дает посредственное разделение, но
высокий выход), или на сортировке клеток (точное
разделение с низким выходом; см. разд. 15.4). Центри¬
фужный элюатриатор (Beckman Coulter) представляет
собой устройство для повышения скорости осаждения
и увеличения выхода и разрешения при разделении
клеток при выполнении разделения в специально
оборудованной центрифуге со специальным ротором
(рис. 15.4) [Lutz et al., 1992]. Суспензия клеток в среде
закачивается в камеру для разделения в роторе при
его вращении. При нахождении клеток в камере цент-
15.3. Методы, основанные на применении антител
277
Рис. 15.2. Устройство для создания градиента. В прозрач¬
ном блоке из твердого пластика вырезаны 2 камеры, соеди¬
ненные между собой тонким каналом, который проходит
через основание камеры и выходит наружу. В наружное
отверстие вставлена трубка с наконечником из нержавею¬
щей стали достаточной длины для того, чтобы доставать до
дна центрифужной пробирки. При закрытом клапане левая
камера заполняется средой с высокой плотностью, правая
камера — средой с низкой плотностью. Клапан 1 открывается,
наконечник из нержавеющей стали помещается на дно про¬
бирки, запускается мешалка, и затем открывается клапан 2.
Когда раствор из левой камеры течет направо, жидкости
смешиваются, и плотность постепенно повышается, пока
смесь течет по трубке
Расстояние от дна пробирки, мм
Рис. 15.3. Градиент, полученный с помощью центрифу¬
гирования. Градиент образуется при центрифугировании
перколла при 20 ООО g в течение 1 ч
робежная сила выталкивает клетки к внешней части
ротора (рис. 15.5). При этом среда прокачивается через
камеру таким образом, чтобы центростремительная
скорость потока находится в равновесии со скоростью
осаждения клеток. Если клетки однородны, то они ос¬
танутся неподвижны, но если они различаются по раз¬
меру, плотности и структуре клеточной поверхности, то
они будут осаждаться при разных скоростях. Благодаря
тому, что седиментационная камера имеет коническую
форму, скорость потока повышается в направлении
внешней части ротора и создается непрерывный гра¬
диент скорости потока. Клетки с различной скоростью
седиментации будут достигать равновесного состояния
в разных участках камеры. Седиментационная камера
освещается источником стробоскопического света, и за
ее содержимым можно наблюдать через специальное
окно. Когда клетки достигают равновесного состояния,
скорость потока повышается, и клетки выкачиваются
в сосуд-приемник. Разделение можно проводить в
полной среде и продолжать культивировать непосред¬
ственно сразу после разделения.
Процедура включает 4 этапа: 1) сборка и стерили¬
зация аппаратуры, 2) калибровка, 3) загрузка образца
и установление равновесных условий и 4) сбор фрак¬
ций. Подробный протокол представлен в инструкции
по работе с элюатриатором (Beckman Coulter). Рав¬
новесие достигается в течение нескольких минут, и
полный цикл занимает 30 мин. За каждый цикл можно
разделить 1x108 клеток, и разделение можно повторять
столько раз, сколько необходимо. Прибор, однако,
достаточно дорогой, и для эффективного разделения
клеток требуется большой опыт работы. С помощью
этого метода можно разделять различные типы клеток
[Teofili et al., 1996; Lag et al., 1996; Yoshioka et al., 1997],
а также клетки на различных стадиях клеточного цикла
[Breder et al., 1996; Mikulits et al., 1997].
15.3. МЕТОДЫ, ОСНОВАННЫЕ
НА ПРИМЕНЕНИИ АНТИТЕЛ
Существует ряд методик, основанных на специфиче¬
ском связывании антитела с клеточной поверхностью.
К ним относятся: иммунный лизис, для которого
используют антитела к нежелательным клеткам, на¬
пример фибробластам в популяции эпителиальных
клеток [Edwards et al., 1980], направленная доставка
цитотоксина с помощью антител [Beattie et al., 1990],
сортировка клеток с активацией флюоресценции (см.
разд. 15.4), иммунный пэннинг и сортировка на маг¬
нитных гранулах с иммобилизованными антителами
(магнитно-активированная сортировка клеток, MACS)
[Saalbach et al., 1997] (см. разд. 15.3.2). Эффективность
этих методов зависит от специфичности выбранных
антител и презентации соответствующих эпитопов на
поверхности живых клеток, которая подтверждается
с помощью иммунологических методов окрашива¬
ния (см. разд. 16.11) или проточной цитометрии (см.
разд. 15.4,16.9.2,21.7.2).
278 ГЛАВА 15. РАЗДЕЛЕНИЕ КЛЕТОК
Из резервуара с
Ротор Крышка центрифуги Окно для наблюдения помощью насоса
Рис. 15.4. Центрифужный элютриаторный ротор (Beckman). Суспензия клеток и жидкость-носитель помещается в центр
ротора и прокачивается в направлении к периферии и далее в периферийную часть камеры разделения. С противоположной
стороны ротора расположен обратный контур для уравновешивания
которые затем можно собрать с помощью механичес¬
кой обработки или мягкой трипсинизации), так и
отрицательный (чтобы удалить ненужные клетки).
15.3.2. Магнитный сортинг
Для магнитной сортировки используют специфиче¬
ские к поверхностным эпитопам клетки антитела,
иммобилизованные на ферритиновых гранулах (Dy-
nabeads производства Dynal; Иллюстрация 23а-с) или
микрогранулах (Miltenyi; Иллюстрация 23d, е). Когда
клеточную суспензию смешивают с гранулами и затем
помещают в магнитное поле (рис. 15.6), клетки, при¬
крепившиеся к Dynabeads, перемещаются к боковой
стенке камеры для разделения. Когда ток отключа¬
ется, клетки и гранулы отсоединяются от стенки, и
далее клетки можно отделить от гранул с помощью
трипсинизации или интенсивного пипетирования.
В системе Miltenyi клетки иммунологически сорби¬
рованы на микропарамагнитных гранулах, которые
могут связываться с ферромагнитными шариками
разделительной колонки при внесении колонки в
магнитное поле (рис. 17.7). С помощью этого метода
разделяют многие клетки, в том числе стволовые,
путем отрицательной сортировки [Bertoncello et al.,
1991], очищают костный мозг от клеток лейкемии
[Trickett et al., 1990] и выделяют эпителий почечных
канальцев [Pizzonia et al., 1991]. Протокол исполь¬
зования Dynabeads доступен на вебсайте Dynal (см.
Приложение III).
15.3.1. Иммунный пэннинг
Для разделения ряда клеток различных типов успешно
используется процесс прикрепления клеток к чаш¬
кам Петри с сорбированными антителами, который
называется иммунный пэннинг [Murphy et al., 1992;
Fujita et al., 2004]. Все многочисленное разнообразие
методик пэннинга проистекает из работы Wysocki and
Sata [1978] с субпопуляциями лимфоцитов. Специфи¬
ческие для каждого типа клеток антитела к эпитопам
клеточной поверхности сорбируются на дне чашки
Петри и затем в чашку добавляют смешанную попу¬
ляцию клеток. Клетки, в отношении которых антитела
специфичны, быстро прикрепляются ко дну чашки.
Остальное затем можно удалить. Можно использовать
как положительный иммунный пэннинг (для того,
чтобы отобрать специфическую субпопуляцию клеток,
Центробежная сила
Центр ротора
<
Край ротора
Направление
вращения
Накопление более крупных t
клеток в области с более
^высокой скоростью г
Накопление более мелких
клеток в области с более
низкой скоростью потока
Центростремительный поток жидкости
Рис. 15.5. Разделительная камера элюатриаторного ро¬
тора
15.3. Методы, основанные на применении антител
279
Рис. 15.6. Магнитный сортинг. Отрицательная сортировка. Коммитированные прогениторные клетки из суспензии костного
мозга связаны с парамагнитными гранулами Dynal с помощью антител к линееспецифическим маркерам. Отрицательные по
этим маркерам стволовые клетки не связаны и остаются в суспензии для дальнейшей сортировки с помощью проточной цито¬
метрии. (я) Внесение пробирки в магнитный штатив. (6) Пробирка сразу оказывается в магнитном поле, (в) Пробирка через
30 с после помещения в магнитный штатив (см. также иллюстрацию 23а)
Использование иммунномагнитных микрогранул
[Gaudernacketal., 1986] (рис. 15.7, Иллюстрация Plate
23d, е) позволяет продолжать культивировать клетки
или проводить дальнейшие процедуры сортировки
без предварительного удаления гранул (Miltenyi).
Поэтому этот метод особенно полезен для положитель¬
ной сортировки. В протоколе 15.2 кратко изложена
инструкция Miltenyi (см. также протокол 23.18D).
Проверьте, нет ли более новой версии протоколов
внесения метки и разделения на вебсайте Miltenyi
(www.miltenyibiotec.com/).
ПРОТОКОЛ 15.2. Магнитно-активированный
клеточный сортинг (MACS)
Схема
Лейкоцитарная масса или другая суспензия смеси
клеток смешивается с микрогранулами с иммоби¬
лизованными антителами, разбавляется и наносит¬
ся на магнитную разделительную колонку. Ассоции¬
рованные с микрогранулами клетки связываются с
ферромагнитными шариками колонки, в то время
как несвязанные клетки выходят из колонки, не за¬
держиваясь. При удалении колонки из магнитного
поля связанные клетки освобождаются от носителя
и выдавливаются из колонки с помощью поршня.
Материалы
Стерильные:
• Буфер: D-PBSA, pH 7.2, содержащий 0,5% БСА
и 2 мМ ЭДТА
• Магнитный сортировщик клеток: MiniMACS,
MidiMACS, VarioMACS или SuperMACS (Mil¬
tenyi)
• Адаптеры для колонки RS+ или VS+
• Колонка для положительной селекции: MS+/
RS+ для клеток в количестве до IxlO7; LS+/
VS+— до IxlO8
• Намагничивающиеся микрогранулы с иммо¬
билизованными антителами к поверхностному
антигену выделяемых клеток
• Пробирка для сбора образца соответствующая
собираемому объему: для MS+/RS+ на 1 мл; для
LS+/VS+ на 5 мл
Протокол
Внесение метки
1. Выделите стандартным методом (см. прото¬
кол 27.1) мононуклеарные клетки из перифе¬
рической крови или приготовьте клеточную
суспензию с помощью трипсинизации или аль¬
тернативных процедур (см. протоколы 12.5,12.6,
12.8 и 13.2).
2. Избавьтесь от мертвых клеток с помощью Фи-
колл-Гипак (см. протокол 12.10).
280
ГЛАВА 15. РАЗДЕЛЕНИЕ КЛЕТОК
3. Удалите агрегаты, профильтровав клетки через
нейлоновое сито с размером пор 30 мкм или
фильтр (Miltenyi, #414:07). (Перед использова¬
нием смочить фильтр буфером.)
4. Промойте клетки буфером и отцентрифугируйте.
5. Ресуспендируйте осадок в 80 мкл буфера на
107 клеток (суммарных) (80 мкл — минимальный
объем, даже если клеток <107).
6. Добавьте 20 мкл микрогранул MACS на 107 клеток.
7. Смешайте и инкубируйте суспензию 15 мин
при 6-12 °С (для меньшего количества клеток
использовать тот же объем).
8. Разбавьте суспензию добавлением 10-20-крат¬
ного объема буфера.
9. Центрифугируйте 10 мин при 300 g.
10. Удалите супернатант и ресуспендируйте клетки,
помеченные микрогранулами в 500 мкл буфера на
1х108 клеток.
Положительная магнитная сортировка:
11. Поместите колонку в магнитное поле магнитного
сортировщика клеток MACS.
12. Промойте колонку: MS+/RS+ — 500 мкл; LS+/
VS+ — 3 мл.
13. Нанесите суспензию клеток на колонку: на MS+/
RS+ — 500-1000 мкл; LS+/VS+ — 1-10 мл.
14. Дайте немеченым клеткам пройти сквозь ко¬
лонку.
15. Промойте колонку буфером: MS+/RS+ — 3 раза
по 500 мкл; LS+/VS+ — 3 раза по 3 мл.
16. Удалите колонку из сортировщика и поместите
ее в пробирку для сбора образцов.
17. Нанесите пипеткой буфер на колонку (MS+/RS+
1 мл, LS+/VS+ 5 мл) и выдавите положительные
клетки поршнем, который поставляется в комп¬
лекте с колонкой.
18. Посчитайте клетки, скорректировать концентра¬
цию в ростовой среде:
а) 1х105-1х106 кл./мл и посеять в культуральный
флакон для первичной культуры.
б) 10-1000 клеток на 1 мл и посеять в чашку
Петри для клонирования.
Парамагнитная микросфера диаметром 50 нм
Антитело к типоспецифическому
клеточному поверхностному Поршень
антигену
Колонка
Колонка-
После удаления
магнита выделяе¬
мые клетки можно
извлечь, выдавли¬
вая их поршнем
оо
•of
|о#о
•А
“)00
Клетки, меченые магнитными части¬
цами, остаются ассоциированными с
ферромагнитными сферами (показа¬
ны черным цветом) в колонке
Немеченые клетки
не задерживаются и
элюируются в токе
жидкости
Смесь фибробластов
и опухолевых клеток
до разделения
Отрицательная
фракция: опухолевые
клетки без примеси
фибробластов
Положительная
фракция
обогащенных
фибробластов
Анти-фибробласт-ФИТЦ
(© Miltenyi Biotec, Germany)
Рис. 15.7. Магнитный сортинг клеток (MACS' Technology). Положительный сортинг. (а) Клетки предварительно инкуби¬
руются с иммобилизованными на парамагнитных микрогранулах MACS антителами к типоспецифическому клеточному по¬
верхностному антигену, (б) После нанесения связаных с микрогранулами клеток на колонку, они удерживаются в магнитном
поле, генерируемом магнитом и матрицей колонки; немеченые клетки выходят из колонки, (в) После освобождения колоний
из магнита клетки, прикрепившиеся к намагниченным микрогранулам, выдавливаются поршнем. (См. также иллюстрацию
23 г, д.) (г) Цитограмма, полученная с помощью проточной цитометрии, распределения клеток в культуре опухолевых кле¬
ток, содержащей фибробласты, до MACS сортировки (верхняя панель), фракция на выходе после MACS сортировки с мик¬
рогранулами с антителами к фибробластам (см. 6), обогащенная фракция фибробластов (нижняя панель) после удаления из
магнитного поля. (г. © Miltenyi Biotec, используется с разрешения производителя) (Анти-фибробласт-ФИТЦ — измерение
интенсивности флюоресценции клеток, меченных ФИТЦ-мечеными антителами к поверхностным антигенам фибробластов;
НЕА125-РЕ — измерение интенсивности флюоресценции клеток, меченных PE-антителами к НЕА 125; НЕА 125 — маркер
опухолевых клеток, РЕ — фикоэритрин)
15.4, Флюоресцентно-активируемый клеточный сортинг
281
15.4. ФЛЮОРЕСЦЕНТНО-АКТИВИРУЕМЫЙ
КЛЕТОЧНЫЙ СОРТИНГ
Разделение клеток'с помощью флюоресценции
[Vaughan & Milner, 1989] основано на однопоточном
прохождении клеток через лазерный луч. Светорас¬
сеяние клеток улавливается одним или несколькими
фотоумножителями и регистрируется (рис. 15.8,15.9).
Если клетки предварительно обработаны флюоресцен¬
тным красителем (например, йодистым пропидием
или хромомицином АЗ для окрашивания ДНК) или
антителами, меченными флюорохромом, то лазер
возбуждает флюоресценцию, которая регистрируется
вторым фотоумножителем. Полученная информация
обрабатывается и выводится на монитор в виде двух¬
мерного (рис. 15.10) или трехмерного графика.
Проточный цитометр представляет собой анали¬
тический прибор, который обрабатывает сигнал от
фотоумножителей для анализа состояния клеточной
популяции (например, для определения процента
клеток в разных фазах клеточного цикла, оценивае¬
мого по сочетанию измерений флюоресценции ДНК
и размера клеток).
Флюоресцентно-активирующий сортер клеток
(FACS; например, FACSAria, производства B-D
Biosciences) представляет собой прибор, который ис¬
пользует флюоресцентный сигнал каждой клетки для
сортировки клеток в одну из двух пробирок для сбора
образцов и в резервуар для слива. Если в специальных
координатах для отображения данных установить
ограничения, то сортировщик клеток разделит клетки
в соответствии с заданными в этих координатах свойс¬
твами (например, по высокому или низкому светорас¬
сеянию, по высокой или низкой флюоресценции) и
направит их в соответствующую пробирку-приемник,
расположенную ниже потока клеток. Поток разбива¬
ется на капли высокочастотной вибрацией проточной
кюветы. Капли, содержащие одиночные клетки со спе¬
цифическими характеристиками, заряжены к моменту
выхода из кюветы. Эти капли отклоняются налево
или направо, в соответствии с их зарядом, так как они
проходят между двумя противоположно заряженны¬
ми пластинами. Заряд подается на короткое время
в промежуток времени после пересечения клеткой
лазерного луча таким образом, что капля, содержащая
одну специфически помеченную клетку, направляется
в приемник. Концентрация клеток в потоке должна
быть достаточно низкой для того, чтобы между клет¬
ками был достаточный интервал и две клетки не могли
попасть в одну каплю [Vaughan & Milner, 1989].
Направляющие пластины
Пробирки для сбора образцов
Трубки для изолирующей жидкости
Трубка для образца
Проточная кювета
Рис. 15.8. Флюоресцентно-активируемый сортер клеток (FACS). Крупным планом изображены проточная кювета и камера
для разделения клеток сортировщика клеток модели FACS IV (Becton Dickinson; см. также рис. 15.9)
282
ГЛАВА 15. РАЗДЕЛЕНИЕ КЛЕТОК
Проточная
кювета
15-кГц вибрационный
излучатель^
Пересечение лучом лазера
Клетки из
резервуара
Изолирующая жидкость
Ламинарный поток
потока клеток
в изолирующей
жидкости
Заряженный
электрод
|азера V- I щ
4 ^ П t
•• /4 + ^
Проходящий свет
прерывание
+ • +
+
Флюоресценция
частиц
Прямое светорас¬
сеяние частиц
ГЛ
'СУ
©
. Поток заряженных
капель
Отклоняющие
-электродные
пластины
Пробирки для
сбора образцов
Рис. 15.9. Проточная цитометрия. Принцип работы проточ¬
ного цитофотометра. Клетки в D-PBSA поступают сверху,
изолирующая жидкость (тоже D-PBSA) закачивается вокруг
потока клеток для создания ламинарного потока внутри
проточной ячейки. Когда поток клеток выходит из ячейки, он
пересекает лазерный луч и генерируемый сигнал приводит
в действие заряженный электрод, заряжая, таким образом,
поток клеток. Затем 15-кГц вибрационный излучатель, за¬
крепленный на проточной ячейке, индуцирует разбивание
потока клеток на капли. Капли, выходящего потока несут
кратковременный заряд и отклоняются электродными плас¬
тинами, расположенными ниже проточной ячейки. Заряд
подается на пластины с достаточной задержкой, с учетом
времени прохождения клеток от пересечения лучом лазера
до вхождения в пространство между пластинами (см. также
рис. 15.8)
Все клетки, обладающие сходными свойствами, со¬
бираются в одну пробирку. Можно установить вторую
систему координат и второй набор ограничений, в этом
случае можно одновременно собирать в другую пробир¬
ку вторую группу клеток, так как при этом изменится
полярность потока клеток и клетки будут отклоняться
в обратном направлении. Все оставшиеся клетки соби¬
раются в центральный резервуар для слива.
Данный метод может быть использован для разде¬
ления клеток в тех случаях, когда какие-либо различия
между клетками можно зарегистрировать по их свето¬
рассеянию (например, размер клетки) или флюорес¬
ценции (например, содержание ДНК, РНК или белка;
ферментативная активность, наличие специфических
Номер канала (интенсивность флюоресценции)
Рис. 15.10. Распечатка, полученная после измерения на
FACS II. Клетки Фрейнда (мышиная эритролейкемия)
фиксировали метанолом в суспензии и окрашивали хро-
момицином АЗ. Затем суспензию смешали с аналогичным
образом зафиксированными и окрашенными эритроцитами
цыпленка и измерили флюоресценцию на FACS II. На рас¬
печатке на вертикальной оси отложено количество клеток,
на горизонтальной оси — номер канала (интенсивность
флюоресценции). Так как интенсивность флюоресценции
прямо пропорциональна количеству ДНК в клетке, по кри¬
вой можно анализировать распределение содержания ДНК
в клеточной популяции. Наиболее низкое содержание ДНК
в эритроцитах цыпленка, которые использовали в качестве
стандарта. Самый большой пик в районе канала 100 соответ¬
ствует клеткам в фазе G1 клеточного цикла. Клетки в области
канала 200, соответственно, находятся в фазе G2 и метафазе
(с удвоенным количеством ДНК на клетку), а клетки между
двумя пиками — в фазе S (синтез ДНК). Пик в канале 250 со¬
ответствует клеткам, в которых содержание ДНК превышает
размерность шкалы или агрегировавшим клеткам. (Любезно
предоставлено профессором B.D. Young.)
антигенов) и может быть применен к разделению
широкого спектра типов клеток. Наиболее широко
метод возможно использовать для гематопоэтиче-
ских клеток [Battye & Shortman, 1991; Pipia & Long,
1997], которые относительно легко дезагрегировать
до необходимой в обязательном порядке клеточной
суспензии, но можно использовать и для солидных
опухолей (например, легкого [Aitken et al., 1991], кожи
[Swope et al., 1997] и кишечника [Boxberger et al., 1997;
см. также протокол 23.17]). Это чрезвычайно эффек¬
тивный прибор, но его производительность ограни¬
чена (допустимый максимум клеток, которые можно
разделить за 1 раз, — приблизительно IxlO7клеток).
(Современные приборы позволяют вести сортировку
со скоростью 7-25 000 кл./с. — Прим. пер.) Хотя более
сложные приборы для разделения клеток очень дорого
стоят (приблизительно 200 000 долларов США) и тре¬
буют штатного высококвалифицированного операто¬
ра, менее дорогие настольные приборы доступны для
аналитического использования (например, приборы
производства Guava Technologies).
15.5. Другие методы
283
15.5. ДРУГИЕ МЕТОДЫ
Другие методы, которые успешно используют для
разделения клеток, слишком многочисленны, чтобы
подробно их описывать. Все они представлены в
табл. 15.1.
Электрофорез в градиенте фиколл a [Platsoucas et
al., 1979] или на висячем листе бумаги; второй метод,
по-видимому, более эффективен и используется для
разделения клеток эпителия канальцев почек [Kreis-
bergetal., 1977].
В аффинной хроматографии используют анти¬
тела [Varon & Manthorpe, 1980; Au & Varon, 1979]
или растительные лектины [Pereira & Kabat, 1979],
иммобилизованные на нейлоновых волокнах [Edel-
man, 1973] или сефадексе (Amersham Pharmacia).
Этот метод представляется полезным для разделения
свежевыделенных клеток крови, но менее подходит для
культивируемых клеток.
Противоточное распределение [Walter, 1975,
1977; Sharpe, 1988] используется для очистки асцит¬
ных опухолевых клеток мыши со средней жизнеспо¬
собностью.
15.6. СОВЕТЫ НАЧИНАЮЩЕМУ
Начинать лучше всего с простых методик, таких как
разделение клеток в градиенте плотности или если
ТАБЛИЦА 15.1. Методы разделения клеток
Метод
Принцип разделения
Оборудование
Примечания
Ссылки
Седиментация при ус¬
Размер клеток
Изготовленные по
Простой метод, но чистота
Miller & Phillips,
корении 1 g
-
заказу делитель¬
ная воронка и пе¬
регородки
разделения и выход неве¬
лики
1969
Изопикническая седи¬
ментация
Плотность клеток
Центрифуга
Простой и быстрый метод
Pertoft & Lau¬
rent, 1982; см.
протокол 15.1
MACS
Антитела к поверх¬
Простой магнит и
Специфичный при наличии
Gaudemack et al.,
ности клеток
проточная камера
высокоспецифичных анти¬
тел к поверхности
1986; см. про¬
токол 15.2
Иммунный пэннинг
Антитела к поверх¬
Чашки с сорбирован¬
Простой, несложная техноло¬
Wysocki & Sata,
ности клеток
ными антителами
гия, если имеются в распо¬
ряжении планшеты, с пред¬
варительно сорбированны¬
ми антителами, но также
зависит от специфичных
антител к поверхности
1978
Элюатриационное цент¬
Размер клеток, плот¬
Специальная цент¬
Быстрый, высокий выход,
Lutz et al., 1992
рифугирование
ность и конфигура¬
ция поверхности
рифуга и элюатри-
ационный ротор
но достаточно сложный
процесс
FACS
Площадь поверхности
клетки, флюоресцент¬
ные маркеры, флюо-
рогенные субстраты
ферментов, многопа¬
раметрический
Проточный цито¬
метр
Сложная технология
Vaughan &
Milner, 1989
Аффинная хромато¬
графия
Поверхностные клеточ¬
ные антигены, повер¬
хностные клточные
углеводы
Стерилизуемое
Элютриация клеток с колон¬
ки сложна, лучше прово¬
дить в неприкрепленной
суспензии
Edelman, 1973
Противоточное распре¬
деление
Сродство компонентов
клеточной поверх¬
ности к фазе раство¬
рителя
Шейкер
Некоторые клетки могут по¬
терять жизнеспособность,
но метод в значительной
степени эффективен для
других клеток
Walter, 1977
Электрофорез в гради¬
енте или на висячем
листе бумаги
Поверхностный заряд
Прибор для электро¬
фореза на висячем
листе бумаги
Kreisberg et
al., 1977;
Platsoucas et
al., 1979
284
ГЛАВА 15. РАЗДЕЛЕНИЕ КЛЕТОК
можно прогнозировать наличие специфического
фенотипа клеточной поверхности, то можно ис¬
пользовать пэннинг на сорбированных чашках или
MACS. Если частота разделения или выход клеток
неудовлетворительны, тогда может возникнуть необ¬
ходимость использовать FACS или элюатриационное
центрифугирование. Элюатриационное центрифуги¬
рование удобно для быстрой сортировки большого
количества клеток, но FACS, возможно, дает более
чистые клеточные популяции, так как селекция идет
по двум или более строгим критериям.
В тех случаях, когда требуется получить суспензию
клеток высокой чистоты, а выбранная культура не
удовлетворяет этим требованиям, возможно, необхо¬
димо использовать двухэтапное фракционирование
(аналогично выделению белков). В таких методиках в
качестве первого этапа используется разделение в гра¬
диенте плотности, в качестве второго этапа — пэннинг
или MACS, и окончательное выделение выполняется
с помощью FACS. Такая последовательность операций
применяется, например, при выделении гематопоэти¬
ческих стволовых клеток [Cooper & Broxmeyer, 1994].
Глава 16
ХАРАКТЕРИСТИКА КЛЕТОК
16.1. НЕОБХОДИМОСТЬ
ХАРАКТЕРИСТИКИ
Существует шесть основных требований к характерис¬
тике клеточной линии:
1) Демонстрация отсутствия перекрестной контами¬
нации (см. разд. 7.10.1,16.3,16.6.2,19.5; табл. 13.2)
2) Подтверждение специфичности происхождения
3) Корреляция с исходными тканями, включающая
следующие характеристики:
а) Идентификация линии дифференцировки, к
которой принадлежат клетки
б) Положение клеток на этой линии (например,
стволовые клетки, предшественники, или диф¬
ференцированное состояние)
4) Определение, являются ли клетки трансформиро¬
ванными или нет
а) Является ли клеточная линия конечной или
постоянной (см. разд. 18.2,18.4)?
б) Проявляет ли она свойства, связанные с малиг-
низацией (см. разд. 18.6)?
5) Выяснение предрасположенности клеточной линии
к генетической нестабильности и фенотипической
вариации (см. разд. 17.11,18.3)
6) Идентификация специфических клеточных линий
внутри одной группы одного и того же происхож¬
дения, селекционированных клеточных штаммов,
гибридных клеточных линий с демонстрацией
свойств, уникальных для этой клеточной линии или
штамма.
16.2. ВЕДЕНИЕ ДОКУМЕНТАЦИИ
И ПРОИСХОЖДЕНИЕ КЛЕТОК
Когда получают новую клеточную линию из первичной
культуры или из уже существующей клеточной линии,
тогда возникает трудность оценки ее потенциальной
ценности. Часто только после определенного периода
использования и распространения клеточной линии
становится ясной важность и полезность клеточной
линии, и с этой точки зрения приобретает особое значе¬
ния детальная информация о ее источнике и развитии.
Однако к этому времени бывает слишком трудно соб¬
рать ретроспективные данные. Поэтому чрезвычайно
важно вести правильные и полные записи начиная с
момента выделения тканей или получения новой кле¬
точной линии, детально описывая источник выделения,
характеристики, все манипуляции с клеточной линией.
Эти записи образуют происхождение (паспорт) линии,
и чем более детальны эти записи, тем более ценна кле¬
точная линия (см. разд. 12.3.11 и 13.7.8).
Этот аспект культивирования клеток стал особенно
важным при широком распространении клеточных
линий в клеточных банках и при личных контактах
между исследовательскими лабораториями и ком¬
мерческими компаниями, при которых клеточные
линии распространяются далеко от своего источни¬
ка. Подтверждение аутентичности клеточной линии
включает характеристику такой клеточной линии при
поступлении, периодически в ходе ее использования,
и подразумевает, что эти данные соотносятся с имею¬
щимися в паспорте линии и дополнят их.
16.3. ПОДТВЕРЖДЕНИЕ АУТЕНТИЧНОСТИ
Характеристика клеточной линии крайне необходима
не только при определении ее функциональных свойств,
но также для подтверждения ее аутентичности. Особое
внимание должно быть уделено возможности перекрест¬
ной контаминации линии с существующими постоян¬
ными клеточными линиями, либо вероятным ошибкам
идентификации в результате неверной маркировки или
путанице при манипуляциях с линиями. Было показа¬
но, что большинство клеточных линий, используемых
в США в конце 1960-х годов, было контаминировано
клетоками HeLa [Gartler, 1967; Nelson-Rees & Flander-
meyer, 1977; Lavappa, 1978]. Это было первым проявле¬
нием большой проблемы, однако дальнейшее использо¬
вание культур клеток показали, что многие люди еще не
знакомы или не желают признавать, что многие широко
используемы линии (например, Hep-2, КВ, Girardi
Heart, WISH, Chang Liver) не являются аутентичными
(см. разд. 16.6.2, 16.6.3 и табл. 13.2). Использование
культур, контаминированных HeLa или другими пос¬
тоянными клеточными линиями, без соответствующих
представлений о признаках контаминации до сих
пор является большой проблемой [Stacey et al., 2000;
Drexler et al., 2003], при этом авторы подчеркивают,
что любая энергично растущая клеточная линия может
дать избыточный рост по сравнению с любой медленно
растущей культурой [Masters et al., 1988; vanHelden
et al., 1988; Christensen et al., 1993; Gignac et al., 1993;
286
ГЛАВА 16. ХАРАКТЕРИСТИКА КЛЕТОК
van Bokhoven et al., 2001a, b; Rush et al., 2002]. Хотя
некоторые из работ, проведенных на таких культурах,
остаются достоверными, например, при исследовании
общих молекулярных процессов, которые происходят
независимо от источника клеток. Однако любые по¬
пытки соотнести поведение клеток с типом тканей, из
которых они, предполагаемо, были выделены, терпят
поражение и не могут рассматриваться как достоверные
в связи с перекрестной контаминацией. К сожалению,
этот факт часто игнорируется редакторами журналов,
учреждениями, предоставляющими гранты, референ¬
тами и исследователями, использующих клеточные
культуры. Характеристические исследования, в том
числе изучение постоянных клеточных линий, сдела¬
ли процесс аутентификации жизненно важным для
подтверждения достоверности (валидации) данных,
полученных на этой культуре.
Природа методов, используемых для характеристи¬
ки, зависит от типа проводимых работ; например, если
легко доступны молекулярные технологии, то предпоч¬
тительны ДНК-фингерпринтинг (см. протокол 16.8),
изучение Д НК-профиля (см. протокол 16.9) или анализ
экспрессии генов (см. разд. 16.7 и протокол 27.8), кото¬
рые в настоящее время наиболее широко используют¬
ся. В то же время цитологические лаборатории могут
проводить хромосомный анализ (см. разд. 16.5) вместе
с FISH и окрашиванием хромосом (см. протокол 27.9),
а лаборатории с оборудованием для иммунологической
экспертизы могут предпочитать использование МНС-
анализа (например, HLA-типирование) одновременно
с исследованием линейно-специфических маркеров.
Комбинация функциональных исследований связана
с вашими собственными интересами, результаты их
должны обеспечивать достоверное подтверждение
аутентичности клеточных линий.
Независимо от возможностей лаборатории в на¬
стоящее время главными стандартными процедурами
идентификации клеточной линии являются ДНК-
фингерпринтинг или исследование профиля ДНК и
множественный изоферментный анализ в агарозном
геле (см. протокол 16.10); если вы не можете или не хо¬
тите использовать проводить эти идентификационные
процедуры сами, то постарайтесь найти коммерческую
компанию, проводящую их.
Некоторые из методов обычно используемые для
характеристики клеточных линий представлены в
табл. 16.1 [см. также Hay et al., 2000].
16.3.1. Видовая идентификация
Хромосомный анализ, обычно называемый кариоти-
пированием (см. протокол 16.7, 27.5), является одним
из наилучших методов для обнаружения различий
между видами. Анализ распределения полос при диф¬
ференцированном окрашивании хромосом может быть
использован для различения человеческих и мышиных
хромосом (см. разд. 16.5), а последующее окрашивание
хромосом с использованием комбинации специфиче¬
ских молекулярных зондов для гибридизации с отдель¬
ными хромосомами (см. разд. 16.5.3 и 27.8.2) улучшат
разрешение и специфичность этого метода. Эти зонды
идентифицируют индивидуальные хромосомные пары
и видоспецифичны. В настоящее время доступность
зондов ограничена несколькими видами, в частности
мышами и человеком, но окрашивание хромосом
позволяет различать человеческие и мышиные хромо¬
сомы при возможной перекрестной контаминации и
образовании межвидовых гибридов. Изоферментный
электрофорез (см. протокол 16.10) также является
хорошим и более быстрым диагностическим тестом,
чем хромосомное окрашивание. Существует простой
набор, позволяющий выполнить эти исследования
(см. протокол 16.10). Комбинация двух методов часто
используется с однозначными достоверными резуль¬
татами [Нау, 2000].
16.3.2. Маркеры дифференцировки
или маркеры ткани
Отдельные органы составлены из тканей, например кожа
состоит из наружного эпидермиса и подлежащей дермы.
Ткани, в свою очередь, состоят из индивидуальных
линий дифференцировки, например дерма содержит
соединительнотканные фиброциты, клетки сосудистого
эндотелия, гладкомышечные клетки, мезенхимальные
клетки кожных бугорков и др.. Можно проследить проис¬
Таблица 16.1. Характеристика клеточных линий и клеточных штаммов
Критерий
Метод
Ссылка
Nfi протокола
Кариотип
Дифференциальное окрашивание
хромосом
Rothfels & Siminovitch, 1958, Roo¬
ney & Czepulkowski, 1986
16.9
Изоферментный анализ
Электрофорез в агарном геле
Hay, 2000
16.12
Антигены клеточной по¬
Иммуногистохимия
Hay, 2000, Burchell et al., 1983,1987
16.13
верхности
Цитоскелет
Иммуноцитохимия с антителами к спе¬
цифическим цитокератинам
Lane, 1982, Moll et al., 1982
16.13
ДНК фингерпринтинг
Рестрикционный анализ, PAGE; зонды
сателлитной ДНК
Hay, 2000, Jeffreys et al., 1985
16.10
ДНК профиль
PCR микросателлитных повторов
Masters et al., 2001
16.11
16.3, Подтверждение аутентичности
287
хождение каждого типа клеток через серию пролифера-
тивных стадий вплоть до стволовой клетки (см. рис. 3.6),
с формированием древообразной структуры. Каждая
«ветка» этого «дерева» может быть определена как
линия дифференцировки, начиная с базальной клетки
эпидермиса, следуя по пути дифференцировки до обра¬
зования зрелого ороговевшего кератиноцита. Некоторые
линии дифференцировки, например миелоидные линии
гематопоэтической дифференцировки, могут ветвиться
на суб-линии (нейтрофилы, эозинофилы, базофилы),
поэтому экспрессия маркеров клеточных линий также
оказывается под влиянием дифференцировки, т. е. связана
с положением клетки на линии дифференцировки (см.
разд. 16.10). Линейные маркеры полезны для установле¬
ния родства отдельных клеточных линий с тканями, из
которых они происходят (табл. 16.1).
Антигены клеточной поверхности. Эти маркеры, в
частности, полезны при разделении гематопоэтических
клеток [Visser & De Vries, 1990], а также эффективны
при отделении эпителиальных клеток от стромальных
элементов при помощи таких антител как анти-ЕМА
[Heyderman et al., 1979], анти-HMFG 1 и 2 [Burchell &
Taylor-Papadimitriou, 1989], а также отделения клеток
нейроэктодермального происхождения (например,
анти-А2В5) [Dickson et al., 1983] от клеток других эм¬
бриональных слоев.
Белки промежуточных филаментов. Эти бел¬
ки относятся к наиболее широко распространенным
маркерам клеточных линий или тканей [Lane, 1982;
Ramaekers et al., 1982]. Кислый глиальный фибрил¬
лярный белок астроцитов (GFAP) [Bignami et al.,
1980] (см. цв. вкладку 116) и десмин [Bochaton-Pial-
lat et al., 1992; Brouty-Boy’e et al., 1992] мышечных
клеток является наиболее специфичным, в то время
как цитокератин является маркером эпителиальных
клеток и мезотелия [Lane, 1982; Moll et al., 1982] (см.
цв. вкладку 11а). Белок нейрофиламентов маркирует
нейроны [Wu & de Vellis, 1987; Kondo & Raff, 2000] и
некоторые нейроэндокринные клетки [Bishop et al.,
1988]. Виментин (см. цв. вкладку 11в), несмотря на то,
что обычно in vivo ограничен клетками мезодермаль-
ного происхождения, может in vitro быть обнаружен и
в других типах клеток.
Продукты дифференцировки и особенности
функциониорвания. Гемоглобин клеток эритроидно-
го ряда, миозин или тропомиозин мышечных клеток,
меланин меланоцитов и сывороточный альбумин ге¬
патоцитов относятся к наиболее известным примерам
специфических маркеров клеточных типов, но, так же
как и маркеры дифференцировки, они зависят от сте¬
пени завершенности дифференцировки.
Транспорт неорганических ионов, а также связанный
с ним перенос воды являются характеристикой абсор¬
бирующего и секреторного эпителия [Abaza et al., 1974;
Lever, 1986]; при монослойном росте некоторые эпители¬
альные клетки формируют куполообразные структуры,
или гемицисты, образование которых в монослое связано
с накоплением воды на нижней поверхности монослоя
[Rabito et al., 1980] (рис. 16.1, см. цв. вкладку12я, б).
Другие специфические функции, которые могут вы¬
ражаться in vitro, включают мышечное сокращение и
деполяризацию мембран нервных клеток.
Ферменты. Для ферментативной характеристи¬
ки могут быть использованы три параметра: 1) кон¬
ститутивный уровень (т.е. в отсутствие действия
индукторов и супрессоров), 2) реактивный уровень
(в ответ на действие индукторов или ингибиторов)
и 3) изоферментный полиморфизм (см. разд. 16.8.1,
16.8.2). Изофермент креатинкиназа ВВ является мар¬
кером для нервных и нейроэндокринных клеток, как
и нейрон-специфичная енолаза. Лактатдегидрогеназа
присутствует в большинстве клеток, но в различных
изоферментных формах. Высокий уровень тирози-
наминотрансферазы, индуцируемой дексаметазоном,
обычно рассматривается как специфичный для гепа¬
тоцитов (табл. 16.2) [Granner et al., 1968].
Рис. 16.1. Куполообразные образования, (а) Купол или гемицист, образованный в эпителиальном монослое в результате
нисходящего транспорта ионов и воды, нижний фокус (на монослое), (б) Верхний фокус (вершина купола) (см. также цв.
вкладку 12а, б)
288
ГЛАВА 16. ХАРАКТЕРИСТИКА КЛЕТОК
Регуляция. Уровень экспрессии многих продук¬
тов дифференцировки находится под регуляторным
контролем факторов окружающей среды, таких как
гормоны, матрикс, соседние клетки (см. разд. 17.7). Так
или иначе, измерение уровня специфических маркеров
линий клеточной дифференцировки может потребо¬
вать предварительной обработки или инкубирования
клеток, например в присутствии гормона, такого
как гидрокортизон, специфических факторов роста
или выращивания клеток на внеклеточном матриксе
определенного типа. Для получения максимальной
экспрессии тирозинаминотрансферазы в клетках пе¬
чени и глютаминсинтетазы в глии требуется предвари¬
тельная индукция дексаметазоном. Глютаминсинтаза
также сдерживается глютамином, поэтому глютамин
должен быть замещен в среде за 48 ч до исследования
[DeMars, 1958].
Клональное соответствие. Хотя многие из мар¬
керов, описанных выше, считаются маркерами клонов,
они могут рассматриваться более правильно как ткане¬
вые маркеры или маркеры клеточных типов, поскольку
они в большей степени являются характеристиками
функционирования клеток, а не источников их про¬
исхождения. Цитокератин встречается в мезотелии
и почечном эпителии, хотя эти ткани происходят из
мезодермы. Нейронспецифическая энолаза и креа-
тинкиназа ВВ экспрессируются в нейроэндокринных
клетках легких, хотя эти клетки, как теперь известно,
происходят из эндодермы, а не из нейроэктодермы,
как это можно было бы предположить в отношении
нейроэндокринного типа клеток.
16.3.3. Уникальные маркеры
Уникальные маркеры включают специфические хромо¬
сомные аберрации (например, делеции, транслокации,
полисомию); антигены главного комплекса гистосов¬
местимости (МНС-антигены), например HLA у челове¬
ка, которые обладают высокой степенью полиморфизма
и могут определяться при помощи ДНК-фингерприн-
тинга (см. протокол 16.8) или изучение профиля ДНК
(протокол 16.9). Ферментативная недостаточность
(например, недостаточность тимидинкиназы (ТК_) и
лекарственная устойчивость (например, устойчивость
к винбластину, обычно соединенная с экспрессией
Р-гликопротеина одним из mrfr-генов, которые кодиру¬
ют выведение белка) не являются истинно уникальны¬
ми, но они могут быть использованы для обнаружения
различий между клеточными линиями из одних и тех
же тканей различных доноров.
16.3.4. Трансформация
Состояние трансформации является одним из главных
элементов цри характеристике клеточной линии и бу¬
дет рассматриваться отдельно (см. гл. 17).
16.4. МОРФОЛОГИЯ КЛЕТОК
Изучение морфологии клеток является самым прос¬
тым и наиболее прямым методом, используемым для
идентификации клеток. Оно имеет некоторые недо¬
Таблица 16.2. Ферменты, являющиеся маркерами
Фермент
Клеточный
Индуктор
Репрессор
Ссылка
Глютамилсинтетаза
Астроглия (мозг)
Гидрокортизон
Глютамин
Hallermeyer & Ham-
precht, 1984
Тирозин аминотрансфераза
Гепатоциты
Гидрокортизон
Granner et al., 1968
Сахароза
Энтероциты
NaBt
Pignata et al., 1994; Va-
chon et al., 1996
Щелочная фосфатаза
Пневмоциты II типа (в легочных
Дексаметазон, оксо-
TGF-p
Edelson et al., 1988;
альвеолах)
статин, IL-6
McCormick & Fresh-
ney, 2000
Щелочная фосфатаза
Энтероциты
Дексаметазон, NaBt
Vachon et al., 1996
Неспецифическая эстераза
Макрофаги
РМА, витамин D
Murao et al., 1983
Ангиотензинпреобразующий
Эндотелий
Коллаген, Matrigel
Del Vecchio & Smith,
фермент
1981
Нейронспецифическая
Нейроны, нейроэндокринные
Hansson et al., 1984
енолаза
клетки
Тирозиназа
Меланоциты
сАМР
Park et al., 1993,1999
DOPA-декарбоксилаза
Нейроны, клетки мелкоклеточной
Dow et al., 1995; Ga-
карциномы легких (SCLC)
zdar et al., 1980
Креатинкиназа ММ
Мышечные клетки
IGF-II
FGF-1,2,7
Stewart et al., 1996
Креатинкиназа ВВ
Нейроны, нейроэндокринные клет¬
Gazdar et al., 1981
ки, SCLC
16.4. Морфология клеток
289
статки, которые должны быть учтены. Большинство
из них связаны со свойством пластичности клеточной
морфологии в ответ на культивирование в различных
условиях. Например, эпителиальные клетки, растущие
в центре конфлюэнтного монослоя, обычно имеют пра¬
вильную полигональную форму, с ясно очерченными
краями, в то время как те же клетки, растущие на краю
монослоя, могут иметь менее правильную, форму,
отличающуюся разнообразием, а при трансформации
могут отрываться от монослоя и принимать фибро-
бластоподобную форму.
В субконфлюэнтном монослое фибробластов, полу¬
ченных выделением из почек хомяка или легких или
кожи человека, клетки имеют мультиполярную или
биполярную форму (см. рис. 16.2а, е, цв. вкладку 8а) и
хорошо распластаны на культуральной поверхности.
В конфлюэнтном монослое клетки биполярны и менее
распластаны (см. рис. 16.26, ж, цв. вкладку 8б, Юг, д).
Они также образуют характерные параллельные ре¬
шетки и завитки, видимые невооруженным глазом (цв.
вкладка 10в). Мышиные ЗТЗ клетки (рис. 16.2г/, ф) при
низкой плотности растут как мультиполярные фибро¬
бласты, но в конфлюэнтном монослое приобретают
эпителиоподобную форму (рис. 16.2ц, ч, цв. вкладка
106). Изменения в составе субстрата [Gospodarowicz
et al., 1978b; Freshney, 1980] и питательной среды
(см. разд. 17.7.2) могут также привести к изменениям
морфологии клеток. Таким образом, сравнительное
изучение клеток должно всегда проводиться на одной
и той же стадии роста и при одинаковой клеточной
плотности на одной и той же среде, и при росте на одном
и том же субстрате.
Термины «фибробластный» и «эпителиальный»
используются в культуре тканей достаточно прибли¬
зительно и часто описывают скорее внешний вид,
чем происхождение клеток. Так, биполярные или
мультиполярные мигрирующие клетки, длина кото¬
рых обычно вдвое больше ширины, будут называться
фибробластными. В то же время полигональные
клетки монослоя, с более правильными контурами
клеток, растущие отдельными островками отдельно
от других клеток, обычно рассматриваются как эпи¬
телиальные (рис. 16.2в, е, и, к, ну о, с). Тем не менее,
когда идентичность клеток не подтверждается, дол¬
жен использоваться термин «фибробласт-подобные»
или «эпителий-подобные» (или эпителиоидные»)
клетки. Клетки, происходящие из карциномы, часто
выглядят как эпителиоидные, но имеют большую
вариабельность морфологии (рис. 16.2//, о, л, м)у не¬
которые мезенхимальные клетки, такие как СНО-К1,
и эндотелиальные клетки также при конфлюэнтном
росте обычно имеют эпителий-подобную морфологию
(рис. 16.2г, т).
Беглый обзор живых культур, преимущественно
при помощи фазово-контрастного микроскопа, яв¬
ляется более полезным, чем редкие исследования
фиксированных и окрашенных культур, поскольку
морфология живых культур дает более общее впечат¬
ление о клеточной морфологии и ее пластичности, а
также обнаруживает различия в грануляции и вакуо¬
лизации, которые возникают в культуре. Нездоровые
культуры часто становятся гранулированными, а затем
в них обнаруживаются вакуоли, окружающие ядро
(см. рис. 13.1).
16.4.1. Микроскопия
Инвертированный микроскоп является одним из са¬
мых важных инструментов в лаборатории культуры
тканей, однако часто он используется неправильно.
Поскольку толщина закрытых культуральных сосу¬
дов делает наблюдение извне затруднительным из-за
большого рабочего расстояния, культуральный сосуд
помещают на предметный столик, освещают сбоку и
рассматривают снизу. Так как толщина стенок куль¬
турального сосуда ограничивает рабочее расстояние,
максимальное увеличение объектива ограничено 40х.
Использование фазово-контрастной оптики, в которой
кольцевой источник света экранируется соответствую¬
щими темными кольцами в объективе и виден только
дифракционный свет, позволяет видеть неокрашен¬
ные клетки с более высокой контрастностью, чем при
обычном освещении. Поскольку это означает, что
интенсивность света более повышена, то для продол¬
жительных наблюдений должен быть встроен фильтр
инфракрасного спектра.
ПРОТОКОЛ 16,1. Использование
инвертированного микроскопа
Схема
. Поместитькулщуру на предметный столик микро-
скоп^вклго^в^йййце^е ^выбрать подходящую
оптическую систему. Сфокусировать освещение на
образце, если необходимо, отрегулировать конден¬
сор и фазовое кольцо.
Материалы
Нестерильные:
• Инвертированный микроскоп (см. разд. 5.2.5)
с фазово-контрастными объективами на 10х и
20х дай 40х и конденсор с соответствуюящми
фазовыми кольцами
* Салфетки для протирания линз
Протокол
1. Удостоверьтесь, что мщсроскоп чист. Протрите
предметный столик 70%-м этанолом и объектив
при помощи специальной салфетки, если это
необходимо.
2. Включите микроскоп, повышая реостатом интен¬
сивность свечения лампы от самой низкой до оп¬
тимальной. Не включайте сразу на максимальное
освещение.
290
ГЛАВА 16. ХАРАКТЕРИСТИКА КЛЕТОК
3. Проверьте положение конденсора и источника
света:
а) Поместите окрашенный препарат, флакон или
чашку на предметный столик
б) Закройте полевую диафрагму.
в) Сфокусируйте изображение ирисовой диа¬
фрагмы.
г) Сцентрируйте изображение ирисовой диа¬
фрагмы в поле зрения.
4. Сцентрируйте фазовые кольца.
а) Если имеется фазовое телескопическое уст¬
ройство, вставьте его на место стандартного
окуляра. Затем сфокусируйте свет на фазовые
кольца и проверьте, чтобы конденсорное коль¬
цо (белое на черном) совпадало с объективным
кольцом (черное на белом).
б) Если фазового телескопического устройства
нет, то замените окрашенный образец неок¬
рашенной культурой (живой или фиксиро¬
ванной), снова наведите на фокус и двигайте
регулятор фазового кольца до получения
оптимума контраста.
5. Исследуйте клетки. Фазово-контрастный объ¬
ектив 10х выявляет достаточное количество
деталей для обычного исследования. Фазово¬
контрастный объектив 4х не даст достаточную
детализацию, а большие увеличения, чем 10х,
ограничивают размеры поля зрения.
а) Обратите внимание на стадию роста (напри¬
мер, редкий, субконфлюэнтный, полный,
плотный монослой).
б) Обратите внимание на состояние клеток
(например, прозрачные и гиалиновые или
гранулированные, вакуолизированные).
6. Проверьте прозрачность среды (например, от¬
сутствие остатков клеток, плавающих гранул,
признаков контаминации). Выберите объектив с
большим увеличением (см. разд. 19.3.1, рис. 19.1я,
в, ду и цв. вкладку 16а, б, в).
7. Сделайте запись ваших наблюдений.
8. Поверните реостат и выключите питание микро¬
скопа.
9. Верните культуру в инкубатор или перенесите в
ламинарный шкаф для дальнейших стерильных
манипуляций.
В культуральных сосудах, взятых прямо из инкуба¬
тора, образуется конденсат на внутренней поверхности
верхней части сосуда и крышки. Этот конденсат может
быть возвращен во флакон путем переворачивания
флакона, осторожным перемещением среды по верх¬
ней части сосуда (не позволяя попадать на горлышко)
и установкой флакона в вертикальное положение на
10-20 с для того, чтобы жидкость стекла вниз. В случае
использования чашек Петри освобождение от конден¬
сата сложнее, и может потребоваться замена крышки на
новую. Если чашки промаркированы правильно, т. е. со
стороны дна, то замена крышки не приведет к каким-то
затруднениям в идентификации культуры. Культуры в
планшетах для микротитрования особенно сложно ис¬
следовать из-за дифракции, вызываемой менисками.
В случае если возникает предположение об изме¬
нении морфологии клеток, полезно иметь записанные
наборы фотографий при различной плотности кле¬
ток (см. рис. 16.2) для каждой клеточной линии (см.
протокол 16.6), приготовленные вскоре после приоб¬
ретения этой линии, и через определенный интервал
времени ее ведения. Фотографирование постоянно
используемых клеточных линий следует выводить на
дисплей, связанный с инвертированным микроскопом.
Сохраненные фотографии могут быть дополнены фо¬
тографиями окрашенных фиксированных препаратов
и сканированными радиоаутограммами, полученными
при ДНК-фингерпринтинге либо цифровыми результа¬
тами исследования профиля ДНК и храниться вместе
с историей ведения клеточной линии в цифровом или
распечатанном виде.
16.4.2. Окрашивание
Применение полихромных красителей, используемых
при окраске мазков крови, таких как краситель Гимза,
является удобным методом для исследований культу¬
ры. Однако при окрашивании мазков крови краситель
Гимза обычно применяется вместе с красителем Мэй-
Грюнвальда, не применяемого в культуре тканей. Сам
по себе краситель Гимза окрашивает ядро в розовый или
фуксиновый цвет, ядрышки в темно-синий, а цитоплаз¬
му в бледный серо-голубой цвет. Он окрашивает клетки,
фиксированные в этаноле или формалине, но не будет
окрашивать препарат, если только в ходе подготовки не
произведена правильная и полная дегидратация.
ПРОТОКОЛ 16.2. Окрашивание по Гимза
Схема
Зафиксировать культуру в метаноле, окрасить ее
непосредственно неразведенным красителем Гимза,
и затем развести краситель 1:10. Промыть культуру,
и исследовать ее во влажном состоянии.
Материалы
Нестерильные:
• D-PBSA
• D-PBSA: метанол, 1:1
• Концентрированный краситель Гимза
• Метанол
• Деионизированная вода
Протокол
Этот протокол предполагает, что используется
монослой клеток, но, начиная с шага 6, можно
16.4. Морфология клеток
291
использовать также фиксированные клетки клеточ¬
ной суспензии (см. разд. 16.4.4).
1. Удалите среду.
2. Промойте монослой D-PBSA, удалите раствор
3. Добавьте 5 мл D-PBSA/метанола на площадь
25 см2. Инкубируйте в течение 2 мин и затем
слейте смесь DPBSA/метанол.
4. Добавьте 5 мл свежего метанола и инкубируйте
10 мин.
5. Слейте метанол и замените его свежим безводным
метанолом. Промойте монослой, затем удалите
метанол.
6. В этом пункте монослой во флаконе может
быть высушен и переложен на хранение или
непосредственно окрашен. Важно, чтобы ок¬
рашивание было выполнено непосредственно
после промывки свежим безводным метанолом,
даже при использовании высушенного флакона.
Если метанол был вылит из флакона за некоторое
время до окрашивания, то вода будет поглощена
остаточным метанолом и ингибирует последую¬
щее окрашивание. Даже «сухие» монослои могут
поглощать влагу из воздуха.
7. Добавьте профильтрованный краситель Гимза,
2 мл на площадь 25 см2, удостоверившись, что
весь монослой покрыт и остается закрытым в
течение всего времени окрашивания.
8. Через 2 мин разведите краситель в 8 мл воды,
мягко покачивайте дальнейшие 2 мин.
9. Смойте краситель водой так, чтобы образующаяся
пена смывалась струей воды и не оставалась на
клетках. Осторожно промойте клетки проточной
водой, пока не исчезнет розоватый оттенок воды,
но клетки останутся окрашенными (обычно
10-20 с).
10. Слейте воду, промойте монослой в деионизованной
воде и исследуйте клетки под микроскопом, пока
монослой все еще влажен. Храните в высушенном
состоянии, перед исследованием увлажните.
Примечание. Окрашивание по Гимза является прос¬
той процедурой, которая дает хороший, высококонт¬
растный полихромный препарат, однако преципитация
красителя может привести к появлению пятнышек на
клетках. Преципитат образуется в результате окис¬
ления при добавлении воды как пена на поверхности
и по всей толщине раствора красителя, в частности
на поверхности предметного стекла. Промывание
красителя восходящим потоком проточной воды уда¬
ляет преципитированные частицы лучше, чем простое
споласкивание проточной водой или другие способы
промывания стекла, поскольку обеспечивает удаление
всех частиц пены из препарата.
Препараты фиксированных клеток могут также
быть окрашены при помощи кристалл-виолета. Крис-
талл-виолет является монохромным красителем и
окрашивает ядро в темно-голубой, а цитоплазму в
светло-голубой цвет. Этот краситель не так хорош для
морфологических исследований, как краситель Гимза,
но метод легок в использовании и может использовать¬
ся повторно. Его удобно применять для окрашивания
клонов при подсчете, поскольку нет проблем с преци¬
питацией, которая есть в случае окрашивания по Гимза,
легче отмывается, позволяя получить светлый фон и
обеспечивая легкость подсчета колоний.
16.4.3. Культуральные сосуды
для цитологии: монослойные
культуры
Ниже приводится список культуральных сосудов, при¬
меняемых для цитологических исследований
1) Обычные 25-см2 флаконы или 5-см чашки Петри
2) Покровные стекла (стеклянные или пластиковые;
см. Приложение II) в многосекционных чашках,
чашках Петри или пробирках Лейтона (рис. 16.3а;
см. также рис. 8.2 и 8.3).
3) Стекла для микроскопа в 9-см чашках Петри или с
прилагаемыми многосекционными камерами (см.
Приложение II; рис. 16.36)
4) Двухмембранные камеры для культур тканей Opti-
Cell (рис. 16.3в).
ПРОТОКОЛ 16.3. Окрашивание
кристаллвиолетом
Материалы
Нестерильные:
• D-PBSA
• D-PBSA/MeOH: метанол 50% в D-PBSA
• Метанол
• Кристаллвиолет
• Фильтровальная воронка и фильтровальная бу¬
мага таких размеров, чтобы слить 5 мл красителя
с каждой чашки
• Бутыль для использованной краски.
Протокол
1. Удалите среду.
2. Промойте монослой D-PBSA, слейте D-PBSA.
3. Добавьте5 мл D-PBSA/MeOH в 25 см2. Оставьте
на 2 мин и затем слейте D-PBSA/MeOH.
4. Добавьте5 мл свежего метанола и оставьте на
10 мин.
5. Удалите метанол. Промойте проточной водой
чашки и позвольте им высохнуть.
6. Добавьте 5 мл кристаллвиолета на 25 см2 и ос¬
тавьте на 10 мин.
7. Поместите фильтровальную воронку в бутылку
для использованной краски.
8. Слейте краситель в фильтровальную воронку.
9. Промойте чашки проточной водой и затем в
деионизованной воде и оставьте чашки до высы¬
хания.
16.4. Морфология клеток
293
294
ГЛАВА 16. ХАРАКТЕРИСТИКА КЛЕТОК
Рис. 16.2. Продолжение
щщ
т
Рис. 16.2. Продолжение
Рис. 16.2. Продолжение
298
ГЛАВА 16. ХАРАКТЕРИСТИКА КЛЕТОК
• Метанол
• Цитоцентрифуга (см. Приложение И)
• Предметные стекла для микроскопа
• Держатели стекол для центрифуги
• Вентилятор
Протокол
1. Покройте стекла сывороткой и промойте.
2. Высушите стекла при помощи вентилятора.
3. Промаркируйте стекла.
4. Разместите стекла в держатели и вставьте держа¬
тели в ротор.
5. Внесите приблизительно 1-2x105 клеток в 200-
400 мкл среды, по крайней мере, в два блока для
образцов (см. инструкцию изготовителя).
6. Включите центрифугу для перемещения клеток
на стекла при 100 g в течение 5 мин.
7. Быстро высушите стекла, поместите их в МеОН
на 10 мин.
Примечание. Некоторые цитоцентрифуги требуют
проведения фиксации in situ.
• Требования безопасности. При использова¬
нии клеток человека или приматов ротор должен быть
герметично закрыт.
Метод падающей капли. Эта процедура выполня¬
ется так же, как в случае приготовления хромосомного
препарата (см. протокол 16.7), но без колцемида и гипо¬
тонического воздействия. Следует тщательно избегать
образования комков клеток, а клеточная концентрация
должна быть такой, чтобы клетки не перекрывали друг
друга. Клетки на краю капли могут разрушаться.
Фильтрация. Этот метод иногда используется в
эксфолиативной цитологии (дальнейшие детали см.
Инструкции производителей Pall Gelman, Millipore,
Sartorius). Этот метод подходит для отбора образцов
из больших объемов с низкой концентрацией.
16.4.5. Микрофотография
Самым простым методом сохранения изображений
на микроскопе является цифровое фотографирование
при помощи CCD-камеры. Выбор по крайней мере
5-мегапиксельной камеры дает разрешение, более чем
достаточное для публикации результатов. Электрон¬
ный вариант хранения изображений повышает удобс¬
тво поиска, обеспечивает возможность их передачи
на другие сайты, форматирование и редактирование.
Изображение, которое представлено на экране, в то
же время при необходимости может быть распечатано
в черно-белом или цветном варианте.
С уменьшением стоимости цифровых камер трудно
оправдать использование пленочных фотоаппаратов.
Тем не менее если вам удобнее использовать фотоплен-
ПРОТОКОЛ 16.5. Фильтрование суспензии
клеток для цитологического исследования
Схема
Осторожно профильтровать суспензию клеток с
низкой концентрацией через вакуумный фильтр.
Промыть клетки и зафиксировать in situ.
Материалы
Нестерильные:
• D-PBSA, 20 мл
• Метанол, 50 мл
• Краситель Гимза
• Клей (DPX или Permount)
• Прозрачные фильтры (например, 25-мм ТРХ
или поликарбонатный, с пористостью 0,5-мкм,
Millipore, Pall Gelman, Sartorius)
• Держатель фильтра (например, Millipore, Pall
Gelman, Sartorius)
• Термос (Millipore)
• Суспензия клеток (106 ячеек в 5-10 мл среды с
20% сывороткой)
• Вакуумный насос
Протокол
1. Соберите фильтрующий комплект (рис. 16.5) с
прозрачным фильтром диаметром 25-мм и диа¬
метром пор 0,5-мкм.
2. Осторожно профильтруйте вакуумным фильтром
суспензию клеток через фильтр, фильтруя оста¬
ток среды до конца.
3. Осторожно добавьте 10 мл D-PBSA, когда объем
суспензии достигнет 2 мл.
4. Повторите шаг 3, когда объем D-PBS А уменьшит¬
ся до 2 мл.
5. Добавьте 10 мл метанола к D-PBSA и продол¬
жайте добавлять метанол, пока чистый метанол
не будет фильтроваться через фильтр.
6. Выключите вакуум до того, как профильтровался
весь метанол.
7. Поднимите фильтр и высушите на воздухе.
8. Окрасьте фильтр красителем Гимза, промойте
водой и высушите.
9. Поместите фильтр на предметное стекло, при
помощи клея DPX или Permount закройте по¬
кровным стеклом с небольшим грузом, придав¬
ливающим покровное стекло.
ку, то существуют два основных размера негативной
и позитивной пленки Polaroid, которые вы можете
использовать: 35 мм, который лучше подходит для
обычной цветопередачи, и большеформатные черно¬
белые фотографии 9x12 см (3,5x4,5 дюйма), которые
хороши для мелкосерийных черно-белых фотографий.
Может быть использована также пленка одноступен¬
чатого процесса, производимая Fudji. В этом случае
вы можете получить немедленный результат без про-
Рис. 16.4. Цитоцентрифуга. Общий вид внутренней части
центрифуги. Ротор и полипропиленовые держатели стекол
помещены в пазы, один держатель в разобранном виде пред¬
ставлен слева (Sakura Finetek)
Рис. 16.3. Культуральные сосуды для цитологии, (а)
Флаконы Nalge Nunc с подвижной стороной; флакон имеет
съемную сторону для обработки, (б) Слайд-камеры Lab-Tek:
съемные пластмассовые камеры на обычном предметном
стекле микроскопа. Имеются одно-, 2-, 4-, 8- и 16-камерные
стекла, (в) Культуральные камеры OptiCell с верхними и
нижними пластмассовыми мембранами, используемые как
для культивирования клеток, так и для наблюдения (а, б
с любезного разрешения Nalge Nunc; с любезного разрешения
доктора Donna Peehl)
цессов обработки пленки и печати снимка. Стоимость
одного фотокомплекта достаточно высока, но при этом
происходит значительная экономия времени. Тем не
менее некоторые фотокомплекты одноступенчатой
Рис. 16.5. Цитологический фильтр. Фильтрующий комп¬
лект для цитологической препаративной работы (например,
Millipore)
Клеточная суспензия
Фильтр
Поддерживающая манжета
Суппорт
Перфорированная пробка
К вакуумному
насосу
Вакуумный флакон
300
ГЛАВА 16. ХАРАКТЕРИСТИКА КЛЕТОК
ПРОТОКОЛ 16.6. Цифровая фотография
на микроскопе
Схема
Установить микроскоп, проверить выравнивание
оптики и сфокусировать объектив на выбранной
области образца. Проверить соединения камеры с
компьютером, перевести световой луч на камеру,
сфокусировать изображение и сохранить его.
Материалы
Стерильные или асептически приготовленные
• Культура для исследования (удостоверьтесь, что
культура является свободной от грязи, например
замените среду в первичной культуре перед фо¬
тографированием)
Нестерильные:
• Инвертированный микроскоп (см. разд. 5.2.5) со
следующими атрибутами: фазово-контрастные
объективы на 10х и 20х или 40х
• Фазовый конденсор с фазовым кольцом для объ¬
ектива 20х или 40х
• Тринокулярная головка или другой порт с адап¬
тером для CCD камеры
• Нейтральные фильтры плотности, 2х и 4х
• Салфетки для линз
• Графический планшет
• Каретка с микрометрической установкой
Протокол
1. Приготовьте культуру для фотографирования:
а) Для культур во флаконе:
i) Перенесите культуру в ламинарный шкаф,
ослабьте крышку, оставьте культуру
охлаждаться.
п) Затяните крышку на флаконе, когда со¬
держание флакона приобретет комнатную
температуру, чтобы избежать любой де¬
формации флакона при охлаждении.
iii) Ополосните средой внутреннюю часть
внутренней поверхности флакона, чтобы
удалить любое уплотнение, которое могло
сформироваться.
iv) Позвольте пленке, образованной средой,
высохнуть при вертикальном положении
флакона.
б) Для чашек и планшетов:
i) Позвольте культуре охладиться до комнат¬
ной температуры в ламинарном шкафу.
й) Замените крышку чашки или планшеты
(удостовериться, что образец помечен со
стороны дна чашки или планшета).
2. Выберите поле зрения и увеличение (4х лучше
для клонов или фотографирования монослоя,
10х — для репрезентативных снимков и 20х — для
детализации клеток), избегая недостаточно хоро¬
ших полей или попадания в поле зрения марки¬
ровки на флаконе. (Всегда маркируйте боковую
сторону флакона или чашки, а не крышку или
основание.)
3. Проверьте фокусировку:
а) Сфокусируйте бинокуляр, отрегулируйте по
глазам, используя окулярную шкалу.
б) Сфокусируйте микроскоп.
4. Подключите монитор, проверьте яркость и контраст
изображения на экране, регулируя интенсивность
света реостатом на микроскопе для черно-белых за¬
писей и с нейтральными фильтрами плотности для
цветных (хотя изображение в дальнейшем может
быть повторно обработано с помощью компьютера,
если цветопередача неправильна).
5. Повторно сфокусируйте микроскоп, если необ¬
ходимо.
6. Сохраните изображение с разрешением, соот¬
ветствующим окончательному использованию
изображения, обеспечивающему оптимальное
электронное хранение (например, 940x740 зани¬
мает 2 МБ на диске и обеспечивает приемлемое
качество изображения при воспроизведении в
размерах 12x18 см).
7. Выключите свет, чтобы избежать перегревания
культуры.
Примечание. Чтобы минимизировать перегрева¬
ние, может быть включен инфракрасный фильтр.
8. Повторите шаги 3-6 с применением каретки с мик¬
рометрической установкой при том же увеличении,
чтобы показать масштаб увеличения. Если это
изображение обработано тем же самым способом,
как другие снимки, эта фотография может быть
использована для создания масштабной шкалы.
9. Возвратите культуру в инкубатор.
10. Запишите название файла в таблицу с названием
культуры, иначе изображения будет трудно найти.
И. Сохраненные изображения могут рассматри¬
ваться, редактироваться и форматироваться при
помощи программ типа Adobe Photoshop и могут
быть распечатаны на высококачественном струй¬
ном цветном лазерном принтере или принтере
Kodak ColorEase.
обработки не дают достаточного качества, относи¬
тельно низкоконтрастны, а цветопередача хуже, чем у
обычных фотопленок. Кроме того, у таких отпечатков
меньший срок хранения.
16.5. ХРОМОСОМНЫЙ СОСТАВ
Хромосомный состав, или кариотип, является одной из
основных характеристик и наиболее точным критерием
для идентификации клеточной линии и связи его с тем
видом и полом животного, от которого они были полу¬
чены. Примеры кариотипов мыши и человека можно
найти на сайте www.pathology.washington.edu/galleries/
16.5. Хромосомный состав
301
Cytogallery/, а крысиный на сайте http://ratmap.gen.
gu.se/Idiogram.html. См. также кариотин человека в
Mitelman [1995] и других млекопитающих — Hsu and
Benirschke [1967], хотя последний был опубликован
прежде, чем была обнаружена исчерченность хромосом.
Хромосомный анализ может также позволять разли¬
чать нормальные и трансформированные клетки (см.
разд. 18.3.1), поскольку количество хромосом наиболее
стабильно в нормальных клетках (кроме мышей, где
хромосомный набор нормальных клеток при культи¬
вировании может достаточно быстро меняться).
ПРОТОКОЛ 16.7. Приготовление
хромосомного препарата
Схема
Зафиксировать клетки, блокированные в метафазе и
набухшие в гипотонической среде. Капнуть клетки на
стекло, окрасить и исследовать (рис. 16.6) [Rothfels и
Siminovitch, 1958; Rooney и Czepulkowski, 1986].
Материалы
Стерильные или асептически подготовленные:
• Культура клеток в log-фазе
• Кольцемид, lxlO-5 М в D-PBSA
• D-PBSA
• Трипсин неочищенный, 0,25%
Нестерильные:
• Гипотонический раствор: 0,04 М. KCL, 0,025 М
цитрат натрия
• Свежеприготовленный фиксатор (уксусный
метанол): 1 часть ледяной уксусной кислоты
плюс 3 части безводного метанола или этанола,
хранить на льду
• Краситель Гимза
• DPX или Permount mountant
• Лед
• Центрифужные пробирки
• Пастеровские пипетки
• Стекла предметные
• Стекла покровные, # 00
• Чашка для стекол
• Низкоскоростная центрифуга
• Вортекс-миксер
Протокол
1. Внесите культуру во флакон 75-см2 при плотности
2х104-5х104 кл./мл (4х103и 1х104 кл./см2) в объ¬
еме 20 мл.
2. Приблизительно через 3-5 дня, когда клетки на¬
ходятся в log-фазе роста, добавьте к среде 0,2 мл
колцемида lxlO-5 М до конечной концентрации
1хЮ"7М.
3. Через 4-6 ч осторожно удалите среду, добавьте
5 мл 0,25% трипсина и инкубируйте культуру
10 мин.
4. Центрифугируйте клетки в трипсине, удалите
супернатант с трипсином.
*
Добавить колхицин
колцемид или
винбластин к клеткам на
стадии log-фазы на 4-6 ч
для блокирования клеток
в метафазе митоза
Трипсинизировать
клетки или стряхнуть
метафазные клетки, если
они слабо прикреплены,
ресуспендировать
в гипотоническом
цитратном растворе при
37 °С на 5-30 мин
Поместить пробирку с
суспензией в холодный
уксуснокислый метанол,
центрифугировать, и
ресуспендировать в
свежем фиксаторе
Повторно
центрифугировать,
ресуспендировать в
свежем фиксаторе
при более высокой
концентрации, капнуть
суспензию на стекло и
быстро высушить
Проверить плотность
клеток и их
распластывание; если
удовлетворительное,
окрасить по Гимза и
закрепить покровное
стекло
Когда клей высохнет,
исследовать под
100х иммерсионным
объективом
Рис. 16.6. Приготовление препарата хромосом. Приготовле¬
ние препарата хромосом из монослойной культуры методом
раскалывания
302
ГЛАВА 16. ХАРАКТЕРИСТИКА КЛЕТОК
5. Ресуспендируйте клетки в 5 мл гипотонического
раствора и оставьте их в течение 20 мин при 37 °С.
6. Добавьте равный объем свежеприготовленного
холодного уксусного метанола, постоянно пере¬
мешивая, затем отцентрифугируйте клетки при
100 g в течение 2 мин.
7. Удалите супернатант, разбейте осадок на вортекс-
миксере, медленно добавляя свежий уксусный
метанол при постоянном перемешивании.
8. Оставьте клетки в течение 10 мин на льду.
9. Центрифугируйте клетки в течение 2 мин при
100 g.
10. Удалите супернатант, содержащий уксусный ме¬
танол, и ресуспендируйте осадок на вортекс-мик-
сере в 0,2 мл уксусного метанола, чтобы получить
суспензию отдельных клеток.
11. Наберите одну каплю суспензии в кончик пасте¬
ровской пипетки и капните примерно с высоты
30 см на холодное предметное стекло. Наклоните
стекло и позвольте капле стечь по стеклу.
12. Быстро высушите стекло над сосудом с кипящей
водой, исследуйте в фазово-контрастном мик¬
роскопе. Если клетки равномерно распределены
и не касаются друг друга, приготовьте большее
количество стекол, используя ту же самую кон¬
центрацию клеток. Если клеток слишком много
и они перекрывают друг друга, то надо развести
суспензию в 2 раза и в 4 раза и вновь сделать
препарат капли. Если клетки при разведении
находятся в удовлетворительной концентрации,
приготовьте большее количество стекол. Если
нет, то разведите суспензию далее и повторите
этот шаг.
13. Окрасьте клетки красителем Гимза:
а) Разместите стекла в кювету для окрашивания,
помещенную над раковиной.
б) Полностью покройте клетки несколькими
каплями профильтрованного красителя Гимза
на 2 мин.
в) Залейте стекла приблизительно 10-кратным
объемом воды.
г) Оставьте стекла для дальнейшей окраски еще
на 2 мин.
д) Отмойте краситель проточной водой.
Примечание. Не сливать краситель со стекол,
поскольку это оставит на стенке пену окислившейся
краски; всегда смывать краситель водой. Даже когда
окрашивание проводится в чашке, краситель никогда
не сливают, он должен быть смыт струей воды.
е) Закончите окраску, промывая каждое стекло
проточной водой, чтобы удалить любой осадок
красителя.
ж) Проверьте окраску под микроскопом. Если
качество удовлетворительно, полностью вы¬
сушите стекло, закройте покровным стеклом
#* 00 при помощи клея DPX или Permount.
Варианты
Блокирование клеток в метафазе. 1) Можно ис¬
пользовать винбластин, 1х10"6М вместо колцемида.
2) Продолжительность блокирования клеток в мета¬
фазе может быть увеличена для того, чтобы увеличить
количество метафазных пластинок на препарате для
подсчета хромосом, или укорочена для снижения
конденсации хромосом и улучшения распознавания
исчерченности (см. разд. 16.5.1).
Сбор митотических клеток. Метафазные клет¬
ки некоторых культур, например СНО или HeLa,
легко отделяются от субстрата при встряхивании
(метод «встряхивания»), не требуя трипсинизации.
Процедура проводится следующим образом: ^до¬
бавить колцемид, 2) осторожно удалить колцемид и
заменить его гипотоническим раствором цитрат/КС1,
3) встряхнуть флакон для того, чтобы отделить от под¬
ложки метафазные клетки до или после инкубации в
гипотонической среде, и 4) фиксировать клетки, как
описано выше.
Гипотоническая обработка. Заменить только
0,075 М КС1 или вместе с 50% раствором HBSS в
дистиллированной воде на гипотонический цитрат,
использованный в протоколе 16.9. Продолжительность
гипотонической обработки может варьировать от 5 до
30 мин для снижения лизиса или улучшения расплас¬
тывания клеток.
Распластывание клеток по стеклу. Возможно, на
этой стадии существует больше возможностей для ва¬
риаций, чем на какой-то еще. Эти вариации призваны
обеспечить большую степень и площадь распласты¬
вания клеток на препарате. Эти вариации включают:
1) изменение высоты падения капли на стекло (закре¬
пите пипетку в штативе и отметьте положение стекла,
проведя пробную серию испытаний с определением
оптимального); 2) высушивание в пламени (высушите
стекло после падения капли прогреванием над пламе¬
нем или выжиганием фиксирующей жидкости пламе¬
нем сразу после распределения капли по стеклу; однако
последний может затруднить последующую идентифи¬
кацию исчерченности); 3) глубокое охлаждение стекла
(поместите стекло на твердый С02 до распластывания
клеток); 4) охлаждение осажденной клеточной суспен¬
зии в холодильнике в течение ночи до того, как капнуть
на стекло; 5) капать на охлажденное стекло (например,
погрузить стекло в холодный этанол и высушить перед
применением) и затем поместить стекло над стаканом
кипящей воды; 6) наклонить стекло или сдуть каплю
по стеклу в то время, как капля растекается (см. также
протокол 27.5, 27.9).
16.5.1. Дифференциальное окрашивание
хромосом
Эта группа методов (см. [see Rooney & Czepulkowski,
1986] была разработана для того, чтобы облегчить
идентификацию отдельных пар хромосом при неболь¬
16.5. Хромосомный состав
303
ших морфологических различиях между парами [Wang
& Fedoroff, 1972,1973]. Для окрашивания по Гимза бел¬
ки хромосом частично расщепляются трипсином, что
приводит к появлению исчерченности при дальнейшем
окрашивании. При окрашивании хинокрином трип-
синизация не требуется. Хромосомная исчерченность
индивидуальна для каждой пары хромосом (рис. 16.7).
Существуют следующие методы выявления хромо¬
сомной исчерченности 1) метод Гимза (использование
только трипсина или, чаще, совместное применение
трипсина и EDTA); 2) Q-окрашивание [Caspersson et
al., 1968] (окрашивание клеток 5%-м (v/v) раствором
дигидрохлорида хинокрина в 45%-й уксусной кисло¬
ты, промывание препарата и заключение препарата в
деионизованной воде при pH 4.5) [Lin & Uchida,1973;
Uchida & Lin, 1974]; 3) С-окрашивание (этот метод поз¬
воляет выделить районы центромеров, фиксированные
препараты предварительно обрабатываются в течение
15 мин 0,2 М НС1 и 2 мин 0,07 М NAOH и затем обра¬
батываются в течение ночи SSC (либо 0,03 М цитрата
натрия, 0,3 М NaCl или 0,09 М цитрат натрия, 0,9 М
NaCl) до окрашивания красителем Гимза [Arrighi &
Hsu, 1974] (см. протокол 27.5).
Методы, которые были разработаны для идентифи¬
кации хромосом мыши и человека, преимущественно
помогают при анализе гибридов мышиных и челове¬
ческих кариотипов. К этим методам относятся: флюо¬
ресцентное окрашивание красителем Hoechst 33258,
вызывающего более интенсивное свечение мышиных
центромеров по сравнению с человеческими [Hilwig &
Gropp, 1972; Lin et al., 1974], щелочное окрашивание
красителем Гимза («Giemsa-И») [Bobrowet al., 1972;
Friend et al., 1976] (см. рис. 16.7e) и гибридизация in
situ с хромосомными красителями (см. ниже и прото¬
кол 27.9).
16.5.2. Хромосомный анализ
Ниже описаны методы, при помощи которых может
быть проанализирован хромосомный набор клетки:
1) Подсчет хромосом. Подсчет количества хромосом
на препарате проводится для 50-100 мазков (хро¬
мосомы должны иметь исчерченность). При помо¬
щи кабельного телевидения или камеры-люцида
можно облегчить процесс. Вы должны постараться
подсчитать все митозы, которые вы увидите, и
классифицировать их а) по количеству хромосом и
б) если подсчет невозможен, то как «почти дипло¬
идные, без возможности подсчета» «полиплоидные
без возможности подсчета». Представьте результа¬
ты в качестве гистограммы (см. рис. 3.9).
2) Кариотип. Сделайте цифровые фотографии при¬
мерно 10-20 препаратов дифференциально окра¬
шенных хромосом. Используя Adobe Photoshope
или аналогичные графические программы, вырежь¬
те отдельные хромосомы и поместите их в новый
файл, где их можно будет развернуть, подрезать,
разместить в линию и рассортировать (рис. 16.8).
Рис. 16.7. Окрашивание хромосом, (а) Выявление дискоа
в хромосомах человека по стандартному методу окраски по
Гимза. (6) Тот же препарат, что и в (я), но окрашенный по
Hoechst 33258. (в) Гибрид человеческой и мышиной клеток,
окрашенный по Гимза при pH 11. Хромосомы человека
менее интенсивно окрашены, чем мышиные хромосомы.
Некоторые хромосомы претерпели транслокацию, образовав
человеческо-мышиный гибрид. (С любезнеого разрешения
R.L. Church.)
Для получения кариотипа следует использовать
программы, позволяющие автоматически анализи¬
ровать изображение (например, Leica CW4000).
Дифференциальное окрашивание хромосом (см.
протокол 27.5) и окрашивание хромосом (см. про¬
токол 27.9) позволяют получить совокупный образ
типичного кариотипа в виде стилизованной картины,
304
ГЛАВА 16. ХАРАКТЕРИСТИКА КЛЕТОК
£8 5{8 ** * “
ОД лл лл •
Приготовить цифровую фотографию
отдельно расположенных хромосом
Выбрать и скопировать каждую
хромосому отдельно
Повернуть хромосому так, чтобы она
располагалась вертикально, короткими
плечами вверх
Вырезать хромосомы и расположить
так, чтобы центромеры находились
на одной линии, рассортировать по
размерам и форме
Отбросить остатки, включая
нежелательные части примыкающих
хромосом
Когда все хромосомы на месте,
установите яркость и контрастность
так, чтобы подавить фон
Рис. 16.8. Препарат кариотипа. Последовательные этапы приготовления картины кариотипа с цифровой фотографии
метафазной пластинки. Клетки китайского хомячка, повторно клонированные из штамма Y-5 Yerganianb and Leonard [1961]
(уксуснокислый орсин)
называемой идеограммой. Идеограммы человека, мыши
и лошади доступны на сайте www.pathology.Washington,
edu/research/cytopages/а крысы — на сайте ratmap.gen.
gu.se/Idiogram.html.
Подсчет хромосом и кариотипирование позволяет
провести видовую идентификацию клеток и при
использовании дифференциального окрашивания
хромосом различить варианты клеточных линий и
хромосомные маркеры. Тем не менее связывание и
кариотипирование являются трудоемким процессом.
Для подтверждения или исключения перекрестной
контаминации культур может быть эффективным
подсчет хромосом с быстрой проверкой морфологии
самой большой хромосомы.
16.5.3. Окрашивание хромосом
С развитием флюоресцентно меченных зондов, которые
связываются со специфическим участком и даже со
специфическим геном на хромосоме, стало возможным
определить локализацию генов, идентифицировать
транслокации и определить видовой источник хромо¬
сом. Этот метод применяется в технологии гибриди¬
зации in situ, которая также может быть использована
для экстрахромосомальной и цитоплазматической
локализации специфических аминокислотных после¬
довательностей, а также для определения местонахож¬
дения специфических видов месенджерной РНК (см.
протокол 27.8 и 27.9).
16.6. Содержание ДНК
305
16.6. СОДЕРЖАНИЕ ДНК
Содержание ДНК может быть измерено по флюорес¬
ценции пропидия иодида при помощи CCD камеры
или в проточном цитофлюориметре [Vaughan & Milner,
1989] (см. разд. 15.4 и 21.2), хотя создание необходимой
суспензии единичных клеток разрушает исходную
топографию образца. Анализ содержания ДНК, в
частности, полезен при характеристике трансформи¬
рованных клеток, которые часто анеуплоидны или
гетероплоидны (см. разд. 18.3).
16.6.1. Гибридизация ДНК
Гибридизация специфических молекулярных зондов с
уникальными последовательностями ДНК (Саузерн-
блоттинг) [Sambrook et al., 1989; Ausubel et al., 1996]
может позволить получить информацию о видоспе¬
цифичных участках, амплифицированных участках
ДНК, или изменении основных последовательностей,
специфичных для данной клеточной линии. Таким
образом, штамм-специфическая генетическая амп¬
лификация, например амплификация гена дигидро-
холатредуктазы (DHFR), может быть определена в
клеточных линиях при селекции по устойчивости
к метотрексату [Biedler et al.,1972]; амплификация
MDR-гена в винбластин-устойчивых клетках [Schoen-
lein et al., 1992]; гиперэкспрессия специфических
онкогенов в трансформированных клеточных линиях
[Bishop,1991; Hames & Glover, 1991; Weinberg, 1989];
а также делеция или утрата гетерозигоности супрес¬
сорных генов [Witkowski, 1990; Marshall, 1991]. Хотя
подобные нарушения могут быть определены при
рестрикционном расщеплении цельной ДНК, метод
ограничен необходимым количеством ДНК и более
общепринято использование ПЦР-метода с приме¬
нением специфических праймеров к интересующей
последовательности, позволяющей проводить де¬
текцию при относительно малом количестве клеток,
что обусловливает его использование в лаборатор¬
ных условиях. В альтернативном методе детекции
специфических последовательностей ДНК методом
гибридизации in situ (см. разд. 27.8) могут быть ис¬
пользованы специальные зонды, применение которых
имеет ряд преимуществ, позволяющих определить
£
ш .—.
> ™
□ О 2 О О» •§
У. м
. 3 S 3 я я 1
Ш
° О
Рис. 16.9. ДНК-фингерпринтинг. Саузерн-блоттинг ДНК клеточных линий, расщепленных при помощи рестрикционного
фермента Hinfl и гибридизованньхе с минисателлитным зондом 33.15 [Jeffreys et al., 1985]. (а) ДНК-фингерпринтинг HeLa
контаминированных клеточных линий и субклонов клеточной линии HeLa cell line. Связывающие участки идентичны в случаях
когда анализировались исходные культуры, полученные из банка клеток (М) и полученные в ходе работы или при передаче
клеток в другие банки (D). Участки фингерпринтинга были стабильны для клеточных линий, но в некоторых случаях были
обнаружены дополнительные полосы (а) (например, HeLa Ohio, INT 407, и Chang Liver). (С любезного разрешения G. Stacey,
NIBSC, UK.) (6) Четыре линии человеческой глиомы—MOG-G-CCM, U-251 MG, SB-18, и MOG-G-UVW (три отдельных
блока)—и удвоение полос контрольной ВНК-21. (С любезного разрешения G. Stacey, NIBSC, UK.)
-23Kb
- 9.5Kb
- 6.6Kb
- 2.3Kb
306
ГЛАВА 16. ХАРАКТЕРИСТИКА КЛЕТОК
топографические особенности и гетерогенность ДНК
в клеточной популяции.
Также возможно пометить клеточные штаммы для
дальнейшей идентификации путем трансфекции ре¬
портерных генов, таких как зеленый флюоресцентный
белок (GFP), люцифераза или Р-галактозидаза [Krotz
et al., 2003], и затем определять его по флюоресценции,
люминесценции или при помощи Саузерн-блоттинг
или путем хромогенного ферментативного исследова¬
ния генного продукта.
16.6.2. Фингерпринтинг ДНК
ДНК имеет области, известные как сателлитная ДНК,
которая, по-видимому, не транскрибируется. Эти об¬
ласти имеют большое количество повторов, их длины
различны, начиная от минисателлитной ДНК, имею¬
щей от 1 до ЗО-kb повторов, до микросателлитной ДНК,
имеющей в повторяющихся последовательностях
только 2-4 основания. Функции этих областей недо¬
статочно понятны, они могут иметь чисто структурную
функцию или могут предоставлять потенциальную
область, кодирующую генетические рекомбинации для
дальнейшей эволюции. Тем не менее независимо от их
функций эти области не являются высококонсерва¬
тивными, поскольку они не транскрибируются, и они
дают начало областям гипервариабельности. Когда
ДНК режется специфическими эндонуклеазами, то
специфические последовательности могут быть поме¬
чены при помощи кДНК, которые гибридизируются
с этими гипервариабельнами участками, либо могут
быть амплифицированы ПЦР-методом со специфи¬
ческими матрицами. Эти зонды были разработаны
Jeffreys et al. [1985] (см. протокол 16.8; Stacey et al.,
1992). Электрофорез показывает варианты в длине
фрагментов сателлитной ДНК (полиморфизм длины
рестрикционных фрагментов (RFLPs), которые явля¬
ются специфичным для того организма, из которого
выделена ДНК. При анализе методом электрофореза
в полиакриламидном геле каждая индивидуальная
ДНК дает специфический гибридизационный учас¬
ток (паттерн), который можно установить методом
ауторадиографии с радиоактивными или флюоресцент¬
ными зондами. Эти участки известны как «отпечатки
пальцев» и специфичны для каждой клеточной линии,
если только от одной особи не получено несколько
клеточных линий или если линии клеток не получены
из одной высоко инбредной линии животных.
ДНК-фингерпринты достаточно стабильны. При
культивировании клеточных линий, полученных из
одного источника, но поддерживаемых в разных лабо¬
раториях в течение многих лет сохраняются очень по¬
хожие фингерпринты. ДНК-фингерпринтинг является
очень эффективным методом в определении происхож¬
дения клеточной линии, если сохранилась исходная
клеточная линия, или ДНК, полученная из нее или от
донорской особи. Этим подчеркивается необходимость
сохранения крови, тканей, или образца ДНК при вы-
ПРОТОКОЛ 16.8. Мультилокусный
фингерпринтинг ДНК клеточных линий
а) Приготовление Саузерн-блотов
ДНК клеточных линий
Схема
Экстракция ДНК из клеток расщеплением при помо¬
щи рестрикционных ферментов иммобилизованных
на нейлоновой мембране методом Саузерн-блоттин-
га [Ausubel et al., 1996,2002], и гибридизация ДНК
с меченой фаговой ДНК фага М13.
Материалы
Стерильные:
• D-PBSA: фосфатно-буферный раствор, без Са2+и
Mg2+ (pH 7,2)
• Hinfl фермент и буфер (например, life Тechnolo-
gies)
Нестерильные:
• Агарозные гели: 0,7% агароза (Тип 1А, Sigma) в
lxTBE, 1 мг/л бромистого этидия
• Требование безопасности. Бромистый
этидий — потенциальный канцероген. Выполняйте
правила безопасности.
• ТВЕ, 5х (основной раствор, pH 8,2): 54 г основно¬
го Трис (Sigma), 27,5 г борной кислоты (Sigma), и
20 мл 0,5 М. EDTA, динатриевой соли (Sigma)
• Загрузочный буфер, 6х: 0,25% (объемных) бром-
фенол-синий, 0,25% (объемных) цианол ксилола,
и 40% (объемных) сахарозы в дистиллированной
воде
• Кислотный промывочный раствор: 500 мл 0,1 М
HCL
• Щелочной промывочный раствор: 500 мл 0,2 М
гидроокись натрия, 0,6 М NaCl
• Нейтрализующий раствор: 500 mL 0,5 М Трис,
pH 7,6,1,5 М NaCl
• SSC, 2х: 1-к-10 разведение основного раствора
20х SSC
• SSC, 20. (основной раствор) 175,3 г NaCl (Analar,
Merck) и 88,2 г цитрата натрия (Analar, Merck)
растворенного в дистиллированной воде до ко¬
нечного объема 1,0 л, при pH 7,2.
Протокол
1. Дважды отмойте в D-PBSA приблизительно
lxlO7 ячеек (полученных соскребанием или
трипсинизацией) и получите осадок клеток в виде
таблетки.
2. Из определенного небольшого количества экстра¬
гируйте высокомолекулярную геномную ДНК,
как описано в шаге 6 этого протокола; проверьте
ее целостность электрофорезом в минигеле. Ме¬
тод экстракции ДНК без применения фенола,
16.6. Содержание ДНК
307
который обычно используется для клеточных
линий, был описано в Stacey et al. [1992].
3. Определите количество ДНК УФ-спектроско-
пией, используя разведение ДНК 1:50 [Sambrook
et al., 1989].
4. Смешайте 5 мкг ДНК с 40 U фермента Hinfl,
3 мкл 10х ферментного буфера и стерильной
дистиллированной водой до конечного объема
30 мкл.
5. Инкубируйте смесь при 38 °С в течение ночи.
Повторно перемешайте, через 1 час микроцент-
рифугируйте гидролизат.
6. Проверьте завершенность расщепления, электро¬
форезом в агарозном минигеле 1 мклгидролизата
в 9 мкл электрофоретического буфера (lxTBE) и
2 мкл 6х загрузочного буфера. Hinfl гидролизат
должен показать полоски низкомолекулярных
фрагментов ДНК (< 5 КБ) без остаточной по¬
лосы геномной ДНК (20-30 КБ).
7. К остатку гидролизата добавьте 6 мкл 6х загру¬
зочной смеси, поместите в аналитический агароз¬
ный гель (0,7% в 1х ТВЕ, 1 мг/л бромистого эти-
дия, по крайней мере, 20 см длиной) параллельно
с маркером расщепления Hindlll и гидролизатом
5 мкг ДНК стандартной клеточной линии (на¬
пример, HeLa). Проведите электрофорез смеси
приблизительно в 3 V/см пока фрагмент Hindlll
с 2,3 КБ не достигнет конца геля (т.е. 17-20х).
Сфотографируйте гель против белой линейки
в ультрафиолетовом трансиллюминаторе для
оценки величины перемещения.
8. Обработайте выделенные фрагменты ДНК в геле
кислотным промывочным раствором (15 мин),
щелочным раствором (30 мин) и, наконец, ней¬
трализующим (30 мин). Затем агарозный гель
может быть перенесен на нейлоновую мембрану
Hybond N, Amersham). Наиболее просто исполь¬
зование капиллярного действия, использую¬
щего 20х SSC буфер, как описано Sambrook et
al. [1989]. Затем промойте мембрану в 2х SSC,
высушите и заверните в специальную упаковку
Saran Wrap (Dow) или ее эквивалент.
9. Зафиксируйте фрагменты ДНК на мембране пу¬
тем экспозиции ДНК в ультрафиолетовом свете
(302 нм) в течение 5 мин, используя UV-трансил-
люминатор (например, ТМ20, UVP Inc).
10. Фиксированные мембраны должны храниться в
темном месте в сухих полиэтиленовых мешках до
гибридизации.
б) Подготовка меченного фаговой
ДНК М13 фага
Схема
Пометить ДНК M13mp9 DNA случайным расши¬
рением праймера [Feinberg и Vogelstein, 1983] и
очистить ДНК на колонке Sephadex.
Материалы
Стерильные:
• Матрица: M13mp9 ssDNA (0,25 нгДамл; ВоеЬ-
ringer Mannheim), растворенный 1:4 в дистилли¬
рованной воде
• HEPES, 1 М., pH 6,6
• DTM реактив: dATP, dCTP, и dTTP, по 100 мкМ
каждого в 250 тМ Трис • HCL, 25 мМ MgCl2 и
50 мМ 2-меркаптоэтанола (pH 8,0)
• Требование безопасности■ Помните, что
2-меркаптоэтанол летуч и ядовит.
• OL реактив: 90 О. D. единиц олигодеоксирибонук-
леотидов в 1 тМ Трис и 1 тМ EDTA (PH 7,5)
• Буфер для метки: LS буфер (100:100:28 1 М
HEPES: DTM: OL)
• Бычий сывороточный альбумин (20 мг/мл, для
использования в молекулярной биологии, Life
Technologies)
• а — [32Р] dGTP (3 Ci/ммоль; Amersham Interna¬
tional Pic)
• Требование безопасности. Помните, что
32Р — источник Р-частиц, обладающих высокой
энергией, действие которых должно быть мини¬
мизировано. Всегда используйте перчатки и 1-см
плексигласовый экран.
• Фермент Кленова (1 мкг/мл, для сиквенса;
Invitrogen)
• Sephadex колонка (например, колонка NICK,
Amersham Pharmacia)
• Буфер для колонок: 10 тМ Трис • HCL, pH 7.5,
I тМ EDTA
Протокол
1. Смешайте 2 мкл растворенного ДНК М13 с
II мкл дистиллированной воды в стерильной
микропробирке.
2. Поместите пробирку в кипящую водяную баню
на 2 мин.
3. Переместите пробирку на лед на 5 мин.
4. Добавьте в пробирку 11,4 мкл LS буфера и 0,5 мкл
BSA.
5. За плексигласовым экраном добавьте в пробирку
10 мкл [32Р] dGTP и 2 мкл фермента Кленова.
6. Перемешайте содержимое пробирки, кратковре¬
менно центрифугируйте.
7. Оставьте пробирку на ночь при комнатной тем¬
пературе за плексигласовым экраном.
8. Аккуратно нанесите содержимое пробирки при
помощи пипетки на колонку Sephadex, предвари¬
тельно уравновесив с 10 мл буферного раствора
для колонок.
9. Применив 2х 400 мкл буферного раствора для ко¬
лонок, соберите элюат со второй порции, 400 мкл,
в микропробирку как очищенную пробу.
308
ГЛАВА 16. ХАРАКТЕРИСТИКА КЛЕТОК
• Требование безопасности. В колонке ос¬
танется высокий уровень радиоактивности, из-за
задержания в ней невключенного нуклеотида
10. Кипятите очищенную пробу в течение 2 мин и
охладите на льду перед смешиванием гибриди-
зационным раствором.
в) Гибридизация
Схема
Гибридизовать меченую ДНК М13тр9 с ДНК кле¬
ток, полученной методом Саузерн-блоттинга; после
тщательной отмывки использовать авторадиогра¬
фию чтобы визуализировать образцы фрагментов,
связавшие последовательности М13 [Westneat и
другие, 1988].
Материалы
Нестерильные:
• Прегибридизационный/гибридизационный рас¬
творы: 0,263 М динатрийфосфат, 7% (объемных)
додецилсульфат натрия, 1 мМ EDTA, 1% (объем¬
ных) V фракции BSA (Roche)
• Раствор для тщательной промывки: SSC, 2х
• SSC, 20х (основной раствор): 175,3 г хлористого
натрия (Analar, Merck) и 88,2 г цитрата натрия
(Analar, Merck) растворенных в дистиллирован¬
ной воде до конечного объема 1 л при pH 7,2
Протокол
1. Добавьте мембраны к 200 мл (до трех мембран)
предварительно нагретого до 55 °С раствора для
прегибридизации в полиэтиленовой коробке для
бутербродов.
2. Покачивайте коробку 4 ч при 55 °С.
3. Переместите мембраны в 150 мл предварительно
нагретого раствора для гибридизации во второй
коробке для бутербродов при 55 °С, добавьте
прокипяченый и охлажденный зонд.
4. Покачивайте коробку при 55 °С в течение ночи,
как минимум 16 ч.
5. Перенесите (до двух мембран) в 1 л раствора для
тщательной промывки и покачивайте смесь при
комнатной температуре в течение 15 мин.
6. Повторите шаг 5.
7. Замените промывочный раствор 1 л предвари¬
тельно нагретого до 55 °С промывочного раствора
плюс 0,1% SDS и покачивайте коробку при 55 °С
в течение 15 мин.
8. Промойте мембраны в lx SSC при комнатной
температуре.
9. Подсушите мембраны на воздухе на бумажном
фильтре, затем оберните их в Saran Wrap и опре¬
делите при помощи счетчика Гейгера-Мюллера
уровень радиоактивности. Примените метод ав¬
торадиографии при -80 °С, позволяющий оце¬
нить время экспозиции пленки для регистрации
X-лучей. Используйте две пленки (например, Fuji
RX) для гибридизованной Саузерн-мембраны в
кассете для авторадиографии.
10. Проявите одну пленку после ночной экспозиции,
для того чтобы обеспечить более точную оценку
требуемого времени экспозиции. Используйте
стандартные реактивы для регистрации Х-лучей
(например, 1:5 (по объему) проявитель CD 15
в течение 5 мин и CD40 фиксатор в течение
5 мин).
делении первичной культуры (см. разд. 12.3.11). Более
того, при предполагаемой перекрестной контаминации
или подозрении на неправильную идентификацию
фингерпринтинг может быть использован для иссле¬
дования клеток и всех потенциальных источников
заражения. ДНК-фингерпринтинг может подтвердить
результаты ранее проведенного изоферментного ана¬
лиза или кариотипирования, [Gartler, 1967;Nelson-Rees
& Flandermeyer, 1977; Lavappa, 1978;Nelson-Rees etal.,
1981], показав, что многие широко используемые кле¬
точные линии заражены линией HeLa (см. разд. 16.3,
7.10.1, 19.5; рис. 16.9 и табл. 13.2). Дальнейший про¬
токол ДНК-финггерпринтинга был разработан Glyn
Stacey (NIBSC, SouthMimms, Herts EN6 3QG, England,
United Kingdom). После публикации первого сообще¬
ния о мультилокусном ДНК-фингерпринтинге Jeffreys
et al. [1985] было разработано множество зондов для
анализа полиморфных локусов, называемых VNTRs
(variable number tandem repeats, вариабельное коли¬
чество тандемных повторов). При мультилокусном
ДНК-фингерпринтинге зонды используются в таких
условиях, которые позволяют проводить перекрестную
гибридизацию с различными семействами ДНК с пов¬
торяющимися последовательностями. Таким образом,
конечный результат представляет собой разнообраз¬
ную информацию из различных частей генома и, таким
образом, метод потенциально чувствителен к измене¬
ниям в составе ДНК генома клеточной линии. Первый
протокол, приводимый здесь, основан на исходной
методике мультилокусного ДНК-фингерпринтинга,
разработанного Jeffreys et al. (1985), который был с
успехом применен в экологических исследованиях кле¬
ток млекопитающих и немлекопитающих животных,
а также растений и микроорганизмов. Пока еще этот
подход требует высокого уровня образования и опыта
проведения электрофореза в агарозном геле. Быстрый
блот-анализ при Саузерн-блоттинге основан на при¬
менении сигнальной системы щелочной фосфатазы,
что позволяет получить результаты анализа за один
рабочий день. Использование в качестве зонда повто¬
ряющихся последовательностей генома фага М13 для
исследования идентичности было продемонстрировано
на животных, растениях и бактериях и таким образом
представляет собой очень широкомасштабную технику
аутентификации, которая имеет дополнительное пре¬
имущество, заключающееся в том, что чистые ДНК-
16.6. Содержание ДНК
309
зонды коммерчески доступны, и при предполагаемом
исследовании клеточных линий имеющих различную
видовую принадлежность, этот метод анализа иден¬
тичности клеточной линии имеет достаточно низкую
стоимость. Коммерческие наборы и системы для
ПЦР-анализа являются быстрым решением вопроса
аутентичности клеточных линий человека, и будут
обсуждаться ниже.
• Требование безопасности. Описанная проце¬
дура включает работу с радиоактивными материалами
и должна проводиться в соответствии с государствен¬
ными и частными требованиями безопасности. До
начала проведения такой работы проконсультируйтесь
в комитете по биологической безопасности.
•
Обсуждение. Этот простой метод фингерпринтин-
га использует ДНК фага М13 (рис. 16.9), который дает
подходящие образцы для фингерпринтинга клеточных
линий широкого круга клеток животных, включая на¬
секомых. Обратите внимание, что некоторые культуры
могут дать фингерпринты с полосами высокого моле¬
кулярного веса, которые плохо различимы. И в этом
случае может быть полезно интерпретировать только
те полосы, которые находятся в диапазоне MB 3-15 kb.
Разрешающая способность данного метода может
быть повышена использованием 0,78 kb фрагмента
Clal-Bsml М13 DNA, содержащего наиболее полезные
зондовые последовательности. Могут применяться
также нерадиоактивно меченные зонды [Moreno,
1990]. Кроме того, для фингерпринтинга могут быть
полезны зонды 33,15 и 33,6 [Jeffreys et al., 1985] и
олигонуклеотидные зонды, например (GTG)5 [Ali et
al., 1986], которые предлагаются компаниями Tepnel
и Fresenius, соответственно. Тем не менее ДНК фага
М13 является наиболее легкодоступным и дешевым
зондом для мультилокусного фингерпринтинга, кото¬
рый может быть с успехом применим для широкого
диапазона видов клеток.
Разнообразные однолокусные зонды и ПЦР-метод
для анализа VNTR также успешно испытан для иден¬
тификации различных культур клеток. Мультиплекс¬
ные PCR-наборы для человеческих клеточных линий
(например, Promega) позволяют получать профили,
специфичные для данного источника происхожде¬
ния культуры клеток. Однако опыт работы с такими
системами показывает, что Саузерн-тест-системы
могут оказаться неспособны обнаружить различия
между клоном и общим источником (Y. Reid, American
Type Culture Collection, личное общение), хотя эти
различия могут быть найдены при помощи мультило¬
кусного ДНК-фингерпринтинга [Stacey et al., 1993].
При использовании методов, основанных на рандо¬
мизированном амплифицированном полиморфном
анализе ДНК, которые используют короткие случайно
выбранные праймерные последовательности могут
возникать проблемы в воспроизведении результатов.
Тем не менее однопраймерный методы, основанные на
мини- или микросателлитных последовательностях,
являются более многообещающими и могут получить
в дальнейшем широкое распространение [Meyer et
al., 1997]. ПЦР-методы также могут применяться
для определения видового происхождения клеточ¬
ной линии [Stacey et al., 1997]. Существуют также и
другие методы идентификации, которые не требуют
использования ПЦР, в том числе изоферментный
анализ (см. разд. 16.8.2) и электрофорез продуктов
рестрикции геномной ДНК в агарозном геле [Stacey
et al., 1997].
Для аутентификации человеческих клеточных
линий существует все возрастающее число компаний,
предлагающих системы идентификации (Приложе¬
ние И). Автор не может ручаться за качество систем,
предоставляемых этими организациями, поэтому очень
важно при взаимодействии с подобными компаниями
задать ряд вопросов, которые позволят вам убедиться
в том, что предлагаемое оборудование соответствует
вашим потребностям. Необходимо узнать следующее:
• Производят ли они предлагаемые системы сами или
только являются посредниками, осуществляющими
продажу?
• В чем специфичность метода, используемого для
индивидуальной идентификации?
• Используются ли генетические маркеры, при помо¬
щи которых работают профессиональные организа¬
ции или другие экспертные центры?
• Имеется ли опыт интерпретации данных для кле¬
точных линий?
• Имеются ли у них специалисты, по работе с этим
методом, для консультации в интерпретации полу¬
ченных данных?
• Имеют ли они аккредитацию соответствующей
профессиональной или правительственной орга¬
низации или формальное членство в какой либо
экспертной группе или организации?
Следует помнить, что крупные организации по сбо¬
ру и коллекции образцов, такие как АТСС (http://www.
atcc.org), ЕСАСС (hpa.org), DSMZ (http://www.dsmz.
de), JCRB (http://cellbank.nibio.go.jp/), также могут
дать консультацию по тестированию и провести его
как сервисная организация.
Мультилокусные методы имеют определенные
возможности для определения изменений в генети¬
ческой структуре во многих локусах генома [Thacker
et al., 1988] и в целом более универсальны для анализа
большого количества видов. Метод фингерпринтинга,
описанный в протоколе 16.10, предлагает дешевый и
несложный путь для проведения качественного конт¬
роля клеточных линий, полученных из различных
видов животных. Такие методы дают возможность
одновременного подтверждения идентичности и ис¬
ключения перекрестной контаминации. Как показано
рядом авторов (например, Stacey et al., 2000; Masters
et al., 2001) существуют важные критерии для того,
чтобы подтвердить сопоставимость полученных дан¬
ных и исключить потерю времени и средств, которые
неизбежны при неправильной идентификации или
перекрестной контаминации культур.
310
ГЛАВА 16. ХАРАКТЕРИСТИКА КЛЕТОК
16.6.3. Анализ профиля ДНК
Поскольку микросателлитные последовательности
достаточно маленькие, то возможно их идентифици¬
ровать и определять количество коротких тандемных
повторяющихся (STR) последовательностей в опре¬
деленных локусах. Мультиплексная система второго
поколения (SGM) исследует 7 различных областей
генома, a SGM Plus® исследует 11, давая разделяющий
потенциал 1:109. Пока это единственная система, кото¬
рая применяется для человеческих линий клеток, но
имеется большой потенциал для получения большого
количества данных, которыми можно было бы обме¬
няться между различными лабораториями [Masters et
al., 2001; Langdon, 2004]. Исследование профиля ДНК
предлагается как сервисное обслуживание множеством
лабораторий, включая LGC и АТСС. Следующий про¬
токол был представлен Amber James (LGC Diagnostics
Dept., Queens Road, Teddington, Middlesex, TW11 0NJ,
England, UK).
• Фосфатный буфер, 0?01 М. (например, таблетки
Sigma PBS, 2 таблетки, разведенные в 400 мл
воды)
• Перед началом процедуры должны быть подго¬
товлены следующие реактивы:
а) Разведенный раствор протеазы: добавить
соответствующее количество растворителя
протеазы к лиофилизированной протеазе
Qiagen®. Раствор является устойчивым до
2 месяцев при хранении при 2-8 °С.
б) Буферные растворы AW1 и AW2: добавить
соответствующее количество абсолютного
этилового спирта в бутылки, содержащие
концентраты AW1 и AW2. Буферные раство¬
ры AW1 и AW2 стабильны при комнатной
температуре в течение года.
в) AL буферный раствор: перед использова¬
нием тщательно перемешать, переворачивая
флакон.
Протокол
(Протокол изготовителя набора с незначитель¬
ными модификациями.)
Если клетки не суспендированы в культуральной
среде, начните с шага 5.
Если осадок был получен из культуральной
среды:
1. Центрифугируйте образцы при 20 000 g в течение
Змин.
2. Используя автоматическую пипетку с легким
нажимом, тщательно удаляющую супернатант,
слейте его.
3. Ресуспендируйте клетки в 200 мкл PBS.
4. Повторите шаги 1 и 2 еще раз.
5. Ресуспендируйте клетки в 200 мкл PBS.
6. Внесите пипеткой по 20 мкл протеазы Qiagen® в
пустые маркированные 1,5-мл микропробирки.
7. Добавьте 200 мкл каждого образца в пробирки,
содержащие протеазу.
8. Добавьте 200 мкл AL буфера (лизирующего бу¬
фера) в каждую пробирку.
9. Перемешайте каждую пробирку при помощи
вортекс-миксера приблизительно 15 с.
10. Инкубируйте образцы при 56 °С в течение
10 мин.
11. После этого осадите содержимое каждой пробир¬
ки центрифугированием приблизительно 3 с при
1000 g.
12. Аккуратно откройте каждую пробирку и добавьте
200мкл абсолютного этилового спирта.
13. Перемешайте при помощи вортекс-миксера
каждую пробирку 15 с и центрифугируйте при
1000 g в течение 3 с для формирования осадка.
14. Аккуратно перенесите смесь в соответственно
маркированную QIAamp® вращающуюся колон¬
ку, помещенную в 2-мл собирающую пробирку.
15. Закройте крышку колонки и вращайте при 6000 g
в течение 1 мин.
ПРОТОКОЛ 16.9. STR-анализ профиля
ДНК клеточных линий
Теоретическое обоснование
Перекрестная контаминация между линиями кле¬
ток — давняя и частая причина научных ошибок и
заблуждений. По оценкам, до 36% клеточных линий
имеет иное происхождение, чем предполагают иссле¬
дователи. STR-анализ профиля ДНК при помощи
SGM Plus™ обеспечивает быстрый и недорогой путь
идентификации STR профиля линии клеток, которая
передается и распространяется между лаборатория¬
ми [Masters et al., 2001].
Схема
DNA извлекается из осадка клеток при помощи
мини-наборов Qiagen® QIAamp®; экстрагированная
ДНК амплифицируется при помощи SGM Plus™ и
затем разгоняется на акриламидном геле при помощи
секвенатора ABI377 DNA, с последующим анализом
при помощи программного обеспечения GeneScan®
Genotyper®.
а) Экстракция осадка
Реактивы и уатериалы
• ДНК-мини-набор QIAamp®
• Микроцентрифужные пробирки, 1,5 мл (напри¬
мер, пробирки Eppendorf Biopure Safelock)
• Центрифуга (например, Eppendorf 5415 D или
подобная спецификация)
• Водяная баня или духовка, способная к поддер¬
жанию температуры 56 °С
• Вортекс-миксер
• Абсолютный этанол
16.6. Содержание ДНК
311
16. Поместите вращающуюся колонку в чистую
собирающую пробирку и слейте пробирку, со¬
держащую фильтрат.
17. Аккуратно откройте крышку вращающейся ко¬
лонки и добавьте 500 мкл буфера AW 1.
18. Повторите шаги 15 и 16.
19. Аккуратно откройте крышку вращающейся ко¬
лонки и добавьте 500 мкл буфера AW2.
20. Закройте крышку и центрифугируйте при
20 000 g 3 мин.
21. AW2 буфер может оказать негативное влияние на
дальнейшие процессы, поэтому желательно заме¬
нить собирающую пробирку и вращать колонку в
течение следующей минуты при 20 000 g, чтобы
гарантировать, что весь AW2 буфер удален.
22. Поместите колонку в соответственно маркиро¬
ванную микроцентрифужную 1,5-мл пробирку.
23. Откройте крышку колонки и добавьте 200 мкл
АЕ буфера (элюирующий буфер).
24. Инкубируйте при 56 °С в течение 1 мин, затем
центрифугируйте при 6000 g в течение следую¬
щей минуты.
25. Определите количество ДНК в каждом образце,
например, при помощи Picogreen (Molecular
Probes).
26. Если образцы не должны быть амплифицированы
немедленно, поместите их на хранение заморо¬
женными.
б) Амплификация при помощи SGM Plus™
Реактивы и материалы
• Компоненты SGM Plus™ PCR смесь (реакцион¬
ная смесь AMPF/STR® PCR, набор праймеров
AMPF/STR® SGM Plus™, ДНК полимераза
Amplitaq gold®)
• Чистая стерильная вода (например, вода для
культуры тканей Sigma)
• Термоциклер (например, A.BI 9700)
Протокол
1. В соответствии с инструкцией к термоциклеру
ABI приготовьте соответствующую смесь для
образцов, которые будут амплифицированы.
На каждый образец потребуется:
а) 21 мкл реакционной смеси AMPF/STR® PCR;
б) 11 мкл набора праймеров AMPF/STR®SGM
Plus™,
в) 1 мкл ДНК полимеразы Amplitaq gold®).
2. На каждый образец разлейте по 30 мкл реакцион¬
ной смеси в индивидуальные PCR пробирки или
в тонкую пластину для микротитрации, окружен¬
ную стенкой, запечатайте пластину адгезивной
фольгой.
3. Выньте образцы из морозильника и разморозьте.
4. Перемешайте образцы.
5. Добавьте приблизительно 1 нг ДНК к соответ¬
ствующей аликвоте SGM смеси Plus™ PCR
SGM, доведите объем чистой стерильной водой
до 20 мкл.
6. Одновременно амплифицируйте образец с из¬
вестным генотипом в качестве контроля (типа
контрольного набора).
7. Поместите образцы на запрограммированный
термоциклер с соответствующими деталями
цикла, указанными ниже, начните процесс.
а) 95 °С в течение 11 мин±5 с
б) 94±1 °С в течение 1 мин± 2 с
в) 59±1 °С в течение 1 мин±2 с
г) 72 ±1 °С в течение 1 мин±2 с
8. Повторите шаги 7(б)-7(г) 28 раз.
9. 60±1 °С в течение 45 мин±5 с.
10. Храните при 4 °С пока не потребуется.
в) Гель-электрофорез с использованием
ДНК секвенатора ABI377
Реактивы и материалы
• ДНК-секвенатор ABI377 (рис. 16.10)
• Материал для подготовки геля: мерный цилиндр,
весы, фильтрующий блок (например, 150-мл
Sartolab), вакуумный насос, гребенки, 2-мм про¬
кладки для геля, магнитная мешалка, мочевина
(например, Fisher), акриламидная подложка (на¬
пример, Bio-Rad 40% Acrylamide/BIS 19:1), чистая
стерильная вода (например, Fisher AR), mixed bed
resin (например, Sigma), пероксидсульфат аммония
(например, Sigma), ЮхТВЕ (например, National
Diagnostics), TEMED (например, Sigma)
• Материалы для гелевой установки: гелевая плас¬
тина и кассетный аппарат, буферная система,
пластина для теплообмена
• Ледяная или электронная холодная пластина
(например, Camlab Ice cube)
• Термоциклер (например, ABI 9700)
• Смесь для внесения образца в гель (загрузоч¬
ная смесь) SGM Plus™: формамид, краситель
декстран синий (входит в набор), GeneScan 500
(GS500 ROX) (входит в набор)
Протокол
1. Приготовьте 4%-й полиакриламидный гель,
смешивая в химическом стакане следующие
компоненты:
а) Мочевина, 18 г
б) Акриламид, 40%, 5,2 мл
в) Вода, 27 мл
г) Mixed bed resin, 0,5 г
2. Залейте гель.
3. В соответствии с технической инструкцией рас¬
положите ABI377 для разгонки геля.
4. Возьмите амплифицированные образцы из
термоциклера и приготовьте загрузочную смесь
следующим образом:
а) формамид 1,2 мкл;
б) GS5QOROX, 0,13 мкл;
312
ГЛАВА 16. ХАРАКТЕРИСТИКА КЛЕТОК
в) загрузочный буфер с декстраном синим,
0,2 мкл.
5. Аликвота 1,5 мкл загрузочной смеси смешива¬
ется в соответствующей пробирке или пластине.
Одновременно приготовьте загрузочную смесь
для двух аллельных контролей (поставляются
в наборе). К соответствующей пробирке или
ячейке добавьте приблизительно 1 мкл ампли-
фицированного PCR продукта или контроля.
Осторожно перемешайте.
6. Чтобы денатурировать ДНК, нагрейте загрузоч¬
ную смесь для геля, содержащую амплифициро-
ванный ПЦР-продукт, приблизительно до 95 °С
примерно на 3 мин, затем немедленно перенесите
на лоток со льдом или электронный блок охлаж¬
дения до остывания до 0 °С.
7. В соответствии с технической инструкцией за¬
пустите секвенатор и загрузите гель начиная с
образцов, которые будут загружены в нечетные
ряды.
8. Гель разгоняется приблизительно 2,5 часа.
г) Анализ результатов исследования
клеточной линии
Материалы
• Программное обеспечение. GeneScan® и Geno¬
typer®
• Компьютер PowerMac
Протокол
Этот раздел охватывает 2 главные области; анализ
полученных данных, собранных при помощи ДНК-
секвенатора с анализирующим программным обеспе¬
чением GeneScan®, и использование программного
обеспечения Genotyper® для того, чтобы присвоить
аллельные обозначения к профилям, предваритель¬
но проанализированным программным обеспечени¬
ем GeneScan®.
1. Откройте изображение геля, отрегулируйте кон¬
траст, убедитесь, что все цвета могут быть ясно
видны.
2. Установите диапазон анализа, включающий
стандартные внутренние пики размера 75-Ьр и
450-Ьр.
3. Восстановите изображение геля.
4. Проследите в автоматическом режиме все резуль¬
таты.
5. Когда прослеживание закончено, проверьте, что
оно выполнено правильно; данные могут тогда
быть получены из каждого ряда для того, чтобы
формировать индивидуальные типовые файлы.
Выберите в меню исследуемые ряды.
6. Программное обеспечение GeneScan® исполь¬
зует два главных контрольных окна, Контроль
Анализа и Контроль Результатов. Контроль Ана¬
лиза появляется, когда проект сначала открыт, и
используется, чтобы определить внутренний
размер стандарта и изменить параметры анализа.
Контроль Результатов может быть выбран, когда
анализ образцов закончен. Это позволяет выбрать,
какие из результатов будут показаны на дисплее,
а также определить формат этих результатов.
7. Используя окно Контроль Анализа, выберите
стандартный размер для каждого анализируемого
образца, размеры пиков следующие: 75,100,140,
150,160,200,250,300,350,400,450.
8. Инсталлируйте соответствующую матрицу.
9. Отметьте мышкой цветные столбцы, помечен¬
ные буквами В, G, Y, и R, чтобы выдвинуть на
первый план все ряды, которые будут проанали¬
зированы. Нажмите на «анализировать», чтобы
начать анализ образцов. Все образцы, которые
проанализированы, будут показаны маленьким
треугольником в соответствующей клетке.
10. Затем проанализированные образцы могут быть
открыты в окне Контроль Результатов.
И. Распечатайте электрофореграмму (EPG) Gene¬
Scan® для каждого образца (рис. 16.11).
12. Затем может быть использовано Программное
обеспечение Genotyper®, которое обработает
результаты, полученные GeneScan®.
13. Все данные от отдельного геля могут быть обрабо¬
таны вместе как отдельный файл Genotyper®.
14. Импортируйте все данные из файла GeneScan®
в матрицу SGM Plus Genotyper®.
15. Промаркируйте пики, нажимая (Apple+ 1).
16. Важно удостовериться, что стандартный размер
правильно распознан в каждом образце.
17. Аллельные контроли должны также приняты во
внимание. Удостоверьтесь, что все представлен¬
ные аллели правильно идентифицированы.
18. Если вышеупомянутое выполнено, проверьте
метки образцов.
19. Проверьте правильность всех обозначений, уда¬
лите любые обозначения, которые могут быть
ошибочными.
20. Как только будет проведена проверка правиль¬
ности всех обозначений, распечатайте результаты
анализа Genotyper® для каждого образца и при¬
ложите к распечатке GeneScan® EPG.
21. Затем может быть сделано сравнение проанализи¬
рованного профиля с опубликованным профилем
каждой клеточной линии.
16.7. РНК И ЭКСПРЕССИЯ БЕЛКА
Клетки со специфическими фенотипическими ха¬
рактеристиками могут быть распознаны анализом
экспрессии гена Нозерн-блоттингом [Sambrook et al.,
1989; Ausubel et al., 1996] с использованием радио¬
активных, флюоресцентных или люминесцентных
исследований. Эта процедура может быть выполнена
16.7. РНК и экспрессия белка
313
Рис. 16.10. Сиквенирование ДНК. Сиквенатор ABI 377,
использующий автоматический электрофоретический
анализатор на полиакриламидном геле для разделения и
количественной амплификации последовательностей STR.
(С любезного разрешения A. James, LGC Promochem.)
на клеточном уровне, как в FISH (см. протокол 27.9),
или in situ гибридизации с радиоактивными зондами
(см. протокол 27.8). Качественный анализ общего
белка клетки показывает различия между клетками,
когда целые клетки или экстракты клеточных мембран
разгоняются на двухмерных гелях [O’Farrell, 1975].
Этот метод дает характерный «отпечаток пальца»,
подобный полипетидной карте гидролизата белка, но
содержит так много информации, что интерпретация
может быть затруднена. Существует возможность
просмотра этих гелей при помощи компьютеризиро¬
ванного денситометра (Molecular Dynamics), который
приписывает количественные параметры каждому
пятну и может нормализовать каждый фингерпринт,
чтобы установить стандарты и интерпретировать по¬
зиционные различия в расположении пятен. Мечение
клеток 32Р, [35S] метионином, или комбинацией 14С
меченых аминокислот, сопровождаемые ауторадио¬
графией, позволяет проводить более легкий анализ, но
этот метод пока не является общераспространенным,
если только в лаборатории не используется постоянно
технология подготовки двухмерных гелей. В настоя¬
щее время количественный анализ фенотипической
экспрессии более доступен при помощи RT-PCR ана¬
лиза транскриптов гена в крупном масштабе методом
микрочипового анализа экспрессии генов (Affyme-
trix)) [Kiefer et al., 2004; Staab et al., 2004]
Пример приведен на цв. вкладке 24, где сравнивают¬
ся три различных типа клеток, для которых этот метод
ясно демонстрирует различия в профиле фенотипиче¬
ской экспрессии.
16.8. АКТИВНОСТЬ ФЕРМЕНТОВ
Специализированные функции клеток in vivo часто
выражаются в активности определенных ферментов,
некоторые из которых могут быть выявлены in vitro
(см. табл. 16.2). К сожалению, активность многих фер¬
ментов утрачивается in vitro (см. разд. 3.4.2, 17.1.1),
и такие ферменты не могут служить маркерами тка¬
невой специфичности, например паренхима печени
теряет аргиназную активность в пределах нескольких
дней культивирования. Однако некоторые линии
клеток экспрессируют определенные ферменты, такие
как тирозинаминотрансфераза в клеточных линиях
гепатомы крысы НТС [Granner et al., 1968]. При по¬
иске специфических маркерных ферментов должны
быть измерены и сравнены конститутивный уровень
и индуцированный уровень активности со сравни¬
тельным исследованием нескольких контрольных
клеточных линий. Глутамилсинтетазная активность,
например, характерная для астроглии мозга, возрас¬
тает в несколько раз, когда клетки культивируются
в присутствии глутамата вместо глутамина [DeMars,
1958; McLean et al., 1986]. Индукция активности фер¬
мента требует специализированных условий для каж¬
дого фермента, и информация об этих условиях может
быть получена из литературы. Общими индукторами
являются глюкокортикоидные гормоны типа декса-
метазона; полипептидные гормоны типа инсулина и
глюкагона; изменения в субстрате или концентрации
продуктов в среде, как в вышеупомянутом примере с
глутамилсинтетазой.
16.8.1. Изоферменты
Активность ферментов можно качественно сравнить
у разных клеточных штаммов, с использованием ви¬
дового ферментативного белкового полиморфизма
и иногда отдельных рас, индивидуумов, и тканей в
пределах вида. Эти так называемые изоферменты, или
изозимы, можно выделить хроматографически или
электрофоретически. Образцы распределения (зи-
мограммы) могут оказаться специфичными для вида
или ткани (рис. 16.12). Nims et al. [1998] обнаружили,
что межвиждовая перекрестная контаминация кле¬
точных культур может быть определена при помощи
изоферментного анализа, если клетки контаминиру-
ющей культуры представляют по крайней мере 10%
общего количества популяции клеток. Среды для
электрофореза включают агарозу, ацетат целлюлозы,
крахмал и полиакриламид. В каждом случае сырой
экстрагированный фермент наносится на одну точку в
геле, и разность потенциалов прикладывается поперек
геля. Различные изоферменты мигрируют с различной
скоростью и могут быть обнаружены позже окраши¬
ванием с хромогенным субстратом. Окрашенные гели
могут читаться непосредственно невооруженным
глазом, фотографироваться и исследоваться позже
или при помощи денситометра. Протокол 16.10 для
изоферментного анализа, использующего агарозный
гель, предоставлен by Jane L. Caputo (American Type
Culture Collection, 10801 University Blvd., Manassas,
Virginia 20110, USA).
314
ГЛАВА 16. ХАРАКТЕРИСТИКА КЛЕТОК
Рис. 16.11. Изучение профиля ДНК. (а) и (б) Клеточные линии HeLa nT47D анализировались при помощи программы Gen-
escan для детекции расположения пиков, обнаруженных секвенатором ABI 377 DNA при гель-электрофорезе, (в) Результаты
анализа клеток HeLa и T47D при помощи Genotyper с определением локализации аллельных типов в отношении размеров
пиков (любезно предоставлено A. James, LGC Promochem)
16.8.2. Изоферментный электрофорез
при помощи Authentikit
Видовая принадлежность источника клеточной линии
может быть определена при помощи гель-электро-
форезной системы Authentikit которая используется
для определения мобильности семи изоферментов
нуклеозидфосфорилазы (NP; Е. С. 2.4.2.1), глкжоза-6-
фосфатаздегидрогеназы (G6PD; Е. С. 1.1.1.49), малатде-
гидрогеназы (MD, Е. С. 1.1.1.37), лактатдегидрогеназы
(LD; Е. С. 1.1.1.27), аспартатаминотрансферазы (AST,
Е. С. 2.6.1.1), манноза-6-фосфат-изомеразы (MPI, Е. С.
5.3.1.8), и пептидазы В (Рер В, Е. С. 3.4.11.4). Эта система
позволяет легко проводить скрининг шести клеточных
линий в одном геле с определением семи генетических
маркеров менее, чем за 3 часа [Нау, 2000]. В большин¬
стве случаев видовая принадлежность источника может
быть определена использованием только четырех из
семи изоферментов: нуклеозидфосфорилазы, глюко-
за-6-фосфатаздегидрогеназы, малатдегидрогеназы и
лактатдегидрогеназы. Аналогично, межвидовая пере¬
крестная контаминация клеточных линий может быть
определена в большинстве случаев с использованием
тех же изоферментов [Nims et al., 1998], хотя пептидаза
В также нужна для анализа контаминации мышиной
клеточной линии клетками хомяка.
Анализ электрофореграмм. Приложить высушен¬
ный гель к форме для документации геля, выровнять
16.9. Антигенные маркеры
315
Аспартатаминотрансфераза Глюкозо-б-фосфатдегидрогеназа Лактатдегидрогеназа Малатдегидрогеназа
со к
* *
Рис. 16.12. Изоферментный электрофорез. Анализ четырех изоферментов при помощи гель-электрофорезной системы
Authentik (Innovative Chemistry), (а) набор из четырех кювет с блоком питания на заднем плане, реактивы справа, три сбор-
но-блочных камеры для геля на переднем плане; (6) лотки для окраски и отмывки геля (Corning); (в-е) электрофореграммы,
полученные в результате анализа (любезно предоставлено АТСС)
участки нанесения образцов, произвести измерения.
Яркая желтая подложка формы разлинована по
миллиметрам и дает максимальную контрастность и
улучшает читаемость малиновых полос на сухом геле
(см. рис. 16.12). Измерьте расстояние миграции фер¬
ментов путем измерения расстояния от середины зоны
аппликации до середины зоны фермента. Запишите
данные измерений расстояний миграции ферментов
для стандартного, контрольного и опытного образцов в
стандартную форму. Перенесите данные в форму Cell I
для заключительной видовой идентификации клеток.
Подробные инструкции для идентификации неизвест¬
ной линии включены в форму. Сохраните форму в
вашей тетради для постоянных записей результатов.
Подробные инструкции по каждому ферменту вы може¬
те найти в руководстве Handbook for Cell Authentication
and Identification, Innovative Chemistry, Inc.
Альтернативные методы выделения. Клеточные
экстракты могут быть приготовлены ультрацентрифу¬
гированием, быстрым троекратным замораживанием
и оттаиванием или обработкой октиловым спиртом.
[Масу, 1978]. Буфер для клеточной экстракции можно
приобрести в Innovative Chemistry, Inc. Контроль и
стандарт можно приготовить с использованием стан¬
дартной процедуры приготовления экстракта (см.
протокол 16.10). Стандарт готовится из линии клеток
мышей L929 (АТСС CCL-1), контроль готовится из
линии клеток человека HeLa (АТСС CCL-2). O’Brien
et al. [ 1980] показали, что образцы могут храниться при
-70 °С до года без существенной потери ферментатив¬
ной активности.
16.9. АНТИГЕННЫЕ МАРКЕРЫ
В результате большого разнообразия и изобилия
антител и наборов для определения, предлагаемых
промышленными компаниями (см. Приложение II),
иммунное окрашивание является наиболее полезным
и доступным методом для характеристики клеточных
линий (табл. 16.3). Однако, независимо от источника
антител, необходимо убедиться в их специфичности
путем использования соответствующего контрольного
материала. Это справедливо как для моноклональных
316
ГЛАВА 16. ХАРАКТЕРИСТИКА КЛЕТОК
ПРОТОКОЛ 16.10. Изоферментный анализ
Схема
Собрать клетки, промыть их D-PBSA, ресуспендиро¬
вать до концентрации 5х107 клеток/мл, приготовить
клеточный экстракт. Хранить при 70 °С. Пригото¬
вить аппарат для геля. Внести 1-2 мкл исследуе¬
мого клеточного экстракта, а также стандартную и
контрольную пробы в агарозный гель. Заполнить
камеру водой, поместить агарозный гель в камеру и
проводить электрофорез в течение 25 мин. Добавить
реагенты для ферментов на 5-20 мин, промыть гель,
высушить и исследовать полученный гель с зонами
локализации фермента.
Материалы
Стерильные или асептически приготовленные:
• Клетки
Экстракты (Innovative Chemistry, Inc.):
Стандартный экстракт L929
Контрольный экстракт HeLa S3
• Буферный раствор для экстракции клеток (Inno¬
vative Chemistry, Inc.) или Triton Х-100 раствор
для экстракции,: 1:15, v/v
• Triton Х-100 в 0,9% NaCl, pH 7,1, содержащий
6,6x10-4 М EDTA (Хранить при 4 °С)
Нестерильные:
• Прибор и реактивы Authentikit (Innovative Chem¬
istry, Inc.):
Пленки агарозного геля, SAB 8,6, по одному на
каждый исследуемый фермент
Вкладыши в лотки для инкубирования по одному
на каждый исследуемый фермент
Лотки для инкубирования
Лотки для окрашивания и промывки
Термостатированные крышки для электрофоре¬
тической камеры и донная часть
Источник постоянного тока (160 V) с таймером
Соединительное устройство
Инкубационная камера/сушка
Магнитная мешалка с 1-дюймовым вкладышем,
Буфер, SAB 8,6
Ферментативные субстраты, по 1 флакону
нуклеозидфосфорилаза (NP)
глюкоза-6-фосфатаздегидрогеназа (G6PD)
малатдегидрогеназа (MD)
лактатдегидрогеназа (LD)
аспартатаминотрансфераза (AST)
манноза-6-фосфат-изомераза (MPI)
пептидаза В (Рер В)
Аппликатор образцов с тефлоновыми наконеч¬
никами
Форма для документации геля
Форма для миграции ферментов
Итоговая форма для анализа идентичности кле¬
ток Cell I. D
• Шприц микролитровый
• Микроцентрифуга (Eppendorf, Brinkmann)
• Пробирки Eppendorf 1,5 мл
• Автоматическая пипетка на 1,0 мл
• Наконечники для автоматической пипетки
• Пипетки, 5,0 мл
• Градуированный цилиндр, 100 мл
• Маркер
• Деионизованная вода
• Дистиллированная вода
• D-PBSA
• Защитные перчатки
• Контейнер для отходов и использованных нако¬
нечников
Протокол
а) Приготовление экстракта:
1. Вырастите клетки в стандартных условиях до
получения 2x107 жизнеспособных клеток.
2. Соберите клетки в соответствии с процедурой,
рекомендуемой для этой клеточной линии.
3. Ресуспендируйте клеточный осадок, полученный
после центрифугирования в D-PBSA, подсчитай¬
те количество жизнеспособных клеток.
4. Центрифугируйте суспензию при 300 g в течение
5-10 мин для осаждения клеток и удалите супер-
натант.
5. Повторите шаги 3 и 4 до общего троекратного
количества отмывок.
6. Добавьте 100 мкл экстрагирующего раствора к
клеточному осадку.
7. Осторожно перенесите суспензию клеток в пи¬
петку малого сечения и суспендируйте покачи¬
ванием суспензии вверх-вниз, пока все клетки не
лизируются.
8. Перенесите лизат клеток в пробирки Eppendorf
(1,5 мл), пипетируйте лизат вверх-вниз несколько
минут, для того чтобы клеточные мембраны от¬
делились и собрались вместе. Центрифугируйте
лизат при максимальном ускорении (9000 g)
течение 2 мин в ультрацентрифуге. Собрать
супернатант, разделите на аликвоты желаемого
объема, храните при 70 °С.
б) Подготовка к работе прибора
для электрофореза
1. Включите инкубационную камеру.
2. Поместите один вкладыш в каждый лоток для
инкубирования.
3. Добавьте 6,0 мл деионизованной воды в каждый
лоток для инкубирования и поместите лотки при
37 °С на 20 мин.
4. Добавьте 95 мл буфера SAB 8,6 в каждую камеру
прибора для электрофореза клеток (общее ко¬
личество 190 мл буфера на одну донную часть).
Этот буфер не должен повторно использоваться,
поскольку значение pH значительно меняется в
ходе электрофоретического процесса.
5. Соедините заполненную донную часть камеры с
соединительным устройством, которое подсоеди¬
16.9. Антигенные маркеры
317
нено к источнику питания с регулируемым напря¬
жением. Источник питания должен быть включен
в заземленную электрическую розетку.
6. Заполните каждую термостатируемую крышку
ячеек для электрофореза 500 мл холодной воды
(4-10 °С). Заполнять следует, держа крышку в
вертикальном положении, иначе вода выльется.
Охладите воду добавлением льда или храните
достаточный запас холодной воды в обычном
холодильнике. Заменяйте воду перед каждым
электрофорезом.
7. Налейте 500 мл деионизованной или дистиллиро¬
ванной воды в каждый лоток для окраски и опо¬
лосните лоток. Эту воду не следует использовать
повторно.
в) Процедура электрофореза:
1. Промаркируйте каждый агарозный гель, который
будет использован. Это можно выполнить при
помощи ярлыков, предоставляемых Innovative
Chemistry Inc. или при помощи маркера, делая
надпись на оборотной стороне геля.
2. Поместите гель в камеру. Сориентируйте гель так,
чтобы лунка для помещения образца располага¬
лась наиболее близко к вам.
3. Промаркируйте каждую лунку для образца, кото¬
рый должен быть проверен (например, стандарт,
контроль, неизвестный 1, неизвестный 2, и т.д.).
На одном геле может быть проверено шесть не¬
известных образцов.
4. Аккуратно снимите пленку агарозного геля с
твердого пластмассового покрытия. Будьте ос¬
торожны, берите гель только за края. Выбросьте
твердое пластмассовое покрытие.
5. Добавьте клеточный экстракт в лунки для образ¬
цов. Используйте диспенсер с присоединенными
тефлоновыми наконечниками, отмеряющими
1 мкл клеточного экстракта в каждую лунку.
Для каждого образца используйте свежий на¬
конечник. Поместите стандартный экстракт в
трек 1, контрольный — в трек 2, а неизвестные
образцы — с 3 по 8. Лунку должна касаться только
капля экстракта, не касайтесь геля наконечником,
чтобы не повредить агарозу. Если требуется 2 мкл
клеточного экстракта, позвольте первой капле
(1 мкл) диффундировать в агарозу прежде, чем
нанесете вторую.
6. Вложите каждую агарозную пленку в крышку
электрофоретической ячейки. Агарозная сторо¬
на должна быть обращена наружу. Сопоставьте
анодную (+) сторону агарозы с анодной стороной
(+) крышки ячейки. Может оказаться необходи¬
мым слегка согнуть пленку агарозного геля, чтобы
вложить его.
7. Соедините крышку ячейки с донной частью.
Черный конец с магнитом внутри должен быть
ближе к источнику тока. Включите напряжение
(160 V) и таймер на 25 мин.
8. Выньте из холодильника флаконы с реактивами-
субстратами для ферментов примерно за 5 мин
до конца процесса электрофореза, оставьте их
подогреваться при комнатной температуре.
9. Добавьте 0,5-1,0 мл деионизованной воды в
каждый флакон с реактивами немедленно перед
использованием. Встряхните, чтобы полностью
растворить реактив.
10. Когда электрофорез окончен, снимите крышку
с донной части и поместите на фильтровальную
бумагу.
г) Процедура окрашивания.
1. Для того чтобы вынуть агарозный гель из крышки
ячейки, возьмите гель за края, согните внутрь и
выньте гель.
2. Поместите пленку агарозного геля на плоскую
поверхность на фильтровальную бумагу гелевой
стороной вверх, так чтобы лунки помещались
напротив вас.
3. Аккуратно промокните остатки буфера с обоих
концов агарозной пленки при помощи безволо-
конной салфетки.
4. Поместите 5-мл пипетку вдоль по нижнему краю
агарозной пленки.
5. Равномерно поливайте восстанавливающий суб¬
страт на агарозную пленку вдоль по ведущему
краю пипетки.
6. Однократным мягким движением толкните пи¬
петку вдоль по поверхности агарозы. Притяните
пипетку к себе и толкните ее поперек поверхности
еще раз. Сверните агарозу начиная с края, удаляя
избыток субстрата в процессе движения. Будьте
осторожны, чтобы не повредить агарозу. Необхо¬
димо проследить, чтобы не было механического
надавливания на гель.
7. Поместите пленку агарозного геля агарозной
стороной вверх в предварительно нагретый лоток
для инкубирования, поставить лоток в инкубатор
на 37 °С примерно на 5-10 мин.
8. После инкубирования промойте пленку агароз¬
ного геля дважды в 500 мл бидистиллированной
или деионизованной воды, каждый раз по 15 мин,
покачивая при помощи магнитной мешалки. Пос¬
ле первых 15 мин выньте гель из воды, удалите
воду, добавьте 500 мл свежей воды и погрузите
в воду гель. Закройте гель, чтобы защитить его
от света. Удостоверьтесь, что гель погружен, а не
плавает на поверхности воды.
9. Выньте гель из воды, поместите на подставку для
сушки в сушильной камере инкубатора или печи.
Сушите 30 мин, или пока агароза не высохнет.
Можно также сушить агарозу при комнатной
температуре в течение ночи.
10. Уберите рабочее место, вылейте буфер из донной
части ячейки, промойте дистиллированной водой.
Вылейте воду из крышек, промойте крышки из¬
нутри, высушите.
318
ГЛАВА 16. ХАРАКТЕРИСТИКА КЛЕТОК
11. Оцените результаты. Полосы стабильны, пленка
может храниться до использования по потребнос¬
ти. Если продолжает развиваться окрашивание
фона, то это означает, что гель недостаточно
промыт.
антител, так и для поликлональной антисыворотки.
Моноклональные антитела высокоспецифичны для
отдельного эпитопа, но специфичность экспрессии
этого эпитопа для отдельного клеточного типа должна
быть подтверждена.
16.9.1. Иммунное окрашивание
Локализация антител обнаруживается по флюорес¬
ценции, для которой антитела конъюгируют с флюо¬
рохромом, например флюоресциин или родамин, или
по отложению продукта преципитации в результате ак¬
тивности пероксидазы хрена или щелочной фосфатазы,
конъюгированной с антителами. Иммунологическое ок¬
рашивание может быть прямым, например специфиче¬
ские антитела сами по себе конъюгируют с флюорохро¬
мом или ферментом и напрямую окрашивают образец.
Обычно, тем не менее, используют непрямой метод, при
котором первичные (специфичные) антитела в своей
нативной форме связываются с антигенами образца,
вслед за этим комплекс обрабатывается вторичными
антителами, индуцированными иммуноглобулином к
первым антителам. Вторичные антитела могут быть ко¬
нъюгированы с флюорохромом [Johnson, 1989] и визуа¬
лизированы при помощи флюоресцентного микроскопа
(см. цв. вкладки 11а, 15г, д и 20в-е)у или пероксидазой
и визуализированы для наблюдения в стандартном
микроскопе после обработки с хромогенным субстра¬
том пероксидазы (см. цв. вкладки 116-г и 12а, б). Для
повышения чувствительности используются различ¬
ные методы, в частности связанные с пероксидазной
активностью. В методе пероксидаза-антипероксидаза
(РАР) дальнейшее амплифицирующее связывание
дополнено реакцией пероксидазы, конъюгированной с
анти-пероксидазеными антителами того же вида, что и
первичные антитела [Jones & Gregory, 1989].
Еще большая чувствительность получена при
использовании биотинконъюгированных вторичных
антител с стрептавидином, конъюгированным с пе¬
роксидазой или щелочной фосфатазой (Amersham)
или вторичными антителами, конъюгированными с
золотом (Janssen) с последующей интенсификацией
серебром (Amersham).
Варианты
Фиксация. В случае окрашивания клеточной по¬
верхности или при наличии антигенов, чувствительных
к фиксатору, необходимо сначала обработать клетки
антителами, а затем произвести последующую фик-
ПРОТОКОЛ 16.11. Непрямая
иммунофлюоресценция
Схема
Зафиксировать клетки, обработать их последова¬
тельно первичными и вторичными антителами.
Исследовать клетки при помощи УФ-света.
Материалы
Стерильные или асептически приготовленные:
• Культура, выращенная на покровном стекле,
предметном стекле со сдвижной камерой или в
полистироловой чашке Петри
Нестерильные:
• Свежеприготовленный фиксирующий раствор 5%-й
уксусной кислоты в этаноле (хранить при 20 °С)
• Первичные антитела, разведенные 1:100—1:1000
в культуральной среде в 10% FBS
• Вторичные антитела, видоспецифично индуциро¬
ванные против первых антител (например, если
первые антитела были получены у кролика, то
вторичные антитела должны быть получены с ис¬
пользованием животных другого вида, например
козы, с получением антител к иммуноглобулину
кролика). Вторичные антитела должны быть ко¬
нъюгированы с флюоресцеином или родамином
• Сыворотка свиньи или иной блокирующий агент
• D-PBSA
• Среда для заключения препарата: 50% глицерин
в D-PBSA, содержащий ингибитор затухания
флюоресценции (Vecta)
Протокол
1. Промойте покровное стекло с клетками в D-PBSA,
поместите в подходящую емкость, например в
24-луночный планшет для 13-мм покровных стекол.
2. Поставьте на 10 мин при -20 °С, добавьте холод¬
ный фиксирующий раствор и оставьте еще на
20 мин.
3. Удалите фиксирующий раствор, промойте пок¬
ровное стекло D-PBSA, добавьте 1 мл нормаль¬
ной свиной сыворотки, оставьте при комнатной
температуре на 20 мин.
4. Промойте в D-PBSA, высушите на фильтро¬
вальной бумаге, поместите, перевернув на каплю
(50 мкл) разведенных первичных антител.
5. Инкубируйте при 37 °С в течение 30 мин или
при комнатной температуре 1-3 ч или при 4 °С
в течение ночи. В последнем случае (при 4 °С)
антитела могут быть разведены 1:1000.
6. Промойте покровное стекло в D-PBSA, перене-
ситеего в каплю вторичных антител, разведенных
1:20, на 20 мин при 37 °С.
7. Промойте покровное стекло в D-PBSA, заклю¬
чите в 50%-й глицерин в D-PBSA с ингибитором
затухания флюоресценции (Vecta).
8. Исследуйте препарат при помощи флюоресцен¬
тного микроскопа.
16.9. Антигенные маркеры
319
Таблица 16.3. Антитела, используемые для распознавания клеточных линий
Тип клеток
Антитела к
Локализация
Специ¬
фичность
Комментарии
Передняя доля гипофиза
hGH
Аппарат Гольджи и секретиру-
емые вещества
Высокая
Астроглия
В-клетки
GFAP
CD22
Промежуточные филаменты
цитоскелета
Клеточная поверхность
Высокая
См. также табл. 22.2
Эпителий молочной же¬
А-лактальбумин,
Аппарат Гольджи и секретиру-
Высокая
Необходима индукция дифференци¬
лезы
казеин
емые вещества
ровки
Колоноректальная и легоч¬
ная аденокарцинома
СЕА
Клеточная поверхность и ап¬
парат Гольджи
Промежу¬
точная
Молекулы клеточной адгезии
Эндотелий
Фактор VIII
Тельца Вейбеле-Паладе
Высокая
Зернистость
Эпителий
V-CAM
Клеточная поверхность
Высокая
Молекулы клеточной адгезии
Эпителий
L-CAM
Клеточная поверхность
Высокая
Молекулы клеточной адгезии
Эпителий
Цитокератин
Промежуточный филамент
цитоскелета
Высокая
Некоторые антитела также окрашивают
мезотелий, специфические антитела
для различных видов эпителия
Эпителий
ЕМА
Клеточная поверхность
Высокая
Некоторые антигены как HMFG
Эпителий
HMFG I & II
Клеточная поверхность
Высокая
Некоторые антигены как ЕМА
Фетальные гепатоциты
AFP
Аппарат Гольджи и секретиру-
емые вещества
Высокая
Экспрессируется также гепатомами
Гематопоэтические стволо¬
вые клетки
CD34
Клеточная поверхность
Высокая
Только в предшественниках, нет в зре¬
лых клетках
Гепатоциты
Альбумин
Аппарат Гольджи и секретиру-
емые вещества
Высокая
Необходима дифференцировка
Незрелые В-клетки
CD20
Клеточная поверхность
Высокая
Нет в плазматических клетках
Мезодермальные клетки
Виментин
Промежуточные филаменты
цитоскелета
Низкая
Экспрессируется также на эпителиаль¬
ных и глиальных клетках в культуре
Мезотелий
Антиген мезотели-
альных клеток
Клеточная поверхность
Высокая
Моноциты и макрофаги
CD14
Клеточная поверхность
Высокая
Миелоидные клетки
CD13
Клеточная поверхность
Высокая
Миоциты
Десмин
Промежуточный филамент
цитоскелета
Высокая
Нервные и нейроэндокрин¬
ные клетки
NSE
Цитоплазма
Промежу¬
точная
Может экспрессироваться на SCLC
Нервные клетки
N-CAM
Клеточная поверхность
Промежу¬
точная
Также экспрессируется на некоторых
SCLC
Нейрональные клетки
Белок нейрофила-
ментов
Промежуточные филаменты
цитоскелета
Высокая
Также окрашивают некоторые SCLC
Олигодендроциты
Основной белок
миелина
Клеточная поверхность
Высокая
Также окрашивают периферические
нейроны
Gal-C
Клеточная поверхность
Высокая
См. также табл. 22.2
Плацентарный эпителий
hCG
Аппарат Гольджи и секретиру-
емые вещества
Низкая
Также экспрессируется на некоторых
опухоевых клетках легких
Эпителий простаты
PSA
Аппарат Гольджи и секретиру-
емые вещества
Высокая
Т-клетки
CD3
Клеточная поверхность
Высокая
Т-клетки и эндотелий
ICAM
Клеточная поверхность
Промежу¬
точная
Сокращения: GFAP, глиальный фибриллярный кислый белок (glial fibrillary acidic protein); EM А, эпителиальный мембранный антиген
(epithelial membrane antigege); HMFG, человеческий глобулярный белок молока (human milk fat globule protein); AFP — альфа- фето-
протеин (a-fetoprotein); CEA — карциноэмбриональный антиген (carcinoembryonic antigen) SCLC, мелкоклеточная карцинома легкого
(small cell lung cuncer); hCG, человеческий хориональный гонадотропин (human chorionic gonadotropin); ICAM, межклеточная молекула
адгезии клеток (intercellular cell adhesion molecule); NSE — нейронспецифическая энолаза (neuron-specific enolase); PSA — простата-
специфический антиген (prostate-specific antigen), Gal — С-галактоцереброзид (galactocerebroside); GRP — гастринвысвобождающий
пептид (gastrin-releasing peptide).
320
ГЛАВА 16. ХАРАКТЕРИСТИКА КЛЕТОК
сацию в соответствии с шагом 2. Когда стеклянная
поверхность используется как субстрат, холодный
ацетон можно заменить на кислый этанол.
Непрямая пероксидаза. Заменить пероксидаза-
конъюгированные антитела на флюоресцентные
антитела на стадии 6 и затем перенести на субстрат
для пероксидазы (диаминобензидин-окрашенный
препарат может быть обезвожен и заключен в DPX,
но этилкарбазол должен быть заключен в глицерин
как в шаге 7).
РЛР. Использовать пероксидаза-конъюгированные
вторичные антитела на стадии 6 и затем перенести
покровное стекло в разведенный комплекс РАР (1:100)
(большинство иммунобиологических компаний-про-
изводителей, например Dako, Vesta) в D-PBSA на
20 мин. Промыть, добавить пероксидазный субстрат,
как для непрямой пероксидазной реакции. Инкубиро¬
вать, промыть и закрыть покровным стеклом.
Маркеры клеточной поверхности. Специфические
антигены клеточной поверхности обычно окрашиваются
в живых клетках (при 4 °С в присутствии азида натрия
для ингибирования пиноцитоза), в то время как внут¬
риклеточные антигены окрашиваются в фиксированных
клетках, иногда необходимо провести легкую трипсини-
зацию для обеспечения доступа антител к антигенам.
HLA-антигены и антигены группы крови могут
быть обнаружены на клетках многих культур человека
и служить полезным специфическими параметром.
В частности, особенно ценным считается Н LA-пол и-
морфизм, особенно когда известен профиль донора
[Espmark & Ahlqvist-Roth, 1978; Stoner et al., 1981;
Pollack et al., 1981].
16.9.2. Иммунный анализ
Исследование клеточного экстракта может быть прове¬
дено при помощи ELISA-метода в микротитрационных
Рис. 16.13. Прибор для иммуноанализа Agilent. Agilent
2100, способный проводить многопараметрический анализ
сложных иммунофлюоресцентно окрашенных образцов,
(а) Отделение для образцов, (б) Держатель для образца.
(С любезного разрешения PromoCell.)
планшетах с покрытыми антителами лунками. Метод
снабжен специальными программами (например, Assay
Designs; R&D) и позволяет автоматически проводить
количественную оценку маркеров и экспрессии бел¬
ковых продуктов. Анализ экспрессии антигена также
может быть автоматизирован и количественно оце¬
нен при помощи проточной цитофлюориметрии (см.
разд. 21.7.2) (B-D Biosciences FACS Star or GuavaTech-
nologies), путем анализа образа потока клеток (Agilent
2100; рис. 16.13) или методом белковых микрочипов с
использованием антител (antibody microarray analysis,
antibody- MAA).
16.10. ДИФФЕРЕНЦИРОВКА
Многие из характеристик, описанных как антигенные
маркеры или активность специфических ферментов,
могут также рассматриваться как маркеры дифферен¬
цировки, и как таковые они могут помочь провести
корреляцию клеточных линий с исходными тканями.
Другие примеры признаков дифференцировки и мар¬
керов специфичных клеточных линий представлены
в гл. 3 (см. табл. 3.1). Соответствующие исследования
этих свойств могут быть получены из ссылок, приве¬
денных в таблице (см. также разд. 17.6 и гл. 23).
в) Интраэпителиальная цервикальная нео-
плазия человека (CIN). Первичная культу¬
ра из цервикальной биопсии субкультивиро-
вана на фидерный слой ЗТЗ, некоторые де¬
генеративные клетки которого сохранились
на препарате вверху справа. Окрашивание
по Гимза. Объектив 10х. (С любезного раз¬
решения M.G. Freshney.)
г) Фибросаркома человека. Первичная куль¬
тура посажена на фидерный слой эмбриональ¬
ного кишечника. Клетки фибросаркомы имеют
веретенообразную форму, клетки фидерного
слоя — многоугольной формы. Окрашивание по
Гимза. Объектив 10х
Цветная вкладка 1. Первичная культура, человеческая
а) Карцинома легкого человека. Разраста¬
ние культуры из первичного эксплантата,
полученного из сквамозно-клеточной кар¬
циномы легкого человека. MOG-L-DAN.
Окрашивание по Гимза. Объектив 10х
б) MOG-GP Астроцитома. Астроглиальные клет¬
ки анапластической астроцитомы. Окрашивание
по Гимза. Объектив 40х
г) Разрастание культуры из почечных каналь¬
цев. Распространение клеток из прилипших
фрагментов после дизаггрегации методом
холодной трипсинизации. Фазовый контраст.
Объектив 10х
д) Почка новорожденной крысы. Первичная культура
из трипсинизированной (холодным методом) почки
новорожденной крысы. Окраска по Гимза. Объектив
10х. (С любезного разрешения M.G. Freshney.)
Цветная вкладка 2. Первичная культура, эксплантат, холодная трипсинизация и коллагеназа
6) Первичный эксплантат.
Клетки мышиной сквамозно-
клеточной карциномы. Фазо¬
вый контраст. Объектив 10х
в) Коллагеназное расщепле¬
ние. Карцинома толстой кишки
человека. Светлопольное осве¬
щение. Объектив 4х
а) Почечные канальцы мыши. Дезаггрегация
почки новорожденной мыши методом холодной
трипсинизации. Соединительная ткань диссо¬
циирована, но фрагменты канальцев и клубочков
остались целыми. Аналогичный препарат полу¬
чен при коллагеназном расщеплении. Нормаль¬
ное светлопольное освещение. Объектив 4х
а) Легкое эмбриона цыпленка. Первичная куль- б) Печень эмбриона цыпленка. Детали
тура легкого13-дневного эмбриона цыпленка см. (а)
через 48 ч после дезаггрегации методом холодной
трипсинизации. Окрашивание по Гимза и фазо¬
вый контраст. Объектив 10х
в) Бедренная мышца эмбриона цыпленка.
Детали культуры как в (а). Обратите внимание
на многоядерные мышечные волокна (красная
стрелка), возникшая в результате слияния мио-
цитов (маленькие веретенообразные клетки
темной окраски). Окрашивание по Гимза, свет¬
лопольное освещение. Объектив 10х
г) Почка эмбриона цыпленка. Детали куль
туры как в (а) Окрашивание по Гимза, свет
лопольное освещение Объектив 10х
Цветная вкладка 3. Первичная культура, эмбрион цыпленка
а) Свежая монослойная субкультура. Клетки б) Переход в log-фазу. Клетки NRK через 48 ч пос-
NRK через 24 ч после пересадки. Фазовый конт- ле пересадки. Фазовый контраст. Объектив 10х
раст. Объектив 10х
в) Середина log-фазы. Клетки NRK через 3 дня
после пересадки, готовы к замене питательной
среды. Фазовый контраст. Объектив 10х
г) Поздняя log-фаза. Клетки NRK через 7 дней
после пересадки, готовы к следующему субкульти¬
вированию. Фазовый контраст. Объектив 10х
Цветная вкладка 4. Фазы ростового цикла
а) Монослой NRK клеток перед трипсинизацией. Фазовый б) Монослой NRK клеток после обработки D-PBSA/
контраст. Объектив 20х EDTA. Предварительное промывание. Фазовый контраст.
Объектив 20х
в) Монослой NRK сразу после удаления трипсина. Фазовый г) Монослой NRK через 1 мин после удаления трипсина.
контраст. Объектив 20х Фазовый контраст. Объектив 20х
д) Монослой NRK через 5 мин после удаления трипсина, е) Полностью дезаггрегированный монослой NRK. Через
Фазовый контраст. Объектив 20х 10 мин после удаления трипсина культура готова к диспергиро¬
ванию и подсчету клеток. Фазовый контраст. Объектив 20х
Цветная вкладка 5. Субкультивирование методом трипсинизации
а) Клоны HeLa. Клоны HeLa-S3 полученные клониро¬
ванием разведением и окрашенные по Гимза через 3 нед.
Масштабный отрезок 5 мм
6) Клон NRK. Маленький клон клеток NRK, клонирова¬
ние разведением. Фазовый контраст. Объектив 4х
г) Карцинома молочной железы на фидерном слое.
Микрофотография колонии эпителиальных клеток
карциномы молочной железы человека. 400 кл./см2,
выращенная на конфлюэнтном фидерном слое клеток
эмбрионального кишечника FHS74Int. Окрашивание по
Гимза. Объектив 10х
в) Карцинома молочной железы, JUW. Клонирование
на пластике. Микрофотография вторичной культуры
из карциномы молочной железы человека. Культура
с плотностью 4000 кл./см2, выращенная на пластике.
Главным образом, фибробласты. Окрашивание по Гимза.
Объектив 10х
д) Действие сыворотки на эффективность культиви¬
рования. Клетки MvlLu, трансфицированные онко¬
геном туе, посеяны на чашки Петри диаметром 6-см
в дозе 500 кл./чашку, фиксированы и окрашены через
2 нед. Слева 10% FBS, справа 20% FBS. Масштабный
отрезок 10 мм. (С любезного разрешения M.Z. Khan.)
е) Действие глюкокортикоидов на эффективность
культивирования. Клетки культуры глиомы человека
раннего пассажа в отсутствие (слева) и присутствие
(справа) 0,25 мкМ дексаметазона. Макрофотография,
общее увеличение приблизительно 10х
Цветная вкладка 6. Клонирование клеток
а) Клон постоянной клеточной линии глиомы MOG-
G-CCM. Эллиптическая и веретенообразная морфоло¬
гия клеток. Окрашивание по Гимза. Объектив 10х
6) Другой клон той же постоянной клеточной линии
глиомы MOG-G-CCM., что и в (а). Эпителиоидная
морфология. Окрашивание по Гимза. Объектив 10х
ш,
W ' '♦ #
0- ь Щ
^ in
щ %
# ■ . #
10 мм
г) Клонированная культура клеточной линии раннего
пассажа. Клетки немелкоклеточной карциномы легкого
человека (NSCLC). Окрашивание по Гимза. Масштаб¬
ный отрезок 10 мм
* *
■ ’ *■ 5 ;"л * fV’. *: , *. , . '
' • *
' * <' v > ' *
* W . V * .
fell'
!
д) Клоны культуры NSCLC человека. Окрашивание
по Гимза. Объектив 4х
т
»' • .
I . ' •
#
•* 4
* •
Ф т •
* » •
0 0
. т • • ф
•
* •- Л *
• *
И
* • # •
♦
•4,.'
• § • •
•. * . *
9 ■
1
% #
W.
.
• f *
. % \ ф ^
$ . *
в) Третий клон постоянной клеточной линии глиомы
MOG-G-CCM. Сквамозно-эпителиоидная морфоло¬
гия. Увеличение такое же, что в (а) и (б). Окрашивание
по Гимза. Объектив 10х
♦
в) Клонированные культуры А2780. Постоянные кле¬
точные линии карциномы яичника человека
Цветная вкладка 7. Клонирование клеток. Морфологическое разнообразие
а) Нормальные эмбриональные легочные
фибробласты человека. Субконфлюэнт-
ный монослой нормальных эмбриональных
легочных фибробластов человека. Окраши¬
вание по Гимза. Объектив 10х
в) Эндотелий пупочного канатика чело¬
века. Клеточная линия, полученная путем
промывания коллагеназой пупочной вены
человека. Окрашивание по Гимза. Объек¬
тив 20х
б) Конфлюэнтный слой фибробластов.
Плотная культура нормальных эмбрио¬
нальных легочных фибробластов человека.
Фазовый контраст. Объектив 10х
г) Меланоциты человека. Вторичная культу¬
ра ТРА-обработанных эпидермальных мела-
ноцитов, выделенных из крайней плоти ново¬
рожденного ребенка (афро-американца). Об¬
ратите внимание на морфологию дендритных
клеток и их удлиненную веретенообразную
форму. В противоположность меланоцитам,
выделенным из крайней плоти новорожден¬
ного ребенка кавказского происхождения,
эти меланоциты обладали высоким уровнем
гранул меланина (фазовый контраст, ув. 320х).
(С любезного разрешения H.-Y. Park.)
Цветная вкладка 8. Конечные клеточные линии
a) HeLa-S3 Цервикальная карци¬
нома человека. Фазовый контраст.
Объектив 40х
« *
•I,
ш ,
фф
ф # #
«
% *,
л? *
» *
*• •
• « , *
# и £ %
• «►
# #
% * ■ *
* *
* ф % 4
* « *
1 *
• * *
* ♦ J _ * i
#
* * Ф
щг
• ‘ ** *
• # * * *
% •*
» ** • Э ^ * %% ;% * • •
%т •* , и>.% * %
^ * Ш* S '% * *
10Qj^Ki
*% «г
б) А549. Аденокарцинома легкого
человека. Окрашивание по Гимза.
Объектив 10х
ей
. >н:ы.
Jfc* ^ «Г ' ^ J :
г* »'%44'WVO //
Ч «Р1
Vw
§т
#
/
100 мкм
в) MOG-G-UVW. Глиома человека.
Окрашивание по Гимза. Объектив
10х
^.
ИнВа§^^
- г Дайтж т& %*,¥ fe-3
п>|рж^ Т'^
Hi*
42 &тйай'' pS #
mvg^WZf ■-%% ж
тШтФ
■ ■ Щ
•-;vrrr
щШш"
Щ
Щ&. 1
£• ,
аЯЫЙЙ ** •
J"
л* •-. .. ■ ;
я^^йв1ШЮгмР®"'.
Ш ШШ>лк -
100 мкм
г) Сасо-2. Колоректальная карци¬
нома человека, образующая купол.
Данная клеточная линия должна быть
субкультивирована прежде, чем она
достигнет этой стадии, пока она еще
субконфлюэнтна. Фазовый контраст.
Объектив 40х
Цветная вкладка 9. Постоянные клеточные линии из опухолей человека
е) Культура клеток почки собаки Madin-
Darby MDCK. Линия эпителиоподобных
клеток из почки собаки. Окрашивание по
Гимза. Масштабный отрезок 100 мкм
ж) Эпителиальная клеточная линия MvlLu из легких
норки. Иммунопероксидазное окрашивание на цитокера¬
тин (первичные антитела АЕЗ). (С любезного разрешения
M.Z. Khan.) Масштабный отрезок 100 мкм
Цветная вкладка 10. Постоянные клеточные линии из нормальных тканей животных
а) СНО-К1. Клон СНО, постоянной клеточной линии,
выделенной из яичника китайского хомячка. Фазовый
контраст. Масштабный отрезок 100 мкм
* % 1*
\ Ч\.
• * •. « » * «•
• »•*
*# , . •
* * « ♦ * ’ » * V* • I»
i • # “ * ф l
»# w ‘ ♦ • w F i •* * *
• ' <u • # . * * 1 •
i, • % <§ * * * v1 i ф
•*
m. m i * A # • # * A
->4*% . • . * v ; v:V%i
ЮОм™, •„/ *
**» •** f * * * * 1 * *
6) Swiss ЗТЗ. Фибробласт-подобные клетки мы¬
шиного эмбриона Swiss. Окрашивание по Гимза.
Масштабный отрезок 100 мкм
в) Окрашенная по Гимза культура ВНК-21 во
флаконе, клон 13. Фибробласты почки новорожден¬
ного хомячка после достижения конфлюэнтности
формируют типичную «воронку», образованную
параллельными лучами из клеток. Окрашивание
по Гимза. Макрофотография. Масштабный отрезок
2 мкм
г) ВНК-21-С13. Фибро¬
бласты почки новорожден¬
ного хомячка, достигшие
стадии конфлюэнтности и
формирующие параллель¬
ные лучи. Окрашивание по
Гимза. Масштабный отрезок
250 мкм
д) Культура ВНК-21-С13 вы¬
сокой плотности. Постконф-
люэнтная культура фиброб¬
ластов почки новорожденного
хомячка, формирующая вто¬
рой монослой более плотно
окрашенных клеток. Окраши¬
вание по Гимза. Масштабный
отрезок 250 мкм
а) Нормальные кератиноциты человека на фидерном слое фибробластов кожи
взрослого человека. Панцитокератин кератиноцитов окрашен в зеленый цвет, а
виментин фибробластов — в красный. Объектив 40х. (С любезного разрешения
Hans-Jurgen Stark.)
б) Глиома человека. Клетки MOG-G-CCM
иммунопероксидазное окрашивание на
белок GFAP. Объектив 10х
в) Стромальные фибробласты молочной железы. Иммунопе¬
роксидазное окрашивание на виментин (коричневый). Объек¬
тив 40х. (С любезного разрешения Valerie Speirs.)
г) Эндотелиальные клетки пупочной вены человека. Им¬
мунопероксидазное окрашивание фактора VIII. Объектив
40х. (С любезного разрешения R.L. Shaw&M. Frame.)
Цветная вкладка 11. Иммунное окрашивание
а) Формирование купола в монослое культуры клеток
аденокарциномы легкого WIL. Фокусировка микроско¬
па на вершине купола. Мозаичное чередование СЕА-по-
ложительных и СЕА-отрицательных клеток. Иммунопе-
роксидазное окрашивание. Объектив 40х
б) Формирование купола в монослое культуры клеток
аденокарциномы легкого WIL. Изображение сфоку¬
сировано на монослое у основания купола. Мозаичное
чередование СЕА-положительных и СЕА-отрица¬
тельных клеток. Иммунопероксидазное окрашивание.
Объектив 40х
в) Культура клеток аденокарциномы легкого
А549, растущая на матригеле. Верхнее фото¬
объектив 4х; нижнее-1 Ох. неокрашенный препа¬
рат, светлопольная микроскопия. (С любезного
разрешения Jane Sinclair.)
г) Культура клеток аденокарциномы легкого А549 на пласти¬
ке «не для культуры тканей». Рост на чашках Петри «не для
культуры тканей» с легочными фибробластами, растущими
на покровных стеклах в центре чашки. Объектив 10х. Светло¬
польная микроскопия. Окрашивание по Гимза. (С любезного
разрешения Valerie Speirs.)
Цветная вкладка 12. Морфологическая дифференцировка эпителиальных клеток
а, б) Гемоглобин в клетках Friend. Окрашивание гемоглоби¬
на бензидином в клетках Friend эритролейкемии: контроль
(а); с добавлением 2% DMSO (б); объектив 100х. (С любез¬
ного разрешения David Conkie.) Окрашивание по Гимза
в, г) Авторадиографическое мечение клеток Friend in situ.
Действие DMSO на индукцию мРНК глобина: контроль (в);
индуцированная 2% DMSO (г). Объектив 100х. (С любезного
разрешения David Conkie.). Окрашивание по Гимза
ш'Щ * •. т
4 * **
% %
%
#
iw V
t
щ
ш.
т
* *
% *
ш
«г е
0' it
Г • у
If . 1'л. ШШ : » L
д) Нормальные глиальные клетки человека. Недифферен¬
цированные нормальные глиальные клетки мозга человека.
Общее увеличение 200х. (С любезного разрешения Margaret
Frame.)
е) Дифференцированные глиальные клетки человека.
Морфологическая дифференцировка нормальных глиальных
клеток, индуцированная фактором созревания глии. Окра¬
шивание по Гимза. Общее увеличение 200х.(С любезного
разрешения Margaret Frame.)
Цветная вкладка 13. Дифференцировка клеток Friend и глиальных клеток человека
а) Контактное ингибирование. Поздняя log-фаза
культуры ВНК21-С13. Клетки стремятся к при¬
обретению параллельной ориентации и не будут
перекрывать друг друга. Микроскоп Olympus СК2.
Объектив 10х
/
в) Формирование фокуса в культуре клеток ЗТЗ.
Монослой клеток Sw3T3, оставленный в состоянии
конфлюэнтности в течение 3 нед, обнаруживаются
фокусы трансформированных клеток, в которых не
происходит контактного ингибирования. Фото ввер¬
ху флакон 75-см2, внизу объектив 4х. Окрашивание
по Гимза
б) Утрата контактного ингибирования. Полиома-
трансформированный клон клеток ВНК21-РД,
в котором отсутствует распознавание клетками
присутствия друг друга и рост происходит случай¬
ным образом с перекрыванием клеток. Микроскоп
Olympus СК2. Объектив 10х
Л ■
* Щ
Jgte ' ‘ W
I • 4 ^ щ
в
%
т
г) Внедрение метки [3Н]-тимидина в клетки легкого нутрии
MvlLu. Клетки легкого нутрии Mvllu были мечены [3H]-TdR и
покрыты радиоавтографической эмульсией (см. протокол 27.3),
а затем окрашены по Гимза. Верхнее фото — контрольная ли¬
ния клеток Mvl; в середине MvlLu, Ml, — линия клеток MvlLu,
трансфицированная онкогеном туе; внизу клетки Т1 с мутант¬
ным геном ras. Клетки Т1 были туморогенными и статистически
достоверно (р < 0,001) имели повышенный уровень мечения
по сравнению с MvlLu. Аналогичный результат был получен с
мечением бромдеоксиуридином [Khan et al., 1991]. Масштабный
отрезок 50 мкм. (С любезного разрешения M.Z. Khan.)
Цветная вкладка 14. Трансформация
б) Клеточная линия WLY глиомы человека. Ин¬
фильтрация монослоя FHS4Int с контактным инги¬
бированием клетками WLY. Нормальные глиальные
клетки не инфильтруют такой конфлюэнтный моно¬
слой (см. цв. вкладку 15а). Объектив 10х. Окраши¬
вание по Гимза
t %, £?***■"
ri
У?
в
' ** J-
а) Нормальная линия клеток FHS4Int эмбрио¬
нального кишечника человека. Монослой клеток,
характеризующихся контактным ингибированием.
Объектив 10х. Гимза
в) Клеточная линия немелкоклеточной кар¬
циномы L-DAN. Инфильтрация нормальных
фетальных легочных фибробластов. Фазо¬
вый контраст. Объектив 40х; г) Непрямая
иммунофлюоресценция на цитокератин.
Миграция L-DAN параллельно фиброблас-
там; д) Миграция L-DAN в фибробластах.
Объектив 100х, непрямая флюоресценция.
Окрашивание на цитокератин
е) Индукция ангиогенеза. Контроль. Грубый экс¬
тракт нормальных глиальных клеток был помещен
на хориоаллантоисную мембрану через 10 дней
инкубации последней. Мембрана была удалена че¬
рез 2 нед. Размеры приблизительно соответствуют
натуральной величине. (С любезного разрешения
Margaret Frame.)
ж) Индукция ангиогенеза. На хориоаллантоисную
мембрану цыпленка через 10 дней ее инкубации был
нанесен грубый экстракт клеток карциномы Walker
256. Мембрана была удалена через 2 нед. Размеры
приблизительно соответствуют натуральной величи¬
не. (С любезного разрешения Margaret Frame.)
Цветная вкладка 15. Свойства трансформированных клеток
а) Контаминированный флакон. По- б) Коагулированная контаминация. Бактериальная контаминация
нижение pH и помутнение среды с небольшим понижением pH
в) Контаминированная бутыль со г) Образцы микробиологического бульона. Контроль чистый
средой. Помутнение с выпадением (слева), положительный контроль мутный (справа), исследуемый
осадка. Дрожжи образец отрицательный (в середине)
д) Микоплазма. Малое увеличение.
Микоплазма контаминирует культу¬
ру, что обнаруживается при окраске
красителем Хехст 33258. Объектив
40х, общее увеличение 180х
е) Микоплазма. Высокое разрешение. Окрашенные красителем
Хехст клетки, инфицированные микоплазмой. Объектив 100х,
общее увеличение 800х
Цветная вкладка 16. Примеры контаминации
мям*Э
♦
ш
О ' ('
о Ф»
1
Щ
! о
«I
О
*•
э
* •
о *
. J
б) Тест МТТ. Микротитрационный планшет с клетками,
окрашенными МТТ, через 24 ч в диапазоне концентраций
VP16. Крайний левый и крайний правый столбцы — пустые,
второй слева и второй справа — необработанные контроли.
С 3 по 8 столбцы — концентрация VP16 с 0 до 10 мкм
в) Клетки НТ29. Клетки Т29, обработанные TRAIL (TNF-
связанный апоптоз-индуцирующий лиганд) в концентрации
0,25 мкм в течение 16 ч, окрашенные акридином оранжевым.
Прикрепившиеся клетки имеют нормальную морфологию.
Объектив 40х. Общее увеличение ~300х. (С любезного раз¬
решения Angela Hague.)
г) Апоптоз клеток НТ29. Клетки, отделившиеся от субстрата
обнаруживают конденсацию хроматина и фрагментацию, что
является признаками апоптоза. Объектив 40х. Общее увели¬
чение ~300х. (С любезного разрешения Angela Hague.)
д) Обмен сестринскими хроматидами (SCE). Необработан¬
ные клетки А2780/Ср70 с добавочной перенесенной челове¬
ческой хромосомой 2(A2780/Cp70+chr2). Объектив ЮОх.
(С любезного разрешения Robert Brown & Maureen Illand.)
e) Индуцированный SCE. Клетки A2780/Cp70, обработанные
10 мкм цисплатина в течение 1 ч, обнаруживается экстенсив¬
ный SCE: темные участки, окрашенные по Гимза, чередуется
между цепями внутри отдельных хромосом. Объектив ЮОх.
(С любезного разрешения Robert Brown & Maureen Illand.)
Цветная вкладка 17. Жизнеспособность и цитотоксичность
а) Цитотоксический тест с высвобождением красителя.
Нафталеновый черный. Стекло гемоцитометра площадью
200 мкм2с жизнеспособными (неокрашенными) и нежизне¬
способными (окрашенными) клетками. Объектив 40х
а) Трансфицрованные мозаичные сфероиды.
Выделенные из клеточной линии глиомы
человека MOG-G-UVW, сфероиды, распре¬
деляются по размеру от 100 до 500 мкм в
диаметре, состоят из смеси клеток, трансфи¬
цированных геном GFP (зеленый) и клеток,
трансфицированных геном NAT. Объектив
40х. (С любезного разрешения Marie Boyd &
Rob Mairs.)
в) Микроносители. Клетки Vero, растущие на
микроносителях. Объектив 10х
г) Гексагональные микроносители. Nunc Micro¬
hex Beads. Общее увеличение 20х. (С любезного
разрешения Nalge Nunc.)
6) Альгинатное инкапсулирование. Изоб¬
ражение клеток, инкапсулированных в
альгинат через 2 ч (i), 3 недели (п) и 4 мес
(iii) in vitro. Внутри альгинатной буси¬
ны обнаруживаются клетки погибшие и
делящиеся, у многих клеточных линий
внутри альгинатной бусины образуются
многоклеточные сфероиды. Масштабный
отрезок 70 мкм. (С любезного разрешения
Tracy-Ann Read & Rolf Bjerkvig.)
Цветная вкладка 18. Сфероиды, инкапсуляция и микроносители
.* тс т* 7 шЖ~Ш* 'tl
а) Культура на вкладыше в фильтрационной лунку.
Клетки аденокарциномы легких человека А549 на по-
ликарбонатном фильтре. Видны отверстия в фильтре
(15 мкм), в частности, в верхнем правом углу. Объектив
4х. Окрашивание по Гимза
* 4%
б) Сокультивирование на вкладыше в фильтрационной
лунке. Эпителиальные клетки эмбриональных легких
человека. На обратной стороне фильтра растут фибро¬
бласты легких. Объектив 4х. Окрашивание по Гимза
в) Со-культура клеток легких. Фокус на вершине.
Эпителиальные клетки эмбриональных легких человека.
Изображение сфокусировано на клетках, покрывающих
фильтр. Объектив 40х. Окрашивание по Гимза
г) Сокультивирование на вкладыше в фильтрационной
лунке. Эпителиальные клетки эмбриональных легких
человека. Изображение сфокусировано на клетках под
фильтром. Объектив 40х. Окрашивание по Гимза
4%
S'
*4
«% ' i* '
4% ■
* 4 V
*
мйк ty~-
V щ
Jp*
а
Ч 'v* 1
:
- Л*.
1 •
V
ф. 4
*4&
* *$>
■Ф #
&
it-г..
0
д) Срез со-культуры. Срез через фильтр с клетками
А549 и фибробластами. Оба клеточных типа были посе¬
яны поверх фильтра, но фибробласты мигрировали через
фильтр, хотя некоторые остались поверх него, видны
три клетки, проникающие через поры. Окраска Н&Е,
объектив 40х
е) Легкие эмбриона цыпленка. Первичная культура
клеток эмбрионального легкого 13-дневного цыпленка,
дизаггрегированного методом холодной трипсинизации.
72 ч после посева при концентрации 1х106кл./мл. Объ¬
ектив 40х. Окрашивание по Гимза
Цветная вкладка 19. Органотипическая культура в фильтрационной лунке
а) Со-культура кератиноцитов и дермальных фибро¬
бластов. Органотипическая культура через 2 нед культи¬
вирования in vitro. Окрашивание Н&Е. Объектив 40х
б) Органотипическая культура имплантированная in
vivo. Органотипическая со-культура in vivo через 3 нед
после трансплантации на мышь nude. Окрашивание Н&Е.
Объектив 40х
в) Интегрин и цитокератин в со-культуре in vitro. Им-
мунофлюоресцентное окрашивание на а-6-интегрин
(зеленый), кератин (красный), ДНК-флюоресценция
(синий). Объектив 40х
г) Интегрин и цитокератин в органотипической культу¬
ре, имплантированной in vivo. Иммунофлюоресцентное
окрашивание на а-6-интегрин (зеленый) и кератин (крас¬
ный), ДНК-флюоресценция (синий). Объектив 40х
д) Инволюкрин и коллаген в органотипической со-
культуре через 2 нед. Со-культура кератиноцитов и
фибробластов с окрашиванием коллагенового геля на
инволюкрин (зеленый) и основной компонент мембраны
коллаген VII (красный). Объектив 40х
е) Инволюкрин и коллаген в органотипической со-
культуре через 3 нед. Со-культура кератиноцитов и
фибробластов с окрашиванием коллагенового геля на
инволюкрин (зеленый) и компонент базальной мембра¬
ны коллаген VII (красный). Объектив 40х
Цветная вкладка 20. Органотипическая культура кожи. (С любезного разрешения Hans-Jwrgen Stark.)
а) Действие контрольного раствора в течение 10 мин
б) Действие 1% раствора SDS в течение 10 мин
‘ ■
ч Щ 9% ' & ,s _ ,
- * ♦
»«» * --fritf а -г
» «•
г
Г. _ о *
. г* « %
• W .»
fl 4# *** Щ
♦? **
5 * * *
•V'
- 9
Jg ■■%'
**'• - V! -
-‘•£Г" ' " -•
?
«Гг». ^
-ч ■ -3 4j * <
Ч . А ^ ^ V ^ ЙЧ
' / »**&•&
§fp
шММ
У
Uii
Tgtf*;
f у/ I0 .ал *%t I
■АМ;ЛаАь&Л'• t
% 4'tf,
в) Действие контрольного раствора в течение 20 мин
г) Действие 1% раствора SDS в течение 20 мин
.JTf
* Г /
It
■ ■. , - '-4'
• ’.З* .о,' .. '
ь-О Л -с-
S, П . » ^
; ** ^ *Г#А
§Ш Ч
-г*
f ’* £^14 •!
- V, Л
Г* №'*•!
а
SkinEthic
д) Действие контрольного раствора в течение 60 мин
е) Действие 1% раствора SDS в течение 60 мин
Цветная вкладка 21. Токсичность в органотипической модели in vitro
а) Стерилизованные бутылки. Эти бутылки были автокла-
вированы с индикатором стерильности Thermalog внутри.
Индикатор становится синим при высокой температуре
и давлении пара. Голубая область движется по полоске с
течением времени стерилизации. Крышка самой крайней
слева бутылки была плотно завернута, по мере расстановки
слева направо крышка все более ослаблена, пока, наконец, у
самой крайней справа бутылки крышка не открыта совсем.
Самая крайняя слева бутылка не стерильна, поскольку пар
не попал в нее. Вторая недостаточно стерильна, поскольку
прокладка в крышке плотно прилипла к горлышку и за¬
печатала его, следующие три стерильны, но коричневая
окраска на индикаторе обозначает, что в конце цикла в
них оставалось остаточное количество жидкости, только
крайняя бутылка справа стерильная и сухая
б) Стандарты pH. Индикатор pH феноловый красный
в стандартном наборе растворов. Крайний слева и
крайний справа неприемлемы и требуют немедленного
действия — замены среды, субкультивирования или
наполнения газом
pH 6,5
pH 7,0
pH 7,4
pH 7,6
д) Разноцветные сосуды для криоконсервирования. Полипропа-
иленовые сосуды для криоконсервирования емкостью 1,0-4,6 мл.
Разноцветные вкладыши в крышки помогают предупредить ошибки
в идентификации
в) Вращающаяся система культуры клеток. Камера вра¬
щается для создания нулевой силы тяжести, так, чтобы
клетки не осаждались и росли в камере в виде агрегатов
г) Культуральная система BelloCell. Клетки растут
на макробусинах в центре камеры, а нижележащая
камера наполнена культуральной средой, которая
поочередно то прогоняется сквозь камеру с буси¬
нами, то возвращается обратно
Цветная вкладка 22. Приготовление среды, культуральные системы и флаконы для криоконсервации
a-в) Магнитный сортинг на системе Dynabeads (Dynal). Отрицательный сортинг; коммитированные клет-
ки-предшественники, выделенные из суспензии костного мозга, связываются с парамагнитными гранулами
Dynal с антителами к маркерам клеточной линии. Клетки, не имеющие линейных маркеров (стволовые),
не связываются и остаются в суспензии, готовыми к сортингу на проточном флуориметре: (а) Помещение
пробирки в магнитный держатель. (б) Пробирка сразу после установки в сортер. (в) Пробирка через 30 с
после начала сортинга
г-д) Магнитный сортинг на системе MidiMacs Separator (Miltenyi Biotec), (г) Колонка, магнит, поддон и
штатив. (д) MidiMacs Separator с колонкой LD. (С любезного разрешения Miltenyi Biotec.)
Цветная вкладка 23. Магнитноактивированный клеточный сортинг
о
г
т—
CNJ
CO
CD
CNJ
CM
CNJ
О
О
O)
■X
Q.
Q_
О
2
CO
CO
OsJ
О
Сг
со
GenBank
accession no.
gene
M92642
COL16A1
AI982638
COL16A1
M91669
COL17A1
X14420
COL3A1
M26576
COL4A1
X05610
COL4A2
Y14690
COL5A2
X15880
COL6A1
M20777
COL6A2
X15882
COL6A2
L02870
COL7A1
X57527
COL8A1
L41162
COL9A3
X17033
ITGA2
X17033
ITGA2
M59911
ITGA3
M59911
ITGA3
X06256
ITGA5
S66213
ITGA6
S66213
ITGA6
X53586
ITGA6
AF032108
ITGA7
M14648
ITGAV
M14648
ITGAV
AF011375
ITGB4
X53587
ITGB4
M35198
ITGB6
Экспрессия транскриптов фибронектина, коллагена и иитегрина. Определена при помо¬
щи микроанализа нуклеотидов в нормальной (NOK), SV40 Т-антиген- иммортализован-
ной (SVpgC2a) и злокачественной (SqCC/Yl) линиях кератиноцитов ротовой полости
человека. Сокращения в наименования GenBank: COL16A1 = коллаген типа XVI, цепь 1
и т.д.; ITGA2 = интегрин 2 субъединица и т.д.; ITGB4 = интегрин, 4 субъединица и т.д.
(по Sarang et al., 2003, ФЕДФ 31: 575-585 с разрешения издателя и автора)
Цветная вкладка 24. Анализ экспрессии генов, Affymetrix Microarry Analysis
Глава 17
ДИФФЕРЕНЦИРОВКА
I
17.1. ЭКСПРЕССИЯ ФЕНОТИПА IN VIVO
Фенотип клеток, которые развиваются и распространя¬
ются в культуре как клеточная линия, часто отличается
от фенотипа клеток, преимущественно доминирую¬
щего в исходной ткани (см. разд. 3.4.2). Эти отличия
объясняются тем, что ш vivo на клетки воздействует
ряд факторов, которые регулируют геометрию, рост и
функции тканей, но подобные факторы отсутствуют в
микроокружении in vitro, (см. разд. 17.1.1).
Дифференцировка — это процесс, ведущий к про¬
явлению фенотипических свойств, характерных для
функционально зрелых клеток этого типа in vivo. Этот
процесс может быть необратимым, как, например,
прекращение синтеза ДНК в ядре эритробласта или
зрелого кератиноцита, или обратимым, как, например,
индукция синтеза альбумина в дифференцированных
гепатоцитах, которая часто утрачивается в культуре, но
может быть реиндуцирована. Дифференцировка, как
она описывается в данном руководстве, представляет
собой комбинацию конститутивнгях (постоянно экс¬
прессируемых без дополнительной индукции) и адап¬
тивных (подверженных положительной и отрицатель¬
ной регуляции экспрессии) свойств, присущих зрелой
клетке. Коммитирование подразумевает необратимый
переход клетки из стадии стволовой в специфическую
линию дифференцировки, завершающуюся формиро¬
ванием клетки, способной проявлять ограниченный
набор свойств, конститутивных или индуцируемых.
Тем не менее концепция коммитирования вызывает
много вопросов, поскольку показано, что некоторые
клетки-предшественники могут превращаться в муль-
типотентные клетки (Kondo & Raff, 2000, 2004; Le
Douarin et al., 2004).
Терминальная дифференцировка предполагает, что
дифференцированная клетка прошла дифференци-
ровку по определенной линии до некоторой точки, в
которой фенотип зрелой полностью выражен и даль¬
нейших изменений не предполагается. В принципе,
это определение не должно исключать такие клетки,
как фиброциты, которые могут превращаться в ме¬
нее фенотипически дифференцированные клетки с
возобновлением пролиферации, но на практике этот
термин имеет отношение к таким клеткам, как нейро¬
ны, миоциты скелетных мышц, кератинизированные
сквамозные клетки, конечная дифференцировка ко¬
торых необратима.
17.1.1. Дедифференцировка
Термин дедифференцировка обычно используется
для описания процесса утраты дифференцированных
свойств клеток некоторой ткани при ее озлокачествле-
нии или при ее росте в культуре. Следует с осторож¬
ностью использовать термин «дедифференцировка»,
который подразумевает комплексный процесс, важный
вклад в него вносят такие факторы, как гибель клетки,
избыточный селективный рост и адаптивный ответ. При
правильном употреблении термин «дедифференциров¬
ка» означает утрату клеткой специфических фенотипи¬
ческих характеристик, присущих зрелой клетке. Процесс
дедифференцировки может быть как адаптивным про¬
цессом, подразумевающим, что дедифференцированный
фенотип может быть восстановлен с использовани¬
ем правильно подобранных индукторов, (см. также
разд. 3.4), так и селективным процессом, подразумеваю¬
щим, что клетка-предшественник была выбрана для се¬
лекции в связи с более выраженным пролиферативным
потенциалом. В любом случае клетка-предшественник
может быть индуцирована к созреванию до полностью
дифференцированной клетки или к дедифференцировке
до стволовой клетки (см. разд. 19.1), в определенных
заданных условиях окружающей среды.
17.1.2. Линейная селекция
Если была выбрана неправильная линия для диффе¬
ренцировки (например, из ткани печени выделены
стромальные фибробласты вместо гепатоцитов), то
никакие индукторы не помогут восстановить необхо¬
димый фенотип. В прошлом такие неудачи ошибочно
приписывались явлению дедифференцировки, однако
настоящей причиной тому является избыточный рост
стромальных фибробластов, вызванный такими фак¬
торами роста, как фактор роста тромбоцитов PDGF
(platelet-derived growth factor) и ингибированием про¬
лиферации эпителиальных клеток трансформирующим
фактором роста-Р (TGF-0).
17.2. СТАДИИ ДИФФЕРЕНЦИРОВКИ
Во взрослом организме существует два главных пути
дифференцировки. В постоянно возобновляющихся
322
ГЛАВА 17. ДИФФЕРЕНЦИРОВКА
тканях, подобных эпидермису, слизистой кишечника и
кроветворной ткани, существует небольшая популяция
тотипотентных и плюрипотентных недифференци¬
рованных стволовых клеток, способных к самовозоб¬
новлению, которые дают начало, в зависимости от
потребностей организма, клеткам-предшественникам,
которые будут делиться и развиваться до стадии
конечной дифференцировки, теряя при этом способ¬
ность к делению (см. рис. 3.6). Этот процесс приводит
к образованию зрелых, дифференцированных клеток,
которые обычно не делятся. Деление компартмента
клеток-предшественников регулируется обратной свя¬
зью, призванной обеспечить правильный объем пула
дифференцированных клеток.
В тканях, которые не обновляются так быстро, но
способны восстанавливаться после нанесения трав¬
мы, в стадии покоя наблюдается невысокий уровень
пролиферации; однако зрелые клетки могут повторно
вступать в деление. В соединительной ткани, например,
клетки типа фиброцитов могут ответить на местное
уменьшение плотности клеток и/или присутствие
одного или большего количества факторов роста
утратой дифференцированных свойств (таких как
способный к синтезу коллагена) и перейти в проли-
феративный цикл. Когда ткань в результате деления
клеток восстановила соответствующую плотность,
деление клеток останавливается и возобновляется
дифференцировка. Этот тип обновления ткани быстр,
потому что относительно большая популяция клеток
включается в процесс.
Неясно, являются ли клетки, которые повторно
входят в цикл деления для восстановления ткани, фе¬
нотипически идентичными основной массе популяции
дифференцированных клеток или они представляют
собой небольшую субпопуляцию обратимо дифферен¬
цированных клеток. Показано, что в печени, которая
регенерирует в ответ на повреждение, зрелые гепа-
тоциты могут вновь вступать в клеточный цикл, в то
время как в скелетных мышцах, в которых терминаль¬
но дифференцированные клетки не могут вступать в
клеточный цикл, регенерация тканей осуществляется
за счет клеток, образующихся из сателлитных клеток,
в покое составляющих латентную субпопуляцию ство¬
ловых клеток (см. разд. 23.3.3). Зрелые фиброциты,
клетки сосудистого эндотелия и глиальные клетки
обнаруживают способность к повторному вступле¬
нию в цикл деления для обеспечения регенерации, но
нельзя исключить также возможность существования
регенеративной субпопуляции внутри общей популя¬
ции клеток.
17.3. ПРОЛИФЕРАЦИЯМ
ДИФФЕРЕНЦИРОВКА
На поздних стадиях дифференцировки интенсивность
клеточного деления снижается, и, в конечном итоге,
оно прекращается. В большинстве клеточных систем
клеточное деление несовместимо с выраженностью
дифференцированных свойств. Опухолевые клетки
могут иногда нарушать это ограничение, например в
меланоме меланин продолжает синтезироваться в ходе
пролиферации клеток. Тем не менее, даже в этих слу¬
чаях, синтез продуктов дифференцировки возрастает
с прекращением деления.
В культуре тканей существует несколько серьезных
последствий этого правила. Поскольку при культивиро¬
вании клеток и тканей размножение и распространение
часто являются главными задачами, то неудивительно,
что большинство клеточных линий не проявляют
полностью фенотипических дифференцированных
свойств. Этот факт был обнаружен много лет назад при
наблюдениях за свойствами органной культуры (см.
разд. 25.2), которая оставалась трехмерной, обладала
высокой клеточной плотностью и характерной архи¬
тектурой и предупреждала диссоциацию и избыточный
селективный рост недифференцированных клеток.
Поэтому, несмотря на значительную ценность
обнаруженных механизмов межклеточных взаимо¬
действий, регулирующих дифференцировку, органная
культура всегда страдала невозможностью получения
большого количества копий идентичных культур, в
частности, если требовалось большое количество кле¬
ток. Гетерогенность образца, которая также является
существенным препятствием для получения фенотипа
тканей, делает проведение итогового биохимического
анализа чистой популяции клеток и их ответа крайне
затруднительными. Однако в последние годы было
осуществлено несколько попыток восстановления
дифференцированного фенотипа в чистой популяции
клеток путем создания специального микроокруже¬
ния и, таким путем, определения индивидуального
влияния, обнаруживаемого по индукции и контролю
дифференцировки. Этот процесс обычно предполагает
прекращение клеточного деления и образование интер¬
активной клеточной популяции высокой плотности,
как в гистотипичной, так и в органотипичной культу¬
ре. Межклеточное взаимодействие стало ключевым
моментом в создании искусственно конструируемых
тканей (см. разд. 25.3.8).
17.4. КОММИТИРОВАНИЕ
И ЛИНИИ ДИФФЕРЕНЦИРОВКИ
Развитие стволовой клетки по специфичному пути
дифференцировки традиционно предполагает быст¬
рое нарастание коммитирования, т.е. приобретение
клеткой структурно-функциональных свойств, ха¬
рактерных для зрелой дифференцированной клетки,
по мере прохождения стадий развития (см. рис. 3.6).
Гематопоэтическая стволовая клетка после коммити¬
рования лимфатической дифференцировки не будет
изменять линию дифференцировки на поздней стадии
и не приобретет характеристики миелоидного или эрит-
роцитарного рада. Аналогично, простейшая нейроэкто¬
дермальная стволовая клетка после коммитирования к
нейрональной дифференцировке не превратится в гли¬
17.4. Коммитирование и линии дифференцировки
323
альную клетку. Таким образом, коммитирование может
рассматриваться как точка между стволовой клеткой
и некоторой специфической стадией клетки-предшес¬
твенника, в которой клетка или ее предшественник
больше не может превращаться в другую линию. Если
такая необратимая стадия коммитирования сущест¬
вует, она должна быть позже, чем предполагали ранее,
поскольку некоторые клетки-предшественники могут
дедифференцироваться в стволовые клетки с мульти-
линейным потенциалом (см. разд. 17.1).
В последние годы было получено много данных о
переходе клеток из одной линии в другую. Возможно,
наиболее существенные из них связаны с регенераци¬
ей хрусталика амфибии с помощью клеток радужной
оболочки глаза [Clayton et al., 1980; Cioni et al., 1986].
Поскольку радужная оболочка представляет собой пол¬
ностью дифференцированный орган, но при этом обес¬
печивает регенерацию хрусталика, это явление было
определено как трансдифференцировка. Однако это
один из немногочисленных примеров, и большинство
данных относилось к опухолевым клеточным системам,
для которых источник популяции опухолевых клеток
может быть неясным. Например, было обнаружено, что
мелкоклеточная карцинома легких может переходить в
сквамозную или крупноклеточную карциному с после¬
дующим обострением после завершения химиотерапии.
Подразумевает ли это, что один клеточный тип, клетки
Кульчицкого [de Leij et al., 1985], которые, как счита¬
ется, дают начало развитию мелкоклеточной карцино¬
мы, изменяет свое коммитирование, или же опухоль
исходно развивалась из мультипотентных стволовых
клеток, и в случае рецидива процесс развивается по
иному пути, — до настоящего времени неясно [Gazdar et
al., 1983; Goodwin et al., 1983; Terasaki et al., 1984]. Ана¬
логично, развитие анапластических сквамозных раков
кожи может дать начало клеткам, которые проявляют
себя как мезенхимальные [Oft et al., 2002].
Клеточная линия К562 была выделена при мие-
лоидной лейкемии, но позже было показано, что одна
способна к дифференцировке эритроидного ряда
[Andersson et al., 1979Ь]. Вместо того чтобы быть ком-
митированным, миелоидный предшественник преобра¬
зуется в эритроидный; опухоль, возможно, развивается
из общей стволовой клетки, которая способна давать
начало как эроитроидной, так и миелоидной линиям
дифференцировки. По некоторым причинам, еще
не известным, постоянные культуры способствуют
преимущественному развитию клеток эритроидной
дифференцировки по сравнению с клетками миело-
идного ряда, наблюдаемыми в исходной опухоли или
ранней культуре. Кроме того, в культуре, полученной
из опухолей, может развиться смешанный фенотип.
Например, в глиоме крысы С6 обнаруживаются при¬
знаки как астроцитов, так и олигодендроцитов, и эти
признаки могут проявляться одновременно в одних и
тех же клетках (Breen & De Velis, 1974).
В целом, тем не менее, такие случаи необычны и
ограничены опухолевыми культурами. Большинство
культур, полученных из нормальных тканей, хотя и мо¬
гут дифференцироваться по различным направлениям,
не меняют линии дифференцировки. Это поднимает
вопрос об истинном статусе клеточных линий, полу¬
ченных из нормальных тканей. Большинство культур
получено 1) из стволовых клеток или ранних клеток-
предшественников, которые могут дифференциро¬
ваться по одному или более направлением (например,
слизистая легких, которая может стать сквамозной
или муцин-секретирующей, в зависимости от способа
стимуляции), 2) поздних клеток-предшественников,
которые сохраняют свою линию дифференцировки, и
3) дифференцированных клеток, таких как фиброциты,
которые могут дедифференцировать и пролифериро¬
вать, но сохраняют свойства линии (см. разд. 3.8). Неко¬
торые культуры мышиных эмбрионов, в общих чертах
называемые фибробластами (например, различные
клеточные линии, обозначаемые ЗТЗ), возможно, более
правильно будет относить к категории (1), поскольку
они могут быть индуцированы к развитию до адипоци-
тов, мышечных клеток, остеоцитов и эндотелия, так же
как и до фиброцитов.
Клеточные линии, возможно, следует рассматри¬
вать как смешанную популяцию стволовых клеток,
клеток-предшественников и дифференцированных
клеток, равновесие между которыми определяется рас¬
творимыми или контакт-опосредованными сигналами
окружающей среды. В наиболее быстро делящихся
клеточных линиях большинство клеток имеет фенотип
предшественников, но может сохранять пластичность
и сдвигаться как в сторону стволовых клеток, так и в
сторону дифференцированного фенотипа, в зависимос¬
ти от полученных сигналов.
В настоящее время описаны примеры дифферен¬
цировки клеток-предшественников (например, клетки
02А, общий предшественник олигодендроцитов и аст¬
роцитов 2-го типа в мозге, которые остаются делящими¬
ся клетками-предшественниками в присутствии PDGF
и bFGF), превращающихся в олигодендроциты в отсут¬
ствие сывороточных факторов роста или в астроциты
2-го типа в присутствии фетальной сыворотки теленка
либо комбинации нейротропного фактора (CNTF) и
bFGF [Raff et al., 1978; Raff, 1990] (см. табл. 23.2). Од¬
нако эти клетки, которые стали предшественниками
олигодендроцитов, могут вновь превращаться в клетки
с фенотипом, подобным стволовой клетке, если подверг¬
нуть их действию BMPs (Kondo & Raff, 2000, 2004).
Миокардиоциты остаются недифференцированными
и делятся в сыворотке и в присутствии bFGF, но диф¬
ференцируются в отсутствие сыворотки [Goldman &
Wurzel, 1992], а культуры зародышевых эмбриональ¬
ных стволовых клеток дифференцируют спонтанно.
Процесс дифференцировки может быть предотвращен
действием bFGF, SCF (фактор стволовых клеток, stem
cell factor), и LIF (фактор ингибирования лимфоцитов,
lymphocyte inhibitory factor) [Matsui et al., 1992]. В этом
случае клетки оставаются в пролиферирующем не
дифференцирующем состоянии.
Следовательно, с разработкой более определенных
сред и идентификацией факторов, индуцирующих
324
ГЛАВА 17. ДИФФЕРЕНЦИРОВКА
дифференцировку, постепенно станет возможным
определить правильный набор факторов, который
будет поддерживать клетки в необходимом состоянии,
подобном стволовой клетке, клетке-предшественнику,
и дифференцированном состоянии.
17.5. ПЛАСТИЧНОСТЬ СТВОЛОВЫХ
КЛЕТОК
Традиционная теория стволовых клеток предпола¬
гает, что на более ранних этапах развития стволовые
клетки обладают большей потенцией. Унипотентная
стволовая клетка — это клетка, которая может дать
начало только одной клеточной линии, например ство¬
ловая клетка в базальном слое эпидермиса, которая
дает начало кератиноцитам. Бипотентная стволовая
клетка способна дать начало двум линиям, например
лимфоидная стволовая клетка может дать начало Т- и
В-лимфоцитам. Мулътипотентная стволовая клетка
дает более чем две линии, например стволовые клетки-
предшественники костного мозга, которые дают начало
гранулоцитам, моноцитам, мегакариоцитам, тучным
клеткам и эритроцитам. Тотипотентпная стволовая
клетка дает начало всем известным клеточным типам.
В гемпатопоэтической системе этот термин может
использоваться для стволовой клетки, которая дает
начало всем клеткам крови, но в своем общем значении
это название подразумевает эмбриональную стволо¬
вую клетку (ES-клетка), которая по доминирующей в
настоящее время теории способна дать начало любой
линии клеток. Порождая внутреннюю клеточную массу
раннего эмбриона, ES-клетка, несомненно должна быть
тотипотентной стволовой клеткой.
Еще одна традиционная концепция предполагает,
что чем выше степень коммитирования, тем выше
вероятность того, что стволовая клетка будет распола¬
гаться в специфической ткани, например гематопоэти-
ческие стволовые клетки расположены в костном мозге,
стволовые клетки энтероцитов — на дне крипт кишеч¬
ника, стволовые клетки кератиноцитов — в эпидермисе.
Вместе с уровнем коммитирования и определенной
гистологической локализацией отмечается снижение
потенции разным направлениям дифференцировки.
Можно ожидать, что, например, стволовые клетки пе¬
чени будут давать только клетки печени (гепатоциты,
клетки желчевыводящих путей). Более того, нельзя
ожидать, что те ткани, которые не регенерируют, на¬
пример нейроны в центральной нервной системе, будут
иметь стволовые клетки. Тем не менее эта довольно
аккуратная концепция линейного коммитирования,
потенции и тканевой локализации постоянно вызывает
вопросы [Vescovi et al., 2002], поскольку есть данные
исследований, которые показывают, что 1) нерегене¬
рирующие ткани, такие как нейроны мозга, содержат
стволовые клетки [Pevny & Rao, 2003] и 2) тканевая
локализация не обязательно означает коммитирование
и снижение потенции стволовых клеток, поскольку
из стволовых клеток печени могут развиться нервные
клетки [Deng et al., 2003], стволовые клетки костного
мозга дают начало миоцитам, [Mangi et al., 2003], из
мышечных стволовых клеток могут развиваться гема-
топоэтические клетки, [Cao et al., 2003], а стволовые
клетки нервной системы могут порождать эндотелий
[Wurmser et al., 2004]. В некоторых случаях диффе¬
ренцировка идет в непредсказуемом направлении,
например, клетки костного мозга развиваются в ге¬
патоциты, кардиомиоциты или нейроны [Alison et al.,
2004; Alvarez-Dolado et al., 2003]. Возможной причиной
этого является слияние клеток [Greco & Recht, 2003],
однако другие авторы утверждают, что слияние клеток
не играет никакой роли [Wurmser et al., 2004].
Такие разработки нарушают традиционную концеп¬
цию, принятую в биологии стволовых клеток, и меняют
понятие о коммитировании линий дифференцировки
(см. также разд. 17.1, 17.4), но они также открывают
бесконечные горизонты для использования биоло¬
гии соматических стволовых клеток в исследовании
регуляции экспрессии генов и тканевой регенерации.
Однако остаются следующие проблемы: 1) определе¬
ние надежных маркеров для выделения такого мень¬
шинства клеточной популяции [Zhou et al., 2001; Cai
et al., 2004]; 2) практические вопросы использования
для регенерации тканей взрослых стволовых клеток,
которые имеют ограничения способности пролифе¬
рации, и 3) разработка условий культивирования для
распространения и дифференцировки, которые так
же эффективны, как те, которые используются для
эмбриональных стволовых клеток. Кроме того, была
обнаружена еще одна категория стволовых клеток в
пупочном канатике. Хотя по своим характеристикам
они относятся к гематопоэтическим, показана их муль-
типотентность [Lee et al., 2004; McGuckin et al., 2004],
поэтому использование этой категории стволовых
клеток может иметь меньшее количество ограниче¬
ний этического плана, чем человеческие ES клетки,
и обеспечит большую долговечность полученных
клеточных линий.
17.6. МАРКЕРЫ ДИФФЕРЕНЦИРОВКИ
Маркеры, которые начинают экспрессироваться на
начальных этапах дифференцировки и сохраняются на
протяжении последующих стадий созревания, обычно
носят название линейных маркеров. Это, например,
белки промежуточных филаментов, такие как цитоке¬
ратины (эпителий) [Moll et al., 1982] или глиальный
фибриллярный кислый протеин (астроциты) [Eng &
Bigbee, 1979; Bignami et al., 1980]. Маркеры зрелого
фенотипа, представляющего конечную стадию диф¬
ференцировки, обычно являются специфическими
клеточными продуктами или ферментами, участвую¬
щими в синтезе этих продуктов, например, гемоглобин
в эритроцитах, сывороточных альбумин в гепатоцитах,
трансглутаминаза [Schmidt et al., 1985] или инволю¬
крин [Parkinson & Yeudall, 2002] в дифференцирую¬
щихся кератиноцитах, глицеролфосфатдегидрогеназа
17.7. Индукция дифференцировки
325
в олигодендроцитах [Breen & De Vellis, 1974] (см.
табл. 3.1). Эти характерные вещества часто появляются
на поздних стадиях дифференцировки, и, возможно, их
экспрессия является обратимой и находится под адап¬
тивным контролем гормонов, питательных веществ,
составляющих веществ матрикса и межклеточного
взаимодействия (см. рис. 17.1).
Поскольку в настоящее время идентифицированы и
секвенированы гены, кодирующие большое количество
продуктов дифференцировки [International Human Ge¬
nome Sequencing Consortium, 2001], стало возможным
обнаружение экспрессии белковых маркеров дифферен¬
цировки с помощью RT-PCR [Ausubel et al., 1996,2002]
и МАА [Kawasaki, 2004], обнаруживающем экспрессию
специфических мРНК (см. цв. вкладку 24). Этот метод
не требует других методов, подтверждающих синтез
конечного продукта. Кроме того, возможно определить
различия при очень низких концентрациях (иди даже
отсутствии экспрессии) и очень высоком уровне экс¬
прессии специфических генных транскриптов. Может
быть произведен одновременный скрининг многих
генов, что дает значительное преимущество перед дру¬
гими методами определения продуктов экспрессии.
Дифференцировка должна рассматриваться как
процесс, связанный с экспрессией одного йли, пред¬
почтительно, более чем одного маркера, характерного
для конечного этапа дифференцировки. Хотя линей¬
ные маркеры полезны для подтверждения клеточной
идентичности, выраженность функциональных свойств
зрелой клетки является наилучшим критерием для
установления терминальной дифференцировки и под¬
тверждения происхождения клеток.
17.7. ИНДУКЦИЯ ДИФФЕРЕНЦИРОВКИ
Существует пять основных параметров, которые ре¬
гулируют процесс дифференцировки (см. рис. 17.1):
межклеточное взаимодействие, взаимодействие клетки
с матриксом, форма клеток и полярность, давление
кислорода и растворимые системные факторы.
17.7.1. Межклеточные взаимодействия
Гомотипичное. Взаимодействие между гомологич¬
ными клетками возникает при высокой клеточной
плотности. Это взаимодействие может включать ком¬
муникацию через щелевые контакты [Finbow & Pitts,
1981], при котором метаболиты, вторичные мессенд¬
жеры, такие как цАМФ, диацилглицерин (DAG), Са2+,
или электрический заряд, могут перемещаться между
клетками. Это взаимодействие, возможно, скорее со¬
гласует экспрессию дифференцировки внутри популя¬
ции сходных клеток, чем инициирует ее. Присутствие
гомотипичных молекул межклеточной адгезии, таких
как CAMs или кадгерин, которые являются кальций-
зависимыми, обеспечивает функционирование другого
механизма, при помощи которого могут взаимодейство¬
вать контактирующие клетки. Эти молекулы адгезии
обеспечивают первоначальное взаимодействие между
похожими клетками через идентичные реципрокно вза¬
имодействующие внеклеточные домены (см. рис. 3.2.1),
которые, видимо, обеспечивают передачу сигнала путем
фосфорилирования внутриклеточного домена [Doherty
etal., 1991; Gumbiner, 1995].
Гетеротипичное. Взаимодействие гетерологич-
ных клеток, например между клетками, имеющими
мезодермальное и эндодермальное или эктодермаль¬
ное происхождение, ответственно за инициацию и
прохождение дифференцировки. В ходе гаструляции
эмбриона и сразу после нее, а также позже в ходе орга¬
ногенеза происходит взаимодействие между клетками,
развивающимися из различных зародышевых листков.
Такое взаимодействие стимулирует дифференцировку
[Yamadaetal., 1991; Hiraietal., 1992; Hemmati-Brivaniou
et al., 1994; Muller et al., 1997]. Например, когда эндо-
дермальные клетки образуют дивертикул кишечника
и пролиферируют внутри прилегающей мезодермы,
тогда мезодерма индуцирует образование альвеоляр¬
ных и бронхиальных путей и сама индуцирует свое
преобразование в эластичную ткань [Hardman et al.,
1990; Caniggia et al., 1991].
МЕЖКЛЕТОЧНОЕ ВЗАИМОДЕИСТВИЕ
Гэмотипичное:
Контакт-опосредованное (CAMS, щелевые
контакты)
Растворимые гомокринные факторы
Гэтвротипичныв:
Растворимые паракринные факторы
Внеклеточный матрикс
Моделирование in vitro: Высокая плотность
(гистотипичное моделирование), рекомбинантные
культуры (органотипичное)
СИСТЕМНЫЕ ИЛИ ЭКЗОГЕННЫЕ ФАКТОРЫ
Естественные: Гормоны, циркулирующие
цитокины и факторы роста, витамины, Са2+
Синтетические: Двумерно полярные соединения,
РМА
Моделирование in vitro: Среда культивированная
со специальными добавками
ДАВЛЕНИЕ КИСЛОРОДА
Моделирование in vitro: Расстояние от
поверхности среды
Концентрация кислорода в газовой стадии
ФОРМА И ПОЛЯРНОСТЬ
Мембранный транспорт
Секреция
Презентация рецептора и рециркуляция
Положение ядра
Моделирование in vitro: Фильтрующие вкладыши
матриксы и складчатость, растягивающая или
сжимающая сила
МАТРИКСНОЕ ВЗАИМОДЕЙСТВИЕ
Внеклеточный матрикс
Коллаген, ламинин, протеогликаны, интегрин;
передача сигналов
Связывание и перенос цитокинов
Моделирование in vitro: продукты матрикса, Matrigel,
внеклеточный матрикс (ЕСМ)
Рис. 17.1. Регуляция дифференцировки. Параметры, контролирующие экспрессию дифференцировки in vitro
326
ГЛАВА 17. ДИФФЕРЕНЦИРОВКА
GM-CSF
V +KGF
Рис. 17.2. Реципрокное паракринное взаимодействие.
Схематическое изображение двойного паракринного пути
регулирования роста кератиноцитов в органотипичной
ко-культуре с фибробластами, влючающего IL-1, KGF, и
GM-CSF, а также их рецепторы [см. также Maas-Szabowski
et al., 2000; Szabowski et al., 2000]. Адаптированный рисунок
приводится no [Maas-Szabowski et al., 2002]
Во что выливаются эти процессы во взрослом
организме — неясно, но есть свидетельства, подтверж¬
дающие, что зрелые клетки эпидермиса сохраняют
реципрокное взаимодействие с подлежащим слоями
дермы. Это взаимодействие, опосредованное дей¬
ствием факторов роста KGF и GM-CSF, а также таких
цитокинов, как IL-la and IL-ip, необходимо для фор¬
мирования кератинизированных чешуек с полностью
перекрестно-связанным кератином (рис. 17.2) [Maas-
Szabowski et al., 2002].
Паракринные факторы роста
Положительное действие. Если взаимодействующие
клетки относятся к различным линиям дифференци¬
ровки, то имеет место действие гетеротипичных па¬
ракринных факторов, например KGF в простате [Planz
et al., 1998], альвеолярный фактор созревания в легких
[Post et al., 1984], и другие потенциальные кандидаты,
такие как IL-6 и онкостатин М при развитии лекгих
[McCormack et al., 1996, McCormick & Freshney, 2000],
фактор созревания глии в мозге, [Keles et al., 1992] (см.
разд. 17.7.1; цв. вкладки 12d и 13е,/), и интерфероны
[см., например, Pfeffer & Eisenkraft, 1991]. Некоторые
факторы роста действуют как морфогены [Gumbiner,
1992], например эпиморфин [Hirai et al., 1992; Radisky
et al., 2003] и фактор роста гепатоцитов /фактор рас¬
сеивания (HGF/SF) [Kinoshita& Miyajima, 2002].
HGF является представителем семейства гепарин-
связывающих факторов роста (HBGF) (см. табл. 10.3),
которые высвобождаются из фибробластов, таких
как MRC-5 [Kenworthy et al., 1992]. Эпиморфин и
HGF вызывают образование трубочек в постоянной
культуре клеток MDCK из почек собаки [Orellana et
al., 1996; Montesano et al., 1997], эпителии слюнных
желез [Furue & Saito, 1997], и эпителии молочной
железы [Soriano et al., 1995]. KGF, который являет¬
ся паракринным фактором, продуцируется фибро¬
бластами кожи и простаты. Он оказывает влияние на
эпидермальную дифференцировку [Aaronson et al.,
1991; Gumbiner, 1992; Maas-Szabowski et al., 2002] и ре¬
гулирует дифференцировку эпителия простаты [Planz
et al., 1998; Thomson et al., 1997]. Такие факторы роста,
как FGF-1, -2, и -3, KGF (FGF-7), TGF-P и активин,
также участвуют в эмбриональной индукции [Jessell
& Melton, 1992]. Оба фактора, HGF и KGF, продуци¬
руются фибробластами, связываясь с рецепторами,
найденными на других клетках (в основном эпите¬
лиальными), и являются классическими примерами
паракринных факторов роста. В некоторых случаях
высвобождение паракринных факторов роста может
находиться под контролем гормонов системного дей¬
ствия. Было показано, что in vivo альвеолярные клетки
II типа в легких производят сурфактант в ответ на
действие дексаметазона. В экспериментах in vitro об¬
наружено, эта индукция синтеза сурфактанта зависит
от связывания стероидов с рецепторами в строме, что
приводит к высвобождению пептида для активации
альвеолярных клеток [Post et al., 1984]. Аналогичным
образом, ответ эпителиальных клеток мышиной про¬
статы на андрогены опосредован стромой [Thomson
et al., 1997]. Было показано, что KGF является по
крайней мере одним из компонентов взаимодействия
в простате [Yan et al., 1992].
Дифференцировка энтероцитов тонкой кишки,
стимулированная гидрокортизоном, также требует
участия подлежащих фибробластов стромы [Kredinger
et al., 1987]. В данном случае происходит модификация
межклеточного матрикса между этими двумя типами
клеток.
[Simon-Assmann et al., 1986]. Дексаметазон-зависи-
мая модификация матрикса также включена в развитие
ответа альвеолярных клеток II типа на действие парак¬
ринных (см. разд. 17.7.3), [Yevdokimova & Freshney,
1997]. Дифференцировка в гематопоэтической системе
находится под контролем нескольких положительно
действующих линейно-специфичных факторов роста,
таких как IL-1, IL-6, G-CSF и GM-CSF (см. табл. 10.3
и 23.3), причем активация последнего также зависит от
матрикса [de Wynter et al., 1993]. Сульфаты гепарина
матрикса также участвуют в активации FGF [Klagsb-
run & Baird, 1991; Fernig &Gallagher, 1994].
Гомокринные факторы являются гомотипичными
паракринными факторами, производимыми в одной
клетке и действующими на соседние клетки той же
линии, а аутокринные факторы стимулируют ту же
самую клетку, которая продуцировала их (см. разд. 3.5
17.7. Индукция дифференцировки
327
и рис. 3.7). Гомокринные и аутокринные факторы, ви¬
димо, неразличимы с практической точки зрения.
Отрицательное действие. Такие факторы, как
MIP-ip, позволяют поддерживать фенотип стволовой
клетки [Graham et al., 1992], a PDGF и FGF-2 иниции¬
руют рост и самообновление клеток-предшественников
0-2А, но ингибирует их дифференцировку [Bogler et
al., 1990]. Другие отрицательные регуляторы диффе¬
ренцировки включают LIF, участвующий в клеточной
дифференцировке ES-клеток [Smith et al., 1988] и TGF-
Р, регулирующий дифференцировку альвеолярных
клеток II [Torday & Kourembanas, 1990; McCormick &
Freshney, 2000; McCormack et al., 1996].
17.7.2. Системные или экзогенные факторы
Физиологические индукторы (табл. 17.1). Различа¬
ют следующие системные физиологические регулято¬
ры, которые вызывают дифференцировку: 1) Гормоны,
которые in vivo образуются в отдаленных органах и
тканях и достигают органа-мишени по сосудистому
руслу (эндокринные факторы). В эту категорию входят
гидрокортизон, глюкагон и тироксин (или трийод¬
тиронин). 2) Витамины, такие как витамин D3 [Jeng
et al., 1994; Rattner et al., 1997] и ретиноевая кислота
[Saunders et al., 1993; Hafny et al., 1996; Ghigo et al.,
1998], поступают в организм из продуктов питания
и могут быть модифицированы в результате мета¬
болизма. 3) Неорганические ионы, в частности Са2+.
Высокая концентрация Са2+ обеспечивает процесс
дифференцировки кератиноцитов [Cho & Bikle, 1997],
(см., например, разд. 23.2.1). Вероятно, это связано с ро¬
лью кальция в клеточном взаимодействии (кадгерины
являются кальций-зависимыми), внутриклеточной пе¬
редаче сигнала и потоке кельция через мембрану в так
называемых «кальциевых волнах», которые передают
сигнал от одной отвечающей клетки к другой, рядом
расположенной, клетке той же линии (см. рис. 3.7).
Наряду с коммуникацией через щелевые контакты и,
возможно, гомокринными факторами и сульфатом ге¬
парина, кальциевые механизмы помогают генерировать
скоординированный ответ.
Другие физиологические индукторы — это па-
ракринные факторы, о которых шла речь выше (см.
разд. 17.7.1). Предполагается, что они действуют прямо,
от клетки к клетке, без участия транспорта по кровенос¬
ной системе. Тем не менее некоторые из этих факторов
(FGF, PDGF, интерлейкины) обнаруживаются в крови,
поэтому они могут играть системную роль.
Нефизиологические индукторы. Rossi and Friend
[1967] обнаружили, что клетки мышиной эритро¬
лейкемии, обработанные диметилсульфоксидом
(DMSO), для производства вируса лейкемии Friend
приобретают красную окраску в результате продук¬
ции гемоглобина (см. цв. вкладку 13а, б), что было
подтверждено возрастанием экспрессии гена глобина
(см. цв. вкладку 13в, г). Впоследствии было показано,
что многие другие клетки — нейробластомы, миеломы
и карциномы молочной железы — отвечают на действие
DMSO дифференцировкой. В список нефизиологичес¬
ких индукторов дифференцировки в настоящее время
внесены и другие вещества: гексаметилен бис-аце-
тамид (НМВА); ЛГ-метилацетамид; бензодиазепины,
действие которых может быть родственно действию
DMSO, и ряд цитотоксических препаратов, таких
как метотрексат, цитозинарабиноза и митомицин С
(табл. 17.2). Бутират натрия тоже был отнесен в список
нефизиологических индукторов дифференцировки,
однако некоторые данные показывают, что бутират
может являться естественным регулятором, например
доказано, что он регулирует в норме поведение кле¬
ток в кишечнике. Механизм действия этих веществ
неясен, но он может быть опосредован изменениями
в текучести мембраны (в частности, это относится к
полярным растворителям, какими являются DMSO,
анестетики и транквилизаторы). Кроме того, их дей¬
ствие может быть связано с взаимодействием с фермен¬
тами, участвующими в передаче сигнала, такими как
протеинкиназа С(РКС) и фосфолипаза D(PLD). Такое
взаимодействие приводит к изменению локализации
из растворимой формы в цитоплазме в эндоплазма-
тический ретикулум, где он активируется. К тому же,
эти вещества могут изменить метилирование ДНК или
ацетилировании гистонов. Индукция дифференциров¬
ки полярными растворителями, такими как DMSO,
может быть фенотипически нормальным явлением, в
то время как индукция цитотоксическими препаратами
может также вызывать экспрессию гена, не связанного
с дифференцировкой [McLean et al., 1986]. Показано,
что активаторы опухолей, такие как миристинфорбо-
лацетат (РМА), вызывают сквамозную дифференци¬
ровку в бронхиальной слизистой, притом что этого не
происходит с бронхиальной карциноме [Willey et al.,
1984; Masui et al., 1986 a, b; Saunders et al., 1993]. Хотя
такие активаторы не являются нормальными регуля¬
торами in vivo, они связываются со специфическими
рецепторами и активируют передачу сигнала, напри¬
мер, путем активации РКС [Dotto et al., 1985].
17.7.3. Взаимодействия клетки с матриксом
Поверхность большинства клеток окружает слож¬
ный матрикс гликопротеинов и протеогликанов,
высокоспецифичный для каждой ткани и даже для
отдельных частей ткани. Reid [1990] показал, что,
изменяя состав искусственного матрикса внесением
различных веществ, можно регулировать экспрессию
генов. Например, добавление матриксного материала,
выделенного из печени, вызывает экспрессию гена
альбумина в гепатоцитах. Более того, было обнаружено,
что коллаген важен для экспрессии функциональных
генов многих эпителиальных клеток [Burwen & Pitelka,
1980; Flynn et al., 1982; Berdichevsky et al., 1992] и для
эндотелия при созревании капилляров [Folkman &
Haudenschild, 1980]. Показано также, что во многих
328
ГЛАВА 17. ДИФФЕРЕНЦИРОВКА
Таблица 17.1. Растворимые индукторы дифференцировки: физиологические
Индуктор
Клеточный тип
Ссылка
Стероидные и родственные Глия, глиома
McLean et al., 1986
им вещества
Легочные альвеолярные клетки II типа
Rooney et al., 1995; McCormick et al., 1995
Гепатоциты
Granner et al., 1968
Эпителий молочных желез
Marte et al., 1994
Миелоидная лейкемия
Sachs, 1978
Ретиноиды
Трахеобронхиальный эпителий
Kaartinen et al., 1993
Эндотелий
Lechardeur et al., 1995; Hafny et al., 1996
Энтероциты (Сасо-2)
McCormack et al., 1996
Эмбриональная карцинома
Mills et al., 1996
Меланома
Lotan & Lotan, 1980; Meyskens and Fuller, 1980
Миелоидная лейкемия
Degos, 1997
Нейробластома
Ghigo et al., 1998
Пептидные гормоны
Меланотропин
Меланоциты
Goding and Fisher, 1997
Тиротопин
Щитовидная железа
Chambard et al., 1983
Эритропоэтин
Эритробласты
Goldwasser, 1975
Пролактин
Эпителий молочной железы
Takahashi et al., 1991; Rudland, 1992; Marte et al., 1994
Инсулин
Эпителий молочной железы
Marte et al., 1994; Rudland, 1992
Цитокины
Фактор роста нервов NGF
Нейроны
Levi-Montalcini, 1979
Фактор созревания глии
Глиальные клетки
Keles et al., 1992; Raff, 1990; Kondo & Raff, 2000,2004
CNTF, PDGF, ВМР2
Эпиморфин
Почечный эпителий
Hirai et al., 1992
Фиброцитарный-пневмо-
Пневмоциты II типа
Post et al., 1984
цитарный фактор
Интерферон-а, р
Клетки А549
McCormack et al., 1996
HL60, Миелоидная лейкемия
Kohlhepp et al., 1987
Интерферон-у
Нейробластома
Wuarin et al., 1991
CNTF
Астроциты 2 типа
Raff, 1990
IL-6, OSM
Клетки А549
McCormack et al., 1996; McCormick & Freshney, 2000
BMP
Клетки ЮТ 1/2
Shea et al., 2003
KGF
Кератиноциты
Aaronson et al., 1991
Эпителий простаты
Thomson et al., 1997; Yan et al., 1992
HGF
Почки (MDCK)
Bhargava et al., 1992; Li et al., 1992
Гепатоциты
Montesano et al., 1991
TGF-P
Бронхиальный эпителий, меланоциты
Masui et al., 1986a; Fuller and Meyskens, 1981
Эндотелии
Меланоциты
Aoki et al., 2005
Витамины
Витамин Е
Нейробластома
Prasad et al., 1980
Витамин D3
Моноциты (U937)
Yen et al., 1993
Миелома
Murao et al., 1983
Остеобласты
Vilamitjana-Amedee et al., 1993
Энтероциты (IEC-6)
Jeng et al., 1994
Витамин К
Гепатома
Bouzahzah et al., 1995
Почечный эпителий
Cancela et al., 1997
Ретиноиды (см. выше)
Неорганические вещества
Са2+
Кератиноциты
Boyce & Ham, 1983
случаях последовательность RGD (аргинин-глицин-
аспарагиновая кислота) в молекулах матрикса способна
взаимодействовать с рецепторами клетки [Yamada &
Geiger, 1997]. Небольшие полипептиды, содержащие
эту последовательность, эффективно блокируют мат-
рикс-индуцированную дифференцировку, что пред¬
полагает необходимость наличия интактных молекул
матрикса [Pignatelli & Bodmer, 1988].
17.7. Индукция дифференцировки
329
Были предприняты относительно успешные попыт¬
ки сымитировать действие матрикса с использованием
синтетических макромолекул. Например, молекулы
поли-О-лизина использовались для активизации роста
аксона нервной клетки в нейрональной культуре (см.
разд. 23.4.1), однако, возможно, требуются еще дли¬
тельные дополнительные исследования специфичнос¬
ти взаимодействия с матриксом. Электростатический
заряд сам по себе, по-видимому, недостаточен для моде¬
лирования более сложных сигналов, продемонстриро¬
ванных для многих различных типов взаимодействия
клеток с матриксом, но изменение заряда, вероятно,
позволяет клеткам прикрепляться и распластываться
на субстрате, и при этих условиях клетки способны
производить свой собственный матрикс.
Было показано, что эндотелиальные клетки [Kin-
sella et al., 1992; Garrido et al., 1995] и многие эпите¬
лиальные клетки дифференцируют более эффективно
на Матригеле [Kibbey et al., 1992; Darcy et al., 1995;
Venkatasubramanian et al.,2000; Portnoy et al., 2004],
матриксном материале, произведенном клетками
саркомы Engelberth-Holm-Swarm (EHS) и нанесен¬
ном чаще всего на ламинин. В качестве подложки
используется также коллаген или протеогликан.
Ряд клеточных линий, например А549 тип II или
клетки аденокарциномы легких Clara, обнаруживают
очевидный морфогенез при росте на Матригеле (см.
цв. вкладку 12в). Этот метод полезен, но имеет ряд
проблем, связанных с внесением других биологичес¬
ких изменений в систему. Тем не менее для решения
некоторых задач требуется определенный матрикс,
который должен быть создан в условиях лаборатории,
если его нет в коммерческой продаже.
Фибронектин, ламинин, коллаген и ряд других
компонентов матрикса могут быть приобретены в
разных компаниях. Тем не менее специфичность мат¬
рикса, возможно, определяется протеогликановыми
функциональными группами, внутри которых имеется
потенциал для широкой вариабельности, в частности
в количестве, типе и распределении сульфатирован-
ных гликозаминогликанов, таких как гепарансульфат
[Femig & Gallagher, 1994].
Внеклеточных матрикс может также играть роль
в модуляции активности факторов роста. Предпо¬
лагается, что протеогликаны матрикса, в частности
гепарансульфатпротеогликаны (HSPGs), могут свя¬
зываться с определенными факторами роста, такими
как GM-CSF [Damon et al., 1989; Luikart et al., 1990],
и делать их более доступными для расположенных
поблизости клеток. Трансмембранные HSPGs могут
также действовать как низко-аффинные рецепторы
для факторов роста и обеспечивать транспорт этих
факторов роста к высокоаффинным рецепторам
[Klagsbrun & Baird, 1991; Fernig & Gallagher, 1994].
Клеткам A549 требуются глюкокортикоиды для ответа
на индукторы дифференцировки, такие как OSM, IL-6,
и среда, кондиционированная фибробластами лег¬
ких. Глюкокортикоид, в данном случае дексаметазон
(DX), стимулирует синтез фракции гепарансульфата
с низкозарядной плотностью, в клетках А549, которая,
как показано, при частичной очистке замещает DX и
активирует OSM, IL-6, и кондиционированную фибро¬
бластами среду [Yevdokimova & Freshney, 1997].
17.7.4. Полярность
и форма клетки
Исследования, проведенные на гепатоцитах [Sattler
et al., 1978], показали, что для полного созревания
необходимо культивировать клетки на коллагеновом
геле с последующим отделением геля от дна чашки при
помощи шпателя или согнутой пастеровской пипетки.
Эти манипуляции индуцируют сокращение геля и пос¬
ледующим изменениям очертаний клетки от расплас¬
тавшейся до кубоидальной или даже колоннообразной.
Одновременно или вслед за изменениями формы, а так¬
же, возможно, для того, чтобы обеспечить доступ к среде
через гель, у клеток развивается полярность, видимая
при использовании электронной микроскопии, при
котором ядро становится асимметрично расположен¬
ным, ближе к базальному концу клетки, формируется
активный комплекс Гольджи и на апикальном конце
клетки происходит секреция.
Похожие процессы установления полярности были
показаны в клетках эпителия щитовидной железы
[Chambard et al., 1983] при помощи комплекта филь¬
трующих ячеек. В данном случае нижняя (базальная)
поверхность образует рецепторы для тиреотропного
гормона (ТТГ) и секретирует трийодтиронин, а верх¬
ний (апикальный) конец высвобождает тиреоглобу-
лин. Исследования на гепатоцитах [Guguen-Guillouzo
& Guillouzo, 1986] (см. разд. 23.2.6) и бронхиальном
эпителии [Saunders et al., 1993] предполагают, что
коллагеновый матрикс не играет существенной роли,
но успех культивирования в фильтрующих лунках
(см. разд. 24.7.4) подтверждает роль контакта нижней
поверхности клеток со средой.
17.7.5. Давление кислорода
Достижение стадии полной кератинизации при сква-
мозной дифференцировке требует расположения
эпидермальных клеток в органотипической конструк¬
ции на поверхности раздела сред воздух/жидкость
[Maas-Szabowski et al., 2002]. Таким же образом рас¬
положение альвеолярных клеток II типа необходимо
для оптимальной дифференцировки II типа [Dobbs
et al., 1997], а эпителий только трахеи на поверхности
воздух/жидкость начнет секретировать слизь, остава¬
ясь сквамозным при росте на дне чашки [Kaartinen et
al., 1993; Paquette et al., 2003]. Суммируя сказанное,
можно сказать, что расположение клеток на границе
раздела жидкой и газовой фаз улучшает газовый обмен,
чрезвычайно облегчая захват кислорода без повышения
парциального давления и риска образования токсич¬
ных свободных радикалов. Тем не менее возможно, что
330
ГЛАВА 17. ДИФФЕРЕНЦИРОВКА
тонкая пленка над культурой имитирует физические
условия in vivo (поверхностное натяжение, недостаток
питательных веществ) так же, как оксигенация. При¬
сутствие D-PBS на апикальной поверхности может в
действительности оказаться предпочтительнее полной
питательной среды [Chambard et al., 1983,1987].
подтверждают наличие взаимной связи между выра¬
женностью дифференцированных свойств качеств и
выраженностью признаков злокачественности, вплоть
до вывода, что индукция дифференцировки может рас¬
сматриваться как способ терапии [Spremulli & Dexter,
1984; Freshney, 1985].
17.8. ДИФФЕРЕНЦИРОВКА
И ЗЛОКАЧЕСТВЕННОСТЬ
Часто отмечается, что при нарастающем развитии рака
гистологическое исследование опухоли обнаруживает
менее дифференцированные клетки, и, с прогности¬
ческой точки зрения, больные с малодифференци¬
рованными опухолями обычно имеют более низкую
выживаемость, чем больные с дифференцированными
формами рака. Кроме того, считается установленным,
что рак является результатом нарушения способности
клеток к нормальной дифференцировке (см. табл. 17.2).
В самом деле, большая часть результатов фундамен¬
тальных исследований была получена на клетках
мышиной лейкемии Friend, клетках миеломы мыши и
человека и нейробластомы. Тем не менее эти данные
17.9. ПРАКТИЧЕСКИЕ АСПЕКТЫ
Понятно, что при созданных правильных условиях
окружающей среды и предполагая, что в культуре при¬
сутствуют соответствующие клетки, дифференцировка
в клеточной культуре достижима частично или даже
полностью. Как общий метод индукции и обеспечения
дифференцировки, противоположный методам клеточ¬
ной пролиферации и размножения, можно предложить
следующий:
1) Выбрать нужный тип клеток при помощи соответ¬
ствующих условий выделения и селективных сред
(см. разд. 10.2.1 и гл. 23).
2) Вырастить клетки до высокой клеточной плотнос¬
ти (>1х105 кл./см2,) на соответствующем матриксе.
В качестве матрикса можно использовать коллаген,
Таблица 17.2. Растворимые индукторы дифференцировки: нефизиологические
Индуктор
Клеточный тип
Ссылка
Двухмерно-полярные компоненты
DMSO
Мышиная эритролейкемия
Rossi & Friend, 1967; Dinnen & Ebisuzaki, 1990
Миелома
Tarella et al., 1982
Нейробластома
Kimhi et al., 1976
Эпителий молочной железы
Rudland, 1992
Гепатоциты
Mitaka et al., 1993; Hino et al., 1999
Бутират натрия
Эритролейкемия
Andersson et al., 1979b
Рак толстой кишки
Velcich et al., 1995
N-метилацетамид
Глиома
McLean et al., 1986
N-метилформамид, диметилформамид
Рак толстой кишки
Dexter et al., 1979
НМВА
Эритролейкемия
Osborne et al., 1982; Marks et al., 1994
Бензодиазепин
Эритролейкемия
Clarke and Ryan, 1980
Цитотоксические препараты
Генистеин
Эритролейкемия
Watanabe et al., 1991
Цитозинарабиноза
Миелоидная лейкемия
Takeda et al., 1982
Митомицин С, антрациклины
Меланома
Raz, 1982
Метотрексат
Колоректальная карцинома
Lesuffleur et al., 1990
Модуляторы передачи сигнала
РМА
Бронхиальный эпителий
Willey et al., 1984; Masui et al., 1986a
Эпителий молочной железы
Wada et al., 1994
Толстый кишечник (НТ29,
Velcich et al., 1995; Pignata et al., 1994
Сасо-2)
Моноциты (U937)
Hass et al., 1993
Эритролейкемия (К562)
Kujoth and Fahl, 1997
Нейробластома
Spinelli et al., 1982
Аббревиатуры: НМВА, Гексаметилен бис-ацетамид (hexamethylene bisacetamide); РМА (ТРА), форбол миристат ацетат (phorbol
myristate acetate).
17.9. Практические аспекты
331
соответствующий коллагену из ткани — источника
клеток. Матрикс может иметь более сложный состав
и быть полученным из тканей или клеток, (см. про¬
токол 8.1), например, Matrigel (см. разд. 17.7.3) или
синтетический матрикс (например, поли-Э-лизин
для нейронов).
3) Перенести клетки в соответствующую среду, кото¬
рая более подходит для дифференцировки чем для
размножения, например для клеток эпидермиса не¬
обходимо увеличение концентрации Са2+до величин
около 3 мМ, для бронхиальной слизистой возраста¬
ние концентрации сыворотки (см. протокол 23.9).
Для других клеточных типов этот шаг может
потребовать различных факторов роста, необходи¬
мых для продолжения клеточной пролиферации и
ответственных за индукцию дифференцировки.
4) Добавить агенты-индукторы дифференцировки, та¬
кие как глюкокортикоиды, ретиноиды, витамин D3;
DMSO; НМВА; простагландины и цитокины, на¬
пример bFGF, EGF, KGF, HGF, IL-6, OSM, TGF-P,
интерфероны, NGF, и меланоцит-стимулирующий
гормон (MSH), в соответствии с типом клеток (см.
табл. 17.1 и 17.2).
5) Добавить взаимодействующий тип клеток в ходе со¬
ответствующей фазы роста (шаг 2 этой процедуры),
индукционной фазы (шаги 3 и 4 данной процедуры),
или обеих фаз. Выбор правильного типа клеток не
всегда очевиден, но фибробласты легкого, необхо¬
димые для созревания легочного эпителия [Post
et al., 1984; Speirs et al., 1991], глиальные клетки
для созревания нейронов [Seifert & Muller, 1984] и
адипоциты костного мозга для гематопоэтических
клеток (см. протокол 23.22) представляют собой
лучше всего охарактеризованные примеры.
6) Культивирование в фильтрационной ячейке [см.
например, Chambard et al., 1983] может быть пред¬
почтительно, особенно для определенных типов
эпителия. В ходе культивирования необходимо
обеспечить доступ базальной поверхности клеток к
питательным веществам и лигандам, возможность
установления полярности и регуляции питательных
веществ и кислорода на апикальной поверхности, пу¬
тем подбора состава и глубины подлежащей среды.
Не все из этих факторов могут потребоваться, по¬
следовательность, в которой эти этапы представлены,
означает лишь определенную степень приоритетности.
Большое значение может также иметь определенный
график работ. Например, матрикс обычно медленно
обновляется, поэтому может быть важным пролон¬
гированное действие матрикс-индуцированных ус¬
ловий, в то время как некоторые гормоны могут быть
эффективны при относительно коротком времени
экспозиции. Более того, ответ на гормоны может за¬
висеть от присутствия соответствующих компонентов
внеклеточного матрикса, плотности клеток или взаи¬
модействия гетерологичных клеток.
Глава 18
ТРАНСФОРМАЦИЯ И ИММОРТАЛИЗАЦИЯ
18.1. РОЛЬ В ХАРАКТЕРИСТИКЕ
КЛЕТОЧНЫХ ЛИНИЙ
Трансформация рассматривается как отдельное собы¬
тие или серия событий, которые определяются генети¬
ческой нестабильностью. Она меняет многие свойства
клеточных линий, включая темпы роста, тип роста
(прикрепленный или суспензионный), продукцию
специализированных продуктов, продолжительность
жизни и туморогенность (табл. 18.1). Таким образом,
трансформация является важным событием, поскольку
эти характеристики важны для доказательства прина¬
длежности клеточной линии при определении того,
происходит ли полученная линия из неопластических
клеток или в линии произошла трансформация при
культивировании. Состояние трансформации являет¬
ся принципиально важной характеристикой, которая
должна быть известна, когда культивируются клетки,
полученные из опухолей. В этом случае необходимо
подтвердить, что клетки получены из неопластической
компоненты опухоли, а не из нормальных фиброблас¬
тов, инфильтрирующих ткань, клеток кровеносных
сосудов или клеток воспаления.
Для подтверждения неопластического происхож¬
дения необходим более чем один критерий, поскольку
большинство из вышеупомянутых характеристик про¬
являются и в нормальных клетках на определенных
стадиях их развития. Исключение составляют анеуп-
лоидия, гетероплоидия и канцерогенность, которые
вместе взятые рассматриваются как неоспоримые по¬
ложительные индикаторы злокачественной трансфор¬
мации. Тем не менее некоторые опухолевые клеточные
линии могут быть почти эуплоидны и нетуморогенны,
поэтому требуются другие критерии.
18.2. ЧТО ТАКОЕ ТРАНСФОРМАЦИЯ?
В микробиологии, где этот термин впервые был ис¬
пользован в таком контексте, трансформация под¬
разумевает изменение фенотипа, которая зависит от
захвата нового генетического материала. Этот процесс
теперь может быть воспроизведен искусственно в
клетках млекопитающих (см. разд. 27.11), он назы¬
вается трансфекцией, или переносом ДНК, для того
чтобы отличать его от трансформации. Трансформация
культивированных клеток подразумевает спонтанные
или индуцированные перманентные фенотипические
изменения, возникшие в результате наследуемых
изменений в структуре ДНК и экспрессии генов.
Хотя трансформация может развиться в результате
инфекции трансформирующим вирусом, таким как
вирус полиомы, либо трансфекции такими генами, как
мутантный ген ras, она также может развиться спон¬
танно после воздействия ионизирующей радиации или
химического канцерогена.
Трансформация ассоциируется с генетической
нестабильностью и тремя основными классами фено¬
типических изменений, один из которых может быть
выражен в одном клеточном штамме: 1) бессмертие,
приобретение бесконечного срока жизни, 2) аберрант¬
ный контроль клеточного роста, утрата клеточной
подвижности в результате контактного ингибирования
подвижности, утрата ограничения клеточной проли¬
ферации при высокой клеточной плотности и утрата
зависимости от прикрепления к субстрату и 3) злока¬
чественность, доказательством которой служит рост
инвазивных опухолей in vivo. Термин трансформация,
обычно используемый здесь, подразумевает все три
этих процесса. Приобретение «бессметрия» само по
себе называется иммортализацией, поскольку оно
может быть достигнуто без значительных нарушений
механизмов контроля роста и злокачественности, ко¬
торые обычно взаимосвязаны.
18.3. ГЕНЕТИЧЕСКАЯ НЕСТАБИЛЬНОСТЬ
Характеристики клеточных линий не всегда оста¬
ются стабильными. В дополнение к уже описанным
фенотипическим изменениям (см. разд. 3.4.2, 17.1.1)
клеточные линии также склонны к генетической
нестабильности. Нормальные, конечные человеческие
клеточные линии обычно генетически стабильны,
но клеточные линии, полученные из других видов,
в частности из мышей, генетически нестабильны и
способны к трансформации. Постоянные клеточные
линии, в частности полученные из опухолей, незави¬
симо от вида, очень нестабильны, что неудивительно,
поскольку именно их нестабильность обеспечивает
возможность мутаций, необходимых для того, чтобы
стать постоянными. Обычно имеют место делеции
или изменения в структуре генов регуляторных ДНК,
таких кшр53. Следовательно, постоянные клеточные
18.3. Генетическая нестабильность
333
Таблица 18.1. Свойства трансформированных клеток
Свойство
Анализ
Протокол, рис. или ссылка
Рост
Иммортализованные
Рост свыше 100 pd*
Kopper & Hajdu 2004; Meeker
and De Marzo 2004
Независимость от субстрата
Клонирование в агаре, может расти в переме¬
шиваемой суспензии
Протоколы 14.4,14.5,13.3
Утрата контактного торможения
Наблюдение под микроскопом
Запись фильма о поведении клеток
Рис. 18.3; цв. вкладка 9
Рост гомологичных клеток в конфлюэнтном
Образование фокусов трансформации
Рис. 18.3
монослое
Снижено ограничение роста при высокой плот¬
Высокая плотность популяции, высокая доля
Протоколы 18.3, 21.10,
ности популяции
делящихся клеток при высокой плотности
клеток
21.11
Низкая потребность в сыворотке
Клонирование при низкой концентрации
сыворотки в среде
Протоколы 21.9, 22.3
Независимость от факторов роста
Клонирование при низкой концентрации
сыворотки в среде
Протоколы 21.9,22.3
Продукция аутокринных факторов роста
Иммуноокрашивание; клонирование при
низкой концентрации сыворотки в кондици¬
онированной среде; рецептор-блокирующие
антитела или пептидные ингибиторы
Протоколы 16.13,21.9,22.3
Продукция трансформирующего фактора роста
Суспензионное клонирование NRK
Протоколы 14.4,14.5
Высокая эффективность посева
Клонирование при низкой концентрации
сыворотки в среде
Протоколы 21.9,22.3
Укорочение периода удвоения популяции
Кривая роста
Протоколы 21.7,21.8
Генетические свойства
Высокий темп спонтанной мутации
Обмен сестринскими хроматидами
Протокол 22.5
Анеуплоидия
Хромосомный набор
Протокол 16.9
Гетероплоидия
Хромосомный набор
Протокол 16.9
Сверхэкспрессия или мутированные онкогены
Саузерн блот, FISH, иммуноокрашивание,
МАА
Ausubel et al., 1996,2002;
протоколы 28.2,16.13;
цв. вкладка 24
Делециции или мутации генов-супрессоров
Саузерн блот, FISH, иммуноокрашивание,
МАА
Ausubel et al., 1996,2002;
протоколы 28.2,16.13;
цв. вкладка 24
Транслокации генов и хромосом
FISH, хромосомное окрашивание
Протокол 28.2
Структурные свойства
Модификация актинового цитоскелета
Иммуноокрашивание
Протокол 16.13.
Утрата связывания фибронектина с клеточной
Иммунноокрашивание
Протокол 16.13.
поверхностью
Модифицированный внеклеточный матрикс
Иммуноокрашивание, DEAE хроматография
Протокол 16.13;
Yevdokimova and Freshney,
1997
Изменение экспрессии генов молекул клеточной
Иммуноокрашивание
Протокол 16.13
адгезии (CAMs, кадгерины, интегрины)
Нарушение клеточной полярности
Иммуноокрашивание, поляризационный тран¬
спорт в фильтрующих лунках
Протокол 16.13; Halleuxand
Schneider, 1994
Неопластические
Туморогенные свойства
Ксенотрансплантация в мышах Nude и
Scid**
Giovanella et al., 1974; Russo
et al.,1993
Ангиогенные свойства
CAM-исследование, фильтрующие ячейки,
продукция VGEF
Цв. вкладка 9; Ment et al.,
1997; Buchler et al., 2004
Усиление секреции протеаз (активатор плазми-
ногена)
Исследование активатора плазминогена
Рис. 18.8; Whur et al., 1980;
Boxman et al., 1995
Инвазивность
Инвазия опухолевых клеток в выделенные и
культивируемые ткани, исследование инва¬
зии в фильтрующих ячейках
Рис. 18.6, 18.7; Mareel et al.,
1979; Brunton et al., 1997
* pd - population doubling -период удвоения популяции
** Scid -severe combine immunodeficite syndrome — тяжелый комбинированный иммунодефицит
334
ГЛАВА 18. ТРАНСФОРМАЦИЯ И ИММОРТАЛИЗАЦИЯ
Номер клона
Рис. 18.1. Клональные вариации. Различия в активности
тирозинаминотрансферазы в четырех субклонах клона 18
штамма H-4-II-E-C3 клеток гепатомы крысы с минимальны¬
ми отклонениями. Клетки клона 18 были выделены, выраще¬
ны, повторно клонированы; клоны второго поколения были
исследованы на тирозинаминотрансферазную активность
с предварительной обработкой культуры дексаметазоном
(ДМ) и без обработки ДМ. Светло-серые столбики — ба¬
зальный уровень; темно-серые столбики — индуцированный
уровень (данные J. Somerville)
штаммы даже после клонирования содержат набор ге¬
нотипов, которые постоянно изменяются.
Существует две основных причины генетической
гетерогенности: 1) высокая скорость спонтанных му¬
таций, которая выше in vitro, видимо, в связи с высокой
скоростью клеточного деления и 2) отсутствие эли¬
минации мутантных клеток, если только эти мутации
не нарушают рост клеток. Минимально-девиантные
клетки печени крыс, при росте в культуре, обладают
конститутивной тирозинаминотрансферазной актив¬
ностью, которая может быть в дальнейшем индуциро¬
вана гексаметазоном, [Granner et al., 1968], но субклоны
клонированных штаммов H4-II-E-C3 [Pitot et al., 1964]
различаются как конститутивным уровнем ферментов,
так и возможностью индукции их активности декса¬
метазоном (рис. 18.1).
18.3.1. Хромосомные аберрации
Для обнаружения генетических нарушений может
быть использован подсчет хромосом (см. рис. 3.9) и
кариотипический анализ. Хотя кариотип мышиных
клеток состоит исключительно из маленьких телоцент¬
рических хромосом, во многих постоянных мышиных
клеточных линиях в результате слияния теломер по¬
является несколько метацентриков (рис. 18.2а; Роберт-
сонианское слияние (Robertsonian fusion)). Более того,
хотя практически каждая клетка у животных имеет
нормальный диплоидный набор хромосом, в культуре
кариотип более вариабелен. В крайних случаях, т.е.
в штаммах постоянных клеточных линий, таких как
Рис. 18.2. Хромосомные аберрации. Примеры аберрантных рекомбинаций: (а) клетки Р2, клон L929 мышиных фибро¬
бластов, обнаруживающий множественные теломерные слияния, две точки хромосомы помечены стрелками; (6) рекомби¬
нантные события между двумя диссимилирующими хромосомами в клетках китайского хомячка Y5. Маловероятно, что эти
клетки выживут
18.4. Иммортализация
335
HeLa-S3 менее чем половина клеток будет иметь тот
же кариотип, т.е. эти линии гетероплоидны (см. также
рис. 3.9). Могут обнаруживаться оба варианта измене¬
ний плоидии и возрастание частоты индивидуальных
хромосомных аберраций (рис. 18.26) [Biedler, 1976;
Croce, 1991]. Вариации количества хромосом встре¬
чаются также в большинстве опухолевых культур
(см. рис. 3.8; протокол 16.9; [Sandberg, 1982]). Число
случаев генетической нестабильности и частота хро¬
мосомных перестроек могут быть определены методом
изучения обмена сестринских хроматид [Venitt, 1984]
(см. протокол 22.5 и цв. вкладку 17Э, е). Некоторые спе¬
цифические аберрации связаны с определенными типа¬
ми злокачественности [Сгосе, 1991]. Первой докумен¬
тированной аберрацией была Филадельфийская хро¬
мосома, обнаруженная при хронической миелоидной
лейкемии (трисомия 13-й хромосомы). Впоследствии
трнаслокации длинного плеча 8-й и 14-й хромосом
были обнаружены при лимфоме Беркитта [Lebeau &
Rowley, 1984]. Некоторые другие лейкемии также
связаны с транслокациями [Mark, 1971]. Менингиомы
часто имеют постоянно присутствующее аберрации, а
при мелкоклеточном раке легкого зачастую обнару¬
живается делеция Зр2 [Wurster-Hill et al., 1984]. Эти
аберрации представляют собой опухолеспецифичные
маркеры, которые могут быть чрезвычайно ценны при
характеристике клеточных линий или подтверждении
новообразований.
18.3.2. Изменение
содержания ДНК
Проточная цитометрия [Traganos et al., 1977] показыва¬
ет, что содержание ДНК опухолевых клеток отражает
хромосомные аберрации, т.е. оно может отличаться
от ДНК нормальных соматических клеток и служить
маркером гетерогенности популяции. Анализ ДНК не
заменяет хромосомный анализ, поскольку клетки с
определяемым нормальным содержанием ДНК могут
иметь анеуплоидный кариотип. При анализе ДНК
также возникают ошибки, поскольку могут взаимно
уничтожаться делеции и полисомия либо могут иметь
место транслокации без общих потерь ДНК.
18.4. ИММОРТАЛИЗАЦИЯ
Большинство нормальных клеток имеют конечный
срок жизни продолжительностью в 20-100 генераций
(см. разд. 3.8.1), но некоторые клетки, преимущест¬
венно полученные от грызунов и большинство клеток
опухолей, могут давать начало постоянным клеточным
линиям с неопределенной продолжительностью жизни.
Клетки грызунов кариотипически нормальны при вы¬
делении и примерно через 12 генераций проходят через
кризис; большинство клеток погибают в ходе кризиса,
но некоторое их количество выживает и характеризу¬
Рис. 18.3. Фокусы трансформации. Монослой нормальных мышиных NIH ЗТЗ фибробластов, обладающих контактным
торможением, оставленный в состоянии конфлюэнтного монослоя на 2 недели, (а) 75-см2 флакон, окрашенный по Гимза (б)
Фазово-контрастное изображение фокусов трансформированных клеток, имеющих избыточный рост по сравнению с нормаль¬
ным монослоем (10х объектив) (см. также цв. вкладку 14в)
Фокусы трансформации ^трансформированная культура ЗТЗ
336
ГЛАВА 18. ТРАНСФОРМАЦИЯ И ИММОРТАЛИЗАЦИЯ
ется ускорением темпов роста и развитием постоянных
клеточных линий.
Если постоянные клеточные линии, полученные из
мышиных эмбрионов (т. е. различные ЗТЗ клеточные ли¬
нии), поддерживаются при низкой клеточной плотности,
что не позволяет образовывать конфлюэнтный слой
при любой продолжительности времени культивирова¬
ния, то они остаются чувствительными к контактному
торможению и ограничению роста плотностью клеток
[Todaro & Green, 1963]. Однако если клеткам позволяют
сформировать конфлюэнтный слой и находиться в нем в
течение продолжительного периода, то образуются фо¬
кусы клеток со сниженным контактным торможением,
которые накапливаются и, в конечном итоге, приобрета¬
ют способность к избыточному росту (рис. 18.3).
Тот факт, что эти клетки неразличимы при низкой
плотности или когда конфлюэнтный монослой только
формируется, предполагает, что они могут возникать de
novo, в результате трансформационных изменений. Эти
клетки приобретают преимущества в росте, и последую¬
щее субкультивирование быстро приведет к избыточному
росту случайным образом изменившихся клеток. Этот тип
клеток часто обнаруживает высокую туморогенность.
18.4.1. Контроль физиологического
старения
Конечный жизненный цикл клеток в культуре регули¬
руется группой из 10 или более доминантно действую¬
щих генов старения, продукты которых негативно
регулируют прохождение клеточного цикла [Goldstein
et al., 1989; Sasaki et al., 1996]. Эксперименты по сомати¬
ческой гибридизации конечных и иммортализованных
клеток обычно дают гибриды с конечным жизненным
циклом, что позволяет предположить, что гены старе¬
ния являются доминантными [Pereira-Smith and Smith,
1988]. Похоже, что один или более из этих генов нега¬
тивно регулируют экспрессию теломеразы [Holt et al.,
1996; Greider & Blackburn, 1996; Smith & de Lange, 1997;
Bryan & Reddel, 1997], необходимой для терминального
синтеза теломерной ДНК, которая в противном случае
постепенно укорачивается в ходе циклов клеточного
деления, до тех пор пока хромосомная ДНК не сможет
больше реплицироваться. Теломераза экспрессируется
в половых клетках и обладает умеренной активностью,
но отсутствует в соматических клетках. Делеции и/или
мутации в генах старения либо сверхэкспрессия или
мутация одного или более онкогенов, которые переве¬
шивают действие генов старения, позволяет клеткам
избежать негативного контроля жизненного цикла и
ре-экспрессировать теломеразу.
Суммируя, можно сказать, что иммортализация яв¬
ляется многоступенчатым процессом, включающим в
себя инактивацию рядя генов-регуляторов клеточного
цикла, таких как Rb и р53. Ген SV40 LTчасто исполь¬
зуется для индукции иммортализации. Известно, что
продукт этого гена, Т-антиген, связывает Rb и р53,
не только обеспечивая большую продолжительность
пролиферативного жизненного цикла, но и ограни¬
чивая активность таких генов-регуляторов ДНК, как
р53, таким образом повышая нестабильность генома
и давая больше шансов образованию новых мутаций,
предпочтительных для иммортализации (т.е. положи¬
тельно регулирующих образование теломеразы или
подавляющих какой-то из ингибиторов теломеразы).
Трансфекция теломеразного гена при помощи регули¬
руемого промотора достаточна для иммортализации
клеток [Bodnar et al., 1998; Vaziri & Benchimol, 1998].
Иммортализация сама по себе не подразумевает раз¬
витие аберрантного контроля клеточного роста и озло-
качествления, поскольку ряд иммортализованных кле¬
точных линий, таких как ЗТЗ и ВНК21-С13, сохраняют
способность к контактному торможению клеточной
подвижности, ограничению пролиферативной актив¬
ности плотностью клеточной популяции, зависимость
от прикрепления к субстрату и не являются тумороген¬
ными. Суммируя, следует заключить, что некоторые
аспекты контроля роста являются нетипичными и что
они участвуют в повышении геномной нестабильнос¬
ти. Более того, иммортализованные клеточные линии
часто утрачивают способность к дифференцировке,
но есть сообщения, касающиеся теломераза-индуци-
рованной иммортализации кератиноцитов [Dickson
et al., 2000] и сателлитных клеток скелетных мышц
[Wootton et al., 2003] без снижения активности р53 и
задержки способности к дифференцировке.
18.4.2. Иммортализация с использованием
вирусных генов
Для иммортализации клеток используют ряд вирусных
генов (табл. 18.2). Было обнаружено, что вирус SV40
может быть использован для иммортализации клеток,
при этом, видимо, геном, ответственным за этот процесс,
является большой Т-ген (17) [Mayne et al., 1996]. Другие
вирусные гены, которые используются для иммортали¬
зации клеток, — аденовирусный ген Е1а [Seigel, 1996],
гены Е6 и Е7папилломавируса человека (HPV) [Peters
et al., 1996; Le Poole et al., 1997], и гены вируса Эпш¬
тейн-Барра (EBV; обычно используется весь вирус)
[Bolton & Spurr, 1996]. Большинство этих генов, ве¬
роятно, действуют путем блокирования ингибирования
прохождения клеточного цикла путем ингибирования
активности таких генов, как CIP-1/WAF-1/p21, Rb,
р53, и р16у что приводит к возрастанию продолжитель¬
ности жизни, снижению контроля ДНК и повышает
возможность дальнейших мутаций. К наиболее широко
используемым в последнее время генам относятся: EBV
для лимфобластных клеток [Bolton & Spurr, 1996],
SV40LT для адгезирующих клеток, таких как фибро¬
бласты [Mayne et al., 1996], кератиноциты [Steinberg,
1996], и эндотелиальные клетки [Punchard et al., 1996],
и hTERT для мезенхимальных стволовых клеток (см.
протокол 18.2) и ряда других клеток. Эндотелиальные
клетки могут быть иммортализованы действием радио¬
активного излучения [Punchard et al., 1996].
18.4. Иммортализация
337
Таблица 18.2. Гены, использованные для иммортализации
Ген
Вставка
Клеточный тип
Ссылка
EBV: ebna, lmpl
инфекция
В-лимфоциты
Bolton and Spun*, 1996; Bouril-
lot et al., 1998; Sugimoto et
al., 2004
SV40LT
Липофекция
Кератиноциты
Steinberg, 1996
Фосфатно- кальциевая трансфекция
Фибробласты
Mayne et al., 1996
Фосфатно- кальциевая трансфекция
Астроглиальные клетки
Burke et al., 1996
Аденовирусная инфекция
Эпителий пищевода
Inokuchi et al., 1995
Микроинъекция
Эндотелий крысиного мозга
Lechardeur et al., 1995
Трансфекция
Эпителий простаты
Rundlett et al., 1992
Трансфекция
Эпителий молочной железы
Shay et al., 1993
Фосфатно-стронциевая трансфекция
Бронхиальный эпителий
De Silva et al., 1996
Фосфатно-стронциевая трансфекция
Мезотелиальные клетки
Duncan et al., 1996
HPV16 Е6/Е7
Ретровирусный перенос
Цервикальный эпителий
Demers et al., 1994
Трансфекция
Кератиноциты
Bryan et al., 1995
Фосфатно-стронциевая трансфекция
Мезотелиальные клетки
De Silva et al., 1994
Фосфатно-стронциевая трансфекция
Бронхиальный эпителии
De Silva et al., 1994
Ретровирусная инфекция
Поверхностный эпителий яичника
Tsao et al., 1995
Ad5 Ela
Эпителиальные клетки
Douglas and Quinlan, 1994
htrt
Трансфекция
Пигментный эпителий сетчатки
Bodnar et al., 1998
Трансфекция
Фибробласты крайней плоти
Bodnar et al., 1998
Трансдукция
Стволовые клетки костного мозга
Simonsen et al., 2002
Трансдукция
Кератиноциты
Dickson et al., 2000
Трансдукция
Миобласты
Wootton et al., 2003
Обычно клетки трансфецируются или ретровирусно
инфицируются иммортализующим геном до того, как
они входят в фазу старения. В результате происходит
увеличение продолжительности времени пролифера-
тивной активности клеточной линии на следующие
20-30 периодов удвоения популяции, после чего
клетки прекращают делиться и входят в кризис. После
критического периода, продолжительность которого
варьирует (до нескольких месяцев), субпопуляция
иммортализованных клеток дает избыточный рост.
Фракция клеток, которые в конечном итоге имморта-
лизованы, может составлять 1x10"5 до IxlO-9
18.4.3. Иммортализация человеческих
фибробластов
Нижеприведенное введение в технику иммортализа¬
ции человеческих фибробластов излагается в сжатом
виде по Mayne et al. [1996]. Несомненно, наиболее
успешным и наиболее часто используемым методом
получения иммортализованных человеческих фиброб¬
ластов проводится путем экспрессии SV40 Т-антигена,
которая не приводит непосредственно к имморта¬
лизации, но инициирует цепь событий, результатом
которых является получение иммортализованной
производной линии, возникающей с низкой вероят¬
ностью, оцениваемой как 1 к 107 [Shay & Wright, 1989;
Huschtcha & Holliday, 1983]. 8У40-трансфицирован-
ные клетки проходят прямую селекцию в соответству¬
ющих условиях культивирования, выжившие клетки
после субкультивирования дают рост до пре-кризисной
популяции 8У40-трансформированных клеток. Эти
клетки постоянно культивируют до тех пор, пока они
не потеряют способность к пролиферации, когда они
неминуемо входят в кризис. Затем культуру необхо¬
димо поддерживать с величайшей тщательностью и
осторожностью; должно быть культивировано доста¬
точно большое число клеток, для того чтобы иметь
достаточную вероятность появления иммортализо¬
ванных производных.
Выбор векторов, экспрессирующих Т-антиген,
зависит от выбора доминантного селектируемого гена-
маркера, который является источником промоторов,
которые обеспечивают экспрессию Т-антигена и сами
альтернативные формы Т-антигена. Хотя, по нашему
опыту, [Mayne et al., 1996], в случае человеческих
фибробластов эффективна селекция для gpt, тем не
менее, G418 (пео) и и гигромицин (hygB) более эф¬
фективны и легче для использования. Большинство
клеточных линий человеческих SУ40-иммортализован-
ных фибробластов были получены при использовании
SV40-вирусов или конструктов, таких как pSV3neo, ко¬
торые экспрессируют Т-антиген с эндогенного промо¬
тора. Мы рекомендуем использовать pSV3neo [Southern
& Berg, 1982; Mayne et al., 1986] для конститутивной
экспрессии Т-антигена. Следует использовать клет¬
ки между 7-ми 15-м пассажами. Трипсинизируйте
клетки за 24-48 ч до трансфекции, концентрация
клеток должна составлять не более 2-2,5х105кл./на
338
ГЛАВА 18. ТРАНСФОРМАЦИЯ И ИММОРТАЛИЗАЦИЯ
ПРОТОКОЛ 18.1. Иммортализация
фибробластов
Материалы
Стерильные
• Буфер HEPES: HEPES, 12,5 мМ; pH 7,12
• 10х CaHEPES: СаС12, 1,25 М; HEPES, 125 мМ;
pH 7,12
• 2х HEPES-забуференный фосфат (2х НВР):
Na2HP04,1,5 мМ; NaCl, 280 мМ; HEPES, 25 мМ;
pH 7,12
• NaOAc: NaOAc, 3 М; pH 5,5
• Раствор EDTA в Трис-буфере (ТВЕ): Tris-HCl,
2 мМ; EDTA, 0,1 мМ; pH 7,12
• Абсолютный этанол
• Среда Игла MEM/15% FCS
• G418 (Invitrogen): 20 мг/мл в буфере HEPES, pH
7,5, стерилизованный фильтрацией через 0,2-мкм
мембрану, хранящийся в малых аликвотах при
-20 °С
• Гигромицин (Roche Applied Science): 2 мг/мл в
сверхчистой воде, стерилизованный фильтрацией
через 0,2-мкм мембрану хранящийся в малых
аликвотах при -20 °С
• SV40 Т-нтиген ДНК
Протокол
1. Оцените количество ДНК, требуемое для транс¬
фекции:
а) Используйте максимум 20 мкг вашего
Т-антигенного вектора (без носителя ДНК)
на каждую чашку и 60 мкг векторной ДНК на
чашку.
б) Включите дополнительно 20 мкг ДНК, по¬
скольку не всегда возможно восстановить
полностью ожидаемое количество после при¬
готовления преципитата.
2. Приготовьте стерильный раствор векторной ДНК
в микроцентрифужной пробирке.
а) Осадите ДНК с одной десятой по объему
частью 3,0 М NaOAc, pH 5,5 и 2,5 объемами
этанола.
б) Хорошо перемешайте, убедитесь, что все внут¬
реннее содержимое пробирки вошло в контакт
с раствором спирта.
в) Оставьте пробирку на льду на короткое время
(5 мин).
г) Центрифугируйте пробирку в микроцентри¬
фуге 15 мин при 15 000 об./мин для получения
осадка.
д) Осторожно выньте пробирку из центрифуги,
откройте в ламинарном шкафу.
е) Удалите супернатант отсосом, стараясь не
взболтать осадок. Убедитесь, что этанол уда¬
лен полностью.
ж) Оставьте осадок в ламинарном шкафу для
культивирования клеток, пока не улетучатся
остатки этанола.
з) Ресуспендируйте осадок ДНК в ТВЕ до ко¬
нечной концентрации 0,5 мг/мл. Может по¬
требоваться использование вортекса для того,
чтобы отделить осадок от стенок пробирки.
и) Инкубируйте пробирку при 37 °С в течение
5-10 мин с периодическим применением вор¬
текса, пока не убедитесь, что осадок полностью
ресуспендирован.
Замечание. Не используйте более высокие
концентрации ТВЕ для ресупендирования вашей
ДНК, поскольку это может нарушить образование
преципитата ДНК.
3. Приготовьте преципитат ДНК с фосфатом кальция.
а) Подсчитайте общий объем требуемого пре¬
ципитата. Конечная концентрация ДНК в
преципитате должна быть 20 мкг/мл, и вам
потребуется по 1 мл на каждую 9-см чашку и
3 мл на каждый флакон с площадью 175-см2.
Помните, что нужно сделать дополнительный
1 мл преципитата, чтобы с достоверностью
получить объем, достаточный для всех ваших
культур, поскольку некоторые потери объема
будут происходить в ходе приготовления пре-
ципитатата.
б) Разведите смесь ДНК/ТВЕ в 12,5 мМ HEPES
до получения 20 мкг/мл в конечной смеси.
в) Добавьте 10х СаС12, один к десяти исходя из
конечного объема.
г) Добавлять раствор по одной капле, перемеши¬
вая, до равного объема 2х НВР. Например, на
9-см чашки при 1 мл/чашку плюс 1 мл допол¬
нительно (т.е. 10 мл смеси)
i) ДНК/ТВЕ 0,5 мл
ii) 12,5 мМ HEPES 3,5 мл
iii) 10, СаС12 1,0 мл
iv) Добавлять по капле к 2х НВР 5,0 мл.
Конечные концентрации в смеси будут составлять:
днк
20 мкг/мл
СаС12
0,125 M
Na2P04
0,75 мМ
NaCl
140 мМ
HEPES
12,5 мМ
pH
7,12
Замечание: при получении фосфатно-кальцие-
вого преципитата ДНК используйте пластиковые
пипетки и пробирки, поскольку преципитат очень
прочно прилипает к стеклу. Для смеси мы рекомен¬
дуем использовать две пипетки в двух портативных
пипетирующих устройствах, одно для пробулькива-
ния стерильного воздуха и второе для аккуратного
добавления ДНК/кальциевой смеси в раствор фос¬
фата; при качественном перемешивании получает¬
ся однородный преципитат. Используйте 1-5 мл
пипетки, в зависимости от желаемого объема пре¬
ципитата. В процессе добавления ДНК/кальциевого
18.4. Иммортализация
339
раствора к раствору фосфата начинает образовывать¬
ся легкий однородный осадок. Этот осадок достаточ¬
но хорошо заметен и дает молочное помутнение по
завершении процесса.
д) После того как весь ДНК/кальциевый раствор
добавлен к фосфату, замените крышку на про¬
бирке, аккуратно переверните пробирку один-
два раза и оставьте пробирку при комнатной
температуре на 30 мин.
е) Важно получить имитацию преципитата без
ДНК Это позволит вам обеспечить эффектив¬
ность выбора условий. Имитация преципитата
может быть получена так же, как описано для
получения истинного преципитата, но ДНК
в этом случае замещается дополнительным
объемом буфера HEPES.
4. Добавьте 1 мл ДНК или имитации преципитата в
каждую чашку или 3 мл в каждый флакон. Убеди¬
тесь, что объем преципитата составляет не более
десятой части общего объема культуральной
среды уже имеющейся на клетках.
5. Оставьте преципитат на клетках как минимум на
6 ч, но не более чем не ночь (16 ч). Для фиброб¬
ластов, полученных от некоторых животных,
действие кальция и фосфата более чем 6 ч может
быть токсичным.
6. Удалите фосфатно-кальциевый преципитат
вакуум-аспирацией. Нет необходимости в пос¬
ледующем промывании клеток, или, по нашему
опыту, в дальнейшей обработке клеток DMSO
или глицерином.
7. Добавьте среду в культуры, инкубировать их,
пока не закончится 48 ч с начала эксперимента.
Затем добавьте в культуры селективные аген¬
ты, в зависимости от вектора, используемого
при трансфекции. Векторы, несущие ген пео,
придают устойчивость к G418 (Генетицин), а
векторы, несущие ген hygb, придают устойчи¬
вость к гигромицину В (Roche Applied Science).
Все фибробласты, по нашему опыту, требуют
100-200 мкг/мл С418или 10-20 мкг/мл гиг-
ромицина В для того, чтобы постепенно убить
клетки за одну неделю.
8. Смените среду на трансфецированных чашках:
а) Разлейте общий требуемый объем среды в
подходящие по размеру стерильные бутылки,
добавьте G418 или гигромицин В из основных
концентрированных растворов до получения
необходимой конечной концентрации.
б) Осторожно вращайте раствор для его переме¬
шивания.
в) Отсосите среду из чашек или флаконов и за¬
мените ее селективной средой.
г) Верните чашки или флаконы в инкубатор.
9. Следите за действием селективной среды путем
ежедневного обследования культуры под микро¬
скопом. При значительном количестве клеток,
отделившихся и погибших, замените среду
свежей селективной средой. Селекция должна
продолжаться все время.
10. Продолжайте замену среды, пока основная
часть клеток не всплывет и не погибнет. Чашки
с имитацией трансфекции, в которых ДНК-
трансфекция не была проведена, через 7-10 дней
не должны содержать жизнеспособных клеток.
Если жизнеспособные клетки сохраняются, то
селекция была проведена неадекватно и может
оказаться необходимым повысить концентрацию
селективного агента.
И. Как только основная часть клеток погибла,
исчезает необходимость в стандартной замене
питательной среды. Клетки следует оставить, не
беспокоя, в инкубаторе на 4-6 недель для того,
чтобы трансфецированные клетки могли нарасти
и сформировать колонии.
12. Как только появились колонии, выделите инди¬
видуальную колонию с использованием колец для
клонирования (см. протокол 14.6) или отберите
несколько колоний вместе путем трипсинизации
чашки или флакона.
13. Замораживайте разлитую на порции суспензию
клеток при первой возможности и регулярно
пересевайте оставшиеся клетки. Как только
трансфектант достигнет достаточного размера и
будет заморожено в жидком азоте достаточное
количество образцов культуры, необходимо обес¬
печить ведение культуры в течение продолжи¬
тельного периода времени до достижения кризиса.
Для того чтобы минимизировать риск заражения
грибами, добавьте амфотерицин В (Fungizone,
Invitrogen) до концентрации 2,5 мкг/мл.
14. Субкультивируйте культуры в обычном порядке,
пока они не потеряют способности к делению. Как
только клетки достигнут кризиса, часто скорость
роста замедляется. Если культуры начинают деге¬
нерировать и деление клеток прекращается, то в
дальнейшем субкультивировании клеток нет необ¬
ходимости. Тем не менее если тяжелый клеточный
дебрис начинает прилипать к остающимся жиз¬
неспособными клеткам, то можно посоветовать
провести трипсинизацию клеток для удаления
дебриса. Как при возвращении всех клеток в тот же
сосуд, так и при использовании меньшего сосуда
необходимо компенсировать клеточную гибель. В
общем, клеточный рост и выживание улучшаются,
если культуры не слишком разрежены. При тер¬
пеливом, тщательном отношении и удовлетвори¬
тельном качестве клеточной культуры вы можете
получить пост-кризисную линию.
15. Когда начинают появляться здоровые клетки,
позвольте колониям вырасти до значительных
размеров до субкультивирования и затем начните
субкультивировать колонии снова. Не подда¬
вайтесь желанию посеять слишком маленькое
количество клеток в большой флакон.
340
ГЛАВА 18. ТРАНСФОРМАЦИЯ И ИММОРТАЛИЗАЦИЯ
16. Заморозьте клетки, разделенные в ампулы, при
первой возможности и продолжайте пополнять
основные запасы в ходе всего использования
культуры.
17. Для того чтобы проверить, является ли пост*
кризисная линия действительно иммортализо-
ванной, мы рекомендуем провести селекцию и
выделение отдельного клона из культуры. До¬
полнительная польза этой процедуры состоит в
достижении гомогенности культуры, полученной
из единичной клетки.
9-см чашку Петри или не более 5,5-6,8x105 клеток на
175-см2 флакон. Клетки должны достигнуть 70-80%
конфлюэнтности, когда они подвергаются трансфек¬
ции, в конечном объеме среды 10 мл/9-см чашку или
30 мл/175-см2 флакон.
Метод фосфатно-кальциевой преципитации ос¬
новывается на образование преципитата ДНК в
присутствии ионов кальция и фосфата. Сначала
ДНК стерилизуется в этаноле и ресуспендируется в
стерильном буфере. Затем тщательно перемешивается
с кальцием, и полученный раствор очень медленно
добавляется, при перемешивании, в раствор фосфата.
Важно заметить, что при образовании преципитата
оптимальный перенос генов осуществляется при ко¬
нечной концентрации ДНК в преципитате 20 мкг/мл.
Объем преципитата ДНК, используемого в культуре,
никогда не должен превышать одну десятую от общего
объема. Необходимо оставить смесь для завершения
процесса в течение 30 мин прежде, чем добавлять к
клеточным культурам.
Посттрансфекционное ведение культуры
1) Уровень селективного агента выбирается так, чтобы
обеспечить мягкое уничтожение в течение пример¬
но недельного периода. Большая часть клеток в
ДНК-обработанных чашках должна погибнуть за
семь дней, а те клетки, которые останутся, явля¬
ются успешно трансфецироваными, т. е. являются
трансфектанатами
2 ) Можно посоветовать заморозить ампулы с клетками
по обычной методике, для того чтобы создать запас
трансфектантов и сэкономить время, которое пот¬
ребуется для восстановления культуры, если она
будет утрачена в результате контаминации.
3) В большинстве случаев кризис является заметным
событием, когда большинство клеток обнаруживает
признаки ухудшения состояния и происходит зна¬
чительное снижение количества жизнеспособных
клеток. В среднем, кризис продолжается примерно
3-6 месяцев, и состояние культуры может ухудшить¬
ся до того, что при обследовании будет обнаружи¬
ваться очень малое количество визуально здоровых
клеток, если они вообще будут обнаруживаться.
4) Как только культура вошла в кризис, вы сможете
достоверно предсказать время вхождения в кризис
для параллельных культур, хранящихся в жидком
азоте. Это позволяет извлечь ампулы с параллель¬
ными культурами из морозильника и получать ряд
флаконов, находящихся в преддверии кризиса. Как
только эти параллельные культуры войдут в кризис
и начнут терять жизнеспособность, содержимое этих
флаконов может быть объединено. В некоторых
случаях во флаконе может быть удовлетворительное
количество клеток, но уровень клеточного дебриса
слишком высок. В этом случае пересейте клетки.
Клетки должны быть трипсинизированы и центри¬
фугированы, затем осадок должен быть возвращен
в исходный флакон, который перед этим можно
промыть несколько раз трипсином для удаления
всех прикрепленных частей клеточного дебриса.
5) Первым признаком выхода клеток из кризиса обыч¬
но является появление одного или более фокусов
относительно здоровых, жизнеспособных клеток
с типичными чертами 8У40-трансформированных
клеток. При субкультивировании эти фокусы рас¬
пространяются, давая здоровую, регенерирующую
культуру. Во многих случаях рано появляющиеся
пост-кризисные клетки растут плохо, но ростовые
свойства улучшаются при дальнейшем субкульти¬
вировании, а субклонирование помогает отобрать
отдельные клоны с наилучшими ростовыми свой¬
ствами. Важно замораживать ампулы с вашими
новыми клетками, как только вы нарастите доста¬
точное для этого количество. Наше рабочее опреде¬
ление иммортализованной культуры предполагает,
что культура может после трансфекции выдержать
как минимум 100 периодов удвоения популяции и
переносит последующее субклонирование.
6) Поскольку появление иммортализованных произ¬
водных в культурах — достаточно редкое событие,
то существенным требованием является проверка
происхождения клеток, подтверждающая соответ¬
ствие исходного источника полученной имморта¬
лизованной культуре, которая, таким образом, не
является результатом контаминации другой им¬
мортализованной культурой, с которой работают в
лаборатории (см. разд. 7.10.1,16.1,16.3,16.6.2,19.5;
табл. 13.2; и протоколы 16.8-16.10).
18.4.4. Теломераза-индуцированная
иммортализация
Теломеры играют существенную роль в стабильности
хромосом и определяют продолжительность клеточно¬
го цикла. Теломераза или терминальная трансфераза
состоит из двух главных субъединиц, РНК-компонен-
ты, (hTR) и белковой каталитической субъединицы
(hTERT). РНК-субъединица повсеместно распро¬
странена как в нормальных, так и в злокачественных
тканях, в то время как hTERT экспрессируется только
в опухолевых тканях, половых клетках и активирован¬
ных лимфоцитах. Предполагается, что первопричина
старения заключается в укорочении теломер, которое
18.4. Иммортализация
341
Рис. 18.4. Совокупное количество периодов удвоения по¬
пуляции (PD) иммортализованных клеток hTERT по срав¬
нению со стареющими контрольными клетками. Совокупное
количество PD было подсчитано для культур человеческих
мезенхимальных стволовых клеток (см. протокол 18.2) и
сравнено со временем роста в культуре клеток с эктопичес¬
кой экспрессией hTERT после ретровирусной трансдукции
(кружочки) и контрольных (нетрансдуцированных) клеток
(квадратики). (С любезного разрешения N. Serakinci.)
сопровождается слиянием теломер и образованием
дицентрических хромосом с последующим апоптозом.
Клетки, трансфицированные теломеразным геном
htrt, приобретают способность к увеличению времени
жизни клеточной линии (см. рис. 18.4), а часть этих
клеток становится бессмертными, но не злокачественно
трансформированными [Bodnar etal., 1998, Simonsenet
al., 2002]. Поскольку значительная доля клонов htrt+
становится бессмертными (иммортализованными),
постольку этот подход представляется многообещаю¬
щим. Хотя функциональность некоторых из этих линий
требует подтверждений, однако имеется несколько
обнадеживающих сообщений о ненарушенной диф¬
ференцировке кератиноцитов [Dickson et al., 2000] и
миоцитов [Wootton et al., 2003]. Предварительное вве¬
дение и протокол 18.2. предоставлены Nedime Serakinci
(Department of Human Genetics, Bartholin Bygningen,
Universitetsparken, 8000, Aarhus C, Denmark). Данный
протокол был успешно использован для мезенхималь¬
ных и нервных линий стволовых клеток человека и
первичных культур.
18.4.5. Трансгенные мыши
Трансгенная мышь линии Immortomouse (H-2Kb-tsA58
SV40 large Т) несет температуро-чувствительный ген
SV40LT. Ряд тканей этой мыши дает начало бессмер¬
тным клеточным линиям, включая эпителий толстого
кишечника [Fenton & Hord, 2004], астроглию мозга
[Noble & Barnett, 1996], мышцы [Ahmed et al., 2004], и
эндотелий сетчатки [Su et al., 2003].
ПРОТОКОЛ 18.2. Иммортализация
мезенхимальных стволовых клеток человека
с помощью теломеразы
Материалы
Основные (стерильные):
• Ростовая среда Дюльбекко в модификации Игла
(DMEM) с высокой концентрацией глюкозы,
4,5 г/л и L-глютамином, 2 мМ, с добавкой 10%
фетальной бычьей сыворотки, 100 ед./мл пени¬
циллина и 100 мкг/мл стрептомицина
• Полибрин, 8 мг/мл
• Универсальные контейнеры
• Культуральные флаконы 25 см2,75 см2
Получение ретровирусного вектора
• Клеточные линии
G13 [Milleretal., 1991]
GP+E-86 [Markovitz, 1988]
• Ретровирусный вектор GCsamhTERT
• ДНК Ам для трансфекции
• Буфер Тх:
HEPES, 0,5 М, pH 7,1 200 мкл
NaCl, 5 М 100 мкл
Na2HP04/NaH2P04,1 М 3 мкл
Сверхчистая вода 1,7 мл
Общий объем 20 мл
• Буфер А:
NaCl (5М) .....*. ...... 600 мкл
EDTA(0,5M) 40 мкл
TrisHCl pH 7,5 (0,5М) ......400 шел
Сверхчистая вода 19 мл
Общин объем 20,04 мл
• СаС1,2,5 М
Трансдукция клеток hMSC
• Клетки hMSC
• Флаконы, 75 см2
• Многолуночные планшеты (6-луночные)
Посттрансдукция
• Материалы для криоконсервации (см. прото¬
кол 20,1)
• Материалы для ДНК-фингерпринтинга (см.
протокол 16.8) или профилинга ДНК см. прото¬
кол 16.9) либо других процедур аутентификации
(см., например, протокол 16.10)
Реактивы для ПЦР
• ДНК, 100 мкг/мл
• ПЦР-буфер 10х (+Mg)
• Сенсорный праймер (2 мкМ)
• Антисенсорный праймер (2 мкМ)
• Смесь dNTP (10 мМ)
• ДНК-полимераза
• Сверхчистая вода
Протокол
Получение ретровирусного вектора
Ретровирусный вектор с геном hTERT объединен
с вирусом лейкемии человекообразной обезьяны
342
ГЛАВА 18. ТРАНСФОРМАЦИЯ И ИММОРТАЛИЗАЦИЯ
гиббона (GALV) и сгруппирован в клеточной линии
PG13 [Miller et al., 1991] в ходе двухшаговой проце¬
дуры. Сначала проводится трансфекция клеточной
линии GP+E-86, при этом используется 20 мкг/мл
ДНК fort (Geron) [Markovitzetal., 1988], полученный
при культивировании супернатант затем использу¬
ется для инфицирования клеток PG13.
1. Посейте в небольшой культуральный флакон
(25 см2) 6,7х105 клеток культуры GP+E-86.
2. Трансфекция клеток GP+E-86:
а) Разлейте в пробирки (универсальные контей¬
неры) по 280 мкл буфера Тх.
б) П]
эиготовьте пробирки с ДНК:
Назва¬
ние кон¬
структа
Концент¬
рация ДНК
15 мкг
ДНК
Буфер А
CaCI,
2,5 М
Общий
объем
Z* мкг/мл
X*
мкл
280-30-Х
30
280
Где XxZ = 15 мкг
в) Постепенно (по одной капле) добавляйте бу¬
фер Тх в пробирки, содержащие раствор ДНК.
Осторожно перемешайте.
г) Инкубируйте при комнатной температуре в
течение 30 мин до образования преципитата.
д) Добавьте свежую среду поверх клеток GP+
Е-86,5 мл на флакон 25-см2.
е) Осторожно добавьте смесь растворов (Тх+
+ ДНК) к клеткам GP+E-86 и инкубируйте
4-6 ч при 37 °С.
ж) Трижды тщательно промойте клетки в
D-PBSA во 25-см2 флаконах.
з) Добавьте свежую среду к клеткам, инкубируй¬
те при 37 °С в течение ночи.
3. Поменяйте среду, покрывающую клетки, добавив
2 мл свежей среды вместо 5 мл, для продукции
вирусов.
4. В тот же день, что и при выполнении шага 3,
посейте клетки PG13, посевная доза lxlO4 / на
каждую лунку 6-луночного планшета.
5. На следующий день соберите супернатант от
трансфецированных клеток GP+E-86 и добавьте
Полибрин до конечной концентрации 8 мкг/мл
(т.е. 1 мкл./мл супернатанта).
6. Пропустите супернатант через 0,45-мкм фильтр
и добавьте 2 мл фильтрата в каждую лунку, со¬
держащую клетки PG13.
7. Центрифугируйте планшеты при 32 °С и 1000 g.
8. Инкубируйте при 37 °С в течение ночи.
9. На следующий день поменяйте среду.
Трансдукция клеток hMSC:
1. Посейте 2х106трансдуцированных клеток PG13
в культуральный флакон 75-см2.
2. На следующий день добавьте 6 мл свежей среды
к клеткам PG13.
3. На следующий день посейте клетки hMSC для
трансдукции в 6-луночный планшет при концент¬
рации приблизительно 2,5х104-7,5х104 кл./мл.
4. Соберите супернатант, добавьте Полибрин до
конечной концентрации 8 мкг/мл, пропустите
супернатант через 0,45-мкм фильтр.
5. Добавьте 2 мл фильтрованного ретровирусно¬
го супернатанта в каждую лунку 6-луночного
планшета.
6. Центрифугируйте планшеты при 32 °С и 1000 g,
инкубируйте при 37 °С в течение ночи.
7. Удалите ретровирусный супернатант и добавьте
свежую среду к клеткам.
Пост-трансдукционное ведение культур:
1. Клетки, которые не включили ген hTERT, должны
начать погибать примерно через 10-12 пассажей;
остающиеся клетки должны представлять собой
клетки, инкорпорировавшие ген hTERT\ и таким
образом они будут селекционированы в ходе
последующих пассажей.
2. Можно посоветовать заморозить ампулы с
клетками обычным путем для формирования
основного запаса трансдуцированных клеток на
различных этапах удвоения популяций (PD),
которые соответствуют кривой роста, для того
чтобы сэкономить время, если культура будет
утрачена в результате контаминации.
3. Используйте следующие формулы для теорети¬
ческого определения PD в каждом субкультиви¬
ровании и установлении PD кривой роста:
PD - In (NKOHe4/NHa4)/ln 2,
где PD количество периодов удвоения популяции,
In — натуральный логарифм, NHa4 — количество
клеток, первоначально посеянных, NK0He4 — общее
количество клеток, выросших в субкультуре.
Пример (для 3,2x10 е клеток, полученных в
результате посева 2х105 клеток):
PD - In (3,2xl06/2xl05)/ln 2
PD = In (16)/ln 2
PD - 2,7726/0,6931 = 4
Если клетки делить 1:4 при каждом субкультиви¬
ровании, то конечное число клеток умножается
в 4 раза при каждом субкультивировании клеток.
Поэтому в приведенном выше примере с 10 субкуль¬
тивированиями NKOIie4 составляет (3,2x10^x4, десять
раз, или 3,4x1012.
Пример (для 3, 2x106клеток полученных
из первоначально посеянных 2x105, после
10 субкультивирований с делением клеток в
отношении 1:4):
PD = In (3,4х1012/2х105)/1п 2
PD = In (1,67х107) In 2
PD - 16,6309/0,6931 - 23
18.5. Абберантный контроль роста
343
4. Очень важно проверять аутентичность иммор-
тализованной клеточной линии в течение всего
периода работы для того, чтобы быть уверенным,
что она получена их исходного начального мате¬
риала и не является результатом перекрестной
контаминации с другими иммортализованными
линиями, с которыми ведется работа в лабора¬
тории (см. разд. 19,5). Это может быть сделано,
например, методом ПЦР против трансгена,
hTERT (см. ниже), ДНК-фингерпринтинга (см.
протокол 16.8), или ДНК-профилинга (см. про¬
токол 16.9). j
ПЦР для hTERT:
1. Разморозьте реактивы для ПЦР, держите их на
льду,
2. Сделайте шаблонную смесь, помните о включе¬
нии положительного контроля (другой hTERT
положительный образец) и отрицательного конт¬
роля (матрица безДНК).
ДНК, 100 мкг/мл. .. 1 мкл (100 нг)
10х ПЦР буфер (+Mg)............ 2 мкл
Сенсорный праймер (2 мкМ)........ 2мкл
Антисенсорный праймер (2 мкМ)2 мкл
dNTP смесь (10 мМ).. 0,4 мкл
ДНК полимераза. 0,1 мкл
Сверхчистая вода 12,5 мкл
Конечный объем „..20 мл (включая ДНК)
3. Поместите пробирки в ПЦР-прибор и включите
программу: Первичная денатурация при 94 °С в
течение 3 мин, денатурация при 94 °С в течение
30 с, отжиг при 59 °С в течение 30 с, амплифика*
ция при 74 °С в течение 1 мин / 30 циклов, затем
74 °С в течение 10 *шй
4. Разгонка продуктов ПЦР в агарозном 1,3-2%
геле.:
18.5. АБЕРРАНТНЫЙ КОНТРОЛЬ РОСТА
Культивированные клетки опухолей, так же как и
культуры, трансформированные in vitro, обнаружива¬
ют аберрантность в факторах, контролирующих рост
культуры, например рост при более высокой плотности
популяции [Dulbecco & Elkington, 1973], образование
клонов в агаре [Freedman & Shin,1974], и рост в конф¬
люэнтном монослое гомологичных клеток [Aaronson et
al., 1970]. Эти клеточные линии демонстрируют также
более низкую зависимость от сыворотки и факторов
роста, обычно формируют клоны с большей эффектив¬
ностью, и в целом приобретают некоторую степень ав¬
тономного контроля роста в результате суперэкспрес¬
сии онкогенов из-за делеции генов-супрессоров. Конт¬
роль роста в данном часто является аутокринным, т. е.
клетки секретируют митогены, к которым сами имеют
рецепторы, либо клетки экспрессируют рецепторы, или
элементы передачи сигналов, которые перманентно
активны и нерегулируемы. Хотя иммортализация не
обязательно подразумевает утрату контроля роста,
многие клетки легко переходят от иммортализации к
аберрантному неконтролируемому росту, возможно,
потому, что иммортализованному генотипу присуща
генетическая нестабильность.
18.5.1. Независимость от прикрепления
к субстрату
Многие свойства, связанные с неопластической транс¬
формацией in vitroy являются результатом изменений
клеточной поверхности [Hynes, 1974; Nicolson, 1976;
Bruyneel et al., 1990], т.е. изменений связываний
лектинов [Laferte & Loh, 1992], изменений гликопро¬
теинов клеточной поверхности [Bruyneel et al., 1990;
Carraway et al., 1992] и молекул клеточной адгезии
[Yang et al., 2004]. Многие из этих изменений могут
коррелировать развитием инвазивности и способнос¬
ти к метастазированию in vivo. При трансформации
фибробластов в результате перестройки интегринов
клетки теряют способность поверхности связываться
с фибронектином, белком LETS (large extracellular
transformation-sensitive, большой внеклеточный бе¬
лок, чувствительный к трансформации) [Hynes, 1973;
Vaheri et al., 1976]. Эта потеря может вносить вклад
в снижение в степень межклеточной и клеточно-суб¬
стратной адгезии [Yamada et al., 1991; Reeves, 1992] и
снижать зависимость деления клеток от прикрепления
и распластывания на подложке. Трансформированные
клетки могут иметь недостаток специфических CAMs
(например, L-CAM), которые, при трансфекции в
клетку, вызывают регенерацию клетки до нормального
неинвазивного фенотипа [Mege et al., 1989], поэтому
они могут рассматриваться как гены-супрессоры
опухолей. Другие CAMs могут быть сверх-экспресси-
рованы, например N-CAM при мелкоклеточном раке
легкого [Patel et al., 1989], при этом внеклеточный
домен подвергается альтернативному сплайсингу
[Rygaard et al., 1992]. Может меняться также экспрессия
и степень фосфорилирования интегринов [Watt, 1991],
потенциально изменяя взаимодействие с цитоскеле¬
том, регуляцию генов транскрипции, адгезию клеток
к субстрату и взаимосвязь между распластыванием
клеток и клеточной пролиферацией [Fata et al., 2004].
В дополнение к этому, утрата межклеточного распоз¬
навания, результат снижения клеточной адгезии, ведет
к дезорганизации роста клеток и утрате контактного
торможения клеточной подвижности и ограничению
клеточного деления плотностью популяции (см.
разд. 18.5.1). Клетки могут расти неприкрепленными
к субстрату как в перемешиваемой суспензионной
культуре, так и суспендированными в полужидкой
среде, такой как агар или метилцеллюлоза. Существует
видимое сходство между изменениями клеточной ад¬
гезии и открепления от тканей, в которых развивается
опухоль и последующим образованием метастазов в
других участках, однако пока не ясна рациональная
344
ГЛАВА 18. ТРАНСФОРМАЦИЯ И ИММОРТАЛИЗАЦИЯ
основа для этого сходства. Возможно, в этом процессе
принимают участие новые механизмы адгезии.
Суспензионное клонирование. Macpherson and
Montagnier [1964] смогли показать, что полиома-
трансформированные клетки ВНК21 могли расти
преимущественно в мягком агаре, в то время как не-
трансформированные клетки клонировались очень
плохо. Впоследствии было показано, что образование
колоний в суспензии часто улучшается после вирусной
трансформации. Ситуация, касающаяся спонтанных
опухолей, менее ясна, несмотря на тот факт, что Shin
с соавторами показали высокую корреляцию между
туморогенностью и суспензионным клонированием в
метилцеллюлозе (Methocel) [Freedman and Shin, 1974;
Kahn & Shin, 1979]. Хотя Hamburger и Salmon [1977]
показали, что многие опухоли человека содержат не¬
большой процент клеток (<1,0%), которые образуют
клоны в агаре, ряд нормальных клеток также будут об¬
разовывать клоны в суспензии [Laug et al., 1980; Peehl &
Stanbridge 1981; Freshney & Hart, 1982] (см. рис. 14.11).
Поскольку среди клеток встречаются нормальные
фибробласты, ценность этого метода для обнаружения
присутствия опухолевых клеток в короткоживущих
культурах из человеческих опухолей сомнительна. Тем
не менее метод сохраняет свою ценность для изучения
неопластической трансформации in vitro с применением
опухолевых вирусов, и он был широко применен Styles
[1977] для изучения механизмов канцерогенеза.
Метод клонирования в суспензии описан в гл. 14
(см. протоколы 14.4 и 14.5). В случае неопластических
клеток особенность применения этого метода связана
с выбором среды для суспендирования. Было предло¬
жено [Neugut & Weinstein, 1979] использовать агар
только для образования клонов наиболее сильно транс¬
формированными клетками, в то время как агароза (в
которой имеется недостаток сульфатированных по¬
лисахаридов) менее избирательна. Montagnier [1968]
показал, что рост нетрансформированных ВНК21
клеток, способных расти в агарозе, но не растущих
в агаре, может быть подавлен добавлением в агарозу
декстрансульфата.
18.5.2. Контактное торможение
Утрата контактного торможения может быть обнару¬
жена морфологически по образованию дизориенти-
рованного монослоя клеток (см. цв. вкладку 14а, 6)
или округлению клеток в фокусах внутри обычного
образца нормальных окружающих клеток (см. рис. 18.3
и цв. вкладку 14в). В культурах человеческой глиомы
дезорганизованные участки роста со снижением плот-
ность-зависимого ограничения роста обнаруживаются
по более высокой плотности популяции, чем в линиях
нормальных глиальных клеток (рис. 18.5) [Freshney
et al., 1980а, Ь]. Поскольку различия в размерах кле¬
ток влияют на плотность культуры, то наилучшим
методом измерения падения плотность-зависимого
Дни
Рис. 18.5. Ограничение клеточной пролиферации в зави¬
симости от плотности. Различия в уровне плато (плотности
насыщения), достигнутого культурами клеток нормального
мозга (кружочки, сплошная линия) и клеток глиомы (квад¬
раты, прерывистая линия). Клетки были посеяны на 13-мм
покровное стекло, через 48 ч покровные стекла были перене¬
сены в 9-см чашку Петри, содержащую 10 мл ростовой среды,
для того чтобы минимизировать фактор истощения среды
ограничения роста является определение индекса
мечения с использованием [3Н]-тимидина при разной
плотности популяции (см. протокол 21.11). Глиома
человека, меченная [3Н]-тимидином в течение 24 ч
в плотной популяции, дает индекс мечения 8%, в то
время как нормальные глиальные клетки дают 2%
[Guneret al., 1977].
ПРОТОКОЛ 18.3. Ограничение клеточной
пролиферации в зависимости от плотности
культуры
Схема
Выращивание культуры до плотности насыщения в
условиях, не ограниченных средой, и авторадиогра¬
фическое определение процента клеток, меченных
[3Н ] -тимидином
Материалы
Стерильные или асептически приготовленные:
• Культуры клеток, готовые к субкультивированию
• Ростовая среда
• Среда для поддержания культуры (без сыворот¬
ки и факторов роста), содержащая 37 КБк/мл
(1,0 мк Ки/мл) [3Н]-тимидина, 74 ГБк/ммоль
(2 Ки/ммол)
• D-PBSA
• Трипсин, 0,25%
• Мкоголуночный планшет, 24-луночный, содер¬
жащий покровные стекла 13 мм в диаметре
18.5. Абберантный контроль роста
345
• Чашки Петри, 9 см (бактериологические, по 1 на
покровное стекло)
Протокол
L Трипсинизируйте клетки и посейте lxlO5 кл./мл
в 24-луночный планшет, 1 мл/лункуг поместите
в каждую лунку покровное стекло 13-мм вдна-
метре.
2. Инкубируйте клетки в С02-инкубаторе с увлаж¬
нением в течение 1-3 дней.
3. nepeHecbnt покровные стекла в 9-см бактериоло¬
гические чашки Петри, каждая содержит 20 мл
среды, поместит! чашки в С02-инкубатор.
4. Продолжайте культивирование, меняя среду
каждые 2 дня, пока клетки не образуют конфлю¬
энтный слой на покровных стеклах.
5. Трипсинизируйте и подсчитывайте клетки с двух
покровных стекол каждые 3-4 дня. Как только
клетки станут располагаться компактно на по¬
кровном стекле, может потребоваться добавить
200-500 ед./мл коллагеназы к трипсинудля того,
чтобы достичь полной диссоциации клеток для
их подсчета.
6. Когда клеточный рост прекратится, т. е. при двух
последовательных подсчетах не обнаружено зна¬
чительного возрастания числа клеток, добавьте
2,0 мл 37 КБк/мл (1,0 МкКн/мл) [3Н]-тимидина,
74 ГБк/ммоль (2 Ки/ммоль) и затем инкубируй¬
те клетки в течение 24 ч.
• Требование безопасности. Манипуляции
с [3Н]-тимидином следует проводить с осторожно¬
стью. Хотя это источник щшкоэнергетичного р-из-
лучения, он включаетсямДНК иможет вызывать
не работайте с [3Н]-тимидином в ламинарном шкафу
с горизонтальным потоком, используйте ламинар¬
ный шкаф д ля работы с препаратами, токсическими
или представляющими биологическую опасность
(см. разд. 7,5.4), уничтожайте жидкие и твердые
радиоактивные отходы в соответствии с правилами
работы с рад иоизотопами, принятыми у вас.
7. Перенесите покровные стекла обратно в 24-лу-
ночный планшет, трипсинизировать клетки для
авторадиографии (см. разд. 27.2).Клетки могут
быть фиксированы в суспензии или в каале на
стекле, как при изготовлении хроьшяшш®
препарата (см. протокол 16.7 без гипотонической
обработки), центрифугированы на стекле при по¬
мощи цитоцетрифуги (см. протокол 16.4), или
задержаны на фильтре при вакуум-фильтрации
(см. протокол 16,5).
Замечание. Культуру с высокой плотностью для
авторадиографии необходимо трипсинизировать, по¬
скольку из-за толщины слоя и слабости проникновения
P-излучения от [3Н]-меченых клеток в подлежащие
слоях они не будут регистрироваться радиочувстви¬
тельной эмульсией, в результате абсорбции Р-частиц
вышележащими клетками (длина среднего пробега
Р-частиц в воде составляет примерно 1 мкм). Если
клетки остаются в монослое при высокой плотности,
то этот шаг можно опустить, а авторадиограмма может
быть приготовлена установкой покровного стекла по¬
верх клеток на предметном стекле.
Анализ. Подсчет количества меченых клеток
представляется как их процентная доля от общего ко¬
личества клеток. Просмотрите авторадиограмму под
микроскопом и подсчитайте общее количество клеток
и долю меченых клеток в репрезентативной части пре¬
парата (см. рис. 21.14).
Вариации. Клетки, синтезирующие ДНК, мо¬
гут быть мечены при помощи бромдеоксиуридина
(BUdR) и затем обнаружены по реакции с антителами
к BUdR-меченой ДНК (Dako). Человеческие клетки,
находящиеся в стадии пролиферации, могут быть
мечены при помощи моноклональных антител Ki67
(Dako) против ДНК-полимеразы или анти-PCNA
против антигена пролиферирующего клеточного
ядра (PCNA, proliferating cell nuclear antigen). Хотя
существует достаточно хорошее соответствие между
[3Н]-тимидином и BudR- меткой, но Ki67 и PCNA
свяжутся с большим числом клеток, поскольку анти¬
ген присутствует в течение всего клеточного цикла
и не ограничен S-фазой. Рост клеток при высокой
плотности в нелимитирующей среде может быть так¬
же достигнут выращиванием клеток в фильтрующей
лунке (B-D Biosciences, Corning Millipore), при этом
следует выбирать диаметр фильтра так, чтобы он был
значительно меньше, чем у чашки (например, 8-мм
фильтр для 24-луночной чашки, Corning Costar) и
подсчета количества клеток в плате, который прово¬
дится при помощи авторадиографии или иммунного
окрашивания с Ki67 или анти-PCNA.
18.5.3. Зависимость от сыворотки
Трансформированные клетки имеют более низкую
зависимость от сыворотки, чем соответствующие им
нормальные клетки [Temin, 1966. Eagle etal., 1970], бла¬
годаря, в частности, секреции факторов роста опухоле¬
выми клетками [Todaro & DeLarco, 1978]. Эти факторы
были описаны как аутокринные факторы роста. В этом
определении подразумевается, что 1) клетка продуци¬
рует фактор; 2) клетка имеет рецепторы к этому факто¬
ру и 3) клетка отвечает на данный фактор вступлением
в митоз. Некоторые из этих факторов могут обладать
несомненной трансформирующей активностью в отно¬
шении нормальных клеток (например, TGF-а)? связы¬
ваясь с EGF рецептором и индуцируя митоз [Richmond
et al., 1985], хотя, в отличие от истинной трансформации
(см. разд. 18.2), данный тип трансформации обратим.
Эти факторы также приводят к тому, что нетрансфор-
346
ГЛАВА 18. ТРАНСФОРМАЦИЯ И ИММОРТАЛИЗАЦИЯ
мированные клетки приобретают трансформирован¬
ный фенотип и начинают расти в суспензии [Todaro
& DeLarco, 1978]. Этот эффект может быть достигнут
обработкой клеток NRK кондиционированной средой
от исследуемых клеток и последующим их клониро¬
ванием в суспензии (см. протоколы 14.2, 14.4, и 14.5).
Опухолевые клетки могут также производить много
гематопоэтических факторов роста, таких как интер¬
лейкины 1, 2, и 3, наряду с колониестимулирующим
фактором (CSF) [Fontana et al., 1984; Metcalf, 1990].
Было высказано предположение [Cuttitta et al., 1985],
что некоторые факторы, такие как гастрин-релизинг-
фактор и вазоактивный интестинальный пептид (VIP)
и человеческий хорионический гонадотропин (hCG),
которые, как до настоящего времени полагали, яв¬
ляются эктопическими гормонами, производимыми
легочной карциномой, могут на самом деле быть ауто-
кринными факторами роста. Аутокринные факторы
роста могут быть обнаружены при помощи иммунного
окрашивания (см. протокол 16.11), но их ценность как
маркеров трансформации ограничена, поскольку мно¬
гие нормальные клетки, например глиальные клетки,
фибробласты и эндотелиальные клетки, продуцируют
аутокринные факторы в ходе пролиферации.
18.5.4. Онкогены
Автономный контроль роста также достигается в транс¬
формированных клетках при действии онкогенов, экс¬
прессируемых как модифицированные рецепторы, на¬
пример продукт онкогена erb-В2 и модифицированный
G-белок, мутантный ген ras или сверхэкспрессия генов,
регулирующая стадии передачи сигнала (например, src
киназа) либо регулирующих транскрипционный конт¬
роль {туеу fos njun) [Bishop, 1991]. Во многих случаях
продукт гена перманентно активен и не может быть
регулирован. Сверхэкспрессия онкогенов может быть
обнаружена методом иммунного окрашивания (см.
протокол 16.11), гибридизацией in situ (см. разд. 27.8),
иммуноблоттингом белкового продукта, RT-PCR для
mRNA, или МАА (см. разд. 16.6.1 и цв. вкладку 24).
В некоторых случаях продукт онкогена (например,
erb-В2, активированный На-ras) можно отличить от
нормального продукта (рецептор EGF и нормальный
ras, соответственно) как качественно, так и количест¬
венно с использованием специфических антител
18.6. ТУМОРОГЕННОСТЬ
Трансформация — это многоступенчатый процесс,
который часто завершается образованием неопласти¬
ческих клеток [Quintanilla et al., 1986]. Однако клеточ¬
ные линии, полученные из злокачественных опухолей,
предположительно уже трансформированные, могут
подвергаться дальнейшей трансформации с возраста¬
ние темпов скорости роста, снижением зависимости
от прикрепления к субстрату, более выраженной ане-
уплоидией и иммортализацией. Это доказывает, что
для злокачественной трансформации требуется некий
порядок шагов, не строго контролируемый и не взаимо¬
зависимый, и не обязательно туморогенный в каждом
индивидуальном случае. Более того, все клеточные
линии, представленные в опухоли, не должны обладать
одними и теми же трансформированными свойствами,
а одинаковый набор качеств не должен быть представ¬
лен в каждой опухоли. Прогрессирование опухоли
может подразумевать экспрессию новых свойств или
исчезновение старых, что может привести к развитию
метастазов или даже спонтанной ремиссии.
Таким образом, существует несколько этапов транс¬
формации, последовательность которых может опреде¬
ляться селективным давлением условий окружающей
среды. In vitro, где существует мало ограничений роста,
трансформационные события не обязательно следуют
в том же порядке, что и in vivo.
18.6.1. Малигнизация
Малигнизация или злокачественность подразумевает,
что в клетках возникла и развилась способность обра¬
зовывать инвазивные опухоли при их имплантации in
vivo изологичному хозяину либо как ксенотрансплан-
тант животному с подавленным иммунитетом. Хотя
развитие злокачественности может быть обнаружено
как дискретное фенотипическое событие, оно часто
сопровождается развитием аберрантного контроля
роста, что позволяет предположить, что некоторые
повреждения, ответственные за аберрантный контроль
роста, также вызывают злокачественность. Одним из
видимых кандидатов, вызывающих такие поврежде¬
ния, является дефицит межклеточного взаимодей¬
ствия, который лишает клетку контроля деления
(ограничения клеточной пролиферации при высокой
плотности культуры) и контроля подвижности (кон¬
тактное торможение). Обычно используются два
подхода для исследования свойств, связанных со зло¬
качественностью: 1) рост в культуре и характеристика
клеток, выделенных из опухолей, и 2) трансформация
in vitro при помощи вируса или химического канце¬
рогена или трансфекция онкогеном, позволяющая
получить клетки, которые были бы туморогенными
и которые можно было бы сравнивать с ^трансфор¬
мированными клетками. Второй подход позволяет
получить трансформированные клоны той же линии,
которые могут рассматриваться как малигнизирован-
ные, и эти клоны могут сравниваться с немалигнизи-
рованными. К сожалению, многие характеристики
клеток, трансформированных in vitro, не были найде¬
ны в клетках, полученных из спонтанных опухолей.
В идеале, должны быть выделены и охарактеризованы
опухолевые клетки и эквивалентные им нормальные
клетки. К сожалению, существует относительно мало
примеров, для которых такой порядок действий был
бы возможен, но даже тогда, когда клетки могут при¬
надлежать одной и той же линии дифференцировки,
18.6. Туморогенность
347
их положение в этой линии не всегда ясно, и, таким
образом, сравнение их не строго оправданно. Хотя
клетки, выделенные из нормальных взрослых тка¬
ней, будут дифференцированными и не способными
к активации, при развитии опухоли они склонны к
утрате дифференцировки и пролиферации. Это будет
отличать популяцию опухолевых клеток, независимо
от степени малигнизации, от популяции нормальных
клеток. Более того, оказалось, что многие, если не
большинство клеток в опухоли, не имеют пролонги¬
рованного времени жизни в культуре, а популяция,
критическая для распространения опухоли и поэтому
являющаяся основной мишенью для химиотерапии,
может быть достаточно маленькой и эквивалентной
- популяции стволовых клеток в нормальной ткани.
До настоящего времени накоплено мало данных,
подтверждающих возможность культивирования
этих клеток, но количество попыток сделать это все
возрастает [Petersen et al., 2003; Takahashi et al., 2003;
Aarti et al., 2004].
18.6.2. Опухолевая трансплантация
Единственным общепризнанным признаком злока¬
чественности является доказательство способности
к инвазивности или метастазированию опухолей in
vivo. Трансплантируемые опухолевые клетки (~1х106),
введенные изогенным хозяевам, будут образовывать
инвазивные опухоли в большой доле случаев, в то
время как нормальные клетки из того же источника в
дозе 1х106не образуют опухолей. Для изучения туморо-
генности человеческих опухолей разработаны модели
с использованием иммуносупрессивных и иммуно-
дефицитных животных. Мыши «nude», генетически
лишенные тимуса [Giovanella et al., 1974], так же как
и тимусэктомированные облученные мыши [Bradley
et al., 1978а; Selby et al., 1980], широко используются
как хозяева для ксенотрансплантантов. Они способны
воспринимать широкий ряд ксенотрансплантантов,
однако имеется много примеров неудачных попыток
привить достаточно точно определенные опухолевые
клеточные линии и опухолевые биопсии, в результате
которых не развивались ксенотрансплантантные опу¬
холи либо, несмотря на способность клеток к инвазии,
не формировались метастазы. Приживаемость клеток
может быть улучшена действием сублетальной дозы
радиации (30-60 Гр), на мышей nude либо использова¬
нием аспленических атимических мышей (scid), а также
путем имплантирования клеток в Matrigel [Pretlow et
al., 1991]. Несмотря на частоту ложноотрицательных
результатов, туморогенез остается хорошим индика¬
тором малигнизации.
18.6.3. Инвазивность
Изучение туморогенеза всегда должны сопровож¬
даться гистологическими исследованиями для до-
12 3 4
Сфероид Сердце эмбриона
опухолевых цыпленка
клеток
Рис. 18.6. Эксперимент с использованием клеток сердца
цыпленка. Опухолевый сфероид (см. протокол 25.2) сокуль-
тивировали вместе с обработанными фрагментами сердца
8-дневного эмбриона цыпленка. 1) Сфероид прикрепляется
к кусочку сердца через несколько часов. 2) Клетка сфероида
начинает проникать в кусочек сердца через 24-48 ч. 3) Опу¬
холевые клетки из сфероида распространяются внутри фраг¬
мента сердца. 4) Сердечные ткани полностью замещены через
8-10 дней [по Mareel et al., 1979]
казательства инвазивности и для подтверждения
гистопатологического сходства индуцированного
новообразования и исходной опухоли. Однако если
клетки не туморогенны или если возможности для
трансплантации отсутствуют либо проведение опы¬
тов на животных нежелательны, то возможно огра¬
ничиться проведением ряда исследований in vitro.
Некоторые виды исследований также предоставляют
возможности использования моделей, позволяющих
получить более точные количественные результаты,
чем эксперименты in vivo.
Хориоаллантоисная мембрана цыпленка. Ис¬
следования на хориоаллантоисной мембране цып¬
ленка (САМ) могут осуществляться на эмбрионе
цыпленка in ovo или на эксплантированной САМ in
vitro. Easty & Easty [1974] показали, что инвазия САМ
может быть показана на органной культуре, позже
другие исследователи попытались с ограниченным
успехом [Hart & Fidler, 1978] сконструировать каме¬
ру для количественных исследований проникнове¬
ния опухолевых клеток через САМ. Преимущество
исследований САМ in ovo заключается в том, что
возможно также одновременно показать ангиогенез
(см. разд. 18.6.4 и цв. вкладку 15е, ж), а последующие
гистологические исследования помогут выяснить,
проникают ли опухолевые клетки в подлежащие слои
базальной мембраны.
Инвазии опухолевых клеток в выделенные куль-
тивируемые ткани. Mareel et al. [1979] разрабо¬
тали модель для инвазии in vitro с использованием
фрагментов сердца куриного эмбриона, совместно
культивированного с перегруппированными в единые
кластеры опухолевыми клетками (рис. 18.6). Обнару¬
жено, что процесс инвазии коррелирует со степенью
злокачественности источника клеток, развивается и
вызывает деструктивные изменения тканей хозяина.
Применение этого метода к клеткам человеческих
опухолей показывает наличие высокой корреляции
348
ГЛАВА 18. ТРАНСФОРМАЦИЯ И ИММОРТАЛИЗАЦИЯ
Культуральная
среда
Внеклеточный матрикс,
т.е. Матригель
Лунка многолуночного Пористый фильтр Инвазивные клетки
культурального Клеточный
планшета
монослои
Хемиоаттрактант,
например, фибробласт-
кондиционированная
среда
Рис. 18.7. Инвазия в фильтрующей ячейке. Клетки, посеянные на нижнюю сторону фильтра, мигрируют через фильтр в бед¬
ный факторами роста Матригель в лунке фильтрующего вкладыша при стимуляции добавлением хемиоаттрактанта, такого
как фибробласт-кондиционированная среда, на верхнюю сторону Матригеля [по Brunton et al., 1997]
между злокачественностью и инвазивностью клеток
[de Ridder & Calliauw, 1990]. Этот метод широко ис¬
пользовался, но его результаты сложно определить
количественно и их интерпретация требует навыков
гистологической работы.
Фильтрующие ячейки. Разработан ряд методов,
основанный на проникновении клеток через фильтры,
покрытые Matrigel или какими-то еще веществами
внеклеточного матрикса (рис. 18.7) [Repesh, 1989;
Schlechte et al., 1990; Brunton et al., 1997; Lamb et
al., 1997]. Степень проникновения в гель или через
дистальные стороны фильтра показывает степень
инвазивности и гистологически определяется по
количеству клеток и расстоянию, которое они пре¬
одолели, либо по подсчету радиоактивных клеток,
предварительно меченных 1251, на дистальной стороне
фильтра или на дне чашки. Этот метод дает количест¬
венные результаты, однако его недостатком является
отсутствие нормальных клеток хозяина в барьере,
обычно связанных с инвазией in vivo. Проникновение
в Matrigel перспективно для оценки степени деграда¬
ции матрикса и отражает продукцию клетками про-
теаз или гликозидаз. Однако неясно, как могут быть
изучены в этой модели клетки, которые не производят
собственных ферментов деградации и обычно исполь¬
зуют протеазы, образуемые в строме.
18.6.4. Ангиогенез
Многие высвобождаемые факторы опухолевых
клеток, включая VEGF [Joukov et al., 1997], FGF-2
[Thomas et al., 1997], и ангиогенин [Hu et al., 1997],
способны к индукции образования сосудов [Folkman,
1992; Skobe & Fusenig, 1998]. Фрагменты опухолей,
осадки культивированных клеток или клеточные
экстракты, имплантированные на поверхность САМ
куриного яйца, вызывают активизацию роста сосудов,
заметную невооруженным глазом через 6-8 дней
(цв. вкладка 9). Поскольку этот тип исследований
недостаточно количественный, для более количе¬
ственных исследований могут быть использованы
методы стимуляция клеточной миграции, [Bagley
et al., 2003], продукции VEGF [Buchler et al., 2004],
или морфогенеза культуры сосудистого эндотелия
[Chen et al., 1997; Ment et al., 1997; Jain et al., 1997] в
фильтрующей ячейке.
150-
о
100
0
1
Л
со
I 50
<
Недифференцированная глиома
Дифференцированная глиома
Нормальная глия
JPTATA RATVAG
ССМ С6
JL
NMBGDU-T
Клеточная линия
Рис. 18.8. Активатор плазминогена (РА). РА продуциро¬
ванный опухолевыми клетками in vitro (единицы измерения
активности выбраны произвольно). Активности РА четырех
культур глиом —JPT, АТА, RAT, и VAG — были выше, чем в
культурах клеток из нормального мозга человека (NMB-C,
GDU-T). Также было обнаружено, что только клетки ССМ
и С6, которые продуцируют дифференциальный маркер
глии — фибриллярный кислый белок, имеют самый низкий
уровень активности РА
18.6. Туморогенность
349
18.6.5. Активаторы плазминогена
Другими продуктами, которые имеют тенденцию к
стимуляции развития трансформированных клеток, яв¬
ляются протеолитические ферменты [Mahdavi & Hynes,
1979], давно связываемые с теориями инвазивного роста
[Liotta, 1987]. Поскольку протеолитическая активность
может быть связана с клеточной поверхностью многих
нормальных клеток и отсутствует во многих опухоле¬
вых клетках, в качестве контроля с применением этого
критерия должны быть использованы эквивалентные
нормальные клетки. Активатор плазминогена (РА) в
некоторых культурах человеческой глиомы выше, чем
в культурах нормальных клеток мозга [Hince & Roscoe,
1980] (рис. 18.8), предварительно показано, что РА
связаны со многими опухолевыми культурами [de Vries
et al., 1995; del Vecchio et al., 1993; Schwartz Albiez et al.,
1991]. PA может быть измерен путем отстаивания фи-
биринового сгустка или выделения свободного раство¬
римого 1251 из [1251] фибрина [Unkless et al., 1974; Strick¬
land & Beers, 1976]. В дополнение к этому, существует
простое хромогенное исследование, разработанное Whur
et al. [1980]. Предполагается, что в случаях некоторых
карцином растворимый урокиназа-подобный РА (иРА)
характерен более, чем тканевой РА (tPA) [Markus et al.,
1980; Duffy et al., 1990], поэтому достаточно информа¬
тивным методом исследования является хромогенное
исследование вместе с зимограммным анализом [Davies
et al., 1993; Boxman et al., 1995] и последующим имму-
ноблоттингом для определения доли каждого типа РА.
Глава 19
КОНТАМИНАЦИЯ
19.1. ИСТОЧНИКИ КОНТАМИНАЦИИ
Поддержание асептических условий является еще
одной из наиболее трудных задач для новичков в
работе с культурой клеток. Проблемы, возникающие
в период начального обучения, могут быть преодоле¬
ны приобретением опыта работы, но в определенных
ситуациях даже наиболее опытные сотрудники будут
страдать от контаминации. Существует несколько
потенциальных путей, приводящих к контаминации
(табл. 19.1), включающих нарушение режима стери¬
лизации растворов, стеклянной посуды и пипеток,
турбуленцию воздуха, наличие твердых частиц (пыль
и споры) в воздухе комнаты, плохое состояние ин¬
кубаторов и холодильников, дефекты ламинарного
шкафа, внесение контаминированных клеточных
линий или биопсийных препаратов и оплошности в
осуществлении стерильных процедур. Последний из
них, надо полагать, наиболее важен.
19.1.1. Основные приемы
стерильной работы
Если используются стерильные реактивы и оборудо¬
вание находится в надлежащем рабочем состоянии,
возможность контаминации зависит от квалифика¬
ции оператора и состояния окружающей среды. Если
уровень квалификации и тщательность работы опера¬
тора высоки и атмосфера чистая (отсутствуют пыль и
сквозняк), то контаминация в результате манипуляций
оператора происходит редко. Если состояние внешней
среды ухудшается (например, в результате строитель¬
ных работ или сезонного повышения влажности) или
техника работы оператора ухудшается (пропускается
одна или более якобы необязательных мер предосто¬
рожности), то вероятность заражения возрастает. Если
и то, и другое происходит одновременно или последова¬
тельно, результаты могут быть катастрофическими.
Давайте рассмотрим такое совпадение событий в гра¬
фической форме (см. рис. 6.1). Результаты работы при
соблюдении надлежащей техники отражены на верхнем
графике. Случайные оплошности изображены в виде
направленных вниз пиков. Нижняя кривая отображает
результаты работы при плохом состоянии внешней сре¬
ды с отдельными случаями спорадического возрастания
риска контаминации, например при случайном открыва¬
нии контаминированной чашки Петри в стерильной зоне
или образование пыли при эксплуатации оборудования.
Только в случае, если две кривые (надлежащей техники
работы и высокого качества состояние внешней среды)
значительно расходятся, совпадение оплошностей в
методологии и загрязнение окружающей среды будут
происходить редко. Однако если происходит прогрес¬
сирующее ухудшение техники или состояния внешней
среды, частота контаминации будет возрастать, а если
ухудшится и то, и другое, то контаминация будет проис¬
ходить регулярно и широко распространяться.
19.1.2. Состояние внешней среды
Совершенно очевидно, что состояние внешней среды в
зоне культивирования клеток необходимо поддержи¬
вать в максимально возможно чистом состоянии, ком¬
ната не должна быть проходной и в ней не должно быть
беспорядка. Следует избегать культивирования клеток
в обычной лабораторной комнате; ламинарный шкаф не
дает достаточной защиты от нестабильного состояния
окружающей среды в типичной лаборатории. Предпоч¬
тительное выделение чистой непроходной зоны — отде¬
льная комната или отдельный блок (см. разд. 4.3,6.2.1).
Оборудование, вносимое в стерильную зону со склада,
поток воздуха от двери, холодильники, центрифуги,
перемещения операторов, — все это повышает риск
контаминации. Мойте поверхности и оборудование в
соответствии с жестким графиком и полностью проти¬
райте все, что вносится в стерильную зону.
Не существует взаимнооднозначного соответствия
между профилактической мерой и причиной конта¬
минации, указанных в таблице; предупредительные
меры взаимосвязаны и могут относиться к нескольким
причинам.
19.1.3. Использование и поддержание
в рабочем состоянии
ламинарного шкафа
Наиболее общий пример неудовлетворительной тех¬
ники — неправильное использование ламинарного
шкафа. Если ламинарный шкаф перегружать буты¬
лями и оборудованием (рис. 6.2, 6.3), то нарушается
ламинарный поток и потенциально биологически
19.1. Источники контаминации
351
Таблица 19.1. Пути контаминации
Путь или причина контаминации
Предотвращение
Процедуры
Манипуляции, пипетирование, дозирование и т. д.
Нестерильные поверхности и оборудова¬
ние.
Брызги жидкости на горле и на внешней
части бутылей и на рабочей поверх¬
ности.
Касание пипетки или удерживание пипет¬
ки слишком низко, касание горлышка
бутылей с внутренней стороны в области
закручивающейся крышки.
Разбрызгивания из емкости для слива.
Осадок пыли или частицы с поверхности
кожи на культуре или бутылях; переме¬
щение рук или инструментов над откры¬
тыми чашками или бутылями.
Рабочие поверхности
Пыль и брызги
Волосы, руки, дыхание, одежда оператора
Пыль с кожи, волос или одежды упала или
занесена потоком воздуха в культуру.
Аэрозольные частицы в процессе разгово¬
ра, кашля, чихания и т.д.
Материалы и реактивы
Растворы
Нестерильные реактивы и среда
Плохие условия хранения (грязь)
Неадекватные методы стерилизации
Плохой поставщик
Стеклянная посуда и завинчивающиеся крышки
Вымойте рабочую зону заранее.
Регулярно протирайте 70%-м спиртом. Не переливайте жидкости через край. До¬
зируйте или переливайте их с помощью пипетки, автоматического дозатора или
устройств для переливания. Если переливания через край нельзя избежать, то:
1) делайте это одним непрерывным движением, 2) удалите бутыль, из который
вы наливали и 3) вытрите брызги.
Держите пипетку выше градуировки.
Не работайте над открытыми сосудами.
Сливайте отходы в стакан с помощью воронки или, предпочтительней, удаляйте
отходы в резервуар с помощью вакуумного насоса.
Не работайте над (в ламинарных шкафах с вертикальным ламинарным потоком
и на открытом месте) или за и над (в ламинарных шкафах с горизонтальным
ламинарным потоком) открытыми бутылями или чашками.
Протирайте поверхность 70%-м спиртом до, в течение и после работы.
Вытирайте брызги немедленно.
Тщательно мойте руки или пользуйтесь перчатками. Носите лабораторную без¬
ворсовую одежду с облегающими манжетами и перчатки, прикрывающие их.
Сведите разговоры к минимуму и отворачивайтесь от рабочего пространства
при разговоре.
Старайтесь не работать при инфекционных заболеваниях носа и горла или но¬
сите маску.
Длинные волосы завязывайте в узел сзади или надевайте шапочку.
Надевайте особую лабораторную одежду. Не используйте лабораторную одежду,
в которой вы работаете в общей лабораторной зоне или в виварии.
Фильтруйте или автоклавируйте растворы перед использованием.
Регулярно мойте и дезинфицируйте места хранения реактивов.
Контролируйте работу автоклава, регистрируя показания термометра или инди¬
катора стерильности (см. Приложение II и протокол 11.5).
Проверяйте целостность фильтров при температуре кипения или проводите
анализ фильтров на присутствие микроорганизмов после использования.
После стерилизации тестируйте все растворы.
Проверяйте растворы; смените поставщика
Пыль и споры в результате хранения
Неудовлетворительная стерилизация (на¬
пример, переполнение камеры автоклава
или слишком большой объем жидкости в
бутылях препятствуют входу пара)
Инструменты, пипетки
Неудовлетворительная стерилизация
Прикрывайте крышки фольгой. Протирайте бутыли 70%-м спиртом перед вне¬
сением в ламинарный шкаф.
Меняйте запас неиспользованной посуды в дальней части полки. Не храните
ничего герметично не закрытого более 24 ч.
Проверяйте температуру загруженного содержимого в течение цикла стери¬
лизации. При автоклавировании крышки пустых бутылей не должны быть
плотно закручены. Правильно заполняйте сушильный шкаф и автоклав (см.
протокол 11.1)
Контакт с нестерильной поверхностью или
нестерильными предметами
Перед использованием стерилизуйте инструменты сухожаровым способом.
Контролируйте работу сушильного шкафа.
Заново простерилизуйте инструменты. (Используйте 70%-м спирт; прожгите в
пламени и охладите инструменты.)
Не беритесь руками за ту часть инструмента или пипетки, которая будет помещена
внутрь культурального сосуда.
Культуральные флаконы и бутыли со средой, находящиеся в использовании
Пыль и споры из инкубатора или холо- Вместо пробок используйте закручивающиеся крышки. Протирайте бутыли перед
дильника помещением в ламинарный шкаф. Упаковывайте планшеты и чашки.
352
ГЛАВА 19. КОНТАМИНАЦИЯ
Окончание табл. 19.1
Путь или причина контаминации
Предотвращение
Плохие условия хранения или инкубации
(грязь).
Среда на внутренней стороне крышки и
брызги на внешних сторонах бутыли
Оборудование и аппаратура
Воздух помещения
Вентиляция, завихрения, сквозняк, пыль,
аэрозоли
Ламинарные шкафы
Повреждение фильтра
Необходимость замены фильтра
Брызги, в особенности внутри ламинар¬
ного шкафа или на нижней рабочей
поверхности.
Сухие инкубаторы
Рост плесени и бактерий на каплях жид¬
кости
Влажные С02-инкубаторы
Рост плесени и бактерий на стенках и пол¬
ках во влажной атмосфере
Споры и т.д., внесенные при принудитель¬
ной циркуляции воздуха.
Другое оборудование
Пыль на цилиндрах, насосах и т.д.
При хранении или инкубации прикрывайте крышки и горло бутылей алюмини¬
евой фольгой. Протирайте флаконы и бутыли 70%-м спиртом перед исполь¬
зованием. Регулярно мойте места хранения и инкубаторы.
Выбраковывайте все бутыли с видимыми брызгами на внешней стороне горлыш¬
ка. Не допускайте проливания жидкостей.
Очистка фильтрованного воздуха.
Снизьте интенсивность движения и постороннюю активность в чистой зоне.
Регулярно мойте пол и рабочие поверхности.
Регулярно проверяйте, нет ли отверстий в фильтре и не нарушена ли герметич¬
ность.
Проверьте герметичность установки фильтра.
Регулярно мойте внутреннюю часть ламинарного шкафа и нижнюю рабочую
поверхность. Дайте спирту проникнуть в щели.
Протирайте любые брызги тампоном с 70%-м спиртом. Регулярно мойте инку¬
батор.
Вымойте инкубатор детергентом, а затем — 70%-м спиртом (см. протокол 19.1).
Открытые чашки Петри помещайте в пластиковые контейнеры с плотно при¬
гнанными крышками (но не закрывайте крышки герметично).
Перед тем как открыть инкубаторы, протирайте их 70%-м спиртом. В воду
добавляйте фунгицид или антибактериальный препарат (но предварительно
проверьте его токсичность).
Проникновение клещей и т. д. в стерильные
контейнеры.
Перед внесением протирайте 70%-м спиртом.
Клещи, насекомые, другие источники инфекции из деревянного инвентаря или установок, из инкубаторов и с мышей и
т.д., принесенных из вивария.
Загерметизируйте все стерильные упаковки.
Избегайте, по возможности, использования деревянного инвентаря, используйте
цельное пластиковое покрытие или рабочие поверхности из нержавеющей
стали.
Если используется деревянный инвентарь, покройте его полиуретановым
лаком или восковой политурой и регулярно мойте дезинфицирующими
растворами.
Не держите животных в лаборатории культуры тканей.
Внесение биологических материалов
Образцы ткани
Образец от инфицированного источника
или занесение инфекции при препари¬
ровании.
Добавляйте антибиотики в растворы для препарирования (см. разд. 12.3).
Все потенциально инфицированные большие образцы ткани погружайте на 30 с
в 70%-й спирт.
Поступающие в лабораторию линии клеток
Контаминация в лаборатории-источнике Работайте с такими линиями клеток отдельно, предпочтительно, в карантине, уже
линии или при транспортировке. после того, как выполнены все стерильные работы. После работы протирайте
рабочую поверхность или ламинарный шкаф 2%-м фенолом в 70%-м спирте и
не работайте там до следующего утра.
В течение двух недель проверяйте наличие контаминации, культивируя культуру
без антибиотиков. (Параллельно сохраните эту же культуру с антибиотиками
на стадии первого пассажа.)
Проверяйте наличие контаминации визуально с помощью фазово-контрастной
микроскопии и окрашиванием микоплазмы красителем Hoechst. Использо¬
вание индикаторных клеток позволит провести проверку на контаминацию
до первого пересева.
19.1. Источники контаминации
353
опасные вещества попадают в комнату. Кроме того,
возрастает риск контакта стерильных пипеток с несте¬
рильными поверхностями бутылей и т. д. В ламинар¬
ный шкаф необходимо вносить только те предметы,
которые непосредственно необходимы для выполне¬
ния текущей операции.
Также ламинарные шкафы должны регулярно об¬
служиваться технически, целостность фильтров, воз¬
духодувов и корпуса должна проверяться, по крайней
мере, дважды в год опытным специалистом. Специа¬
лист также должен проверять герметичность рабочей
области, так как воздух изнутри не должен выходить
наружу, а внешний воздух не должен проникать внутрь.
И то, и другое зависит от скорости циркуляции внут¬
реннего воздуха и внешних завихрений.
19.1.4. Инкубаторы с увлажнением
Одна из основных причин контаминации — это ис¬
пользование инкубаторов с увлажненной атмосферой
(см. разд. 6.6.1). Высокая влажность не требуется, если
не используются открытые сосуды; плотно закрытые
флаконы лучше держать в сухом инкубаторе или
термальной комнате (см. разд. 4.4). Для того чтобы
избавиться от необходимости использовать газовые
баллоны с С02, можно использовать среды для культи¬
вирования при низком содержании С02 (например, на
основе солей Хэнкса, см. разд. 9.3.2). Если же возникла
необходимость заполнения флаконов углекислым га¬
зом, то лучше ввести газ из баллонов или центральной
системы подачи С02, затем плотно закрыть флаконы
и поместить их в обычный инкубатор. Использование
газопроницаемых крышек (см. разд. 8.2.3) минимизи¬
рует риск контаминации, но стоят флаконы с такими
крышками дороже, и степень риска заражения при
содержании таких флаконов в обычной атмосфере
выше, чем в сухом инкубаторе. Для переноса флаконов
в инкубатор без подачи С02, газопроницаемые крышки
можно плотно закрывать второй резиновой крышкой
(B-D Biosciences). Если флаконы с приоткрытыми или
газопроницаемыми крышками содержатся в С02-инку-
баторе, по возможности держите инкубатор сухим, а для
открытых планшетов используйте другой инкубатор.
Если в инкубаторе отсутствует вода, то необходимо
заново откалибровать систему контроля С02, необхо¬
димо контролировать испарение жидкости во флаконах
и, при необходимости, закручивать крышки флаконов
после уравновешивания pH.
Однако во многих случаях необходимо использовать
инкубатор с увлажненной атмосферой. Для того чтобы
снизить риск контаминации, из внутренней камеры
инкубатора необходимо вынуть все доступные детали и
тщательно вымыть. Культуры, хранящиеся в инкубато¬
ре, должны быть помещены в пластиковые контейнеры
(см. разд. 6.6.2). Использование вентилятора в С02-ин-
кубаторе уменьшает время восстановления заданных
значений как С02, так и температуры, но ценой явля¬
ется повышение риска контаминации (см. разд. 5.3.2,
6.6.1); культуры в открытых планшетах лучше культи¬
вировать в неподвижном воздухе и и как можно реже
открывать инкубатор. Наличие сухого инкубатора без
С02 для плотно закрытых флаконов и кратковременных
процедур, таких как трипсинизация, поможет умень¬
шить частоту открывания инкубатора, используемого
для открытых планшетов и клонирования.
Фунгициды. Облицованные изнутри медью инку¬
баторы снижают рост грибов, но обычно они на 20-30%
дороже, чем обычные. Помещение медной фольги в
поддон для воды также ингибирует рост грибов, но
только в лотке, и не защищает стенки инкубатора. Час¬
то используют ряд противогрибковых агентов, среди
которых сульфат меди, рибофлавин, додецилсульфат
натрия (SDS) и 2%-й раствор специализированного
противогрибкового детергента Roccall. Сравнение
колониеобразования в инкубаторах с и без Roccall
показало отсутствие токсического эффекта. Многие из
этих агентов являются детергентами, поэтому важно,
чтобы С02или воздух не проходили через поддон с
водой, иначе жидкость будет пениться. Помните, фун¬
гицид будет только защищать поддон, не используйте
его для обычного мытья!
Мытье инкубаторов. Инкубатор следует регулярно
мыть 10%-м Roccall или аналогичным нетоксичным
противогрибковым моющим средством. Частота мытья
зависит от расположения инкубатора; если он находится
в чистой зоне с фильтруемым воздухом, то мыть его
нужно ежемесячно. В сельской местности следует мыть
чаще, так как там больше спор. Чаще следует мыть и в
случае строительных работ или ремонта. Частота откры¬
вания инкубатора также влияет на повышение контами¬
нации грибами. В рабочем инкубаторе любые брызги не¬
обходимо немедленно вытирать, а контаминированные
культуры удалять сразу же после обнаружения.
ПРОТОКОЛ 19.1. Обработка инкубаторов
Схема
Перенести культуры в запасной инкубатор, выклю¬
чить пустой инкубатор, вымыть его детергентом и
спиртом, включить нагрев и дать инкубатору вы¬
сохнуть. Наполнить водой поддон и возобновить
подачу С02.
Материалы
Нестерильные:
• 2%-й раствор Roccall или аналогичного про¬
тивогрибкового агента в воде для наполнения
поддона
• Запасной С02-инкубатор или пластиковый кон¬
тейнер и герметизирующая лента
• Детергент: 10% Roccall или другой противогриб¬
ковый детергент
• 70%-й спирт
354
ГЛАВА 19. КОНТАМИНАЦИЯ
Протокол
1. Перенесите все культуры в другой С02-инкубатор
или поместите культуры в герметично закрыва¬
ющийся контейнер (например, в эксикатор), за¬
полните их С02и поместите в обычный инкубатор
или термальную комнату.
2. Отключите инкубатор, который будете мыть.
3. Выньте все полки, поддон для воды и съемные
панели инкубатора.
4. Вымойте внутреннюю камеру инкубатора раство¬
ром детергента; постарайтесь промыть все углы и
зазоры.
5. Промойте водой.
6. Протрите внутреннюю камеру инкубатора 70%-м
спиртом.
7. Включите нагрев (но не С02) и оставьте дверь
открытой до тех пор, пока камера не просохнет.
8. Вымойте полки и панели детергентом^ прса*ш|т1г
водой и протрите 70%-м спиртом.
9. Поставьте на место панели и полки в инкубаторе.
10. Поставьте на место поддон и заполните его сте¬
рильной всщой, содержащей 2%-й Roccall.
11. Закройте дверь и возобновите подачу С02.
12. Когда уровень температуры и С02 стабилизиру¬
ется, верните культуры в инкубатор.
Если инкубатор имеет цикл стерилизации, он должен
быть включен после выполнения 9-го шага.
19.1.5. Холодильные камеры
В холодильниках и холодных комнатах также возмож¬
но появление грибковой контаминации на стенках в
увлажненной атмосфере из-за возникновения кон¬
денсата, образующегося каждый раз при открывании
двери и впускании влажного воздуха. Влажный воздух
повышает риск попадания спор на хранящиеся в холо¬
дильнике бутыли; поэтому их необходимо протирать
спиртом перед внесением в ламинарный шкаф (см.
разд. 6.5). Холодильные камеры должны быть чистыми,
каждые несколько месяцев необходимо мыть стенки и
полки дезинфектантами.
19.1.6. Стерильные материалы
Ни стерильный пластик, ни стерильные реагенты не
должны быть источником контаминации, если они
прошли соответствующий контроль качества в фирме-
производителе или у поставщика. Однако нарушение
стерильности может произойти, например, в случае
слишком плотной упаковки при загрузке в стери¬
лизационный шкаф или автоклав, так как при этом
образуются локальные участки недостаточно высокой
температуры, или при повреждении упаковки до или
в процессе хранения, или при низкоуровневой конта¬
минации, которая не была обнаружена при проверке
качества продукта. Избежать этих непредвиденных
обстоятельств сложно, кроме как выполняя надлежа¬
щие процедуры (см. разд. 11.6) и проверяя целостность
всех упаковок.
Критическим является момент появления новых
сотрудников в лаборатории культуры тканей, которые
могут вносить изменения в процедуры стерилизации
(см. гл. 2, Упражнения 3-9), даже если они не будут
выполнять эти процедуры сами. Они также должны
быть осведомлены о различиях между стерильными и
нестерильными материалами и о местах их хранения
(см. разд. 2.2). Единичная ошибка нового сотрудника
может стать причиной ряда проблем, и может пройти
несколько дней, пока эта ошибка будет обнаружена.
19.1.7. Привезенные клеточные линии
и биопсийные образцы
Так как линии клеток и образцы тканей, привезенные в
лабораторию культуры тканей, могут быть контамини-
рованы, полученные клетки должны быть помещены в
карантин (см. разд. 19.1.8) до тех пор, пока не выявится
контаминация, и только после этого полученные клетки
или их производные можно помещать в совместные
хранилища для общего использования (см. разд. 6.2.5).
Всегда, когда это возможно, все линии клеток необходи¬
мо приобретать в банках клеток с хорошей репутацией,
в которых проводится скрининг на контаминацию.
Линии клеток из других источников, так же как и био-
псийный материал от человека или животных, должны
рассматриваться как контаминированные, пока не
будет доказано обратное.
19.1.8. Карантин
Любая культура, заподозренная в наличии контами¬
нации, и любой привнесенный непротестированный
материал должны быть помещены в карантин. Пред¬
почтительно, чтобы для карантина была отведена отде¬
льная комната с ламинарным шкафом и инкубатором,
но если это неосуществимо, можно использовать один
из ламинарных шкафов общего пользования. В этом
случае с подозрительным материалом следует рабо¬
тать в конце рабочего дня, а после работы необходимо
опрыскать и вымыть все поверхности 70%-м спиртом,
с добавлением 2% фенол-содержащего дезинфектанта.
До следующего дня в этом ламинарном шкафу работать
не следует.
19.2. ВИДЫ МИКРОБНОЙ
КОНТАМИНАЦИИ
Культура клеток может быть контаминирована бакте¬
риями, дрожжами, грибами, микоплазмой и изредка
простейшими. Обычно род или вид инфекции не
19.3. Контроль контаминации
355
важны до тех пор, пока заражение не станет часто
повторяться. Необходимо только отметить общие виды
контаминаций (например, из бактерий — палочки и
кокки, дрожжи и т.д.), которые были обнаружены,
место, в котором с культурой последний раз работа¬
ли, и имя оператора. Если какой-то отдельный вид
инфекции часто повторяется, возможно, полезно
идентифицировать ее, для того чтобы найти причину
возникновения. [Более подробно описание процедур
скрининга микробной контаминации описан в Euro¬
pean Pharmacopoeia, 1980; United States Pharmacopeia,
1985; Doyle et al., 1990; Doyle & Bolton, 1994; and Hay
& Cour, 1997.] В целом, быстро растущие микроор¬
ганизмы являются меньшей проблемой, так как они
обычно ясно выражены и легко обнаруживаются,
после чего культура может быть отбракована. Слож¬
ности возникают, когда контаминация скрытая либо
потому, что микроорганизмы слишком мелкие и их
нельзя увидеть в микроскоп, например микоплазма
или медленнорастущие микроорганизмы, которые
можно не обнаружить. Использование антибиотиков
может быть частой причиной скрытой контаминации,
оставшейся необнаруженной (см. разд. 9.4.7).
19.3. КОНТРОЛЬ КОНТАМИНАЦИИ
В табл. 19.1 приведены возможные источники конта¬
минации, а также меры предосторожности, которые
необходимо предпринимать, чтобы избежать конта¬
минации. Однако контаминация происходит даже в
лучших лабораториях, поэтому рекомендуется выпол¬
нение следующих процедур:
1) Проверяйте наличие контаминации визуально и
с помощью микроскопа каждый раз при работе с
культурой. Раз в месяц тестируйте культуры на
микоплазму.
2) Если есть подозрение на то, что культура конта-
минирована, но нет подтверждения in situ, уберите
из ламинарного шкафа или со стола все, за исклю¬
чением подозрительной культуры и контейнера
с Пастеровскими пипетками. Так как при этом
возникает риск для других культур, лучше это
делать после окончания других культуральных
работ. Отберите из культуры образец и поместите
его на предметное стекло. Просмотрите стекло под
микроскопом, желательно, с фазовым контрастом.
Если подтвердится предположение о том, что
культура контаминирована, выбросьте пипетки,
протрите ламинарный шкаф или стол 70%-м спир¬
том, содержащим феноловый дезинфектант, и не
пользуйтесь ламинарным шкафом или столом до
следующего дня.
3) Регистрируйте характер контаминации.
4) Если контаминация новая и не распространилась
на другие культуры, выбросьте культуру, исполь¬
зуемую бутыль со средой и другие реактивы (на¬
пример, трипсин), которые использовались при
работе с культурой. Поместите все эти предметы в
дезинфектант, желательно, в вытяжной шкаф и вне
зоны работы с культурой клеток.
5) Если контаминация новая и широкораспространен¬
ная (а именно, по крайней мере, на две различные
культуры), выбросьте все среды, исходные раство¬
ры, трипсин и т.д.
6) Если ранее уже происходила контаминация такого
же типа, проверьте на контаминацию исходные
растворы а) путем инкубации растворов отдельно
или в питательном бульоне (см. разд. 11.6.2 и цв.
вкладку 16г) или б) поместив раствор в питатель¬
ную агаризованную среду (Oxoid, Difco). Если
результаты (а) и (б) отрицательные, но подозрение
на контаминацию все еще есть, проинкубируйте
100 мл раствора, профильтруйте его через фильтр
с размером пор 0,2 мкм и поместите фильтр в пита¬
тельную агаризованную среду.
7) Если контаминация широко распространена,
мультиспецифична и повторяется, проверьте
а) процедуру стерилизации (например, температуру
стерилизационных шкафов и автоклавов, особенно
в середине процесса стерилизации), б) правила
упаковки и хранения (например, негерметично
закрытую стеклянную посуду следует заново сте¬
рилизовать каждые 24 ч) и в) целостность чистой
комнаты и фильтров ламинарного шкафа.
8) Не делайте попыток лечить контаминированные куль¬
туры, если они не являются невосстановимыми.
19.3.1. Визуально определяемая
микробная контаминация
Характерные признаки микробной контаминации:
1) Внезапные изменения pH. pH обычно снижается
в большинстве случаев бактериальной инфекции
(цв. вкладка 16а), мало изменяется при дрожжевой
контаминации (цв. вкладка 16в), пока контамина¬
ция не станет очень сильной, и иногда повышается
при контаминации грибами.
2) Помутнение среды (см. цв. вкладки 16я-в), иногда
тонкая пленка или пена на поверхности, или пятна
на ростовой поверхности, которые исчезают, когда
флакон встряхивают (цв. вкладка 166).
3) Под микроскопом при малом увеличении (~х100)
в пространстве между клетками видны мерцаю¬
щие гранулы, которые могут быть бактериями
(рис. 19.1а). Дрожжи представляют собой отдель¬
ные круглые или овальные частицы, от которых
могут отпочковываться более мелкие частицы
(рис. 19.1 б). Грибы образуют тонкие нити мицелия
(рис. 19.1в) и иногда плотные скопления спор.
В случае токсичной инфекции наблюдается неко¬
торое ухудшение состояния клеток.
4) Под микроскопом при большом увеличении (~х400)
можно рассмотреть индивидуальные бактерии и
различить палочки и кокки. При этом увеличении
видно, что причиной мерцания, наблюдаемого при
некоторых инфекциях, является движение бакте¬
356
ГЛАВА 19. КОНТАМИНАЦИЯ
Рис. 19.1. Типы контаминации. Примеры микроорганизмов, обнаруженных в контаминированных культурах клеток, (а) Бак¬
терии. (6) Дрожжи, (в) Мицелий гриба, (г) Колонии микоплазмы, растущие в специальном питательном агаре. (д-е) Фотогра¬
фии, сделанные с помощью сканирующего электронного микроскопа, микоплазмы, растущей на поверхности культивируемых
клеток (г-е, любезно предоставлены Dr. М. Gabridge; a-в, объектив 10х) (микоплазму, окрашенную Hoechst 33258 см. на
цв. вкладке 16Э, е)
рий. Некоторые бактерии образуют скопления или
ассоциаты с культивируемыми клетками.
5) Сделав препарат на стекле, можно исследовать
морфологию бактерий при увеличении хЮОО, но
обычно в этом нет необходимости. Бактериальную
инфекцию можно спутать с выпавшими в осадок
компонентами среды и с клеточным дебрисом,
но она отличается регулярной морфологией.
Преципитаты могут быть кристаллическими или
глобулярными или неправильной формы и раз¬
ного размера. Скопления бактерий можно спутать
с выпавшими белками, но много отдельных или
соединенных в цепочки бактерий можно увидеть,
особенно при встряхивании. Если вы сомневаетесь,
поместите образец среды в питательный агар (см.
разд. 11.6.2).
19.3. Контроль контаминации
357
19.3.2. Микоплазма
Наличие заражения микоплазмой (рис. 19Лг-е) нельзя
определить с помощью обычной микроскопии, увидеть
можно только ухудшение состояния культуры. Для
детекции микоплазмы требуется применение методов
флюоресцентного окрашивания, ПЦР, ИФА, иммуно¬
окрашивания, авторадиографии или микробиологи¬
ческие исследования. Флюоресцентное окрашивание
ДНК красителем Hoechst 33258 [Chen, 1977] является
самым простым и наиболее достоверным методом (см.
протокол 19.2) и позволяет выявить микоплазменную
инфекцию в виде окрашенных мелких частиц или ни¬
тей на поверхности цитоплазмы при увеличении х500
(цв. вкладка 163, ё). Ядро культивируемых клеток при
использовании этого метода тоже ярко окрашено, что
является положительным контролем при использо¬
вании этого метода. Большинство других бактерий
тоже выявляется при флюоресцентном окрашивании,
таким образом можно определить наличие инфекции
при низком уровне контаминации или выявить при¬
сутствие очень мелких микроорганизмов, таких как
микрококки.
Протокол
1. Посейте индикаторные клетки в чашки Петри без
антибиотиков таким образом, чтобы конфлюэнт-
ность черр 4-5 дней составила 50-60% (напри¬
мер, 2x10 NRK или 1x10 А549 в 5 мл среды).
2. Добавьте 1,5 мл среды из тестируемой культуры.
3. Инкубируйте культуру до достижения 50-60%
конфлюэнтности.
Примечание. К моменту фиксации культура не
должна достигать конфлюэнтности, иначе окраши¬
вание будет подавляться и дальнейшая визуализация
микоплазмы будет ухудшена.
4. Удалите среду
5. Промойте монослой BSS-PR или D-PBSA и уда¬
лите промывочный раствор.
6. Добавьте свежий BSS-PR или D-PBSA разве¬
денный 50:50 фиксатором, промойте монослой,
удалите промывочный раствор.
7. Добавьте чистый фиксатор, промойте, удалите
раствор.
8. Добавьте свежий фиксатор (-0,5 мл/см2) и фик¬
сируйте 10 мин.
9. Удалите фиксатор.
10. Полностью высушите монослой, если он предна¬
значен для хранения. (Образцы можно собирать
на этой стадии, а окрашивать позднее.)
11. Если вы собираетесь далее проводить окраши¬
вание, смойте фиксатор деионизованной водой
и удалите промывочный раствор.
12. Добавьте Hoechst 33258 в BSS-PR или D-PBSA и
окрашивайте 10 мин при комнатной температуре.
13. Удалите краситель.
14. Промойте монослой водой и удалите промывоч¬
ный раствор.
15. Заключите под покровное стекло в капле забуфе-
ренной глицериновой среды и удалите излишек
с краев покровного стекла.
16. Анализируйте флюоресценцию монослоя, ис¬
пользуя фильтр возбуждения 330/380 нм и ба¬
рьерный фильтр LP 440 нм.
Важно принимать во внимание то обстоятельство,
что микоплазмы не всегда обнаруживают свое присут¬
ствие, влияя на состояние клеток или среды. Многие
микоплазмы, особенно в постоянных клеточных
линиях, растут медленно и не повреждают клетку-
хозяина. Однако они могут влиять на метаболизм
культуры различными способами. Так как микоплазмы
потребляют тимидин из среды, в инфицированных
культурах наблюдается аномальный уровень включе¬
ния [ Н]-тимидина (см. рис. 27.2). Иммунологические
исследования также могут не дать нужного результата
при контаминации микоплазмой, так как вместо по¬
лучения антител к поверхностным антигенам клетки,
получают антитела к белкам микоплазмы. Микоплаз¬
ПРОТОКОЛ 19.2. Выявление микоплазмы
методом флюоресценции
Схема
Культивировать клетки без антибиотиков не менее
недели, внести среду из супернатанта клеткам ин¬
дикаторной культуры, находящимся в начале лога¬
рифмической фазы роста, инкубировать 3-5 дней,
зафиксировать и окрасить клетки, искать флюорес¬
ценцию вне ядра.
Материалы
Стерильные или приготовленные
в асептических условиях:
• Среда из супернатанта, без антибиотиков, пос¬
ле культивирования клеток в течение 7 дней в
монослое или после центрифугирования суспен¬
зионной культуры
• Индикаторные клетки (например, ЗТ6, NRK,
Vero, А549)
• Чашки Петри диаметром 6 см
Нестерильные:
• Краситель Hoechst 33258, 50 нг/мл в BSS без
фенолового красного (BSS-PR) или D-PBSA
• BSS-PR: BSS Хэнкса без фенолового красного
• D-PBSA: PBS Дюльбекко без Са и Mg
• Деионизованная вода
• Фиксатор: свежеприготовленный метанол-уксус-
ная кислота (3:1, на льду)
• Забуференная глицериновая среда для заключе¬
ния препарата (см. Приложение I: Реагенты для
определения микоплазмы)
358
ГЛАВА 19. КОНТАМИНАЦИЯ
мы могут влиять на поведение и метаболизм клеток и
многими другими путями [McGarrity, 1982; Doyle et
al., 1990; Izutsu et al., 1996; Dorazio et al., 1996; Giron et
al., 1996; Paddenberg et al., 1996], поэтому абсолютным
требованием при проведении рутинных периодических
анализов является проверка возможности контами¬
нации всех культур клеток, особенно постоянных
клеточных линий.
Контроль культур на микоплазмы. Внешние
признаки хронической микоплазменной инфекции
включают снижение скорости пролиферации клеток,
снижение насыщающей плотности [Stanbridge &
Doersen, 1978] и слипание клеток при культивирова¬
нии в суспензии [Giron et al., 1996]. Острая инфекция
приводит к общей деградации клеток, при этом могут
оставаться несколько устойчивых колоний, хотя эти
клетки и выросшие из них линии совсем не обязательно
свободны от контаминации, они могут быть хронически
инфицированы.
19.3.3. Окрашивание микоплазмы
флюоресцентными красителями
Культуры окрашивают флюоресцентным красителем
Hoechst 33258, который специфично связывается с
ДНК [Chen, 1977]. Так как микоплазмы содержат
ДНК, их можно легко обнаружить по характерным
флюоресцирующим частицам или нитям на поверх¬
ности клеток и, в случае обширной контаминации, в
окружающих областях. Монослойные культуры клеток
можно зафиксировать и окрасить непосредственно.
При исследовании клеток, растущих в суспензии, после
центрифугирования суспензии, культуральную среду
добавляют к индикаторным клеткам (т. е. к другой мо-
нослойной культуре), гарантированно не зараженным
микоплазмой, но являющимся хорошими клетками-
хозяевами для микоплазмы и хорошо распластанными
в культуре, что позволяет обнаружить присоединившу¬
юся к клеткам микоплазму. Для этого подходят клетки
Vero, ЗТ6, NRK и А549.
Использование индикаторных клеток рекомендует¬
ся в нижеследующем протоколе, так как это помогает
избежать проблем с ложно позитивными результатами,
возникающими из-за дебриса при анализе, проводимом
сразу после размораживания клеток или в первичных
культурах. В случае большого количества дебриса среду
можно профильтровать через стерильный фильтр с раз¬
мером пор 5 мкм или отцентрифугировать при 100 g.
Анализ. Исследуйте внеядерную флюоресценцию.
Микоплазма проявляется в виде флюоресцентных
точек или нитей над цитоплазмой и, иногда, в межкле¬
точном пространстве (см. цв. вкладку 163, ё). Точки по
размеру близки к пределу разрешающей способности
50х объектива (0,1-1,0 мкм) и обычно имеют одинако¬
вые размеры и форму. Не обязательно все клетки будут
инфицированы, поэтому необходимо просмотреть как
можно больше препаратов перед тем, как утверждать,
что культура не инфицирована.
Причиной иногда наблюдающегося слабого од¬
нородного окрашивания цитоплазмы, по-видимому,
является РНК. Эта флюоресценция имеет тенденцию
ослабевать при хранении препарата и при исследовании
на следующий день (после хранения в сухом и прохлад¬
ном месте) обычно не обнаруживается. Этот артефакт
никогда не выглядит как характерные для микоплазмы
точки или нити и легко может быть распознан при
некотором опыте наблюдений.
Если имеются какие-либо сомнения в интерпре¬
тации флюоресцентного теста, его следует повторить
после следующего пассажа тестируемых клеток в
отсутствие антибиотиков. Если результаты остаются
неясными, следует применить другой метод, такой как
ПЦР или ИФА.
19.3.4. Использование ПЦР
для детекции микоплазм
Следующий протокол и введение предоставлены Cord
С. Uphoff и Hans G. Drexler (DSMZ, Braunschweig,
Germany).
Полимеразная цепная реакция (ПЦР) является
очень чувствительным и специфичным методом для
прямого определения микоплазмы в культуре клеток
при низких затратах труда, времени и средств. Метод
отличается простотой, объективностью интерпретации,
воспроизводимостью и документальной обоснован¬
ностью результатов. Опубликованы праймеры как
для обычной, так и для гнездной ПЦР, с узкой или
широкой специфичностью, для микоплазм и различных
видов эубактерий. В большинстве случаев в качестве
мишени используется последовательность 16S рДНК,
так как этот ген содержит области с более или менее
консервативными последовательностями. Этот ген
также дает возможность проведения ПЦР с 16S рДНК
или ОТ-ПЦР (ПЦР с использованием обратной транс-
криптазы) с кДНК 16S рРНК.
Здесь мы описываем, какая смесь олигонуклеотидов
используется для специфического определения мико¬
плазм. Этот подход значительно снижает вероятность
получения ложноположительных результатов из-за
возможной контаминации растворов, используемых
при подготовке образцов и для проведения ПЦР, и из
других материалов, в которые из воздуха могут попасть
бактерии. Одной из основных проблем, связанных с
проведением ПЦР образцов из культур клеток, явля¬
ется неспецифическое ингибирование Год-полимера¬
зы. Для удаления этих ингибиторов мы рекомендуем
обязательно экстрагировать и очищать образцы ДНК
с помощью стандартной фенол-хлороформной экс¬
тракции или с помощью соответствующей колонки
или носителя.
Для подтверждения правильности приготовления
образцов и проведения ПЦР необходимо включать
при проведении ПЦР соответствующие контроли. Они
19.3. Контроль контаминации
359
включают внутренний контроль ДНК для каждого об¬
разца и параллельно положительный и отрицательный
контроли, а также контроль воды. Внутренний конт¬
роль состоит из фрагмента ДНК с теми же праймерами
для амплификации, но с отличающимся от ампликона
содержащих микоплазму образцов размером. Этот
контроль ДНК добавляется в смесь для ПЦР в опре¬
деленном заранее предельном разведении для демон¬
страции чувствительности ПЦР.
ПРОТОКОЛ 19.3. Выявление микоплазмы
с помощью ПЦР
Материалы
• D-PBSA (фосфатно-солевой раствор): 137 мМ
NaCl, 2,7 мМ КС1, 8,1 мМ Na2HP04, 1,5 мМ
КН2Р04, pH 7,2. Автоклавировать 20 мин при
121 °С для стерилизации раствора.
• ТАЕ (Трис, уксусная кислота, ЭДТА), 50х: 2 М
Трис, 5,71% ледяная уксусная кислота (по объе¬
му), 100 мМ ЭДТА. Довести pH до 8,5
• Система для экстракции и очистки ДНК, напри¬
мер фенол/хлороформная экстракция и осажде¬
ние этанолом или набор для экстракции ДНК с
применением ДНК-связывающих матриц
• Амплификатор
• Taq ДНК-полимераза
• Реакционный буфер 6х: 0,09% (вес/объем) бро-
мфеноловый синий, 0,09% (w/v) ксилол цианол
FF, 60% глицерин (v/v), 60 мМ ЭДТА.
• Праймеры (от любого производителя):
5' праймеры (Мусо-5'):
ege
ctg
agt
agt
acg
twe
gc
tgc
Ctg
rgt
agt
аса
ttc
gc
ege
ctg
agt
agt
atg
ctc
gc
ege
ctg
ggt
agt
аса
ttc
gc
3' праймеры
(Myco-
-3’):
geg
gtg
tgt
аса
ага
ccc
ga
geg
gtg
tgt
аса
aac
ccc
ga
(r =
смесь g и a;
w =
смесь t и a)
Исходные растворы праймеров: 100 мкМ в дис¬
тиллированной воде, хранить замороженными при
-20 °С. Рабочие растворы: смесь прямых прайме¬
ров по 5 мкМ каждого (Мусо-5) и смесь обратных
праймеров по 5 мкМ каждого (Мусо-3) в Н20 диет.,
аликвотировать по 25-50 мкл и хранить заморожен¬
ными при -20 °С.
• Внутренний контроль ДНК: Внутренний конт¬
роль приготовить, как описано [Uphoff & Drexler,
2002]. Предельное разведение должно быть оп¬
ределено экспериментально проведением ПЦР с
серией разведений внутреннего контроля ДНК.
• Положительный контроль ДНК: 10-кратное
разведение любого положительного на присут¬
ствие микоплазмы образца, приготовленного как
описано выше.
Смесь дезоксинуклеотидтрифосфатов (dNTPmix):
смесь содержит дезоксиаденозинтрифосфат
(dATP), дезоксицитидинтрифосфат (dCTP),
дезоксигуанозинтрифосфат (dGTP) и дезок-
ситимидинтрифосфат (dTTP) в концентрации
5 мкМ в Н20 диет., хранить аликвоты по 50 мкл
при -20 °С.
Гель агарозный 1?3%-ТАЕ
Протокол
A. Сбор образцов и приготовление ДНК.
1. Перед сбором образцов клеточные линии, тести¬
руемые на присутствие микоплазмы, необходимо
несколько дней культивировать без антибиоти¬
ков, а после разморозки — не менее двух недель.
Это обеспечит титр микоплазм в среде из суперна-
танта, достаточный для обнаружения с помощью
ПЦР анализа.
2. Соберите 1 мл среды из супернатанта прикреп¬
ляющихся клеток или после осаждения сус¬
пензионных клеток. Собранные таким образом
образцы будут содержать некоторое количество
живых или мертвых клеток, что является преиму¬
ществом, так как некоторые штаммы микоплазм
в основном прикреплены к эукариотическим
клеткам или даже проникают внутрь них. Среду
из супернатанта следует хранить при 4 °С в те¬
чение нескольких дней или при -20 °С в течение
нескольких недель. После разморозки образцы
можно использовать немедленно.
3. Среду из супернатанта центрифугируйте при
13 000 g 5 мин, осадок ресуспендируйте в 1 мл
D-PBSA путем перемешивания на вортексе.
4. Суспензию еще раз центрифугируйте и промойте
еще раз D-PBSA, как описано в пунктеЗ.
5. После центрифугирования ресуспендируйте оса¬
док в 100 мкл D-PBSA путем перемешивания на
вортексе и затем 15 мин нагревайте при 95 °С.
6. После лизиса клеток немедленно экстрагируйте
ДНК и очищайте стандартным методом фенол/
хлороформной экстракции и преципитации эта¬
нолом или другими методами.
B. ПЦР-реакция
Следует строго выполнять правила, установленные
для проведения ПЦР, предотвращающие перенос
ДНК: (i) экстракцию ДНК, проведение ПЦР реак¬
ции и электрофорез следует выполнять в местах,
отделенных друг от друга; (ii) все реагенты необ¬
ходимо хранить в малых количествах для того,
чтобы обеспечить постоянный источник незара-
женных реагентов.; (iii) избегайте реамплификаций;
(iv) запасные пипетки, наконечники и пробирки
должны использоваться только для ПЦР, пипетки
360
ГЛАВА 19. КОНТАМИНАЦИЯ
необходимо часто облучать ультрафиолетом; (v) по¬
рядок проведения ПЦР, описанный ниже, необхо¬
димо выполнять точно; (vi) при приготовлении об¬
разцов и проведении ПЦР необходимо использовать
перчатки; (vii) включайте надлежащие контроли,
такие как внутренний, положительный, отрицатель¬
ный и контроль воды.
1. Поставьте каждый тестируемый образец на две
реакции со следующими растворами: Только
образец: 1 мкл dNTPs, 1 мкл Мусо-5,1 мкл
Мусо-3, 1,5 мкл 10х PCR буфера, 9,5 мкл Н20
диет.. Образец и внутренний стандарт ДНК: 1 мкл
dNTPs, 1 мкл Мусо-5,1 мкл Мусо-3,1,5 мкл 10х
PCR buffer, 8,5 хмкл Н20 диет., 1 мкл внутренний
контроль ДНК. Смешайте исходные растворы
для кратного количества образцов, три образца
для реакции без внутреннего контроля ДНК (для
положительного, отрицательного контролей и
контроля воды) и два для внутреннего контроля
ДНК для положительного и отрицательного кон¬
тролей, плюс излишек для пипетирования.
2. Поместите 14 мкл каждого предварительно
смешанного исходного раствора в реакционные
пробирки на 0,2 мл и добавьте 1 мкл Н20 диет, к
водному контролю.
3. Приготовьте смесь Taq ДНК-полимеразы (10 мкл
на реакцию плюс 1 дополнительная реакция —
поправка на пипетирование), содержащую 1х
ПЦР буфер и 1 U Taq полимеразы на реакцию.
4. Уберите все реагенты, используемые для приго¬
товления предварительных исходных растворов.
5. Возьмите тестируемые образцы ДНК и поло¬
жительный контроль ДНК. Не держите в руках
реагенты и образцы одновременно.
6. Добавьте 1 мкл препарата ДНК з одну реакци¬
онную пробирку, не содержащую внутренний
контроль ДНК, и в одну пробирку, содержащую
внутренний контроль ДНК.
7. Для запуска ПЦР перенесите реакционные смеси
(без Taq полимеразы) в амплификатор и запус¬
тите 1 цикл со следующими параметрами: шаг 1:
7 мин при 95 °С, шаг 2: 3 мин при 72 °С, шаг 3:
2 мин при 65 °С, шаг 4: 5 мин при 72 °С.
Во время шага 2 откройте крышку и добавьте
10 мкл Taq полимеразы в каждую пробирку. Для
многих образцов продолжительность этого этапа
может быть увеличена. Открывайте и закрывайте
каждую пробирку по отдельности, чтобы избежать
испарения образцов. Подождите не менее 30 с после
добавления Taq полимеразы в последнюю пробирку,
перед тем как закрыть крышку амплификатора для
того, чтобы температура во всех пробирках урав¬
новесилась и удалился конденсат с крышки перед
продолжением следующего шага цикла.
8. После этого инициирующего цикла выполните
32 цикла нагревания со следующими параметра¬
ми, шаг 1:4с при 95 °С, шаг 2:8 с при 72 °С, шаг 3
16 с при 72 °С плюс 1 с дополнительного времени
в течение каждого цикла.
9. Закончите реакцию последним шагом амплифи¬
кации при 72 °С 10 мин и затем охладите образцы
до комнатной температуры.
10. Приготовьте 1,3% агароза-ТАЕ гель, содержащий
0,3 мкг/мл бромистого этидия. Погрузите гель в
lxTAE и добавьте 12 мкл амплифицированного
продукта (10 мкл реакционной смеси плюс 2 мкл
6х буфера для нанесения) на каждую дорожку и
проводите электрофорез при 10 V/cm.
И. Визуализируйте специфические продукты с
помощью освещения соответствующей лампой
и задокументируйте результаты.
Интерпретация результатов
1) В идеале, все образцы, содержащие внутренний
контроль ДНК, демонстрируют полосу в 986 Ьр.
Если вторая дорожка показывает отсутствие полос
образца и внутреннего контроля, всю процедуру
необходимо повторить.
2) Положительные по микоплазме образцы дают поло¬
су в 502-520 пн, в зависимости от вида микоплазмы
(рис. 19.2)
3) При контаминации реагентов специфической ДНК
микоплазмы или ПЦР-продуктом появляется
полоса в контроле воды и/или в образце с отри¬
цательным контролем. Слабые специфичные для
микоплазмы полосы дают инфицированные куль¬
туры клеток, обработанные антимикоплазменными
агентами или при постоянном применении других
антибиотиков. В этих случаях положительную
Рис. 19.2. Детекция микоплазмы с помощью ПЦР. Флюо¬
ресценция бромистого этидия ПЦР-продуктов инфициро¬
ванных, неинфицированных и контрольных клеток после
электрофореза в 1,3%-й агарозе. ДНК-стандарты: 100, 200,
300,400,500 (сильно окрашенная полоса), 600,700,800,900,
1031, 1200, 1500, 2000 и 3000 пн. Полосы микоплазмы око¬
ло 510 пн. Полоса внутреннего контроля — почти 1000 пн.
(С любезного разрешения Cord Uphoff, DSMZ.)
19.4. Устранение контаминации
361
реакцию дает как остаточная ДНК в культураль¬
ной среде из мертвых клеток микоплазмы, так и
устойчивые к антибиотикам клетки микоплазмы с
очень низким титром.
19.3.5. Альтернативные методы детекции
микоплазмы
Биохимические. Для детекции микоплазменной ин¬
фекции могут быть использованы методы определения
специфичных для микоплазмы ферментов, таких как
аргининдезаминаза или нуклеозидфосфорилаза [см.
Schneider & Stanbridge, 1975; Levine & Becker, 1977].
На этом основан тест по определению токсичности
6-метилпуриндезоксирибозида (Mycotect, Invitrogen).
Микробиологический. Этот очень чувствительный
метод широко распространен при проведении конт¬
роля качества и процедур валидации. Однако он может
быть использован только при наличии специального
оборудования и соответствующей экспериментальной
базы в микробиологии, так как эти микроорганизмы
чрезвычайно привередливы. Кроме того, необходи¬
мо культивировать живую микоплазму в качестве
положительного контроля. Культивируемые клетки
высеваются в питательный бульон для микоплазмы
[Doyle et al., 1990], выращивают 6 дней, после чего
их высевают в специальный питательный агар [Нау,
2000]. Колонии, образовавшиеся через 8 дней, можно
распознать по размеру (-200 мкм в диаметре) и ха¬
рактерной морфологии «яичницы-глазуньи», — плот¬
ный центр с более светлой периферией (рис. 19.1г).
Коммерческие наборы для микробиологического
определения микоплазмы (Mycotrim) можно купить
в «Irvine Scientific».
Хотя культивирование в селективных условиях
и исследование морфологии колоний не дают воз¬
можности идентифицировать виды микоплазмы,
микробиологический метод занимает много времени
и более трудоемок по сравнению с флюоресцентным
окрашиванием. Тестирование микоплазмы в микроби¬
ологической культуре является полезным методом (см.
Приложение И). Специфические моноклональные ан¬
титела в настоящее время позволяют характеризовать
микоплазменную инфекцию.
Молекулярная гибридизация. Молекулярные
пробы, специфичные для ДНК микоплазмы, можно
использовать в Соузерн-блот анализе для детекции ин¬
фекции с помощью стандартного метода молекулярной
гибридизации. Доступен набор для простого односта¬
дийного выделения гибридной ДНК с последующей
детекцией с помощью измерения сцинтилляционным
методом (GenProbe, Fisher Scientific).
Включение [ Н]-тимидина. Одним из методов,
который чрезвычайно успешно используется, является
авторадиография с использованием [ Н]-тимидина
[Nardone et al., 1965]. Культуру инкубируют в течение
ночи с 4 КБк/мл (-0,1 мкКи/мл) [ Н]-тимидина с вы¬
сокой удельной активностью. Далее получают автограф
(см. протокол 27.3). Зерна над цитоплазмой являются
индикатором контаминации (см. рис. 27.2), которая
может сопровождаться снижением включения метки
в ядро из-за поглощения тимидина микоплазмой на
клеточной поверхности.
19.3.6. Вирусная контаминация
Привнесенные клетки, продукты естественного
происхождения, присутствующие в среде, такие как
сыворотка, и ферменты, такие как трипсин, исполь¬
зуемые для субкультивирования, являются потен¬
циальными источниками вирусной контаминации.
Ряд реагентов проходят проверку у производителя
на присутствие ограниченного количества вирусов.
Также предполагают, что крупные вирусы задержи¬
ваются при фильтрации. Но в настоящее время нет
способов избавиться от вирусной контаминации. Луч¬
шим способом избежать ее является гарантия в том,
что продукт получен от не зараженных известными
вирусными заболеваниями животных. Для этого вам
необходимо полагаться на контроль качества выпол¬
ненный поставщиком.
Детекция вирусной контаминации. По-видимому,
лучшим способом определения вирусной инфекции
является проверка с использованием панели антител
методом иммуноокрашивания (см. протокол 16.11)
или ИФА. Альтернативным методом является ПЦР
с подбором соответствующих вирусных праймеров.
Некоторые коммерческие фирмы (BioReliance) пред¬
лагают услугу проверки или тестирования на вирусы.
19.4. УСТРАНЕНИЕ КОНТАМИНАЦИИ
19.4.1. Бактерии, грибы и дрожжи
Наиболее надежный способ избавиться от микробной
контаминации — это выбросить культуру и использо¬
вавшиеся среду и реагенты, так как лечение культуры
может быть неэффективным и привести к развитию
резистентных к антибиотикам микроорганизмов. По¬
пытки деконтаминации должны проводиться только в
экстремальных ситуациях, в карантине и под руковод¬
ством эксперта. В случае неудачи культуру и использу¬
емые реагенты следует проавтоклавировать сразу после
того, как неудача станет очевидной.
Общим правилом, которое должно выполняться
для контаминированных культур, является отбра¬
ковка этих культур. Деконтаминацию следует прово¬
дить только в случае жизненной необходимости для
сохранения линии клеток. В любом случае, полной
деконтаминации трудно добиться, особенно при за¬
ражении дрожжами, и при попытках достичь этого
362
ГЛАВА 19. КОНТАМИНАЦИЯ
можно получить более устойчивые, резистентные к
антибиотикам штаммы.
19.4.2. Устранение микоплазмы
Если в культуре обнаружена микоплазма, выполняется
первое и главное правило для всех форм контамина¬
ции — культура должна быть отбракована, проавто-
клавирована или сожжена. В исключительных случаях
(например, если контаминированная линия незаме¬
нима) можно попытаться провести деконтаминацию
культуры. Деконтаминацию должен проводить только
опытный оператор, и работа должна выполняться в
условиях карантина.
Против микоплазмы эффективны некоторые агенты,
к числу которых относятся канамицин, гентамицин,
тилозин [Friend et al., 1966], полианетол сульфонат
[Mardh, 1975] и 5-бромурацил в сочетании с Hoechst
33258 и облучением ультрафиолетом [Marcus et al.,
1980]. В некоторых случаях могут быть эффективны ко-
культивирование клеток с макрофагами [Schimmelpfeng
et al., 1968], пассирование в животных [Van Diggelen
et al., 1977] и введение цитотоксических антител [Pol¬
lock & Kenny, 1963]. Однако наиболее эффективными
агентами являются тилозин (MP Biomedicals), Агент
для удаления микоплазмы (Mycoplasma Removal Agent,
MR А, также производства MP Biomedicals) [Drexler et
al., 1994], ципрофлоксацин (Bayer) [Mowles, 1988; Hlu-
binova et al., 1994] и ВМ-циклин (BM-Cycline, Roche).
Контаминированные культуры необходимо обраба¬
тывать в соответствии с протоколом 19.3 с использова¬
нием MRA, ВМ-циклина или тилозина в концентраци¬
ях, рекомендованных фирмой-производителем вместо
обычных антибиотиков в DBSS и культуральной среде.
Однако эти операции необходимо проводить только в
случае крайней необходимости, и даже при этом выпол¬
нять их должен опытный специалист в изолированном
месте работы [Uphoff & Drexler 2004]. Гораздо безопас¬
ней уничтожить инфицированные культуры.
• Среда с высоким содержанием антибиотика (см.
Приложение I: Ассортимент сред и табл. 9.4; для
микоплазмы см. разд. 19.4.2)
• Материалы для субкультивирования (см. про¬
токол 13.2)
Нестерильные:
• Микроскоп, предпочтительно с фазово-контраст-
ными объективами 40х и ЮОх
• Материалы для окрашивания микоплазмы (см.
протокол 19.2)
Протокол
1. Аккуратно отберите контаминированную среду.
Если возможно, протестируйте микроорганизмы
на чувствительность к набору индивидуальных
антибиотиков. Если нет — проавтоклавируйте
среду или добавьте гипохлорит (см. разд. 7.8.5).
2. Промойте клетки DBSS (разведение может сни¬
зить количество контаминантов на два порядка
после каждой промывки, если только они не
прикрепились к клеткам):
а) Для монослойных культур: промойте три раза
DBSS, трипсинизируйте и промойте клетки
дважды или более DBSS путем центрифуги¬
рования и ресуспендирования.
б) Для суспензионных культур: промойте пять
раз DBSS путем центрифугирования и ресус¬
пендирования.
3. Посейте клетки в новый флакон в самой низкой
допустимой концентрации.
4. Добавьте среду с высоким содержанием антиби¬
отика и меняйте ее каждые 2 дня.
5. Пассируйте клетки в среде с высоким содержа¬
нием антибиотика.
6. Повторяйте этапы 1-4 в течение трех пассажей.
7. Удалите антибиотики и культивируйте клетки
без них следующие три пассажа.
8. Проверьте культуры с помощью фазово-контраст-
ной микроскопии и окрашивания Hoechst (см.
протокол 19.2).
9. Культивируйте клетки в течение следующих двух
месяцев без антибиотиков, проверяйте клетки
для того, чтобы быть уверенным в элиминации
контаминации (см. разд. 11.6.2).
19.4.3. Устранение вирусной контаминации
Надежных методов элиминации вирусов из культуры
в настоящее время не существует; остается только либо
уничтожить культуру либо смириться с наличием
вирусов.
19.4.4. Персистирующая контаминация
Многие лаборатории страдают от периодической конта¬
минации, которая производит впечатление устойчивой
ПРОТОКОЛ 19.4. Ликвидация микробной
контаминации
Схема
Обработать культуру несколько раз антибиотиками
в высокой концентрации, промывая монослой или
центрифугируя и ресуспендируя суспензионные
клетки. Затем культивировать три пассажа с анти¬
биотиками и три пассажа без. Проверять на конта¬
минацию после каждого пассажа
Материалы
Стерильные:
• DBSS (см. Приложение I: BSS для препариро¬
вания)
19.6. Заключение
363
ко всем средствам защиты, предложенным в табл. 19.1.
Простого решения этой проблемы нет, кроме как сле¬
довать указанным ранее рекомендациям с использова¬
нием логических и аналитических методов, обращать
особое внимание на изменения в методиках, работу
новых сотрудников, новых поставщиков, новое обору¬
дование и неправильное обслуживание ламинарных
шкафов или другого оборудования. Обычно уровень
контаминации повышается при нарушении асептичес¬
кой техники, увеличении количества спор в воздухе,
плохом обслуживании инкубаторов, контаминации в
холодильнике или холодной комнате или дефекты в
работе стерилизационного шкафа или автоклава (либо
неправильная закладка стерилизуемых предметов, либо
нарушение контроля стерилизационного цикла)
Постоянное использование антибиотиков также
способствует развитию хронической контаминации.
Антибиотики ингибируют размножение многих мик¬
роорганизмов, но не убивают их. Микроорганизмы
будут сохраняться в культуре, оставаясь необнару¬
женными в течение долгого времени, но периодически,
при смене условий культивирования, проявляться.
Необходимо культивировать клетки в среде без ан¬
тибиотиков, по крайней мере, хотя бы часть времени
(см. рис. 13.6), а желательно постоянно; в противном
случае скрытая контаминация будет персистировать,
ее происхождение будет трудно установить и изба¬
виться от нее будет невозможно.
Небольшие изменения в методиках, появление
новых сотрудников или увеличение объема работ,
сопровождающееся работой на одном оборудовании
и с одними и теми же материалами большего числа
сотрудников, — все это может влиять на повышение
уровня контаминации. Процедуры необходимо всегда
строго выполнять, даже если их значение не всегда
ясно для оператора и изменения в рутинной работе не
должны происходить по небрежности. Если методики
строго выполняются, от контаминации, может быть,
и не удастся полностью избавиться, но вероятность
ее появления будет снижена и контаминация будет
быстро обнаружена.
19.5. ПЕРЕКРЕСТНАЯ КОНТАМИНАЦИЯ
С развитием методов культивирования клеток поя¬
вились линии клеток с коротким временем удвоения
популяции и высокой эффективностью посева. Хотя
эти свойства делают такие клеточные линии удобными
для экспериментальной работы, они также создают
потенциальную опасность для перекрестного инфи¬
цирования такими клетками других клеточных линий
[Nelson-Rees & Flandermeyer, 1997; Lavappa, 1978; Nel-
son-Rees et al., 1981; Kneuchel & Masters, 1999; Dirks et
al., 1999; MacLeod et al., 1999; van Bokhoven et al., 2001a,
b; Rush et al., 2002; Milanesi et al., 2003; Drexler et al.,
2003] (см. также табл. 13.2). Распространенная пере¬
крестная контаминация многих линий клеток HeLa
и другими быстро растущими клетками в настоящее
время точно установлена [Stacey et al., 2000; MacLeod
et al., 1999], но многие операторы еще не осведомлены
о наличии серьезного риска (см. также разд. 7.10.1,
16.1,16.3,16.6.2; табл. 13.2). Руководитель несет ответ¬
ственность за то, чтобы новые сотрудники осознавали
степень опасности. Редакторы журналов и рецензенты
рукописей и организации, предоставляющие гранты,
прежде чем принимать рукописи или предложения на
выполнение гранта, должны требовать доказательства
аутентификации линий клеток. Если не принять эти
условия, ситуация будет только ухудшаться.
Следующие методики помогают избежать перекрест¬
ной контаминации:
1) Покупать линии клеток в банках клеток с хорошей
репутацией, в которых проводится надлежащая
валидация линий клеток (см. разд. 7.10, 20.2; При¬
ложение И: Банки клеток), или проводить необхо¬
димую аутенфикацию самостоятельно как можно
быстрее (см. разд. 16.3).
2) Не открывать одновременно культуральные флако¬
ны, содержащие разные линии клеток, не открывать
одновременно бутыли со средой для этих клеток.
3) Работать с быстро растущими линиями, такими
как HeLa, отдельно и после работы с другими
культурами.
4) Никогда не использовать одну и ту же пипетку для
разных линий клеток.
5) Никогда не использовать одну и ту же бутыль со сре¬
дой, трипсином и т.д. для разных линий клеток.
6) Не вносить пипетку назад в бутыль со средой,
трипсином и т. д. после того, как она побывала в
культуральном флаконе с клетками.
7) Вначале вносить в культуральный флакон среду и
другие реагенты, и только потом клетки.
8) Не использовать вскрытые пипетки или автома¬
тические пипетки без сменных наконечников для
рутинных работ.
9) Регулярно проверять характеристики культуры и
следить за неожиданными изменениями морфоло¬
гии, скорости роста или другими фенотипическими
свойствами. Перекрестная контаминация или ее
отсутствие могут быть подтверждены с помощью
геномной идентификации (см. протокол 16.8) [Sta¬
cey et al., 1992], определения ДНК-профиля (см.
протокол 16.9), кариотипирования [Nelson-Rees &
Flandermeyer, 1977] (см. разд. 16.5; протоколы 16.7,
27.5,27.9) или изоферментного анализа [O'Brien et
al., 1980] (см. протокол 16.10).
Необходимо постоянно учитывать, что перекрестная
контаминация может произойти и происходит. Нужно
принимать предупредительные меры и регулярно про¬
верять характеристики линии.
19.6. ЗАКЛЮЧЕНИЕ
• Регулярно проверяйте живые культуры на наличие
контаминации с использованием световой и фазо¬
во-контрастной микроскопии, а наличие микоплаз¬
364
ГЛАВА 19. КОНТАМИНАЦИЯ
мы — с помощью флюоресцентного окрашивания
фиксированных препаратов или ПЦР.
• Не добавляйте при рутинных работах всем культу¬
рам антибиотики. Культивируйте по крайней мере
по одному образцу каждой клеточной линии без
антибиотиков не менее двух недель и, желательно,
дольше для того, чтобы проявилась возможная
скрытая контаминация.
• Не делайте попыток деконтаминировать культуру,
за исключением, если эта культура является незаме¬
нимой, и в этом случае проводите деконтаминацию
в условиях строгого карантина.
• Помещайте в карантин все новые линии клеток,
попавшие в вашу лабораторию, до тех пор, пока не
убедитесь в отсутствии контаминации.
• Не пользуйтесь общими средами или другими
растворами для линий клеток или операторов,
периодически проверяйте характеристики клеток
(см. разд. 16.3) для предотвращения перекрестной
контаминации.
• Новые клеточные линии должны быть охаракте¬
ризованы, желательно, с помощью геномной иден¬
тификации или определения ДНК-профиля сразу
после выделения.
Глава 20
КРИОКОНСЕРВАЦИЯ
Поскольку в лаборатории ведется работа с клеточными
культурами клеток, ряд клеточных линий поддержи¬
вается или приобретается при необходимости, если
только в лаборатории не занимаются исключительно
первичными культурами. Различные характеристики
клеточной линии, выявленные при ее исследовании,
добавляются в описание ее происхождения, при этом
каждая информация является ценным материалом.
Будучи уникальной, клеточная линия не может быть
воспроизведена или замещена. В лучшем случае заме¬
щение является дорогостоящей и требующей времени
процедурой. Таким образом, существенным моментом
работы в лаборатории культуры клеток является пре¬
дупреждение потери ценных клеток путем консервации
клеточных линий.
20.1. ПРЕДПОСЫЛКИ МЕТОДА
ЗАМОРАЖИВАНИЯ
Клеточная линия при постоянном культивировании
склонна к вариациям в результате селекции в культуре
ранних пассажей (см. разд. 3.8), старения в конечных
клеточных линиях (см. разд. 3.8.1, 18.4.1), а также
генетической и фенотипической нестабильности (см.
разд. 16.1,18.3) постоянных клеточных линий. Кроме
того, даже в очень хорошо работающей лаборатории
возможна поломка оборудование и контаминация. Пе¬
рекрестная контаминация и неправильная идентифи¬
кация (см. разд. 7.10.1,16.1,16.3,16.6.2,19.5; табл. 13.2)
также постоянно происходят с достаточно высокой
частотой. Таким образом, существует много причин, по
которым требуется замораживание надежного запаса
клеток. Суммируя, можно назвать следующие:
1) Генотипический сдвиг в результате генетической
нестабильности.
2) Старение и проистекающее из этого угасание кле¬
точной линии.
3) Изменение характеристик роста и приобретение
свойств, связанных с малигнизацией.
4) Фенотипическая нестабильность в результате се¬
лекции и дедифференцировки.
5) Контаминация микроорганизмами.
6) Перекрестная контаминация другими линиями.
*7) Неправильная идентификация в результате небреж¬
ной работы.
8) Поломка инкубатора.
9) Экономия времени и материалов, необходимых
для поддержания клеточной линии в период, пока
культура не используется.
10) Необходимость распределения между различными
пользователями.
20.2. ПОЛУЧЕНИЕ КЛЕТОЧНЫХ ЛИНИЙ
ДЛЯ КРИОКОНСЕРВАЦИИ
Существуют определенные требования, которые сле¬
дует выполнить до того, как клеточная линия будет
подготовлена для криоконсервации (табл. 20.1).
Валидация. До криоконсервации должно быть по¬
казано, что клеточная линия свободна от контаминации
(см. разд. 19.3) и аутентична (см. разд. 16.3). Несколь¬
ко ампул вновь полученной клеточной линии может
быть заморожено до завершения процесса валидации
(см. разд. 20.4), но собственно валидация должна быть
проведена до того, как будет замораживаться основной
запас (см. разд. 7.10) или основная масса клеток.
При замораживании. В случае конечной кле¬
точной линии для того, чтобы получить количество
клеток, достаточное для замораживания, культуру
растят примерно до пятого удвоения популяции. Пос¬
тоянные клеточные линии должны быть клонированы
(см. разд. 14.1, 14.3, 14.4), охарактеризованные клоны
отобраны и выращены до концентрации клеток, доста¬
точной для замораживания. Постоянные линии имеют
определенные преимущества; они сохраняют жизне¬
способность неопределенно долго, растут быстрее и
легче могут быть клонированы, однако они обладают
меньшей генетической стабильностью. Конечные кле¬
точные линии обычно диплоидны или близки к этому, и
существует стабильность линии между определенными
пассажами, однако их труднее клонировать, и они могут
случайно погибнуть или трансформироваться.
20.3. ПРИНЦИПЫ КРИОКОНСЕРВАЦИИ
20.3.1. Теоретическое обоснование
замораживания клеток
Оптимальное замораживание клеток для сохранения
максимальной жизнеспособности при восстановле¬
нии после размораживания зависит от минимизации
366
ГЛАВА 20. КРИОКОНСЕРВАЦИЯ
Таблица 20.1. Требования к культуре перед замораживанием
Приобретение Конечная клеточная линия
Постоянная клеточная линия
Стандартизация Среда
Сыворотка (если используется)
Субстрат
Валидация Происхождение
Аутентификация
Т рансформация
Контаминация
Кросс-контаминация и ошибоч-
ная идентификации
Замораживание при ранних пассажах (< 5 субкультур)
Клонирование, селекция, и характеристика; амплификация
Выбор оптимальной среды, и твердо придерживаться этой среды
Выбор партии для использования на всех стадиях (см. разд. 9.6)
Стандартизируют по одному типу и поставщику, хотя необязательно по
размеру или конфигурации
Записи деталей происхождения, истории жизни, и свойств
Проверка характеристик клеточной линии в сравнении с источником и
происхождением (см. разд. 16.1,16.3)
Определение трансформированного состояния (см. табл. 18.1)
Микробиологическое исследование (см. разд. 19.3,11.6.2)
Микоплазмы (см. протоколы 19.2,19.3) Критерии подтверждения иден-
тичности (см. разд. 16.3)
образования внутриклеточных кристаллов льда, а
также предотвращения образования очагов высокой
концентрации солей, возникающих при заморажива¬
нии внутриклеточной воды. Это достигается: а) путем
медленного замораживания, так чтобы позволить воде
покинуть клетку, но не настолько медленно, чтобы
спровоцировать рост ледяных кристаллов; б) путем
использования гидрофильных криопротекторов для
уменьшения содержания воды; в) путем хранения
клеток при самой низкой температуры, какую только
возможно достигнуть, для того чтобы минимизировать
действие высокой концентрации солей на денатурацию
белков в мицеллах внутри льда и г) путем быстрого
размораживания для того, чтобы минимизировать
рост кристаллов льда и появление градиента солей,
образующегося в процессе таяния остаточного внут¬
риклеточного льда.
20.3.2. Концентрация клеток
Показано, что выживание клеток повышается при их
замораживании при высокой концентрации клеток. Это
в основном эмпирическое наблюдение, но, возможно,
связано, в частности, с тем, что уменьшение жизнеспо¬
собности при размораживании требует более высокую
посевную концентрацию; уменьшение жизнеспособ¬
ности может объясняться тем, что при криогенном
повреждении содержимое клетки вытекает. Высокая
концентрация клеток при замораживании также обес¬
печивает достаточное разведение криопротекторов при
размораживании и последующем посеве, позволяющем
избежать центрифугирования (по крайней мере, для
большинства клеток). Количество замороженных кле¬
ток должно быть достаточным для того, чтобы обеспе¬
чить достаточно высокую их посевную концентрацию
при разведении 1:10 или 1:20 при размораживании,
позволяющем развести криопротектор до низких кон¬
центраций. Например, при нормальном субкультиви¬
ровании используется концентрация клеток 1х105/мл,
поэтому при замораживании следует иметь концентра¬
цию клеток 1х107/ш1 среды. После размораживания
клеток 1 мл суспензии следует развести добавлением
среды до 20 мл, что даст концентрацию 5x105 кл./мл
(в пять раз выше обычной посевной концентрации).
Это разведение снизит концентрацию криопротекто¬
ра с 10% до 0,5%; последняя с меньшей вероятностью
будет токсична для клеток. Остаточные количества
криопротектора могут быть разведены при дальнейшем
пересеве, как только культура начнет расти (в случае
суспензионной культуры) или при замене среды после
того, как только клетки прикрепятся к субстрату (для
монослойной культуры).
20.3.3. Среда для замораживания
Клеточную суспензию замораживают в присутствии
криопротекторов, таких как глицерин или диме-
тилсульфоксид (DMSO) [Lovelock & Bishop, 1959].
Показано, что из двух этих криопротекторов более
эффективным является DMSO, возможно потому, что
он проникает в клетки лучше, чем глицерин. Обычно
используются концентрации между 5% и 15%, чаще
всего 7,5% или 10%. Бывают ситуации, при которых
DMSO может являться токсичным или индуцировать
дифференцировку клеток (см. разд. 17.7.2), например
в случае гематопоэтических клеточных линий L5178Y
или HL60 (М. Freshney, личное сообщение). В этих
случаях предпочтительно использовать глицерин или
центрифугировать клетки после размораживания для
удаления криопротектора.
Есть мнение, что клетки должны храниться при 4 °С
после добавления DMSO в среду для замораживания,
однако предварительные эксперименты, проведенные
автором, позволяют предположить, что эта процедура
не повышает выживание клеток после размораживания
и может даже снизить ее (неопубликованные данные),
возможно, в результате проникновения среды внутрь
клеток. Тем не менее этот вопрос еще недостаточно
решен и требует дальнейших экспериментальных ис¬
следований (см. гл. 2, упражнение 17).
DMSO должен быть бесцветным, и его следует
хранить в стеклянном или пропиленовом флаконе,
поскольку он является сильным растворителем и
выщелачивает примеси из резины и некоторых пласт¬
20.3. Принципы криоконсервации
367
масс. Глицерин должен быть не старее одного года,
при более продолжительном хранении он может стать
токсичным.
Возможно использование также других криопро¬
текторов, таких как поливинилпирролидон (PVP) [Su¬
zuki et al., 1995], полиэтиленгликоль (PEG) [Monroy et
al., 1997] и гидроксилэтиловый крахмал (HES) [Pasch
et al., 2000], но общего признания эти криопротекторы
не получили, хотя показано некоторое повышение
выживания при использовании трегалозы [Eroglu et
al., 2000; Buchanan et al., 2004]. Многие лаборатории
также повышают концентрацию сыворотки в среды для
замораживания до 40%, 50% или даже до 100%.
20.3.4. Скорость охлаждения
Большинство культивированных клеток лучше выжи¬
вают, если они охлаждаются со скоростью 1 °С/мин
[Leibo & Mazur, 1971; Harris & Griffiths, 1977]. Воз¬
можно, эта скорость является компромиссом между
быстрым замораживанием, минимизирующим рост
кристаллов, и медленным охлаждением, способс¬
твующим миграции воды из клеток. Вид кривой
охлаждения определяется: а) температурой окружа¬
ющей среды; б) наличием изоляционного материала
вокруг клеток, включая ампулу; в) специфической
теплоемкостью и объемом содержимого ампулы и
г) поглощением теплоты фазового перехода. Когда
клетки замораживают в теплоизолирующем контей¬
нере, помещенном в сверхнизкую температуру замо¬
раживания, как описано в п. (1)-(4), это приводит к
быстрому снижению температуры в начале кривой,
когда перепад температур больше всего, до минимума
в эвтектической точке, некоторому подъему в начале
замерзания, последующему плато, отражающем ла¬
тентное поглощение теплоты фазового перехода, и
затем к более быстрому падению температуры после
окончания замерзания, постепенно достигающей тем¬
пературы замораживающей камеры (рис. 20.1).
Когда клетки помещены в морозильник с жидким
азотом (или на конечной стадии при замораживании в
программируемом морозильнике), температура быстро
падает до -180 °С и -196 °С. Контролируемая скорость
охлаждения может быть достигнута несколькими
способами:
1) Поместить ампулы на вату в пенополистироловую
коробку со стенами толщиной примерно 15 мм. Эта
коробка плюс вата должны обеспечить достаточно
эффективную изоляцию, при которой ампулы будут
охлаждаться со скоростью 1 °С/мин, если коробка
находится при температуре -70 °С или -90 °С в
обычном низкотемпературном морозильнике или
изолированном контейнере с твердым С02.
2) Поместить стержневый держатель для ампул в пено¬
изолируемую трубку со стенками толщиной около
15 мм (рис. 20.2), и поставить их в обычный низко¬
температурный морозильник при -70 °С или -90 °С
или изолированный контейнер с твердым С02.
Минуты
Рис. 20.1. Кривая замораживания. Регистрация падения
температуры в ампуле, содержащей среду, закрепленной
вместе с другими пятью ампулами на алюминиевом стержне,
заключенной в гильзу и помещенной в пенополиуретановую
трубку (см. рис. 20.2) и затем в морозильник при -70 °С
3) Поместить ампулы в узкогорлый морозильник Tay¬
lor Wharton с пробкой (рис. 20.3) и закрыть пробкой
горло морозильника.
4) Поместить ампулы в замораживающий контейнер
Nalge Nunc (рис. 20.4) при температуре -70 °С или
-80 °С.
5) Использовать морозильник с программируемой
скоростью замораживания. 1 °С/мин (рис. 20.5), с
ускоренным замораживанием после достижения
эвтектической точки (см. рис. 20.1).
При использовании любого из первых четырех
методов, например метода изолированной трубки (2)
(см. рис. 20.2), скорость охлаждения будет результатом
среднего арифметического от различных скоростей
на протяжении кривой. При этом не учитывается ги-
Рис. 20.2. Ампулы на стержне. Пластиковые ампулы закреп¬
лены на алюминиевом стержне (внизу), заключены в гильзу
(в середине) и помещены в пенополиуретановую трубку
(в верхней части фото). Теплоизолированная трубка заткнута
с обоих концов хлопковой ватой или другим подходящим
изолирующим материалом
368
ГЛАВА 20. КРИОКОНСЕРВАЦИЯ
морозильника
Поверхность I
жидкого азота I
Рис. 20.3. Пробка для узкогорлого морозильника. Моди¬
фицированная пробка для узкогорлого морозильника, поз¬
воляющая проводить контролируемое охлаждение с разной
скоростью (Taylor Wharton). Показано в разрезе горлышко
морозильника с модифицированной пробкой внутри. Уплот¬
нительное кольцо обычно используется, чтобы поместить
верх ампул в горловой части морозильника. Чем ниже опус¬
каются ампулы, тем быстрее замораживание
перохлаждение в эвтектической точке или удлинение
плато, которое отражает латентное поглощение теп¬
лоты фазового перехода, что может улучшить выжи¬
ваемость клеток. Если выживаемость клеток низкая,
можно изменить среднюю скорость замораживания
(например, изменяя толщину слоя изоляции). Исполь¬
зуйте программируемый морозильник (см. рис. 20.5)
с зондом, определяющим температуру в ампуле, и с
добавлением жидкого азота в морозильную камеру с
необходимой частотой, чтобы обеспечить предвари¬
тельно определенную скорость охлаждения и быстрое
охлаждение после достижения эвтектической точки.
Различные темпы охлаждения в разных фазах кривой
Рис. 20.4. Охлаждающее устройство Nalge Nunc Cooler.
Пластиковый держатель с основанием, заполняемым ох¬
лаждающейся жидкостью. Специфический тепловой эффект
охлаждающего агента в основании изолирует контейнер и
дает скорость охлаждения содержимого ампул примерно
-1°С/мин
Рис. 20.5. Программируемый морозильник. Ампулы по¬
мещаются в изолированную камеру, скорость охлаждения
регулируется впрыскиванием жидкого азота в камеру в ре¬
жиме, определяемом чувствительным датчиком на решетке
с ампулами и предварительно выбранной в пульте управ¬
ления программой (Planer Biomed). (а) Контрольный блок
и морозильная камера (крышка открыта). (б) Увеличенное
изображение замораживающей камеры с четырьмя ампулами,
одна из которых содержит датчик
охлаждения, экспериментально оптимизированные для
конкретных клеток, могут быть запрограммированы
путем внесения соответствующих изменений в кривую.
Программируемое устройство для охлаждения и замо¬
раживания, однако, достаточно дорого по сравнению с
простым оборудованием, описанным выше (1)-(4), и
имеет не очень много преимуществ, если только вы не
предполагаете варьировать скорость охлаждения [см.
Foreman & Pegg, 1979] или изменять вид кривой ох¬
лаждения. При использовании методов изолированных
контейнеров скорость охлаждения пропорциональна
различию в температуре между ампулой и окружа¬
20.3. Принципы криоконсервации
369
ющей атмосферой. Если ампулы помещены в моро¬
зильник при -70 °С, они будут охлаждаться быстро
до температуры -50 °С, но затем скорость охлаждения
значительно снизится (см. рис. 20.1). Таким образом,
время, которое ампулы проводят при -70 °С, должно
быть дольше, чем количество времени, предварительно
предполагаемое для скорости охлаждения 1 °С/мин,
поскольку нижняя часть кривой асимптотична. Безо¬
паснее оставить ампулы при — 70 °С на ночь до того,
как вы перенесете их в жидкий азот. Более того, при
перенесении из замораживающего устройства ампулы
будут нагреваться со скоростью примерно 10 °С/мин.
Критичным является нагревание до -50 °С, поскольку
затем начинается ухудшение состояния клеток, по¬
этому перенос ампул в жидкий азот должен произво¬
диться меньше чем за две минуты.
20.3.5. Морозильные камеры
Хранение в морозильниках с жидким азотом (рис. 20.6,
20.7; см. также разд. 5.5.2) является наиболее удовлет¬
ворительным методом сохранения культивируемых
клеток [Hay et al., 2000]. Замороженные клетки быстро
переносят в морозильную камеру, где они находятся
при температуре -70 °С и ниже. Конструкции моро¬
зильных камер различны по размерам горлышка, через
который осуществляется доступ, по используемой
системе хранения и расположению жидкого азота
(рис. 20.7).
Размеры горлышка. Баллонные системы хранения
обычно имеют узкое горлышко (рис. 20.6а, б, 20.7а),
что позволяет уменьшить скорость испарения жид¬
кого азота, но делает доступ к материалам достаточно
затрудненным. Широкогорлые морозильные камеры
выбираются для удобства доступа и максимального
использования объема, обычно для хранения в раз¬
личных отсеках, однако в этом случае происходит
более быстрое испарение жидкого азота. Тем не менее
можно выбрать относительно узкогорлые морозильные
камеры, использующие систему кассет и отсеков для
хранения. Последние удобнее для контроля со стороны
исследователей, поскольку существует система квад¬
ратных областей, предназначенных для размещения
кассет и лотков, которые располагаются на штативах,
доступ к которым осуществляется аналогично той
системе, что имеется в узкогорлых морозильниках
(рис. 20.6г, 20.76). Эти морозильники обладают умерен¬
ной вместительностью, но удобство в использовании
и сотовая структура расположения кассет делают их
использование предпочтительным для многих иссле¬
дователей (см. ниже).
Система хранения. Имеется два основных типа
хранения, которые используются для 1-мл ампул при
работе с клеточными культурами. Система стержне¬
вых штативов, основанная на опыте хранении спер¬
мы, использует ампулы, прикрепленные на длинных
алюминиевых стержнях, заключенных в картонную
трубку и затем помещенных в цилиндрическую кассету
в морозильной камере. Преимуществом этого метода
является возможность вынуть один набор из шести
ампул, в то время как остальные ампулы останутся в
камере, возможно также вынуть все ампулы с одной
клеточной линией, что ускоряет и упрощает перенос их
из замораживающего устройства в морозильную каме¬
ру на хранение. Штативы могут быть промаркированы
и окрашены, что облегчает их поиск и позволяет изъять
необходимые ампулы, не трогая другие ампулы.
Некоторые исследователи предпочитают исполь¬
зование прямоугольных ящиков, считая такой путь
распределения и поиска ампул более легким. В этом
случае отдельная ампула может быть идентифициро¬
вана по номеру ящика и координатам внутри ящика.
В данном случае, однако, имеет значение размер ящика,
в котором может располагаться от 20 до 100 ампул,
которые одномоментно вынимаются из морозильной
камеры. Кроме того, при необходимости изъятия
нижних отсеков вам потребуется вынимать весь блок.
Загрузка большого количества ампул в ящик должна
производиться поочередно — по одной ампуле за один
раз, в результате чего имеется большой риск длитель¬
ного перегрева хранящихся клеток.
Ампулы. Для экспериментальной работы и в учеб¬
ной лаборатории предпочтительны пластиковые ам¬
пулы, поскольку работа с ними безопаснее и удобнее,
однако в отдельных случаях в хранилищах и клеточных
банках предпочитают работать со стеклянными ампу¬
лами, обеспечивающими более длительное хранение.
При использовании запаянных с соблюдением тре¬
бований герметичности ампул обеспечивается абсо¬
лютная герметичность. Пластиковые ампулы, обычно
полипропиленовые, имеют наиболее общепринятый
объем 1,2 мл (см. цв. вкладку 22Э). Они могут быть
отмечены маркерами или этикетками, которые должны
быть этанол-устойчивы и способны выдержать низкую
температуру морозильника (см. Приложение II, Крио¬
маркеры и Крио-этикетки). Этикетки должны содер¬
жать обозначение клеточного штамма и, желательно,
дату и инициалы исследователя, хотя последние не
всегда могут уместиться на имеющемся пространстве.
Различный цвет колпачков также помогает идентифи¬
цировать ампулы (см. цв. вкладку 22Э).
Помните, что клеточные культуры, которые хра¬
нятся в жидком азоте, могут пережить вас! Они легко
могут, по крайней мере, пережить ваше пребывание в
данной лаборатории. Поэтому запись следует делать
понятной для других и достаточно подробной, чтобы
клетки могли быть использованы другими людьми.
При закручивании крышки пластиковой ампулы
требуется приложить правильно рассчитанное усилие,
поскольку при слишком сильном закручивании может
произойти деформация прокладки. Имеет смысл пот¬
ренироваться с новой партией ампул, чтобы убедиться,
что они: а) закрываются правильно и б) устойчивы к
низкой температуре. Бракованная партия ампул мо-
370
ГЛАВА 20. КРИОКОНСЕРВАЦИЯ
Рис. 20.6. Морозильники с жидким азотом, (а) Узкогорлый морозильник с подготовленными для хранения ампулами на
стержнях в кассетах, (б) Внутренность узкогорлого морозильника: внешний вид кассет, расположенных в центре, как они
должны быть расположены для удобства изъятия. Нормальное расположение хранящихся образцов — под «плечом» моро¬
зильника, как это показано справа вверху, (в) Широкогорлый морозильник с хранящимися образцами в треугольных штативах
(г). Узкогорлый морозильник с хранящимися образцами в квадратных штативах (см. также рис. 20.7). (д) Морозильник боль¬
шой емкости с открытым смещенным портом доступа показывает стержни в кассетах. (е) Морозильники большой емкости в
клеточном банке. На ближнем морозильнике заметны соединения для мониторинга и автоматического заполнения (фото (д)
и (е) любезно предоставлены АТСС)
20.3. Принципы криоконсервации
371
Алюминиевый
3 штативе
ампулами
Пробка для
горлышка
Максимальный
уровень жидкости
для жидкофазного
хранения
Максимальный
уровень жидкости
для газофазного
хранения (короткая
кассета)
Штатив для
ящиков
Пробка для
горлышка
Максимальный
уровень жидкости
для жидкофазно¬
го хранения
Максимальный
уровень жидкости
для газофазного
хранения (корот¬
кая кассета)
Кассета
Жидкий азот
из хранилища
Температурный датчик
Ручка
Вид ящика
сверху
Изолирующая
крышка
Штатив для
хранения
ящиков
Основание
Датчики высокого
и низкого уровня
Максимальный
з уровень жидкости
для жидкофазного
хранения
Максимальный
уровень жидкости
з для газофазного
хранения (короткая
кассета)
Изолирован¬
ные стенки,
перфузиро-
ванные жид¬
ким N2
Предохра¬
нительный
клапан для
выхода газо¬
образного N2
Штатив для
хранения
ящиков
Рис. 20.7. Конструкция азотного морозильника. Четыре основных типа азотных морозильников: (а) Узкогорлый с ампула¬
ми на стержнях в кассетах (большая емкость, низкая скорость выкипания азота), (б) Узкогорлый с ампулами в квадратных
штативах (средняя емкость, низкая скорость выкипания), (в) Широкогорлый с ампулами в треугольных штативах (высокая
емкость, высокая скорость выкипания), (г) Широкогорлый с хранением в ящиках, жидкий азот подводится по трубкам, раз¬
брызгивается через стенки морозильника, режим работы контролируется автоматически по сенсорным датчикам верхнего и
нижнего уровней (в верхней левой части)
жет разрушаться при размораживании. Пластиковые
ампулы большего диаметра выше, чем эквивалентные
стеклянные ампулы, поэтому могут потребоваться
специальные кассеты.
• Требование безопасности. Неопытные иссле¬
дователи должны избегать использования стеклянных
ампул, поскольку существует высокий риск взрыва
при размораживании. Если используются стеклянные
ампулы, они должны быть тщательно и быстро запаяны
в пламени газовой горелки. При слишком долгом запа¬
ивании клетки могут перегреться и погибнуть, а воздух
в ампуле расширится и прорвет дырку в ампуле. Если
ампула запаяна недостаточно герметично, она может
набрать жидкий азот при хранении в жидкой фазе
азотной камеры и впоследствии при размораживании
резко взорвется.
Можно проверить наличие герметичности ампул
путем погружения их в 1%-й раствор метиленового
синего в 70%-м этаноле при 4 °С на 10 мин до замо¬
раживания. Если ампулы недостаточно герметично
закупорены, метиленовый синий попадет в ампулу и
ампулу следует выбросить.
Местонахождение жидкого азота% Если жидкий
азот располагается в основной части морозильной
камеры, то существует возможность выбора между
372
ГЛАВА 20. КРИОКОНСЕРВАЦИЯ
полным заполнением камеры азотом с погружением
в него ампул только частичным заполнением с хра¬
нением ампул в парах азота. Хранение в жидкой фазе
предполагает, что контейнер может быть полностью
заполнен, и таким образом жидкий азот будет доль¬
ше находиться в камере. Однако в этом случае есть
риск попадания азота в ампулы, которые затем при
размораживании могут взорваться. Также в этом слу¬
чае увеличивается вероятность контаминации между
ампулами и переноса загрязнения извне, вносимого
вместе с погружением материала и концентрируемого
при испарении жидкого азота в емкости. При исполь¬
зовании хорошей изоляции и уменьшении испарения
предпочтительно использование газофазного храни¬
лища. При этом уменьшается риск расплескивания
жидкого азота при его переливании через край, когда
что-то погружается в него, а также уменьшается испа¬
рение азота и его попадание в атмосферу комнаты. Тем
не менее существует градиент температуры примерно
в 80 °С от -190 °С у поверхности жидкого азота до
-110 °С у горлышка баллона [Rowley & Byrne, 1992],
хотя конструкция и структура штативной системы
могут снизить этот градиент.
Некоторые морозильные камеры имеют структуру,
в которой жидкий азот наливается между стенками и
не попадает в камеру для хранения. Он пополняется
автоматически при контрольных значениях датчиков
низкого и высокого уровней азота (рис. 20.7г), при
этом испаряющийся азот выпускается через предох¬
ранительный выпускной клапан. Такая конструкция
имеет все преимущества газофазного хранения и, кроме
того, дополнительное преимущество более низкого
расхода жидкого азота и отсутствие температурного
градиента.
Однако уровень азота не является видимым и не
может быть измерен глубиномером, поэтому полная
уверенность в том, что поддерживается нужная тем¬
пература, может быть только при электронном мони¬
торинге (см. ниже). Кроме того, любое препятствие,
вызывающее нарушение поступления жидкого азота в
пространство между стенками, трудно, иногда даже не¬
возможно, удалить, поэтому существенным моментом
является фильтрация жидкого азота, и на этой ступени
не должно быть попадания воды или ее паров в систему,
поскольку лед может блокировать поток азота.
• Требование безопасности. Материалы, пред¬
ставляющие собой биологическую опасность, должны
храниться в газовой фазе, для обучения и демонстра¬
ции также следует использовать газофазное хранение.
Кроме того, если используется жидкофазное хранение
пользователь должен быть осведомлен об опасности
взрыва как стеклянных, так и пластиковых ампул и
должен надевать защитный щиток или очки.
Мониторинг и пополнение уровня жидкого азота.
При расчете затраты средств на азотную морозильную
камеру следует учитывать расходы на его содержание,
которые могут быть достаточно велики и должны быть
определены путем жесткого режима мониторинга и
применением электронных сигнализаторов измене¬
ния уровня азота. Когда азот находится в отсеке для
хранения, то уровень его должен контролироваться
при помощи щупа-глубиномера по крайней мере раз в
неделю с записью данных. Это следует делать также при
наличии автоматического заполнения оборудования,
поскольку эта система может отказать. Когда жидкий
азот полностью отгорожен от камеры для хранения, то
заполнение его производится автоматически, но этот
процесс должен быть также подкреплен предпочти¬
тельно двумя независимыми термосенсорными датчи¬
ками, каждый из которых может дать сигнал тревоги,
если температура на одном из них превысит -170 °С,
а на другом -150 °С.
Если морозильная камера с жидким азотом недо¬
ступна, клетки могут храниться в обычном морозиль¬
нике. Температура в этом морозильнике должна быть
настолько низкой, насколько это возможно. Неболь¬
шое ухудшение состояния клеток было обнаружено
при -196 °С [Green et al., 1967], значительное ухуд¬
шение (5-10% в год) может иметь место при хранении
при -70 °С.
ПРОТОКОЛ клеток
Схмса
Вырастить культуру, до поздней log-фазы, приго¬
товить суспензию клеток высокой концентрации
в среде с криолротектором, разлить определенное
количество в ампулы, и медленно заморозить (см.
рис. 20.8).
Материалы
Стерильные или асептически приготовленные:
• Культура» которая будет заморожена
• Вели монослой: PBSA и 0,25% неочищенного
трипсина
• Ростовая среда. (Сыворотка улучшает выжива¬
ние клеток после замораживания; используйте
до 50%, или даже чистую сыворотку. Если
сыворотка используется с бессывороточными
культурами, то следует отмыть сыворотку после
размораживания.)
• Криопротектор, свободный от примесей (см.
выше): DMSO в стеклянном или полипропиле¬
новом пузырьке или глицерин, свежий и в уни¬
версальном контейнере
• Шприц, 1-5 мл, для размешивания глицерина
(поскольку глицерин вязкий)
• Пластмассовые ампулы, 1,2 мл, предварительно
промаркированные с обозначением клеточной
линии и даты замораживания
Нестерильные:
• Гемоцитометр или электронный счетчик клеток
20.3. Принципы криоконсервации
373
• Стержни и стойки для хранения (стойки могут
уже быть размещены в морозильнике)
• Изолированный контейнер для замораживания:
пенопластовая коробка с ватой или пластмассо¬
вой пеноизоляцией. Пробирка (см. рис. 20.2; или
морозильник с контролируемой скоростью охлаж¬
дения, если он имеется в наличии, см. рис. 20.5)
• Защитные перчатки, нитрильные
Протокол
1. Удостоверьтесь, что культура удовлетворяет
критериям для замораживания (см. табл. 20.1);
проверьте невооруженным глазом и при помощи
микроскопа для определения:
а) признаков здоровой культуры (см.
разд. 13.6.1);
б) морфологических характеристик (см.
разд. 16.4);
в) стадии цикла роста (должна быть позд¬
няя log-фаза перед выходом на плато (см.
разд. 13.6.6);
г) свободы от загрязнения (см. разд. 19.3).
2. Вырастите культуру до поздней log-фазы и, если
вы используете монослойную культуру, трип¬
синизируйте и подсчитайте клетки (см. прото¬
кол 13.2). Если вы используете суспензионную
культуру, подсчитайте и центрифугируйте клет¬
ки (см. протокол 13.3).
3. ресуспендируйте до концентрации 2х106-
2х107 кл./мл.
4. Растворите один из криопротекторов в ростовой
среде, чтобы приготовить среду для заморажи¬
вания:
а) добавьте диметилсульфоксид (DMSO) до
10-20%.
• Требование безопасности. DMSO может
проникать через многие синтетический и естествен¬
ные мембраны, включая кожу и резиновые перчат¬
ки [Horita и Weber, 1964]. Следовательно, любые
потенциально вредные вещества при регулярном
использовании (например, канцерогенные вещества)
могут легко быть перенесены через кожу и даже через
резиновые перчатки. DMSO должен всегда исполь¬
зоваться с осторожностью, особенно в присутствии
любых ядовитых веществ.
Или
б) Добавьте глицерин до 20-30%.
5. Разведите клеточную суспензию 1:1 в среде для
замораживания, чтобы получить приблизительно
1х106-1х107 кл./мл и 5-10% DMSO (или 10-15%
глицерина). Не желательно помещать ампулы на
лед в попытке минимизировать ухудшение со¬
стояния клеток. Нахождение до 30-й минуты при
комнатной температура не вредно при исполь¬
зовании DMSO и полезно при использовании
глицерина.
6. Разлейте клеточную суспензию в предваритель¬
но маркированные ампулы; заверните ампулы,
прилагая адекватное усилие, избегая деформации
прокладки.
7. Поместите ампулы в держатели для хранения в
емкостях с азотом (см. рис. 20.2, 20.6а, б, 20.7а)
или оставьте их незакрепленными для хранения
в ящике (см. рис. 20.6в, г, 20.7б-г).
8. Заморозьте ампулы со скоростью 1 °С/мин одним
из методов, описанных выше (см. разд. 20.3.4).
С применением термоизолированных контей¬
неров это потребует как минимум 4-6 ч после
помещения их при -70 °С при начальной темпе¬
ратуре окружающей среды 20 °С (см. рис. 20.1 и
разд. 20.3.4). Предпочтительно быстрое помеще¬
ние ампул в контейнер при -70 °С.
9. Когда ампулы достигли -70 °С или ниже, про¬
верьте записи использования морозильника
перед удалением ампулы из морозильника на
-70 °С или морозильника с управляемой ско¬
ростью замораживания, и выберите подходящее
местоположение для ампул.
10. Переместите ампулы в морозильник с жидким
N2, предпочтительно не погружая в жидкость,
разместите держатели для ампул и пробирки в
выбранную емкость или индивидуальные ампулы
на определенные места в определенном ящике.
Это перемещение должно быть выполнено быстро
(< 2 мин), так как ампулы могут повторно нагре¬
ваться примерно со скоростью-10 °C/min, и если
температура будет выше чем -50 °С, состояние
клеток будет ухудшаться.
• Требование безопасности. При размещении
ампул в жидком азоте должны использоваться за¬
щитные перчатки и маска для лица
11. Когда ампулы надежно расположены в моро¬
зильнике, заполните соответствующие регист¬
рационные графы в регистрационном журнале
морозильника (см. табл. 20.2 и 20.3).
20.3.6. Протоколирование работы
морозильника
Записи должны обеспечивать а) учет, показывающий,
что находится в каждой части морозильника, б) ин¬
дикацию свободного пространства в морозильнике и
в) указатель клеточных штаммов, описание клеточных
линий: их обозначение, их происхождение, детали ве¬
дения культуры и процедуры замораживания, каковы
были особые характеристики этих процессов и где они
располагаются в морозильнике. Эти записи могут хра¬
ниться в обычном карточном виде либо в компьютерной
базе данных, которая даст последнюю дату хранения
и возобновления запаса. Этот тип данных может быть
связан с отдельными таблицами, в той же базе данных,
используемыми для учета происхождения клеточных
374
ГЛАВА 20. КРИОКОНСЕРВАЦИЯ
Добавить криопротектор DMS0
или глицерин до 10% по объему
Поместить клетки в предварительно
промаркированные ампулы
Закрепить ампулы на алюминиевом штативе, вставить
в гильзу (см. рис. 20.2). Поместить как минимум на
4 часа в морозильник при температуре -70 °С
Гильза
Перенести ампулы в гильзе
в жидкий азот для хранения
Пробка из изолирующего
материала
Теплоизолирующая
трубка
/Трипсинизировать монослой и
ресуспендировать клетки в среде
при 1-10x106/мл
Рис. 20.8. Замораживание клеток. Трипсинизированные
клетки в среде с криопротектором, разливаются по аликво¬
там в ампулы, которые затем закрепляются на алюминиевом
стержне, вводятся в гильзу и затем в изолирующую трубку.
Трубка и ее содержимое помещаются на 4 ч или на ночь при
температуре -70 °С или -80 °С а затем гильза, содержащая
ампулы, переносится в кассеты на хранение в жидком азоте
(см. рис. 20.7а)
линий (табл. 20.2, 20.3; см. также разд. 12.3.10, 13.8).
Материалы, хранящиеся на дисках или ленте, должны
иметь копии на диске или иметь бумажную копию.
Использование компьютерной базы данных требует,
чтобы ответственный за морозильники вел записи в
этой базе. Если введение новых данных должно быть
сделана другими пользователями, то их записи должны
быть доступны для чтения и редактирования, в то вре¬
мя как записи по основным запасам культур клеток и
их распределению должны быть доступны только для
куратора. Либо весь файл может быть доступен для
чтения и внесения изменений куратором с бумажных
носителей — карточек или записей в регистрационной
книге, которые заполняются пользователями.
20.3.7. Размораживание хранившихся
ампул
Когда возникает необходимость, клетки размораживают
и пересевают в относительно высокой концентрации
для более быстрого восстановления культуры. Ампулы
должны быть разморожены так быстро, как это возмож¬
но, для того чтобы минимизировать внутриклеточные
повреждения, вызванные появлением кристаллов льда в
ходе процесса нагревания. Это может быть сделано пог¬
ружением в теплую воду в ведерке или на водяной бане,
но, если ампула находилась при жидкофазном хранении,
согревающая баня должна быть закрыта крышкой на
случай, если ампула протекала и набрала жидкий азот,
который затем при нагревании может взорваться.
После размораживания клеточная суспензия должна
быть медленно разведена, поскольку быстрое разведе¬
ние снижает жизнеспособность. Этот ступенчатый про¬
цесс, в частности, важен при использовании DMSO, в
этом случае быстрое разведение может вызвать острый
осмотический шок и снизить выживаемость вполовину.
Большинство клеток не требуют центрифугирования,
поскольку разведение суспензионной культуры при
пересеве и замещение среды на следующий день для
монослойной культуры достаточно для того, чтобы
удалить криопротектор. Тем не менее некоторые клетки
(часто суспензионные культуры) более чувствительны
к криопротекторам, в частности к DMSO, и требуют
центрифугирования после размораживания. Однако
в этом случае они также должны быть разведены мед¬
ленным добавлением питательной среды.
ПРОТОКОЛ 20.2. Размораживание
замороженных клеток
Схема
Быстро разморозить клетки, медленно развести
их, и пересеять при высокой плотности клеток
(рис. 20.9).
Материалы
Стерильные:
• Культуральные флаконы
• Центрифужные пробирки (если требуется цент¬
рифугирование)
• Среда для роста
• Пипетки, 1 мл, 10 мл
• Шприц и игла 19-g (если вы используете стек¬
лянные ампулы)
Нестерильные:
• Защитные перчатки и маска для лица
• Стерильная вода при 37 °С, слой глубиной 10 см в
чистом, протертом этанолом ведерке с крышкой
• Пинцет
• Этанол 70%
• Тампоны
• Красители: нафталеновый черный (Amido Чер¬
ный), 1%, или Трипановый синий, 0,4%.
Протокол
1. Проверьте регистрационный номер и местополо¬
жение ампулы, которая будет размораживаться.
20.3. Принципы криоконсервации
375
Таблица 20.2. Запись клеточной линии
Клеточная
линия
Дата замораживания
Новая карточка
Место хранения
Вид
Нормальная/
неоплас-
тичная
Взрослая/
феталь-
ная/NB
Микоп- Метод:
лазма:
Дата исследования
Результаты
Инструкции по замораживанию
Режим Криопротектор %
Ткань
Участок
Паспорт /
Основной
№
Аутентификация
Инструкции по оттаиванию
Размораживать быстро до 37°С
Автор
Ссылка
Тип роста
Дата исследования
Метод
Развести до: 5 мл 10 мл
20 мл 50 мл
Нормальное ведение
Центрифугировать для Да/нет
удаления криопротектора?
Особые
характеристики
Частота Посевная
субкуль- концент-
тивиро- рация
вания
Частота Тип
замены
среды
Газовая Буфер
фаза
Агент
Сыворотка
и т.д.
%
pH
Специальные требования
Лицо, заполнившее
Дата
Требования биобезопасности
карту
Любые другие особые условия
2. Соберите все необходимые материалы, приго¬
товьте среду и промаркируйте флакон.
3. Выньте ампулу из морозильника, проверьте по
ярлыку правильность, и если ампула не была
погружена в жидкий азот, поместите ее в стериль¬
ную воду при 37 °С в мензурке, пластмассовом
ведерке, или водяной бане. Если возможно, из¬
бегайте попадания воды на крышку, поскольку
это увеличивает возможность загрязнения.
• Требование безопасности. При выни¬
мании ампулы из морозильника следует надевать
закрытый лабораторный халат и перчатки. Если
ампула хранилась погруженной в жидкий азот, на¬
девайте защитный щиток, или защитные очки, так
же как и закрытый лабораторный халат и перчатки.
Ампулы, включая пластмассовые, хранившиеся в
жидком азоте, могут содержать жидкий азот и, при
размораживании могут взорваться. В этом случае
для размораживания должно использоваться плас¬
тмассовое ведерко с крышкой, чтобы предохранить
исследователя от последствий взрыва.
4. Когда ампула рашорожена, дваж^ прюерьтс
маркировку, чтобы тодт^рда» адентачность
клеток, затем протритетампоном,смотенным 70%-
м этанолом, и вскройте в ламинарном потоке.
5. Перенесите содержимое ампулы в культураль¬
ный флакон при помощи 1-мл пипетки.
6. Медленно добавьте ростовую среду к суспензии
клеток: приблизительно 10 мл за 2 мин, добавляя
капля за каплей в начале, и затем немного быстрее,
постепенно разводя клетки и криопротектор.
Для клеток, которые требуют центрифугирова¬
ния для удаления криопротекторге :
а) Медленно разведите клетки, как в inare 6, но в
центрифужной пробирке или универсальном
контейнере.
б) Центрифугируйте их в течение 2 мин при
100 g.
в) Удалите супернатант среды с криопротек¬
тором.
г) Ресуспендируйте клетки в свежей ростовой
средё.
д) Перенесите во флакон для культивирования.
376
ГЛАВА 20. КРИОКОНСЕРВАЦИЯ
Таблица 20.3. Ведение регистрационных записей по морозильникам
Перечень: Морозильник № Канистра / секция № Пробирка/ящик№.
Клеточный штамм /линия Дата замораживания Заморозил (ФИО)
Число замороженных ампул Количество клеток / ампул В
Среда ростовая Сыворотка Концентрация Среда для замораживания ,
Метод охлаждения
мл
Регистрация размораживания:
Дата размо¬
раживания
Кол-во
ампул
Осталось
ампул
Пересев
Жизнеспособность
Примеча¬
ния
Конц.
Объем
Среда
Исключение
красителя
~% прилипших
за 24 ч
Эффективность кло¬
нирования
7. Осадок в ампуле может быть окрашен нафталено-
вым черным или трипановым синим для опреде¬
ления жизнеспособности (см. протокол 22.3.1).
8. Проверьте после 24 ч:
а) Для прикрепляющихся к субстрату моно¬
слойных культур подтвердите прикрепление
и попытайтесь оценить процент выживания
на основании фотографий клеток при ожи¬
даемой плотности разведения (кл./см2) с
полным выживанием (см. разд. 13.6.1, 16.4.5;
цв. вкладку 4).
б) Для суспензионных культур проверьте внеш¬
ние признаки (прозрачность цитоплазмы,
отсутствие грануляции) и разведите до обыч¬
ной посевной концентрации. Это может быть
сделано более точно, если клетки подсчитаны
и проведена оценка жизнеспособности (см.
разд. 22.3.1), в этом случае клетки могут быть
разведены до обычной посевной концентра¬
ции жизнеспособных клеток.
Данные по оценке жизнеспособности методом пог¬
лощения красителя и другие данные (например, доля
клеток, прилипших к субстрату через 24 ч) должны
быть записаны в соответствующей карте или в файле
для использования при последующих процедурах
размораживания. Одна ампула должна быть разморо-
Вынуть одну ампулу из азотного морозильника и
перенести в воду температурой 37 °С. Если ампула
была погружена в азот при хранении, используйте
стерильную воду в ведерке с крышкой, закрывая
крышку немедленно после внесения в воду
ампулы
После опаивания
промокните ампулу
досуха, затем протрите
70%-м спиртом, откройте
ампулу и перенесите
клетки в культуральный
флакон
Чистов сухое ведерко
Медленно разведите в
культуральной среде
ft
в
Инкубируйте 24 ч
Стерильная вода при 37 °С
Удалите среду и
замените свежей
Рис. 20.9. Размораживание клеток. Ампулы вынимаются
из морозильника и быстро размораживаются в теплой воде
под крышкой, чтобы избежать возможного взрыва ампул,
хранившихся в жидкой фазе
жена после каждого нового замораживания для оценки
успешности процедуры замораживания.
20.4. Планирование и контроль замораживания культур клеток
377
Флаконы для замораживания. Можно замора¬
живать целые флаконы путем выращивания клеток до
поздней логарифмической фазы, добавления 5-10%
DMSO к минимальному объему среды, который полно¬
стью покроет монослой; и помещением флакона в рас¬
ширяющийся полистироловый контейнер с толщиной
стенок 15 мм [Ohno et al., 1991]. Термоизолированный
контейнер помещается в морозильник при -70-90 °С
и замораживается примерно со скоростью 1 °С/мин. В
течение нескольких месяцев выживаемость остается
хорошей, если флакон не вынимается из морозильника.
24-луночные планшеты также можно замораживать
аналогичным образом [Ure et al., 1992] примерно с
150 мкл среды для замораживания на лунку, таким
образом можно заморозить большое количество клонов
в ходе процедуры оценки.
20.4. ПЛАНИРОВАНИЕ И КОНТРОЛЬ
ЗАМОРАЖИВАНИЯ КУЛЬТУР
КЛЕТОК
Как только образуется небольшой избыток клеток,
полученных при первичном культивировании либо
при ведении вновь полученной клеточной линии, под¬
тверждено отсутствие контаминации, несколько ампул
с культурой должны быть заморожены, этот процесс
называется «пробная заморозка». Когда клеточная ли¬
ния успешно наросла или проведена селекция клонов
с определенными характеристиками, их идентичность
и свобода от контаминации подтверждены, следует
провести замораживание посевного стока для хранения
(табл. 20.4).
20.4.1. Контроль за состоянием запасов
клеток в морозильнике
Начальная культура должна быть защищена и не¬
доступна для общего пользования. Когда ампулу
Таблица 20.4. Сбор и хранение клеточных линий
размораживают для проверки жизнеспособности
замороженной начальной культуры и если в ближай¬
шем будущем предполагается использование этой
клеточной линии, клетки могут быть выращены для
распределяемой культуры и вновь заморожены. Эти
ампулы выдаются по индивидуальной потребности.
Индивидуальные пользователи данной культуры при
длительном периоде работы с ней должны затем вновь
заморозить их собственную пользовательскую культу¬
ру, которая должна быть уничтожена по завершении
работы. Пользовательская культура никогда не должна
передаваться другим пользователям, поскольку она не
является достаточно валидированной; новые пользова¬
тели должны затребовать культуру или ампулу из рас¬
пределяемой культуры. Когда распределяемая культура
заканчивается, она может быть пополнена из посевной
культуры. Когда количество ампул посевной культуры
станет менее пяти, она должна быть пополнена прежде,
чем будет использована, с минимальным количеством
пассажей после первого замораживания.
20.4.2. Периодическая замена
культивируемых клеток
Рабочие культуры должны заменяться в морозильнике
через определенные интервалы времени для того, что¬
бы минимизировать действие генетического дрейфа и
фенотипических вариаций. После того как клеточная
линия находилась в кулыуре в течение 2,5 месяцев,
разморозьте следующую ампулу, проверьте характе¬
ристики, удостоверьтесь, что культура свободна от
контаминации (см. разд. 19.3), и размножьте ее, чтобы
заменить имеющиеся клетки.
Уничтожьте существующие культивируемые клет¬
ки, которые разморожены более чем три месяца назад,
и перейдите на новые клетки (рис. 20.10). Повторяйте
этот процесс каждые 3 месяца для клеток, которые
имеют время удвоения популяции (PDT) примерно
24 часа; для клеточных линий с более коротким или
Стадия Источник Количество ампул Распределение Проверка
Замороженная по¬
Автор
1-3
Нет
Происхождение
севная культура
Исходная культура
Нет (только пополнение
или заморожен¬
12
запаса для распреде¬
Жизнеспособность
ная культура
ления культуры)
Аутентичность
Т рансформация
Контаминация
Перекрестная контаминация
Запас для распреде¬
Пробная разморо¬
50-100 (или больше,
Пользователи, включая
Жизнеспособность
ления культуры
женная посевная
культура
если требуется)
другие лаборатории
Контаминация
Перекрестная контаминация
Пользовательская
Запас для распреде¬
20 (5-летний проект, за¬
Жизнеспособность
культура
ления культуры
мещение 4 раза в год)
Контаминация
Перекрестная контаминация
378
ГЛАВА 20- КРИОКОНСЕРВАЦИЯ
Размораживание Использование Проверка
Рис. 20.10. Серийное возобновление культуры. Ампула разморожена, клетки выращены до желаемого количества для обыч¬
ного использования, культура поддерживается в течение 3 месяцев. Через три месяца после первого размораживания новую
ампулу размораживают, выращивают и после проверки характеристик в соответствии с используемой посевной культурой
используют для замены используемой, которая затем уничтожается. Цикл повторяется каждые три месяца, поэтому культура
не остается в использовании долее трех месяцев
долгим PDT может потребоваться, соответственно,
более короткий или долгий период замещения.
20.5. БАНКИ КЛЕТОК
Существует несколько банков клеток (см. Приложе¬
ние II) для безопасного хранения и распределения ва-
лидизированных клеточных линий. Поскольку многие
клеточные линии, особенно гибридомные и другие ге¬
нетически модифицированные клеточные линии, могут
иметь патентные ограничения, необходимо обеспечить
патентные базы данных с ограниченным доступом. Как
правило, предпочтительно получить вашу первичную
посевную культуру из заслуживающего уважения
клеточного банка. Тогда не возникает необходимость
характеризовать и повторно проводить уже сделанный
контроль качества клеточной линии. Более того, чрез¬
вычайно рекомендуем вам передавать ценные культуры
клеток в банк клеток в дополнение к вашему хранению
замороженной культуры, поскольку банк защитит вас
от потери вашей линии и будет отслеживать ее распре¬
деление среди других пользователей. Если вы считаете,
что ваши клетки не должны быть переданы другим
пользователям, они могут быть сохранены в банке с
ограничениями, наложенными на них. Большинство
банков клеток имеют информационный каталог, до¬
ступный on-line, где они выступают в качестве главного
Изолирующая Закрывающийся Сухой лед
а коробка, например полиэтиленовый
пенопластовая
пакетик
Культуральный флакон,
полностью заполненный
средой
из
газом внешняя оболочка
Рис. 20.11. Контейнер для транспортировки клеток. Клетки погружаются в контейнер в замороженном виде в ампуле или
в состоянии растущей культуры, (а) Твердая коробка из плотного картона с изолирующим (пенопластом) полистироловым
упаковочным материалом, содержащим сухой лед и ампулу, закупоренную в пробирку с абсорбирующей ватой. Пробирка
укладывается в закрывающийся полиэтиленовый пакетик, помещаемый в сухой лед. (6) Двустенный пластиковый контейнер
с флаконом, помещенным во внутреннее пространство и фиксированным в нем путем заполнения контейнера газом (Air Box,
Air Packaging Technologies)
Внешняя коробка
прочного картона
Закупоренная
пробирка
Ампула
Абсорбирующая
вата, предохра¬
няющая внутрен¬
нюю пробирку
20.6. Транспортировка клеточных культур
379
источника информации. Существует также несколько
баз данных, которые также могут быть доступны on¬
line, где содержится информация о клеточных линиях,
которые имеются у подписчиков этих баз данных в их
лабораториях (см. Приложение И). Этот тип банков
данных обеспечивает исследователей обширным ма¬
териалом, который потенциально доступен, однако
следует помнить, что клеточные линии, полученные
из других лабораторий, будут значительно отличаться
по большинству характеристик и по особенностям их
культивирования.
20.6. ТРАНСПОРТИРОВКА
КЛЕТОЧНЫХ КУЛЬТУР
Культуры могут быть переданы из одной лаборатории
в другую как в виде замороженных ампул, так и в виде
живых культур. В любом случае:
1) посоветуйтесь с принимающей лабораторией в
отношении того, когда клетки должны быть от¬
правлены;
2) отправьте по факсу или электронной почте ин¬
струкцию, в которой будет указано следующее:
а) что следует сделать по получении;
б) требуемая среда или сыворотка;
в) любые особые добавки;
г) режим субкультивирования;
3) распечатайте спецификацию на клетки и копию
инструкции и наклейте на обратную сторону пакета,
в который будет упакована культура, чтобы получа¬
тель знал, что делать прежде, чем вскроет пакет.
20.6.1. Замороженная ампула
Переправляйте замороженные ампулы в сухом С02
(сухой лед), в толстостенном пенополистироловом
контейнере (рис. 11а). Переносите ампулы из жидкого
азота в сухой С02 возможно быстрее, поскольку ско¬
рость нагревания при комнатной температуре состав¬
ляет около 10-20 °С/мин и не должна подняться выше
-50 °С. Поместите опечатанную полипропиленовую
центрифужную пробирку в сухой С02 в теплоизо¬
лирующей коробке. Выньте ампулу из морозильной
камеры, быстро оберните абсорбирующей бумажной
салфеткой (на случай вытекания) и поместите в по¬
липропиленовую пробирку, закрыв плотной крышкой.
Коробка должна иметь размеры примерно 30x30x30 см
с толщиной стенок 5 см и центральным пространством
около 20x20x20 см. Вам потребуется примерно 5 кг
сухого С02, чтобы заполнить центральное простран¬
ство. Обычно клетки остаются замороженными до
3 дней, если упакованы, как описано; если произошло
их медленное размораживание, их жизнеспособность
быстро падает. Курьер должен быть проинформирован
о том, что клетки находятся в сухом С02. По прибытии
ампула должна быть разморожена и пересеяна, как
обычно (см. протокол 20.2).
20.6.2. Живые культуры
Кроме того, клетки могут быть переданы в виде
растущей культуры. Клетки должны находиться в
середине или в поздней log-фазе; конфлюэнтная или
постконфлюэнтная культура истощит питательную
среду быстрее и может отслаиваться при транспор¬
тировке. Флакон должен быть заполнен до горлышка
средой, обмотан для безопасности вокруг горлышка
растягивающейся непромокаемой адгезивной пленкой
и запечатан в полиэтиленовый пакет. Упакованный
флакон помещается в жесткий контейнер и затем в
пенопластовую или полиэтиленовую противоударную
упаковку. Идеальным для транспортировки флакона с
живой культурой является надувной пакет (Air Pack¬
aging Technologies, Inc). Флакон запечатывается во
внутреннее пространство (рис. 20.116) и фиксируется
за счет наполнения воздухом внешней оболочки. По¬
местите на контейнер этикетки «хрупкий» и большими
буквами НЕ ЗАМОРАЖИВАТЬ!
По получении флакон удаляется из упаковки, тща¬
тельно протирается 70%-м этанолом и открывается в
стерильных условиях. Большая часть среды удаляется
из флакона, оставляется обычное количество куль¬
туральной среды около 5 мл на 25-см2 флакон для
питания клеток. Культура может быть культивируе¬
ма в новой среде, когда она будет готова к пересеву,
но следует сохранить транспортировочную среду на
случай, если возникнут проблемы адаптации к новым
условиям.
Обычно лучше пересылать клетки с курьером, кото¬
рый должен быть проинструктирован как в отношении
содержимого пакета, так и о срочности доставки. При
использовании международной почты потребуется
оформлять прохождение таможенного контроля, по¬
этому компетентный представитель, знакомый с этим
типом информации, сможет помочь быстро провести
посылку через таможенный контроль. Клетки, пересы¬
лаемые в США, должны проходить карантин и конт¬
рольную проверку департаментом сельского хозяйства
(Department of Agriculture), поэтому лучше пересылать
клеточные культуры через клеточные банки ЕСАСС
или АТСС.
Глава 21
КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
Количественный анализ в культуре клеток требуется
для характеристики ростовых свойств различных
клеточных линий для экспериментального анализа
и для установления воспроизводимых условий куль¬
тивирования первичной культуры и поддержания
клеточных линий (см. разд. 13.6). Работа в стерильных
условиях и необходимость мининимизации времени,
которое культура проводит вне инкубатора, часто
приводят к поиску компромисса между скоростью
и точностью. Хотя первичная культура и рутинное
подержание культуры требуют количественного
подхода для подтверждения воспроизводимости
результатов, скорость манипуляций является клю¬
чевым моментом для хорошего выживания и роста
клеток, поэтому в этой ситуации снижение точности
при пипетировании, например, может быть приемле¬
мым в целях более быстрого выполнения процедуры.
Тем не менее в экспериментальных условиях следует
работать как возможно точнее, даже если при этом
действия несколько замедлятся. Хотя при обычном
ведении культуры может быть приемлемой ошибка
±10%, в экспериментальных условиях она должна
быть уменьшена до ±5%. В настоящей главе описыва¬
ются методы подсчета клеток и ряд других подходов,
используемых в количественной оценке процессов
клеточного деления, а также другие методы оценки
клеточного состава — например, ДНК и белка. Методы
исследования цитотоксичности и жизнеспособности
клеток описаны в гл. 22.
21.1. ПОДСЧЕТ КЛЕТОК
Хотя оценка стадий роста культуры может быть
сделана по ее внешнему виду простым наблюдением
при помощи микроскопа, стандартизация условий
культивирования и соответствующие количественные
эксперименты затруднительны для анализа и воспро¬
изведения, если не производится подсчет клеток до,
после, а также, желательно, в ходе проведения каждого
эксперимента.
21.1.1. Гемоцитометр
Концентрация клеточной суспензии может быть оп¬
ределена помещением клеток в оптически плоскую
камеру под микроскопом (рис. 21.1). Подсчитывается
количество клеток внутри определенной площади из¬
вестной глубины (т. е. в определенном объеме), затем
исходя из этой величины высчитывается концентрации
клеток в суспензии.
Протокол 21.1 может быть использован при выпол¬
нении упражнения 12 (см. гл. 2).
ПРОТОКОЛ 21.1. Подсчет клеток
при помощи гемоцитометра
Схема
Трипсинизировать монослойную культуру или
подготовить образец суспензионной культуры,
подготовить стекло для гемоцитометрии и добавить
клетки в счетную камеру. Подсчитать клетки под
микроскопом и определить концентрацию клеток.
Материалы
Стерильные:
• D-PBSA
• Неочищенный трипсин 0,25%
• Ростовая среда
• Желтые наконечники для автоматических пи¬
петок
Нестерильные:
• Автоматическая пипетка 20 мкл или с регулиру¬
емым объемом, 100 мкл
• Гемоцитометр (Improved Neubauer)
• Программируемый счетчик
• Микроскоп
Протокол
1. Подготовка клеток:
а) Для монослойной культуры
i) Трипсинизируйте культуру, как при
обычном субкультивировании (см. про¬
токол 13,2), и ресуспендируйте клетки в
среде примерно до концентрации lxKf/мл.
В случае когда образец подсчитывается в
ходе экспериментального роста, трипсин
не следует удалять, и клетки могут быть
диспергированы в растворе трипсина и
подсчитаны непосредственно в этом раст¬
воре или после разведения 50:50 в среде,
21.1. Подсчет клеток
381
ММfnQwth
*ib*AUU
DefjrhGJmn
ЩШшш?
Поле зрения
объектива 10х
i юлупосереоренная
.поверхность 0,1 мм под Ньютоновские кольца
покровным стеклом
' Покровное стекло /
Клеточная суспензия в пространстве между покровным
стеклом и стеклом
Тройная
параллельная
линия
Рис. 21.1. Использование гемоцитометра. (а) Предметное стекло гемоцитометра (улучшенная модификация Neubauer) и
покровное стекло перед использованием, (б) Придавливание покровного стекла, (в) Внесение клеточной суспензии в счетную
камеру, (г) Продольное сечение гемоцитометра, показывающее положение клеточного образца в камере глубиной 0,1 мм.
(д) Помещение стекла под микроскоп, (е) Увеличенный вид общей зоны подсчета. Центральная область, обведенная пунктир¬
ной окружностью, это зона, которая должна находиться в поле зрения объектива 10х. Эта область составляет квадрат решетки
примерно 1x1 мм. (ж) Микрофотография малого увеличения (1 Ох), показывающая 25 малых (200х200-мкм) квадратов решетки,
составляющих центральную зону, в которе внесены клетки, предварительно обработанные Naphthalene Black (Amido Black),
(з) Микрофотография одного из малых квадратов с высоким разрешением (40х), ограниченного тремя сплошными линиями,
и содержащего 16 самых маленьких (50х 50 мкм) квадратов. Жизнеспособные клетки неокрашенные и прозрачные, с неровным
кольцом, окружающим их; нежизнеспособные клетки темные и не имеют кольца вокруг
382
ГЛАВА 21. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
содержащей сыворотку, если клетки склон¬
ны образовывать агрегаты,
ii) Как следует перемешайте суспензию для
того, чтобы полностью диспергировать
клетки, и перенесите некоторое количество
(~1 мл) в пузырек или универсальный
контейнер,
б) Для суспензионной культуры
i) Тщательно перемешайте суспензию для
диспергирования возможных сгустков.
ii) Перенесите около 1 мл суспензии в пузы¬
рек или универсальный контейнер.
Для данного метода требуется концентра¬
ция как минимум 1x106 кл./мл, поэтому,
возможно, потребуется концентрировать
суспензию центрифугированием (при
100 g в течение 2 мин) и ресуспендировать
ее в измеренном меньшем объеме.
2. Подготовка стекла.
а) Протрите поверхность стекла 70% спиртом,
стараясь не поцарапать полупосеребренную
поверхность.
б) Протрите покровное стекло и, слегка увлаж¬
нив края, прижмите очень осторожно поверх
канавок и полупосеребренной поверхности
для подсчета (см. рис. 21.1). Появление зоны
интерференции (Ньютоновские кольца — ра¬
дужные пятна между поверхностным стеклом
и нижним стеклом, подобные тому, что обра¬
зует масло на воде) показывает, что покров¬
ное стекло прилегает как следует, определяя
глубину счетной камеры.
3. При помощи пипетирования тщательно пере¬
мешайте образец клеток, чтобы диспергировать
любые сгустки, и отберите 20 мкл в наконечник
пипетки.
4. Перенесите клеточную суспензию непосред¬
ственно в счетную камеру гемоцитометра. Для
этого поднесите наконечник пипетки к краю каме¬
ры гемоцитометра и, выпустив каплю, позвольте
ей распределиться в камере под поверхностным
стеклом. Старайтесь избегать недостаточного
или избыточного заполнения камеры, поскольку
в этом случае измерения будут неправильными
в результате изменений поверхностного натяже¬
ния; жидкость должна проникать только через
края желобков.
5. Перемешайте клеточную суспензию, повторно за¬
полнив наконечник пипетки, и заполните вторую
камеру, если она есть.
6. Промокните любые излишки жидкости (не осу¬
шая камеру) и перенесите счетную камеру на
предметный столик микроскопа (см. рис. 21.1).
7. Выберите объектив 10х и сфокусируйте мик¬
роскоп на линиях решетки. Если фокусировка
затруднена из-за малой контрастности, закройте
поле ирисовой диаграммы или сделайте освеще¬
ние слегка наклонным смещением конденсора.
8. Перемешайте счетную камеру так, чтобы поле,
которые вы видите, находилась центральная
часть решетки и самая большая зона, которую вы
видите, была ограничена тремя параллельными
линиями. Эта область составляет 1 мм2. При
стандартном объективе 10х эта область почти
заполняет все поле зрения либо углы решетки
слегка выходят за его пределы (см. рис. 21.1).
9. Подсчитайте клетки, лежащие в пределах 1 мм2-
зоны, пользуясь делением на суб-области (также
ограниченные тремя параллельными линиями) и
единичные клетки сетки, отделенные одиночными
линиями. Подсчитайте клетки, которые лежат на
верхних и левых линях каждого квадрата, но не
учитывайте те, которые лежат на нижних и правых
линиях, ограничивающих каждый квадрат для
того, чтобы избежать повторного подсчета одних
и тех же клеток. Для стандартного субкультиви¬
рования попытайтесь подсчитать клетки при кон¬
центрации 100 и 300 кл./мм2; чем больше клеток
подсчитывается, тем точнее результат подсчета.
Для более точного количественной оценки экспе¬
римента следует подсчитывать 500-1000 клеток
а) Если у вас в распоряжении очень мало клеток
(<100/мм2), подсчитайте один или более до¬
полнительных квадратов (каждый площадью
1 мм2), окружающих центральный квадрат.
б) Если у вас слишком много клеток (>1000/мм2),
подсчитайте только пять малых квадратов,
которые ограничены тройными линиями по
диагонали большого квадрата (1 мм2).
10. Если имеется две камеры, перейдите ко второй
камере и сделайте повторный подсчет. Если нет,
то промойте стекло и повторите подсчет со све¬
жим образцом.
Анализ. Обсчитайте результаты двух подсчетов и
определите концентрацию вашего образца по формуле
с = n/v}
где с — концентрация клеток (кл./мл), п — количество
подсчитанных клеток, v — подсчитанный объем (мл).
Для гемоцитометра Improved Neubauer глубина камеры
составляет 0,1 мм, С учетом того что центральная пло¬
щадь составляет 1 мм2, v равен 0,1 мм3, или 1х10_4мл.
Тогда формула принимает вид:
с = я/10-4, или с = пх 104.
Если концентрация клеток высока и подсчиты¬
вается только пять диагональных квадратов внутри
центральной зоны, площадью 1 мм2(т.е. 1/5 от общей
площади), то это равенство превращается в
с = пх 5 х 104.
21.1. Подсчет клеток
383
Если клеточная концентрация низка, то сосчитайте
десять 1-мм2 квадратов, по пять в каждой камере гемо-
цитометра. Выражение примет вид:
0,1 М лимонной кислоты, содержащей 0,1% кристал -
лвиолета при 37 °С в течение 1 ч и затем подсчитайте
ядра [Sanford et al., 1951].
с = пх 104/Ю или с = пх 103.
Подсчет клеток при помощи гемоцитометра дешев и
дает вам возможность видеть подсчитываемые клетки.
Если клетки были предварительно перемешаны с рав¬
ным объемом витального красителя (см. протокол 22.1;
рис. 21.1ж, з), то в то же время может быть определена
жизнеспособность клеток. (Не забудьте учесть раз-
ведение при окончательном подсчете концентрации
клеток.) Однако процедура достаточно медленна и
может давать ошибку как при взятии образца, так и
при подсчете общего количества клеток. Кроме того,
для этого метода требуется как минимум lxlO6кл./мл.
Большая часть ошибок при этом методе происходит
из-за неправильного взятия образца и переноса клеток
в камеру. Удостоверьтесь, что клеточная суспензия как
следует перемешана до того, как вы переносите обра¬
зец, и не позволяйте клеткам осесть или адгезировать
в наконечнике автоматической пипетки в процессе
переноса их в счетную камеру. Убедитесь также, что
вы получили суспензию единичных клеток, поскольку
агрегаты делают подсчет неточным. Большие агрега¬
ты клеток могут проникать в камеру медленнее, чем
суспензия, или не проходить вовсе. Если в ходе приго¬
товления клеточной суспензии не удается преодолеть
агрегацию клеток (см. табл. 13.4), лизируйте клетки в
Рис. 21.2. Электронный счетчик клеток CASY. Счетчик
клеток CASY 1 (Scharfe Systems), также подходит для опре¬
деления размеров клеток и обнаружения жизнеспособных
и нежизнеспособных клеток, а также одиночных клеток и
агрегатов. (С любезного разрешения Scharfe Systems.)
21.1.2. Электронный подсчет клеток
Основным поставщиком электронных счетчиков кле¬
ток являются компании Beckman Coulter (Coulter Z1 и
Z2) и Scharfe Systems (CASY 1; см. ниже). Оба произ-
K насосу
Объем
образца
вкл/выкл
400 мкм
200 мкм
Считывание результатов:
количество клеток в
отношении к диаметру клеток
(по изменению сопротивления)
Старт-
Трубка с _
отверстиями
Ф ©
©
©
Отверстие _
для образца
©
~Ч
Электроды
©
Мертвая
££5
клетка
Живая
w
клетка
JL
Стакан с
образцом
t
©
Жидкость для счетчика, например D-PBSA или CASY-электролит
Рис. 21.3. Схема работы счетчика клеток CASY. Электро¬
лит (например, D-PBSA или CASY-электролит) в трубке с
отверстиями соединен с насосом и пропускает определенный
объем образца клеток из стакана. Клетки, проходя через от¬
верстие, изменяют поток струи и генерируют сигнал, ампли¬
туда которого пропорциональна объему клетки. (С любезного
разрешения Scharfe Systems.)
384
ГЛАВА 21. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
водителя используют принцип, разработанный Coulter
Electronics (теперь Beckman Coulter) по которому
клетки, прогоняемые через маленькое отверстие, по¬
вышают электрическое сопротивление тока, текущего
через это отверстие пропорционально объему клетки,
что дает серию импульсов, которые сортируются и
подсчитываются.
Электронный счетчик клеток CASY™ (см.
рис. 21.2). В настоящем электронном счетчике имеется
три основных компоненты (см. рис. 21.3): 1) дроссель¬
ная трубка с отверстием, диаметром 150 мкм, связанная
с дозирующим насосом; 2) усилитель, анализирующий
интенсивность импульса, и пересчетное устройство,
соединенное с двумя электродами, одним в дроссельной
трубке и другим — в стаканчике с образцом; 3) аналого-
во-цифровое считывающее устройство, показывающее
результаты подсчета клеток и другие количественные
параметры, такие как объем клеток и распределение
клеток по размерам, в зависимости от модели обору¬
дования (Scharfe Systems).
Когда начат подсчет клеток, измеряемый объем
клеточной суспензии пропускается через отверстие
(см. рис. 21.3). При прохождении каждой клетки через
отверстие она изменяет сопротивление потока жидкос¬
ти, протекающей по трубке пропорционально размеру
клетки. Это изменения сопротивления генерирует
импульс, который усиливается и регистрируется. По¬
скольку размеры импульса пропорциональны объему
клетки (или любой другой частицы), проходящей через
отверстие, генерируется серия сигналов различной ам¬
плитуды. Нижний порог измерения (диаметр клетки)
ограничивается установленным на счетчике для того,
чтобы снизить электронный шум и не учитывать сгус¬
тки клеточного дебриса, регистрируя только клетки
(см. ниже раздел Калибровка). Верхний порог также
устанавливается исследователем или остается постоян¬
ным в зависимости от модели счетчика. Эти пороги
определения устанавливают размеры регистрируемых
частиц. На дисплее счетчика изображается гистограмма
распределения клеток по размерам.
Протокол 21.2. может быть использован вместе с
упражнением 12 (см. гл. 2).
ПРОТОКОЛ 21.2. Электронный подсчет
клеток на основании электрического
сопротивления
Схема
Развести образец клеток в электролите (физиологи¬
ческий раствор или D-PBSA), поместить разведен¬
ный образец под дроссельную трубку и подсчитать
клетки, прогнав 0,5 мл образца через счетчик
Материалы
Стерильные или асептически приготовленные:
• Культура клеток
• D-PBSA
• 0,25% неочищенный трипсин
• Ростовая среда
Нестерильные:
• Стаканчики для подсчета
Протокол
1. Трипсинизируйте клеточный монослой или от¬
берите образец суспензионной культуры. Клетки
должны быть хорошо перемешаны и суспендиро¬
ваны до единичных клеток
2. Разведите образец клеточной суспензии в от¬
ношении 1:50 в 20 мл жидкости для подсчета
в 25-мл химическом стакане или одноразовых
стаканчиках для счетчика. Применение авто¬
матического диспенсера (см. рис. 5.20) ускорит
разведение и повысит воспроизводимость ре¬
зультатов.
Замечание. Быстрое разливание жидкости для
подсчета может вызвать образование пузырей воз¬
духа, которые могут быть оценены как клетки при
прохождении через канал счетчика. Следовательно,
жидкость должна стоять несколько секунд до нача¬
ла подсчета. Если сначала разливается жидкость, а
затем добавляются клетки, то эта проблема будет
минимизирована.
3. Хорошо перемешайте суспензию, поместите ее
под наконечник дроссельной трубки, убедив¬
шись, что канал закрыт и наружный электрод
погружен в жидкость в стаканчике с образцом.
4. Проверьте настройку программы:
а) Выберите порог(и) настройки (минимальный
размер клетки, обычно 7,0 мкм).
б) Объем, который будет подсчитан (обычно
0,5 мл).
в) Необходимые арифметические вычисления
(если используется).
г) Индекс разведения (например, 50 если под¬
считывается 0,4 мл в 20 мл D-PBSA).
5. Проверьте визуальный аналоговый дисплей:
а) убедитесь, что все клетки попадают в пределы
порогов подсчета;
б) проверьте жизнеспособность или клеточ¬
ный дебрис (обнаруживается по падению на
кривой или гистограмме ниже нормального
уровня);
в) проверьте наличие агрегации (обнаруживает¬
ся по регистрации частиц, размеры которых
значительно превышают установленный
диапазон определяемых клеток).
6. Начните процесс подсчета при помощи счетчика.
7. Когда подсчет клеток закончен, распределение
клеток по размерам будет представлено на
аналоговом дисплее (рис. 21.4). Переключение
на цифровую форму даст количество клеток в
миллилитрах.
21.1. Подсчет клеток
385
Диаметр клеток
Рис. 21.4. Аналоговая распечатка результатов подсчета
клеток на электронном счетчике CASY. Сопоставление
числа клеток и их диаметра позволяет провести анализ
распределения клеток по размеру и выявить нижний предел
определения (вертикальная штриховая линия). На распечатке
верхний предел может быть установлен на уровне 30 мкм или
может быть оставлен неопределенным. Пик до вертикальной
пунктирной линии и вертикальной точечной линии представ¬
ляет собой нежизнеспособные клетки, данные могут быть
использованы вместе с данными подсчета жизнеспособных
клеток для определения процента жизнеспособности. (С лю¬
безного разрешения Scharfe Systems.)
Анализ. Если препаративная работа и разведение
выполнены правильно, то концентрация клеток будет
подходящей для использования в качестве начальной
для последующего разведения. В противном случае,
если счетчик установлен на измерение 0,5 мл при раз¬
ведении 1:50, конечный подсчет результата следует ум¬
ножить на 100 для того, чтобы получить концентрацию
исходной клеточной суспензии в кл./мл. Тем не менее
некоторые счетчики автоматически компенсируют
разведение образца, поэтому внимательно изучите
инструкцию к прибору.
Калибровка. Старые счетчики требовали калиб¬
ровки путем подсчета клеток в пробах возрастающей
концентрации начиная с пороговой величины (верх¬
ний предел не устанавливается). График полученных
результатов дает плато, центр которого определяет.
В современных счетчиках с аналоговым дисплеем
нижний порог может быть установлен вручную на
дисплее, обычно эта величина составляет 7,0 мкм,
верхний порог устанавливается на уровне 30 мкм или
оставляется неопределенным. Установление порога на
значении 7,0 мкм может привести к подсчету нежизне¬
способных клеток, в то время как более высокие зна¬
чения пороговой величины (12 мкм на рис. 21.4) будут
исключать большинство нежизнеспособных клеток.
Нежизнеспособные клетки имеют, видимо, меньший
диаметр, поскольку цитоплазматическая мембрана
становится проницаемой, в то время как цитоплазма
сохраняет то же сопротивление, что и электролиты, а
сигнал, возникающий при определении сопротивле¬
ния, генерируется за счет ядра. Обнаружение клеток с
диаметром 30 мкм и выше означает наличие агрегатов,
которые могут быть частично исключены установле¬
нием верхнего предела измерения (например, 25 мкм
или 30 мкм на рис. 21.4) либо использованием ста¬
тистических методов, подходящих для программного
обеспечения данного прибора.
Электронный счетчик клеток Beckman Coulter
Vi-CELL. Этот счетчик использует анализ внешнего
вида сканируемой клетки в оптическом потоке клеток
и определяет различия между живыми и мертвыми
клетками путем их окрашивания Трипаном-синим.
Результаты могут быть представлены на дисплее, где
жизнеспособные клетки обведены зеленым цветом, а
нежизнеспособные — красным. Счетчик также создает
аналоговый график распределения клеток по размерам,
устанавливая верхний и нижний пределы (рис. 21.5).
Этот счетчик должен быть настроен по четырем пара¬
метрам для каждого типа клеток.
Электронный подсчет клеток — быстрый метод, ко¬
торый имеет низкую собственную ошибку, поскольку
подсчитывается большое количество клеток. Хотя
из-за наличия агрегатов, мертвых клеток и частиц деб¬
риса большого размера имеется большая возможность
неверной интерпретации результатов, возможна их
коррекция путем исключения мертвых клеток и агре¬
гатов как на CASY, так и на Vi-CELL, при этом CASY
может вносить программируемую соответствующую
коррекцию по исключению агрегатов. Клеточная сус-
Рис. 21.5. Электронный счетчик клеток Beckman Coulter
Vi-CELL. Неразбавленная клеточная суспензия помещает¬
ся в чашечке в держатель для образца, образец отбирается
в счетчик (справа), смешивается с трипановым синим и
анализируется в потоке клеток. Результаты представлены
на дисплее (слева) в виде графика распределения клеток по
размерам или как вид клеток под микроскопом, где живые
клетки обведены зеленым цветом, а мертвые красным. (С лю¬
безного разрешения АТСС.)
386
ГЛАВА 21. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
пензия должна быть тщательно исследована до начала
разведения и подсчета на счетчике
CASY; Vi-CELL дает возможность визуального на¬
блюдения за клеточной суспензией в ходе подсчета.
Электронные счетчики клеток дороги, но при
правильном использовании удобны и дают большую
скорость и точность при подсчете клеток.
21.1.3. Окрашенные монослои
Бывают случаи, когда клетки не могут быть собраны
для подсчета или когда их слишком мало в суспензии
(например, при низкой концентрации в микротитра-
ционных планшетах). В этом случае клетки могут быть
фиксированы и окрашены in situ, а затем подсчитаны
визуально под микроскопом. Поскольку эта процедура
утомительна и связана с высокой ошибкой оператора,
предпочтительнее использовать метод изотопного
мечения или оценку общего количества ДНК (см. про¬
токол 21.3) или белка (см. протокол 21.4), хотя эти из¬
мерения не могут прямо коррелировать с количеством
клеток — например, если варьирует клеточная плоидия.
Грубая оценка количества клеток в ячейке может быть
получена окрашиванием клеток кристалл-виолетом и
измерением светопоглощения денситометром. Этот
метод также использовался для подсчета количества
клеток на колонию при исследовании клонального рос¬
та [McKeehan et al., 1977]. Окрашивание клеток Кумас-
си-блю, сульфородамином В [Boyd, 1989; Skehan et al.,
1990], или MTT [Plumb et al., 1989] также дает оценку
количества клеток, если допустить, что в стандартном
графике зависимости поглощения от количества клеток
имеется линейная зависимость (см. протокол 22.4).
Окрашивание МТТ имеет еще то преимущество, что
окрашиваются лишь жизнеспособные клетки.
21.2. ВЕС КЛЕТОК
Сырой вес редко используется, если только не имеют
дело с очень большим количеством клеток, поскольку
значительное количество адгерентной внеклеточной
жидкости вносит большую ошибку измерения. Одна¬
ко, по очень грубой оценке, можно считать, например,
что около 2,5x108 клеток HeLa (14-16 мкм в диаметре)
весят 1 г сьфого веса, так же как примерно 8-10x10® кле¬
ток мышиной лейкемии, например, мышиной лимфомы
L5178Y, клеток мышиной эритролейкемии Friend,
миеломы и гибридом (11-12 мкм в диаметре). Около
1,8x108 клеток человеческих диплоидных фиброблас¬
тов (16-18 мкм в диаметре) также весят один грамм
(табл. 21.1). Сухой вес также используется редко,
поскольку соли, выделившиеся из среды, вносят свой
вклад в вес нефиксированных клеток, а фиксированные
клетки теряют часть внутриклеточных низкомолеку¬
лярных веществ и липидов. Тем не менее оценка сухого
веса может быть получена при помощи интерферометра
[Brown & Dunn, 1989].
21.3. СОДЕРЖАНИЕ ДНК
На практике, помимо подсчета количества клеток, наи¬
более полезными количественными методами оценки
клеточного материала являются методы оценки ДНК
и белка. ДНК может быть исследована несколькими
флюоресцентными методами, включая реакцию с
DAPI [Brunk et al., 1979], PicoGreen (набор для иссле¬
дований компании Molecular Probes), либо Hoechst
33258 [Labarca and Paigen, 1980]. Флюоресцентное
излучение Hoechst 33258 при 458 нм возрастает при
взаимодействии зонда с ДНК при pH 7,4 и при высокой
концентрации соли из-за диссоциации белков хромати¬
на. Этот метод обладает чувствительностью 10 нг/мл,
но требует наличия интактной двухцепочечной ДНК.
ДНК также может быть измерена по абсорбции при
260 нм, при этом концентрация 50 мкг/мл дает оптичес¬
кую плотность (O.D.) 1.0. В результате интерференции,
связанной с другими клеточными компонентами, метод
прямого поглощения полезен только для очищенной
ДНК. Протокол 21.3 является относительно простым
и прямым методом определения количества ДНК.
21.4. БЕЛОК
Оценка количества белка и белковый состав клетки
широко используются для оценки общего количества
клеточного материала, в том числе в экспериментах
по клеточному росту, а также как мера экспрессии
специфической активности ферментов, содержания
рецепторов или концентрации внутриклеточных ме¬
таболитов. Количество белка в солюбилизированных
клетках может быть оценено прямо по измерению пог¬
лощения при 280 нм, с минимальным взаимодействием
с нуклеиновыми кислотами и другими веществами.
Поглощение при 280 нм может определить до 100 мкг
Таблица 21.1. Соотношение между размером клетки, объемом и массой
Тип клетки Диаметр (мкм) Объем (мкм3) Количество кл7г х10-6
Теоретическое Измеренное
Мышиная лейкемия (такие как L5178Y 11-12 800 1250 1000
or Friend)
HeLa 14-16 1200 800 250
Диплоидные фибробласты человека 16-18 2500 400 180
21.5. Скорость синтеза
387
ПРОТОКОЛ 21.3. Оценка содержания ДНК с
использованием красителя HOECHST 33258
Схема
Гомогенизировать клетки или ткань в буфере и затем
обработать ультразвуком гомогенизат. Перемешать
аликвоту культуры с красителем Н33258 и измерить
флюоресценцию.
Материалы
Нестерильные:
• Буфер: 0,05 М NaP04; 2,0 М NaCl, pH 7,4, содер¬
жащий 2х10'3М EDTA
• Hoechst 33258: в буфере, 1 мкг/мл если концент¬
рация ДНК выше 100 нг/мл и 0,1 мкг/мл если
ДНК составляет 10-100 нг/мл
Протокол
1. Гомогенизируйте клетки в буфере, ЬсМЯкл./мл в
течение 1 мин, используя гомогенизатор Поттера.
2. Обработать ультразвуком клетки в течение 30 с.
3. Разведите клетки в отношении 1:10 в Hoechst
33258 и буфере.
4. Определите интенсивность флюоресценции при
492 нм с длиной волны возбуждения 356 нм,
используя ДНК тимуса теленка в качестве стан¬
дарта.
белка или примерно 2x105 клеток. Колориметрическое
исследование является более чувствительным, чем из¬
мерение поглощения, и среди прочих методов наиболее
часто используется реакция Брэдфорда с красителем
Кумасси-голубой [Bradford, 1976].
21.4.1. Солюбилизация образца
Поскольку большинство исследований основывается
на последнем колориметрическом этапе, они должны
проводиться в чистых растворах. Клеточный монослой
и клеточный осадок могут быть растворены в 0,5-1,0 М
NaOH с прогреванием до 100 °С в течение 30 мин или
в течение ночи при комнатной температуре. Другим
методом является действие 0,3 М NaOH и 1% лаурил-
сульфата натрия в течение 30 мин при комнатной
температуре. Метод Брэдфорда не зависит от специ¬
фических аминокислот и достаточно чувствителен, для
него требуется 50-100 000 клеток. Краситель Кумасси-
голубой подвергается спектральным изменениям при
связывании с белком в кислотном растворе. Цвет воз¬
никает на определенном этапе после кратковременной
инкубации и может быть измерен в течение 30 мин.
Варианты. Реактивы для метода Брэдфорда
доступны в виде наборов компании Bio-Rad. Для
определения белка могут быть использованы также
сульфородамин В [Skehan et al., 1990] и би-хинная
ПРОТОКОЛ 21.4. Оценка содержания белка
методом Брэдфорда
Схема
Развести белок, перемешать с цветным реагентом
и через 10 мин снять показатели оптической плот¬
ности (ОП).
Материалы
• Лаурилсульфат натрия (SLS, SDS), 3,5 мМ в воде
или 0,3 М NaOH
• Кумасси яркий синий G-250,0,12 мМ (0,01%), в
4,7% этанола и 85% (вес/объем) фосфорной кис¬
лоты: растворить 100 мг Кумасси синего в 50 мл
95% этанола; добавить 100 мл 85% фосфорной
кислоты; и доведите водой объем до 1 л
• Стандартный раствор белка (например, BSA,
10 мкг/мл); при первом проведении исследования
и через 1-2 мес для получения калибровочной
кривой использованием BSA или аналогичного
стандартного белка (1-50 мкг/мл)
Протокол
1. Разведите белок (1 -20 мкг) или клетки (пример¬
но 1х106) в 100 мкл 3,5 мМ SDS в воде или 0,3 М
NaOH.
2. В три различные пробирки внесите 100 мкл SLS
(для контрольного измерения), 100 мкл иссле¬
дуемого раствора белка и 100 мкл стандартного
BSA.
3. Добавьте в каждую пробирку 1,0 мл Кумасси
синего. Перемешайте и оставьте на 10 мин.
4. Проведите измерения на спектрофотометре при
595 нм против SLS.
кислота (ВСА) [Smith et al., 1985]. Чувствительность
этих методов позволяет использовать их для микротит-
рации в планшетах. Набор для микро-ВСА, который
производится компанией Pierce (см. Приложение III),
очень чувствителен и подходит для небольшого коли¬
чества клеток (~1х103).
21.5. СКОРОСТЬ СИНТЕЗА
21.5.1. Синтез ДНК
Измерение параметров синтеза ДНК часто рассмат¬
ривают как репрезентативный метод количествен¬
ной оценки процессов пролиферации (см. также
разд. 22.3.5). [3H]-тимидин([3H]-TdR) или [3Н]-де-
зоксицитидин является обычным предшественником,
который используется в этих исследованиях. Экспози¬
ция с одним из этих препаратов может быть кратков¬
ременной (0,5-1 ч) для оценки скорости синтеза или
длительной (24 ч и более) для того, чтобы измерить
накопление синтезированного ДНК при изначально
388
ГЛАВА 21. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
низкой скорости синтеза (например, в культурах с
высокой плотностью). [3H]-TdR не следует исполь¬
зовать для инкубации более чем в течение 24 ч или
при наличии высокой специфической активности, на¬
пример при наличии радиолитического распада ДНК:
из-за короткой длины пробега P-излучения (~1 мкм)
распадающегося трития происходит высвобождение
энергии внутри ядра, и это вызывает разрушение цепей
ДНК. При продолжительной инкубации или наличии
высокоспецифических активностей требуется исполь¬
зование [14C]-TdR или 32Р.
ПРОТОКОЛ 21.5. Оценка уровня синтеза ДНК
методом включения Н3-тимидиновой метки
Схема
Пометить клетки [3H]-TdR, экстрагировать ДНК,
определить уровень радиоактивности при помощи
сцинтилляционного счетчика
Материалы
Стерильные или асептически приготовленные:
• Клетки на подходящей фазе деления (выращен¬
ные в стеклянных пузырьках или пробирках для
тестирования, если используется метод горячего
2 М РСА; в противном случае используйте обыч¬
ный пластик)
• [3H]-TdR, 0,4 МБк/мл (около 10мкКи/мл),
100 мкл на каждый мл культуральной среды
Замечание: Некоторые среды, например среды Хэма
F10 и F12, содержат тимидин, который, в конечном
итоге, определяет специфическую активность до¬
бавленной метки [3H]-TdR. Следует учитывать этот
фактор при оценке количества изотопа, которое вы
будете добавлять. Хотя 40 кБк/мл (около 1,0 Ки/мл)
изотопа может быть достаточно для большинства
сред, в случае РЮили F12 лучше использовать
0.2 МБк/мл (около 5 мкКи/мл)
Нестерильные:
• Трихлоруксусная кислота (ТХУ) 0,6 М (на льду),
6 мл на 1 мл культуры
• HBSS или D-PBSA, на льду, 2 мл на 1 мл куль¬
туры
• Перхлорная кислота (РСА), 2 М, или 0,3 М NaOH
с 35 мМ лаурилсульфата натрия (SLS, SDS),
0,5 мл на 1 мл
• Метиловый спирт, 1 мл на 1 мл культуры
• Сцинтилляционные пузырьки
• Сцинтиллятор (10х объем перхлорной кислоты
или NaOH/SDS)
Протокол
1. Нарастите культуру до желаемой плотности
(обычно середина log-фазы для максимального
синтеза ДНК или фаза плато для случая ограни¬
чения синтеза ДНК плотностью культуры, см.
разд. 18.5.2).
2. Добавьте [3H]-TdR, 40 КБк/мл (око¬
ло 1,0 мкКи/мл) или 2 МБк/моль (около
50 Ки/моль) в HBSS
3. Инкубируйте клетки 1-24 ч, по необходи¬
мости.
4. Осторожно удалите радиоактивную среду, помес¬
тив ее в соответствующий контейнер для жидких
радиоактивных отходов.
5. Тщательно промойте клетки 2 мл HBSS или
D-PBSА, добавьте холодную 0,6 М ТХУ (на льду)
на 10 мин. Если клетки плохо прикрепляются,
сначала фиксируйте их метиловым спиртом (см.
протокол 21.6).
6. Повторите дважды промывку в ТХУ по 5 мин
каждый раз.
7. Добавьте 0,5 мл 2 М перхлорной кислоты, по¬
местите раствор на горячую плитку при 60 °С
на 30 мин, затем оставьте раствор остывать.
В альтернативном случае можно добавить 0,5 мл
SLS в NaOH, и инкубировать в течение 30 мин
при 37 °С или в течение ночи при комнатной
температуре.
8. Соберите раствор, перенесите в сцинтиллятор и
определите радиоактивность в сцинтилляцион-
ном счетчике
Замечание. Если вы используете метод перхлорной
кислоты для определения белка, осадок может быть
растворен в щелочи (см. протокол 21.4). Для того что¬
бы подтвердить, что результаты оценки синтеза ДНК
отражают количество клеток, должны быть приготов¬
лены реплицированные культуры. Для суспензионных
культур при выполнении шагов 4,5 и 6 осадите клетки
при 100 g в течение 10 мин. Перемешайте клетки в
Вортекс-миксере для разбивания осадка до каждого
промывания (в 0,6 М ТХУ и 2 М перхлорной кислоте)
на шаге 7. Осадите клетки после шага 7 при 1000 g в
течение 10 мин для того, чтобы отделить преципитат
(для оценки белка, если это требуется) от супернатанта
(для сцинтилляционного счетчика).
• Требование безопасности. [3Н]-тимидин
представляет собой биологическую опасность, по¬
скольку вызывает радиационное повреждение ДНК.
Позаботьтесь о том, чтобы избежать случайного по¬
падания в организм через пищеварительный тракт, на
поврежденные участки кожи или через дыхательные
пути в виде аэрозоля. При использовании [3Н]-ти-
мидина работайте в боксе биологической опасности
или на открытой рабочей поверхности, но не исполь¬
зуйте ламинарный бокс с горизонтальным потоком,
поскольку в этом случае аэрозоль будет выдуваться
прямо на вас.
Инкубация с изотопным предшественником может
привести к различным результатам, в зависимости от
условий инкубации и последующих процессов. Инку¬
21.5. Скорость синтеза
389
бация после кратковременного промывания в холод¬
ном (на льду) BSS и перенесения в 0,6 М ТСА позво¬
лит определить включение метки и, при соблюдении
продолжительности эксперимента в несколько минут,
дает прямое измерение однонаправленного потока.
Считается, что при экспериментальном измерении
включения и коротком времени инкубации включение
метки в кислотно-нерастворимые молекулы, такие как
белки и ДНК, минимально. В этих условиях только
кислотно-растворимые молекулы участвуют в процес¬
се экстракции в холодной 0,6 М ТХУ. При длительной
инкубации (2-24 ч) предполагается, что пул предшест¬
венников становится насыщенным. Уровни равновесия
могут быть определены экстракцией холодной ТХУ;
инкорпорация в полимеры может быть измерена экс¬
тракцией с горячей 2 М перхлорной кислотой (ДНК)
и холодным раствором щелочей (РНК) либо горячим
раствором 1,0 М NaOH (белок).
21.5.2. Синтез белка
Колориметрическое исследование позволяет измерить
общее количество белка, присутствующее в данный мо¬
мент. Последующее наблюдение в ходе определенного
периода времени может быть использовано для изме¬
рения накопления или утраты спектра белков (напри¬
мер, синтез-деградация белка), в то время как скорость
синтеза белка может быть определена путем инкубации
клеток с радиоизотопно меченой аминокислотой, такой
как [3Н]-лейцин или [358]-метионин и измерением
(например, сцинтилляционным подсчетом) количества
радиоактивной метки, включенной в кислото-раствори-
мые вещества на 106 клеток или на миллиграмм белка в
течение конкретного периода времени.
• Требование безопасности. Манипуляции с
радиоизотопами следует проводить с осторожностью
и в соответствии с местным руководством, регулиру¬
ющим разрешенные количества, рабочие зоны, мани¬
пуляции и уничтожение радиоактивных веществ (см.
разд. 7.8.5).
ПРОТОКОЛ 21.6. Синтез белка
Материалы
Стерильные или асептически приготовленные:
• Культура клеток, например, 1х104-1х10Рк&/луН'~
ке, в 24-луночном планшете.
• [3Н] лейцин, 2 МБк/мл (около 50 мкКи/мл) в
культуральной среде без сыворотки (специфиче"
ская активность неважна, поскольку она будет оп¬
ределяться по концентрации лейцина в среде).
Нестерильные
• Лаурилсульфат натрия (SLS или SDS), 1%
(35 мМ) в 0,3 М NaOH
• Трихлоруксусная кислота (ТХУ)
• Сцинтилляционные пузырьки
• Пробирки Eppendorf
• Сцинтилляционная жидкость с минимум 10%
допустимым содержанием воды
Протокол
1. Инкубируйте культуру до требуемой клеточной
плотности.
2. Выньте культуру из инкубатора, добавьте пред¬
варительно подогретый раствор радиоизотопа в
среде или сбалансированном солевом растворе с
разведением 1:10 (т.е. 100 мкл на 1 мл на лунку).
3. Верните культуру в инкубатор так быстро, как
возможно.
4. Инкубируйте культуру 4-24 ч.
Замечание. Различные белки образуются с различ¬
ной скоростью. Протокол не имеет в виду какую-то
специфическую группу белков, но относится к обще¬
му белку в быстро пролиферирующих клетках. Когда
проводите анализ синтеза белка в клеточной линии в
первый раз, убедитесь, что скорость синтеза линейна
в течение выбранного времени инкубации. Могут
возникнуть некоторые сложности, связанные с тем,
что пул аминокислот медленно насыщается.
5. Выньте культуру из инкубатора и осторожно уда¬
лите среду из ячеек, перенося ее в контейнер для
жидких радиоактивных отходов (уничтожение
радиоактивных отходов см. разд. 7.7.2).
6. Осторожно промойте клетки холодным раство¬
ром HBSS или D-PBSA.
Замечание: Некоторые монослойные культуры, в
частности некоторые слабо прикрепляющиеся пос¬
тоянные клеточные линии, такие как HeLa-S3, могут
отделяться от стенок лунки при отмывке. В этом
случае после удаления изотопа добавьте метанол для
фиксации монослоя. Оставьте культуру на 10 мин,
осторожно удалите метанол и высушите монослой.
7. Перенесите чашки на лед. Добавьте 0,6 М ТХУ
при 4 °С, на 10 мин для удаления невключенных
предшественников.
8. Повторите шаг 7 дважды, изменив время инкуба¬
ции до 5 мин.
9. Отмойте культуру с метиловым спиртом, высу¬
шите чашки.
10. Добавьте 0,5 мл 0,3 М NaOH, содержащего 1%SLS,
оставьте на 30 мин при комнатной температуре.
11. Смешайте содержимое каждой ячейки и перене¬
сите в отдельные сцинтилляционные пузырьки.
12. Добавьте 5 мл сцинтиллятора и произведите
подсчет на сцинтилляционном счетчике.
Замечание: Предпочтительно использовать сцин¬
тилляторы, способные к биодеградации, например,
Ecoscint, которые, в отличие от толуол- или кси¬
лолсодержащих жидкостей, менее токсичны при
390
ГЛАВА 21. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
манипуляциях и могут быть вылиты в раковину с
большим избытком проточнойзоды, такчтобыуро-
вень радиоактивности упал ниже установленного
законодательством.
При работе с суспензионными культурами, на
шаге 5для удалшшяс^ре^^эЬадите клетки при
21.6. ПРИГОТОВЛЕНИЕ ОБРАЗЦОВ
ДЛЯ ФЕРМЕНТНОГО И
ИММУНОФЕРМЕНТНОГО АНАЛИЗА
Поскольку количество клеточного материала, доступ¬
ного в культуре, часто слишком мало для гомогениза¬
ции, требуются другие методы лизиса, позволяющие
высвобождаться растворимым продуктам и фермен¬
там, которые затем могут быть исследованы. Удобно
поместить культуру в необходимом количестве в
многолуночные планшеты или пробирки для образцов
(см. разд. 22.3.5 и протокол 21.8) или трипсинизиро-
вать партию культуры и поместить аликвоты клеток
в пробирки для исследований. В любом случае клетки
следует промыть в HBSS или D-PBSA, для того чтобы
удалить сыворотку, а затем добавить лизирующий
буфер. Лизирующий буфер следует подбирать в
соответствии с последующими исследованиями, но
если это не является определяющим, то можно ис¬
пользовать 0,15 М NaCl или D-PBS. Если продукты,
концентрация которых будет измеряться, являются
мембранно-связанными, то добавьте в лизирующий
буфер 1% детергента (деоксихолат Na, Nonidet Р40).
Если клетки находятся в осадке, ресуспендируйте их
в буфере при помощи Вортекс-миксера. Проведите
троекратное замораживание-оттаивание путем поме¬
щения в этиловый спирт, содержащий твердый С02
(—90 °С) примерно на 1 мин и затем в воду при 37 °С
на 2 мин (для образцов объемом более 1 мл дольше).
Наконец, осадите пробу при 10 ООО g в течение 1 мин
(например, в центрифуге Eppendorf), и соберите супер-
натант для исследования. Можно также использовать
весь экстракт для изучения ферментативной актив¬
ности, а затем осадить центрифугированием нераст¬
воримую часть, если это необходимо для дальнейших
исследований.
21.7. ЦИТОМЕТРИЯ
21.7.1. Мечение in situ
Флюоресцентное мечение, как прямое с флюоресцент¬
ной меткой (например, Hoechst 33258 для ДНК) или с
конъюгированными антителами для определения анти¬
гена либо с молекулярным зондом (см. протокол 27.9 и
27.8), позволяет измерить количество фермента, ДНК,
РНК, белка, или других клеточных компонентов in situ
при помощи CCD камеры. Этот процесс обеспечивает
проведение как качественного, так и количественного
анализа, но при большом количестве клеток, которые
должны быть сканированы, требует достаточно много
времени.
21.7.2. Проточная цитометрия
Проточная цитометрия клеточной суспензии [Vaughan &
Miller,1989] (см. также разд. 16.9.2; рис. 15.9), в образце
с концентрацией до 1x107 клеток позволяет измерять
различные клеточные компоненты и активности
[Kurtz & Wells, 1979; Klingel et al., 1994]. Этот метод
дает возможность изучать корреляционные взаимо¬
связи этих параметров с другими, такими как размеры
клетки, линия дифференцировки, содержание ДНК и
жизнеспособность клеток [Al-Rubeai et al., 1997].
21.8. УВЕЛИЧЕНИЕ КОЛИЧЕСТВА
ОБРАЗЦОВ ДЛЯ СТАТИСТИЧЕСКОГО
АНАЛИЗА
Поскольку в большинстве случаев культивируемые
клетки могут быть приготовлены в виде однородной
суспензии, необходимость получения большого коли¬
чества повторов для статистического анализа является
редкой. Обычно достаточно получить три повтора,
а для многих простых исследований (например, для
подсчета клеток) достаточно дупликатов. Многие
типы культуральных сосудов могут применяться для
получения повторов экспериментов монослойных
культур (см. разд. 8.2), выбор культуральных сосудов
определяется 1) количеством клеток, требуемых для
каждого образца, и 2) кратностью или типом отбора об¬
разцов. Например, если время инкубации не меняется,
предпочтительно выращивание одинаковых образцов
в многолуночных планшетах, таких как микротитра¬
ционные планшеты или 24-луночные планшеты. Од¬
нако если образцы собираются в течение некоторого
периода времени (например, ежедневно в течение
5 дней), то постоянные манипуляции с планшетами
для ежедневных отборов проб могут ухудшать качество
роста в остальных лунках. В этом случае дубликаты,
образцы лучше готовить в отдельных пробирках или
4-луночных планшетов. Могут быть использованы
плоские стеклянные или специально обработанные
для культуры тканей пластиковые пробирки. Наилуч¬
шими являются пробирки Leighton, поскольку они
обеспечивают плоскую поверхность роста. С другой
стороны, если оптические свойства пробирок неважны,
плоскодонные стеклянные пробирки для образцов и
даже стеклянные сцинтилляционные пузырьки мо¬
гут быть достаточно хорошими контейнерами. Если
используются стеклянные пузырьки или пробирки,
21.9. Клеточная пролиферация
391
они должны быть вымыты и обработаны как стекло
для культивирования тканей (см. разд. 11.3.1); они не
должны быть использованы для тканевой культуры
после сцинтиллятора. Запечатывание большого ко¬
личества пузырьков или пробирок может стать доста¬
точно утомительным, поэтому многие исследователи
предпочитают запечатывать пробирки виниловой
лентой, а не заворачивающейся крышкой. Такие ленты
могут быть разноцветными, что позволяет исполь¬
зовать цвет в качестве кодировки для разных целей.
Адгезивная пленка может быть также использована
для запечатывания микротитрационных планшетов
(см. Приложение И: Приспособление для заклеива¬
ния планшетов). Это позволяет снизить испарение
и контаминацию и дает больше преимуществ перед
другими планшетами. Это также означает, что инди¬
видуальные лунки или ряды могут быть взяты для
получения образцов, без открывания остальной части
планшета. Манипуляции с суспензиями обычно легче,
чем с монослойными культурами, поскольку форма
контейнера и его поверхностный заряд менее важны.
Многократный отбор образцов может быть произ¬
веден из одного и того же флакона с суспензионной
культурой. Этот метод удобнее применять, используя
запечатанную бутыль с силиконизированной резино¬
вой мембраной — ловушкой (Pierce) и применением
шприца и иглы (не забывайте возмещать объем отби¬
раемой культуры равным объемом воздуха).
21.8.1. Получение данных
Анализ данных, полученных на культурах клеток,
не обязательно отличен от анализа результатов, по¬
лученных при других исследованиях. Тем не менее
достаточно легко обеспечить получение большого
количества данных в экспериментах на клеточной
культуре. В частности, при работе с микротитрацион-
ными планшетами исследователь получает несколько
сотен цифр без больших затруднений либо даже
несколько тысяч при использовании роботизирован¬
ных устройств. Манипуляции с этими результатами
будут зависеть от того, как они получены, от условий
эксперимента и ряда других параметров. Хотя подсчет
клеток является приемлемым методом для получения
данных и последующего построения кривой роста для
одной линии клеток при двух-трех наборах условий,
построение большого числа кривых роста при дей¬
ствии комбинации разных факторов роста не является
распространенным методом. Если выбран колоримет¬
рический анализ как конечный этап, то может быть
использовано поглощение (например, МТТ-исследо-
вание; см. протокол 22.4) или флюоресцентное излу¬
чение (например, сульфородамина [Boyd, 1989]), тогда
планшеты исследуются на ридере — автоматическом
считывающем устройстве, и уменьшение количества
данных достигается использованием соответствую¬
щего программного обеспечения. На последнем этапе
радиометрического анализа (например, с [3Н]-тими-
дином) микротитрационные планшеты могут быть
одномоментно протестированы в сцинтилляцион-
ном счетчике (PerkinElmer). Аналогичным образом,
большое количество пробирок с образцами и у или
P-метками может быть автоматически считано путем
роботизированной системы (Beckman Coulter).
21.8.2. Анализ данных
Способность давать большое количество данных обус¬
ловлено возможностью анализа большого количества
репликационных систем, таких как микротитраци¬
онные системы. Как в случае анализа ELISA, шагом,
ограничивающим скорость исследований, является не
этап получения данных, а период их анализа. Таким
образом, важно при выборе параметров измерения
подобрать такую систему культивирования, которая
обеспечит не только получение определенного коли¬
чества данных, но и предполагает методы их обработки.
Самым простым методом является прямое введение
данных в компьютер: в локальную сеть или в персо¬
нальный компьютер, в котором содержатся результаты
выполняемого проекта. Ряд компаний поставляют на
рынок компьютерные программы, которые выводят
на дисплей и анализируют данные, получаемые с
микротитрационных ридеров (не только ELISA). Эти
программы включают титрационные кривые, фермен¬
тативную кинетику и исследование связывания. При
наличии необходимых навыков и с использованием
электронных таблиц для импорта данных, вы сможете
сами установить такой тип программы либо пригла¬
сить консультанта из компании, выпускающей ридеры
планшетов (см. Приложение II).
21.9. КЛЕТОЧНАЯ ПРОЛИФЕРАЦИЯ
Измерение скорости клеточной пролиферации часто
используется для определения реакции клеток на
отдельные стимулы или токсины (см. протоколы 21.7
и 21.8). Количественный анализ кривой роста также
важен при обычном поддержании культуры, поскольку
он является решающим элементом для мониторинга
постоянства культуры и позволяет определить наилуч¬
шее время для субкультивирования (см. разд. 13.7),
оптимальное разведение, и оценить эффективность
посева при различной клеточной плотности. Тести¬
рование среды, сыворотки, новых культуральных
сосудов или субстратов и т.д. также требуют количес¬
твенной оценки.
21.9.1. Планирование эксперимента
Знание стадии роста культуры и параметров ее кине¬
тики является критическим для планирования экс¬
перимента в культуре клеток. Культуры значительно
отличаются друг от друга по многим свойствам в lag-
392
ГЛАВА 21. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
фазе, периоде экспоненциального роста (log-фазе) и
периоде стационарного роста (плато). Таким образом,
важно принимать во внимание не только состояние
культуры, но и стадию начала эксперимента, время
отбора образцов, для того чтобы определить наличие и
интенсивность пролиферации в эти моменты, а также
время удвоения популяции (PDT) и продолжитель¬
ность клеточного цикла. Популяция клеток, которые
выходят на стадию плато, имеет значительно меньшую
долю растущих клеток и разнообразную морфологию,
клетки могут быть более дифференцированными или
более поляризованными. Обычно они имеют тенден¬
цию к секреции большего количества внеклеточного
матрикса, и могут возникнуть сложности при дезагг-
регации. В целом, клеточные культуры более единооб¬
разны в log-фазе, отбор образцов в конце log-фазы дает
самый высокий выход клеток и воспроизводимость.
Важно также учитывать действие продолжитель¬
ности эксперимента на переход из одного состояния
культуры в другое. Добавление фармакологического
препарата в середине экспоненциальной фазы и в более
поздние сроки может привести к различным резуль¬
татам, в зависимости от того, находится ли культура
еще в экспоненциальной фазе, когда ее снимают для
анализа, или уже перешла в фазу плато. Исследование
цитотоксичности при помощи микротитрационных
планшетов (см. разд. 22.4) могут привести к ошибоч¬
ным результатам, если культура достигает фазы плато
в процессе исследования; в то время как клетки в тех
ячейках, в которых плотность выше, достигают плато
и скорость пролиферации снижается. В результате
отмечается значительный сдвиг величины ГО50 (ин¬
гибирование 50% культуры), который тем выше, чем
большее количество ячеек содержит клетки в фазе
плато (см. протокол 22.4). Таким образом, график об¬
работки и отбора образцов требует детального знания
параметров цикла роста.
21.9.2. Цикл роста
Как было описано выше (см. разд. 13.7.2), после суб¬
культивирования клетки развиваются по определенной
схеме, проходя через этапы lag-фазы, экспоненциаль¬
ной или log-фазы и стационарной или фазы плато
(рис. 21.6). Фазы лог и плато дают крайне важную
информацию о клеточной линии, в том числе и время
удвоения клеточной популяции (PDT) в ходе экспо¬
ненциального роста и максимуме клеточной плотности,
достигаемой в фазе плато (т. е. плотности насыщения).
Измерение PDT используется для количественной
оценки ответа клеток на различные ингибиторы или
стимуляторы культур, такие как различия концентра¬
ции питательных веществ, действие гормонов или ток¬
Дни субкультивирования
Рис. 21.6. Кривая роста. Полулогарифмическая кривая возрастания клеточной концентрации (левая ось) и плотности клеток
(правая ось) после субкультивирования. Культуры были посажены в 25-см2 флаконы, ежедневно образцы отбирались для под¬
счета. Можно провести прямую линию через ту часть графика, которая представляет собой экспоненциальную фазу, и получить
время удвоения популяции (PDT) из серединной области этой фазы. Промежуток времени, отделенный точкой пересечения
с линией, экстраполированной от начала экспоненциальной фазы, с линией начальной посевной концентрации — время lag-
фазы, достижение плотности насыщения — выход на плато (по крайней мере, три точки, показывающие неизменную величину
клеточной концентрации без ее возрастания)
21.9. Клеточная пролиферация
393
сических препаратов. Этот показатель также хорош для
контроля культивирования при серийных пассажах и
позволяет оценивать клеточный выход и рассчитывать
фактор разведения при субкультивировании.
Отдельные временные точки недостаточны для мо¬
ниторинга роста культуры, если неизвестны параметры
и форма кривой роста. Снижение количества клеток
после, скажем, 5 дней может быть вызвано снижением
скорости роста некоторых или всех клеток; удлинением
lag-периода, предполагающим адаптацию клеток или
потерей клеток (трудно обнаружить различия между
последними двумя процессами); или снижение плот¬
ности насыщения (см. рис. 21.8). Это не означает, что
кривые роста не представляют ценности. Они могут
быть полезны для испытания сред, сывороток, фак¬
торов роста и некоторых лекарственных препаратов.
Если при мониторинге культуры ответ на воздействие
оказывается полностью охарактеризован, и тип ответа
становится предсказуемым (например, изменения
PDT), то отдельные точки наблюдения могут быть
определены как известные в середине log-фазы. Кривые
роста, в частности, полезны для определения плот¬
ности насыщения, хотя степень роста при плотности
насыщения должна оцениваться по индексу мечения
при помощи [3Н]-тимидина (см. разд. 18.5.2 и прото¬
колы 21.11, 21.12).
Значение PDT, полученное из кривой роста, не
следует путать с продолжительностью клеточного
цикла или временем генерации. PDT представляет
собой интегральную величину, которая применима
ко всей популяции, и описывает суммарный резуль¬
тат широкого спектра скоростей клеточного деления,
включая нулевую, имеющих место в конкретной
культуре. Время клеточного цикла, или время генера¬
ции, измеряется от одной точки клеточного цикла до
следующей такой же точки (см. разд. 21.12) и имеет
отношение только к делящимся клеткам в популяции,
в то время как PDT зависит как от нерастущих, так
и от погибающих клеток. Значения PDT варьируют
от 12-15 ч в быстро растущей культуре мышиных
лейкемических клеток, (например, L1210) до 24-36 ч
во многих прикрепленных постоянных клеточных
линиях и даже до 60 или 72 ч в медленно растущих
конечных клеточных линиях. Новый цикл роста начи¬
нается каждый раз, когда культура пересаживается и
может быть проанализирован более детально, как это
описано в протоколе 21.7. Использование флаконов
для получения кривой роста повышает интенсив¬
ность работы, ограничивая количество репликаций
до двух в день в течение 10 дней, требуя 20 флаконов
и 4 дополнительных флакона для окрашивания или
использования в качестве резервных. Для быстрорас¬
тущих клеточных линий следует выбрать клеточную
концентрацию 2х104 кл./мл; для медленно растущих
конечных клеточных линий — lxlO5кл./мл. После¬
дующее повторение кривой роста с более высокой
и более низкой посевной концентрацией позволит
скорректировать величину посевной концентрации
и установить интервал субкультивирования (см.
ПРОТОКОЛ 21.7. Кривая роста монослоя
во флаконах
Схема
Приготовить флаконы и ежедневно подсчитывать
клетки пока культура не достигнет фазы плато.
Материалы
Стерильные или асептически приготовленные:
• Монослойная клеточная культура, А549, Vero,
или HeLa-S3, в 75-см2 флаконе, на стадии поздней
log-фазы 1
• Трипсин неочищенный, 0,25%, с 10 мМ
EDTA 5 мл
• Ростовая среда с 4 мМ NaHC03 200 мл
• D-PBSA (для предварительной промывки и под¬
счета клеток) 50 мл
• Флаконы, 25 см2 24
Протокол
1. Трипсинизируйте клетки, как обычно, при суб-
культивировании (см. протокол 13.2).
2* Разведите клеточную суспензию до 2х104 кл./мл
в 150 мл среды.
3. Посейте клетки в 24 25-см2 флакона.
4. Закройте флаконы и поставьте в инкубатор при
температуре 37 °С.
Замечание. Для большей простоты при выполне¬
нии этого протокола предусмотрена низко-бикар-
бонатная (4 шМ) среда, но если вы используете
бикарбонатную среду, (например, 23 мМ), то запол¬
ните флаконы 5% СО2 или поместите их, ослабив
крышки или используя газопроницаемые крышки
гСДОчшкубагор.-
5. Через 24 ч возьмите два нервых флакона из ин¬
кубатора, подсчитайте клетки:
а) Полностью удалите среду.
б) Добавьте 2 мл трипсина или смеси трипсин/
EDTA в каждый флакон.
в) Инкубируйте 15 мин.
г) Диспергируйте клетки в смеси трипсин/EDTA
и перенесите 0,4 мл суспензии в 19,6 мл
D-PBSA.
д) Подсчитайте количество клеток в электрон¬
ном счетчике клеток.
6. Повторить отбор образцов через 48 и 72 ч, как в
шаге 5.
7. Если через 72 ч или ранее обнаруживается па¬
дение pH, поменяйте среду (см. протокол 13.1;
цв. вкладку 226).
8. Продолжайте ежедневный отбор образцов для
быстро растущих культур (с периодом удвое¬
ния популяции (PDT) 12-24 h), для медленно
растущих культур (PDT >24 h), снизьте частоту
взятия образцов до 1 раза в 2 дня, пока не будет
достигнута фаза плато.
394
ГЛАВА 21. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
9. Продолжайте менять среду каждые 1,2, или 3 дня
при падении pH.
10. Окрасьте клетки в одном флаконе на 2, 5, 7, и
10 дни (см. разд. 16.4.2).
Замечание: Для подсчета клеток можно использо¬
вать гемоцитометр, однако его использование может
быть затруднено малым количеством клеток в начале
кривой роста. Если вы используете гемоцитометр,
снизьте объем трипсина до 0,5 мл, тщательно диспер¬
гируйте клетки при помощи автоматической пипетки
стараясь не вспенивать трипсин, и перенесите клетки
в гемоцитометр.
разд. 13.7.2). Данный протокол может быть исполь¬
зован вместе с упражнением 16 (см. гл. 2).
Этот тип исследования полезен для сравнения
различных сред, добавок, стимуляторов и ингибито¬
ров роста. Однако для количественного анализа при
одном или нескольких изменяющихся параметрах
предпочтительно использовать многолуночные план¬
шеты. Протокол 21.8 может также быть использован
для 12-луночных планшетов при выполнении упраж¬
нения 16 (см. гл. 2).
ПРОТОКОЛ 21.8. Кривая роста монослоя
в многолуночном планшете
Схема
Подготовить серию многолуночных планшетов с
культурами при трех различных концентрациях
клеток, ежедневно подсчитывать количество клеток
на одном планшете, пока культуры не выйдут на
стадию плато.
Материалы
Стерильные или асептически приготовленные:
• Монослойная клеточная культура, А549, Vero,
или HeLa-S3, в 75-см2 флаконе, на стадии поздней
log-фазы 1
• Трипсин неочищенный, 0,25%, с 10 мМ
EDTA 5 мл
• Ростовая среда, 100 мл, с 26 мМ NaHC03 300 мл
• D- PBSA (для предварительной промывки и
подсчета клеток) 50 мл
• Планшеты, 12-луночные 10
Нестерильные:
• Пластиковые коробки для хранения планшетов.
• С02-инкубатор или С02-источник для продува¬
ния 5% С02
Протокол
1. Трипсинизируйте клетки, как при обычном суб¬
культивировании (см. протокол 13.2).
2. Разведите клеточную суспензию до IxlO5 кл./мл,
ЗхЮ4 кл./мл, и IxlO4 кл./мл, в 25 мл среды для
каждой концентрации.
3. Посейте три 12-луночных планшета, по 2 мл
клеточной суспензии при концентрации
1х104/мл в 4 верхних лунки, по 2 мл клеток,
Зх104/мл, во второй ряд лунок и по 2 мл клеток
с концентрацией 1х105/мл в каждую лунку тре¬
тьего ряда (рис. 21.7). Добавляйте клеточную
суспензию медленно, от центра ячейки, что
позволит предотвратить завихрение по стенкам
лунок. Не встряхивайте планшеты для переме¬
шивания клеток, поскольку циркулярное дви¬
жение среды приведет к концентрации клеток
в центре ячейки.
4. Поместите планшеты во влажный С02-инку-
батор или закупоренные стеклянные коробки
с 5% С02.
5. Через 24 ч возьмите первый планшет из инку¬
батора и подсчитайте количество клеток в трех
ячейках при каждой концентрации.
а) Полностью удалите среду из трех лунок, содер¬
жащих клетки, которые будут подсчитаны.
б) Добавьте0,5 мл смеси трипсин/EDTA в каж¬
дую из трех лунок.
в) Инкубируйте планшет в течение 15 мин.
г) Добавьте 0,5 мл среды с сывороткой, диспер¬
гируйте клетки в смеси трипсин/EDTA/epe-
да и перенесите 0,4 мл суспензии в 19,6 мл
D-PBSA.
д) Подсчитайте клетки в электронном счетчике
клеток.
6. Окрасьте клетки в оставшихся лунках при каждой
плотности клеток (см. разд. 16.4.2).
Замечание: Для подсчета клеток можно использо¬
вать гемоцитометр, однако его использование может
быть затруднено малым количеством клеток в начале
кривой роста. Если вы используете гемоцитометр,
снизьте объем трипсина до 0,5 мл, тщательно дис¬
пергируйте клетки при помощи автоматической пи¬
петки, стараясь не вспенивать трипсин, и перенесите
клетки в гемоцитометр.
7. Повторите отбор образцов через 48 и 72 ч, как в
шагах 5 и 6.
8. Если через 72 ч или ранее обнаруживается па¬
дение pH, поменяйте среду (см. протокол 13.1;
цв. вкладку 226).
9. Продолжите ежедневный отбор образцов для
быстрорастущих культур (с периодом удвое¬
ния популяции (PDT) 12-24 ч), для медленно
растущих культур (PDT >24 ч) снизьте частоту
взятия образцов до 1 раза в 2 дня, пока не будет
достигнута фаза плато.
10. Продолжайте менять среду каждые 1,2, или 3 дня
при падении pH.
21.9. Клеточная пролиферация
395
21.9.3. Анализ кривой роста монослоя
1) Подсчитайте количество клеток на одну лунку, на
мл культуральной среды (см. рис. 21.6) и на см2 рос¬
товой поверхности в лунках следующим образом:
а) Первичный подсчет. Результаты подсчета, полу¬
ченные при помощи электронного счетчика или в
гемоцитометре, — это количество клеток на 1 мл
трипсинизированной культуры.
б) Количество клеток на флакон или лунку. При
использовании трипсина в объеме 1 мл коли¬
чество клеток на флакон или лунку совпадает
с результатом первичного подсчета (см. выше).
При добавлении 2 мл трипсина следует удвоить
результат, при добавлении 0,5 мл — разделить
на 2, полученный результат будет означать ко¬
личество клеток во флаконе или в лунке.
в) Количество клеток на 1 мл культуральной среды
(концентрация клеток). Разделите количество
клеток в лунке или флаконе на объем среды,
использованной при культивировании.
г) Количество клеток на 1 см2поверхности (клеточ¬
ная плотность). Разделите количество клеток
в лунке или флаконе на площадь поверхности
лунки или флакона. Поскольку обычно имеются
незначительные вариации размеров между раз¬
ными производителями оборудования, постольку
следует подсчитать ростовую поверхность имен¬
но для тех флаконов или лунок, которые вы
используете в работе. По грубой оценке, каждая
а
Клетки для , до,, окрашивания
подсчета
Клетки ^ I Клетки
1-го типа 2-го типа
Рис. 21.7. Внешний вид многолуночных планшетов. Внешний вид: (а) 12-луночного планшета с клетками в трех различных
концентрациях и лунками, для подсчета и окрашивания и (б) 24-луночного планшета с дополнительными вариациями для
двух типов клеток
396
ГЛАВА 21. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
лунка в 24-луночном планшете имеет площадь
2 см2, а в 12-луночном планшете — 3,8 см2. Фла¬
коны, рекомендуемые в протоколе 21.7, имеют
площадь поверхности 25 см2.
2 ) Составьте график зависимости клеточной плотности
(кл./см2) и клеточной концентрации (кл./мл) (оба
параметра в логарифмической шкале) от времени
(в линейной шкале) (см. рис. 21.6). Шкалы измере¬
ний по обеим вертикальным осям одинаковы, но не
учитывают фактор, связывающий площадь и объем
среды. В 25-см2 флаконе обычно помещается 5 мл
среды, покрывающей данную площадь, 0,2 мл/см2
или 5 см2/мл среды, поэтому правая ось будет свя¬
зываться с данными на левой оси фактором 5, т. е.
1х105на левой оси будет эквивалентно 2х104 на
правой оси.
3) Определите время lag-фазы, PDT и плотность кле¬
ток на плато (см. рис. 21.6; см. также разд. 21.11.1).
4) Установите соответствующую начальную плот¬
ность для рутинной процедуры пересева из прото¬
кола 21.7. Повторите эксперимент по получению
кривой роста при различных концентрациях клеток,
если это необходимо.
5) Сравните кривые роста при различных условиях
(рис. 21.8) и постарайтесь интерпретировать данные
(см. пояснения к рис. 21.8).
6) Исследуйте окрашенные клетки при различных
плотностях для того, чтобы
а) определить, является ли распределение клеток
по флакону или в лунке единообразным и растут
ли клетки на боковых сторонах лунки;
б) исследуйте различия в клеточной морфологии
при возрастании клеточной плотности;
Дни субкультивирования
Рис. 21.8. Интерпретация кривой роста. Изменения формы
кривой роста могут быть интерпретированы различными
способами, пометки в «легенде» на этом графике объясняют
отличия нормального графика от остальных кривых
в) сравните межклеточные взаимодействия в
нормальных и трансформированных культурах
клеток.
21.9.4. Объем среды, концентрация
и плотность клеток
При использовании многолуночных планшетов и
сравнении результатов с культивированием во фла¬
конах важно помнить, что объем среды, используемой
в многолуночных планшетах, часто пропорционально
выше, чем во флаконе для данной площади поверх¬
ности и плотность клеток будет выше для одной и той
же клеточной концентрации (см. разд. 13.7.3)
Если посеять 5 мл суспензии 2х104кл./мл в
25-см2 флакон, то плотность клеток при посеве будет
4000 кл./см2 (20 000x5-s-25), в то время как если такая
же концентрация клеток посеяна в 2-мл лунку 12-лу-
ночного планшета, то концентрация клеток составит
10 500 кл./см2 (20 000x2+3.8). Эта плотность вдвое
выше, чем во флаконе, и клетки достигнут плато по
крайней мере на 1 день раньше. Если проводится стро¬
гое сравнение, то должно учитываться отношение сре¬
ды с поверхностью культивирования, которое должно
быть одним и тем же, и объем 0,75 мл для одной и той же
клеточной концентрации потребует достижения такой
же клеточной плотности в многолуночном планшете,
что и в 25-см2 флаконе. К сожалению, такой низкий
объем может вызвать неравномерное распределение
клеток в результате искривления мениска жидкости, и
клетки будут распределяться с большей плотностью у
краев лунки. С уменьшением диаметра лунки эта про¬
блема усиливается, поскольку относительная кривизна
мениска возрастает с уменьшением диаметра лунки.
Многолуночные планшеты подходят для сравнения
различных условий роста, среды, сыворотки и фак¬
торов роста или цитотоксинов, но если исследования
кривой роста используются для установления условий
рутинного ведения культуры, то эти исследования
должны проводиться в тех же емкостях, в которых
затем будет проводиться субкультивирование (хотя
различие между 25-см2 флаконом и 75-см2 флаконом
будут минимальным, если объем среды в см2 остается
постоянным). Можно проводить построение кривой
роста в микротитрационных планшетах, например
как контроль для мониторинга количества клеток при
исследовании цитотоксичности (см. разд. 22.3.3). Од¬
нако, поскольку площадь поверхности роста на лунку в
этом случае очень мала, количество клеток будет также
очень малым, по крайней мере в начале эксперимента,
поэтому для подсчета клеток следует группировать
четыре-восемь лунок.
21.9.5. Суспензионные культуры
Кривая роста также может быть получена при росте
клеток в суспензии, обычно без замены среды. Цели
21.9. Клеточная пролиферация
397
исследования в этом случае обычно бывают такими
же, как в случае монослойных культур, т. е. определе¬
ние условий культивирования для рутинного ведения
культуры или исследование различий в условиях роста.
Поскольку в данном случае не требуется трипсиниза-
ция, поскольку из одного и того же сосуда может быть
отобрано несколько проб.
Варианты. Посейте по 20 мл клеточной суспензии
в два 75-см2 флакона для каждой концентрации клеток.
Отбирайте по 0,4 мл культуры из каждого флакона
ежедневно или так, как вам это необходимо. Тщательно
перемешивайте культуру каждый раз перед отбором
образцов, держите флаконы вне инкубатора в течение
минимального промежутка времени. Не меняйте пита¬
тельную среду и не добавляйте питательных веществ в
ходе построения кривой роста.
Помимо того, вы можете использовать флаконы с
перемешиванием (Techne, Integra, Bellco) и ежеднев¬
но отбирать образцы. Если в таких флаконах имеется
ПРОТОКОЛ 21.9. Кривая роста
суспензионной культуры
Схема
Подготовить серию культур при трех различных кон¬
центрациях клеток, подсчитывать клетки ежедневно,
пока культура не достигнет фазы плато
Материалы
Стерильные или асептически приготовленные:
• Суспензионная клеточная культура
• Ростовая среда, 100 мл
• D-PBSA (для подсчета клеток)
• Планшеты 24-луночные
Нестерильные:
• Пластиковые коробки для хранения планшетов
• С02 инкубатор или С02 источник д ля продувания
5% С02
Протокол
1. Добавьте клеточную суспензию в ростовой среде
в лунки планшетов в тех же концентрациях, что
и в случае монослойных культур (см. прото¬
кол 21.8).
2. Отберите по 0,4 мл культуры из трех лунок через
равные интервалы, убеждаясь, что клетки хорошо
перемешаны и полностью дезагрегировали.
3. Подсчитайте образцы на электронном счетчике
клеток (см. разд. 21.1.2) или гемоцитометре (см.
разд. 21.1.1).
4. Подсчитайте клеточную концентрацию в образце
и постройте график зависимости концентрации
клеток в логарифмической шкале от времени в
линейной шкале (см. разд. 21.9.3). Понятие «кле¬
точная плотность» неприменимо к суспензион¬
ной культуре, поскольку клетки не адгезивны
мембранное перекрытие, то отбор образцов может про¬
изводиться без вынесения их из термальной комнаты
путем прокалывания мембраны иглой и отсасывания
образца шприцом.
21.9.6. Фазы цикла роста
Цикл роста (см. рис. 21.6) может быть подразделен на
три фазы:
Lag-фаза (латентная фаза). Эта фаза представ¬
ляет собой промежуток времени после субкультиви¬
рования и пересева, в ходе которого не происходит
видимого увеличения числа клеток. В этот период
адаптации в клетках происходит замена элементов
клеточной поверхности и внеклеточного матрикса,
утраченных при трипсинизации, прикрепление к суб¬
страту, и распластывание по поверхности субстрата.
В ходе последнего процесса происходит восстанов¬
ление цитоскелета как интегральной части процесса
адгезии. Возрастает активность ферментов, таких
как ДНК-полимераза, затем начинается синтез новой
ДНК и структурных белков. Некоторые специализи¬
рованные клеточные продукты могут исчезать и не
появляться вновь до прекращения клеточного деления
при высокой плотности клеток.
Log-фаза (фаза логарифмического роста). Эта
фаза — период экспоненциального нарастания коли¬
чества клеток вслед за окончанием латентной фазы,
и она завершается одним или двумя удвоениями
популяции клеток после достижения стадии конфлю¬
ентного монослоя. Продолжительность фазы логариф¬
мического роста зависит от посевной концентрации
клеток, скорости роста клеток, и плотности клеток,
ингибирующей клеточное деление. В log-фазе фрак¬
ция делящихся клеток высока (обычно 90-100%), и
культура находится в своем наиболее репродуктивном
состоянии. Это оптимальное время для взятия образца,
поскольку популяция наиболее однородна и жизнеспо¬
собность наивысшая. Тем не менее клетки случайным
образом распределены по стадиям клеточного цикла и
для некоторых целей необходимо их синхронизировать
(см. разд. 27.5).
Фаза плато (стационарная фаза). Ближе к за¬
вершению логарифмической фазы культура становится
конфлюэнтной, т. е. вся поверхность, доступная и под¬
ходящая для роста культуры, оказывается занятой клет¬
ками, которые контактируют друг с другом. После этого
скорость роста культуры снижается, и в некоторых слу¬
чаях клеточное деление полностью прекращается после
одного или двух последующих удвоений популяции
(см. разд. 18.5.2). На этой стадии культура переходит
на стадию плато, и фракция делящихся клеток падает
до значений от 0 до 10%. Клетки становятся менее под¬
вижными; некоторые фибробласты становятся особым
образом ориентированными относительно друг друга,
398
ГЛАВА 21. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
образуя типичную сеть клеток. Волны возбуждения
цитоплазматической мембраны уменьшаются, клетка
занимает меньшую площадь поверхности, и меньшая
площадь ее контактирует со средой. Может произойти
относительное повышение синтеза специализирован¬
ных либо структурных белков, может измениться состав
и заряд клеточной поверхности. Прекращение подвиж¬
ности, снижение активных перемещений в мембране
клетки, и прекращение роста клеточной культуры, ко¬
торые происходят по достижении конфлюэнтного слоя,
были впервые описаны Abercrombie и Heaysman [1954].
Совокупность этих процессов получила название кон¬
тактного торможения. Позже было установлено, что
подавления роста нормальных клеток после конфлю-
энции достигается не только в результате контакта с
другими клетками. Оно также связано с уменьшением
степени распространенности клетки [Stoker et al., 1968;
Folkman & Moscona, 1978], нарастанием концентрации
ингибиторов, и истощением питательных веществ, в
частности факторов роста [Dulbecco & Elkington, 1973;
Stoker, 1973; Westermark & Wasteson, 1975] в среде
[Holley et al., 1978]. Это истощение может быть локаль¬
ным в статическом монослое, с образованием размытого
пограничного слоя вокруг клеток [Stoker, 1973], что
может быть преодолено орошением монослоя. Термин
плотностное ограничение (клеточной пролиферации)
обычно используется для того, чтобы подчеркнуть,
что контакты клеток не являются главным ограни¬
чивающим фактором [Stoker & Rubin,1967], а термин
контактное торможение лучше использовать для собы¬
тий, которые являются непосредственным результатом
клеточного контакта (например, снижение клеточной
подвижности и мембранной активности, происходящее
при формировании монослоя с ориентацией клеток в
отношении друг друга.
Культуры нормальных простых эпителиальных и
эндотелиальных клеток прекращают расти по достиже¬
нии конфлюэнтного монослоя и пребывают в состоянии
монослоя. Однако большинство культур, тем не менее,
при регулярном обновлении среды будут продолжать
делиться (хотя и со сниженной скоростью), несмотря
на конфлюэнтность, что приводит к формированию
многослойной клеточной структуры. Эмбриональные
легочные фибробласты и фибробласты кожи взрослого
человека, у которых имеет место контактное ингиби¬
рование подвижности, будут продолжать делиться,
укладывая слои коллагена между клеточными слоями,
до тех пор, пока не будет сформирована многослойная
структура, в оптимальных условиях из шести или семи
слоев [Kruse et al., 1970].
Эти фибробласты сохраняют упорядоченность
параллельных сеточных структур. Таким образом, тер¬
мины плато или стационарная фаза не совсем точно
определяют сущность процесса и должны использо¬
ваться с осторожностью. Культуры, склонные к спон¬
танной трансформации или трансформированные при
помощи вируса или химического канцерогена, обычно
будут достигать более высокой клеточной плотности на
стадии плато, чем их нормальные эквиваленты [Wes¬
termark, 1974] (рис. 21.9). Эта более высокая плотность
клеток связана с более высокой фракцией делящихся
клеток и снижением чувствительности клеточной
пролиферации к плотности клеток. Фаза плато в этом
случае представляет собой стадию равновесия между
Дни субкультивирования
Рис. 21.9. Плотность насыщения. В результате трансформации клеток происходит повышение плотности насыщения суспен¬
зии, в сравнении с эквивалентными нормальными клетками. Это возрастание часто сопровождается укорочением ВУП [данные
получены на клетках нормальной человеческой глии и клеточной линии глиомы; Freshney et al., неопубликованные данные]
21.10. Эффективность культивирования
399
клеточной пролиферацией и гибелью клеток. Рост этих
культур часто является независимым от прикрепления
к субстрату, т. е. они могут быть легко переведены в
суспензионные культуры.
21.9.7. Данные, получаемые
при исследовании кривой роста
Построение кривой роста производится на основании
подсчета числа клеток через определенные интервалы
времени после того, как субкультивирование позволяет
произвести измерение множества параметров, которые
должны быть найдены, чтобы охарактеризовать клеточ¬
ную линию в данных условиях культивирования.
Первый из этих параметров — продолжительность
латентного периода, или lag-время, полученное путем
экстраполяции линии, проходящей через точки фазы
экспоненциального роста, пока она не пересечется с
линией посевной концентрации, т. е. участка линии
кривой роста, соответствующей lag-фазе (см. рис. 21.6).
Затем измеряется время, начиная с момента пересева
до этой точки пересечения. Второй параметр — период
удвоения популяции (PDT), то есть время, необхо¬
димое для культуры, чтобы увеличить количество
клеток в два раза в середине экспоненциальной фазы
или log-фазы роста. Этот параметр не следует путать
с временем генерации или продолжительностью кле¬
точного цикла (см. разд. 21.12), которые определяются
измерением перехода популяции клеток от одной
точки клеточного цикла, до его возвращения к тому
же самому пункту.
Последний из обычно получаемых параметров
цикл роста — уровень плато и плотность насыщения.
Уровень плато это концентрация клеток (клеток/мл
среды) на стадии плато и зависит от типа клеток и
частоты замещения питательной среды. Плотность
насыщения — плотность клеток (кл./см2 поверхности
роста) в стадии плато. Плотность насыщения и уровень
плато трудно определить точно, поскольку устой¬
чивое состояние в стадии плато нелегко достигнуть.
В идеале, культура должна быть перфузирована, без
питательных ограничений или истощения факторов
роста. Разумный компромисс достигается путем вы¬
ращивания клеток на ограниченном пространстве,
скажем, покровного стекла диаметром 15-мм или в
фильтрующей лунке, в чашки Петри с диаметром 9 см
и объемом среды 20 мл, которая заменяется ежедневно
(см. протокол 18.3). При этих условиях ограничение
роста со стороны среды минимально, и плотность
клеток является основным фактором, оказывающим
влияние на параметры кривой роста.
Число клеток при этих условиях является более
точным и воспроизводимым, чем при выходе на плато
при обычных условиях культивирования. Обратите
внимание, что термин плато не подразумевает полное
прекращения клеточного деления, но представляет
собой устойчивое состояние, в котором деление клеток
сбалансировано утратой клеток.
Хотя неправильно говорить о «плотности насыще¬
ния» в суспензионной культуре, не прикреплящиеся
клетки могут выйти на стадию плато из-за истощения
среды. Часто, однако, клетки суспензионной культуры
в стадии плато будут входить в стадию апоптоза доста¬
точно быстро и не смогут даже дать реальное плато.
В случае нормальных клеток, устойчивое состояние
может быть достижимо путем истощения среды — без
дополнительного внесения в среду факторов роста.
В этом случае клетки пересеваются и растут, и плато
достигается без замены среды. Ясно, что условия, ко¬
торые используются в таких случаях для достижения
стадии плато, должны быть тщательно определены.
21.10. ЭФФЕКТИВНОСТЬ
КУЛЬТИВИРОВАНИЯ
Формирование колоний на чашках Петри с плотной
питательной средой при низкой плотности клеток, или
определение эффективности культивирования, являет¬
ся предпочтительным методом для анализа пролифера¬
ции клеток и их выживания (см. также протокол 22.3).
Этот подход показывает различия в скорости роста в
пределах популяции и обнаруживает различия между
изменениями в скорости роста (размер колонии) и
выживании клеток (число колоний). Нужно помнить,
тем не менее, что клетки могут расти по-другому в
виде изолированных колоний при низкой плотности
клеток. В этой ситуации даже при идеальных условиях
в живых останется меньшее количество клеток, и все
факторы, связанные с взаимодействием клеток, будут
потеряны, пока не начнет формироваться колония.
Гетерогенность клонального роста отражает различия
в способности пролиферации клонов в пределах попу¬
ляции, но эти различия могут быть незначительными
в монослое при наличии межклеточных контактов
или большей концентрации и возможности развития
межклеточных взаимодействий. При посеве клеточной
суспензии на чашки Петри в низкой концентрации
(2-50 кл./см2), клетки растут как дискретные колонии
(см. протокол 14.1 и цв. вкладку 6). Число этих колоний
может использоваться, чтобы выразить эффективность
культивирования
N3/ NjXlOO =РЕ,
где N2 — количество сформированных колоний; Nt —
количество посеянных клеток; РЕ -эффективность
культивирования (plate efficiency).
Если может быть подтверждено, что каждая ко¬
лония выросла из отдельной клетки, то этот термин
может быть заменен на эффективность клонирования.
Измерения эффективности клонирования проводятся
путем подсчета числа колоний размерами более неко¬
торого определенного (обычно около 50 клеток) рас¬
тущих на чашке после посева в низкой концентрации.
Этот термин не должен использоваться для характе¬
ристики культуры при восстановлении прикрепляю¬
400
ГЛАВА 21. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
щейся культуры при более высокой плотности клеток.
Выживание в этом случае следует характеризовать
эффективностью посева:
N3/N1xl00=SE,
где N3 — количество прикрепившихся клеток; Nt — ко¬
личество посеянных клеток; SE -эффективность
посева.
SE следует измерять по завершении прикрепления
большинства клеток, но до начала митозов. Этот момент
трудно точно определить, поскольку разрыв между
завершением прикрепления большей части клеток и
началом митотического деления может быть очень
небольшим, и эти события нередко перекрываются.
Тем не менее такой подход позволяет грубо оценить,
например, выживаемость при рутинном ведении куль¬
туры при восстановлении после замораживания, при
формировании первичной культуры и т.д.
Протокол 21.10., разработанный для определения
РЕ, может быть применен при выполнении упражне¬
ния 21 (см. гл. 2), например с небольшими вариация¬
ми, концентрации сыворотки (см. цв. вкладку 6е) и с
применением и без применения фидерного слоя (см.
протокол 14.3).
ПРОТОКОЛ 21.10. Определение
эффективности посева на чашках Петри
Схема
Посеять клетки при низкой концентрации клеток
и инкубировать, пока не образуются колонии (см.
протокол 14.1); окрасить и подсчитать колонии.
Материалы
Стерильные:
• Ростовая среда 400 мл
• Трипсин грубо очищенный, 0,25% 10 мл
• Чашки Петри, 6 см 20
• Пробирки или универсальные контейнеры для
разведений 20
Нестерильные:
• Гемоцитометр или электронный счетчик клеток
• Фиксирующая жидкость:
безводный метанол 100 мл
• D-PBSA 200 мл
• Краситель: кристалвиолет 100 мл
• Фильтровальная воронка и фильтровальная
бумага
Протокол
1. Трипсинизируйте клетки (см. протокол 13.2) для
получения суспензии единичных клеток.
2. Пока клетки трипсинизируются,
а) Пронумеруйте чашки со стороны дна.
б) Отмеряйте среду для последующего разведе¬
ния клеток (рис. 21.10). Должно быть более
чем достаточно среды для трех параллельных
разведений каждой концентрации.
3. Когда клетки приобретут округлую форму и на¬
чнут отделяться:
а) диспергируйте монослой в среде, содержащей
сыворотку или ингибитор трипсина;
б) подсчитайте клетки;
в) разведите клетки до
i) 2х 104/мл в двух 25-см2 флаконах для обыч¬
ного поддержания культуры.
ii) 2х103/мл как наивысшая концентрация для
последующего разведения.
iii) пять дальнейших разведений суспензии (ii)
должны дать 200,100,50,20, и 10 кл./мл.
4. Посейте в чашки Петри клетки в каждой из
концентраций в 5 мл среды (iii). Посейте клет¬
ки в 2 6-см чашки Петри при концентрации
2x103 кл./мл для того, чтобы использовать в
качестве контроля в случае, если клонирование
будет безуспешным (для подтверждения того, что
в исходной суспензии клетки присутствовали, по
крайней мере, при максимальной концентрации).
5. Заполните флаконы 5% С02 и поместите в ин¬
кубатор.
6. Поместите чашки Петри в прозрачную пластико¬
вую коробку и поместите во влажный С02-инку-
батор, желательно ограничить доступ к одной из
чашек и сохранить ее для клонирования.
7. Инкубируйте чашки до появления видимых не¬
вооруженным глазом колоний (1-3 нед.).
8. Окрасьте колонии кристалвиолетом.
а) Удалите среду из чашек.
б) Промойте клетки D-PBSA, слейте промывоч¬
ный раствор.
в) Добавьте 5 мл свежего D-PBSA и затем 5 мл
метанола, осторожно перемешивая (избегая
повреждения колоний).
г) Замените смесь D-PBSA: метанол (50:50) на
5 мл свежего метанола и фиксируйте клетки
в течение 10 мин.
д) Удалите метанол, добавьте профильтрован¬
ный кристалвиолет 2-3 мл на 6-см чашку, убе¬
дившись, что вся поверхность роста покрыта
красителем.
е) Окрашивайте 10 мин.
ж) Удалите краситель, верните его обратно в
бутылку, профильтровав через фильтр.
з) Промойте чашки водой, позвольте высохнуть.
9. Подсчитайте колонии в каждой чашке, исключая
те колонии, в которых имеется менее 50 клеток.
Увеличивающий окуляр может облегчить под¬
счет колоний.
Будет необходимо определить порог, за которым
колонии не будут учитываться. Если большинство
колоний содержат от стадо нескольких тысяч клеток,
то пределом следует считать 50 клеток в колонии.
На практике это фактически естественный предел
21.10. Эффективность культивирования
401
2 мл
I Разведе-
* ние 1:10
■ Под¬
счет и
разве¬
дение
Пересев в два
ювых флакона1
для хранения ЕхЮ
I Разведе-
* ние 1:20
18 мл
[19 мл
Я
Посев в 2 чашки
для контроля ^
по 5 мл с концентрацией
2000 клУмл
0.5Ш 1РазвеДе‘
w ние 1:40
0.2мп
I Разведе-
*ние 1:100
10 ООО клУчашку
0.1 мл
"1 Разведе-
*ние 1:200
Посев в
р
1200/иЛ
100/мп |
м
150/мл I
| 20/мл |
10/мл |
ш^ш
■J
1 по 5 мл с
Посев в
1 по 5 мл с
Посев в
1 по 5 мл с
Посев в
1 по 5 мл с
Посеве 1
I концентрацией
3 чашки
I концентрацией
3 чашки
I концентрацией
3 чашки
I концентрацией
3 чашки
1200 клУмл
1100 клУмл
150 клУмл
[20 клУмл
1
по 5 мл с
,100 клУмл
1000 клУчашку 500 клУчашку 250 клУчашку 100 клУчашку 50 клУчашку
Рис. 21.10. Разведение клеток для клонирования. Предлагаемый режим серийного разведения клеток дает широкий диапазон
плотности посева, подходящий для клонирования клеточной линии в первый раз или для установления линейной зависимости
эффективности посева от посевной (см. рис. 21.11). Впоследствии, когда выбрана подходящая концентрация, клетки могут быть
разведены в несколько шагов до желаемой концентрации ,
видимости невооруженным глазом. Однако, если
колонии очень малы (<100 клеток), то предел со¬
ставляет 16 клеток на колонию. Ниже 16 клеток,
что эквивалентно 4 последовательным клеточным
делениям, было бы затруднительно предполагать
продолжительное клеточное деление
21.10.1. Анализ формирования колоний
Подсчитайте эффективность культивирования (см.
Подсчет во введении к этому разделу) при каждой
посевной плотности. Эффективность культивиро¬
вания должна оставаться постоянной независимо от
плотности посева (т.е. график количества колоний в
зависимости от количества посеянных клеток должен
быть линейным). Тем не менее некоторые клетки могут
не образовывать колоний при очень низкой посевной
концентрации, и эффективность культивирования
снизится. Это влияние можно минимизировать путем
использования фидерного слоя (см. разд. 14.2.3). Если
эффективность культивирования падает при более вы¬
соких концентрациях, то это предполагает агрегацию
клеток или слияние колоний. Посевная концентрация,
которая соответствует линейному участку графика
402
ГЛАВА 21. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
2*
*
3
я
т
(0
X
>5
I
0
1
0
Q)
т
S
1
1000-
10 -
Предполагаемая
эффективность посева—ъу
4/
Реальная
эффективность
посева
Снижение
выживаемости
Агрегация или
слияние
10
100
1000
10000
Количество посеянных клеток
Рис. 21.11. Линейность эффективности посева. Если эффек¬
тивность посева остается постоянной в пределах широкого
диапазона концентраций клеток, кривая линейна (пунк¬
тирная линия), в то время как при низкой выживаемости
при низкой плотности или агрегации клеток при высокой
плотности зависимость эффективности посева от посевной
концентрации нелинейна (сплошная линия)
зависимости эффективности культивирования от ко¬
личества посеянных клеток (рис. 21.11), должна быть
выбрана как рабочая для последующих исследований.
Можно также провести исследование распределе¬
ния колоний по размерам (например, при исследовании
стимулирующей рост клеток активности среды или
сыворотки, см. разд. 11.4.3) путем подсчета количества
клеток в колонии визуально или по данным денсито¬
метрии. Для того чтобы это сделать, при помощи денси¬
тометра измеряют поглощение света после фиксации и
окраски колоний кристалл-виолетом [McKeehan et al.,
1977] или определяют размер колоний на автоматичес¬
ком счетчике колоний (см. разд. 21.10.2).
21.10.2. Автоматический счетчик колоний
Если колонии единообразны по форме и достаточно
дискретны, они могут быть подсчитаны при помощи
автоматического счетчика колоний (см. Приложение II:
Счетчики колоний), которые сканируют планшет при
помощи CCD-телекамеры и анализируют изображение с
получением мгновенного результата подсчета количества
колоний (рис. 21.12). Дискриминатор размеров позволяет
анализировать размеры колоний на основе средней вели¬
чины диаметра колоний, но диаметр колоний не всегда
пропорционален количеству клеток в колонии, поскольку
клетки могут накапливаться в центре колонии.
Хотя автоматические счетчика колоний достаточно
дороги, они могут сэкономить много времени и сделать
подсчет колоний менее субъективным. Тем не менее
они не достаточно хорошо работают, если происходит
перекрывание колоний более чем на 20% или колонии
имеют неправильные контуры.
ШЛнМяМяШмМтНаЛ -fill 1 I
Рис. 21.12. Автоматический счетчик колоний. Автомати¬
ческий счетчик колоний, применяемый для оценки эффек¬
тивности посева и исследований выживаемости (Perceptive
Biosystems)
ПРОТОКОЛ 21.11. Индекс мечения
Н3-тимидином
Схема
Вырастить клетки до соответствующей плотности.
Пометить клетки [3Н]-тимидином в течение 30 мин.
Промыть, фиксировать клетки. Удалить невключив-
шийся предшественник (т.е. 3НТ) и приготовить
авторадиограммы.
Материалы
Стерильные или асептически приготовленные:
• Культура клеток для исследования
• Ростовая среда
• Трипсин неочищенный, 0,25%
• D-PBSA
• [3Н]-тимидин, 2,0 МБк/мл (~50мкКи/мл),
75 ГБк/моль (~2Ки/моль)
• Требование безопасности: Манипуляции
с [3Н]-тимидином должны проводиться с осторож¬
ностью, поскольку он радиоактивен и генотоксичен.
Следуйте принятым Руководствам по его использо¬
ванию (см. разд. 7.7).
• Многолуночные планшеты, содержащие 13-мм
покровные стекла Thermanox (Nalge Nunc).
Нестерильные:
• Гемоцитометр или электронный счетчик клеток
• D-PBSA
• Уксуснокислый метанол (1 часть ледяной ук¬
сусной кислоты на 3 части метанола), на льду,
свежеприготовленный
• Предметные стекла
21.11. Индекс мечения
403
• Среда для заключения препаратов (например,
DPX или Permount)
• Трихоруксусная кислота (ТХУ), 0,6 М, на льду
• Деионизованная вода
• Метанол
Протокол
1. Приготовьте культуру в концентрации 2х104 кл./
мл — 5x104 кл./мл в 24-луночном планшете, со¬
держащем покровные стекла.
2. Позвольте клеткам прикрепиться и начать де¬
литься (48-72 ч), растите до желаемой клеточной
плотности.
3. Добавьте [3Н]-тимидин к среде 100КБк/мл
(около 5 мкКи/мл), и инкубируйте культуры в
течение 30 мин.
Замечание. Некоторые среды, например среды Хэма
F10hF12, — содержат тимидин (см. табл. 9.3,10.1, и
10.2). В этих случаях концентрация радиоактивного
тимидина должна быть увеличена для получения
такой же специфической активности в среде.
4. Удалите среду, содержащую метку, слив ее в
контейнер для жидких радиоактивных отходов.
5. Промойте трижды покровные стекла в D-PBSA.
Приподнимайте покровные стекла со дна лунок,
(не вынимая их полностью) при каждой промыв¬
ке, чтобы удалить изотоп из-под них.
6. Добавьте по 1 мл в каждую лунку раствор
D-PBSA: уксуснокислый метанол, 1:1, затем не¬
медленно удалите его.
7. Добавьте 1 мл уксуснокислого метанола при 4 °С
в каждую лунку, оставьте на 10 мин.
8. Выньте покровные стекла, высушите их при по¬
мощи вентилятора.
9. Приклейте покровные стекла на предметные стек¬
ла при помощи среды для заключения препарат,
клетками наверх.
10. Оставьте на ночь высохнуть.
11. Поместите стекла в 0,6 М ТХУ при 4 °С в чашке
для окрашивания и оставьте на 10 мин. Замените
ТХУ дважды в ходе экстракции, что позволит
удалить невключившиеся предшественники
(3НТ).
12. Промойте стекла деионизованной водой, затем в
метаноле и высушить стекла.
13. Приготовьте авторадиограммы (см. прото¬
кол 27.3).
14. После экспозиции авторадиограммы в течение
соответствующего периода (обычно 1-2 неде¬
ли), обработайте и окрасьте клетки (см. прото¬
кол 27.3), исследуйте под микроскопом с объек¬
тивом 40х.
15. Подсчитайте процент меченых клеток. Чтобы
исследовать репрезентативное поле, следуйте
направлениям сканирования, представленным
на рис. 21.14.
21.11. ИНДЕКС МЕЧЕНИЯ
Клетки, которые синтезируют ДНК, будут включать
метку [3Н]-тимидин (см. разд. 21.5.2.). Процент меченых
клеток, определяемый авторадиографически (см. прото¬
кол 27.3), называется индексом мечения (LI, labeling in¬
dex) [см., например, Westermark, 1974; Macieira-Coelho,
1973]. Измерение LI после 30-60-минутного периода
включения метки с [3Н]-тимидином обнаруживает боль¬
шую разницу между экспоненциально растущими клет¬
ками (LI = 10-20%) и клетками на стационарной стадии
(LI~1%). Было показано, что нормальные клетки MvlLu
имеют более низкий LI при мечении [3Н]-тимидином,
чем их неопластические производные, трансфициро¬
ванные мутантным геном ras неопластических клеток
[Khan et al., 1991] (см. цв. вкладку 14г и рис. 21.13).
Поскольку LI для клеток на фазе плато очень низок,
продолжительность времени мечения [3Н]-тимидином
может быть увеличен до 24 ч для выявления различия
при плотности насыщения.
30
25
20
к
10
5
О
Рис. 21.13. Индекс мечения. Пневмоциты норки, MvlLu,
и онкоген-трансфицированные производные были мечены
[3H]-TdR в течение 1 ч, фиксированы, покрыты авторадио¬
графической эмульсией (см. протокол 27.3), окрашены по
Гимза (см. цв. вкладку 14г), или мечены в течение 1 ч бром-
деоксиуридином (BUdR), фиксированы и окрашены иммуно-
пероксидазой с антителами против BUdR, связанной с ДНК.
Меченые ядра были подсчитаны в процентном соотношении
от общего количества в каждом случае. Mvl Lu -контрольная
клеточная линия; Ml — MvlLu, трансфицированная онко¬
геном тус\ N1 — MvlLu, трансфицированная нормальным
человеческим геном ras; Т1 — MvlLu, трансфицированная
мутантным человеческим геном ras. Т1 -клетки были туморо-
генны и имели статистически боле высокий индекс мечения
(р < 0,001) по сравнению с MvlLu, также отмечаемом при
мечении с бромодеоксиуридином [данные Khan et al., 1991]
MvlLu М1
404
ГЛАВА 21. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
ПРОТОКОЛ 21.12. Определение фракции
пролиферирующих клеток
Схема
Вносить метку в культуру на протяжении 48 ч, пе¬
риодически отбирая пробы для радиоавтографии
Протокол
Следуйте протоколу 21.10, за исключением того, что
при выполнении шага 3 инкубация должна прово¬
диться 15 мин, 30 мин, а затем 1, 2, 4,8, 24 и 48 ч.
21.11.1. Фракция пролиферирующих
клеток
Если клетки метят [3Н]-тимидином в течение различ¬
ных промежутков времени вплоть до 48 ч, то график
зависимости LI от времени представляет собой быстро
возрастающую в течение первых нескольких часов кри¬
вую, которая потом выравнивается с очень небольшим
градиентом практически до плато (рис. 21.15). Уро¬
вень этого плато, соотнесенный с вертикальной осью,
называется фракцией пролиферации культуры — т. е.
долей клеток, находящихся в клеточном цикле во
время мечения.
Анализ. Подсчитайте количество меченых клеток
как процент от общего количества клеток, используя
Рис. 21.14. Направление сканирования препаратов или
чашек. Шаблон сканирования для анализа цитологических
препаратов на стеклах или чашках. Каждая пунктирная линия
представляет собой одно поле зрения микроскопа, большой
круг — протяженность образца (например, покровное стекло,
чашка для культивирования, ячейка, или группа клеток на
стекле). Линии могут быть нарисованы при помощи фломас¬
тера или маркера светлыми прозрачными чернилами
Часы после добавления рЬЦ-тимидина
Рис. 21.15. Фракции роста. Для того чтобы определить фрак¬
цию пролиферирующих клеток, клетки постоянно метятся
[3Н]-тимидином, процент меченых клеток определяется через
определенные интервалы времени методом авторадиографии
(см. текст)
образец сканирования из протокола 21.10. Постройте
график зависимости против времени (см. рис. 21.15).
Замечание. Авторадиограммы с 3Н могут быть
приготовлены, только когда клетки находятся в моно¬
слое, затем, после включения метки, они должны быть
трипсинизированы и препараты должны быть приго¬
товлены методом капли (см. протокол 16.7), поскольку
энергия P-излучения [3Н]-тимидина слишком низкая,
чтобы проникнуть сквозь вышележащие слои клеток.
LI можно определить также путем мечения клеток
бромдиоксиуридином (BUdR), который включается
в ДНК. Это включение может быть обнаружено затем
при помощи иммуноокрашивания с анти- BudR — ан¬
тителами (Dako). Результаты, полученные этим
методом, обычно согласуются с результатами при
использовании [3Н]-тимидина. [Khan et al., 1991] (см.
также рис. 21.13).
21.11.2. Митотический индекс
Митотический индекс это фракция или процент кле¬
ток, находящихся в митозе, определяющаяся путем
подсчета митозов в окрашенных культурах как доля
от общей популяции клеток. Образец сканирования
препарата таков же, как для подсчета меченых ядер
(см. рис. 21.14).
21.11.3. Индекс пролиферации
Ряд антител, таких как Ki67 [Zhu & Joyce, 2004] или
анти-PCNA [Katdare et al., 2004], способны окрашивать
21.13. Клеточная миграция
405
клетки, находящиеся в клеточном цикле. Эти антитела
выработаны против белков, экспрессируемых во время
клеточного цикла и не экспрессируемых в покоящихся
клетках. Некоторые из этих антител направлены про¬
тив ДНК-полимеразы, эпитопы других неизвестны.
Клетки окрашиваются методом иммунофлюоресцен¬
ции или иммунопероксидазой (см. протокол 16.11), и
доля окрашенных клеток определяется цитологически
(см. рис. 21.14) или методом проточной флюоримет-
рии (см. разд. 16.9.2). Этот показатель дает более вы¬
сокий индекс, чем полученный подсчет митотического
индекса или ДНК-мечение, поскольку клетки окра¬
шиваются в ходе всего клеточного цикла. Это также
позволяет определять фракцию роста популяции.
21.12. ВРЕМЯ КЛЕТОЧНОГО
ЦИКЛА
Для определения продолжительности клеточного
цикла (времени генерации) и его составляющих частей
клетки постоянно метят при помощи BUdR и вклю¬
чение метки определяют через различные интервалы
времени методом иммунофлюоресцентной микро¬
скопии или проточной цитометрии. Для проточной
цитометрии клетки также окрашивают иодистым
пропидием и анализируют включение BUdR против
содержания ДНК, что позволяет проследить продви¬
жение клеток в клеточном цикле [Dolbeare & Selden,
1994; Poot et al., 1994].
21.13. КЛЕТОЧНАЯ МИГРАЦИЯ
Клетки в культуре, в частности фибробласты, подвиж¬
ны и могут мигрировать на значительное расстояние
по субстрату, в зависимости от клеточной плотности и
присутствия стимуляторов, таких как факторы роста.
Подвижность характеризуется по активности клеточ¬
ной мембраны, которая визуализируется замедленной
видеосъемкой. Степень подвижности трудно охарак¬
теризовать количественно, но оценка клеточной миг¬
рации может быть осуществлена путем тщательного
анализа последовательности кадров видеозаписи (см.
протокол 27.4) или путем анализа изображения или
треков, оставшихся в результате клеточного фагоци¬
тоза на поверхности чашки, покрытой коллоидным
золотом [Kawa et al., 1997]. Миграция также может
быть исследована путем движения клеток через по¬
ристую мембрану, как при исследованиях хемотаксиса
в камере Бойдена [Schor, 1994] или при изучении в
фильтрующей ячейке (разд. 18.6.3).
Глава 22
ЦИТОТОКСИЧНОСТЬ
22.1. ЖИЗНЕСПОСОБНОСТЬ,
ТОКСИЧНОСТЬ И ВЫЖИВАЕМОСТЬ
Как только клетка переносится из ее нормальной in
vivo окружающей среды, вопрос жизнеспособности, в
частности в отношении экспериментальных исследо¬
ваний, становится фундаментальным. Предыдущие
главы имели отношение к состоянию материалов для
культивирования, связанных с тканями — источника¬
ми происхождения, с характерными количественными
изменениями в росте, и фенотипической экспрессии.
Однако никакие из экспериментальных данных не мо¬
гут считаться приемлемыми, если только не доказано,
что большинство клеток сохраняют жизнеспособность.
Более того, многие эксперименты, которые проводятся
in vitro, имеют единственную цель определить потен¬
циальную цитотоксичность некоторого изучаемого
вещества, в частности если это вещество является
фармакологическим или косметическим препаратом,
и в этом случае необходимо показать, что вещество не¬
токсично, либо, если препарат предполагается исполь¬
зовать как противораковый, цитотоксичность является
существенным моментом его активности.
Новые лекарства, косметика, пищевые добавки, и
т.д. проходят мноочисленные испытания на цитоток¬
сичность до того, как они будут допущены к широкому
использованию. Эти испытания обычно включают
большое количество экспериментов на животных,
хотя в Европе в отношении этих экспериментов раз¬
рабатывается новое законодательство, которое, как
предполагают, будет введено в 2009 г. для средств
местного применения и в 2013 г. для средств общего
действия. Существует значительное давление как со
стороны общественности, так и с экономической точки
зрения требующее проводить, по крайней мере, часть
исследований цитотоксичности на культуре клеток ш
vitro. Введение специализированных клеточных линий
и интерактивных органотипических культур, а также
продолжительное использование долгоживущих куль¬
тур могут послужить обоснованным предложением для
решения этого вопроса.
Токсичность — комплекс явлений, имеющих место
in vivo, среди которых могут быть прямое поврежде¬
ние клеток, как в случае цитотоксического действия
противораковых препаратов, физиологические эффек¬
ты — такие как мембранный транспорт в почках или
нейротоксичность в мозге, воспалительное действие,
как местное, так и общее, и другие системные эффекты.
В настоящее время трудно выявлять системные и фи¬
зиологические эффекты in vitro, поэтому большинство
исследований определяют токсическое действие на
клеточном уровне, или цитотоксичность. Определе¬
ния цитотоксичности разнообразны, в зависимости
от природы исследований, а также от того, убивает
препарат клетки или просто изменяет их метаболизм.
Поскольку может потребоваться, чтобы противо¬
раковый агент уничтожал клетки, то обнаруженное
отсутствие токсичности может привести к проведе¬
нию более тонкого анализа специфических мишеней,
такого как нарушение передачи сигнала в клетке или
межклеточного взаимодействия, результатом которых
может быть развитие воспалительной или аллергиче¬
ской реакции.
Все эти исследования чрезмерно упрощают собы¬
тия, которые они моделируют и измеряют, однако они
применяются благодаря тому, что они дешевы, легко
могут быть количественно охарактеризованы и вос¬
производимы. Тем не менее накапливаются данные,
показывающие, что они не всегда адекватны целям
исследования фармакологических препаратов, которые
действуют на уровне специфических молекулярных
мишеней и точного определения метаболической
регуляции. Приблизительных тесты цитоксичности
пока еще используются, однако существует все возрас¬
тающая потребность применения более тонких тестов,
позволяющих обнаружить метаболические нарушения.
Возможно, наиболее наглядным примером является ин¬
дукция развития воспалительного или аллергического
ответов, которые не предполагают непосредственно
цитотоксического повреждения со стороны аллергена
или воспалительного агента. Этот феномен является
пока наиболее трудновоспроизводимым in vitro.
В этой главе не предполагается определить все тре¬
бования к исследованиям цитотоксичности, многие из
которых могут быть достаточно специализированы в
отношении целей исследования. Вместо этого я сосре¬
доточусь на тех аспектах, которые влияют на клеточных
рост и выживаемость. Под клеточным ростом обычно
подразумевают регенеративный потенциал клеток,
который определяется по клональному росту (см.
протокол 21.10), величине отклонения от расчетной
кривой роста популяции (например, по кривой роста,
см. протоколы 21.7 и 21.8), либо изменению клеточной
массы (общее количество белка или ДНК) или мета¬
22.2. Ограничения in vitro
407
болической активности (например, синтез ДНК, РНК
или белка; МТТ редукция).
22.2. ОГРАНИЧЕНИЯ IN VITRO
Важно то, что любые измерения in vitro могут быть ин¬
терпретированы для предсказания ответа in vivo таких
же или похожих клеток. По крайней мере, различия,
которые существуют между измерениями в условиях
in vivo и in vitro, подробно исследованы.
Фармакокинетика. Измерения токсичности in
vitro обычно предполагает наблюдения за событиями
на клеточном уровне. Например, будет очень затруд¬
нительно воспроизвести комплекс фармакокинетики
лекарственного действия in vitro, и между эксперимен¬
тальными процессами in vitro и in vivo обычно сущест¬
вуют различия во времени действия, концентрации
препарата, скорости изменения концентрации, мета¬
болизма, проницаемости тканей, клиренса и выведе¬
ния. Хотя возможно моделирование этих параметров,
например использование многоклеточных опухолевых
сфероидов для исследования проницаемости препарата
или изменением режима перфузии для воспроизведе¬
ния эффекта изменяющейся концентрации и времени
(СхТ), — большинство исследований концентриру¬
ются на прямом клеточном ответе на прямое действие
цитотоксичного агента, повышая таким образом вос¬
производимость и простоту исследований.
Метаболизм. Многие нетоксичные вещества
становятся токсичными после их метаболизма в пе¬
чени. Кроме того, многие вещества, токсичные in vitro,
могут быть детоксицированы ферментами печени.
Для испытаний in vitro, которые рассматриваются как
альтернатива испытаниями на животных, должно быть
продемонстрировано, что потенциальные токсины
достигают клеток в модели in vitro в той же форме, что
и in vivo. Этот факт для своего подтверждения может
потребовать проведения дополнительных исследо¬
ваний с применением очищенных микросомальных
ферментов печени [McGregor et al., 1988], кокультиви-
рования с активированными гепатоцитами [Guillouzo,
1989; Frazier, 1992], либо генетической модификацией
клеток-мишеней с введением им генов метаболических
ферментов под контролем регулируемого промотора
[Mac et al., 1994].
Тканевой и системный ответы. Природа отве¬
та должна также тщательно учитываться. Ответ на
действие токсина in vitro может быть измерен изме¬
нениями выживания клеток (см. протокол 22.3) или
метаболизма (см. разд. 22.3.4), в то время как главную
проблему in vivo могут составлять тканевой ответ (на¬
пример, воспалительная реакция, фиброз, поражение
почек) или системный ответ (например, лихорадка,
сосудистая реакция). Для повышения эффективности
испытаний in vitro должны быть созданы модели таких
ответов, возможно, с использованием органотипи¬
ческих культур, воссозданных на основе нескольких
клеточных типов и поддерживаемых в специальном
гормон-содержащем окружении. Не следует считать,
что сложено-комплексные тканевые и даже системные
реакции не могут быть смоделированы и воспроизве¬
дены в культуре тканей in vitro. Исследования воспа¬
лительного ответа, тератогенные нарушения, невроло¬
гические дисфункции могут быть выполнимы in vitro,
с учетом понимания межклеточных взаимодействий,
и роли гормонов в их взаимодействии с местными
паракринными и аутокринными факторами.
22.3. ПРИРОДА ИССЛЕДОВАНИЙ
Выбор типа исследования будет зависеть от изучаемого
агента, природы предполагаемого ответа и, в частности,
от клеток-мишеней. Исследования можно разделить на
пять основных классов:
1) Жизнеспособность: непосредственный или быстрый
ответ, такой как изменение мембранной проницае¬
мости или нарушение определенного метаболичес¬
кого пути; коррелирующего с клеточным делением
и выживаемостью.
2) Выживаемость: долговременное сохранение спо¬
собности к самовоспроизведению (5-10 поколений
и более).
3) Метаболизм: исследования, проводимые обычно
при помощи микротитрации, на определение про¬
межуточного метаболита. Параметры метаболичес¬
кого ответа могут быть измерены в ходе (например,
активность дегидрогеназы, синтез ДНК, РНК или
белка), во время или вскоре после окончания дей¬
ствия изучаемого агента. Проведение измерений
через два-три периода удвоения популяции после
экспозиции препарата может в большей степени
отражать потенциал клеточного роста и может кор¬
релировать с выживаемостью клеток.
4) Трансформация: выживаемость клеток в изменен¬
ном состоянии (проявление генетической мутации,
изменения контроля роста, злокачественная транс¬
формация).
5) Раздражение: ответ, аналогичный реакции воспале¬
ния, аллергии или раздражения in mvo; пока трудно
моделируется in vitro, но может оказаться возможным
исследовать раздражение путем мониторинга высво¬
бождения цитокинов в органотипических культурах.
22.3.1. Жизнеспособность
Исследования жизнеспособности проводятся для изме¬
рения доли жизнеспособных клеток после потенциально
травматичной процедуры, такой как первичная дезин¬
теграция тканей, клеточное разделение, криохранение.
Большинство тестов основывается на нарушении мем¬
бранной целостности, измеряемой по проникновению
красителя, для которого в норме клетки непроницаемы
408
ГЛАВА 22. ЦИТОТОКСИЧНОСТЬ
(например, трипановый синий, эритрозин, нафталено-
вый черный) или высвобождению красителя, который
в норме захватывается и остается в жизнеспособных
клетках (например, диацилфлюоресциин или ней¬
тральный красный). Тем не менее это прямой эффект
действия препарата, который не всегда предполагает не¬
посредственную связь с выживаемостью клеток. Более
того, процесс определения включения красителя при¬
водит к переоценке жизнеспособности. Так, например,
90% клеток, размороженных после хранения в жидком
азоте, могут не включать трипановый синий, но только
60% могут быть способны прикрепляться к субстрату
через 24 ч. Обратите внимание, что обычное определе¬
ние жизнеспособности при субкультивировании может
быть неинформативным для трипсинизированных
клеток, поскольку большинство нежизнеспособных
клеток будет утрачено при замене питательной среды
и предварительной промывке перед трипсинизации.
Точное определение жизнеспособного состояния при
субкультивировании требует того, чтобы все клетки
были извлечены из среды и предварительно промыты и
подсчитаны вместе с трипсинизированными. Тем не ме¬
нее жизнеспособность пересеянных клеток будет точно
определяться без этой поправки. Протокол 22.1 может
быть использован вместе с упр. 12 и 17 (см. гл. 2).
ПРОТОКОЛ 22.1. Оценка жизнеспособности
методом отторжения красителя
Принцип
Жизнеспособные клетки — непроницаемы для на-
фталенового черного, трипанового синего, и множес¬
тва других красителей [Kaltenbach et al., 1958].
Схема
Смешайте суспензию клеток с красителем, и иссле¬
дуйте ее при малом увеличении микроскопа.
Материалы
Стерильные или асептически подготовленные:
• Клетки для исследований, флакон для трипси¬
низации, замороженная пробирка с клетками,
предназначенными для размораживания, или
первичная дезаггрегированная культура
• Ростовая среда в соответствии с типом
клеток 20 мл
• Трипсин, 0,25% 5 мл
• D-PBSA 10 мл
Нестерильные:
• Гемоцитометр.
• Краситель для оценки жизнеспособности
• (Например, 0,4% трипановый синий или 1% на-
фталеновый черный в D-PBSA или HBSS) 1-мл
• Пастеровские пипетки
• Микроскоп
• Устройство для подсчета клеток
Протокол
1. Приготовьте суспензию клеток с высокой концен¬
трацией (-1x106 кл./мл), трипсинизацией или
центрифугированием с последующим ресуспен¬
дированием.
2. Возьмите чистое стекло для гемоцитометра и
установите покровное стекло (см. протокол 22.1 и
рис. 21.1).
3. Смешайте одну каплю суспензии клеток с одной
каплей трипанового синего или четырьмя капля¬
ми нафталенового черного.
4. Заполните камеру для подсчета (см. прото¬
кол 22.1).
5. Оставьте стекло в течение 1-2 мин (не дольше,
поскольку состояние жизнеспособных клеток
будет ухудшаться, и они будут окрашиваться).
6. Поместите стекло под микроскоп и, используя
объектив 10х, найдите в поле зрения сетку для
подсчета клеток (см. рис. 21.1).
7. Подсчитайте общее количество клеток и число
окрашенных клеток.
8. Вымойте гемоцитометр и верните в коробку.
Анализ. Подсчитайте процент неокрашенных клеток.
Это число представляет собой процент жизнеспособных
клеток, определенный этим методом. Если на шаге 3
использованы соответствующие объемы клеточной
суспензии и красителя, то этот метод определения жиз¬
неспособности может быть включен в протокол 22.1.
ПРОТОКОЛ 22.2. Оценка жизнеспособности
методом поглощения красителя
Принцип
Жизнеспособные клетки захватывают диацетил-
флюоресцеин и гидролизуют его до флюоресцеина,
для которого мембрана живой непроницаема [Rotman
& Papermaster, 1966]. В результате живые клетки
флюоресцируют зеленым светом; а мертвые клетки
нет. Нежизнеспособные клетки могут быть окрашены
пропидиум иодидом и впоследствии флюоресциро¬
вать красным. Жизнеспособность выражают как про¬
центную долю клеток, флюоресцирующих зеленым.
Этот метод может применяться при CCD анализе или
проточной цитометрии (см. разд. 21.7.2).
Схема
Окрасить клеточную суспензию в смеси пропидия
иодида и диацетилфлюоресцеина, и исследовать
клетки методом флюоресцентной микроскопии или
проточной цитометрии.
Материалы
Стерильные или асептически подготовленные:
• Суспензия клеток
• Диацетат флюоресцеина, 10 мкг/мл, в HBSS
22.3. Природа исследований
409
• Иодид пропидия, 500 мкг/мл
• Нестерильные:
• Флюоресцентный микроскоп
• Фильтры:
• Флюоресцеин: возбуждение 450/590 нм, излуче¬
ние LP 515 нм
• Иодид пропидия: возбуждение 488 пт, излучение
615 нм
Протокол
1. Приготовьте суспензию клеток, как для оценки
жизнеспособности, методом освобождения кра¬
сителя (см. протокол 22.1), но в среде без фенола
красного.
2. Добавьте флюоресцентную окрашивающую
смесь, окрасьте смесь в пропорции 1:10, до ко¬
нечной концентрации 1 мкг/мл диацилфлюо-
ресцеина и 50 мкг/мл иодида пропидия.
3. Инкубируйте клетки при 37 °С в течение
10 мин.
4. Поместите каплю клеток на стекло для микроско¬
па, покройте покровным стеклом, исследуйте клет¬
ки при помощи флюоресцентной микроскопии.
Анализ. Клетки, которые флюоресцируют зеленым,
являются жизнеспособными, в то время как нежиз¬
неспособные светятся красным. Жизнеспособность
может быть выражена как процент клеток, которые
флюоресцируют зеленым, от общего количества клеток.
Окрашенная клеточная суспензия также может быть
проанализирована методом проточной цитометрии
(см. разд. 21.7.2).
При цитотоксичности вы¬
сокой степени или острого
действия цитотоксина, ис¬
пользуйте флаконы в тече¬
ние 1-72 ч в зависимости
от характера цитотоксина и
продолжительности клеточ¬
ного цикла
> Трипсинизируйте монослой, ресус¬
пендируйте и подсчитайте клетки
1 1
Я
—►
ш
&U Л
» J
Для низкой степени цито-
токсического действия или
предполагаемого хрониче¬
ского действия используйте
клонированные культуры
в течение 1-3 недель, в
зависимости от природы
цитотоксина
Проведите серийное разведение
до концентрации 10 и 200 кл./мл
(в зависимости от количества
клеток в контроле)
Инкубируйте 1-3 недели (в зави¬
симости от темпов роста)
Увеличение концентрации цитотоксина
I
Рис. 22.1. Клоногенные исследования монослойных кле¬
ток. Клетки трипсинизируют, подсчитывают и разводят так
же, как в случае клонирования с разведением (см. прото¬
кол 14.1). Испытываемое вещество может быть добавлено
перед трипсинизацией или после клонирования (см. сек¬
цию 22.3.2). Колонии фиксируют и окрашивают, когда они
достигают размера, достаточного для визуального подсчета,
но прежде, чем они начинают перекрывать друг друга
Окрашивание нейтральным красным. Живые
клетки захватывают нейтральный красный из куль¬
туральной среды при его концентрации 40 мкг/мл,
задерживая его в лизосомах. При этом нейтральный
красный не удерживается нежизнеспособными клет¬
ками. Захват нейтрального красного количественно
определяется после фиксации клеток в формалине и
растворении красителя в уксуснокислом этаноле, что
позволяет получить результаты путем считывания
окраски в лунках планшетов на планшетном сканере
ELISA при 570 нм [Borenfreund et al., 1990; Babich &
Borenfreund, 1990]. Нейтральный красный способен
преципитировать, поэтому среду с красителем перед
использованием обычно инкубируют в течение ночи, а
затем центрифугируют. Это исследование не определя¬
ет общее количество клеток, но показывает снижение
поглощения, связанное с утратой жизнеспособных
клеток и полностью автоматизировано.
22.3.2. Выживаемость
Хотя кратковременные исследования удобны и обычно
быстры и легко осуществимы, они выявляют только
погибшие клетки, (т. е. те, которые стали проницаемы
для красителя) в ходе исследования. Однако иногда
клетки, подвергнувшиеся повреждающему действию
(в т.ч. изучения, противораковых препаратов), обна¬
руживают эффект через несколько часов. Природа
исследований, необходимых для проведения измере¬
ния жизнеспособности, в этих случаях должна быть
отлична, поскольку к тому времени, когда будут про¬
изведены измерения, погибшие клетки могут исчез¬
нуть. Таким образом, в ходе долгосрочных испытаний
выживаемость должна быть отражена точнее, нежели
краткосрочными исследованиями токсичности, ре¬
зультаты которых могут быть обратимыми. Выжи¬
ваемость подразумевает сохранение регенеративных
способностей и обычно измеряется эффективностью
посева (см. протокол 21.10). Эффективность посева
позволяет измерить выживаемость путем определения
способности к пролиферации в течение нескольких
поколений, предполагая, что эффективность посева,
достаточно высокая для того, чтобы обеспечить реп¬
резентативный подсчет колоний, позволяет охарак¬
теризовать всю клеточную популяцию в целом. Хотя
определение эффективности посева нельзя считать
идеальным методом, при значении выше 10% резуль¬
таты его являются допустимыми.
410
ГЛАВА 22. ЦИТОТОКСИЧНОСТЬ
ПРОТОКОЛ 22.3. Клоногенный анализ
прикрепившихся клеток
Схема
Подействовать на клетки экспериментальным
агентом в диапазоне концентраций в течение 24 ч.
Трипсинизировать клетки, посеять их в низкой
концентрации клеток, и инкубировать 1-3 недели.
Окрасить клетки (см. цв. вкладку 6а, е) и подсчитать
число колоний (см. рис. 22.1).
Материалы
Стерильные:
• Ростовая среда
• D-PBSA
• Неочищенный трипсин, 0,25%
• Тестируемое химическое соединение, в деся¬
тикратной максимальной концентрации, раз¬
веденное в бессывороточной среде; проверьте
pH и осмоляльность исследуемого раствора, и
отрегулируйте, если необходимо.
• Флаконы, 25 см2
• Чашки Петри, 6 или 9 см, помеченные со стороны
дна
Нестерильные:
• D-PBSA
• Метанол
• Кристаллвиолет 1%
• Гемоцитометр или электронный счетчик клеток
Протокол
1. Приготовьте ряд культур в 25-см2 флаконах:
три для каждой из шести концентраций дей¬
ствующего агента, и три контрольных. Посейте
клетки в концентрации 5х104кл./мл в 4,5 мл
ростовой среды, инкубируйте в течение 48 ч, за
это время культуры разовьются до log-стадии (см.
разд. 21.9.2).
2. Приготовьте последовательное разведение хими¬
ческого соединения до объема 2 мл при 10-крат-
ной концентрации относительно конечной.
а) Если вы проверяете состав впервые, исполь¬
зуйте и 5-кратные разведения в диапазоне
103-105.
б) Если вы можете предполагать приблизитель¬
ную токсичную концентрацию, выберите
более узкий диапазон (10-100).
3. Разведите каждую концентрацию 1:10, добавляя
0,5 мл из 10х концентратов в каждый из трех
флаконов для каждой концентрации, начиная с
контрольной среды и (без добавления агента) и в
прогрессирующей концентрации от самой низкой
до самой высокой.
4. Верните флаконы в инкубатор.
5. Если токсическое действие происходит медленно
или частично обратимо, повторите шаг 3 дваж¬
ды; т.е. подвергните культуры действию агента
в течение 3 дней, заменяя ежедневно среду и
добавляя тот же агент заново. Для быстродей¬
ствующих соединений может быть достаточна
экспозиция в течение 1 ч.
6. Удалите среду из каждой группы из трех флако¬
нов (от самой низкой концентрации действую¬
щего агента до самого высокой) и трипсинизи¬
руйте клетки.
7. Разведите и посейте клетки в чашки Петри при
требуемой для клонального роста плотности (см.
протокол 14.1), разводя все культуры до той же
концентрации, что и контрольная. Начинайте ра¬
боту с флаконов, подвергнутых действию самой
высокой концентрации до самой низкой, и затем
берите контроль.
8. Инкубируйте культуры до формирования ко¬
лоний.
9. Фиксируйте культуры в абсолютном метаноле и
окрашивайте их в течение 10 мин в 1% кристалл-
виолете (см. протокол 16.3).
10. Промойте чашки в проточной воде, дайте стечь и
подсушите, установите в перевернутое положе¬
ние, чтобы высушить.
11. Подсчитайте колонии с > 50 клетками (> 5 гене¬
раций).
Анализ кривой выживания
12. Вычислите эффективность посева при каждой
действующей концентрации.
13. Вычислите относительную эффективность посева:
эффективность посева при каждой концентрации
как доля от контрольной. Это — доля выживания.
14. Постройте зависимость доли выживания в лога¬
рифмической шкале от концентрации в линейной
или логарифмической шкале, в зависимости от
используемого диапазона концентраций (см.
рис. 22.2).
15. Определите 1С50или IC^, которые являются зна¬
чением концентрации цитотоксического агента,
обеспечивающего ингибирование формирования,
соответственно, 50% или 90% колоний.
Поскольку применяется полулогарифмический
график, лучше использовать 1С90, поскольку
это значение, более вероятно, будет падать на
линейную часть кривой, принимая во внимание,
что IC50 имеет тенденцию попадать на колено
кривой, давая менее стабильное значение
16. Проанализируйте кривую на предмет различий
в чувствительности:
а) Наклон кривой и длина колена. Более пологий
наклон и/или более длинное колено свиде¬
тельствует о пониженной чувствительности;
более крутой наклон и/или более короткое ко¬
лено означает повышенную чувствительность.
Как длина колена, так и наклон влияют на
значение 1С50и ICgo, хотя более существенное
различие может наблюдаться для 1С90 (см.
рис. 22.3). IC90часто используется как простая
производная.
22.3. Природа исследований
411
б) Резистентная доля. Доля
ком конца кривой. ^
в) Полная резистентность характ^йзуёп^ от-
сутствиемкакого-то градиента на кривой*
г) Область под кривой: Coboyсупность Кримаи:
выживания может быть сравнена вычисле-
нием области под кривой, но это делается
для определенных целей и математически не
достоверно.
Изменяемые параметры
в определении выживаемости
Концентрация действующего агента. При первой по¬
пытке определить действующую концентрацию должен
быть использован широкий диапазон концентраций в
логарифмической градации (т.е., 1х106М, 1х105М,
1х104М, 1х103и контроль). На основе первой серии экс¬
периментов диапазон концентраций (логарифмический
или линейный) должен быть сужен и в дальнейшем
эксперименты проводятся в этом диапазоне.
Неизменяемые концентрации агентов. Некоторые
условия сложно изменить, например качество среды,
воды или пластика. В этих случаях можно менять кон¬
центрацию сыворотки. Сыворотка может оказывать
маскирующий эффект для веществ с низким уровнем
токсичности. Токсичность можно выявить в отсутствие
сыворотки или при ее низкой концентрации.
Продолжительность воздействия агента. Некото¬
рые агенты действуют быстро, в то время как другие
Концентрация цитотоксина
Рис. 22.2. Кривая выживания. Полулогарифмический гра¬
фик доли выживающих клеток (отношение количества коло¬
ний, формирующиеся из экспериментальных клеток к числу
колоний, формирующихся в контроле) против концентрации
цитотоксина. Типичная кривая имеет «колено», и падение
ICgo в линейном диапазоне кривой. Значение IC50, попадаю¬
щее на «колено», обладает меньшей стабильностью.
1 -
л
ш
к
5 0.01-
0.001
01 23456789 10
Концентрация цитотоксина
Рис. 22.3. Интерпретация кривых выживания. Полуло¬
гарифмический график зависимости выживания клеток от
концентрации цитотоксина. Наклон увеличивается с увели¬
чением чувствительности и уменьшается со снижением чув¬
ствительности, пока кривая не становится полностью плоской
для полностью устойчивых клеток. Частичная резистентность
клеток может быть выражена как стабильная фракция, пока¬
занная на кривой, выравнивающейся к концу.
действуют медленно. При действии ионизирующей
радиации, например, нужно лишь несколько минут,
чтобы достичь требуемой дозы облучения, в то время
как продолжительность действия некоторых антиме-
таболических препаратов, эффективность действия
которых зависит от продолжительности клеточного
цикла, может составлять несколько дней до достиже¬
ния обнаруживаемого эффекта. Продолжительность
воздействия (Т) и концентрация агента (С) связаны,
хотя величина СхТ не всегда является постоянной.
Пролонгированное действие может повышать чув¬
ствительность сверх предполагаемой на основании
соотношения СхТ в результате влияния клеточного
деления и кумулятивности повреждений.
Время воздействия агента. Когда исследуемый агент
растворим и, предположительно, токсичен, следует
использовать схему, описанную в протоколе 22.3, но
когда качество агента неизвестно и можно ожидать как
стимуляции, так и минимального эффекта (например,
более вероятно 20% ингибирование, чем многократное
подавление), то агент может быть введен не в стадии
преинкубации, а в фазе клонального роста. Подтверж¬
дение предполагаемой токсичности, например для ци-
тотоксичных препаратов, требует точных и аккуратных
исследований, с минимальным временем воздействия,
сравнимым с временем действия in vivo, применением
агента до клонального роста. Подтверждение отсут¬
ствия токсичности (например, для водопроводной воды
или нетоксичных фармакологических препаратов)
требует более жестких испытаний, с пролонгированным
длительным временем экспозиции.
412
ГЛАВА 22. ЦИТОТОКСИЧНОСТЬ
Клеточная плотность в ходе воздействия агента.
Плотность клеток в ходе воздействия агента может ме¬
нять ответ клеток на действие агента; например, клетки
HeLa менее чувствительны к алкилирующему агенту
мастину при высокой плотности клеток [Freshney et al.,
1975]. Для определения действия клеточной плотности
на эффект следует варьировать ее на стадии преинку-
бации в ходе воздействия препаратом.
Клеточная плотность при клонировании. Коли¬
чество колоний может падать при высоких концентра¬
циях токсичного агента, однако возможно компенсиро¬
вать этот эффект посевом большего количества клеток
так, что при каждой концентрации сформируется
примерно одинаковое число колоний. Эта процедура
снижает риск влияния на выживаемость клеток низкой
клональной плотности и улучшает статистическую на¬
дежность. Вместе с тем может возрастать вероятность
ошибки, поскольку клетки, подвергнутые действию
более высокой концентрации препарата, будут посеяны
при более высокой клеточной концентрации, и этот
фактор может повлиять на выживаемость. Поэтому
предпочтительно сажать клетки на предварительно
сформированный фидерный слой, плотность которого
составляет (5x103 кл./см2,), что значительно превышает
количество клонируемых клеток. Выполнение этого
этапа позволит надеяться, что клеточная плотность
Концентрация 5-фторурацила, М
Рис. 22.4. Влияние фидерного слоя на долю выживания
клеток. Клетки человеческой глиомы были посеяны на чашки
в присутствии (пунктирная линия) и в отсутствие (сплошная
линия) фидерного слоя после обработки 5-фторурацилом в
различных концентрациях!. При концентрации препарата
1x10-4 М только в присутствии фидерного слоя выявляется
10% резистентная доля. В отсутствие фидерного слоя не¬
большое количество колоний показывает, что резистентная
фракция была неспособна выжить без поддержки
будет постоянной, независимо от клонального выжива¬
ния, которое вносит незначительный вклад в значение
общей плотности клеток. Заметьте, что клонирование
на фидерном слое может иногда выявить резистентную
фракцию клеток, которая не проявляется без исполь¬
зования фидерного слоя (рис. 22.4).
Размер колоний. Некоторые агенты являются ци-
тостатичными (т.е. они ингибируют клеточную проли¬
ферацию), но не цитотоксичными, и в ходе продолжи¬
тельной экспозиции они могут приводить к уменьшению
размеров колоний без снижения количества колоний.
В этом случае размер колоний следует определять
путем измерения оптической плотности [McKeehan et
al., 1977], автоматизированного подсчета колоний или
визуального подсчета количества клеток в колонии.
Для подсчета колоний начальное количество клеток
в колонии (например, 50, как в протоколе 21.9) явля¬
ется абсолютно произвольным, и предполагается, что
большинство колоний значительно превышает это
число. Колонии должны расти до достаточно больших
размеров (>1х103 клеток), когда рост более крупных
колоний станет замедляться, а меньшие, хотя и жиз¬
неспособные, колонии начнут захватываться более
крупными.
Для определения размера колоний окрасьте культуры
до того, как снизится скорость роста крупных колоний,
и подсчитайте все колонии.
Растворители. Некоторые агенты, которые долж¬
ны быть исследуемы, имеют низкую растворимость
в водной среде, и может быть необходимым исполь¬
зовать органический растворитель для того, чтобы их
разводить. Для этих целей обычно используют этанол,
пропиленгликоль и диметилсульфоксид, но они сами по
себе могут быть токсичны для клеток. Следовательно,
для получения растворов нужно использовать мини¬
мальные концентрации растворителей. Агент может
быть растворен, например, в растворителе с высокой
концентрацией, например в 100%-м этаноле, а затем
постепенно разведен в ССР и доведен до конечной
концентрации с учетом разведения в среде. Конечная
концентрация растворителя в среде должна быть <0,5%,
кроме того, следует включить контроль действия рас¬
творителя (т. е. контроль роста клеток при добавлении
той же конечной концентрации растворителя без добав¬
ления испытываемого агента).
Будьте осторожны при использовании органических
растворителей, соприкасающихся с пластиком или ре¬
зиной. Лучше всего для хранения органических раство¬
рителей использовать стекло и использовать пластик,
только когда концентрация растворителя <10%.
Хотя подсчет эффективности является одним из
наилучших методов для исследования степени выжи¬
вания клеток, его следует использовать, только если
эффективность клонирования достаточно высока для
формирования колоний, так чтобы репрезентатив¬
22.3. Природа исследований
413
ность для популяции в целом была достаточно высока.
В идеале, это означает, что в контроле эффективность
посева должна составлять 100. На практике, однако,
это редко осуществимо и в контроле эффективности
посева составляет обычно 20% или менее, что считается
приемлемым.
22.3.3. Анализ, основанный
на клеточной пролиферации
Подсчет клеток через несколько дней после культиви¬
рования также может быть полезен для определения
действия различных компонентов на клеточную про¬
лиферацию, но, по крайней мере на ранних стадиях
исследований, требуется составить полную кривую
роста (см. протоколы 21.7,21.7,21.8 и 21.9), поскольку
подсчет клеток, проводимый в отдельной точке време¬
ни, может быть неоднозначным (см. рис. 21.8, день 7).
Анализ кривой роста с использованием подсчета кле¬
ток осуществим только для относительно небольшого
числа количества образцов, поскольку при широком
скрининге он становится слишком трудоемким. В
случаях, когда исследуется много образцов, может
использоваться подсчет клеток в отдельный момент
времени, например количество клеток через три или
пять дней после экспозиции агента. Время следует
выбирать в log-фазе, предпочтительно в ее середине
исходя из значений для контрольных клеток. Любой
значительный эффект должен быть подтвержден
полной кривой роста в течение всего цикла роста или
какими-то альтернативными исследованиями, таки¬
ми как кривая выживания, полученная при помощи
клоногенных исследований (см. протокол 22.3) или
МТТ-тест (см. протокол 22.4).
22.3.4. Метаболический анализ
цитотоксичности
Исследования эффективности посева связаны с высо¬
кой интенсивностью труда и затратой времени для их
проведения и анализа, в частности когда используется
большое количество образцов и продолжительность
каждого эксперимента может продолжаться от 2 до
4 недель. Более того, некоторые клеточные линии об¬
ладают низкой эффективностью посева, в частности,
нормальные свежевыделенные клетки, поэтому раз¬
работан ряд альтернативных методов определения
цитотоксичности клеток при их высокой плотности.
К этим методам относится метод использования мик-
ротитрационных планшетов (см. разд. 22.3.5). Однако
ни один из этих тестов не измеряет непосредственно
выживаемости. Вместо этого определяют чистое уве¬
личение количества клеток (прирост культуры; см.
разд. 22.3.3), увеличение общего количества белка или
ДНК, либо изменение постоянной метаболической
активности, такой как редукция тетразолиевой соли
формазана, либо синтез белка или ДНК. Выживаемость
в этих случаях определяют как сохранение метаболи¬
ческой или пролиферативной способности клеточной
популяции как целого в течение некоторого времени
после прекращения токсического воздействия. Тем не
менее такие исследования не могут обнаружить разли¬
чия между снижением метаболической или пролифе¬
ративной активностями в отдельных клетках, а также
обнаружить уменьшение количества клеток и, таким
образом, любое новое или исключительное явление,
обнаруженное этими методами, должно быть подтверж¬
дено клоногенными исследованиями выживания.
22.3.5. Микротитрационный анализ
Введение многолуночных планшетов революционизи¬
ровало метод увеличения числа образцов в культуре
тканей. Эти планшеты экономичны в использовании,
годятся для автоматического обслуживания и анализа
и могут обладать хорошими оптическими свойствами.
Наиболее популярны в настоящее время 96-луноч-
ные микротитрационные планшеты, каждая ячейка
имеет размеры области роста 28-32 мм2, вмещает 0,1
или 0,2 мл среды и до IxlO5клеток. Микротитрация
предлагает метод, посредством которого большое
количество образцов может быть исследовано од¬
новременно. Используя этот метод, вся популяция
подвергается действию агента, и впоследствии опре¬
деляется жизнеспособность, обычно путем измерения
метаболических параметров, таких как концентрация
АТР или NADH/NADPH. Большое количество на¬
боров предлагаются компанией Promega (см. При¬
ложение III). Конечной точкой микротитрационного
анализа обычно является оценка количества клеток.
Хотя этот результат может быть достигнут прямо
путем подсчета клеток или методом непрямых мето¬
дов, таких как включение изотопа, оценка жизнеспо¬
собности клеток при помощи МТТ-теста [Mosmann,
1983] широко распространена и рассматривается как
оптимальная методика [Cole, 1986; Alley et al., 1988].
МТТ — это желтый водорастворимый тетразолиевый
краситель, который превращается под действием
живых клеток в красный формазановый продукт,
нерастворимый в водных растворах. На редукцию
МТТ может оказывать влияние ряд факторов [Vistica
et al., 1991]. Процесс исследования, описанный в про¬
токоле 22.4, предоставленном Jane Plumb из Центра
онкологии и прикладной фармакологии (University
of Glasgow, Scotland, United Kingdom), позволит по¬
лучить одинаковые результаты в качестве стандарт¬
ного клоногенного исследования [Plumb et al., 1989].
Он иллюстрирует использование микротитрации в
исследовании противораковых препаратов, но может
быть применим, с небольшими модификациями, для
любых исследований цитотоксичности.
Варианты МТТ-исследования
Другие применения. Похожее исследование мо¬
жет быть использовано для определения клеточной
414
ГЛАВА 22. ЦИТОТОКСИЧНОСТЬ
КЛЕТКА
А
КЛЕТКА
В
| Нулевой ряд (без клеток)
§§§•••#•••••
••••••••••••
••••••••••••
:::::::::::
••••••••••••
••••••••••••
••••••••••••
КЛЕТКА
А
КЛЕТКА
В
•••••••©ООО#
••••••••ООО#
••••••••ООО»
ффффффффоооф
••••••••ооое
ффффффффоооф
^ Нулевой ряд Контроль (только^
(без клеток) растворитель)
КЛЕТКА
А
КЛЕТКА
В
КЛЕТКА
А
КЛЕТКА
В
• ••••
• ••••
• ••••
:::::
• ••••
• ••••
• ••••
• •••
• •••
• •••
• •••
• •••
• •••
• •••
• •••
• ••
• ••
• ••
• ••
• ••
• ••
• ••
• ••
IC^A
ФООООООФФФФФ
фооооооффффф
ФООООООФФФФФ
ф.о.о..о..о.о.о.ф.ф.ф.ф.ф.
ФОООФФФФФФФФ
ФООФФФФФФФФФ
ФОООФФФФФФФФ
ФОООФФФФФФФФ
Установите микротитрацион-
ный планшет, инкубируйте
приблизительно до двойного
удвоения популяции
Увеличение концентрации препарата или токсина
Когда клетки находятся в
фазе экспоненциального
роста, добавьте препарат или
токсин
Удалите препарат и позволь¬
те клеткам восстановиться в
ростовой среде
После двух или трех пери¬
одов удвоения популяции
удалите среду и замените на
среду с МТТ или ХТТ. Через
3-4 ч пропустите через
планшетный сканер, исполь¬
зующее поглощение света,
для получения кривой инги¬
бирования, вычислите Юн
t
ICs*B
Рис. 22.5. Микротитрационное исследование. Стадии
испытания двух различных линий клеток, подвергнутых
воздействию широкого диапазона концентраций одного и
того же препарата, степень выживания которых оценивалась
методом МТТ (см. протокол 22.4). Дальняя левая колонка
не имеет клеток и может использоваться как нулевой ряд, в
установках планшетного сканера. Это исследование проводят
при использовании закрытых пластин, когда все ячейки экви¬
валентны; однако при исследовании планшетов с крышками
имеется риск влияния краевого эффекта, вероятно, из-за
испарения, поэтому лучше оставить крайние левые и крайние
правые колонки незаполненными (т. е., только со средой, как
в протоколе 22.4); некоторые исследователи оставляют также
незаполненными верхний и нижний ряды
радиочувствительности [Carmichael et al., 1987b].
МТТ-тест может быть использован для определения
количества клеток после ряда воздействий, отличных
от воздействия цитотоксичных препаратов, таких как
стимуляция, факторами роста. Однако в этом случае
существенной является необходимость убедиться, что
воздействие само по себе не оказывает действия на
способность клеток к редукции красителя.
Продолжительность воздействия. Как в случае
клоногенного анализа (см. протокол 22.3), некоторые
агенты могут действовать быстрее, и период воздей¬
ствия и восстановления укорачивается. Клетки должны
оставаться в экспоненциальной фазе роста на всем
протяжении исследования (см. разд. 13.7.2 и 21.9.2),
ПРОТОКОЛ 22.4. Оценка цитотоксичности
при помощи МТТ-теста
Принцип
Клетки в экспоненциальной стадии роста подверга¬
ются действию цитотоксического препарата. Про¬
должительность действия обычно определяется как
время, требуемое для максимального повреждения,
но также связано со стабильностью препарата. После
удаления препарата клеткам позволяют развиваться
в течение двух-трех удвоений популяции (PDTS),
чтобы увеличить различие между клетками, которые
остаются жизнеспособными и способны к быстрому
увеличению популяции и теми, которые остаются
жизнеспособными, но не могут культивироваться.
Затем определяют число остающихся в живых
клеток косвенно по уменьшению МТТ-окрашива-
ния. Количество образовавшегося МТТ-формазана
может определяться спектрофотометрически, если
только МТТ-формазан растворен в подходящем
растворителе.
Схема
Инкубируйте монослойные культуры в микро-
титрационных планшетах в выбранном диапазоне
концентраций цитотоксичного препарата (рис. 22.5).
Удалить препарат и заменять питательную среду в
планшетах ежедневно до двух-трех PDTs; затем еще
раз замените питательную среду, и добавьте МТТ в
каждую лунку. Инкубировать планшеты в темноте
в течение 4 ч, затем удалите среду и МТТ. Разведите
нерастворимые в воде кристаллы МТТ-формазан в
DMSO, добавьте буфер, чтобы довести конечное зна¬
чение pH, и измерьте поглощение в автоматическом
спектрофотометрическом планшетном сканере.
Материалы
Стерильные:
• Ростовая среда
• Трипсин (0,25% + EDTA, 1 мМ, в PBSA)
• МТТ: 3-(4,5-диметилтиазол -2-ил)-2,5-дифенил-
тетразолина бромид (Sirma), 50 мг/мл, стерили¬
зованный фильтрацией
• Глициновый буфер Соренсена (0,1 М глицин,
0,1 М NaCl, pH доводится до 10,5 при помощи
1 М NaOH)
• Микротитрационные планшеты (Iwaki)
• Наконечники для автоматических пипеток,
предпочтительно в автоклавируемой коробке для
наконечников
• Чашки Петри (неавтоклавируемые), 5 см и 9 см
или емкость (Corning)
• Универсальные контейнеры или пробирки, 30 мл
и 100 мл
Нестерильные:
• Пластмассовая коробка (чистая пенопластовая
коробка для хранения планшетов)
22.3. Природа исследований
415
• Многоканальная автоматическая пипетка
• Диметилсульфоксид (DMSO)
• DMSO-диспенсер (не обязательно): Labsystems
Microplate Dispenser (Каталожный номер5840 127,
Thermo Electron)
• ELISA-планшетном сканере (Molecular Devices,
SOFTmax PRO; см. также Приложение II)
• Держатель планшета для центрифуги (для роста
клеток в суспензионной культуре)
Протокол
Осаждение клеток:
1. Трипсинизируйте субконфлюэнтную монослой-
ную культуру, соберите клетки в ростовой среде,
содержащей сыворотку.
2. Центрифугируйте суспензию (5 мин при 200 г)
до образования осадка. Ресуспендируйте клетки
в ростовой среде, подсчитайте их.
3. Разведите клетки до 2,5-50х103 кл./мл, в зави¬
симости от скорости роста клеточной линии, в
расчете 20 мл суспензии клеток на микротитра-
ционный планшет.
4. Перенесите суспензию клеток в 9-см чашку Петри,
при помощи многоканальной автоматической пи¬
петки добавьте по 200 мкл в каждую лунку 10 цент¬
ральных колонок плоскодонной 96-луночной
пластины (80 лунок), начиная со 2-й колонки и
заканчивая колонкой 11, таким образом, помещая
0,5-10х103 клетки в каждую лунку.
5. Добавьте 200 мкл ростовой среды в восемь лунок
в колонки 1 и 12. Колонка 1 будет использоваться
как нулевой уровень для планшетного сканера;
колонка 12 поможет поддержать влажность ко¬
лонки Ии минимизирует « краевой эффект».
6. Поместите планшеты в пластмассовую коробку
и перенесите в увлажненную атмосферу, где
инкубируйте при 37 °С в течение 1-3 дней, за
которые клетки перейдут в экспоненциальную
фазу к моменту добавления препарата.
7. Для неадгезирующих клеток приготовьте суспен¬
зию клеток в свежей среде. Разведите клетки до
концентрации до 5-100х103кл.//мл и пересейте
100 мкл из суспензии в круглодонные 96-луноч¬
ные планшеты. Добавьте немедленно препарат в
эти планшеты
Добавление препарата:
8. Приготовьте ряд последовательного пятикратного
разведения цитотоксичного препарата в ростовой
среде для получения восьми концентраций. Этот
набор концентраций должен быть выбран так,
чтобы самая высокая концентрация убивала боль¬
шинство клеток, и самая низкая не действовала
на клетки. Если токсичность препарата известна,
может использоваться меньший диапазон концен¬
траций. Обычно используются три планшета для
каждого препарата, чтобы провести троекратное
определение в пределах одного эксперимента.
9. Для адгезирующих клеток удалите среду из
лунок в колонках от 2 до 11. Это можно сделать
с помощью иглы для подкожных инъекций, при¬
соединенной к вакуумной линии.
10. Замените среду на 200 мкл свежей в восьми лун¬
ках в колонках 2 и 11; эти клетки — контроль.
11. Добавьте цитотоксический препарат к клеткам в
колонках 3-10. Для каждой концентрации препа¬
рата необходимы только четыре лунки, поэтому
ряды А-D могут быть использованы для одного
препарата, а ряды Е-Н — для второго препарата.
12. Перенесите растворы препарата в 5-см чашки
Петри, добавьте 200 мкл в каждую группу из
четырех лунок при помощи автоматической пи¬
петки с четырьмя наконечниками.
13. Верните пластины в пластмассовую коробку и
инкубируйте их в течение определенного пери¬
ода. Для неадгезирующих клеток приготовьте
разведение препарата в два раза выше желатель¬
ной конечной концентрации и добавьте 100 мкл
к 100 мкл клеток в лунках.
Период роста:
14. В конце периода действия препарата удалите
среду их всех лунок, содержащих клетки, и под¬
кормите клетки добавлением 200 мкл свежей сре¬
ды. Отцентрифугируйте планшеты, содержащие
неадгезирующие клетки (5 мин при 200 г) для
получении осадка клеток, затем удалите среду,
используя тонкую иглу, чтобы сохранить осадок
клеток или не удалить осадка клеток.
15. Меняйте среду ежедневно до 2-3 PDTS.
Оценка выживания клеток:
16. Поддерживайте рост заменой 200 мкл свежей
среды в конце роста, добавьте 50 мкл МТТ во все
лунки в колонках 1-11.
17. Оберните планшеты алюминиевой фольгой и
переведите их на 4 ч в увлажненную атмосферу
при 37 °С. Обратите внимание, что 4ч— время
минимальной инкубации, планшеты могут ин¬
кубироваться до 8 ч.
18. Удалите среду и МТТ из лунок (для неадгезиру¬
ющих клеток центрифугированием) и растворите
оставшиеся кристаллы МТТ-формазана добав¬
лением 200 мкл DMSO во все лунки в колон¬
ках 1-11.
19. Добавьте глициновый буфер (25 мкл в лунку) во
все лунки, содержащие DMSO.
20. Измерьте поглощение при 570 нм немедленно,
потому что продукт нестабилен. Используйте
лунки в 1 колонке, которые содержат среду и
МТТ, но не содержат клеток как нулевой уровень
для планшетного сканера.
Анализ МТТ-теста:
21. Постройте график зависимости поглощения
(Y-ось) от концентрации препарата (Х-ось).
416
ГЛАВА 22. ЦИТОТОКСИЧНОСТЬ
22. Вычислите 1С50как концентрацию препарата,
который требуется, чтобы уменьшить поглоще¬
ние до половины контрольного уровня. Среднее
значение поглощения в лунках колонок 2 и 11
используется как контроль. Поглощение, опреде¬
ляемое в колонках 2 и 11, должно быть одинаково.
Иногда они, однако, отличны, и это указывает на
неравномерный посев клеток в планшете.
23. Абсолютное значение величины поглощения
должно быть таким, чтобы значение поглоще¬
ния контроля могло быть сравнимо с опытными
данными, в этом случае данные могут тогда быть
преобразованы в кривую процента ингибирова¬
ния (рис. 22.6), чтобы можно было сравнивать
серию полученных кривых.
а клеточная концентрация в конце работы должна
еще находиться на линейном участке результатов
спектрофотометрического анализа ММТ-теста. При
первом исследовании клеточной линии следует по¬
сеять параллельные чашки для подсчета клеток, чтобы
получить кривую роста (см. протокол 21.8) для опти¬
ческого поглощения МТТ-формазан, чтобы убедиться,
что поглощение пропорционально количеству клеток.
Если кривая роста показывает, что клетки перемеща¬
ются в стационарную фазу, либо поглощение имеет
нелинейную зависимость от концентрации, укоротите
время исследования и приступите прямо к шагу 16
протокола 22.4.
Продолжительность экспозиции связана с количес¬
твом клеточных циклов, которые прошли клетки в ходе
воздействия и восстановления. При более короткой
продолжительности клеточного цикла за время воз¬
действия не только будет быстрее возрастать клеточ¬
ная плотность, но и ответ на препараты, действующие
Концентрация цитотоксина
Рис. 22.6. Кривая ингибирования. Показатель ингибиро¬
вания рассчитывается как процент выживших клеток от
контроля, и составляется график зависимости от концент¬
рации цитотоксина. Типичная сигмовидная кривая, в идеале
1С50 находится в центре изгиба кривой
Кратковременное: PDT < 24 ч
I — ■
t l-ni t ,t ,
Посев Добавление Удаление Оценка
клеток цитотоксина цитотоксина жизнеспособности
\ I ' I' 1
I ——— ■
Долговременное: PDT > 24 ч
I I I I I I I I I I |_
01 23456789 10
Дни
Рис. 22.7. Продолжительность исследования. Стандарт
продолжительности кратковременного и долговременного
исследований. Верхняя диаграмма представляет исследова¬
ние, которое применяется для клетки с PDT < 24 ч, нижняя
диаграмма представляет исследование, которое применяется
для клеток с PDT > 24 ч, хотя возможны промежуточные
варианты
на клеточный цикл, будет обнаруживаться быстрее.
Время клеточного цикла будет влиять на выбор между
кратковременным и долговременным исследованиями
(рис. 22,7, 22.8). При первых попытках провести ис¬
следование может быть желательным брать образцы
ежедневно, в ходе действия препарата и восстановления
клеток. Если ранее получено стабильное значение IC50,
то исследование может быть упрощено.
Конечная точка. Сульфородамин, флюоресцент¬
ный краситель, окрашивающий белок, может также
быть использован для оценки количества белка (в т.ч.
клеточного) в лунках планшета с последующим анали¬
зом на автоматическом планшетном сканере планше¬
тов, регистрирующем флюоресценцию [Boyd, 1989].
Он окрашивает все клетки и не различает живые и
погибшие клетки.
Использование мечения при помощи [3Н]-тимидина
(синтез ДНК), [3Н] лейцина [Freshney et al., 1975] или
[^S] метионина [Freshney & Morgan,1978] (синтез бел¬
ка), либо других изотопов может заменять МТТ-тест.
Количественная оценка может быть достигнута мето¬
дом сцинтилляционной обработки микротитрационных
планшетов. Предлагается два типа сцинтилляционного
подсчета: 1) клеточное содержимое может быть пе¬
ренесено на фильтры с трипсинизацией и на стекло¬
волоконные фильтры при помощи пересасывающего
устройства, и затем фильтры высушиваются, и степень
радиоактивности подсчитывается на сцинтилляцион-
ном счетчике; 2) излучение всех лунок планшета может
одновременно быть обсчитано с применением специ¬
ально адаптированного сцинтилляционного счетчика
(PerkinElmer).
На практике может не иметь значения, какой крите¬
рий используется для определения жизнеспособности
или выживания в конце исследования. Важнее разра¬
ботать оптимальную схему исследования, т. е. продол¬
жительность воздействия препарата и восстановле¬
ния клеток, учет фазы клеточного цикла (клеточная
22.3. Природа исследований
417
Количество клеточных циклов в контроле после момента добавления цитотоксина
Рис. 22.8. Кривая времени падения IC50. Идеальная кривая для действующего агента с прогрессивным увеличением цито¬
токсичности с течением времени, в конечном счете происходит достижение максимального влияния после трех циклов деления
клеток. Не все цитотоксические препараты будут соответствовать этому образцу [см. Freshney at al., 1975]
плотность, скорость роста и т.д.). В кратковременном
исследовании в отсутствие и с минимальным периодом
восстановления, в конечной точке исследования могут
измеряться только жизнеспособные клетки (например,
МТТ); в более продолжительных исследованиях в ко¬
нечной точке измеряются различия между лунками, в
которых было возрастание, и теми, в которых не было
повышения, либо даже было понижение количества
жизнеспособных клеток. Наконец, в монослойных ис¬
следованиях нежизнеспособные клетки будут потеряны
и измеряется возрастание или снижение количества
клеток относительно контрольных лунок, при этом
исследования при помощи МТТ, сульфородамина или
введения изотопов становятся менее важными.
Манипуляции. Ряд манипуляций автоматизирован
при помощи таких приборов, как, например, автодис¬
пенсеры, дилютеры, коллекторы и программируемые
планшетные сканеры планшетов (см. рис. 5.10-5.12),
для снижения времени манипуляций, необходимых
для работы с одним образцом.
22.3.6. Сравнительная оценка
микротитрационного
и клоногенного анализа выживания
Объем среды, требуемый для анализа одного образца
при микротитрации менее чем одна пятнадцатая от
того, что требуется для клонирования, хотя количество
клеток примерно одинаково для обоих методов. Микро-
титрационные исследования также короче по времени
и легче подлежит автоматизации манипуляций, сбора
данных и анализа. Микротитрация, однако, не может
обнаруживать различия между ответом разных клеток
в популяции и степенью выраженности ответа в каждой
клетке, например 50% ингибирование некоторого мета¬
болического параметра могло бы означать, что каждая
клетка ингибируется на 50%, но это становится менее
важно при продолжительном периоде восстановления
клеток, когда относительное возрастание клеточной
пролиферации становится основным критерием выжи¬
вания. Сравнение значений IC50, полученных методом
микротитрации и методом исследования эффективнос¬
ти посева, показало хорошую корреляцию между двумя
методами (рис. 22.9), применяемыми для испытания
противоопухолевых препаратов [Morgan et al., 1983].
В случае IC90 корреляция была не такой выраженной.
Важным характерным свойством микротитрационных
исследований, в частности с фотометрической и радио¬
метрической конечной точкой, является получение
огромного количества данных, часто в готовом для
компьютерного анализа формате (см. разд. 21.8.2). Тем
не менее важно произвести сканирование рядов данных,
включая нулевой ряд, поскольку компьютерный анализ
может использовать различные допущения и коррек¬
ции, когда он имеет дело с аберрантными точками,
которые не являются очевидными, если не произведено
тщательное рассмотрение рядов данных.
22.3.7. Взаимодействие
лекарственных препаратов
Исследование цитотоксичности часто включает в себя
изучение взаимодействия лекарственных препаратов,
совместное действие которых может быть обнаружено
в микротитрационных системах, в которых может быть
одновременно исследовано несколько различных от¬
ношений взаимодействующих препаратов. Анализ ле¬
карственного взаимодействия может быть осуществлен
с использованием изоболограммы для интерпретации
418
ГЛАВА 22. ЦИТОТОКСИЧНОСТЬ
lg IC501 М (микротитрация)
Рис. 22.9. Корреляция между микротитрационным и кло¬
ногенным методами исследования выживания. Проведено
измерение значений 1С50 группы пяти клеточных линий
человеческой глиомы при действии шести препаратов (винк-
ристин, блеомицин, VM-26 эпидофилотоксин, 5-фторурацил,
метил CCNU, митрамицин). Большинство выпадающих то¬
чек было получено на одной линии клетки, что позже было
доказано использованием смеси различных типов клеток.
Пунктирная линия представляет собой регрессию, точки
которой получены с использованием гетерогенной клеточной
линии. Микротитрационная IC^ была получена введением
[35S] метионина [Freshney & Morgan, 1978] [по Freshney et
al., 1982a]
данных [Steel, 1979; Berenbaum, 1985]. Прямолинейный
график предполагает суммирование ответа клеток на
препараты, в то время как криволинейный график
означает синергию, если кривая падает ниже предпола¬
гаемой прямой, и антагонизм, если кривая идет выше.
22.4. ПРИМЕНЕНИЕ ИССЛЕДОВАНИЯ
ЦИТОТОКСИЧНОСТИ
22.4.1 .Скрининг противораковых
препаратов
Скрининг различных веществ для идентификации
новых противораковых препаратов может быть трудо¬
емким и часто неэффективным методом поиска новых
биологически активных соединений. В настоящее
время основным направлением стала разработка и
мониторинг действия препаратов, имеющих конкрет¬
ную мишень действия. Тем не менее продолжаются
попытки улучшить скрининг путем использования
адаптированных, легко автоматизируемых систем, на
основе которых можно быстро определять количество
жизнеспособных клеток методом их окрашивания, на¬
пример методом МТТ [Mosmann, 1983; Carmichael et al.,
1987a; Plumb et al., 1989]. Для дальнейшего сокращения
количества манипуляций инкубацию с МТТ можно
опустить, и конечная точка определяется измерением
количества общего белка с сульфородамином В [Boyd,
1989]. Хотя этот метод быстрее и легче, чем МТТ-тест,
следует помнить, что нежизнеспособные и определенно
неделящиеся клетки еще могут окрашиваться, поэтому
при обнаружении цитотоксической активности ре¬
зультаты исследования должны быть подтверждены
с использованием более надежных методов, таких как
клоногенность или редукция МТТ.
22.4.2. Прогностические исследования
противоопухолевых препаратов
Существует возможность исследования индивиду¬
альной химиочувствительности клеток, выделенных
из опухоли некоего пациента, к противоопухолевым
препаратам с разработкой оптимального режима его
последующего лечения [Freshney, 1978]. Этот метод до
настоящего времени еще не был тщательно отработан,
хотя результаты маломасштабных предварительных
исследований были обнадеживающими [Hamburger
& Salmon, 1977; Bateman et al., 1979; Hill, 1983; Thomas
et al., 1985; Von Hoff et al., 1986]. Требуется лишь раз¬
работать достоверную и воспроизводимую методику
культивирования неопластических клеток, из наиболее
часто встречающихся опухолей (например, опухолей
молочной железы, легких, толстого кишечника), с
тем чтобы приготовление и ведение культуры чис¬
тых опухолевых клеток, способных к пролиферации
сверх обычного количества клеточных циклов, стало
обычной манипуляцией. Поскольку многие из клеток
опухоли имеют ограниченное время жизни, основными
мишенями для химиотерапии являются клоногенные
популяции, с неограниченным репопуляционным
потенциалом [Al-Hajj et al., 2004; Jones et al., 2004].
Разработанные в области стволовых клеток методы
распознавания (см. разд. 23.7) могут сделать выделение
стволовых опухолевых клеток осуществимым. Были
надежды, что клоногенные исследования в мягком
агаре [Hamburger & Salmon, 1977] помогут в выделении
трансформированных стволовых клеток для последу¬
ющих испытаний, однако, несмотря на первоначально
обнадеживающие результаты, выделенные клоны не
обладали долговременной регенеративной способнос¬
тью. Рутинные методы выделения клеточных популя¬
ций с долговременной эффективностью репопуляции
еще не разработаны, но когда это станет возможным
для улучшения специфичности и направленности
действия препаратов, проведение прогностических
исследований противоопухолевых препаратов будет
иметь большее значение. Эти исследования должны
проводиться в большом проценте случаев, желатель¬
но в течение 2 недель после получения биопсийного
материала. Основной проблемой, тем не менее, явля¬
ется организация процесса. Нужно найти достаточное
количество больных с опухолями, для которых точно
выделенные клетки-мишени: 1) будут расти in vitro
достаточно успешно для того, чтобы быть исследован¬
ными; 2) будут отвечать на воздействие и 3) обнаружи¬
ваемый ответ может быть слишком мал, чтобы быть
22.5. Трансформация и мутагенез
419
зарегистрированным. Однако эти проблемы связаны
с любыми исследованиями in vitro, используемыми
в качестве прогностических, поскольку связаны с
достоверностью экспериментальных исследований в
целом. Корреляция низкой эффективности действия
препаратов in vitro с чувствительностью опухоли в
организме больного достаточно высока, но лишь не¬
которые врачи отказываются от введения препарата
на основании отрицательных результатов теста in vitro,
в частности потому, что тот препарат, который прове¬
рялся, возможно, будет использоваться не отдельно, а
в комбинации с другими.
22.4.3. Фармацевтическое тестирование
Ряд фармакологических компаний поддерживают про¬
граммы испытаний in vitro, предполагая, что это может
стать более приемлемой экономически и этически
альтернативой испытаниям на животных. Внедрение
нового законодательства, ограничивающего использо¬
вание тестов на животных, затруднено комплексностью
действия широкого диапазона эффектов, наблюдаемых
in vivo, которые пока не могут быть адекватно воспро¬
изведены in vitro. Однако существует значительное
политическое давление в отношение введения таких
законодательных ограничений, и это определяет
крупномасштабные исследования на выживаемость
для сравнительного анализа эффективности разнооб¬
разных существующих тестов [Knight & Breheny, 2002;
Vanparys, 2002].
22.5. ТРАНСФОРМАЦИЯ И МУТАГЕНЕЗ
Обычно применяемые in vitro тесты для исследования
трансформации включают исследования зависимости
от прикрепления к субстрату (см. протоколы 14.4,14.5),
зависимости клеточной пролиферации от плотности
клеточной суспензии (см. протокол 18.3), и обнаружение
признаков мутагенеза (см. также табл. 18.1). Мутагенез
может быть обнаружен путем регистрации обмена сес¬
тринских хроматид. Эта процедура описана в протоко¬
ле 22.5, предоставленном Maureen Illand и Robert Brown
из Центра онкологии и прикладной фармакологии
(University of Glasgow, Scotland, United Kingdom).
22.5.1. Анализ мутагенеза методом
обмена сестринских хроматид
Обмен сестринских хроматид (sister chromatid exchan¬
ges, SCEs) представляет собой реципрокный обмен
участками ДНК между сестринскими хроматидами в
идентичных локусах в ходе S-фазы клеточного цик¬
ла. Поскольку SCEs является более чувствительным
индикатором мутагенной активности, чем разрывы
хромосом, они становятся основным инструментом в
исследованиях мутагенеза [Latt, 1981].
С изобретением аналога тимидина бромдеоксиури-
дина (BUdR) и его последующего применения в экспе¬
риментах по мечению ДНК, разрешающая способность
метода SCEs значительно возросла по сравнению с
предыдущими методами, которые были основаны на
включении радиоактивно меченых нуклеотидов при
репликации ДНК [Taylor, 1958]. Позже разработанный
Perry and Wolf метод «флюоресценция плюс Гимза»
(FPG) [1974] для подсчета SCEs был усовершенство¬
ван, и впервые была осуществлена стабильная окраска
SCEs. Прежде в ходе подсчета участков обмена нуклео¬
тидами происходило быстрое выгорание флюоресцен¬
тного красителя [Latt, 1981].
Метод FPG включает в себя два существенных
этапа: 1) клетки метятся при помощи BUdR в течение
двух полных циклов деления и затем обрабатываются
кольцемидом (колхизином. — Прим. ред. перев.) для
блокирования деления клеток в метафазе. После экспо¬
зиции BudR ДНК одной хроматиды в каждой хромосо¬
ме содержит бромоурацил в одной цепи, в то время как
ДНК в сестринской хромосоме содержит бромурацил в
обеих цепях; 2) затем из этих клеток изготавливаются
хромосомные препараты, которые окрашиваются флю¬
оресцентным красителем Hoechst 33258, а затем произ¬
водят фотодеградацию при помощи ультрафиолетового
света; затем следует окраска по Гимза. Этот конечный
этап позволяет выявить различные участки включения
бромоурацила в сестринские хроматиды. ДНК, содер¬
жащая бромоурацил, гасит флюоресценцию комплексов
ДНК-Hoechst. Таким образом, хроматида, содержащая
бромоурацил в обеих цепях, слабо флюоресцирует и
слабо окрашивается по Гимза, в то время как хрома¬
тиды, содержащие бромоурацил только в одной цепи,
флюоресцируют более интенсивно. BUdR разрушается
с последующим более темным окрашиванием по Гимза
(см. цв. вкладку 17д, ё). Если имеют место любые SCEs,
то в результате этого метода получают препарат хромо¬
сом, который получил название арлекин-хромосомы.
ПРОТОКОЛ 22.5. Обмен сестринских
хроматид
Схема
Меченные BUdR в течение двух клеточных циклов
клетки, блокированные в метафазе, трипсинизиро¬
вать, инкубировать в гипотоническом буфере и затем
фиксировать. Приготовить препараты клеток, после
обработки Hoechst 33258, и фотодеградировать окра¬
шенный препарат хромосом. Окрасить хромосомы
красителем Гимза, рассмотреть в световой микро¬
скоп, используя иммерсионный объектив.
Материалы
Стерильные:
• D-PBSA
• РЕ: 10 мМ EDTAbPBS
• Трипсин: 0,12% в РЕ
420
ГЛАВА 22. ЦИТОТОКСИЧНОСТЬ
• BUdR (Sigma): 1 мМ концентрат в стерильной
UPW
• Кагуошах: Кольцемид, 10 мкг/мл (Invitroren)
• Ростовая среда
Нестерильные:
• SSC, 2х: разведение 1:10 20х SSC (см. Приложе¬
ние I)
• Гипотонический буфер: 0,075 М KCL
• Буфер Соренсена: фосфатный буфер, 0,066 М.,
pH 6,8 (таблетированный, Merck).
• Метанол: уксусная кислота, 3:1, на льду, свеже¬
приготовленный раствор красителя Гимза 0,76%:
поместите 1 г порошка красителя Гимза (Merck)
в 66 мл глицерина в водяной бане при 56-60 °С
на lV2-2 ч. Охладить раствор, добавить 66 мл
абсолютного этанола
• 3,5% краситель Гимза в буфере Соренсена,
pH 6,8
• Hoechst 33258 (Sirma), 20 мкг/мл, в UPW
• Латекс
• Ксилол
• Среда для заключения DPX (Merck)
• Покровные стекла, 22x15 мм
• Сосуд Coplin
• Штатив для стекол (ThermoElectron)
• Коротковолновая UV лампа в коробке для об¬
лучения
• Требование безопасности. Hoechst 33258 —
канцерогенен; взвешивайте и разводите его в вытяж¬
ном шкафу.
Протокол
Предварительная обработка
1. Посейте клетки в соответствующей плотности
(например, 1х106 клеток в 75-см2 флакон) и ин¬
кубируйте 2 дня при 37 °С.
2. Добавьте BUdR к ростовой среде до конечной
концентрации 10 мкМ.
3. Инкубируйте клетки в темноте при 37 °С в тече¬
ние 48 ч (~2 клеточных циклов).
4. Добавьте кольцемид к клеткам за 1-6 ч до оконча¬
ния инкубирования в зависимости от продолжи¬
тельность времени деления клеток. Для челове¬
ческих клеточных линий конечная концентрация
кольцемида должна составлять 0,01 мкг/мл.
Снятие клеток:
5. Отмойте клетки D-PBSA и трипсинизируйте их
с 1 мл 0,12% трипсина в РЕ.
6. Ресуспендируйте клетки в 10 мл ростовой среды.
Перенесите суспензию клеток в 50-мл центри¬
фужные пробирки.
7. Центрифугируйте суспензию при 1200 g в течение
5 мин.
8. Удалите супернатант, оставляя приблизительно
1,2 мл над осадком. Постукивая по пробирке,
ресуспендируйте осадок.
9. Медленно добавьте 10 мл гипотонического
(предварительно подогретого до 37 °С) и ин¬
кубируйте в течение 10-15 мин при комнатной
температуре.
10. Осадите клетки в настольной центрифуге при
1200 g 5 мин.
И. Удалите супернатант, оставляя 1,2 мл над осад¬
ком, постукивая по пробирке, ресуспендируйте
осадок.
12. Добавьте 10 мл фиксирующего раствора, охлаж¬
денного на льду, вначале капля по капле, хорошо
перемешивая. Оставьте пробирку на льду на
10 мин.
13. Повторите шаги 10-12 еще раз, оставив клетки в
фиксирующем растворе на ночь при 4 °С, чтобы
улучшиться качество препарата.
14. Осадите фиксированные клетки при 1200 g в
течение 5 мин, ресуспендируйте в 3-5 мл смеси
метанол/уксусная кислота.
15. Храните клетки при -20 °С.
Подготовка препарата:
16. Стекла должны быть чистыми и обезжиренными,
поэтому протрите их абсолютным этанолом.
17. Используя короткую стеклянную пастеровскую
пипетку, наберите приблизительно 500 мкл фик¬
сированных клеток.
18. Держа стекло под нисходящим углом и удерживая
пастеровскую пипетку по крайней мере в 15 см
над стеклом, капните 3 капли суспензии на стекло
(см. протокол 16.7).
19. Высушите на воздухе в темноте.
20. Проверьте препарат в фазово-контрастном мик¬
роскопе, чтобы убедиться в том, что метафазные
пластинки, имеющиеся там, равномерно распре¬
делены по препарату и что хромосомы хорошо
разделены.
Арлекин Окраска
21. Погрузите стекла в сосуд ля окрашивания пред¬
метных стекол с красителем Hoechst 33258 при
концентрации 20 мкг/мл на 10 мин. (Используйте
перчатки, так как Hoechst ядовит.)
22. Перенесите препараты в штатив для стекол и
капните 500 мкл 2х SSC на каждый препарат.
23. Закройте препараты покровными стеклами
22x50 мм и зафиксируйте края временным гер¬
метизирующим веществом типа латекса, чтобы
предотвратить загрязнение
24. Поместите препараты в штатив, покровными
стеклами вниз, и поставьте штатив с препарата¬
ми для обработки в коробку с коротковолновым
UV, сохраняя расстояние между препаратами
и источником UV приблизительно 4 см. Чем
дольше препараты подвергаются действию UV,
тем более бледным станет хроматид; облучайте
стекла приблизительно 25-60 мин.
25. Удалите покровные стекла, промойте стекла три
раза по 5 мин в UPW. Закройте штатив алюми¬
ниевой фольгой.
22.5. Трансформация и мутагенез
421
26. Высушите препараты в темноте на воздухе.
27. Окрасьте препараты в сосуде (в емкости для ок¬
рашивания стекол), содержащей 3,5% красителя
Гимза в буфере Соренсена, pH 6,8,3-5 мин.
28. Тщательно ополосните препараты в проточной
воде, дайте стечь, осушить фильтровальной бу¬
магой.
29. Высушите стекла на воздухе на столе в течение
1 ч. Погрузите каждый препарат в Ксилол, на¬
несите 4 капли заключающего вещества DPX
(Merck) на препарат, и заключите под покровное
стекло 22x50 мм, удаляя воздушные пузырьки
с помощью кусочка ткани. (Выполняйте этот
заключительный шаг в вытяжном шкафу, так
как пары ксилола ядовиты. Также используйте
перчатки.)
30. Высушите препараты на воздухе в вытяжном
шкафу в течение ночи.
Анализ:
31. Просмотрите препараты под 40х объективом све¬
тового микроскопа для обнаружения метафаз.
32. Найдите область на стеклах где расположено
большинство метафазных пластинок, и иссле¬
дуйте эту область с иммерсионным объективом
33. Когда не происходит обмена сестринских хро¬
матид, (sister chromatide exchange, SCEs), то
каждая хромосома имеет одну равномерно окра¬
шенную бледную хроматиду и одну равномерно
окрашенную темную хроматиду. Один SCE
происходит, когда обнаруживается одна область
темного окрашивания и на светлой хроматиде
и одна светлая область на темной сестринской
хроматиде (см. цв. вкладку 17д, ё). Каждая точка
неоднородности в окрашенных хромосомах учи¬
тывается как один SCE.
34. Подсчитайте число SCES в клетке, а также число
хромосом в клетке.
35. Большие хромосомы обычно имеют больше SCES,
чем меньшие, частота SCES может варьировать
от клетки к клетке, поэтому более точным пара¬
метром для определения SCE является среднее
количество SCES на хромосому, определяемое
по следующей формуле:
среднее количество SCES в клетке
среднее количество хромосом на клетку
Старайтесь подсчитать приблизительно пятьде¬
сят метафазных пластинок в каждойизутаемой
клеточной линии.
Вариации. Импульсное мечение при помощи BUdR
и последующее окрашивание, как было описано выше,
позволяют обнаружить различия в регионах ранней и
поздней репликации хромосом в ходе клеточного цикла.
Когда клетки метятся при помощи BUdR в поздней
стадии клеточного цикла, ДНК, которая реплициро¬
валась рано, окрасится при помощи краски Гимза в
темный цвет, поскольку внедрится очень мало BudR.
Те регионы, где хромосомы подверглись импульсному
мечению в ранней стадии клеточного цикла и где ДНК
реплицировалась на этих ранних стадиях, Гимза даст
слабую окраску [Latt, 1973].
Кроме того, клетки, которые подверглись посто¬
янному мечению BUdR в ходе только одного клеточ¬
ного цикла, имеют дифференциальное окрашивание
хроматид только в определенных зонах (латеральная
асимметрия) в результате различия содержания тимина
в ДНК [Brito Babapulle, 1981]. После фотодеградации
BUdR для окрашивания SCEs может также быть
использован флюоресцентный краситель акридин
оранжевый. При помощи этого красителя в регионах,
которые имеют двухцепочечные ДНК, обнаруживается
зеленая флюоресценция, и таким образом можно за¬
ключить, что здесь произошло мало включений BudR.
Красная флюоресценция наблюдается в регионах, где
имеется одноцепочечная ДНК, в которую произошло
включение BUdR. Таким образом, красная флюорес¬
ценция эквивалентна светлой окраске с Гимза, а зеленая
флюоресценция эквивалентна темной окраске с Гимза
[Karenberg & Freelander, 1974].
22.5.2. Канцерогенность
Потенциал исследований канцерогенности in vitro
весьма значителен [Berky and Sherrod, 1977; Grafstrom,
1990а, b; Zhu et al., 1991]. Это область, в которой иссле¬
дования in vivo далеки от адекватности; модели слабо
разработаны и исследования требуют недели и даже
месяцы для их проведения. Разработке удовлетвори¬
тельного подхода исследования in vitro препятствуют
1) отсутствие универсального приемлемого критерия
злокачественной трансформации in vitro и 2) присущая
клеткам человека, как основным объектам исследова¬
ния, стабильность.
Большинство общеупотребительных тестов предпо¬
лагают, что канцерогенез в большинстве случаев связан
с мутагенезом (см. разд. 18.3). Это предположение
основывается на исследованиях Ames [1980], где в ка¬
честве объекта использовались бактерии, а активация
осуществлялась при помощи микросомальных фермен¬
тов печени. Этот тест имеет высокую прогностическую
ценность, но, несмотря на это, различия в захвате,
чувствительности и типе клеточного ответа привели
к внедрению альтернативных тестов, использующих
клетки млекопитающих и человека в качестве мишеней,
например исследование обмена сестринских хроматид
(см. разд. 22.5.1).
Некоторые из этих тестов также применяются в
исследованиях мутагенеза, с использованием суспензии
клеток лимфомы L5178Y как клеток-мишеней [Cole
et al., 1990] путем индукции мутаций или реверсии.
Кроме того, они используются в оценке таких цитоло¬
гических событий, как обмен сестринских хромосом
(см. протокол 22.5), как признаков мутагенеза. Другие
исследователи [Styles, 1977] используют трансформа¬
цию как конечный пункт, проводя испытания клоноген-
ности в суспензии (см. разд. 18.5.1) в качестве критерия
422
ГЛАВА 22. ЦИТОТОКСИЧНОСТЬ
трансформации. Критики говорят, что эти системы
используют в качестве объекта клетки, которые уже
частично трансформированы; даже клетки ВНК21-С13,
использованные некоторыми научными работниками,
являются постоянной клеточной линией и не могут
рассматриваться как абсолютно нормальные. Более
того, большая часть раковых опухолей развивается
в эпителиальных тканях и не развивается в клетках
соединительной ткани.
Демонстрация возрастания экспрессии онкогенов
или амплификация, либо возрастание концентрации
продуктов экспрессии измененных онкогенов может
быть достоверным критерием, в некоторых случаях
функционально связанных с канцерогенезом. Теперь
это возможно обнаружить с помощью микроанализа.
Аналогично этому, делеции или мутации в супрессор¬
ных генах также доступны для молекулярного анализа,
в то время как делеции или мутации в генах р53, Rb,
р16, и L-САМ (Е-кадгерин) могут быть признаками
большого количества злокачественных трансформаций.
В настоящее время осуществимо проводить анализ
экспрессии генов, что может стать альтернативой ис¬
пытаний мутагенности и канцерогенности [Nuwaysir et
al., 1999; Desai et al., 2002; Vondracek et al., 2002].
22.6. ВОСПАЛЕНИЕ
В настоящее время растет необходимость исследований
сложных событий на моделях культуры клеток, таких
как воспалительная реакция, развивающаяся в ответ
на фармакологическое воздействие или аппликацию
косметического препарата или ксенобиотика, которые
могут проникать в организм путем ингаляции или с
пищей, и которые могут быть причиной развития аллер¬
гии. Это область, которая находится на ранних стадиях
развития, но имеет многообещающие результаты в бу¬
дущем. Группы защитников прав животных раздражает
неоправданное использование большого количества
животных для испытания новых косметических пре¬
паратов. Эти испытания имеют небольшое значение,
помимо коммерческой прибыли для производителей
косметики, в частности при испытании таких веществ,
как шампуни, применяется тест Дрэйза, при котором
вещество добавляется в глаз кролика. Более важны
клинические исследования, касающиеся возрастающих
аллергических ответов на фармацевтические вещества
и ксенобиотики. Эти реакции недостаточно понятны
и мало контролируемы, главным образом поскольку
отсутствуют простые воспроизводимые тесты in vitro
С начала развития технологии фильтрующих ячеек
(filter well technology) разработано несколько моделей
для изучения реакции кожи и роговицы [Вгаа and
Triglia, 1991; Triglia et al., 1991; Fusenig, 1994b; Roguet
Вкладыш филь- Фильтр
трующей ячейки
Тестируемый
раствор
Монослой -
мишень
Среда
Исследуемые на пред¬
мет реакции клетки-
мишени монослоя (как
в случае стромальных
клеток)
Коллагеновый Исследуемая в отношении растворимых продуктов
гель реакций среда (цитокины, простагландины, N0
Исследуемые на предмет ответной
реакции стромальные клетки (акти¬
вация рецепторов, передача сигнала,
экспрессия генов)
Рис. 22.10. Органотипическое исследование. Гипотетиче¬
ская система исследования для изучения действия исследуе¬
мого вещества на некий слой клеток (например, эпидермаль¬
ных кератиноцитов), при совместном культивировании их с
другим связанным типом клетки (например, фибробласты
кожи в коллагеновом геле) к раздражителю и измерение реак¬
ции по выделению цитокинов (см. также цв. вкладку 21)
et al., 1994; Kondo et al., 1997; Brinch & Elvig, 2001],
использующих оборудование для совместного культи¬
вирования различных типов клеток. В этих системах
предполагается, что взаимодействие аллергена или
раздражающего вещества с первичной мишенью (на¬
пример, клетки эпидермиса) инициирует паракринный
ответ, который вызывает высвобождение цитокинов из
вторичных стромальных компонентов (например, соб¬
ственно кожи) (рис. 22.10). Эти цитокины могут быть
обнаружены и измерены методом ELISA, что позволяет
вести мониторинг степени ответа. Хотя эти исследова¬
ния находятся на ранней стадии развития, некоторые
производители предлагают наборы для обнаружения
ответов на раздражение [Epiderm (MatTek) [Koschier at
al., 1997]; Episkin (Saduc) [Cohen et al., 1997]; SkinEthic
[Brinch & Elvig, 2001; см. цв. вкладку 21]. Протокол для
культуры роговицы, подходящий для моделирования
ответа глаза на раздражение предоставлен Carolyn
Cahn (см. протокол 23.2).
Этот тип систем может быть важной областью иссле¬
дования, с реальной перспективой использования для
скрининга аллергенов в отношении органотипических
культур, созданных на основе клеток кожи отдельных
пациентов. Такие системы могут также быть использо¬
ваны для анализа раздражающего действия на желудоч¬
но-кишечный тракт аллергенов, ответственных за раз¬
витие синдрома раздраженного кишечника. В каждом
случае существует важная возможность исследования
специфических механизмов нарушения межклеточных
взаимодействий, имеющих место при многих аллерги¬
ческих и дегенеративных заболеваниях.
Глава 23
КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ
ТИПОВ КЛЕТОК
Для многих людей клеточные культуры являются прос¬
то субстратом для продукции некоторого количества
клеток, достаточного для экспериментальных или
промышленных целей. Тем не менее отмечается воз¬
растание интереса к специфическим клеточным типам,
используемым для исследования специализированных
процессов и их патологий либо для использования в
клеточной терапии. Генерация специализированной
клеточной культуры может быть получена несколь¬
кими путями: 1) выделением дифференцированных
клеток или тканей для кратковременной нерегенера¬
тивной культуры (см. гл. 25), 2) выделением клеток-
предшественников, наращиванием их в селективных
условиях культивирования (см. ниже и разд. 10.2.1,
14.6) с последующей индукцией дифференцировки
(см. разд. 17.7), или 3) путем выделения стволовых
клеток и создания культуры, которая могла бы как
поддерживаться в виде популяции стволовых клеток,
так и дифференцироваться по одному из нескольких
возможных путей дифференцировки до получения
желательного фенотипа (см. разд. 23.7). В каждом слу¬
чае технологии различны, и настоящая глава призвана
представить некоторые технологии для специфических
типов тканей.
23.1. КЛЕТОЧНЫЕ КУЛЬТУРЫ
СПЕЦИАЛИЗИРОВАННЫХ КЛЕТОК
Имеется много обзоров, посвященных технике куль¬
туры специфических типов клеток [Doyle et al.,
1990-1999; Pollard & Walker, 1990; Freshney et al., 1994;
Leigh & Watt, 1994; Freshney &Freshney, 1996; Ravid
& Freshney, 1998; Haynes, 1999; Federoff & Richardson,
2001; Freshney & Freshney, 2002; Wise, 2002; Vunjak-
Novakovic & Freshney, 2005; см. также табл. ЗА].
Развитие техники иммортализации клеточных ли¬
ний [Freshney & Freshney, 1996] (см. также разд. 18.4)
подразумевало, что станет возможным создание по¬
стоянных клеточных линий на основе конечных линий,
выделенных из ^трансформированных тканей [Chang
et al., 1982; Freshney et al., 1994; Klein et al., 1990; Steele
et al., 1992; Wyllie et al., 1992]. Во многих случаях
дифференцированные свойства бывают утрачены, но
использование регулируемых промоторов (напри¬
мер, температурно-чувствительных) может сделать
возможным восстановление дифференцированного
фенотипа. Получение трансгенных мышей, несущих
крупный Т-ген SV40, открыло новый диапазон воз¬
можностей [Jat et al., 1991; Yanai et al., 1991; Morgan et
al., 1994], поскольку клеточные культуры, выделенные
из этих животных, уже являются иммортализованны-
ми, но сохраняют некоторые дифференцированные
функции. Поскольку эти клетки несут температурно¬
чувствительный промотор иммортализованного гена,
возможно восстановление нормального фенотипа при
температуре 37 °С.
Многие специализированные клетки (напри¬
мер, эпидермальные кератиноциты и меланоциты,
эндотелиальные клетки, гладкомышечные клетки,
фибробласты дермы и меланоциты, а также эпите¬
лий молочной железы) коммерчески доступны (см.
Приложение И). Стоимость их очень высока, но при
увеличении приобретаемой партии стоимость может
упасть. Культуры клеток кожи для цитотоксических
исследований и изучения воспалительных процессов
также можно приобрести в компаниях Episkin (Saduc),
Epiderm (MatTek) и SkinEthic. Эти продукты приготов¬
лены в фильтрующих ячейках путем комбинирования
кератиноцитов с фибробластами дермы и коллагено-
вой подложкой, поддерживаемой нейлоновой сеткой
в так называемом «эквиваленте кожи». Другие ткани,
в частности роговица (SkinEthic), также готовятся
подобным образом для исследования токсичности (см.
протокол 23.2; цв. вкладку 21).
В настоящее время разработан ряд специализиро¬
ванных процедур, и специалисты в разных областях
предложили несколько репрезентативных примеров.
Протоколы в данной главе предполагают, что исследо¬
вателям доступны основные методики и оборудование,
описанные в гл. 4. Соответственно, такое оборудование,
как ламинарный шкаф, инвертированный микроскоп,
водяная баня, настольная центрифуга и пр., не входят
в список материалов.
23.2. ЭПИТЕЛИАЛЬНЫЕ КЛЕТКИ
Эпителиальные клетки ответственны за специфиче¬
ские функции многих органов (например, поляризо¬
ванный транспорт в почках и кишечнике, секреция в
печени и поджелудочной железе, газообмен в легких,
барьерная защита в коже). Эпителиальные клетки
интересны также как модель дифференцировки и ки¬
424
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
нетики стволовых клеток (например, эпидермальные
кератиноциты [Watt, 2001, 2002]) и они относятся к
основным тканям, в которых обычно развивается ра¬
ковый процесс. Следовательно, культуры различных
эпителиальных клеток находятся в фокусе внимания
исследователей в течение многих лет. Главной пробле¬
мой для получения чистой культуры эпителиальных
клеток является избыточный рост стромальных клеток,
таких как фибробласты соединительной ткани и сосу¬
дистый эндотелий. Большинство вариаций методов
имеет своей целью предупреждение такого избыточно¬
го роста путем манипуляций с добавками в среду или
изменение субстрата для культуры (табл. 23.1), кото¬
рый ускоряет рост недифференцированного эпителия
и преимущественно стволовых клеток. Дальнейшие
модификации могут быть направлены на облегчение
дифференцировки эпителия, хотя обычно это проис¬
ходит за счет снижения пролиферации.
Факторы, содержащиеся в сыворотке (многие из
них получены из тромбоцитов), обладают сильной
митогенной активностью в отношении фибробластов,
а также имеют тенденцию к ингибированию пролифе¬
рации эпителия в результате индукции терминальной
дифференцировки. Следовательно, одной из наиболее
значительных задач в выделении и размножении кле¬
точной культуры является разработка селективной
бессывороточной среды (см. разд. 10.2.1) и добавок со
специфическими факторами роста (см. табл. 10.3).
Выделение эпителиальных клеток из донорских
тканей часто лучше всего осуществляется при помощи
коллагеназы (см. протокол 12.8), которая разрушает
стромальные клетки, но сохраняет эпителиальные клет¬
ки в небольших кластерах, позволяя выделять их путем
перенесения на питательный субстрат и поддержания
их жизнеспособности.
23.2.1. Эпидермис
Эпидермис представляет собой одну из наиболее ин¬
тересных моделей эпителиальной дифференцировки
в связи с выраженной эволюционной иерархией гис¬
тологической структуры и еще потому, что он был
одной из первых моделей, в которой использован
фидерный слой ЗТЗ, поддерживающий рост клеток.
Было осуществлено бесчисленное количество попыток
использовать культуру эпидермальных кератиноцитов
как модель дифференцировки, как идеальную тка¬
невый конструкт для органотипической культуры и
исследований межклеточного взаимодействия [Maas-
Szabowski et al., 2002], а также как трансплантацион¬
ный материал для пересадки при ожогах и раневых
повреждениях.
Протокол 23.1 для культуры эпидермальных керати¬
ноцитов был предоставлен Norbert Е. Fusenig и Hans J.
urgen Stark, Division of Carcinogenesis and Differentia¬
tion, German Cancer Research Center, Im Neuenheimer
Feld 280, 6900 Heidelberg, Germany.
В результате развития основных технологий куль¬
туры клеток и нашего понимания требований культуры
различных эпителиальных клеток, в культуре клеток
могут быть выделены и исследованы кератиноциты
Таблица 23.1. Ингибирование избыточного роста фибробластов
Метод
Действующий агент
Ткань
Литературный источник
Селективное открепление
Трипсин
Кишечник плода, сердечная
мышца, эпидермис
Owens et al., 1974; Milo et
al., 1980
Коллагеназа
Корцинома молочной железы
Freshney, 1972; Lasfargues,
1973
Конфлюэнтные фидер¬
ные слои
Мышиные фидерные клетки
ЗТЗ
Эпидермис
Rheinwald and Green, 1975
Фидерные клетки кишечника
плода человека
Эпителий нормальной и ма-
лигнизированной молочной
железы
Карцинома толстой кишки
Stampfer et al., 1980
Freshney et al., 1982b
Селективные ингибиторы
D-валин
Почечный эпителий
Gilbert and Migeon, 1975,
1977
Cis-OH- пролин
Клеточные линии
Kao and Prockop, 1977
Этилмеркуритиосалицилат
Неонатальная поджелудочная
железа
Braaten et al., 1974
Фенобарбитал
Печень
Fry and Bridges, 1979
Антимезодермальные антитела
Сквамозная карцинома
Толстокишечная аденома
Edwards et al., 1980
Paraskeva et al., 1985
Генетицин
Меланоциты, меланома
Levin et al., 1995
Селективная среда
MCDB 153
Эпидермис
Boyce and Ham, 1983
MCDB 170
Молочная железа
Hammond et al., 1984
Низкая концентрация Са2+
Эпидермальные меланоциты
Naeyaert et al., 1991
23.2. Эпителиальные клетки
425
ПРОТОКОЛ 23.1. Эпидермальные
кератиноциты
Схема
Выделить эпидермис из кожи ферментативным
способом, дизаггрегировать кератиноциты, посеять
их в бессывороточную среду или на фидерный слой
клеток, пролиферация которых блокирована.
Материалы
Стерильные:
• Подготовленный 3-дневный фидерный слой (см.
разд. 14.2.3 и протокол 23.4). Облучите клетки
ЗТЗ интенсивностью 30 Грэй, фибробласты че¬
ловека 70 Грэй, предпочтительно в концентри¬
рованной клеточной суспензии.
• FAD: среда для культивирования на фидерном
слое с высоким содержанием кальция; Среда
Хэма F12 и DMEM с добавками по Wu et al.
[1982]: среда Хэма F12 в смеси с DMEM, 1:3, с до¬
бавлением аденина (1,8x10"4 М), холерного токси¬
на (1хЮ~10М), EGF (1-10 нг/л), гидрокортизона
(0,4 мг/мл,1,1 мкМ), 5-10% FCS, пенициллина
(100 U/мл) и стрептомицин (50 мкг/мл).
• Бессывороточная среда для роста кератино¬
цитов (keratinocyte growth medium, KGM):
на основе исходной рецептуры среды MCDB
153, разработанной Boyce and Ham [1983] (см.
табл. 10.1 и Приложение II), дополненная ги¬
пофизарным экстрактом быка с низким содер¬
жанием Са2+(0,03-0,5 мМ), которая может быть
увеличена добавлением СаС12.
• Селективная среда для выделения кератиноцитов
(keratinocyte-defined medium, KDM): бессыво¬
роточная среда (см. Приложение II), основанная
на исходной среде MCDB 153 по Boyce and Ham
[1983]; полностью определенная среда, испытанная
на органотипических культурах [Stark et al., 1999]
• KDM с добавками (supplemented KDM (SKDM)):
а) Взять селективную среду для выделения
кератиноцитов (KDM) без гипофизарного
экстракта (Promocell, Clonetics);
б) добавить: инсулин (5 мг/мл), гидрокортизон
(0,5 мкг/мл), rhEGF (0,1 нг/мл), трансфер¬
рин (20 мкг/мл), 0,1% высокоочищенный
бычий сывороточный альбумин (свободный
от эндотоксинов и жирных кислот, например,
А-8806, Sigma), и L-аскорбиновая кислота
(50 мкг/мл).
в) Довести Ca2+to 1,3 mM.
г) Первичная культура кератиноцитов может
поддерживаться на необработанных чашках
в SKDM, но при низкой пролиферативной
активности. Скорость роста культуры может
быть повышена, если использовать чашки,
обработанные коллагеном (1 типа).
В органотипических культурах на возвышенных
коллагеновых гелях, выращенныхсфибробластами,
эта среда обеспечивает эпидермальный рост и
дифференцировку, сходную с полученной в FAD
[Stark et al., 1999].
• D-PBSA
• EDTA (0.5%; 15 мМ)
• Глицерин (аналитическая степень чистоты)
• Среда FAD с 20% FCS и 10% глицерина
• Ферменты:
а) Термолизин (Т-7902, Sigma)
б) Трипсин (1:250; 0,2 and 0,6%)
в) Диспаза (степеньН, 2,4 U/мл. Boehringer
Mannheim)
• Раствор бетадина (10% иод)
• Ампулы для криоконсервации
• Центрифужные пробирки, 50 мл
• Нейлоновая сетка
• «Cell Strainer» (Becton Dickinson)
• Чашки Петри, бактериологические 10см.
• Скальпели, изогнутые пинцеты
Протокол
Образцы:
1. Получите крайнюю плоть (неонатальную или
ювенильную) — наиболее часто используемый
источник человеческой кожи или образец кожи,
полученный в ходе хирургической операции или
посмертного вскрытия (до 48 ч после смерти).
Кератиноциты, полученные из крайней плоти,
прикрепляются и пролиферируют лучше, чем
клетки, полученные из кожи взрослого человека.
2. Инкубируйте биопсийный образец до 30 мин в
растворе Бетадина (10% иод) для предупрежде¬
ния инфицирования (это не снижает видимым
образом жизнеспособность кератиноцитов).
3. Промойте дважды в D-PBSA по 10 мин.
Отделение эпидермиса
Оптимальная толщина образца получается при рас¬
слоении кожи.
4. Разделите образец кожи при помощи дерматома
Castroviejo (Storz Instrument) на слои толщиной
0,1-0,2 мм. Возможно также использовать под¬
кожные слои ткани, при этом часть дермиса может
быть удалена при помощи кривых ножниц.
5. Рассеките кожу на кусочки размерами 1x2 см при
помощи скальпеля.
6. Промойте ткань от двух до пяти раз в D-PBSA.
7. Инкубируйте ткань с протеазой одним из следу¬
ющих способов:
а) Трипсин: инкубировать плавающие образцы
кожи в 0,6% растворе трипсина в D-PBS А (pH
7,4) в течение 20-30 мин при 37 °С либо (эту
работу, в частности, можно проделать с целым
образцом кожи).
б) Холодный трипсин: инкубировать плавающие
образцы в 0,2% трипсине на льду (4 °С) в те¬
чение 15-24 ч. pH раствора трипсина должен
постоянно контролироваться при помощи
426
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
фенола красного для того, чтобы избежать
сдвига pH, который может привести к изме¬
нению ферментативной активности и снизить
жизнеспособность клеток.
в) Диспаза (степень II): инкубируйте с фермен¬
том 2,4 U/мл в течение 30 мин при 37 °С.
г) Термолизин (0,5 мг/мл) [Germain et al., 1993]:
инкубируйте 12-16 ч при 4 °С.
8. Тщательно контролируйте процесс разделения
эпидермиса. Когда первые признаки отщепления
эпидермиса будут заметными на краю среза об¬
разца кожи, поместите кусочки (дермисом вниз)
в 10-см пластиковую чашку Петри и полейте 5 мл
полной культуральной среды с сывороткой.
9. Отслоите эпидермис при помощи двух изогнутых
пинцетов и поместите его в 50-мл центрифужную
пробирку, содержащую 20 мл полной культу¬
ральной среды.
а) Если разделение производилось при помощи
трипсина, жизнеспособные кератиноциты мо¬
гут быть отделены от эпидермального участка
посредством осторожного пипетирования
и просеиванием через нейлоновую сетку с
ячейками 100-мкм (“Cell Strainer,” B-D Bio¬
sciences).
б) Для получения единичных клеток из эпи¬
дермиса, отделенного при помощи диспазы
или термолизина, требуется дополнительная
инкубация с трипсином (0,2%) и 1,3 мМ EDTA
(0,05%), в течение 10 мин при 37 °С.
10. После расслоения кожи в трипсине осторожно
соскребите рыхло прикрепленные эпидермаль¬
ные клетки на оставшейся дермальной части и
добавьте клетки к эпидермальной суспензии. Это
может привести к более высокому клеточному вы¬
ходу, если чистота популяции кератиноцитов не
является основным вопросом культивирования,
но количество мезенхимальных клеток, загрязня¬
ющих полученную в результате процедуры куль¬
туру кератиноцитов, будет значительно выше.
11. Промойте выделенные эпидермальные клетки
дважды в культуральной среде путем центри¬
фугирования при 100 g в течение 10 мин, под¬
считайте общее количество клеток и количество
жизнеспособных клеток (которые не абсорби¬
руют Трипановый синий, см. протокол 22.1).
Первичная культура
12. Посейте клетки при 37 °С в среду FAD или KGM
при желаемой плотности:
а) В FAD с фидерными клетками (чашки, засе¬
янные на три дня ранее постмитотическими
ЗТЗ клетками [Rheinwald & Green, 1975]
(протокол 14.3) или фибробластами кожи
[Limat et al., 1989]), посеять кератиноциты в
количестве 2-5x104 кл./см2.
б) Для культуры, свободной от фибробластов,
посейте в FAD первичную культуру кератино¬
цитов при высокой плотности (1-5x10s кл./см2)
и поддерживайте ее при высокой плотности
при субкультивировании.
в) Можно, кроме того, посеять клетки при низ¬
кой (1х103кл./см2) и высокой (5х104 кл./см2)
плотности в KGM при низкой концентрации
Са2+ (0,03-0,1 мМ), и субкультивировать в той
же среде.
г) Клетки будут прикрепляться и расти, но с
более низкой скоростью в SKDM, поэтому
эта среда также рекомендуется для экспери¬
ментальных условий, когда требуется низкая
пролиферативная активность.
13. Когда клетки прикреплены (через 1-3 дня), тща¬
тельно сполосните культуру средой для того, чтобы
элиминировать неприкрепившиеся, погибшие и
дифференцированные клетки, и продолжите куль¬
тивирование в среде FAD или KGM. Наслаивание
и медленное падение роста могут быть достигнуты
путем сдвига концентрации Са2+ в KGM.
Субкультивирование
14. Субкультивирование производится следующим
образом:
а) Культивирование в FAD:
i) Инкубировать в 1,3-2,7 мМ (0,05-0,1%)
EDTA в течение 5-15 мин для того, чтобы
инициировать отслоение клеток, которое
заметно по расширению межклеточных
пространств.
ii) Инкубируйте в 0,1% трипсина и 1,3 мМ
(0,05%) EDTA при 37 °С в течение 5-
10 мин, с последующим осторожным пипе*
тированием до полного отделения клеток
б) Культивирование в KGM:
i) не требуется предварительной обработки
EDTA из-за низкой концентрации Са2+
ii) Инкубировать в 0,1% трипсине с 1,3 мМ
EDTA, как при культивировании в FAD.
Криоконсервация:
15. Криоконсервация часто бывает необходима для
поддержания большого количества клеток, по¬
лученных из одного образца ткани; наилучшие
результаты получают, когда используются клетки
из преконфлюэнтной первичной культуры.
а) Трипсинизируйте клетки, как описано выше,
центрифугируйте при 100 g в течение 10 мин.
б) Ресуспендируйте клетки в полной культураль¬
ной среде с сывороткой и подсчитайте.
в) Разаликвотьте по 2х106 кл./мл в FAD с 20%
FCS и 10% глицерином в пробиркидля крио¬
консервации.
г) Инкубируйте для установления равновесия
при 4 °С в течение 1-2 ч.
д) Заморозьте клетки в cells программируемом
аппарате для замораживания (Planer) со
скоростью охлаждения 1 °С/мин. Иначе крио¬
23.2. Эпителиальные клетки
427
консервация может проводиться путем поме¬
щения пробирок в коробки для заморажива¬
ния, заполненные изопропанолом (т.н. Сгуо 1.
С Freezing Container, cat. no. 5100-0001, Nalge
Nunc), который позволяет осуществлять адек¬
ватную скорость охлаждения при помещение
в морозильник с температурой -80 °С.
е) Для восстановления клеток:
i) быстро оттайте пробирки в водяной бане
при температуре 37 °С
ii) разведите клетки культуральной средой в
десять раз
iii) центрифугируйте клетки и ресуспенди¬
руйте их до необходимой концентрации в
выбранной среде, засейте культуральный
сосуд.
большинства стратифицирующихся эпителиев. Сква-
мозный эпителий кожи и выделенные из него эпители¬
альные клетки, — кератиноциты, обычно применяются
для исследования их физиологии и патологии in vitro.
В дополнение к этому, были выделены и культивирова¬
ны в особых условиях клетки слизистой рта [Tomakidi
et al., 1997; Grafstrom, 2002], а также такие структуры
кожи, как волосяные фолликулы [обзор см. Fusenig et
al., 1994]. Осуществлены попытки создания ткани в
культуре и в трансплантате [Limat et al., 1995; Maas-
Szabowski et al.,2000, 2002].
Отделение эпителиального компартмента от подле¬
жащих соединительных тканей обычно проводится пу¬
тем ферментативного расщепления трипсином [см. на¬
пример, Smola et al., 1993], диспазой типа II (2,4 ед./мл,
Boehringer Mannheim [Tomakidi et al., 1997]), или
термолизином [Germain et al., 1993]. Изолированный
эпителий затем диспергируется дополнительным инку¬
бированием в трипсине или механическим пипетирова-
нием, после чего фильтруется через нейлоновую сетку
и суспендируется в бессывороточной среде с низким
содержанием кальция или на фидерном слое клеток
с блокированной пролиферативной активностью в
различных средах с разной рецептурой. Субпопуляция
кератиноцитов с характеристиками стволовых клеток
может быть выделена и благодаря их селективной
адгезии к компонентам базальной мембраны [см.
Bickenbach and Chism, 1998].
Для того чтобы избежать избыточного роста ме¬
зенхимальных клеток, эпителиальный компартмент
должен быть отделен от соединительной ткани и дис¬
пергирован до одиночных клеток, которые затем куль¬
тивируются на средах различного состава. Наиболее
общеупотребительным является метод фидерного слоя,
впервые разработанный Rheinwald & Green [1975],
которые использовали постмитотические (облучен¬
ные или обработанные митомицином-С) клетки ЗТЗ
в качестве мезенхимальных питательных элементов.
Вместо них могут также использоваться первичные
фибробласты, выделенные из кожи или субмукозного
слоя, переведенные в постмитотическое состояние
[Limat et al., 1989; Tomakidi et al., 1997]. Фидерные
клетки обладают широким спектром физиологических
функций [Maas-Szabowski & Fusenig, 1996]. Кроме
того, кератиноциты могут быть выращены без фидер¬
ных клеток в бессывороточной среде с добавлением
экстракта гипофиза или ряда факторов роста и других
добавок, как при низких (0,03-0,06 мМ) концентра¬
циях Са2+ для ингибирования расслоения так и при
нормальных (1,2-1,4 мМ) концентрациях Са2+ для
расслоения и кератинизации [см обзор Holbrook &
Hennings, 1983]. Хотя поддержание субкультуры при
высокой клеточной плотности в присутствии фидер¬
ных клеток (например, облученных ЗТЗ) или культуры
при низкой концентрации кальция в бессывороточной
среде помогает снизить контаминацию фибробластами,
мезенхимальные клетки не полностью элиминируются
при выполнении этих процедур.
Человеческие и мышиные клетки могут культи¬
вироваться на всех трех средах в течение нескольких
месяцев. Мышиные клетки могут быть субкультиви-
рованы только один-два раза, а человеческие клетки
могут быть пересеяны четыре и более раз в FAD и KGM
соответственно.
Характеристика культур кератиноцитов. Куль¬
тивированные клетки должны быть охарактеризованы
для подтверждения их эпидермального (эпителиаль¬
ного) фенотипа, чтобы исключить загрязнение мезен¬
химальными клетками. Это может быть достигнуто,
при использовании цитокератин-детерминированных
антител к эпителиальным клеткам
Контаминация эндотелиальными клетками может
быть идентифицирована антителами против CD31
или антигена родственного фактору VIII. Однознач¬
ное распознавание фибробластов является затрудни¬
тельным, потому что использование антител против
виментина (элемент мезенхимального цитоскелета) не
является специфическим; кератиноциты in vitro могут
начать синтез виментина с различной интенсивностью,
которая зависит от условий культивирования. Для
практической оценки мезенхимального загрязнения
клеток, клетки следует посеять при посевной кон¬
центрации 1-5х102кл./см2) на фидерные клетки, и
морфология клеток 10-14-дневной культуры должна
быть идентифицирована при малом увеличении пос¬
ле фиксации и окраски гематоксилином и эозином
(Н&Е). Более высокочувствительный метод иден¬
тификации загрязнения фибробластами — анализ
экспрессии фактора роста кератиноцитов (KGF) мето¬
дом RT-PCR. Поскольку этот фактор продуцируется
фибробластами, но не продуцируется кератиноцитами,
он представляет собой селективный маркер. Более того,
экспрессия KGF усиливается при сокультивировании
с кератиноцитами, поэтому этот метод позволяет об¬
наружить минимальное загрязнение фибробластами
[Maas-Szabowski et al., 1999]. Отдельные фибробласты
в монослое кератиноцитов могут быть обнаружены ме¬
тодом гибридизации in situ с DNA KGF. Человеческие
фибробласты могут быть идентифицированы и коли¬
428
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
чественно оценены методом фибробласт-специфиче-
ских антител AS02 (Dianova) иммунопероксидазным
методом [Saalbach et al., 1997].
Вариации. Кератиноциты могут быть получены
из внешнего эпителиального влагалища корня волос
(ORS) выдернутых волосяных фолликул скальпа
путем отделения клеток от рассеченного фолликула
[Limat et al., 1989]. Из трех фолликул может быть по¬
лучено до 5x103 клеток при посеве на фидерный слой
фибробластов, по завершении 15-дневной культуры
будет приблизительно 1x106 клеток. Подобно межфол-
ликулярным кератиноцитам, ORS-клетки могут быть
субкультивированы на чашках с фидерным слоем и
заморожены. Эти ORS-полученные кератиноциты,
подобно культуре интерфолликулярных клеток, фор¬
мируют правильный эпидермис при пересадке мышам
Nude и могут быть использованы в клинических целях,
чтобы закрыть хронические раны [Limat et al., 1996].
Кератиноциты можно также вырастить в клональ¬
ной плотности для изучения клональной клеточной по¬
пуляции и их пролиферативного потенциала. Для этой
цели кератиноциты ко-культивируются с облучеными
рентгеновскими лучами ЗТЗ клетками [Rheinwald &
Green, 1975] при сниженной концентрации Са2+ в
фибробласт-кондиционированной среде [Yuspa et al.,
1981], или в определенной бессывороточной среде
[Boyce & Ham, 1983].
Для того чтобы обеспечить условия, более сходные
с условиями in vivo, и изучить регулирование физиоло¬
гии эпителиально-мезхенхимального взаимодействия
кожи, разработаны органотипические со-культуры,
которые получают посевом клеток на коллагеновые
гели (с предварительно посеянными фибробластами)
или кусочки дермы, с подъемом подложки в воздушной
среде [см. обзор Fusenig, 1994а.] В этих улучшенных
условиях роста кератиноцитов проявляется множество
аспектов роста и дифференцирования эпидермиса in
vivoy включая ультраструктурные особенности и нали¬
чие полной базальной мембраны, — свойства, которые
отсутствуют или менее явно варажены в погруженной
в среду культуре на пластике [Smola et al., 1998]. Такие
органотипические культуры могут теперь также быть
созданы и поддерживаться в течение трех недель в
определенной SKDM среде, что позволяет исследовать
молекулярные взаимодействия между эпителиальными
и мезенхимальными клетками, анализировать их без
влияния сыворотки или их неопределенных экстрактов
тканей [Stark et al., 1999].
Такие органотипические культуры также коммер¬
чески доступны (см. рис. 22.10 и разд. 22.6) как в виде
со-культуры с фибробластами, так и в виде монокуль¬
туры кератиноцитов на полиуглеродном фильтрующем
вкладыше (Falcon# 3501), и все более используются
для исследований токсичности и биологической сов¬
местимости изделий, взаимодействующих с кожей in
vitro [Fusenig, 1994b].
Оптимальный рост и дифференцировка изоли¬
рованных кератиноцитов достигаются в условиях ш
vivo, когда клетки трансплантируются как интактные
культуры или в виде суспензии на мышей Nude [см.
обзор Fusenig 1994а]. Это приводит к почти полной
экспрессии значимых белков, дифференцированным
характеристикам нормального эпидермиса и всем био¬
химическим и ультраструктурным характеристикам
полностью кератинизированного эпителия, включая
формирование базальной мемебраны [Breitkreutz et
al., 1997]. В настоящее время доступны коммерческие
комбинированные культуры эпидермиса и стромы,
посаженные на сетчатые фильтры (Episkin, Epiderm,
SkinEthic), которые предлагается использовать в
качестве моделей для исследования раздражающе¬
го действия различных веществ и воспалительного
процесса.
23.2.2. Роговица
В настоящее время проводятся попытки заменить ис¬
следования на глазе кролика (Draize) исследованиями
на культуре эпителия роговицы, поэтому к культуре
роговицы имеется высокий интерес исследователей
[Dart, 2003]. Последующие инструкции и протокол 23.2
для культивирования нормальной роговицы человека
в бессывороточной среде были предоставлены Caro¬
lyn Cahn, Gillette Medical Evaluation Laboratories, 401
Professional Drive, Gaithersburg, MD 20879.
Эпителий роговицы непосредственно контактирует
с внешней окружающей средой и является первой тка¬
нью, которая вовлечена в раневое повреждение глаза.
Наиболее часто в качестве модели для исследования
поверхности человеческого глаза используются модели
животных. Система, которая будет описана ниже, раз¬
работана для токсикологических исследований in vitro,
которые могут дополнить исследования in vivo.
Эпителий роговицы in vivo получает кислород путем
диффузии из пленки слезной жидкости; кроме того,
клетки активно делятся. Эти две характеристики вкупе
с наличием ткани донорской роговицы делают модель
эпителия роговицы хорошим исходным материалом
для создания первичных культур.
Среди источников для получения ткани роговицы
имеется усовершенствованная система банка донорс¬
ких глаз, которая предоставляет донорскую роговицу
для трансплантации. Ткани из этого банка имеют по¬
ниженное качество и могут применяться для исследо¬
ваний после трех дней хранения, поскольку существует
ограниченная жизнеспособность слоя эндотелиальных
клеток in vitro. Хотя эндотелий лабилен, эпителий со¬
храняет генеративные способности в течение продол¬
жительного периода при хранении при 4 °С, что делает
ткани с просроченным сроком годности пригодными
к дальнейшему употреблению в качестве источника
жизнеспособного эпителия.
Немедленно после пассажа клетки приобретают
специфические веретенообразные очертания и реф-
рактильные свойства, сопровождающиеся высокой
миграцией. В течение 7 дней контрольные культуры
23.2. Эпителиальные клетки
429
ПРОТОКОЛ 23.2. Эпителиальные клетки
роговицы
Схема
Ткань роговицы помещают на коллаген и позволяют
клеткам прикрепиться, клетки растут и размножа¬
ются в бессывороточной среде на поверхностях,
покрытых фибронектином-коллагеном.
Материалы
Стерильные и ли асептически приготовленные:
• Бессывороточная среда для кератиноцитов
(KGM, Clonetics), содержащая 0,15 мМ каль¬
ция, человеческий эпидермальный фактор роста
(0,1 нг/мл), инсулин (5 мкг/мл), гидрокортизон
(0,5 мкг/мл), гипофизарный экстракт быка
(30 мкг/мл)
• Минимальная среда Игла
• D-PBSA: фосфатно-буферный раствор Дюльбек¬
ко без Са2+ и Mg2+
• FBS
• Трипсин-EDTA: трипсин, 0,05%, EDTA, 0,5 мМ
• Фибронектин/коллаген (FNC): фибронек¬
тин, 10 мкг/мл, коллаген, 35 мкг/мл, с БСА,
100 мкг/мл в качестве стабилизатора
• Шестилуночный планшет с биологическим пок¬
рытием, предварительно обработанный коллаге¬
ном крысиного хвоста, тип I (B-D Biosciences)
Протокол
Первичные культуры:
1. Поместите донорскую роговицу эпителиальной
стороной вверх на стерильную поверхность и
порежьте на 12 треугольных клинов, используя
однократные разрезы скальпелем, избегая пиля¬
щих движений. Аккуратные манипуляции с рого¬
вицей таким способом снижают повреждение по
отношению к коллагеновому матриксу стромы и
минимизируют высвобождение фибробластов.
2. Переверните каждый сегмент роговицы эпители¬
альной стороной вверх, поместите четыре сегмен¬
та в каждую лунку шестилуночного планшета.
3. Прижмите тщательно каждый сегмент при по¬
мощи пинцетов для того, чтобы убедиться, что
достигнут хороший контакт между тканью и по¬
верхностью, на которой будет происходить куль¬
тивирование. Оставьте подсохнуть на 20 мин.
4. Осторожно поместите по одной капле KGM на
каждый сегмент и инкубируйте в течение ночи
при 37 °С в 5% С02. Хотя донорская роговица,
получаемая из банка глазных трансплантатов,
хранится в среде, содержащей антибиотик (Мс-
Сагеу-Kauffman или Dexsol), все манипуляции
следует проводить в условиях, свободных от
антибиотиков.
5. На следующий день добавьте 1 мл KGM в каждую
лунку. В ходе первоначального периода культи¬
вирования отмечается миграция клеток только с
лимба роговицы. Из центрального региона или
склеры миграции клеток не наблюдается. Из¬
быточный рост фибробластов минимизируется
использованием бессывороточной среды, которая
имеет низкую концентрацию кальция (0,15 тМ)
и это сокращает до минимума повреждение кол-
лагенового матрикса.
6. Через 5 дней после начала эксплантации удалите
сегменты донорской ткани при помощи пинцета
и добавьте Змл среды. После удаления участков
ткани прикрепившиеся клетки продолжают про¬
лиферировать, и в течение 2 недель с момента
установления культуры формируется конфлю¬
энтный монослой, обнаруживающий типичную
морфологию «булыжной мостовой», связанной
с эпителием. Выход клеток составляет примерно
1-5х106 клеток на роговицу.
Размножение культуры
7. В ходе первоначального периода разрастания
производите смену среды культуры дважды в
неделю.
8. При достижении 70-80% конфлюэнтности про¬
мойте клетки в DPBSA и разрушьте слой инкуба¬
цией в растворе трипсин/EDTA в течение 4 мин
при 37 °С.
9. Остановите реакцию действием 10% FBS в
D-PBSA.
10. Промойте клетки (центрифугированием после ре¬
суспендирования в KGM), подсчитайте и посейте
при концентрации 1х104кл./см2на поверхности
для культивирования тканей, покрытые FNC.
11. Инкубируйте культуру при 37 °С в 95% атмосфе¬
ры и 5% С02.
12. Через день после трипсинизации и пересева за¬
мените культуральную среду на свежую.
достигают 70-80% конфлюэнтнсти, продолжая мор¬
фологически сохранять вид «булыжной мостовой»
и, если это возможно, при постконфлюэнтном росте
стратифицируясь на дискретные области. Хотя эпите¬
лий роговицы может быть субкультивирован вплоть
до пяти раз (примерно 9-10 удвоений популяции), в
основном пролиферация происходит между первым и
третьим пассажами. Приблизительный выход из одной
роговицы составляет 1-2x106 клеток. Старение культу¬
ры всегда происходит после пятого пассажа.
Получение постоянной клеточной линии. Разра¬
ботаны клеточные линии эпителия роговицы человека
(НСЕ) с неограниченным временем жизни (см. про¬
токол 18.1), призванные производить большое коли¬
чество клеток для экспериментальных целей.
Долговременное хранение клеток. Клетки могут
храниться замороженными в жидком азоте (см. про¬
токол 20.1).
430
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
Фенотипическое развитие in vitro. Как первич¬
ная культура, так и линии клеток НСЕ сохраняют
фенотипические характеристики эпителия роговицы
in situ. Они продолжают синтезировать коллагеназу и
экспрессировать EGF-рецепторы и специфичные для
роговицы цитокератины, хотя уровень экспрессии
при текущих условиях культивирования ниже, чем
отмечается in situ. В культуре эпителия роговицы на
коллагеновой мембране на жидкогазовом разделе сред
их морфология и барьерные функции в известной сте¬
пени достаточно хорошо сохраняются. Созданы куль¬
туры стратифицирующихся мембран, которые способ¬
ны к ингибированию диффузии Na-флюоресцеина.
Культура на границе раздела жидкой и газовой фаз
может выживать в течение 2 недель in vitro и обычно
используется для исследования раневых повреждений
и механизмов репарации. Более полная трехмерная
тканевая модель может быть сконструирована путем
использования подложки из фибробластов рогови¬
цы в коллагеновом геле (см. разд. 25.3.6). Клетки
роговицы человека, размножающиеся in vitro, могут
послужить подходящей моделью для исследования
основных биологических клеточных механизмов,
а также токсикологических исследований. Хотя
первичные культуры являются адекватными для
исследований in vitro, линии НСЕ с неограниченным
временем жизни предоставляют лучший источник
материала, который к тому же может быть передан
между лабораториями.
23.2.3. Молочная железа
Грудное молоко [Buehring, 1972; Ceriani et al., 1979] и
материал, полученный при редукционной маммоплас-
тике [Stampfer et al., 2002], являются пригодными
источниками для получения нормального эпителия
протоков молочной железы. Метод выделений из
молока дает чистые культуры эпителиальных клеток.
Дезаггрегация в коллагеназе [Speirs et al., 1996] пред¬
почтительна для первичной дезаггрегации, рост на
конфлюэнтных фидерных слоях фетальных клеток че¬
ловеческого кишечника [Stampfer et al., 1980; Freshney
et al., 1982b] подавляет контаминацию стромальными
клетками как нормальной так и малигнизированной
ткани (см. цв. вкладки 6в, г, 156). Оптимизация среды
[Stampfer et al., 1980; Smith et al., 1981; Hammond et al.,
1984] обеспечивает серийные пассажи и клонирование
эпителиальных клеток. Культивирование в коллаге¬
новом геле обеспечивает формирование трехмерной
структуры, которая хорошо коррелирует с гистологией
исходной донорской ткани [Berdichevsky et al., 1992;
Gomm et al., 1997].
Как и в случае эпидермиса, рост эпителиоидных
клеток нормальной молочной железы in vitro стиму¬
лируется холерным токсином [Taylor-Papadimitriou
et al., 1980] и EGF [Osborne et al., 1980]. Гормональная
картина более сложна. Многие эпителиальные клетки
лучше выживают при действии инсулина (1-10 IU/мл)
и гидрокортизона (~1х10'8М), добавленных в среду.
Дифференцировка ацинарного эпителия молочной
железы в органной культуре требует добавления гидро¬
кортизона, инсулина и пролактина [Darcy et al., 1995].
Показано также, что для клеточной культуры требуется
добавление эстрогена, прогестерона и гормона роста
[Klevjer-Anderson & Buehring, 1980].
Дальнейшее введение и протокол 23.3 для получе¬
ния культуры клеток из молока человека предоставлен
Joyce Taylor-Papadimitriou, Cancer Research UK Breast
Cancer Biology Group, Guy’s Hospital, ThomasGuy
House, London SE1 9RT, UK.
Молоко ранней стадии лактации или после отни¬
мания ребенка от груди дает самый высокий выход
клеток и содержит сгустки эпителия, который способен
к пролиферации в культуре [Buehring, 1972; Taylor-
Papadimitriou et al., 1980]. Первичные культуры при
росте на среде с человеческой сывороткой и добавление
гормонов дают клеточные линии, которые имеют огра¬
ниченное количество жизненных циклов, но являются
клоногенными [Stoker et al., 1982]. Эти линии обычно
в конечном итоге оказываются зараженными неэпи¬
телиальными клетками «позднего молока» [McKay &
Taylor-Papadimitriou, 1981].
ПРОТОКОЛ 23.3. Эпителий молочной железы
Схема
Клетки, полученные центрифугированием молока
ранней стадии лактации, выращивают в присут¬
ствии эндогенных макрофагов в обогащенной среде
и могут быть субкультивированы в хелатирующем
растворе смеси протеаз.
Материалы
Стерильные
• Ростовая среда RPMI1640
• Фетальная сыворотка теленка (FBS)
• Человеческая сыворотка (HuS; устаревшая сыво¬
ротка из банка донорской крови, отрицательная
по австралийскому антигену)
• Маточные растворы
а) Инсулин (Sigma), 1 мг/мл в 6 mM НС1
б) Гидрокортизон, 0,5 мг/мл в физиологическом
растворе
в) Холерный токсин (см. Приложение II),
50 мкг/мл в физиологическом растворе.
Примечание. Сыворотка и маточные растворы инсу¬
лина, щдрокортизона, холерного токсина и трипсина
следует хранить при 20 °С.
• Раствор для трипсинизации (TEGPED):
а) EGTA (зтиленгликоль-бис-(0’-аминоэти-
лэфир)-М,М,М,М-тетрауксусная кислота)
(Sigma) 13 mM в D-PBSA. 10 мл
б) EDTA (Sigma), 7 тМ в D-PBSA .... 4 мл
23.2. Эпителиальные клетки
431
в) Трипсин (Difco; B-D Biosciences), 0,2% в
HBSS 4 мл
г) Панкреатин (Difco, B-D Biosciences), 1,0% в
HBSS 2 мл
• Ростовая среда: RPMI 1640, содержащая 15%
FBS; 10% HuS; холерный токсин, 50нг/мл; гид¬
рокортизон, 0,5 мкг/мл; инсулин, 1 мкг/мл
• Пластиковые чашки Nunc, 5 см, или флаконы
25-см2
• Универсальные контейнеры или 20-50-мл цент¬
рифужные пробирки
Протокол
Молоко (2-7-й дни после родов) лучше всего по¬
лучить из районной больницы. Молочная железа
протирается стерильной водой, молоко вручную
сцеживается в стерильный контейнер. Обычно полу¬
чают от 5 до 20 мл от одной пациентки. Порции мо¬
лока объединяются, разводятся средой RPMI1640 в
отношении 1:1 для облегчения центрифугирования
Первичная культура
1. Центрифугируйте разведенное молоко при
600-1,000 g в течение 20 мин. Тщательно удалите
супернатант, оставив некоторое количество жид¬
кости так, чтобы не нарушить осадок.
2. Промойте осевшие клетки два-четыре раза в
RPMI, содержащей 5% FCS, пока супернатант не
будет прозрачным.
3. Ресуспендируйте осадок в ростовой среде, посей¬
те 50 мкл суспензии в 5-см чашку (Nunc) в 6-мл
ростовой среды. Инкубируйте культуру при 37 °С
в 5% С02.
4. Через 3-5 дней замените среду, затем меняйте
среду два раза в неделю. Колонии появляются
примерно через 6-8 дней и распространяются по
чашке, расталкивая макрофаги молока, которые
первоначально появляются и действуют как фи¬
дерный слой.
Субкультивирование клеток молока
5. Инкубируйте клетки в TEGPED (1,5 мл в 5-см
чашке) при 37 °С в течение 5-15 мин, в зависи¬
мости от возраста культуры до получения суспен¬
зии отдельных клеток.
6. Центрифугируйте клетки при 100 g в течение
5 мин и ресуспендируйте в 6 мл среды.
7. Поделите суспензию на три новых 5-см чашки по
2 мл в каждую или в 25-см2 флаконы.
8. Разведите в три раза добавлением 4 мл свежей
среды.
Варианты. В качестве фидерного слоя удобно ис¬
пользовать макрофаги, которые уже присутствуют в
молоке, но они постепенно теряются при размножении
колоний эпителиальных клеток. Тем не менее макро¬
фаги могут быть удалены путем абсорбции на стекле, и
в этом случае могут быть добавлены другие фидерные
клетки. Облученные или обработанные митомицином
клетки ЗТ6 (см. протоколы 14.3, 23.4) обладают самой
лучшей возможностью обеспечения роста [Taylor-
Papadimitriou et al., 1977]. Вместо холерного токсина
могут использоваться аналоги циклической АМР
[Taylor-Papadimitriou et al., 1980], хотя при исполь¬
зования макрофагов в качестве фидерных клеток это
невозможно, поскольку они погибают при действии
этих аналогов АМР.
Использование и применение культуры эпителиаль¬
ных клеток молока. Культура молока позволяет полу¬
чить клетки из полностью функционирующих желез
и позволяет определить фенотипы иммунологических
маркеров [Chang & Taylor-Papadimitriou, 1983]. Клет¬
ки молока успешно трансформируются при помощи
вируса SV40 [Chang et al., 1982] и используются для
генотоксических исследований [Martin et al., 2000].
23.2.4. Шейка матки
Кератиноциты шейки матки могут быть выращены в
серийной культуре при клональной плотности путем
использования модификации [Stanley & Parkinson,
1979] метода, описанного для эпидермальных керати¬
ноцитов [Rheinwald & Green, 1975]. Протокол 23.4 для
культуры эпителиальных клеток из биопсийного мате¬
риала был предоставлен Margaret Stanley, Department
of Pathology, University of Cambridge, UK.
ПРОТОКОЛ 23.4. Цервикальный эпителий
Схема
Суспензия отдельных клеток, полученная фермен¬
тативной дизаггрегацией эпителия из биопсийной
пробы шейки матки, инокулируется в чашки или
флаконы с летально инактивированными клетками
Swiss ЗТЗ и выращивается в среде с добавлением
сыворотки и ростовыми факторами (протокол А)
или на бессывороточной среде для выращивания
кератиноцитов (KGM) без фидерной подложки с ис¬
пользованием модификаций методов, первоначаль¬
но описанных для эпидермальных кератиноцитов
(Boyce & Ham, 1983). Когда колонии кератиноцитов
достигнут размера 1000 клеток или более, культуры
могут быть трипсинизированы и вновь пересеяны с
использованием фидерного слоя фибробластов.
Материалы
Стерильные:
• Транспортная среда: Среда Игла в модифика¬
ции Дюльбекко (DMEM), обогащенная 10%
фетальной сыворотки теленка, с добавлением
100 мкг/мл сульфата гентамицина и 10 мкг/мл
амфотерицина.
• Раствор трипсин/ EDTA для дизаггрегации
эпителиальных клеток:0,25% трипсина (v/v),
432
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
0,25 тМ (0,01%) EDTA как двунатриевая соль с
pH 7,4 в D-DPBSA
• Среда для протокола А: среда для культивиро¬
вания кератиноцитов (КСМ): DMEM с добав¬
лением 10% FBS, 0,5 мкг/мл гидрокортизона и
lxlO'10 М холерного токсина:
а) Не все партии FBS одинаково хорошо обес¬
печивают рост клеток. Следует провести
тестирование образца сыворотки и закупить
большую партию подходящего качества.
б) Эпидермальный фактор роста (EGF): Sigma,
100 \xg разводится в 1-мл стерильной UPW.
Разлить на аликвоты по 100-мкл и хранить
при -20 °С. Рабочий раствор готовится раз¬
ведением 100 мкл EGF с концентрацией
100 мкг/мл в 10 мл среды с сывороткой. Хра¬
нить при 4 °С. Для использования развести в
отношении 1:100 в среде, содержащей сыво¬
ротку до конечной концентрации Юнг/мл.
в) Холерный токсин (СТ) (Sigma): Добавьте
1,18 мл стерильной UPW до 1 мг СТ, чтобы
получить раствор 10“5М. Хранить при 4 °С.
Рабочий раствор: 100 мкл СТ 10~5 М развести
в 10 мл среды с сывороткой. Стерилизовать
фильтрацией и хранить 4 °С. Использовать
при конечной концентрации Ю~10М.
г) Гидрокортизон (Sigma): Растворить 1 мг гид¬
рокортизона в 1 мл 50% этанола в воде (v/v).
Разлить по аликвотам 100-мкл и хранить при
-20 °С. Использовать при конечной концент¬
рации 0,5 мкг/мл.
• Среда для протокола В: среда для роста керати¬
ноцитов KGM (Bio-Whittaker), или Keratinocyte-
SFM (Invitrogen)
• Чашки Петри для культуры тканей, диаметром
6-см и 9-см
• Пинцеты с зубцами
• Искривленные ножницы для иридэктомии
• Одноразовые скальпели лезвие No. 22
• Пипетки
• Центрифужные пробирки
Нестерильные:
• Гемоцитометр
• Источник излучения (рентгеновский или “Со)
Фибробласты Swiss ЗТЗ (Только протокол А):
а) Следует приготовить и заморозить большой
ростковый запас клеток в отдельных ампулах
при концентрации 1x106 клеток. Клетки не
должны быть использованы более чем в тече¬
ние 20 пассажей.
i) Вырастить клетки 3T3s в DMEM с10%
сыворотки теленка в культуральном фла¬
коне 175-см2. Инокулировать клетки при
концентрации 1,5x104 кл./см2. Через два
дня заменить среду. Субкультивировать
каждые 4-5 дней.
ii) Для того чтобы избежать незначитель¬
ной контаминации, поддерживайте один
маточный флакон с клетками в среде без
антибиотика; затем эти клетки используй¬
те при каждом пассаже инокулируя их во
флаконы для получение фидерного слоя.
б) Фидерный слой инактивируется излучением
мощностью 60 Грэй (6000 рад), от рентгенов¬
ского источника или ®°Со. Облученные клетки
(XR-3T3) могут храниться при 4 °С в течение
3-4 дней.
в) В отсутствие источника излучения инактиви¬
руйте клетки митомицином С.
i) Экспозиция клеток ЗТЗ, выросших в мо¬
нослое 400 мкг/мл с митомицином С в
течение 1 часа при 37 °С.
И) Трипсинизируйте обработанные клет¬
ки, ресуспендируйте и дважды отмойте
клеточный осадок свежей средой с сыво¬
роткой, ресуспендируйте до приемлемой
концентрации в полной среде и затем
используйте.
Протокол А: с фидерным слоем ЗТЗ
Первичная культура:
1. Выньте цервикальный биоптат из транспортной
среды, промойте клетки два-три раза в 5 мл сте¬
рильной D-PBSA, содержащей 50 мкг/мл суль¬
фата гентамицина и 5 мкг/мл амфотерицина.
2. Поместите биоптат на стерильную культураль¬
ную чашку эпителиальной поверхностью вниз.
3. Используя одноразовый скальпель, оборудо¬
ванный лезвием № 22, максимально срежьте и
соскребите мышцы и строму, оставляя тонкую
матовую эпителиальную полоску.
4. Тщательно измельчите эпителиальную полоску
при помощи искривленных ножниц для ирид¬
эктомии.
5. Добавьте 10 мл смеси трипсин/EDTA (предва¬
рительно подогретой до 37 °С) к измельченному
эпителию, и перенесите ткань в стерильный стек¬
лянный универсальный контейнер, содержащий
небольшой покрытый пластиком магнит для
магнитной мешалки.
6. Добавьте еще 5-10 мл смеси трипсин/EDTA.
7. Поместите универсальный контейнер на маг¬
нитную мешалку в инкубаторе или термальную
комнату при 37 °С, и медленно перемешивайте в
течение 30-40 мин.
8. Оставьте суспензию при комнатной температуре
на 2-3 мин.
9. Отберите верхний слой отстоявшейся жидкости,
содержащий единичные клетки, и профильтруй¬
те их через сито из пластика или нержавеющей
ткани (Cell Strainer, BD Biosciences) в 50-мл
центрифужную пробирку (см. рис. 12.10).
10. Добавьте 10мл полной среды к фильтрату.
11. Добавьте следующие 15 мл теплой смеси трип¬
син/EDTA к фрагментам ткани в универсаль¬
ном контейнере, и дважды повторите шаги 5-9.
23.2. Эпителиальные клетки
433
Соедините отобранные верхние слои, содержа¬
щие трипсин, открутите на настольной центри¬
фуге при 1000 rpm (80 g) в течение 5 мин.
12. Удалите супернатант, добавьте 10 мл полной сре¬
ды к осадку, энергично ресуспендируйте клетки до
получения суспензии отдельных клеток, подсчи¬
тайте клетки в гемоцитометре. Оцените жизнеспо¬
собность клеток методом окрашивания трипано-
вым синим мертвых клеток (см. протокол 22.1).
13. Разведите суспензию клеток цервикального
эпителия в КСМ, посейте клетки на чашки при
концентрации 2x104 кл./см2 вместе с летально
инактивированными клетками ЗТЗ в концент¬
рации IxlO5кл./см2 (т.е. на IxlO5цервикальных
клеток 5xl05XR-3T3/Ha 60-мм чашку).
14. Инкубируйте культуры при 37 °С в 5% С02.
15. Через 72 ч после первичного посева замените
среду полной средой, содержащей EGF10 нг/мл.
Проверьте культуру микроскопически, чтобы
убедиться в адекватности фидерного слоя. До¬
бавить фидерных клеток если необходимо.
16. Дважды в неделю меняйте питательную среду, в
которой должен содержаться EGF, кроме первич¬
ного посева. Колонии кератиноцитов становятся
видимыми в микроскоп на 8-12-й дни, а нево¬
оруженным глазом — на 14-16-й. В это время
культуры следует пересеять.
Субкультивирование:
17. Удалите среду с клеточного монослоя и удалите
также фидерный слой быстрым ополаскиванием
0,01% EDTA. Дважды промойте D-PBSA.
18. Добавьте предварительно подогретую смесь
трипсин/EDTA, чтобы она покрыла клетки. Ос¬
тавьте культуру при 37 °С, пока кератиноциты
не отслоятся, удостоверьтесь в отделении клеток
при помощи микроскопа. Не оставляйте клетки
в трипсине более чем на 20 мин.
19. Отберите клеточную суспензию из чашки и пере¬
несите в стерильную центрифужную пробирку.
20. Ополосните поверхность роста полной средой,
добавьте суспензию. Перемешайте и распреде¬
лите суспензию при помощи 10-мл пипетки.
21. Центрифугируйте клетки при 80-100 g в течение
5 мин.
22. Удалить супернатант, добавить 10 мл полной
среды, энергично ресуспендировать клетки при
помощи 10-мл. пипетки для получения суспензии
отдельных клеток.
23. Подсчитайте клетки при помощи гемоцитометра.
24. Клетки могут быть пересеяны на инактивирован¬
ные ЗТЗ клетки, как это описано выше, либо за¬
морожены для последующего восстановления.
Протокол В: Бессывороточная среда.
Первичная культура
1. Следуйте шагам 1-11 протокола А для получения
суспензии отдельных клетокв среде, содержащей
сыворотку.
2. Осадите суспензию при 1000 об./мин (80 g) в
течение 5 мин. Удалите супернатант и ресуспен¬
дируйте клетки в 10 мл D-PBSA.
3. Вновь осадите суспензию и отмойте клетки од¬
нократно с D-PBSA. Ресуспендируйте клетки в
10 мл среды KGM (приготовленной в соответ¬
ствии с инструкцией производителя) и посейте
в чашки для культивирования или флаконы при
плотности 2х105кл./см2 (106 клеток на 5-см чашку
Петри, 4х106на 9-см чашку Петри).
4. Инкубировать культуру при 37 °С в 5% С02.
5. Через 72 ч заменить среду; затем менять дважды
в неделю. Культура становится конфлюэнтной в
течение 14-20 дней и в это время должна быть
пересеяна.
Субкультивирование:
6. Следуйте шагам 17-23 протокола А.
7. Осадите суспензию при 80-100 g в течение 5 мин.
Удалите супернатант, ресуспендируйте клетки в
10 мл D-PBSA.
8. Вновь осадите суспензию и однократно отмой¬
те клетки в D-PBSA. Ресупендируйте клетки в
KGM и посейте их на культуральные чашки при
концентрации ^кл./см2 (5x105клеток на 5-см
чашку Петри, 2x106 на 9-см чашку Петри).
9. На этой стадии клетки также могут быть заморо¬
жены для последующего восстановления.
Цервикальные кератиноциты, выращенные как опи¬
сано выше, могут быть использованы для различных
целей, включая исследования папилломавирусного
канцерогенеза, стадий дифференцировки, и изучения
ответа этих клеток на мутагены и канцерогены.
23.2.5. Желудочно-кишечный тракт
Литература по выделению культуры нормального
эпителия из кишечной выстилки не очень обширна,
хотя имеется большое количество публикаций о вы¬
делении постоянных клеточных линий из карциномы
толстой кишки человека (см. разд. 24.8.3). Owens et al.
[1974] смогли культивировать клетки из кишки плода
человека как конечную клеточную линию (FHS 74 Int),
широкое распространение получила клеточная линия
IEC-6 из кишки крысы [Rak et al., 1995; Bedrin et al.,
1997]. Протокол 23.5 основан на методе Booth & O’Shea
[2002]. Приготовление образцов ткани для клеточной
культуры обычно начинается за 1-2 часа после изъятия
от больного. Если это возможно, тщательно порежьте
ткань на мелкие кусочки (1-2 мм) при помощи скаль¬
пеля. Хранение в течение ночи при 4 °С в промывочной
среде (HBSS/PSG, см. ниже) также может обеспечить
успешное выделение клеток.
Крипты могут быть посеяны также на чашки, пок¬
рытые коллагеном или на ЗТЗ фидерный слой. Этот
метод позволит вести рутинное культивирование в
течение примерно одной недели. По истечении этого
434
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
ПРОТОКОЛ 23.5. Выделение
и культивирование клеток крипт
толстого кишечника
Схема
Тщательно измельченную ткань дизагрегируют в
коллагеназе и диспазе, крипты отделяют седимен¬
тацией в сорбите.
Материалы
Стерильные
• HBSS/PSG: сбалансированный солевой раствор
Хэнкса с пенициллином, 100 U/мл, стрептомици¬
ном, 30 мкг/мл, и гентамицином 25 мкг/мл
• Ферментированная расщепляющая смесь: кол¬
лагеназа, тип XI, 150 U/мл, диспаза, 40 мкг/мл,
FBS, 1%, bDMEM
• Ростовая среда: DMEM содержащей 25 мМ
глюкозы, 6,8 мМ пирувата, 0,25 U/мл инсулина,
50нг/мл EGF, 2,5% FBS, и антибиотик как в слу¬
чае HBSS, описанном выше, забуференный до pH
7,4 бикарбонатом натрия в 7,5% С02
• S-DMEM: Ростовая среда с 2% сорбитом (0,11 М),
стерилизованная фильтрацией.
• Температура всех сред должна поддерживаться
37 °С.
• Чашки Петри, 9 см
• Универсальный контейнер или 30-50-мл цент¬
рифужная пробирка
• Многолуночный планшет, 24 лунки, покрытых
коллагеном:
а) Разведите маточный раствор коллагена (Vit-
rogen) в десять раз
б) Инкубируйте чашки с 0,5 мл/лунку в течение
Зч.
в) Удалите излишний Vitrogen и высушите план¬
шеты в ламинарном шкафу
Подготовленные планшеты могут храниться
обернутые адгезирующей пленкой (Saran
Wrap) до 1 недели при 4 °С.
• Флаконы, 25 см2
• Скальпели, держатель #4, лезвие #22
• Пипетки Movette (или аналогичные пластиковые
пипетки с широким диаметром)
Протокол
Выделение крипт из толстого кишечника человека:
1. Ткань д ля культивирования должна быть помеще¬
на в DMEM как можно быстрее после получения
от больного. Предварительные исследования
показывают, что при небольшой продолжитель¬
ности времени между выделением и помещением
в DMEM образец способен выдержать хранение
в течение небольшого периода (до 2 ч) при 4 °С,
более длительное хранение не рекомендуется.
2. Дважды промойте интактную ткань в lx HBSS/
PSG, чтобы удалить любые факторы контами¬
нации.
3. Перенесите ткань в чистую чашку Петри со све¬
жей средой HBSS/PSG.
4. Максимально удалите мышечный слой, измель¬
чите ткань при помощи двух скальпелей.
5. Промойте кусочки измельченной ткани: пере¬
несите их в 25-см2 флакон, содержащий 20 мл
HBSS/PSG и несколько раз пипетируйте при
помощи пипетки Movette.
6. Дайте отстояться содержимому флакона и уда¬
лите HBSS/PSG.
7. Повторяйте процедуру до тех пор, пока раствор
HBSS/PSG не будет оставаться почти прозрач¬
ным. Обычно для этого требуется 5 повторных
промывочных процедур.
8. Перенесите ткань в чашку Петри и удалите ос¬
татки HBSS/PSG.
9. Измельчите ткань при помощи скальпелей, пока
не получите однородную консистенцию. Пере¬
несите измельченную ткань в 25-см2 флакон при
помощи пипетки большого калибра (убедитесь,
что внутренняя часть пипетки увлажнена HBSS/
PSG для предупреждения прилипания).
10. Позвольте суспензии отстояться, удалите избы¬
ток HBSS/PSG.
11. Добавьте 25-30 мл ферментативной расщепляю¬
щей смеси.
12. Инкубируйте при 37 °С в течение 1 ч.
13. Замените расщепляющую смесь:
а) Позвольте кусочкам ткани осесть.
б) Тщательно удалите расщепляющую смесь
из флакона (следя за тем, чтобы не удалить
нерасщепленные ткани).
в) Добавьте свежую расщепляющую смесь и
продолжите инкубацию при 37°С в течение
последующих 1-2 ч.
г) Проверяйте ткань каждые 15-30 мин, чтобы
убедиться, что крипты не подверглись избы¬
точному расщеплению. Выделение завершено,
если примерно 70-80% отдельных крипт ос¬
вободились от окружения.
N.B. Не ждите, пока все крипты станут свободными,
поскольку это может привести к снижению выхода
клеток, вызванного избыточным расщеплением.
Седиментация крипт толстого кишечника
человека:
Выделенные крипты очищают седиментацией в сор¬
бите. Вы можете обнаружить, что крупные агрегаты
жировых/мезенхимальных тканей поднимаются к
верхним слоям в пробирке при седиментации; они
должны быть удалены в первую очередь, поскольку
они могут вызвать проблемы при седиментационном
процессе.
14. Разделите содержимое флакона с ферменти¬
рованной тканью на два стерильных 30-мл
универсальных контейнера и сделайте пометку
«Комплект 1».
23.2. Эпителиальные клетки
435
15. Добавьте S-DMEM до объема 25 мл и удалите
любые плавающие по поверхности частицы жи¬
ровой или мезенхимальной ткани.
16. Позвольте содержимому пробирки осесть в тече¬
ние 1 мин, чтобы отделить все нерасщепленные
ферментативно кусочки ткани.
17. Удалив любые плавающие остатки жировой
ткани на поверхности, перенесите супернатант
из пробирки в две чистых пробирки, пометив их
4 Комплект 2».
18. Оставьте раствор на 30 с.
19. Перенесите содержимое в следующую партию
пробирок и пометьте «Комплект 3».
20. Заполните доверху S-DMEM.
21. Центрифугируйте пробирки третьего комплекта
при 200-300 g в течение 4 мин. (Обычно самая
низкая скорость на стандартной настольной
центрифуге.)
22. Удалите супернатант.
23. Взболтайте осадок потряхиванием и постукива¬
нием по основанию пробирки.
24. Ресуспендируйте крипты еще раз в S-DMEM и
повторите центрифугирование. Это отмывание в
S-DMEM с последующим центрифугированием
может быть повторено до 5 раз, либо пока супер¬
натант не будет чистым.
25. Оставшийся в пробирках 1 и 2 комплекта дебрис
следует исследовать микроскопически на пред¬
мет присутствия крипт. Если обнаруживается
высокая доля крипт, материал можно взболтать
и повторить процедуру седиментации. Если в де-
брисе содержится только неферментированный
материал, его можно выбросить.
26. Крипты готовы к дальнейшему культивирова¬
нию, когда супернатант прозрачен. Для хорошего
роста культуры критическим является исход¬
ная посевная плотность. Обычно лучше всего
рост клеток крипт отмечается при плотности
800-1000 крипт/мл в одной лунке 24-луночного
планшета. Для того чтобы определить число вы¬
деленных крипт:
а) ресуспендируйте изолят в 10 мл ростовой
среды;
б) отберите 100 мкл и добавьте к 900 мкл росто¬
вой среды;
в) возьмите 100 мкл от полученного образца и
подсчитайте количество крипт.
N крипт/мл * N подсчитанных крипт х 100
27. Инкубируйте планшеты при 37 °С в 7,5% С02и
позвольте клеткам прикрепиться к субстрату в
течение 2 дней.
28. Удалите из планшета среду, содержащую все
неприкрепленные клетки, и замените на свежую
среду.
29. Через каждые 5 дней добавляйте 0,5 мл свежей
среды.
периода рост эпителиальных клеток ухудшается и
затем становится превалентным рост контамини-
ровавших культуру фибробластов. Культура плохо
субкультивируется. Были предприняты попытки
применения разнообразных восстанавливающих
агентов, однако пока успехи в субкультивировании
эпителиальных клеток кишечника очень ограничены.
Фибробласты могут быть достаточно легко субкуль-
тивированы и являются потенциальным источником
клеток фидерного слоя.
23.2.6. Печень
Культуры клеток из печени взрослого человека не
экспрессируют полный набор свойств паренхимы
печени, и есть некоторые сомнения в том, что мож¬
но культивировать правильную линию клеток. До
настоящего времени попытки создания пролифери¬
рующей клеточной линии не были особенно успеш¬
ными, хотя функционально активные гепатоциты
могут быть культивируемы в определенных строгих
условиях [Guguen-Guillouzo, 2002]. Некоторые из
наиболее полезных постоянных клеточных линий
с минимальными отклонениями в структуре были
получены из крысиных гепатом Reuber Н35 [Pitot
et al., 1964] и Morris [Granner et al., 1968]. Введение
тирозинаминотрансферазы в эти клеточные линии с
дексаметазоном является хорошей моделью для изу¬
чения регуляции ферментативной адаптации в клетках
млекопитающих [Granner et al., 1968; Reel & Kenney,
1968]. Такие клеточные линии, как Hep-G2, также
были созданы на основе клеток человеческой гепато-
мы и сохраняют некоторые метаболические свойства
нормальной печени [Knowles et al., 1980].
Протокол 23.6 для культуры выделенных гепато¬
цитов человека был предоставлен Christiane Guguen-
Guillouzo, Hopital de Pontchaillou, INSERM U49, Rue
Henri le Guilloux, 35033 Rennes, France. При прове¬
дении перфузии в печень через сосуды и капилляры
с адекватной скоростью потока протеолитические
ферменты, такие как коллагеназа, которые относи¬
тельно нетоксичны, будут разрушать межклеточные
соединения. Разрушение соединительнотканной осно¬
вы произойдет в течение 15 мин, если печень предва¬
рительно промыта от крови и Са2+ путем промывания
бескальциевым буфером [Berry & Friend, 1969]. Гепа¬
тоциты выделяют из клеточной суспензии двумя-тремя
дифференциальными центрифугированиями.
При использовании соответствующих компонентов
биоматрикса для покрытия чашек, например фибро-
нектина, коллагена или Матригеля (см. разд. 8.4.1),
можно полностью избежать добавления FBS.
Анализ. Определите клеточный выход и жизнеспо¬
собность путем подсчета клеток с хорошо сохранившей¬
ся лучепреломляющей формы или теста с трипановым
синим (0,2% w/v; обычно 4-6х108 клеток с жизнеспо¬
собностью около 95%).
436
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
ПРОТОКОЛ 23.6. Выделение гепатоцитов
крысы
Схема
Ввести канюлю в портальную вену или портальную
ветвь, промыть печень бескальциевым буфером в
течение 15 мин, перфузировать печень ферментатив¬
ным раствором (15 мин), собрать и промыть клетки,
подсчитать жизнеспособные гепатоциты [Guguen-
Guillouzo & Guillouzo, 1986].
Материалы
Стерильные:
• Среда Лейбовича L-15
• Среда Хэма F12 или Williams’ Е, обогащенные
0,2% БСА (степень очистки V, Sigma) и 10 мкг/мл
бычьего инсулина (80-100 мл)
• Минимальная среда для культивирования гепа¬
тоцитов (НММ): среда Игла МЕМ, 67,5%, среда
199,22,5% (или William’s Е место этих двух сред),
бычий инсулин, 5 мкг/мл, БСА,1 мг/мл, пируват
20 мМ, пенициллин, 100 U/мл, стрептомицин,
100 мкг/мл, 20% FBS
• HMM/SF: бессывороточная НММ с 1х 10~5М
гемисукцината гегидрокортизона
• Бескальциевый буфер HEPES, pH 7,65:160,8 мМ
NaCl; 3,15 мМ КС1: 0,7 мМ Na2HP0412H20,
33 мМ HEPES, стерилизовать фильтрацией через
0,22-мкм-микропоровый фильтр, хранить при
4 °С (2 месяца)
• Раствор коллагеназы: 0,025% коллагеназа (Sigma,
степень чистоты 1 или Roche #103578); 0,075%
СаС12*2Н20 в бескальциевом буфере HEPES, pH
7,65; подготовка и стерилизация фильтрацией
перед использованием
• Нембутал (Abbott, 5%)
• Гепарин (Roche)
• Пробирки Tygon (внутренний диаметр 3,0 мм;
внешний диаметр 5,0 мм)
• Одноразовые иглы для внутривенного вливания
в вены кожи головы, 20G (Dubemard Hospital
Laboratory, Bordeaux, France)
• Нити для подшивания канюли
• Бутылки с делениями или чашки Петри
• Хирургический инструментарий (заостренные,
кривые и прямые ножницы и зажимы)
• Одноразовые шприцы, 1 и 2 мл
Нестерильные:
• Хронометр
• Перистальтический насос (10-200 об./мин)
• Водяная баня
Протокол
1. Подогрейте промывочный буфер HEPES и
раствор коллагеназы в водяной бане (обычно
примерно до 38-39 °С чтобы получить в печени
37 °С). Оксигенация не обязательна.
2. Установите скорость пропускания в перисталь¬
тическом насосе 30 мл/мин.
3. Анестезируйте крысу (вес 180-200 g) внутрибрю-
шинным введением Нембутала (150 мкг/100 г),
и введите гепарин в бедренную вену (1000IU).
4. Протрите брюшную поверхность тела крысы 70%
этиловым спиртом.
5. Вскройте брюшную полость, наложите незатяну¬
тую лигатуру вокруг портальной вены примерно
в 5 см от печени, введите канюлю в печень, затя¬
ните лигатуру.
6. Быстро вскройте подпеченочные сосуды, чтобы
избежать избыточного давления, и начните пер¬
фузию 500 мл безкальциевого буфера HEPES при
скорости потока 30 мл/мин; удостоверьтесь что
печень белеет в течение нескольких секунд.
7. Перфузируйте 300 мл раствора коллагеназы при
низкой скорости потока 15 мл/мин. в течение
20 мин. Печень становится набухшей.
8. Удалите печень и промойте ее буфером HEPES.
9. После разрушения глиссоновой капсулы распре¬
делите клетки в 100 мл среды Лейбовича L-15.
10. Профильтруйте суспензию через 2 слоя марли
60-80-мкм нейлоновой сетки, оставьте суспен¬
зию на 20 мин для осаждения жизнеспособных
клеток (обычно при комнатной температуре), и
удалите супернатант (60 мл) содержащий дебрис
и мертвые клетки.
11. Промойте клетки три раза низкоскоростным
центрифугированием (50 g в течение 40 с) для
удаления коллагеназы, поврежденных клеток и
непарензиматозных клеток.
12. Суспендируйте выделенные гепатоциты в НММ.
13. Через 2-3 ч, живые клетки прикрепятся к плас¬
тику и начнут распластываться. Удалите мертвые
клетки вместе с верхним слоем среды.
14. После прикрепления клеток удалите FBS и замес¬
тите среду на свежую HMM/SF. В последующем
заменяйте среду каждый день.
Вариации
Выделение гепатоцитов других видов. Основная
двухшаговая процедура перфузии [Seglen, 1975] может
быть использована для получения гепатоцитов из раз¬
личных видов грызунов, включая мышей, кроликов,
морских свинок или сурков путем адаптации объема
и скорости потока перфузионного раствора к разме¬
рам печени. Метод может также быть адаптирован к
печени человека [Guguen-Guillouzo et al., 1982] путем
перфузии доли печени (обычно 1,5 л HEPES буфера
и 1 л раствора коллагеназы при 70 и 30 мл/мин, соот¬
ветственно) или путем взятия биопсии (15-30 мл/мин
в зависимости от размеров образца ткани). Полное
выделение до суспензии единичных клеток может
быть осуществлено путем дополнительной инкубации
с коллагеназой при 37 °С при осторожном перемети-
23.2. Эпителиальные клетки
437
вании в течение 10-20 мин (особенно в случае печени
человека) [Guguen-Guillouzo & Guillouzo, 1986]. Гепа-
тоциты рыбы могут быть получены канюлированием
интестинальной вены и иссечением сердца для избе¬
жания избыточного давления. Перфузия проводится
при комнатной температуре при скорости потока
12 мл/мин.
Поддержание и дифференцировка. Выделенные
паренхиматозные клетки могут поддерживаться в
суспензии в течение 4-6 ч и использоваться в крат¬
косрочных экспериментах. При посеве на питательную
среду с добавлением 1x10-6 М дексаметазона на плас¬
тиковые чашки для культуры (7х105 жизнеспособных
клеток/мл) клетки живут несколько дней. Выживание
в течение нескольких недель может быть достигнуто
посевом клеток в биоматрикс [Rojkind et al., 1980]. Тем
не менее клетки быстро теряют свои специфические
дифференцированные функции. Высокая стабиль¬
ность (2 мес) может быть получена ко-культивиро-
ванием гепатоцитов с эпителиальными клетками
печени крысы, предположительно выделенными из
первичных билиарных клеток [Guguen-Guillouzo et al.,
1983]. Гепатоциты также более стабильны при посеве
на Матригель [Bissell et al., 1987] и способны пройти
один-три клеточных деления, когда они культиви¬
руются в питательной среде с добавлением EGF и
пирувата [McGowan, 1986] или никотинамида [Mitaka
et al., 1991]. Комбинация EGF и DMSO стимулирует
дифференцировку гепатоцитов [Mitaka et al., 1993;
Hino et al., 1999].
23.2.7. Поджелудочная железа
Как ацинарные [Bosco et al., 1994], так и клетки
островков [Vonenet al., 1992; Cao et al., 1997] были
выращены из панкреатической ткани. Были получены
линии, выделенные из «иммортализованных мышей»
[Blouin et al., 1997]. Конверсия аденомы в карциному
привела к повышению интереса к культуре [Perlet al.,
1998]. Протокол 23.7 для культуры ацинарных клеток
поджелудочной железы предоставлен Robert J. Hay &
Maria das Gracas Miranda, American Type Culture
Collection, 10801 University Boulevard, Manassas, VA
20110-2209.
Фракционированные популяции панкреатических
ацинарных эпителиальных клеток инокулировали на
обработанные коллагеном чашки для культуры, позво¬
ляя развиться двумерным агрегатным колониям, кото¬
рые затем изучали [Нау, 1979; Ruoff & Hay, 1979].
Вариации. После прикрепления клеток, F12K-C20
может быть замещена бессывороточной средой МЕМ
с инсулином (Юнг/мл), трансферрином (5 нг/мл),
селеном (9 нг/мл), и EGF (21 нг/мл). Эта среда под¬
держивает рост панкреатических клеток в течение,
по крайней мере, 15 дней без изменения клеточной
морфологии. Предшествующий метод применялся для
ПРОТОКОЛ 23.7. Поджелудочный эпителий
Схема
Диссоциировать клетки ткани поджелудочной желе¬
зы морской свинки, профильтровать суспензию сме¬
шанных клеток через ситовую ткань или нейлоновое
сито, отделить слой суспензии над раствором БСА.
После трех последовательных центрифугирований
и фракционирований взболтать клеточный осадок
и посеять клетки в сосуды для культивирования,
обработанные коллагеном.
Материалы
Стерильные
• Культуральная среда F12K с 20% сывороткой
теленка (F12K-CS20)
• HBSS
• HBSS без Са2+ и Mg2+ (HBSS-DVC)
• D-глюкоза (декстроза) (Difco, No. 0155-174, B-D
Biosciences)
• БСА, фракция V (Sigma)
• Раствор трипсина: 1:250 (Difco, B-D Biosciences)
в 0,25% цитратном буфере; 3 г/л тринатриевого
цитрата; 6 г/л NaCl, 5 г/л D-глюкозы; 0,02 г/л
фенолового красного; pH 7,6.
• раствор коллагеназы: растворимая коллагеназа
(GIBCO) в lx HBSS-DVC (pH 7,2) до конечной
концентрации 1800 U/мл. Доведите pH раство¬
ра до pH 7,2, диализировать с использованием
12-KDa диализной мембраны в течение 4 ч при
4 °С против lx HBSS-DVC, содержащей 0,2%
глюкозы. HBSS удаляют. Этот шаг повторяют
16-18 ч со свежей HBSS-DVC. Фильтруют, сте¬
рилизуют, хранят раствор разлитым по аликво¬
там 10-20 мл при температуре ниже -70 °С.
• Конический флакон, 25 мл (Bellco), силикони-
зированный
• Пипетки, 5 или 10 мл с широким горлышком
(Bellco)
• Делительная воронка Бюхнера
• Диализная установка (Spectro-Por)
• Емкость, 250 мл
• Полипропиленовые центрифужные пробирки,
50 мл
• Ситовая ткань
• Культуральные сосуды, покрытые коллагеном
(Biocoat, BD Biosciences)
Нестерильные:
• Водяная баня с перемешиванием (Backer модель
М5В#11-22-1)
• Центрифуга
Протокол
1. Приготовьте диссоциирующий раствор (смесь
1 части коллагеназы с 2 частями раствора трип¬
сина) и подогрейте до 37 °С.
2. Асептически удалите всю поджелудочную железу
(0,5-1,0 г) и поместите орган в F12K-CS20.
438
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
3. Обрежьте брыжеечные мембраны и другие вне-
органные структуры, измельчите поджелудочную
железу на 1-3-мм фрагменты.
4. Перенесите фрагменты в силиконизированный
25-мл конический флакон в 5 мл предварительно
подогретой диссоциирующего раствора. Переме¬
шивайте раствор в качалочной бане примерно
15 мин при 120 об./мин при 37 °С. Повторите
этот шаг два-три раза со свежим раствором, пока
большая часть тканей не прейдет в диспергиро¬
ванное состояние.
5. После каждой диссоциации позвольте крупным
фрагментам осесть, перенесите супернатант в
50-мл полипропиленовые центрифужные про¬
бирки, содержащие примерно 12 мл холодной
среды F12K-CS20 для нейтрализации диссоци¬
ирующего раствора.
6. Центрифугируйте суспензию при 600 об./мин
в течение 5 мин, и ресуспендировать осадок в
5-10 мл холодной среды F12KGS20. Храните
суспензию клеток на льду.
7. Объедините клеточную суспензию и пропустите
через несколько слоев стерильной фильтрующей
ткани в Бюхнеровской делительной воронке.
Используйте небольшое разрежение путем встав¬
ления ствола делительной воронки в вакуумный
флакон при фильтрации. Отберите аликвоту для
оценки количества клеток.
8. Обычно на этом этапе получают 1-2x10s кл./мг
ткани с 90-95% жизнеспособности.
9. Перенесите слой, содержащий 5х107-5х108 кле¬
ток из полученной суспензии на поверхность
2-4 колонок, содержащих 35 мл 4% холодного
БСАв HBSS-DVC (pH 7,2) в полипропиленовых
50-мл центрифужных пробирках. Центрифу¬
гируйте пробирки при 600 об./мин в течение
5 мин. Этот шаг критичен для достижения хо¬
рошего разделения ацинарных клеток от клеток
островков, протоков к стромы поджелудочной
экезезы. Удалите супернатант и повторите фрак¬
ционирование дважды, каждый раз объединяя и
ресуспендируя осадок в холодной F12K-CS20.
10. Соберите клеточные осадки в 5-10 мл холодной
F12K-CS20. Отберите аликвоту для подсчета
клеток. Получают 2-5х104 кл./мг ткани с жизне¬
способностью 80-95% от общего количества аци¬
нарных клеток. Плотность инокуляции может
составлять Зх105/см2. Клетки прикрепляются и
образуют колонии примерно в течение 72 ч.
исследований на тканях морской свинки и человека
(донорский трансплантат). Добавление облученных
человеческих фибробластов из легких в качестве
фидерного слоя приводило к заметной стимуляции
(вплоть до 500%) включения [3Н]-тимидина и продле¬
вало выживание
23.2.8. Почка
Почки структурно являются комплексным органом, в
котором система нефронов и собирательных трубочек
состоит из большого количества функционально и фе¬
нотипически различимых сегментов. Эта сегментарная
гетерогенность сопровождается многообразием клеток,
которое еще не до конца охарактеризовано. Некоторые
тубулярные сегменты содержат несколько морфологи¬
чески различных клеточных типов. Кроме того, есть
свидетельства возможности быстрых адаптивных из¬
менений в клеточной ультраструктуре, которые могут
коррелировать с изменениями клеточных функций
[Stanton et al., 1981]. Структурная и клеточная гете¬
рогенность представляет дополнительные трудности
специалистам по культурам клеток, при получении
чистой или высокообогащенной клеточной популяции.
Проблема возрастает при работе почек человека, форма
которых и обращение с образцами могут сделать неко¬
торые манипуляции, такие как сосудистая перфузия,
затрудненными или невозможными
В культуре клеток специфических тубулярных
сегментов успешно используется несколько подходов.
Метод градиента плотности является общепримени¬
мым для выделения обогащенных популяций фермен¬
тативно расщепленных тубулярных сегментов, в част¬
ности этот метод эффективен в получении культуры
тубулярных клеток проксимальных отделов канальцев
почек экспериментальных животных [Taub et al., 1989].
Специфические нефроны или сегменты собиратель¬
ных трубочек могут также быть выделены методом
микросечений и затем эксплантированы в субстрат
для культуры. Этот метод, разработанный с использо¬
ванием экспериментальных животных [Horster, 1979],
был применен к культуре человеческих почек [Wilson
et al., 1985] и эпителия стенки кисты поликистозной
почки [Wilson et al., 1986]. Методы иммунодиссекции
[Smith & Garcia-Perez, 1985] и иммуномагнитного
разделения [Pizzonia et al., 1991] также были разрабо¬
таны для выделения специфических типов нефронов
на основе экспрессии типоспецифичных клеточных
эктоантигенов. Эти изящные методы являются мно¬
гообещающими для изучения специфических почеч¬
ных клеточных типов в норме и патологии, однако
они пока применяются практически исключительно
для исследований на экспериментальных животных.
Ограниченная (и непредсказуемая) доступность и
непостоянная форма (например, иссеченный кусок, по¬
врежденная система васкуляризации, длинный период
после нефроэктомии) человеческой донорской почки
повышают значение постепенного ферментативного
расщепления для практических целей при культуре
клеток человеческой почки.
Описан ряд методов для первичной культуры ткани
человеческой почки [Detrisac et al., 1984; States et al.,
1986; McAteer et al., 1991]. Протокол 23.8 и предшест¬
вовавшее введение были предоставлены James McAteer,
Stephen Kempson, and Andrew Evan, Indiana University
School of Medicine, Indianapolis, IN 46202-5120.
23.2. Эпителиальные клетки
439
Первичная культура тканевых фрагментов, извле¬
ченных из внешней коры человеческой почки, пред¬
ставляет собой способ выделения клеток, которые
проявляют много функциональных характеристик
проксимальных трубочек [Kempson et al., 1989].
Постепенное ферментативное расщепление и гру¬
бая фильтрация приводят к получению отдельных
клеток и небольших агрегатов клеток, которые при
посеве с высокой плотностью дают рост гетерогенной
популяции, обогащенной эпителиальными клетками.
Большое количество клеток может быть получено
таким методом, что позволяет использовать его для
установления множественных репликатов первичной
культуры или размножить клетки для последующего
замораживания и хранения. При наличии определен¬
ного опыта функциональные характеристики таких
первично культивированных и субкультивированных
клеток являются воспроизводимыми для почек от
различных доноров, и соответствующие манипуляции
с образцами позволяют получить хорошие культуры
даже от почек с длительным пост-нефроэктомичес-
ким периодом.
Протокол
Диссоциация тканей
1. Отрежьте образцы коры почки толщиной
5-10-мм, промойте фрагменты в охлажденной
основной среде.
2. Извлеките фрагменты наружного коркового слоя
и, используя перекрещенные лезвия, измельчите
ткань до размера 1-2 мм.
3. Перенесите примерно 5 мл фрагментированной
ткани в 50-мл пробирку, содержащую 20 мл теп¬
лого раствора коллагеназа/трипсин. Закрепите
пробирку на инкубируемой платформе кольце¬
вого шейкера при температуре 37 °С.
4. Инкубируйте ткань с осторожным потряхивани¬
ем в течение 1 часа.
5. Удалите и замените раствор ферментов.
6. Затем отберите супернатант через 20 мин после
добавления 20 мл основной среды, содержащей
0,05 мг/мл Dnase, и осторожно гомогенизируйте
при помощи 10-мл пипетки (10 циклов).
7. Разведите собранный супернатант равным объ¬
емом полной культуральной среды и храните
полученную суспензию на льду.
8. Повторите процедуру сбора пять или более раз,
пока фрагменты ткани не будут полностью дис¬
пергированы.
9. Объедините собранные супернатанты и разлейте
на аликвоты в 50 мл пробирки.
10. Центрифугируйте при 100 g в течение 15 мин,
ресуспендируйте каждый осадок в 45 мл полной
питательной среды, содержащей DNase.
11. Профильтруйте суспензию через нитекс-фильтр
(160 мкм).
12. Протокол выделения предполагает выделение
как отдельных клеток, так и их агрегатов. Следо¬
вательно, подсчет клеток невозможен. Для суб-
культивирования или замораживания клеточной
культуры посейте 15 мл в 75-см2 флакон.
Субкультура и криоконсервация. Субкультура
клеток NHK-C ведется обычными методами. Диссо¬
циация при помощи трипсин/ЭДТА дает монодис-
персную суспензию, которая подходит для подсчета
на гемоцитометре. Посейте клетки на чашки Петри для
культуры тканей с плотностью примерно lxlO5 кл./см2.
Криоконсервация проводится обычными методами с
использованием полной среды, содержащей 10% ди-
метилсульфоксида (см. протокол 20.1).
Комментарии и меры предосторожности Опреде¬
ленные вариации этого метода связаны с качеством
образца почки. Важно, чтобы процедуры, применяемые
для получения ткани, были стандартными. Донорская
ткань обычно включает в себя сегменты почки, полу¬
ченные при хирургической операции (нефроэктомии,
например, при почечноклеточной карциноме), и ин-
тактную почечную ткань, которую посчитали непод¬
ПРОТОКОЛ 23.8. Эпителий почки
Схема
Фрагменты ткани извлекают из внешнего коркового
слоя почки, измельчают, отмывают и инкубируют в
качалочной водяной бане в растворе коллагеназа/
трипсин. Ткань периодически пипетируют до обра¬
зования однородной смеси, диссоциирование клетки
фильтруют через фильтр с ячейками ограниченного
размера д ля удаления нерасщепленных фрагментов.
Клетки затем трижды отмывают от ферментов, сус¬
пендируют в среде с сывороткой и сеют на пласти¬
ковую посуду для культивирования.
Материалы
Стерильные:
• Основная среда DMEM/F 12: смесь DMEM и
среды Хэма F12 в отношении 50:50
• FBS (Sterile Systems, Hyclone)
• Полная культуральная среда DMEM/F12 с до¬
бавлением 10% FBS
• BSS
• Коллагеназа (Worthington, тип IV), 0,1%, трипсин
(1:250, Sigma), 0,1% раствор в 0,15 М NaCl
• DNase, 0,5 мг/мл в растворе
• Раствор трипсин/EDTA: трипсин (1:250), 0,1%,
EDTA (Sigma, для культур) 1 мМ
• Скальпели, тканевые пинцеты
• Фильтр нитекс (160 мкм) (Tetko)
• Культуральные чашки, флаконы
• Пробирки стерильные, 50 мл
Нестерильные:
• Кольцевой шейкер (Bellco)
440
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
ходящей для трансплантации (например, в результате
нарушенной сосудистой системы). Эти образцы могут
быть собраны при различных условиях (например,
при наличии или в отсутствие перфузии с различным
периодом тепловой ишемии, или с различной продол¬
жительностью постнефроэктомического периода).
Кроме того, различия в возрасте и состоянии здоровья
донора приводят к тому, что не существует двух экви¬
валентных образцов почки. Хотя не все образцы почки
дают удовлетворительный выход клеток, приведенный
протокол вполне подходит для широкого спектра образ¬
цов, включая свежевыделенные хирургическим путем
и интактные образцы почки, если они находились на
холоде при перфузии в течение примерно 100 ч.
• Требование безопасности. Работа, включаю¬
щая в себя контакты с любыми тканями и биологи¬
ческими жидкостями человека, требует соблюдения
соответствующих мер предосторожности во избежание
контакта с возбудителями, переносимыми кровью (см.
разд. 7.8).
Применение NHK-C клетки главным образом обла¬
дают функциональными характеристиками почечных
проксимальных канальцев, включая специфическую
№+-зависимую транспортную систему неорганическо¬
го фосфата, ингибируемую паратиреоидным гормоном
(РТН) [Kempson et al., 1989]. Они также обладают
флоризин-чувствительной №+-зависимой системой
транспорта гексозы, тип гормональной стимуляции
сАМР, характерный для проксимальных канальцев
(РТН-зависимый, вазопрессин-стимулируемый), и
экспрессируют некоторые ферменты щеточной каемки
проксимальных канальцев (мальтаза, лейцинамино-
пептидаза, гамма-глютамилтранспептидаза). Помимо
того, клетки NHK-C обычно используются для демонс-
транции специфичности ингибитора ко-транспорта
Na+/Pj [Yusufi et al., 1986], а также служат в качестве
модели оксидативного повреждения почечных каналь¬
цев [Andreoli & McAteer, 1990].
23.2.9. Бронхиальный
и трахеальный эпителий
Протокол 23.9 для выделения и культивирования
нормальных эпителиальных бронхиальных и тра¬
хеальных клеток человека, полученных методом
аутопсии, был предоставлен Moira A. LaVeck и John
F. Lechner, National Institutes of Health, Bethesda,
MD, Bayer Diagnostics, P.O. Box 2466, Berkeley, CA
94702, USA.
В присутствии сыворотки нормальные клетки
бронхиального эпителия (NHBE) прекращают де¬
литься и вступают в терминальную дифференцировку.
В бессывороточной среде, оптимизированной для
роста клеток NHBE, в которой не поддерживается
пролиферация легочных фибробластов, происходит
получение чистой культуры клеток NHBE [Lechner &
LaVeck, 1985].
ПРОТОКОЛ 23.9. Бронхиальный
и трахеальный эпителий
Схема
Эта процедура включает выделение фрагментов круп¬
ных воздухоносных путей, помещение их в бессыворо-
точную среду (LHC-9) для того, чтобы инициировать
и поддерживать рост культуры NHBE клеток без
фибробластов; обычно проводится четыре субкуль¬
тивирования и 30 периодов удвоения популяции.
Материалы
Стерильные:
• Культуральная среда:
а) L-15 (Invitrogen)
б) LHC-9 [Lechner & LaVeck, 1985] (см. табл. 10.1)
в) НВ [Lechner & LaVeck, 1985] (см. Приложе¬
ние!)
• Бронхиальная ткань (аутопсийный материал от
доноров, не больных раком).
• Пластиковые чашки для культивирования тка¬
ней (6 и 10 см).
• FN/V/BSA: Смесь человеческого фибронектина
10 мкг/мл (Collaborative Research, B-D Biosci¬
ences), коллагена (Vitrogen 100,30 мкг /мл; Col¬
lagen Corp.), и кристаллического БСА10 мкг/мл
(Вауег) в основной среде LHC (Biosource Inter¬
national) [Lechner & LaVeck, 1985]
• Трипсин, 0,02%, EGTA (Sigma), 0,5 мМ, и по-
ливинилпирролидон (PVP) (USB), 1% раствор
[Lechner & LaVeck, 1985]
• Скальпели No. 1621 (B-D Biosciences)
• Хирургические ножницы
• Полуискривленные щипцы для микродиссекции
• Газовая смесь с высоким содержанием 02 (50%
02,45% N2,5% СО*)
Нестерильные:
• Перчатки (человеческие ткани могут быть конта-
минированы биологически опасными агентами)
• Камера с контролируемой атмосферой (Bellco
№7741)
• Качалочная платформа (Bellco No. 7740)
Протокол
1. Покройте культуральные чашки смесью FN/V/
BSA— 1 мл на 6-см чашку и инкубируйте чашку в
С02-инкубаторе с влажной атмосферой при 37 °С
в течение по крайней мере 2 ч (не превышать 48 ч).
Удалите смесь при помощи вакуум-аспирации и
заполните чашки 5 мл культуральной среды.
2. Асептически иссеченные ткани аутопсийного мате¬
риала легкого от не больных раком доноров в пред¬
шествующие 12 ч помещают в холодную (на льду)
среду Е-15для транспортировки в лабораторию,
где бронхи подвергаются дальнейшему иссечению,
отделяются от периферической легочной ткани.
3. До начала культивирования процарапайте лез¬
вием скальпеля область площадью в один квад¬
23.2. Эпителиальные клетки
441
ратный сантиметр на одном краю поверхности
6-см чашки для культуры
4. Откройте воздуховодные пути (погруженные в
среду L-15) при помощи хирургических ножниц
и порежьте (резкими режущими движениями,
не пилите) ткань при помощи скальпеля на два
кусочка: 2x3 см.
5. Используя скоблящие движения, чтобы предуп¬
редить повреждение эпителия, соберите влажные
фрагменты ткани и поместите их эпителиальной
стороной вверх на очерченную зону 6-см чашки.
Удалите среду, инкубируйте фрагмент при ком¬
натной температуре 3-5 мин — чтобы произошла
адгезия к процарапанной зоне на чашке.
6. Добавьте 3 мл среды НВ в каждую чашку и помес¬
тите их в камеру с контролируемой атмосферой
на качалочную платформу.
7. Заполните камеру газовой смесью с высоким
содержанием кислорода.
8. Вращайте камеру при скорости 10 оборотов в
минуту, заставляя среду постоянно омывать по¬
верхность эпителия.
9. Инкубируйте таким образом фрагменты тканей
при 37 °С, заменяя среду и атмосферу через день
и в последующие 6-8 дней через двухдневный
интервал. Этот этап улучшает последующее
культивирование клеток, поскольку восстанавли¬
вает ишемические повреждения, которые могли
возникнуть во время смерти донора, пока ткань
не была помещена в холодную среду L-15.
10. До культивирования процарапайте семь зон на
поверхности каждой 10-см культуральной чашки
скальпелем, покройте поверхность отмеченных
чашек раствором смеси FN/V/BSA, удалите из¬
быток жидкости, как описано выше.
11. Порежьте влажные фрагменты, восстановленные
после ишемии, на кусочки 7x7 мм, поместите
кусочки эпителиальным слоем наверх на обоз¬
наченные зоны.
12. Инкубируйте кусочки при комнатной темпера¬
туре без среды в течение 3-5 мин, как описано
выше.
13. Добавьте 10 мл среды LHC-9 в каждую чашку, и
инкубируйте в паровом 5%С02-инкубаторе при
37 °С
14. Заменяйте среду на свежую каждые 3-4 дня.
15. Через 8-11 дней инкубации, когда эпителиаль¬
ные клетки перерастут границы эксплантата
более чем на 0,5 см, перенесите эксплантат на
новую культуральную чашку, с процарапанной
зоной и покрытой FN/V/BSA для получения
новой области роста эпителиальных клеток. Этот
шаг можно повторять до 7 раз с высоким выходом
NHBE клеток.
16. Инкубируйте последующий рост клеток в среде
LHC-9 еще 2-4 дня до трипсинизации (с раство¬
ром трипсин/ЕGTA/PVP) и субкультивирова¬
ния для экспериментальных исследований.
23.2.10. Эпителий ротовой полости
Кератиноциты ротовой полости были культивированы
в среде, аналогично LHC-9 и MCDB153, с применени¬
ем чашек, обработанных для лучшего прикрепления
клеток FN/C/BSA. Протокол 23.10 представляет собой
сокращенный протокол, описанный Grafstrom [2002].
ПРОТОКОЛ 23.10. Кератиноциты ротовой
полости
Материалы
Стерильные:
• Транспортная среда Лейбовича L-15 с 100 мкг/мл
гентамицина, 100 U/мл пенициллина, 100 мкг/мл,
Фунгизон, 1 мкг/мл.
• Ростовая среда модифицированная MCDB 153
[Grafstrom,2002]
• Ростовая среда с антибиотиком: ростовая среда с
гентамицином 100 мкг/мл, пенициллин-стрепто-
мицином 100 U/мл и фунгизоном, 1 мкг/мл
• D-PBSA: Дюльбекко PBS без Са2+ и Mg2+
• Трипсин 0,17% в D-PBSA
• РЕТ: трипсин 0,025%; EGTA, 0,5 мМ; поливинил-
пирролидон (PVP), 40 KDa, 1% в D-PBSA
• Скальпели, лезвие #11
• Ножницы
• Пинцеты, 2 пары
• Чашки Петри для культуры тканей, 5 или 10 см
• Чашки Петри не для культуры тканей (для иссе¬
чения тканей) 3,5 или 10 см
• Центрифужные пробирки конические, 15 мл
• Микропипетки, например, Gilson, 100 мкл
• Культуральные чашки, обработанные FN/C/BSA,
5 см (см. протокол 23.9)
Протокол
Первичная культура:
1. Получите материал, — хирургический или ранний
аутопсийный. Поместите в холодную транспорт¬
ную среду и перенесите в лабораторию как можно
быстрее.
2. Поместите ткань в 10-см чашку и ополосните
фосфатно-буферным раствором (D-PBSA).
3. Максимально удалите соединительную ткань,
включая части, содержащие кровь. Если образцы
ткани имеют площадь поверхности около 1 см2,
поделите на меньшие кусочки.
4. Поместите образцы в 3,5-см чашку и добавьте
раствор трипсин 0,17% в таком объеме, чтобы
покрыть образец (1-2 мл) и инкубируйте в те¬
чение ночи при 4 °С.
5. Удерживая каждый образец пинцетом, отслаи¬
вайте и соскребайте эпителий другим пинцетом
в раствор трипсина.
6. Тщательно гомогенизируйте суспензию несколько
раз для дальнейшей дезагрегации клеток, затем пе¬
ренесите суспензию в центрифужную пробирку.
442
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
7. Ополосните чашку D-PBSA, затем добавьте этот
раствор в клеточную суспензию. Используйте
тот же объем для ополаскивания, который вы
использовали при дезагрегации тканей в шаге 4.
8. Отберите аликвоту микропипеткой для опреде¬
ления клеточного выхода.
9. Осадите клетки на 125 g при 4 °С, предпочтитель¬
но на центрифуге с охлаждением, затем ресуспен¬
дируйте клетки в ростовой среде.
10. Разведите суспензию ростовой средой до необ¬
ходимой концентрации и посейте клетки при
5х103/см2на чашки, обработанные FN/C/BSA.
Если клетки получены из аутопсийного мате¬
риала, используйте среду, содержащую антиби¬
отики для первых 3-4 дней культивирования.
11. Замените питательную среду у клеток через 24 ч
для удаления возможного клеточного дебриса и
эритроцитов.
12. Меняйте среду через день.
Субкультивирование:
13. Ополосните однократно клетки D-PBSA.
14. Добавьте раствор РЕТ, так чтобы клетки были
покрыты раствором полностью, например исполь¬
зуйте 3 мл на10-см чашку.
15. Инкубируйте при комнатной температуре, пока
клетки не начнут округляться и/или отрываться
от чашки. Следите за отделением клеток под
микроскопом (процедура обычно занимает
5-10 мин). Скорость отделения может быть повы¬
шена добавлением высокой концентрации трип¬
сина или повышением температуры до 37 °С.
16. Когда большинство клеток отделились, осто¬
рожно постучите по чашке и пипеткой направьте
раствор трипсина на поверхность роста так, чтобы
механически ускорить отделение.
17. Когда клетки отделились, добавьте в чашку 5-
10 мл D-PBSA, чтобы инактивировать действие
трипсина.
18. Перенесите суспензию клеток в центрифужную
пробирку и осторожно гомогенизируйте для по¬
лучения равномерного распределения клеток в
суспензии.
19. Используя 1-мл пипетку, отберите образец клеток
для подсчета и перенесите их в камеру гемоцито-
метра (см. протокол 21.1).
20. Определите концентрацию клеток в суспензии.
21. Осадите клетки центрифугированием в течение
5 мин на 125 g при 4 °С.
22. Удалите супернатант и, аккуратно постукивая по
пробирке, взболтайте осадок.
23. Добавьте ростовую среду по желанию и осторож¬
но размешайте клеточную суспензию пипеткой,
распределите по культуральным сосудам.
24. Поместите все сосуды на лоток. Осторожно
вручную покачивайте лоток с сосудами в разных
направлениях, чтобы быть уверенным, что плот¬
ность клеток в каждом сосуде одинакова (простое
вращение чашек приведет к сосредоточению
клеток в центре чашки). Важно, чтобы полки, на
которых располагаются сосуды, были одинаково
закреплены в инкубаторе, а сам инкубатор не
вибрировал, а также располагался так, чтобы слу¬
чайные сотрясения не нарушали прикрепление и
рост клеток.
25. Инкубировать клетки 4-24 ч до замены среды.
23.2.11. Простата
В ряде публикаций описывается использование кле¬
точных культур из тканей простаты [Cronauer et al.,
1997; Peehl, 2002], при этом особый интерес вызывает
регуляция клеточной пролиферации и дифференци¬
ровки паракринными факторами роста [Yan et al., 1992;
Chopra et al., 1996; Thomson et al., 1997]. Протокол 23.11
для первичной культуры клеток эпителия простаты
крысы предоставлен W.L. McKeehan, Texas А&М
University, 2121 West Holcombe Boulevard, Houston,
Texas 77030-3303, USA. Выделенные эпителиальные
клетки нормальной простаты крысы являются хорошей
моделью для изучения клеточной биологии поддержа¬
ния, роста и функционирования нормальной простаты
в контролируемых исследователем определенных
условиях [McKeehan et al., 1982, 1984; Chaproniere &
McKeehan, 1986]. Нормальные клетки служат в ка¬
честве контрольного типа клеток при исследовании
клеток аденокарциномы простаты. Условия, подобные
описанным здесь, подходят и для первичной культуры
эпителиальных клеток перевиваемых опухолей. Клю¬
чевые характеристики этого метода являются специфи¬
чески поддерживающими для эпителиальных клеток
путем применения улучшенной питательной среды и
гормоноподобных факторов роста [Yan et al., 1992], в
то же время происходит конкурентное ингибирование
роста фибробластов в условиях дефицита или ингиби¬
рующего действия среды.
ПРОТОКОЛ 23.11. Эпителий простаты
Схема
Изъять и подготовить простату для культивирова¬
ния клеток (30 мин). Инкубировать ткань простаты
с коллагеназой (1 ч), собрать отдельные клетки и
небольшие агрегаты клеток (1 ч). Инокулировать
клетки и культивировать до конфлюэнтного моно¬
слоя (7 дней).
Материалы
Стерильные:
• Ростовая среда WAJC404 [McKeehan et al., 1984;
Chaproniere & McKeehan, 1986] (см. табл. 10.1)
• Среда-солевой раствор (MSS) [McKeehan et al.,
1984]:
a) NaCl 12 mM
23.2. Эпителиальные клетки
443
б) КС1 2 мМ
в) КН2Р04 1 мМ
г) HEPES, pH 7,6 30 мМ
д) Глюкоза 4мМ
е) Феноловый красный 3,3 мкМ
• Фетальная бычья или лошадиная сыворотка
• Пенициллин
• Канамицин
• Холерный токсин
• Дексаметазон
• Эпидермальный фактор роста (EGF)
• Овечий или крысиный пролактин
• Инсулин
• Частично очищенный [McKeehan et al., 1984;
Chaproniere & McKeehan, 1986] или очищенный
[Crabb et al., 1986] простатропин (фактор роста
эпителиальных клеток простаты)
• Коллагеназа, тип I (Sigma)
• Чашки Петри (стеклянные или пластиковые),
60 мм
• Ножницы
• Шприц и канюля 14-G
• Флакон Эленмайера, 25 мл
• Проволочное сито, ячейки 1-мм, сделанное так,
чтобы подходить к 50-мл конической центри¬
фужной пробирке
• Коническая центрифужная пробирка, 50 мл
• Нейлоновые фильтры 50,150,100, и 40 мкм, под¬
ходящие для 50-мл коническим пробиркам
• 25-луночные планшеты для культуры тканей
Нестерильные:
• Водяная баня-шейкер при 37 °С
• Центрифуга
• Гемоцитометр или счетчик клеток Coulter
• Крысы, самцы, 10-12 недель
Протокол
1. Асептически выделите желаемую долю простаты и
поместите ее в стерильную 60-мм чашку Петри.
2. Удалите жир с доли и взвесьте его.
3. Добавьте 2 мл коллагеназы: 675 U/мл в MSS, со¬
держащем 100 U/мл пенициллина и 100 мкг/мл
канамицина.
4. Измельчите ткани при помощи ножниц на кусоч¬
ки примерно по 1 мм, Достаточно маленькие для
того, чтобы проходить через канюлю 14-G.
5. Используя шприц и канюлю 14-G, перенесите
измельченные фрагменты ткани в стерильный
25-мл флакон Эрленмайера. Добавьте коллаге-
назу: 1мл на 0,1 г исходного веса ткани.
6. Инкубируйте в течение 1 ч при 37 °С в водяной
бане-шейкере.
7. Отсосите через канюлю 14-G расщепленную
ткань трижды, а затем пропустите суспен¬
зию через грубое (1 мм) металлическое сито,
вставленное в 50-мл пластиковую коническую
пробирку для удаления дебриса и нерасщеплен-
ного материала. Промойте суспензию равным
объемом MSS, содержащего 5% цельной феталь¬
ной сыворотки теленка или лошади.
8. Соберите клетки центрифугированием на 100 g
в течение 5 мин при 4 °С.
9. Ресуспендируйте клеточный осадок в 5 мл MSS
с 5% сыворотки и пропустите суспензию через
нейлоновые фильтры с размером ячеек 250,150,
100 и 40 мкм. Промойте каждый фильтр 5 мл
MSS с 5% сыворотки.
10. Соберите клетки центрифугированием и ресус¬
пендируйте 5 мл питательной среды WAJC404.
11. Подсчитайте клетки и доведите концентрацию до
4х106кл./мл.
12. Поместите 50 мкл, содержащих 2х105 клеток, в
каждую ячейку 24-луночного планшета (площадь
лунки 2 см2), содержащего 1мл среды WAJC404,
10 нг/мл холерного токсина, 1 мкМ дексаметазо-
на, 10 нг/мл EGF, 1 мкг/мл пролактина, 5 мкг/мл
инсулина, 10 нг/мл простатропина, 100 U/мл
пенициллина и 100 мкг/мл стрептомицина.
Частично очищенный источник простатропина
может быть заменен как описано McKeehan et al.
[1984] и Chaproniere и McKeehan [1986].
13. Инкубируйте культуру во влажной атмосфере 95%
атмосферного воздуха и 5% С02при 37 °С. Заме¬
ните среду на 3-й и 5-й дни. Клетки должны быть
близки к конфлюэнтному монослою на 7-й день.
Вариации. Представленная процедура может быть
применима к культуре эпителиальных клеток проста¬
ты человека в модификации, описанной Chaproniere и
McKeehan [1986]. Очищенный фактор роста простатро¬
пин идентичен связывающему гепарин фактору роста
фибробластов первого типа, который ранее назывался
кислый FGF или HBGF-1 [Burgess & Maciag, 1989].
По новой номенклатуре, выработанной в результате
соглашения, этот фактор роста называется FGF-1. При
молекулярной характеристике новых членов семейства
лигандов FGF, так же как и семейства рецепторов FGF
в клетках простаты, было обнаружено, что эпителиаль¬
ные клетки простаты экспрессируют специфические
сплайс-варианты (FGF-R2IIIb) по одному из четырех
FGF-рецепторных генов. Специфический рецептор
распознает FGF-1, а стромальная клетка продуцирует
FGF-7 (также называемый фактором роста кератино¬
цитов), но не продуцирует FGF-2 (ранее называемый
bFGF или HBGF-2), описанный Yan et al. [1992].
Стромальные клетки простаты экспрессируют только
ген FGF-R2, который распознает FGF-1 и FGF-2, но
не FGF-7. Поскольку стромальные клетки отвечают
как на FGF-1, так и на FGF-2, простатропин/FGF-1,
используемый для обеспечения дополнительной се¬
лекции эпителиальных клеток, может быть заменен в
вышеописанной процедуре на FGF-7.
FGF-2 не заменяет FGF-1 или FGF-7. Если в
протоколе используется очищенный FGF-1, то его
активность может быть повышена путем добавления
гепарина в концентрации 10-50 мкг/мл.
444
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
23.3. МЕЗЕНХИМНЫЕ КЛЕТКИ
Данный раздел включает в себя те клетки, которые
происходят из эмбриональной мезодермы и не вклю¬
чает гематопоэтическую систему (см. разд. 23.5).
Мезенхимные клетки включают в себя структурные
(соединительнотканные, мышечные и костные) и
сосудистые (эндотелий и гладкие мышцы кровенос¬
ных сосудов) клетки. В то время как эпителиальные
клетки рассматриваются как паренхима или главные
функциональные клетки органа, соединительноткан¬
ные клетки классифицируются как стромальные или
поддерживающие ткани.
23.3.1. Соединительная ткань
Клетки соединительной ткани обычно считаются «сор¬
няками в саду, взращиваемом культуралыциком». Они
выживают в большинстве случаев при механической
и ферментативной эксплантации и могут быть куль¬
тивированы на многих простых средах, в частности
в присутствии сыворотки. Хотя эти клетки, которые
обычно широко называют фибробластами, могут
быть выделены из многих разнообразных тканей, при
этом подразумевается, что они являются элементами
соединительной ткани, точное распознавание клеток
этого класса не всегда однозначно. Линии фиброб¬
ластов (например, ЗТЗ из мышей) продуцируют и
выделяют в среду коллаген I и III типа [Goldberg,
1977]. Хотя продукция коллагена не ограничива¬
ется фибробластами, синтез типа I в относительно
большом количестве характерен для соединительной
ткани. Тем не менее клетки ЗТЗ могут дифференци¬
роваться до адипоцитов при определенной индукции
[Kuriharcuch & Green, 1978; Smyth et al., 1993], клетки
ЮТ 1/2 могут дифференцироваться до хондроцитов
и кости [Shea et al., 2003] в ответ на морфогенетичес¬
кие белки (BMPs). Показано, что клеточные линии
фибробластов мышиных эмбрионов, такие как группа
клеточных линий ЗТЗ и ЮТ 1/2, представляют собой
примитивные мезодермальные клетки, дифференци¬
ровка которых может быть индуцированна более чем
в одном направлении.
Фибробласты человека, хомяка и цыпленка мор¬
фологически отличаются от мышиных фибробластов,
для них характерна веретенообразная морфологии при
конфлюэнтности, что приводит к разработке парал¬
лельных исследований клеток, отличных от культуры
типа «булыжной мостовой», которую образуют мы¬
шиные фибробласты. Веретенообразные клетки могут
представлять собой более высококоммитированные
предшественники и могут быть более точно определены
как фибробласты. Клетки NIH3T3 могут приобрести
веретенообразную форму, если культивировать при
высокой клеточной плотности. Можно также пред¬
положить, что клеточные линии фибробластов могут
быть получены из васкулярных перицитов, — клеток
кровеносных сосудов, подобных клеткам соединитель¬
ной ткани, но в отсутствие специфических маркеров их
трудно дифференцировать.
Таким образом, ясно, что клеточные линии, обычно
называемые фибробластными, могут быть получены
из эмбриональных и взрослых тканей, но эти линии не
следует рассматривать как идентичные или классифи¬
цировать как фибробласты без достаточного подтверж¬
дения путем экспрессии соответствующих маркеров их
способности дифференцировки в зрелые фиброциты.
Коллаген I типа является одним из таких маркеров.
Антиген Thy I также раньше использовался в ка¬
честве маркера [Raff et al., 1979; Saalbach et al., 1997],
хотя он может также появляться на некоторых гемато¬
поэтических клетках.
Культуры эмбриональных или взрослых фибро¬
бластов могут быть приготовлены путем общеприня¬
тых процедур, таких как первичная эксплантация (см.
протокол 12.4), теплая (см. протокол 12.5) и холодная
(см. протокол 12.6) трипсинизация и коллагеназное
расщепление (см. протокол 12.8).
23.3.2. Жировая ткань
Хотя может быть затруднительно получить культуры
из зрелых жировых клеток, может быть индуцирован¬
на дифференцировка в культурах мезенхимальных
клеток (например, мышиных клеток ЗТЗ-Ll) путем
ведения клеток при высокой плотности в течение не¬
скольких дней [Kuriharcuch & Green, 1978; Vierick et
al., 1996]. Показано, что за индукцию дифференциров¬
ки до жировых клеток отвечает адипогенный фактор
сыворотки. Первичные культуры жировых клеток
могут также быть получены из эпидидимиса крысы;
протокол 23.12 приводится с некоторыми сокращени¬
ями по Quon [1998].
Жировые клетки (адипоциты) — это терминально
дифференцированные, специализированные клетки,
основная физиологическая роль которых классически
характеризуется как энергетический резервуар для
организма. Адипоциты являются хранилищем триг¬
лицеридов при избыточном поступлении энергии и
источником энергии в форме свободных жирных кис¬
лот, высвобождающихся путем липолиза тогда, когда
эта энергия понадобится. Жировые клетки играют
важную роль как активные регуляторы углеводного
и жирового метаболизма [Flier, 1995; Reaven, 1995].
Специфические нарушения жировой ткани могут
вносить непосредственный вклад в патогенез таких
распространенных заболеваний, как диабет, гиперто¬
ния и ожирение [Flier, 1995; Hotamisligil et al., 1995;
Reaven, 1995; Walston et al., 1995; Zhang et al., 1994]. В
протоколе 23.12 дана модификация метода выделения
клеток, описанная Rodbell [1964], оптимизированная
для получения примерно 4 мл плотно расположенных
адипоцитов из скоплений жировой ткани эпидидими-
сов четырех крыс путем флотации дизаггрегированных
клеток после центрифугирования. Данный протокол
может быть изменен для увеличения или уменыие-
23.3. Мезенхимные клетки
445
ния желаемого выхода клеток и может быть также
использован для получения инсулин-реагирующих
клеток, используемых для перемещения ДНК при
электропорации.
ПРОТОКОЛ 23.12. Первичная культура
адипоцитов
Материалы
Стерильные:
• DMEM-A: DMEM, pH 7,4, содержащий 25 мМ
глюкозы, 2 мМ глютамина, 200 нМ (R)-N6-
(1-метил-2-фенилэтил) аденозин (PIA, Sigma),
100 мкг/мл гентамицина и 25 мМ HEPES. Приго¬
товьте аликвоты по 100 мл и подогрейте до 37 °С
перед использованием.
• DMEM-B: DMEM-А с добавлением 7% BSA
• Буфер KRBH: среда Кребса-Рингера, pH 7,4,
содержащий 10 мМ NaHC03,30 мМ HEPES (Sig¬
ma), 200 нМ аденозина (Roche) и 1% (w/v) BSA
(Intergen). Приготовить 1 л буфера KRBH: доба¬
вить 7 г NaCl, 0,55 г КН2Р04,0,25 г MgS04*7H20,
0,84 г NaHC03, 0,11 г СаС12, 7,15 г HEPES, 10 г
BSA, 1 мл 200 мкМ аденозина. Добавить UPW до
1 л. Разделить на аликвоты по 100 мл. Подогреть
до 37 °С, довести pH до 7,4.
• Буфер KRBH-A: KRBH с 5% BSA
• Коллагеназа, 20 мг/мл в 2 мл буфера KRBH,
стерилизованного фильтрацией через фильтр с
порами 0,22-мкм.
• Чашки Петри для культуры тканей, диаметр 6-см
(B-D Biosciences)
• Конические центрифужные пробирки, 50 мл
(Coming)
• Полипропиленовые пробирки, 17 ммхЮО мм
• Флакон из полипропилена с низкой плотностью,
30 мл
• Нейлоновой сито 250 мкм в держателе или филь¬
трующей воронке
• Наконечники для пипеток широкого калибра
200 мкл
• Ножницы: две пары или острые ножницы для
иссечения 10 см; пинцет Перри, 12,5 см.
Нестерильные:
• Крысы самцы Спрэйг-Доули весом 145-170 г,
CD линия (Charles River Breeding Laboratories)
• Пластиковые коробки, наполненные газовой
смесью 70% С02,30% 02
• Гильотина
• Автоматическая пипетка и наконечники на
200 мкл
Протокол
Иссечение жирового скопления эпидидимуса
1. Анестезируйте крыс в пластиковой коробке с
газовой смесью 70% С02,30% 02.
2. Декапитируйте крыс на гильотине и обескровьте.
3. Быстро оботрите тело 70%-м этанолом.
4. Извлеките жировое скопление эпидидимуса,
поддерживая максимально возможный уровень
стерильности.
а) Прорежьте кожу нижней части брюшка при
помощи одной пары ножниц, обнажите брю¬
шину.
б) Используя вторую пару ножниц, откройте
брюшину и пинцетом вытяните яичко.
в) Срежьте жировой скопление эпидидимуса,
стараясь сохранить кровеносные сосуды на
задней стороне.
5. Отправьте ткань в культуральную лабораторию.
Коллагеназное расщепление
и отмывание адипоцитов:
6. Добавьте 4 г жировой ткани (примерно равно
8 жировым скоплениям) в 30-мл флакон из поли¬
пропилена низкой плотности, содержащий 4 мл
буфера KRBH при 37 °С.
7. Измельчите жировые скопления при помощи
ножниц на кусочки примерно по 2 мм в диа¬
метре.
8. Добавьте 1 мл раствора коллагеназы во флакон,
содержащий измельченную жировую ткань,
инкубируйте кусочки в водяной бане-шейкере
при 37 °С примерно 1 ч, пока клеточная смесь не
приобретет консистенцию сливок.
9. После коллагеназного расщепления добавьте во
флакон 10 мл буфера KRBH при 37 °С.
10. Перемешайте клетки во флаконе вращательным
движением, осторожно перелейте клетки через
нейлоновое сито 250-мкм в 50-мл коническую
пробирку.
11. Промойте клетки добавлением 30 мл буфера
KRBH при 37 °С в пробирку и быстро центри¬
фугируйте при 200 g в настольной центрифуге.
Удалите осадок при помощи пипетки. Обратите
внимание, что адипоциты будут плавать на по¬
верхности водного буфера.
12. Повторите отмывание клеток еще два раза до¬
бавлением 40 мл буфера KRBH, центрифугиро¬
ванием и удалением осадка.
13. Дважды отмойте клетки в 40мл DMEM-А при
37 °С.
14. После заключительного отмывания ресуспенди¬
руйте клетки, находящиеся на поверхности среды
в DMEM-А при цитокрите примерно 40%.
Первичная культура адипоцитов:
15. Перенесите по 2 мл суспензии в DMEM-А с
40%-м цитокритом, используя 200-мкл пипетку с
широким диаметром носика в каждую чашку для
культуры тканей (6-см).
16. Поместите клетки во влажный инкубатор при
37 °С с содержанием С025% на 1,5 ч.
17. Добавьте 5 мл DMEM-В в каждую чашку.
446
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
На этой стадии обычно проводится исследование
захвата глюкозы [Quon, 1998] в аликвоте клеток для
проверки их жизнеспособности.
23.3.3. Мышцы
Гладкие мышцы Гладкомышечные клетки получены
культивированием сосудистой ткани [Subramanian et
al., 1991] и затем сокультивированием с сосудистым
эндотелием [Jinard et al., 1997; Klinger & Niklason,
2005]. Протокол 23.13 приводится no Klinger and
Niklason [2005].
ПРОТОКОЛ 23.13. Выделение
и культивирование гладкомышечных клеток
Материалы
Стерильные:
• Среда для гладкомышечных клеток (SMCM):
Модифицированная по Дюльбекко среда Игда
(DMEM) с добавлением 20% фетальной сыво¬
ротки теленка, пенициллин 100 U/мл, стрепто¬
мицин 100 мкг/мл, 0,25% трипсин/EDTA (см.
протокол 12.1 и Приложение I).
• Чашки Петри: 15x2.5 см, 6x1,5 см
• Скальпель с хирургическим лезвием #10
• Ножницы для иссечения
• Пинцет для тканей
Протокол
1. Получите грудную аорту от молодою теленка.
2. Погрузите аорту в раствор Хэнкса.
3. Поставьте на лед до начала выделения клеток.
4. Поместите аорту в чашку Петри 15x2,5-см для
иссечения.
5. Разрежьте аорту продольно при помощи ножниц.
6. Осторожно скребите просветную поверхность
аорты лезвием скальпеля для удаления эндоте¬
лиальных клеток.
7. Рассеките медиальный слой аорть^свободоый от
интимы и адвентициального слоя.
8. Порежьте медиальный слой аорты на сегменты
площадью примерно 1 см2.
9. Поместите медиальный сегмент интимальной
стороной вниз в чашку Петри 6x1,5см.
10. Оставьте сегменты примерно на 10 мин для при¬
крепления клеток к чашке.
11. Добавьте 1 мл SMCM непосредственно на меди¬
альный сегмент в каждую чашку.
12. Поместите мед иальные сегменты в увлажненный
инкубатор при 37 °С и 10% С02на ночь.
13. На следующий день добавьте 5 мл свежего
SMCM в каждую чашку, стараясь не нарушить
прилегание еегмента к чашке.
14. Инкубируйте чашки еще 10 дней, после чего
гладкомышечные клетки должны мигрировать
с сегментов и начать формировать двумерную
культуру.
15. Пересевайте клетки, когда они достигнут стадии
субконфлюэнтного монослоя, используя смесь
0,25% трипсин/EDTA.
Сердечная мышца и трансплантация скелетных
миобластов. Сердечная мышца может быть выделена
методом холодной трипсинизации (протокол 11.6),
при этом кардиомиоциты сохраняют сократительную
способность в течение 3 дней в культуре [Deshpande et
al., 1993]. Хотя кардиомиоциты человека также могут
быть выращены в культуре [Goldman & Wurzel, 1992],
трансплантация сократимых и несократимых клеток
из скелетных мышц представляет собой новую стра¬
тегию для лечения острого инфаркта миокарда; она
была осуществлена на кролике, крысе, свинье, собаке
и овце с доказанным улучшением функции миокарда in
vivo [Leobon et al., 2003; Menasche, 2004; Taylor, 2001].
В свете этих обнадеживающих результатов клеточная
терапия, в особенности клеточная трансплантация,
может быть применима в случае многих патологий
[Bhagavati & Xu, 2004]. Трансплантация аутологичных
скелетных миобластов в постинфарктный рубец недав¬
но была применена к людям [Menasche et al., 2001]. Эти
недавние результаты вызвали новый интерес к фунда¬
ментальным исследованиям молекулярных условий
миогенной пролиферации и дифференцировки in vitro
[Neumann et al., 2003; Mai & Harter, 2003; Anderson &
Wozniak, 2004; Li et al., 2004; Carvalho et al., 2004], a
также быстро развивающийся интерес к культуре ство¬
ловых клеток, дающих миогенные клетки [Bhagavati &
Xu, 2004; Seruya et al., 2004; Bossolasco et al., 2004].
Миогенные клетки из скелетных мышц. В опре¬
деленных условиях, при которых клетки продолжают
проявлять по крайней мере некоторые из своих ха¬
рактерных особенностей, возможно культивирование
миогенных клеток из взрослых скелетных мышц
нескольких видов. Так называемые сателлитные клет¬
ки — это миогенные клетки, частично воспроизводящие
первую ступень дифференцировки скелетных мышц.
Они пролиферируют и беспорядочно мигрируют по
субстрату и затем выстраиваются в линию и, наконец,
подвергаются процессу слияния с формированием
многоядерных миотрубочек [Richler & Yaffe, 1970;
Campion, 1984; Hartley & Yablonka-Reuveni, 1990]. Хотя
с применением трипсинизации может быть проведено
три-четыре пассажа, после дифференцировки (т. е. сли¬
яния) проведение субкультивирования невозможно.
Ниже приведено два различных протокола. Прото¬
кол 23.13 является традиционным методом (расщеп¬
ление и механическая гомогенизация, за которыми
следует посев изолированных клеток). Протокол 23.14
основывается на использовании единичных мышечных
волокон. Для культуры животных тканей могут быть
использованы оба протокола, однако первый может
23.3. Мезенхимные клетки
447
быть также использован для культуры человеческих
клеток. Тем не менее второй протокол дает более
близкое воспроизведение миогенного процесса in
vivo. Первый метод легкоосуществим и дает хорошие
результаты по клеточному выходу и возможностям
дифференцировки, особенно если вы используете
достаточно исходного материала. Однако процент
миогенных клеток, полученных на единицу мышеч¬
ной ткани, очень низок, <1% от всех представленных
клеток [Rosenblatt et al., 1995], что вызывает вопрос о
том, является культура, полученная в результате, дей¬
ствительно репрезентативной для ткани-источника.
Этот метод описан для первичных культур из биопсии
здорового человеческого сердца, которая имеет высокое
содержание миогенных клеток (по крайней мере, 85%
положительны по иммуно-окрашиванию на десмин на
10 день после посева). Он также может быть исполь¬
зован для мышц животных (например, крысы, мыши,
кролика, свиньи или собаки).
Для получения культуры единичных мышечных
волокон, мышцы выделяются индивидуально и ин¬
кубируются в колагеназе для расщепления соедини¬
тельной ткани. Мягкая механическая гомогенизация
затем высвобождает отдельные интактные мышечные
волокна. Мышечные волокна, вместе с ассоциирован¬
ными сателлитными клетками, могут быть получены
из индивидуальных мышц. Это позволяет проводить
сравнение между ответом различных групп мышц на
экспериментальные процедуры или между мышцами
разного возраста или генетического происхождения.
Более того, тяжелые цепи миозина из индивидуальных
мышечных волокон могут быть определены методом
SDS — гель-электрофореза, поэтому можно сравнивать
мышечные волокна с различными типами тяжелых
цепей миозина [Rosenblatt et al., 1996]. Миогенные
культуры могут быть успешно получены из единичного
мышечного волокна, выделенного из мышцы животно¬
го любого возраста, хотя увеличение возраста приводит
к некоторому снижению эффективности культивиро¬
вания. У животных, страдающих хроническими миопа-
тиями, такими как мышечная дистрофия, отмечается
некоторая контаминация фибробластами, возможно
в результате возрастания доли соединительной ткани
[Bockhold et al., 1998].
Протокол 23.14 и часть предшествовавшего вве¬
дения были предоставлены М. Malo, S. Bonavaud, Ph.
Thibert, and G. Barlovatz-Meimon, Facultre des Sciences,
Universite Paris XII, Val deMarne, Avenue General de
Gaulle, F-94010 Creteil, France.
Индукция пролиферации и дифференцировки.
Кривая роста культуры миогенных клеток человека,
полученной в среде Хэма F12 с добавлением 20%
FBS, показывает наличие трех традиционных фаз (см.
разд. 21.9.2): lag- фазу, фазу экспоненциального роста и
плато, которое совпадает с началом слияния. Слияние,
оцениваемое по таким параметрам, как количество ядер,
включенных в мышечные трубочки, или индекса, сли¬
яния (процент ядер, инкорпорированных в мышечные
ПРОТОКОЛ 23.14. Культура миобластов
взрослой мышцы взрослого человека
Схема
Первичная культура может быть легко выращена
в среде Хэма F12 с добавлением 20% фетальной
сыворотки теленка (FBS). Без изменений условий
культивирования эти клетки делятся и дифференци¬
руются путем слияния с образованием многоядерных
мышечных трубочек, что подтверждает принадлеж¬
ность культивируемых клеток к миоцитам.
Материалы
Стерильные:
• Транспортная среда Хэма F12 (Seromed 08101)
без NaHC03 и с 20 мМ HEPES (Serva 25245),
75 мл
• Ростовая среда Хэма с 20% FBS (GIBCO 011-
06290), 200 мл
• Раствор проназы: 0,15% проназы (Sigma Р-6911)
и 0,03% EDTA (Merck 8418) с 20 IU/мл пеницил¬
лина и 20 мкг/мл стрептомицина (0,4% Eurobio
PES 3000), в F12 или DPBS, 100 мл
• D-PBSA, 100 мл
• Нейлоновое сито, 100 мкм
• Скальпели
• Длинные острые ножницы
• Чашки Петри для иссечения, 9 см
• Флаконы, 25 см2,4 шт.
• Центрифужные пробирки, 20 мл, или универ¬
сальные контейнеры, 5 шт.
Нестерильные:
• Гемоцитометр
Протокол
Диссоциация:
1. Обрежьте немышечные ткани с образца, получен¬
ного при биопсии, и промойте в D-PBSA.
2. Порежьте ткань на фрагменты параллельно во¬
локнам, промойте в D-PBSA, взвесьте.
3. Поместите фрагменты параллельно друг другу
в крышку чашки Петри, порежьте на тонкие
цилиндры и затем, наконец, на кусочки объемом
1 мм3, стараясь не разрушить ткань. Последние
разрезы должны быть сделаны в пробирке при
помощи длинных ножниц, старайтесь избегать
разрушения ткани.
4. Промойте в D-PBSA и дайте осесть, удалите су¬
пернатант.
5. Ферментативно разрушьте фрагменты в течение
1 ч при 37 °С в растворе проназы (используйте
15 мл для биоптата весом 1-3 мг), осторожно
встряхивая пробирку через 10-15-мин.
6. После инкубирования гомогенизируйте культу¬
ру при помощи пипетки. Среда должна стано¬
виться все более мутной по мере высвобождения
клеток.
448
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
7. Позвольте фрагментам осесть на дно под дей¬
ствием силы тяжести с образованием осадка Р1
и супернатанта S1.
8. Профильтруйте S1 через нейлоновое сито 100-мкм
в 20-мл центрифужную пробирку. Осторожно
встряхивайте или пипетируйте супернатант для
ресуспендирования клеток.
9. Центрифугируйте пробирку 8-10 мин при 350 g.
Удалите супернатант аспирацией.
10. Очень аккуратно ресуспендируйте осадок, ис¬
пользуя пипетку с резиновой грушей, отмерив
ровно 10 мл ростовой среды и подсчитайте коли¬
чество клеток в гемоцитометре.
И. Разведите суспензию в ростовой среде для
посадки в культуральные сосуды с концентра¬
цией примерно 1,5x104 кл./мл. Из биопсии от
здорового донора получают обычно примерно
1-2х105кл./г.
12. Добавьте 15 мл расщепляющей среды к Р1,
инкубируйте фрагменты в течение 30 мин в
водяной бане при 37 °С, с периодическим встря¬
хиванием.
13. Пипетируйте суспензию, чтобы дезагрегировать
клетки и затем профильтруйте их через нейло¬
новое сито. Промойте фильтр 20 мл ростовой
среды.
14. Центрифугируйте суспензию в течение 8-10 мин
при 350 g, подсчитайте клетки, и посейте, как
описано выше.
15. Перенесите флаконы в инкубатор с влажной
атмосферой с 5% С02на 37 °С.
• Требование безопасности. Остаток био-
псийного материала, все пробирки, планшеты, чашки
и прочие использованные материалы должны быть
обработаны гипохлоритом до уничтожения или
мытья (см. разд. 7.8.5).
Под держание культуры:
16. Аккуратно заменяйте среду через 24 ч после
посева и затем каждые 3-4 дня. Развитие этих
культур происходит главным образом в сторону
дифференцировки. Хронометрирование длитель¬
ности трех фаз человеческих мышечных клеток
показывает продолжительность около 4-6 дней
до достижения пика пролиферации, затем клетки
выстраиваются по одной линии примерно на 8-
й день и около 10-12-го дня отмечается активи¬
зация процесса клеточного слияния. Тем не менее
каждый исследователь должен иметь в виду, что
некоторые клетки могут дифференцироваться
раньше, а другие еще будут пролиферировать,
когда большая часть клеточной культуры уже
вступит в дифференцировку.
Субкультивирование:
1. Осторожно добавьте небольшой объем EDTA к
клеткам и немедленно удалите.
2. Добавьте достаточное количество раствора трип¬
сина (0,25%), так чтобы он покрыл слой кдеток.
3. Когда клетки отделятся от чашки, добавьте
5-10 мл полной ростовой среды, очень осто¬
рожно однократно соберите и слейте культуру
пипеткой и центрифугируйте клетки в течение
10 мин при 350 g.
4. Подсчитайте количество клеток в аликвоте
суспензии и посейте клетки в выбранной кон¬
центрации.
трубочки в отношении к общему числу ядер), начина¬
ется обычно примерно на 8-й день после посева и резко
возрастает примерно на 10-й день. В зависимости от
образца эта хронология может быть сдвинут на один
день раньше или позже.
Таким образом, дифференцировка, выраженная
количеством ядер на миотрубочку/см2, может наблю¬
даться морфологически. Однако процесс дифферен¬
цировки может также быть регистрируем с использова¬
нием биохимических маркеров (таких как саркомерные
белки), ферменты, вовлеченные в дифференцировку
(например, креатинфосфокиназа и ее время-зависи-
мые мышце-специфичные изоформы, либо появление
а-актина [Buckingham, 1992].
Было показано, что семейство генов, наиболее
изученным из которых является MyoD, активирует в
миогенных предшественниках экспрессию гена, спе¬
цифического для мышц [см. обзор Buckingham, 1992].
Дифференцировка миогенных клеток предполагает
как активацию спектра различных генов с соответству¬
ющими изменениями адгезивных свойств клеточной
поверхности [Dodson et al., 1990], так и активацию
урокиназы, активатора плазминогена на клеточной
поверхности и ее рецептора [Quax et al., 1992].
Вариации и применение. Метод эксплантации-ре-
эксплантации был разработан группой исследователей
[Askanas et al., 1990; Pegolo et al., 1990]; надежность
и достоверность его были подтверждены в течение
длительного времени, особенно для исследований
мышечных дефектов.
Когда целью работы является изучение механиз¬
мов дифференцировки, следует делать выбор между
эксплантацией и методами ферментативного расщеп¬
ления с последующей клональной или неклональной
культурой или трехмерным культивированием. Если
целью работ является получение большого количества
клеток для биохимических или молекулярных иссле¬
дований, то ферментативный метод представляется
предпочтительным.
Протокол 23.15 для культуры единичного мы¬
шечного волокна и связанное с ним введение, приве¬
денное выше, предоставлены Т. Partidge & L. Heslop,
Muscle Cell Biology Group, MRC Clinical Sciences
Centre, Hammersmith Hospital, Du Cane Road, London
W12 ONN. Данная процедура имеет ценность, когда
23.3. Мезенхимные клетки
449
она необходима для выделения всех сателлитных
клеток, а также важна, если необходимо избежать
контаминации немиогенными клетками. Метод вы¬
деления единичного мышечного волокна может быть
осуществлен для любой мышцы, которая может быть
выделена с содержанием большой доли неповрежден¬
ных волокон, предпочтительно в виде «сухожилие
к сухожилию». В лаборатории этих сотрудников,
обычно используют мышиные мышцы: длинный раз¬
гибатель пальца (extensor digitorum longus, EDL), a
также пяточную и переднюю большеберцовую (tibialis
anterior, ТА) мышцы.
ПРОТОКОЛ 23.15. Культура отдельных
мышечных волокон скелетных мышц
Схема
Отдельные миофибриллы высвобождаются раз¬
мельчением цельной скелетной мышцы, обрабо¬
танной коллагеназой, после культивирования на
Матригеле.
Материалы
Стерильные:
• Matrigel™ (B-D Biosciences) 1 мг/мл в DMEM
• Среда Игла модифицированная по Дюльбекко
(DMEM, GIBCO, Invitrogen)
• L-глютамин, 200 мМ (Sigma)
• Раствор пенициллина, 104 U/мл, и стрептомици¬
на 10 мг/мл в 0,9% NaCl (Sigma)
• Экстракт куриного эмбриона (MP Biomedicals)
• Лошадиная сыворотка (Invitrogen)
• Фетальная сыворотка теленка (FBS; PAA Labo¬
ratories)
• Посевная среда: DMEM плюс лошадиная сыво¬
ротка 10%, экстракт куриного эмбриона 0,5%,
L-глютамин 4 мМ, пенициллин, 100 U/мл, стреп¬
томицин 100 мкг/мл
• Пролиферационная среда: DMEM плюс 20%
FBS, 10% лошадиная сыворотка, экстракт кури¬
ного эмбриона 1%, L-глютамин 4 мМ, пеницил¬
лин, 100 U/мл, стрептомицин 100 мкг/мл
• Коллагеназа тип I (Sigma)
• Чашки Петри, 9 см
• Модифицированные пастеровские пипетки: пас¬
теровские пипетки отрезаются алмазным стекло¬
резом так, чтобы получились срезы различных
размеров, затем следует оплавить на огне, чтобы
неровные края не повредили миофибриллы
• Многолуночные планшеты, 24 лунки, обра¬
ботанные, 1 мг/мл Matrigel™ (Primaria™ B-D
Biosciences) в DMEM и инкубированные при
37 °С в течение 30-60 мин до формирования геля
Matrigel™ (B-D Biosciences).
Нестерильные:
• Алмазный стеклорез
• Водяная баня-шейкер при 35 °С
• Трансиллюминационный препаровальный сте¬
рео микроскоп FBS (PAA Laboratories)
Протокол
1. Иссеките мышцу немедленно после забоя живот¬
ного, стараясь манипулировать с мышцей, исполь¬
зуя сухожилия и не повреждая миоволокна.
2. Поместите мышцы в 0,2% (w/v) коллагеназу
тип I в DMEM инкубируйте в водяной бане-
шейкере при 35 °С в течение 60-90 мин. Для
мышц большого размера требуется, как правило,
более длительное время обработки, длинная
мышца-разгибатель пальца (extensor digitorum
longus, EDL), выделенная из мыши 6-8-недель
требуется 60 мин.
3. Приготовьте чашки Петри для выделения мио-
фибрилл: промойте чашки сывороткой лошади
для предупреждения прилипания миофибрилл
к поверхности чашки и добавьте 20 мл DMEM.
4. После расщепления коллагеназой перенесите
мышцу в чашку Петри, содержащую DMEM.
5. С использованием трансиллюминационного мик¬
роскопа выделите отдельное мышечное волорю
при помощи модифицированной пастеровской
пипетки. Пипетки также следует смочить 10%-й
лошадиной сывороткой в DMEM для предупреж¬
дения прилипания миофибрилл к стеклу.
6. Как только мышечные волокна выделены, диа¬
метр мышцы уменьшается и следует использо¬
вать пастеровскую пипетку с постепенно умень¬
шающимися размерами отверстий.
7. Как только будут выделены 20-30 миофибрилл,
мышечную массу нужно перенести в свободную
чашку Петри и далее выделять волокна, пока
не будет диссоциировано достаточное их коли¬
чество.
8. Для отщепления волокон, находящихся в серд¬
цевине мышцы большего размера, может потре¬
боваться повторный этап обработки; после того,
как диссоциировали поверхностные волокна,
верните оставшуюся мышечную массу в коллла-
геназу для дальнейшейобработки.
9. Поместите миофибриллы в планшеты для куль¬
туры тканей, предварительно обработанные
Matrigel (Primaria™).
а) Поместите отдельные мышечные волокна в
центре лунки и позвольте им прилипнуть к
Матригелю. Важно убедиться в чистоте под¬
готовительного этапа, важно посеять только
неповрежденные волокна, без участков ги¬
перконтрактур, которые наблюдаются при
загрязнениях адгеренными клетками, часто
ассоциированными с фрагментами микро¬
сосудов.
б) Медленно добавьте в ячейку посевную среду,
стараясь не потревожить волокна.
10. Поместите планшеты в инкубатор при 37 °С и 5%
со2.
450
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
Пролиферация:
Сателлитные клетки начинают мигрировать с ро¬
дительской миофибриллы через 12-24 ч культиви¬
рования. Через 3-4 дня каждое мышечное волокно
будет окружено примерно 50-300 клетками, точное
число клеток зависит от разных факторов, включая
вид мышцы, из которых получают миофибриллы
[Rosenblatt et al., 1996] и возраст мыши-донора
[Bockhold et al., 1998].
11. Когда из миофибриллы мигрирует достаточное
количество клеток, удалите ее из культуры при
помощи пастеровской пипетки, вытянутой в
пламени до очень тонкого конца.
12. Продолжайте культивировать оставшиеся клетки
в той же лунке.
13. Если требуется, проведите субкультивирование
(см. протокол 13.2) и посейте в пролиферацион-
ную среду.
Дифференцировка. Пролиферирующие миогенные
клетки могут быть индуцированы к дифференцировке
до миотрубочек путем снижения содержания лошади¬
ной сыворотки в среде DMEM до 5%.
Миогенность клеток, мигрирующих из мышечных
волокон, может быть исследована методом иммунного
окрашивания на десмин — мышцеспецифичный белок
промежуточных филаментов. По нашим данным, все
клетки, мигрирующие из 98-99% единичных мышеч¬
ных волокон, экспрессируют десмин.
Единичные мышечные волокна могут быть при¬
готовлены из мышц трансгенных мышей Н-2КЬ-
tsA58 для получения миогенных клеток, способных
к многократным циклам деления in vitro [Morgan et
al., 1994]. Трансгенные мыши H-2Kb-tsA58 содержат
температурно-чувствительный мутант tsА58 большого
Т-антигена обезьяньего вируса 40 (SV40) под контро¬
лем индуцируемого промотора (H-2Kb) [Jat et al., 1991].
Белок tsA58 является активным при 33 °С, транскрип¬
ция может быть стимулирована добавлением мышиного
у-интерферона. Миогенные клетки, полученные из этих
мышей, пролиферируют в присутствии у-интерферона
(20 Ед/мл) при 33 °С. При включении по достижении
температуры 37-39 °С в отсутствии у-интерферона как
в культуре, так и при трансплантации in vivo, они диф¬
ференцируются с образованием мышечных волокон, в
быстром и синхронном порядке.
23.3.4. Хрящ
Протокол 23.16 для культивирования клеток из сустав¬
ного хряща был предоставлен Francois Lemare, Sophie
Thenet, and Sylvie Demignot of Laboratoire de Pharma-
cologie Cellulaire de L’Ecole Pratique des Hautes Etudes,
Centre de Recherches Biomredicales des Cordeliers, 15 rue
de L’Ecole deMedecine, 75006 Paris, France.
Хондроциты являются высокоспециализирован¬
ными клетками мезенхимального происхождения,
которые ответственны за синтез, поддержание и де¬
градацию хрящевого матрикса. Большое количество
исследований в области ревматологии сосредоточены
на понимании механизмов, которые индуцируют ме¬
таболические изменения в суставных хондроцитах
при остеоартрозе и ревматоидном артрите. Суставные
хондроциты в культуре являются очень полезным
инструментом, но при культивировании в монослое
они медленно делятся, становятся фибробласто-по-
добными и утрачивают свои биохимические характе¬
ристики [Benya et al, 1977; von der Mark, 1986]. Уже
при первом пассаже отмечается частичный сдвиг от
синтеза коллагена II типа к коллагену I и III типов и
от агрекана к низкомолекулярным протеогликанам.
Одновременно с фенотипическими изменениями
меняется метаболический ответ на интерлейкин-1|3
в процессе субкультивирования [Blanco et al., 1995;
Lemare et al., 1998].
Среди разнообразных методов, используемых
для поддержания фенотипа хондроцитов [см. обзор
Adolphe & Benya, 1992], наиболее многообещающим
представляется метод культивирования на альгинат-
ных бусинах, поскольку показано, что эта система
культивирования приводит к образованию матрикса,
подобного тому, который формируется в естественном
суставном хряще [Нди8е1тапп et al., 1996; Petit et al.,
1996]. Данная система поддерживает экспрессию диф¬
ференцированного фенотипа и также способна восста¬
навливать его в дедифференцированных хондроцитах
[Bonaventure et al., 1994; Lemare et al., 1998]. Еще одним
преимуществом над другими трехмерными методами
является легкость извлечения клеток по завершении
культивирования, что позволяет проводить изучение
экспрессии белков и генов [Lemare et al., 1998].
ПРОТОКОЛ 23.16. Хондроциты
на альгинатных бусинах
Схема
Культивирование на альгинатных бусинах основано
на структурировании суспензии хондроцитов в раст¬
воре альгината в хлористом кальции.
Материалы
Стерильные:
• Для анатомирования сустава и извлечения хряща,
а также ферментативного расщепления хряща
используется среда для иссечения хряща (CDM):
среда Хэма F12 (Invitrogen), с добавлением нетал-
мицина (20 мкг/мл); занкомицина (10 мкг/мл), й
цефтазидима (30 мкг/мл) (все Sigma)
• Ростовая среда: DMEM (глюкоза, 1 г/л)/среда
Хэма F12 (1:1, v/v), гентамицин, 4 мкг/мл, с до¬
бавлением 10% FCS.
Примечание. Следует сравнить различные партии
сыворотки для оценки их способности поддерживать
23.3. Мезенхимные клетки
451
рост хондроцитов, так же как и выражение диффе¬
ренцированного фенотипа (оцениваемой, например,
методом иммуномечения коллагена II типа). Когда
отобрана одна партия, следует закупить ее в таком
объеме, чтобы несколько месяцев не изменять пар¬
тию сыворотки.
• Растворы фермента для расщепления хряща:
а) Трипсин, 0,05%, панкреатический свиной
(Sigma) среде Хэма F12
б) Коллагеназа, 0,3%, тип 1 (Worthington, CLS1)
в среде Хэма F12
в) Коллагеназа 0,06% в среде Хэма F12 + 10%
FCS
Все растворы стерилизовать фильтрацией
через фильтры с диаметром пор 0,22 мкм.
Примечание. Новые партии коллагеназы должны
быть проверены на эффективность расщепления
хряща по сравнению с предыдущей партией.
• Раствор трипсин/EDTA (для субкультивирова¬
ния): 0,1% w/v
• Свиной панкреатический трипсин в PBSA с 5 мМ
(0,02% w/v) EDTA
• Раствор альгината натрия: 1,25% w/v альгината
натрия (Fluka) в 20 мМ HEPES; 0,15 М. NaCl
Подготовка:
а) Используя мешалку с подогревом, сначала
растворите HEPES в 0,15 М. NaCl и доведите
температуру до -60 °С.
б) Добавьте альгинат натрия и продолжайте раз¬
мешивать до полного растворения (> 2 ч).
в) Тщательно доведите pH раствора до 7,4 до¬
бавлением 1 М NaOH; не превышайте pH 7,4,
так как добавление кислоты (HCL или H2S04)
кончается необратимой преципитацией.
г) Разлейте раствор на аликвоты, стерилизуйте
автоклавированием при 121° С в течение
10 мин.
Альтернативная подготовка: Быстро растворите
альгинат натрия в HEPES/NaCL (-10 мин) в мик¬
роволновой печи, тщательно следя за тем, чтобы не
произошло карамелизации
• Раствор желатина: СаС12,102 mM, HEPES, 5 мМ,
PH 7,4
• Раствор для солюбилизации: EDTA, 50 мМ,
HEPES, 10 мМ, pH 7,4
• Стерильный вкладыш для магнитной мешалки
Протокол
Анатомирование:
1. Приготовьте культуры из коленного, плечевого
и бедренного суставов. Эмбрионы или молодые
доноры предпочтительнее, чем взрослые, по¬
скольку они обеспечивают получение больших
количеств клеток и дольше не стареют. Идеально
подходят месячные кролики Fauve Bourgogne
или новозеландские. Однако бедренный сустав,
полученный при хирургической резекции взрос¬
лого человека, также подходит как источник
хондроцитов.
2. Тщательно промойте кусочки тканей в CDM
перед анатомированием. Диссоциация тканей
должна быть начата немедленно. Удалите кожу,
мышцы и сухожилия сустава. Аккуратно отбери¬
те фрагменты хряща от частей сустава, которые
свободны от соединительной ткани.
Диссоциация:
Чтобы предотвратить высыхание хряща, выполни¬
те все этапы диссоциации в 10-см чашке Петри, не
обработанной ТС, содержащей CDM. Количества
растворов фермента, указанные в остальной части
протокола, подходят для плечелопаточного и тазо¬
бедренного суставов одного кролика, но они должны
быть адаптированы к тому количеству хряща, кото¬
рое должно быть выделено.
3. Используя крестообразные скальпели (см. про¬
токол 12.4), измельчите пластинки хряща на
кусочки объемом 1-мм3.
4. Перенесите фрагменты хряща в 30-мл плоскодон¬
ный флакон.
5. Добавьте 10 мл 0,05% раствора трипсина, фраг¬
менты с умеренным перемешиванием на магнит¬
ной мешалке в течение 25 мин в запечатанном
флаконе при комнатной температуре.
6. Удалите раствор трипсина после оседания фраг¬
ментов.
7. Добавьте 10 мл 0,3% раствора коллагеназы и инку¬
бируйте культуру с умеренным перемешиванием
на магнитной мешалке в течение 30 мин в запеча¬
танном флаконе при комнатной температуре.
8. Удалите раствор коллагеназы после осадка фраг¬
ментов.
9. Добавьте 10 мл 0,06% раствора коллагеназы.
10. Перенесите суспензию в 100-мм чашку для
культивирования бактерий. Ополосните флакон
дополнительно 10 мл свежего 0,6%-го раствора
коллагеназы, перенесите раствор в чашку Петри
и инкубируйте при 37°С в инкубаторе с 5% С02.
И. Перенесите суспензию клеток в 50-мл центри¬
фужную пробирку и несколько секунд переме¬
шивайте на вортекс-миксере.
12. Удалите остаточный материал, оставленный пос¬
ле ферментативной обработки, процеживанием
через 70-мкм нейлоновое клеточное сито (B-D
Biosciences; см. рис. 12.10).
13. Центрифугируйте при 400 g в течение 10 мин.
14. Ресуспендируйте клеточный осадок в 20 мл
CDM, и подсчитайте клетки при помощи гемо¬
цитометра.
15. Центрифугируйте клетки при 400 g в течение
10 мин.
Захват
16. Ресуспендируйте клеточный осадок в 1 мл рас¬
твора альгината натрия, затем прогрессирующе
452
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
разведите суспензию в большем количестве рас¬
твора альгината натрия, пока не будет достигнута
клеточная плотность 2х106кл./мл. Это прогрес¬
сивное растворение необходимо, чтобы получить
гомогенную суспензию клеток в альгинате.
17. Быстро выпустите суспензию клеток в каплях
через иглу 21G в желатинизирующий раствор
при умеренной скорости перемешивания на маг¬
нитной мешалке» позвольте альгинату полимери-
зоваться в течение 10 мин, чтобы сформировать
бусины.
18.Промойте бусины Зраза в 5 объемах NaCl,
0,15 М.
19. Разлейте бусины по 5мл (IxlO7 клеток)* в 75-
см2 флаконы, содержащие 20 мл ростовой среды.
20. Инкубируйте культуру в 37 °С в увлажненной
атмосфере 5% С02и 95% воздуха.
Восстановление:
21. Удалите среду.
22. Добавьте 2 объема солюбилизирующего раствора.
23. Инкубируйте культуру 15 мин в 37 °С.
24. Центрифугируйте клетки при 400 g в течение
10 мин.
25. Ресуспендируйте клеточный осадок в ростовой
среде» содержащей 0,06% коллагеназы.
26. Инкубируйте клетки в течение 30 мин при 37 °С
в увлажненной атмосфере 5% С02и 95% воз¬
духа.
27. Центрифугируйте клетки при 400 g в теченйе
10 мин.
28. Ресуспендируйте клеточный осадок в бессыво¬
роточной среде Хэма F12/DMEM и подсчитайте
число клеток в гемоцитометре.
29. Центрифугируйте клетки при 400 g в течение
10 мин.
30. Повторите шаги 28 и 29, без подсчета клеток.
Анализ фенотипа хондроцитов
Коллаген II типа представляет собой 85-90% об¬
щего синтеза коллагена хондроцитами в первичной
культуре. Коллаген II типа является высокоспеци¬
фичным для хондроцитов и рассматривается как их
основной маркер дифференцировки. Экспрессия кол¬
лагена II типа может быть проанализирована методом
непрямой иммунофлюоресценции с использованием
специфических антител, которые, однако, должны
быть тщательно проверены, чтобы убедиться, что не
имеется перекрестного реагирования с коллагеном
I типа, который экспрессируется дедифференциро-
ванными клетками. Иммуноцитохимические исследо¬
вания экспрессии коллагена в культуре хондроцитов
на альгинатных бусинах делают необходимым рассе¬
чение бусин, как это описано в Lemare et al. [1998].
Для более точного анализа фенотипа коллагена при¬
меняется SDS-PAGE и двумерное CNBr-пептидное
картирование очищенного коллагена после включения
[3Н]-пролина [Benya, 1981]. Относительно основных
типов коллагена, SDS-PAGE анализ может выявить
присутствие коллагена III типа или наличие а2(1)
цепи коллагена I. Тем не менее этот метод не может
распознать al-цепи коллагена I и II типов (т.е. не
может служить для распознавания и оценки колла¬
гена I и II), поэтому для подтверждения экспрессии
коллагена II типа необходимо проведение двумерного
CNBr-пептидного картирования. Экспрессия этих
генетически различных типов коллагена может также
быть проанализирована на транскрипционном уровне
Нозерн-блоттингом с использованием общеупотреби¬
тельных методов переноса и гибридизации [Lemare et
al., 1998] или при помощи real-time PCR [Murphy &
Polak, 2004].
Протеогликаны Окрашивание опционовым синим
при кислых pH широко используется как индикатор
присутствия сульфатированного агрекана [Lemare et
al., 1998]. Так же как и при иммуноцитохимическом
исследовании, это окрашивание требует рассечения
бусин. Более точный анализ различных типов синте¬
зированных протеогликанов может быть осуществлен
после внедрения 35S04 [Verbruggen et al., 1990]. Как и в
случае генов коллагена, анализ экспрессии генов агре¬
кана и связанных с ним белков может быть осуществлен
Нозерн-блоттингом [Lemare et al., 1998] или real-time
PCR [Murphy & Polak, 2004].
Вариации и применение. Некоторые исследования
показали, что культура хондроцитов на альгинатных
бусинах представляет собой привлекательную альтер¬
нативную модель хондроцитов [Grandolfo et al., 1993;
Hauselmann et al., 1994,1996; Petit etal., 1996]. Главным
преимуществом этого метода перед другими трехмер¬
ными методами является то, что клетки могут легко
быть извлечены после культивирования, что позволяет
производить исследования экспрессии белков и генов
[Lemare et al., 1998].
Количество доступных клеток часто становится
лимитирующим фактором для изучения метаболизма
хондроцитов, суставной хрящ трудно получить в боль¬
ших количествах, и он имеет низкое содержание клеток.
В этом случае количество доступных хондроцитов
может быть увеличено путем первичного культиви¬
рования клеток в монослое (2 пассажа) и последую¬
щего восстановления дифференцированных свойств
хондроцитов путем культивирования на альгинатных
бусинах в течение 2 недель [Bonaventure et al., 1994;
Lemare et al., 1998]. Одновременно с восстановлением
фенотипа метаболический ответ на интерлейкин-1|3 в
этих клетках восстанавливается на количественном
уровне аналогично тому, который имеется в первич¬
ной монослойной культуре [Lemare et al., 1998]. Это
делает культуру на альгинатных бусинах релевантной
моделью для изучения биологии хондроцитов. Нако¬
нец, каркас, заполненный хондроцитами, введенный в
альгинатный гель, может быть предложен для тканевой
инженерии и трансплантации [Cancedda et al., 2003;
Marijnissen et al., 2002; Sah et al., 2005].
23.3. Мезенхимные клетки
453
23.3.5. Кость
Хотя с костью трудно механически манипулировать,
тонкие срезы, обработанные EDTA и расщепленные
коллагеназой [Bard et al.,1972], дают начало культуре
остеобластов, которые обладают некоторыми функцио¬
нальными характеристиками данной ткани. Для того
чтобы предотвратить избыточный рост фибробластов
без ингибирования остеобластов, обычно используется
антисыворотка против коллагена [Duksin et al., 1975].
Из остеосаркомы получены постоянные клеточные
линии [Smith et al., 1976; Weichselbaum et al., 1976], но
из нормальных остеобластов постоянных клеточных
линий получить пока не удалось. Мезенхимальные
стволовые клетки (см. разд. 23.7.2) могут быть куль¬
тивированы и индуцированы с формированием осте-
оцитов, пригодных для тканевой инженерии [Lennon
& Caplan, 2005; Hofmann et al., 2005].
Протокол 23.17 для культуры костных клеток пре¬
доставлен Edith Schwartz, Departments of Orthopedic
Surgery and Physiology, Tufts University School of Medi¬
cine, 136 Harrison Avenue, Boston, MA 02111.
Культура костных клеток страдает от того, что по
присущей этим клеткам природе манипуляции с этими
тканями затруднены. Тем не менее общепринятая тех¬
ника первичного эксплантата или расщепления в колла-
геназе и трипсине приводит к высвобождению клеток,
которые могут быть пересеяны обычным способом.
ПРОТОКОЛ 23.17. Остеобласты
Схема
В качестве эксплантата культуры используйте фраг¬
менты ткани, достаточно маленькие для того, чтобы
они могли прикрепиться к стенке культурального
сосуда в минимальном количестве среды (см. про¬
токол 12.4). Прикрепившиеся эксплантаты затем
заливаются средой, контролируется рост культу¬
ры. Для дизагрегированной клеточной культуры
используются трабекулы кости, рассеченные на
2-5-мм части и расщепленные в растворе ферментов.
Суспендированные клетки переносятся во флаконы
со средой 12.
Материалы
Стерильные:
• Среда ХэмаР12 с 12% FBS, 2,3 мМ Mg2*, 100 U/мл
пенициллина, и 100 мкг/мл стрептомицина S04
• Раствор трипсина, который обычно используют
для пассажа клеток: растворить 125 мг трипсина
(Sigma, тип XI) в 50 мл раствора Тирода без Са2+и
Mg2+. Доведите конечное значение pH раствора
до 7,0 и простерилизуйте фильтрацией. Разлейте
аликвоты по 5 и 10 мл в пробирки Ругех и храните
при -20 °С
• Расщепляющий раствор для выделения остео¬
бластов
• Раствор А: 8,0 г NaCl, 0,2 г KCL, 0,05 г NaH2P04 •
Н20 в 100 мл UPW
• Раствор В (Раствор коллагеназа/трипсин): рас¬
творите 137 мг коллагеназы (тип I, Worthington
Biochemicals) и 50 мг трипсина (Sigma, тип III)
в 10 мл раствора А. Доведите pH объединенного
раствора до 7,2, и затем доведите UPW объем до
100 мл. Стерилизуйте фильтрацией и разлейте по
10-мл аликвотам, храните при -20 °С
• Чашки Петри, 9 см
• Флаконы для культуры 25 или 75 см2
• Скальпели
• Пинцеты
Протокол А: Культивирование эксплантата
1. Получите экземпляры кости из операционной.
2. Ополосните ткань несколько раз при комнатной
температуре стерильным физиологическим рас¬
твором.
3. Если кость не может использоваться немедленно,
залейте образец стерильной средой Хэма F12,
содержащей 12% FBS, пенициллин, и сульфат
стрептомицина. Кость может храниться в течение
ночи при 4 °С.
4. На следующее утро промойте ткань в растворе,
содержащем пенициллин (100 U/мл) и сульфат
стрептомицина. (100 мкг/мл).
5. Поместите кость в чашку Петри и с использова¬
нием скальпеля и принцета извлеките трабекулы
и поместите их в новую чашку Петри.
6. Добавьте 10 мл раствора Тирода, чтобы он покрыл
выделенные трабекулы. Промойте трабекулы не¬
сколько раз раствором Тирода до удаления крови
и жировых клеток.
7. Предварительно приготовьте 25-см2 флакон, пу¬
тем предварительного инкубирования его с 2 мл
полной среды в течение 20 мин для достижения
равновесия среды с газовой фазой. Доведите pH
по мере необходимости при помощи С02, 4,5%
NaHC03 или НС1.
8. Порежьте трабекулы на фрагменты 1-3 мм.
9. Удалите преинкубационную среду из флакона и
добавьте 2,5 мл свежей среды Хэма F12.
10.Перенесите 25-40 фрагментов трабекул во
флакон.
11. Держа флакон в вертикальном положении,
скользящими движениями переместите частички
эксплантата по основанию флакона при помощи
инокуляционной петли и распределите части
эксплантата равномерно.
12. Оставьте флакон в вертикальном положении в
течение 15 мин в 37 °С.
13. Медленно верните флакон в нормальное горизон¬
тальное положение. Части эксплантата останутся
прикрепленными к основанию флакона.
14. Оставьте флакон в горизонтальном положении
при 37 °С на 5-7 дней. После этого периода
проверьте рост культуры и замените среду во
454
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
флаконе на свежую. Для того чтобы предотвра¬
тить открепление частей эксплантата, медленно
поднимите флакон в вертикальное положение
перед переносом его из инкубатора в ламинар или
к микроскопу.
15. Чтобы поддерживать культуры, заменяйте среду
дважды в неделю.
16. Когда достигнут конфлюэнтный рост, удалите
части эксплантата, трипсинизируйте клеточный
слой, выделите клетки центрифугированием и
отберите их во флаконы или лунки.
Протокол В: Монослойные культуры
из дизагтрегированных клеток
1. Промойте образцы костных трабекул многократ¬
но в растворе Тирода для удаления клеток жира
и крови. Трабекулы извлекаются при помощи
скальпеля и пинцета в стерильных условиях.
2. После максимально возможного сбора костного
материала отмойте остающуюся кровь и жировые
клетки ополаскиванием раствором Тирода три
раза и разрежьте на фрагменты 2-5 мм.
3. Промойте фрагменты трабекул средой F12, со¬
держащей FBS
4. Разместите части кости в маленьком стерильном
флаконе с магнитным вкладышем для мешалки
и добавьте 4 мл расщепляющего раствора. (Это
количество должно покрыть образцы кости.)
5. Перемешивайте раствор, содержащий фрагмен¬
ты кости при комнатной температуре в течение
45 мин.
6. Удалите суспензию высвобожденных клеток,
потому что в полученной суспензии, наиболее
вероятно, содержатся фибробласты.
7. Вновь добавьте 4 мл раствора ферментов для рас¬
щепления фрагментов кости, и перемешивайте
смесь при комнатной температуре 30 мин.
8. Соберите расщепляющий фрагментный раствор,
и центрифугируйте в течение 2 мин при 580 g при
комнатной температуре.
9. После удаления супернатанта суспендируйте
клетки в 4 мл среда Хэма F12 с 20% FBS, и под¬
считайте число клеток.
10. Центрифугируйте суспензию при 580 g в течение
10 мин, ресуспендируйте клетки в 4 мл полной
среды. Эта суспензия станет инокулятом.
И. Предварительно инкубируйте 75-см2 флаконы в
течение 20 мин с 8 мл полной среды F12 до рав¬
новесия с газовой фазой.
12. Удалить преинкубационную среду и добавьте
2млсредыР12.
13. Добавьте 4 мл среды, содержащей суспензию
клеток. Инокулят должен содержать 6000-
10 000 клеток на 1 см2 поверхности.
14. Наконец, добавьте еще 6 мл среды Хэма F12,
чтобы получить полный объем 12 мл.
15. Тем временем добавьте дополнительно 4 мл рас¬
щепляющего раствора к остающимся фрагмен¬
там кости и повторно обрабатывайте в течецие
30 мин. Высвободившиеся клетки соберите, если
необходимо, повторите этот шаг еще несколько
раз. Для большого количества костной ткани,
период ферментативной обработки может быть
увеличен до 1-3 ч. Клеточный индекс подсчи¬
тывается после каждого периода обработки.
Высвобожденные клетки поместите в различные
флаконы.
Субкультивирование:
1. Удалите части эксплантата.
2. Удалите среду, и промыть слой клеток D-PBSA,
0,2 мл/см2.
3. Добавьте трипсин во флакон, 0,1 мл/см2, инку¬
бируйте при 37 °С, пока клетки не ошарятся и не
отделятся друг от друга. Отделение клетки конт¬
ролируйте под микроскопом. Обычно достаточно
10 мин инкубации.
4. Перенесите высвобожденные клетки в центри¬
фужную пробирку с равным объемом среды Хэма
F12 с20% FBS.
5. Центрифугируйте клетки при 600 g в течение
5 мин.
6. Удалите супернатант, и ресуспендируйте клетки
в полной среде осторожным пипетированием.
7. Отделите одну аликвоту суспензии для опреде¬
ления клеточной концентрации и другую для
определения DNA (см. протокол 21.3).
8. Поместите остающиеся клетки во флаконы для
культуры или лунки, которые предварительно
были обработаны для установления равнове¬
сия с газовой фазой. Клетки должны повторно
прикрепиться к стенке емкости для культуры в
течение 24 ч.
23.3.6. Эндотелий
К эндотелиальной клеточной культуре проявляется
большой интерес, поскольку потенциальное участие
эндотелиальных клеток в сосудистых заболеваниях,
восстановлении кровеносных сосудов и ангиоге¬
незе в опухолях весьма значительно. Folkman and
Haudenschild [1980] описали развитие трехмерной
структуры, включающей в себя капиллярную сосу¬
дистую сеть, полученную из чистой эндотелиальной
линии in vitro. Факторы роста, включая фактор ангио¬
генеза, выделенные из культуры клеток Walker 256 in
vitro, играют важную роль в поддержании клеточной
пролиферации и ее выживании, что позволяет фор¬
мироваться вторичным структурам. Эндотелиальные
культуры являются хорошей моделью для исследо¬
вания контактного ингибирования и плотностного
ограничения роста культуры, поскольку клеточная
пролиферация сильно ингибируется при достижении
конфлюэнтного роста [Haudenschild et al., 1976]. Эндо¬
телиальные клеточные культуры также коммерчески
23.3. Мезенхимные клетки
455
доступны (см. Приложение II). Дальнейшее введение
и протокол 23.18 для культуры эндотелиальных клеток
крупных сосудов были предоставлены Dr. Charlotte
Lawson, Royal Veterinary College, Royal College Street,
London NW1 OUT, United Kingdom.
Внутренняя поверхность всех кровеносных сосудов
состоит из одного слоя эндотелиальных клеток. Эндо¬
телий обеспечивает наличие гладкой антикоагулянт-
ной поверхности, участвует в сократительной функции
кровеносных сосудов и действует как проницаемый
барьер, позволяющий осуществляться транспорту
питательных веществ и газов, а также влияет на миг¬
рацию иммунных клеток из крови. Эндотелиальные
клетки могут быть выделены из крупных кровеносных
сосудов, включая человеческую пупочную вену [Jaffe
et al., 1973] и бычью [Booyse et al., 1975] или свиную
[Bravery et al., 1995] аорту. Микрососудистые эндоте¬
лиальные клетки могут быть выделены из адренало¬
вых желез быка, [Folkman et al., 1979], мозга крысы
[Bowman et al., 1981], человеческой кожи [Davison et
al., 1983], человеческой жировой ткани [Kern et al.,
1983], а также из человеческого [McDouall et al., 1996]
и мышиного [Marelli-Berg et al., 2000; Lidington et al.,
2002] сердца.
ПРОТОКОЛ 23.18. Выделение
и культивирование клеток сосудистого
эндотелия
Схема
Инкубирование просвета кровеносного сосуда в
растворе коллагеназы приводит к высвобождению
эндотелиальных клеток, которые могут быть культи¬
вированы в течение нескольких периодов удвоения
популяции на субстрате, покрытом желатином в
подходящей ростовой среде. Для отделения эндо¬
телиальных клеток от дизаггрегировавших частиц
сердечной мышцы используется иммуномагнитное
разделение (см. разд. 15.3.2; рис. 15.6,15.7).
А. Выделение эндотелиальных клеток
из пупочной вены человека (HUVECs)
[Адаптированный метод Jaffe et al., 1973]
Общие материалы для всех протоколов
выделения эндотелиоцитов
Стерильные или асептически приготовленные:
• D-PBSA на льду
• Сбалансированный солевой раствор Хэнкса
(HBSS)
• HBSS/PSG содержащий 300 U/мл пенициллина
• 300 мкг/мл стрептомицина (Sigma) и 50 мкг/мл
гентамицина (Invitrogen)
• Трипсин, 500 мкг/мл, EDTA, 5 мМ (0,2%) (Sigma
кат. номер. Т4174)
• Культуральные флаконы, 25 см2, покрытые 0,5%
желатина
а) Развести 2% раствор желатина типа В, полу¬
ченного из кожи быка (Sigma, G1393) в 4 раза
в D-PBSA
б) Инкубировать во флаконе в течение 20 мин
при 37 °С
в) Удалить желатин непосредственно перед до¬
бавлением клеток
• Скальпель и лезвия No. 22
• Иглы
• Шприцы, 20 мл
• Зубчатый зажим (типа «крокодил»)
• Кровоостанавливающий зажим
• Пинцеты
• острые ножницы
Нестерильные:
• Метанол, 70%
• Бумажные салфетки
• Алюминиевая фольга
• Адгезивная пленка (Clingfilm или Saran Wrap)
• Доска для выделки кожи
• Крепкая нить
Материалы для HUVECs
• Пупочный канатик человека в HBSS/PSG
• Среда роста для HUVEC: среда М199 с солями
Хэнкса, 2 мМ L-глютамина, 25 мМ HEPES, с
добавлением 20% (FBS), 150 ед/мл пенициллин/
стрептомицина и добавки для роста эндотелиаль¬
ных клеток (ECGS) из мозга быка (Roche кат. но¬
мер 1033484,75 мг; используется в концентрации
20 мкг/мл с гепарином 50 мкг/мл или Sigma кат.
номер Е2759,15 мг, используется в концентрации
30 мкг/мл с гепарином 10 мкг/мл)
• Коллагеназа Н: из Clostridium histolyticum (Roche
кат. номер 1074059; 0,5 мг/мл в среде М199. Сте¬
рилизация фильтрацией) 25 мл
• Сыворотка новорожденного теленка (NBS), хо¬
лодная 5 мл
• Адаптеры Люэра (кат. номер 700/180/700) 2
Протокол
Выделение эндотелиальных клеток:
1. Покройте доску для обработки кожи алюминие¬
вой фольгой.
2. Поместите бумажную салфетку на фольгу и про¬
питайте 70% метанолом.
3. Продавите пупочный канатик по всей длине и
удалите любые остатки крови.
4. Отрежьте 2 см или немного больше от одного
конца канатика, включая любые следы от за¬
жима.
5. Введите адаптер Люэра Insert Luer adaptor по
всей длине вены (рис. 23.1).
6. Приколите к разделочной доске иголками кана¬
тик рядом с веной, тщательно следя за тем, чтобы
не проколоть сквозь просвет вены.
7. Используя скальпель и пинцет, отделите артерии
на длину -2 см.
456
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
Рис. 23.1. Вид кровеносных сосудов в пупочном канатике
человека. Черными стрелками указаны пупочные артерии.
Белыми стрелками указаны пупочные вены с введенными
адапторами Люэра
8. Закрепите переходник нитью, используя два
очень прочно затянутых узла.
9. Закройте другой конец канатика при помощи
зажима, влейте в него около 25 мл D-PBSA при
помощи 20-мл шприца присоединенного к пере¬
ходнику. Проверьте наличие любых поврежде¬
ний на вене, раздувшейся от давления D-PBSA,
зубчатыми зажимами зафиксируйте все отверс¬
тия, не пережимая при этом вену.
10. Вылейте из канатика D-PBSA в емкость для от¬
ходов.
11. Отрежьте 2 см от другого конца канатика, введите
и зафиксируйте адаптор как в шаге 8.
12. Используя другой 20-мл шприц, влейте при¬
мерно 25 мл коллагеназы в канатик, пока он не
раздуется.
13. Заверните канатик в пленку Clingfilm, смоченную
метанолом и оставьте на 8 мин при 37 °С.
14. Выдавите колагеназу из канатика и отберите при
помощи шприца.
15. Вылейте коллагеназу из шприцов в пробирку,
содержащую 5 мл NCS.
16. Вставьте шприц с одного конца канатика и, ис¬
пользуя другой шприц, влейте в канатик 25 мл
D-PBSA.
17. Сжимайте или постукивайте канатик по всей
длине несколько секунд, затем отберите D-PBSA
обратно в шприцы.
18. Добавьте содержимое шприцов в пробирку из
шага 15.
19. Центрифугируйте пробирку 10 min, 210 g, 4 °С.
20. Ресуспендируйте осадок в 5 мл ростовой среды
HUVEC.
21. Удалите избыток желатина из флакона и добавь¬
те клетки, полученные на шаге 20.
22. На следующий день удалите среду из флакона.
23. Дважды промойте в D-PBSA и замените на све¬
жую среду HUVEC.
Ведение культуры:
24. Удаляйте половину среды и заменяйте на свежую
три раза в неделю.
25. Пересевайте, разводя клетки в отношении 1:2,
используя смесь трипсин/ЭДТА на флаконы,
обработанные желатином примерно один раз в
неделю.
26. Не используйте дольше шестого пассажа.
В. Выделение эндотелиальных клеток аорты
свиньи (РАЕС)
[Адаптированный метод Bravery et al., 1995]
Материалы для РАЕС
• HBSS/PSGA: HBSS содержащий 300 ед./мл
пенициллин/стрептомицина, 50 мкг/мл гентами-
цина, 2,5 мкг/мл амфотерицина В (GIBCO)
• Свежевыделенная аорта свиньи (асептически
отобранная, если возможно) для транспортиров¬
ки погруженная в HBSS/PSGA
• Ростовая среда РАЕС среда Ml99 с солевыми
добавками Хэнкса, 2 мМ L-глютамина, 25 мМ
HEPES, с добавками 150 ед./мл пенициллин/
стрептомицина, 50 мкг/мл гентамицина, 2,5 мкг/мл
амфотерицина В, 10% FBS, 10% NBBS
• Коллагеназа Н
(как в Материалах для HUVEC) 50 мл
• FBS в 50-мл пробирке 5 мл
Протокол
1. Покройте доску алюминиевой фольгой.
2. Положите аорту на ткань, сбрызнутую метанолом
и, завернув, промокните.
3. На доске, покрытой фольгой, удалите лишние
ткани, оставляя только ветви, но не отрезайте
слишком коротко.
4. Перевяжите ветви ниткой и пережмите зажимом
тонкий конец аорты.
5. Заполните D-PBSA зажмите сверху.
6. Закройте зубчатыми зажимами любые отверстия,
через которые происходит утечка.
7. Слейте D-PBSA.
8. Заполните аорту теплым раствором коллагена¬
зы, перезакрепите зажимы, оберните пленкой,
сбрызнутой метанолом, инкубируйте 8 мин при
37 °С.
9. Бережно массируйте аорту для разрыхления
клеток.
10. Перелейте коллагеназу из аорты в 50-мл пробир¬
ку, содержащую 5 мл FBS
11. Заполните аорту холодным (на льду) D-PBSA,
закрепите зажимом, массируйте, добавьте в про¬
бирку из шага 10, повторите этот шаг.
23.3. Мезенхимные клетки
457
12. Центрифугируйте клетки при 500 g в течение
6 мин при 4 °С.
13. Ресуспендируйте клеточный осадок в 5 мл среды,
поместите в 25-см2 флакон и инкубируйте в тече¬
ние ночи при 37 °С.
14. На следующий день удалите среду из флакона.
15. Дважды промойте D-PBSA и замените на свежую
среду.
Поддержание культуры
16. Трижды в неделю заменяйте среду: если клетки
образуют монослой менее 50% конфлюэнтности,
удаляйте и заменяйте только половину среды;
если монослой клеток достигает 50-80% конф¬
люэнтности, удаляйте и заменяйте всю среду.
17. Когда клетки достигнут 80% конфлюэнтности
монослоя, пересейте, разводя клетки в отноше¬
нии 1:3 с использованием трипсин/ЭДТА.
18. Не используйте долее седьмого пассажа.
С. Выделение эндотелиальных клеток
сердца мыши (МСЕС)
[Адаптированный метод Marelli-Berg et al., 2000]
Материалы для МСЕС
• Сердца мыши в HBSS/PSG (см. протокол 23.16А),
содержащем 300 ед./мл пенициллин/стрептоми¬
цина и 50 мкг/мл гентамицина,5
• Ростовая среда для МСЕС: DMEM с 25 мМ глю¬
козы и 4 мМ L-глютамина, с добавками 10% FBS,
150 ед./мл пенициллина, 150 мкг/мл стрептоми¬
цина и добавки для роста эндотелиальных клеток
(ECGS) из мозга быка
• Среда для неэндотелиальных клеток: DMEM
(1000 мг/л глюкозы), содержащая 15% FCS
• HBSS/PS: HBSS с 150 ед./мл пенициллина
• 150 мкг/мл стрептомицина
• HBSS/FB: HBSS с 1% FBS
• CMF: HBSS без Са2+ и Mg2+ с 150 ед./мл пеницил¬
лина, 150 мкг/мл стрептомицина
• Трипсин/ЭДТА (Sigma)
• FBS
• Коллагеназа А: из Clostridium histolyticum (Roche
кат. номер 103586); используемая концентрация
1 мг/мл в HBSS, свежеприготовленная и стери¬
лизованная фильтрацией 10 мл
• Клеточное сито с ячейками 70 мкм (B-D Biosci¬
ences)
• Чашки Петри, 9 см 2
• Иррелевантные контрольные антитела, напри¬
мер, человеческие ОКТ4 анти-С04 (DAKO)
5 мкг
• Антитела крысы против мышиных клеток
(CD105); клон MY7/18 (Pharmingen, B-D Biosci¬
ences) 5 мкг
• Антитела козы против крысы на микробусинах
(Miltenyi кат. номер130-048-501)
• Блок для разделения MidiMacs (Miltenyi) LS
MACS колонка (Miltenyi 130-042-401)
Протокол
1. Поместите сердца в чашку Петри, содержащую
примерно 5 мл HBSS/PS.
2. Удалите аорту, легочную артерию и другие при¬
датки.
3. Разрежьте сердце пополам и удалите крупные
кровяные сгустки и соединительные ткани.
4. Поместите сердце в новую чашку Петри с CMF,
промойте и замените на свежую среду CMF.
5. Порежьте материал в чашке Петри с помощью
скальпеля.
6. Промойте в CMF путем ресупендирования и
осаждения.
7. Поскребите и соберите ткани боковой стороной
лезвия скальпеля и перенесите в универсаль¬
ный контейнер, содержащий 10 мл подогретого
свежеприготвленного раствора коллагеназы А в
концентрации 1 мг/мл.
8. Инкубируйте в течение 30 мин при 37 °С с пере¬
мешиванием.
9. Добавьте CMF.
10. Диспергируйте клетки, встряхивая ткани и давая
им осесть. Соберите клеточную суспензию и пе¬
ренесите в свежую пробирку.
11. Повторите шаг 10 несколько раз.
12. Центрифугируйте клеточную суспензию при
300 g в течение 8 мин.
13. Осторожно удалите супернатант из клеточного
осадка.
14. Ресуспендируйте клеточный осадок в 5 мл трип¬
син/EDTA, стараясь разрушить любые агрегаты
клеток путем пипетирования и инкубирования в
течение 10 мин при 37 °С.
15. ДобавьтеЮ мл HBSS/FB, содержащего 1% FS, к
дизаггрегированным клеткам для расщепления и
профильтровать через клеточное сито с ячейками
70мкш.
16. Промойте ткань в клеточном сите при помощи
20 мл холодного HBSS/FB.
17. Центрифугируйте клетки фильтрата в течение
8 мин при 300 g.
18. Тщательно отсосите супернатант, удалите, сохра¬
няя клеточный осадок.
19. Добавьте ~5 мкг иррелевантных антител (напри¬
мер, ОКТ4/ОКТ8) к клеточному осадку, диспер¬
гируйте осадок, слегка постукивая по пробирке,
инкубируйте на льду в течение 20 мин, чтобы
блокировать Fc рецепторы.
20. Добавьте к клеточному осадку 5 мкг крысиных
антител к мышиному эндоглину (клон MY7/18),
встряхивая пробирку, и инкубируйте ее при 4 °С
в течение 20 мин.
21. Добавьте 30 мл HBSS и центрифугируйте при
300 g в течение 8 мин при 4 °С.
22. Добавьте 50 мкл бусин с козьими антителами к
иммуноглобулину крысы в 200 мл HBSS.
23. Инкубируйте в течение 15 мин при 4 °С.
458
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
24. Добавьте 30 мл HBSS, промойте и центрифуги¬
руйте при 300 g в течение 8 мин при 4 °С.
25. Поместите колонку MidiMacs на магнит и про¬
мойте в 500 мкл HBSS/1% FCS в соответствии с
рекомендациями производителя.
26.Добавьте Змл HBSS/1% FBCS к клеточному
осадку и перенесите в колонку.
27. Промойте колонку Зх в 1 мл HBSS/FB с после¬
дующей конечной промывкой в 500 мкл ростовой
среды.
28. Удалите колонку и поместите в 15-мл пробирку,
добавьте 4 мл ростовой среды МСЕС, вставьте
поршень, чтобы собрать бусины, связывающие
клетки. Перенесите клеточную суспензию в
25-см2 чашку, покрытую желатином.
29. Для выделения популяции неэндотелиальных
клеток осадите центрифугированием смыв с
колонки и посейте в 25 см2 чашку в DMEM
(1000 мг/л глюкозы) содержащей 15% FBS.
Ведение культуры:
30. Заменяйте среду дважды в неделю; если клетки
образуют менее чем 50% конфлюэнтный моно¬
слой, удаляйте и заменяйте только половину
среды; если клетки достигли 50-80% конфлю¬
энтности монослоя, удаляйте и заменяйте всю
среду.
31. Когда клетки достигнут 80% конфлюэнтности
монослоя, пересейте, разводя клетки в отноше¬
нии 1:3 с применением трипсина/EDTA.
32. Не используйте дольше третьего пассажа.
D. Выделение эндотелиальных клеток
сердца человека (hCEC)
Возможно выделение СЕС из левого желудочка
человека по методу McDouall et al. [1996].
Материалы для НСЕС
• Магнитные бусы, покрытые антителами к челове¬
ческим МНС класса II или человеческим клеткам
CD31 (Dynal)
• Ростовая среда НСЕС: М199 с 4 мМ L-глютами-
на с добавкой 10% FBS, 10% человеческой АВ
сыворотки (мужчины) (Sigma), 150 ед/мл пени¬
циллина, 150 мкг/мл стрептомицина, и ECGS как
в Протоколе А
• Коллагеназа II (Roche), 1 мг/мл
Протокол
1. Следуйте протоколу 23.18С.
2. Проведите расщепление тканей с коллагеназой
II (Roche) как в шаге 7.
3. Используйте магнитные бусы, покрытые ан¬
тителами к человеческому МНС класса II или
человеческим CD31 (Dynal) для выделения эндо¬
телиальных клеток как в шаге С20 и переходите
к шагу С23.
4. Ведение клеток hCEC в ростовой среде НСЕС.
Рис. 23.2. Иссечение обонятельной луковицы. Схемати¬
ческое изображение выделения обонятельных луковиц из
мозга новорожденной крысы. (С любезного разрешения
Susan Barnett.)
Идентификация клеток сосудистого эндотелия.
Сосудистые эндотелиальные клетки являются поли¬
гональными и обладают характерной морфологией
«булыжной мостовой» при культивировании в ста¬
тичных условиях (рис. 23.2). Контаминация фибро¬
бластами может быть легко идентифицирована по их
веретенообразной морфологии при рассмотрении в
фазово-контрастном инвертированном микроскопе.
Сосудистые эндотелиальные клетки могут быть иден¬
тифицированы по продукции фактора VIII [Ноуег
et al., 1973], ангиотензин-преобразующего фермента
[Del Vecchio & Smith, 1981], захвату ацетилирован-
ного липопротеина низкой плотности [Voyta et al.,
1984], присутствию телец Weibel-Palade [Weibel &
Palade, 1964], и экспрессии эндотелий-специфичных
антигенов клеточной поверхности, включая РЕСАМ-1
(CD31) [Parums et al., 1990; Albelda et al., 1990] и эн¬
доглин (CD105) [Gougos & Letarte, 1990]. Сосудистые
эндотелиальные клетки будут образовывать трубки в
Матригеле [McGuire & Orkin, 1987].
Вариации. Клетки эндотелия крупных сосудов мо¬
гут также быть выделены из аорты человека или быка,
из мышечных артерий, включая легочные, коронарные
и из вен по протоколам, аналогичным приведенному
выше. Микрососудистые эндотелиальные клетки могут
быть выделены из кожи [Davison et al., 1983], мозга
[Bowman etal., 1981] и легких [Magee etal., 1994], а так¬
же из специализированных васкулярных полостей ор¬
ганов, включая печень [Shaw et al., 1984], почку [Green
et al., 1992], и лимфатический узел [Ager, 1987].
23.4. НЕЙРОЭКТОДЕРМАЛЬНЫЕ КЛЕТКИ
23.4.1. Нейроны
По-видимому, нервные клетки являются более требо¬
вательными в выборе субстрата, чем большинство дру¬
23.4. Нейроэктодермальные клетки
459
гих клеток. Они плохо выживают на необработанном
стекле или пластике, но при использовании посуды,
обработанной коллагеном или поли-О-лизином, обна¬
руживают избыточный рост нейритов, который также
усиливается при действии полипептида — фактора рос¬
та нервов [NGF; Levi-Montalcini, 1979] и других факто¬
ров, секретируемых глиальными клетками [Marchionni
et al., 1993; Barnett, 2004; Miklic et al., 2004; Wu et al.,
2004]. Клеточная пролиферация не была обнаружена в
культуре большинства нейронов, даже в случае когда
клетки были взяты на эмбриональной стадии развития
организма, когда in vivo обнаруживается большое коли¬
чество митозов. Тем не менее недавние исследования
на эмбриональных стволовых клетках показали, что
некоторые нейроны могут быть простимулированы к
пролиферации in vitro [Thomson et al., 1998] и реколони-
зированы в организме in vivo [Snyder et al., 1992]; такие
нейроны могут быть извлечены даже из пупочного ка¬
натика [Sanchez-Ramos, 2002; Newman et al., 2003].
Протокол 23.19 для монослойной культуры ней¬
ронов мозжечка был представлен Bernt Engelsen and
Rolf Bjerkvig, Department of Cell Biology and Anatomy,
University of Bergen, Zrstadveien 19, N-5009 Bergen,
Norway.
Клетки мозжечка в культуре дают хорошо охаракте¬
ризованную популяцию нервных клеток, которая под¬
ходит для морфологических и биохимических исследо¬
ваний [Drejer et al., 1983; Kingsbury et al., 1985]. Клетки
получают из мозжечка 7-8-дневных крыс, рост не-ней-
рональных клеток предупреждается кратковременным
добавлением в культуру арабинозида цитозина.
• Арабинозид цитозина (Cytostar, Upjohn; порош¬
кообразный)
• Трипсин (тип И): 0,025% в HBSS
• Силикон (Aquasil, Pierce 42799)
• Чашки Петри для культуры тканей, 3,5 см
• Инструменты для иссечения: скальпели, ножни¬
цы, пинцеты
• Пастеровские пипетки и 10-мл пипетки
• Тест-пробирки 12 и 50 мл
Нестерильные:
• Водяная баня
• Силиконизирование Пастеровских пипеток
а) Разведите Aquasil 0,1-1% в UPW.
б) Погрузите в раствор пипетки или пропустите
раствор через пипетку.
в) Просушите пипетки на воздухе 24 ч или не¬
сколько минут при 100°С.
г) Стерилизовать пипетки сухи жаром.
• Обработка культуральных сосудов поли-Ь-лизином
а) Растворите поли-Ь-лизин в UPW (10 мг/л).
б) Стерилизуйте раствор фильтрацией
в) Добавьте 1 мл раствора поли-Ь-лизина в каж¬
дую из чашек Петри (35-мм)
г) Удалите раствор поли-L-лизина через 10-15 мин,
добавьте 1-15 мл культуральной среды.
д) Поместите культуральные чашки в инкубатор
(минимум на 2 ч) до посева клеток
Протокол
1. Иссеките асептически мозжечки крыс и помес¬
тите в HBSS.
2. Измельчите ткань при помощи скальпеля на
маленькие кубики размером примерно 0,5 мм3.
3. Перенесите измельченные ткани в тест-пробирку
(12 мл) и промойте ткани три раза в HBSS. Ос¬
тавляйте пробирки для осаждения кусочков на
дно между промывками.
4. Добавьте 10 мл 0,025% трипсина (в HBSS) к тка¬
ням, инкубируйте пробирку в водяной бане при
37 °С в течение 15 мин.
5. Перенесите трипсинизированные ткани в 50-мл
центрифужную пробирку, добавьте 20 мл росто¬
вой среды, чтобы остановить действие трипсина.
6. Дизагрегируйте ткани тритурацией через сили-
конизированную пастеровскую пипетку, пока не
будет получена суспензия отдельных клеток.
7. Оставьте суспензию клеток в центрифужной
пробирке 3-5 мин, позволяя маленьким сгусткам
тканей оседать на дно. Удалите осадок при помо¬
щи пастеровской пипетки.
8. Центрифугируйте суспензию отдельных клеток
при 200 g в течение 5 мин, отберите супернатант.
9. Ресуспендируйте осадок в ростовой среде, посейте
клетки в концентрации 2,5-3,0х106кл./чашку.
10. Через 2-4 дня (наилучшие результаты обычно
получаются через 2 дня), инкубируйте культуру
с 10 мкМ цитозина арабинозида в течение 24 ч.
11. Регулярно меняйте культуральную среду.
ПРОТОКОЛ 23.19. Гранулярные клетки
мозжечка
Схема
Мозжечки от четырех-восьми новорожденных крыс
порезать на маленькие кубики и трипсинизировать в
течение 15 мин при 37 °С в PBS Хэнкса. Суспензию
клеток посеять в культуральные флаконы или лунки,
покрытые поли-L-лизином
Материалы
Стерильные:
• DMEM с добавками
а) Глюкоза ОмМ
б) L-глютамин 2мМ
в) КС124,5 мМ
г) Инсулин (Sigma 1-1882) 100 мЕд/л
д) р-аминобензоевая кислота (Sigma А-3659)
7 мкМ
е) Гентамицин 100 мкг/мл
ж) Фетальная сыворотка теленка, термоинакти¬
вированная 10%
• Сбалансированный соляной раствор (BSS) Хэн¬
кса с 3 г/л BSA (HBSS)
• Поли-L-лизин, MB. >300 000 (Sigma Р-1524)
460
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
Характеристика нейрональных культур Нейро¬
ны могут быть идентифицированы иммунологически
с использованием нейронспецифических антител к
энолазе или при помощи тетанус-токсина в качестве
нейронального маркера. Контаминация астроцитами
может быть количественно оценена с использованием
глиального фибриллярного кислотного белка в ка¬
честве маркера.
Вариации Суспензия единичных клеток может
быть получена механическим продавливанием через
нейлоновые сита с уменьшающимися ячейками (про¬
токол 12.9) или последовательной трипсинизацией
(например, обработка трипсином в течение 3-5 мин).
Вместо HBSS можно использовать раствор Пака, Креб¬
са или другой буфер с глюкозой.
23.4.2. Глиальные клетки
Были выделены и культивированы глиальные клет¬
ки из мозга птиц, грызунов и человека. Нормальные
линии астроглии взрослого человека экспрессируют
глиальный фибриллярный кислый белок (glial fibrillary
acidic protein, GFAP) [Burke et al., 1996], способны к
высоко-аффинному захвату у-аминомасляной кислоты
и глютамата и имеют высокую активность глютамат-
синтетазы [Frame et al., 1980]. Олигодендроциты
представляют собой не без труда выживающую суб¬
культуру, но клетки-предшественники олигодендро-
цитов зрительного нерва были субкультивированы с
использованием PDGF и FGF2 в качестве митогенов
[Bogler et al., 1990]. Шванновские клетки были куль¬
тивированы с применением холерного токсина как
митогена [Raff et al., 1978; Brockes et al., 1979], однако
в настоящее время обнаружено, что оптимальный рост
получается при действии глиального фактора роста
и форсколина в среде, содержащей 10% сыворотки
[Davis & Stroobant, 1990]. Достаточно успешно были
культивированы и трансплантированы глиальные
клетки для восстановления повреждения спинного
мозга (см. разд. 25.3.8).
Протокол 23.20 разработан для очистки крысиных
ольфакторных капсульных клеток из ольфакторных лу¬
ковиц путем клеточного сортинга (fluorescenceactivated
cell sorter, FACS) и предоставлен Susan Barnett, Divi¬
sion of Clinical Neuroscience, University of Glasgow, CR
UK Beatson Laboratories, Garscube Estate, Bearsden,
G61 1BD, Glasgow, Scotland.
Возможность производить высокообогащенную по¬
пуляцию индивидуальных клеточных типов является
жизненно важной для успешного биологического иссле¬
дования различных элементов, которые контролируют
клеточный рост, дифференцировку и функции. С этой
точки зрения, клеточный сортер является мощным
инструментом для получения обогащенной, хотя от¬
носительно маленькой популяции клеток, выделенной
из гетерогенной клеточной популяции. Первоначально
клеточный сортер предназначался для характеристики
Таблица 23.2. Характеристика глиальных клеток методом
иммуноокрашивания
Клеточный тип
GFAP
GaL-C
04
А2В5
HrF
02-А
-
-
-
+
-
Астроциты I типа
+
-
-
-
-
Астроциты II типа
+
-
-
+
-
Олигодендроциты
-
+
+а
+б
-
ONEC
+/-■
-
+а
-
+
а Зрелые олигодендроциты, лишенные А2В5 и ставшие GalC\
а также 04+.
6 ONECs, утратившие 04 при культивировании.
в GFAP могут быть фиброзные или аморфные. ONECs, при¬
обретшие экспрессию GFAP при культивировании.
и сортировки клеток гематопоэтической системы, для
которой было разработано большое количество клеточ-
но-специфичных антител. С возрастанием доступности
антител, которые характеризуют как глию (табл. 23.2),
так и нейроны, стало возможным перенести эту технику
на область нейробиологии [Abney et al., 1983; Williams
et al., 1985; Trotter & Schachner, 1988; Barnett et al., 1993;
Franceschini & Barnett, 1996].
Ольфакторные луковицы представляют собой
ткань, к которой проявляется высокий интерес, по¬
скольку это единственная ткань центральной нервной
системы млекопитающих, (ЦНС), которая может под¬
держивать нейрональную регенерацию в течение жиз¬
ни [Moulton, 1974; Graziadei & Monti Graziadei, 1979,
1980]. Как только происходит гибель нейрона, — в ходе
нормального клеточного цикла или после поврежде¬
ния, — вновь образованные нейроны рекрутируются из
предполагаемого пула постоянно пролиферирующих
стволовых клеток в ольфакторном эпителии [Schwob,
2002]. Эти нейроны способны образовывать аксоны,
которые реиннервируют ольфакторную луковицу и
восстанавливают обоняние. Полагают, что популяция
глиальных клеток, представленная в обонятельном
нервном слое клеток ольфакторной луковицы, на¬
зываемых ольфакторными капсульными клеткаами
(olfactory ensheathing cells, OECs), поддерживает
или провоцирует рост ольфакторных аксонов в ЦНС
[Doucette, 1984, 1990]. На самом деле, недавние экс¬
перименты показали, что OECs способны к ремиели-
низации экспериментально демиелинизированных
повреждений спинного мозга крыс [Franklin et al., 1996;
Imaizumi et al., 1998], а также что они поддерживают
регенерацию отрезанных аксонов [Li et al., 1997,2003а,
b; Ramon-Cueto et al., 1998]. В настоящее время сущест¬
вует много обзоров, касающихся использования этих
клеток в восстановлении ЦНС. [Franklin & Barnett,
1997,2000; Raisman, 2001; Wewetzer et al., 2002; Barnett
& Riddell, 2004; Barnett & Chang, 2004].
Для того чтобы детально изучать биологию OECs,
необходимо отделить их от других основных типов
клеток, которые составляют ольфакторную луковицу:
нейроны, глиальные клетки ЦНС, олигодендроциты и
астроциты, эндотелиальные клетки, и менингеальные
клетки [Barnett & Chang, 2004].
23.4. Нейроэктодермальные клетки
461
Ольфакторная луковица крысы отвечает крите¬
риям успешного клеточного разделения методом
FACS при котором: 1) она легко образует суспензию
единичных клеток после ферментативной обработки
и 2) мышиные моноклональные антитела 04 [IgM;
Sommer & Schachner, 1981], в конъюгации с анти-
галактоцереброзидными антителами (анти-GalC)
[Ranscht et al., 1982], могут быть использованы для
узнавания OECs среди других клеточных типов, ко¬
торые присутствуют в суспензии [Barnett et al., 1993;
Franceschini & Barnett, 1996].
Антитела 04 распознают сульфатид, полулипид и
проолигодендробластный антиген [Bansal et al., 1992]
и метят многие глиальные клетки центральной и пери¬
ферической нервной системы, включая Шванновские
клетки, олигодендроциты и клетки предшественники
олигодендроцитов 2-го типа и астроцитов (0-2А)
[Gard & Pfeiffer, 1990; Mirskey et al., 1990; Sommer &
Schachner, 1981]. Ольфакторная луковица содержит три
клеточных популяции, которые могут быть помечены
04 антителами: OECs, олигодендроциты и 0-2А пред¬
шественники [Barnett et al., 1993]. В отличие от OECs
олигодендроциты и 0-2 А предшественники также могут
метиться анти-GalC антителами, что позволяет таким
образом проводить негативную селекцию, используя их
как маркеры. Используя эти два типа антител в соеди¬
нении, было показано, что 8-14% 04-положительных
клеток представляли собой OECs и менее чем 1% от
общего количества 04-положительных клетко состав¬
ляли олигодендроциты или 0-2А предшественники.
Более того, характерная сильно разветвленная или
биполярная морфология олигодендроцитов и 0-2А
предшественников, соответственно, позволяет легко
отличить их от плоских фибробласт-подобных OECs.
04-положительные FACS-очищенные OECs могут
быть разделены на два типа клеток, различающихся по
набору нейрональных маркеров — подобные Шваннов-
ским клеткам и астроцитоподобные, [Doucette, 1990;
Franceschini & Barnett, 1996; Wewetzer et al., 2002]. Эти
два типа клеток могут иметь различные функциональ¬
ные роли in vivo. Факторы роста, которые регулируют
их рост и дифференцировку, в настоящее время изу¬
чаются. В присутствии кондиционированной среды
из астроцитов 1 типа (ACM), OECs обладают хорошей
выживаемостью и ростом в течение, по крайней мере,
14 дней. Этот метод для роста астроцитов в культуре
можно найти в Noble and Murrey [1984] и Wolswijk and
Noble [1989]. Дальнейшие эксперименты показали,
что изоформа нейроглина — фактор нейро-дифферен-
цировки-Р — была идентифицирована как фактор в
ACM [Pollock et al., 1999]. Сравнительно недавно была
обнаружена комбинация факторов роста, являющаяся
мощным митогеном для ОЕС, которая включает смесь
ACM, FGF-2, форсколина и нейроглина в 5% фетальной
бычьей сыворотке [Alexander et al., 2002].
Протокол 23.20 описывает быстрый метод очистки
небольшой популяции глиальных клеток из ольфак-
торной луковицы путем использования антител к
клеточной поверхности и FACS. Эти принципы, тем
не менее, могут применяться к очистке фактически
любых глиальных или нейрональных популяций, если
при этом выполняются два главных критерия.
Исследователь может получать суспензию отдель¬
ных жизнеспособных клеток; подходящие типоспе¬
цифичные антитела, пригодные для идентификации
желаемых клеток
ПРОТОКОЛ 23.20. Ольфакторные капсульные
клетки (ОЕС)
Материалы
Стерильные или асептически приготовленные:
• Поли-Ь-лизин (Sigma), основной раствор, 4 мг/мл,
развести 1:300 в стерильной UFW для использо¬
вания в конечной концентрации 13,3 мкг/мл
• Коллагеназа тип I (МР Biomedicals, кат. но-
мер.195109): основной раствор, 2000 Ед/мл в L-15
• Сбалансированный солевой раствор Хэнкса без
Са2+ и Mr2* (HBSS, Invitrogen)
• Среда Лейбовитца L-15 с 25 мкг/мл гентамицина
(МР Biomedicals)
• DMEM/FBS: DMEM (GIBCO) с 5% FBS
• DMEM/BS: DMEM (GIBCO) с добавками (все
компоненты Sigma) [Bottenstein & Sato, 1979]:
а) Глюкоза 25 мМ (4,5 г/л)
б) Гентамицин (Invitrogen) 25 мкг/мл
в) BSA Pathocyte (МР Biomedicals)
0,0286% (v/v)
г) Глютамин 2мМ
д) Бычий панкреатический инсулин
lOpg/мл
е) Человеческий трансферрин 100 мкг/мл
ж) Прогестерон 0,2 мкМ
з) Путресцин 0,10 мкМ
и) L-тироксин .0,45 мкМ
к) Селен 0,224 мкМ
л) 3,3’,5-трийод — L-тиронин 0,49 мкМ
• Моноклональные антитела
04: МАВ345 (Chemicon)
Анти-галактоцереброзид [Ranscht et al., 1982]
(anti-GalC): MAB342 (Chemicon).
• Вторичные антитела: анти-мышиные флюоресце-
ин-меченые IgM и анти-мышиные фикоэритреин-
меченые IgG3 (Southern Biotechnology Associates
или Cambridge Biosciences).
• Флаконы, чашки и покровные стекла, покрытые
13,3 мг/мл поли-Ь-лизина:
а) Инкубируйте по крайней мере 1 ч (максимум
24 ч).
б) Промойте пластик и покровные стекла один
раз в D-PBSA.
в) Высушите на воздухе перед использованием.
• Чашки Петри.
• Полипропиленовые 15-мл конические пробирки
Фалькона с крышками.
• Пинцеты, искривленные и прямые (размер 7).
462
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
• Пробирки полистироловые, 12x75 мм, с круглым
дном и крышкой [Falcon #2058, B-D Biosciences;
эти пробирки годятся как для помещения в них
образцов, так и в качестве собирающих устройств
для FACS].
• Ножницы, искривленные для иссечения.
• Скальпель.
Нестерильные:
• Крысы Спрейг-Доули, 6-8 дней после рождения,
наибольший выход клеток ОЕС получается из
новорожденных крыс, хотя возможно приготов¬
ление из крыс любого возраста.
• Доска для иссечения и булавки.
• Спирт, 70%, в бутыли с аэрозолированием.
• Клеточный сортер с активацией флюоресценции
(FACS; B-D Biosciences; см. разд. 15.4).
Протокол
Приготовление суспензии
диссоциированных ОЕС:
1. Убейте крыс декапитацией. Используйте 15-20 жи¬
вотных для FACS сортинга в течение 2-3 ч чтобы
получить достаточно OECs для 10-15 покровных
стекол (10 000 клеток на одно покровное стекло).
2. Приколите дорзальную сторону головы к доске
для иссечения и обрызгайте 70% этанолом.
3. Погрузите все инструменты для иссечения в 70%
спирт перед использованием и высушите.
4. Удалите с головы кожу, используя острые кри¬
вые ножницы, и сделайте круговой надрез для
удаления верхней части черепа, открывая мозг и
две ольфакторные луковицы на переднем конце
коры возле носа (рис. 23.3).
Рис. 23.3. Клетки сосудистого эндотелия в культуре.
Общий вид монослойной культуры эндотелиальных клеток
аорты человека в фазово-контрастном микроскопе. Увели¬
чение 10х.
5. Используя кривой пинцет, осторожно высвобо¬
дите ольфакторные луковицы и поместите их в
чашку Петри, содержащую каплю L-15 с гента-
мицином.
6. Используя стерильные лезвия скальпеля, измель¬
чите ольфакторные луковицы.
7. Поместите кусочки в маленький флакон, содер¬
жащий 1 мл исходного раствора коллагеназы и
1мл L-15.
8. Инкубируйте при 37 °С в течение 30-45 мин.
9. Центрифугируйте получившуюся суспензию при
100 g в течение 5 мин и удалите супернатант.
10. Ресуспендируйте осадок тканей луковиц в 1 мл
раствора Хэнкса без Са2+ и Mg2+.
11. Глиальные клетки очень хрупкие, поэтому для
получения суспензии одиночных клеток ткани
ольфакторных луковиц должны быть диссоци¬
ированы очень аккуратно, без образования воз¬
душных пузырей. Диссоциацию проводите путем
мягкого пропускания через иглу для подкожного
впрыскивания, сначала 23G, потом 19G
12. Добавьте 5 мл раствора Хэнкса без Са2+ и Mg2+H
вновь центрифугируйте суспензию при 100 g в
течение 10 мин. Удалите супернатант и ресуспен¬
дируйте клетки в DMEM-FBS при концентрации
5x106 кл./мл.
FACS-сортинг OECs:
Для того чтобы максимально удалить 04+ оли-
годендроциты из сортируемой популяции клеток
можно пометить их вторичными антителами к галак-
тоцереброзиду [anti-GalC; Ranscht et al., 1982; Barnett
et al., 1993], это позволяет провести распознавание
и разделение олигдендроцитов и ОЕС. Таким путем
может быть осуществлен двухцветный сортинг, при
котором нежелательные клетки, меченные антитела¬
ми, могут не приниматься во внимание. Сортинг при
помощи FACSVantage занимает 1 час.
13. После диссоциации тканей ресуспендируйте
клетки в концентрации 5x106 в 500 мкл смеси
супернатантов гибридомы антител к 04 и анти-
GalC, инкубируйте 4 °С в течение 30-45 мин в
15-мл центрифужной пробирке. Возможно, сус¬
пензия будет содержать более чем 5x106 клеток,
поэтому приготовьте достаточное количество
пробирок для ряда клеток. Если супернатант
гибридом не будет содержать достаточной кон¬
центрации антител, то повышение концентрации
клеток приведет к видимому снижению процента
клеток 04+. Аналогичное рассуждение примени¬
мо к супернатанту гибридомы анти-GalC. Таким
образом всегда необходимо проводить титро¬
вание антител путем FACS анализа до начала
использования.
14.Через 30-45 мин, дважды промойте клетки
в DMEM/FBS и ресуспендируйте в 500 мкл
DMEM/FBS, содержащей смесь козьих антимы-
шиных флюоресциин-меченых IgM и антимыши-
23.4. Нейроэктодермальные клетки
463
ных фикоэритрин-меченых IgG3 в разведении
1:100. Инкубируйте клетки еще 30 мин при 4 °С.
15. Отмойте клетки дважды в 15 мл DMEM-FCS
путем центрифугирования и ресупендируйте
их до концентрации 2х106 клеток/мл в холодной
(на льду) среде DMEM/FBS в пробирках для
клеточного сортинга (B-D Biosciences 2085).
Храните суспензию клеток на льду. Профиль
FACS антитела-меченых клеток остается ста¬
бильным по крайней мере 3-4 ч при хранении
клеток на льду.
16. Данный протокол описывает метод, использу¬
емый для сортинга клеток на сортере FACStar.
Однако клеточный сортер любого типа дает
сходные результаты. Настройте лазер на волну
излучения 488-нм. Компенсация флюоресценции
должна быть установлена так, чтобы перевести
избыточную флюоресценцию в индивидуальные
каналы флюоресценции. Контрольный образец
клеток, меченный вторичными антителами,
должен быть проанализирован до того, как про¬
водится мечение исследуемых клеток, так чтобы
все неспецифические меченые клетки могли быть
учтены и удалены путем снижения усиления на
фотомножительной трубке.
17. Поместите окно клеточного сортера возле клеток
04+ GalC-. Средний процент клеток 04+ должен
составлять около 10±1%.
18. Отсортируйте 04+ GalC- клетки в полистироло-
вую круглодонную пробирку, содержащую 4 мл
DMEM/FBS.
19. После сортинга перенесите отделенные клет¬
ки в 15-мл центрифужную пробирку, отмойте
пробирку для сортинга средой, добавьте в эту
же центрифужную пробирку, и дополните ее до
верху DMEM/BS. Центрифугируйте при 100 g в
течение 15 мин.
20. Удалите супернатант и ресуспендируйте клетки
в DMEM/FBS. Оптимальная жизнеспособность
и рост клеток для ОЕС могут быть получены при
разведении DMEM/BS:ACM с соотношением 1:1
[Barnett etal., 1993]. ACM получают путем отбора
из монослойной культуры очищенных кортикаль¬
ных астроцитов [Noble & Murrey, 1984], инкуби¬
рованных 48 ч в DMEM/BS. Клетки также могут
быть выращены в ACM (1:5), FGF2 (Юнг/мл) и
форсколине (10 мкМ), растворенных в DMEM/
BS и затем разведенных в отношении 1:1 DM£M
содержащей 10% FBS [Alexander et al., 2002].
Данный протокол дает высокообогащенную попу¬
ляцию 04+ глиальных клеток ольфакторной луковицы
(>97%). Эти клетки могут быть посеяны на обрабо¬
танные поли-Ь-лизином покровные стекла, чашки
Петри или флаконы и использоваться в дальнейших
исследованиях. В настоящее время имеются данные,
подтверждающие прнадлежность этих клеток к OECs,
описанных в электронно-микроскопических исследо¬
ваниях [Doucette, 1984;
Raisman, 1985], исследования продолжаются в
направлении изучения и дальнейшей характеризации
клеток.
23.4.3. Эндокринные клетки
Проблемы культивирования эндокринных клеток
[O’Hare et al.,1978] обусловлены относительно малым
количеством эндокринных клеток в специфических
тканях, в отношении к стромальным и паренхима¬
тозным клеткам. Sato с коллегами [Buonassisi et al.,
1962; Sato & Yasumura, 1966] культивировали функ¬
ционирующие адреналовые и гипофизарные клетки
из крысиных опухолей путем механической дизагре-
гации опухоли [Zaroff et al., 1961] и поддержанием
обычной монослойной культуры. Функциональное
единство клеток поддерживалось путем периодиче¬
ского пересева клеток как перевиваемой опухоли в
крыс [Buonassisi et al., 1962; Tashjian et al., 1968]. Эти
линии в настоящее время полностью адаптированы к
культуре и могут поддерживаться без пассажа через
животных. [Tashjian, 1979] и в некоторых случаях мо¬
гут поддерживаться на полностью охарактеризованных
средах [Hayashi &Sato, 1976].
Фибробласты редуцируются в культуре клеток
панкреатических островков путем обработки этил-
меркуритизозалицилатом [Braaten et al., 1974], клетки
островков очищают также путем центрифугирования в
градиенте плотности [Prince et al., 1978] и путем цент-
рифугальной элютриации [Bretzel et al., 1990]. Клетки
островков, как известно, продуцируют инсулин, но
утрачивают эту способность при культивировании.
Кратковременные клеточные культуры также были
получены из клеток-предшественников протоков
железы, способных к дифференцировке до инсулин-
секретирующих клеток в бессывороточной среде [Gao
et al., 2003].
Из мышей были выделены гипофизарные клетки,
которые производят гормоны гипофиза в течение жиз¬
ни нескольких субкультур [de Vitry et al., 1974]. Хотя
нормальные человеческие гипофизарные клетки выжи¬
вают плохо и большинство культур аденомы гипофиза
человека постепенно утрачивают способность к синтезу
гормонов, некоторые культуры продолжают продуци¬
ровать гормон роста [см., например, Bossis et al., 2004],
трехмерная культура аденомы гипофиза продолжает
секретировать пролактин [Guiraud et al., 1991].
23.4.4. Меланоциты
Для получения культуры, преимущественно состоя¬
щей из меланоцитов, используется высокая зависи¬
мость культуры от субстрата, которая играет роль в
прикреплении и росте клеток на среде с добавлением
гормонов [Naeyaert et al., 1991]. Система помогает
464
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
избежать проблемы контаминации кератиноцитами, в
то время как пролиферация хорошо поддерживается
при минимальном добавлении сыворотки [Gilchrest
et al., 1985; Wilkins et al., 1985] в отсутствие химио¬
терапевтических агентов или промоторов опухоли,
таких как эфиры форбола. Эти вещества негативно
регулируют протеинкиназу С (protein kinase С, РКС)
и, следовательно, тирозиназную активность, сокращая
продукцию пигмента [Park et al., 1993]. При общеупот¬
ребительной дозе добавлении сыворотки (5-20%) рост
меланоцитов значительно лучше, чем описанный в
других системах, а рост фибробластов может быть пре¬
дупрежден снижением концентрации Са2+ до 0,03 мМ
[Naeyaert et al., 1991]. Система, описанная здесь, может
быть в дальнейшем модифицирована удалением из нее
холерного токсина (который провоцирует рост мела¬
ноцитов путем значительного возрастания внутрик¬
леточного уровня сАМР) и дибутирила сАМР. Такая
среда обеспечивает физиологический уровень РКС и
с АМР, что, в свою очередь, позволяет проводить изу¬
чение сАМР-модулирующих агентов [Park et al., 1999]
в условиях, которые оптимальны для исследований
меланогенеза.
Приведенное введение и протокол 23.21 для куль¬
туры меланоцитов человека были предоставлены Нее-
Young Park, Mina Yaar, and Barbara A. Gilchrest, Depart¬
ment of Dermatology, Boston University School of Medi¬
cine, 80 East Concord St., Boston, MA 02118-2394.
ПРОТОКОЛ 23.21. Меланоциты
Схема
Эпидермис, отделенный от небольшого фрагмента
кожи после трипсинизации, диссоциируют в EDTA
и культивируют в бессывооточной среде с добавками
главного FGF (FGF-2).
Материалы
Стерильные:
• D-PBSA
• Фетальная сыворотка теленка (FBS)
• Среда с гормональными добавками для мелано¬
цитов (MHSM):
Основной Объем Конечная
раствор основного концен-
раствора трация
а) Среда 199
(GIBCO 400-1200) 93,1 мл
б) EGF (B-D
Biosciences), 10 мкг/мл 0,1 мл 10 нг/мл
в) Трансферрин
(Sigma), 10 мг/мл 0,1 мл ... 10 мкг/мл
г) Инсулин
(Sigma), 10 мг/мл 0,1 мл ... 10 мкг/мл
д) Трииодтиронин
(Sigma), 1 мкМ 0,1 мл 1 нМ
е) Гидрокортизон
(Calbiochem), 0,28 мМ 0,5 мл 1,4 мкМ
ж) Холерный токсин
(Calbiochem), 0,1 мкМ 1,0 мл 1 нМ
Холерный токсин может быть заменен на экстракт
гипофиза быка (Clonetics), концентрация исход¬
ного раствора 35 мкг/мл, конечная концентрация
0,7нг/мл, и дибутирил сАМР (Sigma), 1 мМ,
конечная концентрация 100 мкМ
з) Главный FGF
(Amgen), 200 нг/мл 5,0 мл 10 нг/мл
• Трипсин/EDTA: трипсин 0.25% в D-PBSA с 0,1%
(3,5 мМ) EDTA (GIBCO 610-5050)
• Соевый ингибитор трипсина (Sigma), 10 мкг/мл
• Чашки для культуры тканей (Falcon, B-D Biosci¬
ences), диаметром 10,6 и 3,5 см diameter
• Пипетки
• Центрифужные пробирки, 15 и 50 мл
Нестерильные:
• Инкубатор с увлажнением (37 °С, 7% С02)
• Водяная баня
• Электронный счетчик клеток или
Протокол
Получение первичной культуры
День 1:
1. Прополощите образец кожи в 70%-м спирте и
затем дважды в D-PBSA.
2. Перенесите ткань в стерильные 10-см чашки,
эпидермисом вниз.
3. При помощи ножниц для иссечения удалите
подкожный жир и глубокие слои дермы.
4. Порежьте оставшуюся ткань на кусочки разме¬
рами 2x2 мм при помощи скальпеля точными
режущими движениями сверху вниз (не исполь¬
зуйте пилящие движение), поднимая лезвие над
тканью.
5. Перенесите фрагменты ткани в 15-мл центри¬
фужную пробирку, заполненную холодным
0,25% трипсином.
6. Инкубируйте фрагменты ткани 1-1,5 ч при ком¬
натной температуре. Для некоторых доноров
инкубирование при 37 °С в течение 1-2 ч после
инкубирования при 4 °С в течение 18-24 ч может
дать более высокий выход меланоцитов.
День 2:
7. Осторожно постукивая по пробирке, отделите от
дна фрагменты, которые прилипли к нему, затем
быстро перелейте содержимое пробирки в 6-см
чашку.
8. Используя пинцет, перенесите фрагменты ткани
по отдельности в сухую 10-см чашку, эпидер¬
мальной стороной вниз. Осторожно подвигайте
каждый фрагмент ткани по чашке. Эпидермаль¬
ная выстилка должна прилипнуть к чашке, что
23.5. Гематопоэтические клетки
465
позволяет точно отделить дерму при помощи
пинцета. Выбросьте фрагменты дермы.
9. Перенесите все эпидермальные фрагменты в
стерильные 15-мл центрифужные пробирки, со¬
держащие 5мл 0,02% (0.7 мМ) EDTA. Старайтесь
поместить каждый эпидермальный фрагмент в
раствор EDTA, а не на пластиковую стенку цен¬
трифужной пробирки.
10. Осторожно встряхивайте пробирку, чтобы ди-
заггрегировать эпидермальные пленки до сус¬
пензии единичных клеток.
11. Центрифугируйте клетки в течение 5 мин при
350 g, а затем отберите супернатант.
12. Ресуспендируйте осадок в бессывороточной
среде MHSM.
13. Подсчитайте клетки при помощи гемоцито¬
метра и инокулируйте 1х106клеток (примерно
2-4х104меланоцитов) на 3,5-см чашку в 2 мл
MHSM содержащего 5% FBS для улучшения
прилипания клеток.
14. Меняйте питательную среду, вновь добавляя
MHSM, содержащую факторы роста и экстракт
гипофиза быка дважды в неделю.
Дни 3-30:
15. В течение 24 ч культуры будут содержать пер¬
вичные кератиноциты с рассеянными меланоци-
тами (рис. 23 Аа). Пролиферация кератиноцитов
должна прекратиться в течение нескольких дней
и колонии меланоцитов начнут формировать¬
ся в течение второй недели. К концу третьей
недели должны остаться только меланоциты
(рис. 22.46, в). В большинстве случаев культуры,
достигшие почти конфлюэнтного состояния, го¬
товы к пересеву через 2-4 недели.
Субкультивирование:
16. Осторожно дважды сполосните культуральные
чашки 0,02% (0,7 мМ) EDTA.
17. Добавьте Змл 0,25% трипсин /0,1% (2,5 мМ)
EDTA и инкубируйте при 37 °С. Обследуйте
чашки под фазово-контрастным микроскопом
каждые 5 мин для обнаружения отделения клеток
от поверхности.
18. Когда большинство клеток отделились, инакти¬
вируйте трипсин соевым инигбитором трипсина
(10 мкг/мл) или 1 мл среды, содержащей 10% сы¬
воротки теленка. (Меланоциты, культура которых
получена в бессывороточных условиях, получают
значительную стимуляцию при «сывороточном
толчке» во время субкультивирования.)
19. Пипетируйте содержимое чашки для того, чтобы
убедиться, что меланоциты полностью отдели¬
лись от чашки.
20. Соберите клеточную суспензию в центрифужную
пробирку и центрифугируйте в течение 5 мин при
350 g.
21. Отсосите супернатант, ресуспендируйте клетки
в бескальциевой среде для меланоцитов, содер¬
жащей 5% FBS, и пересейте на 100-мм чашку в
концентрации клеток 2-4х104. Для хорошего
прикрепления меланоцитов к пластиковой чаш¬
ке (>75%) требуется FBS, однако, если чашки
обработаны предварительно фибронектиом или
коллагеном типа I/I1I, можно не применять FBS
[Gilchrest et al., 1985].
22. Два раза в неделю меняйте среду на бессыворо-
точную или содержащую сыворотку среду для
меланоцитов. Среда, содержащая сыворотку, даст
более высокий выход меланоцитов, но усилит рост
фибробластов. Снижение концентрации кальция
до 0.03 мМ не оказывает действия на пролифе¬
рацию меланоцитов. но ингибирует избыточный
рост фибробластов [Naeyaert et al., 1991].
23.4.5. Подтверждение идентичности
меланоцитов
Культуры меланоцитов могут быть изначально конта-
минированы кератиноцитами и в какое-то другое время
кожными фибробластами. Обе формы контаминации
достаточно редки в установившейся и поддерживаемой
культуре, если она ведется опытным лаборантом или
исследователем, однако является часто встречающейся
проблемой для новичков. Принадлежность культиви¬
руемых клеток к меланоцитам может быть достаточно
точно подтверждена с помощью частого исследования
культуры под фазовым микроскопом, что, в свою
очередь, предполагает, что исследователь знаком с
соответствующей морфологией клеток (рис. 23.46, в).
Более точная идентификация может быть проведена
путем электронно-микроскопического исследования,
DOPA окрашивания или иммунофлюоресцентного
окрашивания с антителами Mel 5 против тирозиназа-
родственного протеина-1.
23.5. ГЕМАТОПОЭТИЧЕСКИЕ КЛЕТКИ
Гематопоэтические клетки были выращены при ис¬
следовании колониеобразующей способности клеток
костного мозга в долгоживущей культуре и как посто¬
янные культуры клеток [Freshney et al., 1994; Klug &
Jordan, 2002].
Под контролем специфических факторов роста ге¬
матопоэтические стволовые клетки CFU-A могут быть
повторно клонированы из агаровых культур (см. прото¬
кол 23.23). Показано, что М1Р-1а является основным
фактором, который поддерживает стволовые клетки в
регенеративном состоянии. Тем не менее большинство
колоний, полученных из суспензии клеток, только од¬
ной линии выживают только как первичная культура,
утратившая эффективность обновления популяции и
не могут быть субкультивированы. Гранулоцитарные
466
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
Рис. 23.4. Культура меланоцитов. а) Культура меланоци-
тов, выделенных из крайней плоти полового члена новорож¬
денного через 1 неделю после инокуляции клеток. Обратите
внимание на колонии кератиноцитов, стратифирующиеся в
центре и формирующие плотные соединения эпителиальных
клеток на периферии. Меланоциты — относительно малень¬
кие темные клетки с отростками, большинство из которых
контактируют с колониями кератиноцитов посредством
выступающих дендритов (наблюдение через фазовый конт¬
раст). б) Десятидневная первичная культура эпидермальных
меланоцитов новорожденного в среде с недостатком ТРА.
У многих клеток отмечается ветвистость дендритов, другие
клетки изменяют морфологию — от биполярности до поли-
гональности (наблюдение через фазовый контраст, х320).
в) Вторичная культура ТРА- обработанных эпидермальных
меланоцитов. Обратите внимание на морфологию дендритов
у клеток, пигментацию и их узкую вытянутую форму
колонии являются наиболее общеупотребительными;
однако при соответствующих условиях могут быть
получены и другие линии (см. протокол 23.23).
Ряд миелоидных клеточных линий был получен
из мышей с лейкемией [Horibata & Harris, 1970], и
было показано, что, подобно некоторым линиям че¬
ловеческим лимфобластоидов [Collins et al., 1977],
они производит глобулиновые цепи и в некоторых
случаях формируют завершенные а- и у-глобулины
(см. разд. 27.10).
Некоторые из этих линий могут быть выращены
в бессывороточной среде [Iscove & Melchers, 1978].
Т-клеточные линии требуют факторов роста Т-клеток
[например, интерлейкин IL-2; Gillis & Watson, 1981],
описаны также факторы роста В-клеток [Howard et al.,
1981; Sredni et al., 1981; см. также Freshney et al.,1994].
Первые человеческие клеточные линии лимфоблас¬
тов были получены культивированием лимфоцитов
периферической крови при очень высокой клеточной
плотности (~ 106/мл), обычно в глубоком слое (>10 мм)
[Moore et al., 1967]. Потом были получены иммортали-
зованные клеточные линии благодаря гену EBV (вирус
Эпштейна-Барра) и в настоящее время эти методы
широко используются [Bolton & Spurr, 1996].
Rossi and Friend [1967] показали, что мышиный
РНК-вирус (“Friend virus”) может вызывать сплено-
мегалию и эритробластозы у инфицированных мышей.
Клеточные культуры, выделенные из измельченной
селезенки этих животных, в некоторых случаях да¬
вали начало постоянным клеточным линиям клеток
эритролейкемии. Все эти линии трансформированы
тем, что в настоящее время определяется как комплекс
дефективных и вспомогательных вирусов, выделенных
из вируса саркомы Молони [Ostertag & Pragnell, 1978,
1981]. Некоторые клеточные линии могут быть полу¬
чены с использованием вируса, который инфективен in
vivo, но не эффективен in vitro. Эти клетки могут быть
пересеяны как солидная или асцитная опухоль мышам
DBA2 или BALB-C
Обработка культуры клеток Friend рядом агентов,
включая DMSO, бутират натрия изобутировую кисло¬
ту, и бисацетамид гексаметилена стимулирует эритро-
идную дифференцировку [Friend et al., 1971; Leder &
Leder, 1975]. Необработанные клетки образуют комп¬
лексы недифференцированных проэритробластов, в
то время как в обработанных клетках обнаруживаются
признак ядерной конденсации, уменьшение размеров
клетки и аккумуляция гемоглобина (см. разд. 17.7)
в меньшем объеме, так что центрифугированный
клеточный осадок имеет красный цвет. Признаки
дифференцировки могут быть продемонстрированы
окрашиванием клеток на гемоглобин с помощью
бензидина (см. цв. вкладку 13а, б) и гибридизацией
с глобин-специфической РНК in situ (см. цв. вклад¬
ку 13в, г). Такие клеточные линии лейкемических
клеток как К562 могут также быть индуцированы к
дифференцировке действием бутирата натрия и геми-
на, хотя DMSO не оказывает индуцирующего действия
[Andersson et al., 1979с].
Макрофаги могут быть выделены из многих тканей
путем сбора клеток, которые прикрепляются к поверх¬
ности при ферментативной дезаггрегации. Тем не менее
выход клеток в этом случае достаточно низок, и в на¬
стоящее время разработан ряд методов, позволяющих
23.5. Гематопоэтические клетки
467
получить большее количество макрофагов. Минераль¬
ное масло или бульон с тиогликолатом [Adams, 1979]
могут быть введены в брюшную полость мыши, через
3 дня перитонеальные смывы будут содержать высокую
долю макрофагов. При необходимости макрофаги мо¬
гут быть очищены за счет способности прикрепляться
к субстрату в присутствии протеаз (см. разд. 14.8.1).
Макрофаги с трудом могут быть субкультивированы в
результате их резистентности к трипсину. Разработаны
методы, использующие гидрофобные пластики (напри¬
мер, чашки Petriperm, Vivascience, Sartorius).
Имеются сообщения о перевиваемых клеточных
линиях макрофагов, главным образом, из мышиных
неопластических образований. Нормальные мышиные
макрофаги не пролиферируют, хотя можно культиви¬
ровать их клетки-предшественники по методу Dexter&
Spooncer (см. протокол 23.22).
23.5.1. Долгоживущие культуры клеток
костного мозга
Последующее введение и протокол 23.22 для долго¬
живущей культуры гематопоэтических клеток кост¬
ного мозга был предоставлен Е. Spooncer, Department
of Biomolecular Sciences, UMIST, Sackville Street,
Manchester M60 1QD, U. K.
При культивировании цельного костного мозга под¬
держиваются отношения между стромой и стволовыми
клетками, в присутствии соответствующих гематопо¬
этических клеток и при наличии этого межклеточного
взаимодействия стволовые клети и специфические
предшественники могут делиться в течение более чем
нескольких недель [Dexter et al., 1984; Spooncer et al.,
1992]. Клетки-предшественники, выделенные из свеже¬
го костного мозга или долгоживущие культуры могут
быть исследованы путем клоногенного роста в мягком
агаре [Heyworth & Spooncer, 1992] или в мышах [Till &
McCulloch, 1961].
ПРОТОКОЛ 23.22. Долгоживущая культура
гематопоэтических клеток костного мозга
Схема
Костный мозг отсасывается и переносится в пита¬
тельную среду и затем выращивается в виде культу¬
ры прикрепившихся клеток в течение 12-30 недель.
Стволовые клетки, а также созревающие и зрелые
миелоидные клетки высвобождаются от прикрепив¬
шихся клеток и переходят в ростовую среду
Предшественники гранулоцитов/макрофагов
могут быть исследованы в мягких гелях (см. прото¬
кол 23.23).
Материалы
Все реактивы должны быть предварительно провере¬
ны на способность поддерживать рост культуры
Стерильные:
• Среда Фишера (Invitrogen), дополненная 50 Ед/мл
пенициллина и 50 мкг/мл стрептомицина и со¬
держащая 16 мМ (1,32 г/л) NaHC03
• Ростовая среда: среда Фишера, как указано выше,
разлитая на аликвоты по 100 мл с добавлением
1 мкМ гидрокортизона сукцината натрия и 20%
лошадиной сыворотки (гидрокортизон сукцина¬
та натрия делают как 1 мМ основной раствор в
среде Фишера и хранятся при -20 °С).
• Шприцы 1 мл, с иглами 21G
• Марлевые тампоны, ножницы, пинцеты
• Флаконы для культуры ткани, 25 см2
Нестерильные:
Пять мышей (С57В1/6Ч DBA/2), Fj-клетки костного
мозга которых хорошо растут в долгоживущей куль¬
туре в отличие от мышей других линий (например,
СВА) [Greenberger, 1980].
Протокол
1. Убейте мышей-доноров методом цервикального
смещения.
2. Протрите 70%-м спиртом шерсть и отрежьте оба
бедра. Соберите 10 бедренных частей в чашку
Петри на льду, содержащую среду Фишера. Одно
бедро содержит 1,5-2,ОхЮ7 клеток.
3. В ламинарном шкафу:
а) Полностью удалите все оставшиеся мышечные
ткани при помощи марлевых тампонов.
б) Удерживая бедренную кость при помощи
пинцета, срежьте коленный конец. Конец иглы
21G должен плотно входить в полость кости.
в) Срежьте другой конец бедренной кости так
близко к концу, как это возможно.
г) Введите конец кости в 100-мл бутылочку с
ростовой средой и всасывая и выдавливая
поршень шприца несколько раз до тех пор,
пока костный мозг вымывается из бедра.
д) Повторите шаги (а)-(г) с оставшимися девя¬
тью костями.
4. Диспергируйте костный мозг до суспензии путем
пипетирования крупных участков костномозго¬
вого стержня через 10-мл пипетку. Нет необхо¬
димости в дезагрегации мелких сгустков.
5. Разлейте 10-мл аликвоты суспензии клеток в
25-см2 флаконы для культуры тканей, постоянно
взмучивая суспензию вращательным движением
для того, чтобы быть уверенным в равномерности
распределения клеток в 10 культурах.
6. Заполните флаконы 5% С02 в воздухе и плотно
заверните крышки.
7. Инкубируйте культуры в горизонтальном поло¬
жении при 33 °С.
8. Еженедельно производите смену среды.
а) Осторожно взболтайте флаконы для суспен¬
дирования слабо прикрепленных клеток.
б) Удалите пипеткой из каждого флакона по 5 мл
ростовой среды вместе с суспензией клеток,
468
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
стараясь не повредить слой прикрепленных
клеток.
в) Добавьте 5 мл свежей ростовой среды в каж¬
дый флакон. Для того чтобы избежать пов¬
реждения, не направляйте струю из пипетки
на клетки.
г) Заполните флаконы газом, верните их в инку¬
батор.
Анализ. Клетки, собранные при смене среды куль¬
туры, могут быть исследованы рядом методов, включая
морфологические исследования, CFC исследования
(см. протокол 23.23), и CFU-S in vivo для стволовых
клеток [Till & McCulloch, 1961].
Вариации. Выращены культуры мышиных эритро-
идов [Dexter et al., 1981 ] и В-лимфоидов [Whitlock et al.,
1984], а также человеческие долгоживущие культуры
[Gartner & Kaplan, 1980; Coutinho et al., 1992].
23.5.2. Анализ колониеобразования
культуры гематопоэтических клеток
Клетки-предшественники гематопоэтических клеток
могут быть клонированы в суспензии в полужидкой
среде в присутствии соответствующих факторов роста
[Heyworth & Spooncer, 1992]. В зависимости от потент-
ности выделенных стволовых клеток могут быть полу¬
чены чистые линии или смеси колоний. Исследования,
используемые для определения колониеобразования
гранулоцитов и макрофагов (GM-CFC), эритроид-
ных бурст-образующих единиц (BFU-E), смешанных
колониеобразующих единиц (CFC-mix), а также
гранулоцитарных, эритроцитарных, макрофагальных
и мегакариоцитарных колоние-образующих клеток
(CFC-GEMM) описаны Testa & Molineux [1993], и
их место в технологии рутинных гематопоэтических
клеточных культур уже хорошо определено [Metcalf,
1990]. Последующее введение и протокол 23.23 осно¬
ваны на Freshney et al. [1994].
Эффективность роста колоний в данном исследо¬
вании возрастает при использовании Метоцеля вместо
агара в качестве полужидкой среды. Данная практика
разработана для более плотных колоний, которые лег¬
че оценить и сосчитать. Поскольку Метоцель является
жидкостью с высокой вязкостью, а не гелем, как агар,
клетки будут оседать сквозь него, хоть и медленно, и
осаждаться на пластиковом дне чашки. Это движение
приведет к тому, что они все расположатся в одной
фокальной плоскости, удобной для последующего
наблюдения. Колонии будут образовываться, если
рост происходит в атмосфере 5% С02 в воздухе, но
для адекватного гематопоэза идеальной является
смесь 10%С02и5% 02в воздухе [Bradley etal., 1978b].
Если инкубатор с газовой смесью недоступен, можно
использовать баллон с газовой смесью. Поместите
чашки в пластиковую коробку с дыркой в крышке.
Запечатайте коробку пластиковой лентой и заполните
газом коробку через дырку, затем ее тоже запечатайте.
Чашка с водой в коробке позволит сохранить влажную
атмосферу.
ПРОТОКОЛ 23^3. Анализ
колониеобразования культуры
гематопоэтических клеток
Схема
Суспендируйте клетки костного мозга в агаре или
метилцеллюлозе, посейте на чашки с соответствую¬
щими факторами роста
Материалы
Стерильные или асептически приготовленные:
• Клетки костного мозга (см. протокол 23.22,
шаги 1-4 для приготовления). Подсчитайте ядро¬
содержащие клетки в гемоцитометре после окра¬
шивания их с метиленовым синим, или лизируйте
клетки Zapoglobin и подсчитайте количество
ядер на электронном счетчике клеток (Beckman
Coulter). Каждая бедренная кость даст примерно
1,0- 1,5х107 клеток.
• Метилселлюлоза, 4000 сР (Fluka)
• Агар Noble (Difco, B-D Biosciences)
• Альфа MEM основной раствор (Invitrogen)
• FBS
• Факторы роста (табл. 23.3), рекомбинантные
(R&D) или из кондиционированной среды
• BSA,10%bD-PBSA
• Чашки Петри, 3 см
• Wehi-cell-кондиционированная среда (Wehi-
СМ): Wehi ЗВ представляет собой миеломоноци-
тарную клеточную линию, которая при культи¬
вировании выделяет в среду IL-3 (мульти-CSF)
[Bazill et al., 1983]. (Метод приготовления конди¬
ционированной среды см. протокол 14.2.).
• Метилцеллюлоза (Methocel)
а) Взвесить 7.2 Methocel добавить ее в 500-мл
колбу, с большим вкладышем для магнитной
мешалки.
Таблица 23.3. Добавки клеток и факторов роста при
исследовании способности к колониеобразованию
Факторы роста /мл
Исследование
IL-6
rMurGM* rIL-36
Частота
встречае-
Еро Клетки мости/105
BFU-E
CFC-mix
CFU-GEMM
100 нг
0,1 нг 1 нг
1 нг
1 нг
2 U 5x104
2 U 5x104
2 U 5x104
40-80
100-180
92-106
а 10% AF1-19T СМ может быть замещено на GM-CSF [Prag-
nell et al., 1988].
6 10% Wehi-CM может быть замещено на rIL-3 [Bazill et al.,
1983].
23.6. Гонады
469
б) Стерилизовать Methocel автоклавированием.
в) Добавить 400 мл стерильной UPW подогретой
до 90 °С чтобы увлажнить метилцеллюлозу.
г) Перемешивать смесь при 4°С в течение ночи
до растворения метилцеллюлозы. Получаем
раствор Methocel 2х. Для более точного разли¬
вания метилцеллюлозы следует использовать
не пипетку, а шприц без иглы.
• Среда Alpha основной раствор
а) Среда Alpha, порошкообразная (GIBCO, упа¬
ковка на 10 л)
б) Витаминный основной раствор МЕМ, 100х,
100 мл
в) Гентамицина сульфат, 200 мг. Перемешивайте
среду на мешалке с подогревом до раство¬
рения, доведите до 3 л добавлением UPW
(не допускайте повышения температуры
выше 37 °С.) До конечной фильтрации через
0,22-мкм фильтр эффективно профильтровать
среду через серию фильтров размерами 5,1,2,
0,8 и 0,45 мкм. Разлейте среду по аликвотам
21-мл, храните при -20 °С.
• Среда Alpha, 2х
а) Основной раствор среды Alpha, 21 мл
б) FBS, 25 мл
в) Глютамин 200 мМ, 1 мл
г) NaHC03,7,5%, 3 мл
Смешать ингредиенты в стерильной посуде, до¬
вести до 37 °С.
Нестерильные:
• Для агара используйте две водяных бани, одну на
37 °С и другую на 55 °С.
• Инкубатор: газовая фаза, 10% С02,5%02,85% N2
ПротоколА: BFU-E, CFC-MIx, и CFU-GEMM
1. Для получения требуемого количества среды для
эксперимента смешайте равные объемы среды
Alpha 2х, в которую добавлен 1% BSA с Метил-
целлюлозой 2х. Храните на холоде, Methocel
более жидкий в холодном состоянии.
2. Получайте культуру в трех копиях, но приготовь¬
те такое количество смеси, чтобы было достаточ¬
но для 4 чашек, поскольку Methocel прилипает к
стенкам пробирок и некоторое количество всегда
теряется.
3. Добавьте в пробирку факторы роста до необхо¬
димой концентрации (табл. 23.3).
4. Добавьте 5х104 клеток.
5. Доведите общий объем до 4,4 мл добавлением
среды 1х, перемешайте на вортекс-миксере.
6. При помощи шприца поместите по 1 мл среды в
3-см чашки Петри (для культивирования тканей).
7. Инкубируйте культуру при 37 °С во влажной
атмосфере с 10% С02и 5% 028-15 дней.
Идентификация колоний
Колонии BFU-E могут быть как единичными,
составленными из очень маленьких клеток, так и
мультицентричными колониями (пачка), каждая с
плотно упакованными очень маленькими розовы¬
ми или красными клетками. Эти колонии содержат
40-80/105 костномозговых клеток
CFC-mix колонии могут быть единичными ком¬
пактными колониями, обычно окруженные клетка¬
ми очень разнообразного размера. Они могут быть
мультицентричными, но клеточная популяция при
этом выраженно гетерогенна. Инцидентность этих
колоний составляет в данном исследовании обычно
100-180/105костномозговых клеток, из них только
примерно 10% будут смешанными колониями, со¬
держащими эритроидные клетки. Если эритроидные
клетки не красные, то может быть затруднительно
для неопытного исследователя точно идентифици¬
ровать колонии. Колонии следует сфотографиро¬
вать, отобрать, провести препаративное центрифу¬
гирование, фиксировать и окрасить по Гимза (10%),
а затем попросить о помощи в идентификации
клеток [Heyworth & Spooncer, 1992].
Протокол В: GM-CFC
1. Рассортируйте 3-см чашки Петри и промарки¬
руйте их (не для культивирования ткани).
2. Приготовьте 0,3% агаровую среду (см. прото¬
кол 14.4) и храните при 37 °С.
3. Приготовьте костномозговые клетки (см. про¬
токол 23.22). Подсчитайте ядросодержащие
клетки после окрашивания метиленовым синим
или лизируйте клетки Zapoglobin и подсчитайте
количество ядер на электронном счетчике клеток
(Beckman Coulter). Каждая бедренная кость даст
примерно 1,0-1,5х 107 клеток.
4. Добавьте 0,1 нг/мл rMurGM-CSF в каждую чашку.
5. Добавьте 1 мл агаровой среды, содержащей
7,5x104 клеток, осторожно покручивая, переме¬
шайте клетки и агар. Оставьте при комнатной
температуре до застывания агара.
6. Другой метод: 0,8% среды Methocel. Обычно этот
метод дает более плотные колонии, которые легче
считать.
7. Поместите чашки в чистую пластиковую коробку.
8. Инкубируйте чашки в течение 6 дней во влажном
инкубаторе при 37 °С в атмосфере 5% С02
9. Используя инвертированный микроскоп, под¬
считайте количество колоний, содержащих более
50 клеток, и выразите результаты подсчета в виде
отношения количество колоний/105 посеянных
клеток.
10. Инцидентность GM-CFC в нормальном костном
мозге составляет 100-120/105 клеток.
23.6. ГОНАДЫ
Зернистые клетки яичника могут поддерживаться и
частично сохранять функциональную активность в
первичной культуре [Orly et al., 1980], но при субкуль¬
470
ГЛАВА 23. КУЛЬТУРЫ СПЕЦИФИЧЕСКИХ ТИПОВ КЛЕТОК
тивировании специфические функции утрачиваются.
Клеточная линия, происходящая из яичника китайско¬
го хомяка [CHO-KI; Као & Puck, 1968], поддерживается
уже много лет, однако ее происхождение до настоящего
времени не идентифицировано. Хотя клетки выглядят
эпителиоидными на некоторых стадиях роста, они
подвергаются фибробласт-подобной модификации,
когда они культивируются в присутствии дибутирил
цикличной АМФ [Ilsie & Puck, 1971].
Клеточные фракции из яичек выделены методом
седиментации, однако сообщений о продолжительно
поддерживаемой культуре этих фракций нет. Линия
ТМ4 представляет собой линию эпителиальных клеток
из мышиных яичек, но их дифференцируемые свойства
не описаны. Клетки Сертоли из яичек также были вы¬
делены в культуре [Mather, 1979].
23.7. СТВОЛОВЫЕ КЛЕТКИ
Культура стволовых клетко была впервые разработана
для гематопоэтических клеток, где осуществлена иден¬
тификация маркеров клеточных линий, а относитель¬
ная легкость дезаггрегации гематопоэтических тканей,
таких как костный мозг, переводит выделение этих кле¬
ток в разряд осуществимых задач (см. протокол 23.22).
Выделение и культивирование стволовых клеток из
плотных тканей более проблематичны, но к настоящему
времени при развитии соответствующих методов до¬
стигнут прогресс в идентификации и культивировании
таких клеток, например, из молочной железы [Welm
et al., 2003]. Одной из проблем является недостаток
маркеров стволовых клеток в плотной ткани, однако и
в этой области имеются опредленные сдвиги [Zhou et
al., 2001; Potten t al., 2003], открывающие возможности
для идентификации и выделения клеток.
23.7.1. Эмбриональные стволовые клетки
Эмбриональные клетки из организмов различных ви¬
дов, в частности мышей, раньше широко использова¬
лись для исследования дифференцировки [Martin &
Evans, 1974; Martin, 1975, 1978; Rizzino, 2002; zur
Nieden et al., 2003], поскольку они могут диффе¬
ренцироваться до ряда различных клеточных типов
(мышцы, кости, нервы и т.д.). Те клетки, которые
прошли пассаж на животных, приводили к формиро¬
ванию тератом, аналогичных спонтанным тератомам
человека. Клетки, выращенные на фидерном слое,
например мышиные фибробласты SCO, будут проли¬
ферировать, но не будут дифференцироваться. Когда
клетки растут на желатине без фидерного слоя или
в неадгерентных пластиковых чашках, образуются
узелки, которые в конечном счете дифференцируют¬
ся. Фактор, ингибирующий лейкемию (LIF), видимо,
является одним из основных регуляторных факторов,
который удерживает клетки в состоянии стволовых
клеток, в то время как ретиноиды, витамин D3 и
плоскостные полярные компоненты индуцируют ли-
ния-специфичную дифференцировку у клеток мыши
и человека [Draper et al., 2002].
В настоящее время получены культуры стволовых
клеток человеческого эмбриона, обнаружены пути их
дифференцировки, что открывает перспективы для их
использовании в восстановлении тканевых поврежде¬
ний [Thomson et al., 1998; Rippon & Bishop, 2004].
23.7.2. Мультипотентные стволовые
клетки взрослого организма
Кроме того, имеется большое количество сообщений
о выделении мультипотентных стволовых клеток из
ряда взрослых тканей, включая костный мозг [Suva
et al., 2004], печень [Deng et al., 2003], головной мозг
[Vescovi et al., 2002; Greco & Recht, 2003], мыщцы [Cao
et al., 2003] и пупочный канатик [Sanchez-Ramos, 2002;
Newman et al., 2003]. Идентификация мультипотент¬
ных стволовых клеток из взрослых тканей открывает
неожиданно обширную область в биологии стволовых
клеток, ранее закрытую из-за существования концеп¬
ции о тканеспецифичности клеточной регенерации.
Это дает начало многим волнующим перспективам
для понимания коммитирования и дифференциров¬
ки взрослых клеток-предшественников, но также
поднимает ряд важных вопросов. Если в головном
мозге существуют стволовые клетки, то почему не за¬
мещаются нейроны? Если существует потенциал для
циркуляции стволовых клеток для репопуляции дру¬
гих тканей, то почему сателлитные клетки скелетных
мышц не замещают кардиомиоциты при повреждении
сердца, если только они не введены искусственно?
В чем биологический смысл, с эволюционной точки
зрения, регенеративной способности и мультипотен-
тности стволовых клеток во многих тканях, которые
никогда не используются? Та концепция, по которой
стволовые клетки могут изменять свою способность
к дифференцировке, но не прямо от одной линии к
другой (например, от астроглиальной клетки до оли-
годендороглии или от эритроидной к миелоидной), а
от производных одного зародышевого листка к про¬
изводным другого зародышевого листка (от таких как
нейроэктодермальные до мезодермальных [Wurmser
et al., 2004] или эндодермальных в нейроэктоджер-
мальные [Deng et al., 2003]), так сильно противоречит
установленной парадигме линейной привязанности,
что многие считают ее причиной атипичного развития
стволовых клеток в эктопичном участке, объясняя
это тем, что введенные стволовые клетки сливаются с
резидентными клетками-предшественниками [ Alvarez-
Dolado et al., 2003; Greco & Recht, 2003]. Тем не менее,
хотя такое слияние клеток принципиально возможно
и играет роль в дифференцировке некоторых эктопич-
ных стволовых клеток, доказательства пластичности
стволовых клеток весьма убедительны [Wurmser et
al., 2004]. В свете данных, доказывающих, что клетки-
предшественники, которые ранее считались линейно
23.7. Стволовые клетки
471
коммитированными, могут претерпевать обратные
стадии и реверсироваться или конвертироваться в
мультипотентные стволовые клетки (см. разд. 17.1),
кажется возможным, что стволовые клетки из одной
ткани могуть превращаться в различные стволовые
клетки с различной потентностью в эктопичном сайте,
возможно, через стадию тотипотентных клеток.
23.7.3. Источники стволовых клеток
и работа с ними
Мышь. Многие из первоначальных клеточных линий
карциномы, такие как F9 и Р19, были выделены из мы¬
шиных тератокарцином, которые затем периодически
трансплантировались под почечную капсулу [Martin &
Evans, 1974]. Линии эмбриональных стволовых кле¬
ток (ES), такие как ES-D3, J1 и R1, были получены
из внутренней клеточной массы преимплантацион-
ного эмбриона [Evans &Kaufman, 1981; Martin, 1981],
клеточные линии эмбрионального листка (EG) были
получены из легочного зародышевого листка [Matsui et
al., 1991; Resnick et al., 1992]. Эти культуры могут быть
культивированы в недифференцированном состоянии
на фидерном слое клеток STO или в присутствии LIF
[Schamblott et al., 2002]. В долгоживущей культуре в
отсутствие LIF или фидерного слоя имеет место спон¬
танная дифференцировка, в частности, если допустить
образование клеточных агрегатов. Дифференцировка
может быть спровоцирована действием ретиноидов,
плоских полярных компонентов, таких как DMSO
или НМВА, дибутирил цикличной АМФ (dbcAMP),
витамина D3, и некоторых цитокинов.
Человек. Клетки человеческой тератокарцино-
мы, такие как NTera-2 [Paquet-Durand et al., 2004] и
ES-клетки [Thomson et al., 1998], так же, как и EG-клет-
ки [Schamblott et al., 1998], могут также быть выращены
в культуре в присутствии LIF, FGF-2, и форсколина
[Schamblott et al., 2002] с возможностью дифферен¬
цировки и значительным потенциалом для тканевой
инженерии [Laslett et al., 2003; Rippon & Bishop, 2004].
Культуры, полученные из спонтанно развившихся
эмбриональных тел, выделенные из EG-культур и де¬
загрегированные при помощи коллагеназы и диспазы,
дают начало EBD- клеточным линиям (embryoid body
derived). Эти линии, видимо, легче поддерживаются и
имеют более длительную продожительность жизнен¬
ного цикла в культуре [Schamblott et al., 2002]. Они
также способны к дифференцировке по ряду различных
направлений и могут иметь значительный потенциал
для тканевой инженерии [Kerr et al., 2003].
Стволовые клетки могут быть выделены из ряда
тканей взрослого человека при помощи FACS или
магнитного сортинга клеток, несмотря на недостаток
линейных маркеров, но благодаря наличию таких мар¬
керов стволовых клеток, как Sca-1 [Kawada et al., 2004;
Oh et al., 2004], Kit [Das et al., 2004; Takahashi et al.,
2004], c_Met [Suzuki et al., 2004], или выбросу Hoechst
33342 [Bhatt et al., 2003].
Глава 24
КУЛЬТУРА ОПУХОЛЕВЫХ КЛЕТОК
24.1. ПРОБЛЕМЫ КУЛЬТИВИРОВАНИЯ
ОПУХОЛЕВЫХ КЛЕТОК
Получение культуры клеток из опухолей, в частности
спонтанных человеческих опухолей, имеет те же про¬
блемы, что и культуры специализированных клеток
из человеческих тканей. Опухолевые клетки должны
быть отделены от нормальных соединительнотканных
клеток, предпочтительно путем использования селек¬
тивной среды, которая поддерживает рост опухолевых
клеток, но не подходит для нормальных клеток. Хотя
разработка селективных сред для нормальных клеток
имеет значительные успехи (см. разд. 10.2.1 и гл. 23),
достижения в опухолевых культурах ограничены рядом
различий как между образцами, так и в самой структуре
образцов опухолевой ткани. Часто удивляет то, что
опухоли, которые образуются in vivo, в результате раз¬
множения клеток, не подчиняющихся регуляторным
сигналам организма, не могут расти in vitro.
Существует ряд возможных причин неспособности
к выживанию некоторых опухолевых культур. Их
требования к наличию питательных веществ могут
отличаться от тех, которыми обладают эквивалентные
им нормальные клетки. С другой стороны, попытки
удалить стромальные клетки могут лишить опухо¬
левые клетки матрикса, питания или необходимых
для выживания сигнальных молекул. Кроме того, в
культуре опухолевых клеток сильное разведение для
обеспечения достаточного количества питательных
веществ на каждую клетку может также привести к
снижению концентрации аутокринных факторов рос¬
та, производимых клетками. Строго говоря, истинные
аутокринные факторы должны быть независимы от
разведения, если они секретируются на поверхности
клетки и действуют на эту же клетку, но, возможно,
некоторые так называемые аутокринные факторы
являются на самом деле гомокринными (см. разд. 3.5),
т. е. они действуют на рядом расположенные подобные
клетки, а не только на саму клетку, высвобождающую
их. Таким образом, требуется близкое взаимодействие
клеток в популяции. Взаимодействие с определенны¬
ми типами стромальных клеток может обеспечивать
паракринные взаимодействия, если строма способна
продуцировать требуемые факторы роста, как спон¬
танно существующие, так и возникающие в ответ на
сигналы от опухолевых клеток, необходимые для их
выживания.
Возможно, что опухолевые клетки не имеют тонкой
зависимости от факторов роста как нормальные клетки
аналогичной ткани, из которой произошла опухоль.
Опухолевые клетки могут продуцировать эндогенные
аутокринные факторы роста, такие как TGF-a, и искус¬
ственное введение экзогенных факторов роста, таких
как EGF, может привести к конкуренции за одни и те
же рецепторы. Более того, ответ опухолевой клетки на
фактор роста или гормон будет зависеть от того, какие
еще факторы роста присутствуют в ткани, некоторые
из которых, возможно, продуцируются самой опухо¬
левой клеткой, и от состояния клетки. Нормальная
клетка, способная к экспрессии супрессоров роста, и
генов старения, может отвечать на эти сигналы иначе,
чем опухолевые клетки, в которых один или более из
этих генов инактивированы или мутантны и в которые
гиперэкспрессируют антагонистические онкогены,
инициирующие неконтролируемый рост.
Поэтому существует много возможных причин
того, что популяция опухолевых клеток иначе, чем
нормальные клетки той же линии, отвечает на пи¬
щевые и митогенные воздействия окружения. Для
подтверждения этого различия требуется больше ин¬
формации об опухолевых клетках, однако в результате
существования гетерогенности опухоли первой задачей
является снижение этой гетерогенности. Имеющиеся
сегодня данные о пищевых потребностях индивидуаль¬
ной опухолевой линии клеток пока не могут успешно
использоваться в терапевтических целях. Поэтому
большой прогресс в терапии может быть достигнут
в результате изучения ответа опухолевых клеток на
воздействие факторов роста и исследования различий
в путях внутриклеточной передачи сигнала.
Возможно, что основная часть клеток в опухоли
имеет ограниченное время жизни в результате ге¬
нетических аберраций, терминальной дифференци¬
ровки, апоптоза и естественного старения, и только
некоторые клетки как аналоги популяции стволовых
клеток в нормальной ткани обладают способностью к
выживанию. Низкая концентрация клеток в культуре
может привести к снижению количества этих клеток,
а также их взаимодействию с другими клетками, так
что выживание их становится невозможным. Клетки
многоклеточных животных, в отличие от прокариот,
плохо выживают при выделении из организма. Даже
опухолевые клетки еще продолжают оставаться эле¬
ментами многоклеточного органа, им может потре¬
24.2. Взятие образцов
473
боваться постоянное клеточное взаимодействие для
выживания. Судьба опухоли для хозяина определя¬
ется неконтролируемой инфильтрацией опухолевых
клеток в ткани и их колониальным ростом, однако
происхождение большей части клеточной популяции
может быть связано с относительно небольшим коли¬
чеством трансформированных стволовых клеток [Jones
et al., 2004; Al-Hajj et al., 2004]. Этот пул стволовых
клеток может быть таким малым, что его разведение
в эксплантате приводит к лишению клеток основных
необходимых условий для выживания, в частности па-
ракринных факторов роста, получаемых от стромаль-
ных элементов, и гомокринных факторов, от других
опухолевых субклонов.
Целью исследователя становится создание правиль¬
ных условий определенного пищевого и гормонального
окружения или, при невозможности этого, в предостав¬
лении некоторого поддерживающего окружения, — не¬
определенного, но тем не менее способного обеспечить
выживание соответствующей или репрезентативной
популяции. Существует тенденция использования
сыворотки и фидерных слоев для поддержания роста
опухолевых клеток, и только некоторые опухоли спо¬
собны расти на бессывороточной среде. Многие транс¬
формированные клетки не ингибируются действием
TGF-p. Поэтому удаление сыворотки из среды исполь¬
зуется для решения главной проблемы ингибирования
роста фибробластов. Адаптация среды, разработанной
для эквивалентных нормальных клеток, пока остается
наиболее логичным подходом к получению линий опу¬
холевых клеток, даже при добавлении минимального
количества сыворотки или кондиционированной среды,
полученной от других клеток [Dairkee et al., 1995].
24.2. ВЗЯТИЕ ОБРАЗЦОВ
Помимо решения проблемы избыточного роста со¬
единительной ткани или сосудистых клеток, которые
стимулируются к инвазии и пролиферации ростом
многих опухолей, получение культуры опухолевых
клеток требует отделения трансформированных клеток
от нормальных клеток эквивалентной ткани, которые
могут иметь аналогичные характеристики. Более того,
хотя любой раздел желудочного эпителия может рас¬
сматриваться как репрезентативный для данной специ¬
фической зоны слизистой желудка, опухолевая ткань, в
зависимости от генетических вариаций и естественной
селекции развития, обычно гетерогенна и состоит из
серий часто разнообразных субклонов, демонстриру¬
ющих значительное фенотипическое разнообразие.
Убедиться в том, что культуры, выделенные из данной
гетерогенной популяции, являются репрезентативны¬
ми, трудно, и никогда нет полной гарантии, если только
для исследования не берется опухоль целиком и выжи¬
ваемость не составляет 100%. Поскольку эти условия
практически недостижимы, на практике культура опу¬
холи представляет собой нечто среднее. Предполагая,
что репрезентативные субпопуляции сохраняются в
культуре и способны к взаимодействию, полученная
культура может быть подобна исходной опухоли.
Проблема селективности приобретает особое
значение, когда образцы берутся из вторичных мета¬
стазов, которые часто растут лучше, но могут не быть
типичными ни для первичной опухоли, ни для других
метастазов. Тем не менее интересно предположить, что
существует некая аналогия между метастатическими
проявлениями и эктопическим развитием стволовой
клетки (см. разд. 23.7.2).
В свете этой многогранной проблемы, стоящей на
пути культивирования опухолей, удивительно, что в
этой области вообще могут быть получены какие-то
достоверные данные. Однако они получены, это может
быть результатом: 1) вышеупомянутой автономии
опухолевой популяции, которая может обеспечить про¬
лиферацию опухолевых клеток в условиях, в которых
невозможно деление нормальных клеток; 2) возраста¬
ния размеров пролиферативного пула, который может
быть больше, чем у большинства нормальных тканей;
3) способности опухоли давать начало новой опухоли
как ксенотрансплантату у мышей с подавленным имму¬
нитетом и возрастанию успеха в выделении культуры
клеток из ксенотрансплантатов и 4) склонности давать
начало иммортализованным постоянным линиям, бо¬
лее выраженной у злокачественно трансформирован¬
ных, чем у нормальных клеток. Это последнее свойство,
более чем какие-то другие, позволяет вести широкие
исследования, выделяя популяции опухолевых кле¬
ток даже с относительно нормальными процессами
дифференцировки, несмотря на неопределенность их
отношений с опухолью, из которой они выделены.
Хотя неопределенность состояния постоянных
клеточных линий сохраняется, тем не менее они явля¬
ются ценным источником клеточных линий человека
для молекулярных и вирусологических исследований.
Вопрос о том, являются они репрезентативными для
прогрессивного развития опухоли, развитие которой
ускоряется в культуре, за счет таинственных популяций
стволовых клеток или это исключительно артефакт,
возникающий in vitro, еще должен быть решен. Полу¬
ченные клеточные линии определенно отличаются от
большинства опухолевых культур ранних пассажей, но
могут еще содержать значительное количество элемен¬
тов генотипа родительских клеток, от которых они про¬
изошли. Их бессмертность, с большей вероятностью,
устанавливается благодаря делеции или супрессии
генов, индуцирующих старение клеток [Pereira-Smith
& Smith, 1988; Goldstein et al., 1989; Holt et al., 1996;
Sasaki et al., 1996] и возрастанию теломеразной актив¬
ности [Bryan & Reddel, 1997; Bodnar et al., 1998], а не
сверхэкспресси генов, имеющих отношение к малиг-
низации как таковой.
24.3. ДЕЗАГРЕГАЦИЯ
Некоторые опухоли, типа человеческой карциномы
яичников, некоторые глиомы и многие трансплантиру¬
474
ГЛАВА 24. КУЛЬТУРА ОПУХОЛЕВЫХ КЛЕТОК
емые опухоли грызунов легко дезаггрегируются чисто
механическими способами типа пипетирования или
разделения на ситах (см. разд. 12.3.8), которые могут
также способствовать минимизации стромальной
контаминации, поскольку стромальные клетки часто
более прочно связаны с волокнистой соединительной
тканью. Многие из обычных человеческих карцином,
однако, являются твердыми или уплотненными, и
опухолевые клетки располагаются среди большого
количества волокон стромы, что делает механическую
дезагрегацию трудной, хотя возможной при соскреба-
нии среза плотной опухоли, успешно используемой для
высвобождения опухолевых клеток в так называемой
технике разбрызгивания (spillage) [Lasfargues, 1973;
Oie et al., 1996].
В большинстве случаев ферментативное разру¬
шение оказывается предпочтительнее по сравнению
с механической дезагрегацией. Хотя трипсин часто
использовался для этой цели, эффективность его про¬
тив волокнистой соединительной ткани ограничена,
и это может уменьшать эффективность отбора опухо¬
левых клеток [Lounis et al., 1994]; было обнаружено,
что неочищенная коллагеназа более эффективна для
нескольких различных типов опухоли [Dairkee et al.,
1997]. Ферментативное разрушение также высвобож¬
дает много стромальных клеток, что требует дополни¬
тельных методов ведения культуры для их устранения
(см. разд. 24.7; рис. 15.7г; цв. вкладку 23). Обработка
коллагеназой может быть проведена в течение не¬
сколько часов или даже дней в полной среде для роста
культуры (см. протокол 12.8).
Обширный некроз — также проблема опухолевой
ткани, с которой обычно не сталкиваются исследо¬
ватели, работающие с нормальной тканью. Обычно
прикрепление к субстрату жизнеспособных клеток поз¬
воляет удалить некротизированные при последующем
пассаже, но если количество некротического материала
велико и с трудом удаляется при иссечении, то для
того, чтобы удалить некротизированные клетки, может
оказаться необходимым использовать разделение в
Ficoll-Hypaque (см. протокол 12.10).
24.4. ПЕРВИЧНАЯ КУЛЬТУРА
Некоторые клетки, например макрофаги, прикрепля¬
ются к субстрату в процессе коллагеназного разруше¬
ния, но они могут быть удалены переносом суспензии
дезагрегированных клеток в свежий флакон, после
удаления коллагеназы. Прикрепившиеся клетки могут
остаться во флаконе. Их можно отдельно культивиро¬
вать, или облучить, или обработать митомицином С с
последующим использованием в качестве фидерного
слоя (см. разд. 14.2.3; цв. вкладки 6г, 11а, 156; прото¬
колы 23.2,23.4). Пересеянные клетки будут содержать
много стромальных клеток (главным образом, фиброб¬
ластов и эндотелиальных клеток), некоторые из кото¬
рых могут быть удалены путем повторного переноса
в свежий сосуд через 2-4 ч, поскольку опухолевые
клетки, в частности кластеры злокачественно изме¬
ненного эпителия, часто требуют большего времени
для прикрепления. Этот метод удаления методом се¬
рийных переносов, вообще говоря, только отчасти дает
успешный результат, но, тем не менее, обычно, требует
селективных условий культивирования для полного
удаления стромальных клеток. Тем не менее он может
иметь то преимущество, что позволяет использовать
стромальные клетки в качестве фидерного слоя.
Для удаления стромальной контаминации раньше
использовался физический метод разделения [Csoka
et al., 1995; Oie et al., 1996; см. также гл. 15], но, в об¬
щем, эти методы подходят, только если клетки будут
использоваться немедленно, поскольку в отсутствие
селективных условий обычно позже развивается стро-
мальный избыточный рост. Тем не менее показано, что
магнитный сортинг (см. разд. 15.3.2 и цв. вкладку 23) с
анти-фибробластными микробусинами (см. рис. 15.7г;
Miltenyi Biotech) является в этом случае эффектив¬
ным методом.
Клонирование как метод очистки имеет определен¬
ные ограничения, поскольку опухолевые клетки в пер¬
вичной культуре часто имеют низкую эффективность
посева (<0,1 %). В качестве метода выделения не только
опухолевых клеток, но и опухолевых стволовых клеток
было предложено суспензионное клонирование [Ham¬
burger & Salmon, 1977]. Однако к тому времени, когда
клон вырастает до достаточно большого количества
клеток, которые можно использовать для исследования
клетки могут подвергнуться значительным изменени¬
ям, и даже стать достаточно гетерогенными в результате
генетической нестабильности. Клонированные изоляты
из опухоли должны быть исследованы в совокупности
и даже в сокультуре для полной и достоверной интер¬
претации данных.
Имеются определенные сложности в получении
и размножении клеточных линий из первичных
клонов, в частности из клонов, выделенных методом
суспензионного клонирования. Может случиться, что
клетки, являясь клоногенными, при этом не будут
стволовыми, либо, будучи первоначально стволовы¬
ми, спонтанно перейдут в зрелое состояние в резуль¬
тате суспензионного роста и утраты регенеративной
способности. Тем не менее клеточные штаммы, кло¬
нированные прямо из опухоли, могли бы быть ценным
материалом для изучения опухолевого клонального
многообразия и взаимодействия. Эти штаммы могут
представлять собой перспективную область буду¬
щих исследований, в частности, при разработке и
использовании селективных условий для выделения
опухолевых стволовых клеток.
24.5. ХАРАКТЕРИСТИКА
Выделение клеток из опухоли может дать нача¬
ло нескольким различным типам линий клеток.
Помимо неопластических клеток, фибробластов
соединительной ткани, клеток сосудистого эндо¬
24.6. Размножение клеточных линий
475
телия и гладкомышечных клеток, инфильтрующие
лимфоциты, гранулоциты и макрофаги, так же как и
элементы нормальной ткани, в которой произошло
новообразование, — все эти элементы могут пережить
эксплантацию. Гематопоэтические компоненты редко
формируют линии клеток, хотя гематопоэтические
клеточные линии были получены из маленькой
клетки карциномы легкого, вызвав серьезное за¬
мешательство, потому что карцинома также имеет
тенденцию образовывать суспензионные культуры,
которые могут экспрессировать миелоидные марке¬
ры [Ruff & Pert, 1984]. Макрофаги и гранулоциты
являются хорошо прикрепляющимися и непролифе¬
рирующими клетками, поэтому они обычно теряются
в субкультуре. Гладкомышечные клетки не делятся
без соответствующих факторов роста и селективной
среды. Поэтому главным потенциальным контами-
нантом опухолевой культуры являются фибробласты,
эндотелиальные клетки и нормальные эквиваленты
неопластических клеток.
Среди этих контаминантов главную проблему со¬
ставляют фибробласты, которые легко растут в куль¬
туре и могут также отвечать на митогенные факторы
опухолевого происхождения. Точно так же эндотели¬
альные клетки, особенно в отсутствие фибробластов,
могут давать ответ на выделенные опухолью факторы
ангиогенеза и легко размножаться. Труднее определить
роль нормального эквивалента клеток, поскольку их
подобие неопластическим клеткам приводит к тому,
что соответствующие эксперименты трудно проана¬
лизировать. Критерии оценки клеток должны быть
выбраны так, чтобы исключить нормальные клетки.
Например, эндотелиальные клетки являются пози¬
тивными по фактору VIII, подвергаются контактному
ингибированию и рост их культуры чувствителен к
ограничению плотностью клеток. Фибробласты имеют
характерную веретеновидную форму, рост их является
ограниченным плотностью популяции (хотя меньшей
степени, чем эндотелиальные клетки), имеют конеч¬
ную продолжительность жизни 50 поколений или
около этого, продуцируют коллаген типа I и строго
диплоидны.
В целом, нормальные клеточные компоненты,
фенотипически эквивалентные опухолевым клеткам,
труднее выделить и устранить. Клетки будут дипло¬
идны, хотя некоторые опухолевые клетки могут быть
близки к диплоидным. Они обычно зависимы от
прикрепления к субстрату и будут иметь конечную
продолжительность жизни, хотя опять же существуют
случаи постоянных клеточных линий нормальных
эпителиальных клеток [Boukamp et al., 1988], и, если
клетки эпителиальные, то они, более вероятно, будут
ингибироваться TGF-fi, содержащимся в сыворотке.
Опухолевые клетки, вероятно, обнаружат генетичес¬
кие отклонения, такие как амплификация онкогенов,
транслокаций, и делеций гена- супрессора которые
можно обнаружить FISH (см. протокол 27.9). Ве¬
роятно также, что опухолевые клетки ангиогенны и
обнаруживают более высокую экспрессию урокиназа-
подобных активаторов плазминогена (иРА), а также
обладают инвазивной активностью (см. разд. 18.6).
В поведенческом аспекте способность неоплас¬
тических клеток расти на предварительно сфор¬
мированном монослое нормальных клеток того же
типа — хороший критерий идентичности для опухо¬
левых клеток и потенциальная модель для их распоз¬
навания. Нормальные клетки также обеспечивают
формирование фидерного слоя, для поддержания
роста опухолевых клеток. Глиома, например, будет
хорошо расти (лучше, чем на пластмассе в некоторых
случаях) на предварительно сформированном моно¬
слое нормальных глиальных клеток [MacDonald et
al., 1985], но их нормальные копии не обладают этим
качеством. Этот может быть верно также для клеток
гепатомы и карциномы кожи.
Нормальные клетки в условиях предельно на¬
сыщенного роста имеют тенденцию к содержанию в
популяции небольшой фракции делящихся клеток
(см. разд. 18.5.2 и 21.11), тогда как неопластические
клетки продолжают быстро делиться после достижения
конфлюэнтности. Поддержание культуры при высо¬
кой плотности может иногда обеспечивать условия
для избыточного роста неопластических клеток (см.
разд. 25.3.6 и протокол 18.3).
24.6. РАЗМНОЖЕНИЕ
КЛЕТОЧНЫХ ЛИНИЙ
Первичные культуры клеток карциномы не всегда
могут быть подвергнуты пересадке с трипсинизацией,
и многие клетки в первичной культуре могут быть
не способны к размножению, из-за генетических или
фенотипических отклонений, конечной дифференци¬
ровки, или недостатка питательных веществ. Однако
некоторые первичные культуры, выделенные из опу¬
холи, могут быть субкультивированы, что открывает
огромные возможности. Существование субкультуры
опухолевых клеток подразумевает, что в субкультуре
нет других клеток, которые имеют избыточный рост.
Возможен вариант, когда исследуемые клетки имеют
более высокую скорость роста, чем загрязняющие их
нормальные клетки, поэтому они могут быть доступны
для клонирования или других методов селекции куль¬
туры (см. разд. 10.2.1,14.6,24.7.2).
Одним из главных преимуществ субкультуры
является возможность ее роста и размножения. На¬
ращенные клетки могут быть заморожены. Реплици¬
рованные культуры используют для характеризации
и исследования определенных параметров, таких как
геномные изменения, изменения в экспрессии генов,
химиочувствительность и инвазивность. Отрицатель¬
ным качеством субкультуры является последующее
развитие с серьезными отклонениями от первона¬
чального фенотипа клеток, выделенных из опухоли,
вследствие свойственной клеткам генетической
неустойчивости и селективной адаптации клеток к
окружению в культуре.
476
ГЛАВА 24. КУЛЬТУРА ОПУХОЛЕВЫХ КЛЕТОК
24.6.1. Постоянные клеточные линии
Одним из главных критериев неопластического про¬
исхождения культуры является способность форми¬
ровать постоянную линию клеток, которая обычно
анеуплоидна, гетероплоидна, нечувствительна к
ограничению роста плотностью клеток, не зависит
от прикрепления к субстрату и часто туморогенна
(см. разд. 18.6.2). Отношения этой клеточной ли¬
нии к первичной культуре и родительской опухоли
трудно оценить, т. к. клетки не всегда типичны для
популяции опухоли. Клетки постоянной линии могут
представлять собой: 1) дальнейшую трансформацию,
стимулируемую адаптацией к культуре, возможную
в соответствии с непостоянными генотипическими
характеристиками опухолевой клетки, или 2) опреде¬
ленную субгруппу или стволовые клетки популяции
опухоли. В настоящее время вторая возможность
кажется более вероятной [Petersen et al., 2003], так
как появление постоянной клеточной линии (часто из
колоний в пределах монослоя) предполагает, что эта
постоянная линия является результатом иммортали-
зации малой субгруппы клеток популяции опухоли,
развившейся благодаря обеспечению соответству¬
ющих условий для их размножения.
Способность формировать постоянные линии
клеток является полезным критерием для подтверж¬
дения злокачественного происхождения, и некоторые
авторы подтверждают что характеристики клеточных
линий (например, туморогенность, гистологические
изменения, химиочувствительность и т.д.) коррели¬
руют с опухолевым происхождением [Tveit & Pihl,
1981; Minna et al., 1983]. В любом случае эти кле¬
точные линии представляют собой полезный экспе¬
риментальный материал, хотя время, требуемое для
их развития, делает непосредственное клиническое
применение трудным. Линии опухолевых клеток
описаны в ряде книг Masters и Palsson [Masters &
Palsson, 1999-2000].
своим успехом продукции пептидных факторов роста
мелкоклеточной формой рака легких, для которых
разработана эта среда. Некоторая доля биопсий, хотя
и не все, будут расти на чистой среде HITES; другие
будут выживать при добавлении низкой концент¬
рации сыворотки (например, 2,5%). Среда HITES
является модифицированной средой RPMI 1640 с
добавлением гидрокортизона, инсулина, трансфер-
рина, эстрадиола и селена. Наиболее важными из
этих компонентов являются, видимо, селен, инсулин
и трансферрин, они встречаются в составе многих
бессывороточных сред (см. табл. 10.2). Группа NCI
также производит селективную среду для аденокар¬
циномы, по общему мнению, подходящую для легких,
толстой кишки и, возможно, многих других карцином
[Brower et al., 1986]. Эта среда также в основе имеет
RPMI 1640, в которую добавлены селен, инсулин и
трансферрин, а также гидрокортизон, EGF, трии-
одтиронин, BSA и пируват натрия (см. табл. 10.2).
Другие селективные среды успешно используются
для карциномы простаты [Uzgare et al., 2004], желч¬
ного пузыря [Messing et al., 1982], и молочной железы
[Ethier et al.,1993].
Другие типы селективных сред основаны на ме¬
таболическом ингибировании роста фибробластов и
не являются специфически оптимизированными для
какого-либо типа опухолей (см. разд. 14.6). Тем не
менее пока не найдены ингибиторы, которые будут
эффективны, за исключением использования моно¬
клональных антител против фибробластов, разрабо¬
танных Edwards et al. [1980] и Paraskeva et al. [1985].
Эти антитела успешно используются в выделении
культуры клеток из рака гортани и толстой кишки.
Показан также положительный эффект использова¬
ния антител для позитивной селекции эпителиальных
клеток или негативном сортинге стромальных клеток
и удалении их из суспензии опухолевых клеток путем
пэннинга или магнитного сортинга (см. разд. 15.3.2,
24.4; рис. 15.7г; цв. вкладку 23).
24.7. СЕЛЕКТИВНАЯ КУЛЬТУРА
ОПУХОЛЕВЫХ КЛЕТОК
Три главных методических подхода в культуре тканей
были адаптированы к культуре опухолевых клеток:
использование селективных сред (см. разд. 10.2.1,
14.6), применение конфлюэнтных фидерных слоев
(см. разд. 24.7.2), и суспензионное клонирование (см.
разд. 14.3,14.8.5).
24.7.1. Селективные среды
Из-за присущих опухолевым клеткам проблем ва¬
риабельности и гетерогенности существует лишь
несколько сред, которые разработаны для них как
селективные. Одной из таких сред является HITES
[Carney et al., 1981; см. табл. 10.2], которая обязана
24.7.2. Конфлюэнтные
фидерные слои
Метод конфлюэнтных фидерных слоев (см. рис. 14.4
и 14.10; цв. вкладку 6в, г), возможно, более чем любой
другой метод, успешно применяется для культиви¬
рования опухолевых тканей разных типов. Smith и
другие [например, Lan et al., 1981] использовали кон¬
флюэнтные фидерные слои эмбриональной тонкой
кишки человека FHS74INT для того, чтобы вырастить
эпителиальные клетки карциномы молочной железы, в
среде, кондиционированной другими клеточными ли¬
ниями. В более поздних сообщениях предполагается,
что использование селективных культур в MCDB 170
является более воспроизводимым подходом [Ham¬
mond et al., 1984].
Фидерные слои мышиных клеток ЗТЗ или эмбри¬
ональные фибробласты STO были успешно использо¬
24.7. Селективная культура опухолевых клеток
477
ваны для культивирования клеток рака груди, толстой
кишки и базалиомы [Rheinwald & Beckett, 1981; Leake
et al., 1987]. На том же основании можно освободить от
любых контаминаций нормальным эпителием клетки
карциномы молочной железы, если посадить их на
конфлюэнтную культуру нормальных клеток эпителия
молочной железы с прекратившимися за счет контакт¬
ного ингибирования ростом (см. протокол 23.3).
ПРОТОКОЛ 24.1. Выращивание клеток
на конфлюэнтном фидерном слое
Схема
Обработать фидерные клетки в середине экспонен¬
циальной фазы митомицином С и пересеять клетки
для получения конфлюэнтного монослоя. Посеять
на конфлюэнтный монослой опухолевые клетки,
отделенные от биопсийного образца коллагеназ-
ным расщеплением или из первичной культуры
трипсином (рис. 24.1). Колонии из эпителиальной
опухоли могут образовываться от 3 недель до 3 ме¬
сяцев. Фибросаркома и глиома не всегда образуют
колонии, но могут инфильтрировать фидерный слой
и постепенно давать сильный рост
Материалы
Стерильные:
• Фидерные клетки (например, ЗТЗ, STO, ЮТ 1/2,
или FHS74Int)
• Митомицин С (Sigma), 1 мг/мл
Замечание: При использовании фидерных клеток
впервые рекомендуется сделать кривую доза-ответ с
митомицином С для того, чтобы быть уверенным, что
выбранная доза позволяет фидерному слою выжить
в течение 2-3 недель, но не дает клеткам в фидерном
слое возможности дальнейшей репликации после
примерно двух периодов удвоения. В этом случае
поступайте следующим образом:
а) Обработайте клетки митомицином С в кон¬
центрации 1-100 мкг/мл в течение ночи (18 ч)
в 25-см2 флаконе
б) Трипсинизируйте клетки и пересейте все со¬
держимое из 25-см2 флакона в 75-см2 флакон
с 20 мл свежей среды.
в) Выращивайте клетки в течение 3 недель, сме¬
няя среду дважды в неделю
г) Окрасьте культуру и проверьте выживание
колоний
• Ростовая среда
• Коллагеназа, 2 ООО Ед/мл, степень очистки CLS
(Worthington) или ее эквивалент
• Трипсин, 0,25%, в PBSA
• Биопсийный образец опухоли или первичная
культура
• Изогнутые пинцеты
• Скальпели с лезвиями #22
• Чашки Петри для иссечения, как для первичной
культуры
Протокол
1. Вырастите фидерные клетки до 80% конфлюэн¬
тности в шести 75-см2 флаконах.
2. Добавьте митомицин С до получения необхо¬
димой конечной концентрации, обычно около
5 мкг/мл.
3. Инкубируйте клетки в течение ночи (-18 ч) с
митомицином С.
4. Удалите среду с митомицином С, промойте мо¬
нослой свежей средой.
5. Вырастите клетки в течение последующих 24-
48 ч.
6. Трипсинизируйте клетки и пересейте их в
25-см2 флаконе при концентрации 5х105 кл./мл
(1х105кл./см2>). Инкубируйте культуру в течение
24 ч.
7. Если вы используете биопсийный материал, об¬
разец следует иссечь и поместить в коллагеназу
на шаге 2
8. Посейте суспензию клеток из биопсии примерно
20-100 мг/флакон, в два 25- см2 флакона, так,
чтобы каждый флакон содержал 6 мл суспензии.
Отберите по 1 мл суспензии из каждого флакона
и добавьте к4 мл среды в оставшихся двух флако¬
нах. Третью пару флаконов следует сохранить как
контроль сохранности фидерного слоя и выжи¬
вания клеток после обработки митомицином С.
Если вы используете первичную культуру, трип¬
синизируйте или отделите клетки в растворе кол¬
лагеназы, конечная концентрация 200 Ед/мл (см.
протокол 12.8), посейте на фидерный слой с кон¬
центрацией 105 кл./мл в двух флаконах и 104 кл./мл
в двух флаконах. Если клетки выделены из глиомы
(цв. вкладка 156) или фибросаркомы (цв. вкладка 1г),
колонии могут не появляться, поскольку клетки опу¬
холи свободно мигрируют среди фидерных клеток.
Выживание клеток этих опухолей будет подтверж¬
дено только путем субкультивирования клеток без
фидерного слоя (к этому времени нормальные клетки
элиминируются).
Важно подтвердить видовую специфичность любой
клеточной линии, выделенной этим методом, для того
чтобы исключить случайную контаминацию устой¬
чивыми клетками из фидерного слоя. Вид исходного
источника клеток может быть подтвержден хромосом¬
ным анализом (см. протокол 16.7) и изоферментным
электрофорезом (см. протокол 16.10), если фидерный
слой имеет иное видовое происхождение, отличное от
первичной культуры. Если фидерные клетки отно¬
сятся к тому же виду, что и используемая первичная
культура, то необходимо исследование профиля ДНК
478
ГЛАВА 24. КУЛЬТУРА ОПУХОЛЕВЫХ КЛЕТОК
Инкубируйте тщательно измель¬
ченные кусочки в коллагеназе в
течение 1-5 дней
Диспергируйте осторожным пи-
петированием, перенесите в про¬
бирку и оставьте для осаждения
на 2-3 мин
Соберите супернатант, центрифу¬
гируйте и ресуспендируйте осадок
и посейте на сформированный
фидерный слой
Ресуспендируйте в среде повто¬
рите осаждение-ресуспендирова-
i ние для отмывания коллагеназы,
I наконец ресуспендируйте в пол¬
ной ростовой среде
Облучите фидерный слой или
обработайте митомицином С
Трипсинизируйте и засейте фла¬
коны или чашки Петри для полу¬
чения конфлюэнтного монослоя
Посейте на сформи¬
рованный конфлюэнт¬
ный слой
Диспергированные клетки Агрегаты («органоиды»)
Инкубируйте 1-3 недели (в зависимости от скорости роста)
Рис. 24.1. Конфлюэнтные фидерные слои. Эпителиальные кластеры после обработки коллагеназой будут формировать ко¬
лонии эпителиальных клеток на конфлюэнтных фидерных слоях, таких как фетальный эпителий тонкой кишки (FHS74Int),
нормальной глии человека или облученные клетки ЗТЗ или STO cells. Диспергированные клетки, даже при контаминации
стромальными клетками, могут также образовывать колонии на конфлюэнтных фидерных слоях со значительным ограни¬
чением избыточного роста стромальных клеток. Селекция проводится в отношении стромальных компонентов, при этом не
затрагивает клеток нормального эпителия
(см. протокол 16.9) или ДНК-фингерпринтинга (см.
протокол 16.8) как фидерного слоя, так и любой куль¬
туры, которая была получена из первичной культуры.
Затем следует провести сравнение результатов с ис¬
следованием биопсии или любой ткани, полученной
от донора.
24.7.3. Суспензионное клонирование
Трансформация клеток in vitro приводит к возраста¬
нию их клоногенности в агаре (см. разд. 14.3, 18.5.1);
показано также, что туморогенность корррелирует
со способностью клонирования в метилцеллюлозе
[Freedman & Shin, 1974] (см. протокол 14.5). По¬
скольку клетки могут быть клонированы в суспензии
прямо из дезагрегированной опухоли [Hamburger &
Salmon, 1977] или, по крайней мере, образовывать
больше колоний, чем нормальные клетки (которые,
возможно, и не будут клонами), постольку суспен¬
зионное клонирование может рассматриваться как
потенциально селективный метод. Тем не менее
эффективность колониеобразования часто бывает
очень низкой (часто <0,1%), а клетки, выделенные из
колоний, размножаются с трудом.
Хотя этот метод не приводит к образованию клеточ¬
ных линий, он использовался раньше для скрининга
лекарственных препаратов в отношении биопсийных
образцов (см. разд. 4.1). FACS или иммуномагнитный
положительный сортинг клоногенных клеток путем
экспрессии маркеров клеток, таких как Sca-1, Kit
[Takahashi et al., 2004], и транспортер ABC [Zhou et al.,
2001] (см. также разд. 23.7.3) и отрицательный сортинг
для более дифференцированных линейных маркеров
может позволить обогатить пул предполагаемых ство¬
ловых клеток, которые могут стать лучшей мишенью
для изучения молекулярных механизмов действия
лекарственных препаратов и их скрининга.
24.7.4. Гистотипическая культура
Отдельно от собственно органной культуры (разд. 25.2),
которая имеет не больше фундаментальных отличий от
опухолевой ткани, чем нормальная ткань, существуют
методы, специфически применимые к опухолевой
культуре: сфероидная культура (см. протокол 25.2)
и культура в фильтрующих колодцах (лунках) (см.
протокол 25.4).
Сфероиды. Нормальные стромальные клетки не
образуют сфероидов или даже не включаются в состав
сфероидов, полученных из опухоли. Таким образом,
культура клеток, выделенная из опухоли, которой
позволяют образовывать сфероиды на неадгезивных
субстратах, таких как агароза (см. протокол 25.2), будет
иметь тенденцию к избыточному росту по сравнению
со стромальными компонентами. Некоторые культуры
из рака молочной железы и мелкоклеточной легочной
карциномы могут дать сфероиды или неправильные
клеточные органоиды, которые всплывают и могут
быть собраны из верхнего слоя среды, при этом стро¬
мальные элементы останутся внизу. Тем не менее сфе¬
роиды или органоиды не всегда быстро появляются в
культуре, и иногда требуются недели или даже месяцы,
чтобы образовались предположительные производные
из малой клеточной популяции в опухоли. В других
случаях, например для карциномы молочной железы,
органоиды могут представлять собой нормальные
эпителиальные компоненты [Speirs, 2004]. Генерация
24.7. Селективная культура опухолевых клеток
479
сфероидов возникает не во всех опухолевых культу¬
рах, но уже описана для нейробластомы, меланомы и
глиомы (см. разд. 25.3.3). Их потенциал для получения
клеточных линий еще не полностью использован,
тем не менее для исследовательских целей большее
внимание уделяется прикрепленным монослоям и
суспензионным колониям в агаре.
Вкладыши для фильтровального колодца. Вкла¬
дыши для фильтровального колодца созданы для того,
чтобы воссоздавать клеточно-матриксное взаимодей¬
ствие в тканях, из которых получены клетки. Таким
образом, они представляют собой идеальную модель
для исследований инвазивности (см. разд. 18.6.3),
ангиогенеза (см. разд. 18.6.4), и других нарушений
клеточных взаимодействий в опухоли.
возможность получения линии «голых» мышей или
имеются препятствия для проведения неонатальной
тимэктомии и облучения, то ксенотрансплантаты
следует рассматривать как первый шаг в генерации
культуры. Хотя при этом может быть получено только
небольшое количество опухолей, выделенную опухоль,
возможно, будет легче вести в культуре и можно будет
сделать несколько попыток выделения культуры с пос¬
ледующим пассажем культуры опухоли через мышей.
Тем не менее следует проявить особую тщательность,
поскольку при выделении клеток фидерного слоя из
мышей следует убедиться, что клеточные линии, вы¬
жившие в итоге, являются человеческими, а не мыши¬
ными. Подтверждение происхождения производится
методами изоферментного и хромосомного анализа
(см. разд. 16.5).
24.7.5. Ксенотрансплантат
Когда культуры получают из опухолей человека, то
недостаток материала и редкая возможность повторить
биопсию затрудняют повторное получение клеток и тем
более исключают многократное получение культуры
одной и той же опухоли. Рост некоторых опухолей у
иммунодепрессивных животных позволил разрабо¬
тать альтернативный подход [Rofstad, 1994], который
позволяет получить значительно большее количество
опухолевой ткани. Было обнаружено, что культуры
могут быть инициированы гораздо легче путем извле¬
чения ее из ксенотрансплантата, перевитого животному
из родительской опухоли, хотя до настоящего времени
неясно, происходит это благодаря доступности боль¬
шего количества материала, обогащению материала
трансформированными клетками, прогрессии опухоли
или модификации опухолевых клеток в организме
гетерологичного хозяина (например, под действием
мышиных ретровирусов).
Используется два главных типа хозяев: генети¬
чески бестимусные «голые», которые являются де¬
фицитными по Т-клеткам [Giovanella et al., 1974], и
неонатально тимэктомированные животные, которые
затем подвергаются облучению и обрабатываются
цитозинарабинозидом [Selby et al., 1980; Fergusson et
al., 1980]. Первый тип хозяев дорог для приобретения
и сложен для содержания, но поддержание опухоли
при этом длительнее. Тимэктомированные мыши
более хлопотны при подготовке, но дешевле и проще
получить их в большом количестве. Тем не менее они
могут вновь стать иммунокомпетентными и, в конеч¬
ном итоге, через несколько месяцев отторгнуть при¬
витую опухоль. Приживаемость опухоли улучшается
путем использования мышей, которые, помимо тимуса,
лишены также селезенки благодаря генетическим из¬
менениям (например, мыши scid) или спленэктомии,
либо путем сублетального облучения «голых» мышей.
Имплантация с использованием фибробластов или
Матригеля также, как сообщают, улучшает прижива¬
емость опухоли [Topley et al., 1993]. Если существует
24.7.6. Характеризация культур
опухолевых клеток
Общие характеристики, которые могут быть использо¬
ваны для идентификации опухолевых клеток в куль¬
туре, описаны в гл. 18 (см. разд. 18.3-18.6 и табл. 18.1).
Хотя эти характеристики часто выражены в постоянных
клеточных линиях, их обнаружение в культурах ранних
пассажей может быть более сложным в результате боль¬
шей гетерогенности. Может потребоваться определение
специфических генетических нарушений, — полисомия,
хромосомные делеции, транслокации, суперэкспрессия
мутагена и мутация или делеция гена опухолевого роста
(см. разд. 27.8.1). Подобным образом, гиперэкспрессия
продуктов онкогена, таких как мутантный ген р53 или
erb-В, может быть обнаружена методами иммунного
окрашивания, приготовления цитологического препа¬
рата либо проточной цитометрии.
24.7.7. Сохранение тканей замораживанием
Часто оказывается затруднительным использовать
весь объем ценного материала, который дает большая
биопсийная масса. В этих случаях можно сохранить
ткани замораживанием.
ПРОТОКОЛ 24.2. Замораживание биопсии
Схема
Измельчить <ш]Щ}^^фаботать кусочки DMSO и
заморозить аликвотывжидком азоте.
Мате/шшт
Стермльнью:
* Биопсия
• Пластиковые ампулы, 1,2 мл (Nalge Nunc)
480
ГЛАВА 24. КУЛЬТУРА ОПУХОЛЕВЫХ КЛЕТОК
• DBSS (см. Приложение I)
• Питательная среда для сбора материала (При¬
ложение I)
• DMSO (самостерилизующийся, если помещен в
стерильный контейнер)
• Инструменты (скальпели, пинцеты, чашкиит.д.,
как для первичной культуры)
Протокол
1. После удаления некротизированной, жировой и
фиброзной тканей измельчите опухоль на кусоч¬
ки примерно 1-2 мм, промойте кусочки в DBSS
как для первичной культуры.
2. Поместите от пяти до десяти кусочков в каждую
ампулу.
3. Добавьте к кусочкам 1 мл ростовой среды, содер¬
жащей 10% DMSO, и оставьте их на 30 мин при
комнатной температуре.
4. Заморозьте ампулы при скорости 1 °С/мин (см.
протокол 20.1) и перенесите их в морозильник с
жидким азотом (чтобы избежать риска взрыва,
не погружайте ампулу в жидкий азот).
5. Для оттаивания ампулы поместите ее в воду с
температурой 37 °С (с соблюдением необходи¬
мых мер предосторожности, см. протокол 20.2).
6. Тщательно протрите ампулу спиртом, откройте
ее, дайте кусочкам осесть, удалите половину
среды.
7. Медленно замените среду на свежую, не содержа¬
щую DMSO. Смешайте осторожным встряхива¬
нием и оставьте на 5 мин.
8. Постепенно замените всю среду на свежую, не
содержащую 0М80,_перенесите кусочки в чашки
Петри и продолжайте работу как с первичной
культурой» но оставляйте в чашке в два раза
больше материала.
24.8. СПЕЦИФИЧЕСКИЕ ОПУХОЛЕВЫЕ
КУЛЬТУРЫ
Общие протоколы, описанные в гл. 12 (см. протоко¬
лы 12.3-12.10), вместе с селективным культивиро¬
ванием (см. разд. 24.7 и гл. 23) обеспечат хорошую
начальную точку для культивирования большинства
типов опухоли. В целом, разумный подход к культуре
опухоли состоит в том, чтобы комбинировать кол-
лагеназное разрушение (например, протокол 12.8) с
тканеспецифичными подходами, описанными в прото¬
колах в гл. 23, с использованием или без использования
фидерного слоя. Хотя для клеток нормальных тканей
часто бывают определены бессывороточные условия
культивирования, пищевые требования и потребности
в факторах роста клеток опухоли могут быть более раз¬
нообразны (см. разд. 24.7.1) и требуют использования
сыворотки. Ниже кратко описаны некоторые специфи¬
ческие примеры опухолевых культур.
24.8.1. Молочная железа
Культура карциномы молочной железы может быть
получена коллагеназным разрушением биопсийного
материала [Leake et al., 1987; Dairkee et al., 1995,1997;
протоколы см. Speirs, 2004] и размножением на фидер¬
ном слое или в MCDB 170. Однако многие из условий,
используемых, чтобы получить культуры из нормаль¬
ной молочной железы (дополнительные факторы роста,
коллагеновое покрытие) могут не быть оптимальными
для карциномы молочной железы [Ethier et al., 1993],
поэтому может потребоваться проверка разнообразных
условий в предварительных попытках культивирова¬
ния, а также подтверждение неопластичной идентич¬
ности культивированных клеток.
Идентификация клеток опухоли молочной железы,
в отличие от нормальных аналогичных клеток, пот¬
ребует подтверждения определенных генетических
повреждений, включая егЬВ-2, с-тш/с, рецептор фак¬
тора роста фибробластов (FGFR) 2 [Ethier et al., 1993;
Ray et al., 2004] и определение повышенного уровня
циклина Е [Willmarth et al., 2004]. Происхождение
клеток может быть подтверждено по наличию цито¬
кератина, ЕМА [Heyderman etal., 1979] и анти-HMFG
1 и 2 типа [Burchell и Taylor-Papadimitriou, 1989];
было предложено считать, что in vitro экспрессия
кератина 19 более вероятна в опухолевых клетках,
чем в нормальных клетках молочной железы [Taylor-
Papadimitriou et al., 1989].
24.8.2. Легкое
Как мелкоклеточная (SCLC), так и не мелкоклеточная
саркома (NSCLC) были успешно культивированы
[Oie et al., 1996; протоколы см. Wu, 2004] с помощью
механического разрушения биоптата в бессывороточ¬
ной селективной среде, — HITEC для SCLC и ACL4
для NSCLC с последующим разделением в градиенте
плотности Ficoll для выделения клеток. Существен¬
ная группа этих линий клеток была накоплена NCI,
некоторые линии доступны через АТСС. Также было
изучено влияние матрикса на экспрессию онкогена и
фактора роста [Pavelic et al., 1992], которые использо¬
вались, чтобы облегчить формирование культуры кле¬
ток карциномы легкого из микрометастазов костного
мозга [Pantel et al., 1995]. Испытание действия хими¬
отерапевтических препаратов на метастазы в мозге из
опухоли легкого in vitro обнаруживает значительную
гетерогенность ответа [Marsh et al., 2004].
Для идентификации клеток SCLC in vitro имеется
ряд маркеров, включая бомбезин-подобную имму¬
нореактивность, DOPA-деккарбоксилазу, N-туе, и
сверхэкспрессию изофермента ВВ креатинкиназы
[Carney et al., 1985; Pedersen et al., 2003], но раз¬
ные варианты SCLC и NSCLC могут не иметь этих
маркеров. Известно, что клетки сквамозного рака
легкого сверхэкспрессируют EGFR, егЬВ-2, и TGF-a
[Piyathilake et al., 2002], показано, что другие клеточ¬
24.8. Специфические опухолевые культуры
481
ные линии клеток NSCLC проявляли нарушенные
свойства in vitro, включая HER2/NEU, ТР53, и К-ras,
которые коррелируют с фенотипом опухоли in vivo, из
которой они были получены [Wistuba et al., 1999].
24.8.3. Желудок
Солидные опухоли рака желудка могут быть разру¬
шены ножницами и пипетированием, что позволяет
высвободить опухолевые клетки из волокон стромы.
Асцит может быть очищен центрифугированием на
Ficoll/метризоат [см. Park et al., 2004]. Возможно
произвести обогащение клетками опухоли путем ме¬
ханического собирания агрегатов опухолевых клеток и
субкультивированием их с очищением от фибробластов
или дифференциальной трипсинизацией. Морфология
клеточных линий разнообразна, диапазон ее распро¬
страняется от хорошо распластанных плоских монос¬
лоев, имеющих вид «гладкой панели», до «булыжной
мостовой», с близко расположенными агрегатами
клеток (рис. 24.2).
24.8.4. Толстый кишечник
Были описаны бессывороточные условия культивирова¬
ния некоторых видов клеточных линий колоректального
рака человека [Fantini et al., 1987; Murakami & Masui,
1980], но эти условия в целом не подходят для недавно
изолированных культур карциномы, которые требуют
присутствия сыворотки. Колоректальная карцинома
была выделена в культуре из биопсий, которые были
взяты как от первичных опухолей, так и из метастазов
[Danielson et al., 1992; Paraskeva&Williams, 1992; Park
and Gazdar, 1996]. Как в случае карциномы легкого, не¬
которые колоректальные опухоли обладают нейроэндок¬
ринными свойствами; имеются сообщения об успешном
использования среды HITES (см. табл. 10.2) для их куль¬
тивирования [Lundqvistetal., 1991]. Для очистки клеток
карциномы для первичного культивирования в обычной
среде (RPMI1640 с 10% FB) использовали плотностное
центрифугирование на Percoll [Csoka et al., 1995].
Протоколы для культуры карциномы толстой киш¬
ки доступны в Whitehead [2004]. Протокол 24.3 взят из
работ Paraskeva и Williams [1992].
Рис. 24.2. Клеточные линии карциномы желудка, (а) Хорошо распластанный монослой клеточной линии SNU-216 карциномы
желудка, (б) Монослой типа «мощеной мостовой» клеточной линии карциномы желудка SNU-484. (в) Более мягкие клетки
образуют слой типа «булыжная мостовая», клеточная линия карциномы желудка SNU-668. Раковые клетки могут расти как
прикрепленная культура, обнаруживая диффузное распространение роста клеток опухоли с веретенообразным или полиго¬
нальным контурами, (г) Клеточная линия карциномы желудка SNU-620. Раковые клетки растут как в прикрепленном виде,
так и в виде плавающих клеточных агрегатов. Воспроизводится по Park et al. [2004]
482
ГЛАВА 24. КУЛЬТУРА ОПУХОЛЕВЫХ КЛЕТОК
ПРОТОКОЛ 24.3. Культура колоректальных
опухолей
Аденомы обычно расщепляются ферментативно, а
карциномы измельчаются хирургическими лезви¬
ями. Если хорошо дифференцированная колорек¬
тальная опухоль не высвобождает раковые клетки,
когда образец порезан на части, она может быть
расщеплена ферментативно.
Материалы
Стерильные:
• Ростовая среда: DMEM, содержащая 2 мМ глю¬
тамина и с добавлением 20% FBS (существенным
является выбор партии сыворотки), гидрокор¬
тизона сукцината натрия (1 мкг/мл), инсулина
(0,2 Ед/мл), 2 мМ глютамина, пенициллина
(100 Ед/мл), и стрептомицина 100 мкг/мл).
• Промывочная среда: ростовая среда с 5% FBS,
200 Ед/мл пенициллина, 200 мкг/мл стрепто¬
мицина, и 50 мкг/мл гентамицина. Гентамицин
добавляют в первичную культуру в течение, по
крайней мере, первой недели, а затем удаляют.
• Раствор для расщепления: DMEM и антибиоти¬
ки, как описано для промывочного раствора, с
коллагеназой (1,5 мг/мл, тип IV, Worthington),
гиалуронидазой (0,25 мг/мл, тип 1, Sigma) и
2,5-5% FBS. Хотя мы используем коллагеназу
Worthington, можно использовать также Sigma
(для культуры тканей).
• Диспаза для субкультуры: приготовьте раствор
диспазы: (нейтральная протеаза, степень 1,
Roche) 1)2 Ед/мл в DMEM, содержащей 10%
FBS, глютамин, пенициллин и стрептомицин.
Простерилизуйте раствор фильтрацией и хра¬
ните при -20 °С. Если при размораживании
образуются хлопья, раствор следует центрифу¬
гировать, отбирается и используется активный
супернатант.
• Флаконы, 25 см2, покрытые коллагеном, тип IV, с
фидерным слоем Swiss ЗТЗ с концентрацией кле¬
ток примерно IxlO4 кл./см2 (см. протокол 23.4)
Протокол
Ферментативное расщепление
1. Промойте образцы опухоли четыре раза в промы¬
вочной среде.
2. Порубите в маленьком объеме той же самой
среды, достаточной только чтобы покрывать
ткани. (Не позволяйте ткани высыхать!) Ткани
измельчают крестообразными хирургическими
лезвиями или острыми ножницами до фрагмен¬
тов приблизительно 1 мм3.
3. После измельчения промойте ткани снова четыре
раза (число промывок может быть изменено с
опытом, в зависимости того, является ли конта¬
минация вероятным фактором) в настольной цент¬
рифуге (300 g, 3 мин) с ресуспендированием.
4. После промывания фрагментов опухоли помес¬
тите их в расщепляющий раствор и инкубируйте
при вращении при 37 °С, обычно в течение ночи
(приблизительно 12-16 ч). Время, на которое
образцы оставляются в растворе, не критическое,
потому что расщепление — мягкий процесс, одна¬
ко важно не допускать, чтобы в ходе процедуры
среда стала кислой. Низкий pH указывает, что
образцы были в среде слишком долго или слиш¬
ком много ткани было помещено в объем расщеп¬
ляющей смеси. Приблизительно 1 см3 опухолевой
ткани помещают в 20-40 мл расщепляющего
раствора.
Неферментативная обработка ткани:
Аденомы почти всегда нуждаются в расщеплении
ферментами. Однако для карцином показано, что
при измельчении ткани опухоли при помощи лезвий
до размеров 1 мм3 маленькие скопления опухолевых
клеток высвобождаются из ткани опухоли в среду.
В этом случае может быть выполнена следующая
процедура:
1. Соберите промывочную среду, содержащую вы¬
свобожденные скопления клеток.
2. Разделите большие и маленькие сгустки, оставив
их оседать в течение нескольких минут в центри¬
фужной пробирке.
3. Удалите супернатант.
4. Поместите оставшиеся части ткани непосред¬
ственно в культуру или осторожно вращайте
ткань 30-60 мин в промывочной среде, чтобы
высвободить более маленькие сгустки, которые
могут тогда быть собраны, посеяны и помещены
в культуру отдельно от остающихся больших
частей ткани.
5. Промойте все образцы три раза перед помеще¬
нием их в культуру.
Стандартные условия для первичной культуры:
1. Инокулируйте трубочки и сгустки клеток, полу¬
ченных из образцов ткани в обработанные колла¬
геном 25-см2 флаконы с 4 мл среды с фидерными
клетками.
2. Инкубируйте при 37 °С в инкубаторе с 5% СО2.
3. Заменяйте среду в культуре дважды в неделю.
Трубочки и клетки начинают прикрепляться к под¬
ложке, эпителиальные клетки мигрируют в пределах
1-2 дней. Больше всего эпителиальных клеток выде¬
ляется из трубочек и маленьких сгустков эпителия
в течение 7 дней, но для прикрепления к подложке
более крупных органоидов может потребоваться до
6 недель, хотя они останутся жизнеспособными все
это время.
Прикрепление эпителия в ходе первичной куль¬
туры и субкультуры более воспроизводимо и эф¬
фективно, когда клетки инокулируют в стандартные
флаконы, покрытые коллагеном (Biocoat, B-D Bio¬
24.8. Специфические опухолевые культуры
483
sciences; см. также протоколы 23.2 и 23.9), и значи¬
тельно лучший рост получен на фвдерном слое кле¬
ток ЗТЗ, чем без него. Когда эпителиальные колонии
размножаются до несколько сотен клеток в колонии,
они становятся менее зависимыми от фидерных
клеток и дальнейшее добавление фидерных клеток
не требуется.
Субкультура и распространение:
Большинство первичных культур колоректальной
аденомы и линии клеток, полученные из аденомы, не
могут быть пассированы при помощи установившей^
ся практики обработки трипсин/EDTA [Paraskeva
et al., 1984,1985]. Дезагрегация клеток аденомы до
суспензии отдельных клеток с 0,1% trypsin в 0,25 мМ
(0,1) % EDTA завершается чрезвычайно бедным рос¬
том. Поэтому вместо этого используется диспаза.
1. Добавьте диспазу к клеточному монослою в объе¬
ме, достаточном для того, чтобы закрыть клетки
(-2,5 мл/25-см2флакон), и оставьте на 40-60 мин
для первичной культуры и 20-40 мин для клеточ¬
ных линий.
2. Как только эпителиальные слои начинают от¬
деляться (они обычно отделяются слоями, а не
отдельными клетками), помогите пипеткой этому
отделению и дезагрегации до меньших скоплений
клеток.
3. Промойте и пересейте клетки в стандартных
условиях для культуры. Может потребоваться
несколько дней для прикрепления скоплений
клеток, поэтому будьте аккуратны при замене
среды.
Контаминация культур колоректальной опу¬
холи фибробластами. При контаминации культуры
фибробластов может быть применен один из нижепри¬
веденных подходов или их комбинация:
1) Физически удалите хорошо заметные колонии
фибробластов путем соскребания тупой стороной
стерильного инструмента (например, клеточный
скребок). Следует не менее шести раз особо тща¬
тельно промыть культуры, чтобы удалить любые
фибробласты, которые могли отделиться от моно¬
слоя, для того чтобы предупредить их повторное
прикрепление.
2) Для некоторых карцином может быть применена
дифференциальная трипсинизация [Kirkland &
Bailey, 1986].
3) При пассажах диспаза предпочтительно (но не
исключительно) удаляет эпителий и оставляет
большую часть фибробластов прикрепившимися к
культуральному сосуду [Paraskeva etal., 1984]. При
субкультивировании клетки, которые были удале¬
ны действием диспазы, могут быть предварительно
инкубированы в пластиковой чашке Петри в тече¬
ние 2-6 ч для того, чтобы позволить осуществиться
первичному прикреплению фибробластов, которые
могли выделиться вмести с эпителиальными клет¬
ками. Сгустки эпителиальных клеток, которые еще
остаются плавать, могут быть перенесены в новый
флакон при стандартных условиях культивирова¬
ния. Этот метод имеет преимущества, поскольку
фибробласты в целом прикрепляются к пластику
намного быстрее, чем сгустки эпителиальных
клеток, поэтому возможен данный этап частичной
очистки.
4) Использование конъюгата анти-Thy-l монокло¬
нальных антител и токсином рицином [Paraskeva et
al., 1985]. Thy-1 антиген представлен в колоректаль¬
ных фибробластах, но отсутствует в эпителиальных
клетках толстой и прямой кишки, поэтому конъ¬
югат убивает фибробласты, контаминировавшие
культуру эпителиоцитов, при этом не повреждая
культуру, независимо от того, выделена она из
аденомы или карциномы.
5) Снижение концентрации сыворотки примерно до
2,5-5%, если фибробласты присутствуют в куль¬
туре в большой концентрации. Стоит помнить, что
нормальные фибробласты имеют конечное число
клеточных циклов in vitro и использование любого
или всех представленных методов будет в конце
концов проводить клетки через большое число
делений и фибробласты погибнут в результате
естественного старения.
24.8.5. Поджелудочная железа
Клеточные линии из первичных или метастатических
опухолей поджелудочной железы были выделены
на среде RPMI 1640 с добавлением FBS. Клеточные
линии были адаптированы к безбелковой среде для
исследования клеточных продуктов [Yamaguchi et
al., 1990] (см. также протокол 23.7). Культура кар¬
циномы поджелудочной железы была использована
для исследования действия генистеина на ангиогенез
[Buchler et al., 2004]. Линии панкреатических клеток
были созданы для изучения аутокринного контроля
роста под действием гастрина [Monstein et al., 2001];
при этом корреляции между экспрессией гастрина и
рецепторами холецистокнинина (ССК), которые в
норме связывают гастрин, не была обнаружена. Ли¬
ния KCI-MOH1 является клоном аденокарциномы
поджелудочной железы после пассажа через мышей
SCID [Mohammad et al., 1999]. Ксенотрансплантатная
опухоль также была использована для установления
клеточной линии НРАС для изучения глюкокортико-
идных рецепторов [Gower et al., 1994]. Протоколы для
культуры карциномы поджелудочной железы путем
разрушения в коллагеназе и посева на чашки, покрытые
коллагеном можно, найти у Iguchi et al. [2004], который
выделил клеточные линии из первичных повреждений,
асцитов и метастазов печени. Две линии из метастазов
печени были получены путем пассажа через «голых»
мышей путем инъекции в селезенку (рис. 24.3).
484
ГЛАВА 24. КУЛЬТУРА ОПУХОЛЕВЫХ КЛЕТОК
Рис. 24.3. Клеточные линии рака поджелудочной железы.
Микрофотографии клеточных линий KP-1N (а) и КР-3 (б)
из метастазов в печень, ксенотрансплантированных в «го¬
лых» мышей, Фазово-контрастный объектив (х125). По
Iguchi et al. [2004]
24.8.6. Яичники
Из опухолей эпителия яичников был получен ряд кле¬
точных линий, например серии линий OAW [Wilson
et al., 1996], OVCAR-3 [Hamilton et al., 1983], A2780,
[Tsuruo et al.,1986], некоторые — в бессывороточной
среде [Jozan et al., 1992]. Другие линии были получены
после коллагеназного разрушения в среде, содержащей
сыворотку, например в среде OSE, содержащей М199 и
MCDB105 в отношении 50:50, с добавлением 15% FBS
[Lounis et al., 1994]. Легче получить генерации культур
из асцитов или легочного экссудата, чем из материала
плотной опухоли [Verschraegen et а1.,2003]. Для очистки
клеток карциномы яичника для получения первичной
культуры использовалось плотностное центрифугиро¬
вание на Percoll, как в случае карциномы толстой кишки
[Csoka et al., 1995]. Протоколы для культуры карцино¬
мы яичника можно найти в работе Wilson [2004].
24.8.7. Простата
Метод с применением матрикса, описанный выше для
легочной карциномы, успешно был применен для выде¬
ления клеточных линий из опухолей простаты [Pantel
et al., 1995]. Для получения культур клеток нормальной
простаты, а также доброкачественных и злокачествен¬
ных опухолей также использовались бессывороточные
культуры [Chopra et al., 1996; Bright & Lewis, 2004]
(см. также протокол 23.11). После иммортализации с
использованием ретровирусных конструктов, кодиру¬
ющих трансформирующие белки Е6 и Е7 герпесвируса
HPV16, можно было получить долгоживущие культу¬
ры [Bright & Lewis, 2004].
Известно, что развитие эпителия простаты нахо¬
дится под паракринным контролем со стороны стро-
мы, секретирующей факторы роста семейства FGF,
включающего KGF [Thomson et al., 1997]. Показано,
что при получении культур из нормальных тканей,
доброкачественной гиперплазии простаты (ВРН) и
карциномы уровень FGF-17 был выше в два раза в
культуре из ВРН [Polnaszek et al., 2004]. Опухолевый
ангиогенез также изучали в культуре из нормальной и
неопластически измененной простаты с использова¬
нием оксида азота [Wang et al., 2003] и добавлением
VEGF [Shih et al., 2003].
24.8.8. Мочевой пузырь
Было показано, что культура карциномы, которая яв¬
ляется поверхностной опухолью, имеет ограниченную
продолжительность жизненных циклов, в то время
как клеточные линии из миоинвазивных опухолей
часто образуют постоянные клеточные линии [Yeager
et al., 1998]. Первичная культура клеток карциномы
переходного эпителия мочевого пузыря обычно ис¬
пользовалась для изучения роли FasL в иммунной
защите от клеток рака мочевого пузыря [Chopin et al.,
2003]. Протоколы для культуры карциномы мочевого
пузыря можно найти в Fu et al. [2004].
24.8.9. Кожа
Меланома. Пигментные клетки кожи могут быть куль¬
тивированы в присутствии соответствующих факторов
роста (см. протокол 23.21), также с определенным
успехом культуры могут быть получены из меланомы
[Creasey et al., 1979; Mather &Sato, 1979a, Ь]. Пер¬
вичные меланомы часто контаминированы фиброб¬
ластами, но возможно проведение клонирования на
конфлюэнтных фидерных слоях нормальных клеток
[см. протокол 24.1; Creasey et al., 1979; Freshney et al.,
1982b]. Клеточные культуры, полученные путем ме¬
ханического разрушения, могут быть освобождены от
фибробластов обработкой 100 мкг/мл Geneticin (G418)
[Halaban, 2004].
Для роста меланоцитов нормальной кожи, дисплас-
тических невусов и метастазов меланомы применяется
среда MCDB 153, с добавлением FGF-2, инсулина,
трансферрина, а-токоферола, экстракта гипофиза
быка, гидрокортизона и 5% сыворотки с добавлением
24.8. Специфические опухолевые культуры
485
также при первых двух пассажах каталазы и РМА
[Levin et al., 1995]. Протоколы для культивирования
клеток меланомы можно найти в Halaban [2004].
Базально-клеточная карцинома. Фидерный
слой клеток ЗТЗ предоставляет возможность куль¬
тивирования базальноклеточной карциномы (ВСС)
кожи [Rheinwald & Beckett, 1981] (см. протокол 23.1).
Oh et al. [2003] использовали первичные культуры
из базальноклеточной карциномы для изучения
ангиогенезного потенциала и обнаружили его кор¬
реляцию с агрессивностью развития опухоли. Было
показано также, что культуры ВСС являются более
чувствительными к цитотоксическому действию ин-
терферонов, чем нормальные кератиноциты [Brysk
et al., 1992].
Сквамозноклеточная карцинома. SCC и эрит-
роплакии были культивированы на фидерных слоях
ЗТЗ [протоколы см. в Edington et al., 2004], что привело
к выделению ценных клеточных линий серии BICR,
представляющих собой различные стадии малигниза¬
ции и иммортализации [Fitzsimmons et al., 2003; Gordon
et al., 2003].
24.8.10. Шейка матки
Культуры доброкачественных и злокачественных опу¬
холей шейки матки могут быть получены из биопсий
методом применения фидерных слоев ЗТЗ, описан¬
ным для нормальных клеток шейки матки [Stanley &
Parkinson, 1979] (см. протокол 23.4). Такие клеточные
культуры используются для изучения хромосомной
нестабильности и интеграции человеческого папилло-
мавируса, который участвует в развитии рака шейки
матки [Koopman et al., 1999].
Протоколы для культуры опухоли шейки матки
можно найти в Stem et al. [2004].
24.8.11. Глиома
Культуры человеческой глиомы могут быть выделены
механическим разрушением, трипсинизацией или кол-
лагеназной обработкой [Westermarketal., 1973; Ponten,
1975; Freshney, 1980; протоколы см. Darling, 2004; см.
также рис. 16.2с и 24.4]. Ряд глиом были культивиро¬
ваны после выделения из грызунов, среди них особого
упоминания заслуживает клеточная линия С6 [Benda
et al., 1968], которая экспрессирует маркер астроци-
тов — глиальный фибриллярный кислый белок — до
98% клеток [Freshney et al., 1980а], но при этом имеет
ферменты глицеринфосфатдегидрогеназу и 2,,3,-цикл
нуклеотидфосфорилазу [Breen & De Vellis, 1974], кото¬
рые являются маркерами олигодендроцитов. Эта линия
является интересным примером клеток-предшествен-
ников, которые одновременно могут созревать по двум
фенотипическим направлениям.
Рис. 24.4. Культуры глиомы человека. Две клеточ¬
ных культуры анапластической астроцитомы человека.
(а) Первичная культура, полученная обработкой коллаге¬
назой, обнаруживает типичные астроциты, которые могут
быть дифференцированными опухолевыми клетками или
реактивной глией, (б) Постоянная клеточная линия MOG-
G-UVW, обнаруживающая один из нескольких морфологи¬
ческих типов, найденных в культурах клеток, полученных
из глиомы. В данном примере клетки имеют плеоморфную
фибробласт-подобную форму; типичная мультиполяр¬
ность, характерная для астроцитов и часто утрачиваемая в
постоянных клеточных линиях, отсутствует (см. также цв.
вкладки 7а, 6, в, 9в и 11 б.)
Культуры глиомы человека обычно используются
для прогностических тестов на химиочувствитель¬
ность [Thomas et al., 1985], а также, реже, в изучениях
ретровирус-опосредованной терапии [Rainov & Ren
2003] и терапии направленного действия с исполь¬
зованием пептидных лигандов, слитных с цитоток¬
синами, селективно действующих на опухоль мозга
[Liu et al., 2003].
24.8.12. Нейробластома
Было выделено несколько линий нейробластомы
(например, SK-N-BE (2) [Biedler and Spengler, 1976],
[Tumilowicz et al., 1970]), которые являются предметом
определенного интереса в связи с их способностью к
486
ГЛАВА 24. КУЛЬТУРА ОПУХОЛЕВЫХ КЛЕТОК
дифференцировке [Dimitroulakos et al., 1994]. Было
обнаружено, что мышиные нейробластомы являются
подходящим субстратом для трансмиссивных губча¬
тых энцефалопатий (TSEs) [Solassol et al., 2003].
24.8.13. Сперматоцитома
Тестикулярные сперматоцитомы были культивиро¬
ваны с использованием клеточного фидерного слоя
STO с последующим добавлением фактора стволовых
клеток (SCF), LIF, и FGF-2, как это применяется в
культуре эмбриональных стволовых клеток [Olie et
al., 1995]. Хотя культуры были гетерозиготны, они не
давали роста культуры герминативных клеток.
24.8.14. Лимфома и лейкемия
Обычная методика получения клеточных линий для
лимфомы и лейкемических клеток затруднена. Мно¬
гие существующие постоянные линии клеток были
выделены из лимфомы Беркитта [Drexler & Minowada
2000]. Показано, что они несут интегрированные гены
ЕВ вируса. Протоколы для культивирования лимфо-
мы-лейкемии можно найти в Drexler [2004].
Глава 25
ОРГАНОТИПИЧЕСКАЯ КУЛЬТУРА
25.1. МЕЖКЛЕТОЧНОЕ
ВЗАИМОДЕЙСТВИЕ
И ФЕНОТИПИЧЕСКАЯ ЭКСПРЕССИЯ
Исторически возникшее расхождение между поддер¬
жанием эксплантированных фрагментов ткани и
культивированием клеток, которые были выделены из
них, привело к развитию методов органной культуры
и клеточной культуры (см. разд. 1.5), при этом клеточ¬
ная культура стала ведущей. В настоящее время, хотя
потенциал использования банков клеточных линий
далек от полного использования, многие исследователи
сходятся на том мнении, что питательные и гормональ¬
ные добавки сами по себе недостаточны для полного
воспроизведения структурной и функциональной
компетенции данной клеточной популяции. Жизненно
важным фактором, не учитываемым при поддержании
клеточной культуры, является межклеточное взаимо¬
действие и сигнальные системы.
25.1.1. Роль клеточной плотности
Межклеточное взаимодействие проявляет себя на
простейшем уровне, когда клеточные культуры до¬
стигают конфлюэнтности, и клетки начинают сильнее
взаимодействовать друг с другом. Это взаимодействие
реализуется в результате контакт-опосредованной
передачи сигналов, формирования межклеточных
контактов, включая щелевидные соединения, и уве¬
личения потенциала обмена гомокринными факто¬
рами (см. разд. 3.5, 17.7.1). Первый значительный
эффект — прекращение подвижности клетки и выход
из клеточного цикла (контактное ингибирование, см.
разд. 18.5.2) в нормальных клетках и снижение клеточ¬
ной пролиферации и возрастание уровня апоптоза а
трансформированных клетках. Если клетки обладают
способностью к дифференцировке, то часто отмечает¬
ся увеличение числа дифференцированных клеток.
Клетки крысиной глиомы С6 обнаруживают увеличе¬
ние процентной доли GFAP-положительных клеток,
(рис. 25.1), секреторные или всасывающие эпите¬
лиальные клетки формируют куполы (см. рис. 16.1;
цв. вкладку 12а, 6), и скелетный симпласт миоцитов
образует отдельные многоядерные миотрубочки.
Нормальный фибробласт, несмотря на контактное
ингибирование, после достижения конфлюэнтности
будет секретировать большее количество коллагена,
и культура будет иметь тенденцию к формированию
многослойной структуры, с верхним слоем более мел¬
ких и более темных клеток, похожих на фиброциты
(см. цв. вкладку 10е).
25.1.2. Реципрокные
взаимодействия
Взаимодействие популяций различных клеток ока¬
зывает кооперативный эффект на их соответству¬
ющий фенотип (см. разд. 17.7.1), а получившиеся в
результате фенотипические характеристики приводят
к развитию новых взаимодействий. Межклеточное
взаимодействие, таким образом, является не еди¬
ничным событием, а каскадом событий. Подобным
образом, экзогенные сигналы инициируют развитие не
единичного события как это происходит в гомогенной
клеточной популяции, а могут вызвать развитие нового
каскада, как результат экзогенно модифицированного
фенотипа одного или более партнеров. Например,
альвеолярные клетки легких синтезируют и высво¬
бождают сурфактант только в ответ на гормональную
стимуляцию расположенных рядом фибробластов
[Post et al., 1984]; аналогичным образом, ответ эпи¬
телия простаты на сигнал от стромы, в свою очередь,
активируется андрогеном, связавшимся со стромой
[Thomson et al., 1997] и вызывающим высвобождение
KGF. Linser and Moscona [1980] отделили Мюллеров¬
ские клетки из нервной ретины от пигментированной
ретины и нейронов и показали, что глюкокортикоид-
индуцированная дифференцировка не осуществляет¬
ся, если Мюллеровские клетки (астроглия) находились
отдельно от нейронов ретины. Присутствие нейронов
из других отделов мозга было не эффективно. Диффе¬
ренцировка эпителия в ответ на компоненты матрикса
часто определяется совместным действием эпителия с
одной стороны и соединительной ткани с другой, как
это показано в случае взаимодействия между эпидер¬
мисом и дермисом in vitro [Fusenig, 1994а; Limat et
al., 1995]. Следовательно, цельная интегрированная
ткань может различным образом отвечать на повсе¬
местно распространенные сигналы не в результате
специфичности сигнала или аффинности рецептора,
а вследствие качества микроокружения, заключающе¬
гося в особенности взаимодействия данного клеточ¬
488
ГЛАВА 25. ОРГАНОТИПИЧЕСКАЯ КУЛЬТУРА
80
75
70
х
л
X
Л
® 60
S
§
с
d 55
£
О
50
45
Рис. 25.1. Действие клеточной плотности на экспрессию
GFAP в клетках Сб. Флаконная культура выращивалась до
различной клеточной плотности, фиксировалась и окраши¬
валась иммунопероксидазой для выявления GFAP, процент
окрашенных клеток подсчитывался исходя из воспроиз¬
водимых результатов подсчета в репрезентативных полях.
(С любезного разрешения R.L. Shaw.)
ного типа с рядом расположенными специфическими
субстанциями. Аналогичным образом, в человеческом
сообществе ответ одного индивидуума на экзогенный
стимул обусловлен как его пространственными и вре¬
менными отношениями с другими индивидуумами,
так и присущими ему внутренними особенностями.
На клеточном уровне недифференцированная нейро¬
нальная клетка может стать нейроном, эндокринной
клеткой или меланоцитом, в зависимости от их оконча¬
тельного расположения, взаимодействия с рядом рас¬
положенными клетками, их ответа, опосредованного
этими соседними клетками, на гормональные стимулы.
По существу, эта преамбула призвана показать, что,
несмотря на то что некоторые клеточные функции, та¬
кие как клеточная пролиферация, гликолиз, дыхание,
транскрипция генов, могут протекать изолированно,
их регуляция, связанная с функционированием мно¬
гоклеточного организма, в конечном итоге зависит от
взаимодействий между клетками соответствующих
линий, от стадии развития данной линии, и от взаи¬
модействия между клетками дифференцированных
линий, находящихся в данном микроокружении. Эта
концепция предполагает, что если вы хотите изучать
Клеток/см2
биологию изолированных клеток или использовать
клетки в качестве субстрата, общепринятый метод
монослойной или суспензионной культуры может
быть адекватен, но если вы хотите выяснить что-то об
интегративных функциях или дисфункциях клеток
in vivoy потребуется гистотипическая или органоти¬
пическая модель.
25.1.3. Выбор модели
Для достижения этой цели существуют два подхода.
Одним из них является принятие клеточного распре¬
деления в изучаемой ткани, эксплантация их и под¬
держание как органной культуры. Второй заключается
в очищении и размножении отдельных клеточных
линий, изучение их по отдельности в условиях взаи¬
модействия гомологичных клеток, их комбинирова¬
ния и обоюдного влияния. Эти подходы дали начало
развитию трех главных типов методов: 1) органная
культура, при которой целый орган или его репре¬
зентативная часть поддерживается в виде небольшо¬
го фрагмента в культуре и сохраняет присущее ему
распределение количественное и пространственное
клеток, представленных в этом фрагменте; 2) гисто¬
типическая культура, при которой распространенные
клеточные линии выращиваются по отдельности до
высокой плотности в трехмерном матриксе, и 3) орга¬
нотипическая культура, в которой клетки различных
линий рекомбинированы в экспериментально опре¬
деленном соотношении и специфических связях для
воспроизведения некоторого компонента изучаемого
органа (рис. 25.2).
Органная культура пытается сохранить исходные
структурные взаимоотношения клеток одного и того
же или различных клеточных типов, а, следовательно,
их интерактивные взаимоотношения для того, чтобы
изучить действие экзогенных стимулов на дальней¬
шее развитие [Lasnitzki, 1992]. Эти взаимоотношения
могут быть сохранены путем эксплантирования ин-
тактной ткани или воспроизведенния ее структуры
путем разделения ее компонентов и последующего их
объединении, как это сделано в ставшими классиче¬
скими экспериментах Grobstein &Auerbach и других
в органогенезе [Auerbach & Grobstein, 1958; Cooper,
1965; Wessells, 1977] (см. также разд. 17.7.1). Органоти¬
пическая культура представляет собой синтетический
подход, в то время как трехмерная культура, имеющая
большую клеточную площадь, воспроизводится из
изолированных (и, предпочтительно, очищенных
и охарактеризованных) клеточных линий, которые
затем комбинируются, после чего исследуется их
взаимодействие и характеризуется их ответ на экзоген¬
ные стимулы. Этими экзогенными стимулами могут
быть регуляторные гормоны, питательные вещества
или ксенобиотики. В каждом случае ответ может
быть отличным от реакции чистого клеточного типа
выращенных изолированно при низкой клеточной
плотности клеток.
25.1. Межклеточное взаимодействие и фенотипическая экспрессия
489
Выделение
клонированием, на
селективных средах
и/или клеточным
сортингом
ГИСТОТИПИЧЕСКАЯ
КУЛЬТУРА
ГЕТЕРОГЕННАЯ
ПЕРВИЧНАЯ
КУЛЬТУРА
Размножение
и посев на желаемый
субстрат. Рост до
высокой плотности
клеток с заменой среды
перемешиванием и
Перфузируемая многослойная
структура, полученная из монослоя
Сфероид или
органоид
Размножение каждой чистой линии отдельно
/ \
Губка или каркас
Объединение на вкладыше
в фильтрационную лунку,
трансмембранном, с матриксом
или без него
Объединение в трехмерном
исследовании в концентрических
капиллярах
4
Перфузированная
капиллярная подложка
Вкладыш в фильтрационную лунку
ОРГАНОТИПИЧЕСКАЯ КУЛЬТУРА
Рис. 25.2. Гистотипическая и органотипическая культура
490
ГЛАВА 25. ОРГАНОТИПИЧЕСКАЯ КУЛЬТУРА
25.2. ОРГАННАЯ КУЛЬТУРА
25.2.1. Обмен питательных веществ и газов
Главным недостатком в тканевой архитектуре в ор¬
ганной культуре является отсутствие сосудистой
системы, ограничивающей размеры (в результате
диффузии) и потенциальную полярность клеток в
органной культуре. Когда клетки культивируются
в плотной массе ткани, газовая диффузия и обмен
питательных веществ и метаболитов осуществляются
только на периферии, и это ограничивает размеры
ткани. Размеры индивидуальных клеток в суспен¬
зии или монослое таковы, что диффузия проходит
быстро, однако при формировании агрегатов клеток
около 250 мкм в диаметре (5000 клеток) рост клеток
начинает ограничиваться диффузией, а примерно при
1,0 мм и более в диаметре (~2,5х105 клеток) часто про¬
является центральный некроз. Для снижения остроты
этой проблемы органные культуры часто помещают на
границу раздела жидкой и газовой фаз для улучшения
газового обмена, при сохранении доступа питательных
веществ. Большинство систем достигают этого путем
использования вкладышей в фильтрующую лунку,
помещая эксплантат на фильтр (см. рис. 25.7,25.8) или
на плотик или гель, обращенный в воздушную среду
(рис. 25.3). Эксплантат, закрепленный на плотном
субстрате, может также аэрироваться путем враще¬
ния культуры, периодически погружая образец то в
жидкую среду, то в газовую фазу [Nicosia et al., 1983;
Lechner and LaVeck, 1985; см. протокол 23.9], или пу¬
тем использования роллерных бутылей или пробирок
(см. протокол 26.3).
Закрепление на плотном субстрате может привести
к развитию разрастания клеток из эксплантата и пос¬
ледующего изменения геометрии образца, хотя этот
эффект может быть минимизирован использованием
несмачиваемых поверхностей. Одним из преимуществ
культуры на газовожидкостной поверхности контакта
является то, что эксплантат сохраняет сферическую
геометрию жидкости, если уровень жидкости под¬
держивается на правильном уровне. Если жидкость
слишком глубока, газообмен ухудшается, если она
слишком мелкая, поверхностное натяжение может
привести к всплыванию эксплантата и простимулирует
разрастание клеток за пределы образца.
Повышенная проницаемость кислорода также мо¬
жет быть достигнута использованием повышения кон¬
центрации кислорода вплоть до использования чистого
кислорода или применения гипербарической оксиге-
нации. Определенные ткани, например щитовидной
железы [de Ridder & Mareel, 1978], простаты, трахеи
и кожи [Lasnitzki, 1992], в том числе новорожденного
или взрослого организма, могут получить пользу от
повышения давления 02, но часто эта польза связана
с риском 02-индуцированной токсичности. Поскольку
простое возрастание давления 02 не улучшает высво¬
бождения С02или обмена питательных веществ и
метаболитов, преимущества повышения концентрации
кислорода могут быть перекрыты другими лимитиру¬
ющими факторами.
25.2.2. Структурное единство
Поддержание структурного единства, вне всяких
сомнений, было и остается главной причиной выбора
органной культуры как метода in vitro перед клеточной
культурой. В то время как клеточные культуры исполь¬
зуют клетки, диссоциированные механическими или
ферментативными методами или выделившиеся за
счет спонтанной миграции клеток, в органной культуре
поддерживаются клеточные связи, существующие в
ткани. Первоначально органная культура была выбрана
для упрощения исследования гистологических харак¬
теристик, но в конечном итоге было обнаружено, что
определенные элементы фенотипической экспрессии
проявляются, только если клетки плотно взаимодей¬
ствуют друг с другом (см. разд. 25.1).
25.2.3. Рост и дифференцировка
Органная _
культура
Микропоровый
фильтр \
из
нержавею-
щей стали
Среда
Чашка
Рис. 25.3. Органная культура. Маленькие фрагменты ткани
на фильтре кладут на вершину решетки из нержавеющей
стали над центральным колодцем чашки с органной куль¬
турой
Существует связь между ростом и дифференцировкой,
так как дифференцированные клетки утрачивают спо¬
собность к делению (см. разд. 17.3). Возможно также,
что прекращение роста независимо от клеточной плот¬
ности может само по себе вносить вклад в индукцию
дифференцировки только путем формирования такого
фенотипа, который делает возможным рецепцию эк¬
зогенных индукторов дифференцировки. В результате
ограничений клеточной пролиферации плотностью
культуры и физических ограничений, накладывае¬
мых геометрией органной культуры, большинство
органных культур не растут или, если растут, то их
пролиферация ограничена внешними слоями клеток.
Следовательно, состояние культуры допускает диффе¬
ренцировку и при данных соответствующих клеточных
25.2. Органная культура
491
взаимодействиях и наличии растворимых индукторов
(см. разд. 17.7) может предоставить идеальные условия
для осуществления дифференцировки.
25.2.4. Ограничения метода
органной культуры
Экспериментальный анализ органных культур зависит
главным образом от гистологических методов, которые
сами по себе не позволяют проводить биохимический и
молекулярный анализ. Биохимический мониторинг тре¬
бует наличия воспроизводимости значений показателей,
которую труднее достигнуть в органной культуре, по
сравнению с распространенными клеточными линиями
в силу особенностей приготовления образцов органной
культуры, некоторых различий в манипуляциях с образ¬
цами и их геометрии, а также вариациях в соотношении
клеточных типов внутри культуры (см. табл. 1.4).
Кроме того, органные культуры приготовить слож¬
нее, чем репликаты культур из распространенных
клеточных линий. Кроме того, отсутствует преиму¬
щество наличия охарактеризованных первоначально
полученных клеточных культур, которым они могут
быть родственны. Органные культуры не могут быть
размножены, и, следовательно, каждый эксперимент
требует использования повторного обращения к
ткани исходного донорского органа, что усложняет
рабочую процедуру. Более того, в качестве реагиру¬
ющей популяции клетки могут быть малой частью
культуры. Для правильного определения типа клеток,
которому приписывают молекулярный ответ на раз¬
дражитель, требуется проведение гистологических
исследований с гистохимическим или иммунохими-
ческим анализом in situ.
Органная культура является более информативным
методом для исследования интегративных взаимоотно¬
шений внутри ткани по сравнению с изолированными
клетками. Можно точно сказать, что будущее понима¬
ние контроля экспрессии генов (и в конечном итоге
поведения клеток) в многоклеточном организме может
оказаться обманчивым, но ограничения, налагаемые
системой органной культуры таковы, что комбини¬
рованные системы чистых клеточных типов могут
принести больше информации на определенной стадии
исследований. Тем не менее нет сомнений, что органная
культура вносит большой вклад в наше понимание
биологии развития и межтканевых взаимодействий.
Применение органной культуры будет продолжаться
до тех пор, пока отсутствуют адекватные комбиниро¬
ванные синтетические системы.
25.2.5. Типы органных культур
Поскольку техника работы с органной культурой
главным образом определяется требованием разме¬
щать ткань так, чтобы обеспечить оптимальных обмен
питательных веществ и газов, в большинстве методов
ткань располагается на границе раздела газ/жидкость
на полужидком гелевом субстрате или агаре [Wolff &
Haffen, 1952], свернувшейся плазме [Fell & Robison,
1929] или плотике из микропорового фильтра, обер¬
точной бумаге для линз или вискозной ткани, располо¬
женной на решетке из нержавеющей стали [Lasnitzki,
1992; рис. 1.3] или прикрепленной к полоске плексиг¬
ласа. Этот тип конфигурации в настоящее время легче
всего достигается путем использования вкладышей в
фильтрационную лунку (см. протокол 25.4). Прото¬
кол 25.1 использует зачаток органа, выделенный из
эмбриона цыпленка, но применим ко многим другим
типам клеток.
ПРОТОКОЛ 25.1. Органная культура
Схема
Иссечь орган или ткань, измельчить до разме¬
ров 1 мм3 или до тонкой мембраны или палочки,
поместить на подложку на границе раздела сред
Bp^yx-q>eAa (например, на вкладыш для фильтра¬
ционной лунки (см. рис. 25.7, 25.8). Инкубировать
во влажном С02-инкубаторе, заменяя среду, когда
это требуется.
Материалы
Стерильные или асептически приготовленные:
• Инструменты для иссечения
• Среда (например, Ml99), с сывороткой или без
• Вкладыш в фильтрационную лунку, обработан¬
ный для нетканевых культур (например, Costar
Transwells поликарбонат #3423, Coming)
• Многолуночные планшеты, 12 лунок (Coming)
Нестерильные:
• Оплодотворетше яйца курицы, 8 дней инку¬
бации
Протокол
1. Поместите вкладыш для фильтрационной лун¬
ки в лунку многолуночного планшета, добавьте
достаточное количество среды до уровня дна
фильтра (~1 мл).
2. Поместите чашки во влажный С02-инкубатор,
позволяя pH среды достичь равновесия при
37 °С.
3. Приготовить ткань, или иссечь целый эмбриональ¬
ный орган (например, бедро ипиголень 8-дневного
эмбриона цыпленка, см. протоколы 12.2 и 12.7).
Ткань не должна иметь толщину в одном изме¬
рении более 1 мм, предпочтительно еще менее
(например, голень 8-дневнош эмбриона цыпленка
может иметь 5 ш в длину, но только 0,5-0,8 мм в
диаметре). Фратент кожи может быть площадью
10 мм2, но только 200 мкм толщиной. Кусочки
тканей, которые должны быть измельчены, чтобы
получить нужный размер, такие как печень или
почки, не должны быть более 1 мм3.
492
ГЛАВА 25. ОРГАНОТИПИЧЕСКАЯ КУЛЬТУРА
4. Для кратковременного хранения иссеченных
тканей (<1 ч) можно использовать HBSS, однако
для более длительного процесса иссечения
пользуйте 50% сыворотку в HBSS, забуференную
HEPES до pH 7,4.
5. Выньте чашки из инкубатора, осторожно пере¬
несите ткани на фильтрь^ Д^вяг аштрацин любой
избыточной жидкостз!^в$|еш£^ с эксплан-
том, обычно лучтмгёего подходит Пастеров¬
ская пипетка, хотя при этом следует особенно
внимательно^ледить за тем, чтобы не проткнута
фильтрУвлажните внутреннюю стороиу ци-
петки средой до аспирации, чтобы предупредить
протыкание фрагмента ткани пипеткой.
6. Проверьте уровень среды, убедитесь, что ткань
смочена средой, но не шлностью в нее погружена,
и верните чаппси в инкубатор.
ды по прежнему покрывает фильтр И ЭКСПЛаНТ, НО
слой среды не на столько глубок, чтобы эксплант
всплыл.
8. Инкубируйте чашки в течение 1-З недель, заме¬
няя среду каждые 2-3 дня и отбирая пробы по
мере необходимости.
Вариации. Большая часть вариаций включает в себя
следующие аспекты:
1) Среда. М199 или CMRL 1066 могут быть исполь¬
зованы с сывороткой или без, для хрящей и костей
можно также использовать среду BGJ [Biggers et
al., 1961].
2) Тип подложки. Органная культура может использо¬
вать в качестве подложки фильтр (например, поли-
карбонатный), положенный на решетку из нержаве¬
ющей стали в чашке для органной культуры (Falcon
#3037, B-D Biosciences; рис. 25.3). Тем не менее
использование вкладыша в фильтрационную лунку
имеет ряд преимуществ в манипуляциях, размерах
образца, материалах и матриксном покрытии (см.
рис. 25.7; табл. 25.1). Различные типы тканей могут
быть комбинированы на разных сторонах фильтра
для изучения их взаимодействий (см. разд. 17.7.1).
Более того, возможно применение маленьких
вкладышей в фильтрационную лунку (например,
6,5 мм, Corning Costar #3423). При этом лунка, об¬
разованная на верхней стороне комплекса фильтров,
формирует мениск из среды, с большой площадью
поверхности, достаточной для газообмена. Можно
также изменить конфигурацию ткани повышением
или понижением уровня среды в чашке и, следова¬
тельно, в лунке. Более глубокий слой среды дает
сферический эксплантат, более мелкий слой дает
плоскую конфигурацию.
3) Давление 02. Эмбриональные культуры обычно
лучше хранятся на воздухе, но ткани эмбрионов
поздних стадий, новорожденных и взрослых орга¬
низмов лучше содержать при повышенном давле¬
нии кислорода [Trowell, 1959; de Ridder & Mareel,
1978; Zeltinger & Holbrook, 1997].
4) Перемешиваемые или статические культуры. Пере¬
мешиваемые культуры малых фрагментов тканей
раньше использовались для конфронтационных
культур при исследовании инвазии [Mareel et al.,
1979; Bjerkvig et al., 1986a, b] (см. протокол 25.5).
5) Качалочные или вращаемые культуры. Ткань при¬
крепляется к субстрату и подвергается поперемен¬
ному воздействию жидкой культуральной среды
и газовой фазы путем помещения культурального
сосуда на раскачивающуюся платформу [см. прото¬
кол 23.9; Nicosia et al.,1983], или прикрепления тка¬
ни к стенке вращающегося флакона или пробирки
(см. разд. 5.3.4 и протокол 26.3).
25.3. ГИСТОТИПИЧЕСКАЯ КУЛЬТУРА
Гистотипическая культура в данном контексте опре¬
деляется как клеточная культура высокой плотности,
близкой к плотности клеток в ткани in vivo. Были
сделаны разнообразные попытки воспроизведения
тканеподобной архитектуры на основе диспергирован¬
ных монослойных культур. Green and Thomas [1978]
показали, что эпидермальные кератиноциты человека
будут образовывать дерматоглифы (т. е. фрикционные
рубцы), если их содержать без пересадки в течение
нескольких недель, a Folkman and Haudenschild [1980]
смогли показать образование капиллярных трубочек в
культуре сосудистого эндотелия в присутствии факто¬
ра роста эндотелия и среды кондиционированной опу¬
холевыми клетками. Когда клетки достигают высокой
плотности, им может недоставать питательных веществ.
Чтобы избежать этого, отношение объема среды к ко¬
личеству клеток должно оставаться примерно таким
же, как в культуре с низкой плотностью клеток. Это
может быть достигнуто посевом клеток на небольшое
покровное стекло в центре большой чашки типа «не
для культуры ткани» или использованием вкладышей
в фильтрационные лунки, которые дают возможность
образования как поляризованных культур с высокой
плотностью клеток, так и гетеротипической комбина¬
ции клеточных типов с последующим воссозданием ор¬
ганотипических культур (см. протокол 25.4). Высокое
отношение среда/клетки может также быть достигнуто
путем перфузии (см. разд. 25.3.2).
25.3.1. Метод геля и губки
Leighton первым продемонстрировал тот факт, что
как нормальные, так и малигнизированные клетки
проникают в целлюлозную губку [Leighton et al.,
1968], обработка коллагеном усиливает этот процесс.
Вместо целлюлозы может быть использован Gelfoam
(желатиновый губчатый матрикс, используемый в
восстановительной хирургии) [Sorour et al., 1975], он
был использован в исследованиях действия механичес¬
25.3. Гистотипическая культура
493
кого растяжения на развитие легких [Liu et al., 1995].
Эти системы требуют гистологического анализа и
ограничены в размерах, подобно органным культурам,
в результате необходимости диффузии газов и пита¬
тельных веществ. Использование трехмерной губки и
гелей значительно повысилось при развитии тканевой
инженерии (см. разд. 25.3.8).
Коллагеиовый гель. Коллагеновый гель (нативный
коллаген, в отличие от денатурированного коллагено¬
вого слоя) представляет собой матрикс для морфоге¬
неза первичных эпителиальных структур. Показано,
что различные типы клеток могут проникать в такой
матрикс с развитием тканеподобной гистологической
структуры. Эпителий молочной железы образует
рудиментарные трубчатые и железистые структуры
при росте в коллагене [Gomm et al., 1997], в то время
как карцинома молочной железы обнаруживает менее
упорядоченный рост [Berdichevsky et al., 1992]. Кле¬
точная линия MDCK почечного эпителия отвечает на
паракринную стимуляцию со стороны фибробластов
продукцией трубчатых структур, но только в колла-
геновом геле [Kenworthy et а1.,1992]. Рост отростков
невритов из нейронов симпатического ганглия, расту¬
щего на коллагеновом геле, соответствует ориентации
коллагеновых волокон в геле [Ebendal, 1976].
Матригель. Matrigel представляет собой коммер¬
ческий продукт, (B-D Biosciences), полученный из
внеклеточного матрикса мышиной саркомы Engel-
breth-Holm-Swarm (EHS), который был использован
для обработки пластика (см. разд. 8.4.1), но также мо¬
жет применяться для образования геля. Он состоит из
ламинина, коллагена, фибронектина и протеогликанов
с рядом связанных с ними факторов роста, хотя он
может быть получен в форме со сниженной концен¬
трацией факторов роста. Обычно его используют в
качестве субстрата для исследований эпителиального
морфогенеза [Larsen et al., 2004], образования капил¬
ляров из эндотелиальных клеток [Jain et al., 1997; Vou-
ret-Craviari et al., 2004], и в изучении злокачественной
инвазии [De Wever et al., 2004]. Тем не менее Matrigel
является комплексным, не до конца изученным мат¬
риксом и может ингибировать некоторые морфогенети¬
ческие события, такие как тубулогенез клеток MDCK,
индуцированный фактором роста гепатоцитов (HGF)
[Williams & Clark, 2003].
25.3.2. Пустотелые волокна
Поскольку при высокой клеточной плотности поступ¬
ление среды и газообмен становятся ограниченными
[Knazek et al., 1972; Gullino and Knazek, 1979] разрабо¬
тали перфузионную камеру на основе пучка пластико¬
вых капиллярных волокон, которая в настоящее время
является коммерчески доступной (см. Приложение II,
половокнистая перфузионная культура). Волокна
являются проницаемыми для газов и питательных
веществ и поддерживают клеточный рост на их внеш¬
ней поверхности. Среда, насыщенная 5% С02, прока¬
чивается насосом через центры капилляров, а клетки
добавляются во внешнюю камеру, окружающую пучки
волокон (см. рис. 8.11; см. также рис. 26.6). Клетки
прикрепляются и растут на внешней стороне пучков
волокон, питаясь за счет диффузии из прогоняемой по
волокнам среды, и могут достичь клеточной плотности
аналогичной таковой в тканях. Различные пластики и
ультрафильтрационные свойства материалов позволя¬
ют регулировать молекулярный вес диффундирующих
веществ, ограничивая его 10, 50 или 100 kDa.
Считается, что клетки этого типа в культуре с
высокой плотностью ведут себя как ткани in vivo. На¬
пример, в таких культурах клетки хориокарциномы
высвобождают больше человеческого хорионического
гонадотропина [Knazek et al., 1974], чем в соответ¬
ствующей монослойной культуре, а клетки карциномы
толстой кишки производят более высокий уровень
СЕА [Rutzky et al., 1979; Quarles et al., 1980]. Тем не
менее существует ряд технологических сложностей в
подключении камер, и они достаточно дорогостоящи.
Более того, взятие образцов из таких камер, определе¬
ние клеточной концентрации также сложно. В целом,
однако, полые волокна представляются идеальной
системой для изучения синтеза и высвобождения
биофармацевтических препаратов, что позволяет в на¬
стоящее время использовать их в полупромышленных
масштабах (см. разд. 26.2.6).
25.3.3. Сфероиды
При культивировании диссоциированных клеток во
вращательном шейкере они могут вновь объединяться
в кластеры. Диспергированные клетки из эмбриональ¬
ных тканей будут высокоспецифичным образом рас¬
пределяться в ходе этой агрегации [Linser & Moscona,
1980]. Показано, что клетки в этих гетеротипичных
агрегатах способны подразделяться на группы с фор¬
мированием тканетипичных структур. Гомотипическая
реаггрегация также происходит достаточно легко. Сфе¬
роиды, которые образуются во вращательном шейкере
или при росте на агаре, используют обычно в качестве
модели для исследований в химиотерапии in vitro
[Twentyman, 1980] и для характеристики злокачествен¬
ной инвазии [Mareel et al., 1980]. Как и в случае орган¬
ной культуры, рост сфероидов ограничен диффузией,
может быть достигнуто стационарное состояние, при
котором клеточная пролиферация во внешних слоях
уравновешивается центральным некрозом (рис. 25.4).
Дальнейшее введение и протокол 25.2 для приготов¬
ления многоклеточных опухолевых сфероидов предо¬
ставлены М. Boyd and R.J. Mairs, Center for Oncology
and Applied Pharmacology, Glasgow University, Cancer
Research UK Beatson Laboratories, Garscube Estate,
Bearsden, Glasgow G61 1BD, Scotland.
Многоклеточные опухолевые сфероиды являются
пролиферирующей моделью бессосудистого метаста-
494
ГЛАВА 25. ОРГАНОТИПИЧЕСКАЯ КУЛЬТУРА
Рис. 25.4. Разделение клеток в сфероидах. Сечение че¬
рез зрелый сфероид диаметром примерно 600-800-мкм.
(а) Ауторадиографически меченый [1251] иоддеоксиуридин
(IUdR), обнаруживает распределение метки на периферии.
(б) Иммунопероксидазное окрашивание с анти-BUdR, дает
похожее распределение метки по периферии сфероида. (С лю¬
безного разрешения Ali Neshasterez.)
зирования. Трехмерная структура сфероидов позво¬
ляет экспериментально изучать различные аспекты
проникновения лекарств и устойчивости опухолевых
клеток к облучению и химиотерапии, которые зависят
от межклеточных контактов. Сфероиды также хорошо
подходят для изучения побочного эффекта при экспе¬
риментальной направленной избирательной терапии
или генной терапии [Boyd et al., 2002, 2004]. Опухоле¬
вые сфероиды человека легче получить из установив¬
шихся клеточных линий или ксенотрансплантатов, чем
из первичных опухолей [Sutherland, 1988]. Из суспен¬
зии единичных клеток (трипсинизированного моно¬
слоя или дезагрегированной опухоли) клетки могут
быть инокулированы в сосуды с магнитной мешалкой
(Techne) с инкубацией, позволяющей образоваться
маленьким агрегатам через 3-5 дней [Boyd et al., 2001 ].
Эта процедура оптимальна; однако большинство кле¬
точных линий не образуют сфероидов таким методом.
Агрегаты могут быть получены иначе: из клеточной
суспензии в стационарных флаконах, предварительно
покрытых агаром. Агрегаты могут быть оставлены в
тех же флаконах или перенесены индивидуально при
помощи пипетки во многолуночный планшет, где про¬
должение роста в течение нескольких недель приведет
к образованию сфероидов максимального размера, при¬
мерно 1000 мкм [Yuhas et al., 1977; Sutherland, 1988].
ПРОТОКОЛ 25.2. Сфероиды
Схема
Трипсинизировать клетки монослоя или дизагре-
гировать первичную ткань и посеять клетки на
субстрат, покрытый агаром. Перенести агрегаты в
24-луночные планшеты для анализа.
Материалы
Стерильные:
• Инертный агар (Noble) (Difco, B-D Biosciences).
• Ростовая среда
• Сверхчистая вода (UPW)
• Трипсин, 0.25%, в PBSA
• Флаконы, 25 см2, или 24-луночные планшеты
• Чашки Петри, 9 СхМ
Замечание. Когда для покрытия субстрата исполь¬
зуется агар, все флаконы, чашки, планшеты должны
быть стерильны, но нет необходимости доводить
стерильность до степени работы с тканевыми куль¬
турами
Нестерильные:
• Pi-насос (см. Приложение II) или эквивалентное
пипетирующее устройство
Протокол
Обработка агаром 25-см2 флаконов:
1. Добавьте 1 г агара Noble в 20 мл UPWb 100-мл в
бутыль из боросиликатного стекла со свободно
завинчивающейся крышкой.
2. Подогрейте агар на водяной бане при 100 °С в
течение 10 мин до полного растворения агара.
3. Немедленно добавьте содержимое бутыли к 60 мл
ростовой среды, предварительно подогретой до
37 °С, разлейте по 5 мл в каждый флакон. Убе¬
дитесь, что в агаре нет пузырьков.
4. Оставьте агар при комнатной температуре
~5 мин, в результате получите флакон, покрытый
1,25% агаром.
Обработка агаром многолуночных планшетов:
1. Добавьте 0,5 г агара к 10 мл UPW, подогрейте,
как описано в шаге 2 для 25 см2 флаконов, а затем
добавьте 40 мл UPW.
2. Поместите 0,5 мл получившегося раствор в
каждую лунку 24-луночного планшета, чтобы
25.3. Гистотипическая культура
495
получить покрытие основания лунок 1% агаром.
Аккуратность и точность манипуляции важны
для того, чтобы правильно проводить фокуси¬
ровку микроскопа при последующем наблюдении
сферридов.
Инициализация сфероидов:
1. Трипсинизируйте конфлюэнтный монослой
(для установленных клеточных линий, см. про¬
токол 13.2) или дезагрегируйте (для плотных
опухолей, см. протоколы 12.5,12.6 и 12.8), чтобы
получить суспензию единичных клеток.
2. Нейтрализуйте трипсин при помощи среды, со¬
держащей сыворотку (если необходимо).
3. Подсчитайте количество клеток, используя элек¬
тронный счетчик клеток или гемоцитометр.
4. Поместите 5x105 клеток в 5 см ростовой среды
в каждый 25-см2 флакон, обработанный агаром,
инкубируйте культуры. Если клетки способны к
формированию сфероидов, через 3-5 дней будут
спонтанно образовываться маленькие (примерно
100-300 мкм в диаметре) агрегаты клеток.
Для последующего роста сфероиды должны быть
перенесены в новые 25-см2 флаконы или 24-луночнй
планшет.
Перенос в 25 см2 флаконы:
1. Перенесите содержимое исходных флаконов в
конические центрифужные пробирки или уни¬
версальные контейнеры.
2. Оставьте сфероиды осаждаться, удалите супер¬
натант с единичными клетками.
3. Ресуспендируйте сфероиды в свежей среде и
перенесите суспензию в новые флаконы, обра¬
ботанные агаром, где рост будет продолжаться
за счет деления клеток во внешнем слое.
Перенос в 24-луночный планшет:
1. Перенесите содержимое каждого из 25-см2 в 6-см
чашку Петри.
2. Добавьте 0,5 мл среды в каждую покрытую ага¬
ром лунку 24-луночного планшета.
3. Выберите отдельные сфероиды выбранного раз¬
мера при малом увеличении (х40) и, используя
пастеровскую пипетку, и Pi-насос или другое
аналогичное устройство при соответствующем
тщательном контроле, перенесите выбранные
сфероиды примерно одинакового диаметра в пок¬
рытые агаром лунки 24-луночного планшета.
4. Поместите планшет в С02-инкубатор.
5. Заменяйте среду в планшете один или два раза в
неделю (заменяя 0,5 мл каждый раз), или добав¬
ляйте 0,5 мл среды (не удаляя среду) один-два
раза в неделю, в результате через 2-4 недели
получите объем 2 мл в каждой лунке.
Анализ. Рост сфероидов в лунках или флаконах
может быть количественно учтен путем регулярного
(2-3 раза в неделю) измерения их диаметра с исполь¬
зованием окуляра-микрометра или координатной сетки
либо, что предпочтительно, путем измерения площади
сечения с помощью сканнера-анализатора изобра¬
жений. Самая точная кривая роста получена путем
выращивания сфероидов в лунках и индивидуального
их мониторинга. Сфероиды также могут использовать¬
ся для клоногенных исследований после обработки
тестируемым агентом. Сфероиды собирают в универ¬
сальный контейнер, отмывают в D-PBSA для удаления
остатков среды и затем инкубируют с трипсином при
37 °С в течение 5 мин. Затем сфероиды дезагрегируют
до суспензии единичных клеток путем пропускания
через иглу шприца, затем подсчитывают. Количество
жизнеспособных клеток оценивается по исключению
трипанового синего (см. протокол 22.1). Клетки затем
могут быть использованы для соответствующих кло¬
ногенных исследований [Boyd et al., 2001, 2004].
Вариации
Трансфецированные мозаичные сфероиды. Сфероиды
могут быть выращены из популяций клеток, которые
были трансфецированы различными генами. Клетки
сначала выращивают в монослое и затем трансфеци-
руются трансгеном и подвергаются селекции транс-
ген-экспрессирующих клеток. Мозаичные сфероиды
формируют добавлением трансфецированных и нетран-
сфецированных монослойных клеток в любых жела¬
тельных пропорциях [Boyd et al., 2002,2004]. Различные
клеточные популяции распределены в получившихся
сфероидах приблизительно равномерными мозаичны¬
ми участками, с поддержанием той же пропорции транс¬
фецированных и нетрансфецированных клеток, какая
была на стадии формирования (цв. вкладку 12а).
Приложения. Сфероиды имеют широкую сферу
применения при моделировании бессосудистого опу¬
холевого роста [Ward & King, 1997], исследовании
роли трехмерной пространственной конфигурации
в экспрессии гена в клеточной популяции [Waleh et
al., 1995; Dangles et al., 1997], а также цитотоксичного
воздействия. Параметры конечной точки воздействия
включают задержку роста, определение доли сферои¬
дов, стерилизованных («вылеченных») в результате
воздействия, формирование колоний в монослое пос¬
ле дезагрегации обработанных сфероидов [Freyer &
Sutherland, 1980, Boyd et al., 1999, 2004].
Важной областью использования сфероидов яв¬
ляется изучение проникновения цитотоксических
препаратов, антител или других молекул, использован¬
ных в направленной избирательной (таргет) терапии
[Sutherland, 1988; Carlsson & Nederman, 1989]. Эта
категория представляет собой особую область приме¬
нения, которая невозможна для суспензии единичных
клеток или монослойной культуры. Сфероиды также
могут быть полезны для изучения гибели клеток под
действием радионуклидов направленного действия
[Mairs & Wheldon, 1996, Boyd et al., 2001, Fullerton et
al., 2004]. Культура сфероидов также применяется в
сравнительных экспериментах по изучению инвазив-
ности сфероидов, полученных из малигнизированных
клеточных популяций, которые выращены в непосред¬
496
ГЛАВА 25. ОРГАНОТИПИЧЕСКАЯ КУЛЬТУРА
ственной близости от нормальных клеточных культур
[de Ridder,1997]. Такие методы также применялись
в неонкологических исследованиях патологических
процессов, таких как исследования ревматоидного
артрита [Ermis et al., 1998]. Мозаичные сфероиды
являются новой вариантной и имеют особое приме¬
нение в исследовании побочного действия. Например,
определенную сложность для генной терапии рака
представляет неэффективность процедур генного
переноса; они вызывают значительные побочные
эффекты в опухолевой популяции, которая не была
успешно трансфецирована. Мозаичные сфероиды от¬
ражают эту ситуацию in vitro и позволяют произвести
оценку различных форм побочного эффекта, таких как
радиационные перекрестные помехи при напрвленном
действии радиоактивных агентов на трансфецирован-
ные клетки [Boyd et al., 2002, 2004].
25.3.4. Вращающиеся камерные системы
Биореактор Miniperm. Перемешивание и аэрация
также может быть достигнуты вращением куль¬
турального сосуда в обычной роллерной бутыли
(см. протокол 26.3) или в двухкамерных системах
(рис. 25.5). Если клеточная суспензия ограничена до
одного небольшого компартмента, то клеточная кон¬
центрация может быть достаточно высокой, поскольку
клетки не разводятся средой. Концентрация продук¬
та (например, антител) накапливается в клеточном
компартменте, в то время как питательные вещества
и продукты отходов диффундируют через полупро¬
ницаемую мембрану в мембранный компартмент и из
него. Примером такого рода оборудования является
система MiniPERMTM, двухкомпартментный ци¬
линдр с клетками в меньшем компартменте и средой в
большем, разделенными полупроницаемой мембраной
(Vivascience, Sartorius; см. рис. 25.5). Он вращается
Клеточная камера Резервуар со средой
Рис. 25.5. Вращающаяся камерная система. Heraeus
Miniperm. Концентрированная клеточная суспензия в левой
камере отделена от камеры со средой полупроницаемой мем¬
браной. Продукт с высоким молекулярным весом остается
вместе с клетками и может быть выделен через порт для
отбора проб, в то время как замещение среды осуществляется
через правый порт. Перемешивание достигается путем враще¬
ния камеры на роллерном стеллаже (Miniperm: Vivascience,
Sartorius AG)
для гарантированного получения смеси, а среда может
быть сохранена или заменена без разрушения клеток
или продукта, хотя главной целью является получение
партии продукта, например моноклональных антител.
Конфигурация камеры и медленное вращение приво¬
дят к образованию агрегатов и таким образом может
повысить получение продукта.
Вращательная система культуры клеток
(RCCS). Заинтересовавшись концепцией клеточного
роста в условиях уменьшенной гравитации в 1980-х гг.,
NASA сконструировала вращательную систему, в ко¬
торой клетки, растущие в суспензии, оказываются под
воздействием смоделированной нулевой гравитации в
медленно вращающейся камере, постоянно изменяю¬
щей вектор седиментации (рис. 25.6). Клетки остаются
стационарными и подвергаются нулевому касатель¬
ному напряжению, что приводит к формированию
трехмерных агрегатов, сфероидподобных структур,
которые могут стать более дифференцированными с
повышением уровня формирования продукта [Мате¬
риалы рабочего совещания NASA по регуляции клеток
и дифференцировке клеток в биореакторах, 1997].
Газообмен происходит в направлении из цилиндра,
содержащего клетки, через центральную силиконовую
мембрану. Когда вращение прекращается, агрегаты
оседают, и среда может быть замещена. Такой биоре¬
актор производится компанией Synthecon, Inc. в форме
одноразового блока RCCS или системы многократ¬
ного использования STLV (медленно вращающийся
латеральный сосуд). В дополнение к первоначальной
цели, для определения действия невесомости на клет¬
ки с применением в космической программе [Freed &
Vunjak-Novakovic, 2002] этот культуральный сосуд
также может использоваться как биореактор для пар¬
тии культуры или тканево-инженерных конструктов
[Dutt et al., 2003; Vunjak-Novakovic, 2005].
25.3.5. Иммобилизация живых клеток
в альгинате
Метод инкапсулирования живых клеток в альгинатных
шариках был широко использован в эксперименталь¬
ных исследованиях, например для исследования гиб-
ридомных клеток при производстве моноклональных
антител [Lang et al., 1995], гормон-продуцирующих
клеток, использованных в модели животных для ле¬
чения сахарного диабета [Soon-Shiong et al., 1992] и
хондроцитов (см. протокол 23.16, цв. вкладку 186).
Альгинат первоначально был выделен из корич¬
невых морских водорослей Laminaria, Macroaysti и
Ascophyllum. Он состоит главным образом из двух
типов моносахаридов: L-глюкуроновой кислоты (G) и
D-маннуроновой кислоты (М). Структурно альгинат
содержит чередующиеся молекулы М и G валентные
катионы связаны между отдельными единицами G, и
инициируют образование растянутой сети альгинатно-
го геля. Альгинатные гели могут формировать шарики,
25.3. Гистотипическая культура
497
белков в организм. Таким образом, такие альгинатные
«биореакторы» могут иметь важный терапевтический
потенциал для лечения ряда болезней, при которых
альгинат может защищать инкапсулированные клетки
от разрушения иммунной системой.
Протокол 25.3 и предшествовавшее введение в аль-
гинатное инкапсулирование предоставлены Тгасу-Апп
Read и Rolf Bjerkvig, Department of Anatomy and Cell
Biology, University of Bergen, Norway.
Рис. 25.6. Вращающаяся система для культуры клеток
Synthecon Rotatory Cell Culture System. В системе Rotary
Cell Culture System™ клетки поддерживаются в суспензии
путем установления скорости вращения цилиндрической
культуральной камеры. Газопроницаемый силиконовый мем¬
бранный сердечник дает клеткам достаточное количество газа,
в то время как отсутствует трение жидкости, вызывающее
формирование пузырей. Биореактор, сконструированный
NASA, можно приобрести в компании Synthecon (см. При¬
ложение III) и широкой сети дистрибьютеров. (а) Камера
многократного использования, (б) Камера одноразового
использования (см. также цв. вкладку 22в)
если капать его малыми порциями в буфер, содержащий
двухвалентные катионы, такие как Са2+. Механическая
прочность, объем, стабильность и пористость коррели¬
руют с содержанием G, так что альгинатные шарики с
высоким содержанием G имеют больший размер пор, в
диапазоне 5-200 нм [Martinsen et al., 1989; Miura et al.,
1986]. Поры таких размеров позволяют идти свободной
диффузии макромолекул из альгинатных шариков и
внутрь них. Иммунная реакция хозяина на альгинат
может быть значительно снижена использованием
альгината при высоких концентрациях G и низких
концентрациях М [Otterlei et al., 1991].
В настоящее время большое количество клеточных
типов могут быть генетически сконструированы для
получения различных специфических белков. Пу¬
тем инкапсулирования клеток в альгинате получено
средство доставки для транспорта рекомбинантных
ПРОТОКОЛ 25.3. Альгинатное
капсулирование
Схема
Трипсинизировать клетки из флакона 75-см2, под¬
считать их и смешать с раствором альгината натрия.
По капле выдавить смесь в раствор хлористого на¬
трия для формирования шариков и культивировать
в суспензии.
Материалы
Стерильные:
• Ростовая среда для выбранных клеток
• D-PBSA
• Трипсин (концентрация соответствует принятой
для субкультуры данных клеток)
• Солевой раствор, содержащий:
а) NaCl 8,0 г
б) D-глюкоза 1,0 г
в) Довести объем UPW до 1л
г) Довести pH до 7,2-7,4 добавлением НС1 или
NaOH
д) Стерилизовать автоклавированием
• СаС12, 0,1 М, содержащий
е) Солевой раствор 500 мл
ж) СаС12 • 2Н20 7,35 г
з) Довести pH до 7,2-7,4 добавлением НС1 или
NaOH
и) Стерилизовать автоклавированием
• Альгинат натрия (PRONOVA™ UP LVG): высо¬
кой чистоты, низкой вязкости, с высоким содер¬
жанием глюкуроновой кислоты
• Стерилизующие фильтры, 0,45 мкм непироген-
ные (Millipore)
Протокол
1. Растворите альгинат в солевом растворе до
концентрации 1,5% и встряхивайте на шейкере
получившийся раствор как минимум 4 часа при
комнатной температуре, пока весь альгинат пол¬
ностью не растворится. Растворенный альгинат
может храниться при 4 °С не более 3 дней.
2. Профильтруйте альгинат трижды через стериль¬
ные непирогенные 0,45-мкм фильтр. Последнюю
фильтрацию следует проводить непосредственно
перед использованием альгината.
498
ГЛАВА 25. ОРГАНОТИПИЧЕСКАЯ КУЛЬТУРА
3. Вырастите клетки до конфлюэнтного слоя.
4. Трипсинизируйте клетки добавлением 3 мл трип¬
сина и подсчитайте их.
5. Центрифугируйте клетки при 900 об./мин в
течение 4 мин и удалите полностью удалите су¬
пернатант с трипсином.
6. Смешайте клетки с альгинатом до концентрации
2x106 кл./мл осторожным ресуспендированием
клеток в альгинате. Очень важно не допускать
образования пузырей воздуха в альгинате, по¬
скольку это может привести к образованию дыр
в альгинатных шариках.
7. Перенесите суспензию клеток в альгинате в сте¬
рильный шприц, с надетой на него иглой 27G.
8. По каплям выпускайте суспензию в стакан,
содержащий 0,1 М СаС12при постоянном пере¬
мешивании жидкости, что инициирует форми¬
рование альгинатных шариков.
9. Оставьте примерно на 10 мин, затем трижды
отмойте в D-PBSA.
10. Последний раз отмойте в ростовой среде.
11. Культивируйте шарики в 175-см2 культураль¬
ных флаконах, содержащих ростовую среду при
37 °С, 100% относительной влажности, и 5% С02
в воздухе.
25.3.6. Вкладыши для фильтрационных
лунок
Вкладыши для фильтрационных лунок представля¬
ют собой коммерциализацию системы для культуры
органов на основе фильтра, которые развивались с
1950-х годов и используются с тех пор в различных
формах. Фильтрующий субстрат представляет собой
микроокружение для изучения межклеточных взаимо¬
действий, расслоения, поляризации и моделирования
тканей. Полярность и функциональное единство мо¬
жет быть установлена по типу эпителия щитовидной
железы [Chambard et al., 1983], кишечника [Halleux &
Schneider, 1994] и почки [Mullin et al., 1997] (см.
разд. 17.7.4). Культуры на фильтрах позволяют воссо¬
здавать многослойный эпидермис [Limat et al., 1995;
Kondo et al., 1997; Maas-Szabowski et al., 2000, 2002].
Другие культуры используются для изучения инвазии
гранулоцитов или злокачественных клеток [McCall et
al., 1981; Elvin et al., 1985; Repesh, 1989; Schlechte et al.,
1990; Brunton et al., 1997]. Одним из самых главных
преимуществ вкладышей в фильтрационные лунки
является возможность рекомбинации клеток при очень
высокой плотности, близкой к тканевой. При этом в
мультирепликативной форме сохраняется доступ к
среде и газообмен. В настоящее время различными
компаниями предлагаются вкладыши в фильтраци¬
онные лунки (см. Приложение II) из разнообразных
прозрачных и полупрозрачных материалов, включая
поликарбонат, PTFE тетрафталат полиэтилена разме¬
рами от 6,5 мм до 9 см, подходящих для 24-, 12- и 6-лу-
ночных планшетов чашек большего размера (рис. 24.5,
24.6; цв. вкладка Plate 13; табл. 25.1). Фильтры могут
быть предварительно покрыты коллагеном, ламини-
ном, фибронектином или Matrigel.
Анализ:
1) Проницаемость: Некоторые эпителиальные (на¬
пример, MDCK и Сасо-2) и эндотелиальные клет¬
ки (например, из пупочной вены) образуют плот-
ПРОТОКОЛ 25.4. Вкладыши
для фильтрационных лунок
Схема
Посеять клетки на вкладыши для фильтрационных
лунок, и культивировать клетки при излишке среды
в многолуночных планшетах.
Материалы
Стерильные:
• Примерно 0,5х106 клеток на см2 фильтра
• Ростовая среда, 1 -20 мл на фильтр (в зависимос¬
ти от емкости принимающей фильтр)
• Вкладыши для фильтрационных лунок
• Многолуночные планшеты для вкладышей: 6-,
12- или 24-луночные
• Пинцеты искривленные
Нестерильные:
• Автоматическая пипетка
Протокол
1. Поместите фильтрационные лунки в планшет
или чашку.
2. Добавьте среду, наклоняя чашку так, чтобы среда
заполнила пространство под фильтром и вытес¬
нила воздух. Добавляйте среду, пока ее уровень
не будет на уровне фильтра (2,5 мл для 6-луноч¬
ного планшета, 1,0 мл для 24-луночного).
3. Установите уровень в чашке и добавьте lxlO6 кле¬
ток в 2 мл среды на верхнюю часть фильтра при
25-мм диаметре фильтра или 5х105в 200 мкл
среды при 6,5-м диаметре фильтра стараясь не
повредить фильтр.
4. Поместите чашку во влажный С02-инкубатор
в защитную коробку (см. разд. 6.6.2). Чрезвы¬
чайно важно избегать встряхивания коробки,
культуры не должны передвигаться в инкубаторе
во избежание расплескивания и последующей
контаминации.
5. Монослои должны установиться на 3-5 дней,
хотя для гистотипической дифференцировки
может потребоваться еще 5-10 дней или больше
(например, для установления поляризационного
транспорта).
6. Культуры могут поддерживаться неопределенно
долго, с замещением среды или перенесением
вкладыша в свежую лунку или чашку каждые
3-5 дней.
25.3. Гистотипическая культура
499
Таблица 25.1. Типы вкладышей в фильтрационную лунку
Изготовитель
Название
Материал
Характеристики
Прозрачность
Пористость (мкм)
Millipore
Millicel
Нитроцеллюлоза
Сетчатый
Непрозрачный
0,45
Полиолефин
Сетчатый
Прозрачный
0,45
Coming Costar
Transwells
Поликарбонат
Абсолютный
Прозрачный
5-8
Полупрозрачный *
0,45-1,0
Falcon (B-D Biosciences)
Inserts
Тетрафталат полиэтилена
Абсолютный
Прозрачный
0,45-3
Nunclon
Anocel
Керамика
Решетчатый
Прозрачный
0,01
Earl-Clay
Ultraclone
Коллаген
Сетчатый
Прозрачный
* Чем выше частота пор, тем ниже прозрачность. Низкопоровый фильтр имеет высокую частоту пор и, соответственно, мень¬
шую прозрачность.
ные межклеточные контакты в течение нескольких
дней после достижения конфлюэнтности. Этот
процесс можно ускорить предварительной обра¬
боткой мембран коллагеном. Трансэпителиальная
проницаемость после этого становится ограничен¬
ной физиологически регулируемым транспортом
через клетки, а межклеточный транспорт падает
до нуля. Это процесс можно регулировать, наблю¬
дая перенос красителя (например люциферина
желтого), или меченого соединения, такого как
[14С] метилцеллюлоза или [14С] инулин, через
мембрану. Проницаемость также можно оценить
по трансэпителиальному электрическому сопро¬
тивлению (TEER).
2) Поляризованный транспорт. Добавление меченой
глюкозы или аминокислоты в верхний компартмент
вкладыша позволит наблюдать транспорт их в ниж¬
ний компартмент, при условии отсутствия обратно¬
го потока. Если клетки обладают р-гликопротеином
или каким-то другим транспортером веществ нару¬
жу, то в нижний компартмент могут быть добавлены
цитотоксин (например, винбластин), который будет
переноситься в верхний компартмент, но не будет
возвращаться обратно.
3) Проникновение клеток через фильтр. Трипсини¬
зируйте и подсчитайте клетки на каждой стороне
фильтра по очереди (трипсинизированные клетки
не проходят даже через фильтр с диаметром пор
8-мкм, поскольку их сферический диаметр в сус¬
пензии превышает эти размеры) или фиксируйте
фильтр, залейте и приготовьте стандартные срезы
для электронной микроскопии или гистологических
исследований. Визуализация тотального препарата
возможна путем заливки и окраски всего препарата
по Гимза на предметном стекле в DPX под покров¬
ным стеклом при давлении, так чтобы расплющить
препарат. Дифференциальный подсчет возможно
провести при последовательной фокусировке мик¬
роскопа на различных оптических плоскостях.
4) Отделение клеток от фильтра и седиментация их
на дно чашки. Подсчитайте клетки после трипсини¬
зации или путем сканирования.
5) Разграничение пространства над и под фильтром.
Подсчитайте клетки как в п. (3), или предваритель¬
но пометьте клетки родамином или изотиоцианатом
флюоресцеина (5 мкг/мл в течение 30 мин в трип-
синизированной суспензии) и измерьте с каждой
стороны фильтра флюоресценцию солюбилизиро¬
ванных трипсинизированных клеток (0,1% SDS в
0,3 N NaOH в течение 30 мин).
6) Клеточная инвазия. Предварительно покройте
фильтр клеточным слоем нормальных фиброблас¬
тов, MDCK либо подобных им клеток. Используя
микроскоп, убедитесь, что достигнуто конфлюэнт¬
ное состояние, и затем посейте EDTА-диссоцииро¬
ванные клетки поверх этого слоя (105-106клеток
на фильтр). Если исследуемые клетки RITC- или
FITC-меченые, то измерьте флюоресценцию, ко¬
торая появится в результате появления клеток под
фильтром.
7) Инвазия через матрикс. Нанесите на фильтр
Matrigel, поместите клетки над Matrigel, и следите
за появлением клеток под фильтром [Repesh, 1989;
Schlechte et а1.,1990]. Можно также посеять клетки
на нижнюю поверхность фильтра, покрыть верх¬
нюю поверхность фильтра Matrigel, и наблюдать
за инвазией клеток в Matrigel при помощи конфо¬
кального микроскопа [Brunton et al., 1997].
Вариации
1) Глубина слоя среды. Толщина слоя среды над филь¬
тром определяет давление кислорода на уровне рос¬
та клеток. Кератиноциты или пневмоциты типа II
из легочных альвеол потребуют меньшей толщины
слоя среды и высокого давления кислорода, в то
время как энтероциты, такие как Сасо-2, будут луч¬
ше расти при более глубоком погружении и более
низком давлении кислорода.
2) Пористость фильтра. Фильтры с диаметром пор
1 мкм позволяют осуществляться межклеточному
взаимодействию и контакту без проникновения
через фильтр. Фильтр с диаметром пор 8-мкм
позволяет живым клеткам проходить сквозь
фильтр. Фильтры с порами 0,2-мкм, по-видимому,
исключают межклеточный контакт. Низкопоровые
фильтры могут быть использованы для изучения
клеточного взаимодействия без непосредственного
контакта клеток.
500
ГЛАВА 25. ОРГАНОТИПИЧЕСКАЯ КУЛЬТУРА
Фильтр Среда или Клеточ-
D-PBSA ный слой
u
Чашка Петри или
лунка многолуночного
планшета
Среда
Взаимодействующий клеточный слой,
например стромальных фибробластов
Коллаген или Matrigel
\jsrTW
Взаимодействующий клеточный слой,
например стромальных фибробластов,
заключенный в коллагене
Коллаген и
1ли Matrigel
Взаимодействующий клеточный слой, например
эндотелиальные клетки под фильтром
Рис. 25.7. Вкладыши в фильтрационную лунку. Схема
гипотетического вкладыша в фильтрационную лунку.
(а) Монослой, выросший на верхней части фильтра, (б) Мо¬
нослой, выросший на матриксе на верхней части фильтра,
(в) Взаимодействующий клеточный слой, добавленный к
нижней стороне фильтра, (г) Взаимодействующий клеточ¬
ный слой, добавленный к матриксу. (Э) Взаимодействующий
клеточный слой, добавленный к нижней стороне фильтра с
матриксным покрытием над ним
3) Взаимодействия клеток через фильтр. Переверните
вкладыш в фильтрационную лунку, поместите его
верхней стороной в чашку Петри и нанесите на
его нижнюю сторону (теперь располагающуюся
сверху) 0,5 мл суспензии клеток с концентрацией
2х106 кл./мл. Поместите крышку поверх чашки
и, слегка касаясь капли клеточной суспензии, до¬
бейтесь, чтобы вся среда протекла через фильтр.
Глубина чашки должна быть примерно на 0,5-1 мм
больше, чем высота вкладыша в лунку, так чтобы
Рис. 25.8. Вкладыши Transwells. Вкладыши в фильтра¬
ционную лунку в 12-луночном планшете, отдельно сбоку
вкладыш (Coming.)
образовалось капиллярное пространство между
крышкой и фильтром. Инкубируйте фильтр в
течение 18 ч. Капиллярность будет удерживать
среду и клетки, пока клетки не осядут на фильтр
и не прикрепятся к нему [Brunton et al., 1997]. На
следующий день переверните фильтр и внесите в
лунку взаимодействующие клетки или Matrigel,
как это описано в протоколе 25.4.
25.3.7. Культуры нейрональных агрегатов
Агрегирующие культуры фетальных клеток мозга
широко используются для изучения дифференци¬
ровки нервных клеток [Seeds, 1971; Trappet al., 1981;
Bjerkvig et al., 1986а]. Агрегирующие клетки проходят
те же стадии дифференцировки, которые наблюдаются
in vivo, что приводит к формированию органоидной
структуры, состоящей из зрелых нейронов, астроцитов
и олигодендроцитов. Также формируются выступаю¬
щие нейропили. В области биологии новообразований
агрегаты могут быть использованы для изучения in
vitro инвазивных процессов клеток опухоли мозга
[Bjerkvig et al., 1986Ь]. Предварительное введение и
протокол 25.5 для агрегирующих культур клеток мозга
предоставлены Rolf Bjerkvig, Department of Cell Biology
and Anatomy, University of Bergen, У Zrstadveien 19,
N-5009, Bergen, Norway.
В течение 20 дней в культуре агрегаты станут сфе¬
рическими и приобретут органоидную структуру.
Анализ. Зафиксируйте образец и залейте его в
парафин или другую среду для гистологических или
электронно-микроскопических исследований. Олиго¬
дендроциты, астроциты и нейроны могут быть иденти¬
фицированы путем электронно-трансмиссионной мик¬
роскопии или иммуногистохимического исследования
локализации основного миелинового белка, кислого
25.3. Гистотипическая культура
501
ПРОТОКОЛ 25.5. Нейрональные агрегаты
Схема
Изъять мозг из плодов крыс на 17-й или 18-й день
созревания (проконсультируйтесь с местным Коми¬
тетом по Биоэтике, см. разд. 7.9.1) и готовить суспен¬
зию единичных клеток мозга. Формировать агрегаты
клеток мозга культивированием в покрытых агаром
лунках многолуночного планшета. Клетки в агрега¬
тах образуют зрелую органоидную структуру мозга
в течение 20 дней культуры.
Материалы
Стерильные:
• Модификация Дюльбекко среды Игла, содер¬
жащая 10% термически инактивированной сы¬
воротки теленка; четырехкратная концентрация
вспомогательных аминокислот; L-глютамин,
2 мМ; пенициллин 100 U/мл; стрептомицин,
100 мкг/мл
• Фосфатно-буферный раствор (PBS) с Са2+ и
Mg2+
• Трипсин, тип II (0,025% в D-PBSA)
• Агар (Difco)
• Многолуночная культура ткани (24-луночные
планшеты; Nalge Nunc)
• Чашки Петри, 10 см
• Тестовые пробирки, 12 мл
• Скальпели, ножницы хирургические пинцеты.
• Флаконы Эрленмайера, 100 мл, 2 шт
• Обработать ячейки агар-содержащей средой сле¬
дующим образом:
а) Приготовьте 3% маточный раствор (3 г агара
в 100 мл D-PBSA) во флаконе Эрленмайера.
б) Подогревайте флакон в кипящей воде, пока
агар не растворится. Поместите пустой флакон
Эрленмайера в кипящую воду и добавьте 10 мл
горячего раствора агара.
в) Медленно добавьте во флакон теплую полную
ростовую среду, пока концентрация агара в
среде не достигнет 0,75%.
г) Добавьте 0,5 мл теплого раствора агара в среде
в каждую лунку многолуночного планшета.
д) Оставьте агар остывать с образованием геля.
Обработанный многолуночный планшет
может храниться в холодильнике в течение
1 недели.
Нестерильные:
• Водяная баня
Протокол
1. Иссеките асептически целый мозг новорожден¬
ного крысенка или плода на 17-й или 18-й день
развития и поместите ткань в 10-см чашку Петри,
содержащую D-PBSA.
2. Используя скальпель, измельчите ткань на мел¬
кие кубики, размером -0,5 см3.
3. Перенесите ткань в пробирку и трижды промойте
в D-PBS А, оставляя ткань оседать на дно пробир¬
ки между промываниями.
4. Добавьте 5 мл раствора трипсина к ткани и ин¬
кубируйте в водяной бане в течение 5 мин при
37 °С.
5. Дезагрегируйте ткань гомогенизацией при помо¬
щи пастеровской пипетки примерно 20 раз.
6. Оставьте ткань для оседания примерно на 3 мин,
перенесите суспензию молочного вида без сгус¬
тков в пробирку, содержащую 5 мл ростовой
среды.
7. Добавьте 5 мл свежего трипсина к недиссоции-
рованной ткани и повторите трипсинизацию и
процедуру диссоциации еще дважды.
8. Центрифугируйте клеточную суспензию при
200 g в течение 5 мин.
9. Отберите и удалите супернатант, ресуспенди¬
руйте клетки, объедините их в 10 мл ростовой
среды.
10. Подсчитайте клетки и добавьте ЗхЮ6 клеток в
1 мл в каждую лунку, покрытую агаром.
11. Поместите многолуночный планшет в С02-инку-
батор на 48 ч.
12. Перенесите агрегаты в стерильные 10-см чашки
Петри и добавьте по 10 мл ростовой среды в
чашку.
13. При помощи пастеровской пипетки перенесите
более крупные агрегаты по одному в лунку, пок¬
рытую агаром.
14. Меняйте среду каждый третий день, тщательно
отбирая старую среду и добавляя новую.
глиального фибриллярного белка и нейрон-специфи-
ческой энолазы, соответственно.
Вариации. Суспензия единичных клеток может
быть получена механическим просеиванием через
стальное или нейлоновое сито [Trapp et al., 1981] (см.
разд. 12.3.8). При помощи ротационного шейкера мож¬
но получить реаггрегирующие культуры. Выберите
необходимую скорость (около 70 об./мин), перенесите
клетки в вортекс, так чтобы увеличить количество
столкновений клеток между собой. Это встряхивание
позволяет предупредить прикрепление клеток к стен¬
кам культурального флакона.
25.3.8. Тканевой эквивалент
и тканевая инженерия
Успехи технологии фильтрационных лунок, которые
поддерживались их коммерческой эффективностью,
привели к их быстрому распространению при разра¬
ботке методов органотипической культуры. Эквива¬
лент кожи был получен путем совместной культуры
эпидермиса и дермы (Mattek Epiderm, Episkin, Skin-
502
ГЛАВА 25. ОРГАНОТИПИЧЕСКАЯ КУЛЬТУРА
Неразлагающиеся
Искусственный
шелк, нейлон,
стекло
ВОЛОКНО
PGA, PLA,
Коллаген
Неразлагающиеся
Силикон, PTFE
(Goretex)
ТРУБКА Биодеградирующие
Коллаген
ПУЧОК МИКРОКАПИЛЛЯРОВ
2-D СЕТЬ
Искусственный
шелк, нейлон,
стекло
Коллаген,
PGA, PLA
3-D СЕТЬ
Искусственный
шелк, нейлон,
стекло
Коллаген,
PGA, PLA
Альгинат
2-D ФИЛЬТР
с
Поликарбонат;
нитрат целлюлозы;
ацетат целлюлозы,
керамика, нейлон
3-D КАПСУЛЫ
О
3-D ГЕЛЬ
Агар, Агароза
Коллаген,
желатин
Гибкая: целлюлоза
Твердая: керамика,
полистирол
3-D ГУБКА
few
Гибкая: коллаген,
желатин (Gelfoam)
Твердая: фосфат
кальция
Рис. 25.9. Каркасы и матрицы. Обзор различных типов каркасов и матриц, используемых в тканевых инженерных конструктах.
Разработано много различных геометрических вариантов, включая линейные волокна или трубки (сверху), двумерные сетчатые
экраны или фильтры (в центре) и трехмерные кубы и сферы (внизу). Хотя использовались неразлагающиеся материалы (слева
в каждом столбике), имеется общая тенденция к применению биодеградирующих материалов (справа в каждом столбике), таких
как коллаген, желатин, полигликолевая кислота (PGA) или полимолочная кислота (PLA) и фосфат кальция
Ethic), с использованием промежуточного слоя кол¬
лагена либо дермальных фибробластов, включенных
в коллаген [Limat et al., 1995; Maas-Szabowski et al.,
2000, 2002]. Модели паракринного контроля роста и
дифференцировки были разработаны на основе клеток,
выделенных из легких, [Speirs et al., 1991], простаты
[Thomson et al., 1997] и молочной железы [Van Roozen-
dahl et al., 1992].
Возможность применения гетеротипической куль¬
туры для изучения межклеточного взаимодействия
открыла широкие перспективы для изучения воспале¬
ния и раздражения in vitro (см. цв. вкладку 21; см. также
разд. 22.6), а также для создания моделей тканевого
взаимодействия с возрастанием соответствия модели и
процессов in vivo [Emura et al., 1997; Gomm et al., 1997].
Конструкция культур тканевых эквивалентов делает
возможной заместительную терапию. Эквиваленты
кожных культур используются при лечении ожого¬
вых повреждений [Hunziker & Limat 1999; Gobet et al.,
1997; Wright et al., 1998]. В настоящее время тканевая
инженерия применяется для создания замещающих
фрагментов во многих органах и тканях, включая
хрящи, костные фрагменты, связки, сердечные и ске¬
летные мышцы, кровеносные сосуды, печень и желч-
25.3. Гистотипическая культура
503
Рис. 25.10. MRI — мониторинг клеток в 3D конструктах, (а) Резервуар со средой и оборудование для подачи газа обеспечивают
конструкты тканево-инженерных хрящей в биореакторах BR1 и BR2. (6) Увеличенное изображение биореактора [см. также
Neves et al., 2003]. (в) Каталка с оборудованием для питания конструктов: резервуары со средой и газовое снабжение, поддержа¬
ние температуры при помощи водяной бани (г). (Э) Снятие показаний приборов с биореактора, соединенного с подачей среды;
помещенного в ЯМР-детектор. Выход может быть визуализирован в двух- и трехмерном изображении MRI (см. рис. 25.11) или
подвергнут спектральному анализу (см. рис. 26.18). (С любезного разрешения A.A. Neves & К. Brindle.)
504
ГЛАВА 25. ОРГАНОТИПИЧЕСКАЯ КУЛЬТУРА
ный пузырь [Atala & Lanza, 2002; Vunjak-Novakovic &
Freshney, 2005].
Как органотипические культуры требуют межкле¬
точного взаимодействия, так и конструкты, применя¬
емые в тканевой инженерии, часто требуют подобных
взаимодействий, например взаимодействия между
эндотелием и гладкомышечными клетками при рекон¬
струкции кровеносных сосудов [Klinger and Niklason,
2005]. В дополнение к биологическим взаимодей¬
ствиям, некоторые конструкты требуют наличия
физических сил. Так, например, скелетные мышцы
нуждаются в механической нагрузке на растяжение
[Shansky et al., 1997; Powell et al., 2002; Shansky et al.,
2005]; кости [Mullender et al., 2004] и хрящи [Seidel et al.,
2004] требуют сжимающего напряжения, а сосудистый
эндотелий в конструктах кровеносных сосудов требует
пульсирующего кровотока [Niklason et al., 2001].
Инженерия тканевых конструктов зависит от не¬
скольких компонентов, в зависимости от типа ткани:
1) Клетки ткани требуемой линии, при этом находя¬
щиеся на пролиферативной стадии клеток-предше-
ственников.
2) Взаимодействующие (дополнительные, интерактив¬
ные) клетки, например дермальные фибробласты
для кожи, гладкомышечные клетки в кровеносных
сосудах, или глиальные клетки в нейрональных
конструктах.
3) Биодеградирующая подложка, например полигли-
колевая кислота (PGA); полимолочная кислота
(PLA), или фосфат кальция (рис. 25.9).
4) Матрикс, например коллаген Matrix, вместо мягких
тканей, или покрывания подложки для улучшения
прилипания клеток.
5) Механический стресс; растягивающий для мышц,
сжимающий для хрящей и костей, пульсирующий
для кровеносных сосудов.
Нервные клетки также используются для тканевой
реконструкции, но в этих случаях чаще используется
клеточная суспензия, чем конструкт. Суспензия клеток
инъецируется в поврежденный участок [Franklin &
Barnett, 1997; Franklin & Barnett 2000; Wewetzer et al.,
2002; Totoiu et al., 2004; Groves et al., 1993].
Рис. 25.11. MRI конструкта хряща. Яркость изображения
пропорциональна клеточной плотности. Расстояние на шкале
в мм. (С любезного разрешения Dr. Kevin Brindle [по Thelwell
et al., 2001].)
25.4. СОЗДАНИЕ ТРЕХМЕРНЫХ
ИЗОБРАЖЕНИЙ КЛЕТОК
(З-Р-РЕКОНСТРУКЦИЯ)
Когда клетки вносятся в подложку в органотипиче¬
ский 3D конструкт, микроскопическое исследование
становится затруднительным. В этом случае использу¬
ются альтернативные методы визуализации состояния
клеток в конструкте. К таким методам относятся ЯМР
(ядерно-магнитный резонанс), если есть возможность
поместить биореактор в ЯМР-детектор (рис. 25.10).
Результат исследования может быть представлен как
магнитно-резонансное изображение (MRI) (рис. 25.11),
может быть проанализирован также эмиссионный
спектр (см. рис. 26.18).
Глава 26
КРУПНОМАСШТАБНОЕ
ПРОИЗВОДСТВО КЛЕТОК
Различие между обычным лабораторным масштабом
производства клеток и широкомасштабным полу¬
чением партии культуры является исключительно
условным, но обычно связывается с таким уровнем
продукции клеток, при котором требуется специаль¬
ная аппаратура и технологии. Хотя количество кле¬
ток lxlO9—1 хЮ10 может быть получено при простом
культивировании на мешалке объемом около 1-10 л
(см. разд. 13.7.5), крупномасштабное получение куль¬
туры в количестве 1 х10и-1х1012 клеток потребует
использования оборудования, от 100 л — лаборатор¬
ного ферментера до полупромышленных опытных
установок объемом от 100 до 1000 л. В промышленных
масштабах используют биореакторы объемом 5000-
20 000 л, но эти вопросы выходят за пределы данной
книги. Термины культивирование с перемешиванием
или спинкультура, являются синонимами, которые
используются для простых систем культивирования
клеток с постоянным перемешиванием в условиях
низкого технического уровня лабораторного обору¬
дования. Термины ферментер и биореактор хотя и не
являются синонимами, но их значения в значительной
мере перекрывают друг друга. Наименование фермен¬
тер получено для микробиологической культураль¬
ной системы, исходно разработанной для бактерий
и дрожжей и первоначально использовавшейся в
качестве лабораторного оборудования объемом при¬
мерно 50-100 л, однако увеличение в масштабах в
соответствии с развитием биотехнологической про¬
мышленности вместе с повышением разнообразия
в создании как монослойных так и суспензионных
культур привело к введению термина биореактор.
В тканевой инженерии этот термин включает куль¬
тивирование компонентов системы в относительно
небольших объемах.
Выбор метода увеличения масштабов культуры
зависит от того, делятся клетки в суспензии или они
должны быть прикреплены к субстрату. Методы,
используемые для суспензионных культур, обычно
проще и могут быть использованы первыми. Моно-
слойные культуры, которые связаны с прикрепле¬
нием к субстрату и, следовательно, с площадью роста
культуры, обычно более сложны, и рекомендуется
приступать к ним позже. Многие методы работы с
монослойными культурами приемлемы также для
суспензионных клеток.
26.1. КРУПНОМАСШТАБНОЕ
ПРОИЗВОДСТВО СУСПЕНЗИОННЫХ
КУЛЬТУР
Крупномасштабное производство клеток суспензи¬
онной культуры, включает в себя, прежде всего, уве¬
личение объема культуральной среды. Необходимо
перемешивание среды, если глубина слоя составляет
более 5 мм, при глубине выше 5-10 см (в зависимости
от соотношения площади поверхности и объема) для
поддержания адекватного газообмена требуется продув
С02и воздуха (рис. 26.1, 26.2). Перемешивание таких
культур лучше производить медленно при помощи
магнита, заключенного в стеклянном маятнике (Techne,
Integra), или при помощи лопасти с большой площадью
■
Рис. 26.1. Большой сосуд для перемешивания. 5-литровый
сосуд Techne 5-L на магнитной мешалке. Обратите внимание
на выступ маятника, который обеспечивает движение его по
кольцеобразному углублению дна флакона. Боковые гор¬
ловины используются для взятия образцов, перфузии С02,
которые могут потребоваться при данном объеме среды (см.
рис. 26.2)
506
ГЛАВА 26. КРУПНОМАСШТАБНОЕ ПРОИЗВОДСТВО КЛЕТОК
Боковое горлышко Другое боковое горлышко для
для подачи 5% С02 добавления клеток, выпускания 5% С02,
Рис. 26.2. Перемешиваемая культура. Схема большого
сосуда для перемешивания, пригодная для культивирования
до 8 л культуры
поверхности (Bellco). Скорость перемешивания долж¬
на быть между 30 и 100 об./мин, что достаточно для
предупреждения осаждения клеток, но не так много,
чтобы силы трения в жидкости могли привести к
повреждению клеток. Когда содержание сыворотки
в среде превышает 2%, следует добавлять пеногаси-
тель Antifoam (Dow Chemical Со.) или Pluronic F68
(Sigma) в концентрации 0,01-0,1%, в частности, при
барботировании. В отсутствие сыворотки может быть
необходимым повысить вязкость среды добавлением
карбоксиметилцеллюлозы (1-2%) (МВ-105).
ПРОТОКОЛ 26.1. Перемешивание 4-литровой
емкости с суспензионной культурой
Процедура приготовления суспензионной культу¬
ры клеток объемом 4 л производится следующим
образом:
Схема
Вырастить пробную культуру клеток и добавить ее в
предварительно подогретый аспиратор среды уже с
установившимся равновесием 5% С02. Медленно пе¬
ремешивать суспензию клеток с пропусканием газа,
пока не будет достигнута необходимая концентрация
клеток, затем собрать клетки.
Материалы
Стерильные или асептически приготовленные:
• Сосуд для стартовой культуры (малый флакон с пе¬
ремешивающим устройством, см. рис. 7.6 и 12.7)
• Ростовая среды, подогретая до 37 °С
• Пеногаситель (силикон: Dow Corning, Merck;
Pluronic F68: Sigma)
• Большая емкость для перемешивания (Bellco, Tech-
пе; см. рис. 26.1 и 26.2); в которой подготовьте:
а) Магнитный стержень, заключенный в стеклянный
маятник или другое подходящее устройство для
перемешивания (см. разд. 26.1), закрепленный на
крышке и перемешивающий суспензию во всем
объеме (например, Techne).
б) Одно впускное отверстие со съемной С02-прони-
цаемой крышкой (например, крышка с цельной
микропоровой мембраной) (см. разд. 8.2.3), или
крышкой с силиконовой диафрагмой с проходя¬
щей через нее иглой широкого диаметра с соеди¬
нением Люэра, присоединенной к стерильному
фильтру (например, Millex).
в) Второе впускное отверстие с трубкой для подклю¬
чения к газовой линии (например, Bellco #1965)
с внутренним диаметром 2-3 мм, которое почти
достигает дна сосуда, но не задевает движущего¬
ся маятника при перемешивании культуры и с
внешним входом, ограничивающимся насадкой
для соединения Люэра, закрытым алюминиевой
фольгой.
г) Простерилизуйте культуральный сосуд в сборе,
но без С02-проницаемой крышки автоклавиро-
ванием при 100 кРа в течение 20 мин, крышку
стерилизуют отдельно.
• Соедините стерильный микропоровый фильтр
диаметром 25-мм, размер пор 0,2 мкм (например,
Millex), с подающей газовой линией перед нача¬
лом использования
Нестерильные:
• Магнитная мешалка
• Источник 5% С02, предпочтительно с изме¬
рителем использованного объема газа, при
-10-30 кПа.
• D-PBSA для подсчета клеток
• Электронный счетчик клеток или гемоцитометр
Протокол
1. Установите начальную культуру (см. прото¬
кол 13.3 и рис. 13.5) в 200 мл среды, посейте ее
с посевной концентрацией 5x104-1x105 кл./мл.
Поместите культуру на магнитную мешалку,
вращающуюся при 60 об./м, инкубируйте куль¬
туру пока не будет достигнута концентрация
5х10э-1х106 кл./мл. (Замечание: не превышайте
концентрацию 1x106 клеток/мл, поскольку клет¬
ки могут войти в апоптоз).
2. Соберите большой сосуд, как показано на
рис. 26.2.
3. Добавьте 4 л среды во флакон.
26.1. Крупномасштабное производство суспензионных культур
507
4. Добавьте 0,4 мл пеногасителя, используя однора¬
зовую пипетку или шприц.
5. Закройте боковые горлышки С02-проницаемой
крышкой.
6. Поместите сосуд на магнитную мешалку в инку¬
батор или комнату с установленной температурой
37 °С.
7. Подсоедините линию, подающую воздух с 5% С02
через стерильный микропоровый фильтр, через
впускное отверстие, снабженное Люэровским
соединением.
8. Включите подачу газа на низкой скорости, при¬
мерно 10-15 мл/мин.
9. Перемешивайте культуру при 60 об./мин.
10. Инкубируйте большой сосуд при перемешивании
примерно 2 ч до установки температурного рав¬
новесия и давления С02.
11. Закройте подачу С02, отсоедините сосуд от линии
подачи С02.
12. Принесите большой сосуд и стартовую культуру
в ламинарный бокс
13. Перенесите стартовую культуру в большой сосуд,
перелив ее одним мягким движением.
14. Верните большой сосуд в комнату с температурой
37 °С или инкубатор.
15. Продолжайте перемешивание с частотой переме¬
шивания 60 об./мин.
16. Подсоедините линию подачи 5% С02, отрегу¬
лируйте газовый поток до 10-15 мл/мин. (Если
нет измерителя скорости потока газа, то скорость
потока оценивается по количеству пузырей.)
17. Инкубируйте культуру в течение 4-7 дней, отби¬
рая образцы каждый день для проверки скорости
деления клеток:
а) Перенесите сосуд в ламинарный бокс.
б) Удалите крышки с боковых горлышек.
в) Отберите 5-10 мл суспензии клеток.
г) Подсчитайте клетки, и проверьте их жизнеспо¬
собность методом исключения красителя.
18. Когда концентрация клеток достигнет желаемого
уровня,
а) Перекройте газ и выключите магнитную ме¬
шалку;
в) Отсоедините сосуд от линии подачи газа;
г) Перенесите культуру в лабораторию, перелей¬
те в центрифужные флаконы
д) Центрифугируйте клетки npnlO&g в течете
10 мин, и соберите клетки (из осадка) или су¬
пернатант, в зависимости от обстоятельств.
Анализ Ежедневно контролируйте скорость роста
клеток. Для наилучшего результата клетки не должны
иметь lag-период более чем 24 часа и должны еще быть
в стадии экспоненциального роста при получении
продукта. Проводите ежедневный подсчет клеток,
проводите итоговый сбор клеток при концентрации
приблизительно lxlO6кл./мл. Суспензионные куль¬
туры имеют тенденцию к выходу в апоптоз, если эта
концентрация превышена.
Вариации.
Добавление среды. Следующие процедуры альтерна¬
тивы для добавления среды в сосуды, объемом больше
чем 5 л:
1) Покупают среду в пакетах (см. Приложение II),
которые можно подвешивать рядом с перемешивае¬
мой емкостью, и, будучи подогретой до 37 °С, среда
бесконтрольно поступает в емкость.
2) Стерилизуют перемешиваемую емкость с предва¬
рительно измеренным объемом UPW и составляют
среду из концентратов in situ. Отметьте уровень
воды на стенке емкости, так, чтобы любая вода, по¬
терянная при испарении в ходе автоклавирования,
могла быть восполнена.
3) Используют автоклавируемую среду (см. Приложе¬
ние И), и автоклавируют ее in situ.
Сбор клеток. Переливание из большой перемеши¬
ваемой емкости может быть трудным, если не имеется
сливного отверстия у основания. Однако такие отвер¬
стия имеют тенденцию засоряться мертвыми клетками,
и лучше избегать применения таких конструкций. Сбор
продукта лучше выполнять при помощи насоса или
вытеснением:
1) Удаляют действующий фильтр от газовой линии и
присоединяют силиконовый шланг или сифон либо
используют перистальтический насос, наклоняя
судно, когда жидкость опускается до уровня осно¬
вания сосуда.
2) Отсоединяют линию подачи 5% С02, заменяют
микропоровый фильтр на гибкую трубку, присоеди¬
няют линию поступления 5% С02 к другому порту,
и выдувают клетки через газовую линию.
26.1.1. Постоянная культура
Если требуется, чтобы клетки сохранились при посто¬
янной концентрации, культура может поддерживаться в
хемостате или биостате. В этом типе системы культуры
клетки выращивают до середины log-фазы (контролиру¬
емой ежедневным подсчетом клеток); измеренный объем
клеток удаляют каждый день, заменяя равным объемом
среды. Кроме того, клетки можно непрерывно удалять
с постоянной скоростью, начиная с середины log-фазы,
добавляя с той же скоростью свежую среду. В последнем
случае потребуется сосуд с перемешиванием с четырь¬
мя портами, два для впуска и выпуска С02, один для
подачи среды и один для выведения среды с клетками,
собираемой в отдельную емкость (рис. 26.3). Скорость
потока среды может быть рассчитана по скорости роста
культуры [Griffiths, 2000], но лучше определить ее эк¬
спериментально последовательным подсчетом клеток
при различных скоростях потока среды. В биореакторе
подсчет клеток может быть выполнен в пробе, взятой
через место соединения с линией подачи С02. Линию
508
ГЛАВА 26. КРУПНОМАСШТАБНОЕ ПРОИЗВОДСТВО КЛЕТОК
СЛИВ ОТХОДОВ ИЛИ БИОРЕАКТОР СОСУД СО СРЕДОЙ И
ЕМКОСТЬ ДЛЯ СБОРА СИСТЕМА ПОДАЧИ
ПОЛУЧЕННОГО ПРОДУКТА
Рис. 26.3. Биостат. Модификация сосуда для суспензионной культуры, представленного на рис. 26.1, с постоянным подве¬
денным впуском свежей среды и выходом клеточной суспензии. Целью такого процесса является скорее поддержание условий
культивирования, чем получение большого количества клеток (см. также рис. 26.17 и 26.18). Собственно первоначальная
культура лучше получается в сосудах, представленных на рис. 26.1 или рис. 26.17
Вентиляционное
отверстие
Вентиляционное
отверстие
Клеточная
суспензия
Охлажденная вода или ледяная баня
Насос
5% со
для
отбора образцов.
Удалить фильтр
и отобрать пробу
шприцом
Водяная ру¬
башка, 37 °С.
подается от
циркулятора
Среда—
Магнитная мешалка
отсоединяют и отбирают образец шприцом с двойным
люэровским адаптером. Скорость потока может регули¬
роваться перистальтическим насосом на входе.
Получение партии клеток в объеме 1-20 л лучше
проводить методом одной партии, описанном в прото¬
коле 26.1. Для мониторинга метаболических изменений
требуется установившийся режим стационарной фазы,
связанный с плотностью клеток, однако в этом случае
выше стоимость среды и выше вероятность контами¬
нации. Тем не менее если работы ведутся в объеме
50-1000 л, то наибольшие трудо- и капиталовложения
производятся на стадии генерации культуры, и метод
одной партии становится более дорогостоящим по вре¬
мени и материалам. Поэтому с точки зрения экономии
времени лучше в этих случаях использовать постоян¬
ные культуры. Суспензионные культуры можно также
перемешивать путем вращения бутылей на роллерных
штативах (см. рис. 26.11), так же как и монослойные
культуры (см. протокол 26.3).
Прикрепленные клетки. Клетки, зависимые от
прикрепления к субстрату, не могут быть выращены
в жидкой суспензии, кроме использования метода
культуры на микроносителях (см. протокол 26.4).
Трансформированные клетки (например, вирус-транс-
формированные или спонтанно трансформированные
постоянные клеточные линии, такие как HeLa-S3)
могут быть культивированы в суспензии. Поскольку
эти клетки сохраняют способность к прикреплению,
необходимо покрыть сосуды для культуры водоот¬
талкивающим силиконом (например, Repelcote), а
концентрация кальция в этом случае должна быть
снижена. Среда S-MEM (Invitrogen, Sigma) является
вариантом среды Игла МЕМ без кальция в составе,
она обычно используется для культивирования клеток
HeLa-S3 и других клеток в суспензии.
26.1.2. Масштаб и комплексность
Стандартная настольная культура с перемешиванием
удовлетворительно обеспечивает работу со средой и
получение партии клеток в объеме до 10 литров. Если,
тем не менее, требуется уделить больше внимания
контролю процесса, то следует использовать контро¬
лируемый ферментер или биореактор. Эти культу¬
ральные емкости регулируют поступление среды и газа
и обеспечивают возможность получения данных по
концентрации кислорода, С02 и глюкозе благодаря на¬
личию соответствующих электродов в культуральном
сосуде. Они контролируются программируемым конт¬
рольным блоком, который записывает и представляет
данные и может быть использован для регулирования
поступления и удаления газа и жидкости, скорости
перемешивания и т.д. (см. разд. 26.3).
26.1.3. Перемешивание и аэрация
Проблемы увеличение масштаба производства клеток
в суспензионных культурах в основном связаны с пе¬
ремешиванием и газообменом. В отличие от простой
перемешиваемой культуры большинство ферментеров
лабораторного масштаба перемешивают среду при по¬
мощи вращающейся турбины или широкой лопасти.
Конструкции таких ферментеров разнообразны, глав¬
ным образом они призваны обеспечить максимальное
движение жидкости при минимальном стрессирую-
щем действии трения жидкости на клетки. Удачные
конструкции обычно используют медленно вращаю¬
щуюся турбину с относительно большой площадью
поверхности лопастей [Griffiths, 2000]. Некоторые
конструкции минимизируют повреждающий эффект
трения жидкости добавлением Pluronic F68, карбокси-
26.1. Крупномасштабное производство суспензионных культур
509
метилцеллюлозы (КМЦ) или поливинилпирролидона
(PVP) [Cherry & Papoutsakis, 1990].
Культуральные пакеты (см. Приложение II).
Для суспензионной культуры клеток можно использо¬
вать пластиковые пакеты со средой. Они газопроницае¬
мы и могут перемешиваться путем переворачивания на
штативах или плоской крутящейся платформе.
Ферментеры с восходящим потоком воздуха.
Крупномасштабные ферментеры часто используют
принцип восходящего движения воздуха (рис. 26.4): 5%
С02 в воздухе продувается насосом в пористое стальное
кольцо в основании центрального цилиндра, пузырьки
воздуха барботируют центральную часть устройства,
унося с собой поток жидкости. Воздух выпускается
затем через верхнюю часть ферментера, а жидкость
опускается на дно цилиндра. Это простое устройство
широко используется в биотехнологической промыш¬
ленности вплоть до объемов в 20 ООО л.
Аэратор культуры Bello Cell. Это необычное
устройство имеет нижнее отделение для среды, которое
выдавливает среду через слой клеток, заякоренных на
Рис. 26.5. Аэратор культуры BelloCell. Клетки выращивают
на матриксном материале, который с другой стороны пог¬
ружен в среду, газовый обмен осуществляется при помощи
прокачивания нижнего отдела устройства
Рис. 26.4. Ферментер с пропусканием воздуха. В фермен¬
тере с пропусканием воздуха биореактор состоит из двух
концентрических цилиндров, внутренний цилиндр короче с
обоих концов, чем внешний, так что образуются внутренняя
и внешняя камеры. Дно внутренней камеры имеет синтериро-
ванное стальное кольцо, через которое пропускается воздух с
5% С02. Пузырьки газа поднимаются, унося с собой клеточную
суспензию. Воздушно-газовая смесь удаляется из верхней час¬
ти емкости, а клеточная суспензия опускается на дно внешней
камеры (модифицированный метод Griffiths [2000])
пористом матриксе, и затем отсасывает среду обрат¬
но в ритме «дыхательных» движений нижней части
(рис. 26.5). Оптимум перемешивания и аэрации опреде¬
ляется минимумом повреждения клеток (Cesco Bioengi¬
neering; Metabios; www.metabios.com/BelloCell.htm).
Перфузионная суспензионная культура. Перфу-
зионные системы на основе полых волокон и мембран
также действуют на принципе компартментализации.
Клетки находятся в компартменте с малым объемом
при высокой концентрации. Среда проходит через
полые волокна внутри клеточного компартмента
(рис. 26.6) или через прилегающие мембранные
структуры (Membroferm, Polymun Scientific; рис. 26.7).
Регуляция газообмена в среде происходит за предела¬
ми культуральной камеры. Одна из версий системы
Membroferm позволяет собирать продукт в третьем
мембранносвязанном компартменте [Klement et al.,
1987]. Подобные компартменты могут также быть
связаны в комплекты для серийной перфузии. Сущест¬
вуют также системы с двойными концентрическими
пространствами с полыми волокнами. В них продукт
собирается во внешнем пространстве, а клетки растут
между двумя концентрическими трубками.
Реакторы с псевдожидким слоем для суспен¬
зионной культуры. Хотя микроносители первона¬
чально были разработаны для монослойных культур
(см. разд. 26.2.4), пористые микроносители могут пре¬
доставлять площадь суспензионным культурам внутри
узких щелей матрикса носителей. В результате более
высокой плотности микроносителей они могут быть
медленно перфузируемы снизу при такой скорости, что
газа
<— Стенка сосуда
Перегородка
Клеточная
суспензия
вереде
Кольцо для
пропускания
газа
Подача газа
Воздух
510
ГЛАВА 26. КРУПНОМАСШТАБНОЕ ПРОИЗВОДСТВО КЛЕТОК
Внесение клеток
Выход
макромолекулярных
продуктов
г
й
&
Микрокапиллярные волокнистые пучки
Перфузия среды
и газообмен
Впуск 5% С02
1 I
Выпускное отверстие
для газа
Резервуар со
средой;
обмен 0г/С02
Рис. 26.6. Перфузия на полых волокнах. (а) графическое
изображение перфузионной системы на полых волокнах.
Среда циркулирует из резервуара в камеру с культурой под
действием перистальтического насоса. Аэрация и обмен С02
регулируются в резервуаре со средой, (б) Картриджи полых
волокон Spectrapor. (в) Картридж с заменяемыми концами
(б, в с любезного разрешения Spectrapor)
их скорость седиментации соответствует скорости по¬
тока. Бусины, таким образом, остаются в стационарной
суспензии, омываемые средой с постоянно заменяемы¬
ми питательными веществами и сбором продукта в ни¬
жележащем резервуаре. Газообмен осуществляется вне
реактора, не требуется механическое перемешивание.
Макропористые бусины или волокнистый матрикс так¬
же раньше использовались для суспензионных культур
в реакторах с неподвижным слоем с перфузией пита¬
тельной средой (CelliGen, New Brunswick Scientific).
26.2. КРУПНОМАСШТАБНОЕ
ПРОИЗВОДСТВО МОНОСЛОЙНЫХ
КУЛЬТУР
Правила, касающиеся использования трансформиро¬
ванных клеток для получения биофармацевтических
препаратов, в настоящее время достаточно мягкие, тем
не менее ряд требований, касающихся клеток, зависи¬
мых от прикрепления к субстрату, сохраняются. Хотя
клетки, которые растут в суспензии, легче поддерживать,
прикрепленные клетки, благодаря их потенциалу к раз¬
витию полярности, могут представлять собой лучшую
модель для посттрансляционной модификации белков
и соответствующих мембранных потоков, ведущих к
секреции белков из клеток в биологически активной
форме. Для зависимых от прикрепления к субстрату
монослойных культур необходимо обеспечить доста¬
точную площадь поверхности субстрата соответственно
количеству клеток и пропорционально объему среды.
Эти требования привели к развитию ряда стратегиче¬
ских подходов, как простых, так и сложных.
26.2.1. Многослойные пропагаторы
(культиваторы)
Система Nunc Сей Factory. Одной из простейших сис¬
тем для линейного увеличения площади монослойных
культур является система Nunc Cell Factory (рис. 26.8;
см. табл. 7.1). Эта система состоит из четырехугольных
блоков, подобных чашке Петри, с общей площадью
поверхности 600-24 ООО см2, взаимно соединенных в
двух соседних углах вертикальными трубками. В ре¬
зультате наличия сквозных отверстий в вертикаль¬
ных трубках среда может протекать между отсеками,
только если блоки расположены на ребре. Когда блок
поворачивают и кладут плашмя, жидкость в каждом
отсеке оказывается изолированной, хотя сквозные
отверстия в соединяющих вертикальных трубках
позволяют осуществляться свободному газообмену.
Данная система имеет то преимущество, что геометрия
и природа субстрата в данном случае не отличаются от
общепринятой чашки Петри. Рекомендуемый метод
использования системы Cell Factory описан в прото¬
коле 26.2 (см. рис. 26.9).
Анализ. Контроль роста клеток в этих камерах
затруднен, поэтому одна плашка оборудована так,
26.2. Крупномасштабное производство монослойных культур
511
Камеры могут быть
соединены в серии
Среда
Клетки
Среда
Полупроницаемые мембраны
Отделение веществ с низким молекулярным весом
Отделение веществ с высоким молекулярным весом
Среда
Перфузия среды
Сбор продукта
Рис. 26.7. Система Membroferm. Трехкамерный мембранный реактор Membroferm [Klement et al., 1987]. Пластинчатая
структура полупроницаемых мембран, основанная на тройных блоках, в которых клетки внутри двойной мембраны питаются
из верхнего отдела через мембрану, проницаемую для низкомолекулярных веществ. Продукты секреции клеток проходят в
нижнюю камеру через мембрану, проницаемую для высокомолекулярных веществ. Каждый базовый блок из трех элементов
может быть соединен с образованием перфузионной системы с отдельными слоями среды и слоев продукта секреции — от¬
дельных или объединенных
ПРОТОКОЛ 26.2. Система NUNC CELL
FACTORY
Схема
Приготовить клеточную суспензию в среде и внести
суспензию в камеры блока положенные на длинный
край (см. рис. 26.9). Повернуть блоки на 90°, а затем
положить блоки горизонтально и заполнить С02.
Запечатать и инкубировать блок.
Материалы
Стерильные:
• Клеточный монослой
• Ростовая среда
• Трипсин, грубо очищенный, 0.25%
• D-PBSA
• Система Nunc Cell Factory, 10-камерная
• Силиконовые трубки и соединители
Нестерильные
• Гемоцитометр или электронный счетчик клеток
и жидкость для подсчета
Протокол
1. Трипсинизируйте клетки (см. протокол 13.2),
ресуспендируйте их, подсчитайте клетки (см.
протоколы 21.1, 21.2), и разведите суспензию до
2x104 кл./мл в 2 л среды.
2. Положите камеру на длинный край, и присоеди¬
ните трубку для доставки среды к разъему: насад¬
ка для соединения Люэра на питающей трубке к
порту на дне системы (см. рис. 26.9).
3. Пропустите клетки и среду через подающую
трубку. Среда во всех камерах достигнет одного
уровня.
4. Поверните камеру на 90° в плоскости монослоя,
так чтобы блок лег на короткий край с входным
портом и питающей трубкой наверху.
5. Отсоедините трубку для доставки среды от резер¬
вуара со средой в месте Люэровского контакта.
6. Поверните камеру на 90°, перпендикулярно
плоскости монослоя, так чтобы камера легла на
основание, а поверхность культуры располага¬
лась горизонтально.
7. Продуйте блок струей воздуха с 5% С02 в течение
5 мин, а затем освободите зажим и выпускное от¬
верстие (камера может быть постоянно заполнена
газом, если это желательно).
8. Перенесите блок в инкубатор, удалив среду из
питающих и вентилирующих портов.
9. Для замены среды (или ее сбора) выполните
шаг 6 в обратном порядке, удалите фильтр на
входящей трубке и затем выполните шаг 4 в об¬
ратном порядке.
10. Протрите соединение Люэра, откройте зажим и
слейте среду.
11. Заменяйте среду как в шагах 2-8.
12. Для того чтобы собрать клетки:
а) Удалите среду как описано в шагах 9 и 10.
б) Добавьте 500 мл D-PBSA и затем удалите
его.
в) Добавьте 500 мл трипсина при 4 °С, и через
30 секунд удалите его
г) Инкубируйте клетки с остатками трипсина в
течение 15 мин.
д) Добавьте среду к клеткам и потрясите камеру
для ресуспендирования клеток.
е) Удалите среду с клеток, как в щагах 9 и 10.
13. Остаток может быть использован для засева
следующей культуры, хотя при использовании
этого метода трудно контролировать посевную
плотность клеток.
чтобы использоваться как экспериментальная куль¬
тура. Предполагается, что эта отдельная плашка будет
вести себя так же, как остальная система. Супернатант
может быть собран неоднократно для выделения ви¬
руса или очистки продукта, выделяемого клетками.
Сбор клеток для анализа зависит от эффективности
трипсинизации.
512
ГЛАВА 26. КРУПНОМАСШТАБНОЕ ПРОИЗВОДСТВО КЛЕТОК
Рис. 26.8. Система Nunc Cell Factory для культуры клеток. Штабелированные поддоны для культуры, каждый площадью
632 см2. (а) Двухкамерная, 1264 см2 и 10-камерная, 6320 см2 системы Cell Factories, (б) 40-камерная система Cell Factory,
25 280 см2. (С любезного разрешения Nunc A/S.)
t
5% СО,
2. Поверните систему на короткую
сторону так, чтобы соединитель¬
ные порты оказались сверху, и
отсоедините от системы подачи
среды
Пружинная клемма
или клапанный
затвор
1. Поставьте систему
Cell Factory на длинную
сторону и внесите клет¬
ки в 2л среды
Коннектор Люэра
соединительная муфта *
с внутренней резьбой
3. Положите на плоскую сторон так,
чтобы культуральная поверхность на¬
ходилась горизонтально, присоедините
фильтр к трубке, подающей среду и
заполните газом, содержащим^ С02
/с
rv
Вентиляционный
клапан
4. Зажмите обе трубки и инкубируйте при
37 °С; примерно через 30 мин ослабьте зажим
[ на линии вентиляции примерно на 30 с
Кожух
Вентиляционный
клапан
Рис. 26.9. Заполнение системы Nunc Cell Factory
Эта техника имеет преимущество простоты, но
может оказаться дорогой, если блоки выбрасываются
каждый раз после получения клеток. Этот метод был
разработан, прежде всего, для сбора супернатанта, но
он также хорош для получения большого количества
клеток (1хЮ8-1хЮ10).
26.2.2. Многоцелевые диски, спирали
и пробирки
Диски, спирали и пробирки использовались для уве¬
личения площади поверхности для роста монослоя, но
немногие из этих систем теперь доступны на коммер¬
ческой основе. Большая часть матриксных или много¬
листных пропагаторов в настоящее время развиваются
в направлении перфузионных систем (см. разд. 26.2.6),
с акцентом на восстановлении продукта. Предлагаемая
компанией Coming на рынок доноголистная (много¬
слойная) перфузионная система, называемая CellCube
(рис. 26.10), является полым полистироловым кубом,
с многочисленными внутренними пластинами, оро¬
шаемыми обогащенной кислородом нагретой средой.
Внутренняя пластина способна к поддержке роста
монослоя на обеих поверхностях.
26.2.3. Роллерные культуры
Если клетки посеяны в круглую бутыль или пробирку,
которая затем вращается вокруг собственной длинной
оси на роллерном штативе (рис. 26.11), то среда, несу-
26.2. Крупномасштабное производство монослойных культур
513
Рис. 26.10. Система для культуры клеток Coming Cell-
Cube. Многолистный клеточный культиватор с допустимой
площадью поверхности роста клеток 6500-см2. (С любезного
разрешения АТСС.)
щая клетки, течет по внутренней части бутыли. Если
клетки не прикрепляются, они будут взбалтываться
катящимся воздействием, но останутся в среде. Если
клетки способны прикрепляться к субстрату, они
постепенно адгезируют к внутренней поверхности
бутыли и начнут расти с формированием монослоя
(рис. 26.12). Эта система имеет три главных преиму¬
щества по сравнению со статической монослойной
культурой: 1) увеличение используемой поверхност¬
ной области для сосуда данного размера; 2) постоян¬
ное, но нежное взбалтывание среды; и 3) увеличенное
отношение площади поверхности среды к ее объему,
что позволяет обеспечить повышенный обмен газа
через тонкую пленку среды над клетками, ненадолго
погружающимися в глубокую часть среды.
Анализ. Мониторинг клеток в роллерных бутылях
может быть затруднен, но обычно возможно увидеть
клетки с помощью инвертированного микроскопа.
Тем не менее для некоторых микроскопов конденсор
должен быть удален, а в других случаях бутыль может
не помещаться на предметный столик. Таким образом,
выбирайте микроскоп с достаточным пространством
на предметном столике. Для повторяемого сбора боль¬
шого количества полученных клеток или для сбора
супернатанта роллерная система является, возможно,
наиболее экономичной, хотя требуется большая интен¬
сивность труда и вложение финансов в приобретение
роллерного стеллажа (см. рис. 26.11).
Вариации
Агрегация. Некоторые клетки могут иметь тен¬
денцию к формированию агрегатов прежде, чем они
прикрепятся к стенкам бутыли. Такое поведение кле¬
ток сложно предотвратить, но можно снизить путем
снижения скорости вращения до 5 или даже 2 об./мин,
пробуя различные типы или партии сыворотки либо
предварительно обрабатывая поверхность бутылей
фибронектином или полилизином (см. разд. 8.4).
Размер. Существуют разнообразные бутыли (размер
500-1800 см2), как стеклянные, так и одноразовые (см.
табл. 8.1; рис. 26.13; Приложение II Роллерные бутыли).
Некоторые бутыли имеют гофрированную внутреннюю
поверхность для увеличения площади поверхности,
доступной для клеточного роста (Coming).
Объем. Объем среды может быть различен. Малый
объем обеспечивает лучший газообмен и может лучше
подойти для нетрансформированных клеток. Транс¬
формированные клетки, которые более анаэробны,
растут быстрее и производят больше молочной кисло¬
ты, для них может быть лучше большой объем среды.
Объемы, представленные в табл. 8.1, означают вмес¬
тимость и могут быть уменьшены вдвое или удвоены
по потребности.
Механические устройства. Для бутылей пред¬
почтительно использовать роллерные стеллажи (см.
рис. 26.1 lb), поскольку они экономично используют
а б
Рис. 26.11. Бутыли для ролерных культур на стеллажах, (а) Малый настольный стеллаж. (б) Большой напольный раздвижной
стеллаж. (С любезного разрешения Bellco.)
514
ГЛАВА 26. КРУПНОМАСШТАБНОЕ ПРОИЗВОДСТВО КЛЕТОК
Пленка среды
Направление вращения
Вращающаяся бутыльч
монослой
Свободно вращающийся ролик
Управляемый ролик
Рис. 26.12. Схема работы роллерной системы культуры.
Клеточный монослой (пунктирная линия) постоянно омы¬
вается жидкостью, но находится в погруженном состоянии
только примерно одну четверть цикла, что обеспечивает
многократный обмен среды и свободный газообмен
ПРОТОКОЛ 26.3. Роллерная бутылочная
культура
Схема
Посеять клеточную суспензию в среду в круглую
бутыль и медленно поворачивать бутыль на рол¬
лерной стойке.
Материалы
Стерильные или асептически приготовленные:
• Среда и диспенсер для среды
• D-PBSA
• Трипсин грудой очистки, 0,25%
• Монослойная культура
• Роллерные бутыли
Нестерильные:
• Гемоцитометр или электронный счетчик клеток
и жидкость для подсчета
• Источник 5% С02
• Роллерная стойка (см. рис. 26.11)
Протокол
1. Трипсинизируйте клетки и посейте их при обыч¬
ной для этих клеток плотности.
Замечание. Газовая фаза в роллерных бутылках
составляет большую часть объема, поэтому при
использовании сред, основанных на солях Хэнкса
и воздушной газовой фазе, (см. табл. 9.1), может
потребоваться добавление в бутылку немного
5% С02 (например, в течение 2 с при скорости
10 л/мин). Если среда С02/НС03-забуференная,
то газовая фаза должна быть продута 5% С02 (при
скорости потока 20 л/мин, 30 с-1 мин в зависи¬
мости от размеров бутыли; см. протокол 13.2).
2. Поместите бутыль на роллерную стойку, и вра¬
щайте ее при 20 об./ч, пока все клетки не прилип¬
нут (24-48 ч).
3. Повысьте скорость вращение до 60-80 об./ч по
мере увеличения клеточной плотности.
4. Для того чтобы произвести смену среды или
собрать среду, перенесите бутыли в стерильную
рабочую зону, удалите среду, как обычно, и заме¬
ните ее на свежую среду (см. протокол 13.1). Для
добавления свежей среды можно использовать
трансфузионную систему (см. рис. 5.7) или пакет
со средой. Если значение объема среды является
критическим, его можно разливать пипеткой
или при помощи перистальтического насоса (см.
разд. 5.2.7).
5. Для того чтобы собрать клетки:
а) Удалите среду, промойте клетки 50-100 мл
D-PBSA, затем удалите D-PBSA.
б) Добавьте 50-100 мл трипсина при 4 °С и вра¬
щайте бутыль в течение 15 с вручную или на
роллерной стойке со скоростью 20 об./мин.
в) Удалите трипсин, инкубируя бутыль в тече¬
ние 5-15 мин, и добавьте среду в бутыль.
г) Встряхивайте и вращайте бутыль, промойте
клетки пипетированием.
пространство и позволяют легко наблюдать за состо¬
янием бутылей. Роллерные барабаны (рис. 26.14), с
другой стороны, требуют больше места для данного
объема культуральной емкости, но могут быть полезны
для большого количества малых емкостей — флаконов
и пробирок. Флаконы и пробирки могут быть также по¬
мещены в цилиндрический держатель с последующим
размещением его на роллерном стеллаже.
26.2.4. Микроносители
Монослойные клетки могут быть выращены на мик¬
робусинах диаметром 90-300 мкм, сделанных из плас¬
тика, стекла, желатина или коллагена [Griffiths, 2000]
Рис. 26.13. Примеры бутылей для роллерных культур.
В центре и слева одноразовые пластиковые бутыли (Falcon,
Corning); справа стеклянная бутыль
26.2. Крупномасштабное производство монослойных культур
515
Рис. 26.14. Барабанный роллерный аппарат. Барабанный
роллерный аппарат используется для роллерной культуры
большого количества небольших объемов (флаконов или про¬
бирок). (С любезного разрешения New Brunswick Scientific.)
(табл. 26.1). Культивирование монослойных клеток
на микроносителях дает максимальное соотношение
площади поверхности, занимаемой культурой, к объ¬
ему среды, которое может доходить до 90 ООО см2/л,
в зависимости от размера и плотности бусин. Такой
тип культивирования имеет дополнительное преиму¬
щество, которое заключается в том, что с клетками в
монослое можно обращаться как с суспензией.
В то время как Nunc Cell Factory позволяет уве¬
личить масштаб производства в соответствии с обще¬
принятой геометрией конструкции (см. разд. 26.2.1),
микроносители требуют значительных отклонений от
обычной конструкции субстрата. Тем не менее это раз¬
личие оказывает относительно небольшое влияние на
микроскопическом уровне (цв. вкладка 18в), поскольку
клетки в этом случае растут на гладкой поверхности на
разделе жидкой и твердой сред, хотя радиус кривизны
бусин оказывает влияние на некоторые клетки, эти
культуры лучше растить на бусинах большого диаметра
или плоских поверхностях (см. цв. вкладку 18г).
Таблица 26.1. Микроносители
Название
Поставщик
Состав
Удельная масса
Диаметр, мкм
DEAE Декстран
Cytodex 1
Amersham Pharmacia
DEAE-декстран
1,03
160-230
Cytodex 2
Amersham Pharmacia
DEAE-декстран
1,04
115-200
Microdex
Dextran Products
DEAE-декстран
1,03
150
Dormacell
JRH Biosciences
DEAE-декстран
1,05
140-240
Пластик
Acrobeads
Galil
Полиакролеин, покрытый оболочкой
1,04
150
Biosilon
Nalge Nunc
ТС-обработанный полистирол
1,05
160-300
Biocarriers
Biorad
Полиакриламид / DMAP
1,04
120-180
Bioplas
Whatman, Cellon
Полистирол
1,04
150-210
Cytospheres
Lux
ТС-обработанный полистирол
1,04
160-230
BioSPEX PlastiSPEX
JRH Biosciences
Полистирол
1,02,1,03,1,04
90-210
Rapidcell P
ICN
Полистирол
1,02,1,03
90-210
Желатин
Ventregel
Ventrex (Bayer)
Желатин
1,03
150-250
Cytodex 3
Amersham Pharmacia
Декстран, покрытый желатином
1,04
130-210
Cultisphere-G
Sigma
Желатин
N/A
120-180
Cultisphere-GL
Sigma
Желатин
N/A
150-330
Cultisphere-S
Sigma
Желатин
N/A
120-180
Стекло
Bioglas
Whatman, Cellon
Стекло, покрытое пластиком
1,03
150-210
BioSPEX GlasSPEX
JRH Biosciences
Стекло, покрытое полистиролом
1,02,1,03,1,04
190-210
Rapidcell G
ICN
Стекло
1,02,1,03
90-210
Ventreglas
Ventrex (Bayer)
Стекло
1,03
90-210
Glass
Sigma
Стекло многократного использования
1,03,1,04
95-210
Целлюлоза
DE-52/53
Whatman
DEAE-целлюлоза
1,03
Волокна
Коллаген или покрытые коллагеном носители
BioSPEX
JRH Biosciences
Полистирол, покрытый коллагеном
1,2,1,03,1,04
90-210
Cytodex 3
Amersham Pharmacia
Коллаген
1,04
130-210
Rapidcell С
ICN
Микроносители, покрытые коллагеном
1,02,1,03
90-210
Biospheres
Whatman, Cellon
Микроносители, покрытые коллагеном
1,02
150-210
516
ГЛАВА 26. КРУПНОМАСШТАБНОЕ ПРОИЗВОДСТВО КЛЕТОК
Основное отличие, имеющееся у систем микроно¬
сителей, состоит в механике манипуляций [Griffiths,
1992]. Существенным является эффективное пере¬
мешивание без взаимного трения бусин, что может
быть достигнуто при помощи вращающегося маят¬
ника (Techne) (см. рис. 26.1; рис. 26.2) или лопасти
(Bellco), так же как в случае суспензионных культур,
вращающихся со скоростью 30 об./мин. Техническая
литература, которая обычно поставляется компаниями-
изготовителями микроносителей, описывает условия,
оптимально позволяющие выращивать культуры, су¬
ществует ряд опубликованных протоколов для систем
с микроносителями [Griffiths, 1992].
ПРОТОКОЛ 26.4. Микроносители
Схема
Посеять клетки при высокой концентрации клеток
и бусины, развести, перемешать и отбирать клетки
по необходимости.
Материалы
Стерильные:
• Ростовая среда
• Микроносители (см. табл. 26.1 и Приложение II)
• Стартовая культура
• Флаконы для перемешивания (см. Приложе¬
ние II)
Нестерильные:
• Магнитная мешалка (Techne, Bellco)
Протокол
1. Суспендировать бусины при 2-3 л в одной трети
конечного объема среды.
2. Трипсинизируйте и подсчитайте клетки. Посейте
клетки. Посейте клетки в суспензию бусин при
концентрации клеток, превышающей нормальную
посевную концентрацию в три или пять раз.
3. Перемешивайте культуру при 10-25 об./мин в
течение 8 ч.
4. Добавьте среду для достижения конечного
объема, обусловленного концентрацией бусин
0,7-1 г/л.
5. Повышайте скорость перемешивания примерно
до 60 об./мин.
6. Если pH падает, то произведите питание культу¬
ры следующим образом: выключите мешалку на
5 мин, позвольте бусинам осесть, затем замените
от одной второй до двух третей среды.
7. Для того, чтобы собрать клетки:
а) Удалите среду.
б) Отмойте клетки путем осаждения
в) Обработайте бусины в смеси трипсин/EDTA.
г) Позвольте бусинам осесть
д) Осадите клетки.
е) Отмойте клетки центрифугированием и ре¬
суспендированием.
Анализ. Подсчет клеток на бусинах может быть
затруднен, так как скорость роста клеток должна быть
проверена определением DNA (см. протокол 21.3);
белка (см. протокол 21.4), если используются небел¬
ковые бусины, или дегидрогеназной активности, с
использованием теста МТТ (см. протокол 22.4) на
образце бусины.
Вариации. Большинство вариацией этого метода
является результатом выбора бусин или конструкции
культурального сосуда и перемешивающего устройства
[Griffiths, 2000]. Плотность бусин изменяется от 1,03 до
1,05 г/см3 и влияет на скорость перемешивания, потому
что для бусин с более высокой плотностью требуется
более высокая скорость. Состав, или покрытие, бусин
также будет влиять на прилипание клеток, более чувс¬
твительные клетки могут предпочитать желатиновые
или коллагеновые бусины, которые растворяются при
действии протеаз. Стеклянные бусины являются са¬
мым легким материалом для обработки и повторного
использования.
26.2.5. Макроносители
Так же как в случае пористых микроносителей, ко¬
торые позволяют клеткам расти в порах бусинки,
существуют пористые макроносители со структурой,
состоящей из ряда различных материалов типа поли¬
молочной кислоты (PLA), полигликолевой кислоты
(PGA), коллагена или желатина (Gelfoam) в разнооб¬
разных конфигурациях (см. рис. 25.9). Они могут быть
загружены клетками и затем помещены в перемешива¬
емый биореактор или быть перфузированы в реакторе
с неподвижным или псевдожидким слоем (рис. 26.15;
рис. 26.16). Макроноситель Fibracel — один из таких
продуктов, разработанных для использования в био¬
реакторе Celligen (New Brunsick Scientific; рис. 26.156)
или в перемешиваемом биореакторе.
26.2.6. Перфузионные монослойные
культуры
Перфузия часто используется, чтобы облегчить замену
среды и восстановление продукта. Система Cellcube,
(Coming-Costar) представляет собой перфузируемый
многолистовой пропагатор одноразового использова¬
ния с площадью поверхности роста культуры от 21 250
до 85 000 см2 (см. рис. 26.10), соединенный с насосами,
оксигенатором и контроллерной системой. Другие пер¬
фузионные системы используют клетки, заякоренные
на макроносителях или бусинах в реакторе с неподвиж¬
ным слоем (см. ниже).
Мембранная перфузия. Многие системы зави¬
сят от технологии мембранного фильтра, в которой
культуральный слой представляет собой плоский,
водопроницаемый лист. Membroferm — это компарт-
26.2. Крупномасштабное производство монослойных культур
517
Верхний слив и
сбор продукта
Стеклянные
или матричные
бусины
Водяная
рубашка
Впускное отверстие
для посева и введения
среды, предварительно
обработанной газом, 37 °С
Рис. 26.15. Реакторы с неподвижным слоем. Клетки растут на поверхности бусин или микроносителей, омываемых средой.
Бусины обычно стеклянные и располагаются в плотном слое на перфорированном основании на дне культурального сосуда
либо, если сделаны из легкого материала, заключены в каркас. При культивировании бусины перестают двигаться и среда
просачивается между ними, (а) Предполагаемая схема реактора. (6) PeaKTopCelligen. (С любезного разрешения New Brunswick
Scientific.)
ментализированная таким способом конструкция, в
которой клетки, поступление среды и выведение про¬
дукта занимают различные мембранные компартменты
(см. разд. 26.1.3; рис. 26.7).
Перфузия на полых волокнах. Существует ряд
перфузионных систем, в которых адгерентные клетки
растут на внешней поверхности перфузируемых пучков
микрокапилляров. Высокомолекулярные продукты
концентрируются во внешнем пространстве вместе с
клетками, в то время как поступление питательных
веществ и удаление метаболитов происходят через
внутренние пространства волокон (см. разд. 25.3.2 и
рис. 26.6). Возможности этой системы для прикреп¬
ленных культур состоят в воссоздании высокой тка¬
нетипичной клеточной плотности, взаимодействии
с матриксом и установлении клеточной полярности.
Все эти свойства могут быть важны для посттранс-
ляционного процессинга белков и экзоцитоза. Хотя
половолоконные системы были первоначально разра¬
ботаны для прилипающих клеток, они также широко
используются для суспензионных культур, таких как
гибридомы (см. разд. 27.10).
Реакторы с неподвижным слоем. Разработаны
также системы со стеклянными или матричными
бусинами (рис. 26.15), со средой, которая через непод¬
вижный слой перфузируется вверх или просачивается
вниз под действием гравитации. Использованная среда
собирается в резервуар. Эти системы детально описаны
Speir& Griffiths [1985-1990] и Griffiths [2000, 2001].
Реакторы с псевдожидким слоем для монослой -
ных культур. В реакторах с псевдожидким слоем
пористые бусины с относительно низкой плотно¬
стью — из керамики, или смеси керамики и натураль¬
ных продуктов типа коллагена — суспендированы
в восходящем потоке среды, когда скорость потока
Верхний слив и
сбор продукта
«■
Плавающие бусины
или микроносители
Водяная
рубашка
Впускное отверстие
для посева и введения
среды, предварительно
обработанной газом, 37 °С
Рис. 26.16. Реакторы с псевдоожиженным слоем. В реакто¬
рах с псевдоожиженным слоем бусины с низкой плотностью
плавают в перфузате, скорость потока которого подобрана
так, чтобы находиться в равновесии со скоростью седимен¬
тации бусин
518
ГЛАВА 26. КРУПНОМАСШТАБНОЕ ПРОИЗВОДСТВО КЛЕТОК
КОНТРОЛЬНЫЙ БЛОК
Периодичность внесения
среды. Добавление NaOH
АНАЛИЗ ОБРАЗЦОВ, fp
ОТБИРАЮТСЯ ИЗ БИО °i
Количество клеток/влажный
вес /биомасса.
Лактат, образование
аммония, утилизация
аминокислот и глюкозы.
Концентрация продукта.
Контаминация.
Аутентификация
Регулятор
температуры
воды в водяной
рубашке
ТОРЫЕ
\ЕАКТОРА
Рис. 26.17. Системы контроля биореактора. Схематическое представление биоректора с перемешиванием с непосредствен¬
ным исследованием проб (слева), передачей сигнала на контрольный блок (слева вверху), в котором хранятся данные и при
помощи которого регулируются условия культивирования в реакторе. Порт для взятия образцов справа отбирает клеточную
суспензию из биореактора для анализа
среды соответствует скорости седиментации бусин
(рис. 26.16), или циркулируют (Cytopilot, Amersham
Biosciences) в камере, подобной ферментеру с восхо¬
дящим движением воздуха (см. рис. 26.4). В то время
как суспензионные клетки попадают и находятся в
пористых бусинах как в ловушках, клетки монослоя
прикрепляются к внешним поверхностям, таким как
в порах бусины или макроносителя.
Микрокапсулирование Альгинат натрия ведет
себя как золь или гель, в зависимости от концент¬
рации двухвалентных катионов. При высокой кон¬
центрации дивалентных катионов он в виде геля
образует полую сферу вокруг клеток в суспензии (см.
протоколы 25.3 и 23.16; цв. вкладку 186). Поскольку
альгинат действует как барьер для молекул с высоким
молекулярным весом, макромолекулы, секретируе-
мые клетками, оказываются заключенными в преде¬
лах везикул, в то время как питательные вещества,
метаболиты и газ свободно проникают сквозь гель.
Продукт и клетки могут быть высвобождены сниже¬
нием концентрации двухвалентных катионов. Эти
гели имеют низкую иммунореактивность и могут
быть введены in vivo.
26.3. КОНТРОЛЬ ПРОЦЕССА
Развитие суспензионных культур контролируется из¬
мерением pH, кислорода, С02, и глюкозы при помощи
соответствующих электродов, которые находятся в
культуре in situ, а также исследованием потребления
питательных веществ типа глюкозы и аминокислот или
увеличением концентрации метаболитов типа молоч¬
ной кислоты и аммиака, и продуктов типа иммуногло¬
булина или гибридом (рис. 26.17). Количество клеток
и значения других параметров, например содержание
АТФ, ДНК и белка, определяют в образцах, получен¬
ных из культуры. Эти данные используются, чтобы
вычислить общее количество биомассы. Температура
среды регулируется предварительным нагреванием
поступающей среды и подогревом водяной рубашки,
который в свою очередь регулируется обратной связью
от датчика температуры. Скорость потока среды также
контролируется и может соответствовать скорости
выхода продукта, если суспензия постоянно выделя¬
ется в режиме биостата (см. разд. 26.1.1). Скорость
перемешивания может регулироваться по вязкости
среды, чтобы уменьшить повреждающее действие
трения жидкости.
26.2. Крупномасштабное производство монослойных культур
519
ю
о
-5
-10
-15
ppm
Рис. 26.18. ЯМР -анализ клеток. Клеточный рост в полых волокнах, анализируемый методом ЯМР. [31Р] ЯМР-спектр кле¬
ток СНО, растущих в ректоре на полых волокнах. Клетки были выращены на макропоровых бусинах в экстракапиллярном
пространстве особым образом сконструированных картриджей полых волокон, которые могут быть приспособлены к 25-мм
диаметру ячейки для проб, ЯМР-спектроскопа. Клетки были представлены при плотности примерно 7х107 кл./мл
Сокращения: РМЕ — фосфомоноэфиры, включая фосфохолин и фосфоэтаноламин; Р, — внеклеточный неорганический
фосфор; ^ (acid) — пул кислого (pH 6,7) внеклеточного в биореакторе; РСг — фосфокреатин; у-АТР — уфосфат АТФ; а-
АТР — а-фосфат АТФ; PDE — фосфодиэфиры; DPDE — дифосфодиэфиры, включая UDP-глюкозу; Р-АТР, Р-фосфат АТФ,
NADH, редуцированный никотинамидадениндинуклеотид. Шкала химического сдвига соотнесена с фосфокреатином при
0,0 ppm. (С любезного разрешения Dr. Kevin Brindle.)
При повышении масштаба производства монослой¬
ных культур клеток, в частности при культивировании
на половолоконных системах или в реакторах с не¬
подвижным слоем, обнаруживается проблема: в этих
условиях невозможно непосредственно наблюдать
клетки и осуществлять контроль развития культуры,
подсчет клеток и определение биомассы становятся
затруднительными. В одном из подходов к этой про¬
блеме [Brindle, 1998; Thelwall, et al., 2001] используется
ядерный магнитный резонанс (ЯМР) для исследо¬
вания содержимого камеры, в которой происходит
развитие культуры. Помещая клетки в перфузируе-
мой половолоконной камере в магнитное поле ЯМР-
спетроскопа, исследователь получает характеристики
ЯМР-спектра, что позволяет идентифицировать и
количественно определять определенные метаболиты
(рис. 26.18). ЯМР-спектроскоп можно также исполь¬
зовать как устройство для получения изображений, с
получением квазиоптического сечения камеры, для
того, чтобы показать распределение клеток и даже
различить размножающиеся и неразмножающиеся
зоны культуры.
Глава 27
СПЕЦИАЛЬНЫЕ МЕТОДЫ
Эта глава включает в себя ряд методов, которые явля¬
ются вспомогательными, зависят от типа клеточной
культуры, но которые не являются существенным
компонентом для начала работы с культурами или
их постоянного ведения. Эти методы представляют
интерес прежде всего для специалистов. Они могут
представлять ценность для некоторых читателей, для
остальных помогут обосновать применение культуры
тканей.
27.1. МЕТОДЫ ВЫДЕЛЕНИЯ
И ИССЛЕДОВАНИЯ ЛИМФОЦИТОВ
Наиболее часто используемым методом выделения
лимфоцитов является флотационное обогащение в
смеси фиколла и метризоата натрия (Ficoll-Hypaque)
[Boyum, 1968а, b], позволяющее отделить лимфоциты
от эритроцитов и сыворотки. Этот метод подходит
для стимуляции лимфоцитов фитогемагглютинином
(РНА) (см. протокол 27.2), хромосомного анализа,
обогащения популяции жизнеспособных клеток (см.
протокол 12.10), а также на первом этапе выделения
различных подклассов лимфоцитов.
ПРОТОКОЛ 27.1. Приготовление лимфоцитов
Схема
Слой цельной цитратной или гепариншированной
крови или плазмы, освобожденные от эритроцитов
методом декстран-активированной седиментации,
нанести на слой Ficoll-Hypaque. После центрифуги¬
рования большая часть лимфоцитов будет обнаружи¬
ваться на границе раздела фаз между Ficoll-Hypaque
и плазмой.
Материалы
Стерильные:
• Проба крови в гепарине или цитрате (концент¬
рация определяется типом контейнера для
сбора крови, исходно содержащего гепарин или
цитрат)
• Чистые центрифужные пробирки или универ¬
сальные контейнеры
• D-PBSA
• Ficoll-Hypaque, доведенный до 1,077 г/см3 (При¬
ложение II)
• Бессывороточная среда
• Шприц с тупоконечной канюлей, пластиковые
Пастеровские пипетки или автоматические пи¬
петки
Нестерильные:
• Гемоцитометр или электронный счетчик клеток
• Центрифуга
Протокол
1. Отберите пробу крови в цитрат-содержащий или
гепаринизированный контейнер и доставьте в
лабораторию.
• Требование безопасности. Человеческая
кровь может быть инфицирована ВИЧ, вирусом
гепатита или другим возбудителем, поэтому мани¬
пуляции с кровью человека должны проводиться
с соблюдением правил предосторожности (см.
разд. 7.8.3).
2. Разведите кровь в D-PBSA в отношении 1:1,
нанесите слой объемом 9 мл поверх 6 мл Ficoll-
Hypaque. Это следует делать в широкогорлых
прозрачных центрифужных пробирках с крыш¬
ками, таких как 25-мл универсальные контейнеры
Sterilin или Nunc, либо в чистых пластиковых
50-мл пробирках Coming.
3. Центрифугируйте суспензию в течение 15 мин
при 400 g (измеренной в центре поверхности
раздела).
4. Осторожно удалите плазму /D-PBSA, не нарушая
слой на границе раздела.
5. Соберите слой на границе раздела при помощи
шприца (с тупоконечной канюлей) или пластико¬
вой Пастеровской пипеткой, разведите его в 20 мл
бессывороточной среды (например, RPMI1640).
• Требование безопасности. При работе с
кровью человека не следует использовать стеклян¬
ные Пастеровские пипетки и шприцы с остроконеч¬
ной иглой. Вместо этого следует использовать 1-мл
автоматическую пипетку или шприц с тупоконечной
канюлей.
27.2, Авторадиография
521
6. Центрифугируйте разведенную отобранную сус¬
пензию клеток при 70 g в течение 10 мин.
7. Сбросьте супернатант, ресуспендируйте осадок
в 2 мл бессывороточной среды. Если потребует¬
ся провести несколько отмывок, например для
удаления сывороточных факторов, повторите ре¬
суспендирование в 20 мл бессывороточной среды
и центрифугирование еще два-три раза, прежде
чем окончательно ресуспендировать осадок в 2 мл
бессывороточной среды.
8. Окрасьте образец полученных клеток метиле¬
новым синим, подсчитайте количество клеток,
содержащих ядра, на гемоцитометре. Можно
также лизировать образец клеток с Zapoglobin
(Beckman Coulter) и подсчитать количество ядер
на электронном счетчике клеток с дроссельной
трубкой диаметром 70-100 мкм.
Лимфоциты будут концентрироваться на поверх¬
ности раздела наряду с некоторым количеством тром¬
боцитов и моноцитов. Здесь также могут быть обнару¬
жены небольшое число гранулоцитов, хотя основная
масса их будет находиться в Ficoll-Hypaque и в осадке,
полученном при выполнении шага 3. Моноциты и оста¬
точное количество гранулоцитов могут быть удалены
из фракции лимфоцитов за счет их преимущественной
адгезии к стеклу (бусины или поверхность флакона)
или пропусканием через нейлоновое сито. Для более
тщательной очистки используйте положительный сор¬
тинг MACS {см. разд. 15.3.2) или FACS {см. разд. 15.4)
с использованием специфических поверхностных мар¬
керов соответствующего подкласса лимфоцитов.
27.1.1. Бласттрансформация
Лимфоциты в очищенной фракции или в цельной
крови могут быть стимулированы при помощи мито-
генов, таких как фитогемагглютинин (РНА), митоген
лаконоса (PWM), или антигена [Berger,1979; Hume
& Weidemann, 1980]. Развившийся ответ может быть
использован для количественного анализа иммуноком¬
петентности клеток. РНА-стимуляция также может
использоваться для получения митозов при хромо¬
сомном анализе периферической крови [Rooney &
Czepulkowski, 1986; Watt & Stephen, 1986] (см. прото¬
кол 16.7).
27.2. АВТОРАДИОГРАФИЯ
Данный раздел имеет своей целью дать представление
о микро-авторадиографии малых молекулярных пред¬
шественников, включающихся на холоде в кислотно¬
растворимые макромолекулы, такие как ДНК, РНК
или белки. В тексте этого разделы упомянуты разные
варианты, которые более подробно освещены в цити-
ПРОТОКОЛ 27.2. ФГА-стимуляция
лимфоцитов
Материалы
Стерильные:
• Среда с 10% FBS или автологической сывороткой
• Фитогемагглютинин (ФГА), 50 мкг/мл
• Пробирки для исследований или универсальные
контейнеры
• Предметные стекла
• Колцемид, 0.01 мкг/мл в BSS
• 0,075 МКС1
Протокол
1* Используя отмытую фракцию лимфоцитов после
шага 7 протокола 27.1, инкубируйте клетки в кон¬
центрации 2х106 кл./мл в среде, 1,5-2,0 см глуби¬
ной, в HEPES или С02-забуференную DMEM,
CMRL 1066, или RPMI1640 с добавлением 10%
аутологической сыворотки или FBS.
2. Добавьте ФГА, 5 мкг/мл (конечная концентра¬
ция) для стимуляции митоза от 24 до 72 ч.
3. Отберите образцы через 24, 36, 48, 60 и 72 ч,
приготовьте мазки или цитоцентрифужные пре¬
параты образцов для определения оптимального
времени инкубации (т.н. пик митотического ин¬
декса).
4. Добавьте колцемид до 0,001 мкг/мл (конечная
концентрация) на 2 ч, в течение которых ожида¬
ется пик митозов, обнаруженный в шаге 3 {Ber¬
ger, 1979].
5. Центрифугируйте клетки после обработки кол-
цемидом, ресуспендируйте осадок в 0,075 М КС1
для гипотонического набухания, дальнейшая
обработка как в случае приготовления препарата
хромосом (см. протоколы 16.7,27.5).
руемой литературе [Rogers, 1979; Stein & Yanishevsky,
1979] (см. также протокол 27.8). Поскольку автора¬
диография широко используется для локализации
материала в электрофоретических блотах и микроско¬
пических препаратах, эти два метода можно разделить
на макро-авторадиографию и микро-авторадиографию,
соответственно (см. Приложение IV: Глоссарий).
Изотопы, которые могут применяться для микроав¬
торадиографии, приведены в списке в табл. 27.1. Низ-
коэнергетичные излучатели (например, 3Н или 55Fe) в
сочетании с тонкой эмульсией дают внутриклеточное
разрешение. Несколько более высокоэнергетичные
источники (например, 14С и 35S) позволяют оценить ло¬
кализацию на клеточном уровне. Высокоэнергетичные
изотопы (например, 1311,59Fe и 32Р) дают низкое разре¬
шение на микроскопическом уровне, но используются
обычно в макроавтографах, хроматограммах и блотах
при электрофорезе ДНК, РНК и белка, для которых
низкоэнергетичные излучатели будут ограничивать
возможность определения включения изотопа. Низкие
522
ГЛАВА 27. СПЕЦИАЛЬНЫЕ МЕТОДЫ
Таблица 27.1. Изотопы, которые могут быть использованы
для авторадиографии
Изотоп
Тип излучения
Энергия (mV)
(средняя)
т1/2
3Н
Р-
0,018
12,3 лет
35Fe
Х-лучи
0,0065
2,6 года
125 J
Х-лучи
0,035; 0,033
60 дней
14С
Р-
0,155
5,57 лет
35S
(3-
0,167
87 дней
45Са
Р-
0,254
164 дня
концентрации высокоэнергетичных источников (14С и
выше) при использовании в толстослойный ядерных
эмульсиях дают треки, которые полезны при определе¬
нии местонахождения небольшого количества высоко¬
энергетичных меченых частиц (например, вирусных час¬
тиц, инфицирующих популяцию клеток или ткани).
При авторадиографии на клеточном уровне чаще
всего используется тритий, поскольку выделяющиеся
(3-частицы имеют в водной среде средний пробег около
1 мкм, что дает очень хорошее разрешение. Тритий-
меченые компоненты обычно дешевле, чем 14С- или
358-меченые эквиваленты, и имеют более длительный
период полураспада. В результате их низкой энергии
излучения, тем не менее, важно, чтобы радиочувстви¬
тельная эмульсия располагалась в непосредственной
близости от образца и между клеткой и эмульсией ни¬
чего не было. Даже в этой ситуации будет регистриро¬
ваться включение изотопа на толщину образца только
1 мкм. (3-Частицы, входя в эмульсию, дают скрытое
изображение в кристаллической решетке галогенида
серебра эмульсии в точке, в которой они останавли¬
ваются и высвобождают энергию. Это изображение
может быть визуализировано как зерна металлического
серебра путем обработки щелочным восстановителем
(проявителя) и последующего удаления оставшегося
неэкспонированного галогенида серебра кислым фик¬
сажем. Скрытое изображение более стабильно при
низкой температуре и безводных условиях, поэтому его
чувствительность (величина сигнала по отношению к
фону) может быть улучшена экспонированием в холо¬
дильнике на слое влагопоглотителя. Эта манипуляция
позволит уменьшить фоновое образование зерен сереб¬
ра под действием температуры.
ПРОТОКОЛ 27.3. Микрорадиография
Схема
Инкубировать культуру клеток с соответствующим
меченым изотопом предшественником (например,
[3Н]-тимидином для мечения ДНК), отмыть, фик¬
сировать и высушить клетки (рис. 27.1). Произвести
необходимые действия (например, удаление невклю-
ченного предшественника). Покрыть образец эмуль¬
сией в темноте и оставить для экспозиции. После
проявления серебряные зерна могут наблюдаться
в зонах, располагающихся над областью включения
метки (рис. 27.2; см. цв. вкладку 14г).
Материалы:
Установка культуры:
Стерильные:
• Клетки
• D-PBSA
• Трипсин
• Ростовая среда
• Покровные или предметные стекла или чашки
Петри (можно использовать чашки не для работы
с культурой тканей, если работа идет на покров¬
ных стеклах) или пластиковые флаконы
Нестерильные:
• Гемоцитометр или электронный счетчик клеток
и сцинтилляционная жидкость
Мечение изотопом:
Стерильные:
• Изотоп
• HBSS
Нестерильные:
• Защитные перчатки
• Контейнер для сбора радиоактивных пипеток
• Контейнер для слива жидких радиоактивных
отходов
Фиксирование и обработка клеток:
Нестерильные:
• Уксуснокислый метанол (1:3, на льду, свежепри¬
готовленный)
• DPX
• Трихлоруксусная кислота (ТХУ), 0,6 N
• Деионизованная вода
• Желатин: 0,2 г в 200 мл UPW; разогретый в
СВЧ-печи в течение 2 мин, охлажденный и про¬
фильтрованный
Получение ауторадиографов:
Нестерильные:
• Светофильтр Kodak Wratten II или аналогичный
• Эмульсия (Amersham Hypercoat Emulsion LM-1,
RPN40)
Замечание: Удобно растопить эмульсию и разлить
по аликвотам в сосуды для погружения, которые
позволяют одновременно манипулировать с несколь¬
кими стеклами одновременно (Amersham). Если
стекла герметично закупорены в темные коробки, то
их можно хранить при 4 °С, пока не потребуются.
• Светонепроницаемые коробки для предметных
стекол (Clay Adams, Raven)
• Силикагель (Fisher)
• Темная виниловая лента (например, электроизо¬
ляционная лента)
• Черная бумага или полиэтилен
27.2. Авторадиография
523
Приготовление препарата меченных изотопом клеток
Монослой
Камера для стекол
\
Трипсинизация
Техника
капли
Суспензия
/ \
Техника Цитоцентрифуга
капли
Ванна 40°
_
■■
Погружение в
расплавленную
авторадиографическую
эмульсию
Удаление
невключенных
предшественников
Фиксация в холодном
уксуснокислом
метаноле
сг
Сушить, расположив
горизонтально,
экспонировать и проявить
Расположить
вертикально для
стекания излишка
Рис. 27.1. Микроавторадиография. Стадии подготовки микрорадиоавтографа из культуры клеток
Замечание. Всю стеклянную посуду и оборудование
следует тщательно промывать и оберегать от изо*
топного заражения. Пластиковые покровные стекла
следует предпочитать стеклянным для минимизации
радиоактивного фона. Будьте особенно тщательны
в предупреждении разбрызгивания и немедленно
удаляйте любые брызги. Носите перчатки и регу¬
лярно их меняйте, например, когда вы переходите
от инкубации (высокий уровень изотопов) к работе
с отмытыми фиксированными препаратами (низкий
уровень изотопов)
Обработка ауторадиографов:
Нестерильные:
• Проявитель: Phenisol (Ilford), 20%
• Стоп-ванна: 1% уксусной кислоты
• Фото-фиксаж: 30% тиосульфат натрия
• Просветляющий агент (Kodak)
• Покровные стекла (#00)
Окрашивание:
Нестерильные:
• Гематоксилин (фильтрованный перед использова¬
нием) или краситель Гимза (см. протокол 16.2)
524
ГЛАВА 27. СПЕЦИАЛЬНЫЕ МЕТОДЫ
Рис. 27.2. Микроавторадиографы. Эта пара фотогра¬
фий — пример включения [3Н]-тимидина в монослой кле¬
ток. Нормальные глиальные клетки были инкубированы с
0,1 мкКи/мл (3,7 кБк/мл), 200 Ки/ммоль (7,4 гБк /мкм),
[ЛН]-тимидин в течение 24 ч, промыты и обработаны, как
это описано в протоколе 27.3. (А) Типичные меченые ядра,
подходящие для определения индекса мечения (см. прото¬
кол 21.11). (В) Подобная культура, инфицированная мико¬
плазмой, показано включение [3Н]-тимидина в цитоплазму
• Фосфатный буфер, 0,01 М, pH 6,5
• Этанол, 50, 70 и 100%
• Histoclear (Fisher)
• DPX (Merck)
Протокол
Подготовка культуры
1. Поместите репликатную монослойную культуру
на покровные стекла (Nunc, Bellco) или чашки
Петри.
2. Инкубируйте культуру при 37 °С, пока клетки не
достигнут необходимой для мечения стадии.
Добавление изотопа:
3. Добавьте изотоп, обычно в диапазоне 0,1-
10 мкКи/мл (-4,0 КБк-0,4 МБк/мл), 100 Ки/
ммоль (~4 ГБк/мкмоль), в течение 0,5-48 ч в
соответствии с требованиями инструкции.
• Требование безопасности. При работе с
радиоизотопами следуйте принятым правилам. По¬
скольку в разных странах эти правила различны, в
настоящем руководстве не приводятся специальные
рекомендации, помимо следующих: носите перчатки,
не работайте в ламинарных шкафах с горизонталь¬
ным воздушным потоком, используйте плоские лот¬
ки с впитывающей тканью для абсорбции случайных
разбрызгиваний, инкубируйте культуры в коробке
или подносе с отметкой, обозначающей работу с
радиоактивностью, уничтожение радиоактивных
отходов производите в соответствии с местным за¬
конодательством (см. разд. 7.7.2).
4. Удалите все среды, содержащие изотоп, тща¬
тельно промойте клетки в BSS, собирая среду и
промывочную жидкость в контейнер для жидких
радиоактивных отходов.
• Требование безопасности. 3Н-нуклеозиды
высокотоксичны, поскольку их конечная локализа¬
ция происходит в ДНК (см. разд. 7.7.1).
Фиксация и обработка клеток:
5. Фиксируйте клетки в уксуснокислом метаноле
на льду в течение 10 мин.
6. Приготовьте стекла:
а) Покровные стекла: поместите покровные
стекла клетками вверх на предметное стек¬
ло с заключающим веществом, таким как
DPX или Permount:
б) Клеточная суспензия (суспензионная куль¬
тура или трипсинизированный монослой):
клетки центрифугируйте на предметное
стекло (см. протокол 16.4) или сделайте
препарат из капли (см. протокол 16.7,
шаги 4-12).
в) Приготовьте несколько дополнительных
контрольных стекол для использования
27.2. Авторадиография
525
при определении оптимальной продол¬
жительности экспозиции. Все препараты
далее будут называться «стекла».
7. Экстрагируйте кислоторастворимые предшест¬
венники (при мечении ДНК, РНК или белка) на
льду 0,6 N ТХУ (3x10 мин), и проведите другие
контрольные экстракции (например, растворение
липидов полярными растворителями или фер¬
ментативное расщепление).
8. Погрузите стекла в раствор желатина на 2 мин.
9. Дайте стечь раствору, держа стекла вертикально,
высушите их (стекла могут храниться при 4 °С в
сухом виде).
Проведение ауторадиографии:
10. Внесите стекла в темную комнату.
11. При красном светофильтре растопите эмульсию
в водяной бане при 46 °С.
12. Осторожно перемешайте эмульсию при помощи
чистого стекла, стараясь не создавать пузырей.
13. Погрузите стекла:
а) Погрузите стекло в эмульсию на 5 с при
46 °С, удостоверьтесь, что клетки покры¬
ты эмульсией. Заметьте, что температура
является критичным моментом, определя¬
ющим толщину слоя эмульсии и, следова¬
тельно, разрешение.
б) Выньте стекло из эмульсии со, дайте стечь
5 с.
в) Погрузите стекло в эмульсию еще раз на
5 с.
г) Выньте стекло из эмульсии, дайте стечь 5 с,
держа его вертикально.
д) Протрите бумажной салфеткой заднюю
часть стекла.
е) Оставьте подсыхать в горизонтальном
положении на бумажной салфетке или
фильтровальной бумаге в течение 10 мин.
ж) Поместите стекла в штатив в светонепро¬
ницаемой коробке и оставьте до полного
высыхания. Не высушивайте стекла в потоке
сухого воздуха, позвольте им высыхать мед¬
ленно во влажной атмосфере во избежание
деформации и трещин эмульсионного слоя.
14. Когда стекла высохнут (2-3 ч), перенесите их в
светонепроницаемую коробку для микроскопи¬
ческих препаратов с влагопоглотителем, таким как
силикагель. Проследите за тем, чтобы не прика¬
саться пальцами к поверхности эмульсии и чтобы
стекла не касались друг друга или осушителя.
15. Опечатайте коробки темной изолентой, обмо¬
тайте их черной бумагой или полиэтиленом и
поместите в холодильник. Данный холодильник
не должен применяться для хранения изотопов.
16. Оставьте коробки при 4 °С в течение 1-2 недель.
Точное время будет зависеть от активности
образца и может быть определено проявле¬
нием контрольных стекол через определенные
промежутки времени. Стекла с продолжительным
временем экспозиции имеют более высокий фон
и склонны к регрессии скрытого изображения.
Лучше повысить активность метки, чем увели¬
чить продолжительность экспозиции.
Проявление ауторадиографов:
17. Верните коробки в темную комнату и при красном
светофильтре вскройте коробки.
18. Оставьте стекла в комнатной атмосфере — при
комнатной влажности и температуре (~2 мин).
19. Приготовьте раствор проявителя при 20 °С.
20. Поместите стекла в проявитель на 2,5 мин при
постоянном осторожном покачивании.
21. Быстро ополосните стекла UPW.
22. Перенесите стекла в фотографический фиксаж
на 5 мин.
23. Промойте стекла в деионизованной воде и затем
поместите их в тиосульфатный осветляющий
агент на 1 мин.
24. Промойте стекла в проточной чистой воде или
пять раз сменяя воду не менее 5 мин.
25. Высушите стекла, исследуйте их на микроскопе,
можно использовать фазовый контраст, поместив
сверху тонкое покровное стекло (#00) смоченное
водой. Удалите покровное стекло, когда вы за¬
кончите исследование стекла и прежде, чем вода
высохнет, в противном случае покровное стекло
прилипнет к эмульсии.
Окрашивание:
26. Для того чтобы окрасить гематоксилином:
а) Окрасьте стекла в свежее профильтрованном
растворе гематоксилина в течение 45 с.
б) Промойте стекла в проточной воде в течение
2 мин.
в) Дегидратируйте стекла в восходящей концент¬
рации этилового спирта: 50,70 и 100% EtOH.
г) Просветлите стекла в Histoclear; заключите в
DPX. Если используются покровные стекла,
используйте #00 с минимумом гистологиче¬
ской среды для заключения, чтобы обеспечить
оптимальное рабочее расстояние для работы с
объективом 100х.
27. Для окраски по Гимза:
а) Нанесите на сухие стекла краситель Гимза на
1 мин.
б) Разведите краску in situ 1:10 в 0.01 М фосфат¬
ного буфера (pH 6,5) на 10 мин.
в) Удалите окрашивающий раствор восходящей
струей воды. Стекла не следует вынимать из
красителя, краску не следует сливать обыч¬
ным способом, поскольку частички краски,
которые располагаются поверх слоя краски,
прилипнут к образцу.
г) Тщательно промойте протонной водой, пока
краска не перестанет выходить из эмульсии,
оставаясь лишь в клетках.
526
ГЛАВА 27. СПЕЦИАЛЬНЫЕ МЕТОДЫ
Анализ
Количественный. Определите специфическую ло¬
кализацию зерен, например, только над ядром или
преимущественно над одним из типов клеток.
Качественный
1) Подсчет зерен. Подсчитайте количество зерен на
клетку, на ядро, или локализующихся еще где-то в
клетке. Количественный анализ возможен только
при низкой плотности зерен — около 5-20 зерен на
ядро, 10-50 на клетку, — чтобы не было перекрыва¬
ния зерен и низкого однородного фона.
2) Индекс мечения. Подсчитайте количество меченых
клеток как долю от общего количества клеток (см.
также протокол 21.11). Для этого анализа плот¬
ность зерен должна быть выше, чем при подсчете
зерен, что облегчит распознавание меченых клеток
(см. цв. вкладку 14г). Если плотность зерен высока
(например, 100 зерен на ядро), то устанавливают
более низкий порог определения, скажем, 10 зерен
на ядро или на клетку. Однако помните, что низкий
уровень мечения, значительно превышающий фо¬
новый, может быть важным, например, при оценке
репарации ДНК.
Микроавторадиография — полезный инструмент
для определения распределения изотопа, включенного
в популяцию клеток, но он меньше подходит для об¬
щего количественного анализа захвата или включения
изотопа, для которого предпочтителен сцинтилля-
ционный подсчет.
Вариации. Изотопы с двумя различными энергиями,
например 3Н и 14С, могут быть выявлены в одном экспе¬
рименте путем нанесения на препарат сначала тонкого
слоя эмульсии, затем слоя чистого желатина и, наконец,
нанесением желатина с вторичным слоем эмульсии
[Rogers, 1979]. Более слабое P-излучение 3Н останав¬
ливается в первом слое эмульсии и желатиновом слое.
В то же время более высокоэнергетичное Р-излучение
14С, имея большую среднюю длину пути, около 20 мкм,
проникнет до верхней эмульсии. Adams [1980] описал
метод приготовления радиоавтографа из чашек Петри
или флаконов. Для этого жидкую эмульсию наливают
непосредственно на фиксированные препараты без
трипсинизации.
Мягкие Р-излучатели можно также обнаруживать
в препарате с помощью световой микроскопии. Для
этого используют очень тонкую пленку эмульсии или
галогенид серебра возгоняют непосредственно на ис¬
следуемый срез [Rogers, 1979].
Вместо радиоизотопных меток в настоящее время
чаще применяют флюоресцентные и люминесцентные
зонды (Amersham Biosciences) (см. протокол 27.9).
Разрешение этих методов часто значительно выше,
количественный анализ их возможен путем примене¬
ния конфокальной микроскопии (Bio-Rad) или CCD
камеры (Dage-MTI), а утилизация реагентов более
безопасна для окружающей среды. Однако оборудо¬
вание для микроскопического исследования дорого
(-50 000-200 000 долларов).
27.3. СЪЕМКА В РЕЖИМЕ
ЗАМЕДЛЕННОГО ВРЕМЕНИ
Съемка в режиме замедленного времени была разрабо¬
тана исходно как кинематографическая техника, при
помощи которой естественный медленный процесс мог
наблюдаться в значительно более высоким темпе. Из¬
начально эта техника требовала использования камеры,
автоматически включающейся по сигналу таймера. На¬
учная кинематография началась с первой кинокамерой,
которую использовал Магеу для регистрации движения
животных в 1888. В 1950-х гг. Michael Abercrombie был
первым исследователем, который применил киносъем¬
ку для исследования тканевых культур клеток строго
количественным способом [Abercrombie & Heaysman,
1954]. Постепенно кинокамеры были заменены на
видеокамеры, и «видеомикроскопия» наилучшим об¬
разом позволила соединить видеосъемку со световой
микроскопией [Inoue & Spring, 1997].
Качество видеоизображения постепенно улучша¬
лось, и цифровая форма постепенно вытеснила 16-мм
пленку. Сама по себе микроскопическая техника оста¬
ется критическим пунктом в этом процессе. Коммер¬
чески доступная микроскопическая техника обычно
требует трудоемкого управления или, по-крайней
мере, полуинтерактивного покадрового анализа запи¬
сей. Автоматический анализ поведения клетки путем
цифровой обработки изображения был достигнут с ис¬
пользованием организмов, имеющих высокое прелом¬
ление [Wessels et al., 1996]. Полностью автоматический
анализ неокрашенных тканевых культур позвоночных
требует специальной микроскопии: цифровая запись
с интерференционного микроскопа с автоматическим
сдвигом фаз [Dunn & Zicha, 1997]. Автоматический
анализ поведения клеток является важным аспектом
съемки в режиме замедленного времени, поскольку
большое количество данных обычно требуется для
исследования явления в этой области с высокой вари¬
абельностью свойств.
Эта предварительная информация и протокол 27.4
для съемки в режиме замедленного времени были пред¬
ставлены Daniel Zicha, Cancer Research UK London Re¬
search Institute, Light Microscopy Laboratory, 44 Lincoln’s
Inn Fields, London, WC2A 3PX, England, UK.
ПРОТОКОЛ 27.4, Видеозщшс1гвэут||ААтном
ствами
соединен сзаписьтающим устро^сгвоы^потн
тельное оборудование
темжраз^у дда культуры клеток;
отдельные кадры снимаютсяяутем включеш*я&вто*
магического регулятора, который также открывает
27.3. Съемка в режиме замедленного времени
527
диафрагму, ограничивая излишнее действие осве¬
щения на клетки.
Материалы:
• Среды: МЕМ с солями Хэнкса, 12 мМ бикарбона¬
та, 10% сыворотки теленка 4 мМ L-глютамина
• Трипсин /EDTA: трипсин (Difco), 0,25% в EDTA,
0,5 мМ
• Покровные стекла, 18x18 мм, No. 1,5 (Merck),
промытые в концентрированной серной кислоте
и дважды прокипяченные в UPW с использова¬
нием тефлоновых держателей для покровных
стекол (Eppendorf), стерилизованные автокла-
вированием
• Чашки Петри, пластиковые, 3,5 см
• Камера для видеозаписи:
а) Предметное стекло толщиной 1 мм с отвер¬
стием 15 мм в диаметре в центре, закры¬
том покровным стеклом #3, приклеенным
УФ-отвердевающим клеем для стекла (South¬
ern Watch and Clock Supplies).
б) Стерилизовать в 70%-м спирте, с последую¬
щим обдуванием чистым сжатым воздухом
в) Восковая смесь: пчелиный воск (Fisher), мяг¬
кий желтый парафин (Fisher), и парафиновый
воск с точкой плавления 46 °С; Fisher), в отно¬
шении 1:1:1
• Микроскоп: Axiovert 135 TV (Zeiss)
• Плексигласовая коробка, позволяющая помес¬
тить в нее станину микроскопа, оборудование,
контролирующее систему подогрева (сконструи¬
рованную по специальному заказу, автоматичес¬
кий регулятор 208 2739, температурный датчик
219 4674, нагревательный элемент 224 565, низко¬
шумовой аккумуляторный вентилятор 583-325;
RS Components, UK)
• Компьютеррегулируемый затвор диафрагмы и
фильтра (Ludl Electronic)
• Камера: RTE/CCD-1300-Y/HS (Princeton Ins¬
truments)
• Компьютер: Macintosh G3
• Программное обеспечение для построения изоб¬
ражения IPlab (Scanalytics), позволяющее запи¬
сывать изображение, полученное микроскопом в
режиме замедленного времени.
Протокол
1. Заранее включите все оборудование, особенно
нагреватель, чтобы установить и стабилизировать
температуру в пределах камеры.
2. Высвободите адгезивную культуру в суспензию,
используя трипсин/EDTA.
3. Поместите примерно 20 000 клеток на покровное
стекло чашку Петри, позвольте клеткам осесть
при 37 °С во влажной атмосфере с 3% С02 в те¬
чение ночи.
4. Установите покровное стекло на камеру для
видеосъемки, которая должна быть заполнена
культуральной средой, оставив маленький пу¬
зырь, чтобы гарантировать равновесие С02 с
солями Хэнкса. Запечатайте покровное стекло
горячей смесью воск/парафин.
5. Используйте соответствующую микроскопиче¬
скую технику, например фазово-контрастную или
флюоресцентную для LD Achroplan 32х/0,4 Ph2
объектив. Объектив с более высоким разрешени¬
ем и с коротким рабочим расстоянием потребует
использования вертикального микроскопа для
наблюдения за адгезивной культурой, распро¬
страняющейся в камере.
6. Подготовьте компьютер к получению изобра¬
жения:
а) Калибруйте размер изображения, используя
координатный микрометр.
б) Выберите время экспозиции; обычно 0,1 с,
достаточное для фазового контраста, прини¬
мая во внимание, что слабая флюоресценция
требует более длительной экспозиции, — 0,4 с
или больше.
в) Выберите интервал времени между кадрами,
номер рамки и название файла. Анализ транс¬
локации клеток при медленном перемещении
(фибробласт-подобные клетки) даст удовлет¬
ворительные результаты при интервале около
5 мин. Наблюдение за клеточной морфоло¬
гией, которая изменяется намного быстрее, по¬
требует интервала примерно в 1 мин или даже
меньше. Также запись транслокации быстрых
клеток, таких как нейтрофилы и лейкоциты,
потребует установления интервала в 1 мин
или менее.
г) Используйте следующий сценарий, чтобы
провести чередование получения фазово-кон-
трастных и флюоресцентных изображений:
i) Произведите сохранение выбранного
названия файла.
ii) Вставьте ярлык START.
iii) Переведите колесо фильтров на флюорес¬
ценцию, и откройте диафрагму дуговой
лампы.
iv) Полностью обеспечьте получение флюо¬
ресцентного изображения, используя
соответствующее время экспозиции.
v) Установить колесо фильтра на фазовый
контраст, и закройте диафрагму дуговой
лампы.
vi) Сохраните флюоресцентное изображение.
vii) Откройте диафрагму галогеновой лампы.
viii) Получите фазово-контрастное изображе¬
ние, используя соответствующую экспо¬
зицию.
ix) Закройте диафрагму галлогеновой лампы.
x) Сохраните фазово-контрастное изобра¬
жение.
xi) Пауза.
xii) Закройте все окна.
528
ГЛАВА 27. СПЕЦИАЛЬНЫЕ МЕТОДЫ
хш) Вернитесь к ярлыку START, еслитояько
xiv) Выйдите из программы,
Отделетефазово-контрастную и фдрооресрент-
нуюзаписи. Поеледовательностикадров могут
7* Сохраните посдедователышсти изображений на
CD диске. ^
• Требование безопасности. 1) Будьте аккурат¬
ны, когда промываете покровное стекла концентриро¬
ванной кислотой. 2) Контролируемый электрический
нагреватель, сконструированный на основе Radio
Spares (RS) компонентов, должен быть как следует
изолирован и заземлен, вентилятор следует распола¬
гать так, чтобы предупредить случайное повреждение
вращающимися лопастями.
Анализ. Для интерактивного измерения положе¬
ния, области и интенсивности окраски изображения
может быть использовано компьютерное программное
обеспечение для создания изображения IPLab. Теория
моментов является хорошей основой для автомати¬
ческого анализа поведения клеток [Dunn & Brown,
1990]. Она может быть применима к флюоресцентным
изображениям для оценки в морфологических иссле¬
дованиях клеточной транслокации с использованием
положения центра тяжести, распределения клеток с
использование площади клетки, интенсивности флюо¬
ресцентного сигнала или коэффициенты формы.
Вариации. Выбор среды зависит от потребностей
исследуемых клеток. Среда с солями Хэнкса имеет то
преимущество, что равновесие pH в этом случае уста¬
навливается автоматически в герметично закрытой ка¬
мере с культурой. Открытая камера может быть сконс¬
труирована так, чтобы чашка Петри располагалась над
отверстием в дне камеры. Такое расположение требует
увлажнения и доставления в камеру смеси воздуха и
С02. Среда на основе среды Хэнкса с 12 мМ гидрокар¬
боната потребует добавления примерно 3% С02, а среда
Дюльбекко МЕМ потребует 10% С02. Открытая камера
требует использования инвертированного микроскопа
и высокого разрешения, которое может быть достигну¬
то, когда используются у-облученные микролуночные
3,5-см чашки (MatTek) No. 1,5 со стеклянным дном,
без предварительного покрытия. В настоящее время
имеются специализированные проточные камеры с
местным подогревом (см. рис. 5.22; Intracell, Royston).
Уже не заслуживают серьезного внимания пленочные
или даже видеометоды регистрации, поскольку циф¬
ровая запись на компьютере обеспечивает большее
удобство и другие преимущества, а именно: обработка
изображения, контрастное выделение контуров, вычи¬
тание фонового «шума» и программное обеспечение
анализа. Альтернативным программным обеспечени¬
ем записи изображения в течение заданного времени
для персонального компьютера является MetaMorph
Imaging Software (Universal Imaging Corporation, PA,
USA). Дифференциальный интерференционный кон¬
траст (DIC) является общей альтернативой фазовому
контрасту деталей при высоком разрешении.
27.4. КОНФОКАЛЬНАЯ МИКРОСКОПИЯ
Съемка в режиме замедленного времени или запись
в режиме реального времени может быть сделана на
видеокамере с высоким разрешением или CCD. Эти
системы позволяют вести запись распределения и пе¬
рераспределения красителя или флюоресцентных проб
внутри клетки. Флюоресцентное изображение может
быть использовано для локализации клеточных орга-
нелл, таких как ядро или комплекс Гольджи [Lippincott-
Schwartz et al., 1991]; измерения колебаний внутрикле¬
точных концентраций кальция [Cobbold & Rink, 1987];
а также проникновение в клетку и перемещение в ней
лекарств [Neyfakh, 1987; Bucana et al., 1990]. Измере¬
ния могут быть проведены в трех измерениях, в то же
время возбуждение и система определения позволяют
проводить визуализацию оптических сечений через
клетку и ее поверхности через короткие промежутки
времени. Конфокальная микроскопия может помочь
визуализировать клетки в трехмерной системе культу¬
ры, как и в исследовании инвазии в фильтрационной
лунке [Brunton et al., 1997].
27.5. СИНХРОНИЗАЦИЯ КЛЕТОК
Для того чтобы исследовать особенности прохождения
клетками клеточного цикла, был разработан ряд мето¬
дов, в соответствии с которыми клеточная популяция
может быть фракционирована или метаболически
блокирована так, чтобы по возвращении к обычным
условиям культивирования клетки смогли находиться
в той же фазе и продолжить синхронное прохождение
по клеточному циклу.
27.5.1. Разделение клеток
Методы разделения клеток были описаны в гл. 15.
Может быть использована седиментация на основе
силы тяжести, [Shall & McClelland, 1971; Shall, 1973],
но метод центрифужной элютриации предпочтителен,
если требуется большое количество клеток (>5х107)
[Breder et al., 1996; Mikulits et al., 1997] (см. рис. 15.4
и 15.5). Клеточный сортинг на основании активации
флюоресценции (см. рис. 15.8 и 15.9) также может
быть использован вместе с нетоксичным обратимым
окрашиванием ДНК, например красителем Hoechst
33342. Выход продукта ниже для этого метода, чем для
центрифужной элютриации (~107 клеток или менее),
но, чистота фракции будет выше.
27.6. Культура амниоцитов
529
Одним из самых простых методов сепарации,
синхронизирующей клетки, является митотическое
стряхивание: клетки в монослойной культуре склон¬
ны к ошариванию в метафазе и откреплению, когда
встряхивают флакон, в котором они растут. Этот метод
хорошо работает для клеток СНО [Tobey et al., 1967;
Petersen et al., 1968] и некоторых линий, производных
от HeLa-S3. Инкубирование клеток при 4°С в течение
от 30 мин до 1 часа может также быть использовано для
синхронизации клеток в клеточном цикле [Rieder &
Cole, 2002] и улучшения выхода продукта при мито¬
тическом стряхивании [Miller et al., 1972].
27.5.2. Блокирование клеточного цикла
Обычно используют два типа блокирования клеточ¬
ного цикла:
1) Ингибирование синтеза ДНК (S-фаза). Агенты,
которые используют обычно для ингибирования
синтеза ДНК, включают в себя тимидин, гидрок-
симочевину, арабинозид цитозина и аминоптерин
[Stubblefield, 1968]. Действие этих агентов вариа¬
бельно, поскольку многие из них токсичны и бло¬
кируют клеточный цикл в других фазах клеточного
цикла, помимо G1, что приводит к ухудшению со¬
стояния клеток. Следовательно, культура будет со¬
держать нежизнеспособные клетки, клетки, которые
блокированы в S-фазе, но жизнеспособны, и клетки,
которые избежали блокирования клеточного цикла
[Yoshida & Beppu, 1990].
2) Трофическое блокирование (Gl-фаза). В этих
случаях сыворотка [Chang & Baserga, 1977] или
изолейцин [Ley & Tobey, 1970] удаляются из
среды в течение 24 ч и затем вновь добавляются,
что приводит к синхронизации клеточного цикла
[Yoshida & Beppu, 1990]. Высокая степень синх¬
ронизации (>80%) может быть достигнута только
в первом цикле деления; на втором цикле степень
синхронизации может быть <60%, а на третьем цик¬
ле распределение клеток по фазам клеточного цикла
может приближаться к случайному. Химическая
блокада часто оказывается токсичной для клеток,
а трофическое блокирование работает не слишком
хорошо для многих трансформированных клеток.
Физические методы фракционирования, возможно,
наиболее эффективные и наименее повреждающие
для клеток (см. разд. 15.2,15.4).
27.6. КУЛЬТУРА АМНИОЦИТОВ
Кариотип эмбриона человека может быть определен
культивированием амниотической жидкости, полу¬
ченной пункцией плодного пузыря, которую можно
провести начиная с 11-й недели беременности, хотя
в большинстве случаев пункцию выполняют между
15-й и 18-й неделями беременности. Врожденные
нарушения обмена, а также связанные с полом или
аутосомные рецессивные и доминантные мутации
могут быть диагностированы главным образом на ос¬
новании исследований тканей плаценты, полученных
из образцов хориональных ворсинок с использованием
прямых методов (например, с использованием некуль¬
тивированного материала). В дополнение к этому, в
настоящее время коммерчески доступны новые методы
(AneuVysion from Vysis), позволяющие обнаружить ос¬
новные трисомии и анеуплоидию половой хромосомы
при помощи некультивируемых амниоцитов. Другие
разработанные подходы обнаруживают трисомии при
помощи молекулярного полиморфизма. Однако боль¬
шинство пренатальных диагнозов синдрома Дауна до
настоящего времени основываются на полном хромо¬
сомном анализе амниотической жидкости, включаю¬
щей культуру клеток.
Метод покровных стекол в протоколе 27.5 был пер¬
воначально предложен Marie Ferguson-Smith, East An¬
glian Regional Genetics Service, Addenbrookes Hospital,
Hills Road, Cambridge, CB2 2QQ England, UK. Позже
он был несколько доработан, альтернативный метод
закрытых пробирок был предложен Mike Griffiths, Re¬
gional Genetics Laboratory, Birmingham Women’s Hos¬
pital, Edgbaston, Birmingham, B15 2 TG, England, UK.
Принцип культивирования амниотической жидкости
основан на выделении клеток центрифугированием и
помещением клеточной суспензии в подходящий куль¬
туральный сосуд. Существует ряд подходов, которые
используют как закрытые, так и открытые системы
для роста клеток в пробирках, флаконах или камерах
с предметными стеклами, либо на покровных стеклах
в чашках Петри. Высококачественные, быстрые ре¬
зультаты могут быть получены при помощи любой из
этих техник, но система закрытой пробирки имеет ряд
преимуществ в надежности, простоте и минимальном
использовании ресурсов.
ПРОТСЖШГ27.5. Культура амниоцитов
А. Культуральная система в закрытой
пробирке
Схема
Йнкубироватьклеточную культуру вшшптшовых
пробирках Лейтона в стандарта ом инкубаторе при
клетки методом трипсин изаттии
Материалы
Стерильные или асегт&ески приготовленные:
• Полная среда;100 млсреды Хэма F10 с добав-
лением 20мМбуфера HEPES, 2 мМ глютамина,
100 и/м#^ншшллина и 100 мкг/мл стрептоми¬
цина е-давдВД иди 2% Ultroser (Invitrogen)
• EkFBS A " '
10 мкг/мл: для получения рабочего
добавив 1-9 мл стерильного D-PBSА,
530
ГЛАВА 27. СПЕЦИАЛЬНЫЕ МЕТОДЫ
затем добавить по 0,1 мл рабочего раствора на
каждый 1 мл среды в культуре для достижения
конечной концентрации 0,1 мкг/мл
• Тимидин: приготовьте маточный раствор 15 мг/мл
в D-PBSA, стерилизуйте фильтрацией. Добавьте
0,1 мл маточного раствора тимидина к 10 мл сре¬
ды. Поменяйте среду на тимидин-содержащую
перед сбором клеток. Конечная концентрация в
среде должна составлять 0,15 мг/мл.
• Бромдеоксиуридин, BUdR: флакон, содержащий
250мг; развести содержимое в 25 мл стерильной
D-PBSA, стерилизовать фильтрацией. Добавить
0,5 мл раствора к 11,5 мл стерильного D-PBSA и
8 мл разведенного раствора колцемида. Разлить
получившийся раствор по аликвотам и хранить
замороженными. Добавить 0,1 мл смеси BUdR-
колцемид к каждому 1 мл культуры. Конечная
концентрация должна составлять 25 мкг/мл
BUdR и 0,04 мкг/мл колцемида.
• Трипсин (Bacto, Difco): Растворите содержи¬
мое флакона в 10 мл UPW, следуя инструкции
производителя для получения 5% раствора
(вес/объем).
• EDTA, 0,5 мМ в изотоническом физиологиче¬
ском растворе (Invitrogen)
• Трипсин/EDTA (ТЕ), раствор для субкульти¬
вирования и сбора клеток: 2 мл разведенного
трипсина (Bacto) в 100 мл EDTA, что дает 0,1%-й
раствор трипсина (вес/объем).
• Универсальные контейнеры для сбора образцов
амниотической жидкости
• Пластиковые одноразовые 10-мл пипетки и
приспособление для пипетирования для уста¬
новления культур
• Пробирки Лейтона (с плоскими ребрами, плас¬
тиковые) и крышки для них (Nunc)
• Шприцы, 20 мл; пластиковые и пластиковые
иглы для перемешивания (Henley Medical) для
питания культур
• Переносящие пипетки/Пастеровские пипетки,
1 мл и 3 мл, пластиковые одноразовые
• Histopaque-1077 (Sigma); модифицированный по
инструкции производителя
Нестерильные:
• Дезинфектанты [например, Virkon (Merck)];
следуйте инструкции производителя
• Гипотонический раствор хлористого калия,
0,075 М КС1 в UPW; добавить 5,574 г хлористого
калия (чда) к 1 л UPW
• Фиксирующий раствор, свежеприготовленный:
3 части метанола (чда) к 1 части ледяной уксус¬
ной кислоты (чда)
• Переносящие пипетки/Пастеровские пипетки,
1 мл, пластиковые, одноразовые
• Предметные стекла, предварительно очищенные
(Berliner Glas KG, Skan, компания-посредник
Richardsons of Leicester)
• Флаконы Coplin
• Раствор перекиси водорода, 5% (по объему): Раз¬
вести 1 часть 30% перекиси водорода в 5 частях
воды
• Физиологический раствор, 0,9% NaCl в UPW
• Раствор трипсина в физиологическом растворе
для определения исчерченности: добавить 1,4 мл
разведенного трипсина (Difco) к 50 мл физио¬
логического раствора, конечная концентрация
0,14% трипсина.
• Буферный раствор, pH 6,8 (Gurr, Merck): доба¬
вить одну таблетку буфера к 1 л UPW.
• Краска Лейшмана (Gurr, Merck), обычно разво¬
дится 1 часть к 4 частям буфера pH 6,8
• Гистологический DPX для заключения препара¬
тов и покровные стекла
• Термостат 60 °С
• Горячая пластина при 75 °С
• Светло-польный и фазово-контрастный вер¬
тикальный микроскоп для оценки полученных
препаратов и исследования хромосомной исчер¬
ченности при хромосомном анализе
Протокол
• Требование безопасности
а) Носите перчатки и лабораторные халаты. Все
манипуляции с образцами следует проводить
в стерильных условиях в микробиологических
боксах II уровня биологической безопасности
(см. разд. 7.8.2).
б) Отработанные среды и гипотонический супер¬
натант (но не фиксирующий раствор) следует
сливать в дезинфицирующий раствор, напри¬
мер гипохлорит или Virkon. Фиксирующий
раствор следует сливать в раствор бикарбоната
натрия, чтобы нейтрализовать кислоту. Через
два часа оба типа отходов могут быть слиты
в обычную канализацию вместе с большим
количеством проточной воды.
в) Бромдеоксиуридин (BUdR) является силь¬
ным мутагеном, его употребление не обяза¬
тельно. Следует ознакомиться с местными
требованиями безопасности (например,
COSHH Control of Substances Hazardous to
Health) до начала работы с этим соедине¬
нием. Растворы и супернатант, содержащие
значительное количество этого вещества,
должны сливаться в опечатывающийся сосуд,
содержащий вермикулит или иной абсорбент
с последующим сжиганием.
г) Протоколы, включающие использование
метанола и уксусной кислоты для фиксации
и приготовления препаратов, должны нахо¬
диться в вытяжном шкафу.
Установление и мониторинг культур в пробирках
1. Предположительно 10-15 мл амниотической жид¬
кости будет доставлено в лабораторию в пласти¬
ковых стерильных универсальных контейнерах.
27.6. Культура амниоцитов
531
2. Используйте стерильные 20-мл шприцы, снаб¬
женные стерильными пластиковыми иглами для
перемешивания (или 10-мл пипетки) для того,
чтобы разделить образец на три пробирки Лейто¬
на. Промаркируйте пробирки идентификацион¬
ными ярлыками, отражающими данные пациента
и тип культуры. Если образец небольшой (объем
менее 5 мл), установите только две культуры.
3. Центрифугируйте пробирки 5 мин при 150 g.
4. Перелейте супернатанты назад в исходный кон¬
тейнер [для биохимических или иммунологичес¬
ких исследований, например для оценки уровня
а-фетопротеина (AFP)], или слейте в емкость с
дезинфицирующим раствором (Virkon).
5. Ресуспендируйте клеточный осадок в 1 мл куль¬
туральной среды в каждой пробирке Лейтона.
6. Инкубируйте при 37 °С.
7. Оставьте культуры в покое на 5-7 дней, чтобы
клетки осели и сформировали колонии
8. Оцените культуры. В зависимости от степени
роста клеток, добавьте 0,5 мл свежей среды или
удалите старую среду и добавьте примерно 1 мл
свежей среды.
9. Впоследствии оценивайте рост клеток по необхо¬
димости (каждые 2-4 дня), заменяя среду, пока
не будет достигнут удовлетворительный рост для
получения желаемого продукта, обычно через
7-12 дней после инициации культур. Если воз¬
можно, накануне сбора клеток поменяйте среду.
Замечания
а) Быстрый рост клеток может быть простиму¬
лирован размножением колонии с использо¬
ванием трипсина (см. Диспергирование и суб¬
культивирование ниже). Культуры, имеющие
избыточный рост, могут быть восстановлены
путем распределения по нескольким дополни¬
тельным пробиркам Лейтона или флаконам,
если требуется большое количество клеток.
б) Образцы амниотической жидкости, содержащие
кровь, могут расти не так хорошо, как чистые.
Одним из методов работы с такими образцами
является отделение некоторой части амнио¬
цитов от красных кровяных клеток с исполь¬
зованием градиентного центрифугирования
(например, Histopaque; см. протокол 12.10).
в) Оценка сильно окрашенных кровью образцов
на 6-7 день обычно невозможна без удале¬
ния среды, содержащей кровь. Среда может
быть собрана в дополнительные пробирки,
которые затем могут быть уничтожены,
если в исходных пробирках отмечается рост
клеток, либо могут инкубироваться, если это
необходимо
г) Дополнительные пробирки могут быть ус¬
тановлены при первой смене среды в любом
случае, когда уничтожение исходной суспен¬
зии нежелательно.
Диспергирование и субкультивирование
10. Предварительно нагрейте примерно до 37 °С по
1,5 мл стерильного раствора ТЕ на каждую про¬
бирку, которую предполагается обработать.
11. Удалите среду из культуральных пробирок, опо¬
лосните один раз 1 мл стерильного раствора ТЕ.
12. Для того чтобы диспергировать культуру:
а) Добавьте 0,2 мл ТЕ к культуре, инкубируйте
культуру при 37 °С в течение 1-2 мин до от¬
деления клеток от стенок пробирок.
б) Просмотрите клетки в инвертированный мик¬
роскоп. Когда клетки перейдут в суспензию,
добавьте 1 мл культуральной среды и верните
пробирки в инкубатор (сыворотка в среде
инактивирует раствор ТЕ).
13. Для субкультивирования:
а) Добавьте 0,5 мл ТЕ в культуру, инкубируйте
ее при 37 °С в течение 1-2 мин до отделения
клеток от стен пробирок.
б) Просмотрите культуру в инвертированный
микроскоп. Когда клетки перейдут в суспен¬
зию, добавьте 1-2 мл культуральной среды
и разделите суспензию между соответству¬
ющим количеством пробирок для субкуль¬
тивирования. Может быть засеяно от 2 до
8 пробирок, в зависимости от первоначальной
плотности клеток. Обычно имеет значение ва¬
риабельность концентрации клеток в каждой
субкультуре, что повышает шансы получить
хороший выход клеток при оптимальной кле¬
точной плотности.
в) Залейте каждую субкультуру свежей средой
до конечного объема 1 мл и инкубируйте
14. Проверьте на следующий день состояние клеток,
если они осели, замените среду.
Стандартная процедура сбора клеток
из пробирок
Сбор клеток может быть проведен с первичной
культуры по стандартной методике, если имеется
резерв клеток. Если остается лишь одна культура,
то сначала следует провести субкультивирование
либо восстановить культуру после основного сбора
продукта.
15. Когда культуры готовы к процедуре сбора про¬
дукта, добавьте в каждую культуру по 0,1 мл
разведенного колцемида.
16. Инкубируйте культуры, пока не образуется до¬
статочное количество ошаренных митотически
делящихся клеток. Обычно это занимает 2-3 ч,
но может составлять полчаса для активных
культур или более 4 ч для медленнорастущих
культур.
17. Сбросьте среду в раствор Virkon, быстро осушите
пробирки бумажным полотенцем.
18. Добавьте 2 мл ТЕ, инкубируйте культуры при
37 °С в течение 3 мин для отделения клеток от
стенок пробирок.
532
ГЛАВА 27. СПЕЦИАЛЬНЫЕ МЕТОДЫ
19. Когда клетки перейдут в суспензию, добавьте
7 мл гипотонического раствора КС1, и оставьте
культуры при 37 °С в течение 5 мин.
20. Центрифугируйте пробирки при 150 г в течение
5 мин.
21. Осторожно слейте супернатант в раствор Virkon.
Обсушите пробирки салфеткой.
Слегка постукивая по стенке пробирки, ресуспен¬
дируйте клетки в небольшом количестве остав¬
шейся жидкости
22. Медленно влейте свежеприготовленный фикси¬
рующий раствор, фиксируйте клетки:
а) Постукивая по стенке пробирки, добавьте пер¬
вые 1-2 мл фиксирующего раствора капля за
каплей, постоянно встряхивая клетки, чтобы
избежать образования комков.
б) Добавить еще 3-4 мл фиксирующего раствора.
23. Центрифугируйте пробирки при 150 g в течение
5 мин.
24. Удалите супернатант, сливая его в раствор бикар¬
боната натрия.
25. Осторожно ресуспендируйте клеточный осадок,
добавьте 5 мл фиксирующего раствора.
26. Замените фиксирующий раствор, повторив
центрифугирование, сливание супернатанта и
добавление раствора (шаги 9-11).
27. Вновь центрифугируйте клетки, слейте суперна¬
тант, ресуспендируйте клеточный осадок в остат¬
ках раствора и приготовьте препараты клеток (см.
ниже Приготовление препаратов).
Замечания
а) Суспензии фиксированных клеток могут хра¬
ниться при -20 °С на любой стадии фиксации.
Перед приготовлением препарата дважды
замените фиксирующий раствор.
б) Восстановление культуры после сбора клеток
может использоваться как альтернатива суб¬
культивированию, когда остается только одна
культура. Этот метод требует применения
стерильного колцемида и стерильного ТЕ на
начальной стадии сбора клеток, которая сле¬
дует после переноса клеток в отдельную цен¬
трифужную пробирку после того, как клетки
отделились от стен пробирки, но до того, как
добавлен гипотонический раствор. В исход¬
ную пробирку сможет быть добавлен свежая
питательная среда; затем на следующий день
среду следует поменять, чтобы удалить ос¬
таточные количества трипсина и колцемида.
При этом клетки уже должны вновь осесть.
в) Если при выходе клеток уровень метафаз
неприемлемо низок, то альтернативной стра¬
тегией может быть следующая:
i) Субкультивируйте клетки во флаконе,
который закреплен под небольшим углом
к горизонтальному положению, пока клет¬
ки не осядут. Наклоняйте флакон таким
образом, чтобы ведущая граница роста
клеток, которые всегда растут с оптималь¬
ной скоростью, проходила по всей ширине
флакона.
ii) Инкубируйте клетки с пониженной кон¬
центрацией кольцемида в течение ночи,
г) Более длинные хромосомы для анализа при
более высоком разрешении могут быть полу¬
чены путем использования тимидиновой синх¬
ронизации или экспонирования с колцемидом
в течение ночи. Тем не менее оба этих подхода
лучше работают на клетках, находящихся в
экспоненциальной фазе роста, что требует
тщательного наблюдения за культурой
Тимидин-синхронизированный сбор клеток
1. Утром того дня, когда предполагается сбор кле¬
ток, замените среду в культурах, используя тими-
дин-содержащую среду (конечная концентрация
0,15 мг/мл).
2. В начале дня отмойте тимидин, используя пред¬
варительно подогретую среду. Слейте среду и
промойте клетки дважды. Добавьте 1 мл среды
без тимидина, что снимет тимидиновый блок.
Время снятия блока зависит от времени, когда вы
предполагаете проводить сбор клеток культуры.
3. Инкубируйте культуру в течение 4 ч, затем до¬
бавьте 0,1 мл раствора колцемида.
4. Инкубируйте культуру в течение еще 2 ч, затем
соберите клети по обычной процедуре (начиная
с шага 2 Стандартная процедура сбора клеток из
пробирок, см. выше).
Сбор клеток после ночного инкубирования
с бромдиоксиуридином и кольцемидом:
1. Замените среду в культурах, в которых предпо¬
лагается сбор клеток утром предыдущего дня.
2. В этот же день добавьте 0,1 мл раствора BUdR-
колцемид в каждый 1 мл культуральной среды
(конечная концентрация в культуре: BUdR,
25 мкг/мл; колцемид, 0,04 мкг/мл).
3. На следующее утро (примерно после 20 ч экс¬
позиции) слейте BUdR-колцемид-содержащую
среду в опечатываемый контейнер, заполненный
вермикулитом. Добавьте в каждую культураль¬
ную пробирку по 2 мл предварительно нагретого
ТЕ, инкубируйте 3 мин до отделения клеток от
стенок.
4. Убедитесь, что клетки перешли в суспензию,
добавьте 7 мл гипотонического раствора 0,075 М
КС1. Инкубируйте 5 мин.
5. Центрифугируйте культуру при 150 g в течение
5 мин.
6. Слейте гипотонический супернатант в контейнер
с вермикулитом.
7. Осторожно ресуспендируйте клеточный осадок,
медленно проведите фиксацию клеток и про¬
должите сбор клеток, как это описано, начиная с
шага 6 ( Стандартная процедура сбора клеток из
пробирок, см. выше).
27.6. Культура амниоцитов
533
Приготовление препаратов:
1. Используйте чистые стекла. Они могут быть
куплены уже в обработанном виде, тем не менее
может оказаться полезным добавить 1 каплю
свежего фиксирующего раствора на каждое
стекло и протереть стекло бумажным полотенцем
непосредственно перед использованием. Если
есть возможность контроля влажности и темпе¬
ратуры, храните чистые стекла при 20-25 °С и
относительной влажности 40-50%.
2. Поместите одну каплю суспензии клеток, на каж¬
дое стекло, капнув суспензию с высоты 1-3 см,
оставьте препарат высохнуть при комнатной
температуре.
3. Исследуйте качество распределения на фазово¬
контрастном микроскопе, если распределение
клеток приемлемо, приготовьте остальные стек¬
ла, регулируя плотность клеток добавлением
новых капель фиксатора, если это необходимо.
4. В некоторых обстоятельствах требуется улуч¬
шить распределение клеток. В этом случае могут
помочь следующие меры:
а) Сначала подышите на стекло, а затем капните
каплю суспензии клеток с высоты 1-3 см.
Оставьте препарат высыхать естественным
образом.
б) Поместите одну каплю суспензии на стекло,
не важно, подышав на него или нет. Оставьте
стекло на несколько секунд, а затем, прежде
чем суспензия клеток успела высохнуть, кап¬
ните каплю свежего фиксатора поверх капли
суспензии.
в) Поместите одну каплю суспензии на стекло, не
важно, подышав на него или нет. Оставьте стек¬
ло на несколько секунд и затем, как только на
высыхающей поверхности появится рябь или
обнаружатся кольца Ньютона, капните каплю
свежего фиксатора. Оставьте высыхать.
г) Если качество распределения остается не¬
удовлетворительным, поместите суспензию
клеток в фиксирующем растворе на морозиль¬
ник и оставьте на ночь, переделяйте препарат
на следующий день.
Выявление G-дисков с помощью трипсина
1. Предварительная обработка препарата может быть
проведена до трипсинизации, но в зависимости от
возраста препарата может быть несущественна.
Для свежих препаратов хорошо работает предва¬
рительная обработка перекисью водорода, кон¬
центрированный раствор Хэнкса хорошо работает
для старых препаратов. Подходящей емкостью
являются флаконы Коплэна (Coplin). Могут быть
использованы также необработанные препараты.
2. Может быть использован любой из следующих
методов обработки:
а) Покройте свежий препарат (после одного часд
высыхания) 5%-й перекиси водорода от 20 с
до 2 мин.
б) Инкубируйте свежие препараты в термостате
при 60 °С в течение нескольких часов или в
течение ночи. Нанесите на препарат 5х раствор
Хэнкса на 5 мин.
в) На стекла, приготовленные день назад или
ранее, нанесите на 60 с буфер рН-6,8
3. Промойте препараты буфером pH 6,8.
4. Нанесите на препараты 0,14%-й трипсин в физио¬
логическом растворе от 3 с до 2 мин
5. Хорошо промойте препараты буфером pH 6,8.
6. Окрасьте препараты красителем Лейшмана све-
жеразведенным буфером pH 6,8 в отношении
1:4 в течение 4 мин.
7. Промойте проточной водопроводной водой,
стряхните остатки воды, аккуратно промокните.
8. Исследуйте исчерченность хромосом в свет¬
ло-польном микроскопе при увеличении 400х.
Если препараты недостаточно окрашены, то
краска может быть удалена буфером pH 6,8 или
метанолом и процедуру окрашивания можно пов¬
торить. Если препараты перекрашены, начните
процедуру вновь на другом препарате, варьируя
время действия трипсина или изменив процедуру
предварительной обработки.
9. Положите препараты на горячую платформу
(75 °С) на несколько минут, чтобы убедиться,
что они полностью высохли, затем заключите их
в DPX под покровное стекло.
Окрашивание хромосом рассматривается позже в
этой главе (см. протокол 27.9).
В. Культуральная система на открытых
покровных стеклах:
Схема
Культивируют клетки на покровном стекле, по¬
мещенном в чашки Петри в инкубатор с 5% С02 в
воздухе и влажностью 95%.
Материалы
Материалы для данной процедуры в большинстве
своем те же, что и для системы культуры в закрытых
пробирках со следующими вариациями:
Стерильные:
• Среда Хэма F10 с L-глютамином, пенициллином,
стрептомицином и FBS и Ultroser G, забуферен-
ная бикарбонатом натрия как для культивирова¬
ния в пробирках
• Пластиковые чашки Петри, 3,5 см, с вентилиру¬
емой крышкой
• Стеклянные покровные стекла, 22 мм, стерили¬
зованные сухим жаром
• Пинцеты
Нестерильные:
• Гипотонический раствор, 0,05 М КС1 в UPW
534
ГЛАВА 27. СПЕЦИАЛЬНЫЕ МЕТОДЫ
• Трипсин, 0,8% в физиологическом растворе для
исследования исчерченности (0,4 мл трипсина
Difco, разведенного в 50 мл физиологического
раствора)
Протокол
Подготовка клеточной культуры
1. Центрифугируйте два образца в универсальных
контейнерах при 150 g в течение 10 мин. Если
образец доставлен только в одном контейнере,
отлейте часть во второй контейнер до центрифу¬
гирования, чтобы избежать риска утери образца
при разбивании контейнера во время центрифу¬
гирования.
2. Поместите стерильные покровные стекла в две
чашки Петри, нанесите на дно и крышку чашек
маркировку, соответствующую образцу.
3. Удалите супернатант при помощи пипетки, оста¬
вив примерно 1 мл жидкости поверх клеточного
осадка.
4. Ресуспендируйте осадок, перенесите суспензию
в чашки Петри.
5. Добавьте 3 мл среды в каждую чашку, инкуби¬
руйте культуры в термостате при 38 °С с 5% С02
в воздухе и повышенной влажностью.
6. Через 5-7 дней проверьте культуры на предмет
клеточного роста, используя инвертированный
микроскоп.
7. Частично замените среду, удалив примерно 2 мл
старой среды при помощи стерильной пастеров¬
ской пипетки.
8. Исследуйте культуру и заменяйте среду два раза
в неделю.
Клетки готовы к сбору, когда имеется достаточное
количество активно растущих колоний подхо¬
дящего размера, обычно на 10- 14-й дни после
инициации культуры.
Сбор клеток с покровных стекол in situ
9. Перенесите покровные стекла в новые чашки Пет¬
ри (предварительно промаркированные) запол¬
ненные средой, за 24 часа до предполагаемого сбора
клеток. Исходные чашки следует сохранить как
резервный источник дополнительных клеток.
10. Добавьте 0,3 мл разведенного колцемида в чаш¬
ки Петри до получения конечной концентрации
0,1 мкг/мл, инкубируйте чашки 2-3 ч.
11. Осторожно удалите среду при помощи пастеров¬
ской пипетки, замените ее на гипотонический
раствор 0,05 М КС1 (3 мл), предварительно по¬
догретого до 37 °С. Инкубируйте 10 мин.
12. Выньте чашки Петри из инкубатора и поместите
их на подходящий поддон, пометив крышку под
чашкой.
13. Добавьте 6 капель фиксирующего раствора, осто¬
рожно спуская по стенке чашки Петри и позволяя
фиксирующей жидкости распространиться по
гипотоническому раствору. Оставьте чашку на
1 мин.
14. Добавьте еще 6 капель фиксирующего раствора
в чашку Петри. Оставьте чашку на 1 мин.
15. Удалите 2 мл супернатанта из чашки Петри, заме¬
ните его на 2 мл свежего фиксирующего раствора,
добавляя раствор осторожно по стенке чашки.
Оставьте чашку на 2 мин.
16. Удалите весь супернатант из чашки Петри и заме¬
ните его на 3 мл свежего фиксирующего раствора,
добавляя раствор осторожно по стенке чашки.
Оставьте чашку на 10 мин.
17. Удалите весь фиксирующий раствор, оставьте
покровные стекла высыхать на воздухе в чашке.
После удаления фиксирующего раствора поддон
может быть поднят с одной стороны, чтобы про¬
стимулировать стекание фиксирующего раствора
с покровных стекол.
Выявление G-дисков с помощью трипсина
18. Оставьте для созревания покровные стекла
(в чашке Петри для идентификации) в термостат
при 60 °С на ночь или на горячую платформу при
75 °С на 2 ч.
19. Нанесите 0,04% раствор трипсина в физиологи¬
ческом растворе и непосредственно перед окра¬
шиванием смешайте 1 часть красителя Лейшмана
с 4 частями буфера pH 6,8.
20. Используя пинцет, поместите покровные стекла в
раствор трипсина, мягко взбалтывая его в течение
нескольких секунд (точное время варьирует в
зависимости от возраста препарата).
21. Промойте покровные стекла буфером, верните их
в чашки Петри.
22. Залейте чашки Петри раствором красителя
Лейшмана на 2-4 мин (в зависимости от качества
красителя).
23. Ополосните покровные стекла буфером, поставь¬
те их на ребро в чашках Петри на абсорбирующую
бумагу, оставьте до высыхания.
24. Стекла могут быть исследованы на предмет ка¬
чества окрашивания до заключения в DPX.
25. Поместите покровное стекло на маркированное
предметное стекло, используя DPX.
27.7. КУЛЬТУРЫ КЛЕТОК
ПОЙКИЛОТЕРМНЫХ ЖИВОТНЫХ
Метод культуры клеток холоднокровных животных
(пойкилотермных) похож на методы, используемые
для теплокровных животных, поскольку, главным
образом, эти методы исходно разрабатывались для мле¬
копитающих животных и птиц. Различия в техниках
первичной культуры заключаются в использовании
различных протеолитических ферментов, таких как
трипсин, ЭДТА и хелатирующих агентов. Эмбриональ¬
ную бычью сыворотку хорошо заменяет гомологичная
сыворотка или гемолимфа (которая более легкодоступ¬
на), но модифицированная рецептура среды может
улучшить клеточный рост. Ряд таких сред доступен
27.7. Культуры клеток пойкилотермных животных
535
через коммерческие компании (см. Приложение II).
Процедуры, используемые для этих сред, во многом
похожи на разработанные для клеток млекопитающих.
Проверьте эти среды и сыворотки, которые в настоя¬
щее время доступны, исследуя их влияние на рост,
эффективность посева и специализированные функ¬
ции (см. разд. 9.6, 10.5). Поскольку разработка сред
для многих линий клеток беспозвоночных животных
проводилась для их эмбриональной стадии развития,
может оказаться необходимым разработать новые ре¬
цепты для неизученного класса беспозвоночных или
изучаемого типа тканей. Большая часть накопленного
опыта до настоящего времени относится к насекомым
и моллюскам.
Два обзора [Vago, 1971, 1972; Maramorosch, 1976]
включают в себя некоторые из ранних разработок в
этой области и некоторые недавние разработки в био¬
технологии, которые были опубликованы Европейским
обществом технологии животных клеток [Jain et al.,
1991; Kloppingeretal., 1991; Speiret al., 1991]. Последние
данные были связаны с применением бакуловирусных
векторов в клеточных линиях насекомых, которые
имеют ряд преимуществ в посттрансляционной моди¬
фикации, обнаруженной на линиях млекопитающих и
без регуляторных проблем, существующих в биофарма-
цевтической отрасли, связанных с выделением клеток
млекопитающих животных и человека.
Культура клеток позвоночных, отличных от птиц
и млекопитающих, также ведется в соответствии с
культуральными процедурами, разработанными для
теплокровных млекопитающих и поэтому в целом нет
больших различий между технологиями культивиро¬
вания. Поскольку это развивающаяся область, следует
пока учитывать определенные основные параметры,
чтобы воспроизводить оптимальные условия для роста
культуры, которые, возможно, потребуется устано¬
вить: например, pH, осмоляльность, (которые могут
варьировать от вида к виду), питательные элементы,
концентрация минералов. Температура может быть
менее важна для жизни, но должна быть фиксирована
в определенном диапазоне температуры окружающей
среды и регулирована в пределах ±0,5 °С; перегревание
может привести к значительному повреждению.
27.7.1. Клетки рыб
Культура клеток рыб становится все более популяр¬
ной в результате растущего коммерческого интереса
к рыбоводству. Протокол 27.6 для культур клеток
рыб полосатого данио приводится по Collodi [1998].
Эти рыбы имеют рад благоприятных характеристик,
которые делают их популярным объектом для исследо¬
ваний, представителем позвоночных, не относящимся
к млекопитающим, удобным для токсикологических
исследований и изучения биологии развития [Powers,
1989; Driever et al., 1994].
Данио достигают половой зрелости примерно в 3 ме¬
сяца, самки производят от 100 до 200 икринок каждую
неделю круглый год. Эмбриогенез заканчивается вне
тела матери в течение 3-4 дней и более, прозрачные
эмбрионы доступны для экспериментальных мани¬
пуляций, включая мечение клеток и метод аблации
[Westerfield, 1993]. Методы in vitro, включающие
культуры эмбриональных клеток также разработаны
для изучения развития рыб. Установлены клеточные
линии и долгоживущие клеточные культуры, иници¬
ированные из бластулы, гаструлы и поздней стадии
эмбриогенеза [Collodi etal., 1992; Ghosh & Collodi, 1994;
Sun et al., 1995a, b; Peppelenbosch et al., 1995].
Линии клеток, полученные от эмбрионов ран¬
ней стадии. Поскольку дифференцировка эмбрионов
полосатого данио происходит в течение гаструляции,
клетки эмбрионов более ранней стадии типа бластулы
являются плюрипотентными [Kane et al., 1992], и ме¬
тоды были разработаны для культуры клеток этих эмб¬
рионов ранней стадии развития. Линия клеток ZEM-2,
полученная из эмбрионов в середине стадии бластулы
эмбрионального развития, растет в культуре в течение
более чем 300 поколений в среде, содержащей низкие
концентрации FBS и сыворотки форели, инсулина,
экстракта эмбриона форели, и среды, кондициониро¬
ванной клетками печени крысы Буффало [Ghosh &
Collodi, 1994].
Линия клеток фибробластов, ZEF, также была полу¬
чена из эмбрионов ранней стадии в среде с добавлением
FBS и FGF. Однажды установленная, линия поддержи¬
валась в среде LDF (см. материалы в протоколе 27.6)
содержащей 10% FBS [Sun etal., 1995а]. Клетки линии
ZEF использовались как фидерный слой для первич¬
ных культур эмбриональных клеток полосатого данио
[Sun et al., 1995а, Bradford et al., 1994а].
Первичные культуры, полученные от эмбрионов
ранней стадии. В дополнение к постоянно растущим
линиям эмбриональных клеток, которые являются
коммерчески доступными, были разработаны методы
получения первичных культур из ранних эмбрионов
данио [Sun et al., 1995b, Bradford et al., 1994a, Ь]. Пер¬
вичные культуры, полученные из ранних эмбрионов
стадии гаструлы, с поддержанием диплоидного набора
хромосом имеют морфологические характеристики
плюрипотентных эмбриональных стволовых клеток
(ES) при росте на фидерном слое фибробластов ZEF
в среде, содержащей FBS, сыворотку форели, экстракт
эмбриона рыб, инсулина, и фактор ингибирующий лей¬
кемию [Sun et al., 1995b; Bradford et al., 1994a].
Линии клеток9 полученные из эмбрионов позд¬
ней стадии. Поздняя стадия развития эмбрионов
данио (20-24 ч после оплодотворения) использует¬
ся для получения трех линий фибробластов: ZF29,
ZF13 [Peppelenbosch et al., 1995], и ZF4 [Driever &
Rangini, 1993]. Линии были получены в среде L-15
Леибовитца (ZF29 и ZF13) или смеси F12 Хэма и
Игла — модифицированной среде Дюльбекко (ZF4)
с добавлением FBS.
536
ГЛАВА 27. СПЕЦИАЛЬНЫЕ МЕТОДЫ
ПРОТОКОЛ 27.6. Культуры клеток эмбриона
полосатого данио
Материалы
Стерильные:
• Основная питательная среда LDF:
• среда Лебовитца L-15,100 мл
• DMEM, 70 мл
• среда Хэма F-12,30 мл
• Селенит натрия 6 мкМ, 200 мкл
• Хранить в холодильнике при 4 °С.
• Первичная среда LDF: основная питательная
среда LDF, с добавками:
• FBS1%
• Сыворотка форели, 0,5%
• Экстракт эмбриона форели, 40 мкг белка/мл
• Инсулин, 10 мкг/мл
• Фактор, ингибирующий лейкемию (LIF), чело¬
веческий рекомбинантный, 10 нг/мл
• рабочая среда LDF: основная среда LDF с до¬
бавками:
• FBS, 1%
• Сыворотка форели (Sea Grow, East Coast Bio-
logicals), 0,5%
• Экстракт эмбриона форели, 40 мкг белка/мл
• Инсулин, 10 мкг/мл
• BRL-кондиционированная среда, 50%
• D-среда: DMEM/F12/10FB: 50/50 DMEM/среда
Хэма F12 с 10% FBS
• Экстракт эмбриона форели:
а) Соберите эмбрионов форели {Shasta Rainbow
или другого вида), 28 дней после оплодотво¬
рения, выращенных при 10 °С; или полосатого
данио, три дня после оплодотворения, содер¬
жавшихся при 28 °С, храните замороженными
при -80 °С.
б) Чтобы приготовить экстракт, разморозьте
эмбрионы (примерно 150 г), гомогенизируйте
в 10 мл LDF в течение 2 мин на льду, исполь¬
зуя гомогенизатор Tissuemizer homogenizer
(Tekmar).
в) Процедите гомогенат через несколько слоев
марли, чтобы удалить хорионы и затем цент¬
рифугируйте гомогенат при 20 000 g в течение
30 мин при 4 °С.
г) После центрифугирования соберите суперна¬
тант, оставляя на поверхности ярко-оранже¬
вый липидный слой.
д) Перенесите супернатант в новую пробирку,
центрифугируйте как в шаге (в).
е) Соберите супернатант, оставляя следы липида,
и затем ультрацентрифугируйте при 100 000 g
в течение 60 мин при 4 °С.
ж) После ультрацентрифугирования соберите
супернатант, оставляя липидный слой. Разве¬
дите супернатант в LDF (1:10) и стерилизуйте
фильтрацией.
з) Экстракт должен пройти через серию филь¬
тров (1,2 мкм, 0,45 мкм и 0,2 мкм).
и) Храните экстракт замороженным при -80 °С
по 0,5-мл аликвотам.
к) При использовании экстракта для культуры
клеток определите концентрацию белка (см.
протокол 21.4) и затем разведите экстракт в
LDF до желательной рабочей концентрации,
л) Храните разведенный экстракт в холодильни¬
ке при 4 °С не более двух месяцев
• Среда LDF, кондиционированная ростом клеток
BRL:
а) Вырастите культуру клеток BRL (АТСС)
при 37 °С в 75-см2 флаконах в среде DMEM/
F12/10FB.
б) Когда кулыура станет конфлюэнтной, заме¬
ните среду FD на LDF с добавлением 2% FBS,
инкубируйте клетки при 37 °С.
в) Через 5 дней удалите LDF, профильтруйте и
храните среду при -20 °С.
г) Добавьте свежую LDF к клеткам BRL, повтори¬
те процесс через 5 дней. Кондиционированная
среда LDF может быть собрана трижды с одно¬
го флакона, прежде чем клеточный монослой
разрушится и нужно будет вновь дожидаться
формирования конфлюэнтного монослоя
• Буфер Хольтфретера:
NaCl, 70 г
КС1,1,0 г
NaHC03,4,0 г
СаС12,2,0 г
UPW, до 1000 мл
Хранить при 4 °С. Рабочий раствор приготавли¬
вается разведением 1:20 в UPW.
• Проназа Е, 0,5 мг /мл в буфере Хольтфретера
(Holtfreter)
• Трипсин, 1%, EDTA, 1 мМ, в D-PBSA
А. Фидерные слои фибробластов
Схема
Готовят фидерные слои фибробластов полосатого да¬
нио со стадии гаструлы [Sun et al., 1995а] путем уда¬
ления хориона в проназе, культивировании клеток в
среде с добавлением FGF и селекцией фибробластов
дифференциальной трипсинизацией
Материалы для фидерных слоев
Стерильные или асептически приготовленные
• Эмбрионы, 8 ч после оплодотворения
• Проназа
• Трипсин
• FBS
• Первичная среда LDF с 10 нг/мл бычьего FGF
(смесь а+ Ь)
• Основная среда LDF с 10% FBS
• Флаконы 25 см2
27.7. Культуры клеток пойкилотермных животных
537
Протокол
1. Соберите примерно 30 эмбрионов (восемь часов
после оплодотворения).
2. Удалите хорион обработкой в проназе [Sun et al.,
1995а].
3. Инкубируйте эмбрионы в трипсине (1 мин) с
осторожным пипетированием для диссоциации
клеток.
4. Добавьте FBS (конечная концентрация 10%)
чтобы остановить действие трипсина.
5. Соберите клетки в осадок путем центрифугиро¬
вания (при 500 g в течение 10 мин).
6. Ресуспендируйте осадок клеток в 5 мл первичной
среды LDF, содержащей FGF перенесите клетки
в 25-см2 флакон.
7. Позвольте клеткам прилипнуть и вырасти до
конфлюэнтной стадии роста при 26 °С.
8. Пересейте культуру в такую же среду.
9. После 2-3 пассажей в культуре будет представ¬
лена смешанная популяция эпителиальных
клеток и фибробластов, поддерживайте клетки
на основной среде LDF с 10% FBS. Селекцию
фибробластов для дальнейшей культуры прово¬
дят дифференциальной трипсинизацией:
а) Обработайте культуру трипсином в течение
1 мин для удаления большинства фиброблас¬
тов, оставьте эпителиальные клетки прилип¬
нуть к пластику.
б) Перенесите фибробласты в другой флакон,
повторите процедуру, когда культура станет
конфлюэнтной.
в) После двух-трех пассажей культура будет
состоять из гомогенной популяции фибро¬
бластов.
10. Приготовьте фидерный слой из фибробластов с
блокированным ростом.
а) Добавьте клетки в соответствующий культураль¬
ный флакон или чашку, позвольте фибробластам
сформировать конфлюэнтный монослой.
б) Добавьте митомицин С, 10 мкг/мл к культуре,
инкубируйте культуру 3 ч при 26 °С.
в) Затем ополосните три раза LDF, культура
фибробластов с заблокированным ростом
может быть использована в качестве фидер¬
ного слоя для культур эмбриональных клеток
полосатого данио.
в. Первичные культуры
Схема
Собирают эмбрионы, удаляют хорион, дезагрегиру¬
ют эмбрионы в трипсине и выращивают первичные
культуры клеток эмбрионов данио со стадии блас¬
тулы и ранней гаструлына фидерном слое эмбрио¬
нальных фибробластов
Материалы для первичных культур:
Стерильные:
• Первичная среда LDF
• Буфер Хольтфретера
• Разведенная хлорная известь, 0,1% в UPW
• Раствор проназы Е
• Фидерные слои эмбриональных фибробластов
• Человеческий рекомбинантный фактор инигиб-
рования лейкемии (LIF), 1 мкг/мл
Протокол
1. Соберите эмбрионы на стадии средней бластулы
или ранней гаструлы, промойте несколько раз
чистой водой.
2. Перенесите эмбрионы в 6-см чашки Петри (50-
100 эмбрионов /на чашку), переместите в лами¬
нарный шкаф и далее работайте в асептических
условиях.
3. Замочите эмбрионы на 2 мин разведенной хлор¬
ной известью и несколько раз промойте в стериль¬
ном буфере Хольтфретера.
4. Удалите хорион инкубированием в 2 мл раствора
проназы Е в течение 10 мин, затем осторожным
помешиванием в чашке Петри отделите эмбрионы
от частично расщепленного хориона.
5. Наклоните чашку, чтобы собрать эмбрионы на
одной стороне, осторожно отберите 1,5 мл рас¬
твора проназы пастеровской пипеткой. Чтобы
предупредить прилипание эмбрионов к чашке,
удерживайте чашку в наклонном положении, так
чтобы эмбрионы оставались суспендированы в
остатках раствора проназы.
6. Осторожно промойте эмбрионы добавлением
2 мл буфера Хольтфретера и аккуратным пере¬
мешиванием.
7. Наклоните чашку и удалите большую часть бу¬
фера Хольтфретера, оставив эмбрионы примерно
в 0,5 мл.
8. Повторите промывание еще два раза.
9. После окончательного промывания оставьте
эмбрионы суспендированными в 0,5 мл буфера
Хольтфретера, добавьте 2 мл раствора трипсина.
10. Инкубируйте эмбрионы в трипсине в течение
1 мин, диссоциируйте клетки осторожным пипе¬
тированием (3-4 раза)/
11. Сразу после этого перенесите суспензию клеток
в стерильные полипропиленовые центрифужные
пробирки, добавьте в каждую пробирку 200 мкл
FBS чтобы остановить действие трипсина.
12. Соберите клетки центрифугированием (при 500 g,
5 мин), ресуспендируйте осадок в первичной сре¬
де LDF (без FBS или сыворотки форели).
13. Посейте клетки при 1х104 кл./см2 на фидерный
слой эмбриональных фибробластов в многолу¬
ночных планшетах или флаконах.
14. Позвольте клеткам прикрепиться к фидерному
слою (-15 мин) затем добавьте FBS и сыворотку
форели. Добавление в среду человеческого реком¬
бинантного LIF (10 ng/мл) обычно используется
чаще, чем BRL-кондиционированная среда [Sun
et al., 1995а, Ь].
538
ГЛАВА 27. СПЕЦИАЛЬНЫЕ МЕТОДЫ
С. Клеточные линии
Материалы для клеточных линий
Стерильные или асептически приготовленные
• Клетки ZEM-2 (или аналогичные)
• Среда LDF
Протокол
1. Растущие культуры, такие как ZEM-2, получен¬
ные из эмбрионов данио ранней стадии, выращи¬
вайте на среде LDF до -70% конфлюэнтности.
2. Инкубируйте культуры при 26 °С атмосфере
окружающей среды.
3. Меняйте среду примерно каждые 5 дней.
4. Субкультивируйте с трипсинизацией (см. прото¬
кол 13.2).
Показано, что сыворотка форели и экстракт эмбрио¬
на также стимулируют рост эмбриональных клеток рыб
других видов [Collodi & Barnes, 1990].
27.7.2. Клетки насекомых
В течение некоторого времени имелся значительный
интерес к культуре клеток насекомых для изучений
контроля пестицидов и токсикологии окружающей
среды. Однако самое большое повышение интереса к
использованию культуры клеток насекомых последо¬
вало за введением бакуловирусов для клонирования
гена [Midgley et al., 1998]. Бакуловирус хорошо раз¬
множается в клетках Sf9, постоянной клеточной линии
полученной из осеннего «походного червя» Spodoptera
frugiperda. Протокол 27.7, разработанный для культуры
клеток Sf9, является сокращенным вариантом метода
Midgley et al [1998]. В этом методе постоянный рост
клеток Sf9 поддерживается в перемешиваемой фляге
(см. протокол 13.3) или сосуде с плоским основанием
с магнитным вкладышем. Перемешивание поддержи¬
вается приблизительно с частотой 80 об./мин, при этом
мешалка не должна быть источником тепла. В идеале,
температура культивирования должна поддерживаться
27 °С, но возможно вырастить клетки без инкубатора в
ПРОТОКОЛ 27.7. Культуры клеток насекомых
Схема
Механически диспергировать клетки монослоя;
поддерживать культуру при 27 °С.
Материалы
Стерильные или асептически приготовленные:
• Клетки: Sf9 [Smith et al., 1983] (АТСС #CRL-
1711) или линии, полученные из капустной пя¬
деницы Trichoplusia ni (Tn368, or BTI-TN-5B1-4;
также известная как «High Five», Invitrogen)
• Среда: EX-CELL 400 Biosciences), содер¬
жащая 2 мМ L-глютамина с добавлением 5%
FBS, 5 мл пенициллин/стрептомицинового
раствора (пенициллин, 50 U/мл; стрептомицин,
50 мкг/мл). Хранить среду при 4 °С в темноте,
подогревайте до комнатной температуры перед
использованием. Клетки Sf9 очень чувствитель¬
ны к изменениям ростовой среды, поэтому при
любых изменениях (например, при использо¬
вании бессывороточной среды EX-CELL 400),
адаптируйте клетки к новой среде постепенным
добавлением ее в течение нескольких дней.
• Диметилсульфоксид (DMSO), 10% в FBS
• Сурфактант Pluronic F68
• Культуральные флаконы
• Флаконы для перемешивания и магнитная ме¬
шалка
• Инкубатор, 27 °С (С02 не требуется)
Протокол
Стандартное ведение культуры:
1. Отделите клетки от стенок флаконов с культурой
осторожным соскребанием или смыванием струей
среды, можно использовать клетки, выращенные
в суспензии в мешалочном флаконе.
2. Подсчитайте клетки на гемоцитометре, опреде¬
лите жизнеспособность методом исключения
трипанового синего или нафталенового черного
(см. протокол 22.1).
3. Посейте клетки в мешалочный флакон в кон¬
центрации 0,5-1х106 жизнеспособных клеток/мл
(20-100 мл в 500-миллилитровый флакон).
4. Инкубируйте клетки при 20-28 °С (оптимальная
температура 27 °С.)
5. Разводите клетки до 0,5-1 х106кл./мл каждые
48-72 ч, или когда их концентрация будет дости¬
гать примерно 4-5x106 кл./мл.
6. Переносите клетки в чистый флакон каждые
3-4 недели.
7. Если клетки образуют сгустки, попробуйте по¬
высить частоту перемешивания и добавьте сур¬
фактант Pluronic F-68 (0,5-1,0%) для снижения
деформации клеток.
Замораживание клеток для хранения
(см. также протокол 20.1):
1. Подсчитайте клетки, центрифугируйте при
1000 об./мин (-200 g) в течение 5 мин.
2. Ресуспендируйте осадок при lxlO7кл./мл 10%
DMSO в FBS.
3. Разлейте клетки по аликвотам, поместите в ам¬
пулы, охлаждайте на льду в течение 1 ч.
4. Упакуйте пробирки в пенопластовый кон¬
тейнер.
5. Медленно заморозьте пробирки (~1 °С/мин) в
течение ночи до -70 °С.
6. Перенесите пробирки в морозильную камеру с
жидким азотом.
27.8. Молекулярная гибридизация in situ
539
7. Для того чтобы восстановить замороженные
клетки, быстро разморозьте ампулы при 37 °С.
(Если клетки хранятся в жидкой фазе, осторож¬
но размораживайте их в закрытом сосуде, чтобы
избежать риска повреждения при возможном
взрыве ампулы.)
8. Перенесите клетки в 25-см2 — флакон, содержа¬
щий 5 мл среды. Наклоните флакон, чтобы клетки
растеклись равномерно.
9. Удалите среду после 2-3 ч, когда большая часть
клеток прикрепится, добавьте 5 мл свежей среды.
10. Оставьте клетки на 2-3 дня для восстановления
до отделения их от стенок, затем перенесите в
флакон для перемешивания или большой плас¬
тиковый флакон, как описано выше.
комнате с постоянной температурой между 20 и 28 °С.
Для этих сред не требуется С02. Клетки могут расти в
стандартных пластмассовых флаконах для культуры
тканей, однако, поскольку клетки прикрепляются к по¬
верхности флакона, для субкультивирования следует
отделять их от стенок, соскребая или смывая клетки
сильной струей среды. Однако этот метод приводит к
гибели большого количества клеток, потому что клетки
сильно прикрепляются к пластмассе, когда растут в
присутствии сыворотки. Клетки должны иметь время
удвоения популяции < 24 ч.
27.8. МОЛЕКУЛЯРНАЯ ГИБРИДИЗАЦИЯ
IN SITU
Гибридизация нуклеиновых кислот используется
обычно для обнаружения определенных нуклеотидных
последовательностей ДНК (Саузерн-блот) или РНК
(Нозерн-блот). Эта техника может рассматриваться
как прикладной цитологический метод работы с фик¬
сированными клетками для определения нуклеотидной
последовательности in situ (см. цв. вкладку 13в, г). Про¬
токол 27.8 для гибридизации in situ был любезно предо¬
ставлен W. Nicol Keith, Centre for Oncology and Applied
Pharmacology, University of Glasgow, Garscube Estate,
Switchback Rd., Glasgow G611BD, Scotland, UK.
27.8.1. Анализ экспрессии генов РНК
методом гибридизации in situ
Молекулярные методы могут быть разделены на две
группы: анализ лизата клеток и анализ in situ. В случае
применения методов анализа лизата (Саузерн-блот и
ПЦР) берется биопсия ткани, которая гомогенизиру¬
ется, для разрушения пространственных отношений
между клетками ткани разрушаются [Murphy et al.,
1995]. Этот процесс ведет к потере информации о
гетерогенности клеток и маленьких субпопуляциях,
ПРОТОКОЛ 27.8. Авторадиографическая
гибридизация in situ
Схема
Гибридизовать радиоактивно меченые зонды с
фиксированными клетками на предметных стеклах.
Затем визуализовать участки гибридизации методом
микроавторадиографии
Материалы
Нестерильные:
• Микроцентрифужные пробирки (например, Ep¬
pendorf)
• Микроцентрифуга (Eppendorf)
Линеаризация плазмидной ДНК:
• Набор для мечения РНК (Amersham, RPN
3100)
• Диэтилпирокарбонат (DEPC; Sigma)
• DEPC-вода: 1 % DEPC в дистиллированной воде
(dH20), автоклавированная
• Фенол-хлороформ-изоамиловый спирт, pH 8
(Sigma)
• Ацетат натрия (NaAc), 3 М, pH 8,0
• Абсолютный спирт, (ЧДА)
• Гликоген, 20 мг/мл (Roche)
• Агароза, 1%, для электрофореза (Invitrogen)
Реактивы и растворы для мечения:
• Дитиотреитол (DTT), 0,2 М (Sigma)
• DTT, 50 мМ: разлить по аликвотам и хранить
при -20 °С
• [35S] UTP (Amersham SJ 603)
• Колонки G50 Sephadex (Amersham Biosciences)
• Буфер для колонок: 0,3 М NaAc, 1 мМ EDTA, 1%
SDS, автоклавированный
• Фенол, pH 5,0 (Sigma)
• Хлороформ-изоамиловый спирт (Biogene)
• Сцинтилляционная жидкость: Ecoscint A (Na¬
tional Diagnostics)
Реактивы и растворы для гибридизации
in situ:
• Histoclear (Fisher)
• NaCl-DEPC: 0,85% NaCl, 1 % диэтилпирокарбонат
(DEPC), автоклавированный
• D-PBSA, 1% DEPC, автоклавированный
• EDTA, 0,5 M: растворить в 1% DEPC в dH20; pH
7,5, автоклавированный
• Буфер для протеиназы К: 1 М Tris НС1, 0,5 М
EDTA, 1% DEPC pH 7,5, автоклавированный
• Основной раствор протеиназы К (Sigma):
20 мг/мл в DEPC/H20; разделить на аликвоты
и хранить при -20 °С
• Формалин
• Триэтаноламин, 0,1 М, с 1% DEPC, автоклави¬
рованный
• Уксуснокислый ангидрид (Sigma)
540
ГЛАВА 27. СПЕЦИАЛЬНЫЕ МЕТОДЫ
• DTT, 1 М: разделить на аликвоты и хранить при
-20 °С
• Смесь для гибридизации, 60%:
Формамид (Fluka), 6 мл
Декстрансульфат, 50%, в 1% DEPC, 2 мл
SSC, 20х, 1 мл
Tris-HCl, 1 М, 100 мкл
Раствор Денхардта, 50х, 200 мкл
SDS, 10%, 100 мкл
tRNA (10 мг/мл; Sigma), 400 мкл
DNA лосося (10 мг/мл; Sigma), 200 мкл
Хранить при -20 °С разлитым по аликвотам объ¬
емом 400-мкл
Моющие растворы и реактивы:
• SSC, 20 (см/ Приложение I; также Invitrogen);
развести до 5х SSC, 2х SSC и 0,1х SSC
• Формамид, 50% в 2х SSC
• Р-меркаптоэтанол (Sigma)
• РНКазный буфер: 0,5 М NaCl, 0,5 М EDTA, 1 М
Tris, pH 7,5
• Маточный раствор РНКазы (Sigma): 10 мг/мл
в DEPC/H2Oj хранить при -20 °С в 400-мкл
аликвотах
• Желатин: 0,2 г в 200 мл dH20; растопить в СВЧ-
печи 2 мин, охладить, профильтровать
Реактивы и растворы для авторадиографии:
См. протокол 27.3.
Протокол
Манипуляции с РНК
Все растворы, применяемые при подготовке зондов
и других манипуляциях вплоть до отмывки после
гибридизации, должны быть свободны от РНКазы.
Растворы следует готовить с DEPC и автоклавиро-
вать в течение 20 мин при 121 °С. Данная процедура
удаляет большую часть РНКаз, но в результате
повсеместной распространенности РНКаз она не
заменяет необходимость обработки растворов, стек¬
ла и пипеток. Целесообразно иметь набор пипеток,
который используется только для работы с РНК, ре¬
гулярная их обработка с DEPC-водой в течение ночи
или использование патентованных анти-РНКазных
средств, таких как RNase-Zap (Ambion), также может
быть полезной. Все стекло должно быть завернуто
в алюминиевую фольгу и автоклавировано перед
использованием. Пластиковые пробирки Eppendorf
могут быть обработаны DEPC-водой или RNase-Zap
перед автоклавированием.
Подготовка зонда:
РНК-зонды (рибозонды)
Подготовка РНК-зондов требует использования
ДНК-матрицы с целевой последовательностью,
возможно получение смыслового и антисмыслово-
го РНК-зондов с включенными радиоактивными
нуклеотидами. Одноцепочечная РНК-зонд является
идеальной, если требуется высокая чувствительность.
Обычно используются зонды длиной 200-1000 пар
нуклеотидов (пн), однако, возможно, оптимальной
является длина 150-200 пн, поскольку проницае¬
мость тканей может быть сниженной при больших
размерах зонда. Для уменьшения размеров зонда при
необходимости может использоваться ограниченный
щелочной гидролиз. Гибриды РНК-РНК или ДНК-
РНК более стабильны, чем их олигонуклеотиды
или ДНК- прототипы (counterparts), что делает их
наиболее популярными зондами.
Промышленно производимые зонды
и контрольные зонды
Коммерчески доступны промышленно производи¬
мые ДНК-матрицы (Ambion) к актину и GAPDH
могут быть использованы для получения РНК-зон¬
дов при их использовании в качестве положительных
контролей, поскольку они участвуют в контроле
геном и экспрессируются повсеместно. Смысловые
зонды обычно используются в качестве негативных
контролей, и лучше использовать их как контроль,
чем «пропускать» пробы. Хорошо охарактеризо¬
ванные образцы опухоли с определенным уровнем
экспрессии РНК могут также использоваться как
положительный контроль при каждом пропускании
(run) препарата. Образцы опухоли разных тканей
также предлагаются разными производителями.
Линеаризация плазмидной ДНК:
1. Используйте 10-20 мкг ДНК в микроцентрифуж-
ной пробирке
2. Добавить 10 единиц рестрикционного фермента
на 1 мкг ДНК, проведите ферментативное рас¬
щепление в соответствие с инструкцией произ¬
водителя фермента.
3. Оставьте реакцию протекать при 37 °С в течение
3 ч или ночи.
Фенол-хлороформная экстракция:
4. Добавьте 400 мкл фенол-хлороформ-изоамило-
вого спирта (pH 8,0), перемешайте на вортекс-
мешалке.
5. Открутите смесь в течение 3 мин при 13 000 об./
мин в микроцентрифуге при комнатной темпе¬
ратуре.
6. Соберите супернатант, перенесите в чистую про¬
бирку, добавьте 10 мкл 3 М NaAc (pH 8).
7. Добавьте 250 мкл 100% этанола (хранить при
-20 °С).
8. Добавьте 1 мкл гликогена, чтобы облегчить пре¬
ципитацию ДНК.
9. Поместите пробирку в сухой лед на 1 ч.
10. Открутите 15 мин при 13 000 об./мин.
11. Удалите супернатант, сохранив осадок.
12. Добавьте к осадку 400 мкл 70% этанола (храня¬
щегося при -20 °С) для отмывания осадка.
13. Открутите 10 мин при 13 000 об./мин.
27.8. Молекулярная гибридизация in situ
541
14. Удалите 70% этанол, высушить осадок на воздухе.
15. Ресуспендируйте осадок в 10-20 мкл DEPC/H20,
в зависимости от количества используемого ДНК
(см. шаг 1), рассчитывая, что конечная концент¬
рация должна составлять 1 мкг/мл.
16. Прогоните 0,5 мкл суспензии на 1% агарозном
геле.
Мечение РНК:
Эта часть процедуры включает в себя включение
радиоактивных нуклеотидов. Используйте набор
Amersham (RPN3100; Amersham Biosciences) в со¬
ответствии с инструкцией на вкладыше со ссылкой
на следующий метод:
1. В микроцентрифужную пробирку внесите:
а) Транскрипционный буфер 5х 4 мкл
б) DTT, 0,2 М 1 мкл
в) HPR1 1 мкл
г) ATP,CTPhGTP 0,5 мкл
д) Линеаризованную ДНК-матрицу
(1 мкг/мл) 1 мкл
е) [^SJUTP 9,5 мкл
ж) РНК-полимераза 2 мкл
2. Смешайте компоненты, инкубируйте раствор
1,5 ч при 37 °С.
ДНКазная экстракция ДНК-матрицы:
3. Добавьте 10 Ед фермента ДНКаза L.
4. Добавьте 1 мкл ингибитора РНКазы.
5. Перемешайте раствор, инкубируйте 10 мин при
37 °С.
Удаление невключенных нуклеотидов:
6. Уравновесьте колонку G50 Sephadex внесением
2 мл колоночного буфера.
7. Добавьте зонд в колонку.
8. Добавьте 400 мкл колоночного буфера, позвольте
буферу протечь сквозь колонку.
9. Добавьте еще 400 мкл колоночного буфера, собе¬
рите элюат в пробирку.
Фенол-хлороформная экстракция:
10. Добавьте 400 мкл фенола (pH 5,0) в пробирку,
перемешайте на вортекс-мешалке, центрифуги¬
руйте 3 мин при 13000 об./мин.
И. Соберите супернатант, перенесите в новую
микропробирку и добавьте 400 мкл хлороформ-
изоамилового спирта. Перемешайте, центрифу¬
гируйте 3 мин при 13000 об./мин.
12. Соберите супернатанат и отберите 1 мкл для
подсчета включения метки.
13. Добавьте 2,5 объема 100% этанола (хранившегося
при -20 °С) к оставшемуся супернатанту.
14. Добавьте 1 мкл гликогена дрожжей для ускорения
преципитации.
15. Поместите пробирку на сухой лед на 30 мин.
16. Центрифугируйте 15 мин при 13 000 об./мин.
17. Удалите спирт и оставьте осадок не поврежденным.
18. Отмойте осадок 70% этанолом, центрифугируйте
10 мин при 13 000 об./мин, удалите этанол.
19. Подсушите осадок на воздухе, ресуспендируйте
в 50 мМ DTT, подсчитав объем 50 мМ DTT сле¬
дующим образом:
а) Добавьте 1 мкл супернатанта, полученного
на шаге 12 к 2-3 мл сцинтилляционной жид¬
кости, определите число импульсов в минуту
(СРМ) на сцинтилляционном счетчике.
б) Объем VDTT = (СРМх400)/6х105
где 400 — объем после фенол-хлороформной
экстракции, 6x105 — требуемое общее число
импульсов в минуту в растворе DTT.
в) Так определяется объем 50 мМ DTT, в кото¬
ром следует ресуспендировать пробу.
20. Отберите 1 мкл и снова подсчитайте СРМ, чтобы
подтвердить активность пробы.
Гибридизация in situ:
Подготовка образца:
Целью настоящего раздела процедуры является
сохранение структуры и морфологии ткани и сохра¬
нение РНК-продуктов. Быстрая обработка образцов
ткани замораживанием или фиксацией в формалине
обеспечивает сохранение РНК. Поперечно свя¬
зывающие фиксирующие жидкости, такие как 4%
параформальдегид и 4% формальдегид, являются
фиксаторами выбора для сохранения и /или доступ¬
ности детекции клеточной РНК продолжительность
фиксации будет зависеть от размеров образца. Более
длительная фиксация обеспечит лучшую сохранность
морфологии ткани, но может уменьшить доступность
для зонда. Парафиновый воск является средой выбо¬
ра для заливки образца. Он позволяет делать срезы
толщиной до 1 мкм и легко удаляется перед гибриди¬
зацией. Поскольку срезы будут проводиться через ряд
растворов, рекомендуется использовать предметные
стекла с предварительной обработкой, удерживающей
препарат на стекле. Замороженные образцы должны
быть охлаждены до -70 °С, и после получения срезов
на криомикротоме они должны быть помещены на
обработанное стекло и зафиксированы.
Подготовка тканей к гибридизации:
Эта стадия обработки ткани перед гибридизацией
требуется для улучшения доступа зонда к целевой
последовательности РНК и снижения неспецифич¬
ного фонового связывания. Образец подвергается
обработке протеазой для повышения доступности
целевой РНК для зонда, особенно если размеры зон¬
да превышают 100 пн. Важно провести последующую
фиксацию в формальдегиде, чтобы предупредить
дезинтеграцию ткани. Неспецифическое связыва¬
ние с аминогруппами снижается ацетилированием
под действием уксуснокислого ангидрида. В ходе
подготовки ткани большое внимание должно быть
уделено защите образа от действия РНКаз. Вся
542
ГЛАВА 27. СПЕЦИАЛЬНЫЕ МЕТОДЫ
стеклянная посуда должна быть обработана, чтобы
удалить какую бы то ни было контаминацию. Все
растворы должны быть обработаны DEPC, в ходе
процедуры оператор должен работать в перчатках.
Манипуляции со срезами тканей должны быть све¬
дены до минимума.
Гибридизация:
Для некоторых пар зонд/целевая последователь¬
ность может быть критичной температура гибри¬
дизации. Формамид в гибридизационном буфере
как дестабилизатор цепи снижает точку плавления
гибридов и обеспечивает снижение температуры
гибридизации. Более низкая температура помогает
сохранить тканевую структуру. В качестве оптималь¬
ной выбрана температура 52 °С. Декстран сульфат в
гибридизационном буфере повышает концентрацию
зонда за счет исключения объема растворителя и
таким образом уменьшает время гибридизации. Хотя
реакция гибридизации практически завершается
через 5—6 ч, удобнее оставить реакционную смесь на
ночь. Концентрация ионов натрия в буфере служит
для стабилизации гибридов.
Постгибридизационная отмывка:
Главной целью постгибридизационной отмывки
является удаление несвязанных и неспецифичес¬
ки связанных зондов путем подбора температуры,
концентрации соли и концентрации формамида.
Использование РНКазы обеспечивает расщепле¬
ние одноцепочечной РНК, не связанной с целевой
последовательностью, но фермент не действует на
связанные комплексы РНК-РНК.
Предварительная обработка парафиновых
срезов:
1. Депарафинируйте срезы действием Histoclear,
Два раза по 10 мин.
2. Регидратируйте, проведя срезы по спиртам нис¬
ходящей концентрации: 100%, 90%, 70%, 50% и
30%, по 10 секунд в каждом.
3. Промойте в 0,85% NaCl и растворе D-PBSA по
5 мин.
4. Обработайте срезы в Proteinase К 7,5 мин: 400 мкл
маточного раствора Proteinase К разведите в
200 мл протеиназного буфера.
5. Промойте в D-PBSA; 3 мин.
6. Обработайте в 4% формалина или 4% парафор¬
мальдегида.
7. Промойте в DEPC-воде в течение 1 мин.
8. Ацетилируйте в 200 мл 0,1 М триэтаноламина
с 500 мкл уксуснокислого ангидрида в течение
10 мин, перемешивая в вытяжном шкафу.
9. Промойте по 5 мин в D-PBSA и 0,85% NaCl.
10. Дегидратируйте, проведя по спиртам восходящей
концентрации: по 10 с в каждом: 30%, 50%, 70%,
90% и 100%.
11. Высушите срезы.
Приготовление зондов и гибридизация:
12. На 20 парафиновых срезов соедините 16 мкл 1М
DTT, 344 мкл 60% Hybridmix и 40 мкл зонда.
Быстро перемешайте на вортекс-мешалке.
13. Денатурируйте зонд при 80 °С в течение 3 мин.
Охладите на льду.
14. Применяйте 20 мкл смеси из шага 12 на каждый
срез, закройте стеклянным покровным стеклом.
15. Проведите гибридизацию раствора при 52 °С в
течение ночи в увлажнительной камере.
Постгибридизационая отмывка:
16. Предварительно подогрейте все рабочие растворы
до требуемой температуры.
17. Отмойте срезы в 200 мл 5х SSC с 250 мкл Р-мер-
каптоэтанола в течение 30 мин при 50 °С.
18. Отмойте в 200 мл 50% формамида и 2х SSC с
1,4 мл Р-меркаптоэтанола в течение 20 мин при
65 °С.
19. Промойте дважды в 200 мл РНКазного буфера в
течение 10 мин каждый раз при 37 °С.
20. Отмойте в 200 мл РНКазного буфера с 400 мкл
раствора РНКазы А в течение 30 мин при
37 °С.
21. Повторите шаг 19, при каждой отмывке увеличив
время до 15 мин.
22. Повторите шаг 18.
23. Отмойте в 200 мл 5х SSC и затем в 200 мл 0,1х
SSC по 15 мин каждая отмывка при 50 °С.
24. Дегидратируйте в серии спиртов восходящей кон¬
центрации: 50%, 70% и 100% по 1 мин в каждом.
25. Высушите на воздухе.
26. Погрузите стекла со срезами в раствор желатина
на 1 мин, затем высушите.
Авторадиография:
См п. Получение ауторадиографов протокол 27.3.
особенно при биопсии опухоли, и представляет собой
результат усреднения изменений. Однако количест¬
венно эти методы могут быть более простыми и более
точными, чем в подходах in situ. При сравнении этих
подходов методы in situ типа РНК-гибридизации in situ
позволяют производить выявить экспрессию гена в ин¬
дивидуальных клетках в пределах их гистологического
контекста [Soder et al., 1997, 1998; Keith, 2003].
Принцип гибридизации in situ основан на специфи¬
ческом связывании меченого нуклеинового кислотного
основания с комплементарной последовательностью в
образце ткани, сопровождаемом визуализацией иссле¬
дования. Этот процесс позволяет как обнаружить, так и
определить локализацию искомой последовательности.
Предпосылками для успеха этой процедуры является
наличие искомых нуклеиновых кислот в образце и
досягаемость этих последовательностей для исследова¬
ния. Образцами, подходящими для in situ hybridization
(ISH), являются клетки культуры и ткани целого ор¬
гана или биопсии.
27.8. Молекулярная гибридизация in situ
543
Анализ
1) Исследуют срезы, используя световую микроско¬
пию в светло- и темно-польном освещении.
2) Отмечают различия при сравнении с положитель¬
ным и отрицательным контролем.
27.8.2. Флюоресцентная гибридизация
in situ (FISH) при анализе генов
и хромосом
Протокол 27.9 для флюоресценции при гибридиза¬
ции in situ (FISH) предоставлен Nicol Keith, Centre
for Oncology and Applied Pharmacology, University of
Glasgow, Garscube Estate, Bearsden, Glasgow G61 1BD,
Scotland, UK.
Флюоресценция при гибридизации in situ — наибо¬
лее прямой путь для определения линейного порядка
генов в хромосомах. Использование хромосома и ген-
специфических зондов может также быть применено
для анализа количественных и структурных отклонений
в пределах индивидуальной клетки. Эти методы имеют
широкий ряд применений при диагностике генетиче¬
ских болезней и идентификации делеций, транслокаций
и амплификации гена в ходе развитие рака [Wiktor &
Van Dyke, 2004; Krupp et al., 2004; Camps et al., 2004].
трансляции, с использованием коммерческих наборов
Roche (раньше Boehringer Mannheim), которая может
бьггь использована д ля внедрения биотина или дипжси-
генина в соответсвии с инструкцией производителя.
Преципитация зонда, содержащего
повторяющиеся последовательности:
Большие космидные зонды часто содержат повто¬
ряющиеся последовательности, которые, не будучи
супрессированы до гибридизации, приводят к по¬
вышению уровня неспецифической гибридизации.
Повторяющиеся последовательности могут быть
супрессированы конкурентным взаимодействием с
немеченой СоиДНК человека, которая обогащена
повторяющимися последовательностями. Реакция с
Cotl ДНК может быть включена в процедуру на ста¬
дии преципитации (шаг (б) настоящего протокола).
а) Для 20-мкл реакции «ник-трансляции», до¬
бавьте к зонду 1 мкл 0,5М EDTA, 2,5 мкл 4,0 М
LiCl, 1 мкл гликогена, от 100- до 1000-кратный
избыток человеческого Cotl DNA (Invitrogen)
и 100 мкл этанола.
б) Поместите на сухой лед на 30 мин или при
-20 °С на ночь для образования преципитата.
в) Осадите на микроцентрифуге в течение 15 мин
для образования осадка ДНК.
г) Отмойте осадок в 70% этаноле.
д) Осадите ДНК и высушить осадок.
е) Ресуспендируйте ДНК в гибридизационном
буфере до концентрации 2-10 нг/мкл.
Преципитация зондов, не содержащих
повторяющихся последовательностей:
Следуйте указанному протоколу, но не включайте в
реакцию преципитации СоЙДНК.
Денатурация зонда:
а) Для зондов, содержащих повторяющиеся пос¬
ледовательности, которые требуют подавле¬
ния с использованием Cotl ДНК, подогрейте
реакционную смесь до 70 °С в течение 10 мин.
Поместите смесь в инкубатор на 37 °С на 1 ч
до нанесения на стекло.
б) Для зондов, не содержащих повторяющихся
последовательностей, подогрейте смесь до
70 °С в течение 10 мин. Охладите реакцион¬
ную смесь на льду в течение 10 мин.
• Буфер для детекции зонда, TBST: 0,05% Tween
20 в 0,1 М Tris, 0,15 М NaCl; pH 7,5
• Формамид, 50%, в lx SSC
• Антитела:
в) Первичные антитела, поликлональная сыво¬
ротка овцы к дишксигенину или биотину, титр
в соответствии с рекомендациями поставщика
(Roche).
г) Вторичные антитела, например, FITC-конъ-
югированные антитела осла против lgG овцы
(Jackson Immunoresearch Inc.)
ПРОТОКОЛ 27.9. FISH с использованием
однокопийных геномных зондов
и хромосомных красителей
Схема
Гибридизовать биотин или дигоксигенин-меченые
зонды с денатурированными хромосомами и про¬
вести детекцию зондов по флюоресцентному окра¬
шиванию с двойными антителами.
Материалы
Нестерильные:
• SSC, 2х (см. Приложение I для SSC, 20х)
• SSC, 1х
• D-PBSA
• Гликоген, 20 мг/мл (Boehringer Mannheim)
• Этанол (EtOH), 70%
• EtOH, 100%
• Фиксатор: метанол: уксусная кислота, 3:1
• РНКаза, 100 мкг/мл в 2х SSC
• Параформальдегид, 1%, в PBSA
• Формамид, 70%, в 2х SSC
• Буфер для гибридизации: формамид (50%), дек-
странсульфат (5%) 2х SSC, ДНК молоки лосося
(500 мкг/мл)
• Меченые зонды:
Большие космидные клоны более всего подходят
для зондов, применяемых для определения одноко¬
пийных генов. Тем не менее можно также использо¬
вать кДНК-зонды. ДНК метят при помощи «ник» —
544
ГЛАВА 27. СПЕЦИАЛЬНЫЕ МЕТОДЫ
д) Разведите антитела в 3% BSA в TBST
• Резиновый латексный клей
• Смесь для заключения препарата: Vectashield;
содержит ингибитор фотообесцвечивания (Vec¬
tor Labs)
Протокол
Приготовление препарата хромосом
и денатурация:
1. Приготовьте клетки с блокированной метафазой
по стандартной методике (см. протоколы 16.7,
27.5), капните фиксированные клетки на стекла.
Обозначьте область распространения капли ал¬
мазным резцом.
2. Повторно фиксируйте клетки в течение 1 ч в све¬
жеприготовленном фиксаторе: метанол: уксусная
кислота, 3:1, высушите стекла на воздухе.
3. Прополощите стекла в 2х SSC в течение 2 мин
при комнатной температуре.
4. Инкубируйте с 100 мкг/мл РНКазы в 2х SSC при
37 °С в течение 1 ч.
5. Прополощите в D-PBSA.
6. Повторно фиксируйте в свежеприготовленном
1% растворе параформальдегида в D-PBSA в
течение 10 мин при комнатной температуре (эта
стадия необязательна).
7. Промойте в D-PBSA.
8. Дегидратируйте в течение 2 мин в большом объ¬
еме 70% этанола и затем 100% этанола. Подсуши¬
те стекла на воздухе.
9. Денатурируйте хромосомы в 70% растворе фор-
мамида в 2х SSC при 70 °С в течение 2-4 мин
(определите оптимальную продолжительность
экспериментально, начав с 2 мин). Убедитесь,
что 70%-й формамид подогрет до 70 °С перед
использованием.
10. Отмойте стекла в нескольких сменах 70%-го эта¬
нола на льду.
11. Дегидратируйте в соответствии с шагом 8, высу¬
шите стекла.
12. Хромосомы готовы к гибридизации.
Гибридизация:
13. Нанесите 10 мкл денатурированного зонда по¬
верх зоны распространения препарата хромосом,
накройте покровным стеклом 22x22 мм.
14. Опечатайте покровные стекла по краям резино¬
вым латексным клеем.
15. Поместите стекла в коробку с влажной атмосфе¬
рой при 37 °С на ночь.
Детекция зонда:
16. Удалите покровные стекла, погрузив в 2х SSC
(при комнатной температуре) и сорвав латекс.
Поместите стекла в сосуд Коплена (Coplin jar).
17. Погрузите стекла, два раза по 10 мин, в 50% рас¬
твор формамида в lx SSC, при 42 °С.
18. Промойте стекла, два раза по 10 мин, в 2х SSC
при 42°С.
19. Прополощите в TBST.
20. Блокируйте неспецифическое связывание инку¬
бированием стекол в 3% растворе BSA в TBST в
течение 30-60 мин при 37 °С.
21. Добавьте первичные антитела. Используйте
100 мкл первичных антител на одно стекло, за¬
тем закройте каждое стекло покровным стеклом
Parafilm.
22. Инкубируйте стекла при 37 °С в течение 1 ч.
23. Промойте стекла в 500 мл TBST в течение
10 мин при комнатной температуре.
24. Добавьтеь на стекла вторичные антитела, конъ¬
югированные с флюорохромом, в 3% BSA/TBST,
в разведении, рекомендованном производителем
или экспериментально определенном.
Замечание. Убедитесь что вы используете правиль¬
ную комбинацию антител, например поликлональ¬
ную сыворотку овцы к дигоксигенину в качестве
первичных антител с последующим действием FITC-
конъюгированных антител осла против IgG овцы.
25. Инкубируйте 30 мин при 37 °С.
26. Промывайте 30 мин в 500 мл TBST при комнат¬
ной температуре.
27. Проведите контрастирующее окрашивание
0,8 мкг/мл DAPI и/или 0,4 мкг/мл иодистого
пропидия в TBST в течение 10 мин. Закройте
покровным стеклом с использованием заключа¬
ющего состава препятствующего выцветанию.
28. Исследуйте стекла при помощи флюоресцентной
микроскопии при помощи соответствующей ком¬
бинации фильтров.
Нуклеиново-кислотные основания метят неи¬
зотопными метками, путем введения нуклеотидов,
модифицированных такими молекулами, как биотин
или дигоксигенин. После гибридизации меченых
зондов с хромосомами производится детекция по¬
следовательностей, которая достигается формирова¬
нием комплексов с антителами, которые распознают
биотин или дигоксигенин в образце. Гибридизация
визуализируется с использованием антител, конъюги¬
рованных с флюорохромами. Флюоресцентный сигнал
может быть обнаружен разными путями. Если сигнал
достаточно силен, может использоваться стандартная
флюоресцентная микроскопия. Однако анализ данных
и их хранение могут быть значительно улучшены при
помощи систем цифрового отображение типа кон¬
фокального лазера, сканирующего микроскопа или
охлаждаемой камеры CCD. Главным преимуществом
флюоресцентной гибридизации in situ (FISH) является
отсутствие радиоактивности, быстрота исследования,
возможность хранения данных и манипуляций с ними
и высокая чувствительность. FISH также показывает
27.9. Слияние соматических клеток
545
точную локализацию сигнала, позволяет одновремен¬
но проводить анализ двух или больше флюорохромов
и обеспечивает количественное и пространственное
распределение сигнала.
Вариации
Хромосомное окрашивание. Красители для окраши¬
вания хромосом коммерчески доступны из множества
источников, включая Invitrogen, Cambio, и Опсог.
Поэтому больше нет необходимости готовить ваши
собственные красители. Протоколы для гибридизации
и детекции изменяются в каждом случае, однако в це¬
лом перед гибридизацией может использоваться раздел
приведенного протокола: а) Подготовка хромосомы
-и денатурация. Гибридизация и детекция могут быть
выполнены согласно инструкциям поставщика. Если
краситель помечен биотином, как в случае красителей
Cambio paint, может применяться раздел (б) (Детекция
зонда) при использовании соответствующих комбина¬
ций антител.
Недавние усовершенствования. Классический кари-
отипический анализ производится методом исследова¬
ния исчерченности хромосомы, с использованием их
дифференциального окрашивания (см. протокол 27.5).
Таким образом, каждая хромосома может быть иденти¬
фицирована по определенному образцу исчерченности.
Однако традиционные методы не могут охаракте¬
ризовать многие сложные хромосомные аберрации.
Недавно были разработаны новые методы кариоти-
пирования, основанные на окрашивании хромосом,
а именно спектральное кариотипирование (SKY) и
мультицветная флюоресценция при гибридизации
in situ (MFISH). Эти методы позволяют проводить
одновременную визуализацию всех 24 человеческих
хромосом с окрашиванием в различные цвета. Кроме
того, визуализация результирующих образцов флюо¬
ресценции в соответствии с программами компьютера
делает эти методы более чувствительными, чем поз¬
воляет человеческий глаз. Эти методы чрезвычайно
успешно применяются при идентификация новых
хромосомных изменений, которые ранее не могли быть
обнаружены традиционными подходами [Wienberg &
Stanyon, 1997; Ried et al., 1997; Macville et al., 1997;
Nordgren, 2003; Lim et al., 2004].
27.9. СЛИЯНИЕ СОМАТИЧЕСКИХ КЛЕТОК
Соматические клетки сливают культивированием
вирусом Сендай или полиэтиленгликолем (ПЭГ)
[Pontecorvo, 1975]. Часть слившихся клеток доходят
до слияния ядер, некоторая доля этих клеток проходит
через митоз, так что оба набора хромосом копируются
вместе и формируется гибрид. В некоторый межвидо¬
вых гибридах например «человек-мышь», один набор
хромосом (человеческий) постепенно был утрачен
[Weiss & Green, 1967]. Таким образом, возможна
генетическая рекомбинация in vitro, и, в некоторых
случаях, возможна также сегрегация. Поскольку про¬
порция жизнеспособных гибридов низка, требуются
селективные среды, чтобы поддержать выживание
гибридов за счет родительских клеток. Используют¬
ся мутанты ТК и HGPRT (см. разд. 27.9.1) из двух
родительских типов клеток, селекция проводится на
среде НАТ (гипоксантин, аминоптерин, тимидин)
(рис. 27.3) [Littlefield, 1964а]. Остаются в живых толь¬
ко клетки, сформированные слиянием двух различных
родительских клеток (гетерокарионы), потому что
родительские клетки и продукты слияния одинаковых
родительских клеток (гомокарионы) дефектны по
тимидинкиназе или гипоксантингуанинфосфорибо-
зилтрансферазе. Родительские клетки и гомокарионы
не могут, следовательно, использовать тимидин или
гипоксантин из среды. Поскольку аминоптерин бло¬
кирует эндогенный синтез пуринов и пиримидинов,
они не способны к синтезу ДНК. Протокол 27.10
для слияния соматических клеток предоставлен Ivor
Hickey, Science Department, St. Mary’s University Col¬
lege, Belfast, Northern Ireland, UK. Хотя во многих ли¬
ниях клеток происходит спонтанное слияние, частота
таких случаев очень низка. Для получения гибридов в
существенном количестве клетки обрабатывают хими¬
ческим стимулятором слияния полиэтиленгликолем
(ПЭГ) [Pontecorvo, 1975]. В этом случае чтобы выде¬
лить клоны гибридных клеток, используются системы
селекции, которые убивают родительские клетки, но
не убивают гибриды.
ПРОТОКОЛ 27.10. Гибридизация клеток
Схтмш
Ддя слияния клеток обеспечивают их шютвый
контакт в суспензии шш монослое. Обрабатывают
клетки кратковременным воздействием PEG для
минимизации гибели клеток, Обычно клеткам дают
время 24 ч для восстановления перед селекцией для
получения гибридов
Материалы
Стерильные:
• PEG 1000 (Merck):
а) Автоклавируйте PEG до перехода в жидкое
состояние и стерилизуйте его
б) Охладите до 37 °С, затем пебремешайте с
равным объемом бессывороточной среды,
предварительно подогретой до 37 °С.
в) Доведите pH примерно до 7,6-7,9, используя
1,0 М NaOH.
г) Храните раствор при 4 °С не более 2 недель.
• Полная ростовая среда
• Бессывороточная ростовая среда
• NaOH, 1,0 М
• Чашки Петри, 5 см
• Универсальные контейнеры
546
ГЛАВА 27. СПЕЦИАЛЬНЫЕ МЕТОДЫ
РОДИТЕЛЬСКИЕ
КЛЕТКИ
СЛИЯНИЕ
КЛЕТКИ,
ОБРАЗОВАННЫЕ
В РЕЗУЛЬТАТЕ
СЛИЯНИЯ
СЕЛЕКЦИЯ В
СРЕДЕ HAT
Родительские
клетки
ВЫЖИВШИЕ
КЛЕТКИ
Устойчивость Устойчивость к
к BUdR 8-азагуанин
Гомокарион
1 i
Гетерокарион
Гомокарион
Родительские
клетки
i i
Гибридный клон
Рис. 27.3. Гибридизация соматических клеток. Селекция гибридных клеток после слияния (см. текст)
Протокол
A. Слияние клеток монослойной культуры:
1. Внесите равное количество двух типов клеток,
которые будут слиты, в 5-см чашку для культуры
тканей. Обычно бывает достаточно использовать
2,5 х105-2,5 х106 клеток каждой из родительских
линий на одну чашку.
2. Инкубируйте смешанную культуру в течение ночи.
3. Подогрейте раствор PEG до 37 °С. Возможно, при
этом потребуется вновь довести pH примерно до
7,6-7,9, используя 1,0 М NaOH.
4. Тщательно удалите среду из культур и однократ¬
но промойте бессывороточной средой.
5. Добавьте 3,0 мл раствора PEG и равномерно
распределите его поверх монослоя клеток.
6. Удалите раствор PEG точно через 1 мин, промой¬
те монослой три раза в 10 мл бессывороточной
среды до возвращения клеток к полной среде.
7. Растите клетки в течение ночи.
8. Добавить селективную среду.
B. Слияние клеток суспензионных культур:
9. Центрифугируйте смесь клеток родительских ли¬
ний: по 4x106 клеток каждой из клеточных линий
при 150 g в течение 5 мин при комнатной темпера¬
туре. Проводите центрифугирование и последую¬
щее слияние в 30-мл пластиковых универсальных
контейнерах или центрифужных пробирках.
10. Ресуспендируйте осадок в 15 мл бессывороточной
среды, повторно центрифугируйте.
11. Отберите среду.
12. Ресуспендируйте клетки в 1 мл раствора PEG
осторожным пипетированием.
13. Через 1 мин разведите суспензию добавлением
9 мл бессывороточной среды и перенесите сус¬
пензию в равных количествах в два универсальны
контейнера или центрифужные пробирки, содер¬
жащие 15 мл бессывороточной среды.
14. Центрифугируйте суспензию при 150 g в течение
5 мин. Удалите супернатант и ресуспендируйте
клетки в полной среде.
15. Инкубируйте клетки в течение ночи при 37 °С.
16. Клонируйте клетки в селективной среде.
Вариации. В литературе встречается описание
большого количества разновидностей методов слияния
с использованием ПЭГ. Хотя процедура, описанная
выше, работает хорошо для клеток мыши, хомяка и
человеческих клеток при межвидовых и внутривидовых
27.10. Продукция моноклональных антител
547
слияниях, но вряд ли она будет оптимальна для всех
клеточных линий. Включение 10% DMSO в раствор
ПЭГ имеет преимущество в снижении вязкости, по
некоторым данным, улучшает слияние [Norwood et al.,
1976]. Также, молекулярный вес используемого ПЭГ
должен быть не более 1000 Да. ПЭГ с молекулярным
весом от 400 до 6000 Да успешно применялся для по¬
лучения гибридов.
Отбор клонов гибридов. Метод отбора, исполь¬
зованный в любом специфическом случае, зависит от
исходного видового происхождения двух родительских
клеточных линий, ростовых свойств клеточных линий,
наличия выбираемых генетических маркеров на обеих
клеточных линиях. Наиболее часто отбираемые в сис¬
теме HAT гибриды: 10~4 М гипоксантина, 6x10~7 М ами-
ноптеринаи 1,6x10-6 М тимидина [Littlefield, 1964а]. Эта
система может использоваться для выделения гибрид¬
ных клеток, полученных слиянием пар клеток мутант¬
ных линий, дефицитных по ферментам тимидинкиназе
(ТК) и гипоксантингуанозинфосфорибозилтрансфера-
зе (HGPRT), соответственно. ТК"-клетки выделяются
экспозицией с BUdR, a HGPRT"-клетки экспозицией с
тиогуанином после процедур, описанных Biedler в гл. 14
(см. протокол 14.9). Когда только одна родительская
клеточная линия несет такую мутацию, тогда селекция
на HAT еще может применяться, если другая линия
клеток не растет или растет плохо в культуре (например,
лимфоциты, стареющие первичные культуры).
Дифференциальная чувствительность к сердечному
гликозиду строфантину является важным фактором в
выборе гибридов клеток грызунов и клетками множества
других видов, включая человеческие. Клетки грызунов
резистентны к концентрациям этого антиметаболита
до 2,0 мМ, а человеческие клетки погибают при 10 мкМ
строфантина. Гибриды намного более резистентны к
строфантину, чем человеческие родительские клетки.
Если HGPRT-дефицитная линия клеток грызуна сли¬
вается с немаркированными человеческими клетками,
то гибриды могут быть выделены в среде, содержащей
HAT и низкие концентрации строфантина.
Хотя существует много сообщений о других систе¬
мах селекции, широко использовались немногие. Мо¬
гут успешно использоваться экзогенные доминантные
маркеры типа резистентности к G418. Следует под¬
черкнуть, что какой бы метод ни использовался, чтобы
изолировать клоны предполагаемых гибридов, должно
быть получено подтверждение гибридной природы
клеток. Для этого используется стандартно проведен¬
ный цитогенетический анализ (см. протокол 16.7) или
молекулярные методы. В некоторых случаях сравнение
числа гибридов с частотой ревертантов может быть
единственным путем этого подтверждения.
27.9.1. Ядерный перенос
Генетические рекомбинантные эксперименты могут
также быть выполнены с изолированными ядрами, но
Приготовьте
антиген
ЧУ
v •
• * *#v
.**,
Проведите слияние
с ТКклетками
мышиной миеломы
Иммунизируйте
мышь
Выделите
селезенку
Диспергируйте
клетки и
посейте на
чашку
Клонируйте в среде HAT
Исследуйте клоны, ГЛ
распространите положительные / \
клоны, испытайте на продукцию/ \
антител и специфичность; / \
заморозьте клонированные / \
линии
Рис. 27.4. Производство гибридом. Схематическая диа¬
грамма получения клонов гибридом, способных к секреции
моноклональных антител
главный интерес в этой технике связан с клонировани¬
ем индивидуальных животных [Копо, 1997; Wolf et al.,
1998]. Фрагменты клеток, содержащие ядро и неболь¬
шой объем цитоплазмы, могут быть получены центри¬
фугированием клеток после обработки цитохалазином
В. Затем производится слияние извлеченных ядер с
реципиентными целыми клетками или безядерными
цитопластами в присутствии ПЭГ (рис. 27.4). Однако в
экспериментах по клонированию животных, чтобы уда¬
лить ядро, используются методы микроманипуляции,
затем ядро одной клетки вводят в оплодотворенную
яйцеклетку первой стадии имплантации, из которой
было удалено существующее ядро.
27.10. ПРОДУКЦИЯ МОНОКЛОНАЛЬНЫХ
АНТИТЕЛ
Моноклональные антитела стали обязательным инстру¬
ментом в исследованиях, диагностике и терапии. С тех
пор как КоЫег и Milstein создали в 1975 г. гибридомную
технологию, во многих различных приложениях метода
моноклональные антитела заменили поликлональные
антитела. Следующее введение и протокол 27.11 были
предоставлены Janice Payne and Tina Kuus-Reichel из
548
ГЛАВА 27. СПЕЦИАЛЬНЫЕ МЕТОДЫ
Hybritech Incorporated, дочерней компании Beckman
Coulter Inc., 7330 Carroll Road, San Diego, С A 92121.
Гибридомы получают слиянием нескретирующих
миеломных клеток с антитела-продуцирующими
В-лимфоцитами в присутствии ПЭГ (рис. 27.4).
Миеломная клетка дефицитна по HGPRT или ТК,
которые необходимы для синтеза ДНК, поэтому она
не может выжить на селективной среде, содержащей
гипоксантин, аминоптерин и тимидин. В-лимфоциты
селезенки, не слившиеся с миеломной клеткой, не мо¬
гут жить в культуре больше нескольких дней. Любая
гибридная клетка В-лимфоцит/миеломная клетка
должна содержать генетическую информацию из обеих
родительских клеток и, таким образом, быть способной
выжить на селективной среде HAT. Такие клетки могут
быть культивированы неопределенно долго и способны
продуцировать неограниченное количество антител.
Супернатант выживших гибридом исследуется на на¬
личие антител методом ELISA. Те гибридомы, которые
выбраны, затем субклонируются, чтобы удостоверить¬
ся, что они продуцируют антитела, специфичные для
отдельного эпитопа. Продукция антител может быть
пропорционально повышена in vivo в виде асцитной
опухоли мышей или in vitro в качестве суспензионной
культуры. Гибридомы также хорошо растут в полых
волокнах и культуральных системах ферментации.
ПРОТОКОЛ 27.11. Продукция
моноклональных антител
Схема
Используя полиэтиленгликоль (PEG), проводят сли¬
яние клеток селезенки иммунизированных мышей
с клетками миеломы (Р3.653). Выделяют колонии
гибридов (гибридомы) в среде HAT. Исследуют су-
пернатанты методом ELISA через 10-14 дней после
слияния (ELISA скрининг, Протокол должен быть
разработан до слияния клеток), наращивают, замо¬
раживают и субклонируют желаемые гибридомы для
достижения моноклональности.
Материалы
Стерильные или асептически приготовленные:
• Мыши (Balb/c или A/J), 6-10 недель
• Миелома Р3.653: эта линия миеломных клеток
АТСС не секретирует иммуноглобулин и хорошо
участвует в слиянии.
• Антиген (оптимальное количество 125 мкг на
мышь, маленькие молекулы могут быть для по¬
вышения антигенных свойств конъюгированы с
гемоцианином (keyhole limpet hemocyanin, KLH,
Sigma).
• Адъюванты (квасцы или адъювант Фрейнда; пол¬
ный или неполный, Sigma): подробное описание
этих и других адъювантов можно найти в Vogel &
Powell [1995].
• Буфер TCD (T-cell depletion): сбалансированный
солевой раствор Хэнкса + 10 мМ HEPES + 0,3%
BSA
• NH4C1,0,16 М
• Антитела Antimouse Thy 1.2 (Accurate). Для
использования разведите в буфере TCD в отно¬
шении 1:500, стерилизуйте фильтрацией.
• Комплемент кролика, Low Тох М (Accurate).
Растворите в 1 мл холодной UPW, разведите в
буфере TCD 1:12, стерилизуйте фильтрацией.
• D-PBSA
• SFM (бессывороточная среда): МЕМ, которая
хранилась при комнатной температуре с неплотно
закрученной крышкой, так чтобы мог выходить
С02; pH должна быть щелочной.
• PEG (полиэтиленгликоль): растопить 10,5 мл
PEG 1450 (Sigma) в водяной бане 56 °С;
добавить 19,5 мл теплой стерильной МЕМ (pH
8,3-8,7); хорошо перемешать; оставить раствор до
достижения равновесного состояния с неплотно
закрученной крышкой на 5-7 до слияния.
• Маточный раствор HAT (100х)(10 мМ гипоксан¬
тина, 40 мкМ аминоптерина, 1,6 мМ тимидина):
1,36 г гипоксантина, 729 мкл аминоптерина, (из
концентрированного раствора, концентрация
25 мг/мл), 0,387 г тимидина, 0,022 г глицина,
разведенного в 4 мл 5 М NaOH + 26 мл UPW.
Доведите до 1 л добавлением UPW. Стерилиза¬
ция фильтрацией.
• Среда HAT: к основной среде, такой как МЕМ
или RPMI, + 10% FBS + 20% среды кондициони¬
рованной клетками селезенки, добавляют маточ¬
ный раствор HAT (конечной разведение 1:100)
• SCM (среда, кондиционированная клетками
селезенки): Препарируйте при помощи иглы
3-5 селезенок интактных (наивных) мышей в
D-PBSA. Подсчитайте клетки, ресуспендируйте
в МЕМ+ 10% лошадиной сыворотки до кон¬
центрации 1х106кл./мл. Перенесите суспензию
во флакон для перемешивания объемом 500-мл,
инкубируйте при 37 °С в 5% С02 в течение 48 ч.
Удалите клетки центрифугированием, храните
супернатант замороженным.
• 8-Азагуанин, маточный раствор, 10 мМ (100.):
1,52 г 8-азагуанина, разведенного в 4 мл 5 М
NaOH + 21 мл UPW. Доведите объем до 1 л до¬
бавлением UPW. Стерилизуйте фильтрацией.
• МЕМ, 10% фетальной сыворотки теленка, 0,1 мМ
8-азагуанина (для поддержания клеток миеломы
Р3.653).
• НТ маточный раствор (ЮОх): 0,408 г гипоксанти¬
на, 0,1161 г тимидина, 0,0067 г глицина, развести
в 2 мл 5 М NaOH + 8 мл UPW. Довести объем
до 300 мл добавлением UPW. Стерилизуйте
фильтрацией.
• НТ среда: НТ маточный раствор развести ЮОх в
основной среде (как для среды HAT, см. выше)
• Шприцы, 1 мл, иглы 25G
27.10. Продукция моноклональных антител
549
• Шприцы, 1 мл, иглы 23G, х 2
• Инструменты для препарирования (ножницы и
пинцеты)
• Чашки Петри, 60x15 мм
• Центрифужные пробирки, 15 мл и 50 мл
• Многолуночные планшеты, 24 и 96 лунки
• Культуральные флаконы
• 100-мл бутыли Nalgene
• Наконечники для пипеток
• Резервуары для автоматических многоканальных
пипеток (100мл, Matrix Technologies; Corning
Costar)
Нестерильные:
• Трипановый синий (Sigma)
• Автоматические 12-канальные пипетки (Matrix
Technologies)
• Инвертированный фазово-контрастный микро¬
скоп
• Система Unopette microcollection system (BD
Biosciences)
• Гемоцитометр
Протокол
Иммунизация:
1. Возьмите кровь мышей для проверки сыворотки
на базальную антигенную реактивность в день
0 перед иммунизацией.
2. Иммунизируйте мышей (А/J или Balb/c) анти¬
геном, в эмульсии с адъювантом Фрейнда или в
смеси с квасцами в отношении 1/10 (по объему),
хорошо перемешанным на вортекс-мешалке. Про¬
ведите три инъекции антигена внутрибрюшинно,
в соответствии со следующим режимом
День
Количество
Адъювант
антигена
0
50 мкг
Квасцы или полный адъ¬
ювант Фрейнда
14
25 мкг
Квасцы или неполный
адъювант Фрейнда
28
25 мкг
Квасцы или D-PBSA
3. Возьмите кровь у мышей на 35-й день, определить
титр антител в сыворотке методом ELISA.
4. После разведения сыворотки 1:30 проведите се¬
рийное разведение сыворотки 1:4 до получения
разведения 1:30 720.
5. Выберите мышей с самым высоким соотношени¬
ем титра сыворотки к антигену для проведения
слияния.
6. Произведите выбранным мышам конечную сти¬
муляцию введением 10 мкг антигена внутривенно
или 25 мкг антигена внутрибрюшинно в течение
3 дней до слияния.
Миелома:
1. Удобно проводить слияние клеток в четверг, так
чтобы мыши получили конечную антигенную
стимуляцию в понедельник.
2. Поддерживайте клетки миеломы Р3.653 в МЕМ
+ 10% фетальной сыворотки теленка + 8-азагуа-
нина.
3. Разводите клетки Р3.653 до 3,5х105 кл./мл каж¬
дый день. Последнее разведение проведите за три
дня до слияния.
Т-клеточное истощение:
1. Возьмите кровь у мышей с подходящим титром
сыворотки и забейте методом цервикальной дис¬
локации.
2. Асептически выделите селезенки, поместите их
в стерильные чашки Петри с 5 мл стерильного
D-PBSA.
3. При помощи двух 1-мл шприцов с надетыми
иглами 23G осторожно препарируйте селезенки.
Грубое разрушение селезенок приводит к высо¬
кой концентрации фибробластов.
4. Перенесите клетки в 15-мл коническую пробирку,
и оставьте для осаждения комков.
5. Перенесите клетки селезенки (без сгустков) в
50-мл коническую пробирку и после разведения
1:100 в Unopette подсчитайте клетки в гемоцито¬
метре.
6. Центрифугируйте клетки при 200 g в течение
8 мин.
7. Для лизиса красных кровяных клеток ресуспен¬
дируйте получившийся осадок в 0,84% NH44C1
(10 мл/на селезенку) и инкубируйте суспензию
при 4 °С в течение 15 мин.
8. Введите в качестве подлежащего слоя к суспензии
клеток селезенки 14 мл лошадиной сыворотки,
центрифугируйте при 450 g в течение 8 мин.
9. Ресуспендируйте получившийся осадок в 50 мл
буфера TCD, центрифугируйте суспензию при
200 g в течение 8 мин.
10. Для Т-клеточного истощения ресуспендируй¬
те получившийся клеточный осадок в раст¬
воре anti-Thy 1,2 при конечной концентрации
1х107 кл./мл.
11. Инкубируйте суспензию при 4 °С в течение
45 мин, и затем центрифугируйте при 200 g в
течение 8 мин.
12. Ресуспендируйте осадок в растворе комплемента
кролика.
13. Инкубируйте суспензию при 37 °С в течение
45 мин и затем центрифугируйте при 200 g в
течение 8 мин.
14. Подсчитайте клетки методом исключения
трипанового синего на гемоцитометре (см. про¬
токол 22.1). Восстановление В-клеток должно
составлять 30-50%.
Слияние:
1. Смешайте клетки миеломы и В-клетки в 50-мл
центрифужной пробирке. Одна процедура сли¬
яния может быть произведена не более чем на
1,2x108 клетках селезенки. Смешайте их с миелом-
550
ГЛАВА 27. СПЕЦИАЛЬНЫЕ МЕТОДЫ
ными клетками Р3.653 в отношении 4:1; таким
образом, максимальное количество клеток Р3.653
на одну процедуру составит 3x107 клеток.
2. Центрифугируйте суспензию при 200 g в течение
8 мин.
3. Взмутите полученный осадок путем потряхива¬
ния и добавляйте 1 мл PEG в пробирку в тече¬
ние^ с.
4. Перемешайте суспензию осторожным прокручи¬
ванием пробирки в течение 75 с.
5. Добавляйте1 мл SFM в течение 15 с, и осторожно
перемешивайте пробирку в течение 45 с.
6. Добавляйте 2 мл SFM в течение 30 с, перемеши¬
вайте содержимое пробирки 90 с.
7. Добавляйте 4 мл среды HAT в течение 30 с, пере¬
мешивайте содержимое пробирки 90 с.
8. Наконец, добавьте 8 мл HAT среды в течение
30 с, перемешивайте содержимое пробирки
90 с.
9. Добавить этот объем (16 мл) в стерильные
бутылки Nalgene, содержащие подсчитанное
количество среды HAT (125 мл, если использу¬
ется максимальное количество клеток). 16 мл
суспензии, содержащей 1,5x108 клеток, полу¬
ченной на шаге 1 данного раздела настоящего
протокола с добавлением 125 мл среды HAT в
бутыли составит 141 мл. Вместе с промывкой на
следующем шаге (шаг 10) общий объем составит
150 мл и даст конечную концентрацию клеток
IxlO6 кл./мл.
10. Ополосните 50-мл коническую пробирку 9 мл
среды HAT, и добавьте этот объем в бутыль.
11. Хорошо перемешайте содержимое бутыли и пе¬
ренесите клетки в стерильный резервуар.
12. Испога>зуя 12-канальную автоматическую пипетку,
посейте клетки по 200 мкл/лунку в стерильный
96-луночный планшет. Конечная концентрация
составит 2x10s кл./лунку.
Селекция гибридом:
1. Замените питательную среду через 5 дней после
слияния, путем аспирации большей части среды
из лунок и добавлением 150-200 мкл свежей
среды HAT на лунку.
2. Производите питание культур два раза в неделю.
3. Через две недели после слияния проведите ск¬
рининг клонов для селекции положительных
гибридом методом ELISA.
4. Через каждые 48 ч повторно исследуйте гибридо¬
мы, которые на предыдущем этапе были выбраны
как положительные.
5. Нарастите наиболее продуктивные гибридомы
в двух лунках 96-луночного планшета в среде,
содержащей 10% FBS и НТ.
6. Повторите исследование клонов, распространив
положительные гибридомы в 24-луночный план¬
шет, и пересейте их в среду, не содержащую НТ,
при этом 2 мл супернатанта культуры следует
взять на исследование. На этом этапе берут объем,
достаточный для того, чтобы провести несколько
селективных испытаний, чтобы убедиться, что ан¬
титела направлены только против интересующего
антигена.
7. Нарастите гибридомы так, чтобы их хранить в
4 лунках 24-луночного планшета, и сохраните их
замораживанием (см. протокол 20.1).
8. Проведите вторичное криоконсервирование пос¬
ле выращивания гибридомы в 75-см2 флаконе
Скрининг. Позаботьтесь о развитии стратегии ск¬
рининга, чтобы получить моноклональные антитела
с желательными характеристиками. Супернатант гиб-
ридомной культуры должен быть исследован так рано,
как только это становится выполнимым, на предмет
иммунореактивности желательных антител. После
первоначальной селекции методом ELISA по реактив¬
ности к иммуногену супернатант распространенной
культуры должен быть проверен в том исследовании,
для которого антитело разрабатывается (Вестерн-блот-
тинг, конкурентное иммуноисследование, проточная
цитометрия, и т.д.). Более детальное обсуждение ELISA
и других иммунологических методов исследования
можно найти в Knott et al [1997].
Субклонирование. Чтобы гарантировать монокло-
нальность, произведите субклонирование гибридом,
которые вас интересуют. Это может быть выполнено
последовательно разведением клеток и посевом эк¬
вивалента 1 клетки в 3 лунки 96-луночного планшета
(см. разд. 14.1) или, сортингом при помощи автома¬
тизированного блока осаждения клеток (ACDU) на
FACStarplus (BD Biosciences) и посевом по одной
клетке в каждую лунку. Субклонирование может быть
выполнено на фидерном слое мышиных спленоцитов.
Фидерный слой готовят, препарируя при помощи
иглы селезенку наивной мыши, затем высевая клетки в
концентрации IxlO6 кл./мл. Затем клетки пересевают в
96-луночный планшет для микротитрации при конечной
концентрации 2x105 кл./лунку. После субклонирования
обычно колонии можно заметить на 5-й день, затем они
должны быть проверены визуально на моноклональ-
ность. Планшеты заполняют свежей средой, начиная
с 7-го дня. Скрининг на положительность гибридом
обычно делается между 10-м и 14-м днями. Выбранные
клоны затем размножаются и замораживаются таким
же образом, как родительская гибридома.
Производство антител. Концентрированные ан¬
титела из интересующих исследователя клонов могут
быть произведены in vivo из асцита в IF А — обрабо¬
танных мышах (Balb/c или nu/nu [Gillette, 1987]) или
in vitro при выращивании суспензионной культуры.
Существуют также половолоконные системы для куль¬
туры клеток (см. Приложение II и разд. 25.3.2, 26.1.3,
26.2.5). Когда гибридомы инокулируют в половолокон¬
ную систему, клетки поддерживаются в определенном
27.11, Перенос ДНК
551
компартменте биореактора, в то время как свежая среда
и продукт жизнедеятельности клеток заменяются. Вы¬
сокие концентрации антител, произведенных клетками,
могут быть выделены из супернатанта в многократно
в разное время.
27.11. ПЕРЕНОС ДНК
Чтобы изучать функцию индивидуальных генов и регу¬
ляторные последовательности, фрагменты ДНК могут
быть субклонированы и затем перемещены в клетки
хозяина разнообразными методами, типа трансфекции,
липофекции и ретровирусной инфекции (рис. 27.5).
Клонированную ДНК часто удобно поддерживать как
часть бактериальной плазмиды, многие плазмиды могут
достигать большого числа копий в течение бактериаль¬
ного роста, таким образом гарантируя обильный запас
ДНК для эксперимента. Плазмидная ДНК, очищается
от бактерий перед использованием. Как только инте¬
ресующая последовательность проходит субклониро¬
вание, она может использоваться как дополнительный
инструмент, например для субклонирования промотор-
ной последовательности. Путем генетических мани¬
пуляций, последовательности промотора могут быть
связаны с репортерным геном (например, P-gal или
CAT), продукты которых (соответственно, Р-галакто-
зидаза или хлорамфеникол ацетилтрансфераза) могут
быть легко обнаружены после трансфекции. Зеленый
флюоресцентный белок (GFP) тихоокеанской медузы
Aequoria victoria, который может использоваться как
репортер, будет флюоресцировать в живых клетках,
освещенных ультрафиолетовым светом, что позволяет
обнаруживать жизнеспособные трансфецированные
клетки на инвертированном микроскопе, что может
использоваться для FACS-сортинга.
Трансфекция может быть временной (транзитор-
ной) или устойчивой. Транзиторная трансфекция
о
‘Ч
г у ^
" J >
О
Экстрагировать Порезать
ДНК рестрикционной
эндонуклеазой
Или экстрагировать РНК и приготовить кДНК 1
" Инкорпорировать
в плазмиду с
выбираемым
маркером
Вырастить
трансфецированные клетки
в селективной среде,
исследовать экспрессию
Трансфецировать в
клетку-реципиент с
фосфатом кальция,
липофекцией или
электропорацией
Клонировать
в бактериях в
селективных
условиях
Рис. 27.5. Перенос ДНК. Изолированные фрагменты
эндонуклеазы ДНК амплифицируют методом клонирова¬
ния генов, добавляют к целым клеткам, и инкорпорируют
липофекцией, электропорацией или ко-преципитацией с
Са3(Р04)2
существует короткое время, и продукт используется
вскоре после трансфекции, эффективность которой
определяется исследованием репортерного гена. ДНК,
используемая для устойчивой трансфекции, содержит
селективный маркер типа пео или hyg В, которым
придают резистентность к G418 (генетицин, аналог
неомицина) или гигромицину, соответственно. Транс¬
фецированные клетки затем подвергаются селекции
длительной экспозицией селектирующего агента,
затем могут быть выделены резистентные клоны (см.
протоколы 14.6 и 14.8).
Ниже представлены два протокола: один для устой¬
чивой трансфекции с использованием электропорации
и один для транзиторного процесса (использование
липофекции). Протокол селекции для устойчивой
трансфекции может быть добавлен к протоколу липо¬
фекции, при условии что конструкт, используемый для
трансфекции, содержит соответствующий селективный
маркер. Перенос ДНК методом копреципитации с
фосфатом кальция и вирусная трансдукция описаны
в гл. 18 (см. протоколы 18.1,18.2).
27.11.1. Копреципитация с фосфатом
кальция
Метод фосфата кальция для введения генов в клетки
млекопитающих был впервые описан Graham and Van
der Eb [1973] и все еще широко используется. В этом
методе экзогенная ДНК смешивается с хлористым
кальцием и затем добавляется в раствор, содержащий
ионы фосфата. Образующийся фосфатно-кальциевый
преципитат ДНК захватывается клетками млекопи¬
тающих в культуре с последующей экспрессией экзо¬
генного гена. Этот метод может использоваться, чтобы
внедрить любую ДНК в клетки млекопитающих для
исследований транзиторной экспрессии или долго¬
срочной трансформации. Эта процедура описана для
иммортализации фибробластов с антигеном SV40 Т
(см. протокол 18.1).
27.11.2. Липофекция
Первоначальный метод опосредованной катионны¬
ми липидами трансфекции ДНК в культивируемые
клетки [Feigner et al., 1987] был улучшен в 1993 г.
заменой монокатионного липидного реагента на поли-
катионный агент Липофектамин [Ciccarone et al., 1993;
Hawley-Nelson et al., 1993]. Метод основан на ионных
взаимодействиях ДНК и липосом с формированием
комплекса, который может доставлять функциональ¬
ную ДНК в культивируемые клетки. Плазмидная
ДНК находится в комплексе, но не инкапсулирована
внутри монослойных липосом размером 600-1200 нм
в диаметре [Mahato et al., 1995а], сформированных
катионными липидами в воде. Преимущества опосре¬
дованной катионными липидами трансфекции перед
другими методами заключаются в более высокой
552
ГЛАВА 27. СПЕЦИАЛЬНЫЕ МЕТОДЫ
эффективности; возможности успешной трансфекции
для широкого круга (более 300 сообщений) эукарио¬
тических клеточных линий, многие из которых устой¬
чивы к другим методам трансфекции; и относительно
низкая токсичность для клеток. Другое преимущество
состоит в том, что основная процедура трансфекции
ДНК может быть приспособлена к трансфекции РНК,
синтетических олигонуклеотидов, белков и вирусов.
Наконец, катионные липосомы могут использоваться
для успешной доставки функциональных генов или
вирусного генома in vivo [Mahato et al., 1995b; Tagawa
et al., 1996; Thorsell et al., 1996]. Неудобством метода
является относительно высокая стоимость реактивов,
которая фактически устраняет крупномасштабное
использование.
Протокол 27.12 представляет собой сокращенный
метод Bichko [1998].
ПРОТОКОЛ 27.12. Транзиторная
трансфекция методом
липофекции
Материалы
Стерильные или асептически приготовленные:
• Катионные липиды
• Липофектамин: состав липосом 3:1 (по объему)
поликатионный липид DOSPA (2,3-диолеилок-
си-К[2(спермин карбоксамид)этил]-М,М-диме-
тил-1 -пропанамин трифторацетат и нейтральный
липид DOPE (Диолеилфосфатидилэтаноламин)
в воде (Invitrogen)
• Липофектин: состав липосом 1:1 (по объему)
катионный липид DOTMA (>1-[1-(2,Здиоле-
илокси)-пропил]-К,М,К-триметиламмония
хлорид) и нейтральный липид DOPE в воде
(Invitrogen)
• Липофект LipofectАСЕ: состав липосом 1:2,5 (по
объему) катионный липид DDAB диметилдиок-
тадециламмония бромид и нейтральный липид
DOPE в воде (Invitrogen)
• Клетки для трансфекции
• ДНК для трансфекции
• Ростовая среда: DMEM (1х) с 10% FBS, пеницил-
лин(100 U/мл), и стрептомицин (100 мкг/мл или
как это требуется для используемых клеток)
• Среда со сниженным содержанием сыворотки:
OPTI-MEM 1 (содержит 2% FBS; Invitrogen)
• Бессывороточная среда: DMEM без сыворотки
и антибиотиков
• D-PBSA
• Солевой буфер:
NaCl, 0,15 М
К2НР04,0,006 М
КН2Р04 0,002 М
• Трипсин, 0,25%, в D-PBSA
• Многолуночные планшеты, 6 лунок, или 3,5-см
чашки Петри
Протокол
A. Трансфекция адгерентных клеток
1. Посейте примерно 1х105-3х105 клеток на лунку
в 6-луночные планшеты в 3 мл ростовой среды.
2. Инкубируйте клетки при 37 °С в С02-инкубаторе
пока клетки не достигнут 50-90% конфлюэнт¬
ности. Этот этап обычно занимает 18-24 ч, или,
по крайней мере, не менее 16 ч.
3. До трансфекции приготовьте растворы ДНК и
липидов в стерильных пробирках:
а) Для получения раствора ДНК разведите
1-2 мкг ДНК в 0,5 мл среды с пониженным
содержанием сыворотки.
б) Для получения раствора липидов разведите
2-25 мкл катионно-липидного реагента в
0,5 мл бессывороточной среды.
в) Соедините два раствора, аккуратно размешай¬
те, инкубируйте при комнатной температуре в
течение 15-45 мин, для обеспечения форми¬
рования ДНК/липидных комплексов.
4. Отмойте клетки один раз в 2 мл бессывороточной
среды.
5. Нанесите ДНК/липидные комплексы на клетки.
В ходе трансфекции следует исключить антибак¬
териальные компоненты.
6. Инкубируйте клетки с комплексами при 37 °С в
С02-инкубаторе в течение 2-24 ч; обычно бывает
достаточно 5-6 ч.
7. После инкубации удалите трансфекционную
смесь с клеток, замените ее на 3 мл полной рос¬
товой среды.
8. Замените ростовую среду на свежую среду через
18-24 ч после начала трансфекции.
9. Исследуйте ростовую среду от трансфецирован-
ных клеток на транзиторную активность гена по
обстоятельствам (см. протоколы 27.14 и 27.15).
B. Трансфекция в суспензии клеток
1. Приготовьте трансфекционную смесь в стериль¬
ных пробирках следующим образом:
а) Разведите 2-5 мкг ДНК в 0,5 мл среды с по¬
ниженным содержанием сыворотки.
б) Разведите 2-20 мкл реагента для катионно¬
липидного реагента в 0,5 мл бессывороточной
среды.
в) Соедините два раствора, осторожно переме¬
шайте получившийся раствор, инкубируйте
его при комнатной температуре в течение
15-45 мин, чтобы обеспечить формирование
ДНК/липидных комплексов.
2. Центрифугируйте суспензию клеток содержащую
примерно 1х106-2х106 клеток, и аспирационно
отберите супернатант.
3. Ресуспендируйте клетки в трансфекционной
смеси, перенесите суспензию в 3,5-см чашку.
4. Инкубируйте клетки в С02-инкубаторе в течение
4-6 ч.
27.11. Перенос ДНК
553
5. В каждую чашку добавьте 0,5 мл ростовой среды,
содержащей 30% сыворотки (большое количество
сыворотки необходимо для того, чтобы защитить
клетки в суспензии, которые более чувствитель¬
ны к токсическому действию составляющих
частей липосом).
6. Инкубируйте чашки в С02-инкубаторе в течение
ночи.
7. Добавьте 2 мл полной ростовой среды в каждую
чашку и инкубируйте чашки в С02-инкубаторе
8. Через 24-72 ч после начала трансфекции, иссле¬
дуйте клетки или среду на предмет активности
гена (см. протоколы 27.14 и 27.15).
27.11.3. Электропорация
ДНК может быть введен в клетки методом электропо¬
рации, когда клетки в высокой концентрации кратков¬
ременно подвергаются действию высокого напряжения
электрического поля в присутствии ДНК, приготовлен¬
ной для трансфекции [Chu et al., 1987]. В мембране кле¬
ток кратковременно образуются маленькие отверстия
[Zimmerman и Vienken, 1982], которые позволяют ДНК
войти в клетки; в некоторых клетках ДНК включается в
геном. Оборудование для электропорации коммерчески
доступно (Bio-Rad). Большинство клеток, невоспри¬
имчивых к химическим методам переноса гена, были
успешно трансфицированы методом электропорации
[Andreason & Evans, 1988; Chu et al., 1987].
Электропорация обычно производится в поле пос¬
тоянной емкости (так обеспечивается постоянная про¬
должительность импульса) с различным напряжением
поля (500-1500 кВ/см) для пилотных исследований. Для
большинства клеток значение параметров, при которых
приблизительно 20-50% клеток остается жизнеспособ¬
ным, после электропорации считается достаточным для
переноса ДНК [Chu et al., 1987; Andreason & Evans, 1988].
Электропорация обычно производится при комнатной
температуре, впоследствии клетки хранят на льду, что¬
бы продлить период времени, когда мембранные поры
остаются открытыми [Andreason & Evans, 1989].
Существуют линейные отношения между концент¬
рацией ДНК, захватом ДНК, и экспрессией репортер-
ного гена [Chu et а1.,1987]. Предполагается, что линеа¬
ризированная ДНК более эффективна для получения
устойчивых трансфектантов, чем суперспиральная
конформация ДНК, возможно, из-за повышенной эф¬
фективности, с которой линейная ДНК встраивается в
геномную ДНК [Potter et al., 1984; Chu et al 1987]. Элек¬
тропорация приводит к интеграции ДНК в низком чис¬
ле копий [Boggs et al., 1986; Toneguzzo et al., 1988], хотя
число введенных копий может быть отрегулировано пу¬
тем изменения концентрации ДНК в суспензии клеток.
Химические методы трансфекции обычно приводят к
интеграции больших конкатемеров, которые могут по
своей природе нарушать функционирование клетки и
приводить к неоднозначным результатам исследования,
включая сверхэкспрессию специфичного гена [Robins
et al., 1981; Kucherlapati & Skoultchi, 1984].
Суспензия клеток легче трансфецируются методом
электропорации, чем прикрепленные клетки, потому
что прикрепленные клетки должны быть отделены
от стенок культурального сосуда. Недостатки метода
электропорации связаны с требованием большего ко¬
личества клеток и ДНК, чем необходимо для переноса
гена химическими методами. Кроме того, при элект¬
ропорации отмечается вариабельность в оптимальных
параметрах между типами клеток. Настоящее введение
и протокол 27.13 являются сокращенным вариантом
метода Cataldo et al [1998]. Протокол описывает ус¬
ловия электропорации, необходимые для устойчивой
трансфекции клона (Y10) мышиных мегакариотиче-
ских клеток линии L8057 [Ishida et al., 1993]. Клетки
L8057-Y10 растут в суспензии и поддерживаются в
среде Хэма F12 с добавлением 10% термоинактивиро¬
ванной FBS, пенициллина (50 U/мл) и стрептомицина
(50 мкг/мл). Здесь представлена общая схема устойчи¬
вой трансфекции клеток L8057-Y10.
ПРОТОКОЛ 27.13. Устойчивая трансфекция
методом электропорации
Материалы
Стерильные или асептически приготовленные:
Экспрессирующие векторы
и приготовление ДНК:
• Линеаризованный вектор pSVTKGH [Selden et
al., 1986]. Этот вектор содержит ген-усилитель
SV40 и тимидинкиназную промоторную после¬
довательность герпес-вируса, которая запускает
экспрессиючеловеческого гормона роста (hGH).
hGH секретируетея непосредственно в куль-
. туральную среду, и обнаруживается методом
радиоиммуноанализа [Ravid et al., 1991].
• Линеаризованный вектор pcDNA3 (Invitrogen).
Этот экспрессирующий вектор содержит энхан-
серно-промоторную последовательность чело¬
веческого цитомегаловируса, расположенный
выше его мультиклонирующего сайта, атаюке
сигнальную последовательность полиаденили-
рования и транскрипционно-терминационную
последовательность гена гормона роста быка
(ЬСН), расположенного ниже мультиклони-
рующео сайта. Вектор рсОКАЗтакже содержит
ген устойчивости к неомицину, что снижает
необходимость одновременной трансфекции
гена антибиотик-резистентности для селекции и
стабильных трансформантов активно экспресси¬
рующих отдельный интересующий исследователя
ген. pcDNA3 также может быть использован как
селективный маркер при одновременной транс¬
фекции с репортерными конструктами, как это
представлено в данном протоколе.
554
ГЛАВА 27. СПЕЦИАЛЬНЫЕ МЕТОДЫ
Культура ткани:
• Клетки L8057-Y10
• Флаконы, 75 см2
• Планшеты шестилуночные
• D-PBSA
• Среда Хэма F12/FB: F12 с глютамином (2 мМ),
пенициллином (50 U/мл), и стрептомицином
(5 мкг/мл), с добавлением 10% FBS (термоина¬
ктивированная); GIBCO #16140-014)
• Geneticin (Invitrogen), 50 мг/мл в D-PBSA (в
зависимости от активности, например если ак¬
тивность партии 731 мкг/мг, разведите 64,4 мг
G418 на mkD-PBSA)
• Селективная среда: F12/FB, содержащая
600 мкг/мл G418
Электропорация:
• Буфер для электропорации [Ravid et al., 1991]:
NaCl, 30,8 мМ
KC1,120,7 мМ
Na2HP04,8,1 мМ
KH2P04,1,46 мМ
MgCl2,5 мМ
• Кюветы Gene Pulser Cuvettes
Нестерильные:
• Прибор Gene Pulser II (Bio-Rad)
• Ретранслятор Емкостного сопротивления Gene
Pulser II Capacitance Extender Plus
Протокол
1. Посейте клетки L8057-Y10 в концентрации
5x10s кл./мл в 75-см2 культуральном флаконе, ин¬
кубируйте флакон при 37 °С в 5% С02в F12/FB.
2. В ходе поздней log-фазы соберите клетки цен¬
трифугированием при 4 °С при 380 g в течение
5 мин.
3. Отмойте клеточный осадок в 10 мл DPBSA,
центрифугируйте при 4 °С при 380 g в течение
5 мин.
4. Отмойте клеточный осадок в 5 мл буфера для
электропорации и подсчитайте клетки, используя
гемоцитометр.
5. Соберите клетки центрифугированием при 4 °С
при 380 g в течение 5 мин.
6. Ресуспендируйте клетки при 1х106кл./0,8 мл
буфера для электропорации.
7. Перенесите 0,8 мл клеток в предварительно ох¬
лажденные кюветы для электропорации.
8. Добавьте 50 мкг линеаризованного pSVTKGH,
вместе с 5 мкг линеаризованного pcDNA3 к
клеточной суспензии (при анализе специфи¬
ческой экспрессии промоторно-репортерных
генов, подходящим отрицательным контролем
является электропорация клеток в отсутствие
плазмидной ДНК).
9. Смещайте ДНК/клеточную суспензию, удержи¬
вая кювету за грани и постукивая по дну. Инку¬
бируйте суспензию на льду в течение 10 мин.
10. Электропорацию суспензии проводят при
400 V/500 mkF, записывая продолжительность
импульса.
11. Удалите кювету из импульсной камеры и помес¬
тите на лед на 10 мин.
12. Перенесите клетки после электропорации в
10 мл F12/FB, промойте кювету средой для
удаления всех клеток и соберите клетки цент¬
рифугированием.
13. Ресуспендируйте клетки в 20 мл F12/FB, вы¬
ращивайте суспензионную культуру в 75-см2
флаконе при 37 °С в 5% С02| для того чтобы
обеспечить экспрессию выбираемого маркерного
неомицинового гена.
14. Через 24-48 ч соберите клетки центрифугирова¬
нием, затем ресуспендируйте в 20 мл селективной
среды.
15. Меняйте селективную среду каждые 2-4 дня в
течение, по крайней мере, 2 недель для того чтобы
удалить обломки погибших клеток и позволить
расти устойчивым клеткам.
16. Проведите исследование экспрессии hGH в су-
пернатанте (см. разд. 27.11.5).
Вариации. Генетицин. Культивируемые клеточные
линии различны по чувствительности к генетицину,
наиболее подходящая концентрация для использо¬
вания должна быть эмпирически определена путем
получения кривой гибели (см. протокол 22.3) на той
клеточной линии, которая трансфецируется. Клетки
должны оставаться в селективной среде в течение
длительного периода роста. Выбор колоний. См. про¬
токолы 14.6 и 14.8.
Транзиторная трансфекция. Используйте кольце¬
вые плазмиды, такие как pCMVP-gal, для того чтобы
заместить pcDNA3. После выполнения шага 10 ресус¬
пендируйте клетки в 2 мл культуральной среды без
селективного агента и культивируйте их в шестилуноч¬
ном планшете в течение трех-четырех дней. Исследуйте
транзиторную экспрессию hGH в супернатанте, приго¬
товьте клеточный лизат для определения активности
Р-галактозидазы.
Экспрессия репортерного гена. В случае транзитор-
ной трансфекции плазмида, содержащая репортерный
ген Р-галактозидазы ко-трансфецируется для того что¬
бы нормализовать экспрессию гена hGH для суммарной
эффективности электропорации. Существуют коммер¬
чески доступные наборы для определения как секреции
hGH (Nichols Institute Diagnostics, #40-2205), так и
активности р-галактозидазы (Promega, #Е2000).
27.11.4. Другие методы переноса ДНК
Ретровирусная инфекция. Ретровирусы обладают вы¬
сокой эффективностью переноса генов и могут инкор¬
порировать большие фрагменты ДНК, чем плазмиды,
спонтанно инфицируя клетки хозяина [Ausubel et al.,
27.11. Перенос ДНК
555
1996; Hicks et al., 1998]. Введенные гены становятся
постоянными. Процесс включения в геном достигается
в ходе нормального клеточного деления, он не пов¬
реждает клетки хозяина и не вызывает генетических
изменений (см. протокол 18.2).
Бакуловирус. Введение геномной последователь¬
ности в бакуловирусы, которые затем размножаются
в клетках насекомых (таких как клетки линии Sf9),
также позволяют клонировать большие участки
(>100 тысяч пар нуклеотидов). Образующиеся белки
проходят посттрансляционную модификацию, кото¬
рая не характерна для прокариотических систем, хотя
существуют отличия от процессинга в клетках мле¬
копитающих. Бакуловирусы не передаются в клетки
млекопитающих, поэтому клетки линии Sf9 не несут
опасности контаминации вирусами млекопитающих;
при этом предполагается, что все добавки к средам, по¬
лученные от млекопитающих (например, FBS), прошли
тщательную проверку перед использованием. Клетки
Sf9 могут быть субкультивированы без трипсина [см.
протокол 26.9; Midgley et al., 1998].
Искусственные хромосомы дрожжей. Искусст¬
венные хромосомы дрожжей (YACs) [Strauss, 1998]
также представляют собой геном, который может
содержать более крупную последовательность ДНК,
чем бактериальные плазмиды, с нисходящим пост-
трансляционным процессингом, таким, который об¬
наруживается в эукариотических клетках, хотя этот
последний аспект, вероятно, имеет значительные
отличия от такового в клетках млекопитающих. Рас¬
пространение в дрожжах также дает высокий выход и
стабильную культуральную систему, ее не так сложно
поддерживать, чем крупномасштабные культуры кле¬
ток млекопитающих или насекомых.
Искусственные хромосомы млекопитающих.
Имеются значительные успехи в применении принци¬
пов YACs к системам клеток млекопитающих [Bridger,
2004], для того чтобы инкорпорировать крупные
нуклеотидные последовательности млекопитающих,
содержащие одну или более структурных и множест¬
венные регуляторные гены в одном конструкте. Такие
конструкты известны как искусственные хромосомы
млекопитающих (MACs) и вводятся в клетки млеко¬
питающих при помощи монохромосомальных методов
переноса [Newbold &Cuthbert, 1998]. Эта технология в
настоящее время привела к созданию искусственных
хромосом человека, открывая новую эру в генной те¬
рапии [Larin & Mejia, 2002; Katoh et al., 2004].
27.11.5. Репортерные гены
Исследования трансфецированных клеток в отношении
репортерных генов, таких как Р-gal или CAT, подтверж¬
дают инкорпорирование конструктов в ДНК, которые
экспрессируются трансфецированной популяцией.
P-gal окрашивание также может быть использовано для
прослеживания судьбы трансфецированных клеток в ис¬
следовании межклеточных взаимодействий [Bradley &
Pitts, 1994]. Протокол 27.14 представляет собой упро¬
щенный метод Bichko [1998], разработанный на основе
Sanes et al. [1986] и модифицированный Invitrogen.
ПРОТОКОЛ 27.14. Окраска In situ
на р-галактозидазу
Материалы
Нестерильные:
• D-PBSA
• Субстрат: X-gaL (Invitrogen), 20 мг/мл в диметил-
формамиде. Хранить при -20 ^С в темноте, в по¬
липропиленовой пробирке не более 6 месяцев.
• Фиксирующий раствор: формальдегид, 1,8%;
глютаральдегид, 0,05% в PBSA. Приготовить
фиксирующий раствор смешиванием 85 мл воды,
10 мл 10х D-PBSA^ 5млформалита<37% раствор
формальдегида) и 0,2 мл глютаральдепзда (25%
раствор). Хранить при 4 °С.
• Красящий раствор: 5 мМ железосинеродисто¬
го калия, 2 мМ MgCl2 в D-PBSA. Хранить при
4 °С.
• Субстрат /красящий раствор 1 мг/мл X-gal в
красящем растворе, приготовить непосредственно
перед использованием.
• Формалин, 10% в D-PBSA
• В данном случае экспрессирующая плазмида для
P-gal будет использоваться как репортерный ген
для трансфекции (например, pCMVpgal [Mac¬
Gregor & Caskey,1989]).
Протокол:
1. Отмойте клетки (например, в 6-луночном план¬
шете) один раз в 2 мл D-PBSA.
2. Фиксируйте клеткав в 1 мл фиксирующего рас¬
твора 5 мин при комнатной температуре.
3. Отмойте клетки дважды в 2 мл D-PBSA.
4. Добавьте в каждую лунку по 1 мл смеси субстрат/
красящий раствор, инкубируйте 2ч и более (в
течение ночи) при 37 °С.
5. Ополосните клетки в каждой лунке добавлением
2 мл D-PBSA. Исследуйте клетки в инвертиро¬
ванном микроскопе, подсчитайте голубые (p-gal
положительные) клетки.
6. Для хранения планшетов фиксируйте клетки в
каждой лунке добавлением 1 мл 10% формалина
в солевом буфере в течение 10 мин при комнатной
температуре, отмыть клетки в буферном растворе,
хранить в буферном растворе при 4 °С.
В качестве исследования репортерного гена при
трансфекции может использоваться экспрессия CAT
(например, pCMVCAT [Boshart et al., 1985]). Прото¬
556
ГЛАВА 27. СПЕЦИАЛЬНЫЕ МЕТОДЫ
кол 27.15 представляет собой упрощенный вариант
метода Bichko [1998] на основе Neumann et al. [1987]
модифицированный Invitrogen.
ПРОТОКОЛ 27.15. Определение активности
хлорамфениколацетилтрансферазы
(CAT ASSAY)
Материалы
Стерильные:
• D-PBSA
• Многолуночные планшеты: 6-луночные, 3,5 см
Нестерильные:
• Трис-буфер: Трис НС1,0,1 М, pH 8,0
• Трис/Тритон: Трис-НС1,0,1 М, pH 8,0,0,1% Три¬
тон Х-100; хранить при 4 °С
• Буфер для разведения CAT: ТрисНС1,0,1 М, pH
8,0,50% глицерин, 0,2% BSA
• Субстрат: 250 мМ хлорамфеникола (GIBCO) в
100% этаноле; хранить в аликвотах при -70 °С.
• [14С]-СоА: [14С] бутирил Коэнзим А (10 мКи/мл;
Amersham Biosciences)
• CAT -фермент стандарты (Invitrogen): пригото¬
вить стандартные растворы 0,2,1,2,4, и10 U/мл в
буфере для разведения CAT. Хранить при 4 °С.
• Жидкий сцинтилляционный коктейль: Есопо-
fluor (Invitrogen)
• Деионизированная дистиллированная вода
(DDW)
• Полипропиленовые сцинтилляционные флако¬
ны, 3,5 мл
• Микроцентрифужные пробирки
• Микроцентрифуга
• Ледяная баня
Протокол
Сбор клеток с 6-луночных 35-мм планшетов:
1. Отмойте клетки один раз в D-PBSA 24-72 ч после
трансфекции.
2. Поставьте чашки на лед, добавьте в каждую лунку
1 мл смеси Трис/Тритон.
3. Заморозьте на 2 ч при -70 °С.
4. Разморозьте планшеты при 37 °С и затем поло¬
жите их на лед.
5. Перенесите клеточные лизаты в микроцентри¬
фужные пробирки, центрифугируйте 5 мин при
максимальной скорости.
6. Соберите супернатант, прогрейте при 65 °С в тече¬
ние 10 мин для инактивации ингибиторов CAT.
7. Центрифугируйте на максимальной скорости
3 мин и соберите супернатант клеточного экстра¬
кта. Храните при -70 °С.
Исследование CAT:
8. Поместите 5-150 мкл клеточного экстракта из
каждого образца в 3,5-мл полипропиленовый
сцинтилляционный флакон добавьте достаточно
большое количество 0,1 М Трис так, чтобы конеч¬
ный объем составлял 150 мкл.
9. В качестве отрицательного контроля используйте
150 мкл 0,lMTris.
10. В качестве положительного контроля:
а) Добавьте 150 мкл 0,1 М Трис в каждый из
5 флаконов.
б) Добавьте 5 мкл каждого из стандартных рас¬
творов CAT во флаконы, чтобы получить стан¬
дартную кривую 1,5,10,20, и 50 mU CAT.
11. К каждому образцу (включая контроли) добавьте
100 мкл следующей смеси:
а) UPW 84 мкл
б) Трис, 0,1 М 10 мкл
в) Хлорамфеникол 1 мкл
г) [14С]-СоА 5 мкл (50 нКи)
12. Закройте крышками образцы и инкубируйте их
при 37 °С в течение 2 ч.
13. Добавьте Змл Econofluor в каждую пробирку,
снова закройте крышками.
14. Перемешайте содержимое, переворачивая про¬
бирки.
15. Инкубируйте пробирки при комнатной темпера¬
туре в течение 2 ч.
16. Подсчитайте образцы в течение 30 с в жидкост¬
ном сцинтилляционном счетчике.
Глава 28
РЕШЕНИЕ ПРОБЛЕМ
Независимо от того, насколько хорошо работает
лаборатория, вместе с новыми методами, новым пер¬
соналом, новым оборудованием или другими новыми
обстоятельствами возникают проблемы, дестабили¬
зирующие повседневный рабочий процесс. Одним из
подходов к решению таких проблем является разра¬
ботка стандартных операционных процедур (СОП)
и неуклонное выполнение принятых протоколов без
каких-либо отклонений. Изменения в протоколах мо¬
гут быть введены только после исчерпывающей оценки
(проверки) возможных последствий. Тем не менее
реализовать этот подход очень трудно, в частности, в
исследовательской среде, где успех и развитие науки
требуют постоянных изменений и постоянного вве¬
дения новых методов. Советы, данные в предыдущих
главах, в основном касались практических аспектов,
инструкций типа «как это делается». Иногда встреча¬
лись замечания по поводу того, что «не надо делать».
Данная глава пытается осветить некоторые возможные
проблемы, а также возникающие трудности и вопросы,
а также, надеюсь, их решение.
28.1. МЕДЛЕННЫЙ РОСТ КЛЕТОК
28.1.1. Проблемы, ограниченные вашими
собственными исходными
культурами
1. Проверьте ваши клетки на предмет контаминации
а) Если вы работаете без применения антибиотиков
(а вы должны работать без них!), в большинстве
случаев контаминация будет визуально заметна,
i) Исследуйте культуру визуально невооружен¬
ным глазом и под микроскопом (см. разд. 28.6;
19.3.1).
и) Уничтожьте контаминированные клетки.
б) Микоплазму нельзя заметить визуально, поэтому
регулярно проверяйте ваши культуры на микро¬
плазмы (см. протоколы 19.2,19.3).
в) Проверьте возможные пути перемещения и пе¬
редачи культур (см. разд. 28.6; 19.1).
2. Проверьте рост ваших клеток в другой среде и сыво¬
ротке (например, различных партий или от разных
поставщиков). Если вы обычно используете сухие
среды или 10х, купите другой исходный материал
или среду 1х. Если в результате обнаружится, что
проблема заключается в среде, см. подраздел о сре¬
дах ниже в настоящем Разделе.
3. Если проблема состоит не в среде, она может быть
связана с вашими клетками.
а) Разморозьте новую ампулу, взятую из морозиль¬
ника. Сравните ее с вашей текущей культурой.
Все манипуляции проводите с культурами па¬
раллельно, но раздельно, во избежание контами¬
нации основного запаса клеток. Делайте это до
проверки или одновременно с пунктами (б)-(г)
проверки (см. ниже).
б) Подсчитайте ваши клетки в субкультуре, по¬
стройте ростовую кривую (см. протоколы 21.7
и 21.8), чтобы сравнить с вашими предыдущими
записями, касающимися этих клеток. Проверьте,
влияют ли на ваши клетки следующие факторы:
i) Плотность посева была слишком низкой для
переноса.
ii) Клетки были субкультивированы слишком
часто.
iii) Клетки оставались на фазе плато слишком
долго до субкультивирования.
в) Были ли изменения в партии трипсина или дру¬
гих диссоциирующих агентов? Проверьте номер
партии и поставщиков.
г) Для оценки жесткости действия диссоциирую¬
щего агента, проверьте:
i) Не была ли продолжительность действия трип¬
сина (или другого агента) слишком долгой?
ii) Не был ли диссоциирующий агент слишком
концентрированным и не имел ли он спе¬
цифической активности, которая была бы
чересчур высокой?
iii) Не был ли инкубатор, использованный для
трипсинизации, слишком теплым?
iv) Не было ли пипетирование при диссоциации
слишком интенсивным?
v) Не были ли клетки чувствительны к EDTA
(если вы использовали EDTA)?
vi) Правильный ли разбавитель для трипсина вы
использовали?
vii) Хорошую ли партию разбавителя для трип¬
сина вы использовали?
28.1.2. Более общие проблемы,
возникающие и у других
сотрудников
Проверьте оборудование и реактивы
коллективного использования
Термальная комната и инкубаторы
1) Температура и стабильность работы контролирова¬
лись неадекватно.
а) Термостаты недоброкачественные.
558
ГЛАВА 28. РЕШЕНИЕ ПРОБЛЕМ
б) Слишком часто открывались инкубаторы.
в) Дверь термальной комнаты оставили открытой.
г) Вентиляторы в системе циркуляции воздуха
сломаны или перегреваются. Проверьте инку¬
баторы при помощи портативных термографов
(см. разд. 5.3.3).
2) Влажность С02-инкубатора недостаточно хорошо
поддерживается
а) Поддон с водой не заполнен.
б) Нарушена герметичность дверей.
в) Слишком частый доступ в инкубатор.
г) Проверьте скорость испарения путем ежеднев¬
ного взвешивания чашки Петри с PBSA.
3) Концентрация С02в С02-инкубаторе плохо конт¬
ролируется
а) Слишком частый доступ в инкубатор.
б) Имеются проблемы с контролем уровня С02.
1. Проверьте pH in situ при помощи чашки Петри со сре¬
дой с предварительно определенным уровнем pH.
2. Проверьте атмосферу в инкубаторе при помощи
С02-тестера (Carborite).
3. Перекалибруйте на индикаторе установку нулевого
уровня и концентрацию С02 в стандартной газовой
смеси.
Среды и реактивы см. разд. 28.2.
28.1.3. Не было ли у вас в лаборатории
каких-либо изменений?
Персонал
1) Персонал, работающий с культурами
2) Персонал, работающий в подготовительной зоне
3) Подготовительные процедуры
4) Курсы и тренинги
5) Распределение места и переполненность помещения
людьми
Материалы
Химические загрязнения
(см. также разд. 28.8)
Среды (см. ниже)
1) Поставщик
2) Партия
3) Тип
4) Хранение
Методы
1) Время инкубации
2) Скорость проведения операций
3) Последовательность операций
4) Месторасположение
5) Масштаб работ
Оборудование
1) Замена оборудования: было ли добавлено какое-то
новое оборудование?
2) Рабочее состояние: возможно, какое-то оборудова¬
ние сломалось?
3) Месторасположение: оборудование обычно исполь¬
зовалось на том же месте или рядом расположенное
оборудование было переставлено?
4) Рабочая температура: работает ли оборудование в
оптимальном температурном режиме и подходит
ли эта температура для ваших образцов?
28.2. СРЕДА
Связаны ли возникшие проблемы только
с вашими собственными запасами или
они касаются общих запасов сред?
Срок ГОДНОСТИ
1. Проверьте номер партии и дату приобретения среды.
Она должна быть не больше 1 года со дня выпуска.
2. Если среда содержит глютамин, она сохраняет ста¬
бильность только в течение 1 месяца при 4 °С.
Условия хранения
1. Проверьте температуру в холодной комнате и холо¬
дильнике, она должна быть не выше 4 °С.
2. Проверьте, хранилась ли среда в темноте или, по
крайней мере, вдали от прямого солнечного света и
источников ультрафиолетового излучения, которые
приводят к разрушению среды.
Адекватность используемой среды
1. Проверьте качество и адекватность среды по срав¬
нению с другой средой (см. разд. 9.6).
2. Купите новую среду 1х, сравните ее с той, которую
вы использовали.
3. Если вы использовали среду 1х, проверьте среду
от другого производителя, проверьте отдельные
добавки к среде (например, сыворотку, факторы
роста, гормоны и т.д.).
Частота замены среды
Проверьте концентрацию клеток и pH. Возможно, pH па¬
дает ниже 7,0 в переделах 48 ч, уровень pH слишком низок
в начале периода роста или концентрация клеток слишком
высока. Кроме того, возможна контаминация среды.
Свойства сред
pH
Проверьте, находится ли pH в интервале между 7,0
и 7,4 в ходе культивирования, нет ли значительных
колебаний pH.
1. Проверьте поступление С02в инкубатор (см. Обо¬
рудование выше).
2. Проверьте регулировку поступления С02 (см. Обо¬
рудование выше).
Если уровень pH слишком высокий,
а) возможно, концентрация С02 в инкубаторе слишком
низкая
б) Концентрация НСО"3в среде слишком высокая
28.2. Среда
559
в) Если вы используете DMEM, следует использовать
также газовую смесь, содержащую 10% С02, если
она приготовлена в соответствии со стандартной
рецептурой.
Осмоляльность\ среда может быть гипер-
или гипотонической
1) Возможно, среда для исходной культуры была не¬
верно составлена.
2) Возможно, неверно добавление одного из компо¬
нентов.
3) Возможно, неверно произведено разведение.
1. Проверьте осмотические свойства на осмометре; она
должна находиться между 270 и 340 мОсмоль/кг.
2. Проверьте подготовительные процедуры.
а) Правильно ли взято количество воды, необходи¬
мое для разведения среды 10х или сухой среды?
б) Не влияет ли добавление реактивов на осмоляль-
ность?
Случайно пропущено добавление
одного из компонентов среды
1. Приготовьте свежую порцию среды (это быстрее,
чем пытаться решить, какой компонент забыт).
2. Возможны, компоненты среды имели низкую рас¬
творимость или произошла преципитация перед
фильтрацией, проверьте растворимость всех ком¬
понентов до фильтрации.
3. Возможно, преципитация произошла при хранении,
убедитесь, что преципитат был растворен путем
перемешивания и подогревания до 37 °С.
4. Если осадок не растворяется, выбросьте всю партию.
5. Если осадок вновь образуется, обратитесь с жалобой
к производителю, или поменяйте поставщика.
Некачественные компоненты
1. Заменяйте по очереди компоненты, каждый раз
используя новый компонент от другого производи¬
теля.
2. Сохраняйте записи номеров партий среды.
Новая партия среды для исходной культуры,
по-видимому, некачественная
1. Сравните партию с предыдущей, если остатки еще
сохранились.
2. Сравните партию с партией от другого поставщика,
если не сохранились остатки предыдущей.
Если среду готовят на SSS, удовлетворительно
ли качество BSS?
1. Проверьте результаты работы других сотрудников,
использующих этот BSS.
2. Проверьте альтернативные источники BSS или
другой состав.
Если среду разводят в воде, удовлетворительно
ли качество воды?
1. Проверьте результаты работы других сотрудников.
2. Сравните со свежей полной средой 1х.
28.2.1. Выбор среды
Производилось ли изменение состава среды
с момента получения клеток?
1. Вернитесь к ранее использованному типу среды
или проведите скрининг различных сред (см.
разд. 11.6.3), если исходная недоступна или не¬
адекватна.
2. Получите среду из того же источника, который был
поставщиком или автором клеточной линии.
3. Сравните компоненты среды с теми, которые вхо¬
дили в среду от поставщика или автора клеточной
линии.
При получении (выделении) новой
клеточной линии:
1. Проведите скрининг нескольких сред (см. разд. 11.6.3)
по нескольким параметрам:
а) Тип среды (см. разд. 9.6).
б) Тип сыворотки (см. разд. 9.6).
в) Концентрация сыворотки (см. протоколы 21.7,
21.8,21.9,21.10, и 22.4).
2. Включите бессывороточную среду в скрининг (см.
разд. 10.5).
а) Селекция на основе критериев культуры тканей,
например MCDB 153 для эпидермальных кера¬
тиноцитов.
б) Проверьте добавление факторов роста (см.
табл. 10.3).
3. Проверьте качество фидерного слоя (см. разд. 8.4.2,
14.2.3) и кондиционированной среды (см. разд. 14.2.2)
и/или качество обработки матриксом (см.
разд. 8.4.1).
28.2.2. Нестабильные реактивы
Глютамин
1. Хранить в замороженном состоянии при -20 °С.
2. Поскольку глютамин разрушается до 50% в течение
3-5 дней при 37 °С, заменяйте среду каждые 3 дня.
3. Используйте стабильные альтернативные компо¬
ненты — например, дипептид Glutamax — с предва¬
рительным тестированием.
Сыворотка
1. Хранить замороженной.
2. Если сыворотка слегка подтаяла, разморозьте ее
полностью, перемешайте и вновь заморозьте.
3. Проверьте партию сыворотки перед использованием.
4. Поочередно используйте разные партии среды, на
случай если плохое качество среды проявляется
медленно. (Возможно, потребуется 2-3 субкульти¬
вирования.)
Другие компоненты
1. Храните при 4 °С.
2. Большинство компонентов сред стабильны в тече¬
ние 1-2 недель при 37 °С.
560
ГЛАВА 28. РЕШЕНИЕ ПРОБЛЕМ
3. Храните добавки в виде малых аликвот, которые
размораживаются и используются однократно, не
замораживайте повторно основные растворы.
Трипсин
1. Храните концентрированные замороженные
основные растворы в виде малых аликвот для
однократного размораживания при одном исполь¬
зовании.
2. Приготовьте раствор из концентрата и храните
разведенный раствор при 4 °С не более 2 недель.
3. Трипсин будет разрушаться, если оставить его при
комнатной температуре более чем на 30 мин.
28.3. ЧИСТОТА СОСТАВЛЯЮЩИХ
ЧАСТЕЙ
28.3.1. Правильно ли работает
система очистки воды?
Качество получаемой воды
1. Исследуйте проводимость воды.
2. Определите общее содержание органического угле¬
рода в воде (total organic content, TOC; оборудова¬
ние Millipore).
3. Проверьте среду, приготовленную разведением
сухой смеси в воде, сравнивания ее со средой 1х.
Режим технического обслуживания
оборудования
1. Когда последний раз заменяли ионообменник?
2. Когда меняли картридж для обратного осмоса?
3. Все ли соединительные трубки чистые? Нет ли
признаков водорослей, грибов или бактерий?
4. Проверьте сосуды для хранения воды на предмет
зарастания водорослями и грибами.
5. Проверьте, не происходит ли переход ионообмен¬
ной смолы в воду.
6. Проверьте химические загрязнения или их оста¬
точные количества в стеклянной емкости тепло¬
обменника.
а) Снимите кожух со стеклянной емкости.
б) Промойте ее 1 М HCI и тщательно прополо¬
щите.
в) Вылейте первую порцию воды после промы¬
вания.
Следы химических веществ
в пластиковых трубках
1. Отсоедините и очистите трубки.
а) Замочите в горячем детергенте.
б) Прополощите и замочите в 1 М НС1.
в) Тщательно прополощите в деионизированной
воде.
г) Восстановите соединения трубок, первую пор¬
цию полученной воды вылейте.
2. Замените трубки на новые инертные промы¬
тые, стерилизованные пластиковые трубки (см.
разд. 11.3.5).
28.3.2. Бикарбонат
Правильна ли концентрация бикарбоната ?
1. Проверьте проводимость или осмоляльность по
сравнению со стандартным раствором.
2. Попробуйте использовать другую партию.
3. Проверьте нет ли следов осадка.
Неправильно приготовленная концентрация
1. Проверьте, не ошиблись ли вы при приготовлении
среды (см. табл. 9.1,9.3,10.1 и 10.2).
2. Проверьте концентрацию С02 в инкубаторе.
28.3.3.Антибиотики
Проблемы могут быть связаны с
1) частотой использования
2) концентрацией
3) номером партии
4) комбинациями препаратов
5) употреблением фунгицидов (например, амфотери-
цин В может быть токсичен)
28.3.4. Сыворотка
Вы используете новую партию?
1. Проверьте контроль качества, проведенный постав¬
щиком.
2. Сравните партию с предыдущей партией или с дру¬
гими партиями.
Концентрация
Слишком низкая? Слишком высокая?
1. Перепроверьте отсутствие токсичности, стимуля¬
цию роста и эффективность посева
а) Постройте кривую роста (см. протоколы 21.7,
21.8,21.9).
б) Проведите исследование клонального роста (см.
протокол 21.10).
2. Попробуйте заменить на
а) новую сыворотку от другого производителя
б) бессывороточную среду (см. разд. 10.5)
в) другую среду с добавлением сыворотки (см.
разд. 10.5.2)
28.4. ПЛАСТИКОВАЯ ПОСУДА
Вы используете новую марку,
тип или партию?
1. Проверьте вашу партию в сравнении с преды¬
дущей.
28.5. Стеклянная посуда
561
2. Попробуйте использовать пластик от другого пос¬
тавщика.
28.5. СТЕКЛЯННАЯ ПОСУДА
28.5.1. Мытье посуды
Имеют ли другие клетки признаки старения или
ухудшения роста? Есть ли проблемы у других ис¬
следователей?
Имеются ли признаки следов загрязнения на стек¬
лянных стенках бутылей для хранения или пипет¬
ках? (см. разд. 28.7.)
Достаточно ли хорошо были прополосканы после
мытья крышки? (Следует добавить несколько мл
BSS, изменение pH покажет присутствие детер¬
гента, оставшегося на крышке.)
Не было ли совместной обработки посуды для куль¬
туры тканей и посуды для химических реактивов?
28.6. МИКРОБИОЛОГИЧЕСКАЯ
КОНТАМИНАЦИЯ
См. также табл. 18.1.
28.6.1. Контаминация, ограниченная
одним исследователем
Спорадическая
Отдельные виды
1. Проверьте правильность приемов асептической
работы оператора (см. разд. 6.3-6.5).
2. Являются ли какая-то среда или реагент уникаль¬
ными для этого исследователя?
3. Проверьте личную чистоплотность и аккуратность
оператора.
а) Моет ли он руки до и после работы с культурой?
б) Меняет ли лабораторный халат?
в) Если у него длинные волосы, завязывает ли он
их сзади?
4. Распорядитесь, чтобы оператор надевал перчатки.
Микоплазма
1. Проверьте, нет ли у других исследователей призна¬
ков контаминации микоплазмой.
2. Удостоверьтесь, что осуществляется программа
отбраковки контаминированных культур.
3. Наиболее частым источником инфекции являются
принесенные из других лабораторий культуры. До
применения их в скрининге для использования
в основной культуральной зоне выдержите их в
необходимом карантине. Ведите журнал записи
поступления культур.
4. Проверьте продукты естественного происхождения
(сыворотка, трипсин) при помощи индикаторной
клеточной линии (см. протоколы 19.2,19.3).
5. Ограничьте использование антибиотиков при
формировании и ведении первичной культуры и в
критических экспериментах.
Мультиспецифическая
контаминация
Может возникнуть в результате грубых нарушений
методов стерильной работы.
1. Убедитесь, что все необходимые процедуры прово¬
дятся правильно (см. протокол 6.1):
а) Рабочее место в ламинарном боксе не загромож¬
дено.
б) Все материалы протираются спиртом перед по¬
мещением их в ламинарный бокс.
в) Все разбрызгивания и загрязнения сразу же
вытираются.
2. Проверьте использование лабораторных халатов:
а) Заменялись ли лабораторные халаты перед на¬
чалом культуральных работ?
б) Пришиты ли пуговицы на халаты и застегнуты
ли они? (Даже просто проходя мимо ламинар¬
ных боксов в незастегнутом халате с развеваю¬
щимися полами, люди могут нарушить ламинар¬
ный поток).
3. Проверьте, нет ли аналогичных проблем у других
исследователей, работающих в том же ламинарном
боксе, возможно, работа его нарушена и он нужда¬
ется в техническом обслуживании.
Повторная контаминация
Отдельные виды
Проблема может быть связана с реактивами или с
клеточной линией.
1. Проверьте, есть ли какие-то реактивы, которые не
используются другими исследователями.
2. Проверьте, используется ли данная клеточная ли¬
ния другими исследователями.
Мультиспецифическая
контаминация
1. Проверьте приемы стерильной работы (см.
разд. 6.3-6.5).
2. Проверьте нестандартные рабочие методики.
3. Проверьте расположение рабочего места: возможно,
рядом располагается слишком много оборудования
или мимо рабочего места идет интенсивное движе¬
ние (см. разд. 6.2)?
4. Не загромождено ли рабочее место в ламинарном
боксе?
5. Не используются ли нестерильные реактивы?
6. Проверьте использование лабораторных халатов
(см. разд. 26.8.1).
Постоянная контаминация
Отдельные виды
Контаминированный раствор
1. Смешайте среду (без антибиотика) 1:1с питатель¬
ным бульоном (например, L-бульон) и инкубируйте
раствор.
562
ГЛАВА 28. РЕШЕНИЕ ПРОБЛЕМ
2. Нанесите среду на кровяной агар. Инкубируйте
чашки, перевернув их вверх дном, с параллельным
«пустым» контролем.
Контаминированная клеточная линия
1. Проверьте клетки окрашиванием по Хехсту (см.
протокол 19.2).
2. Перемешайте клетки с бульоном и инкубируйте.
Мультиспецифическая контаминация
1. Проверьте приемы стерильной работы (см.
разд. 6.3-6.5).
2. Проверьте нестандартные методики на сохранение
стерильности.
3. Проверьте расположение рабочего места: возможно,
рядом располагается слишком много оборудования
или мимо рабочего места идет интенсивное движе¬
ние (см. разд. 6.2)?
4. Проверьте, сколько оборудования и других материа¬
лов находится одномоментно в ламинарном боксе.
Не загромождено ли рабочее место в ламинаре?
5. Проверьте, не используются ли (случайно или пред¬
намеренно) нестерильные реактивы?
28.6.2. Распространенная контаминация
Спорадическая
Отдельные виды
1. Проверьте редко используемые реактивы или среды,
которые могут быть заражены.
2. Проверьте инкубаторы. Помойте, если это необхо¬
димо (см. протокол 19.1).
3. Проверьте, не увеличилось ли количество спор в
воздухе путем экспозиции открытых бактериоло¬
гических чашек с питательной средой в комнате в
нерабочее время.
Мультиспецифическая контаминация
Нарушения стерилизации
1. Проверьте стерилизационные шкафы на предмет:
а) Избыточной загрузки стерилизуемым содержи¬
мым, что препятствует адекватной циркуляции
воздушного потока.
б) Целостности дверных затворов и других от¬
верстий.
в) Температуры и продолжительности цикла сте¬
рилизации (160 °С в течение 1 ч).
2. Проверьте автоклавы на предмет:
а) Избыточной загрузки стерилизуемым содержи¬
мым, что препятствует адекватной циркуляции
пара.
б) Температуры и продолжительности цикла
стерилизации (121 °С в течение 15-20 мин) с
применением эквивалентного образца в качестве
контроля, расположив его в середине загрузки.
в) Следует убедиться, что все пустые сосуды откры¬
ты перед автоклавированием для проникания в
них пара (см. рис. 11.3; цв. вкладку 22а).
Новый сотрудник не соблюдает правил СОП
1. Проверьте уровень работы новых сотрудников при
помощи контролирующего опытного сотрудника.
2. Своевременно проводите тренинги (см. разд. 2.1,
6.3-6.5).
Контаминированные хранилища
(холодные комнаты и холодильники)
1. Вымойте все хранилища.
2. Убедитесь, что стерилизованное оборудование не
хранится на открытых полках.
3. Проверьте цикл использования стерильных запасов.
Повторная контаминация, отдельные виды
Контаминированные реактивы или среда
1. Проверьте частоту использования реактивов раз¬
ными сотрудниками, чтобы сузить проблему до
общеупотребляемых реактивов.
2. Исследуйте наиболее подходящие «кандидатуры»,
инкубируя их с бульоном 1:1.
3. Проверьте инкубаторы на предмет контамина¬
ции, при необходимости вымойте их (см. прото¬
кол 19.1).
4. Проверьте ламинарный бокс.
а) Протрите рабочую поверхность и боковые по¬
верхности рабочей зоны раствором фенолоаль¬
дегидного дезинфектанта в 70%-м спирте.
б) Протрите поверхность под рабочей зоной раство¬
ром фенолоальдегидного дезинфектанта в 70%-м
спирте.
в) Проверьте другие компоненты воздуходувной
системы на предмет щелей и контаминации.
г) Проверьте целостность фильтров при помощи
анемометра или чашек с питательным агаром
(см. разд. 6.4).
д) Проверьте, не являются ли фильтры загрязнен¬
ными или забитыми, например, нет ли перепада
давления на фильтре (см. разд. 6.4).
е) Проверьте соблюдение режима санитарной об¬
работки, измените его, если необходимо.
5. Проверьте график профилактического ремонта и
технического обслуживания оборудования, изме¬
ните его, если необходимо.
Повторная мультиспецифическая
контаминация
Нарушенная стерилизация
1. Проверьте, не перегружаются ли автоклавы и сте¬
рилизационные шкафы.
2. Проверьте, нет ли механических и электрических пов¬
реждений автоклавов и стерилизационных шкафов.
3. Проверьте записи и распечатки циклов стери¬
лизации.
4. Проверьте целостность стерилизационной камеры.
5. Проверьте, выполняют ли новые сорбенты требова¬
ния стандартных протоколов.
6. Проверьте уровень подготовки нового персонала.
7. Проверьте выполнение протоколов в присутствии
опытного сотрудника.
28.7. Химическая конатминация
563
8. Ограничьте использование антибиотиков.
9. Проверьте, не было ли изменений в протоколах
и/или в требованиях к их выполнению.
10. Перепишите СОП так, чтобы они соответствовали
изменившимся условиям.
Зараженные хранилища
(холодильники или холодные комнаты)
1. Вымойте хранилища.
2. Убедитесь, что стерилизованные предметы не хра¬
нятся на открытых полках.
Контаминированный воздух комнаты
1. Проверьте уровень контаминации комнатного воз¬
духа путем оставления открытых чашек с кровяным
агаром в комнатах, когда они не используются, но
вентиляция остается включенной.
2. Проверьте выполнение требований асептики опе¬
раторами.
3. Проверьте целостность вытяжных колпаков.
4. Проверьте качество работы оборудования для по¬
дачи воздуха и
5. Проверьте качество обработки вновь поступающего
оборудования.
6. Не проводятся ли ремонтные работы рядом с мес¬
том работы с культурами? Если да, то постарай¬
тесь улучшить качество изоляции культуральной
лаборатории (например, плотно закрывайте двери,
установите изолирующий экран, запретите проход
через культуральную лабораторию всем сотрудни¬
кам, не работающим в ней, мойте все оборудование
и материалы, поступающие в лабораторию).
28.6.3. Идентификация контаминации
Бактериальная, грибная
1. Проверьте на бактериальную и грибковую контами¬
нацию клетки или среды, используя фазово-конт-
растную микроскопию.
2. Инкубируйте клетки в бульоне.
3. Посейте клетки на кровяной агар и инкубируйте
культуру.
4. Окрасьте по Грамму клетки или проконсультируй¬
тесь у микробиолога.
Микоплазменная
1. Окрасьте культуру по Хехсту а33258 (см. прото¬
кол 19.2).
2. Проверьте уровень цитоплазматического синтеза
ДНК (включение [3Н]-тимидина), с использованием
авторадиографии (см. протокол 27.3).
3. Используйте коммерческие диагностические набо¬
ры для определения микоплазмы (Приложение II
Определение микоплазмы).
Вирусная
1. Проверьте наличие вирусной контаминации, ис¬
пользуя
а) Трансмиссионную или сканирующую электрон¬
ную микроскопию.
б) Иммунное окрашивание флюоресцентными кра¬
сителями или антителами конъюгированными с
пероксидазой (см. протокол 16.11).
в) Исследованием ELISA
г) ПЦР
2. Получением клеточных линий и биологических
реактивов из источников, свободных от вирусного
заражения.
28.6.4. Деконтаминация
1. Уничтожьте все рабочие культуры и не пытайтесь
произвести обеззараживание культур, если только
они могут быть восстановимы.
2. Деконтаминируйте клетки, только если они невос¬
становимы (см. протокол 19.4).
а) Деконтаминацию должен проводить опытный
сотрудник.
б) Работайте в условиях карантина (см. разд. 19.1.8).
28.7. ХИМИЧЕСКАЯ КОНТАМИНАЦИЯ
28.7.1. Стеклянная посуда
Химические остатки после мытья посуды
1. Храните стеклянную посуду для культуральных
работ отдельно от посуды, предназначенных для
химических реактивов.
2. Убедитесь, что не происходит переноса химических
остатков из последней промывки химической посу¬
ды в посудомоечной машине.
3. Проверьте наличие признаков загрязнения посуды
а) визуально;
б) добавлением небольшого объема BSS с феноло¬
вым красным и наблюдением за уровнем pH;
в) добавлением небольшого объема UPW и провер¬
кой электрической проводимости.
4. Клонируйте клетки в стеклянной посуде после сте¬
рилизации сухим жаром.
5. Тщательно выбирайте детергент (см. разд. 11.3.4).
Накопление пыли при хранении
1. Закрывайте горлышки сосудов фольгой при хранении.
2. Храните в зоне, чистой от пыли или в контейнере.
28.7.2. Пипетки
Остатки или засорение после мытья
1. Убедитесь, что пипетки промыты и высушены но¬
сиками вверх.
2. Собирайте пипетки в емкость с детергентом, но
промывайте в чистой воде без добавок.
3. Не разрешайте использовать стеклянные пипетки
для разлива агара.
564
ГЛАВА 28. РЕШЕНИЕ ПРОБЛЕМ
4. Проверьте пипетки после прополаскивания перед
стерилизацией
а) визуальным исследованием;
б) добавлением небольшого объема BSS с феноло¬
вым красным и наблюдением за изменением pH;
в) добавлением небольшого объема UPW и провер¬
кой проводимости.
5. Убедитесь, что ватные пробки были удалены перед
мытьем.
28.7.3. Очистка воды
См. разд. 11.4.1,28.3.1.
28.7.4. Другие реактивы
Загрязнение DMSO растворенным пластиком
или резиной тары
1. Храните DMSO в стеклянных или полипропилено¬
вых емкостях со стеклянными или полипропилено¬
выми крышками.
2. Переливайте DMSO при помощи стеклянной пи¬
петки или автоматической пипетки с полипропи¬
леновыми наконечниками.
Ухудшение качества глицерина
при длительном хранении
1. Покупайте небольшое количество глицерина, кото¬
рое будет использовано в течение 3-6 месяцев.
2. Храните в темных бутылях.
28.7.5. Порошки и аэрозоли
1. Все манипуляции с токсичными химикатами, ко¬
торые образуют порошки и аэрозоли, проводите в
защитном вытяжном колпаке (см. разд. 7.5.4).
2. Избегайте включения тяги при взвешивании порош¬
ков или разливании жидкостей.
3. Контролируйте передвижение людей и перемеще¬
ние оборудования в культуральную лабораторию и
препаративную зону
4. Переодевайтесь в чистый лабораторный халат перед
входом в лабораторию культуры тканей.
28.8. ПЕРВИЧНАЯ КУЛЬТУРА
28.8.1. Низкий выход клеток
в первичной культуре
Первичные эксплантаты не прикрепляются
1. Процарапайте субстрат сквозь эксплантат (см. про¬
токолы 12.4 и 23.9).
2. Прижмите эксплантат покровным стеклом (см.
протокол 12.4).
3. Залейте в сгусток плазмы (см. протокол 12.4).
Незавершенная дезаггрегация
1. Инкубируйте клетки в растворе протеазы в течение
более продолжительного времени.
2. Попробуйте холодную предварительную обработку
перед инкубированием (см. протокол 12.6).
3. Используйте альтернативную или дополнительную
обработку протеазой (см. табл. 13.4; разд. 12.3.6).
Завершенная дезаггрегация
при слабом прикреплении
Плавающие клетки жизнеспособны
1. Обработайте субстрат
а) Покройте пластик коллагеном, фибронектином,
ламинином, поли-Э-лизином или поли-Ь-лизи-
ном (см. разд. 8.4.1).
б) Используйте фидерный слой (см. разд. 8.4.2;
протоколы 14.3, 23.4 и 24.1).
2. Культура может быть неадгерентной, поэтому сле¬
дует растить культуру в суспензии.
Плавающие клетки преимущественно
нежизнеспособны
1. Отрегулируйте концентрацию клеток путем подсче¬
та жизнеспособных клеток.
2. Удалите нежизнеспособные клетки (см. прото¬
кол 12.10).
Плавающие клетки нежизнеспособны,
при этом небольшое количество клеток
прикрепилось к субстрату
Клеточная плотность
Клеточная плотность чересчур низкая, повысьте ее до
1х106 кл./мл.
Плохая клеточная адгезия
Обработайте субстрат (см. разд. 8.4.1, 8.4.2 и протоко¬
лы 14.3, 23.4, 24.1).
Используемые ферменты
слишком токсичны
1. Замените на другую протеазу (см. разд. 12.3.6 и
табл. 13.4).
2. Уменьшите время экспозиции.
3. Используйте холодную предварительную обработ¬
ку протеазой перед инкубированием (см. прото¬
кол 12.6).
Очень кислая среда
1. Проверьте, не заражена ли среда (см. разд. 19.3).
2. Уменьшите концентрацию клеток при посеве или
через 24 часа после посева.
3. Добавьте буфер HEPES, приоткройте флакон.
Неправильно выбранная
или плохо приготовленная среда
1. Проверьте ассортимент сред (см. разд. 9.6; табл. 9.3,
9.6,10.1,10.2 и 10.4).
28.10. Смена среды
565
2. Изучите литературу, касающуюся среды, которую
вы использовали для своих клеток (если вы до сих
пор не сделали этого).
Добавки в среду
1. Используйте разные партии или серии сыворотки
(см. разд. 9.6.2).
2. Замените сыворотку на бессывороточную среду (см.
разд. 10.5).
3. Используйте различные факторы роста (см.
разд. 10.4.3 и табл. 10.3).
4. Используйте другие митогены (например, РМА;
простагландины; гидрокортизон или другие сте¬
роиды; петидные гормоны, такие как инсулин и
трансферрин, см. табл. 10.2).
5. Используйте кондиционированные среды (см.
разд. 9.7.3; протокол 14.2).
28.8.2. Неверно выбраны клетки
Избыточный рост фибробластов
или эндотелия
1. Используйте селективные среды (см. разд. 10.2.1 и
14.6).
2. Используйте селективные субстраты (см. разд. 8.4.1
и 14.8).
3. Используйте селективный фидерный слой (см.
протокол 24.1).
4. Проверьте присутствие перекрестной контаминации
из фидерного слоя или ксенотрансплантата (см.
разд. 19.5; протоколы 16.8,16.9 и 16.10).
Перекрестная контаминация с другими
клеточными линиями
Сравните с другими линиями, которые в настоящее
время находятся в культуре (см. разд. 7.10.1,16.1,16.3,
16.6.2,19.5; табл. 13.2).
28.8.3. Контаминация
1. Проверьте на предмет контаминации (см. разд. 19.3).
2. Предварительно обработайте ткань антибиотиками
(см. Приложение I: Среды и DBSS) или 70% этано¬
лом (см. шаг 7, протокол 12.3).
3. Проведите эрадикацию контаминации, но только
если культура невосстановима (см. разд. 19.4).
28.9. ДИФФЕРЕНЦИРОВКА
28.9.1. Клетки не дифференцированы
1. Примените условия улучшающие дифференциров-
ку среды (см. разд. 17.7).
2. Используйте селективную среду (см. разд. 10.2.1;
табл. 10.1 и 10.2) при выделении клеток и в ходе
наращивания культуры.
3. Растите культуру с соответствующим фидерным
слоем (см. разд. 17.7.1,17.7.3).
28.9.2. Снижение образования
продукта
1. Проверьте уровень экспрессии релевантного гена
с использованием RT-ПЦР или Нозерн-блоттинга
[ Ausubel et al., 1996,2002] либо in situ гибридизации
(см. разд. 27.8).
2. Проверьте экспрессию репортерного гена (исполь¬
зуя RT-ПЦР или Нозерн-блотинг [Ausubel et al.,
1996]; см. разд. 27.12.5).
3. Примените условия, индуцирующие дифференци-
ровку (см. разд. 17.7).
4. Используйте соответствующие селективные среды
для данного типа клеток (см. разд. 10.2.1; табл. 10.1 и
10.2) при выделении клеток и ведении культуры.
28.10. СМЕНА СРЕДЫ
28.10.1. Обычные монослои
pH падает слишком быстро
1. Проверьте на предмет бактериальной контаминации
(см. разд. 19.4.1).
2. Проводите смену среды чаще.
3. Посев клеток проводите при более низкой концент¬
рации.
4. Добавляйте буфер HEPES в среду (см. разд. 9.2.3)
и используйте вентилируемые флаконы (см.
разд. 8.2.3).
pH повышается после смены среды
1. Уменьшите количеств бикарбоната или другого
щелочного компонента в среде.
2. Повысьте концентрацию С02 в среде.
3. Проверьте, нет ли протекания флаконов, инкуби¬
руемых на воздухе, или в манжетном уплотнении
либо в крышке при инкубировании в С02-инку-
баторе.
28.10.2. Клоны
Уровень pH слишком высокий
или слишком низкий
См. разд. 28.12.1, 28.5.
Контаминированные чашки
1. Инкубируйте чашки в специальной коробке (см.
разд. 6.6.2) и протирайте коробку спиртом перед
открыванием.
2. Выбрасывайте крышки от флаконов и чашек, в част¬
ности, если на них есть следы среды. Протирайте
снаружи 70% спиртом.
3. Не используйте вентилятор в инкубаторе.
566
ГЛАВА 28. РЕШЕНИЕ ПРОБЛЕМ
Требования к смене среды
Монослой
Замените среду (см. протокол 12.1) и проверьте через
24 ч.
Суспензия
1. Добавьте среду в агар для культуры.
2. Добавьте среду с Methocel в случае клонов Methocel;
питательные вещества диффундируют через метил-
целлюлозу.
28-11. СУБКУЛЬТУРА
Данный раздел посвящен обсуждению проблемы
низкого выхода клеток или медленного роста после
субкультивирования (см. также разд. 28.1).
28.11.1. Фазы клеточного цикла
при субкультуре
1. Субкультуру проводите на экспоненциальной
или поздней экспоненциальной фазе (см. прото¬
кол 13.2).
2. Убедитесь, что посевная концентрация выбрана
правильно— при слишком низкой концентрации
клетки будут иметь слишком длительную lag-фазу,
при слишком высокой они будут выходить на плато
до того, как будет проведено субкультивирование
(см. разд. 13.7.3).
3. Не проводите субкультивирование на стадии плато,
поскольку клетки на этой стадии склонны иметь
продолжительную lag-фазу. Состояние некоторых
культур, например гибридом, клеток мышиной
лейкемии, будет быстро ухудшаться в фазе плато.
4. Не проводите субкультивирование чаще, чем это
необходимо. Подберите клеточную концентрацию
и интервал субкультивирования, которые наиболее
удобны и дают наиболее воспроизводимый резуль¬
тат (см. разд. 13.7.2).
28.11.2. Старение
1. Проверьте количество поколений, после которых
развиваются признаки старения культуры, клетки
могут быть приближаться к концу конечного жиз¬
ненного цикла (см. разд. 3.8.1,13.7.2,18.4.1).
2. Замените рабочие культуры, как это обычно дела¬
ется путем размораживания (см. разд. 20.4.2).
3. Если необходимо, используйте иммортализацию
(см. протоколы 18.1,18.2).
28.11.3. Среда
Как в случае медленного клеточного роста (см.
разд. 28.1).
28.11.4. Неравномерный рост
Неслучайное распределение клеток
Как в случае клонирования (см. разд. 28.9.4).
Неправильный посев
1. При пипетировании старайтесь не вызвать враща¬
тельного движения суспензии.
2. Не вносите клетки пипеткой в середину чашки без
перемешивания.
3. Не вращайте чашку с целью перемешивания клеток,
перемешивание производите покачиванием слева
направо и вперед-назад либо осторожным пипети-
рованием без завихрений суспензии.
Сотрясения инкубатора
1. Уменыпите частоту доступа в инкубатор.
2. Проверьте надежность и устойчивость расположе¬
ния инкубатора.
3. Убедитесь, что полки имеют горизонтальное распо¬
ложение без угла наклона.
4. Поместите упругую прокладку под флаконы, чашки
и планшеты.
Неравномерное нагревание (см. рис. 8.10)
1. Поместите флаконы или чашки на теплоизоляци¬
онную плитку или металлическую чашку.
2. Проверьте воздух и распределение температуры.
28.12. КЛОНИРОВАНИЕ
См. также Медленный клеточный рост в данной главе.
28.12.1. Низкая эффективность посева
Слишком мало колоний на чашку
1. Повысьте посевную концентрацию.
2. Используйте фидерный слой.
3. Улучшите эффективность посева (см. разд. 14.2).
Колонии слишком диффузные
1. Выберите чашку Петри большего диаметра, чтобы
получить больше пространства для распростране¬
ния колоний.
2. Используйте глюкокортикоид (например, декса¬
метазон, lxlO^-lxlO^M).
Микоплазменная контаминация
1. Проведите скрининг на микоплазмы (см. протоко¬
лы 19.3.3-19.3.5).
2. Произведите эрадикацию микоплазм, но только как
последнее средство, если клеточная линия невоспро¬
изводима (см. разд. 19.4.2; протокол 19.4).
Неаккуратные манипуляции с культурой
1. Не находились ли клетки вне инкубатора слишком
продолжительное время?
28.13. Перекрестная контаминация
567
2. Не было ли время нахождения в растворе слишком
продолжительным?
3. Не испарилась ли среда во время инкубирования?
Среда
Тип среды
1. Выберите богатую среду (например, среда Хэма
F12).
2. Если среда бессывороточная, (например, MCDB 153
для кератиноцитов, см. табл. 10.1 и 10.2), попробуйте
другие бессывороточные среды.
3. С02 является существенным элементом, поэтому
всегда инкубируйте культуру выше минимума в
2% С02.
4. Проверьте испарение путем взвешивания тест-чаш-
ки со средой при тех же условиях в начале и в ходе
периода культивирования.
Если присутствие сыворотки существенно
1. Используйте FBS, которая обычно лучше подходит
для клонирования, чем сыворотка лошади или те¬
ленка.
2. Если FBS уже использовалась, повысьте ее концен¬
трацию
3. Выберите партию на основе эффективности посева
клеток, которые вы используете (см. разд. 11.6.3,
протокол 21.10).
Бедный субстрат
Не изменили ли поставщика пластиковой посуды для
культуры?
1. Проверьте источник поступления пластика и, если
необходимо, измените его.
2. Попробуйте противоположно заряженный плас¬
тик, например, Primaria (BD Biosciences), Cell-
BIND (Corning).
3. Покройте пластик матриксом (см. разд. 8.4.1, про¬
токол 8.1; 17.7.3; протокол 23.9: FN/V/BSA).
28.12.2. Диффузные колонии
1. Обработайте чашки матриксом (см. разд. 8.4.1, про¬
токол 8.1; 17.7.3; протокол 23.9: FN/V/BSA).
2. Используйте глюкокортикоид (например, 1х10-7-
1х10‘5 М дексаметазон) в течение первых 48-72 ч.
3. Используйте фидерный слой (см. протокол 14.3).
28.12.3. Слишком много колоний на чашку
1. Уменьшите посевную концентрацию (кл./мл).
2. Посейте то же количество клеток на чашку большего
размера.
Перекрывающиеся колонии
1. Уменьшите посевную плотность (кл./см2).
2. Растите колонии в течение более короткого времени
(меньше клеток на колонию).
3. Посейте то же количество клеток на чашку большего
размера.
28.12.4. Неслучайное распределение
См. также разд. 28.11.4.
1. Добавьте клетки к среде в бутыли, перемешайте
клеточную суспензию, а затем посейте чашки.
2. Не взбалтывайте чашки при перемешивании клеток
или при пипетировании.
3. Убедитесь, что чашки горизонтальны.
4. Удостоверьтесь, что среда покрывает все дно чашки
равномерно.
5. Убедитесь, что инкубатор не сотрясается.
6. Ограничьте доступ остальных работников к инку¬
батору.
7. Промаркируйте коробку или поднос, содержа¬
щий чашки с пометкой «КЛОНИРОВАНИЕ. НЕ
ТРОГАТЬ!»
8. Поместите коробку или поднос в глубину инкуба¬
тора.
9. Коробку или поднос следует поставить на коврик.
(Мойте коврик регулярно)
28.12.5. Неприкрепляющиеся клетки
Клетки, независимые от прикрепления
к субстрату
1. Клонируйте клетки в агаре (см. протокол 14.4),
агарозе или метилцеллюлозе Methocel (см. прото¬
кол 14.5), нанесенные на подложку в чашках не для
культуры тканей.
2. Добавите факторы роста или добавки к подложке.
3. Используйте чашки для культуры тканей и посейте
фидерный слой до заливки подложки.
Плохо адгезирущие клетки
Клонируйте клетки в Methocel в чашках для культуры
тканей (см. протокол 14.5).
28.13. ПЕРЕКРЕСТНАЯ
КОНТАМИНАЦИЯ
В данном разделе обсуждается проблема перекрестной
контаминации с другой клеточной линией.
28.13.1. Симптомы перекрестной
контаминации
Изменения во внешне виде
Морфология клеток изменяется
Клетки накапливаются и громоздятся друг на друга
при высокой плотности на стадии плато, при этом в
норме в культуре имеет место контактное ингибиро¬
вание роста.
568
ГЛАВА 28. РЕШЕНИЕ ПРОБЛЕМ
Изменение ростовых характеристик
Скорость роста повышается (т. е. укорачивается время
удвоения популяции (PDT).
Клетки растут до более высокой плотности насыщения.
Характеристики клеточных линий
Изменения фенотипических характеристик (см.
разд. 16.1,17.1,18.3).
28.13.2. Предупреждение перекрестной
контаминации
См. также разд. 19.5.
1. Не пользуйтесь одной банкой реактивов вместе с
другими исследователями.
2. Не используйте одну банку реактивов для несколь¬
ких клеточных линий.
3. Не манипулируйте одновременно с более чем одной
клеточной линией.
4. Не возвращайте пипетку в среду после исполь¬
зования или после использования в работе с
клетками.
5. Не используйте автоматическую пипетку для се¬
рийных манипуляций, если только не применяются
наконечники с фильтрами.
6. Манипуляции с быстрорастущими культурами, та¬
кими как HeLa и другие, производите в последнюю
очередь.
7. Проводите аутентификацию клеток перед замора¬
живанием (см. разд. 16.1,16.3).
28.13.3. Элиминация перекрестной
контаминации
1. Уничтожьте культуры!
2. При раннем обнаружении контаминации (напри¬
мер, при наличии признаков смешанной культу¬
ры) и отсутствии возможностей получить чистые
исходные культуры клонируйте клетки (см.
протокол 14.1) и изолируйте колонии, напомина¬
ющие желательные культуры (см. протоколы 14.6
и 14.7).
3. Выполните позитивный и негативный сортинг
методом FACS (см. разд. 15.4) или MACS (см.
разд. 15.3.2).
28.14. КРИОКОНСЕРВАЦИЯ
28.14.1. Плохое восстановление
Скорость замораживания
1. Измените скорость охлаждения (см. разд. 20.3.4),
хотя 1 °С/мин обычно оптимально,
а) Измените толщину стенки если используется
неизолированный контейнер для заморажи¬
вания.
б) Используйте доступный морозильник для изме¬
нения скорости замораживания и формы кривой
роста.
Концентрация клеток
При замораживании
Повысьте концентрацию клеток при замораживании
(оптимальной обычно является концентрация 1х106-
1х107/кл./мл, см. разд. 20.3.2; протокол 20.1).
Размораживание и пересев
1. Разведите культуру медленнее после разморажива¬
ния (см. протокол 20.2) путем постепенного добав¬
ления среды к клеткам.
2. Пересейте клетки в посевной концентрации в
пять раз превышающей нормальную (см. прото¬
кол 20.2).
3. Запасите несколько ампул (вам потребуется цен¬
трифугировать несколько раз для отмывания кон¬
серванта, см. протокол 20.2).
4. Удалите нежизнеспособные клетки (см. прото¬
кол 12.10).
Консервант
DMSO
1. Проверьте, нет ли химического загрязнения DMSO
(пластиком или резиной).
а) Проверьте цвет; DMSO должен быть бес¬
цветным.
б) Если DMSO бесцветный, проведите спектрофо¬
тометрическое исследование и сравните резуль¬
таты со свежим DMSO.
2. Проверьте, способны ли клетки к дифференцировке
после действия DMSO (см. разд. 17.7.2).
3. Центрифугируйте клетки, растущие в суспензии,
чтобы удалить консервант
Глицерин
Глицерин токсичен при хранении в течении нескольких
месяцев на свету (он переходит в акролеин).
1. Разлейте глицерин по аликвотам.
2. Храните глицерин в темноте.
28.14.2. Изменение внешнего вида
культуры после криоконсервации
Ошибочная идентификация
1. Проверьте маркировку ампулы. Если маркировка
неразборчива, уничтожьте ампулу.
2. Проверьте записи (см. разд. 20.3.6).
3. Проверьте аутентификацию (см. разд. 16.3).
Изменились условия с тех пор, как клетки
растились последний раз
1) Появились новые члены лаборатории?
2) Появились новые методы работы?
3) Изменились поставщики сред?
4) Начали использовать новую партию сред?
28.16. Подсчет клеток
569
28.14.3. Контаминация
Негерметичность ампул
Проверьте герметичность: крышка должна быть плот¬
но закрыта, уплотняющая прокладка не должна быть
деформирована.
Контаминация из водяной бани при
размораживании
1. Не заливайте ампулу водой полностью, поместите
ее в поднос только частично погруженной в воду.
2. Размораживайте ампулы в подогреваемой емкости.
3. Тщательно протирайте ампулы после разморажи¬
вания.
• Требование безопасности. Помните, что ам¬
пулы могут взрываться при хранении в жидкой фазе
азота, поэтому при размораживании помещайте ее в
поддон и закрывайте сверху.
Контаминация из жидкого азота
Не погружайте ампулы в жидкий азот полностью. Ис¬
пользуйте для хранения фазу, содержащую пары азота
или перфузируемый кожух морозильника.
28.14.4. Утрата рабочей культуры
Утрата рабочей культуры пользователем
Получите новую рабочую культуру от распределителя
культур (дистрибьютера).
Утрата культуры у дистрибьютера
Вновь нарастите рабочую культуру из исходной куль¬
туры.
Утрата исходной культуры
1. Проверьте надежность контроля за хранением; он
должен проводиться только заведующим.
2. Восстановите запасы из надежного клеточного банка
(см. Приложение II Клеточные банки).
28.15. ГРАНУЛЯЦИЯ КЛЕТОК
Внутриклеточная
Клетки нездоровы?
1. Проверьте кривую роста (см. протоколы 20.7 и 20.8).
2. Проверьте эффективность посева (см. протокол 20.9).
Имеет ли место высокий уровень фагоцитоза?
Проверьте уровень захвата нейтрального красного или
флюоресцентного декстрана (FITC-dextran, Sigma)
при помощи фазового микроскопа при большом уве¬
личении.
Внеклеточная
Единообразный размер частиц
Проверьте на предмет контаминации (см. разд. 19.3).
Различный размер частиц
1. Проверьте преципитацию среды (см. разд. 28.2,
28.2.2).
2. Нет ли преципитации из сыворотки (обычно не
опасной)?
28.16. ПОДСЧЕТ КЛЕТОК
28.16.1. Гемоцитометр
Различные значения при подсчете
Ошибка взятия образца
1. Тщательно перемешивайте клеточную суспензию
перед взятием образца.
2. Клетки должны быть суспендированы до единич¬
ных клеток и не образовывать сгустков (см. прото¬
кол 21.1).
Использование гемоцитометра
1. Убедитесь, что покровное стекло прилегает пра¬
вильно (видны интерференционные кольца; см.
разд. 21.1.1.).
2. Убедитесь, что счетная камера заполнена полно¬
стью, но не переполнена.
3. Достаточным считается количество клеток >200.
Различимость клеток
1. Серебрение в счетной камере не должно повреждать
клетки.
2. Используйте фазово-контрастную оптику.
3. Если фазово-контрастная оптика недоступна, ис¬
пользуйте нецентрованный световой пучок.
4. Исследуйте окраску клеток до их подсчета.
28.16.2. Электронный счетчик
клеток посредством
измерения сопротивления
при пропускании
через дроссель
Вариабельность подсчета
Ошибки взятия образца
1. Тщательно перемешивайте клеточную суспензию
перед взятием образца.
2. Клетки должны быть суспендированы до единич¬
ных клеток и не образовывать сгустков (см. прото¬
кол 21.2).
Подсчет клеток останавливается,
не начинается или идет медленно
(более 25 с)
Забит дроссель
или клеточный канал
1. Проведите промывочный цикл.
2. Проведите цикл промывания дроссельного отвер¬
стия.
570
ГЛАВА 28. РЕШЕНИЕ ПРОБЛЕМ
3. Протрите дроссель кончиком пальца или мягкой
кисточкой.
4. Замочите в детергенте на 1 -18 ч и повторите проце¬
дуру промывания дросселя и трижды промывочный
цикл всего счетчика.
Значения при подсчете ниже, чем ожидалось,
или дроссель часто забивается
Клеточная суспензия агрегирует
1. Энергично диспергируйте клетки пипетированием
исходного образца, разведите образец и продолжите
подсчет.
2. Используйте различные методы дезагрегации (см.
табл. 13.4).
Фоновые значения высокие,
но подсчет клеток не ведется
Электрод не помещен в стакан или отсоединен
1. Поместите электрод в стакан.
2. Проверьте присоединение электрода.
3. Замените электрод, если отсутствует плата с клем¬
мами.
Цикл подсчета не начинается
Забит дроссель
1. Проведите промывочный цикл.
2. Проведите цикл промывания дроссельного отверстия.
3. Протрите дроссель кончиком пальца или мягкой
кисточкой.
4. Замочите в детергенте на 1-18 ч и повторите проце¬
дуру промывания дросселя и трижды промывочный
цикл всего счетчика.
Недостаточно отрицательное давление
1. Проверьте заполненность емкости для слива отходов.
2. Проверьте насос (см. Инструкция, диагностический
тест) и соединения.
3. Если обнаружены поломки в насосе, вызовите ин¬
женера.
Высокий фон подсчета
Проблемы, связанные с интерференцией
от электрических проводов или радиоволн
Существуют проблемы, связанные
с электрооборудованием (двигатели,
флюоресцентные лампы инкубаторы)
1. Проверьте и удалите возможные источники пробле¬
мы путем установки гасителей шумов на оборудова¬
ние. Можно приобрести линейные фильтры, однако
они не всегда эффективны.
2. Проверьте заземление счетчиков, в том числе кор¬
пуса и установочной станины.
Твердые примеси в жидкости для подсчета
1. Приготовьте свежую жидкость.
2. Профильтруйте жидкость через одноразовый
фильтр Millex или аналогичный фильтр.
28.17. ЖИЗНЕСПОСОБНОСТЬ
28.17.1. Морфологические признаки
Грануляция и вакуолизация
(см. также разд. 28.15)
Внутриклеточная грануляция обычно связана с тем,
что клетки нездоровы. Вакуолизация обычно также
показывает, что клетки нездоровы (см. рис. 13.1).
Утрата двойного лучепреломления
Монослойные клетки
Если клетки в норме обладают двойным лучепреломле¬
нием (на краях клетки в фазово-контрастном микроско¬
пе обнаруживается гало), то утрата его обычно связана
с последующей потерей жизнеспособности, например
при высыхании в ходе смены среды в культуре.
Суспензионные клетки
Суспензионные культуры клеток обычно чистые и
прозрачные; грануляция или мутность свидетельствует
о гибели или ухудшении состояния клеток.
1. Поменяйте среду. Монослойные клетки всплывут,
если они нежизнеспособны и таким образом не пот¬
ребуется специальных процедур для их удаления.
2. В случае суспензионных или первично дезагрегиро¬
ванных клеток, обогащение жизнеспособных клеток
производится путем осаждения из на Ficoll-Paque
(см. протокол 12.10).
28.17.2. Определение жизнеспособности
Если имеется промежуточная утрата клеток, связанная
с манипуляциями, проверьте их жизнеспособность ме¬
тодом исключения красителя (см. протокол 22.1) или
включения красителя (см. протокол 22.2) постарайтесь
удалить причину.
а) Жизнеспособность первичных культур обычно
составляет 50-90%.
б) Жизнеспособность клеточных линий обычно
90-100%.
в) Жизнеспособность размороженных клеток обыч¬
но 50-80%.
28.17.3. Цитотоксичность
Если ухудшение состояния культуры продолжается
длительное время, попробуйте обнаружить возможную
причину методом клоногенного исследования (см. про¬
токол 22.3) или микротитрации (см. протокол 22.4).
Глава 29
ЗАКЛЮЧЕНИЕ
Моим намерением было на страницах этого руковод¬
ства описать достаточно подробно основы клеточной
культуры, с обращением к литературе, которая может
потребоваться, только если исследователь решит рас¬
ширить спектр выполняемой работы сверх обычных
методов или прояснить некоторые дополнительные
детали основных приемов и методов. Исходя из опыта
при чтении лекций, в заключение цикла дается неко¬
торое резюме, которое подчеркивает основные пункты,
на которых останавливался лектор. Сейчас я хотел бы
сделать заключение вышеприведенному тексту.
Существуют определенные требования, которые
являются принципиальными для успешной и вос¬
производимой клеточной культуры, они могут быть
определены следующим образом:
• Удостоверьтесь, что ваши инструкции и подготовка
всех, кто работает с вами, происходят из заслужива¬
ющих доверия источников.
• Работайте в чистых помещениях, не перегруженных
оборудованием и сотрудниками, используя для
работы асептические зоны, которые следует обра¬
батывать по окончании работы.
• Для того чтобы избежать переноса контаминации,
включая перекрестную, не делите ни с кем среды,
реактивы, культуры или материалы.
• Не думайте, что ваша работа не подвержена дей¬
ствию микоплазмы, даже если вы никогда не наблю¬
дали ее; регулярно поводите проверку.
• Ведите адекватные записи, в частности отмечай¬
те все изменения в методах, средах, реактивах и
фотографируйте клеточные линии, которые вы
используете.
• Работайте только с теми клеточными линиями, ко¬
торые были получены из соответствующим образом
сертифицированных источников, таких как Меж¬
дународный банк клеток. Не доверяйте остальным
клеточным линиям, даже происходящим от автора,
если только вы не можете подтвердить аутентич¬
ность клеточной линии.
• Как можно более детально ознакомьтесь со свойства¬
ми клеточной линии, которую вы используете — внеш¬
ние признаки, скорость роста и особые характеристи¬
ки — так, чтобы вы могли немедленно отреагировать
на любые изменения, происходящие в культуре.
• Убедитесь, что ваша работа не угрожает вашей соб¬
ственной безопасности или безопасности сотрудни¬
ков, окружающих вас.
• Храните ростковые клеточные линии в жидком
азоте и регулярно заменяйте рабочие линии.
• Защищайте исходные культуры жизнеспособных
клеточных линий и используйте для передачи и рас¬
пределения культуры другие рабочие культуры.
• Старайтесь работать в таких условиях, которые в
точности определены, включая минимальное ис¬
пользование неопределенных добавок к среде, не
вносите никаких изменений в методики по незна¬
чительным поводам.
• Не смешивайте работу с клеточными культурами с
микробиологическими работами
Для того чтобы рассмотреть все увлекательные ас¬
пекты клеточных и тканевых культур, потребовалось
бы написать много томов и изменить цель написания
данной книги. Предоставление всей информации,
достаточной для создания и налаживания работы ла¬
боратории культуры тканей, превышало мои задачи.
Для этого потребовалось бы описать подготовку необ¬
ходимых материалов, разработку некоторых наиболее
важных методов, необходимых для характеристики и
понимания свойств ваших клеточных линий. Эта книга
сама по себе для вас, возможно, окажется недостаточно
полной, но с помощью ваших сотрудников и коллег
из других лабораторий она облегчит вам вхождение в
работу с культурой тканей и сделает ее более приятной
и приносящей удовлетворение.
Приложение I
ПРИГОТОВЛЕНИЕ РЕАКТИВОВ
Данное Приложение содержит список реактивов, ко¬
торые используются в нескольких различных прото¬
колах, использованных в книге. Специализированные
реактивы, используемые только в одном протоколе,
в основном расположены в разделе Материалы этого
протокола
Замечание. Разведения, которые упоминаются
в тексте, 1:10 или 1:100, являются объемными (v/v),
и подразумевается, что конечный объем составляет
10 или 100 частей соответственно.
Агар 2,5%
Агар 2,5 г
UPW 100 мл
1) Кипятить до растворения агара
2) Стерилизовать автоклавированием или кипячением
в течение 2 мин
3) Хранить при комнатной температуре
Амидо Черный
См. Нафтпаленовый черный
Аминокислоты, главные
См. разд. 9.4.1 и табл. 9.3. Предлагаются в виде кон¬
центрата 50х в 0,1 М НС1 такими компаниями, как
Invitrogen, МР Biomedicals, и Sigma.
1) Приготовьте тирозин и триптофан вместе в концент¬
рации 50х в 0,1 М НС1 и оставшиеся аминокислоты
при IOOxbUPW.
2) Развести для использования, как описано в прото¬
коле 11.10.
3) Стерилизовать фильтрацией.
4) Хранить в темноте при 4 °С.
Аминокислоты, второстепенные
Ингредиенты г/л (100х)
L-аланин » 0,89
L-аспарагин Н20 1,50
L-аспарагиновая кислота 1,33
Глицин 0,75
L-глютаминовая кислота 1,47
L-пролин 1,15
L-серин 1,05
Вода 1000 мл
2) Хранить при 4 °С.
3) Использовать при концентрации 1:100.
Антибиотики
См. отдельные заголовки Оксициллин, Стрептомицина
сульфат, Канамицина сульфат, Гентамицин, Мико-
статин.
Бактопептон, 5%
Difco Bactopeptone 5 г
Раствор Хэнкса BSS 100 мл
1) Перемешивать до растворения
2) Разлить по аликвотам в соответствии с использова¬
нием как раствор 1:50, автоклавировать.
3) Хранить при комнатной температуре.
4) Развести 1/10 перед использованием
Буферный раствор Mcllvaines, pH 5,5
На 20 мл На 100 мл
0,2 М Na2HP04 (28,4 г/л) 11,37 мл 56,85 мл
0,1 М лимонной кислоты 8,63 мл 43,15 мл
(21,0 г/л)
Версен
См. EDTA.
Витамины
Подробно представлены в описаниях сред (см. табл. 9.3,
10.1,10.2) и поставляются в концентрациях 100х.
1) Приготавливается индивидуально как основной рас¬
твор 1000-10,ОООх и соединяется по необходимости
с другими концентратами 100х.
2) Стерилизовать фильтрацией.
3) Хранить при -20 °С в темноте.
Гентамицин
Развести до 50 мкг/мл перед использованием.
Гимза краситель
Краситель Гимза можно приобрести в сухом виде и
затем развести его в буферном растворе или воде (см.
протокол 16.2), либо развести в буферном растворе
непосредственно перед использованием. Автор находит
более удачным для культуры клеток первый метод.
1) Стерилизация фильтрацией.
1) Приготовление буфера:
Приложение 1. ПРИГОТОВЛЕНИЕ РЕАКТИВОВ
573
NaH2HP04 • 2Н20 0,01 М 1,38 г/л
Na2HP04*7H20 0,01 М 2,68 г/л
Доведите pH до 6,5.
2) Разведите концентрат красителя Гимза 1:10 в 100 мл
буфера.
3) Профильтруйте раствор через бумажный фильтр
Whatman No. 1.
4) Каждый раз готовьте свежий раствор, поскольку
концентрат дает преципитацию при хранении.
Глюкоза, 20%
Глюкоза 20 г
Раствор Хэнкса до 100 мл
1) Стерилизовать автоклавированием.
2) Хранить при комнатной температуре.
Глютамин, 200 мМ
L-глютамин 29,2 г
Раствор Хэнкса 1000 мл
1) Разведите глютамин в BSS, стерилизуйте фильтра¬
цией (см. протоколы 11.12,11.13).
2) Разлейте по аликвотами и храните при -20 °С.
Глютатион
1) Приготовьте ЮОх раствор (т. е. 0,1 М в HBSS или D-
PBSA), и разведите до 1 мМ перед использованием.
2) Стерилизовать фильтрацией.
3) Разлить по аликвотам и хранить при -20 °С.
Дексаметазон (Merck)
1 мг/мл (ЮОх)
Этот реактив поступает уже стерильным в стеклянных
флаконах.
1) Для разведения добавьте 5 мл воды шприцом во
флакон.
2) После растворения отберите шприцом.
3) Разведите до 1 мг/мл, примерно 2,5 мМ.
4) Разлейте по аликвотам и храните при -20 °С.
5) Для использования разведите раствор до получения
10-50 нМ (диапазон физиологической концентра¬
ции), 0,1-1,0 мкМ (диапазон фармакологических
доз) или 25-100 мкМ (диапазон высоких доз).
Р-метазон (Glaxo) и метилпреднизолон (Sigma) могут
быть приготовлены таким же образом
Забуференный глицерин (для заключения)
См. Микоплазма
Канамицина сульфат (канназин), 10 мг/мл
Канамицин 4,1-г флаконы
BSS Хэнкса 400 мл
1) Добавить 5 мл HBSS из 400-мл бутыли HBSS в
каждый флакон.
2) Оставить на несколько минут для растворения.
3) Отобрать HBSS и канамицин из флаконов и вернуть
раствор в сосуд с HBSS.
4) Добавить еще 5 мл HBSS в каждый флакон, прополос¬
кать и вернуть в бутыль с BSS. Хорошо перемешать.
5) Разлить раствор по 20-мл аликвотам в стерильные
контейнеры и хранить при -20 °С.
6) Тест на стерильность: добавить 2 мл реактива к
10 мл стерильной среды без антибиотиков, инкуби¬
ровать раствор при 37 °С в течение 72 ч.
7) Использовать при концентрации 100 мкг/мл.
Карбоксиметилцеллюлоза КМЦ (СМС)
1) Взвесить 4 г КМЦ и поместить в химический стакан.
2) Добавить 90 мл солевого раствора Хэнкса и перенес¬
ти смесь в кипящую баню, чтобы увлажнить КМЦ.
3) Оставить на ночь при 4 °С для отстаивания.
4) Довести объем до 100 мл раствором Хэнкса.
5) Стерилизовать автоклавированием. КМЦ снова крис¬
таллизуется, но может быть растворена при 4 °С.
6) Для использования (например, для повышения
вязкости среды в суспензионной культуре), исполь¬
зуется 3 мл на 100 мл ростовой среды.
Коллагеназа
2000 Ед /мл в растворе Хэнкса
Коллагеназа Worthington CLS-степени
очистки или эквивалентной
(активность 1500-2000 Ед/мг) 100 000 Ед
Раствор Хэнкса 50 мл
1) Для растворения смеси перемешивать 37 °С в тече¬
ние 2 ч или при 4 °С в течение ночи.
2) Стерилизовать фильтрацией как сыворотку (см.
протокол 11.15).
3) Разделить на аликвоты, так чтобы использовать в
течение 1-2 недель.
4) Хранить при -20°С.
Коллагеназа-трипсин-сыворотка цыпленка
(СТС) [Coon &Cahn, 1966]
Стерильное Объем Конечная
вещество концентрация
Раствор солей без Са2+ и Mg2+-
[Moscona, 1952]
Трипсин, 2,5%, 4 мл 0,1%
Коллагеназа, 1 %, 10 мл 0,1 %
Сыворотка цыпленка 1мл 1,0%
Разлить на аликвоты и хранить при -20 °С.
Колцемид
Колцемид, концентрат ЮОх 100 мг
Раствор Хэнкса 100 мл
1) Перемешивать до растворения.
2) Стерилизовать фильтрацией.
3) Разлить на аликвоты и хранить при -20 °С
• Требование безопасности. Кольцемид токси¬
чен; все манипуляции проводить в вытяжном шкафу в
резиновых перчатках
574
Приложение 1. ПРИГОТОВЛЕНИЕ РЕАКТИВОВ
Красители для оценки жизнеспособности
См. Нафталеновый черный
Трипановый синий поставляется большинством
компаний-поставщиков материалов для культуры
тканей (см. Приложение II).
Кристаллвиолет 0,1%, в 0,1 М лимонной
кислоте
Лимонная кислота 21,0 г
Кристаллвиолет 1,0 г
1) Довести до 1000 мл деионизированной водой.
2) Перемешивать до растворения.
3) Для осветления профильтруйте раствор через филь¬
тровальную бумагу Whatman No. 1.
Кристаллвиолет 0,1% в воде
Кристаллвиолет 100 мг
Вода 100 мл
Фильтровать через бумажный фильтр Whatman No. 1
перед использованием.
Поставляется в готовом виде компанией Merck.
Лактальбумина гидролизат 5% (10х)
Лактальбумина гидролизат 5 г
HBSS 100 мл
1) Подогреть до растворения.
2) Стерилизовать автоклавированием.
3) Использовать в концентрации 0,5%.
Лимонная кислота/кристаллвиолет
См. Кристаллвиолет
МЕМ
См. разд. 9.4 и табл. 9.3.
2-меркаптоэтанол (MW 78)
Основной раствор, 5 мМ 4 мкл
HBSS 10 мл
1) Стерилизовать фильтрацией в вытяжном шкафу.
2) Хранить при -20 °С или каждый раз готовить све¬
жий раствор.
Метоцель (Methocel).
См. Метпилцеллюлоза.
Метилцеллюлоза (1,8%)
1) Взвесить 7,2 г Methocel и добавить в 500-мл бутыль,
содержащую большой вкладыш для магнитной ме¬
шалки.
2) Стерилизовать автоклавированием с ослабленной
крышкой для проникновения пара.
3) Добавить 400 мл стерильной UPW, подогретой до
90 °С для увлажнения Methocel.
4) Перемешивать при 4 °С в течение ночи до рас¬
творения (Methocel будет образовывать плотный
гель, если не перемешивать с помощью мешалки).
В результате получается раствор Methocel 2х, для
использования он должен быть разведен равным
объемом среды 2х по вашему выбору. Более точно
можно набрать объем Methocel, если использовать
шприц без иглы.
5) Перед использованием добавьте клеточную сус¬
пензию в малый объем ростовой среды (см. прото¬
кол 14.5).
Митомицин С
Основной раствор, 10 мкг/мл (50х)
Митомицин 2-мг флакон
1) Внести 20 мл HBSS в стерильный контейнер.
2) Отобрать 2 мл HBSS шприцом и добавить во флакон
с митомицином.
3) Оставить до растворения. Отобрать получившийся
раствор и вернуть обратно в контейнер с HBSS.
4) Хранить в течение 1 недели при 4 °С в темноте (за¬
крыть контейнер алюминиевой фольгой).
5) Для более продолжительного хранения хранить
при-20 °С.
6) Развести до 0,25 мкг/мл для 18- часовой экспозиции
клеток или 20 мкг/мл для 10-часовой экспозиции
(см. протоколы 14.3, 23.4).
• Требование безопасности. Митомицин яв¬
ляется токсичным, все манипуляции с порошкообраз¬
ным митомицином следует производить в вытяжном
шкафу.
мтт
1) Растворить 3-(4,5-диметилтиазолин-2-ил)-2,5-дифе-
нилтетразолия бромид (МТТ) 50 мг/мл в D-PBSA.
2) Стерилизация фильтрацией.
• Требование безопасности. МТТ токсичен;
взвешивать в вытяжном шкафу в перчатках.
Микоплаза, реактивы
Красители (см. Хехст 33258)
Гистологическая среда: глицерин в буфере Mcllvaines
pH 5,5
Для приготовления 40 мл
0,4 М Na2HP04 (5,8 г/л) 11,37 мл
0,2 М Лимонной кислоты (42,0 г/л) 8,63 мл
Глицерин 20,00 мл
1) Добавить Vectashield (Vector) чтобы уменьшить
затухание флюоресценции (см. Инструкцию про¬
изводителя).
2) Проверить pH и довести до 5,5.
Микостатин (Нистатин)
1) Приготовить раствор (100х) с концентрацией 2 мг/мл.
Микостатин 200 мг
Раствор Хэнкса BSS 100 мл
Приложение 1. ПРИГОТОВЛЕНИЕ РЕАКТИВОВ
575
2) Далее по той же методике, что и приготовление
канамицина (см.).
3) Конечная концентрация 20 мкг/мл.
Нафталеновый черный
1) Приготовить раствор 1% в BSS Хэнкса.
Нафталеновый черный 1 g
Раствор Хэнкса BSS 100 мл
2) Максимально растворить краситель.
3) Профильтровать насыщенный раствор через бумаж¬
ный фильтр Whatman No. 1.
Пеногаситель
RD эмульсия 9964,40 (см. Приложение II)
1) Разлить по аликвотам и стерилизовать автоклави¬
рованием.
2) Хранить при комнатной температуре.
3) Развести 0,1мл/л (1:10 ООО).
Пенициллин
(например Crystapen benzylpenicillin [sodium])
1000 000 Ед во флаконе
1) Приготовление как для канамицина, конечная кон¬
центрация 10 000 U/мл.
Crystapen 4 флакона (4 х 106Ед)
HBSS 400 мл
2)Хранить замороженным при -20 °С в аликвотах по
5-10 мл.
3) Использовать при концентрации 50-100 Ед/мл.
Перколл (Percoll)
1) Percoll поставляется стерильным и готовым к упот¬
реблению, он должен быть разведен средой или
HBSS до получения необходимой плотности.
2) Проверьте осмотические свойства. Для доведения
до 290 мОсм/кг потребуется, чтобы разводящая
жидкость была гипо- или гипертонической, поэтому
лучше развести сначала небольшое количество и
проверить осмоляльность и затем рассчитать для
большего объема.
Питательная среда жидкая
См. Инструкцию производителя для приготовления.
См. также Бактопептон и триптозо-фосфатный
бульон.
Стерилизовать автоклавированием
Раствор Хэма F12
См. табл. 9.3.
Раствор HBSS
См разд. 9.3 и Сбалансированный солевой раствор в
данном Приложении.
Раствор Тайрода (Tyrode’s Solution)
NaCl 8,00 г
КС1 0,20 г
CaCl2 0,20 г
Mg2Cl26H20 0,10 г
NaH2P04 Н20 0,05 г
Глюкоза 1,00 г
UPW до 1л
Газовая фаза Воздух
Раствор Хекста (Hoechst) 33258 [Chen, 1977]
2-[2-(4-гидроксифенол)-6-бензимидазоил]-6-(1-метил-
4-пиперазил) бензимидазола тригидрохлорид
1) Приготовить основной раствор 1 мг/мл в D-PBSA
или HBSS без фенолового красного.
2) Хранить раствор при -20 °С. Перед использованием
развести 1:20000 (1,0 мкл в 20 мл) в D-PBSA или
HBSS без фенолового красного при pH 7,0.
• Требование безопасности. Поскольку данное
вещество может быть канцерогенно, все манипуляции
с ним проводите в вытяжном шкафу.
Сбалансированный солевой раствор (BSS)
См. табл. 9.2.
1) Развести каждый компонент отдельно, последним
добавить СаС12.
2) Довести до 1 л.
3) Довести pH до 6,5.
4) Стерилизовать раствор автоклавированием или
фильтрацией. При автоклавировании pH должен
оставаться ниже 6,5, чтобы предупредить преципи¬
тацию фосфата кальция; кальций можно исключить
и добавить позже. Если в состав входит глюкоза,
раствор следует стерилизовать фильтрацией, чтобы
избежать карамелизации глюкозы, либо глюкозу
следует автоклавировать отдельно (см. Глюкоза,
20%, в настоящем Приложении) при высокой
концентрации (например, 20%) и добавлять позже.
Перед автоклавированием отметьте уровень жид¬
кости. Хранить раствор при комнатной температу¬
ре, и если произошло испарение, довести раствор
до метки стерильной UPW перед использованием.
При использовании боросиликатного стекла бу¬
тыль может быть плотно запечатана, в этом случае
испарения не будет.
Сбалансированный солевой раствор Гея
NaCl 7,00 г
КС1 0,37 г
СаС12 0,17 г
MgCl2-6H20 0,21 г
MgS04 • 7 Н20 0,07 г
Na2HP04 • 12 Н20 0,30 г
КН2Р04 0,03 г
NaHC03 2,27 г
Глюкоза 1,00 г
576
Приложение 1. ПРИГОТОВЛЕНИЕ РЕАКТИВОВ
Вода до 1000 мл
С02 5%
Сбалансированный солевой раствор
для иссечения тканей (DBSS)
1) К раствору Хэнкса BSS без бикарбоната, стерилизо¬
ванного автоклавированием, добавить стерильные
растворы:
Пенициллин 250 ед/мл
Стрептомицин 250 мкг/мл
Канамицин 100 мкг/мл
или
Гентамицин 50 мкг/мл
Амфотерицин Б 2,5 цмкг/мл
2) Хранить при -20 °С.
Сбалансированный солевой раствор Хэнкса
(BSS)
См. разд. 9.3 и сбалансированный солевой раствор, в
данном приложении.
Сбалансированный солевой
раствор Хэнкса без фенолового
красного:
Следуйте предыдущей инструкции, но не добавляйте
феноловый красный.
Среды
Основные компоненты многих сред общие, наиболее
часто используемые приведены в главах 9 и 10 (см.
табл. 9.3,10.1, и 10.2), вместе с рекомендуемыми мето¬
дами приготовления. Для тех сред, которые не описаны,
см. Morton [1970], или каталоги фирм-поставщиков
сред (см. Приложение II: Среды).
Среда для биопсийных образцов
Ростовая среда 500 мл
Пенициллин 125 000 ед
Стрептомицин 125 мг
Канамицин 50 мг или
Гентамицин 25 мг
Амфотерицин 1,25 мг
Хранить при 4 °С до 3 недель или при -20 °С в течение
более длительного периода.
Среда HAT
Вещество Концентрация Развести в Молярностъ
(конечный
раствор
ЮОх)
Гипоксантин 136 мг/100 мл 0,05 N НС1 1 х 10"2 М
(Н)
Аминопте- 1,76 мг/100 мл 0,1 N NaOH 4 х 10_5М
рин (А)
Тимидин (Т) 38,7 мг/100 мл HBSS 1,6 х 10~3М
1) Для использования перемешайте равные объемы
трех веществ, стерилизуйте фильтрацией и добавьте
смесь в питательную среду как 3% (v/v).
2) Хранить Н и Т при 4 °С, А при -20 °С.
Среда НВ
К среде CMRL 1066 добавить
Инсулин 5 мкг/мл
Гидрокортизон 0,36 мкг/мл
Р-ретинилацетат 0,1 мкг/мл
Глютамин 1,17 мМ
Пенициллин 50 Ед/мл
Стрептомицин 50 мкг/мл
Гентамицин 50 мкг/мл
Фунгизон 1,0 мкг/мл
FBS 1%
Среда SF12
Среда Хэма F12 (см. табл. 9.3) с добавлением среды
Игла МЕМ, главных аминокислот (2х) и второстепен¬
ных аминокислот (1х) без тимидина, но с фолиевой
кислотой 10х.
Стрептомицин
1) Возьмите 2 мл из бутылки стерильного HBSS
100 мл, добавьте в 1-г флакон стрептомицина.
2) Когда стрептомицин растворится, верните обратно
в бутыль с 98 мл HBSS.
3) Разведите 1:200 для использования. Конечная кон¬
центрация должна составлять 50 мкг/мл.
Трипсин
2,5% (w/v) в 0,85% (0,14 М) NaCl
Раствор трипсина можно приобрести уже готовым к
употреблению. Для самостоятельного приготовления:
1) Приготовьте 2,5% раствор:
Трипсин (например Difco, 1:250) 25 г
NaCl, 0,85% 1л
2) Размешивайте в течение 1 ч при комнатной темпера¬
туре или 10 ч при 4 °С. Если трипсин не разводится
полностью, профильтруйте через бумажный фильтр
Whatman No. 1.
3) Стерилизуйте фильтрацией.
4) Раазлейте по 10-20-мл аликвотам и храните при
-20 °С.
5) Разморозить и развести 1:10 в D-PBSA или РЕ перед
использованием.
6) Хранить разведенный трипсин при 4 °С не более
3 недель.
Замечание. Трипсин может иметь грубую степень
очистки (например, Difco 1:250) или быть очищенным
(Worthington или Sigma Зх перекристаллизованный).
Грубо очищенный трипсин содержит другие протеазы,
которые могут быть важны для клеточной диссоциа¬
Приложение 1. ПРИГОТОВЛЕНИЕ РЕАКТИВОВ
577
ции, но также могут повреждать более чувствительные
клетки. В обычной практике используется грубо очи¬
щенный трипсин, если только не отмечается снижение
жизнеспособности и ухудшение состояния клеток либо
снижение клеточного роста. В этом случае используется
очищенный трипсин, который имеет высокоспецифич¬
ную активность и должен использоваться при пропор¬
ционально более низких концентрациях (например,
0,01 или 0,05%). При использовании грубо очищенного
трипсина, проверяйте на микоплазмы.
Трипсин/EDTA
См. Трипсин, шаг (5)
Трипсин, Версен, Фосфат (TVP)
Трипсин (Difco 1:250) 25 мг (или 1 мл 2,5%)
Фосфатно-буферный раствор, D-PBSA 98 мл
Версен (Двунатриевая соль EDTA (2Н20) 37 мг
Сыворотка цыпленка (MP Biomedicals) 1 мл
1) Смешать D-PBSA и EDTA, автоклавировать смесь,
и хранить при комнатной температуре.
2) Добавить сыворотку цыпленка и трипсин перед
использованием. Если используется сухой трипсин,
стерилизовать фильтрацией перед добавлением к
сыворотке.
3) Разлить по аликвотам и хранить при -20 °С.
Три птозофосфатный бульон
1) Приготовить 10% раствор в HBSS
Триптозы фосфат (Difco) 100 г
Среда Хэнкса 1000 мл
2) Перемешивать до растворения.
3) Разлить по аликвотам по 100 мл и стерилизовать
автоклавированием.
4) Хранить при комнатной температуре.
5) Развести 1:100 (конечная концентрация 0,1%) перед
использованием.
Уксусная кислота/метанол
Добавить 1 часть ледяной уксусной кислоты к 3 частям
метанола. Готовится непосредственно перед употреб¬
лением, хранится на льду.
Фикколл, 20%
1) Высыпать 20 г Ficoll (GE Heathcare) на поверхность
80 мл UPW.
2) Оставить на ночь так, чтобы Ficoll осел и растворился.
3) Довести до 100 мл добавлением UPW.
4) Стерилизовать автоклавированием.
5) Хранить при комнатной температуре.
Фиксирующая жидкость для культуры тканей
См. Уксусная кислота/метанол.
Используйте чистый безводный этанол или метанол
(протокол 16.2), 10% формалин, 1% глютаровый аль¬
дегид или 5% параформальдегид.
Фитогемагглютинин (ФГА, PH А)
1) Приготовить основной раствор 500 мкг/мл (100х)
из лиофилизированной РНА добавлением HBSS в
ампулу шприцом.
2) Разлить по аликвотам и хранить при-20 °С.
3) Развести 1:100 для использования.
Фосфатно-буферный раствор (Раствор
Дюльбекко A; D-PBSA)
См. табл. 9.2 и протокол 11.6.
Фосфатно-солевой буфер Дюльбекко без Са2+
и Mg2+ (D-PBSA) (см. протокол 11.6)
Фосфатно-буферный раствор /EDTA, 10 мМ (РЕ)
1) Приготовить раствор D-PBSA.
2) Добавить EDTA двунитриевую соль, 3,72 г/л и пе¬
ремешать.
3) Разлить, автоклавировать и хранить при комнатной
температуре.
4) Развести D-PBSA/EDTA 1:10 для получения 1 мМ
(наиболее частое употребление) или 1:2 (5 мМ) для
высоко хелатообразующих условий (например,
трипсинизация клеток СаСо-2).
Цитрат натрия/Хлорид натрия (SSC) SSC 20х
1) Приготовить концентрированный раствор SSC:
Тринатриевый цитрат
(дигидратированный) 0,3 М 88,2 г
NaCl 3,0М 175,3 г.
Вода 1000 мл
2) Развести до 1х или 2х по обстоятельствам.
Экстракт эмбриона цыпленка
[Paul, 1975]
1) Выделите эмбрион из яиц, как описано в протоко¬
ле 12.2, и поместите эмбрионы в 9-см чашки Петри.
2) Удалите глаза, используя две пары стерильных
пинцетов.
3) Перенесите эмбрионы в плоскодонные или круг¬
лодонные 50-мл контейнеры по два эмбриона в
каждый контейнер.
4) Добавьте равный объем раствора Хэнкса в каждый
контейнер. Используя стерильную стеклянную
палочку, которая была предварительно раскалена в
пламени горелки и расплющена на конце, раздавли¬
вайте эмбрионы в BSS, пока они не диссоциируют.
5) Оставьте смесь стоять в течение 30 мин при комнат¬
ной температуре.
6) Центрифугируйте смесь в течение 15 мин при
2000 g.
7) Соберите супернатант и, после проверки на сте¬
рильность (см. разд. 11.6.2), разлейте по аликвотам.
Хранить при -20 °С.
Экстракт цыпленка и других тканей может также быть
приготовлен гомогенизацией в гомогенизаторе Поттера
или Уорена [Coon & Cahn, 1966].
578
Приложение 1. ПРИГОТОВЛЕНИЕ РЕАКТИВОВ
1) Гомогенизируйте измельченные эмбрионы с рав¬
ным объемом среды Хэнкса.
2) Перенесите гомогенат в центрифужные, центрифу¬
гируйте при 1000 g в течение 10 мин.
3) Перенесите супернатант в свежие пробирки и цен¬
трифугируйте еще 20 мин при 10000 g.
4) Проверьте на стерильность (см. разд. 11.6.2).
5) Разлейте на аликвоты.
6) Хранить при -20 °С.
ЭДТА (Versene)
1) Приготовьте концентрат 10 мМ, 0,374 г/л в D-
PBSA.
2) Стерилизуйте фильтрацией.
3) Разведите 1:10 или 1:5 для использования при
1,0-2,0 мМ или в исключительных случаях 1:2 для
использования при 5 мМ, разведенный в D-PBSA
или в растворе трипсина в D-PBSA.
ЭГТА
Готовится так же, как ЭДТА, но ЭГТА может быть ис¬
пользована при более высоких концентрациях, потому
что обладает меньшей токсичностью.
PBS
См. Фосфатно-буферный раствор.
D-PBSA
См. Фосфатно-буферный раствор.
РЕ
См. Фосфатно-буферный pacmeop/EDTA.
Приложение II
ИСТОЧНИКИ ОБОРУДОВАНИЯ
И МАТЕРИАЛОВ
В настоящее время существует большое количество компаний-производителей и поставщиков реактивов, обо¬
рудования и материалов, используемых в культуре клеток, поэтому представлены не все возможные источники,
приведены только некоторые поставщики этих продуктов. Адреса поставщиков можно найти в Приложении III.
Список других фирм-производителей представлен в книге BiosupplyNet Source Book, Cold Spring Harbor Laboratory
Press, а также на сайтах www.biosupplynet.com;www.biocompare.com; http //informagen.com/; http //www.martex.
co.uk/laboratorysupplies/index.htm; http //www.biosciencetechnology.com/; www.cato.com/biotech/bio-prod.html;
www.ispex.ca/naccbiologicals.html;www.biospace.com/service_and_supplier.cfm;www.sciquest.com; http //www.coe.
montana.edu/che/CompAlph.htm#A
Наименование материа¬
лов и оборудования
Авидин-FITC
Автоклав настольный,
скороварка
Автоклавируемые среды
Автоклавы
Автоматическая пипетка
Автоматическая пипетка
с репетиром
Автоматические диспен¬
серы
Автоматические пипетки
Gilson
Агар
Агар ЕМ, набор
Агароза
Агароза-мочевина, гель
Агент декальцинирую¬
щий RDO
Агент для уничтожения
микоплазм (MRA)
Поставщик
Vector Laboratories
Astell Scientific; Harvard
Apparatus; LTE; Napco;
Valley Forge
JRH Biosciences; Medi-
atech
Ace; Astell; Bennet; Glob¬
al; Integra; LTE; Preci¬
sion Scientific; SP Indus¬
tries; Steris; VWR
Alpha; BD Biosciences;
Roche Diagnostics;
Corning; Gilson; MP
Biomedicals; Jencons;
Labsystems; Rainin
Brand; Cole-Parmer; Pop¬
per; Radleys
Corning; Genetic Research
Instr.; Gilson; Jencons;
Matrix Technologies;
Michael Smith; MP
Biomedicals; Robbins
Scientific
Bellco; Corning; Gilson;
Schott; Wheaton
Invitrogen; BD Biosci¬
ences (Difco); Oxoid
Plano; Polysciences
Cambrex; FMC Bioprod¬
ucts; Sigma
Applied Biosystems
Apex Engineering
MP Biomedicals
Адаптеры Люэра
Аденин (гидрохлорид)
Аденинмонофосфат db-
cAMP
Адипоциты
Азот мочевины остаточ¬
ный. Реактивы
Азот мочевины, анализ
Аккутаза (Accutase),
Accumax
Акрихина дигидрохлорид
АланинL
Альбумин (БСА)
Альбумин бычий сыво¬
роточный (BSA)
Альбумин крысиный
Альгинат
Аминокислоты
Аминокислоты несуще¬
ственные
Аминокислоты основные
Аминопропилтриэток-
силан
Аминоптерин
Аминоэтанол
Ампулы «малышки*
Bijou
Ампулы, пластик
BD Biosciences (Clay Adams)
Sigma
Sigma
См. Специализированные
клетки
Stanbio Labs
Stanbio Labs
Sigma; TCS; Upstate Bio¬
technology
Sigma
См. Химические реактивы
Sigma
Amersham; Bayer; Bio-
Source; BD Biosciences;
Calbiochem; Cambrex
(Clonetics); MP Bio¬
medicals; Invitrogen;
Intergen; Sigma
MP Biomedicals
ISP; NovaMatrix
JT Baker; Merck; Sigma
Invitrogen; MP Biomedi¬
cals; Sigma
См. Основные аминокис¬
лоты MEM
Fluka
Sigma
См. Этаноламин
Bibby Sterilin
Alpha Laboratories; CLP;
Corning; Fisher Scientif¬
ic; Greiner; Nalge Nunc
580
Приложение II. ИСТОЧНИКИ ОБОРУДОВАНИЯ И МАТЕРИАЛОВ
Ампулы, стекло
Амфотерицин В (Фунги-
зон, Fungizone)
Анализ изображений
Анализ изоферментный
электрофоретический
Анализ изоферментный
электрофоретический,
набор
Анализ хромосом
Анализ цитогенетиче¬
ский
Анемометры
Антибиотики
Антитела CD31-PE
(555446)
Антитела CD34-APC
(555824)
Антитела CD44-FITC
(555478)
Антитела CD45-APC
(555485)
Антитела MUC-1 (му¬
цин)
Антитела р53: D0-1,
РАЬ421, РАЬ240
Антитела вторичные
FITC-конъюгированные
Антитела иммуноглобу¬
линовые кроличьи про¬
тив мыши
Антитела к актину альфа
гладких мышц
Антитела к актину альфа
гладкой мускулатуры
Антитела к альбумину,
кроличьи против кры¬
синого альбумина
Антитела к антигену эпи¬
телиальной мембраны
(ЕМА)
Антитела к виментину
Антитела к десмину мы¬
шиные
Wheaton, Triple Red
Cambrex (BioWhittaker);
Invitrogen; MP Biomed¬
icals; PAA; Sigma
Applied Imaging; Bio-Rad;
Carl Zeiss; Hamamatsu;
Imaging Associates; Im¬
aging Research; Leica;
Molecular Dynamics;
Nikon; Nonlinear Dy¬
namics Perceptive In¬
struments; PerkinElmer;
Scanalytics; Syngene
ATCC; Cell Culture Char¬
acterization Services;
DSMZ; ECACC
Innovative Chemistry
Cell Culture Characteriza¬
tion Services
Cell Culture Characteriza¬
tion Services
Technika; TSI; см. также
Ламинарный шкаф
см. Среды
BD Biosciences (Pharmin-
gen)
BD Biosciences (Pharmin-
gen)
BD Biosciences (Pharmin-
gen)
BD Biosciences (Pharmin-
gen)
Chemicon
Oncogene Science
См. Антитела
Dako Diagnostics
DAKO
DAKO
MP Biomedicals
Dako
Sigma
Sigma
Антитела к кератину 19
(K19)
Антитела к фактору VIII
Антитела к фактору ван
Виллебрандта (фак¬
тор VIII)
Антитела к цитокератину
Антитела козы Alexa
Fluor 488 к мышиному
IgG
Антитела козы тирози-
назные (С-19) против
человека
Антитела кролика про¬
тив человеческого кол¬
лагена I типа
Антитела
Апоптоза ингибиторы
Апротинин (Трасилол,
Trasylol)
Аргинин НС1
Аспарагин
Аспираторы (резервуар)
Ацепромазин
(Acepromazine)
Ацетилхолин (Acetyl¬
choline)
Ацетонитрил (Acetoni-
trile)
Бактерициды для водя¬
ной бани
Бактопептон BD
Баня водяная
Баня водяная качалоч-
ная
Баня для органов
Барабан роллерный
Белки костные морфоге¬
нетические (BMPs)
Dako
Roche Diagnostics
Roche Diagnostics
DAKO; Lab Vision; Santa
Cruz Biotechnology;
Zymed
Invitrogen; Molecular
Probes
Santa Cruz Biotechnology
Biodesign
Amersham; BD Biosci¬
ences; Biopool; DAKO;
Dianova; Invitrogen; MP
Biomedicals; Peprotech;
R&D Systems; Serotec;
Roche Diagnostics; San¬
ta Cruz Biotechnology;
Sigma; Upstate Biotech¬
nology; Vector
MP Biomedicals
Bayer; Serologicals; Sigma
См. Химические реактивы
См. Химические реактивы
Bel-Art; Bibby Sterilin;
Camlab; Coming; In¬
tegra; Kimble-Kontes;
Techmate
Henry Schein
Sigma
TAAB
Guest Medical; Henry
Schein
Biosciences (Difco); Invi¬
trogen
Camlab; Cole-Parmer;
Grant; Polyscience; Sci-
Gene; Thermo Electron
Baker; Grant
Radnoti
Genetic Research Instr.;
New Brunswick
См. Факторы роста
Приложение II. ИСТОЧНИКИ ОБОРУДОВАНИЯ И МАТЕРИАЛОВ
581
Белки, исследование
Белок костный морфоге¬
нетический ВМР-2
Бетадин
Библиотека кДНК
Биобезопасность, про¬
дукция для обеспече¬
ния
Биореактор HARV
Биореактор NASA
Биореакторы
Биореакторы с непод¬
вижным слоем
Биореакторы с псевдо¬
ожиженным слоем
Биосенсоры
Биотин
Блок переключения для
баллонов С02, автома¬
тический
Блок фильтров для
шприца МШех-SV, раз¬
мер пор 5 ед
Блоки биобезопасности
Бокс ламинарный биоло¬
гической защиты
Бокс микробиологичес¬
кой защиты
Бромдиоксиуридин
BUdR
Бульон питательный
Бульон триптозо-фос-
фатный
Бульоны
Бумага фильтровальная
для геля (секвениро-
вание)
Pierce
Wyeth
Bruce Medical
Clontech
Air Sea Atlanta; Altec;
Bel-Art; Cellutech; Cin-
Made; Jencons; Kim¬
berly-Clark; Lab safety
Supply; Saf-T-Pak
Synthecon
Cellon; Synthecon
Alfa Laval; Bellco; Biotech
Instruments; Biovest;
Braun; Cellon; Charles
River; Corning; Genetic
Research Instrumenta¬
tion; Global Medical;
James Glass; KBI; New
Brunswick Scientific;
PerkinElmer; Sartorius;
Sythecon; Vivascience;
Wave Biotech
New Brunswick
Amersham Biosciences
Analytical Technologies; Ap¬
plied Biophysics; CellStat;
Nova Biomedical; YSI
Sigma
Air Products and Chemi¬
cals Inc; Gow-Mac; Lab
Impex; Nuaire; Thermo-
Shandon
Millipore
Cm. Laminar-flow hoods
См. Бокс
микробиологической
защиты
Plas Labs; См. также
Ламинарные шкафы
Sigma; см. также Мече-
ниеДНК клеток BrdU,
набор
BD Biosciences (Difco);
Invitrogen; MP Biomed¬
icals; TCS
BD Biosciences (Difco);
Invitrogen; Oxoid
См. Питательные
бульоны
Bio-Rad
Бусины-маркеры для
центрифугирования в
градиенте плотности
Бутандиона моноксим
(BDM)
Бутыли из жароупорного
стекла
Бутыли
Буфер CHAPS
Буфер Weise
Буфер боратный
Буфер для образцов
Laemmli
Вакуумная консистен¬
тная смазка Edwards
High
Валидация клеточных
линий
Валин
Видеокамера
Видеомагнитофоны
Видеосистема для съем¬
ки в режиме замедлен¬
ного времени
Видео-система съемки в
реальном времени ABI
Prism 7000
Виллин
Винбластин
Витамин В12
Витамины
Витамины, стерильные
растворы
Вкладыши Transwell
Вкладыши в держатели
для фильтров Millicell-
НА
Вкладыши в фильтраци¬
онные лунки
Amersham Biosciences
Sigma
Camlab; Fisher; Schott
Applied Scientific; BD
Biosciences; Bel-Art;
Bellco; Bibby Sterilin;
Caisson; Camlab; Com¬
ing; Fisher; Integra;
Kimble Kontes; Nalge
Nunc; Polytech; Radleys;
Schott; Techmate; VWR
Calbiochem
Merck
Sigma
Bio-Rad
BOC Edwards
ATCC; BioReliance; Cell-
mark; ECACC; LGC
Promochem
См. Химические реактивы
См. CCD-камеры; камеры
цифровые; микроскопы
См. Видео в режиме
замедленного времени
Dage-MTI; Hamamatsu;
Imaging Associates;
см. CCD-камеры;
Видео камеры;
Видеомагнитофоны (см.
также протокол 27.4)
Applied Biosystems
Novacastra Laboratories;
Santa Cruz Biotechnol¬
ogy; Chemicon
Sigma
Sigma
Sigma; См. также Среды
См. Среды
Coming; см. также Вкла¬
дыши в фильтрацион¬
ную лунку
Millipore
BD Biosciences; Bibby
Sterilin; Coming; Integ¬
ra; Millipore; Nalge Nunc
582
Приложение II. ИСТОЧНИКИ ОБОРУДОВАНИЯ И МАТЕРИАЛОВ
Вкладыши для культуры
клеток
Вода для увлажнения
тканей
Вортекс-миксер
Впитывающие салфетки
Benchcote
Вращающаяся колба
Выделение РНК, набор
Газ углекислый С02
Г азогенераторы
Газопроницаемые крыш¬
ки
Газопроницаемые па¬
кеты для культуры
тканей
Газы
Гекса фтор-2-пропанол
(НИР)
Гексамеры рандомизи¬
рованные
Гели полиакриламидные
Гематоксилин
Гематоксилин Харриса
Гемоцитометр
Генетицин (geneticin)
Гентамицин
Гепарана сульфат
Гепарана сульфата про-
теогликаны (HSPGs)
Гепарин
Гепатоциты
Герметик для ткани,
фибриновый Tisseel
VH
Гиалуронизаза
Гибридизация флюорес¬
центная in situ (FISH)
Гидрогели
Гидрокортизон
Гидрокортизон и его
аналоги
См. Вкладыши в филь¬
трационные лунки
Baxter
Gallenkamp; Fisher; gen¬
eral
Whatman
См. Флаконы с
перемешиванием
Qiagen
Air Products; Cryoservice;
Taylor-Wharton
Bioquell; Peak Scientific;
Sartec; Texol
См. Сашки, флаконы,
планшеты для
культуры
См. Пакеты для сред или
пакеты для культур
Air Products; British
Oxygen; Cryoservice;
Matheson Gas; Messer;
T ay lor-Wharton
Aldrich
Invitrogen
Amersham Biosciences;
Bio-Rad
Merck; Sigma
Fisher
Fisher; Omnilab; Thermo
Electron
Invitrogen
Gemini; Invitrogen; Sigma
Sigma
Sigma
Roche; Sigma
См. Специализированные
клетки
Baxter
Sigma; Worthington
Cell Culture Characteriza¬
tion Services (реактивы
см. Красители хромо¬
сомные)
Nektar
Amersham Biosciences;
Merck; Sigma: Upjohn
Biosource; Merck; Sigma
Гидроксил амина гидро¬
хлорид
Гидроксисукцинимид
(NHS)
Гипоксантин
Гипоталамус быка
Гипофиз быка
Гипофиз быка, экстракт
Гипохлорита водный
раствор Clorox
Гипохлориты
Гистидин НС-1Н20
Гистологический нож
Глицерин
Глицерофосфат
Глицин
Глюкагон
Глюкоза
Глюкоза стерильная
Глютамин
Глютамин, стерильный
Глютаминовая кислота
Глютаровый альдегид
Глютатион
Гомогенизатор для мик¬
ротитрационных план¬
шетов
Гомогенизатор тканей
Гормоны
Гофрированный карт¬
ридж для фильтрации
Груша резиновая для пи-
петирования
Гуанидина гидрохлорид
8М
Датчик давления
Датчик изометрической
силы (для культуры
миоцитов)
Датчик кислорода
Дезинфектант Clidox
Дезинфектант Virkon
Дезинфектанты
Pierce
Pierce
Sigma
Pel-Freez
Pel-Freez
Cambrex (Clonetics);
Cascade; Hammond Cell
Technology; Invitrogen;
PromoCell
Polyscience (см. также
Дезинфектанты)
См. Дезинфектанты
См. Химические реактивы
Alabama Research & De¬
velopment
Fisher; Mallinckrodt;
Merck; Sigma; VWR
Invitrogen; Sigma
Sigma
Bedford Laboratories
См. Химические реактивы
См. Среды
См. Химические реактивы
См. Среды
См. Химические реактивы
Fisher Scientific; Plano;
Roth; Sigma
Sigma
См. Измельчитель Mini-
Bead Beater
Kimble-Kontes
Sigma
Pall Gelman
Bel Art; Bellco; Bibby
Sterilin; Cole-Parmer;
Fisher; Jencons Scien¬
tific; VWR
Pierce
Gould; Maxim Medica
Radnoti
Microelectrodes
Tecniplast (Indulab)
Du Pont Animal Health
Solutions; Scientific
Laboratory Supplies
Anachem; BioMedical
Products; DuPont Ani¬
mal Health; Guest
Приложение II. ИСТОЧНИКИ ОБОРУДОВАНИЯ И МАТЕРИАЛОВ
583
Деионизаторы
Деконтаминация (обо¬
рудования и систем
обслуживания)
Дексаметазон
Декстран
Декстран DEAE
Декстроза
Денситометр
Денситометры
Деоксирибонуклеаза
Держатели для фильтров
Дерматотом
Детергент Roccall
Детергенты
Диаминобензидин
Диатилфлюоресциин
Диметилметиленовый
синий
Диметилсульфоксид
(DMSO)
Диметилэтилформамид
Диоксид углерода
Дипептидилпептидаза IV
(CD26)
Диски для клонирова¬
ния, 3 мм
Диспаза
Диспенсеры для горло¬
вины бутыли
Диспенсеры для жид¬
костей
Medical; Hays Chemical
Distribution; Johnson
&Johnson; Lab Impex;
Lab Safety Supply;
Markson LabSales; MP
Biomedicals; National
Chemical; Polyscience;
Sigma; Steris; Tecniplast;
Thomas;
Bamstead Thermolyne;
Bellco; Corning; Dow
Corning; Elga; High-
Q; Millipore; Purite;
U.S. Filter; Vivendi Wa¬
ter Systems; VWR
Anachem; Steris; см. также
Sigma; Merck
Calbiochem; Fisher; Sigma
Amersham Biosciences;
Bio-Rad
Fisher
Mettler Toledo; Parr
Beckman Instruments; Pall
Gelman Sciences; Gilford
Instruments; Helena;
Molecular Dynamics
Sigma
См. Фильтры
Stortz Instruments
(см. также Нож
гистологический)
Henry Schein Rexodent
Alconox; Calbiochem; De-
con; MP Biomedicals;
Pierce
Sigma; Vector
Fisher; Sigma
Polysciences; Sigma
ATCC; JT Baker; Merck;
Sigma
См. Химические реактивы
Air Products; Cryoservice;
Messer; Taylor-Wharton
Biosource; Neomarkers
Sigma
BD Biosciences; Invitro¬
gen; Roche
Brand; Jencons; Fisher;
Polytech
Accuramatic; Barnstead;
Corning; Gilson;
Диссоциация клеток,
агенты
Дистилляторы
Дитиотреитол
Диэтилпирокарбонат
(DEPC)
ДНК Htrt DNA
ДНК матрицы
ДНК полимераза Premix
ДНК тимуса теленка
I типа
ДНКаза
ДНК-фингерпринтинг и
ДНК-профилинг
Емкости
Железа сульфат гепта¬
гидрат Fe2S04.7H20
Железо сернокислое
Жидкость Буэна фикси¬
рующая
Жидкость сцинтилляци-
онная
Жидкость сцинтилляци-
онная Ecoscint
Замедлители угасания
флюоресценции
Заменители сыворотки
Заменители сыворотки
Biotain-MPS
Заменители сыворотки
ТСМ, ТСН
Заменители сыворотки,
CPSR
Jencons; Lawson Mardon
Wheaton; Matrix Tech¬
nologies; Mettler Toledo;
Michael Smith; MP
Biomedicals; Polytech;
Popper; Robbins; Tecan;
Thermo Electron; Zinsser
См. Отдельные фермен¬
ты и заменители трип¬
сина
Corning; Jencons; Steris
Sigma
Sigma
Geron
Ambion
Takara Bio
Sigma
Lome; Serologicals; Sigma;
Worthington
Anglia DNA Bioservices;
ATCC; Cellmark UK;
DNA Bioscience; DNA
Solutions; ECACC; Lab-
oratoryCorporation of
America; LGC Promoch-
em; Orchid-Cellmark;
Verilabs Biosciences; см.
также Аутентификация
См. Аспираторы
См. Химические реактивы
См. Химические реактивы
Sigma
Amersham Biosciences;
BS & S; National Diag¬
nostics; New England
Nuclear (DuPont);
PerkinElmer (Packard)
BS & S; National Diagnos¬
tics; Perkin Elmer
Calbiochem; Citifluor;
Vector
Biosource; Irvine;
Metachem; Protide; cm.
также Специфические
заменители сыворотки
ниже
Cambrex
Celox; MP Biomedicals
Sigma
584
Приложение II. ИСТОЧНИКИ ОБОРУДОВАНИЯ И МАТЕРИАЛОВ
Заменители сыворотки,
Ехсе11-900
Заменители сыворотки,
Ex-cyte
Заменители сыворотки,
ITS Premix
Заменители сыворотки,
Nutridoma
Заменители сыворотки,
Serxtend
Заменители сыворотки,
SIT
Заменители сыворотки,
ТСМ, ТСН
Заменители сыворотки,
Ultroser
Заменители сыворотки,
Ventrex
Заменитель коллагена
Vitrogen
Заменитель сыворотки
Biotain-MPS
Заменитель сыворотки
CPSR
Заменитель сыворотки
Serxtend
Заменитель сыворотки
SIT
Заменитель сыворотки
Ultroser G
Заменитель сыворотки
Ventrex
Зонды молекулярные
PicoGreen
Иглы (для шприцов)
Иглы для биопсии
Идентификация клеточ¬
ных линий
Измельчитель тканей
Mcllwain
Измерение TEER
(трансэпителиальное
электрическое сопро¬
тивление)
Измерители потока газа
JRH Biosciences
Bayer
MP Biomedicals
Roche Diagnostics
NEN (DuPont)
Sigma
Celox; MP Biomedicals
Invitrogen
JRH Biosciences
BD Biosciences; Cohe¬
sion Technology; Col¬
lagen Corp.; см. также
Коллаген
Cambrex (BioWhittaker)
Sigma
NEN (DuPont)
Sigma
Invitrogen; см. также
Заменители сыворотки
JRH Biosciences; см.
также Заменители
сыворотки
Molecular Probes
См. Шприцы
Ranfac; Stille
АТСС; Bioreliance; Cell
Culture Characteriza¬
tion Services; Cellmark;
DSMZ; ECACC; LGC
Promochem; Orchid
Cellmark; см. также
ДНК-профилинг
Campden Instruments
Applied Biophysics; BD
Biosciences; World Pre¬
cision Instruments
Muis Controls; Omega
Engineering; Titan
Enterprises (см. также
Смесители газа)
Измерители расхода
Измеритель скорости
потока воздуха
Изолейцин
Изопротенола гидрохло¬
рид
Изотонический раствор
для счетчика клеток
Изотопы
Иммуноанализатор
Иммуноглобулин IgG
авидин-биотинилиро-
ванный, набор
Иммуноглобулин IgG
биотинилированный
Иммуноглобулин IgG
биотинилированный
анти-мышиный
Иммуноглобулин IgG
осла против кролика
Иммуноглобулин IgG,
крысиный
Иммуноглобулин анти-
кроличий IgG ABC,
пероксидазный набор
Иммуноглобулин анти-
мышиный IgG ABC,
щелочно-фосфатазный
набор для определения
Иммуноглобулины мы¬
шиные против кролика
Инвертированный мик¬
роскоп
Ингибитор трипсина со¬
евый SBTI
Ингибиторы обесцвечи¬
вания флюоресценции
Ингибиторы протеазы
Ингибиторы трипсины
Индикатор стерильности
Thermalog
Индикаторы стериль¬
ности
Индикаторы температу¬
ры, полоски
Индометацин
Инкубаторы
См. Измерители потока
газа
(см. Анемометры)
См. Химические реактивы
Sigma
Beckman Coulter; Scharfe;
см. также
См. Радиоизотопы
Agilent; Guava (см. также
Проточный флюори-
метр)
Dako Diagnostics
Sigma; Vector
Vector
Jackson ImmunoResearch
Sigma
Vector
Vector
Dako
Leica; Nikon; Olympus;
Zeiss
Sigma
См. Замедлители
угасания
флюоресценции
Intergen; Roche Diagnos¬
tics; Sigma
Biosource; Cascade; Sigma;
Serologicals
Bennet; Popper
Appleton Woods; Applied
Scientific; Bennett; Jen-
cons;
См. Индикаторы
стерильности
Sigma
ATR; Barnstead; Bellco;
Binder; Boro Labs; Cam¬
lab; Fisher; Genetic
Приложение II. ИСТОЧНИКИ ОБОРУДОВАНИЯ И МАТЕРИАЛОВ
585
Инкубаторы С02
Инкубаторы для яиц
Инструменты для иссе¬
чения
Инструменты хирурги¬
ческие
Инсулин
Инсулин/трансферрин/
селен (ITS)
Интерлейкины
Иодонитротетразол фио¬
летовый (INT)
Исследование ELISA
Исследование идентич¬
ности
Исследования цитоток¬
сичности
Кабельное телевидение
(CCTV)
Какодилатный буфер
Research Instr.; Global
Medical; Harvard; Ken-
dro; Infers; Lab-Line;
LEEC; LMS; LTE;
Memmert; MP Biomedi¬
cals; Napco; New Bruns¬
wick Scientific; Nuaire;
Omnilab; Precision Sci¬
entific; Robbins; RS Bio¬
tech; Sanyo Gallenkamp;
Scientific Instrument
Centre; SciGene; SP
Industries; Thermo Elec¬
tron; Triple Red; VWR.
Barnstead-Thermolyne;
Boro Labs; Camlab; Fish¬
er; Heinecke; Kendro;
Lab-1 трех; Lab-Line;
LEEC; Memmert; MP
Biomedicals; Napco; New
Brunswick Scientific;
NuAire; Omnilab; Preci¬
sion Scientific; Sanyo
Gallenkamp; SP Indus¬
tries; Thermo-Electron;
Triple Red
G.Q.F. Manufacturing
Cole-Parmer; EMS; Fine
Scientific Tools; Fisher;
Roboz; Swann-Morton;
VWR
См. Инструменты для
иссечения
Cambrex (Clonetics); CP
Pharmaceuticals; Eli
Lilly; Intergen; Invitro¬
gen; Sigma
BD Biosciences; Fisher
Scientific; Invitrogen;
Roche Diagnostics;
Sigma
См. Факторы роста
Sigma
Assay Designs; R & D Sys¬
tems
См. Аутентификация
клеточных линий и
ДНК-фингерпринтинг и
ДНК-профилинг
BioReliance (Invitrogen);
MatTek; Promega; Ski-
nEthic
Dage-MTI; Hamamatsu;
Leica (см. также Каме¬
ры CCD; Мкроскопы)
Plano; TAAB
Калия дигидрогеноорто-
фосфат
Калия ферроцианид
K3Fe (CN)6
Калия ферроци¬
анид тригидрат
K4Fe3(CN)6-3H20
Калия фосфат одноос¬
новный
Калия хлорид КС1
Калия хлорид
Кальций-связывающий-
реагент
Кальций-фосфорный
комбинированный
стандарт
Кальция определение
Кальция хлорид СаС12
Камера для наблюдения
в режиме замедленно¬
го времени
Камера с контролируе¬
мой атмосферой
Камера трансплантаци¬
онная (эпидермис)
Камеры CCD
Камеры
Камеры инкубационные
для микроскопа
Камеры цифровые
Канамицин
Канюли
Карбодиимид, 1-этил-
3-( диметил аминопро-
пил)-ЕОС
Карбоксиметилцеллюло
за (КМЦ)
Кариотипирование
Каркас PGA
Каркасы для тканевой
инженерии
Картриджи перфузион-
ные поликарбонатные
Кассета для диализа
См. Химические реактивы
См. Химические реактивы
См. Химические реактивы
См. Химические реактивы
См. Химические реактивы
См. Химические реактивы
Sigma
Sigma
Sigma
См. Химические реактивы
Bioptechs; Buck Scientific;
Carl Zeiss; Intracel
Bellco; Vineland; NJ
Greiner
BioWorld; Dage-MTI;
Hamamatsu; Leica; Scan-
alytics; Sony; см. также
Микроскопы
Leica; Olympus; Nikon;
Zeiss (смю также
Камеры CCD и Камеры
цифровые)
Buck Scientific; Imaging
Associates
Canon; Kodak; Leica;
Nikon; Olympus; Pola¬
roid (см. также Камеры
CCD; Микроскопы)
Invitrogen; MP Biomedi¬
cals; Sigma
BD Biosciences; Roboz;
Pierce
Fisher; Merck
Cell Culture Characteriza¬
tion Services
Albany International
Albany; Cook Biotech;
EBI; Minucells; Zimmer
Advanced Tissue Sciences
Pierce
586
Приложение II. ИСТОЧНИКИ ОБОРУДОВАНИЯ И МАТЕРИАЛОВ
Катетер для сосу¬
дов пластиковый
Angiocath, 20
Катетеры
Каучук силиконовый,
адгезивный
Каучук силиконовый,
листы
Каучук силиконовый,
пробки
Керамика на основе ко¬
ралла
Керамические палочки и
основы для культуры
Кератиноциты
Кетамин
Кетанест
Кислота аскорбиновая
Кислота аспарагиновая
Кислота липоевая
Кислота пантотеновая
Кислота селеновая
Кислота соляная НС1
Кислота соляная
Кислота трихлоруксус-
ная ТХУ
Кислоты аскорбиновой
2-фосфат
Клетки Balb/c ЗТЗ А31
Клетки HLF
Клетки Swiss ЗТЗ
Клетки бронхиального
эпителия
Клетки костного мозга
стромальные челове¬
ческие
Клетки эндотелиальные
Клеток линии
Клеточные банки
Клеточный скребок
КМЦ
Кожи перфоратор
Коллаген хвоста крысы
Коллаген
BD Biosciences
BD Biosciences
Dow Corning; GE Sili¬
cones
Nusil; Silicone Specialty
Fabricators
Bibby Sterilin
Interpore
Zimmer
См. Специализированные
клетки
Henry Schein; Sigma
Parke-Davis
Sigma; Wako
См. Химические реактивы
Sigma
Sigma
Merck; Sigma
См. Химические реактивы
См. Химические реактивы
См. Химические реактивы
См. Химические реактивы
АТСС
Coriell Institute for Medi¬
cal Research
ATCC
См. Специализированные
клетки
Clonetic-Poietics
См. Специализированные
клетки
См. Cell banks
ATCC; Coriell Cell Repos¬
itories (CCR); ECACC;
DSMZ; HSRRB; JCRB;
Riken
Applied Scientific; Bel-Art;
BD Biosciences; Corn¬
ing; Nalge Nunc; Tech-
mate; Techno Plastic
См. Карбоксиметилцел-
люлоза
Miltey
BD Biosciences
BD Biosciences; Biocolor;
Biodesign; Biomedical
Коллагеназа
Коллагеновая губка Ul¬
trafoam
Коллагеновая губка
Коллагеновый имплант
Zyderm 2
Кольца/цилиндры для
клонирования
Кольцевой шейкер
Кольцевые маркеры
Кольцемид
Компании-поставщики
лабораторного обору¬
дования, основные
Комплекс АР ААР
Комплекс встроенных
фильтров многоразо¬
вого использования
Комплект дисковых
фильтров для стерили¬
зации
Компоненты сыворотки
Ехсе11-900
Компоненты сыворотки
Ex-cyte
Компоненты сыворотки
Nutridoma
Компоненты сыворотки,
смесь ITS
Кондуктометры
Контейнеры для образ¬
цов
Technologies; Biosource
International; Cam¬
bridge Biosciences; Cel¬
lon; Chondrex; Cohesion
Technologies; Collagen
Corporation; CR Bard;
Davol; Invitrogen; In-
amed; Insmed; KeLab;
Lab Vision; MP Biomed¬
icals; Roche; Seikagaku;
Sigma; Stratech; Univer¬
sal Biologicals
Chondrex; Invitrogen;
Lorne; Roche; Serva;
Sigma; Worthington
Davol
Avitene Ultrafoam CR
Bard; Davol; McGhan
Medical
Insmed; McGhan Medical
BelArt; Bellco; Fisher;
Scientific Laboratory
Supplies
Bellco
Nikon
Sigma
Camlab; Cole-Parmer; Fish¬
er; Scientific Instrument
Centre; Thermo Electron;
Triple Red; VWR
DAKO; Vector
См. Фильтры
См. Фильтры
JRH Biosciences
Bayer
Roche Diagnostics
MP Biomedicals
Corning; Mettler Toledo;
Quadrachem; Technika;
Thermo Electron
Alpha Laboratories; Com¬
ing; Bibby Sterilin;
Nalge Nunc; VWR; cm.
также Контейнеры
для транспортировки
образцов
Приложение II. ИСТОЧНИКИ ОБОРУДОВАНИЯ И МАТЕРИАЛОВ
587
Контейнеры для стекол
для микроскопии
Контейнеры для транс¬
портировки
Контейнеры для транс¬
портировки образцов
Контейнеры универсаль¬
ные стеклянные
Контроллеры С02
Контроллеры темпера¬
туры
Конфокальный микро¬
скоп
Коробки для пипеток
Коробки для стекол све¬
тонепроницаемые
Кран трехсторонний за¬
порный
Красители
Красители гистологичес¬
кие
Красители ДНК
Краситель WST-1 инди¬
катор цитотоксичности
Краситель Гимза
Краситель для оценки
жизнеспособности
трипановый синий
Краситель малахитовый
зелёный
Краситель Мэй-Грюн-
вальда, раствор
Краситель прочный
красный TR соль
Краситель прочный фио¬
летовый
Coming; Raymond Lamb;
Raven; Richardsons
Air Packaging Technolo¬
gies; Air Sea Atlanta; Al¬
tec; Bel-Art; Cellutech;
Cin-Made; Jencons;
Kimberly-Clark; Lab
Safety Supply; Nalge
Nunc; Saf- T-Pak
Saf-T-Pak; Nalge Nunc;
Air Sea Atlanta; Cel¬
lutech; Cin-Made Corp
(см. также www.uos.har-
vard.edu/ehs/biobioshi.
shtml и www.ehs.ucsf.
edu/Safety%20Updates/
Bsu/Bsu5.pdf)
Camlab
Air Products; Gow-Mac;
Lab-Line; Lab Impex;
Therma Electron (For¬
ma; Hotpak)
См. Термостаты,
пропорциональные
контроллеры
Biotech Instruments; Lei-
ca; Molecular Dynamics;
Nikon; Zeiss
Bellco; Thermo Electron
Bel-Art; Cole-Parmer;
Raven; Raymond Lamb;
BD Biosciences; Raven
Scientific
Baxter Healthcare; Sher¬
wood-Davis & Geek
Fisher; Merck; Molecular
Probes; MTR; Sigma;
TCS Biologicals
Merck; MTR Scientific;
Sigma
Hoechst; Molecular Probes
Serva
Fisher; Merck; TCS Bio¬
logicals; Sigma
Merck; См. также Среды
Sigma
Merck
SERVA
Sigma
Краситель флюоресцен¬
тный для ДНК Hoechst
33342
Краситель флюоресцен¬
тный для микоплазм
Hoechst 33258
Криомаркеры
Криопробирки
Криофлаконы
Кристаллвиолет
Крысы
Крышки С02-проница-
емые
Крышки проницаемые
Ксилазин
Ксилазина гидрохлорид
Rompun
Ксилен
Культуральная камера
Opticell
Культуральные камеры
Культуральные пробир¬
ки
Культуры клеток специ¬
ализированные
Культуры перфузионные
Кумасси синий R
Лабораторное стекло
Лактальбумина гидро¬
лизат
Лактатдегидрогеназный
тест для оценки жизне¬
способности, набор
Ламинарный ппсаф/бокс
Ламинин
Sigma
MP Biomedicals (набор);
Polysciences; Sigma
GA International
См. Ампулы
См. Ампулы
Merck; Fisher
Charles River; Harlan
Sera-Lab
См. Чашки флаконы
и планшеты для
культуры
См. Газопроницаемые
крышки
NLS Animal Health; Sigma
Bayer Vital
См. Химические реактивы
BioCrystal
См. Стекла для
культуральных камер
См. Чашки флаконы
и планшеты для
культуры
BD Biosciences; Cambrex;
Cascade; Cell Systems;
PromoCell; SkinEthic
Amicon; Bioptechs; Cellco;
Endotronics; Microgon
Sigma
Bellco; Coming; Kimble-
Kontes; Schott; Wheaton
BD Biosciences (Difco);
Invitrogen
Roche Applied Science;
Sigma
AES; Atlas Clean Air;
Baker; Bigneat; Bioquell;
Biosero; Contamination
Control; Envair; Erlab;
Germfree Labs; Kendro;
Kojair; Labcaire; LTE;
Medical Air Technology
(Kendro); MP Biomedi¬
cals; Nuaire; Safelab; SP
Industries; Thermo Elec¬
tron
BD Biosciences; Bio¬
medical Technologies;
Invitrogen; Lab Vision;
Sigma; Stratech
588
Приложение II. ИСТОЧНИКИ ОБОРУДОВАНИЯ И МАТЕРИАЛОВ
Латекс, частицы (стан¬
дарт для измерителей
размеров клеток)
Левамизол
Лейцин
Лента индикаторная для
стерилизации
Лизин НС1
Лимоннокислый свинец
Рейнольда
Линии клеток, базы дан¬
ных
Линия клеток 5637
Линия клеток гепатомы
человека HepG2
Лития бромид
Лития карбонат
Лубрикант силиконо¬
вый, инертный
Лубрикант хирургичес¬
кий Surgilube
Магнитные мешалки
Магния сульфат гепта¬
гидрат MgS04 -7Н20
Магния сульфат
Магния хлорид MgCl2 •
6Н20
Магния хлорид
Макроносители
Маркаин
Маркер для колоний
Маркеры для колоний
Маркеры спирто-нераст-
воримые
Маркеры, ампулы
Марля хирургическая
Марля, сетка
Матригель
Матрикс
Beckman Coulter
Serva
Sigma-Aldrich
См. Индикаторы
стерильности
См. Химические реактивы
EMS; SPI Supplies
АТСС; Coriell; DSMZ;
ЕСАСС; HSRRB; ICLC
DSMZ
См. Клеточные банки
См. Химические реактивы
См. Химические реактивы
Dow Coming
Fougera
Bellco; Camlab; Chemap;
Cole-Parmer; Fisher;
Hanna; Jencons; Lab-
Line; Techne; Thomas;
Wheaton
См. Химические реактивы
См. Химические реактивы
См. Химические реактивы
См. Химические реактивы
New Brunswick
NLS Animal Health
Nikon
Nikon
Radleys
GA International; Radleys
Johnson & Johnson; Ken¬
dall
См. Сетчатые фильтры
и сетки
BD Biosciences; Serva
Accurate Chemical (as¬
says); BD Biosciences;
Biocolor (assays); Biode¬
sign; Biomedical Tech¬
nologies; BioSource;
Cellon; Cook Biotech;
Harbor Bio-Products;
Imperial; Lab Vision;
Matrix Technologies;
NovaMatrix; Pierce; Pro¬
tein Polymer; R&D Sys¬
tems; Sigma; Stratech;
TCS; Tebu-bio
Матрикс ECM
Матрикс EHS (Матри¬
гель, Matrigel)
Матрикс внеклеточный
Машина моющая для
пипеток, сушильное
устройство для пипе¬
ток
Машина моющая для
стекла
Меди сульфат
Меланоциты
Мембранные фильтры
для шприцов
Мембраны Nyaflo
Меркаптоэтанол
Метакрилат, поли(2-гид-
роксиэтил)
Метакрилат: 3-(триме-
токсизилил) пропил-
метакрилат
Метанол
Метилцеллюлоза
Метилцеллюлоза Metho¬
cel
Метионин
Метьризамид
Мечение ДНК клеток
BrdU, набор
Мешалки См. Магнит¬
ные мешалки
Микоплазмы, наборы
для детекции
Микостатин, нистатин
Микроанализ, оборудо¬
вание
Микроденситометр
Микрокрышки, пробир¬
ки микрокапиллярные
Микроманипуляторы
См. Матрикс
BD Biosciences
См. Матрикс
Bel Art; Thermo-Shandon;
Radleys
Burge; Lancer; Miele;
Scientek; Scientific In¬
strument Centre; Steris
Corp.
См. Химические реактивы
См. Специализированные
клетки
См. Фильтры
Gelman Sciences
См. Химические реактивы
Sigma
Sigma
См. Химические реактивы
Dow Coming; Fluka;
Fisher; Sigma; Stem Cell
Technologies
Dow Coming
См. Химические реактивы
Nycomed
Invitrogen (Invitrogen);
Promega; Sigma
ATCC; Gen-Probe; Irvine
Scientific; Metachem;
MP
Bio Whittaker; MP Bio¬
medicals; Invitrogen;
Sigma
Affymetrix; Agilent; Bio-
Rad; Brinkmann; Chemi¬
con; Imaging Research;
Nonlinear Dynamics;
Schleicher & Schuell;
Stratagene; Tecan
См. Анализ изображения,
сканируются
цитометрия
(Drummond) Thermo-
Shandon; Fisher
Brinkmann; Eppendorf;
Leica; Nikon; Steris;
Stoelting
Приложение II. ИСТОЧНИКИ ОБОРУДОВАНИЯ И МАТЕРИАЛОВ
589
Микроносители
Микропипетки
Микропланшеты, гомо-
генизатор-измельчи-
тель MiniBead Beater
Микроскоп электронный
сканирующий (SEM)
Микроскоп электронный
трансмиссионный
Микроскопия электрон¬
ная
Микроскопы
Микроскопы флюорес¬
центные
Микротитрационный
планшетный сканер
Микротитрация, обору¬
дование
Микроцентрифужные
пробирки с фильтром
Митомицин С
Мозг костный
Монитор давления
Мониторинг Real-time
Мониторинг биоэлектри¬
ческий
Мониторинг метаболи¬
ческий
Морозильник с жидким
азотом с регулируемой
скоростью охлаждения
Amersham Biosciences;
Bibby Sterilin; Bio-Rad;
Nalge Nunc; Invitrogen;
MP Biomedicals; Sigma;
SoloHill; TCS
Alpha; Anachem; Applied
Scientific; Bibby Steri¬
lin; Biohit; Brinkmann;
Corning; Elkay; Eppen¬
dorf; Genetic Research
Instr.; Gilson; Jencons;
Lawson Mardon Whea¬
ton; Oxford; Roche;
Thermo Electron
Biospec; Daintree
ElectroScan; JEOL
Philips
Electron Microscopy Ser¬
vices; TAAB; Structure
Probe
Carl Zeiss; Leica; Olympus;
Nikon; Prior
См. Микроскопы
См. Ридеры для
планшетов
Anachem; Berthold; Bio-
Rad; Biospec; BMG
Labtech; Brand; Camlab;
Coming; Dynex; Elkay;
ESA; Genetic Research
Instr.; Gilson; Integra;
Invitrogen; Molecular
Devices; Nalge Nunc;
Nonlinear Dynamics;
PerkinElmer; R &D Sys¬
tems; Robbins; Tecan;
Techmate; Thermo Elec¬
tron; VWR; Whatman;
Zinsser
Coming
Roche Diagnostics; Sigma
Cambrex
Hewlett-Packard
См. Биосенсоры
См. Биосенсоры
См. Биосенсоры
Messer; Nalge Nunc;
Planer; Thermo Electron;
Statebourne; Taylor-
Wharton
Морозильники азотные
Морозильники с жидким
азотом
Морозильники
Морозильники, -20 еС
Морозильники, -70 еС
Мочевина
Муравьиная кислота
Мыши BALB/c nu/nu
Мыши Nude
Мыши SCID
Мышиные антитела
CD105 (555690)
Набор Endofree Maxi
Набор Eukitt
Набор FuGENE 6
Набор LEUCOperm Kit
Набор Qiagen Rneasy
Mini Kit
Набор QIAshredder
Набор QuiAmp DNA
mini kit
Набор RNAseZap®
Набор RNeasy mini kit
Набор Vectastain Elite
ABC Kit
Набор Vectastain Quick Kit
Набор гистологических
красителей Histostain-
SP kit (Streptavidinpe-
roxidase)
Набор для выделения
ДНК
Набор для оценки жиз¬
неспособности Live/
Dead
Набор для растяжения
катетеров
Набор коллагена I типа
ELISA
См. Морозильники;
жидкий азот
Aire Liquide; Bamstead-
Thermolyne; Boro Labs;
Chart Biomed; Cryomed;
Genetic Research; Jen¬
cons; Messer Cryotherm;
Planer; Statebourne;
Taylor-Wharton; Ther¬
mo Electron; VWR.
См. Морозильник с
жидким азотом
с регулируемой
скоростью охлаждения
Bamstead; Fisher; New
Brunswick Scientific
Bamstead; Fisher; New
Brunswick; Nuaire; Pre¬
cision Scientific; Revco;
Thermo Electron
См. Химические реактивы
См. Химические реактивы
Nippon CLEA
Charles River; Nippon
CLEA
Charles River
BD Biosciences (Pharmin-
gen)
Qiagen
Fluka
Roche
Serotec
Qiagen
Qiagen
Qiagen
Ambion
Qiagen
Vector Laboratories
Vector Laboratories
Zymed Laboratories Inc.;
San Francisco; CA;
U.S.A
Qiagen
Molecular Probes
Interlink Baxter Health-
Care
Chondrex
590
Приложение II. ИСТОЧНИКИ ОБОРУДОВАНИЯ И МАТЕРИАЛОВ
Наборы RNAimage
Наборы для исследова¬
ния цитотоксичности
Нагревательные системы
Найл красный
Наконечники для пипе¬
ток, устойчивые к аэ¬
розолям
Насос Pi-Pump
Насос воздуходувного
типа
Насос перистальтичес¬
кий непрерывного дей¬
ствия
Насос шприцевой
Насосы вакуумные
Насосы перистальтичес¬
кие
Насосы перистальтичес¬
кие
Натрия азид
Натрия альгинат
Натрия бикарбонат
Натрия бутират (NaBt)
Натрия гидроокись
Натрия деоксихолат
Натрия лаурилсульфат
(SLS; натрия деокси¬
холат)
Натрия ортованадат
Na3V04
Натрия пируват
Натрия пируват, сте¬
рильный раствор
Натрия селенат
Натрия селенит
Натрия соли
Натрия тиосульфат
Натрия формиат
Натрия фосфат, двуос¬
новный гептагидрат
Натрия хлорид
Натрия хлорид, 0.9%
Натрия цитрат
Нафталеновый черный
Нафтол AS-MX щелоч¬
ной раствор
GenHunter
CellStat; см. также Иссле¬
дования цитотоксич¬
ности
Genetic Research Instr.;
Robbins; VP Scientific
Sigma
Applied Scientific; Bel-art;
Polytech; Radleys
Gorman-Rupp Industries
Cole-Parmer
Harvard Apparatus
BO; Cole-Parmer; Mil¬
lipore; Pall Gelman;
Varian
Altec; Amersham; Cole-
Parmer; Gilson; Michael
Smith; Watson
Altec; Amersham; Cole-
Parmer; Gilson; Michael
Smith; Watson Marlow
Sigma
ISP Alginate
См. Химические реактивы
Sigma
Sigma
Sigma
Sigma
Sigma
См. Химические реактивы
См. Среды
Sigma
Sigma
См. Химические реактивы
Sigma
Fisher
См. Химические реактивы
См. Химические реактивы
Baxter
См. Химические реактивы
R.A. Lamb (см. также
Красители)
Sigma
Нафтол-AS-MX фосфат
Нейлоновые сита, филь¬
тры и марля
Нейтральный красный
Неорганические соли
Ниацинамид
Нигрозин
Нистатин
Нитрат свинца II
Нитрофенила фосфат
Нитрофенол стандарт¬
ный раствор
Нить хирургическая,
дакрон
Нить хирургическая,
дексон
Нить хирургическая,
шелк
Нож-вибратом
Ножницы с пружиной
Ножницы
Нуклеозиды
Оборудование для анес¬
тезии, ветеринарное
Оборудование для сте¬
рильной упаковки
Оборудование для упа¬
ковки транспортаци-
онной
Оборудование для
фильтрации
Одежда защитная
Онкостатин М
Определение размера
клеток
Органотипическая куль¬
тура
Освещение, волоконная
оптика
Осмия тетроксид
Осмометр
Основной фактор роста
фибробластов (bFGF)
Остеогенный протеин 1
(OP-1; ВМР-7)
Serva
См. Фильтры сетчатые
TCS Biologicals
Merck
Sigma
Sigma; TCS Biologicals
Sigma; Invitrogen
См. Химические реактивы
Sigma
Sigma
Davis and Geek
Davis and Geek
Harvard Apparatus
Campden Instruments
Fine Science Tools Inc.;
VWR; см. также
Инструменты для
иссечения
См. Инструменты для
иссечения
Sigma
SurgiVet
Applied Scientific; Buck
Scientific; KNF Corp;
Portex; Portland Plas¬
tics; Roth
См. Контейнеры для
транспортировки
образцов
См. Фильтры
Alexandria Workwear; Lab
Safety Supply; Sigma
См. Факторы роста
Beckman Coulter; Scharfe
ACM-Biotech; MatTek;
Minucells; SkinEthic
Fischer Scientific
Plano; Roth; Sigma
Advanced Instruments;
Analytical @ Technolo¬
gies; Gonotec; Nova Bio¬
medical; Wescor
См. Факторы роста
См. Факторы роста
Приложение II. ИСТОЧНИКИ ОБОРУДОВАНИЯ И МАТЕРИАЛОВ
591
Пакеты биологической
опасности
Пакеты для сред
Пакеты для стерилиза¬
ции
Пакеты и пленка авто¬
клавируемые
Пакеты и пленка нейло¬
новые автоклавируе¬
мые
Пакеты культуральные
Панкреатин
Папаин
Параформальдегид
Пастеровские пипетки
Pastette
Пенициллин
Пенициллин/стрептоми¬
цин
Пеногасители
Пентобарбитал
Пепсин
Пептид RGD: GRGDS:
H-Gly-Arg-Gly-Asp-
Ser-OH
Пептид глицин-аргинин-
глицинаспартат-серин
(GRGDS)
Пептидаза, Дипептидил,
IV (CD26)
Пептиды: RGD, GRGDS,
H-Gly-Arg-Gly-Asp-
Ser-OH
Перекись водорода
(30%раствор)
Перемешивающие фла¬
коны
Перколл
Пермаунт DPX
Перфузия капиллярного
русла
См. Пакеты и пленка
автоклавируемые
American Fluoroseal; Cell
Genix; DuPont; MP Bio¬
medicals; Sigma
См. Упаковка стерильная
Altec; Applied Scientific;
Bibby; Sterilin; Elkay;
Jencons; Lab Safety Sup¬
ply; KNF; Portex
Applied Scientific; KNF
Corp.; Portex
Cell Genix; Du Pont;
Metabios; MP Biomedi¬
cals; PAW BioScience
Products (см. также
Пакеты для сред)
Invitrogen; Sigma
Worthington; Sigma
Electron Microscopy Sci¬
ences; Merck; Sigma
Alpha Laboratories; Bibby
Sterilin; Elkay; Polytech;
Richardsons
См. Среды
См. Среды
Bayer; Dow-Corning;
Merck; Sigma
Abbott
Sigma
Calbiochem
Sigma
Biosource; Neomarkers
Calbiochem
Bellco; Coming; Genetic
Research Instr.; Integra;
Lawson Mardon Whea¬
ton; Techne; см. также
Магнитные мешалки
Amersham Biosciences;
Sigma
м. Красители
См. Половолоконная
перфузионная культура
Перчатки
Перчатки защитные для
криоконсервации
Перчатки нитриловые
Печи, сушильные шкафы
Пинцет изогнутый
Пинцеты
Пинцеты, зажимы
Пипетки
Пипетки Movette
Пипетки автоматические
многоканальные
Пипетки аспирационные
Пипетки и микропипетки
автоматические
Пипетки с широким диа¬
метром
Пиридоксин НС1
Плазма лошадиная
Плазма цыпленка
Планшетный сканер
ELISA
Ansell Medical; Applied
Scientific; Cryomed;
Johnson & Johnson Med¬
ical; Lab Safety Supply;
Radleys; Renco Corp.;
Safeskin; Sentinel Labo¬
ratories; Stoelting; Surgi-
con; VWR Scientific
Taylor-Wharton; Jencons
Ansell; Cole-Parmer; Lab
Safety Supplies; Radleys;
Sentinel; Stoelting
Astell; Barnstead; Cam¬
lab; Dynalab; Harvard;
Kendro; LEEC; LTE;
Memmert; Precision Sci¬
entific; Scientific Instru¬
ment Centre; Thermo
Electron
См. Хирургические
инструменты
См. Инструменты для
иссечения
Fisher Scientific
Alpha Laboratories; BD
Biosciences; Bellco;
Coming; Fisher; Nalge
Nunc; Sterilin
Invitrogen
См. Пипетки и
микропипетки
автоматические
Corning Costar
Alpha Laboratories;
Anachem; Applied Scien¬
tific; Barnstead; Bellco;
Bibby Sterilin; Brand;
Cole-Parmer; Com¬
ing; Daiger; Eppendorf;
Fisher; Genetic Research
Instr.; Gilson; Integra;
Jencons; Lawson Mardon
Wheaton; Matrix Tech¬
nologies; Merck; Messer;
Mettler-Toledo; MP
Biomedicals; Polytech;
Radleys; Rainin; Roche;
Socorex; Thermo Elec¬
tron; Whatman
Bellco
Sigma-Aldrich
MP Biomedicals
Invitrogen; MP Biomedicals
Cm. Plate readers
592
Приложение II. ИСТОЧНИКИ ОБОРУДОВАНИЯ И МАТЕРИАЛОВ
Планшетный сканер
Планшет четырехлуноч¬
ный
Планшеты микротитра-
ционные
Планшеты микротитра-
ционные с удаляемыми
стенками
Планшеты микротитра-
ционные, диспенсеры
для разлива
Планшеты многолуноч¬
ные с биопокрытием
Планшеты многолуноч¬
ные с фильтрующим
дном
Планшеты с питатель¬
ным агаром
Планшеты
Пластик Sylgard
Пластик лабораторный
Пластины многолуноч¬
ные фильтровальные
Платформы качалочные
Пленка для стерилиза¬
ции
Пленка нейлоновая па¬
ропроницаемая
Пленка нейлоновая по¬
лупроницаемая
Плёнка полиамидная
Пленка рентгеновская
Поверхностно-активное
вещество Pluronic F-68
Поддоны и штативы для
морозильника
Позитивный фоторе¬
зист, OCG 825-835
St, Shipley 1813, или
Shipley 1818
Покровные стекла Ger¬
man glass
Bio-Tek; Berthold; Bio-
Rad; BMG; Camlab;
Dynex; Fisher; Merck;
Messer; Molecular De¬
vices; Molecular Dynam¬
ics; PerkinElmer; Tecan;
Thermo Electron.
См. Чашки, флаконы, план¬
шеты для культуры
Applied Biosystems;
см. также Чашки,
флаконы, планшеты для
культуры
Invitrogen
Thermo Electron
BD Biosciences
Nalge Nunc; Techmate;
Whatman
Oxoid
См. Чашки, флаконы
и планшеты для
культуры клеток
Dow Coming
Falcon (BD Biosciences)
Whatman
Bellco
См. Упаковка стерильная
См. Пленка и пакеты
нейлоновые
автоклавируемые
См. Упаковка стерильная,
оборудование
Applied Scientific; Buck
Scientific; KNF Corp.;
Portex; Portland Plas¬
tics; Roth
Eastman Kodak; Fuji
Invitrogen; MP Biomedi¬
cals; Serva; Sigma
См. Морозильники с
жидким азотом
Shipley
Carolina Biological Sup¬
plies
Покровные стекла стек¬
лянные
Поли(2-
гидроксил)метакрилат
(PolyHEMA)
Поливинилпирролидон
Полилизин, поли-D- и
поли-L
Полимиксин В
Полипропиленовые ем¬
кости, 30 мл
Полиэтиленгликольдиак-
рилат (PEGDA)
Полиэтиленгликоль-
диакрилат (PEGDA),
3.4 kDa
Половолоконная перфу-
зионная культура
Праймеры
Праймеры флюоресцен-
тно-Су5-меченые
Препаровальная лупа
Приспособление для за¬
клеивания микротитра¬
ционных планшетов
Приспособление для
заклеивания пластико¬
вых планшетов Mylar
Приспособления для за¬
клеивания планшетов
Пробирки Accuspin
Пробирки круглодонные
с крышками
Пробирки Лейтона
Пробирки пластиковые
Пробирки поликарбо-
натные Nalge Nunc
Пробирки центрифуж¬
ные конические
Пробирки центрифуж¬
ные
Прогестерон в абсолют*
ном этаноле
Chance Propper; Wheaton
Sigma
Calbiochem; Sigma; USB
Biomedical Technologies;
BD Biosciences; Sigma;
Stratech
Invitrogen
Nalge Nunc
Nektar Transforming Ther¬
apeutics; Shearwater
Nektar; Shearwater
Argos; Bellco; Biovest; Fi-
berCell; JM Separations;
Spectrum
Applied Biosciences; Re¬
search Genetics; Riken
Amersham Biosciences
Daigger; Olympus; Leica;
Nikon; Zeiss
См. Приспособление для
заклеивания планшетов
МР Biomedicals; Invitro¬
gen
Anachem; Brandell; Elkay;
Greiner; MP Biomedi¬
cals; Porvair
Sigma Diagnostics
Alpha Laboratories; BD
Biosciences (Falcon);
Bellco; Coming; Kimble-
Kontes
Bellco; Coming; Techno
Plastic
См. Пробирки
центрифужные
См. Пробирки
центрифужные
Alpha; Bibby Sterilin; BD
Biosciences (Falcon);
Coming; Du Pont; Ep-
pendorf; Greiner; Om¬
nilab; Nalge Nunc; Tech¬
no Plastic Products
Sigma
Приложение II. ИСТОЧНИКИ ОБОРУДОВАНИЯ И МАТЕРИАЛОВ
593
Программа компьютер¬
ная BMON
Программа компьютер¬
ная Fast-Track Wave-
maker32 ver.6.6
Программа компьютер¬
ная Fragment Manager
Программа компьютер¬
ная Scion-Image
Программа компьютер¬
ная для испытания
усталостной прочности
связок Wavemaker32
Программа компьютер¬
ная для сбора и хране¬
ния данных Cell Quest
Программа компьютер¬
ная для статистической
обработки SPSS (ver¬
sion 10.0)
Программируемый мо¬
розильник
Пролин
Пролиферация клеток,
исследование
Проназа
Пропидия иодид
Протеаза нейтральная
(Диспаза)
Протеиназа К
Проточные цитометры
Проявители для фотоли¬
тографии Shipley 354,
1813,1818
Проявитель для фотоли¬
тографии и фотогра¬
фии OCG 825-835, 934
Путресцин 2НС1
Радиоизотопы
Распылитель краски
Раствор Krebs-Henseleit
Раствор для счетчика
клеток
Раствор несущественных
аминокислот в МЕМ
Раствор силиконизирую-
щий (Sigmacote)
Раствор солевой забу-
ференный MES
Ingenieurburo Jackel
Instron
Amersham Biosciences
Scion
Instron
BD Biosciences
SPSS
Messer; Planer; Thermo
Life Sciences (Cryomed);
Statebourne
См. Химические реактивы
Lumitech (Cambrex); Pro-
mega; Upstate
Calbiochem; Roche Diag¬
nostics; Sigma
Sigma
Roche Diagnostics
Sigma
Agilent; BD Biosciences;
Beckman Coulter; Ge¬
netic Research; Guava
Technologies
Microchem Corp.
Ciba Specialty; см. также
Химические реактивы
Sigma-Aldrich
Amersham; MP Biomedi¬
cals; PerkinElmer; Sigma
Badger Air Brush Co.
Sigma
Beckman Coulter; Scharfe
См. Среды
Sigma
Pierce
Раствор солевой сбалан¬
сированный без Mg2* и
Са2+ (HBSS)
Раствор солевой сбалан¬
сированный Эрла без
Са2+и Mg2+(CMF)
Раствор солевой Тирода
(TBSS)
Раствор Хэнкса сбалан¬
сированный солевой
(HBSS)
Реагент, блокирующий
пероксидазу
Реактивы биохимичес¬
кие
Регистраторы темпера¬
туры
Ретиноевая кислота
Ретинола ацетат, trans
Рефрактометр
Решетки и чашки для ор¬
ганной культуры
Рибофлавин
Роботизированные сис¬
темы
Родамин В
Роллер-флаконы, плас¬
тик
Роллер-флаконы, стек¬
лянные
Роллер-флаконы, шта¬
тивы
Роторы
Сафранин О
Сахароза Fluka; Sigma
Сбалансированный со- См. Среды
левой раствор, раствор
Хэнкса, Эрла и т. д.
Секвенирование автома¬
тическое, оборудова¬
ние
Секвенирование, набор
Сепарация клеток
См. Среды
Invitrogen
См. Среды
См. Среды
Cytomation; Dako
Calbiochem; Fluka; Merck;
Pierce; Roche Diagnos¬
tics; Sigma; U.S. Bio¬
chemical
См. Термометры с регист¬
рирующим устройством
Sigma
Sigma
Beckman; Cole-Parmer;
Fisher; Technika.
BD Biosciences
Sigma
Beckman Coulter; Bio-
Rad; Cytogration; Gil¬
son; Innovative Cell
Technologies
Sigma
Applied Scientific; BD
Biosciences; Caisson;
Coming; Integra
Bellco
Argos; Bellco; Genetic
Research Instr.; Integra;
Lawson Mardon Whea¬
ton; New Brunswick Sci¬
entific; Robbins;
New Brunswick Scientific
Sigma
ALFred Amersham Biosci¬
ences
Amersham Biosciences
Accurate Chemical &
Scientific; Applied Im¬
mune Sciences; BD Bio¬
sciences;
594
Приложение II. ИСТОЧНИКИ ОБОРУДОВАНИЯ И МАТЕРИАЛОВ
Серебра нитрат
Серии
Сетка нейлоновая Scry-
nel NYHC
Сетчатые и металличес¬
кие фильтры
Сиквенс ДНК
Силикагель
Силикон
Система амплификаци-
онная Superscript
Система культуры кле¬
ток ротационная RCCS
Система культуры
клеток ротационные
(RCCS)
Система охлаждающая
с закрытым горлом и
контролируемым ре¬
жимом охлаждения
Система очистки воды
Система перфузионная
мембранная Membro-
ferm
Система сжатие/растя¬
жение для культуры
миоцитов
Системы с обратным ос¬
мосом
Сита
Сита из нержавеющей
стали
Сита клеточные
Сито с ячейкой 200-мм
Скальпели
Слайд-контейнеры
для эмульсии (Cyto-
Mailer)
BioCarta; Biochrom;
Dynal; Kendro; Miltenyi;
PerkinElmer; Stem Cell
Technologies
См. Химические реактивы
См. Химические реактивы
Merck; см. также
Фильтры нейлоновые
сетчатые
Dynal; Merck; Miltenyi;
Sefar; Stanier; Teledyne
Tekmar; (см. также
Фильтры клеточные и
Сита клеточные)
Geron
Fisher; Merck
Fisher; Merck; Serva;
Sigma
Invitrogen
Synthecon; Cellon
Synthecon; Cellon; Syn¬
thecon; Inc.
Taylor-Wharton
Applied Biosciences; Barn¬
stead; Elga; Genetic
Research Instr.; High-Q;
Millipore; Purite; Triple
Red; U.S. Filter; Vivendi;
Whatman
Polymun Scientific
Instron
Barnstead; Elga; Millipore;
U.S. Filter
Markson; Retsch; cm.
также Сетки, сетчатые
фильтры
BD Biosciences; см. также
Сетчатые фильтры
BD Biosciences; Dynal;
Miltenyi Biotec
BD Biosciences; Tekmar
См. Инструменты для
иссечения
Lab-Tek; Fisher; Statlab
Слайд-флаконы
Смазка силиконовая
Смесители газа
Смесь Pen/Strep Fungi¬
zone
Смешанная перфузион¬
ная система с капил¬
лярным слоем
Соединения Люэра
Сорбитол
Сортер клеточный FACS
Сортер клеточный FAC-
SCalibur
Сортер клеточный
MACS
Сортер клеточный флюо-
ресцентно-активиро-
ванный (FACS)
Сортинг магнитный
Состав герметезирую-
щий для тканей Tissue
Tek-O.T.C
Сосуд культуральный
(STLV, Slow Turning
Lateral Vessel)
Сосуды для культуры
Спектрофотометр для
микропланшетов Spec¬
tra Мах 250
Спирт хлороформизоа-
миловый
Среда 199
Среда CMF-PBS
Среда CMRL-1066
Среда Daigo’s Т
Среда DMEM со стаби¬
лизированным глюта¬
мином
Среда DMEM/F12,
50/50
Среда F12
Среда F12: DMEM
Среда F12H
Среда HEPES
Среда LHC-9
Среда М199
Nalge Nunc; см. также
Слайд-камеры
ВОС Edwards; British
Oxygen; Dow Coming;
Girovac
Gow-Mac; Signal
См. Среды
См. Система перфузион¬
ная половолоконная
Altec; Applied Medical
Technol., Harvard Appa¬
ratus; Popper
Sigma
BD Biosciences; Beckman-
Coulter
BD Biosciences
Dynal; Miltenyi
BD Biosciences
Dynal; Miltenyi
Sakura Finetek
(Raymond Lamb in UK);
Sakura Finetek
Europe
Synthecon; Cellon
См. Чашки флаконы
и планшеты для
культуры
Molecular Devices; см.
также Ридеры для
планшетов
Biogene
См. Среды
См. Среды
См. Среды
Wako
Invitrogen; Biochrom
Biochrom; Cellgro-Mediat-
ech; Invitrogen; Sigma
См. Среды
См. Среды
См. Среды
См. Media
BioSource
См. Среды
Приложение II. ИСТОЧНИКИ ОБОРУДОВАНИЯ И МАТЕРИАЛОВ
595
Среда McCoy’s 5А
Среда MCDB 105
Среда MCDB 153
Среда MCDB-170 and
stocks
Среда МЕМ
Среда МЕМ, 10х
Среда MSCGM
Среда RPMI1640
Среда S-MEM
Среда альфа
Среда бессывороточная
Epi-Life
Среда бессывороточная
Panserin™ 401
Среда Вильямса Е
Среда Гипак-Фиколл
Среда для заключения
Среда для заключения
Vectashield DAPI
Среда для заключения
Vectashield
Среда для заключения
срезов
Среда для заключения
срезов на основе воды
Gel/Mount
Среда для заключения
срезов с флюоресцен¬
цией Cytoseal ТМ 60
Среда для заморажива¬
ния
Среда для криоконсер¬
вации
Среда для роста брон¬
хиального эпителия
(BEGM)
Среда для роста керати¬
ноцитов (KGM)
Среда для роста кле¬
ток скелетных мышц
(SKGM)
Среда для уплотнения
Среда Дюльбекко мо¬
дифицированная по
Iscove’s (IMDM)
Среда Дюльбекко моди¬
фицированная по Игла
(DMEM)
См. Среды
См. Бессывороточные
среды
См. Бессывороточные
среды
См. Бессывороточные
среды
См. Среды
См. Среды
Cambrex
См. Среды
См. Среды
См. Среды
Sigma
CoaChrom
Invitrogen; Sigma
См. Фиколл-Гипак
Fisher
Vector Laboratories
Vector Laboratories
Biomeda; Citifluor; Merck;
Microm; RA Lamb;
Statlab
Biomeda
Microm; Stephens Scien¬
tific; VWR
См. Среда для криокон¬
сервации
MP Biomedicals; Invitro¬
gen; JRH Biosciences;
Sigma
Cambrex (Clonetics; Bio-
Whittaker)
Cambrex; Cascade; Invit¬
rogen; PromoCell; Sigma
Cambrex (Clonetics)
Amersham; Genetic Re¬
search Instr.; MP Bio¬
medicals; Nycomed; Rob¬
bins Scientific; Sigma
См. Среды
См. Среды
Среда заключающая
Histoclear
Среда заключающая
HistoGel
Среда заключающая
Histopaque
Среда замороженная
(DMSO) ORIGEN™
Среда Лейбовитца L-
15 без глютамина
Среда минимальная под¬
держивающая
Среда определенного
состава для кератино¬
цитов (KDM)
Среда определенного
состава для керати¬
ноцитов с добавками
(SKDM)
Среда основная LHC
Среда редуцированная с
сывороткой Opti-MEM
Среда Хэма F-10
(10 мкМ тирозина),
сухая
Среда Хэма F-10
Среда Хэма F12
Среда Хэма F12: DMEM
Среды
Среды бессывороточные
Среды для культуры кле¬
ток насекомых
Fisher
Lab Storage Systems Inc.
Sigma
Origen Biomedical
См. Среды
См. Среды
Cambrex; Cascade; Invit¬
rogen;
См. Среда для роста
кератиноцитов
BioSource
Invitrogen
Invitrogen
См. Среды
См. Среды
См. Среды
ACM Biotech; AES;
ATCC; Biochrom; BD
Biosciences; Biosource;
Caisson; Cambrex; Cas¬
cade; CellGenix; Cell-
Gro-Mediatech; Con¬
naught; Gemini; Genetic
Research Instr.; Medi-
atech; MP Biomedicals;
Irvine Scientific; Invit¬
rogen; JRH; PAA; Paesel
& Lorei; Perbio; Promo¬
Cell; Sigma; StemCell
Technologies; Wako
BD Biosciences; Biosource;
Cascade; CellGenix;
CoaChrom; Roche Di¬
agnostics; Biofluids;
Cambrex (Clonetics);
Cascade Biologicals; Hy-
clone; Hycor; Invitrogen;
Irvine; JRH Biosciences;
Mediatech; Metachem;
MP Biomedicals; PAA;
PromoCell; Roche;
Sigma; Stratech; TCS
Veil Works
BD Biosciences; Bio-
Source; Cambrex;
596
Приложение II. ИСТОЧНИКИ ОБОРУДОВАНИЯ И МАТЕРИАЛОВ
Среды для культуры
тканей
Среды для приготовле¬
ния лимфоцитов
Среды для фосфатного
буфера
Среды на заказ
Среды, солевые раство¬
ры для культуры
Стволовые клетки (вы¬
деление, маркеры, хра¬
нение)
Стекла FlexiPERM (8 ка¬
мер)
Стекла SonicSeal 4-лу-
ночные
Стекла для камер для
культур тканей Lab-
Tek 8 Chamber Tissue
Стекла для культураль¬
ной камеры
Стекла для микроскопии
Стекла многолуночные
Стекла покровные Ther-
шапох
Стекла покровные плас¬
тиковые
Стекла покровные
Стекла предметные
Стеклянные волокна
Стерилизаторы и су¬
шильные шкафы
Стерилизаторы
Стрептавидин-ИТС
Стрептомицин
Hyclone; Invitrogen; МР
Biomedicals; Novagen;
Sigma
См. Среды
Amersham Biosciences;
Cambrex; MP Biomedi¬
cals; Nycomed; Robbins
Scientific; Sigma
Caisson; см. также Среды
См. Среды
Chemicon; Geron;
Metachem; Miltenyi;
Origen; Roche; Universal
Biologicals
Sartorius
Nalge Nunc
См. Стекла для камер
Applied Scientific; Bayer;
BD Biosciences; Bibby
Sterilin; Heraeus;
Metachem; Nalge Nunc;
Stem Cell Technologies
Bellco; Berliner Glas;
H.V. Skan; Laboratory
Sales; Lab-Tek; Microm;
Nalge Nunc; Propper;
Raymond Lamb; Rich¬
ardsons; VWR
См. Камеры для
культивирования,
стекла для
культуральной камеры
Nalge Nunc (см. также
Стекла покровные,
пластик)
Bayer; Coming; МР Bio¬
medicals; Lux; Invitrogen
Bayer; Coming; Fisher;
Invitrogen; Wheaton
См. Стекла для
микроскопии
Millipore; Pall; Whatman
См. Печи, сушильные
шкафы
См. Автоклавы, печи,
сушильные шкафы
Dako Diagnostics
См. Среды
Сульфо-NHS, N-гидрок-
сисульфосукцинимид
Счетчик клеток элект¬
ронный
Счетчики клеток
Счетчики колоний
Сыворотка FCS
Сыворотка быка (отоб¬
ранные партии)
Сыворотка донорская
лошади
Сыворотка донорская
теленка (DCS)
Сыворотка козы
Сыворотка козы норма¬
лизованная
Сыворотка крысы
Сыворотка лошади
Сыворотка рыбы
Сыворотка теленка
Сыворотка фетальная
теленка FBS
Сыворотка фетальная
теленка термоинакти¬
вированная (HIFBS)
Сыворотка цыпленка
Сыворотка, заменители
Сыворотки
Таблетированные сред¬
ства
Таблетки PBS
Термометры с регистри¬
рующим устройством
Термометры электрон¬
ные
Pierce
См. Cell counters
Beckman Coulter; New
Brunswick; Scharfe
BioWorld; BioDu Pont
(NEN Life Sciences);
Cole-Parmer; Don Whit¬
ley; Perceptive Instru¬
ments; PerkinElmer;
Synbiosis
См. Сыворотка
Gemini; Invitrogen; MP
Biomedicals; Sigma; Ser-
aLab
Gemini
См. Сыворотки
Sigma
Vector
Pel-Freeze
См. Сыворотка
East Coast Biologies
См. Сыворотки
См. Сыворотка
Invitrogen; MP Biomedi¬
cals; TCS Biologicals
См. Компоненты сыво¬
ротки
АТСС; Biochrom; Bio-
source; Caisson; Cam¬
brex; Invitrogen; Gemini;
Globepharm; Harlan
Seralab; HyClone; Invit¬
rogen; Irvine; Metachem;
MP Biomedicals; JRH
Biosciences; PAA; Per-
bio; PromoCell; Sero¬
logicals; Sigma; TCS
Biologicals
Johnson & Johnson
Oxoid; См. также Среды
Comark; Cole-Parmer;
Fisher; Harvard; Grant;
Omega; Pierce; Rustrack
Comark; Cole-Parmer;
Fisher; Harvard; Grant;
Labox; Omega; Pierce;
Thermo Electron
Приложение II. ИСТОЧНИКИ ОБОРУДОВАНИЯ И МАТЕРИАЛОВ
597
Термостаты, пропорцио¬
нальные контроллеры
Тест МТТ
Тест ХТТ
Тестирование на микоп¬
лазмы
Тетраметилбензидин
Тефлоновые листы и
тубы
Тиамин-НС1
Тимидин
Тимидин меченный три¬
тием
Тиоглицерин
Тирозин
Токоферол
Толуидин синий
Трансплантат сосудис¬
тый дакроновый
Трансферрин
Трансферрин человече¬
ский
Треонин
Трииодтиронин
Трипсин
Трипсин/ЭДТА
Трипсина заменители
Трипсина заменитель
Trypzean
Триптофан
Трис
Тритон Х-100
Триэтиламин
Трубка диализная
Трубки Pharmed
Трубки силиконовые
Трубки силиконовые для
обмена воздух/С02,
уплотненные платиной
Трубки термоусажива-
ющиеся полиолефи-
новые
Увеличивающие окуля¬
ры
Углерод общий органи¬
ческий (ТОС), оборудо¬
вание для определения
Controls & Automation;
Fisher;
Sigma
Sigma
ATCC; Bionique; ECACC;
DMSZ; Mycoplasma Ex¬
perience
Research Diagnostics
Interplast; Plastim
Sigma
Sigma
NEN Life Sciences
(DuPont)
Sigma
См. Химические реактивы
Sigma
Sigma
Bard
BD Biosciences; Sigma
Sigma
См. Химические реактивы
Sigma
Sigma; Worthington; cm.
также Среды
См. Среды
Hyclone; Innovative Cell
Technologies; Invitro¬
gen; Perbio; Sigma
Sigma
См. Химические реактивы
См. Химические реактивы
Sigma; Fisher
Fisher; Sigma
Cole-Parmer; Chemicon;
Pierce; Serva; Spectrum
Cole-Parmer
Altec; Bibby Sterilin; Dow
Coming; Cole-Parmer;
Nusil; PAW BioScience
Products; Thomas; Wat-
son-Marlow
Cole-Parmer
Appleton Electronics
Bellco; New Brunswick
Scientific
Millipore; Sartec; Thermo-
Electron
Уголь активированный
Ультра-TMB (3,3r, 5,5r-
ттетраметил бензидин )
Ультрамикротом
Универсальные контей¬
неры
Упаковка стерильная,
бумага, полупроницае¬
мая нейлоновая пленка
Уранилацетат
Устройства захвата изоб¬
ражения (в режиме за¬
медленного времени)
Устройство для ионного
напыления
Устройство для ионного
напыления Polaron
SC502
УФ Источники
У Ф-отвердевающий
клей для стекла
Фактор ECGS
Фактор FGF, человече¬
ский рекомбинантный
Фактор GM-CSF
Фактор TGF-pi
Фактор роста EGF ре¬
комбинантный
Фактор роста EGF
Фактор роста HGF/SF,
человеческий, реком¬
бинантный
Фактор роста KGF, че¬
ловеческий рекомби¬
нантный
Фактор роста LIF
Фактор роста NGF
Фактор роста редуциро¬
ванный Matrigel
Фактор роста фиброб¬
ластов
Фактор роста эпидер¬
мальный (rEGF)
Фактор роста эпидер¬
мальный человеческий
(hEGF)
Факторы прилипания
Факторы роста и цито-
кины
Sigma
Research Diagnostics
Leica
Bibby Sterilin; Camlab;
Coming; Elkay; Nalge
Nunc
Applied Scientific; Buck
Scientific; KNF Corp;
Portex; Portland Plas¬
tics; Roth
Plano; SPI Supplies
Scion
Gatan; Fison
Fison
Cole-Parmer; UVP
Summers Optical
См. Факторы роста
См. Факторы роста
См. Факторы роста
R&D Systems; Research
Diagnostics
См. Факторы роста
См. Факторы роста
См. Факторы роста
См. Факторы роста
См. Факторы роста
См. Факторы роста
BD Biosciences; Fisher
Scientific
См. Факторы роста
См. Факторы роста
См. Факторы роста
См. Матрикс
Amersham; Amgen; Aus¬
tral Biologicals; BD
Biosciences; Biodesign;
Biosource; Cambrex;
Cambridge Bioscience;
Collaborative Research
598
Приложение II. ИСТОЧНИКИ ОБОРУДОВАНИЯ И МАТЕРИАЛОВ
Факторы роста стволо¬
вых клеток (SCF)
Фенилаланин
Фенилендиамин (OPD),
таблетки
Фенилизотиоцианат
Феноловый красный
Ферментеры
Фибриновый герметик,
Tisseel VH
Фибробласты
Фибронектин
Фибронектин/коллаген/
БСА
Фибронектин человече¬
ский fibronectin
Фиколл
Фиколл-Гипак
Фильтр нитекс нейлоно¬
вый, 100 мкм
Фильтрация стерилиза¬
цией
Фильтры НТ Tuffryn
Фильтры Tuffryn
Фильтры клеточные
Фильтры стерилизаци¬
онные
Фильтры стерилизаци¬
онные
Фильтры, сита и марле¬
вые тампоны
(BDBiosciences); Gen-
zyme; Invitrogen; MP
Biomedicals; Paesel &
Lorei; Pepro-Tech;
PerkinElmer; Promega;
R&D Systems; Roche;
Serologicals; Sigma;
Stratech; Universal
Biologicals; Upstate Bio¬
technology
См. Факторы роста
См. Химические реактивы
Sigma
Pierce
Sigma
См. Биореакторы
Baxter
См. Специализированные
клетки
Amersham; BD Bioscienc¬
es; Biomedical Technolo¬
gies; Biosource Interna¬
tional; Invitrogen; Lab
Vision; R&D; Sigma;
Stratech; См. также
Матрикс
Biosource International
BD Biosciences (Collab¬
orative Research)
Amersham Biosciences;
MP Biomedicals; Sigma
Amersham Biosciences;
Cambrex; MP Biomedi¬
cals; Nycomed; Sigma
Tekmar-Dohrmann (cm.
также Нейлоновое сито)
См. Фильтры
Pall-Gelman
Pall-Gelman
BD Biosciences; Miltenyi
BD Biosciences; Bibby
Sterilin; Corning; Mil¬
lipore; Nalge Nunc; Om¬
nilab; Pall Gelman; Sar¬
torius; Techno Plastic;
VWR; Whatman.
См. Фильтры,
стерилизации
См. Сетчатые фильтры
Фильтры-насадки на
шприцы
Фильтры-шприцы
Фитогемагглютинин
Флаконы для заморажи¬
вания
Флаконы для культуры
тканей Falcon
Флаконы для культу¬
ры тканей Lifecell
(#420030)
Флаконы для культуры
тканей
Флаконы и мини флако¬
ны сцинтилляционные
Флаконы и чашки Pri¬
maria®
Флаконы и чашки для
культуры тканей, плас¬
тик
Флаконы полистироло-
вые
Флаконы
Флаконы Эрленмайера
Флюоресценция, среды
для заключения
Флюоресценция, среды
для заключения Fluor-
Save
Фолиевая кислота
Формалин забуферен-
ный
Формалина раствор, за-
буференный
Формальдегид
Формамид
Форсуночные порты,
системы Interlink
Фосфокреатин
Фосфоэтаноламин
Фотографические реак¬
тивы, пленки и фото¬
бумага
Фотолитография /фото¬
гравировка
Фотолитография /фо¬
тогравировка, прояви¬
тель MF-319
Microgon; Millipore; Pall-
Gelman; Sartorius; см.
также Фильтры
См. Фильтры
Amersham Biosciences;
Sigma
См. Ампулы
BD Biosciences
Nexell Therapeutics
См. Чашки, флаконы
и планшеты для
культуры
PerkinElmer (Packard)
BD Biosciences (Falcon)
Promo Cell; Sigma
См. Чашки, флаконы
и планшеты для
культуры
См. Чашки, флаконы
и планшеты для
культуры
См. Чашки, флаконы,
планшеты для
культуры
См. Лабораторное стекло
BioMedia; Citifluor; Mi-
crom; Stephens Scien¬
tific; Vector; VWR
Calbiochem
Sigma
Fisher
Sigma
См. Химические реактивы
Merck
BD Biosciences
Sigma
Sigma
Agfa; Eastman Kodak;
Fuji; Ilford
Microchem Corp; Ciba
Specialty Chemicals
Microchem Corp.
Приложение II. ИСТОЧНИКИ ОБОРУДОВАНИЯ И МАТЕРИАЛОВ
599
Фотолитография/ фото¬
гравировка MF-319
Фотопленка Kodak X-
ОМАТ
Фотореактивы просвет¬
ляющие
Фотосенсибилизатор
для фотолитографии
Irgacure 2959
Фотосенсибилизаторы
для фотолитографии
Irgacure 2959
Фторотан
Фунгизон
Фунгициды
Характеризация клеточ¬
ных линий
Хвосты крысиные
Химические реактивы
Хлор
Хлорамфеникол
Хлороформ
Холерный токсин
Холина хлорид
Хондроитинсульфат
Хондроциты
Хромосомные красители
Центрифуги
Центрифугирование в
микротитрационных
планшетах
Циклин-ВМ
Циклический АМФ
Циклоспорин
Цилиндры или лотки для
пипеток
Цинка сульфат гептагид¬
рат
Ципрофлоксацин
Цистеин
Microchem Corp.
Eastman Kodak
Kodak
Ciba Specialty Chemeicals
Ciba Specialty Chemicals
Henry Schein
См. Амфотерицин В
Bio Whittaker; MP Bio¬
medicals; Invitrogen;
Sigma
См. Аутентификация
клеточных линий; ДНК-
фингерпринтинг и
ДНК-профилинт
Biotrol
Calbiochem; Fisher; JT
Baker; Merck; Pierce;
Research Organics;
Roche; Scientific&
Chemical Supplies; Sig¬
ma; USB; Wako
Hays Chemical Dis¬
tribution (см. также
Дезинфектанты)
Sigma
См. Химические реактивы
EMD; List Biological;
Merck; Sigma
Sigma
Sigma
Cm. Specialized cells
Applied Imaging; Cambio;
Vysis
Beckman; DuPont (Sor-
val); Fisher; Kendro;
Thermo-IEC; Life Sci¬
ences International
Beckman Coulter; Thermo
Electron
Roche Diagnostics
Sigma
Novartis; Sigma
Bel Art; Fisher; Nalge
Nunc; Radleys
См. Химические реактивы
Bayer
Sigma
Цистеина гидрохлорида
гидрат
Цистин
Цитобакеты
Цитокины
Цитометр сканирующий
Цитометр
Цитофектин GSV
Цитоцентрифуга
Чашки
Чашки Petriperm
Чашки для культуры на
предметных стеклах
Чашки культуральные,
бактериологические
Чашки Петри Stanzen
Чашки Петри
Чашки с поли-О-лизино-
вым покрытием
Чашки с пониженным
прилипанием, 35 мм
Чашки
Чашки флаконы и план¬
шеты для культуры
Чашки флаконы и план¬
шеты для культуры,
обработанные колла¬
геном
Шейкер ротационный
Шейкер-инкубатор
Шпатели
Шприц Cornwall
Штопфер для автомати¬
ческих пипеток
См. Химические реактивы
См. Химические реактивы
Thermo Electron Corpora¬
tion
См. Факторы роста
Beckman Coulter; Com-
pucyte (см. также
Микроскопы)
См. Проточный
цитометр;
сканирующий
цитометр
Glen Research
Bayer; CSP; Electron
Microscopy Sciences;
Thermo Electron; Sakura
Finetek; Wescor
Combi Ring Dish Renner;
Germany
Sartorius
Vivascience; см. также
Чашки, флаконы,
планшеты для
культуры
BD Biosciences; Fisher
Greiner
См. Чашки, флаконы,
планшеты для
культуры
BD Biosciences (Biocoat)
Coming (Costar)
См. Культуральные
чашки, флаконы и
планшеты
BD Biosciences (Falcon);
Bibby Sterilin; Corning
(Costar); Fisher; Greiner;
Invitrogen; Iwaki; MP
Biomedicals (Lux); Nalge
Nunc; Sarstedt; TPP
BD Biosciences; Coming;
Iwaki Glass
Boeker Scientific; New
Brunswick
Camlab; New Brunswick;
Radleys
Fisher; VWR
Popper; Cole-Parmer
См. Штопфер для
пипеток
600
Приложение II. ИСТОЧНИКИ ОБОРУДОВАНИЯ И МАТЕРИАЛОВ
Штопфер для пипеток
Щелочная фосфатаза.
Материалы для иссле¬
дования
Щелочная фосфатаза.
Набор реактивов для
определения (Red)
Щприцы
ЭГТА
ЭДТА
ЭДТА, раствор стериль¬
ный
Экстракт гипофиза (РЕ)
Экстракт эмбриона цып¬
ленка
Эластаза
Эластаза поджелудоч¬
ной железы
Электрофорез
Элютриатор центрифуж¬
ный
Эмульсия для автора¬
диографии
Эндотелии 1, человече¬
ский, свиной (ЕТ-1)
Bellco; Camlab; Volac
Sigma
Vector
Baxter; BD Biosci¬
ences; Popper(и
основные поставщики
лабораторного
оборудования)
Sigma
JT Baker; Merck; Sigma
См. Среды
Cambrex (Clonetics); Cas¬
cade; Invitrogen; Promo¬
Cell
Accurate Chemical & Sci¬
entific; Invitrogen
Roche Diagnostics; Sigma
Sigma
Amersham Biosciences;
Anachem; Bio-Rad; Invi¬
trogen; Haake; Innova¬
tive Chemistry; Thermo
Electron
Beckman Coulter
Amersham Biosciences
Sigma
Энтеллан, Entellan
Эпидермальный фактор
роста
Эпинефрин
Эпон
Эрленмайера флаконы
для культуры тканей
Эстрадиол
Этанол
Этаноламин
Этидий бромистый
Этикетки для заморажи¬
вания
Этилендиаминтетрааце-
тата двунатриевая соль
Ядерный быстрый крас¬
ный
Яйца куриные
Ярлыки
EDC, 1-этил 3-(3-димети
ламинопропил)карбод
иимид
туо-инозитол
ТРА, 12-О-тетрадекано-
илфорбол-13-ацетат
TRIzol
Tween 20
Tween 80
X-gal: 5-бром-4-хлор-3-
индолил-(3-0-галакто-
пиранозид
Merck
См. Факторы роста
Sigma
Merck; LR-White; Roth
См. Лабораторное стекло
Sigma
См. Химические реактивы
Sigma
Sigma
Computer Imprintable
Label Systems; GA Inter¬
national;
Cm. EDTA
Sigma
Charles River Laboratories
Triple Red; Cm.
также Ярлыки для
криофлаконов
Pierce
Sigma
Sigma
Invitrogen
Sigma
Sigma
Roche
Приложение III
ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ
И ДРУГИЕ ИСТОЧНИКИ
Вначале приводятся номера телефонов, затем номера факсов; два номера, отделенные точкой с запятой означают,
что доступно две линии. Международному коду предшествует знак “+”, который следует заменить соответствующим
кодом выхода на международную телефонную сеть. Продукция упомянута в конце; приводятся только те, которые
упоминаются в настоящем руководстве или связанные с культуральной работой. У авторов не было намерения
давать весь список продукции, предоставляемой поставщиками.
Abbott Laboratories. 100 Abbott Park Road, Abbott
Park, North Chicago, IL 60064-3500, USA. 800-323-
9100; 1-847-937-6100. www.abbott.com. Нембутал.
Accuramatic. 40-42 Windsor Road, Kings Lynn, Norfolk
PE30 5PL, England, UK. 01553 777253.01553 777253.
info@accuramatic.co.uk. www.accuramatic.co.uk. Авто¬
матические диспенсеры, устройства для разлива жид¬
костей, перистальтические разливочные машины.
Accurate Chemical & Scientific Corp. 300 Shames Dr.,
Westbury, NY 11590, USA. +1 516-333-2221; 800-
645-6264. +1516-997-4948. info@accuratechemical.
com. www.accuratechemical.com/. Антитела; сепарация
биомассы; экстракт эмбриона цыпленка; исследова¬
ния межклеточного вещества; клетки HeLa; микро¬
центрифуги.
АСЕ-Autoclave Control Engineering. Unit E, Caven¬
dish Courtyard, Sallow Road, Weldon North, Corby,
Northants.,NNl 5JX, England, UK. +44 (0)1536 206 200.
+44 (0)1536 206 202. sales@aceautoclaves.co.uk. www.
ace-autoclaves.co.uk Крупномасштабные автоклавы;
парогенераторы.
ACM-Biotech GmbH. Josef-Engert Strasse 9, D-
93035 Regensburg, Germany. +49 (0)941 942 743. +49
(0)941 942 7444. techservice@acm-biotech.com, www.
acm-biotech.com. Эндотелиальные клетки; Epiflow;
EPIFLOW: система культивирования клеток с исполь¬
зованием непрерывного обмена среды и постоянного
снабжения газом; гепатоциты; исследования in vitro;
кератиноциты; мембранная перфузия; органотипичес¬
кая культура; селективные бессывороточные среды.
Advanced Instruments Inc. Two Technology Way, Nor¬
wood, MA 02062, USA. 781 320 9000, 800 225 4034.
781 320 8181. mail@aitests.com, www.aitests.com. Ос¬
мометр.
AES Laboratoire. Rue Maryse Basti. e, Ker Lann, CS
17219, F-35172 Bruzcedex, France.+33 (0)2 23 50 12 12.
+33 (0)2 23 50 12 00. aes@aeslaboratoire.com, www.
aeslaboratoire.com. Ламинарный шкаф; среды; боксы
микробиологической защиты.
Aflymetrixlnc. 888-362-2447. +1 408-731-544 \. support©
affymetrix.com; sales@affymetrix.com. www.affymetrix.
сот. Микрочипы для анализа ДНК.
Affymetrix UK. +44 (0) 1628 552550. +44 (0) 1628 552598.
suppofteuwpe@affymetrix.com; saleseurope@affymetrix.com,
www.affymetrix.com. DNA Микрочипы для анализа.
Agilent Technologies. Quantum Analytics, Inc., 363 Vin¬
tage Park Drive, Foster City, С A 94404, USA. 800-227-
9770; 800-992-4199. cag enquiry@agilent.com. www.
chem. agilent.com. Флюоресцентный биоанализатор;
микрочипы для анализа ДНК.
Agilent Technologies UK Ltd. Life Sciences & Chemi¬
cal Analysis Group, Lakeside, Cheadle Royal Business
Park, Stockport, Cheshire SK8 3GR, England, UK.
+44 (0)161 492 7500; 0845 712 5292. 0845 600 8356.
cag enquiry@agilent.com. www.agilent.com/chem/uk.
Флюоресцентный биоанализатор; микрочипы для
анализа ДНК.
Air Packaging Technologies Inc. 25620 Rye Can¬
yon Road, Valencia, CA 91355, USA. +1 661-294-
2222.+1 661-294-0947. llchappell@aol.com. Упаковка
в пакеты с наполнением воздухом; транспортные
контейнеры для клеток.
Air Products & Chemicals, Inc. 7201 Hamilton Blvd,
Allentown, PA 18195, USA. 610-481-4911; 800-654-
4567. 800-880-5204. info@apci.com. www.airproducts.
сот/. C02, автоматический блок переключения для
баллонов с углекислым газом, С02-контроллеры.
Air Products pic. 2 Millennium Gate, Westmere Drive,
Crewe CW1 6AP, England, UK. 0800 389 0202.0193
2 258 502. info@apci.com. wzem.airproducts.co.uk. C02,
автоматический блок переключения для баллонов с
углекислым газом, С02-контроллеры.
Air Sea Atlanta. Atlanta, GA. 1-404-351-8600;. www.
airseaatlanta.com. Контейнеры для транспортировки;
системы безопасности.
Aire Liquide. Division Materiel Cryogenique, Parc Gus¬
tave Eiffel, 8 rue Gutenberg, Bussy Saint Georges 77607,
Marme-la-Valle, Cedex3, France. +33 (0)1 64 76 15 00.
+33 (0)1 64 76 16 99. www.dmc.airliquide.com/. Моро¬
зильники с жидким азотом. Сосуды Дюара.
Aire Liquide America Corp. 2700 Post Oak Blvd., Suite
1800, Houston, Texas 77056, USA. +1 713 624 8000.+1
713 402 2149. www.dmc.airliquide.com/. Морозильники
с жидким азотом. Сосуды Дюара.
602
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
Aire Liquide (UK). См. Borolabs. www.dmc.airliquide.
сот/. Морозильники с жидким азотом. Сосуды
Дюара.
Alabama Research and Development. A Division of Ala¬
bama Specialty Products, P.O. Box 739, Munford, AL
36268, USA. +1 (256) 358-0460. +1 (256) 358-4515.
www.alspi.com/Slicer.htm. Гистологический нож для
тканей Krumdieck.
Albany International Research Co. Albany International
Research Co., PO Box 9114, 111 West Street, Mansfield,
MA 02048-9114, USA. +1 508 339 7300; 800 992 5017.
research@albint.com. xsmw.airesco.com. Платформы для
тканевой инженерии.
Alconox Inc. 30 Glenn Street, Suite 309, White Plains, NY
10603, USA. +1 914 948 4040. +1 914 948 4088. clean-
ing@alconox.com. www.alconox.com. Детергенты.
Aldrich Chemical Co., Inc. 1001 W. St. Paul Ave., Mil¬
waukee, WI 53233, USA. 800-325-3010. 414 273 4979.
www.sigma.sial.com/aldrich. Химические соединения,
биохимические вещества; ЭДТА.
Alexis Corp. (UK) Ltd. P.O. Box 6757, Bingham, Not¬
tingham NG13 8LS, England, UK. +44 (0)1949 836 111.
+44 (0)1949 836 222. alexix-uk@alexis-corp.com. www.
alexis-corp.com. Ангиогенез; антитела: цитокины, ин-
терфероны; рецепторы, передача сигнала; VEGF.
Alexis Corp. (USA). 6181 Cornerstone Court East, Suite
103, San Diego, С A 92121, USA. +1 858-658-0065; 800-
900-0065. +1 858-550-8825; 800-550-8825. axxoraus@
axxora.com. www.alexis-corp.com. Ангиогенез; анти¬
тела: цитокины, интерфероны; рецепторы, передача
сигнала; VEGF.
Alfa Laval. 7DomanRoad, Camberley, Surrey GU15 3DN,
England, UK. +44 (0)1276 63383. +44 (0)1276 685035.
general, uk@alfalaval.com. www.alfalaval.co.uk. Фер¬
ментеры, биореакторы паровые стерилизаторы.
Alpha Laboratories Ltd. 40 Parham Drive, Eastleigh,
Hants S05 4NU, England, UK. +44 (0) 23 8048 3000.
+44 (0) 23 8064 3701. info@alphalabs.co.uk. www.
alphalabs.co.uk/. Ампулы; автоматические пипетки;
центрифужные пробирки; флаконы для заморажи¬
вания; сухие нагреватели блока цилиндров; микро¬
пипетки Pastettes; пипетки; пастеровские пипетки,
приспособление для пипетирования; наконечники
для автоматических пипеток.
Alfa Medical. 59 Madison Ave., Hempstead, NY 11550,
USA. 800-801-9934. +1 516-489-9364. eMail@sterili-
zers.com. www.sterilizers.com/. Полоски-индикаторы
стерильности.
Altec Products Ltd. Bude Business Centre, Bude,
Cornwall, EX23 8QN, England, UK. 0845 359 9000;
+44 1288 357820. 0845 359 9090; +44 1288 357822.
sales@altecweb.com. wbm.altecweb.com. Автоклавиру-
емые пакеты; соединения Люэра; перистальтические
насосы; продукция для обеспечения безопасности;
силиконовые трубки.
Ambion Inc. 2130 Woodward, Austin, ТХ 78744-1832, USA
800 445-1161; +1 (512) 651-0200.+1 (512) 651-0201.
custom@ambion.com. vuww.ambion.com. ДНК-матрицы;
выделение РНК; иРНК; RNAseZap; siRNA; RT-PCR.
Ambion UK. Ermine Business Park, Spitfire Close, Hunt¬
ingdon, Cambridgeshire, PE29 6XY, England, UK. +44
(0)1480 373020; 0800 138 1836. +44 (0)1480 373010.
custom@ambion.com. wx0w.ambion.com. ДНК матрицы;
выделение РНК; RNAi; RNAseZap; siRNA; RT-PCR.
American Association for Cancer Research. 615 Chest¬
nut St., 17th Floor, Philadelphia, PA 19106-4404, USA.
+1 (215) 440-9300. +1 (215) 440-9313. membership@
aacr.org. x0x1m.aacr.org. Общество исследования рака;
журналы, научные встречи.
American Fluoroseal. 431-D East Diamond Avenue,
Gaithersburg, MD 20877, USA. 301.990.1407, 301
990 1472. info@americanfluoroseal.com. www.toafc.
сот/. Мешки для сред.
American Society for Cell Biology. 8120 Woodmont
Avenue Suite 750, Bethesda, MD 20814-2762, USA.
+1 301 347 9300. +1 301/347-9310. ascbinfo@ascb.org.
wx0w.ascb.org/. Журналы, научные встречи, научные
общества.
American Type Culture Collection, (см. АТСС).
Amersham Biosciences. 800 Centennial Ave., P.O. Box
1327, Piscataway, NJ 08855-1327, USA. 800-526-3593.
877-295-8102. cs-us@amersham.com. X0ww4.amersham -
biosciences.com. Антитела; наборы для исследования;
среды для хроматографии; цитокины; DEAE декстран;
уплотняющие среды; стандарты ДНК; электрофорез;
ELISA; эпидермальный фактор роста; фибронектин;
Фиколл-гипак; факторы роста; Hybond N; среда для
приготовления фракции лимфоцитов; маркерные бу¬
сины; микроносители; Перколл; перистальтические
насосы; ФГА; поставки сухих сред; радиоизотопы;
рецепторы; передача сигнала; метризоат натрия;
Ultraser-G; Verax.
Amersham Biosciences. Amersham Biosciences UK Ltd.,
Pollards Wood, Nightingales Lane, Chalfont St. Giles,
Bucks, HP8 4SP, England, UK. 0870 606 1921. +44
(0)1494 498 231; 0800 616 927. orders.gb@amersham.
com. www4.amershambiosciences.com. Продукция см.
Amersham Biosciences (USA).
Amersham Pharmacia Biotech, cm. Amersham Biosci¬
ences.
Amgen, Inc. One Amgen Center Drive, Thousand Oaks,
CA 91320-1799, USA. +1 805-447-1000. +1 805-447-
1010. newproducts@amgen.com. x0x0wAmgen.com. Фак¬
торы роста; FGF.
Amicon, Inc. cm. Millipore.
Amphioxus Cell Technologies, Inc. 11222 Richmond
Ave, Suite 180, Houston, TX 77082-2646, USA.800-
777-2707.281-679-7910. activox@amphioxus.com. хеше).
amphioxus.com. Белки сыворотки человека; клеточные
линии печени человека; гепатоциты; половолокон¬
ные системы культивирования; токсикологические
исследования in vitro.
Anachem Ltd. Anachem House, 20 Charles Street, Luton,
Beds LU2 0EB, England, UK. +44 (0)1582 745 060. +44
(0)1582 745 115. lifescience@anachem.co.uk; sales@
anachem.co.uk.x0x0w.anachem-ltd.com. Деконтаминация;
создание цифровых образов; электрофорез; хранение
в морозильнике; среды; микротитрационные планше¬
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
603
ты; микропробирки; наборы для выделения и очистки
нуклеиновых кислот; пипетки; автоматические пи¬
петки; геметезирующие прокладки для планшетов;
поддоны; перемешивающие флаконы; тампоны для
взятия мазков; наконечники, пробирки.
Analytical®Technologies. Lynchford House, Lynchford
Lane, Farnborough, Hampshire, GU14 6LT, England,
UK. +44 (0)1252 514 711. +44 (0)1252 511 855 info@
analyticaltechnologies.co.uk. www.analyticaltechnolo-
gies.co.uk. Биохимический анализатор; биосенсоры;
коллоид-осмометр; мониторинг: глюкоза, глютамин,
глютамат, лактат; осмометры.
Anglia DNA Bioservices, Ltd. Norwich Research Park,
Colney Lane, Norwich, NR4 7UH, Norfolk, UK. +44
(0)8454 565 365. office@angliadna.co.uk. www.angli-
adna.co.uk. Аутентификация. Генетическая идентич¬
ность, тестирование; ДНК-профилинг.
Ansell Medical. Ansell Medical, 119 Ewell Road, Surbiton,
Surrey KT6 6AL, England, UK. +1 (0)181 481 1804.
+ 1 (0)181 481 1828. www.ansell.com. Нитрильные
перчатки.
Ansell Healthcare. Ansell—Red Bank, 200 Schulz Drive,
Red Bank, NJ 07701, USA +1 732 345 5400. www.ansell.
com. Нитрильные перчатки.
Anton Paar GmbH. Karnter Strasse 322, A-8054 Graz,
Austria. +43316257 0. +43 316257257. density@an-
tonpaar.com; rheology@anton-paar.com. www.anton-
paar.com/. Денситометры, реометры, вискозиметры.
Anton Paar USA Inc. 10215 Timber Ridge Drive, Ashland,
VA 23005, USA. (800)-722-7556; +1 804-550-1051.
804-550-1057. info.us@anton-paar.com. www.antonpaar.
сот/. Денситометры, реометры, вискозиметры.
Apex Engineering Products Corp. 1241 Shoreline Drive, Au¬
rora, IL60504, USA+1 815 436 2200;800 451 6291.+1 630-
820-8886; 815 436 9418. rdo@rdo-apex.com. www.rdo-apex.
com. Декальцифицирующие агенты д ля исследователь¬
ских и научных целей (RDO).
Appleton Electronics. 205 W. Wisconsin Ave., Appleton,
WI54911, USA. (800) 877-8919.001 (920) 734-5172.
www.aedwis.com. Элементы электронного обору¬
дования; полиолефиновые термоусаживающиеся
трубки.
Appleton Woods Ltd. Lindon House, Heeley Rd.,
Selly Oak, Birmingham B29 6EN, England, UK. +44
(0)121 472 7353. +44 (0)121 414 1075. info@appleton-
woods.co.uk. wi0w.appletonwoods.co.uk. Термоиндика¬
торные полоски.
Applied BioPhysics, Inc. 1223 Peoples Ave., Troy, NY
12180, USA. +1 518 276 2165. +1 518 276 2907. www.
biophysics.com. Клеточная адгезия; импеданс клетки;
TEER; трансэпителиальное сопротивление.
Applied Biosciences Corp. P.O. Box 520518 Salt Lake
City, UT 84152, USA. 800-280-7852; 801-485-4988.
801-485-4987. zemw.applied-biosciences.com. Системы
очистки воды.
Applied Biosystems. 850 Lincoln Centre Drive, Foster
City, С A 94404, USA. 800 327 3002; +1 650 638 5800.+
1 650 638 5884. www.appliedbiosystems.com. Праймеры
TaqMan 2X PCR Master Mix.
Applied Imaging. 120 Baytech Drive, San Jose CA
95134-2302, USA. +1 408 719 6400; 800 634 3622.
+ 1 408 719 6401. info@aicorp.com. www.aicorp.com.
Хромосомное окрашивание; анализ изображения;
подсчет окрашенных точек.
Applied Imaging International Ltd. BioScience Centre,
Times Square, Newcastle upon Tyne, NE1 4EP, England,
UK. +44 (0)191 202 3100. +44 (0)191 202 3101. info@
aii.co.uk. www.aicorp.co. Хромосомное окрашивание;
анализ изображения; подсчет окрашенных точек.
Applied Immune Sciences. 5301 Patrick Henry Dr., Santa
Clara, CA 95054, USA. +1 408-980-5812. Иммунное
окрашивание, сепарация клеток.
Applied Medical Technology Ltd. 4 Orwell Furlong,
Cowley Road, Cambridge CB4 0WY, England, UK.
+44 (0)1223 420415. +44 (0)1223 420797. support@
applied-medical.co.uk. www.applied-medical.co.uk/.
Инфузионные насосы; инфузионные системы; кон¬
некторы Люэра и QR.
Applied Scientific. 154 W. Harris Ave., South San Francis¬
co, CA 94080, USA. +1 650 244 9851. +1 650 244 9866.
Антибиотики; мешки для автоклавирования; мешки
биологической опасности; скребки для клеток; клеточ¬
ные сита; стекла для камер; криофлаконы; скрининг
культуры; флаконы; перчатки; среды; микропипетки;
многолуночные планшеты; иглы; пипетки; автомати¬
ческие пипетки; Pi-насосы; роллерные бутыли; липкие
ленты для индикации стерильности; шприцы.
Appropriate Technical Resources Inc. см. ATR.
Argos Technologies Inc. 1141 East Main Street, Suite
104, East Dundee, IL 60118, USA. +1 847 783 0456.
+1 847783 0380. pkneisel@argos-tech.com. www.argos-
tech.com. Оборудования для разлива жидкостей,
половолоконные перфузионные системы культур;
роллерная культура; мешалочная культура; вакуум¬
ный насос.
Astell Scientific Ltd. Powerscroft Road, Sidcup, Kent,
DA14 5DT, England, USA. +44 (0)20 8309 2023.+44
(0)20 8309 2036. sales@astell.com. www.astell.com. Ав¬
токлавы, настольные автоклавы; устройство регист¬
рации данных; стерилизаторы, сушильные шкафы.
АТСС (American Type Culture Collection). 10801 Uni¬
versity Boulevard, Manassas, VA 20110-2209, USA.
+ 1 703-365-2700. +1 703-365-2701. Информация о
продукции: tech@atcc.org\ База данных: help@atcc.
org. www.atcc.org. Клеточные линии; клеточный банк;
фидерные слои; гибридомы; среды; служба детекции
микоплазм; наборы для ПЦР- определения микоп¬
лазм; сыворотка; плазмиды.
Atlas Clean Air Ltd. 176 Lomeshaye Business Village,
Turner Road, Nelson, Lancashire BB9 7DR, England,
UK. +44 (0)1282 447 666. +44 (0)1282 447 789. info@
atlascleanair.com. www.atlascleanair.com. Ламинарные
шкафы; боксы микробиологической защиты.
ATR (Appropriate Technical Resources) Inc. P.O. Box
460, 9157 Whiskey Bottom Road, Laurel, MD 20723,
USA. +1 301 470 2799. +1 410 792 2837. inforeq@
atrbiotech.com. www.atrbiotech.com. Инкубаторы; фер¬
ментеры; шейкеры.
604
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
Austral Biologicals. 125 Ryan Industrial Ct., Suite
207, San Ramon, С A 94583, USA. +1 925 820 8390;
800 433 7105. +1 925 820 6843. sales@australbio.com.
www.australbiologicals.com. Антитела: цитокины; фак¬
торы роста; рецепторы.
Autogen Bioclear. Holly Ditch Farm, Mile Elm, Caine,
Wiltshire, SN11 0PY, England, UK. 0800 652 6744; +44
(0)1249 819008. +44 (0)1249 817266. zemw.autogenbio-
clear.com. Антибиотики. Клетки НЕК 293; MCDB 153;
среды; Primocin; Sebomed.
Axxora LLC. 6181 Cornerstone Court East, Suite
103, San Diego, CA 92121-4727, USA. 800 550 3066;
+1 858 550 8824. +1 (858)550-8825; 1-800-550-8825.
www.biolog. de/usa.html. Ангиогенез; антитела: цито¬
кины; факторы роста, передача сигнала; VEGF.
Baker Co. Inc. P.O. Drawer Е, 161 Gatehouse Road, San¬
ford, ME 04073, USA. +1 207 324 8773; 800 992 2537.
+1 207 324 3869; 800-992-2537. bakerco@bakerco.com.
zemw.bakerco.com. Ламинарные шкафы, боксы микро¬
биологической защиты.
Bard, С. R., Inc. см. C.R. Bard Inc.
Barnstead Thermolyne Corp. 2555 Kerper Boule¬
vard, P.O. Box 797, Dubuque, IA 52004-0797, USA.
800 446 6060; +1 319 589 0538. +1 319-589-0516. mkt@
barnsteadthermolyne.com. zmw.barnsteadthermolyrie.com/.
С02-контроллеры; С02-инкубаторы; диспенсеры; дис¬
тилляторы обратного осмоса; морозильники; терми¬
ческий кожух; плитки; морозильники с азотом; печи;
автоматические пипетки; регистрирующие устройства;
стерилизаторы, мешалки, системы очистки воды.
Baxter Healthcare. One Baxter Parkway, Deerfield, IL
60015-4625, USA. 800-422-9837; +1 847-948-4770;
+ 1 847-948-2000. +1 847-948-3642. www.baxter.com.
Соединения Люэра клапанные; шприцы; наборы
соединительных трубок.
Bayer Corp. Bayer Consumer Care Division, 36 Colum¬
bia Road, P.O. Box 1910, Morristown, NJ 07962-1910,
USA. 800-331-4536. zemw.bayermhc.com. Антибиоти¬
ки; ципрофлоксацин; гистология; приготовление
срезов; элиминация микоплазм; покровные стекла
Thermanox.
Вауегpic. Strawberry Hill, RG14 lJANewbury, Berkshire,
England, UK. +44 (0)1635 563 000. +44 1635 563 300.
medical.science@bayer.co.uk. zsmw.bayer.co.uk/. Антиби¬
отики; ципрофлоксацин; гистология; приготовление
срезов; элиминация микоплазм; покровные стекла
Thermanox.
BCL. См. Roche Diagnostics.
BD Biosciences—Discovery Labware. 2 Oak Park,
Bedford, MA 01730-9902, USA. 800 343 2035.
800 743 6200. labware@bd.com. zemw.bd.com/. Агар, ав¬
томатические пипетки, Бактопептон; бронхиальный
эпителий; исследования на Сасо-2; вкладыши для
культуры клеток; скребки для клеток; колллаген; чаш¬
ки, обработанные коллагеном; стекла для культуры;
фибронектин; фильтры; вкладыши в фильтрацион¬
ную лунку; FACS; флаконы; проточный цитометр; га¬
зопроницаемые крышки; факторы роста; гидролизат
лактальбумина; ламинин; Матригель; микробиоло¬
гические среды; многолуночные планшеты; Natrigel;
питательные бульоны; чашки для органной культуры;
сетки для органной культуры; определение пептидов
peptide TEER; триптоза, пробирки, витронектин.
BD Biosciences. Tullastrasse 8-12, 69126 Heidelberg,
Germany. +49 6221 305 0. +49 6221 305 216. mail@
bdbiosciences.com. www.bd.com/. Продукция см. BD
Biosciences—Discovery Labware.
BD Biosciences Immunocytometry Systems. 2350 Qume
Dr., San Jose, CA 95131-1807, USA. 877 232 8995.
+ 1 408-954-2347. facservice@bdis.com. zemw.bd.com/.
Антитела, сепарация клеток; FACS; проточный ци-
тофлюориметр.
BD Biosciences UK Ltd. Between Towns Road, Cowley,
Oxford, OX4 3LY, England, UK. +44 1865 781 688.+44 1
865 781 578. mail@bdbiosciences.com. zemw.bd.com/. Cm.
BD Biosciences—Discovery Labware.
Beckman Coulter Inc. 4300 N. Harbor Blvd., Box 3100,
Fullerton, CA 92834-3100, USA. 1-800 742 2345;
+1 714 871 4848.800 643 4366; +1 714 773 8283. www.
beckmancoulter.com/. Счетчики клеток; центрифуж¬
ный прибор для отмучивания; центрифуги; деление
клеток по размерам; денситометры; ДНК-зонды;
проточные цитометры; реактивы для иммуногисто¬
химии; латексные частицы (стандарт); манипуляции
с жидкостями, автоматизированное рабочее место;
центрифугирование микротитрационных планшетов;
роботизированные системы для клеточных исследо¬
ваний; сцинтилляционные счетчики; спетрофотомет-
ры (UV/VIS); Vi-CELL.
Beckman Coulter (UK) Ltd. Oakley Court, Kingsmead
Business Park, High Wycombe, Bucks., HP11 1JU, Eng¬
land, UK. +44 (0)1494 441 181. +44 (0)1494 447 558.
beckmancoulter uk@beckman.com. zemw.beckmancoulter.
com. Продукция см. Beckman Coulter Inc.
Becton Dickinson. Cm. BD Biosciences.
Bel-Art Products. 6 Industrial Road, Pequannock,
NJ 07440-1992, USA.+l 973 694 0500; 800 4BEL-
ART.+l 973 694 7199. zemw.bel-art.com. Аспираторная
емкость; мешкодержатели; бутыли; держатели буты¬
лей; оплетенные бутыли; скребки для клеток; кольца
для клонирования; коробки для культур; культураль¬
ные камеры; штативы для культуральных флаконов;
сушильная камера; счетчики для жикостей и газов;
лотки; магнитный скребок для клеток; груша для пи-
петирования; цилиндры для пипеток; автоматические
пипетки; перистальтический насос; поддоны; очки
биобезопасности; сифонное устройство для мытья
пипеток; коробки для предметных стекол; запорные
клапаны; система трубок.
Bellco Glass Inc. 340 Edrudo Rd., Vineland, NJ 08360-
3493, USA. 609 691 1075, 800 257 7043. 609 691 3247.
sales@bellcoglass.com. www.bellcoglass.com. Биореак¬
торы; боросиликатное стекло; кольца для клониро¬
вания; деионизаторы; флаконы для ферментеров;
инкубаторы; магинтные мешалки; увеличивающее
устройство для простмотра; емкости для пипеток;
штопферы для пипеток; поддоны; шейкеры; флаконы
для трипсинизации.
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
605
Bennett Scientific. Тог Green, Croft Road, East Ogwell,
Newton Abbot, Devon, TQ12 6BA, England, UK. +44
(0)1626 369 990. +44 (0)1626 369 992. sales@nbennet-
scientific.com. www.bennett-scientific.com. Автоклавы;
магнитные мешалки; насосы: перистальтический,
пистонный, вакуумный, индикаторы стерильности
Thermalog.
Berthold Technologies. The Priory, High Street,
Redburn, St Albans, AL3 7BR, England, UK. +44
(0)1582 79 1999. +44 (0)1582 79 1937.James.Grand©
Berthold.com. www.Bertholdtech.co.uk. Поглощение
света; биоаналитические инструменты; флюорес¬
ценция; люминесценция; микропланшетный сканер;
Mithras.
Berthold Technologies. Calmbacher Strasse 22,75323 Bad
Wildbad, Germany. +49 (0)7081-177 216.+49 (0)7081-
177 301. Winfried. Erdmann@Berthold.com. www.ber-
tholdtech.com. Поглощение света; биоаналитические
инструменты; флюоресценция; люминесценция;
микропланшетный сканер; Mithras.
Bibby Sterilin Ltd. Beacon Road, Stone, Staffordshire,
ST15 0SA, England, UK. +44 (0)1785 812 121.+44
(0)1785 815 066. bsl@bibby-sterilin.com. www.bibby-
sterilin.com. Аспираторы; мешки биобезопасности;
вкладыши в крышки для бутылей; диспенсеры для
горловин бутылей; бутиловые пробки; стекла для
камер; криофлаконы; чашки; ESCO резина; микро¬
носители Fibra-Cel; фильтры; флаконы; стеклянные
чашки; магнитные мешалки; ручное устройство для
подсчета колоний; бутыли для сред; микропипетки;
кольцевые шейкеры; пипетки; качалочный штатив;
силиконовые трубки; силиконовые стопперы; Si-
lescol; стерилизационные мешки; универсальные
контейнеры.
Bigneat Ltd. 4 & 5 Piper’s Wood Industrial Park, Water-
bury Drive, Waterlooville, Hampshire, P07 7XU, Eng¬
land, UK. +44 (0)23 92 266 400. +44 (0)23 92 263 373.
info@bigneat.com. www.bigneat.com. газовые шкафы
без выводных каналов; ламинарные шкафы; боксы
биологической защиты.
Binder GmbH. Bergstrasse 14, D-78532 Tuttlingen, Ger¬
many. +49 (0) 7461 1792 0. +49 (0) 7461 1792 10. info@
binder-world.com. www.binder-world.com. Инкубаторы:
безводные, C02, с охлаждениемением.
BioCarta. 1-877-641-2355 x227. +1 760-804-1395. info@
biocarta.com. www.biocarta.com/. сепарация клеток;
цитокины; выделение лимфоцитов; бусины для маг¬
нитного разделения клеток; реактив для выделения
лимфоцитов PrepaCyte.
BioCarta Europe. +49 40 525 7030. +49 40 525 70377.
info, europe@biocarta.com. www.biocarta.com/. сепа¬
рация клеток; цитокины; выделение лимфоцитов;
бусины для магнитного разделения клеток; реактив
для выделения лимфоцитов PrepaCyte.
Biochrom AG. РОВ 46 03 09, D-12213 Berlin, Ger-
many.+49 30 77 99 06 0. +49-30 77 10 01 2; 49-30-
77 99 06 66. info@biochrom.de. www.biochrom.de.
Антитела; среда для выделения клеток; среды; сы¬
воротки.
Biocolor Ltd. 10 Malone Road, Belfast BT9 5BN,
Northern Ireland, UK. +44 (0) 1232 459 008. +44 (0)
1232 551 733. info@biocolor.co.uk. www.biocolor.co.uk.
Коллаген; исследования коллагена; Blyscan GAG
исследования; исследования матрикса.
Biocompare, www.biocompare.com. Руководство для
пользователей; справочник по продукции; источники
материалов и оборудования.
BioCrystal, Ltd. 575 McCorkle Blvd., Westerville,
OH 43082-8888, USA. +1 (614) 818-0019.+1 (614)
818-1147. sales@opticell.com. www.opticell.com. Куль¬
туральная камера OptiCell.
Biodesign International. 60 Industrial Park Road, Saco,
ME 04072, USA. 207-283-6500; 888-530-0140.207-283-
4800. info@biodesign.com. zvww.biodesign.com. антитела;
кардиальные маркеры; CD маркеры; коллаген; цито¬
кины; факторы роста; матрикс; сыворотка.
Biofluids. См. Biosource International.
Biogene. BioGene House, 6 The Business Centre, Har¬
vard Way, Kimbolton, Cambs PE28 0NJ, England, UK.
+44 0845 1300 950. +44 0845 1300 960. info@biogene.
com. www.biogene.com/. хлороформ изоамиловый
спирт; реактивы, используемые в молекулярной и
клеточной биологии.
Biohit, Inc. 3535 Route 66, Building 4, P.O. Box 308,
Neptune, NJ 07754-0308, USA.+l 732 922 4900.+1 73
2 922 0557. pipet@biohit.com. www.biohit.com. Микро¬
пипетки, наконечники для пипеток; автоматические
пипетки.
Biohit Oyj. Laippatie 1, 00880 Helsinki, Finland. +358 9
773 861. +358 9 773 86 200. info@biohit.com. www.bio-
hit.com. Микропипетки; наконечники для пипеток;
автоматические пипетки.
Biohit, Ltd. Unit 1, Barton Hill Way, Torquay, Devon
TQ2 8JG, England, UK. +44 1803 315 900. +44 1803 315
530. info@biohit.co.uk. www.biohit.com. Микропипетки,
наконечники для пипеток; автоматические пипетки.
Biomedical Technologies, Inc. 378 Page Street, Stough¬
ton, MA 02072, USA. +1 781 344 9942. +1781 341 1451.
info@btiinc.com. http://www.btiinc.com/. Антитела;
факторы адгезии; матрикс; коллаген; фибронектин;
ламинин; добавки к среде; поли-О-лизин; факторы
роста.
Bionique Testing Laboratories, Inc. Bloomingdale
Rd., RR1, Box 2, Saranac Lake, NY 12983, USA.
+1518 891 2356. +1 518 891 5753. www.bionique.com/.
Исследование на микоплазмы.
Bio-Nobile Oy. P.O. Box 36, Tykist. okatu 4 B, FIN-
20521 Turku, Finland.+35 824 101 100.+35 824 101 123.
pickpen@bio-nobile.com. www.bio-nobile.com. Магнит¬
ная сепарация; счетчик колоний Pickpen.
Bioptechs, Inc. 3560 Beck Road, Butler, PA 16002, USA.
877 LIVE-CELL; +1 724 282 7145. +1 724 282 0745.
info@bioptechs.com. www.bioptechs.com. Культура на
покровных стеклах; подогреваемые чашки с микро¬
наблюдением; перфузионная камера; наблюдение в
замедленном времени.
Bioquell Medical Ltd. 29-31 Lynx Crescent, Weston-
super-Mare, Somerset, BS24 9BR, England, UK.
606
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
+44(0)1934410500. enquiries@bioquellmedical.co.uk.
wwwMoquellmedical.co.uk. Стерилизация перекисью
водорода; ламинар-боксы; боксы биобезопасности;
стерилизаторы.
Bioquell UK Ltd. 34 Walworth Road, Andover,
Hants SP10 5AA, England, UK. 800 220 700; +44
(0)1264 835 835. +44 (0)1264 835 836. enquiries@
bioquell.com. www.bioquell.co.uk. Чистые комнаты;
изоляторы; стерилизация перекисью водорода; газо-
генератоы; ламинарные шкафы; боксы микробиоло¬
гической защиты.
Bio-Rad Laboratories. Life Science Group Div., 2000 Al¬
fred Nobel Dr., Hercules, CA 94547, USA. 510-741-1000;
800 424 6723. 800-879-2289. Isg websupport@biorad.
com. www.bio-rad.com. Хроматография; декстран
DEAE; перенос ДНК; электрофорез; трансфекция;
анализ изображения; микроносители; микропланшет-
ные сканеры; системы микрочипов роботизированная
микротитрация.
Bio-Rad Labs Ltd. Bio-Rad House, Maylands Av¬
enue, Hemel Hempstead, Herts, HP2 7TD, Eng¬
land, UK.0800 181134; +44 (0)20 8328 2000. +44
(0)20 8328 2550. uk.lsg. marketing@bio-rad.com. www.
bio-rad.com. Хроматография; декстран DEAE; перенос
ДНК; электрофорез; трансфекция гена; микроноси¬
тели; ридеры для микротитрационных планшетов;
роботизированная микротитрация.
BioReliance. Invitrogen Bioservices, 14920 Broschart
Rd., Rockville, MD 20850-3349, USA. +1 301 738 1000;
800 553 5372. +1 301 610 2590. info@bioreliance.com.
www.bioreliance.com. Токсикология in vitro; исследо¬
вания цитотоксичности; исследование по контролю
качества; вирусологический скрининг.
BioReliance (UK). Invitrogen Bioservices, Todd
Campus, West of Scotland Science Park, Glasgow
G20 0XA, Scotland, UK. +44 (0)141 946 9999. +44
(0)141 576 2412. www.bioreliance.com. Токсикология
in vitro; исследования цитотоксичности; исследо¬
вание по контролю качества; вирусологический
скрининг.
Biosource International. 542 Flynn Road, Camarillo,
CA 93012, USA. 800 242 0607.805 987 3385. www.bio-
source.com. Антибиотики; покрытие БСА; коллаген;
дипептил пептидаза IV (cd26) клон 202.36; фибро¬
нектин; FNC; гентамицин; факторы роста; HEPES;
гидрокортизон; кератиноциты; среда LHC; среда
MCDB 402; микостатин; среда для нервных клеток;
NR-1; селективна среда; сыворотка; бессывороточная
среда; компоненты сыворотки; пенициллин; стрепто¬
мицин; ингибитор трипсина; витамины.
Biospec. Biospec Products, РОВ 788, Bartlesville, OK
74005, USA. +1-918-336-3363; 800-617-3363. 1-918-
336-6060. info@biospec.com. www.biospec.com. Гомоге¬
низация; отбор проб для микротитрации; MiniBead-
Beater; экстрация РНК.
BioSupplyNet. Cold Spring Harbor Laboratory Press.
www.biosupplynet.com. Руководство для пользовате¬
лей; справочник по продукции; источники материа¬
лов и оборудования.
Biotech Instruments, Ltd. Biotech House, 75a High
Street, Kimpton, Hertfordshire SG4 8 PU, England,
UK. +44 (0)1582 417 295. (+44 0)1582 457491. www.
biotinst.demon.co.uk/. Конфокальный микроскоп;
электрофорез; ферментеры.
Bio-Tek Instruments Inc. Highland Park, PO Box 998,
Winooski, VT 05404-0998, USA. +1 802 655 4740;
888 451 5171. 802 655 7941. labcsr@biotek.com. www.
biotek.com/. Ридеры для планшетов.
Biotrol. Крысиные хвосты.
Biovest International. 8500 Evergreen Boulevard, Min¬
neapolis, MN 55433, United States. +1 763 786 0302.
+1 763 786 0915. accountservices@biovest.com. www.bio-
vest.com. Биореакторы; половолоконная перфузион-
ная культура; крупномасштабное культивирование;
промышленное производство.
Biosero, Inc. 825 S. Primrose Ave, Suite G, Monrovia, CA
91016, USA+1 626-303-0309.+1 626-256-6060. info©
bioseroinc.com. www.bioseroinc.com. Ламинарные шка¬
фы; газовые камеры без выводящих каналов; боксы
биологической защиты.
BMG Labtech GmbH. Hanns-Martin-Schleyer-Str.
10, D-77656 Offenburg, Germany. +49 781 969 68-0.
+49 781 969 68-67. germany@bmglabtech.com. www.
bmglabtech.com. Ридеры для микротитрационных
планшетов; поглощение, флюоресценция, люминес¬
ценция.
BMG Labtech Inc. 2415 Presidential Drive, Building 204,
Suite 118, Durham, NC 27703, USA. +1 919 806 1735.
+1 919 8068526. usa@bmglabtech.com. www.bmglabtech.
com. Микропланшетные сканеры, флюоресценция,
люминесценция, поглощение.
BMG Labtech Ltd. PO Box 73, Aylesbury, HP20 2QJ,
England, UK. +44 (0)1296 336 650. +44 1296 336651.
uksales@bmglabtech.com. xejww.bmglabtech.com. Микро¬
планшетные сканеры, флюоресценция, люминесцен¬
ция, поглощение.
ВОС Edwards. Manor Royal, Crawley West, Sus¬
sex, RH10 9LW, UK. +(44) 1293 528844. +(44)
1293 533453. www.bocedwards.com. Насосы: диафраг¬
мальные; PTFE, вакуумные, силиконовая смазка.
ВОС Edwards USA. 301 Ballardvale Street, Wilmington,
MA 01887, USA. 800 848 9800; +1 978 658 5410. +(1)
978 657 6546. www.bocedwards.com. Насосы: диафраг¬
мальные; PTFE, вакуумные, силиконовая смазка.
Boehringer Mannheim. См. Roche Applied Sciences
Borolabs, Ltd. Unit F The Loddon Centre, Wade
Road, Basingstoke, RG24 8FL, England, UK. +44
(0)870 300 1001. +44 (0)870 300 1004. enquiries@
borolabs.co.uk. www.borolabs.co.uk. С02-инкубаторы;
устройство для регистрации данных; морозильники
с жидким азотом; Ьегистраторы температуры.
Brand GmbH. Postfach И 55, D-97861, Germany. +49
(0) 9342 8080. +49 (0)9342 808 236. info@brand.de.
www.brand.de. Диспенсеры для горловин бутылей;
лабораторное стекло планшеты для микротитрации;
многоканальные автоматические пипетки; пипетки;
автоматические пипетки; приспособления для пипе¬
тирования; штативы; циклические автоматические
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
607
пипетки; ступенчатые автоматические пипетки,
пробирки.
BrandTech Scientific Inc. 11 Bokum Road, Essex, CT
06426-1506, USA. +1-860-767 2562. +1-860-767 2563.
mail@brandtech.com. www.brandtech.com. Cm. Brand
GmbH.
Brandel Corp. 8561 Atlas Drive, Gaithersburg, MD 20877,
USA. 800-948-6506. +1 301-869-5570. sales@brandel.
com. www.brandel.com. Оборудование для герметиза¬
ции планшетов.
Braun В Biotech, Inc. См. Sartorius. Ферментеры.
Brinkmann Instruments, Inc. 1 Cantiague Road, West-
bury, NY 11590-0207, USA. 800 645 3050; +1 516 334
7500.+1 516 334 7506. info@brinkmann.com. zemw.brink-
mann.com. Центрифужные пробирки; градуированные
покровные стекла; оборудование для разлива жидкос¬
тей микрочипы; микрокапилляры; микроцентрифуги;
микроцентрифужные пробирки; микроинъекторы;
микроманипуляторы; микропипетки; наконечники
для пипеток; автоматические пипетки.
British Association for Cancer Research (BACR).
Contact: BACR Secretariat, c/o The Institute of Cancer
Research, McElwain Laboratories, Cotswold Road, Sut¬
ton, Surrey SM2 5 NG, UK. +44 (0) 20 8722 4208. + 44
(0) 20 8770 1395. bacr@icr.ac.uk. zejww.bacr.org.uk. Об¬
щество врачей и исследователей Объединенного ко¬
ролевства Великобритании по исследованию рака.
British Oxygen Co., Ltd. The Priestly Centre, 10 Priestly
Road, The Surrey Research Park, Guilford, Surrey
GU2 5XY, England, UK. 800 111333; +44 (0)1483
579857. zemw.boc.com. Газы; газовые регуляторы; C02;
вакуумные насосы.
British Society for Cell Biology. Контактное лицо
M. Clements, Department of Zoology, Downing St.,
Cambridge CB2 3EJ, England, UK. +44 (0)1223 336 655.
00 44 (0) 1223 353 980. bscb@bscb.org. http://wzem.kcl.
ac.uk/kis/schools/life sciences/biomed/bscb/top.html.
Научное общество с особым интересом к клеточной
и молекулярной биологии.
Bruce Medical Supply. 411 Waverly Oaks Rd., Ste.154,
Waltham, MA 02452, USA. 8002258446; +1781-894-
6262. +1781-894-9519. sales@brucemedical.com. zemw.
store.yahoo.com/brucemedical. Антисептики; Betadine;
пинцеты; растворы для промывания; чашки для об¬
разцов; ножницы.
BS & S (Scotland), Ltd. 5/7 West Telferton, Portobello
Industrial Estate, Edinburgh EH7 6UL, Scotland, UK.
0131 669 2282. 0131 657 4576. bss@telferton.co.uk.
Ecoscint.
Buck Scientific, Inc. 58 Fort Point St., East Norwalk, CT
06855. 203-853-9444; 800-562-5566. 203-853-0569. www.
bucksci.com. Инкубационные камеры д ля микроскопа; ин¬
кубаторы для съемки в режиме замедленной съемки.
Burge Scientific. 6 Langley Business Court, World’s End,
Beedon, Nr. Newbury, Berkshire RG20 8RY, England,
UK. +44 (0)1635 248171. +44 (0)1635 248857. sales@
burge.co.uk. zemw.burge.co.uk/; http://www.arrozemight.
co.uk/burge/index.htm. Моечная машина для лабора¬
торного стекла.
Caisson Laboratories, Inc. 5 West Center, Sugar City,
ID 83440, USA. +1 208 656 0880; 1877 840 0500.
+1 208 656 0888. custserv@caissonlabs.com. www.cais-
sonlabs.com. Изготовление сред на заказ; фетальная
сыворотка теленка; флаконы; media; фосфатно-бу-
ферный раствор; пипетки; роллерные бутыли; трип¬
син; вентилируемые крышки.
Calbiochem-Novabiochem Corp. См. EMD Biosciences
(North America), Merck Biosciences.
Cambio, Ltd. The Irwin Centre, ScotlandRoad, Dry
Drayton, Cambridge, CB3 8AR, England, UK. +44
(0)1954 210 200. +44 (0)1954 210 300.support@cambio.
co.uk. zemw.cambio.co.uk. Хромосомное окрашивание.
Cambrex—Bioresearch Products. 8830 Biggs Ford
Road, P.O. Box 127, Walkersville, MD 21793 0127,
USA. +1 301-898-7025; 800-638-8174. 301-845-8338.
biotechserv@cambrex.com. wzem.cambrex.com/bioprod-
ucts. Агароза; антибиотики; пенициллин, стрепто¬
мицин; ССР: Хэнкса, Эрла; факторы роста; среды:
селективные, бессывороточные; специализированные
клетки: астроциты; В-клетки, кость, гепатоциты, эн¬
дотелиальные клетки, эпидермальные кератиноциты,
женские репродуктивные клетки, почечный эпите¬
лий, легочный эпителий, клетки молочной железы,
меланоциты, предшественники нервных клеток,
остеобласты, клетки простаты, клетки почек, кожи,
гладкомышечные клетки, Т-клетки; витамины.
Cambrex Bio Science Wokingham, Ltd. 1 Ashville Way,
Wokingham, Berkshire RG41 2PL, England, UK. +44
(0)1189 795 234. +44 (0)1189 795 231. sales.uk@cam-
brex.com. zemw.cambrex.com. Cm. Cambrex-Bioresearch
Products.
Cambrex Bio Science Verviers, S. p. r. 1. Parc Industriel
de Petit Rechain, B-4800 Verviers, Belgium. +32-8-732-
1609.+32-8-732-1634. info.europe@cambrex.com. zemw.
cambrex.com/bioproducts. Cm. Cambrex-Bioresearch
Products.
Cambrex. Technical Sales Department, P.O. Box 127,
0245 Brown Deer Road, San Diego, С A 92121, USA.
800-852-5663. 858-824-0826. wzem.clonetics.com.
Cambridge BioScience Ltd. 24-25 Signet Court, New¬
market Road, Cambridge, CB5 8LA, England, UK. +44
(0)1223 316 855. +44 (0)1223 360 732. Tech@cbio.co.uk.
zemw.bioscience.co.uk. Антитела; апоптоз; бакуловирус;
клеточный цикл; коллаген; цитокины; ДНК-исследо-
вания; перенос ДНК; ELISA; проточная цитометрия;
факторы роста; интерлейкин-2; молекулярные зонды;
онкогены; передача сигнала; тетрациклин.
Camlab Ltd. Norman Way Industrial Estate, Over, Cam¬
bridge, CB4 5YE, England, UK. +44 (0)1954 233 100.
+44 (0)1954 233 101. mailbox@camlab.co.uk. www.
camlab.co.uk. Аспираторы; штопферы для автомати¬
ческих пипеток; оплетенные бутыли; центрифужные
пробирки; С02-инкубаторы; контейнеры; система
управления заморожееными запасами; лабораторное
стекло моечная машина для лабораторного стекла;
инкубаторы; магнитные мешалки; плоская медицин¬
ская склянка с крышкой; коробки и шейкеры для
микротитрационных планшетов; печи и сушильные
608
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
шкафы; Парафильм; мешалочные устройства; на¬
конечники для пипеток; автоматические пипетки;
лабораторный пластик; планшетный сканер; шей-
керы для планшетов; штативы; сосуды; качалочные
инкубаторы; стерилизационные и сушильные шкафы;
стерилизационные фильтры; пробирки; мойка и
стерилизация; ультразвуковая баня; флаконы; про¬
мывная склянка; моечные машины.
Campden Instruments. Leicester, UK. 0870 2403702.
UKsales@campdeninstruments.com. www.campden-inst.
сот/. Измельчитель тканей Mcllwain.
Campden Instruments. Lafayette, IN, USA. +1 765
423 1505. USsales@campdeninstruments.com. www.camp-
deninst.com/. Измельчитель тканей Mcllwain.
Carl Zeiss AG. P. О. B. 4041,37030 Gottingen, Germany.
+49 (0) 551 5060 660. +49 (0) 551 5060 464. mikro@zeiss.
de. z0z0wzeiss.de. Получение и обработка флюоресцент¬
ных изображений; конфокальная микроскопия; ана¬
лиз изображений; инкубируемая камера; микроскопы:
флюоресцентные, инвертированные, стерео, верти¬
кальные; с оптическим сечением; фотомикроскопы;
видеосъемка в режиме замедленного времени.
Carl Zeiss, Inc. Microscope Division, One Zeiss Dr.,
Thornwood, NY 10594, USA. +1 914 747 1800;
1800 233 2343. +1 914 681 7446. micro@zeiss.com. www.
zeiss.com/micro. cm. Carl Zeiss AG.
Carl Zeiss Ltd. 15-20 Woodfield Road, Welwyn Gar¬
den City, Hertfordshire, AL7 1JQ, England, UK. +44
(0)1707 871200. +44 (0)1707 330237. wwwzeiss.co.uk.
Продукция см. Carl Zeiss AG.
Cascade Biologies, Inc. 1341 Custer Drive, Portland, OR
97219, USA. 1 800 778 4770. +1 503 292 9521. info@
cascadebio.com. www.cascadebio.com. Клетки эпителия
роговицы; фибробласты кожи; эндотелиальные клет¬
ки; эпидермальные кератиноциты; ростовые добавки
EpiLife; MCDB154; меланоциты; бессывороточные
селективные среды; гладкомышечные клетки; специа¬
лизированные клеточные культуры; трипсин/EDTA;
ингибитор трипсина.
Cascade Biologies, Inc., UK. Mansfield I-Centre, Oakham
Business Park, Hamilton Way, Mansfield, Nottingham¬
shire NG18 5BR, England, UK. +44 (0) 1623 600 815.
+44 (0) 1623 600 816. ukinfo@cascadebio.com. www.
cascadebio.com. Cm. Cascade Biologies, Inc., USA.
Cato Research, www.cato.com/biotech/bio-prod.html.
Интернет-список основных биотехнологических
компаний.
Cell Culture Characterization Services. 4581, Lapeer-
Road, Suite C, Orion Township, MI 48359. +1248-656-
2542; +1248-563-1493. +1 248-656-3899. bhukku@cell-
characterization.com. Аутентификация; хромосомный
анализ; хромосомное окрашивание; кариотипирова¬
ние; изоферменты; FISH.
Cellex Biosciences Inc. См. Biovest International.
CellGenix Technologie Transfer GmbH. Am Flughafen
16, D-79108 Freiburg. +49 761 888 89-330. +49 761
888 89-830. info@cellgenix.com. www.cellgenix.com.
Системы CellGro; цитокины; бессывороточные среды;
культуральные пакеты.
Cellgro. См. Mediatech.
Cellmark Diagnostics. См. Orchid Cellmark.
Cellmark UK. PO Box 265, Abingdon, Oxfordshire,
OX14 1YX, England, UK. +44 (0)1235 528000. cell-
mark@orchid.co.uk. www.cellmark.co.uk. Аутентифика¬
ция клеточных линий. Профили ДНК.
Cellon SA. CELLON S. A., 29 Am Bechler, L-7213 Be-
reldange, Luxembourg. +352 312 313. +352 311 052.
info@cellon.lu. www.cellon.lu/. Биореакторы; коллаген;
культуральные пакеты; CulturSil; Ферментеры; мат¬
рикс, пакеты для сред; NASA, культуральная система
Rotary Cell Culture System; крупномасштабное про¬
изводство клеток.
Cellon UK Ltd. 61 Dublin St., Edinburgh, EN3 6NL, Scot¬
land, UK. +44 (0)7714 332567. +44 (0)1472 852642.
info@cellon.lu. www.cellon.lu/. Биореакторы; коллаген;
культуральные пакеты; CulturSil; ферментеры; мат¬
рикс, пакеты для сред; NASA, культуральная система
Rotary Cell Culture System; крупномасштабное про¬
изводство клеток.
CellStat. 755 Page Mill Road, B-6, Palo Alto, CA 94304,
USA. +1 650 802 0351. www.cellstat.com. Исследование
цитотоксичности; исследование действия лекарс¬
твенных препаратов и метаболический мониторинг;
клеточный мониторинг в реальном времени.
Cellular Products GmbH. Delitzscher Strasse 135,
04129 Leipzig, Sachsen, Germany. +49 (0)341 971 9758.
+49 (0)341 972 5389. info@cellular-products.de. www.
cellularproducts.de. Человеческие цитокины.
CeUutech. Watertown, NY, USA. 800-575-5945. Системы
транспортировки, безопасность отходов.
Celox. См. Protide Pharmaceuticals.
Celtrix Laboratories. Cm. Insmed.
Charles Austen Насосы Ltd. Royston Road, Byfleet,
Surrey, KT14 7NY, England, UK. +44 (0)1932 355277.
+44 (0)1932 351 285. info@charlesausten.com. www.
charlesausten.com. Насосы: диафрагмальные; PTFE,
вакуумные.
Charles River Laboratories. 251 Ballardvale St., Wilm¬
ington, MA 01887. +1 508 658 6000. 800-992-7329;
978 658 7132. comments@criver.com. zmvw.criver.com. Ис¬
следование эндотоксинов. Лабораторные животные.
Charles River (U. К.), Ltd. Manson Rd., Margate, Kent
CT9 4LT, England. +44(0)1843 823575. +44(0)1843
823497. crukcsd@uk.criver.com. zmjw.criver.com/cruk. Ис¬
следование эндотоксинов. Лабораторные животные.
Chart Biomed. Domestic customer service. 800-482-2473;
+1 770-257-12. 8-932-2473; 770-257-1300. www.chart-
biomed.com/. Жидкостные морозильники с жидким
азотом.
Chart Biomed Europe. European Sales and Operation
Director. +44 (0)1932 785570. +44 (0)1932 785 576.
www.chartbiomed.com/. Жидкостные морозильники
с жидким азотом.
Chemap. См. Alfa Laval.
Chemicon Europe. 9 Trident Way, Southall, UB18 7US,
England, UK. +1 909 676 8080; 800 437 7500. +1 909
676 9209. ztmjw.chemicon.com. Cm. Chemicon Interna¬
tional.
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
609
Chemicon International Inc. 28820 Single Oak Drive,
Temecula, CA 92590, USA +1 909 676 8080; 800 437
7500. +1 909 676 9209. www.chemicon.com. Антитела
к цитокинам; диализные системы; исследование с
антителами к цитокинам; GalC; EMA; GFAP; об¬
наружение гипоксии; интегрины; технология мик¬
рочипов; MUC-1; муциновые нейротранспортеры;
онкопротеины; передача сигнала; стволовые клетки;
вилин; вирусы.
Chiron Corp. 4560 Horton Street, Emeryville, CA
94608-2916, USA. +1 510 923-2300. +1 510 923-3376.
communicatioris@chiron.com. www.chinm.com. Цитокины;
фармацевтические препараты, иммуноисследования.
Chondrex Inc. 2607 151st Place NE, Redmond, WA
98052, USA. (425) 702-6365 or (888) CHONDRE.
info@chondrex.com. www.chondrex.com. Коллаген;
антитела к коллагену; коллагеновоке окрашивание;
коллагеназа.
Ciba Specialty Chemicals Corp. 540 White Plains Road,
P.O. Box 2005, Tarrytown, NY 10591-9005, USA.
+1 914 785 2000. +1 914 785 2211. kevin.bryla@cibasc.
com. zemw.cibasc.com. Получение фото- и цифровых
изображений.
Cin-Made Corp. 1780 Dreman Ave., Cincinnati, OH
45223, USA. 513-681-3600. 513-541-5945. zemw.cin-
madepackaginggroup.com. Контейнеры для транспор¬
тировки; биобезопасность.
Clonetics. См. Cambrex.
CoaChrom Diagnostica GmbH. Leo Mathauser Gasse
71, A1230 Vienna, Austria. +43 1699 97 97 0. +43 1699
18 97. office@coachrom.com. www.coachrom.com/.
Антитела; цитокины; эндотоксиновый тест; бессы¬
вороточные среды.
Cohesion Technologies. 2500 Faber Place, Palo Alto,
CA 94303, USA;. +1 650-320-5500. +1 650 320-5522.
admin@cohesiontech.com. zemw.cohesiontech.com. Кол-
лагеновое покрытие Cell Prime; Антитела к фактору
VIII; Vitrogen.
Cole-Parmer Instrument Co. 625 E. Bunker Ct., Vernon
Hills, IL 60061, USA. 847 549 7600, 800 323 4340.
+ 1 847 247-2929. techinfo@coleparmer.com. www.
coleparmer.com/. Устройство для регистрации данных;
сушильная камера; электронный счетчик колоний;
измерительные приборы; pH метры; автоматические
пипетки; насосы; автоматический шприц для непре¬
рывного введения; коробки для стекол; качающаяся
горячая платформа; термометры; системы трубок;
ультразвуковая баня; водяная баня.
Cole-Parmer Instrument Co. Ltd. Unit 3, River Brent
Business Park, Trumpers Way, Hanwell, London,
W7 2QA, England, UK. +44 (0)208 574 7556. +44
(0)208 574 7543. sales@coleparmer.co.uk. zemw.colepar-
mer.co.uk. Cm. Cole-Parmer Co., USA.
Collagen Corp. 2500 Faber Place, Palo Alto, CA 94303,
USA. 415-856-0200.415-856-2238. zemw.collagen.com/.
Коллаген, Vitrogen 100.
Collagen Corp. Cm. Inamed Aesthetics.
Comark Ltd. Comark House, Gunnels Wood Park, Gunnels
Wood Road, Stevenage, Hertfordshire, SGI 2TA, Eng¬
land, UK. +44 (0)1438 367 367. +44 (0)1438 367 400.
salesuk@comarkltd.com. zemw.comarkkd.com. Электрон¬
ные термометры; термометры с регистрирующим
устройством.
Comark Instruments Inc. 9710 SW Sunshine Court, Bea¬
verton, OR 97005, USA. 800 555 6658; +1 503 643 5204.
+1 503 644 5859. sales@comarkUSA.com. www.comarkltd.
com/usa. Электронные термометры; термометры с ре¬
гистрирующим устройством.
CompuCyte Corp. 12 Emily Street, Cambridge, A 02139,
USA. 800-840-1303;+1 617-492-1300.+1 617 577 4501.
www.compucyte.com. Флюорохроматический цито¬
метр.
Computer Imprintable Label Systems Ltd. 2 Southdown-
view Way, Broadwater Business Park, Worthing, West
Sussex, BN14 8BR, England, UK. +44 (0)1903 219 000.
+44 (0)1903 219 111. sales@cils-labels.com. zemw.cils-
labels.com. Ярлыки для автоклавирования; ярлыки
для замораживания, ярлыки для распечатывания на
компьютере, ярлыки для криофлаконов.
Contamination Control Products. 1 Third Ave, Neptune
City, NJ 07753, USA. +1 877 553-2676. +1 732 869-
2999. info@ccpcleanroom.com. zemw.ccpcleanroom.com/.
Ламинарные шкафы; боксы микробиологической
защиты.
Cook Biotech, Inc. 3055 Kent Avenue, West Lafayette, IN
47906, USA. +1 765 497 3355. +1 765 4972361. znvosis@
cookbiotech.com. zemw.cookgroup.com. ECM; внеклеточ¬
ный матрикс; платформа для тканевой инженерии.
Coriell Cell Repository (CCR). 403 Haddon Avenue,
Camden, NJ 08103, USA. 800-752-3805 из US); 856-
757-4848 (из-зи рубежа). 856-757-9737. ccr@coriell.
umdnj.edu. http://locus.umdnj.edu/ccr/. Клеточный
банк; клеточные линии; Архив генетически му¬
тантных клеток человека NIGMS; Архив зрелых
клеток N1 А.
Coming Inc., Life Sciences. 45Nagog Park, Acton,
MA 01720, USA. +1 978 635 2200; 800 492 1110.
+1 978 635 2476. clswebmail@coming.com. zemw.coming.
com/Lifesciences. (См. также Sigma in Europe.) Авто¬
матические диспенсеры; биореакторы; бутыли; куль¬
туральные камеры Cellstack; центрифужые пробирки;
измерители проводимости; культуральные сосуды;
электроды; вкладыши в фильтрационные лунки; фла¬
коны; газопроницаемые крышки; лабораторное стек¬
ло, магнитные мешалки; микропипетки; контейнеры
для микроскопических срезов; микротитрационные
планшеты; мультиповерхностные пропагаторы;
многолуночные планшеты; проницаемые крышки;
Чашки Петри; рН-метры; пипетирующее устройство;
пипетки; роллерные бутыли; вращающиеся флаконы;
устройство для контроля температуры.
Coming B.V. Life Sciences. Koolhovenlaan 12,1119 NE
Schiphol-Rijk, The Netherlands. +31 (0) 20-659-60-
51.+31 (0) 20-659-76-73. cceuml@coming.com. wzem.
coming.com/lifesciences. (См. также Sigma.) Продук¬
ция см. Coming Inc.
Costar. Cm. Coming.
Coulter. Cm. Beckman Coulter.
610
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
CP Pharmaceuticals Ltd. Ash Road North, Wrexham
Industrial Estate, Wrexham LL13 9UF, Wales, UK;
www.cppharma.co.uk/home. htm. Инсулин.
C.R. Bard Inc. Bard Urological Division, 8195 Industrial
Boulevard, Covington, GA 30014, USA. +1 770-784-
6100; 800-526-4455. bob.mccall@crbard.com. www.
bardcontigen.com. Коллагеновая губка.
Cryomed. См. Thermo Electron Corp., Thermo-Haake.
www.therrnols.com. Морозильник с контролируемым
режимом замораживания; перчатки; штативы для
морозильника; морозильники с жидким азотом.
Cryoservice Ltd. Cryoservice Limited, Prescott Drive,
Warndon Business Park, Worcester, WR4 9RH, Eng¬
land, UK. +44 (0)1905 754 500. +44 (0)1905 754 060.
info@cryoservice.co.uk. www.cryoservice.co.uk. C02; газы,
жидкий азот.
CryoTrack. +1 650 305 9406; +1 650 941 6774. info@
cryotrack.com. www.cryotrack.com. Программное
обеспечения для системы контроля замороженных
запасов.
Cytogration, Inc. 113 N. Washington Street, Suite
342, Rockville, MD 20850, USA. +1 301 330 4754;
877 330 4754. +1 301 519 9474. info@cytogration.com.
www.cytogration.com. Роботизированные системы.
Dage-MTI, Inc. 701 N. Roeske Ave, Michigan City,
IN 46360, USA. 219-872-5514. 219-872-5559. info@
dagemti.com. mmw.dagemti.com. CCD камеры; усилите¬
ли изображения; обработка изображения; мониторы,
видеосъемка в режиме замедленного времени.
Daigger. 620 Lakeview Parkway, Vernon Hills, IL
60061, USA. 1-800-621-7193. 1-800-320-7200;. daig-
ger@daigger.com. www.daigger.com. Чашки; флаконы;
нитрильные перчатки; пипетирующее устройство;
лабораторный пластик.
DakoCytomation Inc. 6392 Via Real, Carpinteria, CA
93013, USA. 805 566 6655,800 235 5763.805 566 6688.
customer.service@dakocytomation.com. www.dakocy-
tomation.us. Антитела; реактивы для определения
апоптоза; цитокератины; реактивы для проточной
цитометрии.
DakoCytomation Ltd. Marketing Dept., Denmark House,
Angel Drove, Ely, Cambridge, CB2 4ET, England,
UK. +44 (0)1353 669 911. +44 (0)1353 668 989. www.
dakoltd.co.uk www.dakocytomation.co.uk. Антитела;
реактивы для определения апоптоза; цитокератины;
реактивы для проточной цитометрии.
DakoCytomation Denmark A/S. Produktionsvej 42,
DK-2600 Glostrup, Denmark. 44 85 95 00.44 85 95 95.
contact@dakocytomation.com. zemw.dakocytomation.com.
Антитела; реактивы для определения апоптоза; цито¬
кератины; реактивы для проточной цитометрии.
Damon IEC. См. Thermo Life Sciences.
Davol Inc. 100 Sockanossett Crossroads, Cranston, RI
02920, USA. 800 556 6756. +1 401 946 5379. info@
davol.com. zemw.davol.com. Коллаген; политетрафторэ-
тиленовая сеть.
Day-Impex. См. Keison Products.
Decon Laboratories Ltd. Conway Street, Hove, East Sus¬
sex, BN3 3LY, England, UK. +44 (0)1273 739 241.+44
(0)1273 722 088. mail@decon.co.uk. www.decon.co.uk.
Детергенты; чернила; маркеры, ультразвуковая мо¬
ющая баня.
Deutsche Sammlung von Mikroorganismen und Zellkul-
turen (DSMZ). Mascheroder Weg IB, D-38124 Braun¬
schweig, Germany, contact@dsmz.de. www.dsmz.de.
Клеточный банк; клеточные линии; аутентификация
клеточной линии; исследование на микоплазмы.
Dianova GmbH. Mittelweg 176, D-20148 Hamburg, Ger¬
many. +49 (0)40 450 670. +49 (0)40 450 67 490. info@
dianova.de. zemw.dianova.de. Антитела.
DiaSorin. 1951 Northwestern Avenue, P.O. Box 285,
Stillwater, MN 55082-0285, USA. +1 612 439 9710,800
328 1482.+1 651 351 5669.zemw.diasorin.com/. Кальци-
тонин; эстрадиол; FSH; hGH; остеолацин; пролактин;
трииодтиронин; витамин D.
DiaSorin S. p. A. Via Crescentino, 13040 Saluggia (VC),
Italy. +39.0161.487093. +39.0161.487628. zemw.diasorin.
сот/. Кальцитонин; эстрадиол; FSH; hGH; остеола¬
цин; пролактин; трииодтиронин; витамин D.
Difco. См. BD Biosciences. Агар; микробиологические
среды; бактопептон; гидролизат лактальбумина; пи¬
тательные бульоны; триптозо-фосфатный бульон.
DNA Solutions. (Branches worldwide.) zemw.DNAsolu-
tions.com. Аутентификация; ДНК-профилинг; иссле-
довние идентичности ДНК по судебному запросу.
Doity Engineering Ltd. P.O. Box 25, Isherwood
Street, Rochdale, Lancashire OL11 1ZZ, England,
UK. +44 (0)1706 646 971; +44 (0)1706 345 515. +44
(0)1706 640 454. sales@doity.com. www.doity.com/.
Оборудование для холодных комнат и для термаль¬
ных; стальные штативы для хранения, покрытые
полипропиленом.
Don Whitley Scientific Ltd. 14 Otley Road, Shipley,
West Yorkshire, BD17 7 SE, England, UK. +44 (0)1274
595728. +44 (0)1274 531197. info@dwscientific.co.uk.
zemw.dwscientific.co.uk. Счетчики колоний.
Dow Coming Corp. P.O. Box 0994, Midland, MI 48686-
0994, USA. +1 517-496-4000. +1 517-496-4586. www.
dowcoming.com. Пеногаситель; деионизатор; Methocel;
метилцеллюлоза; системы силиконовых трубок.
Laboratory Corporation of America. 1447 York Court
Burlington, NC 27215, USA. 800-334-5161; +1 336-
584-5171. zemw.labcorp.com/. ДНК-профилинг; уста¬
новление отцовства.
DSMZ. См. Deutsche Sammlung vonMikroorganismen
und Zellkulturen.
DuPont Animal Health Solutions. Chilton Industrial
Estate, Sudbury, Suffolk C010 2XD, England, UK.
+44 1787 377305. +44 1787 310846. biosecurity@gbr.du-
pont.com. zemw.antecint.com. Дезинфектанты Virkon.
Dynal Biotech (USA). 9099 N. Deerbrook Tr., Brown
Deer, WI 53223, USA. 1-800-558-4511. 414-357-4518.
uscustserv@dynalbiotech.com./ zemw.dynalbiotech.com.
Сепарация клеток; MACS; намагничиваемые бусины;
иммуноаффинное клеточное разделение; нейлоновые
сетчатые фильтры.
Dynal Biotech UK. 11 Bassendale Road, Croft Busi¬
ness Park, Bromborough, Wirral, CH62 3QL, UK.
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
611
0800 731 9037; +44 (0)151 346 1234. 0151 346 1223.
ukcustserv@dynalbiotech.com./ www.dynalbiotech.com
См. Dynal Biotech (USA).
Dynalab Corp. 350 Commerce Drive, Rochester NY
14623, USA. 800 828-6595. 716-334-9496. labinfo@
dyna-labware.com. nuww.dynalabcorp.com/. Печи; чашки
для образцов, хранение.
Dynex Technologies Ltd. Columbia House, Colum¬
bia Drive, Worthing, England, UK. 01403 783381.
01403 784397. Микротитрация: планшетные сканеры,
автоматический отбор образцов; дилютеры; диспен¬
серы, миксеры, моющее устройство для планшетов,
программное обеспечение.
Dynex Technologies, Inc. 14340 Sullyfield Cir., Chan¬
tilly, VA 20151-1683, USA. +44 (0) 1903 267555. +44
(0) 1903 267722. dynexuk@btconnect.com. www.dynex-
technologies.com/. Микротитрация: микропланшетные
сканеры, автоматический отбор образцов; дилютеры;
диспенсеры, миксеры, моющее устройство для план¬
шетов, программное обеспечение.
East Coast Biologies, Inc. P.O. Box 489, North Berwick,
ME 03906-0489, USA. +1 207-676-7639. +1 207-676-
7658. info@eastcoastbio.com. www.eastcoastbio.com.
Антитела.
Eastman Kodak Co. Scientific Imaging Systems Div,
343 State St, Rochester, NY 14652-4115, USA. +1 203
786 5600; 800 225 5352. +1 203 786 5694. www.kodak,
com. CCD; цифровая камера цифровые изображения;
камеры к микроскопам; обработка фотографий; фо¬
тографический проявитель и фиксаж; Рентгеновская
пленка.
EBI, L. Р. 100 Interpace Parkway, Parsippany, NJ 07054,
USA. 800-526-2579. ebimedical support@ebimed.com.
www.ebimedical.com/. Матрикс для костного транс¬
плантата; платформа для тканевой инженерии.
ЕСАСС (European Collection of Cell Cultures). CAMR,
HPA Porton Down, Salisbury, Wiltshire, SP4 0JG,
England, UK. +44 (0)1980 612512.+44 (0)1980 611315.
ecacc@hpa.org.uk. www.ecacc.org.uk. Аутентификация;
клеточный банк и помещение в него; клеточные
линии, база данных; характеристика; скрининг на
микоплазмы; валидация.
Econo-med. Trafalgar House, Station Road, Long Sut¬
ton, Spalding, Lincolnshire, PE12 8BP, England, UK.
+44 (0)1406 36 42 42. +44 (0)1406 36 46 76. sales@
economed.com. zejww.econo-med.com. Перчатки нитри-
ловые, виниловые.
Electron Microscopy Services (EMS). 1560 Industry
Road Hatfield, PA 19440, USA. +1 215-412-8400.
+ 1 215-412-8450;. sgkcck@aol.com. www.emsdiasum.
com/ems. Cytotek цитоцентрифуга; инструменты для
иссечения; реактивы для электронной микроскопии;
фиксирующие растворы; лимоннокислый свинец;
ацетат уранила.
Elga Lab Water, US Filter. 10 Technology Drive, Low¬
ell, MA01851, USA.+1 508 887 6300; 800 466 7873 ex
5000.+1 508 887 6266; 800 875 7873. zemw.elgalabwater.
com. Фильтрация; ионный обмен; фотооокисление;
обратный осмос; оборудование для очистки воды.
Elga LabWater. High Street, Lane End, High Wycombe,
Bucks HP14 3JH, England, UK. +44 1494 887 555.
+44 1494 887 837. sandra.welch@veoliawater.com.
zemm.elgalabwater.co.uk. Фильтрация; ионный обмен;
фотооокисление; обратный осмос; оборудование для
очистки воды.
Elkay Laboratory Products. Unit 4, Marlborough Mews,
Crockford Lane, Basingstoke, Hampshire, RG24 8NA,
England, UK. +44 (0)1256 811118. +44 (0)1256 811116.
sales@elkay-uk.co.uk. zemw.elkay-uk.co.uk. Пакеты био¬
безопасности; флаконы для замораживания, однора¬
зовые крышки; фильтрующшие насадки; маркеры;
среды; сосуды; микропипетки; микропипетки; пастет-
ки; пипетки; наконечники для пипеток; пастеровские
пипетки; оборудование для герметизации планшетов;
пробирки для транспортировки; пробирки; системы
трубок; флаконы; универсальные контейнеры.
EMD Biosciences. 10394 Pacific Ctr. Ct., San Diego,
CA 92121, USA. +1 858 450 9600; 800-854-3417.
800 776 0999; +1 858-453-3552. customer.service@em-
dbiosciences.com. wz0w.emdbiosciences.com. Антитела;
биохимические вещества; буферные растворы; хи¬
мические реактивы; холерный токсин; детергенты;
декстран; ЭДТА; сульфат G418; реактивы для опред-
ления сахаров; гигромицин В; иммунохимические
реактивы; ферменты.
EMS. См. Electron Microscopy Services.
Endotronics. См. Biovest International.
Envair Ltd. York Ave, Haslingden, Rossendale, Lancashire
BB4 4HX, England, UK. +44 (0)1706 228416. +44
(0)1706 831957. info@envair.co.uk. zemw.envair.co.uk.
Ламинарный шкаф; CCTV мониторинг; оборудова¬
ние для очистки воздуха стерильных комнат; боксы
микробиологической защиты.
Enzo Life Sciences, Inc. 60 Executive Blvd., Farmingdale,
NY 11735, USA. +1 631 694 7070. +1 631 694 7501;
800 221 7705. zemw.enzo.com. Антитела; т-хромогра-
нин (клон РНЕ-5); микрочипы.
Eppendorf AG. Barkhausenweg 1,22331 Hamburg, Ger¬
many. +49 405 380 10. +49 405 380 1556. eppendorf @
eppendorf.com. zemw.eppendorf.com. Градуированные
покровные стекла; микрокапилляры; микроцентри¬
фуги; микроцентрифужные пробирки; микроинъек-
торы; микроманипуляторы; микропипетки; наконеч¬
ники для пипеток; автоматические пипетки.
Eppendorf UK, Ltd. Eppendorf UK Limited, Endurance
House, Chivers Way, Histon, Cambridge, CB4 9ZR,
England, UK.+44 (0)1223 200 440. +44 (0)1223 ?00 441.
sales@eppendorf.co.uk. zejzem.eppendorf.co.uk.т ко¬
ванные покровные стекла; микрокапшиг
центрифуги; микроцентрифужные пг
роинъекторы; микроманипуляторьг
наконечники для пипеток; автомат
Eppendorf USA. См. Brinkmann.
Erlab DFS SA. Parc D'affaires
27104 Val De Reuil Cedex, F
+33 2 32 09 55 90. Sales@erf
Вытяжные шкафы; лау
кожухи.
612
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
Erlab DFS SA. St Thomas’s House, St Thomas’s Square,
Salisbury, Wiltshire, SP1 1BA, England, UK. +44
(0)1722 341 940. +44 (0)1722 341 950. SalesUK@erlab.
net. wz0w.captair.com. Вытяжные шкафы; ламинарные
шкафы; защитные кожухи.
ESA Inc. 22 Alpha Road, Chelmsford, MA 01824-4171,
USA. 800 959-5095;+1 978 250 7000.+1 978 250-7090.
marketing@esainc.com. z0z0w.esainc.com. Компьютерное
программное обеспечение; микротитрация.
ESACT-UK. Secretary: Alison Stacey, ESACT-UK, PO
Box 117, Saffron Walden, Essex, CB10 1XD, England,
UK. a.stacey@adpro.co.uk. wz0w.esactuk.org.uk. Julian
Hanak, Chairman.
European Collection of Cell Cultures. См. ECACC.
European Life Scientist Organisation. Контактное лицо:
Ingeborg Fatscher, P.O. Box 1151, D-69199 Sandhau-
sen, Germany. +49 6224 925613. +49 6224 925610,11,12.
contact@elso.org. z0z0w.elso.org. Kai Simons, President.
European Society for Animal Cell Culture Technology
(ESACT). Office: PO BOX 1723, 5 Bourne Gardens,
Porton, Salisbury, Wilts SP4 0PL, England, UK.+44 1
980 610 405. +44 1 980 610 405. contact@esact.org, of-
fice@esact.org. www.esact.org. Otto-Wilhelm Merten,
President.
European Society of Toxicology In Vitro (ESTIV). ESTTV
Secretary: Dr Jan van der Valk, NCA, Dept. Animals, Sci¬
ence & Society, P.O. Box80.166, NL-3508 TD Utrecht, The
Netherlands. +3130 253 2163. +3130 253 9227.secretary©
estiv.org. z0z0w.estiv.org. President: R. Combes (UK).
European Tissue Culture Society (ETCS). Secretary:
Petra Boukamp, Deutsches Krebsforschungzentrum, Im
Neuenheimer Feld 280, D-69120 Heidelberg, Germany.
Z0Z0W.etcs. info/etcsmain.html. President: John Masters.
European Tissue Culture Society, UK Branch (ETCS-
UK). Контактное лицо: David Lewis, ECACC, Heath
Protection Agency, Porton Down, Salisbury, SP4 OJG,
UK. +44 (0)1980 612 512. +44 (0)1980 611 315. david.
lewis@hpa.org.uk. z0z0w.ecacc.org.uk/.
Falcon. Cm. BD Biosciences.
FiberCell Systems, Inc. 905 West 7th Street # 334,
Frederick, MD 21701, USA. +1 301-471-1269. +1 301.
865.6375. gcadwell@adelphia.net. zsmw.fibercellsysterns,
com. Половолоконная перфузионная культуральная
система.
Fine Science Tools GmbH. Fahrtgasse 7-13, D-
69117 Heidelberg, Germany. +49 (0)62 21/90 50 50.
+49 (0)62 21/60 00 01. europe@finescience.com. zemw.
finescience.com/. Инструменты для иссечения; хирур¬
гические инструменты.
Fine Scientific Tools. 373-G Vintage Park Drive,
Foster City, CA 94404-1139, USA. 800-521-2109;
+1 650 349 1636. 800-523-2109; 650-349-3729. info@
finescience.com. z0z0w.finescience.com/. Инструменты
для иссечения; хирургические инструменты.
Fisher Scientific Corp. 2000 Park Ln, Pittsburgh, PA
15275, USA. +1 412-490-8300; 800-766-7000. 800-926-
1166. Z0WW 1fishersci.com/. Бутылки из боросиликатного
стекла; КМЦ; центрифуги; химические реактивы;
кольца для клонирования; С02-инкубаторы; контей¬
неры для ядерной фотоэмульсии; покровные стекла;
кристаллвиолет; диацетилфлюоресциин; инструменты
для иссечения; ЭДТА; флаконы Эрленмайера; элект¬
ронные термометры; морозильники; краситель Гимза;
глюкоза; глицерин; гемоцитометры; магнитные мешал¬
ки; микрокрышки (Drummond); Oxoid; пипетки; реф¬
рактометры; стерилизационные и сушильные шкафы;
мешалка с подогревом; липкая лента; термоконтролле¬
ры; регистраторы температуры; вортекс-миксеры.
Fisher Scientific U.K. Bishop Meadow Road, Lough¬
borough, Leicestershire LE11 5RG, England, UK. +44
(0)1509 231166. +44 (0)1509 231893. info@fisher.
co.uk. z0z0w.fisher.co.uk. Продукция см. Fisher Scientific
Corp.
Fison Instruments. Cm. Fisher Scientific.
Fluka Riedel-de Ha. en. Industriestrasse 25, CH-
9471 Buchs SG, Switzerland. +41 (0)81 755 25 11.
+41(0)81 755 28 15. Fluka@sial.com.z0z0w.sigmaaldtich.
com/Brands/Fluka. Биохимические вещества; хими¬
ческие реактивы; ЭДТА; метилцеллюлоза.
Fluka Chemical Corp. См. Sigma-Aldrich, z0ww.sial.com.
Биохимические вещества; химические реактивы;
ЭДТА; метилцеллюлоза.
Fluorochem Ltd. Wesley Street, Old Glossop, Derbyshire
SK13 9RY, England. 01457 868921.01457 869360. en-
quiries@fluorochem.co.uk. zmem.fluorochem.net/. Среда
уплотненная; среда для флотации; флюорохимиче-
ские реактивы.
FMC BioProducts. См. Cambrex. http://zemw.cambreх.
сот/. Сиквенс ДНК, сепарация ДНК, обнаружение
мутаций; сепарация белков; ПЦР.
Forma. См. Thermo Electron Corp. www.thermo.com.
С02-инкубаторы.
Fougera. Melville, NY. z0z0w.fougera.com. Стерильный
хирургический лубрикант Surgilube.
Fresenius AG. D-61346 Bad Homburg v. d. H., Ger-
many.+49 6172 608 0. z0z0w.fresenius-ag.com/. Олиго-
нуклеотидные зонды.
Fuji Medical Systems U.S. A., Inc. Corporate Of¬
fice, 419 West Avenue, Stamford, CT 06902, USA.
800 431-1850; +1 203 324 2000; Customer Service:
800 872-3854; +1 203 602-3609. 203-327-6485. ssg@
fujimed.com. www.fujimed.com. Фотографические
принадлежности: пленка, проявители, фиксирующие
растворы, эмульсии, рентгеновская пленка.
Fuji Photo Film. PO Box 015, Leamington Spa, War¬
wickshire, CV 31 YA, England. +44 (0)1926 335 537.
+44 (0)1926 887 793. z0z0w.fujifilm-europe.com/. Фото¬
графические принадлежности: пленка, проявители,
фиксирующие растворы, эмульсии, рентгеновская
пленка.
G.Q.F. Manufacturing Co. P.O. Box 1552, Savannah, GA
31402-1552, USA. +1 912 236-0651. +1 912 234 9978.
sales@gqfmfg.com. z0z0w.gqfmfg.com. Инкубаторы для
яиц.
GA International. 965 Boul. Cure-Labelle, С. P.41534,
Laval, Quebec, H7V 4A3, Canada. +1 450-973-9420,1-
800-518-0364. +1 450-973-6373. mail@labtag.com. zemw.
labtag.com. Ярлыки для криофлаконов; маркеры.
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
613
GE Heathcare. См. Amersham Biosciences.
GE Silicones. World Headquarters, Wilton, CT 06897.800-
255-8886. www.gesilicones.com/. Клей-герметик для
силиконового каучука.
Gemini Bio-Products. 1301 East Beamer Street, Wood¬
land, С A 95776, USA. 800 543 6464; +1 530 668 3636.
+ 1530 668 3630. www.gembio.com/. Антибиотики;
среды; сыворотка.
Genetic Research Instrumentation Ltd. Gene House,
Queenborough Lane, Rayne, Braintree, Essex
CM77 6TZ, England, UK. +44 (0)1376 332 900. +44
(0)1376 344 724. gri@gri.co.uk. wwwgri.co.uk. Автома¬
тические диспенсеры; пленка для авторадиографии
кассеты; биореакторы; Buchler; морозильники с
регулируемым режимом замораживания; криофла¬
коны; уплотнители среды; проявители и фиксажи;
ферментеры; проточный цитометр Guava; Hotpack
инкубаторы; Labconco; LEEC; среды для получения
фракции лимфоцитов; микропипетки; системы для
микротитрации; морозильники с жидким азотом;
наконечники для пипеток; автоматические пипетки;
охлажденный инкубатор; роботизированное обору¬
дование для разлива жидкостей, роллерные стойки
для инкубатора; роллерный барабан; бокс биобе¬
зопасности; перемешивающие флаконы; системы
очистки воды.
Gen-Probe, Inc. 10210 Genetic Center Drive, San Diego,
CA 92121, USA. 619-546-8000; 800-523-5001.800 288-
3141; 800 342-7441. customerservice@gen-probe.
com. www.gen-probe.com/. Набор для обнаружения
микоплазм.
Germftee Labs, Inc. 11 Aviator Way, Ormond Beach, FL
32174, USA. 800-888-5357; +1 386-677-7742.+1 386-
677-1114. info@germfree.com. vuwwgermfree.com. Мик¬
робиологический бокс биобезопасности; ламинарные
шкафы; защитная камера с перчатками; изоляторы.
Geron Corp. 230 Constitution Drive, Menlo Park, CA
94025, USA. +1 650-473-7700. +1 650-473-7750. info@
geron.com. www.gervn.com. Исследование эмбриональ¬
ных стволовых клеток человека (hESC), руководство
по работе.
GIBCO. См. Invitrogen.
Gilson Inc. 3000 W. Beltline Hwy., PO Box 620027, Mid¬
dleton, WI 53562, USA. 608 836 1551, 800 445 7661.
608 831 4451. sales@gilson.com. www.gilson.com/.
Автоматические диспенсеры; оборудование для
разлива жидкостей, микропипетки; рабочее место
для микротитрации; перистальтические насосы; ав¬
томатические пипетки; роботизированные системы;
вакуумные насосы.
Gilson S.A. S. 19, avenue des Entrepreneurs, BP 145,
F-95400 Villiers le Bel, France. +33 01 34 29 50 00.
+33 01 34 29 50 20. www.gilson.com/. Автоматические
диспенсеры; оборудование для разлива жидкостей
микропипетки; рабоччее место для микротитрации;
перистальтические насосы; автоматические пипетки;
роботизированные устройства; вакуумные насосы.
Glen Research Corp. 22825 Davis Drive, Sterling, VA
20164, USA. +1 703 437 6191;. +1 703 435 9774. sup¬
port@glenres.com. www.glenres.com/. Синтетические
олигонуклеотиды.
Global Medical Instrumentation, Inc. (GMI). 6511 Bun¬
ker Lake Boulevard, Ramsey, MN 55303, UK. 763-
712-8717. 763-497-0176. richard@gmi-inc.com. www.
gmiinc.com. Автоклавы; биореакторы; инкубаторы;
микробиологические боксы биобезопасности; сте¬
рилизаторы; Verax.
Gonotec GmbH. Eisenacher Strasse 56, 10823 Berlin,
Germany. +49 (0)30 7809 588-0. +49 (0)30 7809 588-88.
contact@gonotec.com. www.gonotec.com. Осмометры.
Gow-Mac Instrument Co. 277 Broadhead Rd., Bethlehem,
PA 18017, USA. +1 610 954 9000. +1 610 954 0599.
sales@GOW-MAC.com. http://www.gowmac.com/. Га¬
зоанализаторы; газовые смесители.
Gow-Mac Instrument Co. (Ire) Ltd. Bay К 14a, Industrial
Estate, Shannon, Cty. Clare, Ireland. +353-61-471 632.
+353-61-471 042. sales©GOW-MAC.ie. http://www.
gowmac.com/. Газосмесители; газоанализаторы.
Grace Bio-labs. PO Box 228, Bend, OR 97709,
USA.+1 541 318 1208.+1 541-318-0242; 800-813-7339.
custservice@gracebio.com. www.gracebio.com. Стекла
для культуральной камеры; культура на покровных
стеклах; многолуночные стекла.
Grant Instruments (Cambridge) Ltd. Shepreth, Cam¬
bridge, SG8 6 GB, England, UK.+44 (0)1763 260 811.+1
(0)1763 262 410. info@grant.co.uk. www.grant.co.uk.
Электронные термометры; термометры с регистри¬
рующим устройством; регистраторы температуры;
водяная баня.
Greiner Bio-One GmbH. Maybachstrasse 2, D-
72636 Frickenhausen, Germany.+49 (0)7022 948-0.+49
(0)7022 948-514. info@de.gbo.com. www.greinerbioo-
neinc.com. Чашки; вкладыши в фильтрационные
лунки; флаконы; многолуночные планшеты; обору¬
дование для герметизации планшетов.
Greiner Bio-One Inc. 1205 Sarah St., Longwood, FL
32750, USA. +1 407-333-2800; 800 884 4703.+1 407-
333-3001. info@usgbo.com. www.greinerbiooneinc.com.
Чашки; вкладыши в фильтрационные лунки; фла¬
коны; многолуночные планшеты; оборудование для
герметизации планшетов.
Greiner Bio-One Ltd. Brunei Way, Stroudwater Business
Park, Stonehouse, Glos., GL10 3SX, England, UK. +44
(0) 1453 825 255. +44 (0) 1453 827 277. sales@uk.gbo.
com. www.gbo.com. Чашки; вкладыши в фильтраци¬
онные лунки; флаконы; многолуночные планшеты;
оборудование для герметизации планшетов.
Guava Technologies, Inc. 25801 Industrial Blvd., Hay¬
ward, CA 94545-2991, USA. +1 866 448 2827.+1 510-
576-1500. www.guavatechnologies.com. Настольный
проточный цитометр.
Guest Medical, Ltd. Enterprise Way, Edenbridge, Kent
TN8 6EW, England, UK. 01732 876466.01732 876476.
GuestMedical@B TIntemet.com. www.guestmedical.co.uk.
Аквасан-водяная баня бактериостатическая; устрой¬
ство сбора биологически опасной разлитой жидкости;
гипохлорид таблетированный; автоклавируемые
одноразовые пакеты; глютаровый альдегид.
614
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
Haake Buchler. См. Thermo Electron Corp., Thermo-
Haake. Электрофорез; агент для создания градиента
плотности; боксы для окрашивания.
Hamamatsu System Division. Div. of Hamamatsu Corp.,
360 Foothill Rd., P.O. Box 6910, Bridgewater, NJ 08807,
USA. 908-231-1116. 908-231-0852. www.hamamatsu.
com. CCD-камеры; кабельное TV; анализ изображе¬
ний; видеокамеры; видеомагнитофоны; видеосъемка
в режиме замедленного времени.
Hamamatsu Photonics К.К. 325-6, Sunayama-cho, Hama¬
matsu City, 430 Japan. (81)53 452 2141.(81)53 456 7889.
www.hamamatsu.com. CCD-камеры; кабельное TV; ана¬
лиз изображений; видеокамеры; видемагнитофоны;
видеосъемка в режиме замедленного времени.
Hammond Cell Technology. РО Box 1424, Windsor, CA
95492.707 473 0564.707 473 0564. hctculture@aol.com.
www.hammondcelltech.com. Экстракт гипофиза быка.
Hamo AG. CH-2542 Pieterlen, Switzerland. +41 (0)32 376
0200. +41 (0)32 376 0200. info@hamo.com. www.hamo.
com. Моющие машины для лабораторного стекла;
моющие машины.
Hamo UK Ltd. Spectra House, Boundary Way, Hemel
Hempstead, HP2 7SH, England, UK. +44 (0) 1442 259
735. +44 (0) 1442 219 566. hamo.uk@hamo.com. www.
hamo.com. Моющие машины для лабораторного стек¬
ла; моющие машины.
Hanna Instruments Inc. 584 Park East Dr., Woonsocket,
RI 02895, USA. +1-401-765-7500.+1-401-762-5064.
sales@hannainst.com. www.hannainst.com. Магнитные
мешалки; рН-метры, кондуктометры.
Hanna Instruments. Eden Way, Pages Industrial Park,
Leighton Buzzard, Beds, LU7 8TZ, England, UK.+44
(0)1525 850 855. +44 (0)1525 853 668. salesteam@han-
nainst.co.uk. zejww.hannainst.com/. Магнитные мешалки
рН-метры, кондуктометры.
Harlan Sera-Lab, Ltd. Hillcrest, Dodgeford Ln., Bel¬
ton, Loughborough, Leicester LE12 9TE, England,
UK.01530 222 123. 01530 224 970. hslcsd@harlanuk.
co.uk. wz0w.harlanseralab.co.uk. Антитела; сыворотка.
Harvard Apparatus Inc. 84 October Hill Rd., Holliston,
MA 01746, USA. +1 508-893-8999; 800-272-2775.
+ 1 508-429-5732. bioscience@harvardapparatus.com.
www.harvardapparatus.com. Настольный автоклав;
инкубаторы; печи; стерилизационные шкафы.
Harvard Apparatus Ltd. Fircroft Way, Edenbridge, Kent
TN8 6HE, England, UK. +44 (0)1732 864 001. +44
(0)1732 863 356. sales@harvardapparatus.co.uk. zemw.
harvardapparatus.co.uk. Настольный автоклав; инку¬
баторы; стерилизационный шкаф.
Hays Chemical Distribution, Ltd. Hays House, Millmead,
Guildford, GU2 5HJ, Surrey, England, UK.+44 (0)113
250 5811. +44 (0)113 250 5811. Хлорсодержащий
дезинфектрант Chloros.
Hazelton. Cm. Covance.
Health Science Research Resources Bank (HSRRB).
Rinku-minamihama 2-11, Sennan-shi, Osaka 590-0535,
Japan. + 81-724-80-1655. zemw.jhsf.or.jp. Помещение в
клеточный банк; продажа клеточных линий; клоны
ДНК.
Helena Laboratories. РО Box 752, Beaumont, ТХ
77704-0752, USA. +1 409 842 3714; 800 231 5663.
+1 409 842 6241. wzejw.helena.com/. Денситометры.
Henry Schein Inc. 135 Duryea Road, Melville, NY 11747,
USA. 800-711-6032;+1-631-843-5500.5117.1-800-329-
9109;. custserv@henryschein.com. z0z0w.henryschein.com/.
Roccall.
Henry Schein Ltd. Centurion Close, Gillingham, Kent,
ME8 0SB England, UK. 8700 102 043. 800 413 734.
sales@henryschein.co.uk. z0z0w.henryschein.co.uk. Roccall.
Heraeus. Equipment, cm. Kendro. С02-инкубаторы;
центрифуги; ламинарный шкаф; печи.
Heraeus. Культуральные материалы см. Vivascience
AG. PetriPerm.
Heto-Holten. См. Thermo Electron. Ламинарный шкаф;
боксы микробиологической защиты.
High-QInc. P.O. Box 440, Wilmette, IL 60091, USA. 800-
474-2674;+1-847-256-1231.800-474-2672;+1-847-256-
6114. sales@high-q.com. z0z0w.high-q.com. Дистилляция;
системы очистки воды.
Hotpack. См. SP Industries Co.
HRP, Inc. Cm. Covance Research Products, Inc.
HSRRB. Cm. Health Science Research Resources Bank.
H.V. Skan. Jeremy Drew, 425-433 Stratford Road, Shirley,
Solihull, B90 4AE, England, UK. +44 (0)121733 3003.
44 (0)121 733 1030. info@skan.demon.co.uk. zemw.ber-
linerglas.com. Препараты для микроскопии.
HyClone. 1725 South HyClone Road, Logan, UT 84321-
6212, USA. 800-492-5663; 435-792-8000.800-533-9450;
+1 435-792-8001. info@hyclone.com. z0z0w.hyclone.com.
Эмбриональная сыворотка теленка; среда; сыворотка;
бессывороточная среда, заменители трипсина.
Hyclone Europe S.A. Industriezone III, B-9320 Erembo-
degem-Aalst, Belgium. 53 83 44 04. 53 83 76 38. zexew.
hyclone.com. Эмбриональная сыворотка теленка сре¬
да; сыворотка; бессывороточная среда, заменители
трипсина.
Hycor Biomedicals, Inc. Garden Grove, С A 92841, USA
(800) 382-2527; (714) 933-3000. (714) 933-3220.
ELISA; химически детерминированная среда; бессы¬
вороточные среды.
ICLC Interlab Cell Line Collection. Istituto Nazionale
per la Ricerca sul Cancro с/о С BA, Largo Rosanna
Benzi, 10, Genova (GE), 16132 Italy. +39-0105737 474.
+39-0105737 295. iclc@ist.unige.it. zemw.biotech.ist.unige.
it/cldb. Клеточный банк; банк данных по линиям
клеток.
ICN. См. МР Biomedicals.
IEC. См. Thermo Electron Corp. www.thermo.com.
Центрифуги; С02-контроллеры; С02-инкубаторы;
адапторы для цитоцентрифуги; оборудование для
разлива жидкостей.
Ilford Imaging UK Ltd. Town Lane, Mobberley,
Knutsford, Cheshire, WA16 7JL, England, UK. +44
(0)1565 684 000. +44 (0)1565 873035. www.ilford.com.
Фотографическая эмульсия; проявители; фиксиру¬
ющие растворы; пленка.
Ilford Imaging USA Inc. W. 70 Century Road, Paramus,
NJ 07653, USA. +1 201-265-6000. z0z0w.ilford.com. Фо¬
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
615
тографическая эмульсия; проявители; фиксирующие
растворы; пленка.
Imaging Associates Ltd. 6 Avonbury Business Park,
Howes Lane, Bicester, Oxon 0X26 2UA, England, UK.
+44 (0) 1869 356 240. +44 (0) 1869 356 241. enquiries@
imas.co.uk. www.imas.co.uk. Анализ изображений; ин¬
кубатор для инкубируемой камеры, инкубируемая
камера для микроскопа; микроскопы: конфокальный,
флюоресцентный, инвертированный, стерео, верти¬
кальные; фотомикроскопы; видеосъемка в режиме
замедленного времени.
Imaging Research Inc. Brock University, 500 Glenridge
Avenue, St. Catherines, Ontario, L2S 3A1, Canada.
905 688 2040. 905 685 5861. Цитометр; анализ изоб¬
ражений; флюоресцентные зонды; микроанализ ДНК
и белка.
Inamed Aesthetics. 5540 Ekwill Street, Santa Barbara, CA
93111, USA+1805 683 6761.+1805 967 5839. www.iname-
daesthetics.com. Коллагеновый имплантат; Zyderm 2.
Infors AG. CH-4103 Bottmingen, Basel, Switzerland.
(0)61 425 7700. (0)61 425 7701. Headoffice@inforsht.
com; service@infors-ht.com. www.infors-ht.ch. Инкуба¬
торы; ферментеры; шейкеры.
Infors U.K., Ltd. The Courtyard Business Centre,
Dovers Fam, Lonesome Lane, Reigate, GB-RH2 7QT
Surrey, England, UK. +44 (0)1737 22 31 00. +44
(0)1737 24 72 13. infors.uk@infors-ht.com. www.infors.
ch. Инкубаторы; ферментеры; шейкеры.
Innovatis AG. Meisenstrasse 96, D-33607 Bielefeld, Ger¬
many.+49 (0)521 2997-300.+49 (0)521 2997-285.info©
innovatis.com. www.innovatis.com. Счетчик клеток; под¬
счет клеток в многолуночном планшете; определение
жизнеспособности.
Innovatis Inc. 333 Lancaster Avenue, Westgate V., 207,
Frazer, PA 19355-1823, USA. +1-888-283-1564; +1-
610-889-7319.+1-888-283-1631; +1-610-889-7436.
info@innovatis.com. www.innovatis.com. Счетчик
клеток; подсчет клеток в многолуночном планшете;
определение жизнеспособности.
Innovative Cell Technologies, Inc. 6790 Top Gun St.
#1, San Diego, CA 92121, USA. +1 858 587 1716.
+ 1 858.453-2117. info@innovativecelltech.com. www.
innovativecelltech.com. Диссоциирующие агенты Ac-
cumax; Accutase; проточная цитометрия; роботизи¬
рованная система; замещение трипсина.
Innovative Chemistry, Inc. P.O. Box 90, Marshfield,
MA 02050, USA. +1 781-837-6709. +1 781-834-7325.
www.innovativechem.com/. Набор для изоферментного
электрофортического анализа.
Insight Biotechnology Ltd. P.O. Box 520, Wembley, Mid¬
dlesex, HA9 7YN, England, UK. +44 (0) 20 8385 0303.
+44 (0) 20 8385 0302. info@insightbio.com. www .insight-
bio.com. Антитела; цитокины; ELISA.
Insmed Inc. 800 E. Leigh St., Richmond VA 23219, USA.
+1 804-828-6893. +1 804-828-6894. www.insmed.com/.
Коллагеновый имплантат, Zyderm 2; Vitrogen 100.
Instron. 100 Royall Street, Canton, MA, 02021, USA.1 800
564 8378; 1 781 828 2500. www.instron.com. Система
сжатие/растяжение для культуры миоцитов.
Integra Biosciences AG. Schonbuhlstr. 8, CH-7000, Chur,
Switzerland. +41 (0)81 286 95 30. +41(0)81 286 95 33.
info@integra-biosciences.com. www.integrabiosciences.
com. Аспирационный насос; автоклавы; автомати¬
ческие пипетки; горелка; культуральные флаконы;
диспенсеры; вкладыши в фильтрационную лунку;
фильтровальные флаконы; крышка фильтра; мани¬
пуляции с жидкостями; культура высокой плотности,
флаконы и камеры; оборудование для разлива жид¬
костей, магнитные мешалки; Maxisafe 2000; мемб¬
ранные флаконы; микротитрация; многоканальные
автоматические пипетки; перфузионная мембранная
культуральная система; автоматические пипетки; пе¬
ристальтические насосы и пипетирующие устройства,
соединенные с ними; роллерные бутыли; аппарат для
роллерной культуры; шестилуночная камера; враща¬
ющаяся культура; мешал очная культура; Tecnomouse;
вакуумные насосы.
Integra Biosciences UK. См. Scientific Laboratory Sup¬
plies.
Integra Biosciences (USA). Cm. Argos Technologies.
Intergen Co. Cm. Serologicals.
International Association for Plant Tissue Culture
(IAPTC). Abed A. Watad, Secretary-Treasurer, Depart¬
ment of Ornamental Horticulture, ARO, The Volcani
Center, P.O. Box 6, Bet Dagan 50205 Israel. +972 3
9683500. +972 3 9660589. vcwatad@volcani.agri.gov.il.
http://indycc1.agri.ac.iI/.tzvika/iaptc/ip-home.htm.
Interplast Inc. 100 Connecticut Drive, Burlington, NJ
08016, USA +1 609-386-4990; 1-800-220-1159. 609-
386-9237. www.interplastinc.com/. PTFE; тефлон;
пробирки; пластины, палочки.
Interpore Cross International. 181 Technology Drive,
Irvine, CA 92618-2402, USA. +1 949 453-3200.
+ 1 949 453-3225. www.interpore.com/. Керамика на
коралловой основе.
Intracel, Ltd. Unit 4, Station Road, Shepreth, Royston,
Herts SG86PZ, England, UK. 01763 262680. 01763
262676. Специализированные подогреваемые камеры
для иследований в режиме замедленного времени.
Invitrogen Corp. 1600 Faraday Avenue, Carlsbad,
CA 92008, USA. 800-955-6288; (760) 603-7200.
800 331 2286; +1 760 602 6500. catalog@invitrogen.com.
www.invitrogen.com. Агар; ампулы; антитела; экстракт
эмбриона цыпленка; плазма цыпленка; сыворотка
цыпленка; хромосомное окрашивание; DMEM; F12;
гентамицин; глютамин; факторы роста; Хэнкса сба¬
лансированный раствор (НССР); НССР без Са2+ и
Mg2+ (CMF); HEPES; среда для клеток насекомых; Is¬
cove’s модифицированная среда Дюльбекко (IMDM);
канамицин; L-глютамин; Среда Мак-Кой 5а; среды;
планшеты для микротитрации; минимальная среда-ос
(МЕМ-а); микостатин; фосфатно-буферный раствор;
пенициллин-стрептомицин; Pluronic; рекомбинан¬
тный эпидермальный фактор роста (rEGF); RPMI
1640; сыворотка; бессывороточные среды; трипсин;
трипсин/Э Д Т А.
Invitrogen Ltd. 3 Fountain Drive, Inchinnan Business
Park, Paisley PA4 9RF, UK.+44 (0)141 814 6100;
616
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
0800 269 210 (продажи); 0800 838 380 (сервисное
обслуживание). +44 (0)141 814 6260; 0800 243 485.
euroinfo@invitrogen.com. www.invitrogen.com. См. In¬
vitrogen, Inc.
Irvine Scientific, Inc. 2511 Daimler St., Santa Ana, С A
94303, USA. 800 577 6097; +1 949 261 7800.+1 949 26
1 6522. nucleus@irvinesci.com. www.irvinesci.com. Среда;
набор для детекции микоплазм; сыворотка; бессыво-
роточные среды; компоненты сыворотки.
Irvine Scientific, UK. См. Metachem Diagnostics; Smiths
Medical International.
ISP Alginate. San Diego, С A, USA. ztmw.ispcorp.com/.
Альгинат натрия.
J.T. Baker. Mallinckrodt Baker, Inc., 222 Red School
Lane, Phillipsburg NJ 08865, USA. +1 314-530-2000;
800-354-2050. +1314-530-2328. infombi@mkg.com.
zs3zsjw.jtbaker.com. Держатель для бутылок; буферные
растворы; химические реактивы.
J.T. Baker UK. См. Scientific & Chemical Supplies.
Jackson ImmunoResearch. P.O. Box 9, 872 West Balti¬
more Pike, West Grove, PA 19390, USA.+1 610 869 4024;
800 367 5296. +1 610 869 0171. cuserv@jacksonimmuno.
com. zsjzsjw.jacksonimmuno.com/. Антитела; FITC-конъ-
югированный IgG обезьяны против овцы.
Jackson ImmunoResearch (UK). См. Stratech Scien¬
tific.
James Glass Co. 14 Hanover St., Hanover, MA 02339,
USA.+1 781 829-0967.+1 781.829.2142. sales@james-
glass.com. zsjzsjw.jamesglass.com. Биореакторы; фермен¬
теры; выдувка стекла; лабораторное стекло.
Japanese Cancer Cell Collection. См. Health Science
Research Resources Bank (HSRRB).
Japanese Collection of Research Bioresources. Cm.
JCRB and Health Science Research Resources Bank
(HSRRB).
Japanese Tissue Culture Association (JTCA). +81-3-
5814-5801 .+81 -3-5814-5820. http://jtca.umin.jp.
JCRB. Research Resource Division, National Institute of
Biomedical Innovation, 7-6-8-Saito-Asagi, Ibaraki-shi,
Osaka 567-0085, Japan. (Распределение клеточных ли¬
ний см. HSRRB.) +81 (0)72 641 9811. http://ceUbank.
nibio.go.jp. Клеточный банк; приобретение клеточных
линий; клоны ДНК.
Jencons (Scientific) Ltd. Unit 6, Forest Row Busi¬
ness Park, Station Road, Forest Row, East Sussex,
RH18 5DW, England, UK. +44 (0)1342 826836. +44
(0)1342 826771. uksales@jencons.co.uk. www.jencons.
co.uk. Автоматические диспенсеры; пакеты биобе¬
зопасности; С02-инкубатор; сосуды Дюара; моро¬
зильники с жидким азотом; магнитные мешалки;
микробиологические боксы биобезопасности; микро¬
пипетки; замораживающее устройство контролиру¬
емым режимом с закрытым горлом; автоматические
пипетки; стерилизационная лента; мешалки; Taylor
Wharton.
Jencons Scientific Inc. 800 Bursca Drive, Suite 801, Brid-
geville, PA 15017, USA. 800-846-9959; +1 412-257-8861.
+1 412-257-8809. info@jenconsusa.com. zsjww.jenconsusa.
сот/. Продукция см. Jencons (Scientific) Ltd.
JEOL. 11 Dearborn Road, Peabody, MA 01960, USA.
+1 978 535 5900. +1 978 536 2205. zsjww.jeol.com. Элек¬
тронные микроскопы; сканирующие электронные
микроскопы.
JEOL (UK) Ltd. JEOL House, Silvercourt Watchmead,
Welwyn Garden City, AL7 1LT Herts, England, UK.
+44 (0) 1707 37 71 17. +44 (0) 1707 37 32 54. uk.sales@
jeoleuro.com. zsmjw.jeoleuro.com/. Электронные микро¬
скопы; сканирующие электронные микроскопы.
JM Separations. P.O. Box 6268,5600 HG Eindhoven, The
Netherlands. +31 (0)40 219 7480. +31 (0)40 219 7499.
info@jmseparations.com. www.jmseparations.com.
Половолоконная перфузионная культура Cellmax;
микрокапиллярная перфузия; Triac концентрические
полые волокна.
Johnson & Johnson, Medical, Ltd. Coronation Road, Ascot,
Berks SL5 9EY, England, UK. +44 (0)1344 871 000. +44
(0)1344 872 599. Дезинфектанты; Рекомендуемые таб¬
летки; гипохлорит; протирочный материал; перчатки.
Jouan. См. Thermo Electron Corp. www.thermo.com.
Центрифуги; инкубаторы; печи; боксы микробиоло¬
гической безопасности; водяная баня.
Jouan Nordic A/S. См. Thermo Electron.
JRH Biosciences. 13804 W. 107th Street, Lenexa, KS
66215 USA. +1 913-469-5580; 1 800-255-6032.+1 913-
469-5584. info-us@jrhbio.com. zsjzsjw.jrhbio.com. Автокла-
вируемая среда; партии культуры; пакеты для сред;
сыворотка; бессывороточные среды; Thermo-POW;
Ventrex; витамины.
JRH Biosciences Ltd. Smeaton Road, West Portway, An¬
dover, Hampshire SP10 3LF, UK. +44 (0)1264-333311.
+44 (0)1264-332412. info-eu@jrhbio.com. zsjww.jrhbio.
com. Cm. JRH Biosciences (USA).
KBI BioPharma, Inc. P.O. Box 15579, 1101 Hamlin
Road, Durham, NC 27704, USA. +1 919 479 9898.
+1 919 620 7786. sales@kbibiopharma.com. zsjzsjw.kbibio-
pharma.com. Центрифужный BioReactor; подложка
для клеток высокой плотности.
Keison Products. P.O. Box 2124, Chelmsford, Essex
CM1 3UP, England, UK. +44 1245 600560. +44 1245
6000. info@keison.co.uk. zsjzsjw.keison.co.uk/day-impex.
Сосуды Дюара.
Keison Products, USA. 233 Rogue River Highway #425,
Grants Pass, OR 97527-5429, USA. +1 541 956 9929.
+1 541 610 1646. info@keison.co.uk. zsjww.keison.co.uk/
dayimpex. Сосуды Дюара.
KeLab. KeLab, Karl-Erik Ljung AB, Knipplagatan 10,
414 74 Goteborg, Sweden. +46 (0)31-12 51 60. +46
(0)31-14 82 60. goteborg@kelab-biochem.com. www.
kelabbiochem.com/kontakt.html. Коллаген.
Kendro Laboratory Products. 308 Ridgefield Court,
Asheville, NC 28806, USA. +1 828 658 2711.+1 828 64
5-3368. info@kendro.spx.com. zsjzsjw.kendw.com. Сепа¬
ратор клеток; центрифуги, С02-инкубаторы, инку¬
баторы, ламинарные шкафы; микробиологические
боксы биобезопасности; микроцентрифуги, печи,
морозильники.
Kendro Laboratory Products. Robert-Bosch-Strasse 1,
63505 Langenselbold, Germany. 800-1-536 376 (Sales);
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
617
800-1-112 110 (Service). 800 1112 114.info.de@kendro.
spx.com. wi0w.kendro.com. Сепаратор клеток; центри¬
фуги, С02-инкубаторы, инкубаторы, ламинарные
шкафы микробиологические боксы биобезопасности;
микроцентрифуги, печи, морозильники.
Kendro Laboratory Products. Stortford Hall Park, Bish¬
op’s Stortford, Hertfordshire, CM23 5GZ, England, UK.
+44 (0)1279 82 77 00. +44 (0)1279 82 77 50. info.uk@
kendro.spx.com. www.kendro.com. Сепаратор клеток;
центрифуги, С02-инкубаторы, инкубаторы, ламинар¬
ные шкафы микробиологические боксы биобезопас¬
ности; микроцентрифуги, печи, морозильники.
Kimberley-Clark. 12671 High Bluff Drive, San Diego, С A
92130, USA. 800 255 6401. service@safeskin.com. www.
kcsafety.com/. Нитрильные перчатки.
Kimble-Kontes. Vineland, NJ, USA. +1 (888) 546-
2531 Ex 1; (856) 692-3600 Ex 1. +1 (856) 794-9762.
cs@kimkon.com. www.kimble-kontes.com. Пластиковые
пастровские пипетки; лабораторное стекло, центри¬
фужные пробирки.
Kinetic Biosystems, Inc. См. KBI BioPharma.
KNF Corp. 734 West Penn Pike, Tamaqua, PA.18252,
USA. +1 (570) 386-3550. +1 (570) 386-3703 www.knf-
Corp.com/. Автоклавируемая нейлоновая салфетка,
Kodak Ltd. (См. also Eastman Kodak). P.O. Box 66,
Hemel Hempstead, Hertfordshire, HP1 1JU, UK. +44
(0)1442 261 122. +44 (0)1442 240 609. www.kodak.
co.uk. CCD; цифровые камеры; цифровые изображе¬
ния; просветляющий агент; камеры для микроскопа;
фотографический проявитель; фотографический
закрепитель; рентгеновская пленка.
Kojair (UK) Ltd. 143 Reading Road, Wokingham, Berk¬
shire, RG41 1LJ, England, UK. +44 (0)118 977 0957.
+44 (0)118 977 5302. info@kojair.co.uk. www.kojair.
co.uk. Ламинарный шкаф; блоки микробиологической
защиты.
Lab Safety Supply. PO Box 1368, Janesville, WI
53547-1368, USA. 800 356 0783; +1 608 754 7160.
800 543 9910; +1 608 754 3937. custsvc@labsafety.
com. www.labsafety.com/. Пакеты биобезопасности;
контейнеры для биологически опасных отходов; де¬
зинфектанты; бутылочки для промывания глаз; рес¬
пираторы; нитрильные перчатки; ярлыки; защитная
одежда; контейнер для острых и режущие отходов;
гипохлорит натрия; этикетки.
Lab Vision Corp. 47790 Westinghouse Drive, Fremont,
CA 94539, USA. 800-828-1628; +1 510-991-2800.
+ 1 510-991-2826. LabVision@LabVision.com. www.
labvision.com. маркеры ангиогенеза; антитела:
коллаген, фибронектин, GFAP, ламинин, муцин,
маркеры апоптоза, факторы роста клеточного цикла,
онкогены, герегулин, p21WAFl, MUC2, миелин,
pl6INK4a, BrdU, BUdR, циклин, EGFR, Е-кадгерин,
фибронектин, желудочный муцин, цитокератин, ре¬
цептор ламинина, nm23; концентраторы; цитокины;
внеклеточный матрикс; наборы для экстракции;
инвазионные камеры.
Labcaire Systems, Ltd. 175 Kenn Road, Clevedon, North
Somerset, BS21 6LH, England, UK. +44 (0)1275 793000.
+44 (0)1275 341313. info@labcaire.co.uk. www.labcaire.
co.uk. Блоки микробиологической защиты II класса;
ламинарный шкаф.
Labco Limited (U. К.). Brow Works, Copyground Lane,
High Wycombe, Buckinghamshire, HP12 3HE, United
Kingdom. +44 (0)1494 459741. +44 (0)1494 465101.
sales@labco.co.uk. x0ww.labco.co.uk. Корзины для био¬
логически опасных отходов.
Lab-Line. См. Barnstead Thermolyne Corp.
Labmart (Cambridge), Ltd. 1 Pembroke Avenue, Water-
beach, Cambridge CB5, UK. +44 (0) 1223 441257.+44
(0) 1223 861990. Автоклавы; системы криобиологи¬
ческого хранения; морозильники с жидким азотом;
низкотемпературные морозильники.
Laboratory Impex Systems Ltd. Impex House, 15 Riv¬
erside Park, Wimborne, Dorset, BH21 1QU, England,
UK.+44 (0)1202 840685. +44 (0)1202 840701. sales@
labimpex-systems.co.uk. www.lab-impex-systems.co.uk.
С02-контроллеры; дезинфектанты; холодильники;
морозильники; штативы для морозильников.
Laboratory Sales (UK) Ltd. Units 20-21, Transpennine
Trading Estate, Rochdale OL11 2PX, England, UK. +44
(0)1706 356 444. +44 (0)1706 860 885.sales@lg-uk.com.
x0x0w.ls-uk.com. Покровныые и предметные стекла для
микроскопии; флаконы и уплотняющие вкладыши.
Labox Ltd. The Oval, Hackney Road, London, E29DU,
England, UK. +44 (0)207 613 2400. +44 (0)207 613 3420.
labox.uk@labox.biz. x0x0w.labox.biz. Канистры; коробки,
контейнеры, электронные термометры; лабораторный
пластик; насосы; сифоны; вентильные сифоны.
Labsales. См. Camlab. Температурное устройство регис¬
трации данных; термометр с регистратором.
Lab Storage Systems Inc. P.O. Box 968, St. Peters, MO,
3376, USA. 800 345 4167; +1 636 928 8988.800 45 4117;
+1 636.922.4474. x0x0w.labstore.com. Среда для заклю¬
чения срезов.
Lamb, R.A. См. Raymond A. Lamb.
Lancer UK Ltd. 1 Pembroke Avenue, Waterbeach, Cam¬
bridge CB5 9QR, England, UK. +44 (0)1223 861 665.
+44 (0)1223 861 990. sales@lancer.com. www.lancer.
co.uk. Моечная машина для лабораторного стекла.
Lancer USA Inc. 140 State Road 419, Winter Springs,
FL 32708, USA. +1 407 327 8488. +1 407 327 1229.
sales@lancer.com. wx0w.lancer.com. Моечная машина
для лабораторного стекла.
Lawson Mardon Wheaton. 1501 N. Tenth St., Millville,
NJ 08332-2092, USA 609-825-1100; 800-225-1437.
+1 856-825-1368; +1 856-825-4568. xemw.wheatonsci.
com. Ампулы; боросиликатное стекло; покровные
стекла; диспенсеры; флаконы Эрленмайера; автома¬
тические пипетки Gilson Pipetteman; лабораторное
стекло; оборудование для разлива жидкостей; мик¬
ропипетки; автоматические пипетки; лабораторный
пластик; роллерные штативы; перемешивающие
флаконы: многократного и однократного использо¬
вания.
LEEC Ltd. Private Road, No.7 Colwick Industrial Estate,
NottinghamNG4 2AJ, England, UK. +1 (0)115 961
6222.+1 (0)115 961 6680. sales@leec.co.uk. www.leec.
618
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
co.uk. С02-инкубаторы, механические щетки для
бутылок; стерилизационные шкафы.
Leica Microsystems. 2345 Waukegan Road, Bannock¬
burn, IL 60015, USA. +1 847 405 0123,800 248 0123.+
1 847 405 0164. www.leica-microsystems.com/. Камеры;
CCD камеры; CCTV; конфокальный микроскоп;
анализ изображения; инвертированный микроскоп;
кариотипирование микроманипуляторы; микроско¬
пы; рефрактометры.
Leica Mikrosysteme Vertrieb GmbH. Lilienthalstrasse
39-45, D-64625 Bensheim, Germany. +49 6251 136 0.
+49 6251 136 155.www.leica-microsystems.com/. Каме¬
ры; CCD камеры; CCTV; конфокальный микроскоп;
анализ изображения; инвертированный микроскоп;
кариотипирование; микроманипуляторы; микроско¬
пы; рефрактометры.
LGC Promochem. Queens Road, Teddington, Middlesex,
TW11 0LY, England, UK. +44 (0)20 8943 8480. +44
(0)20 8943 7554. uk@lgcpromochem.com; atcc@lgcpro-
mochem.com. zmejw.lgcpromochem.com. Аутентификация;
клеточные линии; ДНК-профилинг; тренировочные
курсы.
Life Sciences International, см. Thermo Electron Corp.
wz0w.thermo.com. Аварийная сигнализация; С02-ин-
кубаторы; морозильники с регулируемым режимом
охлаждения; регистраторы данных; система регистра¬
ции замороженных запасов; ламинарные шкафы; обо¬
рудование для разлива жидкостей; морозильники с
жидким азотом; низкотемпературные морозильники;
бокс микробиологической защиты; микропипетки;
микротитрация: планшеты, ридеры для планшетов,
моющие устройства для планшетов; магнитный
сортинг; азотные морозильники; программируемые
морозильники; система радиоинформации и аварий¬
ная сигнализация.
Life Technologies. См. Invitrogen.
List Biological Laboratories. 501-B Vandell Way, Camp¬
bell, CA 95008, USA. 800 726-3213; +1 408 866 6363.
+ 1 408 866 6364. info@listlabs.com. www.listlabs.com.
Холерный токсин.
LMS Ltd. The Modern Forge, Riverhead, Sevenoaks,
Kent, N13 2EL, England, UK. +44 (0)1732 451 866.
+44 (0)1732 450 127. sales@lms.ltd.uk. zsmw.lms.ltd.uk.
Настольные автоклавы; инкубаторы с охлаждением;
морозильники; холодильники.
Lome Biochemicals. Reading, Berks., UK. +44 (0)118 934
2400. +44 (0)118 934 2788. info@lomelabs.com. zemw.
lomelabs.com. Коллагеназа; ДНКаза; гиалуронидаза,
трипсин.
LTE Scientific Ltd. Greenbridge Lane, Greenfield, Old¬
ham, OL3 7EN, England, UK. +44 (0) 1457 876 221.
+44 (0) 1457 870 131. info@lte-scientific.co.uk. www.
lte-scientific.co.uk. Автоклавы; настольные автоклавы;
инкубаторы: С02, с охлаждением; сушильные шкафы;
ламинарные шкафы; блоки микробиологической
защиты; сухожаровые шкафы.
Ludl Electronic Products. 171 Brady Ave., Hawthorne,
NY 10532, USA. +1 914 769-6111 x223. +1 914-769-
4759. Cust-Service@ludl.com. zemw.ludl.com/. Электрон¬
ные комплектующие; компьютеризованный затвор;
сменные светофильтры; управляющая система для
съемки в режиме замедленного времени.
Lumitech Ltd. См. Cambrex. Наборы для определения
цитотоксичности; наборы для определения клеточной
пролиферации; наборы для определения контамина¬
ции микоплазмой; люцифераза; набор для определе¬
ния доли аденилатных нуклеотидов.
MA Bioservices. См. BioReliance.
Mallinckrodt-Baker. См. J.T. Baker.
Markson LabSales Inc. 5285 N.E. Elam Young Pkwy., State
A-400, Hillsboro, OR 97124, USA +1 503 648 0762,800
528 5114. +1 503 648 8118. markson@teleport.com. zemw.
markson.com. Реактивы для хроматографии; дезинфек¬
танты; фильтры; молекулярные сита; рефрактометр.
Matrix Technologies Corp. 22 Friars Drive, Hudson,
NH 03051, USA. +1 (0)603 595 0505, 800 345 0206.
+ 1 (0)603 595 0106. info@matrixtechcorp.com. www.
matrixtechcorp.com. Автоматические диспенсеры; ав¬
томатические пипетки с регулируемым расстоянием
между наконечниками, автоматические пипетки;
пипетирующие устройства; роботизированное обо¬
рудование для разлива жидкостей.
Matrix Technologies Corp. Lower Meadow Road,
Brooke Park, Handforth, Cheshire, SK9 3LW, Eng¬
land, UK. 0800 389 4431; +44 (0) 161486 2110. +44
(0) 161 488 4560. info@matrixtechcorp.eu.com. www.
matrixtechcorp.com. Автоматические диспенсеры; ав¬
томатические пипетки с регулируемым расстоянием
между наконечниками, автоматические пипетки;
пипетирующие устройства; роботизированное обо¬
рудование для разлива жидкостей.
MatTek Corp. 200 Homer Avenue, Ashland, MA 01721,
USA. 800-634-9018; +1 508-881-6771. +1 508-879-
1532. information@mattek.com. www.mattek.com/.
Исследование цитотоксичности органотипические
культуры; тканевой эквивалент; трансплантаты.
MBL International Corp. MBL International, 15 В Consti¬
tution Way, Woburn, MA01801,USA.+1 781 939 6964;
1-800 200 5459. +1 781 939 6963. info@mblintl.com.
zemw.mblintl.com/. Антитела; апоптоз; BUdR; клеточ¬
ная пролиферация; рецепторы PCNA (proliferating
cell nuclear antigen, ядерный антиген пролифериру¬
ющих клеток); передача сигнала.
MDH Ltd. См. Bioquell.
Mediatech Inc. 13884 Park Center Road, Herndon, VA
20171, USA. 800 235 5476. zemw.cellgro.com. Автокла¬
вируемая среда (MEM); глютамин; сухая среда Хэма
F12; сбалансированный солевой раствор Хэнкса без
Mg2+H Са2+' (CMF-HBSS); среда для культуры клеток
насекомых; Pluronic; бессывороточная среда.
Medical & Biological Laboratories (MBL) Co Ltd.
Sumitomoshoji Marunouchi Bldg. 5F, 5-10Marun-
ouchi 3 chome, Naka-ku, Nagoya 460-0002, Japan.
+81 52 971 2081. +81 52 971 2337. zemw.mbl.co.jp. Ан¬
титела; апоптоз; BUdR; пролиферация клеток; PCNA;
рецепторы; передача сигнала.
Medical Air Technology (MAT) Ltd. Airology Centre,
Mars Street, Oldham OL9 6LY, England, UK. +44
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
619
(0)161 621 6200.+44 (0)161 624 7547. Sales@medairtec.
сот. www.medicalairtechnology.com/. Ламинарные
шкафы; ламинарные шкафы; боксы микробиологи¬
ческой защиты.
Memmert GmbH. P.O. Box 1720, D-91107 Schwabach,
Germany. +49 (0) 91 229 250. +49 (0) 91 221 4585.
info@memmert.com. www.memmert.com. С02-инкуба-
торы; инкубаторы с охлаждением; стерилизационные
шкафы.
Merck Biosciences, Ltd. Padge Road, Beeston, Not¬
tingham NG9 2JR, England, UK. 800 622 935; +44
(0)115 943 0840. +44 (0)115 9430951. customer, ser-
vice@merckbiosciences.co.uk. www.merckbiosciences.
co.uk. Антитела; пеногаситель; биохимические реак¬
тивы; буферы; КМЦ; химические реактивы; холерный
токсин; Кристалл-виолет; деконтаминирующий агент
Decon; детергенты; дексаметазон; декстран; ДМСО;
ЭДТА; ферменты; G418 сульфат; краситель Гимза;
глюкоза; глицерин; реактивы для исследования саха¬
ров; гидрокортизон; гигромицин В; иммунохимичес-
кие реактивы; меркаптоэтанол; ПЭГ; автоматические
пипетки; ридеры для планшетов; роллерная культура;
силикагель; красители; Трипановый синий.
Merck USA. См. EMD Biosciences.
Merck KGaA. См. VWR. de.vwr.com. Продукция. См.
Merck Biosciences.
Messer Cryotherm. Euteneuen4- D-57548 Kirchen/Sieg,
Germany. +49 27 41/95 85-0. +49 27 41/69 00. info@
cryotherm.de. www.messer-cryotherm.de. Морозильники;
охлаждающие системы с регулируемым режимом;
морозильники с жидким азотом.
Messer UK Ltd. Cedar House, 39 London Road, Reigate,
Surrey RH2 9QE, England, UK. +44 (0)1737 241133.+44
(0)1737 241842. www.messergroup.com. C02; охлажда¬
ющие системы с регулируемым режимом; заморажи¬
вающие системы; сухой лед; газовые смеси; кислород;
азот; морозильники с жидким азотом; калибрацион¬
ные газовые смеси; регуляторы газа; автоматические
пипетки; планшетные сканеры.
MetaBios Inc. 135-Innovation & Development Center,
University of Victoria, R-Hut McKenzie Avenue,
Victoria, British Columbia, V8W 3 W2, Canada. +1
(250) 472-4334. +1 (250) 721-6497. info@metabios.
com. www.metabios.com. Воздуходувные биореакторы;
пакеты для культуры; пакеты для сред.
Metachem Diagnostics Ltd. 29 Forrest Rd., Piddington,
Northampton, NN7 2DA, England, UK. +44-1604-
870370.+44-1604-870194. info@metachem.co.uk. zemw.
metachem.co.uk. Среда; сыворотка; тест-набор на
микоплазмы; бессывороточная среда; компоненты
сыворотки.
Mettler-Toledo GmbH. PO Box VI-400, CH-8606 Grei-
fensee, Switzerland. +41 1944 22 11. +41 1944 31 70.
www.mt.com. Весы, кондуктометры, денситометры,
рН-метры; диспенсеры; автоматические пипетки;
рефрактометры.
Mettler-Toledo Inc. 1900 Polaris Parkway, Colum¬
bus, OH 43240. 800 METTLER; +1 614 438 4511.
+1 614 438 4900. us@mt-shop.com. zemw.mt.com. Весы,
кондуктометры, денситометры, рН-метры; диспенсе¬
ры; автоматические пипетки; рефрактометры.
Mettler-Toledo Ltd. 64 Boston Road, Beaumont Leys,
Leicester, LE4 2ZW, England, UK. 070000 MTSHOP.
0116 236 5500. uk@mt-shop.com. www.mt-shop.com.
Весы; кондуктометры; денситометры, рН-метры; дис¬
пенсеры; автоматические пипетки; рефрактометры.
Michael Smith Engineers Ltd. FREEPOST SCE 7470,
Woking, Surrey, GU21 1BR, England, UK.0800 316 7891.
+44 (0)1483 723 110. zemw.michaelsmith-engineers.co.uk.
Автоматические диспенсеры; измеряющие и диспен-
сирующие насосы; перистальтические насосы.
Microflow. См. Bioquell.
MicroGeN2. См. Texol Products.
Microgon Inc. См. Spectrum. Фильтры; половолоконная
система; стерилизующие фильтры; фильтры-насадки
на шприц.
Microm International GmbH. Robert-Bosch-Str. 49, D-
69190 Walldorf, Germany. +49 6227-836 0. +49 6227-
836 111. zemw.microm.de. Cytoseal 60; стекла для мик¬
роскопа. Среда для заключения стекол.
Miele Inc. 9 Independence Way, Princeton, NJ 08540,
USA. 609 419 9898; 800 843-7231. +1 609 419-4298.
zemw.labwashers.com/. Моечная машина для лабора¬
торного стекла.
Miele Co., Ltd. Abingdon, Oxon, OX14 1TW, England,
UK. 845 3303618. zemw.mieleprofessional.co.uk. Моеч¬
ная машина для лабораторного стекла.
Miles Inc. См. Bayer Corp.
Miles Ltd. Cm. Bayer pic.
Millipore Corp. 290 Concord Rd., Billerica, MA 01821,
USA. +1 978 715 4321; (800) ILLIPORE.(781) 533-
3110. wzem.millipore.com/. Угольный фильтр; деиони¬
зация; электродеионизация; фильтры; держатели для
фильтров; стерилизация фильтрацией; вкладыши в
фильтрационную лунку; молекулярная фильтрация;
насосы; обратный осмос; стерилизация фильтрацией;
Sterivex; сверхчистая вода; очистка воды.
Millipore (U. К.), Ltd. 3&5The Courtyards, Hatters
Lane, Waterford, Hertfordshire, WD18 8YH, England,
UK. 870 900 46 45. 870 900 46 44. wzem.millipore.com/
purecommerce. Cm. Millipore Corp.
Miltenyi Biotec GmbH. Friedrich-Ebert-Strasse 68,
51429 Bergisch Gladbach, Germany. +49 2204 83060.
+49 220485197. macs@miltenyibiotec.de. zemw.Milteny-
iBiotec.com. Антитела: Acl33, CD34; клеточная сепара¬
ция, секреция, анализ секреции цитокинов; сепарация
эпителиальных клеток; сепарация эндотелиальных
клеток; сепарация фибробластов; магнитный сор¬
тинг; магнитные бусины; нейлоновые сита; маркеры
стволовых клеток.
Miltenyi Biotec Inc. 12740 Earhart Avenue, Auburn,
С A 95602, USA. +1 530 888 8871; 800 FOR MACS.
+ 1 530 888 8925. macs@miltenyibiotec.com. www.
MiltenyiBiotec.com. Cm. Miltenyi Biotec GmbH. UK.
0118 944 8000.0118 944 8001. info@moldev.com. zemw.
moleculardevices.com. Микротитрация: планшетные
сканеры, флюориметры, программное обеспечение,
спектрофотометры.
620
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
Miltenyi Biotec Ltd. Almac House, Church Lane, Bis-
ley, Surrey GU24 9DR, England, UK. 01483 799 800.
01483 799 811. macs@miltenyibiotec.co.uk. www.Milt-
enyiBiotec.com. Cm. Miltenyi Biotec GmbH.
Miltex Instruments Co., Inc. 700 Hicksville Road, Beth-
page, NY 11714, USA. 800-645-8000; +1 717 840 9335.
+ 1 717 840 9347. customerservice@miltex.com. www.
miltex.com. Инструменты для иссечения и хирурги¬
ческие инструменты.
Minucells and Minutissue Vertriebs-GmbH. Starenstrasse
2, D-93077 Bad Abbach, Germany. +49 (0)9405 962 440.
+49 (0)9405 962 441. minucells.minutissue@tonline.de.
www.minuiaiemKu.de. Камеры для микроскопа; ор¬
ганотипическая культура; перфузионная культура;
клеточный каркас для культуры; опорная структура
для ткани; курсы по культуре ткани.
Molecular Devices Corp. MaxLine Div., 1311 Orleans
Dr., Sunnyvale, CA 94089, USA 408-747-1700; 800-
635-5577. 4+1 08-747-3602. info@moldev.com. www.
moleculardevices.com. Микротитрация: ридер для
планшетов, флюориметры, программное обеспечение,
спектрофотометры.
Molecular Devices Ltd. 135 Wharfedale Road, Winnersh
Triangle, Winnersh, Wokingham, RG41 5RB, England,
UK. 0118 944 8000. 0118 944 8001. info@moldev.com.
wvm.moleculardevices.com. Микротитрация: ридер для
планшетов, флюориметры, программное обеспечение,
спектрофотометры.
Molecular Dynamics. См. Amersham Biosciences, wvml.
amershambiosciences.com/. Конфокальные микроско¬
пы; денситометры; ридеры для планшетов.
Molecular Probes, Inc. 29851 Willow Creek Road, Eugene,
OR 97402-0469, USA. 800 438-2209; +1 541 335 0338.
+1 541 335-0305. order@probes.com. wvm.probes.com.
ДНК флюорохром; субстраты для ферментов; флюо¬
ресцентные зонды; PicoGreen; красители для оценки
жизнеспособности.
МР Biomedicals, Inc. 15 Morgan, Irvine, CA 92618-2005,
USA. 800.633.1352, +1 949.833.2500; Life sciences
div: 800 854-0530; +1 330 562 1500. +1 949 8595989.
custserv@mpbio.com. www.mpbio.com/. Наборы ам-
нокислот; антибиотики; антитела; ингибитор апоп-
тоза Apotame; автоматические диспенсеры; плазма
цыпленка; цыпленка сыворотка; С02-инкубаторы;
коллаген; среда для криоконсервации; детергенты;
дезинфектанты; Фикол-гипак среды; глютамин;
факторы роста; HEPES; инкубаторы; ламинарные
шкафы; среда для получения лимфоцитов Lympho-
ргер; флаконы с несколькими поверхностями роста;
средство для уничтожения микоплазм (MRA); набор
для обнаружения микоплазм; фосфатно-буферный
раствор; автоматические пипетки; оборудование для
герметизации планшетов; Pluronic F68; пенициллин;
бокс биобезопасности; бессывороточные среды;
стрептомицин; трипсин; сыворотка; Thermanox;
Vitrogen.
МР Biomedicals NV/SA. Domveld 10.1731 Asse Relegem,
Belgium. +32 2 466 00 00; 008000 7777 9999. +32 2
466 26 42. www.mpbio.com/. Cm. MP Biomedicals, Inc.
MTR Scientific, LLC. 9639-122 Dr. Perry Road, Ijams-
ville, MD 21754, USA. +1 301-831-1377. +1 01-874-
1899. info@mtrscientific.com. www.mtrscientific.com.
Сиквенс ДНК; решетка для культуры тканей чело¬
века; красители.
Muis Controls Ltd. 10610-172 Street, Edmonton, Alberta,
T5S 1H8, Canada. +1 780 486 2400. +1 780 486 2500.
wvm.MuisControls.com. Анемометр; расходомер.
MVE Inc. Cm. Chart Biomed.
Mycoplasma Experience. Reigate, UK. +44 (0)1737 226
662. +44 (0)1737 224 751. mexp@mycoplasma-exp.
com. www.mycoplasma-exp.com. Тестирование на ми¬
коплазмы.
Nalge Nunc (Europe) Ltd. Unit la, Thom Business Park,
Hereford HR2 6JT, England, UK. +44 (0) 1432 263933.
+44 (0) 1432 376567. sales@nalgenunc.co.uk. www.nal-
genunc.com. Cm. Nalge Nunc International.
Nalge Nunc International. International Department,
75 Panorama Creek Drive, Rochester, NY 14625, USA.
800-625-4327; +1 716 264 3898. +1 585-586-8987;
+1716 264 3706. nnics@nalgenunc.com. www.nalgenunc.
сот/. Ампулы; стержни; вкладыши для клеточных
культур; клеточные фабрики; клеточные скребки;
слайды для камер; конические пробирки; контейне¬
ры; покровные стекла; криомаркеры, криопробирки;
криофлаконы; культуральные пробирки; чашки;
планшеты для микротитрации с дренажным дном;
фильтры; стерилизационные фильтры, вкладыши в
фильтрационные лунки; флаконы; контейнеры для
замораживания; газопроницаемые крышки; маркеры;
пакеты для сред; микроносители; мультиповерхност-
ные пропагаторы; покровные стекла Thermanox.
Napco. 170 Marcel Dr., Winchester, VA 22602, USA.
800-621-8820. info@napco2.com. www.napco2.com/.
С02-инкубаторы; моечные машины для лаборатор¬
ного стекла.
Napco. См. Precision Scientific/Napco. С02-инкубаторы;
моечные машины для лабораторного стекла.
National Химические реактивы. 1259 Seaboard Indus¬
trial Blvd., Atlanta, GA 30318, USA. 800 237 0263. www.
natchem.com/. Дезинфектанты; иодофор.
National Diagnostics, Inc. 305 Patton Dr., S.W. Atlanta,
GA 30336-1817, USA +1 404 699-2121; 800-526-3867.
+1 404-699-2077. info@nationaldiagnostics.com. www.
nationaldiagnostics.com. Сцинтилляционная жидкость
Ecoscint.
Nektar AL. 490 Discovery Drive, Huntsville, AL 35806,
USA. 800 457 1806. +1 256 533 4805. nektar@al.nektar.
com. vDvm.nektar.com. Гидрогели; полиэтиленгликоля
диакрилат (PEGDA).
Nektar UK. 69 Listerhills Science Park, Campus Road,
Bradford, West Yorkshire, BD7 1HR, England, UK.
+44 (0)1274 305 540. +44 (0)1274 305 570. nektar@
uk.nektar.com. www.nektar.com. Гидрогели; полиэти¬
ленгликоля диакрилат (PEGDA).
NEN Life Sciences. Cm. PerkinElmer.
New Brunswick Scientific Co Inc. Box 4005,44 Talmadge
Road, Edison, NJ 08818-4005, USA.800 631-5417;
+1 732 287 1200. +1 732 287 4222. bioinfo@nbsc.com.
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
621
www.nbsc.com/. Биореакторы; С02-инкубаторы;
сушильный шкаф; ферментеры; ротационный шей-
кер; инкубаторы; увеличивающие устройства для
наблюдения за культурой; качалочные платформы;
роллерная культура; роллерный барабан; роллерные
штативы; масштабирование; шейкер-инкубаторы;
стерилизационный шкаф; ультранизкотемператур-
ные морозильники.
New Brunswick Scientific (U. К.), Ltd. 17 Alban Park,
Hatfield Road, St Albans, Herts, AL4 OJJ, England,
UK.0800 581 331. +44 (0)1727 835 666. bioinfo@nbsuk.
co.uk. xsjfww.nbsuk.co.uk. Cm. New Brunswick Scientific
Co Inc.
New England Biolabs. 32 Tozer Road, Beverly, MA
01915-5599, USA. 800-632-5227;+1 978-927-5054.800-
632-7440; +1978-922-7085. customersewice@neb.com.
xsmw.neb.com. Реактивы для молекулярной биологии;
векторы; передача сигнала.
New England Biolabs (U. К.), Ltd. 73 Knowl Piece,
Wilbury Way, Hitchin, Herts SG4 OTY, England, UK.
+1 (0)1462 420 616; 800 318 486. +1 (0)1462 421 057;
800 435682. info@uk.neb.com. xsjxsjw.neb. uk.com/. Моле¬
кулярная биология, векторы, передача сигнала.
Nichols Institute Diagnostics Div. 1311 Calle Ba-
tido, San Clemente, CA 92673, USA. +1 949-940-7200.
+1949-940-7271. xsjww.nicholsdiag.com/. Иммуноис¬
следования: кость, щитовидная железа, адреналовая
функция.
Nikon Europe В. V. P.O. Box 222,1170 АЕ Badhoevedorp,
The Netherlands. +31 20 4496 222. +31 20 4496 298.
xsjxsjw.nikon-instruments.jp/eng/. См. Nikon Inc.
Nikon Inc. 1300 Walt Whitman Road, Melville, NY
11747-3064, USA. +1 631 547 8500. +1 631 5470306.
biosales@nikonincmail.com. www.nikonusa.com. CCD-
камеры; кольцевой маркер колоний; конфокальный
микроскоп; цифровые камеры; флюоресценция
микроскопы; анализ изображений; инвертированный
микроскопы; микроманипуляторы; микроскопы;
микрофотография; стереомикроскопы.
Nikon Instech Co., Ltd. Parale Mitsui Bldg., 8,
Higashidacho, Kawasaki-ku, Kawasaki, Kanagawa,
210-0005, Japan. +81-44-223-2167.+81-44-223-2182.
xsjw. nikoninstruments.jp/eng/. Cm. Nikon Inc.
Nikon UK Ltd. Nikon House, 380 Richmond Road, Kings¬
ton upon Thames, Surrey KT2 5PR, England, UK.+1
(0)181 541 4440;+1 (0)208 481 6826.+1(0)181 5414584.
xsjxsjw.nikon-instruments.com. Cm. Nikon Inc.
NLS Animal Health. 800-638-8672.+1-888-568-2825.
webadmin@nlsanimalhealth.com. www.nlsanimalhealth.
сот/. Анестетик Marcaine.
Nonlinear Dynamics. Cuthbert House, All Saints,
Newcastle upon Tyne, NE1 2ET, England, UK.+44
(0)191 230 2121. +44 (0)191 230 2131. www.nonlinear.
сот/. Компьютерное программное обеспечение; ана¬
лиз изображений; микрочипы; микротитрация.
Nonlinear USA Inc. 4819 Emperor Blvd, Suite 400, Du¬
rham,NC 27703, USA. +1 919 313 4556. xsjxsjw.nonlinear,
сот/. Компьютерное программное обеспечение; ана¬
лиз изображений; микрочипы; микротитрация.
Nova Biomedical Corp. 200 Prospect Street, Waltham,
MA 02454, USA. 800 458 5813; +1 781 894 0800.
+ 1 781 893 6998. info@novabio.com. www.novabio-
medical.com. Блок анализа биоректора; биосенсоры:
нашатырный спирт; электролиты, глюкоза; глютамин;
глютамат; лактат; осмометры; рН-мониторы.
Nova Biomedical. СЗ-5, Evans Business Centre, Deeside
Industrial Park, Deeside, Flintshire, CH5 2JZ, Wales,
UK. +44 1244 287 087. +44 1244 287080. office@
novabiomedical.co.uk. www.novabiomedical.com. Блок
анализа биоректора; биосенсор: нашатырный спирт;
электролиты, глюкоза; глютамин; глютамат; лактат;
осмометры; рН-мониторы.
Novocastra Laboratories Ltd. Balliol Business Park
West, Benton Lane, Newcastle upon Tyne, NE12 8EW,
UK. +44 (0)191 215 0567. +44 (0)191 215 1152. xsjww.
novocastra.co.uk. Антитела: NC-L-Villin.
NovaMatrix. FMC BioPolymer, Pharmaceutical,
1735 Market Street, Philadelphia, PA 19103, USA.
+ 1 215 299 6420; +1 215 817 6571. +1 215 299 6669.
novamatrixinfo@fmc.com. xsjxsjw.novamatrix.biz, xsjxsjw.fmc.
com. Альгинат; соли хитозана и основания; инкапсу¬
ляция; матрикс; гиалуронат натрия.
NovaMatrix. FMC BioPolymer, Gaustadallreen 21, N-
0349 Oslo, Norway. +47 2295 8638. +47 269 6470. nova-
matrixinfo@fmc.com. xsjxsjw.novamatrix.biz, xsjxsjw.fmc.com.
Альгинат; соли хитозана и основания; инкапсуляция;
матрикс; гиалуронат натрия.
Novartis Pharmaceuticals UK Ltd. Frimley Business
Park, GB- Frimley/Camberley, Surrey GUI6 7SR, Eng¬
land, UK. +44 (0)1276 692 255. +44 (0)1276 692 508.
xsjxsjw.novartis.co.uk. Циклоспорин.
Novartis Institutes for BioMedical Research, Inc.
400 Technology Square, Cambridge, MA 02139, USA.
+1 617 871 8000. +1 617 551 9540. xsjxsjw.us.sandoz.com.
Циклоспорин.
Novo Nordisk A/S. Novo Nordisk A/S, Novo Alle,
2880 Bagsvжгd, Denmark. +45 4444 8888. +45 4449
0555. webmaster@novonordisk.com. www.novonordisk.
com. Соматостатин.
NuAire Inc. 2100 Fembrook Lane, Plymouth, MN 55447,
USA.+1 763 553 1270; 1 800 328 3352. +1 763 553 0459.
nuaire@nuaire.com. xsjxsjw.niiaire.com. С02-анализаторы и
аварийная сигнализация; С02 многоходовой распреде¬
литель; С02-инкубаторы; ламинарный шкаф; низкотем¬
пературные морозильники; бокс биобезопасности.
Nuclepore Corp. См. Coming.
Nunc. См. Nalge-Nunc.
Nusil Silicone Technology. 1050 Cindy Lane, Carpinteria,
CA 93013, USA. +1 805 684 8780. +1 805 566 9905.ste-
veb@nusil.com. xsjxsjw.nusil.com. Адгезивные вещества;
эластомеры; гели; силиконовая смазка.
Nycomed Danmark ApS. Langebjerg 1, P.O. Box
88, DK-4000 Roskilde, Denmark. +45 46 77 11 11.
+45 46 75 66 40. www.nycomed.dk.www.nycomed.no.
Плотностная среда; среда для получения лимфоци¬
тов; метризамид; метризоат натрия.
Nycomed UK Ltd. The Magdalen Centre, Oxford Sci¬
ence Park, Oxford OX4 4 GA, England, UK. +44 1865
622
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
784 500. +44 1865 784 501. list@nycomed.com. www.ny-
comed.com/. Плотностная среда; среда для получения
лимфоцитов; метризамид; метризоат натрия.
Olympus America Inc. 2 Corporate Center Drive P.
O. Box 9058, Melville, NY 11747-9058, USA. 800-446-
5967. +1516 844 5112. olymptis@performark.com. wzvw.
olympusamerica.com/. Конфокальная микроскопия;
цифровые камеры; инвертированный микроскопы;
микрофотография; исследовательские микроскопы;
стереомикроскопы.
Olympus Optical Co. (Europe) GmbH. Wendenstrasse
14-18, D-20097 Hamburg, Germany. +49 40 23 77 30.
+49 40 233 765. main@olympus.uk.com. www.
olympuseuropa.com. Cm. Olympus America, Inc.
Olympus U.K. Ltd. Dean Way, Gt. Western Industrial
Park, Southall, Middlesex, UB2 4SB, England, UK. +44
(0)207 250 0179. +44 (0)207 250 4677. microscope@
olympus.uk.com. http://www.olympus.co.uk/microscopy/.
Конфокальная микроскопия; цифровые камеры;
инвертированный микроскоп; микрофотография; ис¬
следовательские микроскопы; стереомикроскопы.
Omega Engineering, Inc. One Omega Drive, Box
4047, Stamford, CT 06907-4047, USA. 800 848 4286;
+ 1 203 359 1660. +1 203 359 7700. info@omega.com.
www.omega.com. Кондуктометры; электронный тер¬
мометр; анемометр; расходомер; temperature indicator;
рН-метры.
Omnilab. Robert-Hooke-Str. 8, 28359 Bremen, Ger¬
many^ (0)421 175 99-0.+49 (0)421 175 99-300.
info@omnilab.de. www.omnilab.de. Центрифужные
пробирки; С02-инкубаторы; фильтры; флаконы;
гемоцитометры.
Oncogene Research Products. См. EMB Biosciences
(USA), Merck Biosciences (Calbiochem). Ангиогенез;
апоптоз; ELISA наборы; IGF; LIF; PCNA; TGF; TNF;
VEGF.
Oncor, Inc. Marketing Division, 209 Perry Parkway,
Gaithersburg, MD 20877, USA. +1 301 963 3500.
+1 301 926 6129. www.oncor.com. Хромосомное окра¬
шивание.
Orchid BioSciences, Inc. 4390 U.S. Route One, Prince¬
ton, NJ 08540, USA. +1 609 750 2200. +1 609-750-6400.
mail@orchid.com. www.orchid.com. Аутентификация;
ДНК-профилинг; судебная идентификация.
Orchid Cellmark. 20271 Goldenrod Lane, Suite 120,
Germantown, MD 20876, USA. +1 301 428 4980,
800 872 5227. +1 301 428 4877. www.orchidcellmark.
com. Аутентификация; ДНК-профилинг; судебное
исследование; тест на идентичность ДНК.
Origen Biomedical. 4020 S. Industrial Dr., Suite 160,
Austin, TX 78744, USA.+1 512 474-7278. +1 512 617-
1503. sales@origen.com. www.origen.com/. Пакеты для
криохранения; среда для замораживания; хранение
стволовых клеток.
Orion Researchlnc. См. Thermo Electron Corp. www.
thermo.com. Измерительные приборы; растворенный
кислород, pH, температура.
Oxford Instruments Inc. 130A Baker Ave. Exten¬
sion, Concord, MA 01742, USA. +1 978 369 9933.
+1 978 369 6616. info@ma. oxinst.com. www.oxinst.com/.
Микропипетки.
Oxford Instruments pic. Old Station Way, Eynsham,
Witney, Oxon 0X29 4TL, UK. +44 (0) 1865 881 437.
+44 (0) 1865 881 944. info.oiplc@oxinst.co.uk. www.
oxinst.com/. Микропипетки.
OxoidLtd. Wade Road, Basingstoke, Hampshire, RG24 8PW,
England, UK. +44 (0) 1256 841144. +44 (0) 1256 463388.
oxoid@oxoid.com. zvww.oxoid.com/uk.Система, иденти¬
фикации бактерий; Кровяной агар, планшеты; мик¬
робиологический агар, планшеты; питательный агар,
планшеты; питательные бульоны; фосфатно-буферный
раствор, таблетки; триптон-соевый бульон.
Oxoid Inc. 800 Proctor Avenue, Ogdensburg, New York
13669, USA. +1 613 226 1318. +1 613 226 3728. www.
oxoid.com/us. Cm. Oxoid Ltd.
PAA Laboratories GmbH. Haidmanweg9, A-4061, Pasch-
ing, Austria. +43 7229 648 65. +43 7229 648 66. info@
paa.at. z0z0w.paa.at. Антибиотики; факторы адгезии;
БСА; эмбриональная сыворотка теленка; среды; се¬
лективные бессывороточные среды.
PAA Laboratories, Ltd. 1 Technine Guard Ave¬
nue, Houndstone Business Park, Yeovil, Somerset
BA22 8YE, England, UK. +44 (0)1935 411418. +44
(0)1935 411 480. info@paalaboratories.co.uk. wz0w.paa.
at. Продукция см. PAA Laboratories GmbH.
Paar Scientific Ltd.(cM. also Anton Paar). 594 Kingston
Rd., London SW20 8DN, England, UK. 0208 540 8553.
0208 543 8727. paar@psl.anton-paar.co.uk. zsmw.paar-
scientific.com. Денситометры; вискозиметры.
Paesel & Lorei GmbH. & Co. Moselstr. 2b, D-
63452 Hanau, Germany. +49 (0)6181 18 70-0.+49
(0)618118 70-70. info@paesel-lorei.de. zeww.paesel-lorei.
de. Среды; факторы роста; антитела.
Pall Corp. Bio Support Div., 25 Harbor Park Dr., Port
Washington, NY 11746, USA. +1 516-484-3600; 800-
289-7255.+1 (516) 484-3651. custsvc@pall.com. Z0Z0W.
pall.com/laboratory. Фильтры; держатели для фильтров;
фильтрация; фильтр со вставным складчатым фильтру¬
ющим элементом; насосы; вакуумные насосы; фильтра¬
ция микоплазм; ультрафильтрация; денситометры.
Pall Corp. (UK). Havant Street, Portsmouth, Hamp¬
shire, POl 3PD, England, UK. +44 (0)23 9230 3303.
+44 (0)23 9230 2509. www.pall.com/laboratory. Cm.
Pall Corp.
P & T Poultry Supplies and Equipment. Cleeton Cottage
Farm, Cleeton Lane, Cleeton St Mary, Nr Cleobury Mor¬
timer, Shropshire, DY14 0QU, England, UK. +44 (0)1584
890263. info@pandtpoultry.co.uk. www.pandtpoultry.
co.uk/. Инкубаторы для яиц; оплодотворенные яйца.
PAW BioScience Products, Inc. +1 732 842 3939.
+1 732 842 6602. admin@pawbioscience.com. zemw.paw-
bioscience.com. Культуральные пакеты; перфузионный
биореактор с половолоконной системой; силиконо¬
вые системы трубок.
Peak Scientific Instruments. 5424 Sea Edge Dr.,
Punta Gorda, FL 33950-8735, USA. +1 866 647 1649.
+1 866 647 1649. info@peakscientific.com. zeww.peaksci-
entific.com/. Газогенераторы; генераторы азота.
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
623
Peak Scientific Instruments, Ltd. Fountain Crescent,
Inchinnan Business Park, Inchinnan, Renfrewshire,
PA4 9RE, Scotland, UK. +44 (0)141812 8100. +44
(0)141 812 8200. info@peakscientific.com. www.peaksci-
entific.com/. Газогенераторы; генераторы азота.
PelFreeze. См. Dynal Biotech.
Pepro Tech, Inc. Princeton Business Park. 5 Crescent
Ave. Rocky Hill. NJ 08553-0275, USA. (800) 436-9910;
(609) 497-0253. (609) 497-0321. info@peprotech.com;
www.peprotech.com. Антитела; хемокины; цитокины;
ELISA.
PeproTech EC Ltd. Peprotech House, 29 Margra¬
vine Road, London, W6 8LL, England, UK. +44
(0)20 7610 3062. +44 (0)20 7610 3430. info@peprotech.
co.uk. zemw.peprotech.com. Антитела; хемокины; цито¬
кины; ELISA.
Perbio Science UK Ltd. Century House, High Street,
Tattenhall, Cheshire CH3 9RJ, England, UK. +44
(0)1829 771 744.+44 (0)1829 771 644. uk.info@perbio.
com. www.perbio.com. Культуральные пакеты; среды;
пакеты для сред; фетальная сыворотка теленка; за¬
мещение трипсина.
Perceptive Instruments. Blois Meadow Business Centre,
Steeple Bumpstead, Haverhill, Suffolk CB9 7BN, Eng¬
land, UK. 01440 730773. 01440 730630. sales@percep-
tive.co.uk. www.perceptive.co.uk. Счетчики колоний;
анализ изображений.
PerkinEImer LAS (Germany) GmbH. Ferdinand Porsche
Ring 17, 63110 Rodgau-Jugesheim, Germany. 0800 1
81 00 32. 0800 1 81 00 31. de.instruments.perkinelmer.
сот/. Продукция см. PerkinEImer Life Sciences.
PerkinEImer LAS (UK) Ltd. P.O. Box 66, Hounslow
TW5 9RT, England, UK. 0800 896046, 0800 891 715.
0800 891 714. www.PerkinElmer.com. Продукция см.
PerkinEImer Life Sciences.
PerkinEImer Life Sciences. 549 Albany Street, Boston,
MA 02118, USA. 800-762-4000; +1 617 482 9595.
617 482 1380. ProductInfo@perkinelmer.com. www.
PerkinElmer.com. Биореакторы; сепарация клеток;
центрифуги; счетчики колоний; цифровые камеры
для микроскопа; ферментеры; оборудование для по¬
лучения флюорографического изображения; факторы
роста: NGF, bFGF (FGF-2), TGF-a, TGF-P; анализ
изображений; люминометр; изотопные счетчики для
микротитрационных планшетов; микротитрация;
программное обеспечение; молекулярные зонды;
планшетные сканеры; радиоизотопы; сцинтилля-
ционные счетчики; сцинтилляционная жидкость;
заменители сыворотки SerXtend.
Pharmacia. См. Amersham Biosiences.
Pharmingen. 10975 Torreyana Road, San Diego, CA
92121. +1 877 232 8995. +1 858 812 8888. www.
pharmingen.com. Антитела: передача сигнала, ци¬
токины, интерлейкин-2, рецепторы; реактивы для
проточной цитометрии.
Phoretix International. См. Nonlinear Dynamics.
Photometries. 3440 East Britannia Drive, Tucson, AZ
85706, USA. +1 520 889 9933. www.photomet.com/.
CCD камеры и запасные части к ним.
Pierce Chemical Co. 3747 N. Meridan Rd., P.O. Box
117, Rockford, IL 61105, USA. +1 815 968 0747;
800 874 3723. 800 842 5007; +1 815 968 8148. cs@
piercenet.com. www.piercenet.com. Биохимические
вещества; химические реактивы; детергенты; мат-
рикс-покрытые планшеты; пронектин; исследование
белков; флаконы для образцов.
Planer pic. Biomedical Division, Windmill Road, Sun-
bury, Middlesex TW16 7HD, England, UK. +44(0)1932
755 000. +44(0)1932 755 001. www.planer.co.uk/.
Морозильники с жидким азотом; замораживание
клеток; программное обеспечение для контроля
замораживания и регистрации данных; контроль
учета; морозильники с контролируемой скоростью
замораживания.
Plas Labs Inc. 401 East North St., Lansing, MI 48906,
USA. +1 517 372 7177. +1 517 372 2857. PLI@Plas-
Labs.com. www.Plas-Labs.com. Бокс биобезопасности;
защитная камера с перчатками.
Plastim Ltd. Unit 100, Ashville Business Park, Commerce
Road, Staverton, Glos, GL2 9QJ, England, UK. +44
(0)1452 857 733. +44 (0)1452 857 744. www.plastim.
co.uk. Изделия из ПТФЭ: палочки, пластины, про¬
бирки.
Polaroid Corp. Polaroid Corp., North America Sales &
Marketing, 1265 Main St. W2-2C, Waltham, MA 02451,
USA. 800-343-5000.617 386 6271. www.polaroid.com/.
Моментальное фотокамеры; кабельное TV; цифро¬
вые камеры; эмульсии; фотопленка и фотопластины;
камера для микроскопа; фотомикроскопия; камера
Polaroid Land.
Polaroid (U. К.), Ltd. UK Sales & Marketing, 800 The
Boulevard, Capability Green, Luton, LU1 3BA, Bed¬
fordshire, England, UK. +44 (0)1582 409 800. +44
(0)1582 409 801. www.polaroid.com. Cm. Polaroid Corp.
Polyfiltronics. Cm. Whatman. Дисковые фильтры; филь¬
тры; оборудование для фильтрации; держатели для
фильтров; manifolds; микрочипы, микротитрация;
пробирки; системы очистки воды.
Polymun Scientific. Nussdorfer Lande 11, 1190 Vienna,
Austria. +43 1 36006 6202. +43 1 369. 7615. office@
polymun.com. www.polymun.com. Биореактор Mem¬
broferm.
PolyScience. 6600 West Touhy Avenue, P.O. Box 48312,
Niles, IL 60714, USA. 800 229 7569; +1 847 647 0611.
+ 1 847 647 1155. customerservice@polyscience.com.
www.polyscience.com/. Набор для аналитической
химии; холодильные установки; циркуляционный
насос; Clorox; дезинфектанты; глютаровый альдегид;
водяные бани.
Polytech Scientific. 5 Haddonbrook Business Cen¬
tre, Fallodan Road, Orton Southgate, Peterbor¬
ough, Cambridgeshire, PE2 6YX, England, UK. +44
(0)1733 371 724. +44 (0)1733 371 645. sales@polytech-
scientific.com. www.polytechscientific.com. Диспенсеры
для горловин бутылей; бутыли, пластик; камеры для
подсчета клеток; центрифужные пробирки: коничес¬
кие и плоскодонные; криофлаконы; система учета за¬
мороженных запасов; система криохранения; газовые
624
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
горелки; пипетирующее устройство; пипетирующий
насос; Pi-насос; автоматические пипетки; пластико¬
вые пастеровские пипетки; пластиковые клапаны,
чашки для образцов; лента для индикации стериль¬
ности: автоклавы и сушильные шкафы; штативы для
пробирок; универсальный контейнер, 50 мл.
Popper & Sons, Inc. 300 Denton Ave., New Hyde Park,
NY 11040, USA. 888 717 7677; +1 516 248 0300.
888 557 6773; +1 516 747 1188. sales@popperandsons.
com. www.popperandsons.com. Шприц Корнуэлла;
иглы; шприцевой диспенсер повторного цикла; ин¬
дикаторы стерильности; Thermalog; шприцы; много¬
позиционные диспенсеры.
Porvair Sciences. Unit 6, Shepperton Business Park, Gov-
ett Avenue, Shepperton, Middlesex, TW17 8BA, Eng¬
land, UK. +44 (0)1932 240 255. +44 (0)1932 254 393.
www.powair-sciences.com. Оборудование для гермети¬
зации планшетов.
Precision Scientific/Napco. 170 Marcel Dr., Winchester
VA, 22602 USA. (800) 621-8820; +1-540-869-9892.
+ 1 540 869 0130. www.precisionsci.com/lab/another/
napco2.
Автоклавы; центрифуги; С02-инкубаторы, моечные ма¬
шина для лабораторного стекла; ламинарные шкафы;
низкотемпературные морозильники; микробиоло¬
гические боксы биобезопасности; стерилизующие и
сушильные печи; вакуумные насосы.
Princeton Instruments. 3660 Quakerbridge Road, Tren¬
ton, NJ 08619, USA. +1 609-587-9797. +1 609-587-
1970. zsjzsm.princetoninstruments.com/. CCD камеры и
комплектующие.
Prior Scientific Instruments Ltd. 3-4 Fielding Indus¬
trial Estate, Wilbraham Road, Fulbourn, Cambridge,
CB15ET, England, UK. 44 (0)1223 881 711. 44
(0)1223 881 710. uksales@prior.com. www.prior.com.
Микроскопы: флюоресцентные, исследовательские,
стерео; с механизированной платформой; автомати¬
ческий счетчик.
Prior Scientific Inc. 80 Reservoir Park Drive, Rockland,
ME 02370, USA. +1 781 878 8442. +1 781 878 8736.
info@prior.com. Микроскопы: флюоресцентные, ис¬
следовательские, стерео; с механизированной плат¬
формой; автоматический счетчик.
Priorclave Ltd. 129-131 Nathan Way, Woolwich, Lon¬
don, SE28 0AB, England, UK. +44 (0)208 3166620. +44
(0)208 855 0616. sales@priorclave.co.uk. www.priorclave.
co.uk. Автоклавы; стерилизаторы.
Promega Corp. 2800 Woods Hollow Rd., Madison,
WI 53711, USA. +1 608 274 4330, 800 356 9526.800
356 1970; +1 608 277 2601. custsew@promega.com.
www.promega.com. Набор для изучения апоптоза;
набор для изучения клеточной пролиферации;
исследовние цитотоксичности; перенос ДНК; эн¬
дотелиальные клетки; трансфекция гена; факторы
роста; иммортализация; реактивы для молекуляр¬
ной биологии.
Promega U.K. Delta House, Chilworth Recearch Centre,
Southampton S016 7 NS, England, UK. +44 (0)23 8076
0225; 800 378 994. +44 (0)1703 767014; 800 181037.
ukcustsewe@promega.com. zemm.pwmega.com/uk/. Cm.
Promega Corp.
PromoCell GmbH. Sickingenstrasse 63/65, D-69126 Hei¬
delberg, Germany. +49 (0)6221 64934-0; 800 776 66 23.
+49 (0)6221 64934-40; 800 100 83 06. info@pmmocell.
com. zerww.promocell.com/. Адипоциты; хондроциты;
цитокины; фибробласты кожи; ELISA; эндотелиаль¬
ные клетки; эпидермальные кератиноциты; среды; ме¬
ланоциты; клетки назального эпителия; остеобласты;
перфузионная камера; селективные среды; сыворотка;
бессывороточные среды; клетки скелетных мышц;
гладкомышечные клетки; специализированные кле¬
точные культуры; трипсин.
Protein Polymer Technologies Inc. 10655 Sorrento
Valley Road, First Floor, San Diego, С A 92121, USA.
619 558 6064, 800 755 0407. 619 558 6477. info@ppti.
com. zejww.ppti.com. Matrix; Pronectin; бессывороточ¬
ные среды; факторы клеточной адгезии; тканевые
факторы.
Protide Pharmaceuticals, Inc. 1311 Helmo Avenue,
St. Paul, MN 55128, USA. +1 612 730 1500. +1 612
730 8900. info@celox.com. www.protidepharma.com/.
ССР: среда Эрла для криохранения; бессывороточные
среды; компоненты сыворотки; витамины.
Purite Ltd. Bandet Way, Thame, Oxon OX9 3SJ, England,
UK. +44 (0)1844 217 141. +44 (0)1844 218 098. mail@
purite.com. zsjzsjw.purite.com/. Системы очистки воды.
Qiagen Inc. 27220 Tumberry Lane, Valencia, CA 91355,
USA. 800 426 8157; 800-DNA-PREP (800 362 7737).
800 718 2056. zsjzsjw.qiagen.com/. Приготовление ДНК;
приготовление РНК.
Qiagen Ltd. Qiagen House, Fleming Way, Crawley, West
Sussex, RH10 9NQ England, UK. +44 (0)1293 422 911.
+44 (0)1293 422 911. CustomerSewice-uk@qiagen.com.
zsjzsjw.qiagen.com/. Приготовление ДНК; приготовле¬
ние РНК.
Qiagen, GmbH. QIAGEN GmbH., QIAGEN Strasse 1,
40724 Hilden, Germany. 800 79612; +49 (0)2103 29
12000. +49 (0)2103 29 22000. orders-de@qiagen.com.
zsjzsjw.qiagen.com/. Приготовление ДНК; приготовле¬
ние РНК.
QMX Laboratories Ltd. Bolford Street, Thaxted, Essex,
CM6 2PY, England, UK. +44 (0)1371 831 611. +44
(0)1371 831 622. sales@qmxlabs.com.zemw.qmxlabs.com.
Фильтры для шприцов KlarityTM.
Quadrachem Laboratories Ltd. Kingfisher House,
Forest Row Business Park, Forest Row, East Sussex,
RH18 5DW, England, UK. +44 (0)1342 820 820. +44
(0)1342 820 825. enquiries@qclscientific.com. www.
qclscientific.com. Измеритель растворенного кисло¬
рода; кондуктометры; рН-метры.
R & D Systems. 614 McKinley Place, NE, Minneapo¬
lis, MN 55413, USA 800 328 2400; +1 612 379 2956.
800 343 7475. info@mdsystems.com. www.mdsystems,
com. Антитела; цитокины; набор для опредления
эйкозаноидов; ELISA; эпидермальный фактор роста;
фибронектин; факторы роста; интерлейкин-2; мат¬
рикс; матриксные металлопротеиназы; микротитра¬
ция; программное обеспечение.
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
625
R & D Systems Europe. 4-10 The Quadrant, Barton Lane.,
Abingdon, Oxon 0X14 3YS, England, UK. 800 37 34 15;
+44 (0)1235 529 449. +44 (0)1235 533 420. info@RnD-
Systems.co.uk. www.mdsystems.com. Cm. R&D Systems
(USA).
Radleys. Shire Hill, Saffron Walden, Essex CB11 3AZ,
England, UK. +44 (0)1/99 513 320. +44 (0)1799 513 283.
sales@radleys.co.uk. vmw.radleys.co.uk. Спирт-нераст-
воримые маркеры; бутыли; коробки для культуры;
культуральные камеры; воздуходувное стекло; су¬
шильная печь; анемометры; лабораторные тележки;
нитрильные перчатки одноразового использования;
груша для пипеток; цилиндр для пипеток; лоток для
пипеток; автоматические пипетки; Pi-насос; штативы;
димпенсеры; шейкер-инкубатор; сифонная моечная
машина для пипеток; ступенчатые автоматические
пипетки.
Radnoti Glass Technology Inc. 227 West Maple
Avenue, Monrovia, CA91016, USA. 800 428 1416;
+1 626 357 8827. +1 626 303 2998. Desmond@radnoti.
com. www.radnoti.com. Кюветы для органной и ткане¬
вой культур.
Rainin Instrument Co., Inc. Mack Road, PO Box 4026,
Woburn, MA 01888-4026, USA. +1 617 935 3050;
800 472 4646. +1 617 938 1152.pipets@rainin.com. wrww.
rainin.com/. Автоматические пипетки.
Ranfac Corp. 30 Doherty Avenue, P.O. Box 635, Avon,
MA 02322, USA. 800 272 6322; +1 508 588 4400.
+1 508 584 8588. info@ranfac.com. www.ranfac.com/.
Иглы для биопсии.
Raven Biological Laboratories. 8607 Park Dr., Omaha,
NE 68106, USA. 800 728 5702; +1 402 593 0781.+1 40
2 593 0921; +1 402 593 0995. info@ravenlabs.com. wvm.
ravenlabs.com/. Микробиологические среды; коробки
для стекол; индикаторы стерильности.
Raymond A. Lamb LLC. 7304 Vanclaybon Drive, Apex,
NC 27502, USA. +1 919 387 1237. +1 919 387 1736.
sales, na@ralamb.com. www.ralamb. net/information,
html. Покровные стекла; предметные стекла; кассета
для стекол.
Raymond A. Lamb Ltd. Units 4 & 5 Parkview Indus¬
trial Estate, Lottbridge Grove, Eastbourne, East Sussex
BN23 6QE, England, UK. +44 (0)1323 737000. +44
(0)1323 733000.sales@ralamb.com. www.ralamb.co.uk/.
Покровные стекла; стекла для микроскопа; коробки
для стекол.
Recearch Diagnostics Inc. Research Diagnostics
Inc., Pleasant Hill Road, Flanders, NJ 07836, USA.
+1 973 584 7093; 800 631 9384. +1 973 584 0210;. Re-
searchD@aol.com. www.researchd.com/. Ангиопоэтин-1;
антитела; тетраметилбензидин; TGF-pi; ультра- тет-
раметилбензидин.
Recearch Organics Inc. 4353 E. 49th St., Cleveland,
OH 44125-1083, USA. 800 321 0570; +1 216 883 8025.
info@resorg.com. www.resorg.com. Биохимические ве¬
щества; HEPES.
Retsch GmbH. & Co., KG. Rheinische Strasse 36,
42781 Haan, Germany. +49 21 29 55 61-0. +49 21 29
87 02. info@retsch.de. www.retsch.de. Сита.
Revco Scientific, Inc. 308 Ridgefield Ct, Asheville,
NC 28806, USA. 800-252-7100; (Canada) 800-447-
3826;+1 828-658-2711. +1 828 645 3368. sales@revco-
sci.com. www.revco~sci.com. Морозильники ультра-
глубокого замораживания.
Richardsons of Leicester. 112A Milligan Road, Leices¬
ter, LE2 8FB, England, UK. +1 (0)116 283 8604. +1
(0)116 283 7109. sales@richardsonsofleicester.co.uk.
www.richardsonsofleicester.co.uk/. Стекла для мик¬
роскопа (Berliner Glas KG); пастеровские пипетки;
контейнеры для образцов.
Riken. 3-1-1 Koyadai, Tsukuba-shi, Ibaraki 305-0074,
Japan, vmw.brc.riken.go.jp/en/. Клеточный банк; кле¬
точные линии; клоны ДНК; праймеры.
Robbins Scientific (Europe) Ltd. The Exchange,
24 Haslucks Green Road, Shirley, Solihull, West Mid¬
lands, B90 2EL, England, UK. +44 (0)121 744 7445.
+44 0121 744 0775.sales@robsci.co.uk.www.robsci.co.uk.
Автоматические диспенсеры; плотные среды; подог¬
ревающие блоки; средыдля приготовления фракции
лимфоцитов; системы микротитрации; роботизи¬
рованные системы разлива жидкостей; роллерные
стойки для инкубаторов.
Robbins Scientific (Europe) Ltd. См. also Genetic Re¬
search Instrumentation.
Robbins Scientific, USA. Cm. SciGene. wvm.mbsci.com.
Roboz Surgical Instrument Co. Inc. PO Box 10710,
Gaithersburg, MD 20898-0710, USA. 800-424-2984. +1
888 424 3121. info@roboz.com. www.roboz.com. Канюли;
инструменты; стерилизаторы; шприцы; индикаторы
стерильности.
Roche Applied Science (Germany). Sandhofer Str 116,
D-68305 Mannheim 31, Germany. 800-759-4152. Or¬
ders: +49 (0)621/759-4136. www.roche-appliedscience.
сот/. Автоматические пипетки; антитела; наборы для
исследования апоптоза; биохимические вещества; ВМ
цилин; бомбезин хроматогранин; коллаген; коллагена¬
за; DAPI; диспаза; перенос ДНК; DOTAP; DOSPER;
ELISA; ЭДТА; Fugene; факторы роста; GM-CSF; ге¬
парин; интерлейкин-3 (IL-3); липофекция; микропи¬
петки; среда; эрадикация микоплазм; бессывороточная
среда; фактор стволовых клеток (SCF); витамины.
Roche Applied Science (UK). Bell Lane, Lewes, West
Sussex BN7 1LG, England, UK.+44 (0)1273480444;
808 100 9998.808 100 80 60. vmw.roche-applied-science.
com/. Cm. Roche Applied Science (Germany).
Roche Applied Science (USA). P.O. Box 50414, 9115
Hague Rd, P.O. Box 50414, Indianapolis, IN 46250-0414,
USA. 800-428-5433. 800-428-2883. www.rocheapplied-
science.com/. Cm. Roche Applied Science (Germany).
RS Biotech. Laboratory Equipment Division, 4 Mackin¬
tosh Place, South Newmoor, Irvine, Ayrshire, KA11 4JT,
Scotland, UK. +44 (0)1294 222770. +44 (0)1294
2222A8.galaxy@rsbiotech.com. www.rsbiotech.com. C02-
инкубаторы; Galaxy.
RS Components, Ltd. Birchington Road, Corby, Northants,
NN17 9RS, England, UK. +44 (0)1536 444 222. www.rs-
components.com/index.htm. Электронные вентиляторы;
контроллеры подогрева; нагреватели; термисторы.
626
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
Safelab Systems, Ltd. 2 Vines Industrial Centre, Nailsea,
Bristol,BS48 1BG,England,UK. 44(0)1275 855 2Э2.+44
(0)1275 855 131. scdes@safelab.co.uk.wwwsafelab.co.uk.
Ламинарные шкафы; боксы микробиологической
защиты.
Safetec of America Inc. 887 Kensington Ave., Buffalo, NY
14215, USA. +1 716-895-1822; 800-456-7077. +1 716-
895-2969. wwwsafetec.com/. Стерильная комната; лами¬
нарные шкафы; боксы микробиологической защиты.
Saf-T-Pak. 10807-182 Street, Edmonton, Alberta,
T5S1J5, Canada. (780) 486-0211; 1-800-814-7484. (780)
486-0235; 1-888-814-7484. www.saftpak.com/. Транс¬
портировочные контейнеры; системы безопасности.
Sakura Finetek Europe. См. Bayer pic and Raymond
A. Lamb, nmwsakuraeu.com/. Цитоцентрифуга; тка¬
невой капсулирующий состав.
Sakura Finetek U.S. A., Inc. 1750 West 214th Street,
Torrance, CA 90501, USA. 800-725-8723; +1 310-972-
7800. +1 310-972-7888. mail@corp.sakuraus.com. zemw.
sakuraus.com. Цитоцентрифуга; тканевой капсулиру¬
ющий состав.
Santa Cruz Biotechnology Inc. 2145 Delaware Ave.,
Santa Cruz, CA 95060-5706, USA. 800 457 3801;
831 457 3800. +1 831 457 3801 .scbt@netcom.com. zemw.
scbt.com. Антитела: актин, интегрины, кадгерины,
молекулы клеточной адгезии, цитокератин, десмин,
фибронектин, GFAP, виментин; реактивы для про¬
точной цитометрии.
Sanyo Gallenkamp pic. Monarch Way, Belton Park,
Loughborough, Leics LE11 5XG, England, UK. +44
(0)1509 265 265. +44 (0)1509 269 770. sanyo@san-
yobiomedical.co.uk. www.sanyogallenkamp.com/. C02-
инкубаторы; морозильники.
Sanyo Scientific. 1062 Thomdale Avenue, Bensenville,
IL 60106, USA. 630-875-3530; 800-858-8442.630-238-
0074. info@sss.sanyo.com. www.sanyobiomedical.com/.
Инкубаторы; морозильники.
Sarstedt AG & Co. Rommelsdorfer Strasse, Postfach
1220, 51582 Numbrecht, Germany. +49 2293 305 0.
+492293 305 1222. info@sarstedt.com. wwwsarstedt.
сот/. С02-проницаемые крышки; флаконы.
Sarstedt Inc. 1025, St. James Church Road, P.O. Box
468, Newton NC 28658-0468, USA. +1 828 465 400003.
+1 828 465 0718. info@sarstedt.com. iemm.sarstedt.com/.
Флаконы для культуры тканей, С02-проницаемые
крышки.
Sartec Group. Century Farm, Reading Street, Tenterden,
Kent, TN30 7HS, England, UK. +44 (0)1233 758 157.
+44 (0)1233 758 158. sales@sartec.co.uk. wwwsartec.
co.uk. Газогенераторы; анализ уровня общего орга¬
нического углерода (TOC): ТОС анализаторы; ТОС
стандарты; определение чистоты воды.
Sartorius AG. Weender Landstrasse 94-108, D-37075 Got¬
tingen, Germany. +49.551.308.00. +49.551.308. 3289.
www.sartorius.com/. Весы; биореакторы; среда для
хроматографии; дисковые фильтры; ферментеры;
фильтры; держатели фильтров; фильтрационное
оборудование; инвертированный микроскоп; пакеты
для сред; микроскопы; Miniperm.
Sartorius Corp. 131 Heartland Blvd., Edgewood, NY
11717, USA. 516 254 4249,800 368 7178.516 254 4253.
www.sartorius.com/. Весы; биореакторы; среда для
хроматографии; дисковые фильтры; ферментеры;
фильтры; держатели фильтров; фильтрационное
оборудование; инвертированный микроскоп; пакеты
для сред; микроскопы; Miniperm.
Sartorius Ltd. Blenheim Road, Longmead Indus¬
trial Estate, Epsom, Surrey KT19 9QN, England, UK.
+44.1372.737100. +44.1372.720799. sartorius.uk@sarto-
rius.com. wzemsartorius.co.uk. Весы; биореакторы; среда
для хроматографии; дисковые фильтры; ферментеры;
фильтры; держатели фильтров; фильтрационное
оборудование; инвертированный микроскоп; пакеты
для сред; микроскопы; Miniperm.
Scanalytics, Inc. 8550 Lee Highway, Suite 400, Fairfax,
VA 22031-1515, USA. +1 703-208-2230. +1 703-208-
1960. sales@scanalytics.com. zemm.scanalytics.com. Про¬
граммное обеспечение для получения и обработки
изображений; CCD камеры; анализ изображений.
Scharfe Systems. Krammerstrasse 22, D-72764 Reutlin-
gen, Germany. +49 (0)7121 387 86-0.+49 (0)7121
387 86-99. mail@CASY-Technology.com. zemw.CASY-
Technology.com. Счетчик клеток; анализаторы кле¬
точного размера; жидкость для счетчиков; детергент;
чашечки дляобразцов.
Schleicher & Schuell Bioscience. 10 Optical Avenue,
Keene, NH 03431, USA. 800-245-4024. +1 603-357-
7700. custserv@schleicher-schuell.com. zemwschleicher-
schuell.com. Мембраны для блоттинга; стерилизация
фильтрацией; микроанализ.
Schleicher & Schuell BioScience GmbH. Hahnes-
trasse 3, D-37586 Dassel, Germany. +49 5561 791 463.
+49 5561 791 583. salesbio@schleicher-schuell.de. zemw.
schleicherschuell.com. Мембраны для блоттинга; сте¬
рилизация фильтрацией; микроанализ.
Schleicher & Schuell, UK Ltd. Unit 11, Brunswick Park,
Industrial Estate, London N11 1JL, England, UK. +44
(0) 208 361 3111. +44 (0) 208 361 6352. Salesuk@
schleicher-schuell.de. zemm.schleicher-schuell.com. Мем¬
браны для блоттинга; стерилизация фильтрацией;
микроанализ.
Schott AG. Hattenbergstr. 10,55122 Mainz, Germany.+49
(0)6131 66-0.+49 (0)6131 66-2000. info@schott.com.
zemmschott.com. Лабораторное стекло; боросиликатное
стекло; бутыли; бутыли из жаропрочного стекла.
Schott North America, Inc. 555 Taxter Road, Elmsford, NY
10523, USA. +1 914 831 2200. +1 914 831 2201. info@
usschott.com. zemm.schott.com. Лабораторное стекло, бо¬
росиликатное стекло, жаропрочное стекло, бутыли.
Schott Corp. Drummond Road, Stafford, ST 16 3EL, Eng¬
land, UK.+44 (0)1785 223 166. +44 (0)1785 223 522.
info.uk@schott.com. www.schott.com. Лабораторное
стекло, боросиликатное стекло, жаропрочное стекло,
бутыли.
Scientek. 101-11151 Bridgeport Rd., Richmond, ВС V6X
1T3, Canada. 1-866-321-3828;+1 604-273-9094.+1 604-
273-1262. sales@scientek.net. zemm.scientek.net/. Моеч¬
ная машина для лабораторного стекла.
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
627
Scientific & Chemical Supplies. Carlton House, Living¬
stone Road, Bilston, West Midlands, England, UK. +44
(0)190-2402402. +44 (0)190 240 2343. scs@scichem.
co.uk. www.sci-chem.co.uk. Химические реактивы.
Scientific Instrument Centre. Unit 34D Parham Drive,
Boyatt Wodd, Eastleigh, Hants, S050 4NU, England,
UK. +44 (0)23 8061 6821. +44 (0)23 8062 9700. enqui-
ries@sic.uk.com. zemw.sic.uk.com. Инкубаторы; детер¬
генты; моечная машина для лабораторного стекла;
генераторы азота; печи; ультразвуковая баня.
Scientific Laboratory Supplies Ltd. Wilford Indus¬
trial Estate, Ruddington Lane, Wilford, Nottingham,
NG11 7EP, England, UK. +44 (0)115 982 1111. +44
(0)115 982 5275. sales@scientific-labs.com. zemw.scien-
tific-labs.com. Кольца для клонирования; криофлако¬
ны; чашки; флаконы; многолуночные планшеты.
SciGene. 530 Mercury Drive, Sunnyvale, CA 94085,
USA.+1 408-733-7337.+1 408-733-7336. sales@scigene.
com. ztmw.scigene.com/. Инкубаторы; водяная баня.
Scion Corp. Suite H, 82 Wormans Mill Ct, Frederick, MD
21701, USA. +1 301 695 7870. +1 (301) 695-0035. info@
scioncorp.com. zemw.scioncorp.com. Устройство захвата
изображения (в режиме замедленного времени).
Sefar Inc. Фильтрация Division, Moosstrasse 2, CH-
8803 REuschlikon, Switzerland. +41 1 724 65 11.
+41 1 724 15 25. www.scientific-labs.com/. Марлевые
фильтры; сетчатые фильтры, полиэстер, полиамин,
полипропилен.
Sefar Фильтрация USA. 333 S. Highland Ave., Bri-
arcliff Manor, NY 10510, USA. +1 816 452 1520.
+1 816 452 2183. zsmrw.sefar.com. Марлевые фильтры;
сетчатые фильтры: нитекс, полиэстер, полиамин,
полипропилен.
Seikagaku America Inc. 124 Bernard St Jean Drive,
East Falmouth, MA 02536-4445, USA. 888 395 2221;
508 540 3444.+1 508-540-8680. info@acciusa.com. zemw.
seikagaku.com; zemw.acciusa.com. Коллаген I типа.
Sentinel Laboratories Ltd. Units 12-13 Lindfield En¬
terprise Park, Lewes Road, Lindfield, West Sussex,
RH16 2LH, England, UK. +44 (0)1444 484044.+44
(0)1444 484045. brian@sentinel-laboratories.com. zemw.
sentinel-laboratories.com/. Нитрильные перчатки,
защитные маска.
Serologicals Corp. 5655 Spalding Drive, Norcross, GA
30092, USA. +1 678 728-2000. info@serologicals.com.
zemw.serologicals.com. Антитела; апротинин; БСА, хи-
мотрипсин; цитокины; ДНКаза; ELISA; акторы роста;
сыворотка человека; добавки к среде; продукция для
молекулярной биологии; ПЦР; зонды; ингибиторы
протеазы; сыворотка; трипсин.
Serotec Inc. Serotec Inc., 3200 Atlantic Avenue, Suite 105,
Raleigh,NC27604,USA.800-265 7376;+l 919 878 7978.
+1 919 878 3751. serotec@serotec-inc.com. zemwserotec.
com. Антитела: CAMs, integrins, цитокины, EGFr,
фибронектин, факторы роста и цитокины, IL-2, IL-6,
KGF, OSM, VEGF; ELISA.
Serotec Ltd. 22 Bankside, Station Approach, Kidlington,
Oxford OX5 1JE, England, UK. +44 (0)1865 852700.
+44 (0)1865 373 899.serotec@serotec.co.uk. zemwserotec.
com. Антитела: CAMs, интегрины, цитокины, EGFr,
фибронектин, факторы роста and цитокины, IL-2,
IL-6, KGF, OSM, VEGF; ELISA
SERVA Электрофорез GmbH. Carl-Benz-Str. 7, P. О. B.
10 52 60,69115 Heidelberg, Germany. 800 73 78 24 62;
+49 (0)6221 13840-0.+49 (0)6221 13840-10. info@
serva.de. www.serva.de. Альбумины; антибиотики;
коллагеназа; декстраны; диализные системы трубок;
реактивы для лектрофореза; HEPES; латексные час¬
тицы; Pluronic-F68; ПЭГ; силикон.
SGM Biotech, Inc. 10 Evergreen Drive Suite E, Bozeman,
MT 59715, USA.+1 406 585-9535.+1 (406)585-9219.
order@sgmbiotech.com. zemw.sgmbiotech.com. Полоски
для индикации спор; индикаторы стерильности.
Shamrock Scientific Speciality Systems, Inc. 34 Da¬
vis Dr., Bellwood, IL 60104, USA. +1 708 547 9005,
800 323 0249. (800) 248-1907; +1 708 547 902i.sales@
shamrocklabels.com. www.shamrocklabels.com. Лента-
индикатор стерильности; лента, чувствительная к
давлению; ярлыки; лента для нанесения ярлыков;
этикетки; полоски-индикаторы температуры.
Shandon-Scientific. См. Thermo Electron Corp.,
Thermo-Shandon. zemw.thermo.com. Цитоцентрифуга;
C02 автоматическое переключение блоков в балло¬
нах; электрофорез; гемоцитометры; микрокапилляры;
моечная машина для пипеток; сушильное устройство
для пипеток.
Sigma-Aldrich. PO Box 14508, St Louis, М063178,
USA. 800 325 ЗОЮ; +1 314 771 5765. +1 314 771 5757.
OCDOMHC@sial.com. zemw.sigmaaldrich.com. Амино¬
кислоты; антитела; биохимические вещества; БСА;
химические реактивы; кольцемид; коллаген; коллаге¬
наза; дексаметазон; декстран; диацетилфлюоресциин;
дезинфектанты; DMSO; ДНКаза; бромистый этидий;
ЭДТА; формамид; глютамин; глютатион; факторы рос¬
та; среды; заместитель сыворотки MegaCell; соли; бес¬
сывороточные среды; трипсин; Trypzean; витамины.
Sigma-Aldrich Chemie GmbH. Eschenstrasse 5, 82024
Taufkirchen, Germany. 800 51 55 00. 800 64 90 000.
zemw.sigma-aldrich.com. Продукция см. Sigma-Aldrich
(USA).
Sigma-Aldrich Co. Ltd. Fancy Road, Poole, Dor¬
set BH17 7NH, England, UK. + 44 1202 733114;
800 717 181. +44 1202 715460; 800 378 785. ukcustsv@
europe.sial.com. zemw.sigmaaldrich.com. Продукция см.
Sigma-Aldrich (USA).
Signal Instrument Co Ltd. Стандарты House, 12 Doman
Road, Camberley, Surrey, GU15 3DF, England, UK. +44
(0)1276 682 841. +44 (0)1276 691302. Инструменты®
signalgroup.com. zemw.signal-group.com. Смеси газов;
газосмесители.
Signal USA. 355 North York Rd., Willow Grove, PA
19090, USA. 866-SIGN AL-U; +1 215 830 8882. +1 215
830 8922. sales@k2bw.com. www.signal-group.com/.
Смеси газов; газосмесители.
Силиконовый Specialty Fabricators. 3077 Rollie Gates
Drive, PasoRobles, CA93446, USA. 800 394 4284;
805 239 4284.805 239 0523. zemw.ssfab.com/. Силико¬
новая смазка.
628
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
Skan. См. H.V. Skan.
SkinEthic. 45 Rue St Philippe, 06000 Nice, France.+33 4
93 97 77 27. +33 4 93 97 77 28. infos@skinethic.com.
www.skinethic.com. Альвеолярный эпителий; пузыр¬
ный эпителий; культуры роговицы; исследования
цитотоксичности; исследования раздражения; сре¬
да; ротовой эпителий; органотипическая культура;
реконструированный эпидермис человека; специа¬
лизированные клеточные культуры; тканевые экви¬
валенты; вагинальный эпителий.
Smiths Medical International Ltd. Military Road, Hythe,
Kent, CT21 5BN, England, UK. +44 1303 260551.+44 1
303 266761.j.townsend@portex.com. www.smithsmedical.
сот/. Полупроницаемая нейлоновая пленка.
Society for In Vitro Biology (SIVB). SIVB Busi¬
ness Office, 13000-F York Road, #304 Charlotte,
NC 28278, USA. (800) 761-7476; +1 704 588 1923.
+1 704 588 5193. sivb@sivb.org. www.sivb.org. Биоло¬
гическое научное общество с особым интересом к
клеточной культуре и родственным методам.
Socorex ISBA SA. Ch. Champ-Colomb 7, P.O. Box 1024,
Ecublens, Lausanne, Switzerland. +41 21 651 6000.
+41 21 651 6001. socorex@socorex.ch. www.socorex.ch.
Цифровые автоматические пипетки; многоканальные
автоматические пипетки; шаговая автоматическая
пипетка.
SoloHill Engineering Inc. 4220А Varsity Drive, Ann
Arbor, MI 48108, USA. 866 807 3953; +1 734 973 2956.
+ 1 734 973 3029. solohill@ic.net. www.solohill.com.
Микроносители.
Sorvall. Cm. Kendro. http://www.sorvall.com/.
Southern Biotechnology Associates, Inc. P.O. Box
26221, Birmingham, AL 35260, USA. +1 205-945-1774;
800-722-2255. 205-945-8768. info@SouthemBiotech.
com. www.southembiotech.com. Антитела, против мыши
IgGS-фикоэритрин.
SP Industries Co. SP Industries Co., 935 Meams Road,
Warminster, PA, USA. +1 215-672-7800. +1 215 672
7807. hotpack@hotpack.com. wzem.hotpack.com. Авто¬
клавы; С02-инкубаторы; морозильники; ламинарный
шкаф; бокс биобезопасности.
Spectrum Europe B.V. P.O. Box 3262, 4800 Breda, The
Netherlands. +31 76 5719 419. +31 76 5719 772. info@
spectrumeurope.nl. www.spectrumeurope.nl. Cellmax;
системы трубок для диализа; половолоконная пер-
фузионная культура; микрокапиллярная перфузия;
концентрические полые волокна.
Spectrum Medical Inc. 18617 Broadwick Street, Ran¬
cho Dominguez, CA 90220, USA. +1 310 885 4600;
800 634 3300. +1 310 855 4666; 800 445 7330. custom-
erservice@spectrumlabs.com. www.spectrumlabs.com.
Cellmax; системы трубок для диализа; половоло¬
конная перфузионная культура; микрокапиллярная
перфузия; концентрические полые волокна.
SPI Supplies. См. Structure Probe, Inc.
SPSS Inc. 233 S. Wacker Drive, 11th Floor, Chicago, IL
60606. 312.651.3000. www.spss.com/. SPSS (version
10.0) программное обеспечение статистического
анализа.
Staniar, J. & Co. 34 Stanley Road, Whitefield, Manches¬
ter, M45 8QX, England, UK. +44 (0)161 767 1500. +44
(0)161 767 1502. sales@johnstaniar.co.uk. zemw.johnsta-
niar.co.uk/. Марлевые и нейлоновые сита, mesh; сито
из нержавеющей стали.
Stateboume Cryogenics. 18 Parsons Road, Parsons In¬
dustrial Estate, Washington, Tyne & Wear, NE37 1EZ,
England, UK. +44 (0)191 416 4104. +44 (0)191415 0369.
sales@stateboume.com. zemw stateboume.com. Морозиль¬
ник с контролируемым режимом замораживания;
морозильники с жидким азотом; сосуды Дюара.
Statlab Medical Products. 106 Hillside Driv, Lewis¬
ville, TX 75057, USA. +1 972 436-1010; 1-800 442-
3573.+1 972 436-1369. info@statlab.com. zemw .statlab.
сот/. Гистологическая среда для заключения срезов;
контейнер для радиоавтографической эмульсии.
StemCell Technologies France. 29 Chemin du Vieux Chene,
Z. I. R. S. T, F-38240, Meylan, France. +33 (0)4 76 04 75 30.
+33 (0)4 76 18 99 63. info@stemcellfrance.com. zemwstem-
cell.com. Cm. StemCell Technologies Inc.
StemCell Technologies Inc. 808-777 West Broadway,
Vancouver, BC, Canada, V5Z 4J7. +1 604 877 0713;
800 667 0322. +1 604 877 0704; 800 567 2899. infoweb@
stemcell.com. zemw.stemcell.com. Антитела; сепарация
клеток; слайды для камер; атлас колоний; цитокины;
чашки; культура клеток-предшественников гемопо-
этического ряда; интерлейкин-2; среды с метилцел-
люлозой; программное обеспечение; вращательные
флаконы; очистка стволовых клеток; видео.
Steris Corp. 5960 Heisley Rd., Mentor, OH 44060-1834,
USA. +1 440-345-2600; 800-JIT-4-USE. +1 440-350-
7081. www.steris.com. Автоклавы; деконтаминация;
дезинфектанты; сублимационная сушка; моечная
машина для лабораторного стекла; микроманипуля¬
торы; микроинъекторы; дистиллятор; парогенерато¬
ры; Wescodyne.
Steris Corp. STERIS House, Jays Close, Viables, Bas¬
ingstoke, Hampshire RG22 4AX, England, UK. +44
(0)1256 840 400. +44 (0)1276 685662/3. zemwsteris.com.
Автоклавы; деконтаминация; дезинфектанты; субли¬
мационная сушка; моечная машина для лаборатор¬
ного стекла; микроманипуляторы; микроинъекторы;
дистилляторы; парогенераторы; Wescodyne.
Stille АВ. Gordsvogen 14, Box 709, SE-169 27 Solna,
Sweden. 0046 8 588 58 000. 0046 8 588 58 005; info@
stille.se. zemw.stille.se. Иглы для биопсии.
Stille-Sonesta, Inc. 2220 Canton Suite 209, P.O. Box
140957, Dallas, TX 75201, USA. (800) 665 1614;(214)
741 2464.(214)741 2605;. stille@airmail.net. zemwstille.
se/. Иглы для биопсии.
Stoelting Co. 620 Wheat Lane, Wood Dale, IL 60191,
USA. +1 630 860 9700. +1 630 860 9775. physiology@
stoeltingco.com. zemw.stoeltingco.com/physio. Защитные
маски; перчатки, латекс, нитрил; стерилизатор для
инструментов; манипуляторы; Парафильм; контейне¬
ры джля режущих отходов; полоски-индикаторы сте¬
рильности; хирургические инструменты; салфетки.
Stratagene. 11011 N. Torrey Pines Rd., Lajolla, СА92037-
1073, USA. +1 512 321 3321; 800 894 1304. Fax orders:
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
629
+1 512 321 3128; Tech Services: 858 535 0034. tech ser-
vices@stratagene.com. wwwstratagene.com. Библиотеки
клонов генов; микроанализ; ПЦР; векторы.
Stratech Scientific Ltd. Unit 4, Northfield Business Park,
Northfield Road, Soham, Cambridgeshire, CB7 5UE,
England, UK. +44 (0)1353 722 500. +44 (0)1353 727755.
info@stratech.co.uk. www.stratech.co.uk. Антитела: мар¬
керы ангиогенеза, коллаген, фибронектин, GFAP,
ламинин, муцин, маркеры апоптоза, клеточный цикл,
коллаген I; концентраторы; культура на покровных
стеклах; цитокины; чашки; внеклеточный матрикс;
наборы для экстракции; вкладыши в фильтрацион¬
ные лунки; флаконы; факторы роста; инвазионные
камеры; матрикс для обработки; многолуночные
^ стекла; онкогены; чашки Петри; полилизин; бессы¬
вороточные среды, наборы для скрининга.
Structure Probe, Inc. 569 East Gay Street, West Chester,
PA 19380, USA l-(800)-2424-SPI; l-(610)-436-5400.1-
(610)-436-5755. spi3spi@2spi.com. www.2spi.com/. SPI-
Chem; реактивы для электронной микроскопии; Epon
заместитель; реактивы для световой микроскопии.
Stuart Scientific. Holmethorpe Industrial Estate, Redhill,
Surrey, RH1 2NB, England, UK. +44 (0)1737 766 431.
+44 (0)1737 765 952. www.bibby-sterilin.com. Ручной
счетчик колоний.
Summers Optical. Division of EMS Acquisition Corp.,
1560 Industry Road, P.O. Box 380, Hatfield, PA 19440,
USA+1 215 412 8380.+1 215 41 8450;+1 215 412 8451.
sgkcck@aol.com. www.emsdiasum.com/Summers/optical/
cements/cements/uv.html. УФ-затвердевающий клей
для стекла (цемент).
Surgicon, Ltd. Wakefield Rd., Brighouse, W. Yorkshire
HD61QL, England. +44 (0)1484 712 147. +44 (0)1484
400 106. Перчатки; индикаторы стерильности (авто¬
клав).
Swann-Morton Ltd. Owlerton Green, Sheffield, S6 2BJ,
England, UK. +44 (0)114 234 4231. +44 (0)114 231 496.
info@swann-morton.com. xsjww.swann-morton.com. Хи¬
рургические инструменты; инструменты для иссе¬
чения; скальпели, лезвия.
Synbiosis. 97Н Monocacy Blvd., Frederick, MD, 21701,
USA. 603 465 3385.603 465 2291. x0xejw.synbiosis.com/.
Счетчики колоний.
Synbiosis. Beacon House, Nuffield Road, Cambridge, CB4
1TF, England, UK. +44 (0)1223 727 125. +44 (0)1223
727 101. sales@synbiosis.com. x0x0w.synbiosis.com; хеше).
acolytecounter.com. Счетчик колоний Acoltye; счетчи¬
ки колоний; распределение колоний по размерам.
Syngene. Beacon House, Nuffield Road, Cambridge,
CB4 1TF, England, UK. +44 (0)1223 727 123.+44
(0)1223 727 101. eurosales@syngene.com) intlsales© syn -
gene.com. x0x0w.syngene.com. Подсчет колоний; анализ
геля; анализ изображений.
Syngene USA. 5108 Pegasus Court, Suite M, Frederick,
MD 21704, USA. +1 800-686-4407. +1 301-631-3977.
ussales@syngene.com. x0x0w.syngene.com. Подсчет ко¬
лоний; анализ гелей; анализ изображений.
Synthecon, Inc. 8054 El Rio, Houston, TX 77054, USA.
+1 713 741 2582. +1 713 741 2588. rccs@synthecon.com.
x0ww.synthecon.com. Биореактор NASA; вращательная
культуральная камера.
TAAB Laboratories, Equipment, Ltd. 3 Minerva House,
Calleva Park, Aldermaston, Berks, RG7 8NA, England,
UK. +1 (0)118 9817775. +1 (0)118 9817881. sales@
taab.co.uk. www.taab.co.uk. Ацетонитрил; буферные
растворы; инструменты для иссечения; электронная
микроскопия; фиксирующие растворы; глютаровый
альдегид; иммуноцитохимия; какодилат натрия.
Taylor-Wharton Cryogenics. РО Box 568, Theodore, AL
36590-0568, USA +1-251-443-8680; 800 898 2657. +1-
251-443-2209. twsales@harsco.com. xsjxsjw.taylorwharton.
сот/. C02; системы охлаждения с регулируемым ре¬
жимом; морозильники; сосуды Дюара; жидкий азот;
морозильники с жидким азотом.
Taylor-Wharton GmbH. Postfach 14 70, D-25804 Hus-
lum, Germany. 49 48 41 985 0. 49 48 41 985 30. www.
taylor-wharton.com. C02; системы охлаждения с регу¬
лируемым режимом; морозильники; сосуды Дюара;
газы; морозильники с жидким азотом.
Taylor-Wharton UK. См. Taylor-Wharton GmbH.
TCS Biologicals Ltd. Botolph Claydon, Buckingham
MK18 2LR, England, UK. +44 (0)1296 714 222. +44
(0)1296 714 806. sales@tcsgroup.co.uk. xsjww.tcsbiosci-
ences.co.uk/. Стекла для бактериальной культуры;
продукты из крови; сыворотка цыпленка, плазма;
кристаллвиолет; Гимза краситель; гематоксилин; кра¬
ситель Лейшмана; микробиологические красители;
Нейтральный красный; нигрозин; плазма; сыворотка;
флаконы для хранения.
TCS CellWorks Ltd. Park Leys, Botolph Claydon,
Buckinghamshire MK18 2LR, England, UK. +44
(0)1296 713 120. +44 (0)1296 713 122. office@tcscell-
works.co.uk. x0x0w.tcscellworkscatalogue.co.uk/. Accutase;
Accumax; набор для изучения ангиогенеза; брон¬
хиальные клетки; кадгерины; CAMs; хондроциты;
кондиционированная среда; эпителий роговицы;
эндотелиальные клетки; фибробласты; гепатоциты;
интегрины; кератиноциты; лейкоциты; удаление ми¬
коплазм; клетки назального эпителия; остеобласты;
бессывороточные среда; гладкомышечные клетки;
синовиальные клетки.
Tebu-bio APS. Forskersparken CAT, DTU Bygn-
ing 347, 2800 Lyngby, Denmark. +45 45 25 64 01.
+45 45 25 64 03. scandinavia@tebu-bio.com. xeww.tebu-
bio.com. Изучение матрикса; покрытие матриксом.
Tebu-bio Ltd. Unit 7, Flag Business Exchange, Vicarage
Farm Road, Peterborough, Cambs PEI 5TX, England,
UK. +44 (0)1733 421 880. +44 (0)1733 421 882. uk@
tebu-bio.com. x0x0w.tebu-bio.com. Изучение матрикса;
покрытие матриксом.
Tecan Sales Switzerland AG. Cm. strasse 103, CH-
8708 MEannedorf, Switzerland. +41 1(0) 922 89 22.
+41 1 (0)922 89 23. tecan.sales.ch@tecan.com. www.
tecan.com. Сканнеры для микрочипов; микротит¬
рация: ридер для планшетов, диспенсеры, моющие
устройства.
Tecan UK Ltd. Theale Court, 11-13 High Street,
Theale, UK-Reading RG7 5 AH, England, UK. +44
630
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
(0)1189 300 300. +44 (0)1189 305 671. helpdesk-uk@
tecan.com. www.tecan-uk.co. uk. Ридеры для микро-
титрационнных планшетов, диспенсеры, моечные
устройства для планшетов.
Tecan US Inc. P.O. Box 13953, Исследовательски-
eTriangle Park, NC 27709, USA. +1 919 361 5200.
+1 919 361 5201. helpdesk-us@tecan.com. www.tecan.
com. Микротитрация: планшеты, ридеры, диспенсеры,
моечные устройства для планшетов.
Techmate Ltd. 10 Bridgetum Avenue, Old Wolverton,
Milton Keynes, Bucks., MK12 5QL, England, UK. +44
(0)1908322 222. +44 (0)1908 319 941. sales@techmate.
co.uk. www.techmate.co.uk. Ампулы; аспираторы; автокла-
вируемые корзины; бутыли; вкладыши для клеточных
культур; клеточные скребки; центрифужные пробирки;
стекла для камер; конические пробирки; контейнеры;
покровные стекла; криопробирки; маркеры для крио¬
пробирок; криофлаконы; культуральные пробирки; су¬
шильная камера; чашки; дренажное дно, планшеты для
микротитрации; фильтры; стерилизация фильтрацией;
вкладыши в фильтрационную лунку; флаконы; шта¬
тивы для замораживания; газопроницаемые крышки;
ручные вакуумные насосы; пакеты для среды; моечные
устройства для пипеток; лабораторный пластик.
Techne (Cambridge) Ltd. Duxford, Cambridge, CB2 4PZ,
England, UK. 01223 832401. 01223 836838. sales@
techne.com. www.techne.com/. Магнитные мешалки;
перемешивающие флаконы; суспензионная культура;
флаконы для культуры с микроносителями.
Techne Inc. 3 Terri Lane, Suite 10, Burlington, N. J.
08016, USA. +1 609-589-2560; 800-225-9243. +1 609-
589-2571. www.techneusa.com/. Магнитные мешалки;
перемешивающие флаконы; суспензионная культура;
флаконы для культуры с микроносителями.
Technika. 4757 East Greenway Rd., Suite 107B PMB 177,
Phoenix, AZ 85032, USA. +1 480 348 0279. www.Tech-
nika.com. Анемометры; кондуктометры; измерители
растворенного кислорода; рефрактометры.
Techno Пластик Products (ТРР) AG. Zollstrasse 155,
CH-8219 Trasadingen, Switzerland. info@tpp.ch. www.
tpp.ch. Клеточные скребки; centrifuge пробирки;
чашки; фильтры; флаконы; газопроницаемые крыш¬
ки; пробирки Лейтона; многолуночные планшеты;
пипетки; скребки.
Tecniplast UK Ltd. 2240 Parkway, Kettering Venture
Park, Kettering Northants NN15 6XL, England, UK.
info@TecniplastUK.com. www.tecniplast.it. Клетки для
животных и оборудование; дезинфектанты.
Tekmar-Dohrmann. См. Teledyne Tekmar.
Tekto Inc. См. Sefar Фильтрация USA.
Teledyne Tekmar. 4736 Socialville Foster Rd., Mason,
OH 45040, USA. +1 513-247-7000; 800 874-2004.
+1 513-247-7050. sales@tekmar.com. www.tekmar.com.
Нейлоновые сита, нитекс; общий органический уг¬
лерод, определение.
Texol Products Ltd. Myrekirk Road, Dundee, Scot¬
land, DD2 4SX, UK. +44 (0) 1382 618400. +44 (0)
1382 618422. info@texol.co.uk.www.gasgen.co.uk/. Ге¬
нератор азота.
Thermo Electron Corp. 81 Wyman Street, Waltham, MA
02454-9046, USA. +1 781 622 1000. +1 781 622 1207.
Контактное лицо www.thermo.com. Аварийная сигнали¬
зация; центрифуги; С02-контроллеры; С02-инкубато-
ры; кондуктометры; морозильник с контролируемым
режимом замораживания; оборудование для крио¬
презервации; коробки для клеток; цитоцентрифуга;
регистраторы данных; электронные термометры; элек¬
трофорез; система учета замороженных запасов; моро¬
зильники; гемоцитометры; инкубаторы; ламинарные
шкафы; оборудование для разлива жидкостей; моро¬
зильники с жидким азотом; низкотемпературные мо¬
розильники; магнитный сортинг; микробиологические
боксы биобезопасности; микрокрышки (Drummond);
микропипетки; планшеты для микротитрации; реакти¬
вы для микротитрации; диспенсеры; морозильники с
жидким азотом; кольцевые шейкеры; печи; рН-метры;
сушильное устройство для пипеток; моющее устрой¬
ство для пипеток; автоматические пипетки; планшет¬
ный сканер; программируемый морозильник; система
радиоинформации и аварийная сигнализация; бокс
биобезопасности; TOC-анализатор; водяная баня.
Thermo Electron UK. Unit 5, The Ringway Centre, Edi¬
son Road, Basingstoke, Hampshire RG21 6YH, England,
UK.+44 (0)870 609 0203. +44 (0)870 609 9202. sales.
btd.uk@thermo.com. www.therrno.com. Продукция см.
Thermo Electron Corp.
Thermolyne. Cm. Bamstead Thermolyne.
Thomas Scientific. 99 High Hill Rd., P.O. Box 99, Swedes-
boro, NJ 08085, USA. +1 856 467 2000; 800 345 2100.
+1 856 467 3087. value@thomassci.com. www.thomassci.
com. Дезинфектанты; инструменты для иссечения;
ножи для иридоэктомии; магнитные мешалки; мик¬
рошприцы; силиконовые системы трубок; хирурги¬
ческие инструменты.
Titan Enterprises Ltd. Unit 2, 5A Cold Harbour Busi¬
ness Park, Sherborne, Dorset, DT9 4JW, England, UK.
+44 (0)1935 812 790. +44 (0)1935 812 890. info@
flowИзмepumeльныe npu6opu.co.uk. http://www.
flow Измерительные приборы.co.uk/. Анемометр;
расходомер.
ТРР. См. Techo PlasticProducts. www.tpp.ch.
Triple Red Laboratory Technology Ltd. Triple Red
Ltd., Unit D4 Drakes Park, Long Crendon Ind Est.,
Long Crendon, Buckinghamshire, HP18 9BA, England,
UK. +44 (0)1844 218 322. +44 (0)1844 218 332. info@
triplered.com. www.triplered.com. С02-инкубаторы;
ярлыки для криофлаконов; устройство для печата¬
ния этикеток; ярлыки; ламинарный шкаф; системы
очистки воды.
TSI Inc. 500 Cardigan Road, St. Paul, MN, 55126-3996,
USA. 1-800-874-2811; +1 651-490-2811. +1 651-490-
3824. answers@tsi.com. www.tsi.com. Анемометы, счет¬
чики расхода и измеритель скорости воздуха.
U.S. Filter (см. also USF), Ltd. 40-004 Cook Street,
Palm Desert, CA 92211, USA. 800.875.7873 ext. 5000;
+ 1 760-340-0098. +1 760-341-9368. labproducts@us-
filter.com. www.usfilter.com. Деионизация; обратный
осмос; очистка воды.
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
631
Unibioscreen S.A. 40, Avenue Joseph Wybran, 1070 Brus¬
sels, Belgium.+32 (0)25 29 58 34; +32 (0)25 29 58 34.
+32 (0)25 29 59 42. support@unibioscreen.com. www.
unibioscreen.com. Терапия рака; инвазия; ксенотран-
сплантаты.
UniEquip. Fraunhoferstrasse 11, D-82152 Martinsried,
Munich, Germany. +49 (0)89 857 52 00. +49 (0)89 856
13 04. uniequip@t-online.de. www.uniequip.com. Газовая
горелка, зажигаемая инфракрасным излучением.
Universal Biologicals (Cambridge) Ltd. Passhouse Farm¬
house, Papworth St Agnes, Cambridge, CB3 8QU, Eng¬
land, UK. +44 (0) 1480 839 015. +44 (0) 1480 831 912.
info@universalbiologicals.ltd.uk. www.universalbio-
logicals.ltd.uk. Антитела: актин, G белки, нервные
- стволовые клетки, тубулин; коллаген; реактивы для
цитометрии; факторы роста; протеинкиназы.
Upstate Biotechnology. 1100 Winter Street, Suite 2300,
Waltham, MA02451, USA. 800-233-3991, 781-890-
8845. 781-890-8845. info@upstatebiotech.com. www.
upstate.com. Антитела; апоптоз; наборы для исследо¬
вания; набор для изучения клеточной пролифера¬
ции; клеточные сигналы; ферменты; факторы роста;
нейробиология.
Upstate Biotechnology. Upstate House, Gemini Cres¬
cent, Technology Park, Dundee, DD2 1SW, Scotland,
UK.+44 (0)1382 560812. 0800 0190 444. ukinfo@up-
state.com. www.upstate.com. Антитела; апоптоз; наборы
для исследования; набор для изучения клеточной
пролиферации; клеточные сигналы; ферменты; фак¬
торы роста; нейробиология.
USB Corp. 26111 Miles Rd., Cleveland, OH 44128,
USA.+l 216 765 5000; 800 321 9322. 800 535 0898;
+ 1 216 464 5075. customerserv@usbweb.com. www.
usbweb.com. Биохимические вещества; поливинил-
пирролидин.
UVP, Inc. 2066 W 11th St., Upland, CA 91786, USA.
+1 909 946-3197; 800 452 6788. +1 909 946 3597. info@
uvp.com. wisrw.uvp.com. Ртутные лампы; флюоресцен¬
ция; УФ-лампы; УФ-трансиллюминаторы.
UVP, Ltd. Unit 1, Trinity Hall Farm Estate, Nuf-field
Road, Cambridge CB4 1 TG, England, UK. +44(0)
1223-420022. +44(0)1223-420561. uvp@uvp.co.uk.
xsmw.uvp.com. Ртутные лампы; флюоресценция; УФ-
лампы; УФ-трансиллюминатор.
Varian. 3120 Hansen Way, Palo Alto, С A 94304-1030,
USA. +1 650 213 8000. custserv@varianinc.com. xsmw.
varianinc.com. Вакуумные насосы.
Vector Laboratories, Inc. 30 Ingold Road, Burlingame,
CA 94010, USA. 800 227 6666; +1 650 697 3600.+1 65
0 697 0339. vector@vectorlabs.com. xsmw.vectorlabs.com.
ABC набор; щелочная фосфатаза; антитела; биотин/
авидин; ELISA; ферментативный иммуноанализ;
реактивы для проточной цитометрии; ингибиторы,
замедляющие выцветание и гашение флюоресценции;
глюкозы оксидаза; скрининг гибридом; лектины; клей
для стекол; ингибитор затухания УФ-свечения.
Vector Laboratories, Ltd. 3 Accent Park, Blakewell Road,
Orton Southgate, Peterborough PE2 6XS, England, UK.
+44 (0)1733 237 999. +44 (0)1733 237 119. vector@
vectorlabs.co.uk. www.vectorlabs.com. Продукция см.
Vector Labs Inc.
Verilabs Europe. Postbus 1336, 2302 BH Leiden, Neth¬
erlands. +31 (0)900-3628378. info@verilabs.nl. www.
verilabs.nl/. Аутентификация; ДНК-профилинг; ге¬
нетический тест на идентичность ДНК.
Vision Biosystems. Balliol Business Park West, Benton
Lane, Newcastle upon Tyne, NE12 8EW, England, UK.
+44 (0)191 215 4242. +44 (0)191 2154227. sales.eu@
vision-bio.com. www.vision-bio.com. Антитела: NC-L-
Villin.
Vision BioSystems Inc. 700 Longwater Drive, Norwell
MA 02061, USA. +1 781 616 1190. +1 781 616 1193.
sales.usa@vision-bio.com. www.vision-bio.com/. Анти¬
тела: NC-L-Villin.
Vivascience AG. Feodor-Lynen-Strasse 21; 30625 Han¬
nover, Germany. +49 (0)511 524 875-0.+49 (0)511
524 875-19. info@vivascience.com. xsmw.vivascience.de.
Биореакторы; слайды для камер; чашки для культу¬
ры; Flexiperm; Miniperm; многолуночные планшеты;
Quadriperm; Petriperm.
Vivendi Water Systems. High Street, Lane End, High
Wycombe, Bucks HP14 3JH, UK. +44 (0)1494 887 555.
+44 1494 887 837. sandra.welch@veoliawater.com.
xsmw.elgalabwater.com. Фильтрация; ионный обмен;
фотооокисление; обратный осмос; системы очистки
воды.
Volac. См. Camlab; Cole-Parmer.
V&P Scientific Inc. 9823 Pacific Heights Boulevard,
Suite T, San Diego, CA 92121, USA. 800 455 0644;
+1 858 455 0643. +1 858 455 0703. sales@vp-scientific.
com. xsmw.vp-scientific.com/. Подогревающие блоки.
VWR International. 1310 Goshen Pkwy, W. Chester, PA
19380, USA +1 610 431 1700; 800 932 5000.+1 610 436 17
61 .xemw.xmr.com. Автоклавы; бутыли; электрофорез обо¬
рудование; защитные маски; перчатки: латекс, нитрил,
винил; инкубаторы; лабораторные запасы; магнитные
мешалки; микрошприцы; микротитрация: фильтры,
планшеты; морозильники с жидким азотом; рН-метры;
насосы; покровные и предметные стекла; стерилизаци¬
онные фильтры; насадочные фильтры для шприцов;
ультразвуковая баня; системы очистки воды.
VWR International, Ltd. Hunter Boulevard, Magna
Park, Lutterworth, Leics LE17 4XN, England, UK.
0800 22 33 44; +44 (0)1202 669 700. +44 (0)1455 558
586. uk.sales@uk.vwr.com. xsmw.xmr.com. Продукция
см. VWR International, USA
Vysis. 3100 Woodcreek Dr., Downers Grove, IL
60515. USA. 800-553-7042, Ex.l; +1 708 271 7000.
+1 708 271 7008. vysis help@vysis.com. xemw.vysis.com.
Хромосомные красители; молекулярные зонды.
Wako Химические реактивы USA, Inc. 1600 Bellwood
Road, Richmond, VA 23237-1326, USA. +1 804 271
7791.+1 804 271 7791. www.wakousa.com. Антитела;
аскорбиновая кислота; биохимические вещества;
ферменты; среда Диаго Т.
Wallac. См. PerkinEImer.
Watson-Marlow Bredel Насосы Inc. 37 Upton Tech¬
nology Park, Wilmington, MA 01887-1018, USA.
632
Приложение III. ПРОИЗВОДИТЕЛИ ПРОДУКЦИИ И ДРУГИЕ ИСТОЧНИКИ
+ 1 978 658 6168; 800-282-8823. +1978 658 0041.
support @wmbHacocu.com. www.watsonmarlow.com.
Перистальтические насосы; силиконовые системы
трубок.
Watson-Marlow Bredel Насосы Ltd. Falmouth, Corn¬
wall TR11 4BR, England, UK. +44 (0)1326 370 370;
800 0189 844. +44 (0)1326 376 009. info@watson-mar-
low.co.uk. www.watson-marlow.co.uk. Перистальтиче¬
ские насосы; силиконовые системы трубок.
Wave Biotech AG. Ringstrasse 24, CH-8317 Ta-
gelswangen, Switzerland. +41 (0)52 354 36 36. +41
(0)52 354 3646. Контактное лицо via web page. www.
wavebiotech.ch. Биореакторы; культуральные пакеты;
качалочные платформы; волновое перемешивание.
Webb Microtome Ltd. Dr Sim Webb, Webb Mini-Mi¬
crotome, Brisimany, Walnut Hill, Surlingham, Norwich,
NR14 7DQ, England, UK. +44 (0)1603 592 268.+44
(0)1603 592 250.s.f.webb@eclipse.co.uk. www.microtome.
co.uk. Портативный микротом.
Wescor, Inc. 459, South Main Street, Logan, Utah 84321,
USA. 800 453-2725; +1435 752 6011. 801 752 4127.
biomed@wescor.com. www.wescor.com/. Цитоцентри¬
фуга; осмометры; красители для срезов.
Whatman Inc. 9 Bridewell Pi., P.O. Box 1197, Clif¬
ton, NJ 07014, USA. +1 (617) 485-6590; 800-631-
7290.+1 973 773 3991. info@whatman.com. xemw.what-
man.com. Хроматография; фильтрующие заглушки;
фильтровальная бумага; 24-луночные фильтрующие
планшеты; фильтры, стекловолокна, бумага; планше¬
ты с фильтрационными лунками; многоканальные
автоматические пипетки; планшеты для микротит¬
рации; фильтры Nuclepore; автоматические пипетки;
вакуумные насосы.
Whatman pic. 27 Great West Road, Brentford, Middlesex,
TW8 9BW, England, UK. +44<0) 208 326 1740.+44 (0)
208 326 1741. information@whatman.co.uk. xemw.what-
man.plc.uk. Продукция см. Whatman Inc.
Wheaton Science Products. Cm. Lawson Mardon
Wheaton.
Wissenschaftlich-Technische Werkstatten GmbH.
&Co. KG. Dr. — Karl-Slevogt-Strasse 1, D-82362 Weil-
heim, Germany.+49 (0)881 183-0.+49 (0)881 183-420.
info@WTW.com. www.WTW.com. Счетчики колоний;
измерительные приборы: проводимость, кислород,
pH, термометры; анализ воды.
World PrecisionHHCTpyMeHibi. 175 Sarasota Center Bou¬
levard, Sarasota, FL34240-9258, USA. +1 941 371 1003.
+ 1 941 377 5248. wpi@wpiinc.com. www.wpiinc.com.
Вкладыши в фильтрационные лунки; измерители
сопротивления; TEER; измерение трансэпителиаль¬
ного электрического сопротивления.
World PrecisionmiCTpyMeHTbi. Astonbury Farm Business
Centre, Aston, Stevenage, SG2 7EG, England, UK. +44
(0)1438 880025. +44 (0)1438 880026. wpiuk@wpieu-
rope.com. xemw.wpi-europe.com. Вкладыши в фильтра¬
ционные лунки; измерители сопротивления; TEER;
измерение трансэпителиального электрического
сопротивления.
Worthington Biochemical Corp. 730 Vassar Avenue, Lake¬
wood, NJ 08701, USA. 800-445-9603; +1 732 942 1660.
800 368-3108; +1 732 942 9270. office@worthingtonbio-
chem.com. www.Worthington- biochem.com. Коллагеназа;
ДНКаза; гиалуронидаза; трипсин.
WPI. См. World Precisionlnstruments.
WTW Measurement Systems, Inc. 6E Gill St., Wo¬
burn, MA 01801, USA. 800 645 5999; +1 781 569 0095.
+ 1 781-932-3198. info@wtw-inc.com. www.wtw.com/.
Счетчики колоний, измерительные приборы: прово¬
димость, кислород, pH, термометры; анализ воды.
YSI Inc. 1700/1725 Brannum Lane, Yellow Springs,
Ohio 45387, USA. +1 937 767 7241; 800 659 8895.
+1 937 767 8058. Контактное лицо via web page. www.
YSI.com. Биохимические анализаторы, анализаторы;
биосенсоры; мониторинг: холин, глюкоза, глютамат,
глютамин, лактат.
Zeiss. См. Carl Zeiss.
Zimmer Corp. P.O. Box 708, 1800 West Center Street,
Warsaw, IN 46581-0708, USA. 800-613-6131. +1 574-
372-4988. Контактное лицо via webpage, xemwzimmer.
com. Керамические палочки и клеточный каркас для
культуры (тканевая инженерия).
Zinsser Analytic (U. К.), Ltd. Howarth Road, Staffer-
ton Way, Maidenhead, Berks SL6 1AP, England. +44
(0)1628 773 202. +44 (0)1628 672 199. officeuk @zins-
seranalytic.com. xerwwjnnsser-analytic.com/. Автомати¬
ческие пипетки; флаконы; диспенсеры; оборудование
для разлива жидкостей; микротитрация; насосы:
перистальтические, вакуумные.
Zinsser Analytic GmbH. Eschborner Landstrasse 135,
D-60489 Frankfurt, Germany. +49 69 78 91 06-0.
+49 69 78 91 06-80. info@zinsser-analytic.com. xernw.
zinsseraruilytic.com. Автоматические пипетки; флако¬
ны; диспенсеры; разлив жидкостей; микротитрация;
насосы: перистальтические, вакуумные.
Zymed Laboratories, Inc. 561 Eccles Avenue, South San
Francisco, CA 94080, USA. 800-874-4494; 650-871-4494.
+1 650 871 4499. tech@zymed.com. xemw2ymed.c0m. Ан¬
титела: Ki-67, цитокератин, рак молочной железы.
Приложение IV
СЛОВАРЬ УПОТРЕБЛЯЕМЫХ ТЕРМИНОВ
[по Schaeffer, 1990]
Авторадиография. Локализация изотопов в клетках
и срезах тканей (микроавторадиография), а также
в окрашенных электрофоретических препаратах
(блотах) при электрофоретических исследованиях;
достигается путем экспозиции фотографической
эмульсии, помещенной в непосредственной бли¬
зости от образца.
Адаптация. Индукция или подавление синтеза макромо¬
лекул (обычно белков) в ответ на стимул; например,
ферментативная адаптация — изменения фермента¬
тивной активности, обусловленное действием индук¬
тора или репрессора и включающая в себя изменение
скорости синтеза и деградации ферментов.
Аллотрансплантат см. Гомотрансплантат.
Амнноцентез. Пренатальная пункция плодного пу¬
зыря.
Анемометр. Прибор для измерения скорости потока
воздуха.
Анэуплоид. Нарушение гаплоидного числа хромосом
(см. Гаплоид).
Апоптоз. Клеточная гибель путем биологически конт¬
ролируемых внутриклеточных процессов, включая
расщепление ДНК и фрагментацию ядра.
Асептический. Свободный от микробиологического
инфицирования.
Аутокринный. Рецептор-опосредованный ответ клетки
на фактор, продуцируемый этой же самой клеткой.
Аутотрансплантат. Трансплантат, взятый от одной
индивидуально особи и трансплантированный той
же особи.
Биореактор. Культуральный сосуд для получения
клеток в большом количестве, как прикрепленных
к субстрату, так и растущих в виде суспензии.
Биостат. Культуральный сосуд, в котором физические,
физико-химические и физиологические условия,
в т.ч. концентрация веществ, поддерживаются
постоянными. Обычно это осуществляется путем
перфузии, мониторинга и подачи питания в соот¬
ветствии с результатами мониторинга.
Валидация (клеточных линий). Процесс, который
включает в себя аутентификацию, характеризация
и демонстрацию отсутствия контаминации клеточ¬
ной линии.
Вариант (мутант, дивергент) Клеточная линия, экс¬
прессирующая стабильный фенотип, отличный от ро¬
дительской культуры, из которой она произошла.
Вирусная трансформация. Стабильное изменение фе¬
нотипа, вызванное генетическим или наследуемым
действием трансформирующего вируса.
Время генерации. Интервал от одной точки в клеточ¬
ном цикле до такой же точки в следующем цикле.
Отличайте от периода удвоения популяции, которое
определяется путем оценки общего количества кле¬
ток в популяции, и таким образом, среднее значение
времени удвоения популяции, включает эффект
нерастущих клеток.
Время удвоения популяции. Интервал времени, не¬
обходимый для увеличения количества клеток в
популяции в два раза к середине логарифмической
фазы роста.
Гаплоид. Такой набор хромосом, при котором каждая
хромосома представлена один раз, у большинства
высших животных такой набор хромосом представ¬
лен в гаметах; половина набора хромосом, имеющих¬
ся в обычных соматических клетках.
Генетика соматических клеток. Раздел генетики,
изучающий рекомбинацию и сегрегацию генов в
соматических клетках, обычно путем их слияния.
Генотип. Общие генетические характеристики клеток.
Ген-супрессор. Ген, который ингибирует трансформи¬
рованный (малигнизированный) фенотип, обычно
связан с доминантно-негативной регуляцией кле¬
точной пролиферации или клеточной миграции;
часто в трансформированных клетках обнаружива¬
ется мутация или делеция гена-супрессора.
Гетерокарион. Клетка, содержащая два или более
генетически различных ядра; обычно получается
путем слияния клеток.
Гетероплоид. Культура, в которой клетки имеют число
хромосом, отличное от диплоидного и различающе¬
еся друг от друга.
Гибридизация in situ. Связывание специфически
комплементарных нуклеиново-кислотных зондов
с внутриклеточными компонентами; кДНК зонды
для локализации последовательностей мРНК, или
РНК зонды для локализации ДНК (см. Хромосомное
окрашивание). Визуализируется методом радиоизо-
топного мечения и микроавторадиографии, либо с ис¬
пользованием флюорохрома, связанного с зондом.
Гибридная клетка. Одноядерная клетка, которая
получается в результате слияния двух различных
клеток, приводящего к формированию синкариона
(см. Синкарион).
Гистотипичная культура. Культура, сформирован¬
ная в соответствии с морфологией ткани in vivo.
Обычно трехмерная культура воспроизводится
путем восстановления диспергированной клеточной
культуры в результате клеточной пролиферации
634
ПРИЛОЖЕНИЕ IV. СЛОВАРЬ УПОТРЕБЛЯЕМЫХ ТЕРМИНОВ
с образованием многослойности или реагрегации
тканеобразной структуры. Органные культуры не
могут быть размножены, гистотипичные культуры
можно наращивать.
Гликокаликс. Гликозилированные пептиды, белки и
липиды, а также гликозаминогликаны, располага¬
ющиеся на поверхности клетки.
Гомойотермный. Способный к поддержанию по¬
стоянной температуры тела независимо от колеба¬
ний температуры в окружающей среде.
Гомокарион. Клетка, содержащая два или более гене¬
тически идентичных ядра; обычно продукт слияния
клеток.
Гомотрансплантат (аллотрансплантат). Трансплантат,
полученный от генетически отличающегося донора
того же вида, что и реципиент.
Дедифференцировка. Необратимая утрата специали¬
зированных свойств, которые клетки экспрессируют
in vivo. По мере накопления данных, говорящих о
том, что дедифференцировка культуры возникает
путем сочетания селекции недифференцированных
стромальных клеток и дезадаптации, возникшей в
результате отсутствия специфических индукто¬
ров, термин постепенно выходит из употребления.
Будет правильно применять его в отношении
прогрессирующей утраты дифференциальных
морфологических признаков при гистологическом
исследовании, например, опухолевой ткани.
Дезадаптация. Обратимая утрата специфических
свойств в результате отсутствия соответствующих
индукторов (не всегда определенных).
Диплоид. Каждая хромосома представляет собой пару
хроматид, идентичных в аутосомных и женских
половых хромосомах и неидентичных в мужских
хромосомах, в соответствии с хромосомой, номером
хромосомы и морфологией большинства соматиче¬
ских клеток тех видов, из которых эти хромосомы
выделены.
ДНК-профильное исследование. Генетический тер¬
мин для изучения гипервариабельных участков
сателлитных ДНК, в настоящее время главным
образом используется для определения частоты
коротких тандемных повторов (STRs) в микроса-
теллитных ДНК при помощи RT-ПЦР единичного
локуса. Более чувствительный метод, чем мультило-
кусный ДНК-фингерпринтинг, легко поддающийся
количественному определению.
ДНК-фингерпринт. Мультилокусные кДНК-зон-
ды, связанные с гипервариабельными регионами
сателлитной ДНК, вырезанные эндонуклеазами
рестрикции и визуализированные методами ав¬
торадиографии. Участки, специфичные для того
индивидуального организма, из которого была
выделена ДНК.
Зависимость от прикрепления к субстрату. Необхо¬
димость для культуры клеток в прикреплении к
плотному субстрату для выживания и роста.
Злокачественная трансформация. Развитие способ¬
ности к инвазии в нормальную ткань без регуляции
во времени и пространстве; может также приводить
к метастатическому росту (колонизации отдаленных
участков с последующим нерегулируемым инвазив¬
ным ростом).
Злокачественный. Инвазивный или метастатический
(т.е., колонизирующий другие ткани). Термин, ис¬
пользуемый для обозначения типа опухоли, обычно
развивающейся и ведущей к разрушению клеток
хозяина и, в результате, его гибели.
Идеограмма. Распределение хромосом клетки в поряд¬
ке возрастания размера и морфологии для изучения
кариотипа и генетического анализа.
Изотрансплантат. Трансплантат, полученный от доно¬
ра, генетически идентичного или почти идентичного
реципиенту.
Иммортализация. Приобретение клеткой неопределен¬
ной продолжительности жизни. Может быть индуци¬
рована в конечной клеточной линии трансфекцией те¬
ломеразы, онкогенов или большого Т-региона генома
SV40, целого вируса SV40 или вирус Эпштейн-Барра
(EBV). Иммортализация не обязательно является
злокачественной трансформацией, хотя может быть
компонентом злокачественной трансформации.
Индекс разведения. Делитель при разведении клеточ¬
ной культуры при субкультивировании (например,
когда один флакон делят на четыре, или 100 мл до¬
водят до 400 мл, индекс разведения составит 4).
Индукция. Усиление действия, производимое опреде¬
ленным стимулом.
Инфекция (определение, отличное от общеприня¬
того). Перенос геномной ДНК с ретровирусным
конструктом, содержащим последовательность
ДНК в условиях исследования, обычно совместно
с промоторной последовательностью и репортер¬
ным геном, таким как Р-галактозидаза; продукт
инфекции может быть определен окрашиванием с
хромосомным субстратом.
Кариотип. Характерный хромосомный набор клеток.
Карцинома. Опухоль, развившаяся из эпителия, обыч¬
но из клеток эндодермального или эктодермального
происхождения.
Квазиплоид. см. Псевдоплоид.
Клетка-предшественник. Клетка на некоторой стадии
клеточной дифференцировки, которая предполагает
возможность коммитирования к определенному
типу дифференцировки. Ранние стадии будут
пролиферативными, поздние могут быть не проли-
феративными.
Клеточная гибридизация. См. Гибридная клетка.
Клеточная концентрация. Количество клеток в 1 мл
среды.
Клеточная культура. Рост клеток, диссоциированных
из родительских тканей путем спонтанной миграции
или механического или ферментативного расщеп¬
ления.
Клеточная линия. Выросшая культура после первого
субкультивирования.
Клеточная плотность. Количество клеток на1 см2
субстрата.
ПРИЛОЖЕНИЕ IV. СЛОВАРЬ УПОТРЕБЛЯЕМЫХ ТЕРМИНОВ
635
Клеточное слияние. Образование единого клеточного
тела путем слияния двух других клеток, спонтанное
или, чаще, индуцированное действием инактиви¬
рованного вируса Сендай или полиэтиленгликоля.
Клеточный штамм. Охарактеризованная клеточная
линия, полученная селекцией или клонированием.
Клон. Популяция клеток, полученная из одной
клетки.
Количество пассажей. Количество произведенных
пересевов культуры (переносов субкультуры).
Коммитирование. Необратимое развитие из стволовой
клетки до определенного клеточного типа закан¬
чивающееся образованием клетки с возможность
экспрессии ограниченного набора свойств. Детер-
. минация.
Конечная клеточная линия. Культура, которая была
выращена путем субкультивирования, но способна
только к ограниченному числу клеточных генераций
in vitro до гибели клеток.
Конститутивный. Экспрессируемый клетками в отсут¬
ствие внешних регулирующих факторов.
Контактное ингибирование. Ингибирование колеба¬
ний цитоплазматической мембраны и подвижности
клеток, когда клетки находятся в контакте с рядом
располагающимися клетками, как в конфлюэнтной
культуре; часто предшествует, но не обязательно
причинно связано с прекращением клеточного
деления.
Конфлюэнтный монослой. Монослой клеток, в кото¬
ром все клетки контактируют с клетками, располо¬
женными по их периферии и при этом не остается
свободного от клеток субстрата.
Кривая роста. Полулогарифмическая кривая зави¬
симости логарифма количества клеток от времени
роста культуры; обычно делится на lag-фазу (фаза
до начала активного роста), логарифмическую
фазу (период экспоненциального роста) и плато
(постоянное количество клеток, достигается, когда
культура прекращает расти при высокой плотности
клеток).
Ксенотрансплантат. Трансплантация ткани виду,
отличному от того, от которого он выделен; термин
часто используется для описания имплантации
опухоли человека бестимусным (nude) мышам, а
также мышам с подавленным или отсутствующим
иммунитетом.
Культура тканей. Термин возник для обозначения
поддержания жизнеспособности фрагментов тканей
in vitro, но в настоящее время используется как об¬
щепринятый термин, означающий культивирование
эксплантата ткани, органную культуру, культуру
диспергированных клеток, включая культуру
поддерживаемых клеточных линий и клеточных
штаммов.
Купол. Полусферическое или подобное пузырю обра¬
зование на поверхности конфлюэнтного монослоя,
предполагающее наличие ионного транспорта через
монослой и приводящее к накоплению воды под
монослоем.
Ламинарный бокс. Рабочее место с фильтрованным
потоком воздуха ламинарного типа (бестурбулент-
ного), параллельного или перпендикулярного ра¬
бочей поверхности, для поддержания стерильности
рабочего места; параллельный поток называется
обычно горизонтальным, а перпендикулярный
вертикальным.
Ламинарный поток. Поток, который полностью пов¬
торяет форму обтекаемой поверхности, без тур¬
булентности; термин используется для защитных
колпаков или боксов, характеризующихся стабиль¬
ным потоком воздуха над рабочей поверхностью с
минимальной турбулентностью потока.
Лейкемия. Злокачественная болезнь гематопоэтиче-
ской системы с выраженной циркуляцией бластных
форм клеток.
Лимфома. Солидная опухоль лимфоидных клеток.
Липофекция. Трансфекция ДНК путем слияния с
липид-инкапсулированной ДНК.
ЛИФ-фактор (LIF). Фактор, ингибирующий лейке¬
мию, цитокин семейства интерлейкин-6; обычно
используется для ингибирования дифференци¬
ровки и поддержания фенотипа стволовых клеток
в культуре эмбриональных клеток ES.
Логарифмическая фаза (log-фаза). См. Кривая
роста.
Магнитно-активируемый клеточный сортинг (MACS).
Сортинг клеток путем магнитного притяжения на¬
магничивающихся частиц железа, покрытых анти¬
телами, которые связываются со специфическими
антигенами клеточной поверхности.
Макроауторадиография. Распределение радиоизо¬
топов в срезах органов и окрашенных препаратах
(блотах) при электрофорезе, получаемое путем эк¬
спозиции фотографической эмульсии, помещенной
в непосредственной близости от образца, обычно
помещают пленку с блотом в кассету с интенсифи¬
цирующим экраном.
Манометр. Прибор для измерения давления, включает
трубку U-образной формы, содержащую жидкость,
уровень которой в каждой из частей отражает раз¬
ницу давления между концами трубки.
Мезенхима. Рыхлая, часто мигрирующая эмбриональ¬
ная ткань, происходящая из мезодермы, дающая на¬
чало соединительной ткани, связкам, мышцам, гемо-
поэтическим клеткам и т.д. взрослого организма.
Мезенхимальные стволовые клетки (MSCs). Стволо¬
вые клетки, обычно происходящие из костного моз¬
га, обладающие мультипотентными способностями
к дифференцировке до кардиомиоцитов, нервных
клеток, гепатоцитов, а также клеток гематопоэти-
ческого ряда.
Мезодерма. Один из эмбриональных листков, раз¬
вивающийся между эктодермой и эндодермой и
дающий начало мезенхиме, которая, в свою оче¬
редь, дает начало соединительной ткани и т.д. (см.
Мезенхима).
Миелома. Опухоль, развившаяся из миелоидных кле¬
ток; используется для получения моноклональных
636
ПРИЛОЖЕНИЕ IV. СЛОВАРЬ УПОТРЕБЛЯЕМЫХ ТЕРМИНОВ
антител, когда миеломная клетка может продуци¬
ровать иммуноглобулин.
Микроавторадиография. Распределение радиоизо¬
топов в клетках и срезах тканей, визуализируемое
путем экспозиции фотографической эмульсии в
непосредственной близости от образца, обычно
путем погружения в расплавленную эмульсию.
После проявления образец может быть исследован
под микроскопом.
Моноклональное антитело. Антитело, продуцируемое
клоном лимфоидных клеток, как in vitro так и in vivo.
In vitro клон обычно получается из гибрида сенси¬
билизированной клетки селезенки и непрерывно
делящейся клетки миеломы.
Моноклональный. Полученный из одного клона клеток.
Морфогенез. Развитие формы и структуры организма.
Неопластическая трансформация. Преобразование
неканцерогенной клетки в канцерогенную.
Неопластический. Вновь возникший процесс проли¬
ферации клеток, не обусловленный потребностями
организма и приводящий к развитию опухоли.
Онкоген. Ген, который будучи трансфицирован или
инфицирован в нормальную клетку, обычно вызы¬
вает злокачественную трансформацию; обычно по¬
ложительно функционирующий ген, кодирующий
факторы роста, рецепторы, молекулы-передатчики
сигнала или ядерные регуляторы.
Органная культура. Поддержание или рост зачатка
органа, либо части или целого органа in vitro, так что
условия культуры позволяют происходить диффе¬
ренцировке и сохранению строения или функции
данного органа.
Органогенез. Развитие органов.
Органотипический. Гистотипическая культура, со¬
стоящая более чем из одного клеточного типа,
позволяющая создать модель межклеточных взаи¬
модействий, характерных для некоторого органа in
vivo. В данном случае подразумевается реконструк¬
ция из диссоциированных клеток или фрагментов
ткани, в отличие от органной культуры, в которой
структурная организация эксплантированной ткани
сохраняется.
Осмоль. Количество вещества, содержащее 1 моль
осмотически активных частиц.
Осмоляльность, осмотическое давление. Концентра¬
ция осмотически активных частиц в водном раст¬
воре, выраженная в осмоль/кг.
Осмолярность, осмотический свойства Концентрация
осмотически активных частиц в водном растворе,
выраженная в осмоль/л.
Паракринное действие. Действие одной клетки на дру¬
гую, рядом расположенную клетку, опосредованное
некоторым растворимым фактором, без участия
системного кровообращения.
Паренхима. Часть ткани, выполняющая основную
функцию данной ткани, например, гепатоциты в пе¬
чени, в отличие от стромы, такой как фибробласты
соединительной ткани, составляющей опорную
часть ткани.
Паскаль. Единица давления в системе СИ, эквивален¬
тная 1 ньютону на квадратный метр.
Пассаж. Перенос субкультуры клеток из одного куль¬
турального сосуда в другой, обычно, но не всегда,
предполагающий деление на части пролифери¬
рующей клеточной популяции, обеспечивающее
размножение клеточной линии или клеточного
штамма.
Первичная культура. Культура, полученная из клеток,
тканей или органов, взятых непосредственно из
организма, до первого субкультивирования.
Первичный эксплантат. Фрагмент ткани, выделенный
из организма, и помещенный в культуру, таким об¬
разом, чтобы были обеспечены выживание и роста
жизнеспособных клеток за пределы фрагмента.
Плато. См. Кривая роста.
Плоидия. Соотношение обнаруженного количества
хромосом данного типа соматических клеток с
обычным количеством хромосом в нормальных
соматических клетках in vivo. (см. также Гапло¬
идный, Диплодный, Эуплоидныйу Анеуплоидный и
Гетероплоидный).
Плотностное ограничение роста. Ингибирование
митотического деления, связанное с возрастанием
плотности клеток при конфлюэнтном росте.
Плотность насыщения. Максимальное количество
клеток, которые могут расположиться на см2 (в мо¬
нослойной культуре) или в мл (в суспензионной
культуре) в определенных условиях.
Плотность популяции. Количество монослойных
клеток на единицу площади субстрата; для клеток,
растущих в суспензии, плотность популяции иден¬
тична концентрации клеток.
Поддерживающая среда. Среда, обычно без сыворотки
и факторов роста, или с минимальным количеством
сыворотки, создается для поддержания клеток в
жизнеспособном состоянии без пролиферации
(например, для сбора биопсийных образцов или
поддержания клеток на стадии плато без продол¬
жения деления).
Пойкилотермный. Имеющий метаболически не регу¬
лируемую температуру тела, близкую к температуре
окружающей среды.
Постоянная (стабильная) клеточная линия или кле¬
точный штамм. Клеточная линия или штамм, обла¬
дающие способностью к неопределенному количест¬
ву жизненных циклов. Раньше также употреблялись
термины «установленная» и «бессмертная» или
«иммортализованная».
Псевдоплоид. Численно диплоидный хромосомный
набор, имеющий хромосомные аберрации.
Реактив. Вещество (элемент, компонент или смесь),
которое участвует в химической реакции.
Ростовая среда. Среда, используемая для поддержания
клеточной линии; обычно используется основная
среда с добавками, например, сывороткой или фак¬
торами роста.
Сантипуаз. Единица вязкости; 1000 сантипуаз экви¬
валентно 1 Па/с.
ПРИЛОЖЕНИЕ IV, СЛОВАРЬ УПОТРЕБЛЯЕМЫХ ТЕРМИНОВ
637
Саркома. Опухоль, образованная клетками мезо-
дермального происхождения [например, клетками
соединительной ткани мышц (миосаркома), или
кости (остеосаркома)].
Сбалансированный солевой раствор. Изотонический
раствор неорганических солей, примерно воспроиз¬
водящий необходимые физиологические концентра¬
ции; может также содержать глюкозу, но обычно не
содержит других органических компонентов.
Синкарион. Гибридная клетка, которая образуется в
результате слияния ядер, которые она содержит.
Среда поддерживающая. Среда, которая сохранит
клетки в жизнеспособном состоянии без клеточ¬
ной пролиферации (например, бессывороточные
среды или среды с низким содержанием сыворотки,
используемые для культур клеток, зависимых от
присутствия сыворотки).
Среда ростовая. Среда, которая используется при
обычной культуральной работе для поддержания
деления клеток и их накопления.
Среда. Смесь неорганических солей и других пита¬
тельных соединений, способных к поддержанию
выживания клеток in vitro в течение 24 ч.
Старение. Биологически регулируемый процесс утраты
пролиферативного потенциала клеток, связанный с
укорочением теломеров хромосом.
Стволовая клетка. Самая ранняя в линии дифферен¬
цировки клетка, способная стать предшественником
для всех клеток данной линии дифференцировки
при развитии из нее популяции и поддержании этой
популяции. Унипотентная стволовая клетка
даст начало только одному пути дифференцировки,
бипотентная — двум, мультипотентная — более
чем двум, плюрипотентная — нескольким, а то-
типотентная — всем типам дифференцированных
клеток (см. также Эмбриональные стволовые клетки
и Мезенхимальные стволовые клетки).
Строма. Считается, что эта часть ткани выполняет
опорную функцию, как, например, фибробластная
соединительная ткань и ее сосудистая сеть.
Субконфлюэнтный. Менее чем конфлюэнтный моно¬
слой, не весь доступный для роста субстрат покрыт
клетками.
Субкультивирование, см. Пассаж.
Субстрат. Матрикс или плотная подложка, на котором
растет культура.
Суперконфлюэнтный. Монослой, развивающийся
после состояния конфлюэнтности, при котором
все клетки прилегают к субстрату; формирование
многослойности.
Суспензионная культура. Культура, в которой клетки
растут и делятся при суспендировании их в культу¬
ральной среде.
Теломеры. Терминальные регионы хромосом, ко¬
торые предупреждают рекомбинацию с другими
хромосомами и поэтому способны поддерживать
пролиферативную способность клеток. Постепенное
укорочение теломеры имеет место при старении
клетки. В стволовых клетках и некоторых опухо¬
левых клетках длина теломеры поддерживается
постоянной при помощи теломеразы.
Тетраплоид. Двойной диплоидный (или четверной
гаплоидный) набор хромосом.
Тип мощеной мостовой. Тип расположения клеток
в монослое, многоугольные клетки. Правильнее
употреблять термин «эпителиоидные» или «эпите-
лиоподобные клетки».
Трансдифференцировка. Явление, при котором
клетки из одной линии приобретают способность
дифференцироваться в клетки другой линии диф¬
ференцировки.
Трансфекция. Перенос генетического материала из
одной клетки в другую при помощи искусственных
переносчиков; при этом переносится менее чем
все ядро донорской клетки. Трансфекция обычно
достигается путем переноса отдельных хромосом,
участков ДНК или клонированных генов.
Трансформация. Долговременное изменение клеточ¬
ного фенотипа, которое считается возникающим в
результате необратимых генетических изменений.
Может быть спонтанной как при развитии быстро
растущих постоянных клеточных линий из медлен¬
но растущих клеточных линий грызунов ранних
пассажей, либо индуцированной химическим или
вирусным агентом. Обычно дает клеточные линии,
которые имеют более высокую скорость роста, не¬
определенную продолжительность времени жизни
низкую потребность в сыворотке.
Фактор роста. Фактор, выделяемый клетками, ко¬
торый вызывает пролиферацию других клеток;
главным образом, паракринного действия, но может
выделяться в кровь, например, тромбоцитами или
эндотелием.
Фенотип. Совокупность всех экспрессированных
свойств клетки, результат взаимодействия генотипа
с регулирующими условиями окружающей среды.
Ферментативная индукция. Возрастание синтеза
фермента, индуцируемое, например, гормональной
стимуляцией.
Ферментер. Культуральный сосуд для выращивания
клеток в крупных масштабах, часто используемый
для клеток в суспензии; термин сохранился от на¬
звания подобного оборудования, используемого в
микробиологии.
Фибробласт. Пролиферирующая клетка-предшествен¬
ник зрелого дифференцированного фиброцита.
Фибробластный. Напоминающий фибробласты [т. е.
веретенообразная (биполярная клетка) или звезд¬
чатая (мультиполярная) форма]; обычно органи¬
зованная в параллельные ряды при формировании
конфлюэнтного слоя, если имеется контактное
ингибирование. Часто этот термин безоснова¬
тельно используется для недифференцированных
мезодермальных клеток мигрирующего типа или
клеток с выростами, превышающими диаметр ядра
в три раза и более. Более точно называть такие
клетки фибробластоподобными или фиброблас-
тоидными.
638
ПРИЛОЖЕНИЕ IV, СЛОВАРЬ УПОТРЕБЛЯЕМЫХ ТЕРМИНОВ
Фиколл-пак. Уплотняющая среда, приготовленная из
Фиколла (Ficoll) в сочетании с рентгеноконтраст¬
ным йодированным веществом, таким как натрий
амидотризоат.
Флюоресцентная гибридизация in situ (FISH). Свя¬
зывание специфических флюоресцентных зондов
со специфическими внутриклеточными участками
методом гибридизации in situ (см. также Хромосом¬
ное окрашивание).
Флюоресцентно-активируемый клеточный сортер
(FACS). Клеточное разделительное оборудование,
основанное на электромагнитной сортировке. Сус¬
пензии единичных клеток вследствие дисперсии
света или флюоресцентных свойствах отдельных
клеток, обнаруживаемых при лазерном сканирова¬
нии потока отдельных клеток (см. также Флюори-
метр проточный).
Флюориметр проточный. Оборудование, позволяющее
проводить качественный и количественный анализ
отдельных клеток в популяции путем сканирова¬
ния единичных клеток потока при помощи лазера
с различной длиной волн или комплекса лазеров с
разными длинами волн и записью света, который рас¬
сеивается или флюоресценции, которая излучается.
Химически детерминированный. Нечто, полностью
приготовленное из чистых определенных составля¬
ющих (например, среда); отличайте от «бессыворо¬
точной», в состав которой могут вместо сыворотки
входить другие плохо охарактеризованные и нестан-
дартизированные компоненты.
Хроматиды. Парные компоненты, составляющие хро¬
мосому, связанные центромерой.
Хромосома. Комплекс ДНК и нуклеопротеинов, об¬
разующих определенную морфологическую струк¬
туру в ядре, наблюдаемый в метафазе клеточного
деления. Состоит из двух идентичных хроматид,
соединенных в центромере и представленных оп¬
ределенным набором характеристик для каждого
вида организмов.
Хромосомное окрашивание. Использование специфи¬
ческих флюоресцентных зондов для окрашивания
определенных регионов хромосом.
Центромера. Точка соединения двух хроматид в хро¬
мосоме; место прикрепления к веретену деления в
метафазе, телофазе и анафазе клеточного цикла.
Цикл роста. Период роста культуры от момента суб¬
культивирования до вершины логарифмической
фазы, когда культура готова к следующему субкуль¬
тивированию.
Цикличный рост. Рост из низкой клеточной плотнос¬
ти до высокой клеточной плотности с постоянным
периодом субкультивирования, постоянным пов¬
торением ростового цикла с целью поддержания
культуры.
Цитокин. Фактор, выделяемый клеткой, который бу¬
дет вызывать рецептор-опосредованное действие
на пролиферацию, дифференцировку, воспаление
других клеток; обычно паракринного ближнего
действия, реже системного.
Цитостаз. Прекращение клеточной пролиферации.
Цитотоксичность. Клеточное повреждение одного или
более метаболического пути, внутриклеточного
процесса или структур, приводящее к нарушению
функций. Часто, но не всегда, связано с утратой
жизнеспособности.
Число генераций. Количество периодов удвоения
популяций (оцениваемое по разведениям субкуль¬
туры), которое может произойти в культуре после
эксплантации; обязательно содержит примерное
количество генераций в первичной культуре.
Эксплантат. Фрагмент ткани, трансплантированной
из исходного участка, культивируемый в искусст¬
венной среде.
Эксплантация. Выделение тканей для их культуры
in vitro, строго как небольшие фрагменты или с
сопровождающим почечным разрастанием (см.
Первичный эксплантат), но часто используемый как
общий термин для выделения культуры ткани.
Эктодерма. Внешний слой эмбрионального зачатка,
дающий начало эпителию кожи.
Эмбриональная индукция. Взаимодействие (часто
реципрокное) клеток из двух различных гермина¬
тивных слоев, ускоряет дифференцировку.
Эмбриональные стволовые клетки. Тотипотентные
стволовые клетки, выделенные из внутренней кле¬
точной массы эмбриона ранней стадии развития;
может быть культивирована как клеточные линии
с широким диапазоном возможностей дифферен¬
цировки.
Эндодерма. Самый глубоколежащий герминативный
слой эмбриона, дающий начало эпителиальной
компоненте органов, таких как кишечник, печень
и легкие.
Эндокринные факторы. Сигнальные факторы, такие
как гормоны, высвобождающиеся какой-то тканью
и оказывающие действие на отдаленные ткани через
систему кровообращения.
Эндотелий. Эпителиоподобный клеточный слой, вы¬
стилающий пространства в тканях мезодермального
происхождения, таких как кровеносные сосуды, и
происходящие из мезодермы эмбриона.
Эпителиальный. Описанные клетки получены из эпи¬
телия, но термин часто используется более узко для
описания любых клеток многоугольной формы с яс¬
ной четкой границей между клетками. Более точно
последние должны называться эпителиоподобными
или эпителиоидными клетками.
Эпителий. Покровный слой или выстилка из клеток,
как на поверхности кожи или кишечника, обычно
полученный из эмбриональной эндодермы или
эктодермы, но иногда он происходит из мезодермы,
как, например, в почечных канальцах и мезотелии,
выстилающем полости тела.
Эуплоид. Кратное умножение гаплоидного набора
хромосом. Правильная морфологическая характе¬
ристика каждой пары хромосом данного вида, из ко¬
торого произошла клетка, не указана в определении,
но обычно подразумевается. В противном случае мы
ПРИЛОЖЕНИЕ IV, СЛОВАРЬ УПОТРЕБЛЯЕМЫХ ТЕРМИНОВ
639
сказали бы «эуплоид с некоторыми хромосомными
аберрациями».
Эффективность культивирования. Процент клеток,
посеянных при субкультивировании, которые дают
начало колониям. Если считать, что каждая колония
образовывается из одной клетки, то эффективность
посева идентична эффективности клонирования.
Иногда этот термин не совсем верно используют
для описания количества клеток, выживших после
субкультивирования, но в этом случае лучше ис¬
пользовать термин «эффективность посева».
Эффективность посева. Процентная доля внесенных
клеток, которые крепились к субстрату в течение
определенного периода времени (подразумевается
жизнеспособность, но не обязательно пролифера-
тивная активность).
In ovo — «в яйце», обычно в курином яйце.
In vitro. Лат., букв, «в пробирке»; общепринятое
обозначение исследований вне организма хозяина,
например культуры клеток, органные культуры
или препараты органов в питательной среде корот¬
кого времени жизни. Термин также используется
для обозначения биохимических и молекулярных
реакций, которые проводятся в лабораторных про¬
бирках, но в этом случае лучше употреблять термин
«бесклеточные исследования».
In vivo. В живом организме — растении или жи¬
вотном.
Приложение V
ОСНОВНАЯ ЛИТЕРАТУРА
Alberts В., Bray D., Lewis J., Raff М., Roberts К. & Watson J.C.
(2002). The molecular biology of the cell, 4th ed, New York,
Garland.
Ausubel F.M., Brent R., Kingston R.E., Moore D.D., Seid-
man.J.G., Smith J. A., Struhl K. (Eds) (2002). Short Protocols
in Molecular Biology. 5th Edition. Vol. 1 & 2. Hoboken, NJ,
John Wiley & Sons.
Barlovatz-Meimon G. & Adolphe M. (2003). Culture de cel¬
lules aMmales; methodologies, applications. Paris. Editions
INSERM.
Butler M. (ed.), (1999). Mammalian cell biotexhnology, a practi¬
cal approach. Oxford, U.K., IRL Press at Oxford UMveisity
Press.
Davis J.M. (2002). Basic cell cultre, a practical approach. Oxford,
U.K., IRL. Press at Oxford UMversity Press.
Doyle A., GriffitsJ.B. & Newell D.G.(eds.). (1993,). Cell and
tissue culture: Laboratory procedures. Chichester, U.K. John
Wiley & Sons.
Doyle A., Hay R. & Kirsop B.E. (eds.). (1990). Living resources for
biotechnology Cambridge, UK., Cambridge Univercity Press.
Fteshney R.I. & Freshney M.C. (eds.). (2002,). Culture of epithe¬
lial cells. New York, Wiley-Liss.
Freshney R.I. (1999). Freshney‘s Culture of aMmal cells, a mult-
media guide, New York, Wiley-Liss.
Freshney R.I. & Freshney M.G. (1996). Culture of immortalazed
cells. New York, Wiley-Liss.
Freshney R.I., Pragnell C.B. & Freshney M.G. (eds.). (1994).
Culture of hematapoietic cells. New York, Wiley-Liss, Second
in the series «Culture of specialized cells*.
Haynes L.W. (ed) (1999). The neuron in tissue culture, Chichester.
UK. John Wiley & Sons.
Leigh I.M., Lane E.B. & Watt F.M. (eds.). (1994). The keratino¬
cyte handbook. Cambridge, U.K Cambridge Univercity Press.
Leigh I.M. & Watt F.M. (eds.). (1994). Keratinocyte methods.
Cambridge, U.K., Cambridge University Press.
Lodish H., Berk A., Matsudaira P., Kaiser C.A., Kreiger М.,
Scott M.P., Zipursky S.L. & Darnell J. (2004). Molecular cell
biology,5th ed. New York, Scientific American Books. Freeman.
Masters J.R.W. (eds). Animal cell culrure, a practical approach.
3rd ed. Oxford, U.K., IRL Press (2000).
Masters J.R.W. (ed). (1991). Human cancer in primary culture.
London, Kluwer.
Masters J.R.W. & Palsson B. (1999). (eds.). Human cell culture.
Dordrecht, The Netherlands, Kluwer.
Pfragner P. & Freshney R.I. (eds.) (2004). Culture of human tumor
cells. Hoboken, NJ. Wiley-Liss.
Pollack R. (ed) (1981). Reading in mammalian cell culture, 2nd
ed. Cold Spring Harbor, NY, Cold Spring Harbor Laboratory
Press.
Ravid K. & Freshney R.I. (eds.). (1998). DNA tranferto cultured
cells, New York. Wiley-Liss.
Shahar A., de Vellis J., Vemadakis A. & Haber B. (1989). A dis¬
section and tissue culture manual of thenervous system, New
York, Wiley-Liss.
Vunjak-Novakovic G. & Freshney R.I. (eds.). (2005). Culture
of cells for tissue ingineering. Hooboken, N.J., Wiley-Liss (in
press).
Методы культур тканей
Cell preservation technology
Cytotechnology (в настоящее время входит в Methods in Cell
Science)
In Vitro Cell amd Development Biology
Tissue Cilture Research Communiocation (Japanese,)
Клеточная биология
Cell
Cell Biology, International Reports
Cellulat Biology
Cell Growth &Differeniation
Current Opinion in Cell Biology
Euripean Journal of Cell Biology
Experimental Cell Biology
Experimental Cell Research
Journal of Cell Biology
Journal of Celular Phisiology
Journal; of Cell Science
Nature Biotechnology
Nature Cell Biology
Рак
British Journal of Cancer
Cancer Research
European Journal of Cancerand Clinical Oncology
Oncology
International Journal of Cancer
Journal of National Cancer Institute
Список литературы
Aaronson, S. А. & Todaro, G. J. (1968) Development of 3T3-like
lines from Balb/c mouse embryo cultures: Transformation
susceptibility to SV40./. Cell Physiol. 72:141-148.
Aaronson, S. A., Bottaro, D. P., Miki, Т., Ron, D., Finch, P. W.,
Fleming, T. P., Ahn, J., Taylor, W. G. & Rubin, J. S. (1991).
Keratinocyte growth factor A fibroblast growth factor family
member with unusual target cell specificity. Ann, NY Acad.
Sci. 638:62-77.
Aaronson, S. A., Todaro, G. J. & Freeman, A. F. (1970). Human
sarcoma cells in culture: Identification by colony-form¬
ing ability on monolayers of normal cells. Exp. Cell. Res.
61:1-5.
Abaza, N. A., Leighton, J. & Schultz, S. G. (1974). Effects of
ouabain on the function and structure of a cell line (MDCK)
derived from canine kidney; I: Light microscopic observations
of monolayer growth. In Vitro 10:172-183.
Abbott, N.J., Hughes, С. C., Revest, P. A. & Greenwood,J. (1992).
Development and characterisation ol a rat brain capillary
endothelial culrure: Towards an in vitro blood-brain barrier,
J. Cell Sci. 103 (Pt 1): 23-37.
Abercrombie, M. & Heaysman, J. E. M. (1954). Observations on
the social behaviour of cells in tissue culrure; II: “Monolayer¬
ing” of fibroblasts, Exp. Cell Res. 6: 293-306.
Abney, E. R., Williams, B. P. & Raff, М. C. (1983). Tracing the
development of oligodeudrocytes from precursor cells using
monoclonal antibodies, fluorescence activated cell sorting
and cell culture, Dev. Biol. 100:166-171.
Adams, D. C. (1979). Macrophages. In Jakoby, W. B. &
Pastan, I. H. (eds.), Methods in enzymology: vol. 57, cell culture.
New York, Academic Press, pp. 494-506.
Adams, R. L. P. (1980). In Work. T. S. & Burdon, R. H. (eds.):
Laboratory techniques in biochemistry and molecular biology :
Cell culture for biocheists, Amsterdam, Elsevier/North Hol¬
land Biomedical Press.
Adolphe, M. (1984.). Multiplication and type II collagen produc¬
tion by rabbit articular chondrocytes cultivated in a defined
medium. Exp. Cell. Res, 155:527-536.
Adolphe, M. & Benya. P. D. (1992). Different types of cultured
chondrocytes: The in vitro approach to the study of biochemi¬
cal regulation, in Adolphe, М., ed., Bioloqical regulation of the
chondrocytes. Boca Raton, FL, CRC Press, pp. 105-139.
Advisory Committee on Dangerous Parhogens (1995a). Cat¬
egorisation of Biological Agents According to Hazaral and
Categories of Containment. The Stationary Office, P.O. Box
276, London SW8 5DT, England.
Advisory Committee on Dangerous Pathogens (1995b). Protec¬
tion against blood-borne infections in the workplace: HIV
and hepatitis. The Stationery Oflice, P.O. Box 276 London
SW8 5DT, England.
Advisory Committee on Dangerous Pathogens (2003). Infection
at work: Controlling the risks, Department of Health, PC box
777. London SE1 6XH.
Ager, A. (1987). Isolation and culture of high endothelial cells
from rat lymph nodes. J. CeUSci 87 (Pt I): 133-144.
Agy, P. C., Shipley, G. D. & Ham, R. C. (1981). Protein-free me¬
dium for mouse neuroblastoma cells. In Vitro 17:671-680.
Ahmed, I., Collins, C. A., Lewis, M.P., Olsen, I. & Knowles, J. C.
(2004). Processing, characterisation and biocompatibility of
iron-phosphate glass fibres for tissue eugineering. Biomateri¬
als 25 (16): 3223-3232.
Aigner, J., Tegeler, J., Hutzler, P., Campoccia, P. Pavesio, A.,
Hammer, C., Kastcnhauer, B. & Naumann, A. (1998). Car¬
tilage tissue engineering with novel nonwoven structured
biomaterial based on hyaluroMc acid benzyl ester .J. Biomed,
Mater. Res. 42 (2): 172-181.
Aitken, M. L., Villalon, М., Verdugo, P. & Nameroff, M. (1991).
Enrichment of subpopulations of respiratory epithelial
cells using flow cytometry. Am.J. Resp. Cell. Moll. Biol. 4:
174-178.
Albelda, S. М., Oliver, P. O., Rosner, L. H. & Buck, C. A. (1990).
EndoCAM: a novel endothelial cell-cell adhesion molecule,
J. Cell. Biolt 110:1227-1237.
Alberts, B., Bray, D., Johnson, A., Lewis, J., Raff J., Roberts,
K. & Walter, P. (1997). Essential cell biology. New York,
Garland.
Alberts, B., Johnson, A., Lewis, J., Raff, М., Roberts, K. &
Walter, P. (2002), Molecular biology of the cell, 4th ed, New
York. Garland.
Albrecht A. М., Bielder, J. L. & Hutchison, D. J. (1972). Two
different species of dihydrofolate reductase in mammalian
cells differentially resistant to amethopterin and methasquin.
Cancer Res. 32:1539-1546.
Alexander, C. L., FitzGerald, U. F. & Barnett, S. C. (2002).
Identification of growth factors that promote long-term pro¬
liferation of olfactory ensheathing cells and modulate their
antigeMc phenotype. Glia 37:349-364.
Al-Hajj, М., Becker, M. W., Wicha, М., Weissman, I., Clarke, M. F.
(2001). Therapeutic implications of cancer stem cells. Curr
Opin. Genet. Dev., 14:43-47
Ali, S., Muller, C. R. & Epplen, J. T. (1986). DNA finger printing
by oligonucleotide probes specific for simple repeats. Hum.
Genet, 74:239-243.
Alison, М. K., Vig, P., Russo, F., Bigger, B. W., Amofah, E., The¬
mis, M. & Forbes, S. (2004). Hepatic stem cells: from inside
and outside the liver? Cell Prolif 37:1.
Alley, М. C., Scudiero, D. A., Monks, A., Hursey, M. L., CzerwiM-
ski, M. J., Fine, D. L., Abbot, B. J., Mayo, J. G., Shoemaker,
R. H. & Boyd, M. P. (1988). Feasibility of drug screening
642
Список литературы
with panels of human tumour cell lines using a microculture
tetrazolium assay. Cancer Res. 48: 589-601.
Al-Mufti, R., Hambley, H., Farzaneh. F. & Nicolaides, К. H.
(2004). Assessment of efficacy of cell separation techniques
used in the enrichment of fractal erythrohlasts from maternal
blood: triple density gradient vs. single density gradient. Clin
Lab. Haematol 26 (2): 123-128.
Al-Rubeai, M. & Singh, R. P. (1998). Apoptosis in cell culture,
Cur. Opin. Biotechnology. 9:152-156.
Al-Rubeai, М., Welzenbach, K., Lloyd, D. R & Emery, A. N.
(1997). A rapid method for evaluation of cell number and
viability by flow cytometry. Cytotechnology 24:161-168.
Alvarez-Dolado, М., Pardal, P., Garcia-Verdugo, J. М., Fike, J. R.,
Lee, H. O., Pfeffer K., Lois, C., Morrison, S. J. & Alvarez-
Buylla, A. (2003). Fusion of bone-marrow-derived cells with
Purkinje neurons, cardiomyocytes and hepatocytes. Nature
425:968-973.
Ames, B. N. (1980). Identifying environmental chemicals causing
mutations and cancer. Science 204:587-593.
Andersson, J. E. & Wozniak, A. C. (2004). Satellite cell activa¬
tion on fibers: modeling events in vivo an invited review. Can.
J. Phusiol. Pharmacol, 82: 300-310.
Andersson, L. C.,Jokinen, M. & Gahmberg, C. G. (1979c). Induc¬
tion of erythroid differentiation in the human leukaemia cell
line K562. Nature, 278:364-365.
Andersson, L. C., Jokinen, М., Klein, C. & Nilsson, K. (1979b).
Presence of erythrocytic components in the K562 cell line.
Int.J. Cancer. 24: 5-14.
Andersson, L. C., Nilsson, K. A., Gahmberg, C. G. (1979a).
K562 — a human eryrhroleukemic cell line. Int. J. Cancer
23:143-147.
Andreason, G. L. & Evans, G. A. (1988). Introduction and expres¬
sion of DNA molecules in eukaryotic cells by electroporation.
BioTechniques 6: 650-660.
Andreason, G. L. & Evans, G. A. (1989). Optimizaion of electro¬
poration for trarsfection of mammalian cells. Anal. Biochem.
180:269-275.
Andreoli, S. P. & McAteer, J. A. (1990). Reactive oxygen mol¬
ecule-mediated iinjures in endothelial and renal tubular
epithelial cells in vitro. Kidney In. 38: 785-794.
Andzaparidze, O. G. (1908). Clinical experience with vaccines
produced in the human diploid cell line Wl-38. Natl Cancer
Inst Monogr.y 29:477-484.
Antoniades, H. N., Scher, C. D. & Stiles, C. D. (1979). Purifica¬
tion of human platelet-derived growth factor. Proc. Natl.
AcadSci. USA 76:1809.
Aoki, H., Motohashi, Т., Yoshimura, N., Yamazaki, H., Ya-
mane, Т., Panthier, J. J. & Kunisada, T. (2005). Cooperative
and indispensable roles of endothelin 3 and KIT signalings
in melanocyte development. Dev. Dyn. 2005, Mar 14. Online
ahead of publication.
Armati, P. J. & Bonner, J. (1990). A techique for promoting
Schwann cell growth from fresh and frozen biopsy nerve
utilizing d-valine medium. In vitro Cell Dev. Biol, 26:
1116-1118.
Arrighi, F. E. & Hsu, Т. C. (1974). Staining constitutive het-
erohromatin and Giemsa crossbands of mammalian chromo¬
somes, In Yunis J. (ed.), Human cromosome methodology, 2nd
ed, New York, Academic Press.
Artursson, P. & Magnusson, C. (1990). Epithelial transport
of drugs in cell culture II: Effect of extracellular calcium
concentration on the paracellular transport of drugs of dif¬
ferent lipophilicites across monolayers of intestinal epithelial
(Caco-2) cells./. Pharmaceut. Sci. 79: 595-600.
Askanas, V., Bomemann, A. & Engel, W. K. (1990). Immunocy-
tochemical localization of desmin at human neuromuscular
junctions. Neurology 40: 949-953.
Atala, A. & Lanza, R. P. (2002). Methods of tissue Engineering.
San Diego, Academic Press.
Au, A. M.-J. & Varon, S. (1979). Neural cell sequestration on
immunoaffinity columns. Exp. Cell. Res. 120: 269.
Auerbach, R. & Grobstein, C. (1958). Inductive interaction of
embryonic tissues after dissociation and reaggregarion. Exp.
Cell Res, 15: 384-397.
Ausubel, F. М., Brent, R., Kingston, R. E., Moore, D, D, Seid-
man, J. G., Smith, J. A. & Struhl, K. (eds.) (1996). Currentpro-
tocols in Molecular biology, New York, John Wiley & Sons.
Ausubel, F. М., Brent, R., Kingston, R. E., Moore, D. D., Seid-
man, J. G., Smith, J. A. & Struhl, K. (eds.) (eds.) (2002).
Short Protocols in Molecular Biology.5th Edition, Vol. 1 &
2. Hoboken, NJ, John Wiley & Sons.
Babich, H. & Borenfreund, E. (1990). Neutral red uptake. In
Doyle, A., Griffiths, J. B. & Newall, D. G. (eds.), Cell and tis¬
sue culture: Laboratory procedures. Chichester, U.K., Wiley,
Module 4B:7.
Bagley, R. G., Walter-Yohrling, J., Cao, X., Weber, W., Si¬
mons, B., Cook, B. P., Chartrand, S. D., Wang, C., Madden,
S. L. & Teicher, B. A. (2003). Endothelial precursor cells as a
model of tumor endothelium: characerization and comparison
with mature andothelial cells. Cancer Res.63: 5866-5873.
Bakolitsa, C., Cohen, D. М., Bankston, F. A., Bobkov. A. A.,
Cadwell, G. W., Jennings L., Critchley, D. R., Craig. S. W. &
Liddington, R. C. (2004). Structural basis for vinculin activa¬
tion at sites of cell adhesion. Nature 430:583-586,
Balin, A. K., Goodman, B. P., Rasmussen, H. & Cristofaio, V. J.
(1976). The effect of oxygen tension on the growth and me-
tabolisn, of WI-38 cells./. Cell Physiol.89:235-250.
Balkovetz, D. F. & Lipschutz, J. H (1999). Hepatocyte growth
factor and the kidney: It is not just for the liver. Int. Rev.
Cytol. 186:225-260.
Ballard, P. L. (1979), Glucocorticoids and differentiation. Glu¬
cocorticoid Horm. Action, 12:439-517.
Ballard, P. L. & Tomkins, G. M. (1969). Dexamethasone and cell
adhesion. Nature 244:341-345.
Balmforth., A.J., Ball, S. G., Freshney, R. I., Graham, D. I., Mc-
Namee, B. & Vaughan, P. F. T. (1986). D-l dopaminer-gic
and beta-adrenergic stimulation of adenylatcyclase in a
clone derived from the human astrocytoma cell line G-CCM.
J. Neuochem. 47:715-719.
Baltimore, D. (2001). Our genome unveiled, Nature, 409:814-816.
Bansal, R., Stefansson, K. & Pfeiffer, S. E. (1992), Proligoden-
droblasr antigen (POA), a developmental antigen expressed
by A007/04-positive oligodendrocyre progenito to the ap¬
pearance of sulfatide and galactocerebroside./ Neurochem.
58:2221-2229.
Bard, D. R., Dickens, M. J., Smith, A. U. & Sarck, J. M. (1972).
Isolation of living cells from mature mammilan bone. Nature.
236:314-315.
Список литературы
643
Barnes, D. & Sato, G. (1980). Methods for growth of cul¬
tured cells in serum-free is medium Anal. Biochem., 102:
255-270.
Barnes, W. D., Sirbasku, D. A. & Sato, G. H. (eds.). (1984a,). Cell
culture methods for molecular and cell biology. Vol. 1. Methods
for preparation of media, supplements and substrata for serum-
free animal cell culture. New York, Alan R. Liss.
Barnes, W. D., Sirbasku, D. A. & Sato, G. H. (eds.). (1984b,).
Cell culture methods for molecular and cell biology. Vol. 2.
Methods serum-free culture of endocryne system. New York,
Alan R. Liss.
Barnes, W. D., Sirbasku, D. A. & Sato, G. H. (eds.). (1984c,). Cell
culture methods for molecular and cell biology. Vol. 3. Methods
for serum-free culture of epithelial and fibroblastic cells. New
York, Alan R. Liss.
Barnes, W. D., Sirbasku, D. A. & Sato, G. H. (eds.). (1984d,). Cell
culture methods for molecular and cell biology. Vol. 4. Methods
for serum-free culture of neuronal and lymphoid celk. New
York, Alan R. Liss.
Barnett, S. C. & Chung, I. (2004). Olfactory ensheathing cells:
Going solo or in need of a friend? Tends Neurosci. 27 (1):
54-60.
Barnett, S. C. & Riddell, J. S. (2004). Olfactory eusheating cells
(OECs) and the treatment of CNS injury: advantages and
possible caveats./. Anat. 24: 57-67.
Barnett. S. C. (2004). Olfactory eusheating cells: uniqe glial cell
types? J. Neurotrauma. 21: 375-382.
Barnett, S. C., Hutchins, A.-M. & Noble, M. (1993). Purification
of olfactory nerve. Olfactory eusheating cells of the olfactory
bulb. Dev. Biol. 155:337-350.
Bateman, A. E., Peckman, M. J. & Steel, G. G. (1979) Assays
of drug sensitivity for cells from human tumours: In vitro
and in vivo tests on a xenografted tumours. Br. J. Cancer A0:
81-88.
Battye, F. L. & Shortman, K. (1991). Flow cytometry and cell-
separation procedures. Curr. Opin. Immunol. 3: 238-241.
Bazill, G. W., Haynes, М., Garland, J. & Dexter, Т. M. (1983).
Characterisation and partial urification of haemopoietic cell
growth factor in WEHI-3 cell condioned medium. Biochem.
J. 210:747-759.
Beattie, G. М., Lappi, D. A., Baird, A., Hayek, A. (1990). Selective
elimintion of fibroblacts from pancreatic islet monolayers by
basic fibroblast growth factor-saporin mitotoxin. Diabetes.
39:1002-1005.
Beddington, R. (1992). Transgenic mutagenesis in the mouse.
Trends Genet, 8:10.
Bedrin, M. S., Abolafia, С. M. & Thompson, J. F. (1997). Cyro-
skeletal association of epidermal growth factor receptor and
associated signalling proteins is regulated by cell density in
IEC-6 inrestinal cells./. Cell. Physiol. 172:126-136.
Bell, E., Sher, S., Hull, B., Merrill, C., Rosen, S., Chamson, A., As-
selineau, D., Dubertret, L., Coulomb, B., Lapiere, C., Nusgens,
B. & Neveux, Y. (1983). The reconstitution of living skin,
J. Invest. Dermatol. 81: no I Suppl: 2s-10s.
Benda, P., Lightbody, J., Sato, Levine, L. & Sweet, W. (1969).
Differentiated rat glial cell strain in tissue culture. Science,
161:370.
Benders, A. A., G. M. van Kuppevelt, Т. H. М., Oosrehof, A. &
Veecrkamp, J. H. (1991), The biocemical and strutural matu¬
ration of human skeletal muscle cells in culture: The effect of
serum substitute, Ultroser, G. Exp. Cell Res. 195: 284-294.
Benya, P. D. (1981). Two dimensional CNBr peptide patterns of
collagen typesl, II and III. Coll. Relat. Res. 1:17-26.
Benya, P. D., Padilla, S. R., & Nimmi, М. E. (1977). The progeny
of rabbit articular chondrocytes synthesize collagen type I and
III and I trimmer, but not type II: Verification by cyanogen
bromide peptide analysis. Biochemistry 16: 865-872.
Berdichevsky, F., Gilbert, C., Shearer, M. & Taylor-Papadimi-
trou, J. (1992). Collagen-induced rapid morphogenesis of
human mammary epithelial cells: The role of the alpha 2 beta
lintegrin.J. Cell Sci., 102:437-446.
Berenbaum, М. C. (1985). The expected effects of a combi¬
nation of agents: The general solution./. Theor. biol, 114:
413-432.
Berger, S. L. (1979), Lymphocytes as resting cells, in Jakoby, W.
B. & Pastan, I. H. (eds.), Methods in enzymology, vol. 57, Cell
Culture. New York, Academic Press, pp. 486-494.
Berky, J. J. & Sherrod, P. C. (eds.). (1977). Shortterm, in vitro test¬
ing for carcinogenesis, mutagenesis and toxicity. Philadelphia,
Franklin Institute Press.
Bernstein, A. (1975). Differciation of clonal lines of teratocarci-
noma cells: Formation of embryoid bodies in vitro. Proc. Natl
Acad Sci. USA 72:1441-1445.
Bemstine, E. G., Hooper, M. L., Grandchamp, S. & Ephrussi, B.
(1973). Alkaline phosphatase activity in mouse teratoma Proc.
Natl. Acad Sci. USA 70:3899-3903.
Berry. M. N. & Friend, D. S. (1969). High yield preparation
isolated rat liver parenchymal cells: A biocemical and fine
structural study./. Cell. Biol. 43:506-520.
Berthussen. K. (1993). Growth of cells in a new defined protein
free medium., Cytotechnology 11: 219-231
Bertoncello, I., Bradley, T. R. & Watt, S. M. (1991). An improved
negative immunomagnetic selection strategy for the purifica¬
tion of primitive hemopoietic cells from normal bone marrow.
Exp. Hematol. 19:95-100.
Bettger, W. J., Boyce, S. Т., Walthall, B. J. & Ham, R. C. (1981).
Rapid clonal growth and serial passage of human diploid fi¬
broblasts in a lipid-enriched synthetic medium supplemented
with EGF, insulin and dexamethasone. Proc. Natl. Acad. Sci.
USA. 78:5588-5592.
Bhagavati, S. & Xu, W. (2004). Isolation and enrichment of
skeletal muscle progenitor cells from mouse bone marrow.
Biochem Biophys res Commun. 318:119-124.
Bhargava, М., Joseph, A., Knesel, J., Halaban, R., Li, Y.,
Pang, S., Golberg, I., Setter, E., Donovan, M. A., Zamegar, R.,
Faletto, D. & Rosen, E. M. (1992). Scatter factor and hepa-
tocyte growth factor activities, properties, and mechanism.
Cell Crowth Differ. 3:11-20.
Bhatt, R. I., Brown, M. D., Hart, C. A., Cilmore, P., Ramani, V. A.,
George, N.J. & Clarke, N. W. (2003). Novel method for the
isolation and characterisation of the putative prostatic stem
cell. Cytometry 54A (2): 89-99.
Bichko, V. V. (1998). Canonic liposomes. In Ravid, K. & Fresh¬
ney, R. I. (eds.), DNA transfer to cultured cells. New York,
Wiley-Liss, pp. 193-212.
Bickenbach, J. R. & Chism, E. (1998). Selection and extended
growth of murine epidermal stem cells in culture. Exp. Cell
Res. 244:184-195.
644
Список литературы
Biedler, J. L. (1976). Chromosome abnormalities in human tu¬
mour cells in culture. In Fogh, J. (ed.), Human Tumor Cells
In Vitro. New York, Academic Press.
Biedler, J. L. & Spengler, B. A. (1976). A novel chromosomal
abnormality in homan neuroblestoma and anti-folate resistant
Chinese hamster cell lines in culture./. Natl Cancer Inst 57:
683-695.
Biedler, J. L., Albrecht, A. М., Hutchinson, D. J. & Spengler, B. A.
(1972). Drug response, dihydrofolate reductase, and cytoge¬
netics of amethopterin-resistant Chinese hamster cells in vitro.
Cancer Res. 32:151-161.
Biggers, J. D., Gwatkin, R. В. C. & Heyner, S. (1961). Growth of
embryonic avian and mammalian tibiae on a relatively simple
chemically defined medium. Exp. Cell Res. 25:41.
Bignami, A., Dahl, D. & Rueger, D. G. (1980). Glial fibrillary
acidic (CFA) protein in normal neural cells and in pathological
conditions. In Federoff, S. & Hertz, L. (eds.), Advances in cel-
lularneurobiology, vol. 1. New York, Academic Press. Biosafety
in Microbiological and Biomedical Laboratories (1984). Divi¬
sion of Safety, BG31, IC02, NIH, Bethesda, MD.
Birch, J. R. & Pitrt. S. J. (1970). Improvements in a chemical-
lydefined medium for the growth of mouse cells (strain LS)
in suspension ./. Cell Sci. 7: 661-670
Birch, J. R. & Pirt, S. J. (1971). The quantitative glucose and
mineral nutrient requirements of mouse LS (suspension) cells
in chemicalli-defined medium ./. Cell Sci. 8: 693-700.
Birnie, G. D. & Simons, P. J. (1967). The incorpotation of
3H-thymidine and 3H-uridine on stainless steel. Exp. Cell
Res. 46:355-366.
Bishop, A. E., Power, R. F. & Polak, J. M. (1998). Markers for
neuro endocrine differentiation Pathol Res Pract. 1988 Apr;
183 (2): 119-128.
Bishop, J. M. (1991). Molecular themes in oncogenesis. Cell 64:
235-248.
Bissell, D. М., Arenson, D. М., Maher, J. J. & Roll. F. J. (1987).
Support of cultured hepatocytes by a laminin-rich gel: Evi¬
dence for a functionally significant subendothelial matrix in
normal liver. J. Clin. Invest. 79:801-812.
B-jerkvig, R., Laerum, O. D. & Mella, O. (1986a). Glioma cell
interactions with fetal rat brain aggregates in vitro, and with
brain tissue in vivo. Corner Res. 46:4071-4079.
Bjerkvig, R., Steinsvag, S. K. & Laerum, O. D. (1986b). Reaggre¬
gation of fetal rat brain cells in a stationary culture system; I:
Methodology and cell identification. In Vitro 22:180-192.
Blaker, G. J., Birch, J. R. & Pirt, S. J. (1971). The glucose, insulin
and glutamine requirements of suspension cultures of HeLa
cells in a defined culture medium./ Cell Sci. 9: 529-537.
Blanco, F. J., Geng, Y. & Lotz, M. (1995). Differentiation
dependent effects of IL-1 and TGF-b on human articular
chondrocyte proliferation are related to nitric oxide synthase
expression./ Immunol 154:4018-4026.
Blouin, R., Grondin, G., Beaudoin, J., Arita, Y., Daigle, N., Tal¬
bot, B. G., Lebel, D. & Morisset, J. (1997). Establishment
and immunocharacterization of an immortalized pancreatic
cell line derived from the H-2 Kb-tsA58 transgenic mouse.
In Vitro Cell Dev. Biol Anim. 33: 717-726.
Bobrow, М., Madan,J. & Pearson, P. L. (1972). Staining of some
specific regions on human chrosomes, particularly the second¬
ary constriction of no. 9, Nature 238:122-124.
Bochaton-Piallat, M. L., Gabbiani, F., Ropraz, P. & Gabbiani, G.
(1992)Cultured aortic smooth muscle cells from newborn and
adult rats show distint cytoskeletal featres. Differentiation
49:175-185.
Bockhold, K. J., Rosenblatt, J. D. & Partridge, T. A. (1998). Aging
normal and dystrophic mouse muscle: analysis of myogenicity
in cultures of living single fibres. Muscle Nerve 21:173-183.
Bodnar, A. G., Ouellette, М., Frolkis, М., Holt, S. E., Chiu, C.-P.,
Morin, G. B., Harley, С. B., Shay, J. W., Lichsteiner, S. &
Wright, W. E. (1998). Extension of life-span by introduc¬
tion of telomerase into normal human cells. Science 279:
349-352.
Boggs, S. S., Gregg, R. G., Borenstein, N. & Smithies, O. (1986).
Efficient transformation and frequent single-site, single-copy
insertion of DNA can be obtained in mouse erythroleuke-
mia cells transformed by electroporation. Exp. Hematol. 14:
988-994.
Bogler, O., Wren, D., Barnett, S. C., Land, H. & Noble, M. (1990).
Cooperation between two growth factors promotes extended
self-renewal and inhibits differentiation of oligodendrocyte-
type-2 astrocyte (0-2A) progenitor cells. Proc. Natl Acad.
Sci. USA 87:6368-6372.
Bolton, B. J. & Spun*, N. K. (1996). B-lymphocytes. In Fresh¬
ney, R. I. & Freshney, M. G. (eds.), Culture of immortalized
cells. New York, Wiley-Liss, pp. 283-298.
Bonaventure, J., Kadhom, N., Cohen-Solal, L., Ng, К. H., Bour-
guignon,J., Lasselin, C. & Freisinger, P. (1994). Reexpression
of cartilage-specific genes by dedifferentiated human articular
chondrocytes in alginate beads. Exp. Cell Res. 212:97-104.
Booth, C. & O’Shea, J. A. (2002). Isolation and culture of intesti¬
nal epithelial cells. In Freshney, R. I. & Freshney, M. G. (eds.),
Culture of epithelial cells, 2nd ed., Hoboken, NJ, Wiley-Liss,
pp. 303-335.
Booyse, F. М., Sedlak, B. J. & Rafelson, М. E. (1975). Culture of
arterial endothelial cells: Characterization and growth of bo¬
vine aortic cells. Thromb. Diathes. Ahemorrh. 34:825-839.
Borenfreund, E., Babich, H. & Martin-Alguacil, N. (1990). Rapid
chemosensitivity assay with human normal and tumor cells
in vitro. In Vitro Cell Dev. Biol. 26:1030-1034.
Bosco, D., Soriano, J. V., Chanson, M. & Meda, P. (1994). Hetero¬
geneity and contact-dependent regulation of amylase release
by individual acinar cells./. Cell Physiol 160:378-388.
Boshart, М., Weber, F., Jahn, G., Dorsch-Hasler, K., Flecken-
stein, B. & Schaffner, W. (1985). A very strong enhancer
is located upstream of an immediate early gene of human
cytomegalovirus. Cell 41:521-530.
Bossis, I., Voutetakis, A., Matyakhina, L., Pack, S., Abu-Asab, М.,
Bourdeau, I., Griffin, K. J., Courcoutsakis, N., Stergiopoulos,
S., Batista, D., Tsokos, M. & Stratakis, C. A. (2004). A pleio-
morphic GH pituitary adenoma from a Carney complex patient
displays universal allelic loss at the protein kinase A regulatory
subunit 1A (PRKARIA) locus./ Med. Genet. 41:596-600.
Bossolasco, P., Corti, S., Strazzer, S., Borsotti, C., Del Bo, R.,
Fortunato, F., Salani, S., Quirici, N., Bertolini, F., Gobbi, A.,
Deliliers, G. L., Pietro Comi, G. & Soligo, D. (2004). Skeletal
muscle differentiation potential of human adult bone marrow
cells. Exp. Cell Res. 295: 66-78.
Bottenstein, J. E. (1984). Culture methods for growth of neuronal
cells lines in defined media. In Barnes, D. W., Sirbasku, D. A.,
Список литературы
645
Sato, G. Н., eds., Methods for Serum-Free Culture of Neuronal
and Lymphoid Cells, New York, Alan R. Liss, pp. 3-13.
Bottenstein, J. E. & Sato, G. (1979). Growth of a rat neuroblas¬
toma cell line in serum free supplemented medium. Proc. Natl.
Acad. Sci. USA 76:514-517.
Boukamp, P., Petrusevska, R. Т., Breitkreutz, D., Homung, J. &
Markham, A. (1988). Normal keratinisation in a spontane¬
ously immortalised, aneuploid human keratinocyte cell line.
J. Cell Biol 106: 761-771.
Bourillot, P. Y., Waltzer, L., Sergeant, A. & Manet, E. (1998).
Transcriptional repression by the Epstein-Barr virus
EBNA3A protein tethered to DNA does not require RBP-
Jkappa./ Gen Virol. 79: 363-370.
Bouzahzah, B., Nishikawa, Y., Simon, D. & Garr, В. I. (1995)
Growth control and gene expression in a new hepatocellular
carcinoma cell line, Hep40: Inhibitory actions of vitamin K.
J. Cell Physiol. 165:459-467.
Bowman, P. D., Betz, A. L., Ar, D., Wolinsky, J. S., Penney, J. B.,
Shivers, R. R. & Goldstein, G. (1981). Primary culture of
capillary endothelium from rat brain. In Vitro 17:353-362.
Boxberger, H. J., Meyer, T. F., Grausam, М. C., Reich, K., Be¬
cker, H. D. & Sessler, M. J. (1997). Isolating and maintaining
highly polarized primary epithelial cells from normal human
duodenum for growth as spheroid-like vesicles. In Vitro Cell
Dev. Biol. Anim. 33:536-545.
Boxman, D. L A., Quax, P. H. A., Lowick, C. W. G. М., Papapoulos,
S. E., Verheijen,J. & Ponec, M. (1995). Differential regulation
of plasminogen activation in normal keratinocytes and SCC-4
cells by fibroblasts./. Invest. Dermatol. 104:374-378.
Boyce, S. T. & Ham, R. G. (1983). Calcium-regulated differentia¬
tion of normal human epidermal keratinocytes in chemically
defined clonal culture and serum-free serial culture./. Invest.
Dermatol. 81:33s-40s.
Boyd, М., Mairs, R. J., Cunningham, S.H., Mairs, S. C., McClus-
key, A. G., Livingstone, A., Stevenson, K., Brown, М. М.,
Wilson. L., Carilin, S. & Wheldon, Т. E. (2001), A gene
therapy/targeted radiotherapy strategy for radiation cell kill
by [131I] MIBG./ Gen Med 3:165-172.
Boyd, М., Mairs, R. J., Keith, W. N., Ross, S. C., Welsh, P.,
Akabani, G., Owens, J., Vaidyanathan, G., Camithers, R.,
Dorrens, J. & Zalutsky, M. R. (2004). An efficient targeted
radiotherapy / gene therapy strategy utilising human telom-
erase promoters and radioastatine and hamassing radiation-
mediated bystander effects./ Gene Med 6:937-947.
Boyd, М., Mairs, S. C., Stevenson, K., Livingstone, A., Clark, A. М.,
Ross, S. C. & Mairs, R. J. (2002). Transfectant mosaic spher¬
oids: a new model for evaluation of tumour cell killing in
targeted radiotherapy and experimental gene therapy./ Gene
Med 4:1-10.
Boyd, M. R. (1989). Status of th NCI preclinical antitumor drug
discovery screen. Prin. Prac. Oncol. 10:1-12.
Boyd, М., Cunningham, S. H., Brown, М. М., Mairs, R. J. & Whel¬
don, Т. E. (1999). Noradrenaline transporter gene transfer for
radiation cell kill by [131I] meta-iodobenzyl-guanidine. Gene
Ther6:1147-52.
Boyum, A. (1968a ). Isolation of leucocytes from human blood:
A two-phase system for removal of red cells with methylcel-
lulose as erythrocyte aggregative agent. Scand.J. Clin. Lab.
Invest (Suppl 97) 21:9-29.
Boyum, A. (1968b). Isolation of leucocytes from humanblood:
Futher observations-methylcellulose, dextran and Ficoll as
erythrocyte aggregating agents. Scand.J. Clin. Lab. Invest.
(Suppl 97)31:50.
Braa, S. S. & Triglia, D. (1991). Predicting ocular irritation us¬
ing three-dimensional human fibroblast cultures-Cosmetics
Toiletries 106:55-58.
Braaten, J. Т., Lee, M. J., Schewk, A. & Mintz, D. H. (1974) Re¬
moval of fibroblastoid cells from primary monolayer cultures of
rat neonatal endocrine pancreas by sodium ethylmercurithio-
salycilate. Biochem. Biophys. Res. Commun. 61,476-482.
Bradford, C. S., Sun, L. & Barnes, D. W. (1994a), Basic FGF
stimulates proliferatron and suppresses melanogenesis in cell
cultures derived from early zebrafsh embryos. Mol. Mar. Biol.
Biotech. 3: 78-86.
Bradford, C. S., Sun, L., Collodi, P. & Barnes. D. W. (1994b). Cell
cultures from zebrafish embryos and adult tissues./. Tissue
Cult. Methods 16:99-107.
Bradford, M. (1976). A rapid and sensitive method for the quanti¬
fication of microgram quantities of protein utilizing the prin¬
ciple of protein-dye binding. Anal. Biochem. 72:248-254.
Bradley, C. & Pitts, J. (1994). The use of genetuc marking to
assess the interaction of sensitive and multirdrug resistant
cells in mixed culture. Br. J. Cancer 70:795-798.
Bradley. N. J., Bloom, H. J. G., Davies, A. J. S. & Swift, S. M.
(1978a). Growth of human gliomas in immune-deficient
mice: A possible model of pre-clinical therapy studies, Br.
J. Cancer. 38: 263.
Bradley, T. R., Hodgson, G. S. & Rosendaal, M. (1978b). The
effect of oxygen tension on haemopoietic and fibroblast cell
proliferation in vivo.J. Cell Physiol. 97: (Suppl 1): 517-522.
Bravery, C. A., Batten, P., Yacoub, М. H., Rose, M. L. (1995)
Direct recognition of SLA-and HLA-like class II antigens
on porcine endothelium by human T cells results in T cell
activation and release of interleukin-2, Transplantation 60:
1024-1033.
Breder, J., Ruller, S., Ruller, Li., Schlaak, M. & van der Bosch, J.
(1996) Induction of cell death by cytokines in cell cycle-
synchronous tumor cell populations restricted to G1 and G2.
Exp. Сей Res. 223:259-267.
Breen, G. A. M. & De Vellis, J. (1974). Regulation of glycerol
phosphate dehydrogenase by hydrocortisone in dissociated
rat cerebral cell cultures. Dev. Biol, 41:255-266.
Breitkreutz, D., Stark. H.-J., Mirancea, N., Tomakidi, P., Stein-
bauer, H. & Fuseing, N. E. (1997). Integrin and basement
membrane normalization in mouse grafts of human keratino¬
cytes: Implications for epidermal homeostasis. Diffirentiation,
61:195-209.
Breitman, T. R, Kene, B. R., Hemmi, H. (1984). Studies of
growth and dfferenitiation of human myelomnonocytic le-
oukaemia cell lines in serum-free medium. In Barnes, D. W.,
Sirbasku, D. A. & Sato, G. H. (eds,), Methods for serum-free
culture of neuronal and lymphoid ceUs, New York, Alan R. Liss,
pp. 215-236.
Bretzel, R. G., Bonath, К & Federlin, K. (1990). The evaluation of
neutral density separation utilizing Ficoll, sodium diatrizoat
and Nycodenz and centrifugal elutriation in the purification
of bovine and canine islet preparations. Hormone Metab. Res
(Suppl) 25:57-63.
646
Список литературы
Brewer, G. J. (1995). Serum-free B27/Neurobasal medium sup¬
ports differentiated growth of neurons from the striatum,
subsrantia nigra, septum, cerebral, cortex, cerebellum, and
dentate gyrus./ Neurosci. Res, 42:674-683.
Bridger, J. M. (2004) Mammalian artificial chromosomes: modem
day feats of engineering- Isambard Kingdom Brunei style.
Cytogenet. Genome Res. 107: 5-8.
Briggs, R. & King. T. J. (I960)). Nuclear transplantation studies
on the early gastrula (Rana pipiens). I. Nuclei of presumptive
endoderm. Dev. Biol, 1960 Jun 2: 252-270.
Bright, R. K. & Lewis, J. D. (2004). Long-term culture of normal
and malignant human prosate epithelial cells, In Pfragner,
R. & Freshney, R. I (eds.). Culture of human tumorous cells,
Hoboken, NJ, Wiley-liss, pp. 125-144.
Brinch, D. S. & Elvig, S. G. (2001), Evaluation of an in vitro hu¬
man comeal model as alternative to the in vivo eye irratiton
testing of enzymes. Toxicol Lett. 123, suppl. 1:22.
Brindle, К. M. (1998). lnvestigationg the performance of in¬
tensive Mammalian cell bioreactor systems using magnetic
resonance imaging and spectroscopy. Biotech Genet. Eng.
Rev. 15:499-520.
British Standard BS5726 (1992). Microbiological Safety
Cabinets, Parts 1-4. The Stationery Office, P.O. Box 276,
London.
Brito Babapule, V. (1981). Lateral asymmetry in human chromo¬
somes 1,3,4,15 and 16. Cytogenet. Cell Genet. 29:198-202.
Brockes, J. P., Fields, K. L. & Raff, М. C. (1979). Studies on cultured
rat Schwann cells; J. Establishment of purified populations from
cultures of peripheral nerve. Brain Res., 165:105-118.
Brouty-Boye, D., Kolonias, D., Savaraj, N. & Lampidis. T. J.
(1992). Alpha-smooth muscle actin expression in cultured
cardiac fibroblasts of newborn rat. In Vitro Cell Dev. Biol,
28A: 293-296.
Brower, М., Carney, D. N., Oie, H. K., Gazdar, A. F. & Minna, J. D.
(1986). Growth of cell lines and clinical specimens of human
nonsmall cell lung cancer in a serum-free defined medium.
Cancer Res., 46:798-806.
Brown, A. F. & Dunn, G. A. (1989). Microinterferometry of
the movement of dry matter in fibroblasts. J. Cell Sci., 92:
379-389.
Brunk, C. F., Jones, К. C. & James, T. W. (1979). Assay for
nanogram quantities of DNA in cellular homogenates. Anal
Biochem, 92:497-500.
Brunton, V., Ozanne, B., Paraskeva, C. & Frame, M. (1997). A
role for epidermal growth factor receptor, c-Src and focal
adhesion kinase in an in vitro model for the progression of
colon cancer. Oncolene 14: 283-293.
Bruyneel, E. A., Debray, H., de Mets, M. Mareel, М. М., a
Montreul, J. (1990). Altered glycosylation in Madin-Darby
canine kidney (MDCK) cells after transformation by murine
sarcoma virus. Clin. Exp. Metastasis 8: 241-253.
Bryan. D., Sexton, C. J., Williams, D., Leigh, I. M. & McKay, I.
(1995). Oral keratinocytes immortalized with the early region
of human papillomavirus type 16 show elevated expression of
inteleukin 6, which acts as an autocrine growth factor for the
derived T103C cell line. Cell Crowth Differ. 6:1245-1250.
Bryan, Т. M. & Reddel, R. K. (1997). Telomere dynamics and
telomerase activity in in vitro immortalised human cells. Eur.
J. Cancer,.33: 767-773.
Brysk, М. М., Santschi, С. H., Bell, Т., Wagner, R. F., Jr
Tyrin, S. K. & Rajaraman, S. (1992). Culture of basal cell
carcinoma./. Invest. Dermatol. 98: 45-49.
Bucana, C. D., Giavazzi. R, Nayar, R, O’Brian, C. A, Seid, C., Earnest,
L E. & Fan, D. (1990). Retention of vital dyes correlates inversely
with the Multidrug-resistant phenotype of adriamycin-selected
murine fibrosarcoma variants. Exp Cell Res, 190:69.
Buchanan, S. S., Gross. S. A., Acker, J. P., Toner, М., Carpen¬
ter, J. F. & Pyatt, D. W. (2004). Cryopreservation of stem
cells using trehalose: evaluation of the method using a human
hematopoietic cell line. Stem Cell Dev. 13: 295-305.
Buchler, P., Reber, H. A., Buchler, M. W., Friess, H., Lavey, R S. &
Hines, O. J. (2004). Antiangiogenic actavity of genistein in
pancreatic carcinoma cells is mediated by the inhibition of
hypoxia — inducible factor-1 and the down-regulation of
VEGF gene expression. Cancer 100:201-210.
Buckingham, M. (1992). Making muscle in mammals. Trends
Genet., 8:144-149.
Buehring, G. C. (1972). Culture of human mammary epithelial
cells: Keeping abreast of a new method J. Natl. Cancer Inst.
49:1433-1434.
Buick, R. N., Stanisic, Т. H., Fry, S. E., Salmon, S. E., Trent, J. M. &
Krosovich, P. (1979). Development of an agar-methyl cellu¬
lose clonogenic assay for cells of transitional cell carcinoma
of the human bladder. Cancer Res. 39: 5051-5056.
Buonassisi, V., Sato, G. & Cohen, A. I (1962). Hormone-produc-
ing cultures of adrenal and pituitary tumor origin. Proc. Natl.
Acad. Sci. USA 48:1184-1190.
Burchell, J. & Taylor-Papadimitrou, J. (1989). Antibodies to hu¬
man milk fat globule molecules. Cancer Invest, 17: 53-61.
Burchell, J., Durbin, H. & Taylor-Papadimitrou, J. (1983). Com¬
plexity of expression of antigenic deteriminants recognised
by monoclonal antibodies HMFG 1 and HMFG 2 in normal
and malignant human mammary epithelial cells,/. Immunol.
131:508-513.
Burchell, J., Gendler, S., Taylor-Papadimitrou, J., Girling, A.,
Lewis, A., Millis, R. & Lamport, D. (1987). Development
and characterisation of breast cancer reactive monoclonal
antibodies directed to the core protein of the human milk
mucin. Cancer Res. 47:5176-5482.
Burgess, W. H. & Maciag, T. (1989). The heparin-binding fibro¬
blast growth factor family of proteins. Annu. Rev. Biochem.
58:575-606.
Burke, J. F., Price, T. N. C. & Mayne, L. V. (1996). Immortaliza¬
tion of human astrocytes. In Freshney, R. I. & Freshney, M. G.
(eds.), Culture of immortalized cells, New York, Wiley-Liss.
pp. 299-314.
Burrows, М. T. (1912). Rhytmische Kontraktionen der isolierten
Herzmuskelzelle Ausserhalb des Organismus. Munch. Med.
Wochenschr LIX: 1473.
Burwen, S. J. & Pitelka, D. R. (1980). Secretory function of lac-
tating moose mammary epithelial cells cultured on collagen
gels. Exp. Cell Res., 126:249-262.
Butler, M. (1991) Mammalian cell biotechnology. Oxford, IRL
Press at Oxford University Press, Butler. M. & Christie, A.
(1994). Adaptation of mammalian cells to non-ammoniagenic
media. Cytotechnology 15:87-94.
Cable, F. F., Isom, H. C. (1997). Exposure of primary rat hepa-
tocytes in long-term DMSO culture to selected transition
Список литературы
647
metals induces heparocyte proliferation and formation of
duct-like structures. Hepatology 26:1444-1457.
Cai, J., Weiss, M. L., Rao, M. S. (2004). In search of “sternness”,
Exp. HematoL, 32: 585-398.
Calder, C. J., Liversidge, J. & Dick, A. D. (2004). Murine re¬
spiratory tract dendritic cells: isolation, phenotyping and
functional studies .J Immunol. Methods 287: 67-77.
Campion D.G. (1984). The muscle satellite cell — a review. Int.
Rev, Cytol. 87: 225-251.
Camps, J., Morales, C., Prat, E., Ribas, М., Capella, G., Egozcue, J.,
Peinado, M. A. & Miro, R. (2004). Genetic evolution in colon
cancer KM 12 cells and metastatic derivates. Int.J. Cancer.
110:869-874.
Cancedda, R., Dozin, B., Giannoni, P. & Quarto, R. (2003). Tis-
„ sue engineering and cell therapy of cartilage and bone. Matrix
Biol 22:81-91.
Cancela, M. L., Ни, B. & Price, P. A. (1997). Effect of cell density
and growth factors on matrix GLA protein expression by
normal rat kidney cells./ Cell Physiol. 171:125-134.
Caniggia, I., Tseu, I., Han, R. N., Smith, В. Т., Tanswell, K. &
Post, M. (1991). Spatial and temporal differences in fibroblast
behavior in fetal rat lung. Am.J. Physiol. LungMol Cell Physiol.
261: L424-L433.
Cao, B., Zheng, B., Jankowski, R. J., Kimura, S., Ikezawa, М.,
Deasy, B., Cummins, J., Epperly, М., Qu-Petersen, Z. &
Huard, J. (2003). Muscle stem cells differentiate into hae-
matopoetic lineages but retain myogenic potential. Nat. Cell
Biol. 5: 640-646.
Cao, D., Lin, G., Westphale, E. М., Beyer, E. C. & Steinberg, Т. H.
(1997). Mechanisms for the coordination of intercellular calcium
signaling in insulin-secreting cells./ Cell Sci. 110:497-504.
Caplan, A. I. (1991.). Mesenchymal stem cells./ Orthop. Res. 9:
641-650.
Caputo, J. L. (1996). Safety procedures. In Freshney, R. L. &
Freshney M. G. (eds.), Culture ofimmortalzed cells. New York,
Willey-Liss. pp. 25-51.
Carlsson, J. & Nederman, T. (1989). Tumour spheroid technol¬
ogy in cancer therapy research. Eur. J. Cancer Clin. Oncol,
25:1127-1133.
Carmichael, J., DeGraff, W. G., Cazdar, A. F., Minna, J. D. &
Mitchell, J. B. (1987a). Evaluation of a tetrazolsum-based
semiautomated colorimetric assay: Assessment of chemosen-
sitivity testing. Cancer Res. 47: 936-942.
Carmichael, J., DeGraff, W. G., Cazdar, A. F., Minna, J. D. &
Mitchell, J. B. (1987b). Evaluation of a tetrazolsum-based
semiautomated colorimetric assay: Assessment of radiosen¬
sitivity testing. Cancer Res. 47: 936-942.
Carney, D. N., Bunn, P. A., Gardar, A. F., Pagan, J. A. & Min-
n^J. D. (1981). Selective growth in serum-free hormone-
supplemented medium of tumor cells obtained by biopsy from
patients with small cell carcinoma of lung. Proc. Natl. Acad.
Sci. USA 78:3185-3189.
Carney, D. N., Cazdar, A F., Bepler, G., Guccion, J. G., Marangos, P. J.,
Moodt, T. W., Zweig, М. H. & Minna, G. D. (1985) Esrablish-
ment and identification of small cell lung cancer cell lung having
classic and variant features. Cancer Rec, 45:2913-2923.
Carpenter, G. & Cohen, S. (1977). Epidermal growth factor. In
Acton, R. T. & Lynn, J. D. (eds.), Cell culture and its applica¬
tion. New York, Academic Press, pp. 83-105.
Carraway, K. L., Fregien, N., Carraway, K. L. & Cartaway, C. A.
(1992). Tumor sialomucin complexes as tumor antigens and
modulator of cellular interactions and proliferation./ Cell
Sci. 103:299-307.
Carrel, A. (1912). On the permanent lift of tissues outside the
organism./ Exp. Med. 15:516-528.
Carrel, A. & Ebeling, A. H. (1923). Survival and growth of fibro¬
blasts in vitro. J. Exp. Med. XXXVIII: 487.
Cartwright, T. & Shah, G. P. (1994). Culture media. In Davis, M.
(ed.) Basic cell culture, a practical approach. Oxford, UK
IRL., Presss in Oxford University Press, pp. 58-91.
Carvalho, K. A., Cuarita-Souza, L. C., Rebelatto, C. L., Senega-
glia, A. C., Hansen, P., Mendonca, J. G., Сигу, С. C., Francis¬
co, J. C. & Brofman, P. R. (2004). Could the coculture of skeletal
myoblasts and mesenchymal stem cells be a solution for postin¬
farction miocardial scar? Transplantat. Proc. 36:991-992.
Casanova, J. E. (2002). Epithelial cel cytoskeleton and intracel¬
lular trafficking. V. Confluence of membrane trafficking and
motility in epithaelial cell models, Am.J. Physiol. Gastrointest.
Liver. Physiol. 283 (5): G1015-G1019.
Casperson, Т., Farber, S., Foley, С. E., Kudinowski, J., Mod¬
est, E. J., Simonsson, E., Wagh, U. & Zech, L. (1968). Chemi¬
cal differentiation along metaphase chrcomosomes. Exp. Cell
Res. 49:219-222.
Cataldo, L М., Wang, Z. & Ravid, К (1998). Electroporation of DNA
into cultured cell lines. In Ravid, K, A Freshney, R. I. (eds.), DNA
transfer to cultured cells. New York, Wiley-Liss, pp. 55-68.
Cavallaro, U. & Christofori, G. (2004). Cell adhesion and signal¬
ling by cadherins and Ig-CAMs in cancer. Natl Rev. Cancer
4:118-132.
CDC Office of Health & Safety. (1999). Biosafety in Microbio¬
logical and Biomedical Laboratories (BMBL) 4th Edition.
US. Department of Health and Human Services, Centers for
Disease Control and Prevention, and National National Insti¬
tutes of Health, Fourth edition, May, 1999 US Government
Printing Office, Washington: 1999.
Centers for Disease Control. (1988). Update: Universal precau¬
tions for prevention of transmission of human immunodefi¬
ciency virus, hepatitis В virus, and other blood-bome patho-
genes in healthcare settings. MMWR 37:377-382,387,388.
Ceriani, R. L., Taylor-Papadimitriou, J., Peterson, J. A. &
Brown, P. (1979) Characterization of cells cultured fromearly
lactation milks. In Vitro 15: 356-362.
Chambard, М., Mauchamp, J. & Chaband, O. (1987) Synthesis
and apical and basolateral secretion of thyroglobulin by thy¬
roid cell monolayers on permeable substrate: Modulation by
thyroptropin./ Cell Physiol 133:37-45.
Chambard, М., Vemer, B., Gabrion J., Mauchamp J., Bugeia,J. C.,
Pelassy, C. & Mercier, B. (1983). Polarization of thyroid cell
in culture: Evidence for the basolateral localization of the
iodid «pump» and the thyroid-stimulating hormone receptor-
adrnyl cyclase complex./ Cell Biol 96:1172-1177.
Chang, H. & Basegra, R. (1977). Time of replication of genes
responsible for a temperature sensitive function ina cell cycle
specific to mutant from a hamster cell line./ Cell Physiol. 92:
333-343.
Chang, R. S. (1954). Continuous subcultivation of epithelial-like
cells from normal human tissues. Proc Soc. Exp. Biol Med.
87:440-443.
648
Список литературы
Chang, S. Е. & Taylor-Papadimitriou, J. (1983). Modulation of
phenotype incultures frim human milk epithelial cells and
its relation to the expression of a membrane antigen. Cell
Differ12:143-154.
Chang, S. E., Keen, Lane, E. B. & Taylor-Papadimitriou, J. (1982).
Establishment and characterisation of SV40-transformed hu¬
man breast epithelial cell lines. Cancer Res. 42: 2040-2053.
Chapronier, D. M. & McKeehan, W. L. (1986). Serial culture of
adult himan prostatic epithelial cells in serum-free medium
containing low calcium and a new growth factor from bovine
brain. Cancer ResAfr. 819-824.
Chen, С.-S., Toda, K.-I., Maruguchi, Y., Matsuyoshi, N., Horrigu-
chi, Y. & Imamura, S. (1997). Establishment and characterisa¬
tion of a novel in vitro angiogenesis model using a microvascu-
lar endothelial cell line, F-2C, cultured inchemically defined
medium. In Vitro Cell Dev. Biol. Anim. 33:796-802.
Chen, Т. C., Curthoys, N. P., Lagenaur, C. F. & Puschett, J. B.
(1989). Characterisation of primary cell cultures derived
from rat renal proximal tubules. In Vitro Cell Dev. Biol. Anim.
25:714-722.
Chen, T. R. (1977). In situ detection of mycoplasm contamination
in cell culture by fluorescence Hoechst 33258 stain. Exp. Cell
Res. 104:255.
Cherry, R. S. & Papoutsakis E.T. (1990). Understanding and con¬
trolling fluid-mechanical injury of animal cells in bioreactors.
In Spier., R. E. & Griggiths, J.B. Animal Cell Biotechnology,
Vol.4., London. Acad. Press, pp. 71-121.
Cho,J.-K. & Bikle, D. D. (1997). Decrease of Ca-ATPase activity
in human keratinocytes during calcium-induced differentia¬
tion./ CellPhysiol. 172:146-154.
Chopin, D., Barei-Moniri, R., Maille P., Le Frere-Belda, M.
A., Muscatelli-Groux, B., Merendino, N., Lecerf, L., Stop-
pacciario, A. & Velotti, F. (2003). Human urinary bladder
transitional cell carcinomas acquire the functional Fas ligand
during tumor progression. Am.J. Pathol. 162:1139-1149.
Chopra, D. P., Xue-Hu, I. C. (1993). Secretion of alpha-amylase in
human parotid gland epythelial cell in culture./. CellPhysiol.
155:223-233.
Chopra, D. P., Grignon, D. J., Joiakim, A., Mathieu, P. A., Mo-
hamed, A., Sakr, W. A., Powell, I. J., Sarkar, F. H. (1996).
Differential growth factor responses of epathelial cell cul¬
tures derived from normal human prostate, bening prostatic
hyperplasia andprimary prostate carcinoma./. Cell Physiol
169:269-280.
Christensen, B., Hansen, C., Kieler,J. & Schmidt, J. (1993). Iden¬
tity of non-malignant human urigenital cell lines classified
as transformation grade I (TGrl) and II (Tgrll). Anticancer
Res. 13:2187-2191.
Chu, G., Hayakawa, H. & Berg, P. (1987). Electroporation for the
efficient transfection of mammalian cells with DNA. Nucleic
Acids Res. 15:1311-1326.
Ciccarone, V., Hawley-Nelson, P., Gebeyehu, G.,Jessee,J. (1993).
Cationic liposome-mediated transfection of eucariotic cells
high-efficiency nucleic-acid delivery with lipofectin™, Lipo-
fectace™ and Lipofectamine ™ reagents. FASEBJ. 7: Al 131.
Cione, C., Filoni, S., Aquila, C., Bemardini, S. & Bosco, L. (1986).
Transdifferentiation of eye tissue in anuran amphibians:
Analysis of the transdifferentiation capacity of the iris of
Xenopus laevis larvae. Differentiation. 32:215-220.
Clarke, G. D. & Ryan, P. J. (1980). Tranquilizers can block mi-
togenesis in 3T3 cells and induce differenciation in Friend
cells. Nature 287:160-161.
Clayton, М., Bower, D. J., Clayton, P. R., Patek, С. E., Ran¬
dall, F. E., Sime, C., Wainright, N. R. & Zehir, A. (1980).
Cell culture in the investigation of normal and abnormal dif¬
ferentiation of eye tissues. In Richards, R. J. & Rajan, К. T.
(eds.), Tissue culture in medical research (II). Oxford, U.K.,
Pergamon Press.
Cobbold, P. H. & Rink, T. J. (1987). Fluorescence and biolumi¬
nescence measurement of cytoplasmic free Ca2+ Biochem. J.
248:313.
Cohen, C., Roguett, R., Cottin, М., Olive, C., Leclaire, J. & Rou-
gier, A. (1997). The use of Episkin, a reconstructed epider¬
mis, in the evaluation of the protective effect of sunscreens
against chemical lipoperoxidation induced by UVA./. Invest.
Derematol.iOS: 77.
Cohen., S. (1962). Isolation of a mouse submaxillary gland protein
accelerating incisor eruption and eyelid opening in the new¬
born animal./. Biol. Chem. 237:1555-1562.
Cole, J., Fox, М., Gamer, R. C., McGregor, D. B. & Thacker, J.
(1990). Gene mutation assays in cultured mammalian cells.
In Kirkland, D. J. (ed.), Basic mutagenicity tests: UKEMS
recommended procedures, Part I (revised). Cambridge, U.K.,
Cambridge University Press.
Cole, R. J. & Paul, J. (1966). The effect of erythropoetin on
haem system in mouse yolk sac and cultured foetal liver cells.
/. Embriol. Exp. Morphol. 15: 245-260.
Cole, S. P. C. (1986). Rapid chemosensitivity testing of human
lung tumour cells using the MTT assay. Cancer Chemother.
Pharmacol. 17: 259-263.
Collen, D., Stassen,J. М., Marafino, B. J. Jr., Builder, S., DeCock, F.,
Ogez, J., Tajiri, D., Pennica, D., Bennett, W. F., Salwa, J. et al.
(1984). Biological properties of human tissue-type plasmino¬
gen activator obtained by expression of recombinant DNA in
mammalian cells./ Pharmacol. Exp. Ther. 231:146-152.
Collins, С. H. & Kennedy, D. A. (1999). Evolution of microbio¬
logical safety cabinets. Br. J. Biomed. Sci. 56:161-169.
Collins, S. J., Gallo, R. C. & Gallagher, R. E. (1977). Continuous
growth and differentiation of human myeloid leukaemic cells
in suspension culture. Nature 270:347-349.
Collodi, P. (1998). DNA transfer to blastula-derived cultures from
zebra fish. In Ravid, K. & Freshney, R. I. (eds.), DNA transfer
to cultured cells, New York, Wiley-Liss, pp. 69-92.
Collodi, P., Barnes, D. W. (1990). Mitogenic activity from trout
embryos. Proc. Natl. Acad. Sci. USA 87:3498-3502.
Collodi, P., Kamei, Y., Ernst, Т., Miranda, C., Buhler, D. R. &
Barnes, D. W. (1992). Culture of cells from zebrafish (Bra-
chidanio rerio) embryo and adult tissues. Cell Biol. Toxicol.
8:43-61.
Compton, С. C., Warland, G., Nakagawa, H., Opitz, O. G. &
Rusgi, A. K. (1998). Cellular characterization and successful
transfection of serially subcultured normal human esophageal
kerationocytes./. Cell. Physiol. 177:274-281.
Coon, H. D. (1968). Clonal cultures of differentiated rat liver
cells./ Cell. Biol. 39:29a.
Coon, H. D. & Cahn, R. D. (1966). Differentiation in vitro. Ef¬
fects of Sephadex fractions of chick embryo extract. Science
53:1116-1119.
Список литературы
649
Cooper, P. D., Burt, A. M. & Wilson, J. N. (1958). Critical effect of
oxygene tension on rate of growth of animal cells in continu¬
ous suspended culture. Nature 182:1508-1509.
Cooper, S. & Broxmeyer, H. (1954). Purification of murine
granulocyte-macrofage progenitor cells (CFC-GM) using
counterflow centrifugal elutriation. In Freshney, R. I., Prag-
nell, I. B. & Freshney, M. D. (eds.), Culture of haempopoetic
cells. New York, Willey-Liss, pp. 223-234.
Corielli, L. L., Tall. M. G. & Gaskill, H. (1958). Common antigenes
in tissue culture cell lines. Science. 128:198.
Cou, J. Y. (1978). Establishment of clonal human placental cells
synthesizing human chorionadotropin. Proc. Natl. Acad. Sci.
USA. 75:1854-1858.
Courtenay, V. D., Selby, P. I., Smith, I. E., Mills, J. & Peckham, M. J.
" (1978). Growth of human tumor cell colonies from biopsies
using two soft-agar techniques. Br. J. Cancer 38: 77-81.
Coutinho, L. H., Gilleece, М. H., de Wynter, E. A., Will, A. &
Teasta, N. G. (1992). Clonal and long-term cultures using
human bone marrow, In Teasta, N. G. & Moulineux, G. (eds.),
Haemopoesis: A practical approach. Oxford, U.K., IRL Prss at
Oxford University Press, pp. 75-106.
Crabb, I. W., Armes, L. G., Johnson, С. M. & McKeehan, W. L.
(1986). Characterization of multiple forms of prostatropin
(prostate epithelial growth factor) from bovine brain. Bio¬
chem. Biophys. Res. Commun. 136:1155-1161.
Creasey, A. A., Smith, H. S., Hackett, A. I., Fukuyama, K., Ep¬
stein, W. L. & Madin, S. H. (1979). Biological properties of
human melanoma cells in culture. In Vitro 15:342.
Croce, С. M. (1991). Genetic approaches to study of the
molecular basis of human cancer. Cancer Res. (Suppl) 51:
5015s-5018s.
Cronauer, М. V., Eder, I. E., Hittmair, A., Sierek, G., Hobisch, A.,
Culig, Z., Thunhur, М., Bartsch, G. & Klocker, H. (1997). A
relible system for the culture of human prostatic cells. In Vitro
Cell Dev. Biol. Anim. 33:742-744.
Crouch, E. C., Strone, K. R., Bloch, M. & McDivitt, R. W. (1987).
Heterogeneity inthe production of collagens and fibronectin
by morphologically distinct clones of a human tumor cell
line: Evidence for intratumoral diversity in matrix protein
biosynthesis. Cancer. Res. 47: 6086-6092.
Cruickshaank, C. N. D. & Lowbury, E. J. L. (1952). Effect of ati-
biotics on tissue culture of human skin. Br. Med.J. 2:1070.
Csoka, K., Nygren, P., Graf, W., Pahlman, L., Glimelius, B. &
Larsson, R. (1995). Selective sensitivity of solid tumors to
suramin in primary cultures of tumor cells from patients. Int.
J. Cancer 63:356-360.
Cuttitta, F., Carney, D. N., Mulshine, J. М., Moody, T. W., Fe-
dorko, J., Fichler, A. & Minna, J. D. (1985). Bombesin-like
peptides can function as autocrine growth factors in human
small-cell lung cancer. Nature 316:823.
Dairkee, S. H., Deng, G., Stampfer, M. R., Waldman, F. M. &
Smith, H. S. (1995). Selective cell culture of primary breast
carcinoma. Cancer. Res. 55: 2516-2519.
Dairkee, S. H., Paulo, S. C., Traquina, P., Moore, D. H., Lju-
ing, В. M. & Smith, H. S. (1997). Partial enzymatic degrada¬
tion of stroma allows enrichment and expansion of primary
breast tumor cells. Cancer. Res. 57:1590-1596.
Damon, D. H., Lobb, R. R., D’Amore, P. A. & Wagner, J. A.
(1989). Heparin portentiate the action of acidic fibroblast
growth factor by prolonging its biological half-life. J. Cell.
Physiol 138: 221-226.
Danen, E. H. & Yamada, К. M. (2001). Fibromecti, integrins, and
growth control./ Cell. Physiol. 189:1-13.
Dangle, V., Femenia, F., Laine, V., Berthelmy, М., LeRhun, D.,
Poupon, M. F., Levy, D. & Schwartz-Comil, I. (1997). Two
and three-dimensional cell structures govern epidermal
growth factor function on human bladder carcinoma cell lines.
Cancer. Res. 57: 3360-3364.
Danielson, K. G., McEldrew, D., Alston, J. Т., Roling, D. B.,
Damjanov, I., Daska, I. & Spinner, N. (1992). Human colon
carcinoma cell lines feom the primary tumopr and a lymph
node metastasis. In Vitro Cell Dev. Biol 28A: 7-10.
Darcy, К. М., Shoemaker, S. F., Lee, P.-P. H., Vaughan, М. М.,
Black., J. D. & Ip, М. M. (1995). Prolactin and epidermal
growth factor regulation of the proliferation of normal rat
mammary epithelial cells in three dimensional primary cul¬
ture./. Cell Physiol 163: 346-364.
Darling, J. L. (2004). In vitro culture of malignant brain tumors. In
Pfragner, R. & Freshney, R. I. (eds.) Culture of human tumor
cells. Hoboken, NJ, Wiley-Liss, pp. 349-372.
Darnel, J. E. (1982) Variety in the level of gene control in eu-
caryotic cells. Nature 297:365-371.
Dart, J. (2003). Comeal toxicity the epithelium and stroma in
iatrogenic and factitios disease. Eye 17:886-892.
Das, A. V., James, J., Zhao, X., Rahnenfuhrer, J. & Ahmad, I.
(2004). Identification of c-Kit receptor as a regulator of adult
neural stem cells in the mammalian eye: interaction with
Notch signaling. Dev. Biol 273 (1): 87-105.
Davies, B., Brown, P. D., East, N., Crimmin, M. J. & Balk-
wili, F. R. (1993). A synthetic matrix metalloproteinase bur-
denand prolonging survival of mice bearing ovarian carcinoma
xenografts. Cancer. Res. 53:2087-2091.
Davis. J. B. & Stroobant, P. (1990). Platelet-derived growth
factors and fibroblast growth factors are mitogens for rat
Schwann cells./. Cell. Biol 110:1353-1360.
Davison. P. М., Bensch, R. & Karasek, M. A. (1983). Isolation and
long-term serial cultivation of endothelial cells from microves¬
sels of the adult human dermis. In Vitro 19:937-945.
Dawe, C. J. & Potter, M. (1957). Morphologic and biologic pro¬
gression of a lymphoid neoplasm of the mouse in vivo, and in
vitro. Am.J. Pathol. 33:603.
Degos, L. (1997). Differentiation in acute promyelocytic leuke¬
mia: European experience./ Cell Physiol 173:285-287.
De Larco, J. E. & Todaro, G. J. (1978). Epithelioid and fibroblas-
toid rat kidney cell clones: Epidermal growth factor receptors
and the effect of mouse sarcoma virus transformation./ Cell.
Physiol. 94:335-342.
De Leij, L., Poppema, S., Nulend, I. К., Haar, A. Т., Schwander, E.,
Ebbens, F., Postmus, P. E. & The, Т. H. (1985). Neuroendo¬
crine differentiation antigene on human lung carcinoma and
Kulchitski cells, Cancer. Res. 45: 2192-2200.
Del Veechio, P. & Smith, J. R. (1981). Expression of angiotensin
converting enzyme activity in culturated pulmonary artery
endothehal cells./ Cell. Physiol 108:337-345.
Del Vecchio, S., Stoppell, M. P., Carriero. М. V., Fonti, R.,
Massa, O., Li, P. Y., Botti, G., Cerra, М., d’Aiuto, G., Es-
positi, G., Salvatore, M. (1993). Human urokinase receptor
concentration in malignant and benign breast tomours by in
650
Список литературы
vitro quantitative autoradiografy: Comparison with urokinase
levels. Cancer. Res., 53: 3198-3206.
DeMars, R. (1958). The inhibition of glutamine of glutamiyl
transferase formation in cultuere of human cells. Biochem.
Biophys. Acta 27:435-436.
Demers, G. W., Halbert, C. L. & Galloway, D. A. (1994). Elevated
wild-type p53 protein levels in human epitheliil cell lines
immortalized by the human papillomavirus type 16 E7 gene.
Virogy. 198:169-174.
Deng, J., Steindler, D. A., Laywell, E. D. & Petersen, В. E. (2003).
Neural trans-differentiation potential of hepatic oval cells in
the neonatal mouse brain, Exp. Neurol. 182: 373-382.
Dennis, C. (2003). Developmental biology: Synthetic sex cells.
Nature. 424:364-366.
Dennis, C., Gallagher, R. & Campbell, P. (2001). The Humane
Genome. Nature 209: 813-858.
Department of Health and Social Security. (1986). Good Labora¬
tory Practice: The United Kingdome Compliance Programme
London. HMSO.
De Ridder, L. (1997). Autologous confrontation of brain tumor
derived spheroids with human dermal spheroids. Anticancer
Res. 17:4119-4120.
De Ridder, L. & Calliauw, L. (1990). Invasion of human brain
tumor in vitro. Relationship to clinical evolution./. Neurosurg.
72:589-593.
De Ridder, L. & Mareel, M. (1978). Morphology and ^-concen¬
tration of embryonic chick thyroids cultured in an atmosphere
of oxygen. Cell Biol. Int. Rep. 2:189-194.
Desai, К. V., Kavanaugh, C. J., Calvo. A. & Green, J. E. (2002).
Chipping away at breast cancer: insights from microarray
studies of human and mammary cancer. Endoc. Relat. Cancer.
9:207-220.
Deshpande, A. K., Baig, M. A., Carleton, S., Wadgoankar, K. &
Siddiqui, M. A. Q. (1993). Growth factors activate cardio¬
genic differentiation in avian mesodermal cells. Mol. Cell
Differ. 1:269-284.
De Silva, R., Moy, E. L. & Reddel. R. R. (1996). Immortaliza¬
tion of human branchial epithelial cells. In Freshney, R. I. &
Freshney M.G. (eds.). Culture of immortalized cells, New York,
Wiley-Liss, pp. 121-144.
De Silva, R., Whitaker, N.J., Rogan. E. M. & Reddel, R. R. (1994).
HPV-16 E6 and E7 genes, like SV40 early region genes, are
insufficent for immortalization of human mesothelial and
bronchial epithelial cells. Exp. Cell Res. 213:418-427.
Detrisac, C. J., Sens, M. A., Garvin, A. J., Spicer, S. S. & Sens, D. A.
(1984). Tissue culture of human kidney epithelial cells of
proximal tubule origin. Kidney Int. 25: 383-390.
De Vitry, F., Camier, М., Czemichow, P., Benda, P., Cohen. P &
Tixier-Vidal, A. (1974). Establishment of a clone of mouse
hypothalamic neurosecretory cells sinthesizing neurophysin
and vasopressin. Proc. Natl. Acad. Sci. USA 71:3575-3579.
De Vonne, T. L. & Mouray, H. (1978). Human a2-macroglobulin
and its antitrypsin and antithrombin activities in serum and
plasma. Clin. Chem. Acta 90:83-85.
De Vries, J. E., Dinjens, W. N. М., De Bmyne, G. K., Ver-
spaget, H. W., van der Linden, E. P. М., de Bruine, A. P.,
Mareel, М. М., Bosman, F. T. & ten Kate, J. (1995). In vivo and
in vitro invasion in relation to phenotypic characteristics of hu¬
man colorectal carcinoma cells. Br. J. Cancer 71: 271-277.
De Wever, O., Westbroek, W., Verloes, A., Bloemen, N.,
Bracke, М., Gespach, C., Bruyneel, E., Mareel, M. (2004).
Critical role of N-cadherin in myofibroblast invasion and
migration in vitro stimulated by colon-cancer-cell-derived
TGF-p or wounding./ Cell. Sci. 117:4691-4703.
De Wynter, E., Allen, Т., Coutinho, L., Flavell, D., Flavell. S. U. &
Dexter, T. J. (1993). Localisation of granulocyte macrophage
colony — stimulating factor in human long-term bone marrow
cultures./. Cell Sci. 106: 761-769.
Dexter, T. J., Spooncer, E., Simmons, P. & Allen, T. D. (1984).
Long-term marrow culture: An overview of technique and
experience. In Wright, D. G. & Creenberger, J. S. (eds.),
Longe-term bone marrow culture. New York, Alan R. Liss
Kroc Foundation Series 18, pp. 57-96.
Dexter, Т. М., Allen, T. D., Scott, D. & Teich, N. M. (1979).
Isolation and caracterisation of a bipotential haemopoietic
cell line. Nature 277:417-474.
Dexter, Т. М., Testa, N. G., Allen, T. D., Rutherford, S. & Seolnick,
E. (1981). Molecular and cell biological aspects of erythropoi-
esis in long-term bone marrow culture. Blood 58: 699-707.
Dickson, M. A., Hahn, W. C., Ino, Y., Ronfard, V., Wu ,J. Y., Wein¬
berg, R. A., Louis, D. N., Li, F. P., Louis, D. N., Li, F. P., A Rhe-
inwald, J. C. (2000). Human keratinocytes that express hTERT
and also bypass a pl6 (INK4a)-enforced mechanism that limits
life span become immortal retain normal growth and differen¬
tiation characteristics. Mol. Cell. Biol. 20:1436-1447.
Dickson, J. D., Flanigan, T. P. & Kemshead, J. T. (1983). Mono¬
clonal antibodies reacting specifically with the cell surface of
human astrocytes in culture. Biochem. Soc. Trans. 11: 208.
Dimitroulakos, J., Squire, J., Pawlin, G. & Yeger, H. (1994).
NUB-7: A stable- 1-type human neuroblastoma cellline ind-
ducible along N-and S-type cell lineages. Cell Growth Differ.
5:373-384.
Dinnen, R. & Ebisuzaki, K. (1990). Mitosis may be an obligatory
route to terminal differentiation in the Friend erythroleuke-
mia cell. Exp. Cell Res., 191:149-152.
Dirks, W. G., MacLeod, R. A. F. & Drexler, H. C. (1999). ECV304
(endothelial) is really T24 (bladder carcinoma): cell line
cross-contamination at source. In Vitro Cell Dev. Biol. Anim.
35:558-559.
Dobbs, L. G. & Gonzalez, R. F. (2002). Isolation and culture of
pulmonary alveolar epithelial type II cells In Freshney, R. I. &
Freshney, M. G. (eds.), Culture of epithelial cells, 2nd ed.,
Hoboken, NJ, Willey-Liss, pp. 278-301.
Dobbs, L. G., Pian, М., Dumars, S., Maglio, М., Allen. L. (1997).
Maintenance of the differentiated type II cell phenotype
by culture with an apical air surface. Am. J. Physiol. 273:
L347-L354.
Dodson, М. V., Mathison, B. A. & Mathison, B. D. (1990). Effects
of medium and substratum on ovine satellite cell attachment,
proliferation and differentiation. In Vitro Cell Dev. Biol. 29
(1): 59-66.
Doering, С. B., Healey, J. F., Parker, E. Т., Barrow, R. T. & Lol-
lar, P. (2002). High level expression of recombinant porcine
coagulation factor VIII./. Biol. Chem. 277:38345-38349.
Doherty, P., Ashton, S. V., Moore, S. E. & Walsh, F. S. (1991).
Morphoregulatory activities of NCAM and N-cadherin can be
accounted for by G-protein-dependent activation of L-andN-
type neuronal Ca2+-channels. Cell. 67: 21-33.
Список литературы
651
Dolbeare, F., Selden, J. R. (1994). Immunochemical quantitation
of bromodeoxyuridine: Application to cell-cycle kinetics.
Metod Cell. Biol. 41:297-316.
Dorazio, J. A., Cole, В. C. & Steinstreilein, J. (1996). Mycoplas-
ma-arthritidis mitogen up-regulates human NK cell activity.
Infect. Immun. 64 (2): 441-447.
Dotto, G. P., Parada, L. F. & Weinberg, R. A. (1985). Specific
growth response of ras-transformed embryo fibroblasts to
tumour promoters. Nature 318:472-475.
Doucetre, J. R. (1984). The glial cells in the nerve fibre layer of
the rat olfactory bulb. Anat. Rec. 210: 285-391.
Doucette, J. R. (1990). Glial influences on axonal growth in the
primary olfactory system. Glia 3:433-449.
Douglas, J. L. & Quinlan, M. P. (1994). Efficient nuclear localiza¬
tion of the Ad5 El A12S protein is necessary for immortaliza¬
tion but not cotransformation of primary epithelial cells. Cell
Growth Differ. 5:475-483.
Douglas, W. H. J., McAteer, J. A., DelPOrco, R. T. & Phelps. D.
(1980). Visualization of cellular aggregates cultured on a three-
dimentional collagen sponge matrix. In Vitro 16:306-312.
Dow, J., Lindsay, G. & Morrison, J. (1995). Biochemistry:
molecules, cells and the body. Workingham, U.K. Addison
Wesley.
Doyle, A. & Bolton, B. J. (1994). The quality control of cell
lines and the prevention, detection and cure of contamina¬
tion. In Davis, J. M. (ed.), Basic cell culture, a practical ap¬
proach. Oxford, U.K., IRL Press at Oxford University Press,
pp. 242-271.
Doyle, A., Criffiths, J. B. & Newail, D. G. (eds.) (1990-1999).
Cell and tissue culture: ilaboratory procedures. Chichester,
U.K., Wiley.
Doyle, A., Morris, C. & Mowel, J. M. (1990) Quality control. In.
Doyle, A., Hay., R. & Kirsop, В. E. (eds.). Animal cells, liv¬
ing resources for biotechnology. Cambrige, U.K. Cambridge
Univercity Press, pp. 81-100.
Draper, J. S., Pigott, C., Thomson, J. A. & Andrews, P. W. (2002).
Surfice antigens of human embrionic stem cells: changes upon
differentiation in culture./. Anat. 200: 249-258.
Drejer, J., Larsson, О. M. & Schousboe, A. (1983). Characteriza¬
tion of uptake and release processes for and d-and /-aspartate
in primary cultures of astrocytes and cerebellar granule cells.
Neurochem. Ref. 8:231-243.
Drexler, H. G., Dirks, W. G., Matsuo, Y. & McLeod, R. A. (2003).
False leukemia-lymphoma cell lines: an update on over
500 cell lines. Leukemia 17:416-426.
Drexler, H. G., Gignac, S. М., Hu, Z.-B., Hopert. A., Flecken-
stein, E., Viges. M. & Uphoff, С. C. (1994). Treatment of
mycoplasma, contamination in a large panel of cell cultures.
In Vitro Cell Dev. Biol. 30A: 344-347.
Drexler, H. G. (2004), Establishment and culture of human leu¬
kemia-lymphoma cell lines In Pfragner. R. & Freshney, R. I.
(eds)., Culture of human tumor cells. Hoboken. NJ. Wiley-Liss,
pp. 319-348.
Drexler, H. C. & Minowada, J. (2000). Human leukemia-lyom-
phona cell lines: historical perspective, state of the art and
future prospects. In Masters, J. R. W. & Palsson, B. O. (eds.),
Human cell culture, Vol. Ill Cancer cell lines, Part 3: Leuke¬
mia and lymphomas. Dordrecht, The Netherlands, Kluwer,
pp. 1-88.
Driever, W. & Rangini, Z. (1993) Characterisation of a cell line
derived from zebra-fish (Brachydanio rerio). In Vitro CeU Dev.
Biol. Anim.29A: 749-754.
Driever, W., Stemple, D., Schier, A. & Solnica-Krezel, L. (1994),
Zebrafish: Genetic tools for studying vertebrate development.
Trends Genet. 10:152-159.
Duffy, M. J., Reilly, D., O’Sullivan, C., O’Higgins, N., Fen-
nelly,J. J. & Andreasen, P. (1990). Urokinmase-plasminogen
activator, a new and independent prognostic marker in breast
cancer. Cancer. Res., 50:6827-6821.
Duksin, D., Maoz, A. & Fuchs, S. (1975). Differential cytotoxic
activity of anticollagen serum on rat osreoblasrs and fibro¬
blasts in tissue culture. Cell 5:83-86.
Dulbecco, R. (1952). Production of plaques in monolayers of tis¬
sue cultures by single particles of an animal virus. Proc. Natl.
Acad. Sci. USA 38:747.
Dulbecco, R. & Elkington J. (1973). Conditions limiting multi¬
plication of fibroblastic and epithelial cells in. dense cuiltures.
Nature 246:197-199.
Dulbecco, R. & Freeman, G. (1959). Plaque formation by the
polyoma virus. Virology 8:396-397.
Dulbecco, R., Vogt, M. (1934) Plaque formation and isolation
of pure cell lines with poliomyelitis viruses./. Exp. Med. 199:
167-182.
Duncan, E. L., De Silva, R. & Reddel, R. R. (1996). Immortaliza¬
tion of human mesothelial cells. In Freshney, R. I. & Fresh¬
ney, M.G. (eds.). Culture of immortalized cells. New York,
Wiley-Liss. pp. 239-258.
Dunham, L. J. & Stewart, H. L. (1953). A survey of transplant¬
able and transmissible animal tumors. / Natl Cancer Inst.
13:1299-1377.
Dunn, G. A. & Brown, A. F. (1990). Quantifying cellular shape
using moment invariants. In Alt, W. & Hofman, G. (eds.)
Biological motion: Lecture in biomathematics 89, Berlin
Springer-Verlag, pp. 10-34.
Dunn, G. A. & Zicha, U. (1997). Using the DRIMAPS system of
interference microscopy to study cell behavior, In Celis, J. F.
(ed.), Handbook of cell biology, 2nd ed. New York, Academic
Press, pp. 44-53.
Dunn, М. E., Schilling, K. & Mugnaini, F. (1998). Development
and fine structure of murine Purkinje cells in dissociated cere¬
bellar cultures: Neuronal polarity. Anat. Embryol. 197:9-29.
Dutt, K., Harris-Hooker, S., Ellerson. U., Layne, D., Kumar, R.
and Hunt, R. (2003). Generation of 3D retina-like structure
from a human retinal cell line in a NASA bioreactor. Cell
Transplant. 12: 717-731.
Eagle, H. (1955). The specific amino acid metabolism of mamma¬
lian cells (stain L) in tissue culture./ Biol. Chem. 214:839.
Eagle, H. (1959) Amino acid metabolism in mammalian cell
cultures. Science 130:432.
Eagle, H. (1973). The effect of environmental p H on the growth
of normal and malignant cells./ Cell Physiol. 82:1-8.
Eagle, H., Foley, G. E., Koprowski, H., Lazarus, H., Levine, F. M. &
Adams, R. A. (1970). Growth characteristics of virus-trans-
formed cells./ Exp. Med. 131:863-879.
Earle, W. R., Schilling, E. L., Stark, Т. H., Straus, N. P.,
Brown, M. F. & Shelton, E. (1943). Production of malignancy
in vitro; IV: The mouse fibroblast cultures and changes seen
in the living cells./ Natl. Cancer Inst, 4:165-212.
652
Список литературы
Easty, D. М., Easty, G. С. (1974). Measurement of the ability
of the cells to infiltrate normal tissues in vitro. Br. J. Cancer
29:36-49.
Ebendal, T. (1976). The relative roles of contact inhibition and
contact guidance is orientation of axons extending on aligned
collagen fibrils in vitro. Cell Resy 98:159-169.
Echarti, C. & Maurer, H. R. (1989). Defined, serum-free culture
conditions for the GM-microclonogenic-assay using agar-
capillaries. Blut 59:171-176.
Echarti, C. & Maurer, H. R. (1991). Lymphokine-activated
killer cells: determination of their tumor cytolytic capacity
by a clinogenic microassay using agar capillaries J. Immunol.
Methods 143:41-47.
Edelman, G. M. (1973). Nonenzymatic dissociations; B: Specific cell
fractionation of chemically derivatized surfaces, In Kruse, P. F.,
Jr. & Paterson, М. K., Jr. (eds.), Tissue culture methods and
applications. New York, Academic Press, pp. 29-36.
Edelson, J. D., Shannon, J. H. & Mason, R. J. (1988). Alkaline
phosphatase: A marker of alveolar type II cell differentiation,
Am. Rev. Respir. Dis. 138:1268-1275.
Edington. K, G., Berry, I. J., O’Prey, М., Bums, J. E., Clark, L. J.,
Mitchell, R., Robertson, G., Soutar, D., Coggins, L. W. &
Parkinson. E. K. (2004). Multistage head and neck sqna-
mous cell carcinoma, In Pfragner, R. & Freshney, R. I. (eds.),
Culture of Human Tumlour Cells. Hoboken, NJ. Wiley-Liss,
pp. 261-288.
Edwards, A. М., Silva, F., Jofre, B., Becker, М. I. & De
loannes, A. E. (1994). Visible light effects on tumoral cells in
a culture medium enriched with tryptophan and riboflavin,
J. Photocem. Photobiol. 24:179-186.
Edwards, R. G. (1996). The history of assisted human conception
with especial reference to endocrinology. Exp. Clin Endocrinol.
Diabetes 104 (3): 183-204.
Edwards, P. A. W., Easty, D. M. & Foster, C. S. (1980). Selective
culture of epithelioid cells from a human squamous carcinoma
using a monoclonal antibody to kill fibroblasts, Cell Biol Int
Rep. 4:917-922.
Eisinger, М., Lee, J. S., Hefton, J. М., Darzykiewicz, A.,
Chiao, J. W., Deharven, E. (1979). Human epidermal cell
cultures-growth and differentiation in the absence of dermal
components or medium supplements. Proc. Natl. Acad. Sci.
USA, 76:5340.
Elliget, K. A. & Lechner, J. F. (1992). Normal human bronchial
epithelial cell cultures. In Freshney, R. I. (ed.), Culture of
epithelial cells. New York, Wiley-Liss, pp. 181-196.
Elvin, P., Wong, V. & Evans, C. W. (1985). A study of the adhe¬
sive, locomotory and invasive behaviour of Walker 256 car¬
cinosarcoma cells, Exp Cell. Biol. 53:9-18.
Emura, М., Katyal, S. L., Ochiai, A., Hirohashi, S. & Singh, G.
(1997). In vitro reconstitution of human respiratory epithe¬
lium. In Vitro Cell Dev. Biol. 33: 602-605.
Enders, J. F., Weller, T. J. & Robbins, F. C. (1949). Cultivation of
Lansing strain of poliomyelitis in cultures of various human
embryonic tissues, Science 109:85.
Eng, L. F. & Bigbee, J. W. (1979). linmunochemistry of nervous-
system specific antigenes. In Aprison, М. H. (ed.). Advances
in neurochemistry. New York, Plentum Press, pp. 43-98.
Engel, L. W., Young, N. A., Tralka, T. S., Lippman, М. E., O’Brien,
S. J. & Joyce, M. J. (1978). Establishment and characteriza¬
tion of three new continuous cell lines derived from breast
carcinomas. Cancer Res, 38:3352-3364.
Eppig. J J. & O’ Brien, M, J. (1996). Development in vitro of
mouse oocytes from primordial follicles. Biol. Reprod., 54:
197-207.
Epstein, M. & Barr, Y. M. (1964). Cultivation in vitro of human
lymphoblasts from Burkitt’s malignant lymphoma. Lancet
1:252.
Ermis, A., Muller, B., Hopf, Т., Hopf, C., Remberger, K., Just-
en, H. P., Welter, C. & Hannselman, R. (1998). Invasion
of human cartilage by cultured multicellular spheroids of
rheumatoid synovial cells — a novel in vitro model system for
rheumatoid arthritis./. Rheumatol. 23: 208-213.
Eroglu, A., Russo, M. J., Bieganski, R., Fowler, A., Cheley, S.,
Bayley, H. & Toner, M. (2000). Intracellular trehalose im¬
proves the survival at cryopreserved mammalian cells. Nat.
Biotechnol. 18:163-167.
Espmark, J. A. & Ahlqvist-Roth, L. (1978). Tissue typing of cells
in cultures; I: Distinction between cell lines by the various
patterns produced in mixed haemabsorption with selected
multiparous sera .J. Immunol. Methods 24:141-153.
Ethier, S. P., Mahacek, M. L., Gullik, W. J., Frank, T. S. & We¬
ber, B. L. (1993). Differential isolation of normal luminal
mammary epithelial cells and breast cancer cells from pnimary
and metastatic sites using selective media. Cancer. Res. 53:
F.627-635.
European Commitee for Standardisation. (1999). Biotechnol¬
ogy performance criteria for microbiological safety cabinets
[W191]. CEN/TC233 Biotechnology, Central Secretariat,
European Committee for Srandardisation, Rue de Stassart
36, B-1050 Brussels, Belgium.
European Pharmcopoeia. (1980). Biological tests, 2nd ed., Part I,
Vol. 2. Maisonneuve, S. A., France.
Evans, Т., Rosenthal, E. Т., Youngblom, J., Distel, D. & Hunt, T.
(1983). Cyclin: a protein specified by maternal mRNA in sea
urchin eggs that is destroyed an each cleavage division. Cell
33:389-396.
Evans, M. J. & Kaufman, М. H. (1921). Establishment in cul¬
ture of pluripotent cells from mouse embryos. Nuture 292:
154-156.
Evans, V. & Bryant, J. C. (1965). Advances in tissue culture at the
National Cancer Institute in the United States of America. In
Ramakrishan, С. V. (ed). Tissue culture. The Hague, W. Junk,
pp. 145-167.
Evans, V. J., Bryant, J. C., Fioramonti, М. C., McQuilkin, W. Т.,
Sanford, К. K. & Earle, W. R. (1956). Studies of nutrient media
for tissue С cells in vitro, I: A protein-free chemically defined
medium for cultivation of strain L cells. Cancer Res. 16:77.
Evans, V. J. & Sanford, К. K. (1978). Development of defined
media for studies on malignant transformation in culture, In
Katsuta, H. (ed.), Nutritional Requirements of Cultured Cells.
Baltimore, University Park Press, pp. 149-192.
Fantini, J., Galons, J. P., Abadie, B., Canioni, P., Cozzone, P. J.,
Marvali, J. & Tirard, A. (1987). Growth in serum-free me¬
dium of human colonic adenocarcinoma cell lines on micro-
carriers: A two-step method allowing optimal cell spreading
and growth. In Vitro 23: 641-646.
Fata, J. E., Werb, Z. & Bissell, M. J. (2004). Regulation of mam¬
mary gland branching morphogenesis by the extracellular
Список литературы
653
matrix and its remodelling enzymes. Breast Cancer Res, 6:
1-11.
Federoff, S. (1975). In Evans. V. J., Perry, V. P. & Vincent, М. M.
(eds.). Manual of the Tissue Culture Association 1: 53-57.
Federoff, S. & Richardson, A. (2001). Protocols for neural celll
culture Clifton, NJ, Humana Press.
Feinberg, A. P. & Vogelstein, B. (1983). A technique for radio-
labeling DNA restriction fragments to high specific activity.
Anal. Biochem. 132: 6-13.
Feigner, P, Gadek, Т., Holm, М., Roman, R., Chan, H. W.,
Wenz, М., Northrop, J. P., Ringold, G. M. & Danielsen,
M. (1987). Lipofection: A highly efficient, lipid-mediated
DNA-transfection procedure. Proc. Natl. Acad. Sci. USA. 84:
7413-7417.
Fell, H. B. & Robinson, R. (1929). The growth, development and
phosphatase activity of embryonic avian femora and limb buds
cultivated in vitro. Biochem. J. 23: 767-784.
Fenton, J. I. & Hord, N. G. (2004). Flavonoids promote cell
migration in nontumorogenic colon epithelial cells differing
in Ape genotype: Implications of matrix metalloproteinase
activity. Nutr. Cancer, 48:182-188.
Fergusson, R. J., Carmichael, J. & Smyth, J. F. (1980). Human
tumor xenoprafts growing in immunodeficient mice: A. use¬
ful model for assessing chemotherapeutic agents in bronchial
carcinoma. Thorax 41: 376-380.
Femig, D. G., Gallagher, J. T. (1994). Fibroblast growth factors
and their receptors: An information, network controlling tis¬
sue growrh, morphogenesis and repair. Prog. Growth. Factor
Res. 5:353-377.
Finbow, М. E., Pitts, J. D. (1981). Permeability of junctions
between annual cells, Exp Cell. Res. 131:1-13.
Fiorino, A. S., Diehl, A. М., Lin, H. Z., Lemischka, I. R. &
Reid, L. M. (1998). Maturation-dependent gene expression
in a conditionally transformed liver progenitor cell line. In
Vitro Cell Dev. Biol. Anim. 34: 247-258.
Fischer, A. (1925). Tissue culture. Studies in experimental mor¬
phology and general physiology of tissue cells in vitro. London,
Heineman.
Fischer, G. A. & Sartorelli, A. C. (1964). Development, main¬
tenance and assay of drug resistance, Methods Med. Res, 10:
247-258.
Fisher, H. W., Puck. Т. T. & Sato, G. (1958). Molecular growth
requirements of single mammalian cells: The action of fetuin
in promoting cell attachment of glass. Proc. Natl. Acad. Sci.
USA. 44:4-10.
Fitzsimons, S. A., Ireland, H., Barr, N. I., Cuthbert, A. P., Go-
ing, J. J., Newbold, R. F. & Parkinson, F. K. (2003). Human
squamous cell carcinomas lose a mortality gene from chromo¬
some 6ql 4.3 to ql5. Oncogene 22:1737-1746.
Flier, J. S. (1995). The adipocyte: storage depot or node on the
energy information superhighway? Cell 80:15-18.
Flynn, D., Yang, J., Nandi, S. (1982). Growth and differentiation
of primary cultures of mouse mammary epythelium embedded
in collagen gel. Differentiation. 22:191-194.
Fogh, J. (1977). Absence of HeLa cell contamination in 169 cell
lines derived from human tumors, J. Natl. Cancer. Inst. 58:
209-214.
Fogh, J. & Trempe, G. (1975). in Fogh, J. (ed.), Human tumor cells
in vitro. New York, Academic Press, pp. 115-159.
Folkman, I. & Moscona, A. (1978). Role of cell shape in growth
control. Nature 273:345-349.
Folkman, I., Haudenschild, С. C. & Zerter, B. R. (1979). Long¬
term culture culture of capillary endotelial cells. Proc. Natl.
Acad. Sci. USA 76:5217-5221.
Folkman, J. (1992). The role of angiogenesis in tumor growth.
Sem. Cancer Biol. 3: 65-71.
Foikman, J. & D’Amnore, P. A. (1996). Blood vessel formation:
what is its molecular basis? Minireview. Cell. 87:1153-1155.
Folkman, J. & Haudenschild, C. (1980). Angiogenesis in vitro.
Nature 288:551-556.
Fontana, A., Hengamer, H., de Triholet, N. & Weber, E. (1984).
Glioiblastoma cells release interleukin 1 and factors inhib¬
iting interleukin 2-mediated effects. J. Immunol, 132 (4):
1837-1844.
Food and Drug Administration. (1992). Federal Register. 21 Code
of Federal Regulations, Part 58. Office of the Federal Register,
National Archives and Records, Washington, DC
Foreman, J. & Pegg, D. E. (1979). Cell preservation in a pro¬
grammed cooling machine: The effect of variations in super¬
cooling. Cryobiology 16:318-321.
Foster, R. & Martin, G. S. (1992). A mutation in the catalytic
domain of pp60v-src is responsible for the host — and tem¬
perature-dependent phenotype of the Rous carcinoma virus
mutant tsLA33-l. Virology 187:145-155.
Frame, М., Freshney, R. I., Shaw, R. & Graham, D. I. (1980)
Markers of differentiation in glial cells. Cell Biol. Int. Rep.
4: 732.
Franceschini, I. A. & Barnertt, S. C. (1996). Low-affinity NGF-
receptor and E-N-CAM expression define two types of olfac¬
tory nerve ensheathing cells that share a common lineage.
Dev. Biol. 173: 327-343.
Franklin, R. J. M. & Barnett, S. C. (1997). Do olfactory glia have
advantages over Schwann cells for CNS repair? J. Neurosci
Res. 50:1-8.
Franklin, J. М., Gilson, J. М., Franceschini, I. A. & Barnett, S. С
(1996). Schwann cell-like myelination following transplanta¬
tion of an olfactory-nerve-ensheating-cell line into areas of
demyelination in the adult CNS. Glia 17: 217-224.
Franklin, R. J. M. & Barnett, S. C. (2000). Olfactory ensheating
cells-and CNS regeneration — the sweet smell of success?
Neuron 28:1-4.
Frazier, J. M. (1992) In vitro toxicity testing. New York, Marcel
Dekker.
Fredin, B. L, Seiffert, S. C. & Gelehrter, T. D. (1979). Dexa-
methasone-induced adhesion in hepatoma cells: The role of
plasminogen activator. Nature 277:312-313.
Freed, L. E. & Vunjak-Novakovich, G. (2002). Spaceflight bio-
reactor studies of cells and tissues. Adv. Space Biol. Med. 8:
177-195.
Freedman, V. H. & shin, S. (1974). cellulaar tumorogenicity in
nude mice: Correlation with cell growth in semi-solid medium.
Се11Ъ: 355-359.
Freshney, M. G. (1994). Colony-forming assays for CFC-GM,
BFU-E, CFC-GEMM, and CFC-mix. In Freshney, R. I., Prag-
nell, I. B. & Freshney, M. G. (eds.) Culture of haempopoetic
cells. New York, Willey-Liss.
Freshney, R. I. (1972). Tumour cells disaggregation in collage-
nase. Lancet.2:488-489.
654
Список литературы
Freshney, R. I. (1976). Separation of cultured cells by isopycnic
centrifugation in metrizamide gradients. In Rick-wood, D.
(ed.). Biological separation. London amd Washington, Infor¬
mation Retrieval, pp. 123-130.
Freshney, R. I. (1980). Culture of glioma of the brain. In Tho¬
mas, D. G. T. & Graham, D. I. (eds.), Brain tumours: scientific
basis, clinical investigation and current therapy. London, But-
terworths, pp. 21-50.
Freshney, R. I. (1985). Induction of differentiation inneoplastic
cells. Anticancer Res., 5:111 -130.
Freshney, R. I. (ed.). (1992). Culture of epithelial cells. New York,
Willey-Liss.
Freshney, R. I. & Freshney, M. G. (eds.) (1996)/ Culture of im¬
mortalized ceUs. New York, Willey-Liss.
Freshney, R. I. & Hart, E. (1982). Clonogenicity of human glia
in suspension. Br. J. Cancer 46:463.
Freshney, R. I. & Morgan, D. (1978). Radioisotopic quantitation
inmicrotitration plates by an autoradiographic method. Cell
Biol. Int. Rep. 2:375-380.
Freshney, R. I., Celik, F. & Morgan, D. (1982a). Analysis of
cytotoxic and cytostatic effects. In Davis, W., Malvoni, C. &
Tannenberg, St. (eds.) The control of tumor growth andits
biological base, Forchritte in der Onkologie, Band 10. Berlin.
Akademie-Verlag, pp. 349-358.
Freshney, R. I., Hart, E. & Russel, J. M. (1982b). Isolation and
purification of cell cultutre from human tumours. In Reid, E.,
Cook, G. M. W. & Moore, D. J. (eds.), Cancer cell organelles;
methodological surveys (B): Biochemistry, Vol.2 Chichester,
U.K. Horwood, pp. 97-110.
Freshney, R. I., Morgan, D., Hassanzadah, М., Shaw, R. &
Frame, M. (1980a). Glucocorticoids, proliferation and the
cell surface. In Richards, R. J. & Rajan, К. T. (eds.). Tissue
culture in medical research (II). Oxford U.K. Pergamon Press,
pp. 125-132.
Freshney, R. I., Paul,J. & Kane, I. M. (1975). Assay of anticancer
drugs in tissue culture Conditions affecting their ability to
incorporate 3H-leucine after drug treatment. Br. J. Cancer.
41:857-866.
Freshney, R. I., Pragnell, I. B. & Freshney, М., (eds.) (1994).
Culture of haempopoetic cells. New York, Willey-Liss.
Freshney, R. I., Sherry, A., Hassanzadar, М., Freshney, М., Cril-
ly, P. & Morgan, D. (1980b). Control of cell proliferation in
human glioma by glucocorticoids, Br. J. Cancer 41:857-866.
Freshney, R. I. (2002). Other epithelial cells. In Freshney, R. I.
(ed.) Culture of epithelial cells. 2nd ed. Hoboken, NJ, Wiley-
Liss., 402-436.
Freshney, R. I. & Freshney, M. G. (2002). In Freshney, R. I. (ed.)
Culture of epithelial cells. 2nd ed. Hoboken, NJ, Wiley-Liss.,
Chapters 6,10,11,12,13.
Freyer, J. P. & Sutherland, R. M. (1980). Selective dissociation
and characterisation of cells from different regions of multicell
tumour spheroids. Cancer Res.40:3956-3965.
Friend, C., Patuleia, М. C. & Nelson, J. B. (1966). Antibiotic effect
of tylosine on a mycoplasma contamination in a tissue culture
leukemia cell line. Proc. Soc. Exp. Biol. Med. 121:1009.
Friend, К. K., Dorman, B. P., Kucherlapati, R. S. & Ruddle, F. H.
(1976). Detection of interspecific translocations in mouse-
human hybrid by alkaline Gimsa staining. Exp. Cell Res. 99:
31-36.
Fround S.J. (1999). The development, benefitsand disadvantages
of serum-free media. Dev. Biol. Stand. 99:157-166.
Fry., J., Bridges, J. W. (1979). The effect of phenobarbitone on
adult rat liver cells and primary cell lines. Toxicol Lett. 4:
295-301.
Fu, V. X., Schwarze, S. R., Reznikoff, C. A. & Jarrard, D. F.
(2004)/ The establishment and characterization of bladder
cancer cultures in vitro. In Pfragner, R. & Fresshney, R. I.
(eds.) Culture of human tumor cells. Hoboken, NJ, Wiley-Liss
pp. 832-839.
Fujita H., Asahina, A., Mitsui, H. & Tamaki, K. (2004). Lang-
erhans cells exhibit low responsiveness to double-stranded
RNA, Biochem Biophys. Res. Commun. 319:832-839.
Fuller, В. B. & Meyskens, F. L. (1981). Endocrine responsiveness
in human melaniocyte and melanoma cells in culture./. Natl.
Cancer Inst 66:799-802.
Fullerton, N. E., Boyd, М., Mairs, R. J., Keith, W. N., Alder-
wish, O., Brown, М. М., Livingstone, A. & Kirk, D. (2004).
Combining a targeted rediotherapy and gene therapy ap¬
proach for adenocarcinoma of prostate. Prostate Cancer
Prostatic Dis. 7:355-363.
Fume, M. & Saito, S. (1997) Aynergistic effect of hepatocyte
growth factor and fibroblast growth factor-1 on the branching
morphogenesis of rat submandibular gland epithelial cells.
Tissue Cilt. Res. Commun. 16:189-194.
Fusenig, N. E. (1994a). Epithelial-mesemchymal interactions
regulate keratinocyte growth and differentiation in vitro. In
Leigh, I. М., Lane, E.B. & Wat. F. M. (eds.) Keratynocyte
handbook, Vol. 1. Cambridge, U.K. Cambridge University
Press, pp. 71-94.
Fusenig, N. E. (1994b). Cell ulture models: reliable tools in
pharmacotoxicology? In Fusenig, N. E. & Graf, H. (eds.)
Cell culture in pharmaceutical research. Berlin & Heidelberg,
Springer-Verlag, pp. 1-7.
Fusenig, N. E., Limat, A., Stark, H.-J. & Breitreutz, D. (1994).
Modulation of the differentiated phenotype of keratynocytes
of the hair follicle and from epidermis. J. Dermatol. Sci.l:
142-151.
Gao, R., Ustinov, J., Pulkkinen, M. A., Lundin, K., Korsgren, O. &
Otonkoski, T. (2003). Characterization of endocrine progeni¬
tor cells and critical factors for their differntiation in human
adult pancreatic cells culture. Diabetes 52:2007-2015.
Gard, A. L. & Pfeffer, S. E. (1990). Two proliferative stages of
the oligodendrocyte lineage (A2B5+04-and 04GalC-) under
different mitogenic control. Neuron 5: 615-625.
Gardner, D. K. & Lane, M. (2003) Towards a single embryo trans¬
fer. Reprod. Biomed. Online 2003 June 6 (4): 470-481.
Garrido, Riese, H. H., Aracil, M. & Perez-Aranda, A. (1995).
Endothelial cell differentiation into capillary-like structures
in response to tumor cell conditioned medium: a modified
chemotaxis assay. Br. J. Cancer. 71: 770-775.
Gartner, S. M. (1967). Genetic markers as tracers in cell
culture.2nd Decennial Review Conference on Cell, Tissue and
Organn Culture. NCI Monograph. 26. pp. 167-195.
Gaudemack, Т., Leivestad, Т., Ugelstad, J. & Thorsby, E. (1986).
Isolation of pure functioanlly active CD8+ T cells. Positive
selection with monoclonal antibodies directly conjugated to
monosized magnetic microspheres. J. Immunol Methods 90:
179-187.
Список литературы
655
Gaush, С. R., Hard, W. L. & Smith, T. F. (1966). Characterization
of an established line of canine kidney cells (MDCK). Proc.
Soc. Exp. Biol. Med. 122:931-933.
Gazdar A.F., Carney, D. N. & Minna J.D. (1983). The biology of
nonsmall cell lung cancer. Semin. Oncol. 10:3-19.
Gazdar, A. F., Carney, D. N., Russel, E. K., Sims, H. L., Bay-
lin, S. B., Bunn, P. A., Guccion, J. G. & Minna, J. D. (1980).
Establishment of continuous clonable cultures of small cell
carcinoma of the lung which have amine precursor uptake and
decarboxylation properties. Cancer. Res. 40:3502-3507.
Gazdar, A. F., Zweig, M. N., Carney, D. N., Van Steirteghen, A. C.,
Baylin, S. B. & Minna, J. D. (1981) Levels of creatine kinase
and its BB isoenzyme in lung cancer specimens and cultures.
Cancer. Res. 41:2773-2777.
Germain, L, Rouabhia, М., Guignard, R., Carrier, L., Bou-
vard, V. & Auger, F. A. (1993). Improvement of human
keratinocyte isolation and culture using thermolysin, Bums
19:99-104.
Gerper, P., Whang-Peng. J. & Monroe, J. H. (1969). Transfor¬
mation and chcromosome changes induced by Epstein-Barr
virus in normal human leukocyte cultures. Proc. Nad. Acad.
Sci. USA 63 (3): 740-747.
Gey, G. O., Coffman, W. D. & Kubicek, М. T. (1952). Tissue cul¬
ture stuies of the proliferative capacity of cervical carcinoma
and normal epithelium. Cancer Res. 12: 364-365.
Ghigo, D., Priotto, C., Migliorino, D., Geromin, D., Franchino, C.,
Todde, R., Coetamegna, C., Pescarmona, G. & Bosia, A.
(1998). Retinoic acid-induced differenciation in a human
neuroblastoma cell line is associated with an increase in nitric
oxide syntesis./. Cell Physiol. 174:99-106.
Ghosh, D., Collodi, P. (1994). Culture of cells from zabrafish
(Brahydanio rerio) blastula-stage embryos. Cytotechnology
14:21-26.
Ghosh, D., Danielson, К. C., Alston, J, T. & Heyner, S. (1991)/
Functional differential of mouse uterine epithelial cells grown
on collagen gels or reconstituted basement membranes. In
Vitro Cell Dev. Biol., 27 A: 713-719.
Giard, D. J., Aaronson, S. A., Todaro, G. J., Arnstein, P.,
Kersey, J. H., Dosik, K. & Parks, W. P. (1972). In vitro culti¬
vation of human tumors: Establishment of cell lines derived
from series of solid tumors./. Natl. Cancer Inst 51:1417.
Gignac, S. М., Steube, K., Schleithoff, J. W., MacLeod, R. A.,
Quentmeier, H. & Drexler, H. G. (1993). Multiparameter
approach in the identification of criss-cintaminated leukemia
cell lines. Leukemia Lymphoma 10 (4-5): 359-368.
Gilbert, S. F. & Migeon, B. R. (1975). d-Valine as a selective
agent for normal human and rodent epithelial ells in culture.
Cell 5:11-17.
Gilbert, S. F. & Migeon, B. R. (1977). Renal enxzymes in kid¬
ney cells selected by d-valine medium. J. Cell. Physiol. 92:
161-168.
Gilchrest, B. A., Albert, L. S., Karassik, R. L. & Yaar, M. (1985).
Substrate influences human epidermal melanocyte attach¬
ment and spreading in vitro. In Vitro Cell Dev. Biol. 21:114.
Gillete, R. W. (1987). Alternatives to pristane priming for as¬
citic fluid and monoclonal antibody production./. Immunol.
Methods 99: 21 -23.
Gillis, S. & Watson, J. (1981). Interleukin-2 dependent culture
of cytplytic T cell lines. Immunol. Rev. 54:81-109.
Gimbrone, M. A., Jr., Cotran, R. S. & Folkman,J. (1974). Humna
vascular endothelial cells in culture, groth and DNA synthesis.
J. Cell. Biol. 60:673-684.
Giovanella, В. C., Stehlin, J. S. & Williams, L. J. (1974). Hetero-
transplantation of human malignanr tumorin «nude» mice; II:
Malignant tumors induced byinjection of cell cultures derived
from human solid tumors./. Natl. Cancer Inst 52: 921.
Giron, J. A., Lange, M. & Baseman, J. B. (1996). Adherence, fibro-
nectin-binding and induction of cutoskeleton reorganization
in cultures human-cells by Mycoplasma penetrans. Infect.
ImmunolM: 197-208.
Gjerset, R., Yu., A. & Haas, M. (1990). Establishment of continu¬
ous cultures of T-cell acute lymphoblastic leukemia cells at
diagnosis, Cancer Res. 50:10-14.
Glavin, G. B., Szabo, S., Johnson, B. R., Xing, P. I., Morales, R. E.,
Plebani, M. & Nagy, L. (1996). Isolated rat gastric mucosal
cells: Optimal conditions for cell harvesting, measures of
viability and direct cytoprotection./. Pharmacol. Exp. Thera¬
peutic 276:1174-1179.
Gluzman, Y. (1981). SV40-transformed simian cells support the
replication of early SV40 mutants. Cell. 23:175-182.
Gobet, R., Raghunnath, М., Altermatt, S., Meuli-Simmen, C.,
Benathan, М., Dietl, A. & Meuli, M. (1997). Eficacy ofcul-
tured epithelial autigrafts in pediatric bums and reconstrutive
surgery. Surgery 121: 546-661.
Goding, C. R. & Fisher, D. E. (1997). Meeting review-regula-
tion of, melanocyte differentiation and growth. Cell Growth
Differ. 8:935-940.
Goldberg, B. (1977). Collagen synthesis as a marker for cell type
in mouse 3T3 lines. Cell 11:169-172.
Goldman, В. I. & Wurzel, J. (1992). Effects of subcultivation and
culture medium on differentiation of human fetal cardicac
myocytes. In Vitro Cell Dev. Biol. Anim. 28A: 109-119.
Goltstein, S., Murano, S., Benes, H., Moerman, E. J., Jones, R. A. &
Thweatt, R. (1989)Studies on the molecular genetic basis
of replicative senescence in Werner syndrome and normal
fibroblasts. Exp. Gerontol.2A (5-6): 461-468.
Goldwasser, E. (1965) Erythropoetin and the differentiation of
red blood cells. Fed. Proc. 34:2285-2292.
Gomm, J. J., Coope, R. C., Browne, P. J. and Coombes, R. C.
(1997) Separated human breast epithelial and myoepithelial
cells have different growth factor requirement in vitro but can
reconstitute normal breast lobuloalveolar structure./. Cell
Physiol. 171:11-19.
Good, N. E., Winget, G. D., Winter. W., Connolly, T.N. Iza-
wa, S. & Singh, R. М. M. (1966). Hydrogen ion buffers and
biological research, Biochemistry 5:467-477
Goodwin, G., Shaper, J. H., Abezoff, M. D., Mendelsohn, G. &
Baylin, S. B. (1983). Analysis of cell surface proteins de¬
lineates a differentiation pathway linking endocrine and
nonendocrine human lung cancers. Proc. Natl. Acad. Sci. USA
80:3807-3811.
Gordon. К. E., Ireland, H., Roberts, М., Steeghs, K., McCaul, J. A.,
MacDonald, D. G. & Parkinson, E. K. (2003). High levels of
telomere dysfunction bestow a selective disadvantage dur¬
ing the progression of human oral squamous cell carcinoma.
Cancer Res. 63:458-467,
Gospodarowicz, D., Rudland, P., Lindstrom, J. & Benirschke, K.
(1975). Fibroblast growth factor: its localization, purification,
656
Список литературы
mode ot action, and physiological significace. Adv. Metab.
Disord. 8: 301-335.
Gospodarowicz, D. (1974). Localization of fibroblast growth
factor and its effect alone and with hydrocortisone on 3T3
cell growth. Nature 249:123-127.
Gospodarowicz, D. & Moran, J. (1974). Growth factors in mam¬
malian cell cultures. Annu. Rev. Biochem. 45:531-558.
Gospodarowicz, D., Delgalo, D. & Vlodavsky, I. (1980), Permis¬
sive effect of the extracellular matrix on cell proliferation in
vitro. Proc. Natl. Acad. USA 77:4094-4098,
Gospodarowicz, D., Greenburg, G. & Birdwell, C. R. (1978b).
Determination of cell shape by the extracellular martix and
its correlation with the control in cellular growth. Cancer
Res. 38:4155-4171.
Gospodarowicz, D., Greenburg, G., Bialecki, H. & Zetter, B. R.
(1978a). Factors involved in the modulation of cell prolifera¬
tion in vivo and in viro: The role of fibroblast and epidermal
growth factors in the proliferative response of mammalian
cells. In Vitro 14:85-118
Goto, S., Miyazaki, K., Funabiki, T. & Yasumistu, H. (1999).
Serum-free culture conditons for analysis of secretory pro-
teinases during myogenic differentiation of mouse C2C12
myoblasts. Anal Biochem. 272:135-112.
Cougos, A. & Letarte, M. (1990)/ Biol Chem 265:8361-8364.
Cower, W. R., Risch, R. М., Godellas, С. V. & Fabri, P. J. (1994).
HPAC, a new human glucocorticoid sentensive pancreatic
ductal adenocarcinoma cell line. In Vitro Cell Dev. Biol.30A:
151-161.
Grafstrom, R. C. (1990a). In vitro studies of aldehyde effects
related to human respiratory carcinogenesis. Mutation Res.
238:175-184.
Grafstrom, R. C. (1990b). Carcinogenesis studies in human
epithelial tissues and cells in vitro: Emphasis on serum-free
culture conditions and transformation studies. Acta Physiol.
Scand. 140:93-133.
Grafstrom, R. C. (2002). Human oral epithelium. In Fresh¬
ney, R. I. & Freshney, M. G. (eds.), Culture of epithelial cells,
2nd ed. Hoboken, NJ. Wiley-Liss, pp. 195-255.
Graham, F. L. & Van der Eb, A. J. (1973). A new techinque for
the assay of infectivity of human adenovirus 5 DNA. Virology
52:456-461.
Graham, F. L., Smiley, J., Russell, W. C. & Naim, R. (1977).
Characteristics of a human cell line transformed by DNA from
human adenovirus type./ Gen. Virol. 36: 59-72.
Graham, G. L., Freshney, M. G., Donaldson, D. & Pragnell, I. B.
(1992). Purificarion and biochemical characrerisation of
human and minute stem cell inhibitors. Growth Factors 7:
151-160.
Crampp, G. E., Sambanis, A. & Stephanopoulos, G. N. (1992). Use
of regulated secretion in protein production from animal cells:
An overview. Adv. Biochem. Eng. Biotechnol. 46: 35-62.
Grandolfo, М., d’Andrea, P., Paoletti, S., Martina, М., Silves-
trini, G., Bionucci, E. & Vittur, F. (1993). Culture and dif¬
ferentiation of chondrocytes entrapped in alginate beads.
Calcif Tissue Int. 52:131-138.
Granner, D. K., Hayashi, S., Thompson, E. B. & Tomkins, G. M.
(1968). Stimulation of tyrosine aminotransferase synthesis
by dexamethasone phosphate in cell culture./. Mol. Biol. 35:
291-301.
Graziadei, P. P. C. & Monti Graziadei, G. A. (1979). Neurogenesis
and neuron regeneration in the olfactory system of mammals;
I: Morphological aspects of differentiation and structural
organisation of the olfactory sensory neurons./ Neurocytol.
8:1-18.
Craziadei, P. P. C. & Monti Graziadei, G. A. (1980). Neurogenesis
and neuron regeneration in the olfactory system of mammals;
III: Deafferentation and reinnervation of the olfactory bulb
following section of the/i/a olfactoria in rat./. Neurocutol. 9:
145-162.
Greco, B. & Recht, L. (2003). Somatic plasticity of neural stem
cells: Fact or fancy?/. Cell. Biochem. 88: 51-56.
Green, A. E., Athreya, B., Lehr, H. B. & Coriell, L. L. (1967).
Viability of cell cultures following extended preservation in
liquid nitrogen. Proc. Soc. Exp. Biol. Med. 124:1302-1307.
Green, D. F., Hwang, К. H., Ryan, U. S. & Bonrgoignie, J. J.
(1992). Kidney IntAi: 1506-1516.
Green, H. & Kehinde, O. (1974). Sublines of mouse 3T3 cells that
accumulate lipid. Cell 1:113-116.
Green, H. & Thomas, J. (1978). Pattern formation by cultured
human epidermal cells: D evelopment of curved ridges resem¬
bling dermatoglyphs. Science 200:1385-1388.
Green, H., Kehinde, O. & Thomas, J. (1979). Growth of cultured
human epidermarl cells into multiple epithelia suitable for
grafting. Proc. Natl. Acad. Sci. USA 76: 5665-5668.
Creenberger, J. S. (1980). Self-renewal of factor-dependent
haemopoietic progenitor cell lines derived from long-term
bone marrow cultures demonstrate significant mouse strain
genotypic variation./. Supramol. Struct. 13:501-511
Greene, L, A. & Tischler, A. S. (1976). Establishment of a nor¬
adrenergic clonal line of rat adrenal pheochromocytoma cells
which respond to nerve growth factor. Proc Natl Acad Sci
USA. 73:2424-2428.
Greider, C. W. & Blackburn, E. H. (1996). Telomeres, telomerase,
and cancer. Sci. Am., February 1996.
Griffiths, B. (2001). Scale-upof suspension and anchorage-depen-
dent animal cells. Mol. Biotechnol. 17: 225-238.
Griffiths, B. (2000). Scaling up of animal cell cultures. In Mas¬
ters, J. R. W. (ed.)> Animal Cell Culture, a Practical Approach,
Oxford, U.K., Oxford University Press, pp. 19-67.
Griffiths, J. B. & Pirt, G. J. (1967). The uptake of amino acids by
mouse cells (Strain LS) during growth in batch culture and
chemostat culture: The influence of cell growth rate. Proc. R.,
Soc. Biol. 168:421-438.
Griffiths, J. B. (1991). Products from animal cells. In Butler, M.
(ed.), Mammalian cell Biotechnology, a prarctical approach,
Oxford, U.K., IRL Press at Oxford University Press,
pp. 207-235.
Grizzle, W. E. & Polt, S. H (1988). Gudelines to avoid personal
contamination by infective agents in research laboratories
that use human tissue./ Tissue Cult Methods 11:191-200.
Gross, S. K., Lyerla, T. A., Williams, M. A. & McCluer, P. H.
(1992). The accumulation and metabolism of glycosphingo-
lipids in primary kidney cell cultures from beige mice. Moll
Celll Biochem 118:61-66.
Groves, A., Barnett, S. C., Franklin, R. J. М., Crang, A. J.,
Mayer, М., Blakemore, W. & Nobel, M. (1993). Repair of
demyelinated lesions by transplants of purified 0-2 A progeni¬
tor cells. Nature. 362:453-455.
Список литературы
657
Gugel, Е. А. & Sanders, J. Е. (1986). Needle-stick transmission of
human colonic adenocarcinoma New Engl.J. Med. 315,1487.
Guguen-Guillouzo, C. (2002). Isolation and culture of animal and
human hepatocytes. In In Freshney, R. I., and Freshney, M. G.
(eds.), Culture of epithelial celb. 2nd ed. Hoboken, NJ, Wiley-
Liss, pp. 337-379.
Guguen-Guillouzo, C. & Cuillouzo. A. (1986). Methods for
preparation of adult and fetal hepatocytes. In Cuguen-Cuil-
louzo, C. & Cuillouzo, A. (eds.) Isolated and cultured hepato¬
cytes. Paris. Les Editions INSERM, John Libbey Eurotext,
pp. 1-12.
Guguen-Guillouzo, C., Campion, J.P. Brissot, P., Glaise, D.,
Launois, B., Bourel, M. & Cuillouzo. A. (1982). High yield
preparation of isolated human adult hepatocytes by enzymatic
^ profusion of the liver. Cell Biol. Int. Rep.6: 625-628.
Guguen-Guillouzo, C., Clement. B., Baffet, G., Beaument, C.,
Morel-Chany, Ciise, E., Glaise, D. & Guillouzo, A. (1983).
Maintenance and reversibility of active albumin secretion by
adult rat hepatocytes co-cultured with mother liver epithelial
cell type. Exp Cell. Res. 143:47-54.
Cuilbert, L. I. & Iscove, N. N. (1976). Partial replacement of serum
by selenite, transferrin, albumin and lecitin in haemopoetic
cell cultures. Nature 263:594-595.
Guillouzo, A. М. A. (1989). Methodes in vitro enpharmacotocoxi-
cologie. Les Editions INSERM 170: 200.
Guiraud, J. М., Beuron, F., Sion, B., Brassier, G., Faivre, J., Thei-
ulant, M. L & Duval, J. (1991). Human prolactin producing
pituitary adenomas in three dimensional culture. In Vitro Cell
Dev. Biol. 27a: 188-190.
Gullino, P. M. & Knazek, R., A. (1979). Tissue culture on artifi¬
cial capillaries. In Jakoby, W. B. & Pastan, I. (eds). Methods
in enzymology, Vol. 58: Cell Culture. New York. Academic
Press, pp. 178-184.
Gumbiner, B. (1992). Epithelial morphogenesis. Cell 69:
385-387.
Gumbiner, В. M. (1992). Signal transduction of betacatenin. Curr.
Opin Cell Biol., 7:634-640.
Gumbiner, B. & Simons, K. (.1986). A functional assay for pro¬
tein involvedinestablishing an epithehal occluding barrier:
Identification of a uvomorulin-like polypeptide./. Cell. Biol.
102:457-468.
Guner, М., Freshney, R. I., Morgan, D., Freshney, M. G., Tho¬
mas, D. G. T. & Graham, D. I. (1977). Effects of dexamytasone
and betamethasone on in vivo cultures from human astrocy¬
toma Br. J. Cancer 35:439-447.
Gupta, K., Ramakrishnan, S., Browne, P. V., Solovey, A. & Heb-
bel. R. P. (1997). A novel technique for culture of human
dermal micro vascular endothelial cells under either serum-
free or serum-supplemented conditions: Isolation by panning
and stimulation with vascular endothelial growth factor. Exp
Cell. Res. 230:241-251.
Gustafson, C. J., Eldh, J. & Kratz, G. (1998) Culture of human
urothelial cells on a cell-free dermis for autotransplantation.
Eur. Urol. 33:503-506.
Hafny, В. E. L., Bourre, J.-M. & Roux, F. (1996). Synergistic
stimulation of gamma-glytamyl transpeptidase and alkaline
phosphatase activities by retinoic acid and astroglial factors
in immortalised rat brain microvessel endothelial cells ./. Cell.
Physiol.i67:451-460.
Halaban, P. (2004). Culture of melanocytes from normal, benign,
and malignant lesions. In Pfragner, R. & Freshney, R. I.
(eds.) Culture of human tumor celb. Hoboken, NJ, Wiley-Liss
pp. 289-318.
Hallermeyer, K. & Hamprecht, B. (1994). Cellular heterogenec-
ity in primary cultures of brain cells revealed by imnnuno-
cytochemical localisation of glutamyl synthetase. Brain Res.
295:1-11.
Halleux, C. & Schneider, Y.-J. (1994). Iron absorption by Caco-
2 cells cultivated in serum-free meddium as in vitro model of
the human intestinal epithelial barrier./. Cell Physiol 158:
17-28.
Ham, R. G. (1963). An improved nutrient solution for diploid Chi¬
nese hamster and human cell lines. Exp Cell. Res. 29: 515.
Ham, R. G. (1965). Clonal growth of mammalian cells in a
chemically defined synthetic medium. Proc. Natl Acad. Sci.
USA 53:288.
Ham, R. G. (1984). Growth of human fibroblasts in serum-free
media. In Barnes, D. W., Sirbasku, D. A. & Sato, G. H. (eds).
Cell culture methods for molecular and cell biology. Vol. 3. New
York Alan R. Liss, pp. 249-264.
Ham, R. G. & McKeehan, W. L. (1978). Development of improved
media and culture conditions for clonal growth of normal
diploid cells. In Vitro 14:11-22.
Ham, R. G. & McKeehan, W. L. (1979). Media and growth re¬
quirements. In Jakoby, W. B. & Pastan, I. H. (eds.),. Methods
in enzymology; vol.58: Cell culture. New York, Academic Press
pp. 44-93.
Hamburger, A. W. & Salmon, S. E. (1977). Primary bioassay of
human tumor stem cells. Science 197:461-463.
Hamburger, A. W., Salmon, S. E., Kim. М. B., Trent, J. М.,
Soehnlen, B., Alberts, D. S. & Schmidt. H J. (1978). Direct
cloning of human ovarian carcinoma cells in agar. Cancer.
Res. 38:3438-3444.
Hames, B. D. & Glover, D. M. (1991). Oncogenes. Oxford, UK
IRL Press at Oxford University Press.
Hamilton. Т. C., Young, R. C., McKoy, W. М., Grotzinger, K. R.,
Green, J. A., Chu, E. W., Whang-Peng, J., Rogan, A. М.,
Green, W. R. & Ozols, R. F, (1983). Characterization of a
human ovarian carcinoma cell line (NIH: OVCAR-3) with an¬
drogen and estrogen receptors. Cancer Res. 43:5379-5389.
Hamilton, W. G. & Ham, R. G. (1977). Clonal growth of Chi¬
nese hamster cell lines in protein-free media. In Vitro, 13:
537-547.
Hammond, S. L., Ham, R. G. & Stampfer, M. R. (1984). Serum
free growth of human mammary epithelial cells: Rapid clonal
growth in defined medium and extended serialpassage with
pituitary extract. Proc. Natl. Acad. Sci. USA 81:5435-5439.
Hanks, J. H. & Wallace, R. E. (1949). Relation of oxygen and
temperature in the preservation of tissues by refrigeration.
Proc. Soc. Exp. Biol Med. 71:196.
Hansson, B., Ronnback, L., Persson, L. I., Lowenthal, A.,
Noppe, М., Ailing, C. & Karlsson, B. (1984). Cellular com¬
position of primary cultures from cerebral cortex, striatum,
hippocampus, brain-stem and cerebellum. Brain Res. 300:
9-18.
Hardman, P., Klement, B. J. & Spooner, B. S. (1990) Growth
and morphogenesis of embryonic mouse organs on Biopore
membrane. In Vitro Cell Dev. Biol. 26:119-120.
658
Список литературы
Harrington, W. N. & Godman, G. С. (1980). A selective inhibitor
of cell proliferation from normal serum. Proc. Natl. Acad. Sci.
USA. 77:423-427.
Harris, H. & Watkins, J. F. (1965). Hybrid cells from mouse and
man: Artificial heterokaryons of mammalian cells from differ¬
ent species. Nature 205: 640-646.
Harris, L. W. & Griffiths, J. B. (1977). Relative effects of cooling
and warming rates on mammalians cells during the freeze-
thaw cycle. Cryobiology 14: 662-669.
Harris, M. (1959). Essential growth factor in serum dialysate for
chick skeletal muscle fibroblasts. Proc. Soc. Exp. Biol. Med.
102:468.
Harrison, R. G. (1907). Observations on the living developing
nerve fiber. Proc. Soc. Exp. Biol. Med. 4:140-143.
Hart. I. R. & Fidler, I. J. (1978). An in vitro quantitative assay for
tumor cell invasion Cancer. Res. 38:3218-3224.
Hartley, R. S., A Yablonka-Reuveni, Z. (1990). Long-term
maintenance of primary myogenic culture on a reconstited
basement membrane. In Vitro Cell Dev. Biol. 26:955-961.
Hashimota, N. (2004). Stem cell systems in skeletal muscle.
Tanpakushitsu Kakusan Koso 49: 741-748.
Hass, R., Gunji, H., Hirano, М., Weichselbaum, R. & Kufe, D.
(1993). Phorbol ester-induced monocytic differentiation
is associated with G2 delay and down regulation of cdc25
expression. Cell Growth Differ. 4:159-166.
Hassell, J.R. Robey, P. G., Barrach, H. J., Wilczek, J., Ren-
nard, S. I. & Martin, G. R. (1980). Isolation of a heparan
sulfate-containing proteoglycan from basement membrane.
Proc. Natl. Acad. Sci. USA 77:4494-4498.
Haudenschild, С. C., Zahniser, D., Folkman, J. & Klagsbrun, M.
(1976). Human vascular endothelial cells in culture, Exp Cell.
Res. 98:175-183.
Hauschka, S. D. & Konigsherg, I. R. (1966). The influence of col¬
lagen on the development of muscle clones Proc. Natl. Acad.
Sci. USA 55:119-126.
Hauselmann, H. J., Fernandes, R. J., Mok, S. S., Schmid, Т. М.,
Block, J. A., Aydelotte, М. B., Kuettner, К. E., Thonar, E. J.-M. A.
(1994). Phenotypic stability of bovine articular chondrocytes
after long-term culture in alginate beads .J. Cell Sci. 107:
17-27.
Hauselmann, H. J., Masuda, K., Hunziker, E. B., Neidhart, М., Mok,
S. S., Michel, B. A. & Thonar, E. J.-M. A. (1996). Adult human
chodrocytes cultured in alginate form a matrix similar to native
human cartilage. Am.J. Physiol Cell Physiol. 40: C742-C752.
Hawley-Nelson, P., Ciccarone, V., Gebevehu, G.,Jessee, J. (1993).
A new polycationic liposome reagent with enhanced activity
for transfection. FASEB 7: A167.
Hay, E. D. (ed.). (1991). Cell biology of extracellular matrix. New
York, Plenum Press.
Hay, R. J. (1979). Identification, separation and culture of mam¬
malian tissue cells. In Reid, E. (ed.), Methodological surveys in
biochemistry f Vol. 8. Cell Populations. London. Ellis Horwood,
pp. 143-160.
Hay, R. J. (2000). Cell, line preservation and characterization,
In Masters, J. R. W. (ed.). Animal cell culture, a practical
approach. Oxford, U.K., IRL Press at Oxford University
Press, pp. 95-148.
Hay, R. J. & Cour, I. (1997). Testing of microbial contamination:
Bacteria and fungi. In Doyle, A., Griffits, J. B. & Newall, D. G.
(eds.) Cell and tissue culture: Laboratory procedures. Chiches¬
ter, U.K., Wiley, Module 7 A: 2.
Hay, R. J. & Strehler, B. L. (1967). The limited growth span of
cell strains isolated from the chick embryo. Exp. Gerontol.
2:123.
Hay, R. J., Caputo, J. & Macy. M. L. (1992). Establishing of
verifying cell line identity, In ATCC quality control methods
for cell lines, 2nd ed. Rockville, MD. American Type Culture
Collection, pp. 52-66.
Hay, R. J., Miranda-Cleland, М., Durkin. S. & Reid, Y. A.
(2000). Cell line preservation and authentification. In Mas¬
ters, J. R. W. (ed.), Animal cell culture. Oxford University
Press, Oxford, pp. 69-103.
Hayashi, I. & Sato, G. H. (1976). Replacement of serum by hor¬
mones permits growth of cells in a defined medium. Nature
259:132-134.
Hayflick, L (1961). The establishment of a line (WISH) of hu¬
man amnion cells in continuous cultivation. Exp Cell. Res.
23:14-20.
Hayflick, L. & Moorhead, P. S. (1961). The serial cultivation of
human diploid cell strains. Exp Cell. Res. 25:585-621.
Haynes, L. W. (1999). The neuron in tissue culture. Chichester,
U.K., John Wiley & Sons.
Heald, K. A., Hail, C. A. & Downing, R. (1991). Isolation of
islets of Langerhans from the weanling pig. Diabetes Res.
17:7-12.
Health and Safety Commission (1985). Approved Code of Prac¬
tice «The Protection of Persons Against Ionising Radiation
from An Work Activity». HMSO, London.
Health and Safety Commission (1991a). Safe working and preven¬
tion of Infection in clinical Laboratories HMSO Publications,
P.O. Box 276, London, SW8 5DT, England.
Health and Safety Commission (1991b). Safe working and pre¬
vention of Infection in clinical Laboratories — Model Rules
for Staff and Visitors, HMSO Publications, P.O. Box 276,
London, SW8 5DT, England.
Health and Safety Commission (1992). Genetically Modified
ed Organisms (Contained Use) Regulations: SI 1992/3217
HMSO, ISBN 0-11-025332-9.
Health and Safety Commission (1999a) Carcinogens ACOP and
Biological agents ACOP. Control of Substances Hazardous
to Health Regulations, Approved Codes of Practice, L5 HSE
Books, HMSO Publication Centre, P.O. Box 276, London
SW8 5DT England.
Health and Safety Commission (1999b) Control of Substances
Hazardous to Health Regulations. Statutory Instruments.
No.437, HSE Books, Sudbury, U.K.
Health Services Advisory Committee (1992) Safe Disposal of
Clinical Waste. HSE Books, Sudbury, U.K.
Heffelfinger, S. C., Hawkins, H.H. Barrish, J., Taylor, L. & Dar¬
lington. G. (1992). SR HEP-1: A human cell line of endothe¬
lial origin. In Vitro Cell Dev. Biol. 28A: 136-142.
Heldin, С. H., Westermark, B. & Wasteson. A. (1979). Plate¬
let-derived growth factor: Purification and partial characre-
rization. Proc. Natl. Acad. Sci. USA 76:3722-3726.
Hemmati-Brivaniou, A., Kelly, O. G. & Melton, D. A. (1994).
Follistatin, an antagonist of activin, is expressed in the Spe-
mann organizer and displays direct neuralizing activity. Cell
IT. 283-295.
Список литературы
659
Heyderman, Е., Steele, К. & Ormerod, М. G. (1979). A new
antigen on the epithelial membrane: Its immunoperoxidase
localisation in normal and neoplastic tissue / Clin. Pathol
32:35-39.
Heyworth, С. M. & Spooncer, E. (1992). In vitro clonal assays
for murine multiipotential and lineage restricted myeloid
progenitor cells, in Testa, N. G. & Molineux, G. (eds.), Hae¬
mopoiesis: A practical approach. Oxford, U.K., IRL Press at
Oxford University Press, pp. 37-54.
HHS Publication No. (CDC) 93-8395. 3rd Edition (1993),
Biosafety in Microbiological and Biomedical Laboratories,
U.S. Department of Health and Human Services, Public
Health Service, Centers for Disease Control and Prevention,
and National Institutes of Health. U.S. Covemment Printing
Office, Washington.
Hicks, G. G., Chen, J. & Ruley, H. E. (1998). Production and use
of retroviruses. In Ravid. K. & Freshney, R. I. (eds.), DNA
transfer so cultured cells, New York, Wiley-Liss, pp. 1-26.
Higuchi, К (1977). Cultivation of mammalian cell lines in serum-
free chemically defined medium, Metod Cell Biol 14:131.
Hill, В. T. (1983). An overview of clonogenic assays for human
tumour biopsies. In Dendy, P. P. & Hill, В. T. (eds.), Human
tumour drug sensitivity testing in vitro. New York, Academic
Press, pp. 91-102.
Hilwig, I. & Gropp, A. (1972). Staining of constitutive hetero-
chromatine in mammalian chromosomes with a new fluoro-
chrome. Exp. Cell. Res. 75:122-126.
Hince, T. A. & Roscue, J. P. (1980). Differences in pattern and
level of plasminogen activator production between a cloned
cell line from an ethylnitrosourea-induced glioma and one
from normal adult rat brain./. Cell. Physiol 104:199-207.
Hino, H., Tateno, C., Sato, H., Yamasaki, C., Katayama, S., Ko-
hashi, Т., Aratani, A., Asahara, Т., Dohi, K. & Yoshizato, K.
(1999). A long-rerm culture of human hepatocytes which
show a high growth potential and express their differentiated
phenotypes. Biochem. Biophys. Res. Commun. 256:184-189.
Hirai, Y., Takebe, K., Takashina, М., Kobayashi, S. & Takeichi, M.
(1992). Epimorphin: A mesenchymal protein essential for
epithelial morphogenesis. Cell 69:471-481.
Hlubinova, K., Feldsamova, A. & Prachar, J. (1994). Evaluation
of two methods for elimination of mycoplasma. In Vitro Cell
Dev. Biol. 30A: 21-22.
Hofmann, S., Kaplan. D., Vunjak-Novakovic, G., Meinel, L.
(2005). Tissue engineering of bone. In Vunjak-Nova¬
kovic. G. & Freshney, R. I. (eds.) Culture of cells for tissue
ingineering Hoboken, NJ, Wiley-Liss.
Hoheisel, D., Nitz, Т., Franke, H., Wegener, J., Hakvoort, A.,
Tilling, T. & Galla, H. J. (1998). Hydrocortisone reinforces
the blood-brain properties in a serum free cell culture system.
Biochem. Biophys. Res. Commun. 247:312-315.
Holbrook, K. A. & Hennings, H. (1983). Phenotypic expression
of epidemal cells in vitro: A review./. Invest. Dermatol. 81:
lls-24s.
Hollenberg, M. D. & Cuatrecasas, P. (1973). Epidermal growth
factor: Receptors in human fibroblasts and modulation
of action by cholera toxin. Proc. Natl. Acad. Sci. USA 70:
2964-2968.
Holley, R. W., Armour. R. & Baldwin, J. H. (1978). Density- de¬
pendent regulation of growth of BSC-1 cells in cell culture:
Growth inhibitors formed by the cells. Proc. Natl. Acad. Sci.
USA 75:1864-1866.
Holt, S. E., Shay, J. W. & Wright, W. E. (1996). Refining the
telomere-telomerase hypothesis of ageing and cancer. Nat.
Biotechnol. 14:836-839.
Hoober, J. K., Cohen, S. (1907). Epidemial growth factor. I. The
stimulation of protein and ribonucleic acid synthesis in chick
embryo epidermis. Biochem. Biophys. Acta 138: 347-356.
Hopps, H., Bemheim, В. C., Nisalak, A., Tjio, J. H. & Smadel. J. E.
(1963). Biologic characteristics of a continuous cell line
derived from the African green monkey. J. Immunol 91:
416-424.
Horibata, K. & Harris, A. W. (1970). Mouse myelomas and lym¬
phomas in culture. Exp. Cell Rev. 60:61-77.
Horita, A. & Weber, L. J. (1964), Skin penetrating property ot
drugs dissolved in dimethylsulfoxide (DMSO) and other
vehicles. Life Sci 3:1389-1395.
Hornsby, P. J., Yang, L., Lala, D. S., Cheng, C. Y. & Salmons, B.
(1992). A modified procedure for replica plating of mamma¬
lian cells allowing selection of clones based on gene expression.
Biotechniques 12:244-251.
Horster, M. (1979). Primary cultura of mammalian neephron
epithelia: cpi rbelia: requirements for cell outgrowth and prolf-
eration from defined explanted nephron segments. Pflugers
Arch 382:209-215.
Hotamisligil, G. S., Arner, P., Caro, J. F., Atkinson, R. L. &
Speigelman, В. M. (1995). Increased adipose tissue expres¬
sion of tumor necrosis factor-а in human obesity and insulin
resistance./. Clin. Invest. 95:2409-2415.
Howard, М., Kessler, S., Chused, T. & Paul, W. E. (1981). Long
term culture of normal mouse В lymphocytes. Proc. Nad. Acad.
Sci. USA 78:5788-5792.
Howie Report. (1978) Code of practice for prevention of infec¬
tion in clinical laboratories and post-mortem moms London.
Stationery Office.
Hoyer, L. W. de los Santos, R. P. & Hoyer, J. R. (1973). Anti¬
hemophilic factor antigen: localization in endothelial cells
by immunofluorescence microscopy, /. Clin. Invest. 52:
2737-2744.
HSE (1990). A guide to the Genetically Modified Organisms
(Contained. Use) Regulations 1992 (as amended), NSE Books,
ISBN 0 7176 1186 8.
Hsu T.C. & Benirschke, K. (1967). Atlas of mammalian chromo¬
somes. Vols. 1-4. New York, Springer.
Hu, G.-F., Riordan, J. F. & Valee, B. L. (1997) A putative angio-
genin receptor in angiogenin-responsive human endothelial
cells Proc. Natl. Acad. Sci. USA 94:204-209.
Hughes, S. E. (1996). Functional characterization of the spon¬
taneously transformed human umbilical vein endothelial cell
line ECV304: Use in an in vitro model of angiogenesis. Exp.
Cell Res. 225:171-185.
Hull, R. N., Cherry, W. R. & Weaver, G. W. (1976). The origin
and characteristics of a pig kidney cell strain, LLC-PKI. In
Vitro 12:670-677.
Human Cytogenetic Nomenclature: See Intemationsl System for
Human. Cytogenetic Nomenclature.
Hume, D. A. & Weidemann, M. J. (1980). Mitogenic Lympho¬
cyte transformation. Amsterdame Elsevier/North Holland
Biomedical Press.
660
Список литературы
Hunziker, Т. & Limat, А. (1999). Cultured keratinocytes grafts.
CurrProbl Dermatol, 27: 57-64.
Huschtscha, L. L. & Hollyday, R. (1983) Limited and unlimited
growth of SV40-transformed cells from human diploid MRC-
5 fibroblasts./ Cell. Sci.63:77-99.
Hynes, R. O. (1983) Alteration of cell-surface proteins by viral
transformation and by proteolysis. Proc. Natl. Acad. Sci. USA.
70:3170-3174.
Hynes, R. O. (1974) Role of cell surface alterations in cell
transformation: The importance of proteases and cell surface
proteins. Cell. 1:147-156.
Hynes, R. O. (1992). Integrines: Versatility, modiulation, and
signaling in cell adhesion. Cell. 69:11-25.
Hyvogen, Т., Alakuijala, L., Anderson, L., Khomutov, A. R.,
KhomutovR. M. & Eloranta, T. O. (1988). 1-Amino-qxy-
3-aminopropane reversivbly prevents the proliferation of
cultutred babty hamster kidney cells by interfering with
polyamine synthesis./. Biol. Chem. 263:1138-1144.
Ido, H., Harada, K., Futaki, S., Hayashi, Y., Nishiuchi, R., Nat-
suka, Y., Li, S., Wada, Y., Combs, A. C., Ervasti, J. M. & Seki-
guchi, K. (2004). Molecular dissection of the alpha-dystrogly-
can-and integrin-binding sites within the globular domain of
human laminin-10./. Biol. Chem. 279:10946-10954.
Iguchi, H., Mizumoto K., Kono, A. & Takiguchi, S. (2004). Pan¬
creatic cancer-derived cultured cells: genetic alterationsand
application to an experimental model ofpancreatic cancer
metastasis. In Pfragner, R. & Freshney, R. I. (eds.) Culture of
human tumor cells. Hoboken, NJ, Wiley-Liss., pp. 81-96.
Ikonomou, L., Schneider, Y. J. & Agathos, S. N. (2003). Insect
cell culture for industrial production of recombinant proteins.
Appl. Microbiol. Biotechnol. 62:1-20.
Illmensee, K. & Mintz, B. (1976) Totipotency and normal differ¬
entiation of sisngle teratocarcinome calls cloned by injection
into blastocyts. Proc. Natl. Acad. Sci. USA. 73:549-553.
Ilsie, A. W. & Puck, Т. T. (1971). Morphological transformation
of Chinese hamster cells by dibutil adenosine cycline 3’-5’-
monophosphate and testosterone. Proc. Natl. Acad. Sci. USA.
2:358-361.
Imaizumi, Т., Lankford, K. L., Waxman, S. G., Greer, C. A. and
Kocsis (1998). Transplanted olfactory ensheasting cells
remyelinate and enhance axonal conduction in the demy¬
elinated dorsal columns of therat spinal cord./ Neurosci 18:
6176-6185.
Inamatsu, М., Matsuzaki, Т., Iwarani, H. & Yoshizato, K. (1998).
Establishment of rat dermal papula cell lines that sustain the
potency toinduce hair follices from a follicular skin./ Invest.
Dermatol. Ill: 767-775.
Inokuchi, S., Handa, H., Imai, Т., Makuuchi, H., Kidokoro, М.,
Tohya, H., Aizawa, S., Shimamura, K., Ueyama, Y., Mi-
tomi, T. & Sawada, Y. (1995). Immortalisation of human
oesophagal epithelial cells by a recombinant SV40 adenovirus
vector. Br. J. Cancer. 71:819-825.
Inoue, S. & Spring, K. R. (1997) Video microscopy.2nd ed. new
York and London, Plenum Press.
International Human Genome. Sequencing Consortium (2001)
Initial sequencing and analysis of the human genome. Nature.
409:860-921.
International System for Human Cytogenetic Nomenclature
(1978). Report of the Standing Commettee on Human Cytoge¬
netic Nomenclature. Washingtone, DC, National Foundation
of the March of Dimes.
Ireland, G. W., Dopping-Hepenstal, P. J., Jordan, P. W. &
O’Neill, С. H. (1989). Limitation of substratum size alters
cytoskeletal organization and behaviour of Swiss 3T3 fibro¬
blasts. Cell Biol. Int. Rep. 126:121-126.
Iscove, N. N., Guillbert, L. W. & Weymann, C. (1980). Complete
replacement of serum in primary culture of erythropoetin-de-
pendent red cell precursors (CFU-E) by albumin, transferrin,
iron, unsaturated fatty acid, lecitine and cholesterol. Exp Cell.
Res. 126:121-126.
Iscove, N. & Melchers, F. (1978). Complete replacement of
serum by albumin, transferrin and soybean lipid in cultures
of lipopolysacharide-reactive В lymphocytes. J. Exp. Med.
147:923-933.
Ishida, Y., Levin, J., Baker, G., Stenberg, P., Yamada, Y., Sa¬
saki, H., Inoue, T. (1993). Biological and biochemical char¬
acteristics of murine megacaryoblastic cell line L8057. Exp.
Hematol. 21:289-298.
Itagaki, A. & Kimura, G. (1974). TES and HEPES buffers in
mammalian cell cutures and viral studies: Problems of carbon
dioxide requirements. Exp Cell. Res. 83:351-360.
Iyer, V. R., Eisen, М. B., Ross, D. Т., Schuler, G., Moore, Т.,
Lee, J. C. F., Trent., J. М., Staudt, L. М., Hudson, J., Jr., Bo-
guski, M. S., Lashkari, D., Shalon, D., Botstein, D. & Brown,
P. O. (1999). The transcriptional program in the response of
human fibroblasts to serum. Science. 283:83-87.
Izutsu, К. Т., Fatherazi, S., Belton, С. М., Oda, D., Cart¬
wright, F. D. & Kenny, G. E. (1996). Mycoplasm orale infec¬
tion affects К and Cl currents in the HSG salivary gland cell
line. In Vitro Cell Dev. Biol. Anim. 32: 361-365.
Jacobs, J. P. (1970). Characteristics of a human diploid cell des¬
ignated MRC-5. Nature 227:168-170.
Jacobs, J. P., Garrett, A. J. & Meron, R. (1979). Characteristics of
a serially propagaterd human diploid cell designated MRC-9.
/. Biol Stand. 7:113-122.
Jaffe, E. A., Nachman, R. L., Becker, G. C. &Ninick, C. R. (1973).
Culture of human endotelial cells derived from umbillical
veins./ Clin. Invest. 52: 2745-2756.
Jain, D., Ramasubramamanyan, K., Gould, S., Lenny, A., Cande-
lore, М., Tota, М., Strader, C., Alves, K., Cuca, C., Tung, J. S.,
Hunt, G., Junker, B., Buckland, В. C. & Silberklang, M.
(1991). In Sspeir, R. E., Griffits, J. B. & Meignier, B. (eds.)
Production of biologicals from animal cells in culture. Oxford,
U.K., Butterworth-Heinemann, pp. 345-351.
Jain, R. K., Schlenger, K., Hockel, M. & Yuan, F. (1997). Quanti¬
tative angiogenesis assays: Progress and problems. Nat. Med.
3:1203-1208.
Janssen, M. L., van Leewen, М. B., Scholtmeijer, K., van Koo-
ten, T. G., Dijkhuizen, L. & Wosten, H. A. (2003). Coating
with genetic engeneering hydrophobin promotes growth
of fibroblasts on a hydrophobic solid. Biomaterials. 23:
4847-4854.
Jat, P. S., Noble, M. D., Ataliotics, P., Tanaka, Y., Yannoutsos,N.,
Larsen, L. & Kiossis, D. (1991). Direct Derivation of confi-
detially immortal cell lines from an H-2 Kb-tsA58 transgenic
mouse. Proc. Natl. Acad. Sci. USA 88: 5096-5100.
Jeffreys, A. J., Wilson, V. & Thein, S. L. (1985). Individual specific
«fingerprints» of human DNA. Nature. 316: 76-79.
Список литературы
661
Jeng, Y.-J., Watson, С. S. & Thomas, M. L. (1994). Identification
of vitamin D-stimlated phosphatase in I EC-6 cells, a rat small
intestine crypt cell line. Exp. Cell Res. 212:338-343.
Jenkins, N. (ed.) (1992) Growth factors, a practical approach.
Oxford, U.K., IRL Press at Oxford University Press.
Jessell, Т. M. & Melton, D. A. (1992). Diffusible factors in verte¬
brate embryoMc induction, Cell 68:257-270.
Jin, D. I., Lee, S. H., Choi, J. H., Lee, J. S., Lee, J. E., Park, K. W.,
Seo, J. S. (2003). Targeting efficiency of a-l,3-galactosyl
transferase gene in pig fetal fibroblast cells. Exp. Mol Med
35:572-577.
Jinard, F., Sergent-Engelen, Т., Trouet, A., Remade, C. &
Schneider, Y.-J. (1997). Compartment coculture of porcine
arterial endothelial and smooth muscle cells on a microporous
membrane. In Vitro Cell Dev. Biol. AMm. 33:92-103.
Johnson, G. D. (1989). Immunofluorescence. In Catty, D. (ed.).
Antibodies. Vol. II: A practical approach. Oxford, U.K., IRL
Press ar Oxford UMvercity Press, pp. 179-200.
Jones, E. L., Matsui, W. H. & Smith, B. D. (2004), Cancer stem
cells: are we missing the target? J. Natl. Cancer Inst. 96:
583-585.
Jones, E. L. & Gregory, J. (1989). Immunoperoxidase methods, In
Catty, D. (ed.). Antibodies. Vol. II: A practical approach. Oxford,
U.K., IRL Press ar Oxford UMvercity Press, pp. 155-177.
Joukov, V., Kaipainen, A., Jeltsch, М., Pajusola, K., Olofsson, B.,
Kumar, V., Eriksson, U. & Alitalo, K. (1997). Vascular endo¬
thelial growth factors VEGF-B and VTGF-C./. Cell Physiol.
173:211-215.
Jozan, S., Roche, H., Cheutin, J., Carton, M. & Salles, B. (1992).
New human ovarian cell line OVCCRl/sf in serum-free me¬
dium. In Vitro Cell Dev. Biol. 28 A: 687-689.
Kaartinen. L., Nettesheim, P., Adler, К. B. & Randell, S. H. (1993).
Rat tracheal epithelial cell differentiation in vitro. In Vitro Cell
Dev. Biol. 29A: 481-492.
Kahn, P. & Shin, S.-L. (1979). Cellular tumorigeMcity in nude
mice: Test of association among loss of cell-surface fibrone-
crin, anchorage independence and tumor-forming ability.
J. Cell Biol. 82:1.
Kaltenhach, J. P., Kaltenbach, M, H. & Lyons, W. B. (1958).
Mgrosin as a dye for differentating live and dead ascites cells.
Exp Cell. Res. 15:112-117.
Kaminska, B., Kaczmarek, L. & Grzelakowska-Szrabert, B. (1990).
The regulation of G0-S transition in mouse T-lymphocytes
by polyamines. Exp Cell. Res. 191: 239-245.
Kane, D. A., Warga, R. M. & Kimmel, С. B. (1992). Mitotic
domains in the early embryo of the zebrafish. Nature 360:
735-737.
Kao, F. T. & Puck, Т. T. (1968). Genetics of somatic mammalian
cells; VII: Induction and isolation of nutritional mutants
in Chinese hamster cells, Proc. Natl. Acad. Sci. USA 60:
1275-1281.
Kao, W.-Y. & Prockop, D. I. (1977). Proline analogue removes
fibroblasts from cultured mixed cell populations Nature 266:
63-64.
Karenberg, J. R. & Freelnader, E. F. (1974). Giemsa techMque
for the detection of sister chromatid exchanges. Chromosoma
48:355-360.
Katdare, М., Osborne, М., Telang, N.-T. (2004). Soy isoflavone
geMstein modulates cell cycle progression and induces
apoptosis in HER-2/neu oncogene expressing human breast
epithehal cells. Int.J. Oncol. 21:809-815.
Katoh, М., Ayabe, F., Norikane, S., Okada, Т., Masumoto, H., Hor-
ike, S., Shirayoshi, Y. & Oshimira, M. (2004). Construction of
a novel human artificial chromosome vector for gene delivery.
Biochem. Biophys. Res. Commun. 321: 280-290.
Kawa, S., Kimura, S., Hakomori, S. & Igarashi. V. (1997). Inhibi¬
tion of chemotactic motility and trans-endothelial migration
of human neutrophils by sphingosin 1-phosphate. FEBS Lett.
420:196-200.
Kawada, H. Fujita. J. Kinjo, K., Matsuzaki, V., Tsuma, М., Mi-
yatake, H., Mugurumma, Y., Tsuboi, K., Itabashi, Y., Ikeda, Y.,
Ogawa, S., Okano, H., Hotta, Т., Ando. K. & Fukuda, K.
(2004). Nonhematopoietic mesenchymal stem cells can be
mobilized and differentiate into cardiomiocytes after myo¬
cardial infarction. Blood 104:3581-3587.
Kawasaki. E. S. (2004). M icroarrays and the gene expression
profile of a single cell. Ann NY Acad Sci 1020:92-100.
Kedinger, М., Simon-Assmann, P., Alexandre. F. & Haffen, K.
(1987). Importance of a fibroblastic support for in vitro differ¬
entiation of intestinal endodermal cells and for their response
to glucocorticoids. CeU Differ. 20:171-182.
Keen, M. J. & Rapson, N. T. (1995), Development of a serum-
free culture medium for the large scale production of a
recombinant protein from a Chinese hamster ovary cell line.
Cytotechnology 17:153-163.
Keilova, H. (1948). The effect of streptomycin on tissue cultures.
Experimentia 4:483.
Keith, W. N. (2003). In situ analysis of telonmerase RNA gene
expression as a marker for tumor progression. Methods Mol.
Med. 75:163-176.
Keles, G. E., Berger, M. S., Lim. R. & Zaheer, A. (1992). Ex¬
pression of glial fibrillary acidic protein in human medul-
loblastoma cells treated with recombinant glia maturation
factor-beta. Oncol. Res. 4:431-437.
Kelley, D. S., Becker, J. E. & Potter, V. R. (1978). Effect of insu¬
lin, dexamethasone and glucagon on the aminoacid transport
ability of four rat hepatoma cell lines and rat hepatocytes in
culture. Cancer. Res. 38:4591-4601.
Kempson, S. A., McAteer, J. A., Al-Mahrouq, H. A., Dousa, T. P.,
Dougherty, G. S. & Evan, A. P. (1989), Proximal tubule char¬
acteristics of cultured human renal cortex epitelium J. Lab.
Clin. Med. 113: 285-296.
Kenworthy, P., Dowrick, P., Baillie-Johnson, H., McCann, B.,
Tsubouchi, H., Arakaki, N., Daikuhara, Y. & Warn, R. M.
(1992). The presence of scatter factor in patterns with meta¬
static spread to the pleura. Br. J. Cancer 66: 243-247.
Kern, P. A., Knedler, A. & Eckel, R. H. (1983). Isolation and
culture of microvascular endothelium from human adipose
tissue./ Clin. Invest 71:1822-1829.
Kerr, D. A., Llade, J., Shamblott, M. J., Maragakis, N. J.,
Irani, D. N., Crawford, Т. О., Krishnan, C., Dike, S., Gear¬
hart, J. D. & Rothstein, J. D. (2003) Human embryonic germ
cell derivatives facilitate motor recovery of rats with diffuse
motor neurone injury./ Neurosci, 23: 5131-5140.
Khan, M. Z., Spandidos, D. A., Kerr, D. J., McNicol, A. М.,
Lang, J. C., de Ridder, L. & Freshney, R. I. (1991). Oncogene
transfection of mink lung cells: effect on growth characteris¬
tics in vitro aid in vivo. Anticancer Res. 11:1343-1348.
662
Список литературы
Kibbey, М. С., Royce, L. S., Dym, М., Baum, В. J. & Klein-
man, H. K. (1992) Glandular-like morphogenesis of the
human submandibulartumor cell line A253 on basement
membrane components. Exp Cell. Res. 198:343-351.
Iscove, N. & Melchers, F. (1978). Complete replacement of
serum by albumin, transferrin and soybean lipid in cultures
of lipopolysacharide-reactive В lymphocytes. J. Exp. Med.
147:923-933.
Ishida, Y., Levin, J., Baker, G., Stenberg, P., Yamada, Y., Sa¬
saki, H., Inoue, T. (1993). Biological and biochemical char¬
acteristics of murine megacaryoblastic cell line L8057. Exp.
Hematol. 21:289-298.
Itagaki, A. & Kimura, G. (1974). TES and HEPES buffers in
mammalian cell cutures and viral studies: Problems of carbon
dioxide requirements. Exp Cell. Res. 83:351-360.
Iyer, V. R., Eisen, М. B., Ross, D. Т., Schuler, G., Moore, Т.,
Lee, J. C. F., Trent., J. М., Staudt, L. М., Hudson, J., Jr., Bo-
guski, M. S., Lashkari, D., Shalon, D., Botstein, D. & Brown,
P. O. (1999). The transcriptional program in the response of
human fibroblasts to serum. Science. 283:83-87.
Izutsu, К. Т., Fatherazi, S., Belton, С. М., Oda, D., Cart¬
wright, F. D. & Kenny, G. E. (1996). Mycoplasm orale infec¬
tion affects К and Cl currents in the HSG salivary gland cell
line. In Vitro Cell Dev. Biol. Anim. 32:361-365.
Jacobs, J. P. (1970). Characteristics of a human diploid cell des¬
ignated MRC-5. Nature 227:168-170.
Jacobs, J. P., Garrett, A. J. & Meron, R. (1979). Characteristics of
a serially propagaterd human diploid cell designated MRC-9.
J. Biol Stand. 1:113-122.
Jaffe, E. A., Nachman, R. L., Becker, G. C. & Ninick, C. R. (1973).
Culture of human endotelial cells derived from umbillical
veins .J. Clin. Invest. 52:2745-2756.
Jain, D., Ramasubramamanyan, K., Gould, S., Lenny, A., Cande-
lore, М., Tota, М., Strader, C., Alves, K., Cuca, C., Tung, J. S.,
Hunt, G., Junker, B., Buckland, В. C. & Silberklang, M.
(1991). In Sspeir, R. E., Griffits, J. B. & Meignier, B. (eds.)
Production of biologicals from animal cells in culture. Oxford,
U.K., Butterworth-H^inemann, pp. 345-351.
Jain, R. K., Schlenger, K., Hockel, M. & Yuan, F. (1997). Quanti¬
tative angiogenesis assays: Progress and problems. Nat. Med.
3:1203-1208.
Janssen, M. L., van Leewen, М. B., Scholtmeijer, K., van Koo-
ten, T. G., Dijkhuizen, L. & Wosten, H. A. (2003). Coating
with genetic engeneering hydrophobin promotes growth
of fibroblasts on a hydrophobic solid. Biomaterials. 23:
4847-4854.
Jat, P. S., Noble, M. D., Ataliotics, P., Tanaka, Y., Yannoutsos, N.,
Larsen, L. & Kiossis, D. (1991). Direct Derivation of confi-
detially immortal cell lines from an H-2 Kb-tsA58 transgenic
mouse. Proc. Natl. Acad. Sci. USA 88:5096-5100.
Jeffreys, A. J., Wilson, V. & Thein, S. L. (1985). Individual specific
«fingerprints» of human DNA. Nature. 316: 76-79.
Jeng, Y.-J., Watson, C. S. & Thomas, M. L. (1994). Identification
of vitamin D-stimlated phosphatase in IEC-6 cells, a rat small
intestine crypt cell line. Exp. Cell Res. 212:338-343.
Jenkins, N. (ed.) (1992) Growth factors, a practical approach.
Oxford, U.K., IRL Press at Oxford University Press.
Jessell, Т. M. & Melton, D. A. (1992). Diffusible factors in verte¬
brate embryoMc induction, Cell 68:257-270.
Jin, D. I., Lee, S. H., Choi, J. H., Lee, J. S., Lee, J. E., Park, K. W.,
Seo, J. S. (2003). Targeting efficiency of a-l,3-galactosyl
transferase gene in pig fetal fibroblast cells. Exp. Mol Med
35:572-577.
Jinard, F., Sergent-Engelen, Т., Trouet, A., Remade, C. & Schnei¬
der, Y.-J. (1997). Compartment coculture of porcine arterial
endothelial and smooth muscle cells on a microporous mem¬
brane. In Vitro Cell Dev. Biol. AMm. 33:92-103.
Johnson, G. D. (1989). Immunofluorescence. In Catty, D. (ed.).
Antibodies. Vol. II: A practical approach. Oxford, U.K., IRL
Press ar Oxford UMvercity Press, pp. 179-200.
Jones, E. L., Matsui, W. H. & Smith, B. D. (2004), Cancer stem
cells: are we missing the target? J. Natl. Cancer Inst. 96:
583-585.
Jones, E. L. & Gregory, J. (1989). Immunoperoxidase methods,
In Catty, D. (ed.). Antibodies. Vol. II: A practical approach.
Oxford, U.K., IRL Press ar Oxford University Press,
pp. 155-177.
Joukov, V., Kaipainen, A., Jeltsch, М., Pajusola, K., Olofsson, B.,
Kumar, V., Eriksson, U. & Alitalo, K. (1997). Vascular endo¬
thelial growth factors VEGF-B and VTGF-C./. Cell Physiol.
173:211-215.
Jozan, S., Roche, H., Cheutin, J., Carton, M. & Salles, B. (1992).
New human ovarian cell line OVCCRl/sf in serum-free me¬
dium. In Vitro Cell Dev. Biol. 28A: 687-689.
Kaartinen. L., Nettesheim, P., Adler, К. B. & Randell, S. H. (1993).
Rat tracheal epithelial cell differentiation in vitro. In Vitro Cell
Dev. Biol. 29A: 481-492.
Kahn, P. & Shin, S.-L. (1979). Cellular tumorigeMcity in nude
mice: Test of association among loss of cell-surface fibrone-
crin, anchorage independence and tumor-forming ability.
J. Cell Biol. 82:1.
Kaltenhach, J. P., Kaltenbach, M, H. & Lyons, W. B. (1958).
Mgrosin as a dye for differentating live and dead ascites cells.
Exp Cell. Res. 15:112-117.
Kaminska, B., Kaczmarek, L. & Grzelakowska-Szrabert, B. (1990).
The regulation of G0-S transition in mouse T-lymphocytes
by polyamines. Exp Cell. Res. 191:239-245.
Kane, D. A., Warga, R. M. & Kimmel, С. B. (1992). Mitotic
domains in the early embryo of the zebrafish. Nature 360:
735-737.
Kao, F. T. & Puck, Т. T. (1968). Genetics of somatic mammalian
cells; VII: Induction and isolation of nutritional mutants
in Chinese hamster cells, Proc. Natl. Acad. Sci. USA 60:
1275-1281.
Kao, W.-Y. & Prockop, D. I. (1977). Proline analogue removes
fibroblasts from cultured mixed cell populations Nature 266:
63-64.
Karenberg, J. R. & Freelnader, E. F. (1974). Giemsa techMque
for the detection of sister chromatid exchanges. Chromosoma
48:355-360.
Katdare, М., Osborne, М., Telang, N.-T. (2004). Soy isoflavone
genistein modulates cell cycle progression and induces
apoptosis in HER-2/neu oncogene expressing human breast
epithehal cells. Int.J. Oncol. 21:809-815.
Katoh, М., Ayabe, F., Norikane, S., Okada, Т., Masumoto, H., Hor-
ike, S., Shirayoshi, Y. & Oshimira, M. (2004). Construction of
a novel human artificial chromosome vector for gene delivery.
Biochem. Biophys. Res. Commun. 321:280-290.
Список литературы
663
Kawa, S., Kimura, S., Hakomori, S. & Igarashi. V. (1997). Inhibi¬
tion of chemotactic motility and trans-endothelial migration
of human neutrophils by sphingosin 1-phosphate. FEBS Lett.
420:196-200.
Kawada, H., Fujita. J., Kinjo, K., Matsuzaki, V., Tsuma, М.,
Miyatake, H., Mugurumma, Y., Tsuboi, K., Itabashi, Y.,
Ikeda, Y., Ogawa, S., Okano, H., Hotta, Т., Ando. K. & Fu-
kuda, K. (2004). Nonhematopoietic mesenchymal stem cells
can be mobilized and differentiate into cardiomiocytes after
myocardial infarction. Blood 104: 3581-3587.
Kawasaki, E. S. (2004). M icroarrays and the gene expression
profile of a single cell. Ann NY Acad Sci 1020:92-100.
Kedinger, М., Simon-Assmann, P., Alexandre. F. & Haffen, K.
(1987). Importance of a fibroblastic support for in vitro differ¬
entiation of intestinal endodermal cells and for their response
to glucocorticoids. Cell Differ. 20:171-182.
Keen, M. J. & Rapson, N. T. (1995), Development of a serum-
free culture medium for the large scale production of a
recombinant protein from a Chinese hamster ovary cell line.
Cytotechnology 17:153-163.
Keilova, H. (1948). The effect of streptomycin on tissue cultures.
Experimentia 4:483.
Keith, W. N. (2003). In situ analysis of telonmerase RNA gene
expression as a marker for tumor progression. Methods Mol.
Med. 75:163-176.
Keles, G. E., Berger, M. S., Lim, R. & Zaheer, A. (1992). Ex¬
pression of glial fibrillary acidic protein in human medul-
loblastoma cells treated with recombinant glia maturation
factor-beta. Oncol. Res. 4:431-437.
Kelley, D. S., Becker, J. E. & Potter, V. R. (1978). Effect of insu¬
lin, dexamethasone and glucagon on the aminoacid transport
ability of four rat hepatoma cell lines and rat hepatocytes in
culture. Cancer. Res. 38:4591-4601.
Kempson, S. A., McAteer, J. A., Al-Mahrouq, H. A., Dousa, T. P.,
Dougherty, G. S. & Evan, A. P. (1989), Proximal tubule char¬
acteristics of cultured human renal cortex epitelium J. Lab.
Clin. Med. 113:285-296.
Kenworthy, P., Dowrick, P., Baillie-Johnson, H., McCann, B.,
Tsubouchi, H., Arakaki, N., Daikuhara, Y. & Warn, R. M.
(1992). The presence of scatter factor in patterns with meta¬
static spread to the pleura. Br. J. Cancer 66:243-247.
Kern, P. A., Knedler, A. & Eckel, R. H. (1983). Isolation and
culture of microvascular endothelium from human adipose
tissue./ Clin. Invest 71:1822-1829.
Kerr, D. A., Llade, J., Shamblott, M. J., Maragakis, N. J.,
Irani, D. N., Crawford, Т. O., Krishnan, C., Dike, S.,
Gearhart, J. D. & Rothstein, J. D. (2003) Human em¬
bryonic germ cell derivatives facilitate motor recovery
of rats with diffuse motor neurone injury./. Neurosci, 23:
5131-5140.
Khan, M. Z., Spandidos, D. A., Kerr, D. J., McNicol, A. М.,
Lang, J. C., de Ridder, L. & Freshney, R. I. (1991). On¬
cogene transfection of mink lung cells: effect on growth
characteristics in vitro aid in vivo. Anticancer Res. 11:
1343-1348.
Kibbey, М. C., Royce, L. S., Dym, М., Baum, B. J. & Klein-
man, H. K. (1992) Glandular-like morphogenesis of the
human submandibulartumor cell line A253 on basement
membrane components. Exp Cell. Res. 198: 343-351.
Kiefer, J., Alexander, A. & Farach-Carson, М. C. (2004). Type I
collagen-mediated changes in gene expression and function of
prostate cancer cells. Cancer. Treat. Res. 118:101-124.
Kimhi, Y. H., Palfrey, C. & Spector, I. (1976). Maturation of
neuroblastoma cells in the presence of dimethyl sulphoxide.
Proc. Natl. Acad. Sci. USA 73:462-466.
Kingsbury, A., Gallo, V., Woodhams, P. L. & Balazs, R. (1985)
Survival, morphology and adhesion properties of cerebellar
interneurons cultured in chemically defined and serum-
supplemented medium. Dev. Brain Res 17:17-25.
Kinoshita, Т., Miyajima, A. (2002). Cytokine regulation of liver
development. Biochem. Biophys. Acta 1592:303-312.
Kinsella, J. L., Grant, D. S., Weeks, B. S. & Kleinman, H. K.
(1992). Protein kinase С regulates endothelial cell tube for¬
mation on basement membrane matrix, Matrigel. Exp Cell.
Res. 199:56-62.
Kirkland, S. C. & Bailey. I. G. (1986). Establishment and charac¬
terisation of six human colorectal adenocarcinoma cell lines.
Br. J. Cancer 53:779-785.
Klagsbrun, M. & . Baird, A. (1991). A dual receptor system is
required for basic fibroblast growth factor activity. Cell 67:
229-231.
Klein, B., Pastink, A., Odijk, H., Westerveld, A. & van der Eb, A. J.
(1999). Transormation and immortalization of diploid xeroderma
pigmentosum fibroblasts. Curr Opin Biotecknol 191:256-262.
Kleinman, H. K., Philp, D. & Hoffman, M. P. (2003). Role of the
extracellular cell matrix in morpogenesis. Curr Opin Biotech-
nol. 14:526-532.
Kleinman, H. K., McGoodwin, E. B., Rennard, S. I., Martin, G. R.
(1979). Preparation of collagen substrates for cell attachment:
Effect of collagen concentration and and phosphate buffer.
Anal Biochem 94:308-312.
Kleinsmith, L J. & Pierce, G. B. (1964). CancerResTA: 1544-1551.
Klement, G., Scheirer, W. & Katinger, H. W. (1987). Construc¬
tion of a large scale membrane reactor system with different
compartments for cells, medium and product. Dev. Biol. Stand.
66:221-226.
Klevjer- Anderson, P. & Buehrig, G. C. (1980). Effect of hormones
on growth rates of malignant and nonmalignant human mam¬
mary epithelia in cell culture. In Vitro 16:491-501.
Klingel, S., Rothe, G., Kellermann, W. & Valet, G. (1994). Flow
cytometric determination of cysteine and serine proteinase
activities in living cells with rhodamine 110 substrates. Metod
Cell. Biol. 41:449-459.
Khnger, R. Y. & Niklason, L. E. (2005). Tissue engineered blood
vessels. In Vuhjak-Novakovic, G. & Freshney, R. I. (eds.),
Culture of celb for tissue ingineering Hoboken, NJ, Wiley-Liss
(in press).
Kloppinger, М., Fertig, G., Fraune, F. & Miltenburger, H. G.
(1991). High cell density perfusion culture of insect cells
for production of baculovirtus and recombinant protein. In
Speir, R. E., Griffiths, J. B. & Meignier, B. (eds.), Production
of biologicab from animal celb in culture. Oxford, U.K. But-
terworth-heinemann, pp. 470-474.
Klug, C. A. & Jordan, С. T. (2002). Hematopoetic stem cell proto¬
cob Clifton, N.J. Humana Press.
Knazek, R. A., Gullino, P., Kohler, P. O. & Dedrick, R. (1972).
Cell culture on artificial capillaries: An approach to tissue
growth in vitro. Science 178: 65-67.
664
Список литературы
Knazek, R. A., Kohler, Р. О. & Gullino, P. M. (1974). Hormone
production by cells grown in vitro on artificial capillaries. Exp
Cell. Res. 84: 251.
Knedler, A. & Ham, R. G. (1987). Optimized madium for clonal
growth of human microvascular endothelial cells with mini¬
mal serum. In Vitro 23 (7): 481-491.
Kneuchel, D. J. & Masters, J. R. W. (1999). Bladder Cancer. In
Masters, J. R. W. & Palsson, B. (eds.). Human cells culture,
Vol. I, Dordreht, Kluwer, pp. 213-230.
Knight. D. J. & Breheny, D. (2002). Alternatives to annual testing in
the safety evaluation of products. Altem. Lab. Anim, 30:7-22.
Knott. C., L., Kuus-Reichel, K., Liu, R. & Wolfert, R. L. (1997).
Development of antibodies for diagnostic assays. In Price, C. &
Newman, D. (eds.), Principles and practice of immunoassay.
2nd ed. New York, Stockton Press, pp. 36-64.
Knowles, В. B., Howe, С. C. & Aden, D. P. (1980). Human hepato¬
cellular carcinoma cell lines secrete the major plasma proteins
and hepatitis В surface antigen. Science 209:497-499.
Kohler, G. & Milstein, С (1975). Continuous cultures of fused cells se¬
creting antibody of predefined specificity. Nature 256:495-497.
Kohlhepp, E. A., Condon. М. E. & Hamburger, A. W. (1987).
Recombinant human interferon-a enhancement of retinoic
acid induced differentiation of HL-60 cells. Exp. Hematol,
15:414-418.
Kondo, S., Kooshesh, F. & Sauder, D. N. (1997). Penetration of
keratinocyte-derived cytokines into basement membrane.
J. Cell. Physiol. 171:190-195.
Kondo, T. & Raff, M. (2000). Oligodendrocyte precursor cells
reprogrammed to become rnultipotential CNS stem cells.
Science 289:1754-1757.
Kondo, T. & Raff, M. (2004). Chromatin remodeling and histone
modification in the conversion of oligodendrocyte precursors
to neural stem cells. Genes Dev 18: 2963-72.
Kono, T. (1997). Nuclear transfer and reprogramming Rev.
Reprod. 2: 7480.
Koopman, L. A., Szuhai, K., van Eendenhurg, J. D., Bezrook-
ove, V., Kenter, С. C., Schuuring, E., Tanke, H. & Fleuren, G. J.
(1999). Recurrent integration of human papillomaviuses 16,
45 and 67 near translocation breakpoints in new cervical
cancer cell lines. Cancer Res 59:5615-5624.
Kopper, L. & Hajdu, M. (2004). Tumor stem cells. Pathol. Oncol.
Res. 10: 69-73.
Koren, H. S., Handwerger, B. S. & Wunderlich. J. R. (1975).
Identification of macrophage-like characteristics in a murine
tumor cell line J. Immunol 114:894-897.
Korenberg, J. R., Chen, X. N., Adams, M. D. & Venter, J. C.
(1995). Toward a cDNA map of the human genome. Genomic
29:364-370.
Kosehier, F. J., Roth, R. N., Wallace, K. A., Curren, R. D. &
Harbell, J. W. (1997). A comparison of three-dimensional hu¬
man skin models to evaluate the dermal irritation of selected
petroleum products. In Vitro Toxicol, 10:391-405.
Kreisberg, J. L., Sachs, C., Pretlow, T. G. E. & Me Guire, R. A.
(1977). Separation of proximal tubule cells from suspensions
of ran kidney cell by free-flow electrophoresis ./. Cell. Physiol.
93: 169-172.
Krotz, F., Sohn, H. Y., Gloe, Т., Plank, C. & Pohl, U. (2003). Mag-
netofection potentiates gene delivery to cultured endothelial
cells./ Vase Res. 40:425-34.
Krupp, W., Geiger, K., Schobe, R., Siegert, G. & Froster, U. G.
(2004). Cytogenetic and molecular cytogenetic analyses in
diffuse astrocytomas. Cancer Genet. Cytogenet. 153:32-38.
Kruse, R. H., Puckett, W. H. & Richardson, J. H. (1991). Biologi¬
cal safety cabinetry. Clin. Microbiol. Rev. 4: 207-241.
Kruse, P. F., Jr., Keen, L. N. & Whittle, W. L. (1970). Some
distinctive characteristics of high density perfusion cultures
of diverse cell types. In Vitro 6: 75-78.
Kucherlapati, R. & Skoultchi, A. (1984). Introduction of puri¬
fied genes into mammalian cells. CRC Crit. I Rev. Biochem.
16:349-379.
Kujoth, G. C., Fahl, W. E. (1997). C-sis/platelet-derived growth
factor-В promoter requirements for induction during the
12-O-tetradecanoylphorbol- 13-acetate-mediated megakaryo-
blastic differentiation of K562 human erythroleukemia cells.
Cell Growth Differ. 8:963-977.
Kuriharcuch, W. & Green, H. (1978). Adipose conversion of
3T3 cells depends on a serum factor. Proc. Natl. Acad. Sci.
75:6107-6110.
Kurtz, J. W. & Wells, W. W. (1979). Automated fluorometric
analysis of DNA, protein, and enzyme activities: Application
of methods in cell culture. Anal. Biochem. 94:166.
Labarca, C. & Paigen, K. (1980). A simple, rapid, and sensitive
DNA assay procedure. Anal Biochem. 102: 344-352.
Laferte, S. & Loh, L. C. (1992). Characterizationof a family
of structurally related glycoproteins expressing beta 1-6-
branched asparagines-linked oligosaccharides in human colon
carcinoma cells. Biochem. J. 283:192-201.
Lag, М., Becher, R., Samuelsen, J. Т., Wiger, R., Refsnes, М., Huit-
field, H. S. & Schwarze, P. E. (1996). Expression of CYP2B1
in freshly isolated and proliferating cultures of epithelial rat
lung cells. Exp. Lung Res. 22: 627-649.
Lamb, R. F., Hennigan, R. F., Katsanakis, K. D., Turnbull, K.,
MacKenzie, E. D., Bimie, G. D. & Ozanne, B. W. (1997).
AP-1-mediated invasion requires increased expression of the
hyaluronan receptor, CD44. Mol. Cell Biol. 17:963-976.
Lan, S., Smith, H. S. & Stampfer, M. R. (1981). Clonal growth
of normal an malignant human beast epithelia./. Surg. Oncol.
18:317-322.
Lane, E. B. (1982). Monoclonal antibodies provide specific intra¬
molecular markers for the study of tonofilament organisation.
J. CeUBol. 92:665-673.
Lang, M. S., Hovenkamp, E., Saelkoul, H. F., Knegt, P. & van
Ewijk, W. (1995). Immunotherapy with mononclonal antibod¬
ies directed against the immunosuppressive domain of pl5E
inhibits tumour growth. Clin. Exp. Immunol. 102:468-475.
Langdon, S. P. (2004). Characterization and authentication
of cancer cell lines: an overview. Methods Mol. Med. 88:
33-42.
Lange, W., Brugger, W., Rosenthal, F. М., Kanz, L. & Linde-
mann, A. (1991). The role of cytokins in oncology. Int. Cell
Cion. 9:252-273.
Larin, Z. & Mejia, J. E. (2002). Advances in human artificial
chromosome technology. Trends Genet. 18:313-319.
Larsen, М. C., Brake, P. B., Pollenz, R. S & Jecoate, C. R. (2004).
Linked expression of Ah receptor, ARNT, CYP1 Al, and CY-
P1В1 in rat mammary epithelia, in vitro, is each substantially
elevated by specific extracellular matrix interactions that
precede branching morphogenesis. Toxicol. Sci. 82:46-61.
Список литературы
665
Lasfargues, Е. Y. (1973). Human mammary tumors. In Kruse, P.,
Paterson, М. K. (eds):« Tissue Culture Methods and Applica¬
tions.» New York, Academic Press, pp. 45-50.
Laslett, A. L., Filipczyk, A. A. & Pera, M. F. (2003). Character¬
ization and culture of human embryonic stem cells. Trends
Cardiovasc. Med. 13: 295-301.
Lasnitzki, I. (1992). Organ culture. In Freshney, R. I. (ed.),
Animal cell culture, a practical approach. Oxford, U.K., IRL
Press at Oxford University Press, pp. 213-261.
Latt, S. A. (1973). Microfluorometric detection of DNA replica¬
tion in human metaphase chromosomes. Proc. Natl. Acad. Sci.
USA 70:3395-3399.
Latt, S. A. (1981). Sister chromatid exchange formation. Annu.
Rev. Genet. 15:11-55.
Laug, W. E., Tokes, Z. A., Benedict, W. F. & Sorgente, N. (1980).
Anchorage independent growth and plasminogen activa¬
tor production by bovine endothelial cells .J. Cell Biol. 84:
281-293.
Lavappa, K. S. (1978). Survey of ATCC stocks of human cell lines
for HeLa contamination. In Vitro 14 (5): 469-475.
Law, L. W., Dunn, Т. B., Boyle, P. J. & Miller, J. H. (1949).
Observations on the effect of a folic acid antagonist on
transplantable lymphoid leukemia in mice. J. Natl. Cancer
Inst. 10:179-192.
Leake, R. E., Freshney, R. I. & Munir, I. (1987). Steroid responses
in vivo and in vitro. In Green, B. & Leake, R. E. (eds.), Steroid
hormones, a practical approach, Oxford, U.K., IRL Press at
Oxford University Press, pp. 205-218.
Lebeau, М. M. & Rowley, J. D. (1984). Heritable fragile sites in
cancer. Nature 308: 607-608.
Lechardeur, D., Schwartz, B., Paulin, D. & Scherman, D. (1995).
Induction of blood-brain barrier differentiation in a rat brain-
derived endothelial cell line. Exp. Cell Res. 20:161-170.
Lechner, J. F. & LaVeck, M. A. (1985). A serum free method for
culturing normal human bronchial epithelial cells at clonal
density .J. Tissue Cult. Methods 9:43-48.
Lechner, J. F., Haugen, A., Autrup, H., McClendon, I. A.,
Trump, B. F. & Harris, С. C. (1981). Clonal growth of epi¬
thelial cells from normal adult human bronchus. Cancer Res.
41:2294-2304.
Leder, A. & Leder, P. (1975). Butyric acid, a potent inducer of
erythroid differentiation in cultured erythroleukemic cells.
Cell 5:319-322.
Le Douarin, N. М., Creuzet, S., Couly, G., Dupin, E. (2004).
Neural crest cell plasticity and its limits. Development 131:
4637-50.
Lee, M. W., Choi, J., Yang, M. S., Moon, Y. J., Park, J. S.,
Kim, H. C., Kim, Y. J. (2004). Mesenchymal stem cells from
cryopreserved human umbilical cord blod. Biochem. Biophys
Res. Commun. 320:273-8.
Lee, D. R., Kaproth, М. T. & Parks, J. E. (2001). In vitro produc¬
tion of haploid germ cells from or frozen-thawed testicular
cells of neonatal bulls. Biol. Reprod. 65:873-878.
Lee, Т. H., Baik, M. G., Im, W. B., Lee, C. S., Han, Y. М., Kim, S. J.,
Lee, К. K. & Choi, Y. J. (1996). Effects of EHS matrix on
expression of transgenes in HC11 cells. In Vitro Cell Dev. Biol.
Anim. 32:454-456.
Lehle, K., Buttstaedt,J. & Bimbaum, D. E. (2003). Expression of
adhesion molecules and cytokines in vitro by endothelial cells
seeded on various polymer surfaces coated with titaniumcar-
boxonitride.y. Biomed. Mater. Res. 65A: 393-401.
Leibo, S. P., Mazur, P. (1971) The role of cooling rates in low-
temperature preservation. Cryobiology, 8:447-52.
Leibovitz, A. (1963). The growth and maintenance of tissue cell
cultures in free gas exchange with the atmosphere. An.J. Hyg.
78:173-183.
Leigh, I. M. & Watt, F. M. (1994). Keratinocyte methods. Cam¬
bridge, U.K., Cambridge University Press.
Leighton, J. (1991). Radial histophysiologic gradient culture
chamber rationale and preparation. In vitro Cell Dev. Biol.
27 A: 786-790.
Leighton, J., Mark, R. & Rush, G. (1968). Patterns of three-di¬
mensional growth in collagen coated cellulose sponge: Carci¬
nomas and embryonic tissues. Cancer Res. 28: 286-296.
Lemare, F., Steimberg, N., Le Griel, C., Demignot, S. & Adolphe,
M. (1998). Dedifferentiated chondrocytes cultured in algi¬
nate beads: Restoration of the differentiated phenotype and
of the metabolic response to intereukin-ip./. Cell Physiol.
176: 303-313.
Lennon, D. & Caplan, A. (2005). Mesenchymal stem cells. In
Vunjak-Novakovic, G. & Freshney, R. I. (eds.), Culture of cells
for tissue engineering. Hoboken, NJ, Wiley-Liss (In press).
Leobon, B., Garcin, I., enasche, P., Vilquin, J. Т., Audinat, E. &
Charpak, S. (2003). Myoblats transplanted into rat infarcted
myocardium are functionally isolated from their host. Proc.
Natl. Acad. Sci. USA 100:7808-7811.
Le Poole, I. C., van den Berg, F. М., van den Wijngaard, R. М.,
Galloway, D. A., van Amstel, P. J., Buffing, A. A, Smiths, H. L.,
Westerhof, W. & Das, P. K. (1997). Generation of a human
melanocyte cell line by introduction of HP 16 E6 and E7 genes.
In Vitro Cell Dev. Biol. Anim. 33:42-49.
Le Roith, D. & Raizada, М. K. (eds.) (1989). Molecular and cel¬
lular biology of insulin-like growth factors and their receptors.
New York, Plenum.
Lesuffleur, Т., Barbat, A., Dussaulx, E. & Zweibaum, A. (1990).
Growth adaptation to methotrexate of HT-29 human colon
carcinoma cells is associated with their ability to differentiate
into columnar absorptive and mucus-secreting cells. Cancer
Res. 50:6334-6343.
Lever, J. (1986). Expression of differentiated functions in kidney
epithelial cell lines. Min. Elec. Metab. 12:14-19.
Levi-Montalcini, R. (1966). The nerve growth factor: its mode of
action on sensory and sympathetic nerve cells. Harvey Lect.
60:217-259.
Levi-Montalcini, R. C. P. (1979). The nerve growth factor. Sci.
Am. 240:68.
Levin, D. B., Wilson, K., Valadares de Amorim, G., Webber, J.,
Kenny, P. & Kusser, W. (1995). Detection of p53 mutations
in benign and dysplastic nevi. Cancer Res. 55:4278-4282.
Levine, E. M. & Becker, B. G. (1977). Biochemical methods for
detecting mycoplasma contamination. In McGarrity, G. Т.,
Murphy, D. G. & Nichols, W. W. (eds.), Mycoplasma infection
of cell cultures. New York, Plenum Press, p. 87-104.
Ley, K. D. & Tobey, R. A. (1970). Regulation of initiation of
DNA synthesis in Chenese hamster cells; II; Induction of
DNA synthesis and cell division by isoleucine and glutamine
in Gj-arrested cells in suspension culture./. Cell Biol. 47:
453-459.
666
Список литературы
Li, Y., Decherchi, P. & Raisman, G. (2003a). Transplantation of
olfactory ensheathing cells into spinal cord lesions restores
breathing and climbing./ Neurosci. 3: 727-731.
Li, Y., Foster, W., Deasy, В. М., Chan, Y., Prisk, V., Tang, Y.,
Cummins, J. & Huard, J. (2004). Transforming growth fac-
tor-betal induces the differentiation of myogenetic cells into
fibrotic cells in injured skeletal muscle: a key event in muscle
fibrogenesis. Am.J. Pathol. 164:1007-1019.
Li, Y. М., Schacher, D. H., Liu, Q., Arkins, S., Rebeiz, N., Mc-
Cusker, R. H., Jr., Dantzer, R. & Kelley, K. W. (1997). Regula¬
tion of myeloid growth and differentiation by the insulin-like
growth factor I receptor. Endocrinology 138: 362-368.
Li, A. P., Roque, М. М., Beck, D. J. & Kaminski, D. L. (1992).
Isolation and culturing of hepatocytes from human livers.
J. Tissue Cult. Methods 14:139-146.
Li, Y. et al. (2003b). Transplanted olfactory ensheathing cells
promote regeneration of cut adult rat optic nerve axons.
J. Neurosci. 23:2000-2002.
Lidington, E. A., Rao, R. М., Marelli-Berg, F. М., Jat, P. S., Has-
kard, D. O. & Mason, J. C. (2002). Am.J. Physiol. Cell Physiol.
282: C67-C74.
Liebermann, D. & Sachs, L. (1978). Nuclear control of neurite in¬
duction in neuroblastoma cells. Exp. Cell Res. 113:383-390.
Lillie, I. H., MacCallum, D. K., Jepsen, A. (1980). Fine structure
of subcultivated stratified squamous epithelium grown on
collagen rafts. Exp. Cell Res. 125:153-165.
Lim, G., Karaskova, J., Vukovic, B., Bayani, J., Beheshti, B., Ber-
nardini, М., Squire, J. A. & Zielenska, M. (2004). Combined
spectral karyotyping, multicolor banding, and microarray
comparative genomic hybridization analysis provides a detailed
characterization of complex structural chromosomal rearrange¬
ments associated with gene amplification in the osteosarcoma
cell line MG-63. Cancer Genet. Cytogenet. 153:158-164.
Limat, A., Breitkreutz, D., Thiekotter, G., Klein, E. C., Braa-
then, L. R, Hunziker, T. & Fusenig, N. E. (1995). Formation of
a regular neo-epidermis by cultured human outer roor sheath
cells grafted on nude mice. Transplantation 59:1032-1038.
Limat, A., Hunziker, Т., Boillat, C., Bayreuther, K. & Noser, F.
(1989). Post-mitotic human dermal fibroblasts efficiently
support the growth of human follicular keratinocytes./. Invest.
Dermatol. 92:758-762.
Limat, A., Mauri, D. & Hunziker, T. (1996). Successful treatment
of chronic leg ulcers with epidermal equivalents generated
from cultured autologous outer root sheath cells .J. Invest.
Dermatol. 107: 128-135.
Lin, С. C. & Uchida, I. A. (1973). Fluorescent banding of chromo¬
somes (Q-bands). In Kruse, P. F. & Patterson, М. K. (eds.),
Tissue culture methods and applications. New York, Academic
Press, pp. 778-781.
Lin, M. A., Latt, S. A. & Davidson, R. L. (1974). Identification of
human and mouse chromosomes in human-mouse hybrids by
centromere fluorescence. Exp. Cell Res. 87:429-433.
Linser, P. & Moscona, A. A. (1980). Induction of glutamine
synthetase in embryonic neural retina-localization in Muller
fibers and dependence on cell interaction. Proc. Natl. Acad.
Sci. USA 76:6476-6481.
Liotta, L. (1987). The role of cellural proteases and their inhibi¬
tors in invasion and metastasis: Introductionary overview.
Cancer Metastasis Rev. 9: 285-287.
Lippincott-Schwartz, J., Glickman, J., Donaldson, J. G., Rob¬
bins, J., Kreis, Т. E., Seamon, К. B., Sheetz, M. P. & Klaus-
ner, R. D. (1991). Forskolin inhibits and reverses the effects
of Brefeldin A on Goldi morphology by a с AMP-independent
mechanism./. Cell Biol.112:567.
Littlefield, J. W. (1964a). Selection of hybrids from matings of
fibroblasts in vitro and their presumed recombinants. Science
145:709-710.
Littlefield, J. W. (1964b). Three degrees of guanylic acid pyro-
phosphorylase deficiency in mouse fibroblasts. Nature 203:
1142-1144.
Litwin, J. (1973). Titanum disks. In Kruse, P. F. & Patter¬
son, М. K. (eds.), Tissue culture methods and applications. New
York, Academic Press, pp. 383-387.
Liu, T. F., Cohen, K. A., Willingham, М. C., Tatter, S. B.,
Puri, R. K. & Frankel, A. E. (2003). Combination fusion
protein therapy of refactory brain tumors: demonstration of
efficacy in cell culture./ Neurooncol. 65:77-85.
Liu, L., Delbe, J., Blat, C., Zapf, J. & Harel, L. (1992). Insulin
like growth factor binding protein (IGFBP-3), an inhibitor
of serum growth factors other than IGF-I and -II./. Cell
Physiol. 153:15-21.
Liu, М., Xu, J., Souza, P., Tanswell, B., Tanswell, A. K. & Post, M.
(1995). The effect of mechanical strain on fetal rat lung cell
proliferation: Comparison of two- and three-dimensional
culture systems. In Vitro Cell Dev. Biol. Anim. 31:858-866.
Lopez-Casillas, F., Wrana, J. L. & Massague, J. (1993). Betagly-
can presents ligand to the TGF-p signaling receptor. Cell 73:
1435-1444.
Lotan, R. & Lotan, D. (1980). Stimulation of melanogenesis in
a human melanoma cell line by retinoids. Cancer Res. 40:
33-45.
Lounis, H., Provencher, D., Godbout, C., Fink, D., Milot, M.-J. &
Mes-Masson, A.-M (1994). Primary cultures of normal and
tumoral ovarian epithelium: A powerful tool for basic molecu¬
lar studies. Exp. Cell Res. 215:303-309.
Lovelock, J. E. & Bishop, M. W. H. (1959). Prevention of freez¬
ing damage to living cells by dimethyl sulphoxide. Nature
183:1394-1395.
Lowe, К. C., Davey, M. R. & Power, J. B. (1998). Perfluoro-
chemicals: their applications and benefits to cell cultur. Trends
Biotechnol. 16:272-277.
Luikart, S. D., Maniglia, C. A., Furcht, L. Т., McCarthy, J. B. &
Oegama, T. R. (1990). A heparan sulphate-containing fraction
of bone marrow stroma induces maturation of HL-60 cell in
vitro. Cancer Res. 50:3781-3785.
Lundqvist, М., Mark, J., Funa, K., Heldin, N. E., Morstyn, G., Wed-
del, B., Layton, J. & Oberg, K. (1991). Characterisation of a
cell line (LCC-18) from a cultured human neuroendocrine-dif-
ferentiated colonic carcinoma. Eur. J. Cancer 12:1662-1668.
Lutz, M. P., Gaedicke, G. & Hartmann, W. (1992). Large-scale
cell separation by centrifugal elutriation. Anal. Biochem. 200:
376-380.
Maas-Szabowski, N., Stark, H. J. & Fusenig, N. E. (2000). Kera¬
tinocyte growth regulation in defined organotypic cultures
through IL-1-induced KGF expression in resting fibroblasts.
J. Invest. Dermatol. 114:1075-1084.
Maas-Szabowski, N., Stark, H. J. & Fusenig, N. E. (2002). Cell
interaction and epithelial differentiation. In Freshney, R. I. &
Список литературы
667
Freshney, М. G. (eds.), Culture of epithelial cells. 2nd ed. Hobo¬
ken, NJ, Wiley-Liss, pp. 31-63.
Maas-Szabowski, N. & Fusenig, N. E. (1996). Interleukin-1-
induced growth factor expression in postmitotic and resting
fibroblasts./ Invest, Dermatol. 107:849-855.
Maas-Szabowski, N., Fusenig, N. E. & Shimotoyodome, A. (1999).
Keratinocyte growth regulation in fibroblast co-cultures via a
double paracrine mechanism./. Cell Sci. 112:1843-1853.
McDonald, С. М., Freshney, R. I., Hart, E. & Graham, D. I.
(1985). Selective control of human glioma cell proliferation
by specific cell interaction. Exp. Cell Biol. 53:130-137.
Mace, K., Gonzalez, F. J., McConnel, I. R., Gamer, R. C., Avanti, O.,
Harris, С. C. & Pfeifer, A. M. A. (1994). Activation of promuta¬
gens in a human bronchial epithelial cell line stably expressing
human cytochroe P450 1A2. Mol. Carcinogen. 11:65-73.
MacGregor, G. & Caskey, C. (1989). Constmction of plasmids
that express. E. coli beta-galactosidase in mammalian cells.
Nucleic Acids Res. 17:2363-2365.
Maciag, Т., Cerondolo, J., Isley, S., Kelley, P. R. & Forand, R.
(1979). Endothelial cell growth factor from bovine hypo-
thalamus-identification and partial characterization. Proc.
Natl. Acad. Sci. USA 76:5674-5678.
Macieira-Coelho, A. (1973). Cell cycle analysis; A: Mammalian
cells. In Kruse, P. F. & Patterson, М. K. (eds.), Tissue cul¬
ture methods and applications. New York, Academic Press,
pp. 412-422.
Macklis, J. D., Sidman, R. L. & Shine, H. D. (1985). Cross-linked
collagen surface for cell culture that is stable, uniform, and opti¬
cally superior to conventional surfaces. In Vitro 21:189-194.
MacLeod, R. A., Dirks, W. G., Matsuo, Y., Kaufmann, М.,
Milch, H., Drexler, H. G. (1999). Widespread intraspecies
cross-contamination of human tumor cell lines arising at
source. Int.J. Cancer 83:555-63.
Macpherson, I. (1973). Soft agar technique. In Kruse, P. F. &
Patterson, М. K. (eds.), Tissue culture methods and applica¬
tions. New York, Academic Press, pp. 276-280.
Macpherson, I. & Bryden, A. (1971). Mitomycin С treated cells
as feeders. Exp. Cell Res. 69:240-241.
Macpherson, I. & Montagnier, L. (1964). Agar suspension culture
for the selective assay of cells transformed by polyoma vims.
Virology 23: 291-294.
Macpherson, I. & Stoker, M. (1962). Polyoma transformation of
hamster cell clones-an investigation of genetic factors affect¬
ing cell competence. Virology 16:147.
Macville, М., Veldman, Т., Padilla-Nash, H., Wangsta, D.,
O’Brien, P., Schrock, E. & Ried, T. (1997). Spectral keratyp-
ing, a 24-colour FISH technique for the identification of chro¬
mosomal rearrangements. Histochemistry 108: 299-305.
Macy, M. (1978). Identification of cell line species by isoenzyme
analysis. Manual Am. Tissue Cult. Assoc. 4:833-836.
Magee, J. C., Stone, A. E., Oldman, К. T. & Guice, K. S. (1994).
Am.J. Physiol. Lung Mol. CellPhysiol. 267: L433-L441.
Mahato, R. I., Kawabata, K., Nomura, Т., Takakura, Y. &
Hashida, M. (1995a). Physicochemical and pharmacokinetic
characteristics of plasmid DNA/cationic liposome complexes.
J. Pharmaceut. Sci. 84:1267-1271.
Mahato, R. I., Kawabata, K., Takakura, Y. & Hashida, M. (1995b).
In vivo disposition characterictics of plasmid DNA complexed
with cationic liposomes./. Drug Targeting 3:149—157.
Mahdavi, V. & Hynes, R. O. (1979). Proteolytic enzymes in
normal and transformed cells. Biochim. Biophys. Acta 583:
167-178.
Mairs, R. I. & Wheldon, Т. E. (1996). Experimental tumour thera¬
py with targeted radionuclides in multicellular tumour spher¬
oids. In Hagen, U., Jung, H. & Streffer, C. (eds.,), Radiation
research, 1985-1995 (Proceedings of the 10th International
Congress of Radiation Research, Wurzbrg, Germany).
Mai, A. & Harter, M. L. (2003). MyoD is functionally linked to the
silencing of a muscle-specific regulatory gene prior to skeletal
myogenesis. Proc. Natl. Acad. Sci. USA 100:135-1739.
Maltese, W. A. & Volpe, I. J. (1979). Induction of an oligoden-
droglial enzyme in C-6 glioma cells maintained at high density
or in serum-free medium./. Cell Physiol. 101:459-470.
Management of Health and Safety at Work Directive 89/391/
EEC.
Management of Health and Safety at Work Regulations. (1999).
Statutory Instmment 1999 No. 3242. The Stationery Office
Limited, ISBN 0 11 085625 2.
Mangi, A. A., Noiseux, N., Kong, D., He, H., Rezvani, М., Ing-
wall, J. S. & Dzau, V. J. (2003). Mesenchymal stem cells modi¬
fied with Akt prevent remodeling and restore performance of
infracted hearts. Nat. Med. 9:1195-1201.
Maniatis, T,, Hardison, R. C., Lacy, E., Lauer, J., O’Connel, C.,
Quon, D., Sim, G. K. & Efstradiadis, A. (1978). The isolation
of structural genes from libraries of eukaryotic DNA. Cell 15:
687-701.
Maramorosch, K. (1976). Invertebrate tissue culture. New York,
Academic Press.
Marchionni, M. A., Goodearl, A. D., Chen, M.S. Bermingham-Mc-
Donogh, O., Kirk, C., Henricks, М., Danehy, F., Misumi, D.,
Sudhalter, J., Kobayashi, K., Wroblewski, D., Lynch, C.,
Baldassare, М., Hiles, I., Davis, J. B., Hsuan,J.J.,Totty,N. F.,
Otsu, М., McBumey, R. N., Waterfield, M. D., Stroobant, P. &
Gwynne, D. (1993). Glial growth factors are alternatively
spliced erbB2 ligands expressed in the nervous system. Nature
362:312-318.
Marcus, М., Lavi, U., Nattenberg, A., Ruttem, S. & Markowitz, O.
(1980). Selective killing of mycoplasmas from contaminated
cells in cell cultures. Nature 285:659-660.
Mardh, P. H. (1975). Elimination of mycoplasmas from cell
cultures with sodium polyanethol sulphonate. Nature 254:
515-516.
Mareel, М. М., Bmyneel, E. & Storme, G. (1980). Attachment of
mouse fibrosarcoma cells to precultured fragments of embry¬
onic chick heart. Virchows Arch. В Cell Pathol. 34:85-97.
Mareel, М., Kint, J. & Meyvisch, C. (1979). Methods of study
of the invasion of malignant СЗН-mouse fibroblasts into
embryonic chick heart n vitro. Virchows Arch. В Cell Pathol.
30:95-111.
Marelli-Berg, F. М., Peek, E., Lidington, E. A., Stauss, H. J. &
Lechler, R. I. (2000)./ Immunol. Methods 244: 205-215.
Marh, J., Tres, L.L. Yamazaki, Y., Yanagimachi, R. & Kierszen-
baum, A. L. (2003). Mouse round spermatids developed in
vitro from preexisting spermatocytes can produce normal
offspring by nuclear injection into in vivo-developed mature
oocytes. Biol. Reprod. 69:169-176.
Maijnissen, W. J., van Osch, G. J., Aigner, J., van der Veen, S. W.,
Hollander, A. P., Verwoerd-Verhoef, H. L. & Verhaar, J. A.
668
Список литературы
(2002). Alginate as a chondrocyte-delivery substance in
combination with a non-woven scaffold for cartilage tissue
engineering. Biomaterials 23:1511-1517.
Mark, J. (1971). Chromosomal characteristics of neuroenic tu¬
mours in adults. Hereditas 68:61-100.
Markovitz, D., Goff, S. & Bank, A. (1988). A safe packaging
line for gene transfer separating viral genes on two different
plasmids./ Virol 62:1120-1124.
Marks, P. A., Richon, V. М., Kiyokawa, H. & Rifkind, R. A.
(1994). Inducing differentiation of transformed cells with
hybrid polar compounds: a cell cycle-dependent process. Proc.
Natl. Acad. Sci. USA 91:10251-10254.
Markus, G., Takita, H., Camiolo, S. М., Corsanti, J., Evers, J. L. &
Hobika, J. H. (1980). Content and characterization of plas¬
minogen activators in human lung tumours and normal lung
tissue. Cancer Res. 40: 841-848.
Marsh, J. W., Donovan, М., Burholt, D. R., George, L. D. & Kom-
blith, P. L. (2004). Metastatic lung disease to the central
nervous system: in vitro response to chemotherapeutic agents.
/ Neurooncol. 66: 81-90.
Marshall, C. J. (1991). Tumor suppressor genes. Cell 64:313-326.
Marte, В. М., Meyer, Т., Stabel, S., Standke, G. J. R., Jaken, S.,
Fabbro, D. & Hynes, N. E. (1994). Protein kinase С and mam¬
mary cell differentiation: Involvement of protein kinase С
alpha in the induction of beta-casein expression. Cell Growth
Differ. 5:239-247.
Martin, F. L., Cole, K. J., Williams, J. A., Millar, В. C., Harvey, D.,
Weaver, G., Grover, P. L. & Phillips, D. H. (2000). Activation
of genotoxins to DNA-damaging species in exfoliated breast
milk cells. Mutat. Res. 470:115-124.
Martin, G. R. (1975). Teratocarcinomas as a model system for the
study of embryogenesis and neoplasia. Cell 5: 229-243.
Martin, G. R. & Evans, M. J. (1974). The morphology and growth
of a pluripotent teratocarcinoma cell line and its derivatives
in tissue culture. Cell 2:163-172.
Martin, G. R. (1978). Advantages and limitations of teratocarci¬
noma stem cells as models of development. In Johnson, М. H.
(ed.), Development in mammals, Vol. 3 Amsterdam, North-
Holland Publishing, p. 225.
Martin, G. R. (1981). Isolation of a pluripotent cell line from
early mouse embryos cultured in medium conditioned by
teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 78:
7634-7638.
Martinsen, A., Skj&k-BrHek, G. & Smidsrod, O. (1989). Aginate
as immobilization material; 1: Correlation between chemi¬
cal and physical properties of alginate gel beads. Biotechnol.
Bioeng. 33:79-89.
Maskell, R. & Green, M. (1995). Applications of the comet assay
technique. Int. Micr. Lab. 6: 2-5.
Massague, J., Cheifetz, S., Laiho, М., Ralph, D. A., Weis, F. M. &
Zentella, A. (1992). Transforming growth factor-beta. Cancer
Surveys 12:81-103.
Masson, E. A., Atkin, S. L. & White, М. C. (1993). d-valine se¬
lective medium does not inhibit human fibroblast growth in
vitro. In Vitro Cell Dev. Biol. Anim. 29A: 912-913.
Masters, J. R. W. & Palsson, B. (eds.) (1999-2000). Human cell
culture, Vol. I-IV, Dordrecht, Kluwer.
Masters, J. R., Bedford, P., Kearney, A., Povey, S. & Freanks, L. M.
(1988). Bladder cancer cell line cross-contamination: Identifi¬
cation using a locus-specific minisatellite probe. Br.J. Cancer
57 (3): 284-286.
Masters, J. R. W., Thomson, J. A., Daly-Bums, B., Reid, Y. A.,
Dirks, W. G., Packer, P., Toji, L. H., Ohno, Т., Tanabe, H., Ar-
lett, C. F., Kelland, L R., Harrison, М., Virmani, A, Ward, Т. H.,
Ayres, K. L. & Debenham, P. G. (2001). STR profiling pro¬
vides an international reference standard for human cell lines.
Proc. Natl. Acad. Sci. USA 98:8012-8017.
Masui, Т., Lechner, J. F., Yoakum, G. H., Willey, J. C. & Har¬
ris, С. C. (1986b). Growth and differentiation of normal and
transformed human bronchial epithelial cells. Cell Physiol
(Suppl) 4: 73-81.
Masui, Т., Wakefield, L. М., Lechner, J. F., LaVeck, M. A.,
Spom, М. B. & Harris, С. C. (1986a). Type beta transforming
growth factor is the primary differentiation-inducing serum
factor for normal human bronchial epithelial cells. Proc. Natl.
Acad. Sci. USA 83:2438-2442.
Mather, J. (1979). Testicular cells in defined medium. In Ja-
koby, W. B. & Pastan, I. H. (eds.), Methods in enzymology;
Vol. 57: Cell culture. New York, Academic Press, pp. 103.
Mather, J. P. (1998). Making informed choices: Medium, serum,
and serum-free medium; how to choose the appropriate me¬
dium and culture system for the model you wish to create.
Methods Cell Biol. 57:19-30.
Mather, J. P. & Sato, G. H. (1979a). The growth of mouse mela¬
noma cells in hormone supplemented, serum-free medium.
Exp. Cell Res. 120:191.
Mather, J. P. & Sato, G. H. (1979b). The use of hormone supple¬
mented serum-free media in primary cultures. Exp. Cell Res.
124:215.
Matsui, A., Zsebo, K. & Hogan, B. L. M. (1992). Derivation of
pluripotential embryonic stem cells from murine primordial
germ cells in culture. Cell 70: 841-847.
Matsui, Y., Toksoz, Nishikawa, S., Williams, D., Zsebo, K., Ho¬
gan, B. L. (1991). Effect of steel factor and leukemia inhibi¬
tory factor on murine primordial germ cells in culture. Nature
353:750-752.
Mayne, L. V., Price, T. N. C., Moorwood, K. & Burke, J. F. (1996).
Development of immortal human fibroblast cell lines. In
Freshney, R. I. & Freshney, M. G. (eds.), Culture of immortal¬
ized cells. New York, Wiley-Liss, pp. 77-93.
Mayne, L. V., Priestly, A., James, M. R. & Burke, J. F. (1986).
Efficient immortalisatin and morphological transformation
of human fibroblasts with SV40 DNA linked to a dominant
marker. Exp. Cell Res. 162:530-538.
McAteer, J. A., Kempson, S. A. & Evan, A. P. (1991). Culture of
human renal cortex epithelial cells. J. Tissue Cult. Methods
13:143-148.
McCall, E., Povey, J. & Dumonde, D. C. (1981). The culture of
vascular endothelial cells on microporous membranes. Thromb.
Res. 24:417-431.
McCormack, S. A., Viar, M. J., Tague, L. & Johnson, L. R. (1996).
Altered distribution of the nuclear receptor гаг (p) accom¬
panies proliferation and differentiation changes caused by
retinoic acid in Caco-2 cells. In Vitro Cell Dev. Biol. Anim.
32:53-61.
McCormick, C. & Freshney, R. I. (2000). Activity of growth fac¬
tors in the IL-6 group in the differentiation of human lung
adenocarcinoma. Br. J. Cancer 82:881-890.
Список литературы
669
McCormick, С., Freshney, R. I. & Speirs, V. (1995). Activity of
interferon alpha, interleukin 6 and insulin in the regulation of
differentiation in A549 alveolar carcinoma cells. Br.J. Cancer
71: 232-239.
McDouall, R. М., Yacoub, M. & Rose, M. L. (1996). Isolation,
culture, and characterisation of MHC class II-positive mi-
crovascular endothelial cells from the human heart. Microasc.
Res. 51:137-152.
McFarland, D. C., Liu, X., Velleman, S. G., Zeng, C., Coy, C. S. &
Pesall, J. E. (2003). Variation in fibroblast growth factor
response and heparin sulfate proteoglycan production in
satellite cell populations. Comp. Biochem. Physiol. С Toxicol.
Pharmacol. 134:341-351.
McGamty, G. J. (1982). Detection of mcoplasmic infection of cell
cultures. In Maramorosch, K. (ed.), Advances in cell culture,
Vol. 2. New York, Academic Press, pp. 99-131.
McGowan, J. A. (1986). Hepatocyte proliferation in culture. In
Guillouzo, A. & Guguen-Guillouzo, С (eds.), Isolated and
cultured hepatoctes. Paris, LesEditions Inserm, John Libbey
Eurotext, pp. 13-38.
McGregor, D. B., Edwards, I., Riach, C. J., Cattenach, P., Mar¬
tin, R., Mitchell, A. & Caspry, W. J. (1988). Studies of an
S9 based metabolic activation system used in the mouse
lymphoma L51768Y cell mutation assay. Mutagenesis 3:
485-490.
McGuckin, C. P., Forraz, N., Allouard, Q., Pettengel, R. (2004).
Umbilical cord blood stem cells can expand hematopoietic and
neuroglial progenitors in vitro. Exp. Cell Res. 295: 350-9.
McGuire, P. G. & Orkin, R. W. (1987). Lab. Invest. 57:94-105.
Mcllwrath, A., Vasey, P., Ross, G. & Brown, R. (1994). Cell
cycle arrests and radiosensitivity of human tumour cell lines:
Dependence on wild-type p53 for radiosensitivity. Cancer
Res. 54:3718-3722.
McKay, I. & Taylor-Papadimitriou, J. (1981). Junctional com¬
munication pattern of cell cultured from human milk. Exp.
Cell Res. 134:465-470.
McKeehan, W. L. (1977). The effect of temperature during
trypsin treatment on viability and multiplication potential
of single normal human and chicken fibroblasts. Cell Biol.
Int. Rep. 1:335-343.
McKeehan, W. L. & Ham, R. G. (1976a). Stimulation of clonal
growth of normal fibroblasts with substrata coated with basic
polymers ./. Cell Biol. 71: 727-734.
McKeehan, W. L. & Ham, R. G. (1976b). Methods for reducing
the serum requirement of growth in vitro of non-transformed
diploid fibroblasts. Dev. Biol. Standard 37:97-98.
McKeehan, W. L. & McKeehan, K. A. (1979). Oxocarboxylic
acids, pyridine nucleotide-linked oxidoreductases and serum
factors in regulation of cell proliferation./. Cell Physiol. 101:
9-16.
McKeehan, W. L., Adams, P. S. & Rosser, M. P. (1984). Modified
nutrient medium MCDB 151 (WJAC401), defined growth
factors, cholera toxin, pituitary factors, and horse serum
support epithelial cell and suppress fibroblast proliferation
in primary cultres of rat ventral prostate cells. In Vitro 18:
87-91.
McKeehan, W. L., Adams, P. S. & Rosser, M. P. (1984). Direct
mitogenic effects of insulin, epidermal growth factor, cholera
toxin, unknown pituitary factors and possibly prolactin, but
not androgen, on normal rat prostate epithelial cells in serum-
free primary cell culture. Cancer Res. 44:1998-2010.
McKeehan, W. L., Hamilton, W. G. & Ham, R. G. (1976).
Selenium is an essential trace nutrient for growth of
WI38 diploid human fibroblasts. Proc. Natl. Acad. Sci. USA
73:2023-2027.
McKeehan, W. L., McKeehan, K. A., Hammond, S. L. &
Ham, R. G. (1977). Improved medium for clonal growth of
human diploid cells at low concentrations of serum protein.
In Vitro 13:399-416.
McLean, J. S., Frame, М. C., Freshney, R. I., Vaughan, P. F. T. &
Mackie, A. E. (1986). Phenotypic modification of human
glioma and non-small cell lung carcinoma by glucocorticoids
^nd other agents. Anticancer Res. 6:1101-1106.
Medical Research Council (2002). Human Tissue and Biological
Samples for Use in Research; Operational and Ethical Guide¬
lines. Available from MRC External Communications +44
(0)2076 376 422 or online at www.mrc.ac.uk.
Meeker, A. K. & De Marzo, A. M. (2004). Recent advances in
telomere biology: implications for human cancer. Curr. Opin.
Oncol. 16:32-38.
Mege, R. М., Matsuzaki, F., Gallin, W. J., Goldberg, J. I., Cum-
mingham, B. A. & Edelman, G. M. (1989). Construction of
epithelial sheets by transfection of mouse sarcoma cells with
cDNAs for chicken cell adhension molecules. Proc. Nad. Acad.
Sci. USA 85: 7274-7278.
Melera, P. W., Wolgemuth, D., Biedler, J. L. & Hession, C. (1980).
Antifolate-resistant Chinese hamster cells: Evidence from
independently derived sublines for the overproductions of
two dihydrofolate reductases encoded by different mRNAs.
J. Biol. Chem. 255:319-322.
Menasche, P., Hagege, A. A., Scosin, М., Pouzet, B., Desnos, М.,
Duboc, D., Schwartz, K., Vilquin, J. T. & Marolleau, J. P.
(2001). Myoblast transplantation for heart failure. Lancet
357:279-280.
Menasche, P. (2004). Skeletal myoblast transplantation for car¬
diac repair. Expert Rev. Cardiovasc. Ther. 2: 21-28.
Ment, L. R., Stewart, W. B., Scaramuzzino, D. & Madri, J. A.
(1997). An in vitro three-dimensional coculture model of
cerebral microvascular antiogenesis and differentiation. In
Vitro Cell Dev. Biol. Animal 33:684-691.
Merten, O. W. (1999). Safety issues of animal products used in
serum-free media. Dev. Bol. Stand. 99:167-180.
Messing, E. М., Fahey, I. L., deKemion, I. B., Bhuta, S. M. &
Bubbers, I. E. (1982). Serum-free medium for the in vitro
growth of normal and malignant urinary bladder epitheial
cells. Cancer Res. 42: 2392-2397.
Metcalf, D. (1969). Studies on colony formation in vitro by mouse
bone marrow cells. I. Continuous cluster formation and rela¬
tion of clusters to colonies./. Cell Physiol. 74:323-332.
Metcalf, D. (1990). The colony stimulating factors. Cancer 65:
2185-2195.
Meyer, W., Latouche, G. N., Daniel, H. М., Thanos, М., Mitch¬
ell, T. G., Yarrow, D., Schonian, G. & Sorrel, Т. C. (1997).
Identification of pathogenic yeasts of the imperfect genus
Candida by polymerase chain reaction fingerprinting. Elec¬
trophoresis 18:1548-1549.
Meyskens, F. L. & Fuller, В. B. (1980). Characterization of the
effects of different retinoids on the growth and differentiation
670
Список литературы
of a human melanoma cell line and selected subclones. Cancer
Res. 40:2194-2196.
Michalopoulos, G. & Pitot, H. C. (1975). Primary culture of pa¬
renchymal liver cells on collagen membranes: Morphological
and biochemical observations. Exp. Cell Res. 94:70-78.
Michler-Stuke, A. & Bottenstein, J. (1982). Proliferation of glial-
derived cells in defined media ./. Neurosci. Res. 7: 215-228.
Midgley, C. A., Craig, A. L, Hite, J. P. & Hupp, T. R. (1998). Baculo-
virus expression and the study of the regulation of the tumor sup¬
pressor protein p53. In Ravid, K. & Freshney, R. I. (eds.), DNA
transfer to cultured cells. New York, Wiley-Liss, pp. 27-54.
Miklic, S., Juric, D. M. & Caman-Krzan, M. (2004). Differences
in the regulation of BDNF and NGF synthesis in cultured
neonatal rat astrocytes. Int.J. Dev. Neurosci. 22:119-136.
Mikulits, W., Dolznig, H., Edelmann, H., Sauer, Т., Deiner, E. М.,
Ballou, L., Beug, H. & Mullner, E. W. (1997). Dynamics of
cell cycle regulators: Artefact-free analysis by recultivation
of cells synchronized by centrifugal elutriation. DNA Cell
Biol. 16:849-859.
Milanesi, E., Ajmone-Marsan, P., Bignotti, E., Losio, M. N.,
Bemardi, J., Chegdani, F., Soncini, M. & Ferrari, M. (2003).
Molecular detection of cell line cross-contaminations using
amplified fragment length polymorphism DNA fingerprinting
technology. In Vitro Cell Dev. Biol. Animal 39:124—130.
Miller, A. D., Garcia, J. V., von Suhr, N., Lynch, С. М., Wil¬
son, C. & Eiden, М. V. (1991). Construction and properties
of retrovirus packaging cell based on gibbon abe leukemia
virus./ Virol. 65: 2220-2224.
Miller, G., Lisco, H., Kohn, H. I., Stitt, D. & Enders, J. F. (1971).
Establishment of cell lines from normal adult human blood
leukocytes by exposure to Epstein-Barr virus and neutraliza¬
tion by human sera with Epstein-Barr virus antibody. Proc.
Soc. Exp. Biol. Med. 137:1459-1465.
Miller, G. G., Walker, G. W. R. & Giblack, R. E. (1972). A rapid
method to determine the mammalian cell cycle. Exp. Res. 72:
533-538.
Miller, R. G. & Phillips, R. A. (1969). Separation of cells by veloc¬
ity sedimentation./. Cell Physiol. 73:191-201.
Mills, K. J., Volberg, Т. М., Nervi, C., Grippo, J. F., Daw¬
son, М. I. & Jetten, A. M. (1996). Regulation of retinoid-in¬
duced differentiation in embryonal carcinoma PCC4, azalR
cells: Effects of retinoid-receptor selective ligands. Cell Growth
Differ. 7:327-337.
Milo, G. E., Ackerman, G. A. & Noyes, I. (1980). Growth and
ultrastructural characterization of proliferating human ke¬
ratinocytes in vitro without added extrinsic factors. In Vitro
16: 20-30.
Minna, I. D., Carney, D. N., Cuttitta, F. & Gazdar, A. F. (1983).
The biology of lung cancer. In Chabner, B. (ed.), Rational
basis for chemotherapy. New York, Alan R. Liss.
Miskey, R., Dubois, C., Morgan, L. & Jessen, K. R. (1990). 04
and A007 sufatide antibodies bind to embryonic Schwann
cells prior to the appearance of galactocerebroside: Regulation
by axon-Schwann cell signals and cyclic AMP. Development
109:105-116.
Mitaka, Т., Norioka, K.-I. & Mochizuki, Y. (1993). Redifferentia¬
tion of proliferated rat hepatocytes cultures in LI5 medium
supplemented with EGF and DMSO. In Vitro Cell Dev. Biol.
29A: 714-722.
Mitaka, Т., Sattler, C. A., Sattler, G. L., Sargent, L. M. & Pi¬
tot, H. C. (1991). Multiple cell cycles occur in rat hepatocytes
cultured I the presence of nicotinamide and epidermal growth
factor. Hepatology 13: 21-30.
Mitelman, F. (eds.) (1995). ISCN. An international system for
human cytogenetic nomenclature, Basel, S. Karger.
Miura, Y., Akimoto, Т., Kanazawa, H. & Yagi, K. (1986). Synthe¬
sis an secretion of protein by hepatocytes entrapped within
calcium alginate. Artif. Organs 10:460-465.
Mohammad, R. М., Li, Y., Mohamed, A. N., Pettit, G. R., Adsay, V.,
Vaitkevicius, V. K., Al-Katib, A. M. & Sarkar, F. H. (1999).
Clonal preservation of human pancreatic cell line derived from
primary pancreatic adenocarcinoma. Pancreas 19:353-361.
Moll, R., Franke, W. W. & Schiller, D. L. (1982). The catalog of
human cytokeratins: Patterns of expression in normal epithe-
lia, tumours and cultured cells. Cell 31:11-24.
Monroy, B., Honiger, J., Darquy, S. & Reach, G. (1997). Use of
polyethyleneglycol for porcine islet cryopreservation. Cell
Transplant. 6:613-621.
Monstein, H. J., Ohlsson, B. & Axelson, J. (2001). Differential
expression of gastrin, cholecystokinin-A and cholecystoki-
nin-B receptor mRNA in human pancreatic cancer cell lines.
Scand./. Gastroenterol. 36:738-743.
Montagnier, L. (1968). Correlation entre la transformation des
cellule BHK21 et leur resistance aux polysaccharides acides
en mileu gelifie. CRAcad. Sci. D 267:921-924.
Montesano, R, Matsumoto, K., Nakamura, T. & Orci, L. (1991).
Identification of a fibroblast-derived epithelial morphogen as
hepatocyte growth factor. Cell 67:901-908.
Montesano, R., Soriano, J. V., Pepper, M. S. & Orci, L. (1997).
Induction of epithelial branching tubulogenesis in vitro./. Cell
Physiol. 173:152-161.
Moore, G. E., Gemer, R. E. & Franklin, H. A. (1967). Culture of
normal human leukocytes./. Am. Med. Assoc. 199:519-524.
Moreno, R. F. (1990). Enhanced conditions for DNA fingerprint¬
ing with biotinylated M13 bacteriophage./. Forensic Sci. 35:
831-837.
Morgan, J. E., Beauchamp, J. R., Pagel, C. N., Peckham, М., Atali-
otis, P., Jat, P. S., Noble, M. D., Farmer, K. & Prtridge, T. A.
(1994). Myogenic cell lines derived from transgenic mice
carrying a thermolabile T antigen: a model system for the
derivation of tissue-specific and mutation-specific cell lines.
Dev. Biol. 162:486-498.
Morgan, J. E., Freshney, R. I., Darling, J. L., Thomas, D. G. T. &
Celik, F. (1983). Assay of anticancer drugs in tissue culture:
cell cultures of biopsies from human astrocytoma. Br.J. Cancer
47: 205-214.
Morgan, J. E., Moore, S. E., Walsh, F. S. & Partridge, T. A. (1992).
Formation of skeletal muscle in vivo from the mouse C2 cell
line./ Cell Sci. 102:779-787.
Morgan, J. G., Morton, H. J. & Parker, R. C. (1950). Nutrition of
animal cells in tissue culture; I: Initial studies on a synthetic
medium. Proc. Soc. Exp Biol. Med. 73:1.
Morton, H. J. (1970). A survey of commercially available tissue
culture media. In Vitro 6: 89-108.
Moscona, A. A. (1952). Cell suspension from organ rudiments of
chick embryos. Exp. Cell Res. 3: 535.
Moscona, A. A. & Piddington, R. (1966). Stimulation by hydro¬
cortisone of premature changes in the developmental pat¬
Список литературы
671
tern of glutamine synthetase in embryonic retina. Biochem.
Biophys. Acta 121:409-411.
Mosmann, T. (1983). Rapid colorimetric assay for cellular growth
survival: Application to proliferation and cytotoxicity assays.
J. Immunol. Methods 65:55-63.
Moulton, D. G. (1974). Dynamics of cell populations in the olfac¬
tory epithelium. Ann. NY Acad. Sci. 237: 52-61.
Mowles, J. (1988). The use of ciprofloxacin for the elimination of
mycoplasma from naturally infected cell lines. Cytotechnology
1:355-358.
Muirhead, E. E., Rightsel, W. A., Pitcock, J. A. & Inagami, T.
(1990). Isolation and culture of juxtaglomerular and renomed-
ullary interstitial cells. Methods Enzymol. 191:152-167.
Mullender, М., El Haj, A. J., Yang, Y., van Duin, M. A., Bur¬
ger, E. H. & Klein-Nulend, J. (2004). Mechanotransduction
of bone cells in vitro: mechanobiology of bone tissue. Med.
Biol. Eng. Comput. 42:14-21.
Muller, U., Wang, D., Denda, S., Meneses, J. J., Pedersen, R. A. &
Reichardt, L. F. (1997). Integrin alpha 8 beta 1 is critically
important for epithelial-mesenchymal interactions during
kidney morphogenesis. Cell 88: 603-613.
Mullin, J. М., Marano, C. W., Laughlin, К. V., Nuciglio, М., Ste¬
venson, B. R. & Soler, A. P. (1997). Different size limitations
for increased transepithelial paracellular solute flux across
phorbol ester and tumour necrosis factor treated epithelial
cell sheets./. Cell Physiol. 171: 226-233.
Munthe-Kaas, A. C. & Seglen, P. O. (1974). The use of metri-
zamide as a gradient medium for isopycnic separation of rat
liver cells. FEBS Lett. 43:252-256.
Murakami, H. (1984). Serum-free cultivation of plasmacyto¬
mas and hybridomas. In Barnes, D. W., Sirbasku, D. A. &
Sato, G. H. (eds.), Methods fo serum-free culture of neuronal
and lymphoid cells. New York, Alan R. Liss, pp. 197-206.
Murakami, H. & Masui, H. (1980). Hormonal control of human
colon carcinoma cell growth in serum-free medium. Proc. Natl.
Acad. Sci. USA 77:3464-3468.
Murao, S., Gemmell, M. A. & Callighan, M. F. (1983). Control
of macrophage cell differentiation in human promyelocytic
HL-60 leukemia cells by 1,25-dihydroxyvitamin D3 and phor¬
bol-12-myristate-13-acetate. Cancer Res. 43:4989-4996.
Murphy, C. L. & Polak, J. M. (2004). Control of human articular
chondrocyte differentiation by reduced oxygen tension./ Cell
Physiol. 19:451-459.
Murphy, D. S., Hoare, S. F., Going, J. J., Mallon, E. E.,
George, W. D., Kaye, S. B., Brown, R., Black, D. M. &
Keith, W. N. (1995). Characterization of extensive genetic
alterations in ductal carcinoma in situ by fluorescence in situ
hybridization and molecular analysis./. Natl. Cancer Inst. 87:
1694-1704.
Murphy, S. J., Watt, D. & Jones, G. E. (1992). An evaluation of
cell separation techniques in a model mixed cell population.
J. Cell Sci. 102:297-798.
Naeyaert, J. М., Eller, М., Gordon, P. R., Park, H.-Y. & Gil¬
chrest, A. (1991). Pigment content of cultured human mela¬
nocytes does not correlate with tyrosinase message leel. Br.
J. Dermatol. 125:297-303.
Nardone, R. М., Todd, G., Gonzalez, P. & Gaffney, E. V. (1965).
Nucleoside incorporation into strain L cells: Inhibition by
pleuropneumonia-like organisms. Science 149:1100-1101.
National Research Council. (1989). Biosafety in the laboratory:
Prudent practices for the handling and disposal of infectious
materials. Washington, DC, National Academy Press.
National Sanitation Foundation. (1983) Standard 49: Class II
(laminar flow) biohazard cabinetry. Ann Arbor, Michigan.
Nelson, J. B., ed. (1960). Biology of the pleuropneumonia-like
organisms. Ann. Nw York. Acad. Sci. 79:305.
Nelson-Rees, W. A., Daniels, D. & Flandermeyer, R. R. (1981).
Cross-contamination of cells in culture. Science 212:446-452.
Nelson-Rees, W. & Flandermeyer, R. R. (1977). Inter- and intra-
species contamination of human breast tumor cell lines HBC
and BrCa5 and other cell cultures. Science 195:1343-1344.
Neugut, A. I. & Weinstein, I. B. (1979). Use of agarose in the
determination of anchorage-independent growth. In Vitro
15:351.
Neumann, Т., Heuschka, S. D. & Sanders, J. E. (2003). Tissue
engineering of skeletal muscle using polymer fiber arrays.
Tissue Eng. 9:995-1003.
Neumann, J., Morency, C. A. & Russian, K. O. (1987). A novel
rapid assay for chloramphenicol acetyltransferase gene-ex-
pression. Biotechniques 5:444.
Neves, A. A., N., Medcalf, Brindle, K. (2003). Functional assess¬
ment of tissue-engineering meniscal cartilage by magnetic
resonance imaging and spectroscopy. Tissue Eng. 9:51-62.
Newbold, R. F. & Cuthbert, A. P. (1998). Mapping human senes¬
cence genes using microcell-mediated chromosome transfer.
In Ravid, K. & Freshney, R. I. (eds.), DNA transfer to cultured
cells. New York, Wiley-Liss, pp. 237-264.
Newman, М. B., Davis, C. D., Kuzmin-Nichols, N. & Sanberg, P. R.
(2003). Human umbilical cord blood (HUCB) cells for central
nervous system repair. Neurotox Res. 5:355-68.
Neyfakh, A. A. (1987). Use of fluorescent dyes as molecular probes
for the study of multidrug resistance. Exp. Cell Res. 174:168.
Nichols, W. W., Murphy, D. G. & Christofalo, V. J. (1977).
Characterization of a new diploid human cell strain, I MR-90.
Science 196: 60-63.
Nichola, N. A. (1987). Hemopoietic growth actors and their
interactions with specific receptors./. Cell Physiol. (Suppl.)
5:9-14.
Nicholson, G. L. (1976). Trans-membrane control of the recep¬
tors on normal and tumor cells; II: Surface changes associated
with transformation and malignancy. Biochim. Biophys. Acta
458:1-72.
Nicosia, R. F. & Ottinetti, A. (1990). Modulation of microvas-
cular growth and morphogenesis by reconstituted basement
membrane gel in three-dimensional cultures of rat aorta:
A comparative study of angiogenesis in Matrigel, collagen,
fibrin, and plasma clot. In Vitro Cell Dev. Biol. 26:119-128.
Nicosia, R. F., Tchao, R. & Leighton, J. (1983). Angiogenesis-
dependent tumor spread in reinforced fibrin clot culture.
Cancer Res. 43:2159-2166.
Nielsen, V. (1989). Vibration patterns in tissue culture vessels.
Nunc Bulletin 2 (May 186; rev March 1989). Roskilde, Den¬
mark, A/S Nunc.
Niklason, L. E., Abbott, W., Gao, J., Klegges, B., Hirschi, К. K.,
Ulubayram, K., Conroy, N., Jones, R., Vasanawala, A., Sanz-
giri, S. & Langer, R. (2001). Morphological and mechanical
characteristics of engineered bovine arteries./. Vase. Surg.
33 (3): 628-638.
672
Список литературы
Nilos, R. М. & Makarski, J. S. (19780. Control of melanogenesis in
mouse melanoma cells of varying metastatic potential./ Natl.
Cancer Inst. 61: 523-526.
Nims, R. W., Shoemaker, A. P., Bautemschub, M. A., Rec, L. J. &
Harbell, J. W. (1998). Sensitivity of isoenzyme analysis for
the detection of interspecies cell line cross-contamination. In
Vitro Cell Dev. Biol. Anim. 34: 35-39.
Noble, M. & Barnett, S. C. (1996). Production and growth of
conditionally immortal primary glial cell cultures and cell
lines. In Freshney, R. I. & Freshney, M. G. (eds.), Culture of
immortalized cells. New York, Wiley-Liss, pp. 331-366.
Noble, M. & Murrey, K. (1984). Purified astrocytes promote the
in vitro division of a bipotential glial progenitor cell. EMBO
J. 3:2243-2247.
Nordgren, A. (2003). Hidden aberrations diagnosed by interphase
fluorescence in situ hybridization and spectral karyotyping
in childhood acute lymphoblastic leukemia. Leuk. Lymphoma
44:2039-2053.
Norwood, Т. H., Zeigler, C. J. & Martin, G. M. (1976). Dimethyl
sulphoxide enhances polyethylene glycol-mediated somiatic
cell fusion. Somatic Cell Genet. 2: 263-270.
Nurse, P. (1990). Universal control mechanism regulating onset
of M-phase. Nature 344:503-508.
Nuwaysir, E. F., Bittner, М., Trent, J., Barrett, J. C. & Af-
shari, C. A. (1999). Microarrays and toxicology: the advent
of toxicogenomics. Mol Carcinog. 24:153-159.
Obata, Y., Kono, T. & Hatada, I. (2002). Gene silencing: matura¬
tion of mouse fetal germ cells in vitro. Nature 418:497.
O’Brien, S. J., Shannon, J. E. & Gail, М. H. (1980). Molecular
approach to the identification and individualization of human
and animal cells in culture: Isozyme and allozyme genetic
signatures. In Vitro 16:119-135.
O’Farrell, P. H. (1975). High resolution two-dimensional elec¬
trophoresis of proteins./. Biol. Chem. 250:4007-4021.
Office of Nuclear Regulatory Research, see U.S. Nuclear Regula¬
tory Commission.
Oft, М., Akhurst, R. J. & Balmain, A. (2002). Metastasis is driven
by sequential elevation of H-ras and Smad2 levels. Natl. Cell
Biol. 4: 487-494.
Oh, С. K., Kwon, Y. W., Kim, Y. S., Jang, H. S., Kwon, K. S.
(2003). Expression of basic fibroblast growth factor, vascular
endothelial growth factor, and thrombospondin-1 related to
microvessel density in nonaggressive basal cell carcinomas./
Dermatol. 30:306-13.
Oh, H., Chi, X., Bradfute, S. B., Mishina, Y., Pocius, J., Mi¬
chael, L. H., Behringer, R. R., Schwartz, R. J., Entman, M. L. &
Schneider, M. D. (2004). Cardiac muscle plasticity in adult
and embryo by heart-derived progenitor cells Ann. NY Acad.
Sci. 1015:182-189.
Oh, Y. S., Kim, E. J., Schaffer, B. S., Kang, Y. H., Binderup, L.,
MacDonald, R. G. & Park, J. H. (2004). Synthetic lowcal-
caemic vitamin D3 analogues inhibit secretion of insulin-like
growth factor II and stimulate production of insulin-like
growth factor-binding protein-6 in conjunction with growth
suppression of HT-29 colon cancer cells. Mol. Cell Endocrinol.
183:141-149.
O’Hare, M. J., Ellison, M. L.,Nerville, A. M. (1978). Tissue culture
in endocrine research: Perspectives, pitfalls, and potentials.
Curr. Top. Xp. Endocrinol. 3:1-56.
Ohmichi, H., Koshimizu, U., Matsumoto, K. & Nakamura, T.
(1998). Hepatocyte growth factor (HGF) acts asa mesen-
chyme-derived morphogenic factor during fetal lung develop¬
ment. Development 125:1315-1324.
Ohno, Т., Saijo-Kurita, K., Miyamoto-Eimori, N., Kurose, Т.,
Aoki, Y. & Yosimura, S. (1991). A simple method for in situ
freezing of anchorage-dependent cells including rat liver
parenchymal cells. Cytotechnology 5:273-277.
Oie, H. K., Russel, E. K., Carney, D. N. & Gazdar, A. F. (1996).
Cell culture methods for the establishment of the NCI series
of lung cancer cell lines./ Cell Biochem. Suppl. 24:24-31.
Olie, R. A., Looijenda, L. H. J., Dekker, М. C., de Jong, F. H., van
Dissel-Emiliani, F. M. F., de Rooij, D. G., van der Holt, B. &
Oosterhuis, J. W. (1995). Heterogeneity in the in vitro
survival and proliferation of human seminoma cells. Brit.
J. Cancer 71:13-17.
Olsson, I. & Ologsson, T. (1981). Induction of differentiation in
a human promyelocytic leukemic cell line (HL-60). Exp. Cell
Res. 131:225-230.
Operational Guidelines for Ethics Committees That Review
Biochemical Research (2000). World Health Organization,
Geneva, TDR/PRD/ETHICS/2000.1.
Orellana, S. A., Neff, C. D., Sweeney, W. E. & Avner, E. D. (1996).
Novel Madin Darby canine kidney cell clones exhibit unique
phenotypes in response to morphogens. In Vitro Cell Dev. Biol.
Animal 32:329-339.
Organisation for Economic Co-operation and Development
(2004). The application f the principles of GLP to in vitro
studies. OECD Series on Principles of Good Laboratory
Practice and Compliance Monitoring, No. 14; ENV/JM/
MONO (2004)26.
Orly, J., Sato, G. & Erickson, G. F. (1980). Serum suppresses
the expression of hormonally induced function in cultured
granulosa cells. Cell 20: 817-827.
Osborne, С. K., Hamilton, B., Tisus, G. & Livingston, R. B. (1980).
Epidermal growth factor stimulation of human breast cancer
cells in culture. Cancer Res. 40:2361-2366.
Osborne, H. B., Bakke, A. С & Yu,J. (1982). Effect of dexametha-
sone on НМВА-induced Friend cell erythrodifferentiation.
Cancer Res. 42: 513-518.
Osborne, R., Durkin, Т., Shannon, H., Doman, E. & Hughes, C.
(1999). Performance of pen-fronted microbiological safety
cabinets: the value of operator protection tests during routine
servicing./. Appl. Microbiol. 86:962-970.
Ostertag, W. & Pragnell, I. B. (1978). Changes in genome com¬
position of the Friend virus complex n erythroleukaemia cells
during the course of differentiation induced by DMSO. Proc.
Natl. Acad. Sci. USA 75:3278-3282.
Ostertag, W. & Pragnell, I. B. (1981). Differentiation and viral
involvement in differentiation of transformed mouse and rat eiy-
throid cells. Curr. Topics Microbiol. Immunol 94/95:143-208.
Otterlei, М., Ostgaard, K., Skjik-Bnick, G., Smidsred, O., Soon
Shiong, P. & Espevik, T. (1991). Induction of cytokine pro¬
duction from human monocytes stimulated with alginate.
/. Immunother. 10: 286-291.
Owens, R. B., Smith, H. S. & Hackett, A. J. (1974). Epithelial cell
culture from normal glandular tissue of mice. Mouse epithelial
cultures enriched by selective trypsinisation./. Natl. Cancer
Inst. 53: 261-269.
Список литературы
673
Paddenberg, R., Wulf, S., Weber, A., Heimann, P., Beck, L. &
Mannherz, H. G. (1996). Intemucleosomal DNA fragmen¬
tation in cultured cells under conditions reported to induce
apoptosis may be caused by mycoplasma endonucleases.
Europ.J. Cell Biol 71:105-119.
Pantel, K., Dickmanns, A., Zippelius, A., Klein, C., Shi, J.,
Hoechtlen-Vollmar, W., Schlimok, G., Weckermann, D.,
Obemerder, R., Fanning, E. & Rietmuller, G. (1995). Estab¬
lishment of micrometastatic cell lines: A novel source of tumor
cell vaccines./. Natl. Cancer Inst. 87:1162-1168.
Paquet-Durand, F., Tan, S. & Bicker, G. (2004). Turning tera-
tocarcinoma cells into neurons: rapid differentiation of NT-
2 cells in floating spheres. Brain Res. Dev. Brain Res. 142:
161-167.
Paquette, J. S., Tremblay, Bernier, P. V., Auger, F. A., Lavio-
lette, М., Germain, L., Boutet, М., Boulet, L. P. & Goulet, F.
(2003). Production of tissue-engineered three-dimensional
human bronchial models. In Vitro Cellular £ Developmental
Biology-Animal. 39: pp. 213-220.
Paraskeva, C. & Williams, A. C. (1992). The colon. In Fresh¬
ney, R.I. (ed.), Culture of epithelial cells. New York, Wiley-
Liss, pp. 82-105.
Paraskeva, C., Buckle, B. G. & Thorpe, P. E. (1985). Selective
killing of contaminating human fibroblasts in epithelial
cultures derived from colorectal tumors using an anti-Thy-1
antibodyricin conjugate. Br. J. Cancer 51:131-134.
Paraskeva, C., Buckle, B. G., Sheer, D. & Wigley, С. B. (1984).
The isolation and characterisation of colorectal epithelial
cell lines at different stages in malignant transformation
from familial polyposis coli patients. Int. J. Cancer 34:
49-56.
Pardee, A. B., Cherington, P. & Medrano, E. E. (1984). On de¬
ciding which factors regulate cell growth. In Barnes, D. W.,
Sirbasku, D. A. & Sato, G. H. (eds.), Methods for serum-free
culture of epithelial and fibroblastic cells. New York, Alan
R. Liss, pp. 157-166.
Park, H. Y., Perez, J. М., Laursen, R., Нага, M. & Gilchrest, B. A.
(1999). Protein kinase С-beta activates tyrosinase by phos-
phorylating serine residues in its cytoplasmic domain./. Biol.
Chem. 274:16470-8.
Park, H. Y., Russakovsky, V., Ohno, S. & Gilchrest, B. A. (1993).
The P isoform of protein kinase С stimulates melanogenesis
by activating tyrosinase in pigment cells./. Biol. Chem. 268:
11742-11749.
Park, J. G. & Gazdar, A. F. (1996). Biology of colorectal and gas¬
tric cancer cell lines./ Cell Biochem. Suppl. 24:131-141.
Park, J.-G., Ku, J.-L., Kim, H.-S., Park, S.-Y. & Rutten, M. J.
(2004). Culture of normal and malignant gastric epithelium.
In Pfagner, & Freshney, R. I. (eds.), Culture of human tumor
cells, Hoboken, NJ, Wiley-Liss, pp. 23-66.
Parker, R. C., Castor, L. N. & McCulloch, E. A. (1957). Altered
cell strains in continuous culture. Special publications. NY
Acad. Sci. 5:303-313.
Parker, R. C. (1961). Methods of tissue culture, 3rd ed., London,
Pitman Medical, p. 47.
Parker, R. C., Healy, G. M. & Fisher, D. C. (1954). Nutrition of
animal cells in tissue culture. VII. Use of replicate cell cul¬
tures in the evaluation of synthetic media. Canad.J. Biochem.
Physiol. 32: 306.
Parkinson, E. K. & Yeudall, W. A. (2002). The epidermis. In
Freshney, R. I. (ed.), Culture of epithelial cells, 2nd ed New
York, Wiley-Liss, pp. 65-94.
Parums, D. V., Cordell, J. L., Micklem, K., Heryet, A. R.,
Getter, К. C., and Mason, D. Y. (1990)./ Clin Pathol 43:
752-757.
Pasch,J., Schiefer, A., Heschel, I., Dimoudis,N. & Rau, G. (2000).
Variation of the HES concentration for the cryopreservation
of keratinocytes in suspensions and in monolayers. Cryobiol¬
ogy 41:89-96.
Patel, K., Moore, S. E., Dickinson, G., Rossell, R. J., Beverley, P.,
Kemshead, J. T. & Walsh, F. S. (1989). Neural cell adhesion
molecule (NCAM) is the antigen recognised by monoclonal
antibodies of similar specificity in small-cell lung carcinoma
and neuroblastoma. Int.J. Cancer 44:573-578.
Paul, J. (1975). Cell and tissue culture. Edinburgh, Churchill
Livingstone, pp. 172-184.
Paul, J., Conkie, D. & Freshney, R. I. (1969). Erythropoietic cell
population changes during the hepatic phase of erythropoiesis
in he foetal mouse. Сей Tissue Kinet. 2:283-294.
Pavelic, K., Antonie, М., Pavelic, L., Pavelic, J., Pavelic, Z. &
Spaventi, S. (1992). Human lung cancers growing on extra¬
cellular matrix: Expression of oncogenes and growth factors.
Anticancer Res. 12:2191-2196.
Peat, N., Gendler, S. J., Lalani, N., Duhig, T. & Taylor-Papad¬
imitriou, J. (1992). Tissue-specific expression of a human
polymorphic epithelial mucin (MUC1) in transgenic mice.
Cancer Res. 52:1954-1960.
Pederson, N., Mortenson, S., Sorensen, S. B., Pederson, M. W.,
Rieneck, K., Bovin, L. F. & Poulsen, H. S. (2003). Transcrip¬
tional gene expression profiling of small cell lung cancer cells.
Cancer Res. 6 (8): 1943-1953.
Peehl, D. M. & Stanbridge, E. J. (1981). Anchorage-independent
growth of normal human fibroblasts. Proc. Natl. Acad. Sci.
USA 78:3053-3057.
Peehl, D. M. & Ham, R. G. (1980). Clonal growth of human
keratinocytes with small amounts of dialysed serum. In Vitro
16:526-540.
Peehl, D. M. (2002). Human prostatic epithelial cells. In Fresh¬
ney, R. I. & Frehney, M. G. (eds.), Culture of epithelial cells,
2nd ed., Hoboken, NJ, Wiley-Liss, pp. 171-194.
Pegolo, G., Askanas, V. & Engel, W. K. (1990). Expression of
muscle-specific isozymes of phosphorylase and creatine
kinase in human muscle fibers cultured aneurally in serum-
free, hormonally/chemically enriched medium. Int. J. Dev.
Neurosci. 8:299-308.
Peppelenbosch, M. P., Tertoolen, L. G. J., DeLaat, S. W. &
Zivkovic, D. (1995). Ionic responses to epidermal growth
factor in zebrafish cells. Exp. Cell Res. 218:183-188.
Pereira, М. E. A. & Kabat, E. A. (1979). A versatile imunoadsor-
bent capable of binding lectins of various specificities and
its use for the separation of cell populations./ Cell Biol. 82:
185-194.
Pereira-Smith, O. & Smith, J. (1988). Genetic analysis of indefi¬
nite division in human cells: Identification of four complemen¬
tation groups. Proc. Natl. Acad. Sci. USA 85: 6042-6046.
Perl, A.-K., Wilgenbus, P., Dahl, U., Semb, H. & Christofori, G.
(1998). A causal role for E-cadherin in the transition from
adenoma to carcinoma. Nature 392:190-193.
674
Список литературы
Perry, P. & Wolf, S. (1974). New Giemsa method for the differ¬
ential staining of sister chromatids. Nature 251:156-158.
Pertoft, H. & Laurent, Т. C. (1982). Sedimentation of cells in
colloidal silica (Percoll). In Pretlow, T. G. & Pretlow, T. P.
(eds.), Cell separation, methods and selected applications,
Vol. 1. New York, Academic Press, pp. 115-152.
Peters, D. М., Dowd, N., Brandt, C. & Compton, T. (1996). Hu¬
man papilloma virus E6/E7 genes can expand the lifespan
of human comeal fibroblasts. In Vitro Cell Dev. Biol. Animal
32: 279-284.
Petersen, O. W., Gudjonsson, Т., Villadsen, R., Bissell, M. J. &
Ronnov-Jessen, L. (2003). Epithelial progenitor cell lines as
models of normal breast morphogenesis and neoplasia. Cell
Prolif. 36 Suppl 1:33-44.
Petersen, D. F., Anderson, E. C. & Tobey, R. A. (1968). Mitotic
cells as a source of synchronized cultures. In Prescott, D. M.
(ed.), Methods in cell physiology. New York, Academic Press,
pp. 347-370.
Petit, B., Masuda, K., d’Souza, A., Otten, L., Pietryla, D., Hart¬
mann, D. J., Morris, N. P., Uebelhart, D., Schmid, Т. M. &
Thonar, E. J.-M. A. (1996). Characterization of crosslinked
collagens synthesiezed by mature articular chondrocytes
cultures in alginate beads: Comparison of two distinct matrix
compartments. Exp. Cell Res. 225:151-161.
Pevny, L. & Rao, M. S. (2003). The stem-cell menagerie. Trends
Neurosci. 26:351-359.
Pfeffer, L. M. & Eisenkraft, B. L. (1991). The antiproliferative
and antitumour effects of human alpha interferon on cultured
renal carcinomas correlate with the expression of a kidney-
associated differentiation antigen. Interferons Cytokines 17:
30-31.
Pfagner, R. & Freshney, R. I. (eds.) (2004). Culture of human
tumor cells, Hoboken, NJ, Wiley-Liss, Chapters 1,2,3,5,14.
Phillips, P. D. & Cristofalo, V. J. (1988). Classification system
based on the functional equivalency of mitogens that regulate
WI-38 cell proliferation. Exp. Cell Res. 175:396-403.
Pignata, P. D. & Maggini, L., Zarrilli, R., Rea, A. & Acqua-
viva, A. M. (1994). The enterocyte-like differentiation of the
Caco-2 tumour cell line strongly correlates with responsive¬
ness to cAMP and activation of kinase A pathway. Cell Growth
Differ. 5:967-973.
Pignatelli, M. & Bodmer, W. F. (1988). Genetics and biochem¬
istry of collagen binding-triggered glandular differentiation
in a human colon carcinoma cell line. Proc. Natl. Acad. Sci.
USA 85:5561-5565.
Pipia, G. G. & Long, M. W. (1997). Human hematopoietic pro¬
genitor cell isolation based on galactose-specific cell surface
binding. Nat. Biotechnol. 15:1007-1011.
Pitot, H., Periano, C., Morse, P. & Potter, V. R. (1964). Hepato¬
mas in tissue culture compared with adapting liver in vitro.
Natl. Cancer Inst. Monogr. 13: 229-245.
Piyathilake, C. J., Frost, A., Manne, U., Weiss, H., Bell, W. C.,
Heimburger, D. C. & Grizzle, W. E. (2002). Differential
expression of growth factors in squamous cell carcinoma
and precancerous lesions of the lung. Clin. Cancer Res. 8:
734-744.
Pizzonia, J. H., Gesek, F. A, Kennedy, S. М., Coutermarach, B. A.,
Bacskal, B. J. & Friedman, P. A. (1991). Immunomagnetic
separation, primary culture, and characterisation of cortical
thick ascending limb plus distal convoluted tubule cells from
mouse kidney. InVitro Cell Dev. Biol. 27 A: 409-416.
Planas-Silva, M. D. & Weinberg, R. A. (1997). The restriction
point and control of cell proliferation. Curr. Opin. Cell Biol.
9:768-772.
Planz, B., Wang, Q., Kirley, S. D., Lin, C. W. & McDougal, W. S.
(1998). Androgen responsiveness of stromal cells of the human
prostate: Regulation of cell proliferation and keratinocyte
growth factor by androgen./. Urol. 160:1850-1855.
Platsoucas, C. D., Good, R. A. & Gupta, S. (1979). Separation of
human lymphocyte-T subpopulations (T-mu, T-gamma) by
density gradient electrophoresis. Proc. Natl. Acad. Sci. USA
76:1972.
Plumb, J. A., Milroy, R. & Kaye, S. B. (1989). Effects of the pH
dependence of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltet-
razolium bromide-formazan absorption on chemosensitivity
determined by a novel tetrazolium-based assay Cancer Res.
49: 4435-4440.
Pollack, M. S., Heagney, S. D., Livingston, P. O. & Fogh, J.
(1981). HLA-A, В, С & DR alloantigen expression on forty-
six cultured human tumor cell lines .J. Natl. Cancer Inst. 66:
1003-1012.
Pollack, R. (1981). Readings in mammalian cell culture, 2nd ed. Cold
Spring Harbor, NY Cold Spring Harbor Laboratory Press.
Pollard, T. D. & Borisy, G. G. (2003). Cellular motility driven by as¬
sembly and disassembly of actin filaments. Cell 112:453-465.
Pollard, J. W. & Walker, J. M. (eds.). (1990). Animal ceUculture:
Methods in molecular biology, 5. Clifton, NJ, Humana Press,
pp. 83-97.
Pollock, G. S., Franceschini, I. A., Graham, G. & Barnett, S. C.
(1999). Neuregulin is a mitogen and survival factor for old-
factory bulb ensheathing cells and a related isoform is pro¬
duced by astrocytes. Eur. J. Neurosci. 11: 769-780.
Pollock, M. F. & Kenn, G. E. (1963). Mammalian cell cultures
contaminated with pleura-pneumonia-like organisms; III:
Elimination of pleura-pneumonia-like organisms with specific
antiserum. Proc. Soc. Exp. Biol. Med. 112:176-181.
Plnaszek, N., Kwabi-Addo, B., Wang, J. & Ittmann, M. (2004).
FGF 17 is an autocrine prostatic epithelial growth factor
and is upregulated in benign prostatic hyperplasia. Prostate
60:18-24.
Pontecorvo, G. (1975). Production of mammalian somatic cell
hybrids by means of polyethylene glycol treatment. Somat.
Cell Genet. 1:397-400.
Ponten, J. (1975). Neoplasic human glia cells in culture. In:
Fogh, J. (ed.), Human tumor cells in vitro. New York, Plenum,
pp. 175-185.
Ponten, J. & Westermark, B. (1980). Cell generation and aging
of nontransformed glial cells from adult humans. In Fe-
doroff, S. & Hertz, L. (eds .)> Advances in cellular neurobiology,
Vol. 1. New York, Academic Press, pp. 209-227.
Poot, М., Hoehn, H., Kubbies, М., Grossmann, A., Chen, Y. &
Rabinovitch, P. S. (1994). Cell-cycle analysis using continu¬
ous bromodeoxyuridine labeling and Hoechst 33358-ethidium
bromide bivariate flow cytometry. Methods Cell Biol. 41:
327-340.
Post, М., Floros, J. & Smith, В. T. (1984). Inhibition of lung
maturation by monoclonal antibodies against fibroblast-
pneumocyte factor. Nature 308: 284-286.
Список литературы
675
Potten, С. S., Booth, С., Tudor, G. L., Booth, D., Brady, G., Hur¬
ley, P., Ashton, G., Clarke, R., Sakakibara, S. & Okano, H.
(2003). Identification of a putative intestinal stem cell and
early lineage marker; musashi-1. Differentiation 71:28-41.
Potter, H., Weir, L. & Leder, P. (1984). Enhancer-dependent
expression of human k immunoglobulin genes introduced
into mouse pre-B lymphocytes by electroporation. Proc. Natl.
Acad. Sci. USA 81:7161-7165.
Powell, C. A., Smiley, B. L., Mills, J. & andenburgh, H. H.
(2002). Mechanical stimulation improves tissue-engineered
human skeletal muscle. Am. J. Physiol. Cell Physiol. 283:
C1557-C1565.
Powers, D. A. (1989). Fish as model sytems. Science 246:
352-357.
Pragnell, I. B., Wright, E. G., Lorimore, S. A., Adam, J., Rosendaal,
М., deLamarter, J. F., Freshney, М., Eckmann, L., Sproul,
A. & Wilkie, N. (1988). The effect of stem cell proliferation
regulators demonstrated with an in vitro assay. Blood 72:
196-201.
Prasad, K. N. & EdwardsPrasad, J., Ramanujam, S. & Sakamoto,
A. (1980). Vitamin E increases the growth inhibitory and
differentiating effects of tumour therapeutic agents on neu¬
roblastoma and glioma cell in culture. Proc. Soc. Exp. Biol.
Med. 164:158-163.
Preissman, A., Weismann, R., Bucholz, R., Werner, R. G. &
Noe, W. (1997). Investigation on oxygen limitations of
adherent cells growing on macroporous microcarriers. Cyto-
technology 24:121-134.
Pretlow, T. G. & Pretlow, T. P. (1989). Cell separation by gradient
centrifugation methods. Methods Enzymol. 171:462-482.
Pretlow, T. G., Delmoro, С. М., Dilley, G. G., Spadafora, C. G. &
Pretlow, T. P. (1991). Transplantation of human prostatic carci¬
noma into nude mice in Matrigel. Cancer Res. 51:3814-3817.
Prince, G. A., Jenson, A. B., Billups, L. C. & Notkins, A. L. (1978).
Infection of human pancreatic beta cell cultures with mumps
virus. Nature 271:158-161.
Proceedings-NASA bioreactors workshop on regulation of cell
and tissue differentiation (1997). In Vitro Cell. De. Biol. Anim.
33:325-405.
Provision and use of work equipment (1995). Directive 89/355/
EEC and 95/63/EC.
Provision and use of work equipmet. (1992). Health and Safety
Executive, Broad Lane, Sheffield S3 7HQ England.
Puck, Т. T. & Marcus, P. I. (1955). A rapid method for viable
cell titration and clone production with HeLa cells in tissue
culture: The use of X-irradiated cells to supply conditioning
factors. Proc. Natl. Acad. Sci. USA 41:432-437.
Puck, Т. Т., Cieciura, S. J. & Robinson, A. (1958). Genetics
of somatic mammalian cells; III: Long term cultivation of
euploid cells from human and animal subjects./. Exp Med.
108:945-956.
Punchard, N., Watson, D., Thomson, R. & Shaw, M. (1996).
Production of immortal human umbilical vein endothelial
cells. In Freshney, R. I. & Freshney, M. G. (eds.), Culture of
immortalized cells. New York, Wiley-Liss, pp. 203-238.
Qwarles, J. М., Morris, N. G. & Leibovitz, A. (1980). Car-
cinoembryonic antigen production by human colorectal
adenocarcinoma cells in matrix-perfusion culture. In Vitro
16:113-118.
Quax, P. H., Frisdal, E., Pederson, N., Bonavaud, S., Thibert, P.,
Martelly, I., Verheijen, J. H., Blasi, F. & Barlovatz-Mei-
mon, G. (1992). Modulation of activities and RNA level of
the components of the plasminogen activation system during
fusion of human myogenic satellite cells in vitro. Dev. Biol.
151:166-175.
Quintanilla, М., Brown, K., Ramsden, M. & Balmain, A. (1986).
Carcinogen specific mutation and amplification of Ha-ras
during mouse skin carcinogenesis. Nature 322: 78-79.
Quon, M. J. (1998). Transfection of rat adipose cells by electro¬
poration. In Ravid, K. & Freshney, R. I. (eds.), DNA transfer
to cultured cells. New York, Wiley-Liss, pp. 93-110.
Rabito, C. A., Tchao, R., Valentich, J. & Leighton, J. (1980). Ef¬
fect of cell substratum interaction of hemicyst formation by
MDCK cells. In Vitro 16:461-468.
Radisky, D. C., Hirai, Y., Bissell, M. J. (2003). Delivering the
message: epimorphin and mammary epithelial morphogenesis.
Trends Cell Biol. 13:426-34.
Raff, М. C. (1990). Glial cell diversification in the rat optic nerve.
Scince 24:1450-1455.
Raff, М. C., Abney, E., Brockes, J. P. & Homby-Smith, A. (1978).
Schwann cell growth factors. Cell 15:813-822.
Raff, М. C., Fields, K. L., Hakomori, S. L., Minsky, R.,
Pruss, R. M. & Winter, J. (1979). Cell-type-specific markers
for distinguishing and studying neurons and the major classes
of glial cells in culture. Brain Res. 174:283-309.
Rainov, N. G. & Ren, H. (2003). Clinical trials with retrovirus
mediated gene therapy-what have we learned?J. Neurooncol.
65:227-36.
Raisman, G. (1985). Specialized neuroglial arrangement may
explain the capacity of vomeronasal axons to reinnerveate
central neurons. Neuroscience 14:237-254.
Raisman, G. (2001). Olfactory ensheathing cells-another miracle
cure for spinal cord injury? Nat. Rev. Neurosci. 5:369-75.
Rak, J., Mitsuhashi, Y., Erdos, V., Huang, S.-N, Filmus, J. & Ker-
bel, R. S. (1995). Massive programmed cell death in intestinal
epithelial cells induced by three-dimensional growth condi¬
tions: Suppression by mutant c-H-ras oncogene expression.
J. Cell Biol. 11:1587-1598.
Ramaekers, F. C. S., Puts, J. J. G., Kant, A., Moesker, O.,
Jap, P. H. K. & Vooijs, G. P. (1982). Use of antibodies to in¬
termediate filaments in the characterization of human tumors.
Cold Spring Harbor Symp. Quant. Biol. 46:331-339.
Ramon-Cueto, A., Plant,. W., Avila, J. & Bunge, М. B. (1998).
Long-distance axonal regeneration in the transected adult
rat spinal cord is promoted by olfactory ensheathing glia
transplants./. Neurosci. 18:3803-3815.
Ranscht, B., Clapshaw, P. A., Price, J., Noble, M. & Seifert, W.
(1982). Development of oligodendrocytes and Schwann cells
studied with monoclonal antibody against galactocerebroside.
Proc. Natl. Acad. Sci. USA 79:2709-2713.
Rathjen, P. D., Lake, J., Whyatt, L. М., Bettess, M. D. &
Rathjen, J. (1998). Properties and uses of embryonic stem
cells: Prospects for application to human biology and gene
therapy. Reprod. Fertil. Dev. 10: 31-47.
Rattner, A., Sabido, O., Mssoubre, C., Rascle, F. & Frey, J.
(1997). Characterization of human osteoblastic cells: Influ¬
ence of the culture conditions. In Vitro Cell Dev. Biol. Anim.
33: 757-762.
676
Список литературы
Ravid, К. & Freshney, R. I. (1998). DNA transfer to cultured celb.
New York, Wiley-Liss.
Ravid, K., Doi, Т., Beeler, D. L., Kuter, D. J. & Rosenberg, R. D.
(1991). Transcriptional regulation of the rat platelet factor 4
gene: Interaction between an enhancer/silencer domain and
the GATA site. Mol. Cell Biol. 11: 6116-6127.
Ray, М. E., Yang, Z. Q., Albertson, D., Kleer, C. G., Wash¬
burn, J. G., Macoska, J. A. & Ethier, S. P. (2004). Genomic
and expression analysis of the 8pll-12 amplicon in human
breast cancer cell lines. Cancer Res. 64:40-47.
Raz, A. (1982). B16 melanoma cell variants: Irreversible in¬
hibition of growth and induction of morphologic differen¬
tiation by anthracycline antibiotics ./. Natl. Cancer Inst. 68:
629-638.
Reaven, G. M. (1995). The fourth musketeer-from Alexandre
Dumas to Claude Bernard. Diabetologia 38: 3-13.
Reddel, R., Ke, Y., Gerwin, В. I., McMenamin, M. G., Lech-
ner, J. F., Su, R. Т., Brash, D. E., Park, J. B., Rhim, J. S. &
Harris, С. C. (1988). Transformation of human bronchial
epithelial cells by infection with SV40 of adenovirus-12
SV40 hybrid virus or transfection via strontium phosphate
coprecipitation with a plasmid containing SV40 early region
genes. Cancer Res. 48:1904-1909.
Reed, S. I. (2003). Ratchets and clocks: the cell cycle, ubiquity-
lation and protein turnover. Nat. Rev. Mol. Cell Biol. 4(11):
855-864.
Reel, J. R. & Kenney, F. T. (1968). «Superinduction» of tyrosine
transaminase in hepatoma cell cultures: Differential inhibition
of synthesis and turnover by actinomycin D. Proc. Natl. Acad.
Sci. USA 61:200-206.
Reeves, М. E. (1992). A metastatic tumour cell line has greatly
reduced levels of a specific homotypic cell adhesion molecule
activity. Cancer Res. 52:1546-1552.
Reid, L. M. (1990). Stem cell biology hormone/matrix synergies
and liver differentiation. Curr. Opin. Cell Biol. 2:121-130.
Reid, L. M. & Rojkind, M. (1979). New techniques for cultur¬
ing differentiated cells. Reconstituted basement membrane
rafts. In Jakoby, W. B. & Pastan, I. H. (eds.) Methods in
enzymology, vol 57, cell culture. New York, Academic Press,
pp. 263-278.
Reitzer, L. J., Wice, В. M. & Kennel, D. (1979). Evidence that
glutamine, not sugar, is the major energy source for cultured
HeLa cells ./. Biol. Chem. 254:2669-2677.
Relou, I. A., Damen, C. A., van der Schaft, D. W., Groenewe-
gen, G., Griffioen, A. W. (1998). Effect of culture conditions
on endothelial cell growth and responsiveness. Tissue Cell
30:525-30.
Repesh, L. A. (1989) A new in vitro assay for quantitating tumor
cell invasion. Invas. Metast. 9:192-208.
Resnick, J. L, Bixler, L. S. & Donovan, P. J. (1992). Long term
proliferation of mouse primordial germ cells in culture. Nature
359:550-551.
Rheinwald, J. G. & Beckett, M. A. (1981). Tumorigenic keratino-
cyte lines requiring anchorage and fibroblast support support
cultured from human squamous cell carcinomas. Cancer Res.
41: 1657-1663.
Rheinwald, J. G. & Green, H. (1975). Erial cultivation of strains
of human epidermal keratinocytes: The formation of keratin¬
izing colonies from single cells. Cell 6: 331-344.
Rheinwald, J. G. & Green, H. (1977). Epidermal growth factor
and the multiplication of cultured human keratinocytes.
Nature 265:421-424.
Richard, O., Duittoz, A. H. & Heor, Т. K. (1998). Early, middle,
and late stages of neural cells from ovine embryo in primary
cultures. Neuroscience Res. 31: 61-68.
Richler, C. & Yaffe, D. (1970). The in vitro cultivation and dif¬
ferentiation capacities of myogenic cell lines. Dev. Biol. 23:
1-22.
Richmond, A., Lawson, D. H., Nixon, D. W. & Chawla, R. K.
(1985). Characterization of autostimulatory and trans¬
forming growth factors from human cells. Cancer Res. 45:
6390-6394.
Rickwood, D. & Bimie, G. D. (1975). Metrizamide, a new density
gradient medium. FEBSLett. 50:102-110.
Ried, Т., Liyanage, М., du Manoir, S., Heselmeyer, K., Auer, G.,
Macville, M. & Schrock, E. (1997). Tumor cytogenetics
revisited: Comparative genomic hybrization and spectral
karyotyping./ Mol. Med. 75:801-814.
Rieder, CL. & Cole, R. W. (2002). Cold-shock and the mam¬
malian cell cycle. Cell Cycle. 1:169-175.
Rindler, M. J., Chuman, L. М., Jr. (1979). Retention of differen¬
tiated properties in an established dog kidney epithelial cell
line (MDCK)./. Cell Biol. 81:635-648.
Rippon, H. J. & Bishop, A. E. (2004). Embryonic stem cells. Cell
Prolif. 37: 23-34.
Rizzino, A. (2002). Embryonic stem cells provide a powerful and
versatile model system. Vitan Horm. 64:1-42.
Robertson, К. M. & Robertson, C. N. (1995). Isolation and growth
of human primary prostate epithelial cultures. Methods Cell
Sci. 17:177-185.
Robins, D. М., Pipley, S., Henderson, A. S. & Axel, R. (1981).
Transforming DNA integrates into the host chromosome.
Cell 23: 29-39.
Rodbell, M. (1964). Metabolism of isolated fat cells: Effects of
hormones on glucose metabolism and lipolysis./. Biol. Chem.
239:37-380.
Rodriguez, A. М., Elabd, C., Delteil, F., Astier, J., Verno-
chet, C., Saint-marc, P., Guesnet, J., Guezennec, A., Amri, E. Z.,
Dani, C. & Ailhaud, G. (2004). Adipocyte differentiation of
multipotent cells established from human adipose tissue.
Biochem. Biophys. Res. Commun. 315: 255-263.
Rofstad, E. K. (1994). Orthotopic human melanoma xenograft
model systems for stydies of tumor angiogenesis, patho¬
physiology, treatment sensitivity and metastatic pattern. Br.
J. Cancer 70:804-812.
Rogers, A. W. (1994). Techniques, of autoradiography, 3d ed.
Amsterdam, Elsevier/North-Holland Biomedical Press.
Reguet, R., Cohen, C. & Rougier, A. (1994). A reconstituted
human epidermis to assess cutaneous irritation photoirrita¬
tion and photoprotection in vitro. In vitro Skin Toxicol. 10:
141-149.
Rojkind, М., Gatmaitan, Z., Mackensen, S., Giambrone, M. A.,
Ponce, P. & Reid, L. M. (1980). Connective tissue biomatrix:
Its isolation and utilization for long-term cultures of normal
rat hepatocytes./. Cell Biol. 87: 255-263.
Rooney, D. E. & Czepulkowski, В. H. (eds) (1986). Human cy¬
togenetics, a practical approach. Oxford, U.K., IRL Press at
Oxford University Press.
Список литературы
677
Rooney, S. A., Young, S. L. & Mendelson, C. R. (1995). Mole-
sular and cellular processing of lung surfactant. FASEB J. 8:
957-967.
Rosenblatt, J. D., Lunt, A. I., Parry, D. J. & Partridge, T. A.
(1995). Culturing satellite cells from living single muscle fiber
explants. In Vitro Cell. Dev. Biol. 31: 773-779.
Rosenblatt, J. D., Lunt, F. I., Parry, D. J. & Partridge, T. A. (1996).
Phenotype of adult mouse muscle myoblasts reflects their
fiber type of origin. Differentiation 60:39-45.
Rosenfeld, M. A., Yoshimura, K., Trapnell, В. C., Yoneyama, K.,
Rosenthal, E. R., dalemenas, W., Fukayama, М., Bargon, J.,
Stier, L. E., Stratford-Perricaudet, L., Perricaudet, М., Gug-
gino, W. B., Pavirani, A, Lecocq, J.-P. & Crystal, R. G. (1992).
In vivo transfer of the human cystic fibrosis transmembrane
conductance regulator gene to the airway epithelium. Cell
68:143-155.
Rosenman, S. J. & Gallatin, W. M. (1992). Cell surface glyco-
conjugates in intercellular and cell-substratum interactions.
Semin. Cancer Biol. 2:3 57-366.
Rossi, G. B. & Friend, C. (1967). Erythrocytic maturation of
(Friend) virus-induced leukemic cells in s spleen clones. Proc.
Nad. Acad. Sci. USA 58:1373-1380.
Rothblat, G. H. & Morton, H. E. (1959). Detection and possible
source of contaminating pleuropneumonia-like organisms
(PPLO) in culture of tissue cells. Proc. Soc. Exp. Biol. Med.
100:87.
Rothfels, К. H. & Siminovitch, L. (1958). An air drying technique
for flattening chromosomes in mammalian cells growth in
vitro. Stain Technol. 33:73-77.
Rotman, B. & Papermaster, B. W. (1966). Membrane properties
of living mammalian cells as studies by enzymatic hydrolysis of
fluorogenic esters. Proc. Natl. Acad. Sci. USA 55:134-141.
Rous, P. & Jones, F. A. A. (1916). A method of obtaining suspen¬
sions of living cells from the fixed tissues, and for the plating
out of individual cells .J. Exp. Med. 23: 555.
Rowley, S. D. & Byrne, D. V. (1992). Low-temperature storage of
bone marrow in nitrogen vapor-phase refrigerators decreased
temperature gradients with an aluminum racking system.
Transfusion 32750-754.
Rudland, P. S. (1992). Use of peanut lectin and rat mammaru
stem cell lines to identify a cellular differentiation pathway
for the alveolar cell in the rat mammary gland./. Cell Physiol.
153:157-168.
Ruff, M. R. & Pert, С. B. (1984). Small cell carcinoma of the lung:
Macrophage-specific antigens suggest hematopoietic stem cell
origin. Science 225:1034-1036.
Rules and guidance for pharmaceutical manufacturers and distrib¬
utors. (1997). The Stationery Office, Ltd., London, U.K.
Rundlett, S. E., Gordon, D. A. & Miesfeld, R. L. (1992). Char¬
acterisation of a panel of rat ventral prostate epithelial cell
lines immortalized in the presence or absence of androgens.
Exp. Cell Res. 203: 214-221.
Ruoff, N. M. & Hay, R. J. (1979). Metabolic and temporal studies
on pancreatic exocrine cells in culture. Cell Tissue Res. 204:
243-252.
Rush, L. J., Heinonen, K., Mrozek, K., Wolf В .J., Abdel-Rah-
man, М., Szymanska, J., Peltomaki, P., Kapadia, F., Bloom¬
field, C. D., Caligiuri, M. A & Plass, C. (2002). Comprehensive
cytogenetic and molecular genetic characterization of the Tl 1
acute myeloid leukemia cell line reveals cross-contamination
with K-562 cell line. Blood 99:1874-1876.
Russo, J., Calaf, G., Russo, I. H. (1993). A critical approach to
the malignant transformation of human breast epithelial
cells with chemical carcinonogens. Crit. Rev. Oncog. 4 (4):
403-417.
Russo, A. A., Tong, L., Lee, J. O., Jeffrey, P. D. & Pavletich, N. P.
(1998). Structural basis for inhibition of the cyclin-dependent
kinase cdk6 by the tumour suppressor pl61NK4a. Nature
395: 237-243.
Putzky, L. P., TomitaJ. Т., Calenoff, M. A. & Kahan, B. D. (1979).
Human colon adenocarcinoma cells; III: In vitro organoid
expression and carcino-embryonic antigen kinetics in hollow
fiber culture./. Natl. Cancer Inst. 63:893-902.
Rygaard, K., Moller, C., Bock, E. & Spang-Thomsen, M. (1992).
Expression of cadherin and NCAM in humen stall cell lung
cancer lines and xenografts. Br. J. Cancer 65:573-577.
Saalbach, A., Aust, G., Haustein, U. F., Herrmann, K. & An-
deregg, U. (1997). The fibroblast-specific MAb AS02: a novel
tool for detection and elimination of human fibroblasts. Cell
Tissue Res. 290:593-599.
Sachs, L. (1978). Control of normal cell differentiation and the
phenotypic reversion of malignancy in myeloid leukaemia.
Nature 274:535-539.
Safe Use of Work Equipment. Provision and use of Work Equip¬
ment. Provision and Use of Work Equipment Regulations.
(1998). Approved Code of Practice and Guidance, L22, HSE
Books, ISBN 0 7176 1626 6.
Safe working and the prevention of infection in clinical laboratories.
(1991). Health and Safety Commission, HMSO Publication,
P.O. Box 276, London, SW8 5DT, England.
Sager, R. (1992). Tumor suppressor genes in the cell cycle. Curr.
Opin. Cell Biol 4:155-160.
Sax, R., Thonar, E. & Masuda, K. (1991). Tissue engineering of
articular cartilage. In Vinjak-Novakovic, G., Freshney. R. I.
(eds.), Culture of cells for tissue engineering. Hoboken, HJ,
Wiley-Liss (In press).
Saier, M. N. (1984). Hormonally defined, serum free medium for
a proximal tubular kidney epithelial cell line, LLC-PKI. In
Bames, WD (ed.,), Methods for serum free culture of epithelial
and fibroblastic cells. New York, Alan R. Liss, pp. 25-31.
Sambrook, J., Fritsch, E. F. & Maniatis, T. (1989). Molecular
cloning: A laboratory manual, 2d ed. Use of recombinant
Cold Spring Harbor, NY, Cold Spring Harbor Laboratory
Press, 3 vols.
Sanchez-Ramos, J. R. (2002). Neural cells derived from adult
bone marrow and umbilical cord neityblood./. Neurosci. Res.
69:880-893.
Sandberg, A A. (1982). Chromosomal changes in human cancers:
Specificity and heterogeneity. In Owens, A H., Coffey, D. S. &
Baylin, S. B. (eds.), Tumour cell heterogeneity. New York,
Academic Press, pp. 367-397.
SanesJ.R., Rubenstein, J. L. & Nicolas, J. F. (1986). Use of re¬
combinant retrovirus to study post-implantation cell lineage
in mouse cinbryos. EMBOJ. 5:3133-3142.
Sanford, К. K., Earle, W. R. & Likely, G. D. (1948). The growth in
vitro of single isolated tissue cells./ Natl. Cancer Inst. 11:773.
Sanford, К. K., Westfall, В. B., Fioramonti, М. C., Mc-
Quilkin, W. Т., Bryant, J. C., Peppers, E. V. & Earle, W. R.
678
Список литературы
(1955). The effect of serum fractions on the proliferation of
strain L mouse cells in vitro./. Natl. Cancer. Inst. 16: 789.
Sarang, Z., Haig, Y., Hansson, A., Vondracek, М., Wamgard, L, Graf¬
strom, R. C. (2003). Microarray assessment of fibronectin, col¬
lagen and integrin expression and the role fibronectin-collagen
coating in the growth of normal, SV40 T-antigen-immortalized
and malignant human oral keratinocytes. ATLA, 31:575-585.
Sasaki, М., Honda, Т., Yamada, H., Wake, N. & Barrett, J. C.
(1966). Evidence for multiple pathways to cellular senescence.
Cancer Res. 54: 6090-6093.
Sato, G. H. & Yasumura, Y. (1966). Retention of differentiated
function in dispersed cell culture. Trans. NY acad. Sci. 28:
1063-1079.
Sattler. G. A., Michalopoulos, G., Sattler, G. L. & Pilot, H. C.
(1978). Ultrastructure of adult rat hepatocytes cultured on
floating collagen membranes. Cancer res. 38:1539-1549.
Sauners, N. A., Bemacki, S. H., Vollberg, Т. M. & Jetten, A. M.
(1993). Regulation of transglutaminase type-I expression in
squamous differentiating rabbit tracheal epithelial, cells hu¬
man epidermal keratinocytes — effects of retinoic acid and
phorbol esters Mol. Endocrinol. 7:387-398.
Scanlon, E. F., Hawkins, R. A Fox, W. W. & Smith, W. S. (1965).
Fatal homotransplanted melanoma (a case report). Cancer
12:782-789.
Schaeffer, W. I. (1990). Terminology associated with cell, tissue
and organ culture, molecular genetics. In Vitro Cell Dev. Biol.
26:97-101.
Shaeffer, K., Herrmuth, H., Mueller, J., Coy, D. H., Wong, H. C.,
Walsh, J. H., Classen, М., Schusdziarra, V. & Schepp, W.
(1997). Bombesin-like tides stimulate somatostatin release
from rat fundic D cells in primary culture. Am. J. Physiol.
Castrointest. Liver Physiol. 273: G686-G695.
Schamblott, M. J., Axelman, J., Stemeckert, J., Christoforou, N.,
Patterson, E. S., Siddiqi, M. A., Kahler, H., Ifeanyi, L. A. &
Gearhart, J. D. (2002). Stem cell culture: pluripotent stem
cells. In Methods of tissue engineering, Atala, A. & Lanza, R. P.,
Eds., San Diego, Academic Press, pp. 411-420.
Schamblott, M.J., Axelman, J., Wang, S., Bugg, E. М., Little¬
field, J. W., Donovan, P. J., Blumethal, P. D., Huggins, G. R. &
Gearhart, J. D. (1998). Derivation of pluripotent stem cells
from cultured human primordial germ cells. Proc. Natl. Acad.
Sci. USA. 95:13726-13731.
Scher, W., Holland, J. G. & Friend, C. (1971). Hemoglobin syn¬
thesis in in murine virus-induced leukemic cells in vitro; I:
Partial purification and identification of hemoglobins. Blood
37:428-437.
Schimmelpfeng, 1., Langenberg, U. & Peters, I. M. (1968). Mac¬
rophages overcome mycoplasma infection of cells in vitro.
Nature 285:661.
Schlechte, W., Brattain, M. & Boyd, D. (1990). Invasion of extra¬
cellular matrix by cultured colon cancer cells; Dependence on
urokinase receptor display. Cancer Commun. 2:173-179.
Schlessinger, J., Lax, I. & Lemmon, M. (1995). Regulation of
growth factor activation by proteoglycans: What is the role
of the low affinity receptors? Cell 83: 357-360.
Schmidt, R., Reichert, U., Michel, S., Shrott, B. & Boullier, M.
(1985). Plasma membrane transglutaminase and comified
envelope competence in cultured human keratinocytes. FEBS
Lett. 186:204.
Schneider, E. I. (1975). A simple biochemical technique for the
detection of mycoplasma contamination of cultured cells.
Methods Сей Biol. 10: 278-290.
Schoenlein, P. V., Shen, D.-W., Barrett, J. Т., Pastan, I. T. & Got-
tesman, М. M (1992). Double minute chronosomes carrying the
human multidrug resistance 1 and 2 genes are generated from
the dimerization of submicroscopis circular DNAs in colchi¬
cine-selected KB carcinoma cells. Mol. Biol. Cell 3:507-520.
Schor, S. L. (1994). Cytokine control of cells motility modula¬
tion and mediation by the extracellular matrix. Prig. Growth
Factor Res. 5:223-248.
Schousboe, A., Thorbek, P., Hertz, L. & Krogsgaard-Larsen, P.
(1997). Effects of GAB A analogues of restricted conformation
on GAB A transport in astrocytes and brain cortex slices and
on GAB A receptor binding./. Neurochem. 33:181-189.
Schulman, H. M. (1968). The fractionation of rabbit reticulo¬
cytes in dextran density gradients. Biochim. Biophys. Acta
148:251-255.
Schwartz Albiez, R., Heidtmann, H.-H., Wolf, D., Schirrma-
cher, V. & Moldenhauer, G. (1991). Three types of human
lung tumour cell lines can be distinguished according to
surface expression of endogenous urokinase and their capacity
to bind exogenous urokinase. Br. J. Cancer 65:52-57.
Schwob, J. (2002). Neuronal regeneration and the peripheral
olfactory system. Anat. Rec. 269:33-48.
Scott, W. N., McCool, K. & Nelson, J. (2000). Improved method
for the production of gold colloid monolayers for use in the
phagokinetic track assay for cell motility. Anal. Biochem.
287:343-344.
Scotto, K. W., Biedler, I. L. & Melera, P. W. (19860. Amplification
and expression of genes associated with multidrug resistance
in mammalian cells. Science 232:751-755.
Seeds, N. W. (1971). Biochemical differentiation in reaggregating
brain cell culture. Proc. Natl. Acad. Sci. USA 68:1858-1861.
Seglen, P. O. (1975). Preparation of isolated rat liver cells. Meth¬
ods Cell Biol. 13:29-83.
Seidel, J. O., Pei, М., Gray, M. L., Langer, R., Freed, L. E. &
Vunjak-Novakovic, G. (2004). Long-term culture of tissue
engineered cartilage in a perfused chamber with mechanical
stimulation. Biorheology 41:445-458.
Seifert, W. & Muller, H. W. (1984). Neuron-glia interaction in
mammalian brain: Preparation and quantitative bioassay
of a neurotropic factor (NTF) from primary astrocytes. In
Barnes, D. W., Sirbasku, D. A. & Sato, G. H. (eds.), Methods
for serum free culture of neuronal and lymphoid cells. New
York, Alan R. Liss, pp. 67-78.
Seigel, G. A. (1996). Establishment of an E la-immortalised reti¬
nal cell culture. In vitro Cell Dev. Biol. Anim. 32: 66-68.
Selby, P. J., Thomas, M. J., Monaghan, P., Sloane, J. & Peck-
ham, M. J. (1980). Human tumour xenografts established
and serially transplanted in mice immunologically deprived
by thymectomy, cytosine arabinoside and whole-body irradia¬
tion. Br. J. Cancer 41:52.
Selden, R. F., Howie, К. B., Rowe, М. E., Goodman, H. M. &
Moore, D. (1986). Human growth hormone as a reporter gene
in regulation studies employing transient gene expression.
Mol. Cell Biol 6:3173-3179.
Seruya, М., Shah, A., Pedrotty, D., du Laney, Т., Melgiri, R.,
Mckee, J. A., Young, H. E. & Niklason, L. E. (2004). Clonal
Список литературы
679
population of adult stem cells: life span and differentiation
potential. Cell Transplant. 13:93-101.
Shah, G. (1999). Why do we still use serum in the production of
biopharmaceuticals? Dev. Biol. Stand. 99:17-22.
Shall, S. (1973). Sedimentation in sucrose and Ficoll gradients
of cells growth in suspension culture. In Kruse, P. F. & Pat¬
terson, М. K. (eds.), Tissue culture methods and applications.
New York, Academic Press, pp. 198-204.
Shall, S. & McClelland, A. J. (1971). Synchronization of mouse
fibroblast LS cells growth in suspension culture. Nat. New
Biol. 229:59-61.
Shansky, J., Chromiak, J., Del Tatto, M. & Vandenburgh, H.
(1997). A simplified method for tissue engineering skeletal
muscle organoids in vitro. In Vitro Cell Dev. Biol. Anim. 33:
659-661.
Shansky, J., Ferland, P., McGuire, S., Powell, C., DelTatto, М.,
Nackan, М., Hennessey, J. & Vandenburgh, H. H. (2005). Tissue
engineering human skeletal muscle for clinical applications. In
Vunjak-Novakovic, G. & Freshney, R. I. (eds.), Culture of cells
for tissue engineering. Hoboken, NJ, Wiley-Liss (in press).
Sharpe, P. T. (1988). Methods of cell separation. Amsterdam,
Elsevier.
Shaw, R. G., Johnson, A. R., Schulz, W. W., Zahlten, R. N. &
Combes, B. (1984) Hepatology 4:591-602.
Shay, J. W. & Wright, W. E. (1998). Quantitation of the fre¬
quency of immortalization of normal human mammary
diploid fibroblasts by SV 40 large T antigen. Exp. Cell Res.
184:109-118.
Shay, J. W., Wright, W. E., Brasiskyte, D. & der Haegen, A.
(1993). E6 of human papillomavirus type 16 can overcome the
Ml stage of immortalization in human mammary epithelial
cells but not in human fibroplasts. Oncogene 8:1407-1413.
Shea, С. М., Edgar, С. М., Teinhom, T. A., Louis, C. & Gersten-
feld, L. C. (2003). BMP treatment of C3H10T1/2 mesen¬
chymal stem induces both chondrogenesis and osteogenesis.
J. Cell. Biochem. 90:1112-1127.
Shiga, М., Kapila, Y. 1., Zhang, Q., Hayami, T. & Kapila, S. (2003).
Ascorbic acid induces collagenase-1 in human periodontal
ligament cells but not in MC3T3-E1 osteoblast-like cells:
potential association between collagenase expression and
changes in alkaline phosphatase phenotype. J. Bone Miner.
Res. 18: 67-77.
Shih, S. J., Dall’Era, M. A., Westphal, J. R., Yang, J., Sweep, C. G.,
Gandour-Edwards, R. & Evans, C. P. (2003). Elements
regulating angiogenesis and correlative microvessel density
in benign hyperplastic and malignant prostate tissue. Prostate
Cancer Prostatic Dis. 6:131-137.
Shipley, C. D. & Ham, R. G. (1983). Multiplication of Swiss 3T3
cells in a serum-free medium. Exp. Cell Res. 146:249-260.
Simon-Assmann, P., Kedinger, M. & Haffen, K. (1986). Immu-
nocytochemical localization of extracellular matrix protoin
in relation to rat intestinal morphogenesis. Differentiation.
32: 59-66.
Simonsen, J. L., Rosada, C., Serakinci, N., Justesen, J., Stenderup,
K., Rattan, S. I., Jensen, T. G. & Kassem, life-span and main¬
tains the osteogenic potential of human bone marrow stromal
cells. Nat. Biotechnol. 20:560-561.
Sinha, М. K., Buchanan, C., Raineri-Maldonado, C., Khazanie, P.,
Atkinson, S., DiMarchi, R. & Caro, J. F. (1990). IGF-II recep¬
tors and IGF-II-stimulated glucose transport in human fat
cells. Am.J. Physiol. 258: E534-E542.
Skehan, P., Smoreng, R., Scudie D., Monks, A., McMahon, JVis-
tica, D., Warren, J. Т., Bokesch, H., Kenney, S. & Boyd, M. R.
(1990). New colorimetric cytotoxicity assay for anticancer-
drug screening./ Natl. Cancer Inst. 82:1107-1112.
Skode, M. & Fusenig, N. E. (1998). Tumorigenic conversion of
immortal human keratinocytes through stromal cell activa¬
tion. Proc. Natl. Acad. Sci. USA 95:1-6.
Smith, A. D., Datta, S. P., Smith, G. H., Campbell, P. N., Bent¬
ley, R. & Mckenzie, H. A. (eds.). (1997). Oxford dictionary
of biochemistry and molecular biology, Oxford, Oxford Uni¬
versity Press.
Smith, A. G., Heath, J. K., Donaldson, D. D., Wong, G. G.,
Moreau, J., Stahl, M. & Rodgers, D. (1988). Inhibition of plu-
ripotential stem cell differentiation by purified polypeptides.
Nature 336: 688-690.
Smith, H. S., Lan, S., Geriani, R., Hackett, A. J. & Stampfer, M. R.
(1981). Clonal proliferation of cultured non-malignant and ma¬
lignant human breast epithelia. Cancer Res. 41:4637-4643.
Smith, H. S., Owens, R. B., Hiller, A. J., Nelson-Rees, W. A. &
Johnston, J. O. (1976). The biology of human cells in tissue
culture; I: Characterization of cells derived from osteogenic
sarcomas. Int.J. Cancer 17:219- 234.
Smith, M. D., Summers, M. D. & Frazer, M. J. (1983). Produc¬
tion of human beta interferon in insect cells infected with a
baculovirus expression vector. Mol. Cell Biol. 3:2156-2165.
Smith, P. K., Krohn, R. I., Hermanson, G. Т., Mallia, A. K., Gart¬
ner, F. H., Provenzano, M. D., Fujimoto, E. K., Goeke, N. V.,
Olson, B. J. & Klenk, D. C. (1985). Measurement of protein
using bicinchoninic acid. Anal. Biochem. 150:235-239.
Smith, S. & de Lange, T. (1997). TRFL, a mammalian telomeric
trotein. Trends Genet. 113: 21-26.
Smith, W. L. & Garcia-Perez, A. (1985). Immunodissection: Use
of monoclonal antibodies so isolate specific types of renal cells.
Am.J. Physiol. 248: F1-F7.
Smola, H., Stark, H.-J., Thiekotter, G., Mirancea, N., Krieg, T. &
Fusenig, N. E. (1998). Dynamics of basement membrane for¬
mation by keratinocyte-fibroblast in organotypic skin culture.
Exp. Cell Res. 239:399-410.
Smola, H., Thiekotter, G. & Fusenig, N. E. (1993). Mutual induc¬
tion of growth factor gene expression by epidermal-dermal
cell interaction./. Cell Biol. 122:417-429.
Smyth, M. J., Rodney, L. Sparks, R. L., Wharton, W. (1993).
Proadipocyte cell lines: models of cellular proliferation and
differentiation./ Cell Sci. 106:1-9.
Snyder, E. Y., Deitcher, D. L., Walsh, C., Arnold Aldea, S.,
Hartwieg, E. A., Cepko, C. L. (1992). Multipotens neural cell
lines can engraft and participate in development of mouse
cerebellum. Cell 68,33-51.
Soder, A. I., Going, J. J., Kaye, S. B. & Keith, W. N. (1998). Tu¬
mour specific regulations of telomerase RNA gene expression
visualized by in situ hybridization. Oncogene 16:979-983.
Soder, A. I., Hoare, S. F., Muir, S., Going, J. J., Parkinson, E. K. &
Keith, W. N. (1997). Amplification, increased dosage an in
situ expression of the telomerase RNA gene in human cancer.
Oncogene 14:1013-1021.
Solassol, J., Crozet, C. & Lehmann, S. (2003). Prion propagation
in cultured cells. Br. Med. Bull. 66: 87-97.
680
Список литературы
Sommar, I. & Schachner, М. (1981). Cells that are 04-antigen-
positive and 01-antigen-negative differentiate into Ol anti¬
gen-positive oligodendrocytes. Neurosci. Lett. 29:183-188.
Soon-Shiong, P., Feldman, E., Nelson, R., Komtebedde,J., Smid-
srod, O., Skjak-Br®k, G., Espevik, Т., Heintz, R. & Lee, M.
(1992). Successful reversal of spontaneous diabetes in dogs
by intraperitoneal microencapsulated islets. Transplantation
54:769-774.
Sordillo, L. М., Oliver, S. P. & Akers, R. M. (1998). Culture of
bovine mammary epithelial cells in d-valine modified medium:
Selective removal of contaminating fibroblasts. Cell Biol. Int.
Rep. 12:355-364.
Soriano, V., Pepper, M. S., Nakamura, Т., Orci, L. & Montesano, R.
(1995). Hepatocyte growth factor stimulates extensive devel¬
opment of branching duct-like structures by cloned mammary
gland epithelial cells ./. Cell Sci. 108:413-430.
Sorieul, S. & Ephrussi, B. (1961). Karyological demonstration
of hybridization of mammalian cells in vitro. Nature 190:
653-654.
Sorour, O., Raafat, М., El-Bolkainy, N. & Mohamad, R. (1975).
Infiltrative potentiality of brain tumors in organ culture.
J. Neurosurg. 43: 742-749.
Soule, H. D., Maloney, Т. М., Wolman, S. R., Teterson, W. D.,
Brenz, R., McGrath, С. М., Russo,J., Pauley, R.J.,Jones, R. F. &
Brooks, S. C. (1990). Isolation and characterization of a spon¬
taneously immortalized human breast epithelial cell line,
MCF-10. Cancer Res. 50:6075-6086.
Soule, H. D., Vasquez, J., Long, A., Albert, S. & Brennan, M.
(1973). A human cell line from a pleural effusion derived from
abreast carcinoma./ Natl Cancer Inst. 51:1409-1416.
Southam, С. M. (1958). Homotransplantation of mammalian cells
to antibiotic resistance with a bacterial gene under control of the
SV40 early region promoter./. Mol. App. Genet. 1:327-341.
Southern, P. J. & Berg, P. (1982). Transformation of mammalian
cells to antibiotic resistance with a bacterial gene under con¬
trol of the SV40 early region promoter./. Mol. App. Genet.
1:327-399.
Southgate, J., Masters, J. W. R. & Trejdosiewicz, L. K. (2002). In
Freshney, R. I. & Freshney, M. G. (eds), Culture of epithelial
cells, 2nd ed. New York, Wiley-Liss, pp. 383-399.
Speir, R. E., Griffiths, J. B. & Meignier, B. (eds). (1991). Produc¬
tion of biological from animal cells in culture. Oxford, U.K.,
Butterworth- Heinemann.
Speir, R. & Griffiths, J. B. (1985-1990). Animal cell biotechnology.
London, Academic Press, 4 vols.
Speirs, V. (2004). Primary culture of human mammary tumor
cells. In Pfagner, R. & Freshney. R. I. (eds.), Culture of human
tumor cells. Hoboken, NJ, Wiley-Liss, pp. 205-219.
Speirs, V., Green, A. R. & White, М. C. (1996). Collagenase III:
A superior enzyme for complete disaggregation and improved
vitability of normal and malignant human breast tissue. In
Vitro Cell Dev. Biol. Anim. 32: 72-74.
Speirs, V., Ray, K. P. & Freshney, R. I. (1991). Paracrine control
of differentiation in the alveolar carcinoma, A549, by human
foetal lung fibroblasts. Br. J. Cancer 64:693-699.
Spinelli, W., Sonnenfeld, К. H. & Ishii, N. (1982). Effects of
phorbol ester tumor promoters and nerve growth factor on
neurite outgrowth in cultured human neuroblastoma cells.
Cancer Res. 42: 5067-5073.
Splinter, T. A. W., Beudeker, M. & Beek, A. V. (1978). Changes
in cell density induced by isopaque. Exp. Cell Res. Ill:
245-251.
Spooncer, E., Eliason, J. & Dexter, Т. M. (1992). Long-term mouse
bone marrow cultures. In Testa, N. G. & Molineux, G. (eds.),
Haemopoiesis: a practical approach. Oxford, U.K., IRL Press
at Oxford University Press, pp. 57-74.
Spremulli, E. N. & Dexter, D. L. (1984). Polar solvents: A novel
class of antineoplastic agents./ Clin. Oncol. 2: 227-241.
Sredni, B., Sieckmann, D. G., Kumagai, S. H., Green, I. &
Paul, W. E. (1981). Long term culture and cloning of
non-transformed human B-lymphocytes. J. Exp. Med. 154:
1500-1516.
Stacey, G. N., Bolton, B. J. & Doyle, A. (1993). Multilocus
DNA fingerprinting used for definitive isolation of HeLa
contamination in cell lines and determination of genetic
diversity amongst HeLa cell clones. In Vitro Cell Dev. Biol.
29 A: 123 A.
Stacey, G. N., Bolton, B. J., Morgan, D., Clark, S. A. & Doyle, A.
(1992). Multilocus DNA fingerprint analysis of cell-banks:
Stability studies and culture identification in human B-
lymphoblastoid and mammalian cell lines. Cytotechnology
8:13-20.
Stacey, G. N., Hoelzl, H., Stehenson, J. R. & Doyle, A. (1997).
Authentication of animal cell cultures by cultures by direct
visualization of repetitive DNA, aldolase gene PCR and iso¬
enzyme analysis. Biologicals 25:75-85.
Stacey, G. N., Masters, J. R. М., Hay, R. J., Drexler, H. G., Mac¬
Leod, R. A. F. & Freshney, R. I. (2000). Cell contamination
leads to inaccurate data: we must take action now. Nature
403:356.
Stampfer, М., Halcones, R. G. & Hackett, A. J. (1980). Growth
of normal human mammary cells in culture. In Vitro 16:
415-425.
Stampfer, M. R., Yaswen, P. & Taylor-Papadimitriou, J. (2002).
Culture of human mammary epithelial cells. In Fresh¬
ney, R. I. & Freshney, M. G. (eds.), Culture of epithelial cells,
2nd ed. Hoboken, NJ, Wiley-Liss, pp. 95-135.
Stanbridge, E. J. & Doerson, C.-J. (1978). Some effects that my-
coplasmas have upon their injected host. In McGarrity, G. J.,
Murphy, D. G. & Nichols, W. W. (eds.), Mycoplasma infection
of cell cultures. New York, Plenum Press, pp. 119-134.
Stanley, M. A. & Parkinson, E. (1979). Growth requirements of
human cervical epithelial cells in culture. Int.J. Cancer 24:
407-414.
Stanley, M. A. (2002). Culture of human cervical epithelial cells.
In Freshney, R. I. & Freshney, M. G. (eds.), Culture of epithe¬
lial cells, 2nd ed. Hoboken, NJ, Wiley-Liss, pp. 138-169.
Stanners, C. P., Eliceri, G. L. & Green, H. (1971). Two types of
ribosome in mouse-hamster hybrid cells. Nat. New Biol. 230:
52-54.
Stanton, B. A., Biemesderfer, D., Wade, J. B. & Giebisch, G.
(1981). Structural and functional study of the rat distal
nephron: Effects of potassium adaptation and depletion.
Kidney Int. 19:36-48.
Stark, H. J., Baur, М., Breitkreutz, D., Mirancea, N. & Fuse¬
nig, N. E. (1999). Organotypic keratinocyte cocultures is
defined medium with regular epidermal morphogenesis and
differentiation./ Invest. Dermatol. 112: 681-691.
Список литературы
681
States, В., Foreman, J., Lee,J. & Segal, S. (1986). Characteristics
of cultured human renal cortical epithelia. Biochem. Med.
Metab. Biol. 36:151-161.
Steel, G. G. (1979). Terminology in the description of drug-
radiation interactions. Int. J. Radiat. Oncol. Biol. Phys. 5:
1145-1150.
Steele, M. P., Levine, R. A., Joyce-Brady, M. & Brody, J. S. (1992).
A rat alveolar type II cell line developed by adenovirus 12
SE1A gene transfer. Am.J. Resp. Cell Mol. Biol. 6: 50-56.
Stein, H. G. & Yanishevsky, R. (1979). Autoradiography. In
Jakoby, W. B. & Pastan, I. H. (eds.), Methods in enzy¬
mology; vol. 57: Cell culture. New York, Academic Press,
pp. 279-292.
Steinberg, M. L. (1996). Immortalization of human epidermal
keratinocytes by SV40. In Freshney, R. I. & Freshney, M. G.
(eds.), Culture of immortalizes cells. New York, Wiley-Liss,
pp. 95-120.
Stem, P., West, C. & Burt, D. (2004). Culture of cervical car¬
cinoma cell lines. In Pfragner, R. & Freshney, R. I. (eds.),
Culture of human tumor cells. Hoboken, NJ, Wiley-Liss,
pp. 179-204.
Steube, K. G., Grunicke, D., Drexler, H. G. (1995). Isoenzyme anal¬
ysis as rapid method for the examination of the species identity
of cell cultures. In Vitro Cell Dev. Biol. Anim. 31:115-9.
Stewart, С. E. H., James, P. L., Fant, М. E. & Rotwein, P. (1996).
Overexpression of insulin-like growth factor-II induces accel¬
erated myoblast differentiation./ Cell Physiol. 169: 23-32.
Stoker, M. G. P. (1973). Role of diffusion boundary layer in
contact inhibition of growth. Nature 246:200-203.
Stoker, M. G. P. & Rubin, H. (1967). Density dependent inhibi¬
tion of cell growth in culture. Nature 215:171-172.
Stoker, М., O’Neill, C., Berryman, S. & Waxman, B. (1968).
Anchorage and growth regulation in normal and vims trans¬
formed cells. Int.J. Cancer 3:683-693.
Stoker, М., Perryman, M. & Eeles, R. (1982). Clonal analysis
of morphological phenotype in cultured mammary epithe¬
lial cells from human milk. Proc. R. Soc. Lond. set. B. 215:
231-240.
Stoner, G. D., Katoh, Y., Foidart, J.-M., Tmmph, B. F., Stei-
nert, P. & Harris, С. C. (1981). Cultured human bronchial
epithelial cells: Blood group antigens, keratin, collagens and
fibronectin. In Vitro 17:577-587.
Strange, R., Li, F., Fris, R. R., Reichmann, E., Haenni, B. &
Burn, P. H. (1991). Mammary epithelial differentiation in
vitro: Minimum requirements for a functional response to
hormonal stimulation. Cell Growth Differ. 2: 549-559.
Strangeways, T. S. P. & Fell, H. B. (1925). Experimental studies
on the differentiation of embryonic tissues growing in vivo
and in vitro. I. The development of the undifferentiated limb-
bud (a) when subcutaneously grafted into the post-embryonic
chick and (b) when cultivated in vitro. Proc. Roy. Soc. London.
ser.B 99:340-366.
Strangeways, T. S. P. & Fell, H. B. (1926). Experimental studies
on the differentiation of embryonic tissues growing in vivo
and in vitro. II. The development of the isolated early embry¬
onic eye of the fowl when cultivated in vitro. Proc. Roy. Soc.,
London, ser. В 100: 273-283.
Strauss, W. M. (1998). Transfection of mammalian cells with
yeast artificial chromosomes. In Ravid, K. & Freshney, R. I.
(eds.), DNA transfer to cultured cells. New York, Wiley-Liss,
pp. 213-236.
Strickland, S. & Beers, W. H. (1976). Studies on the role of plas¬
minogen activator in ovulation: In vitro response of granulose
cells to gonadotropins, cyclic nucleotides, and prostagland-
ings./ Biol. Chem. 251: 5694-5702.
Stryer, L. (1995). Biochemistry, 4th ed. New York, W.H. Freemann,
p. 505.
Stubblefield, E. (1968). Synchronization methods for mammalian
cell cultures. In Prescott, D. M. (ed.), Methods in cell physiol¬
ogy. New York, Academic Press, pp. 25-43.
Styles, J. A. (1997). A method for detecting carcinogenic organic
chemicals using mammalian cells in culture. Br. J. Cancer
36:558.
Su, X., Sorenson, С. M. & Sheibani, N. (2003). Isolation and
characterization of murine retinal endothelial cells. Mol. Vis.
1:171-178.
Su, H. Y., Bos, T. J., Monteclaro, F. S. & Vogt, P. K. (1991). Jun
inhibits myogenic differentiation. Oncogene 6:1759-1766.
Subramanian, М., Madden, J. A. & Harder, D. R. (1991). A
method for the isolation of cells from arteries of various sizes.
/ Tissue Cult Methods 13:13-20.
Subramanian, S. V., Fitzgerald, M. L. & Bemfield, M. (1997).
Regulated shedding of syndecan- 1 and -4 ectodomains by
thrombin and growth factor receptor activation./. Biol. Chem.
272:14713-14720.
Suggs, J. E., Madden, М. C., Friemann, M. & Edgell, C. — J. S.
(1986). Prostacyclin expression by a continuous human cell
line derived from vascular endothelium. Blood 4:825-829.
Sugimoto, М., Tahara, H., Ide, T. & Fumichi, Y. (2004). Steps
involved in immortalization and tumorigenesis in human
Blumphoblastoid cell lines transformed by Epstein-Barr vims.
Cancer Res. 64:3361- 3364.
Suh, H., Song, M. J. & Park, Y. N. (2003). Behavior of isolated rat
oval cells in porous collagen scaffold. Tissue Eng. 9:441-20.
Sun, L., Bradford, C. S. & Barnes, D. W. (1995a). Feeder cell
cultures for zebrafish embryonal cells in vitro. Mol. Mar. Biol.
Biotech. 4:43-50.
Sun, L., Bradford, C. S., Ghosh, C., Collodi, P. & Barnes, D. W.
(1995b). ES-like cell cultures derived from early zebrafish
embroyos. Mol Mar. Biol. Biotech. 4:193-199.
Sundqvist, K., Liu, Y., Arvidson, K., Ormstad, K., Nilsson, L.,
Toftgard, R. & Grafstrom, R. C. (1991). Growth redulation
of serum-free cultures of epithelial cells from normal human
buccal murosa. In Vitro Cell Dev. Biol. 27A: 562-568.
Sutherland, R. M. (1998). Cell and micro environment interac¬
tions in tumour microregions: The multicell spheroid model.
Science 240:117-184.
Suva, D., Garavaglia, G., Menetrey, J., Chapuis, B., Hoffmeyer, P.,
Bemheim, L. & Kindler, V. (2004). Non-homatopoietic hu¬
man bone marrow contains Long-lasting, pluripotential
mesenchumal stem cells./. Cell Physiol 198:110-118.
Suzuki, A., Nakauchi, H. & Taniguchi, H. (2004). Prospective
isolation of multipotent using flowcytometric cell sorting.
Diabetes. 53:2143-2152.
Suzuki, Т., Saha, S., Sumantri, C., Takagi, M. & Boediono, A.
(1995). The influence of polyvinylpyrrolidone on freezing
of bovine IVF blastocysts following, biopsy. Cryobiology 32:
505-510.
682
Список литературы
Swope, V. В., Supp, А. P., Babcock, G. F. & Boyce, S. Т. (1997).
Regulations of pigmentation in cultured skin substitutes
sorting of melanocytes and keratinocytes./. Invest. Dermatol.
109:289-295.
Sykes, J. A., Whitescarver, J., Briggs, L. & Anson, J. H. (1970).
Separation of tumor cells from fibroblasts with use of discon¬
tinuous density gradients./ Natl. Cancer Inst. 44:855-864.
Tagawa, М., Yokosuka, O., Imazeki, F., Ohto, M. & Omata, M.
(1996). Gene expression and active virus replication in the
liver after injection of duck hepatitis В virus DNA into the
peripheral vein of ducklings./. Hepatol. 24:328-334.
Takahashi, H., Yanagi, Y., Tamaki, Y., Muranaka, K., Usui, T. &
Sata, M. (2004). Contribution of bone-morrow-derived cells
to choroidal neovascularization. Biochem. Biophys. Res. Com-
тип.320:372-375.
Takahashi, K. & Okada, T. S. (1970). Analysis of the effect of
«conditioned medium» upon the cell culture at low density.
Dev. Growth Differ. 12:65-77.
Takahashi, K., Mitsui, K. & Yamanaka, S. (2003). Role of Eras
in promoting tumour-like properties stem cells. Nature 423:
541-545.
Takahashi, K., Suzuki, K., Kawahara, S. & Ono, T. (1991). Effects
of lastogenic hormones on morphological development and
growth of human breast epithelial cells cultivated in collagen
gels Jpn.J. Cancer Res. 82: 553.
Takeda, K., Minowada, J. & Bloch, A. (1982). Kinetics of appear¬
ance of differentiation-associated characteristics in MI1, a line
of human myeloblastic leukaemia cells, after treatment with
TRA, DMSO, or Ara-C. Cancer ResA2:5152-5158.
Tarella, C., Ferrero, D., Gallo, E., Luyca Pagliardi, G. & Rus-
cetti, F. W. (1982). Induction of HL-60 cells by dimethyl-
sylphoxide: Evidence for a stochastic model not linked to the
cell division cycle. Cancer Res. 42:445-449.
Tashjian, A. H., Yasamura, Y., Levine, L, Sato, G. H. & Parker, M.
(1968). Establishment of clonal strains of rat pituitary tumor cells
that secrete growth hormone. Endocrinology 82:342-352.
Taub, M. (1984). Growth of primary and established kidney cell
cultures in serum-free media. In Barnes, D. W., Sirbasku, D. A.,
Sato, G. H., eds., Methods for Serum-Free Culture of Epithelial
and Fibroblastic Cells, New York, Alan R. Liss, pp. 3-24.
Taub, M. L., Yang, S. I. & Wang, Y. (1989). Primary rabbit proxi¬
mal tubule cell cultures maintain differentiated functions
when cultured in a hormonally defined serum-free medium.
In Vitro cell Dev. Biol. 25:770-775.
Taylor, D. A. (2001). Cellular cardiomyoplasty with autologous
skeletal myoblasts for ischemic heart disease and heart failure.
Curr. Control. Trials Cardiovasc. Med. 2:208-210.
Taylor, J. H. (1958). Sister chromatid exchanges in tritium labeled
chromosomes. Genetics. 43:515-529.
Taylor-Papadimitriou, J., Purkiss, P. & Fentiman, I. S. (1980).
Choleratoxin and analogues of cyclic AMP stimulate the
growth of cultured human epithelial cells. J. Cell Physiol.
102:317-322.
Taylor-Papadimitriou,J., Shearer, M. & Stoker, M. G. P. (1977).
Growth requirement of human mammary epithelial cells in
culture. Int.J. Cancer 20:903-908.
Taylor-Papadimitriou, J., Stampter, М., Bartek, J., Lewis, A.,
Boshell, V., Lane, E. B. & Leigh, I. M. (1998). Keratin ex¬
pression in human mammary epithelial cell cultured from
normal and malignant tissue: relation to in vivo phenotypes
and influence of medium./. Cell. Sci. 94:403-413.
Tedder, R. S., Zuskerman, A. М., Goldstone, A. N., Hawkins, A. E.,
Fielding, A., Briggs, E. М., Irwin, D. C., Heptonstall, J. &
Brink, N. S. (1995). Hepatitis В transmission from contami¬
nated cryopreservation tank. Lancet 346:137-140.
Temin, H. M. (1966). Studies on carcinogenesis by avian sarcoma
viruses; III: The differential effect of serum and polyanions
on multiplication of uninfected and converted cells./ Natl
Cancer Inst. 37:167-175.
Teofili, L; Rutella, S., Pierelli, L., Ortu La Barbera, E., di Mario, A.,
Menichella, G., Rumi, C. & Leone, G. (1996). Separation
of chemotherapy plus G-CSF-mobilized peripheral blood
mononuclear cells by counterflow centrifugal elutriation: In
vitro characterization of two different CD34 cell populations.
Bone Marrow Transplant. 18:421-425.
Terasaki, Т., Kameya, Т., Nakajima, Т., Tsumuraya, М., Shimo-
sato, Y., kato, K., Ichinose, H., Nagatsu, T. & Hasegawa, t.
(1984). Interconversion of biological characteristics of stall
cell lung cancer cells depending on the culture conditions.
Gann 75:1689-1699.
Testa, N. G. & Molineux, G. (eds). (1993). Haemopoiesis. Oxs-
ford, U.K., Oxsford Universsity Press.
Thacker, J., Webb, М. B. & Debenham, P. G. (1988). Finger¬
printing cell lines; Use of human hypervariable DNA probes
to characterise mammalian cell cultures. Somat. Cell Mol.
Genet. 14: 519-525.
Thelwall, P. E., Neves, A. A. & Brindle, К. M. (2001). Measure¬
ment of bioreactor perfusion using dynamic contrast agent-
enhanced magnetic resonance imaging. Biotechnol. Bioeng.
75: 682-690.
Thomas, D. G. Т., Darling, J. L., Paul, E. A., Mott, Т. C.,
Codlee, J. N., Tobias, J. S., Capra, L. G., Collins. C. D.,
Mooney, C., Bozek, Т., Finn, G. P., Arigbabu, S. O., Bul¬
lard, D. E., Shan-non, N. & Freshney, R. I. (1985). Assay of
anti-cancer drugs in tissue culture: Relationship of relapse free
interval (FRI) and in vitro chemosensitivity in patients with
malignant cerebral glioma. Br. J. Cancer 51:525-532.
Thomas, S., Gray, E. & Robinson, C. J. (1997). Response of HU-
VEC and EAhy926 and fibroblast growth factors. In Vitro
Cell Dev. Biol. Anim. 33:492- 494.
Thompson, E. B., Tomkins, J. M. & Curran, J. F. (1966). Induction
of tyrosine a-ketoglutarate transaminase by steroid hormones
in a newly established tissue culture cell line. Proc. Nad. Acad.
Sci. USA 56: 296-303.
Thompson, L. H. & Baker, R. M. (1973). Isolation of mutants of
cultured mammalian cells. In Prescott, D. (ed.), Methods in cell
biology, Vol. 6. New York, Academic Press, pp. 209-281.
Thomson, A. A., Foster, B. A. & Cunha, G. R. (1997). Analysis
of growth factor and receptor mRNA levels during develop¬
ment of the rat seminal vesicle and prostate. Development
124: 2431-2439.
Thomson, A. W. (ed.). (1991). Cytokine handbook. London,
Academic Press.
Thomson,J. A., ltskovitz-Eldor,J., Shapiro, S. S., Waknitz, M. A. &
Swiergiel, J. J. (1998). Embryonic stem cell lines derived from
human blastocysts. Science 282:1145-1147.
Thorsell, A., Blomqvist, A. G. & Heilig, M. (1996). Cationic lipid-
mediated delivery and expression of prepro-neuro-peptide Y
Список литературы
683
cDNA after intraventricular administration in rat: Feasibility
and limitions. Regul. Peptides 61: 205-211.
Till, J. E. & McCulloch, E. A. (1961). A direct measurement of
the radiation sensitivity of normal mouse bone marrow cells.
Radiat. Res. 14:213-222.
Tobey, R. A., Anderson, E. C. & Peterson, D. F. (1967). Effect of
thymidine on duration of G1 in Chinese hamster cells./. Cell.
Biol. 35:53-67.
Todaro, G. J. & DeLarco, I. E. (1978). Growth factors pro¬
duced by sarcoma virus-transformed cells. Cancer. Res. 38:
4147-4154.
Todaro, G. J. & Green, H. (1963). Quantitative studies of the
growth of mouse embryo cells in culture and their develop¬
ment into established lines ./. Cell. Biol. 17:299-313.
Tomakidi, P., Fusenig, N. E., Kohl, A. & Komposch, G. (1997).
Histomorphological and biochemical differentiation capacity
organotypic co-cultures of primary gingival cells ./. Periodont.
Res. 32:388-400.
Toneguzzo, F., Keating, A., Glynn, S. & McDonald, K. (1988).
Electrical field mediated gene transfer: Characterization of
DNA transfer and patterns of integration in lymphoid cells.
Nucleic Adds Res. 16:5515-5532.
Topley, P., Jenkins, D. C., Jessup, E. A. & Stables, J. N. (1993).
Effect of reconstituted basement membrane components on
the growth of a panel of human tumour cell lines in nude mice.
Br. J. Cancer 67:953-958.
Torday, J. S. & Kourembanas, S. (1990). Fetal rat lung fibroblasts
produce a TGF-P homologue that blocks type II cell matura¬
tion. Dev. Biol. 13:35-41.
Totoiu, М. O., Nistor, G. I., Lane, Т. E. & Keirstead, H. S. (2004).
Remyelination, axonal sparing, and locomotor recoveiy follow¬
ing transplantation of glial-committed progenitor cells into the
MHV model of multiple sclerosis. Exp. Neurol. 187:254-265.
Tozer, В. T. & Pirt, S. J. (1964). Suspension culture of mammalian
cells and macromolecular growth promoting fractions of calf
serum. Nature 201:375-378.
Traganos, F., Darzynkiewicz, Sharpless, T. & Melamed, M. R.
(1977). Nucleic acid content and cell cycle distribution of five
human bladder cell lines analyzed by flow cytofluorometry.
Int.J. Cancer 20:30-36.
Trapp, B. D., Honegger, P., Richelson, E. & Webster, H. de F.
(1981). Morphological differentiation of mechanically dis¬
sociated fetal rat brain in aggregating cell cultures. Brain
Res. 160:235-252.
Trickett, A. E., Ford, D. J., Lam-Ро Tang, P. R. L. & Vowels, M. R.
(1990). Comparison of magnetic particles for immunomag-
netic bone marrow purging using an acute lymphoblastic
leukaemia model. Transpl. Proc. 22:2177-2178.
Triglia, D., Braa, S. S., Yonan, C. & Naughton, G. K. (1991). Cyto¬
toxicity testing using neutral red and MTT assays on a three-
dimensional human skin substrate. Toxic. In Vitro 5:573-578.
Trotter, J. & Schachner, M. (1988). Cells positive for the 04 sur¬
face antigen isolated by cell sorting are able to differentiate
into oligodendrocytes and type-2 astrocytes. Dev. Brain Res.
46:115-122.
Trowell, O. A. (1959). The culture of mature organs in a synthetic
medium. Exp. Cell. Res. 16:118-147.
Troyer, D. A. & Kreisberg, J. I. (1990). Isolation and study of
glomerular cells. Methods Enzymol. 191:141-152.
Tsao, М. C., Walthall, В. I. & Ham, R. G. (1982). Clonal growth of
normal human epidermal keratinocytes in a defined medium.
J. Cell. Physiol. 110:219-229.
Tsao, S.-W., Mok, S. C., Fey, E. G., Fletcher, J. A., Wan, T. S. K.,
Chew, E.-C., Muto, M. G., Knapp, R. G. & Berkowitz, R. S.
(1995). Characterisation of human ovarian surface epithe¬
lial cells immortalized by human papilloma viral oncogenes
(HPV-E6E70RFs). Exp. Cell. Res. 218:499-507.
Tsuruo, Т., Hamilton, Т. C., Louis, K. G., Behrens, В. C.,
Young, R. C. & Ozols, R. F. (1986). Collateral susceptibil¬
ity of adriamycin-, and melphalan-, and cisplatin-resistant
human ovarian tumor cells to bleomycin./ря./. Cancer Rea.
77:941-945.
Tumilowicz, J. J., Nichols, W. W., Cholon, J. J. & Greene, A. E.
(1970). Definition of a continuous human cell line derived
from neuroblastoma. Cancer. Res. 30: 2110-2118,
Turner, R. W. A., Siminovitch, L., McCulloch, E. A. & Till, J. J.
(1967). Density gradient centrifugation of hemopoietic col¬
ony-forming cells./. Cell. Physiol. 69:73-81.
Tuszynski, М. H., Roberts, J., Senut, М. C., U, H. S. & Gage, F. H
(1996). Gene therapy in the adult primate brain: lntraparen-
chymal grafts of cells genetically modified to produce nerve
growth factor prevent cholinergic nieuronal degeneration.
Gene Therapy 3:305-314.
Tveir, К. M. & Pihl, A. (1981). Do cells lines in vitro reflect
the properties of the tumours of origin? A study of lines
derived from human melanoma xenografts. Br. J. Cancer 44:
775-786.
Twentyman. P. R. (1980). Response to chemotherapy of
EMT6 spheroids as measured by growth delay and cell sur¬
vival. Eur.J. Cancer 42:297-304.
U.S. Departament of Health and Human Services (1993). Bio¬
safety in microbiological and biomedical laboratories, 3d ed.
Pttbhcation (CDC) 93-8395, Centers for Disease Control.
US Govt. Printing Office, Washington, DC.
U.S. Nuclear Regulatory Commission (1997). Draft regulatory
guide DG-0006. Guide for the preparation of applications
for commercial nuclear pharmacy Licenses. Office of Nuclear
Regulatory Research, U.S. Nuclear Regulatory Commission,
Washington, DC 20555.
Uchida, I. A. & Lin, С. C. (1974). Quinacrine fluorescent patterns.
In Yunis, J. (ed.) Human chromosome methodology, 2d ed. New
York, Academic Press, pp. 47-58.
United States Pharmacopeia. (1985). Sterility tests, 21st re¬
vision. United States Pharmacopeial Convention, Inc.,
pp. 1156-1160.
Unkless, I., Dano, K., Kellerman, G. & Reich, E. (1974). Fibri¬
nolysis associated with oncogenic transformation Partial
purification and characterization of cell factor, a plasminogen
activator./. Biol. Chem. 249:4295-4305.
Uphoff, C.C. & Drexler, H. G. (2002) Comparative PCR analysis
for detection of mycoplasma infections in contionuous cell
lines. In Vitro Cell Dev. Biol. Animal 38: 79-85.
Uphoff C.C. & Drexler, H. G. (2004). Elimination of Mycoplasma
from infected cell lines antibiotics. Methods Mol. Med. 88:
327-334.
Ure, J. М., Fiering, S. & Smith, A. G. (1992). A rapid and efficient
method for freezing and reco vering clones of embryonic stem
cells. Trends Genet. 8: 6.
684
Список литературы
Uzgare, A. R., Xu, Y. & Isaacs, J. T. (2004). In vitro culturing and
characteristics of transit amplifying epithelial cells from hu¬
man prostate tissue./. Cell. Biochem. 91:196-205.
Uzgare, A. R., Xu, Y. & Isaacs, J. T. (2004). In vitro culturing
and characteristics of transit amplifying epithelial cells from
human prostate tissue./. Cell. Biochem. 91:196-205.
Vachon, P. H., Perreault, N., Magny, P. & Beallieu, J.-F. (1996).
Uncoordinated transient mosaic patterns of intestinal hydro¬
lase expression in differentiating human enterocytes./. Cell.
Physiol. 166:198-207.
Vago, C. (ed). (1971). Invertebrate tissue culture, Vol. 1. New
York. Academic Press.
Vago, C. (ed). (1972). Invertebrate tissue culture, Vol. 2. NewYork
Academic Press.
Vaheri, A., Ruoslanti, F., Westermark, B. & Ponten, J. (1976).
A common cell-type specific surface antigen in cultured hu¬
man glial cells and fibroblasts: Loss in malignant cells./. Exp.
Med. 143: 64-72.
van Bokhoven, A., Varella-Garcia, М., Korch, C. & Miller, G. J.
(2001a). TSU-Prl and JCA-1 cell are derivatives of T24 blad¬
der carcinoma cells and are not of prostatic origin. Cancer.
Res. 61:6340-6344.
van Bokhoven, A., Varella-Garcia, М., Korch, C., Hessels, D. &
Miller, G. J. (2001b). Widely used prostate carcinoma cell
lines share common origins. Prostate 47:36-51.
Van Diggelen, O., Shin, S. & Phillips, D. (1977). Reduction
in cellular tumorigenicity after mycoplasma infection and
elimination of mycoplasma from infected cultures by passage
in nude mice. Cancer. Res. 37:2680-2687.
Van Helden, P. D., Wild, I. J., Albrecht, C. F., Theron, E.,
Thomley, A. L. & Hoal-van Helden, E. G. (1988). Cross-
contamination of human esophageal squamous carcinoma
cell lines detected by DNA fingerprint analysis. Cancer. Res.
48:5660-5662.
Vanparys, P. (2002). ECVAM and pharmaceuticals. Ahem Lab
Anim., 30: Suppl 2:221-3.
Van Roozendahl, С. E. P., van Ooijen, B. Klijn, J. G. М.,
Claasen, C., Eggermont, А. М. М., Henzen-Logmans, S. C. &
Foekens, J. A. (1992). Stromal influences on breast cancer cell
growth. Br. J. Cancer 65:77-81.
Varga Weisz, P. D. & Barnes, D. W. (1993). Characterization of
human plasma growth inhibitory activity on serumfree mouse
embryo cells. In Vitro Cell Dev. Biol. 29A: 512-516.
Varon, S. & Manthorpe, M. (1980). Separation of neurons and
glial cells by affinity methods. In Fedoroff, S. & Hertz, L.
(eds.), Advances in cellular neurobiology, Vol. 1. New York,
Academic Press, pp. 405-442.
Vaughan, A. & Milner, A. (1989). Fluorescence activated cell
sorting. In Catty, D. (ed.), Antibodies; Volume II: A practical
approach. Oxford, U.K., IRL Press at Oxford University
Press, pp. 201-222.
Vaziri, H. & Benchimol, S. (1998). Reconstitution of telomer¬
ase activity in. normal human cells leads to elongation of
telomeres and extended replicative life-span. Curr. Biology
8:279-282.
Velcich, A., Palumbo, L., Jarry, A., Laboisse, C., Racevskis, J. &
Augenlicht, L. (1995). Patterns of expression of lineage-specif-
ic markers during the in vitro-induced differentiation of HT29
colon carcinoma cells. Cell Growth Differ. 6:749-757,
Venitt, S. (1984). Mutagenicity testing, a practical approach.
Oxford, U.K., IRL Press.
Venkatasubramanian, J., Sahi, J., Rao, М. C. (2000). Ion transport
during growth and differentiation. Ann N Y Acad Sci. 2000,
915:357-72.
Verbruggen, G., Veys, E. М., Wieme, N., Malfait, A. М., Gijsel-
brecht, L., Nimmegeers, J., Almquist, K. F. & Broddelez, C.
(1990). The synthesis and immobilization of cartilage-specific
proteoglycan by human chondrocytes in different concentra¬
tions of agarose. Clin. Exp. Rheumatol. 8:371-378.
Verschraegen, C. F., Hu, W., Du, Y., Mendoza, J., Early, J.,
Deavers, М., Freedman, R. S., Bast, R. S., Jr., Kudelka, A. P.,
Kavanagh, J. J. & Giovanella, В. C. (2003). Establishment
and characterization of cancer cell cultures and xenografts
derived from primary or metastatic Mullerian cancers. Clin.
Cancer. Res. 9:845-852.
Vescovi, A., Gritti, A., Cossu, G. & Galli, R. (2002). Neural stem
cells: plasticity and their transdifferentiation potential. Cells
Tissues Organs 171:64-76.
Vierick, J. L., McNamara, P. & Dodson, М. V. (1996). Prolifera¬
tion and differentiation of progeny of ovine unilocular fat
cells (adipofibroblasts). In Vitro Cell Dev. Biol. Anim. 32:
564-572.
Vilamitjana-Amedee, J., Bareile, R., Rouais, F., Caplan, A. I. &
Harmand, M. F. (1993). Human bone marrow stromal cells
express an osteoblastic phenotype in culture. In Vitro Cell
Dev. Biol. 29A: 699-707.
VisserJ.W. & De Vries, P. (1990). Identification and purifica¬
tion of murine hematopoietic stem cells by flow cytometry.
Methods Cell Biol. 33:451-468.
Vistica, D. Т., Skehan, P., Scudiero, D., Monks, A., Pittman, A. &
Boyd, M. R. (1991). Tetrazolium-based assays for cellular
viability: A critical examination of selected parameters
formazan production. Cancer Res. 51:2515-2520.
Vlodavsky. I., Lui, G. M. & Gospodarowicz, D. (1980). Morpho¬
logical appearance, growth behavior and migratory activity of
human tumor cells maintained on extracellular matrix versus
plastic. Cell 19:607-617.
Vogel, F. R. & Powell, M. F. (1995). A compendium of vaccine
adjuvants and excipients. In Powell, M. F. & Newman, M.
(eds.), Vaccine desigh: The subunit and adjuvant approach.
New York, Plenum Publishing, pp. 141-228.
von Briesen, H., Andreesen, R., Esser, R., Brugger, W., Meichsner,
C., Becker, K. & Rubsamen-Waigmann, H. (1990). Infection
of monocytes/macrophages by HIV in vitro. Res Virol. 141:
225-231.
Von der Mark, K. (1986). Differentiation, modulation and dedif¬
ferentiation of chondrocytes. Rheumatology 10:272-31 5.
Vondracek, М., Weaver, D., Sarang, Z., Hedberg, J. J., Willey, J.,
Wamg&rd, L. and Grafstrom, R. C. (2002). Transcript pro¬
filing of enzymes involved in detoxification of xenobiotics
and reactive oxygen in human normal and Simian virus 40 T
antigen-immortalized oral keratinocytes. Int.J. Cancer 99:
776-782.
Vonen, B., Bertheussen, K., Giaever, A. K., Florholmen, J. &
Burhol, P. G. (1992). Effect of a new synthetic serum replace¬
ment on insulin and somatostatin secretion from isolated rat
pancreatic islets in long term culture./. Tissue Cult. Methods
14:45-50.
686
Список литературы
Wessels, D., Titus, М. & Soil, D. R. (1996). A Dictyostelium
myosin I plays a crucial role in regulating the frequency of
pseudopods formed on the substratum. Cell Motil. Cytoskel-
eton 33: 64-79.
Westerfield, M. (1993). The zebrafish book: A guide for the
laboratory use of zebrafish (Brachydanio reio). Eugene, OR,
University of Oregon Press.
Westermark, B. (1974). The deficient density-dependent growth
control of human malignant glioma cells and virus-trans¬
formed glialike cells in culture. Int.J. Cancer 12: 438-451.
Westermark, B. (1978). Growth control in miniclones of human
glial cells. Exp. Cell. Res. Ill: 295-299.
Westermark, B., Ponten, J. & Hugosson, R. (1973). Determi¬
nants for the establishment of permanent tissue culture lines
from human gliomas. Acta Pathol. Microbiol. Scand. A 81:
791-805.
Westermark, B., Wasteson, A. (1975). The response of cultured
human normal glial cells to growth factors. Adv Metab Disord.,
8:85-100.
Westneat, D. F., Noon, W. A., Reeve, H. K. & Aquadro, C. F.
(1988). Improved hybridisation conditions for DNA finger¬
prints probed with M13. Nucleic Acids Res. 16: 4161.
Wewetzrer, K., Verdu, F., Angelov, D. N. & Navarro, X. (2002)
Olfactory ensheathing glia and Schwann cells: two of a kind?
Cell Tissue Res. 309:337-345.
Whitehead, R. H. (2004). Establishment of cell lines from colon
carcinoma. In Pfragner, R. & Freshney. R. I. (eds.), Culture of
human tumor cells. Hoboken, NJ, Wiley-Liss, pp. 67- 80.
Whitlock, C. A., Robertson, D. & Witte, O. N. (1984). Murine В
cell lymphopoiesis in long term culture./. Immunol. Methods
67:353-369.
Whur, P., Magudia, М., Boston, I., Lockwood, J. & Williams, D. C.
(1980). Plasmninogen activator in cultured Lewis lung car¬
cinoma cells measured by chromogenic substrate assay. Br.
J. Cancer 42:305-312.
Wienberg, J. & Stanyon, R. (1997). Comparative painting
of mammalian chromosomes. Curr. Opin. Genet. Dev. 7:
784-791
Wiktor, A. & Van Dyke, D. L. (2004). Combined cytogenetic
testing and fluorescence in situ hybridization analysis in the
study of chronic lymphocycic leukemia and multiple my¬
eloma. Cancer Genet. Cytogenet. 153:73-76.
Wiktor, T. J., Fernandes, М. V., Koprowski, H. (1964). Cultiva¬
tion of Rabies Virus in Human Diploid Cell Strain WI-38.
J. Immunol. 93:353-66.
Wilkenheiser, K. A., Vorbroker, D. K., Rice, W. R., Clark, J. C.,
Bachurski, C. J., Oie, H. K. & Whitsertt, J. E. (1991). Pro¬
duction of immortalized distal respiratory epithelial cell
lines from surfactant protein C/simian virus 40 large tumor
antigen trans-genic mice. Proc. Natl. Acad. Sci. USA 90:
11029-11033.
Wilkins, L. Gilchrest, B. A., Szabo, G., Weinstein, R. & Maciag, T.
(1985). The stimulation of normal human melanocyte pro¬
liferation in vitro by melanocyte growth factor from bovine
brain./. Cell Physiol. 122: 350.
Willey, J. C., Moser, С. E., Jr., Lechner, J. F. & Harris, С. C.
(1984). Differential effects of 12-O-tetradecanoylphorbol-
13-acetate on cultured normal and neoplastic human bron¬
chial epithelial cells. Cancer Res. 44: 5124-5126.
Williams, M. J. & Clark, P. (2003). Microscopic analysis of the
cellular events during scatter factor/hepatocyte growth
factor-induced epithelial tubulogenesis. /. Anat. 203:
483-503.
Williams, B. P., Abney, E. R. & Raff, M.C. (1985). Macroglial
cell development in embrionic rat brain: Studies using mono¬
clonal antibodies, fluorescence-activated cell sorting and cell
culture. Dev. Biol. 112:126-134.
Williams, G. M. & Cunn, J. M. (1974). Long-term cell culture of
adult rat liver epithelial cells. Exp. Cell Res. 89:139-142.
Willmarth, N. E., Albertson, D. G. & Ethier, S. P. (2004).
Chromosomal instability and lack of cyclin E regulation in
hCdc4 mutant human breast cancer cells. Breast Cancer Res.
6: R531-R539.
Wilson, A. P., Dent, М., Pejovic, Т., Hubbold, L. & Rodford, H.
(1996) Characterisation of seven human ovarian tumour cell
lines. Br. J. Cancer 74:722-727.
Wilson, A. P. (2004). The development of human ovarian epithe¬
lial tumor cell lines from solid tumors and ascites. In Culture of
human tumor cells, Ed. Pfragner, R., Freshney, R. I., Hoboken,
NJ, Wiley-Liss, pp. 145-178.
Wilson, P. D., Dillingham, M. A., Breckon, R. & Anderson, R. J.
(1985). Defined human renal tubular epithelia in culture:
Growth, characterization, and hormonal response. Am.
J. Phyciol. 248: F436-F443.
Wilson, P. D., Schrier, R. W., Breckon, R. D. & Gabow, P. A.
(1986) A new method for studying human polycystic kidney
disease epithelia in culture. Kidney Int. 30:371-378.
Wise, C. (2002). Epithelial cell culture protocols. Clifton, NJ,
Humana Press.
Wistuba, I. I., Bryant, D., Behrens, C., Milchgrub, S., Vir-
mani, A. K., Ashfaq, R., Minna, J. D. & Gazdar, A. F. (1999).
Comparison of features of human lung cancer cell lines and
their corresponding tumors. Clin. Cancer Res. 5:991-1000.
Witkowski, J. A. (1990). The inherited character of cancer-an
historical survey. Cancer 2: 229-257.
Wolf, D. P., Meng, L., Ely, J. J. & Stouffer, R. L. (1998). Recent
progress in mammalian cloning./ Assist. Reprod. Cancer. 15:
235-239.
Wolf, E. T. & Haffen, K. (1952). Sur une methode de culture
d’organces embryonnaires in vitro. Tex. Rep. Biol. Med. 10:
463-472.
Wolswijk, G. & Noble, M. (1989). Identification of an adult-spe¬
cific glial progenitor cell. Development 105:387-400.
Wootton, М., Steeghs, K., Watt, D., Munro, J., Gordon, K., Ire¬
land, H., Morrison, V., Behan, W. & Parkinson, E. K. (2003).
Telomerase alone extends the replicative lifespan of human
skeletal muscle cells without compromising genomic stability.
Hum. Gene Therapy 14:1473-1487.
Work with Ionising Radiation. Ionising Radiation Regulations
(1999). Approved Code of Practice and Guidance, L121, HSE
Books, ISBNO 7176 1746 7.
Wright, K. A., Nadire, К. B., Busto, P., Tubo, R., McPher¬
son, J. M. & Wentworth, В. M. (1998). Alternative delivery
of keratinocytes using a polyurethane membrane and the
impli- cations for its use in the treatment of full-thickness
bum injury. Bums 24: 7-17.
Wu, D. K. & de Vellis, J. (1987). The expression of the intermedi¬
ate filament-associated protein (NAPA-73) is associated with
Список литературы
687
the stage of terminal differentiation of chick brain neurons.
Brain Res. 421:186-93.
Wu, H., Friedman, W. J. & Dreyfus, C. F. (2004). Differential
regulation of neurotrophin expression in basal forebrain as¬
trocytes by neuronal signals./. Neurosci. Res. 76: 76-85.
Wu, R. (2004). Growth of human lung tumor cells in culture.
Culture of human tumor ce11sy Ed. Pfragner, R., Freshney, R. I.,
Hoboken, NJ, Wiley-Liss, pp. 1-21.
Wu, Y. J., Parker, L. М., Binder, N. E., Beckett, M. A., Sinard, J.
H., Griffiths, С. T. & Rheinwald, J. G. (1982). The mesothelial
keratins: A new family of cytoskeletal proteins identified in
cultured mesorhelial cells and nonkeratinizing epithelia. Cell
31:693-703.
Wuarin, L., Verity, M. A. & Sidell, N. (1991). Effects of in-
terferon-gamma and its interaction with retinoic acid on
human neuroblastoma differentiation. Int. J. Cancer 48:
136-141.
Wurmser, A. E., Nakashima, K., Summers, R. C., Toni, N.,
D’amour, K. A., Lie, С. C. & Gage, F. H. (2004). Cell fu¬
sion-independent differentiation of neural stem cells to the
endothelial lineage, Nature 430:350-356.
Wurster-Hill, D., Cannizzaro, L. A., Pettengill, O. S., Sorenson,
G. D., Cate, С. C. & Maurer, L. H. (1984). Cyrogenetics of
small cell carcinoma of the lung. Cancer Genet. Cytogenet.
13:303-330.
Wyllie, F. S., Bond, J. A., Dawson, Т., White, D., Davies, R. &
Wynford-Thomas, D. (1992). A phenotypically and karyo-
typically stable human thyroid epithelial line condition¬
ally immortalized by SV40 large T antigen. Cancer Res. 52:
2938-2945.
Wysocki, L. J. & Sata, V. L. (1978). «Panning» for lymphocytes:
A method for cell selection. Proc. Natl. Assad. Sci. USA 75:
2844-2848.
Yaffe, D. (1968). Retention of differentiation potentialities dur¬
ing prolonged cultivation of myogenic cells. Proc. Natl. Assad.
Sci. USA 61:477-483.
Yamada, К. M. & Geiger, B. (1997). Molecular interactions in cell
adhesion complexes. Curr. Opin. Cell Biol. 9: 76-85.
Yamada, Т., Placzek, М., Tanaka, H., Dodd, J. & Jessell, Т. M.
(1991). Control of cell pattern in the developing nervous
system: Polarizing activity of the floor plate and notochord.
Cell 64:635-647.
Yamadaguchi, N., Yamamura, Y., Koyama K., Ohtsuji, E.,
Imanishi, J. & Ashihara, T. (1990). Characterization of new
pancreatic cancer cell lines which propagate in a protein-free
chemically defined medium. Cancer Res. 50: 7008-7014.
Yan, G., Fukabori, Y., Nikolaropoulost, S., Wang, F. & McKee¬
han, W. L. (1992). Heparin binding keratinocyte growth
factor is a candidate stromal to epithelialcell ahdromedin.
Mol. Endocrinol. 6: 2123-2128.
Yanai, N., Suzuki, M. & Obinata, M. (1991). Hepatocyte cell
lines established from transgenic mice harboring tempera¬
ture-sensitive simian virus 40 large T-antigen gene. Exp. Cell
Res. 197:50-56.
Yang, J., Mani, S. A., Donaher, J. L., Ramaswamy, S., ltzyk-
son, R. A., Come, C., Savagner, P., Gitelman, I., Richard¬
son, A. & Weinberg, R. A. (2004). Twist a master regulator
of morphogenesis, plays an essential role in tumor metastasis.
Cell 117:927-39.
Yasamura, Y., Tashjian, A. H. & Sato, G. (1966). Establishment
of four functional clonal strains of animal cells in culture.
Science 154:1186-1189.
Yeager. T. R., DeVries, S., Farrard, D. F., Kao, C., Nakada, S. Y.,
Monn, T. D., Bruskewits, R., Stadler, W. М., Meisner, L. F.,
Gilchrest, K. W., Newyton, M. A., Waldman, F. M. & Rezni-
koff, C. A. (1998). Overcoming cellular senescence in human
cancer pathogenesis. Genes Dev. 12:163-174.
Yen, A., Coles, M. & Varvayanis, S. (1993). 1,25-dihydroxy
vitamin D3and 12-O-tetradecanoyl phorbol-13-acetate
synergistically induce monocytic cell differentiation: FOS
and RB expression./. Cell Physiol. 156:198-203.
Yeoh, G. С. Т., Hilliard, C., Fletcher, S. & Douglas, A. (1990). Gene
expression in clonally derived cell lines produced by In vitro
transformation of rat fetal hepatocytes: Isolation of cell lines
which retain liver-specific markers. Cancer Res. 50:75-93.
Yerganian, G. & Leonard, M. J. (1961). Maintenance of normal
in situ chromosomal features in long-term tissue cultures.
Science 133:1600-1601.
Yevdokimova, N. & Freshney, R. I. (1997). Activation of paracrine
growth factors by heparan sulphate induced by glucocorticoid
in A549 lung carcinoma cells. Brit.J. Cancer 76:261-289.
Yoshida, M. & Beppu, T. (1990). In Doyle, A., Griffiths, J. B. &
Newell, D. G., Cell and tissue culture: Laboratory procedures.
Chichester, U.K., John Wiley & Sons. Module 4E2.
Yoshioka, М., Nakajima, Y., Ito, Т., Mikami, O., Tanaka, S., Mi¬
yazaki, S. & Motoi, Y. (1997). Primary culture and expression
of cytokine mRNAs by lipopolysaccharide in bovine Kupffer
cells. Vet. Immunol. Immunopathol. 58:155-163.
Young, H. F., Duplaa, C., Romero-Ramos, М., Chesselet, M. F.,
Vourc’h, P., Yost, M. J., Ericson, K., Terracio, L., Asahara, Т.,
Masuda, H., Tamura-Ninomiya, S., Detmer, K., Bray, R. A.,
Steele, T. A., Hixson, D., el-Kalay, М., Tobin, B. W., Russ, R. D.,
Horst, M. N., Floyd, J. A., Henson, N. L., Hawkins, К. C.,
Groom, J., Parikh, A., Blake, L., Bland, L. J., Thompson, A. J.,
Kirincich, A., Moreau, C., Hudson, J., Bowyer, F. P. 3rd,
Lin, T. J. & Black, A.C. Jr. (2004). Adult reserve stem cells
and their potential for tissue engineering. Cell. Biochem.
Biophys. 40:1-80.
Yuhas,J. М., Li, A. P., Martinez, А. О. & I. adman, A. J. (1977). A
simplified method for production and growth of multicellular
tumour spheroids (MTS). Cancer Res. 37:3639-3643.
Yuspa, S. H., Koehler, B., Kulesz-Martin, M. & Hennings, H. (1981).
Clonal growth of mouse epidermal cells in medium with reduced
calcium concentration./ Invesrt Dermatol. 76:144-146.
Yusufi A.N. K., Szczepanska-Konkel, М., Kempson, S. A., Mc-
Ateer, J. A. & Dousa, Т. P (1986) Inhibition of human renal
epithelsal Na+/Pi cotransport by phosphonoformic acid.
Biochem. Biophys. Res. Commun. 139: 679-686.
Zaroff, L., Sato, G. H. & Mills, S. E. (1961). Single-cell platings
from freshly isolated mammalian tissue. Exp. Cell Res. 23:
565-575.
Zeltinger, J. & Holbrook, K. A. (1997). A model system for long¬
term serum-free suspension organ culture of human fetal
tissues: Experiments on digits and skin from multiple body
regions. Cell Tissue Res. 290: 51-60.
Zeng, C., Pesall, J. E., Gilkerson, К. K. & McFarland, D. C. (2002).
The effect of hepatocyte growth factor on turkey satellite cell
proliferarion and differentiation. Poult Sci. 81:1191-1198.
688
Список литературы
Zhang, Y., Proenca, R., Maffei, М., Barone, М., Leopold,
L. & Friednman, J. M. (1994) Positional cloning of the
mouse obese gene and its human homologue. Nature 372:
425-432,
Zhou, S., Schuetz, J. D., Bunting, K. D., Colapietro, A.-M., Sam-
path, J., Morris, J. J., Lagutina, I., Grosveld, G. C., Osawa, М.,
Nakauchi, H. & Sorrentino, B. P. (2001). The ABCtransporter
Bcrpl /ABCG2 is expressed in a wide variety of stem cells and
is a molecular determinant of the side-population phenotype.
Nat. Med. 7:1028-1034.
Zhu, C. & Joyce. N. C. (2004). Proliferative response of comeal
endothelial cells from young and older donors. Invest. Oph¬
thalmol. Vis. Sci. 45:1743-1751,
Zhu, S. Y., Cunningham, М. I., Gray, Т. E. & Nettesheim, P.
(1991)Cytotoxicity, genotoxicity transfoming activity of
4-(methylnitrosamino)-1 -(3-pyridyl)-1 -butanone(NNK) in
rat trachea epithelial cells. Mutation Res. 261 (4): 249-259
Zimmermann, H., Hillgartner, М., Manz, B., Felien, P., Brun-
nenmeier, F., Leinfelder, U., Weber, М., Cramer, H., Sch¬
neider, S., Hendrich, C., Volke, F. & Zimmermann, U. (2003).
Fabrication of homogeneosly cross-linked, functioual alginate
microcapsules validated by NMR-, CLSM- and AFM-imag-
ing. Biomatenals 24: 2083-2096.
Zoubiane, G. S., Valentijn, A., Lowe, E. Т., Akhtar, N., Bagley, S.,
Gilmore, A. P. & Streuli. С. H. (2003). A role for the cyto-
skeleton in prolactin-dependent mammary epithelial cell
differentiation./. Cell Sci. 117: 271-280.
zur Nieden, N. I., Kempka, G. & Ahr, H. J. (2003). In vitro differ¬
entiation of embryonic stem cells into mineralized osteoblasts.
Differentiation 71:18-27.
Zwain, I. H., Morris, P. I. & Cheng, C. Y. (1991). Identification
of an inhibitory factor from a Sertoli clonal cell line (TM4)
that modulates adult rat Leydig cell line steroidogenesis. Mol.
Cell Endocrinol. 80:115-126
Предметный указатель
Аберрантный контроль роста 343
Автоклавируемые среды 199
Автоматические пипетки 90
Автоматический диспенсер 100
Автоматический счетчик колоний 402
Авторадиография 521
Активаторы плазминогена 349
Альгинат 496
Анализатор размера клеток 93
Анализ профиля ДНК 310
Ангиогенез 348
Антибиотики 158,252
Антигены клеточной поверхности 287
Апоптоз 62
Асептическая зона 73,85
- обслуживание 73
Асептические методы 106
Аспирационный насос 87
Аутентичность 138
Аутокринная сигнальная система 66
Аутокринные факторы 326
Аффинная хроматография 283
Аэратор культуры BelloCell 509
Базальная пластинка 60
Бакуловирус 555
Белки промежуточных филаментов 287
Бессывороточные среды
- недостатки 168
- преимущества 168
- приготовление 44,179
- разработка 178
- селективные 149
Бикарбонат 197,560
Биологическая опасность 129
Биопсийный материал человека 216
Биореактор 505
Биоэтика 136
Бипотентная стволовая клетка 324
Бласттрансформация 521
Блокирование
- клеток в метафазе 302
- клеточного цикла 529
Блот-анализ при Саузерн-блоттинге 308
Буферизация 151
Валидация 137
Вентилирование 143
Вентиляция 72
Весы 98
Внеклеточный матрикс 60,146
Возраст культуры 235
Время генерации 393
Вставные фильтры 145
Вторичная культура 235
Вторичная фильтрация по направлению
струи 209
Выбор бессывороточной среды 171
Выбор детергента 184
Выбор клеточной линии 240
Выбор среды и сыворотки 160
Выделение клонов 265
Выживаемость 407,409
Газы 125
Гемоцитометр 104,380
Генетицин 554
Генетическая нестабильность 332
Гетероплоидия 70
Гетероплоидные линии 335
Гетеротипическая паракринная система 66
Гибридизация 305,308
Гидролизат аминокислот 163
Гипотоническая обработка 302
Гистотипическая культура 25,35,478,488
Глютамин 559
Гомокринная передача сигнала 66
Гомокринные факторы 326
Гомотипическая паракринная передача
сигнала 66
Горелка 110
Гормоны 158
Деадаптация 65
Дедифференцировка 32,64,321
Дезагрегация 224,227
Дезинфектанты 105
Дезинфекция окуриванием 136
Десмосомы 59,61
Диализ 207
Диспенсеры 90
Дифференциальное окрашивание хромо¬
сом 302
Дифференцировка 63,287,321
ДНК-фингерпринтинг 286
- мультилокусный 308
Дозирование 89
Желатин 145
Жидкий азот 126
Жизнеспособность 230,407
Завинчивающиеся крышки 183
Зависимость от подложки 34
Замена среды 242
Замораживание клеток 52,372
Идентификация культуры клеток 71
Излучение
- от высокоэнергетичных источников 127
- ионизирующее 126
- от меченых реагентов 127
Изопикническая седиментация 274
Изоферментный анализ 55,316
Изоферментный электрофорез 314
Изоферменты 313
Иммортализация 69,332
Иммунный анализ 320
Иммунный пэннинг 278
Инвертированный микроскоп 88
Ингибирование пролиферации 62
Ингибиторы 160
Индекс мечения 403
Индекс пролиферации 404
Индекс разведения 235,248
Индукция дифференцировки 63
Инкубаторы 78,94,114
Инкубирование 73
Искусственные хромосомы дрожжей 555
Карантин 73,78
Кариотип 300,303
Кариотипирование 286
Клеточная адгезия 58
Клеточная культура 25,34
Клеточная линия 51,68
Клеточная плотность 395
Клеточная подвижность 61
Клеточная пролиферация 62,398
Клеточно-субстратное взаимодействие 58
Клеточный цикл 62
Клеточный штамм 34,235
Клональное соответствие 288
Клонирование в агаре 262
Клонирование в метилцеллюлозе 264
Клонирование в монослое 56
Клонирование клеток 255
Коллаген 145
Коллагеназа 226
Коллагеновый гель 493
Кольца для клонирования 266
Коммитирование 321
Компоненты для бессывороточного куль¬
тивирования 170
Кондиционирование 145
Кондиционированная среда 163,260
Кондуктометр 100
Конечные линии 235
Контактное ингибирование 61
Контактное торможение 344,398
Контаминация 138,167
- виды микробной контаминации 354
- вирусная 361
- мультиспецифическая 561
- повторная 561
- постоянная 561
- распространенная 562
- устранение 362
Контроль, проверка качества и хранение
сред 208
Контроль безопасности 120
Контроль клеточной пролиферации 62
Конфиденциальности требования 134
Конфлюэнтная культура 68
Конфлюэнтные фидерные слои 476
Конфокальная микроскопия 528
Концентрация действующего агента 411
690
Предметный указатель
Концентрация клеток 243,395
Концентрация сыворотки 57
Копреципитация с фосфатом кальция 551
Краситель Гимза 49, 50, 290
Краситель Мэй-Грюнвальда 290
Кривая роста 162,210
Криоконсервация
- контейнеры для криоконсервации 102
- культивируемых клеток 51
Критерии субкультивирования 244
Ксенотрансплантат 479
Культивирование
- в атмосфере С02119
- в суспензии 250
- на альгинатных бусинах 450
- с перемешиванием 505
- эффективность 399
Культура амниоцитов 529
Культуральные пакеты 509
Культура ткани 25
Куриный эмбрион 215
Ламинарное оборудование 77
Ламинарные шкафы 72,73,85,129
Ламинарный поток 112
Латентная фаза 397
Липофекция 551
Личная гигиена 108
М-фаза 62
Магнитная мешалка 96
Магнитная мешалка с подогревом 99
Магнитно-активированный клеточный
сортинг (MACS) 279
Магнитный сортинг 278
Макроносители 516
Малигнизация 346
Маркерные гранулы 274
Маркеры
- антигенные 315
- дифференцировки 286,324
- ткани 286
- уникальные 288
Маркировка клеточной культуры 240
Матригель 146,493
Межклеточное распознавание 58
Межклеточные контакты 59
Мембранная перфузия 516
Метаболизм 407
Металлические субстраты 148
Метод градиента плотности 438
Метод иммуномагнитного разделения 438
Метод микросечений 438
Метод падающей капли 298
Метод холодной трипсинизации 223
Методы иммунодиссекции 438
Метотрексат-резистентность 270
Микоплазма 357,561
- обнаружение 52
Микрокапсулирование 518
Микроносители 514
Микротитрационные планшеты 257
Микротитрационный анализ 413
Микротрубочки 61
Микрофотография 298
Митотический индекс 404
Многослойные пропагаторы 510
Моечная машина 100
Молекулы клеточной адгезии 58,219
Монослойная культура 34,291
Монослойные клоны 267
Морозильники 101
МТТ-тест 413
Мультилокусный ДНК-фингерпринтинг
308
Мультипотентная стволовая клетка 324
Мышиный эмбрион 212
Нагреватели 79
Направленная миграция 61
Неадгезивные субстраты 148
Необратимый переход в стадию диффе¬
ренцировки 62
Неорганические элементы 160
Неравномерный рост 144
Нестабильность клеточных линий 32
Нозерн-блоттинг 312
Номер пассажа 235
Оборудования количество72
Одноразовые фильтры 200
Одноразовый пластик 139
Ожоги 126
Окрашивание
- клеточной культуры 49
- кристаллвиолетом 291
- по Гимза 290
Онкогены 346
Опухолевая трансплантация 347
Органические добавки 158
Органная культура 25,33,488
Органотипическая культура 27,488
Осмометр 100
Осмотическое давление 152
Отбор и анализ проб 143
Оттаивание замороженных клеток 52
Отходов уничтожение
- биологически опасных 136
- радиоактивных 127
Оценка риска 120
Очиститель воды 96
Паракринные сигнальные молекулы 66
Паракринные факторы роста 326
Параллельные культуры 254
Паровой стерилизатор (автоклав) 98
Пассаж 34,235
Пенообразование 154
Первичная документация 231
Первичная культура 55,211,217
Первично эксплантируемая культура 34
Первичный подсчет 395
Первичный эксплантат 218
Первое субкультивирование 235
Передача клеточных сигналов 66
Переливание 112
Перенос ДНК 332
Перенос жидкостей 37,38
Перфузионная суспензионная культура 509
Перфузия на полых волокнах 517
Пипетирование 37,38,89,110
Пипетки 104
Плато фаза 399
Плотностное ограничение 398
Плотность клеток 243
Плотность насыщения 399
Плотные контакты 59
Поверхностное натяжение 154
Подготовительная зона 73
Подложки проницаемые 145
Подсчет клеток в гемоцитометре 45
Подсчет хромосом 303
Подтверждение аутентичности 285
Полилизин 145
Полная питательная среда 154
Полные питательные среды 154
Получение первичной культуры 67
Получение постоянных клеточных ли¬
ний 34
Полые волокна 145
Пользовательская культура 377
Помещение для мытья посуды 81
Помещение для стерильных манипуля¬
ций 77
Порошкообразные среды 197
Порядок поддержания культуры 241
Посевной сток 377
Постоянная клеточная линия 235
Постоянный клеточный штамм 235
Поспрансфекционное ведение культуры 340
Препаровальная лупа 93
Приготовление питательной среды 40
- концентрата 44
- из порошка 45
Приготовление сред 81
Приготовление стандартных растворов 43
Прикрепление и рост клеток 139
Прикрепление эксплантатов 218
Проверка культуральных свойств 209
Продукты дифференцировки 287
Промежуточные филоменты 61
Противоточное распределение 283
Протирание поверхностей 109
Проточная цитометрия 282
Проточные цитометры 104
Прямой микроскоп 102
Пустотелые волокна 493
Раздражение 407
Размораживание замороженных клеток 374
Распластывание клеток по стеклу 302
Распределяемая культура 377
Реакторы
- с неподвижным слоем 517
- с псевдожидким слоем для монослой¬
ных культур 517
- для суспензионной культуры 509
Регистратор температуры 95
Репликативные колонии 268
Репортерные гены 555
Ретровирусная инфекция 554
Реципрокная передача сигнала 61
Реципрокные взаимодействия 487
рН-метр 99
Роллерные культуры 512
Роллерные штативы 96
Рост субкультуры 46,47
Саузерн-блоты ДНК клеточных линий 306
Сбалансированные солевые растворы
151,154,193
Сбор митотических клеток 302
Сдерживающая среда 242
Селективная адгезия 272
Селективное открепление 272
Сервисные тележки 88
Сигнальные молекулы 66
Скорость седиментации 275
Смена среды в монослойной культуре 41
Составление сред 149
Предметный указатель
691
Сосуды для культивирования клеток 139
Специализированная среда 198
Спинкультура 505
Спорадическая контаминация 561
Среда Игла в модификации Дюльбекко 149
Среда Хэма 149
Среда Хэма F12 211
Стадия сливного роста 68
Стандартная лабораторная практика
(GLP) 113
Стандартная процедура (СОП) ИЗ, 120
Стандартные операционные процедуры 120
Старение культуры 68
Стационарная фаза 397
Стекло 139
Стеклянные пипетки 183
Стерилизационные фильтры 105
- многократного использования 190
Стерилизация 97,187
- воды 42
- методом фильтрации 199
- среды 194,199
- стеклянной посуды 42
- фосфатно-буферного раствора для
среды Дюльбекко 43
Стерильная зона 106
Стерильность 163
Стимуляция эффективности посева 257
Субкультивирование 235,243
Субкультура 34
- клеток, растущих в суспензии 250
Субстрат 139
Суспензионная культура 34,142
Суспензионное клонирование 261
Сушильные шкафы 97
Сушка пипеток 96
Сфероиды 478,493
Счетчик клеток 92
Сыворотка 159,203,559
- недостатки 164
Тепловая инактивация 163
Термальная комната 78
Терминальная дифференцировка 321
Термостаты 79
Тестирование сыворотки 162
Техника ведения культуры ткани 37,39
Типы культуры ткани 33
Типы первичных культур клеток 211
Ткани животных 136
Ткани человека 136
Тотипотентная стволовая клетка 324
Транзиторная трансфекция 551,554
Трансгенные мыши 341
Трансдифференцировка 323
Трансмембранные протеогликаны 59
Трансмемранные протеогликаны 59
Трансфекция 332
Трансформация 69,288,332,407
Трансформация in vitro 69
Требование безопасности 83, 85, 92, 113,
183,189,207,212,216,298,306
Требования конфиденциальности 134
Трехмерные матриксы 147
Трипсин 221,560
Трипсинизация 221
Тромбоцитарный фактор роста 62
Туморогенность 346
Унипотентная стволовая клетка 324
Уровни биологической защиты 129
Устранение микоплазмы 362
Фаза логарифмического роста 397
Фазы сливного роста 61
Факторы роста 158,160
Ферментеры 505
- с восходящим потоком воздуха 509
Ферменты 287
Фидерные слои 57,146,261
- селективные 273
Фильтрация 298
Фильтры многократного использования 200
Флюоресцентно-активируемый клеточ¬
ный сортинг 281
Фосфатно-кальциевая преципитация 340
Характеристика клеточных линий 54
Химическая токсичность 124
Холодильники 101
Хранение 82
Хромосомные аберрации 334
Хромосомный анализ 303
Хромосомный препарат 301
Центрифуга 89
Центрифугирование 292
Центрифужный элютриатор 104
Цикл роста 248
- фазы 397
Цилиндры для пипеток 87
Циркуляция воздуха 79
Цитометрия 390
Цитоскелет 61
Цитотоксичность 57,406
Цифровая фотография на микроскопе 300
Человеческий биопсийный материал 131
Экспрессия белка 312
Экспрессия репортерного гена 554
Электропорация 553
Электрофорез 283
Эмбриональный экстракт 163
Эндокринные сигнальные молекулы 66
Эффективность клонирования 399
Эффективность посева 162,210
DHFR-амплификация 270
ELISA-метод 320
Gl-фаза 62
Lag-фаза 397
Log-фаза 397
МЕМ Игла 211
S-фаза 62
Учебное издание
Фрешни Р. Ян
КУЛЬТУРА ЖИВОТНЫХ КЛЕТОК
Практическое руководство
Ведущий редактор канд. биол. наук В. В. Гейдебрехт
Редактор канд. биол. наук О. П. Кисурина
Художник Н. А. Новак
Технический редактор Е. В. Денюкова
Корректор Д. И. Мурадян
Компьютерная верстка: О. А. Пелипенко
Подписано в печать 22.07.10. Формат 60x90/8.
Уел. печ. л. 86,5. Тираж 1500 экз. Заказ 0-1178.
При участии ООО «ЭМПРЕЗА»
Издательство «БИНОМ. Лаборатория знаний»
125167, Москва, проезд Аэропорта, д. 3
Телефон: (499)157-5272
e-mail: binom@Lbz.ru, http://www.Lbz.ru
Отпечатано в полном соответствии с качеством
предоставленного электронного оригинал-макета
в типографии филиала ОАО «ТАТМЕДИА» «ПИК «Идел-Пресс».
420066, г. Казань, ул. Декабристов, 2.
E-mail: idelpress@mail.ru