/



























































































































































































































Автор: Копылев Б.А.
Теги: производство различных неметаллов, металлоидов и их соединений химия фосфорная кислота фосфаты
Год: 1981




Текст
I Б. А. Копылев
технологи:
экстракционной ФОСФОРНОЙ
КИСЛОТЫ
6П7.Й
Кбб
УДК 601.634.2
Копылев Б. А.
Технология экстракционной фосфорной кислоты.— 2-е изд., перераб. — Л.: Химия, 1981,— 224 с., ил.
Рассмотрены'-свойства сырья, полупродуктов и кислот фосфора. Изложены физико-химические основы процессов; описаны технологические схемы; приведены материалы по технике безопасности, утилизации побочных продуктов, обезвреживанию стоков и выбросов в атмосферу. Переработано в соответствии с последними литературными данными и результатами экспериментальных исследований (1-е изд. вышло в 1972 г.). . .
Предназначена для инженерно-технических и научных работников промышленности фосфорных удобрений. Полезна • аспирантам и студентам химико-технологических вузов.
224 'Стр-.-Ц'Э табл., 40 рис., список литературы 27'3 ссылки.
Рецензент: Кандидат технических наук В. Н. Белов
„ 31404-037
К---------—- 37.81.2802020000
050(01)—81
© Издательство «Химия», 1981
Б. А. Копы лев
ТЕХНОЛОГИЯ ЭКСТРАКЦИОННОЙ ФОСФОРНОЙ ' кислоты
Издание второе, переработанное
Ленинград-«Химия», Ленинградское отделение-1981
ОГЛАВЛЕНИЕ
Предисловие................................................. ®
Введение................................................... ?
Литература.................................................. И
Глава I. Фосфатное сырье.................................... 12
1.1. Фосфатные минералы . . .'............................. 12
1.2. Характеристика фосфатов различных месторождений...... 15
Апатиты......................................... 15
Фосфориты . . . . . . ....................... 16
1.3. ' Обогащение фосфатного сырья......................... 20
1.4. Облагораживание (обезмагнивание) доломитизированных фосфоритов и карбонат-апатитовых руд....................... 22
. Литература.......................- . .................... 26
Глава II, Физико-химические основы экстракции фосфорной кислоты 28
II.1. Взаимодействие основных частей фосфатов с серной и фосфорной кислотами........................................... 28
11.2. Равновесие в реакционных системах.................... 30
Система H2SO4—Н3РО4—Н2О..................• . . . 30
Система СаО—Р2О5—Н2О......................... . . 31
Система СаО—Р2О3—NH3—Н2О.......................*35
Система MgO—Р2О5—Н2О.......................... 36
Система MgO—СаО—Р2О3—Н2О....................... 37
Система MgO—Р2О5—SO3—Н2О ....................... 39
Система Ме2О3—Р2О3—Н2О.......................... 40
II.3. 'Скорость разложения фосфатов серной и фосфорной кислотами 41
Разложение серной кислотой...................... 41
Разложение фосфорной кислотой . ................ 43
II.4. Кристаллизация сульфата кальция из фосфорнокислых растворов ................................................ 47
Модификации сульфата кальция................... 47
Растворимость сульфата кальция в серной кислоте . . 50
, Растворимость сульфата кальция в фосфорной кислоте .................................................. 50
3
Растворимость сульфата кальция в фосфорной кислОТё, 'содержащей различные вещества...................... 51
Фазовые превращения кристаллогидратов сульфата кальция ' в фосфорнокислых растворах ....... 53
Скорость фазового перехода кристаллогидратов суль-' фата кальция в фосфорнокислых растворах .... 57
^Гранулометрическая характеристика кристаллов сульфата кальция . . ............................. 61
Литература..................................................... 64
Г лава 111. Сернокислотная экстракция фосфорной кислоты .... 67
III.1. Общие положения . ...................................... 67
ПГ.2. Аппаратура процесса................................„ . 68
Транспортирование и хранение .фосфатного сырья . . 69
. Дозирование фосфатного сырья и кислоты ...... 70
Аппараты для разложения фосфата и кристаллизации сульфата- кальция...................,......... 74
Фильтрование экстракционной пульпы................... 81
Дополнительное оборудование......................... 86
III.3. Различные технологические схемы...........................91
111.4. Дигидратный способ....................................... 99
Условия проведения процесса . . ..................... 99
Производство фосфорной кислоты из апатитового концентрата .......................................... 101
Производство фосфорной кислоты из фосфоритов . 106
Перспективные направления развития дигидраткого способа . .................................... 112
III.5. Полугцдратный способ.....................................119
Физико-химические основы процесса.................- . 119
’ Технологические схемы и режимы ......................124
Технико-экономические показатели'....................132
111:6. Двустадийные способы........... . .................. 136
Полугидратно-дигидратиый способ.....................'136
Дигидратио-полугидратный способ......................139
III .7. Получение концентрированной фосфорной кислоты . . . . . 140
Условия проведения процесса...................... . . 140
' Ангидритный (безупарочный) процесс................... 149
Литература»................................................... 153
Глава IV. Экстракция фосфорной кислоты азотной и соляной
кислотами и с использованием органических растворителей .... 156
IV .1. Азотнокислотный способ разложения фосфатов ............157.
Физико-химические основы процесса................... 157
4
Технологические схемы и режимы ........ 161
.Производство фосфорной кислоты.....................162
Комплексная азотнокислотная переработка апатитового концентрата с вымораживанием нитрата кальция ............................................ 163
1V .2. Солянокнслотный способ разложения фосфатов ...... 167
I V.3. Экстракция фосфорной кислоты органическими растворителями . ...................................................... 168
Литература . . . ...............................' . . . 171
Глава V. Полифосфорные кислоты .............................. J-73
V. I. Безводная жидкая фосфорная кислота.......................174
V.2 . Суперфосфорная кислота................................. 176
V.3. Ультрафосфорная кислота................................ - 185
V. 4. Экстракционно-термическая полифосфорная кислота .... 187
Литература.................................................... 190
Глава VI. Улавливание и утилизация соединений фтора...........- 192
V I.1. Фазовое распределение фтора по стадиям различных процессов ...............:...................................... 192
VI .2. Методы поглощения и осаждения соединений фтора . . .' . 195
Абсорбционные способы . -........................... 196
Экстракционные способы . т .... ....................197
Ионообменные способы ............................... 198
Адсорбционные способы.........................• . . 199
Осадительные методы..................................199
VI. 3. Технологические схемы и режимы..........................202
Абсорбция соединений фтора из газовой фазы . . . 202
Поглощение соединений фтора при экстракции -фосфорной кислоты . .-............................... 204
VI.4 . Получение различных соединений фтора....................210
Литература................................................ . 214
Глава VII. Основные положения техники безопасности в производстве экстракционной фосфорной кислоты...........................216
7 J
ПРЕДИСЛОВИЕ
За период, прошедший после выпуска первого издания' этой книги (1972 г.), значительно возросло в стране производство экстракционной фосфорной кислоты, являющейся полупродуктом при получении концентрированных и сложных фосфорсодержащих туков. Достигнут определенный прогресс в совершенствовании и интенсификации применяемых методов и технологических процессов.
В связи с резким ростом масштабов производства актуальной стала проблема фосфатного сырья и вовлечения в переработку бедных, неиспользовавшихся до сих пор, видов фосфатов различных-месторождений. Наиболее экономичным и перспективным методом в этом отношении /является комплексное использование сырья с извлечением из сырья попутных веществ в качестве дополнительных товарных продуктов. В первую очередь это относится к выделению соединений фтора, стронция, магния, редкоземельных элементов и др.
Исследования и опытные разработки в области' химии и технологии экстракционной фосфорной кислоты за это время также значительно расширились.
Обобщение многочисленных и многообразных новых сведений, наряду с сохранением основных материалов первого издания, не утративших своего значения, и притом при сокращении объема книги оказалось весьма сложной задачей и автор благодарен В. Н. Белову за тщательный просмотр рукописи и сделанные при этом полезные замечания и рекомендации.
Автор признателен товарищам за благожелательное отношение, замечания и пожелания, высказанные по материалам, помещенным в первом издании этой книги, и советы и рекомендации по дальнейшему ее улучшению будут приняты с благодарностью.
ВВЕДЕНИЕ
Фосфор — элемент, достаточно распространенный в природе. Содержание его в земной коре (кларк) составляет 0,08—0,1'2% (масс.) или —'0,07% от общего числа атомов, звмной коры. Он образует разные окислы и кислоты, гидриды (фосфины), галогениды, сульфиды и оксисульфиды, нитриды и фосфорнитролгалогениды, а также многочисленные фосфорорганические соединения [1].
Все кислородные кислоты фосфора — низшие и высшие — содержат РОН-группы, которые отщепляют ион водорода;' по Р—Н-связям диссоциация отсутствует.
Кислородными кислотами с высшей степенью окисления фосфора являются: ортофосфорная кислота Н3РО4, называемая обычно фосфорной, и продукты ее дегидратации (или гидратации Р2О5) —'пиро-, Триполи- и тетраполифосфорные кислоты. Из перечисленных в природе и технике наиболее важна фосфорная кислота; конденсированные (полимерные) фосфорные кислоты, . в том числе полифос-форные Нп+гРОзп+i —'С линейным строением фосфат-аниона, а также метафосфорные (НРОз) с циклическим строением фосфат-аниона и ультрафосфорные кислоты с разветвленной сетчатой структурой менее важны.
В чистом безводном виде фосфорная кислота представляет собой бесцветные призматические кристаллы, плавящиеся при 42,35 °C. Она образует водные растворы любых концентраций. При 0,5°С в воде растворяется 78,7% Н3РО4, а при 29,3 °C — конгруэнтно плавится гидрат Н3РО4-0,5Н2О, содержащий 91,6% Н3РО4 (рис. 1).
Фосфорная кислота имеет большое значение как один из важнейших компонентов питания растений. Она играет большую роль в метаболизме (обмене веществ и энергии) животных. Под последним, как известно, понимают совокупность химических реакций в живых клетках, обеспечивающих организм веществами и энергией для его жизнедеятельности. В результате этого, производные фосфора содержатся в костях, мозге, крови и тканях организмов человека и живот-
7
+ 40
Рис. 1. Фазовая диаграмма системы ' НзРО4—Н2О.
+ 20
-80
Е |.-40 с
13^ _ гл
о о
" 424'
' - Жидкость +Н3РО4
29,3'
'Жидкость +Н3РО4- //2Н2О
„ , 6'2,5%H3POt~l®
Н3РО^-//2Н2р^еЭ 9:,6%H3pp4 ,
О а
о
44 X
о «ь
X
о
X +
£ X
I
-100'------------------------------
О 20 40 60 80 100
, НзРО4,% , , '
10 .20 40 60 72,45
ных [2]. Весьма отрицательно (заболевание животных рахитом, малокровием и др.) сказывается на состоянии организма пониженное содержание в рационе питания соединений фосфора или введение их в неусвояемой форме.
Важными добавками в корм скота являются кормовые фосфаты — соли фосфорной кислоты: дикальцицфос-фат, обесфторенный природный фосфат, монокальцийфосфат, диаммонийфосфат, динатрийфосфат и фосфаты .карбамида.
РгО5,%
Кормовые фосфаты должны растворяться в 0,4%-ной НС1 и содержать по отечественным стандартам фтора — в пределах. 0,2—0,3%; других вредных примесей не более: мышьяка — 0,012 %, тяжелых металлов — 0,008%, металлического железа — 0,01 %.
Фосфорсодержащие минеральные удобрения используют для обеспечения питания растений фосфором и,, кроме того, они служат для пополнения запасов его в п'очве. Верхний пахотный Слой почвы хорошего плодородия глубиной 23— 25 см содержит Р2Оя в ~1,5 раза меньше, чем азота, и в 10—12 раз, чем К2О [3].
Фосфорные удобрения не только способствуют повыше-, нию урожайности различных сельскохозяйственных культур, но и придают растениям зимостойкость и устойчивость к другим неблагоприятным- климатическим условиям, создают условия для более быстрого созревания урожая в районах с коротким вегетационным периодом, повышают питательность выращиваемых культур и оказывают благоприятное воздей- . ствие на соотношение между колосом и стеблем, плодом и’ ботвой. Это особенно важно в противодействие азотным удобрениям, стимулирующим рост стеблей и ботвы за счет плодов. Они также благоприятно действуют на почву, способствуя ее структурированию, развитию почвенных бакте-^ рий, изменению растворимости других содержащихся в почве веществ и подавлению некоторых образующихся вредных органических веществ.
8
В «Основных направлениях экономического и социального развития СССР на 1981 — 1985 годы и на период до 1990 года» намечено довести производство минеральных удобрений до 150—155 млн. т. (36—37 млн. т питательных^, веществ), а поставки сельскому хозяйству — не менее 115 млн. т (26,7 млн. т питательных веществ) и химических кормовых добавок — до 5 млн. т. Одновременно предусматривается, что производство туков должно увеличиваться за счет фосфорных концентрированных и сложных удобрений. Эти продукты составляют ~85% от всех фосфорных соединений, потребляемых в разных отраслях народного хозяйства. Остальные ~ 15% приходятся на технические фосфаты, производимые для нужд промышленности, 'и др.
Ортофосфорная кислота служит прежде всего для получения высококачественных безбалластных фосфорных и сложных удобрений — фосфатов кальция, аммония, калия, нитроаммофосов,. нитроаммофоски и сложносйешанных удобрений в твердом и жидком виде, а также кормовых фосфатов.
Различные соли фосфорной кислоты широко применяют во многих отраслях промышленности, в строительстве, разных областях техники, в коммунальном хозяйстве и быту, для защиты от радиации, для умягчения воды, борьбы с котельной накипью и изготовления различных моющих средств.
Фосфорная кислота, конденсированные кислоты и дегидратированные фосфаты служат катализаторами в процессах дегидратирования, алкилирования и полимеризации углеводородов.
Особое место занимают фосфорорганические соединения как экстрагентыгпластификаторы, смазочные вещества, присадки к пороху и абсорбенты в холодильных установках. Соли кислых ^лкилфосфатов используют как поверхностноактивные вещества, антифризы, специальные удобрения, антикоагулянты латекса, для повышения огнестойкости и др. Кислые алкилфосфаты применяют для экстракционной переработки урановорудных щелоков'[4].
В настоящее время наряду с ортофосфорной кислотой-выпускают также суперфосфорную (полифосфорную) кислоту, представляющую собой смесь орто-, пиро-(Н4Р2О7), Триполи- (Н5Р3О10), тетраполи-(НбР4О|3) и других вплоть до октаполи- (HioPeChs) и нона-(НцРя02я) полифосфорных кислот. Суперфосфорная кислота (76% Р2О5 или 105% Н3РО4) содержит 47—49% Р2С>5 в виде ортофосфорной, 40—42% в виде пирофосфорной, 8—12% в виде триполифосфорной и ~1% в виде тетраполифосфорн&й кислот ([5, 6].
Исходным сырьем для производства фосфорной кислоты служат природные фосфаты, содержащие нерастворимые в воде фосфаты кальция.
9
Фосфорную кислоту из природных фосфатов получают
двумя основными методами — термическим и кислотным. Первый заключается в высокотемпературном восстановлении фосфатов и возгонке в электрических печах элементарного фосфора, который затем окисляют до фосфорного ангидрида, образующего при гидратации фосфорную кислоту, назы-" ваемую в этом случае термической.
Кислотный метод основан на вытеснении сильными кислотами (в основном серной) фосфорной кислоты из фосфатов. Полученная таким способом фосфорная кислота называется экстракционной.
Главное преимущество термического способа по сравнению с экстракционным — возможность переработки любых видов сырья, в том числе и. низкокачественных фосфоритов, и. получении концентрированной кислоты высокой чистоты [5]. Однако он связан с расходованием значительных количеств электроэнергии (12—18 МВт-ч на 1 т фосфора или 7—8 MiBt-ч на 1 т P2Os). Поэтому термическая кислота до-' роже экстракционной примерно в два раза (в расчете на 1 т Р2О5), причем стоимость только электроэнергии (и притом по льготному тарифу) составляет половину и больше (т. е. 75—85 руб. ф7р) всей себестоимости экстракционной кислоты. Кроме того, дефицит электроэнергии в общем, балансе народного хозяйства и необходимость использования
ее во все возрастающих количествах в промышленности, сельском хозяйстве и быту ограничивает масштабы развития производства фосфорной кислоты термическим способом.
Доля термической фосфорной кислоты в общем потреблении ее для производства удобрений в США и других странах не превышает 5—6%. В США в 1977 г. было переработано 47,25 млн. т фосфатного сырья (на 5,8% больше, чем в 1976 г.) ([8]. При этом использование фосфатов для производства экстракционной фосфорной кислоты увеличилось на 14% с соответственным уменьшением сырья в производстве фосфора термическим методом.
Рост мирового потребления экстракционной и термической кислоты за последние 15 лет характеризуется следующим образом (в млн. т Р2О5) [5]:
1965 1970 1975 1980
Экстракционная кислота 4.5 8.8 12,0 15,3
Термическая кислота 1.4 1,7 2,6 3,6
в том числе для удоб- 0,2 0,3 0,7 1,1
рений
, Производимая термическая кислота в основном расходуется для получения технических и кормовых фосфатов и лишь в небольших количествах для получения высококонцентрированных удобрений.
10
В Советском Союзе на основе фосфоритов Каратау (в Казахской ССР) создана мощная промышленность элементарного желтого фосфора и термической кислоты.
В настоящее время наиболее распространено производство фосфорной кислоты экстракцией ее из фосфатов серной кислотой. Этим способом производят основную массу фосфорной кислоты, перерабатываемой на удобрения. В значительно меньших количествах для этих же целей используют фосфорную кислоту, экстрагируемую азотной кислотой. По-видимому, азотнокислотная экстракция фосфорной кислоты в ближайшее время может получить большое распространение вследствие возможности комплексной и безотходной переработки этим методом различных видов фосфатного сырья.’
ЛИТЕРАТУРА
1. Везер Ван Дж. Фосфор и его соединения. Пер. с англ. М., ИЛ, 1962. СЧ]2. Ваггаман В. Фосфорная кислота, фосфаты и фосфорные удобрения.
Пер. с англ. М., Госхимиздат, 1957.
3. Прянишников Д. Н. Избр. соч, Т. 7. Агрохимия. М., Се.тьхозгиз, 1963.
4. Tut hill Е. J. — Ind. Eng. Chem. Proc. Dess, and Dev,, 1967, v. 6, № 3, p. 314.
Сщ5. Термическая фосфорная кислота, соли и удобрения на ее основе/Под ред. Н. Н. Постникова. М., Химия, 1976.
6. Соколовский А. А., Унанянц Т. П. Краткий справочник по минеральным удобрениям. М., Химия, 1977.' *
7. Производство фосфора в. СССР и за рубежом. Обзорная инф. Сер. Фосфор и его соединения. М., НИИТЭХИМ, .1978.
8. Garrand L.—Mining Eng., 1978, v. 30, № 5, p. 535—536.
Глава I
ФОСФАТНОЕ СЫРЬЕ
Фосфор находится в земной коре в виде минералов большей частью химически стойких, нерастворимых в воде и почвенных растворах. Они встречаются повсеместно и часто в виде крупных скоплений.
1.1. ФОСФАТНЫЕ МИНЕРАЛЫ
' В природе известно свыше 120 фосфатных минералов. Наиболее распространены и имеют промышленное значение минералы фосфоритовой и апатитовой групп [1, 2], основной из* которых в последней — фторапатит Caio(P04)6F2 (табл. 1.1). .
К апатитам относятся минералы с общей формулой Сац^РСМбКг с элементарной кристаллической ячейкой из 42 частиц (где R —фтор, хлор и гидроксил). Некоторая
Таблица [.Г
Состав апатитовых минералов
Минерал ' • Содержание, % Массовые отношения
р2О5 СаО О X О о СаО/РгОб | 1 СО2/Р2О8 F/P2O5
Фторапатит Ca10(PO4)eF2 42.24 55.58 3,77 — 1,32 0,09
Хлорапатит Сац» (РО4)вС12 40,91 55,72 6,81 — 1,39 — —
Г идрсжсила!пат'ит Са10(РО4)в(ОН)2 42,40 55,88 — — 1,32 — —
Карбонатапатит Саю'Р5СО2з(0Н)з 35,97 56,79 — 4,46 1,59 0,-12 —
Франколит' 37,14 56,46 3,44 3,54 1,52 0,09 0,09
Курскит Са1оР<,аС1,2022,8р2 (ОН)1>2 34,52 56,86 3,85 5,35 1,64 0,16 0,41
12
часть ионов Са в апатитах обычно замещена ионами Sr, Ёа, Mg, Мп, Fe, а также трехвалентных редкоземельных элементов (р. з. э.) в сочетании со щелочными металлами.
Другие апатитовые минералы являются «продуктами замещения части фосфора углеродом (карбонатапатит, а также франколит и курскит 1[1, 3—5]; в качестве заместителей могут выступать элементы полуторных соединений (железо, алюминий), а также щелочные металлы. В результате получаются такие апатитовые минералы, как крандаллит Са(А1, Fe)3(PO4)2(OH)5-5H2O и милизит Ca0,5(Na, К)о,5 (А1, Fe)3(PO4)d (ОН) 415 .1,5НйО.
Важнейшими другими минералами, содержащими фосфор, являются амблигонит LiAl(PO4)F, бирюза СиА1б(РО4)4(ОН)8-5Н2О, вавеллит 4А1РО4-2А1 (ОН)3-9Н2О, вагнерит Mg2PO4F, варисцит А1РО4-2Н2О, вивианит Fe3(P04)2-8H2O, ксенотим YPO4, либетенит Си2(РО4)ОН, литиофилит Li (Мп, Fe) PO4, монацит (Се, La, Оу) РСи, отэнит Ca(UO2)2(PO4)2-8H3O, мироморфит РЬз(РО4)'3С1, триплит (Мп, Fe)2PO4F, трифилин1 Li (Fe, Мп)РО4.
Некоторые из этих минералов служат источником получения целевых редкоземельных элементов, урана^и т. д., а получаемые соединения фосфора при их переработке представляют. собой побочный продукт.
Из апатитов наиболее распространен фторапатит, меньше— гидроксил апатит и еще реже — хлорапатит. Апатиты входят в состав многих изверженных пород. Вкрапленные в них или сопутствующие им другие минералы магматического происхождения имеют кристаллическое строение [5]. К ним относятся [6, 7]: нефелин (Na, К)AlSiO4-nSiO2, пироксены [эгирин NaFe(SiO3)2 и эгирин-авгит Ca(Mg, Fe, Al) (SiO3)], титаномагнетит Fe3O4-FeTiO2, ильменит FeTiO3, сфен CaTiSiOs, полевые шпаты, слюда, эвдиалит и др.
Месторождения осадочных фосфоритов образовались в результате выветривания фосфатных пород, уноса их реками в моря, взаимодействия с другими породами и отложения как в рассеянном состоянии, так и в виде крупных скоплений. В общем балансе осадочных фосфатов кальция некоторую долю занимают фосфорные соединения органического происхождения (раковины, кости); последние под влиянием геологических и химических процессов сконцентрировались во многих местах земной коры.
Различают следующие группы и типы фосфоритов. В зависимости от условий накопления фосфатного вещества фосфориты подразделяются на три группы:
хемогенные, образованные химическим путем;
органогенные, образовавшиеся за счет скопления погибших организмов или органических выделений;
13
вторичные, т. е. руДопроЯвлениЯ, прошедшие стадию размыва и последующей концентрации.
По вещественному составу фосфориты подразделяются на четыре типа. *
I. Желваковые (Вятско-Камские месторождения, Актюбинские и др.).
II. Ракушечниковые (месторождения Эстонии и Ленинградской обл.).
III. Микрозернистые (месторождения Каратау).
IV. Остаточно-метасапатические (Белкинское и Телекское месторождения), образовавшиеся в результате сложных вторичных превращений фосфатных пород.
В подавляющем большинстве фосфориты состоят из фторапатита, реже—из карбонатапатита и в небольшом количестве в них находится изоморфная смесь фторгидроксилапа-тита. Кроме фторапатита, в фосфатное вещество" некоторых фосфоритов входят также франколит (штаффелит) и курс-кит [1].
Фосфоритные руды отличаются от апатитовых высокой дисперсностью содержащихся в них фосфатных минералов [5] и тесным срастанием их с сопутствующими минералами— примесями. Фосфат встречается как в виде коагулировавшего геля, близкого к аморфному, так и в явно кристаллической форме, хотя имеются многочисленные промежуточные группы.
Вследствие’высокой дисперсности фосфатного . вещества фосфориты, в отличие отхапатитовых руД, обладают большей удельной поверхностью и растворяются в кислотах быстрее апатита. Содержащаяся в некоторых из них Р2О5 извлекав ется кислыми почвенными растворами и усваивается растениями. Поэтому Особенно легко разложимые фосфориты, в частности желваковые и в меньшей степени ракушечниковые, применяют после размола в виде фосфоритной муки как дешевое удобрение на кислых почвах.
Фосфоритные, а также апатиткарбонатные и апатитсили-катные руды, помимо фосфатного вещества, содержат значительные количества других минералов [2]: глауконита (R2O+RO)R2O3-4SiO2-2HiO (где R2O — Na^O и К2О, RO — MgO, СаО и FeO, R2O3 —Рег03 и А12О3); лимонита ' 2Fe(OH)3*Fe2O3; кальцита СаСО3; доломита CaCO3-MgCO3; магнезиальных силикатов (например, форстерита Mg2SiO4); каолина A12O3-2SiO2-2H2O; пирита FeS2, полевых шпатов, кварца, гранита и других, а также небольшие количества органического вещества.
1.2. ХАРАКТЕРИСТИКА ФОСФАТОВ РАЗЛИЧНЫХ МЕСТОРОЖДЕНИЙ
Апатиты
Советский Союз располагает крупнейшими месторождениями апатита на Кольском полуострове. Мощность рудного тела, простирающегося здесь на большой площади, достигает глубины 200 м и более. Апатиту сопутствует в основном нефелин. В среднем хибинская руда содержит 10—30% Р2О5 в зависимости от зон залегания разрабатываемых пластов.
Важнейшие месторождения апатита в Хибинах — Куэль-порское, Кукисвумчоррское, Юкспорское и Расвумчоррское. Кукисвумчоррское месторождение начали разрабатывать открытыми методами на созданном комбинате «Апатит» в 1930 г. В настоящее время действуют рудники как на. Кукис-вумчоррском, • так и на Юкспорском и- Расвумчоррском месторождениях. Работы ведутся открытым и подземным способами. Апатито-нефелиновая руда с рудников поступает на обогатительные фабрики, где флотацией выделяют из нее апатитовый концентрат, содержащий 39,4—40% Р2О5.
Объем производства концентрата на комбинате «Апатит» составил (млн. т): 1965 г. — 7,5; 1967 г. — 8,7; 1970 г.— 14,5; 1975 г. — 15,33 и в 1980 г. (план) — 19,0 [8].
Увеличение добычи руды, по-видимому, в. дальнейшем возможно в небольших размерах из-за ограниченности запасов и соображений эксплуатации этого уникального месторождения в течение достаточно длительного периода.
Кольский апатитовый концентрат — лучшее в мире сырье для производства фосфорной кислоты и удобрений. Он частично экспортируется в страны СЭВ и некоторые другие.
• Промышленное значение имеет т^кже Ковдорское месторождение апатито-магнетитовых руд (Мурманская обл.). После извлечения из них магнитной сепарацией железного концентрата остаются хвосты, содержащие 11,5—12,0% Р2О5 [9]. Из' последних дальнейшим обогащением могут быть получены концентраты с содержанием до 34—38% Р2О5 (табл. 1.2|. Однако вследствие трудности отделения магниевых соединений в них находится до 7% и более MgO в виде, главным образом, форстерита 2MgO-SiO2; это крайне затрудняет переработку концентрата кислотными методами, в частности, экстракцией серной кислотой.
, Малое содержание в продукте ионов фтора свидетельствует о замещении их в решетке апатита ионами гидроксила [10].
В СССР имеются некоторые другие залежи апатитовых-руд в Джамбульской, Иркутской (Белозиминское — в Западном Прибайкалье), Кемеровской (Белкинское) и других об-
15
Таблица 1.2
Химический состав Ковдорской исходной рядовой руды и продуктов ее переработки
Компоненты Среднее содержание компонентов, %
исходная железная руда хвосты магнитной сепарации апатитовый концентрат
Р2О5 7,09 11,5—12,5 34,1—38,8
СаО 13,72 28,7—29,5 47,5—53,0
MgO 12,55 18,6—20,9 2,0—7,0
SiO2 9,07 15,8-1’6,2 0,5—5,1
A12O3 3,15 1,2—-3,2 0,1—0,4
со2 . 9,10- 12,0—13,4 1,3—6,9
Fe 31,52 (Fe2O3) 2,5—3,4 0,3-0,7 (FejOaJ
FeO 12,33 1,7—2,4 0,2—0,3
TiO2 Н. О. 0,1—0,2 Н.О.
МпО н. о. 0,2—0,4 0,03—0,05
s 0,17 0,1—0,3 н. о.
iKaO+Na^O И. О. 0|,7—<Ы 0,3—0,4
ZrO2 Н. О. 0,1—0,18 н. о.
F Н. О. 0,1—0,3 0,5—0,83
ластях, в Бурятской АССР (Ошурковское), Якутской АССР (Селигдорское). Из них на базе Белозиминского и Ошурков-ского месторождений предполагается строительство предприятий по производству фосфатного сырья [в].
В последнее время- открыто месторождение апатитов в районе оз. Ессей Красноярского края. Апатитоносные образования широко распространяются, от озера к юго-востоку Таймырского полуострова до бассейна р. Котуй и Оаймеча.
Новый массив апатитоносных пород на юге, Якутии открыт в бассейне реки Хани — зоне Байкало-Амурской магистрали.
Ошурковское месторождение близ Улан-Уде—одно из наиболее изученных [11, с. 104] —простирается от Байкала до Тихого океана и насчитывает огромные запасы фосфатного сырья (более 1 млрд. т). На его базе строится Забайкальский апатитовый комбинат — крупнейшее на востоке страны горнодобывающее предприятие. Химическая переработка этого сырья может обеспечить фосфорными удобрениями сельскохозяйственные районы Сибири и Дальнего Востока. Флотацией руды, содержащей ~4% Р2О5, получен концентрат с содержанием 34,5—37,1% РаО5 при степени извлечения ее 79,3—86,3% [12].
Фосфориты
Фосфориты составляют в отечественных фосфатных ресурсах 75—80%, поэтому их значение в общем балансе фосфатного сырья исключительно велико.
, - 16
В Советском Союзе насчитывается более 200 месторождений фосфоритов с общими выявленными запасами свыше 16 млрд. т. Однако большинство из ник маломощные, а крупные месторождения состоят из бедных песчанистых, желваковых и ракушечниковых фосфоритов.
Крупные залежи пластовых фосфоритов в Союзе были открыты в 30-х гг. в Южном Казахстане в горном хребте Ка-ратау. Они превосходят по своим запасам и мощности пла-
стов все известные аналогичные месторождения.
В бассейне Каратау выявлено около 45 месторождений. Важнейшие из них — Чулактау, Аксай, Джанатас, Кокджон и Коксу. Имеются и другие значительные месторождения — Герес, Учбас, Акджар и Тьесай. По своим запасам (~5 млрд, т) бассейн представляет собой вторую (после Хибин) сырьевую базу фосфатов в стране [13].
Пласты фосфоритов имеют темно-серый цвет различных оттенков. Руда состоит из микроскопических зерен фосфатного вещества и оолитов, сцементированных фосфатом.или карбонатом, и мелкокристаллического апатита — продукта метаморфизации фосфатов органического происхождения. Руда содержит в зависимости от месторождения и залегания пласта, в %: РгО5—16—26; СО2 — 5—19; MgO — 1,5—4; SiO2—15—35; Fe2O3— 0,6—1,6 (в том числе 0,1—0,7% FeS2 и 0,3—'1,2% FeO); А120з—1,5-J2,7; F— 1,5—2,4; Na2O — ^0,3—0,9; К2О —0,5—0,7; МпО —0,1—0,7; Н2О —0,8—1,7 и прочие.
w Третьей по значимости сырьевой базой является Актюбин-^ская группа месторождений. Они представлены желваковы-ми фосфоритами, простирающимися на большой площади с общим запасом не менее 1 млрд, т [14]. Из них наиболее важное — Чилисайское, на базе которого намечается построить рудник мощностью 950 тыс. т фосфоритного концентрата, содержащего 24% Р2О3, с последующим увеличением его мощности до 5 млн. т [8].
Другие выявленные и предварительно оконтуренные месторождения: Богдановокое (с разведанными запасами в ~100 млн. Т), Коктюбинское (с запасами в 250 млн. т). Руды содержат в среднем 12—>14% Р2О5; до 5% R2O3; 3—4% карбонатов и 30—65% нерастворимого остатка.
Из Брянских месторождений наиболее важны Полпин-ское, Сеннинское, Подбу'жское и Слободско-Каторецкое. Добыча руды ведется преимущественно открытыми методами.
Большие залежи фосфоритов сосредоточены в Вятско-Камских и Егорьевских месторождениях. Суммарные запасы первых составляют-~ 1 млрд т. Фосфатное вещество сосредоточено в желваках, свободно залегающих без цементации
в песчанистой, слегка глинистой породе. Чистые желваки, 20~32% P2°s>и 2>5-1з’8%
2- И73 Т.-‘-хннческея. . I 17
библаот^жа I
от
WONS
R2O3, а руда — 21—27% Р20б и 4,5—11,5% R2O3; она легко поддается обогащению избирательным дроблением с грохочением и обмывкой песка.
Егорьевское месторождение, расположенное в Московской области, простирается на площади до ~950 км2. Запасы его исчисляются примерно в 700 млн. т. Однако вследствие длительной и интенсивной эксплуатации срок дальнейшего использования этих залежей ограничен. В руде содержится 20—25% Р2О5, 5—15% R2O3 и 10—25% нерастворимого остатка.
Месторождения в Тульской, Курской, Калужской, Рязанской и других областях менее мощны. Второстепенное значение в последнее время имеют также фосфориты украин- ских месторождений — Подольское и Кролевецкое (правобе- | режная Украина) и Славяноко-Изюмское (Донбасс). -1
Ракушечные (ободовые) фосфориты находятся в относительно больших количествах в Эстонской ССР и Ленинградской области [1; 15, с. 32]. Месторождения образовались в. результате скопления фосфатных раковин.
Важнейшим из них является Кингисеппское в Ленинградской области (балансовые запасы ~200 млн. т), Маарду и Тоолсе в Эстонской ССР. Дополнительно к горнохимическому предприятию в Маарду предполагается строительство рудника на базе месторождения Тоолсе. Добываемая руда содержит 7—10% Р2О5 и сильно загрязнена кварцевым песком и другими примесями.
Фосфориты Кингисеппского месторождения состоят из: слабо ожелезненного кварцевого песка и обломков раковин брахиопод [15, с. 32]. Руда содержит 70—75% кварца, 15— 25% фосфатов, ~1% карбонатов (преимущественно доломитов) и небольшие количества глауконита и других минералов. Содержание Р2О5 в отдельных образцах колеблется от 5—6 до 15%.
Эстонские и кингисеппские фосфориты имеют весьма дисперсную структуру. Они состоят из рыхлых агрегатов (0,1 — 0,15 мкм) мельчайших пористых частиц. Руды обогащают избирательным дроблением и флотацией с получением концентрата, содержащего от 20—22 до 30% Р2О5. На базе Кингисеппского месторождения» работает Кингисеппское производственное объединение (КПО) «Фосфорит», выпускающее ~ 1,7 млн. т концентрата (в усл. ед.) в год. Это единственное в мире предприятие, перерабатывающее столь бедные фосфориты 'в высококачественный концентрат-.
Месторождение ракушечниковых фосфоритов Тоолсе является одним из крупных и перспективных [16]. В руде содержится от 0,5 до 12,7% Р2О5. Основными минеральными примесями являются кварц, доломит и пирит [17], а также другие железистые минералы. Руда достаточно хорошо под
18
дается обогащению флотацией с получением концентратов, содержащих 28—34% Р2О5 при степени извлечения ее 70— 85% [11, с. 78; 18, 19].
Из разведанных месторождений в Сибири наиболее перспективно Таштагольское (Кемеровская область), представленное пластовыми карбонатными фосфоритами. Флотацией руды двух разведанных участков — Нимзасского и Белкин-ского — получен концентрат, содержащий, в %: Р2О5— 29,3; СаО—48,2; MgO — 6,5; СО2 —5,1; Fe2O3—1,2; А12О3 —0,6; F — 2,5 и SiO2 — 3,5.
В районах Западного Прибайкалья обнаружены залежи пластовых вторичных (остаточных) фосфоритов. Наибольший интерес представляет Сарминское месторождение. В руде содержится 16—18% РгО5.
Залежи фосфоритов в Красноярском крае (важнейшие — Телекское, Тамалыкское и Сейбинское) относятся как к желваковым песчанистым, так и к вторичным остаточным. Руды Телекского месторождения представлены остаточно-метасоматическими фосфоритами; они содержат, в %: Р2О5 — 12; СаО — 19,2; MgO — 4,3; СО2 — 8,1; Те2О3 — 5,3; А12О3 — 11,1 и SiO2 — 38,3. Руда Тамалыкского месторождения содержит, в %: Р2О5—НО—23; СаО — 27—35; R2O3—-2—3; MgO — 1—1,5 и SiO2 — 25—45.
Руда Сейбинского месторождения состоит из 22—35% гидроксилкарбонатапатита, 40—47% кварца, 10—15% каолинита и 7—15% гидрогематита [20]. Она содержит 9—15% Р2О3.
Фосфоритные руды Удско-Шанторского месторождения (Хабаровский край) содержат 7—15% Р2О5 и 0,2—0,5% MgO. Из них получены концентраты с содержанием 30— 33% РгО5 при степени извлечения 78—188% [21].
Мировые запасы фосфатов, пригодных для разработки современными средствами, оцениваются в 150 млрд, т (~20—23 млрд, тРД), из которых 80% составляют фосфориты, в том числе в СССР —27% от мировых запасов промышленного значения [22]. Мировая добыча природных фосфатов в настоящее время составляет ~ 100 млн. т в год (в расчете на товарный продукт, удовлетворяющий требованиям мирового рынка, содержащий не менее 30% Р2О5) и непрерывно увеличивается в среднем на 1,5—^2% в год [23].
До последнего времени потребность в фосфатном сырье в СССР покрывалась в основном апатитовым концентратом. Дальнейший рост потребности в фосфатном сырье необходимо покрыть за счет соответствующего увеличения добычи -фосфоритов, в первую очередь бассейна Каратау, а также Актюбинского (Чилисайского), Белозиминского и других месторождений, Ошурковских, Ковдорских и других апатитов.
.2* 19
Расчетами, выполненными в ЛенНИИГипрохиме, показано, что за счет запасов сырья бассейна Каратау при ускоренном его развитии возможно обеспечить перспективную потребность страны в фосфатном сырье в И и 12 пятилетках на 35% (в 10 пятилетке — на 26%) [24, с. 1]. При этом доля от общего количества сырья, направляемого на термическую переработку, уменьшится от 56% в 1980 г. до, 30% в 2000 г. Соответственно доля сырья, используемого для сернокислотной экстракции, возрастет по фосфоритной муке от 28% в 1980 г. до 60% в 2000 г. и уменьшится по фосконцен-трату (с учетом производства двойного суперфосфата) от 16% в 1980 г. до 10% в 2000 г. ' _
1.3. ОБОГАЩЕНИЕ ФОСФАТНОГО СЫРЬЯ
Пригодность фосфатного, сырья для кислотной переработки определяется, как известно, содержанием в нем не только Р2О5, но и разлагаемых кислотами примесей — карбонатов кальция и магния, окисей железа, алюминия и др. При этом массовое отношение Fe2O3: Р2О5 в сырье не должно превышать 7—'10%, MgO :T2Os 7—8%.
Для выделения из фосфатных руд фосфорсодержащего минерала и максимального отделения пустой породы применяют как первичную обработку их (например, грохочение и отмывку), так и последующее вторичное обогащение—в основном флотацию [25]. Последняя может быть применена и в качестве первичного, т. е, самостоятельного способа обогащения.
Желваковые руды отделяют из тонкодисперсной пустой породы (с размерами менее 0,5 мм) грохочением или отмывкой. Остающийся материал (класс +0,5 мм) содержит до 22-—25% Р2О5, который иногда и составляет так называемый мытый продукт. Аналогичным образом подвергают первичной обработке ракушечниковые руды. Так, при дроблении и измельчении низкосортной руды Маарду, содержащей всего 5—10% Р2О5, происходит практически классификация основных минералов [26] с накоплением фосфатов в классе —0,5+0,25 мм, содержащем 26^-27% Р2О5.
Однако при неравномерности состава руды отдельных участков требуется подбор специальных условий их обогащения [26].
Из общих запасов разных типов фосфатного сырья бассейна Каратау (без учета фосфатов в кремнистом сырье) высококачественные руды составляют ~10%, в том числе низкомагниевые — всего ~2,2—2,5% [15, с. 5; 27].
В Советском Союзе фосфатное сырье с рудников выпус-' кают в виде флотационных концентратов (хибинский апатитовый концентрат, каратауский, егорьевский, кингисеппский и другие флотационные концентраты), мытых фосфоритов 20
f
(вятский, егорьевский, актюбинский) и первичных фосфоритных концентратов, получаемых избирательным дроблением и отсевом — курский (щигровский), брянский (полпинский), , эстонский и др.
В настоящее время флотациоАный метод обогащения фбс-фатных руд является наиболее эффективным.
Получаемый на Кировской апатито-нефелиновой обогатительной фабрике флотационный апатитовый концентрат отличается однородностью и содержит 39,4—40% РзОб- Это наиболее концентрированное фосфатное сырье в мире. Согласно ГОСТ 5.1183—72 апатитовый концентрат должен содержать не менее 39,4% Р2О5 (в пересчете на сухое вещество) и не более 1% влаги (летом не более 1,5%); остаток на сите 0,05 мм "(1600 отв/см2) должен составить не более 14%.
Флотационным обогащением некоторых фосфоритов (кингисеппских, марокканских и других) получают из руды достаточно богатые концентраты. Отделение кремнистых минералов от фосфатных обычно не представляет затруднений и осуществляется для песчанистых желваковых и ракушечниковых фосфоритов при пом'ощи анионных собирателей в присутствии щелочных реагентов — регуляторов. Однако трудно поддаются флотации фосфориты, содержащие большое количество карбонатов и состоящие из мелкокристаллических частиц фосфатов. Так, для фосфоритов Каратау даже отделение кремнистых минералов осложняется из-за необходимости тонкого измельчения руды вследствие высокой дисперсности включенного в фосфат халцедона. Особенно затруднительно отделение карбонатов, имеющих близкую фло-тир’уемость с фосфатами. Поэтому эффективное использование месторождения является предметом непрерывных изыс-' каний [28]. -
Производственным объединением «Каратау» выпускается товарная руда, которая'для термической переработки (по ОСТ 6-25-19—74) должна содержать не менее 21% РгО5 Для класса +'10 и +70 мм, а для кислотной переработки богатая фосфоритная мука должна содержать не менее 28% Р2О5 (ТУ 6-25-5—73). .
Для кислотной переработки фосфатное сырье получают как путем сухого размола высококачественных (богатых) руд, так и флотационным обогащением менее богатых (рядовых) фосфоритных руд. При этом даже из фосфоритов со средним содержанием 23,3% Р2О5 и 3,6% MgO при существующих методах обогащения получают флотационный кон- центрат, содержащий только 27,9% Р2О5 и 2,45% MgO. Сте-. пень извлечения Р2О5 во флотационный концентрат не превышает 63—65%, т. е. при обогащении теряется до 35% фос
21
фатного вещества. Хвосты обогатительной фабрики, содержащие 16—18% Р2О5 и 4—6% MgO, не используют.
Неэкономичность и неэффективность обогащения рядовой руды, а также весьма небольшие запасы богатой руды об-, условливают необходимость изыскания путей широкого использования для химической переработки руд, содержащих 23,5% Р2О5 (и меньше) и 3—3,5% MgO (и больше).
Флотация ракушечниковых оболовых фосфоритов протекает более успешно вследствие физической расчлененности частиц основных их минералов—фосфата и кварца. Из весьма бедной кингисеппской руды . (6,5—8,5% Р2О5), получают флотационный концентрат, содержащий 28—30% Р2О5. В чистом фосфатном веществе раковин концентрация Р2О5 составляет 34,5—36,5%.
В лабораторных условиях были достигнуты удовлетворительные результаты при флотации фосфоритов Красноярского [20] и Хабаровского краев [21].
В стадии лабораторных исследований [29] находится изучение обогащения бедных фосфоритных руд Каратау, содержащих карбонаты, путем обжига их при 950—1000 °C,' гашения обожженного продукта водой й отделения тонкой суспензии гашеной извести (и гидроокиси магния) от более крупных частиц фосфорита.
1.4. ОБЛАГОРАЖИВАНИЕ (ОБЕЗМАГНИВАНИЕ) ДОЛОМИТИЗИРОВАННЫХ ФОСФОРИТОВ
И КАРБОНАТ-АПАТИТОВЫХ РУД
Очевидно, наиболее полное удаление- карбонатов, а также эффективное обогащение любых бедных фосфоритных руд возможно (наряду с усовершенствованием механических и флотационных методов) с применением химических способов— обработкой руды растворами кислот или солей J3, 30].
Химическое обогащение с целью удаления карбонатов может быть применено к шламам, образующимся при измельчении фосфоритов, и вследствие высокой дисперсности не поддающимся флотации.
Доломит, содержащийся в фосфоритах, растворяется в разбавленной (2—5%-ной) H2SO4 с большой скоростью с переходом части магния, а также некоторого (незначительного) количества фосфата в раствор [31]. Полученный в лабораторных условиях остаток (концентрат) из фосфорита Ка-рат'ау (Чулак-Тау) содержал 29—30% Р2О5 и всего 0,5— 1,0% СО2. Но при этом происходит образование разбавленных й отбросных растворов, а также потеря фосфатного вещества в результате перехода его в раствор.
Между тем обезмагнивание магнийсодержащих руд (месторождений Каратау, Ковдора и’др.), составляющих ~30% всех отечественных запасов, является весьма важным как в 22 ,
плане подготовки фосфатного сырья для дальнейшей, переработки, так и вследствие возможности получения водо- или цитраторастворимых магниевых удобрений, спрос на которые не удовлетворяется.
Многолетними исследованиями и опытными работами, проведенными совместно рядом организаций (ЛТИ им. Ленсовета, ЛенНИИГипрохимом, КПО «Фосфорит»), определены пути проведения процесса по циркуляционной схеме (с получением сульфата или аммонийфосфата магния в качестве продукта) или по «прямой»-схеме — с регенерацией образующихся разбавленных растворов известковым молоком (с получением «магнезий»)..
Показано, что сернокислотная экстракция фосфорной кислоты из доломитизированных фосфоритов с предварительным их обезмагниванием может быть проведена весьма экономично с высокими техническими показателями (стр. 115 и сл.).
Эффективность процесса селективного растворения карбонатов зависит в основном от температуры и так называемой «действующей» 1концентрапии кислоты в реакционной зоне, которая определяется концентрацией и нормой исходной кислоты, отношением Т:Ж в обрабатываемой суспензии, продолжительностью и интенсивностью перемешивания. Суммирующим показателем этих условий является значение pH процесса. .
При низких температурах (20—40 °C) скорости разложения карбонатов и фосфатов различаются ненамного. С повышением температуры скорость . разложения карбонатов возрастает так, что почти вся кислота й* короткое время взаимодействует с ними. В результате этого масса кислоты, реагирующей с фосфатом, весьма невелика, что обусловливает незначительное разложение, фосфатной части с минимальными выходами (потерями) Р2О5 в жидкую фазу. Наибольшая селективность достигается 'в интервале 80—95 °C при pH 1,5—4,5 (в зависимости от природы фосфата), концентрации исходной кислоты 5—93%, норме ее 75—85% от стехиометрического количества в расчете на карбонаты, отношении 2^Ж:Т^5 и продолжительности 5—30 мин. Имеющиеся в фосфатном сырье Каратау примеси не влияют на степень и скорость обезмагнивания его. Лишь в диапазоне pH 1,5— 2,5 небольшая часть соединений полуторных окислов переходит в жидкую фазу. Содержащиеся в сырье соединения кремния в виде тонкодисперсных частиц кварца ’и халцедона практически не переходят в раствор. В отличие от этого при обезмагнивании ковдорского апатита, содержащего форстерит Mg2SiO4, в образующихся растворах содержится до 0,3—0,5% SiO2 (в растворенном и коллоидном состояниях).
Образующиеся растворы обезмагнивания содержат 0,5— 2% MgO и 0,01— 0,05 Р2О5 (в зависимос.ти от сырья).
23
При этих условиях из исходного сырья извлекается 60— 75% MgO, 65—®0% СО2 и не более 0,5% Р2О5. Например, полученный из рядовой муки Каратау (состава, в %: Р2О5— 23—24; СаО —41—42; MgO —3—3,5; СО2 —8—8,5; R2O3 — 3,0—3,3; SiO2—15—16 и F — 2) обезмагненный фосфорит (в пересчете на сухой продукт) содержит, в %: MgO—1—0,7; СО2 — 2—1,4 и SO3 — 7 и при практически таком же, как и в исходном сырье, содержании Р2О5 (23—24%), СаО (41— 42%X'R2O3 (3%), SiO2 (16%) и F (2%).
Ойзмагнивание доломитизированных фосфоритов по циркуляционной схеме изучено применительно к разным реакционным средам — циркулирующим растворам с выделением из части его в качестве продукта растворенного компонента {27].
В сульфатно-аммиачном процессе растворителем карбонатов является серная кислота, а оборотным раствором служит 10—20%-ный раствор (NH4)2SO4. Оборотный раствор регенерируют из образовавшегося раствора с выделением из него соединений магния добавкой к нему фосфорной кислоты и аммонизацией с образованием магнийаммонийфоюфата [29, с. 4] .
По сернокислотному способу суспензию фосфорита в оборотном растворе сульфата магния (10—25% MgSO4) обрабатывают серной кислотой и образующуюся продукционную часть раствора сульфата магния отводят из цикла для получения товарного сульфата магния. Видоизменение этого процесса— обработка суспензии фосфорита в 10—25%-ном растворе MgSO4 при Т:Ж=1:2—4 отходящим сернистым газом, содержащим 0,05—1% SO2 и 8—20% кислорода при 70—90 °C в течение 5—30 мин [27]. В этих условиях растворенная двуокись серы (сернистая, кислота) интенсивно окис-. ляется в жидкой фазе в присутствии каталитических примесей фосфата (Fe2O3, МпО2 и др.) с образованием серной кислоты.
Наиболее интересен сернокислотный метод как по селективности, так п в плане использования одной и той же кислоты для последующей переработки обезмагненного продукта в фосфорную кислоту.
Процесс может быть рационально использован для обез-матнивания фосфоритов, содержащих от 2 до 10% MgO (9—45% доломита) и от 15—18 до 28% Р2О5. Но при увеличении содержания доломита в фосфорите увеличивается и количество расходуемой кислоты. При обработке фосфорита Каратау, содержащего 23—24% Р2О5 и 3—4% MgO, расход серной кислоты составляет 0,1—0,15 т на 1 т обезмагненного продукта.
В прямой, открытой схеме отпадает надобность в циркуляции оборотных растворов, а это исключает различные тех-24
нологические трудности (транзитная перекачка больших масс раствора, возможное накопление приме'сей и т. п.). В данном случае раствор, получаемый в результате обезмагнива-ния, обрабатывают известковым молоком с выделением ив него сульфата кальция и гидроокиси магния и регенерацией жидкой фазы, которая возвращается в процесс для сусйпензи-рования фосфата.
Сущность метода заключается в обработке водной суспензии фосфатов при Ж:Т=1—4:1 5—93%-ной H2SO4 при 60—90 °C в течение 5—15 мин с переводом магниевых соединений в раствор в виде сульфата. Карбонатный кальций при этом связывается в сульфат в виде гипса и остается в составе фосфорита. Известно, что исходные фосфориты плохо фильтруются из-за тонкодисперсной структуры основного вещества и содержания большого количества нерастворимого остатка в виде шлама и кремнезема. При сернокислотном обезмагнивании фосфоритов выделяющийся в твердую фазу гипс взаимодействует со шламовыми частицами кремнезема, что обусловлено разным знаком заряда их поверхностей. В результате этого значительно улучшается' (в 5—10 раз и больше) фильтруемость обезмапненных фосфоритов.
Концентрация MgO в растворе составляет 0,5—2%. В осадке в зависимости от нормы кислоты остается 0,5—'1 % MgO. Степень извлечения MgO составляет 70—85%.
Селективность процесса определяется значением pH среды, которое зависит от природы фосфатного сырья. Оптимальный диапазон pH для фосфоритов различных месторождений лежит в интервале 2,5—4. При этом потери РгО5 не превышают 0,5% (отн.); селективность процесса составляет не менее 99,5%.
Жидкую и твёрдую фазы разделяют на вакуум-фильтрах (например, дисковых) без промывки. Производительность фильтрования определяется природой сырья и значением pH среды; при обогащении фосфоритов Каратау и Кингисеппа она равна 600—1000 кг/(м2-ч), считая^ на сухой осадок.
Растворы; содержащие 0,5—2% MgO в виде сульфата магния, направляют на нейтрализацию 5—10%-ным известковым молоком с получением оборотной воды, содержащей 0,006 MgO и 0,08% СаО, и смеси гидроокиси магния с гипсом [24, с. 22]. Процесс протекает при 60—80 °C в течение 15—30 мин при норме гидроокиси кальция 105—110% от стехиометрической в расчете на содержание MgO. Осаждение смеси гидроокиси магния и гипса протекает со скоростью 1—2 м/ч.
Выделять гидроокись магния из ее смеси с гипсом можно гидравлической классификацией [32] в отстойниках непрерывного действия, а также в гидроциклонах (размер частиц гидроокиси магния 0,05—0,8 мкм, а кристаллов гипса 0,1 —
25
0,4x0,05—0,1 мм). При этом образуется продукт, содержащий 80—85% Mg(OH)2, и пригодный для защитного покрытия трансформаторной стали. Такая магнезия по действующим прейскурантам оценивается в ~700 руб/т. Потребность одного металлургического завода в магнезиальном покрытии составляет ~ 1000 т MgO/год.
При сушке сгущенной суспензии (Ж:Т=3—5:1) при 100—105 °C в аппарате с кипящим сдоем с отдувкой гидроокиси магния, уносимой с газом и отделенной в фильтрах или циклонах, продукт содержит 45—50% MgO. Цена на техническую гидроокись магния (магнезию) для шлакоси-таллов по ТУ 60 86-62-52—73 равна 435 руб/т (45% MgO). Потери MgO с гипсом составляют 15—20%.
Образующийся в процессе разделения смеси гипс, кром-е некоторого количества окиси магния, не содержит других примесей и может быть использован для строительных целей.
Нейтрализовать растворы можно аммиаком при добавке фосфорной кислоты с образованием магний аммонийфосфата. Однако это представляется менее целесообразным по сравнению с осаждением гидроокиси магния известковым молоком.
Облагороженный концентрат, содержащий 23—28% Р2О5 и 0,5—1% MgO, является вполне доброкачественным, (отношение в нем MgO к Р2О5 составляет 2—5%), пригодным для кислбтной переработки в концентрированные фосфорсодержащие удобрения. В частности, при обезмагнивании кингисеппского флотационного концентрата, используемого в качестве вторичного фосфата в производстве-двойного-суперфосфата, возможно сэкономить до 15—47% расходуемой в процессе фосфорной кислоты, получаемой из апатитового концентрата. Это может дать заводу двойного суперфосфата, мощностью 1—1,5 млн. т (в усл. ед.), экономию в несколько миллионов рублей.
Дополнительное обогащение обезмагненного фосфорита физико-механическими методами (флотацией, в тяжелых суспензиях и т. п.) и отделением фосфатной части его от балластных веществ—нерастворимого остатка и сульфата кальция — может позволить получить из фосфоритов высококачественные концентраты, соответствующие содержанию полезного вещества требованиям зарубежных стандартов.
ЛИТЕРАТУРА
1. Бушинский Г. И. Апатит, фосфорит; вивианит. М., Изд. АН СССР, 1952.
2. Гиммельфарб Б. М. Агрономические руды. М., Изд. АН СССР, 1938. 3. Чепелевецкий М. Л., Бруцкус Е. Б. Суперфосфат. Физико-химические основы производства. ЛА., Госхимиздат, 1958.
4. Кореньков Г. Л., Устинова Н. А. Горнохимическое сырье зарубежных стран. Л., Химия, 1965.
26
5. Вольфкович С. И., Гришпан Л. Б., Шехтер А. Б. — ДАН СССР, 1952, т. 85 ,Ks 1, с. 137. ►
6. Ферсман А. Е. Апатит. Т. 3. Хибинские апатиты. Л., ОГИЗ, 1931.
7. Фивег М. П„ Шубин А. П. Апатиты. Л., ОНТИ, 1937.
8. Кожевников А. О. — Хим. пром., 1977, № 3, с. 166.
9. Горбунов Г. И.—В кн.: Проблемы минерального сырья. М., Наука, 1975, с. 59,-
10. Пирогов В. И. и др.—Хим. пром., 1971, № 3, с. 185.
-11. Обогащение гориохимических руд. Труды ГИГХС. Вып. 20. М-, 1973.
12. Смирнов Ю. М., Селиванова И. С. — Хим. пром., 1974, № 3, с. 208— - . 210.
13. Мельникова М. В. — В кн.: География химической промышленности. М.,'Мысль, 1967, с. 50;
14. Гиммельфарб Б. М.— Изв. АН СССР. Сер. геол., 1962, № 11, с. 32; Милецкий Б. Е.^ Железко В. И. — Вести. АН КазССР, 1964, № 3 (228), с. 18.
15. Геология месторождений фосфоритов. Труды ГИГХС. Вып. 26. М., 1974.
-16. Мустыйги 3. — Изв. АН СССР. Химия. Геология, 1970, т. 19, № 1, с. 57.
17. Наливкина А. И., Горлова В. М. Промышленность горнохимического сырья. М., НИИТЭХИМ, 1974, вып. 2, с. 3; Раудсепп Р. — Изв. АН ЭССР. Химия. Геология, 1975, т. 24, № 2, с. 137; Смирнов Ю. М., Цу-цульковский В. Я-, Юркова П. А. — Труды ГИГХС, 1971, вып. 15, ч. II, с. 38.
18. Юркова Л. А., Селиванова И. С., Смирнов А. И. — Хим. пром., 1970, №11, с. 829.
19. Цуцульковский В. Я-, Юркова Л. А., Наливкина А. И., Смирнов А. И. — Там же, 1972; № 9, с. 683.
20. Колмогорова В. И. — Там же, 1965, № 11, с. 29.
21. Алехин А. М., Петрунина С. И., Кожевников А. О., Рахманова Р.,И. — Там же, 1973, № 11, с. 835—838.
22. Зверев А. С., Файзуллин Р. М., Фахрутдинов Р. 3. и др. Геологиче- ские методы поисков и разведки месторождений неметаллических полезных ископаемых.' М., ВИЭМС, 1975; Минеральное ресурсы промышленно развитых капиталистических и развивающихся стран на начало 1973 г. М., Изд. Всес. геол, фонда, 1974, с. 3.31—333.
23. Stephenson G. L. — Eng. a. Mining J., 1972, № 3. р. 157—159.
24. Фосфорная промышленность. Научно-техн. реф. сб. НИИТЭХИМ, ЛенНИИГипрохим. Вып. 3. М., 1979.
25. Ратобыльская Л. Д., Бойко Н. - Н., . Кожевников А. О. Обогащение фосфатных руд. М., Недра, 1979. 262 с.; Глембоцкий В. А. Физнко-химия флотационных процессов. М., Недра, 1972; Глембоцкий В. А., Классен В. И. флотация. М., Недра, 1973.
26. Зачураев В. Г., Ранц Т. Ф., Шувалова Н. К. — Хим. пром., 1975, № 10, с. 768—770.
27. Переработка фосфоритов Каратау. Промышленные методы и лабораторные разработки/Под ред. М. Е. Позина, Б. А. Копылева. В. Н. Белова и В. А. Ершова. Л., Химия, 1975.
28. Мухтаров М. А., Колиев Б. В., Ургалиев М. М. — Хим. пром., 1975, №'5, с. 351—357.
29. Баскакова М. И,, 'Иоаниди М. А. — Там же, 1976, № 9, с. 671—672; Вейдерма М. А., Вескимяэ X. И., Куусик Р. О.— Там же, 1974, № 4, с. 270—273. - -
30. Черняк А. С. Химическое обогащение руд. Изд. 2-е. М., Недра, 1976.
31. Чепелевецкий М. Л. и др. — В кн.: Сообщения о научно-технических работах НИУИФ, 1957, № 2, с. 15.
32. Ризе Д. Ф., Кулькова Т. Ф., Коробицын А. С. — Хим. пром.. 1972, № 7, с. 524—525.
Г лава II
ФИЗИКО-ХИМИЧЕСКИЕ ОСНОВЫ ЭКСТРАКЦИИ ФОСФОРНОЙ КИСЛОТЫ
П.1. ВЗАИМОДЕЙСТВИЕ ОСНОВНЫХ ЧАСТЕЙ ФОСФАТОВ С СЕРНОЙ И ФОСФОРНОЙ КИСЛОТАМИ
При разложении фосфатов серной кислотой наряду с фосфорной кислотой образуется практически нерастворимый сульфат кальция:
Ca5F (РО4) 3+5H2SO4 -> 5CaSO4+3H3PO4+HF
В случае смешения фосфата с серной кислотой даже средних концентраций (начиная с ~30% H2SO4) образуется густая малоподвижная пульпа, не "поддающаяся разделению. Поэтому разложение фосфата проводят в присутствии некоторого количества циркулирующей продукционной фосфорной кислоты и возвращаемых в процесс промывных растворов (так называемого раствора разбавления). В результате этого вначале фосфат реагирует в той или иной степени с фосфорной кислотой, содержащейся в растворе разбавления;
Ca5F (РО4) з+пН3РО4 -> 5Са (Н2РО4)2+ •
4-i(n—7)H3PO44-HF
Затем образовавшийся Са(Н2РО4)2 взаимодействует с серной кислотой в присутствии фосфорной кислоты: . .
С а (Н^РО4) 2-|- H2S О4+тН3 РО4
->-CaSO4+ (m-f-2) Н3РО4
, В действительности механизм процесса более сложен в соответствии с особенностями смешанных растворов.
Сульфат йальция может быть выделен в форме дигидратз CaSO4-2H2O (гипса), полугидрата CaSO4-0,5H2O или безводного CaSO4 (ангидрита)*. В зависимости от этого процесс можно.вести дигидратным, полугидратным или ангидритным способами при разных температурах с получением фосфорной кислоты разной концентрации. Количество воды, вводимой в систему, определяется не столько степенью овод-нения выделившегося сульфата кальция, сколько отмывкой
* В дальнейшем сульфаты кальция для краткости будем называть дигидратом (или гипсом), полугидратом и ангидритом.
28
фосфорной кислоты из осйдка и необходимостью создания определенных текучести пульпы и концентрации кислоты.
В настоящее время наиболее распространен дигидратный экстракционный процесс, осуществляемый при 70—>80 °C с получением фосфорной кислоты, содержащей 24—32% Р2О5.
Содержащиеся, в природных фосфатах силикаты (нефелин, глауконит, каолин) разлагаются с образованием сульфатов И кремниевой кислоты, например:
,Na2O К2О • А120а- 2S1O2+5H2SO4 —>
^Na2SO4+K2SO4+Al2(SO4)3+2SiO2+5H2O
Выделившаяся кремниевая кислота реагирует с растворенным фтористым водородом, образуя кремнефтористоводородную кислоту
6HF+SiO2^H2SiF6+2H2O
которая частично выделяется в газовую фазу в виде эквимолекулярной смеси 2HF и SiF4. Тетрафторид кремния образуется из H2SiF6 в присутствии избытка кремниевой кислоты в растворе:
2H2SiF6-HSiO2-> 3SiF4+2H2O
Над растворами фосфорной кислоты давление паров соединений фтора относительно невелико (меньше, чем над растворами серной кислоты). В газовую фазу при экстракции, например, дигидратным способом выделяется 6—10% фтора от его общего количества [1, 2].
Соединения фтора, уносимые с газами, абсорбируются .водой с образованием раствора H2SiF6 .
. (п+2) H2O4-3SiF4 2H2SiF6+SiO2 • /гН2О
Из других примесей наибольшее влияние на процесс экстракции оказывают карбонаты кальция и магния и соединения полуторных окислов (железо, алюминий) [2].
Карбонаты и силикаты кальция и магния легко разлагаются серной кислотой:
CaMg(>CO3)2+2H2SO4^CaSO4+MgSO4+i2H2O+2CO2
. Mg2SiO4+2H2SO4^2MgSO4+SiO2+2H2G| Ca2SiO4+2H2SO4 2CaSO4+SiO2+2H2O
При этом увеличивается расход серной кислся'ы,-идущей на разложение балластных (нефосфатных) веществ, и уменьшается выход свободной (активной) кислоты. При содержании в сырье ~28% MgO от количества фосфорного ангидрида вся фосфорйая кислота полностью связывается в дигидрофосфат магния, и выход ее фактически равен нулю.
Соединения алюминия (нефелин, каолины) и железа (гематит, гетит, лимонит, бурый железняк) растворяются в ре
29
акционной смеси и -переходят в фосфаты алюМини-я и железа А12Оз+Н25О4+Са(Н2РО4)2-^2А1-РО4+'Са5О4+ЗН2О Fe2O3-|-H2SO4+Ca (Н2РО4) 2 = 2FePO4-{-JCaSO4-]-3H2O
образующие пересыщенные растворы, из которых медленно выделяются гидраты фосфатов железа и алюминия — FePO4-2H2O, НзРе(РО4)2-2,5Н20, А1РО4-2Н2О и АМНРСМзХ Х«Н2О. Поэтому в присутствии окислов трехвалентных ме-
таллов и осо-бенно железа выход фосфорной кислоты в жидкую фазу уменьшается. При 70—80 °C и содержании в рас-
творе 25—32% Р2О5 и 2—3% SO3 ^растворимость фосфатов железа невелика (2% в расчете на Fe2O3). Таким образом, „ „ л 2-100
при содержании Fe2O3 в исходном сырье не более
25.--32
яг8—6% от общего количества Р2О5 фосфаты железа полностью остаются в растворе. Это предопределяет требования к исходному сырью для максимального извлечения Р2О5.
Оптимальные режимы экстракции фосфорной кислоты из природных фосфатов дигидратным, полу-гидратным или ангидритным методами, обеспечивающие максимальную степень извлечения фосфорного ангидрида и выделение хорошо фильтрующихся и отмываемых от кислоты кристаллов сульфата кальция, достигаются выбором соответствующих физико-химических условий процессов и аппаратурного их оформления. Важнейшие из них: температурные и концентрационные условия, определяющие растворимость в образующихся системах, пересыщение растворов сульфатом кальция и выделение его в твердую фазу (кристаллизацию), а также смешивание реагентов и продолжительность их взаимодействия. _ . .
П.2. РАВНОВЕСИЕ В РЕАКЦИОННЫХ СИСТЕМАХ
Система H2SO4—Н3РО4—Н2О
Допускают [3], что кислоты в этой системе образуют комплекс эквимолекулярного состава H4PO4+-HSO4~.
Плотность тройных систем сильно зависит от содержания воды и в меньшей степени от соотношения в них., кислот [4]. Если воды в системе ~20% (мол.), плотность изогидратных растворов практически не зависит от соотношения кислот.
Зависимость вязкости от концентрации указывает, что ассоциированные комплексы, имеющиеся в двойных системах, в системе H2SOj— Н3РО4—Н2О диссоциируют при смешении [4]. Максимум вязкости в двойной системе H2SO4— Н2О, соответствующий составу H2SO4-H2O, обнаруживается в тройной системе. Добавление Н3РО4 вызывает разрушение гидрата, й в концентрированных растворах, где фосфорной
30
кислоты больше, чем серной, он отсутствует полностью. Прй 75°C увеличение содержания серной кислоты от Одо 10,5% в растворах с концентрацией 73—76% Н3РО4 понижает давление пара от 12600 до 2500 Па (95—34,2 мм рт. ст.) [5, с. 195].
Показано [5, 6], что тепловой эффект смешения 62%-ной Н3РО4 и 94%-ной H2SO4 с образованием смесей, содержащих 58—62% Н3РО4 и 1—6,5% H2SO4, соответствует (в ккал/г смеси) численному значению концентрации (в %) серной кислоты в смеси. Например, для смеси, содержащей 1,99% H2SO4, тепловой эффект смешения равен 2,0 ккал/г смеси.
Система СаО—Р2О5—Н2О
Данные по растворимости в системе в настоящее время имеются в области температур от 0 до 152 °C [7]. Растворимость при 25 °C в системе СаО—Р2О5—Н2О в диапазоне концентраций, представляющих интерес для экстракционного процесса, представлена на рис. II. 1. Кривой FiF соответ-' ствуют растворы, равновесные с Са(Н2РО4)2; кривой FB— равновесные с Са(Н2РО4)2-;Н2О, а кривой ВО — равновесные с СаНРО4. Составы отдельных твердых фаз также приведены на рис. II. Ь Эвтонические растворы составов F или В насыщены Са(Н2РО4)2 и Са(Н2РО4)2-Н2О или Са(Н2РО4)2-Н2О и СаНРО4.
В области низких концентраций Р2О5 (0—30 мг/л) изотерма растворимости при 25 °C состоит из трех ветвей, соответствующих растворам, равновесным с СаНРО4-2Н2О, Саз(РО4)2-0,5Н2О или Са5(РО4)зОН.
С повышением температуры растворимость Са(Н2РО4)2 и Са(Н2РО4)2-Н2О увеличивается, а СаНРО4 уменьшается. В диапазоне температур 25—100 °C содержание СаО в растворах, находящихся в моновариантном равновесии с твердыми фазами Са(И2РО4)2 и Са(Н2РО4)2-Н2О, возрастает от ~0,2 до 1,34%, а в растворах, равновесных* с Са(Н2РО4)Х ХН2О и СаНРО4, почти не изменяется и составляет 5,75% при 25 и 5,54% прй 100 °C. Содержание Р2О5 в растворе в первом случае уменьшается от 61,66 до 56,18%, а во втором — увеличивается от 24,2 до 40,27%.
Инвариантные равновесия в системе СаО—Р2О5—Н2О устанавливаются при 2'5 °C и содержании в жидкой фазе 23,50% Р2О5 и 5,8% СаО . относительно твердых фаз Са(Н2РО4)2-Н2О-|-СаНРО4~|-СаНРО4-2Н2О; при 36 °C и содержании в жидкой фазе 0,14% РгО5 и 5,81% СаО относительно —' СаНРО4+Са’НРО4-2Н2О--|-(.Саз(РО4)2-|Н2О; при 152°C и содержании в растворе 53,0% Р2О5 и 5,6% СаО относительно — Са (Н^РО4)2+Са (HdPO4)2-H2O+CaHPO4. -
31
Рис. II.1. Изотерма растворимости при 25 °C и поля кристаллизации в системе СаО—Р2О3—Н2О.
Твердые фазы: ЛГ- Са (Н2РО4)2; АГ — Са(Н2РО4)2-Н2О; R--СаНРО4; Р —СаНРО4-2Н2О; Q — Са3(РО4)2-Н2О; S,—Са3(РО4)3ОН.
Узловые точки в системе СаО—Р2О5—НгО — это точки перехода, а не конечные точки кристаллизации различных фосфатов. Объясняется это инконгруэнтностыо фосфатов кальция относительно насыщенных' ими водных растворов. Так, луч растворения Са(Н2РО4)2-Н2О (см. рис. 1Ы, прямая ОЛ4) лежит вне поля кристаллизации Са(Н2РО4)2. Вначале он проходит через поле кристаллизации СаНРО4, а зап тем через поле смесей Са (Н2РО4)2-Н2О с СаНРО4.
Линии одинакового отношения СаО к Р2О5 в системе являются одновременно и лучами испарения, и линиями постоянных значений степени нейтрализации свободной фосфорной кислоты. В области ненасыщенных растворов эти линии характеризуют изменение концентрации раствора при изотермическом испарении его до пересечения с кривой насыщения. Если луч испарения пересекает линию насыщения раствора дикальцийфосфатом, то при дальнейшем выпаривании в твердую фазу выделится СаНРО4. Состав раствора при этом будет изменяться по линии OiS (рис. II.2), а состав системы — по линии того же значения постоянного отношения СаО и Р2О5. В точке В состав раствора остается постоянным до тех пор, пока выделившийся СаНРО4 не перейдет в Са(ЩРО4)2. После этого при дальнейшем выпаривании состав раствора будет изменяться по кри/ой BQ, и в~ твердую фазу выделится Са(Н2РО4)2-Н2О. Количество твердой
32
и жидкой фаз в системе по окончании выпаривания (или в любой другой момент) определяется отношением отрезков ME к МА, а доля выделившихся кристаллов — отношением ME к АЕ.
Лучи степени нейтрализации фосфорной кислоты можно нанести на диаграмму по значениям отношения СаО: P2Os. Так, при полной (100%-ной) нейтрализации первого атома водорода фосфорной кислоты с образованием Са(НгРО4)2 массовое отношение СаО: Р2О5=0,3943. При любом другом отношении СаО к Р2О5 (С:Р), меньшем 0,3943, степень нейтрализации z (в %) равна:
Р-0,3943
где С и Р — содержания СаО и Р2О5 в системе, %.
. Фигуративная точка системы, образующейся при взаимодействии фосфата! с фосфорной кислотой, определяется на диаграмме пересечением луча растворения фосфорная* кислота — гидроксилапатит и луча той или иной степени нейтрализации' Лучи растворения Я/?* PQR или TSS^H (см. рис. 11:2) получают соединением точки на ординате, отвечающей концентрации исходной фосфорной кислоты, например, точки Н, Р или Т с точкой R, которая соответствует составу гидроксилапатита. Замена фторапатита гидроксил апатитом допустима, если не учитывать образования фтористого водорода.
Степень нейтрализации в пульпе (системе), соответствующую разложению .фосфата кислотой в данный момент при тех или иных условиях, определяют Отношением содержания
70 г
Гг<~>5, уо
СаО, %
Рис. П.2. Физико-химический анализ еостава продуктов взаимодействия апатита и фосфорной кислоты в системе СаО—Р2О5—Н2О^
3-5473 . I 33
Р2О5, связанной в виде Са (tbPO^H'^o, связ). к сумме содержания связанной и свободной РгОДРрюв своб) '
* • р ^2^5 связ
2= ------------------------100
р2°5связ' р20освоб
В случае обработки апатита фосфорной кислотой степень нейтрализации связана со степенью разложения сырья К (в %)
- ЗЗЗАК ~ AK+ 72,5n
где A — содержание P2O5 в исходном апатите, %; «— масса 100%-ной Н3РО4, расходуемой на 100 кг апатита, юг. Отсюда:
__ 72,5/гг
~ А (333 — z)
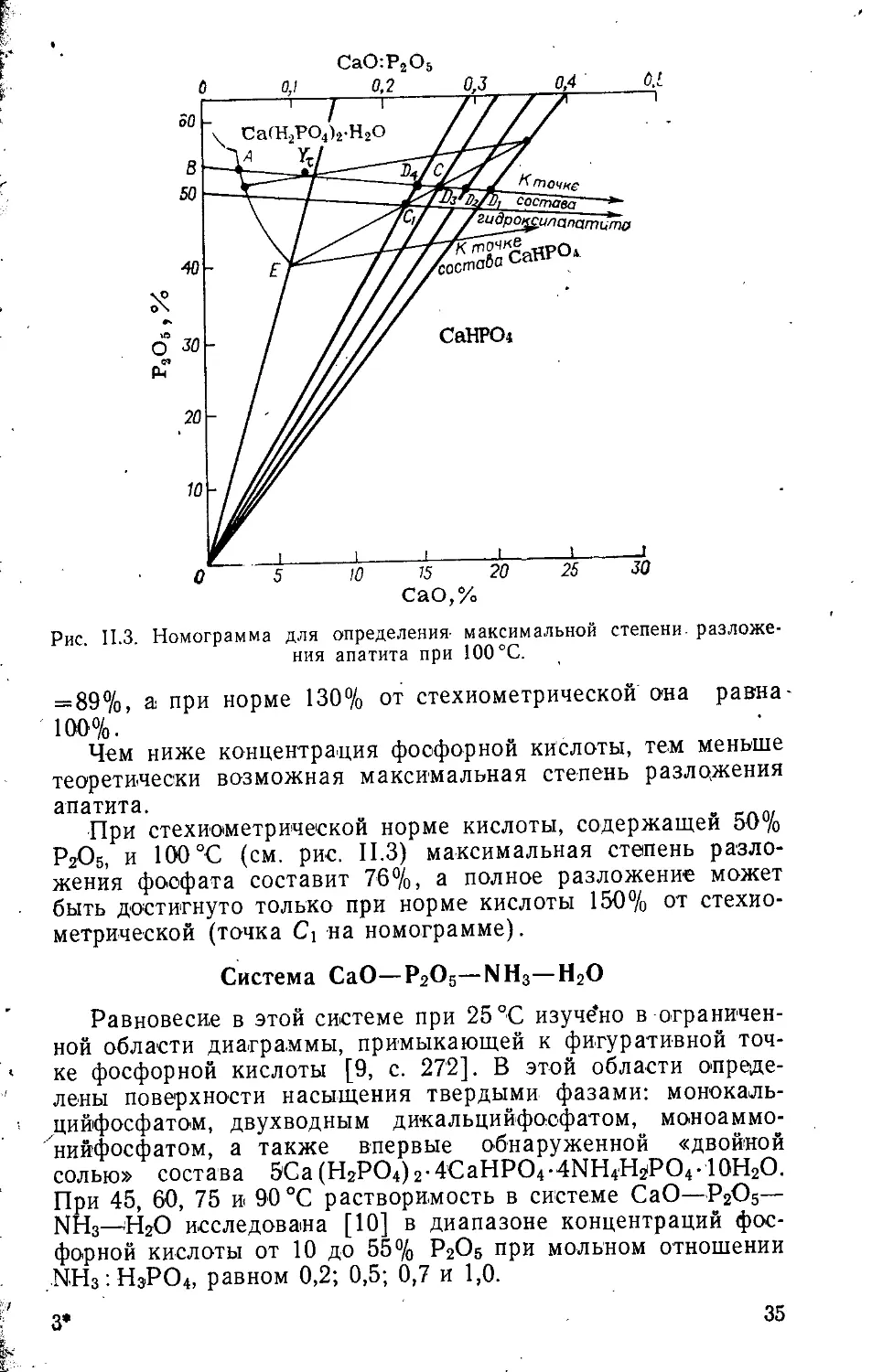
Весьма удобным представляется графический метод расчета максимальной (теоретической) степени разложения фосфорита фосфорной кислотой [8]. В его основе лежит предположение, что разложение фосфата прекращается в момент начала выделения кристаллов СаНРО4, экранирующих поверхность частиц фосфата, что тормозит диффузию к ней кислоты.
Прямая BD на рис. II.3 является отрезком прямой, описывающей растворение гидроксилапатита в фосфорной кислоте. Точки пересечения (Z>i, D2, D3, D4) прямых постоянного отношения СаО : Р2О5 с лучом, отвечающим растворению апатита, указывают на состав системы при разложении апатита фосфорной кислотой концентрации 54% РгО5, взятой в количествах 100, НО, ГЗО и 150% от стехиометрической нормы по реакции получения однозамещенного фосфата кальция. Точка Ут, характеризующая состав прореагировавшей пульпы, по мере протекания реакции смещается по направлению к точке Ь4.
Степень разложения фосфатов (гидроксилапатита) определяется отношением отрезков Ут к BDi. Если состав прореагировавшей смеси отвечает точке А, жидкая фаза становится насыщенной Са(Н2РО4)2-Н2О. В этом случае степень разложения апатита составляет (BA/BDi) • 100= 12%. Когда состав жидкой фазы достигает эвтонической точки Е, она становится равновесной с твердыми фазами Са(НгРО4)2Х ХНаО и СаНРО4 и дальнейшее разложение прекращается. Таким образом, максимальная степень разложения при стехиометрической норме Н3РО4 и заданных условиях составит: (iBEfBDi) -100 = 83%. При повышении нормы НаРО4 до 110% степень разложенйя фосфата соответствует (BC/BD?) • 100 =
34
Рис. II.3. Номограмма для определения- максимальной степени- разложения апатита при 100 °C.
= 89%, а при норме 130% от стехиометрической она равна 100%.
Чем ниже концентрация фосфорной кислоты, тем меньше теоретически возможная максимальная степень разложения апатита.
При стехиометрической норме кислоты, содержащей 50% Р2О5, и 100°C (см. рис. II.3) максимальная степень разложения фосфата составит 76%, а полное разложение может быть достигнуто только при норме кислоты 150% от стехиометрической (точка С] на номограмме).
Система СаО—Р2О5—NH3—Н2О
Равновесие в этой системе при 25 °C изучено в ограниченной области диаграммы, примыкающей к фигуративной точке фосфорной кислоты [9, с. 272]. В этой области определены поверхности насыщения твердыми фазами: монокальцийфосфатом, двухводным дикальцийфосфатом, моноаммонийфосфатом, а также впервые обнаруженной «двойной солью» состава 5Са (H2PO4)2-4CaHPO4-4NH4H^PO4- ЮН20. При 45, 60, 75 и 90 °C растворимость в системе СаО—Р2О5— NH3—Н2О исследована [10] в диапазоне концентраций фосфорной кислоты от 10 до 55% Р2О5 при мольном отношении N-H3: Н3РО4, равном 0,2; 0,5; 0,7 и 1,0.
3*
35
MgO, %
Рис. II.4. Изотермы растворимости при 25, 80 и 130 °C в системе MgO—P2Os—
Н2О [9].
Присутствие фосфатов аммония в растворе расширяет область кристаллизации дикальцийфосфата и уменьшает поле кристаллизации монокальций-фосфата. При 45 и 60 °C дикальцийфосфат находится в равновесии с растворами концентрации менее 30% Р2О5, содержащими от 1 до 3% NH3.' Из растворов, содержащих 30—39% РгО5 при 45 °C и 29,4—49,2% Р2О5 при
60 °C, выделяются в твердую фазу фосфаты кальция и мо-нраммонийфосфат. Выделение двузамещенного фосфата кальция и моноаммонийфосфата наблюдается'при 75 °C из растворов, содержащих до 49% Р2О5 и 6% NH3, а при 90°C —до 54% Р2О5 и 6,7% NH3. При больших концентрациях в этих условиях кристаллизуются монокальций- и мо-ноаммонийфосфат.
Система MgO—Р2О5—Н2О
На рис. II.4 представлены' изотермы растворимости в системе MgO—Р2О5—Н'2О.- Растворимость MgHPO4-3H2O практически не зависит от температуры, но сильно возрастает с увеличением содержания Р2О5 в растворе. Так, в растворе, равновесном с MgHPO4-3H2O и содержащем 1,3% Р2О3, растворимость MgO’ равна 0,4%, а при концентрации 39,4% Р2О5- 9,6%.
Растворимость Mg(H2PO4)2-4H2O, Mg(iH2PO4)2;'2'H2O й Mg(H2iPO4)2 возрастает с повышением температуры и увеличением содержания Р2О5 в растворе.
Если сравнить растворимость при 25 °C в системах MgO—Р2О5—НгО и СаО—P^Os—Н2О (рис. II.5, последняя система'—кривая ОВС) видно, что фосфаты магния характеризуются значительно большей растворимостью ,(что. справедливо и при других температурах) в растворах фосфорной' кислоты, чем фосфаты кальция. В растворе, насыщенном Mg(H2PO4)2-4H2O MgHP04-3H2O, при содержание 33,1% Р2О5 находится 8,3% MgO [0,207 моль Mg(H2PO4)2 в 100 г раствора]. Аналогичный раствор в системе СаО—Р2О5—Н2О, насыщенный Са^НяРО^г-'НяО и СаНРО4, при той же кон-
36 •
Рис. II.5. Изотермы растворимости при 25 °C и поля кристаллизации в системе MgO— Р2О5-Н2О.
Твердые фазы: 7—Mg(H2PO4)2;
II — Mg (Н2РО4) 2 • 2Н2О; III — V»
Mg(H2PO4)2-4H2O; IV — °-MgHPO4H2O. о
л"
центрации Р2О5, сод'ер-Зт 3,9% СаО [0,07 моль (Н2РО4У2 в 100 г раствора].
Разница в растворимостях однозамещенных фосфатов магния и кальция еще больше увеличивается с ростом концентрации фосфорной кислоты. При 25 °C и содержании в растворе, 33,1% Р2О5 растворимость Mg(l^PO^2 в 3 раза больше, чем Са(Н2РО4)2;. при содержании в растворе 38% Р2О5— в 3,15 раза, а в растворе с концентрацией 59,2— £>9,6% Р2О5 — в 20 раз.
Мономагни'йфосфат Mg(H2PO4)2 более устойчив в водных растворах, чем Са(Н2РО4)2 (из-за большей растворимости). Водные растворы Са(Н2рО4)2 при 25°C устойчивы лишь в очень разбавленных1 растворах, содержащих не боле$ 1% соли. При этой же температуре водные растворы Mg(H2PO4)2 устойчивы вплоть до содержания в них 28,7 г Mg(iH2PO4)2 в 100 г Н2О, что соответствует 100%-ной ней-
трализации первого иона водорода в растворе, содержащем 14,4% Р2О3 и 4,1% 'MgO. '
Система MgO CaO—Р2О Н2О
Изотерма растворимости этой системы при 80 °C (рис. II.6) характеризуется пятью полями насыщения: MgHPO4-3H2O, |Mg(H2PO4)2-2H2O, Mg(H2PO4)2, Са(Н2РО4)2 Н2О и СаНРО4. Растворы находятся в равновесии в узловых точках: Qj —
, с MgHPO4-3H2O, Mg(HsPO4)2-2H2O ’и Са(Н2РО4)2-Н2д;
Q2 —с MgHPO4-3H2O, СаНРО4 и Са(Н2РО4)2-Н2О; Q3 — с Mg(*H2PO4)2-2H2O, Mg(H2PO4)2 и Са (H2PQ4)2-HaO.
Поля кристаллизации фосфатов магния на диаграмме весьма малы, что объясняется большой растворимостью фосфатов магния и малой растворимостью фосфатов кальция, еще более уменьшающейся в присутствии соединений магния. Таким образом, фосфаты кальция высаливаются из раствора под влиянием фосфатов магния. Так, при добавлении MgO к моновариантному раствору в системе СаО— Р2О5—Н2О, равновесному с Са(Н2РО4)2-Н2О и Са(.НРО4), растворимость СаО уменьшается от .5,7% в точке В в от
37
сутствие MgO (см. рис. П.|6).до 0,7% в точке фг при насыщении раствора.
Приведенная изотерма при 80 °C позволяет определять составы насыщенных растворов лишь на пограничных линиях, разделяющих отдельные поля насыщения. Например, кривая BQ2 может быть использована для определения по одному какому-нибудь компоненту состава раствора, равновесного с Са (Н2РО4)2-Н2О и СаНРО4.
При экстракции фосфорной кислоты из фосфатов большое значение имеет характеристика растворов, составы которых расположены внутри полей кристаллизации одной соли. Для этого пользуются дополнительной^ величиной, определяющей состояние системы внутри поля насыщения раствора одной, солью — суммой концентраций солей в растворе или растворимостью СаО в растворах с одинаковой концентрацией MgO.
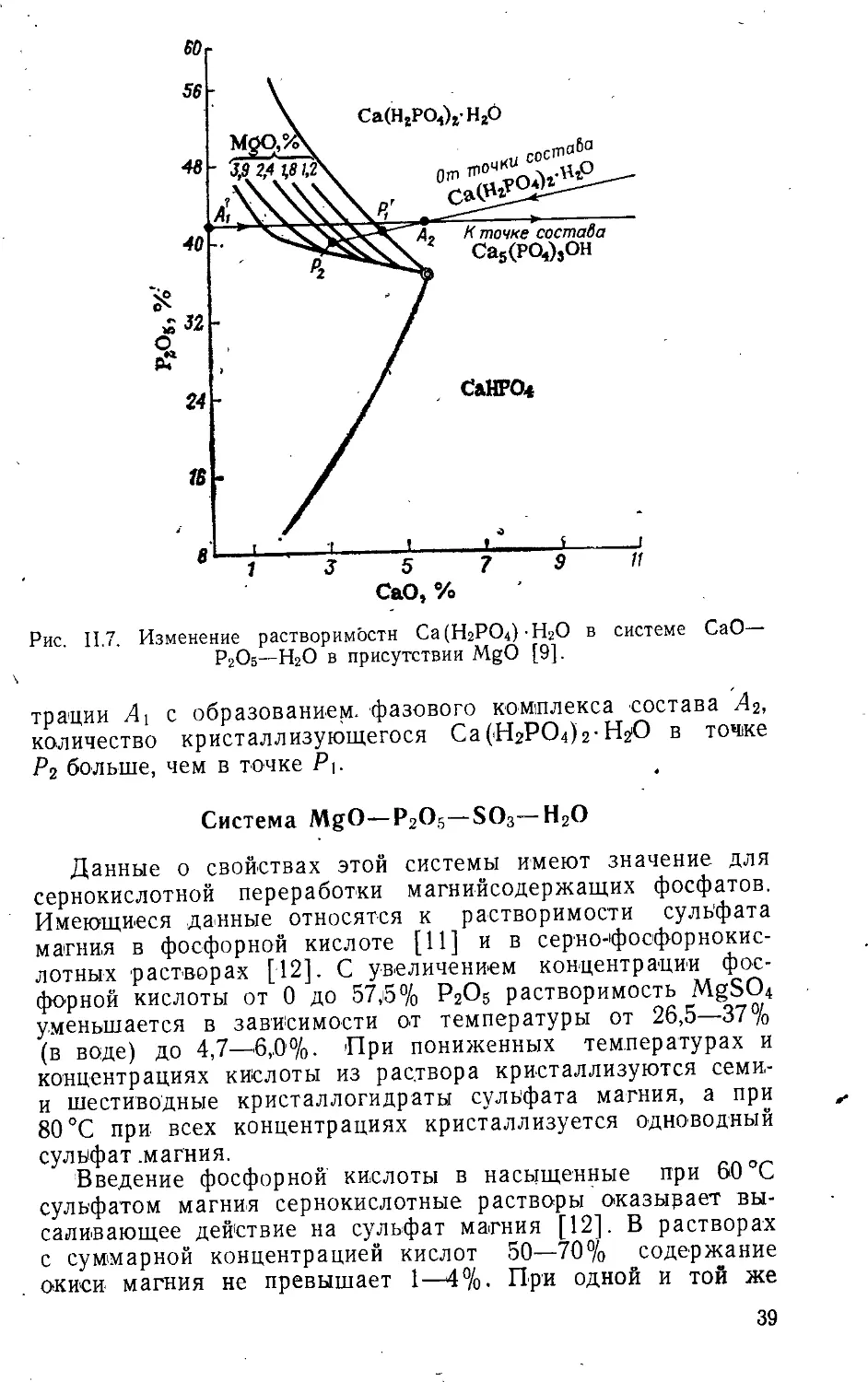
На рис. II.7 показаны изолинии (по MgO) растворимости -Ca(H2PO4)2-H2O в растворах, содержащих разные количества MgO (1,2—3,9%), не насыщенных солями магния [9]. Как видно, с увеличением содержания MgO в растворе уменьшается растворимость Са(Н2РО4)2-Н2О и увеличивается доля выкристаллизовавшегося Са (Н2РО4)2-Н2О. При взаимодействии, например, Са5(РО4)гОН и кислоты концен-
Pz°S, %
Рис. II.6. Изотерма растворимости при 80 °C и поля кристаллизации в v системе MgO—СаО—Р2О5—Н2О [10].
Тверды'е фазы: / — Са(Н2РО4)2Н2О; II— СаНРО4; III— Mg(H2PO4)2X Х2Н2О; IV — Mg(H2PO4)2; V — MgHPO4 • ЗН2О.
38
60
СаО, %
Рис. II.7. Изменение растворимости CafHjPOO -Н2О в системе СаО— Р2О5—Н2О в присутствии MgO [9].
трации At с образованием, фазового комплекса состава А2, количество кристаллизующегося Са(НгРОДг-НгО в точке Р2 больше, чем в точке Рь
Система MgO—Р2О5—SO3—Н2О
Данные о свойствах этой системы имеют значение для сернокислотной переработки магнийсодержащих фосфатов. Имеющиеся данные относятся к растворимости сульфата магния в фосфорной кислоте [11] и в серно-фосфорнокислотных растворах [12]. С увеличением концентрации фосфорной кислоты от 0 до 57,5% Р2О5 растворимость MgSO4 уменьшается в зависимости от температуры от 26,5—37% (в воде) до 4,7—6,0%. При пониженных температурах и концентрациях кислоты из раствора кристаллизуются семи-и шестиводные кристаллогидраты сульфата магния, а при 80 °C при всех концентрациях кристаллизуется одноводный сульфат .магния.
Введение фосфорной кислоты в насыщенные при 60 °C сульфатом магния сернокислотные растворы оказывает высаливающее действие на сульфат магния [12]. В растворах с суммарной концентрацией кислот 50—70% содержание окиси магния не превышает 1—4%. При одной и той же
39
суммарной концентрации кислот растворимость сульфата магния уменьшается с увеличением доли серной кислоты. Из растворов с начальным содержанием 60,5 и 42,6% H2SO4 кристаллизуется -моногидрат сульфата магния в виде мельчайших кристаллов. Из растворов с. исходными концентрациями 11,7 и 20% H?SO4 вначале в твердую фазу выделяется шестиводный кристаллогидрат, а по мере увеличения доли фосфорной кислоты происходит его -обезвоживание до три- и моногидратного сульфата магния.
Система Ме2О3—Р2О5—Н2О
Соединения трехвалентного железа в процессе экстракции переходят в труднорастворимый фосфат FePO4-2HiO. В то же время фосфаты двухвалентного железа, вследствие их хорошей растворимости, аналогично фосфатам магния, нейтрализуют кислоту и высаливают фосфаты кальция '{13].
Известны комплексные соединения Fe3+ с фосфорной кислотой: НзРе(РО4)2-2,5Н2О и НбРе(РО4)3, которые реагируют с другими веществами как одноосновные кислоты, образуя монозамещенные соли. При 80 °C РеРО4-2НгО находится в равновесии с растворами, содержащими до ~41% Р2О5 [13]. В более концентрированных растворах (41—52% Р2О5) равновесной твердой фазой будет FeHs(PO4)2-2,5H2O. При концентрации кислоты ниже 15% Р2О5 растворимость фосфата железа резко уменьшается до десятых сотых долей процента. Поэтому при отмывке фосфорной кислоты,из осадка ранее растворенный фосфат железа будет осаждаться. С повышением температуры от 70 до 80 °C растворимость средней соли уменьшается и увеличивается выделение ее в твердую фазу. >. -
С увеличением содержания серной кислоты в. растворе растворимость фосфата железа увеличивается тем больше, чем выше концентрация кислоты. При прибавлении к раствору, соответствующему двойной точке кристаллизации среднего и кислого фосфата, ,15 г SO3/100 г Н2О (в растворе) он становится очень вязким. При более высоких концентрациях SO3 масса образует гель.
Равновесие в системе Р2О5—Fe2O3—Н2О устанавливается весьма медленно, особенно при растворении гидроокисных форм железа и в отсутствие в осадке твердой фазы, равновесной-с раствором. Даже в растворах, равновесных с одной . твердой фазой и в присутствии ее в ос’адке, когда длительность процесса значительно сокращается, в некоторых случаях равновесие достигается лишь в течение 4—6 мес.
Растворимость фосфатов алюминия больше, чем фосфа-, тов железа. В системе А12О3—РгО5—Н2О наиболее стабилен А1РО4-2Н2О.. Растворимость его при 80 °C в растворе, содер-
40
г' жащем 30% Р2О5, составляет 5% А12О3 (растворимость FePO4-2<H2O — 1% Fe2O3). В области, низких концентраций кислоты (до ~5% Р2О3) выделена метастабилыная фаза неопределенного состава. Из растворов, содержащих больше ~5% Р2О5, кристаллизуется также в метастабильном состоянии А12(НРО4)з-7Н20, который с течением времени переходит в средний фосфат с выделением фосфорной кислоты. Имеются сведения о растворимости в фосфорной кислоте фосфатов лантана при 25 [14] и.80°C [15], фосфатов церия [16] и гадолиния [17]. Растворимость фосфатов лантана в фосфорной кислоте, содержащей 10,3—42,7% Р2О5, пои 25 °C составляет 0,04—0,19% La2O3 и уменьшается при 80°С до 0,01—0,053% Ьа2О3.
Незначительная растворимость фосфата лантана в фосфорной кислоте по сравнению с .фосфатами алюминия и железа объясняется более основным характером лантана и большим его ионным радиусом, чем у алюминия и железа (0,122 против 0,057 и 0,067 А) [15].
П.З. СКОРОСТЬ РАЗЛОЖЕНИЯ ФОСФАТОВ СЕРНОЙ И ФОСФОРНОЙ кислотами
Разложение серной кислотой
Скорость разложения фосфата серной кислотой в присутствии раствора разбавления (фосфорной кислоты) зависит не толцкр1 от активности ионов водорода, но и от степени пересыщения жидкой фазы твердым продуктом реакции — сульфатом кальция.
Общий вид зависимости степени' разложения фосфата за определенное время (изохрона) от концентрации серной кислоты в смеси изображается кривой с двумя максимумамй (рис. II.8). Так как скорость взаимодействия фосфата с серной кислотой велика, то можно предположить, что начальная действующая концентрация серной кислоты в жидкой фазе пульпы определяется временем ее подачи, интенсивностью перемешивания (гомогенизацией) системы, и объемом раствора разбавления. Например, в простейшем виде можно допустить, что при подаче 100 ч. 1(ма'сс.) H2SO4 для разложения 400 ч. (мас'с.) апатита в 400 ч.
(Масс.) Н3РО4 (отношение Ж:Т = 2,5:1 в расчете на сухой
Рис. II.8. Общий вид зависимости степени разложения фосфата от концен-. трации исходной серной кислоты (изохрона) [18].
Концентрация H2SO4
41
фосфогипс) в течение 1 ч начальная концентрация H2SO4 в смеси будет 0,3% (три полном смешении в течение 1 мин) и 3% ('при полном смешении в течение 10 мин). В действительности же за 10 мин прореагирует много кислоты и, следовательно, усредненная фактическая концентрация серной кислоты в зоне реакции будет во много раз меньше. Кроме того, в разных местах объема концентрация кислоты будет зависеть от скорости потоков.
Очевидно,' что фосфат разлагается сильно разбавленной серной кислотой, и скорость процесса максимальна при некоторой оптимальной концентрации серной кислоты в смеси с фосфорной (см. первый максимум на рис-. 11.8).
В области концентраций серной кислоты в смеси, меньших оптимальных, разложение фосфата замедляется, вероятно, вследствие недостаточной активности ионов водорода., а при больших концентрациях — вследствие торможения реакции при экранировании поверхности зерен фосфата коркой выделяющегося сульфата кальция;
Непосредственные данные о скорости разложения фосфатов смесями фосфорной кислоты с небольшими количествами (0,1—0,3%) серной кислоты при соотношениях жидкости к фосфату, применяемых в производственных условиях, отсутствуют.
Показано [19], что фосфорит Флориды разлагается при Ж:Т=50:1 и 65—85 °C в растворах фосфорной кислоты концентрации 25—35% Р2О5, содержащих до <^1,5% H2SO4, практически полностью (на 99,5—99,9%) в течение 1 — 15 мин. По степени пересыщения (определяемой отношением фактических и равновесных произведений моляльных концентраций Са2* и SO42~) меньше 2,5 сульфат кальция выделяется в объеме жидкости и не препятствует разложению фосфата; если же степень пересыщения более 2,5, то сульфат кальция -откладывается на поверхности зерен фосфата, затормаживая его дальнейшее разложение.
Приближенно о скорости процесса можно судить также по данным взаимодействия фосфатов с серной кислотой большей концентрации (30—50%) как в отсутствие, так и при наличии в начальной смеси фосфорной кислоты. При массовом соотношении фосфата к кислоте (Ж:Т), равном .1 :4, разложение подольского фосфорита (32,8 Р2О5) с частицами <53 мим 30%-ной H2SO4 при 70°C завершается полностью за 15 мин [18]. Скорость реакции значительно увеличивается с повышением температуры даже цри использовании фосфата грубого помола и 50%-ной H2SO4 (рис. II.9). Однако в присутствии фосфорной кислоты скорость растворения фосфата серной кислотой несколько уменьшается, по-видимому, вследствие уменьшения активности ионов водорода.
42
Рис. II.9. Зависимость степени разложения фосфата серной кислотой от времени и температуры (размер частиц фосфорита 105—150 мкм).
Скорость разложения фос-' фата зависит от его природы и прямо пропорциональна поверхности зерен минерала. Разложение крупнозернистого апатита практически линейно зависит от времени, потому
что в этом Случае суммарная
(поверхность зерен изменяется медленно. При разложении по-лидиоперсной фосфатной муки линейная зависимость отсутствует. Мелкие зерна обладают большой общей поверхностью и реакция протекает вначале с большой скоростью. Крупные зерна с относительно малой суммарной поверхностью реагируют медленно, что приводит' к постепенному
уменьшению скорости растворения апатита к концу реакции.
Известно, что осадочные фосфориты, содержащие фторапатит в виде мельчайших зерен (<1 мкм), разлагаются с большой скоростью.
При предварительном смешении фосфата с раствором разбавления разложение его в той или иной степени протекает в результате взаимодействия с фосфорной кислотой.
Разложение (фосфорной кислотой
Скорость разложения фосфатов зависит, в первую очередь, от количества применяемой фосфорной кислоты и фазового состава образующейся смеси. В соответствии с этим' различают:
растворение фосфата в большом количестве кислоты, достаточном для образования гомогенного (не считая нерастворимых примесей) раствора;
взаимодействие фосфата с фосфорной кислотой в незагустевающей пульпе, когда количество выделившихся кристаллов продукта реакции относительно невелико по сравнению с количеством жидкой фазы; это происходит при растворении фосфата в разбавленной или в большом избытке концентрированной фосфорной кислоты, необходимом для образования Са(НгРО4)2, но недостаточном для создания гомогенного раствора; ' • ,
взаимодействие фосфата с концентрированной фосфорной кислотой (50—55% Р20г), взятой в количестве, равном или несколько большем (на 5—10%), чем необходимо для полу
43
чения Са(Н2РО4)2, с образованием сплошного твердого монолита (этот процесс, имеющий значение в производстве двойного суперфосфата [1], здесь не рассматривается).
Разложение фосфатов без кристаллизации твердой фазы. Скорость разложения фосфата фосфорной кислотой с образованием гомогенного раствора моно'кальцийфосфата, в избытке кислоты определяется законами диффузии. Она пропорциональна концентрации ионов водорода, размеру реагирующей поверхности, интенсивности перемешивания и т. д. По мере нейтрализации свободной кислоты и накопления в растворе ионов кальция вплоть до образования насыщенного раствора скорость процесса уменьшается. Однако скорость разложения фосфата достаточно велика при использовании кислоты относительно небольшой концентрации. При разложении крупнозернистого отобранного апатитового концентрата фосфорной кислотой концентрации 20% Р2О5 , при Ж:Т = 20:1 при 80 °C фосфат полностью переходит в раствор за ~3 ч (рис, 11.10).
Температурный коэффициент скорости и растворения апатита до достижения - насыщения в относительно разбавленных растворах фосфорной кислоты находится в пределах 1,3—>1,5, а в растворах, содержащих 51,5—53,5% Р2О5 — 1,68—1,73 ,[20].
Скорость разложения фосфорита Каратау в большом объеме фосфорной кислоты (40 г фосфорита! в 200 мл кдс-лоты) значительно больше, чем апатита и также увеличивается с повышением температуры. При 60 °C уже через 30 мин степень разложения фосфорита кислотой, содержащей 29% РгО5 (40% Н9РО4), составляет 99,86%, тогда как
Рис. 11.10. Зависимость степени разложения апатитового концентрата фосфорной кислотой от времени и температуры [13]: Фосфорная кислота содержит 20% Р2О5; Ж:Т=20: 1.
Рис. II_.11. Зависимость активности ионов водорода (кривая /) и вязкости (кривая 2) насыщенных растворов системы СаО—Р2О5—Н2О от концентрации фосфорной кислоты при 40 °C.
44
при 20 6iC она достигает 98,32% лишь за 1,5 ч. Температурные коэффициенты скорости разложения фосфорита в кислоте, содержащей от 7,2 до 22% Р2О5, изменяются от 1,28 до 1,34.
Нейтрализация фосфорной кислоты соединениями магния, кальция, полуторных окисЛов и некоторых других резко уменьшает скорость разложения фосфата. В фосфорной кислоте, нейтрализованной карбонатом магния на 30 и 50%, скорость разложения апатита при 40 °C и соотношении Т : Ж—4 цЗО уменьшается в 3 и 14 раз [21].
Скорость разложения (и скорость растворения за 1 ч) ! ’апатита фосфорной кислотой разных концентраций при массовом отношении кислоты к апатиту, превышающем на 15— 2'5% теоретическое (равновесное) отношение [22], возрастает при увеличении концентрации исходной кислоты лишь до определенного предела (до -~46% РгО5). При росте, же содержания РзО5 в кислоте до 48 и 50% степень разложе-' ния апатита уменьшается. Это объясняется, по-видимому, изменением активности ионов водорода и вязкости раство--ров кислоты (рис. II..11).
При содержании в кислоте 26—46% РгО5 активность^ ионов водорода возрастает в большей степени, чем вязкость,' и скорость разложения увеличивается. При содержании в растворе 48—50% Р2О5 и более скорость разложения уменьшается вследствие торможения процесса, когда увеличение вязкости раствора превалирует над возрастанием, активности ионов водорода.
Известно, что в присутствии солей сильных электролитов активность водородных ионов в растворе увеличивается. Опытами растворения кингисеппского фосфорита фосфорной кислотой концентрации 45% Р2О5 (при Т:Ж=1:50) при 70°C показано [£3], что при добавке от 10 до 30% (по отношению к стехиометрической норме на СаО в растворе) серной кислоты скорость процесса увеличивается в 1,75— 2 раза, а при добавке от 20 до 40% сульфата калия или аммония — в 1,5—1,8 раза [23, с. 60]. Скорость растворения апатита в аналогичных условиях фосфорной кислотой кон-ч центрации 50% Р2О5 при 75—85 °C, содержащей 10% NH4C1 или NH41NO3, увеличивается в 1,4—1,5 раза [23, с. 65].
При внесении в насыщенный фосфатами кальция раствор некоторого количества серной кислоты он становится ненасыщенным и разложение фосфата ускоряется до насыщения системы вновь [24].
Разложение фосфатов с кристаллизацией продуктов реакции в незагустевающей пульпе. Стадии растворения апатита в насыщенных растворах приведены на рис. 11.12. Последовательные процессы пересыщения и кристаллизации изображены в виде зигзагообразных кривых А\В при 100 °C
45
Рис. 11.12. Схема разложения апатита в насыщенных растворах системы СаО—Р2О5—Н2О [9].
и ab при 25 °C. Полностью процесс разложения фосфата при 25 °C до образования фазового комплекса состава С может быть представлен ломаной линией сСЬ, т. е. движением по1 лучу растворения аС и по лучу кристаллизации СЬ.
В незагустевающей пульпе выделяющиеся кристал-
лы продукта реакции и зерна фосфата разровнены, и последние (при достаточном количестве жидкой фазы и интенсивном перемешивании) не покрываются изолирующей (шламовой) коркой вплоть до полного их растворения. При этом скорость процесса определяется, главным образом, скоростью кристаллизации твердой фазы из пересыщенного раствора, так как пересыщение жидкой фазы достигается быстро и,
как неравновесное состояние системы, является следствием разности скоростей растворения и кристаллизации. В свою очередь, чем больше пересыщение раствора, тем больше движущая сила и скорость кристаллизации и тем меньше размеры выделяющихся кристаллов.
Наиболее пересыщены монокальцийфосфатом концентрированные растворы фосфорной кислоты из-за / их высокой вязкости, а также (хотя и ц меньшей степени) разбавленные растворы, по-видимому, из-за меньшей вефоятности образо-зования в них кристаллических зародышей <[25, с. 31]. Повышение температуры приводит к уменьшению пересыщения монокальцийфосфатом концентрированных растворов, увеличению его в сильно разбавленных растворах и практически мало сказывается на пересыщении фосфорнокислых растворов средних концентраций.
Скорость кристаллизации фосфатов кальция из пересыщенных растворов зависит не только от пересыщения, но и от диффузионных условий, определяемых концентрацией, вязкостью, температурой кислоты, перемешиванием и т. п.
При 25 °C пересыщенные растворы достаточно устойчивы и не разлагаются в течение длительного времени. Из растворов, содержащих 34—39% Р2О5, кристаллизация Са(Н2РО4)2 в периодических условиях опытов протекает в течение нескольких суток. С повышением температуры уве-
46
, личивается средняя скорость и степень кристаллизации за • определенное время.
Из пересыщенного раствора, содержащего 55,5% Р2О5, монокальцийфосфат при 25 °C начинает выделяться через '5 мин, но скорость кристаллизации весьма мала (10— 20' мт/мин) и проходит через максимум. С повышением температуры от 25 до 100 °C скорость кристаллизации увеличивается в десятки раз и максимум ее (140 мг/мин) достигается в течение 2—3 мин.
При разложении фосфата в незагустевающей пульпе в непрерывных условиях кристаллизации твердой фазы значительно ускоряется, так как реагенты в каждый последующий ( момент поступают в ранее прореагировавшую массу с кри-tсталлами (затравка).
С повышением температуры от 50 до 70 °C степень разложения апатита увеличивается тем больше, чем больше норма кислоты. При норме термической кислоты (концентрации 45% Р2О5) 400 и 500% повышение температуры от 50 до 60°C приводит к увеличению степени разложения апатита за 2 ч от 86,3 до 96,1 и 98,5%.
Применение термической фосфорной кислоты, содержащей 40,5—41% Р2О5 при норме ее 700—450%’ (степень нейтрализации жидкой фазы в конце процесса 20—30%) приводит при 100 42 к разложению апатита за 1 ч на 92—98,1% [26, с. 170]. При степени нейтрализации кислоты 35—40%, достигаемой уменьшением нормы кислоты до ~380 и 330%, разложение апатита кислотой, содержащей 42% Р2О5, в тех же условиях составляет 78,6 и 72%. При норме кислоты (55% Р2О5) 200—250% от стехиометрии возможно при 80 °C достигнуть разложения фосфорита на 96—98% за 90 мин [27].
11.4. КРИСТАЛЛИЗАЦИЯ СУЛЬФАТА КАЛЬЦИЯ ИЗ ФОСФОРНОКИСЛЫХ РАСТВОРОВ
Модификации сульфата кальция
Выделение твердых фаз в системе CaSO4—Н3РО4—Н2О [дигидрат (гипс) CaSO4-2H2O, полугидрат CaSO4-0,5H2O и безводный CaSO4 — ангидрит]' в стабильном или мета-стабильном состояниях зависит от температуры и концентрации фосфорной кислоты [28].
Гипс образует кристаллы моноклинной системы в пластинчатой, столбчатой, игольчатой и волокнистой формах. Кристаллическая структура гипса характеризуется слоистой решеткой, в которой слои Са2+ и SO42- разделены молекулами воды.
47
Наличие в кристаллах гипса после обработки их горячей водой избыточного количества воды (по сравнению со стехиометрической формулой Са8О4-2НгО) объясняют возникновением деформаций в его кристаллической структуре, приводящих к волокнистым расщеплениям в кристаллах [29, 'С. Г] •
Полугидрат кристаллизуется в гексагональной сингонии и существует в виде а- и p-разновидностей, из которых первая представляет .собой хорошо образованные кристаллы < (игольчатые, призматические или ромбовидные) с шелковистым блеском. При выделении сульфата кальция из растворов (в том числе и фосфорнокислых) осаждением или нагреванием гипса в растворах NaCl, MgSO4, HNO3 и других он почти всегда образуется в виде а-полугидрата.
Из жидкой фазы p-полугидрат выделяется при нагрева- ' нии а-полугидрата в 2%-ном растворе NaOH. В отличие от а-полугидрата p-полугидрат характеризуется большей дисперсностью, меньшим совершенством кристаллической структуры.
Методом ИК-спектроскопии установлено, что при дегидратации гипса при 170 °C независимо от выдержки образцов в течение 3'0—180 мин образуется р-полугидрат [29, с. 4]. Последний при 200 °C и выдержке 180 мин на 50% переходит в ангидрит. а-Полугйдрат уже за 60 мин при' ,170 °C заметно превращается-в ангидрит, но структурная его перестройка, сопровождаемая небольшим экзотермическим эффектом, происходит при 200—220 °C.
Естественная разновидность безводного Сульфата кальция (нерастворимый у-ангидрит) кристаллизуется в ромбической системе с образованием мелких прямоугольных, ромбовидных, ромбовидно-пирамидальных и призматических кристаллов [30].
Нагреванием при 95—100 °C а-полугидрата в вакууме в течение 1,5—2 ч или p-полугидрата при 100—102 °C и атмосферном давлении в течение 18—20 ч можно получить так называемые а- и р-обезвоженные полугидраты [31]. На воздухе они быстро притягивают влагу и переходят в исходное состояние. По своей гигроскопичности а- и р-обезвоженные полугидраты превосходят СйС12 и H2SO4.
При длительном обезвоживании а- и р-полугидратов под разрежением (13—'130 Па; 0,1—1 мм рт. ст.) при 100 °C или нагревании а- и р-обезвоженных полугидратов до 220—240 и 340—380 °C -при атмосферном давлении образуются две модификации так называемого растворимого безводного сульфата кальция [32], имеющие кристаллическую структуру ангидрита — а- и р-ангидриты — и обладающие повышенной растворимостью в воде по сравнению с естественной модификацией ангидрита—у-ангидритом (рис. 11.13).
48
Рис. 11.13. Растворимость различных модификаций сульфата кальция в воде [32]:
1— CaSO4-2H2O; 2 — y-CaSO4; <4 3- a-CaSO4; 4 — 0-CaSO4; 5— й a-CaSO40,5H2O; 6 — 0-CaSO4X Я X0,5H2O; 7 — а-полугидрат обезвоженный; 8 — fj-пблугидрат обез- a воженный. о
Полугидрат, полученный при сернокислотном разложении апатита, дегидратируется в три стадии [33]. При 40—440 °C удаляется гигроскопическая влага, в интервале 160—<210 °C а-полугидрат переходит в а-обезвоженный полугидрат, который при 230—280 °C превращается в ангидрит.
В зависимости от условий растворения или образования сульфата кальция в жидкую фазу вначале переходит та или иная модификация с повышенной растворимостью, которая метастабильна относительно наименее растворимой — стабильной — фазы. Последовательность выделения из раствора тех или иных фаз и скорость перехода метастабильной фазы в стабильную зависит от степени пересыщения раствора и условий перехода его в стабильное состояние.
Если количество сульфата кальция в момент образования его в жидкой фазе превышает растворимость стабильной’ •фазы (что практически наблюдается почти при всех условиях взаимодействия серной кислоты с фосфатом), то в твердую фазу переходит метастабильная форма, сульфата кальция. В дальнейшем выделившаяся ранее метастабильная твердая фаза превратится в стабильную.
, В процессе сернокислотной экстракции фосфатов вначале выделяется а-полугидрат [9, 34], более плотный и грубодисперсный, чем p-полугидрат, и переходящий затем в стабильную для данных условий форму сульфата.
Скорость фазового превращения одной модификации в' другую определяется концентрацией и скоростью растворения метастабильной фазы, а также степенью пересыщения рАствора и скоростью его разложения. Максимальная концентрация и время достижения ее в водных системах p=iCaSO4-0,5H2O при взаимодействии реагентов за первые 5—120 с зависит от Ж:Т суспензии, гранулометрического состава твердой фазы и температуры [35].
4—5473
49
Растворимость сульфата кальция в серной кислоте
В системе CaSO4—H2SO4—Н2О сульфат кальций выделяется в твердую фазу в виде всех трех модификаций — ангид-.рита, полугидрата и дигидрата.
Максимальная растворимость сульфата кальция при 50 °C наблюдается в 5—110%-ной H2SO4 и равна ~ 0,4%. Растворимость сульфата кальция уменьшается с разбавлением , H2SO4 до ~0,2%- (т. е. до растворимости его в чистой воде) и до -~0,015%_ в растворе, содержащем 50% H2SO4. В области концентраций до 16,7% H2SO4 единственная твердая фаза—дигидрат, Твердые фазы CaSO4-2H2O и CaSO4 образуются в растворах, содержащих до 41% H2SO4, а фазы CaSO4-0,5H2O и CaSO4 — в области концентраций >41% H2SO4.
Последующие исследования с применением не только изотермического, но и кинетического методов [36], позволили получить данные также о растворимости твердых фаз (полугидрата и дигидрата при повышенных температурах), су-ществующ’их в течение короткого времени. С повышением температуры максимумы на кривых растворимости смещаются в область более концентрированных растворив.
Растворимость сульфата кальция в фосфорной кислоте
Растворимость трех форм сульфата.кальция в фосфорной кислоте изучена при различных температурах [28].
Наибольшая растворимость гипса при 25 °C, равная 0,7% CaSO4, соответствует растворам фосфорной кислоты, содержащим 18,3—'18,8% Р2О5. С увеличением содержания• Р2О5 в растворе до 55% растворимость дигидрата становится равной 0,17.% CaSO4.
Растворимость полугидрата в фосфорной кислоте, содержащей 41,13—51,15% Р2О5, значительно больше, а ангидрита в этих же условиях меньше, чем растворимость дигидрата.
С повышением температуры от 25 до 80 °C растворимость гипса увеличивается, причем максимум его -растворимости наблюдается для обеих температур в почти одинаковой области концентраций кислоты ( — 18—25% Р2О5). Раствори-хмость полугидрата изменяется аналогично растворимости гипса — возрастает с повышением температуры.
Растворимость метастабильного полугидрата при 25— 80 °C в разбавленных растворах фосфорной, кислоты (6,9— 28,2% НзРО4), определенная кинетическим методом, максимальна при концентрации- -~20% Н3РО4 и составляет <•—'1,6—1,66% CaSO4 (в — 2 раза больше чем гипса) [37, с. 6].
5°
fe
С повышением концентрации фосфорнокислотного раствора от 45 до 54% Р2О5 при 120°C растворимости CaSO4X Х0,5Н2О и ' CaSO4 уменьшается от 1,06 до 0,78% CaSO4-0,5H2O и от 0,57 до 0,32% CaSO4. Увеличение температуры от 80 до 120 °C-приводит к увеличению растворимости полугидрата в ~1,5 раза, а ангидрита в ~2 раза [37, с. 11].
Растворимость сульфата кальция
•в фосфорной кислоте, содержащей различные вещества
С увеличением содержания в растворе серной кислоты растворимость всех трех форм сульфата кальция уменьшав ется. Наиболее резкое изменение растворимости дигидрата наблюдается при содержании в растворе 3—3,5% H2SO4 [38]. При 70 °C растворимость .дигидрата в фосфорной кислоте, содержащей 25% Р2О5, составляет 1,2% CaiSO4, в присутствии 3% H2SO4 —0,46% CaSO4, и при содержании в растворе 6% H2SO4--0,26%. В присутствии ионов кальция растворимость сульфата кальция в фосфорной кислоте уменьшается еще в большей степени [39].
Насыщенные растворы в системе СаО—Р2О5—SO3—Н2О [40] практически не отличаются по своему составу от растворов системы СаО—Р2О5—НгО [41]. Это объясняется весьма малой растворимостью сульфата кальция, еще более уменьшающейся с увеличением концентрации раствора.
Наличие в. фосфорной кислоте, содержащей 45%' Р2О5, при 60 °C примесей А12О3 или Fe2Os, приводит к некоторому уменьшению растворимости полугидрата. Присутствие в частично нейтрализованной окисью кальция фосфорной кислоте до 1% AI2O3 или Fe2O3 не влияет на растворимость полугидрата.
Растворимость гипса и полугидрата в растворах фосфорной кислоты, содержащих сульфат магния, уменьшается в тем большей мере, чем больше MgO в растворе и при 2% MgO растворимость обоих кристаллогидратов меньше в несколько раз, чем в отсутствие магния.
В растворах фосфорной кислоты (до 20% Р2О5), содержащих кремнефтористоводородную кислоту, растворимость дигидрата-возрастает с увеличением концентрации последней (рис. 11.14).
Повышенные концентрации в растворе Н3РО4 (до 55%) и H2SiF6. (более 12%, в расчете на F) ведут к частичному разложению последней [26, с. 32; 39, с. 1714].
; H2SiF6+2H2O SiO2-H6HF
В результате, реакции сульфата кальция и плавиковой . кислоты в твердую фазу выделяется CaF2 и растворимость .4* - - 51
/
Рис. 11.14. Растворимость гипса в смесях фосфорной и кремнефтористоводородной кислот при 70 °C [39, с. 1714].
сульфата уменьшается вследствие появления в жидкой фазе эквивалентного количества сво-> бедной серной кислоты; В сме-и 20 40 сях> содержащих 3% F' (в виде
Содержание Р2О3,% HF) и более 55о/о н3РО4, в твердую фазу выделяется двухводный фторид кальция. Одновременно возможно выделение в твердую фазу также CaSiF6 вследствие малой степени гидролиза его в смеси кислот [26, с. 32], а также образование фторсульфатов кальция с иной растворимостью, чем CaSO4.
Содержание в фосфорной кислоте до 1% H2SiF6 (в расчете на F) не влияет на растворимость полугидрата. В частично нейтрализованной окисью кальция фосфорной кислоте растворимость полугидрата в присутствии H2SiF6 несколько увеличивается.
В смесях фосфорной и азотной кислот растворимость сульфатов кальция больше, чем в фосфорнокислых, и меньше, чем в азотнокислых растворах.
С увеличением доли азотной кислоты в смеси кислот от 0 до 100% растворимость сульфата кальция либо возрастает монотонно, например при 20 °C в пределах' значений растворимости в фосфорной и азотной кислотах в отдельности [42], либо. при 50 и 60 °C изменяется экстремально., возрастая (от растворимости в фосфорной кислоте) до максимального значения, соответствующего определенному отно-, шению кислот в смеси, а затем убывая до значения растворимости в азотной кислоте.
Известно, что труднорастворимые в воде вещества становятся более растворимыми в присутствии хорошо растворимых солей. В соответствии с этим изыскиваются возможности увеличения растворимости сульфата кальция в фосфорной кислоте введением различных добавок.
В присутствии в фосфорной кислоте нитрата аммония наибольшее увеличение (в ~ 2,5 раза) растворимости дигидрата сульфата кальция наблюдается при 60 °C в растворе, содержащем 31,2% Р2О5 и 31% NH4NO3 (1,64% CaSO4 по сравнению с 0,67 в отсутствие NH4NO3). При более высоких температурах растворимость увеличив'ается в несколько мрныпей степени (в 1,4—1,5. раза) [43, 44].
При 80 °C в растворе, содержащем 20—25% Р2О5 и 15—' 25% NH4NO3, растворимость полугидрата, сульфата кальция больше в 2—3 раза, чем в фосфорной кислоте.
> 52
В присутствии в растворах фосфорной кислоты моноаммонийфосфата растворимость как гипса, так и полугидрата выше, чем в чистой фосфорной кислоте. При 75 °C, 20% Р2О5 и 2,85% NH4+ растворяется 1,84% CaSO4 по сравнению с 1,30% в отсутствие ионов аммония, т. е. больше в ~1,4 раза.
В присутствии сульфатов аммония,- калия или* натрия растворимость дигидрата сульфата кальция в фосфорнокислых растворах уменьшается в 5—7 раз, чем в их отсутствие [45]. Наиболее резкое уменьшение растворимости наблюдается при малых концентрациях солей (до 4—5%)- При этом природа катиона не оказывает существенного влияния на характер изменения растворимости сульфата кальция.
Фазовые превращения кристаллогидратов сульфата кальция в фосфорнокислых растворах
На рис. 11.15 показана [9] растворимость различных модификаций сульфата кальция'в растворах фосфорной кислоты при различных температурах [28; с. 521; 34].
В растворе, содержащем 30% P2Os, растворимость наиболее растворимого полугидрата при 50 °C сравнительно велика: /—1,2% CaSO-t. При 80°C и содержании в кислоте 45—50% Р2О5 наиболее растворим гипс (рис. 11.15). Однако он весьма быстро разлагается с образованием полугидрата.
Рис. 11.15. Политермы растворимости кристаллогидрата сульфата кальция в растворах фосфорной кислоты [9].
53
Поэтому в концентрированных растворах фосфорной кислоты, содержащих 45—50% Р2О5, при 80 °C вначале образуется метастабильный полугидрат, который затем переходит в ангидрит [34].
Степень фазового перехода полугидрата в ангидрит в растворах фосфорной кислоты концентрации от 45 да 56% Р2О5 в большей мере возрастает во времени с повышением температуры от 80 до 120 °C, чем с ростом концентрации кислоты. В указанном диапазоне концентраций кислот при 80 °C за 6 ч она изменяется от 9,3 до 12,3%, а при 120 °C от 93,0 до 99,9%.
В фосфорной кислоте, содержащей <30% Р2О5, превращение при 80 °C первоначально выделяющегося полигидра-. та в стабильный ангидрит [28] происходит через промежуточное оводнение до гипса,, которое в аналогичных растворах при 25 °C протекает весьма быстро. (1—3 мин).
Пересечение кривых растворимости гипса и ангидрита при 25 °C, соответствующее их равновесию, происходит при содержании в кислоте 33,75% Р2О5, когда стабильны равновесные твердые фазы — гипс и ангидрит. Пересечение кривых растворимости гипса и полугидрата при 25 °C отвечает раствору, содержащему 49% Р2О5. Однако эти фазы мета-стабильны и с течением времени переходят в стабильный ангидрит.
Пересечение кривых растворимости гипса и полугидрата при 80 °C для указанного превращения происходит при содержании в растворе 33% Р2О5 (33,3% Р2О5 согласно вычислениям на основании тензиметрических данных [36, с. 2754]).'
Давление пара, при 25°C в системах CaSO4-2H2O^
CaSOH-2H2O и CaSO4-2H2O^ CaSO4- 6,5H2OH-l,5H2O равно 2287 и 1210 Па (17,2 и 9,1 мм рт. ст.). Этим значениям давления пара отвечают растворы фосфорной кислоты, содержащие 34,30 и 48,99% Р2О3.
Вследствие своих дегидратирующих свойств фосфорная кислота тем больше снижает температуру превращения кристаллогидратов," чем выше ее концентрация. Например, превращение гипса в нолугидрат в системе CaSO4—Н2О происходит при 107 °C. В присутствии фосфорной кислоты, содержащей 33% Р20б, это превращение протекает при 80 °C, а в растворе, содержащем ~49% Р2О5,—при 25°C (табл. III).
На рис. 11.16 показана проекция диаграммы i—[Р2О5] растворимости в системе СаО—Р2О5—SO3—Н2О. Линия аб разделяет области двух стабильных, а .линии cd двух мета-стабильных фаз. На этих линиях одновременно сосуществуют соответствующие кристаллогидраты.
Выше и правее линии ab стабильна твердая фаза — ангидрит. В области abed первоначально выделяющийся из
54 ...
Таблица,*!/!
Условия фазовых превращений сульфата кальция
Температура, °C Концентрация кислоты (% Р2О5) для фазового превращения
CaSO4-2H2O^ CaSO4-0.5H2O+ . -Н,5Н2О CaSO4-2H2O-t -tCaSO4+2H2O.
25 49,00 33,75
40 46,40 . 26,50
60 40,90 11,04
66 — 0
80 33,0 —
107 0 —
раствора полугидрат переходит в более устойчивый, но также метастабильный-гипс, который затем превращается в стабильный ангидрит.
Ниже,и левее линии ab стабильная твердая фаза—гипс — наименее растворима в этих условиях. Увеличение растворимости сульфата кальция здесь наблюдается в ряду: гипс->^_ -* ангидрит-»-полугидрат. В соответствии с этим в области kab метастабильный ангидрит непосредственно оводняется до гипса, а метастабильный полугидрат предварительно дегидратируется с последующим оводнением до гипса. По этим же причинам в области выше и правее линии cd превращение метастабильных гипса и полугидрата в стабильный ангидрит происходит для первого случая через полугидрат, а для второго —' непосредственно.
Скорость взаимного превращения метастабильных фаз в области диаграммы abed (рис. 11.16) и метастабильных в стабильную фазу зависит от температуры, концентрации раствора, растворимости сульфата кальция и т. д.
Данные о превращении кристаллогидратов сульфата кальция в растворах фосфор ной кислоты получены с применением веществ, не содержащих примесей. Количество фосфорной кислоты и сульфата кальция в опытах определялось растворимостью СаО в системе СаО—Р2О5—Н2О и стехиометрическим количеством серной кислоты, взятой для осаждения ионов кальция. .
В- условиях производства экстракционной фосфорной кислоты равновесие между фазами не достигается, поскольку процесс протекает за ограниченное время. Реакционная среда, помимо основных реагентов, содержит разные примеси. Отношение между жидкой и твердой фазами в пульпе значительно меньше, чем при выделении сульфата кальция из насыщенного или почти насыщенного раствора фосфата кальция в фосфорной кислоте.
55
Рис. 11.16. Схема превращения кристаллогидратов сульфата кальция в фосфорной кислоте при различных температурах: •
П —CaSO40,5H2O; A —CaSO4; Г —CaSO4-2H2O.
Рис. 11.17. Области существования кристаллогидратов сульфата, кальция в условиях производства фосфорной кислоты сернокислотным методом [48].
При наличии в кислоте примесей изменяется растворимость кристаллогидратов (стр. 51) и в некоторых условиях возможно образование твердых растворов сульфата И'фосфата кальция [40, 46]. Вследствие этого изменяется устойчивость метастабильных фаз и условия существования их и стабильных фаз.
В присутствии в растворе избыточного количества СаО сверх содержания ее в виде CaSO4, а также при наличии соединений магния расширяется область кристаллизации гипса (например, до 35% Р2О5 при 80°C и до 37% Р2О5 при 70°C). Присутствие в растворе серной кислоты усиливает его дегидратирующие свойства, вследствие чего расширяется область фазового перехода полугидрата в ангидрит и уменьшается область стабильного гипса.
В присутствии в растворе фосфорной кислоты ионов аммония или калия равновесный фазовый переход полугидрата в гипс смещается в область более высоких температур и концентраций [47]. Например, при мольном отношении NH3: Н3РО4 = 0,5 равновесному переходу кристаллогидратов отвечает при 70°C концентрация 44,2% Р2О5 (вместо 34% Р2О5 в отсутствие NH3), а при 80 и 90°C — 43,2 и 40,5% Р.2О5 (вместо 29,56 и 24,5% Р2О5). В'присутствии ионов калия повышение равновесной концентрации кислоты меньше, чем в присутствии ионов аммония.
В условиях, близких к производственным, область существования гипса значительно расширяется. Одновременно появляется достаточно широкая область существования мета- .
56
стабильного полугидрата при высоких температурах и концентрациях кислоты (рис. 11.17). Для сравнения пунктиром на рис. П.17 показана граница равновесной области существования стабильного гипса.
Область существования гипса в производственных условиях (т. е. в течение определенного времени при соответствующем отношении Ж: Т в пульпе, составе кислоты и •других параметрон, характеризующих технологический режим) простирается в'крайних точках: до ~ 105 °C (в воде) и до ~50% РгО5 (при 0°С), а полугидрата до ~250 °C и 50—60% РгО5 и более (см. рис. 11.17),
Скорость фазового перехода кристаллогидратов сульфата кальция в фосфорнокислых растворах
Превращения метастабильных фаз, связанные с уменьшением концентрации сульфата кальция в пересыщенном растворе и переходом его в стабильное состояние, зависят не только от температуры и концентрации кислоты, но и от условий выделения кристаллогидратов (перемешивания, соотношения и состава жидкой и твердой фаз,, наличия примесей, затравки и т. п.).
Равновесие кристаллогидратов сульфата кальция со стабильной фазой устанавливается сравнительно быстро в концентрированных растворах и при высокой температуре. В фосфорной кислоте (40—50% P20s) при 90—110 °C (выше кривой cd, см. рис. 11.16) выделяющийся вначале полугидрат быстро переходит в стабильную фазу — ангидрит. Но в этих условиях наиболее растворим гипс (см. рис. 11.15), который должен был бы выделяться раньше полугидрата.'По-видимому, скорость его перехода в CaSO4-0,5H2O так велика, что в твердой фазе даже через очень короткое время после начала реакции гипса уже нет. В то же время CaSO4-0,5H2O превращается в ангидрит с измеримой скоростью: при 90 °C и содержании в кислоте 50% Р2О5 фазовый переход в растворе становится заметным через 2—3 ч и заканчивается в течение 21 ч.
.Скорость дегидратации полугидрата резко возрастает с ростом температуры выше 100 °C. Степень превращения полугидрата в ангидрит в растворе фосфорной кислоты-концентрацией 45% Р2О5 с повышением температуры от 110 до 120qC увеличивается за 6 ч в ~2 раза (с 45,8 до 93%).
t Образовавшийся. в области abed (ом. рис. 11.16) дигидрат существует в виде метастабильной формы в разбавленных растворах (при содержании Р2О5<10%) в течение нескольких месяцев или в течение нескольких суток (при концентрации 25% Р2О5). Поэтому в обычных условиях получения фосфорной кислоты (25—32% Р2О5) отделяемый от
57
жидкой фазы осадок содержит сульфат кальция в форме метастабильного гипса. Относительно большая устойчивость метастабильного гипса и весьма малая, скорость перехода его в стабильную твердую фазу (ангидрит), по-видимому, объясняется торможением процесса дегидратации вследствие большого содержания воды в разбавленных растворах. В таких же растворах преимущественно протекает оводнение полугидрата, а не дегидратация дигидрата.
Продолжительность полного превращения полугидрата в гипс уменьшается почти в 10 раз при понижении концентрации фосфорной кислоты от 28,5 до 10% Р2О5.
Понижение температуры кислоты одного и того же состава в области метастабильного существования гипса (области- abed) приближает систему к области растворов, равновесных с гипсом (область ниже ab). Это приводит к ускорению фазового перехода полугидрата в гипс. Так, в фосфорной кислоте, содержащей 25% Р2О5, полное превращение полугидрата в гипс при 60°C происходит за 2 ч, т. е. почти в 3 раза скорее, чем при 80°iC.
Фазовый переход полугидрата сульфата кальция в гипс происходит в слабых растворах фосфорной кислоты (до 25% Р2О5) практически полностью в диапазоне температур до 95—100 °C. Так, при 95 °C в растворе концентрацией 25% Р2О5 (при Ж:Т=6:1), хотя скрытый период кристаллизации задерживается на ч, фазовый переход- полугидрата в гипс за 4 ч завершается на 92%, т. е. практически без гипсования в дальнейшем.
На рис. 11.18, а показаны изохроны скрытых периодов перехода полугидрата в гипс и ангидрит, а на рис. 11.18, б — изохроны кристаллизации гипса и ангидрита (без учета длительности скрытых периодов превращения). Длительность кристаллизации различных фаз в процессе превращения полугидрата различна в областях II и III и IV и V (см. рис. 11.18, а). .
Сведения о -влиянии других веществ и примесей, содержащихся в фосфорной кислоте, на скорость фазовых переходов кристаллогидратов сульфата кальция весьма ограничены, хотя в этом направлении непрерывно ведутся, исследова,-ния.
В фосфорной кислоте с концентрацией 15—35% Р2О5, содержащей серную кислоту (до 8% H2SO4), скорость перехода полугидрата в гипс уменьшается с увеличением концентрации Р2О5 и уменьшением содержания H2SO4 в растворе. При одной и той же концентрации Р2О5 процесс ускоряется при увеличении содержания H2SO4. Превращение полугидрата и гипса в ангидрит ускоряется с увеличением концентрации обеих кислот. '
58
Рис. 11.18. Изохроны скрытых периодов перехода полугидрата в гипс и ангидрит (а) и изохроны кристаллизации гипса и ангидрита (б) в фосфорно’й кислоте.
Соединения стронция (3,0—7,0% SrO) или трехвалентного церия (1,8—2,8% Се2О3), внесенные в раствор искусственно или с фосфатным сырьем, приводят к замедлению гидратации полугидрата в фосфорнокислотных- растворах, содержащих 10—20% Р2О5 и 5% H2SO4 в ~20 раз, чём в их отсутствие, но полная гидратация требует не более 6—7 ч [49].
Если в фосфорной кислоте (10—35% Р21О5) находится HaSiFe, то продолжительность фазового перехода также зависит от концентрации Р2О5. При 70 °C в кислоте, содержащей 10—35% Р2О5, гипс не изменяется в течение 4 недель [39, с. 1714]; в кислоте, содержащей 40% Р2О5— в течение 1 суток, а при концентрации 45% Р2О5 — 2 ч.
Дегидратация гипса и -полугидрата в растворе Н3РО4 ускоряется в присутствии свободной H2SO4 и замедляется при наличии Mg2+, Са2+, Al3+, Fe-3-+ [49]. Присутствием солей магния в фосфорной кислоте, получаемой из фосфоритов Каратау, можно объяснить тот факт, что гипс достаточно устойчив в кислоте, содержащей 34—36% Р2О5.
Описанные выше результаты исследования скорости фазового превращения кристаллогидратов сульфата кальция могут быть использованы с той или иной степенью приближения для оценки состава выделяющихся фаз и в реальных технических процессах. Например, при экстракции фосфорной кислоты, содержащей ~25% Р2О5, при 80 °C твердая фаза должна состоять из гипса. Скрытый период превращения полугидрата в гипс в .кислоте, не содержащей примесей (см. рис. 11.18, а), равен 6 ч, а время кристаллизации гипса (см. рис. 11.18,6) —3,5 ч. Таким образом, продолжительность всего процесса образования кристаллов гипса составляет 9,5 ч. В действительности, как известно, экстракция такой фосфорной кислоты завершается за меньшее время вследствие непрерывного течения процесса из-за наличия зародышей кристаллов гипса и ускоряющего действия примесей, содержащихся в кислоте. Гипс в стабильный при 80°C ангидрит в условиях экстракционного процесса не переходит, так как только скрытый период его измеряется сутками.’
С повышением температуры до 90—100 °C в разбавлен- • ных фосфорнокислотных растворах концентрации до ~20— 22% Р2О5 переход полугидрата в гипс протекает еще с большей скоростью и в практических условиях полугидрат может быть обнаружен в этом случае лишь в самый начальный период процесса.
В растворе, содержащем 35—45% Р2О5, при 80—95 °C время скрытого перехода полугидрата в гипс измеряется многими часами и сутками (в зависимости от концентрации и температуры кислоты). Поэтому твердой фазой при экс-60
тракции фосфорной кислоты в. указанных условиях будет полугидрат, и процесс определяется как полугидратный. При температурах выше 100—105 °C и концентрации кислоты более 50% Р2О5 твердая фаза состоит из ангидрита, который образуется довольно быстро из выделяющегося вначале полугидрата.
Содержание Р2О5 в образующемся осадке в результате захвата им ионов НРО42^ весьма зависит от скорости фазового перехода полугидрата сульфата кальция в гипс и уменьшается с увеличением длительности превращения, когда система меньше отклонена от равновесного состояния.
Гранулометрическая характеристика кристаллов сульфата кальция
Форма и размеры выделяющихся кристаллов зависят От их свойств (растворимости и комплексообразования, структуры и т. п.), температуры и концентрации кислоты, степени пересыщения и условий снятия пересыщения. Кривые зависимости пересыщения фосфорнокислых растворов сульфатом кальция от концентрации кислоты располагаются симбатно кривым растворимости гипса и ангидрита. Максимальная разность концентраций (в расчете на СаО) в пересыщенных и насыщенных фосфорнокислых растворах составляет ~0,2% СаО (при 80 °C и содержании 49,7% Р2О5), а минимальная — 0,03% СаО (при 25 °C и содержании 45,6% Р2О5). Растворимость равна соответственно 0,176 и 0,10% СаО.
На рис. 11,19 представлено изменение во времени концентрации сульфата кальция в пересыщенном растворе, содержащем 45,6% Р2О5 при -60 °C (лабораторные условия). В этих условиях стабильная твердая фаза — ангидрит, первая метастабильная фаза — гипс, а вторая—полугидрат. Как видно, при переходе от первой метастабильной фазы ко второй наблюдается как бы вторичное пересыщение раствора ('максимум.на кривой).
' Размеры выделяющихся кристаллов сульфата кальция при различных условиях изучены в большей мере при образовании гипса и в меньшей — при образовании полугидрата и ангидрита. С повышением температуры от 30 до 80 °C длина кристаллов гипса увеличивается в 10—45 раз, а ширина в 3—
CaSO4-2H2O
CaSO4* 0»5HjO f i 1 111 i-i
1 J 5 7 9 11 13 )S
Время,ч
Рис. 11.19. Изменение во времени концентрации сульфата кальция (в расчете на СаО) в пересыщенном растворе, содержащем 45,6% Р2О5 при 60 °C.
£ б
? 0,3 a 0,276 S. 0,238
3 V
61
5 раз [50]. С изменением концентрации кислоты от 25 до 34% РгО5 продольные и поперечные- размеры кристаллов значительно уменьшаются. Большое влияние на .форму кристаллов оказывают содержащиеся в кислоте примеси [13, 51].
Увеличение содержания СаО в 'реакционной пульпе вы-, зывает уменьшение размеров кристаллов при возрастании степени их неоднородности [50]. Большие кристаллы образуются при норме кислоты 90 и 110% от ее стехиометрического количества.
На рост кристаллов гипса оказывает влияние соотношение между концентрациями Са2+ и SO42- в растворе по сравнению с их стехиометрическими значениями в CaSO4. При избытке Са2+ гипс образуется в виде тончайших игл длиной 30—80 мкм; при избытке SO42- их размеры достигают 100'мкм в ширину и несколько сотен микрометров в длину. Для получения крупнокристаллического однородного осадка необходимо, чтобы массовое отношение SO42-: СаО в жидкой фазе было по возможности постоянным и равным 1,5—3. Оптимальным является содержание в растворе 1—2,5% SO3 и 0,35—0,79% СаО.
При небольшой' концентрации SO3 примеси Fe3+ и А13+-способствуют образованию более широких и коротких (изометрических) кристаллов гипса и полугидрата [52]. Образование кристаллов по форме, близкой к изотермической, происходит при содержании в растворе от 0,2 до' 1,5% R2O3, что. обуславливается уменьшением длины кристаллов почти в 1,5 раза; ширина же их практически не меняется. Благоприятное влияние на форму и размеры кристаллов сульфата кальция оказывают ионы SiF62~ в сочетании с ионами А13+.
С увеличением содержания алюминия в системе уменьшается доля фтора, связанного с кремнием, и увеличивается доля фтора, связанного в жидкой фазе в несиликатные комплексы. При этом увеличивается количество крупных кристаллов в гипсе за счет уменьшения мелких [53]. Оптимальное отношение в растворе фтора, не связанного с кремнием, больше или равно двум. Минимальное содержание алюминия, связанного с фтором, в растворе составляет ~0,2% [54]. Совместное влияние ионов алюминия и фтора, по-ви-димбму, обусловлено равновесием в системе 6F-—SiF62-— A1F2+ (или A1F63-).
В присутствии в растворе соединений р. з. э. (церия, нио-дима и др.) фракционный состав гипса изменяется в сторону увеличения мелких кристаллов, что проявляется уже при их концентрации ~ 0,003—0,005 % [55]. При содержании в растворе 0,15—0,18% 'Р> з. э. подавляется эффективность дей-$2
ствия на кристаллизацию гипса модификатора — фторалю-мината (0,5—0,6% А1 и 1,2—1,5% F).
Хорошо отфильтровывающийся сульфат кальция образуется в присутствии 1—<2% (по отношению к Р2О5) сульфатов магния, цинка, железа, никеля и меди, а также ионов аммония и нитрата. При содержании в растворе 0,5—>1,5% NH4+ значительно увеличиваются размеры кристаллов гипса, исчезают кристаллы шламовых размеров (<20 мкм), повышается изометричностъ частиц.
При наличии в растворе большого количества кремние--вой кислоты гипс кристаллизуется в виде тончайших игл. Кроме того, выделяющийся при экстракции фосфорной кислоты из некоторых фосфоритов илистый осадок кремниевой кислоты сильно затрудняет отделение жидкой фазь( от твердой.
О размерах кристаллов полугидрата и ангидрита, образующихся в концентрированных растворах фосфорной кислоты при повышенных температурах, имеются весьма ограниченные сведения. Известно, что при непрерывном проведении процесса в лабораторных условиях полугидрат кристаллизуется' в форме небольших шестигранных призм размером 20— 30 мкм, собранных в крупные агрегаты, а ангидрит— в виде широких утолщенных пластинок [56]. Крупные компактные агрегаты кристаллов полугидрата хорошо ’ отделяются от раствора. Ангидрит, наоборот, выделяется из раствора, содержащего избыток серной кислоты (3—5% SO3) в виде относительно крупных кристаллов, образующих пластины неправильной квадратной формы.
При. длительности процесса 4—6 ч из растворов, содержащих 35—43% Р2О5, при 75—85 °C выделяются агрегаты полугидрата, имеющие размеры от 75 до 200—300 мкм; они состоят из отдельных мелких кристаллов в 10 мкм.
Отличие гранулометрической характеристики кристаллов полугидрата и гипса, по-видимому, объясняется.более высокими относительными скоростями роста и более низкими относительными скоростями образования зародышей полугидрата, чем гипса.[57].
'Кристаллы полугидрата длиной до 150 и шириной до 15— 20 мкм выделяются в фосфорнокислом растворе, содержащем <~25—33% Р2О5 и 15—25% HNO3. Такие кристаллы отделяются фильтрованием от раствора; представляющего собой смесь азотной (-~50% HNO3) и концентрированной фосфорной кислоты (~45—65% Р2О5) со скоростью, в 3—6 раз большей, чем дигидрат в производстве кислоты, содержащей 30—132% Р2О5.
Образование кристаллов ангидрита связано с дегидратацией первоначально выделяющегося полугидрата, В раство-
63
ре, содержащем 50% Р2О5, при 90 °C перекристаллизация начинается только через 6 ч и заканчивается более, чем через 30 ч (стр. 57);
В кислоте, содержащей >47% Р2О5 и 5% H2SO4, дегидратация полугидрата при 100 °C заканчивается в течение 20—40 мин. Образующиеся кристаллы ангидрита имеют прямоугольную форму с размерами (15—30) X (6—40) мкм. С понижением концентрации H2SQ4 до 3% продолжительность полного превращения полугидрата при 100 °C увеличивается до 80—85 мин. При этом получаются значительно более крупные кристаллы, имеющие также прямоугольную форму, с размерами, например, (30—40) X (6—8) мкм. В этих условиях образуется раствор с относительно небольшим пересыщением ангидритом, что обуславливает выделение более крупных кристаллов.
В присутствии ионов кальция выделяются мелкие и неразвитые по форме кристаллы ангидрита, хотя фазовый период протекает очень медленно. Для полного превращения полугидрата в ангидрит в этом случае требуется от 150 до 600 мин, что зависит от нормы серной кислоты, использованной для осаждения сульфата кальция.
С повышением температуры дегидратация полугидрйта ускоряется,'и соответственно уменьшаются размеры кристал-, лов ангидрита. При 120°С в растворе, содержащем 3—3,5% H2SO4, продолжительность процесса меньше в 2—3 раза, чем при 100 °C, и составляет 30—40 мин вместо 80—420 мин.
ЛИТЕРАТУРА
1. Позин М. Е. Технология минеральных солей. Т. II. 4-е изд. Л., Химия, 1974.
2. Соколовский А. А., Яшке Е. В. Технология минеральных удобрений ’и кислот. М., Химия, 1970.
3. Щукарев С. А., Баличева Т. Г., Борча К. Я- — ЖНХ, 1963, т. 8, с\ 1437.
4. Владимирова В. И., Крпылев Б., А., Машовец В. П, — Тезисы докладов научно-технической конференции ЛТИ им. Ленсовета. Л., Химия, 1965, с. 57.
5. Жданова М. В. и др. — В кн.: Новые исследования по технологии минеральных удобрений/Под ред. М. Е. Позина и Б. А. Копылева. Л., Химия, 1970, с. 192.
6. Жданова М. В.,'3инюк Р. Ю., Копылев Б. А. — Тезисы докладов научно-технической конференции ЛТИ им. Ленсовета. Л., 1968, с. 15.
7. Соколовский А. А. — Изв. вузов. Химия п Хим. технол., 1963, т. 6, № 3, с. 91—97.
8. Орехов И. И. — Хим. пром., 1972, № 4, с. 272—й75.
9. Чепелевецкий М. Л., Бруцкус Е. Б. Суперфосфат. Физико-химические основы производства. М., Госхимнздат, 1958.
10. Орехов И. И„ Слободкина Г. Л. — ЖНХ, 1972, т. 17, с. 829—832.
11. Бектуров А. Б., Ержанова Р. С., Литвиненко Е. И. — Изв. АН КазССР. Сер. хим., 1968, № 1, с. 1—7.
64
12. Костылева Л. В., Баранович 3. М. — В кн.: Технология минеральных удобрений. Межвуз. сб. трудов. Л., ЛТИ им. Ленсовета, 1978, с. 29— 33 - '
13. Чепелевецкий М. Л. и др. — Труды НИУИФ, 1937, вып. 137.
11. Тананаев И. В., Чудинова И. И. — ЖНХ, 1962, т. 7, с. 2285.
15. Василенко И. А., Чепелевецкий М. Л. — Там же, 1957, т. 2, № 10, с. 2486.
16. Ульянов А. И., Казакова Т. И. — Изв. АН СССР. ОХН, 1963, т. 11, №6, с. 1181.
17. Тананаев И. В., Петушкова С. М.— ЖНХ, 1964, т. 9, с. 1094.
18. Позин М. Е., Копылев Б. А., Тен Се-ден. — Труды ЛТИ им. Ленсовета, 1956, вып. 36, с. 5. —
19. Gilbert R. L., Moreno Е. С. — Ind. Eng. Chem., Proc. Des. a. Deve-lopm., 1965, v. 4, № 4, p. 368.
20. Позин M. E., Копылев Б. А., Жильцова Д. Ф. — ЖПХ, 1959, т. 23, № 10, с. 2164.
21. Южная Е. В. — Автореф. канд. дисс. М., НИУИФ, 1956.
22. Позин М. Е., Копылев Б. А., Кыршев И. П. — ЖПХ, 1963, т. 36,
№ 6, с. 1175.
23. Труды СЗПИ. № 6. Химия и хим. технол., Л., СЗПИ, 1969.
. 24. Орехов И. И., Власова Т. Л. — Хим. пром., 1975, № 10, с. 755—
757.
25. Позин М. Е., Копылев Б. А. Новые методы получения минеральных удобрений. Л., Госхимиздат, 1962.
26. Исследования по химии и технологии удобрений, пестицидов и солей, М., Наука, 1966.
27. Вольфкович С. И. и др. — ЖПХ, 1976, № 2, с. 284—288.
28. Белопольский П. А., Таперова А. А., Шульгина М. И. — Там же, 1939, т. 12, № 1, с. 3; Таперова А. А., Шульгина М. И. — Там же, 1945, ’ . т. 18, № 9—10, с. 521.
29. Промышленность минеральных удобрений и серной кислоты. Реф. сб. НИУИФ. М., НИИТЭХИМ, 1976. Вып. 1, 29 с.; вып. 3, 107 с.
30. Яржемский Я.' Я'. Микроскопическое изучение галогенных пород. М., Наука, 1966.
31. Белянкин Д. С. Избранные труды. Т. 1. М., Изд. АН СССР, 1956.
32; Здановский А. Б., Спиридонов Ф. П. — ЖДУК 1967, т. 40, № 5, с. 1152.
33. Кармышев В. Ф. — Тгм же, 1977, т. 50, № 5, с. 1157—1158.
34. Таперова А. А., Шульгина М. И. — Там же, 1950, т. 23, № 1, с. 32.
35. Волощенко К. А. и др. — Там же, 1976, т. 49, № 1, с. 3—5.
36. Здановский А. Б., Власов Г. А., Сотников Л. И. — ЖНХ, 1968, т. 13, № 9, с. 2552, 2754; Здановский. А. Б., Спиридонов Ф. П. — Там же, . 1966, т. 11, № 1, с. 20; Здановский А. Б., Власов Г. А. — Там же, 1968, т. 13, № 9, с. 2552.
37. Технология минеральных удобрений. Новые пути получения. Л., изд. ЛТИ им. Ленсовета, 1973.
38. Куртева О. И., Бруцкус Е. Б. — Сборник аспирантских работ. М., НИУИФ, 1959, с. 15; 1963, с. 12.
39. Куртева О. И., Бруцкус Е. Б. — ЖПХ, 1961, т. 34, № 8, с. 1714; Позин М. Е., Копылев Б. А., Ядацкая Я- К. — Изв. вузов. Химия и хим. технол., 1964, т. 7, № 5, с. 699; Токарев Г. И., Копылев Б. А., Варшавский В. Л. — ЖПХ, 1969, т. 42, № 1, с. 12.
40. Campbell А. П., Coots J. 117— Ind Eng. Chem., 1948, v. 40, № 7, p. 1294.
41. Elmore K. L., Farr T. D. — Ibid., 1940, v. 32, № 4, p, 580.
; 42. Вильциц E. Л. и dp. — ЖПХ, 1968,.t. 41, № 9, c. 1883.
43. Морозова Г. А., Копылев Б. A. — Изв. вузов. Химия и хим. технол., 1972, т. 15, вып. 7, с. 975.
44. Дмитревский Б. А., Копылев Б. А., Морозова Г. А. — В кн.: Переработка фосфоритов ^аратау. Л., Химия, 1975,. с. 189—197.
5-5473 65
45. Копылева Т. В., Власова Т. J1., дрехов И. И. — ЖПХ, 1976, № 2, с. 483—485.
46. Чепелевецкий М. Л., Хамский Е. В. — Там же, 1959, т. 32, № 5, с. 948.
47. Орехов И. И. и др. — В кн.: Физико-химический анализ жидких систем. Тезисы докладов пятого совещания (5—7 июня 1973 г.). Каунас, 1973, с. 327—328.
48,- Lehrecke Л.— Chem. Fabr., 1933, № 50, р. 505; Sanfourshe А.— Ind. ’ chim., 1934, № 244, р. 331; № 245, р. 406.
49. Лозин М. Е. и др.— ЖПХ, 1976, т. 49, № И, с. 2361—2366. .
50. Любченко Т. В., Копылев Б. А., Лозин М. Е. — Там же, 1967, т. 40, № 10, с. 2225.
51. Воскресенский С. К, Милованова С. К- — Сообщения о научно-технических работах НИУИФ, 1957, № 2, с. 9; Милованова С. К. Научнотехн. информ, бюлл. НИУИФ, 1957, № 9, с. 49.
52. Копылев Б. А., Любченко Т. В., Лозин М. Е. — ЖПХ, 1969, т. 42, № 11,. с. 2431.
53. Glabisz U., Gabriel Л. — Przem. chem., 1973, t. 52, № 4, s. 283—296.
54. Gabriel Л., Glabisz U. — Ibid., 1976, t. 55, № 3, s. 148—151
55. Glabisz U„ Gabriel /7. — Ibid., 1974, t. 53, № 6, s. 350—353.
56. Воскресенский С. К., Ионасс A. A. — Труды НИУИФ, 1940, вып. 153,
с. 92, 94.
57. Amin А. В., Larson М. А. — Ind. Eng. Chem., Proc. Des. a. Developm., 1968, v. 7, № 1, p. 133.
Глава lit
СЕРНОКИСЛОТНАЯ ЭКСТРАКЦИЯ ФОСФОРНОЙ кислоты
‘ III.1. ОБЩИЕ ПОЛОЖЕНИЯ
Экстракция фосфорной кислоты [1—5] серной кислотой заключается в разложении фосфата серной кислотой и выделении сульфата кальция в присутствии раствора разбавления (циркулирующей фосфорной кислоты), разделении получаемой пульпы и. отмывке фосфорной кислоты от осадка.
Физико-химические условия и конструктивное оформление разложения фосфата должны обеспечить максимальное (почти полное) извлечение фосфорного ангидрида из фосфата и получение кислоты с максимально возможной (при данном режиме) концентрацией. -
Для успешного разделения образующейся суспензии необходимо, чтобы в процессе экстракции сульфат кальция выделялся в виде хорошо отфильтровывающихся и легко промываемых кристаллов.
Экстракция фосфорной кислоты из природных фосфатов и отделение ее от сульфата кальция (фильтрование пульпы) —'Это взаимосвязанные процессы единой сложной химико-технологической системы. Нарушение режима одного из них приводит к ухудшению показателей другого [6; 7, с. 18]. Тесная их связь проявляется не только при изменении тех- 7 нологических параметров, но и зависит от условий конструктивного оформления каждого. Ухудшение кристаллизации сульфата кальция (вследствие нарушения сульфатного режима, недостаточного перемешивания и т. п.) в экстракторе приводит к недоотмывке кислоты из осадка и изменению концентрации фильтратов при фильтровании. В свою очередь забивание коммуникаций и фильтровальной ткани ведет к нарушению водного и теплового балансов в экстракторе.
С целью поддержания необходимого температурного режима в экстракторах пульпу охлаждают, обдувая ее поверхность воздухом или барботируя его, или же используют вакуум-испарители. Охлаждение пульпы за счет испарения воды при продувке воздухом или в вакуум-испарителях при-б* • • 67
водит, кроме тбго, к увеличению концентрации Р2С>5 в жидкой фазе.
В качестве затравки для кристаллизации применяют часть циркулирующей продукционной пульпы; количество последней позволяет регулировать также и температуру экстракционной массы (пульпы). На некоторых установках (обычно небольшой мощности) экстракцию проводят без циркуляции пульпы.
Выделяющиеся при разложении фосфата (и частично при фильтровании пульпы) газообразные фторсодержащие сое-, динения, а также пары воды улавливают в абсорбционной системе (см. гл. VI).
Часто при производстве кислоты с концентрацией, недостаточной для получения удобрений, ее упаривают. Этим же методом из обычной экстракционной фосфорной кислоты получают суперфосфорную кислоту.-
И 1.2. АППАРАТУРА ПРОЦЕССА
Аппаратура для производства экстракционной фосфорной кислоты в основном не зависит от способа выделения сульфата кальция. Различие технологических режимов сказывается главным образом на соотношениях между отдельными аппаратурными элементами, а также и на выборе конструкционных материалов, защите от коррозии и величине и организации потоков.
Наиболее распространен дигидратный режим, усовершенствование которого позволяет получать из апатитового концентрата или высококачественных фосфоритов при 65—80 °C фосфорную кислоту с содержанием 30—32% Р2О5. Из других фосфоритов этим способом получают кислоту, содержащую 20—27% Р2О5 (в зависимости-от состава фосфата).
Фосфорную кислоту с содержанием более 30—32% Р2О5 получают по высокотемпературному режиму (90—100 °C и выше) с выделением в твердую фазу полугидрата или ангидрита, Полугидратный процесс в настоящее время широко используется в промышленности.
Ангидритный процесс, протекающий при температурах выше 100 °C с применением концентрированной кислоты, осложняется сильной коррозией аппаратуры, трудностью отделения мелких кристаллов; кроме того, для поддержания автотермичности процесса из-за недостаточной экзотермич-ности его необходим подогрев реагентов. Помимо этого возникают большие трудности при отмывке фосфорной кислоты из осадка (вследствие напряженного водного баланса). Поэтому ангидритный процесс до сих пор не вышел из стадии небольших опытных масштабов,
68
Производство экстракционной фосфорной кислоты состоит из следующих основных стадий: дозирования фосфатного сырья и серной кислоты, разложения фосфата смесью серной и фосфорной кислот (экстракции), охлаждения циркулирующей пульпы, фильтрования экстракционной пульпы, абсорбции фторсодержащих газов (гл. VI) и концентрирования (упаривания) /фосфорной кислоты.
Транспортирование и хранение фосфатного сырья
В качестве фосфатного сырья в СССР используют тонко-измельченный апатитовый концентрат, а также фосфоритную муку Каратау.
Поступающее на завод фосфатное сырье выгружают из железнодорожных вагонов и с помощью ленточных транспортеров, шнеков, элеваторов или пневматическим способом подают на склад, а из него — в расходный бункер, а затем в бункер дозатора [8].
Фосфатное сырье большей частью транспортируют в специальных саморазгружающихся вагонах (апатитовозах) бункерного типа или в пневматических цистернах (пневморезервуарах). Саморазгружающийся 60-тонный цельнометаллический вагон бункерного типа с двухсторонней боковой выгрузкой может- быть загружен в течение 6—7 мин и раз-. -гружен с применением вибраторов за 8—40 мин.
Из вагонов фосфатное сырье выгружают в прирельсовые скреперные траншеи, расположенные по обе стороны железнодорожного пути. Две траншеи вмещают по 1500 т фосфатного сырья и имеют такую длину, которая позволяет одновременно разгружать пять 60-тонных саморазгружаюг ,щих!ся вагонов. Траншеи опорожняют скреперами емкостью 1 м3, подающими сырье в устройства для отделения случайных примесей (щелевые бункера или другие) и для транс-,портировки на оклад (элеваторы и ленточные конвейеры). Сырье на новых заводах хранят в складах силосного типа, которые, хотя и требуют больших капитальных вложений, но обеспечивают'улучшение условий труда и сокращение, потерь сырья.
Склады силосного типа устраивают в виде трех или.шести железобетонных башен (силосов) внутренним диаметром 1 Г,5 м и высотой 21,4 м, общей вместимостью от 10 000 до 20000 т, что обеспечивает запас сырья на 10—45 суток. Сырье в силосах во избежание зависания материала аэрируют сжатым воздухом, который подводят под - пористые плиты, помещенные в днище силоса.
- Для подачи сырья из скреперных траншей в силосы и : внутрицехового перемещения его часто еще используют ленточные и винтовые конвейеры и элеваторы. Это обуславли
69
вает большое число пересыпок (перегрузок) материала и сопровождается значительной запыляемостью помещений и. потерями сырья.
Более рационален пневмотранспорт сырья [8, 9]. Для этого силосные склады оборудуют сдвоенными стационарными камерными пневмонасосами с верхней выгрузкой и автоматическим управлением, производительностью 50 т/ч при подъеме на 40 м. Пневматический транспорт требует большего расхода электроэнергии, чем конвейерный, но позволяет улучшить условия труда и уменьшить потери сырья.
Дозирование фосфатного сырья и кислоты
От точности и равномерности подачи материалов зависят концентрация реагирующих компонентов, форма и размеры выделяющихся кристаллов сульфата кальция, степень извлечения фосфорной кислоты и т. п.
Серная кислота со склада сернокислотного цеха поступает в сборники-, предназначенные для хранения сменного запаса. Из них ее центробежными насосами " подают в напорный бак, откуда через дозаторы кислота поступает в экстрактор.
Наиболее сложно точно дозировать твердые материалы; для этого существуют различные методы. Фосфатное сырье со склада подают в один или несколько цеховых запасных бункеров, вместимость которых достаточна для обеспечения суточной потребности цеха. В бункерах установлены сигнализаторы верхнего и нижнего уровня. В нижней (конической) части бункера находится аэрирующее устройство, содержащее пористые плитки иди трубки с соплами, через которые поступает воздух, служащий для предотвращения зависания материала.
Из запасного бункера фосфат через шнековый или тарельчатый питатель, элеватор и систему транспортирующих шнеков подают в питательный бункер, производительность которого несколько больше, чем расход фосфата. Это обеспечивает полную загрузку питательного бункера и поступление фосфата в дозатор под постоянным напором.
Весь транспортный узел сблокирован так, что остановка любого механизма влечет за собой немедленную остановку всех других. Это исключает возможность завала фосфатом и поломки механизмов при случайной остановке одного из них.
Из питательного бункера фосфат поступает в дозатор, а из него—в экстрактор через скребковый конвейер, который служит одновременно затвором, препятствующим попаданию в дозатор паров воды и газообразных фторсодержащих соединений, выделяющихся в экстракторе.
70
Дозаторы фосфатного сырья [8, первая ссылка]. По принципу действия дозаторы бывают объемными и весовыми. Первые не реагируют на изменение массы материала, приводящее к нарушению точности дозировки, но они более удобны, чем весовые, при изменении массы подаваемого материала.
Чаще всего фосфат дозируют с помощью весовых дозаторов. Простейший ив них (пойдометр), снабженный механическим питателем (шибером и заслонкой), представляет собой ленточный транспортер, натянутый на барабаны (рис. III.1, а). Рабочая часть транспортера опирается на весовой ролик, который связан системой рычагов с весовым коромыслом. С помощью регулирующей заслонки устанавливают расход фосфата, поступающего на ленту дозатора из бункера, /ак,. чтобы коромысло весов с грузом слабо колебалось около среднего положения. При отклонении от этого положения регулировку производят заслонкой. Системы с механическими питателями обладают большой инерционно- -стью, а значит малой точностью дозировки (±1—2%). При автоматизации дозатора точность дозирования достигает ±0,5—4%.
Рис. III.1. Ленточный весовой дозатор с механическим (а) и вибрационным (б) питателями:
/ — ленточный транспортер; '2— бункер питателя; 3 — горизонтальный шибер; 4 — вертикальный шибер; 5 — заслонка; 6 — коромысло весов; 7—ролик весов; 8 — опора; 9 — электродвигатель; 1'0 — вибрационный питатель; // — вибратор; 12— электрический регулятор.
71
Большую точность дают ьссосие дозаторы с вибрационными питателями (рис. III.1,6) вследствие их незначительной- инерции. Ленточный транспортер такого дозатора одним своим концом подвижно закреплен на ошоре, а другим опирается на весы. Материал подают на ленту при помощи электровибрационного питателя и уравновешивают на ленте грузом, перемещающимся по коромыслу весов. Электрический регулятор питателя действует так, что подача материала увеличивается при малой нагрузке и уменьшается при избытке дозируемого материала.
.Подачу материала ленточными дозаторами с вибрацион-
ными питателями регулируют также изменением скорости и движения ленты.
В некоторых применяемых дозаторах количество дозируемого фосфата измеряют по скорости вращения .лентопротяжного механизма.' Количество материала на ленте регулируют заслонкой, связанной с весоизмерительными элементами. Во избежание нарушения дозировки фосфата в результате плохого истечения сырья или.зависания его в бункере предложено электромеханическое устройство, связывающее массу материала и скорость ленты 1[ 10, с. 10].
Для дозирования высокотекучей фосфоритной муки Каратау наиболее пригодны порционные весы периодического действия, которыми через установленные промежутки времени (например, 40—45 с) отвешивают отдельные порции
муки.
Аналогичным способом действуют и объемные ковшевые дозаторы, ковши которых периодически заполняют определенным количеством материала, направляемым на разложение. Более совершенен
Фоссрат
объемный барабанный дозатор (рис. Ш.2), состоящий из цилиндрического корпуса, в котором' помешен вращающийся на валу барабан со сменными стаканами. По мере вращения барабана материал из бункера заполняет отдельные стаканы, которые затем попеременно совпадают с питающей течкой, а материал высыпается..
Равномерность и точность подачи весовыми дозаторами обеспечивается при хорошей сыпучести, по-
Рис. Ш.2. Схема барабанного дозатора: 1 — корпус; 2—барабан; 3 — сменные стаканы; 4, 6 — ребра; 5 — течка дозатора;
7 — привод; 8 — вал.
72
8 сборник В экстрактор кислоты
Рис. III.3. Щелевой дозатор с подвижной переливной воронкой: / — корпус; 2 — уплотнение; 3 — подвижная переливная воронка; 4— расходомерная щель; 5 — профиль щели.
стоянстве состава и свойств материалов. Объемные дозаторы отличаются большей простотой конструкции и надежностью в эксплуатации. Однако настройка их на заданную производительность и регулирование ее в процессе'работы весьма затруднительны.
Северодонецким филиалом НИИХиммаша предложена методика выбора оптимального типа объемных питателей для сыпучих химических’ материалов с учетом их физико-химических свойств, условий эксплуатации и технико-экономических показателей дозаторов [11].
Дозаторы серной кислоты. Для дозирования серной кис-доты служат расходомеры различной конструкции — диафрагменные, сифонные и другие. Наиболее распространены щелевые дозаторы, представляющие собой прямоугольную коробку с перегородкой, в которой имеется вертикальная щель определенного профиля (так называемая щель равного.расхода). Кислота, поступающая в расходомер из напорного бака, устанавливается перед щелью на том или ином уровне, определяющем расход кислоты (рис. III.3). В расходомере устанавливается уровнемер с дистанционной, передачей показаний его_ на контрольный щит. Расход кислоты регулируют при помощи автоматического клапана или вентиля.
<В сифонном дозаторе количество вытекающей кислоты регулируют сифоном, который можно устанавливать на различной высоте. Равномерность расхода .кислоты достигается постоянным уровнем жидкости в расходомере и неизменным положением сифона.
Диафрагменный дозатор — бак с четырьмя трубами. По одной из них, помещенной в предохранительную трубу, кислота поступает в дозатор. Другая—переливная — служит для поддержания определенного уровня жидкости в баке. Подача кислоты должна несколько превышать ее расход, и избыточное количество кислоты сливается по переливной
73
Рис. III.4. Шайбовый дозатор:
1 — напорный бак; —2 — запорный вентиль; 3 — дозирующая шайба.
трубе со* съемным стаканом. Из дозатора кислота вытекает в экстрактор по двум сливным трубам, каждая из которых снабжена диафрагмой—достоянной или сменной. Через трубу с постоянной диафрагмой в экстрактор поступает основное количество кислоты (немного больше требуемого). Остальное ее количе-
ство протекает через трубу со съемной диафрагмой, перестановкой которой регулируют подачу кислоты.
Шайбовый дозатор (рис. III.4) —разновидность диафрагменного — более прост по устройству и надежен в эксплуатации. Он состоит из напорного бака, запорного вентиля и дозирующей шайбы. При одном и том же уровне в напорном баке количество киёлоты, проходящей через шайбу с калиброванным отверстием, постоянно. При изменении расхода кислоты устанавливают другую шайбу, отградуированную на соответствующий расход кислоты. В небольших пределах регулировать расход кислоты можно и при одной и той же шайбе, изменяя уровень жидкости в напорном баке.
Аппараты для разложения фосфата и кристаллизации сульфата кальция
Разложение фосфата и кристаллизация сульфата кальция обычно протекают в одних и тех же аппаратах — экстракторах. Общий их объем определяется необходимым временем пребывания в них реакционной массы (пульпы), а также производительностью системы.
Экстракторы — вертикальные цилиндрические или прямоугольные резервуары большой вместимости, снабженные пропеллерными или турбинными мешалками, вращающимися с частотой 400—'600 об/мин. Они оборудованы вытяжными трубами для газов и паров.
Устойчивые металлические и неметаллические материалы защищают экстракторы, мешалки и трубопроводы от коррозии при действии горячих кислот и других примесей, а также от эррозии, т. е. истирания перемешиваемой пульпой [1—5]. Так, поверхность стальных цилиндрических реакторов покрывают листовым свинцом или полиизобутиленом или же футеруют плиткой из плавленого диабаза в два слоя 74
и кислотоупорным кирпичом толщиной 130 мм (днище аппарата—кирпичом толщиной 250 мм). Железобетонные прямоугольные аппараты футеруют кислотоупорным силикатным и углеродным кирпичом. Кроме того, для футеровки применяют графитовые блоки. В процессе работы футеровка покрывается тонким слоем сульфата кальция, который .защищает ее от истирания.
Крышки резервуаров — разъемные, что облегчает их монтаж. Защищают их гуммированием или футеровкой кислотоупорным материалом.
Газоходы изготовляют из черного металла и гуммируют. Наконечники труб- для барботажа охлаждающего воздуха изготовляют из специальной стали. Трубопроводы и насосы для перекачки пульпы и фосфорной кислоты, валы и лопасти мешалок выполняют из стали ЭИ-943, а для разбавленных кислот—из сталей ЭИ-448 и Х18Н10Т или 0Х23Н28МЗДЗТ. Трубопроводы монтируют с уклоном из коротких участков труб (на фланцах) во избежание отложений сульфата кальция и для удобства разборки, промывки и чистки.
Части трубопроводов, недоступные для чистки, изготовляют из полиэтилена (поверхность его не покрывается осадком), а также из армированной резины.
На некоторых заводах имеются установки небольших масштабов, а также установки, используемые в опытных целях, состоящие из нескольких одномешальных цилиндрических реакторов небольшой вместимости (20—50 м3), соединенных в батарею [12]. Производительность батареи с общим рабочим объемом ~140 м3 составляет ~5 т/ч апатита или ~47 т Р2О5 в сутки (15—16 тыс. т Р2О5 в год).
• На современных заводак большой мощности устанавливают одно- и двухбаковые многомешальные аппараты с интенсивным местным перемешиванием пульпы. В конструкциях экстракторов с объемом 1000 м3 и более стремятся совместить быстрое выравнивание концентраций реагирующих веществ с интенсивным теплообменом путем применения внутренней и внешней циркуляций, секционирования и других приемов [13].
Батарейные (многореакторные) одно- и многомешальные экстракторы. Батарейные экстракционные системы состоят обычно из четырех последовательно расположенных реакторов с общим полевым объемом 320—340 м3 (при объеме каждого 80—85 м3).
Цилиндрические реакторы диаметром 6950 и высотой 4000 мм изготовлены из стали со слоем резины в 5 мм и футерованы в полкирпнча кислотоупорным силикатно-алюми-ниевым и в четверть кирпича—углеродистым материалом. Каждый реактор разделен на два отделения кирпичной пе-75
регородкой, имеющей в нижней части отверстия для передвижения пульпы. Экстракторы сообщаются между собой переточными желобами. В каждом отделении установлено по две двухъярусных мешалки с 12 лопастями каждая и частотой вращения 1500 об/мин.-Мешалки длиной 3230 мм изготовлены из нержавеющей стали; вал мешалок гуммирован.
Эффективность работы батарейных систем и условия их эксплуатации зависят от способа их соединения, интенсивности перемешивания, охлаждения пульпы й других факторов [13].
При верхних перетоках пулыпй из одного одномешалыно-го аппарата в другой увеличивается вероятность проскока непрореагировавших частиц сырья через систему вдоль поверхности пульпы. При установке в аппаратах карманов с нижним перетоком для пульпы в нижней их части осаждаются, а затем затвердевают частицы, что требует частой чистки вертикальных перетоков.
Последовательно-параллельное соединение отдельных аппаратов позволяет при использовании шиберов, расположенных в перетоках между реакторами, ремонтировать аварийный экстрактор без остановки всей системы.
На рис. Ш.5 показана схема батареи, состоящей из четырех многомешальных секционированных аппаратов. В каждом экстракторе установлена диаметральная перегородка с нижними перетоками для пульпы, в результате чего пульпа, поступающая по внешним верхним- перетокам в первую секцию экстрактора, может переходить в следующую его секцию по нижним перетокам. Исходные компоненты (фосфатное сырье, серная кислота и раствор разбавления) подают в первую секцию, а продукционную пульпу откачивают На. фильтрацию погружным насосом из последней секций.
!ешальная система (НИУИФ, ибульский суперфосфатный за-и реакторов с общим рабочим объемом 137 м3 с одной мешалкой в каждом аппарате. Суточная мощность системы по дигид-ратному режиму составляет 45 т Р2О5 из апатитового концентра-
Многореакторная од ЛенНИИГипрохим и Д вод — ДСЗ) состоит из ,1
Рис. III.5. Батарея из^ четырех многомешальных секционных экстракторов: 1 — диаметральная перегородка; 2 — нижний переток в перегородке; 3 — внешний верхний переток; 4 — мешалка; 5 — погружной насос; 6 — отвод пульпы; 7 — подача фосфата.
76
та, а при работе по пдлугидрйтному режиму имеет удвоенную производительность [10, вып. 6, с. 13].
Многореакторная многомешальная система (НИУИФ, ЛенНИИГипрохим, ВПО «Минудобрение» — г. Воскресенск) состоит из 4 реакторов (не секционированных) с общим рабочим объемом 350 м3 и. общим количеством мешалок 13. Она'работает на апатитовом концентрате по полугидратно-* му режиму, с суточной производительностью 250 т Р2О5.
Удельные энергетические мощности при работе по полугидратному режиму составляют на первой системе 0,39 кВт/м3 и 14,4 кВт/м Р2О5, а на второй — 0,68 кВт/м3 и 31 кВт/м Р2О5.
В многореакторной многомешальной системе фирмы JCC (Швеция) с общим рабочим объемом 575 м3 и суточной производительностью 200 кг Р2О5 (из апатитового концентрата по дигидратному режиму) установлены 4 аппарата, снабженные 20 мешалками. В системе фирмы Ргауоп (Бельгия), состоящей из 4 реакторов, разделенных каждый на четыре секции с 4 мешалками, общий рабочий объем составляет 332 м3, а суточная производительность при переработке апатитового концентрата в дигидратном режиме-составляет 166 т Р2О5. При суточной производительности, равной 10'00 т Р2О5, об_-щий объем системы из 4 реакторов, имеющих 11 секций и 1'1 мешалок, составляет 2300 м3.
Батарейные или многореакторные системы громоздки в эксплуатации и для больших мощностей не нашли широкого применения.
Одно- и двухбаковые многомешальные экстракторы. Секционированные цилиндрические аппараты обычно разделяются перегородками на четыре секции с нижними и верхними перетоками для пульпы (рис. Ш.6, а). Исходные реагенты подают в первую секцию сверху; во вторую секцию пульпа переходит по нижнему перетоку и т. д. Из последней секции пульпу отводят через верхний переток погружным насосом на циркуляцию и фильтрование. В перегородке, разделяющей первую и четвертую секции, перетока нет.
Прямоугольный секционированный экстрактор с 8 основными квадратными и 2 вспомогательными секциями имеет рабочий объем 730 м3 (рис. Ш.6, б). В перегородках, разделяющих отдельные секции, . расположены чередующиеся нижние и верхние перетоки, так что пульпа совершает''зигзагообразный путь. Фосфатное сырье, серную кислоту, раствор разбавления и циркулирующую пульпу подают в первые секции. Из двух последних вспомогательных секций отводят пульпу на рециркуляцию и фильтрование.
Рассматриваемый экстрактор представляет собой железобетонный прямоугольный аппарат с плоским дном; его размеры: 25,2X11,2x6,45 м; изнутри он футерован в два слоя
77
Ct
Рис. Ш.6. Схема экстракторов: а— цилиндрический с четырьмя секциями; б — прямоугольный; в — цилиндрический без перегородок; г — двухбакбвый без перегородок.
кислотоупорным силикатным и углеродным кирпичом. Экстрактор разделен перегородками из кислотоупорного кирпича на 10 секций — отделений, из которых восемь — рабочих, девятая — карман для установки насосов, а десятая — распределительная емкость, откуда охлажденную 4 вакуум-ис-парителе пульпу направляют частично на фильтрование, а в основном — в первую секцию.
Одни перегородки между рабочими секциями имеют отверстия для .перетока пульпы из предыдущего отделения в последующее, а другие перегородки имеют меньшую высоту для перетока пульпы по верху. Вследствие этого пульпа передвигается по отделениям экстрактора по синусоиде. Каждая рабочая секция снабжена двухъярусной мешалкой (с шестью лопастями в каждом ярусе) высотой 5,4 м и диа-' метром 1,5 м, изготовленной из нержавеющей стали ЭИ-943. Гуммированный вал мешалки вращается с частотой 70— 72 об/мин. Иногда в каждой секции устанавливают по три лопастных или пропеллерных мешалки с удельной мощностью электродвигателя 0,81 кВт/м3 объема реактора. Первые четыре секции (по ходу пульпы) имеют объем 100 м3 каждая, а четыре последние— 150 м3.
В экстракторах с внутренней рециркуляцией пульпы отсутствуют циркуляционные центробежные насосы, что упрощает конструкцию трубопроводов. Одновременно отпадает
78,
надобность в вйкуум-испарйтеле и затратах на Перекачку больших количеств циркуляционной пульпы на барометрическую высоту (более 10 м). Пульпу в этом случае охлаждают обдувом поверхности ее воздухом (через специальные патрубки в крышке экстрактора), подаваемом вентилятором, который отсасывает одновременно газообразные фторсодержащие соединения.
Внутреннюю рециркуляцию пульпы можно создать гидродинамическим взаимодействием потоков друг с другом и с цилиндрической поверхностью стенки резервуара [14]. Для этого в центре экстрактора помещают пропеллерную мешалку, расположенную в диффузоре (внутренняя зона перемешивания), а по периферии несколько турбинных мешалок (рис. III.6, в). При вращении пропеллерной и турбинных мешалок в разных направлениях у стенок экстрактора и в центральной его части, а также вокруг периферийных мешалок образуются замкнутые потоки, имеющие обратные направления. В результате этого время циркуляции частиц фосфата, поступающих в центр экстрактора, достаточно для практически полного их разложения.
Экстрактор указанной конструкции при внутреннем диаметре 13 м и высоте заполнения 3,18 м имеет рабочий объем 425 м3. Один из недостатков такого экстрактора—возможность накопления осадка на дне его в местах, не охватываемых сферой действия мешалок.
Применяют также экстракторы с внутренней циркуляцией— трех- или четырехсекционированные— с вертикальными перегородками и нижними и верхними перетоками. Внутренняя циркуляция пульпы создается нагнетателями пропеллерного типа, расположенными в центре аппарата. Экстракторы с внутренней циркуляцией позволяют оптимизировать температурный режим, быстро устранять' пересыщения и чрезмерный избыток сульфат-ионов в жидкой фазе суспензии; кроме того,, в них можно увеличить степень извлечения фосфорной кислоты на Г—4,5%.
Рабочий объем цилиндрических аппаратов ограничен длиной вала перемешивающего устройства, определяющей высоту слоя пульпы; последний во избежание чрезмерного роста изгибающих напряжений не должен превышать 3,5— 4 м. По этой причине высота аппаратов составляет 4 м, диаметр 12—14 м, а рабочий объем ~450 м3. Лишь в прямоугольном аппарате, в котором одиночная мешалка располо-, жена в центре квадрата, гидродинамические условия ее работы весьма благоприятны; длина вала составляет ~5,4 м, а высота уровня перемешиваемой пульпы — 4,6 м. Поэтому экстракторы большой вместимости сооружают с увеличенной Площадью их горизонтального сечения, а перемешивающие
79
устройства располагают та'к, чтобы зоны эффективного перемешивания.перекрывали в'сю площадь днища.
На типовую систему производительностью ПО—140 тыс. т Р2О5 в год (при' среднем удельном объеме экстрактора -—'2,0—2,5 м3 на 1 т Р2О5 в сутки) аппарат должен иметь рабочий объем — 850 м3, что обеспечивается установкой двух (спаренных) цилиндров (рис. III.6, г) или одного прямоугольного экстрактора.
Однореакторные. многосекционные аппараты разработаны НИИХиммашем для переработки апатита по дигидратному режиму с рабочим объемом в 440 м3 и суточной- производительностью 166 т Р2О5 (1 реактор, 4 секции, 13 мешалок, съем 15,7 кг Р2О5/м3-ч); НИУИФом и ЛенНИИГипро-химом — для полугидратаого процесса на апатите с рабочим объемом 800 м3 и суточной производительностью 420 т РгО5 (1 реактор, 7 секций, 8 мешалок, съем — 22 кг Р2О5 с 1 м3/ч).'
В последнее время за рубежом разработаны для экстракции фосфорной кислоты из фосфористой муки Того и Флориды дигидратным способом однореакторные одномешаль-ныё экстракторы с рабочими объемами 400 и 700 м3 и однореакторный одномешальный экстрактор на 700 м3 (без учета объема аппарата, используемого в качестве гидрозатвора); работающий под вакуумом (фирмы Gulf-Design, США), с внутренней циркуляцией; мощность аппарата 200 тыс. т P2Q5 в 1 г. Разработан и двухреакторный' экстрактор, один из реакторов которого работает под вакуумом (фирмы Kellogg-Lopker, США); общий рабочий объем 400 м3 и мощность 100 тыс. т Р2О5. Он представляет собой безмешальный аппарат с внешней циркуляцией и тангенциальным' перемешиванием.
В настоящее время широкое распространение получили экстракторы с рабочим объемом 900 м3, состоящие из двух круглых корпусов, соединенных перетоком. Размеры корпуса: диаметр 13,3,- высота 9,276 м. .Каждый корпус имеет 8 винтовых мешалок, расположенных по периферии, и одну пропеллерную. Скорость вращения винтовых мешалок п=1Г2 об/мин; а пропеллерных п=140 об/мин. Мощность ме- ’ шалок 55 кВт каждая.
. Для дигидратного процесса* переработки апатита НИУИФом и НИИХиммашем разработаны двухбаковые мно-гомешальные аппараты с общим объемом ИЗО м3, суточной производительностью 450 т Р2О5 при съеме 16,7 кг Р2О5 с 1 м3/ч (2 реактора, 8 секций, общее число мешалок 18). Для этого же процесса существуют аппараты с общими объемами 900 и 1400 м3 с суточными производительностями 470 и 1000 т Р2О5 при съемах 22 и 30 кг Р2О5 с 1 м3/ч,
80
Аналогичные аппараты для экстракции фосфорной кис- лоты в дигидратном процессе из фосфоритной муки Каратау и других месторождений имеют рабочий объем 850 и ИЗО м3 - с суточной производительностью 223 и 300 -т Р2О5 при съеме 10,7—10,9 кг Р2О5 с 1 м3/ч. Резкое уменьшение в этом слу-' чае производительности системы (в ~2 раза и более) объ-; ясняется особенностями экстракции фосфорной кислоты из фосфоритов — интенсивной инкрустацией аппаратуры и ком-: муникаций и необходимостью частых остановок для их чист- ки (см. стр. 111).
Дйухбаковые реакторы, по-видимому, являются аппаратами, в наибольшей степени соответствующими технологиче-’ ским особенностям процесса экстракции фосфорной кислоты. , В так называемых расчлененных экстракторах (многореак- -; торных или многосекционных) степень пересыщения жидкой фазы пульпы сульфатом кальция в первом аппарате или первой секции, куда подается фосфатное сырье, весьма ве-' лика [15]. Это приводит к затруднениям при разложении ( сырья (шламование) и получении хорошо отфильтровываю-щи'хся кристаллов. В отличие от этого в двухбаковом экс-, тракторе с циркуляционным перемешиванием в первом ап-; парате ликвидируется пересыщение, достигается гомогенность пульпы и изотермичность процесса, а во втором—>про-; исходит дозревание пульпы с завершением формирования : кристаллов гипса и полугидрата с оптимальными размерами ; и фильтрующими свойствами.
В связи с широким развитием экстракции фосфорной кислоты из низкосортного сырья НИИХиммашем планируется . разработка высокоинтенсивного оборудования, в частности железобетонных герметичных реакторов вместимостью до 2000 м3 [10, вып. 12, с. 21]. Такие экстракторы оснащают высокоинтенсивными перемешивающими устройствами с приводами КРП; пульпа в них охлаждается воздухом, пена, гасится механически. В этих условиях отпадает надобность в : вакуум-испарителях для охлаждения пульпы, теплообменниках, насосах 9ПХП, вакуум-насосах ВВН-50 ц другом технологическом оборудовании. •
Фильтрование экстракционной пульпы
Отделение и промывку фосфогипса производят на ленточных, конвейерно-лотковых и карусельных вакуум-фильтрах. Одно из главных требований к фильтрам — обеспечение хорошей отмывки гипса от фосфорной кислоты при наименьшем расходе воды.
В старых схемах для фильтрования применяли барабанные вакуум-фильтры. В этих условиях необходима репуль-пация (размешивание) осадка с водой и промежуточными 6-5473 81
i •
растворами для промывки его. Основные недостатки этих схем—।громоздкость, сложность обслуживания ['16], трудность защиты от коррозии и малая поверхность фильтрования.
В настоящее время для фильтрования фосфорной кислоты используют высокопроизводительные вакуум-фильтры наливного типа — ленточные, конвейерно.-лотковые и, в особенности, карусельные. Они позволяют совмещать основную операцию — фильтрование — с промывкой осадка; что уменьшает число фильтратов. Ленточный фильтр состоит из двух барабанов, укрепленных на горизонтальной сварной металлической станине, на которых натянута бесконечная лента [16, 17]. Один из барабанов — ведущий — соединен с приводным механизмом, другой — ведомый, натяжной. Их изготовляют из стали с гуммированной поверхностью.
Ленту с рифленой поверхностью выполняют из специальных сортов резины. Она может двигаться со скоростью 3— 8 м/с. Плоские борта ленты на верхнем горизонтальном участке ее движения специальными направляющими отгибаются вверх; в результате лента принимает форму желоба, наполняемого суспензией и промывными водами. При движении ленты верхняя ее ветвь скользит по столу фильтра-, состоящему из золотниковой решетки, вакуум-камер и боковых направляющих. Лента на наружной поверхности имеет ребра с промежуточными канавками, образующими дренажную полость. На ребра укладывают фильтровальную ткань. Осевые сквозные отверстия в ленте соединены с отверстиями золотниковой решетки, по которым фильтрат проходит в вакуум-камеру.
Фильтровальную ткань крепят на ленте резиновыми жгутами (шнурами), уложенными в продольных пазах ленты. Зоны фильтрования, промывки и подсушки осадка разграничивают резиновыми или матерчатыми перегородками, свободно подвешенными над лентой на рейках. Лента поддерживается опорными роликами. Натягивают ее винтовым устройством, смонтированным на оси натяжного барабана и на раме фильтра.
Суспензия поступает на фильтр из лотка и по мере движения ленты происходит отделение из нее жидкой фазы и промывание остающегося на ленте осадка. Промытый и подсушенный осадок перемещается к ведущему барабану, где осадок при повороте ленты растрескивается, отделяется от ткани и сбрасывается в транспортирующее устройство. Освобожденную от осадка фильтровальную ткань промывают водой (регенерируют). Промывную венду, содержащую некоторое количество осадка {10—30 г/л), собирают в корыте и удаляют.
82
Ленточные вакуум-фильтры отличаются высоким съемом фильтрата и осадка с единицы фильтрующей поверхности (вследствие отложения в первую очередь крупных частиц осадка), легкой регенерируемостью фильтрующей ткани, простотой конструкции, легкостью защиты от коррозии и удобством обслуживания. Однако на этих фильтрах недостаточно разделены отдельные фазы фильтрования и промывки осадка, что увеличивает расход воды на промывку и уменьшает концентрацию продукционной фосфорной кислоты. Кроме того, они имеют относительно небольшую производительность вследствие ограниченной площади фильтрующей поверхности. Общая длина фильтра с поверхностью фильтрования 10 м2 составляет 13,36 м. Удобство фильтрования и промывки полугидрата — преимущества при использовании ленточных вакуум-фильтров в полугидратном процессе по сравнению с другими фильтрами.
' Хорошее разделение фаз при фильтровании достигается применением конвейерно-лотковых фильтров;—кроме того, при этом уменьшается расход воды для промывки [16]. Фильтр состоит из ряда прямоугольных лотков с бортами, смонтированных в виде непрерывной ленты. Борта (радиальные стенки) соседних лотков (путчей) —общие, в результате чего образуется .непрерывное кольцо из фильтровальных ячеек. Эти фильтры изготавливают диаметром до 4 м при поверхности-фильтрования до 12 м2.
Наибольшими достоинствами обладает получивший широкое распространение карусельный вакуум-фильтр (рис. III.7) [13, 16]. Он состоит из 24 отдельных лотков длиной 1,9 м, шириной 0,9 м у внутреннего и 1,2 м у наружного концов и глубиной 0,2 м. На их днищах уложена фильтровальная ткань. Лотки установлены на каретках с колесиками, движущимися по круговым рельсам. С помощью двух шайб, образующих головку фильтра,—подвижной (вращающейся вместе с лотками) и неподвижной—' фильтраты отсасываются в соответствующие вакуум-сборники через эластичные шланги, соединяющие лотки с головкой фильтра. Обе шайбы головки имеют ряд отверстий. Подвижная шайба вращается по неподвижной, и отверстия одной поочередно совпадают с отверстиями другой. При этом происходит попеременное сообщение с соответствующими сепараторами (и вакуум-насосами), соединенными с отверстиями неподвижной шайбы. Так происходит последовательное отделе^ ние первого и других (промывбчных) фильтратов.
После того как осадок прошел зоны фильтрования и промывки, каждый лоток порознь автоматически опрокидывается для выгрузки лепешки осадка. В этот момент отверстия в обеих шайбах головки совпадают с линией подачи сжатого воздуха от вентилятора: осадок отдувается и он попадает в
б*
83
Фильтрат 2
промывку ткани
• Рис. III.7. Схема карусельного вакуум-фильтра.
бункер. После промывки ткани сильной струей воды лоток вновь возвращают в первоначальное рабочее положение, соединяют с вентилятором; при этом происходит подсушка фильтровальной ткани; затем лоток снова заливают пульпой.
Цикл работы каждого лотка состоит из фильтрования, обезвоживания осадка, двух или более промывок осадка с промежуточным его обезвоживанием, разгрузки фосфогипса и промывки ткани. Во время фильтрования, промывки и обезвоживания осадка лоток соединен с источником вакуума; при разгрузке осадка, сопровождаемой опрокидыванием лотка — с источником сжатого воздуха и в период промывания ткани — с атмосферой. Суспензия и промывная жидкость поступают равномерно по всей длине фильтро-. вальной перегородки лотка из специальных дозирующих устройств. Поверхность фильтрования равна 40—100 м2.
Благодаря тому что отдельные лотки изолированы друг от друга, можно получить концентрированный основной фильтрат, что позволяет также производить многоступенчатую противоточную промывку минимальным количеством воды. -
Для фильтрования применяют ткани ив синтетических волокон— лавсановые (арт. 136), хлориновые, полипропиленовые нетканые (арт. 931506) и др., которые обладают механической прочностью и химической стойкостью в фосфорной кислоте, содержащей 20—45% Р2О5 и более при 80—90 °C.
После первой зоны фильтрования осадок сульфата кальция содержит значительное количество (до 50%) фосфорной 84
кислоты, которую отмывают вначале промывными растворами, а затем теплой водой, подаваемой на последнюю зону фильтра. Промытый осадок с влажностью 18—35%, содержащий незначительное 'количество фосфорной кислоты ,(~1% от общего ее количества), почти на всех заводах направляют в отвал по канатной дороге, вывозят на автомашинах или разбавляют водой (до отношения Ж:Т=10— 12:1) и откачивают насосами в шламоотстойники. В последних твердая масса осаждается, а осветленную жидкую фазу возвращают в производство на разбавление очередной партии осадка.
Утилизация больших количеств сульфата кальция, образующихся в производстве фосфорной кислоты, не получила до сих пор широкого распространения. В настоящее время . подготовлены к промышленному использованию различные способы переработки сульфата кальция (1фосфогипса и фос-фополугидрата) с получением следующих продуктов: высокопрочного гипсового вяжущего и изделий из него; серной кислоты и извести; серной кислоты и цемента; гранулированного фОсфогипса. в качестве минерализатора при производстве цемента и добавки при помоле цементного клинкера; средства, используемого при мелиорации солонцовых почв; сульфата аммония и карбоната кальция для известкования кислых почв.
Фильтрат после промывки осадка водой (четвертый при трехступенчатой промывке и четырехфильтратной схеме фильтрования), содержащий 5—7% Р2О5, используют для промывки осадка в третьей зоне. В свою очередь фильтрат >с третьей зоны (третий фильтрат) направляют на промывку во вторую зону, откуда полученный второй фильтрат подают в качестве раствора разбавления в экстракционную систему. Все фильтраты по выходе из фильтра поступают в соответствующие сепараторы, в которых происходит отделение паровоздушной смеси от. жидкости. Затем фильтраты стекают в сборники, откуда их насосами откачивают по назначению.
Паровоздушная смесь, выделившаяся в сепараторах, поступает в барометрический конденсатор, орошаемый водой, откуда конденсат вместе с охлаждаемой водой стекает через гидрозатвор в канализацию. Воздух из барометрического конденсатора отсасывают вакуум-насосом.'
Описанная схема фильтрования экстракционной пульпы связана с установкой фильтров (и экстракторов) на барометрической высоте (11—12 м) и неравномерной работой обычных насосов при недостаточном заполнении барометрических труб. В отличие от этого в так называемой каскадной схеме экстракционная система и вакуум-фильтровальная ' аппаратура расположены на небольшой высоте и материалы
85
передвигаются самотеком от подачи пульпы до вывода в отвал отфильтрованного и промытого осадка [49]. При этом значительно снижаются не только затраты энергии, но и сокращается высота зданий и обслуживающих площадок.
Фильтраты перекачивают при помощи вертикальных 'вакуум-насосов, присоединенных непосредственно к сепараторам. Последние одновременно служат и сборниками фильтратов. Паровоздушную смесь из сепараторов отсасывают мокрым (водокольцевым) вакуум-насосом. Однако сооружение и эксплуатация технологических систем каскадного типа связаны с необходимостью создания серии типоразмеров на-сОсов для перекачки жидкостей и пульп при разрежении [20].
Газообразные фторсодержащие соединения, выделяющиеся в различных аппаратах, отсасывают по вытяжным трубам вентилятором и улавливают в поглотительной системе с получением кремнефтористоводородной кислоты (см. стр. 192),
Дополнительное оборудование
В производстве экстракционной фосфорной кислоты нормальная работа основного технологического узла экстрактор—фильтр обеспечивается совершенством’ конструкций и высокими эксплуатационными показателями многочисленного дополнительного оборудования, начиная с подачи сырья и кончая отгрузкой продукции.
Воздух, используемый в системе пневмотранспорта сырья, перед выбросом в атмосферу очищается от уносимой им пыли в аспирационной системе, состоящей обычно из циклона, рукавного фильтра и вентилятора1 [8, первая ссылка]. Циклоны обычно устанавливают по одному на каждый силос конструкции НИИОГаз — ЦН-15 диаметром 0,4—0,5 м с общей высотой 4,5 м и гидравлическим сопротивлением 0,9—1 кПа (90—100 мм вод. ст.). Температура поступающих в циклон газов должна быть на 15—20 °C выше температуры -конденсации влаги из газа. Это предотвращает налипание на внут-’ ренней поверхности циклона влажной пыли.
Рукавные фильтры служат для очистки газа после циклона. Из изготовляют из шерстяных, хлопчатобумажных и синтетических 'тканей, и имеют они по несколько секций, поочередно отключаемых для разгрузки пыли. Фильтры типа МФЦ имеют автоматическое устройство для встряхивания и обратной продувки фильтровальной ткани. Для встряхивания рукавов используют электродвигатель А02-32-6, мощностью 2,2 кВт и частотой вращения 1000 об/мин.
Весьма эффективна и удобна очистка пылевыбросов в мокрых пылеуловителях (скрубберах) с возвратом образую-86 /
щейся загрязненной пылью жидкости в технологический процесс.
Воздух просасывается через аспирационную систему с помощью вентилятора, например типа ВД-42 производительностью 25 000 м3/ч и напором 2 кПа (200 мм вод. ст.); для привода вентилятора в действие служит электродвигатель А02-82-8 мощностью 30нВт с частотой вращения 1700 об/мин. (Указанные характеристики, взяты в качестве примера и для разных заводов могут существенно изменяться по типам и характеристикам.)
вентиляторы [21] используют не только для просасыва-ния воздуха; их применяют для отсоса газов, выделяющихся в экстракторе; для отдувки лепешки и осушки ткани при фильтрации и др. Приводим некоторые данные о наиболее часто используемых типах вентиляторов:
Ц9-67 № 3; производительность 1500 м3/ч; напор 2 кПа (200 мм вод. ст.); привод от электродвигателя АО-54-4 мощностью 4,5 кВт; частота вращения п=4500 об/мин;
Ц9-57; производительность 3000 м3/ч; напор 1,5 кПа (150 мм вод. ст.); привад от электродвигателя А0-62-4 мощностью ГО кВт; п =4460 об/мин;
В.МН-15; производительность 50 000 м3/ч; напор 5,8 кПа (580 мм вод. ст.); привод от электродвигателя мощностью 160 кВт;
В Д-4 3,5; производительность 30000 м3/ч; напор 3,2 кПа (320 мм вод. ст.); привод от электродвигателя мощностью 100 кВт; п=4000 об/мин;
ВНЖ-43,5; производительность 40 000 м3/ч; напор 5,2 кПа (520 мм вод: ст.); привод от электродвигателя мощностью Г25 кВт; п —1000. об/мин.
Корпус и рабочее колесо вентиляторов изготовляют из нержавеющей стали 0323Н28МЗДЗТ (ЭИ-943). В некоторых случаях турбину (рабочее колесо) делают открытого типа с двумя опорами на концах вала для уменьшения отложений осадков и облегчения очистки турбины.
Насосы. В производстве фосфорной кислоты транспортируют большие массы серной и фосфорной кислот, фильтраты, растворы' кремнефтористоводородной кислоты, а также пульпу сульфата кальция в фосфорной кислоте. Для этого используют центробежные консольные насосы [22] и на.сосЫ погружного типа [23].
Первые (табл. III.1) применяют для подачи, кислот, не содержащих значительных количеств твердых взвесей, серной и кремнефтористоводородной кислоты, а также воды, потребляемой в разных целях. Их обычно устанавливают на одном уровне со сборниками и работают они под заливом. Регулирование работы насосов достигается дросселированием в напорном трубопроводе.
87
Таблица III.I !
Характеристики некоторых консольных центробежных насосов [8, вторая ссылка]
Марка * Сталь Производительность, м3/ч Напор, м Мощность электродвигателя, кВт
ЗХ-4И-1 Х23Н28МЗДЗТЛ 45 . 90 40
ЗХ-6И-1 То же 45 54 22
ЗХ-9И-1 5Х20Н25МЗД2ТЛ 45 = 7- 31 13
4АХ-ЗИ-1 Х23Н28МЗДЗТЛ 45 54 40
4АХ-5И-1 То же 45 31 22
4АХ-6И-1 5Х20Н25МЗД2ТЛ 45 21 10
5АХ-5И-1 Х23Н28МЗДЗТЛ 90 40 55
* Частоты вращения у насосов марок ЭХ — 3000 об/ 5АХ — 1,500 об/мин. мин, 4АХ и
Практика работы на отдельных предприятиях требует непрерывного- усовершенствования насосио-го хозяйства и применения в ряде случаев новых типов и конструкций.
При перекачке фосфорной кислоты продолжительность работы консольных насосов составляет 2000—2500 ч. Но для перекачивания растворов кремнефтористоводородной кислоты срок их службы сокращается в 2,0—2,5 раза. 'Низкую продолжительность работы в этих условиях (800—1000 ч) имеют также гуммированные насосы ЗХ-9Р и 6Х-9Р. g то же время срок службы графитопластовых насосов ЗХ-9Г-2а, ЗХ-42Г-2а. (производительность 45 м3/ч) и 4Х-Г2Г-2а. (производительность 90 м3/ч) при перекачивании растворов HzSiFg составляет более 6 мес.
Погружные насосы применяются при под-аче пульпы из экстрактора на охлаждение в вакуум-испаритель (например, 9ПХП-9И, производительность 600 м3/ч), для подачи пульпы ’ на фильтр (например, 5ПХП-9И, производительность 120 м3/ч) и при перекачке из сборников продукционной-кислоты и фильтратов (например, ЗПХП-5Н производительность 60 м3/ч).
Насосы, используемые для перекачки фосфорной* кислоты, изготавливаются только- из высококачественной нержавеющей стали. Например, насосы ЗПХП-5И (производительность 45 м3/ч) с напором ЗГ м изготовляют из стали Х23Н28МЗДЗТЛ, а насосы 5ПХ-9И-7 — из стали 0Х23Н28МЗДЗТЛ.
При эксплуатации насосов возникают нередко значительные трудности из-за их конструктивных недостатков, а также нарушения правил обслуживания. Например, в некоторых случаях в погружных насосах 9ПХП-Г2И выходит из строя 88
нижний подшипник и в него попадает агрессивная среда [24]. В результате увеличения давления на сальниковый узел циркуляционных насосов 0ХГ8-55И (на стадии выпаривания кислоты) охлаждающая жидкость проникает в подшипник и выводит его из строя. Это можно устранить повышением давления запорной воды в сальниковом уплотнении, заменой некислотоустойчивой сальниковой набивки на кислотоустойчивую (УСФ-19).
Несоответствие производительности насосов реальным материальным потокам приводит к необходимости их замены или установки электродвигателей с другой мощностью. Низкое качество резиновых чулок на винтовых насосах типа 1В80 приводит к преждевременному выходу их из строя.
Вакуум-насосы [25] предназначены для отсасывания воздуха, отходящего, после очистки газообразных потоков из ва-куум-испарителя и из вакуум-фильтра.
Широкое распространение в производстве экстракционной фосфорной кислоты получили водокольцевые вакуум-насосы типов КВН и ВВН, а также струйные вакуум-насосы (эжекторы).
Водокольцевые насосы просты по конструкции, быстроходны, непосредственно соединены с электродвигателем, мало засоряются и способны создать разрежение до 90—96%. В связи с возможностью проскока с отсасываемым воздухом кислых капель и брызг> они дол'жны быть изготовлены из нержавеющей стали.
Реже применяют эжекторы, представляющие собой трубу Вентури с соплом, через которое подается рабочий газ или пар, засасывающий при адиабатическом расширении удаляемые газы или. воздух. Они просты по устройству, по обладают низким к. п. д. и высоким расходом пара. Для создания высокого вакуума требуется установка трех—четырех последовательно работающих эжекторов.
Водокольцевые вакуум-насосы марок ВВН-3 (ГОСТ 10889—64) и КВН-8, имеют производительность (при условиях всасывания) 0,05 и 0,66 м3/с при разрежении. 70%, предельное разрежение — 90 и 82%, мощность электродвигателя 7,5 и 2,8 кВт, частота вращения — 24,3 с-1.
Для перемешивания пульпы применяют лопастные, пропеллерные и турбинные мешалки, изготавливаемые, как правило, из спецстали 0Х23Н28МЗДЗТЛ [26]. На старых заводах небольшой мощности еще встречаются лопастные меФалки с частотой вращения 30—40 об/мин, .вал и лопасти 'которых изготовлены из углеродистой стали и. гуммированы. „Кроме того, на лопасти одевают сменные резиновые чулки. •’При хорошем обслуживании такие мешалки работают без ремонта до полугода.
89
Пропеллерные мешалки, подобные гребному винту или винту самолета, имеют частоту вращения 100 об/мин и более.
‘Современные экстракторы (объемом 740 м3 и больше) снабжены высокочастотными (120—400 об/мин) турбинными мешалками, изготовленными из опецстали указанной выше марки и приводимыми каждая во вращение через редуктор от мотора. Большие мешалки, установленные в рабочих секциях,. приводятся во вращение от электродвигателя АО2-9Г-4, мощностью 76 кВт с частотой вращения 1000 об/мин. Малая турбинная мешалка приводится во вращение от электродвигателя АО-62-8 мощностью 7 кВт, снабженного редуктором' ВО-1П-7/75:11000 и частота вращения ее ЮОО об/мин.
Во всех случаях, применяют мешалки с выносными индивидуальными приводами (со стандартными электродвигателями и типовыми редукторами), устанавливаемыми на крышке аппарата. В качестве приводов применяют цилиндрические редукторы, с планетарными зубчатыми передачами. Конструкция их 'такова, что устанавливать их можно только вертикально. В зависимости от диаметра вала мешалки и потребляемой мощности высота мотора-редуктора со стойками и расположенными в ней муфтами достигает 1,5— 2,0 м.
Вращающиеся валы мешалок уплотняют при помощи сальников, торцевых и бесконтактных уплотнений. Универсальные типовые уплотнения типа УТ изготовляются из нержавеющей стали Х18НЮТ. Для смазки и охлаждения служит проточная вода, циркулирующая в полости кожуха' уплотнителя, или нейтральная смазка типа НТ. Для защиты ротора и подшипников привода'от коррозионной среды в верхнюю часть привода подают инертный газ.
Отношение диаметра аппарата (секции) к размеру перемешивающего устройства (лопасти, пропеллера, турбины) не превышает .3—4. Этим определяется количество мешалок и их расположение в аппарате. Определены также нормализо-' ванные значения соотношений между расстоянием мешалки от основания аппарата и ее диаметром, между шириной лопаток турбинных мешалок и их диаметром и др. (ММ 5874-66, РТМ 144-66 и РТМ 145—66).
Работу мешалок характеризуют интенсивностью и эффективностью. Последние зависят от типа и размеров мешалок, частоты вращения, а также от физико-химических свойств перемешиваемых пульп (вязкости, плотности, содержания твердой фазы и др.) и конструктивных особейностей аппарата — наличия внутренних устройств, формы днища и т. д. Под интенсивностью перемешивания понимают время достижения требуемого технологического результата (гомогенизации пульпы, степени разложения фосфата, изотермич-.
90
ности среды и др.)' при одной и той же частоте вращения для сравниваемых устройств. Эффективность мешалки определяется минимумом затрат энергии для достижения заданного технологического процесса.
В секционированных многомешальных аппаратах требуемый эффект (циркуляция пульпы и гомогенное распределение потоков) достигается как в результате. взаимодействия создаваемых отдельными мешалками потоков со стенками, так и за счет сложения потоков и контуров движений, создаваемых отдельными мешалками. При противоположном направлении вращения соседних перемешивающих устройств создаваемые отдельными мешалками потоки складываются, что приводит к интенсивной циркуляции внутри экстрактора. При вращении в одном направлении создаваемые отдельными'мешалками потоки разрушаются, что может вызвать осаждение твердой фазы.
II 1.3. РАЗЛИЧНЫЕ ТЕХНОЛОГИЧЕСКИЕ СХЕМЫ
В отечественной и зарубежной технике применяют разнообразные схемы производства фосфорной кислоты, различающиеся масштабами, используемыми конструкциями аппаратуры, ее компоновки, затратами энергии-и другими характеристиками.
Независимо от режима—дигидратного или полугидрат-ного — аппаратурное оформление процесса должно удовлетворять одинаковым требованиям, обеспечивающим полноту разложения сырья и кристаллизацию сульфата кальция в условиях минимального пересыщения им жидкой фазы.
Помимо физико-химических параметров (температура, концентрация, присутствие в растворе H2SiF6, ионов А13+ и других, уменьшающих степень пересыщения раствора сульфата кальция) эффективность процесса определяется точностью и синхронностью дозировки реагентов, рациональным распределением потоков фосфата и серной кислоты, созданием оптимального концентрационного и температурного полей в системе путем охлаждения массы реакционной смеси, интенсивного ее перемешивания и т. д.
При недостаточно интенсивном перемешивании пульпы на дне экстрактора накапливается осадок, что может привести к уменьшению (на 10——15%) реакционного объема аппарата и сокращению времени реакции. Во избежание забивания осадком нижние перетоки в секционированном аппарате должны иметь большую протяженность и значительное поперечное сечение.
Газы, выделяющиеся в производстве фосфорной кислоты, улавливают на различных стадиях процесса, а кислые стоки нейтрализуют или регенерируют..
91
На рис. III.8 показана одна из широко распространенных схем производства фосфорной кислоты. Фосфат разлагают в экстракторе с рабочим объемом 730 м3, а пульпу отфильтровывают на карусельном вакуум-фильтре с поверхностью 80 м2. Суточная производительность одной технологической нитки при переработке апатитового концентрата по дигид-ратному режиму, при отношении в пульпе Ж: Т= (Г,6—4,7) :1 составляет 330 т Р2О5 или 37 т/ч апатитового концентрата. Эти данные и все приводимые ниже относятся к дигидрат-ному процессу. При работе полугидратным способом они должны быть увеличены на ~25—30%. Продолжительность пребывания пульпы в экстракторе составляет 7—7,5 ч.
Отвешенный автоматическими весами апатитовый концентрат (35—38 т/ч) поступает в первое отделение экстрактора. Серную кислоту. (33—35 т/ч в расчете на моногидрат) с концентрацией 92,5% (или 75%-ную H2SO4), разбавленную до 55—57% и охлажденную в графитовых холодильниках до 50—70 °C, подают через распределительную коробку в первые три отделения экстрактора, Сюда направляют также раствор разбавления, содержащий 22—24% Р2О5 (55— 57 м3/ч) и имеющий плотность 1,24—4,26 г/см3. Его получают-смешиванием части основного фильтрата (оборотного раствора) со вторым фильтратом/
Подача в первые три секции охлажденных потоков серной кислоты (до 55—57 ЧС), раствора разбавления (до 50— 55 °C) и в первую секцию циркулирующей пульпы (до 65— 70 °C) отводит теплоту реакции и поддерживает оптимальные температуры реакционной массы в экстракторе (в первом его отделении 70—74 °C, в восьмом 68—72 °C и в десятом^ 65—69°C). Циркулирующая пульпа охлаждается в ва-куум-испарителе при разрежении 76—84 кПа (570—6'30 мм рт. ст.) на 3—5 °C и возвращается на экстракцию черев десятую распределительную секцйю экстрактора в количестве 1900—2000 т/ч (кратность циркуляции 12—1'3).
Вакуум-испаритель—стальной, гуммированный цилиндрический аппарат диаметром 4,5 и высотой 0,6 м с коническим футерованным днищем. Пульпу в него подают двумя вертикальными погружными насосами (производительность каждого 600 м3/ч при напоре 12 м), установленными в девятой секции — кармане экстрактора. Насосы, изготовленные из нержавеющей стали, имеют высоту 3,5 и диаметр 1,0 м. Они приводятся во вращение с частотой 750 об/мин электродвигателем мощностью' 90 кВт.
. *По выходе из испарителя охлажденная пульпа поступает по барометрическому трубопроводу в десятую распределительную секцию экстрактора. Отсюда часть пульпы (455— 160 т/ч) откачивают погружным насосом на вакуум-фильтр. Количество пульпы, поступающей на фильтр, регулируют 92
Ватмоссреру
Апатит
.'23
/2
30
Ча абсорб
32
10
17
20
9
или в отвал
А ' 42
42
53
47
4S
Рис. III.8. Схема производства экстракционной фосфорной -кислоты: 1— склад апатита; • 2— затворы; 3, 21 — циклоны; 4, 22 — рукав-
ные фильтры; 5, 25 — винтовые питатели; 6— компрессор; 7, 24, 26, 44, 45—бункеры; 8, 38, 39 — ваку-ум-насосЫ; 9 — хранилище серной кислоты; 10, 17, 20, 31, 36, 47, 53 — погружные насосы; 11, 14, 15 — напорные баки; 12 — смеситель; 13, 16 — холодильники; 18, 19 — сборники серной кислоты; 23 — вентилятор;
27 — ленточный дозатор; 28 — экстрактор; 29, 32 — распределительные коробки; 30— испаритель; 33 — промывная башня; 34, 41 — барометрические конденсаторы; 35 — циркуляционный бак; 37, 42, 52 — бакн для конденсата; 40— карусельный фильтр; 43 — сепаратор; 46 — ленточный транспортер; 48 — бак для фосфорной кислоты; 49—51 — сборники фильтратов.
J[-18 19
74
12
21' 22; 2
24
25
О и fr
I
На пер
еработку
автоматическим индукционным расходомером, установленным на .пульпопроводе и связанным с уровнем пульпы в экстракторе. Избыток пульпы из распределительного отделения перетекает в восьмую секцию экстрактора.
Одновременно с водой в паровую фазу вакуум-испарите-ля выделяются и соединения фтора, которые отмывают в промывной башне, орошаемой циркулирующим • раствором 2%-ной H2SiF6. Промывная башня имеет высоту 1'2,5 и диаметр 3,9 м. Кислоту 'разбрызгивают через форсунки, установленные в верхней части башни в четыре ряда, по семь форсунок в каждом ряду. Затем пары воды конденсируются в поверхностном конденсаторе (диаметр 1,6 и высота 7,8 м), охлаждаемом водой (600—700 м3/ч). После этого паровоздушная смесь вакуум-насосами выбрасывается в атмосферу. Окончательно газообразные фторсодержащие соединения из аппаратуры всей системы—из сборников фильтратов, экстрактора, распределительных коробок, после вакуум-насо-сов, а также напорных баков оборотных растворов улавливают в абсорберах Вентури, абсорберах с плавающей насадкой (АПН) и других (см. гл. VI).
Фильтрование пульпы и отмывку фосфорной кислоты из осадка проводят при разрежении 66—.800 кПа (500—ООО мм рт. ст.). Карусельный вакуум-фильтр с поверхностью фильтрации 80 м2 имеет высоту 4,75 и диаметр 15,64 м. Он приводится во вращение электродвигателем., мощностью 5,5 кВт.
Вакуум на вакуум-испарительной установке и в системе вакуум-фильтра создают вакуум-насосы типа ВВН-50-Х, изготовленные из нержавеющей стали марки ЭП-567.
На некоторых отечественных заводах применяют систему воздушного охлаждения реакционной массы в экстракторе. В данном случае пульпа, в экстракторе охлаждается за счет насыщения отсасываемого из экстрактора воздуха водяными парами при прохождении его над -поверхностью пульпы. Количество отсасываемого из экстрактора воздуха определяется количеством теплоты, снимаемой в экстракторе, и степенью насыщения воздуха водяными парами. Для системы экстракции мощностью^ НО—14.0 тыс. т Р2О5 в год количество отсасываемого воздуха составляет 1'00—Г50 тыс. м3/ч.
За рубежом фосфорную кислоту получают по той или иной технологической схеме с использованием различной аппаратуры. В ОША наиболее распространены [30] способы фирмы Ргауоп и Dorr-Olivar. Способ бельгийской фирмы Ргауоп используют* в США с 1950 г. Фосфорит разлагают как в серии экстракторов, так и в одном миогосекционном аппарате. Способ фирмы Ргауоп с одним экстрактором широко применяют в США, Франции и других странах.
Производительность технологической линии с одним бетонным прямоугольным экстрактором (ЙЗХ10Х6 м) с рабо-94 - '
чим объёмом 820 м3, футерованным кислотоупорным кирпй-чом и графитовыми блоками, составляет 450 т Р2О5 в сутки. При отношении Ж:Т=(1,65—l,7):il на суточное производство 1 т Р2О5 необходимо 1,83 м3 рабочего объема экстрактора, 0,56 м2 производственных помещений с объемом 4,64 м3.
В Англии применяют также цилиндрические четырехсекционные реакторы диаметром 12 м и высотой 6 м, изготовленные из железобетона с толщиной наружной стенки 350 мм. Реактор покрыт внутри натуральным каучуком - и футерован углеродистым кирпичом на фурановой смоле. Суточная производительность реактора 220 т Р2О5.
Метод фирмы Dorr—Oliver с одним экстрактором конструкции типа «цилиндр в цилиндре» используют в США [27J. Экстрактор состоит из двух цилиндров—наружного диаметром- 14,9 м и внутреннего диаметром 4,9 м. Суточная производительность экстрактора — 295 т Р2О5. Фосфат, оборотная фосфорная кислота с фильтра и 94%-ная H2SO4 поступают в кольцевое пространство экстрактора, образованное внутренней поверхностью наружного цилиндра'и наружной поверхностью внутреннего цилиндра. Смесь серной и оборотной фосфорной кислот впрыскивают форсункой. Время нахождения пульпы в кольцевом пространстве ~8 ч; охлаждают ее воздухом. Из кольцевого пространства пульпа перетекает во внутренний цилиндр (центральную секцию) через отверстия в нижней его части, где она находится в течение ~4 ч. Из внутреннего цилиндра пульпа поступает в сборник, откуда ее направляют на фильтрование.
В последнее время в США расширяется производство фосфорной кислоты по способу Kellogg—Lopker [31] с применением безмешальных экстракторов (рис. III.9). Реакционная система состоит из двух аппаратов — растворителя и вакуум-иопарителя. В растворитель поступает фосфат, пеногаситель, а из испарителя — сильно разбавленная в циркулирующем потоке пульпы' серная кислота. Концентрацию свободной серной кислоты поддерживают в пределах 2— 2,5%. Фосфат разлагается в растворителе при атмосферном давлении, Ж:Т=(1,5—2):4 и 70—75°C в течение 2—2,5 ч. Циркуляцию пульпы между растворителем и испарителем' создает осевой насос; в испаритель пульпа поступает тангенциально. Сюда же вбрызгивается серная кислота, которая немедленно диспергируется в массе реакционной пульпы. В испарителе происходит отвод теплоты реакции за счет испарения воды. Он соединен с барометрическим конденсатором, в котором конденсируется большая часть водяных паров, и вакуум-насосом, откачивающим несконденсировавшие-ся пары и газообразные фторсодержащие соединения в абсорбционную систему.
95
о
I . .IE ПГ
Рис. III. 9. Схема фирмы Kellogg—Lopker:
I — подготовка руды; II— реакционная система; III—'фильтрация; I — молотковая мельница; 2— фосфат;. 3 — элеватор; 4 — грохот; 5 — хранилище-бункер; 6 — питатель; 7 — пеногаситель; 8— растворитель-экстрактор; 9 — испаритель; 10 — скруббер на выходной линии; 11— к вакуум-системе; 12 — подача серной кислоты; 13 — сброс в атмосферу; 14 — циркуляционный насос; 15—насос, питающий фильтр; 16—фильтр; 17—подача воды; 18— отжатый гипс; 19 — продукционная фосфорная кислота;
20 — циркулирующая фосфорная кислота.
♦' Достоинство (процесса—возможность использования сырья грубого помола и точного регулирования и контролирования кристаллизации гипса. При тщательном контроле за концентрацией серного ангидрида, подачей реагента и температурным режимом (разница температур пульпы в экстракторе и испарителе не более 2 °C) степень разложения фосфата составляет 98,5—99,5%, а сульфат кальция выделяется в виде крупных однородных кристаллов.
Пульпу на фильтрование отводят насосом из циркуляционной линии в питающую емкость фильтра; фильтрование происходит на ленточных фильтрах при суточной производительности до 700 т Р2О5 или на карусельных фильтрах — при мощности 1000—1(500 т Р2О5. При трехкратной промывке осадка он содержит ~2'5% влаяи и не более 0,5% Р2О5.
Указывают [28], что за счет простоты оборудования эксплуатационные расходы уменьшаются на ~40% по сравнению с обычными системами. При этом образующиеся осадки являются рыхлыми, легко удаляемыми за Г2 ч циркулирующей горячей водой при остановке системы 1 раз в 4 недели.
Технологический выход Р2О5, т. е. степень перехода. Р2О5 из сырья в фосфорную кислоту ((Квых) составляет (без учета механических потерь) 95—96% для апатитового концентрата и 71—94% для разных сортов фосфорита. Его опре-- деляют (в %) по общему содержанию Р2О5 в отбросном осадке сульфата кальция (фосфогипсе)
' 96
^hx-ioo- Г - too
Р2О5 фосф
где Р р2о5 ОбЩ — общее содержание Р2О5 в осадке (фосфогипсе), %; Г — выход сухого осадка на единицу фосфата , (для дигидратного процесса — гипсовое число), ч. (масс.); А’205фосф — содержание Р2О5 в сухок фосфате, %.
Технологический выход обычно на 2—>3% ниже коэффициента извлечения Р2О5 в раствор (Лизвл)- Это объясняется недостаточной отмывкой фосфогцпса или фосфополугидрата от фосфорной кислоты.
Коэффициент извлечения Р2О5 в раствор (в %) определяется по содержанию водонерастворимой Р2О5 в осадке:
(/'п п - - Рр ) 1'
1 2'5 оощ г2иа водораств^ <
А113ВЛ - IM Р ’
1'2°5 фосф
В зависимости от фосфатного сырья Лизвл находится в пределах 95—99%.-
Коэффициент отмывки фосфогипса или фосфополугидрата (Котм, в %), характеризует потери водорастворимой РгО5 в отбросном осадке:
Рр.,о_ г'100
Ко™ - 100 -
^Р2О5 фосфЛи-вл
•100
В оптимальных условиях КОТм = 97—99%.
Очевидно, что технологический выход (в %) определяется, в свою очередь, значениями Дизвл и Котм по формуле:
Квых — КизвлКотм/1'00
Переход Р2О5 в раствор (Кизвл) не однозначно характеризует степень разложения фосфата из-за протекания вторичных процессов (выделение фосфатов железа из пересыщенных растворов и др.). Поэтому КраЗЛ (коэффициент, характеризующий степень разложения фосфата) определяют по отношению СаО: SO3 в осадке. Если вся содержавшаяся в фосфате СаО связана с SO3 (или в осадке имеется SO3 более, чем связано с СаО), то фосфат разложился нацело — весь кальций фосфата превращен в сульфат кальция.
Следовательно, при отношении СаО: SO3 в осадке, 'равном или меньшем 0,7 (0,7 — отношение молекулярных масс СаО и SO3 в Ca^Qt), коэффициент разложения фосфата составляет 1.00%. Большее, чем 0,7 отношение СаО : SO3 свидетельствует о недоразложении фосфата
z ' -4>азл = г °'" -- Ю0
£caO/\SO3
где Ссао и Cso3 — содержания СаО, и SO3 в осадке, %.
7-5473 ' 97
Стехиометрическую норму серной кислоты, т. е. количе ство массовых частей моногидрата H2SO4 на 100 ч. (масс.) фосфата (GK)*, определяют из общего содержания СаО i фосфате
^|см= 1,75С',„
где С'сао — содержание СаО в фосфате, %.
Если концентрация серной кислоты С (в %), а коэффи циент избытка ее (т. е. отношение фактического количествг к стехиометрическому) составляет а, то действительный рас ход кислоты G'K составит:
О'/ = 1,75ССаэ~- • 100
Кратность циркуляции пульпы N определяют из отноше ния
N = F цирк/Е ОТВ, где Ецирк и Еотв — количество пульпы, циркулирующей и отводимой на фильтрацию.
Обычно задают значение N и псГ нему вычисляют количество циркулирующей пульпы.
Количество воды W, необходимое для отмывки фоофогип-са, определяют по рассчитанным или установленным экспе-' рименталыно исходным показателям с использованием уравнения материального баланса
1004-S+ Г=Ф+1 оог+ FK+ G
где S— расход исходной серной кислоты; Ф — количество продукционной фосфорной кислоты; WK — содержание жидкой фазы в отбросном промытом осадке; G —количестве выделяющихся газов (SiF4 и СОг) и водяного пара.
Количество продукционной фосфорной кислоты определяют с учетом./СВЬ1Х, установленного опытным путем
р
ф __ р2°5 фосф д.-
4 р ^ВЫХ
^2^5 ф.’ к-ты
-где Рр2о5ф к_ты-содержание Р2О5 в фосфорной кислоте, %.
При отсутствии опытных данных G следует вычислить из баланса
100+5+/? = EOTB+G,
где R—количество раствора разбавления для поддержания заданного отношения Д:Т в пульпе (n); FOTB= 100Г(/г-|-1).
Баланс- фильтрования и промывки фосфогинса составляют по экспериментальным данным.
* Все соотношения приводятся из расчета массовых частей вещества, приходящихся на 100 ч. (масс.) фосфата.
9?
1П.4. ДИГИДРАТНЫЙ СПОСОБ »
Условия проведения процесса
Технически фосфорную кислоту впервые получили более 100 лет назад разложением низкокачественных фосфоритов, содержащих значительные количества соединений трехвалентных металлов, разбавленной (5—10%-ной) серной кислоты, в которой соединения железа и особенно алюминия переходят в раствор в незначительной степени.
Раствор с концентрацией 8—10% Р2О5 упаривали до содержания в нем ~40%* Р2О5. При* разложении фосфатной породы более концентрированной (30-МО%-ной) серной кислотой выделяются игольчатые кристаллы гипса. Они удерживают значительное количество жидкой фазы и плохо промываются. Вследствие этого потери Р2О5 велики.
Существенным шагом вперед в производстве фосфорной кислоты был переход к установкам непрерывного действия и разбавление 75 и 93%-ной H2SO4 не водой или слабыми промывными водами, а раствором фосфорной кислоты, т. е. проведение процесса с применением раствора разбавления. В этих условиях выделяютс-я ромбические кристаллы гипса, которые хорошо фильтруются и отмываются.
В настоящее время дигидратным способом производят фосфорную кислоту с содержанием 20—25% Р2О5 (обычно из низкосортного сырья — бедных фосфоритов) и 30—32% Р2О5 (из высококачественного сырья — апатитового концентрата и др.).
Как было указано выше (см. стр. 54), гипс устойчив только при соприкосновении с растворами фосфорной кислоты, над которыми давление пара больше, чем над твердым CaSO4-2H2O‘при данной температуре. В области устои- ' чивого существования гипс выделяется из растворов фосфорной кислоты непосредственно в' виде первично кристаллизующейся фазы. Но эта область расположена при относительно низких температурах (см. рис. 11.16 и 11.17), так что гипс находится в равновесии с раствором фосфорной кислоты, содержащей ~20% Р2О5 при £<50°С, а при концентра-_щии ~30% Р2О5— при ^<35 °C. При наличии примесей, в частности свободной H2SO4, устойчивая область гипса сокращается в направлении понижения температуры его равновесия с растворами. .
> В этих условиях скорость разложения фосфата мала, и вследствие большой вязкости кислоты отделение ее от осадка весьма затруднительно. Поэтому экстракцию ведут при более высоких температурах, т. е. когда дцгидрат метаста--билен и кристаллизуется из первоначально выделившегося полугидрата.
s 7* _ > . 99
Практическое значение имеют температуры выше 60 °C. При более низких температурах гидратация чполугидрата идет медленно. При 65—70 °C в растворах, содержащих до 32—33% Р2О5, полугидрат быстро превращается в- гипс (стр. 57).
Если, например, при 80 °C в растворах, содержащих 10— 25% Р2О5, превращение тТолугидрата в гипс происходит в течение 1—5 ч, то при концентрации 32% Р2О5 в течение значительно более длительного времени в твердой фазе находится полугидрат, а затем появляется даже, ангидрит. Поэтому получение такой кислоты дигидратным способом возможно при температуре несколько ниже 80 °C. Кроме того, при содержании в кислоте бол Ре 32—33% Р2О5 уменьшаются размеры выделяющихся кристаллов гипса. Это затрудняет фильтрование кислоты и отмывку осадка.
Крупные изометричные и однородные кристаллы дигидрата образуются при незначительном (20—40%) пересыщении раствора; При колебании температуры выделяются неоднородные по размерам кристаллы. Образованию более крупных кристаллов способствует поддержание в растворе небольшого избытка сульфат-ионов (плюсовый режим). При наличии в растворе избытка ионов кальция выделяются очень тонкие игловидные кристаллы [29].
На рис. III. 10 кривой ААг условно представлено изменение растворимости сульфата кальция в растворе фосфорной кислоты по мере ее экстракции. Область пересыщения раствора сульфатом кальция с избытком сульфат-ионов, в которой образуются кристаллы гипса с оптимальной структурой, показана схематически (штриховая кривая). Она распространена более вправо и менее влево от прямой jMiMj — линии стехиометрических отношений СаО: SO3 в растворе (соответствующих сульфату кальция). Правее линии ЛЕИ] расположены растворы, содержащие избыток сульфат-ионов, а левее — избыток ионов кальция.
Содержащиеся в высококачественном фосфатном сырье (польском апатитовом концентрате, флоридских, алжирских, марокканских и аналогичных других фосфоритах) незначительные количества примесей практически не влияют на
л
Рис. Ш.10. Схема , изменения растворимости сульфата кальция в растворах фосфорной кислоты (^4^41) в зависимости от концентрации серной кислоты:
ММ;—линия стехиометрических отношений СаО : SO3 в растворе; штриховая кривая ограничивает область пересыщенные растворов (при небольшом избытке сульфат-ионов), из которых выделяются изометрические кристаллы гипса.
100
описанные выше условия выделения дигидрата сульфата из фосфорной кислоты.
Однако при использовании бедных фосфоритов, содержащих менее 30% РгО5 и значительные количества соединений железа и алюминия, магния, щелочных металлов и других, растворимого кремнезёма, rfepacTBopHMoro остатка и органических веществ, существенно изменяется состав образующейся фосфорной кислоты и ее свойства, а также фильтрующие свойства получаемой суспензии (пульпы). Поэтому при одном и том же практически аппаратурном оформлении технологические режимы осуществления процесса, его интенсивность и надежность, а также качество получаемой кислоты (концентрация Р2О5, содержание примесей и т. д.) существенно отличаются для разных видов сырья.
Производство фосфорной кислоты из апатитового концентрата
Из апатитового концентрата дигидратным способом в настоящее время производят фосфорную кислоту, содержащую (в %): Р2О5 — 38—32; SO3—1,8—2,8; СаО —0^2—0,4;
ЁегОз — 0,8—Г, 1; AI2O3 — 0,5—4,0 и F—1,5—1,9, а также некоторые количества взвешенного гипса. Процесс осуществляют в весьма больших масштабах. Суточная производи- ' тельность действующих цехов составляет 300—1000 т Р2О5 при производительности одной технологической нитки до 300 т Р2О5 и более. Изыскиваются способы увеличения суточной производительности аппаратов до 1000 т Р2О5 и более (см. стф. 81).
Показатели технологического режима. Температуру реакционной массы поддерживают в начале экстракции 72— 75 °C, иногда 75—77 °C, а в конце 65—68 °C. В диапазоне температур 68—75 °C обеспечивается рост кристаллов и выделение гипса оптимальной структуры. Этому способствует также использование затравки — твердой фазы, находящейся вциркулирующей продукционной пульпе. .
Кратность циркуляции определяется отношением количества возвращаемой в процесс продукционной пульпы (например, 1600 т/ч) к количеству отводимой на фильтрование (например, 160 т/ч). Циркуляция большого количества пульпы (при кратности циркуляции 10—12) .позволяет не только поддерживать оптимальный температурный, режим в экстракторе, но в качестве затравки приводит также к уменьшению пересыщения жидкой фазы сульфатом кальция и обеспечивает рациональное отношение SO3: СаО в растворе.
'Серную кислоту (моногидрат) расходуют из расчета 100% от стехиометрической нормы, необходимой для разложения фосфатной части сырья, а также реагирующих с кис
101
лотой примесей. Подачу серной кислоты и апатитового концентрата регулируют так, чтобы концентрация ее в жидкой фазе пульпы (в свободном виде) составляла 1,5—2,5%. Массовое отношение жидкой и твердой фаз (Ж: Т) в пульпе находится в пределах (2,5—3):1. Она создается соответствующим 'Количеством раствора разбавления (с концентрацией -22-24% Р2О5 и плотностью 1,24—1,26 г/см3) —смеси второго фильтрата с частью продукционной кислоты — и рассчитывается по отношению к количеству образующётюся ди-гидрата сульфата кальция (гипсу). Из 1 т апатитового концентрата образуется 1,6 т гипса (гипсовое число).
Пульпа находится в реакторах 6—7 ч. За это время апатит разлагается на 97,5—98%. Отфильтровывают пульпу и промывают осадок на карусельном вакуум-фильтре (стр. 83) по четырехфильтратной схеме. Образующиеся фильтраты содержат Р2О5: первый (основной) 30—32%, второй 22—25%, третий 14—18% и четвертый 4—6%. Фильтр с рабочей поверхностью фильтрования 80—100 м2 работает с производительностью 700—750 кг/(м2-ч) сухого осадка.
Образующийся слой фосфогипса толщиной 50—60 мм окончательно промывают нагретой до 55—70 °C водой, которую падают насосом через дозатор с постоянным уровнем и диафрагму на фильтр. Средний расход воды на промывку фосфогйпса ~ 1200 л/т сухого осадка или ~ 750—800 кг/т апатитового концентрата. Степень отмывки фосфорной кис-, лоты из осадка составляет 99%. Промытый фосфогипс содержит 40—42% влаги. В осадке после основного (первого) фильтрования содержится жидкой фазы 50—52%,’После второй 48—50% и после третьей ~45% жидкой фазы.
В сухой массе фосфогипса в среднем находится, - %: СаО — 39,6; SO3 — 35,8; P2Os— 1.1 (в‘-том числе водораство- . римый Р2О5 —0,25); Fe2O3 — 0,08; А12О3 — 0,02; К —0,03; Na—J0,Г; нерастворимый остаток —7,0. Удельная поверхность фосфогйпса 0,48—0,25 м2/т.
На 1 т Р2О5 образуется 5,8—6 т-влажного фосфогйпса.
• . 5’~=""Для Производства 1 т Р2О5 в сутки необходимо 2,2 м5' ~ реактора, 0,7 м2 производственных помещений, объем которых 585 м3.
С 1 м3 экстрактора снимают 18—20 кг Р2О5/ч, а с 1 м2 Поверхности фильтра — 180—200 кг/ч.
Усовершенствование и рационализация отдельных элементов процесса на разных предприятиях позволяет не только стабилизировать технологический режим, но и повысить его эффективность. Это, помимо улучшения механического обслуживания аппаратуры., относится к оптимизации как - ' разложения фосфатного сырья и кристаллизации сульфата кальция, так и фильтрования пульпы.
102
Например, при пространственном разделении подачи фосфата и серной кислоты с предварительным разбавлением последней циркулирующей пульпой в первых секциях экстрактора и поступлением фосфата в последующие одну-две секции увеличивается степень разложения сырья и размеры выделяющихся кристаллов дигидрата. Разложение апатитового концентрата серной кислотой, предварительно разбавленной всем количеством циркулирующей пульпы до концентрации 2—10% H2SO4, протекает практически нацело в течение не более 4 ч, а производительность фильтрации фосфо-гипса при трехкратной промывке составляет более 1500 кг/ч с 1 м2 поверхности фильтра [30].
Интенсификация процесса может быть достигнута путем оптимизации раздельно осуществляемых в 'двух реакторах разложения фосфата и кристаллизации сульфата кальция [5]. Фосфатное сырье разлагают в первом реакторе серной и оборотной фосфорной кислотами в присутствии циркулирующей пульпы при 65—70 °C и небольшом избытке SO3 в жидкой фазе пульпы. Во втором реакторе, предназначенном в основном для снятия остаточного пересыщения жидкой фазы сульфатом кальция, добавляется небольшое количество серной кислоты при несколько меньшей интенсивности перемешивания пульпы и температура поддерживается на 5—15 °C ниже, чем в первом. В результате’этого на второй стадии образуются более однородные кристаллы дигидрата, что приводит к увеличению на 15% производительности фильтров и реакторов. Выход Р2О5 при этом возрастает на 0,5—0,8%.
В целях регулирования кристаллообразования сульфата кальция при экстракции используют сточные растворы (от производства криолита), содержащие- ~0,1% A1F3 и 0,1 — 0,2% NaF, 'а также отбросные загрязненные фосфатными соединениями растворы кремнефтористоводородной кислоты.
Материальный баланс. В представленном ниже материальном балансе не учтены механические потери, которые составляют обычно 3%. Он также не включает количество циркулирующей пульпы, которое в процессе не изменяется.
Материальный баланс экстракции на 1 т апатитового концентрата
Приход Количество, !КГ ' Расход Количество, кг
Апатитовый концентрат (39,4% Р2О5) 1000" Фосфорная, кислота (32% Р2О5) 1181
в том .числе: в том числе:
р2о5 394 Р2О5 378
СаО 320' СаО 4
фтор 30 SOs 22,9
н4о 10 фтор - 18
103
др оч и е 46 . Н2О 737,3
Серна'я кислота (93% H3SO4) в том числе: , прочие Фосфогипс (влажный) в том числе: 26,5 2667
SO3 742,9 Р2о5 16
НгО Вода (для разбавления 235,6 СаО SO3 207 6
кислоты и промывки фосфогишса) 2173,7 фтор Н2О прочие Фтористые газы в том числе: фтор прочие Испарившаяся вода: при фильтрации в вакуум-испарителе 165 13'12 17 6,0 2.1 32 264
Всего 4152,2 Всего 415'2,2
Выход фосфорной кислоты составляет (378 : 394) -100« «96%. Увеличить его можно в основном за счет повышения степеней разложения апатита в процессе экстракции и отмывки фосфорной кислоты из фосфогипса. Апатид разлагается не полностью при нарушении режима работы экстракторов —1 уменьшении продолжительности пребывания пульпы в экстракторе при зарастании, перетоков в перегородках гипсом, нарушении температурного режима и недостаточной интенсивности и перемешивания пульпы.
Повышение температуры пульпы объясняется недостаточной кратностью циркуляции пульпы (при забивании всоса на погружном насосе или пульпопровода к ва^ум-испари-телю) и низким разрежением в вакуум-испарителе. Большое значение имеет также соблюдение необходимой концентрации SO3 и точная дозировка поступающих реагентов. Недостаточное перемешивание пульпы, помимо уменьшения степени разложения апатита, приводит к засорению перетоков и снижению температуры пульпы.’
Плохое отмывание фосфорной кислоты из фосфогипса происходит тогда, когда на последнюю стадию подают недостаточное количество воды с низкой температурой. Это может произойти из-за незначительного разрежении и недоброкачественного состояния фильтровального полотна (закупоривания пор, наличия повреждений, ткань плохо промыта и не просушена).
Увеличение степени отмывки фосфорной кислоты из фосфогипса достигается при использовании 93 %-ной HgSCh (вместо 75%-ной) для экстракции фосфатов. Это позволяет улучшить баланс воды в технологическом процессе и ввести большее количество воды для промывки гипса. При этом уменьшаются потери фосфорной 'кислоты с фосфогипсом, идущем в отвал, и облегчаются процессы фильтрования и
104
промывания (однако увеличение концентрации серной кислоты не изменяет концентрации получаемой фосфорной кислоты, которая предопределяется оптимальными условиями кристаллизации гипса, рассмотренными выше).
Большое значение имеет распределение основных компонентов и, примесей сырья по отдельным стадиям производства и получаемым продуктам. Это предопределяет качество (чистоту) продукционной кислоты, степень утилизации образующихся побочных продуктов—фтористых и других соединений, а также обеспечение санитарных условий труда.
Переход основных компонентов апатитового концентрата (Р2О5, СаО и фтора) в конечные продукты можно оценить приближенно по данным, приведенным выше.
Обычно (по средним данным; см. также стр. 192) фтористые соединения, содержащиеся в апатитовом концентрате, на 70—80% остаются в жидкой фазе и на ~Ф0% удаляются с осадком. В газовую фазу выделяется незначительное количество фтористых соединений (10—15%). В отмытом фосфогипсе содержится небольшое количество фтора (0,07— 0,2%) в виде неразложившегося апатита, а также водорас-твопимых соединений в неотмыто<й фосфорной кислоте.
Распределение примесей по фазам сильно зависит от со-’ става применяемого фосфатного сырья, в особенности от содержания в нем кремнезема, окислов щелочных и трехвалентных металлов. В фосфорной кислоте (31,8% Р/Эб), полученной из фосфорита, содержащего, в %; Р2О5— 34,42; СаО —49,4; SiO2 —5,08; R2O3— 1,5; Na2O — 0,35; SO3 — 0,76; F—'3,52; CO2 — 3,41, остается только 25—30% F от общего его количества в сырье; при этом ~30%F выделяется в газовую фазу и 40—45% осаждается с гипсом [31]. Большее выделение фтора в газовую фазу в этом случае и меньший переход его в кислоту, чем при использовании апатитового концентрата, объясняется значительным содержанием кремнезема при ограниченном количестве окислов щелочных металлов в сырье. Соответственно этому распределяется по фазам и кремнезем.
Основная масса кремнезема, содержащегося в сырье, переходит на 85—90% в фосфогипс. В газовую Фазу выделяется в вице кремнефтористых соединений ~10% и в продукционной кислоте остается 3—5% SiO2 от первоначального его количества в сырье.
Технико-экономические показатели. На 1 т Р2О5 в готовой кислоте расходуется обычно 2,64—й,7 т апатитового концентрата и 2,4—2,6 т моногидрата серной кислоты.
Стоимость сырья на производство фосфорной кислоты экстракционным способом, составляет 70—80% от общих затрат. В зависимости от стоимости сырья, а также степени, его использования себестоимость экстракционной фосфорной
105
Таблица III.2
Сравнительная калькуляция себестоимости производства экстракционной фосфорной кислоты из апатитового концентрата (на 1 т Р2О5)
Затраты Завод № 1 Завод № 2
1 расход, т 1 цена за 1 т, руб. сумма расходов, ру J- расход, т цена за 1 т.. руб. * сумма расходов, руб.
Сырье: апатитовый концеи- 2,61 19,52 51,53 2,71 21,80 59,08
трат (39,4% Р2О5) серная кислота 2,44 17,51 42,73 2,58 • 16,05 41,36
(100%-ная H2SO4) Вспомогательные мате- — — — 1,64
риалы Отходы — —- . — — — 1,36
Всего за вычетом отхо- — — 94,26 — — 100,72
«ов Энергетические затраты: электроэнергия, кВт-ч 128 0,014 1,80* 132 0,0138 1,82
пар, Мкал 0,184 4,81 0,89 0,164 4,75 0,78
вода, м3 40,3 0:0064 0,26 47,2 0,013 0,61
сжатый воздух, тыс./м3 0,67 2,13 1,43 —
Зарплата с начисления- — — 0,69 — — 1.12
ми . Амортизация — — 6,42 — 6,50
Цеховые расходы -t- — 5,60 — — 5,70
Общезаводские расходы — — 2,27 — — 1,34
Итого себестоимость 113,62 118.59
кислоты „колеблется от ~ 110 до 120 руб. и выше ' на 1 т
Р2О5. . '
В табл. III.2 приведена калькуляция себестоимости' экстракционной фосфорной кислоты (в пересчете на 1 т Р2О5) для двух заводов с мощностью первого в два раза большей, чем второго.
Производство фосфорной кислоты из фосфоритов
При использовании фосфоритов, в состав которых входит значительное количество нерастворимых примесей (кремнезем, силикаты и другие), получают более разбавленную кислоту (20—25% Р2О5), чем из апатитового концентрата, вследствие необходимости применения для отмывки осадка большого количества воды. При наличии в фосфоритах растворимых соединений магния, железа, алюминия и других
106
последние также переходят в кислоту и отрицательно сказываются на кристаллизации сульфата кальция.
Растворимость фосфата железа невелика (менее 2% при 70—80 °C в 16—20%-ной Н3РО4), и вначале образуются пересыщенные растворы, из которых постепенно осаждаются фосфаты железа (см. гл. II). В результате этого часть экстрагированной Р2О5 теряется. В присутствии 2—3% сульфат-ионов фосфорнокислые растворы, содержащие фосфаты железа, устойчивы длительное время, если они получены из природного фосфата, в котором массовое соотношение Fe2O3: РА <0,1'2 [31].
Фосфаты алюминия обычно не выделяются в твердую фазу вследствие их большей растворимости в фосфорной кислоте и меньшего содержания окиси алюминия в фосфатном сырье, чем окислов железа. Предварительное прокаливание фосфоритов при 850—4'050 °C уменьшает скорость растворения Fe2O3 и А12О3 в фосфорной и серной кислотах.
При переработке различных фосфоритов расход фосфорита (в расчете на 1 т Р2О5 в кислоте) составляет от 3,3 до 6 т, а серной кислоты от 2,6.до 4,1 т. Расходные коэффициенты при переработке фосфоритов больше, чем при переработке-апатита: по фосфа-ту в 1,5—2,3 раза (ню РА, содержащейся в фосфате, в 1,02—1,27 раза) и по серной кислоте в 1,2— 1,7 раза [в зависимости от количества примесей на 1 ч. (масс.) РА в фосфате].
Использование традиционных методов экстракции, аналогичных применяемым для богатых видов сырья, для переработки бедных фосфоритов приводит к снижению мощности установки на 30—45% и соответственному увеличению капитальных вложений и эксплуатационных затрат [32].
Особенно значительные трудности возникают при переработке магнийсодержащих фосфоритов, вследствие осложнения кристаллизации сульфата кальция, высокой степени нейтрализации получаемой кислоты и снижения качества образующихся удобрений.
Затраты на переработку магнезиального фосфатного сырья в экстракционную, кислоту (и затем в аммофос) возрастают от 110—120 руб. на 1 т Р2О5 при отношении MgO: P20f равном 6%, до 220;—230 руб. при отношении, равном 20% [10, вын. 9, с. 4]. • -
Суммарные приведенные затраты на производство аммофоса при оценке стоимости фосфатного сырья в размере 50 руб /т Р2О5 составляют в зависимости от отношения MgO к Р2О5 в сырье, равного 6 и 20%, соответственно 260—270 и ~400 руб /т Р2О5. В последнем случае приведенные затраты на производство аммофоса из экстракционной кислоты и из термической, становятся соизмеримыми.
107
В отечественной промышленности освоено производство фосфорной кислоты из фосфоритов Каратау и разработана технология применительно к чилисайским фосфоритам. Ведутся лабораторные и опытные исследования экстракции фосфорной кислоты из фосфоритов вятского месторождения и др.
Фосфориты Каратау, в том числе и флотационный их концентрат содержат значительное 'количество нерастворимого остатка и соединений магния..При полном переходе последних в кислоту в виде сульфата магния концентрация сульфатов в жидкой фазе пульпы будет больше, чем это необходимо для выделения кристаллов сульфата кальция оптимальных размеров и формы. Регулирование концентрации сульфат-ионов возможно путем распределения подачи серной кислоты по разным точкам системы.
При этом в первом реакторе процесс протекает при содержании в жидкой фазе пульпы 2—2,5% SO3, обеспечивающем образование крупных кристаллов сульфата кальция. Затем разлагается оставшийся фосфорит, и гипс откладывается на образовавшихся в первом реакторе крупных кристаллах сульфата кальция. При обработке фосфорита вна-' чале оборотным раствором при 30—60 qC Стечение 5—20 мин, а затем серной кислотой возможно осуществить процесс при отсутствии в жидкой фазе чрезмерного избытка сульфат-ионов [33].
В 1969 г. был введен в эксплуатацию первый крупный завод по экстракции фосфорной кислоты из фосфоритов Каратау. Первоначально технологический режим был установлен в расчете на переработку флотационного концентрата или богатой фосфоритной муки (см. стр. 21). Процесс протекает в цилиндрическом четырехсекцйонном экстракторе вместимостью 600 м3 (с рабочим объемом .400 м3), снабженном'четырьмя турбинными и одиннадцатью лопастными мешалками. Оптимальная температура (75—80 °C) обеспечивается подачей в первую секцию экстрактора циркулирующей экстракционной пульпы, охлажденной в вакуум-испари-теле. Сюда же поступает фосфорит. В первую и вторую секции ’ экстрактора подают охлажденную 55— 65%-ную H2SO4, а также раствор разбавления, состоящий из части продукционной кислоты (первого фильтрата с содержанием 23% Р2О5) и второго фильтрата (10—12% Р2О5). Массовое отношение жидкой и твердой фаз (Ж:Т) в пульпе равно (2,5—3):4 (в расчете на дигидрат). Экстракцию ведут при концентрации свободной серной кислоты в жидкой фазе пульпы 3—4%. При. взаимодействии реагентов в течение 5—7 ч из фосфорита извлекается 95—96% Р2О5. Съем Р2О5 с 1 м3 реактора составляет 9—10 кг/ч. *
108
Съем кислоты с. 1 м2 поверхности фильтра составляет 100—1И0 кг Р2О5/ч; съем сухого дигидрата с 1 м2 поверхности фильтра ~600 кг/ч. При расходе ~4200 кг воды на 1 т сухого осадка коэффициент отмывки из него фосфорной кислоты составляет 98%. Таким образом, выход Р2О5 в кислоту (продукт) составляет 94%. Образующиеся четвертый (1% Р2О5) и третий (7—<8% Р2О5) фильтраты используют для промывки осадка во второй и первой зонах.
Из флотационного концентрата Каратау, содержащего 28% Р2О5 и ~2% MgO (с остатком на сите в ТОО меш не более 10—42%), получается кислота состава, в %: Р2О5 — 23—25; SO3 —3,8—4; Fe2O3 — 1,4—1,5; MgO—1,5—1,9; F — 1,6—1,8 [34, 35].
Наличие во флотационном концентрате органических реагентов затрудняет ведение технологического процесса из-за выбросов пены на крышку экстрактора [35]. В результате этого нагрузка на экстрактор по сырью снижается на .25— 50%.
В последующем для получения экстракционной кислоты в промышленных условиях перешли на богатую фосфоритную муку состава, в %: РгО5— 28,0; СаО — 43,4; R2O3 — 2,0—2,05; СО2 —5,3; MgO — 2,45; Na2O —0,4; К2О — 0,55; Н2О — 0,48 и F — 2,9. Образующаяся фосфорная кислота содержит в среднем, в %: Р2О5— 22,4; SO3 — 3,1; СаО — 0,5; MgO — 2; R2O3 — 0,9 и F — 2,1.
Описанный технологический процесс положен в основу разработанной НИУИФом системы экстракции фосфорной' кислоты из .богатой фосфоритной муки Каратау (28% РгО5), мощностью 136 тыс. т Р2О5 в год [36]. Она состоит из двух (спаренных) экстракторов с рабочим объемом каждого 850 м3 и из трех карусельных вакуум-фильтров с полезной фильтрующей поверхностью 80 м2 каждый.
В дальнейшем в 1971—4972 гг. НИУИФом, Алмалыкским химическим и Джамбульским суперфосфатным заводами процесс был усовершенствован со значительным увеличением производительности системы.
По новому усовершенствованному режиму температура в экстракторе поддерживается в интервале 90—.100 °C, -а концентрация SO3 в пульпе снижена до 1,8—2,5% (вместо 3,0— 3,5%) [36, 37]. Это обеспечивает протекание процесса при концентрации 22—25% Р2О5 в области существования мета-стабильного дигидрата и вследствие увеличения растворимости сульфата кальция кристаллизацию последнего из раствора с меньшей степенью пересыщения. В результате этого гипс выделяется,в твердую фазу в виде крупных однородных и изометричных кристаллов, фильтрующихся с производительностью 750—780 кг/ч с 1 м2'поверхности фильтра. Этому способствует также уменьшение с повышением температуры
109
вязкости жидкой фазы пульпы й промывка осадка водой с 1 температурой 80—85 °C (вместо 50—60 °C по обычному ре- ’ жиму). ' ' \
'При переработке по этому режиму богатой фосмуки (28% Р2О5) часовой съем Р2О5 с 1 м2 поверхности фильтра увеличивается в ~1,3 раза (до 130—140 вместо 100—110), производительность фильтрования также_ увеличивается в ~ 1,3 раза, степень разложения сырья и выхода РгО6—на •. 1%. '
Вследствие соответственного увеличения производительности оказывается возможным на системах, запроектированных для богатой фосмуки, мощностью 136 тыс. т Р2О5 достичь этой же производительности на усовершенствованном (горячем) режиме и при использовании рядовой руды (24,5% Р2О5 и 3,0% MgO). При этих условиях степень разложения рядового сырья 95—96%, коэффициент отмывки гипса 97—98% и коэффициент выхода Р2О6—92—94%. Съем Р2О5 с 1 м2 поверхности фильтра составляет 110—. 120 кг/ч (вместо' 130—140 кг/ч при переработке богатой руды, содержащей 28% Р2О5 и 2,2 % MgO/.
Продукционная фосфорная кислота содержит, в %: Р2О5 — 20—2’2; SO3 —2—2,5; СаО — 0,3—0,6; MgO—1,8— 2,3; Fe2O30,5—0,7; А12О3 — 0,7—1,0% и F— 1,4—4,7.
На производство фосфорной кислоты требуется (на 1 т Р2О5) фосфатного сырья Каратау (100% Р2О5) —1,12—1,195 т, серной кислоты (моногидрата) — 3,3—3,6 т, электроэнергии— 270—290 кВт-ч, воды— 200—250 м3 и фильтровального полотна — 0,15—0,18 м2. Расходные коэффициенты, на получение из этой кислоты 1 т аммофоса (43% Р2О5 и 11% N) составляют 0,447 т (100% Н3РО4), 0,144 т NH3 (100%), 8500 м3 природного газа [37].
Переработка бедных забалансовых руд Каратау прямой сернокислотной экстракцией, по-видимому, возможна путем дальнейшего снижения концентрации Р2О5 в жидкой фазе пульпы по мере уменьшения Р2О5 и увеличения содержания примесей в руде. Повышение при этом температуры процесса до 1'00—<105°C приводит к значительному удалению фтористых соединений в виде SiF4, что способствует улучшению кристаллизации и фильтрования фосфогипса. Выход Р2О5 в кислоту достигает 90% при степени разложения сырья 85— 90% и отмывке фосфогипса на 98—99% [38]. Так, разложе-нией руды Каратау состава, в %: Р2О5—18,9; MgO — 3,1;
.СаО —32,8; А12О3 —2,1; РегО3 —1,85; SiO2 — 25,2 и F — 2,0.— в лабораторных и опытных условиях при 100—Г05 °C, Т: Ж=,1:(2,5—3), 2—3% SO3 в пульпе в- течение 4—6 ч получена кислота, содержащая 16—18% Р2О5, 3—5% SO3 и 0,5—0,6% F. Производительность фильтрования фосфогипса составила 500—700 кг/(м2-ч) в расчете на' сухой отмытый.
НО
осадок [37]. Но полученная в этих условиях кислота почти полностью нейтрализована и может быть использована для производства только низкокачественных удобрений.
Несмотря на существенные усовершенствования, производство фосфорной кислоты из рядовых фосфоритов Каратау— трудный ^.процесс, осложняемый интенсивной инкрустацией трубопроводов подачи пульпы на карусельный вакуум-фильтр, рабочих органов фильтра — головки, фильтровальных сеток, рессиверов, барометрических груб и коммуникаций первого *и второго фильтратов, а также хранилищ фосфорной кислоты. Это приводит к необходимости остановки системы для чистки (производимой в основном вручную) на 12 ч и более через каждые 7—10 суток работы и относительно кратковременному сроку межремонтной эксплуатации оборудования [10, вып. 10, с. 15].
Отложения, образующиеся в барометрической трубе и головках фильтра, состоят в основном (50—65%) из кремнефторидов калия (преимущественно) и натрия, а также соединений железа в виде FeP_O4-2H2O и РеНз(РО4)2-2,5Н2О (3—11,5% Fe2O3), магния, кальция (в виде сульфата) и др. Увеличенное содержание сульфата кальция наблюдается в отложениях на фильтровальной ткани (до 50—60%) и в хранилищах кислоты (30—40%).
Чилнсайские фосфориты отличаются, помимо низкого содержания Р2О5, большим количеством карбонатов полуторных окислов и нерастворимого осадка. В одиннадцатой пя-• тилетке, кроме основного сырья (хибинский апатит и фосфориты Каратау, содержащие около 24% Р2О5), намечается освоение чилисайского флотационного концентрата, содержащего 22—24% Р2О5.
Фосфорная кислота, полученная из чилисайского флотационного концентрата, характеризуется [36, первая ссылка; . .39] пониженной концентрацией Р2О5 (20—22%); кроме того, для экстракции необходима повышенная норма серной кислоты (103—105% от стехиометрической, считая на СаО в сырье). С увеличением нормы серной кислоты от 100 до 105—106% (от стехиометрии) степень извлечения Р2О5 увеличивается на 2—3% (от 96 до 98,5%). Это объясняется тем, что образующиеся при разложении фосфаты железа в присутствии серной кислоты (2,5—3,5”% SO3) остаются в жидкой фазе длительное время в пересыщенном состоянии. В этих условиях в кислоту переходит 80—85% Fe2O3, содержащейся в сырье. При недостаточном содержании SO3 (до 2%) увеличивается количество фосфатов железа в фосфогипсе, что приводит к уменьшению степени извлечения Р2О5 в раствор.
Оптимальными условиями процесса являются (помимо указанной выше концентрации и нормы серной кислоты):
111
температура пульпы в экстракторе 80—82 °C, продолжительность обработки пульпы 6 ч, массовое отношение Ж: Т в пульпе 2: 1, расход воды для промывки фосфогипса 1000 кг на 1 т сухого осадка при температуре ее 80—85 °C. При этом коэффициент разложения концентрата составляет 95— 97%, коэффициенты отмывки Р2О5 из фосфогипса — 98 и выхода Р2О5 в кислоту — 94—95%. Съем Р2О5 с 1 м3 экстрактора—>10—11 кг/ч, с 1 м2 поверхности фильтра 85 кг/ч при производительности фильтрования 600 кг/(м2-ч) в расчете на сухой осадок (с учетом гипсового числ'а, равного 1,4 т/т фосфата).
Получаемая кислота плотностью 1300 кг/м3 содержит, в %: Р2О5 — 21— 22; SO3 —2,5—3,5; СаО — 0,4; MgO —0,7; Ре^Оз — 0,9; AI2O3—1,2 и F—1,2. На основании результатов полузаводских испытаний составлены исходные данные для проектирования цехов экстракционной фосфорной кислоты из чилисайских флотоконцентратов [39]. При мощности производства 136 тыс. т Р2О5 в год себестоимость кислоты оценена в 178,2 руб. в расчете на 1 т Р2О5. При этом удельные капиталовложения в производство (с учетом, общезаводского хозяйства) составляют 1043,5 руб., а приведенные затраты — 303,4 руб.
Переработка полученной кислоты в аммофос, содержащий 46% Р2О5 и 1;1% N, связаны с дополнительными удельными капиталовложениями, равными 505,0 руб. на 1 т аммофоса, а приведенные затраты на 1 т Р2О5 в продукте составляют 388,2 руб. Полная себестоимость Г т аммофоса (натура) равна 137,0 руб. При оптовой цене 178 руб/т прибыль предприятия составляет 41 руб на 1,т аммофоса.
Перспективные направления развития дигидратного способа
В общефцикле переработки природные фосфатов в удобрения или технические соли производство экстракционной фосфорной кислоты является промежуточным технологическим процессом. Недостаток его — образование кислоты невысокой- концентрации и необходимость ее упаривания, а также трудности, возникающие при переработке бедных видав сырья.
В настоящее время накопились многочисленные и всесторонние данные о свойствах модифицированных фосфорнокислых растворов и поведении в них кристаллогидратов сульфата кальция (стр. 56), а также сведения о селективном растворении составных частей фосфатного сырья. Они послужили основанием различных предложенных способов экстракции фосфорной кислоты из апатитового концентрата и фосфоритов Каратау, в сочетании с последующим ее использованием, или с предварительным обезмагниванием бед-
112
ных доломитизированных фосфоритов для кислотной переработки. - .
Применительно к производству аммофоса разработано в лабораторных и опытных условиях получение концентрированных фосфорнокислых растворов, содержащих 39—40% Р2О5 и 1,9—3,0% NH3 [40, 41]. При получении нитрофосфат-ных растворов в производстве нитроаммофоса или нитроаммофоски представляет интерес разработанный в лабораторных условиях способ экстракции фосфорной кислоты в присутствии нитрата аммония (стр. 52). Предварительная подготовка для экстракции доломитизированных и других магнийсодержащих фосфатов путем и>х обезмагничивания (стр. 22) разработана в лабораторных и опытных масштабах.
Получение концентрированных фосфорнокислых растворов. Сущность способа заключается в разложении фосфатного сырья серной кислотой с применением в качестве раствора разбавления частично аммонизированной фосфорной кислоты [40, 41]. При частичной нейтрализации фосфорной кислоты аммиаком [до мольного отношения NH3: Н3РО4 (а) меньше 1] происходит повышение активности воды по сравнению с чистыми фосфорнокислыми растворами [42]. Это обуславливает увеличение диапазона температур и концентраций кислоты, в котором сосуществуют метастабильные полугидрат и дигидрат сульфата кальция (стр. 56). Например, при 70 °C-равновесному переходу гипс—полугидрат в чистой фосфорной кислоте соответствует концентрация кислоты 36,5% Р2О5, в фосфорной кислоте, нейтрализованной до значения. а=0,3—40,6, а в кислоте, нейтрализованной до «=0,5 — 42,0% Р2О5.
Исследованием ф42] процесса на лабораторной установке полунепрерывного действия установлены оптимальные условия его проведения: температура разложения фосфорного сырья 80 °C, мольное отношение NH3: Н3РО4 в растворе разбавления а = 0,3 (2—2,5% NH3), отношение Ж:Т в пульпе 3:1, количество H2SO4— стехиометрическое в расчете на СаО, продолжительность экстракции 6 ч.
Получаемые растворы содержат 38—40% Р2О5. Степень извлечения Р2О5 из апатита составляет 97%, эффективность отмывки 99% и технологический выход 96%. Процесс протекает в устойчивом дигидратном режиме с выделением в осадок гипса. Производительность фильтрования гипса достигала 900 кг/(м2-ч).
Опытами экстракции из апатитового концентрата фосфорной кислоты концентрации 30% Р2О5 на установке непрерывного действия в условиях, имитирующих промышленные, показано [43], что в присутствии в растворе ионов аммония
8—5473
113
кристаллизация гипса происходит с образованием более крупных, (с размерами 300—800 X 50—200 мм) кристаллов и большей однородностью их гранулометрического состава, чем в их отсутствие. В результате этого продолжительность фильтрования пульпы, уменьшается на ~30%.
Содержание аммиака в промытом фосфогипсе при отмывке его от кислоты «а. 98—99% составляет не более 0,001—0,003%, что в несколько раз ниже требований суще-
Г- ствующих санитарных норм.
С Преимущества описанного-процесса, помимо получения
₽ в дигидратном режиме кислоты повышенной концентрации
(38—40% Р2О5) и соответственного повышения (на 25— 30%) производительности систем, заключается также в эко-; номии на упаривании растворов, значительном. уменьшении простоев оборудования на чистку (стр. 117) и снижении коррозии аппаратуры вследствие частичной .нейтрализации фосфорной кислоты. Экономия только на упаривании кислоты от 38—40% Р2О5 до 54% Р2О5 (или остаточной влаги 25%) по сравнению с кислотой концентрации 28—30% Р2О5 может составить ~10—45 руб. на 1 т Р2О5.
Экстракция фосфорной кислоты в присутствии нитрата аммония. В основе процесса лежит увеличение растворимости при 60—80 °C как дигидрата, так и в особенности пюлу-- , - гидрата в фосфорной кислоте, содержащей нитрат аммония (стр. 52). Наибольшая растворимость обоих кристаллогидратов наблюдается в растворах, содержащих ~10—4'5% Р2О5 и 20—30% NH4NO3. Количество вводимого' раствора нитрата аммония определяется составом исходного фосфатного сырья и заданным соотношением Р2О5 и азота в готовом удобрении -(например, в пределах от 1:1 до 1:2).
Применение нитрата аммония в виде раствора приводит к интенсификации разложения фосфатного сырья. Одновременно увеличиваются размеры выделяющихся кристаллов гипса. Это приводит к повышению производительности фильтрования пульпы и к существенному увеличению степени отмывки фосфорной кислоты из осадка [44].
При обработке апатита при 80 °C 60 %-ной H2SO4 при норме ее 102—105% от стехиометрического количества в присутствии 50—60%-ного раствора NH4NO3 при Ж:Т=-= 2,5:1 степень разложения его уже в течение двух часов достигает 98%. Размеры кристаллов выделяющегося гипса: Длина 180—340 мкм и ширина 24—30 мкм.
Степень разложения фосфорита Каратау при использовании оборотных растворов, содержащих помимо нитрата аммония, 8—'10% Р2О5, в указанных выше условиях составляет 95—198%'. Производительность фильтрования фосфогипса в расчете на сухой отмытый осадок составляет ~ 1000 кг/ч с 1 м2 поверхности фильтра в 1 ч [45].
114
Процесс можно проводить следующим образом. В экстрактор подают апатитовый концентрат или рядовую фосфоритную муку (содержащую 24,5—26,5% Р2О5, ' 3—3,6% MgO и 2,5—3,0% КгОэ), оборотный раствор и 50—65%-ный раствор NH41NO3. Фосфат обрабатывают при 75—80 °C, отношении Ж:Т в .пульпе, равном (2,5—3):1, и содержании свободной серной кислоты в жидкой фазе 1—3% в течение 2—4 ч. Отделенный на вакуум-фильтре осадок фосфогипса промывают в две-три ступени — вначале промывными растворами, а затем горячей (75—80 °C) водой. В промытом осадке содержится 34—38-% влаги и всего 0,4—0,6% Р2О5 и 0,03—0,05% нитрата. Часть первого фильтрата, соответствующая вводимому в процесс количеству Р2О5 с фосфатом и NH4NO3 с раствором нитрата аммония, отводят в качестве продукта. Последний содержит. 14—15% Р2О5 и 30— 32% NH4NO3. Его подвергают аммонизации, сушке и грануляции с получением конечного азотнофосфорного удобрения.
Второй фильтрат полностью возвращается на экстракцию. Смесь его с частью первого фильтрата и с раствором нитрата аммофоса представляет собой раствор разбавления.
Экстракция фосфорной кислоты из доломитизированных (карбонатных) фосфатов с их предварительным обезмагни--ванием. Выше (сир. 23) были, описаны различные методы облагораживания (обезмагнивания) магнийсодержащих фосфатов.
Из них наибольший интерес представляет сернокислотный метод как по- причине селективности удаления из сырья соединений магния и двуокиси углерода при минимальном переходе в раствор пятиокиси. фосфора, так и сочетания процесса подготовки сырья с последующей сернокислотной переработкой обезмагненного продукта.
Экстракция фосфорной кислоты, из обезмагненного фосфорита практически завершается в течение не более трех часов (т. е. в два раза быстрее, чем при разложении исходного фосфорита аналогичного состава) с получением и мало 'загрязненной (поддающейся упариванию), и более концентрированной кислоты, содержащей 26—28% Р2О5 (вместо 20—22%) при коэффициенте разложения 96—98%.
- Производительность фильтрования фосфогипса, полученного из обезмагненного фосфорита, составляет 1200 вместо 500—600 кг/(м2-ч) из необезмагненного.
Процесс разработан совместно ЯТИ им. Ленсовета, Лен-НИИгипрохимом и КПО «Фосфорит», в лабораторных и опытных условиях. Ниже приводится технологический режим переработки рядовой фосфоритной муки Каратау (23—24% Р2О5 и 3—3,5% MgO) с предварительным обезмагнива-нием ее.
8Ф 115
Сущность способа заключается в предварительной обработке водной суспензии фосфоритной руды [Ж : Т= (2-—4) ;4] серной кислотой концентрации 75—93% H2SO4, взятой в стехиометрическом или несколько мёныпем (70—80% от стехиометрии) количестве из расчета разложения карбонатов, в течение 5—15 мин при 80—90 °C, отделении фильтрованием жидкой фазы от твердой (первая стадия) и последующей переработки последней в фосфорную кислоту (вторая стадия), а маточного раствора в гидроокись магния (стр. 25).
На первой стадии соединения магния, содержащиеся в исходном фосфорите, перехрдят в раствор в виде сульфата магния, а карбонат кальция в виде сульфата кальция (гипса) ’входит в состав твердой фазы. Концентрация сульфата магния в растворе составляет 0,5—2% MgO. В осадке остается 0,5—1% MgO в зависимости’ от нормы кислоты и состава исходного фосфорита.
Обработку суспензии фосфорита проводят в реакторе, снабженном лопастной многоярусной мешалкой. Реагенты подают со скоростью, обеспечивающей поддержание в жидкой фазе рН = 2—4 (в зависимости от свойств руды).
Вытекающую из реактора пульпу фильтруют на вакуум-фильтре под разрежением (67—80)-103 Па (500—600 мм рт. ст.) при 70—80 °C без промывки. Производительность фильтрования составляет 600—800 кг/ч (в пересчете на сухой неотмытый осадок) с 1 м2 поверхности фильтра.
. Облагороженный (обезмагненный) фосфорит с влажностью 15—25%, содержащий, в %: Р2О5— 23—25; MgO — 0,5—1,0; СаО —41—42; SO3 — 6,5—7,5> СО2 — 1,8—2,2; R2O3 — 3—3,5; F—1,8—2,3 и нерастворимого осадка — 16— 17, направляют на экстракцию фосфорной кислоты.
Отделенный от твердой фазы раствор, полученный после обезмагнивания фосфорита, содержит 0,5—4 % MgO, 0,02— 0,05^о Р2О5 и до 3—5 г/л твердых взвешенных частиц. Его обрабатывают известковым молоком, в результате чего выделяют в твердую фазу гидроокись Магния и сульфат кальция. В жидкой фазе находится 0,07—0,08% СаО и 0,006% MgO, которую в качестве оборотной воды возвращают в процесс. Обработку раствора обезмагнивания ведут при 60— 80°С, рН = 10—И, молярном отношении CaO:MgO = 0,9— 1,0 в течение 20—60 мин. Известковое молоко применяют с концентрацией 5—40% СаО в количестве 105—-110% от стехиометрического в расчете на содержание MgO в растворе. Расход окиси кальция составляет 0,12—0,1'3 т на 1 т обез-магненного фосфорита. Степень осажденйя гидроокиси магния составляет 99,7—99,9%.
Полученную разбавленную суспензию (Ж:Т=10:1) сгущают -в отстойнике (или гидроциклоне) до отношения 116
Ж:Т=(3—5)?1. Скорость отстаивания твердой фазы — Mg(OH)2 и CaSO4-2H2O,— равная 1—1,5 м/ч, увеличивается до 2—3 м/ч в присутствии 0,001—0,002% полиакриламида. Сгущенную суспензию сушат с классификацией (отдувкой гидроокиси магния) в аппарате кипящего слоя при температуре на выходе из аппарата 100—105 °C. В готовом магнезиальном продукте содержится 45—47% MgO 1[в твердой фазе суспензии ~20% Mg(OH)2 или 14—15%.-MgO] и 1% влаги.. Продукт удовлетворяет требованиям ТУ 60-86-62-52—7^, на техническую магнезию (гидроокись магния).
Обезмагненный. фосфорит поступает в экстрактор, куда •подают оборотный раствор, а также серную кислоту. Экстракцию ведут при 75—80 °C, концентрации в жидкой фазе 1,5—2,5% SO3, Ж:Т= (2—2,5) Я, в течение 3—4 ч. При этих условиях степень разложения фосфорита-составляет 98%, коэффициент отмывки 98%, степень извлечения в кислоту 96%. Расход Р2О5 фосфорита на 1 т Р2О5 в кислоте равен 1,06, расход серной кислоты с учетом обезмагнивания — 3,5—3,7 т, в том числе на экстракцию'—2,55—2,65 т моно*-гидрата. Съем' P20s с 1 м3 экстрактора составляет 29,5— 30 кг/ч, а с 1 м2 поверхности фильтра 165—170 кг/ч. Фосфагипс (гипсовое число 1,3) фильтруется с производительностью 1200—1300 кг/ч сухого промытого осадка с 1 м2 [7, с. 1'1].
Получаемая фосфорная кислота содержит, в %: Р2О5— 24—28; SO3 —1,5—2,0; MgO —0,9—4,1; СаО —0,3—0,4; R2O3—1,7—1,8; F—1,2—1,3; SiO2 — 0,2—0.3 и взвешенных твердых частиц—1,5—2,0.
Переработка растворов обезмагнивания на магнезию по ориентировочным расчетам увеличивает суммарный объем капиталовложений по отделению обезмагнивания в 1,5 раза, потребность в электроэнергии и других энергетических затратах— в 1,5 раза, численность рабочих—в 1,4 раза. В то же время использование обезмагненного фосфорита позволяет увеличить производительность цеха экстракционной фосфорной кислоты в ~1,5 раза или уменьшить, соответственно!, капиталовложения на сооружение экстракционной системы. При этом переработка раствора обезмагнивания в товарную магнезию приводит к4 существенному снижению расходов на производство фосфорной кислоты. Принимая потери MgO с гипсом при сушке и разделении магнезиально-гипсовой смеси в аппарате с кипящим слоем, равными 15%, на 1 т Р2О5 возможно получить 0,14—0,16 т гидроокиси магния, содержащей 45% MgO. При цене на техническую гидроокись магния (магнезию) по ТУ 60-86-62-52—73 в 435 руб. за Г т стоимость реализации дополнительного продукта составляет ~60 руб./т Р2О5.
117
Себестоимость кислоты, полученной прямой экстракцией из рядовой руды Каратау (24,5% Р2О5 и 3% MgO), определена в 140,79 руб./т Р2О5. Таким образом, по сравнению с. производством- экстракционной кислоты из рядовой руды использование обезмагненного фосфорита приводит к снижению стоимости кислоты в ~1,7 раза.
Очевидно, что процесс рентабелен и при получении менее ценных сортов магнезий (например, с содержанием-32% MgO, оцениваемой в 380 руб./т и др.), а также магниевых удобрений — сульфата магния, магц^йаммонийфосфата и т. п.
В лабораторных и опытных условиях отработан дигид-ратный процесс экстракции фосфорной кислоты также из обезмагненного Ковдорского апатита [7, с. 14] состава, в %: Р2О5 — 37,2; СаО —50,7; MgO — 0,76; SO42- —6,7; Fe2O3 — 0,7; А120з — 0,1; SiO2—1,8; F — 0,79. При этом степень разложения фосфата через 3—3,5 ч составила 98—98,8%, съем сухого, отмытого осадка при фильтровании—1'250— 1400 jcr/ (м2-ч).
Полученная кислота близка по составу (в %: Р2О5 — 28—30; СаО — 0,2—0,4; MgO —. 0,5—0,6; SO3 — 1,9—2,2; Fe2O3— 0,3—0,4; F — 0,3—0,4 и SiO2 — 0,15—0,2) к кислоте из хибинского апатитового концентрата, но больше содержит окиси магния и меньше фтора, полуторных окислов и двуокиси кремния. Показатели же процесса выше, чем при переработке хибинского, апатита.
Рассмотренный пример подготовки' доломитизированного фосфатного сырья к экстракции с предварительным его обез-магниванием является лишь частным элементом большой народно-хозяйственной задачи, относящейся к комплексному использованию всех компонентов минерального сырья. В этих условиях стоимость дополнительно производимых продуктов может оказаться настолько существенной, что распространенное’ мнение об обязательном удорожании переработки фосфатного сырья по мере снижения содержания в нем Р2О5 и увеличения содержания примесей должно будет подвергнуться основательному пересмотру (или вовсе полному исключению).
Заслуживает внимания то обстоятельство, что проблему комплексной переработки всех фосфатных руд связывают даже с.извлечением урана из экстракционной фосфорной кислоты [46]. Во всей произведенной в 1976 г. в мире экстракционной" кислоте содержалось ~3000 т растворенной U3O8, а к концу века количество окиси урана в кислоте может возрасти до 8000 т/г.
В США разработаны два процесса извлечения урана из кислоты. Считают, что стоимость Извлечения урана из фосфорной кислоты может составить ~ 30—35 долл, за 1 кг 118
U3O8 на заводе с суточной мощностью в 1000 т Р2О5. При этом возможно получить дополнительный доход в 7—8 млн. долл, при существующей цене 90 долл, за 1 кг U3O8 и содержании ~0,45 кг U3O8 на 1 т Р2О5, что характерно для многих фосфатных руд.
III.5. ПОЛУГИДРАТНЫЙ СПОСОБ
В отличие от дигидратно!го полугидратным методом получают кислоту, содержащую 35—48% P/V Это позволяет увеличить мощность действующих цехов в 1,3—1,5 раза и несколько уменьшить количество отхода — сульфатного остатка.
В значительной мере успехи, достигнутые, в области изучения и освоения полугидратного метода, основаны на технических усовершенствованиях и достижениях производства экстракционной фосфорной кислоты дигидратным ' методом.
Оба процесса протекают с выделением твердых фаз —дигидрата и полугидрата сульфата кальция в метастабильном состоянии, но резко отличающихся по своей растворимости, устойчивости, размерам и форме кристаллов.
Успешное осуществление процесса полугидратным методом [47] возможно при выделении достаточно стабильных кристаллов полугидрата, обеспечивающих максимально полное отделение фосфорной кислоты от осадка и не гидратирующихся в процессе промывки водой на фильтре и при дальнейшей транспортировке и хранении.
Физико-химические основы процесса
Температурные условия процесса зависят от концентрации получаемой кислоты и в области метастабильного существования полугидрата в системе CaSO4—H^PCU—Н2О (см. рис. 11.16) заключены в относительно небольших пределах. При концентрации кислоты, от 35 до 50% Р2С>5 верхний диапазон температур изменяется от ПО до ~90°C (рис. III. 11). Нижний диапазон, начиная с 70X3, не представляет практического интереса, особенно при содержании в кислоте более 37—40% Р2О5, вследствие малой с'корости реак-
Рис: III.11. Влияние температуры и концентрации фосфорной кислоты на выделение полугндрата.
119
ции и затруднений, связанных с отделением осадка от кислоты большой вязкости.
Если предположить, что увеличение вязкости кислоты по мере роста ее концентрации элиминируется повышением температуры по линии ВС. (см. рис. Ш.’11), то рабочая область температур и концентраций кислоты при'полугидратном процессе может быть представлена заштрихованной площадью АВС. Вследствие малой скорости фазового перехода полугидрата в' ангидрит при условиях, соответствующих разграничивающей их линии, сама экстракция может быть проведена при несколько более высоких температурах, например, по линии АС', без образования в пульпе ангидрита. При длительном выдерживании пульпы (например, при остановке системы) следует избегать высоких температур.
При малой растворимости и медленном растворении полугидрата последний долго дегидратируется и при 115— 125 °C в концентрированной фосфорной кислоте. Например, в фосфорной кислоте, содержащей 48% Р2О5 и 2,9—3,3% СаО, не было обнаружено перехода полугидрата в ангидрит при 115 °C в течение 4 ч, а при Г25°С в течение 3 ч (более длительных наблюдений не производилось). В то же время при отсутствии СаО в кислоте полугидрат в этих условиях переходит в ангидрит через 1—'1,5 ч.
Как было указано (стр. 63), из растворов, содержащих более 35% Р2О5, при температурах выше 75 °C, т. е. в областях АВС или АВС', выделяются компактные агрегаты полугидрата с размерами от 75 до 200—300 мкм. Они образованы из плотно сросшихся отдельных кристаллов в ~ 10— 40 мкм, хорошо' отфильтровываются^ и характеризуются относительно малой влагоемкостью.
Присутствие в растворе 0,5—0,6% фтористых и кремне-фтористых соединений приводит к резкому уменьшению размеров кристаллов и замедлению фильтрования в 2 раза. Увеличение содержания фтористых соединений до 1% замедляет фильтрование в 5 раз. Появление' кристаллов полугидрата игольчатой формы вызвано торможением роста граней призмы при быстром росте вершинных граней вследствие избирательной адсорбции примеси на гранях растущего! кристалла. Но совместное присутствие в растворе примесей соединений алюминия и ионов фтора вызывает (при содержании до 2% А120з и 0,4—0,5% F") образование более изомет-ричных кристаллов с лучшими фильтрующимися свойствами, чем в. отсутствие примесей. Если в жидкой фазе пульпы находится не менее 0,8—1% А120з, то этот эффект наблюдается и при содержании 0,7—1,2% F' в виде SiF62~, F- и A1F2+, особенно при интенсивном перемешивании реагентов.
Весьма важно, но и значительно сложнее, регулировать свойства полугидрата (стабильность, скорость гидратации и 120
схватывание), особенно в процессе его промывки, а также при транспортировании и хранении. При промывке осадка, отделенного от продукционной кислоты, он соприкасается со все более разбавленными растворами фосфорной кислоты и с той или иной скоростью переходит в гипс. Полное превращение полугидрата в гипс в растворах, содержащих 10—18% Р2О5, завершается в течение 1 ч (см. стр. 58). При содержании в кислоте 25% Р2О5 фазовый переход заканчивается в течение 1,5—2 ч. При совместном присутствии примесей Ре^Оз, AI2O3, SiO2 и других оводнение полугидрата ускоряется в несколько раз и заканчивается в течение 10—15 мин. Аналогично и еще в большей степени на процессе гидратации сказывается влияние затравки.
В то же время агрегаты полугидрата, выделенные при 70—100 °C из кислоты концентрации 35—40% Р2О5, весьма медленно гидратируются в разбавленной фосфорной кислоте (’2—3% Р2О5). При 20 °C и массовом отношении Ж:Т = 4: 1 оводнение или степень гидратации полугидрата составила 1'5—20% через 3 ч и 30—40% через 24 ч. В присутствии затравки дигидрата (не менее 30% по отношению к полугидрату) скорость гидратации увеличивается.
С увеличением содержания серной кислоты в растворе (в исследованном диапазоне от 1,8 до 7,4% SO3) гидратация полугидрата замедляется и уже при концентрации 4,65% SO3 в кислоте, содержащей 33,8% Р2О5, в течение 6 ч практически не идет.
Максимальная скорость гидратации в растворах фосфорной кислоты концентрации 5—10% Р2О5 наблюдается при отношении Ж: Т в гидратной пульпе от 6 : 1 до 4: 1. В кислоте, содержащей 22% Р2О5, оводнение полутидрата ускоряется с увеличением отношения Ж: Т в гидратируемой пульпе и при Ж: Т=’1О: 1 достигает 65% за 6 ч по сравнению с 25—30%, когда отношение Ж: Т= (3-н4) :1.
'Скорость гидратации полугидрата в значительной степени зависит и от условий его получения [48, 49]. Чем выше концентрация фосфорной’ кислоты, в которой происходило выделение полугидрата, тем медленнее происходит его оводнение в разбавленных растворах [49]. Влияние концентрации кислоты оказывается тем сильнее, чем больше она удалена от пограничной линии (см. рис. Ш.Н), разделяющей поля гипса и полугидрата.
Различное время полной гидратации полугидрата, выделенного из фосфорнокислых растворов разной концентрации, по-видимому, связано с поверхностными явлениями на границе раздела кристалл—раствор, в частности, с появлением пленки фбефорной кислоты, тормозящей растворение кристаллов полугидрата.
121
Гидратация полугидрата сульфата кальция в магни,йсо-дер'жащих фосфорнокислых растворах концентрации 15— 20% Р2О5 при 80 °C протекает практически полностью за короткое время Дменее 4 ч) [50]. В растворе, содержащем до 20% Р2О5, 3% MgO и 6% SO42- (т. е. в эквивалентном количестве на связывание MgO) практически полная гидратация полугидрата сульфата кальция при 80 °C завершается через 4 ч. В аналогичных условиях в отсутствие серной кислоты полугидрат гидратируется только на ~15%.
Данные рис. III.12, а показывают, что примеси HzSiFg и Fe2O3, содержащиеся в растворе, из которого кристаллизуется полугидрат, весьма сильно..уменьшают его стабильность. Уже при содержании 1% SiF62- или Ре20з время появления первых кристаллов гипса уменьшается от 30 (в отсутствие примесей) до ~ 16—18 мин. Напротив, в присутствии в растворе 1% F~ продолжительность скрытого периода^ гидратации полугидрата увеличивается до ~41.мин.
Полный переход полугидрата, выделенного в присутствии 1,5% F", в гипс происходит за 92 мин, что в. 1,9 раза превышает . время фазового перехода полугидрата, осажденного
время, мин
из чистого фосфорнокислого раствора.
По-видимому, изменение скорости гидратации выделяющегося полугидрата в присутствии изученных примесей связано с изменением свойств пленки-фосфорной кислоты '(концентрации, вязкости, адгезии и т. п.), образующейся при этом на . поверхности кристаллов полугидрата. В условиях, обеспечивающих максимальную сохранность пленки (Отсутствие перемешивания, малые значения Ж:Т), характерных, напри-
Рис. Ш.12. Влияние примесей в растворе на стабильность (скрытый период кристаллизации) полугидрата (а) и продолжительность перехода полугидрата в гипс (б):
Условия гидратации: Ж:Т=6:1: температура 60 °C и содержание Р2О5 в жидкой фазе суспензии —
5%.
122
мер, для процесса промывания водой осадка полугидрата на ленточном или карусельном вакуум-фильтре, действие примесей должно быть наиболее ярко выражено.
Хотя время соприкосновения на фильтре осадка с разбавленными растворами весьма незначительно! (0,3—0,5 мин), все-таки остающееся на фильтре после каждого цикла (оборота) некоторое количество осадка будет гидратироваться; это же приводит к загипсовыванию ленты в течение сравнительно короткого времени.
Относительно более регулируемой является гидратация полугидрата, промытого водой, которая в основном. определяется концентрацией жидкой фазы фильтровальной лепешки и ее влажностью (табл. Ш.З).
Осадок, содержащий 40% жидкой фазы, затвердевает через 6 ч после начала гидратации, а фильтровальная ле-’ пешка с влажностью 20% не твердеет и-за 1'0—15 суток.
-При хранении полугидрата в куче на открытом воздухе поверхность его покрывается слоем рассыпчатого порошка толщиной 1—2 см. Под ним образуется твердая корка толщиной 1—2 см, которая препятствует проникновению атмосферной влаги внутрь кучи. Поэтому .полугидрат внутри кучи очень медленно гидратируется внутренней влагой без образования связи между отдельными кристаллами гипса. В результате этого масса внутри кучи не твердеет, а представляет собой рассыпчатый порошок. При хранении полугидрата в помещении при 15—25 °C осадок также не затвердевает.
Очевидно, что полугидрат после оТмывки и отжима до влажности не более 20% можно транспортировать в сухом
Таблица Ш.З
Влияние содержания жидкой фазы л осадке на степень гидратации полугидрата
Начальное содержание кристаллизационной воды в твердой фазе, осадка 6,76%; температура гидратации 25 °C, концентрация РгО5 в жидкой фазе лепешки 5%; препарат полугидрата содержит: 60—80% изотермических, 1'0—20% деизотермических агрегатов и 10—20% призматических монокристаллов.
Продолжительность гидратации полугидрата, ч Степень гидратации полугидрата (в %) при влажности фильтровальной лепешки, % Продолжительность гидратации полугидрата, ч Степень гидратации полугидрата (в %) при влажности фильтровальной лепешки, %
20 40 20 | 40
2 10 16 6 ' <-...33
4 16 23 10 171 4G
123
виде к месту хранения или переработки, не опасаясь его затвердевания.
Влагоемкость осадка определяется фильтрующими свойствами полугидрата и режимом фильтрования. В условиях нормальной работы фильтровальная лепешка отмытого и отжатого полугидрата содержит менее 20% жидкой фазы.
Для обезвоживания осадка с предотвращением гидратации и твердения (схватывания) его при 23—25 °C в водных суспензиях (при отношении Ж:Т = 3:1) обрабатывают разными добавками: преимущественно гидроокисью кальция, карбонатами кальция, натрия и. др. -Показано [51], что для обеспечения существенной стабилизации полугидрата сульфата кальция с помощью водных растворов-ингибиторов необходима отмывка, лепешки от фосфорной кислоты до содержания не более 0,3—0,5% свободной Р2О5.
Стабилизация полугидрата гидроокисью кальция связана с нейтрализацией содержащейся в нем свободной фосфорной кислоты [52]. Рентгенофазовых! анализом образца- полугидрата сульфата кальция, обработанного известковым молоком, установлено наличие на его поверхности как СаНРОг •2НгО, так и CaSO4-2H2O [10, вып. 1, с. 15]. По-видимому, образующаяся корка обусловливает ингибирование пошугид-рата.
Стабилизация полугидрата различными щелочными добавками в пульпе (Т : Ж= 1 '• 5) при 20 и 70—75 °C в присутствии 5 кг NH3, МагО или СаО на- 1 т Р2О5 в продукционной кислоте достигается в большей степени гидроокисью кальция и карбонатом аммония и в меньшей — содой и известняком [10, вьвп. 4, с. 1]. В первом случае гидратация полугидрата не происходит в течение по крайней мере 7 ч.
Технологические схемы и режимы
♦
В СССР освоено и налажено в промышленных масштабах производство полугидратным методом фосфорной кислоты. концентрации 45—48 и 35—38% Р2О5. Кислоту концентрации 45—48% -Р2О5 в небольших масштабах производят по способу, разработанному в ЛТИ им. Ленсовета совместно с Винницким химическим комбинатом и ЛенНИИГипрохимом [47]. В основе способа получения 35—38%-ной кислоты на типовых промышленных установках лежат разработки, проведенные в НИУИФе, Лен-НИИГипрохиме, Воскресенском ПО «Минудобрение», иа Красноуральском медеплавильном комбинате и др.
В настоящее время в Советском Союзе ~30% от всего количества кислоты производится полугидратным способом.
В ранних лабораторных исследованиях НИУИФа [53] опыты получения кислоты, содержащей 35—43% Р2О5, по-124
лугидратным способом проводились при 75—85 °C. Степень разложения апатитового концентрата смесью серной (92— 94% H2SO4) и фосфорной кислот за 5—6 ч составляла 96— 97%. В этих условиях полугидрат выделяется в виде крупных, хорошо фильтрующихся агрегатов. Производительность фильтрования в лабораторных условиях с промывкой осадка. по пяти- и шестиступенчатым схемам составляла 1900— 3000 кг/(м2-ч) сухого осадка. Хотя эти данные были ориентировочными и получены в условиях фильтрования, отличающихся от промышленных, но они указывали на принципиальную возможность выделения полугидрата такой структуры, которая позволяет вести достаточно эффективное фильтрование его. Однако условия отмывки полугидрата и дальнейшей его способности к оводнению и гипсованию не были изучены.
По способу, предложенному в НИУИФе [54], апатитовый концентрат разлагают смесью 92—93%-ной H2SO4 и оборотной концентрированной фосфорной кислоты при 94— 95 °C с получением кислоты, содержащей 43—48% Р2О5 и 0,7—1,3% SO3. Процесс протекает при интенсивном выделении фторсодержащих газообразных соединений (более 50% от общего содержания в сырье) и степени разложения апатита', равной 97,5%.
В настоящее время фосфорную кислоту концентрации 35—37% Р2О5 из апатитового концентрата полугидратным методом по способу НИУЙФа и Воскресенского ПО «Минудобрения» получают на ряде предприятий. Для предварительного смешения серной и фосфорной кислот применяют высокоскоростной смеситель конструкции Воскресенского ПО «Минудобрения» и НИИХиммаша. Он представляет собой аппарат типа «труба в трубе», выполненный из нержавеющей стали. Во внутреннюю трубу сверху под'ают разбавленную охлажденную серную кислоту с температурой не более 60 °C, которая распределяется форсункой, помещенной на конце трубы. В наружную трубу тангенциально подводят оборотную фосфорную кислоту. Кислоты смешиваются на выходе из трубы над поверхностью пульпы в экстракторе.
Разложение апатитового концентрата ведут при 90—• 97 °C, Ж : Т (2ч-2,5) П в течение' 4—6 ч при стехиометрической норме серной кислоты в расчете на СаО в фосфате. Степень разложения апатита составляет 97—98%, степень отмывки, Р2О5 из осадка полугидрата 99%, при расходе воды 500—550 кг/т сухого осадка с температурой не ниже 70°C. При этом съем Р2О5 с 1 м3 объема экстрактора составляет '2'6—27 кг/ч, а с 1 м2 поверхности фильтра — 360— 400 кг/ч. Производительность фильтрования фосфополугид
125
рата равна 1200—1300 кг с 1 м2/ч (в расчете на сухой отмытый осадок).
Процесс ведут преимущественно с применением цилиндрических реакторов. Прямоугольные железобетонные экстракторы с рабочим объемом 740 м3 с годовой производительностью 140 тыс. т Р2О5 представляют большие затруднения в эксплуатации. Экстрактор разделен внутри перегородками на восемь рабочих секций и две дополнительных, изнутри покрыт антикоррозионной защитой. Крышка экстрактора защищена листом из нержавеющей стали.
В зоне газовой фазы все секции сообщаются между собой. Над перегородками между нечетными и четными секциями устанавливается специальное устройство в виде вала с лопастью, при вращении которого снимаются отложения геля кремниевой кислоты, образующегося в результате интенсивного выделения фторсодержащих газов в узлах между' крышкой экстрактора и перегородками.
В первой по четвёртую и в восьмой секциях установлены • турбинные мешалки с интенсивной! циркуляцией, в пятой, шестой и седьмой секциях—-винтовые мешалки:
Выделяющиеся в экстракторе фторсодержащйе газы и водяные пары протягиваются вентилятором из четвертой секции экстрактора в абсорбционную систему.
Образующуюся пульпу из девятой секции отводят погружными насосами в вакуум-испарительную установку. Охлажденная на 3—5 °C пульпа из испарителя по барометрическому трубопроводу перетекает в распределительную десятую секцию экстрактора. Основное количество охлажденной пульпы из десятой секции поступает в первую секцию экстрактора, а остальное—на фильтр.
Водяные пары и фторсодержащие газы, выделяющиеся при кипении пульпы в вакуум-испарителе, проходят ловуш-. ку-'брызгоуловитель, орошаемый водой при помощи форсунки, газоход (для предотвращения зарастания осадком), абсорбционную башню, циклон-сепаратор, поверхностные конденсаторы, а затем вакуум-насосом несконденсировавшиеся пары и газы выбрасываются через выхлопную трубу в атмосферу.
Фильтрование пульпы с четырехкратной промывкой осадка производится на карусельном вакуум-фильтре с поверхностью 80 м2. Осадок промывают водой с температурой не менее 80 °C в количестве ~700 кг на 1000 кг апатита.
Промытый и подсушенный воздухом фосфополугидрат выгружают в бункер, откуда вывозят самосвалами в отвал, например, в выработанные карьеры. В фбсфополугидрате со-~ держится не более (в %): общей, воды — 25; общей Р2О5— 1,5; водорастворимой Р2О5 — 0,6; F — 0,5; СаО — ~28—30;
126
S03 — 40—42; FeaOg —0,05; AlA —6,02; K — 0,02 и Na— 0,08. ' '
Получаемая фосфорная кислота содержит 36,5—37% Р2О5, 1,1—4ДГ/100 мл SO3, —2,0% F.
В другой экстракционной системе, работающей по полу-гидратному режиму, с годовой мощностью 70 тыс. т Р2О5 процесс осуществляется в одном цилиндрическом реакторе, разделенном радиальными перегородками на четыре секции. В первую секцию экстрактора подают 92,5—94%-ную H2SO4 и раствор разбавления, содержащий 25—27% Р2О5. В ре-' зультате этого происходит разбавление серной кислоты.
Апатитовый концентрат поступает во вторую секцию. Для ~ лучшего созревания кристаллов полугидрата сульфата кальция ~20% от общей массы серной кислоты подают в тре-. тыо секцию экстрактора. Из второй секции эоктрактора пульпу погружным насосом откачивают в вакуум-испаритель, работающий под разрежением не ниже 46,5 кПа (350 мм рт. ст.). Охлажденная пульпа стекает в первую секцию экстрактора^
Отходящая из вакуум-июпарителя паровоздушная смесь последовательно проходит через брызгоуловитель, абсорбционную башню, .межтрубное пространство поверхностных конденсаторов (в трубное пространство1 подается охлаждающая вода), барометрический конденсатор, ловушки и, наконец, вакуум-насосом через маслоотделитель выбрасывается в атмосферу. Во избежание отложения кремнезема' и других осадков на стенках труб поверхностного конденсатора их непрерывно отмывают чистой водой.
Пульпу отфильтровывают на карусельном вакуум-фильтре с поверхностью 40 м2, с трехкратной противоточной промывкой осадка. Осадок промывают водой, подогретой до 80 °C и выше. В продукционной кислоте содержится 35— 36% Р2О5. Промытый осадок поступает в бункер, откуда смывается в бак гидрошлама, куда дозатором вводят известковое .молоко [55]. Нейтрализованную суспензию из бака гидрошлама откачивают на расстояние 1—2 км в гипсо-. накопитель (площадью —-100 га), где происходит отстаивание твердой фазы.
В способе КМК—УПИ' (Красноуральского медеплавиль-- ного. комбината и Уральского политехнического института) фосфорную кислоту полугидратным методом [56] получали вначале в многореакторной системе, ранее работавшей по ( дигидратному, а позднее —по дигидратно-полугидратному ; режиму [57].
Из технологической схемы дигидратного процесса, были ( исключены узлы охлаждения серной кислоты и пульпы в ва-[ куум-испарителях или в экстракторах. Сырьем служит апа-[ титовый концентрат и 75—94%-ная H2SO4. Экстракцию про-
127
водили на четырех системах, состоящих каждая из четырех каскадно расположенных аппаратов, при 95—97 °C с получением кислоты, содержащей 30—37% P2Q5 и 1 —1,5 F.
.Пульпу отфильтровывают на ленточных вакуум-фильтрах каждый с рабочей поверхностью 10 м2 при разрежении 0,04—0,06 МПа (0,4—0,6 ат) с применением лавсановой или хлориновой ткани. Осадок на фильтре промывают подогретой до 60—80 °C водой. Производительность фильтра1 800— 1200 кг/(м2-ч) сухого осадка; толщина осадка зависит от нагрузки и достигает 25—60 мм. После предварительной ре-пульпации водой до значения Т:Ж=1:'(7—Г2) осадок насосами выбрасывают в отвал. Вследствие недостаточной стабильности .полугидрата происходит загипсовывание фильтровальной ткани и выход ее из строя уже через 7—9 суток.
В дальнейшем процесс был подвергнут существенному усовершенствованию и интенсификации, в результате чего все применявшиеся ранее 16 реакторов были заменены одним цилиндрическим четырехсекционным экстрактором с рабочим объемом* 150 м3 [58]. Экстракцию проводят при 104*^-108 °C — температуре кипения пульпы (в испарительном режиме), отношении Ж:Т= (2—3):1 в течение 2—3 ч.
Экстрактор представляет собой стальной аппарат, защищенный двумя слоями полиизобутилена, слоем диабазовой плитки и кислотоупорного, кирпича. Пульпу перемешивают двух- или трехъярусными мешалками, изготовленными из стали ЭИ-943. В первой секции экстрактора, большей по объему, установлены две мешалки, в остальных—дю одной. Надежность химической защиты экстрактора обеспечивается образованием на его стенках, днище и перегородках плотного «гарнисажного» слоя сульфата кальция и кремнефторидов толщиной до 60 мм в результате скорости вращения мешалки, равной 2,1—3,4 м/с на конце лопасти. Слой этот служит тепловой изоляцией экстрактора и защитой футеровки от эрозии твердых частиц пульпы.
Создание в зоне реакции необходимой минимальной концентрации реагентов (стр. 51) достигается предварительным разбавлением в отдельности исходной серной кислоты, а также апатитового концентрата в циркулирующей пульпе и части оборотной кислоты. Во взаимодействие приводятся разбавленные потоки реагентов, полученные в специальных смесителях.
Выделяющиеся в экстракторе, фторсодержащие газы (в количестве 30—50% от содержащегося в сырье фтора) с водяными парами абсорбируются в скрубберах.
Съем Р2О5 составляет 100—:130 кг/ч с 1 м5 рабочего объема. С 1 м2 поверхности фильтра получают 300—340 кг Р2О5. Выход Р2О5 в продукт равен 95—96%. Получаемая фосфорная кислота, содержит 34—37% PsOs- '
128
Производство кислоты концентрации 37—40% Р2О5 возможно при использовании для экстракции купоросного Масла в условиях, исключающих кристаллизацию сульфата кальция в виде мелких кристаллов ангидрита. Для этого необходимо вести экстракцию при 90—400 °C и содержании 0,5—1,5% свободной серной кислоты в жидкой фазе пульпы [59]. С увеличением концентрации Р2О5 в жидкой фазе пульпы уменьшается величина допускаемой концентрации серной кислоты.
В промышленных опытах процесс проводили при отношении в пульпе Ж: Т= (2—3):1 в течение 4—7 ч. При этом содержание в растворе 1—4,6% фтора не влияет отрицательно на кристаллизацию полугидрата. Производительность фильтрования пульпы составила 1000—4400 кг с 1 м2/ч в расчете на сухой отмытый фосфополугидрат.
Процесс протекает со степенью извлечения в раствор 96—98% Р2О5 при степени отмывки фосфорной кислоты из осадка на 97—89%.
При получении на промышленной установке фосфорной кислоты концентрации 40—45% Р2О5 процесс проводили [60] при 90—95 °C, содержании в жидкой фазе пульпы 600— 65О> г/л Р2О5, 1,0—1,6% фтора, 0,4 —1,0% свободной серной кислоты, отношении Ж:Т=(2—3):1. В этих условиях выход Р2О5 составил 90,0—95,8%, а удельный съем сухого фосфополугидрата— 850—4100 кг/(м2-ч). При концентрации в растворе более 2,5% H2SO4 степень разложения апатита и производительность фильтрования осадка резко уменьшается.
В ЛТП им. Ленсовета (Кафедра технологии неорганических веществ), начиная с 50-х гг., были выполнены многочисленные и многосторонние исследования, в результате которых были получены новые данные и расширены имеющиеся сведения (см. гл. II) о скорости растворения фосфатов в кислотах, растворимости, пересыщении и кристаллизации твердых фаз в образующихся системах и др, [61—©3]. Полученные данные послужили основанием для выбора оптимальных режимов и технологических условий производства фосфорной кислоты, содержащей 45—48% Р2О5, полугидратным способом [47, с. 14; 63].
Отличительная черта этого способа (ЛТИ им. Ленсовета, Винницкого химического комбината и ЛенНИИГипрохима), используемого уже в течение более 10 лет, — практически полное разложение апатита в избытке фосфорной кислоты при одновременном удалении из реакционной массы кремнефтористых соединений и обработке полученной монокальцийфосфатной пульпы серной кислотой. Процесс осуществляется в каскаде из 4—5 экстракторов с фильтрованием пульпы на ленточных вакуум-фильтрах,'
9—5473
129
Апатитовый концентрат разлагают в 3,5—4-кратном избытке фосфорной кислоты ® расчете на образование Са^НгРСМг при 90—95X3 в течение 1,2—1,7 ч. Избыток фос- -форной кислоты, содержащей 45—48% Р2О5, создают введением в реактор первого фильтрата — части продукционной кислоты^ и фосфорнокислотнополугидратной пульпы. Степень ; разложения апатита 98,5—99%.
Разложение апатита избытком концентрированной фосфорной кислоты протекает с достаточной скоростью уже при 60—'70 °C. С повышением температуры до 90—95 °C увели- • читается концентрация СаО в жидкой фазе пульпы от 2,5— 2,7 до ~4,3°/о и отношение Ж‘-Т в пульпе, а также уменьшается вязкость раствора и пульпы. Соотношение между фосфорной кислотой, поступающей с первым фильтратом и циркулирующей пульпой, зависит от отношения Ж:Т в пульпе. При Ж:Т = 3:1 с пульпой вводится в ~2,5 раза больше кислоты, чем с первым фильтратом.
•В фосфорной кислоте, применяемой для разложения апатита, должно находиться не более 0,4% свободной H2SO4. В присутствии больших количеств свободной серной кислоты уменьшается степень разложения апатита и- выделяющийся при этой стадии сульфат кальция плохо, фильтруется. Однако при содержании в кислоте ~4% А12О3 и 1% F’- диапазон концентрации свободной H2SO4 может быть расширен до 1,0—4,4 %.
Для удаления переходящего в раствор SiF62~ к апатиту добавляют немного соды, в результате чего ~50% содержащегося в апатите фтора осаждается в’виде Na2SiF6. С газами удаляется 35% фтора. При интенсивном перемешивании с внутренней циркуляцией реагентов процесс может быть осуществлен без добавления соды.
Образующуюся в первой стадии монокальцийфосфатную пульпу подают на вторую стадию—экстракцию фосфорной кислоты. Серную кислоту для взаимодействия с Са(Н2РО4)2 подают в смеси со вторым фильтратом, содержащим 34— 35% Р2О5, в виде раствора , регенерации. Разложение Са(Н2РО4)2 и кристаллизация полугидрата происходят при ' 95—400 °C. В жидкой фазе пульпы поддерживают «нулевой» или «минусовый» сернокислотный режим (практически от- 1 сутствует свободная серная кислота) при отношении Ж: Т от (2,5: 1) .до (3:1).
Создание необходимых градиентов концентраций и тем- ператур с предотвращением образования местных пересыще-’ ний в основном определяется условиями смешения реагентов. Лучшие результаты были достигнуты при подаче циркулирующей полугидратной пульпы из последнего экстрактора- непосредственно на стадию разложения апатита и противоточном смешении серной кислоты, разбавленной вторым
130
фильтратам, с обрабатываемой пулытой. Это позволяет максимально уменьшить пересыщение жидкой фазы ионами кальция и снизить концентрацию серной кислоты при разбавлении ее в прореагировавшей зоне.
При указанном технологическом режиме полугидрат выделяется в виде шарообразных изометрических компактных агрегатов диаметром до 100 мкм, хорошо фильтрующихся и стабильных при промывке водой.
Часть прореагировавшей пульпы (суспензии полугидрата в фосфорной кислоте, содержащей 45—48% Р2О5) направляют на стадию разложения фосфата, а часть отводят на фильтрование. Предварительно последнюю часть пульпы можно направить в гидроциклон для сгущения до значения Ж:Т = 2-: 1. Фильтруют и промывают полугидрат на ленточном фильтре по четырехстадийной, схеме (одно основное фильтрование и три промывки). Первый фильтрат частично отводят в виде продукции, а остальную массу его подают на разложение апатита.
Осадок отмывают от фосфорной кислоты на 98% при расходе 800 кг Н2О/т апатита и разрежении 0,045—0,05 МПа (0,45—0,5 ат). После этого осадок содержит ~47,5% влаги. После первой зоны фильтрования в осадке находится .30—35% жидкой фазы. Воду подают нагретой до 60—80 °C. Фильтрование и промывку продолжают 1,2—1,5 мин, а производительность фильтра достигает 1300—4500 кг/(м2-ч) сухого осадка. Нагрузка фильтра по зонам (в % от общей фильтрующей поверхности) распределяется следующим образом: в первой зоне — 5р—60, во второй — 20—30, в третьей и четвертой — по 10.
При фильтровании и промывке осадка фильтровальная ткань (лавсан на шелку) не гипсуется, и срок ее службы’ достигает 10 суток. На второй и третьей зонах фильтра откладывается незначительное количество осадка (в основном осадок состоит из' кремнефторидов), легко удаляемое, при периодической (1 раз в. 7—10 дней) промывке системы горячей водой. Промытый осадок полугидрата устойчив при хранении: он не схватывается в течение весьма длительного времени в закрытом помещении цеха, на открытой площадке и на шламовом поле.
Получаемую фосфорную кислоту, содержащую (в %): Р2О5— 45—47; СаО —0,1—0,2; F — 0,3—0,5; SQ3 — 0,2—0,4 и R2O3— 1,2, используют для производства кормовых фосфатов и удобрений. В отличие от обычной экстракционной фосфорной кислоты (неупаренной и упаренной), из нее не выделяется со временем значительных, количеств примесей и ее мо’жно без.труда транспортировать в цистернах.
С реакторов обеих стадий .процесса снимают 29— 30 кг/(м3-ч) Р2О5. Необходимая поверхность фильтрования 9* _ . 131
составляет 2,8—'2,9 м3/ч на 1 т PsO5. Кратность циркуляций пульпы, которая определяется отношением количества пульпы, возвращаемой на разложение апатита и отбираемой на фильтрование, составляет ~2:1.
Виже приведен примерный материальный баланс процесса по стадиям, (в кг) на 1 т апатитового концентрата (с учетом циркулирующих потоков):
На разложение апатита 1000 Со стадии разложения /
апатитовый концен- апатита
трат пульпа 14 457
первый фильтрат 2625 газы и водяные 168
циркулирующая 11 000 пары
пульпа Со стадии кристаллиза-
На кристаллизацию ции полугидрата 16 600
серная кислота • 965 пульпа
92,5% H2SO4) водяные пары 101
второй фильтрат 1279 От стадии фильтрова-
5 фосфорнокислотная 14 457 ния 790
пульпа продукционная кис-
На фильтрацию лота
вода на промывку 824 полугидрат 1700
v пульпа 5600 первый фильтрат на 2625
разложение апатита
второй фильтрат на 1279
кристаллизацию испаряется воды 30
Технико-экономические показатели
На производство фосфорной кислоты (1 т 100%-ной РгО5) концентрации 35—37% Р2О5 расходуется апатитового концентрата от 1,07 до 1,17 т (100% Р2О5), серной кислоты (моногидрата) от 2,50 до 2,65 т, электроэнергии от 105 до 150 кВт-ч, воды 16—50 м3, фильтровального полотна 0,06— 0,12 м2. Выход Р2О5 в кислоту 90—94% при степени извлечения 97—98% и коэффициенте отмывки 98—98,5% и остальных «хозяйственных» потерях 2,5—6%.'
С повышением концентрации продукционной кислоты до 45—48% Р2О5 выход Р2О5 в кислоту снижается до 85,5— 87%, а расходные коэффициенты на 1 т Р2О5 в кислоте составляют: апатита—1,12—.1,15 т (100% Р2О5), серной кислоты 2,6—2,9 т (100% H2SO4), электроэнергии 315— ! 340 кВт-ч, воды 16—20 м3 и фильтровального полотна 0,5— 0,7 м2. ,
Как видно из табл. III.4, фактическая цеховая себестоимость кислоты концентрации 35—37% Р2О5 в расчете на 1 т Р2О5 составляет 125,17 руб. Сопоставляя с табл. III.2 калькуляции себестоимости 1 т Р2О5 в кислоте концентрации 28—32% Р2О5, полученной дигидратным способом, можно заключить (без учета неизменных элементов — расходов на
132
Таблица Ш.4
Примерная калькуляция себестоимости производства экстракционной фосфорной кислоты концентрации 35—37% Р2О5 полугидратным методом из апатитового концентрата (на 1 т Р2О5)
Затраты Расход Цена за единицу/ руб. Сумма расходов/ руб.
Сырье апатитовый концентрат (39,4Р/ 2,850 17,93 51,00 Р2О5), т серная кислота (100,% H2SO4), т 2,533 24,25 61,43 Вспомогательные материалы известь негашеная (100% СаО), 0,053 18,97 1,01 т фильтровальная ткань, м2 0,049 4,75 0,23 Отходы кремнефтористоводородная кис- 0,025 18,00 0,45 лота (в пересчете на фтор), т Всего за вычетом отходов —- — 114,12 Энергетические затраты электроэнергия, кВт-ч 144,0 0,01324 Г,91 пар, Гкал 0,259 5,96 1,54 вода, м3 35,89 0,0063 0,23 сжатый воздух, тм3 0,085 2,04. 0,17 Зарплата с начислениями, руб — — 0,97 Амортизация и содержание оборудо- — — 5,32 вания, руб. Цеховые расходы, руб. — — 1,81 Цеховая себестоимость, руб. — — 125,17
сырье и материалы, ‘которые возрастают при полугидратном способе независимо от цены на них вследствие увеличения, расходных коэффициентов), что эффективность полугидрат-ного процесса определяется увеличением производительности системы, т. е. ее интенсивности; приводящей к уменьшению других затрат (энергетических, заработной платы с начислениями и т. п.). Затраты на эти статьи составляют, кроме общезаводских расходов, по дигидратному способу 17,09 н 16,53 для двух заводов (см. табл. III.2), а по полугидрат-ному способу—11,95 руб. Соответственно экономия, достигаемая в последнем случае, составляет 4—5 руб. на 1 т Р2О5, и при производительности 140 тыс. Р2О5 — 0,5—0,7 млн. руб. В основном экономический эффект полугидратного процесса (даже без учета снижения расходов на упарку кислоты до концентрации 54% Р2О5) обусловлен увеличением производительности труда в основном за счет снижения цеховых расходов и частично — уменьшения относительных расходов на амортизацию оборудования в интенсивных условиях его работы. Интенсификация, достигаемая при полугидратном
133
режиме, намного превышает усложнение эксплуатационных ; условий по сравнению с дигидратным процессом.
Так, в системах, работающих по дигидрйтжому режиму^ > смена фильтровальной ткани производится один раз в 25 суток, чистка трубопроводов карусельных фильтров — один р,аз-в год, промывка горячей водой трубопроводов продукционной ф'осфорной кислоты — через 2—3 месяца.
При полугидратном процессе замену фильтровальной ткани, чистку оборудования и промывку трубопроводов (в ТОМ : числе и промывку поддона фильтра и чистку буикеров пблу-гидрата под филцтром й на тракте при его удалении) приходится производить через 7—ФО дней работы'.
В соответствии с этим проектные и плановые режимы '» работы дигидратных "(мощностью ПО тыс. т Р2О5) и полу-гидратных (мощностью 140 ТЫС. Т Р2О5 в год) приняты с учетом отечественного и зарубежного опыта, соответственно, следующие [64]: рабочее время 330 дней по 24 ч (7920 ч) и 300 дней по 24 ч (7200 ч), планово-предупредительный ремонт—г20 дней по 24 ч (480 ч) и'50 дней по 24 ч (1200 ч)7 капитальный ремонт в обоих случаях—15 дней по 24 ч (360 ч).
Таким образом, рабочее время работы системы при полугидратном режиме сокращается на 720 ч (или 9—10%), а время на ППР увеличивается на 720 ч (или на 150%), т. е. в 2,5 раза. При дигидратном режиме на ППР при -ремонте и подготовке оборудования к очередному рабочему пробегу выделяют ~40 ч в неделю, а в полугидратном процессе — 24 ч через каждые шесть дней.
Режим‘времени работы полугид ратных систем (Для апатитового концентрата) почти такой же, какой .принят при дигидратной переработке богатой фосфоритной муки (28% Р2О5) Каратау, для которой, из-за повышенного загипсовы-вания аппаратуры и коммуникаций,- рабочее время составляет 305 дней по 24 ч (7320 ч), ППР—-45 дней по 24 ч (1080 ч), капитальный ремонт—15 дней по 24' ч (360 ч). В этих_условиях ремонт и чистку оборудования проводят ,в течение 18 ч после 5 дней работы.
В то же время интенсивность производства кислоты тем или иным способом является сложной категорией, определяемой не только, как это обычно делается, по степени выхода Р2О5 и удельным съемам Р2О5 с единицы реакционной аппаратуры (экстрактора и фильтра), но и рядом других параметров — режимом времени работы, надежностью и устойчивостью процесса и т. п.
Предложена [65] попытка оценки совокупной интенсивности (7абс), отражающей технологические, аппаратурные и эксплуатационные условия работы системы, по формуле:
7абс =1КвыхКвпКконц?э?ф
134
Здесь КВых — степень извлечения Р2О5, %; Квр — коэффициент использования времени, учитывающий рабочее время системы (тр — сутки пробега системы за 365 дней), т. е. Гр/365; Яконц — коэффициент, учитывающий эффективность от повышения концентрации кислоты по сравнению с общепринятой (30% Р2О5); значение Дконц можно рассчитать на оснований соотношения стоимостей упаренной . кислоты (54% Р2О5), полученной из дигидратной (30% РгО5) и полу-гидратной (42% Р2О5) и составляющих соответственно 152,05 и 139,50 руб. (данные НИУИФ); q3 — удельный съем с экстрактора, кг Р2О5/(м3-ч); <?ф —удельный съем с фильтра, 1КГ Р2О5/(м2-ч).,
Приблизительно при одних и тех же значениях Квых, с/ф, Квр и Ккыщ абсолютные интенсивности двух систем, работающих, по полугидратному процессу мощностью 140 и 280 тыс. т Р2О5, составляют для первой 4589 и для второй 6215 [65],.т. е. отличаются на 35,5%. Значение 6215 получается в основном за счет более интенсивной работы системы большей единичной мощности, равной соответственно 470 и 1000 т РгО5 в сутки. Удельный съем Р2О5 составляет в первом случае 22, а во втором — 30 кг P20s/(m3-4), т. е. отличаются на ту же величину (36%).
Таким образом, понятие совокупной интенсивности, во-первых, не отражает уменьшения удельных капиталовложений, затрат на обслуживание, а также влияния других факторов, присущих установкам повышенной единичной мощности, и, во-вторых, по-видимому, сводится в неявном виде к эффективности, достигаемой на укрупненных системах. ,
На отечественных заводах мощность большинства систем, работающих на апатитовом концентрате по дигидратному режиму, составляет 110 тыс. т P20s. Разрабатываются и проектируются системы мощностью 140, 280-—350 тыс. т РгО5 как в дигидратном, так и пол угидратном режимах, как оптимальные по снижению удельных капитальных* и экс-'плуатационных расходов на 1 т Р2О5.
.Что касается эффективности полугидратных-систем., то, помимо увеличения на них производительности, дальнейшее усовершенствование процесса и аппаратурного его оформления позволит увеличить рабочее время их эксплуатации, уменьшить затраты на чистку и профилактический ремонт оборудования. Это связано также с установлением оптимальной концентрации кислоты, производимой полупидрат-ным методом (которая, по-видимому, находится в диапазоне 35—38% Р2О5 или около него), других технолотических параметров, а также конструктивного оформления процесса с применением в первую очередь.высококачественных марок нержавеющей стали.
135
-
Так, ,при раздельном разложении, фосфатного сырья серной и оборотной фосфорными кислотами в первом реакторе при 85—90 °C при небольшом избытке SO3 в жидкой фазе пульпы и завершении кристаллизации полугидрата во втором реакторе при несколько пониженной температуре и добавлении небольшого количества серной кислоты произво- : дительность реакторов и фильтров, как и при дигидратном процессе (стр. 103), увеличивается на 10—15%, а выход Р2О5— на 0,5—0,8% [5].
Указывают, что при НО °C полугидратный процесс можно вести в интервале концентраций 32—50% Р2О5 в течение 2,5—4 ч. При содержании в жидкой фазе пульпы 45— 50% Р2О5 и 0,5—1,3% SO3 степень разложения апатитового концентрата составляет 94—96%, а отмывки фосфополугид-рат.а-г98—99% [66].
В конструктивном отношении для лолугидратного процесса оптимальным может оказаться, по-видимому, экстрактор из Двух сдвоенных цилиндров, изготовленный из стали марки 0Х23Н28МЗДЗТ (ЭИ-943) с футеровкой (стр. 81) и наружной перекачкой кислоты насосами, изготовленными из сплава Н54Х22М7С4Д4Л [67]. Для изготовления реактора, очевидно, возможно использовать и другие марки стали, например 0Х21Н-21М4Б [68].
III.6. ДВУСТАДИЙНЫЕ СПОСОБЫ
Сложность стабилизации полугидрата и зависимость ее . как от постоянных, так и переменных (случайных) параметров обусловливает затруднения в регулировании промышленного процесса производства кислоты в полугидратном режиме, особенно на установках большой мощности. Это вызвало за рубежом и в Советском Союзе интерес к двустадийным способам получения фосфорной кислоты, позволяющим повысить на 2,0—2,5% (абс.) степень извлечения
Р2О5, полуДить концентрированную кислоту (до 55%) и использовать сульфат кальция без дополнительной очистки для производства строительных материалов.
На первой стадии процесс ведут с выделением в твердую фазу полугидрата или дигидра.та, а во второй стадии перекристаллизовывают полугидрат в дигидрат или дигицрат в полугидрат.
Полугидратно-дигидратный способ
В данном случае на первой стадии выделяется относительно устойчивый полугидрат, не обводняющийся в процессе экстракции. Во второй стадии полугидрат, не отделенный или после отделения от жидкой фазы, перекристаллизовы
136
вают в дигидрат с выделением крупных, хорошо образованных и быстро фильтрующихся кристаллов.
(Преимущества указанного способа — максимальная (до 98,5%) степень извлечения из сырья фосфорной кислоты в раствор при минимальном расходе серной кислоты и получение гипса высокого качества, содержащего не больше 0,3% ' . общей Р2О5 (вместо обычных 0,5—4,5%) и 0,02—0,08% водорастворимой Р2О5. Это объясняется предотвращением замещения сульфа1-ионо1в фосфат-ионами в кристаллической решетке осадка и высвобождением ионов НРО42-, которые удерживались (адсорбировались) на поверхности первоначально выделившихся частиц твердой -фазы, так как полугидрат предварительно переходит в жидкую фазу. По этому способу работают некоторые заводы в Японии и в других странах с максимальной суточной мощностью 200 т (60— 65 тыс. т в 1 год) Р2О5 (кислота содержит 30—32% Р2О5 • ;[27, 47].
Для выделения негидратирующегося при экстракции по- лугидрата процесс проводят при повышенных температурах (90—95 °C).
Гидратация полугидрата и кристаллизация дигидрата при различных условиях описаны' в гл,- II. Ниже приведены некоторые дополнительные сведения перекристаллизации по- .лугидрата в дигидрат (и частично дигидрата в полугидрат).
Фтор-ионы, в отличие от всегда присутствующих в кислоте кремнефторид-ионов, способствуют оводнению полугидрата. В то же время гидратация полугидрата протекает бы-; стрее, если он получен из фосфорита, содержащего небольшие количества фтористых соединений (~0,3%) и окислов ' трехвалентных металлов (~0,8%): фтор в этом случае, ве-. роятно, связан в несиликатный комплексный ион. В присут-% ствии серной кислоты концентрация фтор-ионов увеличива-I ется за счет распада кремнефторид-ионов, и при избытке полуторных окислов гидратация ускоряется тем в большей ! степени, чем больше содержание серной,кислоты в растворе [10, вып. 4, с. 3]. Она ускоряется также'при добавлении к фосфату или в пульпу, активного кремнезема (реагирующего с фтором с образованием SiFe2-) и одновременном добавлении ионов калия или натрия, обусловливающих выделение . из раствора фторсиликатов последних.
; Перекристаллизация полугидрата ускоряется в присут-, ствии минеральных кислот — серной, азотной, соляной и их натриевых солей, сульфатов лития, аммония, хлорида же-’ лева и других. При этом в кислых средах процесс проте-( кает активйее, чем в нейтральных и щелочных, а технический фосфополупидрат, получаемый в производстве фосфор-. ' ной кислоты, оводняется значительно медленнее, чем искус-; ственно приготовленный «чистый» полугидрат [63,^69]. Гид
137
ратаЦия тонкодиоперсного полугидрата происходит интенсивнее, чем крупнокристаллического, при сопоставимых содержаниях в осадке фосфорной кислоты [70].
Скорость гидратации полугидрата зависит и от многих других факторов—содержания в осадке воднонераствери-мой Р2О5, захваченной Р2О5 в присутствии в растворе привнесенных из апатитового концентрата соединений стронция и церия. При содержании последних в растворе в количестве 3,8—<7,0% SrO или 1,8—2,8% Се2О3 гидратация замедляется в ~20 раз по сравнению с чистыми растворами, но полностью завершается в течение не более 6—7 ч [70]. Большая скорость превращения полугидрата в дигидрат наблюдается при 50—60°C и содержании в растворе 5—8% H2SO4 вслед-, ствие, по-видимому, максимальной скорости растворения и растворимости кристаллогидратов в этих условиях..
Хорошо фильтрующиеся кристаллы дигйдрата (размером 5X150 мкм) выделяются - при содержании в кислоте 27— 31% Р2О5, 2,5% SO3 и продолжительности гидратации 5— 9 ч. При ускоренной гидратации выделяются-длинные и тонкие кристаллы, обладающие худшей фильтруемостью. Для ускорения перехода полугидрата в гипс при одновременном образовании крупных кристаллов изометрической формы перекристаллизацию ведут в присутствии зародышей мелких кристаллов гипса. Их получают, добавляя немного измельченного природного, фосфата к' пульпе, содержащей полугидрат и фосфорную кислоту, в условиях "-стабильности гипса.
Сокращение длительности перекристаллизации полугид- -рата' в гипс достигается также тем, что процесс не доводят до конца. Степень гидратации (моль Н2О/моль CaSO4) составляет обычно 1,8—1,9 (вместо 2,0 в CaSO4-2H2O).
В практических условиях перекристаллизацию полугидрата в гипс ведут при 40—65 °C в двух и более кристаллизаторах с рециркуляцией пульпы, содержащей гидратированные кристаллы. Продолжительность кристаллизации гипса в трех кристаллизаторах составляет ~12 ч. Температуру поддерживают в первом кристаллизаторе 60, во втором — 55 и в третьем — 50 °C. При этом средняя степень гидратации твердой фазы-соответствует составу: в первом кристаллизаторе CaSO4-1,68Н2О, во втором — CaSO4-1,86Н2О и в третьем — CaSO4- 1.94Н2О.
Часть пульпы, вытекающей из последнего кристаллизатора, возвращают в первый кристаллизатор, а другую часть фильтруют на вакуум-фильтре непрерывного действия с двумя или тремя промывками лепешки гипса.
В другом варианте полугидратного-дигидратного процесса полугидрат отфильтровываю^ от кислоты концентрации более 40% РгО5 и затем его перекристаллизовывают в гипс
138
в растворе серной кислоты и разбавленном растворе фосфорной кислоты. После отфильтровывания гипса раствор, содержащий серную и фосфорную кислоты, используют для промывки осадка' полугидрата. после первого фильтрования, а затем подают на стадию экстракции.
Имеются сведения [7'1] об использовании этого метода на заводах, работающих по способу «Ниссон» (фирма «Тойо—Менка»), Указывают [72], что этим методом возможно перерабатывать любые типы фосфатов и получить кислоту с концентрацией 45—50% Р2О5 и гипс высокого качества. Система Тойо—Менка производительностью 510 т/сутки Р2О5 (с концентрацией 42% Р2О5) состоит (по рекламным данным) из пяти экстракторов и четырех кристаллизаторов с общим объемом 4026 м3 (в 3—4 раза, большим, чем в обычных дитидратных системах) и двух карусельных фильтров Эймко с рабочей поверхностью 129 и 88 м2 и удельным съемом всего'2,3 т Р2О5 с 1 м2 поверхности фильтра в сутки (в 1,75—2 раза меньше, чем в дигидратном режиме).
Дигидратно-полугидратный способ
Перекристаллизацию дигидрата в полугидрат рассматривают в качестве одного из путей перевода действующих систем с дигидратного на подугидратный режим [73]. В статических условиях (добавлением в экстрактор с дигидрат-ной пульпой упаренной фосфорной кислоты при одновременном повышении температуры до 95—98 °C) при содержании в кислоте 38—42% Р2О5, 2% SO3 и 98 °C дигидрат полностью переходит в полугидрат за 1,5—4 ч. При непрерывном ведении процесса с подачей сырья и отводом полугидратной пульпы на фильтрование время перекристаллизации сокращается в ~1,5—2 раза.
Образующийся полугидрат выделяется в виде плохо фильтрующихся тонких нитевидных кристаллов длиной ГООО—'2000 мкм и шириной 3—Ю мкм.
Исследования фильтрования полугидрата, полученного дегидратацией реактивного CaSO4-2H2O в растворах фосфорной кислоты, содержащих разные примеси, показали, что только повышение температуры от 80 до 105 °C приводит к уменьшению времени фильтрования в ~2 раза [10, вып. 3, с. 7]. С увеличением концентрации фосфорной кислоты от 40 до 55% РгО5 длительность фильтрования при 95 °C увеличивается в 4—5 раз. Рост содержания А12О3, Fe2O3 и SiF62- до 2% в растворе увеличивает время фильтрования в ~2 раза.
Процесс может быть проведен следующим образом.
'Природный фосфат разлагают смесью серной и фосфорной кислот при 50—7'0 °C с получением кислоты концентра-
139
цией до 39% РгО5. Из полученной пульпы отделяют ('без промывки гипса) декантацией продукционную кислоту (3'3— 38% Р2О5). Отстоявшуюся гипсовую суспензию-смешивают в дегидраторе с концентрированной серной кислотой или смесью ее с фосфорной кислотой. В жидкой фазе суспензии содержится 20—30% Р2О5 и 10—20% H2SO4. При этом температура повышается до 75—85О|С, что .способствует быстрой перекристаллизации дигидрата в полугидрат, протекающей со скоростью, значительно превышающей скорость обратного перехода. В этих условиях промытый полугидрат содержит всего ~0,'2% общей Р2О5 и в том числе 0,01 % водорастворимой, а также 0,1% F.
По-видимому, в определенных условиях может стать интересным совмещение в одном технологическом цикле ди-гидратного и полугидратного процессов. В этом случае апатитовый концентрат разлагают в две ступени без перекристаллизации кристаллогидратов сульфата кальция. В первой ступени часть апатита обрабатывают в дипидратном режиме при 75 °C, остальные в полугидратном Испарительном режиме при 100—1105 °C в присутствии раствора разбавления— первого фильтрата, содержащего ~32% Р2О5.
Пульпу, получаемую в первой ступени с концентрацией в жидкой фазе 28—30% Р2О5, смешивают с пульпой из второй ступени (в кислоте 38—40% Р2О5). Смешанную пульпу направляют на фильтрование. За счет интенсивного разложения части апатита и увеличения концентрации продукционной кислоты от 28—29 до 31—32% РгО5 возможно ’этим способом увеличить выпуск фосфорной кислоты, производительность труда и оборудования на типовой житие мощностью 105—110 тыс. т Р2О5 в гОд на 20—125%.'
III.7. ПОЛУЧЕНИЕ КОНЦЕНТРИРОВАННОЙ ФОСФОРНОЙ КИСЛОТЫ
Условия проведения процесса
Теоретически выпаривание чистых растворов может привести к получению очень концентрированной кислоты-, так как над фосфорной кислотой любых концентраций (вплоть до 98% Н3РО4) пар содержит практически только воду. Выпаривание воды до остаточного содержания 53—55% Р2О5 не сопровождается дегидратацией фосфорной кислоты и образованием фосфорного ангидрида в неорто-форме. Однако процесс осложняется сильной коррозией аппаратуры и выделением примесей, содержащихся в кислоте. Горячая фосфорная кислота оказывает сильное корродирующее действие на большинство известных металлов, сплавов н силикатшуке-рамическмх материалов. Коррозия резко усиливается при (выпаривании раствора, содержащего 2—5% свободной 140
H2SO4. Поэтому к такой кислоте, .поступающей на выпаривание, целесообразно добавлять раствор Са^Н^РООг в количестве, необходимом для уменьшения содержания H2SO4 до 1 % и меньше.
Отечественные марки графита (ППГ, АРВУ, ПРОГ, МПГ-6) являются высококорровионнюстойкими материалами до 150°C в разбавленной и концентрированной (85% НЭРО4 или 61,5% PaOs)-фосфорной кислоте [74]. Выделяющиеся в процессе упаривания осадки могут забивать аппаратуру, в результате чего резко снижается ее производительность.
С увеличением концентрации и понижением температуры фосфорной кислоты уменьшается растворимость содержащихся в ней примесей. Все это приводит к непрерывному увеличению количества' выделяющихся осадков как при упаривании кислоты, так и при последующем ее охлаждении (остывании). Количество и состав выделяющихся осадков зависят от.условий получения фосфорной кислоты. С увеличением в ней свободной H2SO4 уменьшается осаждение солей кальция (вследствие снижения их содержания в. исходной кислоте) и усиливается выделение солей железа и алюминия. Осадки состоят обычно из сульфатов и фосфатов кальция, железа и алюминия, фторида и кремнефторида кальция (табл. III.5).
Осадок, выделяющийся при стоянии исходной фосфорной кислоты (28—32% Р2О5), состоит главным образом из сульфата кальция- и кремнефторида натрия, а также небольшого', количества комплексных кремнефторидов. При добавлении к кислоте солей натрия количество кремнефторидов в осадке увеличивается. Основным компонентом отложений, образующихся в "первой и второй зонах вакуум-фильтра, является NajSiFe (60—80%) [75]; остальное—сульфат кальция и комплексные соединения фосфатов равных элементов. Твердые вещества, содержащиеся в упаренной кислоте (50— 54% РгО5), состоят ив ангидрита, полугидрата и смеси изоморфных однозамещенных ортофосфатов железа, алюминия и кальция вероятного состава: (Fe, AQsKHuCPO^g^HjO.
Таблица II 1.5
Состав осадков, выделяющихся из экстракционной и упаренной фосфорной кислоты
Кислота Содержание в осадке, %
Р2О6 Са so*- Al Fe F SiO2
Экстракционная, поступающая 1,9 14,8 38,9 0,3 0,2 19,9 10,3 на упаривание (32% Р2О5) Упаренная из концентратора 6,8 12,9 29 0 5,1 0,3 22,0 5,3 (54% Р2О5) Упаренная после хранения 38,9 3,3 4,7 1,5 9,6 12,9 6,1 (54% Р2О6)
141
Добавление солей калия к этой кислоте увеличивает степень осаждения соединений железа и, в некоторой степени, соединений алюминия. Состав отложений, образующихся на элементах аппаратуры при концентрировании кислоты, в основном такой же, какой выделяется из упаренной кислоты. Содержание примесей в сырье • и' экстракционной кислоте обусловливает возможную конечную концентрацию ее при упаривании.
Кислота, содержащая 53-=55.% Р2О5, может быть получена из относительно мало загрязненных фосфатов — апатитового концентрата или обогащенных высокосортных фосфоритов.
Кислота, полученная из низкокачественного фосфорита и упаренная до концентрации 40—45% Р2О5, может после охлаждения превратиться в густую кашеобразную массу. Кислота, полученная из неббогащенных фосфоритов Каратау, вследствие содержания значительного количества солей магния, может быть упарена только до концентрации 37— 38% Р2О5. Дальнейшее концентрирование ее приводит к образованию весьма вязкой загустевающей массы, что объясняется выделением тонкодисперсных, трудно оседающих кристаллов фторида магния, которые образуются при разложении содержащегося в кислоте кремнефторида' магния при температуре выше 80 °C.
Вязкость кислоты, полученной из фосфорита. Каратау, концентрации 43—45% Р2О5 при. 20 °C равна вязкости кис- , лоты, полученной из апатитов, концентрации 52,5% Р2О5 и составляет ~30 сП [76, № 1].
При упаривании кислоты из фосфоритов Каратау до 40— 42% Р2О5 из нее при 30 °C выделяются относительно небольшие количества осадка.; упаренная, до 55% Р2О5 содержит большое количество плохо фильтрующегося белого коллоидного осадка, содержащего в числе других соединения полуторных окислов и окиси кальция [76, № 6].
Остывшая кислота концентрации 54% Р2О5 представляет собой неподвижную густую массу.
Температура упаривания кислоты, полученной из чили-сайских фосфоритов, в интервале концентраций от 20 до 51% Р2О5 и от 51 до 53% Р2О5 повышается по сравнению с .чистыми растворами, соответственно, на 15—20 и 25— 33°C [76, № 1].
Вязкость упаренной кислоты резко увеличивается и кислота концентрации >45% Р2О5 она представляет собой при остывании густую незастывшую массу. В диапазоне концентраций 40—42% Р2О5 она достаточно подвижна и транспортабельна.
.По мере увеличения концентрации фосфорной кислоты возрастает давление пара растворенной в ней F^SiFe и зна-142
ЧйТельная ее часть удаляется в виде смеси SiP4 и HF. Прй выпаривании кислоты до 55—57% Р2О5 в газовую фазу удаляется ~85% содержащегося в ней фтора, который улавливают в абсорбционной системе (стр. 206).
На первых, мало производительных чреновых свинцовых установках кислоту упаривали периодическим путем несколько* суток.
Попытка применения свинцовых трубчатых или змеевиковых выпарных аппаратов, обогреваемых парсим, не увенчалась успехом вследствие быстрого отложения на греющих трубах шламовой корки и ухудшения теплопередачи.
В настоящее время экстракционную фосфорную кислоту, выпаривают в основном в аппаратах с теплопередачей через греющую поверхность, обогреваемую паром (или другими теплоносителями). В небольших масштабах используют установки с непосредственным обогревом топочными газами *•> [63; 76, 'Н. П. Попов]. Вакуум-выпарные аппараты применяют с выносной греющей камерой, с графитовыми нагревательными трубками, с внутренними! вертикальными свин-‘ цо'выми трубами или плёночные. Упаривание кислоты непосредственным обогревом осуществляют в барботажных концентраторах-камерах из кислотоупорного, материала; оно Оказалось особенно эффективным в производстве суперфос-фо.рной кислоты (см. гл. V, стр. 182)7
Выпарка фосфорной кислоты под разрежением протекает при пониженной температуре (70—90°C). Это. благоприятно в коррозийном отношении и позволяет использовать дешевый отбросный пар сернокислотного производства.
За рубежом, (в США и других странах) весьма распро-\ странены однокорпусные вакуум-выпарные аппараты С’внут-. »ренней или выносной греющей камерой. Корпус выпарного аппарата (высотой 4,5 и диаметром 1,95 м) изготовляют из v гуммированной стали. Графитовые нагревательные трубки ’ г "устанавливают вертикально. Пар под давлением 0,07— 0,7 МПа (0,7—7 ат) проходит в межтрубном пространстве, а внутри трубок циркулирует кислота. Аппарат работает при разрежении 86—93 кПа (650—700 мм рт. ст.); трубопроводы сокового пара изготовляют из стали и гуммируют.
Наиболее распространены выпарные аппараты с выносной нагревательной камерой, в которых процесс проводят при интенсивной принудительной циркуляции кислоты и автоматическом контроле режимных параметров (рис. III.13). : Эффективное уменьшение инкрустации греющих трубок до-у стнгнуто при упарке кислоты по циркуляционной схеме, в ' которой циркулирующая упаренная концентрированная фас-L- форная кислота смешивается с исходной разбавленной. Растворимость примесей в упаренной кислоте значительно мень-* ше; чем в исходной. Поэтому при смешении примеси, содер-
143
Рис. III.13. Вакуум-вьшарной аппарат
с интенсивной циркуляцией:
1 — подогреватель; 2 — испарительная камера; 3 — конденсатор; 4 — сборник-отстойник.
жащиеся в разбавленной кислоте, кристаллизуются. Их. отделяют в 1 отстойнике, а- осветленную кислоту направляют в выпарной аппарат. В результате этого примеси, содержащиеся в исходной кислоте, вы- деляются в основном не в выпар- -ном аппарате, а при смешении ее с концентрированной кислотой. #
Фактически по этому способу
упаривают продукционную кислоту (т. е. сконцентрированную до конечной концентрации) и несколько повышают ее концентрацию до желаемого значения. Меньшую часть ее отводят в качестве продукта, а большую после отстаивания примесей направляют на концент-
рирование.
Процесс проводят и так, что в выпарном аппарате получают кислоту с конечной концентрацией, соответствующей готовому продукту. Меньшую часть полученной кислоты отводят как готовый продукт, а основную ее массу после смешения с разбавленной кислотой и отделения выделившихся примесей возвращают в цикл. Кратность циркуляции (т. е. отношение циркулирующей кислоты к массе продукта) составляет 100—(150. >
В результате отделения в отстойнике части примесей отложение солей на греющих элементах при выпаривании резко уменьшается. Образующиеся небольшие отложения (накипь) удаляют 1 раз в 3 дня для кислоты из фосфоритов или 1 раз в 10 дней для кислоты из апатитового концентрата (аппарат останавливают и промывают горячей водой или содовым раствором). Пульпу на нижней части отстойника откачивают шламовым насосом в экстракторы получения фосфорной кислоты.
Иногда для облегчения очистки аппаратуры кислоту упаривают до концентрации на 2—в % больше заданной, и осадок после смешения со слабой кислотой не отделяют. Вместо отстойника под аппаратом устанавливают мешалку для поддержания твердых примесей во взвешенном состоянии. Оса-> док вместе с кислотой циркулирует через аппарат, но не отлагается на греющих трубках вследствие большой скорости
144
циркуляции. После 60—70 ч работы аппаратуру промывают под разрежением в течение 8—12 ч горячей подкисленной водой при повышенной скорости циркуляции.
В некоторых системах (например, французской фирмы Speischime) вместо трубчатых подогревателей применяют обогревательные пластинчатые элементы из рифленых панелей (с размерами 4550X1575X892 мм) из прокатного листа нержавеющей стали толщиной 12 мм, покрытые слоями эбонита (толщиной 5 мм) и резины (толщиной 6 мм). Поверхность теплообмена подогревателя—172 м2. В обогревательные элементы подают пар под давлением 0,3 МПа (3 ат) (133°C), подогреватель соединяют со стальным гуммирован нием вакуум-испарителем, имеющим высоту 8,23 и диаметр 3,35 м.
Производительность установки, состоящей из трех параллельно работающих выпарных аппаратов, была рассчитана на 340 т РгО5 в сутки (по выпаренной воде 16,83 т/ч) при концентрировании кислоты от 32 до 54% Р2О5. Однако уже в первый период эксплуатации они оказались малопригодными вследствие инкрустации греющих поверхностей пластинчатых теплообменников, интенсивной их коррозии и забивки промежутков между пластинами рыхлым осадком, состоящим из сульфата кальция,, кремнефторидов натрия и калия и фосфатов полуторных окислов. Кроме того, из испарителей вместе с паром уносилось много брызг фосфорной кислоты, которая полностью переходила в кремнефтористую кислоту.
Более удобны графитовые блочные теплообменники, представляющие собой блоки с просверленными каналами для кислоты и пара (поверхность нагрева 158—228 м2). Концентрирование кислоты производят при разрежении 89 кПа (670 мм рт. ст.). При мощности производства в 100—'110 тыс. т Р2О5 в год для концентрирования фосфорной кислоты от'содержания 32 до 54% .Р2О5 устанавливают три выпарных, аппарата, работающих параллельно.
' При замене пластинчатых теплообменников из нержавеющей стали блочными графитовыми, увеличении частоты вращения осевого циркуляционного; насоса с 700 до 1000 об/мин, установке ловушки в циркуляционном контуре для предотвращения забивки теплообменника и других усовершенствованиях производительность вакуум-вышарных установок повысилась на 10—43% по сравнению с проектной [77].
Процесс ведут следующим образом (рис. III.14). Исходную фосфорную кислоту, содержащую 28—30% Р2О5, 2,0— 3,8% SO3, 1,57—1,73% F, подают из бака-хранилища в выпарной аппарат через трубу, соединяющую теплообменник и испаритель. В последнем поддерживается температура 85—90°C при разрежении 89—92 кПа (670—690 мм рт. ст.), 10—5473 145
Рис. Ш.14. Усовершенствованная вак.уум-выпарная установка:
1 — теплообменник; 2— испаритель; 3, 6 — циркуляционный насос; 4 — брызгоуловитель; 5 — рециркуляционный бак; 7 — промывная башня; 8 — поверхностный барометрический конденсатор; 9 — двухступенчатое ваку-ум-эжекторное устройство; 10—-барометрический затвор.
создаваемом двухступенчатым пароэжекторным устройством. Упаренная кислота, содержащая 51—53% Р2О5, 3,7—4,4% SO3, 0,'6—0,8% F, самотеком частично выводится из испарителя в хранилище; остальную основную массу возвращают, в циркуляционный контур. При помощи осевого циркуляционного насоса она поступает в графитовый теплообменник, обогреваемый паром, в количестве 10—14 т/ч с избыточным давлением 0,18—0,25 МПа .(1,8—'2,5 ат). Теплообменник с общей поверхностью нагрева 228 м2 представляет собой греющую камеру, состоящую из 13 графитовых блоков с 308 проходными каналами диаметром 35 мм. Кислота протекает по каналам со скоростью 2,7 м/ч. Коэффициент теплоотдачи теплообменника при загрязненной поверхности составляет 150Q—2100 кДж/(м2-ч). Осевой циркуляционный насос производительностью 3000 м3/ч с напором 3,2 м и частотой вращения 1000 об/мин выполнен из стали типа 0Х23Н28МЗДЭТ. Газопаровая смесь, отсасываемая из испарителя пароэжекторным устройством, отделяется от капель фосфорной кислоты и соединений фтора, а очищенный водяной пар конденсируется в поверхностном конденсаторе.
Производительность одного вакуум-выпарного аппарата (каждого из трех в установке для концентрирования кислоты) составляет 5,5—6,8 т/ч 100%-ной Р2О5 или 8,9—9,8 т/ч выпаренной воды. 'Расход пара с избыточным давлением 0,4—0,5 МПа (4—5 ат) на пароэжекторную установку равен 0,8 т/ч, расход воды на поверхностный конденсатор 760—800 м3/ч. Благостей с 1 м2 поверхности нагрева графитового теплообменника — 39—45 кг/ч.
146
Из-ва инкрустаций каналов установку останавливают после 5—7 дней работы на 8—12 ч для промывки и чистки. Промывают аппарат 2—4%-ным раствором кремнефтористоводородной кислоты. Чистка теплробменников весьма трудоемка и затруднительна вследствие хрупкости графита.
Обычно за 5—6-суточный пробег коэффициент теплопередачи снижается на 625—420 кДж/(м2 ч • К). При обработке всего выпариваемого потока затравочными кристаллами снижение коэффициента теплопередачи через 12 суток составило ~100 кДж/(м2-ч-К), т. е. в 3—4 раза меньше [77].
Осмотр трубок теплообменника, а также данные изменения коэффициента теплопередачи свидетельствуют, что скорость накипеобразования составляет не более 0,25—0,3 мм в месяц (вместо 0,2—0,5 мм в 160 трубках, 1—5 мм в 100 трубках и полностью забитых 48 трубок — через 7 суток работы по обычному режиму). На этом основании сделано заключение, что при обработке кислоты затравочными кристаллами, (когда отложения при выпаривании сосредоточены в объеме жидкости), срок работы аппарата между промывками может быть продлен до 2—3 месяцев.
Упаренная фосфорная киблота имеет плотность 1,65— 1,75 г/см3 и содержит, в %: Р2О5 — 52—54; СаО — 0,1— 0,3; SO3 — 3,4—4,2; FesOs — 0,7—0,75; А12О3 — 0,55—0,6; MgO—' 0,02—0,04; SiO2 — 0,35—0,4; F —0,4—0,5; Na2O—<0,02 и K2O —0,06.
На 1 т Р2О5 необходимо выпарить ~1,3 т воды и израсходовать 1,6—'1,7 т греющего пара.
Полузаводскими испытаниями установлена возможность упаривания в вакуум-выпарном аппарате с принудительной циркуляцией и вынесенной зоной кипения неосветленной не-обеСфторенной кислоты, полученной из чилисайокого флотационного концентрата, до концентрации 40—43% Р2О5. При скорости циркуляции в трубках греющей камеры 2,5—3 м/с срок работы аппарата между промывками определяется примерно в течение 3—4 суток [40, выл. 3, с. 5].
Накипь после промывки аппарата кремнефтористоводородной кислотой содержала, в %: Н2Ообщ— 9,32; Р2О5общ — 8,43; Р2О5во-дн — 6,18; SO3 — 33,55; СаО — 22,85; MgO—1,02; Fe2O3 — 0,45; R2O3—3,28; F — ’2,1; нерастворимый остаток — 25,5 и SiO2pac^ —0,46.
- К недостаткам циркуляционной системы следует отнести ' необходимость перекачивания больших масс выпаренной кислоты, довольно частую остановку системы для ее чистки и получение загрязненного продукта (5—18% твердых частиц в упаренной кислоте).
Применяемые иногда схемы с рециркуляцией шлама позволяют получить осветленный готовый продукт. Для этого из упаренной кислоты отделяют на центрифуге нераствори
10*
147
мые частицы и возвращают их в отстойник-питатель, куда ' подают поступающую на концентрирование разбавленную кислоту. Некоторое количество продукта из центрифуги (10—<35%) направляют в питательный бак установки (в качестве зародышей кристаллизации). Шлам из -отстойника перекачивают в экстрактор или на вакуум-фильтр, а жидкость— в выпарной аппарат. Осветленная кислота содержала ~О,'6°/о твердых частиц.
Дальнейшее осаждение примесей при хранении кислоты после центрифугирования можно предупредить нагреванием ее в течение 10—15 мин при 120—150 °C под давлением 0,2— 0,5 МПа- (2—5 ат), не допуская концентрирования кислоты. Затем кислоту следует охладить до 50—70 °C для уменьшения коррозии. Из обработанной таким образом кислоты шлам не выделяется при перевозке и хранении в течение 2—4 месяцев.
Инкрустация греющей поверхности существенно уменьшается при регулировании кристаллизации- выделяющихся при-. месей (в частности, сульфата кальция) в вакуум-выпарны-х аппаратах, оборудованных в циркуляционном контуре специальной емкостью для - кристаллизации -основной массы примесей и укрупнения их частиц.,
Исходную фосфорную кислоту вводят в циркуляционную трубу. В выносном кипятильнике примеси частично выделяются в виде зародышевых кристаллов и во взвешенном состоянии поступают в испаритель.
Сконцентрированный раствор стекает из испарителя по центральной циркуляционной трубе в расположенный иод испарителем кристаллизатор. Здесь кристаллизация продолжается на ранее выпавших зародышах, в результате чего примеси выделяются в виде крупных кристаллов. По мере дальнейшего роста кристаллов наиболее крупные из них опускаются на- дно аппарата, откуда их выводят и отфильтровывают от кислоты.
Для этого из кристаллизатора отводят на фильтр часть пульпы в количестве, соответствующем получаемой продук--ционной кислоты. Влажные кристаллы возвращают в экстракторы разложения фосфата для использования уносимой ими фосфорной кислоты. Основную массу пульпы после смешения со свежей кислотой вновь возвращают в цикл.
В’ зависимости от производительности процесс можно вести как на однокорпусных, так и на многокорпусных установках. Суточная производительность установки из трех пог следовательно соединенных аппаратов составляет 360 т РгО5 (2'2 т”выпаренной воды в 1 ч). Подобные системы могут работать 30 сут до- остановки для удаления отложений.
-Капиталовложения в вакуум-выпар ной аппарат с кристаллизатором на 25% больше, чем в обычный. Но это уве-
личение окупается уменьшением продолжительности остановок аппаратуры для очистки и облегчением операций хранений продукционной кислоты и ее использования. С увеличением суточной производительности системы от 150 до 450 т Р2О5 (При концентрировании кислоты от 30 до 54% Р2О5) удельные капитальные затраты (в расчете на 1 т Р2О5) снижаются на 25%, а эксплуатационные—на 10—12%.
На, упарииание кислоты от 30 до 54% Р2О5 расходуется (!в расчёте на 1 т Р2О5 и в зависимости от пр о ив води тел внести) 10—15 кВт-ч электроэнергии, 1,8—>1,9 т пара* и 40— 75 м3 воды при 18 °C.
Независимо от применяемого метода (вакуум-выпарки или непосредственного нагрева) концентрирование фосфорной кислоты—дорогостоящий процесс.
Для получения из кислоты, содержащей 30% Р2О5, продукта с концентрацией 50% Р2О5 необходимо' удалить 1333 кг воды на 1 т Р2О5. Это связано с расходом 1700 кг пара при вакуум-выпарке или 100 кг нефти в аппаратах непосредственного обогрева кислоты топочными газами. В то же время паровая выпарка, фосфорной кислоты под вакуумом при определенных условиях (использование отброс-_ ного пара, переработка относительно чистых растворов и т. д.) может быть дешевле, чем выпарка горячими газами, на она неудобна из-за коррозии и эрозии. Вследствие инкрустации поверхностей нагрева графитовые теплообменники необходимо периодически промывать, что связано с потерей 20—25% рабочего времени.
Концентрирование фосфорной кислоты в аппаратах с непосредственным нагревом горячими (топочными) газами, подаваемыми со стороны, барботирующими через слой кислоты или получаемыми погружным горением топлива в жидкости, может быть осуществлено в течение длительного времени — месяцами— без чистки. Но в этом случае очистка отходящих газов от фтора и тумана кислоты до санитарных норм усложняется, окупаясь лишь при производстве особых сортов кислоты — суперфосфорной и других (стр. 183).
Ангидритный (безупарочный) процесс
Фосфорную кислоту с выделением сульфата кальция в виде.стабильного ангидрита можно экстрагировать в области температур и концентраций кислоты, расположенной выше линии cd (см. рис. II.16), разграничивающей поля существования метастабильного полугидрата и ангидрита..
Повышение температуры и увеличение концентрации кислоты' способствует выделению в твердую фазу ангидрита, устойчивого на всем протяжении процесса, экстракции. Это
149
происходит в растворах, содержащих. 50—60% Р2О5 выше 100°C (при концентрации 35—37%'Р2О5 выше 120°C). Применение более высоких температур по сравнению с полугидратным процессом, позволяющее увеличить интенсивность оборудования, а также выделение меньшего количества
твердого отхода наряду с получением высококонцентрированной кислоты (>50% Р2О5) и выделением основной мас-. сы фтора в газовую фазу, составляют определенные преиму-
щества ангидритного процесса..
Основные трудности возникают при кристаллизации в концентрированной кислоте крупных, хорошо фильтрующихся агрегатов ангидрита, а также при отмывке фосфорной кислоты из отжатого осадка (вследствие напряженного водного баланса). Кроме того, процесс осложняется необходимостью подогревания реагентов (теплоты реакции недостаточно для поддержания автотермического режима). В различных разрабатываемых способах указанные затруднения пытаются разрешить равными методами. По-видимому, про-
ведение процесса в автотермических условиях возможно при разложении фосфата Олеумом или смесью его с купоросным маслом с получением кислоты концентрации 48—53% Р2О5.
Кислота концентрации >50% Р2О5 может быть получена обработкой комплексного пюлифосфата (образуется при нагревании до 200 °C смеси природного фосфата и 70%-ной Н3РО4) смесью концентрированной H2SO4 и 60%-ной Н3РО4.
Выщелачивание гранулированного суперфосфата серной кислотой в противотоке могло бы представить интерес для производства фосфорной кислоты, содержащей ~40% Р2О5, в аппаратуре суперфосфатных заводов. Однако сведений о его промышленном применении не имеется.
Заслуживает внимания обработка камерного простого суперфосфата концентрированной серной кислотой. По нашим наблюдениям, в известных условиях серная кислота вытесняет из влажного осадка фосфорную кислоту, практически не смешиваясь с ней. Это позволяет заместить фосфорнокислую жидкую фазу суперфосфата серной кислотой, которую возможно отмыть от ангидрита водой (1в сочетании с промежуточными промывными растворами).
Представляет интерес производство фосфорной кислоты полугидратно-ангидритным способом, по которому шлам (полугидрат), полученный в результате предварительного разложения фосфата до отделения от жидкой фазы-, обрабатывают серной кислотой. Ангидрит отделяют от жидкой фазы и промывают водой.
До сих пор не вышли из стадии опытно-промышленной разработки зарубежные способы Норденгрена, Пита и клинкерный. Некоторые работы в этой области проводились так
же в СССР (НИУИФ, Институт химии АН КазССР). По способу НИУИФа [78] процесс ведут при 105—112 °C в присутствии избытка серной кислоты в жидкой фазе реакционной пульпы (6—6,5% SO3) с получением кислоты, содержащей 42—4/% Р2О5 и 4,5—5,5% SO3. В этих условиях ангидрит выделяется в виде хорошо фильтрующихся агрегатов. Выход Р2О5 в продукт составляет ~95% при степени разложения апатита 97% и отмывке (в пять стадий) фосфорной кислоты от ангидрита на 98%. Степень выделения фтора в газовую фазу составляет 70—75% от его содержания в апатитовом концентрате.
В Швеции осуществлен в небольших производственных масштабах способ Норденгрена с получением кислоты, содержащей 40—50% Р2О5. Выделяющийся ангидрит образует агрегаты, фильтрующиеся в 3—5 раз быстрее, чем крупные одиночные кристаллы. Агрегированию кристаллов способствует длительная периодическая загрузка реагентов и предварительное подогревание кислоты, а также нейтрализация положительно заряженных кристаллических ядер отрицательно заряженными сульфат-ионами (аналогично агломерации ионов при осаждении коллоидных частиц электролитами) . Поэтому осаждение ангидрита серной кислотой производят из геля Са(Н2РО4)2, образующегося при предварительной обработке фосфорита фосфорной кислотой при 85— 90 °C в течение 0,5 ч. При внесении геля в смесь серной и фосфорной кислоты, содержащую 40—45% Р2О5 и нагретую до 140—150 °C, в первый момен! образуются частицы сульфата кальция коллоидных размеров. При дальнейшем взаимодействии с серной кислотой.в реакторах в течение 2—4 ч при 125—135 °C выделяется основная масса ангидрита при одновременной агломерации кристаллических частиц.
После окончания агрегирования кристаллов ангидрита и одновременного доразложения фосфата пульпу охлаждают до 90—<100°C в последнем реакторе обдувкой воздухом и направляют на фильтрование с четырехкратной промывкой. Промытый шлам-ангидрит достаточно стабилен, и его можно хранить навалом во влажном виде. Поэтому и периодическая чистка установки более проста, чем при дигидратном способе. Это объясняется почти полным выделением фтористых соединений в газовую фазу при разложении фосфата, вследствие чего исключается (или протекает в очень небольшой степени) образование твердого осадка двойных солей фтористых соединений и сульфата кальция..
В кислоте, получаемой ангидритным способом, содержит-~0,5% фтора, т. е. в -~3 раза меньше, чем в кислоте,.получаемой дигидратным способом. Она содержит также меньшее количество сульфата кальция, так как растворимость его во всех формах в кислоте с концентрацией 40—50%
151
Р2О5 меньше, чем в кислоте с концентрацией 30—33% Р2О5. Кроме того, -ангидритный процесс менее длителен вследствие того, что агломерация кристаллов происходит значительно быстрее их «выращивания».
В способе Пита' фильтрующие кристаллы .ангидрита образуются при перекристалливрции предварительно выделенного полугидрата. Фосфорит разлагают при 95—120°C концентрированной фосфорной кислотой, содержащей не менее 40% Р2О5, с образованием раствора Са(Н2РО4)2. Последний затем обрабатывают серной кислотой -с выделением полугидрата, который в присутствии затравки кристаллов ангидрита перекристаллизовывают в дегидраторе в хорошо фильтрующийся ангидрит. 4
Полугидрат перекристаллизовывают в ангидрит (на 70— 100%) в присутствии затравки в течение 1—4 ч при, 120— 130 °C и содержании в жидкой фазе 40—55% Р2О5 и 1—4% H2SO4. Плоские пластинки затравочных кристаллов ангидрита с размерами от 13X6 до 40X13 мим готовят в отдельном аппарате и вносят одновременно с фосфатом в количестве 0,01—2% от массы1 фосфата. В кислоту . переходит ~90% Р2О5 фосфата при расходе на промывку 0,3—0,6 ч. (масс.) воды на 1 ч. (масс.) осадка. Указывают, что в присутствии моноаммонийфосфата (0,1—,0,2% NH3) размеры кристаллов ангидрита увеличиваются до (<60—100) X (15— 25) мкм [79]. .
Клинкерный способ известен с 1950 г. (фирма Davison Chemical Corporation, США), но промышленного применения не получил из-за трудностей, связанных с высокотемпературной обработкой кислых загустевающих пульп (при 300 °C и выше) и получением легко выщелачиваемого клинкера. Суть способа состоит в получении тестообразной массы смешением фосфата с 98%-ной H2SO4, подогретой до 120—150°C, прокаливанием массы в течение 30 мин при 2'00—250°C с образованием клинкера, который затем подвергают водному выщелачиванию. При этом в газовую фа- ’ зу выделяется-до 90% фтора (~-60% при смешении и ~25% при прокаливании1) сит общего его количества в сырье.
_ Полученный продукт содержит, в %: Р2О5 — 53—55; SO3—1,7—1,8; F — 0,1—0,20 и R2O3—'~2,5 и пригоден непосредственно как для производства двойного суперфосфа- ’ та, так-и кормовых.средств.
По способу TVA (Tennesse Valley Autority, США) фосфат обрабатывают олеумом (90—НО °C) в смесителе (без прокаливания реакционной массы) и последующим извлечением фосфорной кислоты из образующегося горячего материала.
152
Продукционная фосфорная кислота содержит 50—60% Р2О5, 0,2—0,6% фтора, 2,6% тонкодцсперсного ангидрита и некоторое количество соединений железа и алюминия, а также сульфат-ионов.
ЛИТЕРАТУРА
1. Позин М. Е. Технология минеральных солей. Т. II. Л., Химия, 1974.
2. Slack.А. V. Phosphoric Acid. Pt. 1, New York, 1968.
3. Nyges R. Phosphoric Acid by the Wet Process. New York, 1967.
4. Соколовский А. А. Технология минеральных удобрений. M., Химия, 1966.
5. Соколовский А. А., Унанянц Т. П. Краткий справочник по минеральным удобрениям. М., Химия, 1977.
6. Белов В. И.— Труды ЛенНИИГипрохнм, 1976, выл. 23, с. 63—79.
7. Фосфорная промышленность. Научно-техн. реф. сб. НИИТЭХИМ, ЛенНИИГи.прохим. Вып. 3. М., 1979.
8. Копылов В. А. и др. Производство двойного суперфосфата. М., Химия, 1976; Разумов И. М. Псевдоожижение и пневмотранспорт сыпучих материалов, М., Химия, 1972.
9. Морозов Э. И., Плюхин Д. С., Летичевский Н. Н.— Хим. пром., 1971, № 10, с. 771—773.
10. Промышленность минеральных удобрений и серной кислот.ы. Вып. 4.' М., НИИТЭХИМ, 1978, с. 10—12.
И. Модестов Б. С., Стрелецкий Г. Ф., Трунова Н. И. — Хим. пром., 1978, № 3, с. 219—221. '
12. Ворошин В. А. Производство экстракционной фосфорной кислоты. М., Химия, 1967.
13. Обработка жидких сред/Под ред. В. И. Мельникова. Труды НИУИФ, Вып. 211. М„ 1969.
14. Мельников В. И. — Труды НИИХиммаш, 1954,-№ 16, -с. 105.
15. Гинзбург Э. Н.-—Хим. пром., 1977, № 4, с. 289—292.
16. Жужиков В. А. Фильтрование. Теория и практика разделения суспензий. 3-е изд. М., Химия, 1971.
17. Гинзбург Э. Н., Мельников В. И. Метод расчета основных технологических параметров ленточного вакуум-фильтра. М., НИУИФ, 1961.
19. Воскресенский С. К., Зотов Б. Г., Серебряная Р. М. — Хим. пром., 1969, № 10, с. 745.
20. Гинзбург Э. pl. и др. — Труды НИУИФ, 1971, вып. 215, с. 33.
21. Центробежные вентиляторы/Под ред. Т. С. Маломаха. М., Машиностроение, 1975.
22. Центробежные насосы типа X (А; Д;.К; Е; И; Л).. Каталог. М., 1974.
23. Центробежные химические погружные , насосы типа ХПА, ХПАО, ПХП, Каталог. М., 1974.
24. Винников А. Я. и др. — Хим. пром., 1978, № 7, с. 51-6—517.
25. Борзенков Л. 3. Вакуум-насосы в химической промышленности. М.. Машиностроение, 1964.
26. А'льперт Л. 3. Основы проектирования химических установок. М., Высшая школа, 1976; Штербачек 3., Тауск П. Перемешивание в химической промышленности/Под ред.. И. С. Павлушепко. Л., Госхим-издат, 1963.
27. Найшуллер Т. М., Ромашова Н. Н. — Хим. пром, за рубежом, 1968, т. 5 (65), с. 30.
28. Chem. Age Int., 1972, v. 105, № 2778. p. 11:—'12.
29. Любченко T, В., Копылев Б. А., Позин М. Е. — ЖПХ, 1967, т. 40, № 10, с. 2225.
30. А. с. 611880 (СССР).
31. Воскресенский С. К. — В кн.: Исследования по производству мине-. ральных удобрений. М., НИУИФ, 1957, с. 33.
153
32. Вольфкович С. И., Забелешинский Ю. А.— Хим. пром., 1977, № 2, с. 112—114.
33. А. с. 513930 (СССР). . ' .
34. Борисов В. М., Кармышов В. Ф., Кувшинников И. Н. и др. — Хим.
- пром., 1973, Ns 2, с. 114—117; Борисов В. М., Серебряная Т. М,, До-холова А. Н. и др. — Там же, 1975, Ns 7, с. 508—509.
35г Хамидов В. А. — Хим. пром., 1972, № 2, с. 108.
36. Кармышов В. Ф. Химическая переработка низкосортных' фосфоритов в фосфорные и комплексные удобрения. Обзорная инф. Серия Минеральные удобрения и серная кислота. М., НИИТЭХИМ, НИУИФ, 1975, с. 7—23; А. с. 390017 (СССР).
37. Алешин А. М.; Борисов В. М., Кармышов В. Ф. и др. — Хим. пром., 1976, № 7, с. 510—512.
38. Павлинов Р. В., Шляпинтох Л. П., Назаров Е. А. и др. — Труды ЛенНИИГипрохим, 1976, вып. 26, с. 18—25.
39. Кармышов В. Ф., Самигуллина Л. И., Харитонов А. Б. и др. — Хим. пром., 1975, № 11, с. 834—837.
40. А. с. 431142 (СССР).
41. Орехов И. И. и др. — Хим. пром., 1976, № 1, с. 30—31.
42. Слободкина Г. Л., Орехов И. И., Свердлова В. П. — Труды СЗПИ, 1972, Ns 21. с. 8—12.
43. Schraider J., Gorecki H., Szczygiol I. — Przem. chem., 1977, № 1, s. 28—32.
44. A. c. 379552 (СССР).-
45. Кутфитдинов P. H„ Копылев Б- А., Позин M. E.—’Тезисы докладов научно-технической конференции молодых специалистов. Л., Лен-. НИИГипрохим, 1971, с. 39.
46. Hurst Е. I., Arnold W. D., Ryon A. D. — Chem. End., 1977, v. 84, № 1, p. 56—57. . .
47. Новые исследования по технологии минеральных удобрений/Под ред. М. Е. Позина и Б. А. Копылева. Л., Химия, 1970.
48. Белопольский А. П., Таперова А. А., Шульгина М. Н. — ЖПХ, 1939, Ns 1, с. 3.
49. Хамский Е. В., Чепелевецкий М. Л. — Там же, . 1958, т 37, Ns 7, с. 976; 1959, т. 32, Ns 5, с. 948.
50. Лаптева Т. Г., Копылев Б. А., Позин М. Е. — В кн.: Технология минеральных удобрений (Физико-химические и прикладные исследования"). Вып. 5. Л., ЛТИ им. Ленсовета, 1976, с. 34—39.
51. Лаптев В. М., Копылев Б. А., Варшавский В. Л. — В кн.: Техноло- гия минеральных удобрений. Новые пути получения. Л., ЛТИ им. Ленсовета, 1973, с. 31—39.
52. А. с. 548566 (СССР).
53. Воскресенский С. К., Ионасс А. .4, —Труды НИУИФ, 1940, вып. 153, с. 92. '
54. Воскресенский С. К-, Серебряная Р. М. — Хим. пром., 1966, № 12, с. 32
55. Позин М. Е. и др. — Там же, 1977, Ns 10, с. 765—766.
56. Гриневич А. В., Семенов А. Н., Вильнянский Я. Е. — Там же, 1969, Ns 11. с. 838.
57. Семенов А. Н. и др. — В кн.: Рационализаторские предложения, внедренные в химической промышленности. М., НИИТЭХИМ, 1967, Ns .8.
58. Поплаухин А. С., Гриневич А. В., Кожин В. Г. — Хим. пром., 1978, Ns 6, с. 430—43.3.
59. Гриневич А. В., Вильнянский Я. Е., Гафаров М'. Н. — Там же, 1972, -Ns 3,'с. 194—195. '
60. Гриневич А. В. и др. —Там же, 1972, Ns 7, с. 547.
61. Копылев Б. А. — Автореф. докт. дисс. Л., ЛТИ им. Ленсовета, 1962.
62. Позин М. Е., Копылев Б. А. Новые методы получения минеральных удобрений. Л., Госхимиздат, 1962.
154
63. Копылев Б: А: Технология экстракционной фосфорной кислоты. Л., Химия, 1972..
64. Гинзбург Э. п.— Хим. пром., 1976, № 7, с. 512—514.
65. Гинзбург Э. Н.— Там же, 1973, № 3, с. 195—197. *
66. Кармышов В. Ф. и др. — ЖПХ, 1975, т. 48, № И, с. 2345—2347. ‘
67. Бозин Н. А., Широкова Н. В., Мосолова А. В. — Хим. пром., 1975, № 11, с. 852—853.
68. Бозин Н. А. и др. — Там же, 1971, № 7, с. 528—529. .
69. Кармышов В. Ф., Фролова Н. Г. — ЖПХ, 1976, т. 49, № 12, с. 2626— 2629.
70. Позин М. Е. и др. — Там же, 1976, № 11, с. 2361—2366. •'
71. Phosph, a. Potassium, 1976, № 83, р. 48—49.
72. Technocrat, 1975, v. 8, № 2, р. 75.
73. Фролова Н. Г. и др. — ЖПХ, 1979, т. 52, № 1, с. 18—22.
74. Антонов А. М., Фокин М. Н., Воробьева Н. Ф. — Там же, 1977, т. 50, № 10, с. 2271—2274.
75. Варшавский В. JI. и др. — Хим. пром., 1974, № 5, с; 602—604.
‘76. Борисов В. М. и др. — Там же, 1977, № 1, с. 28—29; № 6, с. 440—
441; Попов Н. П. Выпарные аппараты в производстве минеральных удобрений. Л., Химия, 1974.
77. Коробанов В. Н. и др,-—Хим. пром., 1978, № 5, с. 358—359.
78. Воскресенский С. К. и др. — Труды НИУИФ, 1971, вып. 215, с. 3.
79. Костанян А. К, Зинюк Р. Ю., Грамота Т. Б. — Тезисы докладов XI Всесоюзной межвузовской конференции по технологии неорганических веществ и минеральных удобрений. Ч. 1. Новочеркасск, 1978, с. 150.
Глава IV
ЭКСТРАКЦИЯ ФОСФОРНОЙ кислоты АЗОТНОЙ И СОЛЯНОЙ КИСЛОТАМИ И С ИСПОЛЬЗОВАНИЕМ ОРГАНИЧЕСКИХ РАСТВОРИТЕЛЕЙ
Экстракция фосфорной кислоты, из фосфатов серной кислотой (несмотря на ее широкое распространение и усовершенствования) обладает существенными недостатками: большой расход серной кислоты (2,5—3,1 т моногидрата на 1 т Р2О5) и необходимость перерабатывать или складировать значительное количество отхода— ф осфагипс а (4,5—6,0 т на 1 т Р2О5, в пересчете на сухое вещество). Поэтому непрерывно изыскиваются возможности экстрагирования фосфорной кислоты другими неорганическими, веществами — азотной, соляной, фтористо- и кремнефтористоводородной кислотами, а также с применением органических растворителей.
Основным затруднением при разложении фосфатов азотной или соляной кислотой является отделение фосфорной кислоты от хорошо растворимых нитрата и хлорида кальция. При использовании кремнефто-ристой или кремнефтористоводородной кислот .выделяется осадок, легко отделяемый фильтрованием. Однако в этом случае регенерация кйс-' лоты требует применения высоких температур, но зато можно осуществлять процесс без дополнительных реагентов — кислот, используя фтор, содержащийся в- сырье.
Из указанных способов азотнокислотный с последующим вымораживанием четырехводного нитрата кальция является наиболее экономичным. По этому способу работают заводы ,за рубежом и в настоящее время строится и проектируется 'нисколько заводов большой мощности в СССР.
Перспективность этого метода определяется возможностью комплексной переработки фосфатного сырья (по безотходной технологии) с получением, помимо удобрений, соединений стронция, фтора, р. з. э., кормовых продуктов, карбоната кальция (для известкования кислых почв Нечерноземья), а также относительно малой чувствительности к качеству природных фосфатов.
‘156 ’ •
iv.l. АЗОТНОКИСЛОТНЫЙ СПОСОБ РАЗЛОЖЕНИЯ ФОСФАТОВ
Физико-химические основы процесса
*> -
При разложении фосфатов азотной кислотой получаю? азотндкислотную вытяжку, состоящую в основном из раствора нитрата кальция в фосфорной кислоте [1]:,
CasF (РО4) э+ IOHNO3 -> ЗН3РО4+5Са (NO3) 2+HF
В раствор также переходят продукты взаимодействия -азотной кислоты с примесями,, содержащимися в фосфатах (нитрат кальция, магния, железа, алюминия и др.) Фтори- • стый водород, выделяющийся при разложении фосфата, образует с кремнеземом, содержащимся в сырье, кремнефтористоводородную кислоту.
В образующейся сложной многокомпонентной системе окислы железа и алюминия, нитраты и другие соединения реагируют с выделяющейся фосфорной кислотой при дальнейшей ее переработке с образованием малорастворимых фосфатов, что приводит к потере Р2О5.
Поэтому фосфориты, содержащие более 12% ЕеаО3 (от .количества Р2О5), непригодны для азотнокислотной переработки без предварительного обогащения (стр. 20). Примеси А1гО3, хотя и менее вредны., чем Fe2O3, но также нежелательны [2].
В отличие от экстракции серной кислотой при азотнокис-лотной экстракции р. з. э., содержащиеся в апатитовом концентрате в количестве 0,9—0,95%-, почти полностью переходят в раствор. Выделить их (на 80—90% от -общего количества в апатитовом концентрате) можно при частичной нейтрализации раствора аммиаком до pH = 2—2,5. При этом происходит нейтрализация всей свободной азотной кислоты и ~50% первого иона водорода фосфорной кислоты. В полученном сухом осадке содержится ~65% фосфатов р. з. э., в том числе' ~30% фосфата- церия. Выход фосфатов р. з. э. из 1 т апатитового концентрата составляет 10—11 кг.
Полное разложение фосфатов достигается применением стехиометрического количества азотной кислоты. При этом получается фосфорнокислый раствор, насыщенный соединениями кальция,- Расход азотной кислоты (норму) вычисляют, исходя из содержания СаО в апатите или СаО и MgO в фосфоритах. По мере разложения фосфата,- нейтрализации азотной кислоты и накопления в растворе продуктов реакции процесс постепенно замедляется. Поэтому для обеспе- -чения постоянной скорости разложения применяют некоторый избыток азотной кислоты.(в зависимости от назначения и последующего использования вытяжки).
157
। Таблица IV. 1
Примерный состав растворов (в %), подучаемых разложением фосфатов 47%-ной HNO3 при норме ее 120% от стехиометрического количества
Компоненты Сырье Компоненты Сырье
апатитовый концентрат фосфориты Каратау апатитовый концентрат фосфориты Каратау
HNO3 5,92 '5,71- Mg(NO3)2 3,16
Н3РО4 >1в„52 9,53 A1(NO3)3 — 0,56
Ca(NO3)2 37,78 312„10 H2SiF6 0,912 0,88
Fe(NO3)3 0,45 0,89 Н2О 40,94 47,17
Ce(NO3)3 0,47 —•
В результате разложения фосфатов-азотной кислотой образуется пульпа, твердая фаза которой состоит главным образом из нерастворимых соединений кремнезема, а также нитратов стронция. Жидкая фаза пульпы — азотнокислотная вытяжка----содержит различные количества кислот и нит-
ратов (табл. IV.J). Массовое отношение Ж:Т в пульпе зависит от .содержания в сырье нерастворимых примесей и колеблется от (90—95) :4 (при разложении апатитового концентрата) до (50—10) :1 (при разложении фосфоритов).
'Взаимодействие фосфатов с азотной кислотой протекает в кинетической области [3] с достаточно большой скоростью [4].
Степень разложения фосфата (отношение количества Р2О5, перешедшего в раствор, к количеству Р2О5 в исходном сырье), достигаемая в течение 2 ч при 50°C, практически равна при количестве 100% (от стехиометрии) норме применяемой кислоты [5]. При меньшей продолжительности процесса, например 30 мин, и норме кислоты 95—100% от стехиометрического количества степень разложения фосфата меньше на 1,5—'2,5 %.'С увеличением температуры и концентрации кислоты скорость разложения возрастает, так что при одной и той же продолжительности процесса степень разложения фосфата соответственно увеличивается.,
Некоторое уменьшение выхода фосфорной кислоты' в раствор относительно нормы кислоты- объясняется частичным разложением фосфата до монокальцийфосфата:
Са3 (РО4) 2 :+4HNO3 Са (HdPO4) 3+2Ca;(NO3) 2 •
Степень перехода различных компонентов в раствор в среднем составляет, в %: Р2О5, СаО, MgO и Се2О3— 98—99; F — 95 и Fe2O3 — 70.
Взаимодействие фосфатов с азотной кислотой обычно завершается за 1—2 ч. Скорость процесса- практически не за
158
висит от концентраций применяемой исходной кислоты, рай-ной ~45—55% HNO3. Но с увеличением начальной концентрации кислоты уменьшается количество вводимой воды и увеличивается содержание компонентов в растворе. При норме кислоты 110—115% от стехиометрического1 количества изменение концентрации ее от 46 до 55% HNO3 приводит к увеличению в образующихся растворах содержания: P2OS от 9 до 11%, HNO3 от *5 до 7% и Са(МО3)2 от 38 до 42%.
Повышение температуры сопровождается ускорением разложения фосфата азотной кислотой вследствие уменьшения вязкости раствора и улучшения условий диффузии реаген--тов. Однако при температурах выше 50 °C увеличивается коррозия аппаратуры', а также количество выделяющихся окислов азота в газовую фазу [6, с. 131; 7, с. 1196]. Поэтому обычно процесс ведут при 45—50 °C. Для поддержания указанной температуры за счет теплового эффекта реакции азотная кислота должна поступать с температурой ~3'0°С. Температуру регулируют подогревом или охлаждением кислоты в теплообменнике, а также при помощи нагреваемых или охлаждаемых элементов внутри реактора.
Добавка мочевины в азотнокислотную вытяжку 5 количестве 4 кг на 1 т апатита снижает выделение окислов азота в газовую фазу в 50---75 раз 1[7, с. 2161]. Но при этом резко ухудшаются условия разделения твердой и жидкой фаз азотнокйслотной вытяжки.
Получение фосфорной кислоты вымораживанием из азот-н©кислотной вытяжки нитрата кальция основано на изменении его растворимости от температуры [5, 8].
Растворы, полученные разложением апатита 55, 52, 50 и 47 %-ной HNO3, на диаграмме системы СаО—Р2О5—М2О5— Н2О находятся в поле кристаллизации Са (NO3)2-4H2O. Теоретический выход этой соли при охлаждении до 5 °C азотно-кислотной вытяжки, полученной с применением 30%-ного избытка 55%-ной HNO3, составляет 71,5%, а стехиометрическим количеством 47%-ной^ННО3 — 30%. В последнем случае выход соли можно повысить до 49%, если использовать 10%-ный избыток HNO3. Для выделения в твердую фазу большего количества Ca(NO3)2-4H2O требуется охладить раствор до —5—115 °C.
В присутствии избытка азотной, а также фосфорной кислот растворимость нитрата кальция еще больше подавляется, и степень выделения его при охлаждении увеличивается [6, с. 63; 9];
Скорость и степень кристаллизации нитрата кальция увеличиваются при использовании повышенной концентрации ' (60% HNO3) исходной кислоты [10], особенно в присутствии затравки Ca(NO3)2-4H2O в количестве 10—20 г • на
159
100 г вытяжки. В присутствии ретурного раствора фосфорной кислоты (100—200 г на 100 г вытяжки) с добавлением 25—30 г безводного нитрата кальция степень выделения Са (МОз)2-4Н2О достигает 86% при-{-3 °C и ~90% при. —7 °C [Н]. Несколько меньший эффект оказывает добавка в азотнокислотную вытяжку безводного нитрата кальция в ’отсутствие фосфорной кислоты .[12].
Более сложным является действие примесей, содержащихся в сырье,.— соединений магния, полуторных окйслов и других. При содержании в растворе примесей до 1,5—2% MgO или А12О3 кристаллизация нитрата кальция несколько увеличивается [13]. Аналогичным образом действует и содержащаяся до 1% в растворе Fe2O3, но при 2,5% Fe2O3 степень выделения нитрата кальция уменьшается на 26% (отн.). Таким образом, при ограниченном содержании Fe2O3 в фосфорите (с учетом ее неполного .перехода в раствор) по-литермическая кристаллизация нитрата кальция может быть использована и для переработки фосфоритов, содержащих значительные количества'примесей.
Добавление хлорида калия в азотнокислотную вытяжку на стадии ее охлаждения оказывает существенное влияние на состав .выделяющегося осадка за счет конверсии КС1 в KNO3 [14]. При этом хлор остается в жидкой фазе, а твердая фаза представляет собой полупродукт для получения бесхлорного сложного удобрения.
Качество получаемого удобрения, определяемое долей водорастворимой Р2О5 от общего ее количества, зависит от" степени удаления из раствора нитрата кальция. Почти полное осаждение указанной соли достигается разложением фосфатов концентрИррванной азотной кислотой (60—70% . HNO3) и охлаждением полученного раствора до —40, — 15 °C.
Однако при низких температурах с увеличением концентрации раствора резко возрастает и его вязкость, что затрудняет кристаллизацию нитрата кальция. В этих 'условиях образуются весьма устойчивые пересыщенные растворы [15]. Поэтому кристаллизацию нитрата кальция предпочитают вести в диапазоне температур от 5 до —5 °C. В связи с трудностью регулирования процесса зачастую выделяющиеся кристаллы налипают на охлаждающую поверхность,. которую необходимо часто очищать.
При быстром охлаждении растворы нитрата кальция могут существовать в пересыщенном состоянии длительное время [15, 16], а в присутствии затравки кристаллизация из таких растворов протекает весьма интенсивно [16]. Это позволяет провести процесс непрерывным путем в две стадии— при предварительном охлаждении раствора и после-160
дующей кристаллизации из него нитрата кальция в регулируемых условиях.
Раствор, полученный после отделения кристаллов нитрата кальция, содержит разные количества фосфорной кислоты и остаточной соли (в зависимости от концентрации азот- • ной кислоты, условий экстракции и кристаллизации).
Технологические схемы и режимы
Растворы, получаемые вымораживанием нитрата кальция из азотнокислой вытяжки фосфатов, используются, преимущественно для производства нитрофосфатных удобрений. В зависимости от условий ведения процесса и степени выделения нитрата образующиеся продукты содержат водорастворимую Р2О5 в разных количествах — от 40 до 95% Г’17]
При осаждении остаточного кальция, содержащегося в маточном растворе в количестве 6—7% СаО (полученном охлаждением вытяжки, образующейся разложением фосфата 120%-ной нормой 56,7%-ной HNO3 до —5, —10°С), серной кислотой или сульфатом аммония содержание СаО в фильтрате после отделения сульфата кальция не превышает 40,5-0,6% [18].
Нитрофоску, содержащую 75% Р2О5 в водорастворимой форме (от общего ее количества), можно получить разложением апатитового концентрата 55%-ной HNO3 (110% от стехиометрии) при добавлении в вытяжку 5% H2SO4 (от массы вытяжки) и охлаждении ее до 10 °C [19]. При обра-гботке в аналогичных условиях фосфорита Каратау образуется нитрофос, содержащий ~30% питательных веществ с соотношением N:P2O5=1:1 при 60% водорастворимой Р2О5 от общей ее массы.
Комбинированием охлаждения вытяжки — политермиче-ской кристаллизации нитрата кальция—с доосаждением остаточного кальция серной кислотой можно получить кон- дициониую фосфорную кислоту.
В течение последних 20—25 лет за рубежом [20] были разработаны различные процессы производства нитрофосфатных удобрений с высоким содержанием воднораствори-мой пятиокиси фосфора. Помимо политермической кристаллизации нитрата кальция (Norsk Hydro and Chemo-projekt/Bamag) к ним относятся также экстрагирование органическими растворителями из азотнокислотной вытяжки фосфатов фосфорной кислоты (Typpi Оу) и осаждение кальция из нитрата в виде сульфата (Stamicarbon, Cherpico, Foster Wheeler и другие).
Сметные затраты на сооружение основного производства нитрофосфатных заводов суточной производительностью 1400 т продукта (|в натуре) (28:14:0) оцениваются в сред-11-5473 161
'нем от 5 до 10 млн. долл. "[20]. При этих условиях стоя- I мость одной томны продукта (28:14:0) может составить I ~25 долл., т. е. ниже, чем при производстве удобрений на ! i основе сернокислотной экстракции, если цена на серу пре- ; вышает 5 долл, за тонну. j
Производство-фосфорной кислоты j
Товарная фосфорная кислота экстракцией из фосфата азотнокислотным методом может быть получена по одному' из различных вариантов следующим образом (подробнее см. [21]).
Фосфат разлагают 50—60%-ным HNO3, подогретой • до 70—80°C и взятой с 10—15%-ным избыткам .(из расчета на окись кальция фосфата). Теплоты, выделяющейся при реакции, достаточно для нагревания смеси до температуры ки- . пения (~il25°C). Выделяющиеся в реакторе газы и пары поступают в абсорбционное отделение, где поглощаются раствором соды.
После выхода из реактора азотнокислотная вытяжка поступает в систему кристаллизации нитрата кальция, состоящую из двух или трех кристаллизаторов. Кристаллизацию нитрата кальция ведут в две стадии: на первой раствор охлаждают до ~18°С водой и выделяют ~60% от общего содержания нитрата кальция, а на второй раствор охлаждают до —10 °C рассолам или испаряющимся аммиаком.
Для удаления оставшегося нитрата кальция раствор, полученный после отделения на центрифуге нитрата кальция, обрабатывают вначале водной суспензией карбоната бария, а затем серной кислотой. В результате образуется фосфат кальция и нерастворимый в фосфорной кислоте нитрат бария. При этом нитрат-ионы на 98—99% выводятся из раствора.
Отделенный нитрат бария поступает на конверсию в NH4NO3 и ВаСО3 в результате взаимодействия с аммиаком и двуокисью углерода под-давлением. Карбонат бария нерастворим в концентрированном -растворе аммиачной селитры; его отфильтровывают, промывают и возвращают на стадию окончательного удаления нитрата-кальция. Полученную фосфорную кислоту, содержащую ~5% кальция от первоначального его количества, обрабатывают серной кислотой. После отделения гипса фосфорная кислота содержит 27—35% Р2О5.
При определенных условиях (использование вырабатываемой на месте азотной кислоты, комбинирование производства с выпуском сложных удобрений, использование отбросного карбоната кальция) применение метода азотнокислотной экстракции может быть экономичнее сернокислот-
162
Кого. Например, себестоимость фосфорной кислоты (в пересчете на 1 т Р2О5), полученной из фосфорита, содержащего 36% Р2О5, при мощности установки 30 тыс. т кислоты (54%, Р2О5) на 10—12% меньше при использовании для экстракции азотной, чем серной кислоты. Капитальные вложения в производство фосфорной кислоты методом азотнокислотной и сернокислотной экстракции, ио-видимому, одинаковы. В первом случае они выше на ~1,5%, что меньше допускаемой точности расчетов капитальных вложений.
Комплексная азотнокислотная переработка апатитового концентрата с вымораживанием нитрата кальция
Апатитовый концентрат помимо основных компонентов (РхО5, СаО и F) содержит в среднем 2,2—2,8% SrO и 0,8— 1,0% Ln2O3 (окислы р. з. э.).
При сернокислотной экстракции фосфорной кислоты основная масса стронция и р. з. э. соосаждается с сульфатом кальция, из которого в настоящее время они не могут, быть извлечены экономичными способами. В отличие от этого азотнокислотной переработкой возможно попутное выделение из апатитового концентрата 70—80% стронция и р. з. э. При этом стронций находится в'основном в нерастворимом остатке (в количестве 20—22%), а фосфаты р. з. э. осаждаются при Нейтрализации раствора до pH 1,6—4,9 [22] (см. схему । [23]).
Апатит разлагают в каскаде из нескольких реакторов 54%-ной HNOj при норме 110—120% от стехиометрии (в расчете на содержание СаО и SrO в апатите) при 50—55 °C в течение 1,5 ч. Степень разложения апатита составляет 99—199,5%. В образующейся суспензии отношение Т:Ж составляет около — 4 : 40 [23].
Твердую фазу, содержащую в основном безводный нитрат стронция ( — 60% от'общей ее массы) и нерастворимый остаток апатита, выделяют путем сгущения в аппарате непрерывного действия с последующим фильтрованием сгущенного продукта на вакуум-фильтре. Удельная нагрузка при сгущении равна — 0,25 м3/(м2-ч). В осветленном растворе остается —0,24% твердой фазы.
Нитрат стронция -перерабатывают в карбонат стронция; целевой продукт соответствует требованиям ГОСТ.2821—75 на карбонат стронция.
Осветленный раствор охлаждают в кожухотрубном теплообменнике до -28—30 °C, а затем в каскаде из трех кристаллизаторов непрерывного действия холодильным 'рассон лом да —15 °C. В опытном кристаллизаторе объемом 200 л, производительностью 200. л/ч и поверхностью теплообмена
11*
163
Принципиальная схема комплексной переработки апатитового концентрата [23]
Апатит Азотная кислота
NPK — удобрения
164
Г м2 (5 трубок диаметром 38 X 20 мм <и длиной 1,7 м) коэффициент теплопередачи при охлаждении отфильтрованного раствора составил 1050 Вт/(м2-К). Кристаллизатор работает в течение 4—6 ч, а затем его кратковременно (до 2— 3 мин) пропаривают паром, подаваемым вместо холодильного раствора [23, 24].
Суммарное время пребывания суспензии в кристаллизаторах 3—4 ч. Степень выделения нитрата кальция составляет 85—90%. Основная масса кристаллов нитрата кальция имеет размер 0,3—1 мм (90% от всей массы).
Маточный раствор после отделения нитрата кальция на вакуум-фильтре поступает в каскад реакторов для осаждения кремнефторида натрия путем обработки раствора содой при 25—30 °C в течение 1 ч. При расходе соды 110— 150% от стехиометрии степень выделения фтора из раствора составляет 75—80%. Осадок кремнефторида натрия отделяется на отстойной центрифуге.
Для осаждения концентрата' р. з. э. обесфторенный раствор нейтрализуют газообразным аммиаком до pH = 1,2— 1,4. Полнота’осаждения составляет ~80% (по отношению к количеству р. з. э. в апатите), содержание их в промытом высушенном концентрате 25%.
Нитрофосфатный раствор после отделения концентрата р. з. э. содержит, в %: Р2О5—~20; СаО — 2; NO3~—15; F“ — 0,3 и р. з. э. — 0,1. Выход фосфора в нитратнофосфат-ный раствор составляет ~96% по отношению к апатиту. При нейтрализации раствора аммиаком до pH = 5 и последующей его упаркой, корректировкой соотношения питательных .веществ, путем добавки нитрата аммония и хлорида калия получают удобрение — нитроаммофоску, в которой содержание Р2О5 води составляет 78—80%, отношение N : Р2О5: 1К2О = 1:1:1, суммарное содержание питательных веществ 48—50%.
При необходимости повышения доли водорастворимых форм фосфата до 96% нитрофосфатный раствор перед нейтрализацией аммиаком должен быть освобожден от остаточного кальция обработкой, например, сульфатом аммония с последующим отделением фосфогипса;
Четырехводный нитрат кальция, полученный из азотнофосфорнокислотных растворов, подвергают конверсии в нитрат аммония и карбонат кальция путем обработки газооб; разными NH3 и СОг. Степень конверсии составляет 99,9% при суммарном времени пребывания суспензии в реакторах 45 мин и нормах для NH3 и СО2 102% и 106% от стехиометрии. Производительность фильтрования карбоната кальция составляет 1,2 т/(м2-ч) по сухому осадку. Средняя влажность осадка 30%, концентрация получаемого раствора нитрата аммония 50%.
165
Комплексная переработка апатита с кристаллизацией нит-' рата кальция обеспечивает извлечение фосфора в нитрбфос-фатный 'раствор на 96%,•фтора во фторид кальция—'65%, стронция в карбонат—’65%, р. з. э. в их концентрат — 80%,' кальция в карбонат — 97%. Процесс отработан на укрупненной опытной установке мощностью 100 кг/ч и использован при проектировании строящихся заводов.
Дальнейшее усовершенствование отдельных узлов схемы обеспечивает эффективное их решение также применительно к фосфоритам Каратау й других месторождений. Известно, что отделение шлама от азотнокислотной вытяжки Кольского апатита, фосфорита Каратау и других фильтрованием протекает очень медленно вследствие коллоидной структуру осадка. При внесении в массу азотнокислотной вытяжки 4,5—6,5% гипса (от веса суспензии) шлам практически полностью адсорбируется на поверхности кристаллов гипса [25]. Это объясняется разноименным зарядом гипса и шлама, в результате чего происходит гетерокоагуляция тон<коди1С1Персны1Х частиц шлама на кристаллах гипса. При этом производительность фильтрования неотмытого влажного! осадка, полученного из фосфорита Каратау, увеличивается до 1200—6000 кг С* 1 м2 поверхности фильтра, в 1 ч (в зависимости от условий процесса) вместо 15— 30 кг/(|м2-ч) .при непосредственном фильтровании шлама из азотнокислотной вытяжки.
Аналогичные результаты получены при совместном отделении нерастворимого остатка с нитратом кальция. Но последний в этом случае загрязнен кремнеземом, что в некоторой степени осложняет его дальнейшую переработку конверсией в СаСОз и NH4NO3.
Двухступенчатое вымораживание нитрата кальция позволяет выделить его из азотнокислотной вытяжки на (Ю—95%; при значительном сокращении энергозатрат на охлаждение растворов [18]. Сущность разработанной схемы заключается только в проведении второй стадии при —45°C. Холодный сгущенный осадок (после отделения раствора, направляемого на дальнейшую переработку) .смешивается с отделенной от нерастворимого остатка и нитрата стронция и охлажденной до 28—30 °C азотнокислотной вытяжкой. На этой стадии температура смеси становится равной 0—10 °C и из поступающей азотнокислотной вытяжки осаждается 60—70% нитрата кальция. Последний отводят на конверсию вместе с нитратом кальция, выделившимся во второй стадии и прошедшим через «теплую» ветвь. Производительность фильтрования выделившихся кристаллов размерами 0,5—4 мм по влажному . неотмытому осадку составляет 5000—5500 кг/(м2-ч). Количество раствора, охлаждаемого 166 . *
до —(110—15)42, в 2—3 раза меньше, чем при одноступенчатой кристаллизации.
Во в'торой ступени выделяется смесь четырех- и двухводного нитрата кальция, которая сгущается в отстойнике обычного типа (скорость отстаивания 0,5—1 м/с) до отношения Ж:Т = 0,5: 1 и возвращается на первую ступень кристаллизации. Осветленный раствор со второй ступени, содержащий (гари разложении апатита 57,5 %-ной HNO3) 23—24% Р2О5 и 1,6—2,0% СаО, направляется на дальнейшую переработку.
Увеличение степени выделения нитрата кальция из азотнокислотной вытяжки может быть достигнуто при введении в вытяжку перед ее охлаждением нитрата аммония [6, с. 63] в количестве ~20% от массы фосфатного сырья или безводного нитрата кальция в количестве 30—>50% от апатита [11, 12]. В этих условиях степень выделения нитрата кальция при О'и —10°С составляет 80—83%' (вместо 65— 70%), что обеспечивает получение конечного продукта — нитрофоски, содержащей 48% питательных веществ, в том чйсле 50—60% Р2О5 в водорастворимой форме,
Этот метод представляется весьма важным для фосфоритов Каратау, в которых отношение СаО: РгО5>1,3 (соответствующее отношение в апатите). В этом случае в азотно-кислотной вытяжке, полученной разложением фосфорита 65—70%-ной HNO3, содержится больше нитрата кальция и в результате этого возникает возможность получения из фосфоритов Каратау нитрофоски без затрат серной кйслоты и по упрощенной азотнокислотной технологии.
При вымораживании из азотнокислотной вытяжки рядовой фосфоритной муки Каратау ~80% нитрата кальция и доосаждении остаточного кальция сульфатом аммония нерастворимый остаток фосфогйпса легко отделяется вместе с фосфогипсом, возвращаемым на стадию разложения в виде сгущенной пульпы. После конверсии фосфогигаса и возвращения в процесс раствора сульфата аммония нерастворимый остаток фосфорита отводится из процесса вместе с карбонатом кальция.
В этом случае получение из фосфорита Каратау сложных удобрений - типа нитроаммофоски осуществляется без применения серной кислоты по комплексной безотходной схеме.
I
IV.2. СОЛЯНОКИСЛОТНЫЙ СПОСОБ РАЗЛОЖЕНИЯ ФОСФАТОВ
Разложение фосфатов соляной кислотой, аналогично азотнокислотному разложению, протекает с образованием растворимых веществ:.
Cai3F (РО4) зф 10НС1 -> 3H3PO4+5CaC12+HF
167
Содержащиеся в фосфоритах карбонаты кальция и магния и соединения полуторных окислов переходят в хлориды. При переработке апатитового концентрата в раствор переходят также хлориды р. з. э. Находящийся в апатитовом (концентрате нефелин практически разлагается полностью с образованием хлоридов натрия, калия и алюминия" с выделением в твердую фазу кремниевой кислоты,' реагирующей с фтористоводородной кислотой.
В зависимости от содержания в сырье нерастворимого остатка образуется пульпа с массовым отношением Ж:Т = = 90:1 (|при переработке апатитового концентрата) и (20—50):4 (при переработке фосфоритов).
Скорость разложения фосфатов растворами соляной кислоты достаточно велика. При 20—30 °C из вятских прокаленных фосфоритов ГО %-ной' НО извлекается в раствор уже за 1 ч 97—99% Р2О5, одновременно лишь до 27% соединений трехвалентных металлов переходит в раствор. С увеличением концентрации и температуры кислоты скорость разложения увеличивается.
Получаемые растворы имеют состав, близкий к равновесному. При обработке при 40 °C вятских прокаленных фосфоритов 10%-ной НО образуется раствор плотностьк) 1,165—1,170' г/см3, содержащий, в %: Р2О5 — 4,5—4,7; СаО —6,9—7,1; О —8,7—9,1; Fe2O3—0,3--0,35; А12О3 — 0,08—0,10, и F—0,40—0,45.
Соединения р. з. э., переходящие из апатитового концентрата на 70—80% в раствор, могут быть выделены в виде фосфатов путем нейтрализации при 45—50 °C известью или известковым молоком 25—50% первого иона, водорода фос-‘форной кислоты [21]. Из образующейся разбавленной пульпы с отношением Ж:Т=(250—300) :1 сгущением и фильтрованием отделяют осадок, содержащий в сухом виде до 40% окислов р.'з. в.
Экстракция фосфорной кислоты! из фосфатов соляной кислотой имеет ограниченное значение. Она применяется в производстве желатины для обработки обезжиренных костей. Получаемые разбавленные растворы нейтрализуют известняком или известковым молоком для выделения преципитата. '
IV.3. ЭКСТРАКЦИЯ ФОСФОРНОЙ кислоты ОРГАНИЧЕСКИМИ РАСТВОРИТЕЛЯМИ
Метод селективной экстракции широко применяют в нефтехимии, коксохимии, при извлечении рассеянных и редких элементов из растворов, полученных азотнокислотным разложением природных руд, а. также при получении урана высокой чистоты. Нитраты трехвалентных р. з. э. хорошо, экс-168
трагируются растворами трибутилфосфата в углеводородах, Например .в керосине. < .
Для очистки водного раствора уранилнитрата UOZ(NO3)2 от примесей его обрабатывают в сильнокислой среде (Зн. HNO3) 20%-ным раствором трибутилфосфата в керосине. При этом ураиилнитрат переходит в органическую фазу, а примеси, остаются в водной.
Разделение жидких смесей селективной экстракцией основано на различии растворимости веществ в двух несме-шивающихся жидких фазах; Это позволяет применить селективную экстракцию в самых разнообразных случаях, на>-пример для концентрирования теплочувствительных или агрессивных растворов (вместо их выпаривания) в производстве удобрений (в частности, нитрата калия из хлорида калия и азотной кислоты) и для получения фосфорной кислоты из солянокислотной, а также азотнокислотной вытяжки фосфатов.
При обработке солянокислотной вытяжки фосфатов — водного раствора фосфорной, соляной кислот и хлорида кальция—(бутанолом или изопентанолом вся фосфорная кислота и часть соляной переходят в спиртовую фазу. Хлорид кальция и основная масса соляной кислоты остаются в водной фазе, из которой отгоняют хлористый водород, а хлорид кальция идет в отброс. При отгонке органического растворителя из неводной фазы получают фосфорную кислоту (55—58% R2O5), менее загрязненную примесями, чем при сернокислотной экстракции'.
Процесс осуществляют следующим образом. Солянокислотную вытяжку фосфатов, после отделения нерастворимых примесей, подают на экстракцию в, многоступенчатый смесительно-отстойный экстрактор. Навстречу фосфорнокислот-ному раствору в экстрактор поступает растворитель (обычно бутанол). В результате взаимодействия растворителя с-вытяжкой в спиртовую фазу переходит вся экстр агаров ан-'ная фосфорная кислота, а> также избыточная (свободная) соляная.кислота, некоторые количества соединений кремния, и фтора (1в зависимости от'их содержания в сырье) и немного хлорида кальция и воды.. Водная фаза содержит в основном хлорид кальция и .незначительные количества соляной кислоты1 и органического растворителя.
После разделения несмешивающихся жидких фаз получают неводный экстракт (раствор фосфорной и соляной кислот в экстрагенте) и рафинат — водный раствор хлорида кальция. Экстракт промывают водой для отделения фосфорной и соляной кислот от экстрагента, который возвращают в процесс. Образующийся при этом водный раствор смеси кислот дистиллируют, в результате чего регенерируется соляная кислота и выделяется концентрированный раствор,
169
фосфорной кислоты. Предварительно (до промывки)- из экстракта выделяют фтористые соединения солями, натрия или калия.
Получаемая фосфорная кислота имеет розовый цвет и содержит 80% Н3РО4 (58% Р2О5). В ней содержится незначительное. количество (0,01—0,05%) иоНов некоторых тяжелых металлов (железа, хрома, урана) и 0,3% ионов кальция (без дополнительной промывки).
На производство 1 т Р2О5 ('в виде 80 %-ной кислоты) при-мощности 15—30 т Р2О5 в сутки требуется: НС1 (в расчете на 100%) 1,8—2 т; растворителя (спирт) —5 м3, воды тех-иологической — 18 м3, воды для охлаждения — 2’00 м3, электроэнергии — 150 кВт-ч и пара — 6 т’.
Фосфорную кислоту из азотнокислотной вытяжки фосфатов можно получать экстракцией органическими растворителями (кетонами, спиртами, гликолями).
При разложении фосфатов слабой азотной кислотой в присутствии трибутилфосфата можно получить концентри-рованные растворы, из которых нитрат кальция мож.ет быть выделен политермической кристаллизацией при более высоких температурах по сравнению с обычно, применяемыми [26,- с. 2143].
При экстракции фосфорной кислоты изоамиловым спиртом из растворов, содержащих нитраты кальция, магния и азотную кислоту, коэффициент распределения фосфорной кислоты (отношение концентрации ее в органической и водной фазах, в моль/л) и степень извлечения ее в ~1,3— 2 раза больше, чем при экстракции Н3РО4 из бинарных растворов [26, с. 273]. Присутствие в растворе нитрата кальция способствует увеличению, а 2—4% азотной кислоты и нитрата магния—уменьшению коэффициента распределения фосфорной кислоты.
При комбинировании кристаллизации нитрата, кальция из азотнокислотной вытяжки с селективной экстракцией органическими растворителями вначале из нее выкристаллизовывают' С a (NO3) 2-4НгО.. Из маточного раствора, полученного после центрифугирования кристаллов, экстрагируют амиловым спиртом Н3РО4 и непрореа1гировав‘шую (избыточную) HNO3. Из водной фазы отгоняют спирт, а из органической, содержащей Н3РО4, HNO3 и некоторое количество воды, с помощью вторичного растворителя (например, бутилацетата) экстрагируют HNO3. Водная фаза представляет собой конечный продукт, содержащий, в %: Р2О5— ~55; H1NO3 — 0,05; СаО — 0,075; MgO —0,004; и следы К2О3 и SO3.
В процессе, разработанном фирмой Typpi Оу (Финляндия), азотнокислотную вытяжку фосфата после отделения нерастворимого остатка обрабатывают третичным амиловым спиртом, как весьма безопасным и высоко эффективным в 170 -
условиях процесса ([20]. В результате этого в органическую фазу переходит фосфорнаякислота и часть азотной кислоты. После экстракции органическую фазу нейтрализуют аммиаком с выделением в твердую фазу фосфата и нитрата аммония. После их отделения растворитель снова возвращают в процесс, а осадок перерабатывают в удобрение типа 26 : 26 :0 или при добавлении калийной соли—в нитроаммофоску 18:18:18. Водную фазу, содержащую нитрат кальция, перерабатывают в нитрат аммония и карбонат кальция высокой чистоты.
На производство 1 т нитроаммофоски марки 18:18:18 требуется: 0,468 т апатита (39% Р2О5); 0,814 т азотной кислоты (100% HNO3); 0,267 т аммиака (100% NH3); 0,.300 т хлорида калия (60% КгО); 0,189 т углекислого газа; 0,225 т пара (0,75 МПа — 7,5 ат) и 0,217 т (0,025 МПа — 2,5 ат); 50 кВт-ч электроэнергии; 52 м3 воды для охлаждения и 0,2 м3 технологической воды; 30 кг топлива и 2,0 кг растворителя.
ЛИТЕРАТУРА
1. Набиев М. Н. Азотнокислотная переработка фосфатов. Т. 1. Теоретические основы азотнокислотнои- переработки фосфатов. Ташкент, Фан, 1976, с. 367.
2. Клевке В. А., Поляков Н. Н., Арсеньева Л. 3. Технология азотных удобрений. М., Госхимиздат, 1963.
3. Пархомовск'ий В. Л., Доливо-Добровольский В. В. — ЖПХ, 1971, т. 44, № 7, с. 1633—1635.
4. Гольд инов 'А. Л., Абрамов О. Б., Шишканов А. П. — Там же, 1978, т. 51, № 7, с. 1474.
б. Вольфкович С. И., Соколовский А. А. — Успехи химии, 1974, т. 43, № 3, с. 564—583.
6. Технология минеральных удобрений. Новые пути получения. Вып. 4.
• Л., ЛТИ им. Ленсовета, 1973, с. 131—139.
7. Больдинов А. Л., Абрамов О. Б., Шишканов А. П. — ЖПХ, 1977, т. 50, № 6, с. 1196—1197; 1978, т. 51, К° 10, с. 2161—2164.
8. Марголис Ф. Г., Унанянц Т. П. Производство комплексных удобрений. М., Химия, 1968, с. 203.
9. Дмитревский Б. А., Позин М. Е., Копылев Б. А.— ЖПХ, 1971, т 44, № 9, с. 1934—1940.
10. Позин М. Е., Копылев Б. А., Ван Ли-шэн.— Там же, 1961, т. 34, № 5, с. 994—1001.
11. Акимов Л. И. и др. — В кн.: Технология минеральных удобрений. Новые пути получения. Вып. 5. Л., ЛТИ им. Ленсовета, 197б/с.'99— 103.
12. Акимов Л. И. и др. — ЖПХ, 1976, т. 49, № 8, с. 1677—1681.
13. Акимов Л. И., Морозова Г. А., Дмитревский Б. А. — В кн.: Технология минеральных удобрений. Межвуз. сб. трудов. Л.; ЛТИ им. Ленсовета, 1978, с. 71—77.
14. Матвеева Н. А., Орехов И; И., Панов В. П. — ЖПХ, 1978, т. 51 №-1, с. 178—180.
15. Позин М. Е., Копылев Б. А., Ван Ли-Шэн. — Там же, 1960 т. 33 № 12, с. 2675.
16. Позин М. Е. — Там же, 1961, т, 34, № 11, с. 2384.
171
17. Абашкина Т. Ф; Методы азотнокислотной переработки фосфатного сырья в сложные удобрения. Обзорная информация НИУИФ. М., НИИТЭХИМ, 1977.
18. Морозова Г. А., Вундервальд Л. Н. — В кн.: Технология минеральных удобрений. Межвуз. сб. трудов. Л., ЛТИ им. Ленсовета, 1977, с. ПО— 117.
19. Орехов И. И., Матвеева Н. А., Любченко Т. В. — Хим. яром., 1977, № 1, с. 24—26.
20. Сравнительные стоимости производства удобрений, полученных посредством процесса нитрофосфата и обычного процесса на основе сёры. Второй межрегиональный симпозиум по удобрениям ООН. Киев, 21/IX—1/Х — 1971 г. VD/WG 99/23.
21. Копылев Б. А. Технология экстракционной фосфорной кислоты. Л., Химия, 1972.
22. Абашкина Т. Ф. и др. — Хим. пром., 1977, № 12, с. 902—9Q4.
23, Гольдинов А. Л. и др. — Там же, № 9, с. 673—675.
24. IJoctHUKoe В. А. и др. — Там же, 1978, № 12, с. 927—929.
25. Позин М. Е., Копылев Б. А., Кутфитдинов Р. Н. — Научно-техническая конференция ЛТИ им. Ленсовета. Краткие сообщения. Л., 1970, с. 14—15.
26. Панов В. П., Смирнова Л. Я., Орехов И. И. — ЖПХ, 1976, т. 49, № 10, с. 2143—2147; 1975, т. 48, tfs 2, с. 273—277.
Глава V
ПОЛИФОСФОРНЫЕ кислоты
К полифосфорным кислотам относятся [1]: безводная жидкая суперфосфорная, ультрафосфорная, астрофосфорная и Другие кислоты, содержащие, наряду с ортофосфорной, различные количества кислот в неорто-форме. Различаются они содержанием Р2О5, соотношением лолиформ и физикохимическими свойствами,, обусловленными составом исходной ортофосфорной кислоты и методом, а также условиями их получения. Полифосфорные кислоты получают или дегидратацией ортофосфорной кислоты при 200—280 °C, или растворением в кислоте фосфорного ангидрида.
По мере увеличения концентрации Р2О5 в полифосфор-ной кислоте содержание в. ней орто-формы уменьшается, а содержание других форм (пиро-, Триполи-, тетраполи- и некоторых других) увеличивается, достигает максимума, а затем уменьшается. В табл. V.1 приведен состав термической
Таблица V.1
Состав термической полифосфориой кислоты
Общее содержание РаО5, % Содержание Р2О5 в виде кислот, %
Н3РО4 (орто) Н4Р2О7 (пиро) НбРзОю (Триполи) (И1ГОП -edioi) £rO6d9H HtPsOis (пеита-поли) Н8Р6О)9 (гексаполи) Н9Р7О22 (гептаполи) HioP-gOas (октаполи) Нл+2РпО3п+1 (высшие)
72,41 94,37 5,6 - __ __
73,86 80,2 19,7 — — — — — — —.
75,30 74,70 24,17 1,13 — — —. —— —.
76,75 47,50 41,87 12,10 — -JL — — —
79,20 23,30 43,80 26,60 6,10 — — ——й
80,52 14,62 32,73 21,15 13,86 5,73 — 12,2 —.
82,54 10,20 33,40 21,50 14,30 8,60 11,9 — 5,12 —-
85,04 . 5,04 33,80 24,60 15,30 9,00 2,83 2,83 — 2,2
85,46 0,46 10,10 20,30 6,90 6,20 5,10 7,25 — 43,95
178
g*
te-
r fe
fe
is
' полифосфорной кислоты [2], полученной упариванием teip* Г мической фосфорной кислоты.
*г Различные сорта выпускаемых полифасфсхрных кислот | содержат по крайней мере треть РгО5 в виде полиформ.
Переход ортофосфорной кислоты в полиформы возможен при содержании в смеси >63% РгО5. Соответственно тех-L .ничеокая полифосфорная кислота может иметь концентра-• цию от ~65 до 100% Р2О5. Однако практически, в соответ-Г ствии с требуемыми свойствами, ее выпускают с содержа»-нием от 69—70 до 82—90% Р2О5.'
f Собственно полифосфорной кислотой называют почти < безводную фосфорную кислоту с концентрацией 69—70% ' РгО5 и молярным отношением Н2О : Р2О5 несколько большим 3:1.
.Суперфосфорной кислотой называют продукт, содержащий 72—76% Р2О5 с отношением H^OiPgOs от 3:1 до 2,4—2,6:1, а ультрафосфорной—смеси с. содержанием Р2О5 83% и более.
V.I. БЕЗВОДНАЯ ЖИДКАЯ ФОСФОРНАЯ КИСЛОТА
Полифосфорная или. безводная фосфорная кислота — специфический продукт, отличающийся от концентрированной фосфорной кислоты (50—57% Р2О5) и от суперфосфорной кислоты (76% Р2О5). не-только составом, но и свойствами; цвет ее — от темно-коричневого до зеленого.
В табл. V.2 представлены состав и некоторые свойства двух образцов безводной фосфорной кислоты, полученной упариванием концентрированной экстракционной кислоты, из различных фосфоритов США.
Добавка небольших количеств азотной (~3%) и'ли серной (5%) в исходную чистую ортофосфорную кислоту приводит к снижению температур образования полифосфорных
Таблица V.2
Состав и свойства некоторых образцов безводной фосфорной кислоты [3]
It
Кислота Содержание,. % Плотность при 15,8 °C, г/см3 Вязкость при 66 °C, сПз Коррозия мягкой стали при 52 °C мм/год Температура замерзания, °C •
р2о5 О. - о (Z) ♦ «О о С1 0- А12О3
Экстракционная I 53,0- 0,9 2.3 1,3 2,1 1.7 16 12,5 —23
Безводная I 68,4 0,4 3,0 1,7 2,7 2,0 1000 5 4,4
Экстракционная II 52,6 0,7 3,1 1,4 1,3 1,7 16 12,5 -25
Безводная II 69,1 0,3 4,1 1,8 1,7 2,0 400 0,38 32
к
I 174 . .
Таблица V.3
Влияние. добавки серной кислоты на свойства безводной фосфорной кислоты (69% Р2О5) [3]
Свойства Содержание серной кислоты, %
0 5 1 10 15 ' 20 25
Температура замерзания,. 21 —1 -1 -1 -1 -1
°C Вязкость при 27 °C, сП- 3700 1070 700 440 420 420
Коррозия мягкой стали при 52 °€, мм/год —• — — -0,28 1,12 2,41
кислот [4]. Соответственно увеличивается степень дегидратации при одинаковой температуре. При этом действие серной кислоты больше, чем азотной.
С увеличением содержания серной кислоты температура замерзания и вязкость кислоты уменьшаются, а коррозион-*. ное ее действие на сталь увеличивается (табл. V.3).
Ан^огично серной кислоте добавка аммиака к безводной фосфорной кислоте еще больше влияет на температуру ее замерзания. Так, безводная фосфорная кислота (69% Р2О5 и 5% примесей) замерзает при температуре. выше 21 °C. При наличии 0,55% NH3 она затвердевает ниже 15,6 °C, в присутствии 1% NH3 — при 13 °C и содержании 2,7% NH3— ниже О °C.
На кривой зависимости температуры замерзания фосфорных кислот от молярного отношения Н2О к Р2О3 (т. е. от содержания Р2О5) имеется несколько экстремальных точек. Даже в узком д’иапазрнееконцентрацйй от 65 до ~75% Р2О5 (в пределах отношения Н2О: Р2О5='2,4—3,8) растворы характеризуются разными температурами замерзания. В то же время вследствие склонности к переохлаждению безводная фосфорная кислота остается жидкой в течение длительного времени при температуре ниже точки замерзания. Например, кислота концентрации 73,43% Р2О3 (в виде Н3РО4— 62,81%, а^Н+РгО?—10,62 Р?О5) не кристаллизуется в диапазоне температур от 23 °C до —15 °C в течение свыше 1. месяца [5]. Кристаллизация кислоты наступает лишь при внесений в нее зародышей. Кроме того, для предотвращения кристаллизации кислоты при колебаниях окружающей температуры „к ней можно, добавить небольшое количество аммиака или серной кислоты.
Растворы с концентрацией, соответствующей отношению Н2О: Р2О5 = 3,3-3,5, оказывают слабое корродирующее действие на мягкую сталь.
Состав безводной кислоты и содержание полимерных кислот обусловливают возможность растворения в ней самой или продуктах, полученных при ее переработке, при
175
месей металлов) которые находятся в исходном растворе. Способное раствориться в кислоте количество примесей тем больше, чем меньше в безводной кислоте отношение Н2О: Р2О5 и, соответственно, чем больше количество полимеров. Так, при отношении Н2О:Р2О5=3,2 образуются растворимые 'внутрикомплекеные соединения железа («хелаты») с содержанием в кислоте 4% Ре20з, а при отношении НгО5: Р2Оз = 3,0 — с содержанием 7% Fe2O3. Аналогичным образом сказывается уменьшение отношения Н2О ; Р2О5 и на растворении других примесей.
V.2. СУПЕРФОСФОРНАЯ КИСЛОТА
Суперфосфорная кислота — наиболее распространенная из полифосфорных кислот. Нередко под названием суперфосфорной кислоты' выпускают растворы, содержащие до 83% Р2О5. По аналогии с олеумом, супер фосфорную кислоту называют также фосфолеумом. Но в отличие от ^еума, она может быть получена упариванием водных растворов фосфорной кислоты и ее дегидратацией.
Полученная термическим путем суперфосфорная кислота.— вязкая окрашенная в цвет янтаря прозрачная жидкость. Продукт, полученный из экстракционной кислоты, имеет более темную окраску, чем исходная кислота; зеленую, если ее производят из осветленной очищенной кислоты, и черную—при исходной темной кислоте. Светлую зеленую суперфосфорную кислоту из экстракционной кислоты получают при предварительном врокаливании фосфата и , ' длительном отстаивании полученной кислоты.
Состав суперфосфорной кислоты, полученной термическим способом, почти не отличается от равновесного распределения полиформ в системе Н2О—РгО5. Экстракционная супер фосфорная кислота содержит, наряду с орто-, пиро*- и другими кислотами^, некоторые количества их солей, а также примеси из исходной кислоты. ,
При одном и том же содержании Р2О5 в растворе концентрация полифосфорных кислот в экстракционной суперфосфорной кислоте больше, чем в термической. Это объясняется влиянием примесей, связывающих некоторое количество свободной воды, что эквивалентно увеличений) концентрации Р2О5 в системе. В присутствии воды происходит гидролиз суперфосфорной кислоты с образованием ортофосфорной. Практически (без учета примес'ей) суперфосфорная кислота содержит от 69 до 74% Р2О5. ,
В продукте, содержащем 72% Р2О5 и полученном из экстракционной кислоты, находится от 35 до 50% кислот в по-. лиформе.
176
Хотя суперфосфорная кислота замерзает при более высокой температуре, чем. ортофосфорная, но при концентрации 72—75% Р2О5 образует эвтонические растворы с низкой температурой замерзания. Такие растворы не кристаллизуются при комнатной температуре. Иногда для понижения точки замерзания суперфосфорной кислоты к ней добавляют от 1 до 20% серной кислоты или от 1 до 2% -аммиака (стр. 75). Помимо устранения опасности кристаллизации добавка серной кислоты приводит к уменьшению вязкости кислоты и облегчает ее перекачку и транспортировку. Обычно суперфос-форную кислоту с целью уменьшения ее вязкости перекачивают и хранят IB нагретом виде.
Количество примесей, находящихся в упаренной экстракционной фосфорной кислоте (53—54% Р2О5), как правило, мало по сравнению с количеством, необходимым для насыщения ими суперфосфорной кислоты. Это предотвращает выделение твердых отложений как из самой кислоты, так и при получении из нее различных продуктов [6].
Добавление 2—8°/o 'H2S04 к полифосфорной кислоте концентрации 68,7—75',5°/о Р2О5 увеличивает содержание в ней полиформ [7, с. 102].
Поведение общих примесей при получении суперфоюфор.-. ной кислоты весьма сложно. Например, в кислоте с концентрацией 53% Р2О5 при отношении Н^О : PiO5 = 7,0 и содержании ~4—6% примесей последние сперва находятся в пересыщенном состоянии, а затем при хранении или транспортировании кислоты’ выделяются в твердую фазу. Если же упаривать кислоту до отношения Н2О : РгОб = 3,6 : 1, то растворимость примесей резко падает, и степень осаждения их в этих условиях настолько велика, что может привести к загустеванию и твердению основной массы кислоты!.
Экстракционная фосфорная кислота, содержащая более 0,3% MgO, непригодна для получения суперфосфорной кислоты из-за возможности шламообразования.
Дальнейшее упаривание кислоты приводит к образован нию полифосфорных кислот, по достижении концентрации, соответствующей мольному отношению Н?О: РгО5=2,75, примеси переходягт в растворенное (стабильное) состояние. Это указывает на. трудности, которые могут возникнуть при упарке кислоты. При недостаточной упарке кислоты возможно выделение из жидкой фазы твердых образований.
С повышением температуры кислоты до 50—60 °C уменьшается ее вязкость, что также облегчает кристаллизацию примесей. Кроме того, вязкость суперфосфорной кислоты зависит от ее концентрации и состава фосфатйого сырья. При одной и той же концентрации вязкость экстракционной су-пеффосфорной кислоты в несколько раз больше термической.
12-5473 1 177
Большое влияни.е на вязкость кислоты оказывают взвешенные примеси нерастворимых твердых веществ; с увеличением их содержания в-кислоте от 0,1 до 4,5% вязкость кислоты увеличивается в 10—20 раз.
Вязкость кислоты, полученной выпариванием под вакуумом, меньше, чем кислоты., полученной выпариванием под атмосферным давлением, вследствие уменьшения поли-форм.
* При 40 °C суперфосфорная кислота (полученная упариванием экстракционной фосфорной кислоты из апатитового концентрата в две стадии с отделением осадка после первой стадии декантацией), содержащая 70,7—74,6% Р2О5 [соответственно, 53,0—55,(8% Р2О5 (орто), 3,07—2,43% SO3, 2,11—2,33% R2O3, 0,69—0,76% РегО3] имеет кинематическую вязкость от 283 до 2010 сСт [7, с. 92].
Состав и свойства суперфосфорной кислоты изменяются при попадании в нее влаги вследствие гидролиза поликислот и перехода их в орто-форму. Скорость разложения поли-фосфорных кислот зависит от начальной концентрации, pH и_ температуры. При комнатной температуре-в кислоте концентрации 105% Н3РО4 (76% PjOs) полное превращение полиформ в орто-форму наступает через 17 суток; при 50 °C —за 1ч.
Суперфосфорная кислота., вследствие образования с примесями (соединения железа и алюминия) растворимых комплексных соединений, предотвращает ретроградацию фосфорного ангидрида в почве. На ее основе можно получать высококонцентрированные жидкие и твердые удобрения [8], а также удобрения замедленного (регулируемого) действия фЭ, 10] и кормовые средства [Нф.
В полузаводских условиях разработан способ получения сложных удобрений нейтрализацией аммиаком полифосфорной кислоты концентрации 74—76% Р2О5 при 180—210 °C в течение 2,5 ч [12].
Вязкость расплавов тетрапирофосфата! аммония в при-' сутствии нитрата или ортофосфата аммония в диапазоне температур 182—195 °C снижается в 5,5—1Г раз. При этом на снижение вязкости большее влияние оказывает нитрат аммония ([13]. Затраты на хранение, транспортирование суперфосфорной кислоты в ~1,3—1,5 раза меньше, чем термической фосфорной кислоты.
Полифосфатные соединения гидролизуются в почве до^ ортофосфатов очень медленно [14, вып. 4, с. 5] и растения успевают использовать фосфорный ангидрид раньше, чем произойдет его ретроградация. Растворимость полифосфата аммония При 25 °C в воде составляет 169,8 г/л, в 2 %-ном •растворе лимонной кислоты ~1-75 г/л [14, вып. И, с. 11 ].
178
Полифосфорные кислоты способны также .соединяться С микроэлементами почвы, переводя 'их в формы, способные усваиваться растениями.
Концентрированные жидкие удобрения, содержащие 10— 11% азота и 30—-35% PsO5, получают нейтрализацией суперфосфорной кислоты1 аммиаком при 70 °C до pH = 6—6,3. По- -лучаемый раствор весьма стоек при О °C [15].
При'добавке калийных солей к раствору полифосфатов аммония получают комплексные жидкие удобрения состава N-WV-К2О (в %): 10—10—10 или 5—15—10.
Суперфосфорная кислота также способствует стабилизации суспендированных удобрений и ^позволяет получать их в более текучем и концентрированном виде (типа 15—45— 15 или 10—30—40).
Нейтрализацией суперфосфорной кислоты аммиаком под давлением 0,2—0,4 МПа (2—4 ат) при 170 °C можно получить расплавленный полифосфат аммония, содержащий ~ 16% азота и 60% Р2О5. Он легко гранулируется в смеси с рециркулируемой мелкой фракцией.
Представляет интерес разработанный в лабораторных и " опытных условиях одностадийный (безупарочный) процесс аммонизации полифосфорной кислоты' в присутствии растворов аммиачной селитры при атмосферном' давлении' [16]. При этом одновременно достигаются большие значения степени аммонизации кислоты и получение продуктов с широ- . ким диапазоном отношений и концентраций питательных веществ с влажностью, не превышающей 1%, без дополни- • тельных затрат на, их упарку и сушку.
При взаимодействии суперфосфорной кислоты с карбамидом или плавом, полученным при его синтезе, образуются практически негигроскопичные продукты, содержащие 34% азота и 35% РгО5 или 46% азота и 30% Р2О5 (в зависимости от соотношения в исходной смеси N: Р2О5). Обработкой природных фосфатов суперфосфорной кислотой, содержащей >70% Р2О5, можно получить двойной (тройной) суперфосфат (50—54% Р2О5).
Суперфосфорная кислота,, содержащая всего 0,1—0,3% F, пригодна для производства. кормовых фосфатов и изготовле-' нця зубных цементов. Обычно эти продукты получают из более дорогой термической кислоты.
В органической химии суперфосфорная кислота; (и дру-. гие полифосфорные кислоты) находят широкое применение для проведения реакций дегидратации, конденсации, циклизации, полимеризации и других, в производстве аминов, ке--тонов, олефинов и т. п. ,
Несмотря на то, что суперфосфорная кислода.—.мвнёё1 ? агрессивна по отношению к большинству материалов, чем обычная фосфорная кислота, хранение ее в емкостях из 12* / .179
обыкновенной стали считают невозможным. Некоторые ингибиторы, например иодистый натрий и другие, заметно снижают коррозионное действие кислоты только при обычной температуре.
При хранении в хранилищах, контейнерах и транспортировании кислоты в цистернах их изготовляют из стали марок Х18Н10Т, Х17НЗМ2Т и оборудуют рубашкой или змеевиком, а другие емкости изготовляют из углеродистой стали, футерованной графитом или другими кислотоупорными неметаллическими материалами [2].
Экстр акционная суперфосфорная кислота менее агрессивна, чем термическая. Для хранения и перевозки 'ее до 45 °C применяют обогреваемые цистерны из бутилового и неопренового каучука, иногда из углеродистой стали с поливиниловым покрытием. При температуре 90—95 ’С кислоту транспортируют в утепленных цистернах из специальной стали.
Производство суперфосфорной кислоты развивается весьма быстрыми темпами, и в 1975 г. оно в США составило ~ 1350 тыс. т (в расчете на 100% PgOs)-
Термическую суперфосфорную кислоту производят двумя способами [2, 6]. По первому — фосфорным ангидридом, полученным сжиганием элементарного фосфора, насыщают ограниченное количество воды [17]; достигают этого .рециркуляцией суперфосфорной кислоты пониженной концентрации. Указанный процесс представляет интерес при сжигании элементарного фосфора в местах потребления концентрированных жидких удобрений, получаемых из суперфосфорной кислоты.
По другому способу в обычную термическую фосфорную кислоту вводят твердый фосфорный ангидрид до образования кислоты определенной концентрации.
Широкому применению термической суперфосфорной кислоты препятствует ее высокая стоимость. В то же время, если экстр акционная фосфорная кислота не очень загрязнена примесями, то полученная из нее упариванием суперфосфорная кислота практически обладает теми же свойствами, что и термическая. Более того, совместное присутствие в исходной кислоте сульфатов, соединений фтора и окислов трехвалентных металлов способствует резкому увеличению общего содержания полиформ при более низкой общей концентрации Р2-О5.
Сведение к минимуму или вообще исключение осаждения примесей при упаривании экстракционной кислоты достигается применением исходной кислоты, полученной из обогащенных фосфатов, (апатитового концентрата и других), не содержащих соединений магния, и предварительным ее отстаиванием.. 'Продолжительность упаривания при 240— 180
280 °C 10—20 мии. Процесс ведут в барботажных аппаратах с выносными или погружными горелками или на вакуум-выпарных установках.
Оба способа имеют свои преимущества и недостатки. Выпарка раствора С применением непосредственного нагрева горячими- газами осложняется потерями тумана Р2О5, а вакуум-выпарка — отложением солей на преющих элементах. Наиболее распространены барботажные аппараты с выносными пли погружными горелками.
'Предложено получать суперфосфорную кислоту непосредственным нагреванием кислоты перегретым паром под разрежением, топочными газами в пневмотрубе, а также радиационным обогревом кислоты, образующей пленку ![18].
Производство суперфосфорной кислоты в барботажных аппаратах, сопровождающееся дегидратацией ортофосфор-ной кислоты с образованием полифосфорных соединений, Связано с уносом отходящими газами значительных количеств фосфорной кислоты в виде брызг и тумана. Помимо этого необходимо регулировать температуру жидкости и продолжительность ее обработки во избежание образования нерастворимых циклических полифосфатов.
Улавливание туманообразной фосфорной кислоты связано с большими трудностями. Унос брызг и тумана фосфорной кислоты с газами из выпарного аппарата тем больше, чем выше концентрация продукта [19]:
Концентрация продукта, 50 55 60 65
% Р2О,
Потери Р2О5 от посту- 4,7 6,3 8,1 9,3
лающей кислоты, %
Образование тумана фосфорной кислоты можно уменьшить снижением температуры газов, поступающих в концентратор, до 600—4000 °C и длительности нагревания кислоты не более 20 мин во избежание чрезмерного образования нерастворимых соединений. Но в большинстве случаев получаемая экстракционная суперфосфорная кислота все-таки загрязнена нерастворимыми примесями.
Предложены различные способы механического отделения нерастворимых примесей. Например, при концентрировании упаренной экстракционной кислоты в двух аппаратах с погружным горением в две стадии [18] в первом аппарате получают кислоту, содержащую 67—70% Р2О5. Затем* из нее отфильтровывают примеси, а во втором аппарате осветленную кислоту упаривают до содержания 76—80% Р2О5. В полученном продукте содержится не более 0,2% нерастворимого остатка и менее 0,01 % F.
В НИУИФе отработано в полупромышленных масштабах производство суперфосфорной кислоты, содержащей 73—
181
76% Р2О5, с минимальным выделением осадка в процессе концентрирования кислоты в барботажном аппарате [20]'. Упарка осуществляется в две стадии — от 30 до 53—57% Р2О5 и затем — до 73—76% Р2О5 (см. ниже).
Полученная’ кислота содержит, в %: Р2О5 (общ) 72,5 75,5; (орто-форма 30—35)’; R2O3 — 1,8—2,5; SO3 — 2,5—4 и F — 0,05—0,15. Количество дегидратированных форм в кислоте составило, в %: пирофосфорной— 56; триполифосфор-ной— 2'4; тетраполифосфорной—15 и пентаполифосфор-ной — 5. При указанных условиях 1 м3 газа содержал 1 — 3 г f и 1—4 г H2SO4 при 0 °C и атмосферном давлении. В газовую фазу выделялось 80% соединений-фтора и 20% . H2SO4 от их содержания в исходной упаренной кислоте, содержащей 53—55% Р2О5. Брызги и туман фосфорной кислоты в отходящих газах составляли 7—8 г/м3 при 0 °<С и- атмосферном давлении (до 2% от упариваемой кислоты).
Интенсивность работы выпарного аппарата характеризовалась по выпаренной воде—'370 кг/ч, влагосъему с зеркала испарения — 2000 кг/(м2-ч), удельному расходу теплоты — 6400 Дж/кт.
При упаривании осветленной фосфорной кислоты состава, в %: Р2О5—43—57; SO3 — 0,9—3,5; F — 0,2—0,7; R2O3 — 1,0—1,68; СаО — 0—0,05 и нерастворимого остатка. 0—10,05% (при температуре газов в барботажной трубе 650—900 °C, отходящих газов 245—265 °C, исходной кислоты 8—10 °C и полифосфорной кислоты 270—350 °C и продолжительности пребывания кислоты в концентраторе 8—72 мин) получали полифосфорную кислоту со степенью полимеризации 45— 56%, содержащую, в %: Р2О5(об1ц) — 71— 75; Р2О5 (орто-) — 28—<44; SO3—1,0—4,4; Fo6iu — 0,04—0,18; R2O3— 0,04—0,11 и нерастворимого остатка — 0,04—0,43 [14, вып. 7, с. 9].
Термическое превращение ортофосфорной кислоты в присутствии фосфатов алюминия отличается от обезвоживания термической и очищенной экстракционной фосфорной кислоты [6, 21]. При 300—340 °C происходит дегидратация А1(Н^РО4)з с образованием аморфного труднорастворимого триполифосфата алюминия А1Н2Р3О10. При 390—<405 °C в твердую фазу выделяется и метафосфат алюминия.
Для получения прозрачной суперфосфорной кислоты следует упаривать кислоту со скоростью, при которой температура процесса остается, ниже температуры кристаллизации твердой фазы.
Потери фосфорного ангидрида устраняют оборудованием эффективной установки очистки газов, отходящих из концентратора.
На рис. V.<1 показана схема второй ступени двухступенчатого производства, суперфосфорной кислоты концентрации 72—76% Р2О5, содержащей 52—60% полиформ (от общего 182
Рис. V.l. Полузаводская установка для концентрирования упаренной экстракционной фосфорной кислоты до полифосфорной:
1— бак, фосфорной кислоты (53—56% Р2О5); 2 — погружной насос; 3— напорный бак; 4 — топка; 5 — концентратор; '6 — регулятор уровня; 7 — сборник-охладитель полифосфорной кислоты; 8, 15, 19 — брызгоуловнте-ли; 9—аппарат распыливающего типа (APT); 10, 16, 20 — циркуляционные сборники; 11 — форсунка, вмонтированная в газоход; 13, 18 — скрубберы Вентури; 12, 17—холодильники; 14 — вентиляторы.
количества фосфорного ангидрида), производительностью 800 кг/ч Р2О5 с улавливанием тумана фосфорной кислоты в насадочных башнях, в аппаратах распыливающего типа (APT) и скрубберах Вентури [22, 23].
Исходную кислоту (25—30% Р2О5) выпаривают в концентраторе первой ступени объемом 3 м3, представляющем собой четырехугольную емкость, футерованную кислотоупорным кирпичом с коническим днищем.-На крышке аппарата смонтирована топка с барботажной трубой, изготовленной из стали 0Х'23Н28МЗДЭТ и охлаждаемой воздухом. В нижней части трубы крепится наконечник из той же стали, находящийся при работе аппарата под слоем кислоты. Наконечник снабжен водоохлаждаемой рубашкой и погружен в кислоту на глубину 500 мм, что обеспечивает унос нерастворимых примесей с кислотой.
На первой стадии кислота упаривается дымовыми газами с температурой 900—1000 °C, проходящими по барботажной-трубе со скоростью 65—75 м/с, до концентрации 53—55% Р2О5. Концентрированную кислоту с температурой 115— 118 °C выводят из аппарата-через сборник в хранилище-отстойник. Выделяющиеся на первой ступени в газовую фазу соединения фтора и туман фосфорной кислоты, в количестве 7,5 г F и 1,5 г Р2О5 на- 1 м3 сухих газов, с температурой 118—'120°‘С, пройдя брызгоуловитель,- поступают в абсорбционную систему. Очищенные газы вентилятором выбрасываются в атмосферу. .
Упаренная кислота из отстойника (состава, в %: Р2О5 — 53—55; SO3 — 2—2,5; Робщ —0,3; R2O3 — 1,3—4,5; СаО— 0,049 и нерастворимый остаток 0—0,05) поступает в аппарат распыливающего типа (APT) для подогрева и частичного
183
концентрирования (на 1—2% P2OS) за счет теплоты газов, отходящих от концентратора второй ступени [22, 23].
'Подогретая упаренная кислота из аппарата APT через напорный бак поступает в концентратор второй ступени, где происходит 'образование полифоофорной кислоты при взаимодействии с топочными газами, поступающими при 900 °C. Последние проходят в кислоту по графитовой барботажной трубе, погруженной в слой жидкости на глубину 30—100 мм, со скоростью 65—70 м/с.
Получаемая по-лифосфорная кислота с температурой 290—300°C (состава, в %: Р2О5 (общ> — 72—76; Р2О5(орто_)— 30-J35; 'R2O3—1,8—2,0; SO3 —3,0—4,0; F —0,05—0,15 и не,-растворимый остаток — 0,08—0,13) через гидрозатвор поступает в сборник-охладитель, откуда перекачивается в хранилище.
Концентратор второй ступени объемом 1,8 м3 изготовлен из стального- кожуха, футерован свинцовым листом и слоем графитовых блоков. На крышке аппарата, футерованной кислотоупорным бетоном, установлена топка для сжигания природного газа. Для промышленных аппаратов рекомендуется [22] устанавливать несколько барботажных труб в отдельных кольцевых отсеках для равномерного распределения тепловой нагрузки в объеме кислоты- [2'3].
Интенсивность работы полупромышленного аппарата не отличается существенно от приведенной выше (стр. 183) и составляет: 2250 кг/ч по съему выпаренной воды 1 м2 поверхности кислоты или 250 кг/ч с 1 м3 концентратора при ,удельном расходе теплоты (с учетом использования теплоты отходящих газов в APT), равном 4800 кДж/кг.
Оптимальные технологические и конструктивные параметры концентраторов могут быть определены на основе минимизации суммы приведенных эксплуатационных и капитальных затрат [24].
Для получения суперфосфорной кислоты используют также вакуум-выпарные аппараты с выносным кипятильник ом или аппараты с падающей пленкой. Для 'цагревания применяют пар высокого давления '[до ~3 ‘МПа: (~30 ат)] или даутерм (смесь дифенила и окиси дифенила) при 240°C (в-последнем случае не требуется пара 'высокого давления).
Выпарка исходной кислоты с концентрацией 54% Р2О5 протекает при усиленной -искусственной циркуляции; это препятствует выделению 'значительного количества осадков. Тем не менее накапливающуюся накипь необходимо через каждые 4—5 дней смывать в течение '12 ч разбавленной серной кислотой.
Вакуум-выпарные установки для получения суперфосфорной кислоты, содержащей 70—75% Р2О5, имеют суточную 184
производительность 50—150 т Р2О5 (1—3 т/ч выпаренной воды).
Более эффективно протекает выпарка кислоты в пленочном1 аппарате [6], представляющем собой трубчатый ваку-ум-аппар'ат. Кислота в нем проходит внутри труб, со стенок которых она стекает тонкой пленкой. Пар проходит параллельно внешней стороне труб. Несконденсированная часть пара поступает в двухсекционный эжектор. Суточная производительность аппарата 2'00 т Р2О5 (3,8 т/ч выпаренной воды). Кислота отделяется в нижней части испарителя и направляется в приемник, откуда часть ее перекачивают для рецикла, а другую часть после охлаждения—в хранилище готового продукта.
По сравнению с первым вакуум-испарителем вакуум-концентратор пленочного типа более дешев, количество циркулирующей жидкости в нем меньше, он проще в регулировании, его реже нужно промывать (I раз в 15 дней) для удаления загрязнения, да и сама установка.более мощна.
V.3. УЛЬТРАФОСФОРНАЯ КИСЛОТА
Ультрафосфорная. кислота (УФК), содержащая 83% Р2О5 и более, в отличие от обычной фосфорной кислоты такой же концентрации, при комнатной температуре находится в жидком состоянии. Кроме того1, в ней очень мило мета-фосфорной кислоты, что исключает образование нерастворимых соединений.
Получаемый продукт содержит 83—98 и даже более 100% Р2О5 (в том числе до 80% в полиформе) и I —15% примесей солей (концентрация Р2О5 рассчитана на чистую кислоту).
Значительное 'содержание фосфора в ультрафосфорнои кислоте можно объяснить образованием низших окйслов фосфора (например, Р2О3) в зоне нагревания при повышенной температуре. Возможно, что в присутствии низших окис-лов фосфора метафосфорная, три- или тетраполифосфорная кислоты могут иметь некоторые новые формы с высокой пептизирующей епособйостью.
Общая формула УФК HnrPnOn(5+r)/2 (где г — молярное отношение Н2О: Р2О5 (г<1; при г=1 состав' отвечает Н3РО3, при г = 0 — твердый P^Os) позволяет определять молярный состав кислот, получаемых при разных условиях. В чистом виде УФК при обычных условиях —прозрачные, стеклообразные продукты, весьмб. гигроскопичные. Свойства образующихся продуктов зависят от условий их получения [25].
На рис. V.2 показана зависимость 'вязкости от концентрации Р2О5 для супер- и ультрафосфорнои кислот, полученных из упаренной-экстракционной кислоты, содержащей 9%
185
Р«О(,%(<$ез учета примесей)
Рис. V.2. Зависимость вязкости супер- и ультрафосфорной кислот от содержания P2Os при 25 °C.
Рис. V.3. Температура кипения ультрафосфорной кислоты разной .концентрации при различном содержании примесей.
примесей. Как видно, максимум соответствует той концентрации кислоты (82—83% Р2О5), кз которой нри дальнейшем упарйвании раствора можно получить продукт .в жидком виде. Содержащиеся-в растворе твердые примеси образуют стойкую суспензию, и продукт можно перекачивать при комнатной температуре.
Благодаря тому, что твердые частицы в ультрафосфорной кислоте не.коагулируют и не осаждаются, жидкие концентрированные удобрения, полученные из нее, более устойчивы, чем продукты, приготовленные из суперфосфорной кислоты.
С увеличением содержания примесей в исходной кислоте от 5 до 12% температура кипения продукта разной концентрации уменьшается (рис. V.3). Наличие примесей в растворе позволяет дегидратировать ортофосфорную кислоту при более низкой температуре, чем при их отсутствии.
Для получения ультрафосфорной кислоты исходную экстракционную фосфорную-кислоту (неударенную или упаренную), содержащую до 15% примесей, обрабатывают в течение короткого времени при 320—'400 °C топочными газами, поступающими в жидкость с температурой от 800 до 1000°C, образующимися в выносной камере сжигания, установленной над концентратором. Быстро движущийся поток горячих га-, зов смешивается с жидкостью, которая в высокотурбулент-ном состоянии частично уносится с газом в верхнюю часть аппарата. Газожидкостную смесь отводят из верхней части концентратора в сепаратор для отделения брызг продукционной кислоты. Газ, отходящий из сепаратора, проходит скруббер с плавающей насадкой, где из него улавливаются водорастворимые примеси.
186
Продукционную кислоту удаляют из концентратора через охлаждаемую водой переливную трубу в сборник, откуда перекачивают ib хранилище готовой продукции.
Ультрасульфофосфорная кислота, содержащая 8'0—’100% Р2О5 (без учета концентрации примесей),.5—10% SO3 в виде сульфогрупп и 5—15% примесей (соединения трехвалентных металлов, фтора и другие), менее агрессивна, чем экстракционная кислота, и более, текуча, чем супер фосфорная кислота. При содержании 5% H2SO4 в кислоте концентрации 78% Рй05(общ) степень превращения орто-форм в"полифор-мы за 90 мин при 340°C увеличивается на ~7% (с 79,8 до 87,4%) |[26].
Ультрасульфофосфорную кислоту можно получить упариванием экстракционной кислоты, к которой добавлено необходимое количество концентрированной серной кислоты при 230—340 °C в течение 10 мин.
При получении ультранитрофосфорной кислоты до обработки экстракционной кислоты, к ней добавляют. HNO3 до содержания ~0,5% азота. При этом происходит окисление органических соединений и переход низших цитратнонерастворимых соединений фосфора в циэратнорастворимые. Одновременно улучшается цвет осветленного продукта.
Для получения ультранитросульфофосфорной кислоты к исходной кислоте, кроме HNO3, добавляют H2SO4 до содержания 5—10% SO3.
V.4. ЭКСТРАКЦИОННО-ТЕРМИЧЕСКАЯ ПОЛИФОСФОРНАЯ КИСЛОТА
Комбинирование экстракционного способа производства фосфорной кислоты с термическим представляет собой один из реальных путей широкого использования бедных фосфоритов Каратау и других месторождений для получения высококачественных удобрений и кормовых фосфатов [2, 27]. При этом из различных методов (смешение слабой экстракционной кислоты с термической, растворение твердой Р2О5 в экстракционной кислоте и др. [27]) наиболее экономичным является так называемый барботажно-испарительный {14, вы!п. 4, с. 10]. По ЭТему способу, продукты сжигания элементарного фосфора до пятиокиси с температурой 1500°C приводятся во взаимодействие со слабой экстракционной кислотой, упариваемой за счет теплоты поступающих газов.
В производстве термической кислоты одной и той же концентрации (например, 54% Р2О5) выделяется в ~2,5 раза больше теплоты по сравнению с расходом теплоты в экстракционном процессе (соответственно 1’0 и 4 млн. кДж на 1 т Р2О5). .
187
9
По своим физико-химическим свойствам экстракционно-термическая полифосфорная кислота (ЭТПФК)4 занимает промежуточное положение между термической и экстракционной кислотами при равных концентрациях [2]. Однако । плотность и вязкость ее несколько больше при использовании экстракционной кислоты из каратауского фосфорита, чем из апатитового концентрата [27,ус. 127]. При соответствующих равных концентрациях Р2О5 плотность ЭТПФК, полученной с использованием кислоты из фосфорита Каратау, немного выше плотности термической полифосфорной кислоты (ТПФК), а вязкость ее в 4—5 раз больше, чем тпфк.
Вязкость ЭТПФК концентрации 74,85% Р2О5 (содержащей, в %: MgO — 0,65; СаО—'0,18; Ре20з— 0,12; AI2O3 — 0,34; SO3 — 0,81; As •—0,0019 и F —0,10), полученной в лабораторных условиях с применением экстракционной кислоты из фосфорита Каратау (содержащей, в %: Р2О5—19,0; MgO —2,5; F—1,2; SO3 — 4,8; SiO2 —0,6; RgO3 — 1,5), равна 461,1 сП при 40 °C и 67,9 сП при 80 °C [28].
В вависимости от концентрации исходной экстракционной кислоты и получаемого продукта доля Р2О5 в последнем за счет сгорания фосфора составляет от 30 до 50%.
Экстракционно-термическую фосфорную кислоту можно получить из экстракционной фосфорной кислоты в башне гидратации теплообменной испарительной установки получения термической кислоты [27]. При этом газ поступает в башню гидратации охлажденным на 40—50%> т. е. при 800—900 °C, и процесс может быть существенно осложнен отложением примесей.
Более перспективен разработанный в НИУИФе совмещенный экстракционный и термический процесс по так называемому барботажно-испарительному методу (рис. V.4), при котором 75—80% энергетических затрат на получение продукта компенсируется за счет теплоты сжигания фосфора.
Расплавленный фосфор сжигают в двух форсунках циклонной камеры. Продукты сгорания, содержащие 230—250 г Р2О5/м3 при 1500 °C, проходят по охлажденной барботажной трубе со скоростью 4'0—60 м/с и поступают в камеру упаривания, куда подают одновременна экстракционную фосфорную кислоту концентрации 19—22% (или 29—30%) Р2О5. В результате концентрирования в камере разбавленной кислоты и растворения в ней фосфорного ангидрида образуется полифосфорная кислота концентрации 72—80% Р2О5, которую через промежуточную емкость направляют на склад.
Отходящие из камеры упаривания газы, содержащие туман фосфорной кислоты, водяные пары, соединения фтора проходят через каплеуловитель и электрофильтр или волок-, «истый фильтр, в которых улавливается туман фосфорной
a
Рис. V.4. Схема установки для получения экстракционно-термической по-лифосфорной кислоты:
1—хранилище фосфора; 2 — хранилище экстракционной фосфорной кис-1 лоты; 3— барботажная труба; 4 — выпарная камера; 5 — воздуходувка;
6 — каплеуловитель; 7 — сборник продукционной кислоты; 8—насос; 9— газоход; 10— электрофильтр; 11 — волокнистый фильтр; 12— сборник кислоты; 13, 16, 20, 2/ —насосы; 14, /7 —скрубберы для улавливания фтора; 15, 18, 23 — сборники H2SiFe; 19 — теплообменник; 22 — труба Вентури; 24 — вентилятор.
кислоты. Отделяемая в этих аппаратах полифоофорная кислота через сборник присоединяется к основному потоку продукции. Затем .газы, освобожденные от тумана фосфорной кислоты и содержащие водяные пары и соединения фтора, проходят через насадочный и полый скрубберы и скруббер Вентури, орошаемые циркулирующей кремнефтористоводородной кислотой. Образующаяся в процессе кремнефтористоводородная кислота выводится из цикла для переработки на фториды. Очищенные газы вентилятором выбрасываются в атмосферу.
Описанный процесс позволяет резко сократить энергетические затраты на. получение товарной продукции. При совмещении типовых установок производства -термической й экстракционной фосфорной кислоты, единичная мощность комбинированной системы при выпуске кислоты .концентрации 72 и 22’% Р2О5 в исходной экстракционной кислоте увеличивается в 1,75 раза по сравнению с мощностью экстракции и в 2,33 раза по сравнению- с мощностью производства термической кислоты. Это следует из соотношений (на 1 т Н3РО4) экстракционной — 0,571—и термической — 0,429 — кислот в получаемом продукте. Соответственно этому почти в такой же мере снижаются эксплуатационные затраты и удельные капиталовложения [28].
Как видно из проектной калькуляции себестоимости ЭТПФК в расчете на. 1 т Н3РО4 концентрации 72% Р2О5, себестоимость t т продукта равна ~ 170 руб.:
189
Статьи затрат Расход Цена еди- ницы, руб. Сумма, РУб-
Основные материалы, т: -
желтый фосфор 0,1354 . 700,00 94,78
экстракционная фосфорная кис- 1,8818 . 30,8 57,96
лота (22% Р2О5)
Итого I 152,74
Энергетические затраты:
электроэнергия, кВт ч 42,6 0,0104 0,44
вода, м3:
свежая 1,5 0,007 0,01
оборотная 160 0,058 0,93
химически очищенная 2,7 0,025 0,07
пар, т 0,12' 2,59 0,31
воздух сжатый, нм3 200 0,003 0,60
Итого II 2,36
Основная и дополнительная заработ- 1 — 0,33
ная плата производственных рабо-
чих в начислением
Амортизация основных средств,-зда- — —. 0,06*
ний, сооружений, оборудования
Итого 155.,49
Цеховые расходы: 1,24
всего цеховая себестоимость 156,73
Общезаводские расходы:
% от цеховой себестоимости 8,62
общезаводская себестоимость — — 165,35
Коммерческая себестоимость ' — 169,60
В то же время три средней себестоимости! 1 т фосфорита 700 'руб. (по лучшим данным заводов за 1977 г. [29]) расходы только по. фосфору ,на. производство термической поли-, фосфорной кислоты* концентрации 72% Р2О5 составляет 22Q руб.. (при оптовой цене 1 т кислоты 73% Р2О5, равной 223 руб.).
ЛИТЕРАТУРА
1. Ван Везер Д. Фосфор и. его соединения. Пер. с англ./Под ред. А. И. Шерешевского. М., ИЛ, 1962. 678 с.
2. Термическая фосфорная кислота, соли и удобрения иа ее основе/Под ред. Н. Н. Постникова. М., Химия, 1976. 336 с.
3. Joang D. С., Scott С. В. — Chem. Eng. Progr., 1963; v. 59, № 12, p. 80.
4. Беглов Б. M., Борухов И. А., Джураев Ф. X. — ЖПХ, 1977, т. 50, № 2, с. 460—463.
5. Михайлин . А. Д. и др. — Там же, 1975, т. 48, № 7, с. 1435—1438.
6. Dumon R.— Chim. et ind., 1964, v. 92, № 3, p. 232—240.
7. Промышленность фосфорных удобрений. Труды НИУИФ Вып 215. М„ 1971.
8. Кочетков В. Н. и др. — ЖПХ, 1977, т. 50, № 9, с. 1913—1917.
9. Рило И. П., Лозинский А. А. —Там же, № 10, с. 2164—2172,
190
10. Беглов Б. М.— Автореф. докт. дисс. Ташкент, АН УзССР, 1975.
И. Зотова К. С., Паниди Е. В.— В кн.: Проблемы химии и химической технологии. М., Наука, 1977, с. 240—252.
12. Жданов Ю. Ф. а др. — Труды НИУИФ, 1973, вып. 221, с. 161—175.
13. Смирнова И. М., Лепилина Р. Г., Позин М. Е.—ЖПХ, 1976, т. 49, № 8, с. 1672.
14. Промышленность минеральных удобрений и серной кислоты. Реф. сб. НИУИФ. М„ НИИТЭХИМ, 1978.
15. Малахова-И. И. и др. — Хим. пром., 1975, № 10, с. 752—755; Кочетков В. И: и др. — Там же, 1976, № 2, с. 117—118.
' 16. Сыркин Л. И. и др. — Там же, 1972, № 8, с. 594—597.
17. Михайлин А. Д. и др. — Там же, 1977, № 7, с. 513.-
18. Попов И. П., Лыков М. В., Леушева А. Г. — Труды НИУИФ, 1971, вып. 219, с. 35—53.
19. Slack А. V. Phosphoric Acid. V. 1, part 2. New York, Intersciense Publ. Inc., 1958.
20. Попов H. П. и dp. — В кн.: Промышленность минеральных удобрений и серной кислоты. Реф. инф. М., НИУИФ, 1969 Вып. 1, 1969; вып. 7, 1971.
21. Борисов В. М„ Ажикина Ю. В., Ларик И. С. — Хим. пром., 1969, № 4, с. 277; Щегров Л. Н., Печковский~В. В., Борисов И. В. — ДАН БССР, 1967, т. 11, № 9, с. 816.
22. Серебряная Р. М. и др. — Хим. пром., 1973, № 9, с. 676- 678.
23. Попов И. П. Выпарные аппараты в производстве минеральных удобрений. Л., Химия, 1974, с. 126.
24. Бугрин В. П. и др. — Хим. пром., 1976, № И, с. 864—867. •
25. Михайлин А. Д. и др. Труды НИУИФ. 1975, вып. 226, с 31—40.
26. Беглов Б. М. и др. — ЖПХ, 1977, т. 50, № 12, с. 2633—2636.
27. Переработка фосфоритов Каратау. Промышленные методы и лабо-раторйые -разработки/Под ред. М. Е. Позина, Б. А. Копылева, В. Н. Белова, -В. А. Ершова, Л., Химия, 1975.
28. Михайлин А. Д. и др. — Хим. пром., 1976, № 2, с. 113—115.
29. Туров Ю. Я. и др. Производство фосфора и карбида кальция. Обзорная инф. ЛенНЙИГипрохим. Серия Фосфор и его соединения. М., НИИТЭХИМ, 1978, с. 1—23.
tлава VI
УЛАВЛИВАНИЕ И УТИЛИЗАЦИЯ СОЕДИНЕНИЙ ФТОРА
В фосфатных рудах содержание фтора по отношению к Р2О5 колеблется от 8 до 12%. По сравнению с разведанными мировыми запасами плавикового шпата — порядка 65 млн. Ca.F2, являющегося в настоящее время главным видом сырья для производства важнейших соединений фтора, запасы фтора в фосфатном сырье достигают 2800 млн. т (в пересчете на 'CaF2).
Улавливание и утилизация фтора в производстве экстракционной фосфорной кислоты определяются санитарными требованиями, экономическими, а также экологическими соображениями [1, 2].
VI.1. ФАЗОВОЕ РАСПРЕДЕЛЕНИЕ ФТОРА ПО СТАДИЯМ РАЗЛИЧНЫХ ПРОЦЕССОВ
Распределение фтора по фазам —газовой, жидкой (кислоте) и твердой (фосфогипсе) в производстве экстракционной фосфорной кислоты зависит от природы фосфатного сырья, его состава, условий проведения процесса (дигидратным, полугидратным методом), концентрации получаемого продукта и т. п. При аналогичных условиях для разных заводов имеющиеся сведения неоднозначны.
При дигидратном методе переработки апатитового концентрата выделение фтора в газовую фазу составляет (здесь и ниже в расчете от поступающего с сырьем) на стадии экстракции от 1—1,5 [3] до 7% [4, с. 67], при вакуум-охлажде-нии — от 3 [4, с. 67] до 7% '[3], и фильтровании 0,3—'0,5%; в кислоту переходит 78—80%, а в фосфогипсе остается 13— 17%'фтора (в расчете на сухой продукт).
В полугидратном способе производства концентрированной фосфорной кислоты, содержащей 45—48% Р2О5, в газовую фазу переходит 30—45% F в зависимости! от температуры и других условий. Основное 'количество его удаляется при разложении апатита фосфорной кислотой или при смешении серной и оборотной фосфорной кислот.
192
При практически одинаковой температуре (90—97 °C) повышение концентрации кислоты от 35 до 45% P20s, приводит к реэ/ому повышению степени выделения фтора. Ниже приведены данные, полученные при осуществлении, процесса. разными способами: с разделением, стадий (стр. 130) разложения апйтит.а и кристаллизации сульфата ' кальция (без введения соды) и без разделения обеих стадий (стр. 125) |[5]. В первом случае при получении кислоты концентрации 35,5и 45% Р2О5 в газовую фазу переходит фтора 10 и 42% (*в фосфорную кислоту 73 и 29%, а в фосфополугидрат 17 и 29р/о). Во втором случае {5, 6] при получении кислоты 35—38 и 40—44% P20s в газовую фазу переходит фтора 13—18 и 34—35% (в фосфорную кислоту 62—70 и 40—-44%, а. в фосфополугидрат 17—22 и 22—25%).
При работе со смесителем ВХК-НИИХиммаш (стр. 125) использование башенной серной кислоты с получением -кислоты с концентрацией 35-^38% Р2О5 основное количество1 фтора (47—80% от общего его выхода в газовую фазу) выделяется в вакуум-испарителе [6].
При получении кислоты (42—47% Р2О5 и 4,5—5,5% SO3) ангидритным методом при 105—112 °C в опытных полупромышленных условиях в газы выделяется до 70—75% F от количества его, введенного с апатитовым концентратом [4, с. 3].
В процессе упаривания кислоты, полученной дигидратным методом, до ~54% Р2О5 из нее удаляется ~80%, а при упаривании до 60% Р2О5— 88—90% F. Степень выделения соединений фтора при получении полифосфорной кислоты зависит от концентрации конечного продукта и исходной кислоты. При содержании в полифосфорной кислоте 73% Р2О5 степень выделения соединений фтора составляет 90—93% (если исходная кислота содержала 30% Р2О5); из кислоты, предварительно упаренной до 48—50% Р2О5, ~ 90%; из кислоты, содержащей ~60% Р2О5, — 70% [7, с. 83].
В производстве фосфорной кислоты концентрации 22—24 и 23—27% Р2О5 дигидратным способом из фосфоритов Кара-тау при 90—95 °C в газовую фазу выделяется [8, с. 24] 8—13 и 19% F (в жидкой фазе остается 76—85 и 80%, а в фосфо-гипсе. 6—13 и 11% F от его содержания в исходном сырье). В пблугидратно^ способе получения из фосфорита кислоты/ содержащей 33—35 и 30—40% Р2О5, три 92—100 °C в газовую фазу выделяется [8, с. 24] 13—17 и 23—24% F (в жидкой фазе остается 48—54 и 44—47%. и в фосфополугидрате 29—32% F).,
Показано 1[9, № 7], что упариванием^ полученной при 90— 95 °C из фосфорита Каратау (месторождения Кок-Джон) фосфорной кислоты (состава, в %: 'Р^О5 — 20—22; СаО — 0,7—0,9; MgO— 1,7—1,8; R2O3 — 1,0—4,4; F— 1,2—1,5 и 13-5473 193
S03 — 2—2,5) в присутствии активного кремнезема — кизельгура (150—300% от стехиометрии на фтор) —до 50 и 52% Р2О5 степень обесфторивания увеличивается на 15— 25% (от 45 и 55% в отсутствие кизельгура до 69 и 77% в присутствии 300% кизельгура). При упаривании кислоты до 40 и 45% Р2О5 (в отсутствие и в присутствии кизельгура) степень обесфторивания составляет ~25 и 35—40 %,. Упаривание неосветленной кислоты (содержащей 1,3% нерастворимого остатка), полученной из чилисайских фосфоритов, от 20,9 до 52,5% Р2О5 приводит к обесфториванию ее на 29% [9, № 6], т. е. больше на, 10%, чем из осветленной, из которой удаление фтора составляет 19%.
При добавлении к фосфорной кислоте, полученной полугидратным методом из фосфоритов Каратау, серной в количестве 105—>120% от стехиометрии в расчете на полуторные окислы, окись кальция и другие примеси, остаточное содержание фтора в процессе ее упаругвания уменьшается [9, № 11 в ~ 1,5 раза (от 0,74—0,92 до 0,51—0,62%).
Количество фтора, переходящее в газовую фазу, тем больше, чем меньше в фосфате окислов трехвалентных и щелочных металлов, а также чем выше температура и. больше концентрация кислоты. Переходу фтора в газовую фазу способствует также интенсификация перемешивания реакционной массы, а кроме того, увеличение разрежения в системе и количества просасываемого воздуха.
Обработкой исходной кислоты (29—30% Р2О5) или предварительно упаренной (до 45—55% Р2О5) перегретым паром с температурой до 500 °C или смесью его с воздухом в течение 30—50 мин возможно достичь степени обесфторивания, равной 85—95% [10]. В обесфторенной кислоте из апатита или из богатых руд Каратау остается всего 0,05—0,08% F (в среднем до 0,1 % F).
Максимальное извлечение фтора (на 93,5—98%) при прочих одинаковых условиях упаривания кислоты достигается при введении в систему кизельгура в количестве от 100 до 70% от стехиометрии на разложение H2SiF6 [11]. Последний, помимо разложения H2SiF6 до SiF4, по-видимому, действует разрушающим образом на появляющуюся в процессе упарки монофторфосфорную кислоту H2PO3F [12, с. 1371].
В газах, выделяющихся в производстве экстракционной фосфорной кислоты дигидратным методом, содержится В основном S1F4. При упаривании экстракционной фосфорной кислоты, а также, по-видимому, частично при производстве кислоты полугидратным методом фтор выделяется преимущественно в виде почти эквивалентной (при выпаривании кислоты) или неэквивалентной (при полугидратном процессе) смеси SiF4 с HF. Это объясняется превращениями соединений фтора при изменении концентрации фосфорной кис
194
лоты. В кислоте, содержащей 29—30% Р2О5, 'находится в. основном SiFg2- и некоторое количество F". При упаривании ее до 38% Р2О5 (степень обесфторивания 10—12%) в газовую фазу выделяется преимущественно SiF4 и небольшое количество HF. Мольное отношение F: SiO2 составляет 4,15— 4,35. Дальнейшее упаривание до 48—56% Р2О5 сопровождается увеличением этого мольного отношения до 5,8 в газовой фазе и 8,9 в остающейся жидкой фазе [12, с. 1371]. .Объясняется это- тем, что кремнефториды разлагаются быстрее и полнее, чем фториды, и в жидкой фазе увеличивается относительное количество последних.
В образующейся при водной абсорбции газов кремнефтористоводородной кислоте мольное отношение F: SiO2 ниже стехиометрического и составляет от 5 до 5,8. Это связывают [1-2, с. 1374] с возможностью образования, наряду с кремнефтористоводородной, также пентафторкремниевой кислоты-HSiF5.
В дальнейшем при получении полифосфорных кислот фториды при повышенной температуре и концентрации кислоты разрушаются с выделением фтористоводородной кислоты в газовую фазу. Количество ее в газовой фазе особенно возрастает при использовании для получения полифосфорной кислоты предварительно упаренной кислот-Ы' до 54—56% Р2О5, что связано с необходимостью усиления защиты оборудования от коррозии.
VI.2. МЕТОДЫ ПОГЛОЩЕНИЯ И ОСАЖДЕНИЯ СОЕДИНЕНИЙ ФТОРА
Как было указано выше, соединения фтора выделяются в газовую фазу практически, на всех стадиях производства фосфорной кислоты. В процессе экстракции выделяется сравнительно небольшое количество их (1,5—2% от общего количества в сырье) из экстрактора, 3—<6% при вакуум-охлаждении, 0,25—0,4% при фильтровании, и некоторое количество— из оборотных сборников фосфорной кислоты, промежуточных емкостей и хранилищ— и притом в весьма разбавленном состоянии, в среднем около- 0,5—0,7 г/м3.
Газы, отходящие из экстракторов производства кислоты концентрации 30—32% РгОд, содержат ~2,5 г/м3, при охлаждении воздухом пульпы в экстракторах—0,18— 0,34 г/м3 (по сравнению с 8,5—9 г F/м8 из барабанных концентраторов фосфорной кислоты).
Поглощение фтррсодержащих газов в производстве фосфорной кислоты—.цесьма трудный абсорбционный процесс вследствие низкой их концентрации, загрязненности туманом фосфорной кислоты, выделения кремнезема, значительного коррозионного воздействия на аппаратуру и высоких сани-
13*
195
тарных требований. Допустимое содержание соединений фтора. в воздухе производственных помещений составляет 0,5 мг/м3, а в воздухе населенных мест — 0,02 мт/м3 (см. #
пл. VII).
Абсорбционные способы
Поглощение фторсодержащих тазов, выделяющихся в процессе экстракции, предусматривает главным образом санитарные цели и осуществляется на некоторых заводах с использованием речной вады. Образующиеся стоки на системе производительностью 55 тыс. т Р2О5 в год в количестве 170— 230 м3/ч содержат от 30 до 100—150 мг F/л, а суточный сброс фтора составляет 800—1000 кг 1[13]. Усовершенствование процесса позволило сократить количество- сточных вод до-15—20 м3/ч, которые затем обрабатывают на -стадии нейтрализации. Указывают [13] на возможность полной ликвидации стоков путем использования воды, отходящей со станции нейтрализации, после их фильтрования и отстаивания в отстойнике.
Для санитарной очистки газов в качестве поглотителей могут быть использованы также щелочные растворы соды, гидроокиси кальция, аммиака и т. п. Степень очистки газа, содержащего 0,2—0,7 г F/м3, Д9—3,5 %-ным раствором Na2CO3 или 0,2—0,5 %-ным известковым молоком составляет 95—99% (водой 86—90%) [14].
Абсорбционные методы с 'использованием в качестве поглотителя циркулирующей кремнефтористоводородной кислоты, отводимой частично в виде продукта, применяют для улавливания газов, выделяющихся при упаривании кислоты. В некоторых случаях мо-Гут быть использованы растворы соды, -поташа, аммиака, фторида аммония и др.
Весьма существенно- изыскание растворителей, действующих селективно по- отношению к HF и SiF4. В этом отношении представляют перспективный интерес исследования по скоростной абсорбции фтористого водорода из его смеси с SiF4 (в единичной капле) в течение ~ 0,2—0,25 с [2, 15] с применением в качестве абсорбентов воды, водных растворов KF и КС1, KjHF2 и др. Изучены также абсорбционные свойства концентрированной серной и фторсульфоновой кислот И их смесей [16], которые, поглощая фтористый водород, не ' реагируют с четырехфтористым кремнием. При весьма хорошей растворимости HF в концентрированной серной кислоте (96,5% H2SO4) десорбция его осложняется образованием стойкого комплекса H2SO4-HF и необходимостью поэтому использовать оборотную серную кислоту, содержащую 50% (мол.) фтористого водорода.
196
В отличие от этого в системе HSO3F—H2SO4 азеотропные точки отсутствуют, т. е. использовать смесь фторсульфоновой и серной кислот для абсорбции фтористого водорода 'выгоднее. Процесс протекает в одну стадию и отличается большей емкостью по фтористому водороду и, кроме того, менее кор-розионио опасен.
Заслуживает внимания использование в качестве селективного абсорбента 50%-ного раствора бифторида калия KF-HF, при помощи которого вначале из смеси, содержащей водяные пары, HF и SiF4, абсорбируется SiF4 при 105— ПО °C с образованием KzSiF6 /[17]. Из остающейся смеси водяных паров и фтористого4 водорода последний извлекается в первой стадйи при 140—160 °C смешанным раствором би- и трифторида калия KF-2HF’, образующимся во второй стадии абсорбции при ПО—115 °C при взаимодействии газов паровой смесй. с 50%-ным раствором KF-HF. Из полученного в первой стадии трифторида калия отгоняется HF при 300—350 °C, который перерабатывается в конечный продукт. Остающийся при этом бифторид калия идет на приготовление основного абсорбционного раствора — 50%-ного раствора 'KF-HF. Недостатки метода заключаются в трудностях отделения KjSiFe от вязкого раствора, в применении высоких температур с образованием малотекучи^ расплавов и, возможно, в необходимости предварительной подготовки исходной смеси путем высокотемпературного гидролиза.
Экстракционные способы
Эти способы, предназначенные для обесфторивания жидкой фазы— кислоты, известны давно [2]. Для извлечения фтора из экстракционной фосфорной кислоты могут служить такие органические растворители, как кетоны, альдегиды, эфиры, алифатические и ароматичеокие-спирты. Наиболее селективны по отношению к гексафторсиликат-иону, присутствующему и основном в экстракционной кислоте, доступны и в то же время относительно дешевы третичные амины и фосфиноксиды [18]. В качестве разбавителей могут быть^ис-пользованы керосин, бензол,. толуол, трибутилфосфат и др.
Степень извлечения фтора за один контакт фаз составляет при использовании дигидратной фосфбрной кислоты 16—20; при обработке полугидратной 39—41 и 72—74% упаренной кислоты. В полученной фосфорной кислоте фтора содержится менее 0,2%. Потери экстрагента — триал-киламина, обусловленные растворимостью и механическим уносом при очистке 1 т упаренной кислоты, определяются в Количестве- 0,05—0,08% от исходного количества.
197
Фтор из органической фазы реэкстрагируют 10%-ным раствором NH4OH. Полагают [18], что стоимость такой очистки может обойтись в 6—8 руб. на 1 т Р2О5.
В отличие от других методов, в которых в органическую фазу извлекается фосфорная кислота, выделяемая затем в очищенном виде лишь на 50%, описанный способ имеет определенные преимущества. Однако и в этом случае процесс представляется достаточно сложным, связанным с регенерацией экстрагента, его очисткой и обработкой рафината.
Ионообменные способы
Улавливание соединений- фтора из газов и особенно выделение их из фосфорной кислоты при помощи различных сорбентов — ионообменных смол АВ-17-8, АПМ и др.— является предметом многих исследований.
В лабораторных и модельных условиях показано, ’что сорбцией соединений фтора (HF и SiF4) из. газов ионитом АВ-17-8 достигается очистка газа на 90%. Остаточная концентрация фтора в газе составляет ~20—30 мг/нм3 [7, с. 42]..Процесс изучен в укрупненных масштабах применительно к доочистке фторсодержащих газов с использованием фильтра диаметром 450 мм и высотой 1600 мм, загруженного 5 кг смолы АВ-47, производительностью 80 м3/ч [8,. с. 77]. Высота сл-оя смолы составляла 100—120 мм. Содержание фтора на входе в фильтр в среднем составляло ~ 10 мт/м3 с колебаниями от 6 до 15 мг/м3 (в том числе 25— 50% в виде тумана). Выходящий из сухого фильтра газ содержал в среднем 1,17 мг F/м3 при степени' улавливания его, равной 88%.
Лучшие результаты достигнуты в режиме орошения фильтра водой.. При начальном среднем содержании в газе 24,3 мг F/м3 (в том числе 11,9 мг/м3 в виде тумана) общая степень очистки составила, 93,3% (остаточное содержание фтора 1,62 мг/м3).
Регенерацию смолы производили 1%-ным раствором NaOH после прохождения через колонну ~20 тыс. м3 газа.
Применение анионитов для извлечения фтора из экстракционной фосфорной кислоты затруднено вследствие ма-лой*их емкости и сложности регенерации [19].
Представляет интерес очистка кислоты & основном от катионов, что облегчает ее дальнейшую обработку. Ионный обмен широко разрабатывается для получения ‘кислоты реактивной чистоты применением сульфокатионитов КУ-2 и фосфорсодержащих катионитов типа КФ-11 и СФ-5 [20].
Предварительной обработкой осветленной экстракционной фосфорной кислоты (21,5% Р2О5), полученной из фосфоритов Каратау, катионитом КУ-2 из нее возможно полно-198
етью удалить СаО (при первоначальном содержании 0,58%) и существенно уменьшить (от 1,89 до- 0,14%) содержание MgO ['2'1, вып. 5, с. 26]. После катИонирЬвания -кислота содержала, в %: Р2О5.— 20,8; MgO — 0,14; Fe2O3 —-0,6; SO3 — 0,64; F—1,96 и R2O3—1,2. Концентрация последних четырех компонентов практически оставалась неизменной.
Очищенная от двухвалентных катионов фосфорная кислота поддается упариванию без затруднений до 60—65%, Образующийся при этом в небольшом количестве осадок быстро оседает. Одновременно существенно увеличивается степень обесфторивания кислоты, составляя 83 и 91% при упаривании кислоты до 55 и 65% Р2О5. Количество выделившегося фтора в этих условиях равно ~75 и 82 кг на 1 т Р2О5. Однако сравнительно малая емкость катионита и отсутствие сведений о его -регенерации, активности его после регенерации, образовании -неиспользуемых стоков не позволяют судить о .применении метода в к-рупнопромышленных масштабах.
Адсорбционные способы
Для -санитарной очистки газов от соединений фтора изучено поглощение их при фильтровании через твердые поглотители: активированные' угли, крупно- и- мелкопористые силикагели -(КСК и ШСК) и инертные по отношению к фтору -гранулы полипропилена [22]. Степень очистки при высоте слоя-050 мм с содержанием фтора на входе 0,2—0,2:5 г/-м3 на силикагеле ШСК и полипропилене составляет 90—94%. Содержание соединений фтора после очистки не превышает 15—30 мг/м3. .
Недостатком этих способов, так же как и при Использовании ионообменных смол, является .забивка фильтра двуокисью кремния и резкое в результате этого возрастание сопротивления его через 20—30 ч работы. Это объясняют образованием в жидкой фазе пента- и гексафторкремниевых кислот и- геля кремниевой кислоты. Для предотвращения забивания фильтра необходимо периодически его промывать растворами фторида и бифто-рида калия или очищать с одновременным орошением этими растворами.
Осадительные методы
В'основе этих методов лежит осаждение из экстракционной фосфорной кислоты малорастворимых кремнефторидов натрия или калия. Издавна на суперфосфатных заводах кремнефтористоводородную, кислоту, получаемую водной абсорбцией отходящих газов, перерабатывают в кремнефторид натрия, используемый в качестве инсектицида или дефоли
199
анта или как исходный полупродукт для производства различных соединений фтора.
Кремнефторид натрия из экстракционной фосфорной кислоты, полученной из апатитового концентрата, осаждают на ряде промышленных предприятий. Выделение при этом фтора. [2] на 60—80% (при некоторых условиях и на 90%) из экстракционной кислоты, .помимо увеличения выхода его и упрощения технологии по сравнению с упариванием, позволяет облегчить производство аммофоса и других .фосфорсодержащих удобрений и повысить их качество.
Растворимость кремнефторида натрия при 50 °C с увеличением- концентрации Р2О5 в кислоте от 25 до 48% Р2О5 уменьшается в отсутствие Na2SO4 от 0,9 до 0,4% и от 0,105 до 0,044%, если в растворе присутствует 5% Na2SO4. Аналогично, с увеличением концентрации кислоты от 23 до 48% Р2О5 растворимость K2SiF6 уменьшается в отсутствие K2SO4 от 0,501 до 0,480% и от 0,046 до 0,035% в присутствии 5% K2SO4![23],
Обработка экстракционной кислоты (30,2—30,6% Р2О5 и 1,8—1,9% F) из апатитового концентрата солями щелочных металлов (натрия, калия) в количестве 125—150% от стехиометрической нормы в расчете на H2SiF6 при 50 °C в течение 50 мин приводит к обесфториванию кислоты на 90— 93%. Содержание фтора в аммофосе, полученном из обес-фторенной кислоты, не превышает 0,5—0,6%, а по концентрации водорастворимой Р2О5 и питательных веществ значительно превышает качество продукта, получаемого- из не-обесфторенной кислоты [24].
Для осаждения фтора из фосфорной кислоты (30% Р2О5 и 1,9'8% F) оптимальный расход осадителя (с учетом дальнейшей переработки кремнефторидов в разные продукты и возвратом солей натрия и калия на осаждение фтора) составляет для солей натрия и калия 120—125% и 104—405% от стехиометрического количества в расчете на H2SiF6 [8, с. 77]. В фильтрате с учетом проскока твердой фазы содержится 0,31—0,35% F. При этих условиях минимальная себестоимость фтора во- фторсиликате натрия или калия составляет при использовании NaCl или КС1 ~37 и 43 руб/т, а максимальная ~62 руб/т — при осаждении фторсиликата калия поташом [23].
'Основной недостаток- процесса— образование мелких труднофильтрующихся кристаллов со значительным проскоком твердой фазы через фильтровальную ткань (лавсан-30, лавсан-144, ТФ и др.), а также получение низкокачественных продуктов — кремнефторида натрия или калия. .
Циркуляция суспензии -кремнефторида в фосфорной кислоте с кратностью, равной единице (т. е. в равных объемах циркулирующей пульпы и раствора, поступающего на осаж-200
дение), позволяет увеличить размеры выделяющихся .кристаллов кремнефторида натрия от 3,5—5 до 25 мкм. При этом скорость осаждения твердой фазы возрастает в 5— 7 раз, а производительность фильтрования в ~2 разд [2'5].
Но при использовании регенерированного осадителя — сульфата натрия (полученного прокаливанием кремнефторида натрия с сульфатом аммония) — происходит непрерывное (от цикла к циклу) снижение степени обесфторивания кислоты в результате накопления в регенерированном продукте балластных примесей, в частности сульфата кальция в виде ангидрита i[21, вып. 1, с. 7].
Способ осаждения фтора из кислоты имеет особенно важное значение в производстве фосфорной кислоты из бо-.гатых и рядовых фосфоритов Каратау и других месторождений, в связи с трудностями упаривания и малой эффективностью обесфторивания при ее концентрировании.
Обесфторенная на 80—90.% кислота из рядовой руды- Каратау (20—23,2% Р2О5, 1,9—2,1% F и 4,2—3,1% MgO), упаривается при меньшей температуре и с большей интенсивностью до 50—52% Р2О5, чем необесфторенная до 42—43% Р2О5 [26]. Количество выделяющегося при этом осадка в ~3 раза меньше (0,6% от массы упаренной кислоты). Оса-Док состоит в основном, из сульфата кальция в виде полугид-рата и ангидрита, кремнефторида и частично фторида магния. Введение осадителей на стадий экстракции' кислоты не влияет на основные показатели процесса и позволяет получить кислоту, содержащую ~0,2% F при использовании в качестве осадителя солей калия и ~0,4—'0,45—при осаждении солямилнатрия [27, с. 47].
При разложении фторсодержащего гипса 1%-ным раствором H2SO4 при 350—400 °C в течение 1,5 ч в газовую фазу выделяется 80—85% F [27, с. 46]; при использовании 35%-ной H2SO4 в количестве 120% (от стехиометрического на' содержание фтора) при 250 °C степень выделения фтора за 30 мин увеличивается до 98,5—99;5% [2'8]. Выщелоченный из осадка водой раствор сульфата натрия возвращается в процесс осаждения фтора из кислоты. При этом степень осаждения фтора возвратным Осадителем не изменяется [28]. . '
Опытами, проведенными с применением, двух образцов кислот — Алмалыкского химического завода (20,3—22,7% Р2О5, 2,2—2,6% MgO, 1,5—4,8% F) и опытного цеха Воскресенского филиала НИУИФа (20,4—21,5% Р2О5, 0,87—0,95% MgO; 1,75—'1,9% F), уточнены нормы осадителя, температурные и другие условия осуществления процесса, а также качество получаемого'.продукта [29]. С-увеличением нормы осадителя (сульфата натрия или калия) от 80 до 160% (от стехиометрического количества в расчете на H2SiF6) степень
201
обесфторивания за 40 .мин. при 70 °C увеличивается от 60— 65 до 75—80%. Образующийся кремнефторид натрия содержит нё~более 73,7% основного продукта при использовании неосветленной кислоты. Содержание Na2SiF6 в продукте увеличивается до 85—87% при предварительном отделении взвесей из кислоты.
VI.3. ТЕХНОЛОГИЧЕСКИЕ СХЕМЫ И РЕЖИМЫ
На заводах экстракционной фосфорной кислоты во всех случаях улавливают соединения фтора из газов, выделяющихся на всех стадиях процесса, а на некоторых предприятиях, помимо этого, обесфторивают получаемую кислоту осаждением из нее преимущественно кремнефторида натрия, (реже — калия).
Поглощение-фтор.а из газовой фазы определяется не столько степенью его утилизации, которая являетея достаточно высокой, но главным образом достигаемой эффективностью в санитарном отношении. Потери фтора при абсорбции составляют ~2% от всего фтора, выделившегося в газовую фазу. Но выбросы его в воздушную среду вследствие неполноты поглощения значительны как в абсолютном отношении; так и по концентрации, превышающей в некоторых случаях в 3—5 раз предельно допустимую по санитарным нормам (8—10 мг/м3). Поэтому обычно фтор абсорбируют либо в одну ступень — промывкой газа щелочным раствором, преимущественно известковым молоком, циркулирующим через станцию нейтрализации, либо в две ступени—с предварительной водно-кислотной промывкой газа с получением продукционной кремнефтористоводородной кислоты.
Осаждение фтора из экстракционной кислоты является весьма перспективным не только по соображениям, указанным выше (стр. 192), но и вследствие возможности облегчения санитарной очистки газов, а также упрощения технологического процесса получения конечного продукта (.исключается предварительное образование кремнефтористоводородной кислоты).
Абсорбция соединений фтора йз газовой фазы
Для поглощения фторсодер-жащих гадов применяют вы-, сокоинтенсивные абсорберы., эффективно работающие в условиях частичного выделения геля и присутствия в газе тумана кислоты. К таким аппаратам относятся полые шли струйные абсорберы, аппараты распыливающего типа (APT), а также абсорберы Вентури.
Полые или струйные абсорберы орошаются поглотительной жидкостью, распыливаемой форсунками, установленны-202 z
ми в несколько рядов вертикально или горизонтально. Газ, поступающий в нижнюю часть абсорберов, взаимодействует с тонкораспыленным поглотителем, направленном по сече-:нию или вдоль аппарата. Такие аппараты- называют также полыми распылительными абсорберами и используют их обычно в качестве первых по потоку промывных скрубберов.
Аппарат распиливающего типа APT (рис. VI.1) .представляет собой колонну диаметром' 300 мм и высотой 2900 мм, в верхней части которой имеется конусный распределитель и распылительный конус. Поглотительная жидкость, Стекая с конуса, .взаимодействует с поступающими на очистку газами при высокой их линейной скорости (20 м/с). Благодаря этому создается большая поверхность контакта фаз, обеспечивающая высокую эффективность процессов абсорбции и теплообмена. Далее газожидкостной поток проходит цилиндрическую камеру и в сепараторе газопаровая смесь отделяется от жидкости.
Основной, недостаток полых распиливающих абсорберов и аппаратов APT — недостаточное использование объемного' пространства вследствие малого заполнения его факелом распыленной жидкости. '
Более компактными и весьма интенсивными являются абсорберы Вентури (рис. VI.2). Разбавленную кремнефтори-< стовбдородную кислоту подают форсунками в сужение меж-
Рис. VI.Г. Аппарат распиливающего типа: 1—конусный распределитель; 2—-распылительный конус; 3— корпус испарителя; 4 — сепаратор.
Рис. VI.2. Схема абсорбции фтористых газов в аппарате Вентури: / — абсорбер Вентурн; 2 — форсунка; 3 — сборник; 4 — отстойник; 5 — центробежный насос.
203
ду двумя .конусами абсорбера, где создаются большие скорости газового потока. При установке абсорбера Вентури непосредственно на сборнике кислоты последний'одновременно служит и брызгоулавливателем и отстойником. В сборник подают необходимое количество воды, и из него отводят образующуюся кремнефтористоводородную кислоту.
Скрубберы Вентури могут быть также использованы для улавливания тумана, фосфорной кислоты вместо громоздких, и дорогих электрофильтров.
Для улавливания фторсодержащих газов на некоторых заводах применяются абсорберы с плавающей насадкой (АПН) и абсорберы пенного типа, характеризующиеся высокой степенью очистки (до 97—98%) [30].
Абсорбционные аппараты, газоходы, вентиляторы, сборники и т. и. изготовляют из нержавеющей. стали или гуммируют и футеруют кислотоупорными материалами.
Поглощение «соединений фтора при экстракции фосфорной кислоты
Фторсодержащие газы, выделяющиеся при разложении фосфатов, по вытяжным трубам экстракторов и других аппаратов отсасывают вентилятором п протягивают через несколько последовательно установленных абсорберов. При содержании в газе тумана Р2О5 их иногда предварительно пропускают через фильтры или другие аппараты.
Газы, выделяющиеся из экстракторов и вакуум-испари-телей, в дигпдратном процессе получения фосфорной кислоты абсорбируют в одно- или двухступенчатых полых башнях или абсорберах Вентури, а в полугидратном процессе — в пенном аппарате или аппарате с плавающей насадкой или сочетании одного с другим.
Абсорберы орошают водой или возвратной слабой кремнефтористоводородной кислотой, в результате чего образуется раствор, содержащий 5—10% H2SiF6 и иногда 0,1—0,5% Р2О5. Вода поступает из напорного бака через дозирующий расходомер в сборник или в последний (по ходу) абсорбер. Образующаяся кремнефтористоводородная кислота, содержащая взвесь SiO2, поступает из первой (по ходу газа) ступени абсорбции в сборник, откуда ее насосом направляют в отстойник для осветления.
Разбавленные растворы, получаемые при \абсорбции газов в отдельных абсорберах из экстракторов, вакуум-испа-рителей, фильтров и баковой аппаратуры в дигидратном ’ процессе, используют или (частично) для промывки фильтрующей ткани, или возвращат в экстракторы. Более концентрированные растворы, образующиеся при поглощении соединений фтора из газов в совмещенной абсорбционной си-. стеме с циркуляцией растворов или в полугидратном процес
204
се, могут быть использованы для производства кремнефторидов или фторидов. .
В производстве дигидратным методом фосфорной кислоты, используемой, например, для получения аммофоса, соединения фтора улавливают большей частью водой (свежей или оборотной) в абсорберах АПН, установленных в отдельности на линиях отвода газов из экстрактора, вакуум-испарителя, баковой аппаратуры и на стадии фильтрования. Образующуюся слабую кремнефтористоводородную кислоту нейтрализуют известковым молоком, осадок отфильтровывают и сбрасывают на шламовое поле. Отфильтрованную жидкость (регенерированную воду) возвращают в процесс на орошение абсорберов. При содержании в поступающих газах фтора 0,12—0,26 г/м3, в отходящих газах после очистки содержится 40—30 мг F/m3. ' .
По другой схеме газы из экстрактора, работающего по дигидратному режиму, поступают по газоходу длиной ~>15 м в двухступенчатый абсорбер Вентури с диаметром горлови-. ,ны каждого 500 мм. Абсорберы снабжены каждый 15 форсунками щелевого типа с площадью истечения ~44 мм2. Пройдя обе ступени, газы поступают на санитарную очистку, откуда вентилятором выбрасываются в атмосферу. Газы поступают на абсорбцию с температурой от 52 до 64 °C и выходят из последней ступени абсорбера с температурой от 39 до 45 °C [31].
Оба абсорбера орошаются в замкнутом цикле с отводом избытка жидкости из второй ступени на первую и выводом образующейся кремнефтористоводородной кислоты из первой ступени. Абсорбционную систему подпитывают на второй ступени из абсорбера санитарной очистки раствором, содержащим, в мг/л: F — 84—85; SiO2— 25—27 и P2Os —6,0— 6,5. Скорость газа в горловине абсорбера равна 15,5 м/с при скорости истечения жидкости в форсунках 15,5 и 17,3 м/с и манометрическом давлении перед форсунками 0,25 и 0,31 МПа (2,5 и 3,0 ат); удельный расход жидкости в первой и второй ступени равен 3,9 и 4,35 л/м3.
Степень поглощения фторсодержащих газов в первой ступени составляет 75—80%, во второй — 60—65%. Общая степень извлечения составляет 90,5% (с колебаниями от 70— 83 до 93—<96%) при средней концентрации фтора в газе на входе в первую ступень ~300 мг/м3, на' выходе из нее 53 мг/м3 и после второй ступени 21,5—22,3 мг/м3. Растворы, циркулирующие во второй и первой ступенях, содержат, в. мг/л:, F 105 и -200; SiO2 —-8,5 и 43,5 и Р2О5 — 6,5 и 6,9.
;В некоторых дигадратных системах газы, из экстр акторов, вакуум-сборников и баковой аппаратуры собирают в один коллектор и направляют непосредственно на санитарную очистку водой.
205
Газы йз вдкуум-иопарителя проходят через промывную башню — абсорбер распиливающего типа. Последний орошается циркулирующим раствором 2 %-ной HaSiFe, распиливаемым механическими форсунками при плотности орошения 8 м3/(м2-ч). Температура в абсорбере та же, что и в вакуум-испарителе (65—67°C). Степень улавливания фтора в APT составляет ~95%. Из абсорбера APT газн проходят барометрический конденсатор;' затем их направляют на санитарную очистку.
Из зонта, установленного над фильтром, паровоздушная смесь вакуум-насосом просасывается через барометрический конденсатор, где очищается от фтора на 75%; затем смесь подают в систему санитарной очистки.
При совмещении описанной системы очистки газов в процессе производства кислоты концентрации 30—32% Р2О5 с улавливанием фторсодержащих соединений в процессе упаривания ее до 54% Р2О5 возможно получить 10%-ный раствор HsSiFg в качестве продукта с возвратом всех образующихся конденсатов в экстрактор. Ниже приведен баланс фтора для указанного процесса в расчете на 1 т апатитового концентрата [32, с. 290]:
Приход, кг Расход, кг
С апатитовым концен- 30,00 С фосфогипсом 5 „01
тратом 0,35 С продукционной фос- 2,80
С раствором из абсор- фориой кислотой (54%
бера санитарной очи- - Р2О5)
стки 0,94 С продукционной крем- 22,18 .
С раствором после По- нефтористоводородиой
глощения фтора из КИСЛОТОЙ 0,26
вакуум-испарителя 0,08 С газами из экстракте-
С раствором после по- ра, вакуум-сборников
глощения фтора из га- и др. 0,96
зов фильтрации и про- С газами из вакуум-ис-
мывки фосфогйпса 1,00 парител-я 0,12
С раствором после по- С газами из вакуум-
глощения фтора, вы- фильтра 1,03
деляющегося при упа- С газами после образо-
_ ривании кислоты * ваиия H2SiF6 (за вычетом потерь после санитарной обработки)
Потери с газами после 0,01
санитарной .очистки
Всего 32,37 Всего - , 32,37
Схема поглощения соединений фтора, .выделяющихся в процессе упаривания фосфорной кислоты, в аппаратах поверхностного типа, показана на рис. Ш.4. Отсасываемые из испарителя водяные пары очищают в центробежном брыз-гоуловителе от капель фосфорной кислоты. Соединения фто
205
ра выделяются в промывной башне — Лолой абсорбере; снабженном форсунками, через которые производится орошение башни 10— 12%-ной HjzSiFe. Промывная башня, изготовленная из стали, гуммирована, имеет диаметр 3125 мм и высоту 9000 мм. Она орошается циркулирующим растворам (10—12%-ной H2SiF6) с плотностью орошения 33— 35 м3/(м2>ч) при скорости сокового пара 3,5—4,5 м/с. Башню орошают циркуляционным насосом производительностью 265 м3/ч, создающим напор 40 м кислоты. Насос работает от электродвигателя мощностью 7.4 кВт с частотой вращения 1450 об/мин.
Фтор в башне абсорбируется при условиях, близких к применяемым в иепарителе (температуре 85—95 °C н разрежении ~9-104 Па, или ~680 мм рт. ст.). Вакуум в системе выпарка-абсорбция создается двухступенчатым пароэжек-. торным устройством, работающим при подаче 0,8 т/ч пара под давлением 0,4—0,5 МПа (4—5 ат).
Очищенный от соединений фтвра водяной пар конденсируется в поверхностном барометрическом конденсаторе, выполненном из стали 0Х23Н28МЗДЗТ, с поверхностью теплообмена 340 или 200 м2, образованной 836 или 496 трубками диаметром 30x2 мм и длиной 5000 мм. Расход воды нй конденсатор составляет 750—850 м3/ч. Внутренние поверхности остальной аппаратуры.— баков, емкостей, трубопроводов и др. — гуммированы.
Получаемая 10—12%-.ная H2SiF6 содержит всего 0,01— 0,04% Р2О5. Выход кислоты (100% H2SiF6) составляет 50— 70 кг на 1 т 100%-ной Р2О5.
В производстве фосфорной кислоты концентрации от 35—38 до 40—42% Р2О5'полу гидратным способом фторсодержащие газы улавливают в зависимости от особенностей проведения процесса экстракции по-разному. При отсутствии предварительного смесителя серной кислоты с оборотной фосфорной соединения фтора абсорбируют раздельно из газов, отводимых из экстрактора, вакуум-испарителя и от вакуум-фильтра, вакуум-сборников и другой аппаратуры. Образующиеся При абсорбции газов из карусельного фильтра и из вакуум-испарителя 2—4%-ный и 4—6%-иый раствор H2SiF6 подают на закрепление в систему абсорбции газов из экстрактора..
В системах, работающих с предварительным, смешением кислоТ, газы из смесителя, пройдя брызгоуловитель, где происходит отделение брызг серной н фосфорной кислот, очищаются .от соединений фтора в абсорбере с плавающей насадкой, а затем после санитарной очистки выбрасываются в атмосферу. Газы, выделяющиеся в других аппаратах, поглощаются так же, как описано выше с применением полых
207
бйрубберов -скоростного типа, абсорберов с плавающей насадкой и т. п.
Ниже приводится описание схемы абсорбции фторсодержащих газов в проектируемых новых системах производства кислоты концентрации 40—45% Р2О5 полу-гидратным способом в двух спаренных двухсекционированных экстракторах ' с предварительным смешением серной и оборотной фосфорной кислот, поступающих в первый корпус. Часть серной кислоты' (20% от общего ее количества) подается во второй корпус экстрактора.
Смеситель скоростного действия установлен непосредственно на крышке первого корпуса экстрактора и выделяющиеся в нем газы, присоединяются к потоку, отводимому от экстрактора. Поддержание в экстракторе температуры 98— 100 °C обеспечивается системой воздушного охлаждения пульпы.
По этой схеме предусматриваются три системы абсорбции соединений фтора (по увеличению концентрации кремнефтористоводородной кислоты): от карусельного вакуум-фильтра, от баковой аппаратуры и от экстрактора.
Газы, отсасываемые вентилятором от зонта, установленного над фильтром, очищаются от соединений фтора в пенном абсорбере. Сюда поступают также газы, отсасываемые вакуум-насосами от фильтра, которые предварительно проходят -через барометрические конденсаторы. Абсорбер орошается слабой ~0,5—1%-ной ^SiFe, подаваемой насосом из циркуляционного сборника. После абсорбции га-зы с температурой 4-0—50 °C, пройдя брыз-гоуловитель, выбрасываются вентилятором в выхлопную трубу. .
Рассчитанное количество -0,5—'1%-ного раствора H2SiF6' ив сборника передают на абсорбцию соединений фтора из газов от баковой аппаратуры. Пополнение абсорбционного цикла производится водой.
Все емкостное об орудование находится под вытяжкой, , создаваемой специальным вентилятором. Газы, собираемые в общий коллектор, очищают в пенном аппарате, орошаемом циркулирующей 1—2%-ной H2SiF6.
Из экстрактора газовоздушная смесь эвакуируется с помощью двух хвостовых вентиляторов. Для очистки газов от брызг пульпы на крышке экстрактора в местах выхода- газа устанавливают расширители, где за счет резкого снижения скорости потока происходит выпадение брызг с образованием сплошной жидкости, собираемой в сборнике. Затем для очистки от фторсодержащих соединений газы проходят через два последовательно установленных струйных абсорбера и Два четырехполочных абсорбера с подвижной кольцевой , насадкой (АПКН). Последние могут работать как последовательно, так и параллельно.
208
Две верхние секции (полки) абсорберов АПКН орошаются циркуляционной 2—3%-ной H2SiFe, часть которой перекачивается в нижние секции. Пополнение верхних-абсорбционных секций абсорберов АПК'Н производится 1—2 %-ной НгЗтРб-йз .цикла абсорбции газов баковой аппаратуры.
Нижние секции абсорберов орошаются 10—42%-ным раствором H2SiF6, как и струйные абсорберы, откуда продукционное количество кислоты откачивают на переработку. Пополнение орошения производится за счет 2—3%-ного раство-ра H^SiFe из верхних секций абсорберов АПКН. . Пройдя брызгоуловители, газы из баков и экстрактора выбрасываются вентиляторами в атмосферу.
При двухстадийном (двухступенчатом) производстве по-лифосфорной кислоты (или концентрировании кислоты; до 53—54% Р2О5) в барботажнбм концентраторе (стр. 183) в первой стадии отходят газы, содержащие 7,5 г F/м3 и ~1,5 г/м3 тумана Р2О5. Газы очищают в абсорбционной системе, состоящей из двух последовательно соединенных гуммированных насадочных башен и изготовленного из фаоли-та скруббера Вентури. В^первой башне, заполненной графитовыми кольцами, для орошения используют циркулирующую 10—'12%-ную НгБШб с температурой 65—70 °C. Отходящие из башни газы с температурой 70 °C содержат 1,0 г Р2О5/М3 и 0,5 г F/м3 при степени поглощения Р2О5 — 30 и F—93%. Получаемая кислота, отводимая из циркулирующего потока, содержите—42% H^SiFe и 0,15 % Р2О5.
Вторая башня является конденсационной. Она также заполнена графитовыми кольцами, орошается водой с температурой 12—'15 °C и служит в основном для охлаждения отходящих газов до 20—30 °C. После конденсационной башни в 1 м3 газов находится 0,9 г Р2О5 и0,3 г F при степени улавливания Р2О5 — 11 и F — 33%.
Затем газы направляют в скруббер Вентури, орошаемый слабой фосфорной кислотой (4% Р2О5), охлаждаемой в графитовом теплообменнике; здесь они окончательно очищаются от примесей и вентилятором йыбрасываются в атмосферу. На, выходе из скруббера. Вентури газы содержат 0,03 г PsOs/m3 и 0,02 г F/м3; при степени поглощения Р2О5 — 98 и F —95%.
Газы, отходящие от концентратора второй ступени с температурой 220 °C, проходят последовательно через брызгоуловитель, аппарат APT (изготовлен из стали 0Х2ЭН28МЗДЗТ), орошаемый циркулирующей предварительно упаренной и охлажденной до 85—90 °C кислотой, н абсорбционную систему. Последняя состоит из форсунки, вмонтированной в. газоход, где тазы охлаждаются от 98 °C (на выходе из APT) до 70 °C, и двух последовательно соединенных фаолитовых-скрубберов Вентури, в которых проис
14-5473 209
ходит очистка газов от тумана фосфорной и серной кислот й соединений 'фтора. Скрубберы орошаются циркулирующей фосфорной кислотой концентрации 10—25% Р^О5 с температурой 50 °C — для первого и 40 °C — для второго. Газы выходят из второго скруббера Вентури с температурой 56 °C и вентилятором выбрасываются в атмосферу. При содержании в газах на входе в APT, в г/м3: Р^О5 — 15; соединений фтора — 3; тумана SO3—3 — концентрация очищенного газа на выходе из второго скруббера Вентури составляет, в г/м3: Р2О5 — 0,15; F — 0,01, SO3 — 0,04.
V1.4. ПОЛУЧЕНИЕ РАЗЛИЧНЫХ СОЕДИНЕНИЙ ФТОРА
Образующуюся при улавливании фторсодержащих газов преимущественно водно-абсорбционными методами кремнефтористоводородную кислоту перерабатывают в кремнефториды и фториды.
Потребность народного хозяйства в рйвличйых фторнеор-ганичеоких продуктах в 1975 г. составила, в тыс. т: кремнефторидов натрия—41,0, калия — 9,0, аммония—>2,0 и магния—2,2; фторидов натрия (содовый)—31,0, натрия (кристаллический) —6,0 и магния — 0,6; фторида-бнфторида аммония— 8,0; плавиковой кислоты (30—40%-ной) — 12,1 <[33].
Переработка кремнефтористоводородной кислоты связана со значительными потерями фтора на различных стадиях процесса. Получающиеся при экстракции и упаривании фдс-форной кислоты частично разбавленные растворы (0,5—2% H2SiF6) нейтрализуют и сбрасывают в отвал. В производстве упаренной фосфорной кислоты степень выделения фтора в газовую фазу составляет в среднем ~60—65% (при начальной концентрации 6—8 г/м3), нз которых обычно теряется 30—40% в основном в виде неиспользованной H2SiF6 {34]. Съем фтора составляет с единицы продукции 50—55 пег на 1 т Р2О5.
Технология различных соединений фтора, в частности из. кремнефтористоводородной кислоты, а также исследования в этом направлении освещены в литературе достаточно подробно [1, 2, 35]. Ниже приводятся краткие принципиальные характеристики отдельных продуктов и процессов.
кремнефторид натрия является во многих случаях конечным продуктом переработки фтора в производстве фосфорной кислоты и служит для получения многих других соединений фтора; применяется в сельском хозяйстве (в качестве инсектицида); в строительной и лепкой промышленности, для фторирования воды, при переработке цветных металлов н др. Его получают взаимодействием неосветленной кремнефтористоводородной кислоты с раствором поваренной соли или сульфата натрия. Образующаяся пульпа в гидроцикло-210
Мделйется от геля кремниевой кислоты, который ухо-, дит вместе с соляной (или серной) кислотой в сливы, направляемые на нейтрализацию. Сгущенную пульпу' ((пески) подвергают центрифугированию. Отфугованный (продукт высушивают. Общий выход фтора в готовый продукт составляет 92%.
Выше (стр. 199) были приведены условия обесфторива-ния экстракционной фосфорной кислоты осаждением из нее кремнефторида натрия (или калия). Этим методом получают продукт, содержащий 93—96% основного вещества при общем выходе фтора из апатитового концентрата ~60% [36]. Фосфорную кислоту, содержащую 1—3 г/л твердой взвеси, отстаивают от шлама фосфогипса в сгустителе Дорра диаметром 24 м. Осветленную кислоту (содержание твердых частиц ~0,2 г/л) обрабатывают 16—20%-ным Na^Os (140—450% от стехиометрии) в течение 60 мин прй 60— 70°C (летом) и 50—55 °C (зимой). Остаточное содержание фтора в кислоте равно 4—6 г/л (в исходной 12—18 г F/л). Кремнефторидная пульпа (Т:Ж=1:30) сгущается сначала в сгустителе Дорра (до Т:Ж=’1:4—6), затем в конусных сгустителях (до Т:Ж=’1:1,5—2,5), после чего репульпиру-ется и подвергается двухступенчатому гидроциклонирова-нию.
Кремнефторид калия применяют в производстве -титана, циркония и электролитического силумина. Его получают аналогично кремнефториду натрия или конверсией пульпы последнего с хлоридом калия.
Кремнефторид аммония используют в качестве антисептика для обработки древесины. Кроме того, он может быть весьма эффективным исходным сырьем для производства фтористого водорода и плавиковой кислоты. Его получают нейтрализацией 10—20%-ного раствора EhSiFe аммиаком или аммиачной водой при 2—5 °C до остаточной кислотности меньше 1%. Полученный раствор упаривают и выкристаллизовывают'кремнефторид аммония, который отделяют фильтрацией или центрифугированием от маточного раствора, возвращаемого в процесс. При использовании кремнефтористоводородной кислоты, содержащей некоторое количество фосфорной, маточный раствор аммонизируют до pH=9—10 и после выделения геля кремниевой кислоты и фосфата аммония возвращают в процесс.
Фторид-бифторид аммония применяют главным образом для извлечения редких металлов из руд, в цветной металлургии, в производстве строительных материалов и других. Его получают преимущественно при улавливании соединений фтора оборотным раствором NH4F. При этом происходит образование. при взаимодействии с HF бифторида аммония (NH4HF2), а с SiF4— кремнефторида аммония. При подще-14* ' 211
лачивании раствора аммиачной водой (iNH^zSiFe разлагается с образованием NH4F и SiO2-H2O.
После отделения геля кремниевой кислоты фильтрацией оставшийся раствор упаривают с получением готового продукта, содержащего ~20% NH4F и ~80% NH4HF2. Одновременно осадок геля дополнительной промывкой и фильтрацией перерабатывают в «белую сажу», используемую в качестве наполнителя в производстве резиновых изделий.
Большую часть (70—80%) фторидов натрия, алюминия и криолита (NasAlF'e) производят на криолитовых заводах и используют в производстве металлического алюминия. Фторид натрия в значительных количествах применяют для приготовления антисептических паст.
Фторид натрия в основном получают обработкой плавиковой кислоты содой. Для заводов фосфорной кислоты и фосфорсодержащих удобрений представляют интерес суспензионный и термический епособы, пригодные для переработки Na2SiF6 или H2SiFe. В первом способе Na2SiF6 реагирует с содой в присутствии небольшого количества воды [37], при 70 °C и перемешивании в течение 30 мин [38]. Образующийся продукт содержит ~75% NaF. Во втором способе Na2SiF6 прокаливают при 580—620 °C [39]. Выделяющийся SiF4 абсорбируется водой, а образующийся H2SiF6 перерабатывают поваренной солью в кремнефторид натрия, возвращаемый в процесс.
Фторид натрия высокой чистоты (99,5% NaF) можно, получить предварительной обработкой раствора H2SiF6 аммиачной водой. Гель кремниевой кислоты отделяют от раствора NH4F. К последнему добавляют NaCl в присутствии N1H4OH. В результате этого выделяется NaF, который отфильтровывают, промывают водой и сушат.
Фторид алюминия и криолит, обычно производимые из растворов HF, возможно также получать из H2SiFg, Na2SiF6 или бифторида и фторида аммония [I, 2]. Использование для этого H2SiF6 основано на растворении технической гидроокиси алюминия в 10—115%-ном растворе H2SiF6 при 90— 100 °C. После отделения геля SiO2-H2O раствор AIF3 перерабатывают в готовый продукт.,При добавлении NaF к раствору A1F3 выделяется криолит, который отфильтровывают и высушивают.
Фторид кальция, синтетический, образующийся из отходящих фторсодержащих газов производства фосфорной кислоты, представляет интерес,в плане консервации фтора при избыточном его выделении из фосфатного сырья по сравнению с текущей его потребностью. Вследствие практической нерастворимости в воде и возможности длительного хранения это соединение может заменить плавиковый шпат при производстве, плавиковой кислоты и, следовательно, элементарного 212
фтора. Наиболее простым способов является нейтрализация HjSiFe мелом Или известняком [Зв] с образованием эквимолекулярной смеси 3CaF2 и SiO2. Для отделения SiO2 из смеси ее обрабатывают раствором фторид-бифторида аммония. В- раствор переходит кремнефторид и фторид аммония, от которого отфильтровывают фторид кальция. Маточный раствор нейтрализуют аммиачной водой с образованием NH4F и SiO2 из (NH4)2SiF6. Раствор NH4F после отделения SiO2 упаривают до образования NH4HF2 и NH3, который улавливают и возвращают в процесс в виде аммиачной воды. Видоизменением этого процесса является получение CaF2 непосредственно из фторидов аммония [2] —продуктов улавливания фторсодержащих газов сухнм и мокрым способами. В первом случае взаимодействие карбоната кальция с би-- фторидом или фторидом аммония в количестве 110% от стехиометрии [40] протекает при 170—'180 °C. Во втором случае раствор фторида аммония реагирует при 70—80 °C с карбонатом кальция. Выделяющийся в обоих способах аммиак возвращают в процесс.
При обработке раствора фторида аммония с концентрацией 82 r.F/л карбонатом кальция (в количестве 100—107% от стехиометрии) при 70 и 80 °C степень конверсии фторида аммония за 30-^40 мин составляет соответственно 95—97 и 98—99% [41]. Производительность фильтрования CaF2 составляет 5—6 т/(м2-ч) в расчете на сухой продукт. В продукте, полученном с применением 100%-ной нормы высококачественного карбоната (не более 1,2% SiO2), содержится 95—97% основного вещества.
При использовании в качестве исходного материала Na2SiF6 (выделенного из фосфорной кислоты) предложено [41] его вначале обработать аммиачной водой с получением раствора фторида аммония с выделением в осадок NaF' и SiO2. Раствор NH4F используют для получения CaF2, а осадок выщелачивают водой с получением раствора NaF', который переводят в GaF2 и Na2CO3, направляемый на получение Na2SiF6.
Фтористый водород (безводный) используют в производстве фреонов, фторопластов, элементарного фтора, в атомной промышленности и др. Плавиковую кислоту, обычно получаемую из плавикового шпата, применяют в основном для производства фторидов, потребляемых в металлургии алюминия. Некоторое количество ее получают из фтористого водорода. ’.
В последние годы в Советском Союзе и за рубежом ведутся интенсивные разработки методов получения фтористого водорода и плавиковой кислоты из соединений фтора, образующихся в производстве фосфорной кислоты и фосфорсодержащих удобрений, в первую очередь кремиефтористо-
213
водородной кислоты, а также из фторида и бифторида аммония, кремнефторидов натрия, аммония и др. [2, 42].
- По-видимому, сернокислотное разложение кремнефтористоводородной кислоты с последующим выделением фтористого водорода из парогазовой смеси — одно из наиболее простых в технологическом отношении. Как было указано выше (стр. 196), при взаимодействии раствора HzSiFg с концентрированной серной кислотой (93% H2SO4) при 100— 150 QC образуется фторсульфоновая кислота HSO3F', связывающая HF, a SiF4 выделяется в газовую фазу. Последний перерабатывают в H2SiF6, например, гидролизом с выделением SiO2, который после фильтрации и отмывки сбрасывается в отвал.
Из фторсульфоновой кислоты выделить HF можно нагреванием и продувкой воздуха. Затем охлаждением газовоздушной смеси до — 20 °C из нее конденсируют продукт — жидкий фтористый водород. Отработанную серную кислоту с концентрацией 65—70% H2SO4 можно использовать в процессе экстракции фосфорной кислоты.
Этот способ, по-видимому,, может быть использован непосредственно на заводах производства экстракционной фосфорной кислоты. По другим соображениям, вероятно, заслуживающим внимания, на заводах фосфорной кислоты и удобрений следует выпускать кристаллический кремнефторид аммония 1(33] или натрия или, в крайнем случае, концентрированную кермнефтористоводородную кислоту, а переработку в различные фторнеорганические продукты производить на специализированных заводах.
ЛИТЕРАТУРА
7 1. Позин М. Е. Технология минеральных солей. Ч. II. Л., Химия, 1974.
Ц 2’ Гамин Н' П’’ Зайцев В. А., Серегин М. Б. Улавливание и переработ-
v ка фторсодержащих газов. М., Атомиздат, 1975.
; . 3. Зайцев В. А. и др. — Хим. пром., 1971, № 8,, с. 593—597.
* .'-4. Производство фосфорных удобрений. Труды НИУИФ. Вып. 215. М„ .
1971.
5. Позин М. Е. и др. — ЖПХ, 1976, т. 49, № 12, с. 2593—2603.
6. Новиков А. А. и др. — Хим. пром., 1975, № 11, с. 843—844.
7. Выделение и улавливание фтористых соединений при производстве фосфорных удобрений. Труды НИУИФ. Вып. 220. М., 1971.
8. Очистка газов и сточных вод при производстве фосфорсодержащих удобрений. Труды НИУИФ. Вып. 228. М., 1976.
9. Борисов В. М. и дц. — Хим. пром., 1977, № 7, с 511—513; № 6, с. 440—441; № 1, с. 28—29.
10. Вольфкович С. И. и др. — ЖПХ, 1977, т. 50, № 1, с. 3—5.
11. Борисов В. М. и др. — Хим. пром., 1976, № 11, с. 839—840.
- -- 12. Сенатова Г. И., Копылов В. А., Позин М. Е. — ЖПХ, 1976, т. 49, . № 6, с. 1371—1374; 1374—1375.
13. Хрипунов Н. Ф., Абрамович А. Я., Копылов В. А. — Хим. пром., 1975,
Л № 2, с. ПО—112.
-S ' 14. Сенин В. Н. и др, — Там же, 1976, № 10, с. 755—756.
S 214 .
15. Сенин В. Н. и др, — Труды МХТИ им. Д. И. Менделеева, 1969, вып. 60, с. Т41—143.
16. Громов Б. В. и др. — Хим. пром., 1976, № 4, с. 286—288.
17. Галкин Н. П. и др. — Там же, Ns 9, с. 673—675.
18. Опарин А. В. и др. — Тезисы докладов V Всесоюзного симпозиума по химии неорганических фторидов. М., Наука, 1978, с. 219.
19. Шрамбан Б. И., Кочеткова В. В. — Хим. пром., 1973, Ns 2, с. 151— 152.
20. Филатова Л. Н. и др. — Там же, 1976, Ns 6, с. 438—439.
21. Фосфорная промышленность. Реф. сб. ЛенНИИГипрохим. Вып. 5. М., НИИТЭХИМ, 1977.
22. ’ Архипова Л. И. и др. — Хим. пром., 1973, Ns 2, с. 128—130.
23. Зайцев В. А. и др. — Там же, 1974, № 7, с. 523—526.
24. Алексеева Л. И. и др. — Там же, 1976, № 7, с. 515—519.
25. Антосиков А. И. и др. — Там же, 1977, Ns 11, с. 854—859.
26. Миркина Л. И. Автореф. канд. дисс. Л., ЛТИ им. Ленсовета, 1972.
27. Тезисы докладов IV республиканской конференции по химии природ-"ных солей н удобрений. Алма-Ата — Джамбул, 1977.
28. Копылев Б. А. и др. — В кн.: Химия и химическая технология. Вып. 21. Алма-Ата, Изд-во КазГУ, 1977, с. 154—159.
29. Копылов В. А., Токмакова Т. А., Борисов В. М. — Хим. пром., 1979, № 1, с. 26—28.
30. Позин М. Е., Мулленов И. П., Тарат Э. Я. Пениые газоочистители, теплообменники и абсорберы. Л., Госхимиздат, 1959; Пенный режим и пенные аппараты/Под ред. И. П. Мухленова и Э. Я. Тарата. Л„ Химия, 1977.
31. Гурова Н. М„ Митюишна М. К, Чуприлина Р. П. — Хим. пром., 1978, Ns 6, с. 437—438.
32. Расчеты по технологии неорганических веществ/Под ред. М. Е. Лозина. 2-е изд. Л., Химия, 1977.
33. Антипенко Г. Л.; Марков С. С.— Хим. пром., 1974, Ns 9, с. 683—686.
34. Ниязбекова Б. С. — В кн.: Промышленность минеральных удобрений и серной кислоты. Реф. сб. НИУИФ. Вып. 5. М., НИИТЭХИМ, 1978, с. 9—11.
35. Труды УНИХИМ. Л., Химия, 1968, вып. 27, с. 146; 1975, вып. 35— 36, с. 95.
36. Антосиков А. И. и др. — Хим. пром., 1977, Ns 2, с. 118—119.
37. Богачев Г. Н., Г узь С. Ю. Производство криолита, фтористого алюминия и фтористого натрия. М.—Л., Мёталлургиздат, 1940.
38. Габриелова М. Г-, Субботина О. П., Томилова М. Д. — В кн.: Исследования по химии и технологии удобрений, пестицидов и солей. М., Наука, 1966, с. 246.
39. Вольфкович С. Габриелова М. Г. — Хим. пром., 1951, Ns 3, с. 72— 75. -
40. Шапиро Л. Д. и др.— В кН.: Технология минеральных удобрений. Межвуз. сб. трудов Л., ЛТИ им. Ленсбвета, 1979, с. 48. -
41. Зайцев В. А. и 6р. — Хим. пром., 1978, Ns 12, с. 908—911.
42. Комаров В. И., Быков В. И., Житова Е. С. Методы получения фтористого соединения. М„ НИИТЭХИМ, 1977.
Глава VII
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕХНИКИ БЕЗОПАСНОСТИ В ПРОИЗВОДСТВЕ ЭКСТРАКЦИОННОЙ ФОСФОРНОЙ кислоты
Основа безопасной работы в фосфорнокислотном производстве (помимо знания технологического процесса, прави-л обслуживания аппаратуры, личной и технической культуры, высокой трудовой, дисциплины и сознательности) — безусловное соблюдение установленных государственными законами специальных требований и норм по обеспечению нормальных условий труда и предотвращению несчастных случаев. В производстве экстракционной фосфорной кислоты выделяются вредные газы, в нем используют агрессивные растворы и протекает оно при повышенных температурах в сложной .аппаратуре, обслуживаемой транспортными средствами, приводимыми в действие электрическими двигателями. Оно размещено в помещении, которое возводится -в соответствии с требованиями, обеспечивающими чистоту, нор" мальную освещенность, легкий доступ к отдельным, агрегатам и их элементам, отсутствие загроможденности и т. п.
По характеристике сырья, процессов и готовой продукции технология экстракционной фосфорной кислоты' относится к вредным производствам. В атмосферу производственных помещений могут выделяться фторсодержащие газы, пары фосфорной кислоты, фосфатная пыль и сернистый газ. Эти вещества оказывают вредное действие на организм человека, и их выделение .в атмосферу рабочих помещений должно немедленно ликвидироваться. ' .
Ниже приведены предельно допустимые концентрации (ПДК) веществ, применяемых и образующихся в производстве.
Относительно токсических свойств веществ следует сказать, что апатитовый концентрат и фосфоритная мука в пылевидном состоянии вызывают раздражение органов дыхания. При длительном систематическом вдыхании развивается хроническое заболевание — силикоз. Токсическое действие фосфатного сырья усугубляется наличием в нем соединений фтора.
216
Вещества я материалы В воздухе рабочей зоны производствен-ных помещений, мг/м3 В атмосферном воздухе населенных мест
максимальная разовая, мг/м3 среднесуточная, мг/м3 '
Серная кислота, серный ан- '1 0,3 0,-1. гидрид Сернистый ангидрид '10 0,5 0,15 Фосфорный ангидрид 1 0,Р5 0,05 Фтористый водород 0,5 0,5 (фторео- 0,01 (фторсо- держащие держащие соединения) соединения) Фосфорная кислота, 28—42% Р2О5 1 (в пересчете — 0,15 (на Р2О5) на Р2О5) Кремнефтористоводородная — 0,0Й (в пере- кислота - счете на фтор) Апатитовый концентрат 6 — 0,5
Для предотвращения попадания пыли в атмосферу подача фосфата в производственные помещения обычно осуществляется пневмотранспортом, оборудованным соответствующими устройствами для очистки транспортирующего воздуха.
Основное условие, обеспечивающее безаварийную работу пневмотранспорта, — поддержание заданного давления воздуха [~0,6 МПа (~6 ат)] на пневмотрубопроводах и насосах. Превышение давления может привести к разрыву коммуникаций и созданию аварийной обстановки.
При транспортировании сырья механическим транспортом в целях уменьшения пыления течки устанавливают на минимальной высоте с углом наклона, обеспечивающим оптимальные скорости падения материала на ленты транспортера^ Все точки (места) пылеобразования должны быть максимально герметизированы и снабжены укрытиями с местными отсосами, совмещенными с технологической вытяжкой.
Узел приема и дозировки фосфатного сырья выделяется в отдельное помещение.
При образовании запыленной -атмосферы необходимо пользоваться индивидуальным средством защиты — противо-пылевым респиратором ШБ-'l типа «Лепесток», респиратором Ф-45 или другим.
Серная кислота (концентрированная) при попадании на кожу вызывает сильные, долго не заживающие ожоги. При попадании кислоты на большие участки поверхности, кожи образующиеся ожоги весьма опасны (иногда с летальным исходом). Пары серной кислоты раздражают и обжигают слизистую верхних дыхательных путей и поражают легкие.
217
При попадании на кожу серной кислоты обожженное место необходимо немедленно обильно промыть сильной струей воды, чтобы удалить с поверхности полностью кислоту, смочить 5%-ным раствором соды и смазать вазелином. Недостаточная и медленная промывка поврежденного места может усилить ожог вследствие выделения теплоты при разбавлении кислоты и последующей нейтрализации ее содой. В помещении цеха во многих местах устанавливаются гидранты и бачки с содовым раствором. При сильных ожогах после принятия первых мер помощи следует обратиться в здравпункт. На тело наиболее вероятно попадание кислоты вблизи насосов, сборников, холодильников, кислотопроводов и вентилей арматуры. Поэтому при их обслуживании необходимо пользоваться предохранительными очками, а ремонт их и набивку сальников производить в резиновых сапогах, фартуках и перчатках.
Сернистый ангидрид может выделяться из серной кислоты. Он вызывает раздражение кожи, слизистых глаз, носоглотки и верхних дыхательных путей. При накоплении в глухих непроветриваемых местах (мешках) значительной концентрации его в воздухе (3'0—60 мг/м3) возможны острые отравления, вызывающие отек легких и расширение сердца. Признаками даже легкого отравления служат головокружение и раздражение слизистых оболочек — щипание в носу, чихание, позывы к кашлю. Сильные отравления вызывают общую слабость, одышку, сильный кашель; возможна по- , теря сознания.
Для предупреждения выделения в цехе сернистого газа все емкости, трубопроводы и коммуникации на тракте серной кислоты должны' быть герметизированы.
Соединения фтора, выделяющиеся в газовую фазу в процессе экстракции, упаривания кислоты, а также из рабочих растворов, сильно раздражают слизистую верхних дыхательных путей и оказывают общее отравляющее действие на организм. При высоких концентрациях появляется раздражение слизистой глаз и носоглотки, а в дальнейшем — медленно заживающие изъязвления. Фторсодержащие соединения действуют на центральную -нервную систему и вызывают удушье, иногда рвоту, колики, заболевания органов пищеварения, десен и зубов.
Предельно допустимая концентрация в воздухе рабочей зоны производственных помещений газов, содержащих HF, равна 0,5 мг/м3.
При отравлений фторсодержащими газами первая помощь заключается в ингаляции содовым' раствором, При тяжелых отравлениях необходимы покой и тепло. '
Для предупреждения попадания газов в атмосферу все
218
емкости трубопроводы и коммуникации должны быть гер-метиэированы.
Кремнефтористоводородная кислота—протоплазматический яд, приводящий к нарушению кальциевого и фосфорного обмена в организме. При острых отравлениях поражает центральную нервную систему. При попадании на кожу вызывает жжение и воспаление кожи, а также появление синих пятен на ногтях.
Фосфорная кислота и полифосфорные кислоты1 раздражающе действуют на слизистую верхних дыхательных пу-тей, вызывая воспаление слизистой глаз и носоглотки, носовые кровотечения, сухость в носу и глотке/крошение зубов. При попадании (в особенности горячих кислот) на кожу образуются слабые, но долго не заживающие ожоги. Для предотвращения ожогов при эксплуатации и особенно при производстве ремонтных работ необходимо пользоваться защитными средствами—очками, резиновыми рукавицами, проти-вокислотной спецодеждой.
Попавшую на тело кислоту следует немедленно смыть обильным колйчеством воды (1но не сильной струей) и смочить пораженные места 5%-ным раствором перманганата калия. В случае поражения глаз необходимо тщательно их промыть водой и обратиться к врачу.
Экстракционная пульпа при повышенной температуре действует на организм аналогично фосфорной кислоте, но в несколько более сильной форме, вследствие содержания в ней частично 'серной кислоты, а также твердой фазы — фосфогипса. Наряду с химическими ожогами она может причинить и термический ожог.
Фосфогипс также действует раздражающе на слизистую дыхательных путей, вследствие содержания в нем остаточной фосфорной кислоты и соединений фтора.
К работе в цехах экстракционной фосфорной кислоты допускаются только лица, прошедшие медицинский осмотр и сдавшие технический минимум по данному производству.
Для каждого рабочего места установлены рабочие инструкции и инструкции по технике безопасности. Точное и сознательное выполнение всех указаний и требований инструкций является необходимым условием и гарантией безопасной работы.
Поступающие на работу проходят в отделе техники безопасности завода вводный инструктаж об основных опасностях и вредностях, могущих возникнуть на предприятии, а также о правилах безопасности поведения на территории -• предприятия. В цехе проводится первичный инструктаж об опасностях и вредностях .производства и общих правилах техники безопасности в цехе. По указанию начальника цеха работник направляется в распоряжение начальника смены,
219
.‘ от которого он .получает инструктаж непосредственно на рабочем месте. Начальник смены проводит повторный инструктаж одни раз в три месяца. Проверка знаний правил техни-' ки безопасности и инструкций проводится не реже одного раза в полугодие для всех работающих в цехе. Лица, не сдавшие экзамена по технике безопасности или нарушающие правила безопасной работы, от работы отстраняются.
Рабочие места оборудованы плакатами и инструкциями по технике безопасности. Все работающие в цехе один раз в шесть месяцев проходят медицинский осмотр.
Работники цеха ^обеспечиваются спецпитанием, спец--одеждой н индивидуальными средствами защиты. Приступая к работе, необходимо иметь при себе противогаз, защитные очки, респиратор и резиновые перчатки.
С целью создания нормальных и безопасных условий труда в цехе оборудована приточная и вытяжная вентиляция для подачи в рабочие помещения свежего воздуха и удаления вредных выделений. Фланцы кислотопроводов в местах прохода снабжены .предохранительными кожухами. Все емкости серной, фосфорной, кремнефтористоводородной кислоты и пульп снабжены' герметическими крышками и местной вытяжной вентиляцией. Вращающиеся части агрегатов имеют ограждения, выполненные в виде решетки или сетки. Вся.электрическая аппаратура герметически укрыта и заземлена. Запрещается производить работу' при отсутствии ограждений У движущихся частей оборудования, обслуживаемых площадок, лестниц и т. .п.
При ремонте оборудования надо соблюдать особую осторожность и следить за строгим' соблюдением правил безопасной работы обслуживающим персоналом — ремонтниками, монтажниками, строителями.
Запрещается производить ремонт оборудования на ходу, а также ремонт и чистку аппаратов, находящихся под давлением кислоты, воздуха, газа или вакуумом. Перед ремонтом аппараты отключают от питающих трубопроводов, промывают, отключают от выпускных трубопроводов и сушат. Остатки кислоты нейтрализуют известью. Ремонтные и очистные работы внутри аппаратов, разрешаются только после их проветривания и в присутствии дублера, находящегося снаружи аппарата. При остановках на ремонт оборудования с электроприводом электродвигатель должен быть обесточен и на пускателе вывешен плакат: «Не включать — работают люди». Электрооборудование разрешается ремонтировать только работникам электр о служб.
В случае разлива серной или фосфорной кислот их необходимо нейтрализовать известью или известковым молоком, находящимися на расстоянии не менее 15—20 м от наиболее опасных мест.
220
Отбор проб кислоты или пульпы, а также переливайиё их производится в защитных очках и резиновых перчатках. Осмотр кислотопроводов, аппаратов и емкостей с кислотой или пульпой выполняется также в резиновых перчатках и защитных очках. ,
Все работающие в фосфорнокислотных цехах должны уметь оказать первую медицинскую самопомощь и взаимопомощь при ожогах кислотой и других несчастных случаях.
Борис Аронович Копылев
ТЕХНОЛОГИЯ ЭКСТРАКЦИОННОЙ ФОСФОРНОЙ кислоты
Редактор В. А. Станкевич Ю' И. Васильев
Техн, редактор 3. Е, Маркова
Корректор Л. С. Александрова
ИБ № 860
*
4
4
Сдано в набор 18.09.81. (Подписано в печать 30.10.81.
М-22633. Формат бумаги бОхЭО1/^.
Бумага тип. № 2. Литерат. га р нит. Высокая печать. Усл. печ. л. 14,0.
Усл. кр.-отт. 14;25. Уч.-изд. л. 14,6.
Тираж 2900 экз. Зак. 5473.
Цена 75 коп. Изд. № 1572.
Ордена «Знак Почета» издательство «Химия», Ленинградское отделение.
191186, г. Ленинград,'Д-186. ^Невский пр., 28.
Межвузовская типография (3) СППО-2 Ленуприздата.
1980’13,, Ленинград, Московский пр., 26.