/








































































































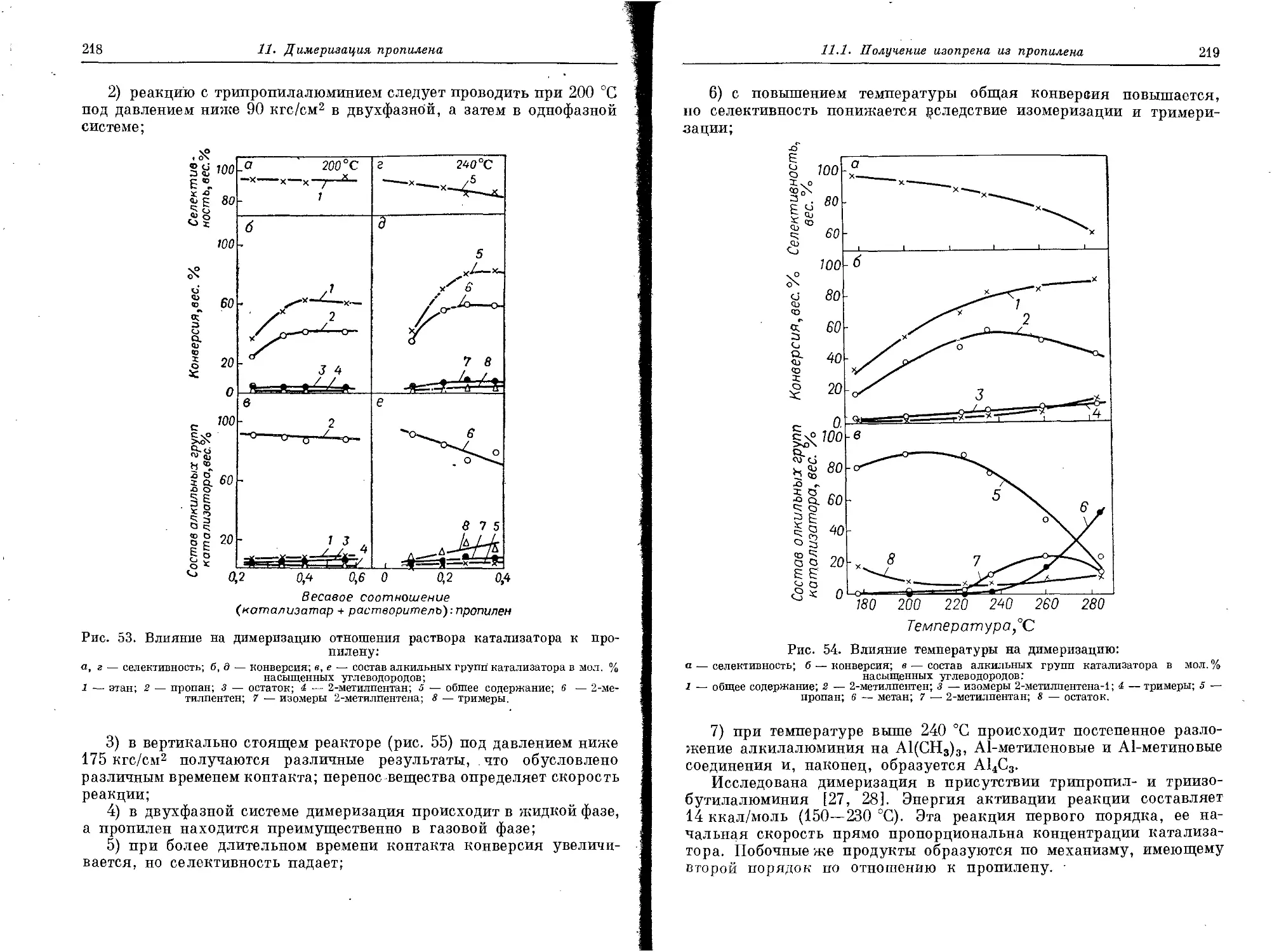











































































Автор: Андреас Ф. Гребе К.
Теги: производство органических веществ органические соединения химическая промышленность
Год: 1973




Текст
PBOnLEMHtMlls
von
DR. FRIEDRICH ANDREAS
und
DR. KARLHEINZ GRORE
A
AK. AD EMIE • VERLAG • BERLIN
1969
Ф. АНДРЕАС, К. ГРЕБЕ
ХИМИЯ
И ТЕХНОЛОГИЯ
ПРОПИЛЕНА
Перевод с немецкого В. Н. Тихомировой и Э. 3. Черниной
Под редакцией 3. Н. ПОЛЯКОВА
ЛЕНИНГРАД ИЗДАТЕЛЬСТВО «ХИМИЯ»
ЛЕНИНГРАДСКОЕ ОТДЕЛЕНИЕ • 1973
УДК 661.715.3 + 547.313.3
А65
Андреас Ф., Гребе К.
А65 Химия и технология пропилена. Л., «Химия»,
1973.
368 стр., 75 рис., 90 табл., список литературы 2115
ссылок.
В книге собраны обширные сведения о химии пропилена
и его производных, из которых особый интерес представляют
акрилонитрил, окись пропилена и полипропилен. Подробно
описаны методы получения, свойства и области применения этих
продуктов, представлены технологические схемы производства,
дан обзор производственных мощностей и потребления в ряде
зарубежныхстран. Приведена обширная библиография.
Книга предназначена для научных и инженерно-технических
работников нефтехимической промышленности, а также промыш-
ленности пластмасс, химических волокон, синтетического кау-
чука, лаков и красок. Она может быть также полезна препода-
вателям, аспирантам и студентам вузов.
0253-1
050(01)—
© Издательство «Химия», 1973
ПРЕДИСЛОВИЕ РЕДАКТОРА ПЕРЕВОДА
Технологические процессы на основе пропилена в по-
следнее время приобретают все больший интерес для специа-
листов-химиков в связи с необычайно быстрым развитием этой
отрасли химической промышленности. Однако обобщающая
литература по этому вопросу практически отсутствует.
Предлагаемая вниманию читателей книга Ф. Андреаса и
К. Гребе в сжатой форме освещает широкий круг вопросов
химической технологии пропилена. По существу, монография
представляет собой систематизированный обзор современных
промышленных процессов, сырьем для которых служит про-
пилен. Достаточно подробно и полно освещены промышлен-
ные процессы получения собственно пропилена.
Особенно ценной является попытка провести экономи
ческую оценку существующих промышленных методов полу-
чения пропилена и разнообразных продуктов на его основе
В книге отсутствует глубокое и детальное описание химических
реакций, но весьма выпукло показана роль пропилена как
сырья для самых разнообразных химических синтезов. Боль
шой интерес для широкого круга специалистов представляют
данные по объемам производства ряда Продуктов на основе
пропилена в различных странах.
3. Поляков
•ПРЕДИСЛОВИЕ АВТОРА
Бурное развитие промышленности органической химии за по-
следние 35 лет основано на использовании низших олефинов (эти-
лен, пропилен, бутен и т. д.), получаемых в больших количествах
в качестве побочных продуктов при переработке нефти, для синтеза
соединений различного молекулярного веса и состава. Результатом
этого является постоянно растущий спрос на олефины во всех про-
мышленных странах. Таким образом, ранее нежелательные побоч-
ные продукты стали ценным нефтехимическим сырьем.
Постоянно увеличивающийся спрос на этилен способствовал
целевой разработке новых методов получения этилена из нефтяного
сырья. Это позволило производить необходимое количество этилена
во всех индустриально развитых странах. Характерно также бы-
строе увеличение спроса на пропилен.
Настоящая книга призвана дать всем специалистам, занимающимся
данными проблемами, представление о развитии «химии пропилена»
и помочь в быстром и по возможности полном определении перспек-
тив развития этой важной отрасли нефтехимии.
Авторы будут очень признательны всем коллегам за советы,
пожелания, а также критические замечания.
Лейна, май, 1969
Ф. Андреас
К. Гребе
ВВЕДЕНИЕ
В 1795 г. путем отщепления воды от этанола с помощью кон-
центрированной серной кислоту был получен этилен [1], ставший
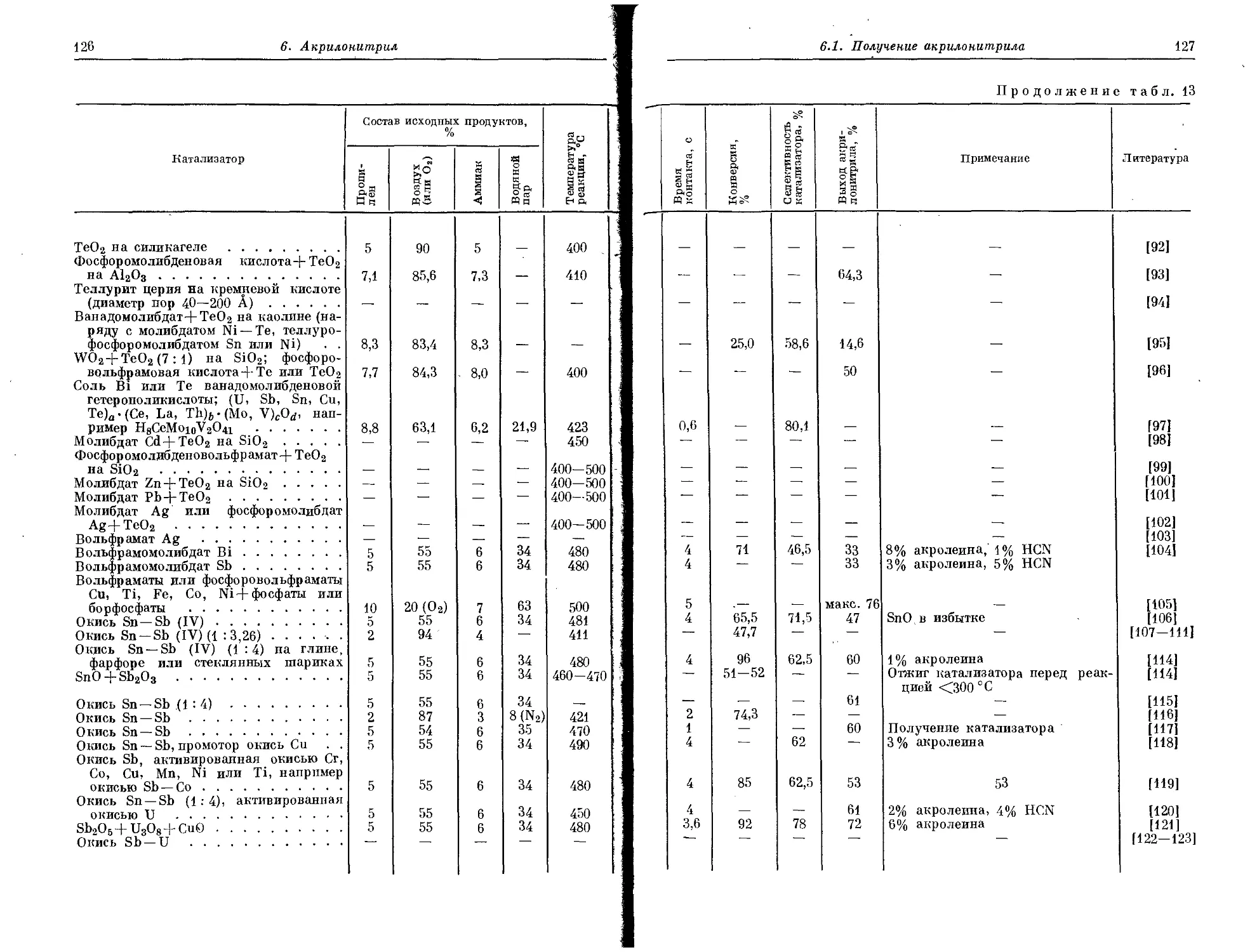
первым соединением ряда олефинов. Благодаря способности обра-
зовывать с хлором жидкий продукт, он получил название «масло
голландских химиков» [2], от которого впоследствии было образо-
вано наименование всего ряда простых ненасыщенных алифатиче-
ских /углеводородов.
Лишь спустя 50 лет Рейнольдс [3] выделил очередной гомолог
этого ряда — пропилен. Он наблюдал его при пропускании сивуш-
ного масла через накаленные трубки. Несмотря на то что для полу-
чения пропилена и были разработаны специальные методы, в част-
ности из пропилового и изопропилового спиртой, еще несколько
последующих десятилетий пропилен оставался лишь лаборатор-
ным продуктом и промышленных процессов получения пропилена
не существовало.
1. ПОЛУЧЕНИЕ ПРОПИЛЕНА
В начале XX в. в период, когда объем переработки нефти не-
прерывно возрастал для удовлетворения увеличивающейся потреб-
ности в топливе, возникла необходимость в очистке образующихся
побочных газов (абгазрв). При этом в больших количествах выде-
лялся пропилен, правда, еще сильно загрязненный различными
примесями.
Пропилен при очистке смесей отработанных газов сравнительно
легко поглощается 80-—90%-ной серной кислотой. При этом обра-
зуется изопропилсульфат, который затем переходит в изопропило-
вый спирт. Это привело к созданию первого нефтехимического про-
дукта. В конце 20-х годов американская фирма Standard Oil Со.
ввела в действие первую установку по производству изопропилового
спирта. С этого времени постоянно рос интерес к пропилену [5—31,
119, 124]. '
Так, американская фирма Carbon and Carbide Chemicals в период
с 1934 г. по 1939 г. разработала 12 новых продуктов из пропилена
[4], в то время кдк из этилена только 6.
В течение последних 30 лет разработаны следующие методы
получения пропилена:
выделение пропилена из нефтезаводских и крекинг-газов;
образование пропилена при синтезе Фишера—Тропша и при по-
лучении полукоксовых и коксовых газов;
получение пропилена из углеводородов Са—С4 и высших угле-
водородов.
1.1. ВЫДЕЛЕНИЕ ИЗ НЕФТЕЗАВОДСКИХ И КРЕКИНГ-ГАЗОВ
При чисто физической разгонке нефти и природного газа оле-
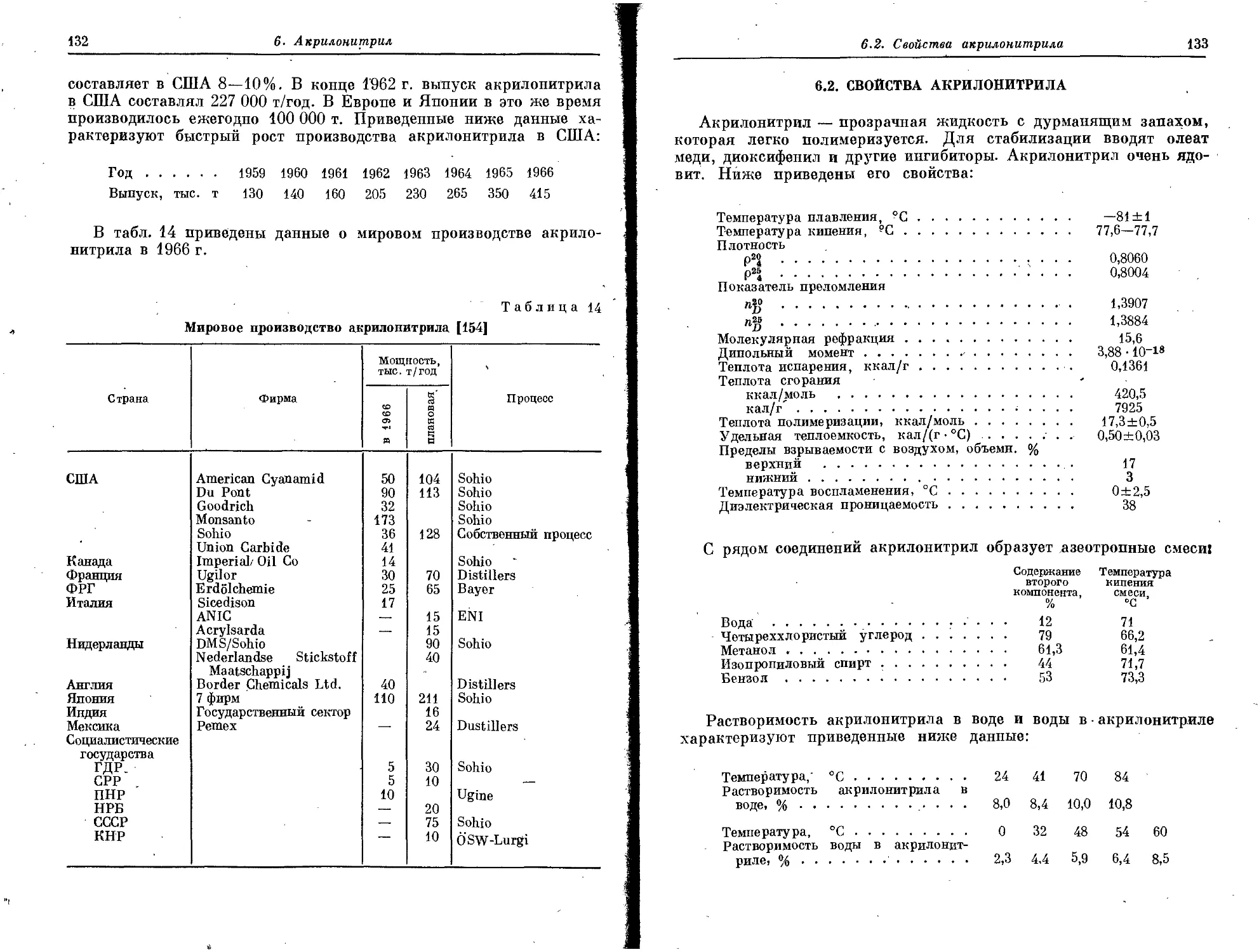
фины отсутствуют. На современных нефтеперерабатывающих заво-
дах олефинсодержащие абгазы образуются при производстве высоко-
качественного бензина путем риформинг- и крекинг-процессов.
Газы термического (I) и каталитического (II) крекинга имеют разный
состав (в %) в зависимости от метода работы установок:
Водород ..............
Метан.................
Этан' ................
Этилен ...............
П ропан...............
П ропилен.............
I II I й
8,0 29,6 Бутан 5,0 3,0
33,4 18,0 Изобутан 1,0 7,5
22,1 8,0 Бутилен 8,0 5,2
2,1 4,0 Изобутилен 1,8 1,7
7,0 15,0 Соединения С5 и выс-
7,0 8,0 шие углеводороды . . — —
1.2. Образование при синтезе по методу Фишера — Тропша
9
Общий выход газов при смешанном крекинге достигает 10—12,
при крекинге в газовой фазе 20—25, при термическом риформинге
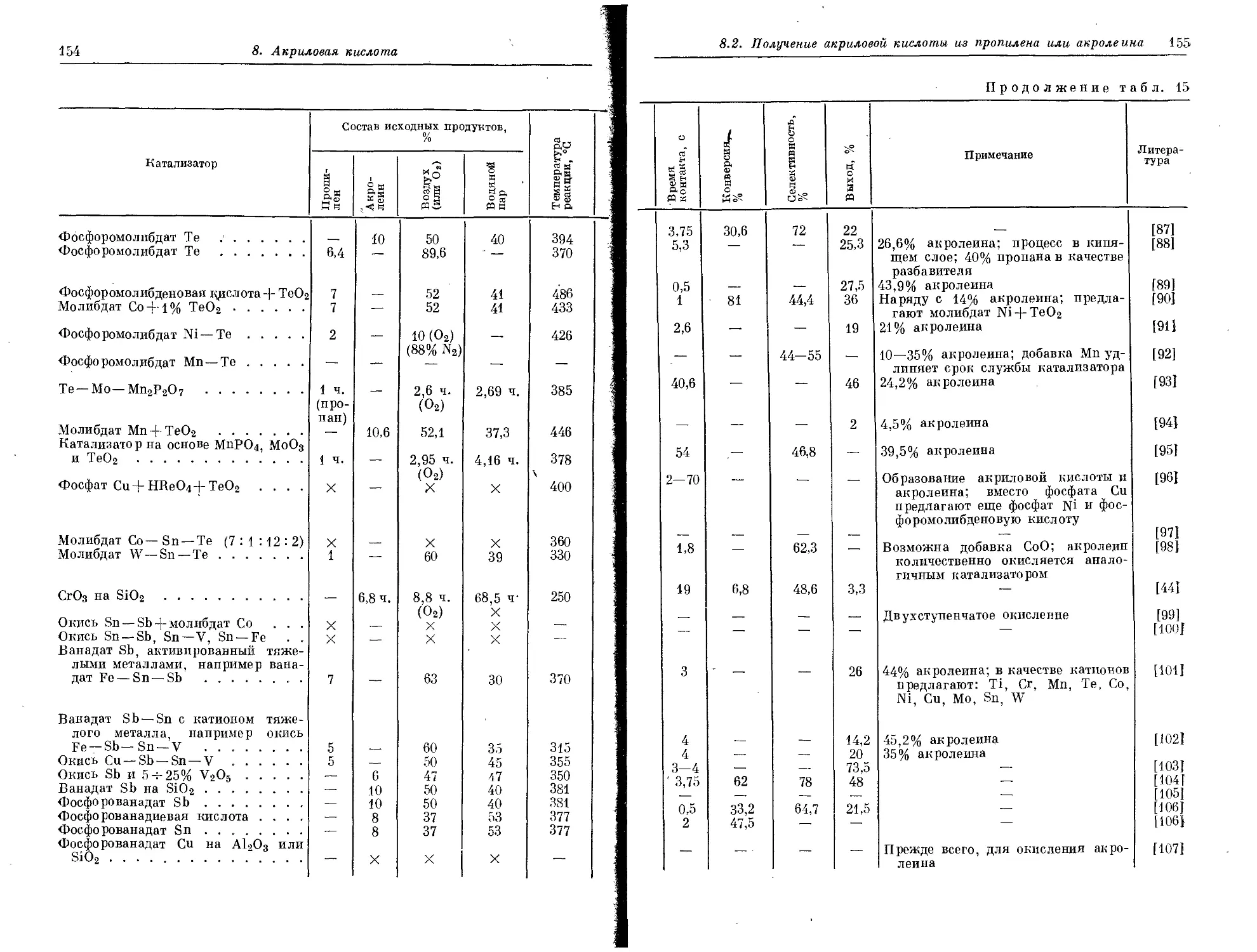
15—20 и при каталитическом крекинге 8—12 вес. % от исходного
продукта.
Обычно выход газа составляет 4,5—5,5 вес. % от общего коли-
чества сырой нефти, поступающей на нефтеперерабатывающий завод.
Газовая смесь [32], образующаяся на современном нефтеперераба-
тывающем заводе, как правило, имеет следующий состав (в мол., %):
Инертные газы
(N2, О2, СО) ... . 4,1
Водород............... 6,1
Метан................ 39,1
Этан ................ 17,5
Этилен ............... 7,3
Пропан...............9,4
П ропилен........... 8,9
Бутаны.......... . : 2,6
Пентаны ......... 1,4
Сероводород......... 3,0
Двуокись углерода . . 0,6
Ниже описано получение пропилена на современном нефтепере-
рабатывающем заводе [33].
Нефтезаводской газ компримируют, охлаждая при этом после
каждой ступени сжатия. Кислые газы, в основном H2S и СО2, аб-
сорбируют этаноламином и затем газ промывают щелочью. После
охлаждения и дальнейшей компрессии проводят обезвоживание
(раньше с помощью А12О3, в настоящее время на молярных ситах).
Методом низкотемпературного фракционирования смесь разде-
ляют на этан, этилен, пропан, пропилен и топливный газ. Этан
и пропан подвергают дальнейшему крекингу в трубчатых печах
в присутствии водяного пара для получения этилена и пропилена.
После компрессии и охлаждения газы снова направляют на уста-
новку для разделения газов. Ацетилен удаляется путем каталити-
ческого гидрирования либо .из общего количества нефтезаводского
газа, либо только из этиленовой фракции. Разделение пропана и
пропилена осуществляется дистилляцией или, если это целесообразно,
проведением со смесью ряда реакций. Стоимость установки для
производства 90 000 т этилена и 43 000 т пропилена из нефтезавод-
ских газов составляет 9,9 млн. долларов, цена 1 фунта этилена
и пропилена 0,0241 доллара.
1.2. ОБРАЗОВАНИЕ ПРИ СИНТЕЗЕ ПО МЕТОДУ ФИШЕРА — ТРОПША
При синтезе Фишера—Тропша как побочный продукт образуется
пропилен. Его количество зависит от катализатора и условий реак-
ции. При нормальном давлении и температуре реакции 175—210 °C
на кобальтовых катализаторах фракция С3—С4 содержит до 43%
олефинов [35]. Аналогичное содержание олефинов наблюдается
при 100 °C и среднем давлении 10 кгс/см2.
10
1. Получение пропилена
1.4. Получение из соединений С3
11
При циркуляционном режиме работы, когда время пребывания!
сырья в реакторе значительно ниже, содержание олефинов увели-!
чивается до 65% [37]. Использование железных катализаторов спо-1
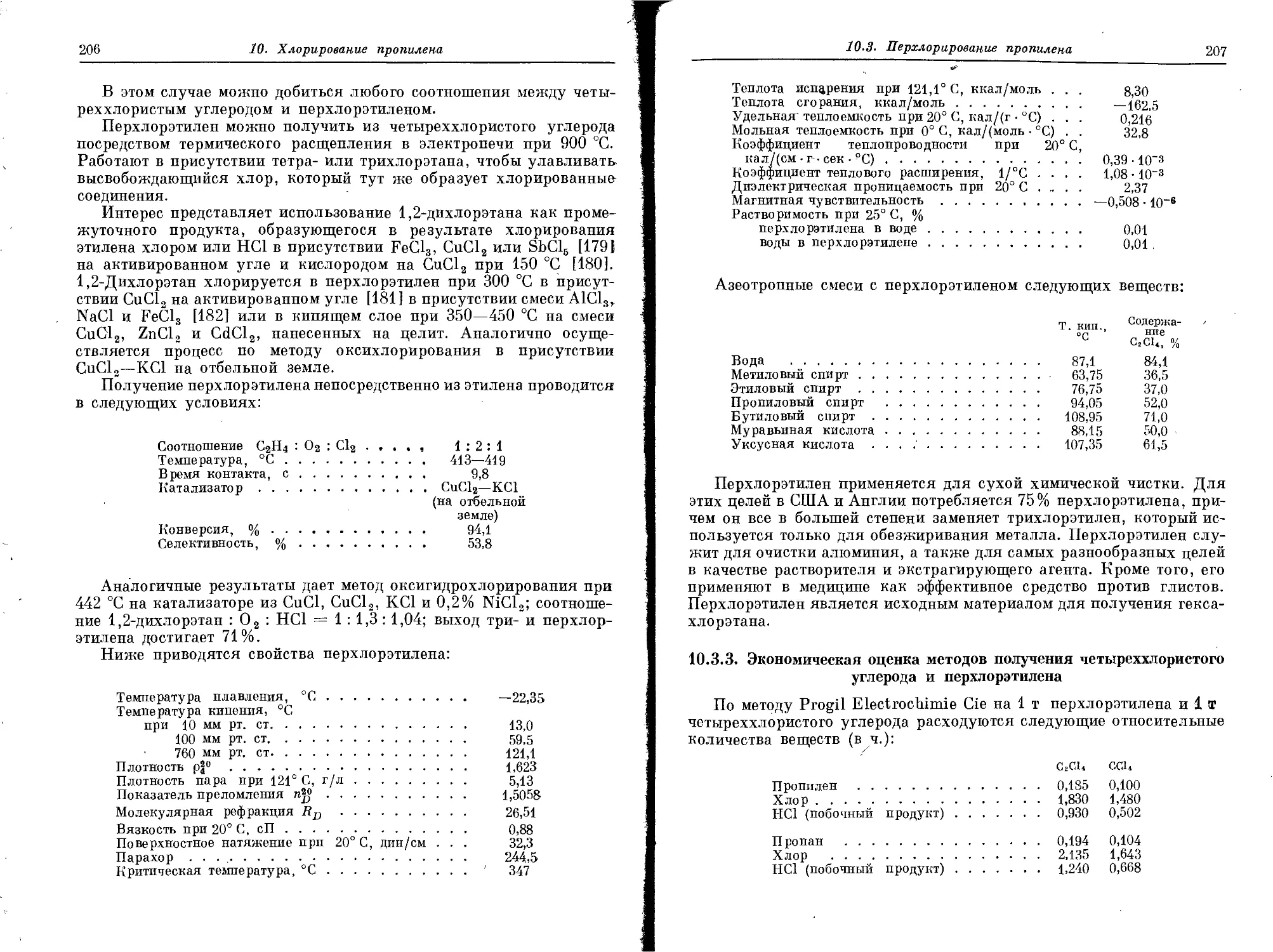
собствует дальнейшему повышению содержания олефинов. Присут-1
ствие железного порошка, суспендированного в масле, через которое]
пропускают газ синтеза, приводит к образованию во фракции С3—С4|
75—80% олефинов при 250 °C и давлении 20 кгс/см2. По литератур-]
ным данным продукты, образующиеся при синтезе Фишера — Тропша!
на железном катализаторе при максимальной температуре 225 °C]
и давлении 10 кгс/см2 (процесс I. G. Farbenindustrie), имеют сле-
дующий состав (в вес. %):
Метан................ 8,9
Этан ................ 4,3
Этдлен .............. 3,0
Пропан .............. 1,4
Пропилен............. 8,9
Бутан................ 2,8
Бутилены............. 5,2
Фракция С5........... 6,5
Фракция Св—Сю .... 16,7
Фракция Сц—Ст7 .... 9,3
Парафиновый гач (неочи-
щенный парафин) ... 5,9
Высокоплавкий парафин 13,6
Низшие спирты......... 4,0
Высшие спирты ........ 1,5
Сложные эфиры......... 7,1
1.3. ВЫДЕЛЕНИЕ ИЗ ПОЛУКОКСОВЫХ И КОКСОВЫХ ГАЗОВ ’
При полукоксовании каменного угля при 500—600 °C образуется]
примерно 10% газа, содержащего 1—8% пропилена. При коксо-]
вании при 1000 °C получается 25% газа, но пропилена в нем только]
0,36% [36]. После выделения из коксового газа водорода в оста-]
вшемся сжиженном газе (рур-газоль) содержание олефиновой фрак-i
ции С3—С4 доходит до 54%, и он может служить источником полу-
чения пропилена. J
1.4. ПОЛУЧЕНИЕ ИЗ СОЕДИНЕНИЙ С3 I
Большую роль играет дегидрирование этана и бутана в этилен]
и бутилен. Дегидрирование пропана в промышленном масштабе]
осуществляется незначительно, так как пропилен, образующийся]
совместно с другими углеводородами при других процессах, в част-1
пости при пиролизе, полностью покрывает потребность в данном]
продукте в большинстве промышленных стран. Поэтому термическое!
и каталитическое дегидрирование пропана описывается вкратце. 1
Правда, пропилен, получаемый путем каталитического дегидриро-]
вания пропана, дешевле образующегося при пиролизе. |
1.4.1. Термическое дегидрирование пропана
В промышленном масштабе осуществляется только термическое
дегидрирование этана и изобутана, которое приводит к получению
соответственно этилена и изобутилена.
При кратковременном нагревании пропана до высоких температур
образуется лишь небольшое количество пропилена, так как реак-
ции крекинга (1) и (2) протекают лучше, чем реакция дегидриро-
вания (3):
С3Н8 > СН4+С2Н4—16,1 ккал/моль (1)
2С3Н8—> С2Нв+С3Нв+СН4 (2)
С3Н8 С3Н8 + Н2 —30,0 ккал/моль (3)
Решающую роль при термическом крекинге пропана играет
реакция (1).
Образование большого количества этилена объясняется тем,
что для разрыва связи С—С нужно значительно меньше энергии
(62,5 ккал/моль) [38], чем для разрыва связи G—Н (87 ккал/моль
для первичной связи, 85,8 ккал/моль для вторичной и 83 ккал/моль
для третичной). Реакция дегидрирования является равновесной,
при повышении температуры равновесие сдвигается вправо.
Представленные на рис. 1 теоретические соотношения равно-
весия между пропаном и пропиленом не могут осуществиться из-за
реакции крекинга [39]. В табл. 1 сравниваются результаты дегид-
рировайця различных парафинов С2—С4.
Таблица 1
Равновесная концентрация олефинов при дегидрировании
парафиновых углеводородов С2—С4 при разных температурах
Температура, °C Содержание олефинов, вес. %
Этилен Пропилен Бутилен Изобутилен
427 1,6 6,5 11,4 12,5
450 2,4 9,1 15,2 16,4
500 4,9 16,4 24,5 26,3
525 — — 29,2 28,7
550 — 26,1 34,3 35,9
• 575 — 31,1 — —
600 15,2 35,3 ' 41,9 42,7
727 34,5 47,3 48,7 48,7
12
7. Получение пропилена
Обширные исследования чисто термического дегидрирования
пропана, проведенные Фреем и сотрудниками [40, 41], полностью
подтвердили приведенные данные. Ниже сопоставлены полученные
ими результаты по составу газов (в вес. %) при опытах в кварцевых
трубках при 575 °C, нормальном давлении и различном времени
контакта:
74 с 4 мин
Азот..............0,5 0,7 Этилен
Водород..........4,1 8,0 Пропан .
Метан ............4,6 11,7 Пропилен
• Этан.............0,5 3,2
74 с
4,0
81,8
4,5
4 мин
8,2
58,9
9,1
Хотя содержание пропилена при значительном увеличении вре-
мени контакта и повышается, тем не менее одновременно все силь-
нее проявляются нежелательные побочные реакции, так что выход
Температура, °C
Рис. 1. Расчетная равновесная кон-
центрация при дегидрировании га-
зообразных парафиновых углеводо-
родов [39]:
1 — изобутан; 2 — бутан; з — пропан;
4 — этан.
Рис. 2. Зависимость конверсии про-
пана от времени и температуры пиро-
лиза [120].
(в пересчете на используемый пропан) понижается. На рис. 2 по-
казана конверсия пропана при термическом дегидрировании [120].
При промышленных масштабах проведения термического дегид-
рирования (крекинге) пропана в этилен выход пропилена можно
повысить путем изменения условий реакции до соотношения эти-
лен : пропилен =1:1.
1.4.2. Каталитическое дегидрирование пропана
При получении пропилена путем дегидрирования пропана не-
обходимо применение специальных катализаторов для того, чтобы
в кратчайшее время преодолеть сравнительно высокую энергию
1.4. Получение из соединений С3
13
связи С—Н (87,3 ккал/моль). Эффективный катализатор существенно
ускоряет процесс дегидрирования. При этом количество нежела-
тельных побочных продуктов становится незначительным, так как
побочные реакции протекают медленнее. Ниже приводятся условия
процесса и состав продуктов, получаемых при каталитическом (I)
и термическом (II) дегидрировании пропана:
I п I п
Параметры процесса: Выход, мол. %:
Температура, ?С .... 592 600 Водород .............. 18,5 50,0
Давление, мм рт. ст. . . 600 760 Метан................. 33,5 1,0
Время контакта, с . . 252- 2,7 Этан .................. 7,2 0,0
Конверсия, вес. % . . 25,0 25,0 Этилен ............... 20,2 0,0
Пропилен.............. 20,5 49,0
/ При нека-алитическом процессе высокую энергию связи С—Н
можно преодолеть, увеличив время контакта при высоких темпе-
ратурах, что, однако, ведет к убыстрению реакций разложения.
Для каталитического дегидрирования пропана применяются
катализаторы: Cr2O3, МоО3, V2O5, ТЮ2 и СеО2 [42]. Из них наилуч-
шим образом зарекомендовал себя Сг2О3, который используется
для уменьшения рекристаллизации на носителе у-А12О3. В про-
мышленности работаю^ при температурах, превышающих 500 °C.
Ниже указан выход Пропилена (в объемн. %), полученный при
600 °C, нормальном давлении и разном времени пребывания про-
панЩна катализаторе (90% Сг2О3, 10% А12О3) [13]:
2,7 с 8,9 с 17,5 с 36,0 с
Водород 20,5 24,0 27,5 29,0
Метан 0,5 3,0 13,5 19,0
Этан 0,0 5,0 7,5 29,0
Этилен . 0,0 0,3 0,2 1,0
Пропан 59,0 41,0 32,0 6,0
Пропилен 20,0 21,0 14,0 9,0
Отсюда следует, что дегидрирование без побочных реакций воз-
можно лишь при низких значениях конверсии и коротком времени
пребывания пропана на катализаторе.
Протекание реакции С3Н8 # С3Нв + Н2, сопровождающееся уве-
личением объема, указывает на окончание процесса дегидрирования.
Поэтому понижение давления способствует образованию пропилена
при дегидрировании, заканчивающемся реакцией равновесия. При-
веденные ниже данные характеризуют влияние давления на про-
цесс дегидрирования бутана в бутен при 527 °C [43]:
Давление, кгс/смЗ............... 0,01 0,1 1 10 100
Конверсия бутана в бутен, вес. % 97 80 38,5 13 4
14
7. Получение пропилена
Новаковский [44] на основании обстоятельного изучения ката-
литического дегидрирования пропана рекомендует применение ка-
тализатора следующего состава: 93,5% А12О3, 5% Сг203 и 15%
, К2О. При работе в кварцевом реакторе и объемной производитель-
ности катализатора 300 л/ч (в расчете на пропан) он рекомендует
температуру реакции 610—660 °C, а при работе в металлическом
реакторе и объемной производительности катализатора 400—700 л/ч
оптимальной температурой будет 570—600 °C.
Для приготовления катализаторов А12О3 пропитывают водным
раствором СгО3, Cr(NO3)3, (NH4)2Cr2O7 или (NH4)2CrO4 [45]. Осо-
бенно активны совместно осажденные катализаторы Сг2О3 — А12О3
[46].
Катализатор с Сг2О3 при 575 °C дает 95% пропилена, выход
остается постоянным в широком диапазоне, однако конверсия со-
ставляет только 25,8% [47].
1.5. ПОЛУЧЕНИЕ ПРОПИЛЕНА ИЗ ДРУГИХ
ГАЗООБРАЗНЫХ УГЛЕВОДОРОДОВ
1.5.1. Получение и? этилена и этана
При нагревании этилена в присутствии кислорода до 377—600 °C
[48] наряду с другими соединениями (в основном С4) получают
пропилен.
Пропилен образуется также при нагревании зтана до 800—
880 °C [49, 50] в основном в результате взаимодействия первона-
чально образующегося бутилена с этиленом. Максимальный выход
(91%) получают в атмосфере кислорода, при времени контакта 3 с
и температуре реакции 490 °C [51].
1.5.2. Получение из бутана и изобутана
При промышленном циролизе бутана происходит расщепление
его на этилен и зтан, а также на пропилен и метан. Дегидрирование
до бутилена или бутадиена происходит в гораздо меньшем масштабе
по сравнению с образованием пропилена. Это становится понятным
при рассмотрении теплового эффекта отдельных реакций:
С4Н10 —> С3Нв + СЩ-17,6 ккал/моль
С<Н10 —>- С2Н4+С2Нв-17,0 ккал/моль
С4Н10 —► С4Н8 + Н2 —30,0 ккал/моль
Фролих [52] исследовал состав конечного газа при пиролизе
бутана в зависимости от температуры. Он установил, что общее
содержание олефинов будет максимальным при 690 °C, в то время
как максимальный выход пропилена наблюдается уже при 650 °C.
1.6. Получение пропилена путем пиролиза углеводородов
15
Первичные продукты пиролиза бутана [53] имеют следующий
состав (в моль/100 моль прореагировавшего бутана):
600 °C 650 °с
Метан и пропилен.................... 48,5 48,0
Этан и этилен........................34,5 37,7
Водород и бутилен................... 16,0 12,3
Пропан .............................. 0,0 11,0
В полученной при пиролизе бутана фракции С3 почти отсутствует
пропан. Это является большим преимуществом, так как отпадает
необходимость в разделении пропана и пропилена и можно получать
очень чистый пропилен.
В литературе описаны различные разновидности пиролиза бутана;
среди них особый^ интерес представляет каталитический метод при
температуре 600 °C в присутствии катализатора SiO2 — ZrO2 —
AI2O3 [54]. Кроме того, описаны методы пиролиза в присутствии
кислорода [55] или водяного пара [56].
В промышленности хорошо зарекомендовал себя процесс пиро-
лиза бутана в реакторе с кварцевым теплоносителем. В результате
пиролиза 100 кг бутана при 943 °C наряду с другими продуктами
получается 44,1 кг этилена и 12,5 кг пропилена; конверсия составляет
91%. Если при пиролизе основное значение придается пропилену,
то целесообразно проводить процесс в трубчатой печи.
Термическое дегидрирование изобутана [57] также осуществлено
в промышленных масштабах. В этом процессе наряду с 50% изобути-
лена получается 25% пропилена (конверсия 20—30%, температура
650—730 °C, давление 5,2—6,6 кгс/см2). При температуре реакции
600—650 °C получают даже 63 мол. % изобутилена и 34,5—36 мол. %
пропилена [53, 58].
1.6. ПОЛУЧЕНИЕ ПРОПИЛЕНА ПУТЕМ ПИРОЛИЗА УГЛЕВОДОРОДОВ
Пиролиз углеводородов, таких, как этан, бутан, бензин, керосин
и другие нефтяные фракции, превратился в один из самых совре-
менных и экономичных методов получения олефинов, которые при-
обрели такое большое значение в промышленности органической
химии [59]. Процесс производства газообразных олефинов на крупно-
тоннажных пиролизных установках обходится дешевле, чем их вы-
деление из нефтезаводских газов.
В процессе пиролиза в зависимости от температуры, типа ката-
лизатора и продолжительности крекинга могут происходить разно-
образные изменения химической структуры молекул [60]:
1) углеводороды с неразветвленной цепью расщепляются на
низкомолекулярные осколки, причем расщепление, в зависимости
16
1. Получение пропилена
от условий, происходит в разных местах молекулы; большая часть
осколков имеет при этом ненасыщенный характер;
2) в результате отщепления атомов водорода цепи и кольца
дают ненасыщенные соединения;
3) циклические парафины переходят в ароматические соединения;
4) от разветвленных циклических углеводородов отщепляются
боковые цепи с образованием олефинов;
5) ароматические соединения разрываются с образованием боль-
шого количества кокса;
6) разветвленные линейные углеводороды большей частью пере-
ходят в нафтеновые и ароматические;
7) углеводороды с' прямой цепью изомеризуются в разветвлен-
ные цепи;
8) низкомолекулярные осколки полимеризуются или конденси-
руются в высокомолекулярные углеводороды измененной струк-
туры.
Количество энергии, необходимое для разрыва связи С—С,
понижается с увеличением длины цепи углеводорода и составляет
при расщеплении бутана на этан и этилен 32 ккал/моль, декана
на пентан и пентен 29 ккал/моль, эйкозана на декан и децен
12 ккал/моль. Расщепление олефинов требует большей затраты
энергии, например, для превращения бутилена в этилен нужно
41 ккал/моль, а децена в пентен 30 ккал/моль.
Основные факторы, определяющие ход пиролиза — сырье, про-
должительность процесса, температура и давление [60].
Сырье (крекинг-сырье). Высокомолекулярные соединения рас-
щепляются легче низкомолекулярных, причем парафины нормального
строения отличаются наибольшей склонностью к расщеплению;
далее следуют изопарафины, олефины, нафтены и ароматические
углеводороды.
Исходное сырье может быть неодинаковым в разных странах.
Например, в США для производства этилена предпочитают брать
этан, получаемый из дешевых природных газов Техаса. В ФРГ
и Японии, где достаточно бензина и не хватает природного газа,
оправдало себя использование бензина [66].
Ниже указан выход, по'лучаемый при пиролизе этана и бензина
(в т):
Этан Бензин Этан Бензин
Расход исходного Бутилены .... — 35 000
сырья 129 500 513 000 Бензол — 28 800
Продукты пиролиза: Толуол Ксилол — 17 500 6 500
Этилен 100 000 100 000 Топливо, горючее — 86 900
Водород, метан ... 26 600 90 000 Остаточные масла — 25000
Пропилен — 90 000 Прочее и потери 3000 16 700
Бутадиен — 15 500
1.6. Получение пропилена путем пиролиза углеводородов
17
Температура. Ниже приведена
ных углеводородов (в °C):
температура расщепления различ-
Этан............. 820—850
П ропан ......... 750—820
Легкий бензин
(фракция 100 °C) 760—790
Бензин (фракция
160 SC)......... 750—800
Сырая нефть . . . 730—760
При невысоких температурах (~410 °C) связи С—С разрываются
прежде всего в середине молекулы; с повышением температуры места
разрыва перемещаются на конец
цепи, при этом образуются длин-
ноцепные олефины и низкокипя-
щие продукты, кроме того, все
в большем количестве отщепляется
водород в результате реакции
дегидрирования. При температуре
выше 550 °C начинается образо-
вание ароматических углеводоро-
дов, которое достигает максимума
при 700—900 °C, при этом также
образуются углекислый газ и кокс.
Выше 1000 °C углекислый газ
и кокс будут единственными ко-
нечными продуктами.
Продолжительность пиролиза.
Пиролиз является эндотермиче-
ским процессом, требующим по-
стоянного подвода тепла, что не-
избежно увеличивает продолжи-
тельность процесса и способствует
появлению вторичных реакций.
При низких температурах это вы-
Рис. 3. Зависимость состава газа
пиролиза от температуры при пиро-
лизе пропана:
1 — сумма олефинов; 2 — этилен; з — во-
дород; 4 — пропилен.
ражается в интенсивной изомеризации и образовании низкомолеку-
лярных олефинов, при средних температурах усиливаются реакции
дегидрирования (образование диолефиновых углеводородов) и аро-
матизации, при повышенных температурах дополнительно полу-
чаются газ и кокс.
Давление. При низком давлении (1—2 кгс/см2) — крекинг-про-
цесс в паровой или паро-жидкостной фазе — образуются более не-
насыщенные углеводороды одновременно с газом и коксом. При вы-
соком давлении (до 70 кгс/см2) — жидкофазный крекинг — меньше
образуется газа и ненасыщенных соединений.
Основы расщепления парафинов на олефины описаны в многих
работах [61—64]. Герхольд [65] подробно изложил механизм реак-
ции расщепления газообразных и жидких углеводородов. На рис. 3
представлена зависимость' состава продуктов пиролиза пропана от
2 Заказ 399
Таблица 2
Характеристика процессов пиролиза для производства этилена и пропилена [29]
Установки пиролиза Сырье Условия реакции Состав продуктов пиролиза
Тип нагрева Нагреватель, нагре- вающее средство Наименование процесса Температура, °C Давление, кгс/см2 Время контакта, с Теплоноситель, ка- тализатор Газообразные Жидкие Выход твердых продуктов, %
Компоненты Содержание, % Выход, % Характери- стика Выход, %
Термические процессы
Прямой Трубча- Stone- Пропан 790 3,5 0,9 — Олефины 36,1 / 15,7 3,0 «й
тая печь Webster с2 25,7 | 81,3 «С ©.
Сз 8,3 % й ©
uodu
илена
Косвен- Грану- Phillips Этан 960— 1,35 0,12 Корунд Олефины 58,0 В ос- 6,1 0,8
ный лир О- Petro- —835 с2 56,2 | 92,5 новном
ванные leum Сз 1,0 арома-
подвиж- (процесс тиче-
ные с галеч- ские
агенты — ным теп-
шарики лоноси-
То же — телем) Пропан 820 — 1,1 Кварц Олефины 50,8 — 6,3 0,3
, галька Socony с2 37,5 j. 97,2
Vac. Oil С3 12,2
(термо-
Фор)
То же — Farb- Мазут 580 1,2 Кокс Олефины 7,0 75,0 10,0
кокс werke (35 :1) с2 1,8 | 14,0
* Hoechst С3 0,5
То же — Lurgi- Пропан 825— 1,5 — Кварц Олефины 55,0 T. кип. 8,5 0,6
песок Ruhrgas 760 с2 39,0 | 90,9 > 200 °C,
С$ 12,0 6,2% 1.6.
Бензин, Олефины 42,0 T. кип. 39,0 —
сырая с2 23,0 j 58,0 <200 °C, ©
нефть Сз 13,0 17,0% «й «С гъ
Косвен- Порош- Процесс Этан 1000 — 0,01 Кокс Олефины 63,8 — 5,0 2,7 3 й
ный кооб- Лавров- (20 : 1) с2 59,2 | 92 3
разные ского — Сз 4,0 Ъ
подвиж^ Брод- 3
ные ского й
агенты ©
(кокс) ©
Газооб- Kellogg Пропан 690 1-3 0,8 15 : 1 Олефины 65 В ос- 10,5 0 3 <й
разные с2 44,0 | 88 новном 3
агенты — сз 16,1 арома-
пар тиче- 3
ские й
Бензин 630 1-3; 1,0 12 : 1 Олефины 40 . То же 33,0 0 © £
с2 22,5 j 66 й Со
Сз 12,0 .3
Дизель- 575 1-3 1,0 8 :1 Олефины 40 » 43,0 0 «й ©
ное топ- с2 21,3 j 56 ©
ливо Сз 14,6 ©
Косвен- То же — East- Пропан 1980 1,2 0,005 n2 + co2 Олефины 35 I RD 18,0 0 ©
ный горючие man с2 30,0 z OU ©
газы ©
Бензин 2000 1,2 0,005 n2 + СО2 Олефины 50 — 25,0 0
с2 40,0 | 70
С3 4,0
а со
Продолжение табл. 2 g
Установки пиролиза Сырье Условия реакции Состав продуктов пиролиза
Тип нагрева Нагреватель, нагре- вающее средство Наименование процесса Температура, °C Давление, кгс/см4 Время контакта, с Теплоноситель, катализатор Газообразные Жидкие Выход твердых продуктов, %
Компоненты Содержание, % Выход, % Характери- стика Выход, %
Прямой Косвен- ный Непод- вижный катали- затор — медная прово- лока Порош- кооб- разный подвиж- ный ка- тализа- тор — SiO2 Катарол Tsutsumi Бензин Легкое масло Сырая нефть Те 650 640 950— 800 рмокап 1,5 1,5 0,3 шлити 3 30—60 tecKue про Медь Медь SiO2 цессы Олефины С2 С3 Олефины сз Олефины с2 Сз 26,7 11,6 10,6 26,4 11,6 10,9 57,6 23,4 34,2 j 62,4 | 49,4 | 70,1 В ос- новном арома- тиче- ские То же Арома- тиче- ские 25% 37,0 50,0 29,2 1. Получение пропилена 2 И Ч ° О 4 °
Порош- кооб- разный подвиж- ный ка- тализа- АзНИИ— Алиев Остаток от пере- гонки нефти 700 1,2 12 — Олефины с2 Сз 27,6 15,7 8,7 | 47,3 Т.кип. <200 °C, 26,0%; т. кип. > 200 °C, 13,4% 39,4 13,3
Прямой тор — кокс Непод- Auto- Этан 680 850— 1,2 Окисл 0,6 8 гтелън 1,0 яе процес ai2o3 н- Олефины с2 (<з :ы 19,6 10,0 7,8 | 36,2 Т. кип. < 200 9С, 11,0%; т. кип. > 200 -°C, 26,0% 37,0 26,8 6. Получение пропилена пу
вижный грану- лятор — шарики Грану- лиро- ванные подвиж- ные агенты — кокс therm Bartho- lome Auto- therm (BASF) Сырая нефть 900 725 1,0 + О2 09 : сы- рая нефть — = 0,4 = 1 Олефины с2 сз со2+со углево- дороды 45,0 23,0 12,5 4 49,3 16,6 1109,1 Т. кип. <200 °C, 14,0%; т. кип. > 200 °C, 4,0% Вода 6,5% 24,4 4,5 (2,0) тем пиролиза углеводородов
22
1. Получение пропилена
со
сб
W
*5
ю
сб
Оптимальные виды сырья Для различных процессов пиролиза [59]
чхфэн ЕЕСкЧЭ 1 I+++ III 11+ 1+
НОХВХЭО 1 I+++ III 11+ 1+
0ЯИЕП01 0ОНЧ1Г01ОЯ 1 I+++ III 11+ 1+
О0ИКПО1 эончггэеий 1 I+++ III I++ II
ниенад + 111+ 11 1 ++1 II
ф ф £ Я S Р* »• О ь S ж о М >» Е-< 2 t=t 3 я е о s. m о ф sa g я k •s ? 8 + 1 + 1 + H—1—F g H—F 1 1 1 §' S' g о ft 8
HBiig g' + ++ 1 + +++ 1 1 + 1 & II s* Й
нвпосТц 3 g S i + ++1+ +++ 1+1 f +1 e g з
нвхе § 5 § i + ++i+ +++ ill | +i tq О
Характеристика процесса пиролиза | • • • • • • о • • . ... . x—s g сб • • Й • • • ... CQ • 5 • § • • • . • • • g H ~ • g •< -Я о Q • я • • Я .'Д Й -CO о cu н з> к g 8 н С.-— о g -я 2 . Й М ей -S И & S § И— И й ’яд- я,’ч >. S g к • 2 • -а -а | 5-0 & & 3 g § • “ е.е- п $ н и -а • s < « • ОД© И и ф 5 22 о D S • **5 ё§||зез|^з g.|g § ЙИЯ ё g ®3»*>г*аОфОрчф св*®03 S S =§g §§•§§§«&§ яз а и н о S я. о ° Я о Я’—'И К Й S «Й < <
1.6. Получение пропилена путем пиролиза углеводородов
23
температуры. В табл. 2 и 3 охарактеризованы современные про-
цессы крекинга и пиролиза [59].
Различные процессы крекинга отличаются друг от друга видом
и способом подвода тепла, температурой расщепления, временем
контакта и исходным сырьем. Целью всех методов является по воз-
можности дешевое получение необходимых для современной нефте-
химии продуктов, в частности олефинов и ароматических углеводо-
родов.
В зависимости от техники подвода тепла для расщепления про-
цессы пиролиза подразделяются следующим образом:
1) пиролиз в трубчатых печах;
2) процесс с неподвижным теплоносителем (например, регене-
ративный крекинг-процесс);
3) процесс с подвижным гранулированным теплоносителем (на-
пример, флюид-процесс, процесс с подвижным катализатором);
4) процесс с газообразным теплоносителем (расщепление в го-
могенной среде, например пиролиз в паровой фазе, пиролиз окисле-
нием, автотермическое расщепление, пиролиз дымовыми газами);
5) процесс с жидким теплоносителем.
Далее кратко описываются отдельные процессы крекинга, при
которых в больших или меньших количествах наряду с основным:
продуктом получают и пропилен.
1.6.1. Пиролиз в трубчатых печах
Пиролиз в трубчатой печи (рис. 4) — наиболее распространенный:
процесс термического расщепления легких и средних углеводо-
родов.
Установки являются развитием перегонных аппаратов, только
подвод тепла осуществляется таким образом, чтобы исходный про-
дукт быстро нагревался и необходимое время оставался при высокой
температуре. Исходный продукт предварительно нагревается в тепло-
обменнике вне печи до ~300—350 °C и вводится в зону конвекции
печи, где подогревается горячими отработанными газами до ~400—
500 °C. Затем он попадает в зону излучения и достигает окончатель-
ной температуры пиролиза благодаря непосредственному обогреву
труб от сжигания газового или котельного топлива. При пиролизе
пропана температура достигает 780—800 °C, для легкого бензина
достаточно 720—750 °C. Трубы изготовляют из высоколегированных
хромоникелевых сталей, в наиболее теплонапряженных местах
применяют сплавы меди или хрома.
Недостатком пиролиза в трубчатых печах является периодиче-
ское прерывание цикла для выжигания кокса, что к тому же препят-
ствует использованию высококипящих углеводородных фракций.
(Разработаны специальные крекинг-процессы, работающие с обра-
зованием кокса, причем попеременно в одних камерах идет-
24
1. Получение пропилена
образование, а в других удаление кокса.) Недостатком является также
ограничение температуры внутри труб (не выше 830 °C) и непро-
должительный срок их службы вследствие высокого теплового
напряжения. Тем не менее трубчатые печи получили широкое рас-
пространение благодаря простоте обслуживания.
В последнее время наметилась тенденция к повышению темпе-
ратуры реакции и значительному уменьшению времени контакта
при пиролизе в трубчатых печах 1122, 123]. Этому способствовала,
Рис. 4. Радиационно-конвекционная трубчатая пи-
ролизная печь [121].
в частности, разработка жаропрочных высококачественных сталей
(например, сплав Incoloy Alloy 800, выдерживающий 100 000 ч
работы при 1050 °C). Пиролизные печи из таких сталей выдерживают
длительную высокую тепловую нагрузку. Выход этилена по срав-
нению с выходом пропилена в обычных трубчатых печах крекинга
выше (до 34,5%).
Айзенлор [125] приводит зависимость выхода различных про-
дуктов пиролиза от параметров процесса:
Параметры процесса:
Температура на входе, СС .... 580 596 598
Температура на выходе^ °C ... 780 807 830
Соотношение пар/бензин, кг/кг • . 0,7 0,7 0,7
Время контакта, с 0,5 0,5 0,5
Скорость пара, м/с Тепловое напряжение поверхности 300 300 300
нагрева, ккал/(м2.ч) 60 000 60 000 60 000
Температура камеры сгорания, °C 1000—1100 1000—1100 1000—1100
1.6. Получение пропилена путем пиролиза углеводородов
25
Выход продуктов, вес. %:
Метан ....................
Этилен ...................
Пропилен .................
Продукты С4 (суммарно) . .
Бутадиен..................
Пиролизный бензин ....
Котельное топливо.........
Соотношение этилен/пропплен
До сих пор известны следующие кратковременные и высокотем-
пературные процессы пиролиза:
1) кратковременный крекинг фирмы Lummus (метод SRT);
2) крекинг фирмы Kellogg
(Н SC-метод);
3) пиролиз фирмы Furnace
von Selas;
4) пиролиз в многозонной
печи фирмы Foster—Wheeler;
5) сверхизбирательный кре-
кинг фирмы Stone and Webster
(метод U SC);
6) кратковременный пиро-
лиз фирмы Lurgi.
1.6.2. Пиролиз в реакторе
с кварцевым теплоносителем
(процесс фирмы Phillips
Petroleum Со.)
Использование косвенных
теплоносителей позволяет пе-
редать за одно и то же время
значительно большее количе-
ство тепла, что заметно повы-
шает выход продукта в единицу
времени и на единицу объ-
ема [66].
В процессе фирмы Phillips
Petroleum Со. (рис. 5) приме-
12,1 15,4 17,2
22,9 27,6 29,7
18,3 19,1 16,8
12,6 11,8 10,0
4,3 5,0 4,9
27,0 18,9 16,7
1,3 1,6 2,8
0,8 0,68 0,56
Рис. 5. Схема пиролиза в реакторе
с гранулированным подвижным тепло-
носителем:
НЯЮТСЯ корундовые шарики 1 — подогреватель; г — реактор; 3 — элеватор,
или кусочки кварца диаметром
~4 см, которые нагреваются в подогревателе до 800—1000 °C, мед-
ленно падают по трубе и подаются в реактор. Состав исходной реак-
ционной смеси (I) и пиролизного газа (II) (в мол. %) при пиролизе
бутана приведен ниже (конверсия бутана 91%) [67]:
26
1. Получение пропилена
• I II I II
Водород . . . . . — 16,6 Пропилен '. ... . — 6,7
Метан .... 30,5 Бутан 94,6 3,5
Этан .... . . — 2,0 - Изобутан 4,7 0,3
Этилен . . . . . — 37,3 Бутилен — л 0,5
Ацетилен . . . . — 1,2 Бутадиен — 0,6
Пропан . . . .. . 0,3 0,1 Другие углеводороды 0,4 0,7
в противотоке при температурах 900—
Циролиз проводится
980 °C (на входе) и 795—825 °C (на выходе). Конверсия достигает
95% при времени контакта 0,1—0,3 с. Охлажденные шарики под
действием
воздуха
собственного веса падают навстречу потоку горячего
подогреватель, и образовавшийся кокс (~1%) там сго-
рает. Выход продукта в ре-
акторе с кварцевым тепло-
носителем на 25% превышает
выход в трубчатом реакторе.
в
Рис. 6. Схема
процесса «Hoechster Кокет»
[69]:
1 — коксоотделитель; 2 — коксоразделитель; 3 —
промежуточная емкость; 4 — подогреватель кокса;
•5 — зона смешивания; 6 — реактор; 7 — бункер-
подъемник; 8 — горячая газодувка; 9 — циклон;
10 — колонна резкого охлаждения и отгонки лег-
ких фракций; 11 — ректификационная колонна.
с
1.6.3. Пиролиз в реакторе
коксовым теплоносителем
(процесс фирмы
Farbwerke Hoechst)
Фирма Farbwerke Ho-
echst в 1956 г. разработала
[66—70] процесс непрерыв-
ного пиролиза сырой нефти,
остатков перегонки нефти,
бензина и др., который по-
лучил наименование «НоесД-
ster Кокет» (рис. 6).
Шарики кокса диаметром
3—15 мм подаются транспор-
тирующим газом (азотом) из
бункера подъемника на вы-
соту ~75 м в коксоотдели-
тель. Отделенный от кокса
транспортирующий газ воз-’
вращается через циклон в
бункер подъемника для гру-,
бого и тонкого разделения. Газ нагревается до ~600 °C шариками
кокса, которые потом направляются в Подогреватель и накаляются
там до ~740 °C. Предварительно нагретая сырая нефть впрыски-
, вается в зону смешивания, и в реакторе идет пиролиз при ~690 °C.
Такая температура устанавливается для сырой нефти или дистил-
лятов легкого масла, в то время как расщепление остатков от пере-
гонки нефти происходит при ^—580 °C.
1.6- Получение пропилена путем пиролиза углеводородов
27
В период расщепления на шариках кокса оседает дополнительное
количество кокса, из-за чего размер частиц увеличивается. Дро-
бильная установка с последующей воздушной сепарацией снова
производит необходимое количество частиц с нужными размерами,
которые опять подаются в установку. Кокс не нужно обновлять-
в течение всего цикла, так как образующиеся коксовые шарики
вполне заменяют истирающиеся частицы кокса.
Выход продуктов реакции (в вес. %) при пиролизе сырой нефти
[70] при 760 °C без расщепления С3/С3 (I) и'с расщеплением C2/Cs
(II) приведен ниже (соотношение сырая нефть : теплоноситель =
= 1 : 30, предварительный нагрев нефти — до 380 °C):
I II I II
Окись углерода од 0,1 Углеводороды С4 . . 6,0 6,2
Водород 0,4 0,9 Олефины С2—С4 (сум-
Метан 12,1 12,5 марно) 37,2 43,7
Этан 6,8 — Крекинг-газ .... 58,0 57,5
Этилен 17,7 23,8 Легкий крекинговый
Ацетилен .... 0,1 0,1 дистиллят .... 18,7 19,2
Пропан 1,1 — Тяжелый крекинго-
Пропилен .... 13,7 13,9 вый дистиллят . . 20,8 20,8
Кокс 2,5 2,5
1.6.4. Крекинг сырой нефти в кипящем слое
(процесс фирмы BASF)
Проведение реакций крекинга в кипящем слое требует наруж-.
ного подвода тепловой энергии, необходимой для пиролиза. Здесь
возможны два пути: либо теплоносителе постоянно подогревается
в другой части системы—генераторе, либо часть сырой нефти сжи-
гается и дает в результате автотермического обогрева кипящий слой
[71-75].
Для первого способа фирма Badische Anilin-und Sodafabrik
разработала установку для пиролиза в кипящем слое (рис. 7)',
которая была введена в эксплуатацию в 1959 г. %
В реакторе частицы нефтяного кокса диаметром 0,1—1 мм под-
держиваются в кипящем слое подаваемой снизу смесью пара с не-
большие количеством кислорода (температура 500 °C). Поверх
распределительной решетки для газового потока впрыскивается
сырая нефть при 300—400 °C. В реакторе устанавливается темпера-
тура 720 °C. Частицы нефтяного кокса, величина и вес которых
непрерывно возрастают во время процесса, отводятся из реактора
снизу. Отходящие газы охлаждаются в циклоне до 300 °C впрыском
кубовых остатков из дистилляционной колонны, а летучие продукты
фракционируются в колонне.
28
1. Получение пропилена
Рис. 7. Схема окислительного пиролиза на гра-
нулированном подвижном коксе (пиролиз в ки-
пящем слое по методу фирмы BASF):
1 — реактор; г — циклон; з — холодильник; 4 — ректи-
фикационная колонна; 5 — сепаратор.
Сырая нефть
Рис. 8. Схема флюид-процесса фирмы BASF:
1 — регенератор; 2 — реактор; з — дозирующие устройства;
4 — циклоны; 5 — устройство для резкого охлаждения; в —
сепаратор; 7 — рекуператор; 3 — перегонная колонна; Й —
холодильнику 10 — отстойник.
1.6. Получение пропилена путем пиролиза углеводородов
29
При крекинге сырой нефти в кипящем слое [73] имеет место
следующий выход продуктов (I — в кг/т сырой нефти, II —
в объемн. %):
I ы
Водород................................ 8 15,8
Метан................................ 120 29,3
Этан.................................. 45 5,9
Этилен .............................. 230 32,3
Ацетилен .............................. 1 0,1
Пропан ................................ 7 0,6
Пропилен............................. 125 11,7
Углеводороды С4 ...................... 60 4,3
Итого . . . 596 100
Углекислый газ...................... 395
Окись углерода ..................... 100
Легкая нефть........................ 140
Фракция нафталина..................... 40
Кокс ................................. 45
Вода ................................. 64
Потери ............................... 20
Всего . . . 1400
Количественный баланс свидетельствует об относительно вы-
соком содержании пропилена. Присутствие серы в сырой нефти
существенно не сказывается на ходе процесса. При введении в ки-
пящий слой щелочных солей в качестве каталитических эффективных
соединений связанная сера полностью переходит в сероводород.
Особыми преимуществами отличается автотермический метод,
когда в комплексном производстве одновременно работает установка
по производству аммиака (на 1 т С2Н4 можно выработать 2 т NH3).
Недостатком является потребность в чистом кислороде, который
должен быть приготовлен заранее. Когда хотят обойтись без ки-
слорода, то работают по первому варианту. В этом случае следует
применить инертный теплоноситель, из которого нужно выжечь
кокс в регенераторе (флюид-процесс).
При флюид-процессе [76] эндотермические реакции крекинга
и экзотермическое сгорание проводятся раздельно. Благодаря этому
можно работать с воздухом вместо кислорода (рис. 8). Одновременно
можно использовать и более легкие нефтяные фракции, так как
в данном случае производство нефтяного кокса не является опре-
деляющим.
Из реактора постоянно отводится определенная часть насыщен-
ного сажей и нефтяным коксом теплоносителя, который подается
в регенератор подогретым воздухом и регенерируется при 900 °C
за счет частичного сгорания остаточного нефтепродукта. В самом
реакторе температура достигает 760—790 °C, время контакта ~1 с.
30
1. Получение пропилена
Выходящие из реактора газы пропускаются через циклон и затем
резко охлаждаются до 250 °C тяжелым маслом.
Выход продуктов на этой установке (в кг/т сырой нефти)
составляет:
Этан
Этилен
30—45 Пропилен ..........100—115
220—250 Бутилен + бутадиен 50—60
Пиролизный бензин . .175—225
Очищенный крекинг-газ имеет примерно следующий состав
(в объемн. %):
Окись углерода .... о,5
Водород........... 14
Метан...................33
Этан . . ............... 5
Этилен....................34
Углеводороды С3...........10
Углеводороды С4.........4
Остаточный газ после разделения высших углеводородов может
служить полупродуктом для получения синтез-газа (1,3 т NH3/t
сырой нефти).
1.6.5. Пиролиз в реакторе е песком в качестве теплоносителя
(процесс фирмы Lurgi)
Впервые процесс пиролиза нефти и бензина с использованием
раскаленного песка был применен в 1958 г. фирмой Erdolchemie
GmbH, Dormagen. Затем были пущены 4 установки, одна из которых
работает на здводе Лейна II (ГДР). Вначале установка для кре-
кинга песком, разработанная фирмой Lurgi в содружестве с фирмой
Farbenfabriken Bayer, предназначалась только для крекинга нефти.
Позднее, благодаря благоприятной ситуации на. нефтяном рынке,
смогли использовать бензиновые фракции с температурой кипения
40-165 °C.
С помощью простой технологической схемы (рис. 9) можно
кратко пояснить метод. После нагрева в подогревателе до 350—
400 °C сырье пиролиза впрыскивают вместе с перегретым паром
в реактор 7 с кипящим слоем, состоящим из кварцевого песка с диа-
метром песчинок 0,4—1,2 мм. В результате контакта с горячими
дымовыми газами и прямого обогрева горящим мазутом песок на-
каляется до 1000 °C и пневмотранспортом через сборник 5 по-
дается в реактор, где его температура составляет ~850 °C. Сырье
пиролиза нагревается в реакторе до необходимой температуры,
время контакта ~0,3—0,5 с. Ниже приведена температура нагрева
различных,видов сырья (в °C):
Этан ......... 820—850
Пропан......... 750—825
Легкий бензин
(т. кип. 100 °C) 760—790
Тяжелый бензин
(т. кип. 160 °C) . . 750—800
Нефть . ....... 730—760
1.6. Получение пропилена путем пиролиза углеводородов 31
Таблица 4
Результаты крекинга бензина, получаемые при разных условиях
на установке фирмы Lurgi
Показатели процесса I , II Ш IV V VI
Характеристики сырья: Плотность при 20 °C, г/см? 0,702 0,701 0,691 0,689 0,697 0,708
Пределы кипения, °C . . 41—162 43-165 36-151 36-153 44—165 44-162
Состав сырья, объемн. %: Парафины 73-86 73-86
Нафтены Ароматические угле во до- — — 10—20 10-20 — —
роды — — 4-5 4-5 — —
Олефины — '— 0—0,2 0—0,2 —
Параметры процесса:
Количество сырья, т/ч . . Средняя температура рас- 8,3 8,1 8,4 7,9 7,6 7,6
щепления, °C Количество реакционного 719 720 721 735 747 770
водяного пара, вес. % (в расчете на сырье) . . 39 46 41 45 47 66
Выход, вес. % (в расчете
на сырье):
Этилен 16,2 18,6 21,0 24,7 25,1 28,4
Пропилен . '. 14,8 15,2 14,9 15,0 13,5 10,9
Олефины С< И бутадиен Сумма олефиновых фрак- 10,6 10,4 8,6 8,3 7,7 5,6
ций С2—С4 41,6 44,2 44,5 48,0 46,3 44,9
Прочие газовые фракции 16,6 16,9 20,0 22,4 22,6 24,8 .
Крекинг-бензин, 200 °C . . 40,0 35,3 30,1 25,0 25,9 25,0
Тяжелое масло 1,0 2,5 2,8 3,7 4,9 4,0
После резкого охлаждения бензином продукты реакции пода-
ются через циклон 3, где отделяется песок, в котел-утилизатор
(служащий для выработки нужного количества пара) и затем в воз-
душный холодильник 2 для охлаждения выделенных продуктов
до 150 °C. Капельки тумана, присутствующие в остаточном газе,
выделяются электростатически или в мультициклоне 1. Легкую нефть,
кипящую при ~30 °C, перегоняют, тяжелую нефть возвращают
снова в процесс в качестве мазута для подогрева. Образующийся
при пиролизе кокс осаждается на песке и сгорает во время нагрева.
Преимуществом процесса является его гибкость, возможность
применения очень высоких температур, хорошая теплопередача
в кипящем слое и, наконец, легкость удаления образующегося кокса.
32
1. Получение пропилена
Потеря песка из-за истирания составляет ~0,15.% от всего коли-
чества песка, циркулирующего в течение 1 ч. Эрозию стенок можно
значительно уменьшить, установив по возможности невысокую
скорость подачи песка в пневматическом напорном трубопроводе,
так что внутреннюю каменную облицовку напорного трубопровода
Нагретый воздух
Рис. 9. Схема некаталитического пиро-
лиза с косвенным обогревом и подвиж-
ным гранулированным теплоносителем
(крекинг фирмы Lurgi):
1 — мультициклон; 2 — воздушный холодиль-
ник; з — циклон; 4 — камера подогрева сырья;
5 — сборник; 6 — нагреватель; 7 — реактор
для пиролиза.
нужно обновлять лишь спустя
4—5 лет.
В табл. 4 сопоставлены ре-
зультаты, получаемые при про-
цессе Lurgi в различных экс-
плуатационных условиях. Из
таблицы видно, что с повыше-
нием температуры и количества
пара увеличивается выход эти-
лена. Особое значений для
предотвращения вторичных ре-
акций имеет резкое охлажде-
ние выходящих из реактора
газов.
1.6.6. Пиролиз в трубчатой
печи (процесс фирмы Kellogg)
тельно нагревают в трубчатой
В разработанном фирмой
Kellogg Со. (Нью-Йорк) про-
цессе (рис. 10) используют в ка-
честве сырья для пиролиза
преимущественно тяжелую бен-
зиновую фракцию или газойль.
Исходный продукт предвари-
печи до 590—680 °C в при-
сутствии небольшого количества водяного пара. При этом уже про-
исходит некоторый крекинг. Выходящий из трубчатой печи
исходный продукт смешивается с водяным паром и подогревается
в отдельно стоящем подогревателе до ~930 °C, после чего в изоли-
рованной трубе проводится пиролиз (время контакта не должно
превышать 1с).
Пар обеспечивает пиролиз необходимым теплом и должен умень-
шить вторичные реакции путем снижения парциального давления
углеводородов. Кроме того, добавка пара сильно сокращает выделе-
ние кокса. После окончания пиролиза газы попадают в котел-ути-
лизатор для выработки пара высокого давления. Там они сразу
охлаждаются до 200—260 °C. Благодаря очень хорошему тепло-
обмену процесс, потребляющий так много пара, технически прием-
лем. Из котла-утилизатора газы направляются в ректификационную
колонну, где отделяется кубовый остаток, применяемый в качестве
1.6. Получение пропилена путем пиролиза углеводородов
33
Таблица 5
Выход газообразных продуктов (в вес. % в расчете на сырье)
при пиролизе тяжелого бензина и газойля в паровой фазе
(процесс фирмы Kellogg)
Продукты пиролиза Тяжелый бензин Газойль
Режим с вы- сокой пропу- скной спо- собностью режим с низ- кой пропу- скной спо- собностью Режим с вы- сокой пропу- скной спо- собностью Режим с низ- кой пропу- скной спо- собностью
Водород Метан Этилен Этан . Пропилен Пропан 0,4 6,2 ИЗ 1 «о * 3,0 а 10,5 * 0Л 96 0,8 10,9 22,5 1 пл * 2,4 / 90 . 12,0 1 97 * 0,4 J 9/ 0,2 6,1 ^о}78’5* 19-6 } 92 * 0,5 ’ 9,0 М3}87* ад}96*
* Содержание олефинов в
этан-этиленовой или пропан-пропиленовой франции (в %).
топлива. Из верхней части колонны выходят водяной пар, легкие
углеводороды и-газ. Часть углеводородов вместе с водяным паром
конденсируется и исполь-
зуется для орошения ко-
лонны, часть — отводится на
разделение. Раз попадает
в компрессорную установку
и перерабатывается там
обычным способом. В табл. 5
приведен выход при пиро-
лизе (82—86]. Результаты
свидетельствуют о повыше-
нии выхода этилена при уже-
сточении условий пиролиза.
1.6.7. Пиролитический
крекинг «термофор»
Пиролитический крекинг
«термофор» [87—90], разра-
ботанный фирмой Socony-
Vacuum Oil Со., можно срав-
нить с Процессом в печи
с кварцевым теплоносите-
лем (см. рис. 5), только он
отличается большим разно-
3 Заказ 399
Рис. 10. Схема некаталитического пиро-
лиза в паровой фазе (процесс фирмы
Kellogg):
I — печь для получения перегретого пара; 2 —
холодильник; 3 — реактор пиролиза; 4 — гене-
ратор пара (525—675 °C);
34
1. Получение пропилена
образней. Холодная иди предварительно подогретая смесь
углеводородов, причем исходным сырьем могут быть как газооб-
разные, так и жидкие углеводороды, вместе с водяным паром
подается навстречу инертному материалу, например гальке
определенного размера (противоточный процесс). Инертный ма-
териал является теплоносителем. В настоящее время используют
спекшийся корунд благодаря его большому теплопоглощению.
Время контакта составляет 1—2 с, температура теплоносителя
колеблется в диапазоне 810—840 °C в зависимости от исходного
продукта. Выходящий из реактора теплоноситель элеватором на-
правляется в нагреватель, где освобождается от осажденного кокса
путем выжигания и одновременно подогревается до температуры,
необходимой в реакторе.
В табл. 6 перечислены продукты, получаемые при пиролитиче-
ском крекинге термофор. Выход пропилена в данном процессе не-
большой.
Таблица 6
Результаты пиролиза [различных видов сырья при пиролитическом
крекинге термофор
Показатели процесса Этан Этан Пропан Нефть из Ми- чигана Нефть из Slaug- hter Duggan Нефть из Мирандо
Характеристики сырья:
Плотность при 20 °C, г/см3 .... — , — — 0,856 0,870 0,926
Пределы кипения, °C . — — — 105-535 66—493 116— 490
Параметры процесса:
Температура кирпича, °C 830 966 820 843 816 816
Время контакта, с 2,3 0,12 1,2 — — —
Выход, вес. % расчете на сырье);
Водород 5,7 5,7 2,0 0,9 0,7 0,7
Метан 12,8 10,2 26,2 14,8 15,3 13,0
Этан 18,2 11,6 3,5 3,5 4,5 3,0
Этилен 49,2 56,2 37,5 28,8 17,9 12,5
Ацетилен 0,7 3,8 0,0 — — —
Пропан 1,1 0,7 7,9 0,7 0,6 0.6
Пропилен 0,7 1,0 12,2 6,4 8,0 6,8
Бутан . . . 1,0 0,3 0,0 0,9 • 1,2 0,2
Бутилен 1,8 0,4 1,1 1,4 1,7 2,9
Бутадиен 1,6 3,2 2,3 2,5 1,0 1,6
Углеводороды С5 и выше ...... 6,7 6,1 6,8 37,4 45,5 27,4
Кокс 0,5 0,8 0,5 2,7 3,6 1,7
1.6. Получение пропилена путем пиролиза углеводородов
35
1.6.8. Процесс Лавровского — Бродского
Советские ученые Лавровский и Бродский [91—92] разрабо-
тали крекинг в кипящем слое (рис. 11), подобный процессу
фирмы Lurgi, только теплоносителем служат частицы кокса. Кок-
совые частицы нагреваются в подогревателе горячими отработан-
ными газами, которые получают
сжиганием смеси нефти с воз-
духом в топочной камере, и
направляются в реактор вместе
с водяным паром. Непосред-
ственно перед входом в реактор
подводится сырье (газообраз-
ные или легкоиспаряющиеся
углеводороды),’ которое дви-
жется в прямотоке с коксовыми
частицами. После выхода из
реактора частицы кокса пне-
вмотранспортом возвращаются
в подогреватель.
При использовании этана
в качестве исходного продукта
температура на входе в реак-
тор составляет ~1 ООО °C, время
контакта 0,01—0,02 с, соотно-
шение кокс : исходный продукт
20 : 1, конверсия свыше
90%, содержание этилена 60—
65%. Выход пропилена состав-
ляет только 1—5%, так как
данный процесс предназначен
главным образом для перера-
ботки этана в этилен.
Рис. 11. Схема некаталитического пи-
ролиза в кипящем слое с порошкооб-
разным теплоносителем (метод Лавров-
ского — Бродского)
1 — циклон; 2 — реактор; з — сепаратор;
4 — подогреватель для теплоносителя; 5 —
компрессор.
1.6.9. Автотермический пиролиз по Бартоломе
Давно известный метод автотермического дегидрирования этана
в этилен (рис. 12) усовершенствован в настоящее время для дегид-
рирования природных газов [93]. В реакторе с керамической футе-
ровкой теплоносителем являются фарфоровые шарики. Газовая
смесь из этана и пропана вводится в реактор вместе с чистым
кислородом и сжигается не до конца при 850—900 °C. Давление
0,6 кгс/см2, время контакта 1 с. При этом получаются следу-
ющие продукты: этилен, пропилен, метан, окись и двуокись
углерода.
3*
36
1. Получение пропилена
Преимущество данного процесса состоит в том, что кокс обра-
зуется только в очень ограниченном количестве. Недостатками
являются относительно низкий
ПР°Лам Кис^д
Рис. 12. Схема окислительного (авто-
термического) пиролиза:
1 — подогреватели; 2 — реактор; з — холо-
дильник.
выход олефинов и значительное
потребление чистого кислорода.
1.6.10. Катарол-процесс
В 1941 г. в Англии Вейц-
манн с сотрудниками [94—97]
разработал катарол-процесс
с медным катализатором. Уста-
новка работает на фирме Pet-
rochemicals Ltd. (Англия). Испа-
ренный и перегретый исходный
материал — в основном сред-
няя нефтяная фракция (т. кип.
100—260 °C) — подвергается
термическому крекингу в труб-
чатом реакторе при 400—
500°С; давление 2,5—4,5 кгс/см2,
время контакта 3 с. Продукты
крекинга поступают без разде-
ления в цилиндрический, за-
полненный медными спиралями
трубчатый реактор.
При 650 °C, времени кон-
такта 0,5—2 мин и объемной
производительности катализа-
тора 0,3—0,4 л/ч происходит
сильное образование аромати-
ческих углеводородов в результате дегидрирования нафтенов и
цикло конденсации олефинов. Метод мало применяется для полу-
чения олефинов, он больше . служит для производства основных
ароматических веществ.
Зависимость состава продуктов реакции от исходного сырья —
нефти из Техаса (I), парафинового тяжелого бензина из Ирана (II),
парафинового керосина из Ирана (III) — приведена ниже:
I
Характеристики сырья:
Плотность при 20 ° С, г/см3 . . 0,799
Пределы кипения по Энглеру
(5-95%), °C.................' 90-205
п ш
0,756 0,796
113-185 175-261
1.6. Получение пропилена путем пиролиза углеводородов
37
Выход, вес. % (в расчете на
Водород ..................
Метан.....................
Этан . . .................
Этилен ........... .......
Пропан ...................
Пропилен .................
Бутан.....................
Бутилен ..................
Жидкие продукты,..........
Результаты свидетельствуют
для получения пропилена.
сырье):
0,5 0,9 0,5
18,3 24,0 13,7
6,5 9,6 7,4
7,4 11,6 11,6
9,6 10,6 10,9
1,9 1,3 1,4
1,3 0,8 0,5
4,8 4,5 1,4
50,0 37,0 50,0 '
о непригодности данного процесса
1.6.11. Процесс фирмы Tsutsumi
Процесс фирмы Tsutsumi (рис. 13) — это метод в кипящем слое
[98—100]. Он служит для крекинга тяжелых нефтяных фракций,
в частности керосина (пределы кипения 200— 300 °C), в вакууме
Рис. 13. Схема каталитического пиролиза с ка-
тализатором в кипящем слое (процесс фирмы
Tsutsumi):
1 — пиролизная печь; 2 — регенератор; з — реактор.
38
1. Получение пропилена
(при остаточном давлении 200 мм рт. ст.). В подогревателе катали-
затор — гранулированная двуокись кремния с высокой удельной
поверхностью — накаляется до ~1200 °C. Сырая нефть поступает
в реактор в противотоке с перегретым водяным паром (соотношение
примерно 1,0 : 1,3) при 450 °C. При температуре пиролиза 850—
900 °C керосин дает 70% газообразных продуктов, содержащих
около 50—60% олефинов. Кроме того, образуется 28% жидких
продуктов с .содержанием ароматических углеводородов 25%. Во
избежание образования кокса в реакторе поддерживается избыток
водорода. Процесс дает хороший выход, но затраты высоки.
Был разработан также вариант процесса со стационарным слоем-
Результаты, полученные при пиролизе керосина (т. кип. 200—310 °C)
на пилотной установке [100], представлены ниже:
Параметры процесса:
Исходное количество сырья, кг........."8240
Расход пара кг........................ 4010
Затрата теплоты, ккал...................9 • Ю6
Циркуляция катализатора, кг/ч .... 200
Выход газообразных продуктов, вес. % 70,1
Водород ............................... 1,1
Метан и некоторое количество этана . . 11,4
Этилен................................ 34,2
Пропилен.............................. 23,4
Выход жидких продуктов, вес. % . . . 29,2
Бензол............‘.................... 5,8
Толуол................................. 2,9
Ксилол ................................ 2,9
Фракция ароматических углеводородов
(т. кип. 150—200° С) ................ 7,0
Нафталин , 3,8
Тяжелое масло.......................... 6,8
1.6.12. Процесс АзНИИ — Алиева
В Советском Союзе разработан процесс специально для крекинга
высококипящих нефтяных фракций. Катализатором является по-
рошкообразный нефтяной кокс, оказывающий сильное дегидриру-
ющее воздействие. Поэтому в газообразных продуктах содержится
значительная доля алкенов. Процесс протекает в кипящем слое при
700 °C и небольшом давлении (~2,2 кгс/см2). Исходный продукт
впрыскивается вместе с водяным паром. Объемная производитель-
ность катализатора составляет 4—5 л/(л • ч), время контакта ко-
леблется в диапазоне 5—12 с. Реактор и генератор кокса снабжены
обогревательными рубашками. Оба агрегата нагреваются снаружи,
т. е. косвенно. Специальные камеры сжигания вырабатывают не-
обходимую для процесса теплоту.
Выход этилена и пропилена превышает 25%, ацетилен при этом
процессе не образуется.
1.6. Получение пропилена путем пиролиза углеводородов 39
В настоящее время все в большем объеме используются катализа-
торы на молекулярных ситах в пиролизных установках и установ-
ках крекинга в кипящем слое. В США уже сейчас половина крекинг-
установок работает с молекулярными ситами [101—103].
1.6.13. Процесс Коппере — Хаше — Вульфа
Данный метод является высокотемпературным пиролизом и при-
меняется прежде всего для выработки ацетилена и этилена. Диапазон
рабочих температур 900—1600 °C [104—109].
При методе Коппере—Хаше—Вульфа для достижения высоких
температур используется принцип регенеративной печи. В печи,
заполненной огнеупорным кирпичом, получают нужную темпера-
туру реакции, сжигая горячий газ с подогретым избыточным коли-
чеством воздуха. Спустя 0,5—2 мин камера переключается, подо-
гретый углеводород вводится в систему вместе с водяным паром.
Менее чем через 0,03 с продукты реакции выходят из печи, охла-
ждаются до ~370 °C и подвергаются дальнейшему резкому охла-
ждению путем орошения водой.
Соотношение ацетилена и этилена можно сильно изменить, варьи-
руя температуру печи [70]. В качестве исходных продуктов исполь-
зуются только газообразные углеводороды. Результаты пиролиза
этана (I) и пропана (II) при различных условиях приведены ниже:
I II и II
Параметры процесса:
Температура, °C . . . 954 1010 1240 1343
Давление, кгс/см2 . . . 1,05 1,05 1,05 0,63
Расход пара, кг/кг
исходного газа 0 4,4 6,2
Выход продуктов, вес. %:
Этилен . . . 50,7 34,3 32,6 13,4
Ацетилен . . . 4,1 2,1 16,1 28,3
Пропанпропилен ». . . . -3,0 8,4 — —
Результаты свидетельствуют об очень низком-выходе пропилена
при данном процессе.
1.6.14. Высокотемпературный пиролиз (процесс фирмы
Farbwerke Hoechst)
В 1960 г. фирмой Farbwerke Hoechst был разработан высоко-
температурный пиролиз [104, 110—113], работающий на остаточ-
ном газе от процесса «Hoechster Кокег». Газ, в основном метан
и водород, сжигается для получения требуемых высоких температур
в атмосфере чистого кислорода в металлической горелке, охлаждае-
мой водой под давлением. Температуру пламени понижают до нуж-
ного предела распылением вторичного пара в камеру сжигания,
40
1. Получение пропилена
а затем впрыскивают легкий бензин крекинга. В последующем
реакторе с паровым охлаждением крекинг-газ находится 0,002—
0,003 с, здесь он резко охлаждается водой. Общий выход этилена
и, ацетилена составляет 50—54%, соотношение ацетилен : этилен —
в пределах 80 : 20 и 30 : 70.
1.6.15. Синтез ацетилена по способу фирмы BASF
Синтез ацетилена по способу фирмы Badische Anilin- und Soda-
fabrik [114—116] протекает в горелке, где частично сжигаются
подогретые до 600 °C угле-
Крекинг-газ в0д0р0дЫ1 в частности метан,
и недостаточное количество
кислорода. Крекинг-газы
резко охлаждаются впры-
скиваемой водой.
Азот
Сырая
нефть
Кисло-
род
Воздух
Рис. 14. Схема высокотемпературного кре-
кинга фирмы BASF:
1 •— подогреватель; 2 — реактор; 3 — котел-ути-
лизатор; 4 — скруббер; 5 — циклон; 6 — холо-
дильник; 7 — водоотделитель; 8 — абсорбер для
углеводородов С4; 9 — отпарная колонна для угле-
водородов С4.
с4.
1.6.16. Метод разложения
нефти при высоких
температурах (процесс
фирмы BASF)
Данный метод (рис. 14)
служит прежде всего для
производства ацетилена и
синтез-газа из сырой нефти
[117]. Обогащенное кисло-
родом пламя горит под слоем
нефти в реакторе. Образую-
щиеся при -—1500 °C в ре-
сгоранйя и крекинга горячие газы тут же
тяжелым маслом, температура которого
зультате частичного
резко охлаждаются
250 °C. Основное количество образующейся сажи улавливается
тяжелым маслом и вместе с жидкими продуктами Подается в по-
гружную горелку.
Примерно 10% теплоты сгорания исходного продукта пропадает,
6% расходуется на подогрев и испарение, 80—85% используется
на реакцию крекинга, из них 18% — на получение ацетилена. Со-
став крекинг-газа мало зависит от сырья и незначительно изме-
няется с давлением. На 1 т ацетилена приходится 3,5 т NH3. Ниже
приведен выход продуктов реакции (в объемн.%):
Окись углерода . . . 40—45
Углекислый газ . . . 7—9
Азот ...............0,5—1
Водород..............23—30
Метан...............£4—5
Этилен............. 6—7
Ацетилен........... 7—8
Пропилен ............. 1—2
Высшие олефины .... 0—5
Высшие ацетилены . . . 0,5—1
1.6. Получение пропилена путем пиролиза углеводородов
41
1.6.17. Современные промышленные установки США
В США в 1965 г. около 88% потребляемого пропилена получали
на крекинг-установках по производству бензина, 12% — на кре-
кинг-установках по производству этилена. В других странах доля
пропилена, производимого на крекинг-установках по производству
этилена, значительно выше, так как там меньше каталитических
крекинг-установок. Пропилен до сих пор никогда не производился
как главный продукт процесса, он является исключительно побоч-
ным продуктом [30].
Состав исходного пропилена (в вес. %) при высокотемпературном
(I) и среднетемпературном (II) крекинге указан ниже [118]:
I и
Фракция С2 . . 0,25 0,25
Пропан .... 2,55 4,80
Пропилен . . . 92,60 93,60
I И
Пропадиен .... 1,80 0,50
Метилацетилен . . 2,50 0,55
Фракция С4 ... 0,30 0,30
Как уже упоминалось, пропилен образуется в качестве побоч-
ного продукта почти при всех методах пиролиза для получения эти-
лена. Также в больших количествах пропилен вырабатывается при
разделении газовой смеси, образующейся на нефтеперерабатыва-
ющих заводах при крекинге и риформинге.
Приведенные в конце книги обзорные таблицы потребления и
производственных мощностей пропилена в различных странах сви-
детельствуют о повсеместном увеличении выпуска пропилена. Од-
нако несмотря на широкие возможности использования пропилена
для производства различных продуктов (см. схему возможных пре-
вращений пропилена), в большинстве промышленных стран наблю-
дается избыток пропилена из-за постоянного роста производства
этилена, при котором обязательно образуется пропилен. В США
в 1963 г. избыток пропилена составил —8 млн. т. при потреблении
пропилена в нефтехимии 1,54 млн. т. Избыточный пропилен был
использован в США большей частью в качестве топлива. Тем не
менее, предполагают [31], что после 1975 г. в США может оказаться
дефицит пропилена.
ЛИТЕРАТУРА
1. Deimann et al., Crells Chemische Annalen, № 2, 195, 310, 430 (1795).
2. Ibid., № 2, 200 (1795).
3. Reynolds, Liebigs Ann. Chem., 77, 118 (1851);
A. Cahours, Compt. rend. T., 21, 143—145 (1850).
4. US Tariff. Commission report, Census of production, Synthetic organic Che-
micals.
5. W. Grimme, Brennstoff-Chem., 33, 40—42 (1952).
6. Kirk — Othmer, Encyclopedia of Chemical Technology, N. Y., v. 11,
1953, p. 153.
42
1. Получение пропилена
7. Р. Ferrero, Ind. chim. beige, 20, 247—256 (1955).
8. Sh. Noguchi, J. Soc. org. synth. Chem., 13, 395—405 (1955).
9. R. С о о m a n s, Metano (Padova), 9, 317—322 (1955).
10. H. И. Попов a, Tp. Вост.-Сибирск. филиала АН СССР, сер. хим.,
№ 4, 37-57 (1956).
11. L. W. Hatch, Petroleum Refiner, 38, № 8, 107—114 (1959).
12. R. F. Messing, J. W. Bradley, Chem. Eng., 67, № 18, 60—70
(1960).
13. M. F о u re z, Ind. chim. beige, 25, 649—656 (1960).
14. A. F. M i 11 i d ge, J. Inst. Petroleum, 46, № 443, 353—366 (1960).
15. P. W. Sherwood, Ind. Chemist, 36, 497—502, 542—546, 595—598 (1960).
16. J. W. Bradley, Chem. Eng., 36, № 4, 88—92 (1958).
17. Ullman n, Enzyklopadie der technischen Chemie, Miinchen, Bd. 10,
1958, S. 136-148.
18. Chem. Eng. News, 41, № 19, 92—105 (1963); Chem. Industrie, 15, 551—554
(1963).
19. N. J. Lewis, Oil Gas J., 61, № 23, 202—206 (1963).
20. Chem. Eng. News, 44, № 24, 34—36 (1966); Chem. Industrie, 18, 544 (1966).
21. I. В a t h о г у, E. Foldes, Magyar Kem. Lapja, 20, 119—124 (1965).
22. P. W. Sherwood, Chim. et Ind. (Milano), 85, 401—408, 576-587 (1961).
23. J. land, Ropa Uhlie, 8, № 2, 39—43 (1966).
24. G. G e i s e 1 e r et al., Chem. Technik, 11, 656—661 (1959).
25. P. W. Sherwood, Ind. Chemist, 38, 55—58 (1962).
26. F. Wetter, Rev. Assoc, franc, techn. petrole, № 179, 37—51 (1966).
27. R. D о b г о w о 1 s k i, Przemysl Chem., 45, 457—460 (1966).
28. Г. Ф. Б о p и с о в и и и др., Хим. пром., № 8, 561—566 (1963).
29. Е. Boye, Chemiker-Ztg., 85, № 9, 302—306 (1961).
30. R. В. S t о b a u g h, Hydrocarbon Processing, 46, № 1, 143—154 (1967).
31. N. E. Ockerblom, Chem. Industrie, 19,'757—60 (1967).
32. E. O. Curtiss, Chem. and Ind. (Aug.), 18—23 (1953).
33. Petroleum Processing, № 6, 91—93 (1956).
34. E. E g 1 о f f, Royal Inst, of Chem. Symposium, St. Andrews, 1947.
35. F. Martin, E.Weingaertner in Winnacker — Weingaertner
«Chemische Technologie — Organische Technologies I, Munchen, 1952, S. 835.
36. W. Scheer, Feuerungstechnik, 29, 283 (1941).
37. F. A s i n g e r, Chemie und Technologie der Paraffinkohlenwasserstoffe,
Berlin, 1956, S. 122.
38. H. A. Skinner, Trans. Faraday Soc., 41, 645—662 (1945).
39. K. S. Pf itzer, J. Chem. Phys., 5, 473—479 (1937).
40. F. E. Frey, H. J. H e p p, Ind. Eng. Chem., 25, 441—449 (1933).
41. F. E. Frey, D.F. Smith, ibid., 20, 949 (1928).
42. A. V. Grosse, V. N. Ipatieff, Ind. Eng. Chem., 32, 268—272
(1940).
43. К. К. Ke a r b у in Chemistry of Hydrocarbons, N. Y., v. 2, 1955, p. 226.
44. L. Nowakowski, J. N e p t e r, Przemysl. Chem., 40, 696—698
(1961).
45. V. N. Ipatieff, Ber. dtsch. chem. Ges., 34, 3589 (1901); 35, 1047 (1902);
ЖРФХ0, 34, 182 (1902); 40, 500 (1908).
46. V. I. К'о m a r e w s k у, С. H. Riesz, Oil Gas J., 42, № 7, 90—93
(1943).
47. Пат. США 2184234, 35, 1934.
48. H. H. Storch, J. Am. chem. Soc., 56, 376 (1934), 57, 2601 (1935).
49. C. G. S i 1 с о k s, Proc. Roy. Soc., A 233, 465—479 (1956).
50. A. M. Б p о д с к и й и др., Кинетика и катализ, 5, № 1, 49—59 (1964).
51. Пат. США 2000964, 1933.
52. Р. К. F г о h 1 i с h, Р. J. W i е z е v i с h, Ind. Eng. Chem., 27, 1055—
1062 (1935).
1.6. Получение пропилена путем пиролиза углеводородов
43
53. М. Neuhaus, L. F. Marek, ibid., 24, 400—402 (1932).
54. В. S. G г е е n s f е 1 d е г, Н. Н. V о g е, Ind. Eng. Chem., 37, 516 (194'5).
55. С. Ф. В а с и л ь е в, Н. А. Л а п и д е с, Новости нефтяной и газовой
техники. Нефтепереработка и нефтехимия, № 4, 22—25 (1961).
56. С. Ф. Васильев и др., Труды института горных ископаемых АН СССР,
№ 16, 59-65 (1961).
57. Р. М. Arn ol d, Oil Gas J., 44, Ns 9, 87-89 (1945).
58. L. F. M а г e k, M. Neuhaus, Ind. Eng. Chem., 25, 516—519 (1933).
59. N. C. D e b i e, V. S c h о г г, Brit. Chem. Eng., 9, Ns 1, 24—31 (1964).
60. U 11 m a n n, Enzyklopadie der technischen Chemie, Berlin—Miinchen,
Bd. 6, 1955, S. 625-634.
61. F. A s i n g e r, Chemie und Technologie der Monoolefine, Akademie Verlag,
Berlin, 1957.
62. G. E g 1 о f f, The Reactions of Pure Hydrocarbons, N. Y., 1937.
63. Ch. Ellis, The Chemistry of Petroleum Derivatives, N. Y., 1937.
64. P. W. Sherwood, Petroleum Refiner, 30, Ns 9, 220—225 (1951).
65. M. Gerhold, Erdol u. Kohle, 9, 24—29, 93—98, 157-162, 228-233
(1956).
66. H. Holzrichter, Chem. Industrie, 15, 290 (1963);
M. О. К i 1 p a t r i c k et al., Oil Gas J., 53, Ns 1, 162—165 (1954);
Petroleum Refiner, 33, Ns 4, 171—174 (1954).
67. F. A s i n g e r, Einfuhrung in die Petrolchemie, Berlin, 1958, S. 63.
68. K. Winnacker, Chemie-Ing.-Techn., 27, 399 (1955).
69. H. Krekeler, Petroleum Refiner, 34, Ns 10, 139—141 (1955).
70. H. К a m p t n e r, Erdol u. Kohle Erdgas Petrochemie, 14, 346—354 (1961).
71. A. S t e i n h о f f e г, O. Frey, Chemie-Ing.-Techn., 32, Ns 12, 782—
788 (1960).
72. A. Steinhoffer, ibid., 36, Ns 9, 894—898 (1964).
73. A. S t e i n h о f f e r, ibid., 32, Ns 12, 787 (I960).
74. A. Steinhoffer, Erdol und Kohle, 16, Ns о—1, 540—547 (1963).
75. A. Steinhoffer, Petroleum Refiner Hydrocarbon Processing, 42,
Ns 7, 119—124 (1963).
76. E. Th. H e г p e r s, Gas. Warme .International, 16, 308 (1967).
77. Chemical Eng., 66, Ns 13, 37 (1959).
78. Chemical Eng., 66, Ns 17, 66 (1959).
79. W. Bayer, Erdol und Kohle Erdgas Petrochemie, 15, 799—802 (1962).
80. P. S ch ma If el d, Petroleum Refiner Hydrocarbon Processing, 42, Ns 7,
145-148 (1963).
81. P. Schmalfeld, Erdol und Kohle Erdgas Petrochemie, 14, Ns 7, 537—
541 (1961).
82. С. C. King, J. Warburton,
83. С. С. К i n g, "
123 (1951).
84. Petroleum Processing, Ns 2, 87—88 (1956).
85. Oil Gas J., 54, Ns 35, 113—114 (1956).
86. Petroleum Refiner, 36, Ns_ll, 246 (1957).
" ’ ' Potas,
Oil Gas J., 51, Ns 31, 92—94 (1952).
J, Warburton, Petroleum Refiner, 30, Ns 7, 122—
A. E.
Petroleum Eng., 19, Ns 12, 43—
A. E.
A. E.
Oil Gas J., 47, (26.8), 104 (1948).
Petroleum Refiner, 27, Ns 9, 468
P о t a s,
P о t a s,
87. S. С. E a s t w о о d,
46 (1948).
88. S. С. E a s t w о о d,
89. S. С. Eastwood,
(1948).
90. P. W. S h e r w о о d,
В. K. A m e г i c et al., Process for continuous Thermocontact Treatment
Petroleum (London), 19, Ns 161, 309 (1956).
91.
of Oil Stock on Coke, Proc. 5th World Petrol. Congr. 5/30—6/5, 1959.
92. К. П. Л а в p о в с к и й, A. M. Б p о д с к и й, ДАН СССР, 72, 745—
748 (1950).
93. Е. В a rt ho 1 о me, Н. Nonnenmacher, Cracking of Gaseous
44
1. Получение пропилена
Hydrocarbons by partial Oxydation, Proc. 5th World Petrol. Congr. 5/30—
6/5 (1959).
94. Petroleum Refiner, 28, № 4, 130 (1949).
95. Ibid., 31, № 7, 154 (1952).
96. Oil Gas J., 50 (20.12), 201 (1951).
97. M. R u h e m a n n, H. S tojiner, Petroleum (London), 11, 112 (1949).
98. S. Tsutsumi, J. Fuel Soc. Japan, 34, 356 (1955). -
99. S. Tsutsumi, ibid., 35, '560 (1956).
100. S. Tsutsumi, Petroleum Refiner, 36, № 9, 287 (1957).
101. Oil Gas J., 65, № 22, 91 (1967).
102. Ibid., 64, № 46, 194—197 (1966).
103. Ibid., 65, № 6, 101—112 (1967).
104. H. Kamptner, Erdol und Kohle Erdgas Petrochemie, 16, № *6—1,
547-551 (1963).
105. R. L. Hasche, Chem. Metallurg. Eng., № 7, 78—83 (1942).
106. G. L. Fleming, Chem. Eng. Progr., 52, № 6 , 249 (1956).
107. P. W. Sherwood, Petroleum, № 9, 309—312 (1956).
108. I. F. Farnsworth et al., Ind. Eng. Chem., 47, 1517—1522 (1955).
109. H. Kamptner, Erdol und Kohle Erdgas Petrochemie, 14, 349 (1961).
110. K. Winnacker, Kunststoffe, 47, 397—403 (1957).
111. Chem. Week от 18.5.1957.
112. U 11 m a n n, Enzyklopadie der technischen Chemie, Miinchen, Bd. 10,
1958, S. 146—147.
113. H. K. Kamptner et al., Hydrocarbon Processing Petroleum Refiner,
45, № 4, 187—193 (1966).
114. H. Sachsse, Chem.-Ing.-Technik, 21, 129 (1949).
115. E. Bartholome, ibid., 26, 253—258 (1954).
116. U 11 m a n n, Enzyklopadie der technischen Chemie, Munchen, Bd. 10,
1958, S. 133.
117. E. Th. H e r p e r s, Gas Warme International, 16, 306—307 (1967).
118. A. D. P 1 a i s t о w e, M. R. W у n n, European Chemical News, Sup-
plement, 9, (23.2), 10—13 (1966).
119. Marshall Sittig, Chemicals from Propylen, N. Y., 1965.
120. F. W. Sullivan et al., Ind. Eng. Chem., 27, 1027—1033 (1935).
121. K. Thormann, Chem. Apparatur, 27, № 17, 113, 131 (1940).
122. R- A. D u с к w о r t h, Chem. Process Engn., №2,67—77 (1968).
123. К. H. Eisenlohr et al., Erdol Kohle Erdgas Petrochemie, 20, № 2,
82—89 (1967).
124. F. Andreas, K. Grobe, Chem, Technik, 20, 151 — 159, 289—297
(1968).
2. ВЫДЕЛЕНИЕ И СВОЙСТВА ПРОПИЛЕНА
2.1. ВЫДЕЛЕНИЕ ПРОПИЛЕНА ИЗ ПРОДУКТОВ, ОБРАЗУЮЩИХСЯ
ПРИ РАЗЛИЧНЫХ ПРОЦЕССАХ
Для разделения олефинсодержащих газовых смесей на фракции
с определенным числом атомов углерода или для получения очищен-
ных олефинов существуют три принципиально различных способа:
1) низкотемпературная ректификация под давлением;
2) абсорбция;
3) адсорбция.
Ректификация под давлением — наиболее распространенный и
хорошо зарекомендовавший себя метод разделения реакционных
газов, образующихся на нефтеочистительных установках, на фрак-
ции с определенным числом атомов углерода. Поскольку в боль-
шинстве реакционных смесей содержатся еще водород и метан,
ректификация под давлением осуществляется при низких темпера-
турах (рис. 15, см. в конце книги). (Более подробно этот процесс
был уже описан в разделе 2.1.)
При абсорбционном методе можно использовать .более низкое
давление и более высокие температуры. Газовая смесь под давле-
нием в противотоке контактирует с поглотительным маслом, в кото-
ром растворяются все углеводороды, имеющие 2 и более атомов
углерода. Метан и водород при этом не абсорбируются и выводятся
с установки. Затем газообразные углеводороды выделяются из
поглотительного масла и разделяются ректификацией, что после
удаления водорода и метана не представляет значительных труд-
ностей. Освобожденное от газообразных углеводородов поглоти-
тельное масло возвращается на установку. Выделение газов из по-
глотительного масла можно провести таким образом, что при этом
уже будет иметь место разделение на фракции с определенным чи-
слом атомов углерода. Дальнейшее разделение на отдельные компо-
ненты путем перегонки не представляет труда. Часто получаемая
при фракционировании чистота уже достаточна для последующей
переработки. Абсорбционный метод обладает большими достоин-
ствами для концентрирования газов с небольшим содержанием оле-
финовых углеводородов.
Более новым методом выделения метана и водорода с одновре-
менным фракционированием остаточных углеводородов является
гиперсорбция, непрерывный процесс адсорбции — десорбции на
46
2. Выделение и свойства пропилена
компрессоры; 2 — осушители; 3 — деметанизатор; 4 — деэтанизаторы; 5 — абсорбер; в — адсорберы.
2.2. Получение чистого пропилена
47
активированном угле. Он требует относительно больших сооружений,
но зато можно обойтись без компрессионных установок и установок
глубокого охлаждения.
Адсорбционный метод хорошо подходит для поглощения или
выделения микрокомпонентов.
На рис. 16 показана принципиальная схема важнейших промыш-
ленных методов разделения газовых смесей на нефтеперерабатыва-
ющих заводах и крекинг-установках.
В соответствии с растущим влиянием полимеризации и других
процессов, требующих применения концентрированных олефинов,
низкотемпературная ректификация под давлением приобрела за
последнее время гораздо большее значение в мировом масштабе,,
чем низкотемпературная абсорбция. К тому же вредные примеси,
мешающие дальнейшей переработке, легче удалить из нефтехимиче-
ских первичных продуктов, чем из готовых продуктов.
2.2. ПОЛУЧЕНИЕ ЧИСТОГО ПРОПИЛЕНА
При получении пропилена [1—4] из нефтезаводских газов вы- •
деляют преимущественно смесь пропана с пропиленом, содержащую
около 40—60% пропилена. При разделении крекинг-газов путем
низкотемпературной ректификации под давлением получается про-
пиленовая фракция с содержанием пропилена от 80 до 95%.
Большая часть химических синтезов на основе пропилена (по-
лучение изопропилового спирта, получение окиси пропилена мето-
дом хлоргидринирования, оксосинтез,алкилирование, олигомеризация
и т. д.) может быть проведена со смесями пропан-пропилен. Для
некоторых же синтезов (например, получение полипропилена, со-
полимера этилена с пропиленом, акрилонитрила, акролеина, аллил-
хлорида) необходим пропилен высокой степени чистоты. Применяемые
при получении полипропилена катализаторы отравляются содержа-
щимися в пропилене кислородом, окисью углерода и углекислым
газом, а также соединениями серы и водой. Кристалличность и мо-
лекулярный вес полимеров сильно изменяются под влиянием
посторонних олефинов.
По американским данным [1] стоимость очистки пропилена очень
высока. В США она составляет 3 цента на 1 кг мощности установки,
причем используемая пропиленовая фракция имеет обычно следу-
ющий состав (в %):
Углеводороды С, .................... 2—3
Пропан.............................. 35—53
Углеводороды С4 .................... 3—4
Пропплен............................ Остаток
до 100%
48
2. Выделение и свойства пропилена
Очищенная пропиленовая фракция содержит около 99,5—99,7%
пропилена и следующие примеси (в млн-1):
Кислород ............... 4
Сероуглерод..............2
Углекислый газ..........1
Этан .............. 50
Пропан............. 0,3%
Существует три способа получения чистого пропилена:
1) экстрактивная дистилляция;
2) двухколонная дистилляция;
3) рекомпрессионная дистилляция.
2.2.1. Экстрактивная дистилляция
При экстрактивной дистилляции [5] (процесс Distex) введение
третьего компонента так изменяет соотношение показателей отно-
сительной летучести пропана и пропилена, что можно успешно
осуществить разделение. Применяемые для этого растворители
должны быть полярными. Ниже приведены такие растворители
и достигаемые при их введении сдвиги соотношений летучести при
28 СС:
Содержание С3Н» в смеси, мол. % Соотношение летучести СзН,/С,Н.
100 0,83—0,87
40 1,03
15—18 1,19
12-15 1,23
26-34 1,29
14-19 1,35
1 15—25 1,72
Без растворителя...............
Ацетон ........................
Фурфурол ......................
Фурфурол с 4% воды.............
Акрилонитрил ..................
Ацетонитрил . . ...............
Акрилонитрил—ацетонитрил (1:1)
Соотношение летучести становится обратным, поток с высоким
содержанием пропана выходит из головной части колонны. Несмотря
на эффективное разделение процесс Distex не может быть применен
в промышленном масштабе, так как отделение экстрагирующего
агента требует дополнительных затрат. Тем не менее он имеет боль-
шое значение при разделении фракции С4 на отдельные компоненты.
2.2.2. Двухколонная дистилляция
При дистилляции на двух колоннах [1] после удаления соедине-
ний серы путем промывки щелочью и этаноламином необходимо
полностью удалить фракцию С2. С этой целью применяется колонна
с 40 тарелками, на которой можно понизить содержание фракции
С 2 с 2% до 150 млн-1. Фракционирование оставшейся смеси пропан —
пропилен, состоящей из 43% пропилена, 54% пропана и 3% фрак-
ции С4, проводится на двух последовательно соединенных колоннах,
2.2. Получение чистого пропилена
49
первая из которых имеет 62 тарелки, а вторая 78. Чистый пропилен
высушивается алюмосиликатом кальция (молекулярные сита) до
6,8 кгс/см2, И “С
Пропан
4,1 кес/см2,
2 °C
содержания воды 10 млн"1.
Регенерацию осушителя
можно осуществлять на-
гретым природным газом.
2.2.3. Рекомпрессионная
дистилляция
Рекомпрессионная дис-
тилляция [2] (рис. 17, а)
осуществляется под давле-
нием 7—14 кгс/см2. Для
нее требуется всего одна
колонна с 80—100 тарел-
ками. Исходный пропилен,
содержащий менее 2%
Пропилен
Рис. 17. Схема фракционирования пропилена
для полимеризации:
а — фракционирование при низком давлении, ком-
прессия головного продукта; б — фракционирование
при низком давлении, компрессия кубового продук-
та; в — фракционирование при высоком давлении;
J — ректификационная колонна; 2 — колонна; S —
сборник дистиллята; 4 — отпарная колонна.
Рис. 18. Число теоретических
тарелок для разделения смеси
пропан — пропилеи, содержа-
щей 40 (1) и 60 (2) мол. % про-
пилена (минимальное флегмо-
вое число 1,2).
фракций С2 и С4, легко можно получить на нефтеперерабатывающих
I заводах и химических установках. Если используется 90%-ный
пропилен, образующийся при крекинге бутана, то чистый пропилен
можно получить на колонне с 70 тарелками, работающей при 21 кгс/см2.
Однако предварительную и последующую обработку следует про-
водить так же, как при дистилляции на двух колоннах.
По данному методу в США работают установки фирмы Sun Oil
(мощностью 60 000 и 90 000 т/год чистого пропилена).
4 Заказ 399
50
2. Выделение и свойства пропилена
Другие методы получения пропилена основаны на фракциони-
ровании при низком давлении и компрессии продукта из нижней
части колонны (рис. 17, б), а также на фракционировании при вы-
соком давлении (рис. 17, в).
Рис. 18 дает представление о необходимом числе теоретических
тарелок для получения 99%-ного чистого пропилена (коэффициент
использования 0,9).
В 1958 г. в США мощности по производству очень чистого про-
пилена составляли 22 700 т, а в 1962 г. — 386 000 т. В других стра-
нах производство 99%-ного пропилена также постоянно увеличи-
вается. Дистилляция является наиболее распространенным методом.
2.2.4. Разделение пропана и пропилена на молекулярных ситах
В последних патентах описано разделение пропана и пропилена
на молекулярных ситах. Для этого рекомендуется цеолит А [6]
и адсорбенты с диаметром входов в поры 5 А [7]. Во втором случае
в качестве десорбера предлагается парафин С6—С7.
2.2.5. Разделение с помощью серной кислоты
Экстракцию пропилена из смеси пропан — пропилен и извлече-
ние изобутилена из смеси углеводородов С4 можно осуществить
адсорбцией в серной кислоте. Однако в промышленности этот метод
применяется фирмой Standard Oil [8] только при получении изо-
бутилена. Описано [9] введение пропана и пропилена после сжи-
жения в 75%-ную серную кислоту при 40 °C. При этом пропилен
удается удержать в виде гидросульфата, который позже может
быть удален путем гидролиза водой при 80 °C.
2.2.6. Экономическая эффективность получения высокочистого
пропилена
Ниже рассмотрена стоимость концентрирования пропилена ди-
стилляцией при различном давлении; из фракции С3 с соотношением
СаН6 : С3Н8 = 1:1 получили 22 700 фициент использования 0,9) [1]: т 99 %-ного пропилена (коэф-
Давление перегонки, кгс/см2 . . . 7 14 ,21 21 *
Фактическое число тарелок .... 70 90 120 70
Расстояние между тарелками, см . . 46 41 46 46
Диаметр колонн, м 1,68 1,83 1,98 1.68
Мощность компрессора, л- с. ... 1100 500 — —
Расход пара, т/ч 0 0 6100 5200
Расход электроэнергии, кВт .... 20 25 35 20
* В последней графе приведена вычисленная стоимость при использова-
НИИ 90% пропилена.
2.3. Свойства пропилена
51
Затраты на выделение, цент/л С3Н6
прямые...................... 0,40 0,37 0,46
косвенные................... 0,25 0,22 0,21
Капиталовложения для производства
продукции на 1000 долларов . . . 720 636 520
0,28
0,12
288
Отсюда следует, что общие расходы на получение 99%-ного
пропилена примерно на 0,5 цент/фунт меньше расходов на получе-
ние 95%-ного этилена, однако цена может колебаться в зависимости
от местных условий. Затраты на очистку 95 %-ного пропилена соста-
вляют лишь 60% от расходов, необходимых для получения чистого
пропилена из фракции, содержащей наряду с 50% пропана только
50% пропилена.
2.3. СВОЙСТВА ПРОПИЛЕНА
Ниже приведены физические свойства пропилена [10—13]:
Температура плавления, °C ........................... —185
Температура кипения, °C.............................. —47,70
Тройная точка, °C.................................... —185,25
Плотность р
при—150 °C......................................... 0,729
-100 °C '..................................... 0,671
—50 °C....................................... 0,6116
—47 °C..............................: ... . 0,6095
20 °C...................................... 0,5139
25 °C...................................... 0,5053
Плотность пара по отношению к воздуху................ 1,49
Вязкость, сП
при —185 °C........................................... 15
150 °C....................................... 1,29
—ио °C......................................... 0,44
-100 °C........................................ 0,37
Критическая температура
°C.................................................. 91,9
К................................................. 365,1
Критическое давление, кгс/см2........................... 46,4
Критический объем, г/мл............................... 0,233
Мольный объем (жидкий пропилен), мл/моль
при 20 9С.......................................... 81,88
25 °C........................................... 83,27
Теплота плавления
ккал/моль ........................................ 0,7176
ккал/г.............................................. 16,7
Теплота испарения при —47 °C
ккал/моль ......................................... 4,402
к ал/г........................................... 104 ,62
Теплота образования (газ) при 25 °C, ккал/моль . . . 4,879
Энергия образования, ккал/моль....................... 14,990
Энтропия при 25 °C, кал/(°С • моль)................... 63,80
4*
52
2. Выделение и свойства пропилена
Теплота сгорания при 25° С и постоянном давлении
ккал/моль................................*......... 460,428
кал/г............................................. 10 942,3
Удельная теплоемкость, ккал/(кг-°С)
при —150 °C ...................................... 0,501
—100 °C ..................................... 0,498
—50 °C....................................... 0,520
Удельная теплоемкость при 25 РС, кал/(моль • °C) . . 15,27
Пределы взрываемости в смеси с воздухом при20?С
и 760 мм рт. ст., объемн. %
верхний ............................................ 11,1
нижний ............................................. 2,0
Температура вспышки при 18 °C на воздухе, °C
теоретическая ............................. 2200
действительная . 1935
Температура воспламенения (VDE 0173), гС . . . . . . 540
Растворимость при 20 9С и 1 кгс/см2, мл С3Нв/100 мл
растворителя
в воде................................................ 44,6
этаноле ....................................... 1250
уксусной кислоте.............................
Давление паров, мм рт. ст.
при —112,11 °C . . . 10 при —48,28
—93,70 °C ... 50 -47,99
-84,12 .... 100 —47,70
—69,46 .... 250 —41,48
—56,50 .... 500 —31,55
Постоянные уравнения Antoine lg р = А — B/(C-[-t)
Л ............................................
524,5
740
750
760
. 1000
. . 1500
6,8196
785,00
247,00
В ....................................
С .....................................
ЛИТЕРАТУРА
1. J. A. S h е г г е d, J. R. F a i г, Ind. Eng. Chem., 51, № 3, 249—52 (1959).
2. R. A. Labine, Chem. Eng., 67, № 11, 78—81 (1960).
3. P. W„ Sherwood, Petroleum, 27, № 5, 226—228 (1964).
4. P. W. Sherwood, Brennstoff-Chem., 43, 209—212 (1962).
5. Пат. США 2588056, 1952.
6. Пат. США 3076636, 1963.
7. Франц, пат. 1303028, 1962.
8. Petroleum Refiner, 36, № 11, 302 (1957).
9. Франц, пат. 865398, 1949.
10. К i г k-0 t h m е г, Encyclopedia of Chemical Technology, Bd. 11, N. Y.,
1953.
11. U 11 m a n n, Enzyklopadie der technischen Chemie, Bd. 10, Munchen —
Berlin, 1958, S. 49.
12. R. W. Gallant, Hydrocarbon Processing Petroleum Refiner, 44, № 8,
127—133 (1965).
13. L. W. C a n j a r et al., ibid., 44, .№ 9, 219-222 (1965).
3. ИЗОПРОПИЛОВЫЙ СПИРТ
Изопропиловый спирт (изопропанол, пропанол-2, диметилкар-
бинол) II] был открыт в 1855 г. М. Бертло [2] при обработке
пропилена концентрированной серной кислотой и разложении
продукта реакции водой.
Возможны, следующие реакции взаимодействия пропилена с сер-
ной кислотой и водой:
CH3CH=CH2 + H2SO4—► (CH3)2CHOSO3H
(CH3)2CHOSO3H+H2O —> CH3CHOHCH3 + H2so4
Изопропилсульфат, реагируя с пропиленом, дает диизопропил-
сульфат:
CH3CH=CH2+(CH3)2CHOSO3H —> [(CH3)2CH]2SO4
Диизопропилсульфат снова образует с водой изопропилсульфат
и изопропиловый спирт:
I(CH3)2CH]2SO4+H2O —> (CH3)2CHOSO3H + CH3CHOHCH3
Кроме того, изопропиловый спирт реагирует с диизопропил-
сульфатом с образованием диизопропилового эфира и изопропил-
сульфата:
[(CH3)2CHJ2SO4+CH3CHOHCH3 —> (CH3)2CHOSO3H + (CH3)2CHOCH(CH3)2
В данном случае нужно учитывать образование некоторых по-
бочных продуктов, например, диизопропилового эфира, полимеров.
Реакция пропилена с серной кислотой до сих пор остается
основой крупнотоннажного производства изопропилового спирта.
Первая пилотная установка, работающая по разработанной
в США Эллисом [3] технологии, была пущена в эксплуатацию в
1919 г. фирмой Melco Chemical Со. Уже в 1920 г. начала работать
первая промышленная установка фирмы Standard Oil Со. (Нью-
Джерси) на нефтеперерабатывающем заводе в Бейвей [4]. Поэтому
изопропиловый спирт (петрохол) можно рассматривать как первый
нефтехимический продукт.
Данный технологический процесс до сих пор применяется для
промышленного производства изопропилового спирта, хотя он имеет
ряд недостатков (например, высокая коррозия аппаратуры).
54
3. Изопропиловый спирт
Со времени сооружения первых промышленных установок по-
требление изопропилового спирта непрерывно растет, так как этот
низкокипящий спирт находит разнообразное применение благодаря
дешевому способу изготовления [5, 105].
3.1. ПОЛУЧЕНИЕ ИЗОПРОПИЛОВОГО СПИРТА
3.1.1. Получение из углеводов
В небольших количествах изопропиловый спирт получают на-
ряду с бутанолом при брожении углеводов [6].
3.1.2. Гидратация пропилена серной кислотой в жидкой фазе
В литературе [7—20] описаны основы реакции пропилена с сер-
ной кислотой, в частности влияние давления, температуры, концент-
рации кислоты и продолжительности процесса. Особый интерес
Время, ч
Рис. 19. Зависимость концентрации
алкилсульфата от концентрации ки-
слоты и продолжительности аб-
сорбции:
1 _ 75%-ная H2SO4; 2 — 85%-ная H2SO4;
3 —95%-ная H2SO4.
Рис. 20. Зависимость поглощения про-
пилена от продолжительности абсорб-
ции и концентрации кислоты при 20 РС
и давлении 9 кгс/см2:
1 — 75%-ная H2SO4; 2 — 85%-ная H2SO4; 3—
95%-ная H2SO4.
представляют исследования Шульца с сотрудниками [21, 22].
Полученные ими результаты приведены на рис. 19—23. Пропилен
лишь незначительно абсорбируется 60—75%-ной серной кислотой
даже при давлении 8 кгс/см2 и продолжительном времени пребы-
вания в аппарате.' При этом абсорбирующая фракция будет пол-
ностью изопропилсульфатом. 85%-ная серная кислота при 20 °C
и 8 кгс/см2 абсорбирует спустя 6 ч максимально 1,68 моль С3Н6/моль
H2SO4. Лишь 76% прореагировавшего пропилена переходит в изо-
пропилсульфат, остатком будет диизопропилсульфат (наряду с не-
большими количествами побочных продуктов). 95%-ная серная
кислота поглощает пропилен очень быстро. В данном случае абсорб-
3.1. Получение изопропилового спирта
55
ция практически не зависит от давления; скорость абсорбции опре-
деляется в первую очередь концентрацией кислоты, а давление
и температура имеют второстепенное значение.
Энтелис и сотрудники [23, 24] исследовали кинетику гидратации
пропилена и определили константы
Казарновский [25] изучал за-
кономерности экстракции с по-
мощью лабораторных моделей
промышленной установки. Он
нашел, что при использовании
75 %-ной серной кислоты при
65—70 °C образуется 25,3% сво-
бодной серной кислоты, 32,9%
изопропилсульфата, 25,6% изо-
пропилового спирта, 9,2% воды,
2,8% диизопропилового эфира и
2,6% полимера.
Многочисленные исследования
гидратации пропилена серной
кислотой привели к двум различ-
Концентрация Нг5О<,,%
Рис. 21. Зависимость поглощения
пропилена от концентрации кислоты
при 20 ?С:
1 — 2 ч, 8 кгс/см2; 2 — 1ч, 8 кгс/см2;
3 — 0,5 ч, 8 кгс/см2; 4 — 1ч, 5 кгс/см2;
S — 1 ч, 2 кгс/см2.
ным методам:
1) метод концентрированной
кислоты;
2) метод разбавленной кислоты.
Первый метод аналогичен ис-
пользуемому для этилена. Пропи-
лен или газы, содержащие пропилен, абсорбируются под давлением
в 94%-ной серной кислоте при 20 °C. Образующаяся смесь моно-
и диизопропилсульфатов омыляется после разбавления водой, затем
изопропиловый спирт и диизопропиловый спирт, являющиеся по-
бочными продуктами, отгоняются водяным паром. Разбавленную ки-
слоту регенерируют и возвращают в процесс.
По методу разбавленной кислоты применяют 70%-ную сер-
ную кислоту, температуру 60 °C, давление ~26 кгс/см2. Присоеди-
нение пропилена происходит одновременно с омылением только что
образовавшегося изопропилсульфата. Во второй колонне, рабо-
тающей при пониженном давлении, отгоняют спирт и эфир, а серную
кислоту передавливают в первую колонну, работающую под давле-
нием. Примущество данного метода — более удобный отвод тепла
и отсутствие регенерации кислоты.
Метод концентрированной кислоты. Газы стабилизации с кре-
кинг-установок с содержанием 20—24% пропилена вначале отмывают
в скрубберах от сероводорода [26]. Затем удаляют высшие
углеводороды фракционной перегонкой и концентрируют пропилен
до минимум 50%. Показано [27], что абсорбцию пропилена можно
существенно улучшить с помощью абсорбционного масла, большей
56
3. Изопропиловый спирт
частью газойля. Абсорбционное масло облегчает переработку низко-
концентрированного пропилена. Кроме того, оно значительно сни-
жает склонность к полимеризации при использовании высококон-
центрированного пропилена.
Рис. 22. Зависимость поглощения пропилена
от давления:
а - - 75%-ная H2SO4, 20 °C; 6—75%-ная H2SO4,50 °C;
в — 85%-ная H2SO4, 20 °C; г — 85%-ная H2SO4, 35 °C;
0 — 95%-ная H2SO4, 20 °C; е — 85%-ная H2SO4, 50 °C;
Продолжительность абсорбции-----2 ч;-----1 ч.;
— • — •— 0,5 ч.
Абсорбция проводится в противотоке в экстракционной колонне
(рис. 24) с 8 тарелками под давлением 8—10 кгс/см2 в 92%-ной
серной кислоте при 20 °C. На каждой тарелке имеется слой абсорб-
ционного масла. После экстракции давление серной кислоты пони-
жается, в отстойнике отделяется экстракционное масло и снова
подается в колонну. Экстракт содержит около 1,1—1,3 моль
С3Нв/моль H2SO4. Расход кислоты составляет —12 кг на 10 л
100%-ного изопропилового спирта, расход пропилена — около
39 м3 (теоретически 32 м3).
3-1. Получение изопропилового спирта
57
Содержание пропилена,
моль СзНв/моль H3SO4
Рис. 23. Зависимость плотности
абсорбционных растворов от
содержания пропилена:
1 — 75%-ная' H2SO4; 2 — 85%-ная
H2SO4.
Температура на отдельных тарелках экстракционной колонны
не должна превышать 20 °C, иначе возможны значительные потери
пропилена -из-за полимеризации.
Из отстойника экстракт поступает в освинцованный реакцион-
ный аппарат с турбомешалкой (гидролизатор),-~дде разбавляется
водой до получения 40%-ного раствора, который перемешивается
приблизительно в течение 1 ч при 50 °C. После 4-часового пребы-
вания в аппарате без перемешивания спирт и образующийся диизо-
пропиловый эфир экстрагируются
водяным паром. Пары спирта и
эфира промываются 1 % растворйм
едкого натра. После разбавления
водой до концентрации 15% конден-
сат выдерживают несколько дней
для отделения полимеризата. При
ректификации вначале выделяют
~2% легкокипящих компонентов,
во второй колонне изопропиловый
спирт концентрируют до 91,3%-ной
азеотропной смеси, кипящей при
80,4 °C. Выход изопропилового
спирта составляет 85—90% по отно-
шению к исходному пропилену.
Для получения безводного изо-
пропилового спирта азеотропную
смесь обезвоживают бензолом или
диизопропйловым эфиром, реже —
диэтиловым эфиром. Находящийся в сборнике и разделенный на 2 слоя
головной продукт частично возвращается в верхнюю часть колонны
(верхний слой из изопропилового спирта и бензола), частично по-
дается в колонну для концентрации (нижний слой из водного изо-
пропилового спирта) для регенерации остатков изопропилового
спирта и бензола. Безводный изопропиловый спирт (>99%) отго-
няется снизу колонны.
Все соприкасающиеся с разбавленной серной кислотой детали
аппаратов выполняются либо из свинца, либо из меди. После про-
мывки 1 % раствором едкого натра сталь — наиболее подходящий
материал для изготовления емкостей, труб и колонн.
Экономические расчеты показали, что при методе концентриро-
ванной кислоты около 22% всех расходов приходится на потребле-
ние серной кислоты и ее регенерацию и 8 % — на охлаждение в про-
цессе реакции. Стремление улучшить данный способ с целью
снижения расхода кислоты и стоимости ее регенерации привели
к разработке метода разбавленной кислоты.
Метод разбавленной кислоты. По методу разбавленной кислоты
[28—35] пропилен абсорбируется под давлением 25 кгс/см2 при
58
3. Изопропиловый спирт
бескислотный
пропан
t NaOHi
1 — скруббер; 2 — абсорбер; 3 — охлаждающие змеевики; 4 — холодильник для пропана; 5 — маслоотстойник; 6 — дрос-
сельный клапан; 7 — разделительный резервуар; 8 — гидролизатор; 9 — смотровое стекло; 10 — холодильник; 11 — холо-
дильник для гидролизата; 12 — отстойник; 13 — колонна для промывки щелочью; 14 — экстракционная колонна; 15 —
емкость для сырого спирта; 1в, 17 — колонны для отбора спирта.
3-1. Получение изопропилового спирта
59
65 °C в 70%-ной серной кислоте (рис. 25). При этом диизопропил-
сульфат не образуется, а изопропиловый спиртполучают сразу
без последующего разбавления водой.
В абсорбционную бестарельчатую колонну, заполненную 70%-ной
серной кислотой, нагнетается предварительно компримированный
крекинг-газ, содержащий пропилен. Серная кислота поглощает
Пониженное
Рис. 25. Схема непрерывного получения изопропилового спирта по методу
разбавленной кислоты:
1, в, и — испарители; 2 — абсорбер (25 кгс/сма); 3 — сборник для смеси пропилсульфата
и серной кислоты; 4 — скруббер для пропана; 5, 9, 1г — холодильники; в — колонна для
отгонки спирта; 7 — скруббер; ю — сборник; 13 — резервуар для 70%-ной серной кислоты;
14 — вакуумный пароструйный насос.
1 моль С3Нв/моль H2SO4. Крекинг-газ с первоначальным содержа-
нием пропилена около 20—25% абсорбируется до остаточного со-
держания 5—6%.
Остаточный газ экстрагируется в колонне с колпачковыми та-
релками свежей 70%-ной серйой кислотой, выходящей из колонны
для омыления, до остаточного содержания пропилена 1%. Затем
серная кислота с 0,2% пропилена проходит через бестарельчатую
колонну. В отгонной колонне и колонне для омыления к смеси сер-
ной кислоты и изопропилсульфата добавляется вода до получения
60%-ного раствора, а изопропилсульфат омыляется. 85%-ный
изопропиловый спирт отгоняется. С низа колонны выводится 70 %-ная
серная кислбта, которую можно сразу употреблять для следующего
цикла.
3.1.3. Другие процессы гидратации в жидкой фазе
В небольших количествах изопропиловый спирт можно получать
периодическим способом из пропилена [36—39]. Абсорбция осуще-
ствляется 75%-ной серной кислотой при 45 °C под давлением
60 3. Изопропиловый спирт
в автоклаве с мешалкой. Отстоявшуюся эмульсию серной кислоты
после окончания реакции разбавляют при охлаждении до 30%,
а затем гидролизуют продуванием водяного пара при 85, 90 и
100 °C. Этот процесс можно проводить также для гидратации бутилена.
Известна также гидратация пропилена 0,1—15%-ной серной
кислотой в медных аппаратах под давлением при высокой темпера-
туре (150-300 °C). [40].
Применение некоторых катализаторов значительно ускоряет
процесс сернокислотной гидратации. Для этой цели используются
соли железа, кобальта, никеля, меди, платины, серебра [41, 42],
а также соединения висмута [43, 44]. Сульфат серебра [45 , 46]
и соли меди [47 — 49] сильно ускоряют гидролиз сложных эфиров
серной кислоты. Рекомендуется применять в качестве катализаторов
галогениды бора или бораты в соединении с сульфатами никеля
и других тяжелых металлов [50]. Необходимые для этого реакцион-
ные условия определены Поповым [51]. При высоком давлении
и высокой температуре каталитическое действие проявляют сульфаты
органических оснований, например изопропиламина, анилина, наф-
тиламина, хинолина [52], а также сульфаты и галогениды цинка,
магния, бериллия [53] и алюминия [54]. Соли алюминия обладают
каталитическим действием при высоком давлении и низких темпе-
ратурах в водном растворе. Наконец, следует упомянуть еще крем-
невую или борвольфрамовую кислоту и их соли [55], однако про-
цессы с ид участием протекают при 200—300 °C под давлением уже
в газообразной фазе.
Кроме серной кислоты можно применять для гидратации и раз-
бавленную фосфорную кислоту при 165—290 °C и высоком давлении
[56]. Такое же действие при 120 °C и среднем давлении оказывает
90%-ная фосфорная кислота [57] либо с добавкой окиси никеля
или хлорида цинка, либо нанесенная на SiO2 [58]. Предлагается
также смесь равных объемов 99,5%-ной серной кислоты и ледя-
ной уксусной кислоты после разбавления ее водой [59]. Воз-
можно применение 20—30%-ной фтористоводородной кислоты. Про-
цесс проводят преимущественно при 90—120 °C и давлении выше-
атмосферного [60].
Совсем недавно предложено использовать перфоторкарбоновую '
кислоту при 150—200 °C и среднем давлении [61, 62].
*
3.1.4. Гидратация пропилена в газовой фазе на катализаторах
Как уже упоминалось, классический метод получения изопро-
пилового спирта в жидкой фазе имеет ряд недостатков. Так, потери
кислоты довольно значительны, а расходы на ее регенерацию су-
щественны. Кроме того, большие затруднения вызывает коррозия
оборудования. Поэтому были проведены многочисленные опыты по
прямому присоединению воды к олефинам на неподвижном слое
3.1. Получение изопропилового спирта
61
катализатора. В результате в 1948 г. фирма Shell Chemical Со.
(США) ввела в эксплуатацию первую установку по гидратации эти-
лена, работающую по данному методу с применением фосфорной
кислоты на цеолите (кизельгуре). С 1951 г. на фирме Imperial Che-
mical Industries Ltd. в Вилтоне (Англия) работает установка по
производству изопропилового спирта методом прямой гидратации
в паровой фазе (рис. 26).
Рис. 26. Схема получения изопропилового спирта прямой гидратацией:
1 — скруббер; 2 — хранилище разбавленного изопропилового спирта; 3 — реактор.
Реакция непосредственного превращения олефинов в спирт,
является равновесной, протеканию реакции способствуют низкая
температура, высокое давление и высокое соотношение пар : оле-
фины:
RCH=CH2 + H2O •—> RCH(OH)CH3
С точки зрения термодинамики для достижения максимальной
конверсии нужно работать при таком давлении, которое лишь не-
значительно ниже точки насыщения. Если это условие выполнено,
температуру можно варьировать в широком диапазоне, так как она
относительно мало влияет на состояние равновесия.
Свободная энтальпия гидратации пропилена в газовой фазе
составляет — 8900 ккал/моль + 37,5т (Т — ЗОО4-6ОО К), а эн-
тальпия реакции ДЯ — около 9000 кал/моль. Равновесие можно
сдвинуть вправо повышением давления, поскольку реакция связана -
с уменьшением объема [63].
Из многих возможных катализаторов реакции прямой гидратации
олефинов для промышленного применения рекомендуются только два:
1) фосфорная кислота на носителе;
2) окись вольфрама с промотором.
Гидратация на вольфрамсодержащих катализаторах. Первые
опыты по получению изопропилового спирта из пропилена путем
гидратации в газовой фазе относятся к 1929—1930 гг. [64, 65].
62
3- Изопропиловый спирт
Фирма Shell Development Со. получила патент на гидратацию про-
пилена при 300 °C в присутствии металлов платиновой группы,
а также Au, Ag, Си, Fe, Ni, Со, Сг, Та, V, W, Мо и Мп. Особыми
преимуществами отличается комбинация CuO 4- WO3 на активиро-
ванном угле.
Во время второй мировой войны тщательно исследовали вольфрам-
содержащие катализаторы. Наиболее подходящей оказалась окись
вольфрама, активированная окисью цинка и нанесенная на активный
силикагель. При соотношении пропилен : водяной пар = 1 : 10,
температуре 230—240 °C, давлении 200—250 кгс/см2 и 50%-ной
конверсии пропилена за проход можно получить конечный выход
изопропилового спирта 95%. Однако выход в единицу времени на
единицу объема будет очень низким: 0,8 кг изопропилового спирта
на 1 л катализатора в 1 день [66].
Низшие окиси вольфрама (WO, WO2, W4OXI, W2O5) неплохо
зарекомендовали себя в качестве катализаторов [67—72]. W2O5
можно получить путем восстановления WO3 этиловым или изопро-
пиловым спиртом при 250 °C. Конверсия на этом катализаторе до-
стигает 6,5% при 230 °C, 10% при 250 °C и 14,5% при 270 °C. Воз-
можна добавка графита в качестве восстановителя WO3, имеющей
недостаточную каталитическую активность [73].
Сульфиды вольфрама и никеля могут успешно применяться
вместо окиси вольфрама [74]. В этом случае получают до 43,7%
изопропилового спирта при 270 °C и 256,6 кгс/см2.
По литературным данным на нитридах вольфрама или молиб-
дена [75] при 250 °C, давлении 120 кгс/см2 за 12 ч получают из
80 г пропилена и 540 г водяного пара 54 г изопропилового спирта,
в то время как 36 г пропилена в реакцию не вступают.
Кроме окиси вольфрама целесообразно использовать и кремне-
вольфрамовую кислоту [76]. Оптимальные условия работы катализа-
тора: температура 175—190 °C и давление около 20 кгс/см2. Ана-
логичное действие оказывают борновольфрамовая кислота и подоб-
ные гетерополикислоты [77].
Рунге с сотрудниками [78, 79] провели в 1952—1953 гг. обшир--
ные исследования по определению наиболее подходящих катализа-
торов для гидратации пропилена. С этой целью были изучены кислые
катализаторы, такие, как серная кислота, нафталинсульфокислота,
фосфорная кислота, кислые фосфаты, окись вольфрама без промо-
тора и носителя, а также на различных носителях, например на
активированном кислотой монтмориллоните. Показано, что серная
кислота не подходит из-за нестойкости, а фосфатные катализаторы
отличаются незначительной активностью. Фосфорные кислоты на
носителях проявляют при средней крепости кислоты максимальную
каталитическую активность, причем наилучшим носителем явля-
ется крупнопористый силикагель. Выход в единицу времени на
единицу объема составил 0,52 кг изопропилового спирта на 100 мл
3-1. Получение изопропилового спирта
63
катализатора в 1 день. Конверсия пропилена при повышенных тем-
пературе и давлении достигала 5,1%. Монтмориллонит характери-
зуется растущей каталитической эффективностью с увеличением
кислотности. Самыми оптимальными катализаторами были окиси
вольфрама с промоторами.
Результаты, полученные Рунге при гидратации пропилена в га-
зовой фазе, представлены в табл. 7. Из таблицы видно, что конвер-
сия пропилена увеличивается при повышении давления и соотно-
шения вода : пропилен. Однако уровень нужного давления зависит
от уровня температуры, так как для достижения максимальной кон-
версии давление должно лежать лишь немного ниже точки насыще-
ния на основании законов термодинамики. .Высший предел темпе-
ратуры опять же зависит от активности катализатора.
Таблица 7
Зависимость конверсии пропилена в газовой фазе от давления,
температуры и содержания воды в исходном газе
(термодинамическое равновесие)
Темпера- тура, °C Давле- ние, кгс/смг Содержа- ние воды, % Конвер- сия, % Темпера- тура, °C Давле- ние, кгс/см2 Содержа- ние воды % ^Конвер- сия, %
100 1 50 8,9 250 81,0 50 22,5
150 1 50 2,8 300 175,0 50 23,3
200 1 50 1,0 275 202,0 30 18,3
250 1 50 0,4 275 152,0 40 21,2
100 2,0 50 . 17,5 275 121,0 50 . 22,9
150 9,7 50 20,2 275 101,0 60 24,0
200 31,7 50 21,5 275 86,6 70 25,0
Был сделан вывод о том, что природа и активность катализатора —
определяющие факторы для данного процесса.
Рунге исследовал 120 различных катализаторов гидратации
и нашел, что самым оптимальным является окись вольфрама с про-
мотором. Катализаторы этого типа сохраняют активность и через
1000 ч работы. На них легко достигается конверсия 8,8% и выход
94% при 260—320 °C и давлении 80—200 кгс/см2. В реакционной
смеси содержится до 34% спирта. Неожиданным оказалось образо-
вание из пропилового спирта наряду с диизопропиловым эфиром
и полимерами значительного количества примесей.
Опытные данные, полученные Рунге при эксплуатации пилотной
установки, подтвердили, что окись вольфрама с промотором является
наилучшим катализатором. Для промышленного использования
хорошо зарекомендовали себя также следующие катализаторы:
20% окиси вольфрама и 5% окиси цинка на особо подготовленном
64
3- Изопропиловый спирт
носителе из силикагеля [79] и 5—50% окиси хрома в сочетании
с синей окисью вольфрама [80].
Гидратация иа молибденсодержащих катализаторах. Хотя во мно-
гих патентах предлагается использование молибденсодержащих ка-
тализаторов для прямой гидратации пропилена, подобные катали-
заторы менее эффективны по сравнению с вольфрамсодержащими?
Имеются сообщения о катализаторе, получаемом путем восстано-
вления фосформолибденовой кислоты или ее аммониевой соли. [81].
Кроме того, для гидратации пропилена в газовой фазе успешно
используется восстановленная окись молибдена при 271 °C и 256,6—
258,4 кгс/см2 [82]. Наконец, применяют катализатор, состоящий
из 10% МоО3 на носителе из 75% SiO2 и 25% А12О3; на этом ката-
лизаторе при 260 °C конверсия достигает 30,6% [83].
Гидратация на фосфорсодержащих катализаторах. Гидратацию
пропилена можно проводить и на фосфате бора при 200.°C и 100 кгс/см2.
Выход составляет 98—99%, но конверсия очень низка [84]. Про-
цесс проводят под давлением в жидкой фазе; в газовой фазе резуль-
таты должны быть еще лучше.
Были предложены фосфаты или сама фосфорная кислота на самых
различных носителях. Так, в качестве носителя рекомендуется
активированный уголь, работающий при 100—350 °C и 20—100 кгс/см2
[85]; носитель из силикагеля (при 170—180 °C и 8,5—10 кгс/см2
выход составляет 96%, конверсия 5,7%); носитель из диатомита
повышенной прочности (конверсия 4%), а также различные носи-
тели, содержащие SiO2, A12(SO4)3 и А12О3 [86—89].
Гидратация нц алюминий- и кремнийсодержащих катализаторах.
Гидратация пропилена на катализаторе, состоящем из 90% SiO2
и 10% ALO3, происходит при 310 °C и 105,46 кгс/см2, селективность
составляет 99%, конверсия достигает 12% при объемной скорости
1-5 ч-1 [90].
Фирма Montecatini Soc. предлагает катализаторы на основе
гидроокиси алюминия, на которых проводят гидратацию при 250 °C
и 250 кгс/см2 [91]. Активность катализаторов на основе А12О3 —
— SiO2 усиливается предварительной обработкой смесью H2S — Н2;
при пропускании 1 объема газовой смеси в соотношении С3Н6:
: водяной пар = 15 : 1 на 1 объем катализатора в 1 ч получают
при 271 °C и 268 кгс/см2 конверсию 45%; селективность 98% [92].
Гидратация на ионитах. Использование ионитов для гидратации
пропилена дает хорошие результаты. При 70,3 кгс/см2 на кислой
ионообменной смоле, состоящей из 88—96% стирола и 4—12% ди-
винилбензола со свободными группами сульфоновой, серной или
фосфорной кислоты, например Дауэкс-8 X 50, конверсия при
171 °C составила 34,7% (селективность 55%), при 177 °C — 25,4%
(селективность 71%) [93—95]. При этом побочными продуктами
были диизопропиловый эфир «1%), ацетон (1—2,5%) и полимеры
(10—35%). Для удаления полимеризующихся соединений пропилен
I
3-2. Свойства изопропилового спирта
65
можно пропустить предварительно через смесь, состоящую из 90%
SiO2 и 10% А12О3, а затем — через ионит [96].
Кайзер [97] провел обширную работу по определению оптималь-
ных условий гидратации на ионитах. Он исследовал зависимость
между соотношением вода : олефины, давлением и временем кон-
такта на ионитах Амберлит-15 и Амберлит IR-120. Было показано,
что на ионитах можно достичь таких же значений конверсии и се-
лективности, как при гидратации на неорганических катализато-
рах. Максимальная конверсия составляла 72,9% при объемной
скорости жидкости 0,6 ч"1 и селективности 96,4%. Ниже будет
показано, что реакция протекает по псевдопервому порядку иму-
щественно зависит от давления и температуры.
Гидратация другими методами. Кроме уже названных катализа-
торов в литературе описаны системы состава TiO2 и Sb2O5, TiO2
и А12О3 и TiO2 и Fe2O3, которые дают после 4-часовой реакции при
270 °C и 250 кгс/см2 до 7,8% изопропилового спирта [98].
Воздействие спокойных электростатических разрядов (~20 000 В)
также вызывает гидратацию [99].
В заключение следует упомянуть косвенную гидратацию, при
которой олефины вступают в реакцию со смесью из СО и Н2 в при-
сутствии соответствующих катализаторов [100]', например, они
реагируют с СО и водяным паром в присутствии хлористого родия
и пиридина [101].
3.1.5. Экономическая оценка методов гидратации
в жидкой и газовой фазе
Жидкофазная сернокислотная гидратация пропилена [102] по-
зволяет изготовлять 30—40%-ный пропилен, и в этом заключается
преимущество метода. Процесс осуществляется при низком давле-
нии и высокой степени превращения, изопропиловый спирт полу-
чается более ^высокой концентрации, чем при газофазной гидратации.
Недостатком является применение серной кислоты и связанные
с этим проблемы коррозии, а также необходимость концентрирования
(упарки) возвращаемой в процесс кислоты и, наконец, высокий
расход кислоты. Тем не менее, на сегодняшний день жидкофазная
гидратация считается более экономичной по сравнению с газофазной.
3.2. СВОЙСТВА ИЗОПРОПИЛОВОГО СПИРТА
Ниже приведены свойства изопропилового спирта [103]:
Температура плавления, °C....................... —98,5
Температура кипения, РС......................... 82,4
Плотность р|°................................... 0,789
Показатель преломления п|° 1,37723
5 Заказ 399
66
3- Изопропиловый спирт
Вязкость при 20 РС, сП............................... 2,43
Поверхностное натяжение при 20 °C, дин/см . . . 21,7
Критическая температура, РС......................... 234,9
Критическое давление, кгс/см2 ........................ 53
Теплота плавления при температуре текучести
кал/г ........................................... 21,08
ккал/моль ........................................ 1,284
Теплота испарения при температуре кипения, кал/г 160
Теплота испарения при 25 °C, ккал/моль.......... 11,05
Теплота образования, ккал/моль....................... 76,4
Теплота сгорания,
кал/г............................................. 7970
ккал/моль......................................... 478,9
Удельная теплоемкость, кал/(г • °C).................. 0,563
при 30 9С......................................... 0,677
50 °C................................./ . "0,740
Коэффициент теплопроводности при 0рС,
кал • см/(сек • см2 . °с)........................ 3,633 . щ-4
Коэффициент теплового расширения, 1/°С.......... 0,001075
Нижний предел взрываемости в смеси с воздухом,
объемн. % 2,5
Температура вспышки (в закрытой аппаратуре), рС 15
Температура самовоспламенения, РС..................... 456
Диэлектрическая проницаемость при 20 °C .... 26
В литературе [104] приведен обзор двойных и тройных азеотроп-
ных смесей.
3,3. ПРИМЕНЕНИЕ ИЗОПРОПИЛОВОГО СПИРТА
Основное количество изопропилового спирта используется для
получения ацетона, однако оно уменьшается из-за серьезной кон-
куренции кумольного метода производства ацетона. Если раньше
из изопропилового спирта получали 95% ацетона, то в 1963 г.
в США таким путем было получено только 72,6%, а 20,4 % ацетона
уже производили через кумол.
Изопропиловый спирт нашел широкое распространение в ка-
честве растворителя для жиров, натуральных и синтетических смол,
нитролаков (в сочетании с другими растворителями), алкалоидов,
протеина, хлорофилла и пр. Он используется и как составная часть
детергентов (жидкие мыла).
Наряду с применением для синтеза ацетона изопропиловый спирт
употребляют для синтеза различных сложных эфиров (например,
изопропилацетата — растворителя лаков), для введения изопро-
пиловой группы в другие соединения (тимол, изопропилфенол).
Ксантогенат изопропилового спирта является важным флотацион-
ным агентом. Изопропилат алюминия используется для восстано-
вления альдегидов по методу Меервейн — Понндорфа.
Изопропиловый спирт заменяет этиловый во многих косметиче-
ских и фармацевтических препаратах. Но он допущен только для
Литература
67
наружного употребления, например дезинфекции; применение длн
лекарств и пищевых продуктов запрещено.
В большом количестве изопропиловый спирт используется для
улучшения качества горючего. При применении бензина в карбю-
раторе двигателя может наступить обледенение при температуре
от —8 до 4-13 °C и относительной влажности воздуха 60—100%,
затрудняющее включение и выключение двигателя.
Для устранения этого нежелательного явления достаточно до-
бавить 1,5—3% изопропилового спирта.
В настоящее время изопропиловый спирт находит применение
в качестве антиобледенителя для крыльев и пропеллеров самолетов,
а смесь этиленгликоля и изопропилового спирта устраняет обле-
денение стартовых дорожек и взлетно-посадочных полос на аэро-
дромах.
ЛИТЕРАТУРА
1. L. F. Hatch, Isopropylalkohol, N. Y., 1961.
2. M. Berthelot, Ann. China, et phys., 43, № 3, 391 (1855).
3. C. Ellis, The Chemistry of Petroleum Derivatives, v. 11, N. Y., 1937,
p. 346.
4. C. Ellis, Chem. Metallurg. Eng., 23, 1230 (1920).
5. Chem. Industrie, 17, 118 (1965).
6. Пат. США 1725083, 1927; англ. пат. 293015, 1928; 401284. 1932; франц,
пат. 743530, 1932; пат. США 2420998, 1947.
7. В. Т. Brooks, Chem. Rev., № 2, 369 (1925).
8. В. Т. Brooks, Ind. Eng. Chem., 27, 282 (1935).
9. С. O. G u r m e, Chem. Metallurg. Eng., 25, 1049 (1921).
10. H. C. F u 1 1 e r, ibid., 29, 588 (1923).
11. S. D. К i r k p a t r i k, ibid., 33, 402 (1926).
12. W. W. Clough, C. O^Johns, Ind. Eng. Chem., 15, 1030 (1923).
13. В. В. Пигулевский, H. В. Рудакова, Труды опытно-исслед.
завода Химгаз, Материалы по крекингу и химической переработке его
продуктов, вып. 3, 1936, стр. 234—260.
14. С. С. X а й н, С. А. Н а з а р о в, там же, стр. 455—474 (1936).
15. В. С. Г у т ы р я и др., Азерб. нефт. хоз., № 7, (1936).
16. В. С. Г у ты р я, В. Л. Б уйн ицк а я, ЖПХ, 10, 882—886 (1937).
17. Refiner, 18, 83 (1939).
18. В. Т. Brooks, J. W. Humphrey, Refiner, 13, 248 (1934).
19. В. Т. Brooks, J. W. Humphrey, Refiner, 11, 224 (1932).
20. В. В. Пигулевский и др., Труды VI Всесоюзного Менделеевского
съезда по теоретической и прикладной химии, Харьков, 1932; т. 2, вып. 1,
Киев, 1939, стр. 711—715.
21. R. G. S с h u 1 t z е et al., Erdol und Kohle, 6, 687 (1953).
22. R. G. S c h u 1 t z e et al., Erdol und Kohle, 8, 402—406 (1955).
23. С. Г. Э н т e л и с и др. ДАН СССР, 114, 848-851 (1957).
24. С. Г. Э и те л и с и др., ДАН СССР, 134, 856—859 (1960).
25. С. Н. К а з а р н о в с к и й и др., Труды по химии и хим. технол., 1,
431—438 (1958).
26. F. A s i n g е г, Chemie und Technologie der Monoolefine, Berlin, 1957,
S. 581—584; I. G. P a r k, С. M. В e a m e r, in Kirk — О t h m e r,
Encyclopedia of Chemical Technology, v. 11, N. Y., 1953, p. 185—187.
27. Пат. США 1365043, 1921.
5*
68
3- Изопропиловый спирт
28. Пат. США 2473224, 1949.
29. Франц, пат. 799076, 1936; пат. США 1988611, 1935.
30. Англ. пат. 652624, 1951; пат. ФРГ 938547, 1956; пат. США 2541673, 1950.
31. Англ. пат. 715483, 1954; пат. США 2609400, 1954.
32. W. Н. S h i f f 1 e г et al., Ind. Eng. Chem., 31, 1101 (1939).
33. В. T. Brooks, ibid., 27, 286 (1935).
34. Франц, пат. 1135432, 1957; 1377276, 1964.
35. Англ. пат. 830369, 1960.
36. W. Grimme, Angew. Chem., 60, 213^(1948).
37. W. Grimme, Bergbau-Arch., 9, 114 (1948).
38. M. A. M a t he ws, L. W. W о о d, В. I. О. S. Target, Nr. C22 Final
Re pt. Nr. 13 und 22.
39. E. Reinmuth, Chemiker-Ztg., 76, № 11/12, 248 (1952).
40. Герм. пат. 681268, 1939.
41. Франц, пат. 695707, 1930.
42. Англ. пат. 978926, 1965; инд. пат. 92082, 1964.
43. Франц, пат. 799704, 1935.
44. Англ. пат. 324897, 1930; франц, пат. 662968, 1930.
45. Пат. США 2050444, 1934.
46. Е. К. Ремиз, ЖПХ, 9, 705—706 (1936).
47. Пат. США 1995908, 1932; франц, пат. 695707.
48. Пат. США 2139953, 1936.
49. Пат. США 2210316, 1937.
50. Пат. США 2135455, 1935.
51. В. А. Попов, Труды СНО Горьковского политехнического ин-та им.
А. А. Жданова, № 1, 90—94 (1957).
52. Пат. США 2036317, 1934.
53. Пат. США 2107515, 1933.
54., Пат. США 2148288, 1936; герм. пат. 640556, 1932; 681268, 1934.
55. Франц, пат. 865020, 1937; пат. США 2162913, 1935.
56. F. М. М a j е w s k i, L. F. Marek, Ind. Eng. Chem., 30, 205 (1938).
57. Пат. США 2107194, 1933.
58. D. A. S. 1249845, 1967; Jap. A. S. 5373/66.
591 Герм. пат. 567117, 1930.
60. Пат. США 2583413,' 1952.
61. Пат. США 2858331, 1958.
62. Пат. США 2830091, 1958.
63. U 11 m a n n, Enzyklopadie der technischen Chemie, Munchen, Bd. 14,
1963, S. 391.
64. Пат. США 1999620, 1930; англ. пат. 335551, 1929.
65. Герм. пат. 569019, 1930.
66. К. К ammerme ye г, G. В. Carpenter, FIAT Final Rept. 968
Р. В., 78, 277 (1947).
67. Англ. пат. 622937, 1949; пат. США 2536738, 1951.
68. W. Н u n t е г, А. С. Т а у 1 о г, Brit. Int. Object. Subcommittee, Final
Rept. 1558, 22.
69. Англ. пат. 646284,1950; франц, пат. 951860, 952096, 1949; пат. США 2683753,
1954.
70. Англ. пат. 680865, 1952; пат. США 2725403, 1955.
71. Англ. пат. 727665, 1952.
72. Англ. пат. 699088, 1953; пат. США 2751420, 1956.
73. Бельг, пат. 611845, 1962.
74. Англ. пат. 996780, 1965; D. A. S. 1249844, 1967; пат. США 3006970, 1961.
75. Пат. ФРГ 922585 1955.
76. J. Muller, Н. J. W a t е г m a n n, Brennstoff-Chem., 38, 357 (1957).
77. Франц, пат. 865020, 1937; пат. США 2162913, 1965.
78. F. R u n g е et al., Brennstoff-Chem., 34, 330—333 (1953).
Литература
69
79. Пат. ГДР 12356, 1956.
80. Франц, пат. 1481464, 1967.
81. Пат. США 2825704, 1958.
82. Пат. США 2995609, 1958.
83. Пат. США 3076036, 1963.
84. Герм. пат. 574275, 1932; пат. США 1977634, 1932; англ. пат. 378865.
85. Пат. США 2051144, 1933; 2232610, 1937.
86. М. А. Да лип и др., Хим. пром., 385—387 (1959).
87. Пат. США 2496621, 1950.
88. Франц, пат. 1385123, 1965.
89. N. О h t a, J. Chem. Soc. Japan, Ind. Chem. Sect., 51, 141 (1948).
90. Англ. пат. 692800, 1953; пат. США 2658924, 1953.
91. Инд. пат. 68947, 1962; итал. пат. 611444, 1959.
92. Англ. пат. 966917, 1965; пат. США 3129253, 1964.
93. Англ. пат. 788883, 1958; 809318, 1959; пат. США 2813908, 1957; 2861045,
1958.
94. D. A. S. 1210768, 1965.
95. Пат. США 3140322, 1964.
96. Пат. США 2845463, 1958.
97. J. R. Kaiser et al., Ind. Eng. Chem., Prod. Res. Development, 1, 296—
302 (1962).
98. Англ, пат. 710006, 1954; 718723, 1954; пат. США 2769847, 1956,
99. Англ. пат. 695076, 1953.
100. Пат. США 2671814, 1954.
101. Пат. США 3020314, 1962.
102. Р. W. Sherwood, Ind. Chemist, 36, 499 (1960).
103. Kirk — Othmer, Encyclopedia of Chemical Technology, N. Y., 1953,
p. 182—183; Ullma nn, Enzyklopadie der technischen Chemie, Bd. 14,
Munchen, 1963, S. 390.
104. M. L e c a t, Tables azeotropiques, Brussel, 1949; В e i 1 s t e i n, Hand-
buch der Organischen Chemie, Berlin, Bd. III/l, 1958, S. 1446.
105. Chem. Eng. News, 41, № 19, 99 (1963).
it. ОКИСЬ ПРОПИЛЕНА
Окись пропилена была открыта Озером [1], когда он обрабатывал
1-хлорпропанол-2 едким кали:
С1СН2СН(ОН)СН8 Н2С---------СНСН3
Аналогичный результат получил Крассуский [2] при действии
на 1-хлорпропанол-2 окиси свинца и воды. В литературе имеется ряд
работ по синтезу окиси пропилена [3—9].
В настоящее время окись пропилена получают двумя путями:
через пропиленхлоргидрин и прямым окислением пропилена в окись
пропилена. В промышленном масштабе окись пропилена производили
до сих пор только через хлоргидрин.
Данный метод остается пока наилучшим, тдк как он дает хороший
выход (85—90%); при прямом окислении выход пропилена дости-
гает только ~35%. Кроме того, при переходе от хлоргидринного
метода к прямому окислению при получении окиси этилена можно
использовать освободившиеся установки для производства окиси
пропилена.
4.1. ПОЛУЧЕНИЕ ОКИСИ ПРОПИЛЕНА
ЧЕРЕЗ ПРОПИЛЕНХЛОРГИДРИН
Метод получения окиси пропилена путем присоединения хлорно-
ватистой кислоты к пропилену, впервые подробно описанный Мар-
ковниковым [10], до сих пор является самым распространенным.
Протекающие при этом реакции можно записать следующим обра-
зом:
С12+Н2О НОС1 + НС1
.—► С1СН2СН(ОН)СН,
2СН2=СНСН3 + 2НОС1—
1-> СН2(ОН)СНС1СН3
С1СН2СН(ОН)СНз + СН2(ОН)СНС1СН3+ Са(ОН)2 —►
—> 2Н2С—СНСН3 + СаС12 + Н2О
О
Мольное соотношение а- и p-пропиленхлоргидринов составляет
9 : 1..
4.1. Получение окиси пропилена через пропиленхлоргидрин
71
В результате воздействия элементарного хлора на пропилен
и хлоргидрин в качестве побочных продуктов образуются ди хлор-
пропан и дихлордиизопропиловый эфир:
СН2С1СН(ОН)СН3 + С12 —> СН2С1СН(ОС1)СН3
СН2С1СН(ОС1)СН3+СН2=СНСН3 —> СН2С1СН(СН3)-О-СН(СН3)СН2С1
СН2=СНСН3 + С12—> СН2С1СНС1СН3
Поскольку присоединение хлора идет значительно быстрее,
чем присоединение хлорноватистой кислоты [11], нужно по возмож-
ности подавлять реакцию хлора с пропиленом. Для этого взаимодей-
ствие хлора с водой следует проводить в отдельной колонне. Собст-
венно хлоргидринирование происходит в следующей второй колонне.
Большое значение имеет хороший контакт между жидкой фазой
и углеводородом. Целесообразно выбрать двухкамерную установку.
Недиссоциированная кислота всегда присоединяется по двойной
связи. В присутствии буферного бората (pH = 10) хлоргидрин
практически не образуется, между тем как в кислой среде (pH =
= 4,7) наступает хлоргидринирование. Увеличение диссоциации
равносильно уменьшению выхода хлоргидрина.
Так как степень диссоциации слабых кислот возрастает пропор-
ционально температуре, то хлоргидринирование при повышенных
температурах идет, в принципе, хуже.
Константа диссоциации НОС1 при 25 °C равняется 1,05-10*7.
Если определить с помощью термодинамических характеристик
показатель константы диссоциации для 65 °C, то можно установить
изменение только на одну десятую степени, которое так мало, что
не имеет практического значения.
По теории реакция взаимодействия должна проходить с образо-
ванием 1 моль хлоргидрина на 1 моль НС1. Однако аналитическое
исследование процесса свидетельствует о избытке соляной кислоты,
получающейся по следующему механизму:
СН2С1СН(ОН)СН3 + С12 —► СН2С1СН(ОС1)СН3 + НС1
СН2С1СН(ОС1)СН3-|-СН2=СНСНз —> СН2С1СН(СН3)-О-СН(СН3)СН2С1
• Соотношение пропиленхлоргидрина и соляной кислоты зависит
от температуры и концентрации хлоргидрина, соотношения пропи-
'лен: хлор, концентрации пропилена в исходном газе и от аппаратур-
ного оформления процесса. С повышением температуры растет из-
быточное количество соляной кислоты, которое может достигнуть,
например, при 75—80 °C примерно 50%. При 40 °C избыток коле-
блется в пределах 6—10% при концентрации пропиленхлоргидрина
50 г/л. Омыление пропиленхлоргидрина происходит так же, как
72
4. Окись пропилена
омыление этиленхлоргидрина, однако оно протекает примерно в 20 раз
быстрее.
Уменьшение констант скорости во время реакции обмена можно
объяснить протеканием вторичных реакций. Действительно, окись
пропилена реагирует с образованием промежуточного соединения,
которое затем медленнее омыляется. Это нужно учитывать при про-
мышленном производстве и быстро удалять образовавшуюся окись
пропилена из реакционного раствора [12].
При промышленном получении процессе лучше всего идет в реак-
торе с двойной камерой, который применяется для превращения
этиленхлоргидрина [13, 14]. При сравнимых рабочих условиях
выход в таком реакторе составляет 87,5% пропиленхлоргидрина
с одновременным образованием 11,0% пропилендихлорида и 1,5% ди-
хлордиизопропилового эфира. В противоположность этому реактор
с одной камерой дает только 69,2% продукта, одновременно 21,6%
прореагировавшего пропилена высаждается в виде пропиленди-
хлорида, а 9,2% превращается в дихлордиизопропиловый эфир.
Это свидетельствует о несомненном превосходстве двухкамерной
системы для процесса превращения пропилена в хлоргидрин. В пер-
вой камере такой установки хлор растворяется вместе с подаваемой
свежей водой в циркулирующем пропиленхлоргидрине, разбавлен-
ном водой. После введения хлора- смесь подается во вторую камеру,
где попадает в поток пропилена, и реакция заканчивается.
При хлоргйдринировании пропилена нельзя избежать образова-
ния побочных продуктов, так как в растворе хлоргидрина наряду
со свободной НОС1 имеются еще свободные НС1 и С12. Реагируя
с пропиленом, С12 вызывает образование 1,2-дихлорпропана, а в ре-
зультате реакции с пропиленхлоридрином получается дихлордиизо-;
пропиловый эфир.
Как и в случае этиленхлоргидрина, для подавления побочных
реакций желательно работать при температуре ниже 50—60 °C.
При этих условиях этилендихлорид можно в значительной степени
вывести из верха колонны газовым потоком и предотвратить образо-
вание второй фазы в реакторе. При реакции превращения пропилена
более тяжелый дихлорид не позволяет работать с чистым пропиленом,
что было бы выгодно. Тем не менее, дихлорид можно отогнать во время
реакции обмена при 50—60 °C, использовав поток углеводорода,
содержащий более 45% пропилена. Не вступивший в реакцию газ
содержит инертные газы: метан, этан, пропан или азот. При началь-
ном контакте с пропиленовым потоком водная фаза должна содержать
не более 0,5 г/л хлора [12].
Концентрация самого раствора хлоргидрина также не должна
значительно повышаться, так как это способствует протеканию
побочных реакций (рис. 27, 28).
Из этого следует, что концентрацию продукта нельзя привести
к экономическому показателю при однократном прохождении через
4.1. Получение окиси пропилена через пропиленхлоргидрин
73
аппарат и приходится использовать вышеописанную циркуляцион-
ную систему.
Концентрация пропиленхлоргидрина на выходе из реактора вли-
яет на конечный результат в гораздо большей степени, чем при реак-
ции обмена этилена. При 45 °C и концентрации хлора 0,15 г/л в слу-
чае с пропиленом получается 97% качественного продукта, если
Концентрация С3Н7ОС1, г/л
Рис. 27. Влияние избытка НС1 на
образование пропиленхлоргидрина:
1 — 1 г/л хлора, 45 °C; 2 — 0,15 г/л хлора,
45 °C; 3 — 1 г/л хлора, 35 "С; 4 — 0,15 г/л
хлора, 35 °C.
Рис. 28. Зависимость выхода пропилен-
хлоргидрина в пересчете на С12 от ко-
нечной концентрации пропиленхлорги-
дрина:
1 — 0,15 г/л хлора, 35 °C; 2 — 0,15 г/л хлора,
45 °C; 3 — 1 г/л хлора, 35 °C; 4 — 1 г/л хлора
45 °C.
общая концентрация пропиленхлоргидрина ниже 5 г/л. При концент-
рации пропиленхлоргидрина 50 г/л выход снижается до 89,5%, а при
70 г/л выход примерно на 7 %- ниже теоретически вычисленного
(87%). Понижение, температуры до 35 °C приводит к незначительному
увеличению выхода. Дальнейшее уменьшение температуры не по-
вышает выхода, а при 15 °C наблюдаются потери продукта из-за
затруднений с аппаратурным оформлением.
При двухкамерной системе для производства хлоргидрина нужен
реакционный объем порядка 1,2 м3/(т-сут).
Омыление пропиленхлоргидрина в окись пропилена происходит
в основном аналогично образованию окиси этилена. Однако здесь
можно использовать только известковое молоко, содержащее не бо-
лее 1% MgO, иначе- будет преобладать образование пропионового
альдегида.
Скорость образования окиси пропилена значительно ниже (при-
мерно в 10 раз), чем при производстве окиси этилена. Однако пока-
зано, что образование пропилена в основном замедляется присут-
ствием продукта в реакционной среде. Поэтому для ускорения
и
4. Окись пропилена
реакции необходимо максимально быстро выделять продукт из
системы по мере его образования.
Описан [16] двухколонный реактор для получения 93,5% про-
пиленхлоргидрина и 6% 1,2-дихлорпропана. Предложено также
вводить хлор и пропилен в водную хлорноватистую кислоту в раз-
личных местах колонного реактора [15, 17]. Рекомендуется и после-
довательное включение нескольких реакторов в каскад, причем
в первый реактор загружают воду и хлор, а в последующие вводят
реакционную смесь из предыдущего реактора и пропилен. Такой
режим работы дает хороший выход [18]. Описан метод одновремен-
ного воздействия хлора, воды и пропилена друг на друга [19—21].
Обзор лабораторных опытов по получению пропиленхлоргидрина
сделан Мейем и Франке [9]. Необязательно использовать чистый
пропилен для хлоргидринирования, даже лучше разбавлять его
пропаном [22—25]. Предложен способ проведения реакции пропи-
лена с хлором даже в 20—80%-ной серной кислоте при 10—11 °C
[26].
Окись пропилена можно получать и совместным хлоргидриниро-
ванием этилена и пропилена, так как образующийся пропиленхлор-
гидрин омыляется в известковом молоке примерно в 20 раз быстрее,
чем этиленхлоргидрин [27].
Показатель pH имеет решающее значение для направленного
отщепления НС1 от пропиленхлоргидрина [28]. При pH = 9 4- 12
образуется окись пропилена, при pH = 94-7 — пропиленглико ль,
в очень кислой среде — ацетон. Увеличение мольного соотношения
Са(ОН)2 : окись пропилена от 1 : 1 до 2 : 1 повышает выход пропи-
ленгликоля, уменьшает выход окиси пропилена и не влияет на выход
карбонильных соединений. Оптимальные параметры производства
окиси пропилена приведены ниже [28]г
Температура, РС.......................... 100
Концентрация Са(ОН)г, г/л ............... 200
pH.................................... 9-12
Подвод тепла, ккал/моль C3H7OCI .... 500
Концентрация C3H7OCI.................... Высокая
При этих условиях выход продуктов реакции составляет (в мол. %):
Окись пропилена.........................., 91
П ропиленглнколь........................... 7
Карбонильные соединения.................. 2
Схема установки для получения окиси пропилена методом хлор-
гидрирования показана на рис. 29. Для получения хлорноватистой
кислоты в нижнюю часть колонны 3 с кислотостойкой облицовкой
вводят газообразный хлор.
4.1. Получение окиси пропилена через пропиленхлоргидрин
75
Газообразный пропилен, смешанный в незначительном избытке
с циркулирующим газом, подается в другом месте колонны 3. Из
колонны часть реакционного раствора отводится для дальнейшей
переработки в дегазационную колонну 4 с аналогичной кислотостой-
кой облицовкой. Большая часть однако циркулирует через перепуск-
ное устройство. В верхнюю часть перепускной трубы подается све-
жая вода в таком количестве, чтобы концентрация хлоргидрина
в колонне не превышала 40 г/л, а содержание свободного хлора было
Рис. 29. Схема установки по производству окиси пропилена:
1 — промывная колонна с содовым раствором; 2 — компрессор; •') — ко-
лонна для хлоргидрина; 4 — дегазационная колонна; 5 — гидролизер; в —
дефлегматор; 7 — конденсатор для неочищенной окиси пропилена; 8 —
емкость для неочищенной окиси пропилена; 9 — тарельчатая колонна; 10.—
конденсатор для очищенной окиси пропилена.
не выше 0,3 г/л. Температура реакции в колонне должна поддержи-
ваться в пределах 40—45 °C. Отходящие с верха колонны 3 и дегаза-
ционной колонны 4 непрореагировавшие пропилен и пропан вместе
с хлористым водородом, смешанным с дихлорпропаном, направля-
ются под небольшим вакуумом в куб промывной колонны 1, заполнен?
ной кольцами Рашига. Подаваемый в верхнюю часть промывной ко-
лонны раствор соды нейтрализует реакционную смесь и одновременно
поглощает дихлорпропан. Нейтрализованная смесь пропан — про-
пилен возвращается через компрессор 2 в колонну 3.
Для предотвращения накопления инертных газов осуществляется
постоянный сброс газа перед подачей циркулирующей газовой смеси
на реакцию. Если содержание пропилена в газе, вводимом в колонну
3, не превышает 40 объемн. %, то получают оптимальный результат.
Из дегазационной колонны 4 реакционная смесь поступает в гид-
ролизер 5 через перепускное устройство, где добавляется 10—20%-
ное известковое молоко. Гидролизер представляет собой горизон-
76
4. Окись пропилена
тальный стальной резервуар, снабженный перегородками. В каждую
ячейку, образованную перегородками, подается пар и реакционная
смесь нагревается до 60—80 °C. При этом происходит моментальное
превращение пропиленгидрина в окись пропилена, которая испа-
ряется и вместе с водяным паром и дихлорпропаном поступает в де-
флегматор 6, где часть водяного пара конденсируется и возвращается
в цикл. В холодильнике 7 дистиллят охлаждается, после чего напра-
вляется для хранения в промежуточную емкость 8.
В ректификационной колонне 9 с 50 теоретическими тарелками
фракционируют сырую окись пропилена. Из верхней части выходит
при 34 °C 98 %-ная окись пропилена, а в кубе колонны остаются вода,
дихлорпропан с небольшими примесями дихлордиизопропилового
эфира, пропионовый альдегид и пропиленгликоль. Дихлорпропан
из куба колонны периодически удаляют осушкой хлоридом кальция.
Выход окиси пропилена составляет около 80% по отношению к ис-
ходному хлору. Установка работает непрерывно и, несмотря на
небольшие размеры аппаратов, имеет высокую производительность.
4.2. ПОЛУЧЕНИЕ ОКИСИ ПРОПИЛЕНА ПРЯМЫМ ОКИСЛЕНИЕМ
Если получение окиси этилена с давних пор осуществляли пря-
мым окислением на серебряных катализаторах, то современное про-
изводство окиси пропилена все еще базируется на классическом
методе хлоргидрирования. Выход окиси пропилена при прямом оки-
слении пропилена до сих пор остается незначительным, хотя работы
по исследованию этого процесса начались свыше 30 лет назад:
1935 г. — Societe Francaise de Catalyse [321 — каталитическое
окисление в присутствии золота, сплавов серебро — золото и зо-
лото — серебро — марганец;
1936 г. — Пигулевский (СССР) [36] — окисление пропилена над
катализатором NH4VO3;
1950 г. — Union Carbide, Linde Air — окисление пропилена
без катализатора в башнях с кольцами Рашига;
1952 г. — General Aniline and Film Corp. — окисление смеси
олефинов в кипящем слое в присутствии серебра, сплавов серебро —
платина и серебро — золото — марганец;
1954 г. — Celanese Corp, of America — окисление олефинов в па-
ровой фазе или смесей олефинов и парафинов с помощью катализа-
торов;
1957 г. — National Research Corp. — окисление смеси олефинов
с пропионатом марганца.
Опыты по исследованию прямого окисления пропилена в окись
пропилена с получением хорошего выхода продолжаются и в на-
стоящее время. Хотя эти поиски не принесли пока желаемого ре-
зультата, тем не менее можно надеяться, что когда-нибудь процесс
прямого окисления с высоким выходом будет освоен.
4.2. Получение окиси пропилена прямым окислением
77 ''
4.2.1. Окисление пропилена в жидкой фазе под давлением
Первые опыты по прямому окислению пропилена в окись про-
пилена в жидкой фазе были описаны в 1957 г. в американском
патенте [38]. Окислению подвергали либо чистый пропилен при
21,1 кгс/см2 и 175—225 °C, либо смесь пропилена и пропана (1 : 1)
при 52,7 кгс/см2 и 160—180 °C в присутствии пропионата марганца,
растворенного в бензоле. Во избежание гидролиза окиси пропилена '
в пропиленгликоль необходимо работать с инертным растворителем.
По отношению к израсходованному пропилену выход составил 40%
окиси пропилена и 20% пропиленгликоля наряду с кислотами, окисью
углерода и метилформйатом. Добавка пропана повышает выход,
хорошее действие оказывает также добавка нитробензола, подавля-
ющего полимеризацию во время окисления. В качестве растворите-
лей используют соединения, которые инертны к кислороду и окиси
пропилена, не смешиваются с водой и растворяют большие количе-
ства пропилена: бензол, изооктан, неопентан, изододекан и т. д.
Можно проводить окисление в толуоле при повышенных температурах
(210—230 °C) при введении водного фосфатного буфера (pH = 6,5)
[39].
Хорошему окислению пропилена в ароматических углеводородах
способствует добавление к реакционной смеси Na2CO3 [40] или
К2СО3 [41] для нейтрализации образовавшихся кислот. По первому
методу при конверсии 12,5% получают 28,8 мол. % окиси пропилена
и 18 мол. % пропиленгликоля наряду с кислотами и эфирами. Для
инициирования реакции рекомендуется вводить соединения с карбо-
нильными или карбоксильными группами, например пропионовый
альдегид или ацетальдегид. По второму методу. [41], благодаря спе-
циальной конструкции реакционной камеры, получают высокую
конверсию (94,2%) и высокий выход на единицу объема в единицу
времени: 100 г/ч окиси пропилена и 50 г/ч пропиленгликоля.
Имамура с сотрудниками [42, 43] провел обширные исследования
жидкофазного окисления пропилена. При 145— 220 °C и давлении
2—10 кгс/см2 нафтенат марганца является наилучшим по селектив-
ности катализатором для образования окиси пропилена. Индукцион-
ный период сокращают повышением парциального давления кисло-
рода и концентрации пропилена, а также добавкой ранее получен-
ного оксидата.. Увеличение температуры реакции оказывает анало-
гичное действие (продолжительность индукционного периода при
145 °C составляет 6 ч, при 220 °C — 1 мин). Окисление протекает
по радикальному механизму, обрывают цепь перекисные соединения
типа СН2=СН—СН2О2. Ниже 190 °C энергия активации составляет
16,1 ккал/моль, выше 190 °C — 0 ккал/моль. Скорость поглощения
кислорода не зависит от его парциального давления (при 2—5 кгс/см2)
и пропорциональна количеству растворенного в бензоле пропилена
в 1,3—1,4 степени, добавка пропана не оказывает никакого влияния.
78
4. Окись пропилена
При 10—15 %-ной конверсии наивысшую селективность для
реакций получения окиси пропилена и пропиленгликоля достигали
при давлении 22—30 кгс/см2, получая 700 г окиси пропилена и про-
пиленгликоля на 1000 г израсходованного пропилена [44].
Пропилен в растворе можно превращать в окись пропилена и без
катализатора [45]. На рис. 30 представлена схема такой установки.
В реактор 2, заполненный нагретым до нужной температуры раство-
ром пропилена в углеводороде, вводят кислород или воздух. Выходя-
щая из реактора смесь охлаждается в теплообменнике 3 до 10—20 °C.
Окись
1 — подогреватель; 2 — реактор; 3 — теплообменник;
4 — флорентийский сосуд; 5 — экстрактор; в—отстойник.
После теплообменника кислые фракции (жирные кислоты и т. д.),
получающиеся при окислении, нейтрализуются водным раствором
едкой щелочи. Во флорентийском сосуде 4 водный щелочной экст-
ракт отводится снизу. Верхний слой, содержащий раствор углеводо-
родов, подается в экстрактор 5 для экстрагирования водой при 10—
20 °C. По меньшей мере 50—70% окиси пропилена и пропиленгли-
коля поглощается водой. Водный экстракт выводится из экстрактора
и фракционируется в колоннах для получения чистой окиси пропи-
лена и пропиленгликоля. Участвовавший в реакции углеводород
возвращается через отстойник 6 в подогреватель 1. Таким же образом
непрореагировавший газ возвращается в процесс. Все агрегаты уста-
новки за исключением ректификационных колонн работают под
давлением.
Процесс характеризуется следующими параметрами:
Давление, кгс/см2'...................................... 57
Температура, 8С..................................... 167—179
Подача кислорода, объемн. ............................. 216
Подача пропилена, вес. ч............................... 233
4.2. Получение окиси пропилена прямым окислением
79
Подача з%-ного раствора К2СО3, вес. ч............... 1437
Подача воды на экстракцию при 10 9С, вес. ч......... 5400
Конверсия, %
кислорода ...................................... 99,8
пропилена«...................................... 94,2
Выход окиси пропилена и пропиленгликоля, % ... 46,3
Содержание окиси пропилена, %........................ 70
Окись пропилена можно выделить путем экстрагирования водой
под давлением при 20 °C [45].
Окисление без давления при 200 °C можно проводить во многих
растворителях (диметилфталат, силиконовое масло, фторуглеводо-
роды, дибутилсебацинат и пр.), но конверсия составляет только
1,0—1,5%/ч, селективность не выше 55—60% [46].
Для ркисления под давлением рекомендуются нитрилы, напри-
мер ацетонитрил [47] (при 200 °C и 51 кгс/см2 выход окиси пропилена
составляет 18%). При использовании в качестве растворителей по-
лиацилированных сложных эфиров, например пропиленгликоль-
диацетата, конверсия через 5 мин достигает 20%; при этом полу-
чается смесь следующего состава (в мол. %):
Окись пропилена . . . 44,6
Ацетальдегид ......... 4,2
Метиловый спирт . . . 10,2
Метилацетат v......... 1,8
Ацетон .............. 2,2
Муравьиная кислота . . 0,4
Уксусная кислота . . . 16,5
Вода ..................12,6
Для окисления в жидкой фазе предложены самые различные ка-
тализаторы, например разлагающиеся при нагреве соли Со, Си,
Мн, V или Сг с карбонатом или ацетатом свинца или бария в качестве
промотора [49]. После 10-минутного окисления при 200 °C и давле-
нии 56 кгс/см2 в бензоле из 465 г пропилена в присутствии таких
катализаторов получают следующие количества (в г) окиси пропилена
С3Н6О и пропиленгликоля С3Н8О2:
с»н«о с3н,оа
Со(СН3СОО)2 .............. 265 61
. РЬСО3 .................... 190 73
Со(СН3СОО)2+РЬСО8 . . . '. 312 62
Со(СН3СОО)2+ВаСО3 .... 307 65
’ Мп(СН3СОО)2'+Ва(СН3СОО)2 298 60
При окислении под давлением в бензоле рекомендуется добавлять
фталоцианин кобальта, никеля и особенно меди [50] (при 175 °C
через 10 мин получают 40,1% окиси пропилена и 2,2% пропиленгли-
коля).
Предложен еще ряд растворителей: эфир борной кислоты в при-
сутствии ацетата ртути (180 °C, 3 кгс/см2) [51]; катализаторы кобальта,
например каприлат Со, и частично смешанный с водой раствори-
тель, например смесь хлорбензола и ацетона (при 150 °C и 49 кгс/см2
получают 62,4% окиси пропилена наряду с пропиленгликолем и дру-
гими продуктами) [52].
80
4. Окись пропилена
Как уже упоминалось, добавки альдегидов сильно снижают
индукционный период и, следовательно, температуру реакции. На-
пример, при совместном окислении пропилена и ацетальдегида при
100^°С и 80 кгс/см2 получили следующие результаты [53]:
Конверсия, %
пропилена 43
ацетальдегида ...» 88
Выход, мол. %
окиси пропилена . . 57
уксусной кислоты . . 70
Окисление пропилена в метилацетате в присутствии ацетальде-
гида [54] при 200 °C привело к получению 49,4 мол. % окиси про-
пилена. Ультрафиолетовое облучение и перекисные алкильные
и ацильные соединения ускоряют окисление в присутствии ацеталь-
дегида [55]. Добавки перекиси водорода или алкилгидроперекисей
в присутствии солей Си, Мп, N1 или Со также ускоряют окисление
[56].
В некоторых патентах предлагается проводить окисление про-
пилена в окись пропилена перекисными соединениями по аналогии
с эпоксидированием по Прилежаеву. Рекомендуются следующие
перекисные соединения: альдегидмоноперацетаты (например, ацет-
альдегидмоноперацетат) при 70 °C [57]; надуксусная кислота в аце-
тоне и уксусной кислоте или в метилале под давлением и при 30—
80 °C [58]; перекись mpem-бутила в mpem-бутиловом спирте при
80 °C и 28 кгс/см2 в присутствии нафтената Мо (выход окиси пропи-
лена 86% [59]) или МоО3 при 1ГО °C (селективность 86,7% [601);
аралкилгидроцерекиси в присутствии катализатора Mo, V или W
(при использовании гидроперекиси кумола и нафтената Мо конверсия
при 90 °C через 1 ч составляет 92,7%, селективность 98% [61]);
образующаяся из ацетальдегида надуксусная кислота без катализа-
тора [62, 63] или в присутствии СоС12, FeCl3 или CuBr2 (при 56—
200 °C и 31—42 кгс/см2 образуется максимально 73,5% окиси про-
пилена) [64].
Можно применять предварительное переокисление циклогекса-
нола в присутствии СаСО3 и перекиси циклогексанона при 120 °C,
а также реакцию с пропиленом в присутствии фталевого ангидрида
[65]. Наконец, можно окислять пропилен совместно с органическим
соединением, способным образовывать гидроперекись (насыщенный
углеводород, алкилбензол, карбонильное соединение), например,
при 80 °C и 50 кгс/см2 совместно с ацетальдегидом, который перехо-
дит в уксусную кислоту[66]. Прямое эпоксидирование пропилена
осуществляют с помощью Н2О2 при 50г-70 °C в присутствии уксусной
кислоты и кремнефосфорной кислоты [67].
На фирме Monsanto Со. работает пилотная установка, где пропи-
лен окисляется надуксусной кислотой в смесь окиси пропилена
и пропиленгликоля, выход 60%.
4.2. Получение окиси пропилена прямим окислением
81
4.2.2. Окисление пропилена в газовой фазе
До сих пор не удалось окислить пропилен в окись пропилена
на серебряных катализаторах с таким же хорошим выходом, как в слу-
чае превращения этилена в окись этилена. При 130—260 °C и вре-
мени контакта 0,6—6 с получали менее чем 0,07% окиси пропилена
[29, 30]. Основными продуктами реакции были СО2 и вода. Введение
промоторов должно улучшать выход. Для этого рекомендуются до-
бавки СиО к катализатору из Ag2O; в этом случае при 160—180 °C
получается окись пропилена, при повышении температуры — акро-
леин [31]. Были предложены катализаторы на основе Ag/Au и
Ag/Au/Cu [32].
Катализаторы на основе AgO/BaO2 на различных носителях
теоретически должны дать конверсию 36% после непродолжительной
обработки хлором при 220 °C, однако выход составил только 18%
[37]. Хорошее действие оказывают катализаторы серебра при работе
в кипящем слое при 220—260 °C с промоторами и без них [34].
Катализаторы окиси Bi — Р — Мо приводят к образованию
небольшого количества окиси пропилена наряду с акролеином [35].
Ванадаты не способствуют образованию окиси пропилена из пропи-
лена [36]. В ряде патентов описано прямое окисление пропилена
в смесь различных продуктов, в том числе в окись пропилена. При
330—370 °C под давлением до 5 кгс/см2 и при конверсии 12,8% вы-
ход окиси пропилена составил 24 мол. % наряду с другими продук-
тами [33]. При 315 °C в исходном газе находилось только 2,7% окиси
пропилена наряду с 3,4% ацетальдегида и 0,7% акролеина. Хорошее
действие оказывала добавка галогенсодержащих соединений.
По данным фирмы Farbenfabriken Bayer [68] в окислительный
реактор, состоящий из подогревательного устройства, реакционной
камеры емкостью 0,8 л и холодильника, при 9 кгс/см2 подают каждый
час 20 м3 газовой смеси с содержанием 45 объемн. % пропилена
и 5 объемн. % кислорода (остальное составляет преимущественно
азот, окись и двуокись углерода). В подогревателе поддерживается
температура 280—300 °C, в реакционной камере она повышается до
430—450 °C. В результате реакции окисления содержание кислорода
падает до 0,5—1 объемн. %. Выходящая из окислительной печи
реакционная смесь охлаждается и промывается водой под да-
влением.
Из оставшихся газообразных фракций, содержащих наряду
с пропиленом кислород, азот, окись и двуокись углерода, выделяют
окись углерода и превращают ее в двуокись, которую затем удаляют
промывкой щелочью. В оставшиеся газы добавляют отработанный
пропилен с кислородом, и смесь снова подают через компрессор в оки-
слительный реактор. При дистилляции экстракционной воды при
нормальном давлении на 1000 кг израсходованного пропилена полу-
чают следующие продукты окисления (в кг):
6 Заказ 399
82
4. Окись пропилена
Окись пропилена .... 270
Окись этилена......... 24
Ацетальдегид .........330
Пропионовый альдегид 25
Акролеин................73
Ацетон .................16
Смесь вусококипягцих ве-
ществ .............. 62
Выход в единицу времени на единицу объема для окиси пропилена
составляет 400—450 г/(л-ч).
Аналогичный метод описан в американском патенте [69]. Про-
пилен и смесь пропилена с пропаном окисляли при 340 °C (затем
температура повышалась до 450—510 °C); получали 8—11,5 мол. %
окиси пропилена наряду с пропионовым альдегидом, акролеином
и гидроксиацетоном. В качестве разбавителя предложено исполь-
зовать для окисления водяной пар [70], что дает при 215—260 °C
19,7% смеси окиси пропилена и пропиленгликоля. Радиационное
облучение повышает выход спиртов, альдегидов и окиси пропилена
при окислении пропилена воздухом [71]. Окись пропилена наряду
с другими продуктами получается также и при окислении пропана
[72]. На фирме ICI (Англия) работает опытная установка по прямому
окислению пропилена [73].
Всеобщее внимание привлекли публикации и патенты [74—84],
в которых описан метод одновременного получения окиси пропилена
и стирола. В этом процессе исходными продуктами являются
этилбензол и пропилен. Этилбензол при 130 °C и давлении около
3,5 кгс/см2 в присутствии катализатора нафтената молибдена под
воздействием кислорода превращается в гидроперекись а-этилбен-
зола. При одноразовом проходе конверсия достигает 13%, а выход—
84%. Из гидроперекиси а-этилбензола при взаимодействии с про-
пиленом при 110 °C в присутствии нафтената молибдена образуются
фенилметилкарбинол и окись пропилена. В результате отщепления
воды от фенилметилкарбинола получается стирол. При использова-
нии в качестве катализатора двуокиси титана и температурах 182—
282 °C (оптимально 200—250 °C) выход стирола составляет 80—95%.
В процессе протекают следующие реакции:
СН2-СН3
ООН
I
сн—сн3
О2; 130 °C; 3,5 кгс/сма ,
ООН
I
СН-СН3
+ сн2=снсн3 —>
4.2. Получение окиси пропилена прямым окислением
83
О
С---СН-СН3
но °с
ОН
сн=сн2
I
TiO2, 182-282 °C
+ н20
Подобные результаты получают при использовании гидропере-
киси кумола (образуется а-метилстирол). Этот метод подробно описан
Ландау (74].
Реакция между' гидроперекисью и пропиленом дает лишь незна-
чительное количество окиси пропилена. Применение катализатора '
существенно улучшает положение. Ниже приведены результаты вза-
имодействия гидроперекиси а-этилбензола и пропилена при 110 °C
в присутствии 0,002 моль/моль ROOH нафтенатов различных метал-
лов (селективность -95 моль окиси пропилена/моль прореагировав-
шего пропилена):
Нафтенат.....................Мо
Конверсия, %.................97
Селективность, моль окиси
пропилена/моль прореаги-
ровавшей ROOH ...............71
W Ti Nb Re Та
83 54 22 25 100
65 55 20 23 10
Из молибденовых соединений можно употреблять как раствори-
мые, так и нерастворимые. Кроме гидроперекиси а-этилбензола
применяют гидроперекись кумола, гидроперекись пгрепг-бутила и др.
Реакция протекает по ионному механизму. Оптимальные условия
реакции:
Температура, °C ................................... 80—130
Продолжительность, ч................. 2—3
Давление, кгс/см2 ...................... 17,5—70
Соотношение олефины : гидроперекись (2 4-6) : 1
Концентрация катализатора, моль/моль
ROOH .............................. 0,001—0,006
Ниже приведена схема совместного получения окиси пропилена
и стирола:
Возвратный этилбензол
6*
лен 4? катализатор
84
4. Окись пропилена
Установки для комбинированного синтеза окиси пропилена и сти-
рола будут построены в США (мощность 80 000 т) [82] и в Испании
(мощность 24000 т) [83].
Фирма Arco — Halcon внедрила в производство аналогичный
метод получения mpem-бутилового спирта и окиси пропилена с ис-
пользованием гидроперекиси mpem-бутила [84].
4.3. СВОЙСТВА ОКИСИ ПРОПИЛЕНА
Ниже приведены физико-химические свойства окиси пропилена:
Температура плавления, ?'С ...................... —112,1
Температура кипения, 9С.............................. 34,2
Плотность р2° ...................................... 0,8313
Показатель преломления п^............................ 1,363
Дипольный момент, D............................. . 2,00
Вязкость при 25 9С, сП .............................. 0,28
Абсолютная вязкость) сП ............................ 0,38
Оптическое вращение (+)-формы [а]|£, 9.............. 11,6
Критическая температура, °C ....................... 209,1
Критическое давление, кгс/см2 ...................... 50,2
Теплота испарения в точке кипения, ккал/моль ... 6,6
Теплота сгорания (жидкости) при постоянном объеме
ккал/моль....................................... 453,1
кал/г ........................................... 7771,3
Удельная теплоемкость, кал/(г • °C) . .......... 0,51
Коэффициент теплового расширения, 1/еС......... 0,00151
Пределы взрываемости в смеси с воздухом, объемн. %
верхний ...»..................................... 36,8
нижний ........................................ 2,8
Температура вспышки, °C ..................... . . —44
Давление пара окиси пропилена при различных температурах
приведено в работах [85, 86].
Окись пропилена смешивается с обычными органическими рас-
творителями неограниченно. В воде при 20 °C растворяется 40,5 вес. %
окиси пропилена, а в окиси пропилена растворяется при 20 °C
12,8 вес. % воды. При более низких температурах выкристаллизо-
вывается гидрат С3НвО-16Н2О (точка затвердевания — 3 °C). При
повышенном давлении образуются азеотропные смеси с минималь-
ной температурой кипения, например при 3,1 кгс/см2 с 0,1 % воды
и при 5,2. кгс/см2 с 0,2% воды.
Химические свойства окиси пропилена подобны свойствам окиси
этилена. Она тоже реагирует с соединениями, имеющими активные
атомы Водорода, например с водой дает пропиленгликоль. Гидрата-
ция окиси пропилена легко идет при обычной температуре в присут-
ствии щавелевой кислоты в качестве катализатора, которую впослед-
ствии легко можно выделить в виде оксалата кальция. Реакция про-
4.3- Свойства окиси пропилена
85
Таблица 8
Скорость гидролиза окиси этилена и окиси пропилена при 20 °C
Концентрация окиси олефина, г/100 мл pH Л-10’, моль/(л-мин)
Окись пропилена Окись этилена
0,1 2,22 10 2 .
0,3 1,82 32 6
0,5 1,64 52 10
0,7 1,54 73 13
текает в кислом растворе почти в 5 раз быстрее, чем в случае с окисью
этилена. В табл. 8.указаны скорости гидролиза окисей этилена и про-
пилена.
Во всех других реакциях окись пропилена в большинстве слу-
чаев более инертна по сравнению с окисью этилена. Соотношение
образующихся при гидролизе моно-, ди- и тригликолей зависит
от соотношения вода : окись пропилена (табл. 9).
Таблица 9
Зависимость состава продукта гидролиза от соотношения
вода: окись пропилена
Соотношение вода : окись пропилена Концентра- ция раствора окиси пропи- лена, г/100 мл Конверсия окиси пропилена в гликоли, % Содержание гликолей в продукте гидролиза, вес. %
моно- Ди- три- МОНО- ДИ- три
5 :1 48 60,0 26,0 14,0 63,5 24,0 12,5
9 : 1 31 73,5 21,0 5,5 76,0 19,0 5,0
12 : 1 26 79,0 18,0 3,0 81,0 16,5 2,5
20: 1 18 87,0 12,0 1,0 88,5 . 10,5 1,0
25 :1 15 90,0 9,0 1,0 91,0 8,0 1,0
Благодаря несимметричному строению молекулы окись пропилена
обладает большими реакционными возможностями. Если полученная
из пропилена и хлорноватистой кислоты смесь хлоргидрина состоит
из 95% СН3СН(ОН)СН2С1 и 5% СН3СНС1СН2ОН, то в результате
присоединения соляной кислоты и окиси пропилена получают 75%
СН3СН(ОН)СН2С1 и 25% СН3СНС1СН2ОН. При реакции окиси
пропилена с водным аммиаком образуется, напротив, только изо-
мерный моноизопропаноламин CH3CH(OH)CH2NH2 и большее или
меньшее количество ди- и триизопропаноламина в зависимости
от соотношения исходных продуктов.
При каталитическом присоединении спиртов и фенола в резуль-
тате действия щелочью почти всегда образуется первичный моно-
86
4. Окись пропилена
эфир пропиленгликоля CH3CH(OH)CH2OR. Кислотная или неката-
литическая реакция дает кроме того и вторичный эфир пропиленгли-
коля СН3СН(ОК)СН2ОН.
Полимеризация окиси пропилена приводит к образованию поли-
пропиленгликоля. Изомеризация окиси пропилена в пропионовый
альдегид не представляет затруднений на таких катализаторах, как
квасцы, Cr2O3 — CdCl2 — CdO, Cr2O3 — WO3 — Fe2O3 или Cr2O3 —
WO3. При использовании Li3PO4 [87—96] или Li3AsO4 [94] можно
перегруппировать окись пропилена в аллиловый спирт. Остальные
катализаторы для превращения окиси пропилена в аллиловый
спирт и пропионовый альдегид были исследованы Полковниковой
[97] (табл. 10).
Таблица 10
Результаты превращения окиси пропилена в аллиловый спирт
на различных катализаторах
Катализатор Температур а перегруппи - ровки, °C Йремя контакта, с Конверсия, % Выход, % Соотноше- ние аллило- вый спирт : пропионовый альдегид
аллиловый спирт пропионовый альдегид
UO3 400 32 5,4 42 13 3,2 : 1
UsOg 400 30 33 16 7 2,3: 1
МоО3 350 28 21 18 5 3,6: 1
CraO3+SnO2 •350 26,5 25 52 7 7,4:1
Cr2O3+NiO 300 35 31 48 17 2,8:1 1
СеО2 400 8 11 12 18 0,7:1
СоО 400 10 10 35 11 3,2:1 |
FeO 400 22,5 90 11 20 0,5 : 1 - ]
В4С 400 12 19 14 28 0,5:1 '
Кокс 400 17,5 19 31 15 2:1
4.4. ПРИМЕНЕНИЕ ОКИСИ ПРОПИЛЕНА
Обзор применения окиси пропилена в США [98] для получения
различных веществ приведен ниже (в тыс. т.):
1959 1960
Пропиленгликоль............................ 59 70,3
Полипропиленгликоль и продукты присоеди-
нения окиси пропилена для производства
пенополиуретана ........................; 20,4 31,8
Полипропиленгликоль в качестве тормозной
жидкости............................... • • 9,05 9,5
Поверхностно-активные вещества ...... 5,45 5,9
Прочие соединения (изопропаноламин, дипро-
пиленгликоль и др.) ...................... 11,3 12,7
Итого . . . 105,2 130,2
4.4. Применение окиси пропилена
87
Основное количество пропиленгликоля идет для производства
полиэфиров [98] (в тыс. т):
1959 1960
Полиэфиры................................ 19,5 24,5
Пластификаторы........................... 2,7 2,7
Пластификаторы для целлофана............. 8,2 9,1
Тормозная жидкость ........................... 4,5 4,5
Фармацевтические и косметические средства . . 15,0 15,9
Прочие цели (например, увлажнение табака) 5,0 5,5
На экспорт............................... 6,7 6,7
Итого ... 61,6 68,9
Полипропиленгликоль (диапазон молекулярных весов 400—
2000) [99], получаемый полимеризацией окиси пропилена в щелоч-
ной или кислой среде, является важным промежуточным продуктом
для производства пенополиуре-
танов, алкидных смол, эмуль-
гаторов, деэмульгаторов, сма-
зочных средств, тормозных
жидкостей. Дипропиленгликоль
отдельно и вместе с диэтилен-
гликолем используется при по-
лучении типографских красок
и в качестве гидравлической
жидкости с низкой температу-
рой затвердевания. Он обла-
дает незначительной токсич-
ностью по сравнению с эти-
ленгликолем, что позволяет
применять его при изготовлении
фармацевтических и косметиче-
ских средств, а также пищевых
продуктов. Смесь полиэтилена
с полипропиленгликолем яв-
ляется интересным исходным ве-
ществом для получения неионо-
генных детергентов и специаль-
Рис. 31. Схема получения триизопро-
паноламина с образованием моно- и
диизопропаноламина:
1 — добавление моноизопропаноламина; 2 —
аммиак; 3 —добавление диизопропаноламина;
4 — моноизопропаноламин; 5 — диизопропа-
ноламин; 6* — триизопропаноламин.
ных смазочных масел.
Как уже упоминалось, ступенчатая реакция окиси пропилена
с аммиаком дает при 50—100 °C моно-, ди- и триизопропаноламины
[100], причем их равновесный выход зависит от соотношения окись
пропилена : водный аммиак. Скорость реакции образования моно-
и диизопропаноламинов аналогична скорости образования моно- и
диэтаноламинов, а триизопропаноламин получается значительно
медленнее триэтаноламина. Добавление моно- и диизопропаноламинов
/
88
4. Окись пропилена
к реакционной смеси для дальнейшего обмена приводит к преимуще-
ственному образованию триизопропаноламина (рис. 31). Вводимое
количество моно- и диизопропаноламинов очень высоко и пропорцио-
нально количеству свободного аммиака и моноизопропаноламина [13].
Изопропаноламины — интересные промежуточные продукты для
синтеза красителей, фармацевтических средств и, при известных
условиях, полиэфирных амидов.
Возможность изомеризации окиси пропилена с помощью Li3PO4
или Li3AsO4 в аллиловый спирт [101] и в пропионовый альдегид
[102] использована в промышленности (фирма Olin Mathieson Che-
micals Ltd. разработала на. основе этого метода процесс синтеза гли-
церина).
В последнее время все большее значение приобретает полимери-
зация окиси пропилена в окись полипропилена.
Сополимеризация окиси пропилена с другими ненасыщенными
эпоксисоединениями (эпоксибутадиен, аллилглицидиловый эфир,
окись винилциклогексена и пр.) в присутствии катализатора при-
водит к получению каучукоподобных полимеров [103, 104], которые
широко применяются.
ЛИТЕРАТУРА
1. Oser, Liebigs Ann. Chem. Spl., 1, 255 (1861).
2. К. К pa с су с кий, ЖРФХО, 34, 287—315 (1902).
3. Н. Schrader, Angew. Chem., 42, 541—546 (1929).
4. G. К i d d о о, Chem. Eng., 59, № 9, 149—168 (1952).
5. H. T о k u n o, Yukagaku, 14, № 8, 454—462 (1965); C. A., 64, 672h (1966).
6. H. Shingu, ibid., 8, 355—361 (1959).
7. J. В i g e о n, L'Ind. chim., 40, № 433, 221 (1953).
8. A. C. F у v i 1, Chem. and Ind., № 10, 384-388 (1964).
9. A. M а у, К. H. F r a n k e, Chem. Technik, 12, 59—60 (1960).
10. W. M ar ko wn iko w, Z. Chemie, 423 (1870); Liebigs Ann. Chem., 153,
251 (1870); A. M i c h a e 1, J. Prap. Chem., 60, № 2, 454 (1899).
11. G. С. I s г a e 1, J. Chem. Soc. (London), 1950, 1282—1286.
12. Англ. пат. 292066, 1927.
13. M. P. Ferrero et al., Ind. Chim. beige, 19, 133 (1954).
14. К. E. Murray, J. Council'Sci. Ind. Res., 17, 213—221 (1944).
15. L. Smith, S. S k у 1 e, Acta chem. Scand., 4, 39 (1950); 5, 1415—1416
(1951).
16. Франц, пат. 982969, 1951.
17. Франц, пат. 983910, 1951.
18. Англ. пат. 1016359, 1966; D. A. S. 1168406, 1963; франц, пат. 1357443,
1964; англ. пат. 974462, 1964; D. A. S. 1156394, 1963.
19. В. С. Э т л и с, Л. Н. Г р о б о в, ЖПХ, 32, 874—877 (1959).
20. D. A. S. 1063140, 1959.
21. Бельг, пат. 630446, 1963; D. A. S. 1211140, 1966; пат. США 3277189, 1966.
22. Пат. США 2566355, 1951.
23. Н. Т о k u п о et al., Yukagaku, 12, 97—102 (1963); реф. С. А., 59, 7453а
(1963).
24. Англ. пат. 738171, 1955.
25. Бельг, пат. 633518, 1963.
26. Па*. США 2436591, 1948.
Литература
89
27. D. Porret, Helv. chim. Acta, 30, 701—706 (1947); пат. США 2417685,
1947.
28. J. Myskowsij et al., Chem. stosowana, Ser. A10, 325 (1966).
29. Я. Б. Г о p о x а в а т с к и й, М. Я. Рубаник, Укр. хим. ж., 24,
63-67 '(1958).
30. Б. В. Суворов и др. ДАН СССР, 172, 1096-1098 (1967).
31. Пат. США 2985668, 1961.
32. Англ. пат. 451130, 1936; франц, пат. 785149, 1935.
33. Бельг, пат. 592446, 1960.
34. Пат. США 2600444, 1952; 2628965, 1953.
35. С. С. М с С a i n, G. W. G о d i n, Nature, 202, 692—693, 1964.
36. В. В. Пигулевский, Е. Я. Яржемская, Труды опытно-ис-
след. завода Химгаз. Материалы по крекингу и химической переработке
его продуктов, вып. 3, 1963, стр. 178—183. s
37. I. К a m i у a, Kogyo Kagaku Zasshi, 64, 1513—1514 (1961).
38. Англ. пат. 786301, 1957; 917926, 1936; D. A. S. 1043311, 1958; пат.. США
2780634, 2780635, 2784202, 1957.
39. Пат. США 2824119, 1958.
40. Англ. пат. 950669, 1964; D. A. S. 1192172, 1965; франц, пат. 1376471, 1964.
41. Франц, пат. 1334158, 1963.
42. J. Imamur a et al., Tokyo Kogyo Shikensho Hokoku, 59, № 10, 447—
457 (1964).
43. J. I m a m u r a et al., Kogyo Kagaku Zasshi, 67, № 7, 1026—1031 (1964).
44. F. L a n о s et al., Chim. et Ind. (Paris), 91, 47—56 (1965).
45. D. A. S. 1192171, 1965.
46. F. W. Meadow, K. U. I ng о 1 d. Can. J. Chem. Eng., 42, № 2, 86—
87 (1964).
47. Пат. США 3210380, 1965.
48. Бельг, пат. 628810, 1963; франц, пат. 1424689, 1966.
49. Англ. пат. 933548, 1963; D. A. S. 1150060, 1963; пат, США 3071601, 1963.
50. Японск. пат. 22575, 1965.
51. Пат. США 3210381, 1965.
52. D. A'. S. 1242588, 1967; франц, пат. 1386354, 1965.
53. Niederld. A. S. 6504592, 1965.
54. Франц, пат. 1367762, 1964.
55. Англ. пат. 963430, 1964.
56. Франц, пат. 1323787, 1963.
57. Англ. пат. 735974, 1955.
58. Англ. пат. 900836, 1962.
59. Бельг, пат. 641452, 1964.
60. Niederld. A. S. 6402137, 1964.
61. Niederld. A. S. 6500118, 1965.
62. D. A. S. 1240515, 1967.
63. D. A. S. 1225175, 1966.
64. Франц, пат. 1377981, 1964.
65. Бельг, пат. 638162, 1964; D. A. S. 1212057, 1966.
66. Франц, пат. 1397611, 1965.
67. Франц, пат. 1292304, 1961.
68. Бельг, пат. 623552, 1963; D. A. S. 1161551, 1964.
69. Англ. пат. 718603, 1954; пат. США 2689253, 1954.
70. D. М. Newitt, Р. S. М е и е, J. Chem. Soc. (London), 1946, 97—100.
71. Англ. пат. 882863, 1961.
72. Пат. США 3132156, 1964.
73. Chem. Industrie, 17, 309 (1965).
74. R. Landau et al., Proc. 7th World Petrol. Congr., Mexiko, v. 5, 1967,
p. 67—72.
75. Бельг, пат. 657838, 1966; Niederld. A. S. 6515037, 1966.
90
4. Окись пропилена
76. Франц, пат. 1462198, 1966.
77. Франц, пат. 1462490, 1966. .
78. Франц, пат. 1460573, 1966.
79. Франц, пат. 1495694, 1967.
80. Франц, пат. 1464239, 1967.
81. Франц, пат. 1487076, 1967.
82. Chem. Eng., 73, № 23, 102—103 (1966).
83. Chem. Eng. News, 45, № 16, 17 (1967); 43, № 43, 40-42 (1965).
84. European Chemical News (London), 11 № 284, 34 (1967).
85. T. Я. Bott, H.N. Sadler, J. Chem. Eng. Data, 11, 25 (1966).
86. R. W. Gallant, Hydrocarbon Processing, 46, № 3, 143—150 (1967).
87. L. G. Lu nds ted t et al., Ind. Eng. Chem., 43, 728—730 (1951).
88. П. Г. С e p г e e в и др. Хим. наука и пром., 2, 133 (1957).
89. Пат. США 2986585, 1961.
90. Англ, пат. 902953, 1962.
91. Канад, пат. 690646, 1964; D. A. S. 1197077, 1965; франц, пат. 1271563_
1962; швейц, пат. 403742,1966.
92. D. A. S. 1251308, 1967; франц, пат. 1344311, 1964.
93. Пат. США 3238264, 1966.
94. Пат. США 3209037, 1965.
95. Франц, пат. 1316720, 1963.
96. Франц, пат. 1496221, 1966.
97. А. Г. Полковников а, Л. А. Иванова, Хим. пром., 42, № 1,
16—19 (1966).
98. Chem. Eng. News, 38, № 41, 20 (1960).
99. Kirk — О th me r, Encyclopedia of Chemical Technology, N. Y., v. 7,
1951, p. 261—262.
100. Пат. США 1988225, 1935.
101. Пат. США 2426264, 1947.
102. Пат. ЩПА 2435460, 1948; 2521170, 1949.
103. Е. Е. Gruber et al., Ind. Eng. chem. Product Res. Development, 3,
194—199 (1964); Rev. gen. caout. plast., 41, 1811—1817 (1964).
104. L. Marker et al., Kautschuk, Gummi, Kunststoffe, 19, 337 (1966).
5. АКРОЛЕИН
Акролеин [1—4] — простейший ненасыщенный альдегид, обла-
дающий очень хорошей реакционной способностью благодаря нали-
чию двух функциональных групп (альдегидной группы и двойной
связи).
Акролеин был открыт примерно 100 лет назад Редтенбахером
при сухой перегонке жиров [5]. В течение многих лет существовал
лишь один метод получения акролеина — перегонка глицерина
в присутствии водоотщепляющих веществ [6—7]. Более полувека
работали над внедрением данного процесса в жидкой и газовой фазе
в промышленность, используя самые различные катализаторы (бор-
ную или фосфорную кислоту, фосфаты, окись алюминия и др.).
Однако высокая стоимость глицерина и низкий выход (не более
50%) не позволили применить этот процесс в промышленном мас-
штабе.
Получение акролеина в результате пиролиза при 500—550 °C
диаллилового эфира, являющегося промежуточным продуктом син-
теза аллилового спирта из аллилхлорида [8—10], ограничилось
только пилотной установкой мощностью 100 т/год, которая работала
на фирме Shell в 1946—1948 гг.
Получение, свойства и применение акролеина подробно описаны
в литературе [1—3]. В 1962 году появилась монография Смита об
акролеине и его производных [4].
Акролеин можно получить:
1) из глицерина
СН2(ОН)СН(ОН)СН2ОН СН2=СНСНО + 2Н2О
I
2) из диаллилового эфира
СН2=СНСН2-О-СН2СН=СН2 -515°£> СН2=СНСНО + СН2=СНСН2
3) из аллилового спирта [И]
СН2=СНСН2ОН °” •200°сч> СН2=СНСНО
92
5. Акролеин
4) из тетрагидрофурфурилового спирта [12]
СН2
н2с---сн2 А]Р01 350оС н2с/ \сн
I I А1Р-.°Л> 35°. с-> । || +н20
Н2Сч уСН-СН2ОН Н2С. уСН
о о
сн2
н2с/ Хен Rno—
I II — СН2=СНСНО + СН2=СН2 (выход 85%)
Н2С\ /СН
. о
5) из формальдегида и ацетальдегида
НСНО + СН3СНО СН2=СНСНО + Н2О + 19,5 ккал
6) из пропилена [26]
СН2=СНСН3 °2’ 1!атализат°Р-> сн2=снсно
Применение в промышленности нашли только два последних ,
метода, которые поэтому будут описаны более подробно.
5.1. СИНТЕЗ АКРОЛЕИНА ИЗ ФОРМАЛЬДЕГИДА
И АЦЕТАЛЬДЕГИДА
Метод был разработан фирмой Degussa, которая с 1942 г. произ-
водит акролеин в промышленном масштабе. С 1955 г. фирма Union
Carbide также получает акролеин по этому методу.
Формальдегид в виде 30%-ного водного раствора пропускается
вместе с эквимолекулярным количеством ацетальдегида при 300—
320 °C через силикагель, пропитанный 10% раствором силиката
натрия. Степень превращения составляет 45—52%, а выход дости-
гает 70—80% [13—15]. Непрореагировавшие альдегиды отделяются
перегонкой от акролеина и снова возвращаются в процесс. На рис. 32
изображена схема установки по синтезу акролеина из формальдегида
и ацетальдегида.
Чистый ацетальдегид и 30% водный раствор формальдегида
поступают из сборников 1 и 2 в смесительный резервуар с мешалкой
3 и затем в испаритель 4. Отсюда пары альдегида направляются через
перегреватель 5 в трубчатую контактную печь 6. Перегреватель
может обогреваться маслом, дифенильной смесью или газом. Пары
реакционной смеси через конденсатор 7 поступают в колонну 10.
После конденсатора 7 происходит сброс давления и газы дроссе-
лирования промываются водой для удаления остатков альдегида.
5-1. Синтез акролеина из формальдегида и ацетальдегида 93
производства акролеина
Рис. 32. Схема г^?—------------- ~—
из формальдегида и ацетальдегида:
1, 2 — сборники; з — смесительный резервуар;
4 — испаритель; ' - ...
тактная
Вода также поступает в колонну 10 на перегонку. Образующийся
в колонне 10 дистиллят с содержанием ацетальдегида и кротоно-
вого альдегида вводится в верхнюю часть колонны 11, где отгоняется
ацетальдегид, а снизу отводится акролеин. Отстой из колонны 10,
непрореагировавший 10%-ный формальдегид, подается в колонну
9 для перегонки под давлением. В результате получается концент-
рированный раствор формальдегида. Реакционная вода и вода из
раствора формальдегида сте-
кает снизу. Непрореагиро-
вавшие альдегиды возвраща-
ются через сборники 1 и 2
снова в цикл.
является
силикагель, пропитанный
10% раствором силиката на-
трия, оптимальные темпера-
туры для этого катализатора
303—320 °C. Поскольку об-
разование акролеина проте-
кает экзотермически, под-
вод тепла осуществляется
в трубчатой печи.
Смолообразование во вре-
мя реакции, которое нельзя
полностью ликвидировать,
вызывает необходимость пе-
риодической регенерации
катализатора примерно через
150 ч. Регенерация прово-
дится водяным паром и воздухом в
5 — перегреватель; в — кон-
печь; 7 — конденсаторы; з — промыва-
тель; 9—11 — колонны.
течение 24 ч при 500—550 °C.
Степень превращения в течение одного реакционного периода
в среднем для формальдегида составляет 50—52%, а для ацетальде-
гида 44—46%. При эквимолекулярных количествах формальдегида
и ацетальдегида выход в единицу времени на единицу объема до-
стигает 3,5 моль/(ч-л катализатора). Выход от теоретического до-
стигает 72—75% в расчете на формальдегид и 72—82% в расчете
на ацетальдегид.
В качестве побочных продуктов образуются метиловый спирт,
кротоновый альдегид и отработанный газ, которые можно без труда
удалить. Для устранения полимеризации акролеина при разделении
смеси нужно непрерывно подавать в перегонные колонны 10 и 11
ингибитор. Добавка 0,1% фенольного ингибитора (гидрохинон или
пирокатехин) делает акролеин устойчивым в течение года и исклю-
чает образование твердых осадков.
Предложен ряд других конденсационных катализаторов: ацетат
свинца [16], карбонат лития на силикагеле ‘ [17], двуоким титана
94
5. Акролеин
и КОН на SiO2 [18], катионсодержащий диатомит [19], очень ста-
бильный катализатор, получаемый кальцинированием смеси МоО3
и MgO с окисью цинка и другими веществами в качестве промоторов
[20]. При исследовании кинетики конденсации [21—23] показано,
что наиболее подходящим катализатором является CsOH.
5.2. ПОЛУЧЕНИЕ АКРОЛЕИНА ПРЯМЫМ ОКИСЛЕНИЕМ ПРОПИЛЕН А
В 1936—1939 гг. впервые установили [24], что при окислении
пропилена в присутствии сульфата ртути в сернокислом растворе
получается очень незначительное количество акролеина.
Окисление пропилена в акролеин на катализаторах окиси меди
(I) [26, 181] и молибдата висмута проводится в промышленном мас-
штабе. О механизме реакции до сих пор известно лишь то, что первой
ступенью реакции является окислительное дегидрирование.
Исследования пропилена, меченного 13С, показали, что при окисле-
нии окисью меди (I) вначале отщепляется атом водорода от группы СН3
с образованием симметричного промежуточного продукта, который
может окисляться с обоих концов [182]:
СН2=СН-СН3 —г- СН2—СН—СН2 —
—> сн2=сн-сн сн2=сн-сно
Адамс и Яннингс [79] проводили опыты с дейтерированным про-
пиленом, который они окисляли в акролеин на окиси меди (I) и мо-
либдате висмута. Механизм окисления пропилена одинаков для
обоих катализаторов. Водород или дейтерий отщепляли от метильной
группы и затем еще раз удаляли водород или дейтерий от одного
из концов. Тем не менее осталось неясно, по какому механизму про-
исходит присоединение.
Адамс с сотрудниками [183] изучали кинетику окисления про-
пилена на катализаторах молибдата висмута. Они нашли, что по
отношению к пропилену реакция будет первого порядка и не зависит
от кислорода и других продуктов. Энергия активации составляет
при 350—500 °C около 20 ккал/моль. Молекулярный водород не
влияет на образование акролеина и не окисляется. Наилучшая селек-
тивность в отношении образования акролеина достигается при ис-
пользовании катализаторов молибдата висмута при 490—520 °C.
Побочными продуктами будут угольная кислота, формальдегид
и ацетальдегид.
5.2.1. Окисление пропилена на медьсодержащих катализаторах
В 1942 г. американские ученые обнаружили, что при пропуска-
нии пропилена через окись меди (I) и селенид серебра на асбесте
при 295 °C образовывалось значительное количество акролеина [25].
5.2. Получение акролеина прямим окислением Пропилена 95
Это привело к разработке нового процесса в 1946—1947 гг. фирмой
Shell Development Со. [26]. Пропилен вместе с воздухом и водяным
паром пропускали при 370—400 °C и небольшом давлении в присут-
ствии 0,03 мол. % изопропилхлорида через окись меди (I) на кар-
биде кремния. Максимальный выход акролеина равнялся 51%. По-
вышение давления кислорода увеличивает выход акролеина до 68—
81% [27]. В промышленном масштабе конверсия пропилена соста-
вляет 14% при 368 °C и объемном соотношении пропилен : водяной
пар : кислород = 4,4 : 4,7 : 1 в присутствии 0,4% окиси меди (I)
на карбиде кремния. Выход акролеина колеблется в пределах 65—
85% [28].
Образование акролеина происходит одновременно с полным оки-
слением до двуокиси углерода и воды, при этом катализаторы с вы-
соким содержанием меди способствуют окислительной деструкции.
Побочными продуктами реакции будут формальдегид, ацетальдегид,
окись углерода, органические кислоты, карбонильные соединения
и полимеры.
Изучение механизма реакции показало, что Си2О является эф-
фективным катализатором, в то время как СиО приводит к полному
окислению пропилена в СО2, а металлическая медь не активна. Из
окисей металлов Си2О является единственным катализатором, кото-
рый окисляет избирательно [29]. Катализаторы V2O5, WO3, МоО3
и Сг2О3 на пемзе (особенно V2O5 и WO3) хотя и давали значительное
количество акролеина, но реакция протекала неселективно [30].
Оптимальным количеством катализатора — окиси меди, нанесенной
на окись алюминия, силикагель, пемзу или карбид кремния, — яв-
ляется 1—1,5% Си. При более высоких концентрациях на катализа-
торе присутствовала частично металлическая медь, способствующая
образованию СО2 [31—35]. Независимо от первоначальной формы
образуется совершенно определенная смесь Си — Си2О — СиО [36—
40], которая прежде всего зависит от соотношения пропилен : ки-
слород. Например, при используемом в промышленности соотно-
шении С3Н6 : О2 = 7,5 : 1 и 1,5% СиО на карбиде кремния полу-
чается смесь из 70% Си2О и 30% СиО. При соотношении С3Н6 : О2 =
= 30 : 1 уменьшается выход акролеина из-за образования меди.
Наиболее подходящими носителями являются пористое стекло
[41], пемза и особенно карбид кремния [39, 42]. Рекомендуется при-
менение пористой меди при'300 °C (конверсия 2,5%, выход 54%),
конверсия возрастает с увеличением поверхности катализатора [43].
Обычная окись алюминия не может быть носителем, так как способ-
ствует образованию СО2 и воды, тем не менее предложен катализатор
из 1,5% СиО на неактивированной окиси алюминия, действие кото-
рого можно улучшить добавкой Fe2O3, ThO2 или МоО3 [44]. Про-
моторами для катализатора Си2О (на глиноземе или SiC) могут быть
10~4 — 10"6 моль брома, иода, бромистого или йодистого водорода,
бром- или иодсодержащих углеводородов. Максимальный выход
96
5. Акролеин
акролеина (73%) получали при 391 °C и добавке иода [45]. Аналогич-
ное действие оказывает добавление к катализатору СпО на силика-
геле галогенида меди, особенно СиВг2 (выход 12,5%, селективность
30—40%) [46] и смеси щелочных окисей и галогенидов (например,
Na2O и NaCl) [47].
Медьсодержащие катализаторы известны как эффективные до-
бавки к селену или селенсодержащим соединениям (работы амери-
канской фирмы Distillers Со. Ltd.). Исследования советских авторов
[48] показали, что селен является переносчиком кислорода, при этом
Se и SeO2 вместе с СиО и Си2О образуют окислительно-восстано-
вительную систему [49], а образование СО2 подавляется. В качестве
катализаторов рекомендуются CuO, CuSO4 и CuSeO3 на А12О3,
SiC, пемзе, стеклянном порошке, силикагеле, которые обрабатыва-
ются парами селена. Для катализаторов, содержащих 12—16% Си,
оптимальными условиями будут: температура 325 °C, время контакта
2—2,5 с; при этом достигается выход 81—82%, конверсия 90—95%.
В 1949 г. появился первый патент фирДы Distillers Со. Ltd.
[50], в котором предложен катализатор из Ag2Se и СиО на асбесте
(350 °C, выход 15%) и катализатор СиО — Se (выход 12%) [51].
Газообразный пропилен можно также пропустить перед окислением
через селен. Незначительное количество унесенных селеновых ча-
стичек достаточно для каталитического воздействия. При 320 °C
получали 77% акролеина на СиО — А12О3 (конверсия 84%) [52].
Катализатор СиО на силикагеле повышает выход акролеина с 3,5
до 23% при наличии следов селена или SeO2 [53—54]. Кроме СиО
предложены окиси других металлов [55] и различные соли меди
, (хромат, молибдат, сульфат, вольфрамат и ванадат) ц присутствии
селена [56]. Применение CuSO4 — А12О3 дает 62% акролеина. Для
регулировки процесса можно вначале пропускать исходный про-
дукт через неактивированную СиО при 1000 °C и лишь после этого —
/ - через активный катализатор [57].
Аналогично селенсодержащим соединениям рекомендуют также
сернистый газ, CuS, Ag2S, Sb2S3 и Fe2S3 в качестве промоторов для
. катализаторов, содержащих силикат меди [58].
В патентах и публикациях фирмы Montecatini Soc. рекомендуется
проводить окисление пропилена в акролеин в трубчатых реакторах,
состоящих из медных или покрытых медью труб. В оптимальных усло-
виях (390 °C, 4 кгс/см2, время контакта 0,5 с) селективность 91%,
конверсия 78% [59—62].
Разновидностью метода является окисление пропилена в акролеин
с катализатором СиО на носителе в кипящем слое [63].
При окислении пропилена в акролеин на медьсодержащих ката-
лизаторах энергия активации в присутствии водяного пара соста-
вляет 20 ± 1 ккал/моль [64], в отсутствие водяного пара 30 ккал/моль
(36 ± 2,5 ккал/моль) [65, 66]. Для образования СО2 достаточно
энергии активации 35 и соответственно 36 ккал/моль. Состояние
5.2. Получение акролеина прямым окислением пропилена 97
меди оказывает некоторое влияние на катализатор. Если доля Си (II)
составляет 60—70% от общего количества меди, то энергия активации
равняется 17—18 ккал/моль, при наличии только 20—50% Си (II)
энергия активации снижается до И—12 ккал/моль [67]. Количество
меди, необходимое для получения селективных катализаторов, за-
висит от материала носителя [68].
Окисление пропилена в присутствии СиО на SiC — реакция
первого порядка по отношению к кислороду и нулевого порядка
по отношению к пропилену [69]', поэтому скорость окисления воз-
растает с увеличением концентрации кислорода [64]. Селективность
образования акролеина повышается с ростом концентрации пропи-
лена [64—66]. Водяной пар является лучшим разбавителем по
сравнению с пропаном или азотом (при конверсии 6% оптимальный
выход 70%) [70—71]. Образование СО2 уменьшается при введении
водяного пара. Тем самым повышается и селективность; оптимальная
концентрация пропилена будет 10% [72]. Лучше всего действует
добавка 40% водяного пара (при 340—400 °C), выше этого показателя
катализатор становится нестойким [73].
Введение металлов, обладающих меньшей электроотрицатель-
ностью по сравнению с СиО и поэтому действующих как доноры
электронов (щелочные или щелочноземельные металлы), повышает
активность и снижает селективность. Электронные акцепторы (С1_,
SO4-, S, Р), напротив, увеличивают селективность и уменьшают
активность [74, 75].
Окисление пропилена на медьсодержащих катализаторах из-
учалось рядом авторов [76—78]. Гетерогенные медные катализаторы
зарекомендовали себя лучше, чем гомогенные [81].
Исследование окисления пропилена в присутствии Си2О и
Bi2(MoO4)3 показало, что механизм окисления в обоих случах оди-
наков: вначале отщепляется атом Н от группы СН3, затем второй
атом Н [79]. Акролеин всегда является промежуточным продуктом
при последующем окислении до СО2 и воды [80].
Метод прямого каталитического окисления пропилена на медных
катализаторах был внедрен в промышленности лишь в 1959—1960 гг.
фирмами Shell Chemical -Со. (мощность установки 15 000—
20 000 т/год) и Union Carbide Со. Поэтому сегодня классический син-
тез акролеина из формальдегида и ацетальдегида, бывший до 1959 г.
основным методом промышленного производства акролеина, счи-
тается устаревшим.
Вначале катализатором для этой реакции служила окись меди
(I), нанесенная в количестве 1—2% на карбид кремния. Данный
катализатор очень селективен в отношении окисления пропилена
в акролеин. В зависимости от состава реакционной смеси и условий
реакции в катализаторе устанавливается динамическое равновесие:
Си ~Си2О СиО
7 Заказ 399
98
5. Акролеин
Из этих трех форм окись меди (I), по литературным данным,
является специфическим, селективно действующим катализатором
для окисления пропилена в акролеин. Окись меди (II) оказывает
каталитическое действие на реакцию полного окисления пропи-
лена в СО3, а металлическая медь неактивна.
Промывная Остаток
вода
Рис. 33. Схема производства акролеина, методом окисления про-
пилена:
Г — трубчатый реактор; г — обогревательная система; з — холодильник; 4, —
скруббер; 5 — колонна для удаления СОг; в — газодувка; 7—9 — дистилля-
ционные колонны.
При окислении пропилена в основном возможны следующие
реакции:
СН2=СНСН3 + О2 —> СН2=СНСНО + Н2О + 88,2 ккал
СН2=СНСН3+О2 —> СН3СНО + НСНО + 79,9 ккал
СН2=СНСН3 + 0,5О2-> СН3СН2СНО + 54,0 ккал
, СН2=СНСН3 + ЗО2 —> ЗСО + ЗН2О +259,9 ккал
СН2=СНСН3 + 4,5р2 > ЗСО24 ЗН2О + 462,7 ккал
В незначительном количестве образуются, кроме того, органиче-
ские кислоты, карбонильные соединения и полимеры.
На рис. 33 представлена схема производства акролеина путем
окисления пропилена.
5.2. Получение акролеина прямым окислением пропилена 99
воздухом вводится после
отделитель
И газодувку 6
Рис. 34. Зависимость от давления об-
щего выхода акролеина (/) и выхода
в единицу времени на единицу реак-
ционного объема (2).
Пропилен вместе с кислородом или
карбонизационной башни в цикл через
в следующий отделитель. В по-
догревателе смесь нагревается
до 250—350 °C и поступает
в заполненный катализатором
трубчатый реактор 1. Для под-
держания постоянной темпера-
туры реакции имеется специ-
альная система 2. Образую-
щаяся смесь охлаждается в хо-
лодильнике 3 до 50—80 °C и
направляется дальше в скруб-
бер 4, где продукты реакции
поглощаются водой. Промывная
вода из скруббера 4 поступает
в перегонную колонну 7, где
отгоняются образующиеся про-
дукты окисления. Вода, вытекающая из низа колонны, содер-
жит небольшое количество кислот (акриловой, уксусной и др.).
После нейтрализации вода снова возвращается для промывки в скруб-
бер 4. В колоннах 8 и 9 осуществляется дистилляция акролеина.
Выходящие из скруббера 4 остаточные газы содержат наряду
с непрореагировавшим пропиленом также азот, кислород, окись
Рис. 35. Зависимость
состава продуктов оки-
сления пропилена от да-
вления:
1 — акролеин; 2 — ацеталь-
дегид; з — окись пропилена;
4 — пропионовый альдегид;
5 — ацетон.
и двуокись углерода. Для удаления двуокиси углерода смесь газов
пропускают через колонну для декарбонизации 5, после чего снова
используют в реакционном цикле.
Влияние давления на процесс получения акролеина из пропи-
лена показано на рис. 34 и 35.
Выделить чистый акролеин из реакционной смеси не просто.
По литературным данным при процессе фирмы Shell получают в
7*
100
5. Акролеин
качестве дистиллята в перегонной колонне смесь следующего со-
става (в вес. %):
Акролеин...........' 80—90
Ацетальдегид .... 3—10
Пропионовый альде-
гид ..............0,5—3
Ацетон ...............2—5
Высококипящие вещест-
ва .................1—2
Удаление минимального количества пропионового альдегида,
имеющего почти такую же температуру кипения, что и акролеин
(пропионовый альдегид 49 °C, акролеин 50 °C), удается осуществить
только экстракционной перегонкой с водой при повышенном давле-
нии во второй колонне.
5.2.2. Окисление пропилена на катализаторе
фосфоромолибдате .висмута (метод Sohio)
Потребность современной нефтехимии в акрилонитрилу, получае-
мом непосредственно из пропилена путем каталитического окисления,-
привела к разработке еще одного, более экономичного синтеза ак-
ролеина из пропилена. Лучшие результаты дал катализатор фосфоро-
молибдат висмута на кремневой кислоте. г
В 1960 г. американская фирма Sohio [82] ввела в строй первую
промышленную установку для производства акрилонитрила по
данному методу. При этом получали и акролеин в промышленном
масштабе.
Таблица 11
Различные методы синтеза акролеина из пропилена
Фирма Катализатор Темпера- тура, °C Время кон- такта, с Объемное соот- ношение про- пилен : кисло- род :водяной пар Конверсия, % 0s- W О И 3 и
Shell Cu2O на SiC или A12O3; иод в ка- честве промотора 370-400 0,2 4,4 : 1 : 4,7 14 ** 68-81
Sohio Фосфоромолибдат висмута на крем- невой кислоте 425-480 1,6 1 : 9,3 * : 3,5 56 ** 70
Distillers СиО на А12О3, си- ликагеле или пем- зе; селен в каче- стве промотора 320-350 2-2,5 2% пропилена 90- 95 з* 81—82
Monte- catini Покрытые медью трубчатые спира- ли 390' 0,5-5 (5 кгс/см2) 21 : 4 : 25 78 91
* Воздух.
“ Процесс с рециркуляцией, конверсия указана за 1 проход.
•* Для получения высокой конверсии необходимо высокое соотношение кислород : про-
пилен .
5.3,- Свойства акролеина
101
В течение последних лет был разработан ряд промышленных ме-
тодов получения акролеина (табл. И) [83, 84]. В табл. 12 приведен
обзор описанных в литературе катализаторов для окисления про-
пилена в Акролеин.
5.3. СВОЙСТВА АКРОЛЕИНА
Акролеин обладает следующими свойствами (см. также [3]):
Температура плавления, °C ........................... —86,95
Температура кипения, °C .......................... 52,69
Плотность р^° ......................>............... 0,8389
Плотность пара по отношению к воздуху при 20 °C / . 0,8402
Показатель преломления ............................. 1,4017
Вязкость при 20 °C, сП................................ 0,393
Критическая температура, К............................ 510
Критическое давление, кгс/см2........................ -51,58
Критический объем, см3/моль .......................... 189
Теплоты испарения
кал/г ............................................. 129,5
кал/моль . . ..................................... 7260
Теплота образования (газ) при 25 °C, ккал/моль . . . —17,79
Энергия образования (газ) при 25 °C, ккал/моль . . . —12,86
Теплота сгорания (жидкость) при 25 ®С, ккал/г . . . 6,95
Удельная теплоемкость (жидкость) при 17—44 °C,
ккал/(г • ®С) ..................................... 0,511
Удельная теплоемкость (газ), кал/(моль • °C)
при 300 ........................................ 16,07
5Q0 ......................................... 22,97
700 27,86
Коэффициент теплового расширения при 205С, 1/°С 0,00143
Пределы взрываемости в смеси с воздухом, объемн. %
верхний ............................................ 31
нижний ........................................... 2,8
Температура воспламенения в закрытом сосуде, РС . • —25
Электропроводность при 10 °C, Ом*1.................1,55 • Ю"7
На рис. 36 показано равновесие жидких смесей акролеина и воды.
Рис. 36. Равновесие жидких смесей акроле-
ина и воды:
1 — мол. ч.; 2 — вес. ч.
Концентрация акролеина*
вес. или мол.ч.
102
5. Акролеин
Катализаторы для окисления
Состав газа % Температура
Катализатор
реакции,
S Воздух Водяной °C
I» (или Ог) пар
Й s
Окиси металлов п переходных элемен-
тов (Fe, Со, Ni, Си, Zn, В, Р, Сг, Мо, W, V), например CUO + P2O5 на
силикагеле 9 36 55 480
CUO+BPO4 Fei-ToBit-toPo-eMoisOsa-gK например, 3 32 65 460
Fe7Bi2PMoi2O52 на SiO2 12,7 6,5 (О2) 5,0 400
Окись Bi—Мо—В—Si—Р — —
Фосфоромолибдат Bi (Bi9PMoi2O52) • 9 36 55 445
Молибдат Bi 9 36 55 445
Cu2-i0Co2_i0(Mo, W, V)12(P, Si)o,5-2040-70 — — —
Молибдат Bi на SiO2 . . 15 85 (на- — 395
сыщен- ный но-
Молибдат Bi дяным паром)
10 50 40 452
Молибдат Bi + Те (TeO2) 10 50 40 452
Bi2O3 • 2МоОз на корунде — — — 500
Молибдат Bi на SiO2 — — — 380
Молибдат Bi 10 16 (О2) — 500
Молибдат Bi + CuO
6Bi2O3-I6MOO3 и 7Bi2O3 • I6MOO3 . . . — —
Молибдат Bi + As2O3 на SiO2 13 87 — 480
CuaO на SiC+молибдат Bi на SiO2 10 25 65 420
MoO3 или Bi2O3 с H3PO4 или H6BO3 __ — — 375
Bio, 5-isPo-5^0i гОзв-76 8 61 31 427
Bio,5-i8Po-5Mo1203e-76 на кислой отбель-
ной земле, пемзе, силикагеле, диато- мите, цеолите 1 ч. 5-8 ч. 3-10 ч. 410-480
Молибдат Bi и фосфоромолибдат Bi 8,7 52,2 39,1 445 3
Фосфоромолибдат Bi на SiO2 .... 10 59 31 430
Фосфоромолибдат Bi на силикагеле — —. — —
Фосфоромолибдат Bi+HBr 5,5 16,5 78 400
5.3. Свойства акролеина
103
Таблица 12
пропилена в акролеин
Конвер- сия % Селек- тивность катали- затора, % Выход акролеина % Примечание Литература
Время контак- та, с
12 [851
4 ч 22 — — — 185]
0,1-2 94,4 89 83,7 Метод непрерывный, разбави- тель СО или СО2 [86]
3,1 56,9 71,9 41 — [87]
3,1 95 73,1 70 — [88]
— — — — — [891
— 75-76 80 60-61 Давление 2,5 кгс/см2 [90]
4 70 73 18,9 51 — [91]
— 45 — — В кипящем слое [92]
— 70 — — Описано разделение 193]
— 37 43 16 Наряду с 50% акрилонитрила добавляется 10% NH3 и 64% Не [94]
— — — — — [95]
— — — — — |96|
— — — 49 — 1971
— 30 75 22,5 Двухступенчатый реактор с Сп2О в первой части п мо- либдатом Bi во второй [98]
— — — Незначи- тельный — [99]
5 3,2 51,2 1,6 Процесс в кипящем слое, в ре- акторы помещены пластины пз окиси Sb —U (8 : 1) [100]
1,8 65,1—70,4 72,5-97 49,9-63,3 [101]
10 — — 26,2 (через 8,5 ч) Циркуляционный процесс [102]
— 60 70 42 Описано получение из Н3РО4, МоО3, Bi(No3)3 [ЮЗ]
-— — — — Лабораторные исследования [104
1,78 1 78 Обработка катализатора газо- образным НВт [105]
104
5. Акролеин
Катализатор Состав газа, % Температура реакции, °C
Пропи- лен Воздух (или Оз) Водяной пар
Комбинация молибдата Bi (I система) и 80% МоО3-Р2О5, 6,2% Bi2O3, 3,8% МоО3+9,5% SiO2 (II система) . . . 490/3,8
Фосфоромолибдат Bi 1 Я. 1 я. (О2) — КГС/СМ2, 467/3,5 КГС/СМ2 425
Молибдат Bi, молибдат Со, фосфат Си,
ванадат Си, вольфрамат Си и др. — — — —
Молибдат Са—Bi 33 17 (О2) 50 450
Молибдат щелочноземельных металлов — — — —
Молибдат Bi + MgO, А12О3 или Fe2O3 Про- — 500
Медьсодержащий катализатор (первый реактор) и вольфрамат или молибдат Bi, Ni, Со пан 350/7
Н8[Х(Мо2О7)6] + Bi2O3; X—редкозе- мельный металл, например додекамо- либдатоцериевой кислоты кгс/см2; 375— —500/6-7 КГС/СМ2 450
Фосфоромолибдат Bi + H2SOa или суль- фаты, молибдат Bi+H2SOi 14 67 ' 19 430
Боромолибдат Со ..... — — — —
Фосфоро- или кремнемолибдат Си—Bi
(Мо : Си : Р : Si = 1 : 0,5 : 0,1 + Bi2O3) 53 12 (О2) 35 380— 500
(Си, Ni, Cr)B(Si, Р)(Мо, W, V)i2 - 040-eo,
например Cu9PMoi2O47,5 на SiC . . . — — — —
N l0-20,;°10-15i' cl,0-"I:il0Il-4 • • Po.i-2Moi2035-85, например Ni10FeBiPMoi2O57
CuO + кремнемолибденовая кислота . . 10 50 40 440
Фосфоромолибдат Fe—Bi — — — —
Молибдат Fe + Cr2O3 8 66 26 380
Молибдат Fe + Ti — — — —
Фосфоромолибденовая кислота+As2O3 — — — • —
Фосфорованадат Сг—Bi, например
Ст (VO3)3 • CrPO4 Bi2O3 25 25 50 —
5.3. Свойства акролеина
105
Продолжение табл. 12
Время контак- та, с Конвер- сия, % Селек- тивность катали- затора, % Выход акролеина, % Примечание Литература
— — — 50 (I си- стема) 24,3 (II система) Метод получения акриловой кислоты [106]
1 60 74 44,5 Процесс в кипящем слое; во- дяной пар за счет теплоты реакции [107]
. — — — [1081
4 — — 67 — - - |Ю9|
— —. — 76 — [ПО]
— — — — — [111]
— 85 — 69—70 Два последовательно включен- ных реактора [112]
— — — —, — [ИЗ]
— 73 86 63 Описано получение [1141
— — Высо- кая 52-60 Высокий выход в единицу вре- мени на единицу объема /115]
0,12 — 52-60 — Высокий выход в единицу вре- мени на единицу объема [116]
— — — — — [117]
— — — — — [118]
10 40 40 16 — |119|
—- — — 79 — [120]
8 82,5 (че- рез 4 ч) 52,6 43,4 15,6% акриловой кислоты [121]
— — — — Образуется также уксусная кислота [121а]
—~ — — — Преимущественно для произ- водства акриловой кислоты [122]
— 7,6 67,2 5 — [123]
106
5, Акролеин
Катализатор Состав газа, % Температура реакции, °C
Пропи- лен Воздух (или О г) Водяной пар
Арсеномолпбдат Bi, ванадовольфрамат Bi, комбинации Bi—Sn—As—Мо—
-v-w-в . . Bi (VO3)3 BiPOd Bi2O3, фосфорована- — — — 440
дат Bi — — — 475
Cu2O + молибдат или вольфрамат . . . — — 300-360
Молибдат Си — — — —
Фосфороборванадат — — — 475-650
Фосфорованадомолибдат — — — —
Ванадат Bi, фосфорованадат Bi ... — — — 400-450
Bii2PWi2O5g,5 13 13 (О2) 20 500
Ванадат Ag, Sn или Bi В1гО3+ кремневанадовольфрамовая ки- 10 49 41 480
слота 10,5 57 32,5 430
ТеО2
TeO2 + 5%Re2O7 С0М004 • ТеО2 HReOa (вместо СоМоО4 — — — 345
также молибдат или фосфат Ni и Си) — — — 350-425
Молибдат Bi + TeO2(Te) 10 50 40 452
СиМоО4+теллурат Си — — — 400
Молибдат Мп + ТеО2 10,6 52,1 37,3 525
Мп2Р2О7 • МоО3 • ТеО2 Фосфоромолибдат Bi-TTeO2 на TiO2 16 (про- пан) 41 43 385
или силикагеле 10,0 52,5 37,5 - 480
Фосфоромолибдат Bi + TeO2 15 71 14 480
ТеО3+6МоО3 и TeO3 + 6WO3 и др. . . МоО3 + Сг2О3 пли WO3+ТеО2+окись 13 13 (О2) 20 485
As, Sb или Bi — — — 400
Фосфоромолибдат +1% Те или ТеО2 Фосфорованадиевая или фосфоровольф- 12 18 (О2) 70 486
рамомолибденовая кислота+ ТеО3 ТеО2, активированная С12, Вг2 или 12 15 71 14 400
на бентоните — — — _—
(Bi, Te)0.2_ls(Ce, Th)(Mo, W, V)12O37-M Cu6_9(Te, Se)0,5-i,e(P, Sf)0-i(Mo, W)12-' — — — 400—600
• O40-60 — — — —
Теллуромолпбдат Ni — — — —
Sb2O44-SnO2 = l : (3-7-50) ....... 11,7 48,2 40,1 360
5.3' Свойства акролеина
107
Продолжение табл. 12
Время контакта, с Конвер- сия, % Селек- тивность катали- затора., % Выход акролеина, % -Примечание Литература
— — 85 —z .— [124]
— 74,8 — 9,8 Высокий выход в единицу нре- [125]
мени на единицу объема
.— — — — По сравнению с Си2О повы- [126-
шейная селективность 127]
— — — — Преимущественно для получе- [128]
ния акриловой кислоты
.— —• — — [129]
— —- — —— Ц30|
— — — — |131|
— 56 59 33 54% аргона в качестве инерт- [132]
ного газа
— 8,6 — — — [133]
— Незна- — — — [134]
читель-
нал
— •— —. —. — [135]
— 9,5 —• — — [136]
7-38 55—100 —. — — [137]
— 70 73 51 3% акриловой кислоты [91]
— — — — — 11381
5 15- 27 4 .— Ц39|
40,6 — — 24,2 46% акриловой кислоты [140]
5 43 73 31,4 [141]
1,25 38 84 32 — [142]
1 — — — Добавка 54% Аг [143]
— 32 89 28,5 — [144]
2,5 — — 43,9 27,5 акриловой кислоты [145]
1,25 55 91 50 — [146]
0,05—20 9 — >60 — [147] [148]
— 69 —. — [149]
— 90 —- —. — [150]
4 '— —• 78,2 — [151-
153]
108
5. Акролеин
Катализатор Состав газа, % Температура реакции, °C
। Пропи- лен Воздух (или О2) Водяной пар
Sb2O4-SnO2 на SiO2 — — — —
Sb2O4 • SnO2 на графите 10 70 ' 20 452
Sb2O4 • SnO2 на глине, фарфоре, стек-
лянных шариках 10 56,5 33,5 497
Sb2O4 • SnO24- Sb2O3 10 50 40 400
Sb2O4 • SnO2 с добавлением солей Ca,
Fe (III) или Ba 10 50 40 480
Sb2O4 SnO2 на SiO2 (из этилсиликата) — — — —
Sb2O4 SnO2+окись Bi, Ni или Co . . — — — —
Sb2O4 • SnO2 (3,26 : 1) .' 10,6 49,6 39,8 411
Sb2O4SnO2 . . . : — — 439-524
440-519
439-483
Sb2O4 • SnO2 10 50 40 467
Sb2O4 SnO2 — — —
Окись Sb —U +окись металла (напри-
мер, CuO) 5 55 34 480
Sb2O3+окиси металлов побочных
групп, например Sb203• U3O3 .... 8 62 30 480
Sb2OS+Fe2O3 —.— — — —
Окись Sb —Мп — — — —
Ванадат Sb, фосфат Sb и Bi2O3 на SiC 20 20 60 475
Молибдат Sb + окись Sn, Ti или W . . .— -— — —
Фосфорованадат Sn — — — —
Окись As с добавкой окисей других
металлов, например As2O5 • Fe2O3 . . 9 77 14 390
Окись Мо пли другого тяжелого ме- *
талла+B2O3+As2O5 на SiO2 .... 10 45 45 370-380
Молибдат Fe — Cl или молибдат С1 +
+ As2O5 .— — — —
Вольфрамовая кислота-)-As2O5 .... — — — —
Арсенованадат Си на ТЮ2 10 . 49 41 419
Фосфоромолибдат -)- As2O5 14 71 15 400
Титанат Sb + окись W, Мо или As . . 10 50 40 450
Фосфат Ag, Fe или Си 12 18 (О2) 70 640
Окись Си —Au на SiC 24 46 30 300
5.3. Свойства акролеина
109
Продолжение табл. 12
Время контакта, с Конвер- сия, % Селек- тивность катали- затора, % Выход акролеина, % Примечание Литература
1 I 1 1 1 1 о~ i 1 1 «= 1 1 I 1 1 с| I I I 1 75 3,1 15 80-90 13,4 14,4 64 50 42 28 61 68 59 29 36 89 64 97 39,4 39,8 61 67,7-71,8 32 17 30,9 6 51 1,7 4 80-85 „ 5,2 57 . 32 40 24 67 Улучшенная механическая ус- тойчивость Улучшенная механическая устойчивость в результате добавки Sb2O3 Улучшенная механическая ус- .тойчивость Патент на получение катали- затора Запатентован процесс с тремя последовательно включае- мыми слоями катализатора Обработка реактора антимо- нитом Na Быстрое охлаждение реакци- онных газов Прежде всего для получения акрилонитрила Особенно для окисления бу- тилена Многоступенчатое окисление 37% ацетальдегида [154] [155] [156] [157] [158] [159] [160] [161] [162] [108] [163] [164] [165- 166] [167] [168] [169] [170] [171] [172] [1731 [174] 175] [176] [177] 178] [179] 1801
110
5. Акролеин
5.4. ПРИМЕНЕНИЕ АКРОЛЕИНА
Изменение сырьевой базы из-за внедрения нового процесса по-
влияло и на цены. Если перед 1960 г. акролеин стоил еще 46 цент/фунт,
то теперь можно считать, что цена снизилась по меньшей мере до
20 цент/фунт и ниже. Самое большое количество акролеина идет
в настоящее время на синтез глицерина в отсутствие хлора, когда
акролеин гидрируется в аллиловый спирт, который затем перекисью
водорода превращается в глицерин. Установка для производства
глицерина фирмы Shell имеет мощность 20 000 т/год. Аналогичные
мощности имеют промышленные установки фирмы Degussa, Union
Carbide и т. д. Прочее потребление акролеина оценивалось в 1961 г.
в США примерно в 1000 т/год.
Наряду с производством аллилового спирта для синтеза глице-
рина одной из основных областей применения акролеина до сих
пор был синтез метионина. При взаимодействии акролеина с метил-
меркаптаном в присутствии катализаторов (пиридин, ацетат меди,
пиперидин, этилат натрия) в результате ряда превращений обра-
зуется метионин [184]:
сн2=снсно -HaS--> ch3sch2ch2cho N;’
---> CH3SCH2CH2CH(NH2)CN--► CH3SCH2CH2CH(NH2)COOH
Несмотря на большую реакционную способность акролеина
не удалось полимеризовать его в продукт, представляющий интерес
для промышленности. Возможности промышленного использования
акролеина тщательно изучал Шульц с сотрудниками [185]. Реакция
акролеина с многофункциональными соединениями ведет к интерес-
ным конденсационным полимерам, среди которых особенно известны
полиацетали с многоосновными спиртами. При полимеризации
продукта конденсации с пентаэритритом (торговая марка «ультра-
лев»)
уО—СН2\ /СН2-ОХ
сн2=сн-сн/ ;сн-сн=сн2
\о-сн/ \сн2-о/
получаются вещества с хорошей прочностью на разрыв, высокой
ударной прочностью, стойкостью к истиранию [186]. Но эти продук-
ты обладают, к сожалению, невысокой теплостойкостью (65—104 °C).
Фирмы Shell Development Со. и Union Carbide Chemical уже сей-
час производят низкомолекулярные продукты из акролеина в боль-
шом масштабе [187]. Так, в результате гидролиза продукта димери-
зации акролеина [188, 189] и его дальнейшего гидрирования обра-
зуется 1,2,6-гексантриол, применяемый в качестве пластификатора
и составной части при производстве алкидных и полиэфирных смол.
О большой реакционной способности акролеина свидетельствует
схема, приведенная на стр. 111.
н,
С12
HOCl
02
Н;О
-----> Аллиловый спирт
------>- Пропионовый альдегид >- Пропиловый спирт
а, Р-Дихлорпропионовый альдегид [190]
НС1
а-Хлоракролеин —>• а-Хлоракрилатные смолы
Глицериновый альдегид [191]
Акриловая кислота [192]
> 1,3-Пропандиол [193]
снон
Н2С
Н2С
CH2
I [194]
СНСНО
О
ЗЗз—> Р-Пиколин [195]
СН2
СН2 ОНС-(СН2)3-СНОНСНО
Реакция Тищенко
Аллилакрилат —
[196]
Н2С
ноне
снсно
Акролеин
сн2
Димеризация
НС
НС
о
Реакция Дильса—
—Альдера
о
сн2
СНСНО
,сн2
нс^
нс.
hcZH2
нс.
нго
[197]
сн2
СН2
НС
C-R
сн2
CH-OR
НС
СН2
I [198]
C-R
О
сн2
НС CHOR
О
—> Глутаровый альдегид [199]
Полимеризация^ долиакролеин [200]
Сополим_еризация__> Смоды [2Q1]
CH.SH.NH.CN^ MeTI10HI1H l202]
GO(NHg)g или CS(NHs),^ Смоды [203]
C(CHaOH)i к0нденсаци0Н1Шй продукт [190]
112
5. Акролеин
ЛИТЕРАТУРА
1. Н. Schulz, Н. Wagner, Angew. Chem., 62, 105—118 (1950). 1
2. S. A. Ballard et al., Proc. 4th World Petrol. Congr., Sect. IV, 1955,
p. 141-154.
3. H. Г. Коральник, Сборник научно-исслед. работ Ташкентского
текст, ин-та, № 17, Химия и химическая технология высокомолекулярных
соединений, вып. 1, 1964, стр. 5—83.
4. С. W. S m i t h, Acrolein, New York — London, 1962.
5. J. Redtenbacher, Liebigs Ann. Chem., 47, 114 (1843).
6. E. F i s c h e r, I. T a f e 1, Ber. dtsch. chem. Ges., 20, 3388 (1887).
7. H. A d k i n s, W. H. H a r t u n g, Org. Syntheses, vol. 6, 1926, p. 1.
8. F. G. Watson, Chem. metallurg. Eng., 54, № 12, 106—109 (1947).
9. Пат. США 2309576, 1943.
10. Angew. Chem., B20, 303 (1948); F. G. Watson, Chem. Eng., 54, № 12,
107—109 (1947).
И. П. В. Симаков, В. А. Покровский, ДАН СССР, 63, 143—
145 (1948).
12. J. G. В г е m п е г et al., I. Chem. Soc. (London), 1946, 1018—1022.
англ. пат. 547334, 1942.
13. Пат. США 2245582, 2246037, 1941.
14. Шведск. пат. 101032, 101145, 1938; пат. США 2288306, 1942.
15. Т. Ishikawa, Rept. Govt. Chem. Res. Inst (Tokyo), 50, 387—390 (1955).
16. Пат. США 2294955, 1942.
17. T. Ishikawa, T. К a m i о, Yuki Gosei Kagaku Kyokaoshi, 20, 56—
61 (1962); T.. I s h i k a w a, T. К a m i o, Tokyo Kogyo Shikensko Hokoku,
58, 40-47 (1963).
18. Пат. ПНР 45675, 1962.
19. Пат. ПНР 45582, 1962.
20. D. A. S. 1203243, 1965.
21. St. В a s i n s к у, St. Malinowski, Roczniki Chem. (Ann. Soc.
Chim. Polonorum), 38, 635—641 (1964).
22. H. J ablcz ynska — Jedrzejewska et al., ibid., 33, 965—
973 (1959).
23. St. Malinowski et al., Chim. Ind. (Paris), 85, 885—896 (1961); St.
Malinowski, St Basinsky, Bull. Acad, poldn. Sci., Ser. Sci.
chim., 11, 55—61 (1963); ср. также J.,Chem. Soc. (London), Sect. B, 1013—
1020 (1967).
24. Пат. США 2197258, 1936; 2334091, 1938; 2270705, 1939.
25. Пат. США 2383711, 1942.
26. Пат. США 2486842, 1946; 2451485, 1947
27. Пат. США 2608585, 1951.
28. Англ. пат. 640383, 1950.
29. Л. Я. М а р г о л и с и др. ЖОХ, 26, 1368 (1956).
30. О. В. И с а е в и др., ЖОХ, 29, 1522 (1959).
31. Н. И. П о п о в а и др. Изв. Сибирского отделения АН СССР, серия хим;
наук, 7, 40 (1957).
32. О. В. И с а е в и др., ДАН СССР, 119, № 1, 104 (1958).
33. О. В. И с а е в и др., там же, 124, № 4, 858 (1959).
34. Я. Б. Г о р о х о в а т с к и й и др. ЖПХ, 36, № 12, 2725—2728 (1963).
35. Л. П. Шаповалова и др., Укр. хим. ж., 28, № 9, 1031 — 1036 (1962).
36. Н. И. Попов а, Е. В.'В е р м е л ь, ДАН СССР, 124, № 4, 842 (1959).
37. Н. И. П о и о в а и др, Изв. вузов (Иваново). Химия и хим. технол., 2,
926—929 (1959).
38, Е. Е. В е р м е л ь, Материалы конференции молодых научных сотруд-
ников Восточно-сибирского филиала Сибирского отделения АН СССР,
Благовещенск, № 3, I960, стр. 35—38.
Литератора
113
39. Ю. Д. К е р н о с, Б. Л. Молдавский, ЖПХ, 33, 2593—2597 (1960).
40. О. В. И с а е в и др., ДАН СССР, 124, 858-860 (1959).
41. Я. Б. Гороховатский и др., Кинетика и катализ, 3, 230—236
(1962).
42. Н. И. П о и о в а и др., Труды Всесоюзного совещания по химической
переработке нефтяных углеводородов в полупродукты для синтеза волокон
и пластических масс, Баку, 1957, стр. 191—195; Н. И. Попова,
Е. В. В е р м е л ь. Изв. Сибирского отделения АН СССР, № 9, 74—85
(1957).
43. Я. Б. Гороховатский и др., Кинетика и катализ, 3, 133—138
(1962).
44. Н. И. П о п о в а и др., Изв. Сибирского отделения АН СССР, № 12,
78—82 (1960).
45. D. A. S. 1001673, 1957; голл. пат. 95094; пат. США 2879300, 1959.
46. Т. I s h i k a w а, Т. К a m i о, Tokyo Kogyo Shikensho Hokoku, 59,
№ 5, 228—233 (1964).
47. D. A. S. 1240063, 1967; франц, пат. 1345265, 1963.
48. Б. Д. К p у ч а л о в и др., Кинетика и катализ, 3, 247—251 (1962).
49. N. К о m i n a m i et al., J. chem. Soc.. Japan, Ind. Chem. Sect. (Kogyo
Kagaku Zasshi), 65, 1510-1533 (1962).
50. Англ. пат. 625330, 1949.
51. Англ. пат. 658240, 1951; пат. ФРГ 877450, 1953.
52. Англ. пат. 655210, 1951; 727318, 1954; 875160, 1961; пат. США 2670381,
1954.
53. Франц, пат. 1325538, 1963.
54. Англ. пат. 658179, 1951; авт. свид. СССР 159515; Бюлл. изобр., № 1, 13
(1964).
55. Англ. пат. 678557, 1951; пат. ФРГ 806440, 1951.
56. Англ. пат. 694354, 1953.
57. Англ. пат. 706552, 1954.
58. Пат. США 3009960, 1958.
59. М. A g a m е n none, Petrol. Mater. Prima Ind. Chim. Mod., Comun.
Giornata Chim. (Milano). 1961, p. 209—225.
60. M. A g a m e n n о n e, Chim. e Ind. (Milano), 4, 875—884 (1961).
61. G. Marullo, M. Agamennone, Chim. e. Ind. (Milano), 46, 376—
378 (1964).
62. Англ. пат. 847564, 1960; D. A. S. 1103911, 1961; итал. пат. 580541, 1958.
63. Авт. свид. СССР 136728, 1961; Бюлл. изобр., № 6, 19 (1961).
64. Я. Б. Гороховатский, Е. Н. Попова, Кинетика и катализ,
5, 134—143 (1964).
65. В. М Б е л о у с о в и др. Кинетика и катализ, 3, 221—229 (1962).
66. В. М. Бе лоусов и др., ДАН СССР, 137, 1396—1398 (1961).
67. Н. И. П о п о в а, К. П. Ж д а н о в а, Изв. Сибирского отделения АН
СССР, № 12, 48-52 (1961).
68. Я. Б. Гороховатский и др., ЖПХ, 36, 2725—2728 (1963).
69. Н. И. Попова и др., Синтез и свойства мономеров. Сборник работ
12-й конференции по высокомолекулярным соедииеииям, Институт нефте-
химического синтеза АН СССР, 1962, стр. 178—183.
70. А. М. Г а р и и ш и др., Кинетика и катализ, 3, 257—260 (1962).
71. Т. Ishikawa, Tokyo Kogyo Shikensho Hokoku, 55, 82—86 (1960).
72. E. H. Попова, Я. Б. Гороховатский, ДАН СССР, 145, 570—
572 (1952).
73. А. Г. П о л к о в н и к о в а и др., Кинетика и катализ, 3, 252—256 (1962).
74. Е. К. Е н и к е е в и др., Кинетика и катализ, 1, 431—439 (1960).
75. Л. Я. М а р г о л и с и др., Кинетика и катализ, 3, 181 —188 (1962).
76. s. Z. Roginski, Radioisotopes Sci. Res., Proc. Intern. Conf., Paris,
v. 2, 1957, p. 1—20; 'реф. C. A., 54, 56f (1960).
8 Заказ 399
114
5. Акролеин
77. О. В. И с а е в и др., ДАН СССР, 129, 141—144 (1959).
78. А. Г. Полковниковаи др., Нефтехимия, 3, № 2, 246—253 (1963).
79. R. С. A d a m s, Т. J. Jennings, J. Catalysis, 2, № 1, 63—68 (1963).
80. О. А. Г.оловинаи др., ДАН СССР, 142, 619—622 (1962).
81. Н. И. П о и о в а и др., Изв. Сибирского отделения АН СССР, № 8,
78-82 (1961).
82. F. Veatch et al., Petroleum Refiner, 41, № 11, 187—190 (1962).
83. Chem.-Ing.-Techn., 34, 334 (1962); World Petroleum, 31, 60 (1960).
84. P. w. Sherwood, Chem. Rdsch. (Solothurn), 14, 394 (1961); Brennstoff-
Chem., 38, 164 (1959).
85. Англ. пат. 839808, 1960; D. A. S. 1079615, 1960.
86. D. A. S. 1125901, 1133358, 1137427, 1962.
87. D. A. S. 1223813, 1966.
88. Англ. пат. 821999, 1959; пат. США 2941007, 1960.
89. Франц, пат. 1362890, 1964.
90. Канад, пат. 677630, 1964; D. A. S. 1146046,1963; швейц, пат. 406178, 1966.
91. Англ. пат. 963610, 1964.
92. О. В. Исаев и др., Хим. пром. 41, 71—72 (1965).
93. Канад, пат. 677762; D. A. S. 1160837, 1160858, 1155109, 1964; пат. США
3162514, 1964; ср. англ. пат. 1013379, 1965, 1016101, 1966.
94. Бельг, пат. 611378,1962; дополнение к франц, пат. 1281927; доп. пат. 80776,
1963.
95. Франц, пат. 1476377, 1967.
96. Авт. свид. СССР 143385, 1962; Бюлл. изобр., № 24, 15 (1961).
97. Англ. пат. 881335, 1961.
98. Бельг, пат. 618222, 1962; D. A. S. 1251301, 1967; пат. США 3177257, 1965.
99. Бельг, пат. 620834, 1963; D. A. S. 1134979, 1962; швейц, пат. 406177, 1966.
100. D. A. S. 1223358, 1966; Франц, пат. 1375538, 1964.
101. Бельг, пат. 642946, 1964; D. A. S. 1219917, 1966; франц, пат. 1393374,
1965.
102. Англ. пат. 963611, 1964.
103. F. Veatch et al., Proc. Intern. Congr. Catalysis, Paris, v. 2, 1960,
p. 2647—2658; C. A. 55, 24547e (1961).
104. S. Serban, Rev. Chim. (Bukuresti), 18, № 2, 65 (1967).
105. Англ. пат. 1018944, 1965; франц, пат. 1351913, 1964.
106. Англ. пат. 939713, 1963.
107. Англ. пат. 912686—9126988, 1962.
108. Бельг, пат. 633024, 1963.
109. Франц, пат. 1639893, 1964.
НО. Англ. пат. 985009, 1965.
111. Авт. свид. СССР 176878, 1965; Бюлл. изобр., № 24, 17 (1965).
112. Бельг, пат. 618223, 1962; англ. пат. 986853, 1965; пат. США 3159680, 1964.
113. Англ. пат. 1010423, 1965; D. A. S. 1197441, 1965; пат. США 3173957, 1965.
114. Англ. пат. 1038274, 1966; D. A. S. 1207367, 1965.
115. Ю. Д. К е р н о с, Б. Л. Молдавский, ЖПХ, 33, 2593—2597 (1960),
116. J. Obloj et al., Przemysl chem., 42, № 7, 359—361 (1963).
117. Франц, пат. 1366300, 1964.
118. Niederld. A. S. 6512930, 1966.
119- Франц, пат. 1372357, 1964.
120. Бельг, пат. 613157, 1962.
121. Франц, пат. 1410564, 1965.
121а. Англ. пат. 1019426, 1965.
122. Пат. США 3190913, 1965.
123. Англ. пат. 1015180, 1965; голл. пат. 110366, 1964.
124. Niederld. A. S. 6506246, 1965.
125. Австрал. пат. 255563, 1961; англ. пат. 950686,-1964; D. A. S. 1160838, 1964;
пат. ГДР 44914, 1966; франц, пат. 1307222, 1963.
Л итература
115
126. Н. И. Попова и др., Кинетика и катализ, 2, 916—919 (1961).
127. Л. М. Калибердо и др., там же, 3, 237—240 (1962).
128. Авт. свид. СССР 131347, 1960; Бюлл. изобр., № 17, 20 (1960).
129. Пат. США 3029288, 1962; франц, пат. 1494432, 1967.
130. Англ. пат. 1017599, 1965; пат. США 3230248, 1966.
131. Бельг, пат. 611002, 1962.
132. D. A. S. 1161552, 1964.
133. Англ. пат. 873712, 1959.
134. Пат. США 3260753, 1966.
135. Пат. США 2648638, 1953; пат. США 2753377, 1956.
136. Пат. США 2662921, 1953.
137. Niederld. A. S. 6500643, 1965.
138. Франц, пат. 1494431, 1967.
139. Англ. пат. 989401, 1965; пат. США 3345406, 1967.
140. Niederld. A. S. 6508659, 1966; ср. также Niederld. A. S. 6500418, 1965;
франц, пат. 1491653, 1967.
141. Франц, пат. 1380884, 1964; англ. пат. 1014121, 1965.
142. Niederld. A. S. 6511989, 1966.
143. Англ. пат. 1001505, 1965; франц, пат. 1342963, 1963.
144. Англ. пат. 973375, 19₽4; D. A. S. 1197442, 1965; пат. США 3168572,
1965.
145. Англ. пат. 967485, 1964; франц, пат. 1342962, 1963; пат. США 3192259,
1965.
146. D. A. S. 1253696, 1967.
147. Пат. США 2643269, 1963.
148. О. A. S. 1243664, 1967; франц, пат. 1372397, 1964.
149. Франц, пат. 1367801, 1964.
150. Англ. пат. 990831, 1965.
151. Бельг, пат. 587605, 1960; англ. пат. 864666, 1961; инд. пат. 76718, 1960.
152. Англ. пат. 906328, 1962; франц, пат. 1299139.
153. Англ. пат. 962841, 1964; доп. пат. 83569, 83126, 1964.
154. Бельг, пат. 633502, 1963.
155. Бельг, пат. 632411, 1963; доп. пат. 84114, 1965.
156. Франц, пат. 1360915, 1964.
157. Бельг, пат. 630159, 1963; англ. пат. 961361, 1964; ср. англ. пат. 1011599,
1965.
158. Бельг, пат. 614364, 1962; доп. пат. 81246, 1963.
159. Франц, дополнит, пат. 84020, 1965.
160. Франц, дополнит, пат. 83722, 1965.
161. Англ. пат. 904602, 1962; ср. англ. пат. 1011599, 1965.
162. Бельг, пат. 616979, 1962.
163. Бельг, пат. 632663, 1963.
164. Англ. пат. 1007929, 1965; белы. пат. 651114, 1967; Niederld. A. S. 6411912,
1965.
165. Англ. пат. 991085, 1965.
166. Пат. США 3198750, 1965.
167. Англ. пат. 973338, 1964; D. A. S. 1232552, 1967.
168. Пат. США 3257474, 1966.
169. Голл. пат. 106994, 1963.
170. Niederld. A. S. 6515741, 1966.
171. D. A. S. 1221208, 1966; франц, пат. 1496072, 1967; Niederld. A. S. 6513729,
1966.
172. Бельг, пат. 617523, 1962.
173. Франц, пат. 1438499, 1966.
174. Jap. A. S. 2134, 1965.
175. Англ. пат. 972403, 1964.
176. Франц, пат. 1364810, 1964.
8*
116
5. Акролеин
177. Niederld. A. S. 6511736, 1966.
178. Англ. пат. 993921, 1965; франц, пат. 1395072, 1965.
179. Niederld. A. S. 6500728, 1965.
180. Японск. пат. 23646, 1965.
181. О. В. Исаев, Л. Я. Марголис, Кинетика и катализ, 1, 237
(1960).
182. Н. Н. V о g е et al., J. Catalysis, 2, 58 (1963).
183. C. R. A d a m s et al., J.' Catalysis, 3, 379—386 (1964).
184. Пат. США 2485236, 1949; J. R. Catch et al., J. Chem. Soc. (London),
1947, 1609; Nature, 159, 578 (1947); E, Pierson et al., J. Am. Chem.
Soc., 70, 1450 (1948).
185. R. C. S c h u 1 z e t. al .,1 Kunststoffe, 47, 303 (1957); 50, 568 (1960); Mak-
romol. Chem., 29, 190 (1959); 21, 227 (1956); 30, 39 (1959); 24, 141 (1957);
32, 181, 197, 209 (1959); 28, 58, 157 (1958); 20, 162 (1956); 17, 62 (1955);
Angew. Chem., 76, 357—364 (1964); 69, 163 (1957).
186. Пат. ФРГ 837289, 852301, 855162, 1952; 881108, 1953.
187. H. J. S a n d e г s et al., Ind. Eng. Chem., 50, 854—858 (1958).
188. K. Adler, E. R u d e n, Ber. dtsch. chem. Ges., 74B, 920—926 (1941).
Пят СТПА 9fi4Q?Q7 40^4
190’ H. S. Sanders,’ Ind. Eng. Chem., 50, 854-860 (1958); англ. пат. 526122,
1940.
191. E. Weldmann, V. Prey, Mh. Chem., 84, 543—550 (1953); англ,
пат. 763467, 1956.
192. Пат. США 2744928, 1956; пат. ФРГ 869950, 1953; франц, пат. 1088680,
1955.
193. Пат.'ФРГ 922166, 1955.
194. Пат. США 2434110, 1948.
195. Пат. США 2520097, 1950.
196. Пат. США 2516627, 1950; англ. пат. 740005, 1955.
197. Пат. ФРГ 874313, 1953.
198. R. I. Langley, W. S. Emerson, J. Am. Chem. Soc., 72, 3079-
(1950).
199. C. W. Smith et al., J. Am. Chem. Soc., 74, 2018 (1952).
200. R. C. Schulz, Kunststoffe, 48, 257— 261 (1958).
201. Пат. США 2881088, 1959; англ. пат. 751934, 1956; пат. ФРГ 962824, 1957;
Франц, пат. 871009, 1941.
202. С. D. Durd, L. L. Gershbein, J. Am. Chem. Soc., 69, 2328 (1947);
J. R. Catch, J. Chem. Soc. (London), 1947, 1609; англ, пат. 605311,.
1945; пат. США 2485236, 1945.
203. Герм. пат. 733099, 740422, 1938; 745422, 1939; 740894, 1940; 748842, 1942;
пат. США 2307742, 1942.
6. АКРИЛОНИТРИЛ
Акрилонитрил был открыт Моро в 1893 г. [1]. Он получал его
из этиленциангидрина (I) или из амида акриловой кислоты (II)
путем отщепления воды с помощью Р2О5Г
CH2(OH)CHCN—
I
ch2=chconh2—
II
ch2=chcn
—112^
Промышленное использование акрилонитрила началось в 1930 г.,
когда из него был получен стойкий к маслам и горючему искусст-
венный каучук (буна N или пербунан) [2].
Производству и применению акрилонитрила посвящен ряд об-
зорных работ [3—21].
6.1, ПОЛУЧЕНИЕ АКРИЛОНИТРИЛА
Постоянно растущий спрос на акрилонитрил — сырье для полу-
чения высококачественного полимера пербунана N — вызвал мно-
гочисленные исследования по созданию промышленного экономич-
ного метода производства акрилонитрила.
6.1.1. Получение акрилонитрила через окись этилена
Первым промышленным методом явилось получение акрилонит-
рила через этиленциангидрин:
Н2С-—CH2 + HCN
ch2-ch2-cn-—>-CH2=CH-CN+H2O
ОН
Жидкая синильная кислота реагирует с жидкой окисью этилена
при 50—60 °C в присутствии едкого натра и диэтиламина в качестве
катализаторов [8, 22]. Этиленциангидрин очень легко образуется
по данному способу.
Синильную кислоту можно получить при действии 90 %-ной
серной кислоты на 25% раствор цианида натрия. Гораздо дешевле
118
6, Акрилонитрил
и проще с технической точки зрения производить синильную кислоту
дегидратацией формамида:
HCONH2 HCN
Дегидратация [23, 24] этиленциангидрина проводится каталити-
чески в жидкой фазе при температуре 200—280 °C или в газовой фазе
с активной окисью алюминия в качестве катализатора. При работе
в жидкой фазе стальную емкость для дегидратации, снабженную
мощной мешалкой, заполняют наполовину техническим этилен-
циангидрином и 3%-ной окисью магния или бокситом и постепенно
нагревают до 280 °C. Воду и образующийся акрилонитрил непрерыв-
но отгоняют. В зависимости от степени отгонки обоих компонентов
вводят дополнительное количество этиленциангидрина. Спустя 40 ч
реакцию прерывают для очистки реакционного котла. Затем дегид-
ратация продолжается. Для осуществления непрерывного процесса
нужно иметь несколько реакторов.
Сырой акрилонитрил отделяется от воды и перегоняется с добав-
ками метиленового синего и парафениленового синего в качестве
«стабилизаторов. При периодической перегонке вначале получают
азеотропную смесь акрилонитрил —вода. Смесь разделяют и слой
акрилонитрила возвращают в перегонный куб для удаления следов
воды. Выход чистого акрилонитрила в пересчете на исходный циан-
гидрин составляет 75—78%. Во время перегонки нужно строго
исключать медь.
6.1.2. Получение акрилонитрила из ацетилена
и синильной кислоты
В 1942 г. на смену получению акрилонитрила через этиленциан-
тидрин пришел технически более совершенный метод производства
из ацетилена и синильной кислоты:
^тт 80—90°С; CU2CI2-1- NH4CI <-1ТТ - го .
CH=CH + HCN---------------------> CH2=CHCN+43,1 ккал
Первая промышленная установка была введена в строй фирмой
Bayer Leverkusen [2]. Новый метод значительно удешевил произ-
водство акрилонитрила, который с тех пор получил широкое при-
менение в промышленности.
Первые успешные опыты были проведены в 1939 г. Курцом [25]
на фирме Bayer Leverkusen. Курц вводил оба компонента — си-
нильную кислоту и ацетилен — при 75 °C в концентрированный вод-
ный раствор Си2С12 и NH4C1 (катализатор Ньювленда). Он получал
выход акрилонитрила до 85%. На 1 моль ацетилена нужно 0,8 моль
синильной кислоты. Побочными продуктами являются ацетальдегид,
моно- и дивинилацетилен. После промывки и перегонки получают
чистый акрилонитрил.
6.1. Получение акрилонитрила
11&
6.1.3. Получение акрилонитрила из пропилена, аммиака
(или окиси азота) и кислорода
В 1949 г. фирма Allied Chemical and Dye Corp. [30] сообщила
о прямой реакции превращения пропилена, аммиака и кислорода
в акрилонитрил согласно уравнению:
CH2=CHCH3 + NH3 + 3/2O2 Окис.ь-М°^ CH2=CHCN + 3H2O
Спустя несколько лет фирма Distillers Comp., Ltd. (Эдинбург)
опубликовала сведения о процессе превращения акролеина в акри-
лонитрил реакцией с аммиаком и кислородом. Акролеин получали
окислением пропилена на воздухе. Это двухступенчатый метод по-
лучения акрилонитрила в отличие от одноступенчатого процесса
фирмы Allied Chemical and Dye Corp. Однако оба процесса дают очень
незначительный выход, поэтому они не нашли практического при-
менения.
В начале 1959 г. появился известный теперь во всем мире про-
цесс Sohio американской фирмы Standard Oil Со. [32, 33], в кото-
ром используются те же исходные материалы — пропилен, аммиак,
и кислород воздуха — и получается акрилонитрил в результате
50 %-ной конверсии пропилена и аммиака. Уже в 1960 г. фирма
пустила в Лайме крупнотоннажную установку.
Акрилонитрил в процессе Sohio получается каталитическим оки-
слением пропилена на катализаторе фосфоромолибдате висмута
в присутствии аммиака:
CH2=CHCH3 + NH34-3/2O2 —> CH2=CHCN + 3H2O
Для повышения выхода в процессе Sohio в реактор вводят во-
дяной пар.
Многие фирмы перешли на процесс Sohio или разработали ана-
логичные процессы. Из них особый интерес представляет метод
фирмы Du Pont с использованием NO, по которому уже работает
крупнотоннажная установка [34—36]:
4СН2=СНСН3 +6NO —> 4CH2=CHCN+N2 + 6H2O
Далин с сотрудниками [37] подробно изучил и описал влияние
параметров реакции аммоноокисления в присутствии катализатора,
который он называл «окислы металлов переменной валентности».
Было показано, что при температурах ниже 350 °C синтезируется
незначительное количество акрилонитрила. Наилучший результат
получается при температуре ~450 °C, времени контакта примерно
2 с, мольном соотношении компонентов пропилен : аммиак : кисло-
род : водяной пар = 1 : 1 : 1,8 : 1. Конверсия пропилена за один
проход составила 70%. Выход акрилонитрила 65—68%.
Реакция аммонокисления для пропилена является реакцией
первого порядка, а для аммиака и кислорода — нулевого порядка.
120
6. Акрилонитрил
Это означает, что соотношение NH3 : С3Н6 должно составлять по
меньшей мере 1, а соотношение О2 : С3Н6 — минимально 1,5. При
более низких соотношениях появляются значительные потери и на-
блюдается низкая конверсия из-за образования акролеина. Повыше-
ние концентрации NH3 и О2 мало сказывается на стехиометрическом
отношении. Что касается количества пропилена в исходном газе,
то его концентрация находится в линейной зависимости от скорости
Инертный
газ
: Легкие
примеси
«его
§
а.
5 а
Г
to Высококипящие
г примеси
Х§
С О.
ч *
0QQ
Акрило-
нитрил
К
2
4
4
НгО Высококипящие
примеси
Рис. 37. Общая схема получения акрилонитрила:
1 — реактор; 2 — регенерационная колонна; з — сепаратор;
4 — система .окончательной очистки акрилонитрила.
реакции при данном давлении и температуре. Максимальная кон-
центрация определяется содержанием примесей других компонентов
в исходном газе и, следовательно, экономическими соображениями
в отношении чистоты используемого пропилена.
Существуют различные промышленные методы аммонокисления,
ниже кратко описываются некоторые из них.
Процесс Sohio (фирма Standard Oil Со.). Вначале катализа-
тором процесса был концентрированный (примерно 50—60%-ный)
фосфоромолибдат висмута на двуокиси кремния. Позднее стали ис-
пользоватр молибдат висмута. Применяемые в промышленности
катализаторы основываются, главным образом, на молибдене и вис-
муте [38].
Воздух используется в качестве цкислителя. Соотношение кисло-
род : пропилен варьируется в пределах от 1 : 1 до 2 : 1, а соотно-
шение аммиак : пропилен — от 1:1 до 2,5 : 1. На каждый объем
пропилена добавляют приблизительно один объем пара. Реакцию
проводят при 425—510 °C, давлении 2—3 кгс/см2 и времени контакта
~15 с в реакторе с кипящим слоем (рис. 37).
6.1. Получение акрилонитрила
121
Выходящий из реактора продукт промывается водой, а образо-
вавшийся раствор подвергают азеотропному и обычному фракциони-
рованию. Поскольку может произойти реакция между непрореаги-
ровавшим аммиаком и акрилонитрилом в жидкой фазе, нужно чтобы
промывная вода имела температуру ниже 15 °C, а последующая от-
гонка проводилась быстро.
На 1 кг исходного пропилена получается 0,73 кг акрилонитрила,
0,11 кг ацетонитрила и 0,13 кг HGN. Дальнейшие исследования ката-
лизатора и усовершенствование процесса могут привести к повыше-
нию выхода акрилонитрила до 1,01 кг на 1 кг пропилена. Регенера-
ция части цианистого, водорода, являющегося побочным продуктом,
может повысить выход.
Новейшие установки процесса Sohio работают с катализатором
на основе окиси урана (катализатор 21), эффективность которого*
выше на 20%.
Процесс фирмы Imperial Chemical Industries Ltd. В этом процессе
в качестве катализатора используется висмут-ванадийвольфрамат
на двуокиси кремния. Предпочтительнее проводить процесс в кипя-
щем слое. В этом случае облегчается контроль температуры при эк-
зотермической реакции. Обычно температура реакции достигает
450— 470 °C. По сообщениям фирмы ICI при мольном соотношении
в исходной смеси пропилен : кислород (воздуха) : аммиак : водя-
ной пар = 1:1,7: 1,07 : 1,03 1 кг пропилена дает 0,3 кг акрило-
нитрила и 0,09 кг ацетонитрила на каждый цикл. Окончательные
данные о выходе в этом процессе до сих пор не получены.
В табл. 13 содержится литературный обзор всех катализаторов
для получения акрилонитрила из пропилена.
Процесс фирмы Е. I. du Pont de Nemours. В данном случае про-
пилен вступает в реакцию с окисью азота, которую получают в ре-
зультате частичного окисления аммиака воздухом. Получаемая при
этом смесь, состоящая из 15% окиси азота и 83—84% азота, употре-
бляется для'реакции с пропиленом. В результате образуется акрило-
нитрил. Процесс можно рассматривать как двухступенчатый ва-
риант метода аммонокисления.
Катализатором является серебро (при известных условиях с про-
мотором из окиси кальция) на двуокиси кремния. Без промотора
реакция протекает при 460—500 °C, с промотором — при 420—
460 °C.
Наилучший результат достигается при избыточном количества
пропилена. Так, исходная смесь из 4% окиси азота, 1.2% пропилена
и 84% неактивных веществ дает при 420—455 °C и времени контакта
5,1 с акрилонитрил с конверсией 10% за один проход и выходом 90%
по отношению к окиси азота.
Кроме окиси азота в качестве разбавляющего газа применяют
(по экономическим соображениям) смесь пропилена (-—15%) с про-
паном. Обычно при использовании избытка пропилена и исходных
122
6. Акрилонитрил
Катализаторы для получения
Катализатор Состав исходных продуктов, о/ /о Температура реакции, °C
Пропи- лен Воздух (ИЛИ О г) Аммиак Водяной пар
МоО3 на А120з 11,1 26,9 3,5 58,5 480
МоО3(15%) на А12О3 30 70 10 — 500
Окись Мо на А120з; соли Na пли К в качестве промотора 11,5 31,3 3,9 53,5 480
МоО3 или Сг20з на SiO2 — —• — — —
Молибдат Bi на SiO2 — — — — —
10,7% МоО3, 12,7% Bi2O3 на 76,7% SiO2 62 — 38 — 377
Молибдат Bi на кусках гранита . . . 10 80 10 — 470
Молибдат Bi 9 14 (О2) 9 — 500
Молибдат Bi; промотор 1—5% ВаО и 1-10% SiO2
Молибдат Bi; промотор СаО или SrO Молибдат Bi; промотор окись Be, Li, Na или Ag 9 73 9 9 480
— — — 465
Молибдат Bi; промоторы окись Be, Са, Sr, Ba, Zn, Cd, Li, Na или Ag . . . — — 400-500
Молибдат Pbo,o8-o,75 Bio,1-0,в — — — — —
Молибдат Bi с добавкой небольшого количества Fe2O3 и P2O5 ....... 10,5 79 10,5 — 500
Молибдат и фосфоромолибдат Bi, Sn, Sb, вольфрамат Bi 7,4 76,4 7,4 8,1 455
Молибдат Bi; промотор окиси В, Р или Мп ... .
Молибдат, фосфоромолибдат Bi, Sn, Sb и др — — — —
Молибдат Sn, пер молибдат — .— — — —
Молибдат Sn — — — — —
Фосфоромолибдат Bi на корунде . . . 9,5 71,5 9,5 9,5 465
Фосфоромолибдат Bi — — — — —
18,3% Bi2O3, 14,8% МоО3, 6,3% Fe2O3 и 0,9% Р2О5 на SiO2 8 56 8 28 450
Фосфоромолибдат Fe — Bi 7,5 57 7,5 28 400
Фосфоромолибдат Fe; промотор NiO 6,5 53 6,5 34 425
Кремнефосфоромолибдат Bi 7,7 76,9 7,7 7,7 482
Соли фосфорных и борфосфорных кис- лот Ti, Мп, Fe, Со, Ni, Си, Zn, Ag, Cd, Sn, Ce, Bi, Pb, Th или U, нап- ример фосфат Си на кизельгуре . . 11 54 2 33 480—490
6.1. Получение акрилонитрила
123
Таблица 13
акрилонитрила из пропилена
Время । контакта, с 1 Конверсия, % Селективность катализатора, % Выход акри- лонитрила, % Примечание Литература
3,2 8,1 — — [39, 401
— — — 22,2 — [41]
3,9 23 35,2 8,2 — [42]
— — — [43]
— — 80 - — — [44, 45]
— 80 — — Примесь насыщенного водяным паром воздуха [46]
—— — 70 — [47,48,154]
— 37 50 18,5 Наряду с .43% акролеина; добав- ка 59% Не и 9% Н2 [49]
77 .— — [50-52]
— — — 60 — [53]
— — — 64 Неподвижный катализатор [54]
То же [55]
— — — - — — [56]
1,3 83,8 77,5 65 Повышенная механическая ус- тойчивость [57]
— 65 . 61,1 40 Добавка HCN к исходному про- дукту, уменьшение образова- ния HCN [581
— — — — — [59]
— — — — — [60-61]
.— — — — — [62]
— — — — — [63]
— 60 — — 4% ацетонитрила [64]
— — Добавка О2 к исходному продук- ту для улучшения срока служ- бы катализатора [65]
— — .— 74,3 Катализатор в кипящем слое [66]
— — — 69 — [67]
5,5 — — 61,3 1,6% ацетонитрила, 26,3% HCN [68]
7 — -— 74,8 Катализатор в кипящем слое [69]
1 3,75 18 80 14,4 10% ацетонитрила, 4% HCN [70]
124
6. Акрилонитрил
Катализатор Состав исходных продуктов, Температура реакции, °C
Пропи- лен Воздух (или О2) Аммиак Водяной пар
Молибдат, фосфоромолибдат, фосфоро- вольфрамат и вольфрамат Bi на ВРО4, например .10% фосфоромолибдата Bi на ВРО4 9 42,5 6 42,5 550
Фосфоромолибдат Bi на 1% Н3ВО3 + + SiO2 9 ' 73 9 9 454
Бормолибдат Sn (также с добавкой В2О3, МоО3, окиси В-Mo, окиси Sn—В или молибдата Sn)
Фосфоромолибдат Bi на фосфоромолиб- дате титана 6,7 49,5 4 39,8 520
Окись Си, Ti, Fe, Со или Ni на ВРО4, фосфоромолидат Си на ВРО4 .... 8 40 7 45 515
Молибдат Bi или вольфрамат Bi на А1РО4 10,5 79 10,5 — 480
Молибденованадат Bi 29 42 (О2) 29 — 450
Вольфрамованадат Bi на SiO2 .... 14,8 76,3 3,2 5,7 400—460
Вольфрамат Bi 8 76 16 — 570
Ванадат Bi .... 24 24 (О2) 28 24 450
Bi2O3+додекамолибденоцериевая кис- лота 9,5 71,5 9,5 9,5 485
Соли молибденоцериевой или молибде- новой кислоты La или Th — — — — —
В12Оз+гексамолибденохромовая кис- лота 9,5 71,5 9,5 9,5 495
Молибдат Со 10 50 6 34 447
Арсеномолибдат Bi на SiO2 или SiC 11,4 65,2 23,4 — 480
Аз2О3 + ванадомолибдат Bi, ванадо- вольфрамат Bi, вольфрамомолибдат Bi или молибдат Bi 14,7 75,2 3,1 7,0 430-490
Молибдат Со + ТеО2 (1:1) Молибдат Со+ 50% ТеО2 .2 94 4 — 400
10 50 ' 6 34 400
Арсеновольфрамомолибдат — — — — —
(Bi, Te)0.i-i8(Ti, Zr) МО12О38-75; (Bi, Te)0.i-i8(Mn, Co, Ni)Mo8O39- 75; (Bi, Te)011_i8(Cr, Al, Те) Moi2O39-75 например Bi4(TiMoi2O40)3 8,2 61 7,3 23,5 470
TeO2 • MoO3 —Mn2P2O7 (5 : 15:3) . . • 10,5 31 13,4 45 440
Фосфоромолибдат Bi + TeO2 + HReO4 (100:30: 1) на SiO; Cu2P2O7+TeO2 + + HReCU на SiO2 — — — — 400-450
6.1. Получение акрилонитрила
125
Продолжение табл. 13
Время контакта, с Конверсия, % Селективность катализатора, % Выход акри- лонитрила, % Примечание Литература
1,9 42 63,8 27 17,2% ацетонитрила, 10,2% HCN [71]
7,6 58 83,3 48 Катализатор в кипящем слое [72]
— — — — — [73]
1 55 66 36 5% ацетонитрила, 13% HCN [711
— 40,5 69 28 10% ацетонитрила, 5% HCN [74]
57 88 50 — [75]
— — — — 76]
— — — — Катализатор в кипящем слое 77]
0,2 89 72 64 — 78]
— — 20 — [79]
— — — 47,9 — [80]
— — — — — [811
— 82,6 — • — [82]
4,1 35,5 50,1 17,8 — [831
9 — 70 — 8% ацетонитрила, подвод О-2 во [841
время регенерации катализа-
тора
16 — [85]
— 54,8 58,6 32,1 — [861
— 93,5 58 53 — [86]
— — — — [87]
0,6 84,8 - — 5,9% ацетонитрила; 2,6% акро- [88]
леина
38 100 86,4 84,4 Использование специального ре- [89]
актора
10 84,1 65,5 55 — [90-91]
126
6. Акрилонитрил
Катализатор Состав исходных продуктов, % Температура реакции, °с
1 Пропи- лен Воздух (или О2) Аммиак Водяной пар
ТеО2 на силикагеле 5 90 5 400
Фосфор омолибденовая кислота-]-ТеО2 на AI2O3 7,1 85,6 7,3 410
Теллурит церия на кремневой кислоте (диаметр пор 40—200 А)
Ванадомолибдат+ТеО2 на каолине (на- ряду с молибдатом Ni —Те, теллуро- фосфоромолибдатом Sn или Ni) 8,3 83,4 8,3
WO2-]-TeO2 (7 : 1) на SiO2; фосфоро- вольфрамовая кислота+Те или ТеО2 7,7 84,3 . 8,0 400
Соль Bi или Те ванадомолибденовой гетерополикислоты; (U, Sb, Sn, Си, Те)а.(Се, La, Th)6-(Mo, V)cOd, нап- ример H8CeMoi0V2O41 8,8 63,1 6,2 21,9 423
Молибдат Cd+TeO2 на SiO2 — — — 450
Фосфоромолибденовольфрамат+ ТеО2 на SiO2 400-500
Молибдат Zn + TeO2 на SiO2 — — — — 400-500
Молибдат Pb-j-TeO2 — — — 400- 500
Молибдат Ag или фосфоромолибдат Ag+TeO2 — 400- 500
Вольфрамат Ag — — — — —
Вольфрамомолибдат Bi 5 55 6 34 480
Вольфрамомолибдат Sb 5 55 6 34 480
Вольфраматы или фосфоровольфраматы Си, Ti, Fe, Со, Ni-f-фосфаты или борфосфаты 10 20 (О2) 7 63 500
Окись Sn—Sb (IV) 5 55 6 34 481
Окись Sn —Sb (IV) (1 : 3,26) ...... 2 94 4 — 411
Окись Sn —Sb (IV) (1 :4) на глине, фарфоре или стеклянных шариках 5 55 6 34 480
SnO + Sb2O3 5 55 6 34 460-470
Окись Sn — Sb (1 : 4) 5 55 6 34
Окись Sn — Sb 2 87 3 8(N2) 421
Окись Sn —Sb 5 54 6 35 470
Окись Sn — Sb, промотор окись Си . . 5 55 6 34 490
Окись Sb, активированная окисью Ст, Со, Си, Мп, Ni или Ti, например окисью Sb — Со 5 55 6 34 480
Окись Sn —Sb (1:4), активированная окисью U 5 55 6 34 450
Sb2O5~J~ UgOg+ CU0 5 55 6 34 480
Окись Sb—U — — — — —
6.1. Получение акрилонитрила
127
Продолжение табл. 13
Время контакта, с Конверсия, % Селективность катализатора, % Выход акри- лонитрила, % Примечание Литература
— — — — — [92]
— — — 64,3 — [93]
— __ — — — [94]
— 25,0 58,6 14,6 — [95]
— — — 50 — [96]
0,6 — 80,1 — [97]
— — — — [98]
— — — — — [99]
— — — — — [100]
— — — [101]
— — — — — [102]
— — — — [ЮЗ]
4 4 71 46^5 33 33 8% акролеина, 1% HCN 3% акролеина, 5% HCN [104]
5 — — макс. 76 - [1051
4 65,5 71,5 47 SnO в избытке [106]
—- 47,7 — — — [107-111]
4 96 62,5 60 1% акролеина [114]
51—52 — — Отжиг катализатора перед реак- цией <300 °C [114]
— — — 61 — [115]
2 74,3 — — — [116]
1 — — 60 Получение катализатора [117]
4 62 — 3 % акролеина [118]
4 85 62,5 53 53 [119]
4 — — 61 2% акролеина, 4% HCN [120]
3,6 92 78 72 6% акролеина [121]
[122-123]
128
6. Акрилонитрил
Катализатор Состав исходных продуктов, о/ /0 Температура реакции, °C
Пропи- лен Воздух 1 (или Ог) 1 К I <i В одяной пар
Окись Sb—U; промотор окись BirSn,
Pt, As, W, P, Mg, -например
USb^gOn^H-2,5.% MgO на SiO2 . . .
Окись Fe —Sb+1,73% Re2O7 на SiO2
Ванадат Sb —Bi —Fe................
Окись Mn —Sb......................
Окись Mn —Sb на SiC ..............
Окись Fe — Sb ....................
Ванадат Sb .......................
Ванадат Sn на SiO2 или А120з ....
Фосфорованадат Sn на SiO2; активато-
ры—окись Ba, Al, Ti, W, Sb, Cr, Pb
Ванадат Sn на SiO2 . . ...........
V2O5 на SiO2 •....................
Ванадат Sb —Cr ...................
Станнат Co, Cu, Ni, Mn или Cr ...
Вольфрамат Sn на SiC..............
Боромолибдат Sn на SiO2 ..........
CuO, V2O5, МоОз фосфоромолибденовая
кислота, Bi2O3, Fe3O4 или TeO2, Se,
SeO2 или селенитами (особенно
Ag2SeO3), например CuO + фосфоро-
молибденовая кислота на SiO2 • • •
Cu2O + Se на МоО3 ................
CuO + воздух, насыщенный селеном . .
CuO на пемзе-J-МоОз на А120з . . . .
Триполифосфат- Си..................
Силикат Ni, или Со.................
Фосфат Fe или арсенофосфат Fe ...
Окись As-J-окись Fe, Bi, Al, Sn, Zr
Cr, Co, например арсенат Fe (III) • •
Гексамолибденохромовая кислота+
-}-As2O5 на SiO2..................
Гексамолибденоцериевая кислота+
' +AS2O3, фосфоромолибденовая кис-
лота 4-As2O5........................
Додекамолибденоцериевая кис лота+
+ MnO + As2O5 ...................
7 86 7 1 1 1. 470 450
7 86 7 — 427
7 86 7 — 427
— — — — —
— .— — — —
14 а 14 28 500
6,5 " 77,4 6,6 9,5 520-540
9,5 71,5 9,5 9,5 540
9 68,5 13,5 9 540
4,8 ' 54,5 5,9 34,8 458
10 90 90 90 480
— —' — — 622
6,3 47 6,3 40,4 510
65 25 (О2) 10 — 330
— — — — 400
2,3 97,7 97,7 — 400
— — — — 450
350-450
— — — — 250—400
10,5 16 10,5 63 480
8,9 80,4 10,7 — 480
8,9 80,4- 10,7 — 340
9 73 9 9 500
— — — — —
9 73 9 9 472
6.1. Получение акрилонитрила 129
Продолжение табл. 13
Время контакта, с Конверсия, % Селективность катализатора, % Выход акри- лонитрила, % Примечание Литература
79,5 [124]
— 70 — — — [125—1271
.— — — — — 128]
3 — — 48,6 — 129]
3 — — 61,9 20% ацетОяитрила 130] '
.— — — — —— 130]
— 15 11 — — 131]
3,5 92 36 33,8 — [132-133]
4-5 95-98 67 65 1% ацетонитрила, 10% HCN [134]
2 — — 23 13% ацетонитрила, [135]
2 — — 31 7 % ацетонитр ил а
.— 65 91 59 — [136]
4 85 37,5 32 — [137]
—- 42,5 64 27,2 Исходный продукт пропан, О2 и [138]
NH3
1,3 82,8 66,5 55 4,6% ацетонитрила, 7,8% HCN [139]
75-80 ( 74,3 56-60 1,4% ацетонитрила, 1,2% акро- [140]
леина; полезна добавка LiCl
— — — — Двухступенчатый синтез в двух- [141]
камерной печи
— — — 1,65 Описано приготовление Se [142]
— — — 37,5 Сырье — пропилен или акролеин; [143]
двухступенчатый синтез в двух-
камерной печи
10 — — — — [144]
— — — — Необходима предварительная вое- [145]
становительная обработка ката-
лизатора; побочный продукт —
пропиовитрил
— 55 — — [146]
5,8 83,5 67,4 CR 3 Процесс в кипящем слое [147]
6,2 46,7 89,5 41.8 То же
2,0 38,3 60,5 23,2 12,5% ацетонитрила [148]
— — — — — [149]
1,6 — 42,6- — [150]
9 Заказ 399
130
6. Акрилонитрил
Состав исходных продуктов,
%
Катализатор
Фосфат Nb или Та; промотор — окись
Си, Ag, Fe, Со, Ni, Се, Pb, Bi, Мо
или W, например Nb2O5-|-CuO на ВРО4
Ванадомолиодат Си и пары Be ... .
Окись Мо или W на А12О3...........
43,8 4,4 43,8 520-530
разбавляющих газов продукт реакции имеет следующий состав
(в объемн. %):
Пропилен 37 Азот ................51
Пропан............... 7 Акрилонитрил ...... 5
Затруднения возникают с регенерацией. После охлаждения и про-
мывки водой с целью удаления акрилонитрила остаточные газы
еще богаты непрореагировавшим пропиленом, а содержание в них
азота и пропана слишком высоко для проведения регенерации. По-
этому реакционные газы подвергают экстракционной перегонке
с бензонитрилом или акрилонитрилом в качестве растворителя.
При этом в верхней части колонны почти полностью удаляются весь
азот и избыточный‘пропан.
6.1.4. Другие процессы получения акрилонитрила
Проводились опыты по получению акрилонитрила путем катали-
тического дегидрирования пропионитрила [26]
CH3CH2CN -тц-2-> СН,=С11СА .
но выход был так мал, а конечный продукт содержал столько при- j
месей, что метод не получил дальнейшего развития. ?
То же самое относится к получению акрилонитрила [27] из
CH2C1CH2CN с помощью фенолята натрия.
Оригинальностью отличается способ получения акрилонитрила
окислением аллиламина в воздухе при 500 °C [28]:
CH2=GHCH2NH2+2O2—> ch2=chcn ’
Но в промышленности этот метод не получил признания.
6.1. Получение акрилонитрила
131
Продолжение табл, 13
Время контакта, с Конверсия, % Селективность катализатора, % Выход акри- лонитрила, % Примечание Литература
40 78 31 6,9% ацетонитрила, 8.2% HCN [151]
— — — 40,3 Исходный продукт —пропан, О2 и NH3 [152]
— — — — Исходный продукт—пропан [153]
С точки зрения экономики интерес представляет разработанный
в 1955—1960 гг. синтез из ацетальдегида и синильной кислоты через
нитрил молочной кислоты [29]:
CH3CHO + HCN —> CH3CH(OH)CN CH2=CHCN
6.1.5. Экономическая оценка методов получения акрилонитрила
Капиталовложения для процессов аммоноокисления пропилена
значительно ниже, чем, например, для метода получения акрило-
нитрила из ацетилена и синильной кислоты. Так, для процесса Sohio
годовая стоимость 1 т акрилонитрила составляет 340 долларов в от-
личие от 840—1200 долларов для ацетиленового метода.
Расходы на сырье также существенно снижаются. Применение
смеси пропилен — пропан способствует дальнейшей экономии средств.
Даже при содержании пропилена ниже 40% получают хорошие
результаты.
При оптимальных условиях аммоноокисления на 1 кг пропилена
приходится следующий выход продуктов (в кг) [38]:
Акрилонитрил ............................ 1,01
Ацетонитрил ............................. 0,09
Синильная кислота........................ 0,08
Использование большого избытка пропилена дает наилучшие
результаты. Внедрение аммоноокисления в промышленность пони-
зило цены на акрилонитрил с 32 цент/фунт в 1961 г. до 14,5 цент/фунт
в 1962 г.
Распространение процесса аммоноокисления в промышленности
привело к резкому росту производства, которое достигло 44% в пе-
риод 1961—1962 гг. Ежегодный прирост выпуска акрилонитрила
9*
132
б- Акрилонитрил
составляет в США 8—10%. В конце 1962 г. выпуск акрилонитрила
в США составлял 227 000 т/год. В Европе и Японии в это же время
производилось ежегодно 100 000 т. Приведенные ниже данные ха-
рактеризуют быстрый рост производства акрилонитрила в США:
Год............ 1959 1960 1961 1962 1963 1964 1965 1966
Выпуск, тыс. т 130 140 160 205 230 265 350 415
В табл. 14 приведены данные о мировом производстве акрило-
нитрила в 1966 г.
Таблица 14
Мировое производство акрилонитрила [154]
Страна Фирма Мощность, тыс. т/год Процесс
в 1966 плановая
США American Cyanamid 50 104 Sohio
Du Pont 90 ИЗ Sohio
Goodrich Monsanto Sohio 32 173 36 128 Sohio Sohio Собственный процесс
Канада Union Carbide Imperial/ Oil Co 41 14 Sohio
Франция Ugilor 30 70 Distillers
ФРГ Erdolchemie 25 65 Bayer
Италия Sicedison 17
ANIC —. 15 ENI
Нидерланды Acrylsarda DMS/Sohio — 15 90 Sohio
Англия Nederlandse Stickstoff Maatschappij Border Chemicals Ltd. 40 40 Distillers
Япония 7 фирм 110 211 Sohio
Индия Мексика Государственный сектор Pemex 16 24 Dustillers
Социалистические государства ГДР. 5 30 Sohio
СРР 5 10 —
ПНР НРБ 10 20 Ugine
СССР — 75 Sohio
КНР — 10 OSW-Lurgi
6.2. Свойства акрилонитрила
133
6.2. СВОЙСТВА АКРИЛОНИТРИЛА
Акрилонитрил — прозрачная жидкость с дурманящим запахом,
которая легко полимеризуется. Для стабилизации вводят олеат
меди, диоксифенил и другие ингибиторы. Акрилонитрил очень ядо-
вит. Ниже приведены его свойства:
Температура плавления, °C.....................
Температура кипения, 9С.......................
Плотность
Показатель преломления
W ............................................
......................................
Молекулярная рефракция .......................
Дипольный момент .............................
Теплота испарения, ккал/г ................ .
Теплота сгорания
ккал/моль ....................................
кал/г..............................; ... .
Теплота полимеризации, ккал/моль..............
Удельная теплоемкость, кал/(г • °C)....... . .
Пределы взрываемости с воздухом, объема. %
верхний ................................... .
нижний.....................................
Температура воспламенения, °C.................
Диэлектрическая проницаемость.................
-81 ±1
77,6-77,7
0,8060
0,8004
1,3907
1,3884
15,6
3,88 • 10-18
0,1361
420,5
7925
17,3±0.5
0,50±0,03
17
3
0 ±2,5
38
С рядом соединений акрилонитрил образует азеотропные смеси»
Содержание Температура
второго кипения
компонента, смеси,
% °C
Вода .......................... 12 71
Четыреххлористый углерод............ 79 66,2
Метанол ............................ 61,3 61,4
Изопропиловый спирт ................ 44 71,7
Бензол ............................. 53 73,3
Растворимость акрилонитрила в воде и воды в • акрилонитриле
характеризуют приведенные ниже данные:
Температура," °C...................
Растворимость акрилонитрила в
воде, %................... • . .
Температура, °C..................
Растворимость воды в акрилонит-
риле, %..........................
24 41 70 84
8,0 8,4 10,0 10,8
0 32 48 54
2,3 4.4 5,9 6,4
60
8,5
134
6. Акрилонитрил
Давление пара
указано ниже:
акрилонитрила при различных температурах
Температура,
°C
-16,0
-14,5
-7,5
0,15
2,3
4,2
15,8
Давление,
мм рт. ст.
12,5
13,7
21,1
31,4
34,8
38,9
69,5
Температура,
°C
20,0
30,9
' 32,0
48,0
60,0
72,0
78,8
Давление,
мм рт. ст.
86,9
139,7
146,2
260
405
620
771
6.3. ПРИМЕНЕНИЕ АКРИЛОНИТРИЛА
Благодаря высокой реакционной способности акрилонитрил
служит исходным материалом для многочисленных органических
синтезов [155]. Первой большой областью применения акрилонит-
рила была промышленность синтетического каучука. В 1930 г. были
разработаны сополимеры с бутадиеном, которые появились на рынке
в 1934 г. под торговым наименованием пербунан. Основные типы —
пербунан (72% бутадиена, 28% акрилонитрила) и пербунан экстра
(60% бутадиена и 40% акрилонитрила). Наличие нитрильной
группы придает вулканизату стойкость к маслам и алифатическим
углеводородам, однако с увеличением количества акрилонитрила
ухудшается упругость.
В настоящее время основное количество акрилонитрила идет
на производство полиакрилонитрила, являющегося исходным про-
дуктом для полиакрилонитрильных волокон. Полимеризация акри-
лонитрила для выработки волокна протекает в большинстве случаев
радикально в присутствии окислительно-восстановительных систем,
например персульфатов с дисульфитами, дитионитами, тиосульфа-
тами и др. [156]. Переработка в волокна всегда происходит в рас-
творе. Формование волокна из расплава неосуществимо из-за высо-
кой температуры плавления, превышающей температуру разложе-
ния. Хорошим растворителем при формовании волокна является
диметилформамид [157, 158]. Из других растворителей, правда,
не получивших широкого распространения, следует назвать диметпл-
ацетамид, лактон, нитрилы оксикислот, циклические сульфоны,
динитрилы, триамид гексаметилфосфорной кислоты [159] и др.
Обычно формование волокна происходит по сухому способу, так как
метод высаждения не получил признания из-за трудностей при реге-
нерации диметилформамида. Полиакрилонитрильные волокна вошли
в обиход под торговыми наименованиями дралон, долан, редан,
орлон, акрилан, крезлан, вольприла, куртель, нитрон, кашмилон
и ДР-
Литература
135
HgS, NazS
Акрилонитрил
н2о
Сам по себе полиакрилонитрил не представляет большого
интереса. Необходимость улучшения свойств полистирола, прежде
всего повышения атмосферостойкости, стойкости к растворителям
и ударной вязкости, привело к созданию ударопрочного полисти-
рола — сополимеров на основе акрилонитрила, бутадиена и стирола
(АБС) [160], стирола и акрилонитрила (САН), значение которых
постоянно растет.
Разнообразно использование акрилонитрила при цианоэтилиро-
вании [161]. Он может присоединяться как к органическим, так
и к неорганическим соединениям:
Иминодипропионитрил, иминотрипропионитрил
Тподипропионптрил
Дицианоэтиловый эфир
Акрилонитрил взаимодействует также со спиртами (в том числе
и высокомолекулярными: поливиниловым спиртом, крахмалом, цел-
люлозой и др.), аминами, амидами и др. Особое признание получило
циано этилирование некоторых натуральных веществ. Присоедине-
ние или замещение галогенов (после гидрирования в пропионитрил)
с последующим омылением имеет некоторое значение при синтезе
гербицидов а,а,р-трихлорпропионовой кислоты и а,а-дихлорпро-
пионовой кислоты.
В последнее время описаны методы получения нитрила адипино-
вой кислоты восстановительной димеризацией, например, электро-
литическим путем. Он является исходным материалом обоих компо-
нентов найлона 6,6 — гексаметилендиамина и адипиновой кислоты.
Полиакриламид, получаемый гидролизом полиакрилонитрила, при-
меняется как средство для очистки воды и сточных вод, для обога-
щения урановых минералов.
В заключение следует назвать синтезы р-аланина, дилаурил-
тиодипропионовой кислоты (антиоксиданты для полипропилена),
пимелиновой кислоты.
Литература
1. Ch. Moureu, Ann. chinlie, 7, № 2, 187—191 (1894).
2. О. Bayer, Angew. Chem., 61, 229—241 (1949).
3. A. J. W e i t h, Chem. Eng. News, 31, 2763—2765 (1953).
4. Sh. Wake, High Polymers (Jap.), 1, 47—54 (1944).
5. В. С. M. Dorset, Textile Mfrg., 80, 87—91 (1954).
6. J. Mayor, Ind. chimique, 44, 98—100, 275—278 (1957); 45, 43-45 (1958).
7. M. A. Matthews, BIOS Final Rept., № 757, ср. О. T. S. Rept. P. B.
178/3 u 47 716; L. H. Smith, German Synthetic Fibre Development,
О. T. S. Rept P. B. 7416, S. 548 (1946).
8. R. F. M e r r i n g, R. L. James, Chem. Ind. Weekblad, 68, № 2,
19-24 (1951).
136
6. Акрилонитрил
9. R. L. Hanke, J. C. McNally, P. B. 34813 (FIAT Final Report
83b), The Production of Acrylnitrile in the I. G. Farbenindustrie Plants of
Ludwigshafen, Hills and Leverkusen, Technical Industrial Intelligence Branch,
U. S. Dept, of Commerce, July 18, 1946.
10. С. С. Бобко в, Хим. наука и пром., 2, 34—45 (1947).
И. Р. W. She г w о о d, Ind. Chemist, 34, 361—364 (1958).
12. Р. . W. Sherwood, Melliand Textilber., 43, 1183—1184 (1962).
13. K’ u n. Y u a n C h ’ i u, Hua Hsuch Tung Pao, № 5, 32—38 (1962).
14. P. W. Sherwood, Petro/Chem. Eng., 33, 610 (1961); 34, № 10, 48—
51 (1962).
15. P. W. Sherwood, Ind. Chemist, 39, 242—245 (1963).
16. M. S i 11 i g, Acrylonitril, N. Y., 1965.
17. Ch. F r u n z a, S. M a t a c h e, Rev. Chim. (Bucuresti), 16, № 3, 134—
138 (1965).
18. И. К. Колчин, К. А. Гуськов, Хим. пром., № 12, 881—887
(1965).
19. S. S е г b a n, G. G о i d e a, Rev. Chim. (Bucuresti), 16, № 12, 537—542
(1965).
20. Б. В. С у в о p о в и др., Успехи химии, 34, 1526—1549 (1965).
21. М. R. Lichtenberger, Rev. assoc, franc. techn. petrole, № 183,
29—35 (1967). 4
22. Герм. пат. 561397, 570031, 577686; F. Erlenmeyer, Liebigs Ann.
Chem., 191, 273Д1878).
23. Герм. пат. 299682, 1915; 679256, 1932; франц, пат. 729952, 1931.
24. Герм. пат. 496372, 1925; пат. США.2374051, 1941.
25. Р. Ku,rtz, Petroleum Refiner, 32, № И, 142—143 (1953); Liebigs Ann.
Chem., 572, 23, 24, 38 (1951); герм. пат. 728767, 1939.
26. Пат. США 2385552, 1941; герм. пат. 51834, 1935.
27. Франц, пат. 805563; итал. пат. 393682, 1941.
28. L. М. Р е t е г s et al.. Ind. Eng. Chem., 40, 2046—2053 (1948); пат. США
2375016, 1940.
29. R. Burns et al., Chem. Soc. (London), 1935, 400—406; пат. США 2183357,
1939; 2417748, 1943; 2417749, 1943; англ. пат. 424885, 1933.
30. Пат. США 2481826, 1949.
31. Англ. пат. 709337, 1954; пат. ФРГ 897560, 1953; пат. США 2691037, 1954.
32. F. Ve at ch et al., Chem. Eng. Progress, 56, № 10, 65—67 (I960).
33. Австрал. пат. 243791, 1963; белы. пат. 571200, 1958; англ. пат. 867438,
1960; канад. пат. 687664, 1964; D. A. S. 1127351, 1962; франц, пат. 1222460;
голл. пат. 120493, 1965; пат. США 2904580, 1959.
34. Англ. пат. 729013, 1955; пат. США 2736739, 1956.
35. Пат. США 3023226, 1962.
36. Пат. США 3118927, 1964.
37. М. А. Далини др., ДАН СССР, 145,1058—1060 (1962).
38. F. У е a t с h et al., Hydrocarbon Processing, 41, № 11, 187—190 (1962).
39. Англ. пат. 965173, 1964.
40. Бельг, пат. 577691, 1959; англ. пат. 848924, 1960; франц, пат. 1219512, 1959;
пат. США 3086041, 1963.
41. И. А. Коршунови др., Труды по химии И хим. технол., 2, 445—449
(1962).
42. D. A. S. 1162830, 1964.
43. D. A. S. 1241437, 1967.
44. Англ. пат. 977114, 1965; канад. пат. 684867, 1964; франц, пат. 1288404, 1962;
австр. пат. 224098, 1962.
45. Англ. пат. 978267, 1964; канад. пат. 684354, 1964; D. A. S. 1138753, 1962;
франц, пат. 1308958, 1963; Австр. пат. 226671, 1963.
46. Канад, пат. 684355, 1964; франц, пат. 1288403, 1288405, 1962; австр. пат.
224099, 1962; швейц, пат. 392493, 1965; пат. США 3153665, 1964.
Литература
137
47. R. Schonbecl et al., Chem.-Ing.-Techn., 38, 701—705 (1966).
48. R. S ch о nbe ck, Hydrocarbon Processing, 46, № 8, 124—126 (1967).
49. Бельг, пат. 611378, 1962.
50. Пат. США 3186955, 1965.
51. Пат. США 3280166, 1966.
52. Англ, пат. 981005, 1965; инд. пат. 90913, 1963; пат. США 3230246, 1966.
53. Австр. пат. 248410; 1966.
54. Бельг, пат. 621685, 1961; франц, пат. 1323299, 1963; австр. пат. 228188, 1963.
55. Австр. пат. 235257, 235264, 1964.
56. Австрал. пат. 262794, 1965; австр. пат. 225689, 1963.
57. Пат. ГДР 32976, 1964; франц, пат. 1367764, 1964.
58. Англ. пат. 884437, 1961; D. A. S. 1180739, 1964; швейц, пат. 404640,
1966; пат. США 3050546, 1964.
59. Англ. пат. 965200, 1964.
60. D. A. S. 1249257, 1249258, 1967.
61. Англ, пат, 933493, 1963.
62. Бельг, пат. 623669, 1963; франц, пат. 1365889, 1963; пат. США 3009943,
1962.
63. Бельг, пат. 684603, 1967.
64. Англ. пат. 967879, 1964; пат. ГДР 35390, 1965; франц, пат. 1325486, 1963;
австр. пат. 226672, 1963.
65. Пат. США 3200141, 1965; ср. швейц, пат. 385192, 1965.
66. Бельг, пат. 611239, 1961; канад. пат. 679979, 1964; D. A. S. 1243175, 1967.
67. Айгл. пат. 977311, 1964; D. A. S. 1242599, 1967; австр. пат. 225688, 1963;
пат. США 3135783, 1964.
68. Бельг, пат. 616585, 1962; D. A. S. 1243176, 1967.
69. Голл. пат. 300817, 1965.
70. D. A. S. 1197446, 1965.
71. Австрал. пат. 251950, 1964; англ. пат. 957022, 1964; D. A. S. 1206887, 1965;
франц, пат. 1269382, 1961; австр. пат. 225687, 1963; пат. США 3280167,
1966.
72. Бельг, пат. 631437, 1963..
73. Пат. США 3309395, 1967; японск. пат. 7215, 1966.
74. D. A. S. 1206886, 1965.
75. Франц, пат. 1343725, 1963.
76. Eu. Guccione, Chem. Eng., 72, № 6, 150—152 (1965).
77. Англ. пат. 904418, 1962.
78. Австрал. пат. 260108, 1965; англ. пат. 925495, 943374, 1963; канад. пат.
677634, 1964; франц, пат. 1301077, 1963-
79. Англ. пат. 913832, 1962; франц, пат. 1355561, 1962.
80. Англ. пат. 984370, 1965; франц. \пат. 1311683, 1963; пат. США 3262962,
1966; 3316182, 1967.
81. Пат. США 3226421, 1966.
82. Англ. пат. 1012225, 1965; франц, пат. 1329910, 1963.
83. Пат. США 3153085, 1964.
84. Бельг, пат. 595544, 1961; англ. пат. 885442, 1961; ср. англ. пат. 972403,
1963; франц, пат. 1335166, 1963.
85. Англ. пат. 929650, 1963.
86. Канад, пат. 685241, 1964; англ. пат. 874593, 1959; D. A. S. 1165015, 1964.
87. Франц, пат. 1474661, 1967.
88. Бельг, пат. 637628, 1964; франц, пат. 1384635, 1963.
89. Niederld. A. S. 6500697, 1965; белы. пат. 686093, 1966.
90. Niederld. A. S. 6500644, 1965; пат. США 3290354, 1966.
91. Пат. США 3322687, 1967.
92. Англ. пат. 937380, 1963; канад. пат. 683419, 1964; D. A. S. 1070170, 1961,’
франц, пат. 1292673; инд. пат. 73281, 1962; пат. США 3164628; 1965.
93. Англ. пат. 948014, 1964; D. A. S. 1247300, 1967.
138
6. Акрилонитрил
94. Франц, пат. 1505213, 1505215, 1967.
95. Пат. США 3164626, 1965.
96. Англ, пат 909907, 1962; D. A. S. 1248642, 1967; пат. США 3161671, 1964.
97. Бельг, пат. 636449, 1965; англ. пат. 1016031, 1965; ср. Chem. Industrie,
19, 624 (1967).
98. Jap. A. S. 7772/66, 1966.
99. Jap. A. S. 7773/66, 1966.
100. Jap. A. S. 7774/66, 1966.
101. Jap. A. S. 7854/66, 1966.
102. Jap. A. S. 7855/66, 1966.
103. Канад, пат. 623383, 1961.
104. Австрал. пат. 243315, 1964; D. A. S. 1165584, 1964; пат. ГДР 36656, 1964.
105. D. A. S. 1211155, 1966.
106. Англ. пат. 876446, 1959; канад. пат. 677475, 1964; D. A. S. 1203757, 1965,
107. Англ. пат. 904602, 1962; пат. США 3152170, 1964.
108. Франц, пат. 1360915, 1964.
109. Англ* пат. 967134, 1964.
НО. Англ. пат. 961357, 1964.
111. Англ. пат. 968924, 1964.
112. Англ. пат. 997490, 1965.
113. Англ. пат. 976008, 1964.
114. Англ. пат. 978520, 1964.
115. Англ. пат. 1007685, 1965.
116. Англ. пат. 897226, 1962; канад. пат. 684273, 1964; франц, пат. 1272308,
1962; пат. США 3094552, 1963.
117. Англ. пат. 933070, 1963; D. A. S. 1231675, 1967; франц, пат.' 1293088, 1962.
118. Бельг, пат. 661104, 1965.
119. Бельг, пат. 646937, 1964; пат. США 3340291, 1967.
120. Niederld. A. S. 6408649, 1965.
121. Бельг, пат. 655056, 1965; Niederld. A. S. 6411912, 1965.
122. Бельг, пат. 633450, 1963; англ. пат. 971038, 1964; D. A. S. 1205502, 1965;
ср. пат. США 3308151, 1967.
123. Nitrogen, 45, № 1, 44 (1967); Oil Gas J., 64, № 47, 143 (1966).
124. Бельг, пат. 641142, 1964.
125. Бельг, пат. 641143, 1964; пат. США 3338952, 1967.
126. Бельг, пат. 616585, 1962; 622025, 1963; 648052, 1964; пат. США 3198750,
1965.
127. J. Takeo, М. S u m i о, Bull. Japan Petrol. Inst., 2, 76 (1960).
128. Японск. пат. 3616/66, 1966.
129. Пат. США 3200081, 1965.
130. Англ. пат. 983755, 1965.
131. Бельг, пат. 632797, 1963.
132. Франц, пат. 1365889, 1964; австр. пат. 242678, 1965.
133. С. Р. Рафиков и др., ЖОХ, 32, 839 (1962).
134. Niederld. A. S. 6410205, 1965; белы. пат. 636567, 1965.
135. Франц, пат. 1355561, 1964.
136. Niederld. A. S. 6516563, 1966.
137. Niederld. A. S. 6404666, 1964.
138. Пат. США 3118928, 1964.
139. Франц, пат. 1396962, 1965; пат. США 3309395, 1967.
140. Англ. пат. 964613, 1964; D. A. S. 1205527, 1965.
141. Англ. пат. 875160, 1961.
142. Jap. A. S. 12107, 1962.
143. ' И. А. Коршунов и др., Труды по химии и хим. технол., 2, 445—449
(1962).
144. Бельг, пат. 612136, 1962; франц, пат. 1255121, 1960.
145. Jap. A. S. 19471, 1961.
Литература
139
146. Бельг, пат. 615298, 1962; 628287, 1963; англ. пат. 943374, 1962; франц, пат.
1366301, 1963.
147. Бельг, пат. 617523, 1962; англ. пат. 1019372, 1965.
148. Пат. США 3293280, 1966.
149. Пат. США 3287394, 1966.
150. Пат. США 3293279, 1966.
151. D. A. S. 1211154, 1966.
152. Англ. пат. 913832, 1962; франц, пат. 1255121, 1962.
153. Франц, пат. 1462595, 1966.
154. F. Schonbeck, Osterreich. Chemiker-Ztg., 68, 79—87 (1967).
155. D. W. McDonald et al., Hydrocarbon Processing Petroleum Refiner,
40, № 7, 149—154 (1961).
156. W. C. Thomas, Fortschr. Hochpolymerenforschg., 2, 401—441 (1961).
157. Герм. пат. 915034, 1942.
158. Пат. США 2404713—2404728, 1943.
159. Пат. США 2487859, 1948; 2641585, 1950; 2643986, 1951.
160. Пат. США 2439209, 1946.
161. Н. А. В ruson, Organic Reactions, v. V, N. Y., 1949, p. 79.
7. АЦЕТОН
к
Ацетон, один из простейших и важнейших кетонов, обнаружен
впервые в 1595 г. Либавиусом при сухой перегонке ацетата свинца.
Но лишь в 1832 г. Дюма и Либих точно определили его природу
и состав. До 1914 г. ацетон получали почти исключительно прй
коксовании древесины, но повышенный спрос на него во время пер-
вой мировой войны очень быстро привел к созданию новых методов
производства.
7.1. ПОЛУЧЕНИЕ АЦЕТОНА
7.1.1. Старые методы
Самый старый метод промышленного производства ацетона
заключался в сухой перегонке ацетата кальция, получающегося
при нейтрализации известью древесного уксуса, который образуется
при коксовании древесины [1]. Сейчас этот метод уже не находит
применения, так как ацетон в этом случае содержит слишком много
примесей, а исходный материал дефицитен.
Известны также способы получения ацетона путем бактериаль-
ного расщепления углеводов (крахмала, сахаров, мелассы), причем
в качестве побочных продуктов образуются бутиловый или этиловый
спирт [2—4]. Ацетон и бутиловый спирт получаются в мольном
соотношении от 2 : 1 до 3 : 1.
В настоящее время этот метод в США еще занимает определенное
положение для получения некоторого количества ацетона.
В Германии был разработан технологический процесс производ-
ства ацетона на основе уксусной кислоты [5]. При 400 °C через
контакты из церия пропускали уксусную кислоту:
2сн3соон 400 °с’ Се-> сн3сосн3+со2+н2о
Такой ацетон отличается особой чистотой.
Ацетон производят также из ацетилена прямым синтезом:
2СН^СН + ЗН2О > СН3СОСН3-|-СО2-|-2Н2 + 40 ккал/моль
Ацетилен вступает во взаимодействие с водяным паром при
450 °C в присутствии катализаторов (в частности ZnO или Fe2O3 —
ZnO) [6].
7.1. Получение ацетона
141
В США фирма Celanese Corp, производит ацетон наряду с дру-
гими кислородсодержащими соединениями путем окисления смеси
бутан — пропан небольшим количеством чистого кислорода при
330—370 °C и 7—10 кгс/см2. На долю ацетона приходится 5—7%
общего количества оксидата [7]. Более высокий выход получают
при использовании изобутана.
7.1.2. Получение из изопропилового спирта*
До настоящего времени основное количество ацетона получают
все еще дегидрированием изопропилового спирта [13 J:
СН3СН(ОН)СН3 —> СН2СОСН3 + Н2-15,9 ккал/моль
Дегидрирование протекает при 35Q—400 °C в присутствии таких
катализаторов, как сплав железо — медь — цинк [8], окись цинка
или окись цинка с 4,5% углекислого натрия [9], медь, свинец
и т. д. [10].
Водород
Рис. 38. Схема установки по производству ацетона:
1 — компрессор для водорода; 2 — система регенерации катализатора; 3 — испари-
тель; 4 — камера сгорания; 5 — емкость для сырого ацетона; 6 — емкость для рас-
твора ацетона в воде.
Упрощенная схема установки для производства ацетона из
изопропилового спирта [11] приведена на рцс. 38. Изопропиловый
спирт обрабатщвается водородом при высоких температурах, причем
1 объем водорода поглощает примерно 1 объем паров изопропилового
спирта. Смесь при 380 °C проходит через кожухотрубный реактор
с трубами из хромоникелевой стали, в котором находится катализатор
* Получение ацетона из кумола будет описано в главе о синтезе кумола.
142
7. Ацетон
в виде кусочков пемзы, пропитанной ацетатом цинка (~7%
ZnO). Максимальная конверсия изопропилового спирта составляет
около 97% [12], время контакта 1 с. Реакционные газы охлаждают,
образующийся 20%-ный ацетон концентрируют и очищают перегон-
кой. Водород промывают от ацетона в противотоке воды, концентра-
ция водорода ~99%. Регенерацию катализатора проводят через
10 суток, выжигая загрязнения смесью азота с 2% кислорода. Срок
службы катализатора около 6 месяцев.
В последнее время все чаще получают ацетон путем окисления
изопропилового спирта воздухом; при этом образуется также пере-
кись водорода:
СН3СН(ОН)СН3+О2 —> СН3СОСН3 + Н2О2
Этот метод играет определенную роль при производстве глице-
рина в отсутствие хлора.
7.1.3. Метод прямого окисления пропилена
Разработанная Шмидтом [15—17] и внедренная в промышлен-
ности технология прямого синтеза ацетальдегида из этилена
CH2=CH2 + PdCl+H2O —> CH3CHO + Pd + 2HC1
может служить также для непосредственного получения ацетона
из пропилена. В этом случае пропилен (или богатая пропиленом
смесь газов) под действием раствора катализатора PdCl2 и СнС12
в соляной кислоте (рис. 39) превращается в ацетон; восстановлен-
ный катализатор снова окисляется воздухом. При этом протекают
следующие реакции:
СН2=СНСН3+ PdCl3+H2O —> CH3COCH3 + Pd+2HC1
2CuCl2+Pd — -* 2CuCl+PdCl2
2CuC1 + 2HC1 + i/2O2—> 2CuCl2+ H2O
СН2=СНСН3Н-1/2О2 --> CH3COCH3
Выход составляет 92—94% при 90—120 °C и давлении
9—12 кгс/см2. В качестве побочных продуктов получается 0,5—1,5%
пропионового альдегида и ~2% моно- и дихлорацето нов. Предпола-
гают, что реакция идет через комплекс [PdCl2(OH)C3He]', который
в присутствии воды гидролитически расщепляется на ацетон, палла-
дий и соляную кислоту [18]. Агентами повторного окисления будут
СнС12 или FeCl3, а также смеси обоих соединений [19].
Реакцию можно проводить при комнатной температуре, но повы-
шенные температуры предпочтительны [20]. В большинстве случаев
pH раствора составляет 3—4. Давление также благоприятно влияет
на ход реакции. Обычно работают с очень разбавленным раствором
7.2. Свойства ацетона
143
PdCl2 (частично вместе с ацетатом меди) [21, 22]. Использование
концентрированного раствора PdCl2 ускоряет реакцию.
Модифицированные катализаторы акролеина находят применение
и при прямом окислении пропилена в ацетон. Катализаторы на
основе МоО3 или Bi2O3 в присутствии Н3РО4 или Н3ВО3 при 375 °C
дают наряду с ацетальдегидом, уксусной кислотой, формальдегидом,
этилацетатом и акролеином также и ацетон [23, 24], причем хорошее
действие оказывает добавка Ag [25]. Можно использовать фосфоро-
молибдат Bi [26] на А12О3 и фосфоромолибденовую кислоту на
Sid2 (260 °C, 1 с) [27].
Рис. 39. Схема получения
ацетона (фирма Aldehyd
GmbH/Hoechst Uhde Са):
1 — реактор; 2 — регенератор;
3 — выделение сырого ацетона;
4—дегазатор; 5 — колонна для
дистилляции.
Вышеназванные методы еще не применяются для промышленного
производства ацетона. В промышленности в основном принято дегид-
рирование дешевого и легкодоступного изопропилового спирта.
Кроме того, ацетон образуется в большом количестве в виде побоч-
ного продукта при синтезе кумола.
7.2. СВОЙСТВА АЦЕТОНА
Ацетон характеризуется следующими свойствами:
Температура плавления, °C ...................
Температура кипения, РС......................
Плотность
(? ......................................
Р1!.......................................
О 30
Р 4.......................................
Плотность пара по отношению к воздуху . . .
Показатель преломления n'ff..................
Дипольный момент, D.........................
Поверхностное натяжение при 20 РС, дин/см . .
Критическая температура, РС .................
Критическое давление, кгс/см2................
Критическая плотность, кг/м®.................
Теплота плавления при 95 РС, ккал/моль . . . .
Теплота испарения, кал/г
при 56,3 рС...............................
0 °C.................................
Энергия образования, ккал/моль ..............
-94,9
56,5
0,81378
0,79705
0,77986
2,0025
1,35998
2,80
47
0,268
1,366
125,28
140,5
56,2
144
7. Ацетон
Энтропря, кал/моль ..................................... 47,8
Теплота сгорания, ккал/моль............................. 430,9
Мольная теплоемкость, калДмоль • °C).................... 29,9
Пределы взрываемости, %
верхний............................................ 15,3
нижний .............................................. 1,6
Температура воспламенения, °C........................ —17
Диэлектрическая проницаемость......................... 21,4
Давление паров, мм рт. ст.
при 20 °C .... . 179,63 60 °C............. 860,48
30 °C . . 281,00 80 °C.............. 1611,05
40 °C... 420,15 100 9С.............. 2797,27
50 °C... 620,86 140 °C.............. 6974,43
7.3. ПРИМЕНЕНИЕ АЦЕТОНА
Объем производства аЦетона в США различными методами пока-
зан ниже (в тыс. т):
1963 1966
Дегидрирование (нз изопропилового
спирта).............................. 393 393
Синтез из кумола.......................110 146
Ферментация............................ 23 23
Окисление пропана и бутана ............ 16 16
48% ацетона в США потребляется для синтеза различных про-
дуктов, в ФРГ это количество составляет только 5%, правда, 45%
падает на пластмассы (полиметилметакрилат, бисфенол А).
Разнообразны продукты, получаемые при конденсации ацетона
в щелочной среде [28]. При температуре 10—20 °C в метанольном
растворе ацетон димеризуется в присутствии небольших количеств
щелочи в диацетоновый спирт, из которого получаются следующие
вещества:
Н2, 100 °C,
2СН3СОСНз (СН3)2С(ОН)СН2СОСН3 ЗООкгс/см* (СНа)2С(ОН)СН2СН(оН)бн2
Диацетоновый спирт - Гексиленгликоль
100°С,
0,05%Н3РО4 ‘
(СН3)2СНСН2СН(0Н)СН3
Метилизобутилкарбинол
Н2, 104 “С
100 кгс/см*, Ni
СН3ОН, щелочь
i
(СН3)2 С = СНСОСН3
Окись мезитила
110°С,
Pd,
1 кгс/см*
I
i
-> (СН3)2С(ОСН3)СН2СН(ОН)СН3
Метиловый эфир гексиленгликоля
(СН3)2СНСН2СОСН3
Метилизобутилкетон
7.3- Применение ацетона
145
Возможные превращения изофорона
146
7. Ацетон
Гексиленгликоль добавляется преимущественно к топливу.
Окись мезитила, образующаяся при отщеплении воды, способна
вступать в различные реакции присоединения, например с метанолом
в присутствии небольших количеств щелочи. При гидрировании
окиси мезитила получают метилизобутилкетон — очень важный
растворитель. Гидрирование в жестких условиях дает метилизобу-
тилкарбинол. Метилизобутилкетон и метилизобутилкарбинол явля-
ются очень хорошими растворителями для поливинилхлорида,
сополимеров винилхлорида, производных целлюлозы, хлорирован-
ного каучука и т. д. В большинстве случаев по растворяющей спо-
собности они превосходят сложные эфиры.
При каталитической конденсации ацетона в присутствии осно-
ваний при 200 °C наряду с'та к называемыми изоциклитонами обра-
зуется изофорон, который также является основой разнообразных
синтезов (см. схему на стр. 145). Сам изофорон до сих пор занимает
исключительное положение как растворитель виниловых лаков;
он придает лакам горячей сушки блеск и прочность. Мировое про-
изводство изофорона в 1967 г. составляло 16 тыс. т. При осторож-
ном гидрировании получают 3,3,5-триметилциклогексанон, который
идет на получение перекисей и служит растворителем. Образую-
щийся в результате полного гидрирования 3,3,5-триметилциклогек-
санол является важным компонентом специальных пластификаторов,
особенно во взаимодействии с длинноцепными алифатическими моно-
и дикарбоновыми кислотами. Но еще большее значение придают
продукту его окисления азотной кислотой — а,а,у-триметиладипи-
новой кислоте. Эту кислоту этерефицируют в специальные пластифи-
каторы и превращают через диметиловый эфир в 2,2,4-триметилгек-
сандиол-1,6 путем энергичного гидрирования. Кроме того, кислоту
можно превратить гидрированием динитрила в 2,2,4-триметилгек-
саметилендиамин.
Другой путь получения диамина из изофорона заключается в
действии на него цианистого водорода и образовании нитрила изо-
форона — 3,3,5-триметил-5-цианоциклогексанона, который при спе-
циальных условиях можно восстановить в 1-аминометил-1,3,3-
триметил-5-аминоциклогексан (изофорондиамин).
Диамины легко перевести в диизоцианаты и далее использовать
в качестве отвердителей эпоксидных смол. Особенно велика их
роль для изготовления прозрачных полиамидов. Кислотными компо-
нентами будут алифатические и ароматические дикарбоновые кис-
лоты. Полиамиды идут на изготовление смол, связующих для лаков,
клеев, высококачественных пластмасс.
ЛИТЕРАТУРА
1. U 1 1 m a n n, Enzyklopadie der technischen Chemie, Munchen, Bd. 1, 1951,
S. 106.
2. Пат. США 1329214, 1920.
Литература
147
3. J. Н. Northrop et al., Ind. Eng. Chem., 11, 723 (1919).
4. W. H. P e t e r s о n et al., Ind. Eng. Chem., 13, 757 (1921).
5. Герм. пат. 298851, 1916.
6. Англ. пат. 299048, 1928; 313897, 1928; 330350, 1929; 472093, 1936.
7. R. Е. Meyer, Oil. Gas J., 54, Ks 7, 82 (1956).
8. Пат. США 1952702, 1933.
9. Пат. США 1895516, 1933.
10. Пат. США 1895528, 1895529, 1933.
11. J. A. Mathews, L. W. Wood, BIOS Target C22 Final Rept. Nr. 131
item. Nr. 22.
12. H. J. Kolb, R. L. Burwell, J. Am. chem. Soc., 67, 1084 (1945).
13. Англ. пат. 173539, 1920; пат. США 1365035, 1497817, 1918.
14. U 1 1 m a n n, Enzyklopadie der technischen Chemie, 3. Aufl., Munchen —
Berlin, 1953, S. 38.
15. J. S m i d t et. al., Angew. Chem., 71, 176—182 (1959).
16. W. H a f n e r et al., Chem. Ber., 95, 1575—1581 (1962).
17. J. S m i d t, H. К г e к e 1 e r, Erdol und Kohle, 16, (6—1), 560—563
(1963).
18. J. S m i d t, Chem. and Ind., 1962, 54—61.
19. D. A. S. 1142593, 1963; англ. пат. 884962, 1961; D. A. S. 1061767, 1960.
20. D. A. S. 1183488, 1964.
21. D. A. S. 1145602, 1963.
22. Англ. пат. 878777, 1961.
23. D. A. S. 1197070, 1965.
24. Бельг, пат. 620834, 1963.
25. Франц, пат. 1397639, 1965.
26. Пат. США 2874191, 1959.
27. Итал. пат. 640465, 1962.
28. К. Schmitt, Chem. Industrie, 18, № 4, 204—210 (1966).
10*
8. АКРИЛОВАЯ КИСЛОТА
Акриловая кислота [1—6] впервые была получена Редтенбахе-
ром [7] окислением акролеина на воздухе. Интенсивные исследова-
ния акрилатов и полиакрилатов [8] привели в 1927 г. к созданию
метода промышленного производства акрилатов фирмой Rohm
und Haas Со. [9—12].
8.1. Получение акриловой кислоты различными способами
Существует семь промышленных методов получения акриловой
кислоты и акрилатов. Если более старые методы базируются в основ-
ном на ацетилене, то в будущем решающую роль будут играть про-
цессы, где исходным продуктом является пропилен.
До сих пор важнейшим остается метод, основанный на работах
Реппе о присоединении СО и воды или спиртов к ацетилену при
каталитическом воздействии Ni(C0)4. Синтез акрилатов протекает
при 35—45 °C под давлением стехиометрически по уравнению:
4C2H2+4ROH + Ni(CO)4+2HCl > 4CH2=CH-COOR +NiCl2 + H2
Образующийся NiCl2 снова превращается в Ni(C0)4 (метод-фирмы
Rohm und Haas Со.).
Процесс фирмы Dow Badische Chemical Со., освоенный с 1960 г.,
также базируется на трудах Реппе. Ацетилен реагирует с СО и Н2О
при 220 °C и давлении 100 кгс/см2 в тетрагидрофуране в присутствии
растворимых соединений никеля и дает акриловую кислоту, которая
впоследствии этерифицируется [13].
На фирме Celanese Corp, в 1958 г. внедрен разработанный фир-
мой В. F. Goodrich Со. метод, основанный на кетене [14—21].
Кетен, получаемый пиролизом уксусной кислоты или ацетона, пре-
вращается в p-пропиолактон при 10—20 °C в растворителе, напри-
мер диалкиловом эфире, в присутствии А1С13 или других катализа-
торов Льюиса:
Alai, СН2—С=О
СН2=С=О + НСНО ---| I
СН2-0
Р-Пропиолактон переводится в акриловую кислоту в газовой
фазе в присутствии фосфорной кислоты при 150—180 °C [22]. Акри-
ловую кислоту можно получать также при 150 °C путем термической
8.2. Получение акриловой кислоты из пропилена или акролеина 149
деполимеризации поли-0-пропиолактаиа [23—24], который обра-
зуется при длительном хранении 0-пропиолактона (полимеризация
последнего ускоряется кислотами, основаниями или солями).
Предложен новый усовершенствованный способ прямого получе-
ния акрилатов [25, 26]. Кетен взаимодействует при 70—110 °C
с метилалем с образованием метилового эфира 3-метоксипропионовой
кислоты в присутствии BF3, TiF4, HPFe, BF3-2(C2H5)2O и других
катализаторов. В результате пиролиза этого сложного эфира при
190—220 °C в присутствии кислоты выделяется метилакрилат [27, 28].
Процесс фирмы Union Carbide Corp, основывается на окиси
этилена, которая превращается в этиленциангидрин либо при тем-
пературах выше 90 °C без катализатора [29], либо при 55—60 °C
в присутствии NaOH, CaO, MgCO3[30], цианидов щелочноземельных
металлов или третичных аминов [31 ]. Этиленциангидрин реагирует
при 150 °C с 75 —80%-ной серной кислотой и спиртами и дает акри-
латы [17, 32—33]:
HOCH2CH2CN+ROH + H2SO4 •*—> CH2=CHCOOR + NH4HSO4
Фирма Diamond Alkali Со. предложила метод прямого присо-
единения СО к окиси этилена с образованием акриловой кислоты при
120— 250 °C и давлении 200—500 кгс/см2 в присутствии Со2(СО)8 [34L
Для фирм, производящих акрилонитрил, интерес представляет
метод его окисления при ПО °C [35—37]:
H2C=CHCN+H2SO4+H2O —> H2C=CHCONH2 • H2SO4
H2C=CHCONH2 H2SO4+ ROH £ ch2=chcoor + NH1HSO4
По этому или подобному методу работают французская фирма
Ugilor SA, японские фирмы Tokai Gas and Chemical Co. и Toyo
Koatsu Industries Ltd. (опытная установка), американская фирма
Sohin Chemical Co. (опытная установка).
До сих пор не внедрены в промышленность следующие методы
получения акрилатов: альдольная конденсация формальдегида с уксус-
ной кислотой в паровой фазе на катализаторах из цеолита Са при
375—385 °C [38—39]; взаимодействие формальдегида, спиртов и
уксусной кислоты [40]; реакция формальдегида и эфиров в присут-
ствии солей щелочных металлов карбоновых кислот [41], метабора-
тов Na или К [42] и цеолитов [43].
8.2. ПОЛУЧЕНИЕ АКРИЛОВОЙ КИСЛОТЫ ИЗ ПРОПИЛЕНА
ИЛИ АКРОЛЕИНА
Акриловую кислоту можно получить из пропилена либо одно-
ступенчатым прямым окислением, либо двухступенчатым окислением
вначале до акролеина, который или выделяют, или окисляют в акри-
ловую кислоту во втором реакторе (рис. 40). В табл. 15 приведены
150
8. Акриловая кислота
Катализаторы для получения
Катализатор Состав исходных продуктов, % Температура реакции, °C
Пропи- лен Акро- леин Воздух (или Os) Водяной пар
МоО3 на SiO2 13,5 43,2 43,2 376
Молибдат Со — 10 54 36 35Г
Молибдат Со — X X х —
Молибдат Со — X X х —
Молибдат Со — 3-5 25-30 40 330—385
Молибдат Со — 10 60 30 360
Молибдат Со с добавками — X X х —
Молибдат Со с добавками — X X X —
Молибдат Со с добавкой Ni .... .— X X
Молибдат Со; окись Sb —Sn (наряду с окисью Sb — Sn—»V и окисью
Sn—V) X — X X —
Фосфоромолибденовая кислота . . — 20 10 60 425
Фосфоромолибденовая кислота . . — 10,5 26,3 63,2 468
Молибдат Bi - 9.5 61,8 28,7 400
Bi2O3; фосфоромолибдат+Fe203 . . 26 — 27 46 —
Фосфоромолибдат Bi X X х
Молибдат Bi (I ступень) 1 Ч. —> 1,5 ч. (О2) 5,8 ч. 490/3,8 КГС/СМ2
Фосфоромолибдат Bi (II ступень) — X X X 467/3,5 КГС/СМ2
Фосфоромолибдат + 0,1—5°% As2O5
на SiO2 1 Ч. .— 1,5 ч. 6 ч. 514
Молибдат Co + As2O5 10 — 50 40 450
Молибдат Bi—Со — 10 54 36 350
Молибдат Bi —Со . . X — X (О2) X 420-480
8.2. Получение акриловой кислоты из пропилена или акролеина 151
акриловой кислоты из пропилена
Таблица 15
Время контакта, с Конверсия, % Селективность, % Выход, % Примечание Литера- тура
19,8 32,3 70 22,5 [44]
3,9 68 85,5 58,2 — [45]
— — 76,5 — — [46]
— 55-70 60-80 — — [47]
1-30 52-89 63-84 — — [48]
3,6 60 73 43,8 Улучшение механической устойчи- вости в результате добавки молиб- дата Мп или Cd [49]
— — — — Добавка 0,1—10% молибдата Мп, Cr, Cd, Sn, Al, Zn, Sb, U или Th [50]
— —. — — Аналогично [50], получение катали- затора при добавке NH3 или амина [51]
— — — — — [52]
— — — 41 Двухступенчатый метод; I ступень— на окиси Sb —Sn; 2 ступень—на молибдате Со [53]
15 22,5 85 19,2 — [54]
— — 85,3 — Улучшение механической устойчи- вости носителем SiC [55]
3,2 — — 30,3 Наряду с этим предлагается молиб- дат W, Ni, Ti, Fe, V, Sn, Sb и Се [56]
— — — — Добавка 0,2 объемн. % НВт, тем самым понижается выход акроле- ина по сравнению с выходом ак- риловой кислоты [57]
— — — — — [54]
0,4 72,3 — — Двухступенчатый процесс; I ступень: 50% акролеина, 2,7% акриловой кислоты [58]
0,5 87 — - 23,6 II ступень: 24,3% акролеина [58]
0,5 23,9 Наряду с 24,1% акролеина [59]
36 47 44,6 21,0 — [60]
5—8 70 72-95 — Добавка 5% ЕИгОз повышает селек- тивность молибдата Со [61, 62]
— — — — Исследования кинетики реакции [63]
152
8. Акриловая кислота
Катализатор Состав исходных продуктов, % Температура реакции, °C
Пропи- лен , Акро- леин Воздух (или О А Водяной пар
Фосфоромолибдат Li и окись As . . 14 — 71 15 400
Молибдат Sn на фарфоре 1 я. 1 я. (О2) 2 я. 340
Окись Sn —Мо (3: 1) — — — —
Молибдат Sb—Sn или окись Sb — Sn —Мо (9 : 3 : 12) X X X 435
Фосфоромолибдат S’n — 8,3 41,7 50 425
Соли V или Се кремнемолибденовой или оловомолибденовой кислоты — 22 56,7 21,3 350
Молибдат Sn + As2O5 .. . 10 — 50 40 ' 406
Молибдат Ti —Sn —Со — X X X —
Молибдат Fe 13,3 — 33,4 53,3 270
Молибдат Fe, Ст, V и Ti ..... — X X X —
Фосфоромолибдат Fe — Ст 7 я. —. 60 я. 23 я. 380
Фосфоромолибдат Fe—Bi 1 я. — 1,5—Зя. (О2) 8,2 (О2) 3—40 я. 350-450
Fe7Bi2PMoi2O59 на SiO2 ...... 7,3 3,6 — 400
FeCo0,3Nii2BiPMoi2O5e — 1 я. 8 я. 15 я. 340
Молибдат Ti на A12O3 — 10 50 40 275
Фосфо ро-или боромолибдат W+10% активатора — — — —
В'2,5-751Рз-571МО23-229в(Н2О)п_25п . . — 10 51 60 427
Борофосфомолибдат Bi на SiO2 . . Борофосфоромолибдат Fe, Ei ... — X х X 365
— х х X 365
Sn5_i2Sb5_12B3o-i2oP30-12oMOi2(JA: . . — 4,5 4,5 (О2) 15,0 390
Боромолибдат Со 5,3 — 75,1 (N2) 42,7 52 425
Борофосфаты тяжелых металлов, например окись Си, Bi, В, Р . . 1,1 я. — 8,25 ч 6 ч. 440—460
Молибдат Со + теллурид Си (0,5—1 %) 10 48 42 438
Теллуромолибдаты тяжелых метал- лов, например окись Ti—Те—Мо 10,7 — 59,3 . 30 400
8.2. Получение акриловой кислоты из пропилена или акролеина 153
Продолжение табл. 15
Время контакта, с Конверсия, % Селективность, % Выход, % Примечание Литера- тура
— 70 50-70 — Наряду с 10—15% акролеина; улуч- шенная селективность в резуль- тате добавки Li2O [64]
— — — — Исследования реакции [65]
— — — — При соотношении SnO2 : МоО3 —3 : 1 наилучшая селективность [66]
2,5 85 63 53,5 — [67]
12 60 60 36 Предложен фосфоромолибдат Sb [54]
5 74,8 — — - [68]
4 — — 18,4 Наряду с 11,9% акролеина [69]
— — — — — [70]
— — — 2,9 Наряду с 9,5% акролеина и 36,5% уксусной кислоты [71]
— — — — — [72]
8 82,5 15,6 12,9 Наряду с 52,6% акролеина [73]
— — — 60 — [74]
— — — 54,3 53 ч. СО2, 19,9 ч. СО и 8 ч. Н2 в качестве разбавителя [75]
4,5 80 62,5 50 Общий состав: Fe1_sCo0-7Nig_i4 Bii_gPMo12O45 [76]
— 29 58 16,8 Предложен молибдат Ti —W И вольфрамат Ti [77]
— — — — Активатор: окись Fe, Мп, Cr, Bi или V [78]
.— 43,7 46 20 — [79]
— 46 56 , 25,8 — [80]
— 81 75 61 — [80]
3,2 87,3 80,2 70 — [81]
0,44 32 42 13,5 Кроме того, боромолибдат Ni, фос- форомолибдат Со и молибдат Со —V [82, 83]
1 40-45 40-42 — В качестве катионов предлагают: Си, Ag, Fe,.Co, Sb, Bi, Mo, VV, U, Pb, Sn, Ce, Th, Nb, Та, V или Cr [84]
2,9 27 43 11,6 33% акролеина [85]
9,3 75 18,5 13,6 69% акролеина; в качестве катио- нов предлагают Ti, Bi, Sn, Sb, W, V, Cr, Cu, Ni, Mn [86]
154
8. Акриловая кислота
Катализатор Состав исходных продуктов, % Температура реакции, °C
Пропи- лен Акро- леин Воздух (или О 2) Водяной пар
Фосфоромолибдат Те . 10 50 40 394
Фосфоромолибдат Те 6,4 — 89,6 — 370
Фосфоромолибденовая крслота + ТеО2 7 — 52 41 486
Молибдат Со + 1% ТеО2 7 — 52 41 433
Фосфоромолибдат Ni—Те 2 — 10 (О2) (88% N2) — 426
Фосфоромолибдат Мп—Те — — — — —
Те—Мо—Мп2Р2О7 1 ч. (про- пан) — 2,6 ч. (О2) 2,69 ч. 385
Молибдат Мп + ТеО2 — 10,6 52,1 37,3 446
Катализатор на основе МпРОд, МоО3
и ТеО2 1 Ч. — 2,95 ч. (О2) 4,16 ч. 378
Фосфат Cu + HReO4-J-TeO2 .... X — X X 400
Молибдат Со— Sn —Те (7:1:12:2) 360
Молибдат W—Sn — Те 1 — 60 39 330
СгОз на SiO2 — 6,8 ч. 8,8 ч. (О2) 68,5 ч- 250
Окись Sn — Sb 4-молибдат Со . . . —
Окись Sn — Sb, Sn—V, Sn — Fe . . Ванадат Sb, активированный тяже- X — X X —
лыми металлами, например вана-
дат Fe —Sn—Sb 7 — 63 30 370
Ванадат Sb — Sn с катионом тяже- лого металла, например окись
Fe —Sb—Sn —V 5 — 60 35 315
Окись Си —Sb —Sn —V 5 — 50 45 355
Окись Sb и 5 4-25% V2O5 — 6 47 47 350
Ванадат Sb на SiO2 — 10 50 40 381
Фосфорованадат Sb — 10 50 40 381
Фосфо рованадиевая кислота . . . . — 8 37 53 377
Фосфорованадат Sn — 8 37 53 377
Фосфорованадат Си на А12О3 или
SiO2 — X X X —
8.2. Получение акриловой кислоты из пропилена или акролеина 155
Продолжение табл. 15
1 Время контакта, с Конверсия^ % Селективность, % Выход, % Примечание Литера- тура
3,75 30,6 72 22 — [87]
5,3 — — 25,3 26,6% акролеина; процесс в кипя- щем слое; 40% пропана в качестве разбавителя [88]
0,5 — — 27,5 43,9% акролеина [89]
1 81 44,4 36 Наряду с 14% акролеина; предла- гают молибдат Ni + ТеОг [90]
2,6 — — 19 21% акролеина [91]
— — 44-55 — 10—35% акролеина; добавка Мп уд- линяет срок службы катализатора [92]
40,6 — — 46 24,2% акролеина [93]
— — — 2 4,5% акролеина [94]
54 46,8 —- 39,5% акролеина [95]
2-70 — — — Образование акриловой кислоты и акролеина; вместо фосфата Си предлагают еще фосфат Ni и фос- форомолибденовую кислоту [96]
— — — — — [97]
1,8 — 62,3 — Возможна добавка СоО; акролеин количественно окисляется анало- гичным катализатором [98]
19 6,8 48,6 3,3 — [44]
— — — — Двухступенчатое окисление [99]
— — — — — [100]
3 ' — — 26 44% акролеина; в качестве катионов предлагают: Ti, Cr, Мп, Те, Со, Ni, Си, Мо, Sn, W [101]
4 4 — — 14,2 20 45,2% акролеина 35% акролеина [1021
3-4 — — 73,5 — [103]
‘ 3,75 62 78 48 — [104]
— — — — — [105]
0,5 33,2 64,7 21,5 — [106]
2 47,5 — — — [106]
— — - — — Прежде всего, для окисления акро- леина [107]
156
8.- Акриловая кислота
Катализатор Состав исходных продуктов, % Температура реакции, °C
Пропи- лен Акро- 1 леин Воздух (или О2) Водяной пар
Фосфорованадат Мп . Арсено ванадат Си; окись Си —As— -V (1:3:2) Фосфороарсенованадат Си; окись Си— As—Р—V(1: 2 : 2 : 2) . . . . Молибдат 'V; окись V—Мо (1:1) Молибдат VO и добавки V2O5, ванадат Ag или Ag2O . . . . Фосфоровольфрамовая кислота . . Фосфофомолибденовольфрамовая ки- слота 10 10 X X 22 10,4 6,3 X 49 49 56,7 54,2 9,1 41 41 X 21,3 35,4 84,6 386 450 350 371 420
данные о катализаторах для одно- и двухступенчатого процесса
получения акриловой кислоты. Из всех окисей металлов наилучшим
катализатором для окисления акролеина является МоО3 [115]. j
Рис. 40. Схема прямого окисления пропилена
в акриловую кислоту:
I, 2 — реактор; 3 — холодильник; 4 — блок очистки.
Описаны также методы окисления пропилена или акролеина
в акриловую кислоту в паровой или жидкой фазе.
Окисление акролеина в жидкой фазе можно проводить в щелоч-
ном растворе с использованием СнО или Сн(ОН)2 [116], Ag2O [117]
или катализаторов СнО—Ag2O [118]; максимальный выход 98%.
Окисление протекает уже при комнатной или несколько повышенной
8.2. Получение акриловой кислоты из пропилена, или акролеина
157
Продолжение табл. 15
Время контакта, с Конверсия, % Селективность, % Выход, % Примечание Литера- тура
— — — То же [108]
9,5 50 10 5 5% акролеина
2,5 54 4 2 24% акролеина [109]
— — 66,9 — — [110]
5 74,8 26,2 19,6 В качестве добавок рекомендуют ге- терополикислоты Се—Мо, молиб- дат Ag, Fe (II) или Fe (III) [111]
— — — — — [112]
9,3 84,3 50,2 42,4 — [ИЗ]
— — 53 — Предложена фосфор омолибденована- довольфрамовая кислота [114]
температуре [119], но может идти и при 4 °C на катализаторах
Ag [120].
Окисление акролеина в жидкой фазе происходит в присутствии
нафтената Со и хелатов Со [121, 122], ацетата Мп или Ni в уксусной
кислоте при 200 °C и давлении 20 кгс/см2 в присутствии НВт [123].
Уже при 20—34 °C акролеин окисляется ванадиевой кислотой в
уксусной кислоте [124].
Эффективными промоторами окисления акролеина кислородом
являются триалкил- или триарилфосфаты. В присутствии стеарата
Ni и трибутилфосфата акролеин окисляется в акриловую кислоту
в бензоле при 65 °C и 6 кгс/см2 с конверсией 28% и селективностью
87,5% [125]. Промоторами при окислении акролеина кислородом
в жидкой фазе, могут быть и ароматические нитросоединения, напри-
мер, возможно окисление- при 50 °C и 5 кгс/см2 в гексане в присутст-
вии нитробензола [126, 127]. При 75 °C смесь пропан — пропилен
окисляется с образованием окиси пропилена или акриловой кислоты
[128]. Предложен целый ряд катализаторов для окисления акроле-
ина в бензоле: молибдат Си (при 50 °C и давлении кислорода
10 кгс/см2 получают 67% акриловой кислоты); молибдат Ti (62%),
молибдат Со (64%), смесь молибдатов [129], иод [130]. Возможно
окисление под давлением и без добавки катализатора (при 25—30 '“С
и давлении кислорода 5 кгс/см2 конверсия 32%) [131].
Перекись водорода окисляет акролеин в акриловую кислоту в
присутствии вольфрамовой кислоты [132], гетерополикислоты на
158
8. Акриловая кислота
основе вольфрама (например, селено-вольфрамовой кислоты) [133]
или SeO2 [134]. Вместо перекиси водорода можно использовать и
органические гидроперекиси в присутствии SeO2 [135] или СгО3
[136]. Можно проводить комбинированное окисление пропилена,
например, окислить его в акролеин на катализаторе фосфорована-
домолибдате, а затем перевести перекисью водорода в акриловую
кислоту в присутствии SeO2 [137].
Катализатором при окислении пропилена кислородом в газовой
фазе может служить НВт; при 224 °C получают наряду с аллилбро-
мидом смесь кислот, в том числе и акриловую кислоту [138].
Существует метод одновременного получения акрилонитрила,
акролеина и акриловой кислоты [139]. Реакционная смесь из окиси
азота и водяного пара, получаемая при 800—900 °C из смеси
NH3—Н2О—О2 на сите Pt—Rh, взаимодействует с пропиленом при
400—420 °C. Наконец, пропилен можно окислять смесью Se—NH3
с одновременным образованием акрилонитрила [140].
8.3. ТЕОРЕТИЧЕСКОЕ РАССМОТРЕНИЕ РЕАКЦИИ ОКИСЛЕНИЯ
ПРОПИЛЕНА ИЛИ АКРОЛЕИНА В АКРИЛОВУЮ КИСЛОТУ
Активность различных окисей в процессе каталитического окис-
ления акролеина убывает в следующем ряду [115]:
МоО3 > V2O5 > WO3 > SeO2 > ТеО2 > Nb2O5 > Та2О5 > СгО»
Для каталитического окисления подходят только катализаторы
с электроотрицательностью выше 2,93. Неактивные окиси Со2О3
и РЬО2 можно сделать активными, вводя Н3РО4. Опыты на V2O5 [141]
свидетельствуют об активирующем воздействии сильно электроот-
рицательных добавок (Н3РО4, H2SO4, МоО3, Н3ВО3, ТеО2, WO3)
на окисление акролеина, в то время как Cr2O3, TiO2 и SnO2 индиф-
ферентны. Наилучшие результаты дает система V2O5—Н3РО4 (при
500 °C выход акриловой кислоты составляет 72%).
Изучение окисления пропилена на катализаторе V2O5—А1203
при 240—320 °C свидетельствует о понижении выхода акриловой
кислоты с повышением температуры, оптимальная температура
240—260 °C (выход 16—18%) [142]. Выход других кислот (пропио-
новой, уксусной) уменьшается, а количество акролеина (2—6%)
остается постоянным по всему диапазону температур. С возрастанием
температуры резко уменьшается образование побочных продуктов.
Кинетику окисления пропилена над молибдатом Со—Bi исследо-
вали при 420—480 °C [63]; конечные продукты — акролеин, акрило-
вая кислота, СО и СО2.
Лазукин [66] и Холявенко [65], определяя активность SiO2,
МоО3 и молибдата Sn для окисления пропилена в диапазоне темпе-
ратур 380—480 °C, пришли к выводу, что максимальное количество
8.4. Свойства и применение акриловой кислоты
159
альдегида —90% (в основном акролеин наряду с 10—20% ацеталь-
дегида) получается при окислении над МоО3; при использовании
SnO2 селективность в отношении образования альдегида составляет
только 20—45%. Оптимальная температура для получения альдегида
430—450 °C, в присутствии окисей кислоты не образуются. Напротив
для получения акриловой кислоты хорошо зарекомендовал себя
смешанный катализатор окиси Sn—Мо при соотношении Sn : Мо =
= 3:1, оптимальная температура 340 °C. Наибольший выход
акролеина наблюдается при соотношении Sn : Мо = 9:1 или 1 : 9.
При повышении температуры выход акролеина повышается, а выход
акриловой кислоты понижается. Для совместного производства
акролеина и акриловой кислоты самой подходящей температурой
будет 360° С, концентрация пропилена 20—30%, соотношение про-
пилен : кислород : водяной пар = 1:1:2.
8.4. СВОЙСТВА И ПРИМЕНЕНИЕ АКРИЛОВОЙ КИСЛОТЫ
Ниже представлены свойства акриловой кислоты:
Температура плавления, °C................................ 13
Температура кипения, °C.................................. 141
Плотность
рД.................................................. 1,062
р22................................................ 1,060
Показатель преломления, wg............................. 1,4224
Теплота плавления при 13° С, ккал/моль........... 2,66
Теплота испарения при 13,6° С, ккал/моль................ 8,9
Теплота образования, ккал/моль...................... 89,8
Теплота сгорания, ккал/моль.......................... 329
Диэлектрическая проницаемость ....................... 5,6 • 10~5
Давление пара, мм. рт. ст.
при 0 °C............... 2,35 100 °C.............249
20 °C............. 7,76 120 °C.............475
40 °C..............22 141 РС.............760
60 °C..............54
Акриловая кислота является в основном промежуточным про-
дуктом для производства эфиров акриловой кислоты, из которых
важнейшими являются метилакрилат, этилакрилат, бутилакрилат
и 2-этилгексилакрилат. Ниже приводятся данные о потреблении
перечисленных продуктов в США в 1965 г. (в тыс. т) [5]:
Этилакрилат ............................... 45,4
2-Этилгексилакрилат ....................... 13,6
Метилакрилат................................ 6,8
Бутилакрилат................................ 4,5
Прочие акрилаты (среди них прежде всего изо-
бутил- и изодецилакрилат)................... 2,3
160
8. Акриловая кислота
Выпуск акриловой кислоты составляет 4500 т и распределяется Я
следующим образом: 1360 т — для производства эфиров и солей Я
акриловой кислоты (полиакрилаты аммония и натрия), 1.780 т — Я
используется в текстильной промышленности, при бурении нефтя- Я
ных скважин, в производстве коагулянтов. В первую очередь акри- 1
ловая кислота и ее соли [143] идут на изготовление водораство- 1
римых полимеров и сополимеров, которые применяются в качестве
замасливателей, аппретур, связующи^, загустителей, диспергаторов, ?
Для этой цели служат также и сополимеры с акрилатами. -
45% акрилатов в виде полимерных дисперсий или растворимых 1
полимеров расходуется на производство красок для внутренних
и наружных покрытий. Покрытия отличаются стойкостью к истира- ;
нию, быстро сохнут , и не желтеют (сополимер акриловой кислоты ’
с этил- и метилакрилатами, сополимер стирола с бутилакрилатом,
сополимеры винилацетата с бутил- и 2-этилгексилакрилатами). j
Дисперсии составляют 80% всех покрытий, содержащих акрилаты. ]
Лаки на основе растворимых акрилатов получили признание :
для окраски бытовых приборов и кузовов автомобилей методом ’
распыления. Лаки горячей сушки содержат менее 50% акрилатов, <
а лаки холодной сушки в основном состоят из акрилатов. Для
лаков горячей сушки используют также стирол, меламиновые и |
эпоксидные смолы. Значение этих лаков в будущем сильно воз- 1
растет.- . I
19% акрилатов используется в текстильной промышленности, 1
где. они часто заменяют крахмал или резину. Акрилатные дисперсии 1
придают материалу прочность к стирке и не желтеют в отличие от I
более дешевых винильных эмульсий. Они пригодны для текстильных 1
клеев и кэширования пенопластов. 65% всего производимого коли- |
чества метилакрилата находит применение при производстве поли- |
акрилонитрильных волокон. 6% метил- и этил акрилатов в сочета- |
нии с метилметакрилатом и стиролом расходуется на получение |
блескообразователей в средствах для натирки полов и высококаче- 1
ственных сапожных кремах; акрилаты частично вытеснили воск, |
ранее применявшийся для этих целей. 1
Еще 6% акрилатов (преимущественно в форме дисперсий) ис- 1
пользуются для отделки кожи. Акрилатные дисперсии повышают I
эластичность и прочность склеивания покровного слоя с основой. |
При этом метиловый эфир, дающий мягкие пленки, употребляется |
прежде всего для облицовки кожи, а бутиловый эфир — для обра- 1
ботки тяжелой кожи. Распространение искусственной кожи (напри- 1
мер, марки корфам фирмы Du Pont) неминуемо вызовет увеличение 1
потребления акрилатов. 1
В бумажной промышленности США находят сбыт остальные 6% -I
производимых акрилатов, а в Европе эта промышленность является 1
основным потребителем акрилатов. Главным образом они употрёбля- J
ются для мелования бумаги и картона, а также для получения 1
8.4. Свойства и применение акриловой кислоты,
161
св
Ч
ю
сС
Ь
Свойства некоторых полиакрилатов
S ф не раство- ряющие пленку , ' З" , 1 g^o3gg«H g g 1S о § а « ййно® ° а « ч ф « S 5 * £- < S ф < >> ч
6н S а о и 6н и 03 а растворяющие- пленку - ® 1 нч i-T Ф “ ф 3 Ф J* S а Й » Ф ® >_Г 3 Ф В i-Г ^S&Sg|gS ф И « ° ° ® - ® g S S 2 ® S g rt ° ИЙОфч'-аЗ Яйл®® я2и?ЯчЯ& и 21? ® й я 2 s ® 8 ЗЙФкЕйё5- а Я я ф й Й Й я ф ® § о й й О * * s О ч ч 2 я я ф 5* ©лссь- ©лссф ч да >> Я >в< Я § Ч сб ръ Ч сС Ръ О Q да О S U Я
□о ‘нинеяохг -яэхэ BcKxBdanwajj О О cq о со 1 1 1 •!• -1- 1 О CQ К7 Т-1 vr 1 1
эинэйоилопойод £ 2. а 2 о ® ® S и ? ¥ »о £ S й § И S Да О * К ° »
atqdeed ви qxooHbodix i S а а ® £5 и g® й да ж о/ jp |« S Да о И ° и н
эинэжнховд ® hO о $ И я й 2 я . Ч ф о s 0 3 Н а
ИЯНЭЕП ЧИОКЙЭЯ!, 1 ° « м § а ё * ® а о а Да ° 2 °S
ЧХ00ИЦЭ1СН 1 + + +
odXxedaimax цоихшшоя ndn HirodaiTOiftf ей ия -нэки aHHBaoeedgo + + + +
Полиакрилат « ч и . S . н • к. а s ® . а. и . £ . Sa® ч и « ч • St, S н Ч S g Я ® S •£ >. ' ® Ч Я r% я я я ч о ч ч S ч § о1-4 о о ч о да КСК К
И Заказ 399
162
8. Акриловая кислота
покрытий и кэширования бумаги. Наконец, 3,5% акрилатов исполь-
зуется в производстве клеев. Этил-, бутил- и 2-этилгекси'лакрилат
часто в комбинации со стиролом, винилацетатом или виниловыми
эфирами, являются составными частями клеев, идущих, например,
на изготовление клейких лент. Особое значение придается сополи-
меру этилакрилата и этилена, обладающему свойствами эласто-
меров.
В табл. 16 приведены свойства некоторых полиакрилатов.
Наиболее широкое распространение получили следующие сопо-
лимеры акрилатов:
сополимеры с винилхлоридом — акрилаты здесь действуют как
«внутренние пластификаторы»;
сополимеры с небольшим количеством акрилонитрила — улучше-
ние стойкости акрилатов к большинству растворителей;
сополимеры с акриловой кислотой — незначительное содержание
акриловой кислоты повышает полярность акрилатов и тем самым
адгезию и способность водных дисперсий к загустеванию;
сополимеры с амидами, например JV-метилоламидом, меламином,
аминами, эпоксисоединениями, хлоргидрином, хлорированными угле-
водородами и другими мономерами, содержащими реактивные
группы, являются основой клеев и лаков холодной и горячей сушки.
ЛИТЕРАТУРА
1. Т. Р. F о г b a t h, Chem. Eng., 69, № 5, 96—98 (1962).
2. Marshall Sittig, Acrylic acid and Esters, N. Y., 1965.
3. J. Mayor, Ind. chimique, 51, 343—346 (1964).
4, Europa-Chemie, № 12, 12 (1967).
5. A. Theurer, Chem. Industrie, 19, № 6, 345—348 (1967).
6 Chem. Eng. News, 45, № 7, 48—50 (1967).
7. J. Redtenbacher, Liebigs Ann. Chem., 47, 125 (1843).
8. 0. Rohm, Dissertation «Uber Polymerisationsprodukte der Acrylsaure»,
Tubingen, 1901; cp. Her. dtsch. Chem. Ges., 34, 427, 573 (1901).
9. Герм. пат. 262707, 1912.
10. Герм. пат. 295340, 1915.
11. Герм. пат. 656421, 1928.
12. Герм. пат. 660634, 1928.
13. W. Repp е, Chemie und Technik der Acetylen — Druck — Reaktionen,
Weinheim, 1951, S. 94—104.
14. T. L. G г e s m a n et al., J. Am. Chem. Soc., 70, 998 (1948).
15. Пат. США 2356459, 1941.
16. Пат. США 2844622, 1958.
17. Р. W. Sherwood, Ind. Eng. Chem. 54, № 10, 37—42 (1962).
18. Пат. США 2469704, 1949.
19. Пат. США 2469110, 1949.
20. Пат. США 2469690, 1949.
21. Пат. США 3069433, 1962.
22. Пат. США 3176042, 1965.
23. Пат. США 2361036, 1941.
24. Пат. США 3002017, 1961.
25. Пат. США 3049560, 1962.
Литература
163
26. Пат. США 3134807, 1964.
27. Пат. США 2980730, 1961.
28. Пат. США 3115522, 1963.
29. Пат. США 2390519, 1945.
30. Пат. США 2459430, 1949.
31. Пат. США 2453602 1948.
32. J. Gordon, Hydrocarbon Processing Petroleum Refiner, 40, № 9, 251—
260, (1961).
33. Пат. США 2890101, 1959. .
34. Пат. США 3024275, 1962.
35. European Chemical News, 5, № 113, 30 (1964).
36. Пат. США 2759016, 1956.
37. Пат. США 3067241, 1962.
38. J. F. V i t с h а, V. A. Sims, Ind. Eng. Chem. Product Res. Develop-
ment, 5, 50—53 (1966).
39. Пат. США 3247248, 1966.
40. Пат. США 3100795, 1963.
41. Пат. США 3051747, 1962.
42. Пат. США 3089901, 1963.
43. Пат. США 3089898, 1963.
44. Пат. США 2881213, 1953.
45. Англ. пат. 878802, 1961.
46. Франц, пат. 1299468, 1962; пат. ВНР 150183.
47. Англ. пат. 915799, 915800, 1963.
48. Франц, пат. 1393175, 1965.
49- Англ. пат. 924532, 1963.
50. Австрал. пат. 253809, 1964.
51. Франц, пат. 1331932, 1963.
52. Пат. США 3322693, 1967.
53. Англ. пат. 1036957, 1034914, 1966; франц, пат. 1399498, 1505085, 1967.
54. Англ. пат. 822140, 1959; D. A. S. 1245363, 1967; пат. США 2881212, 1959.
55. Бельг, пат. 625912, 1963.
56. Англ. пат. 903034, 1962; D. A. S. 1253703, 1967.
57. Пат. США 3293290, 1966.
58. Англ. пат. 939713, 1963.
59. Австрал. пат. 260939, 1965; белы. пат. 616690, 1962; англ, пат, 968056,
1964; франц, пат. 1320461, 1963.
60. Бельг, пат. 630896, 1963.
61. Б. В. К и р ь я н, К. И. Гробова, Труды по химии и хим. технол.,
№ 3, 210-214 (1965).
62. Авт. свид. СССР 184839, 1966; Бюлл. изобр., № 16, 30 (1966).
63. И. М. Барышевская и др., Укр. хим. ж., 33, 984—985 (1967).
64. Франц, пат. 1433572, 1966.
65. К. М. X о л я в е н к о и др., сб. «Катализ и катализаторы», АН УССР,
вып. 2, 64—71 (1966).
66. В. И. Л аз у кин и др., там же, № 2, 50—64 (1966); реф. С. А., 66,
75620у (1967).
67. Бельг, пат. 614404, 1962; пат. США 3259652, 1966.
68. Бельг, пат. 620549, 1963.
69. Бельг, пат. 630896, 1963; англ. пат. 961468, 1964; пат. США 3280182, 196.6.
70. Японск. пат. 21328/65, 1965.
71. Бельг, пат. 622753, 1963; англ. пат. 1014584, 1965; канад. пат. 698243, 1964;
франц, пат. 1346534, 1964.
72. Англ. пат. 953763, 1964.
73. Франц, пат. 1410564, 1965.
74. D. A. S. 1242817, 1967; франц, пат. 1315258, 1963; пат. США 3264347,
1966.
11*
164
8. Акриловая кислота
75. Бельг, пат. 611961, 1962.
76. Франц, пат. 1462565, 1966; Niederld. A. S. 6515836, 1966.
77. Англ. пат. 1007405, 1965; франц, пат. 1392718, 1965.
78. Франц, пат. 1366580, 1964.
79. Бельг, пат. 625848, 1963; англ. пат. 964639, 1964; пат. США 3172203, 1965.
80. Бельг, пат. 636618, 1964; японец, пат. 3695/66, 1966.
81. Франц, пат. 1446972, 1966; Niederld. A. S. 6512010, 1966.
82. Англ. пат. 893077, 1962; пат. США'3065264, 1963.
83. Пат. США 3087964, 1963.
84. Англ. пат. 967241, 1964; D. A. S. 1204659, 1965; франц, пат. 1289710,1962,
85. Франц, пат. 1470788, 1967; Niederld. A. S. 6604795, 1966.
86. Франц, пат. 1447982, 1966.
87. Англ. пат. 1015415, 1965; Niederld. A. S. 6500362, 1965.
88. Niederld. A. S. 6513697. 1966.
89. Франц, пат. 1342962, 1963; пат. США 3192259, 1965.
90. Франц, пат. 1335423, 1963.
91. Англ. пат. 1037002, 1966.
92. Бельг, пат. 686094, 1966.
93. Niederld. A. S. 6508659, 1966.
94. Англ. пат. 989401, 1965.
95. Niederld. A. S. 6500418, 1965.
96. D. A. S. 1244147, 1967.
97. Англ. пат. 1035147, 1966. .
98. Франц, пат. 1425871, 1966.
99. Англ. пат. 996898, 1965.
100. Франц, пат. 1429477. 1967.
101. Niederld. A. S. 6603943, 1966.
102. Бельг, пат. 659233, 1965; Франц, пат. 1471983, 1967; Niederld. A. S. 6515215,
1966.
103. Франц, пат. 1412880, 1965.
104. Niederld. A. S. 6405674, 1964.
105. Бельг, пат. 648235, 1967; англ. пат. 991836, 1965; японец, пат. 5604/66,
1966.
106. Англ. пат. 1031821, 1967; D. A. S. 1252663, 1967; франц, пат. 1380324,
1964; японец, пат. 5446/66, 1966.
107. Пат. США 3238254, 1966.
108. Пат. США 3238253, 1966.
109. Франц, пат. 1364810, 1964.
110. Франц, пат 1387693, 1965; ср. англ. пат. 1007353, 1965.
111. Бельг, пат. 620549, 1963.
112. Пат. США 2881214, 1959.
ИЗ. Пат. США 3021366, 1962.
114. Бельг, пат. 610392, 1962.
115. N. Kominami, Н. Nakajima, J. Chem. Soc. Japan, Ind. Chem.
Sect. (Kogyo Kagaku Zasshi), 69, 233—236 (1966).
116. Бельг, пат. 670431, 1966; Niederld. A. S. 6513085, 1966.
117. Бельг, пат. 656905, 1965.
118. Бельг, пат. 657745; 1965.
119. Пат. США 2930801, 1960.
120. Г. Н. Козел, М. И. Фарберов, ЖПХ, 39, № 9, 2101—2108,
1966.
121. Пат. США 3155719, 1964.
122. А. М i s о п о et. al., J. Chem. Soc. Japan, Ind. Chem. Sect. (Kogyo Ka-
gaku Zasshi), 69, 2129—2133 (1966).
123. Англ. пат. 904304, 1962; пат. США 3271447, 1966.
124. Пат. США 2341339, 1944.
125. Англ. пат. 1058328, 1967; Niederld. A. S. 6508269, 1965.
Литература
165
126. Франц, пат. 1397151, 1965; Niederld. A. S. 6408316, 1965.
127. Франц, пат. 86214, 1966; дополнит, пат. 1397151.
128. Франц, пат. 86815, 1966; дополнит, пат. 1401176.
129. Франц, пат. 1346534, 1964.
130. Пат. США 3114769, 1963.
131. Пат. ФРГ 869950, 1953.
132. П. Г. Сергеев, Л. М. Букреева, ЖОХ, 28, 101—103 (1958).
133. Пат. США 2744928, 1956.
134. С. W. Smith, R. Т. Holm, J. Org. Chem., 22, 746—748 (1957).
135. Голл. пат. 86505, 1957.
136. Бельг, пат. 645825, 1966.
137. Пат. США 3230248, 1966.
138. Голл. пат. 62760, 1949.
139. Niederld. A. S. 6516585. 1966.
140. D. A. S. 1229073, 1966.
141. N. Kominami, Н. Nakajima, J. Chem. Soc. Japan, Ind. Chem.
Sect. (Kogyo Kagaku Zasshi), 69, № 2, 237—239 (1966).
142. Б. В. Суворов и др., ДАН СССР, 172, № 5, 1096-1098, 1967.
143. Герм. пат. 623404, 1930.
144. U 1 1 m а пп, Enzyclopadie der technischen Chemie, Bd. 14, Munchen,
1963, S. 271.
9. ОКСОСИНТЕЗ ПРОПИЛЕНА
При оксосинтезе, называемом также реакцией Релена или гидро-
формилированием, окись углерода и водород присоединяются по
двойным связям, образуя альдегиды или спирты:
—* RCH2CH2CHO + H2 —> ЯСН2СН2СН2ОН
RCH=CHa4-CO + H2— СНО СН2ОН
—* RCHCH3 + H2 —> RCHGH3
Реакция, которая известна теперь во всем мире под названием
оксосинтеза, была открыта в 1938 г. Реленом [Ц. На первых про-
мышленных установках длинноцбпные олефины С14 — С17 превра-
щали в альдегиды, а затем в спирты с целью получения сырья для
синтетических моющих средств (например, установка фирмы Ruhr-
chemie мощностью 10 000 т в Холтене). Низшие олефины (такие, как
этилен и пропилен) лишь после 1945 г. стали применять для оксосин-
теза в промышленном масштабе. В настоящее время во многих стра-
нах имеются промышленные установки оксосинтеза пропилена [2].
Вначале для оксосинтеза использовали гетерогенные катализа-
тбры кобальта, применяемые также при синтезе по методу Фишера —
Тропша. Позднее перешли на растворимые в реакционной среде
катализаторы кобальта (нафтенат, олеат, стеарат, сульфат) [3—4].
Наиболее эффективными катализаторами оксосинтеза, вероятно,
являются Со2(СО)8 и НСо(СО)4, которые легко образуются при воз-
действии на кобальт окиси углерода и водорода под давлением при
повышенных температурах. Механизм реакции оксосинтеза тол-
куется по-разному. Особенно глубоко изучали эту реакцию Натта [51,
Мартинс [6], Марко [7] и Хек [8].
Можно представить механизм реакции оксосинтеза следующим
образом:
[Со(СО)4]2+Н2 4=± 2НСо(СО)4
HCo(CO)4+RCH=GH2 НСо(СО)3 •RCH=CH2 + CO
НСо(СО)3 • RCH=CH2-|-HCo(CO)4-> RCH2CH2CHO + [Co(CO)3]2
2[Со(СО)3]2 4=2- [Со(СО)3]4
[Со(СО)3]4+4СО -► 2[Со(СО)4]2
9. Оксосинтез пропилена
167
Оксосинтез превратился в один из важнейших процессов нефте-
химической промышленности. Это, в первую очередь, объясняется
широкими возможностями процесса. Как известно из литературы,
практически все олефины или олефинсодержащее сырье могут
взаимодействовать с окисью углерода и водородом. Благодаря этому
в промышленности производят целый ряд альдегидов и спиртов
С3 — С20. Продукты реакции при использовании в качестве сырья
высокочистого олефина представляют собой сложную смесь, так как,
с одной стороны, одновременно образуются всегда несколько изомер-
ных альдегидов и спиртов, а, с другой стороны, постоянно протекают
побочные реакции. Большое значение придается поэтому подбору
нужных условий реакции для уменьшения нежелательных побочных
реакций.
После второй мировой войны процесс распространился во многих
странах. В США в конце 1963 г. мощности оксосинтеза составляли
почти 300 000 т.
Английская фирма ICI [9] увеличила мощность своей установки
оксосинтеза в Билленгеме с 170 000 т в 1965 г. до 270 000 т. в 1967 г.
Остальные крупнотоннажные установки приходятся на западногер-
манские фирмы BASF, Chemische Werke Hills, Ruhrchemie, француз-
скую фирму Kuhlmann и итальянские фирмы Montecatini и Celene.
В Японии фирма Nissan Chemical Industries Ltd. купила за пол-
миллиона долларов процесс французской фирмы Kuhlmann для
установки производительностью 10 000 т/год.
Оксосинтез выгодно отличается от других синтезов спиртов тем,
что условия процесса можно варьировать без больших изменений
аппаратуры.
Основная доля производимых при оксосинтезе продуктов прихо-
дилась в США в 1962 г. на полученные из пропилена соединения
С4 (в тыс. т):
Соединения С4
Соединения С5
150 Этилгексанон.................36
9 Соединения С8—С23 .... 59
Среди соединений С4 первое место занимает бутиловый спирт,
хотя производство его оксосинтезом сопряжено с некоторыми труд-
ностями: до сих пор наряду с 60% бутилового спирта образуется
40% изобутилового спирта:
2СН3СН=СН8+2СО + 4Н 2—
—*СН3СН2СН2СН2ОН
СНзСНСНз
СН2ОН
Систематические исследования образования изомеров [10—13]
свидетельствуют о том, что соотношение альдегида (I) к а-метилаль-
168
9. Оксосинтез пропилена
дегиду (П) у алифатических а-олефинов зависит от температуры и
парциального давления СО:
I—* RCHoCH2CHO
RCH=CH2+CO + H2- I
I—> rchch2
1
CHO
II
Это показано на примере гидроформилирования пропилена при
180 °C HI, 13]:
Растворитель...............
Парциальное давление СО,
мм рт. ст..................
Соотношение масляного и изо-
масляного альдегидов . . ,
Толуол Толуол Гексан Гексан
146 7 102 102
4,18 2,19 4,0 1,6
Содержание высших продуктов конденсации в общем количестве
оксопродуктов в опытах с толуолом составило 1%.
Были проведены обширные работы по изысканию оптимальных
условий оксосинтеза [14—16]. Опыты при 150 °C, общем давлении
180—190 кгс/см2 (СО+Н2) и продолжительности реакции 1 ч пока-
зали, что повышение концентрации кобальта от 0,1 до 0,8 вес. %
не приводит к заметному изменению скорости реакции, соотношения
масляного и изомасляного альдегидов и доли высококипящих фрак-
ций. При увеличении времени реакции до 3 ч количество высоко-
кипящих компонентов, напротив, значительно возрастает с повыше-
нием содержания кобальта:
*
Содержание Со, вес. % ...............0,1 0,8
Содержание высококипящих фракций % . . 20 40
Кобальт является, следовательно, каталитическим ускорителем
образования высококипящих продуктов (в форме сильнокислого
карбонильного водорода кобальта).
Увеличение содержания кобальта сдвигает соотношение масля-
ного и изомасляного альдегидов несколько в сторону масляного
альдегида:
Содержание Со, вес. %................0,1 0,8
Соотношение масляного и изомасляного
альдегидов ...........................1,52 1,74
При повышении давления газа до 200 кгс/см2 скорость реакции
возрастает, дальнейшее увеличение давления не вызывает увеличе-
9. Оксосинтеа пропилена
169
ния скорости реакции. Соотношение масляного и изомасляного
альдегидов также изменяется с изменением давления:
Давление, кгс/см2 .............................. 100 200 300
Соотношение масляного и изомасляного альдегидов 1,1 1,4 1,8
Повышение температуры отрицательно сказывается на соотно-
шении альдегидов и содержании высококипящих фракций:
Температура, °C.................... 120 140 160 180
Соотношение масляного и изомасляного
альдегидов ........................ 1,9 1,6 1,23 0,85
Содержание высококипящих фракций, % 45 — — 48
Таким образом, при повышенных температурах масляный альде-
гид скорее подвергается последовательным реакциям, чем изомасля-
пый альдегид.
Значительная доля высококипящих побочных продуктов при
оксосинтезе пропилена образуется из бутиловых спиртов, которые
получаются при гидрировании масляных альдегидов, в результате
их взаимодействия с альдегидами при ацеталировании. Наблюда-
ются также альдольная конденсация, дегидратация, тримеризация
и другие реакции, вызываемые в основном сильной кислотой НСо(СО)4.
В оксосмеси обнаружены бутиловый и изобутиловый спирты,
2-этил-4-метилпентеналь-2, 2-этилгексеналь-2, 2-этил-4-метилпента-
нол и 2-этилгексанол.
Опыты, проведенные с различными растворителями (толуолом,
кетонами и ацеталями) показали, что растворители мало влияют
Таблица 17
Производство в США оксоспиртов из пропилена и его производных
(в тыс. т/год)
Спирт 1963 г. 1968 г.
Всего Оксосинте- зом Всего Оксосин- тезом
Бутиловый спирт 147,5 44 185 92
2-Этилгексанол 80 41 ИЗ 68
Изооктиловый спирт * . . 36 36 54 54
Дециловый спирт 34 34 43 43
Тридециловый спирт . . . 2,3 2,3 4,5 4,5
* Изооктиловый спирт получают окислением гептена, который, в свою очередь, обра-
зуется в результате алкилирования пропилена бутиленом.
170
9. Оксосинтез пропилена
на образование масляного и изомасляного альдегидов [15]. Скорость
реакции также не меняется. Повышение содержания растворителя
улучшает конверсию.
Американской фирме Shell удалось получить при оксосинтезе
пропилена масляный и изомасляный альдегиды в соотношении
10 : 1 (добавка трибутилфосфина). Это послужило толчком к исполь-
зованию оксосинтеза для получения бутилового спирта. Оксосинтез
имеет также огромное значение при синтезе масляного альдегида,
который является исходным веществом для производства масляной
кислоты, 2-этилгексанола, триметилпропана и бутиламинов.
Рис. 41. Схема установки оксосинтеза:
I — гидроформилирование; II — перегонка масляного альдегида; III —
гидрирование; IV — перегонка бутанола. 1 — реактор; 2 — регенера-
ция катализатора.
Табл. 17 дает представление о производстве оксоспиртов на основе
пропилена в США в 1963 и 1968 гг. [17].
Оксосинтез проводится в 3 стадии. Ниже указаны типичные
условия реакции:
Синтез (превращение олефина в альдегид)'.
Давление, кгс/см2 .................................... 201—251
Температура, °C....................................... 170—175
Соотношение СО : Н2................................ 1,2 : 1
Содержание катализатора Со в расчете на исходный
олефин, %.......................................... 0,5—1
Отношение смеси СО/Ня к исходному олефину .... 1000 : 1
Конверсия олефина, %............................... 90—95
Регенерация кобальта:
Температура, °C....................................... 180
Давление, кгс/см2.................................... 21
Гидрирование образующегося альдегида:
Температура, °C.......................г.............. 150
Давление, кгс/см2.................................... 101
9. Оксосинтез пропилена
171
172
9. Оксосинтез пропилена
На рис. 41 и 42 приведены схемы установки оксосинтеза.
Катализатор, олефины, а также смесь окиси углерода с водоро-
дом подаются в реактор для оксосинтеза. Поскольку реакция про-
текает с сильным выделением тепла (34,4 ккал/моль олефинов),
реактор необходимо охлаждать.
Выходящая из реактора реакционная смесь обрабатывается в
сепараторе для кобальта водяным паром при 180 °C и 21 кгс/см2
после выделения окиси углерода и водорода, направляемых в ре-
цикл. Освобожденные от окиси углерода продукты реакции гид-
рируются в спирты в гидрогенизаторе и затем направляются на рек-
тификацию.
В настоящее время известно несколько вариантов процесса
оксосинтеза, которые отличаются друг от друга в основном способом
производства и восстановления катализатора. Наиболее распрост-
раненными методами являются:
1) метод с неподвижным катализатором;
2) суспензионный метод;
3) солевой метод (катализатор—нафтенат кобальта).
При солевом методе себестоимость производства масляного
альдегида самая низкая — 82 единицы, при методе с неподвижным
катализатором — 100 единиц, а при суспензионном методе — даже
120 единиц [18—19]. По литературным данным в США наибольшее
распространение приобрел солевой метод с катализатором нафтена-
том кобальта.
9.1. АЛЬДОКС-ПРОЦЕСС
Модифицировав оксосинтез [20—23], можно получать из низко-
молекулярных олефинов высокомолекулярные спирты. В данном
случае образующиеся при оксосинтезе промежуточные альдегиды
превращаются в высшие спирты путем ди-, тримеризации и т. д.
Для этого метода применяется обычный для оксосинтеза катализатор
кобальта. В случае димеризации добавляют небольшое количество
цинксодержащего соединения.
Усовершенствованный процесс оксосинтеза, разработанный аме-
риканской фирмой Esso Research and Engineering Co., получил
название альдокс-процесса. В Японии с 1964 г. работает установка
альдокс-процесса [25], на которой ежегодно из пропилена произ-
водится 6000 т 2-этилгексанола и 3000 т бутилового спирта. В 1965 г.
мощность этой установки была доведена до 10 000 т/год 2-этилгек-
санола и 5400 т/год бутилового спирта.
Из 90%-ного пропилена получают 45 вес. % октилового спирта
при следующих условиях:
Давление, кгс/см2.................... 211
Температура, °C...................... 149—175
9.2. Свойства и применение масляных: альдегидов
173
Концентрация катализатора, % (по от-
ношению к загрузке)
Со (в форме олеата)............... 0,3
Zn ............................... 0,2
Соотношение СО : Н2..................... * 1,25—1,45
Нагрузка, объем жидкости/(объем
реактора • ч)......................... 0,6
На рис. 43 изображена схема установки альдокс-процесса.
Рис. 43. Схема установки альдокс-процесса:
1 — реактор для оксосинтеза; 2 — аппарат для деметаллизации; з — от-
стойник; 4 — колонны; 5 — подогреватель; в—печь для гидрирования.
9.2. СВОЙСТВА И ПРИМЕНЕНИЕ МАСЛЯНЫХ АЛЬДЕГИДОВ
Ниже приведены свойства масляного (I)
альдегидов:
и изомасляного (II)
Температура плавления, °C
Температура кипения, °C
Плотность
р-т • • ...............
('2 ...................
Р2?....................
Р2|....................
P’S ...................
Рй.....................
Р25....................
Показатель преломления
„20
nD....................
„26
nD.....................
„20
“а.....................
..................
I II
-99 -65,9
74,78 64,2—64,6
1,075 0,8349
0,8223 . —
0,8040 0,7904
0,7988 —
0,7484 —
— 0,7972
— 0,7879
1,3807 1,3730
1,3750 —
1,3811 1,3709
1,3914 1,3817
174
9. Оксосинтез пропилена
I
Дипольный момент (пар), D......................... 2,12
Вязкость сП
при 20 °C..................................... 0,43
18,35 °C ................................ 0,4575
72,2 °C................................... 0,2696
Теплота испарения при 74,8° С, кал/г . . . 104,4
Теплота сгорания при постоянном давлении,
ккал/моль.................................. 599,90*
Нижний предел взрываемости в смеси с воз-
духом, % 2,5
Температура вспышки, °C . . . .'........... -Юн—12—22
Температура воспламенения, °C................... 210—230
Диэлектрическая проницаемость при 74,8 ?С 10,78
Растворимость
масляного альдегида в воде
при 0 °C............................... 8,7
20 °C .............................’ 7,1
40 °C ................................... 5,4
воды в масляном альдегиде
при 0 °C..................................... 3,0
20 °C.................................... 3,2
40 °C.................................... 3,4
* Для изомасляного альдегида.
Формула давления пара масляного альдегида (в диапазоне тем-
ператур —15 -=—j-80 °C);
1g Р = 7,959 —1768,379/7*
Масляный альдегид образует азеотропную смесь с 6%, воды тем-
пература кипения смеси 68 °C. Изомасляный альдегид растворяется
при 20 °C в 9 объемах воды и образует азетропную смесь с 5% воды,
температура кипения смеси 60,5 °C.
Важнейшими производными продуктами масляного и изомасля-
ного альдегидов являются бутиловый и изобутиловый спирты.
Поскольку выделение альдегидов из реакционной смеси затруднено
и сопровождается значительными потерями, продукты гидрофор-
милирования сразу же гидрируют. Для этого смесь окиси углерода
с водой вытесняют чистым водородом и затем проводят гидрирование
при 190—200 °C и 200 кгс/см2. Образующиеся спирты являются
наиболее распространенными растворителями, для смол и лаков.
Их фталаты и фосфаты находят все большее применение в качестве
пластификаторов.
Окисление масляного альдегида в присутствии солей марганца
дает масляную кислоту [24], которая применяется для этерифика-
ции целлюлозы в бутират и ацетобутират целлюлозы.
Конденсация масляного альдегида с формальдегидом в присутст-
вии известкового молока дает 1,1,1-триметилолпропан, который
часто заменяет глицерин и пентаэритрит и является важным компо-
нентом в производстве алкидных смол и полиуретанов.
Литература
175
При альдольной конденсации масляного альдегида получают
2-этилгексанол, один из важнейших компонентов пластификаторов.
Конденсация с поливиниловым спиртом приводит к образованию
поливинилбутираля, пленку из'которого применяют для изготовле-
ния комбинированных стекол (триплексов) и который используют
также для получения вспомогательных средств в текстильной про-
мышленности. При восстановительном аминировании получают бутил-
амин.
Изомасляный альдегид гидрируют в изобутиловый спирт, а также
в изомасляную кислоту. Конденсация с формальдегидом приводит
к образованию диметилдиметилолметана:
(СН3)2СНСНО + 2НСНО + 2Н2О —> (СН3)2С(СН2ОН)2 + НСООН
В результате альдольной конденсации через изобутираль обра-
зуется 2,2,4-триметилпентандиол-1,3. Недавно было предложено •
дегидрирование в метакриловый альдегид [25];
ЛИТЕРАТУРА
1. Герм. пат. 849548, 1938; франц, пат. 860289; пат. США 2327066, 1943.
2. Chem. Eng., 57, № 2, 111 (1950); Petroleum Refiner, 31, № 1, 149 (1952).
3. Герм. пат. 877330, 1942.
4. Англ. пат. 736875, 1955; пат. США 2695315, 1950.
5. G. N a 11 a et al., Chim. Ind. (Milano), 37, 6 (1955).
6. A. R. M a r t i n s, Chem. and Ind., 1954, 1536.
7. L. M a r k о et al., Brennstoff-Chem., 44, 184—187 (1963).
8. R. F. H e c k, D. S. Breslow, 2 erne Congres international de Catalyse,
Paris, 1960; J. Am. Chem. Soc., 83, 4023—4027 (1961).
9. Chem. Industrie, 17, № 6, 309 (1965).
10. U 11 m a n n, Enzyclopadie der technischen Chemie, Bd. 13, Munchen —
Berlin, 1962, S. 65.
11. V. L. H u g h e s, I. К i r s c h e n b a u m, Ind. Eng. Chem., 49, 1999—
2003 (1957).
12. P. Pino et al., Chim. Ind. (Milano) в печати;
U 1 1 m a n n, Enzyclopadie der technischen Chemie, Bd. 13, Munchen,
1962, S. 65.
13. P. Pino et al., Chem. and Ind., 1962, 1400.
14. V. Macho, Chem. Zwesti, 16, 667—672 (1962).
15. V. Macho et al., ibid., 13, 343-347 (1963).
16. Пат. ЧССР 111947, 1964.
17. Chem. Eng. News, 41, № 19, 104 (1963).
18. Д. M. P у д к о в с к и й и др., Хим. пром., № 8, 652—658 (1959).
19. Д. М. Р у д к о в с к и й и др., Хим. пром., № 5, 335—338 (1961).
20. Пат. США 2811567, 1957.
21. Англ. пат. 912974, 1962; D. A. S. 1190928, 1965.
22. Chem. Eng., 68, № 25, 70— 71 (1961).
23. Chem. Industrie, 16, 302 (1964).
24. Пат. США 2456549, 1947.
25. Ch. W. Н а г g i s, H. S. Y о u n g, Ind. Eng. Chem., Product Res. Deve-
lopment, 5, 72—75 (1966).
10. ХЛОРИРОВАНИЕ ПРОПИЛЕНА
В зависимости от реакционных условий хлорирование пропилена
дает самые различные конечные продукты.
Хлорирование при низких температурах (до 250 °C) ведет пре-
имущественно к присоединению хлора по двойной связи, как это
обычно имеет место в случае олефинов с прямыми цепями:
СН2=СНСН3+ С12 —► СН2С1СНС1СН3
Горячее хлорирование (при 500—550 °C) дает главным образом
замещенные продукты, например аллилхлорид:
СН2=СНСН3+С12 —> СН2=СНСН2С1+НС1
Перхлорирование пропилена при 450—550 °C ведет к присоеди-
нению хлора и замещению, а также к отщеплению хлорированной
молекулы. В зависимости от условий хлорирования из смеси про-
пан — пропилен образуются перхлорэтилен и четыреххлористый
углерод (процесс Progil-Electrochimie Solvay, Франция) [1]:
СзН„ + (6 + п/2)С12 -> ЗСС14 + пНС1
2СзНп+ (6 + п)С12 —> ЗС2С14+2гаНС1
10.1. ПРИСОЕДИНЕНИЕ ХЛОРА ПО ДВОЙНОЙ СВЯЗИ
Дихлорпропан, образующийся при температурах ниже 250 °C
в результате присоединения хлора, служит главным образом как
растворитель и средство для очистки. Для производства дихлор-
пропана не требуется специальных установок или цехов, так как
при получении окиси пропилена методом хлоргидринирования или
при синтезе аллилхлорида этот продукт образуется как побочный
в количествах, превышающих потребность в нем. С целью исполь-
зования избыточных количеств дихлорпропана были проведены
многочисленные исследования по превращению этого продукта в
аллилхлорид путем дегидрохлорирования [2].
10.2. Замещающее хлорирование
177
10.2. ЗАМЕЩАЮЩЕЕ ХЛОРИРОВАНИЕ
t
10.2.1. Получение и характеристика аллилхлорида
Метод горячего хлорирования. Как уже было сказано, при хло-
рировании олефинов с прямой цепью при температурах до 250 °C
в результате присоединения хлора к двойной связи образуются
преимущественно дихлориды.
К совершенно иным продуктам приводит хлорирование олефинов
с разветвленной цепью, например изобутилена, который содержит
третичный углеродный атом. Здесь замещающее хлорирование про-
исходит с сохранением двойной связи уже при —40 °C без заметного
присоединения:
СН2=С-СНз + С12 —► СН2=С-СН2С1 + НС1
I I
СН3 СН3
Замещающее хлорирование неразветвленных олефинов с сохра-
нением двойной связи впервые удалось осуществить Стюарту и Вей-
денбауму [3] на пентене-2. Однако выход был очень низким.
Замещающее хлорирование олефинов с прямой цепью проходит
успешно только при достаточно высоких температурах. Исследова-
ния Гролля [4—5] (Shell Development Со.), а также Флемминга [6]
показали, что в этом случае осуществимо замещающее хлорирование
с хорошим выходом и возможно использование этой реакции в про-
мышленности. В 1942—1943 гг. в Германии (Оппау) были пущены
полупромышленные установки, а в 1948 г. в Хьюстоне (Техас)
введена в действие первая крупная промышленная установка по
производству аллилхлорида, перерабатываемого далее в глицерин.
При проведении горячего хлорирования в промышленных условиях
необходим точный контроль температуры, давления и концентра-
ции [7—9].
С повышением температуры реакция присоединения хлора посте-
пенно сменяется реакцией замещения (табл. 18).
Таблица 18
Зависимость характера реакции хлорирования от температуры
Средняя темпе- ратура реакции, °C Мольное соотно- шение пропи- лен : хлор Содержание хлора в продук- тах присоеди- нения, % Содержание хлора в продук- тах замещения, % Конверсия, г С1
100 см3«мин
210 3,44 : 1 74,7 25,3 0,081
320 6,03 : 1 22,5 77,5 0,200
400 6,34 : 1 3,8 96,2 0,550
510 6,32 : 1 1,3 98,7 4,060
590 6,60 : 1 0,3 99,7 10,900
12 Заказ 399
178
10. Хлорирование пропилена
Рис. 44. Технологическая схема получения аллилхлорида:
— емкость для сырого пропилена; 2 — емкость для сухого пропилена; 3 — отделитель; 4 — сушиль-
|ые башни; 5 — подогреватель; 6 — смесительное сопло; 7 — реакционные печи; в — холодильники;
। — аппарат для предварительного фракционирования; 10 — циркуляционные насосы; и — абсорбер
НС1; 12 — аппарат щелочной промывки газов; 13 — компрессор; 14, 15 — перегонные колонны.
10-2. Замещающее хлорирование
179
Ниже кратко описывается промышленный метод хлорирования
олефинов (рис. 44). В специальном смесительном сопле, помещенном
в подогревательную печь, перемешивают до получения однородной
смеси чистый пропилен, нагретый примерно до 350—400 °C, и чистый
безводный, неподогретый хлор. Во избежание накопления хлора и
связанного с этим избыточного хлорирования пропилен пропускают
через два боковых отвода, а хлор — через главную трубу. Затем
реакционная смесь, содержащая пропилен и хлор (лучше всего
в отношении 5:1), поступает в реактор, представляющий собой
стальной резервуар. Благодаря выделяющемуся при хлорировании
теплу в реакторе устанавливается температура 500—530 °C:
СзН6 + С12 —> С3Н5С1 + НС1 + 26 ккал
Время пребывания реакционной смеси в горячей зоне от 2 до 3 с.
Увеличение времени пребывания нежелательно, так как аллилхло-
рид заметно разлагается при повышенных температурах, а продукты
разложения, особенно сажа, загрязняют установку.
Выделившийся при реакции клареновый углерод действует как
катализатор хлорирования. Реактор долаген работать почти на мак-
симальной мощности. Выходящая из реактора реакционная смесь
содержит аллилхлорид, непрореагировавший пропилен, ненасыщен-
ные моно- и дихлориды, хлористый водород и небольшое количество
высоко хлорированных продуктов (табл. 19). При точном соблюде-
нии- температурного режима насыщенные дихлориды образуются
в небольших количествах, так как 1,2-дихлорпропан, например,
снова разлагается уже при 500 °C.
После охлаждения этих продуктов до 50—120 °C можно прово-
дить обработку. Для этого имеется два способа.
Экстракция.' Образовавшийся хлористый водород вымывается
водой в выложенной кирпичом башне. При этом давление паров
хлоридов настолько высоко, что конденсация еще не происходит.
После водной промывки газ имеет в среднем следующий состав:
Вес. % Мол. %
Аллилхлорид...................... 24,8 15,8
2-Хлорпропен-1.................. 0,7 0,4
Дихлорпропан+дихлорпропилен . . 4,0 1,8
Пропилен......................... 70,5 82,0
В отделителе при 7—10 °C конденсируется до 50% хлоридов
(сырой продукт I). Оставшиеся хлориды абсорбируются из пропи-
лена углеводородами, например октаном (сырой продукт II), а затем
отгоняются из абсорбционной жидкости. Насыщенный октаном
неабсорбированный пропилен выходит из абсорбера и после проточ-
ного охлаждения рассолом снова возвращается в цикл. На трех-
четырех специальных колоннах (лучше всего из никеля) из про-
дуктов I и II периодически или непрерывно выделяется чистый
аллилхлорид.
12*
180
10. Хлорирование пропилена
, Таблица 19
Перечень возможных продуктов, образующихся при горячем
(500—530 °C) хлорировании пропилена ,
Соединение Формула Темпера- тура - кипения, °C Pi0 7120 D Средний выход, %
Аллилхлорид или З-хлорпропен-1 . . . СН2=СН-СН2С1 44,6—44,9 0,9374 1,415 83,0
2-Хлорпропеи-1 . . . . СН2=СС1—СНз 22,5 0,9093 1,397 2-2
1-Хлорпропен-1 (цис) н\ /С1 ЧУ II- 32,0-32,5 (Pl6) 0,932 1,4055 0,4
1-Хлорпро пен-1 (транс) н/с\сн3 СК /Н II 36,7-37,0 0,930 1,4054 0,4
Пропилхлорид .... /С\ Hz хсн3 СН8-СН2-СН2С1 46 0,8919 1,3892 0,5
Изопропилхлорид . . . СН3—СНС1—сн3 36,5 0,8616 1,3811 0,7
1,3-Дпхлорпропен-1 низкокипящий .... СНС1=СН-СН2С1 104,1 1,225 1,469 1,0
высококипящий . . . СНС1=СН—СН2С1 112,1 1,226 1,475 6,5
3,3-Дихлорпропен-1 . . СН2=СН-СНС12 84,4 1,170 — 0,7
2,3-Дихлорпропен-1 . . СН2=СС1-СН2С1 93,8 1,211 1,4603 1,2
1,2-Дихлорпропан . . . СН2С1-СНС1-СН3 96,6 1,156 1,4388 2,6
Трихлорпропаны и выс- шие хлориды .... — — — — 1,0
Конденсационный метод. Этот метод считается лучшим и потому
внедрен в промышленность. Реакционные газы направляются прямо
из реактора, где проводилось хлорирование, в так называемый
форфракционатор, где в результате охлаждения жидким пропиленом
при —40 °C конденсируются органические хлориды. Пропилен
вместе с образовавшимся при реакции хлористым водородом посту-
пает в HCl-абсорбер, где хлористый водород поглощается водой.
Пропилен после щелочной промывки возвращается в цикл [10, 11].
Зависимость среднего выхода продуктов (в расчете на израсходо-
ванный пропилен) от температуры показана на рис. 45.
Наиболее оптимальным оказалось соотношение пропилен : хлор =
= 5 : 1. При меньшем соотношении увеличивается содержание нена-
сыщенных дихлоридов, при более высоком соотношении, хотя и
повышается выход аллилхлорида, но существенно увеличивается
нагрузка на систему циркуляции, особенно на абсорбер, что при-
водит к росту эксплуатационных расходов.
10.2. Замещающее хлорирование
181
Очень важно применять высокочистый пропилен и, особенно,
очень чистый и безводный хлор. Любая органическая примесь хлори-
руется, что вызывает повышение расходов хлора в результате обра-
зования побочных продуктов. Также не следует использовать смеси
пропана с пропиленом, так как это приводит к образованию трудно-
отделяемых монохлоридов [12].
Подогреватель пропилена, смесительное сопло и реактор могут
быть выполнены из обычной стали, так как в местах контакта с хло-
ром образуется защитная пленка из кларенового углерода. НС1-
Абсорбер лучше всего изготавливать из кирпича, пропитанного-
силикатом натрия, или из стойкого
к химическим воздействиям камня.
Пригодны также игурит и керамика.
Перегонная аппаратура может быть
выполнена из материалов хастеллой
А и дурихлор, но чаще употребляют
монельметалл или никель. Метод
горячего хлорирования за последние
годы в основном не изменялся, но
появилось множество вариантов кон-
струкции реактора. При этом стре-
мились снизить образование про-
Рис. 45. Влияние температуры
реакции на выход продуктов го-
рячего хлорирования пропилена
при мольном соотношении пропи-
лен : хлор=4 :1 (заштрихована об-
ласть оптимальных температур).
1 — аллилхлорид; 2 — 1,2-дихлорпро-
пан; 3 — высококипящие продукты;
4 — легкокипящие продукты.
дуктов присоединения при смешении
пропилена с хлором. Например,
сконструирован реактор типа цик-
лона, позволяющий работать с более
низким соотношением пропилен: хлор
(3 : 1) [13—15]. В этот реактор оба
газа вводятся раздельно по каса-
тельной к противоположным сторо-
нам циклона. Предложены также
реакторы шарообразной, эллипсоидной и других форм, обеспечи-
вающие хорошее смешение без мертвых пространств и выходы
не менее 80% [16—18] .
Для улучшения теплового баланса предлагается реактор с двой-
ными стенками, в котором отработанные газы быстро нагревают
свежий газ до оптимальной температуры 382—494 °C. В таком реак-
торе при 485 °C и времени контакта 2,13 с получается 79,8 мол. %
аллилхлорида и 3,04 мол. % 1,2-дихлорпропана [19].
Следующая возможность улучшить смешение компонентов реак-
ции состоит в том, чтобы впрыскивать в камеру для хлорирования
хлор по оси, а пропилен по касательной [20]. Наконец, предлагается
установка для хлорирования, на которой работает вертикальный
реактор и применяются агенты теплопередачи [12].
Были проведены расчеты по методу градиента с целью оптимиза-
ции температуры, давления, состава впрыскиваемого продукта и
182
10. Хлорирование пропилена
времени реакции. Если хлорирование проводится в присутствии
1,2-дихлорпропана, то увеличивается выход аллилхлорида [23].
Другие методы. Аллилхлорид получают также пиролизом 1,2- ди-
хлорпропана [24]. При этом образуется 55—70% аллилхлорида,
3.0—40% смеси цис- и транс-изомеров 1-хлорпропена-1 и 5% 2-хлор-
пропена-1.
При 250—400 °C в присутствии различных катализаторов [25]
преимущественного образования аллилхлорида не наблюдалось, при
560—640 °C выход аллилхлорида достигал примерно 60% [26].
В качестве катализаторов пиролиза 1,2-дихлорпропана в аллилхло-
рид предложены ВаС12, СоС12, NiCl2, ZnCl2, МнС12, FeCl3 и CuCl2
на активированном угле, силикагеле или А12О3 [27].
Можно осуществлять также хлорирование пропана или 1-хлор-
пропана при 500—700 °C в присутствии катализатора [28]. Наконец,
при горячем хлорировании пропилена можно использовать вместо
С12 смесь НС1 и О2 или воздуха (катализатор Lid, благородные
металлы, или соединения теллура на пемзе) [29].
Простой диаллиловый эфир, образующийся как побочный про-
дукт при превращении аллилхлорида в аллиловый спирт, может
быть снова переведен в аллилхлорид с 88%-ным выходом посредст-
вом пропускания с НС1 над CuCl при 15—40 °C и под давлением [30]
или при 55 °C [31].
Свойства и применение аллилхлорида. Ниже приведены свойства
аллилхлорида:
Температура плавления °C.............................—136,4
Температура кипения, °C
при 744 мм рт. ст............................... 44,5
751 мм рт. ст.................................. 44,9
760 мм рт. ст................................. 45,1
772,5 мм рт. ст.............................. 46,7
Плотность
PS ..................................................... 0,9637
Pis.................................................... 0,9442
pF...................................................... 0,9379
PF.......................................................0,9311
PF...................................................... 0,9245
Показатель преломления
п^...........................................................1,4188
ng.......................................................1,4154
. ng.......................................................1,4130
Дипольный момент, D..................................... 2,02
Вязкость при 30° С, П................................ 0,00336
Критическая температура, °C............................. 240,7
Теплота испарения, кал/моль............................. 6940
Теплота сгорания, ккал/моль............................. 440,8
Удельная теплоемкость (пара), кал/(г • °C)
при —17,8 °C........................................ 0,222
37,8 °C ..................................... 0,230
93,3 °C ..................................... 0,224
149 °C....................................... 0,212
10.2. Замещающее хлорирование
183
Пределы взрываемости в смеси с воздухом, объемн. %
верхний........................................... 11,51
нижний............................................ 3,28
Температура воспламенения, °C...................... —26,7
Диэлектрическая проницаемость
пар при 19° С и 760 мм рт. ст. ................ 1,0128
жидкость при 19,5° С.............................. 7,3
Растворимость при 20° С, вес. ч./100 вес. ч.
воды в аллилхлориде '.............’.............. 0,08
аллилхлорида в воде.............................. 0,36
Формула давления пара р (в мм рт. ст.) и значения давления
пара при различных температурах приведены ниже:
2142 R
Р = 20,89279 - - 4,41821g Т
t, с р, мм рт. ст. (, “С Р, мм рт. ст.
0 119,3 35 533,1
5 151,8 40 639,3
10 191,3 45 761,5
15 238,8 50 901,1
20 295,5 362,5 55 1060,0
25 441,3
30
Ниже приведены температуры кипения и содержание аллил-
хлорида в двойных азеотропных смесях со следующими веществами::
т. кип., °C Содержа- ние алилхло- рида, вес, % т. кип., °C Содержа- ние алилхло- рида, вес. %
Вода 43,0 97,8 Пропцонитрлл 44,95 80
Пентан 35,5 28 Метилаль . . 41,4 20
Циклопеитан .... 44,3 63 Ацетон . . . 44,6 90
Метиловый спирт . . 39,85 90 Муравьиная кислота 44,4 92,5
Этиловый спирт . . 43,5 95 Этилформиат 45,2 90
Изопропиловый спирт 45,1 98 Сероуглерод . 41,2 50
Аллилхлорид получил довольно большое значение благодаря
легкости получения и высокой реакционной способности. Он явля-
ется исходным продуктом при введении аллиловой группы в другие-
соединения: сложные аллиловые эфиры (например, аллилфталат),
аллиламины, аллилизотиоцианат (искусственное горчичное масло
CH2=CHCH2NCS). В результате реакции обмена аллилхлорида
с тиомочевиной образуется так называемый тиозинамин, применяе-
мый в фотографии. Однако большая часть аллилхлорида исполь-
зуется для получения эпихлоргидрина и глицерина. Некоторое-
Возможности получения глицерина Из Пропилена
10-2. Замещающее хлорирование
185
количество аллилхлорида перерабатывается в аллиловый спирт,
а также в аллиловый крахмал.
При реакции обмена аллилхлорида с аммиаком (целесообразно
проводить эту реакцию в автоклаве с мешалкой при 100 °C под
давлением) в зависимости от добавляемого количества аммиака
получается моно-, ди- или триаллиламин [147]. Преимущественно
получают моноаллил амин, и в этом случае избыток хлористого
аммония оказывает самое благоприятное действие [148]. Моно-
аллиламин можно синтезировать также путем гидролиза соляной
кислотой аллилового горчичного масла, полученного из аллил-
хлорида под действием роданидов щелочных металлов или аммония
[149]. Моно- и диаллиламины являются промежуточными продук-
тами для химических синтезов.
В последнее время приобрел значение аллиловый крахмал, обра-
зующийся при взаимодействии аллилхлорида или аллилбромида
с крахмалом в присутствии щелочи [150]. Аллиловый крахмал,
иногда вместе с каучуком, применяется для получения покрытий,
клеев и пластмасс [151]. Аллилсахароза, являющаяся продуктом
реакции сахарозы и аллилхлорида, после полимеризации тоже
может быть использована для производства покрывных материа-
лов [152].
Аллилхлорид используется для синтеза аллилсиликонов [153]
и аллетрина — синтетического средства для борьбы с вредными
насекомыми [154].
Аллилхлорид полимеризуется в присутствии катализаторов Фри-
деля — Крафтса [155]. Полимеры применяются для пропитки бумаги,
дерева и других материалов. В результате полимеризация в присут-
ствии радикальных катализаторов (перекиси бензоила) получа-
ются полимеры, прйменяемые в качестве пластификаторов, клеев
и смазок, а также для получения лаков и пропиток [156]. При нагре-
вании аллилхлорида с водорастворимым полисульфидом [157],
а также 1,2,3-трихлорпрбпаном [158] образуются продукты подоб-
ные тиоколам.
Смесь из дихлорпропана и дихлорпропенов, получаемая как
побочный продукт при горячем хлорировании пропилена, широко
употребляется под названием «Д-Д» в качестве ценного средства для
окуривания почвы с целью борьбы с нематодами. (250—500 кг/гектар)
[159-160].
На схеме показаны различные пути синтеза эпихлоргидрина и
глицерина из аллилхлорида.
10.2.2. Получение и характеристика эпихлоргидрина
Получение эпихлоргидрина. Как видно из схемы (см. стр. 184),
получение эпихлоргидрина является важной промежуточной сту-
пенью при синтезе глицерина. Эпихлоргидрин впервые был
186
10. Хлорирование пропилена
синтезирован в 1854 г. [32] при взаимодействии глицерина с хло-
ристым водородом:
СН2-СН-СЩ+2НС1 —-12° °G-> СН2-СН-СН2 + 2Н2О
I - I I III
ОН ОН ОН С1 ОН С1
СН2-СН-CH2 + NaOH 6°~8° °G-> CH2-CH-CH2 + NaCl + H,O
III I \/
Cl ОН Cl Cl о
Этот метод получил наибольшее распространение.
Эпихлоргидрин образуется из аллилхлорида после присоедине-
ния хлорноватистой цислоты через дихлоргидрин и дальнейшего
отщепления соляной кислоты известковым молоком [33]:
2СН2=СНСН2С1 +2НОС1 > СН2-СН-СН2 (30%)- 1 1 1 С1 ОН С1 —СН2—СН-СН2 (70%)- 1 1 1 CI CI он +Са(ОН)2 г
20-40. °C, рН=3 4- 5 -СаС12> н2о ’
—> 2Н2С—СН-СН2С1
V
Технологическая схема промышленного метода получения эпи-
хлоргидрина из аллилхлорида изображена на рис. 46. Для получе-
ния дихлоргидрина аллихлорид вводят в реакцию обмена с хлорно-
ватистой кислотой в водной фазе. Поскольку аллилхлорид плохо
растворяется в воде (при 20 °C в воде растворяется только 0,36 вес. %
аллилхлорида), необходимо принимать особые меры, чтобы воспре-
пятствовать прямому контакту хлора и аллилхлорида. В противном
случае в результате присоединения хлора образуется слишком
большое количество трихлорпропана.
Чтобы не допустить непосредственного соприкосновения хлора
с аллихлоридом, хлорноватистую кислоту получают в отдельной
башне и работают с большим разбавлением и при низкой температуре.
Это делаетсй для того, чтобы введенный хлор по возможности без
остатка перешел в хлорноватистую кислоту:
С12 + Н2О —► HOG1+HC1
Хлорноватистую кислоту получают в башне с кислотоупорной
облицовкой путем непрерывного введения 1—2%-ного раствора
едкого натра и хлора. Образовавшаяся кислота выходит из верхней
части башни, затем при тщательном смешении реагирует с аллил-
хлоридом. При этом происходит хлоргидрирование. Из процесса
постоянно выводится реакционная смесь в количестве, равном
объему выходящей из башни хлорноватистой кислоты.
10-2. Замещающее хлорирование
187
Реакционная смесь пропускается через термодиффузионное раз-
делительное устройство, где отделяются трихлорпропан и тетра-
Рис. 46. Технологическая схема получения эпихлоргидрина и глицерина на
промышленной установке:
а — хлорирование пропилена:
1 — нагреватель (от 20 до 400 °C); 2 — реактор; 3 — фракционная колонна; 4 — абсорбер;
5 — промывная колонна; в — сушильная башня; 7 — система из трех колонн для перегонки
аллилхлорида.
б — хлоргидрйрование аллилхлорида:
1 — растворитель щелочи; 2 — приготовление НОСГ, з — реактор; 4 — отбор трихлорпро-
пена и тетрахлорпропилового эфира; 5 — аппарат для отщепления НС1; в — колонна азео-
ропной дистилляции; 7 — сепаратор; 8— система из двух колонн для обезвоживания и пере-
гонки эпихлоргидрина;
в — омыление эпихлоргидрина:
1 — подогреватель (10 о—180 °C, 10 кгс/см’); 2 — нейтрализатор; з — колонна для пере-
гонки глицерина.
хлордиизопропиловый эфир. После смешения дихлОргидрина в аппа-
рате с мешалкой с 15%-ным известковым молоком в реакционной
колонне осуществляется превращение в эпихлоргидрин и отгоняется
азеотропная смесь с водой. Водный слой возвращается в реакцион-
ную колонну, а сырой эпихлоргидрин дистиллируется в другой
колонне. При такой технологии выход составляет более 90%.
188
10. Хлорирование пропилена
Разработан метод непосредственного получения эпихлоргидрина
из аллилхлорида, минуя промежуточную стадию образования гли-
цериндихлоргидрина. Он состоит в окислении аллилхлорида пере-
кисными соединениями. Однако этот метод до сих пор не внедрен
в промышленность. В литературе указаны следующие окислители
для этой цели: надуксусная или надпропионовая кислота [34],
перекись водорода в присутствии WO3 [35], кислород и ацетальде-
гид [36], пероксикарбоксиминокислота [37], ароматические нитро-
соединения [38], а также каталитическое окисление воздухом на
окиси серебра. Окись серебра, нанесенная на губчатый алюминий,
предварительно активируется пропусканием над ней водорода и
азота [39].
Современное мировое производство эпихлоргидрина оценивается
в 300 тыс. т. Исходным продуктом почти всегда служит аллихлорид.
Свойства и применение. Ниже приведены свойства эпихлор-
гидрина:
Температура плавления, °C . .
Температура кипения, °C . . .
Плотность
Ро ........................
PS ........................
PF.........................
. >50
Pso........................
Показатель преломления
...............................
.......................
Вязкость, П
при 0° С ......................
25° С .................
Поверхностное натяжение, дип/см
при 12,5° С...............
31,0° С ...............
89,0° С ...............
Теплота сгорания, кал/г ....
Температура воспламенения, °C
Диэлектрическая проницаемость
при 21,5° С....................
Электропроводность
при 25° С, ом"1.см-1 ....
--57.2
116,11
1,2040
1,2031
1,1732
1,1633
1,43805
1,43580
0,0156
0,0103
39,13
35,48
27,72
4524.4
40,5
20,8
34 • IO’9
О растворимости воды в эпихлоргидрине см. в работе [40], об
азеотропных смесях с различными растворителями — в работе [41].
Эпихлоргидрин — химически очень активное соединение, высо-
кой активностью обладают содержащиеся в нем эпоксигруппа и атом
хлора. Поэтому эпихлоргидрин приобретает все большее значение,
как промежуточный продукт органической химии. Наряду с приме-
нением для синтеза глицерина эпихлоргидрин употребляется в боль-
шом количестве для производства эпоксидных смол, которые полу-
10.2. Замещающее хлорирование
189
чают взаимодействием дифенилолпропана, синтезируемого из ацетона
и фенола, с эпихлоргидрином. Производство эпоксидных смол непре-
рывно увеличивается. Рассчитывают, что в 1980 г. в США выпуск
их достигнет 80 тыс. т. Эпоксидные смолы производятся также
в Англии, Голландии, ФРГ, Швейцарии, Бельгии, Франции, Япо-
нии, ЧССР и СССР. Кроме того, эпихлоргидрин находит применение
в производстве ионообменных смол.
Недавно эпихлоргидрин стали применять для получения хлор-
гидринового каучука:
г-сн2-сн-о--
СН2С1 . п .
С этой целью эпихлоргидрин полимеризуют с алкилалюминием
в присутствии хелата металла, иногда вместе с окисью этилена [43,
44]., Хлоргидриновые каучуки разработаны фирмой Hercules Pow-
der (США).
Гомополимер поступает в продажу под названием Гидрин 100,
а сополимер с окисью этилена — под названием Гидрин 200 (с недав-
них пор Херклор X и Херклор Ц). По данным фирмы, эти типы гид-
ринов должны обладать такой комбинацией свойств, какой до сих
пор не было ни у одного из синтетических каучуков. По жаростой-
кости и сопротивлению действию озона и других окислителей Гидрин
100 и Гидрин 200 равны этилен-пропиленовым сополимерам. По мас-
лостойкости они приближаются к нитрильному каучуку, а по газо-
проницаемости соответствуют бутилкаучуку.
При реакции эпихлоргидрина с фенолами или спиртами полу-
чаются простые глицидные эфиры, применяемые для различных
целей в качестве активных растворителей или как стабилизаторы
для галогенсодержащих полимеров.
Способность к поликонденсации продуктов взаимодействия эпи-
хлоргидрина и аммиака используется для получения высокомоле-
кулярных смол.
10.2.3. Получение и характеристика аллилового спирта
Получение. Аллиловый спирт образуется в результате гидро-
лиза аллилхлорида [46]. Этот продукт имеет очень большое значение.
В лабораторных условиях омыление аллилхлорида целесообразно
осуществлять с применением водного раствора гидроокиси кальция
в автоклаве с мешалкой при 150 °C. В промышленности (рис. 47)
омыление проводится 5—10% раствором едкого натра при 150 °C
под давлением 1.3—14 кгс/см2 [8, 47, 48]. В этих условиях выход
достигает 85—95%. Побочные продукты — это в основном простой
диаллиловый эфир (5—10%), а также непрореагировавшие хлоро-
прены и высококипящие полимеры аллилового спирта и пропионо-
вого альдегида. Количество побочных продуктов можно уменьшить,
190
10. Хлорирование пропилена
1 — реактор; 2 — подогреватель; з — колонна для отгонки с водяным паром аллилового спирта из водного раствора; 4 — обез-
воживающая колонна с диаллиловым эфиром в качестве носителя; 5 — аппараты для водной промывки диаллилового эфира; в—
колонна для очистки аллилового спирта перегонкой; 7 — циркуляционные насосы.
10.2. Замещающее хлорирование
191
проводя омыление под давлением с максимально низкой концентра-
цией едкого натра и добавляя по возможности небольшое количество
карбоната натрия.
Ниже показано влияние концентрации шелочи на гидролиз
аллилхлорида в аллиловый спирт:
Концентрация NaOH, %......................... 10 5 2,5
Температура, °C............................. 158 157 158
Выход алилового спирта, %................. 82,5 87,5 < 93,5
Выход диаллилового эфира, %................. 11,5 7,6 3,0
Количество непрореагцровавшего аллилхлори-
да, % ....................................... 6,0 4,9 3,3
Метод, который может в будущем приобрести значение, состоит
в изомеризации окиси пропилена в аллиловый спирт в присутствии
литийфосфатных катализаторов [49]; другая возможность получить
аллиловый спирт без введения хлора состоит в избирательном гид-
рировании акролеина на Cd—Zn-катализаторе, нанесенном на
пемзу [50].
Свойства и применение. Свойства аллилового спирта приведены
ниже:
Температура плавления, °C...........................—129
Температура кипения, °C............................ 96,9
Плотность
pF.................................•.................. 0,8564
р|°................................................... 0,8520
pF . . ,........................................... 0,8476
Показатель преломления
п=»........................................................1,4133
ng.....................................-...............1,4111
Дипольный момент, D.................................... 1,63
Вязкость, П
при 15° С..........................................0,01486
30° С.......................................0,01072
Критическая температура, °C............................ 271,9
Теплота испарения, кал/моль ........................... 9550
Теплота сгорания, ккал/моль............................ 442,4
Удельная теплоемкость, кал/(г-°С)
пар при 20° С................-.................... 0,58
жидкость при 20,5—95,5° С......................... 0,665
П ределы взрываемости в смеси с воздухом, объемн. %
верхний .................................. 18,0
нижний .......................................... 2,5
Температура воспламенения, °C..................... . 22,2
Диэлектрическая проницаемость
при 16,2° С....................................... 20,3
Формула давления пара аллилового спирта р (в мм рт. ст.)
1g Р= 32,62580- 7,94975 1g Т
192
10. Хлорирование пропилена
Азеотропная смесь с водой содержит 72,3% вес. % аллилового
спирта, ее температура кипения 88,89° С.
Аллиловый спирт применяется как средство для дезинфекции
почвы, для борьбы с нематодами, а также как консервант для сухой
кройяной плазмы.
Однако более важно его значение как промежуточного продукта
для синтеза других веществ. При полимеризации аллилового спирта
образуется водорастворимый полимерный спирт, который может
быть использован для синтеза сложных полиэфиров (особое значение
приобрел диаллилфталат). Полимер диаллилфталата пригоден для
получения термо- и дуропластов. Он используется также для произ-
водства лаков и пр- Находят применение и другие сложные эфиры:
эфиры циануровой, малеиновой, фумаровой и акриловой кислот.
При действии двуокиси серы на аллиловый спирт в присутствии
растворителей и перекисей образуются полимерные аллилсульфоно-
вые кислоты — промежуточные продукты для производства вспомо-
гательных средств для текстильной промышленности и пластифика-
торов [51].
С недавних пор аллиловый спирт приобрел значение как проме-
жуточный продукт для синтеза глицерина без применения хлора,
причем глицерин получается в результате одностадийной реакции
обмена с перекисью водорода:
СН2=СНСН2ОН+Н2О2 —w-3’-—~70 ° с-> СН2(ОН)СН(ОН)СН2ОН
10.2.4. Получение и характеристика глицерина
Глицерин был открыт в 1779 г. Шееле [52] при омылении оливко-
вого масла.
Получение из продуктов не нефтехимического происхождения.
До развития методов искусственного синтеза глицерин получали ще-
лочным омылением масел и жиров. При омылении образуется смесь
мыла (натриевых солей жирных кислот) с водным раствором глице-
рина. Мыла высаливают хлористым натрием, глицерин получают из
раствора путем повторного сгущения и кристаллизации осажденного
хлористого натрия. Полученный 80 %-ный темный глицерин очищается
перегонкой и обработкой активированным углем.
Метод [53—58], который не служит для получения чистого гли-
церина, а дает скорее смесь глицерина с другими гликолями, основан
на углеводах как исходных веществах (крахмал, древесная мука
и сахар, особенно тростниковый). Из углеводов в результате гидро-
лиза получают сначала гексозы, которые затем гидрируют в 40—
50% водном растворе в присутствии 6% никеля под давлением водо-
рода 300 кгс/см2 и при температуре, повышающейся от 80 до 180 °C.
После выпаривания реакционная смесь — глицероген — состоит
примерно из 35—40% глицерина, 25—30% пропиленгликоля, 5—10%
этнленликоля, 1—6% воды и гекситов.
10.2. Замещающее хлорирование
193
Ферментативный метод получения глицерина не внедрен в про-
мышленность, Лишь в Германии во время первой мировой войны
большие количества глицерина наряду с ацетальдегидом получали
при дрожжевом брожении гексоз в присутствии сульфита натрия.
Данный метод не выдерживает конкуренции в сравнении с более рен-
табельным способом получения глицерина из дешевого сырья, напри-
мер из мелассы.
Получение глицерина из пропилена. Замена мыла синтетическими
моющими средствами привела к недостатку глицерина. Поэтому
фирма Shell Chemical Со. построила в 1948 г. в Хьюстоне (Техас)
первую установку по производству синтетического глицерина в не-
сколько стадий [59—60]:
Пропилен
4
Аллилхлорид
+
Глицерин- Аллиловый
дихлоргидрин спирт
Эпихлоргидрин Глицеринмонохлоргидрин
I I
Глицерин
Существует бесхлорный промышленный метод, по которому рабо-
тает пока только одна установка фирмы Shell Chemical Со. в Норко
(Луизиана):
Пропилен----► Акролеин -->- Аллиловый спирт -—> Глицерин
Третий метод, реализованный в масштабе пилотной установки,
разработан фирмой Olin Mathieson Chemical Со.:
Пропилен --> Пропиленхлоргидрин---> Окись пропилена ->
---> Аллиловый спирт-> Глицерин
В данном случае расход хлора значительно меньше.
Синтез через аллилхлорид. Синтез глицерина через аллилхлорид
можно осуществить в двух вариантах:
1) аллилхлорид переводится хлорноватистой кислотой в глицерин-
дихлоргидрин, который реагирует с известковым молоком, образуя
эпихлоргидрин, а эпихлоргидрин под действием едкого натра пре-
вращается в глицерин (см. рис. 46);
2) аллилхлорид посредством кислого или щелочного гидролиза
омыляется в аллиловый спирт, который в водном растворе с хлором
превращается в смесь глицеринхлоргидринов, а из этой смеси в резуль-
тате обработки ее водным раствором щелочи образуется глицерин.
13 Заказ 399
194
10. Хлорирование пропилена
Выбор какого-либо из этих двух методов определяется экономиче-
скими соображениями.
Наконец, глицерин можно получить также омылением глицерин-
хлоргидрина непосредственно раствором едкого натра. Однако эко-
номически целесообразно сначала переводить дихлоргидрин в эпи-
хлоргидрин, используя для этого более дешевое известковое молоко
(см. стр. 186).
Предварительно очищенный перегонкой эпихлоргидрин омыляется
10—15% раствором едкого натра [61]. Перегонка необходима для
того, чтобы позже, при выпаривании раствора глицерина, не образо-
вывались известковые соли. Получается 5—10% глицериновый рас-
твор, который выпаривают так же, как подмыльный щелок при про-
изводстве мыла.
Если омылять эпихлоргидрин при 65—100 °C и во время реакции
добавлять глицеринхлоргидрины, то конечная концентрация глице-
рина повысится [62]. Исследования Иосино [63] показывают, что
выход глицерина увеличивается с понижением концентрации едкого
натра, однако этот результат нельзя использовать в промышленности,
так как конечная концентрация глицерина получается очень низкой.
Уменьшение выхода глицерина объясняется полимеризацией проме-
жуточного продукта глицидола и образованием полиглицеринов
(полиглицидола). С увеличением pH эти побочные реакции прояв-
ляются сильнее. Во избежание получения сильно щелочных раство-
ров теперь проводят гидролиз эпихлоргидрина при повышенных
температурах (150—180 °C) с карбонатами щелочных металлов и под
давлением [64—65]. Можно осуществлять гидролиз на основных ани-
онообменных смолах [66] или проводить комбинированный кислотно-
щелочной гидролиз с 0,1 н. НС1 при 80—105 °C и 30 % раствором соды
Предложено [68] гидролизовать глицериндихлоргидрин натрон-
ной известью или NaOH таким образом, чтобы образовывалась только
часть эпихлоргидрина, а остальное количество сразу же гидролизо-
валось бы в глицерин [69—72].
Синтез через аллиловый спирт. Как уже было сказано, глицерин
можно получать и из аллилового спирта, причем несколькими путями:
1) через хлоргидрин;
2) посредством окисления перекисью водорода.
При хлоргидрировании аллилового спирта лучше всего работать
с разбавленным (5%-ным) водным раствором при 15 °C. Оказалось,
что количество получаемого глицерина тем больше, чем меньше кон-
центрация аллилового спирта. О зависимости выхода глицерина от
концентрации аллилового спирта см. в работе [73].
Образуется смесь а- и |3-глицеринмонохлоргидринов и аф-глице-
риндихлоргидрина. Реакция ускоряется ионами водорода и хлора,
образующимися при взаимодействии хлора с водой, и поэтому проте-
кает быстрее, чем при применении чистой хлорноватистой кислоты.
10.2. Замещающее хлорирование 195
Мясковский [74] указывает следующие оптимальные условия
образования глицеринхлоргидрина из водного' раствора аллилового
спирта в присутствии хлора:
Концентрация аллилового спирта, % ... . 4,5—5,5
Температура реакции, °C.................50—60
Продолжительность реакции, с............ 5
При этих условиях выход глицеринмонохлоргидрина составляет
88%, глицериндихлоргидрина — 9%.
Из хлоргидринов путем щелочного омыления получают разбавлен-
ный водный раствор глицерина, который перерабатывают в 99%-ный
глицерин. Выход глицерина в расчете на аллиловый спирт соста-
вляет 90%. Продукт омыления содержит около 93,5% глицерина,
1,5% непрореагировавших хлоргидринов и 3,5% высококипящих
соединений (полиглицеринов).
Для омыления лучше брать не едкий натр, а соду. Раньше рабо-
тали с 10% растворами едкого натра и 1% раствором соды [75, 76].
Теперь рекомендуется увеличивать концентрацию содового раствора.
Мясковский предлагает применять растворы из 75% соды и 25%
едкого натра [77]. Описано также применение одной только соды,
но в этом случае надо проводить гидролиз при повышенной темпера-
туре, т. е. с 10% раствором соды при 200—220 °C [78], а с 20% —
при 150 °C [79-80].
Гидролиз а-глицеринмонохлоргидрина под давлением осуществим
и с водой при 135 °C и 25 кгс/см2 [81]. Сильноосновные анионообмен-
ные смолы (Амберлит ИРА 400, Дауэкс 2) предлагаются в качестве
гидролизующих агентов [82]. В этом случае для гидролиза образовав-
шегося глицидола необходима дополнительная обработка каталити-
ческими количествами катионообменных смол (Амберлит ИР 112
и ИР-120, Дауэкс 50) при 50 °C. Мягкий гидролиз можно осуществить
путем постепенного добавления оснований при повышении темпера-
туры [83].
Другой путь получения глицерина из аллилового спирта состоит
в хлорировании последнего. Соответствующие исследования проводи-
лись в 1948 г. Ингом [84]. Он получил при хлорировании в водной
соляной кислоте 72% глицериндихлоргидрина. Такой же выход
указан в одном из американских патентов [85], где описано проведе-
ние хлорирования дополнительно и в темноте. Наконец, хлорирова-
ние удается осуществить и в присутствии соды [86]. Правда, этот
метод не нашел признания из-за сложности и неудовлетворительного
выхода продукта.
Получение глицерина из пропилена без применения хлора. В миро-
вом масштабе хлор является дефицитным сырьем, поэтому были
предприняты попытки уменьшить расход хлора при синтезе глице-
рина или вовсе обойтись без него. Фирмой Shell Chemical Со.
13*
196
10. Хлорирование пропилена
был разработан такой метод, но до сих пор он применен только на
одной крупной установке в Норко (Луизиана). Отдельные стадии
этого метода следующие [87—88]:
Пропилен ---> Изопропиловый спирт-> Ацетон + П2О2
Акролеин ---> Аллиловый спирт ----> Глицерин
Ацетон
Рентабельность этого метода в большой мере зависит от потреб-
ности в ацетоне, получающемся в количестве 2 моль на.1 моль гли-
церина.
Одна из важнейших и сложнейших стадий в этом методе — гид-
рирование акролеина в аллиловый спирт. Для этого очищенный
акролеин вместе с 2—3-кратным мольным количеством изопропило-
вого спирта при 391—401 °C пропускают над катализатором MgO—
ZnO. При этом образуется 77,4% аллилового спирта [89—90[. Пред-
лагаются и другие катализаторы [91]: ZnO—CaO, MgO—Та2О5,
Fe2O3—MgO, MgO—Cu2O. На этой стадии рекомендуется также при-
менять в качестве катализатора алюминийалкоголят вторичного
спирта (реакция проводится по Мервейну — Понндорфу [92]).
При получении аллилового спирта выход достигает 84,2—92,1%.
Частичное окисление изопропилового спирта в ацетон при одно-
временном образовании перекиси водорода можно осуществить
в жидкой и в газовой фазе.
Окисление в жидкой фазе. При окислении вторичных спиртов в жидкой
фазе [93] перекись водорода, добавляемая к свежему изопропиловому спирту
в концентрации 0,5—1%, действует как инициатор реакции. Реакция протекает
при 90—140 °C и давлении кислорода 2,5 кгс/см2. Конверсия кислорода дости-
гает 80—95%. Выход можно увеличить, а время окисления сократить, если до-
бавить немного ледяной уксусной кислоты [94] или солей тяжелых металлов
[кобальта, железа (III), никеля, марганца, хрома] [95], разумеется, в присут-
ствии стабилизаторов перекиси водорода (кислородных кислот олова, сурьмы,
кремния, алюминия или их щелочных солей) [96]. Такой же эффект дости-
гается при облицовке реактора станнатом или фосфатом натрия [97—98]. Реак-
ция ускоряется также под действием УФ-облучения (стабилизатор TiO2) [99].
Перед дистилляцией продукт реакции следует разбавить водой, чтобы исклю-
чить реакции между перекисью водорода и органическими соединениями. Аце-
тон и изопропиловый спирт удаляются вместе с легкими фракциями. Остается
6—10% водный раствор Н2О2, содержащий еще небольшие количества спиртов,
альдегидов и кетонов [100—102]. Если удовлетвориться сравнительно низкой
конверсией спирта (15%), то можно получить выход перекиси водорода в преде-
лах 87-93%.
Обширйые исследования по образованию Н2О2 при окислении изопропило-
вого спирта были проведены Бурхардтом и другими [106]. Скорость реакции
увеличивалась с повышением температуры, но выход Н2О2 падал. Лучшими
инициаторами реакции оказались Н2О2, перекись трет-бутила и mpem-бутил-
10-2. Замещающее хлорирование
197
пербензоат. Окисление нужно проводить с некоторым избытком кислорода,
так как в отсутствие кислорода даже скорее, чем в его присутствии, может
произойти реакция между изопропиловым спиртом и Н2О2, что вызовет умень-
шение выхода.
Димерная и тримерная перекись ацетона и другие перекиси были иденти-
фицированы с помощью тонкослойной хроматографии как побочные и вероятные
промежуточные продукты этой реакции окисления.
Окисление в газовой фазе. Об окислении изопропилового спирта в газовой
фазе впервые было сообщено в 1949 г. [103]. Реакция проводится при 460 °C
и отношении воздух : изопропиловый спирт =10: 7 в присутствии 3 ч. водя-
ного пара или при 400 РС и отношении воздух : изопропиловый спирт =1:1
[104]. Достигается конверсия кислорода порядка 60—80%, причем время пре-
бывания изопропилового спирта в реакторе составляет от 0,5 до 1 с. Реактор
должен быть облицован кислотоупорной эмалью. Очень важно быстрое охлажде-
ние реакционной смеси до температуры ниже 100 °C. Лучше всего это дости-
гается впрыскиванием воды в смесь. Выход перекиси водорода при окислении
в газовой фазе достигает около 70%, выход ацетона — 90%. При оптимальных
условиях (420 °C, время контакта 50—80 с) получаются следующие результаты:
Конверсия, %...................40
Выход, %
перекись водорода ...... 80
ацетон .......................90
метиловый спирт...........1—2
глиоксаль...................1—2
формальдегид-]-ацет альдегид . . 5%
Для промышленности выгоднее более короткое время контакта и более
низкая конверсия.
Гидроксилирование аллилового спирта перекисью водорода в гли-
церин [107] катализируется различными соединениями тяжелых
металлов: V2O5, СгО3, солями Fe(II), OsO4, вольфрамовой кислотой,
Na2WO4, фосфоровольфрамовой кислотой, фосфоромолибденовой кис-
лотой и др. [108—109]. При гидроксилировании в промышленных
условиях хорошо зарекомендовала себя вольфрамовая кислота.
Окисление аллилового спирта осуществляется 2М раствором Н2О2
при 60—70 °C в присутствии 0,2% катализатора, причем лучше всего
брать соотношение аллиловый спирт : Н2О2 = 1:1 [110]. Выход
глицерина в расчете на аллиловый спирт составляет 80—90%.
Для улучшения выхода предлагается проводить окисление в не-
сколько стадий: а) в две стадии при 40 °C и 70 °C [111] или при 50 °C
и 70 °C [112]; б) три стадии с NaHWO4 при 30—45 °C, 40—60 °C
и 70—90 °C (выход 90%) [113]; в) в нескольких реакторах с постепен-
ным ступенчатым повышением температуры от 50 до 70 °C [114].
Можно также во время реакции ступенчато изменять величину pH.
Сначала работают при pH =3,5 и 45 °C. Во 2-й фазе процесс идет
при комнатной температуре, при этом частично образуется глицидол.
Затем при омылении в результате подкисления до pH = 2[115] или На-
гревания до 70 °C образуется глицерин [116]. Гидролиз можно осу-
ществить и путем нагревания до 145 °C реакционной смеси, окислен-
ной при 45 °C [117].
198
10- Хлорирование пропилена
С целью повышения активности катализатора можно нанести
вольфрамовую кислоту на гель А12О3 или SiO2 или на активирован-
ный уголь [118]. Можно ввести в реакцию свободную WO3, суспенди-
рованную в уксусной кислоте [119]. (О регенерации WO3 см. в ра-
боте [120].) Окисление в присутствии изо- или гетерополивольфрамо-
вых [121] и особенно фосфоровольфрамовых [122] кислот (а также
бериллий- или марганецвольфрамовой кислоты [123]) проходит при
70 °C с выходом 89—90%. В качестве катализаторов предлагаются
также гетерополикислоты, содержащие Мо, Cr, S, Se или Те [124].
Окись OsO4 оказалась таким же хорошим катализатором, как и воль-
фрамовая кислота. Уже при 30 °C без ускорителей или в присутствии
ускоряющих реакцию солей марганца или церия [125] она дает гли-
церин с выходом 74—90%. Так же можно превращать акролеин
в глицериновый альдегид [126]. Используя муравьиную кислоту
в качестве переносчика гидроперекиси (предположительно в форме
пермуравьиной кислоты), можно обходиться без дополнительного
катализатора. В результате 10-дневной реакции при 30 °C или при
нагревании до 46 °C образуется 82—94% глицерина [127—128].
Аналогично можно проводить взаимодействие аллилового спирта
с Н2О2 и аллилформиатом (75—80%) [129]. Применение изопропил-
формиата в качестве растворителя, по-видимому, основано на сход-
ном течении реакции [130]. Подобный характер имеет и реакция
эпоксидирования аллилового спирта с перуксусной кислотой в уксус-
ную кислоту при одновременном гидролизе в глицерин [131—132].
Теоретическое изучение гидроксилирования аллилового спирта
показывает, что в качестве катализаторов пригодны только те окиси
(например, WO3, OsO4 [133], МоО3, V2O5, [134]), которые могут
образовывать перкислоты, проявляющие затем окисляющее действие.
Так, H2WO5 служит переносчиком кислорода и при других реакциях
окисления, например при окислении акролеина в акриловую кис-
лоту [135].
В одном из патентов [136] предлагается прямой путь непосред-
ственно от пропилена к глицерину:
„ О2, 5 50 °C (резкое охлаждение)
Пропилен------------------------------> Аллилгидроперекись----->
WO3 или 0s04, 20-100 °C „
--->------------------------> Глицерин (75%)
Недавно вступившая в строй установка по производству глице-
рина мощностью 18 500 т/год (фирмы Food Machinery and Chemical
Corp, в Бэйпорте, Техас) работает по новому методу, отраженному
на следующей схеме [205, 206]:
. о2
Ацетальдегид----> Перуксусная кислота-----г н 0
--------------------------------------------> Глицидол —----> Глицерин
Окись пропилена --------------------------------------------> Аллиловый спирт-!
10-2. Замещающее хлорирование
199
Установка на 12 000 т/год фирмы DaicelLtd. (Отака), очевидно,
будет работать по тому же принципу.
Экономические исследования [137] приводят к выводу, что бес-
хлорный метод более выгоден, особенно если перекись водорода полу-
чается на том же предприятии и если обеспечен необходимый сбыт
ацетона.
Свойства и применение глицерина. Ниже приведены свойства
глицерина:
Температура плавления, °C......................... 18,18
Температура кипения, °C........................... 290,5
Плотность
РЙ................................................... 1,26557
f>!§................................................. 1,26362
РУ................................................... 1,26201
Показатель преломления п™................................ 1,47399
Дипольный момент, D..................................... 2,56
Вязкость, сП
при 20° С ......................................... 1499
25° С ......................................... 945
30° С .......................................... 624
Поверхностное натяжение, дин/см
при 0° С . . .............................. 88,0
20° С......................................... 63,4
90° С......................................... 58,6
150° С......................................... 51,9
202° С......................................... 45,3
Теплота плавления при 18 °C, ккал/моль..................4,414
Теплота испарения, ккал/моль
при 55° С................................... 21,1
165° С......................................... 18,74
Теплота образования при 18 °C и 760 мм рт. ст.,
ккал/моль.............................................—157,9
Теплота сгорания, ккал/моль
при постоянном объеме.............................. 396,8
постоянном давлении........................... 397,2
Удельная теплоемкость, кал/(г-°С)
при —250° С........................................ 0,0471
—200° С . . '.................................... 0,115
0° С......................................... 0,540
50° С......................................... 0,600
100s с......................................... 0,669
Температура вспышки в закрытом сосуде, °C............. 177
Температура горения, °C............................... 204
Температура воспламенения на воздухе при 760 мм
рт. ст., °C........................................ 520
Диэлектрическая проницаемость
при —140° С........................................ 2,8
—1°С............................................ 3.6
21° С......................................... 15,3
40° С......................................... 33,1
50° С......................................... 35,8
94,9° С....................................... 30,5
225° С........................................ 25,3
200
10. Хлорирование пропилена
Ниже приведены температуры кипения глицерина при различных
давлениях [138]:
р, мм рт. ст. Т. кип., °C р, мм рт. ст. Т. кип.,
760 290.0 50 203,62
500 274,23 30 190,87
300 256,32 20 181,34
100 222,41 10 161,11
70 212,52 5 152,03
Далее указано давление паров глицерина при различных темпера-
турах [139]:
t, °C Р, мм рт. СТ. t, °C р, мм рт. ст.
125,5 1 208,0 60
153,8 5 220,1 100
167,2 10 240,0 200
182,2 20 263,0 400
198,0 40 290,0 760
Другие физические характеристики смотрите в следующих лите-
ратурных источниках: вязкость глицерина при различных темпера-
турах — [140]; термическое расширение глицерина и его водных
растворов, показатели преломления водных растворов глицерина
при 20 °C — [141]; температуры кипения водных растворов глице-
рина при 760 мм рт. ст. — [142]; температуры застывания и плот-
ность водных растворов глицерина — [143]; вязкость водных рас-
творов глицерина — [144]. Физические характеристики глицерина
приведены также в работе [145].
Основным назначением глицерина является использование его
для получения нитроглицерина — важнейшего' взрывчатого веще-
ства. Вместе с фталевым ангидридом глицерин применяется для полу-
чения глифталевых смол, он также используется для производства
хинолина, бензантрона и ализарина.
В косметической промышленности глицерин служит основой ма-
зей, вводится также в состав средств для ухода за кожей рук и зуб-
ных паст. Применяют его для приготовления напитков, экстракции
запахов, для увлажнения табака. В текстильной промышленности
глицерин используют для получения шлихтовочных масс и аппретур.
В технике глицерин применяется в нагревательных системах,
как охлаждающая жидкость, затворная жидкость, антифриз, сма-
зочное средство, пластифицирующий агент, передатчик давления
в гидравлических системах, при изготовлении электролитных кон-
денсаторов, копировальных чернил, гектографных масс, штемпельных
и печатных красок, в соединении с РЬО используется как замазка.
Во многих случаях глицерин стараются заменять другими веще-
ствами, более подходящими для специальных целей. К таким заме-
10-3- Перхлорирование пропилена
201
цителям относятся этиленгликоль, 1,2- или 1,3-пропиленгликоль,
полигликоли, триметилолэтан, триметилолпропан, гексантриоды,
пентраэритрит и другие многоатомные спирты, полиглицерин и т. д.
В качестве пластификаторов для сложных и простых эфиров
целлюлозы применяются триацетат и трибутират -глицерина. Моно-
фосфаты используются в фармацевтической промышленности. Хлор-
гидрины, которые являются промежуточными продуктами в произ-
водстве глицерина, имеют значение также для других синтезов.
Полиглицерины, получаемые при нагревании глицерина со щело-
чами до температур выше 200 °C, служат вместо глицерина для полу-
чения шлихтовочных масс и аппретур, в качестве пластификаторов,
смазочных средств, а также для производства косметических препа-
ратов и алкидных смол.
10.3. ПЕРХЛОРИРОВАНИЕ ПРОПИЛЕНА
Метод перхлорирования [161—167] пропилена и других подоб-
ных углеводородов был изучен для того, чтобы разработать дешевый
и не зависящий от производства ацетилена способ получения перхлор-
зтилена — весьма перспективного растворителя.
Высокотемпературное (450— 700 °C) хлорирование низкомолеку-
лярных алифатических углеводородов, главным образом метана,
этана, пропана, бутана, изобутана, этилена и пропилена, а также
их хлорпроизводных, проходит уже не как чистая реакция замеще-
ния, а большей частью как «расщепляющий» и «строящий» крекинг.
В случае метана преобладает соединение обломков Сх с образованием
перхлорэтилена, в случае пропанов и пропиленов — расщепление
с образованием четыреххлористого углерода и перхлорэтилена,
в случае этанов и этиленов в зависимости от условий реакции могут
получаться различные продукты [183—186]. ч
Первоначально для метода высокотемпературного хлорирования
применяли метан и продукты его хлорирования. При этом метан,
а позднее и природный газ, хлорировали в избытке хлора при 450—
700 °C до образования четыреххлористого углерода. В процессе
реакции большая часть метана превращается в перхлорэтилен (побоч-
ные продукты — гексахлор этан, гексахлорбензол) [187, 188]. С повы-
шением температуры увеличивается выход перхлорэтилена, при не-
котором подъеме парциального давления хлора, напротив, увеличи-
вается выход тетрахлоруглерода [189]. Введение инертного газа
(N2) способствует переходу четыреххлористого углерода в перхлор-
этилен [190]. \
Каталитическое хлорирование метана в кипящем слое на мате-
риале с большой поверхностью проводится при несколько более
низких температурах, оптимально при ~360 °C [191]. Хлорирова-
нием метана в присутствии большого избытка четыреххлористого
202
10. Хлорирование пропилена
углерода при 500—700 °C можно добиться 96%-ного выхода перхлор-
этилена [192]. Эти методы получили большое промышленное значе-
ние, с тех пор как в качестве сырья стали использовать С3-углеводо-
роды.
Соответствующие установки работают с 1954 г. на фирме Рго-
gil — Electrochimie Cie. в Понт де Клэ (мощность 35 тыс. т/год)
и на фирме Pechiney Cie. в Сент-Обэне (мощность 8 тыс. т/год), послед-
няя по методу Scientific Design Со. [193], при котором можно попе-
ременно использовать С4—С4-углеводороды.
Рис. 48. Технологическая схема получения четы-
реххлористого углерода и перхлорэтилена путем
перхлорирования смеси пропан — пропилен:
1 — реактор; 2 — подогреватель.
По методу Scientific Design Со. газообразные хлор, углеводоров
и четыреххлористый углерод вводятся в реактор при 500—650 °C.
Процесс проводится без катализатора и без подвода тепла извне.
Выходящий газ резко охлаждается 21—36 %-ной соляной кислотой,
не абсорбированные при этом газы пропускаются через НС1-абсорбер,
дающий 20%-ную соляную кислоту, которая снова возвращается
в цикл. Газы, выходящие из HCl-абсорбера, подаются на установку
для регенерации хлора. Продукты реакции после закалочного аппа-
рата направляются в отстойник. Верхний слой представляет собой
соляную кислоту, а из нижнего слоя дистилляцией выделяются
четыреххлористый углерод и хлор [194], возвращаемые в цикл.
С верха последней колонны выделяется чистый перхлорэтилен.
При этом методе практически нет потерь.
Установка фирмы Progil—Electrochimie Cie. (рис. 48) работает
с фракцией пропан-пропилен и хлором, в которых ограничивается
содержание кислорода. Два реактора установлены последовательно.
Хлор подается только в первый реактор, а С3-фракция распределяется
на оба реактора. Температура регулируется количеством разбави-
теля — четыреххлористого углерода и охлаждением реакторов.
В первом реакторе поддерживается температура 450 °C, во втором —
550 °C (возможен и вариант с тремя реакторами, в которых устана-
вливаются температуры 460, 530 и 560 °C) [195—196]. Выход перхлор-
ч
10-3- Перхлорирование пропилена
203
этилена может изменяться в пределах от 30 до 65%; четыреххлори-
стый углерод и перхлорэтилен получаются очень чистыми. На 1 т
перхлорэтилена расходуется 0,185 т пропилена и 1,83 т хлора, при
этом получается 0,93 т НС1; на 1 т четыреххлористого углерода рас-
ходуется 0,10 т пропилена и 1,48 т хлора и образуется при этом
0,502 т НС1.
Известны и другие варианты перхлорирования пропилена или
пропана. Например, при перхлорировании в кипящем слое в качестве
разбавителей предлагаются гексахлорэтан, четыреххлористый угле-
род и перхлорэтилен [197], в качестве закалочных средств (охлади-
телей) .пригодны четыреххлористый углерод и хлористый этилен.
В последнем случае получается особенно высокий выход: 55,8%
четыреххлористого углерода, 44% перхлорэтилена и всего 0,2%
побочных продуктов [198].
Разработан двухстадийный метод: хлорирование и пиролиз [199,
200]. В качестве катализаторов используются FeCl3 при 425—525 СС
[201], СнС12—ВаС12 на активированном угле, иногда в присутствии
солей кобальта, никеля или церия в качестве активаторов (промото-
ров) [202]; рекомендуется также фотохимическое инициирование
[203]. Смолообразования во время пиролиза можно избежать исполь-
зованием четыреххлористого углерода и перхлорэтилена в качестве
разбавителей [202]. Чтобы добиться оптимального баланса хлора,
образующийся при пиролизе хлор вводят в реакцию обмена со све-
жим углеводородом и пиролизуют образовавшуюся смесь хлориро-
ванных углеводородов при 425—525 °C [204].
10.3.1. Получение и характеристика четыреххлористого углерода
Четыреххлористый углерод (тетрахлорметан) был впервые полу-
чен Дюма в 1840 г. (из метана и хлора) и Рэйнольтом (из метилхлорида
и хлора). Позднее четыреххлористый углерод начали получать хлори-
рованием сероуглерода. Этот метод стал рентабельным после того,
как были найдены оптимальные условия взаимодействия S2C12 и CS2
в присутствии железа [168]:
CS2 + 3C12 —> CC14-|-S2C12
, 2S2Cl2-]-CS2-> CCI4+6S
Четыреххлористый углерод можно получить и из метана путем
его хлорирования или из хлорированных метанов, образующихся
при хлорировании метана. Первая промышленная установка для
хлорирования метана, пущенная в 1923 г. фирмой Farbwerke Hoechst,
давала четыреххлористый углерод наряду с метилхлоридом, метилен-
хлоридом (главным продуктом) и хлороформом в различных соотно-
шениях. Чтобы добиться полного хлорирования до четыреххлори-
стого углерода, хлораторы соединили каскадом, а хлор и продукты
хлорирования метана направили противотоком.
204
10. Хлорирование пропилена
Выше описан метод высокотемпературного хлорирования при-
родного газа или продуктов неполного хлорирования метана. Пря-
мой синтез четыреххлористого углерода из составляющих его эле-
ментов технически еще невозможен.
' Ниже приведены свойства четыреххлористого углерода:
Температура плавления, °C...................... —22,9
Температура кипения, °C
при 10 мм рт. ст............................... —20,7
100 мм рт. ст............................ 22,5
760 мм рт. ст............................ 76,7
10 кгс/см2................................ 176,0
Плотность р24°.................................... 1,592
Показатель преломления п^ ........................ 1,4607
Молекулярная рефракция BD ......................... 26,51
Динамическая вязкость, кгс • с/см2
жидкости при 20° С.............................. 99,45 • 10~®
пара при 76,7° С............................ 1,175 -10’®
Поверхностное натяжение при 20° С, кгс/см . . . 27,295-10"®
Парахор.................... . . ............. 219,0
Критическая температура, °C........................ 283,1
Критическое давление, кгс/см2...................... 46,5
Критическая плотность, г/см3....................... 0,558
Теплота плавления, ккал/моль....................... 0,577
Теплота испарения при 76,7° С, ккал/моль .... 7,04
Энергия образования, ккал/моль................. —33,8
Энтропия, кал/(?С • моль)........................... 52
Удельная теплоемкость, ккал/(кг-°С)
жидкости при 20° С . ........................... 0,2028
пара при 76,7° С........................... 0,1440
Коэффициент теплопроводности, ккал/(м • ч • °C)
жидкости при 20° С.............................. 0,1017
пара при 76,7° С...........................’• 0,0069
Коэффициент теплового расширения, 1/°С • • 4,5 10~4
Диэлектрическая проницаемость.................. 2,25
Магнитная чувствительность.....................-0,429-10“®
Растворимость, г/кг
ССЦ в воде при 25° С........................ 1,16
воды в СС14 при 20° С....................... 0,08
Температура кипения и содержания четыреххлористого углерода
в двойных азеотропных смесях со следующими веществами:
Т. кип., сск, %
°C
Вода.......................... 66 95,9
Метиловый спирт............... 55,7 79,4
Этиловый спирт................ 65,1 84,1
Изопропиловый спирт........... 68,95 82,0
Муравьиная кислота............ 66,65 81,5
Ацетон........................ 56,1 11,5
Нитрометан ................... 71,3 83,0
Ацетонитрил................... 65,1 83,0
1,2-Дихлорэтан................ 75,3 78,4
Четыреххлористый углерод является очень хорошим растворите-
лем для смол, масел, жиров, восков, пеков, битумов и др. Благодаря
10.3- Перфорирование пропилена
205
этому свойству и негорючести главной областью применения его
является химическая очистка. Кроме того, четыреххлористый угле-
род служит для обезжиривания прецизионных металлических деталей,
а также в быту как средство для удаления пятен и для ухода за по-
лами. Вследствие негорючести он используется для тушения огня.
Четыреххлористый углерод применяется также для борьбы с вреди-
телями зерна, для дезинсекции'почв и как растворитель для природ-
ных и синтетических средств борьбы с насекомыми-вредцтелями.
В химической промышленности четыреххлористый углерод служит
исходным материалом для получения фторхлорметанов, использу-
емых в качестве хладоагентов.
В резиновой промышленности он применяется для набухания
и растворения сырой резины и в качестве разбавителя для S2C12
при холодной вулканизации.
10.3.2. Получение и характеристика перхлорэтилена
Перхлорэтилен (четыреххлористый этилен) был впервые получен
Фарадеем в результате отщепления хлора от гексахлорэтана. Ранние
промышленные методы получения перхлорэтилена основывались
главным образом на синтезе из ацетилена, который сначала превра-
щали в четыреххлористый этан (процесс Wacker). Были разработаны
следующие процессы производства перхлорэтилена из тетрахлор-
этана:
1) пиролиз или отщепление НС1 слабыми основаниями с получе-
нием трихлорэтилена, хлорирование в пентахлорэтан в присутствии
FeCl3 и повторный, пиролиз при 220—300 °C на катализаторе ВаС12
или СиС12 (носитель — активированный уголь) [169] или отщепление
НС1 с помощью Са(ОН)2 [170];
2) дегидрирование тетрахлорэтана в атмосфере хлора при 300 °C
в присутствии ВаС12 или СпС12 на активированном угле [171] или
в атмосфере кислорода в присутствии CuCl2, ZnCl2 или СаС12 на
пемзе или целите [172];
3) хлорирование трихлорэтилена хлором при 300 °C на активиро-
ванном угле [173].
При прямом хлорировании ацетилена выделяется большое коли-
чество тепла(117 ккал/моль), поэтому необходимо разбавлять ацети-
лен и хлор инертными газами [174] или осуществлять пламенную
реакцию между ацетиленом и 3—3,5-кратным количеством хлора при
750—950 °C (80—85% перхлорэтилена) [175].
Очень перспективным кажется метод Soc. d’Ugine, при котором
протекают следующие реакции [176]:
С2С14+2С12 6Я0^700°с^ 2СС14
С2С14 + С12 . 200 Ч- 2 50 °C, акт. уголь^ СзС1в
3C2C]e+C2H2 4C.CU + 2HC1,
206
10. Хлорирование пропилена
В этом случае можно добиться любого соотношения между четы-
реххлористым углеродом и перхлорэтиленом.
Перхлорэтилен можно получить из четыреххлористого углерода
посредством термического расщепления в электропечи при 900 °C.
Работают в присутствии тетра- или трихлорэтана, чтобы улавливать
высвобождающийся хлор, который тут же образует хлорированный
соединения.
Интерес представляет использование 1,2-дихлорэтана как проме-
жуточного продукта, образующегося в результате хлорирования
этилена хлором или НС1 в присутствии FeCl3, CuCl2 или SbCl5 [1791
на активированном угле и кислородом на СнС12 при 150 °C [180].
1,2-Дихлорэтан хлорируется в перхлорэтилен при 300 °C в присут-
ствии СиС12 на активированном угле [181] в присутствии смеси А1С13,
NaCl и FeCl3 [182] или в кипящем слое при 350—450 °C на смеси
CuCl2, ZnCl2 и CdCl2, нанесенных на целит. Аналогично осуще-
ствляется процесс по методу оксихлорирования в присутствии
СиС12—КС1 на отбельной земле.
Получение перхлорэтилена непосредственно из этилена проводится
в следующих условиях:
Соотношение С2Н4 : О2 : CI2 . . . , , 1:2:1
Температура, °C......................... 413—419
Время контакта, с....................... 9,8
Катализатор..........................CuCl2—КС1
(на отбельной
земле)
Конверсия, %................ . . . . 94,1
Селективность, %..................... 53,8
Аналогичные результаты дает метод оксигидрохлорирования при
442 °C на катализаторе из CuCl, CuCl2, КС1 и 0,2% NiCl2; соотноше-
ние 1,2-дихлорэтан : О2 : НС1 = 1 : 1,3 :1,04; выход три- и перхлор-
этилена достигает 71%.
Ниже приводятся свойства перхлорэтилена:
Температура плавления, °C.......................... —22,35
Температура кипения, °C
при 10 мм рт. ст.......................... 13,0
100 мм рт. ст.............................. 59,5
760 мм рт. ст.............................. 121,1
Плотность р|° ...................................... 1,623
Плотность пара при 12Г С, г/л..................... 5,13
Показатель преломления п™......................... 1,5058
Молекулярная рефракция RD ........................ 26,51
Вязкость при 20° С, сП.............................. 0,88
Поверхностное натяжение при 20° С, дин/см . . . 32,3
Парахор . . ........................................ 244,5
Критическая температура, °C......................’ 347
10.3. Перхлорирование пропилена
207
Теплота испарения при 121,1° С, ккал/моль . . . 8,30
Теплота сгорания, ккал/моль.................... —162,5
Удельная' теплоемкость при 20° С, кал/(г - °C) . . . 0,216
Мольная теплоемкость при 0° С, кал/(моль - °C) . . 32,8
Коэффициент теплопроводности при 20° С,
кал/(см • г • сек • РС)......................... 0,39 • 10'3
Коэффициент теплового расширения, 1/°С .... 1,08 • 10“3
Диэлектрическая проницаемость при 20° С........ 2,37
Магнитная чувствительность.....................—0,508 • 10~6
Растворимость при 25° С, %
перхлорэтилена в воде......................... 0,01
воды в перхлорэтилене...................... 0,01 .
Азеотропные смеси с перхлорэтиленом следующих веществ:
Т. кип., °C Содержа- ние C2CI4, %
Вода 87,1 84,1
Метиловый спирт 63,75 36,5
Этиловый спирт 76,75 37,0
Пропиловый спирт 94,05 52,0
Бутиловый спирт 108,95 71,0
Муравьиная кислота 88,15 50,0
Уксусная кислота ....... 107,35 61,5
Перхлорэтилен применяется для сухой химической чистки. Для
этих целей в США и Англии потребляется 75% перхлорэтилена, при-
чем он все в большей степени заменяет трихлорэтилен, который ис-
пользуется только для обезжиривания металла. Перхлорэтилен слу-
жит для очистки алюминия, а также для самых разнообразных целей
в качестве растворителя и экстрагирующего агента. Кроме того, его
применяют в медицине как эффективное средство против глистов.
Перхлорэтилен является исходным материалом для получения гекса-
хлорэтана.
10.3.3. Экономическая оценка методов получения четыреххлористого
углерода и перхлорэтилена
По методу Progil Electrochimie Cie на 1 т перхлорэтилена и 1 т
четыреххлористого углерода расходуются следующие относительные
количества веществ (в ч.):
с2сц cch
Пропилен ............................ 0,185 0,100
Хлор................................. 1,830 1,480
НС1 (побочный продукт)............. 0,930 0,502
Пропан .............................. 0,194 0,104
Хлор ................................ 2,135 1,643
НС1 (побочный продукт)............. 1,240 0,668
208
10. Хлорирование пропилена
Детальное сравнение различных методов получения четыреххло-
ристого углерода и перхлорэтилена затруднительно, поскольку
влияющие факторы едва ли допускают обобщение. Решающую роль
играет наличие того или иного сырья в том месте, где работает уста-
новка.
Превращение трихлорэтилена в перхлорэтилен целесообразно
только в тех случаях, когда трихлорэтилен нельзя использовать
как таковой. Методы, основанные на ацетилене и других углеводоро-
дах как исходных веществах, всегда дают хлористый водород в ка-
честве побочного продукта. Такие процессы проводятся иногда в не-
сколько стадий и при повышенных температурах. Выход хлористого
водорода повышается при применении в качестве сырья ацетилена,
поэтому рентабельность процесса зависит от использования хлори-
стого водорода. Это осуществляют получением из НС1 хлора по
методу «Deacon».
Получение перхлорэтилена из ацетилена останется промышлен-
ным методом, даже если вместо него все в большем масштабе будут
применяться и другие углеводороды, в частности С3-фракция.
ЛИТЕРАТУРА
1. S. A. Mille г, Chem. Process Eng., 47, 268 (1966).
2. D. A. S. 1210801, 1966.
3. T. D. Stewart, B. J. We i de nba urn, J. Am. Chem. Soc., 58,
98 (1936).
4. Пат. США 20077382, 1937; 2130084, 1938; 2321472, 1943; герм, пат.736315,
1936. ,
5. Н. Р. A. G г о 11, G. Hearne, Ind. Eng. Chem., 31, 1530 (1939).
6. Герм. пат. 720079, 740986, 1938.
7. Е. С. W i 11 1 a m s, Chem. Metallurg. Eng. 47, 834 (1940); 48, 87 (1941).
8. E. C. W i 11 i a m s, Trans. Am. Inst. Chem. Eng., 37, 157 (1941).
9. A. W. Fairbairn, H. A. Cheney, A. J. Cheriniavsky, Chem.
Eng. Progress, 43, 280 (1947).
10. Бельг, пат. 611644—611646, 1962.
11. D. A. S. 1211151, 1215691, 1966.
12. Герм. nai. 720545; 1937; пат. США 2410647, 1938.
13. J. Dykyj et al., Chem. prumysl., 11, 281 (1961).
14, P. Kluco vsky, J. D у k у j, Acta Chim. Acad. Hung., 36, № 1—4,
145 (1963).
15. Пат. ЧССР 94473, I960.
16. Голл. пат. 84704, 1957.
17. Англ. пат. 762153, 1956.
18. Пат. США 2763699, 1956.
19. Англ. пат. 901680, 1962. .
20. Пат. США 3054831, 1962.
21. D. A. S. 1114184, 1961.
22. L. A. R е е d, W. F. S t е v е n s, Can. J. Chem. Eng., 41, № 4, 182 (1963).
23. D. A. S. 1147219, 1963.
24. Пат. США 1477047, 1919; 2207193, 1937.
25. Е. G а 1 i t z е п s t е i п, С. W о о I f, J. Soc. Chem. Ind., 69, 298 (1950).
26. E. T. M с В e e et al., Ind. Eng. Chem., 33, 180 (1941).
27. Бельг, пат. 572902, 1959.
Литература
209*
28. Англ. пат. 495900, 1937.
29. Пат. США 2966529, 1960.
30. Пат. США 2494034, 1950.
31. Chem. Eng. Progress, 43, 280 (1947).
32. М. Berthelot, S. de L u c a, Ann. Chimie, [3], 41, 299 (1854).
33. D. L. Y a b г о f f, J. Anderson, Proc. 3d World Petrol. Congress,
Section V, 1950, p. 22.
34. Англ. пат. 784620, 1957; пат. США 3150154, 1964.
35. Англ. пат. 754359, 1956; пат. США 2786854, 1957.
36. Франц, пат. 1376471, 1964.
37. Пат. США 3053856, 1962.
38. Niederld. A. S. 6408316, 1965.
39. Kiryu Т., Komatsu И., Mem. Fac. Technol. (Tokyo) Metropol-
Univ., 12, 963 (1962); С. A, 59, 7019h (1963).
40. P. Leone, M. В e n e 11 i, Gazz. chim. Ital., 52 (11), 77 (1922).
41. L. A. H о r s 1 e y, Ind. Eng. Chem., Anal. Ed., 19, 508 (1947).
42. Chem. Industrie, 16, 484 (1964).
43. E. J. Vandenberg, Rubber Plastics Age, 46, № 10, 1139 (1965); Rev-
gen. caoutch. plast., 43, 76 (1966).
44< W. D.W i11 i s et al., Rubber World, 153, № 1, 88 (1965);
P. Fournier, Rev. gen. caoutch. plast., 43, 1469 (1966).
45. Chem.. Industrie, 17, № 10, 583 (1965); пат. ГДР 55828, 1967; D. A. S„
1237313, 1967.
46. Allylalcohol, San Francisco, 1946.
47. Пат. США 2072016, 1936.
48. Пат. США 2318033, 1939.
49. Пат. США 2426264, 1947; англ. пат. 603815, 1948; пат. США 2479632, 1945р
1917179, 1933, 1945;
П. Г. Сергеев, Л. М. Букреева, А. Г. Полковников а,.
ЖПХ, 31, 1415 (1958);
L. G. Lundstedt et al., Ind. Eng. Chem., 43, 728 (1951);'
П. Г. Сергеев, Л. M. Букреева. А. Г. Полковников a,.
Хим. наука и пром., 2, 133 (1957); пат. США 2986585, 1961; англ. пат.
902953, 1962; канад. пат. 690646, 1964; D. A. S. 1197077, 1965; швейц.
пат. 403742, 1966; франц, пат. 1271563, 1962; 1344311, 1964; доп. франц.
пат. 79905, 1963;
, Пат. США 3238264, 1966; 3209037, 1965; франц, пат. 1316720, 1963.
50. Голл. пат. 56516, 1940; герм. пат. 858247, 1952; англ. пат. 734247, 1955
пат. США 2763696, 1956; белы. пат. 623031, 1963;
S. К unichika, Sh. Oka, М. S a k a i, Bull. Inst. chem. Res., Kyoto
Univ., 44, № 3, 215 (1966);
Niederld A. S. 6409479, 1966; авт. свид. СССР 175928, 1965; Бюлл-
изобр., 42, № 21, 12 (1965);
С. Д. Мехтиев, О. А. Нариманбеков, 5, 237 (1965).
51. Франц, пат. 894673, 1943; пат. США 2198936, 1938.
52. К. W. Scheele, Crells. Chem. J., 4, 190 (1779).
53. Герм. пат. 541362, 1927.
54. Герм. пат. 892590, 1941.
55. Пат. США 2852570, 1958.
56. D. A. S. 1077203, 1960.
57. D. A. S. 1077204, I960.
58. G. N a t t a, R. R i g a m о n t i, E. В e a t i, Bet. dtsch. chem. Ges., 76,
643, 655 (1943).
59. L. S. C a s 1 i n, OibGas J., 47, № 20, 88, 128 (1948); J. W. Cu tcheon,
Soap, sanit. Chemical, 24, № 9, 37, 44 (1948).
60. D. P. T h о г n t о n jr., Petroleum Processing, 3, 931 (1948).
61. Petroleum Refiner, 32, № 11, 138 (1953).
14 Заказ 39 9
'210
10. Хлорирование пропилена
62. Голл. пат. 83256, 1956.
63. М. Y о s h i п о et al., Yukagaku, 13, № 11, 582 (1964); 14, № 3, ИЗ; № 4,
182 (1965).
64. Англ. пат. 768779, 1957.
65. Голл. пат. 93903, I960.'
66. J. V i г i о t, Bull. Assoc. Franc. Techniciens Petrole, 151, 17 (1962).
67. Пат. ЧССР 104347, 1962.
68. Пат. США 2858345, 1958.
69. Пат. США 2873298, 1959.
70. Пат. США 2605293, 1952.
71. Франц, пат. 1328311, 1963.
72. D. A. S. 1156774, 1964.
73. Е. С. Williams, Trans. Am. Inst. Chem. Eng., 37, 157 (1941).
74. J. Myszkowski, A. Z. Zielinski, Przemysl. chem., 44, № 5,
249 (1965).
75. E. C. W i 11 i a m s, Chem, Metallurg. Eng., 47, 835 (1940).
76. Пат. США 2318032, 1939.
77. J. Myszkowski, A. Z. Zielinski, E. Laskowska, Prze-
mysl. chem., 44, № 10, 565 (1965).
78. Англ. пат. 940284, 1963.
79. Англ. пат. 889613, 1962.
80. K.-Sh. Chia, T.-H. К u, Ch.-T. Chou, Union Ind. Res, Inst. Rept.
(Taiwan), 37, 15 (1958).
81. Пат. США 2311741, 1940.
82. Англ. пат. 971633, 1964.
83. Англ. пат. 773436, 1957.
•84. Н. R. Ing, J. Chem. Soc., London, 1948, 1395.
85. Пат. США 3037059, 1962.
86. Пат. ФРГ 840235, 1952.
87. Petroleum Refiner, 34, № 12, 160 (1955).
88. М. Mugdan, D. Р. Y о u n g, J. Chem. Soc., London, 1949, 2988,
89. Англ. пат. 619014, 1949.
90. Голл. пат. 65211, 1950.
91. Пат. США 2767221, 1956.
92. Англ. пат. 755600, 1956; пат. США 2779801, 1957.
93. Англ. пат. 708339, 1954; голл. пат. 76939, 1954; белы. пат. 199726, 1956;
пат. США 2871104, 1959.
94. Пат. ФРГ 935303, 1955.
95. Англ. пат. 751508, 1956; пат. ФРГ 962069, 1957.
*96. Англ. пат. 731238, 1955.
97. Бельг, пат. 525653, 1956; голл. пат. 83355, 1956,
98. Пат. США 2871101, 2871102, -2871103, 1959.
•99. Пат. США 2910415, 1959.
100. Р. W. S h е г w о о d, Chem.-Ing.-Techn., 32, 461 (1960).
101 Англ. пат. 760605, 1956; пат. ФРГ 962065, 1957; голл, пат. 86525,
1957.
102. Пат. ФРГ 940349, 1956.
103. Пат. США 2479111, 1949.
104. М. Baccareda, С. Pedrazzini, Riv. combustibili, 8, 417 (1954).
105. A. R. В u г g е s s. C. F. С u 11 i s, E. J. N e w i t t, J. Chem. Soc.
(London), 1961, 1884; A. R. В u r g e s s, J. Appl. Chem., 11, № 7, 235
(1961).
107. Герм. пат. 863492, 1940.
108. Герм. пат. 890943, 892589, 908135, 1943.
109. Пат. США 2414385, 1947.
110. С. Д. Мехтиев, А. А. Бахши-Заде, G. Я. Мехтиев, Азерб.
хим. ж., № 5, 47 (1961).
Литература
21f
111. Авт. свид. СССР 166009, 1964; Бюлл. изобр., 41, № 21, 12 (1964)
112. Пат. США 2731502, 1956.
113. Пат. ПНР 48251,. 1964.
114. Англ. пат. 725375, 1955.
115. D. A. S. 1212056, 1966.
116. Авт. свид. СССР 107762, 1957; Бюлл. изобр., № 8, 16 (1957).
117. Пат. США 2862978, 1958.
118. Пат. США 2776301, 1957.
119. Англ. пат. 654764, 1951.
120. D. A. S. 1198806, 1965.
121. Пат. США 2838575, 1958.
122. Т. Mazonski, D. Gasztych, W. Zielinski, Przemysl. Chem.
41, 251 (1962).
123. Пат. США 2773909, 1956.
124. Пат. США 2754325, 1956.
125. Пат. ФРГ 907774, 1954.
126. Пат. США 2718529, 1955.
127. Пат. США 2500599, 1950.
128. Пат. США 2739173, 1956.
129. Англ. пат. 771346, 1957; голл. пат. 83175, 1956.
130. Англ. пат. 799753, 1958.
131. Англ. пат. 917747, 1963; D. A. S. 1222028, 1966; франц, пат. 1287162, 1962..
132. Т. S u z и к i, Y. М i z u m u г a, J. Chem. Soc. Japan, Ind. Chem. Sect.,
69, 434 (1966).
133. Л. В. С у л и м а, ЖОХ, 1, 75 (1965).
134. Англ. пат. 730431, 1955.
135. П. Г. С e p г e e в, Л. M. Б у к p e e в а, ЖОХ, 28, 101 (1958).
136. D. A. S. 1158951, 1963.
137. А-. S. В a n с i u, Rev. chim., Bucuresti, 13, 628 (1962).
138. С. V. Rechenberg, Einfache und fraktionierte Destination, Leip-
zig, 1963.
139. D. R. S t u 11, Ind. Eng. Chem., 39, 520 (1947).
140. G. T a m m a n n, W. H e s s e, Z. anorg. allg. Chem., 17, 1060 (1932);
V. V a n d, Research, 1, 44 (1947).
141. C. S. M i n e r, N. N. D a 1 t о n, Glycerol, New York, 1953.
142. E. Schlenker, Das Glycerin, Stuttgart, 1932.
143. L. B. Laue, Ind. Eng. Chem., 17, 294 (1925);
W. L. В о s a r t, A. S n о d d у, там же, 19, 506 (1927).
144. M. L. S h e 11 у, Ind. Eng. Chem., 24, 1060 (1932).
145. U 1 1 m a n n, Enzyclopadie der technischen Chemie, • Bd. 8, Munchen —
Berlin, 1957, S. 194.
146. C. S. M i ne r, N. N. D a 1 t о n, Glycerol, New York, 1953.
147. Пат. США 2216548, 1938.
148. L. M. P e t e r s, К. E. M a r p 1 e, Ind. Eng. Chem., 40, 2046 (1948).
149. Org. Syntheses, Coll., 2, 24 (1943).
150. Пат. США 2405973, 2406369, 1944.
151. Франц, пат. 954646, 954702, 1947.
152. М. Z i е f, Е. Y а п о v s к у, Ind. Eng. Chem., 41, 1697 (1949).
153. Пат. США 2443741, 1944.
154. Chem. Metallurg. Eng., 57, № 6, 111 (1950); Manufact. Chemist pharmac..
fine chem. Trade J., 21, 343 (1950);
Chem.-Ing.-Techn., 22, 338 (1950).
155. Пат. США 2263654, 1938; 2331869, 1941.
156. Герм. пат.706510, 1938; Р. D. В а г t 1 е t t, R. Altschul, J. Am.
Chem. Soc., 67, 816 (1945);
пат. США 2533425, 1946.
157. Пат. США 2259470, 1939.
14*
212
10. Хлорирование пропилена
158. Пат. США 2485107, 1943.
159. Chem. Eng. News, 25, 2712 (1947).
160. Пат. США 2424520, 1947.
161. S. А. М 1 1 1 е г, Chem. Process Eng., 47, 268 (1966).
162. R. Tanaka, Yuki Gosei Kagaku Kyokai Shi, 20, 482 (1962); C. A.,
58,' 2355a (1963).
163. W. U. S e i 1 e r, Chem. Eng. News, 38, № 22, 124 (1960).
164. J. Kraft, R. T h e r m e t, L. P a r v i, Chim. Ind. (Paris), 83, 557
(1960).
165. J. J. Lukes, Chem. Eng. Progr., 54, № 3, 75 (1958).
166. С. H. Chilton, Chem. Eng., 65, № 9, 116 (1958).
"467. S. A. M i 11 e r, Chem. Process Eng., 48, № 4, 79 (1967).
168. Герм. пат. 72999, 1893.
169. Герм. пат. 846847, 1942.
170. Герм. пат. 901774, 1940.
171. Герм. пат. 679389, 1935; пат. США 2342100, 1940.
172. Герм. пат. 757142, 1939.
173. Пат. ФРГ 806456, 1949.
174. Герм. пат. 725276, 1937.
175. Герм. пат. 734024, 1940.
176. Англ. пат. 749408, 1956; пат. ФРГ 935965, 1956; франц, пат. 1073631, 1954;
англ. пат. 788828, 1958; доп. франц, пат. 69061, 1958.
177. Пат. США 2305821, 1938.
178. Австрал. пат. 166909, 1948.
179. Герм. пат. 298931, 1915; 442342, 1917.
180. Пат. США 2644846, 1948.
181. Пат. США 1947491, 1933.
182. Пат. США 2034292, 1933.
183. Е. Т. М с В е е et al., Ind. Eng. Chem., 33, 176 (1941).
184. W. Hirschkind, Ind. Eng. Chem., 41, 2749 (1949).
185. Пат. США 2442323, 1948.
186. Пат. США 2442324, 1948.
187. U 11 m a n n, Enzyclopadie der technischen Chemie, Bd. 5, Miinchen,
1954, S. 412.
188. Бельг, пат. 449037, 1943.
189. Франц, пат. 1406470, 1965; Niederld. A. S. 6506177, 1965.
190. И. Stegner, Przemysl. Chem., 42, № 6, 306 (1963).
191. Англ. пат. 710244, 1953; пат. США 2806768, 1957.
192. Англ. пат. 772126, 1957; 794408, 1958.
193. D. A. S. 1228245, 1966; пат. США 2746998, 1956.
194. Франц, пат. 1356229, 1964.
195. Англ. пат. 819987, 1959.
196. D. A. S. 1074027, 1960.
197. Франц, пат. 1147756, 1957; 1493131, 1967.
198. Пат. США 2857438, 1958.
199. Е. Т. М с В е е, L.W. Devaney, Ind. Eng. Chem., 41, 803 (1949).
200. Франц, пат. 1373709, 1964.
201. Англ. пат. 960022, 1964.
202. Пат. США 2919296, 1959; франц, пат. 1166211, 1958.
203. Австрийский пат. 164479, 1949.
204. Бельг, пат. 614382, 1962; франц, пат. 1292994, 1962.
205. Oil Gas J., 66, № 14, 80 (1968).
206. Hydrocarbon Processing, 47, № 4, 17 (1968).
11. ДИМЕРИЗАЦИЯ ПРОПИЛЕНА
11.1. ПОЛУЧЕНИЕ ИЗОПРЕНА ИЗ ПРОПИЛЕНА
Изопрен [1—12] был впервые получе-н в 1860 г. в результате сухой
перегонки каучука [13]. Вскоре после того как стало известно, что
изопрен является основным элементом структуры натурального кау-
чука, были предприняты попытки его синтеза. Работа эта задержалась
на некоторое время, так как удалось получить качественный синте-
тический каучук на основе бутадиена, например стирол-бутадиено-
вый и стирол-акрилонитрильный. Однако позже эти типы перестали
удовлетворять возросшим требованиям и для специальных примене-
ний до сих пор требуется натуральный каучук.
Исследования в этой области достигли значительного размаха
благодаря работам Циглера и Натта, которым удалось осуществить
стереоспецифическую полимеризацию диенов. На этой основе перво-
начально был получен в промышленном масштабе полибутадиен.
Указанныеработы открыли также путь к получению более дешевого
полиизопрена, а это, в свою очередь, дало стимул к поискам рента-
бельного синтеза изопрена. Совместные исследования фирм Goodyear
Tire & Rubber Со. и Scientific Design Со., проводимые с 1960 г.,
привели к разработке промышленного метода, известного под назва-
нием «метод Goadyear-SD».
Первая установка для работы по этому методу мощностью
20 000 т/год была построена в 1961 г. в Бомоне (Техас). Сырьем слу-
жил пропилен, поступавший в больших количествах с нефтепере-
рабатывающих заводов. На этой установке вырабатывали полиизо-
прен по цене 25 цент/фунт, т. е. дешевле, чем натуральный каучук.
Исследованы также и некоторые другие возможности синтеза
изопрена. Ниже кратко изложены известные пути синтеза изопрена,
представляющие интерес для использования в промышленности [10 ].
1. Конденсация метилэтилкетона с формальдегидом, гидрирова-
ние полученного 2-метилбутанол-1-она-3 в 2-метилбутиленгликоль-1,3
и дегидратация последнего над фосфорным катализатором с образо-
ванием изопрена [14]:
СН3СОСН2СН3 + СН2О —> СН2ОНСНСОСН3 —*
I
СНз
-->• СН2СНСНСН3 -----+ СН2=С—сн=сн2
I I I ’2нг0 I
ОН I ОН СНз
СНз -
214
11. Димеризация пропилена
2. Конденсация ацетона с ацетиленом в присутствии натрия
с образованием метилбутинола, который гидрируется в метилбутенол.
Последний переходит в изопрен при отщеплении воды [15—17]-
СН3
CHsCH + CH3COCH3 —t СН=С-С-СН3 —+
I
ОН
сн3
I
--> СН2=СН-С-СН3 —СН2=С-СН=СН2
। ° _И аМ “ ।
он сн3
3. Конденсация формальдегида с изобутиленом в диметилдиоксан
и разложение последнего на изопрен, формальдегид и воду (метод
Institute Francaise du Petrole) [18, 19]:
CH3 CH3
I . I
CH2=C-CH3 + 2HCHO >• H2C-C-CH3 >
H2C \
—> CH2=CH-C=CH2 + H2O + HCHO
4. Дегидрирование изопентана через изопентен в изопрен (метод
Shell Chemical Corp.) [20—22]:
СН3СНСН2СН3 —СН2=С-СН=СН2
I -2Нг I
СНз СН3
5. Из пропилена получают изопрен в четыре стадии (рис. 49):
1) димеризация пропилена
2СН2=СН-СН3 —> СН2=С-СН2СН2СН3
I
СН3
2) изомеризация метилпентена
СН2=С—СН2СН2СН3 —> СН3—С=СН—СН2СН3
I I
сн3 сн3
3) термическое расщепление (пиролиз, деметанизация) метил-
пентена
СН3—С=СН-СН2СН3 —> СН2=С—сн=сн2 + сн4
I I
СН3 СНз
4) экстрактивная перегонка.
11.1: Получение изопрена из пропилена
215
Для димеризации пропилена [23] предлагаются в основном две
группы катализаторов:
1) алюминийорганические соединения (катализаторы Циглера —
Натта); '
2) щелочные металлы (или соединения, из которых легко обра-
зуются щелочные металлы) на различных носителях.
Углеводороды > Cg
Рис. 49. Схема получения изопрена:
1, 4, 8, 16 — насос; 2 — реактор; 3,6 — вентиль; 5 — дегазационная
башня; 7, 9, 15, 17—20 — колонны; 10 — сборник; 11 — печь кре-
книга; 12 — система охлаждения; 13 — емкость; 14 — компрессор.
В промышленности наибольшее значение получили соединения
первой группы, особенно трипропилалюминий.
11.1.1. Димеризация пропилена с алюминийорганическими
соединениями
Из исследований Циглера вытекает, что под влиянием A1R3
олефины вступают в реакцию олигомеризации. Например, пропилен
димеризуется в присутствии трипропилалюминия [5а], растворен-
ного в инертном растворителе. Это экзотермическая реакция, проте-
кающая при 150—250 °C (оптимально 200—210 °C) и под давлением
200 кгс/см2, конверсия достигает 60—95%. Главным продуктом диме-
ризации в данном случае является 2-метилпентен-1:
СН3СН=СНг+(С3Н7)3А1 —> СН3СН2СН2СН(СН3)СН2А1(С3Н7)2 —>
—> СН3СН2СН2(СН3)С=СН2 + (С3Н7)2А1Н
(С3Н7)2А1Н + СН3СН=СН2 —> А1(С3Н7)з
216
11. Димеризация пропилена
Исходный пропилен должен быть как можно лучше очищен от
полярных соединений и кислорода, так как окисление одной группы
в алкоксигруппу дезактивирует катализатор димеризации. Поэтому
Рис. 50. Зависимость равновесия смесей различных углеводородов с гексаном
от температуры (в скобках приведены температуры кипения углеводородов
при 760 мм рт. ст.):
1 — 3,3-диметилбутен-1 (41,2 °C); 2 — 4-метилпентен-1 (53,9 °C); 3 — гексен (63,5 °C); 4 —
2,3-диметилбутен-2 (73,2 °C); 5 — 2-этилбутен-1 (64,7 °C); 6 —чис-гексен-З (66,4 °C), транс-
гексен-3 (67,1 °C); 7 — 2,3-диметилбутен-1 (55,7 °C); 8—чис-4-метилпентен-2 (56,3 °C),
транс-4-метилпентен-2 (58,6 °C); 9— З-метилпентен-1 (54,1 °C); 10 — грас-гексен-2 (68,8 °C),
транс-гексен-2 (67,9 °C); 11 — 2-метилпейтен-2 (67,3 °C); 12 — 2-метилпентен-1 (60,7 °C);
13 — чнс-З-метилпентен-2 (70,5 °C), транс-З-метилпентен-2 (67,6 °C).
в ряде публикаций рекомендуется постоянно регенерировать катали-
заторы процесса. Хорошо зарекомендовали себя катализаторы: три-
пропилат алюминия [24], тетрапропилат титана + триэтилалюминий
[25] и трипропилалюминий -f- трипропилат алюминия + ацетилаце-
тонат никеля в присутствии фенилацетилена (для продления срока
службы катализатора). При использовании таких катализаторов
можно рассчитывать на выход 53% 2-метилпентена-1 и 41% 2-метил-
пентена-2 [26].
Были изучены условия реакции и определены следующие опти-
мальные условия (рис. 50—54) [5]:
1) для достижения высокой селективности нужно применять
катализатор при низкой концентрации, время контакта должно быть
коротким;
Давление, кгс/см2
Рис. 51. Влияние давления на димеризацию:
« — селективность; б, в — конверсия (о — течение вверх; х — течение вниз); г, д — состав
алкильных групп катализатора в мол. % насыщенных углеводородов (г — течение вверх,
& — течение вниз);
J — общее содержание; 2 — тримеры; з — 2-метилпентен-1; 4 — изомеры 2-метилпентена-Г,
5 — остаток; в — этан; 7 — пропан.
Время реакции, мин
Рис. 52. Влияние времени пребывания в реакторе на димеризацию:
а, г — селективность; б, б — конверсия; в, е — состав алкильных групп катализатора в мол.
% насыщенных углеводородов;
1 — всего; г — 2-метилпентен-1; з — изомеры 2-метилпентена-1; 4 — тримеры; 5 — 2-метил-
пентан; в — метан; 7 — пропан; 8 — остаток.
218
11. Димеризация, пропилена
2) реакцию с трипропилалюминием следует проводить при 200 °C
под давлением ниже 90 кгс/см2 в двухфазной, а затем в однофазной
системе;
Рис. 53. Влияние на димеризацию отношения раствора катализатора к про-
пилену:
а, г — селективность; б, д — конверсия; в, е — состав алкильных групп катализатора в мол. %
насыщенных углеводородов;
1 — этан; 2 — пропан; 3 — остаток; 4 — 2-метилпентан; 5 — обшее содержание; 6 — 2-ме-
тилпентен; 7 — изомеры 2-метилпентена; 8 — тримеры.
3) в вертикально стоящем реакторе (рис. 55) под давлением ниже
175 кгс/см2 получаются различные результаты, что обусловлено
различным временем контакта; перенос вещества определяет скорость
реакции;
4) в двухфазной системе димеризация происходит в жидкой фазе,
а пропилен находится преимущественно в газовой фазе;
5) при более длительном времени контакта конверсия увеличи-
вается, но селективность падает;
11.1. Получение изопрена из пропилена
219
6) с повышением температуры общая конверсия повышается,
по селективность понижается ^следствие изомеризации и тримери-
зации;
Рис. 54. Влияние температуры на димеризацию:
а— селективность; б — конверсия; в — состав алкильных групп катализатора в мол. %
насыщенных углеводородов:
1 — общее содержание; г — 2-метилпентен; 3 — изомеры 2-метилпентена-1; 4 — тримеры; 5 —
пропан; 6 — метан; 7 — 2-метилпентан; 8 — остаток.
7) при температуре выше 240 °C происходит постепенное разло-
жение алкилалюминия на А1(СН3)3, Al-метиленовые и А1-метиновые
соединения и, наконец, образуется А14С3.
Исследована димеризация в присутствии трипропил- и триизо-
бутилалюминия [27, 28]. Энергия активации реакции составляет
14 ккал/моль (150—230 °C). Эта реакция первого порядка, ее на-
чальная скорость прямо пропорциональна концентрации катализа-
тора. Побочные же продукты образуются по механизму, имеющему
второй порядок по отношению к пропилену.
220
11. Димеризация пропилена
До сих пор обширные исследования проводились в основном
только с алюминийорганическими катализаторами [5а]. В присут-
ствии трипропилалюминия, очевидно, протекают различные реакции
(поскольку оказалось, что во время димеризации алкильные группы
К холодильнику.
Катализатор v
tПропилен
на алюминии заменяются про-
пильными, то в качестве катали-
затора можно взять любой другой
алюминийалкил). Прежде всего
происходит образование такой
цепи:
А1-С-С-С+С=С—С
—► >А1—С—С—С—С—С
Z I
С
Эта реакция может произойти
еще раз, причем образуется цепь:
\ai-c—С-С-С-С—С—С
С с
Наряду с этим возможна дис-
социация образованного на первой
стадии изогексилалюминия:
Далее, А1—Н реагируем непо-
средственно с пропиленом, обра-
Рис. 55. Реактор для димеризации:
1 — термоэлементы; 2 — реакционный сосуд,
заполненный стеариновым бисером (емкостью
115 мл); 3 — трубка для термоэлемента.
11.1. Получение изопрена цз пропилена
22 f
Комбинация двух последних реакций дает:
С
С
Эта брутто-реакция воспроизводит реакцию вытеснения. Оказа-
лось, что вытеснение происходит гораздо быстрее, чем дальнейшее’
образование цепи из изогексилалюминия в изононилалюминий.
Становится ясно, почему во время олигомеризации избирательно
образуется 2-метилпентен-1.
Рассмотрев распределение относительных зарядов в металлорга-
нической молекуле и в молекуле пропилена, можно понять, как.
образуется цепь:
\ai-h —> \ai-c-c-c
8- 8+
с=с
^>А1-С-С-С > \a1-C-C-C-C-C
с
8- 8+
с=с
I
с
В результате некоторых отклонений получаются два вида изо-
меров:
1) изомеры со структурной формулой, отличающейся от структур-
ной формулы 2-метилпентена-1; они образуются во время реакции
образования цепи вследствие аномального присоединения пропилена
к ^>А1—Н или А1—С—С—С (побочные реакции);
2) изомеры с такой же структурной формулой, как у 2-метил-
пентена-1; они образуются благодаря тому, что ^>А1—Н обладает
еще некоторым химическим сродством к 2-метилпентену-1, причем:
присоединение может происходить следующим образом:
С
С
При диссоциации этого гексилалюминия может получаться
2-метилпентен-2. Следовательно, алкилы алюминия могут вызвать-
222
11. Димеризация пропилена
Таблица 20
Металлорганические катализаторы димеризации пропилена
Катализатор Темпера- тура, °C Давле- ние, кгс/см2 Литера- тура
Триэтилалюминий в декалине 300 250 [29, 30]
Триэтил- или трибутилалюминий 225—250 120-200 [31]
Трипропилалюминий 180 — [32]
Трипропил алюминий Триизобутилалюминий (при 180 °C конверсия — — [33]
70—75%, 80,7% 2-метилпентена-1) 180—250 — [34]
Тригексилалюминий 180 120 [35]
Триалкилалюминий Триалкилалюминий в присутствии соединений никеля и платины (например, триизобутил- — — [36]
алюминий и ацетилацетонат никеля) .... 250 130 [37]
Диэтилалюминийхлорид Различные алкилорганометаллические соедине- ния: триэтилалюминий, диэтилбериллий, ди- фенилбериллий, А1Нз, диметилалюминийгид- рид, трифенилалюминий, тригексилалюминий, 290 60-95 [38]
LiAlH$, LiAl-тетраэтил, NaAl-тетрафенил . . Соединения MeRn (где Ме=Ве, Al, Са, In; R = H, 200 350 [39]
алкил, арил) Алкилы А1=Ве, Ga, In (например, трипропил- — — [40]
алюминий) Алкиллцтий, ариллитий и свободный щелочной 180 120 [41]
металл — — [42]
-А1Н3 — — [43]
Алкилхлорид и натрий Триэтилалюминийтрихлорид+ацетилацетонат ни- келя трифенилфосфор, бис-я-аллилникель, 150 — [44]
л-аллилникельхлорид Три этил алюминийтрихлорид + диизопропил сали- 60 10 [45]
цилат никеля Соединения АМеН3 [где А = щелочные металлы, Me = Zn, Sn, Cd, Pb;R = алкил; например, от —20 до +20 [46]
KZn (С2Н5)3[ ... Смесь щелочного металла и MeR2 (где Me = Zn, Cd, Hg; R = алкил, арил) (К, Rb, Cs) (Cd, 160 110 [47]
Zn, Sn, Cd) R3 100-250 — [48]
смещение двойных связей. Селективность (в %) в отношении различ-
ных олефинов следующая [5а]:
2-Метилпентен-1 . . 88
4-Метилпентен-1 . . 2
4-Метилпентен-2... 1,2
н-Гексен...........5,2
2-Метилпентен-2... 3,6
11.1. Получение изопрену из пропилена
223:
В табл. 20 приводится краткий перечень других металлорганиче-
ских, в частности алюминийорганических, соединений, которые
рекомендуются как катализаторы димеризации пропилена.
11.1.2. Димеризация пропилена в присутствии катализаторов
на основе щелочных металлов
В 1962 г. благодаря исследованиям British Petroleum Со., Ltd.
стало известно, что соединения щелочной металл — графит, имеющие-
пластинчатую структуру, являются отличными катализаторами
димеризации пропилена [49—52]. Различные типы этих соединений
ведут себя по-разному, но самый пригодный из них NaC64. Ниже
показаны некоторые из этих катализаторов и основные продукты,,
получаемые при реакции димеризации:
XClttllUUCl" — Ц
тура, С Главный продукт
NaC40.............. 150 4-Метилпентен-2
NaC64 ....... <140 4-Метилпентен-1
NaCea.......... 140—160 4-Метилпентен-2
NaCe4 ................... >160 2-Метилпентен-2
КС24, зв, 4в, во .... 150—160 2-Метилпентен-2
КС8,16, 24 ............ 100—160 4-Метилпентен-1
В частности, получаются гексеновые смеси следующего состава-
(в %):
кс„ кс„ NaC„ NaC,
4-Метилпептен-1 . . . 85,8 1,6 51,3 21
4-Метилпентен-2 . . . 6,3 16,3 43,9 56
2-Метилпентен-2 . . . 0,6 62,7 3,8 18
2-Метилпептен-1 . . . 3,0 12,0 — 3
и-Гексен 3,8 8,4 — 2
Примечание: Условия реакции при использовании КС,, и КС,,:
160° С, 80-110 кгс/см2; NaC,4: 126° С, 110 кгс/см2; NaCx: 160° С,
130 кгс/см2.
Реакция длится несколько минут. При температурах выше 200 °C
в присутствии NaC84 получаются преимущественно тримеры и тетра-
меры, димеров образуется меньше.
Сами щелочные металлы так же, как их гидриды, обладают высо-
кой активностью и поэтому часто предлагаются как катализаторы,,
причем на самых различных носителях. В табл. 21 дается обзор
таких катализаторов; особенно перспективной кажется комбинация
натрия с карбонатом калия в качестве носителя.
Димеризация в присутствии К, ВЬ или Cs ведет в основном к об-
разованию 4-метилпентена-1, который медленно изомеризуется в 4-
метилпентен-2. Димеризация же в присутствии Na приводит
224
-11. Димеризация пропилена
Таблица 21
Катализаторы димеризации пропилена
Катализатор Носитель Темпе- ратура, °C Давле- ние, кгс/см2 Примечание Литера- тура
К, Rb или Cs Дисперсия в гептане. 300 — Главный продукт 4-метилпентен-1 [53]
К, Rb или Cs Дисперсия в углеводоро- дах, простых эфирах, ами- нах, NH3 и др- 200 5-300 Конверсия 60— 84%; —60% 4- метилпентена-1 [54, 55]
К Дисперсия в парафине 150 41 — [56]
К Активаторы — длинноцеп- ные алифа- тические спирты, кис- лоты, амины или меркап- таны [57]
К Дисперсия в бензоле 150 — — [58]
к + кон Дисперсия в парафиновом масле 170—173 60 Главный продукт 4-метилпен- тен-Г [59]
КН, РЬН или — 120—125 6-500 То же [60]
CsH
КН Дисперсия в минеральном масле 150-198 — Конверсия 59,5% [61]
_К + металл- органическое соединение Дисперсия в дизельном ' топливе 180 180—200 Главный продукт 4-метилпентен-1 [62]
(например,
бутиллитий).
К, Rb или Cs и металлор- Хлористый на- трий 180 — Выход 28%, селек- тивность 81% [63]
ганическое соединение MeR2 (напри- мер, диэтил- ртуть)
и алифатиче- ский амин Активирован- ный уголь 150 Конверсия 43,6%; 43% 4-метилпен- тена-1, 47,9% 4-метилпентена- [64]
Реакционный продукт из К и К2Ог Нет 190 30 2 79% 4-метилпен- тена-1 [65]
11.1. Получение изопрена из пропилена
225
Продолжение табл. 21
Катализатор Носитель Темпе- ратура , °C Давле- ние, кгс/см’ Примечание Литера- тура
Реакционный продукт из К и простого эфира (напри- мер, анизола) Нет 150 •— Конверсия 70%; 78% 4-метилпен- тена-1, 16% 4- метилпентена-2 [66]
Na или NaOH Активирован- ный уголь 140—145 t 42 Селективность >90%; 85% ме- тилпентенов [67]
Щелочной ме- талл Фторид щелоч- ного металла 150-200 20-110 Главный продукт 4-метилпентен-1 [68]
к СаН2, NaBH4, LiAlH4 и др. 150 37 37,6% 4-метилпен- тена-2, 52% 4- метилпентена-1, 7,2% 2-метил- пентена-2 [69]
Na или Li К2СО3 160 120 — [70, 71]
Na К2СО3 160 140 Главный продукт 4-метилпентен-1 [72]
Na К2СО3 170 НО Исходные продук- ты этилен и бу- тены [73]
Na К2СО3 160 НО Димеризация про- водится в при- сутствии других парафинов [74]
Na К2СО3 160 119 Главный продукт 4-метилпентен-1 [75]
Na или Li К2СО3 160 НО — [76]
Na к2со3 155 105 Na растворяют в NH3, добавляют К2СО3, и смесь выпаривают; 70% 4-метилпен- тена-1, 18% 4- метилпентена-2 и др. [77]
NaH К2СО3 140 105 Конверсия 99%; 76,5% 3- и 4-ме- тилпентена-1, 10,8% 4-метил- пентена-2, 8,2% «-гексена и др. [78]
Na К2СО3 153 103 Удаление «-алке- нов путем поли- меризации А1С13 [79]
K, Rb или Cs К2СО3 на MgO 175 70 Конверсия 74%; 35% 4-метилпен- тена-1, 44% 4- метилпентена-2 и др. [80]
15 Заказ 399
226 11. Димеризация пропилена Продолжение таб
Катализатор Носитель Темпера- тура, °C Давле- ние, кгс/см2 Примечание Литера- тура
к к К, КН или ка- лийорганиче- ское соедине- ние LiOH + KNH2 LiOH К или knh2 LiOH + К в жидком NH3 LiOH + KNH2 Органическое соединение Na, К, Rb, или Cs А12О3 Mg2SiO4 А120д Y-A12O3 y-ai2o3 A12O3 y-ai2o3 Нет 150 180 65-205 50-300 151 200-206 180-205 85 70 10-100 5-200 70 55-60 30 85% 4-метилпенте- на-1 Конверсия 80%; селективность 90%; 47% 4-ме- тилпентена-1, 40% 4-метилпен- тена-2, 7% 2-ме- ' тилпентена-2, 5% н-гексена и ДР- Главный продукт 2-метилпентен-2 (максимум 68,8%) Конверсия 48%; селективность 96% Конверсия 33%; селективность 75% Конверсия 31 % С фенилнатрием 65% 2-метилпен- тена-2; с амил- калием 75,6% 4- метилпентена-1 [81] [82] ; [83] [84] [85] ; [86] [87] [88]
к установлению термодинамического равновесия метилпентенов [89].
Механизм реакции, ведущей к образованию в основном 4-метилпен-
тена-1, объясняется следующим образом:
СН2=СН-СН3 —> [СН2—СН—СН2]“Ме+
+СН2=СН-СН8 '
--> СН2=СНСН2СНСН2Ме+
СН3
---> СН2=СНСН2СНСН3 + [СН2—СН—-СН2]~Ме+
СН3
11.1.3. Димеризация пропилена на различных катализаторах
Для димеризации пропилена можно применить также каолин
[90] или двуокись кремния — окись алюминия [91—92] типа цео-
лита. Для получения максимальных количеств димеров необходимо'
11.1. Получение изопрена из пропилена 227
работать при высоких температурах (>• 288 °C) и нейтрализовывать
катализатор, промывая его содовым раствором. При температуре
454 °C и давлении 3,5 кгс/см2 достигается низкая конверсия (3,5%)
и высокая селективность (95%). Добавлением воды или низших спир-
тов удается повысить конверсию без значительного уменьшения
селективности [93]. Рекомендуется также добавлять окись, никеля
к катализатору А12О3—SiO2 (316 °C, 13 кгс/см2) [94] или жирные
кислоты С4 —С4 к катализатору М&О—SiO2 (310—450 °C) [95].
WO3 или восстановленные окиси вольфрама> (также в присутствии
NiO) тоже годятся для димеризации пропилена, при этом в основном
образуется 2-метилпентен-2 (277—282 °C, 43,2 кгр/см2) [95а].
При комнатной температуре в присутствии 90—92%-ной H2SO4
возможна сравнительно селективная димеризация главным образом
в 4-метилпентен-1 [96]; также действует BF3-2H2O [97], причем
в этом случае пропилен димеризуется уже при 4—12 °C с конверсией
81,3% и селективностью 93,1%. Довольно избирательную димериза-
цию пропилена можно осуществить с Н3РО4 на медном катализаторе,
активированном Н3РО4 [98]. При повышенных температурах (270—
305 °C, 270—410 кгс/см2) [99] в качестве катализатора для преимуще-
ственной димеризации пропилена можно применять фосфорную кис-
лоту на системе SiO2—МоО3 в присутствии водорода, в этом случае
реакция возможна в более мягких условиях (290—315 °C, 14 кгс/см2).
В качестве катализаторов рекомендуются галогениды платиновой
группы [100], причем галогениды Ru и Rh действуют уже при темпе-
ратуре ниже 100 °C, соли Pt, Pd, Os и Ir требуют температур от 130
до 225 °C.
Наконец, предлагается нагревать олефины до 325— 350 °C в при-
сутствии серы или серу- или галогенсодержащего органического
соединения (например, l'-бромгексана), но при этом образуются
в значительном количестве три- и тетрамеры [101].
Недавно для димеризации пропилена была предложена окись
кобальта на активированном угле [102, 103].
Ц.1.4. Изомеризация метилпентена
Вторая стадия метода получения изопрена из пропилена состоит
в изомеризации 2-метилпентена-1 в 2-метилпентен-2 [5в]:
СН2=С— СН-СН3 —► СН3-С=СН-СН3
I I
СНз СН3
Изомеризацию проводят с применением кислотных катализаторов.
Требуемые температуры лежат в области от 150 до 300 °C, скорость
подачи жидкого углеводорода от 0,5 до 15,0 л/(ч-л катализатора).
Применяя катализаторы с оптимальной эффективностью, можно
избежать нежелательных побочных реакций, например полимериза-
ции. При высокой конверсии достигаются выходы до 99%.
15*
228
11. Димеризация пропилена
Катализаторы изомеризации можно повторно активировать окис-
лительной регенерацией.
Опираясь на вычисленные отношения термодинамического равно-
весия для различных гексеновых изомеров в области от 300 до 1000 К
(рис. 50), Баас и сотрудники показали, что для достижения макси-
мальной конверсии 2-метилпентена-1 в 2-метилпентен-2 в каждый
проход следует поддерживать как можно более низкую температуру.
Исследования Эммета [105] подтвердили, что подобную изомериза-
цию легко осуществить в мягких условиях со слабокислыми катали-
заторами [10] и что сдвиг двойных связей при этом проходит очень
селективно. Эти результаты подтверждаются и другими авторами.
Описан метод, по которому можно изомеризовать 2-метилпентен-1
при комнатной температуре с 50% раствором серной кислоты, полу-
чив при этом равновесную смесь 2-метилпентена-1 и 2-метилпентена-2
[107]. ,
Казанский и сотрудники [110] испытывали в качестве катализа-
тора сдвига двойной связи А12О3, активированную пропусканием
воздуха при 450 °C. При 80 °C 2-метилпентен-1 быстро дает 62%
2-метиленпентена-2. Гексен-1, напротив, при 80 °C никак не реагирует.
По Олдхему [111] и Фречу [106] можно при 80—260° С селективно
превратить 2-метилпентен-1 в 2-метилпентен-2, используя молекуляр-
ные сита 5 А. В табл. 22 показано, что для изомеризации можно
применять как жидкие катализаторы, так и катализаторы на носи-
теле.
Некоторые катализаторы изомеризации
Таблица 22
Катализатор Температу- ра, °C Характеристика процесса Литература
50%-ная H2SO4 20-25 Продукты реакции: 2-ме- тилпентен-1 и 2-метил- пентен-2 [Ю7]
60—70%-ная H2SO4 ... 0—15 — [103]
Н3РО4 на пемзе .... 130 — [35]
А120з 200 — [109]
Силикагель 20—25 Большая продолжитель- ность реакции [109]
SiO2—А12О3 — — [37]
А12О3 активированнаяо 80 — [110]
Молекулярные сига 5 А 80-260 — [112]
25% А12О3, 75% Si02o 170 Конверсия >80% [5в]
Молекулярные сита 4 А 200 Конверсия 70% [5в]
A12(SO4)3.H2O 150 Конверсия >70% [5в]
Трехокись вольфрама . . 50-400 Изомеризация 2- и 4-ме- тилпентена-1 [112]
11.1. Получение изопрена из пропилена
229
При тщательном проведении опытов на лабораторной аппаратуре
(рис. 56) Баасу и сотрудникам удалось полностью подтвердить ре-
зультаты своих предшественников. Ниже дается краткое описание
этой маленькой лабораторной уста-
новки.
2-Метилпентен-1 нагревается до
кипения в колбе 1-емкостью 100 мл;
электрическая нагрузка обогрева-
тельной рубашки определяет ско-
рость прохождения гексеновой смеси
через реактор. Трубчатая печь 3 на-
гревает перекисную трубку (диаметр
2 см), состоящую из двух частей:
подогревателя 2 (длина 10—15 см)
и реакционной части 4 (длина 15—
20 см). Температура измеряется тер-
мометром 5. Пары гексенов охла-
ждаются в холодильниках 6 и 7,
а конденсат стекает через устрой-
ство для взятия образцов жидкости
8, капельницу 9 и холодильник 10
к распределителю барботирующего
пара с мерным цилиндром и круглой
колбой 13 емкостью 100 мл. Гексены
можно улавливать в этой колбе
или в калиброванном мерном ци-
линдре, с помощью которого можно
измерять «пространственную ско-
рость» по отношению к холодной
жидкой гексеновой смеси и к запол-
ненному катализатором объему ре-
актора. Наконец, в круглой колбе
можно улавливать весь продукт.
Оставшиеся в реакторе гексены мож-
но удалить слабой струей азота,
колбе 13.
Рис. 56. Лабораторная установка
для изомеризации:
1, 13 — круглые колбы; г — подогре-
ватель; з — трубчатая печь; 4 — реак-
тор; 5 — термометр; в — сосуд Дьюа-
ра, охлаждаемый смесью СО2 — спирт;
7 — шариковый холодильник; S — ус-
тройство для взятия образцов жидко-
сти; 9 — капельница; 10 — холодиль-
ник; 11 — отвод для удаления воздуха
через пробку, заполненную ватой; 12—
соединитель с мерным цилиндром и
круглой колбой.
улавливая их в
^руглой
Кроме того, на этой установке можно проводить опыты по рецир-
куляции. Для этого надо оборудовать паукообразный распределитель
коленчатым отводом и вентиляционным устройством. Тогда холо-
дильник 10 можно будет поворачивать на 180° вокруг оси холодиль-
ников 6 и 7 и присоединять к перекисной трубке реактора. Теперь
маленькая колба 13 укрепляется в том месте, где до того был коленча-
тый отвод. Таким путем можно заставить гексены рециркулировать;
они возвращаются в колбу. 13 через холодильник 10. С помощью
капельницы 9 можно получить представление о скорости рециркуля-
ции. Шприцем для инъекций можно брать из отверстия 8, закрытого
230 11. Димеризация пропилена
резиновой пробкой, небольшие пробы жидкости и анализировать их
на хроматографе.
По окончании опыта продукты снова собираются в колбе 1. Из
этой колбы фракция с помощью микродистилляционной аппаратуры
отгоняется при кипении до 75 °C, а затем анализируется на газохро-
матографе (колонна с диметилсульфоланом). На основании данных
анализа вычисляют состав изомерной смеси. В колбе 1 после отгонки
остаются полимеры и, в зависимости от количества образовавшихся
полимеров, гексены, представляющие собой остаток в дистилляцион-
ной аппаратуре. Определяют показатель преломления (ид) этих
продуктов. Установлена графическая зависимость между показате-
лем преломления и соотношением полимера и изомера в синтетиче-
ских остаточных смесях. Пользуясь "графиком, можно определять
содержание полимеров в остатке. Зная состав изомера и количество
образовавшихся полимеров, можно вычислить конверсию — отноше-
ние образовавшегося продукта к введенным веществам.
Для опытов применяли 2-метилпентен-1 со степенью чистоты не
менее 90%, содержащий в качестве примесей гексены (иногда неболь-
шое количество 2-метилпентена-2). При расчетах это учитывалось.
Ниже перечислены исследованные катализаторы:
Содержание
’ А1,О,.
вес, %
Кизельгур — окись алюминия (405 м2/г) 13,3
Кизельгур — окись алюминия.............. 25
Кизельгур (163 м2/г; 0,23% С1),........; 100
Алюмосиликат кальция (4А и 5А) . . . . ' —
А12(8О4)з • Н2О (нагрет в течение 24 ч до
180° С) ............................. 29,8
Катализатор, состоящий из 25% А12О3 и SiO2 был приготовлен
по данным Циапетта и Плакка. Все катализаторы на основе системы
кизельгур — окись алюминия перед употреблением нагревали при-
мерно в течение 8 ч До 500—550 °C при пропускании воздуха. Моле-
кулярные сита перед употреблением также нагревали при пропуска-,
нии воздуха в течение 5 ч до 400—450 °C. Катализатор A12(SO4)3
приготавливали путем медленного (24 ч) нагревания до 180 °C
A12(SO4)3-18H2O; при 180 °C стабильный гидрат содержит одну
молекулу кристаллизационной воды. Из всех катализаторов были
отпрессованы пилюли (размеры круглой пилюли 3 X 2,5 мм); раз-
меры молекулярных сит: 1 -(-2 X 2-4-5 мм («пробитые градом»).
Результаты опытов отражены на рис. 57—59, из которых видно,
что изомеризацию двойной связи вполне возможно осуществить так,
чтобы не произошла ни полимеризация, ни другая изомеризация.
На такую очень селективную изомеризацию влияет главным образом
место разветвления в углеродных цепях по отношению к двойной
связи. Этим объясняется и тот факт, что для реакции при мягких
г
11.1. Получение изопрена из пропилена
231
условиях достаточно катализаторов со слабокислыми свойствами.
Катализаторы крекинга, обладающие сильнокислыми свойствами,
Рис. 57. Влияние объемной скорости на
изомеризацию 2-метилпентена-1 в 2-ме-
тилпентен-2:
о
о — у-А1203; х — молекулярное сито 5 А.
способствуют' протеканию реак-
ции полимеризации 2-метилпен-
тена-1. Исследование дистил-
лята показало, что эта полиме-
Время рециркуляции, мин
Рис. 58. Влияние продолжительно-
сти рециркуляции на изомеризацию
2-метилпентена-1 в 2-метилпентен-2
с помощью катализатора крекинга:
1 — полимеризация; 2 — изомеризация.
ризация ограничивается почти исключительно димеризацией гек-
сена. Реакции изомеризации, не представляющие собой целена-
правленного сдвига двойной связи, приобретают важное значение
только при температурах
выше 200 °C.
После длительного упо-
требления эффективность
катализатора уменьшает-
ся. Это может быть вы-
звано тем, что поверхность
катализатора частично по-
крывается полимером. Ка-
тализатор можно восста-
Рис. 59. Влияние продолжительности исполь-
зования катализатора на изомеризацию 2-ме-
тилпентена-1 в 2-метилпентан-2:
о
1 — молекулярное сито 5 А, 180 °C, объемная ско-
рость 5,1; 2 — у-А120з, 180 °C, объемная скорость
6,3; 3 — 25% А12О3 на SiO2, 150 °C, объемная
скорость 6,2.
новить путем тщательного
выжигания полимера.
Работы Бааса и сотруд-
ников свидетельствуют
о том, что 2-метилпен-
тен-1 можно очень легко
изомеризовать в 2-метил-
пеитен-2, применяя для этого слабокислые катализаторы-
Реакцию удается осуществить в мягких условиях, достигая
высоких значений конверсии при исключительно высокой селек-
тивности.
232
11. Димеризация пропилена
11.1.5. Пиролиз метилпентена
Третьей стадией современного синтеза изопрена является пиро-
лиз 2-метилпентена-2 в изопрен и метан. Для этого' требуются высо-
кие температуры и короткое время контакта. Благодаря резонансу
изопрен сравнительно стабилен. При удачном подборе условий ре-
акции можно избежать образования ароматических веществ и неже-
лательных промежуточных продуктов типа ацетилена.
Рис. 60. Схема лабораторной установки для пиролиза:
1 — сборник с дистиллированной водой; 2 — бюретки для реактивов с воронками для запол-
нения; 3 — фильтры; 4 — расходомеры жидкости; 5 — подогреватель; (> — подогревательная
труба из нержавеющей стали, заполненная стружкой из нержавеющей стали; 7 — смеситель;
8 — реактор; 9 — тигельная печь; 10 — холодильник Либиха (максимальная температура
70 °C); 11 — медная трубка, обмотанная нагревательной проволокой; 12 — газопровод, обмо-
танный нагревательной лентой; 13 — водоотделитель (температура 40 °C); 14 — сушильная
башня с ВаО (температура 40 °C); 15 — водосборник; 1в — буферная емкость; 17 — ртутный
затвор; 18 — баллон для проб газа; 19 — восьмиходовой кран с трубкой для проб газа в тер-
мостате при 40 °C; 20 — колонка для газо-жидкостной хроматографии; 21 — катарометр
в термостате при 40 °C; 22 — впрыск жидкости; 23 — сигнал катарометра на измерительный
щит и регистрирующий прибор; 24 — кран прецезионной регулировки; 25 — осушитель; 2 в —
открытый жидкостной манометр; 27 — счетчик пузырей; 28 — подогреватель для нагревания
азота-разбавителя. (В подогревателе, смесителе и в реакторе имеются термоэлементы платина/
платина—родий).
Пиролиз 2-метилпентена-2 в изопрен проводится в крекинг-печи.
Для того чтобы добиться превращения олефинов с хорошими выхо-
дами и с минимумом побочных реакций, в качестве катализатора
применяют бромистый водород, а в качестве разбавителя — пар.
Пиролиз 2-метилпентена-2 проводится при температурах 650—800 °C
и времени контакта от 0,05 до 0,3 с. Изопрен, метан, другие газы
и непрореагировавший 2-метилпентен-2 разделяются ректификацией.
2-Метилпентен-2 снова возвращается в пиролизную печь.
Наиболее эффективными оказались кислые катализаторы. Среди
прочих рекомендуются у-А12О3 и НВг. Применяемый бромистый
водород выделяют на резко охлаждаемых системах (квенч-системах)
и тоже возвращают в пиролизную печь.
11-1- Получение изопрена из пропилена
233
Баас с сотрудниками [5с] тщательно исследовал эту реакцию
и в полупромышленном масштабе. На рис. 60 схематически изобра-
жена аппаратура, в которой при пиролизе 2-метилпентена-1 (по-
лучен димеризацией пропилена под действием A1R3; степень чистоты
98,9%), 2-метилпентена-2 (получен изомеризацией 2-метилпенте-
на-1; степень чистоты ^>98,5%) и З-метилпентена-2 (получен дегид-
рированием З-метилпентанола-З) они получили различные количег
ства изопрена (табл. 23).
$
Результаты пиролиза гексенов
Таблица 23
Параметры процесса Номера опытов
I II III IV V
Пиролиз 2-м етилпентен а-1*
Температура, °C 750 800 800 800 800
Время контакта, с 0,02 0,01 0,01 0,02 0,02
Конверсия, мол. % 19,6 17,7 24,1 53,3 52,9
Выход изопрена, % *. 18,4 18,1 16,9 14,6 15,7
Пиролиз 2-м етилпентен а-2
Разбавление — —
Температура, РС 650 700
Время контакта, с . . . . 0,10 0,01
Конверсия, мол. % ... 11,4 12,8
Выход изопрена, % ... 60,3 60,2
N2; N2; — — — Н2О; н2о
1 : 3 1 : 3 1:3 1: 3
750 750 760 760 800 700 800
0,005 0,02 0,02 0,15 0,04 0,06 0,005
4,8 38,8 45,8 93,4 74,3 12,1 22,2
61,2 57,3 52,3 34,7 47,8 62,5 60,0
Пиролиз 3-метилпентен а-2*
Температура, °C............................
Время контакта, с..........................
Конверсия, мол. % ................ . .
Выход изопрена, % .........................
750 800 800 800 800
0,02 0,005 0,01 0,02 0,015
20,6 6,0 16,6 48,7 39,0
71,0 81,6 74,9 60,1 65,2
* Пиролиз 2-метйлпентена-1 и з-метилпентена-2 проводили без разбавления.
Данные табл. 23 показывают, что до пиролиза целесообразно
сначала изомеризовать 2-метилпентен-1 в 2-метилпентен-2. Еще
лучше было бы использовать в качестве исходного материала 3-метил-
пентен-2, но, к сожалению, его трудно получить.
Коэффициент полезного действия этого метода производства изо-
прена определяется в первую очередь последней стадией, т. е. де-
метанизацией. Перед последней стадией выход изопрена достигает
около 65 мол. %. Опыты показывают, что расщепление происходит
точно или примерно по реакции первого порядка, когда имеет место
разрыв одной или нескольких С— С-связей. Однако энергии
234
11. Димеризация пропилена
активации, относящиеся к этим процессам первого порядка, значи-
тельно меньше, чем энергии диссоциации С—С-связей.
Такие экспериментально найденные закономерности теоретиче-
ски оказываются вполне понятными, если исходить из предположе-
ния, что реакции разложения проходят через цепные радикальные
реакции [113]. Реакция пиролиза состоит из трех частей: иницииро-
вания, роста цепи (цепная реакция) и обрыва цепи. При реакции
инициирования образуются радикалы, которые, отщепляя водород,
вызывают множество следующих друг за другом цепных реакций.
Эти цепные реакции состоят из двух частей: отщепления водо-
рода и стабилизации радикалов, образовавшихся после отщепления
водорода. В частности, реакции отщепления водорода влияют на
окончательный состав продукта пиролиза. В результате рекомбина-
ции радикалов происходит обрыв цепи и цепные реакции прекраща-
ются. Все это можно проиллюстрировать следующей схемой:
инициирование
М —> 2R
рост цепи
R + M —>• RH + R' (отщепление водорода)
R' —> М'4-R (стабилизация радикала)
R + M —> RH + R'
R' -->• M'-J-Rht. д.
Обрыв цепи
R + R' —> М'
Очень часто возникновение начальных реакций ложно объясняют
столкновением со стенкой реактора. Возможно, что инициирование
происходит бимолекулярно, как это предполагают в случае пиро-
лиза этана [114]:
М + Х—> 2R + X
причем X может быть равен М.
Непосредственно за каждой начальной реакцией быстро следуют
цепные реакции, которые благодаря образованию новых радикалов
сами обеспечивают дальнейший рост цепи. Соотношение между ро-
стом цепи и инициированием (средняя длина цепи) очень велико.
В схеме окончание выбрано произвольно. Эта реакция может про-
ходить также на стейке реактора или тримолекулярно:
R + R'+ (молекула газа) —► М
По данной схеме можно вычислить в стационарном состоянии
что рассматриваемый пиролиз протекает как реакция первого по-
рядка. Однако это зависит от выбранной схемы пиролиза.
11.1. Получение изопрена из пропилена
235
11.1.6. Свойства и применение изопрена
Ниже показаны свойства изопрена;
-%
Температура плавления, -С.........................•
Температура кипения, °C
при 760 мм рт. ст..................................
1 мм рт. ст...............................
10 мм рт. ст..............................
Плотность
PF..............................................
P’F ......................................
pF..............................................
Показатель проломления
Дипольный момент, D
в гексане ......................................
—145,95
34,076
-79,8
-51,6
0,6805
0,7732
0,6754
1,42160
1,41852
0,15
газ.................................... 0,38
Поверхностное натяжение при 20° С, дин/см. 16,9
Теплота плавления при 15° С, кал/г . . ... 16,8
Теплота испарения при 34° С, ккал/моль..... 6,7
Теплота сгорания при 25° С, ккал/моль
газ..........' .................. . 760,83
жидкость......................................... 754,28
Мольная теплоемкость, кал/(9С • моль)
при —23,169 С (газ)........................... 19,57
25,3° С (твердый и жидкость)............... 36,63
1227 ?С (газ) ............................. 60,30
Диэлектрическая проницаемость
при —75° С.................................... 2,343
25° С...................................... 2,098
Несмотря на то, что после второй мировой войны во всех про-
мышленных странах были значительно увеличены мощности по
производству синтетического каучука и мировое производство син-
тетического каучука начало достигать уровня производства натураль-
ного каучука, поиски нового синтетического каучука продолжались.
Это произошло потому, что имеющиеся синтетические каучуки по
своим свойствам не вполне соответствовали натуральному каучуку.
В этой ситуации открытие цис-i ,4-полиизопрена — стереоспеци-
фического каучука, практически не отличимого от натурального
продукта, — явилось важным шагом вперед в развитий технологии
каучуков. Этот новый каучук был синтезирован при использовании
металлорганических соединений Циглера (органических соединений
лития). Разработка дешевой технологии производства обеспечила
полный успех новому продукту.
236
11. Димеризация пропилена
11.2. П0ЛИ-4-МЕТИЛПЕНТЕН-1
В результате димеризации пропилена на специальных цинковых
катализаторах (160 °C, 100—150 кгс/см2) образуется преимущест-
венно 4-метилпентен-1 [47, 115, 116]:
СН2=СН-СН2-СН-СН3
I
СНз
Выход,
вес. %
Выход,
вес, %
4-Метилпентен-1
4-Метилпентен-2
57 2-Метилпентен-1 ... 1
36 2-Метилпентен-2 ... 3
Фирма British Petroleum Со. разработала метод получения моно-
меров. Путем полимеризации фирма ICI получила из 4-метилпенте-
на-1 новый пластик [117, 1181:
'-СН2—СН-
СН2СН(СН3)2 J л
Этот пластик производится в больших количествах и поступает
в продажу под названием ТРХ. Плотность его 0,83 г/см3, ниже чем
у всех известных термопластов, температура плавления ~240 °C.
Изготовленные из этого материала прессованные детали сохраняют
стабильность формы при температуре до 200 °C. Кроме того, пластик
ТРХ прозрачен. Светопроницаемость достигает 90%, т. е. несколько
меньше, чем у плексигласа (у полиметилметакрилата 92%). Недо-
статком является деструкция под действием света. Поэтому нестаби-
лизированный ТРХ пригоден только для применения в закрытых
помещениях. Этот материал стоек ко многим химическим средам,
сильные кислоты и щелочи не разрушают его, однако он растворяется
в некоторых органических растворителях, например в бензоле, че-
тыреххлористом углероде и петролейном эфире. Ударная прочность
нового термопласта такая же, как у высокоударопрочного полисти-
рола. Диэлектрические свойства тоже хорошие (диэлектрическая
проницаемость 2,12).
Несмотря на высокую температуру плавления, ТРХ можно
перерабатывать обычными методами экструдирования, литья под
давлением и выдувания. Полимер тиксотропен и обладает хорошей
текучестью. Тиксотропия объясняется внутренним пластифициру-
ющим действием изобутильных боковых цепей.
Указанные свойства обеспечивают многообразные возможности
применения нового продукта. Так, стабильность формы при высоких
температурах позволяет изготовлять из ТРХ стерилизующиеся хи-
рургические инструменты. Благодаря этому же свойству, а также
высокой прозрачности пленки ТРХ пригодны в качестве упаковоч-
ного материала для пищевых полуфабрикатов.
11.з. Содимеризация пропилена
237
11.3. СОДИМЕРИЗАЦИЯ ПРОПИЛЕНА
11.3.1. Содимеризация с этиленом
Содимеризация пропилена с этиленом при образовании изопен-
тенов осуществляется взаимодействием триэтилалюминия (как ис-
точника этилена) с пропиленом в алифатических или ароматических
углеводородах, служащих растворителями [120]. Основной продукт
реакции — 2-метилбутен-1. Реакция проводится преимущественно
при 100—180 °C и под давлением 13—65 кгс/см2, продолжительность
реакции от 30 мин до 6 ч, соотношение триэтилалюминий : пропи-
лен = 1:3 4-8. Наряду с основным продуктом образуются бутены
и гексены; 2-метилбутен-1 отделяется от них фракционированием.
Можно также осуществить содимеризацию путем пропускания
смеси этилена с пропиленом при ~140 °C над катализатором на
основе КН и проциленом в гептане (91 % пентена-1) [121] или над
триэтилалюминием (при 220—300 °C, 80—150 кгс/см2) [122]. Ката-
лизатором может служить система алкилалюминий тетраалкилти-
танат [123].
2-Метилбутен-1 из пропилена и триэтилалюминия синтезируется
в Японии на пилотной установке [124].
В продуктах реакции пропилена с триэтилалюминием содержится
углеводород, который высвобождается в результате реакции с Со,
Pt или Ni [125].
Модифицированная содимеризация осуществляется взаимодей-
ствием пропилена с триэтилалюминием при 130—140 °C с добавле-
нием этилена и коллоидного никеля и при нагревании до 160—170 °C.
Выделяющийся при этом 2-метилбутен-1 может быть дегидрирован
в изопрен при 700 °C на катализаторе MgO — Fe2O3 [126, 127].
2-Метилбутен-1 можно перевести в изопрен путем дегидрирования.
Однако этот метод синтеза едва ли сможет получить промышленное
значение, так как триэтилалюминий не только действует как ката-
лизатор, но и сам вступает в реакцию.
11.3.2. Содимеризация с изобутиленом
Содимеризация пропилена с изобутиленом была впервые изучена
Ипатьевым [128]. Образующиеся при этом изомерные гептены можно
путем оксосинтеза перевести в изооктиловый спирт, представляющий
собой важный компонент пластификаторов. Неизвестно, можно ли
этот метод реализовать в промышленном масштабе.
Оптимальные условия реакции получения изомерного гептена
по Ипатьеву: 135 °C, 38 кгс/см2, 375 л газовой* смеси/(л-ч). В ка-
честве катализатора предлагается тот же катализатор, что и для
олигомеризации пропилена, — фосфорная кислота на диатомовой
земле (кизельгуре).
238
11. Димеризация пропилена
При введении в реакцию эквимолярных количеств обоих газов
конверсия достигает 93%, причем пропилен реагирует легче, чем
бутилен. Полученная смесь состоит на 40—45% из гептенов, на 10—
15% из октенов и-других олефинов. Гептеновая фракция содержит
в основном 2,3-диметилпентен. В 1 ч на 1 л катализатора образуется
1040 см3 полимерного продукта [129].
Пропилен и бутилен можно сополимеризовать также при 190 °C.
ЛИТЕРАТУРА
1. J. J.Garmon. W. E.Morrow, V. J.Anhorn Chem. Eng. Progr.,
61, № 6, 57 (1965). .
2. H. J. Os terh of, Trans. Inst. Rubber Ind., 41, № 2, T53 (1965). ’
3. Oil in Canada, 13, № 19, 28 (1961).
4. C. J. В a a s,
C. J. В a a s,
5b. C. J. В a a s,
C. J. В a a s,
5d. C. J. В a a s,
6. P. — ’
7. И.
8. V.
9. M.
10.
5а.
5с.
Chem. Weekblad, 57, 389 (1961).
Brennstoff-Chem., 45, 161 (1964).
' 45, 258 (1964).
" 295 (1964).
J. C. Vlugter,
J. С. V 1 u g t e r,
J. С. V 1 u g t e r,
J. С. V 1 u g t e r,
J. С. V 1 u g t e r,
W. Sherwood, Ind. Eng. Chem., 55, № 3, 29 (1963).
Я. Тю.ряев и др., Хим. пром., 1963, 647.
J.Anhorn et al., Chem. Eng. Progr., 57, № 5, 41 (1961).
Suzuki, Nenryo Kyokaishi, 41, 114 (1962); C., 15017 (1962).
Landau, G. S. Schaffel, A. C. Depre z, Erdol Kohle-Erd-
Brennstoff-Chem
Brennstoff-Chem.,
Brennstoff-Chem., 45, 321 (1964).
45,
11.
12.
13.
14.
R.
gas — Petrochem., 16, № 7, 754 (1963).
Chem. Week (6.5.), 73, 78' (1961).
R. B. S t о b a u g h, Hydrocarbon Processing, 46, № 7, 149 (1967).
C. G. W i 11 i a m s, Jahresber. Fortschr. Chem., 495 (I860).
Англ. пат. 320362, 1928.
Jahresber. Fortschr. Chem., 495 (1860).
15. Герм, пат., 246241, 1910.
16. M. de M a 1 d e, Chim. e. Ind. (Milano), 45, № 6, 665 (1963).
17. G. Coppa-Zuccari, Chim. et Ind., Paris, 85, № 3, 363 (1961);
J. Morrison, Oil Gas Intern., 7, № 10, 56 (1967).
18. A. G i r a u d, Rev. gen. caoutchouc plast., 38, 1598 (1961).
19. F. С о u s s e m a n t, M. H e 11 i n, Erdol u. Kohle, 15, 274, 348 (1962).
20. R. W. King, Hydrocarbon Processing, 45, № 11, 189 (1966).
24. Chem. Age, 84, 898 (1960).
22. A. A. di Giacomo et al., Chem. Eng. Progr., 57, № 5, 35 (1961).
23. Д. Я. M у ч и н с к и й, В. Н. 3 л о т ч е н к о, Ю. Ф. С е м е н о в, Изв.
АН ТуркмССР, Сер. физ.-техн., хим. н геол, науки, № 2, 133 (1965).
24. , Франц, пат. 1419670, 1965.
25. Итал. пат. 586452, 1957.
26. D. A. S. 1178419, 1964.
27. В. С. Ф е л ь д б л ю м и др., Нефтехимия, 3, 13, 20 (1963).
28. В. С. Ф е л ь д б л ю м и др., Нефтехимия, 5, 493 (1965).
29. Англ. пат. 876680, 1961.
30. Англ. пат. 853187, 1960.
31. С. И. К р ю к о в и др., Изв. вузов. Химия и хим. технол., 7, 821 (1964).
32. Франц, пат. 1269563, 1961.
33. Франц, пат. 1356224, 1964. „
34. В. С. Ф е л ь Д б л ю м и др., Нефтехимия, 4, 257 (1964).
35. Франц, пат. 1250416, 1958.
36. Авт. св. СССР 107121, 1957; Бюлл. изобр., № 6, 75 (1957).
37. Англ. пат. 840028, 1960; пат. США 3104269, 1963.
38. Англ. пат. 896822, 1962.
Литература
239
39. Англ. пат. 775384, 1957.
40. Австрал. пат. 247253, 1963; англ. пат. 975454, 1964.
41. Бельг, пат. 575326, 1959; англ. пат. 831249, 1960; франц, пат. 1250416,
1269563, 1961; австр. пат. 212295, 1960.
42. Англ. пат. 979877, 1965; пат. США 3278634, 1967.
43. Пат. США. 3076047, 1963.
44. Пат. США 3219723, 3219724, 1965.
45. Г. Ewers, Angew. Chem., 78, 593 (1966); Франц, пат. 1497673, 1967.
46. Франц, пат. 1385503, 1965; В. С. Фельдблюм и др., Нефтехимия, 7,
379 (1967).
47. Англ. пат. 914144, 1962; пат. США 3182096, 1965.
48. Пат. США 3094573, 1963.
49. Австрал. пат. 256952, 1962; англ. пат. 912822, 19622; D. A. S. 1215131, 1966;
франц, пат. 1284453, 1284455, 1962; австр. пат. 231414, 1964; пат. США
3084206, 1963; австр. пат. 226659, 1963.
50. Австрал. пат. 256758, 1965; белы. пат. 614921, 1962; англ. пат. 912823—
912826, 1962; франц, пат. 1284456, 1284457, 1301836, 1962; пат. США
3325559 1967
51. Англ. пат. 903014, 1962; англ. пат. 932342, 1963.
52. Франц, пат. 1315324, 1963.
53. Пат. США 2986588, 1961.
54. Англ, пат; 868945, 1961-.
55. Пат. США. 3075027, 1963.
56. Франц, пат. 1410776, 1965.
57. Англ. пат. 889268, 1962; D. A. S. 1227446, 1966.
58. Англ. пат. 824917, 1959.
59. Пат. США 3255272, 1966.
60. Англ. пат. 978590,' 1964.
61. Франц, пат. 1356267, 1964.
62. Франц, пат. 1364460, 1964; пат. США 3340323, 1967.
63. Пат. США 3094573, 1963; 3311673, 1967. i
64. Бельг, пат. 641774, 1964.
65. Пат. США 3185745, 1965.
66. Японск. пат. 20373, 1965.
67. Бельг, пат. 618384, 1962; франц, пат. 1359070, 1964.
68. Пат. США 3104271, 1963.
69. Пат. США 3275706, 1966.
70. Бельг, пат. 611534, 1962.
71. Бельг, пат. 616564, 1962.
72. Бельг, пат. 619715, 1962; англ. пат. 991705, 1965; франц, пат. 1318568, 1963;
пат. США 3260770, 1966.
73. Бельг. Пат. 614789, 1962.
74. Англ. пат. 1003576, 1965.
75. Франц, пат. 1343359, 1963.
76. Бельг, пат. 631742, 1963.
77. Франц, пат. 1382602, 1964.
78. Франц, пат. 1377787, 1964.
79. Niederld A. S. 6509224, 1966; франц, пат. 1501379, 1967.
80. Англ. пат. 1011354, 1965; Niederld A. S. 6402598, 1964.
81. Пат. США 3175020, 1965.
82. Англ. пат. 1012077, 1965; Niederld A. S. 6402587, 1964.
83. Англ. пат. 993035, 1965; D. A. S. 1201329, 1965; пат. США 3175201, 1965;
D. A. S. 1244155, 1967.
84. D. A. S. 1248640, 1967; пат. США 31636’12, 1964.
85. Пат. США 3148157, 1964.
86. Пат. США 3128318, 1964.
87. Пат. США 3154595, 1964; D. A. S. 1253263, 1967.
2 40
11. Димеризация пропилена
88. Англ. пат. 933700, 1963.
89. A. W.Shaw et al.; J. Org. Chem., 3286 (1965).
90. Пат. США 2482008, 1949.
91. Англ. пат. 935288, 1963; франц, пат. 1244497, 1961; D. A. S. 1201331, 1965.
92. Пат. США 2404056, 1944; 2430137, 1946.
93. Франц, пат. 1244499, 1961; пат. США 3047644, 1962; D. A. S. 1218430, 1966.
94. Пат. США 2507864, 1950.
95. Лат. США 3133127, 1964. '
95а. Бельг, пат. 632055, 1963; пат. США 3268617, 1966,
96. В. Т. В г о о s, J. Am. Chem. Soc., 56, 1998 (1934).
97. Англ. пат. 933235, 1963.
98. Пат. США 3246046, 1966.
99. Англ. пат. 940682, 1963; пат. США 2446619, 1948.
100. Англ. пат. 849393, I960; пат. США 3013066, 1961; франц, пат. 1499833,1967.
101. Франц, пат. 1364531, 1964.
102. В. G. S с h u 1 t z, J. М. S с h и с к, В. S. W i 1 d i, J. Catalysis, 6,
385 (1966).
103. Франц, пат. 1406158, 1965.
104. F. D. R о s s i n i u. a., Selected Values of Physical and Thermodynamic
Properties of Hydrocarbons and Related Compounds, Pittsburgh, 1953.
105. H. P. E m m e 11 et al., Advances in Catalysis and related Subjects, Bd. 6,
New York, 1958, p. 98.
106. Швейц, пат. 398547, 1966.
107. F. C. Whit.more et al., J. Am. Chem Soc., 72, 51 (1950).
108. Пат. США 3268616, 1966.
109. H. К о c h, Н. van R а а у, Brennstoff-Chem., 32, 161 (1951).
НО. Б. А. Казанский, И. В. Гостунская, Н. Б. Добросер-
до в а, ДАН СССР, 130, 82 (1960).
111. Англ. пат. 886716, 1962.
112. Пат. США 3268617, 1966.
113. F. О. R i с е, К. F. Н е г z f е 1 d, J. Am. Chem. Soc., 56, 284 (1934).
114. L. Kiichler, H. T h e i 1 e, Z. physik. Chem., B42, 359 (1939).
115. Chem. Industrie, 17, 266 (1965).
116. Chem Eng. News, 43, № 13, 44 (1965).
117. L. A. Muirhead, Rubber and Plastics Age, 40, 501 (1959).
118. R. D. D e a n i n, SPE J., 23, № 2, 39 (1967).
119. Chem. Eng. News, 44, № 46, 39 (1966).
120. Пат. США 3080411, 1963.
121. Японск. пат. 26887/65, 1965.
122. D. A. S. 1218431, 1966.
123. Англ. пат. 856288, 1960.
124. Petrol. Chem. Eng., 37, № 1, 16 (1965). '
125. Англ. пат. 837350, 1960.
126. Японск. пат. 2662, 1959.
127. Proc. 7th World Petrol. Congr., Vol. 5, 1967, p. 107.
128. N. I p a t i e f f, R. E. S c h a a d, Ind. Eng. Chem, 37, 362 (1945).
129. B. Blouri, Chim. et Ind., Paris, 71, 706 (1954).
12. ОЛИГОМЕРИЗАЦИЯ ПРОПИЛЕНА
В этом разделе рассматривается в основном три- и тетрамериза-
ция пропилена.
Известно, что олефины можно олигомеризовать термически —
нагреванием до повышенных температур под давлением, а также ка-
талитически — при мягких условиях. Термически легче всего олиго-
меризуется этилен, а каталитически — изобутилен. Однако терми-
ческая олигомеризация до сих пор не приобрела значения в нефте-
химической промышленности, этот метод применяется в небольшом-
объеме только для получения пояимер-бензина.
Каталитическая олигомеризация пропилена может быть осущест-
влена как в жидкой, так и в газовой фазе. Оба метода применяются
в промышленности.
12.1. ТЕРМИЧЕСКАЯ ОЛИГОМЕРИЗАЦИЯ
Первые опыты по термической полимеризации пропилена [1]
были проведены Ипатьевым [2], который показал, что при высоком
давлении и 330—370 °C образуется полимер следующего состава
в %):
Парафины....................... 8
Олефины....................... 26
Циклоолефины.................. 22
Ароматические вещества не были найдены. Более обширные ис-
следования на различных олефинах [3] позволили определить оп-
тимальные условия получения олигомеров (с областью кипения бен-
зина). В табл. 24 приведены результаты, полученные с пропиленом.
Эти результаты свидетельствуют о том, что при повышении да-
вления количество образовавшегося полимера почти не увеличивается.
Но при этом удается значительно увеличить выход продукта в еди-
ницу времени на единицу объема. Температура и время контакта
очень мало влияют на термическую олигомеризацию.
Промышленный метод термической олигомеризации крекинг-га-
зов был разработан фирмами Pure Oil Со. и Alco Products Inc. (ме-
тод Alco) [4]. По методу Alco фракция С3 — С4 нагревается до 480—
540 °C под давлением 50 кгс/см2. При этом реагируют только олефины,
16 Заказ 399
242
12. Олигомеризация пропилена
Таблица 24
Оптимальные условия получения олигомеров пропилена
Давление, кгс/см2 Температура, °C Время контакта, мин Выход жидкости, вес. % Выход бен- зина, объемн. % Октановое число
35 450 5,7 16,3 ' 80,3 78
70 430 22,6 46,9 73,1 87
140 430 16,9 64,0 69,5 75
140 450 10,6 62,8 66,2 80
210 400 27,3 62,6 59,5 75
210 430 ‘ 16,4 61,0 63,4 . 74
210 450 8,7 61,2 67,1 74
а парафины не изменяются. При последующем пиролизе при
700 °C непрореагировавшая парафиновая часть вовлекается в олиго-
меризацию (метод Multiple Coil).
12.2. КАТАЛИТИЧЕСКАЯ ОЛИГОМЕРИЗАЦИЯ
12.2.1. Олигомеризация пропилена в паровой фазе
на фосфорнокислых катализаторах
Важнейший способ олигомеризации пропилена заключается в про-
пускании его над фосфорной кислотой, нанесенной на кизельгур,
активированный уголь или асбест. Собственно катализатор состоит
из смеси каталитически активных орто- и пирофосфорной кислот.
При температуре выше 240—260 °C эти кислоты дегидратируются
в каталитически неактивную метафосфорную кислоту. Чтобы вос-
препятствовать дегидратации кислот и тем самым максимально
продлить срок службы катализатора, нужно добавить к газовой сме-
си 2—10% водяного пара (лучше всего 5%). Это делается при тем-
пературе олигомеризации 300 °C [5]. Не рекомендуется значительно
увеличивать содержание пара в смеси, так как при этом может ухуд-
шиться твердость катализатора, кроме того, это может вызвать не-
которое гидратирование в изопропиловый спирт.
В качестве носителя для катализатора лучше всего применять
кислые соединения, так как при использовании нейтральных носи-
телей, например активированного угля, могут образоваться алкил-
фосфаты, которые затем улетучиваются. Наиболее употребительны
в этой роли кизельгур и асбест; однако в этом случае усиливается
образование кремнефосфатов (правда они нелетучи). На эти носители
можно наносить до 75% фосфорной кислоты.
г
12.2. КаталипШческая олигомеризация 243
Метод с применением фосфорной кислоты был разработан амери-
канской фирмой Universal Oil Products Со. (метод UOP, 1933) [6—
13]. Производство приобрело широкий размах во время второй
мировой войны с целью получения ценного горючего для самолетов.
В качестве катализатора применяется фосфорная кислота' (62—
65% Р5О5) на диатомовой земле или на асбесте. Активированный уголь
почти не применяется по вышеуказанным причинам. Катализатор
приготавливается нагреванием 74—76%-ной Н3РО4 до 260 °C (смесь
орто- и пирофосфорной кислот 65 : 35) и смешиванием с асбестом.
После охлаждения до 20—30 °C и сушки при 150 °C катализатор
готов к употреблению.
Катализатор нужно восстанавливать после пропускания 100-крат-
ного количества полимера (примерно через 60 рабочих дней). Ре-
генерация осуществляется посредством контролируемого окисления
смесью воздуха с отработанным газом при 480—540 °C и последу-
ющей гидратацией образовавшейся метафосфорной кислоты при
285 °C. Менять катализатор необходимо после пропускания 700- мак-
симум 1300-кратного объема продукта полимеризации [14, 15].
. Компания Universal Oil Products разработала два метода олиго-
меризации пропилена:
1) метод камерной печи под низким давлением;
2) метод камерной печи под высоким давлением.
Метод камерной печи под низким давлением. При реализации
этого метода в промышленном масштабе предварительно нагретая
до 170—210 °C смесь газов под давлением 14—20 кгс/см2 проходит
через ряд контактных печей, в которых фосфорнокислый катализатор
находится в виде кусочков или пилюль. Для получения полимер-бен-
зина исходным продуктом служит фракция С3—С4. Исходная смесь
с высоким содержанием олефинов поступает сначала в башню с наи-
менее активным катализатором. В конце процесса загружается башня
с самым активным катализатором. Такой способ обеспечивает мак-
симальную конверсию и хороший температурный контроль. Реакция
олигомеризации экзотермична (теплота реакции 16,5 ккал/моль про-
пилена [16]), поэтому температура в печи поднимается на 60—65 °C.
Постепенное ослабление активности катализатора компенсируется
за счет непрерывного повышения его температуры.
Поступающие в печь газы должны быть свободны от серы и азота.
Это достигается путем предварительной щелочной и водной промывки
[17]. Конверсия олефинов исходной смеси достигает 90%. Выходящие
газы охлаждаются и обрабатываются затем абсорбционным маслом
(высококипящей полимерной фракцией); неконденсирующаяся часть
выходит из абсорбера; растворившаяся часть разгоняется в стаби-
лизирующей колонне (при 24,5 кгс/см2) и в колонне для перегонки
под давлением (при 7 кгс/см2). Полимер-бензин образуется в виде
средней фракции, а абсорбционное масло остается в нижней части
колонны. Можно работать также без промежуточной абсорбции.
16*
244
12. Олигомеризация пропилена
В этом случае выходящий из реакции продукт освобождается в спе-
циальной колонне от пропана и разгоняется как обычно.
Метод камерной печи под высоким давлением. Подобные уста-
новки (полимеризация Midget) работают при 250 °C и давлении
35 кгс/см2. Они применяются главным образом для небольших про-
изводств (3500—3000 м3/ч крекинг-газа) и не требуют значительного
ухода [18—19]. Используется газ из стабилизатора крекинг-уста-
новок, содержащий около 2/3 полимеризующихся олефинов. Выход
достигает 90%.
12.2.2. Олигомеризация пропилена с образованием тримера
и тетрамера на фосфорнокислых катализаторах
Направленная полимеризация с образованием ди-, три- и тетра-
меров проводится в мягких условиях [20]. С повышением темпера-
туры тенденция ненасыщенных углеводородов к полимеризации
уменьшается, а при повышении давления — усиливается. Поэтому
полимеры
Рис. 61. Технологическая схема получения тетрамера пропилена
из смеси пропан — пропилен:
1 — колонна для щелочной промывки; 2 — колонна для водной промывки;
з — сборник; 4 — катализаторная камера; 5 — колонна для удаления про-
пана; 6 — циркуляционная колонна; 7 — колонна для очистки.
низкие температуры способствуют образованию димеров и, возможно,
тримеров, повышение температуры ведет к повышению степени по-
лимеризации; при 500 °C и низком давлении преобладает деполиме-
ризация.
При применении в качестве катализатора ортофосфорной кислоты
следует поддерживать температуру на уровне 130—205 °C, при более
высоких температурах (особенно выше 300 °C) возникают явления
гидрополимеризации (одновременное образование высших парафинов
наряду с олефинами) и гетерополимеризации (образование высших
олефинов, у которых число атомов углерода не соответствует крат-
ному от С3).
12.2. Каталитическая олигомеризация
245
Метод получения пропиленового тетрамера был разработан
первоначально в Германий. Реакция проводилась под давлением
40—60 кгс/см2. Использовали смесь из 75% Н3Р04 и 25% Н4Р2ОГ
[21]. Получалось 15—20% гексанов и ноненов, 35—40% тримера
пропилена, 29% тетрамера и 16% высших олефинов. Теперь работают,
как и при получении полимер-бензина, по принципу камерной печи
с фосфорной кислотой на носителях, например кизельгуре, отбель-
ной земле или асбесте (рис. 61). В этом процессе особенно важно
поддерживать постоянную температуру в печи.
Для получения однородной фракции пропиленовых тетрамеров
смешивают исходный продукт — смесь пропана с пропиленом —
с димерной и тримерной частью из циркуляции и полученную смесь
доводят до реакции при 170—220 °C и давлении 14—42 кгс/см2.
Температура в различных местах слоя катализатора поддерживается
постоянной путем вдувания пропана. Реакционные продукты разде-
ляются перегонкой и тетрамер извлекается как фракция, кипящая
при 177—230 °C (или при 188—200 °C при повышенных требованиях
[14]). Ниже указан расход исходных продуктов и выход реакцион-
ных продуктов, получаемых на установке UOP при конверсии про-
пилена 92,3% [15]:
«1/плш объемы.
м’/день 0/
/в
Пропан-иропиленовая фракция................. 95 100 *
Израсходованный исходный продукт........... 51,5 45,6
Полимер для добавки к бензину.............. 0,3 0,3
Тетрамер пропилена......................... 28,5 24,3
Высококипящие полимеры как остаток .... 1,3 1,1
* 49,3% пропилена.
Пропилен, используемый для тетрамеризации, должен быть как
можно лучше очищен от изобутилена.
12.2.3. Олигомеризация пропилена различными методами
на фосфорнокислых катализаторах [22—32]
В качестве носителей рекомендуются каолин [33], кремний,
карбид кремния, мрамор и стекло, [34], силикагель [35], фуллерова
земля после обработки ее галогенидами бора или алюминия [36],
кварц [37]. Носители чаще всего служат для усиления активности
катализатора. В качестве добавок, способствующих олигомеризации,
рекомендуются соли меди [38, 41, 43] и кальция [38], фосфаты ами-
нов и аммония [39, 40], никелевые соли [41, 42] и соединения марган-
ца [44].
Для получения более узких фракций, имея в виду определенный
олигомер, предлагается осуществлять конверсию в несколько стадий:
246
12. Олигомеризация пропилена
[45, 46}. С целью увеличения доли низших олигомеров с температу-
рой кипения 400 °C можно проводить олигомеризацию в присутствии
насыщенного углеводорода (например, цетана) [47].
В литературе [48] описана возможность автоматического упра-
вления тетрапропиленовой установкой с помощью вычислительной
машины.
Обширные исследования олигомеризации в присутствии жидкой
фосфорной кислоты проведены Ипатьевым [49], соответствующие
промышленные способы разработаны Calif. Res. Corp. Работают с квар-
цевым носителем, кислота удерживается на нем в жидком виде
(катализатор в форме жидкостной пленки), при восстановлении
пленка вымывается и наносится новая [50, 51].
12.2.4. Олигомеризация пропилена на различных катализаторах
Олигомеризация пропилена возможна и на других кислых ката-
лизаторах кроме фосфорной кислоты (табл. 25).
Таблица 25
Катализаторы олигомеризации пропилена
Катализатор Характеристика процесса Литература
Минеральные кислоты Серная кислота 205—315 °C 99%-ная при 25 °C 96%-ная при 50 °C [52] [53]
Фосфат кремния — [54]
фосфорная кислота, Al2O3-SiO2/Al2O3 Двухступенчатый метод [55]
Тетраборгипофосфорная кислота В4О7(Н3Р2О6) 2 [56]
Al 2О 3— Si О 2 (алюмосиликат) 120 °C, 32 кгс/см2 [57]
Al2O3^SiO2 180—220 9С [58]
А12О3—SiO2 Инициатор Сг2О; 170 °C для полу- чения тетрамера, 190 °C трпмера и 210 °C димера [59]
Активированная отбельная земля — [60]
А12О3-Сг2О3 (6%)-SiO2 Вместо Сг2О3 в качестве инициато- ров предлагаются: NiO, ThO2, Fe2O3, MnO2, UO3, V2O5, WO3, MoO2, MoO3; последние дают в ос- новном высокомолекулярные со- единения [61]
BF3 Инициирование с помощью TiO2 [62]
BF3-H3PO4, bf3-hpo3, 2BF3—H4P2O7 на акти- вированном угле Лучше всего BF3—НРО3 [63]
BF3—H3PO4 100 9С; от три- до гексамера [64]
BF3—H3PO4 60—70 9С, в основном тетрамер [65]
12.3- Продукты олигомеризации пропилена
247
Продолжение табл. 25
Катализатор 4 Характеристика процесса Литература
HF—BF3 4—60 °C, давление; в основном те- [66]
ZnCl, тра- и пентамеры Главным образом тример [67—69]
AlGlg — [70]
А1Вг3 От —50 до 100 °C [71]
А1Вг3—НВг — [72, 73]
Al 2( SO4) 3/силикагель — 74]
Т i О 2 / си лик аге л ь — 75]
TiCl3 95—315 °C 76]
Cu2P2О7 150 °C (метод Kellogg) [77-79]
Кремневольфрамовая кис- 150—160 °C, давление [80]
лота Окиси переходных метал- МоО3, WO3, UO5, Сг2О3, NiO, FeO, [81]
лов на алюмосиликате Молекулярные сита ZnO [82]
12.3. ПРОДУКТЫ ОЛИГОМЕРИЗАЦИИ ПРОПИЛЕНА
При полимеризации пропилена в присутствии хлористого алюми-
ния [70] (70 °C, нитрометановый раствор) получаются продукты с бо-
лее высокой средней степенью полимеризации, чем при реакции
в присутствии фосфорной кислоты. Число атомов С у этих продуктов
более 24 и степень разветвления больше. Образующийся при этом
тример идентифицирован как З-изопропилгексен-2:
СН3-СН=С-СН2-СН2-СН3
I
СН(СН3)2
Фракция С12, полученная в присутствии А1С13, представляет
собой вероятно, 3-изопропил-4-этилгёптен-2:
СН(СН3)2
СН3-СН=С-С-СН2-СН2-СН3
I
С2Н5
Высшие олигомеры образуются из первичных димеров в резуль-
тате изомеризации двойных связей.
При полимеризации с жидкой фосфорной кислотой (полугидрат
метафосф'орной кислоты) при 160—170 °C и под давлением 50—
60 кгс/см2 Террес [83] получил следующие продукты:
СН3СН2СН2СНСН3 СН3СН2СН2СНСН2СНСН3
I I I
сн3 сн3 сн3
2-метилпентан 2,4-диметилгептан
248
12. Олигомеризация пропилена
СН3СН2СН2СНСН2СНСН2СНСН3 СН3СН2СН2СНСН,СНСН,СНСН2СНСН3
III I I ‘I I
сн3 сн3 сн3 сн3 сн3 сн3 сн3
2,4,6-триметилнонан 2,4,6,8-тетраметилундекан
СН3СН2СН2СНСН2СНСН2СНСН2СНСН2СНСН3
I I I I I
сн3 сн3 сн3 сн3 сн3
2,4, 8,10-пентаметилтридекан
Более высокая степень олигомеризации недостижима. Опыты
с твердыми и жидкими фосфорнокислыми катализаторами дают оли-
гомеры с подобной структурой. Правда, Кох и другие [84] пришли
к иным результатам, чем Террес. Они нашли в тримерной фракции:
СН2=ССН2СН,СН2СН2СН2СН3 СН3СН=ССН2СН2СН,СН2СН3
I I '
сн3 сн3
2-метилоктен-1 З-метилоктен-2
СН3СН2СН=ССН2СН2СН3
с2нБ
4-этилгептен-З
сн3с=ссн2сн,сн2сн3
I I
сн3 сн3
2,3- диметилгептен-2
СН3СН=ССН2СН2СН3
СН2СН2СН3
З-пропилгексен-2
сн3с=снснсн.,сн2сн3
I I
сн3 сн3
2,4- ДИметилгептен-2
СН3СНСН2С=СН2СН,СН3
I I
сн3 сн3
2,4-диметилгептен-4
СН3СН=ССН2СН2СН-СН3
I I
сн3 сн3
3,6-диметилгептен-2
сн3с=снсн2снсн.2сн3
I I
сн3 сн3
2,5- ДИметилгептен-2
СН3СН,С=СНСН2СН2СН3
| I
СНз сн3
3,6-диметилгептен-З
сн3
СН3СН=ССНСН2СН2СН3
I
сн3
3,4-диметилгептен-2
сн3
I
СН3СН2СН=ССНСН2СН3
сн3
4,5 - диметилгептен- 3
с2нБ
СН3СН=ССНСН2СН3
СН3
3-метил-4-этилгексен-2
сн3
I
сн3сн=сснсн2сн3
С2Нб
3-этил-4-метилгексен-2
12.4. Применение олигомеров пропилена
249
Несмотря на мягкие условия реакции, в тетрамерной фракции
содержалось (в %):
Фракция С12.........50 С13..............17
С10....... ^14.................
Сц........29
Исследования Петрова с ZnCl2 показали, что образуются такие
соединения [67, 85, 86]: '
СН3С=ССН2СНСН3 СН2=ССН2СНСН2СН2СН3 ,
III II
СН3СН3 сн3 сн3 сн3
2,3,5-триметилгексен-2 2,4-диметилгептен-1
СН3С=СНСН2СН2СНСН3 СН3СН2СН=СНСН2СН2СНСН3
сн3 сн3 сн3
2,6-диметилгептен-2 7 -метилоктен- 3
Исследования, проведенные с твердым фосфорнокислым катали-
затором, показали, что олигомеризация пропилена при концентра-
ции его свыше 3,1 моль на 20 г катализатора проходит как реакция
первого порядка [87]. Опыты с фосфорной кислотой на силикагеле
[88] показали, что доля димеров и тримеров увеличивается с повы-
шением содержания воды. Результаты эти истолковываются так:
на первой стадии происходит отложение пропилена на кислом ката-
лизаторе и образование сложных эфиров фосфорной кислоты, кото-
рые затем реагируют с пропиленом. В результате образуются ионы
карбония, которые присоединяются к другим молекулам олефина
или путем отдачи одного протона стабилизируются на анионах фос-
форной кислоты; при этом выделяются олигомеры.
Найденная зависимость [89] между кислотностью алюмоси-
ликатного катализатора и скоростью олигомеризации показывает, ч
что важное значение для олигомеризации имеет кислотная функция.
12.4. ПРИМЕНЕНИЕ ОЛИГОМЕРОВ ПРОПИЛЕНА
Первоначально методы олигомеризации были разработаны для
того, чтобы получить компоненты добавок для горючих с высоким
октановым числом. Однако в этом случае исходили не из чистых
фракций С3, а из фракций С3 — С4, и получали олигомеры с нужным
интервалом температур кипения (неселективная полимеризация).
Среди продуктов селективной олигомеризации господствующее по-
ложение занимает тетрапропилен. Несколько лет назад он служил
компонентом алкилирования для ароматических веществ при полу-
чении изододецилбензола, который после сульфирования и нейтра-
лизации переводился в алкиларилсульфонаты. До 1961—-1962 гг.
такие продукты были важнейшими исходными веществами для полу-
чения моющих средств. Однако в дальнейшем применение тетрапро-
пилена для этой цели было запрещено, так как недостаточная биоло-
250
12. Олигомеризация пропилена
тическая разрушаемость получаемого из него алкиларилсульфоната
приводила к сильному загрязнению сточных вод.
Тример, получаемый как побочный продукт при производстве
тетрапропилена, применяется как компонент для синтеза неионо-
генных моющих средств. Для этого фенол алкилируют тримером про-
пилена и на полученный продукт действуют окисью этилена.
ЛИТЕРАТУРА
1. Я. Т. Э й д у с и др., Усп. хим., 22, 838 (1953).
2. V. N. I р a t i е f f, Н. Р i n е s, Ind. Eng. Chem., 28, 684 (1936).
3. F. W.Sulli van jr., R. F. Ruthruff, W. E. Kuehtzel, Ind.
Eng. Chem., 27, 1072 (1935).
4. M. В. С о о k, H. R. Swanson, C. R. W a g n e r, Refiner, 14, № 11,
506 (1935); Oil Gas J., 34, № 26 , 56 (1935).
5. A. E. D uns t an, D. A. H о we s, J. Inst. Petrol. Technol, 22, 347 (1936).
6. Пат. ФРГ 886302, 1948.
7. Пат. США 2163275, 1936.
8. Пат. США 2202104, 1936.
9. Герм. пат. 876990, 1943.
10. Пат.' США 1933513, 1935.
И. Пат. США 2020649, 1936.
12. Пат. США 1993512, 1935.
13. Пат. США 2018065, 1936.
14. Е. К. Jones, Petroleum Refiner, 33, № 12, 186 (1954).
15. F. L. Re sen, Oil Gas J., 52, № 46, 203 (1954); 53, № 46, 163 (1955).
16. С. M. F о n t a n a, G. A. К i d d e r, J. Am. Chem. Soc., 70, 3745 (1948).
17. Пат. США 2843639, 1958.
18. V. N. I p a t i e f f, В. В. С о r s о n, E. E g 1 о f f, Ind. Eng. Chem., 27,
1077 (1935).
19. F. F. Lob an, Oil Gas J., 52, № 19, 108, 138, 139 (1953).
20. Пат. США 2572724, 1949; англ. пат. 675816, 1952.
21. FIAT № 1141, Final Rept., J. D. Brandner et al., Synthetic Deter-
gents and related Surface Active Agents in Germany.
22. Англ. пат. 696568, 1953.
23. Пат. США 2500307, 1950.
24. Пат. США 2814655, 1957.
25. Пат. США 2486533, 1949.
26. Пат. США 3041386, 1962.
27. Пат. США 3046316, 1962.
28. Пат. США 2755324, 1956.
29. Д. Я. M уч и н с к и й, Изв. АН ТуркмССР, Сер. физ.-техн., хим. и геол,
науки, № 6, 58 (1960).
30. Пат. ФРГ 939149, 1956.
31. Пат. США 2517720, 1950.
32. Пат. США 2952428, 1952; англ. пат. 681042, 1952.
33. Пат. США 2586852, 1952.
34. Пат. США 2619512, 1952.
35. О. Я. Ш к е и д з е и др., Нефтепереработка и нефтехимия, № 3, 28 (1966).
36. Пат. США 2596497, 1952.
37. Пат. США 2770665, 1956.
38. Пат. ФРГ 885701, 1948.
39. Пат. США 2537282, 1946.
40. Пат. США 2826622, 1958.
41. Англ. пат. 710537, 1954.
Литература
251
42. Пат. США 2778804, 1957.
43. Пат. США 2618614, 1952.
44. Пат. США 2692241, 1964.
45. Пат. США 2728804, 1955.
46. Пат. США 2802890, 1957.
47. Пат. США 3256359, 1966.
48. Н. tilde г, Chem.-Ing.-Techn., 34, 133 (1962).
49. V. N. I patieff, Ind. Eng. Chem., 27, 1067 (1935).
50. Пат. США 2852579, 1958.
51. U 1 1 m a n n, Enzyclopadie der Technischen Chemie, Bd. 6, Miinchen —
Berlin, 1955, S. 665.
52. Пат. США 2870217, 1959.
53. H. 0 h t s u к a, N. T о m i t a, H. Sato, Mem. Fac. Eng. Hokkaido Univ.,
8, № 1, 23 (1950); C. A., 46, 6075f (1952).
54. Франц, пат. 1268601, 1961.
55. Англ. пат. 969403, 969404, 1964.
56. Пат. США 2559576, 1951.
57. Англ. пат. 945021, 1963; пат. США 3137739, 1964.
58. Г. М. П а н ч е н к о в и др., Изв. АН ТуркмССР, Сер. физ.-техн., хим.
и геол, науки, № 2, 109 (1960).
59. Г. М. П а н ч е н к о в и др., № 3, 97 (1961); пат. США 3188360, 1965.
60. Е. J. Houdry et al., Refiner and Nat. Gasoline Manufacturer, 17, 574
(1938).
61. Англ. пат. 790195, 790196, 1958.
62. Пат. США 2766312, 1956.
63. А. В. Топчиев, Я. М. П а у ш к и н, ДАН СССР, 58, 1057 (1947).
64. Англ. пат. 727944, 1955.
65. Пат. США 2816944, 1957.
66. Пат. США 2528876, 1950.
67. A. Petrow, Fette-Seifen-Anstrichmittel, 57, 798 (1955).
68. Л. И. Ан ц у с, А. Д. Петров, ДАН СССР, 70, 425 (1950).
69. О. L. В г a n d е s, W. A. G г u s е, A. L о wyt Ind. Eng. Chem., 28,
554 (1936); J. Inst. Petr. Techn., 22, 256a (1936).
70. E. T e r r e s et al., Erdol und Kohle, 12, 547 (1959).
71. Пат. США 30716347''1963.
72. Пат. США 2525787, 1950.
73. С. М. F о n t a n a, Cationic Polymerisation and related Complexes, 1952,
121; C. A., 48, 5625d (1954).
74. Англ. пат. 993008, 1965.
75. Пат. США 2458818, 1949.
76. Пат. США 3193596, 1965.
77. Petroleum Refiner, 33, № 9, 193 (1954).
78. J. D. W a d d е 11, Oil Gas J., August, 1941.
79. J. H. S t e f f e n s, M. V. Z i m m e r m a n, H. J. L ai turi, Chem.
Eng. Progr., 45, 269 (1949).
80. J . J. V e r s t a p p e n, H. J. W a t e r m a n, J. Inst. Petr. Techn., 41,
№ 383, 343 (1955).
81. A. Clark, Ind. Eng. Chem., 45, 1476 (1953).
82. C. J. N о r t о n, Chem. and Ind., 258 (1962).
83. E. T e r r e s, Brennstoff-Chem., 34, 355 (1953).
84. H. Koch et al., Brennstoff-Chem., 44, 331 (1963)1
85. Д. Я. M у ч и н с к и й и др., Изв. АН ТуркмССР, Сер. физ.-техн., хим.
и геол, науки, (2), 133 (1965).
86. Л. И. Ан ц ус, А. Д. Петров, Изв. АН СССР, ОХН, 599 (1950).
87. М. R е р a s, Chem. Przmysl., 15, № 9, 543 (1965).
88. М. Rep as, Chem. Techn., 17, № 4, 222 (1965).
89. O. J о h n s о n, J. Phys. Chem., 59, 827 (1955).
13. АЛКИЛИРОВАНИЕ ПРОПИЛЕНА
Реакция парафиновых углеводородов с олефинами была открыта
•30 лет назад и с тех пор известна под названием «алкилирование».
Наибольшее значение эта реакция получила при взаимодействии
олефинов, содержащихся в газах нефтеочистки, с изопарафинами.
Она осуществляется с целью получения высокооктановых топлив.
Для алкилирования используются главным образом изопарафи-
ны — изобутан и изопентан, а из олефинов — этилен, пропилен
[1—4] и бутилен.
Алкилирование можно проводить термическим или каталитиче-
ским путем. Промышленное значение получил пока только каталити-
ческий метод, при котором употребляются в основном серная кислота
и фтористый водород и меньше — хлористый алюминий (в частности
при алкилировании этилена).
Значение алкилирования постоянно возрастает, так как в про-
мышленных странах полимер-бензин все больше заменяется алкили-
рованным бензином [6].
13.1. ТЕРМИЧЕСКОЕ АЛКИЛИРОВАНИЕ
Термическое алкилирование было впервые подробно изучено
фрейем [7—9]. Парафины могут при определенных условиях реаги-
ровать с олефинами при повышенных температурах, когда крекинг
еще не наступает. Особенно хорошо проходит алкилирование под
давлением, при этом парафины нормального и изомерного строения
реагируют почти с одинаковой скоростью. Термический метод был
внедрен в промышленность специально для получения неогексана
(2,2-диметилбутана) [7] взаимодействием этилена с изобутаном:
СН3 СН3
СН3-СН+СН2=СН2 —> СН3—С-СН2-СН3
I I,
сн3 сн3
Оба компонента начинают реагировать при 500 °C. Изобутан
вводится в большом избытке. Образующиеся продукты перечислены
ниже:
13-2. Каталитическое алкилирование 253
-------9--------------------------------------------
Параметры реакции:
Отношение этилен : изобутан, % 11,8 : 88,2 25,6 74,4
Давление, кгс/см2................. 315 315
Температура, 'С................... 515 505
Время реакции, мин................ 2,2 4
Выход бензина, % от всего коли-
чества продукта.................. 14,8 35,4
Выход жидких продуктов, вес. %
Изопентан (этилен, изобутан) . . 5,0 3,1
и-Пентан.......................... 5,0 2,7
и-Гексан.......................... 5,2 2,7
Неогексан........................ 42,1 . 31,3
2,3-Диметилбутан ................ 17,5 9,9
2-Метилпентан.................... 17,5 9,9
и-Гексан......................... 17,5 2,9
Гептены ......................... 2,4 2,0
Гептаны .......................... 4,1 4,1
Октены............................ 3,2 2,9
Октаны .......................... 12,7 13,7
Углеводороды Сд и выше .... 5,9 26,2
Термическое алкилирование проходит по механизму радикаль-
но-цепной реакции, поэтому добавка агентов образования радикалов
(например, хлорорганических соединений) вызывает понижение тем-
пературы реакции с 500 до 400 °C.
Алкилирование пропилена изобутаном проводили при 400 °C
под давлением 280—1050 кгс/см2 в присутствии 1,2,3-трихлорпро-
пана и 1,2-дихлорпропана [10]. В результате получались 2,2-диметил-
пентан и 2-метилгексан. С повышением давления образуется больше
2-метилгексана, что свидетельствует об уменьшении относительной
скорости реакции третичного атома углерода. Другими катализа-
торами термического алкилирования под давлением являются тет-
раэтилсвинец [11] и перекиси (например, перекись бензоила [12],
перекись тпрет-бутила [13]).
13.2. КАТАЛИТИЧЕСКОЕ АЛКИЛИРОВАНИЕ
13.2.1. Алкилирование с серной кислотой
Алкилирование с серной кислотой в качестве катализатора по-
лучило значение как способ производства алкилированного бензина,
при этом изобутан алкилируется С3 — СБ-олефинами.
Известны три метода алкилирования с серной кислотой:
1) каскадный метод с автоохлаждением — процесс Kellogg;
2) метод вытекания с охлаждением — процесс Stratford Engi-
neering;
3) метод «тайм-танк».
254
13. Алкилирование пропилена
Последний метод сравнительно старый, теперь, вероятно, таких
установок уже не строят. Для двух остальных процессов строятся
установки мощностью 10 000—20 000 т алкилата в год.
Каскадный метод с автоохлаждением (процесс Kellogg).
На рис. 62 изображена схема установки, работающей по этому
методу [2, 28—30]. Богатый олефином продукт после предваритель-
ной обработки проходит теплообменник и попадает в каскадный
Рис. 62. Схема установки каскадного метода с автоохлажде-
нием (процесс М. W. Kellogg Со.):
I — блок предварительной обработки сырья; 2 — теплообменник; з — система
автоохлаждения; 4 — колонна для удалении пропана; 5 — колонна для от-
гонки изобутапа; в — блок обработки реакционных продуктов; 7 — каскад-
ный реактор.
реактор, в котором смесь серная кислота— изобутан течет слева на-
право. Обычно устанавливается 2—3 каскада. Время прохождения
через каскады от 30 до 50 мин. Поток изобутана и кислоты подается
в низ реактора. В отдельных каскадах, оборудованных скоростными
мешалками, все три компонента реакции хорошо перемешиваются
и начинают реагировать. На выходе из реактора кислота и смешан-
ный с изобутаном алкилат разделяются. Большая часть кислоты
возвращается в реакцию, а смесь алкилата и изобутана попадает
в работающую под давлением колонну для удаления бутана. Непро-
реагировавший изобутан отбирается с верха колонны и тоже воз-
вращается в процесс. Алкилат собирается в нижней части колонны.
В колонне для перегонки под давлением отгоняется непрореагиро-
вавший пропан. Продукт из нижней части этой колонны возвращается
на установку вместе с пропиленовой фракцией.
Метод вытекания с охлаждением (процесс Stratford Engineering).
При этом методе [29, 31—34] (рис. 63) работают только с одним реак-
13-2. Каталитическое алкилирование
255
тором. Свежая кислота, све-
жий углеводород и циркули-
рующая эмульсия (кислота и
углеводород) загружаются в
один конец горизонтального
реактора (рис. 64). Реакцион-
ная смесь интенсивно переме-
шивается скоростной мешалкой.
Благодаря охлаждению изобу-
таном в реакторе устанавли-
вается температура 2 °C. Рабо-
чее давление достигает 5—
6 кгс/см2. Время контакта 20—
30 мин. Концентрация кислоты
около 88—90%. Лучше рабо-
тать с 94—95 %-ной кислотой,
но это невозможно из-за
слишком больших потерь ки-
слоты.
Алкилирование всегда со-
провождается процессами по-
лимеризации, которые подавля-
ются по мере увеличения избыт-
ка изопарафина. Изобутан — са-
мый важный компонент парафи-
нового алкилирования — содер-
жится в природных газах и
в газах нефтеочистки в гораздо
меньшем количестве, чем бутан.
Поэтому фракции бутана часто
изомеризуют в изобутан в при-
сутствии хлористого алюминия.
Кроме того, можно алкилиро-
вание объединить с полимери-
зацией, так как вследствие
распада олефинов изобутан
оказывается уже сконцентри-
рованным. Фракция С4 может
быть очень насыщена изобу-
таном также в результате ката-
литического крекинга [14].
Для алкилирования изо-
бутана бутиленом требуется
90%-ная серная кислота. При
этом выделяется 85%-ная ки-
слота, в присутствии которой
256
13. Алкилирование пропилена
можно алкилировать изобутан пропиленом, получая иэогептаны |
[15, 16]. После этого кислота еще разбавляется и в таком виде ;
используется для алкилирования изопентана Св- и С7-олефинами,
а также для селективного вымывания диолефинов. После этого ки-
слота регенерируется. Регенерация серной кислоты определяет j
минимальную мощность алкилирования, обеспечивающую рента-
бельность установки. На меньших установках выгоднее работать
только с фтористым водородом [17]. (При отсутствии обработки по-
тери катализатора в присутствии фтористого водорода значительно
уменьшаются [18].)
Рис. 64. Реактор Stratco для алкилирования.
В табл. 26 рассматриваются результаты алкилирования изопара-
фин — олефин, полученные на примере системы изобутан — бути-
лен, но распространяющиеся, конечно, и на систему изобутан — про-
пилен.
С повышением отношения изобутан : олефин увеличивается вы-
ход алкилата, а расход кислоты уменьшается. В промышленности
определено оптимальное соотношение изопарафин : олефин = 5:1,
но вследствие возвращения в реакцию изобутана устанавливается
более высокое соотношение. Реакция длится около 5 мин при тем-
пературе 0—10 °C и концентрации серной кислоты 98—100% (для
С4-углеводородов). Для пропилена ойтимальная температура не-
сколько выше (10—15 °C), так как в этом случае работают лишь с
90%-ной серной кислотой. Иногда температуру поднимают до 16—
32 °C, так как образовавшийся изопропилсульфат стабилен при
пониженных температурах [19]. Обычно объемное отношение серной
кислоты к смеси углеводородов равно примерно 1 : 1, в этом случае
углеводородная смесь эмульгируется в кислоте, благодаря чему
получают алкилаты лучшего качества. Скорость перемешивания
13-2. Каталитическое алкилирование
257
Таблица 26
Выход и качество алкилата, полученного в присутствии серной кислоты
при использовании различных олефинов [25—27]
Показатели 1 Пропилен 40% пропи- лена, 60% 1 бутилена Бутилен Амилен
Выход алкилата, объемн. % в расчете на: олефин 178 174 172 160
алкилированный авиационный бензин (т. кип. 170 °C) 150-60 156-62 153—65 140—49
Расход изобутана, л/л олефина 1,275 1,17 1,106 0,965
Расход 98%-ной серной кислоты, кг/100 л алкилата 21-23 11,5— 4,8—7,2 11,5
Качество алкилата для авиационного бензина: Октановое число'по Research 89-92 15,5 93—95 94—97 92-93
Октановое число по ASTM 87-90 91—93 92-95 90-92
Октановое число по Research с добавкой 1,5 см3 тетраэтилсвинца 100,5— 102,5- 103- 102—
102,5 104,5 105,5 103
является важным фактором', обеспечивающим максимальный выход
и оптимальное качество продукта. Поэтому наилучшие результаты
достигаются при работе с хорошими мешалками [19].
Рентабельность процесса определяется главным образом расхо-
дом кислоты. При алкилировании пропиленом он довольно велик.
Чтобы уменьшить потребность в серной кислоте, можно осуществить
реакцию обмена образовавшегося изопропилсульфата с изобутаном
и бутиленами; при этом образуется алкилат [20—22]. В одном реак-
торе производится алкилирование в токе пропилена, а в другом —
в токе бутилена [23].
Самый высококачественный алкилат с минимальным расходом
кислоты получается при реакции изобутана и олефина в отношении
1:1. Потери возникают в результате полимеризации олефинов
и самоконденсации изобутана при одновременном образовании про-
пана [24].
13.2.2. Алкилирование с фтористым водородом
Первоначально фтористый водород применяли как катализатор
алкилирования в ароматическом ряду [3, 35—43], а теперь он имеет
большое значение как катализатор алкилирования в алифатическом
ряду. Это объясняется большим расходом серной кислоты, особенно
при алкилировании в системе пропилен — изобутан. Здесь расход
17 Заказ 399
258
13. Алкилирование пропилена
кислоты в 3,5 раза больше, чем при алкилировании в системе бути-
лен — изобутан. При использовании фтористого водорода такого
различия нет.
Октановое число продуктов алкилирования системы пропилен —
изобутан приближается к 90, выход составляет 1,7 объема алкилата
на 1 объем пропилена и 1 объем изобутана [44]. На 1 кг HF обра-
зуется 840 л алкилата. Недостатком процесса является слабое вза-
имодействие ф?ористого водорода с олефинами (в результате
щелочную промывку
Рис. 65. Схема процесса алкилирования с HF (Phillips
Petroleum Со.):
1 — главная ректификационная колонна для отгонки HF;
2 — отпарная колонна для отгонки HF; в — декантатор-от-
стойник; 4 — отстойник-реактор; 5 — колонна для отгонки
бутана.
образуются около 0,01% алкилфторида), что может привести к по-
тери фтора и появлению примесей в алкилате (правда, эти примеси
не вызывают коррозии). Остаточный фтор можно отщепить в форме
HF пропусканием алкилата над CaF2 или бокситом при 175—250 °C,
возможно также экстрактивное отделение [45].
Алкилирование в присутствии фтористого водорода проводится
при 20—30 °C; при более низкой температуре происходит преиму-
щественно полимеризация, кроме того, присоединяется заметное
количество фтора. При более высоких температурах образуются
большие количества высококипящих высокоалкилированных^ угле-
водородов. Время контакта 5—15 мин, оптимальное соотношение
изобутан : пропилен = (1 -4-4) : 1. Фтористый водород вводится,
по возможности, в безводной форме (максимальное содержание воды
1,5%). Эту форму легко получить азеотропной перегонкой, поскольку
азеотропная смесь 37% HF и 63% воды кипит только при 120 °C.
Наличие воды недопустимо из-за опасности коррозии и усиления
присоединения фтористого водорода к олефинам.
13.2. Каталитическое алкилирование
259
На практике хорошо себя зарекомендовали следующие методы
алкилирования.
Метод Phillips Petroleum Со. [46—49]. Смесь олефина и изобу-
тана турбулентно перемешивается с фтористым водородом при —20 °C
(рис. 65). В ходе реакции температура в трубчатом реакторе повы-
шается до 0—5 °C. При использовании пропилена или амиленов
требуются более высокие температуры (10—32 °C). Время контакта
Рис. 66. Схема алкилирования в системе изобутан — олефины (Universal
Oil Products Со.):
1 — реактор; 2 — смеситель-отстойник; 3 — резервуар для хранения свежей HF; 4 — отде-
литель; 5 — конденсатор; 6, 12 — приемник; 7 — нагреватель; 8 — регенератор кислоты;
9 — отделитель смолы; 10 —отстойник-декантатор; 11 — колонна для удаления пропана;
13 — отделитель HF.
всего 20—40 с. Плавиковая кислота циркулирует, алкилат отде-
ляется и фтористый водород после охлаждения снова добавляется
к исходному продукту. Для случая, когда применяется смесь про-
пилена с бутиленом, разработан специальный вариант [50], при
котором сначала в реакцию при пониженной температуре вводится
бутилен. По окончании алкилирования, когда реакционная смесь
нагревается, происходит реакция с пропиленом.
Метод Universal Oil Products Со. [51, 52]. Исходные продукты
перемешиваются (рис. 66), отношение фтористый водород : углеводо-
род = 1,42 :1, температура реакции 32—43 °C, время контакта 17 с.
Потеря HF достигает 1 кг на 1790 л алкилата. Раньше для этого
метода применяли горизонтальные реакторы Stratco [53] и верти-
кальные цилиндрические реакторы [54, 55]. Самая крупная установка
UOP производит 1300 м3 алкилата в день [50]. Описан метод кон-
троля процесса на установках алкилирования [56—58].
17*
260
13- Алкилирование пропилена
В опытах, осуществленных в Советском Союзе [59—61], были
определены следующие оптимальные условия: отношение (пропилен-]-
+ изобутан) : HF = 1 : 0,6 -4-1; отношение пропилен : изобутан =
= 1 : 12,5, температура реакции 25 °C.
13.2.3. Алкилирование в присутствии
различных катализаторов
В первую очередь следует назвать алкилирование в присутствии
хлористого алюминия. Эта реакция изобутана с этиленом для полу-
чения 2,3-диметилбутана. Так же могут реагировать и смеси этилена
с пропиленом [50]. Метод алкилирования пропилена с изобутаном
не дает никаких преимуществ по сравнению с методом с H2S04
или HF. Действительно, активным катализатором является ком-
плекс из А1С13, НС1 и органического вещества. Запатентованы спе-
циальные комплексные системы керосин — А1С13 — НС1 [62] и
А1С13 — диэтиловый эфир или А1С13 — диметиловый эфир [63].
В литературе часто встречаются сведения о применении трехфто-
ристого бора, чаще всего в виде комплекса; но этот метод, кажется,
еще не осуществлен в промышленном масштабе. Рекомендуются также
BF3 на А12О3 [64] и HBF4 [65]. Эти катализаторы применяются
главным образом для того, чтобы сначала диспропорционировать
смесь из этилена, пропилена и бутилена, а затем провести алкилиро-
вание. Запатентованы также такие катализаторы, как комплексные
соединения BF3-H2O [66], BF3 — алкилфторид [67], специальные
комплексные системы с фосфорными кислотами (BF3, H3P04), BF3-
•Н4Р2О7 [68—70]) и, наконец, фторбораты цинка и меди [64]. Раз-
работаны специальные катализаторы для низкотемпературного ал-
килирования. Это катализаторы на основе железа, BF3 и HF(FeF2-
•BF3) [71-73].
Таблица 27
Катализаторы алкилирования
Катализатор Характеристика процесса Лите- ратура
Бромистый алюминий Температура реакции, 30—70 °C [74]
Бромистый алюминий Алкилирование сопровождается изо- 175]
меризацией
В2О3 на А12О3 Температура реакции 200 °C, под [76]
давлением
Платина на А12О3 250 °C, 7 кгс/см2 177]
Вода 455 °C, 32 кгс/см2 [78]
Натриевые соли диолефинов или — [79]
ацетиленов и пр.
у-О б лучение Температура реакции 300—400 °C, [80]
55 кгс/см2
у-Облучение 36 ?С, 30 кгс/см2 [81]
13-3- ^Оптимальные условия алкилирования
261
В табл. 27 перечислены и другие катализаторы, также предла-
гаемые для алкилирования.
13.3. ОПТИМАЛЬНЫЕ УСЛОВИЯ АЛКИЛИРОВАНИЯ
Хотя реакция между олефином и изобутаном в присутствии кис-
лого катализатора проходит очень быстро [82, 83], время контакта
в промышленных условиях выдерживают в пределах 5—20 мин.
Это делается для того, чтобы добиться более высокого отношения
изобутан : олефин в кислой фазе и, таким образом, не допустить
полимеризации олефина или образования сложных эфиров. После
предварительного насыщения кислой фазы изобутаном (раствори-
мость 0,1% в 99,5%-ной H2S04 [84], в промышленных условиях рас-
творимость 0,4%) можно сократить время реакции, особенно при
турбулентном перемешивании в трубчатых реакторах.
Растворимость изобутана в HF выше, чем в H2S04 и, наоборот,
олефины хуже растворяются в HF, чем в H2S04. Ниже показана
взаимная растворимость изобутана и HF:
Температура, °C...........
Растворимость, %
изобутана в HF . . . .
HF в изобутане . . . .
0 5 10 15 50 60
1,95 2,1 2,2 2,4 3,6 4,0
0,22 0,25 0,29 0,32 0,87 1,2
Относительная скорость абсорбции некоторых олефинов в кон-
центрированной серной кислоте:
Этилен........ 0,002 Бутен-2........ 2
Пропилен .... 0,6 Изобутилен . . . 30—40
Бутен-1........ 1
Для обеспечения хорошего контакта между компонентами реак-
ции, высокой скорости циркуляции и хорошей теплопередачи тре-
буется интенсивное перемешивание.
Качество полученного алкилата будет лучше, а расход кислоты
меньше, если работать с эмульсией углеводорода в кислоте, а не
наоборот. Обычно при низких температурах получаются алкилаты
лучшего качества. Реакции алкилирования в присутствии H2S04
особенно чувствительны к температуре. Оптимальный температурный
предел от 5 до 13 °C, при более низких температурах повышается
вязкость H2S04, при более высоких увеличивается расход H2SO4,
а качество алкилата ухудшается. Оптимальные температуры алкили-
рования с HF от 27 до 43 °C.
Для алкилирования пропилена лучше всего применять высоко-
концентрированную H2S04 [87]. HF целесообразно вводить в кон-
центрации 86—91%.
262
13- Алкилирование пропилена
13.4. ПРИМЕНЕНИЕ АЛКИЛАТОВ
Алкилаты из олефинов и изобутана применяются в основном
в качестве высокооктановых топлив. Потребление этого продукта
постоянно растет; в этой области наблюдается все усиливающаяся
тенденция к замене полимер-бензина алкилированным бензином.
ЛИТЕРАТУРА
1. F. Albright, Chem. Eng., 73, № 14, 119 (1966),
2. F. Albright, там же, 73, № 17, 143 (1966).
3. F, Albright, там же, 73, № 19, 205 (1966).
4. F. Albright, там же, 73, № 21, 209 (1966).
5. Oil Gas J„ 64, 58 (1966).
6. Chem. Eng., 73, № 13, 98 (1966); Oil Gas J., 65, № 16, 130 (1967).
7. F. E. Frey, H. J. H e p p, Ind. Eng. Chem., 28, 1439 (1936).
8. G. G. О b e r f e 11, F. E. Frey, Refiner and Nat. Gasoline Manufac»
turer, 18, 486, 503 (1939).
9. H. J. H e p p, F. E. Fre y, Ind. Eng. Chem., 45, 410 (1953).
10. J. E. К n a p, E. W. С о m i n g s, H. G. Dr ickamer, Ind. Eng.
Chem., 46, 708 (1954).
11. Голл. пат. 60768, 1948.
12. Пат. США 2519072, 1950.
13. Пат. США 2909581, 1959.
14. Oil Gas J., 54 (21.3), 166 (1955).
15. Пат. США 2417251, 1947.
16. Англ. пат. 591544, 1948.
17. Chem. Eng., 61, № 1, 114 (1954).
18. Petroleum Refiner, 30,’ № 9, 151 (1951).
19. J. О. I v e r s о n, L. Schmerling, Advances in Petroleum Chemistry
and Refining, New York, 1958.
20. Пат. США 3227774, 3227775, 3234301, 1966.
21. Oil Gas J., 65, № 1, 48 (1967).
22. Пат. США 3083247.
23. Пат. США 2967208, 1961.
24. J. Е. Hofmann, J. Org. Chem., 29, 1497 (1964).
25. S. R. S t i 1 e s, Petroleum. Refiner, 34, № 2, 103 (1955).
26. E. C. Oden, W. J. В u s c h jr-, G. R. J ones, Petroleum Refiner, 29,
№ 4, 103 (1950).
27. E. С. О d e n, W. J. В u s c h, Ind. Eng. Chem., 42, 2524 (1949).
28. Пат. США 2859259, 1958.
29. Hydrocarbon Processing Petroleum Refiner, 43, № 9, 165 (1964).
30. S. R. Stile s, Petroleum Refiner, 34, № 2, 103 (1955).
31. A. R. G о 1 d s b y, D. H. P u t n e y, Petroleum Refiner, 34, № 9, 148
(1955).
32. Пат. США 3027242, 1962.
33. D. H. P u t n e у, О. W e b b, Petroleum Refiner, 38, Ks 9, 166 (1959).
34. Пат. США 3175023, 1965; пат. США 3121126, 1964.
35. С. В. L i n n, A. V. G г о s s e, Ind. Eng. Chem., 37, № 10, 924 (1945).
36. F. E. F г e y, Chem. Met. Eng., 50, № 11, 126 (1953).
37. D. P. T h о r n t о n jr., Petroleum Processing, 6, 488 (1951).
38. C. G. G e r h о 1 d et al., Trans. Am. Inst. Chem. Eng., 39, 793 (1943).
39. G. M. W e b b, W. S. G a 1 1 a w a y, Petroleum Refiner, 33, № 9, 201
(1954); Oil Gas J., 53, № 46, 166 (1955).
Литература
263
40. J. A. S с о 11, R.M. Coop er, Oil1 Gas J., 44, № 47, 204 (1946); Pet-
roleum Refiner, 26, 180, 280, 354 (1947).
41. F. L. R es en, Oil Gas J., 56 (24.11), 73 (1958).
42. Пат. США 2430333, 1947.
43. Пат. США 2436483, 1948.
44. Natl. Petrol. News, 34, R-131 (1942).
45. Пат. США 3204011, 1965.
46. Пат. США 3080438, 1963.
47. Пат. США 3169152, 1965.
48. Пат. США 3169153, 1965.
49. Пат. США 3233007, 1966.
50. Hydrocarbon Processing Petroleum Refiner, 43, № 9, 165 (1964).
51. Пат. США 3249650, 1966.
52. К. F о г г у, С. S с h г a g е, Hydrocarbon Processing Petroleum Refiner,
45, № 1, 107 (1966).
53. A. R. Orr, Petroleum Refiner, 34, № 7, 165 (1955).
54. E. К. J ones, Petroleum Refiner, 38, № 7, 157 (1959).
55. G. W. G. McDonald, Hydrocarbon Processing Petroleum Refiner, 41,
№ 3, 137 (1962).
56. Пат. США 3002818, 1962.
57. Пат. США 2881235, 1959.
5$. Пат. США 2416395, 1947.
59. Н. И. П л о т к и и а, В. Г. П л го с м и н, Изв. Сибирского отделения
АН СССР, № И, 17 (1958).
60. В. Г. Телегин и др., Труды ВНИИ нефтехимических процессов, № 3,
187 (1960).
61. Н. И. П л о т к и н а, Г. В. П л го с м и н, Труды Института химии АН
СССР, Уральск, филиал, № 4, 59 (1960).
62. Пат. США 2488602, 2488603, 1949.
63. Пат. США 2897248, 1959.
64. Пат. США 2945907, 1960.
65. Пат. США 3236912, 1966.
66. Cl. S р е г a n t a, Gh. Andrescu, Petrol, si Gaze, Bukarest, 7, 639 (1956).
67. Пат. США 2653982, 1953.
68. А. В. T о п ч и e в, Я. М. П а у ш к и н, ДАН СССР, 62, 641 (1948).
69. А. В. Т о п ч и е в, Я. М. П а у ш к и н, Нефтяное хоз., 25, № 6, 54 (1947).
70. А. В. Топчиев и др., ДАН СССР., 58, 815 (1947).
71. D. A. S. 1139821, 1962.
72. Пат. США 2830958, 1958.
73. Пат. США 2886535, 1959.
74. Пат. США 2539350, 1951.
75. Англ. пат. 861797, 1961.
76. Пат. США 2972642, 1961.
77. Бельг, пат. 626447, 1963.
78. Пат. США 2460719, 1949.
79. Пат. США 2834818, 1958.
80. Р. J. L и с с h е s i, С. Е. Н е a t h, J. Am. Chem. Soc., 81, 4770 (1959).
81. Англ. пат. 861183, 1961.
82. J. E. Hofmann, A. Schriesheim, J. Am. Chem. Soc., 84, 953
(1962).
83. J. F. M о s b y, L. F. A 1 b r i g h t, Ind. Eng. Chem. Product Res. Deve-
lopment, 5, 183 (1966).
84. C. R. C u p i t et al., Petro/Chem. Engineer, December 1961, S. 47; Januar,
1962, p. 49.
85. H.S. Davis, J. Am. Chem. Soc., 50, 2780 (1928).
86. H. S. D a v i s, Schuler К., J. Am. Chem. Soc., 52, 721 (1930).
87. Пат. США 3133975, 1964.
14. КУМОЛЬНЫЙ СИНТЕЗ ФЕНОЛА
Синтез фенола и ацетона из бензола и пропилена через кумол
и его гидроперекись [1—13] был впервые описан Хоком [14]. Пер-
вая промышленная установка мощностью 8000 т была пущена
в 1953 г. на фирме Shawinigan Со. в Монреале (Канада). С тех пор
появилось много новых установок.
14.1. алкилирование бензола пропиленом
Кумол (изопропилбензол) был впервые получен в 1841 г. [20]
при перегонке n-куминовой кислоты с гидроокисью кальция или
бария. До 1942 г. кумол не имел промышленного значения, но во
время второй мировой войны его стали применять как компонент
авиационного бензина с высоким октановым числом и очень низкой
температурой затвердевания (—96 °C).
В лабораторных условиях кумол получали взаимодействием
бензола с изопропилхлоридом [21] или изопропилбромидом [22]
по реакции Фриделя—Крафтса в присутствии хлористого, алюми-
- ния, бромистого алюминия или алюминиевых стружек в струе НС1.
При промышленном синтезе тоже исходят из пропилена, который
вводится в реакцию с бензолом тоже по Фридель—Крафтсу в при-
сутствии различных катализаторов. Эта реакция была впервые
описана Берри [23], работавшим при 70 °C в присутствии А1С13.
Для промышленного синтеза кумола предлагаются два основных
пути:
1) алкилирование в жидкой фазе в присутствии А1С13, H2SO4
или HF;
2) алкилирование в паровой фазе на твердых катализаторах
фосфорная кислота — кизельгур [16].
14.1.1 . Алкилирование в жидкой фазе
В промышленности алкилирование бензола пропиленом в жидкой
фазе обычно осуществляется в присутствии серной кислоты [24—
30]. Ниже описан процесс работы на установке фирмы Petroleum
Ind. Maatschappij. Используется пропилен, не содержащий этилен,
что необходимо во избежание образования этилсерной кислоты.
Бензол же с содержанием незначительного количества тиофена
вполне пригоден.
14.1. Алкилирование бензола пропиленом
265
При температуре 20 °C лучше всего применять 90%-ную кислоту,
при 40 °C — 88%-ную, а при 50 °C —86%-ную. Кислоту с концент-
рацией свыше 90% не следует применять, так как в этом случае
возможно сульфирование бензола — единственная побочная реак-
ция при алкилировании.
Для обеспечения жидкой фазы алкилирование проводится под
давлением 11,5 кгс/см2 при 30—40 °C. Мольное отношение про-
пилен : бензол = 1:5, объемное отношение углеводород : H2SO4 =
= 1:1. При интенсивном перемешивании алкилирование оканчи-
вается через 20—30 мин, конверсия пропилена достигает 100%.
На 1 объем H2SO4 можно получить 12—16 объемов сырого кумола.
При промышленном синтезе кумола смесь пропан — пропилен
вводится в эмульсию бензола и серной кислоты и одновременно
непрерывно удаляется некоторое количество алкилатной эмульсии.
После отделения серной кислоты, которая большей частью снова
подается на алкилирование (регенерируется только часть серной
кислоты), это количество алкилатной эмульсии заменяется новой
серной кислотой.
Сырой алкилат промывается 5% раствором едкого натра и затем
в присутствии этого раствора отгоняется пропан (170 °C, 14 кгс/см2).
Для омыления сложных эфиров серной кислотой (из пропилена
и серной кислоты) требуется высокая температура. Во второй колонне
под давлением 1,7 кгс/см2 отгоняется бензол и, наконец, в третьей
колонне получают чистые фракции кумола. Ниже приведены ре-
зультаты, полученные на промышленной установке [24]:
Исходные продукты:
Бензол (21,3% свежего, 42>2% циркуляцион-
ного), вес. %.......................... 63,5
Фракция пропан —пропилен, вес. % . . . . 34,7
Концентрация Н2§04, %....................88—90
Концентрация непрерывно добавляемой
H2SO4, %............................... 90
Условия реакции:
Мольное отношение бензол : пропилен
первый реактор.........................6,2:1
второй реактор.......................5,2:1
Время контакта, мин
первый реактор......................... 35
второй реактор.......................... 30
Объемное отношение смеси H2SO4 : углеводо-
род .................................... 1:1
Температура, °C.............-............30—40
Давление, кгс/см2.....................: . 11,5
Результаты алкилирования;
Конверсия пропилена, %................... 99
Выход сырого кумола, объем/объем 90%-ной
H2SO4 ...................................10—12
266
14. Кумольный синтез фенола
Содержание H2SO4 в израсходованной кисло-
те, % .................................81—82
Содержание воды в кислоте, %.............11—12
Содержание чистого кумола в сыром кумоле,
% ...................................... 95
Алкилирование бензола пропиленом можно осуществить и в при-
сутствии А1С13 при нормальном или повышенном давлении [31].
Исходным продуктом и здесь может служить тиофенсодержащий бен-
зол [32]. Для достижения оптимальных величин конверсии необ-
ходимо работать в несколько влажной среде. При использовании
А1С13 можно отметить следующие недостатки:
1) исходная смесь пропан — пропилен не должна содержать
этилена;
2) образующийся алкилат имеет высокую степень диспропор-
ционирования и изомеризации;
3) при работе в присутствии небольших количеств влаги обра-
зуется НС1, следствием чего является усиление коррозии и увели-
чение расхода катализатора;
4) реакционный продукт и отработанные газы нужно промывать
щелочью.
В присутствии А1С13 алкилирование осуществляется труднее,
чем в присутствии H2SO4. Имеется целый ряд предложений по улуч-
шению технологии и увеличению выхода кумола [33—36], а также
по удлинению срока службы катализатора [37].
Несмотря на указанные трудности, способ с использованием
хлористого алюминия находит все более широкое применение (рис. 67).
Реакция происходит практически без давления при 50—70 °C в реак-
ционных башнях высотой 15 м. В этих условиях имеет место ката-
литическое переалкилирование, поэтому высокоалкилированные про-
дукты целесообразно снова возвращать в процесс. Оптимальное
отношение бензол : пропилен составляет примерно 4,5 4-4,7:!, выход
98% в расчете на бензол, расход катализатора 0,025 г/кг кумола.
Если нужно получить полиизопропилбензолы, то в промышлен-
ных условиях чаще применяют хлористый алюминий, так как при
этом увеличивается выход полиалкилбензолов [38—40]. Фракция
диизопропилбензола содержит преимущественно м- и «-соединения.
Оба зти соединения можно перевести в изо- [42] и терефталевую
кислоту [42—47]. Это делается путем непрерывного окисления
воздухом или окислителями. 1,3,5-Триизопропилбензол может быть
превращен во флороглюцин (через тригидроперекись путем ки-
слотного расщепления) [48, 49]. Аналогично можно из 1,3- и 1,4-
диизопропилбензола получить резорцин [50] и гидрохинон [51].
Хлористый алюминий годится и для того, чтобы перевести поли-
изопропилбензолы в кумол реакцией диспропорционирования после
добавления бензола [52, 53].
14.1. Алкилирование бензола пропиленом
267
Axuiopodpo____
ОН HOUIDUUOQ 4
268
14. Кумольный синтез фенола
Энергия активации ионного алкилирования, которая в отсутствие
катализаторов составляет 21,4 ккал/моль [54], в присутствии А1С13
достигает в зависимости от конверсии 3,4—4,3 ккал/моль [55].
Фтористый водород также является хорошим катализатором
алкилирования. В его присутствии можно употреблять пропилен
с содержанием этилена, так как последний значительно более инер-
тен. Например, при алкилировании бензолом при комнатной тем-
пературе и нормальном давлении в присутствии фтористого водо-
рода смесь, содержащая наряду с этаном и метаном 19% пропилена
Рис. 68. Схема получения кумола из бензола и смеси пропан-пропилен:
1 — колонна для щелочной промывки; 2 — колонна для водной промывки; 3 — ап-
парат для катализа; 4 — колонна для удаления пропана; 5 — бензольная колонна;
в — колонна для очистки; 7 — емкость для сырца.
и 77% этилена, дает только изопропилбензол. Если работают под
давлением 20—35 кгс/см2, то остаточный газ может превратиться
в этилбензол [56—61]. Реакция проходит по первому порядку [62].
14.1.2 . Алкилирование в паровой фазе
Алкилирование бензола пропиленом в паровой фазе можно про-
водить при тех же условиях, что и олигомеризацию пропилена,
применяя фосфорную кислоту в качестве катализатора.
Во время второй мировой войны большая часть кумола в США
производилась по этому методу, разработанному Universal Oil
Products Со. и перенятому другими фирмами (рис. 68). Метод
применяется при олигомеризации пропилена и использует в ка-
честве катализатора фосфорную кислоту на носителях [24, 63—
66]. В данном случае этот метод пригоден потому, что для получения
авиационного бензина не нужен очень чистый кумол; последний
можно использовать вместе с трудноотделяемыми побочными про-
дуктами.
Для алкилирования в паровой фазе применяется катализатор
из 62—65% Р2О5 и 25% SiO2 (на кизельгуре или асбесте) [67].
14,1. Алкилирование бензола пропиленом
269
'%
При условии добавления небольших количеств воды для гидратации
катализатора продолжительность его жизни достигает 700 ч; 1 кг
катализатора дает около 750—800 л кумола. Исходный бензол
и пропилен должны быть очищены от серы.- Содержание тиофена
в бензоле допускается до 0,14%. Бензол предварительно очищается
серной кислотой.
Ниже приведены условия работы и результаты, полученные
на промышленной установке для алкилирования бензола пропиленом
в газовой фазе.
Исходные продукты:
Бензол (17,98% "свежего, 34,56% циркуляционного), % 52,54
Фракция пропан—пропилен (содержание пропилена
20%), %............................................ 47,46
Мольное отношение бензол: пропилен (для разбавлен-
ного пропилена выше)..................................3,05 :1
Вода для гидратации катализатора, вес. %............ 0,06
Условия реакции:
Температура, °C...................................... 255
Эффективность катализатора, л продуктами • л)........ 3,5
Давление, кгс/см2.................................... 25
Результаты алкилирования:
Конверсия пропилена, %................................. 94
Содержание чистого кумола в сыром кумоле, % .... 88,3
Выход чистого кумола, л/л бензола................... 1,54
Выход кумола, л/кг катализатора...................... 780
И при этом методе взятый пропилен может содержать этилен,
который в данных условиях почти не реагирует.
Оптимальная температура реакции 250 °C, давление 28—35 кгс/см®.
При реакции выделяется 23,4 ккал/моль тепла, конверсия про-
пилена достигает 95%.
По методу Shell Oil Со. очищенный бензол и смесь пропан —
пропилен смешиваются друг с другом и через испаритель пода-
ются в реактор. Максимальная температура реактора 300 °C, он
работает под давлением 18—28 кгс/см2. Тепло реакции отводится
с помощью наружного охлаждения. Выходящие пары после охла-
ждения подаются для удаления пропана в колонну, которая рабо-
тает при 200 °C и 12 кгс/см2. Продукт из нижней части колонны
попадает в колонну для отгонки бензола, а оставшаяся часть про-
дукта подается в колонну для перегонки и получения чистого ку-
мола.
Подробное исследование механизма реакции проведено Колес-
никовым и сотрудниками [68].
270
14. Кумольный синтез фенола
14.1.3 . Алкилирование в присутствии
различных катализаторов
Сравнительные опыты по алкилированию с различными катали-
заторами [131—135] показали, что по сравнению с А1С13, H2SO4,
А1С12 • HSO4, А1С12 • Н2РО4 и BF3 • Н2О самый активный ката-
лизатор — это BF3 • Н3РО4. В других опытах [136] было найдено,
что при тех же условиях самым активным соединением является
А1С13.
Пайнес [137, 138] показал, что алкилирование бензола пропи-
леном возможно и без катализаторов; процесс проводили при 350—
500 °C и 70 кгс/см2 в медной трубке.
Для того чтобы вернуть в процесс полиизопропилбензолы, обра-
зующиеся при получении кумола, их вместе с бензолом подвергают
диспропорционированию. Подобная реакция осуществима в при-
сутствии обычных катализаторов алкилирования: А1С13 [139—
141], H2SO4 [141], Н3РО4 [141], HF [142], силикат алюминия
при 400 °C [143, 144] и силикат Al, Mg или Zr [145]. Были про-
ведены исследования по определению равновесия' между бензолом,
кумолом и полиизопропилбензолами [146]. Термодинамические ис-
следования алкилирования бензола пропиленом [148] показали,
что при низких температурах образование диизопропилбензола до-
минирует над образованием кумола, только при температурах выше
220—230 °C условия становятся противоположными (см. также
аналогичные исследования с толуолом [147]).
В табл. 28 дан обзор описанных в литературе катализаторов
для получения кумола.
Т а б л и ц а 28
Катализаторы, применяемые при получении кумола
Катализатор Характеристика процесса Литература
Н3РО4 Алкилирование в жидкой фазе [69]
BF34-HF 40—45 9С, давление [70]
BF3 + изопропиловый 30 ?С [71]
спирт
BF3 45 9С, 23 кгс/см2 [72]
BF3-1,6H2O 45 9С [73]
BF3 на у-А12О3 121 ?С, 37,4 кгс/см2, особенно при- годен для этиленсодержащего про- пилена [74]
BF3 на модифицированном —. [75]
А12О3- SiO2
BF3 на SiOj-MgO 20—250 ?С, 200 кгс/см2 [76]
BF3 на у- или 0-А12О3 Модификация А12О3 с помощью BF3 [77]
14.1. Алкилирование бензола пропиленом
271
'Продолжение табл. 28
Катализатор Характеристика процесса Литература
BF3 на А12О3 Модификация соединениями серы (на- 178] -
пример, тиофеном)
BF3 с ZrO2; А12О3 + —. 179]
~h ZrO2 4“ AlgVa 4"
+ BnOo 1
BF3 Процесс протекает в присутствии [80]
воздуха и пентана
bf3-h3po4 — [81-83]
bf3-h2po3f — [84]
bf3.h20 (11,3%) + — [85]
+ BF3-H3PO4
BF3+ H2SO4 —. [86]
Борофосфорная кислота 275 РС, 175 /кгс/см2 [87]
Металлический Al Процесс протекает в присутствии [88]
гептилбромида
A1C13 + этилхлорид 200 РС, давление [89]
A1CL в нитрометане 0-40 °C- [90]
A1C1„.H2PO4 Очень эффективный катализатор [91]
А1С13 + H3PO4 — [92]
A1C13 + триэтилалюминий — [93]
Хлористый алкилалюми- В присутствии следов кислорода [94] -
ний + TiCl4 или ZrCl4
Алкилалюминийхлорид В присутствии этилена [95]
Комплекс А1Вг3 — угле- — [96]'
водород
A12(SO4)3 на SiO2 120—170 РС [97]
Триизобутилалюминий + 68—78° С; жидкофазный метод в гек- [98]
+ TiCl4 на кислом А12О3 сане
A1,OS, SiO2 или ZnO в при- 300-350 °C, 100 кгс/см2 Н2 [99]
сутствии НС1
А1РО4 на алюмосиликате — [100]
А12О3 Активация с помощью СС14; 1335 С, I101J
3,5 кгс/см2
А13О3 Модификация BF3 и соединениями [102J
серы (например, тиофеном)
Алюмосиликат 150—200 РС, давление [103]
350-450 рС [104—107]
— [108—109]
300 РС, 10 кгс/см2; алкилирование [110]
в псевдокипящем слое
20-80 °C [111-112]
SiO2-Al2O3 и НС1 250—260 °C, давление [ИЗ]
Бентонит — [114]
Монтмориллонит — глино- — [115]
зем
МонтмОриллбнит или фул- Предварительная кислая обработка . [116]
лерова земля
Силикат алюминия Активирование добавкой 13% ред- [Н7]
ких земель; 200—350 РС
272
14. Кумольный синтез фенола
Продолжение табл. 28
Катализатор Характеристика процесса Литература
MgCl, или FeCl„ на А1„О3 300—350 РС, 100 кгс/см2 Н2 [118]
FeCl3 Комнатная температура [119]
Окиси или сульфиды воль- фрама + фосфорная или кремневая кислота 260—280 РС, давление; в присут- , ствии 0,1—1% воды [120]
Окись вольфрама с добав- 280 9С, 48 кгс/см2 [121]
кой других окисей (ZnO, TiO2, Р2О5, Со3О4)
ZnCl2 на А12О3 и других 200 ?С [122, 123]
носителях
Смесь из TiCl2 и TiCl3 на А12О3 — [124]
Галогениды тяжелых ме- 50 рС; жидкофазный метод [125]
таллов в солянокислом растворе
Mg(II2PO4)2, Са(Н2РО„)2, 180—350 9С, давление [426]
Zn(H2PO4)2, Cu2P2O7 или Ag4PaO7 на кизельгуре [127]
N а-цеолит Активирование хлористым пропилом
Са-цеолит 250—300 9С [128]
Кислая ионообменная смо- ла Основа — сульфированная смола или сульфированный уголь [129]
Пятифтористый фосфор — ИЗО]
14.2. ОКИСЛЕНИЕ КУМОЛА
Первые тщательные исследования окисления кумола [149] —
второй стадии синтеза фенола и ацетона из кумола — были прове-
дены в 1944 г. При действии ультрафиолетового света [14, 150]
удалось превратить кумол на 7 % в а-гидроперекись кумола (сокра-
щенно КМГП). Эти опыты были затем развиты и доведены до про-
мышленной стадии фирмами Distillers Go., Ltd. (Эдинбург) и Her-
cules Powder Со. (Вилмингтон). В промышленность внедрены в ос-
новном два способа: окисление в эмульсии и окисление в чистом
кумоле.
14.2.1. Окисление кумола в эмульсии
Предназначенный для окисления кумол должен быть максимально
очищен от тиофена и изопропилтиофена. Поэтому в промышленности
употребляется 99,9%-ный кумол.
Окисляется только часть кумола. Непрореагировавший кумол
отделяется и непрерывно возвращается в реакцию. Предварительно
он подвергается гидрирующей рафинации с целью гидрирования
14.2. Окисление кумола
273
образовавшегося а-метилстирола. Для облегчения окисления свеже-
введенный кумол тоже рафинируется. В данном случае рекомен-
дуется обработка раствором едкого натра [151,152, 155, 267], едкого
кали или твердой содой [153, 154], перегонка над натрием [151],
обработка активированной окисью алюминия [155], а также комби-
нированная кислая и щелочная промывка [156].
Предварительно очищенный кумол окисляется до содержания
КМГП 25%. Окисление проводится в резервуарах с мешалкой [157,
158] или в реакторных башнях высотой 8—10 м воздухом, вдува-
емым в агрегаты снизу, при температурах 110—130 и под давле-
нием 5—10 кгс/см2. К реакционной смеси нужно добавлять неболь-
шое количество щелочи, чтобы воспрепятствовать разложению КМГП
под действием кислот (например, муравьиной, бензойной), следы
которых образуются во время окисления. Опасность взрыва в газо-
вой камере на окислительной установке (пределы взрываемости
смеси паров кумола с кислородом 1,5—43 объемн. % кумола) устра-
няется поддержанием содержания водяного пара в газовой фазе
10—20% [159, 160]. Количество воды в окислительном растворе
в 2—3 раза превышает количество кумола. К водному раствору
добавляются эмульгаторы, чтобы окисление протекало в эмульсии.
Эти эмульгаторы резко ускоряют окисление и увеличивают выход
[161]. Особенно хорошо действуют анионактивные эмульгаторы:
стеарат натрия [157, 162—168], рицинолеат натрия [165], каприлат
натрия [167], бентонит [169, 265], теопол [157], лаурилсульфат
натрия и неионные эмульгаторы [170]. Катионактивные эмульгаторы,
напротив, сначала тормозят окисление и ускоряют его только после
преодоления начальной стадии реакции (индукционного периода)
[171]. Кумол можно окислить и без добавления специальных эмуль-
гаторов, так как введенные щелочи (разбавленный едкий натр [172—
174], сода [175, 176, 269], гидроокись кальция [177, 178], окись
кальция [275]), окиси щелочных или щелочноземельных металлов,
а также щелочные соли слабых кислот [178, 180] сами оказывают
эмульгирующее действие.
Величина pH раствора должна быть в пределах 8,5—10,5, так
как при более высоких величинах выход КМГП уменьшается вслед-
ствие разложения, а в нейтральной или кислой области самооки-
сление идет слишком медленно. Большое значение для выхода имеет
отношение кислотной фазы к углеводородной части. Скорость реак-
ции повышается с увеличением степени разбавления кумола. С дру-
гой стороны, при этом уменьшается выход в единицу времени на
единицу объема. Как уже было сказано, оптимальное отношение
составляет 3 : 1 [158, 163].
По окончании окисления реакционная смесь подается в отстой-
ник и разделяется. Углеводородный слой или направляется непо-
средственно на кислое расщепление, или предварительно концентри-
руется в присутствии соды, пока содержание КМГП не достигнет
18 Заказ 399
274
14. Кумольный синтез фенола
70—90%. Концентрирование можно проводить путем одно- или много-
ступенчатой ректификации под вакуумом или атмосферным давле-
нием, в обычной колонне или выпариванием в тонком слое. Пере-
гонка при отсутствии кислоты препятствует преждевременному рас-
щеплению гидроперекиси.
14.2.2. Гомогенное окисление кумола
Гомогенное окисление кумола осуществляется при 100—130 °C;
добавляется небольшое количество содового раствора или твердой
соды, чтобы уловить кислые побочные продукты [181, 182] и создать
атмосферу водяных паров. Часто окисление проводят при различ-
ных температурах, например при 120 °C (до 10% КМГП), при
117 °C (до 15% КМГП), при 115 °C (до 20% КМГП) и при 110 °C
(до 27% КМГП) [183], давление при этом составляет 4—6 кгс/см2.
Предлагается также работать при постоянной температуре и пони-
жающемся давлении кислорода [184]. Быстрое окисление является
достоинством этого метода [185].
На фирме Phenolchemie GmbH [5] окисление проводят без до-
бавления щелочи при 130 °C и 6 кгс/см2 до концентрации КМГП
15%, причем устанавливается величина pH 3,5—4,0. Короткое
время контакта в значительной степени препятствует образованию
побочных продуктов [186]. Процесс осуществляется в реакторе
с мешалкой емкостью 5—10 м3. Продукт реакции концентрируется
при 90—100 °C и давлении 90 мм рт. ст. до 20% (отгонка формаль-
дегида и муравьиной кислоты) и при 90—100 °C и давлении 500 мм
рт. ст. до 60%. После освобождения главного продукта второй сту-
пени от захваченной КМГП его можно сразу же возвратить в про-
цесс [187].
14.2.3. «Сухое» окисление кумола
Третий метод [188—190] разработан Bataaf Petroleum Maats-
chappij. Очищенный кислотой кумол окисляется кислородом в мед-
ных башнях при 120—130 °C. Медь (в форме колец) при специальных
условиях протравливается азотной кислотой. После этого медь
действует как ускоритель окисления (уменьшение индукционного
периода) и одновременно как стабилизатор для образующейся КМГП.
Охлаждение не требуется, так как тепловой эффект реакции соста-
вляет всего 100 ккал на 1 кг образовавшейся КМГП.
Подобный метод [191, 192] был опробован в лабораторном мас-
штабе. Окисление проводилось в присутствии меди, активирован-
ной дымящейся HNO3 или О2 при 200—300 °C (спирали, опилки,
медь Ренея), а также окисей или солей меди.
14.2. Окисление кумола
275
14.2.4. Ускорение окисления кумола добавками
Перекисные соединения добавляются с целью сокращения или
полной ликвидации индукционного периода, имеющего место при
самоокислении кумола без добавок, особенно при окислении чис-
того кумола. Принято добавлять 0,2—1% КМГП из предыдущих
загрузок [193—198], еще лучше вводить натриевую соль КМГП
[193, 199] в количестве 0,05—0,5%. Можно также работать в при-
сутствии стабилизирующих добавок, например тпретп-бутилата на-
трия [195], BaSO4 [196], карбоната или стеарата натрия [197],
формиата, оксалата или бензоата щелочного или щелочноземель-
ного металла [198].
Кроме КМГП в качестве добавок предлагаются ВаО2 [200],
Н2О2 [201—203], а также озон [204, 205], однако эти соединения
наряду с окислением ускоряют и разложение, так что полезное
действие эти добавок находится под сомнением.
В табл. 29 дан обзор добавок, рекомендуемых для ускорения
окисления и увеличения выхода КМГП. Неизвестно, получили ли
эти добавки какое либо промышленное значение, поскольку они
в той или иной мере наряду с окислением кумода ускоряют и раз-
ложение КМГП.
, Таблица 29
Добавки, вводимые при окислении кумола
Добавка Характеристика действия добавки Литература
Стеарат кобальта Сравнительно сильно ускоряет раз- ложение, образуется в больших количествах а-кумиловый спирт [164J [194, 206]
Нафтенат или стеарат церия Лучше, чем стеарат кобальта [207-209]
Стеарат меди — [210]
Нафтенат марганца В основном образуется а-кумиловый спирт (особенно в щелочном рас- творе) В щелочном растворе, незначитель- ное увеличение выхода КМГП [211]
Соли кобальта (II) или (III) [212]
Комплексы типа Хороший эффект [213]
[Co.(NH3)e]X3
Растворимые в кумоле соли — [214]
тяжелых металлов
Окиси металлов, в част- — [215, 274]
ности РЬО2
Фталоцианин Си, Zn, Mg, а также Mn, Fe(II) и Со Сильно ускоряют , окисление и не- значительно — разложение, хуже фталоцианин Mn, Fe (II) и Со (образование а-кумилового спир- та). 217
18*
276
14. Кумольный синтез фенола
Продолжение табл. 29-
Добавка Характеристика действия добавки Литература
Фталоцианин Mg Без разложения окисление с фтало- цианином Си при 100—105 9С, с фталоцианином Со при 80—85 ?С, с фталоцианином Ni при 85—90 ?С [218]
Благоприятное действие, хороший эффект достигается при очистке фталоцианинов [219]
Фталоцианин V Исследования механизма реакции по- казывают, что радикалы, образу- ющиеся при разложении КМГП, действуют как ускорители [220]
Фталоцианин Си Добавка гидроокиси или карбоната щелочного металла ускоряет окис- ление [221, 274];
Фталоцианин Ni Через короткое время катализ затор- маживается адсорбцией перекиси а-метилстирола на катализаторе [222]
Полифталоцианин Си Не лучше, чем мономерный фтало- цианин [223]
Полифталоцианин Си, по- литетрацианоэтилен, по- лиакрилонитрил Органические полупроводники, осо- бенно полиакрилонитрил, действу- ют как слабые катализаторы [224]
NaHCO3 — 226]
СаСО3 Сокращение индукционного периода [227]
Хелатные полимеры Си, Cd, Мп и Ni Хелатные полимеры Go, Fe, Zn неактивны [225]
Cu-Полихелат а-тиоамидов и дитиокарбонат натрия; Мп-полихелат 5,5'-мети- ленбиссалицилальдегида, Ni- и Мп-бискетонхелаты Добавка хелатов со структурой типа энзимов; наиболее эффективный Cu-хелат а-тиопиколинамидодифе- нила [228, 229]
НВт + соль щелочного или щелочноземельного ме- талла и органической кис- лоты, например НВт + + бензоат натрия [230]
НВт + бензойная кислота — [231, 232]
Этилкапронат натрия и дру- гие Na-соли разветвлен- ных жирных кислот — [233, 234]
Мононатриевая соль дикар- боновой кислоты, напри- мер адипинат натрия — [235]
Си- и Ag-соли органических кислот, например ацетат, бензоат, стеарат или наф- тенат серебра, формиат или ацетат меди [236]
F
14.2. Окисление кумола
277
Продолжение табл. 29
Добавка Характеристика действия добавки Литература
Соли слабых кислот пере-
ходных металлов, напри-
мер формиат железа (II)
Соли Си, Hg, Sn, Pb, Bi,
Fe, Co или Ni, например
mpem-бутилбензоат свин-
ца
Соли ртути (II)
Трилон Б
Натриевая соль этилендиа-
минтетрауксусной кисло-
ты
Станнат, плюмбат, висму-
тат, антимонат натрия
Аммиак
Амины, например пиридин
Первичный амин третичного
алкана
Гидроокиси тетраалкилам-
мония
KNO3 на А12О3
КС1О4, КС1О3 и другие
галогениды
Различные неорганические
соли, например NaCl,
BaSO4, СаС12, NaBr,
KNO3, СаСО3
Р-Кетоэфиры, например
сложный ацетоуксусный
эфир
Р-Дикетоны, а-арилкетоны,
например фенилацетон
Фенол
Формальдегид
Метиловый спирт
Азобис(изобутиронитрил)
NH3 стабилизирует КМГП
Лучше, чем другие щелочи
Кальцинирование при 550° С
Фенол должен ускорять окисление
кумола
Формальдегид является антиинги-
битором окисления кумола
Должен действовать лучше, чем до-
бавка КМГП
[237, 2381
[239]
[240]
[241]
[242]
[243]
[244]
[245]
[246]
[247]
[248]
[249]
[250]
[251]
[252]
[253]
[254]
[255]
[256, 283]
14.2.5. Фотохимическое окисление кумола
Первые опыты по фотохимическому окислению были проведены
Хоком [10]. Обобщенные сведения по этой теме дают Говард [257]
и Дюруп [258]. Последний касается также вопросов радиохимиче-
ского окисления.
Скорость окисления увеличивается пропорционально интенсив-
ности облучения в степени 1/2 [258]. Фотохимическое окисление
278
14. Кумольный синтез фенола
происходит только в присутствии сенсибилизаторов, из которых
наиболее эффективны антрахинон и некоторые его производные
(259, 260]. Рекомендуется также TiO2 и окиси других металлов
(261]. Исследования показывают, что выход обычно не увеличи-
вается, повышается только скорость окисления [262].
Были проведены опыты по окислению кумола под действием
у-облучения [263, 264]. Во избежание разложения КМГП окисление
нужно проводить при температуре ниже 70 °C (энергия активации
6,7 ккал/моль).
14.2.6. Исследование процесса окисления кумола
Кучер и другие [265] нашли, что скорость окисления повышается
с увеличением pH и количества воды. Эмульгатор в водной фазе
повышает растворимость кислорода, кумола и КМГП. Реакция
.окисления инициируется в этой же фазе [265, 266]:
RH + O2 —> R.-j-HO2
R* + O2 —> ROO. + RH —> ROOH + R.
Перекисные радикалы, очевидно, образуют комплексы с водой
1268]. Одновременно стеарат натрия увеличивает стабильность
КМГП [267].
Как и при окислении чистого кумола, скорость окисления уве-
личивается по мере повышения давления, но в последнем случае
влияние давления значительно больше. Однако это компенсируется
тем, что скорость окисления в эмульсии априори больше, чем ско-
рость окисления чистого кумола [270—272]. Давление выше
10 кгс/см2 уже не оказывает никакого влияния на окисление. Если
окисление проводится в присутствии металлической меди, то ско-
рость окисления не зависит от давления [188].
Изучение ингибирования реакции окисления показало, что при
окислении в жидкой фазе самым сильным ингибитором является
фенол, при щелочном окислении в эмульсии сильнее действует
п-бензохинон [267], так как фенолят натрия ингибирует значительно
слабее, чем фенол. Кроме того, очень сильно затормаживают оки-
сление изопропилтиофен, гидроксикислоты (например, гидрокси-
бензойные кислоты) [273], а-метилстирол [275], ацетофенон, 2-ме-
тил-2-фенилоксиран [290]. В начале реакции ингибиторы действуют
сильнее, чем позже, когда уже образуется достаточное количество
КМГП [274].
Поскольку разложение КМГП в процессе реакции усиливается,
то при окислении чистого кумола выход КМГП проходит через мак-
симум [275]. Скорость же окисления постоянно увеличивается
^пропорционально концентрации гидроперекиси в степени 1/2 [276]).
Наблюдаемый в начале реакции период ингибирования является
14.2. Окисление кумола
219
функцией чистоты кумола: сначала должны быть разрушены име-
ющиеся следы ингибиторов [277].
Кажущиеся константы скорости (составленные из констант
скорости инициирования, роста и обрыва цепи) окисления кумола
были определены Белтрамом [278]:
Температура, °C............... 100 110 120
К-106
некатализируемая реакция 7,37 13,49 22,1
реакция, катализируемая
NaOH ...................... 7,62 14,30 23,0
Отсюда вычислена кажущаяся энергия активации: при отсут-
ствии катализатора Е = 15,9 ккал/моль, а в присутствии NaOH
Е = 16,3 ккал/моль. Изучение термического разложения КМГП,
происходящего и во время окисления, показало, что при 120 °C
образуются следующие продукты разложения [279]: фенол, ацето-
фенон, кумол, кислоты (среди них муравьиная, бензойная).
В процессе окисления кумола наблюдается еще образование
а-кумилового спирта (а-гидроксиизопропилбензола) из КМГП —
первичного продукта окисления [280]. Для объяснения образования
побочных продуктов составлена схема реакции [281]. Предполагают,
что сначала возникает радикал кумила, из которого и образуются
в дальнейшем побочные продукты [282—283]:
а-кумиловый спирт
Исследования энергии активации Е разложения при различных
условиях дали следующие результаты (для реакции роста цепи
Е = 8,6 ккал/моль [284]):
_ - р
Добавка ккал/моль
Без добавки [284] 30,4
Кумол [285] .... 31
Кумол [286].......... 41
Добавка ккал/моль
Кумол [287] .... 27
Метиловый спирт
[285].............. 31
Стирол [285] .... 20
Агент образования
радикалов [285] • . 6,5
Бензойная кислота
[286]............... 29
280
14. Кумольный синтез фенола
При изучении термического разложения КМГП при 110—160 °C
в бензоле [287J были найдены следующие продукты разложения
(в мол. %):
Двуокись углерода . . 8,0
Метан.................30,0
Этилен.................. 3,0
Ацетофенон............49,0
Бензойная кислота . . 7,4
Дифенил.............. 2,9
а-Метилстирол........ 2,3
Фенол................ 0,9
а-Кумиловый спирт . . 37,0
Вода.................. 60,0
Распад КМГП происходит в соответствии с уравнениями:
Начало
ROOH—> RO- + «ОН
Рост цепи
RO-+RH —> ROH + R.
OH« + RH —> H2O + R.
R-+ROOH---> ROH+RO.
Обрыв цепи
2R---> R-R
В бензоле радикалы почти не реагируют, значительно сильнее —
в кумоле (RH). Влияние температуры на состав продуктов разло-
жения показано ниже (в мол. %):
110 °C 137°С 160 °C
Метан .... 27 39 52
Ацетофенон .... 30 50 63
Бензойная кислота . . . . 0,9 2,4
Дикумил .... 21 26 29
а-Метилстирол .... 0 2,5 0
а-Кумиловый спирт . . . . .... 141 130 106
Вода 31,9 25,6
Изучение разложения КМГП при температурах, при которых
разложение еще не проявляется (60 °C), ив присутствии солей ко-
бальта (II) показало, что это реакция первого порядка [2511, не за-
висящая от концентрации КМГП. Растворитель же влияет на ско-
рость. Под действием солей кобальта КМГП разлагается с образо-
ванием радикалов [288 J.
В водном 0,1 н. растворе H2SO4 скорость разложения в 7 раз
выше, чем в водном 0,1 н. растворе NaOH, и в 60 раз выше, чем
в воде. Добавка тенсида повышает скорость разложения в кислой
и щелочной среде [289].
Действие ингибиторов окисления основывается на их способ-
ности отдавать кислород; благодаря этому улавливаются перекис-
ные радикалы. Опыты по окислению кумола в присутствии антиокси-
дантов [290 , 291] показали, что количество определяемых продуктов
14,3- Разложение гидроперекиси кумола на фенол и ацетон
281
разложения (ацетофенона, кумилового спирта, а-метилстирола и
2-метил-2-фенилоксирана) зависит от интенсивности антиокисляющего
действия.
Карноитский [294] описывает механизм радикального кислого
и щелочного разложения КМГП (при щелочном разложении обра-
зуется а-кумиловый спирт и высвобождается кислород).
При высокотемпературном (500 °C) разложении кумола найдены
следующие продукты разложения: формальдегид, стирол, а- и
Р~метилстирол, бензальдегид, ацетофенон, метилбензилкетон.и вода
[292].
Подробные сведения о применении КМГП приведены в работе
[293].
14.3. РАЗЛОЖЕНИЕ ГИДРОПЕРЕКИСИ КУМОЛА
НА ФЕНОЛ И АЦЕТОН
Метод окисления кумола и кислого расщепления КМГП был
разработан с целью получения фенола. Рентабельность метода
зависит от возможностей сбыта образующегося при этом ацетона,
14.3.1. Промышленные методы разложения
с применением кислот
Разложение КМГП обычно осуществляется концентрированной
серной кислотой. В промышленности применяется несколько мето-
дов (см. рис. 67).
По методу, разработанному Distillers Со. [297] и Hercules Pow-
der Со. [298] раствор КМГП концентрируется перегонкой в вакууме
до 75%, щелочь действует при этом как стабилизатор. Кислое рас-
щепление производится непрерывно в растворе ацетона при 60—
65 °C, теплота реакции (400 ккал/кг КМГП) отводится через кипя-
щий ацетон. Отношение ацетон : КМГП = 1,5 : 1, серная кислота
добавляется в количестве 0,1—2%.
По методу PhenoIchemie GmbH 60%-ную КМГП разлагают
40 %-ной H2SO4 при соотношении концентрат КМГП : кислота =
= 1:4. Температура реакции поддерживается на уровне 40 °C,
время контакта составляет от 90 до 120 с [299]. Кислота после
отделения может быть снова возвращена в реакцию. Органическая
фаза промывается 2% раствором едкого натра [300]. Продукт реак-
ции путем разгонки разделяется на сырой ацетон, кумол и фенол.
Сырой ацетон очищается в специальной колонне после промывки
раствором едкого натра’ [301]. Кумол отгоняется с водяным паром.
Затем для разрушения а-метилстирола проводится очистка гидриро-
ванием при 140 °C и давлении 10 кгс/см2 на никелевых катализато-
рах [302].
282
14. Кумольный синтез фенола
Для удаления окиси мезитила фенол обрабатывается серной
кислотой и после нейтрализации перегоняется [303, 304]. Остаток
сырого фенола крекируется при 270—290 °C, причем после перегонки
можно получить дополнительные количества фенола и а-метилсти-
рола. Для дальнейшей очистки эти продукты снова направляются
на главную ректификационную колонну [305].
В промышленности разложение гидроперекиси кумола осуще-
ствляется только серной кислотой, но в различных вариантах.
Гетерогенное расщепление: а) избытком 10 %-ной H2SO4 под да-
влением 120 °C [157, 306]; б) 60%-ной H2SO4 при 60 °C [307, 308];
в) 20-кратным избытком H2SO4 при 30 °C [309]; г) водяным паром
и 10—25%-ной H2SO4 при 110—120 °C [310]. Гомогенное расщепле-
ние: для этого применяются небольшие количества концентрирован-
ной серной кислоты (0,2—1%); предварительно вводится в избытке
ацетон, чтобы его парами отводить реакционное тепло [311—317].
Для отведения тепла можно вводить в избытке фенол [318—320]
(температура реакции 55—65 °C) или проводить разложение в смеси
фенола с ацетоном [321—323].
Разложение на твердом катализаторе проводят серной кислотой
на пемзе (температура реакции ниже 50 °C) [324]. В качестве ката-
лизаторов для кислотного разложения КМГП предложены также:
перхлорная кислота [325], хлористый водород [326], сера, селен,
фосфор или мышьяк (низкая конверсия) [327], двуокись серы [328,
329], политионовые кислоты [330],CuSO4 [331], NiSO4, трихлорметан-
сульфоновая кислота [334], n-толуолсульфокислота, фосфорная
кислота, кислые силикаты [335—338], а также кислые катион-
обменные смолы [183, 195, 256, 339, 340] и цеолиты [341]. При до-
бавлении перекиси водорода можно перевести а-кумиловый спирт
в фенол и ацетон [342 , 343], но этот метод сравнительно дорог.
Математическая модель разложения описана Лопатиным [344].
14.3.2. Обработка продуктов реакции
Для удаления серной кислоты смесь фенола с ацетоном можно
обработать СаСО3 [345—347], основным анионобменным соединением
[348], или слабой органической кислотой [349]. Разложение
и дистиллятивную обработку реакционных продуктов можно про-
- водить непрерывно [350—352], осуществима также экстрактивная
очистка фенола [353—356] и рафинация на силикате алюми-
ния [357].
ч Кумол, полученный при разложении КМГП, можно очищать
рафинирующим гидрированием на системе палладий — уголь [358],
на MoS2 или WS2 [359], а также на кобальт-молибденовом катали-
заторе на А12О3. При этом одновременно улучшается качество фенола
[360, 361]. С целью очистки кумол можно также обрабатывать ед-
14.3. Разложение гидроперекиси -кумола на фенол и ацетон
283
ким натром под давлением [362]. а-Кумиловый спирт можно снова
восстановить в кумол [363].
Вследствие нежелательной конденсации фенола с «-метилстиро-
лом и а-кумиловым спиртом при разложении КМГП образуются
смолы, для удаления которых проводят: а) реакцию остатка с кон-
центрированной серной кислотой и гидрирующее расщепление
при 350 °C и давлении 50 кгс/см2 на кобальт-молибденовом катали-
заторе (носитель А12О3) с образованием фенола и различных угле-
водородов [364—365]; б) сульфирование остатка серной кислотой
и связывание формальдегида катионобменными соединениями [366];
в) термическое расщепление остатка при 240—400 °C с получением
добавочного количества фенола [367].
14.3.3. Влияние кислот на разложение гидроперекиси кумола*
Кислотное разложение КЙГП является’ реакцией первого по-
рядка по отношению к КМГП и одновременно к концентрации водо-
родных ионов [294]:
—d [КМГП]
dt
К2 [Н+]
Этот вывод был подтвержден
причем были определены энергии
жения КМГП:
опытами в
активации
присутствии кислот,
Е кислотного разло-
Кислота Е,
ккал/моль
Хлорная [368]........................ 19
п-ТоЛуолсульфоновая [369]............. 21,3
Бензолсульфоновая [370] ............. —
Серная [368]......................... 17
Серная в ледяной уксусной [371] .... 20*
* В этом случае энергия активации может
быть уменьшена до 16,1 ккал/моль добавлением аце-
тона [371].
Разложение замедляется добавкой воды [322, 371]; ацетон,
ацетофенон и уксусный ангидрид ускоряют его. При разложении
серной кислотой в ледяной уксусной кислоте реакция становится
автокаталитической, начиная от содержания КМГП 0,02 моль и выше.
284
14. Кумольный синтез фенола
14.4. ЭКОНОМИЧЕСКАЯ ОЦЕНКА КУМОЛЬНО-ФЕНОЛЬНОГО
МЕТОДА
Ниже сопоставлены экономические показатели установки Ра-
шит — Хукер на 20 000 т (выход 90%) и кумольно-фенольной уста-
новки такого же масштаба (выход 84%); цены указаны в пфеннигах
за 1 кг:
Куиоль- но-феноль- ный иетод Процесс Рашиг— Хукер
Сырье (бензол, пропилен) 39,5 28.05
Катализаторы, добавки, энергетические расходы 13,1 13,75
Расходы на оплату труда, содержание обору- дования, общие расходы 8,05 9,4
Общие расходы на переработку 60,65 51,2
Кредит на ацетон , 20,15 40,50 —
Списание со счетов и уплата процентов . . . . 10,6 51,1 12,6 63,8
Сравнение на основе американских цен в таких же отношениях
действительно и для Европы. Решающим фактором является спрос
на ацетон и наличие дешевого пропилена (получаемого, например,
в качестве побочного продукта на установках по производству
этилена). Метод Dow представляет интерес только в том случае,
если толуол значительно дешевле бензола.
ЛИТЕРАТУРА
1. Р. W. Sherwo о d, Petroleum Processing, 8, 1348 (1953).
2. Р. W. Sherwood, Petroleum Eng., 30, № 6, 32 (1958).
3. H. H о c k, H. К г о p f, Angew. Chem., 69, 313 (1957).
4. E. G о t o, Kagaku, 10, 541 (1955).
5. H. Kropf, Chem.-Ing.-Techn., 36, 759 (1964).
6. J. Aleman Vega, N. L. Manzanares Bonnail, Rev. cienc.
apl. Madrid, 8, 234 (1954).
7. J. A. О’С о n n о r, Chem. Eng., 58, № 10, 215 (1951).
8. J. P о s p i s i 1, Chemie, Prag., 9, 885 (1957).
9. В. Г. С о л о n e ц к и й и др., Хим. пром., 42, № 7, 509 (1966).
10. Ind. Chemist, 36, № 5, 215 (1960).
11. Н. Н о ck, Erdol und Kohle, 14, 892 (1961).
12. П. Г. С e p г e e в, Б. Д. К р у ч а л о в, Хим. пром., 1957, 201.
13. Б. Д. К р у ч а л о в, Б. И. Г о л о в а н е н к о, Одновременное производ-
ство ацетона и фенола, М., 1963.
14. Н. Hock, S. Lang, Вег. dtsch. chem. Ges., 76, ИЗО (1943); 77, 257 (1944).
15. P. W. Sherwood, Petroleum Refiner, 32, № 1, 97 (1953).
16. P. W. S h e r w о о d, Ind. Chemist, 30, 25, 71 (1954); Erdol und Kohle, 7,
23, 156 (1954).
17. A. T о m i t a, Yuki Gosei Kagaku Kyokai Shi, 14, 20 (1956).
18. E. K. J о n e s, Petroleum Refiner, 33, № 12, 186 (1954).
Литература
285
19. Н. J. S h u i к i fl, N. А. Р о z d n j а к, Bull. Acad. Sci. USSR, Div. Chem.
Sci. 713 (1957) (Engl. Ausgabe).
20. Ch. Gerhardt, A. Cahours, Liebigs Ann. Chem., 38, 88 (1841).
21. C. Radziewanowski, Ber. dtsch. chem. Ges., 28, 1137 (1895).
22. G. G u s t a v s о n, Ber. dtsch. chem. Ges., 11, 1251 (1878).
23. T. M. Berry, E. E. R e i d, J. Am. Chem. Soc., 49, 3148 (1927).
24. S. H. M c A 11 i s t e г, J. A n d e r s о n, E. F. В u 1 1 a r d, Chem. Eng.
Progr., 43, № 4, 189T (1947) (Trans. Am. Inst. Chem. Engrs).
25. Англ. пат. 486520, 1936.
26. Пат. США 1994249, 1932. |
27 N V. Ipatieff, В. В. С о г s о n, Н. Р i n е s, J. Am. Chem. Soc., 58,
919 (1936); ЖОХ, 6, 1519 (1936).
28. H. L. Wunderly, F. I. S о w a, J. A. N ie u wlan d, J. Am. Chem.
Soc., 58, 1007 (1936).
29. Пат. США 2143493, 1937.
30. Пат. США 2572701, 1951.
31. Пат. США 1953702, 1929.
32. В. Г. Рав дин, Нефтепереработка и нефтехимия, (9), 29 (1965); (12),
31 (1963).
33. Англ. пат. 794570, 1958.
34. Н. Н. Лебедев и др., Труды МХТИ им. Д. И. Менделеева, (23), 52
(1956).
35. Авт. свид. СССР 130041, 1960; Бюлл. изобр., № 14, 15 (1960).
36. Sh. Iwai, J. Chem. Soc. Ind. (Japan), 45, Suppl. binding, 97 (1942); C. A.,
45, 2430b (1951).
37. T. Mazonski, D. Gasztych, Przemysl. chem., 41, 137 (1962).
38. Пат. США 3109037, 1963.
39. A. N e w t о n, J. Am. Chem. Soc., 65, 320 (1943).
40. E. П. Б а б и н, В. П. Ma p штупа, ЖФХ, 37, 656 (1963).
41. Франц, пат. 1103464, 1954.
42. Франц, пат. 1088332, 1953.
43. Бельг, пат. 536664, 1955.
44. Франц, пат. 961030, 1948; Франц, доп. пат. 59916, 1949.
45 D. A. S. 1004604, 1954; 1005505, 1007761, 1007762, 1008279, 1955; 1079046,
1958; 1087589, 1955; 1117558, 1959.
46 J Р. Fortuin et al., 5. Welterdolkongress, vol. IV, Paper 18,
1959.
47. R. van Heide n, E. C. Kooyman, Rec. trav. chim. Pays-Bas, 89,
57, 1237, 1257 (1961).
48. Англ. пат. 751598, 1952.
49 F. S e i d e 1, M. Schulze, H. В a 11 z, J. prakt. Chem., 3, (4), 278
(1956); пат. ГДР 12239, 1955.
50. Англ. пат. 641250, 1947.
51. Франц, пат. 1079454, 1953.
52 G. Zollner, I. Wurdits, I. Marton, Magyar Kem. Lapja, 9, 199
(1954).
53. T. Fir la, Rozniki Chem., 14, 87 (1934).
54. H. И. Ш и p к о в, К. P. P у с т а н о в, ЖФХ, 32, 219 (1958).
55. Я. M. П а у ш к и н, И. Г. Ми р га л ее в, Изв. вузов Нефть и газ, (2),
77 (1962).
56. J. Н. Simons, S. А г с h е г, J. Am. Chem. Soc., 60, 2952 (1938).
57 Пат. США 2423470, 1938.
58. Пат. США 2408173, 1943.
59 W. S. Calcott, J. М. Tinker, V. Weinmayr, J. Am. Chem. Soc.,
61, 1010, 1939.
60. F. E. С о n d о n, M. P. M a t u s z a k, там же, 70, 2539 (1948).
61. Англ. пат. 782084, 1957.
286
14. Кумольный синтез фенола
62. К. Г. Плюснин и др., Труды Института химии АН СССР, Уральск,
филиал, (4), 3 (1960).
63. Пат. США 2382318, 1942.
64. Е. G и с с i о n е, Chem. Eng., 70, № 9, 92 (1963).
65. Англ. пат. 769383, 1957.
66. Б. Д. К р у ч а л о в, П. Г. С е р г е е в, Хим. наука и пром., 1,287 (1956).
67. Chem. Eng. Progr., 43, 190 (1947).
68. И. М. Колесников, Г. М. П а н ч е н к о в, Труды Московского ин-та.
нефтехимической и газовой промышленности, 37, 85 (1962).
69. Пат. США 2713600, 1955.
70. Пат. США 2408753, 1943.
71. Пат. США 2425839, 1942.
72. Пат.- США 3197522, 1965.
73. Пат. США 3046315, 1962.
74. D. A. S. 1148220, 1963.
75. Пат. США 3068301, 1962.
76. Пат. США 3050570, 1962.
77. Пат. США 2939890, 1960.
78. Пат. США 3086998, 1963.
79. Пат. США 3054834—3054836, 1962.
80, Пат, Сшл 248S752, 1349.
81. Пат. США 2412595, 1948.
82. Я.М.Паушкин, А.В. Топчиев, ЖПХ, 21, 1065 (1948).
83. Я. М. П а у ш к и н, А. В. Т о п ч и е в, Л. И. С е р г а ч е в а, ДАН СССР,
64, 81 (1949).
84. А. В. Т о п ч и е в, В. В. А н д р о н о в, ДАН СССР, 111, 365 (1956);
113, 1076 (1957).
85. А. В. Т о п ч и е в, М. В. К у р а ш о в, Я. М. П а у ш к и н, ДАН СССР,
93, 839 (1953).
86. S. J. S 1 a n i n a, F. J.Sowa, J. A. N ieuwland, J. Am. Chem. Soc.,
57, 1547 (1935).
87. Пат. США 2652434, 1953.
88. Н. И. Шуйкин, Н. А. Поз дня к, Т. П. Д о б р ы н и я а, Изв. АН
СССР, Сер. хим., 7, 1244 (1966).
89. Пат. США 2403785, 1943.
90. Пат. США 2385303, 1941.
91. Е. П. Б а б и и, Л. И. М а р ы ш к и н а, Нефтехимия, 4, № Г, 26 (1964).
92. Пат. США 2467326, 1949.
93 М. Ishikawa, A. Kogure, Kogyo Kagaku Zasshi, 67, № 11, 1890 (1964).
94. Пат. США 3129255, 3129256, 1964.
95. Англ. пат. 997303, 1965.
96. Пат.. США 2438211, 1948.
97 Y.Ogino, I. Kawakami, Kogyo Kagaku Zasshi, 68, 45 (1965).
98. Пат. США 3031514, 1962.
99. Пат. США 2357978, 1941.
100. Франц, пат. 1341111, 1963.
101. Бельг, пат. 621879, 1962.
102. Пат. США 3120958, 1965.
103. Пат. США 2419599, 1942.
104. Ю. Г. Мамедалиев, Изв. АН СССР, ОХН, 1946, 458.
105 А. А. О’К е 11 у, J. Kellett, J.Plucker, Ind. Eng. Chem., 39,
154 (1947).
106. Пат. США 2410111, 1939.
107. Пат. США 2448160, 1946.
108. W. С е 11 е г et al., Przemysl. Chem., 42, № 8, 430 (1963).
109 Г. M. Панченко в, И. М. Колесников, Изв. вузов Нефть и газ,
5, 55 (1959).
Литература.
287
110. Ю. Г. Мамеда лиев, В. С. Алиев, С. А. Султанов, ДАН
АзербССР, 14, 681 (1958).
111. С. А. Султанов, Ю. Г. Мамедалиев, А. Б. Тартерян, Сбор-
ник трудов Азербайджанского НИИ по переработке нефти, выл. 3, 1958,
стр. 296.
112. А. В. Т о п чие в и др., ДАН СССР, 130, 587 (1960).
ИЗ. Пат. США 2408167, 1944.
114. Пат. США 2881227, 1959.
115. Пат. США 2930819, 1960.
116. Пат. США 2945072, 1960.
117. X. М. Ми на ч е в, Я. И. И с а к о в, ДАН СССР, 170, № 1, 99 (1966).
118. Пат. США 2329585, 2402847, 1941.
119. W. М. Р о 11 s, L. L. С а г р е n t е г, J. Am. Chem. Soc., 61, 663 (1939).
120. Франц, пат. 1307006, 1963.
121. Бельг, пат. 610802, 1962.
122. Н. И. Ш у й к и н, Н. А. П о з д н я к, Изв. АН СССР, ОХН, 1957, 697.
123. А. Б. Кучка ре в, ДАН УзбССР, 1, 21 (1957).
124. Пат. США 2965686.
125. Англ. пат. 870772, 1961.
126. Пат. США 2290211, 1940; 2387948, 2412229, 1942.
127. И. М. Колесников, Г. М. Панч е н ко, В. А. Т р е т ь я к о в а,
ЖФХ, 40, № 6, 1426 (1966).
128. X. М. Миначев й др., Нефтехимия 5, № 5, 676 (1965); 6, 53 (1966).
129. Франц, пат. 1335216, 1963.
130. Пат. США 2767230, 1956.
131. А. В. Топчиев и др. Труды Всесоюзного совещания по химии и пере-
работке углеводородов и полупродуктов для синтеза волокон и пластиче-
ских масс, Баку, 1957, стр. 51.
132. Я. М. Паушкин, М. В. К у р а ш о в, Изв. АН СССР, ОХН, 1956,
1006.
133. А. В. Т о п ч и е в и др., Труды Московского института нефтехимической
и газовой промышленности им. И. М. Губкина, 24, 269 (1959).
134. Я. М. Па ушкин и др., ДАН СССР, 91, 1141 (1953).
135. И. М. К о л е с н и к о в, И. Г. М и р г а л е е в, Я. М. П а ушкин,
Хим. пром., 3, 174 (1964).
136. С. В. Завгородний, Т. Б. Консовская, Изв. вузов. Химия
и хим. технол., 4, 1, 128 (1961).
137. Пат. США 2758140, 1956.
138. Н. Pines, I. Т. А иг i g о, J. Am. Chem. Soc., 79, 4958 (1957).
139. T. F i г 1 a, Rozniki Chem,, 14, 87 (1934).
140. Франц, пат. 1020129, 1953.
141. Англ. пат. 763179—763183, 1956.
142. Пат. США 2420073, 1942.
143. W. М. К u t z, В. В. Corson, Ind. Eng. Chem., 38, 761 (1946).
144. Пат. США 2589057, 1952.
145. Англ. пат. 791317, 1958.
146. А. В. Т о п ч и е в, Я. М. П а у ш к и н, М. В. К у р а ш о в, ДАН СССР,
88, 849 (1953).
147. Н. М. Колесников и др., Азерб. хим. ж., 6, 66 (1965).
148. Н. А. Смирнова и др., Азерб. нефт. хоз., 45, № 11, 37 (1966).
149. V. J.Karnojitzky, Chim. Ind., Paris, 93, № 1, 56 (1965)^
150. G. A. R u s s e 1 1, J. Am. Chem. Soc., 78, 1047 (1956).
151. Y. Tsunoda, Y. Matsumoto, Tokai Denkyoku Qiho, 17, № 1, 17;
№ 2, 20 (1956); C. A., 51, 8032hi (1957); Tokai Technol. J., 16, 22 (1955);
C. A., 50, 13770g (1956).
152. Англ. пат. 999441, 1965.
153. Англ. пат. 695717, 1953; пат. США 2619509, 1952,
288
14. Кумольный синтез фенола
154. Пат. США 2621213, 1952.
155. Пат. США 2629744, 1953.
156. Пат. США 2633476, 1953.
157. G.P. Armstrong, R. Н. Н а 11, D.C. Quinn, J. Chem. Soc. (Lon-
don), 666 (1950).
158. Австрал. пат. 138871; англ. пат. 610293, 1948; пат. ФРГ 924449, 1953.
159. Англ. пат. 653761, 1951; пат. ФРГ 819092, 1949; 899501, 1951; 699047, 1953.
160. Англ. пат. 681990, 1952.
161. Р. В. К у ч ер и др., Сборник научных работ Института фиа.-орг. химии
АН БССР, 8, 22 (1960).
162. G. Р. А г m s t г о n g, R. Н. Н а 11, D. С. Quinn, Nature, 164, 834
(1949).
163. Австрал. пат. 143548; англ. пат. 630286, 1949; пат. ФРГ 926426, 1953;
пат. США 2632772, 1949.
164. Т. Kato, Kogyo Kagaku Zasshi, 66, № 2, 209 (1963).
165. H. Osmolsk i, Przemysl Chem., 9, 142 (1953).
166. T. Takahashi, J. Chem. Soc. Japan, 57, 363 (1954).
167. P. В. К у ч e p и др., ДАН СССР, 117, 638 (1957).
168, Пат. США 2547938, 1951.
169. Р. В. К у ч е р и др., Доповиди АН УкрССР, 42 (1957).
170. Японск. пат. 7019, 1961.
171. Р. В. К у ч е р и др., Колл, ж., 21, 309 (1959).
172. Пат. США 2663740, 1953.
173. Пат. США 2632773, 1953.
174. С. А. Султанов н др., Азерб. нефт. хоз., 36, № 7, 34 (1957).
175. Пат. США 2681936, 1954.
176. Англ. пат. 823325, 1959.
177. Пат. США 2632774, 1953.
178. A. Di eric hs, Е. Р г е u, Freiberger Forsch.-Hefte, А51, 109 (1956).
179. Y. Tsunoda, К. Matsumoto, Kogyo Kagaku Zasshi, 62, 1555
(1959).
180. Инд. пат. 66197, 1960.
181. Англ. пат. 676770, 1952.
182. Англ. пат. 653761, 1951; белы. пат. 498028; пат. ФРГ 819092, 1949-
183. J. А. О’ Connor, Chem. Eng. News, 215 (1951).
184. Англ. пат. 922379, 1963; франц, пат. 1287252, 1962; итал. пат. 613419,
613420, 1964.
185. D. A. S. 1131674, 1962.
186. Франц, пат. 1322469, 1963.
187. D. A. S. 1134376, 1962; 1175236, 1964.
188. J.P. Fortuin Н. J. Waterman, Chem. Eng. Sci., 2, 182 (1953);
Spec. Suppl., 3, 60 (1954).
189. P. W. S h e r w о о d, Petroleum Refiner, 33, № 12, 144 (1954).
190. J. H. de В oer, J. P. Fortuin, H. J. W a t e r m a n, Koninkl. Med.
Akad. Wetenschap., Proc., 61B, 170 (1958); C. A., 53, 2144e (1959).
191. Z. Leszynski, Przemysl Chem., 37, 30 (1958).
192. Z. Leszynski, Przemysl. Chem., 43 , 255 , 321 (1964).
193. О. А. Колмаков и др., Труды по химии и хим. технол., 1, № 1, 36
(1958).
194. Р. Т. Н u a n g, М.-К. Kuo, P.-F. Tan, Hua Kung Hsueh Рао, 1, 50
(1959); С. А., 58, 4397а (1963).
195. Англ. пат. 676770—676772, 1952.
196. Итал. пат. 663421, 1964.
197. Англ. пат. 758934, 1956.
198. Пат. США 2681937, 1954.
199. Англ. пат. 922379, 1963.
200. Sh. Yamada, Kogyo Kagaku Zasshi, 60, 65 (1957).
Литература
289
201. В. Д. Ен алев и др., Сб. научных работ Института физ.-орг. химии
АН БССР, 8, 126 (1960).
202. Р. В. К у че р и др., ЖФХ, 35, 2322 (1961).
203. Р. В. К у ч е р и др., ДАН СССР, 132, 1348 (1960).
204. Англ. пат. 731002, 730896, 1955.
205. G. W a g п е г , I. prakt. Chem., 27, № 5—6, 297 (1965).
206. Т. К a t о, Kogyo Kagaku Zasshi, 66, № 2, 209 (1963).
207. Англ. пат. 745128, 1956.
208. Англ. пат. 712708, 1954.
209. Пат. ФРГ 889443, 1953.
210. Р. G е о г g е, Е. К. R i d е а 1, A. J. В. R о b е г t s о n, Proc. Roy. Soc.,
London, А185, 288, 297 (1946).
211. Т. Kato, Kogyo Kagaku Zasshi, 66, 399 (1963).
212. Р. В. Кучер, ЖФХ, 33, 617 (1959).
213. Японск. пат. 2372, 1955.
214. Англ. пат. 665897, 665898, 1962.
215. Н. Н о с к, Н. К г о р f, J. prakt. Chem., 6, № 4, 120 (1958).
216. Н. Н о с k, Н. К г о р f, J. prakt. Chem., 9, № 4, 173 (1959).
217. D. A. S. 1128847, 1962.
218. H. К г о p f, Liebigs Ann. Chem., 637, 73, 110 (1960).
219. D. A. S; 1041960, 1958; 1076132, 1960.
220. H. К г о p f, Tetrahedron Letters, 577 (1962).
221. H. H о c k, H. К г о p f, J. prakt. Chem., 13, 285 (1961).
222. H. Kropi, Erdol und Kohle, 15, 78 (1962).
223. С. В. Рогинский и др., ДАН СССР, 148, 118 (1963).
224. С. 3. Рогинский и др., Каталитические реакции в жидкой фазе.
Труды Всесоюзной конференции, Алма-Ата, 1962, стр. 334.
225. _Н. П. К р е й е р, Г. М. Ал и кина, М. Г. Т ро и ц кая, там же,
стр. 342.
226. Пат. США 2577768, 1951.
227. Пат. США 2613227, 1952.
228. Авт. свид. СССР 168265, 1965; Бюлл. изобр., 42, № 4, 22 (1965).
229. Н.П. К р е й е р, Научные основы подбора и производства катализаторов;
Сибирск. отд. АН СССР, 1964, стр., 218.
230. Англ. пат. 838029, 1960.
231. Франц, пат. 1032010, 1953.
232. Пат. США 2973310, 1961.
233. Пат. ГДР 25265, 1963; франц, пат. 1218577, 1958; австр. пат. 214912, 1959.
234. Франц, доп. пат. 76585, 1962.
235. Англ. пат. 895622, 1962.
236. Англ. пат. 760367, 1956.
237. Пат. США 2655545, 1953.
238. Р. В. К у че р и др., Изв. вузов. Химия и хим. технол., 4, 971 (1961).
239. Niederl. A. S. 6504559, 1965.
240. Англ. пат. 719895, 1954; голл. пат. 78094, 1955.
241. К. Maruyama, R. Goto, Т. Ко minami, Nippon Kagaku Zasshi,
85, № 2, 145 (1964).
242. Японск. пат. 6361, 1957; пат. США 2861107, 1958.
243. Niederl. A. S. 6408468, 1965.
244. Пат. США 2632026, 1953.
245. Англ. пат. 713138, 1954; голл. пат. 77402, |955; пат. США 2743086, 1956.
246. Пат. США 3160668, 1964.
247. Англ. пат. 741507, 1955.
248. Франц, пат. 1392364, 1965.
249. Jap. A. S. 5919, 1957.
250. Пат. США 2776999, 1957.
251. Англ. пат. 729132, 1955; пат. США 2674629, 1954.
19 Заказ 399
290
14. Кумольный синтез фенола
252. Пат. США 2792424—2792426, 1957.
253. Англ. пат. 097776, 1953; пат. США 2697121, 1954.
254. Пат. ФРГ 928230, 1953; пат. США 2680139, 1954.
255. D. A. S. 1000024, 1958.
256. О. А. Колмаков и др., Труды по химии и хим. технол., 1, № 3, 693
(1958).
257. J. A. Howard, J. С. Robb, Trans. Faraday Soc., 59, Tl. 7, (487), 1590
(1963).
258. M. D u r u p, Actions chim. et biol. radiations, 4, 18 (1958); C. A., 53, 881c
(1959).
259. A. E t i e n n e, A. le Berre, Compt. rend., 249, 105 (1959).
260. Франц, пат. 1239348, 1960.
261. Пат. США 2955996, 1960.
262. A. le Berre, Bull. Soc. chim. France, 1933 (1959).
263. Пат. США 3113085, 1963.
264. M. D u r u p et al., Proc. U. N. Intern. Conf. Peaceful Uses At. Energy, 2nd,
Geneva, 29, 143 (1958).
265. P. В. Кучер и др., сб. «Окисление углеводородов в жидкой фазе»,
АН СССР, 1959, стр. 212.
266. С. Д. К а з ь м и н, Р. В. К у и е р, Нефтехимия, 3, № 3, 371 (1963),
267. В. А. Симонов, М. С. Немчов, ЖОХ, 32, 2914 (1962).
268. Р. В. К у ч е р и др., ЖПХ, 35, 170 (1962).
269. Р. В. К у ч е р и др., Укр. хим. ж., 27, 658 (1961).
270. С. Д. Казьмин, ЖПХ, 35, 422 (1962).,
271. Р.-Т. Huang, J.-Ch. Yang, Hua Hsueh Hsueh Pao, 29, № 3, 159 (1963);
C. A., 59, 9861c (1963).
272. В. Л. Антоновский и др., ЖПХ, 37, 2453 (1964).
273. В. А. С и м о н о в, М. С. Немч ов, ЖОХ, 32, 3179 (1962).
274. V. J. Karnojitzky, Chim. Ind., Paris, 93, 56 (1965).
275. В. А. Ш у ш у н о в, Б. А. Р е д о ш к и н, Ю. Д. Г о л у б е в, ЖПХ,
35, 832 (1962).
276. С. Д. К а з ь м и н, Р. В. К у ч е р, Кинетика и катализ, 2, 422 (1961),
277. Б. В. Е р о ф е е в, Т. И. С о р о к о, ЖПХ, 33, 903 (1960).
278. Р. Beltrame, Chim. е Ind., Milano, 41, 202 (1959).
279. Б. А. Р е д о ш к и н, В. А. Ш у ш у н о в, Химия перекисных соедине-
ний, ИОНХ, АН СССР, 1963, стр. 207.
280. В. Л. Антоновский и др., там же стр. 219.
281. В. Л. А н т о н о в с к и й, Б. Я. М а к а л е Ц, ДАН СССР, 140, 1072,
282. Н. S. Blanchard, J. Am. Chem. Soc., 81, 4548 (1959).
283. H. S. В 1 a n c h a г d, J. Am. Chem. Soc., 82, 2014 (1960).
284. С. Д. Казьмин, P. В. К у ч e p, ЖОХ, 31, 3171 (1961).
285. J. W.L.Fordham.H.L. Williams, Canad. J. Res., 27B, 943 (1949);
C. A., 44, 3775c (1950).
286 Y. Tsunod a, K. Matsumoto, T. Kato, Tokai Denkyoku Giho, 19,
№ 1, 41 (1958); C. A., 52,. 17154d (1958).
287. G. H. Twigg et al., Erdol und К oh 1 e, 15, 74 (1962).
288. E. А. Кузьмина и др., Химия перекисных соединений ИОНХ АН
СССР, 1963, стр. 231.
289. Р. В. К у ч е р, А. И. Ю р з е н к о, Колл, ж., 18, 555 (1956).
290. W. G. L 1 о у d, R. G. Z i m m e r m a n, Ind. Eng. Chem. Product Res.
Development, 4, 180 (1965).
291. W. G. Lloyd, R. G. Z i m m e r m a n, A. J. D ie t z le r, ibid., 5,
327 (1966).
292. Франц, пат. 1404636, 1965.
293. T. D. M a n 1 у, Ind. Chemist, 32, 271 (1956).
294. V. Karnojitzky, Chim. Ind., Paris, 93, № 5, 529 (1965).
295. Chem. Eng., 66, № 23, 94 (1959).
Литература
291
296. G. С а р г а г a, G. Lemetre, Chim. е Ind., Milano, 42, 974 (1960)
297. Пат. ФРГ 861251, 1950.
298. Бельг, пат. 500289.
299. D. A. S. 1112527, 1959.
300. D. A. S. 1128859, 1957.
301. D. A. S. 1128848, 1958.
302. D. A. S. 1134361, 1960.
303. D. A. S. 1126887, 1959.
304. Авт. свид. СССР 164297, 1964; Билл, иаобр., 41, № 15, 19 (1964).
305. D. A. S. 1121621, 1959.
306. Н. Osmolski, Przemysl. Chem., 9, 142 (1953).
307. Англ. пат. 629429, 626095, 1947.
308. Англ. пат. 689734, 1953.
309. Англ. пат. 754862, 1956.
310. Англ. пат. 748565, 1956; голл. пат. 76525, 1954.
311. Англ. пат. 684039, 1952; пат. США 2663735, 1949.
312. Пат. США 2957921, 1960.
313. Англ. пат. 748287, 1956.
314. Англ. пат. 802524, 1958.
315. Англ. пат. 791759, 1958; пат. США 2904592, 1959.
316. Итал. пат. 572864, 1958.
317. Англ. пат. 930161, 1963.
318. Пат. США 2993407, 1961.
319. Франц, пат. 1037198, 1953.
320. Англ. пат. 721700, 1955; пат. США 2761877, 1956.
321. Англ. пат. 846516, 1960.
322. G. Burtzlaff et al., J. prakt. Chem., 28, № 5—6, 305 (1965).
323. В.Л. Антоновский, Л. M. Дягилева, Химия перекисных соеди-
нений, ИОНХ АН СССР, 1963, стр. 226.
324. Англ. пат. 754783, 1956.
325. Англ. пат. 735229, 1955; пат. США 273527, 1956.
326. Англ. пат. 942982, 1963.
327. Англ. пат. 704121, 1954; пат. США 2668859, 1954.
328. Англ. пат. 673576, 1952; пат. США 2626281, 1953.
329. Голл. пат. 77510, 1955.
330. Jap. A. S. 6360, 1957.
331. Пат. США 2671809, 1954; англ. пат. 736715, 1954.
332. Пат. США 2889368, 1959.
333. Англ. пат. 803480, 1958.
334. Пат. ЧССР 87048, 1957.
335. Z. Leszczynski, Przemysl. Chem., 44, № И, 626; № 9, 488 (1965).
336. S. С а г г a, Chim. е Ind., Milano, 45, № 9, 1087 (1963).
337. Р. В. К у я е р, В. А. С о р о к а, ЖПХ, 34, 1573 (1961).
338. Z. Leszczynski, Е. Treszczanowicz, Przemysl. Chem., 13,
664 (1957); W. С е I 1 е г, там же, 13, 701 (1957); Z. Leszczynski
et al., там же, 13, 703 (1957).
339. Пат. ПНР 42393, 1959.
340. Ю. А. Ш л я п н и к о в и др., Труды по химии и хим. технол., 2, № 1,
13 (1959).
341. Е. Г. Б ер ез к о в а, К. В. Т о п ч и е в а, Л. И. П и г у с о в а, Кине-
тика и катализ, 5, 903 (1964).
342. Jap. A. S. 13824, I960.
343. Пат. ФРГ 930637, 1955.
344. А. Л о п а т и н, Л. С. Саркисьянц, Д. Н. Ф р п д, Хим. пром., 43,
671 (1967).
345. В. Г. Солонецкий и др., Хим. пром., 42, 509 (1966); Р. Е. Г у р о -
вич, Г. Л. М а т и н а, там же, 42, 661 (1966).
19*
292
14. Кумольный синтез фенола
346. Англ. пат. 819026, 1959.
347. Итал. пат. 625785, 1961.
348. Авт. свид. СССР 118415, 1959; Бюлл. пзобр., № 5, 17 (1959).
349. Англ. пат. 768715, 1957.
350. Пат. США 3205272, 1965.
351. Пат. ЧССР 101816, 1961.
Г» А Ч 1-109179 10е. 3
353' Англ. пат. 649286, 1951; пат. США 2663743, 1963.
354. Англ. пат. 756408, 1956; пат. США 2744143, 1956.
355. Пат. США 2744144, 1956.
356. Франц, пат. 1503079, 1967.
357. Франц, пат. 1501638, 1967.
358. Бельг, пат. 672051, 1966.
359. Франц, пат. 1484288, 1967.
360. Авт. свид. СССР 166709, 1964; Бюлл. изобр., 41, № 23, 26 (1964).
361. Ю. Н. EonaipoB и др., Нефтепереработка и нефтехимия, 3, 23 (1967).
362. Итал. пат. 663418, 1964.
363. Пат. США 3337646, 1967.
364. А. Б. Воль-Эпштейн и др., Нефтепереработка и нефтехимия,
сб. 10, 37 (1965); Нефтехимия, 7, № 2, 191 (1967).
365. D. A. S. 1219035, 1966; англ. пат. 931377, 1962; франц, пат. 1289952, 1962.
366. Е. А. К р а в ц о в а, Е. В. П е т р о в а, ЖПХ, 39, № 8, 1831 (1966).
367. Англ. пат. 670444, 670445, 1952.
368. Y. Tsunoda, I. Kato, Nippon Kagaku Zasshi, 80, 575 (1959).
369. F. Н. Seubold jr., W. E. V a u g h a n, J. Am. Chem. Soc., 75, 3790
(1953).
370. В. А. Ш у ш у н о в, В. П. М а с л е н н и к о в, ДАН СССР, 176, № 6,
1355 (1967).
371. В. А. Шушунов и др., Труды по химии и хим. технол., 1, № 1, 50,
55 (1958).
15. ПОЛИПРОПИЛЕН
Развитие производства полиолефинов началось одновременно
в Германии и в Англии. В Германии сначала была изучена и успешно
осуществлена полимеризация изобутилена в полиизобутилен. В Ан-
глии с 1930 фирма ICI занималась полимеризацией этилена [5].
В 1938 г. там была пущена первая установка по получению поли-
этилена высокого давления. Очень скоро за ней последовали и дру-
гие установки, на которых этилен полимеризовался под высоким
давлением.
В 1955 г. Циглер [6—8] опубликовал первые результаты опытов
по полимеризации этилена под нормальным давлением в присут-
ствии металлорганических соединений. Позднее эти катализаторы
в модифицированной форме стали применять для полимеризации
различных виниловых соединений.
За период с 1950 г. по 1960 г. в области полимеризационных
процессов с применением специально разработанных катализаторов
Циглера и Натта была открыта новая глава, представляющая зна-
чительный теоретический и практический интерес. Речь идет о сте-
реоспецифической полимеризации. Различные стереоизомерные по-
лимеры, полученные на основе одного и того же мономера в зависи-
мости от хода полимеризации могут значительно различаться по
физическим свойствам (температуре плавления, кристалличности,
механическим свойствам и т. д.).
Из пропилена [1—4] стереоспецифической полимеризацией в за-
висимости от условий реакции могут быть получены следующие
продукты:
СН3 СН3 СН3 СН3 СН3
Н ] н I Н I Н | Н |
| Н I Н I Н I Н I н
н н н н н
Изотактические полимеры
СН3 Н СН3 Н СН3
Н | Н I Н I Н | Н I
| /Сх I ХСЧ I I /С. I хсч
ЧУ I ЧУ | ЧУ I ЧУ | ЧУ | \
| Н I СН3| Н I CHgl Н
н н н н н
Синдиотактические полимеры
294
15. Полипропилен
CHg Н CHg CHg H
H I H I H I H I H I
| H I CHgl H I H I CHg
H H H H H
Атактические полиме&ы
Стереоспецифическая полимеризация проходит тремя этапами:
а) инициирование, б) рост цепи, в) обрыв цепи.
Очень хорошим катализатором для полимеризации пропилена
оказалась система R3A1 -ф TiCl3 — катализатор, разработанный Циг-
лером и Натта. В этой системе хлор в TiCl3 — собственно катализа-
торе — является агентом переноса цепи, а гидрид алюминия (или
алкилалюминий) — сокатализатором.
Для объяснения стереоспецифической полимеризации Натта
предложил мостиковый механизм:
Цепь Цепь Цепь
I I I
СН.-Х сн—х сн-х
I I I
СП, сн2 . сн2
\тк' >А1/ +сн2=сн-х -----> ^>Ti< \ai<--> >А1<--->
R | в" | W
сн2=сн сн2=сн
I I
X X
Цепь Цепь
I I
СН-Х сн-х
сн2 сн2
+ I I
сн2-сн СН-Х сн-х
I I I
XX _ сн2 сн2
--> \?ь, /СН2-CH— Цепь > \fi'' \а1/ > Чп,/ /А1\^
'R-А1— " RZ V
В 1954 г. Натта [10—18] с большим успехом использовал подоб-
ные системы для полимеризации пропилена. Уже в 1957 г. фирма
Montecatini пустила первую промышленную установку по получению
полипропилена.
Вскоре в США появилась первая установка Hercules Powder Со.,
а в ФРГ в сотрудничестве с Циглером была построена первая уста-
новка на фирме Farbwerke Hoechst AG.
Полипропилен [19 — 45] наряду с хорошими физическими и хи-
мическими свойствами, присущими всем полиолефинам, обладает
такой температурной стойкостью, какой не достигал ни один из
15.1. Получение полипропилена
295
известных стойких к коррозии термопластов. Температура размягче-
ния полипропилена около 140 °C. Все это делает этот материал очень
перспективным для химического машиностроения. Аппараты и
установки из полипропилена имеют температурный предел приме-
нения до 100 °C и выше.
15.1. ПОЛУЧЕНИЕ ПОЛИПРОПИЛЕНА
15.1.1. Катализаторы полимеризации пропилена
Из многих предложенных комбинаций катализаторов в промыш-
ленности закрепилась система Al (С2Н5)3 — TiCl3. Каталитическое
действие TiCl3 зависит главным образом от его кристаллической
структуры (табл. 30). В промышленности применяются в основном
а-, у- и 6-формы.
Таблица 30
Кристаллические модификации TiCl3
Модифи- кация Кристалли- ческая структура Цвет Получение X арактеристика
а Гексаго- Фиоле- Восстановление TiCl4 Высокая стереоспеци-
нальная товый с водородом или TiCl4 с титаном при 400 ?С фичность (80 —90% нерастворимой в гептане части)
р Линейная Корич- невый Восстановление TiCI4 с водородом в элек- трической дуге или разложение CH3TiCl3 в углево- дородах Умеренная стереоспе- цифичность (40— 50% нерастворимой в гептане части)
т Кубиче- ская Фиоле- товый Нагревание р-формы в течение 2—3 ч до 250—300 °C или раз- ложение CH3TiCl3 в отсутствие рас- творителей Так же активен, как а-форма
б Частично неупоря- доченная фиоле- товый Восстановление TiCl4 с металлическим А1, причем конечный продукт еще содер- жит А1С13 Стереоспецифичность такая же или луч- ше, чем у о^формы, высокая скорость реакции (15]
Применение TiCl3 вместо TiCl4 дает следующие преимущества:
1) повышение кристалличности твердого полипропилена;
2) увеличение температур размягчения и плавления;
296
15. Полипропилен
3) повышение прочности при растяжении (в 3 раза) и жесткости;
4) уменьшение относительного удлинения при растяжении;
5) снижение проницаемости для газов и паров.
Кроме весьма эффективной системы А1(С2Н5)3 — TiCl3, описано
немало других каталитических систем [35]. Ниже приведены важней-
шие из них:
TiCi4 + А1С13 + P(OR)3 [48]
Li [49]
TiCl3 + Li [49]
TiCl3 + NaC6H0 + PO(NR2)3 [50]
TiCl4 + A1R3, ROAr [51]
TiCl3 + A12(C2H6)3C13 + Дибутиловый эфир, эпихлоргидрин [45]
TiCl4 + A1(C2H5)3 + AI(C2H5)2C1 [52]
TiCI3 + AI(C2H5)3 + Алканоламин [53]
Ti(CeH5)2 (ци«ло-С6Н6)2 + A1R3 [54]
TiCI3+Al2Alk3X3 + (N, P, As, Sb)R, [55]
TiCl4 + A1(C2H5)2C1 + Cd (C2H5)2 [56]
TiCl4 + Al + Ti(OR)4 [57]
TiX4 + Al + Pt или Pd [58]
A12Alk3Xg Al [59]
TiCl4 + NaAlk + Cd(Alk)2 [60]
TiCl4 или TI4 + A1C2H6C12 + A1(C2H6)3 [61]
TiCl4 + Al + MoO3 [62]
TiCl4 + A1(C2H6)3+ Si2H2O3 [63]
Ti(OR)4 + A1(C2H5)2C1 [64]
CaTiF6 + AlAlk2Cl или AlAlkX2 [65]
V2O6 + Al + A1C13 [66]
MoS2 + AlAlk3 [67]
TiCl4 + A1C2H5C12 + InCl3 [68] '
TiCl3 + C2(C6H6)6 [69]
V(OR)3 + AlAlk3 [70]
TiCI3 + Ti(ifUK4o-C6H5)2Cl2 + Na [71]
Боксит + O2 [73]
Ti(OR)4 + SbCl5 + Na [72]
TiCl4 + Al + NaF [74]
TiCl4 + GeAlk3H [75]
Fe+ O2 [76]
TiCI4 + Na + A1(CN)3 [77]
TiCl4 + AlAlk3 + SiCl4 [78]
TiCL + C [79]
IrCI4 + A1(C2H6))2C1 [80]
TiCl3 + A1(C2H6)3 + Углеводород с кислым Н-атомом [81]
ZrH4 + HC1 [82]
TiCI4 + Al + NAlk*X [83]
TiOg/SiOj + AlAIk3 [84]
TiCI3 + Al2Alk3Cl3 + KC1 [85]
TiCl3 + A1(C2H5)2C1 + NaCl [86]
TiCl4 + Li [87]
MoO3 + A12O3 + NaAIk [88]
TiCl4 + AlAlk(OR)X [89]
Сплав Ti — Al + Алкилгалогенид [90]
TiCl4 + Ионизирующее излучение [91]
TiCl3 + AlAlkj + Ультразвук [92]
TiCl4 + Al и A1C1, + NaF [93]
TiCl4 + AlAlk2OR [94]
15-1. Получение полипропилена
297
a-TiCl3 + Zn(C2H5)2 или Ве(С2Н5)2 [95]
TiCl3 + А1(С2Н6)3 [96]
Ацетилид Си, Ag или Hg [97]
МоО3 + А12О3 + NaAIk[98]
VC14 + А1А1к»С1 + Анизол [100]
FeCl3 + А1А1к2С1 [99]
TiCl3 + Al(C2H6)2Br [101]
TiCl4 + Na [102]
TiCl4 + A1(C2H6)3 [103]
TiCl4 + (iwo-C4H8) [104]
Ti(OR)4 + AlAlkX2 [105]
Ti(OB)4 + Al2Alk3X3 [106]
TiCl4 + Sn2Alk*O [107]
CrCl3 + AIAlk3 [108]
TiCl2 [109]
Al2Fe [110]
TiCl4 + Al + AICI3 [111]
TiCl3 + Al [109]
TiCl4 + К + AICI3 [112]
TiCl3 + A1C2H6CI2 + (R,N)3PO, R2O, R3N, R2S, R3P [113]
ZrCL + Al или Ti [114]
TiCl4, ZrCl4 + Fe, Co или Ni [114]
15.1.2. Полимеризация пропилена
Для полимеризации применяется очень чистый пропилен (>99 %),
не содержащий соединений серы, воды и метилацетилена [9]. Про-
пилен подвергают перегонке на нескольких колоннах и сушке с по-
мощью молекулярных сит.
В настоящее время полипропилен производится методом низкого
давления. Молекулярный вес полипропилена не зависит от давления
[16].
Полимеризацию проводят обычно в области температур 50 — 100 °C,
когда образуется полимер, не растворяющийся в реакционной среде.
Температура реакции оказывает большое влияние на структуру
полимера, причем в присутствии TiCl4 достигается иной эффект,
чем в присутствии TiCl3. Время полимеризации колеблется от 0,5
до 10 ч [17]. По окончании периода инициирования скорость реак-
ции не зависит Ют длительности полимеризации [18]. Она также не
зависит от концентрации алюминийорганического соединения. Ско-
рость реакции существенно зависит от концентрации TiCl3 и пропи-
лена, температуры реакции и способа получения TiCl3 (TiCl3 не дол-
жен содержать TiCl4, так как в противном случае увеличивается со-
держание атактического полипропилена).
В качестве растворителей для полимеризации обычно применяют
насыщенные углеводороды, например гексан, гептан и др.; они слу-
жат одновременно осадителями для образовавшегося полипропи-
лена. В системах, где полипропилен растворяется, образуется глав-
ным образом аморфный полимер [46].
В результате интенсивного перемешивания получается более
высокомолекулярный продукт [47].
298
15. Полипропилен
15.1. Получение полипропилена
299
Растворители, употребляемые при полимеризации пропилена,
должны быть тщательно очищены от катализаторных ядов (воды,
карбонильных соединений, перекисей, азот- и серусодержащих
соединений) и агентов обрыва цепи, например алкилхлоридов, оле-
финов и ароматических соединений. Катализатор применяется либо
в виде раствора, либо в виде суспензии. Раствор катализатора обычно
приготавливают партиями.
Для проведения полимеризации (рис. 69) чистый пропилен,
суспензию катализатора и разбавитель подают в реактор. Смесь
при перемешивании нагревается до 50—100 °C, при этом давление
поднимается максимум до 5 кгс/см2. Добавляемое количество ката-
лизатора (0,25—0,50 вес. % от взятого растворителя) зависит в из-
вестной мере от степени чистоты мономера и растворителя. К ката-
лизатору добавляется примерно равное количество активатора.
Во время полимеризации образовавшийся полипропилен выпа-
дает в осадок. На большинстве установок концентрация пропилена
в углеводороде подбирается так, чтобы прореагировавший раствор
содержал около 20—$0% осажденного твердого вещества. В раздели-
тельной колонне отгоняется непрореагировавший пропилен и часть
растворителя. Остается суспензия полипропилена в растворителе.
Растворитель после перегонки или возвращается прямо в реактор,
или еще раз перегоняется перед повторным использованием. Отогнан-
ный Пропилен конденсируется, перегоняется и снова возвращается
в реактор. Суспензия полипропилена пропускается через промежу-
точный сборник и центрифугу, где полипропилен освобождается
от остаточного растворителя. Разбавитель отсасывается, тоже очи-
щается на колонне и возвращается в реакцию. Отделенный на центри-
фуге сырой полипропилен суспендируется в низших спиртах (в .ме-
тиловом или изопропиловом). Для разложения содержащегося еще
в полипропилене катализатора к растворителю добавляется соляная
кислота. Затем суспензия спирт — пропилен центрифугируется,
спирт освобождается путем перегонки от остатков катализатора
и разбавителей. После промывки водой, сушки, выдержки и добавки
антиоксидантов полипропилен готов для дальнейшей переработки.
15.1.3. Экономическая оценка производства полипропилена
Хотя цена пропилена ниже, чем многих других мономеров, поли-
пропилен имеет самую высокую цену. Ниже указана стоимость про-
изводства некоторых полимеров (в центах за 1 кг):
Мономер Полимер Расходы на полимериза- цию
Поливинилхлорид . . . 12 22 10
Полистирол .... . . 12 20 8
Полипропилен . . . . 6 31 25
300
15. Полипропилен
Высокая стоимость полипропилена объясняется большим рас-
ходом пропилена (1,2—1,3 т/т), сложностью технологии и приме-
нением сложной системы катализатора.
Высокий расход пропилена обусловлен образованием примерно
10% атактического полимера наряду с 90% желаемого изотактиче-
ского. Для снижения выхода атактического полипропилена необ-
ходимо добиваться повышения стереоспецифичности катализатор-
ных систем. Кроме того, атактический полимер, очень похожий по
своим свойствам на полиизобутилен, следует по мере возможности
использовать для какой-либо полезной цели.
15.2. СВОЙСТВА ПОЛИПРОПИЛЕНА
Свойства полипропилена в большой степени зависят от молеку-
лярного веса, молекулярно-весового распределения и кристалличе-
ской структуры полимера. Поэтому продукты, полученные в раз-
личных условиях, обладают и разными свойствами. Ниже приведены
средние показатели некоторых свойств полипропилена:
Плотность, г/см3 .................................0,90—0,91
Температура плавления кристаллитов, SC............170—175
Температура превращения второго порядка, 9С . . . —35
Температура хрупкости, ®С......................... от —10
до +20
Температура размягчения по Вика, °C
, 1 кг ............................... ; . . . . 140—155
5 кг........................................... 85—105
Прочность, кгс/см2
при растяжении (50 мм/мин) ....................... 300—360
разрыве................................... 300—400
Удлинение, %
при растяжении....................................... 5—15
при разрыве .................................... 500—700
Модуль упругости, кгс/см2
при растяжении..................................... 12—18000
при изгибе ....'............................... 9—15000
Твердость
цо Роквеллу (шкала В)........................... 85—95
по Шору (шкала Д)............................ 70—75
Твердость при вдавливании шарика, кгс/см2
через 10 с........................................ 650
через 60 с........................................ 580
Ударная прочность по Изоду, кгс • см/см2.......... 80
Ударная вязкость по Изоду, кгс • см/см2 .......... 4—6,5
Удельная теплоемкость при 23 РС, кал/(г • °C) .... 0,46
Коэффициент теплопроводности, кал/(см • с • °C) . . 3,3-10“4
Коэффициент линейного расширения, 1/°С............ 0,00018
Абсорбция воды через 24 ч, % ..................... 0,03
Удельное поверхностное сопротивление, Ом..........—5 • 101*
15.2. Свойства полипропилена 301 -
Удельное объемное сопротивление, Ом • см ............1015—1017
Диэлектрическая проницаемость (при 106 Гц) .... 2,0—2,1
Тангенс угла диэлектрических потерь
при 10е Гц ......................................... 0,0002—
0,0003
105 Гц ........................................ 0,0009
104 Гц ........................................ 0,0019
103 Гц ........................................ 0,0019
102 Гц ........................................ 0,0025
Термические свойства изотактического полипропилена описаны
в работе [115]. Приведенные ниже характеристики различных типов
полипропилена с примерно одинаковой степенью изотактичности
иллюстрируют влияние молекулярного веса на свойства полипро-
пилена [33]:
Характеристическая вяз-
кость, дл/г.............. 2,5
Показатель текучести рас-
плава, г/Ю мин........... 9,8
Предел текучести, кгс/см2 400
Модуль Юнга (при 1 Гц)
£-108, дин/см2............. —
Прочность при ударном из-
гибе по Изоду при 20 °C,
кгс • см/см2 ............ 3,1
Содержание экстрагируемой
части, %.................. 2
3,0 3,0 4,2 5,0 5,5 9,3
7,4 3,4 1,7 1,0 0,4 0,1
380 390 368 365 330 336
71 65 63 59 60 53
4,0 5,1 6,8 8,3 10,0 15,8
2 3 4 3 5 2
Важное значение имеет химическая стойкость полипропилена
[116]. При комнатной температуре он устойчив в водных растворах
солей, мыл и моющих средств, разбавленных и концентрированных
минеральных кислотах и щелочах, растворах перекисей, раститель-
ных и минеральных маслах, в спиртах. В углеводородах и хлори-
рованных углеводородах полипропилен набухает, в сильно концент-
рированных окислителях -(например, олеум, дымящая азотная кис-
лота, бромистый водород, отбеливатели) — разлагается. Раствор
иода и перманганата калия окрашивает полипропилен.
15.3. ПРИМЕНЕНИЕ ПОЛИПРОПИЛЕНА
Полипропилен благодаря ряду ценных свойств, не присущих
ранее известным термопластам, активно вытесняет многие виды
полимеров и находит все новые области применения. Ниже сравни-
302
15. Полипропилен
ваются некоторые свойства полипропилена со свойствами других
полимеров:
Плотность, г/см3 .........
Прочность при растяжении,
кгс/см2 .......................
Модуль упругости Е 105, кгс/см2
Ударная прочность с надрезом
(при 20—25 °C), см-кгс/см2 . .
Температура размягчения по
Вика, °C.....................
Усадка при плавлении, % . . .
Полипро- пилен Найлон 6 Полиэти- лен высокой плот- ности . Полиэтилен низкой плотности
0,90—0,91 1,13-1,14 0,93-0,97 0,91-0,93
250-400 500- 700 200-350 100-150
0,1-0,15 0,04-0,35 0,04-0,1 0,01-0,03
10-26 2,5-25 5,5-33 88
150 210-220 120-128 88—90
1-2 1-4 1-4 2-5
Полипропилен перерабатывается обычно литьем под давлением,
прессованием и экструзией. В промышленности перерабатывается
почти исключительно изотактический полипропилен. Методы пере-
работки полипропилена в общем схожи с методами переработки
полиэтилена высокой плотности.
Предметы, изготовленные из полипропилена, сохраняют форму
при температуре до 150 °C, однако при 140 °C модуль прочности
составляет только 10% от этой характеристики при комнатной тем-
пературе, так что практически температура эксплуатации не пре-
вышает 135 °C.
Применение полипропилена при низких температурах ограничи-
вается сравнительно высокой температурой хрупкости (от —10 до
4-20 °C). Ударная вязкость достаточно высока для большинства
назначений. С другой стороны, имеются возможности улучшения
ударной вязкости при низких температурах (модификация каучуком
или полиизобутиленом, блочная сополимеризация с 2—10% этилена).
Если подвергнуть нестабилизированный полипропилен действию
кислорода при повышенных температурах, то материал окисляется,
происходит деструкция цепи и полимер очень быстро становится
хрупким. Нормальная стабилизация антиоксидантами позволяет
непрерывно применять полипропилен при 120 °C в течение 100 дней,
путем специальной стабилизации можно увеличить стабильность до
2 лет при 120 °C и до 60—80 дней при 140 °C. Особенно эффективная
светостабилизация полипропилена достигается добавкой 1—2% га-
зовой сажи. Склонность к растрескиванию под напряжением у поли-
пропилена выражена значительно слабее, чем у полиэтилена, и по-
этому обычно не учитывается.
Из полипропилена изготовляют изделия самого различного на-
значения. В некоторых областях полипропилен служит заменителем
металла, дерева и стекла [117]. Из-за более высокой по сравнению
с полиэтиленом стоимости полипропилен применяли до сих пор
в тех случаях, когда требовались повышенная жесткость, лучший
блеск и более высокая стабильность формы. Полипропилен легко
15.3. Применение полипропилена
303
прессуется, поэтому из него изготовляют прессованные 'изделия,
тонкостенные и обладающие очень высокой устойчивостью формы.
Материал перерабатывается при температурах от 200 до 280 °C.
Температура переработки должна быть как можно ниже, так как
по сравнению с полиэтиленом полипропилен менее стабилен.
При получении прозрачных изделий экструдированный материал
нужно быстро охладить, чтобы образующиеся кристаллы были как
можно мельче. При медленном охлаждении образуются более круп-
ные сферолиты [118].
Переработка полипропилена литьем под давлением не предста-
вляет особых трудностей. Изготовленные отливки мало или совсем
не коробятся. Тенденцию к усадке можно уменьшить повышением
давления и увеличением времени подпитки.
Пленки из полипропилена лучше всего изготовлять на экстру-
дерах с широкощелевым мундштуком, так как при выдувании полу-
чаются менее блестящие и более мутные пленки. Свойства полипро-
пиленовой пленки можно значительно улучшить вытяжкой при
10—20 СС, т. е. ниже точки плавления кристаллитов; особенно боль-
шое значение приобрела биаксиально вытянутая пленка. Ниже при-
ведены свойства полипропиленовых пленок, полученных методом
экструзии рукава с раздувом (I) и экструзией (II):
г
Плотность, г/см3 ........................... 0,900—0,908
Прозрачность................................От плохой
до довольно
хорошей
Прочность при растяжении..................... Высокая
Сопротивление разрыву, кгс/см3 ............. 300—700
Прочность при разрыве образца с надрезом,
кгс/см .................................... 20— 500
Прочность при мгновенном растяжении,
кг-см/см2 .................................... 20—300
Предел текучести, кгс/см2 ................... 200—400
Ударная вязкость.............................. Довольно
хорошая
Удлинение, %
при растяжении............................... 5—20
при разрыве................................. 100—800
Проницаемость для водяных паров (24 ч, тол-
щина 0,05 мм), г/м2.............................. —
Температура сварки, ?С......................... 140—175
Температура эксплуатации, РС................от —15 до
+130
II
0,885—0,908
От довольно
хорошей
до отличной
Высокая
300-700
90-450
20-300
200—450
Довольно
хорошая
5—20
' 100-800
1,1
140-175
от —15 до
+ 130
По физико-механическим свойствам и цене полипропиленовая
пленка может конкурировать с целлофаном. Однако нужно было
разработать специальные методы сварки при нагреве (короткое
время, низкая температура, высокое давление) [122—123]. Пленки
применяются в первую очередь как прозрачный упаковочный ма-
териал для пищевых продуктов и текстильных товаров, а также как
304'
15. Полипропилен
оберточный материал. В промышленности полипропиленовая пленка
используется для изоляции и для квитирования других материалов.
Для изготовления труб применяется полипропилен с очень
низким показателем текучести расплава, причем работают при
температурах 240—250 °C. Полипропиленовые трубы выдерживают
окружные напряжения от .60 до 80 кгс/см2. Усталостная прочность,
вероятно, средняя между усталостной прочностью полиэтилена
низкого давления (50 кгс/см2) и непластифицированного поливинил-
хлорида (100 кгс/см2); трубы из полипропилена становятся хрупкими
при 0 °C. Особый интерес может представить применение этих труб
для подачи жидкостей при повышенных температурах.
Из полипропилена можно экструдировать и. листы, предназнача-
емые для облицовки емкостей. Методом вакуум-формования из поли-
пропилена изготовляют корпуса приборов, чемоданы и др. Пред-
меты, изготовленные методом формования с последующим раздувом
(например, емкости, бутылки), при очень тонких стенках обладают
большой твердостью и стабильностью формы [124].
Методом прядения из расплава из полипропилена очень легко
получать волокна и отдельные нити [125—129]. Большое значение
здесь имеет конструкция сопла. Выдавленный жгут тотчас охлажда-
ется в водяной ванне. Неориентированные^ первоначально нити
ориентируются при 150 °C посредством вытягивания в 8 раз. Однако
в кипящий воде нить может дать большую усадку. Во избежание
этого нить дополнительно обрабатывают, например, нагревают
в течение нескольких секунд до 150 °C в натянутом состоянии.
Обычно для получения волокна применяют полимер с не очень
большим молекулярным весом; высокомолекулярные продукты ис-
пользуются только для производства волокна с высокой текстильной
вязкостью.
Ниже сравниваются свойства некоторых полимерных волокон
Полиэтилен высокого давления Полиэтилен низкого давления Полипро- пилен Найлон 6
Плотность, г/см3 . . . Разрывная длина, 0,92-0,93 0,95-0,96 0,90-0,91 1,14
гс/денье Прочность при разрыве, 1-3 5-8 5-9 4-9
кгс/см2 Удлинение при разры- 1000—3000 4500-6500 6500 5000—8500
ве, % Модуль жесткости, 25-50 10—20 15-25 15-30
кгс/см2 1500-3500 5000-10000 10000-13000 30000-55000
Усадка , при 100 °C, % Температура размяг- 26-60 5-10 10—15 5-10
яения, °C 85-120 125-135 150-160 210—220
Высокое отношение прочность : вес, отличная химическая стой-
кость и высокая гибкость — все эти свойства благоприятствуют
применению полипропилена для получения нитей технического
Литература
305
назначения (например, для производства морских канатов). Изго-
товляют из полипропилена также фильтровальные ткани для хими-
ческой промышленности, покрывала, ковры. До сих пор еще труд-
ность представляет изменение цвета; поэтому материал окрашивают
пигментами и выделывают из него только изнаночную сторону ков-
ров. Наряду с красителем добавляют -соли металлов.
Полипропилен с высоким содержанием наполнителя (асбест,
тальк, окись цинка, каолин и др.) обладает улучшенной стойкостью
к высоким температурам. Полипропилен можно вспенивать. Вспе-
ненный полипропилен является хорошим звукоизоляционным мате-
риалом, например для оболочек телефонных и телевизионных ка-
белей [131]. Для повышения прочности полипропилен армируют
стекловолокном [132, 133]. Разработан способ получения пленок
для изготовления мешков [134].
ЛИТЕРАТУРА
1. К. Mehnert, Н. Becher, Kunststoffe, 55, 438 (1965).
2. Е. М. Honeycutt, Chem. Eng. Progr., 61, № 8, 88 (1965).
3. К. Vesely, Osterreich. Chemiker-Ztg., 68, № 3, 91 (1967).
4. M. Farber, Am. Dyestuff Reporter, 55, P536 (1966).
5. Англ. пат. 471590, 1937.
6. К. Ziegler et al., Angew. Chem., 67, 541 (1955).
7. Пат. ФРГ 878560, 1950.
8. E. G r a h m s, E. G a u b e, Angew. Chem., 67; 548 (1955).
10. G. N a 11 a, J. Am. Chem. Soc., 77, 1708 (1955).
11. G. N a 11 a, J. Polymer Sci., 16, 143 (1955).
12. G. N a 11 a, Atti Acad. naz. Lincei, Mem. (VIII), Bd. 4, II Section, 19,
61 (1955).
13. G.Natta, P. С о r r a d i n i, ibid., 19, 73 (1955).
14. G. N a 11 a, Makromol. Chem., 35, 93 (1960).
15. G. N a 11 a, Chem. e Ind., Milano, 42, 1207 (1960).
16. G. N a 11 a, SPE-J, 15, № 5, 373 (1959).
17. R. A. Labine, Chem. Eng., 67, № 13, 96 (1960).
18. G. N a 11 a, J. Polymer Sci., 34, 21 (1959).
19. T. О. J. Kresser, Polypropylen, New York, 1960.
20. M. S i 11 i g, Polyolefin Resin Processes, Houston, Tex. 1961.
2i. А. В. Топчиев, Б. А. Кренце л ь, Полиолефины. Новые синтети-
ческие материалы, Москва, 1958, New York, 1962.
22. N. G. G а у I о г d, Н. F. М а г k, Lineare und stereoregulare Additions-
polymere, Нью-Йорк, 1959.
23. J. M. Go p pel, Brit. Plastics, 32, 207 (1959).
24. A. V. G a 1 a n t i, Ch. L. M a n t e 11, Polypropylen, Fibers and Films,
New York, 1965.
25. Д. Я. M у ч и н с к и й, Л. А. П о т о л e в с к и й, Полимеризация про-
пилена, М., 1965.
26. J. Cech, Chemie, Prague, 9, 897 (1957).
27. М. Ottolenghi, С. Crespi, Modern Plastics, 35, № 7, 89 (1958).
28. G. Bier, Kunststoffe, 48, 354 (1958).
29. T. Inoue, Yuki Gosei Kagaku Kyokai Shi, 17, 205 (1959).
30. I. Obi о j et al., Przemysl. Chem., 38, 78 (1959).
31. A. Gumbold t, H. Schmidt, Chemiker-Ztg., 83, 636 (1959).
32. С. E. Farnsworth, C. W. Virgin, Chem. Eng. Progr., 55, Ns 11,
166 (1959).
20 Заказ 399
306
15. Полипропилен
33. G. W. Raamsdonk, Kunststoffe, 51, 269 (1961).
34. Plastics, London, 26, № 285, 157; № 286, 107 (1961).
35. M. S i 11 i g, Petroleum Refiner, 40, X» 3, 129 (1961).
36. M. Comp os tella et al., Angew. Chem., 74, 618 (1962).
37. G. Szlekys-Gorecka, Polymeri-Tworzywa wielkoczasteczkowe, 9, 293;
411, 458 (1964).
38. G. Campbell, Plastics, London, 29, № 321, 59, 71; № 322, 52 (1964).
39. B. G i 1 о d o, Kim. Sanayi, 12, № 60, 176 (1964); C. A., 62, 13317c (1965).
40. W. R. О ’ D о n n e 1, Am. Dyestuff Reporter, 54, № 25, 109 (1965); 54,
P-1060 (1965).
41. О. P e s t a, Plaste und Kautschuk, 12, 682 (1965).
42. P. Pest a, Kunststoff-Rdsch., 12, 461 (1965).
43. H. W. Haines jr., Ind. Eng. Chem., 55, № 2, 30 (1963).
44. T. Kakagi, S. Yoshioka, Plastics Age, Osaka, 11, № 9, 51 (1965);
C. A., 63, 18349f (1965).
45. H. I к e g a m i et al., Kogyo Kagaku Zasshi, 69, № 2, 321 (1966).
46. A. A. Harban, E. Field, H. N. Friedlaender, 15th Annual
Tech. Conference of SPE, New York, Jan. 1959.
47. P. W. Sherwood, Industrial Chemist, 36, 430, 48. Пат. США 2962487, 1960. 595 (1960).
49. Пат. США 2962472, 1960. 50. Пат. США 2958688, 1960. 51. Пат. США 2956992, 1960; пат. ГДР 16980, 1959. 52. Англ. пат. 795182, 1958; пат. США 2954367. 53. Англ. пат. 1035207, 1966. 54. Пат. США 2952670, 1960. 55. Пат. США 2951066, 1960. 56. Японск. пат. 25910 (1965). 57. Пат. США 2948712, 1960. 58. Пат. США 2945845, 1960. 59. Пат. США 2945018, 1960. 60. Пат. США 2940962, 1960. 61. Пат. США 2938019, 1960. 62. Пат. США 2938018, 1960. 63. Пат. США 2938000, 1960.
64. Пат. США 2936302, 1960. 65. Пат. США 2935500, 1960. 66. Пат. США 2935498, 1960. 67. Пат. США 2935497, 1960. 68. Пат. США 2935496, 1960. 69. Пат. США 2933483, 1960. 70. Пат. США 2933482, 1960. 71. К. Az um a et al., Kogyo Kagaku Zasshi, 68, № 7, 1245 (1965)..
72. Пат. США 2930785, 1960. 73. Пат. США 2928814, 1960. 74. Пат. США 2925410, 1960. 75. Пат. США 2925409, 1960. 76. Пат. США 2925408, 1960. 77. Пат. США 2921056, 1960. 78. Пат. США-2920062, 1960. 79. Пат. США 2919266, 1959. 80. Пат. США 2918458, 2918459, 1959. 81. Пат. США 2914515, 1959. 82. Пат. США 2913442, 1959. 83. Пат. США 2912424, 1959. 84. Пат. США 2912421, 1959. 85. Пат. США 2909511, 1959.
Литература
307
86. Пат. США 2909510, 1959.
87. Пат. США 2908670, 1959.
88. Пат. США 2908669, 1959.
89. Пат. США 2905661 г 1959.
90. Пат. США 2905646, 1959.
91. Пат. США 2903404, 1959.
92. Пат. США 2899414, 1959.
93. " Пат. США 2899413, 1959.
94. Пат. США 2898328, 1959.
95. А. П. Ф и р с о в и др., Высокомол. соед., 6, 352 (1964).
96. Пат. США 2893984, 1959.
97. Пат. США 2890212, 1959.
98. Пат. США 2887471, 1959.
99. Пат. США 2882263, 1959.
100. A. Zambelli, G. Natta, I. Р a s q и о n, J. Polymer Sei., C, 4,
411 (1963).
101. I. L. Mateo, R. Garzon, J. L. Yufieste, Rev. Plasticos Mod.,
16, N 103, 1 (1965).
102. Пат. США 2881156, 1959.
103. Пат. США 2874153, 1959.
104. Wm. J. Bailey, E. T. Yates, J. Org. Chem., 25, 1800 (1960).
105. Пат. США 2833755, 1958. 1
106. Пат. США 2824090, 1958.
107. Пат. США 2820778, 1958.
108. J. F. Gillespie, J. W. L. Fordham, Ind. Eng. Chem., 51, № 11,
1365 (1959).
109. Пат. США 3050471, 1962.
НО. Пат. США 3138578, 1959.
111. Пат. США 874215, 1953.
112. R. van Н е 1 d е n et al., Tetrahedron Letters, 12, 24 (1959).
113. H. W. С о о v e r jr., F. В. I о у n е г, J. Polymer Sei., АЗ, 2407 (1965).
114. Т. J. Miranda, G. F. D’A 1 е 1 i о, Proc. Indiana Acad. Sci., 75, 73
(1965); C. A., 66, 18899x (1967).
115. R. G. G r i s k e y, N. Waldman, Modern Plastics, 43, № 8, 160,
218 (1966).
116. P. P a r r i n i, L. Pinto, Materie plast. Elastomeri, 31, 268, 1207, 1244
(1965).
117. Mod. Plastics, 38, № 12, 72 (1961).
118. P. W. O. W i j ga, Rubber J. and Internat. Plastics, 134, № 19, 754 (1958).
119. Mod. Packaging EncycL, 149 (1960).
120. Mod. Plastics, EncycL, 696, 700 (1959).
121. H. Mark, Chem. Industrie, 16, 701 (1964).
122. K. Richard, G. Diederich, E. Gaube, Kunststoffe, 49, 671 (1959).
123. R. L. van В о s k i r k, Mod. Plastics, 37, № 9, 41 (1960).
124. P. C. Webbs, Plastics, 54 (1965).
125. В. С. M. Dorset, Textile Manufacturing, 87, 503 (1961).
126. E. Z. Cohen, Am. Dyestuff Reporter, 51, № 16, 43 (1962).
127. J. E. M с I n t у r e, R. J. B. Marsden, Rev. Textile Progr., 16, 50 (1964).
128. P. van О о s t, Chemiefasern, 16, № 6, 480 (1966).
129. Mod. Textiles Mag., 43, № 12, 21, 46 (1962).
130. V. L. Ehrlich, Mod. Textiles Mag., 39, № 11, 59 (1958); Text. Res. J.,
29, 209, 679 (1959).
131. F. S с a 1 a r i, Materie plast. Elastomeri, 28, 570 (1962).
132. Plastics, 31, № 344, 719 (1966).
133. Plastica, 5, 190 (1966).
434. О. P e s t a, Osterreich. Chem.-Ztg., 68, № 3, 95 (1967).
20*
16. СОПОЛИМЕРЫ ПРОПИЛЕНА С ЭТИЛЕНОМ
Синтез этих новых высокомолекулярных продуктов [1—26,
135] оказался возможным благодаря работам Циглера и Натта по
применению металлорганических смешанных или координационных
катализаторов. Такие катал из аторные системы позволили наряду
с полимеризацией зтилена и пропилена осуществить и стереоспеци-
фическую полимеризацию 1,4-бутадиена и изопрена с получением
стереокаучуков. По своей пространственной структуре эти новые
продукты очень похожи на натуральный каучук и обладают многими
ценными свойствами этого каучука.
Затем путем сополимеризации этилена и пропилена [27] удалось,
получить исключительно интересный пластик со свойствами эласто-
мера. Для этого использовали металлорганические катализаторные
системы, например состоящие из титановых и ванадиевых соединений
и органических соединений бериллия, цинка или алюминия. Эти
этилен-пропиленовые сополимеры, известные под названием ЭПР,
при статистическом распределении мономерных элементов по мак-
ромолекуле представляют собой аморфные вещества, по внешнему
виду похожие на невулканизированный натуральный каучук. Од-
нако эти полиолефиновые каучуки, как и натуральный каучук,
приобретают ценные механические свойства только после вулкани-
зации.
Поскольку этилен-пропиленовые сополимеры имеют насыщенный
характер, то их нельзя вулканизовать с помощью обычно исполь-
зуемых в резиновой промышленности систем из серы и ускорителей
вулканйзации. Правда, эти сополимеры можно сшивать органическими
перекисями, но перекисная сшивка применима не во всех случаях.
Нужно было так модифицировать зти продукты, чтобы оказалась
возможной вулканизация с помощью традиционных систем сера —
ускоритель.
Было найдено, что двойные связи, необходимые для реакции
сшивки, можно создать, вводя в структуру сополимера многократно
ненасыщенный алифатический или циклоалифатический углеводород.
Полученные таким образом терполимеры поддаются вулканизации
серой, как и натуральный каучук. В отличие от чистых сополимеров
эти продукты называются ЭПДМ или ЭПТ.
В 1954 г. Циглер и Монтекатини [29] подали заявку на патент
по сополимеризации этилена и а-олефинов, Натта [28] впервые
в 1955 г. описал получение аморфных сополимеров. При подробном
Сополимеры пропилена с этиленом
309
изучении сополимеризации были испытаны и различные комбинации
этилена с пропиленом. Оказалось, что в присутствии обычных катали-
заторных систем Циглера при сополимеризации зтилена с небольшим
Содержание мономера, мол.
Рис. 70. Влияние состава смеси этилен — пропилен на сополимеризацию
(растворитель бензин с температурой кипения бензина 60—90 ?С, темпера-
тура 50 ?С, давление 1 кгс/см2):
.....скорость полимеризации;--- вязкость (0,1 % раствор в декалине, 135 °C); —-
плотность; а — нерастворимая часть; б — растворимая часть.
Рис. 71. Влияние состава смеси этилен — пропилен на сополимеризацию (рас-
творитель бензин с температурой кипения 60—90 ?С, температура 50 ’С, да-
вление 1 кгс/см2):
. . . . содержание нерастворимой части; -— содержание пропилена (по содержанию СН«-
групп, определенному ИК-спектральным анализом).
количеством пропилена, или наоборот, образуется много кристал-
лического полимера и мало аморфного. Напротив, при равномер-
ном соотношении компонентов получается в основном раствори-
мый аморфный сополимер. Чем богаче пропиленом исходная газооб-
разная смесь, тем выше ее растворимость в диспергаторе.
310
16. Сополимеры пропилена с этиленом
Несмотря на высокую концентрацию мономера, скорость поли-
меризации в присутствии одного и того же катализатора уменьшается
с увеличением отношения пропилен : этилен (рис. 70). Молекуляр-
ный вес полимера при этом тоже уменьшается. Добавка пропилена
влияет на содержание растворимой части в меньшей степени, чем
добавка этилена (рис. 71). Нерастворимые полимеры, богатые эти-
леном и пропиленом, кристалличны, а растворимые — аморфны
или мало кристалличны.
В 1962 г. появились первые сообщения [30—32] о применении
диена как третьего компонента сополимеризации и о возможности
вулканизации такого каучука серой.
16.1. ТЕРМОПЛАСТИЧНЫЕ СОПОЛИМЕРЫ
Нестатистической сополимеризацией пропилена с небольшим
количеством этилена удается получить сополимеры [34], которые
сохраняют характерные для полипропилена твердость, жесткость,
Рис. 72. Влияние добавки пропилена При полимеризации эти-
лена на свойства полимеров:
1 — показатель текучести расплава, 190 °C; г — удлинение при разрыве;
3 — твердость при вдавливании шарика, 60 с; 4 — плотность; 5 — предел
прочности при растяжении.
температуру размягчения, стабильность размеров и хорошие пока-
затели крипа, обладая при этом высоким сопротивлением растрески-
ванию под,напряжением и жаростойкостью. На рис. 72 показана за-
висимость свойств сополимера от состава. Ударная вязкость при
комнатной и пониженных температурах и показатель текучести рас-
плава даже улучшаются.
Подобные сополимеры особенно пригодны для переработки литьем
под давлением, они применяются в больших количествах вместо
чистого полипропилена [35]. Начиная с 1960 г., фирма ICI выпу-
скает в продажу такие сополимеры под названием «пропатен-копо-
16-2. Эластомерные сополимеры,
311
лимер ГВМ-101» (преимущественно для литья под давлением кор-
зин для бутылок и других емкостей) и «пропатен-кополимер ГВМ-102»
(для изготовления молочных, пивных и лимонадных бутылок методом
экструзии с раздувом [36]. Разработки в этом направлении ведет
главным образом фирма ICI Ltd. (следует ожидать значительного
увеличения производства [134].
16.2. ЭЛАСТОМЕРНЫЕ СОПОЛИМЕРЫ
16.2.1а Катализаторы и условия сополимеризации
Для получения сополимеров применяются растворимые и не-
растворимые в углеводородах системы катализаторов. Ниже указаны
системы, нерастворимые в углеводородах:
А1(С2Н5)з+Т1С14* [37-39]
А1(иао-С4Н9)3+Т1С14 [40—41] А1(С6Н1з)з+Т1С14** [37-42] А1(С6Н13)3+Т1С13** [37] А1(С2Н5)2С1+Т1С1з [28-43] А1(С6Н13)3+Т1С12 [37] А1(СвН13)з+УС1з** [44]
A1(C2H5)2C1+VC13 Т1С13/А1С13+Комплексообразователь (эфир, тиоэфир, третичный амин или фосфин) — диэти- ловый эфир VC14+HA1C12 VC14+H3A1-N(CH3)3 VC14+HA1C12 - N(CH3)3 VCl4+HAlBrN(CH3)2 VC14+H2A1N(CH3)2 [45] [46] [47] 48] 48] 48] 48]
* Кристалличность сополимера 10%.
** Аморфные сополимеры.
В качестве катализаторных систем, растворяющихся в углеводо-
родах, предложены следующие:
Al(uao-C4H9)3-)- VC14 A1(C6H13)3+VC14 [49, 50] [51—54]
(AlH)(uao-C4H9)2+VCl4 [55]
A1(C2H5)2C1+VC14 [56]
Al2(C2H5)3Cl3-j-VC14 [57]
Al(w3o-C4He)3+VOCl3 [58-62]
A1(C6H13)3+VOC13 [63-65]
A1(C2H5)2C1+VOC13 [56, 66, 67]
Al2(C2H5)3Cl3-j-VOCl3 [57, 68]
A12(C2H5)3C13+VO(OC2H6)C12 [68, 69
A1(C2H5)2C1+VO(OC2H5)C12 [69, 70
А12(С2Н5)3С13-[-УО(иго-С4Н9)з [68, 71
A1(C2H5)2C1+VO(C5H7O2)2 [70, 72
Al(C2H5)2Cl+V(C5HrO2)3 [73]
Al(C2H5)2Cl-[-VCl(C5H ?O2)2 72]
A12(C2H5)3C13+VO(C5HjO2)2 [74]
А1г(СгН5)зС]3+У(С5Н7О2)з [74]
312
16. Сополимеры пропилена с зтиленом
При гомо полимеризации этилена и пропилена, как и при их со-
полимеризации различные системы катализаторов ведут себя по-раз-
ному. Если А1(С6Н13)3 TiCl3 и A1(CGH13)3 VC13 [44] дают
при гомополимерйзации кристаллические полимеры, то при сополи-
меризации в присутствии этих систем образуется аморфный продукт.
Напротив, системы А1(С6Н9)3 -]- VOC13 [64], А1(С6Н9)3 + VC14 [51]
и А1(С2Н5)2С1 -]- V(C5H7O2)3 [73] дают аморфные полимеры как при
гомо-, так и при сополимеризации. Для образования аморфного
продукта алкильная цепь в алюминийорганических соединениях
должна иметь определенную минимальную длину. Рцдкционная спо-
собность различных катализаторных систем при сополимеризации
тоже различна [28, 37, 4р]:
rC2H« rC3H,
Al(CeH13)3+TiCl4 33,36 0,032
Al(C6H13)3+TiCl3 15,72 0,110
Al(C6H13)3+TiCl2 15,72 0,110
A1(C6H13)3+VC13 5,61 0,146
A1(C6H13)3+VC14 7,08 0,088
A1(C6H13')3+VOC13 17,95 0,065
A1(C2H5)2C1+V(C5H7O2)3 15,0 0,04
A1C2H5C12+VO(OR)C12 17,5 0,05
Максимальная активность катализатора достигается при отно-
шении Al : Me = 1 : 2. В качестве растворителя алифатические угле-
водороды пригодны больше, чем ароматические. Температуры поли-
меризации лежат в области от 0 до —30 °C, катализатор получают
также преимущественно при этих температурах. Если хотят ввести
растворимые в углеводородах катализаторы при температурах выше
О °C, то добавляют комплексообразующие агенты, например простые
эфиры, тиоэфиры, третичные амины или фосфины, содержащие по
•крайней мере один разветвленный алкильный остаток или арома-
тическое кольцо.
Старение катализаторных систем по разному влияет на сопо-
лимеризацию. При введении системы А1(С6Н13)3 VOC13 или
А1(С6Н13)3 4- VC13 состав сополимера при старении катализатора
почти не изменяется [37, 44, 51, 63, 73]. Если же используется
А1(С6Н13)3 -]- V(C5H7O2)3, то при старении катализатора уменьшается
вязкость сополимера [73].
Изменение концентрации катализатора почти не влияет на состав
сополимера, однако уменьшение концентрации VC14 вызывает уве-
личение характеристической вязкости. Это показано на примере
сополимеризации в присутствии системы А1(С6Н13)3 + VC14 в геп-
тане:
Концентрация VCLC Ю4, моль/л.................... 12,7 8.44 5,65 3,75
Время полимеризации, мин........................... 4 6 8 10
£т)] при 135 °C в тетралине....................... 2,14 2,83 3,90 4,44
16.2. Эластомерные сополимеры
313
Выход сополимера проходит через максимум; во многих случаях
это зависит от отношения Al : V; при использовании системы
А1(С6Н13)3 + VC14 это отношение составляет 2,5 [75]. Скорость по-
лимеризации тоже проходит через максимум независимо от величины
отношения Al : V [48]. Ниже показаны оптимальные отношения
Al : V при, сополимеризации с некоторыми катализаторными систе-
мами:
VCh-p А1НС12 + Диэтпловый эфир..................... 2,5—3
VCI4+А1НС12 + Триэтиламин........................... 1,8
VCI4+ НА1Вг • (СН3)а .............................. 1,25
TiCEi-h А1НС12 + Диэтиловый эфир.................... 1,5
VCl4-)-H2AlN(CH3)2.............................. 0,8-1,0
TiC14+HaAlN(CH3)a .................................. 1,0
VCI4+А1Н3+ Триметиламин ........................ 0,6—0,8
TiCh-]-АШ3 + Триметиламин........................... 0,5
V(C5H7O2)3 + A1(C2H5)2C1 [73] ............... . 3,5
Продолжительность полимеризации не влияет на состав сополи--
меров. Однако через некоторое время катализатор, особенно рас-
творимый, теряет активность. Для восстановления активности ка-
тализатора можно добавлять гексахлорциклопентадиен [75]. Таким
образом удается в несколько раз увеличить срок действия катализа-
тора. Реактивацию можно осуществить и нагреванием катализатор-
ной системы [76].
Молекулярный вес этилен-пропиленового сополимера регули-
руется добавкой диалкилцинка [77] в результате обмена алкильного
радикала. Молекулярный вес сополимеров можно регулировать,
и добавкой водорода [78], который гидролитически расщепляет ак-
тивный катализаторный комплекс.
16.2.2. Промышленные методы сополимеризации
Сополимеризацию можно проводить так же, как полимеризацию,
пропилена (см. рис. 69). При периодическом методе реакцию прово-
дят в автоклаве, куда при —65 °C сначала вводят жидкий пропилен,
а затем подают этилен под таким давлением, чтобы газ был нужного,
состава. Оба компонента могут быть растворены в гептане, циклогек-
сане или бензоле. Компоненты катализатора подают отдельно в виде,
растворов в углеводородах. Полимеризация продолжается примерно
10—40 мин, после чего ее прекращают добавкой спирта. Для удале-
ния соединений ванадия и алюминия реакционную смесь обрабаты-
вают кислотами. После очистки добавляют антиоксиданты для ста-,
билизации сополимера.
При непрерывном методе сополимеризации [79] применяется
катализаторная система А1(пзо-С4Н9) + VOC12 в бензольном рас-
творе; отношение Al : V = 4:1. Смесь этилен-пропилен (1 : 3,45) и
раствор катализатора подаются одновременно. Реакция, происходит-
при температуре 39 °C под давлением 5 кгс/см2.
314
16. Сополимеры пропилена с этиленом
16.2.3. Вулканизация этилен-пропиленового каучука
Вулканизация перекисями. Перекисная вулканизация осуще-
ствляется свободными радикалами [80], образующимися в резуль-
тате нагревания смеси с добавленными в нее перекисями до 130—
150 °C. Эти свободные радикалы отнимают водород у полимерных
цепей. Объединение возникших таким образом полимерных радика-
лов ведет к желаемой сшивке цепей. Образующиеся мостики — это
связи углерод — углерод:
R-O-O-R —> 2RO.
2RO. + 2PH > 2P.4-2ROH
Р.Ц-Р- —> Р-Р
где Р = полимер.
Перекисная вулканизация может быть ускорена за счет одно-
временного добавления серы [81]. Сера реагирует с полимерным
радикалом, образуя персульфениловый радикал, который может
присоединяться по месту двойной связи, возникшей в результате дис-
пропорционирования полимерного радикала. При этом образуется
серный мостик. Судя по расходу 3 атомов серы на 1 моль перекиси,
можно сделать вывод о наличии трисульфидных мостиков [82];
предполагается также наличие пентасульфидных мостиков.
Сре^и перекисей для вулканизации этилен-пропиленового кау-
чука самой важной является перекись дикумила [80, 83—93]. Вул-
канизация проводится обычно при 155 °C и продолжается 45 мин.
Чтобы добиться эффективной дополнительной вулканизации можно
произвести кратковременное (—1—2 мин) нагревание до 200 °C.
Температура должна быть 145 °C, так как перекись дикумила ста-
бильна при более низких температурах. Ниже показаны свойства
продуктов, вулканизованных 4.вес. ч. перекиси дикумила (I); 4 вес.ч
перекиси дикумила и 1 вес. ч. серы (II); 4 вес. ч. перекиси дикумила,
2 вес. ч. серы, 10 вес. ч. сурика и 2 вес. ч. хинондиоксима (III):
I
Условия сополимеризации:
Температура, °C...................... 160
Время вулканизации, мин................ 30
Характеристика сополимеров:
Прочность при растяжении, кгс/см2 . . . 189,1
Напряжение при 300%-ном удлинении,
кгс/см2............................... 92,1
Удлинение при разрыве, % . . . 320
и ш
154 154
40 40
233,4 234,8
79,9 96,7
420 415
Для вулканизации предлагаются и другие перекиси: перекись
дибензоила [84, 93, 94]; 2,5-диметил-2,5-бис(бензоилперокси)гексан
[84]; трет-бутилперацетат [84]; бутиловый эфир 4,4-бис(трет-бу-
тилперокси)валериановой кислоты [84]; трет-бутилпербензоат [84,
93]; 2,5-диметил-2,5-бис(трет-бутилперокси)гексан [84]; 2,5-диме-
16.3- Эластомерные терполимеры
315
тил-2,5-бис(трет-бутилперокси)гексин-3 [84]; ди-трет-бутилпере-
кись [84, 92, 93, 95—97]; кумил-трет-бутилперекись [75]; тетра-
хлор-трет-бутилперекись [96, 98]; пероксифталевая кислота [99];
бис [4,4-ди(трет-бутилперокси)циклогексил] пропан [100]; метило-
вый эфир 4,4-бис(трет-бутилперокси)валериановой кислоты [100];
кетальные перекиси [10] и т. д.
Степень перекисной сшивки можно регулировать добавкой к ис-
ходной смеси следующих веществ: аморфного полипропилена или
тройных сополимеров пропилена [82], хинондиоксима, динитробен-
зола, дифенилгуанидина, дивиниладипината, триаллилцианурата,
ди аллилфталата, хинона, полибутадиена («бутон 150»), этилендиметил-
акрилата [84], н-динитрозо-ТУ-метиланилина (Эластопар), дибензо-
ил-п-хинондиоксима, м-финиленбисмалеинимида [102]. Ленас [103]
предлагает ряд моно- и полифункциональных мономеров и полиме-
ров, пригодных в качестве соагентов при перекисной вулканизации..
Вулканизацию можно ускорить добавкой следующих соединений:
железных солей различных органических кислот (фталевой, стеари-
новой, нафтеновой, 2-этилкапроновой, щавелевой), FeCl2, FeCl3,
А1С13, а также комбинаций Fe2O3 с указанными кислотами. Для
улучшения свойств этилен-пропиленового каучука лучше всего
применять печную сажу [80, 104]. В качестве вспомогательных
средств вводят пластификаторы (ароматические масла), силикагель,
глину (последнюю с целью адсорбции примесей).
Другие методы вулканизации. Вулканизацию основаниями можно
осуществлять путем прививки кислотных групп (малеиновый ангид-
рид, малеиновая кислота и т. д.) на этилен-пропиленовый каучук.
Вулканизация хлорированных этилен-пропиленовых сополимеров.
Этилен-пропиленовые сополимеры легко можно хлорировать [105].
Сополимер с 40% хлора мягок, а с 30% еще гибок. Вулканизовать,
такие продукты можно серой и тетраметилтиурамдисульфидом в при-
сутствии ZnO; после добавки меркаптобензтиазола [106] достигается
полная вулканизация и дополнительное улучшение свойств. В ре-
зультате бромирования этилен-пропиленового каучука тоже полу-
чается отлично вулканизуемый продукт [107]. Для вулканизации
галогенированных сополимеров предлагаются также ZnO, полити-
олы + ZnO, дитиокарбаматы, тритиокарбонаты и т. д. [108]. Не-
достатком вулканизованных хлорированных продуктов является их
пониженная стойкость к озону, связанная с образованием двойных
связей во время хлорирования в результате дегидрохлорирования.
16.3. ЭЛАСТОМЕРНЫЕ ТЕРПОЛИМЕРЫ
16.3.1. Проведение терполимеризации
Для осуществления вулканизации этилен-пропиленовых сополи-
меров серой при сополимеризации предложено вводить третий ком-
понент, а именно диен, чтобы ввести в сополимер двойные связи.
316
16. Сополимеры пропилена с этиленом
Концевая двойная связь алифатических диенов вступает в реакцию
сополимеризации, а находящаяся внутри молекулы остается для
последующей вулканизации. Предпочитают вводить диены с одной
концевой двойной связью, так как при применении диенов с двумя
концевыми двойными связями возможна частичная циклизация
Этилен
Пропилен
Растворитель на
регенерацию .
Возврат непро-
реагировавшия
мономеров
Пар
Катализатор
Растворитель
Суспензия
сополимера
Рис. 73. Схема получения этилен-пропиленового по-
лимера по методу U. S. Rubber Со.:
1 — дегазатор; 2 — промывная башня; з — коагулятор.
и вызванное этим загрязнение эластомера нерастворимой фракцией.
У циклических диенов двойная связь в бицикле наиболее активная.
Ниже перечислены диены, предложенные в качестве третьего
компонента для этилен-пропиленовых терполимеров:
Пентадиен-1,4 [30]
2-Метилпентадиен-1,4 [30]
Гексадиен-1,5 [30]
Гексадиен-1,4 [30]
2-Метилгексадиен-1,5 [30]
Декатриен-1,4,9 [109]
11-Этилтридекадиен-1,11 [30]
6-Метилгептадйен-1,5 [30]
Циклогептадиен-1,4 [109]
^ис,!ргс-Циклооктадиен-1,5 [108
110-113]
ц uc ,!f ис-Метилциклоокта-
Диен-1,5 [114]
Дициклопентадиен [30, 110, 115—121]
4,7,8,9-Тетрагидроинден [109] -
Норборнен [110]
2-Метиленнорборнен [30, 122]
Норборнадиен [110]
Алкил-2-норборнадиен (алкил
С8-Са4) [108]
5-Этилиденнорборнен [116]
4-Винилциклогексен [117]
Циклооктатетраендимер [117]
Бицикло[3.2.0]гептадиен-2,6 [109]
1,2-Дивинилциклобутан [109]
1,2,4-Тривинилциклогексан [109]
Для терполимеризации можно использовать те же системы ката-
лизаторов, что и для сополимеризации этилена с пропиленом. Воз-
можность успешного введения третьего мономера в присутствии ка-
тализаторной системы А1(А1к)3 -ф- VOG13 уменьшается в такой по-
следовательности:
Норборнены > 11-этилтридекадиен-1,11 > 5-метилгептадиен-1,5
> пентадиен-1,4 > изопрен
16.4. Свойства и применение этилен-пропиленовых сополимеров 317
1,3- Диены, например изопрен или бутадиен, при нормальных
условиях терполимеризации оказываются неактивными.
Успешно проведенная в промышленных условиях сополимериза-
ция в присутствии катализаторной системы А1(А1к)3 -ф- VG13 с цис,
грс-октадиеном или дициклопентадиеном в качестве третьего
компонента описана Бренноном [123] (рис. 73). ; ,,
В качестве компонентов катализатора при терполимеризации
применяются в основном VC14 и VOG]3, а также А1(С2Н5)2С1 и
А12(С2Н5)3С13 [114].
16.3.2. Вулканизация этилен-пропиленовых терполимеров
Вулканизация терполимеров проводится так же, s как вулкани-
зация диенового каучука. В качестве активаторов рекомендуются
CdO и особенно ZnO [32] или ZnO и стеариновая кислота [124].
Получается вулканизованный продукт с хорошими механическими
свойствами. Применяемые ускорители вулканизации перечислены
ниже:
2-Меркаптобензтиазол [32, 108, НО,
125]
2-Меркаптобензтиазолин [32]
А,А-Диэтилтиокарбамид-2-меркап-
тобензтиазол [32]
Тиурамсульфид [32,126]
Тетраметилтиурамдисульфид [108,
НО, 111, 115, 118]
Тетраметилтиураммоносульфид [32.
125]
Дитиокарбаматы [32,111,126]
Пентаметилендитиокарбамин—кис-
лый пиперидин [32]
Дипентамегилентиурамтетрасуль-
фид [111]
Наиболее употребительно совместное применение тетраметил-
тиурамдисульфида и 2-меркаптобензтиазола с ZnO и S [127]. Быст-
рее всего вулканизуются терполимеры, полученные с циклоокта-
диеном-1,5. Терполимеры, полученные в.присутствии гексадиена-1,4,
2-метиленнорборнена и дициклопентадиена, вулканизуются медлен-
нее [100]. Самая быстрая из всех известных вулканизующих систем
содержит в качестве одного из компонентов теллурдиэтилдитиокар-
бамат [128].
16.4. СВОЙСТВА И ПРИМЕНЕНИЕ ЭТИЛЕН-ПРОПИЛЕНОВЫХ
СОПОЛИМЕРОВ И ТЕРПОЛИМЕРОВ
Вязкоэластические свойства этилен-пропиленовых сополимеров
зависят от химического состава сополимера, распределения моно-
меров (влияние этого фактора наблюдается только при наличии
кристаллических цепных сегментов), среднего молекулярного веса
и молекулярно-весового распределения [129].
Состав сополимеров влияет главным образом на динамические свой-
ства (например .упругое восстановление) и температуру превращения
318
16. Сополимеры, пропилена с этиленом
второго порядка (точка перехода от каучукоподобного к стекло-
образному состоянию). По свойству упругого восстановления
этилен-прцпиленовый каучук похож на натуральный и превосходит
стирол-бутадиеновый и еще больше бутилкаучук.
Ниже сравниваются свойства этилен-пропиленовых терполиме-
ров (ЭПТ) и других типов каучуков:
ЭПТ Нату- ральный каучук Бута- диен- стироль- ный каучук Бутил- каучук Полибу- тадиен
Прочность при разрыве, кгс/см2 210 285 265 140 150
Удлинение при разрыве, % . . . Напряжение при 300%-ном удли- 450 600 550 600 500
нении, кгс/см2 130 90 105 75 80
Твердость по Шору Эластичность, % 65-70 60 62 65 60
при 22° С 45 43 41 8 49
75° С. 50 57 56 36 58
Структурная прочность, кгс/см Упругое восстановление при 20 °C, 15 32 14 11 14
% 50 55 40 20 30
При минимальном упругом восстановлении и увеличении содер-
жания пропилена температура кристаллизации сдвигается линейно
в сторону пониженных температур (при содержании 72% этилена
минимум составляет —50 °C, при содержании 30% этилена — при-
мерно -22 °C) [130].
Молекулярный 'вес этилен-пропиленового каучука не должен
быть слишком высоким, так как очень высокомолекулярные про-
дукты трудно перерабатываются; оптимальными являются каучуки
с вязкостью по Муни от 30 до 50. Полимеры с высоким молекулярным
весом можно перерабатывать, добавив к ним пластифицирующие
минеральные масла. Молекулярно-весовое распределение должно
быть очень узким, ибо в противном случае существенно ухудшаются
динамические свойства. Сополимеры с отрегулированным молеку-
лярным весом и узким молекулярно-весовым распределением хорошо
перерабатываются на смесителях (легко поглощают наполнители,
обладают достаточной клейкостью, поддаются экструзии в калибро-
ванные профильные детали).
Плотность нерулканизованного этилен-пропиленового каучука
0,86—0,87, температура стеклования около —95 °C и температура
хрупкости примерно —100 °C.
Этилен-пропиленовый каучук растворяется в ароматических,
алифатических и хлорированных углеводородах, обладает отлич-
ными электроизоляционными свойствами, особенно после вулкани-
зации. По газопроницаемости этот каучук можно сравнить с натураль-
ным каучуком.
16.4. Свойства и применение этилен-пропиленовых сополимеров
319
Этилен-пропиленовые сополимеры отлично совмещаются с добав-
ками масел, особенно парафиновых и нафтеновых, являющихся эф-
фективными пластификаторами.
Ниже указана рецептура смеси для получения этилен-пропиле-
нового вулканизата, применяемого для электроизоляции (в вес. ч.):
Этилен-пропиленовый
каучук 404 ........ 100
Сажа.................10
Глинозем.............125
Полиэтилен ...... 10
Стеарат свинца .... 2,5
Эластопер............0,5
Петролатум............. 5
Окись цинка ........... 5
Перекись кумила 40 9С 7
Сера .................0,3
Далее приведены физико-механические, тепловые и электриче-
ские свойства вулканизованного продукта:
Плотность, г/см3 .................................... 0,86
Сопротивление растяжению, г/см2................... 56,25
Относительное удлинение, % ........................... 620
Напряжение при 300%-ном удлинении, гс/см2 .... 23,20
Твердость по Шору А................................... 64
Температура стеклования, °C........................... —94
Коэффициент теплопроводности, кал/(см • с • 9 С) . . . 8,5 • 10~4
Электрическая прочность, В/мм...................... 28 103
Удельное сопротивленце*, Ом • см..................20,4-10й
Удельное поверхностное сопротивленце, Ом............ 5 1018
Коэффициент мощности * 0,5
Диэлектрическая проницаемость *....................... 3,7
Тацренс угла диэлектрических потерь при 1000 Гц 1,5 10"*
* После выдержки образца в воде при 75 °C объемное электрическое
сопротивление составило 1,4-10“ Ом-см, коэффициент мощности 2,3, ди-
электрическая проницаемость 4,2.
Очень важным свойством вулканизованных этилен-пропилено-
вых каучуков и этилен-пропиленовых терполимеров является отлич-
ная стойкость к озону, поскольку оба продукта не содержат двойных
связей.
Вулканизованный терполимер набухает в различных углеводоро-
дах и хлорированных углеводородах, но обладает отличной стой-
костью к действию полярных растворителей. Терполимер стоек
к кислотам и щелочам, однако мало стоек по отношению к окисля-
ющим веществам, например азотной и хромовой кислотам.
Ниже приведены рецептура вулканизационной смеси и электри-
ческие свойства вулканизованного терполимера:
Рецептура, вес. ч.:
Терполимер .............................. 100
Окись цинка ............................... 5
Стеариновая кислота ....................... 1
Сера....................................... 1
Тетраметилтиурамдисульфид .............. 0,75
Теллурдиметилдитиокарбамат................ 1,5
320
16. Сополимеры пропилена с этиленом
Электрические свойства:
Удельное объемное сопротивление, Ом-см . . 2,4-1015
Электрическая прочность, В/мм .............. 3 • 104
Диэлектрическая проницаемость при 1000 Гц 2,5
Тангенс угла диэлектрических потерь при
1000 Гц................................... 0,002
Температура хрупкости - вулканизованного этилен-пропилено-
вого полимера лежит около —68 °C, а температура стеклования —
около —60 °C.
Натта [129] изучал свойства терполимеров в зависимости от со-
держания третьего компонента. С увеличением количества несопря-
женного диена уменьшалась характеристическая вязкость невулка-’
низованного каучука, вязкость по Муни снижалась до минимума
при содержании двойных связей 2,2%, а затем снова повышалась.
У вулканизованного продукта предел прочности при растяжении
не уменьшался, удлинение при разрыве снижалось, напряжение
при 300%-ном удлинении увеличивалось, а сила упругого восстано-
вления оставалась постоянной.
По свойствам вулканизованный этилен-пропиленовый каучук
и этилен-пропиленовый терполимер можно сравнить с лучшими сор-
тами синтетического каучука. На первом плане стоит стойкость
к старению, обусловленная насыщенным характером продукта и со-
храняющаяся при повышенных температурах, отличная озоностой-
кость, значительная химическая стойкость. Даже при длительном
действии озона в повышенной концентрации ухудшения свойств
не наблюдается.
Ниже дано сравнение свойств этилен-пропиленового терполи-
мера со свойствами других сортов каучука (этилен-пропиленовый
каучук++ гораздо лучше, + лучше, = равноценен, — хуже)
[120а, 131]:
Нату-
ральный
каучук
Бутадиен-
стироль-
ный
каучук
Бутил-
каучук
Поли-
бутадиен
Удлинение при растяжении
Сопротивление дальнейшему
разрыву.....................
Упругое восстановление . . .
Динамическое поведение . . .
Поведение при низких темпе-
ратурах ....................
Жаростойкость...............
Стойкость к растворителям
Стойкость к старению ....
Озоностойкость.............
Сопротивление истиранию . .
Электрические свойства . . .
Литература
321
Этилен-пропиленовые сополимеры и терполимеры применяются
главным образом в автостроении (покрытия педалей, коврики)
и в машиностроении, для изготовления кабельных оболочек, для
производства прорезиненных материалов, транспортерных лент и рем-
ней, Шлангов с внутренним слоем, губчатой и ячеистой резины.
Применение для автопокрышек еще ограничено, так как клейкость
при конфекционировании и прилипание к полиэфирному и полиамид-
ному корду и к стальной проволоке оставляет желать лучшего.
Однако уже были изготовлены шины на 100% из этилен-пропилено-
вого терполимера и, можно ожидать, что в будущем эта область
приобретет гораздо большее значение. Из этого материала, вероятно,
будут изготовляться шины для легковых автомобилей (в грузовых
машинах при трении шины разогреваются слишком сильно для
этилен-пропиленового каучука). Особенно подходящим материалом
для производства шин кажется этилен-пропилендициклопентадиено-
вый терполимер с высокой вязкостью, низкой степенью ненасыщен-
ности и большим содержанием серы (наполнитель — сажа САФ)
[132].
ЛИТЕРАТУРА
1. G. N a 11 a, Rubber Plastics Age, 38, 495 (1957).
2. Chem. Eng. Progr., 55, № 6, 16 (1959).
3. W. L. C a rri ck et al., J. Am. Chem. Soc., 82, 1502 (1960).
4. L. В e r t i, Chim. e Ind., Milano, 43, 644 (1961).
5. G. C r e s p i, Chim. e Ind., Milano, 43, 769 (1961).
6. L. 0. Amberg, A. E. Robinson, Ind. Eng. Chem., 53, 368 (1961).
7. W. W. С г о u ch, С. M. Tucker, Oil Gas J., 60, № 36, 101 (1962).
8. F. Ze p per nick, Gummi-Asbest-Kunststoffe, 14, 1006, 1922 (1961).
9. W. S. Fedor, Chem. Eng. News, 40, № 11, 88 (1962),
10. H. E. H a x о jr., W. R. Bingham, W. G. Whitehouse, Rubber
Age, 94, 255 (1963).-
11. A. R. Gardner, Product Eng., 34, № 9, 46 (1963).
12. J. M. M i t c h e 11 et al., Rubber Age, 94, 427 (1963).
13. E. Schmidt, Kautschuk, Gummi Kunststoffe, 17, 8 (1964).
14. H. M. С e и д о в, И. A. A p у т ю н о в, М. А. Д а л ин, Изв. вузов,
Нефть и газ, 8, № 10 (1965).
15. R. R. Garrett, Rubber Plastics Age, 46, 915 (1965).
16. Shu-ko Chiao, Hua Hsueh Tung Pao, 4,193 (1965); C. A., 63, 10149d
(1965).
17. F. I n n e s, Rubber J. Intern. Plastics, 147, № 7, 30; № 8, 31 (1965); C. A.,
63, 16581h (1965). •
18. G. Alliger, F. C. W e i s s e r t, Ind. Eng. Chem., Int. Ed., 57, № 8,
61 (1965).
19. J. E. Hauck, Materials Design. Eng., 62, № 1, 101 (1965).
20. W. Cooper, Encycl. Polym. Sci. Technol., 5, 406 (1966); C. A., 66, 76681f
(1967).
21. R. G e r m a n, R. H a n k, G. V a u g h a n, Kautschuk, Gummi Kunst-
stoffe, 19, № 2, 67 (1966).
22. Г. П. Белов, Каучук и резина, № 2, 38 (1964).
23. Е. Е. Schroeder, Chem. Eng., .73, № 24, 132 (1966).
24. D. J. A n g i е г, R. J. Lauria, Rev. gen. caoutchouc Plastiqrues, 43,
691 (1966).
21 Заказ 399
322
16. Сополимеры пропилена с этиленом
25. St. Z а 1 w е г t, J. О Ы о j, Polimery, И, № 2, 61 (1966); С. А., 65, 13917е
(1966).
26. Р. Р. Brown, Rubber Wld, 153, № 5, 78 (1966).
27. G. Bier, Angew. Chem., 73, 186 (1961).
28. Итал. пат. 554803, 1955.
29. Бельг, пат. 538782, 537164, 1954.
30. Е. К. Gladding et al., Ind. Eng. Chem. Product Res. Development,
1, № 2, 65 (1962).
31. G. N a 11 a, G. M a z z a n t i, Rubber Age, 45, 636 (1961).
32. J. I. Verbang, M. S. F a w c e 11, E. J. G о 1 d b e r g, Ind. Eng.
Chem., Product Res. Development, 1, № 2, 70 (1962).
33. Chem. Industrie, 19, № 4, 190 (1967).
34. E. C. A. Horner, Trans. Plastics Inst., 35, № 116, 415 (1967).
35. Modern Plastics, 47, Oktober (1965).
36. Chem. Industrie, 15, 478 (1963); Plaste u. Kautschuk, 14, 178 (1967).
37. G. N a 11 a et al., Chim. e Ind., Milano, 40, 896 (1958).
38. Итал. пат. 581418, 1957.
39. H. M. С e и д о в и др., Азерб. хим. ж., 4, 37 (1963).
40. Англ. пат. 851002, 1957.
41. Бельг, пат. 614644, 1962.
42. Итал. пат. 602970, 1957.
43. С. Bier, L. Lehmann, J. Lengering, Makromol. Chem., 44, 347
(1961).
44. G. N a 11 a, et al., Chim. e Ind., Milano, 40, 717 (1958).
45. Бельг, пат. 631164, 1963.
46. Бельг, пат. 631995, 1962.
47. W. Marconi et al., Chim. e Ind., Milano, 46, 1131 (1964).
48. W. Marconi et al., там же, 46, 1287 (1964). '
49. Англ. пат. 898261, 1957.
50. Итал. пат. 615556, 1959.
51. G. М a z z a n t i et al., Chim. e Ind., Milano, 39, 825 (1957).
52. G. N a 11 a et al., там же, 45, 125 (1960).
53. Франц, пат. 1353179, 1962.
54. Канад, пат. 690531, 1959.
55. Итал. пат. 621604, 1959.
56. Англ. пат. 857938, 1959.
57. R. Е. С u n if i n g h a m, J. Polymer Sci., A3, 3157 (1965).
58. Итал. пат. 587 666, 1967.
59. Пат. США 3147230, 1964.
60. Пат. США 3047551, 1962.
61. R. J. Kelly et al., Ind. Eng. Chem., Product Res. Development, 1, 210
(1962).
62. E. J u n g h a v i v a n, A. G о u 1 d t, C. Bier; Makromolekulare Chem.,
58, 18 (1962).
63. G. N a 11 a et al., Chim. e Ind., Milano, 39, 733 (1957).
64. G. Mazzanti et al., там же, 39, 743 (1957).
65. Англ. пат. 856736, 1960.
66. Англ. пат. 886368, 1959.
67. Франц, пат. 1374265, 1963.
68. Франц, пат. 1370358, 1962.
69. Франц, пат. 1359982, 1962; итал. пат. 638656, 1962.
70. Пат. США 3153023, 1964.
71. Франц, пат. 1367508, 1962.
72. Итал. пат. 638375, 1962.
73. G. N a 11 a et al., Ind. Polymer Sci., 51, 411 (1961).
74. R. E. C u n n i n g h a m, J. Polymer Sci., Al, № 4, 1203 (1966).
75. A. G u m b о 1 d t et al., Markomolekulare Chemie, 101, 229 (1967).
Литература
323
76. G. N a 11 a et al., J. Polymer Sci., 51, 429 (1961).
77. G. N a 11 a et al., Chim. e Ind., Milano, 43, 871 (1961).
78. G. N a 11 a et al., там же, 41, 519 (1959).
79. Итал. пат. 587666, 1957; франц, пат. 1295575, 1960; англ. пат. 933551,
1963.
80. G. N a 11 a et al., Rubber Plastics Age, 42, № 1, 53 (1963).
81. L. O. Amberg, A. E. R о b i n s о n, Rubber Plastics Age, 42, 875,
1211 (1961).
82. A. E. R о b i n s о n et al., Ind. Eng. Chem., Product. Res. Development,
1, № 2, 78 (1962).
83. S. B. R oh inson, G. I. Z i a г n i k, Rubber Plastics Age, 43, № 10,
1117 (1962).
84. L. P. Lenas, Ind. Eng. Chem., Product Res. Development, 3, № 4, 269
(1964).
85. M. D. S t e m m e t, Kautschuk und Gummi, 14, 146, 154 (1961).
86. Англ. пат. 887763, 1959.
87. Пат. США 3041321, 1963.
88. Пат. США 3047552, 1963.
89. Бельг, пат. 617498, 1961.
90. Бельг, пат. 617191, 1961.
91. В. Н. Р е й х и др., Каучук и резина, 20, № 6, 1 (1961).
92. L. Т. Е Ь у, J. V. F u s с о, Rubber Age, 91, 949 (1962).
93. Итал. пат. 587681, 1959.
94. Бельг, пат. 896598, 1957.
95. Англ. пат. 898264, 1959.
96. Англ. пат. 898264, 1959.
97. Англ. пат. 852035, 1960.
98. Итал. пат. 606492, 1959.
99. Итал. пат. 587681, 1959.
100. A. Valvassori, G. Sartori, Rev. gen. caoutchouc plastiques,
43, № 11, 1458 (1966).
101. L. P. Lenas, Ind. Eng. Chem., Product Res. Development, 5, № 2,
138 (1966);
102. R. J. Kelly et al., Rubber Chem. TechnoL, 35, № 4, 1101 (1962).
103. L. P. Lenas, Ind. Eng. Chem., Product Res. Development, 2, № 3,
202 (1963).
104. L. O. Amberg, A. E. Robinson, Rubber Plastics Age, 42, № 7,
875 (1961).
105. M. Bruzzone, G. Crespi, Chim. e Ind., Milano, 42, 1226 (1960).
106. G. Crespi, M. В г trz zone, Preprints A. C. S. Div. J. Petrol. Chem.,
6, № 4, 105 (1961).
107. Англ. пат. 883731, 1958.
108. Бельг, пат. 631580, 1964.
109. G. Sartori, A. Valvassori, G. Mazzanti, Mater, plast. Elasto-'
meri, 32, № 1,3 (1966).
110 G. Sartor , A. Valvassori, M. Farina, Atti Acad. naz. Lincei,
Rend Classe Sci. Fis., Mat. Nat., 35, № 6, 563 (1963).
111. G. N a 11 a et al., Rubber Chem. TechnoL, 36, № 4, 988 (1963).
112. Бельг, пат. 633763, 1963.
113. G. N a 11 a et al., Chem. e Ind., Milano, 45, № 6, 651 (1963).
114. A. Valvassori et al., J. Polymer Sci., C, № 16, 23 (1967).
115. Пат. США 3000866, 1963.
116. Chem. Industrie, 19, № 3, 122 (1967).
117. Niederl. A. S. 6511068, 1966.
118. Бельг, пат. 622040, 1962.
119. Бельг, пат. 636343, 1963.
120. L. О. Amberg, В. F. Brown, Rubber World, 147, № 6, 52 (1962).
21*
324
16. Сополимеры пропилена с атиленом
121. Chem. Age, 89, № 2272, 146 (1963).
122. Англ. паг. 953662, 1964.
123. Р. J. Brennan, Chem. Eng., 72, № 14, 94 (1965).
124. Англ. паг. 934742, 1963.
125. G. N a 11 a et al., Proc. Rubber Technol., 4th Conf., London (Preprint),
1962 S. 12.
126. I. D’. Roche, Rubber Age, 93, № 6, 921 (1963).
127. P. P. В г о w n, Rubber World, 153, № 5, 78 (1966).
128. J. В i g g s, T. С о 1 с 1 о u g h, J. T. К e h n, Rev. gen. caoutchouc
plastiques, 43, 1631 (1966).
129. G. N a 11 a et al., Rubber Chem. Technol., Rubber Rev., 36, 1583 (1963).
130. G. N a 11 a, G. С r e s p i, Rev. gen. caoutchouc plastiques, 37, 1003 (I960).
131. A. G u m b о 1 d t, 01, Z. fur Mineralolwirtschaft, 4, № 2, 50 (1966).
132. R. W. К ip die, S. van der Burg, Rubber Age, 98, № 4, 65 (1966).
133. G. N. F о s t e r, N. W a 1 d m a n, R. G. G r i s к e y, Mod. Plastics,
43, № 9, 245 (1966).
134. Chem. Industrie, 20, 81 (1968).
135. N. H. Phung, G. S. Bhargava, G. Lefebre, Rev. Inst, franf.,
Petrole^Ann. Combustibles liquides, 22, № 4, 642 (1967).
17. ДИСПРОПОРЦИОНИРОВАНИЕ ПРОПИЛЕНА
(ТРИОЛЕФИНОВЫЙ МЕТОД)
В 1964 г. впервые было установлено [1], что пропилен можно
диспропорционировать в этилен и С4-олефины (наряду с высшими
олефинами). В качестве катализатора для этой реакции была взята
пропитанная Мо(СО)в или W(CO)e окись алюминия А12О3, которая
затем активировалась при 540—580 °C. Кроме того, эффективными
катализаторами являются МоО3 на А12О3 и активированный катали-
затор на основе 3,4% СоО, 11,0% МоО2 и 85,6% А12О3 [2—4].
Ниже показан состав продуктов и параметры процесса диспро-
порционирования в присутствии катализатора СоО-MoOs-AljOg
при 160 °C, давлении 31,5 кгс/см2 и меняющейся нагрузке:
Параметры процесса;
Объемная скорость .....................
Конверсия, %...........................
Селективность (С2 + С4), % ........
Состав продукта, вес. %:
Этилен ................................
Бутен-1........................... . .
траке-Бутен-2..........................
1{ис-Бутен-2...........................
Углеводороды С6 и выше.................
4,0 8,5 14,5 33,8 62,0 110,0
41,0 42,9 37,2 26,2 17,3 6,9
86,6 94,1 91,2 93,6 95,9 98,0
23,7 29,8 32,4 30,2 35,0 35,5
6,6 3,6 1,7 1,9 0,9 0,5
37,0 39,9 36,9 40,0 36,0 38,0
19,3 20,8 20,2 21,5 24,0 24,0
13,4, 5,9 8,8 6,4 4,1 2,0
Далее показано влияние температуры
на диспропорционирова-
ние пропилена:
Параметры процесса:
Температура, °C.......................
Конверсия, %..........................
Селективность (С2 + С4), %............
Состав продукта, вес. %:
Этилен................................
Пропилен .............................
Пропан ...............................
Бутен-1...............................
тракс-Бутен-2.........................
^ис-Бутен-2...........................
Изобутан .............................
Пентен-1..............................
Пентен-2..............................
2-Метилбутен..........................
Пентан ...............................
Углеводороды С3 и остаток ............
95 160 205 300
17,9 37,9 42,6 14,6
93,9 91,4 86,2 58,9
6,0 10,0 11,6 1,9
82,1 62,8 57,4 85,4
Следы 0,1 0,2 1,5
0,1 1,9 3,4 2,5
7,3 14,6 13,9 2,7
3,4 7,5 7,8 1,5
— Следы Следы 0,1
— Следы 0,2 0,2
— 0,8 1,7 1,1
— 0,4 0,5 0,2
— 0,1 Следы —
1,1 1,8 3,3 2,9
326
17. Диспропорционирование пропилена
В последующие годы метод был разработан до стадии промышлен-
ного внедрения; в разработке участвовали фирмы Phillips Petroleum
Со. и British Petroleum Со., Ltd.
Наряду с катализаторами на основе молибдена и вольфрама,
к которым, кроме вышеназванных, относятся также MoS2 и WS2 [5],
был предложен катализатор Ве2О, на А12О3 [6—9]. При использо-
вании этого катализатора уже при 25 °C и времени контакта 1,8 с
конверсия пропилена достигает 34,7%; при этом образуется 11,8%
Рис. 74. Схема установки триолефинового про-
цесса (Phillips Petroleum Со.):
1 — реактор для диспропорционирования; 2 — колонна
для регенерации пропана; 3 — ректификационная ко-
лонна; 4 — колонна для очистки бутена.
этилена’и^22,4% бутена-2, что соответствует селективности 98,5%.
Значительно меньше конверсия при применении в качестве носителя
SiO2, TiO2, ZrO2, ThO2 или SnO2.
Увеличив время реакции, можно синтезировать на СоО + МоО3+
+ А12О3 (при 210 °C и 31,5кгс7см2) высшие олефины (вплоть до
С1вН32), пригодные для получения биологически разрушаемого
исходного сырья для моющих средств [10]. Продукты диспропор-
ционирования могут быть использованы также для алкилирования
с помощью BF3/HF [11].
Для получения высших олефинов можно диспропорционировать
и другие олефины, например бутены. Так, в присутствии катализа-
тора СоО + МоО3 + А12О3 из бутена при 163 °C после получасовой
реакции получена смесь, состоящая из 23,2 ч. этилена, 32,2 ч. про-
пилена, 26,1ч. пентена, 17,2 ч. гексена, 1,2 ч. гептена и 0,1ч. ок-
тена [12].
С середины 1966 фирма Shawinigan Chemicals Ltd. строит в Ва-
реннесе (Квебек) крупнотоннажную промышленную установку дис-
пропорционирования пропилена (рис. 74), с тем чтобы превращать
Литература
327
имеющийся еще на мировом рынке избыток пропилена в бутены
и этилен, на которые существует большой спрос.
Исходный пропилен должен быть очнь чистым (>99,5%), ни
в коем случае не должен содержать азотных, фосфорных и серных
соединений и ацетиленов. Этот метод дает выход в единицу времени
на единицу объема около 100; катализатор, о котором подробных
сведений не имеется, необходимо регенерировать каждые 2—10 дней.
Исходным продуктом могут служить также и смеси пропан — про-
пилен. При использовании чистого пропилена конверсия составляет
43—44%, селективность 94—98%. После перегонки получаются
очень чистые продукты: 99,8%-ный этилен и 96,4%-ный бутен-2
(наряду с 3,46% бутена-1). Бутен-2 можно либо подвергнуть алки-
лированию, либо дегидрировать в бутадиен. В настоящее время
бутен-2 в основном и используется для получения бутадиена. Дегид-
рирование можно осуществлять термически или лучше каталитиче-
ски (выход 76,9%) [13]; присутствие бутена-1 при этом нежелательно
[14—16].
При осуществлении диспропорционирования пропилена в промы-
шленных условиях [16, 17] выгодно соединить установку триолефи-
нового метода с нефтепиролизной установкой. В этом случае выход
этилена при низкотемпературном крекинге должен повыситься на
57%, при среднетемпературном крекинге — на 40%, а при высоко-
температурном — на 28%. При такой комбинации пиролизной и три-
олефиновой установок (мощность 100 000 т/год) цена на этилен сни-
зилась бы с 12,4 до 5,3 пфеннигов за 1 кг.
Необходимо однако подчеркнуть, что имеющийся теперь избыток
пропилена, как предполагают, в будущем значительно сократится
и триолефиновый процесс потеряет свое значение.
ЛИТЕРАТУРА
1. R. L. В а и k s, G. С. Bailey, Ind. Eng. Chem., Product. Res. Develop-
ment, 3, 170 (1964).
2. Пат. США 3261879, 1966.
3. Niederl. A. S. 6410487, 1965.
4. Niederl. A. S. 6400549, 1964.
5. Пат. США 3340322, 1967.
6. Niederl. A. S. 6610196, 1967.
7. Niederl. A. S. 6511659, 1966.
8. Niederl. A. S. 6605328, 1966.
9. Niederl. A. S. 6611840, 1967.
10. Пат. США 3296330, 1967.
12. Пат. США 3236912, 1966.
13. Пат. США 3345285, 1967; англ. пат. 1050558, 1966.
14. Hydrocarbon Processing, 46, № 11, 232 (1967).
15. European Chemical News, il, № 264, 34 (1967).
16. P. H. J о h n s о n, Proc. 7th World Petrol. Congr., vol. 5, S. 247.
17. P. W. Sherwood, Gas- und Waserfach, Ausg. Gas- Erdgas, Miinchen,
108, 936 (1967).
ПРИЛОЖЕНИЕ
ПРОИЗВОДСТВО, МОЩНОСТИ И ПОТРЕБЛЕНИЕ ПРОПИЛЕНА
И ПРОДУКТОВ НА ЕГО ОСНОВЕ
Таблица 1
Мощности по производству пропилена в США на конец 1966 г. [1]
I—получение на установках по производству бензина;
II—получение на установках по производству этилена.
Фирма ( Источник । получения Мощность, тыс. т Фирма Источник получения Мощность, тыс. т
Ashland Oil and Refining I 59 Phillips Petroleum Со. I и II 81
Co. Shell Chemical Co. I и II 127
Atlantic Richfield Co. I 114 I и II 145
Amoco Chemicals Corp. I 181 I 59
I 50 Signal Oil and Gas Co. I 41
Chevron Chemical Co. I 72 Sinclair Petrochemicals,Inc. I 100
Cities Service Oil Co. I 145 I 72
Clark Oil and Refining Co. I 32 Sinclair-Koppers II 45
Dow Chemical Co. I и II 27 Skelly Oil Co. I 41
II 123 Sohio Chemical Co. I 50
II 23 Sun Oil Co. I 136
Du Pont de Nemours II 27 Suntide (Sunray D-X) Co. I 32
II 90 Texaco, Inc. I 63
El Paso Products Co. II 27 Texas City Refining Co. I 45
Enjay Chemical Co. I и II 436 Texas Eastman Co. II 45
Enjay Chemical Co. I 200 Tidewater-Oil Co. I 113
B. F. Goodrich Chemical II 41 Union Carbide Corp. II 23
Co. II 45
II 23
Gulf Oil Corp. II 59 II 91
I 118 II 45
I и II 200 II 23
Jefferson Chemical Co. II 68 Union Oil I 63
Marathon Oil Co. I 32 Суммарное поступление с I 7160
Mobil Chemical Co. I и II 236 бензиновых установок
Monsanto Co. II 136 Суммарное поступление с II 1000
Petroleum Chemicals Inc. (Cites Service) II 27 этиленовых установок
II 23 Общее производство I -и II 8160
Petro Gas Producing Co. II 2,7 1
Приложение
32»
Таблица 2
Методы получения пропилена в США [4, 6]
Метод производства Выпуск пропилена, тыс. т
1965 г. 1970 г. 1975 г.
Термический крекинг 1100 1100 1100
Каталитический крекинг 5700 6200 6200
Расщепление этана 90 150 230
Расщепление пропана 540 1150 770
Расщепление нефти 320 500 900—1800
Таблица 3-
Потребление пропилена в США [2—9]
Продукт на основе пропилена Потребление пропилена, тыс. т
1961 г. 1962 г. 1963 г. 1964 г. 1965 г. 1966 Г. 1970 Г. 1975 г. 1980 г.
Акриловая кислота — — — — — — 45,4 90,8 200
Акрило- 16 21 65,8 106,6 242,7 265,4 458,1 648,6 1000
• Масляный альдегид 86 93 93 115,7 142,9 179,2 222,3 315,3 400-
Кумол 53 73 97,5 131,5 165,6 183,7 294,8 519,4 700-
Додецен 218 235 335,7 283,5 145,2 163,3 181,4 129,3 140
Нонен 86 99 79,4 88,5 122,5 133,8 156,4 186,0 230-
Эпихлор- гидрин 18 20 22,7 27,2 31,8 38,6 61,2 97,5 130
Этилен- пропилено- вый каучук — — 2,3 4,5 4,5 9,1 40,8 63,5 100 ‘
Глицерин 39 35 49,9 56,7 61,2 63,5 79,4 83,9 90
Гептен 14 16 186,0 299,4 331,3 353,8 353,8 340,2 350
Изопрен — 18,1 36,2 43,1 49,9 56,7 56,7 60
Изопропи- ловый спирт 485 508 564,7 580,6 591,9 621,4 764,3 873,2 1000-
Полипро- пилен 46 69 104,3 142,9 199,6 294,8 587,4 936,7 1500
Окись пропилена 152 165 206,4 238,1 254,0 299,4 408,2 612,4 800
Прочее — — — — — — 4,5 45,4 90
Суммарно для нефте- химии 1213 1334 1825,8 2111,4 2336,3 2655,9 3717,4 4998,9 6790
Для улуч- шения качества бензина 5000 5000 5900
330
Приложение
Таблица Потребление пропилена в Японии
Продукт на основе пропилена Потребление пропилена, тыс. т
1965 г. 1966 Г. 1968 г. 1970 г.
Ацетон, изопропиловый спирт Акрилонитрил Алкилбензол - Октиловый спирт Полипропилен Окись пропилена Прочее . . 52 173 30 38 78 43 43 72 236 34 39 100 44 63 110 284 34 50 173 59 71 125 360 34 64 280 75 91
Таблица 5 j г Потребление пропилена в Западной Европе [11]
Продукт на основе пропилена Потребление пропилена, тыс. т
1964 г. 1970 г. 1975 г.
Моющие средства 230 100 < 20
Изопропиловый спирт 200 260 305
Окссйспирты 75 95 НО
Кумол 160 215 260
Окись пропилена 81 125 165
Полипропилен 55 370 860
Три- и перхлорэтилен 25 25 25
Глицерин 33 45 65
Эпоксидные смолы и акрилаты .... 18 30 40
Изопрен 13 40 60
Этилен-пропиленовый каучук и прочее 19 30 70
Акрилонитрил — 90 200
Таблица 6
Мощности по производству пропилена в 1967 г. [12—15]
Страна Фирма Мощность, тыс. т
ФРГ Badische Anilin — und Sodafabrik Caltex Deutschland GmbH Deutsche Erdol AG Erdolchemie GmbH Esso AG Hamburg Ghemische Werke Hills AG Sholven Chemie AG Saarland-Raffinerie GmbH 120 185 50 120 (1969 г. — 350) 110 (1970 г. — 200) 38 130 90
Приложение
331
Продолжение табл. 6
Страна Фирма Мощность, тыс. т
ФРГ Rheinische Olefinwerke GmbH 300
1 Франция Union Rheinische Braunkohlenstoff- AG Ammoniac Sarro-Lorrain 50 140
Antar Petroles de 1’Atlantique Compagnie Francaise de Raffinage Esso Standard S. A. F. Naphtachimie S. A. 15 110 150 (1969 г.—280) 104
Великобритания Usines Chimiques de Mazingarbe Rhone-AlpeS Compagnie de Raffinage Shell-Berre Totalchimie British Celanese Ltd. 12 140 90 25 100 (1971 г.) 18,4
British Hydrocarbon Chemicals Ltd. 200 (1971 г.) 330 80 345 178,5 40 12 60 25 157 37 40 300 (1970) 140 90 17
Италия Бельгия British Hydrocarbon Chemicals Ltd. Esso Chemical Ltd. Imperial Chemical Industries, Ltd. Shell Chemical Co., Ltd. Shell Refining Co., Ltd. Asfalti, Bitumi, Cementi e Derivati SpA Anic, SpA Celene, SpA Monteshell Petrochimica Rumianca, SpA Sicedison, SpA Montecatini Edison SpA Societa Industrial Catanese Societa Italiana Resine Cobenam, S. A.
Нидерланды Societa Chimique des Derives du Pet- role (Petrochim) S. A. Gulf Oil Raffinerie 200 150 (1970)
Дания Shell Nederland Chemie N. V. Dow Chemical N. V. DSM Chemicals (Staatsmijnen Lim- burg) Maersk Refinery 190 250 (1969) 85 45
Швеция Svenska Esso A. B. 120 (1970)
Испания Etileno 75 (1969)
- Empresa Nacional (Calvo Sotelo) de Combustibles Liquidos у Lubri- cantes, S. A. Union Espanola de Explosives 100 (1969) 24
Австрия Industries Quimicas Asociadas, S. A. Petrochemie Schwechat 25 70 (1970)
Австралия Altona Petrochemical Co. Pty. Ltd. 15
Канада Shell Refining Dow Chemical of Canada Ltd.
332
Приложение
Продолжение табл. 6
Страна fe Фирма Мощность, тыс. т
Канада Колумбия Индия Япония Южная Африка Турция Венесуэла ГДР Аргентина Польша СССР Бразилия Румыния Пуэрто-Рико Болгария ЧССР Imperial Oil Ltd. Sha winigan Chemicals Ltd. Shell Canada Ltd. Empresa Colombina de Petroleos Herdillia Chemicals Ltd. National Organic Chemical Industries Daikyowa Sekiku Kagaku К. K. Idemitsu Sekiyu Kagaku К. K. Japan Olefin Chemicals Kasei Mizushima К. K. Maruzen Sekiyu Kagaku К. K. Mitsubishi Yuka К. K. Mitsui Sekiyu Kagaku Kogyo К. K. Sumitomo Chemical Co. Tonen Sekiyu Kagaku К. K. Nippon Petrochemicals Co. South African Coal, Oil and Gas Corp. Petkim Petrokhimiya A. S. Institute Venezolano de Petroquimia Leuna-Werke ««Walter Ulbricht» VEB Erdolverarbeitungswerk Industries Petroquimicas Argentines Koppers S. A. Yacimientos Petroliferos Fiscales Petroquimica Argentina S. A. Polimex Техмапшмпорт Union Carbide do Brasil S. A. Petroquimica Uniao S. A. Нефтехимический комбинат Питешты Union Carbide Caribe, Inc. Phillips Puerto Rico Corp. Commonwealth Oil/PPG 100 125 10 50 20 22 75,6 30 105 72 176 172 200 175 154 15 20 82 18 140 (1970) 12 8 10 52,4 26 30 30 130 (1970) 100 (1972) 51 31,5 270 53 30 (1971)' 35 30
Югославия Чили Южная Корея Финляндия Organsko Kemijska Industrija (OKJ) Petroquimia Dow Corea Oil Co. Neste Oy 20 43,1 75 (1971)
Приложение
333
•л одет 3720 1100 1 1 О 1 1 1 - 240 О «чч 1 1 S ° 1 № 1
•Л 8961 3400 820 1 1 1 1 1 1 [ I 1 1 ю сч 1 [
•л Д961 3110 730 457 00 ео 1 1 1 СО 1 1 1 1 1 1 СО ю
•л 9961 2660 610 360 311 1 314 1 г-- 120 о ю сТ 1 1 ]
•л 9961 2360 460 251 260 250 1 1 о «чч 100 I л СО 1 1 со 1
•л V96I 1620 333 240 252 180 6 1 CSJ О CD CSJ CD ci ю «чч 1 1 1
•л 8961 1524 270 220 1 163 150 1 I 1 1 1 1 1 1 1
•л 8961 1334 105 176 1 152,5 О 1 I 1 1 1 1 1 1
•л 1961 1213 63,5 142 1 126 133 1 I 1 [ 1 1 1 1 1
•л 0961 1124 ю сч 113 1 113,2 1 ю 1 1 1 1 1 1 1 !
•л 6961 902 о I 107,6 1 1 1 1 1 I 1 1 1 1
•л 8961 862 ео О ю 1 58,5 СО 1 1 1 1 1 1 1 1 1
•л Д961 815 i О ео 32,5 1 1. 1 1 1 1 1 1 1 1 1
•л 9961 594 1 00 1 СЧ 1 1 1 1 1 1 1 1 1 1
•л 896Г 553 1 00 1 1 1 1 1 1 1 1 1 1 1 1
Страна США Япония . . . ФРГ Великобрита- ния . . . . Франция . . . Италия .... Канада .... Австрия ; . . . Нидерланды Индия .... Бразилия . . Бельгия . . . Испания . . . Аргентина . . Мексика . . .
334
Приложение
Таблица 8
Мощности по производству изопропилового спирта в 1967 г.
Страна Фирма Мощность, тыс. т
США* Enjay 218
Shell Chemical Со. 82
150
23
Union Carbide 59
112
127
Аргентина Carboclor Industries Quimicas 12
Yacimientos Petroliferos Fiscales 8
1,2
Австралия Shell Chemical Pty. Ltd. 15
Бразилия Rhodia Industries Quimicas e Textels 4,2
Канада Shawinigan Chemicals Ltd.
Франция , Naphtachimie S. A. 30
Cie. Francaise de Produits Chim. Shell 70
ФРГ Rheinpreussen AG. 70
Hibemia-Chemi e 30
Badische Anilin — und Sodafabrik 6
Scholven-Chemie 50
Италия Monteshell Petrochimica SpA 15
Япония Nippon Petrochemicals Co. Ltd. 13
Мексика Petroleos Mexicanos 22,7
Нидерланды Shell Nederland Chemie N. V. 120
Испания Union Espanola de Explosives S. A. 15
Industries Quimicas Asociados 15
Великобритания British Celanese Ltd. 20
British Hydrocarbon Chemicals Ltd. 30,6
Imperial Chemical Industries Ltd. ' 45
Shell Chemical Co. Ltd. 130
25,5
BP Chemicals 50
Индия Union Carbide India Ltd. 12
* Данные по США приведены на конец 1966 г.
Таблица 9
Производство изопропилового спирта в различных странах (в тыс. т)
f* fi si fi Si
Страна >4< CD 00 О 04 co CD о
Ci Ci 01 Ч^ es О чЧ a> чН Ci 4^ Ci Ci чЧ Ci Ci
США 390 440 466 535 580 665 680 700 720 795 902
ФРГ — — — — 30 — 60 60 — — —.
Велико- британия — — — — — 123 121 130 — — —
П риложение
335
Таблица 1О
Потребление изопропилового спирта в США [2] (в тыс. т)
Назначение 1963 г. 1968 Г. Назначение 1963 г. 1968 Г
Ацетон Растворители Химикаты 340 82 52 386 95 59 Лекарственное сырье и фармацевтические' про- дукты ? . Прочее Экспорт 34 54 13 50 54 18
Таблица 1£
Мощности по производству акрилонитрила в 1967 г. [1, 17,z 18]
Страна Фирма Мощность, тыс. т Метод получения
США* American Cyanamid Со. 54 Sohio
Du Pont 91 Пропилен 4- NO
82 Sohio
Goobrich 20 SPhio
Monsanto 145 Sohio
56 C2H2 4- HCN
Sohio 91 Sohio
Union Carbide 41 Этиленциангидрин;
Канада Imperial Oil Co. 14 Sohio
Франция Ugilor 30 Distillers
Ugilor 30 Ugilor-BP
St6. Chimique de Carbonnages 40 —
ФРГ Erdolchemie GmbH 85; Bayer/Sohio
130 (1970)
BASF 25 Sohio.
Knapsack Griesheim AG 25 C2H2 4- HCN
Индия Indian Government 16 C2H2 4- HCN
Италия Acrilsarda, Rumianca SpA 25 Sohio
Anic 16 SNAM-Progetti
Societa Industrial Catanese (Sin- cat) Societa Edison-Azienda Chimica 65 (1968) Edison
17 Sicedison
(Sicedison)
-336
Приложение
Продолжение табл. И
Страна Фирма ' Мощность, тыс. т Метод получения
Япония Asaki Kasei Kogyo К. К. 103 (1970) Asahi/Sohio
Kassei Mizushima К. К. 46,8 Sohio
(Mitsubishi)
Mitsui Sekiyu Kagaku 21,6 Sohio
Kogyo К. K.
Nitto Chemical Industry Co. 32 Sohio
Sumitomo, Chemical Co. 60 (1970) Sohio
Showa Denko К. K. 25 Sohio
Toyo Koatsu Industries Co. 10 Sohio
11 Sohio
Мексика Petroleos Mexicanos 26 Distillers
Нидер- DMS Netherlands/Sohio 45 Sohio
ланды Nederlandse Stikstof Maatschap- 40 —
pij N. V.
Велико- Border Chemicals Ltd. 120 Distillers
Британия Monsanto Chemical Ltd. 68
Южная Chung-Ju Fertilizer 12
Корея Corea Oil Co. 26,7 —
ГДР VEB Erdolverarbeitungswerk 20 Sohio
Chemische Werke Buna 15 C2H2 HCN
Болгария Technoimport 22 Sohio
Румыния Industrialimport 22 Sohio
СССР Техмашимпорт 50 Sohio — Asahi
Техмашимпорт 25 Distillers/Ugine
Польша Polimex 10 Ugine
Китай China National Technical Import 10 OSW-Lurgi
Турция Petkim Petrokimya 5,5 —
Испания Paular 20 (1969) Sohio
Пуэрто- Commonwealth Oil Refining 45 Sohio
Рико
Австрия Osterreichische Stickstoffwerke (1970) bsw
Пакистан —. 5 Sohio
Египет — 6 —
* Данные по США приведены на конец 1966 г.
Таблица 12
Производство акрилонитрила (из ацетилена и из пропилена)
в разных странах (в тыс. т)
Страна 1961 г. 1962 г. 1963 г. 1 1964 г. 1965 г. 1966 г. 1967 г. 1968 г. । 1970 г. 1
США 135 166 206,5 269,5 350 422 500 530 600
Япония 25 29 — 78 153 166 190 210 —
Франция .... — 10 — — 18 25 — — 70
Италия — — — — — — 32 — —
Приложение
337
Таблица 15
Потребление акрилонитрила в Японии [19] (в тыс. т)
Назначение 1965 г. 1966 г. 1967 г. 1969 г. 1971 г.
Волокно 87 93 123,8 169,4. 201,3
Полимеры АБС и САН . . . 6 10 13,5 19,7 25,9
Нитрильный каучук 3 4 4 5,2 6,0
Прочие 8 9 14,1 16,6 19,3
Экспорт 47 41 15 12 12
Всего 151 157 170,4 222,9 264,5
Таблица 14
Мощности по производству окиси пропилена и пропиленгликоля
в 1967 г. [1, 17]
Страна Фирма Мощность, тыс. т
США * Бельгия Канада Франция ФРГ Италия Япония Нидерланды Испания Швеция Великобритания Oxiran Corp, (метод стирол — окись пропилена) Celanese Chemical Со. Dow Chemical Со. Jefferson Chemical Co., Inc. Olin Mathieson Chemical Co. Union Carbide Corp. Wyandotte Chemicals Corp. Carbochimique S. A. Marles-Kuhlmann-Wyandotte S. A. Dow Chemical of Canada Ltd. . Marles-Kuhlmann-Wyandotte S. A. Naphtachimie S. A. Progil-Electrochimie BASF Erdolchemie GmbH Farbwerke Hoechst AG Chemische Werke Hiils Celene SpA Montecatini Asahi Denka Kogyo К. K. Asahi Glass Co. , Ltd. Mitsui Kagaku Kogyo К. K. Showa Denko К. K. Shunan Sekiyu Kagaku К. K. Nippon Soda Dow Chemical International N. V. Olin Quimica Alcudia S. А. (метод стирол — окись пропилена) Мо och Domsjo Aktiebolag Imperial Chemicals Ltd. 75 5 55 9 27 90 36 86 36 6 30 9 27 7 30 45 10 12 20 2 8,4 11.5 14 27 9 7,2 30 8 24 3 35
22 Заказ 399
338
П риложение.
Продолжение табл. 14
Страна Фирма Мощность, тыс. т
Великобритания Pfitzer Ltd./BP Chemicals 15
Shell Chemical Co. Ltd. 35
Аргентина Dow Quimica Argentina S. A. 10
* Данные по США приведены на конец 1966 г.
Таблица 15
Производство окиси пропилена в разных странах (в тыс. т)
Страна 1958 г. I960 г. 1962 г. 1964 г. 1965 г. 1966 г. 1967 г. 1968 г. [ 1970 г.
США 59 140 185 258 274 323 372 427 445
Япония — . 2,9 13,3 33,9 — 87,8 87,8 87,8 —
Франция .... 1,5 4,6 9,1 14 17 .— .— — —
ФРГ — -— — 33 — 50 -— — —-
Италия — — — 8 — — — — —
Великобритания — — — 26 — .— — — —
Бельгия .... -— 3 — 4 — — — — —
Таблица 16
Мощности по производству пропиленгликоля в 1967 г.
Страна Фирма Мощность, тыс. т
США ФРГ Бельгия Канада- Франция Италия Япония Celanese Chemical Со. Dow Chemical Со. Jefferson Chemical Co., Inc. Olin — Mathieson Chemical Corp. Union Carbide Corp. Wyandotte Chemicals Corp. BASF Erdolchemie GmbH Chemische Werke Hills AG Carbochimique S. A. Chemcell Ltd. Dow Chemical of Canada Ltd. Naphtachimie S. A. Celene SpA Asahi Denka Kogyo К, K. Asahi Glass Co., Ltd. Mitsui Kagaku Kogyo К. K. Showa Denko К. K. 3 50 50 15 40 10 8 24 27 11 5 6,0 2,4 2,4 6,5
П риложение
339
Продолжение табл. 16
Страна Фирма МОЩНОСТЬ, ТЫС. т
Мексика Policies S. A. de С. V.
Швеция Мо och Domsjo Aktiebolag 5 ’
Великобритания ICI Ltd. Shell Chemical Ltd. 13
Аргентина Dow Quimica Argentina S. A. ' 10
Таблица 17
Потребление окиси пропилена в США [20, 21] (в тыс. т)
Назначение 1959 г- I960 г. 1965 г. 1966 г. 1971 Г.
Пропиленгликоль 59 57 97 100 113
Полигликоли и полиэфиры для получения полиуретанов . . 20 32 94 126 285
Полипропиленгликоль как тор- мозные ЖИДКОСТИ 9 9,5 10 10 11
Поверхностно-активные веще-
ства 5,5 6 9 11 20
Прочее (в том числе изопропа-
ноламины) 11 42 72 57 25
Всего 105 147 282 304 454
Таблица 18
Потребление окиси пропилена в Японии [22] (в тыс. г)
Назначение 1966 г. 1967 Г. 1968 г. 1969 г. 1970 г.
Пропиленгликоль Полипропиленгликоль .... Прочее 13,6 34,6 0,6 15,2 38,8 0,7 16,9 43,2 0,8 18,7 47,2 0,9 20,8 51,3 1,0
Всего 48,8 54,7 60,9 66,8 73,1
22*
340
П риложение
Таблица 19
Потребление пропиленгликоля в Японии [22] (в тыс. т)
Назначение 1966 г. 1967 г. 1968 г. 1969 г. 1970 Г.
Ненасыщенные полиэфирные СМОЛЫ 8,0 9,1 10,3 11,6 13,1
Другие промышленные цели 1,8 2,0 2,2 2,4 2,6
Зубной порошок ...... 0,56 0,59 0,62 0,65 0,68
Косметика 1,0 1,1 1,21 1,33 1,46
Пищевая промышленность . . 1,5 1,65 1,82 2,0 2,2
Медицина 0,36 0,4 0,44 0,48 0,53
Увлажнение табака 1,0 1,05 1,1 . 1.15 1,2
Различные фармацевтические препараты 0,39 0,43 0,47 0,52 0,57
Всего 14,61 16,32 18,16 20,13 22,34
Таблица 2
Потребление полипропиленгликоля в Японии [22] (в тыс. т)
Назначение 1966 г. 1967 г. 1968 г. 1969 г- 1970 г.
Гибкий поропласт 28,0 30,8 33,6 36,4 39,2
Жесткий поропласт 1,02 1,32 1,74 2,28 2,94
Лаки 0,75 0,9 1,05 1,2 1,32
Эластомеры 0,02 0,04 0,13 0,23 0,38
Прочее 2,4 2,7 3,1 3,5 3,9
Общее потребление внутри стра-
ны 32,19 35,76 39,62 43,61 47,74
Экспорт 2,4 3,0 3,6 3,6 3,6
Всего 34,59 38,76 43,22 47,21 51,34
Таблица 21
Мощности по производству кумола в 1967 г. [30]
Страна Фирма Мощность, тыс. т
США Gulf Oil Со. 140
200
Monsanto Со. 90
Texaco Inc. 64
Clark Oil and Refining Corp. 54
Marathon Oil Co. 68
Coastal States Petrochemical 45
Приложение
341
Продолжение табл. 21
Страна Фирма Мощность, тыс. т
США DX-Sunray Oil 43
Shell Chemical 36
Chevron 36
Skelly Oil 36
Hercules, Inc. 27
Amoco Chemical 23
Dow Chemical Co. 55
Бельгия Societe Chimique des derives du petrole 30
Канада British American Oil Co. Ltd. 50
Франция Atlantique Progil Electrochimie 80
Rhone-Poulenc 100
ФРГ Chemische Werke Hills AG. 90
BASF 10
Farbenfabriken Bayer AG . 20
Scholven-Chemie 50
Индия Herdilia Chemicals Ltd. 14,5
Италйя Societa Italiana Catanese 100 (1969)
Societa Italiana Resine
Societa Italiana fenolo acetone 70
Япония Mitsui Sekiyu Kagaku Kogyo К. K. 60
Великобритания British Hydrocarbon Chemicals Ltd. 115
ГДР Leuna-Werke «Walter Ulbricht» 45 (1969)
Советский Союз — 95
— 83
— 25
Румыния Завод «Георге Георгиу-Деж» 50
ЧССР — 45
Нидерланды Gulf Oil Raffinaderij 150 (1970)
Таблица 22
Производство кумола в разных странах (в тыс. т)
Страна 1956 г. со 1960 г. 1962 г. 1964 г. 1 1965 г. 1966 г. 1967 г. оо со 05 | 1970 г.
США 73 79 118 145 250 300 448 613 795 758
ФРГ — — 140 —
Великобритания — — — — 60 — — — — —
Франция .... — — 31,8 44,5 136,5 150 — — — —
Канада — — 9,5 __ — — . — — — —
Италия — —- — — 50 — — — —
Бельгия .... — — — — 15 — — — — —
342
П риложение
Таблица 23
Мощности кумольно-фенольных установок в разных странах
Страна Фирма Мощность no производст- ву фенола, тыс. т Мощность по производст- ву ацетона, тыс. т
США Allied Chemical Corp. 113 62
California Chemical Corp. 23 13,5
У Clark Oil and Refining Corp. 23 13,5
Hercules, Inc. 23 13,5
Monsanto Co. 63 37
Shell Chemical Co. 23 13,5
Union Carbide Co. 125 74
Skelly Oil Co. 23 13,5
U. S. Steel Co. 90 54
Канада Shawinigan Chemicals Ltd. 44 26
Франция Progil Electrochimie 20 12
Италия Rhone — Poulenc Societa Italiana Resine 50 30
Societa Italiana fenolo acetone 40 24
Societa Edison Chimica 130 80
Великобритания ICI Ltd. 20 12
Hydrocarbon Chemicals Ltd. (1970) 40 24
ФРГ Phenolchemie GmbH 156 96
Индия Herdilia Chemical Ind. 15 9
Япония Mitsui Sekiyu Kagaku Kogyo 36 21
Советский Союз 45,7 27
40,6 24
41,2 24
40,6 24
30 18
45 27
60 36
45 26
ЧССР Slovnaft 22 13
Польша — 30 18
Румыния Завод «Георге Георгиу-Деж» 33 19,5
Болгария — 30 18
Югославия — 6 3,5
ГДР Leuna-Werke «Walter Ulbricht» 30 18
Бразилия Rhodia Industrias Quimica e Textels 45,4 27
Таблица 24
Производство фенола в разных странах (в тыс. т)
Страна 1960 г. ' • 1962 г. 1963 Г. 1964 г. 1965 г. 1966 г. 1967 г. 1968 г. 1970 г.
США 331 351 401 482 532 578 650 720 880
Япония — —. — — 84 99 118 — —
Приложение
343
Продолжение табл. 24
Страна 1960 г. 1962 г. 1 1963 г. 1964 г. 1965 г. 1966 г. 1967 г. 1968 г. 1970 г.
ФРГ 102 106 124 13Q 156 165 175 190
Великобритания — — — 47,2 48,3 77,2 81 — —
Италия — — — — 105 115 120 120 —
Франция .... 54,0 72 Д 83 89 93,6 — — — —
Испания .... — — 2,2 2,2 2,2 2,3 — — —
Нидерланды . . . — — — — 24 — — — —
Таблица 25
Расширение мощностей и производства по отдельным методам
синтеза фенола в США [31, 32] (в %)
Метод Мощность Производство
1965 г. 1959 г. 1965 г. ‘
Сульфирование 20 27 23
Хлорирование 19 30 22
Метод Рашита 20 20 21
Кумольно-фенольный синтез 32 23 29
Окисление толуола . . . 3 — 1 5
Окисление циклогексана 4 — Г 5
Таблица 26
Потребление фенола в США [9, 33] (в тыс. т)
Назначение 1962 г. 1963 г. 1968 г. 1970 г. 1980 г.
Фенольные смолы 201 182 295 285 455
Алкилфенолы 24 36 40 41 84
Бисфенол А 23 27 33 45 100
Капролактам 23 27 33 36 45
Адипиновая кислота 16 16 20 21 23
Очистка нефти 19 20 25 26 27
2,4-Д 1 11 14 18
Пентахлорфенол J 10 11 24 14 18
Пластификатор 14 21 29
Салициловый эфир ... .1 А7 11 14 18
Экспорт I 21 21 18
Прочее J - И 27 45
344
Приложение
Таблица 27
Потребление фенола в Японии [34] (в тыс. т)
Назначение 1966 г. 1967 г. 1968 г. 1969 г. | 1970 г. 1
Фенольные смолы 29 33 37,4. 42,4 47,9 1
Алкилфенолы 7,1 9.4 10,8 12,4 14.3
Бисфенол А 4,4 5.3 5,8 6,4 7.0
Найлон 34 18,4 12 — —
Пентахлорфенол 2,8 2,9 2,9 2,9 2,9 ;
Прочее . 10,2 11,0 11,0 12,9 13,9 |
Экспорт 2' 2,5 3 3,5 4
Всего 89,5 82,5 82,9 80,5 90,0 ;
Таблица 28
Мощности по производству ацетона [35]
Страна Фирма Мощность, тыс. т i Метод
' США* Аргентина Бразилия Канада Франция Shell Chemical Corp. Union Carbide and Chem. Co. Allied Chemical Corp. Humble Oil (Enjay) Co. Puplickers Industries Co. Tennessee Eastman Co. Monsanto Chemical Co. Celanese Corp, of America Chevron Chemical Co. Clark Oil and Refining Co. Skelly Oil Co. Hercules Inc. Carboclor Industrias Quimicas S. A. I. C. Rhodia-Industrias Quimicas e Textels Shawinigan Chemical Ltd. Shell Canada Ltd. Naphtachimie S. A. ProgiLElectrochimie 95 68 45 59 54 9 74 64 50 23 20 36 16 16 14 14 14 10,5 4,2 26 15 12 Кумол Изопропиловый спирт Изопропиловый спирт Изопропиловый сппрт Изопропиловый спирт Изопропиловьш сппрт Кумол Кумол Изопропиловый сппрт Ферментация Изопропиловый сппрт Кумол Окисление углеводоро- да Кумол Кумол Кумол Кумол Изопропиловый сппрт Изопропиловый сппрт Кумол Изопропиловый сппрт Изопропиловый спирт Кумол
П риложение
345
Продолжение табл. 28
Страна Фирма Мощность, НВТ, т Метод
Франция Rhone-Poulenc 30 Кумол
Ugine
Cie Francaise de Produits 30 Изопропиловый спирт
ФРГ Phenolchemie GmbH 96 Кумол
Rheinpreussen AG 96 Изопропиловый спирт
Индия Herdilla Chemicals Ltd. 9 Кумол
10 Ферментация
Италия Societa Italiana fenoloacetone 24 Кумол
Societa Edison Chimica 80 Кумол
Япония Daikyowa Sekiku Kagaku К. K. 27 Ферментация
Kasei Mizushima К. K. 15 Изопропиловый спирт
Mitsui Sekiyu Kagaku 21 Кумол
Kogyo К. K.
Nippon Petrochemicals 26,5 Изопропиловый спирт
Co Ltd.
Kyowa Yuka Co. 60,3 /4 Q79\ Окисление пропилена
Мексика Quimica General S. A. 10 Изопропиловый спирт
Испания Union Espanola de Explosives 13 Изопропиловый спирт
S. A.
Industrias Quimicas Asociados 8 Изопропиловый спирт
Великобритания British Hydrocarbon 40 Кумол
Chemicals Ltd.
ICI Ltd. 30 Изопропиловый спирт
12 Кумол
Shell Chemical Co. Ltd. 25,5 Изопропиловый спирт
Courtaulds Ltd. 14,3 Изопропиловый спирт
Distillers Co., Ltd. 37,8 Изопропиловый спирт
Советский Союз — 27 Кумол
— 24 Кумол
— 24 Кумол
— 24 Кумол
— 18 Кумол
— 27 Кумол
— — Кумол
— 36 Кумол
— 27 Кумол
ЧССР Slovnaft 13 Кумол
Польша — 18 Кумол
Румыния Завод «Георги Георгиу-Деж» 19,5 Кумол
Болгария — 18 Кумол
Югославия — . 3,5 Кумол
ГДР Leuna-Werke «Walter Ulbricht>> 18 Кумол
Нидерланды Shell Nederland Chemie N. V. 40 Изопропиловый спирт
Данные По США приведены на 1967 г.
346
П риложение
Таблица 29
Производство ацетона в разных странах (в тыс. т)
Страна 1958 г. 1960 г. 1962 г. 1963 Г. 1964 г. 1965 г. 1966 г. 1967 г. 1968 г. СП
США .... Великобрита- 280 345 385 427,2 457,2 505 562 626 680 780
НИЯ .... — — - 103,2 116,9 117,7 124,5 130
Франция . . 31,4 49,0 57,0 63,0 69,3 72,0 76,3 100
Япония . . . Индия . . ФРГ .... 8,1 11,1 17,5 0,5 26,2 0,9 34,0 0,6 55 59’0 0,7 83,0 96,0 11 12 20
Италия . . . — 30
Испания . . — ' 1
Мексика . . Пакистан . . — —• — 7 — — — 9
Таблица 30
Потребление ацетона в США [2, 9] (в тыс. т)
Назначение 1963 г. 1968 г. 1970 г. 1980 г.
Метилизобутилкетон 77 110 136 168
Метилизобутилкарбинол 25 26 27 32
Метилметакрилат 59 88 120 254
Бисфенол А Изофорон, гексиленгликоль, окись мези- 9 11 15 35
типа, диацетоновый спирт Растворители (всего) ......... 33 36 36 40
100 115 122 155
для лаков и красок 45 48 ' —
ацетатцеллюлозы 18 18 — —
виниловых смол 20 29 —
ацетилена • • •, 17 20
Фармацевтические продукты 20 29 36 50
Экспорт 15 8 9 9
Прочее 5Q 48 41 50
Всего 394 471 542 793
П риложение
347
Таблица 31
Мощности по производству эпоксидных смол
Страна Фирма Мощность, тыс. т
Австралия Shell Chemical (Australia) Pty Ltd.
Канада Shell Canada Ltd. —
ФРГ Rheinische Olefinwerke GmbH 4
Ruhrchemie AG —
Dow Chemical Co. —
Япония Asahi-Ciba-Ltd. 4
Mitsubishi Yuka К. K. 8
Нидерланды Shell Nederland Chemie N. V. 10
Швейцария CIBA Ltd. 8
Великобритания Shell Chemicals UK 8
США Celanese Corp. 11,3
CIBA Products 22,7
Dow Chemical Co. 15,9
Reichhold Chemicals Ltd. 4,5
Shell Chemical Co. 31,8
Union Carbide Corp. 11,3
Мексика Productora Quimica de Jalisco S. A. 1,0 -
Бразилия Cloroquimica SA (1969)
Франция Prochal (Pechiney-Saint-Gobain)
ГДР Leuna-Werke «Walter Ulbricht» 1
Таблица 32
Производство и сбыт бисфенола А, эпоксидных смол
и поликарбонатов в США [37] (в тыс. т)
Год Производство бисфенола А Сбыт эпоксидных смол Сбыт поликарбонатов
1958 13,9 18,7
1960 22,9 30,0 0,2
1962 28,2 39,2 1,8
1963 32,8 39,6 3,6
1964 42,2 47,7 5,0
1965 44,1 54,1 6,9
1966 54,1 60,0 8,2
1970 84,0 90,0 23—30
Таблица 33
Мощности по производству глицерина
Страна Фирма Мощность, тыс. т Метод
США Atlas Chemical Ind. Dow Chemical Co. 11,5 32 Аллилхлорид Аллилхлорид
348
П риложение
Продолжение табл. 33
Страна Фирма Мощность, тыс. т Метод
США Olin Mathieson Chemical 18 Окись пропилена
Shell Chemical Co. 50 16 Аллилхлорид Акролеин
F. M.C. 18,5 Аллиловый спирт -|- надук- сусная кислота
Нидерланды Shell Nederland Chemie N. V. 25 Аллилхлорид Эпихлоргидрин
Советский Союз T ехмашимцорт 60 Аллилхлорид
— 11 Аллилхлорид
Италия ANIC SpA 48
ГДР Rumianca SpA 5 Аллилхлорид
Leuna-Werke «Walter Ulbricht» 1,2 Аллилхлорид
Франция Solvay und Cie. 6 Аллилхлорид
Япония Showa Denko К. K. 8,8 Аллилхлорид
Польша Daicel Ltd. 12 Аллиловый спирт 4- надук- сусная кислота
— 13 Аллилхлорид
ФРГ Deutsche Solvay-Werke (1970) Аллилхлорид
Таблица 34
Производство глицерина в США
Производство, тыс. т О СЗ 1О со ОС
со со со со со со СО со
0) 05 05 05 05 05 05 05
Синтетический глицерин .... 65 63 72 81 91 93 104 114
Общее производство глицерина . 136 111 137 148 157 163 172 180
Таблица 35
Потребление глицерина в США [9] (в тыс. т)
Назначение 1963 г. 1970 г. 1980 г.
Алкидные смолы 35 36 36
Целлофан 27 23 18
Лекарственное сырье, косметика . . . 27 41 57
Увлажнение табака 21 30 43
Пищевая промышленность 13 16 23
Взрывчатые вещества 9 9 И
Прочее И 15 25
Всего 143 170 213
П риложение
349
Таблица 36
Мощности по производству оксоспиртов
Страна Фирма Мощность, тыс. т Конечный продукт
ФРГ Япония Нидерланды Пуэрто-Рико Великобритания Австралия США Италия Австрия Франция Польша Румыния Швеция Советский Союз Испания ЧССР Болгария Корейская Народ- ная республика Канада BASF Farbwerke Hoechst AG Erdolchemie GmbH Chemische Werke Huis AG Ruhrchemie Daikyowa Sekiku Kagaku K. K. Japan Butanol Mitsubishi Kasei Kogyo К. K. Mitsubishi Kasei Kogyo К. K. Kasei Mizushima Toneri Sekiyu Kagaku К. K. Chisso Corp. Nissan Petrochemicals Konam N. V. Union Carbide Caribe Inc. W. R. Grace und Co. ICI Ltd. Shell Chemicals UK CSR Chemical Air Products and Chemicals, Inc. Enjay Chemical Co. Shell Chemical Co. Texas Eastman Co. Tidewater Oil Co. Dow Badische Co. Union Carbide Corp. 1 Union Carbide Corp, j Celene SpA Montecatini Edison SpA Petrochemie Schwechat AG Courrieres Kuhlmann Oxochimie Mo och Domsjo BASF Espanola SA Chemicke Zavody Korea Industrial Equipment BASF Canada Ltd. 150 140 12 120 150 25 12 30 7,9 38 12 5,4 42 40 25 ИЗ 20 50 35 56 20 91 100 38 30 45 (1969) 80 45 56 20 28 (1970) 20 (1969) 30 (1971) 4 45 (1971) Бутанол Бутанол Бутанол Бутанол Бутанол Октанол Бутанол Бутанол Октанол Октанол Октанол Октанол Бутанол Бутанол Бутанол Октанол Бутанол Бутанол, октанол Спирты Бутанол, октанол Бутанол Октанол Бутанол Спирты Октанол Октанол Октанол Бутанол, октанол Бутиральдегид Бутанол, октанол С5— С13-спирты Октанол Бутанол Бутанол, октанол Бутанол, октанол Октанол Бутанол, октанол Бутанол Октанол Бутанол, октанол
350
II риложение
Таблица 37
Мощности по производству акриловой кислоты и акрилатов
Страна Фирма Мощность, тыс. т Метод
Канада Франция ФРГ Япония Великобритания Италия США Chemcell Ltd. Ugilor Ugilor BASF Rohm und Haas Mitsubishi Yuka К. K. Japan Acrylic Chemical Co. Osaka Gas Japan Catalytic Chemical Ind. Co. Tokai Gas and Chemical Co. Border Chemicals Ltd. Lennig Chemicals Ltd. Montecatini Celanese Corp. Rohm und Haas Co. Union Carbide Corp. Dow-Badische Chemical Corp. B. F. Goodrich Chemical Co. Minnesota Mining and Manufac- turing Co. 10 8 28 50 2 6,5 20 6 6 10 6,3 15 15 6 2 15,9 36,3 18,1 90 20,4 18,2 2,3 1,4 Пропиолактон Акрилонитрил Пропилен Ацетилен Ацетилен . Пропилен Ацетилен Пропилен Пропилен Пропилен Акрилонитрил Пропилен Пропилен / Ацетилен Пропиолактон Ацетилен Ацетилен Пропилен Этиленциангидрин Ацетилен
Таблица 38
Производство и потребление акриловой кислоты,
этил- и 2-этилгексилакрилата * в США [36] (в тыс. т)
Год Акриловая кислота Этилакрилат 2-Этилгексилакрилат
Произ- водство Потреб- ление Произ- водство Потреб- ление Произ- водство Потреб- ление
1959 0,3 5,6
1960 — 0,5 — 7,4 — —
1961 5,5 1,0 28,4 8,9 — —
1962 8,0 1,9 32,6 10,9
1963 11,2 2,2 36,2 13,6 7,0 6,3
1964 14,7 2,0 41,2 16,4 10,3 9,3
1965 15,8 4,5 45,4 45,5 — 13,6
. * В 1965 г. потребление метилакрилата составило 6,8 тыс. т, бутилакрилата 4,5 тыс. т,
прочих акрилатов 2,3 тыс. т.
Приложение
351
Таблица 39
Потребление акрилатов в США £36J
Назначение Потреб- ление, тыс. т 'Назначение Потреб- ление, тыс. т
Лакокрасочные материалы (включая латексные краски и отделочные средства) . . Текстильная промышленность Средства для ухода за полами 32,7 13,6 4,5 Средства для дубления кожи Обработка бук аги Клеи Прочее 4,5 4,5 2,7 10,1
Таблица 40 Мощности по производству олигомеров
Страна, Фирма Мощность, тыс. т
Канада Франция ФРГ Италия Тринидад Великобритания США Мексика Imperial Oil Ltd. Cie Francaise de Raffinage Esso Standard S. A. F. Esso AG Chemische Werke Hills AG Societa Industrial Catanese Soc. Montecatini-Edison Societa Italiana Resine Texaco Trinidad, Inc. Grange Chemicals Ltd. Esso Chemical 4 Apple River Chemical Co. Continental Oil Co. Enjay Chemical Co. Gulf Oil Corp. Marathon Oil Co. Sun Oil Co. Texaco Inc. Union Oil Co. of California Pemex 36 (9 + 27) 42 35 50 14 35 14 20,4 10 30 11 24
Таблица 41 Мощности по производству додецилбензола
Страна Фирма МОЩНОСТЬ, тыс. т
Аргентина Франция Yazimientos Petroliferos Fiscales Esso Standard S. A. F. Chevron Chemical Co. S. A. Francaise Petrosyn- these CER 15 32 . 35
352
Приложение
Продолжение табл. 41
Страна Фирма Мощность, тыс. т
ФРГ Chemische Werke Hills AG 60
Rheinpreussen AG 25
Иран National Petrochemical Co. of Iran 10
Италия Societa Italiana Resine 50
Sicedison SpA 30
Япония Mitsubishi Yuka К. K. 32
Nippon Petroleum Detergent Co. Ltd. 30
Sumitomo Kagaku Kogyo К. K. 15
Nissan Petrochemical Co. —
Toyo Koatsu Industries Co. 20
Мексика Pemex 30,9
Pemex 27,6
Бразилия Empresa Carioca de Produtos Quimicos 12—15
Великобритания Grange Chemicals Ltd. 24
Shell Chemical Co., Ltd.
Венесуэла Quimica Venoco 10
Южная Африка Karbochem (Pty) Ltd. 7,7
США Allied Chemical Corp.
Continental Oil Co. —
Monsanto Co. 70
Турция Petkim Petrokimya 10 (1969)
Колумбия Empresa Colombiana de Petroleos 16,5
Пакистан Fanji Foundation 5
Таблица 42
Мощности по С3 — С4-алкилированию в США и Канаде в 1967 г. [39]
Фирма Мощность, тыс. т Метод
США
American Oil Со. 144 HF
Lion Oil, Div. of Monsanto Co. 210 H2SO4
Atlantic Richfild Co. 420 НдЬОд
Cult' Oil Corp. 150 HF
Mobil Oil Corp. 252 HF
Phillips Petroleum Co. 630 H2SO4,
Shell Oil Co. 390 H2SO4
432 H2SO4
Signal Oil and Gas Co. 90 H2SO4
Standard Oil Co. of California 324 H2SO4
270 H2SO4
Texaco Inc. 1 ‘ 264 H2SO4
Union Oil Co. of California -зоо H2SO4
Tidewater Oil Co. 300 H2SO4
Приложение
353
Продолжение табл. 42
Фирма Мощность, тыс. т Метод
Standard Oil Со. of Calif. 228 HaSO4
American Oil Co. 318 HaSO4
Clark Oil and Refining Co. 308 HF
Marathon Oil Co. 156 HaSO4
Mobil Oil Co. 144 HF
Shell Oil Co. 1200 HaSO4
Texaco, Inc. 150 H2SO4
324 HaSO4
Union Oil Co. of Calif. 96 HF
American Oil Co. 930 HaSO4
Cities Service Oil Co. 252 HaSO4
Indiana Fann Association, Inc. 60 ‘HF
Mobil Oil. Co. 180 HaSO4
Sinclair Refining Co. 300 HaSO4
American Oil Co. 96 HaSO4
Apco Oil Corp. 132 HF
Century Refining Co. 48 HF
CRA, Inc. 111 HF
CRA, Inc. 94 HF
Derby Refining Co. 120 HF
Mobil Oil Co. 238 HF
National Cooperative Refinery ASS 150 HaSO4
Phillips Petroleum Co. 396 HF
Skelly Oil Co. 396 HF
Cities service Oil Co. 1020 HaSO4
Continental Oil Co. 264 HaSO4
Humble Oil and Refining Co. 1350 HaSO4
Murphy Oil Corp. 96 HF
Shell Oil Co. 720 H2SO4
Tenneco Oil Co. 240 HF
Bay Refining Co. 45 HF
Leonard Refineries, Inc. 72 HF
Marathon Oil Co. 150 H3SO4
Mobil Oil Co. 204 HaSO4
Great Northern Oil Co. 222 HaSO4
Northwestern Refining Co. 150 HF
Gulf Oil Corp. 216 H2SO4
Standard Oil Co. (Kentucky) 510 HaSO4
American Oil Co. 276 H2SO,
Continental Oil Co. 16 HF
Humble Oil and Refining Co. 156 HF
Chevron Oil Co. 500 HaSO4
Hess Oil and Chemical Corp. 180 h2so4
Humble Oil and Refining Co. 640 h2so4
Mobil Oil Co. 138 HF
Texaco Inc. 180 H2SO4
Continental Oil Co. 90 HF
Shell Oil Co. 90 HF
Mobil Oil Corp. 147 H2SO4
American Oil Co. 144 HF'
Ashland Oil and Refining Co. 240 HF
23 Заказ 399
354
Приложение
Продолжение табл. 42
Фирма Мощность, тыс. т Метод
Gulf Oil Corp. 238 HF
306 HF
Standard Oil Co. of Ohio 372 H2SO4
Sun Oil Co. 360 h2so4
Union Oil Co. of Calif. 86 HF
108 H2SO4
Bell Oil and Gas Co. 180 HF
Champlin -Petroleum Co. 120 HF
Continental Oil Co. 346 HF
Midland Cooperatives, Inc. 80 HF
Sunray DX Oil Co. 300 HF
Sunray DX Oil Co. 150 H2SO4
270 HF
Texaco Inc. 150 H2SO4
Atlantic Richfield Co. 450 h2so4
Gulf Oil Corp. 480 h2so4
Sun Oil Co. 600 H2SO4
United Refining Co. 66 H2SO4
Delta Refining Co. 96 HF
American Oil Co. 1640 H2SOt
American Petrofina Co. of Texas 90 HF
Atlantic Richfield Co. 164 HF
Chevron Oil Co. 210 H2SO4
Coastal States Petrochemical Co. 72 HF
Cosden Oil and Chemical Co. 240 HF
Crown Central Petroleum Corp. 228 HF
Gulf Oil Corp. 1102 H2SO4
Hess Oil and Chemical Corp. 180 HF
Humble Oil and Refining Co. 1500 H2SO4
La Gloria Oil and Gas Co. 144 H2SO4
Marathon Oil Co. 99 H2SO4
Mobil Oil Corp. 1060 h2so4
Phillips Petroleum Co. 540 HF
360 HF
Pontiac Refining Corp. 114 HF
Shamrock Oil and Gas Corp. 270 H2SO4
Shell Oil Co. 360 h2so4
162 HF
Signal Oil and Gas Co. 270 H2SO4
Southwestern Oil and Refining Co. 120 HF
Suntide Refining Co. 216 HF
Texaco, Inc. 72 H2S04
90 h2so4
900 h2so4
Texas City Refining, Inc. 210 HF
Union Oil Co. of Calif. 298 H2SO4
American Oil Co. 300 h2so4
Beeline Refining Co. 56 HF
Phillips Petroleum Co. 96 HF
Mobil Oil Corp. 198 HF
Shell Oil Co. 510 h2so4
П риложение
355
Продолжение табл. 42
Фирма Мощность, тыс. т Метод
Texaco Inc. 180 H2SO4
Murphy Oil Corp. 66 HF
American Oil Co. 78 H2SO4
Sioux Oil Co. 48 HF
Канада
Imperial Oil Ltd. 102 H2SO4
114 HF
British American Oil Co. Ltd. 63 H2SO4
Pacific Petroleum Ltd. 42 HF
Imperial Oil Ltd. 66 HF
BP Refinery Canada Ltd. 120 H2SO4
British American Oil Co. Ltd. 180 HF
Regent Refining (Canada) Ltd. 60 H2SO4
Canadian Petrofina Ltd. 90 h2so4
Shell Canada Ltd. 90 HF
Texaco Canada Ltd. 120 H2SO4
Таблица 43
Мощности по производству полипропилена в 1968 г. [1]
Страна Фирма Мощность, тыс. т
США* Австрия Франция ФРГ Италия Alamo Industries, Inc. (Diamond Alcali Co.) Avisun Corp. (Amoco Chemicals) Enjay Chemical Co. (Standard Oil Co. of New J ersey) Hercules Powder Co. Novamont Corp. (Montecatini Edison) Rexall-El Paso Chemical Co. Shell Chemical Co. Texas Eastman Co. Monsanto Co. Danubia Petrochemie AG Naphtachimie S. A. Ste . Normande de Matieres Plastiques Hibernia Chemische Werke Hills AG/Scholven Chemie BASF Scholven-Chemie AG Farbwerke Hoechst AG Esso Chemie Schwab Hall Gaswerke Monteshell Petrochimica SpA ANIC Polymer SpA 32 115 68 165 36 34 54 40 23 20 60 10 20 24 3 13 36 35 13 50 25 40 (1970) 40
23*
356
Приложение
Продолжение табл. 43
Страна Фирма Мощность, тыс. т
Япония Нидерланды Испания Швеция Великобритания Китай Польша Пакистан Южная Корея ЧССР Австралия Канада * Данные по С11Ь> Chisso Sekiyu Kagaku К. К. Mitsubishi Yuka К. К. Mitsui Kagaku Kogyo К. К. Shin Nippon Chissoshiryo К. К. Sumitomo Kagaku Kogyo К. К. 1 Sumitomo Kagaku Kogyo К. K. J Tokuyama Soda К. K. Nippon Petrochemicals Co. Ltd. Ube Kosan К. K. Showa Denko К. K. Rotterdamse Polyolefinen Maatschappij N. V. Paular S. A. Dow-Uniquesa S. A. Svenska Esso A. B. ICI Ltd. Shell Chemical Co., Ltd. National Technical Import Fanji Foundation Korea Oil Co. Slovnaft Hoechst Chemicals (Australia) Ltd. Shell Co. of Australia Ltd. Imperial Oil Ltd. приведены на середину 1969 г. 110 (1970) 110 (1969) НО (1970) 26 ПО (1970) Iff 55 (1970) 30 35 30 (1970) 18 8 3 90 (1970) 15,3 5 20 (1968) 5 20 30 (1971) 27
Таблица 44
' Производство полипропилена в разных странах (в тыс. т)
Страна 1959 г. 1960 г. 1961 г. 1962 г. 1963 г. СО о 1965 г. 1966 г. 1967 г. 1968 г. 1970 г.
ГТП А 9 16,5 46 65 89,3 122,6 169,7 251 280 360 500
Япония .... ФРГ . . . 3 5,5 5 21,2 39,5 10 57 22 99,7 170,2 261 40 240
Италия . . . 4 5 7 — — 20 26 35 42 — —
Великобритания Нидерланды — — — — 2 11 5 19,5 9,5 26 — 4
Польша . . . — — — — — — к
Индия .... , — — — — — —
Австрия . . . — — — — —
Франция . . . — — — — — 16
Пакистан . . — — — — — — — 5 10 18
Испания . . . — — —
П риложение
357
Таблица 45
Потребление полипропилена в США [25—28] (в тыс. т)
Назначение UT5 СО О
со со СО со СО
о. С5 05 05 05 05
Литье под давлением 23,5 45 70 95 125 215
Волокно и мононити 9,5 25 41 52 94 147
Пленки и листы 8 18 25 27 25 54
Формование раздувом 1,8 2 4 2 7
Изоляция проводов и кабелей .... 0,5 1 2 2 5
Трубы и профили 1 I2 2,3 5 5 9 1 9 1 8
Экструзионное покрытие 1 — — —
Экспорт .' 7 — 11 14 23 23 34
Прочее (пенопласт и др.) 4 13,6 5 12 8 6
Всего ..... 47 117,2 163 222 290 476
Таблица 46
Потребление пропилена в Японии [23, 24] (в тыс. т)
Назначение 1965 г. 1966 г. 1967 г. 1968 г. 1969 г. 1970 г.
Литье под давлением 26 40 38,5 46 55 66
Ровница (пряжа) . . . — 7,1 12 21 27 35
Пленки 19,2 25,7 27 35 44 53
Переработка раздувом 1,5 1,6 3 ’ 4 5 6,5
Волокно 9 18,1 20 25 30 36
Экспорт 1,2 2,5 2 3 4 5
Прочее 1,3 1,5 2,5 3 4 5
Всего 58,2 96,5 105 137 169 .206,5
Таблица 47
Развитие производства и потребления полипропилена [29]
Страна или группа стран Производство, тыс. т Потребление, тыс. т
1960 г. 1965 г. 1968 г. 1965 г. 1968 г. 1970 г. 1975 г.
США Южная и Центральная Америка 16,5 170 290 150 3 330 19 475 40 900 100
358
Приложение
Продолжение табл. 47
Страна или группа стран Производство, тыс. т Потребление, тыс. т
1960 Г. 1965 г. 1968 г. 1965 г. 1968 г. 1970 г. 1975 Г,
Страны Европейского экономи- ческого сообщества .... 5,1 63 150 50 130 210 450
Страны Европейской ассоциации
свободной торговли .... 0,4 24 95 30 90 150 320
Остальные страны Западной Ев-
ропы t —- ♦ 3 11 20 50
Социалистические государства Европы ' ♦ 4 30 70 220
Япония . 57 185 50 120 180 360
Прочие .страны мира .... — 1 80 15 70 135 350
Всего в мире 22 315 305 800 800 1280 2750
* Попадает в рубрику «Прочие страны мира».
Таблица 48
Мощности по производству этилен-пропиленовых сополимеров
Страна Фирма Мощность, тыс. T
США* Dow Chemical Со. Humble Oil and Refining Co. Du Pont de Nemours u. Co. Enjay Chemical Co. U. ’S. Rubber Co. Copolymer Rubber Co. 10 32 22,7 17,5 25
ФРГ Chemische Werke Hiils AG 20 (1969)
Италия Monteshell Petrochimica SpA 15 - (1969 : 40)
Япония Mitsubishi, Yuka К. K. (Japan EPR Co.) Sumitomo Chemical Mitsui Petrochemical Ind. 10 22
Нидерланды Канада DMS Netherlands, Chemical Division 10 (1969 : 33) 10 (1969)
Австрия Danubia Petrochemie 15
* Динамика производства зтилен-пропиленового каучука в США:
1970
50
Год...................... 1962 1963 1964 1965 1966
Выпуск, тыс. т........... 2 5 8 15 25
Приложение
359
Таблица 49
Мощности по производству перхлорэтилена
Страна Фирма Мощность, тыс. т Сырье
Аргентина Electroclor S. А. Природный газ
Австралия ICI of Australia and New Zealand Этилен
Ltd.
Канада Dow Chemical of Canada Ltd. Пропан
Франция Pechiney-Saint Gobain 20 Пропилен, дихлор-
пропан
Progil-E lectrochimie 15 Пропилен
Италия Rumianca SpA 10 Природный газ
Sincat ' 20 Этилен
Япония Asahi-Penn Chemical Jugen Kaisha 10 Этилен
Kanta Denko 3
Южная Африка Sumitomo Chemical Co. African Explosives and Chemical 16,8 -
Ind. Ltd.
США Detrex Chemical 11,3 Ацетилен
Diamond Alcali Corp. 22,7 Ацетилен
Dow Chemical Co. 31,7 Природный газ
» » » 13,6 Природный газ
» » » 32,7 Природный газ
Du Pont de Nemours u. Cie. 27,2 Ацетилен
Ethyl Corp. Frontier Chemicals Co. 8,2 Этилен
Pittsburgh Plate Glass Co. 20,4 Природный газ
» » » » 2 Природный газ
Vulcan Materials Природный газ
Hooker Chemicals 3,2 . Ацетилен
Stauffer Chemical Co. 20,4 Пропилен
» » » » Пропилен
Мексика Pemex 6,6 Природный газ
Швеция Mo och Domsjo 20
Нидерланды Koninklijke Nederlandse Zoutin- 40 Пропилен
dustrie
Советский Союз Техмашимпорт 20 Пропилеи
Таблица 50
Мощности по производству изопрена
Страна Фирма Мощность, тыс. т Метод
США Enjay Chemical Со. » о » Goodyear Tire and Rubber Co. Shell Chemical Co. » » » 4,5 4,5 49,5 45 18 Крекинг Крекинг Димеризация пропиле- на Дегидрирование изо- пентена Дегидрирование изо- пентена
360
П риложение
Продолжение табл. 50
Страна Фирма Мощность, тыс. т Метод
США ФРГ В еликобритания Goodrich-Gulf Chemicals Inc. Chemische Werke Hiils AG British Petroleum Co. ICI Ltd. 60 10 2 2 . Дегидрирование изо- пентена (?) Дегидрирование изо- пентена Димеризация пропиле- на Димеризация пропиле- на (для получения полиметилпентена)
Таблица 51
Потребление пропилена в Европе для синтеза
органических веществ [41] (в тыс. т)
Страна 19 65 г. 1967 г.
ФРГ 280 398
Австрия . . . 5 10
Франция . . . 218 292
Италия . . . 170 190
Нидерланды 112 135
Швеция . . . 2 3
Всего 787 1028
Т а < блица 52
Потребление пропилена в Западной Европе для оксосиитеза [41]
Мощности, тыс. т Потребление, тыс. т
Страна после увели-
' имеющиеся увеличения имеющееся чения мощ- ностей
ФРГ 534 644 320 390
Франция 48 60 29 36
Великобритания 86 110 52 66
Италия 110 110 66 66
Нидерланды 76 76 46 46
Австрия 50 50 30 30
Всего 904 1050 543 634
П риложение
3 61
Таблица 53
Потребление пропилена в Западной Европе для получения
изопропилового спирта и акрилонитрила [41]
Изопропиловый спирт Акрилонитрил
Потребление Потребление
Мощности, пропилена, Мощности, пропилена,
тыс. т тыс. т тыс. т тыс. т
Страна ее я Я > к Л *5
" « й> Я ’ "к и к Й
I ^Я Я я 2 о я В после ч личени МОЩНО! в ^Я ' © Я Я 2 о Е о Я Я я В ' S о Я и Я £ § о Е з © Я © Я Я 2
• Я Я Я Я
ФРГ 136 136 116 116 92 155 98 164
Франция ....... Великобритания .... Италия 100 100 85 85 85 100 90 106
225 15 225 15 191 13 191 13 40 32 220 72 42- 34 233 76
Нидерланды 130 130 110 110 0 85 0 90
Испания 15 28 13 24 —
Всего 621 634 528 539 249 632 264 669
Таблица 54
Потребление пропилена в Западной Европе [41] для получения
кумола и окиси пропилена
Кумол Окись пропилена
Мощности, тыс. т Потребление пропилена, тыс. т Мощности, тыс. т Потребление пропилена, тыс. т
Страна
• ВЭЭИЙ1О1Э1 "к щ И ч £ О Я я о СУ © В © после уве- личения мощностей я о О) я в © после уве- личения я 8 © S 2 © эсле уве- ичения эщностей
я я я я Я Я 2
Бельгия . 30 30 12 12 30 30 27 27
ФРГ 160 210 62 82 97 117 88 106
Франция 180 180 70 70 42 41 37 37
Великобритания .... 115 115 45 45 45 80 41 72
Италия 170 270 66 105 38 40 35 36
Нидерланды 0 150 0 59 30 3 30 27 27
Швеция — — — — О 0
Испания — — — — 0 24 0 22
Всего 655 955 255 373 284 365 258 330
362
Приложение
Т а б л и ц а 55
Потребление пропилена в Западной Европе в 1968 г. [41] (в тыс. т)
Страна Оксосинтез 1 Изопропиловый спирт Кумол Акри.П1итрИл Окись пропилена Полипропилен * Всего
Бельгия 0 0 10 0 . 22 (0) 32
ФРГ 256 93 50 78 70 (30) 577
Франция 23 68 56 72 30 (10) 249
Великобритания 42 153 36 34 33 (26) 298
Италия 53 10 53 27 28 (35) - 171
Нидерланды 37 88 0 0 22 (5) 147
Австрия 24 - 0 0 0 0 (6) 24
Швеция 0 0 0 0 2 . (3) 2
Испания • . о - 10 0 0 0 (5) 10
Всего 435 422 205 211 207 1480
* Собственная оценка авторов.
Газ'с высоким содержанием Щ
Газ с высоким содержанием СЩ
II
Гидроочищенный бензин
Котельное топливо
О
Q
Рис. 15. Схема получения пропилена из пропана и бензина:
1 — печь для пиролиза бензина; 1а — узел закалки газа пиролиза; 2 — колонна для охлаждения маслом; з — отделитель котельного топлива; 4 — колонна для охлаждения
компрессии и щелочной очистки газов; 7 — осушители; 8 — колонна отгонки метана; 9 — колонна отгонки этана; Ю — аппарат для удаления ацетилена; 11 — узел колон а вторично^ от
гонки метана; 13 — колонна отгонки этилена; 14 — охлаждение этилена; is — печь для пиролиза этана и пропана; 16 — колонна отгонки пропана; 17 — колонна отгонки пропадиена и метилацет ена, в уда
ление легкокипящих фракций; 19 — колонна отгонки бутана; 20 — система для гидроочистки, стабилизации и повторной перегонки.
Заказ 399
КНЫЕ ПРЕВРАЩЕНИЯ ПРОПИЛЕНА
Алкидные Триметш па Полимеа Метак] Растворитель Пластификатор | + 1 Метакр смолы 1 Изобутилацетат альд Дибутилфталат | юлпро- f Изобутиловый а' Бутиловый спирт спирт акрилат жлаты иловый егид Алкидные смолы t Пластификатор Диметилдиметилол- 1 метан | 2-Этилгексилфталат
. Масляный ал ьд еги д И зом ас ля ны 2-Этилгексиловый й альдегид спирт
Оксосинтез
— ... Пропилен
Алкилирование
Этилен
Бутилен
1 [ Изобутаи |
4- [етыреххлористый углерод V Перхло])этилен 1 И зогептан I
1 Растворитель Растворитель V Антидетони рующее го рючее
Бутилен
Гептен-1
Изооктиловый спирт
Бензол
Кумол
Гидроперекись
кумола
Бутадиен
Изооктилфталат
Пластификатор
Растворитель Растворитель 1 Эпоксидные смолы 1 1
изобутилкарбинол —1 Гексилег Диацетоно I ГЛИКОЛЬ 1 зый спирт t Ацетон 1 Эпихлоргидрин 4 Дифенилолпропан 1 фенол 1 Пластмассы 1 1 Полиакрилаты 1 Акрилаты 4 Акриловая кислота 1 1 Синтетическ< -- ^2:'• --- Полиакри • Акрило эе волокно Г . лонитрил нитрил
Окисление
I
Пропилен
Полимеризация
Трипропилен Тетрапропилен
ш-1 1 + Фенол 1 1 Дециловый спирт 1 Бензол f 1 + Фенол 1 Тридециловый спирт
эн-2 1 Нонилфенол 1 Фталевый ангидрид 1 i Додецилбензол 1 4* Додецилфенол 1 Тридецилсульфат 1
1 Окись этилена 1 Пластификаторы 1 Алкил (арил) —сульфо- Окись этилена Моющие средства
ВОЗМОЖНЫЕ ПРЕВРАЩЕНИЯ ПРС
Растворитель Изопропилацетат Ацетон Эпоксидные смолы Дифенило лпро- пан t Растворитель Полиуретаны, полиэфиры Пропиленгли- коль Окись пропилена Пластмассы t Ацетобутцрат целлюлозы Масляная кислота Алкидные смолы Триметилолпро- П ласти Дибутг Бутило!
па н
Пропиленхлор-
гидрин
Изопропиловый спирт Масляный альдегид
t Присоединение —
t Пропилен
| |
Хлорирование
1,2-Дихло рпропан
Аллилхлорид
Растворитель
Дихлоргидрин
глицерина
Эпихлоргидрин
Аллиловый спирт
Глицерин
Эпихлоргидрин
Четы рех х л о ри стый
углерод
Растворитель
Перхлорэт
Р аствори
Полиуретаны, полиэфиры
Пропиленгликоль
Окись пропилена
Эпоксидные
смолы
Глицерин
Каучук
Аллиловый спирт
Акролеин
Акриловая кислота
Растворитель Растворитель
I I
Метилизобутилкетон Метилизобутилкарбинол
1 _ |
I
Окись мезитила
Раствор>
Гексилент
I
Диацетонов!
Окисление
1
Пропилен
Полимеризация
Трипропи
Полипропилен
I
Пластмассы
Сополимер этилена с
пропиленом
1
Каучук
4-М ети лпен тен-1
I
Поли-4-метилпентен-1
I
Пластмассы
2-Метилпентен-1 Фенол I
+ 2-Метилпен тен-2 1 1 Нонилфенол
1 1
Изопрен Окись этилена
т
Полиметакрилат
Метакрилаты
Метакриловый
альдегид
Алкидные смолы
Диметил диметилол-
метан
Пластификатор
2-Этилгексилфталат
Иаомасляный альдегид
2-Эти лгексило вый
спирт
t
Оксосинтез
Алкилирование
Бутилен
Гептен-1
Изооктиловый спирт
I
Изооктилфталат
Пластификатор
Бензол
I
Кумол
Гидроперекись
кумола
J i
Этилен Бутилен
I
Бутадиен
смолы
Пластмассы
Синтетическое волокно
ЛИТЕРАТУРА
1. R. В. Stobaugh, Hydrocarbon Processing, 46, № 1, 143 (1967).
2. Chem. Eng. News, 41, № 19, 92 (1963).
3. Chem. Eng. News, 44, № 24, 34 (1966).
4. A. Theurer, Chem. Industrie, 19, № 11, 757 (1967).
5. Chem. Industrie, 15, № 9, 551 (1963).
6. N. E. О с к e r b 1 о о m, A. P. S t u a r t, Hydrocarbon Processing, 46,
№ 5, 225 (1967).
7. Oil Gas J., 65, № 16, 130 (1967).
8. Chem. Eng. News, 45, № 18, 30 (1967).
9. R, N. Ricklers, Future Chemicals Growth Patterns, Noyes Develop-
ment Corp.
10. Gummi-Asbest-Kunststoffe, 20, № 6, 685 (1967).
11. R. N. Rickies, Future European Chemical Growth, Noyes Develop-
ment Corp.
12. Chem. Eng. News, 42, № 31, 118, 132 (1964).
13. Chem. Eng. News, 42, № 35, 76, 86 (1964).
14. Chem. Eng. News, 42, № 39, 97, 101, 107 (1964).
15. K. W. Schneider, Chem. and Ind., 42, 1764 (1967).
16. Oil Gas J., 64, № 36, 144 (1966).
17. Oil Gas International, 6, № 10, 54, 121-(1966).
18. R. S c h 6 n b e c k, Osterreich. Chem.-Ztg., 68, 79 (1967).
19. Chem. Industrie, 19, 128 (1967); Qummi-Asbest-Kunststoffe, 20, № 11, 1239
(1967).
20. Chem. Eng. News, 38, № 41, 20 (I960).
21. Chem. Eng. News, 73, № 23, 102 (1966).
22. T. C. Ponder, Hydrocarbon Processing, 45, № 12, 176c (1966).
23. Chem. Industrie, 18, 713 (1966).
24. Gummi-Asbest-Kunststoffe, 20, 860 (1967).
25. Chem. Industrie, 18, 325(1966).
26. Kunststoffe, 56, 379 (1966).
27. Kunststoffe, 53, 266 (1963).
28. Gummi-Asbest-Kunststoffe, 20, 1014 (1967).
29. G. Brauer, Plaste u. Kautschuk, 14, 534(1967).
30. ВИКИ, Москва, 19, № 24, 4 (1967).
31. Chem. Eng. Progr., 48, № 1, 31 (1967).
32. Oil Gas J., 64, № 1, 83 (1966).
33. Chem. Industrie, 16, 395 (1964).
34. Gummi-Asbest-Kunststoffe, 20, 685 (1967).'
35. Marktinformationen, 11, № 28, 7 (1967).
36. Chem. Industrie, 19, 345 (1967); Chem. Eng., 45, № 7, 48, 53 (1967).
37. Chem. Industrie, 19, 349 (1967).
38. W. Stobaugh, Hydrocarbon Processing, 46, № 7, 149 (1967).
39. Oil Gas J., 65, № 14, 183 (1967).
40. European Chemical News, Supplementband 1.3. 1968, Ethylen, Propylen
and their derivatives.
41. Chem. Industrie, 20, № 8, 537 (1968).
СОДЕРЖАНИЕ
Стр.
Предисловие редактора перевода .......... 5
Предисловие автора ....................... 6
Введение ................................. 7
1. Получение пропилена
1.1. Выделение из нефтезаводских и крекинг-газов................. 8
1.2. Образование при синтезе по методу Фишера — Тропша ... 9
1.3. Выделение из полукоксовых и коксовых газов.................. 10 ;
1.4. Получение из соединений С3................................. 10'
1.4.1. Термическое дегидрирование пропана..................... 11
1.4.2. Каталитическое дегидрирование пропана.................. 12
1.5. Получение пропилена из других газообразных углеводородов 14
1.5.1. Получение из этилена и этана .......................... 14
1.5.2. Получение из бутана и изобутана...........;............ 14
1.6. Получение пропилена путем пиролиза углеводородов .... 15
1.6.1. Пиролиз в трубчатых печах............................. 23
1.6.2. Пиролиз в реакторе с кварцевым теплоносителем (процесс
фирмы Phillips Petroleum Со.)................................. 25
1.6.3. Пиролиз в реакторе с коксовым теплоносителем (процесс
фирмы Farbewerke Hoechst).........................'........... 26
1.6.4. Крекинг сырой нефти в кипящем слое (процесс фирмы BASF) 27
1.6.5. Пиролиз в реакторе с песком в качестве теплоносителя
(процесс фирмы Lurgi)......................................... 30
1.6.6. Пиролиз в трубчатой печи (процесс фирмы Kellogg) ... 32
1.6.7. Пиролитический крекинг «термофор»..................... 33
1.6.8. Процесс Лавровского — Бродского ...................... 35
1.6.9. Автотермический пиролиз по Бартоломе.................. 35
1.6.10. Катарол-процесс ..................................... 36
1.6.11. Процесс фирмы Tsutsumi.............................. 37
1.6.12. Процесс АЗНИИ — Алиева .............................. 38
i 1.6.13. Процесс Коппере — Хаше — Вульфа ...................... 39
1.6.14. Высокотемпературный пиролиз (процесс фирмы Earbwerke
Hoechst).................................................... 39
1.6.15. Синтез ацетилена по способу фирмы BASF............... 40
1.6.16. Метод разложения нефти при высоких температурах (про-
цесс фирмы BASF).............................................. 40
1.6.17. Современные промышленные установки США.............. 41
Литерат ура ............................................ 41
2. Выделение и свойства пропилена
2.1. Выделение пропилена из продуктов, образующихся при различ-
ных процессах................................................ 45
2.2. Получение чистого пропилена............................. 47
2.2.1. Экстрактивная дистилляция .......................... 48
2.2.2. Двухколонная дистилляция ........................... 48
2.2.3. Рекомпрессионная дистилляция ....................... 49
2.2.4. Разделение пропана и пропилена на молекулярных ситах 50
2.2.5. Разделение с помощью серной кислоты................. 50
Содержание
2.2.6. Экономическая эффективность получения высокочистого
пропилена ...........................(.................. 50
2.3. Свойства пропилена..................................... 51
Литература .............................................. 52
3. Изопропиловый спирт
3.1. Получение изопропилового спирта........................ 54
3.1.1. Получение из углеводов............................. 54
3.1.2. Гидратация пропилена серной кислотой в жидкой фазе 54
3.1.3. Другие процессы гидратации в жидкой фазе........... 59
3.1.4. Гидратация пропилена в газовой фазе на катализаторах 60
3.1.5. Экономическая оценка методов гидратации в жидкой и газо-
вой фазе............................................ . 65
3.2. Свойства изопропилового спирта......................... 65
3.3. Применение изопропилового спирта....................... 66
Литература ............................................... 67
4. Окись пропилена
4.1. Получение окиси пропилена через пропиленхлоргидрин .... 70
4.2. Получение окиси пропилена прямым окислением............ 76
4.2.1. Окисление пропилена в жидкой фазе под давлением ... П
4.2.2. Окисление пропилена в газовой фазе................. 81
4.3. Свойства окиси пропилена............................... 84
С4. Применение окиси пропилена............................. 86
Литература ............................................... 87
5. Акролеин
5.1. Синтез акролеина из формальдегида и ацетальдегида...... 92
5.2. Получение акролеина прямым окислением пропилена .... 94
5.2.1. Окисление пропилена на медьсодержащих катализаторах 94
5.2.2. Окисление пропилена на катализаторе фосфоромолибдате
висмута (метод Sohio).................................... 100
5.3. Свойства акролеина ................................... 11
5.4. Применение акролеина ...........................: . 110
Литература .............................................. 112
6. Акрилонитрил
6.1. Получение акрилонитрила .............................. 117
6.1.1. Получение акрилонитрила через окись этилена....... 117
6.1.2. Получение акрилонитрила из ацетилена и синильной кислоты 118
6.1.3. Получение акрилонитрила из пропилена, аммиака (или
окиси азота) и кислорода k............................. 119
6.1.4. Другие процессы получения акрилонитрила........... 130
6.1.5. Экономическая оценка методов получения акрилонитрила 131
6.2. Свойства акрилонитрила ............................... 133
6.3. Применение акрилонитрила.............................. 134
Литература .............................................. 135
7. Ацетон
7.1. Получение ацетона .................................... 140
7.1.1. Старые методы ................................... 140
7.1.2. Получение из изопропилового спирта................ 141
7.1.3. Метод прямого окисления пропилена................. 142
7.2. Свойства ацетона ..................................... 143
7.3. Применение ацетона...................-................ 144
Литература .............................................. 146
366
Содержали е
8. Акриловая кислота
8.1. Получение акриловой кислоты различными способами .... 148
8.2. Получение акриловой кислоты из пропилена или акролеина 149
8.3. Теоретическое рассмотрение реакции окисления пропилена
или акролеина в акриловую кислоту.......................... 158
8.4. Свойства и применение акриловой кислоты................... 159
Литература ................................................ 462
9. Оксосинтез пропилена
9.1. Альдокс-процесс ...................................... .172
9.2. Свойства и применение масляных альдегидов.............. 173
Литература .................................................. 175
10. Хлорирование пропилена
10.1. Присоединение хлора по двойной связи.................. 176
10.2. Замещающее хлорирование .............................. 177
10.2.1. Получение и характеристика аллилхлорида............... 177
10.2.2. Получение и характеристика эпихлоргидрина............. 185
10.2.3. Получение и характеристика аллилового спирта . 189
10.2.4. Получение и характеристика глицерина.................. 192
10.3. Перхлорирование пропилена ........................... 201
10.3.1. Получение и характеристика четыреххлористого углерода 203
13.3.2. Получение и характеристика перхлорэтилена ...... 205
10.3.3. Экономическая оценка методов получения четыреххлори-
стого углерода и перхлорэтилена......................... 207
Ли тература ...........................................• • 208
11. Димеризация пропилена
11.1. Получение изопрена из пропилена......................... 213-
11.1.1. Димеризация пропилена с алюминийорганическими соеди-
нениями ............................................ .... 215
11.1.2. Димеризация пропилена, в присутствии катализаторов
на основе щелочных металлов ............................. 223
11.1.3. Димеризация пропилена на различных катализаторах 226
11.1.4. Изомеризация метилпентена ........................... 227
11.1.5. Пиролиз метилпентена ................................. 232
11.1.6. Свойства и применение изопрена....................... 235
11.2. Поли-4-метилпентен-1 .......’.....................’. . 236
11.3. Содимеризация пропилена ................................. 237
11.3.1. Содимеризация с этиленом.............................. 237
41.3.2. Содимеризация с изобутиленом.......................... 237
Литература ................................................... 238
12. Олигомеризация пропилена
12.1. Термическая олигомеризация .............................. 241
12.2. Каталитическая олигомеризация............................ 242
12.2.1. Олигомеризация пропилена в паровой фазе на фосфорно-
кислых катализаторах .................................... 242
12.2.2. Олигомеризация пропилена с Образованием тримера и тет-
рамера на фосфорнокислых катализаторах................... 244
12.2.3. Олигомеризация пропилена различными методами на фос-
форнокислых катализаторах................................ 245
12.2.4. Олигомеризация пропилена на различных катализаторах 246
12.3. Продукты олигомеризации пропилена........................ 247
12.4. Применение олигомеров пропилена.......................... 249
Литература .................................................. 250
Содержание
367
13. Алкилирование пропилена
13.1. Термическое алкилирование . . .’...................... 252
13.2. Каталитическое алкилирование ........................ 253'
13.2.1. Алкилирование с серной кислотой................... 253
13.2.2. Алкилирование с фтористым водородом............... 257
13.2.3. Алкилирование в присутствии различных катализаторов 260
13.3. Оптимальные условия алкилирования..................... 261
13.4. Применение алкилатов ........................... ' . 262
Литература .............................................. 262
14. Кумольный синтез фенола
14.1 Алкилирование бензола пропиленом....................... 264
14.1.1. Алкилирование в жидкой фазе................... 264
14.1.2. Алкилирование в паровой фазе ................... 268
14.1.3. Алкилирование в присутствии различных катализаторов 270
14.2. Окисление кумола ..................................... 272
14.2.1. Окисление кумола в эмульсии................... . 272
14.2.2. Гомогенное окисление кумола...................... 274
14.2.3. «Сухое» окисление кумола.......................... 274
14.2.4. Ускорение окисления кумола Добавками.............. 275
14.2.5. Фотохимическое окисление кумола .................. 277
14.2.6. Исследование процесса окисления кумола........... 278
14.3. Разложение гидроперекиси кумола на фенол и ацетон ..... 281
14.3.1. Промышленные методы разложения с применением кислот 281
14.3.2. Обработка продуктов реакции....................... 282
14.3.3. Влияние кислот на разложение гидроперекиси йумола 283
14.4. Экономическая оценка кумольно-фенольного метода .... 284
Литература ............................................... 284
15. Полипропилен z
15.1. Получение полипропилена ..................... . . 295
15.1.1. Катализаторы полимеризации пропилена ........ 295
15.1.2. Полимеризация пропилена ......................... 297
15.1.3. Экономическая оценка производства полипропилена . . 299
15.2. Свойства полипропилена .Г............................. 300
15.3. Применение полипропилена ............................. 301
Литература ............................................... 305
16. Сополимеры пропилена с этиленом
16.1. Термопластичные сополимеры............................ 310
16.2. Эластомерные сополимеры . . . .->.................... 311
16.2.1. Катализаторы и условия сополимеризации............ 311
16.2.2. Промышленные методы сополимеризации............... 313
16.2.3. Вулканизация этилен-пропиленового каучука......... 314
16.3. Эластомерные терполимеры . . ......................... 315
16.3.1. Проведение терполимеризации....................... 315
16.3.2. Вулканизация этилен-пропиленовых терполимеров .... 317
16.4. Свойства и применение этилен-пропиленовых сополимеров и
терполимеров............................................. 317
Литература ............................................ 321
17. Диспропорционирование пропилена (триолефиновый метод)
Литература .............................................. 327
Приложение
Производство, мощности и потребление пропилена и продуктов на его
основе ..................................,.......... .328
Литература .....................................л ... . 363