/























































































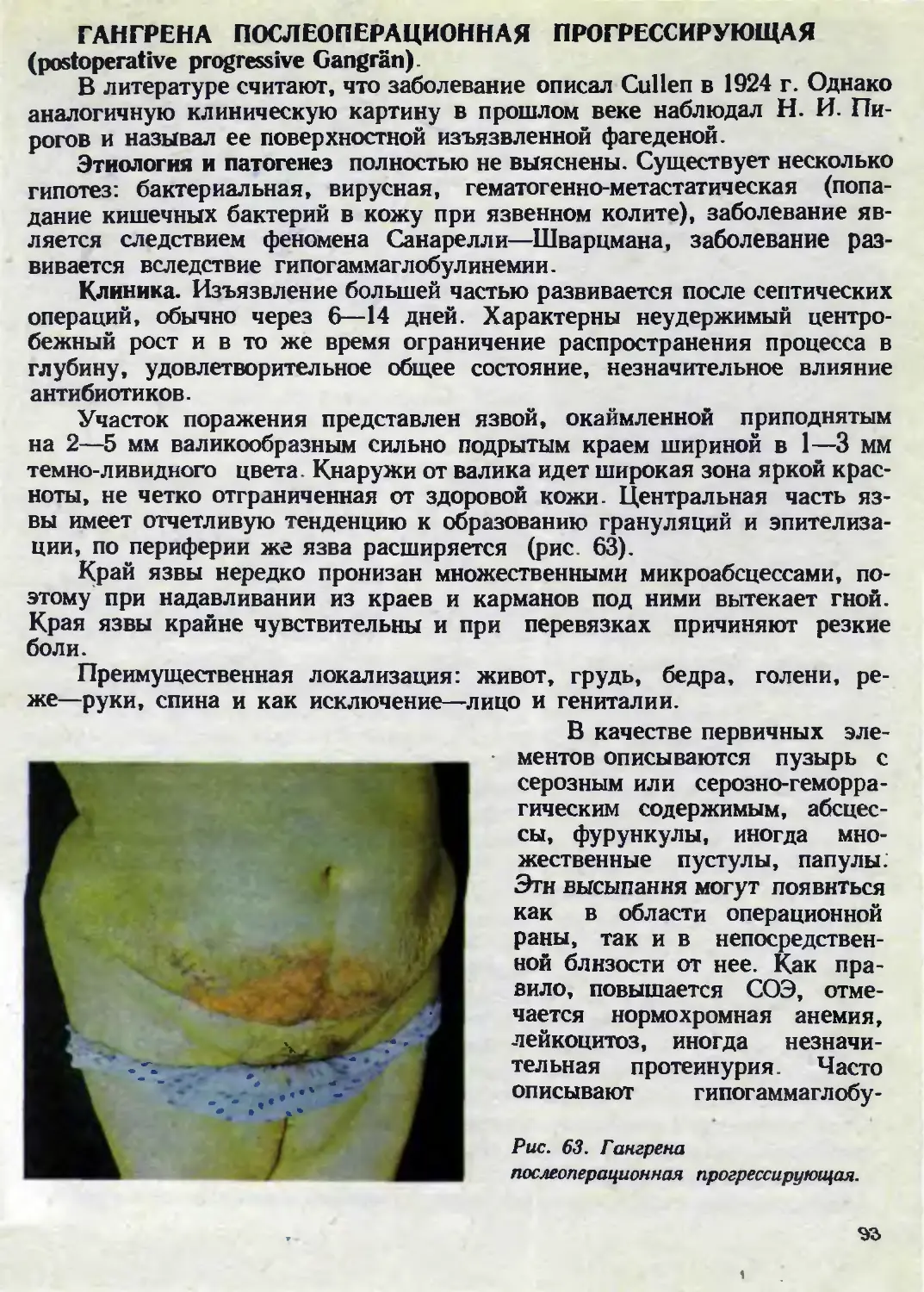





















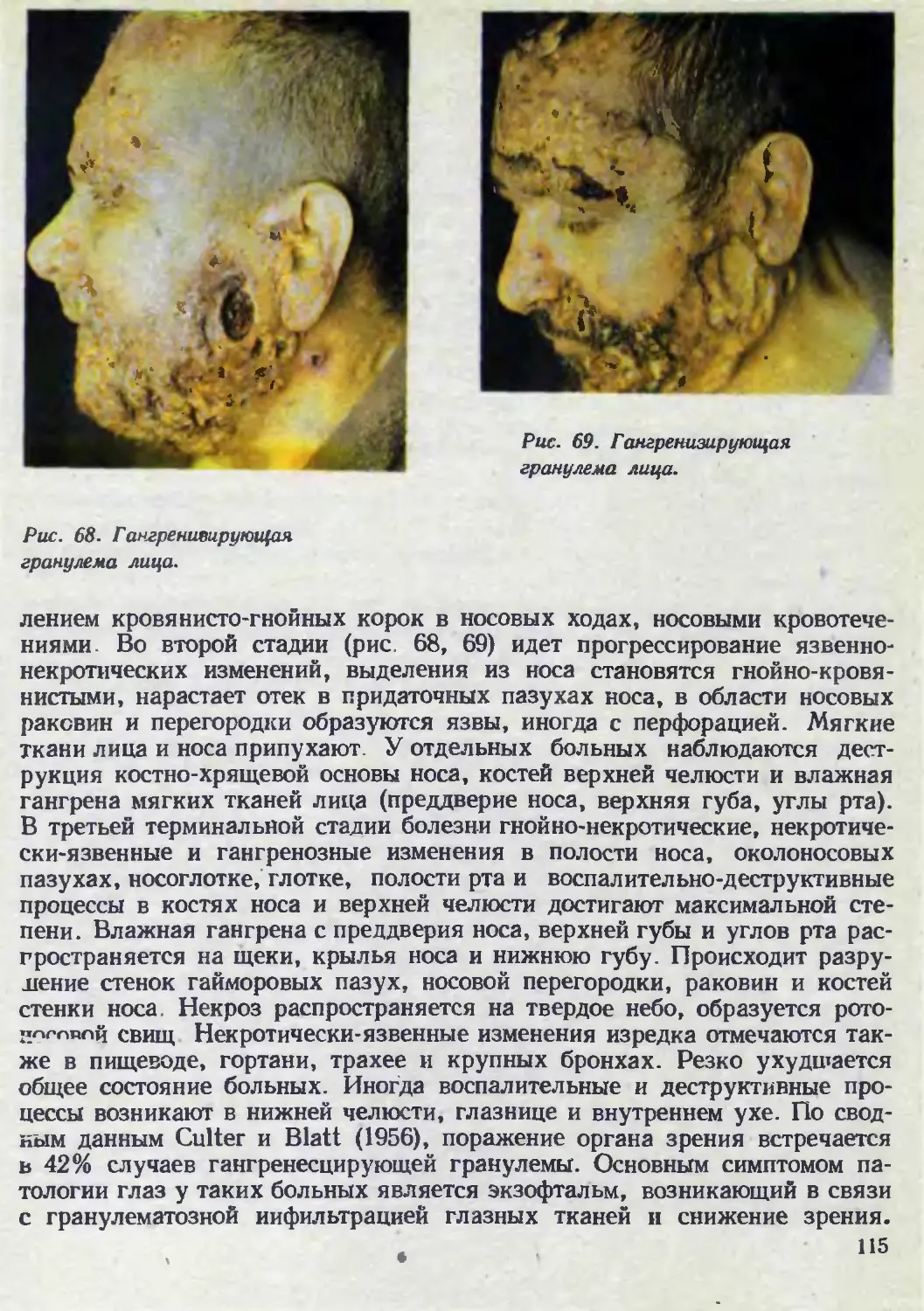








































































































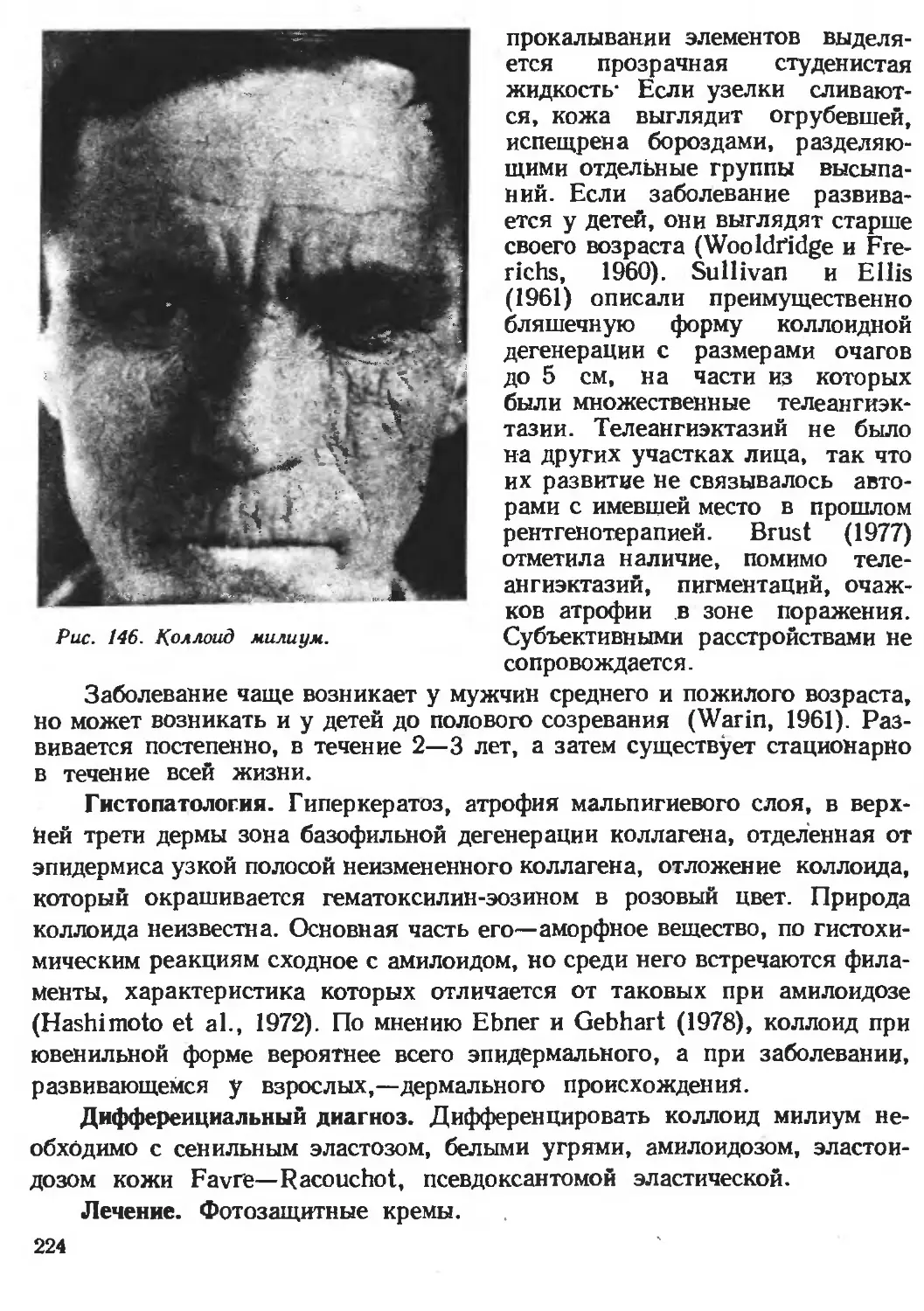




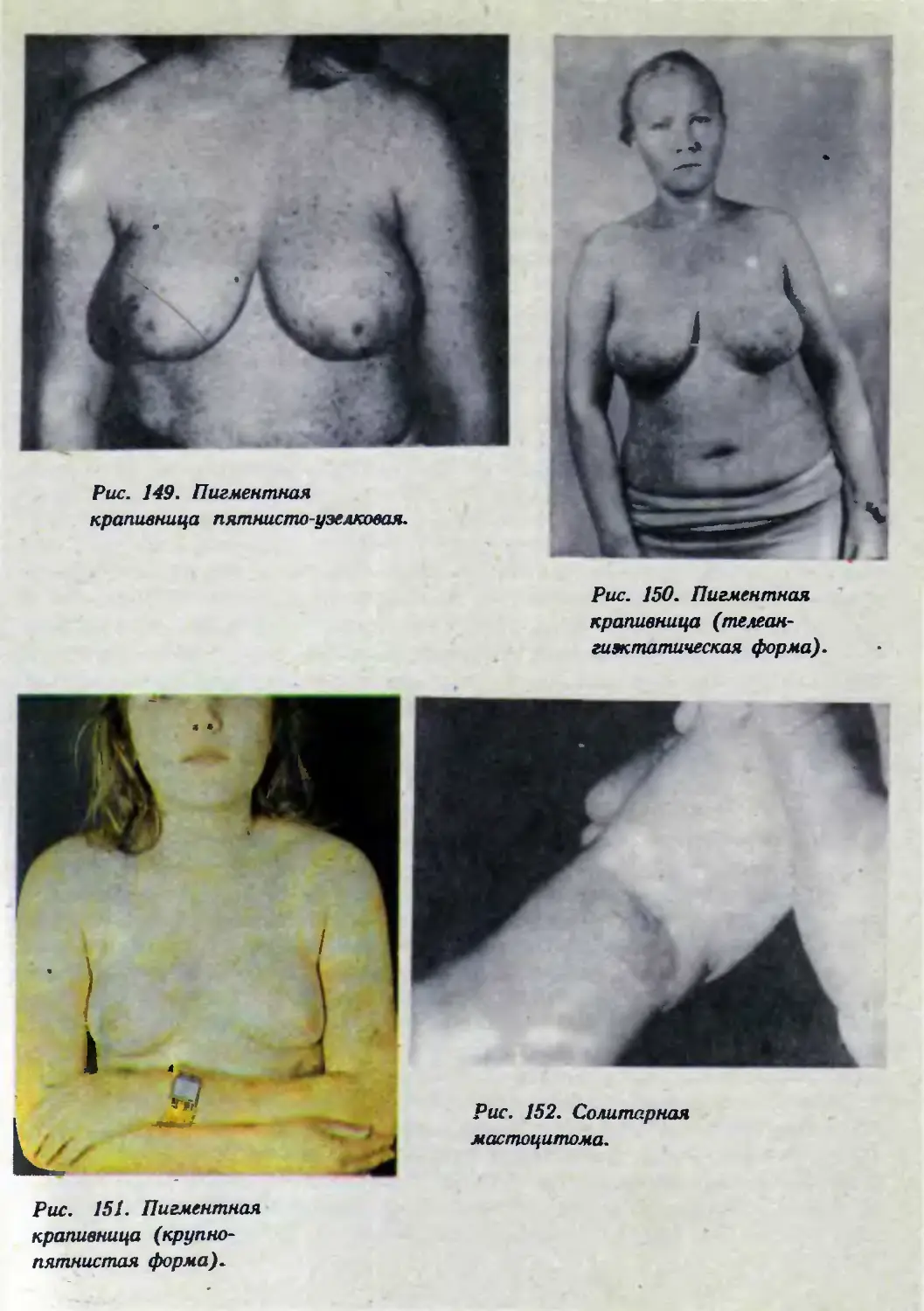
























































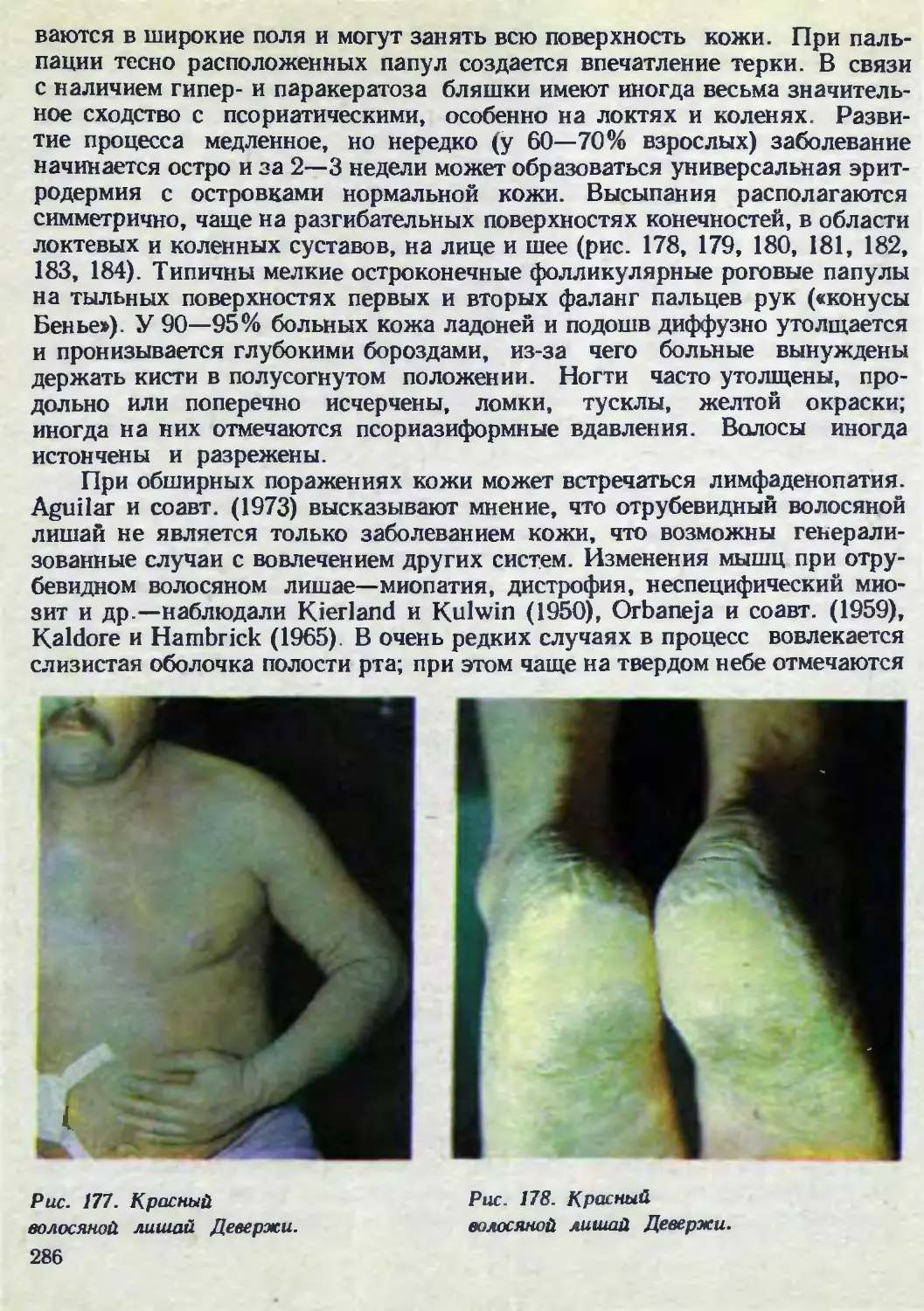

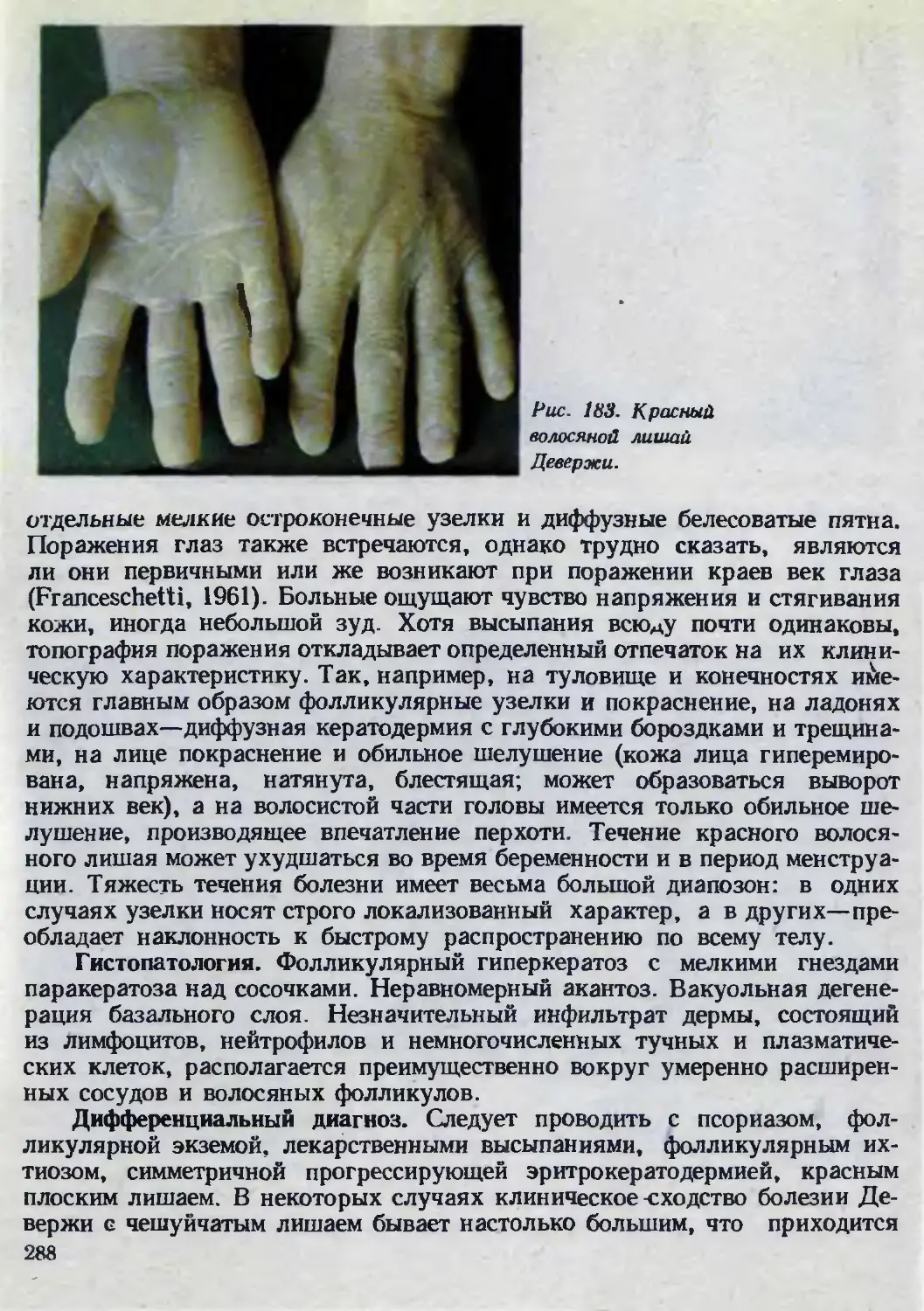















































































































































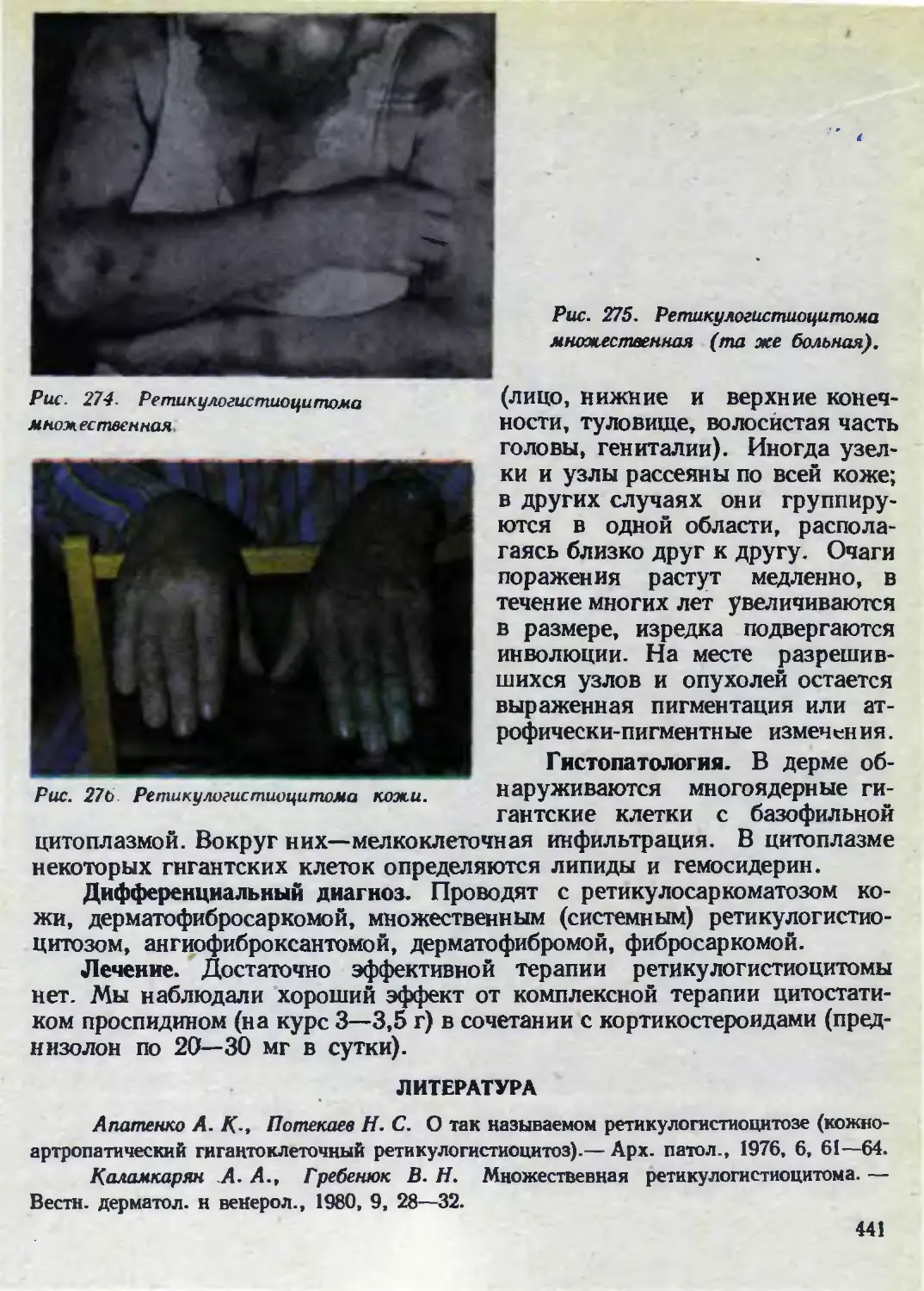






























































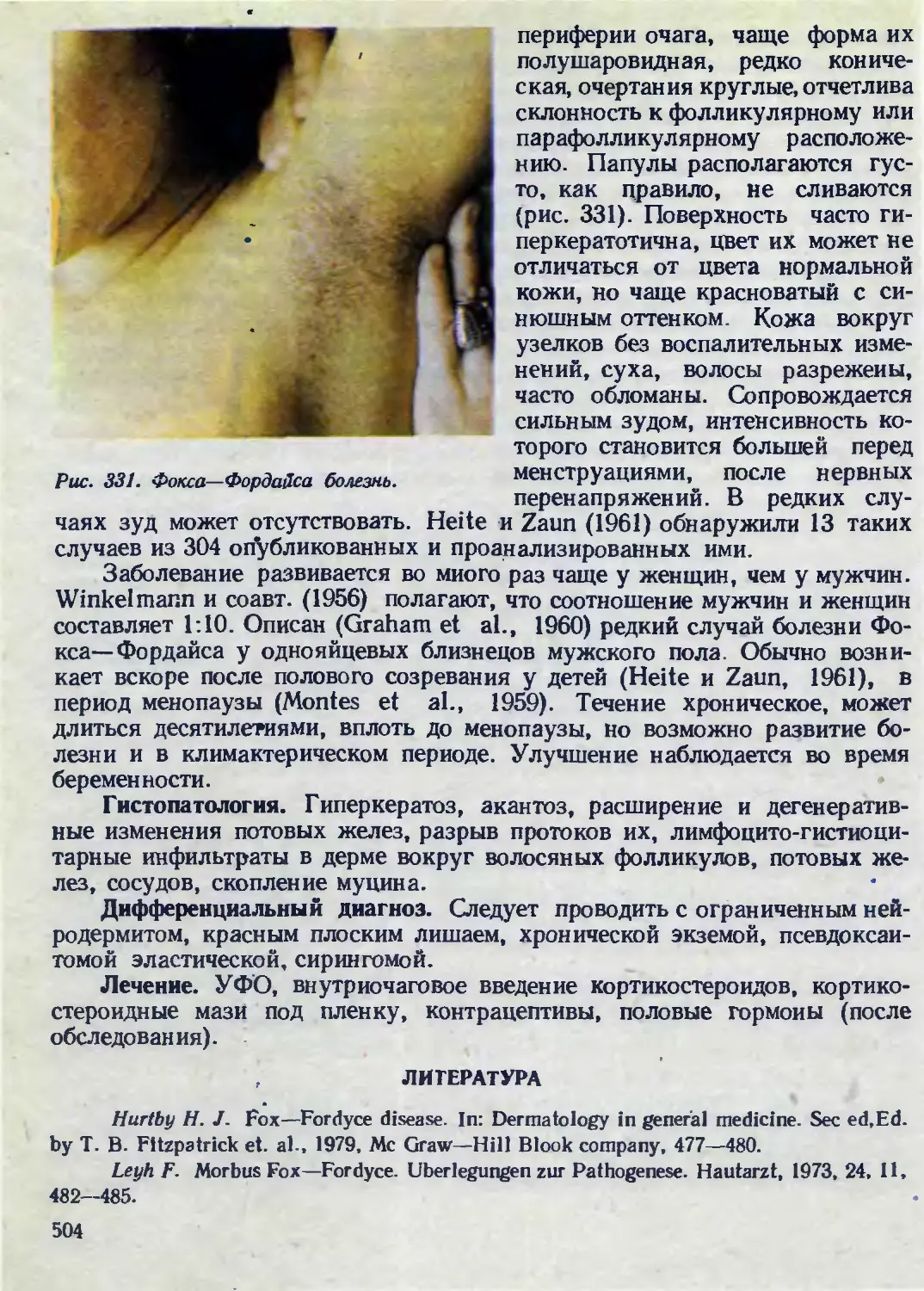






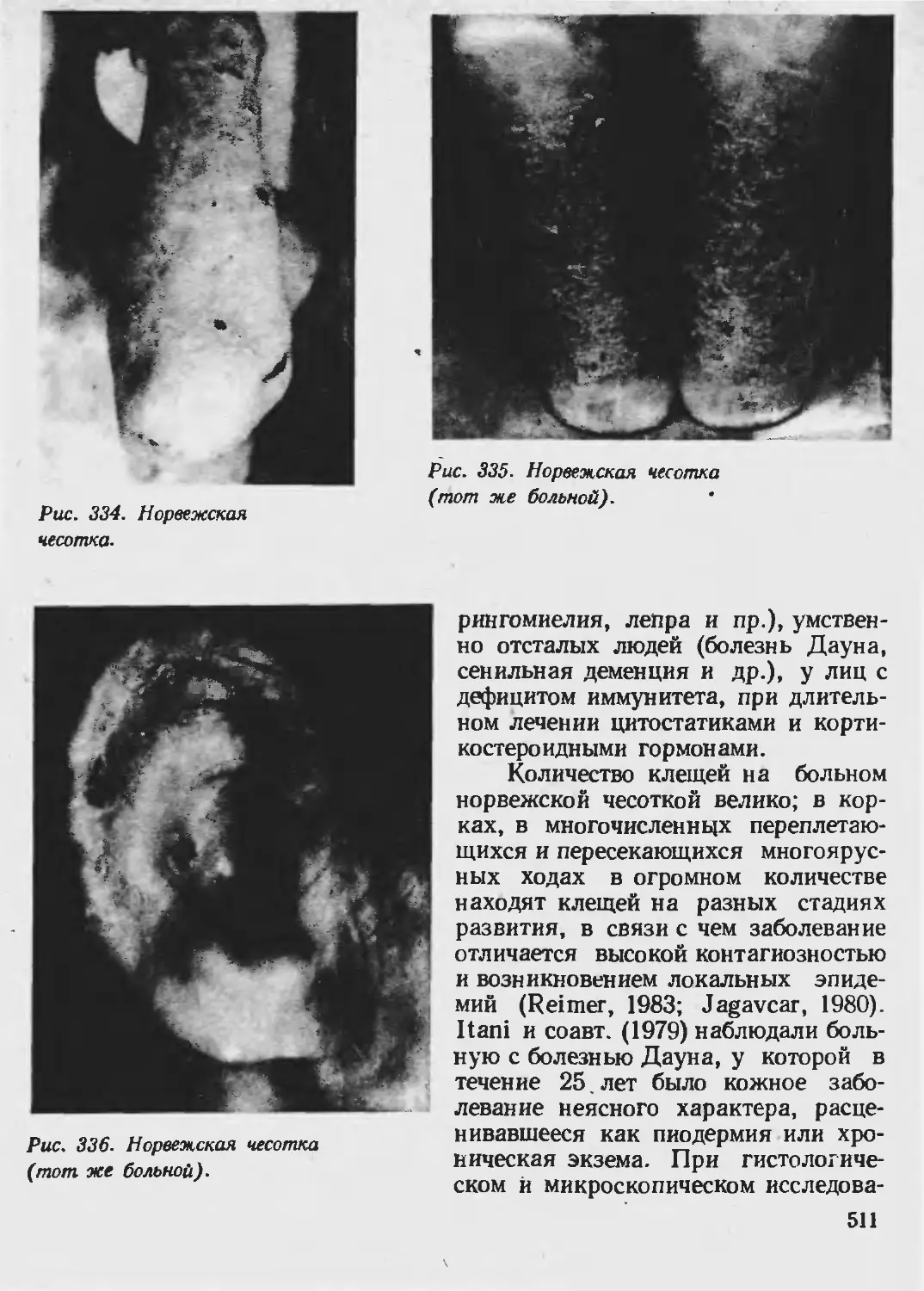























































Автор: Каланкарян А.А. Мордовцев В.Н. Трофимова Л.Я.
Теги: венерология дерматология
ISBN: 5-540-00713-1
Год: 1989




Текст
А. А.Коламкарян
В. Н. Мордовцев
А. Я. Трофимова
КЛИНИЧЕСКАЯ
ДЕРМАТОЛОГИЯ
РЕДКИЕ И
. И7ИЧНЫЕ
ДЕРМАТОЗЫ
А. А. Каломкорян
В. H. Мордовцев
А. Я. Трофимова
КЛИНИЧЕСКАЯ
ДЕРМАТОЛОГИЯ
РЕДКИЕ И
АТИПИЧНЫЕ
ДЕРМАТОЗЫ
ЕРЕВАН «АЙАСТАН» 1989
ББК 55.8
К 170.
Издается по рекомендации Научного
Совета по дерматологии и венерологии
Академии медицинских наук СССР и
Всесоюзного научно-медицинского общества
дермато-венерологов
Рецензенты: член-корр. АМН СССР, профессор А. Ф. ЗАХАРОВ
доктор медицинских наук, профессор А. А. АНТОНЬЕ В
Каламкарян А. А. и Др.
К 170 Клиническая дерматология: Редкие и атипичные
дерматозы /А. А. Каламкарян, В. Н. Мордовцев,
Л. Я- Трофимова. — Ер.: Айастан, 1989 567. с.: ил.
Книга посвящена описанию относительно редко встре-
чающихся дерматозов или атипичных форм распространения и
тяжело протекающих заболеваний кожи.
Основное внимание уделено детальному описанию клиники.
Достаточно полно представлены и современные взгляды на этио-
логию и патогенез того или иного дерматоза. Особенности гисто-
логического строения даются в объеме, необходимом для прове-
дения дифференциальной диагностики.
Монография содержит обширный иллюстративный мате-
риал.
Рекомендуется дерматологам и врачам других клиниче-
ских специальностей.
9405000000
701 (01)—89
123—87
ISBN 5-540 00713-1
ББК 55. 8
© Издательство «Айастан,» 1УвУ
ВВЕДЕНИЕ
Диагностика болезней кожи нередко представляет значительные труд-
ности из-за большого количества клинических форм (около 2000), а также
потому, что для их обозначения применяется масса различных терминов-
синонимов.
Не умаляя значения лабораторных, в том числе и новейших, диагности-
ческих методов, в дерматологии, как ни в одной медицинской дисциплине,
рещающую роль мы отводим клинической диагностике. Если распространен-
ные, часто встречающиеся дерматозы достаточно освещены в руководствах,
монографиях, атласах, изданных в нашей стране, то до сих пор нет книги,
посвященной редким и атипично протекающим заболеваниям кожи. Знаком-
ство же с ними необходимо для подготовки квалифицированных специалистов.
Опыт показывает, что именно в этих случаях делается наибольшее количест-
во диагностических ошибок, следовательно и нерациональных терапевтиче-
ских рекомендаций.
В настоящей книге мы постарались по мере возможности восполнить
этот пробел.
В основу книги положен опыт, накопленный мной и моими коллегами в
отделе дерматологии Центрального кожно-венерологического института
Минздрава СССР за последние 15 лет. ч
Изложение материала не ограничивается описанием клинической кар-
тины заболеваний. Нозологические формы иллюстрированы оригинальными,
в значительной мере цветными фотографиями. Кроме того, сжато изложе-
ны современные представления об этиологиии, патогенезе, о патоморфологи-
ческих, в том числе ультраструктурных изменениях кожи. Даны краткие
рекомендации по лечению и профилактике.
Каждая статья снабжена указателем (современной) отечественной и
зарубежной литературы. z
Полагаем, что данная монография, помимо предоставления врачам
практических знаний, может способствовать дальнейшему научному изу-
чению причин клинического полиморфизма дерматозов, совершенствованию
их классификации.
3
Мы надеемся, что книга будет полезной не только для дерматологов,
но и для врачей других специальностей, нередко сталкивающихся в своей
работе с кожными изменениями при заболеваниях внутренних органов,
нервной системы, желез внутренней секреции и др.
Авторы с благодарностью примут все замечания и предложения по
улучшению этого издания.
АДЕНОМА САЛЬНЫХ ЖЕЛЕЗ СИММЕТРИЧНАЯ (Adenoma sebaceum
symmetricum).
Синонимы: туберозный склероз, синдром Бурневиля—Прингля.
Bourneville описал в 1880 г. туберозный склероз головного мозга.
Симметричную аденому сальных желез описал Pringle в 1890 г.
Этиология и патогенез. Туберозный склероз относят к комплексным
порокам развития, которые наследуются аутосомно-доминантно. Так как
способность производить потомство у больных сильно понижена, то пере-
дача более чем в двух поколениях редка (Simon et al., 1979).
Клиника. Изменения кожи обычно появляются в детском или
юношеском возрасте. Узелки величиной от булавочной головки до горошины
располагаются симметрично в носо-щечных складках, на подбородке, в
околоушных областях. Узелки округлой, овальной формы, уплощенные,
коричневато-красного цвета, обычно тесно прилежат друг к другу, иногда
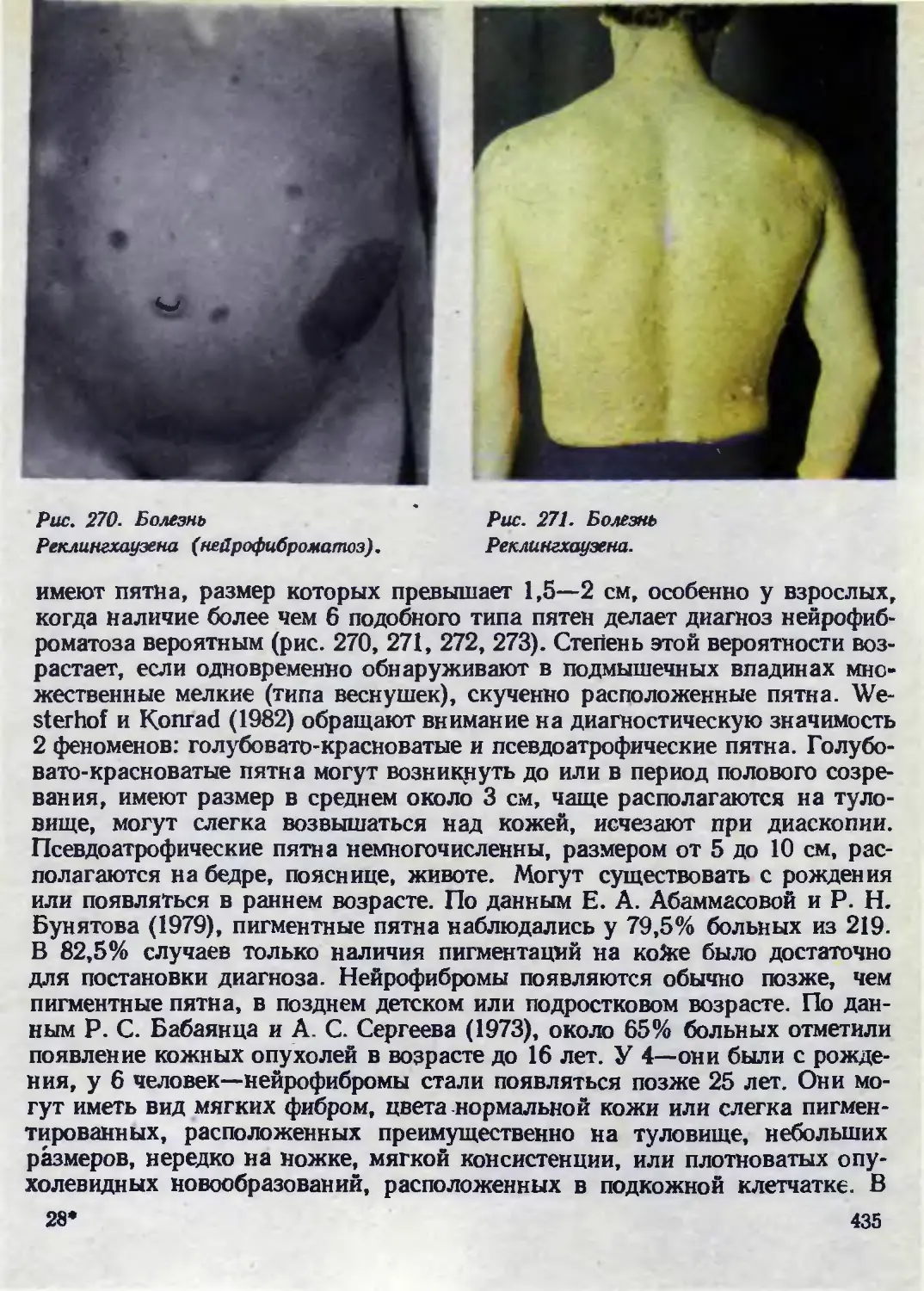
сливаются, выстоят над окружающей кожей (рис. 1, 2). Поверхность их
гладкая, нередко с телеангиэктазиями. Часто встречаются под- и около-
ногтевые фибромы, гипертрофические изменения десен. Следствием очагов
глиоза головного мозга и йтрофии коры являются эпилепсия и умствен-
ное недоразвитие.
В полную развернутую картину болезни входят также внутрицереб-
ральные обызвествления, опухоли сетчатки, гамартомы и кисты почек,
рабдомиомы сердца, гамартомы печени, щитовидной железы, яичек, ки-
стозные изменения скелета. Имеются типичные для туберозного склероза
легочные изменения, рентгенологически—сотовидная структура, милиар-
ная пятнистность и др. Клиническими симптомами таких изменений яв-
ляются нарастающая одышка, кровохарканье, спонтанный пневмоторакс
(Hoffmann и Schreiner, 1980).
Прогноз определяется изменениями в головном мозге, внутренних
органах. При поражении легких смерть наступает от спонтанного пнев-
моторакса с недостаточностью правого сердца.
5
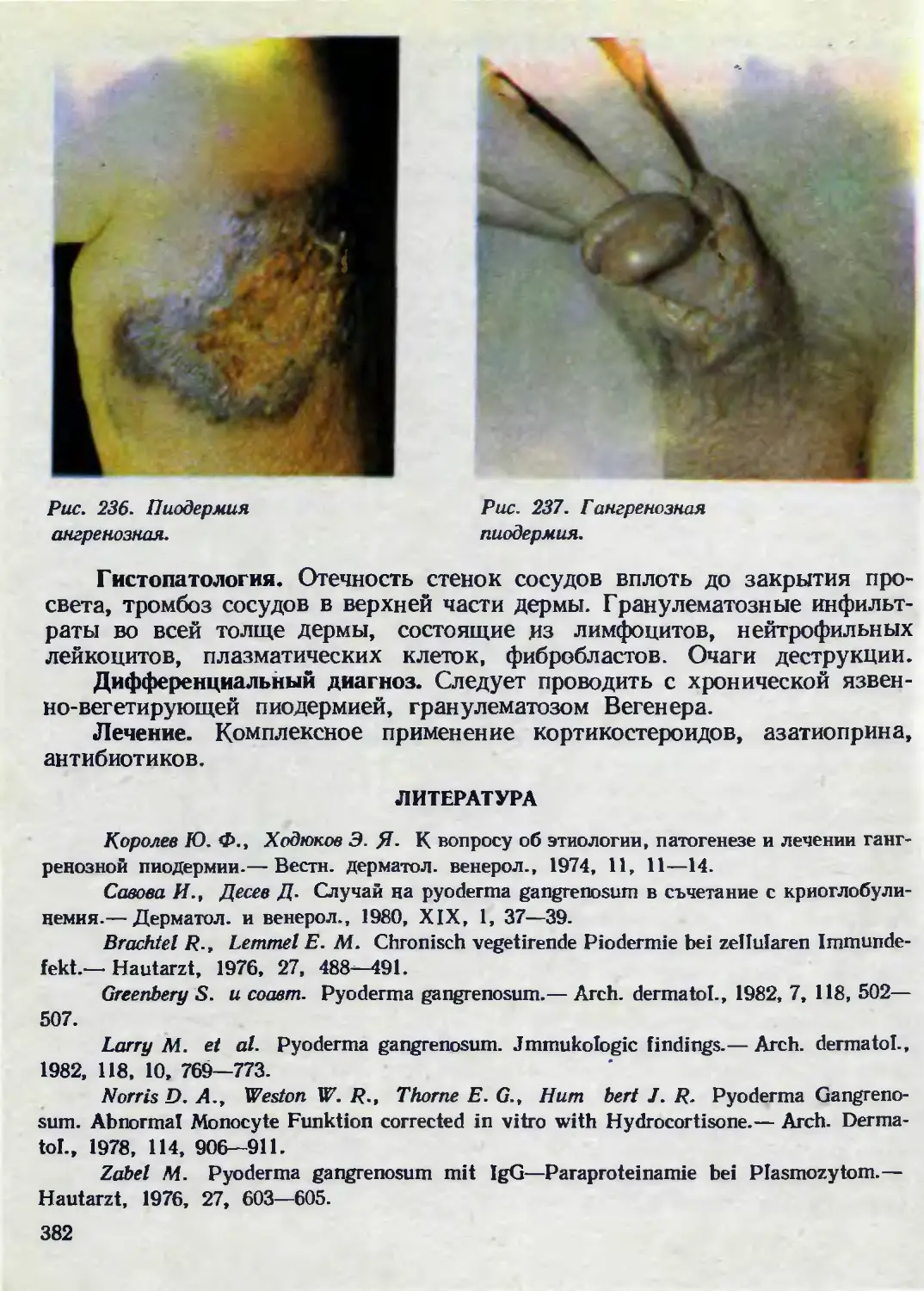
Рис. 1. Болезнь
Б урневиля-Прингля.
Рис. 2. Аденома сальных
желез симметричная.
Гистопатология. Гистологическая структура соответствует ангиофиб-
роматозу. Избыточное развитие сальных желез, гиперплазия капилляров.
Nickel и Reed (1962) на основании изучения 74 наблюдений установили
атрофические изменения сальных желез за счет перифолликулярной про-
лиферации коллагена.
Дифференциальный диагноз. Проводят с epithelioma adenoides cysticum.
Лечение: крупные узелки на лице подвергают электрокоагуляции,
криодеструкции.
ЛИТЕРАТУРА1
Hoffmann Ch., Schreiner W.—D. Rezidivierender Spontanpneumothorax Symptom des
Morbus Bourneville—Pringle.—Hautarzt, 1980, 31, 2, 91—95.
Simon In. N., KelemenJ., Szorenyi A. et. al.. Ein seltener Mosaizismus der Chromo-
som-10—Trisomie bei einer Patientin mit Bournevielle—Pringle—Syndrome.—Hautarzt,
1976, 30, 6, 292—294.
АКНЕ ФУЛЬМИНАНС (acne fulminans).
Впервые на эту разновидность акне обратили внимание Burns и Col-
ville (1959), выделив ее в отдельную нозологию под названием «акне кон-
глобата с септицемией». У мальчика 16 лет, страдающего в течение двух
лет вульгарными акне, внезапно повысилась температура (до 38—39°),
появились боли в суставах, общее недомогание, слабость. Огромные дозы
пенициллина не оказали эффекта. При повторном исследовании кровь была
стерильной. В дальнейшем эта нозология описывалась под самыми раз-
ными названиями: «акне конглобата и артрит», «акне конглобата и септи-
1 В списке Литературы здесь и Далее приведены источники в основном за последние
10 лет.
6
Рис. 3. Акне фульминанс.
Рис. 4. Акне фульминанс.
цемия», «острое фебрильное язвенное акне конглобата с полиартралгией»,
«острое фебрильное язвенное акне с лейкемоидной реакцией». Kligman
(1975) предложил заменить эти громоздкие названия на «акне фульминанс»
(от лат. fulmen—молния), который подчеркивает внезапное начало, тяжесть
заболевания и исключает упоминание об акне конглобата, имеющем дру-
гое течение, симптоматологию и прогноз.
В отечественной литературе есть лишь сообщение А. А. Каламкаряна
и Р. С. Топурия (1984) с описанием двух случаев акне фульминанс. За ру-
бежом имеется до 25 подобных описаний (Kelly и Burns, 1971; Strom et al.,
1973; Lippert и Post, 1975; Goldschmidt et al., 1^77, Engber и Marino, 1980;
Capdevila et Melo, 1981 и др.).
Этиология и патогенез. Заболевают почти исключительно молодые
мужчины, страдающие умеренно выраженными вульгарными угрями. Боль-
шинство исследователей рассматривают акне фульминанс как острейшую
экзацербацию вульгарных угрей, сопровождающуюся внезапным нару-
шением общего состояния, высыпанием множественных болезненных пу-
стуй, повышением температуры (38—39°), артралгией, лейкоцитозом
(20000—30000), повышением СОЭ [40—90 мм/час]. Бактериальная флора
(стафилококки, стрептококки, микрококки, протеус, кандида), по всей
вероятности, не является причиной данного заболевания.
Нет доказательств наличия иммунных дефектов или септицемии как
факторов патогенеза этой болезни.
7
I
Клиника. Заболевание встречается главным образом у мужчин моло-
дого возраста (17—20 лет), хотя наблюдается также и у женщин. Вне-
запное появление болезненных пустул, изъязвляющихся папул и узлов
является характерной клинической особенностью акне фульминанс.
Множественные, покрытые корочками, изъязвляющиеся поражения с гной-
ным и геморрагическим эксудатом располагаются на спине, груди, боко-
вых сторонах шеи и плечах (рис. 3, 4). Относительно пощаженное лицо
и бросающееся в глаза отсутствие комедонов являются характерными
чертами этого заболевания. Заживание очагов поражения часто сопровож-
дается образованием множества рубцов, в том числе и келоидных. Сустав-
ные симптомы (боли чаще в коленных и плечевых суставах) обычно средней
тяжести, но имеются сообщения об акне фульминанс с тяжелой персисти-
рующей артралгией. Ревматоидный фактор отрицательный. Постоянно
отмечаются лейкоцитоз (до 15—30 тыс.) и резкое повышение СОЭ (до 50—
90 мм/час.).
Прогноз благоприятный.
Гистопатология. Инфильтрат из полиморфноядерных лейкоцитов,
вторгающийся в фолликулярный аппарат и вызывающий его разрушение.
Дифференциальный диагноз. Его проводят с хронической пиодер-
мией и акне коиглобата.
Лечение. Успех лечения определяется обязательным назначением
кортикостероидных препаратов (преднизолон 30—40 мг в сутки) в ком-
бинации с антибиотиками и нестероидными противовоспалительными сред-
ствами (индометацин). Под влиянием указанной терапии в течение бли-
жайших дней наступает резкое улучшение общего состояния, норма-
лизуется температура, прекращаются боли в суставах, постепенно регрес-
сируют кожные высыпания.
Hartman и Piewig (1983) наблюдали хороший результат от применения
13-цисретиноевой кислоты (изотретиноин) по 1 мг/кг в сутки. В тече-
ние нескольких недель.
ЛИТЕРАТУРА
Каламкарян. А. А., Топурия Р. С. Акне фульминанс.— Вест, дерматол. и венерол.
1984, 7, с. 34—37.
Capdeoila Е., Melo S. Acne fulminans.— Aetas Dermato—Sif., 1981, v. 72, No 3—4,
p. 149—152.
Engber P., Marino C. Acne fulminans with prolonged pofyarthrafgia.— Int. ,J. Der-
matol., 1980, v. 19, No 10, p. 567—569.
Goldschmidt H., Leyden J., Stein K- Acne fulminans. Investigation of aucute febfile
ulcerative acne.— Arch. Dermatol., 1977, v. 133, No 4, p.444—449.
Hartmann K., PlowigG. Acne fulminans. Tratamento de—11 pacientes com о acido
13—cis—retinoico. An Bras dermatai., 1983, 58, 1, 3—10.
Haneke E. Levamisolbehandlung der Acne fulminans.—Z. Hautkr., 1981, Bd. 56, Hf.17
S. 1160—1166.
8
Lipper H., Post В. Schwere AcneformenT mit ungewohn lichem klinischen Verlauf.—
Hautarzt, 1975, Bd. 26, Hf. 10, S. 532—534.
Strause J. Acne fulminans.—In: Dermatology in general medicine. New York, 1979,
p. 453—454.
АКРОГЕРИЯ СЕМЕЙНАЯ (acrogeria familiaris).
Синоним: синдром Готтрона.
Впервые описал Gottron в 1941 году под названием «семейная акро-
терия». Заболевание относится к очень редким дефектам, в мировой ли-
тературе опубликовано всего 19 случаев (De Groot и Woerdeman, 1980).
Этиология и патогенез. По мнению Laurent и соавт. (1975), семейная
акрогерия является следствием нарушения структуры и функции фибро-
бластов, а по мнению Sunter и Sitzmann (1978)—нарушения коллагенового
обмена. Gottron (1941), Laugier с соавт. (1959) считают ее локализованным
вариантом детской прогерии. В то же время некоторые другие авторы
(Batschvaroff et al.. 1961; Martinez et al., 1965; Sunter и Sitzmann, 1978)
выделяют как самостоятельную нозологическую форму. Семейные случаи
акротерии (Базыка А- П., 1979; Gottron, 1941; Butenand и Christophers, 1970;
Rosenberg, 1976; De Groot и Woerdeman, 1980), столь редкого заболева-
ния позволяют думать о его генетической обусловленности. Batschvaroff
и соавт. (J96J) видят причину заболевания в пониженной функции гипофиза.
Как показывает анализ литературы, многие авторы считают целесообраз-
ным выделить акрогерию в самостоятельную нозологическую форму, рас-
сматриваемую ими как наследственное заболевание соединительной ткани
с поражением дермы, передающееся по рецессивному типу.
Клиника. Чаще заболевание наблюдается у женщин (15—женщин,
4—мужчин). Проявляется оно либо при рождении, либо в первые годы
жизни ребенка. Акрогерия Готтрона отличается от других атрофий кожи
локализазацией патологического процесса на кистях и стопах, остальная
часть кожи—нормальной толщины. Лишь в редких случаях описаны од-,
новременная атрофия кожи в области предплечий, лица, особенно кон-
чика носа. Кожа тыла кистей и стоп тонкая, сухая, дряблая, сморщен-
ная, желтоватого цвета, полупрозрачная. У части больных одновремен-
но истончается подкожная жировая клетчатка в этих областях. Паль-
цы конусовидно истончены, кисти маленькие (акромикрия). В ряде случаев
описывают «просвечивающие» вены на теле, легкое образование кровопод-
теков и рубцов. Из 6 больных старше 19 лет—3 имели elastosis perforans.
Волосы обычно не изменены, описывают различные аномалии ногтей, вклю-
чая онихогрифоз.
Прогноз для жизни благоприятный, в отношении излечения неопреде-
ленный.
Гистопатология. Гистологическая картина вариабельна, часто обнару-
живают атрофию дермы, но жировой клетчатки не всегда. Основной патоло-
гический процесс локализуется в дерме, затрагивая эластические и колла-
геновые волокна, которые могут рассматриваться как дегенеративные.
Коллагеновые волокна обычно слабо окрашены. Эластические волокна
9
фрагментированы, комкообразны, неравномерны. Сосуды субпапиллярного
отдела расширены с обычными эндотелиальными клетками. Эпидермис
мало изменен, имеется небольшая его атрофия, часто обнаруживают мела-
нин в базальном слое, особенно в биоптатах с кисти. При электронно-
микроскопическом исследовании обнаружен так называемый псевдоэласти-
ческий материал. Он состоит из аморфных масс с неровными краями, с
темным^ и светлыми участками и небольшим количеством гранулярного
материала. Резко выражены изменения фибробластов—отечная вакуо-
лизированная цитоплазма и гранулофиламентозные образования (Lau-
rent et al., 1975).
Дифференциальный диагноз. Его проводят с детской прогерией, с синд-
ромом Huriez (1963), который проявляется с младенческого возраста в
виде атрофии кожи тыла кистей и склеродермоподобных изменений паль-
цев. х
Лечение. Кремы с витаминами А и Е.
ЛИТЕРАТУРА
Базыка А. П., Акрогерия Готтрона.— Вести, дерматол. и венерол., . 1979, 2, 31—36.
De Groot W., Woerdeman M. Familial acrogeria (Gottron).—Brit. J. Dermatol.,
1980, 103, 2, 213—223.
АКРОДЕРМАТИТ СТОЙКИЙ ПУСТУЛЕЗНЫЙ АЛЛОПО (acroder-
matitis continua suppurativa Hallopeau).
Синонимы: стойкий дерматит Крокера, персистирующий дерматит Сет-
гона.
Hallopeau в 1890—1897 гг. впервые описал это заболевание.
Этиология и патогенез не выяснены. Содержимое фликтен и пустул
часто стерильное, кровь также стерильна, что противоречит инфекционной
природе болезни. Vidal, Audry рассматривают это заболевание как трофо-
невроз, сам Аллопо—как особую локализацию герпетиформного импетиго
Гебры. Сторонниками того же взгляда были А. И. Поспелов (1905), Tourai-
пе (1943). Некоторые авторы (Scog, 1958; Solterman, 1958; Lapiere, 1958;
Baker и Ryan, 1968; Braverman et al., 1972; Oosterling et al., 1978) рассмат-
ривают генерализованный пустулезный псориаз Цумбуша, стойкий акро-
дерматит Аллопо и герпетиформное импетиго Гебры как один процесс.
Г. И. Мещерский (1931), Р. А. Капкаев и Л. В. Белова (1978) полагают,
что акродерматит Аллопо близок к герпетиформному дерматиту Дюринга.
А. А. Боголепов (1931) считал акродерматит синдромом, зависящим от
разных причин. По М. Г. Мгеброву, акродерматит представляет собой
пиодермию. Мы, как Mdslein (1959) и др., считаем стойкий акродерматит
самостоятельной нозологической формой. Mensing et al. (1983) рассматри-
вают акродерматит как результат дефицита цинка.
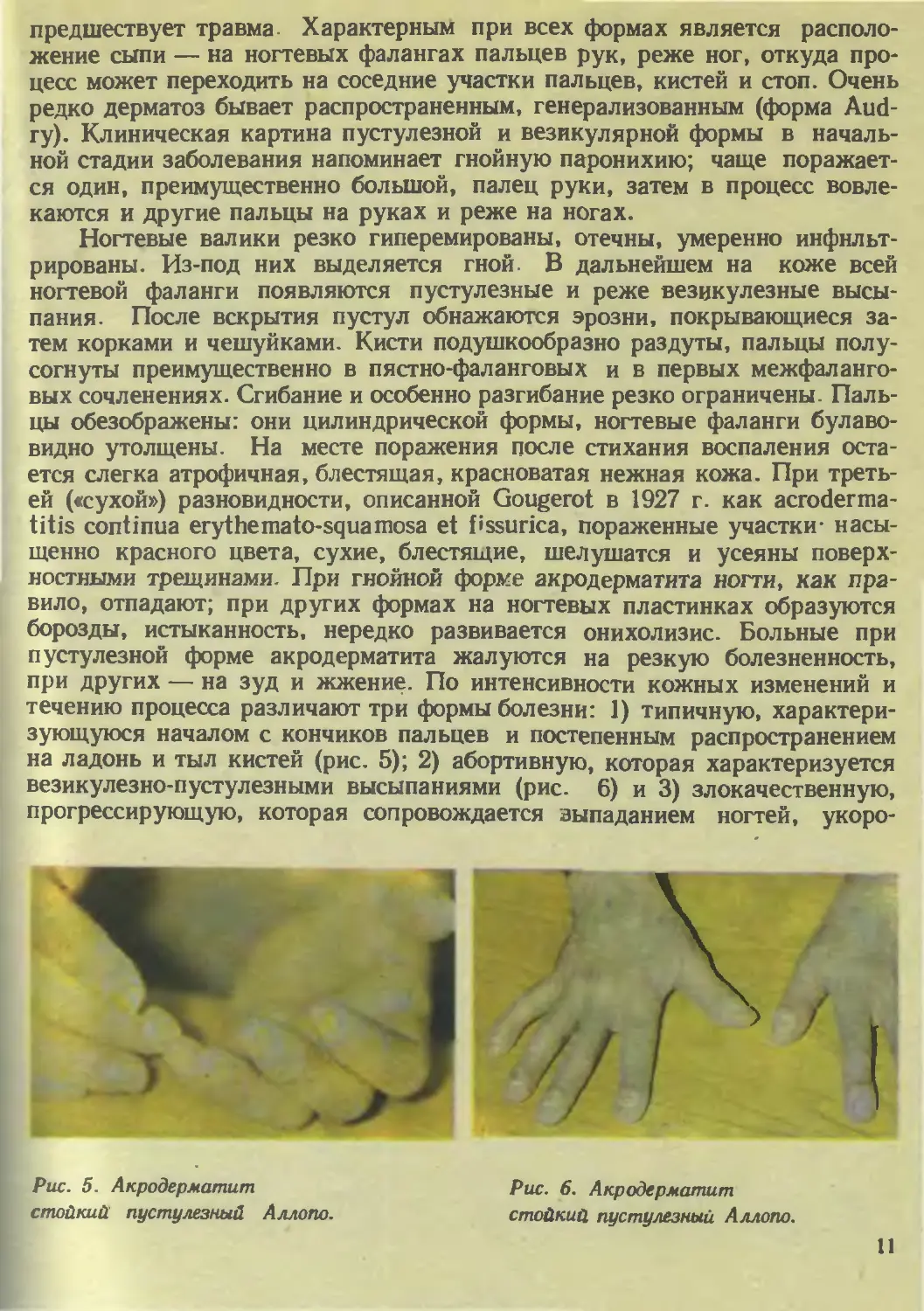
Клиника. Клинически различают следующие формы этого дерматоза:,
пустулезную, везикулезную и эритемо-сквамозную. Заболевание может
начаться в любом возрасте. Чаще болеют мужчины. В последние годы
акродерматит стал чаще встречаться у детей. Началу заболевания нередко
10
предшествует травма. Характерном при всех формах является располо-
жение сыпи — на ногтевых фалангах пальцев рук, реже ног, откуда про-
цесс может переходить на соседние участки пальцев, кистей и стоп. Очень
редко дерматоз бывает распространенным, генерализованным (форма Aud-
гу). Клиническая картина пустулезной и везикулярной формы в началь-
ной стадии заболевания напоминает гнойную паронихию; чаще поражает-
ся один, преимущественно большой, палец руки, затем в процесс вовле-
каются и другие пальцы на руках и реже на ногах.
Ногтевые валики резко гиперемированы, отечны, умеренно инфильт-
рированы. Из-под них выделяется гной. В дальнейшем на коже всей
ногтевой фаланги появляются пустулезные и реже везикулезные высы-
пания. После вскрытия пустул обнажаются эрозии, покрывающиеся за-
тем корками и чешуйками. Кисти подушкообразно раздуты, пальцы полу-
согнуты преимущественно в пястно-фаланговых и в первых межфаланго-
вых сочленениях. Сгибание и особенно разгибание резко ограничены. Паль-
цы обезображены: они цилиндрической формы, ногтевые фаланги булаво-
видно утолщены. На месте поражения после стихания воспаления оста-
ется слегка атрофичная, блестящая, красноватая нежная кожа. При треть-
ей («-сухой») разновидности, описанной Gougerot в 1927 г. как acroderma-
titis continue erythemato-squamosa et fissurica, пораженные участки- насы-
щенно красного цвета, сухие, блестящие, шелушатся и усеяны поверх-
ностными трещинами. При гнойной форме акродерматита ногти, как пра-
вило, отпадают; при других формах на ногтевых пластинках образуются
борозды, истыканность, нередко развивается онихолизис. Больные при
пустулезной форме акродерматита жалуются на резкую болезненность,
при других — на зуд и жжение. По интенсивности кожных изменений и
течению процесса различают три формы болезни: 1) типичную, характери-
зующуюся началом с кончиков пальцев и постепенным распространением
на ладонь и тыл кистей (рис. 5); 2) абортивную, которая характеризуется
везикулезно-пустулезными высыпаниями (рис. 6) и 3) злокачественную,
прогрессирующую, которая сопровождается выпаданием ногтей, укоро-
Рис. 5. Акродерматит
стойкий пустулезный Аллопо.
Рис. 6. Акродерматит
стойкий пустулезный Аллопо.
11
Pui 7 iep чатит
стойкий пустулезный Аллопо
Рис. 8. Акродерматит
стойкий пустулезный Аллопо.
чением фаланг, мутиляцией пальцев, распространением процесса на весь
кожный покров, напоминая по течению герпетиформное импетиго Гебры.
Прогноз для жизни благоприятный, однако заболевание склонно к
рецидивам, отличается упорством к терапии.
Гистопатология. В эпидермисе паракератоз, акантоз с удлинением
эпидермальных отростков. Характерным является образование так назы-
ваемой спонгиоформной пустулы Kogoj, расположенной в мальпигиевом
слое. В дерме — воспалительный инфильтрат, содержащий большое коли-
12
чество нейтрофилов. Kogoj (1962) подчеркивает отсутствие гистологических
различий между пустулезным псориазом и акродерматитом Аллопо.
Дифференциальный диагноз. Его проводят с пустулезным псориазом,
герпетиформным импетиго Гебры, пустулезным бактеридом Эндрюса, гер-
петиформным дерматитом Дюринга, пиодермией.
Лечение. Антибиотики, одни и в сочетании с кортикостероидами, суль*
фаниламиды, жидкость Кастеллани, мази, содержащие кортикостероиды и
антибиотики, мазь с мехлорэтамином (Notowicz и Stolz, 1978).
ЛИТЕРАТУРА
Волошин Р. Н. Случай стойкого суппуративного акродерматита Галлопо. Вести.
Дерматол. и венерол., 1982, 960—62.
КОЯамкарян А. А., Трофимова Л. Я, Акимов В. Г., Волошин Р. Н. К вопросу
о пустулезном псориазе, герпетиформном импетиго и акродерматите Галлопо.—Вес^н.
Дерматол. и венерол., 1980, 5, 40—44. '
Капкаев Р. А., Белова Л. В. Акродерматит хронический Галлопо.— Вест, дерма-
тол. и венерол., 1978, 2, 54—57.
Donala R., Lowe N. Mutilating acrodermatitis continua (Hallopeau's acrodermati-
tis).—Arch. Dermatol., 1978, 114, 9, 1385—1386.
Menslng et. al. Zinkmangel — induzierte acrodermatofos. Z. Hautkr., 1983, 10, 58,
723—732.
АКРОДЕРМАТИТ ЭНТЕРОПАТИЧЕСКИЙ (acrodermatitis enteropat-
hica).
Синоним: синдром Danbolt—Closs
Этиология и патогенез. Акродерматит энтеропатический является
редким аутосомно-рецессивным заболеванием, связанным с дефицитом в
организме цинка, возможно за счет нарушения его всасывания в кишечнике
(Moynahan и Grupper, 1979). Предполагалась роль в развитии акродер-
матита энтеропатического кандида—инфекции, эндокринных (гипотиреои-
дизм, гипопаратиреоидизм, диабет, недостаточность коры надпочечников)
и иммунных нарушений (в первую очередь недостаточность клеточного им-
мунитета), целиакии изменений обмена липидов, аминокислот (Фандеев
Л. И. и Сердюкова Г. И. 1962; Julius et al* 1973; White et al., 1973;
Neldner, 1974; Zinsburg et al., 1976; Коляденко В. Г. и Шупенько Н. М.,
1981). Но, по всей вероятности, в большинстве случаев эти изменения яв-
ляются вторичными, зависящими главным образом от тяжести течения
заболевания.
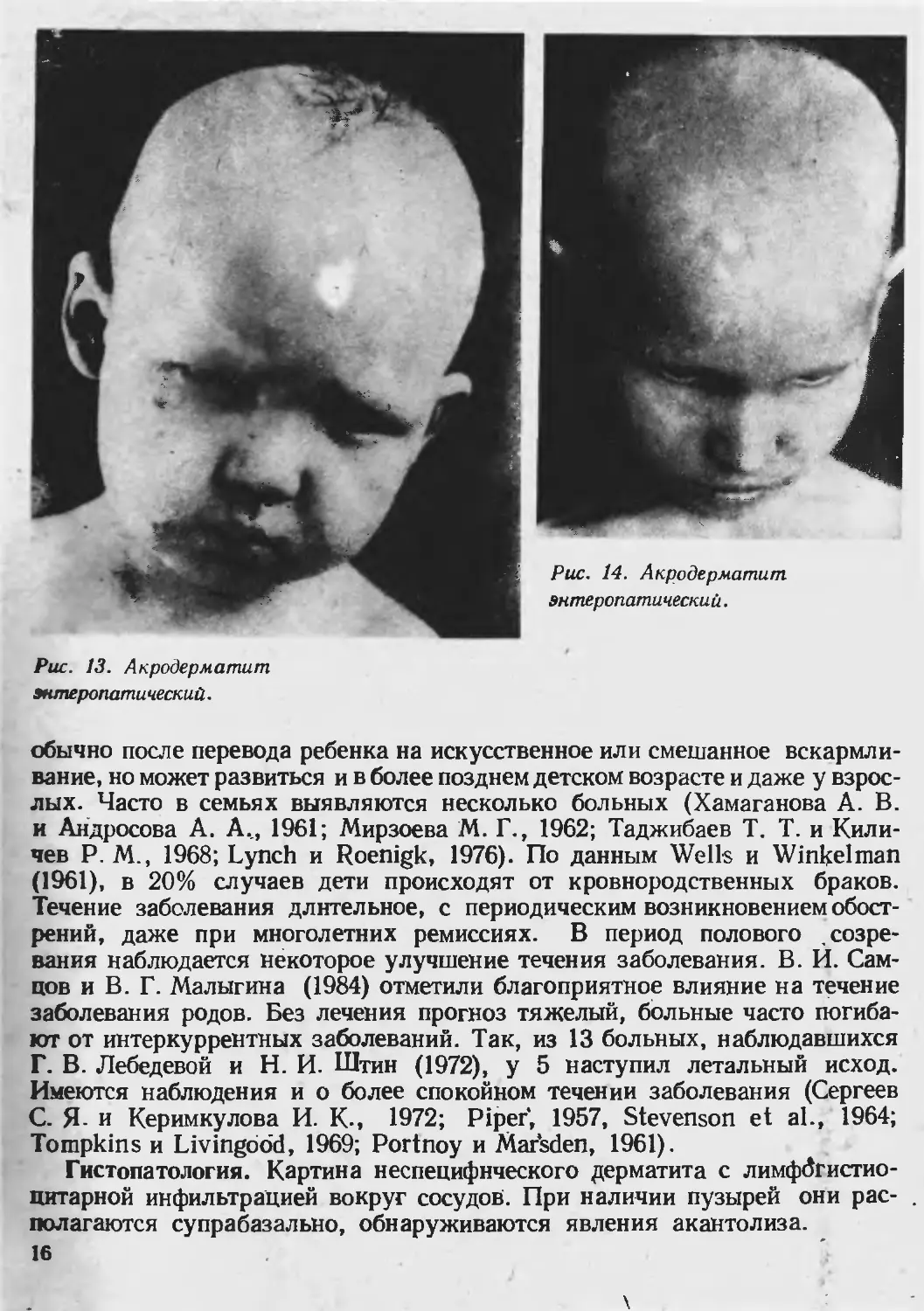
Клиника. Основными симптомами являются кожные высыпания, ало-
пеция и диарея. Сыпь полиморфна, может располагаться по всему кож-
ному покрову, но характерным, является первоначальное появление высы-
паний периорифициально (вокруг рта, носа, глаз, в перианальной области),
на локтях и стопах, затем в области коленных и локтевых суставов, в круп-
ных складках, на бедрах и ягодицах. Туловище и волосистая часть головы
поражаются реже. Вначале возникают эритематозные, везико-пустулезные
13
Рис. 10. Акродерматит
энтеропатический.
Рис. 9. Акродерматит
энтеропатический.
или буллезные высыпания, которые сливаются в обширные очаги пора-
жения, нередко мокнущие, часто эрозированные, покрытые серозными и
серозно-гнойными корками и чешуйко-корочками (рис. 9). Если эксудация
нерезко выражена, а шелушение значительное, что обычно бывает при дли-
тельном существовании заболевания, очаги приобретают сходство с псо-
риатическими, тем более что они, как правило, довольно резко ограниче-
ны (рис. 10). Часто при локализации высыпаний на кистях и стопах раз-
виваются паронихии (рис. 11, 12). Сыпь, располагающаяся вокруг круп-
ных суставов, как правило, не носит эксудативного характера, обычно
здесь обнаруживаются небольших размеров красноватые, слегка шелуша-
щиеся лихеноидные элементы. В процесс вовлекается не только кожа, но и
полуслизистые и слизистые: блефарит, конъюнктивит с фотофобией, глоссит,
стоматит и др. (рис. 13). Характерно выпадение волос, которое может до-
стигать степени тотального облысения (рис. 14), дистрофические измене-
ния ногтей. Диарея резко выражена и приводит к истощению детей. Име-
ются описания случаев (Portnoy и Marsden, 1961), когда заболевание про-
14
Рис. 11. Акродерматит
янтеропатический.
Рис. 12. Акродерматит
энтеропатический.
и волос (Stevenson et
текало без диареи, а также без поражения ногтей i_ “1______
al., 1964). Больные отстают в росте, но это, по мнению Deffner и Perry
(1973), —вторичный признак, зависящий от возраста ребенка, тяжести
болезни и времени начала лечения. Может быть нарушено психическое
развитие детей, они становятся плаксивыми, раздражительными. По дан-
ным Heite и Ody (1966), кожные изменения наблюдаются у 100% больных,
выпадение волос—у 97%, паронихии—у 97%, дистрофий ногтей—у 96%,
диарея—у 91 %,задержка роста—у78%,обнаружение Candida albicans—у
69% больных. Заболевание возникает преимущественно в грудном возрасте,
15
Рис. 13. Акродерматит
s иперопатический.
обычно после перевода ребенка на искусственное или смешанное вскармли-
вание, но может развиться и в более позднем детском возрасте и даже у взрос-
лых. Часто в семьях выявляются несколько больных (Хамаганова А. В.
и Андросова А. А., 1961; Мирзоева М. Г., 1962; Таджибаев Т. Т. и Кили-
чев Р. М., 1968; Lynch и Roenigk, 1976). По данным Wells и Winkelman
(1961), в 20% случаев дети происходят от кровнородственных браков.
Течение заболевания длительное, с периодическим возникновением обост-
рений, даже при многолетних ремиссиях. В период полового .созре-
вания наблюдается некоторое улучшение течения заболевания. В. И. Сам-
цов и В. Г. Малыгина (1984) отметили благоприятное влияние на течение
заболевания родов. Без лечения прогноз тяжелый, больные часто погиба-
ют от интеркуррентных заболеваний. Так, из 13 больных, наблюдавшихся
Г. В. Лебедевой и Н. И. Штин (1972), у 5 наступил летальный исход.
Имеются наблюдения и о более спокойном течении заболевания (Сергеев
С. Я- и Керимкулова И. К., 1972; Piper, 1957, Stevenson et al., 1964;
Tompkins и Livingoo'd, 1969; Portnoy и Mar’sden, 1961).
Гистопатология. Картина неспецифнческого дерматита с лимфдгистио-
цитарной инфильтрацией вокруг сосудов. При наличии пузырей они рас-
полагаются супрабазально, обнаруживаются явления акантолиза.
16
Дифференциальный диагноз. Следует проводить с псориазом, детской
экземой, буллезным эпидермолизом.
Лечение: препараты цинка, энтеросептол, общеукрепляющие средства.
ЛИТЕРАТУРА
В. Г. Коляденко, Н. М. Шупенько. Энтеропатический акродерматит (болезнь Дан-
бол га—Клосса). Вести, дерматол. и венерол., 1981, 3, 51—53.
Самцов В. И., Мальгина В. Г. Энтеропатический акродерматит взрослых. Лечение
окисью цинка. Вест. Дерматол. и венерол. 1984, 9, 39—41.
Ginsburg R., Robertson A., Michel В. Acrodermatitis enteropathica.— Arch, derma-
tol., 1976, 112, 5, 653—660.
Moynahan E. J., Grupper C. Acrodermatitis enteropathica and other zinc—deficien-
cy disordes—in Dermatology in general medicine. Sec. ed. Ed. T. B. Fitzpatrick et. al., N. Y.,
1979, 1371—1375.
Neldner К- H., Hagler L., Wise W. R., Stif el F. B., Lufkin E. G., Herman R. H.
Acrodermatitis enteropathica, Arch dermatai., 1974, 110, 5, 711—*721.
АКРОКЕРАТОЗ ВЕРРУЦИФОРМНЫЙ Г0ПФА (acrokeratosis ver-
ruciformis Hopf).
Этиология и патогенез. Акрокератоз верруциформный является ред-
ким дерматозом с аутосо!мно-доминантным типом наследования. Niedelman
и Me Kusick (1962) описали 24 больных, членов одной семьи в четырех поко-
лениях. Самостоятельность его признается не всеми. Так, Krause и Eh-
lers (1969) считают, что клиническое сходство проявлений фолликулярного
дискератоза Дарье и акрокератоза верруциформного, сочетание их у од-
ндго и того же больного, обнаружение при акрокератозе верруциформном
супрабазально расположенных щелей и дискератоза свидетельствуют про-
тив самостоятельности этого дерматоза. Waisman (1960) полагал, что акро-
кератоз верруциформный без других проявлений—forme fruste болезни
Дарье. Niordson и Sylvest (1965) наблюдали у одного больного болезнь
Дарье, семейную доброкачественную пузырчатку и верруциформный акро-
кератоз, причем еще несколько членов семьи страдали верруциформный
акрокератозом. Herndon и Wilson (1966) наблюдали в четырех поколениях
одной семьи 7 больных акрокератозом верруциформным, 2—фолликуляр-
ным вегетирующим дискератозом и 3—с недифференцированным типом
аномалии ороговения. Это дало основание авторам рассматривать оба со-
стояния как проявление общих генетических нарушений, выраженных в
различной степени. И. М. Щербаков (1964) полагает, что верруциформ-
ный кератоз, видимо, является вариантом эпителиального невуса.
Клиника. Высыпания располагаются преимущественно на тыльной
Поверхности кистей и стоп, где обнаруживаются мелкие полигональных
очертаний папулы, сходные с бородавками (рис. 15, 16). Поверхность их
гладкая или чаще покрыта гиперкератотическими наслоениями, верру-
коЗна. Цвет их мало отличается от нормальной кожи или слегка красновато-
коричневатый. При слиянии отдельных папул могут возникать небольшие
веррукозные бляшки. Наряду с этим сыпь может располагаться на пред-
2 Клиническая дерматология 17
Рис. 15. Акрокератоз
Гопфа.
Рис. 16. Акрокератоз
Г опфа.
плечьях, локтях и коленях. У больных, наблюдавшихся В. И. Кечкером
и Ю.С Эрдманом (1960), отдельные высыпания располагались на воло-
систой части головы, а у больной, описанной Л. П. Цыркуновым (1960),
была поражена кожа затылка, ладоней, передние края подмышечных впа-ч
дин, внутренние поверхности бедер, подколенные впадины, область ахил-
лова сухожилия Не исключено, впрочем, что в данном случае было соче-
тание акрокератоза верруциформного и болезни Дарье. Описано (Mina и
Scheremetjeva, 1962) полосовидное расположение бородавчатых высыпаний
на руках и ногах, весьма напоминающих по клинической картине линей-
ный невоидный дерматоз. Изменения ладоней и подошв наблюдаются не-
постоянно, они могут быть в виде нерезко выраженного диффузного утол-
18
щения рогового слоя, точечного кератоза, прерывистых папиллярных ли-
ний. Может наблюдаться дистрофия ногтей, лейконихия.
Заболевание существует с рождения или развивается в раннем детском
возрасте, в редких случаях—у взрослых (Schueller, 1972). Течение неоп-
реде. енно длительное.
Гистопатология. Акантоз, гиперкератоз, гипергранулез, папиллома-
тоз, а также супрабазальные щели, явления дискератоза.
Дифференциальный диагноз. Его необходимо проводить с бородавками,
верруциформной дисплазией Левандовского—Лютца, фолликулярным веге-
тирующим дискератозом Дарье (не всегда легко).
ЛИТЕРАТУРА
Krause W., Ehlers J., Ober die Beziehung zwischen Akrokeratosis verruciformis Hopf
und Dyskeratosis follicularis vegetans Darier.— Hautarzt, 1969, 20, 9, 397—403.
АКРОПАТИЯ ПСЕВДОСИРИНГОМИЕЛИТИЧЕСКАЯ (acropathia
pseudo-syringomyel ica).
Синонимы: псевдосирингомиелитическа'я язвенно-мутилирующая и де-
формирующая акропатия; синдром Thevenard—Brown, синдром Bureau-
Barriere
Эта редкая болезнь описывается под различными названиями в зави-
симости от преобладания тех или иных разнообразных кожно-трофических
нарушений
Различают две формы псевдосирингомиелитической акропатии: семей-
ную (описана впервые Thevenard и Browns в 1942 году), наследуемую по
рецессивно-доминантному типу, возникающую в детстве, поражающую
нижние и верхние конечности и сочетающуюся с другими врожденными
уродствами; приобретенную (описана Bureau и Barriere в 1953 году), про-
являющуюся у мужчин среднего возраста поражением только нижних
конечностей, при этом наследственный фактор и врожденные аномалии от-
сутствуют.
В отечественной и зарубежной литературе имеются единичные описа-
ния (Каралицкий Е.М., 1970; Големба П. Й. и соавт., 1973; Коляденко
В. Г. и соавт., 1982; Dereux, 1970; Gebnart et al., 1971; Bossi и Peirone,
1972; Kind, 1976; Diem et al., 1975; Harman, 1977 и др.) этого заболевания.
Этиология и патогенез. Этиология ненаследственной (приобретенной)
формы псевдосирингомиелитической акропатии неизвестна; патогенез за-
болевания окончательно не выявлен. Признавая в патогенезе этого забо-
левания ведущую роль изменений вегетативной нервной системы, многие
авторы полагают, что основой заболевания является, по-видимому, хро-
ническая алкогольная интоксикация элементов периферической нервной
системы (нейротоксический механизм;, а предрасполагающими фактора-
ми — неблагоприятные условия внешней среды (охлаждение, хронические
травмы, давление обуви, патология печени, витаминная недостаточность,
метаболические нарушения и др.).
Novotny (1964) описал псевдосирингомиелитическую акропатию, харак-
2* 19
Рис. 17. Акрспати.ч
псечдосирингом'. -лип:: ческа ч.
Рис. 18. Акрстатич
псевдссирингомие штическая.
Рис. 19. Акропатия
псевдосирингомиелитическая.
теризовавшуюся поражением симпатических узлов, и отметил в них де-
генерацию клеток и фиброз.
В обширной группе трофических нарушений нижних конечностей
псевдосирингомиелитическая акропатия, имея некоторые черты сходства
с другими дистрофическими акропатиями, все же обладает определенными
и характерными свойствами, которые позволяют выделить ее в особую
нозологическую форму.
Клиника. Практически заболевают только мужчины, большей частью
в возрасте от 40 до 60 лет. Мы наблюдали двух больных в возрасте 53 и
68 лет В большинстве случаев поражаются обе нижние конечности За-
болевание начинается с гипергидроза и появления очагового подошвен-
ного гиперкератоза чаще всего в точках опоры у основания 1 и V пальцев,
а также на пятке, где затем образуются глубокие безболезненные округ-
лые язвы, дно которых покрыто некротическим налетом, а края отвесные,
ровные, плотные (рис. J7, 18). Язвы постепенно углубляются — вплоть
до кости (рис 19). Постепенно деформируются стопы и пальцы, развива-
ется акроостеолиз, ограничивается подвижность в суставах. Рентгенологи-
чески определяется остеопороз костей стоп Выражены вегетативные рас-
стройства: кожа стоп синюшная, влажная, ногти деформированные.
Характерно диссоциированное расстройство чувствительности кореш-
кового характера в дистальных отделах нижних конечное! й: температур-
ная и болевая чувствительность i счезают, а глубокая сохраняется. Дви-
гательная функция не нарушается Ахилловы рефлексы не вызываются
У некоторых больных отмечается снижение тонуса мышц нижних конеч-
ностей
Заболевание протекает длительно, торпидно, обостряясь часто зимой,
с трудом поддается лечению
Гистопатология. Морфологически определяются дегенеративные из-
менения в задних корешках и задних столбах спинного мозга, обнаружи-
ваются поражения поясничных симпатических узлов.
Дифференциальный диагноз. Его проводят с сирингомиелией, обли-
терирующим эндартеринтом, полиневритом, лепрой, табесом, диабетиче-
ской ангиопатией, акросклеротической формой склеродермии.
Лечение. Из-за неясности этиологии и патогенеза специфических ме-
тодов лечения этого заболевания нет.
ЛИТЕРАТУРА
Коляденко В. Г. и др. Псевдосирингомиелитическая язвенномутилирующая и де"
формирующая акропатия.— Вести, дерматол. и венерол., 1982, 2, 55—59.
Diem Е., Wolf G., Oppolzer R. Zur Kenntnis der nicht-familiaren sogenannten spo-'
radischen Acropathia ulceromutilans der unteren Extremitaten (Bureau—Barriere—Synd-
rom).—Z. Hautker., 1975, Hd. 50, Hf. I, S. 13—24.
АМИЛОИДОЗ КОЖИ (amyloidosis cutis).
Синонимы: амилоидный лихен, локальный диссеминированный узел-
ковый амилоидоз кожи.
Амилоидоз (амилоидная дистрофия) представляет собой нарушение
обмена, выражающееся в образовании и отложении в тканях амилоида.
Новевшими исследованиями установлено, что амилоид является гликопро-
теином, в котором фибриллярные и глобулярные белки тесно связаны с
полисахаридами (Серов В. В., Шехтер А. Б., 1981). Существуют четыре те-
ории патогенеза амилоидоза: теория диспротеиноза (или парапротеино-
за), иммунная теория, теория клеточной локальной секреции и мутаци-
онная теория. И. А. Шамов (1972), В. В. Серов и соавт. (1974), В. Н. Ару-
тюнян, Г. А Еганян (1982) полагают, что общим механизмом, позволяющим
понять клиническое разнообразие амилоидоза, являются мутации, ведущие
к образованию клона клеток мезенхимального происхождения — амило-
идобластов, синтезирующих фиблиллярный белок амилоида, возможно, и
полисахариды. Общепринятой классификации амилоидоза нет.
Анализ литературных данных (Юкелис И. М., 1929; Серов В. В. и
соавт., 1974; Шапошников О. К. и соавт., 1976; Brownstein и Helwig, 1970;
Gottschalk et al., 1975; Lever и Schaumburg-Lever, 1975; Goertier et al.,
1976; Breatnach, 1979; Noren etal., 1983, и др.) и собственные клинико-мор-
фологические наблюдения позволяют различать четыре основные формы
амилоидоза.
1. Первичный системный амилоидоз (Lubarsch, 1929), который возни-
кает «первично» без предшествующего заболевания. Поражаются преиму-
щественно органы мезодермального происхождения. Первичный систем-
ный амилоидоз и множественные миеломы, сочетающиеся с амилоидозом,
имеют кожные проявления в 10—40% случаев Кожные изменения при пер-
вичном системном амилоидозе полиморфны (петехии, пурпуры, папулы,
узлы, опухоли, бляшки, склеродермоподобные изменения, пойкилодер-
МИЯ, алопеция, желтоватые ксантомэподобные бляшки и пузырные вы-
сыпания). Более частыми являются пурпурозные высыпания, однако бо-
лее характерны папулы и склеродермоподобные бляшки. Описаны пузыр-
ные высыпания, которые встречаются чрезвычайно редко (Каламкарян
А. А., 1980). Beacham и соавт. (1Й80) описали больного 74 лет, у которого
наряду с желтовато-коричневой инфильтрацией на коленях, локтях и
ягодицах имелись пузыри, наполненные желтоватой жидкостью. При
биопсии костного мозга выявлено 42% плазмоцитов, что позволило поста-
вить диагноз первичного амилоидоза. В ряде случаев возникают «плотные»
отеки, когда кожа не собирается в складку, лицо становится амимичным.
Гистологически обнаруживается отложение амилоида в коже и подкожной
клетчатке, главным образом внеклеточно вокруг сосудов и в их стенках.
Brownstein и Helwig (1970), Rubinow и Cohen (1978), Westermark и Sten-
kist (1973) указывают, что примерно у 50% больных первичным системным
амилоидозом в нормальной на вид коже обнаруживается амилоид.
2. Вторичный системный амилоидоз развивается при хронических
нагноительных процессах (туберкулез легких, хронический нефрозо-ие-
фрит, хроническая пиодермия, системная красная волчанка, ревматоид-
ный артрит и др.). Амилоид откладывается в основном в паренхиматозных
органах и, как правило, не сопровождается кожными поражениями.
22
3. Первичный местный амилои-
доз кожи—поражается исключитель-
но кожа. О первичном (изолирован-
ном) амилоидозе кожи сообщил в
1921 г. Konigstein. Guttmann (1923—
—1928) изучил и описал его под
названием amiloidosis cutis localis
nodularis et disseminata, a Freudent-
hal (1923—1925)—под названием li-
chen amyloidosus. Выработка ами-
лоида происходит в самой коже фи-
бробластами кожи (.Ebner, 1968;
Rotermuhd и Klingmuller, 1970;
Shapiro et al., 1970; Brownstein и
Hashimoto, 1972; Goodmanr. et al.,
1972; Breathnach et al., 1982; Pype.
1982. и др.).
К настоящему времени в миро-
вой литературе описано немногим
более 130 случаев, первичного мест-
ного амилоидоза кожи, в том числе
семь случаев семейного локализован- *
ного амилоидоза кожи (Andrade et
al., 1970; Sagher et al., 1963; Vasily
et al., 1978 и др.). Заболевание на-
блюдается у лиц обоего пола, но
чаще у пожилых мужчин. Первичный
Рис. 20. Первичный амилоидоз кожи.
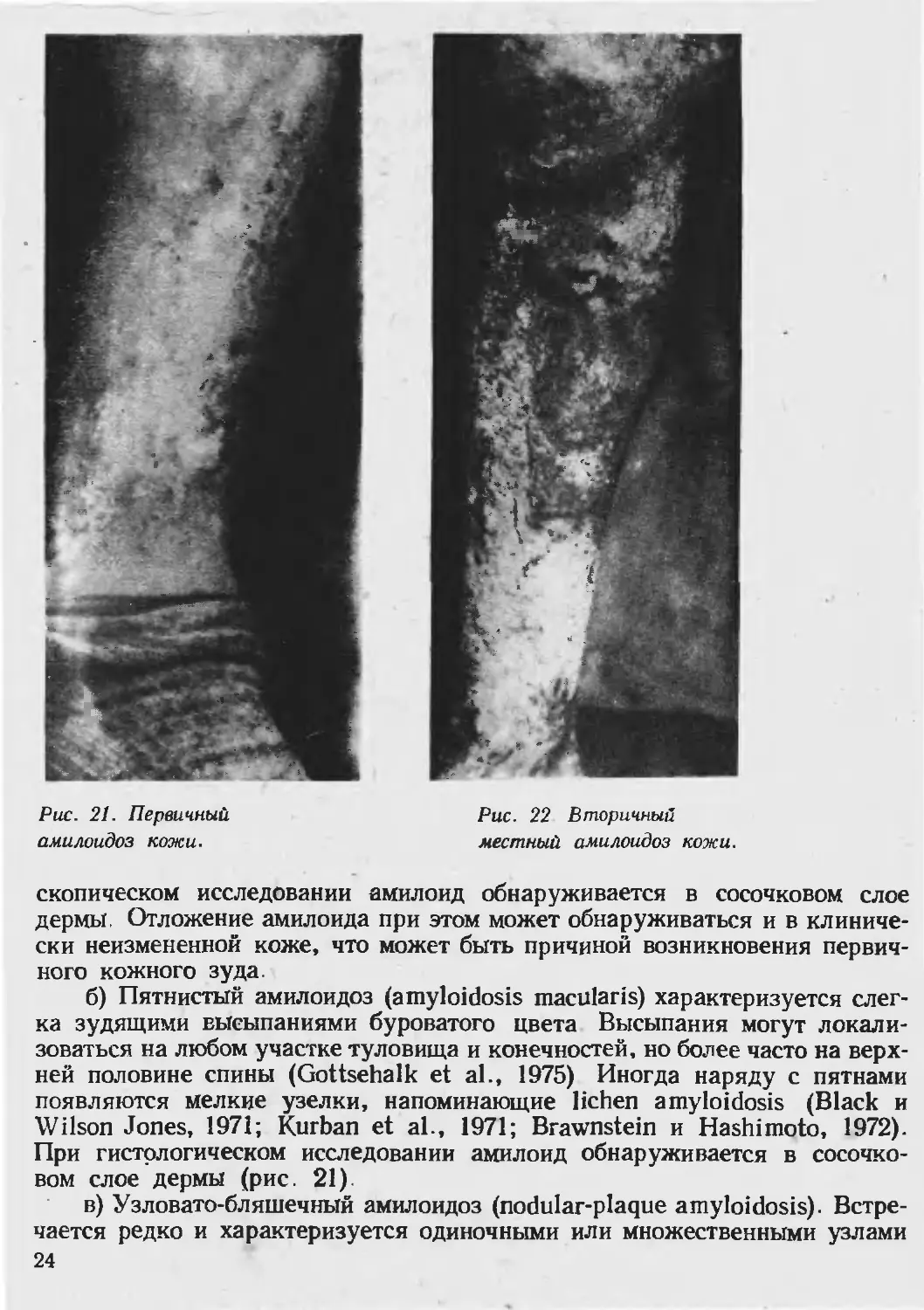
местный амилоидоз кожи встречается в трех клинических вариантах:
узелковый, пятнистый и узловато-бляшечный (рис. 20).
а) Узелковый амилоидоз (lichen amyloidosus) чаще локализуется на
голенях (рис. 20), но иногда на других участках кожного покрова—на го-
ловке полового члена (Weitzner et al., 1970), в области венечной борозды
(Degos et al., 1961), вокруг наружного отверстия уретры (Friedmann, 1967).
В этих трех случаях не было проявлений амилоидоза на других участках
кожи. На передней поверхности обеих голеней строго симметрично распо
ложены многочисленные величиной от булавочной головки ДО горошины
полушаровидные блестящие, полупрозрачные узелки, тесно прилегающие
друг к другу, но не сливающиеся и разделенные узкими бороздками здо-
ровой кожи. Некоторые узелки конической формы и связаны с волосом,
другие имеют в центре роговые шипики. Окраска папул бледно-розовая
или буровато-синюшная, поверхность гладкая или покрыта плотно сидя-
щими мелкими грязно-серыми чешуйками. Кожный покров на месте высы-
паний уплотнен, с трудом берется в складку. Местами на голенях имеются
линейные расЧесы, кровянистые корочки. Беспокоит сильный, мучитель-
ный зуд пораженных участков. В результате расчесов возникает резко
выраженная лихенификация. При гистохимическом и электронно-микро-
2.3
Рис. 21. Первичный
амилоидоз кожи.
Рис. 22 Вторичный
местный амилоидоз кожи.
скопическом исследовании амилоид обнаруживается в сосочковом слое
дермы. Отложение амилоида при этом может обнаруживаться и в клиниче-
ски неизмененной коже, что может быть причиной возникновения первич-
ного кожного зуда.
б) Пятнистый амилоидоз (amyloidosis macularis) характеризуется слег-
ка зудящими высыпаниями буроватого цвета Высыпания могут локали-
зоваться на любом участке туловища и конечностей, но более часто на верх-
ней половине спины (Gottsehalk et al., 1975) Иногда наряду с пятнами
появляются мелкие узелки, напоминающие lichen amyloidosis (Black и
Wilson Jones, 1971; Kurban et al., 1971; Brawnstein и Hashimoto, 1972).
При гистологическом исследовании амилоид обнаруживается в сосочко-
вом слое дермы (рис. 21).
в) Узловато-бляшечный амилоидоз (nodular-plaque amyloidosis). Встре-
чается редко и характеризуется одиночными или множественными узлами
24
и бляшками размерами от вишни до лесного ореха, которые, сливаясь,
образуют бугристые бляшки различной величины. Некоторые очаги по-
ражения носят характер бородавчатых образований с роговыми наслоения-
ми и чешуйками на поверхности. Локализация—главным образом разги-
бательные поверхности голеней Высыпания сопровождаются зудом, иног-
да весьма мучительным с наличием расчесов и лихенифнкации кожи. Гисто-
логически обнаруживается диффузное отложение амилоида в коже и под-
кожной жировой клетчатке, в стенках сосудов, мембранах потовых желез
и жировых клеток.
4. Еюрнчный местный амилоидоз кожи (amyloidosis cutis localis se-
cundaria. рис 22) характеризуется отложением амилоида в коже, поражен-
ной до зтогс другими дерматозами,—красным плоским лишаем, хрониче-
ским нейродермитом, в области язв и рубцов кожи и др. Вторичный ами-
лоидоз кожи обычно клинически не определяется, но иногда проявляется
в виде узелков, бляшек и узлов. В редких случаях амилоид нахсдят при
некоторых кожнъ.х опухолях—эпителиоме, болезни эоуена, актиническом
кератозе и др. (MacDonald и Black, 1980) Hashimoto и Brownstein (1973)
подтвердили это электронно-микроскопическими исследованиями. Важную
диагностическую ценность имеют красочные реакции на амилоид, в том
числе окраска красным коего, метиловым фиолетовым и тиофлавином Т
или S. Окраска) красным гснп достаточно специфична.
Прогноз для Ж1 эки благоприятный, течение хроническое.
Ди( фзрггциалы ын / нагие . Его следует проводить с микседематоз-
ным лиленом, узчэьзтым веррукозным нейродермитом, веррукозным крас-
ным лишаем.
Лечение. Резохин (делагил) длительно по 0,5 г на день. Местно—мази,
содержащие кортикостероиды, деготь, борную кислоту, ихтиол, АСД; че-
репицеобразные повязки чз лейкопластыря. Рентгенотерапия по методике,
применяемой для лечения хронической лихенифицированной экземы При
ограниченных очагах показана лазеротерапия (Ракчеев А. П. и Капкин
В. В., 1980). С. И. Полищук проводил лечение 5%. унитиолом внутри-
мышечно (по 5 мл 20 инъекций), местно применял 80—90% раствор диме-
ксида.
ЛИТЕРАТУРА
Арутюнян В. М., Еганян Г. А. О роли нарушения иммунологического гомеостаза
в патогенезе приобретенного амилоидоза.— Иммунология, 1982, 6, 40—43.
Беляев Н. В., Грацианов Д. А., Зайцева Т. А. Локализированный амилоидоз
кожи.— Вести. Дерматол. и венерол.. 1978, 1. 59—62.
Серов В. В., Шамов А. И. Амилоидоз.— М.: Медицина, 1977. Полищук С. И. Ло-
кализованный амилоидоз кожи.— Вёстн. дерматол. и веиерол., 1982, 3, 56—58.
Шапошников О. К., Самцов В. И., Разнатовский И. М. Случай первичного си-
стемного амилоида.— Вестн. дерматол. и венерол., 1976, 12, 48—53.
Breathnach S , Wilkinson J., Black M. Systemic amyloidosis with an underlying
lymphoproliferative disoder.—Clin. Exp. Dermatol., 1979, 4, 4, 495—499.
25
Beachman В., Greer К., Anderws В., Cooper P. Bullous amyloidosis.— J. Amer.
Acad. Dermatol-, 1980, 3, 5, 506—510.
Breathnach S. et al. Localized cutaneous amyloidosis: dermal amyloid deposits do
not bind antibodies to amyloid A protein, prealbumun of fibronectin.— Brit. J. Dermatol.,
1982, 107, 4, 453—459.
Chapel T., Birmingham D., ' Malinowski J. Nodular ultralesional hemorrhage in
primary systemic amyloidosis.—Arch. Dermatol., 1977, 113, 9, 1248—1249.
Goertiler E. et. al. Amyloidosis cutis nodularis.— Hautarzt, 1976, 27, 1, 16—25.
MacDonald D., Black M. Sescndary localized cuta leous amyloidosis in mela ;ocytic
naevi.— Brit. J. Dermatol., 1980, 103, 5, 553—556.
Keren P., Westermark P., Cornwell G., Murdoch W. Immunofluorescence and his-
tochemical studies of focalized cutaneous ar.n luidusis.—B-rit. J. Dermatol., 1983, 108, 3,
277—285.
Rubinow A., CohenA. Sein involvement in generalized amyloidosis.— Ann. Intern.
Med., 1978, 88, 4, 781—785.
Visaly D., Bhatia Sh., Uhliti S. Familial primary cutaneous amyloidosis.— Arch.
Dermatol., 1978, 114, 8, 1173—1176.
АНГИОКЕРАТОМА (angiokeratoma).
Эпителиальные выбухания, образованные субэпидермальными рас-
ширениями капиллярных полостей, сопровождающие_л реактивными изме-
нениями эпидермиса.
Группу апгиокератом составляют: ангиокератома Mibelli, ангиоке-
ратома мошонки (вульвы) Fordyce, кожные изменения при болезни Fabry,
ограниченная невиформная ангиокератома, солитарная папулезная ангио-
кератома
АНГИОКЕРАТОМА МИБЕЛЛИ (angiokeratoma Mibelli).
Описана Мибелли в 1889 г.
Этиологий и патогенез. Наблюдается у лиц с повышенной чувствитель-
ностью к холоду, склонностью к озноблениям, что приводит к асфиксии и
нарушению капиллярного кровообращения Gertler (1970), Hundeiker
(1978) полагают, что заболевание развивается на врожденной ангионевро-
тической основе. Пенетрантность его слабая. И Л Петрова и соавт. (1978)
также высказывают предположение о генетической природе ангиокера-
томы Мибелли, в связи с уменьшением содержания полового хроматина в
ядрах эпителиальных клеток слизистой оболочки щеки и повышения греб-
невого счета (дерматоглифика) у обследованной ими пациентки. Schnyder
(1954), Bean (1958), Hauss (I960) относят ангиокератому Mibelli к ангиоке-
ратотическим невусам. С. Т. Павлов (1926), Pautrier и Levg (1927) и др.
придают значение туберкулезной интоксикации.
Клиника. Начинается в препубертатном периоде, чаще у девочек.
Высыпания в виде мягких темно-красных, лиловатых узелков диаметром
1—5 мм, исчезающих при диаскопии. Располагаются симметрично на тыле
пальцев рук или ног, иногда под ногтями. Более редкой локализацией
являются локти, колени, кончик носа, ушные раковины. Поверхность
узелков шероховатая, гиперкератотичная, иногда покрыта бородавчатыми
26
разрастаниями.. Как правило, имеются гипергидроз ладоней и подошв,
акроасфиксия А. В. Устиновский (1926) различает три стадии ангиокера-
томы: 1) vascularis; 2) naeviformis; 3) verrucosum. Заболевание может
остановиться на любой из них. Течение хроническое с ухудшением в холод-
ное время года. Редко наблюдается спонтанное разрешение высыпаний.
Дерматоз иногда имеет семейный характер. При капилляроскопии выяв-
ляют расширение восходящих и нисходящих колен ‘капилляров, сужение
окружающих сосудов.
Прогноз благоприятный.
Гистопатология. Гиперкератоз, акантоз, неравномерная пролифера-.
ция эпидермальных отростков. Расширение капилляров в сосочках и под-
сосочковой сети. Сосочки могут приподниматься каверноподобными, на-
полненными кровью полостями, выстланными эндотелием. Покрываю-
щий их эпидермис истончен.
Дифференциальный диагноз. Его проводят с другими формами ангио-
кератом. .с геморрагическим ангиоматозом Рендю-Ослера, с гемо - и лим-
фангиомами, озноблениями.
Лечение. Содержание рук и ног в тепле. Удаление крупных образова-
ний с помощью жидкого азота, электрокоагуляции.
ЛИТЕРАТУРА
Капкаев Р. А., Камзолова К- П., Данильянц Е. И. Ангиокератома Мибелли.—
Вести, дерматол. и венерол., 1976, 2. 68—70.
Петрова И. Л. Меморский В. П., Ильянкова Г. Т. Ангиокератома Мибелли.—
Вести, дерматол. и в»нерол., 1978. 3, 52—54.
Hundeiker М. Systematik der angiektatiscben und angiokeratotischen Navi.— Hau-
tarzt, 1978, 29, 10, 511-517.
АНГИОКЕРАТОМА ТУЛОВИЩА ДИФФУЗНАЯ ФАБРИ (angioke-
ratoma corporis diffusum Fabri).
Независимо др\ч >т друга заболевание описали в 1898 г. Fabri и An-
derson.
Этиология и патогенез. Кожные проявления являются симптомом
редкого врожденного, сцепленного с Х-хромосомой рецессивного заболе-
вания, обусловленного отсутствием или уменьшением активности лизо-
сомального энзима, который участвует в катаболизме сфинголипидов-
церамидтригексозидазы (а-галактозидазы). Расщепление сфинголипидов,
являющихся нормальной составной частью мозга, других органов и экстра-
целлюлярных жидкостей, прерывается на определенном этапе. Это ведет
к накоплению церамидтригексозида в мозге, почках, мышце сердца, в стен-
ках сосудов и др. органах (Галлер Г. и соавт., 1979; Видершайн. Г. Я-,
1980; Gokel и Hubner, 1982).
В отделе дерматологии ЦКВИ (Трофимова И. Б. и Айвазян А. А.,
1983) в течение многих лет наблюдался больной, у которого с
четырехлетнего возраста имелась развернутая симптоматика болезни Фаб-
ри с обильными высыпаниями на коже сосудисто-узелкового характера
27
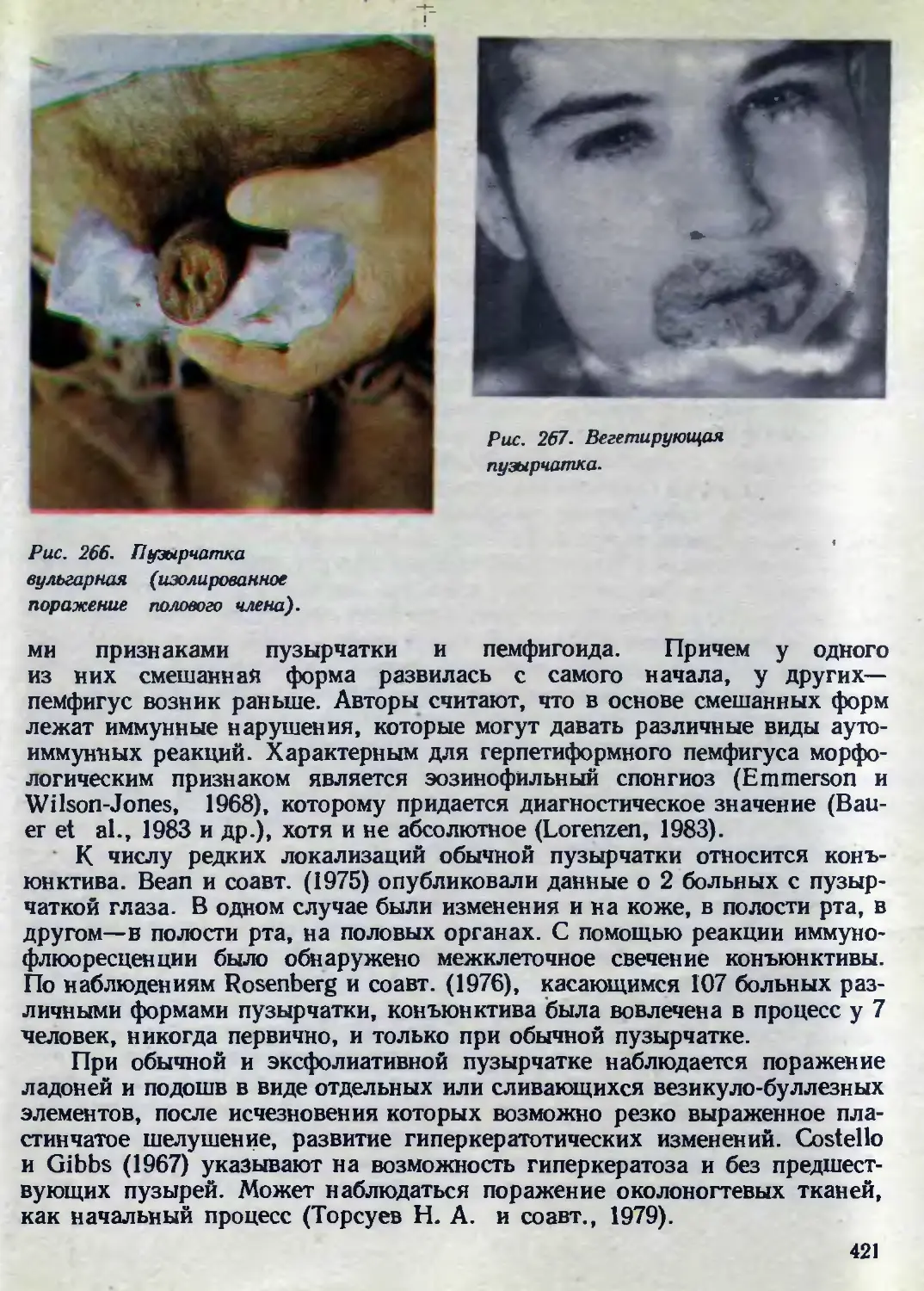
темно-красного цвета. Клинический диагноз подтвержден энзимологиче-
скими исследованиями. Активность а-галактозидазы у больного практиче-
ски отсутствовала (0,95 нмоль/мг белка/ч), у его здорового брата—59
нмоль/мг белка/ч, у отца—53 нмоль/мг белка/ч.
Клиника. Заболевание обычно наблюдается у мужчин, однако имеются
сообщения о болезни Фабри у гетерозиготных женщин (Wadskow et al.,
1975; Hachimoto et al., 1976). У гомозиготных мужчин картина болезни
бывает развернутой и обычно приводит к смерти до 40 лет. У гетерозигот-
ных женщин чаще наблюдаются слабые проявления заболевания. Харак-
терными для них являются помутнения роговицы.
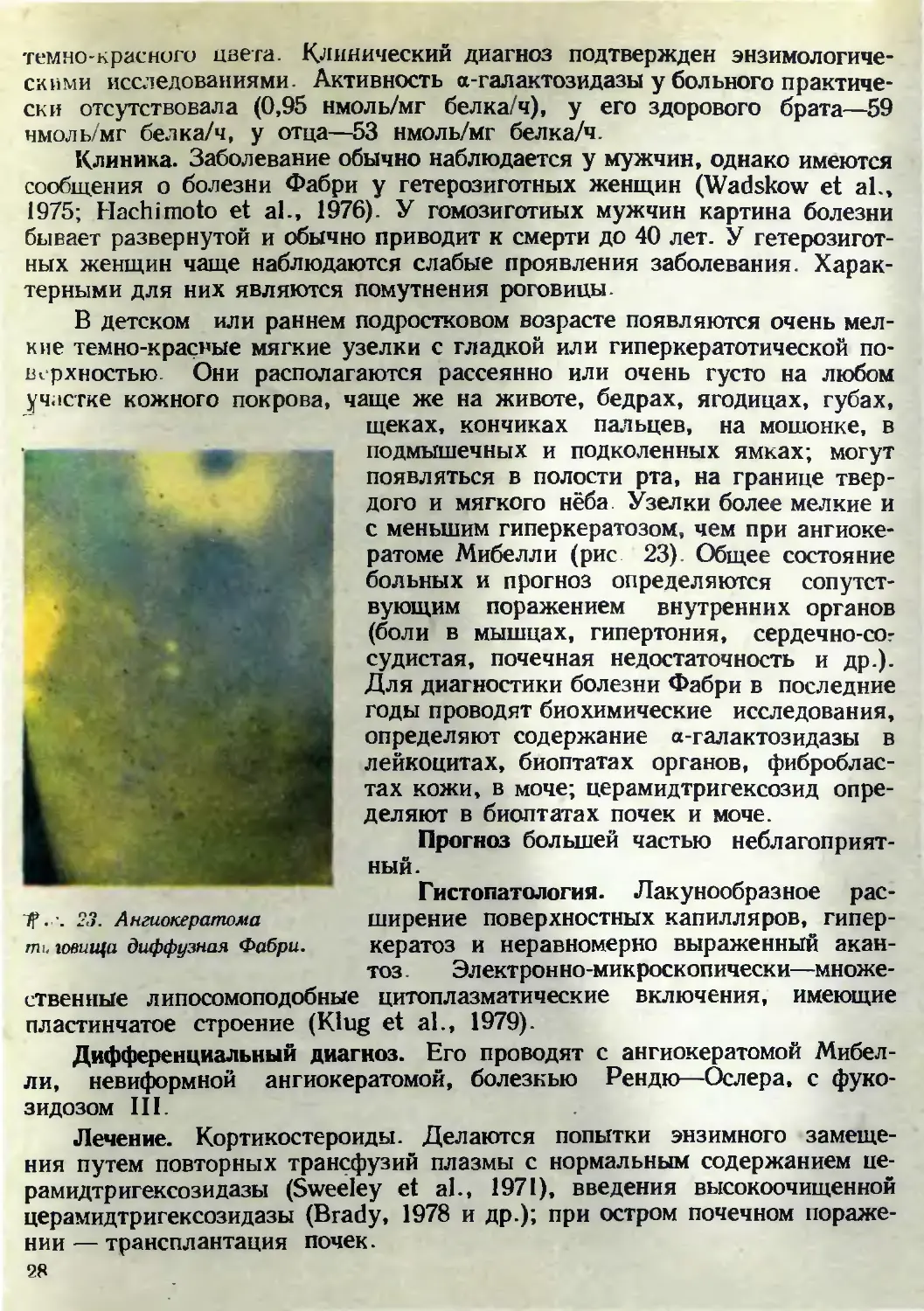
В детском или раннем подростковом возрасте появляются очень мел-
кие темно-красные мягкие узелки с гладкой или гиперкератотической по-
верхностью. Они располагаются рассеянно или очень густо на любом
участке кожного покрова, чаще же на животе, бедрах, ягодицах, губах,
щеках, кончиках пальцев, на мошонке, в
ТР..-. 2,3. Ангиокератома
т говиша диффузная Фабри.
подмышечных и подколенных ямках; могут
появляться в полости рта, на границе твер-
дого и мягкого нёба. Узелки более мелкие и
с меньшим гиперкератозом, чем при ангиоке-
ратоме Мибелли (рис 23). Общее состояние
больных и прогноз определяются сопутст-
вующим поражением внутренних органов
(боли в мышцах, гипертония, сердечно-сог
судистая, почечная недостаточность и др.).
Для диагностики болезни Фабри в последние
годы проводят биохимические исследования,
определяют содержание а-галактозидазы в
лейкоцитах, биоптатах органов, фиброблас-
тах кожи, в моче; церамидтригексозид опре-
деляют в биоптатах почек и моче.
Прогноз большей частью неблагоприят-
ный.
Гистопатология. Лакунообразное рас-
ширение поверхностных капилляров, гипер-
кератоз и неравномерно выраженный акан-
тоз. Электронно-микроскопически— множе-
ственные липосомоподобные цитоплазматические включения, имеющие
пластинчатое строение (Klug et al., 1979).
Дифференциальный диагноз. Его проводят с ангиокератомой Мибел-
ли, невиформной ангиокератомой, болезнью Рендю—Ослера, с фуко-
зидозом III.
Лечение. Кортикостероиды. Делаются попытки энзимного замеще-
ния путем повторных трансфузий плазмы с нормальным содержанием це-
рамидтригексозидазы (Sweeley et al., 1971), введения высокоочищенной
церамидтригексозидазы (Brady, 1978 и др.); при остром почечном пораже-
нии — трансплантация почек.
28
ЛИТЕРАТУРА
виоершайн I- Я- биохимические основы гликозидозов.— М.: Медицина, 1980.—
288 с.
Галлер Г., Ганефельд М-, Яросс В. Нарушения липидного обмена. Диагностика.
Клиника. Терапия.— М.: Медицина, 1979.— 192 с.
Трофимова И. Б. Айвазян А. А. К вопросу о диффузной ангиокератоме Фабри.
Вести, дерматол.' и венерол., 1983. 1, 54—57.
Bernheimer. Angiokeratoma corporis diffusum universale Fabry.— Hautarzt, 1978,
29, 10, 554—554.
Brady R. O. Spingolipidosis.— Ann. Rev. Biochem., 1978, 47, 687—713.
Dooretzky L., Fisher В. K- Fucosidosis.— Int. J. Dermatol., 1979, 18, 3, 213—216.
Hashimoto K., Lieberman Ph., Lamkin N. Angiokeratoma Corporis Diffusum (Fabry
Disease). A Lysosomal Disease.—Arch. Dermatol., 1976, 112, 1416—1423.
Klug H., Zabel K-, Eoens U. Morbus Fabry: Klinische, biochemische und electro-
nenmikroskopische Untersuchungenl.— Derm. Mschr., 1979, 165, 1, 46—54.
Wadkoo S., Andersen V-, Kobayasi T. et al. On the diagnosis of Fabry’s disease.—
Acta Dermatovener., 1975, 55, 5, 363—366.
Gokel J., Hubner G. Zur Morphologie des M. Fabri—zwei ungewohnliche Beobach-
tungen, —Verb. Dtsch. Ges. Pathol. 66 Tag., Gottingen, 1—5 Juni, 1982, Stuttgart, NewJork,
1982, 303—307.
АНГИОКЕРАТОМА МОШОНКИ (ВУЛЬВЫ) (angiokeratoma scroti
s. vulvae).
Описана Fordyce в 1895 г.
Этиология и патогенез. Является пороком развития. Генетическая
обусловленность неясна. Характер доброкачественный.
Клиника. Ангиокератома мошонки встречается относительно часто,
обычно появляется в среднем возрасте, хотя описаны случаи более раннего
появления. Ангиокератома вульвы встречается редко (Verbov и Manglab-
ruks, 1978). На мошонке, реже на половом члене, редко на больших поло-
вых губах, в области бедер появляются отдельные или множественные узел-
к и буро-красного цвета, диаметром 2—5 мм с гладкой или гиперкератотиче-
ской поверхностью. Иногда больные жалуются на зуд, однако это не харак-
терно. При почесывании ангиокератомы могут кровоточить и даже быть
причиной небольшой анемии (Singh, 1974). Различные варианты заболева-
ния-приведены на рисунках 24, 25, 26.
Прогноз благоприятный.
> Гистопатология. Лакунарные расширения капилляров в верхней части
дермы. Эндотелий уплощен. Эпидермис над ними может быть истончен,
иногда в состоянии умеренно выраженного акантоза.
Дифференциальный диагноз. Проводят с сенильными ангиомами,
другими ангиокератотическими изменениями.
Лечение. Наиболее крупные или кровоточащие ангиокератомы могут
быть подвергнуты диатермокоагуляции.
29
ЛИТЕРАТУРА
Verbov J., Ma.'.glabruks К. Angiokeratoma of vulva. — Dcrmatologia., 1978, 156-
5, 296—298.
АНГИОКЕРАТОМА ТУЛОВИЩА ОГРАНИЧЕННАЯ НЕВИФОРМ-
НАЯ (angiokeratoma corporis circumscriptum naeviforme).
Синоним: ангиокератотический невус.
Заболевание описал Fabry в 1915 году.
Этиология и патогенез. Заболевание относят к порокам развития,
формирующимся в эмбриональном периоде. Оно не связано с энзимными
нарушениями и отделено от диффузной ангиокератомы туловища Fabry.
Клиника. Изменения кожи большей частью имеются уже при рожде-
нии, но изредка могут появляться в детском иди даже во взрослом возрасте.
Hundeiker (1978) наблюдал появление невиформной ангиокератомь в пу-
30
Рис. 27. Ангиокератома
туловища ограниченная
невиформная.
бертатном периоде. Она может существовать самостоятельно или быть
частью синдрома, например, синдрома Клиппеля—Тренонея—Вебера (Nodi,
1960; Schnyder, 1963; Schimpf и Wehberg, 1969 и др ), синдрома Штурге—
Вебера—Краббе (Dammert, 1965 и др.). Может сочетаться с кавернозной
гемангиомой (Scholz и Sebastian, 1971). Knoth и соавт (1963) описали со-
четание у одного пациента невиформной ангиокератомы с naevus flamnieus
и подложной кавернозной гемангиомой, у двух других больных — с под-
кожной кавернозной гемангиомой,
капиллярной и кавернозной эпиду-
ральной гемангиомой, располагав-
шейся на уровне V—VII грудных
позвонков. Эти наблюдения свиде-
тельствуют о том, что невиформная
ангиокератома может быть частным
проявлением системных пороков
развития сосудистой системы
Встречается редко. Располагает-
ся на нижних, реже на верхних ко-
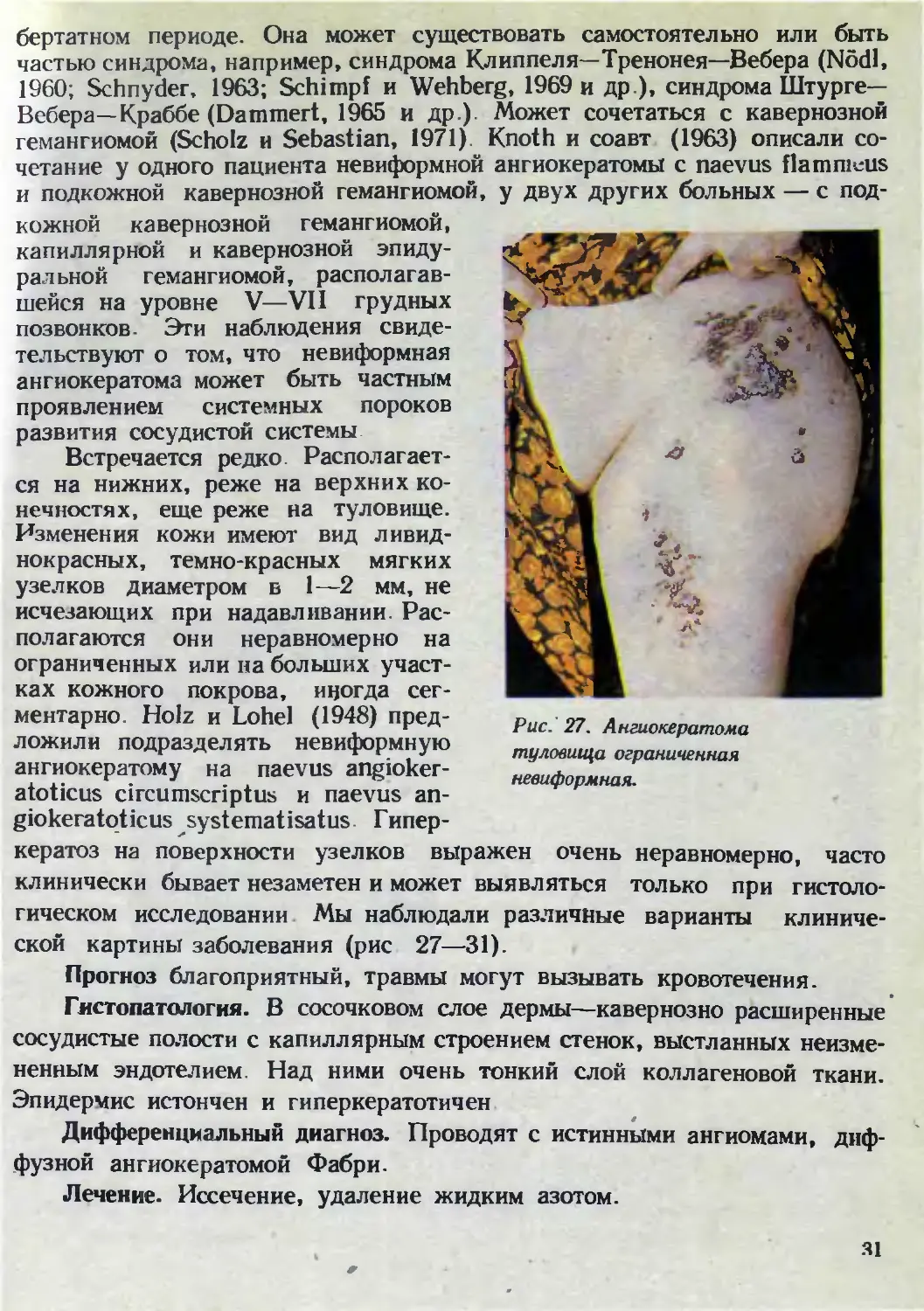
нечностях, еще реже на туловище.
Изменения кожи имеют вид ливид-
нокрасных, темно-красных мягких
узелков диаметром в 1—2 мм, не
исчезающих при надавливании. Рас-
полагаются они неравномерно на
ограниченных или на больших участ-
ках кожного покрова, иногда сег-
ментарно. Holz и Lohel (1948) пред-
ложили подразделять невиформную
ангиокератому на naevus angioker-
atoticus circumscriptus и naevus an-
giokeratoticus systematisatus. Гипер-
кератоз на поверхности узелков выражен очень неравномерно, часто
клинически бывает незаметен и может выявляться только при гистоло-
гическом исследовании Мы наблюдали различные варианты клиниче-
ской картины заболевания (рис 27—31).
Прогноз благоприятный, травмы могут вызывать кровотечения.
Гистопатология. В сосочковом слое дермы—кавернозно расширенные
сосудистые полости с капиллярным строением стенок, выстланных неизме-
ненным эндотелием. Над ними очень тонкий слой коллагеновой ткани.
Эпидермис истончен и гиперкератотичен
Дифференциальный диагноз. Проводят с истинными ангиомами, диф-
фузной ангиокератомой Фабри.
Лечение. Иссечение, удаление жидким азотом.
Я1
Рис. 29. Ангиокераг.ома
туловища ограниченная невифермная.
Рис. 28. Ангиокератома
туловища ограниченна
невиформная.
Рис. 31. Ангиокератома
невиформная
ЛИТЕРАТУРА
Рис. 30. a -гиокерагпома
туловища ограниченная
невиформная.
Hundeiker М. Systematik der angiektatischen und angiokeratotischen Navi. —Haut-
arzt, 1978, 29, 511—517.
Odeh F. Angiokeratoma corporis circumscriptum naeviforme mit \ngiodysplasie
(Klippel-Trenaunay-Weber Syr drom). — Z. Hautkr., 1979, 54, 15, 701—706.
32 ' *
ЛИТЕРАОУРА
АНГИОКЕРАТОМА СОЛИТАРНАЯ ПАПУЛЕЗНАЯ (angiokeratoma
solitarum papillosum).
Синонимы: тромботическая ангиэктазия, капиллярная аневризма ко-
жи.
Этиология и патогенез. В основе заболевания лежат субэпидермаль-
ные расширения капилляров сосочков дермы, обусловленные предрас-
положением; часть эктазий, преимущественно окруженных эпидермисом,
тромбированы. В развитии папулезной ангиокератомы может играть роль
травма.
Клиника. Папулезная ангиокератома выглядит как ежевикоподоб-
ная опухоль. Она может появляться в любом возрасте. Kalkoff (1974) пред-
ложил назвать ее множественная тромботическая ангиэктазия для того,
чтобы оттенить клинические особенности и для отличия от невиформной
ангиокератомы. Он считает название ангиокератома неудачным потому,
что кератоз клинически может быть не выражен. Эго—более поздние, часто
множественные манифестации, размер которых зависит от тромботических
изменений, экстравазатов, эритроцитов и гемосидериновой пигментации.
Папулезная высыпная ангиокератома появляется в виде плоской или
клумбообразной черноватой опухоли, окаймленной узелками. Поверхность
ее обычно грубозернистая, неправильно деформированная с равномерной и
крапчатой пигментацией. Локализация может быть различной. Из 116
случаев Imperial и Helwig (1967) в 7% очаги располагались на голове, в
13%—на руках, в 56%—на ногах, в 11%—на мужских гениталиях У
83% пациентов были одиночные очаги, у 17%—множественные. У боль-
ных с множественными очагами они располагались в локтевых сгибах,
на затылке, тыле кистей. Среди 5 больных (4 женщины, 1 мужчина), наблю-
давшихся Kalkoff (1974), у двух папулезная ангиокератома располагалась
на пальцах рук. Цвет варьирует от красного до фиолетового, коричневого
или черного. Величина от 2 до 10 мм в диаметре. Нередко вокруг папулез-
ной ангиокератомы наблюдается коричневатый ободок, что может созда-
вать сходство с меланомой.
После иссечения отдельных очагов рецидивы, как правило, не насту-
пают. На других участках кожи новые очаги могут появляться.
Гистопатология. Расширение капилляров сосочкового слоя дермы в
форме лакун; акантоз со значительным удлинением эпидермальных отрост-
ков, которые, охватывая лакуны, симулируют образование «внутриэпидер-
мальных» кровяных кист, примерно в 1/3 случаев в эктазиях имеются
тромбы, организовавшиеся или находящиеся в стадии организации. В
подсосочковом слое дермы сосуды не изменены.
Дифференциальный диагноз. В первую очередь его нужно проводить
с меланомой, что имеет наибольшее значение для практики; с ангиогистио-
цитомой, ангиофибромой, пиогенной гранулемой.
Лечение: хирургическое иссечение, диатермокоагуляция.
3 Клиническая дерматология 33
ЛИТЕРАТУРА
Kalkoff К. W. Zur Differentialldiagnoee Angtektasia eruptiva thrombotica (syn. An-
giokeratoma, Thrombosed Angioma, L’angiome noir) und malignes Melanom. — Derm,
Mschr., 1974, 160, 8, 621—630.
АНГИОМАТОЗ НАСЛЕДСТВЕННЫЙ СЕМЕЙНЫЙ ГЕМОРРАГИЧЕ-
СКИЙ (angiomatosis haemorrhagica hereditaria et familiaris).
Синонимы: болезнь Ослера—Рендю—Вебера, наследственная геморра-
гическая телеангиэктазия.
Rendu (1896) описал сочетание множественных телеангиэктазий с но-
совыми кровотечениями. До него Rabington (1865) первым сообщил о чле-
нах одной семьи, страдавших рецидивирующими кровотечениями из носа,
a Legg (1876) описал семейные телеангиэктазии. Osler (1901) выделил на-
следственные геморрагические телеангиэктазии в самостоятельный синдром.
Этиология и патогенез. Аутосомно-доминантное заболевание. Имеются
сообщения о семьях, в которых оно обнаружено в нескольких поколениях
(Камышанский О. А. и соавт., 1979 и др.). В основе его лежит врожден-
ная слабость мезенхимы. Hapes (1909), И. А. Кассирский, Г. А. Алексеев
(1970) установили дефект мышечного и эластического слоев сосудов.
Клиника. Встречается редко; болеют как мужчины, так и женщины.
Первые симптомы болезни чаще появляются во втором десятилетии жизни
и выражаются частыми повторными кровотечениями, обычно из носа. В
дальнейшем на коже и на слизистых оболочках появляются телеангиэк-
тазии, паукообразные, сосудистые невусы и мелкие ангиомоподобные об-
разования величиной от булавочной головки до 0,5—1 см в диаметре. Они
располагаются на лице, преимущественно в скуловых областях, носогуб-
ных складках, на лбу, подбородке, ушных раковинах, в меньшей степени—
на открытой части груди, руках, где они чаще локализуются на сгибатель-
ной поверхности пальцев, под ногтями- Слизистая оболочка рта (особенно
губ и языка), носа вовлекаются почти постоянно. Сосудистые узелки могут
быть также в легких, желудочно-кишечном тракте, в мочевом пузыре и в
других органах — мягкой мозговой оболочке, ретине, печени, селезенке,
почках, и др. Сосудистые узелки, телеангиэктазии склонны к кровотечени-
ям, особенно в пубертатном периоде, во время беременности, инфекцион-
ных заболеваний. Повторны^ кровотечения могут приводить к развитию
анемий; сильные кровотечения могут быть опасны для жизни. В связи с
частыми кровотечениями из языка, губ следует проявлять большую осто-
рожность при стоматологических манипуляциях, даже при чистке зубов.
Свертываемость крови, количество тромбоцитов, ретракция сгустка, время
кровотечения при болезни Рендю—Ослера нормальные. Спектр клинических
проявлений заболевания может быть весьма разнообразен и зависит от
степени и характера вовлеченных в процесс органов. Патологические из-
менения с возрастом усиливаются.
Прогноз сомнительный из-за опасности больших кровотечений.
Гистопатология. В герхней части дермы (слизистой оболочки)—мно-
жественные мешковидные расширения капиллярных сосудов.. В них на-
рушена эластическая архитектоника.
о4
Дифференциальный диагноз. Его следует проводить с телеангиэкта-
зиями при циррозе печени, гемофилии, стромбоцитопенической пурпурой,
болезнью Фабри.
Лечение. Симптоматическое.
ЛИТЕРАТУРА
Виноградов А. В. Дифференциальный диагноз внутренних болезней, М., 1980,
г. 2, 607—608.
Камышинский О. А., Кирсанова Н. А., Шабловская Л. М. Болезнь Рендю—Осле-
ра в четырех поколениях.— Тер. арх., 1979, 12,105—106.
АНЕТОДЕРМИЯ (anetodermia).
Синонимы: пятнистая атрофия кожи, пятнистая эритематозная атро-
фодермия.
Анетодермия (anetos—пустой)—своеобразная разновидность атрофии
кожи, нередко существующая одновременно с другими формами идиопатиче-
ской атрофии кожи.
Анетодермия эритематозная (anetodermia erythematosa) впервые опи-
сана и подробно изучена Jadassohn в 1891 г. В отечественной литературе
имеются лишь единичные сообщения об этой форме атрофии кожи (Воро-
нов Д. Л., 1927; Ландесман А. У. и Эйнох А. И., 1930; Арешев В. Г.,
1926; Болдырева А. М., 1928; Беньмович Е. Б. н Машкиллейсон Л. Н.,
1928; Торсуев Н. А., 1941).
Этнология и патогенез. Большинство авторов указывает на причин-
ную роль эндокринных (щитовидная железа, яичники) и нервных нару-
шений (Акопджанянц С., 1929; Пашков Б. М., 1931; Торсуев Н. А., 1941;
Oppenheim, 1906; Pautrier, 1952, Van Brabandt, 1982 и др.).
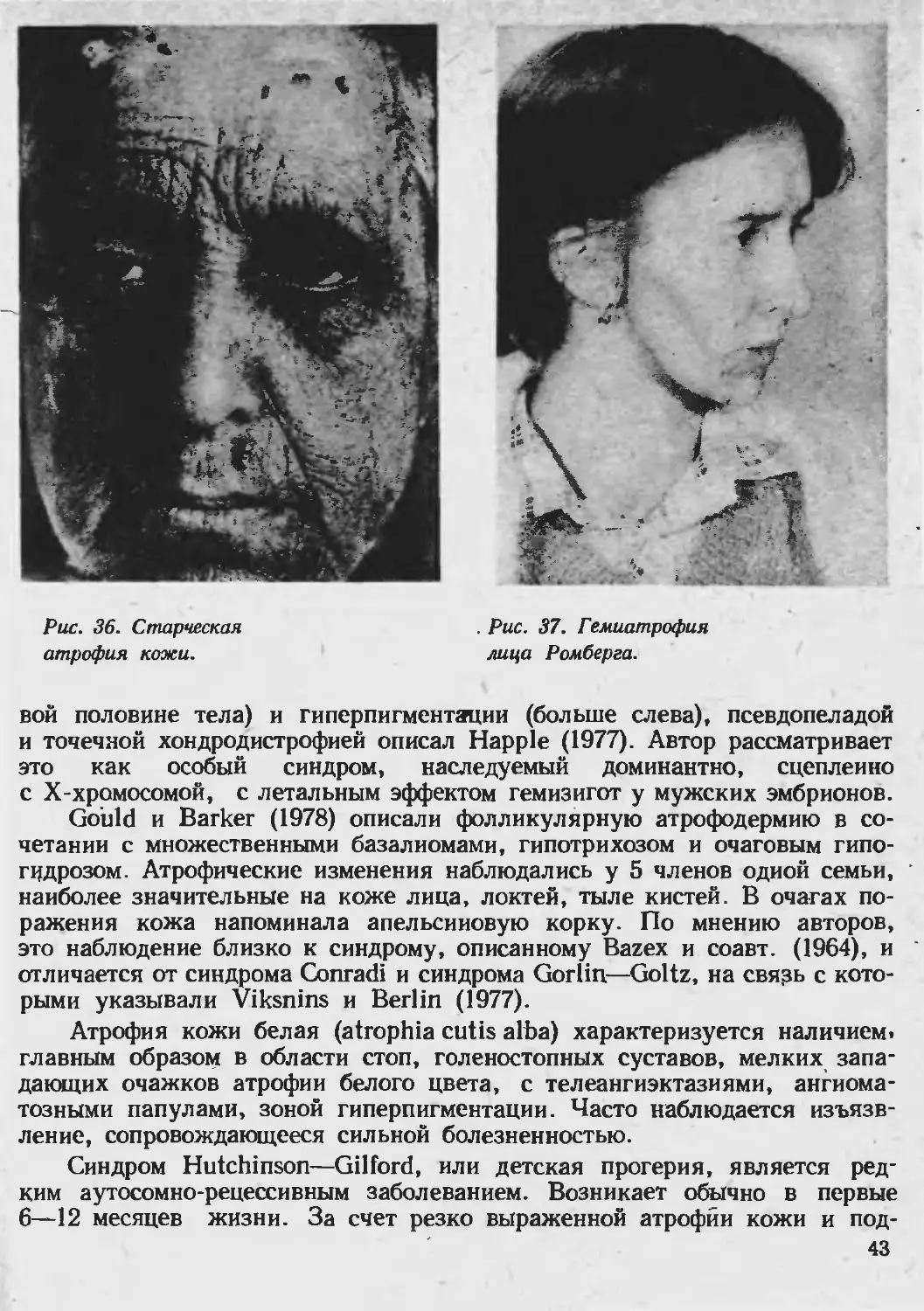
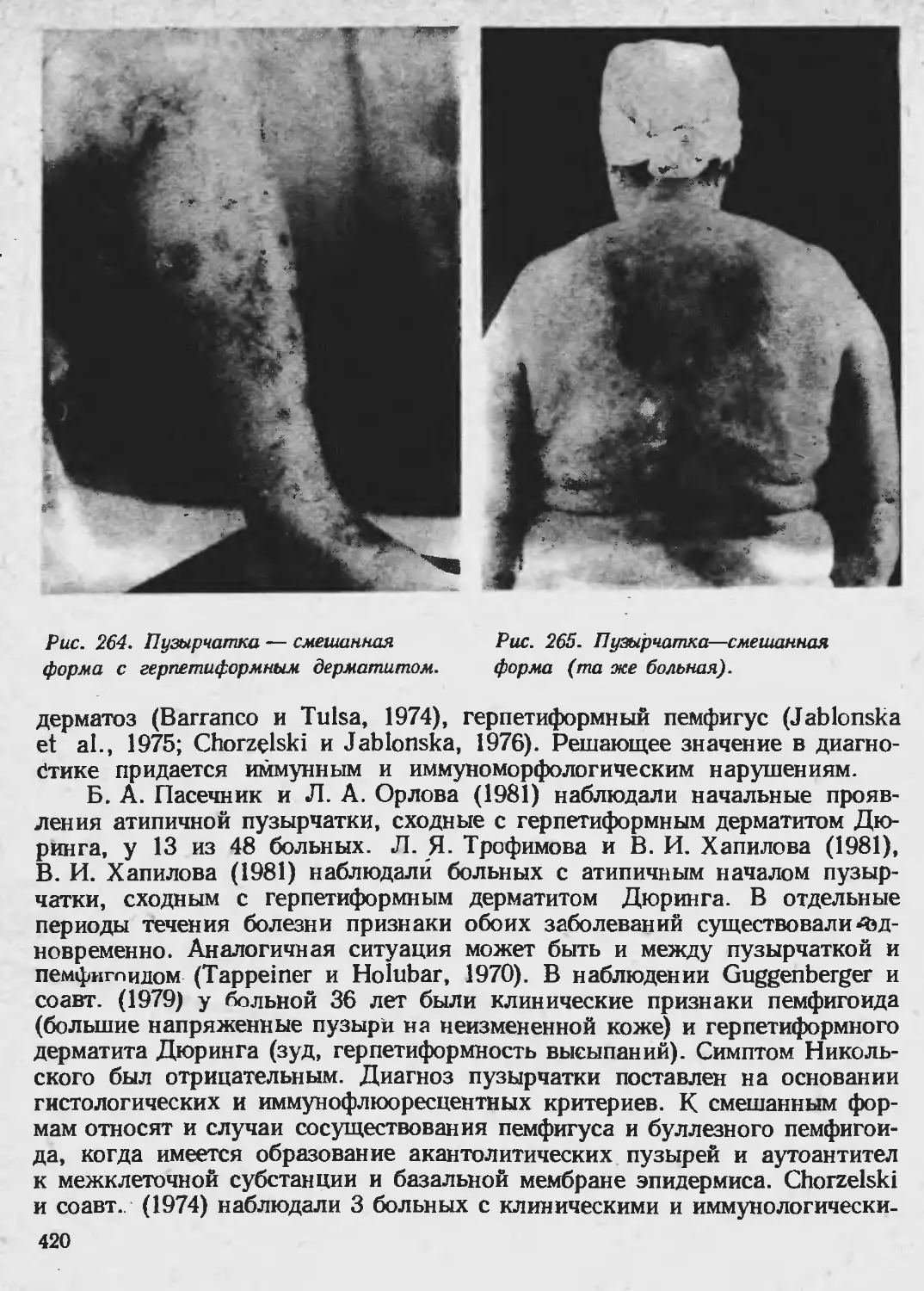
Клиника. Классический тип анетодермии Ядассона характеризуется
появлением единичных или множественных пятен ливидно-розового или
желтовато-розового цвета, неправильно округлых очертаний до 1—2 см в
диаметре, локализующихся чаще иа туловище, верхних конечностях и
лице. Постепенно, без каких-либо субъективных ощущений на месте воспа-
лительного пятна развивается атрофия, которая начинается в центре пят-
на. Кожа на этих местах становится бледной, морщинистой, нацрминая
смятую папиросную бумагу; очаг слегка выступает над уровнем окружаю-
щей кожи (рис. 38). В дальнейшем, на местах первичных элементов остают-
ся белые блестящие, рубцевидные пятна, которые могут переходить в
мягкие грыжеподобные выпячивания кожи, дающие при пальпации впе-
чатление пустоты (палец легко «проваливается» вглубь). Болезнь поражает
чаще женщин преимущественно молодого возраста—от 20 до 30 лет (Mil-
ler, 1979). Данные параклинических лабораторных исследований остаются
в пределах нормы.
Д. Л. Воронов (1927) демонстрировал на заседании Московского об-
щества дермато-венерологов больную 35 лет анетодермией в сочетании с
идиопатической прогрессирующей атрофией кожи нижних конечностей и
поясницы. Тип анетодермии Швеннингера и Буцци был описан этими ав-
торами под названием «множественные доброкачественные опухолевидные
разрастания кожи». Клинически он проявляется грыжеподобно выпячи-
з* 35
вающимися очагами атрофии, преиму-
щественно на спине и верхних конеч-
ностях. Подчеркивается «опухолепо-
добность» и голубовато-белый цвет
очагов атрофии. В отличие от клас-
сического типа анетодермии Ядассона
атрофические очаги значительно боль-
ше выстоят над коружающей кожей,
на их поверхности могут быть телеан-
гиэктазии, всегда отсутствует первая
воспалительная стадия. Chapol et al.
(1977) описали больную 83 лет с узло-
ватым первичным амилоидозом кожи,
у которой имелись несколько ането-
дермических поражений типа Швен-
нингера и Буцци
Исключительно редко анетодер-
мия развивается на месте волдырей
(уртикарный тип Pellizzari). Высыпа-
ния, как правило, не сопровождаются
субъективными ощущениями, образу-
ются мешочкоподобные выпячивания,
при их пальпации палец как бы про-
Рис. 32. Анетодермии. валивается в пустоту, после чего по-
верхность бляшки остается на неко
торое время слегка вдавленной. Течение анетодермии хроническое.
Гистопатология. Главные изменения локализуются в дерме, эпидер
мне поражается мало. Основной гистологический признак—гибель эласти-
ческой, а иногда и коллагеновой ткани (Беньямович Е. Б. и Машкиллей
сон Л. Н., 1928). В поверхностных слоях дермы незначительный инфиль-
трат из лимфоцитов, единичных плазматических клеток и фибробластов.
Подкожная жировая клетчатка не вовлекается в процесс. Н. А. Торсуев
(1941) обнаружил утолщение соединительнотканной оболочки нервных
пучков. Colombo и соавт. (1974) при ультраструктурном исследовании ане
тодермии типа Швеннингер-Буцци обнаружили изменения в эластиче
ской ткани. Коллагеновые волокна оставались неповрежденными, но от-
мечались большие вариации их диаметров в зоне сильных поражений
(3000—12000 А). В эластике были обнаружены структурные аномалии,
которых не было вне очага сильного повреждения.
Дифференциальный диагноз. Проводят с рубцовыми атрофиями,
атрофическими родимыми пятнами, вторичными пятнистыми атрофиями
кожи, развивающимися на местах предшествующих других дерматозов.
Лечение. При активных прогрессирующих формах анетодермии ре
комендуются общеукрепляющие средства, витамины А, Е. Reiss et al.
(1973) сообщили о хороших терапевтических результатах, достигнутых
при лечении анетодермии аминокапроновой кислотой.
36
ЛИТЕРАТУРА
Miller IF., Ruggles C., Rist T. Anetoderma.— Intern. J. Dermatol., 1979, 18, 1,
43—45.
Van Brabandt S. Anetoderma of Jadassohn. Dermatologica (Basel), 1982; 165, 5,
513—514.
АТРОФИЯ КОЖИ (atrophia cutis)
Этнология и патогенез. Гетерогенная группа дерматозов, общим для
которых является выраженное в разной степени истончение эпидермиса и
дермы, а в некоторых случаях и подкожной клетчатки и глубже лежащих
тканей, первичного или вторичного происхождения, этиология и патогенез
которых неодинаковы при разных формах и в большинстве случаев недо-
статочно выяснены. При старческой атрофии кожи—это процесс физиоло-
гический, хотя и может быть ускорен неблагоприятным влиянием различ-
ных факторов (избыточная длительная инсоляция, истощение, эндокрин-
ные нарушения и др.) В основе лежит нарушение трофики, преобладание
процессов диссимиляции, снижение активности цитоплазматических фермен-
тов При полосовидной атрофии значение имеют обменные (ожирение) и
гормональные (в первую очередь синдром Иценко—Кушинга) нарушения,
беременность, острые и хронические инфекции, особенно туберкулез, тиф,
скарлатина. Hauser (1958) рассматривает полосовидные атрофии кожи как
проявление олигосимптомного синдрома Кушинга. Описано развитие поло-
совидной атрофии кожи после наружного применения под окклюзионную
повязку кортикостероидных мазей или при внутриочаговом введении кор-
тикостероидов (Epstein et al., 1963; Chernosky и Knox, 1964; Kikuchi и
Horikawa, 1974 и др.;. Идиопатическую прогрессирующую атрофию кожи
связывают этиологически со снирохемой из рода Borrelia, которая
передается с укусами клещей. Белая атрофия кожи (atrophie blanche Mi-
lian) рассматривается как результат поражения поверхностных сосудов
кожи (Gray et al., 1966; StevanoviC, 1974 и др.). Сосудистые изменения мо-
гут лежать в основе и пятнистой атрофии кожи (Newmann и Vacatco, 1959;
Cramer, 1963). В качестве основных причин возникновения прогрессирую-
щей гемиатрофии лица указываются (Ehlers, 1961; Harnack, 1962 и др.)
нарушения трофики, обусловленные изменениями нервной системы (поро-
ки развития, инфекции, энцефалиты, черепно-мозговые травмы, повреж-
дения симпатических ганглиев, особенно шейных, органические и сосуди-
стые изменения мозга, опухоли).
Атрофодермия Пазини—Пьерини чаще считается разновидностью по-
верхностной склеродермии (Машкиллейсон Л. Н., 1965; Федорова Е. Г.,
1978; Брайцев А. В. и Марзеева Г. И., 1973; Jablonska и Szczepanski,
1962, и др.), чем собственно атрофией кожи (Разнатовский И. М., 1967
и др.). В развитии некоторых форм атрофии основную роль играют наслед-
ственные факторы. Это касается прежде всего таких заболеваний как синд-
ромы Werner, Rothmund, Thomson, Huriez, Hutohinson—Gilford. В случаях
детской прогерии, описанных Д. Д. Соколовым (1955), предполагалась
роль сифилитической инфекции. Наследственности придается значение и
37
в случаях червеобразной атрофии кожи, но в ее развитии важна роль и
гормональных нарушений. Особыми вариантами атрофии кожи являются
вызванные экзогенными факторами, включая такие, как генерализованная
или локальная атрофия, индуцированная - кортикостероидами, актиниче-
ские, хронические лучевые повреждения кожи. В развитии стероидной
атрофии кожи основное значение имеют сосудосуживающий эффект,корти-
костероидов, особенно выраженный у фторсодержащих препаратов, тормо-
жение пролиферативно-репаративных процессов, синтеза волокнистых струк-
тур дермы, усиление дистрофических изменений коллагеновых и эласти-
ческих волокон. Вторичные атрофии могут быть следствием самых раз-
ных по своей природе заболеваний (красная волчанка, пиодермия, тубер-
кулез кожи, третичный сифилис и др.).
Клиника. Общим для всех форм атрофий является истончение кожи
и в той или иной степени подкожной клетчатки, а для некоторых из них
(например, для, гемиатрофии прогрессирующей лица, линейной склеро-
дермии типа coup de sabre) и более глубоких тканей. Кожа теряет свою
эластичность, становится вялой, сухой, нередко густо покрыта телеанги-
эктазиями, пигментируется. На участках атрофии возможно развитие
злокачественных новообразований. Полосовидная атрофия кожи (atro-
phia cutis striata) обычно наблюдается у лиц женского пола, но встречается
и у мужчин, возникает е период полового созревания, при беременности,
ожирении. Обычно располагается на животе, бедрах, молочных железах,
пояснице в виде продольных узких полос сначала розовато-синюшного,
затем белесоватого цвета. В редких случаях атрофические полосы могут
располагаться по всему кожному покрову, включая и лицо. Иногда изъ-
язвляются.
Своеобразную полосовидную атрофию (stria migrans) описали Shelley
и Cohen '1964), расположенную поперек спины как след от удара плетью.'
Строфические изменения начались с позвоночника, где было пятно, выз-
ванное инородным телом черного цвета, сходное с татуировкой, и посте-
пенно распространялись в латеральном направлении и в течение 18 ме-
сяцев, заняв всю ширину спины. В качестве возможной причины указы-
вается подъем тяжестей. Сходную форму атрофии наблюдал Carr (1966),
но связывает ее развитие с приемом кортикостероидов.
Локальные атрофии кожи от кортикостероидных мазей развиваются
преимущественно у детей и молодых женщин, как правило, при нерацио-
нальном (длительно, многркратно в течение дня) применении фторсодер-
жащих кортикостероидов (синалар, фторокорт и др.). Эти изменения мо-
гут происходить в эпидермисе или дерме, быть ограниченными, диффузны-
ми или в виде полос (striae rubrae distensae), реже наблюдается глубокая
атрофия, чаще после инъекций, чем от наружного применения (рис. 33).
Кожа выглядит старческой, мелкоскладчатол, напоминает папиросную
бумагу, легко травмируется. За счет просвечивающих сосудов и расшире-
ния капилляров, что наблюдается при более выраженном истончении ко-
жи и более глубоком процессе, кожа приобретает ливидный оттенок (ги-
beosis steroidica), особенно значительный в тех случаях, когда кортико-
38
Рис. $3. Атрофия
ко^и^линейная.
стероидные препараты назначены по
поводу дерматозов, признаком кото-
рых являются телеангиэктазии (на-
пример, розацеа). С нюшный цвет в
очагах атрофии может поддерживать-
ся и за счет вторичной воспалитель-
ной реакции, обусловленной противо-
спалительным действием фтора. Эти
изменения наиболее часты на личе,
шее, верхней части груди. Могут быть
обратимы, • но полностью только при
поверхностной атрофии, захватываю-
щей эпидермис (Schopf, 1972). Очаго-
вые, в том числе полосовидные атро
фии, по данным Goldman и Kitzmiller
(1973), вызванные кортикостероид-
ными мазями, наблюдаются периа-
нально, в подмышечных впадинах,
на кистях. Существенна роль дли-
тельного применения кортикостероидов в развитии так называемой спон-
танной псевдорубцовой звездчатой атрофии кожи (Goldman et al., 1975),
располагавшейся чаще на тыле кистей и разгибательной поверхности
предплечий.
Braun-Falco и Baida (1970), Schopf (1972) считают, что речь идет при
этом не о псеьдорубцовых изменениях, а о настоящих рубцах, которые
развиваются не спонтанно, а под влиянием травмирования атрофически
измененной кожи. Наблюдается этот вид атрофии преимущественно у по-
жилых л_?п--й Schopf описал развитие очгговой атрофии кожи, сходной
с анетодермией после трехнедельного смазывания очагов разноцветного
лишая мазью, соде эжгшей флуоцинолон-ацетоних С точки зрения Marge-
scu (1983), для уменьшения вероятности развития атрофических изменений
более рациональным было бы применение кортикостероидных мазей в ве-
чернее время, когда пролиферативная активность клеток эпидермиса мини-
мальна. Глубокие атрофии развиваются обычно после подкожных, внутри-
очаговых инъекций кортикостероидных препаратов В их зоне может одно-
временно наблюдаться уплотнение, что придает, особенно при наличии
ливидной окраски, сходство со склеродермией (Nasemann, 1971). Чаще (Na-
semann (1970) атрофические изменения этого типа наблюдаются в области
кистей, локтевых суставов, волосистой части головы. Они могут развиться
через несколько месяцев после окончания лечения, особенно у молодых
лиц (рис. 34). Поэтому Nasemann не рекомендует проводить это лечение
цетям до 12 - летнего возраста без особых на то показаний. По наблюдениям
автора, атрофические изменения глубоких тканей разрешались бесследно
через 1—3 года.
А. Л. Машкиллейсон (1965) рекомендует внутриочаговое введение кор-
гикостероидов при ряде дерматозов, но только в том случае, если обычные
39
методы оказались неэффективными. Описано (Schopf, 1972) развитие атро-
фии подкожной клетчатки у четырехлетней девочки, которой ежедневно
по поводу келоида в течение 6 месяцев под окклюзионную повязку на ночь
применялась фторсодержащая кортикостероидная мазь. Пораженная ко-
нечность была в объеме меньше здоровой' на 4 см. После отмены лечения
состояние значительно улучшилось.
Особым вариантом кортикостероидной атрофии является универсаль-
ная (cutis praesenilis medicamentosa), когда весь кожный покров истон-
чен, приобретает синюшные оттенки за счет просвечивающих сосудов,
множественных телеангиэктазий. Нередки кровоизлияния (Winer et al.,
1962). Впервые эта форма атрофии описана Foldvari (1961). Обычно раз-
вивается при длительном пероральном применении кортикостероидных
препаратов. Описано (Иванов Н. А., 1963 и др.) развитие рубцовых изме-
нений на месте разрешившихся пузырей в процессе кортикостероидной
терапии больной пузырчаткой.
Развитию атрофических изменений кожи при идиопатической про-
грессирующей атрофии кожи (atrophia cutis idiopathica progressiva) пред-
шествует отечно-воспалительная стадия. Поражаются в основном конеч-
ности, но в процесс может вовлекаться и туловище. Постепенно воспале-
ние стихает, окраска приобретает синюшные тона, кожа истончается, по
внешнему виду напоминая папиросную бумагу (симптом А. И. Поспелова),
пигментируется, появляются множественные телеангиэктазии. За счет
выраженного уменьшения подкожной клетчатки просвечивают крупные
сосуды. Наряду с атрофическими изменениями могут быть склеродермо-
подобные, фиброзные уплотнения. Возможно изъязвление, при многолет-
нем существовании—развитие злокачественных опухолей. В очагах иди-
опатической атрофии кожи могут развиваться лимфопролиферативные за-
болевания, лимфоцитома, лимфосаркома и другие опухолевые процессы
(Knoth, 1960 и др.).
При атрофодермии Пазини—Пьерини (atrophodermia idiopathica Ра-
sini—Pierini) поражается наиболее часто кожа туловища, особенно спины
вдоль позвоночника, где обнаруживаются немногочисленные, обычно
крупные слегка западающие участки, вначале розовато-синюшные, затем
гиперпигментированные, округлых или овальных очертаний, как прави-
ло, без уплотнения и сиреневого кольца эритемы по периферии, свойствен-
ного склеродермии (рис. 35). Одновременно могут быть типичные очаги
поверхностной склеродермии, которые развиваются в зоне атрофически
измененной кожи или на внешне неизмененных участках кожного покрова
(Кее et al., 1960). Возникает чаще у лиц женского пола в юношеском или
молодом возрасте. После медленного прогрессирования остается в стацио-
нарном состоянии. Weiner и Gaut (1959) наблюдали атрофодермию Пази-
ни—Пьерини у 2 братьев. У обоих через 6 недель на месте биопсии разви-
лись гипертрофические рубцы.
Поздние рентгеновские повреждения кожи характеризуются очагами
истончения кожи, дисхромией, множественными телеангиэктазиями, пой-
килодермией, часто ограниченными кератозами, которые склонны к пере-
40
рождению. Landhaler et al. (1983) иа
блюдали выраженную рентгеиодерму
у 6,5% больных, леченных мягкими
рентгеновскими лучами по поводу
базалиом век. Могут развиваться в
процессе лучевой терапии, после дли-
тельного воздействия малых доз рент-
генозских лучей, особенно при на-
рушении техники безопасности с ис-
точниками ионизирующей радиации.
Для псевдопелады Брока харак-
терно наличие на волосистой части
головы очагов рубцевидной атрофии
с алопецией, различных, обычно не-
равномерно округлых очертаний, час-
то сливающихся между собой. По
их периферии может наблюдаться ие-
большия перифолликулярная воспа-
Рис. 34. Атрофия
кожи стероидная.
лительная реакция. Наиболее вероятно, что это не самостоятельное забо-
левание, а конечный этап многих дерматозов, в первую очередь красного
плоского лишая. Ronchese (1966) указывает на близость псевлопелады к
идиопатической прогрессирующей атрофодермии Пазини—Пьерини и счи-
тает, что ее следовало бы рассматривать как форму этой атрофодермии,
локализующуюся на голове.
Кожа при старческой атрофии (рис. 36) суха, истончена, морщиниста,
покрыта мелкоотрубевидными чешуйками, нередко пигментирована, осо-
бенно на открытых участках. Окраска приобретает цианотический оттенок,
усиливающийся за счет просвечивающих сосудов. Нередко больные стра-
дают от мучительного зуда. Своеобразным вариантом актинически инду-
цированной атрофии кожи является cutis rhomboidalis nuchae, клинически
проявляющейся выраженной складчатостью кожи, ромбовидной за счет
пересечения косо идущих глубоких борозд. Нередко сочетается с коме-
донами, множественными кистами, эластоидозом в области лица (синдром
Favre—Racoushot).
Прогрессирующая гемиатрофия лица (hemiatrophia faciei progressiva)
клинически характеризуется постепенной, как правило, односторонней
атрофией кожи, подкожной клетчатки, а также мышечной и костной тка
ии, захватывающей скуловую область и нижнюю челюсть, кожу лба
(рис. 37). В редких случаях может распространиться на шею и область
плеча, туловище. Отмечается западение, асимметрия лица, кожа в оча
гах поражения может быть уплотнена, но чаще истончеьа,
суха, наблюдается дисхромия. Брови и ресницы выпадают. Раз
вивается обычно в детском или юношеском возрасте, изредка
у взрослых. Может сочетаться с полосовидной склеродермией еп coup de
sabre, особенно если начинается с детства. Кожа в этих случаях уплотнена,
спаяна с подлежащими тканями, часто пигментиоована. Поражение рас-
41
полагается в виде полосы,
захватывающей волосистую
часть головы, кожу лба, но
может спускаться и на ниже-
лежащие участки лица, пора-
жая иногда и слизистую по-
лость рта (Harnack, 1962 и
др.). Атрофические измене-
ния развиваются вторично.
По мнению Harnack (1962),
сочетание этих заболеваний
может свидетельствовать о
том, что они имеют сходство
не только по клиническим,
но и по этиологическим и па-
тогенетическим параметрам.
Основными симптомами
червеобразной атрофии кожи
(atrophodermia vermiculata)
являются симметричные (в
казуистических случаях —
унилатеральные, Rozum et
al., 1971) густо расположен-
ные очажки атрофии на ли-
це, преимущественно на ще-
ках (как исключение, на
Рис. 35. Атрофодермия
Пазини—Пьерини. .
других участках тела) глубиной около 1 мм, а также фолликулярные
роговые пробки, акнеиформные, милиумп'добные высыпания, встречаю-
щиеся по краю и больше в начале заболевания, разделенные узкими по-
лосками нормальной кожи, как бы вкрапленные в нее, что придает оча-
гам «изъеденный», сетчатый характер, сходство с пораженной червоточи-
ной поверхностью дерева или медовыми сотами (рис. 38;. Воспалительная
реакция наблюдается не во всех случаях, более выражена в начальной
стадии. Kooij и Venter (1959) наблюдали эчаги червеобразной атрофии
кожи не только на лице, шее, но и на конечностях, кроме того, обнаруже-
ны болезнь Дауна, врожденный порок сердца. Заболевание развивается в
пред- и пубертатном периоде, реже позднее. Rand и Baden (1983) объе-
диняют червеобразную атрофодермию вместе с ульэритемой и keratosis
follicularis spinulosa decalvans в одну группу—keratosis pilaris atrophi-
cans, учитывая, что эти сочетания имеют такие общие признаки, как
фолликулярный гиперкератоз, воспаление и атрофию. Распространенную
фолликулярную атрофодермию в сочетании с точечным гиперкератозом
и гипергидрозом ладоней и подошв, фолликулярным кератозом наблюда-
ли Braun-Falco и Margescu (1967).
Фолликулярную атрофодермию на тыле кистей и предплечий в соче-
тании с полосовидно расположенными очажками атрофии (больше на пра-
42
Рис. 36. Старческая
атрофия кожи.
Рис. 37. Гемиатрофия
лица Ромберга.
вой половине тела) и гиперпигментации (больше слева), псевдопеладой
и точечной хондродистрофией описал Happle (1977). Автор рассматривает
это как особый синдром, наследуемый доминантно, сцеплеино
с Х-хромосомой, с летальным эффектом гемизигот у мужских эмбрионов.
Gould и Barker (1978) описали фолликулярную атрофодермию в со-
четании с множественными базалиомами, гипотрихозом и очаговым гипо-
гвдрозом. Атрофические изменения наблюдались у 5 членов одной семьи,
наиболее значительные на коже лица, локтей, тыле кистей. В очагах по-
ражения кожа напоминала апельсиновую корку. По мнению авторов,
это наблюдение близко к синдрому, описанному Bazex и соавт. (1964), и
отличается от синдрома Conradi и синдрома Gorlin—Goltz, на свя.зь с кото-
рыми указывали Viksnins и Berlin (1977).
Атрофия кожи белая (atrophia cutis alba) характеризуется наличием*
главным образом в области стоп, голеностопных суставов, мелких запа-
дающих очажков атрофии белого цвета, с телеангиэктазиями, ангиома-
тозными папулами, зоной гиперпигментации. Часто наблюдается изъязв-
ление, сопровождающееся сильной болезненностью.
Синдром Hutchinson—Gilford, или детская прогерия, является ред-
ким аутосомно-рецессивным заболеванием. Возникает обычно в первые
6—12 месяцев жизни. За счет резко выраженной атрофии кожи и под-
43
кожной клетчатки. Ребенок имеет
старческий вид. Кожа становится
тонкой, складчатой, пигментиру-
ется, особейно на открытых,
участках тела, сквозь нее про-
свечивают вены. Волосы резко
редеют, вплоть до тотальной ало-
пеции, ногти истончены, зубы
дистрофичны. Лобные бугры уве-
личены, отмечается черепно-лице-
вая диспропорция, уменьшение
подбородка, выпуклые глаза, что
придает лицу птичье выражение.
Могут быть склеродермоподобные
изменения, чаще на туловище и
бедрах, дистрофические изменения
костей с переломами, атрофии
мышц, периартикулярный фиброз.
В зоне склеродермоподобных из-
менений выявлена гиалинизация
соединительной ткани, выражен-
ное уменьшение подкожно-жиро-
вой клетчатки. Культуру фибро-
Рис. 38. Атрофия бластов получить не удалось
кожи вермикулярная. (Fleischmajer и Nedwich, 1973).
Короткие ключицы, заостренные
пальцы, coxa valga, гипогонадизм; может быть отставание в психическом
развитии (Соколов Д. Д., 1955). Больные рано погибают, в среднем не
доживая до 15 лет, за счет тяжелого атеросклероза.
Гистопатология. Истончение эпидермиса и дермы, дегенерация кол-
лагеновых и эластических волокон. При червеобразной атрофодермии в
эпидермисе роговые пробки и кисты, при идиопатической прогрессирующей
атрофии—в начальной стадии выраженный полосовидный, преимуществен-
но лимфоцитарный инфильтрат, отделенный от эпидермиса узкой полоской
соединительной ткани, в поздних—фиброз.
Дифференциальный диагноз. Его необходимо проводить от склеродер-
мии.
Лечение: витамины, препараты, улучшающие трофику (компламин,
продектин, андекалин и др.), при идиопатической прогрессирующей атро-
фии—антибиотики.
44
ЛИТЕРАТУРА
Ризнашоы:кий И. М. Идиопатическая атрофодермия Пазини—Пьерини. Вести,
дерматол. и венерОл. 1975, 5, 55—58.
Gould D. J-. Barker D. J. Follicular atrophoderma with multiple basal cell carcino-
mas (Bazex). Brit. J. dermatol., 1978, 99, 4, 431—335
Goldman L., Kitzmiller K- W- Perianal atrophoderma from topical corticosteroids.
Arch, dermatol., 1975, 107, 4, 611—612.
Happle R. DermatoTogische Leitsymptome einer Sonderform des Chondrodysplasia
punctata. Hautarzt, 1977, 28, Suppl, J. 2, 260—263
Kikuchi J., Horikawa S. Periiumphatic atrophy of the skin. Arich. dermatol., 1974,
109, 4, 558—559.
Rand-R., Haden H. P. Keratosis follicularis spinulosa decalvans. Report of two ca-
sesand literature review. Arch, dermatol., 1983, 119, 1, 22—26.
БАЗАЛИОМА (basalioma)
Синоним: epithelioma basocellulare.
Этиология и патогенез. Согласно Гистологической классификации
ВОЗ (1980), заболевание представляет собой базально-клеточный рак,
медленно растущий и редко метастазирующий, возникающий в эпидермисе
или волосяных фолликулах, клетки которых сходны с базальными клет-
ками эпидермиса. Однако отнесение базалиомы к раку кожи весьма услов-
но, так как очень редкий переход ее в базальноклеточную карциному на-
блюдается, как правило, под действием сильных канцерогенов, прежде
всего рентгеновских лучей. По мнению А К Апатенко (1973), базалиому
следует рассматривать не как рак или доброкачественное новообразование,
а как особого рода опухоль с местнодеструктивным ростом. Hundeiker и
Petres (1968) полагают, что базалиома занимает промежуточное положение
между невусами и карциномами: с одной стороны, как и рак, она может
быть вызвана различными канцерогенами, с другой—отсутствует склон-
ность к метастазированию. Вопрос о гистогенезе базалиомы не решен.
Многие (Головин Д И., 1958, Вихерт А. М. и соавт., 1973 и др.) придер-
живаются дизонтогенетической теории развития базалиом из первичного
эпителиального зачатка, который может дифференцироваться в направ-
лении различных структур. Lever (1983), ранее придерживающийся этой
теории, допускает возможность, высказанную рядом авторов (Pincus,
1953; Kint, 1974 и др.), возникновения базалиомы из плюрипотентных кле-
ток эпидермиса, которые обладают эволюционными возможностями в сто-
рону волосяных структур, сальных и апокринных желез. Об этом, по его
мнению, может свидетельствовать развитие опухоли после инсоляции и
рентгеновых лучей. О том, что базалиома разнородна, высказывали свое
мнение Gottron (1964), А. К, Апатенко (1973) и другие авторы. Так, А. К-
Апатенко считает, что базалиомы могут развиваться из всех эпителиаль-
ных структур кожи, в том числе из эмбриональных зачатков и пороков
развития. Приведенные материалы показывают, что положения, выдви-
нутые Krompecher в 1903 году о гистогенетической неоднородности база-
лиомы, сохранились до настоящего времени.
45
Рис. 40. Базалиома.
Рис. 39. Базалиома.
Рис 41. Базалиома.
Провоцирующую роль, ПОМИМО
интенсивной инсоляции и лучевой
энергии, могут играть ожоги, длитель-
ный контакт с нефтяными продукта-
ми, дегтем, прием препаратов мышь-
яка. Л. П. Цыркунов (1985) наблю-
дал множественные базалиомы лица
у больной, длительно работавшей с
асфальтом. В патогенезе придается
значение утрате клеток опухоли спо-
собности к кератинизации. Указыва-
ется на тесную связь базалиомы с со-
единительно-тканной стромой, харак-
тер которой, по мнению А. К. Апатеи-
ко, влияет на структуру, особенно таких вариантов как склеродермоподоб-
ная базалиома, фиброэпителиома, трихобазалиома. Большое значение в раз-
витии базалиом придается иммунным нарушениям. Генетические факторы
наибольшую роль играют при множественных базалиомах, наблюдающих-
ся при синдроме Gorlin—Goltz. Считалось, что базалиома развивается
обычно на неизмененной коже. По мнению Gertler (1965), подобное ут-
верждение является неправомерным, так как при этом не учитывается
тот факт, что развитию базалиомы могут предшествовать клинически
46
неопределяемые микроизменения кожи, прежде всего такие, как кол-
лоидная старческая дегенерация и атрофия. Именно этим, как считает
агтор, и объясняется наиболее Частая локализация базалиомы на лице.
Имеются описания возникновения ее на фоне туберкулезной волчанки
(Ehring nGattwinkel, 1974), рентгеновской кожи (van Dijk и Mali, 1979),
не усов (Sigal и Saunders, 1967), рубцов после прививок, ожогов
(Мгшкиллейсон Н А. и соавт., 1980; Reed и Wilson-Jones, 1968; Zelikson,
1967; Almeida Goncalves, 1966; Goughan et al., 1969; Gordon, 1974), псо-
риаза (Ельнин В. Д. и Основина Н. Г., 1980), в том числе после фотохи-
миотерапии (Moler и Howitz, 1976), дерматофибромы и других патологиче-
ских состояний (Ganowitz и Goldstein, 1964; Goette и Helwig, 1975; Rotte-
leur et al., 1983).
Клиника. Наиболее часто (рис. 39, 40, 41) базалиома имеет вид оди-
ночного ограниченного плотноватого образования полушаровидной формы,
чаще округлых очертаний, незначительно возвышающегося над уровнем
кожи, в центре которого обычно имеется небольшое западение, покрытое
тонкой, плотно прилегающей чешуйко-корочкой; край валИКООбрйЗНО утол-
щен, в его зоне могут быть множественные телеангиэктазии. При легком
натяжении видно, что периферический валик состоит из миниатюрных
опухолевидных узелковоподобных элементов («кожных жемчужин»). Опу-
холь медленно увеличивается в размерах, может быть небольшая болез-
ненность и кровотечение после удаления корочки. При изъязвлении воз-
никает вначале поверхностная, а с течением времени—более глубокая не-
заживающая язва (ulcus rodens) с гладким или мелкозернистым дном,
скудным серозно-гнойным отделяемым. Ulcus rodens встречается чаще у
мужчин, располагается на верхних 2/3 лица, прежде всего на пограничной
зоне щек и виска, в ретроаурикулярной области. Б. А. Беренбейн и В. Ю.
Уджуху (1976) описывают следующие варианты опухолевой (узелковой)
базалиомы: крупноузловатый, конглобированный, бородавчатый, опухоле-
во-язвенный. Реже встречается первично язвенная базалиома, а также
такие поверхностно распространяющиеся формы базалиомы, как рубцую-
щаяся и педжетоидная. Рубцующаяся базалиома (рисунки 42, 43) epit-
helioma basocellulare planum cicatricans) имеет вид медленно увеличиваю-
щегося очага поражения, чаще полициклических очертаний, состоящего
из центральной рубцевидной части и периферической зоны, образованной
опухолевидными элементами, в которой могут быть эрозии, изъязвления,
наслоения корок, изредка ограниченные очажки экзофитного роста. Пед-
жетоидная, или эритематозная базальноклеточная эпителиома (рисунок
44) epithelioma basocellulare pagetoides) характеризуется наличием слегка
уплотненных очагов поражения, одного или нескольких, красновато или
красновато-коричневатого цвета с неровной поверхностью, покрытой че-
шуйками или корочками. Края резко отграничены от здоровой кожи, слег-
ка приподняты в виде тонкого валика из очень мелких опухолевидных
элементов. В центре обнаруживаются атрофические изменения, рубчики,
иногда поверхностные изъязвления, покрытые кровяной корочкой, телеан-
гиэктазии; неравномерность окраски, рассеянные пигментации. Очаг
47
может принимать псориазиформный, экзематоидный вид, быть схожим с
дискоидной красной волчанкой. Рост опухоли очень медленный. Предпоч-
тительная локализация — туловище и лицо. Пигментированная разно-
видность базальноклеточной эпителиомы (epithelioma basocellulare pigmen-
tosum) отличается от обычной формы наличием точечной или сетевидной
Рис. 44. Педжетоидная
эпителиома.
коричневатой пигментации опу-
холи, занимающей всю ее по-
верхность или распределяющей-
ся неравномерно по краю. Рас-
полагается чаще на туловище
или на лице. При локализации
на лице может иметь большое
сходство с ограниченным пре-
канцерозным меланозом Дюбрея
(Friihwald, 1959). Растет мед-
ленно, обычно не вызывает
деструктивных изменений, Wi-
ner и Fevin (1961) наблюдали
пигментную базалиому, развив-
шуюся на волосистой части
головы в зоне веррукозного не-
вуса. По течению и гистологи-
ческому строению пигментная
базалиома не отличается от
обычной. К более редким вариантам базалиомы относится скле-
родермиформная (epithelioma basocellulare sclerodermiforme) и веге-
тирующая (epithelioma basocellulare vegetans). Склеродермиформная
базалиома имеет вид небольшого четко отграниченного очага пора-
48
жения с уплотнением в основании, более значительным по пери-
ферии, плоского или слегка возвышающегося, желтовато-беловатого
цвета. Периферический валик из опухолевидных элементов обычно отсут-
ствует. В центре могут выявляться атрофические изменения, телеангиэк-
тазии, дисхромия, но изъязвление бывает редко. Располагается чаще на
лице. Freeman и соавт. (1973) выделяют три типа базалиомы со склерозиро-
ванием: сходный с поверхностной склеродермией; кольцевидный с уплот-
нением в центре и нодулярной периферией; нодулярный с зонами склеро-
за, обычно в основании. Вегетирующая базалиома представляет собой
узловатое образование, значительно выступающее над кожей. Поверхность
ее эрозирована, бородавчата, легко кровоточит, отчетлива склонность к
распаду. Может достигать значительных размеров. Curry и соавт. (1977)
описали гигантские базалиомы с экзофитным ростом, вегетирующие, до
20 см в диаметре, с метастазами в регионарные лимфоузлы. Grande и соавт.
(1982) описали подкожный вариант базалиомы, по-видимому, развившейся
после неполного удаления существовавшей ранее нодулярной формы. Встре-
чаются и первично глубокие формы базалиомы (Шанин А. П., 1969), рас-
полагающиеся чаще на туловище, конечностях, а также на нижней губе
и половых органах. Обладают быстрым ростом, дают метастазы в регионар-
ные лимфоузлы. Мы наблюдали развитие базалиомы на очаге красного
плоского лишая (рис. 45).
В большинстве случаев (до 95%, по Curry et al., 1977) базалиома рас-
полагается на голове или шее. Из 611 больных, наблюдавшихся Bonniger
и Konz (1979), в 82,9% случаев опухоль была в области головы и шеи,
11,9%—на туловище, 2,1%—на верхних и 3,1%—на нижних конечностях.
На губах, в отличие от спиналиомы, базалиома встречается не часто. Из
105 случаев рака этой локализации, по данным Gaiseuhauer (1970), в 25,8%
была базалиома, 74,2%—спиналиома. Располагается в большинстве слу-
чаев на верхней губе (Пашков Б. М. и соавт., 1970). По данным Schubert
(1984), базалиома в области нижней губы наблюдалась у 0,5% больных.
В области ногтей базалиома может быть сходной с хронической паронихией
(Hc.'fman, 1973). Редкой локализацией являются подошвы (Berger et al.,
1966, Lewis et al., 1965) и особенно ладони (Hymen и Barsky, 1965; John-
son, 1960). На ладонях и подошвах обычно периферический венчик из
«раковых жемчужин» не выявляется, в связи с этим базалиома имеет вид
шелушащихся, гранулематозных, гиперкератотических или эрозивных оча-
гов (Costello и Jibs, 1964). Базалиомы обычно одиночны (в 75,8% случаев
по данным Sebastian и Smolz, 1973), но могут быть и множественными,
чаще—это поверхностные формы. Количество их у одного больного может
быть различным. Так, у 196 больных, наблюдавшихся Moller и соавт. (1975),
было 1100 базалиом. У 3 больных, не включенных в эту выборку,
количество опухолей было трудно сосчитать. Аналогичные больные наблю-
дались также нами (рис. 46). По мнению Epstein (1974), при множествен-
ных базалиомах выше риск развития новых опухолей после лечения.
Заболевание развивается примерно с равной частотой у мужчин и
женщин (Jung et al., 1968; Kleine-Natrop et al., 1969), обычно у лиц по-
4 Клиническая дерматология 49
жилого возраста. Имеются описания базалиом и у детей (Maron, 1963).
Течение базалиомы, как правило, хроническое, многолетнее. Обычно опу-
холь растет медленно, оставаясь локализованной чаще, на стадии ulcus
rodens. Лишь в относительно редких случаях язва распространяется вглубь
и по поверхности, инфильтрирует и вызывает значительные разрушения
подлежащих тканей (ulcus terebrans). Течение изменяется, становится
агрессивным и, если не будет проведено лечение, прогноз может быть тя-
желый из-за опасных кровотечений или кахексии (Ильин И. И. и соавт.,
1978). Описано периневральное распространение базалиомы (Hanke et al.,
1983), встречающееся редко, обычно при рецидивах. Считается общепри-
нятым, что классическая базалиома не метастазирует (Kreuzer и Zandes,
1965; Albrecht et al., 1977; Schimpf, 1979; Cranmer et al., 1970). По мне-
нию Steigleder (1975), исключения из этого настолько редки, что в таких
случаях прежде всего возникает сомнение в правильности диагноза. Ука-
зывается, что метастазирует так называемая метатипическая базалиома,
промежуточная или смешанная по своей гистологической структуре (Gert-
50
ler, 1965; Kleiman, 1968 и др.)- Большое значение в развитии метатипиче-
ского рака придается рентгеновскому облучению (ВОЗ, 1980; Gottron,
1964). По мнению Schimpf (1979), в случаях метастазирующей базалиомы
речь идет скорее не о переходном раке, а о переродившейся базальнокле-
точной карциноме. Hundeiker и Brehm (1971) допускают исключительно
редкую вероятность метастазирования обычной базалиомы при прораста-
нии опухоли в сосудистую стенку. Costanza et al. (1974) обнару-
жили в литературе 90 случаев метастазирующих базалиом. В боль-
шинстве это были крупные опухоли, длительно существующие, подвергаю-
щиеся лечебным вмешательствам (Beck et al., 1983 и др.). Обычно мета-
стазы—в регионарные лимфоузлы, редко в висцеральные органы (легкие,
печень, кости). По мнению Mikhail и соавт. (1977), достоверных случаев
описания метастатической базальноклеточной карциномы—78 (включая и
2 их наблюдения). Одна из наиболее частых причин метастазирования—
неполное удаление.
Гистопатология.. Эпидермис слегка атрофичен, иногда изъязвлен, раз-
растание опухолевых базофильных клеток, сходных с клетками базального
слоя, но не имеющих межклеточных связей, с большим овальным или
продолговатым ядром со скудной цитоплазмой, в виде глубоко проникаю-
щих в дерму разветвляющихся тяжей и гнезд. Митозы встречаются в не-
большом количестве. Но описаны случаи с резко выраженной ядерной
атипией. Строма богата волокнистой субстанцией, клетками (плазматиче-
ские клетки, лимфоциты, гистиоциты), иногда может быть фиброзной. В
непосредственной близости к опухоли соединительная ткань подвергается
слизистой дегенерации. По мнению Getzrow (1966), характер реакции стро-
мы имеет существенное значение для определения степени малигнитета
опухоли. При более благоприятных, поверхностных формах отчетлива
реакция стромы в виде палисадообразно расположенных соединительно-
тканных клеточных элементов, образования мукополисахаридов, эластиче-
ских волокон вокруг опухолевых клеток, при агрессивном течении—реак-
ция стромы скудная. При метатипической форме клетки имеют более
эозинофильную цитоплазму, полигональные очертания, отсутствует тен-
денция к палисадообразному расположению инфильтрата (ВОЗ, 1980;
Cramer, 1970).
Zerchin и Rahbari (1975) описали адамантиноидный вариант базалио-
мы, клинически и биологически неразличимый от обычной формы, гистоло-
гически напоминающей адамантиному.
Дифференциальный диагноз. Дифференцировать базалиому необходи-
мо с кератоакантомой, спиноцеллюлярной эпителиомой, шанкриформной
пиодермией, твердым шанкром, сенильным кератозом, туберкулезной вол-
чанкой, педжитоидную’эпителиому—с красной волчанкой, болезнью Боуэ-
на.
Лечение. Хирургическое, лучевое, колхаминовая, проспидиновая ма-
зи.
4*
51
ЛИТЕРАТУРА
Беренбейн Б. А., . Уджуху В. Ю. -К вопросу о наружном лечении базалиом кожи
проспидином. Вести, дерматол. и венерол., 1976, 9, 55—59.
Ильин И. И., КарповскаяО. Г., Михеева А. П. О базалиомах с деструктируклцим
ростом. Вести, дерматол. и венерол. 1978, 1. 56—59.
Апатенко А. К- Эпителиальные опухоли и пороки развития кожи. М., Медици-
на, 1973.
Л. П. Цыркунов. Множественная базалиома у асфальтировщика. Вест, дерматол.
и венерол. 1985, 2, 48—51
“ing Н. Ein modifiziertes Verfahren der mikroskopisch kontroflierten Chirurgie
fur die Entfernung von Basaliomen.— Derm. Mschr., 1982, 168, 7, 488—493.
Beck H. J., Andersen J., Birkler N. E., Ottosen P. D. Giant basal cell carcinoma
with metastasis and secondary amyloidosis: report a case Acta derm, venerol. (Stockh), 1983,
63, 6, 564—567. ,
Bonniger F., Konz B. Ergebnisse dermatochirurgiseher Basaliombehandlung In: Ope-
rative Dermatologie, Springer—Verlag. Berlin. Heidelberg—New—York, 1979, 201—206.
Curry M. C., Montgomery R. K-, Winkelmann. Giant basal cell carcinoma. Arch.
Derm., 1977, 113, 3, 316—319.
Hanke C. W., Wolf R. L. HochmanS.A., O’Brain J. J. Perineural spread of basal
cell carcinoma.— J. derm suig. oncol., 1983, 9, 9, 742—747.
Grande D. J., Whitaker D. S., Roranda F. C. Subdermal basal cell carcinoma.—
J. Derm. Surg. Oncol., 1982, 8, 9, 779—781.
GormbeyD. E., Hifsch P. Aggresive basal cell carcinoma of the scalp.— Arch. Derm.,
1978, 144, 5, 782—783. v
Kleinberg C., Penetrante R. B., Milgrom H., Pickren J. W., Metastatic basal cell
carcinoma of the skin.— J. Am. Acad. Derm., 1982, 7, 5, 655—659.
Moller R., Howitz J. Methoxalen and multiple basal, cell carcinoma.— Arch. Derm.,
1976, 112, 7, 1613—1614.
Mikhail J. R., Nims L. P., Kelly A. P., DitmarsD. M., Eybr W. R. Metastatic
basal cell carcinoma.— Arch. Derm., 1977, 133, 9, 1261—1269.
Moller R., Nielsen A., Reymann F. Multiple basal ceff carcinoma and internal malig-
nant tumors.— Arch. Derm., 1975, 111, 4, 584—585.
Rotteleur G., Cheoallier L. M., Piette F., Bergcend H. Basa. ce.. carcinoma over-
ling histiocytofibroma,— Acta derm., venero.. (Stokh.) 1983, 63, 6, 567—571.
Schubert H., Haufgikeit und Loka.ization von Basa.iomen im Kopf Ha.s—Bereic.
Derm., Mschr., 1984, 170, 7, 453—456.
Schimpf A. Verwi.dertes Basa.iom, ma.ignes Basa.ze-.karcinom. In: «Nicht entzund-
iche Dermatosen. III. B. Bosartige Geschwu.ste. Leukamie.— Hrsg. Gottron H. A., Kor-
ting J. N., Springer—Ver.ag 1979, 219—224.
52
БАЗАЛЬНОКЛЕТОЧНОГО НЕВУСА СИНДРОМ (basocellulamevus syn-
dromtm)
Синоним: синдром Gorlin—Goltz
Этиология и патогенез. Заболевание представляет собой генетически
детерминированную ассоциацию базальноклеточных невоидных эпителиом
с разнообразными пороками развития скелета, глаз, нервной системы,
реже других органов и тканей. Сочетание кожных и мозговых аномалий
дало основание некоторым авторам (Herberg и Wiskemann, 1963, Hermans
et al., 1965, van Dijk и Sanderink, 1967 и др.) назвать этот синдром «пятым
факоматозом.» Наследуется аутосомно-доминантно (Gerber, 1965; В. Анд-
реев и Н Златков, 1977). Имеются указания на возможность рецессив-
ного наследования. Отмечено возникновение базалиом после травм, при-
вивок, инъекций (Panizzon et al., 1979), ожогов (Zann, 1981). Указы-
вается на возможность их развития под воздействием факторов, обладаю-
щих канцерогенным эффектом: УФО, мышьяк, деготь (Reymann, 1975).
Клиника. Обычно множественные (иногда до сотен) высыпания мел-
ких опухолевидных элементов, сходных с базалиомой, цвета нормальной
кожи или желтовато-коричневатые, округлых очертаний, полушаровидной
или слегка уплощенной формы, незначительно возвышающиеся над уров-
нем кожи (рис. 47). Поверхность их гладкая, могут быть телеангиэктазии,
просвечивающие милиумиподобные образования. Стечением времени раз-
меры высыпаний постепенно увеличиваются, обычно до периода полового
созревания больных, и могут достигать 1,5 см в диаметре и более. Могут
быть и другие кожные изменения, в частности эпидермоидные кисти, милиа,
фибромы, веснушки, комедоны, ладонно-подошвенный гиперкератоз, иног-
да в сочетании с мелкими опухолевидными элементами (Howell и Mehregan,
1970). Характерным признаком считается наличие множественных мелких
углублений величиной с булавочную головку на ладонях и подошвах, под
которыми иногда обнаруживается базально-клеточная эпителиома (Ho-
well и Freeman, 1980). На основании анализа 75 опубликованных случаев
этого синдрома van Dijk и Sanderink представили следующую частоту
ассоциированных пороков развития: зубные кисты выявлены у 61 больного;
аномалии ребер—у 36; гипертелоризм—у 28; эпителиальные и сальноже-
лёзистые кисты, липомы—у 26; «дискератоз» ладоней и подошв—у 18; каль-
цификация falx cerebri—у 15; сколиоз и кифоз —у 15; белые угри и коме-
доны—у 11; аномалии пястных и запястных костей—у 9; позднее развитие
зубов—у 9; spina bifida—у 7; изменения глаз—у 6; мозговые аномалии—
у 6; фибромы и кисты яичника—у 4; лимфатические мезентериальные ки-
сты—у 3; волчья пасть, заячья губа—у 2; киста ductus thyreoglossus—у
одного больного; множественные кисты длинных костей—у одного; средин-
ный носовой синус—у одного больного. Авторы подчеркивают, что этим
не исчерпывается все многообразие возможных пороков развития, кото-
рые могут наблюдаться при этом синдроме, учитывая, что большинство
больных—дети, и значительная часть из них была обследована не полно-
стью. У одной из больных, 42-летней женщины, van Dijk и Sanderink (1967)
обнаружили двурогую матку, у второй, 11-летней девочки,—волчью пасть,
53
Рис. 47. Синдром
базальноклеточных
невусов.
заячью губу, кавернозную геман-
гиому, двустороннюю паховую гры-
жу, готическое небо, вдавленную
грудину, крыловидную лопатку,
скрюченный мизинец, genu valgum.
И по данным более поздних наблюде-
ний и публикаций (Happle et al.,
1971; Zaun, 1981; Howell и Anderson,
1982, Schaarschmidt, 1983), наиболее
частым, помимо базальноклеточных
невусов, проявлением этого синдрома
являются одонтогенные кисты, кото-
рые встречаются чаще на нижней
челюсти. Наблюдаются также изме-
нения глаз (гипертелоризм, катарак-
та, врожденная слепота и др.) Happle
et al. (1971) описали пигментную
ретинопатию со вторичной катарак-
той обоих глаз, рецидивирующие
кровоизлияния в хрусталик левого
глаза. Из поражений нервной системы, помимо кальцификации мозговых
оболочек, отмечены умственная отсталость, изменения ЭЭГ, врожденная
гидроцефалия, частичная агенезия corpus callosum, опухоль мозжечка г
Fitzpatrick и Thomson (1982) указывают, что при синдроме Горлина имеется
6 основных и около 38 дополнительных клинических признаков. Наиболее
частая локализация—лоб, веки, нос, щеки, но высыпания могут быть
на туловище, в подмышечных впадинах, на волосистой части головы.
В относительно редких случаях базальноклеточная карцинома распо-
лагается на ладонях (Taylor и Wilkins, 1970); она, как полагают, могла
развиться в зоне точечных углублений.
Имеются описания линейного унилатерального базальноклеточного
невуса с комедонами и атрофией (Carney, 1952, Bleiberg и Brodkin, 1969,
Burck et al., 1984 и др.), который рассматривается как дерматоз, отличный
от синдрома Gorlin—Goltz
Заболевание развивается примерно с равной частотой у лиц обоего
пола, в раннем детском возрасте, но может существовать с рождения. В
то же время описано появление кожных опухолей и у взрослых, даже в
77-летнем возрасте, у негра, наблюдавшегося Martin и Waisman (1978).
Вариации возраста к началу заболевания могут быть значительны и в пре-
делах одной родословной. Так, в семье, описанной Meyswan et al. (1982),
у матери высыпания появились в 30 лет, у ее дочери—с рождения, у сы-
на—в пятилетием возрасте. У всех была срединная фистула носа. С воз-
растом количество и размеры опухолей увеличиваются. Может наблю-
даться тенденция к самопроизвольному регрессированию отдельных эле-
ментов (Telle, 1965), но это бывает редко. Обычно с возрастом течение ухуд-
шается. Указывается (Anderson, 1967; Geppard, 1983), что с рождения су-
54
шествуют костные аномалии, карциномы же могут появляться в различ-
ные возрастные периоды.
Описано сочетание и с другими опухолями, в частности семиномой
(Zaun, 1981), раком матки (Batschward и Minkov, 1967), фибросаркомой
верхней челюсти (Reed, 1968), амелобластомой иижней челюсти (Happle,
1973), хронической железодефицитной анемией (Torok et al., 1982).
Различают две стадии: невоидную и неопластическую. Невоидная
фаза продолжается долго, десятилетиями, но затем внезапно может перей-
ти в неопластическую, в период которой из-за значительного разрушения
под.ежащей ткани может наступить летальный исход (Berendes, 1971).
Прогноз поэтому неблагоприятный, так как часто наступает превращение
опухолевидных элементов в ulcus rodens с деструктивным ростом. Усиление
роста некоторых элементов может наступить внезапно, при спокойном и
стационарном течении других. Появление изъязвления—неблагоприятный
прогностический признак. Повышенную склонность к злокачественному
перерождению невоидных базалиом Happle et al. (1971) связывают с хро-
мосомными аберрациями (повышенный разрыв и обмен хроматидами),
А. А. Каламкарян и соавт. (1983)—и с иммунными нарушениями. Schon-
ning (1968) описал развитие спиналиомы в области кисты верхней челюсти.
Гистопатология. В дерме ограниченные гнезда и тяжи из клеток, сход-
ных с клетками базального слоя эпидермиса, иногда с наличием кистопо-
добных пространств, с небольшой инфильтрацией из фибробластов и лим-
фоцитов. Обращается внимание на значительную разницу между клиниче-
скими проявлениями и их гистологическим строением, на идентичность
гистологической картины с наблюдаемой при базальноклеточном раке
(Howell и Саго, 1959). Особенно велика склонность к перерождению в ul-
cus rodens высыпаний на шее и лице, в первую очередь на веках и коже
носа, в связи с чем опухолевидные элементы, располагающиеся в этой
зоне, рекомендуется удалять с профилактической целью (Howell и Саго,
1959).
Дифференциальный диагноз. Дифференцировать синдром базально-
клеточного невуса. необходимо от пигментных невусов, фолликулярного
дискератоза Дарье, себоцистоматоза, сирингомы, трихобазалиомы.
Лечение. Удаление наиболее крупных и интенсивно растущих эле-
ментов, проспидиновая мазь (50%).
ЛИТЕРАТУРА
Андреев В., Златое Н. Наследствени и врождени болести и синдроми в дерматоло-
гнята. София. Медицина и физкультура, 1977
Каламкарян А. А., Капкин В. В., Богатырев И. И. и др. Синдром Горлина.
Вести, дерматол. и венерол., 1983, 1, 4—9.
Fitzpatrick Р., Thompson G. Got.in’s syndrome on nevoid basa. ce.. carcinoma. Cn-.
nad. Med. Ass. J., 1982, 127, 6, 645—670.
Leppard B. J., Skin cysts in the basal cell naevus syndrome.— CTin exp.dermatol.,
1983, 8, 6, 60S—612.
55
Martin S., Waisman M. Basal cell nevus syndrome in a black patient.— Arch. Derm.,
1978, 114, 9, 1356—1357.
Meysman L., Speelman G., oan Ruttinghe R. Gorlin syndrome with nasal fistula.—
Dermatologies, 1982, 165, 5, 522—524.
• Reyman F. Multiple basalcell carcinomas of the skin.— Arch. Derm., 1975, Ш, 7,
877—879.
Schaarschmidt H. Zur Differentialdiagnose des Basalzellnavussyndroms.— Derm.
Mschr., 1983, 169, 5, 294—299.
Torok J., Camisa C., Williams H. T4ie nevoid basal—cell carcinoma syndrome re-
sulting in chronic iron-deficiency anemia. J. dermatoL surg. oncol, 1982, 8, III, 960—963.
БАЛАНИТ КСЕРОТИЧЕСКИЙ ОБЛИТЕРИРУЮЩИЙ (balanitis xe-
rotica obliterans).
Синоним: послеоперационный ксеротический облитерирующий ба-
ланит.
Заболевание впервые описал A. Stuhmer в 1928 г.
Этиология и патогенез. Этиология дерматоза точно не установлена.
Предполагается связь с механической, термической и химической травмой;
заболевание может возникать при ряде инфекционных и вирусных заболе-
ваний (пневмония, ревматизм, ангина, рецидивирующий генитальный гер-
пес, грипп и др ), при болезнях крови, эндокринных нарушениях (диабет,
микседема), нервно-психических заболеваниях, аллергических реакциях
(применение некоторых лекарственных средств), старческих дистрофиче-
ских и атеросклеротических заболеваниях и др.
Stuhmer (1928) считал, что эта форма стоит в тесной связи с предшест-
вующей операцией обрезания по поводу фимоза. Подобный случай описа-
ли А. Я- Прокопчук и соавт. в 1936 г.
Особо надо остановиться на вопросе о взаимоотношении ксеротиче-
ского баланита со склеротическим и атрофическим лихеном и краурозом
полового члена. Нам кажется недостаточно обоснованной позиция тех
авторов (Layman и Freeman, 1944; Layman, 1951; Vander Ploeg, 1983; Le-
ver и Schaumburg-Lever, 1983), которые отождествляют вышеупомянутые
три дерматоза. Мы поддерживаем точку зрения о нозологической самостоя-
тельности этих дерматозов, высказанную недавно Hayman (1983).
Ксеротический баланит имеет ряд существенных отличий от крау-
роза и склероатрофического лихена, из которых главными являются ост-
рое начало, выраженные воспалительные явления.
Клиника. Заболевание встречается относительно редко, может воз-
никнуть в любом возрасте (от 32 до 85 лет), но чаще в возрасте свыше 50
лет.
Начинается с появления гиперемии и отека головки полового члена,
реже внутреннего листка крайней плоти и венечной борозды. В дальней-
шем кожа отдельных участков головки покрывается сероватым налетом,
после удаления которого обнажаются эрозированные поверхности, четко
очерченные, обрамленные белесоватым ободком—остатками некротизи-
рованного эпителия. При бурном прогрессе число эрозий увеличивается,
56
Рис. 48. Ксеротический
баланопостит.
Рис. 49. Ксеротический
баланопостит.
они захватывают большую поверхность голоаки полового члена; из пре-
пуциального мешка появляется обильное выделение серозной или серозно-
геморрагической жидкости (рис. 48, 49). При длительно существующем
процессе, а также при усилении воспалительных явлений может возникнуть
фимоз, реже парафимоз (Pasieczny, 1977). В отделе дерматологии ЦКВИ
мн недавно наблюдали пациента 46 лет, у которого вследствие сильного
отека головки полового члена развились субэпидермальные пузыри с гемор-
рагическим содержимым, с образованием множественных болезненных
эрозий и трещин, увеличением паховых лимфатических узлов. Губки на-
ружного отверстия мочеиспускательного канала стекловидно отечны,
ярко-красного цвета; нередко воспалительно-атрофический процесс за-
канчивается уретральным стенозом. При затяжном течении ксеротиче-
ского баланита, вследствие увеличения объема головки полового члена,
нередко образуются трещины, которые часто изъязвляются с последующим
рубцеванием.
При длительном течении заболевания из-за постоянного раздраже-
ния и уплотнения ткани образуются трещины и ссадины, ведущие к смор-
щиванию и потере эластичности внутреннего листка крайней плоти и кожи
головки полового члена. Открытие головки болезненно, затруднено из-за
спаек между головкой и крайней плотью. Больные жалуются на боли,
зуд, парестезии, болезненные эрекции, болезненный coitus.
Прогноз благоприятный, за исключением случаев сращения уретраль-
ного отверстия с внутренним листком препуциального мешка. В очень
57
редких случаях может развиться рак (Grutz, 1947; Post et al., 1975; Wal-
lace, 1971).
Гистопатология. Незначительная атрофия эпидермиса, участки пара-
кератоза, акантоз. В сосочковом и подсосочковом слое—разной степени
инфильтрат из лимфоцитов, плазматических клеток и полиморфноядерных
лейкоцитов; сосуды несколько расширены и окружены клетками инфильт-
рата.
Дифференциальный диагноз. Его следует проводить с лейкоплакией,
болезнью Боуэна, эритроплазией Кейра, красным плоским лишаем, скле-
роатрофическим лихеном, склеродермией, краурозом полового члена,
хроническим ограниченным плазмоцитарным баланопоститом (Zoon, 1952),
с первичным сифилитическим баланитом (Foil шапп, 1930).
Лечение. В остром периоде—примочки (Sol. argenti nitrici 0,5%; Sol.
Resorcini 2% и др.), кортикостероидные кремы и мази. Мы наблюдали у 9
больных выраженный эффект от лечения гелий-неоновым лазером (10—18
облучений). По стихании острых воспалительных явлений проводят бужи-
рование уретрального канала, рассечение сращений между головкой по-
лового члена и крайней плотью. Во избежание рецидивов показана, опера-
ция кругового обрезания.
ЛИТЕРАТУРА
Hyman A. Balanitis xerotica obliterans.— In: Clinical Dermatology. Philadelphia,
1983, 1—4—27:12.
Lever W., Schaumburg-Lever G. Histopathology of the Skin.— Philadelphia: Lip-
pincott company, 1983, Chapter 16, p. 271—289.
Vander PloegD. Diseases of the penis.— In: Cfincal dermatology. Philadelphia, 1983,
4, 28, 4:1.
Pasiecmy T. The treatment of balanitis xeorotica obliterans with testosterone pro-
pionate ointment.— Acta Dermato—VenereoL, 1977, v. 57, N3, p.275—277.
Post R-, Janner M. Lichen sclerosus et atrophicus penis.— Z. Hautkr., 1975, v. 50,
p. 675-681.
БЕХЧЕТА СИНДРОМ (Behcet syndromum)
Синоним: тройной синдром.
Впервые описан турецким дерматологом Behcet в 1937 г. Touraine
(1950) считает этот синдром идентичным описанному им большому афтозу;
Robinson и McCrumb (1950) рассматривают его как вариант многоформной
эксудативной эритемы.
Этиология и патогенез. Высказывались предположения о вирусной
природе, которые пока не получили подтверждения. Denman et al. (1979),
Williams и Lehner (1977), Lira (1983) важную роль отводят иммунным и
аутоиммунным нарушениям (обнаружение циркулирующих антител к сли-
зистой оболочке полости рта и др.). Семейные случаи заболевания, нали-
чие болезни в нескольких поколениях одной семьи (Goolamali et al., 1976,
Aronsson и Tegner, 1983) позволяют предполагать участие генетических
механизмов.
58
Клиника. Болеют лица обоего пола, мужчины чаще, чем женщины,
преимущественно в возрасте от 20 до 40 лет. При синдроме Бехчета пора-
жаются многие органы и ткани, но наиболее частыми проявлениями яв-
ляются орогенитальные язвы (рис. 50, 51), кожные поражения, артрал-
гия, неврологические поражения и тромбоз сосудов. Основные симптомы
по своей частоте распределяются следующим образом: афтозный стоматит
(у 90—100% больных), язвы на гениталиях (у 80—90% больных), глазные
симптомы (у 60—85% больных). Далее по частоте следуют тромбофлебиты,
артриты, высыпания на коже, поражения нервной системы и др. Изменения
ротовой полости характеризуются появлением афтозных молочницеподоб-
ных эрозий и язв, имеющих различные очёртания. Они появляются на
языке, мягком и твердом небе, нёбных дужках, миндалинах, щеках, деснах
и губах и сопровождаются сильными болями. Обычно высыпания начи-
наются с ограниченного болезненного уплотнения слизистой оболочки,
на котором затем образуется сначала поверхностная, покрытая фибриноз-
ным налетом, а затем кратерообразная язва с небольшой гиперемией вок-
руг. Язва может увеличиваться в размерах до 2—3 см в диаметре. У дру-
гих больных процесс начинается как обычная поверхностная афта, ио
спустя 5—10 дней в основании такой афты появляется инфильтрат, а сама
афта превращается в глубокую язву. После заживления остаются мяг-
кие, поверхностные, гладкие рубцы. Одновременно может существовать
3—5 очагов поражения. Иногда изъязвления афтозного характера бывают
на слизистой оболочке носа, в гортани, пищеводе, желудочно-кишечном
тракте. Поражения на гениталиях состоят из мелких пузыоьков, поверх-
59
ностных эрозий и язв, изредка с подрытыми краями. На половых органах
у мужчин язвы располагаются преимущественно на мошонке, у корня
полового члена :и на внутренней поверхности бедер. Очертания неправиль-
ные, размеры достигают 2—4 см в диаметре. Дно язв неровное, покрытое
серозно-гнойным налетом, часто резко выражена болезненность. У женщин
на больших и малых половых губах в большом количестве обнаруживаются
болезненные при пальпации язвы величиной от горошины до размеров
трех-пятикопеечной монеты. Иногда на коже туловища и конечностей
наблюдаются высыпания, в виде узловатой и многоформной эксудативной
эритемы, узелков, гнойничков, акнеиформных и геморрагических элемен-
тов и др У 15—25% больных с синдромом Бехчета встречается поверхно-
стный рецидивирующий тромбофлебит нижних конечностей (Dawling,
, 1961; Nazzaro, 1966; Haim, 1975 и др.). Godeau etal. (1980) на основании
анализа 35 историй болезни указывают на частоту при синдроме Бехчета
кардиоваскулярных осложнений. Они встречаются в 46% случаев (веноз-
ные тромбозы, поражения легочных артерий, инфаркт миокарда, перикар-
дит и др.). По мнению Haim (1968, 1975), синдром Бехчета—системное за-
болевание, основу которого составляет васкулит.
Обычно заболевание сопровождается тяжелым общим состоянием
больного, повышением температуры, сильной головной болью, общей сла-
бостью, поражением суставов, параличами черепно-мозговых нервов, ме-
нинго-энцефалитом и др. Периодически наступает улучшение и даже спон-
танная ремиссия, которая через несколько недель или месяцев сменяется
рецидивом.
Прогноз нередко неблагоприятный. При поражении глаз может раз-
виться слепота. При длительном течении заболевания наступает инвали-
дизация больных. Особенно неблагоприятен прогноз при поражении
центральной нервной системы—летальность достигает 30% (Pallis и Fudge,
1956; Sygel и Larson, 1965; Schottland et al., 1963; Kiernan et al., 1978;
Wiliams et al., 1977 и др.).
Гистопатология. При гистологическом исследовании краев язвы сли-
зистой оболочки выявляется воспалительный инфильтрат, состоящий из
лимфоидных клеток, плазмоцитов, гистиоцитов и нейтрофильных лейко-
цитов, а также обилие сосудов с разрыхленным набухшим эндотелием.
При исследовании очагов поражения кожи гениталий отмечается резко
выраженный отек дермы, обилие сосудов, вокруг которых беспорядочно
располагаются эритроциты. Клеточный инфильтрат состоит из лимфоци-
тов, полинуклеаров, гистиоцитов и фибробластов. При электронно-микро-
скопическом исследовании-в' глубоких отделах дермы выраженные изме-
нения посткапйллярных венул, наблюдаются очаги некроза и склероза.
(Clausen, Bierring, 1983).
Дифференциальный диагноз. Его проводят с пузырчаткой, окуло-
генито-уретральным синдромом Рейтера, буллезной эксудативной много-
формной эритемой, афтозным стоматитом, с острой язвой Чапина—Липшю-
ца, афтозно-язвенным фарингитом, рецидивирующими рубцующимися глу-
бокими афтами (periadenitis .mucosa necrotica Sutton).
60
Лечение. Рекомендуется комбинированное лечение: кортикостерои-
ды, антибиотики, противокоревой гамма-глобулин, цитостатики (проспи-
дин и др.). Nethercot и Lester (1975) наблюдали благоприятный эффект от
циклофосфамида и азатиоприна. Витамины группы В, аскорутин, препа-
раты кальция и др. Mizshima et al. (1977), Graham-Brown и Sarkany
(1980) наблюдали выраженный эффект от приема колхицина по 0,5 мг 2
раза в день в течение 4—6 недель Учитывая важную роль васкулита в
патологии, в терапию рекомендуется включать антикоагулянты. Наружно
—полоскания дезинфицирующими растворами (борной кислоты, риванола,
ромашки), кортикостероидные мази, 10% синтомициновая эмульсия.
ЛИТЕРАТУРА
Aronsson A., Tegner Е. Behcet's syndrome in two brothers.— Acta Dermato—Vener.,
1983, 63, I, 73—74.
Clausen J., Bierring F. Involement of post-capillary venules in Behcet’s disease an
electronmicroscopic study.— Acta Dermato—Vener., 1983, 63, 3, 191—197.
Denman A., Goolomani S., Salo A. et. al. Lymphocyte and chromosomal abnorma-
lities in Behcet’s syndrome.— Brit. J. Dermatol., 1979, 101, I, 23—24.
Godeau P., Wechler B., Maaouni A. et. al. Manifestations cardio-vasculaires de la
maladle de Behcet.— Ann. Dermatol. VenereoL, 1980, 107, 8—9, 741—747.
Golamali S., ComaishJ., Hassanych F., Stephens A. Familial Behcet’s syndrome.—
Brit. J. Dermatol., 1976, 95, 12, 637—642.
Graham-Brown R., Sarcany J. Failure of colchicine and fibrinolytic therapy in Beh-
cet’s disease.— CLIN. Exp. Dermatol., 1980, 5, 1, 87—92-
Haim S. Behcet’s disease: etiology and treatment.— Dermatologica, 1975, 150, 3,
163—168.
LimS., Haw Ch., Kim N., Fusaro R. Abnormalities ol T—cell subsets in Behcet’s
syndrome.— Arch. Dermatol., 1983, 119, 4, 307—310.
БОУЭНА БОЛЕЗНЬ (morbus Bowen).
Этиология и патогенез. Это заболевание рассматривается или как пред-
раковый дерматоз, так как оно длительно сохраняет свой интраэпителиаль-
ный характер, или как внутриэпидермальная карцинома, ибо прогресси-
рование процесса у всех больных приводит к развитию инвазивного рака
(Венкеи и Шугар, 1962; Schimpf, 1979 и др.) Lever (1975) рассматривает
болезнь Боуэна как шиповидноклеточную внутриэпидермальную карци-
ному и подчеркивает, что отнесение ее к преканцерозам обусловлено не
морфологическими данными, а клиническим течением. Melzer (1961) счи-
тает, что болезнь Боуэна не является самостоятельным дерматозом, а пред-
ставляет собой с самого начала внутриэпидермальный спиноцеллюлярный
рак со смешанной бовеноидной, педжетоидной и базальноклеточной струк-
турой, который развивается под влиянием эндо- или экзогенных кан-
церогенов. Придается особое значение инсоляции, старческим изменениям
кожи, особенно в случаях локализации болезни Боуэна на открытых уча-
стках тела, а также хронической интоксикации мышьяком.
61
Подчеркивается (Schimpf, 1979), что среди факторов, способствующих
возникновению болезни Боуэна, большее значение сейчас придается мышь-
яку, чем это делалось раньше. Предполагается даже (Graham и Helwig,
1963), что все случаи болезни Боуэна—следствие интоксикации мышьяком.
Особое подозрение, если очаги множественны. Роль травмы, хронического
воспаления и других раздражителей, вирусов пока неясна. На возможную
роль вирусной инфекции обращается особое внимание в случаях множест-
венных очагов в области половых органов у лиц молодого возраста, отли-
чающихся клинически от классической формы болезни Боуэна, но гисто-
логически представляющих собой спиналиому in situ (Berger и Hori, 1978,
Mortimer et al., 1983).
Gross и соавт. (1983) обнаружили вирус человеческой папилломы
16 ДНК у 8 из 10 больных бовеноидным папулезом и только у одного из
5 человек, страдавших болезнью Боуэна. Особенностью этой формы явля-
ется спонтанное регрессирование или излечение под влиянием консер-
вативной терапии. Описано развитие болезни Боуэна на фоне самых разно-
образных предшествующих заболеваний и изменений кожи: порокератоз
Мибелли (Coskey и Mehregan, 1975; ороговевающая киста (Shelly и Wood,
1981); эпидермодисплазия Левандовского— Лютца (Vulcan et al., 1979)
в процессе фотохимиотерапии больных псориазом (Hoffman et al., 1979),
ангиоретикулез Капоши (Королев Ю. Ф., 1984), актинический кератоз
и др-
Клиника. Имеется один, реже несколько медленно растущих по пе-
риферии, резко ограниченных, шелушащихся или покрытых корками оча-
гов поражения, красновато-коричневатого цвета, округлых, овальных или
реже полициклических очертаний (рис. 52). Так как инфильтрация, как
правило, небольшая, бляшки плоские или незначительно возвышаются
над кожей, но могут быть и нодулярные образования, обычно в какой-то
из зон. Располагаться они могут на любом участке кожного покрова, а
также на полуслизистых и слизистых, но наиболее часто поражается ли-
цо, туловище и конечности (Schimpf). По данным Oldenbiirger (1980), очаги
болезни Боуэна наиболее часто располагаются на голове (46%), затем на
ладонях (14%),'половых органах (10%). Слизистые и полуслизистые по-
ражаются примерно в 10% случаев. Поверхность очагов неровная, зерни-
стая, что выявляется особенно отчетливо после удаления чешуек и корок.
При выраженном шелушении поверхность может быть псориазиформной,
сходной с актиническим кератозом. Наблюдаются экземоподобные, боро-
давчатые, гиперкератотические проявления (Венкеи и Шугар, 1962). Ги-
перкератоз может достигать степени кожного рога. Периферический край
более активен, плоский или несколько приподнят, имеет более интенсив-
ную окраску. Описаны кольцевидные очаги (Sartoris et al., 1982). В зоне
очага поражения могут сохраняться небольшие участки клинически не-
измененной кожи; изъязвление наблюдается обычно при многолетнем су-
ществовании болезни. Чаще оно бывает в аногенитальной области и сви-
детельствует об инвазивном росте. Здесь же могут быть и вегетирующие
формы Изъязвление характерно также для болезни Боуэиа, располагаю-
62
щейся в области кистей и стоп.
В связи с этим необходимо
производить гистологическое
исследование во всех случаях
длительно существующих яз-
венных изменений, • особенно
инфильтрированных, покрытых
корками. Описаны множествен-
ные, преимущественно папулез-
ные высыпания в области по-
ловых органов (бовеноидный
папулез), которые встречаются
у лиц молодого возраста, име-
ющие красновато-коричневатый
или синюшный цвет, доброка-
чественное течение, но гистоло-
гически представляющие рак in
situ. У 12 из 34 больных, на-
блюдавшихся Wade и соавт.
(1979), обнаруживались, кроме
того, кондиломы, генитальные
бородавки, у 1/3—herpes sim-
plex.
Gross и соавт. (1985) выделяют 3 клинических варианта бовеноидного
папулеза: пятнистый, папулезный и сходный с лейкоплакией. Предпола-
гается вирусное происхождение заболевания (вирус человеческой папил-
ломы). Могут встречаться пигментированные формы (Lee et al., 1971; Wa-
gers et al., 1973; Fisher et al., 1982; Scarborough et al., 1982), имеющие не-
которое клиническое сходство с меланомой. При их наличии требуется
обследование для исключения рака шейки матки (Parent et al., 1983).
На слизистой полости рта болезнь Боуэна проявляется в виде обычно
одиночных, плоских, слегка запавших, иногда лихеноподобных, сходных
с лейкоплакией, но чаще папилломатозных очагов поражения (Машкил-
лейсон А. Л., 1967, 1970). Преимущественно папилломатозный характер
носят очаги поражения в области век (Berger, 1971). Поражается чаще
верхнее веко (Me Callum et al., 1975). Имеются сообщения о локализации
болезни Боуэна в области роговицы и конъюнктивы (Maier и Calugaru,
1977), ладоней (Wagers et al., 1973) ногтевого ложа (Nogitaet aL, 1983) на
головке полового члена (Happle, 1972).
Обычно болезнь не сопровождается субъективными расстройствами,
иногда бывает зуд или жжение.
Заболевание развивается обычно у людей старше 40 лет примерно с
одинаковой частотой у мужчин и женщин. Течение хроническое, прогрес-
сирующее с неизбежным, по данным Венкеи и Шугар, через неопределенный
промежуток времени (иногда исчисляющийся десятилетиями) развитием
спиналиомы, недифференцированной, слабо или совсем не ороговевающей,
63
которая быстро метастазирует. Несколько иные данные по этому поводу
привела Oldenbiirger (1980). Не было обнаружено особой склонности к
развитию карциномы и раннему метастазированию. Так, из 100 больных,
со сроком заболевания от 4 недель до 18 лет, перерождение наступило у 4.
Болезнь Боуэна, располагающаяся на слизистых, протекает значительно
быстрее, с ранним образованием инь зивной карциномы. Так, у одного из
4 больных, наблюдавшихся А. Л. Машкиллейсоном (1967), через несколь-
ко месяцев после возникновения болезни в полости рта развился рак гор-
тани. При локализации болезни Боуэна у женщин в ано-генитальной об-
ласти необходимо обследование для исключения опухоли внутренних по-
ловых органов. Считается (Graham и Helwig, 1959), что при болезни Боуэ-
на чаще чем среди населения, наблюдаются карциномы внутренних орга-
нов, которые развиваются через 5—10 и более лет после возникновения
изменений на коже. К сходному выводу пришли Peterka с соавт. (1961).
По мнению Callen и Headington (1980), наличие любого типа интраэпидер-
мального рака может служить важным маркером раковой опухоли внутрен-
них органов. Это, по мнению Belisario (1971), дает основание предполагать,
что речь идет при этом о канцерогенном эффекте мышьяка Но это мнение
поддерживается не всеми. Так, Anderson с соавт. (1973) при системном ана-
лизе 207 случаев не обнаружили достоверного различия между ожидаемой
и наблюдаемой частотой опухолей внутренних органов.
Гистопатология. Акантоз с проникающими в дерму удлиненными эпи-
телиальными тяжами, пролиферация атипичных шиповидных клеток, их
вакуолизация, особенно в верхних слоях, наличие больших округлых кле-
ток с эозинофильной протоплазмой, большим гиперхромным почковидным
или овальным ядром (клетки Bowen), часто встречающиеся фигуры митоза,
комкообразование ядер внутри многоядерных эпидермальных гигантских
клеток, дискератоз, образование иногда истинных «роговых жемчужин».
Базальный слой сохранен. При его нарушении развивается картина ти-
пичной спиноцеллюлярной карциномы. В дерме хроническая воспалитель-
ная реакция из лимфоцитов, гистиоцитов, плазматических клеток.
По мнению Seifi и Mizuno (1964), гигантские клетки возникают или
вследствие патологического деления дискератотических клеток или фаго-
цитоза дискератотических продуктов. Hofmann и Wilsch (1973; описали
как особый вариант болезни Боуэна длительное сосуществование в очаге
типичных для болезни Боуэна гистологических изменений и псевдоэпи-
телиальной гиперплазии по периферии опухоли с постепенным злокачест-
венным перерождением и метастазированием. Возможно сочетание гисто-
логических признаков болезни Боуэна и экстрамаммарной болезни Пед-
жета (Peralta et al., 1983).
Дифференциальный диагноз. Дифференцировать необходимо с база-
лиомой, сенильным кератозом, псориазом, туберкулезом кожи, хрониче-
ской экземой, бугорковым сифилисом, болезнью Педжета.
Лечение: хирургическое иссечение, электрокоагуляция, лучи Рент-
гена.
64
ЛИТЕРАТУРА
Машкиллейсон А. Л. Болезнь Боуэна на слизистой оболочке полости рта и красной
каймы губ. Вести, дерматол. и венерол. 1967, 8, 34—39
Машкиллейсон А. Л. Предрак красной каймы губи слизистой оболочки рта. М. Ме-
дицина, 1970.
Королев Ю. Ф. Аигиоретикулез Калоши, осложненный лимфолейкозом, керато-
акантомой и болезнью Бовена. Вести, дерматол. и венерол. 1984, 1[ 35—37.
Berger В. W., Hori J. Multicentric Bowens disease of genitalia. Arch. Derm., 1978,
114, II, 1698—1699.
Callen J. P., Headington J. Bowen’s and non-Boweh’s squamous intraepidermal
neoplasia of the skin.— Arch. Derm., 1980, 116, 4, 422—426.
Coskey R. J., Mehregan A. Bowen disease associated with porokeratosis of Mibelli.—
Arch. Derm., 1975, III, II, 1480—1481.
Graham J. H., Helwig E. B. In: «Dermatology in general Medicine» Text---book
and Atlash. Ses. ed. Ed. T. B. Fitzpatrick et a!., 1979.
Gross G., Hagedorn M., Jkenberg H. et al. Bowenoid papulosis. Arch, dermatol.,
1985, 121, 7, 858—863.
Fischer J. B., Greer К- E., Walker A. N. Bowen’s disease mimicking melanoma.—
Arch. Derm., 1982, 118, 6, 444—445.
Lever W. F.,. Schaumburg—Lever T. Histopathology of the skin. 5 ed. Philadelphia,
Toronto, 1975, 473—475.
Maier N., Galugaru M. Modificari cutanate in boala Bowen oculare (epiteliomul intra-
epitelial corneo-conjunctival).— Dermato—Venerologie, 1977, 25, 2, 135—128.
Mikhail J. R. Bowen disease and squamous cell carcinoma of the nail bed.— Arch.
Derm. 1974, 110, 2, 267—271.
Me Callum D. J., Kinmont P. D. C., Wynn D. et al. Intraepidermal carcinoma
of the egelid margin.— Brit. J. Derm., 1975, 93, 3, 239—258.
Mortimer P. S., Sonnex T. S., Dawber R. P. Cryotherapy for multicentric pigmen-
ted Bowen’s disease.— Clin. exp. dermatol., 1983, 8, 3, 319—322.
Nogiia T., Ohyama K., Onot. Bowen’s disease of the nail bed. Dermatologies, 1983.
167, 217—219.
Parent D. Lejeuene F., Ledoux M., Achten G. Maladie de Bowen pigmentee mul-
ticentrique associee a un cancer du col uterin. Ann. Dermatol. Venereol., 1983, 110, 53—57.
Peralta О. C., Barr R. Y., Romansky S. G. Mixed carcinoma in situ: an immuno-
histochemical study.— J. cutan. pat hoi., 1983, 10, 350, 358.
Sartoris S., Ghidella P. E., Maragnan P. etal. Malattia die Bowen anulare.— Chron
Derm., 1982, 13, 4, 335—337.
Shelly W. B., Gray Wood M. Occult Bowen’s disease in keratinous cysts.— Brit. J.
Derm., 1981, 105, I, 105—108.
WadeT.R., KopfA.U., Ackerman В. Bowenoid papulosis of the genitalis.— Arch.
Derm., 1979, 115, 3, 306—308-
БЛУМА СИНДРОМ (Bloom syndromum)
Синоним: наследственная телеангиэктатическая эритема.
Этиология и патогенез. Синдром Блума является заболеванием, на-
следуемым по аутосомно-рецессивному типу. На наследственный характер
5 Клиническая дерматология 65
заболевания впервые указал Szalay (1963). По данным German (1974),
из 49 больных с синдромом Блума 27 были евреи-ашкенази. 31 больной из
этих 49 был выявлен в Северной или Средней Америке, 10—на европейском
континенте (из них только один еврейской национальности), 7—в Израиле,
один родился в Малайзии в европейской семье, его брат—в Германии
после возвращения родителей из Малайзии.
Клиника. Изменения на коже развиваются в раннем детском возрасте,
но могут существовать с рождения. Наиболее характерным признаком,
определяющим одно из названий синдрома, является стойкая телеангиэк-
татическая эритема на щеках и в области носа, располагающаяся нередко
в виде «бабочки», что придает очагам поражения некоторое сходство с крас-
ной волчанкой. Реже подобные изменения обнаруживаются на ушных ра-
ковинах и на тыле кистей. Эритема светочувствительна, в связи с чем забо-
левание ухудшается в летнее время. Интенсивность красноты варьирует
значительно, обычно она более выражена у лиц мужского пола. На губах
и нижних веках могут быть шелушение, везикулезные, буллезные высы-
пания с последующим наслоением корок, рубцевидные изменения. Реже
обнаруживаются глоссит, фолликулярный кератоз, пигментации и дру-
гие изменения кожи. Могут наблюдаться ихтиозиформные изменения
(Thomas, 1980). Из общих проявлений синдрома Блума в первую очередь
обращает на себя внимание нарушение роста. Дети рождаются с низким
весом, который, по данным German (1974), составляет в среднем для маль-
чиков 2094 г, для девочек—1841 г, отстают в росте. Взрослые, за редким
исключением, не выше 150 см (German, 1974). Части тела развиты про-
порционально, за исключением головы, которая обычно меньшего размера;
лицо—узкое и удлиненное, нос относительно крупный, челюсти несколько
недоразвиты. Умственное развитие обычно не страдает. Но Braun-Falco
и Margescu (1966) описали больного 10 лет с синдромом Блума, у которого
обнаруживались умственная отсталость, уменьшенный объем черепа, ги-
пертелоризм, разреженные брови и ресницы, аномалии зубов, грубые цир-
кулярные складки кожи на шее, диссеминированный фолликулярный ке-
ратоз, складчатость кожи на тыле пальцев, ренальный несахарный диабет.
Имеются сообщения и о других дефектах (гипертрихоз, acanthosis nigri-
cans, аномалии развития пальцев, конская стопа и др.), но вероятнее всего
сочетание их с основными признаками синдрома Блума не является зако-
номерным. Судя по имеющимся публикациям, половое созревание как
правило наступает в обычные сроки. Однако, по данным German (1974),
ни у одного из трех женатых мужчин не было детей, у всех обследованных
им мужчин было недоразвитие яичек. В большинстве случаев содержание
иммуноглобулинов снижено, слабо выражена у больных склонность к
формированию аллергических реакций замедленного типа. Нарушен про-
лиферативный ответ лимфоцитов на воздействие митогенов (Hiitteroth et
al., 1975). Вследствие иммунных нарушений больные синдромом Блума
предрасположены к респираторным и желудочно-кишечным инфекциям.
С возрастом подверженность к инфекциям уменьшается. Значительно по-
вышен у них риск развития злокачественных новообразований и лейке-
66
мий. Так, по данным German et al. (1977), из 71 больного с синдромом Блу-
ма у 6 развилась лейкемия, у 2—лимфосаркома, у 4—различные формы
рака внутренних органов. Средний возраст больных, в котором диагности-
ровано то или иное злокачественное заболевание, составлял 20 лет. Описа-
на (Dick., 1979) норвежская чесотка, развившаяся у 13-летнего больного
с синдромом Блума, как следствие иммунодефицита.
При цитогенетическом исследовании выявляются различные хромо-
сомные аберрации, из которых наиболее характерными являются неста-
бильность хромосом, разрыв их, преимущественно в центромерной облас-
сти, и обмен хроматидами гомологичных хромосом (Chaganti et al., 1974,
Kuhn et al., 1979). Считается, что синдром Блума чаще развивается у маль-
чиков, но? возможно, что эта разница объясняется и тем, что у девочек
из-за менее выраженных клинических проявлений заболевание не всегда
диагностируется (Landan et al., 1966). Течение хроническое, прогноз для
выздоровления неблагоприятный. До введения в практику антибиотиков
многие больные, по-видимому, погибали от интеркуррентных заболеваний.
При определении прогноза всегда следует учитывать повышенный риск
развития у больного острого лейкоза или рака.
Гистопатология. Уплощение эпидермиса, расширение сосудов верх-
ней части дермы, диффузные полосовидные и незначительные периваску-
лярные инфильтраты (Lever, 1975). Описаны в то же время изменения,
несколько напоминающие дискератоз при болезни Боуэна (Katzenellen-
bogen и Laron, 1960).
Дифференциальный диагноз. Дифференцировать синдром Блума не-
обходимо от синдрома Rotmupd—врожденной пойкилодермии Thomson,
телеангиэктатической атаксии, синдрома Cockayne, врожденного дискера-
тоза.
Лечение. Фотозащитные кремы, общеукрепляющие средства, гормо-
нальные препараты (по показаниям). Диспансерное наблюдение.
ЛИТЕРАТУРА
Суворова К- Н., Антоньев А. А. Наследственные дерматозы, М., 1977.
7?. S. К. Chaganti, Schonberg S., German J. A manyfold increase in sister chroma-
tid exchanges in Bloom’s syndrome lymphocytes.— Proc. Nat. Acad. Sci., 1974, 71, II, 4508—
4512.
German J. Bloom’s syndrome 11. The prototype of human genetic disorden predispo-
sing to chromosome instability and cancer.— In. Chromosomes and Cancer, Ed. by J. German,
1974, 601—617.
German, J. Bloom, D. Passarge E. — Bloom’s syndrome V. Surveillance for cancer
in affected famillies.— Clinical genetics, 1977, 12, 162—168.
Kuhn E. M., Therman E. Chromosome breakage and rejoining of sister chromatils in
Bloom’s syedrome.— Chromosoma, 1979, 73, 3, 275—286.
Dicken С. H., Dewaid G., Gordon H. Sister chromatid exchanges in Bloom’s syndro-
me.—Arch. Derm., 1978, 114, 5, 755—750.
5* 67
Dick F., Burgdorf W. H., Gentry W. C Norwegian scabies in Bloom’s syndrome.—
Arch. Dermatol., 1979, 115, 2, 212—213.
Hutteroth T. H.,~ Liturio S. D., German G. Abnormal immune responses of Bloom’s
syndrome lymphocytes in vitro.— J. ciin. invest., 1975, 56, I, 1—7.
л Thomas P. Internistisch-padiatrische und genetische Befunde beim Bloom-syndrom.—
Z. Hautkrkh., 1980, 55, 22, 1479—1485.
БРОМОДЕРМА (bromoderma).
Бромодерма—лекарственный дерматоз (токсидермия), развивающийся
в результате применения бромистых препаратов. Бромодерма, особенно
вегетирующая, в настоящее время считается редким заболеванием.
Pini (1895) описал тяжелую бугристую сыпь, вызванную употребле-
нием брома, и назвал ее бромодермой. Первая русская работа о вегети-
# рующей бромодерме принадлежит С. Ф. Проскурякову (1910). Затем по-
явились работы многих других отечественных и зарубежных авторов (Вер-
силова М. А., 1913; Павловская-Карышева К- А., 1916; Сорока Е. И.,
1931; Крапивинцев П. Н., 1935; Goos, 1971; Parish и Polin, 1974; Millins
и Rogers, 1978; Smith и Scheen, 1978; Carney, 1981 и др.).
Этиология и патогенез. Существуют различные теории, объясняющие
причины и механизмы возникновения бромистых дерматозов. По мнению
многих авторов, в качестве главной причины следует считать повышенную
чувствительность организма к лекарственному веществу. Еще в 1935 г.
П. Н. Крапивинцев высказывал взгляд на бромодермию и иододермию
как на своеобразную аллергическую реакцию организма. Бромодерма воз-
никает большей частью после приема внутрь больших доз бромистых со-
лей, особенно бромистого калия, вследствие его кумуляции.
Parish и Polin (1974) описали 38-летнюю женщину, у которой на 6 ме-
сяце беременности появились везикулярные элементы на коже обеих
голеней с последующей пустулизацией и некротизацией. При тщательном
сборе анамнеза было выяснено, что в течение 3 месяцев больная прини-
мала препарат нервин, содержащий бромистый аммоний. Сывороточный
уровень бромида оказался равен 86 мг%. Принимая во внимание легкость
проникновения ионов брома через плацентарный барьер, авторы преду-
преждают о возможности бромистого отравления плода в случаях бромо-
дермы у беременных.
Клиника. В результате приема препаратов брома могут возникать как
осложнение кожиые высыпания самого разнообразного типа: эритематоз-
ные, уртикарные, папуло-пустулезные, везикулезные, буллезные, бородав-
чатые и папилломатозные, акнеиформные и др. Во многих случаях встре-
чаются разные комбинации указанных поражений, однако принято выде-
лять три типа бромодермы: генерализованные сыпи, бромные угри и ту-
берозную (узловатую) бромодерму.
Генерализованные сыпи бывают чаще у детей; оии имеют вид пятни-
стых обычно быстро исчезающих высыпаний. Редко представляются в ви-
де пузырьков и пузырей (большей частью при одновременном приеме бро-
ма и йода).
68
Бромистые угри (acne bromica), являющиеся наиболее частой и ти-
пичной формой бромодермы, проявляются в виде фолликулярных пустул
размером от булавочной головки до чечевицы и обильных розовато-фио-
летовых узелковых элементов, которые усеивают поверхность кожи ли-
ца, спины и конечностей. Часто пустулы и узелки окаймлены ярко-крас-
ным воспалительно-отечным ободком. После заживления могут оста-
ваться поверхностные рубчики буро-фиолетового цвета.
Бромодерма туберозная, вегетирующая (bromoderma tuberosum vege-
tans) может быть вызвана самыми разнообразными препаратами брома,
принимаемыми долго и в значительных дозах. Она свойственна главным
образом молодому возрасту и поражает преимущественно женщин. Эта
сыпь развивается одновременно в нескольких местах, но в первую очередь
на нижних конечностях, несколько реже на верхних конечностях и еще
реже на лице. На голенях и бедрах возникают обычно немногочисленные
ограниченные узловатые и опухолевидные бляшки фиолетово-красного
цвета, возвышающиеся над уровнем кожи на 0,5—1,5 см. Величина узлов
от горошины до голубиного яйца, они покрыты кровянисто-гнойными,
довольно плотно сидящими корками. После снятия корок обнажается
язвенная, бугристая поверхность, на которой могут развиваться бородав-
чато-сосочковые разрастания. Очаги поражения располагаются или от-
дельно, или сливаются, образуя обширные участки поражения величиной
с ладонь взрослого. Опухоль мягка на ощупь. При легком сдавливании с
боков, с освобожденной от корок вегетирующей поверхности, вытекает
обильный гной. Вся опухоль производит впечатление мягкой губки, про-
питанной гиоем. Болезненность очагов поражения относительно невелика.
В литературе приводятся отдельные наблюдения (Smith и Scheen, 1978),
когда сначала образуются крупные пузырные поражения на голенях, ко-
торые затем превращаются в глубокие кратерообразные (эктимоподобные)
язвы и вегетирующие (фунгоидные) бляшки с элевирующим папиллома-
тозом по периферии. Подобные буллезные высыпания, сопровождающиеся
вегетациями, могут симулировать вегетирующую пузырчатку. Следует
подчеркнуть, что различные формы бромодермы наблюдаются нередко
одновременно у одного и того же больного. Видимые слизистые поражаются
очень редко. Болезнь протекает благоприятно и довольно быстро поддается
лечению, оставляя после себя атрофические рубцы и пигментации.
Гистопатология. При узловато-туберозной бромодерме наиболее ха-
рактерны значительный папилломатоз, мощный акантоз, веррукозная псев-
докарциноматозная гиперплазия, состоящая из лейкоцитов, плазмати-
ческих клеток, гистиоцитов и эозинофилов.
Дифференциальный диагноз. Следует проводить с веррукозным ту-
беркулезом кожи, сифилисом, вегетирующей пузырчаткой, глубоким бла-
стомикозом и гангренозной пиодермией. Ключом к диагностике бромодер-
мы служат соответствующие анамнестические указания о приеме бромисто-
го препарата, повышенное содержание ионов брома в сыворотке крови и
обнаружение брома в моче.
69
Лечение. Лечение бромодермы состоит прежде всего в прекращении
приема бромистых препаратов. Терапевтические мероприятия обычно
сосредоточиваются на усилении выделения из организма брома. Прини-
мают внутрь 10—20 г. поваренной соли в сутки, 10—20% раствор хлори-
стого кальция 3 раза в день по 1 столовой ложке, антибиотики (пеницил-
лин, метациклин, ампициллин и др.), ванны с марганцовокислым калием,
влажные антисептические повязки, мази и кремы, содержащие антибио-
тики.
ЛИТЕРАТУРА
Baker И. Drug reactions.— In: Textbook of dermatology) Ed. A. Rook, D. Kilkin-
son, F. Ebling. Oxford-London, 1979, vol. I, p.llll—1449.
Millins J., Rogers R. Furosemide as an adjunct in the therapy of bromism and bro-
moderma.— Dermatologica, 1978, 156, 2, 111—119.
Parish L., Polin J. Bromoderma in pregnancyl.— Dermatologica, 1974, 148, 4, 247—
252.
Smith S., Scheen R. Bromoderma.— Arich. Dermatol., 1978, 114, 3, 458—459.
ВАСКУЛИТ КОЖИ АЛЛЕРГИЧЕСКИЙ (vasculitis cutis aliergica)
Этиология и патогенез. Аллергические васкулиты кожи полиэтиоло-
гичны по своей природе и как правило не носят специфического характе-
ра. В их патогенезе основную роль играют аллергические реакции, под-
тверждением чему является обнаружение в коже иммунных комплексов,
патоморфологические признаки по типу феномена Артюса, Санарелли—
Шварцмана, положительные пробы на различные аллергены. У больных
выявляются также изменения клеточных и гуморальных факторов имму-
нитета. Характерно наличие у большинства больных сосудистых антиге-
нов, у многих—стрептококкового антигена, положительные реакции на
введение аутологичных лимфоцитов сыворотки крови (Домасева Т. В.,
1973; Шапошников О. К- и соавт., 1977, 1980 и др.). Обнаруживается ци-
тотоксический эффект сыворотки крови на кожу экспериментальных жи-
вотных, изменение в системе биогенных аминов (Кулага В. В., 1971).
Повышенная чувствительность наиболее часто обусловлена инфекцион-
ными агентами, в первую очередь пиококками (особенно стрептококками),
медикаментами, особенно антибиотиками, сульфаниламидами, анальгетиками
(Рахманов В. А. и Иванов О. Л., 1967; Скрипкин Ю. К. и соавт. 1969;
И. Б. Трофимова, 1976; Шапошников О- К. и соавт., 1983, и др.). Могут быть
причиной аллергических васкулитов и другие агенты, в том числе эпидер-
мальные, пыльцевые и бытовые аллергены (Антоньев А. А. и соавт., 1978).
Облегчают развитие заболевания переохлаждения, эндокринные (особен-
но сахарный диабет), нейротрофические, микроциркуляторные нарушения,
патология внутренних органов (особенно печени), интоксикации и дру-
гие неблагоприятные воздействия.
Клиника. Описано множество клинических вариантов васкулитов ко-
жи, объединяемых в отдельные группы в зависимости от глубины пораже-
ния, рода и калибра измененных сосудов, характера гистологических из-
70
менений, степени системности процесса и других параметров (Попов Л.,
1963; Павлов С. Т. и Шапошников О. К-, 1966; Арутюнов В. Я- и Голем-
ба П. И., 1966; Шапошников О. К. и Деменкова Н. В., 1974 и др.). Опыт
показывает, что далеко не всегда наблюдаемые у больных проявления па-
тологического процесса соответствуют тому или иному варианту кожного
васкулита, рассматриваемого как отдельная нозологическая единица. На
разных этапах течения заболевания клиническая картина может существен-
но изменяться и приобретать черты, свойственные другой форме (например,
появление некроза, ранее отсутствующего). Ю. Я- Ашмарин (1972), рас-
сматривая вопросы классификации васкулитов, отмечает, что наряду с
клиническими формами, значительно отличающимися друг от друга, есть
такие формы, проявления которых укладываются в рамки индивидуаль-
ных колебаний у разных больных, что может быть при любом заболевании.
Мы полагаем, что деление иа многообразные клинические формы васкули-
тов является в большой мере условным, и те или иные особенности клиники
васкулита, послужившие основанием для разграничения отдельных ва-
риантов, определяются, при равном экзогенном воздействии, состоянием
макроорганизма в данном отрезке времени. Поэтому практически более
рационально делить васкулиты кожи не на отдельные нозологические
единицы, а только на такие общие группы, как поверхностные и глубокие
васкулиты и по течению—на острые и хронические. Именно эти параметры
наиболее существенны для определения лечебной тактики, особенно если
невозможно провести детальное обследование для уточнения этиологиче-
ского фактора и патогенетических механизмов. Важным аргументом в
пользу этой точки зрения является в значительной мере близость патогене-
за и морфологических проявлений васкулитов кожи,-трактуемых как раз-
ные формы. Острые формы васкулитов кожи встречаются в терапевтиче-
ской и педиатрической практике (геморрагический васкулит, острая узло-
ватая эритема). Дерматологи чаще имеют дело с хроническими вариантами,
как правило протекающими без признаков системности. В каждом же кон-
кретном случае диагноз может быть детализирован с учетом предполагае-
мого этиологического фактора (инфекционно-аллергический, токсико-ал-
лергический васкулит, медикаментозный и т. д.), характера клеточного
инфильтрата (лейкоцитарный, мононуклеарный) выраженности кариоре-
ксиса, типа изменения сосудов (эксудативный, пролиферативный, некро-
тический) и других параметров. Высыпания как правило полиморфные,
состоят в различных сочетаних из петехиальных, эритематозных, эритема-
то-уртикарных, эритемато-узелковых элементов, частью с геморрагическим
компонентом, поверхностных некрозов, изъязвлений. Могут возникать
пузыри, пузырьки, в том числе с геморрагическим содержимым. Размеры
высыпаний обычно небольшие. Близко расположенные некротические очаж-
ки могут сливаться. Сыпь располагается чаще на ногах, реже поражаются
руки, ио в процесс может вовлекаться и кожа туловища (рис. 53, 54, 55).
Цвет высыпаний зависит от длительности их существования—при их по-
явлении они более яркой окраски, затем приобретают синюшные, бурова-
тые тона. На месте регрессировавших элементов может оставаться пигмеи-'
71
Рис. 53. Аллергический
некротический васкулит
кожи.
тация, если имелись изъязвления—рубцы, нередко оспеновидные. Субъек-
тивные расстройства обычно незначительны, может быть зуд, жжение,
болезненность, в основном при некротических изменениях. Болеют чаще
взрослые. Наиболее частой формой глубоких васкулитов является хрони-
ческая узловатая эритема.
Заболевание протекает длительно, нередко годами, ремиссии чере-
дуются с обострениями, чаще в холодное время года. Общее состояние, в
отличие от острых форм, значительно не нарушается, но может наблюдать-
ся небольшое повышение температуры, головная боль, недомогание, об-
щая слабость, артралгии, у отдельных больных—альбуминурия, гемату-
рия.
Гистопатология. Отечность и набухание эндотелия, отложение фибри-
ноидных масс в стенках и вокруг сосудов (при наличии пролиферации эндо-
телия, сушение просвета сосудов), кариорексис, тромбообразование, экстра-
вазаты, лейкоцитарная инфильтрация, в ранних стадиях с большим ко-
личеством нейтрофилов, эозинофилов, в поздних—мононуклеаров, при
длительном течении обнаруживается отложение гемосидерина.
72
Дифференциальный диагноз. Следует проводить с папуло-некротиче-
ским туберкулезом, таксидермиями, острой формой парапсориаза.
Лечение. Антигистаминные средства, препараты кальция, витамины
(С, Р), рутин, при наличии инфекции—антибиотики, распространенном,
тяжело текущем процессе—кортикостероиды, цитостатики (фосфазин).
ЛИТЕРАТУРА
Антоньев А. А., Ройтбурд М.Ф., Мутина Е. С. Рутштейн Л. Г. Некоторые
вопросы патогенеза, клиники и лечения аллергических васкулитов. Вести, дерматол. и
венерол., 1978, II, 7—10.
Иванов О. Л., Насонов Е. Л. Циркулирующие иммунные комплексы у больных
васкулитами кожи. Веста, дерматол. и венерол. 1983, 10, 11—14.
Трофимова И. Б. К вопросу о патогенезе и лечении дермальных и гиподермальиых
васкулитов. Дисс. канд. М. 1976.
Шапошников О. Д., Деменкова Н. В. Сосудистые поражения кожи. Л. Медицина,
1974. .
Шапошникове. К.., Домасева Т. В., Зубжицкий Ю. Н. Антигенные (ангиотропные)
особенности стрептококка при аллергических васкулитах кожи. Веста, дерматол. и вене-
рол. 1980, 9, 22—28.
Шапошников О. Д., Домасева Т. В., Стогов Ю. Н., Походзей И. В., Дарпенко-
ва Н. И. Васкулиты пиококковой природы. ВеСтн. дерматол. и венерол. 1983, 2.
Шапошников О. К., Домасева Т. В. Внутрикожные пробы с аутологичными лим-
фоцитами и сывороткой при аллергических сосудистых заболеваниях кожи. Вестн. Дерма-
тол. и венерол. 1977, 38, 3—7.
Шапошников О. Д., Домасева Т. В. О значении иммунного комплекса в механизме
развития аллергических васкулитов кожи. Вести, дерматол. и венерол. 1975, 4, 3—6.
Шапошников О. Д. Аллергические васкулиты кожи. В кн. «Дифференциальная
диагностика кожных болезней.» Под ред. А. А. Студницииа. М. Медицина 1983, 286—304.
ВЕРНЕРА СИНДРОМ (Werner syndromum)
Синоним: прогерия взрослых. Заболевание описал Werner в 1904 году.
Этиология и патогенез. Полагают, что в основе заболевания лежит
дефект обмена соединительной ткани (Pavlik и Когр, 1971; Santoianni et.
al., 1978). Большая роль отводится эндокринным нарушениям, в частности
дисфункции гипофиза. Центральные нервно-сосудистые повреждения при-
водят к трофическим изменениям. Заболевание наследуется аутосомно-
рецессивно. Greither (1955), изучивший около 75 случаев, описанных в
литературе, отметил, что в 20% случаев родители больных были кровными
родственниками.
Клиника. Заболевание чаще наблюдается у мужчин, может встречать-
ся у нескольких членов одной семьи (Rabbiosi и Borroni, 1979). Начинается
обычно в возрасте 20—30 лет, имеет прогредиентное течение. Больные низ-
кого роста с маленькими тонкими руками, вследствие атрофических изме-
нений в подкожной клетчатке и мышцах. Изменения кистей весьма сходны
73
со склеродактилией. Черты лица заостренные, «птичий» нос. Волосы рано
седеют, становятся тонкими, выпадают (рис. 56). Развивается катаракта.
Характерен хриплый высокий голос. Часты дистрофические изменения
ногтей, гиперкератоз подошв. Кожа на лице, на дистальных участках рук
и ног становится бледной истонченной, «натянутой». При малейшей трав-
ме появляются изъязвления, трофические язвы. Почти всегда наблю-
дается гипогенитализм. Нередко больные страдают диабетом, атероскле-
розом сосудов. Имеется нарушение обмена кальция—остеопороз, каль-
цинаты в коже и других тканях; психическая эффективность; в части слу-
чаев—умственная отсталость. Имеются сообщения о частом сочетании
синдрома Вернера со злокачественными новообразованиями. Djawari et al.
(1980) объясняют это имеющимся у больных иммунодефицитом. Описа-
но сочетание синдрома Вернера с псориатической эритродермией (Бутов
Ю. С. и соавт., 1977), с несращением твердого неба (Балявичене Г. Р.,
1980). Продолжительность жизни обычно не превышает 40 лет. Больные
погибают от злокачественных опухолей, сердечно-сосудистых нарушений
и Др )
Гистопатология. Истончение эпидермиса, атрофия придатков кожи.
Выраженные дистрофические изменения эластических волокон.
Дифференциальный диагноз. Проводят со склеродермией, акрогерией,
синдромом Ротмунда.
Лечение симптоматическое.
ЛИТЕРАТУРА
Балявичене Г. Р. Синдром Вернера в сочетании с частичным несращением твердого
нёба.— Вести, дерматол. и венерол., 1980, 6, 55—58.
Бутов Ю. С., Пономарев Б. А., Сасиков Б. М. Сочетание псориатической эритро-
дермии и синдрома Вернера у одного больного.— Вести, дерматол. и венерол., 1977, 3,
68—70.
Djawari D., Lukaschek Е., Jecht Е- Altered cellular immuniti in Werner’s syndro-
me.— Derma to togica, 1980, 161, 4, 233—237.
Rabbiosi G., Borroni G. Werner’s syndrome. Seven casses in one lamily.— Dermatoo-
gica, 1979, 158, 5, 355—360.
' Santoianni P., Aula F., Lembo G. Syndrome di Werner. Considerazioni clinico-
genetiche su di un caso.— Ацп. Ital. Derm. Clin, sper., 1978, 32, 143—152.
ВИСКОТТА-ОЛДРИЧА СИНДРОМ (Wiskott-Aldrich syndromum)
Синдром Вискотта—Олдрича —редкое семейное, наследственное забо-
левание (наследуется по рецессивному, сцепленному с Х-хромосомой типу),
характеризующееся геморрагическим синдромом, хронической экземой и
повторными инфекционными заболеваниями.
Впервые описан Wiskott в 1937 году, в дальнейшем более детально
Aldrich в 1954 году. С тех пор описано около 170 случаев. В .отечественной
литературе имеются публикации в педиатрической литературе о больных
синдромом Вискотта—Олдрича (Матвеев М. П., Денисенко Г. С., 1968;
74
Шабалов Н. П. и соавт., 1975; Ожегов А. М.» Мякишева Л. С., 1979),
в дерматологической литературе подобных описаний нам найти не удалось.
Патогенез. Этот синдром связывают с наследственными нарушениями
в системе клеточного и гуморального иммунитета. При изучении соотно-
шения отдельных классов иммуноглобулинов сыворотки крови выявлены
характерные изменения, заключающиеся в увеличении уровня JgA, JgD
и JgE, уменьшении концентрации JgM. Waldmann и соавт. (1972) обнару-
жили повышения уровня JgE в крови больных синдромом Вискотта-
Олдрича в 30—33 раза. Как проявления недостаточности клеточного им-
мунитета, вероятно, можно рассматривать довольно частое возникновение
злокачественных новообразований при этом синдроме; почти у 10% боль-
ных развиваются злокачественные лимфоретикулярные опухоли (Bailow
et al., 1973).
В патогенезе иммунной недостаточности ведущая роль отводится
снижению функции вилочковой железы. Иммунный дефицит характери-
зуется рецидивирующими инфекциями, тромбоцитопенией и склонностью
к кровотечениям.
Тромбоцитопения является составной частью болезни, в основе кото-
рой лежит качественная неполноценность тромбоцитов: тромбоциты дефект-
ны структурно, функционально и метаболически (Baldin, 1972; Kuramoto
et al., 1970, Ormerod, 1985 и др.). ।
Клиника. Заболевание характеризуется клинической триадой: реци-
дивирующая тромбоцитопения с геморрагией (пурпура, петехии, экхи-
мозы, желудочно-кишечные и урогенитальные кровотечения, кровотечения
из слизистых оболочек, цереброменингеальные геморрагии), хроническая
рецидивирующая экзема и склонность к инфекциям (гнойный отит, пиодер-
мия, пневмония, менингит, энтероколит, рецидивирующий астмоидный
бронхит). Синдром возникает у мальчиков обычно в раннем детском возра-
сте, иногда у новорожденных (Erttmann et al., 1983) или в первые месяцы
жизни ребенка. Однако известны единичные случаи синдрома, отмеченного
в возрасте 14—15 лет (Wolff, 1967, Yorgensen, 1972).
Экзематозный компонент синдрома является специфичным и обычно
ранним (обычно на 2-м или 3-м месяце) признаком, имеет упорный рециди-
вирующий характер с наиболее частой локализацией на лице, голове, конеч-
ностях и ягодицах. На коже упомянутых участков имеются обильная пят-
нисто-папулезная и везикулезная сыпь преимущественно геморрагическо-
го характера с множественными расчесами и петехиальными кровоизлия-
ниями. Часто имеются кожные высыпания, напоминающие атопический
дерматит (Mackie., 1978). Экзематоидиый дерматит может персистировать
и становиться генерализованным (эксфолиативная эритродермия). В ред-
ких случаях высыпания описываются как себорейные или по типу нейро-
дермитоподобной экземы с расчесами. Часто развиваются фурункулы и
абсцессы, резистентные к терапии.
Почти у всех больных наблюдается гепато- и спленомегалия. В мие-
лограмме—падение числа мегакариоцитов; в крови—эозинофилия, то-
75
ксические изменения нейтрофилов, лимфопения и резко выраженная тром-
боцитопения .
Гистопатология. Исследование кожи из очагов поражения устанавли-
вает наличие неспецифических воспалительных изменений.
Прогноз. Все дети погибают в течение первых 3—7 лет, лишь один
больной дожил до 15 лет (Yorgensen, 1972). Причиной смерти больных были
генерализованные инфекции, развитие тромбо-геморрагического синдрома
(кровотечений, кровоизлияния в жизненно важные органы), злокачествен-
ных новообразований и др.
Лечение. Оно малоэффективно, но в части случаев применение корти-
костероидов, трансфузия свежей крови, эритроцитной массы, плазмы дают
временное уменьшение явлений кровоточивости и экзематозных проявле-
ний. В настоящее время трансплантация костного мозга является бо-
лее успешным методом восстановления гематологических нарушений
(Perry et al., 1980; Erttmann et al., 1983). Спленэктомия вызывает лишь
наибольшее увеличение тромбоцитов. С целью борьбы с инфекцией назна-
чают антибиотики, а-глобулин.
В отделе дерматологии ЦКВИ мы наблюдали мальчика 14 лет, который
болен с рождения, когда появились гнойный отит и эксудактивный диатез.
В возрасте 1 года •> месяцев выявлена тромбоцитопения с геморрагическим
синдромом. В последующем многократно переносил гнойный отит, фурун-
кулез, бронхопневмонии. Отмечалась постоянная склонность к кровоте-
чениям (кровавая рвота, кровотечения носовые, кишечные, почечные).
Почти постоянно (курсами) получал преднизолон по 20—25 мг в сутки и
антибиотики с незначительным улучшением. На коже внутренних поверх-
ностей плеч, на пояснице, боковой поверхности туловища обильная розо-
вая мелко-папулезная сыпь, покрытая серозно-геморрагическими короч-
ками. В области ануса разрастания остроконечных кондилом. От ануса
к мошонке тянется большая язва, длиной 4 см с ярко-красным дном, уме-
ренным серозно-геморрагическим отделяемым. Местами кожные высы-
пания носят псориазиформный характер. Ногтевые пластинки в форме
часовых стекол. Гепатомегалия, лимфаденопатия. Резкая тромбоцито-
пения (10—15 тыс.), эозинофилия (до 17%). В Институте иммунологии (Мо-
сква) установлена комбинированная иммунологическая недостаточность,
JgM—67мг%, JgA—192 мг%, JgG—720 мг%. Erok—72%—750. На основа-
нии клинико-лабораторных данных установлен синдром Вискотта—Олдрича.
ЛИТЕРАТУРА
Ожегов А. И., Мякишева Л. С. Семейный случай синдрома Вискотта—Олдрича.—
Педиатрия. 1979,'8, 66—68.
Шабалов Н. П., Рахленко Е. И., Козбан Н. А. Синдром Вискотта—Олдрича.—
Педиатрия, 1974, 2, 78—79.
Baughman R. Wiskott-Aldrich syndrome. In: Clinical dermatology, ed. Demis, Dob-
son, McGrire, Harper a. Row, publishers, Philadelphia, 1983, v. 3, 13—7, 1—3.
76
Mackie R. et al. Wisco tt—Aldrich syndrome with pa tia response to transfer factor.—
Brit. J. Dermatol., 1978, 98, 567—571. .
Holmberg L. et al. A prenata. study of fetal platelet count and size with application
to fetus et risk for Wiskott—Aldrich Syndrome.— J. pediatr., 1983, 102, 3, 773—776.
Erttmann R., Landbleck G. Wiskott-Aldrich Syndrome.—Mschr. Kinderheilk,
1983, 131, 8, 524—527.
Perry G. et al. The Wiskott-Aldrich Syndrome in the United States and Canada (18.12—
1979).—J. pediatr., 1980, 97, 1, 72—78.
ВОЛОСАТИК (myasis linearis)
Синонимы: ползучая болезнь, мигрирующая линия.
Этиология и патогенез. Заболевание относится к группе редких па-
разитарных дерматозов. В данном разделе приведены не только поражения
кожи, вызванные личинками мух (миазы), но и личинками гельминтов
(larva migrans), учитывая большое клиническое сходство между larva mig-
rans и волосатиком, развившемся от проникновения в кожу личинок же-
лудочного овода. Миазы вызываются в основном личинками овода лошадей
или крупного рогатого скота (Hypoderma bovis, Hypoderma lineatum; Ga-
sterophilus haemorrhoidalis; G. interstinalis (Mahal, 1974). Наиболее частой
причиной larva migrans являются личинки гельминтов, паразитирующих
у кошек и собак (Ankylostoma braziliense, A. caninum, Uncinaria stenocep-
hala и др.), стронгиляты крупного рогатого скота (strongyloididae sp. р.)
шистосоматиды водоплавающих птиц (Алексеева М. И., 1980). В редких
случаях заболевание может быть обусловлено личинками гельминтов, свой-
ственных человеку (strongiloides stercoralis и др.). Развивается в основном
у людей, занятых уходом за лошадьми и крупным рогатым скотом, свя-
занных с земледельческими работами (обработка и удобрение почвы), а
также у детей, контактирующих с почвой, песком или купающихся в воде,
зараженной личинками в инвазионной фазе. Допускается возможность
непосредственного откладывания мухами яиц на коже или волосах чело-
века с последующим развитием личинок и проникновением их в эпидер-
мис (Заречная С. Н. и Крашкевич К- В., 1981; Madjarov, 1967)
Клиника. Клиническая картина заболевания, обусловленного личин-
ками нематод и желудочного овода лошадей (сем. Gasterophilidae), сходна,
что делает затруднительной возможность высказать суждение только по
клиническим проявлениям о виде возбудителя (Haufe, 1964; Morgan, 1964).
По мнению же Madjarov (1967), заболевание, вызванное личинками нематод,
может протекать более тяжело. Это касается изменений кожи (возможность
развития везикулезных, буллезных высыпаний, пиодермии), но главное—
общих реакций: головная боль, тошнота, озноб. По-видимому, это зависит
еще и от количества личинок, проникших в кожу. Так, у 8-летнего маль-
чика, описанного Haufe и соавт. (1964), с larva migrans, вызванным Stron-
gyloides stercoralis, помимо кожного зуда, других нарушений общего со-
стояния не было. После извлечения личинки заболевание регрессирова-
ло. Миазы, вызванные личинками овода лошадей и нематод, клинически
характеризуются наличием непрерыной эритемато-отечной валикообраз-
77
Рис. 56. Волосатик.
v ной полосы, протяженность ко-
торой и очертания зависят от
длительности нахождения в
коже личинки и направления
ее передвижения (рис. 56).
Личинка передвигается доволь-
но быстро. Особенно быстрым
считается распространение по-
лос, обусловленных личинками
стронгилоидеса, отсюда и наз-
вание—Larka currens, (Arthur и
Shelley, 1958). Согласно данным
Э. П. Тихомировой и А. Ф. Про-
хорова (1982), личинка продви-
галась в коже на 12 см за 1
час. При этом может возникать
причудливый рисунок: изгибы,
зигзаги, полициклические фи-
гуры. У одного из больных,
описанных Katz и соавт. (1965;,
было около 200 активных оча-
гов. В наблюдении А. А. Ка-
ламкаряна (1954) полос было
настолько много, что их тру-
дно было подсчитать. Smith
и соавт. (1976) описали строн-
гилоидоз кожи с локализа-
цией в области ягодиц, клинически сходный с центробежной кольцевид-
ной эритемой. Субъективные расстройства—зуд, жжение. По данным
Peters и Kramer (1966), миазы, вызванные личинками мух рода Hypoder-
ma, протекают с более выраженными воспалительными изменениями на
коже (а иногда и более или менее тяжелыми общими явлениями), чем обу-
словленные личинками рода Gasterophilus. Внедрение в кожу личинок под-
кожного овода крупного рогатого скота (Hypoderma bovis) и пищеводника
(Hypoderma lineatum) сем. Hypodermatide не сопровождается развитием
непрерывных полос, так как личинки мигрируют в подкожной клетчатке.
Передвигаются личинки быстро—до 12 см за 12 часов (Morgan et al., 1964),
обычно в направлении верхней части туловища и головы, где появляется
отечность, уплотнение красновато-синюшного цвета, сопровождающееся
болезненностью, исчезающие через несколько дней, чтобы возникнуть
вновь по ходу миграции. Клиническая картина может напоминать ангио-
невротический отек, узловатую эритему. При выходе личинок через кожу
возникает фурункулоподобный инфильтрат, на верхушке с серозной жид-
костью (Morgan et al., 1964). Личинки могут пенетрировать в глаз, что
вызывает резкую болезненность, обширные отеки, кровотечение, а также
может сопровождаться повреждением зрительного нерва, отслойкой сет-
га.
чатки, слепотой. Волосатик существует до тех пор, пока з кож&живут ли-
чинки. Миазы, наблюдающиеся в европейских странах, протекут более
благоприятно, чем в странах с жарким климатом. Наступает самоизлече-
ние, так как личинки не находят в коже условий для завершения жизнен-
ного цикла, а мигрируют очень редко и поэтому через недели или месяцы
погибают (Stone и Mall ins, 1964; Katz et al., 1965). \
Гистопатология. В дерме—инфильтрация из нейтрофилов и эозино-
филов вокруг сальных желез и волосяных фолликулов; иногда опреде-
ляется ход личинки в верхних слоях эпидермиса.
Дифференциальный диагноз. Дифференцировать волосатик необходи-
мо от искусственно вызванной крапивницы (urticaria factitia), чесотки.
Лечение. Извлечение личинки иглой под контролем лупы после про-
светления кожи вазелиновым маслом, методы, применяемые для лечения
больных чесоткой, противоглистные средства.
ЛИТЕРАТУРА
Алексеева. М. И. Larva migrans. БЭМ, 3-е изд., 1980, 12,'325—326.
Заречная, С. Н. Крашевич. К- В. Миазы. БЭМ, 3-е изд., 1981, 15, 133—136.
Тихомирова, Э. П. Прохоров. А. Ф. Феномен Larva migrans при стронгилоидозе.
Вести, дерматол. и венерол. 1982, 8, 62—63.
ВОЛЧАНКА КРАСНАЯ (lupus erythematodes).
Этиология и патогенез. Придается значение иммунным нарушениям,
генетическим факторам, вирусной инфекции, нейроэндокринным расстройст-
вам, экзогенным воздействиям. Одной из основных гипотез развития яв-
ляется аутоиммунная, согласно которой в организме образуются. раз-
личные аутоантитела и иммунные комплексы, откладывающиеся в стенках
мелких сосудов и под базальной мембраной эпидермиса. Причина их по-
явления неизвестна. Помимо возможной роли медленной вирусной ин-
фекции, изменения антигенов кожи под влиянием ультрафиолетовых лу-
чей и других неблагоприятных воздействий предполагаются соматиче-
ские мутации в стволовых лимфоидных клетках у генетически предрас-
положенных лиц. О значительных иммунных нарушениях, особенно при
системной красной волчанке, свидетельствуют угнетение Т-лимфоцитов
(Т-супрессоров) и гиперактивность В-клеток, ассоциация с другими ау-
тоиммунными заболеваниями, в том числе с герпетиформным дерматитом
Дюринга (Tomas и Daniel, 1983 и др.), пемфигоидом (Clayton et al., 1982 и
др.). В. К- Подымов (1981) придает основное значение в развитии заболе-
вания недостаточной активности N-ацетилтрансферазы и ингибированию
лизилоксидазы. Видимо, это может быть одним из факторов, больше спо-
собствующих развитию системнЬй красной волчанки, вызванной лекарст-
венными препаратами, с более высоким риском у медленных ацетиляторов.
О роли генетических факторов свидетельствуют семейные случаи, в
том числе у однояйцевых близнецов (Griinhagen, 1952; Steagal et al., 1962;
Laurence et al., 1982) ассоциация красной волчанки с некоторыми анти-
генами тканевой совместимости (HLA В„ HLA В8 HLAA10, HLAA18
79 .
и др-)» обнаружение иммунологических ’феноменов красной волчанки у
клини’. « здоровых родственников больных (Андреева Н. В.иПолилов
М. И./ 1961; Ефимочкина Т. К„ 1974; Steagal et al., 1962 и др.). Cubela
(1983/ из 35 обследованных семей в 8 обнаружил по 2 больных, причем в
6—/о были родственники 1 степени родства, в том числе в 2 семьях в 2
голеижях, в одном случае сын и мать страдали системной красной вол-
ков, ь другом—сын системной, отец—дискоидной. Генетическая пред-
пл.о^енность, по-видимому, неодинакова у больных дискоидной и
системной красной волчанкой. На это может указывать, в частности, раз-
личие в распределении у больных этими формами антигенов тканевой сов-
местимости. Например, выявлена ассоциация антигена В8 с системной
красной волчанкой и у женщин с поздним (после 40 лет) началом дискоид-
ной, антигена В,—с дискоидной красной волчанкой у женщин и у мужчин
с возрастом начала болезни в пределах 15—39 лет (Millard et al., 1977).
Эти данные, по мнению Millard и соавт. (1977), Rowell (1979), могут свиде-
тельствовать о наличии предрасположенности только к дискоидной или
системной красной волчанке или к обеим формам. Больные дискоидной
красной волчанкой, трансформировавшейся в системную, или системной с
дискоидными очагами имеют предрасположенность к обеим формам. Ука-
зывается, что у женщин, больных красной волчанкой, имеющих генотип
HLAB8, заболевших в интервале от 15 до 39 лет, риск трансформации в
системную красную волчанку повышен. Доля таких больных среди лиц с
дискоидной красной волчанкой составляет около 5%. Хотя это только
эмпирическая оценка, но и она важна, так как дает основание для реко-
мендации о характере диспансерного наблюдения для лиц с различным
генотипом, страдающих дискоидной красной волчанкой. К сожалению,
пока мы имеем очень мало маркеров, которые бы характеризовали тот или
иной тип предрасположенности. Кроме того, данные об ассоциации крас-
ной волчанки с различными антигенами тканевой совместимости неодно-
родны. Prystowsky и Gilliam (1975) не обнаружили связи между этими
антигенами и дискоидной красной волчанкой. Aguello и соавт. (1983) ука-
зывают на высокую частоту дискоидной красной волчанки у лиц с генети-
чески детерминированным низким уровнем С4 компонента комплемента.
Douglas и соавт. (1976) наблюдали развитие высыпаний, сходных с систем-
ной красной волчанкой, при недостаточности Cj. Предполагают (Burch и
Rowell, 1970; Hammerstein, 1972), что при красной волчанке возможно
наследование, сцепленное с X-хромосомой.
Из нейроэндокринных нарушений основное значение придается гипер-
эстрогении и гипофункции коры надпочечников (Скрипкин Ю. К., 1979).
На роль вирусной инфекции указывают данные об обнаружении тубуло-
ретикулярных структур, преимущественно в эндотелии дермальных со-
судов, фибробластах, капиллярах, почечных клубочках, напоминающих
парамиксовирусы (Hashimoto и Thompson, 1970, Haustein, 1972 и др.),
циркулирующих антител к различным типам вирусов. В. А. Гребенников
и, В. Е. Гавриленко (1984) придают значение активации кининовой систе-
мы с повышением уровня свободных кининов. Провоцирующими развитие
80
Рис. 57. Геморрагическая
красная волчанка.
Рис. 58. Гипертрофическая
красная волчанка.
или обострение заболевания являются в первую очередь гиперинсоля-
ция и другие неблагоприятные метеовлияния (ветер, холод), травмы, ле-
карственные препараты (гидралазин, прокаинамид, гризеофульвин, изо-
ниазид, контрацептивы, пеницилламин, резерпин, метилдофа, антибиотики
и др.), хронические инфекции (туберкулез, гепатит и др.).
Клиника. Обычно различают 2 формы: дискоидную (ограниченную и
диссеминированную) и системную (острую, подострую и хроническую)
красную волчанку. Большинством (Смелов Н. С. и соавт., 1961; Лелис
И. И., 1962; Рахманов В. А. и Иванов О. Л., 1964; Скрипкин Ю. К-,
1979; Tufanelli и Dubois, 1964 и др.) признается патогенетическая близость
дискоидной и системной красной волчанки. На это указывает возможность
перехода дискоидной формы в системную, что наблюдается, по данным
разных авторов, у 2—20% больных (Tufanelli и Dubois, 1964; Tufanelli,
1972), высокая частота (около 85%) поражения кожи при системной крас-
ной волчанке, причем у значительной доли больных (28,6%, по данным
Tufanelli и Dubois, 1964) с обнаружением типичных очагов дискоидной
красной волчанки, сходство гистологических критериев и иммунологиче-
ских феноменов, хотя и разная их выраженность и частота выявляемости.
Так, иммунные комплексы при дискоидной красной волчанке выявляются
только в очаге поражения, а при системной—и в клинически неизменен-
ненной коже, редко по сравнению с системной обнаруживаются LE-клет-
ки.
6 Клиническая дерматология 81
В типичных случаях наличие таких основных симптомов дискоидной
красной волчанки как эритема, фолликулярный гиперкератоз и рубце-
видная атрофия позволяют без больших затруднений поставить правиль-
ный диагноз, особенно если очаги-располагаются на лице в виде «бабочки».
Однако неравномерная выраженность указанных признаков, а также та-
ких, как инфильтрация, телеангиэктазии, изменение пигментации, при-
водит к большому разнообразию клинической картины, в том числе к раз-
витию атипических форм, требующих для своего распознавания опреде-
ленного опыта. К числу таких форм относятся центробежная эритема, вер-
рукозная или вегетирующая, пигментная, гиперкератотическая, гипертро-
фическая или тумидная, глубокая красная волчанка и др. По данным
Л. Н. Машкиллейсона и соавт. (1937), из 1500 больных, наблюдавшихся
с 1921 по 1934 гг. в Московском институте кожного туберкулеза, у 1162 че-
ловек была дискоидная красная волчанка, у 167—центробежная эритема;
у 61—диссеминированная красная волчанка (включая подострую) у 5—
острая красная волчанка; у 38—себорейная, у 6—пигментная; у 8—вер-
рукозная и папилломатозная; у 5—розацеаподобная; у 5—роговая и гип-
совидная; у 3—тумидная; у одного—геморрагическая (данные о 39 боль-
ных не приведены). В. Я- Арутюнов (1961) указывает, что из 200 больных
опухолевидная красная волчанка была у одного больного; центробежная—
у 15, фолликулярная—у одного, телеангнэктатическая—у одного, роза-
цеаподобная—у одного, инфильтративная—у 2 больных. По данным И. А.
Липского (1961), касающимся 180 больных, дискоидная красная волчанка
была у 82,2%, центробежная—у 10,6%, веррукозно-папилломатозная—у
2,2%, тумидная—у 0,5%, диссеминированная хроническая—у 1,7%.
Поражение слизистых, помимо рта, было у 2 больных, век, чаще нижних,—
у 8 человек.
Особенностью центробежной эритемы ( 1. е. superficial is) по сравнению
с типичной дискоидной является отсутствие или малая выраженность ги-
перкератоза, поверхностный характер без инфильтрации и развития атро-
фических изменений. Может быть небольшое шелушение, точечные гемор-
рагии (Лелис И. И., 1962). Помогают распознаванию наличие четких гра-
ниц, симметричность расположения на лице, рецидивирующий характер.
Среди 370 больных И. И. Лелис (1962) наблюдал центробежную эритему
в 3,2% случаев. По его мнению, центробежная эритема может быть од-
ним из начальных симптомов хронической системной красной волчанки,
а также ценным диагностическим признаком, встречающимся у 75% боль-
ных, при системной красной волчанке вообще. Как своеобразную форму
центробежной эритемы И. И. Лелис (1970) рассматривает chilblain-lupus,
когда имеется выраженный цианотический компонент в очагах поражениях,
располагающихся на носу, щеках, ушных раковинах, кистях, с обостре-
нием в холодное время года. По наблюдениям Millard и Rowel( (1978),
локализация в области носа встречается редко. Авторы сообщают о высо-
кой частоте ознобленной красной волчанки (11,3% от общего числа обсле-
дованных больных дискоидной формой заболевания). Все были женщины.
Очаги располагались преимущественно на тыле пальцев кистей и стоп, в
82
области подошв, особенно на пятках, икроножных мышц, реже на локте-
вых и коленных суставах. Обычно очаги ознобления появляются через
несколько лет после того, как развилась типичная дискоидная красная
волчанка, преимущественно на лице, ио могут возникать одновременно или
ранее. У 2 из 17 больных они были изолированными. Отличается резистент-
ностью к терапии. Трансформация в системную красную волчанку наблю-
далась чаше при одновременном появлении признаков дискоидной крас-
ной волчанки и очагов ознобления, а также при комбинации с высыпания-
ми типа многоформной эксудативной эритемы. Красная волчанка иногда
(Rowell, 1979) может иметь розацеаподобный вид, когда на лице обнару-
живают на фоне светочувствительной эритемы высыпания множественных
папулезных элементов, но, в отличие от розацеа, без пустулизации. И. И.
Лелис (1970) считает, что розацеаподобная форма красной волчанки по
существу не отличается от центробежной эритемы. У лиц, страдающих
себореей, поверхностные очаги красной волчанки покрыты рыхлыми на-
слоениями серовато-желтых чешуек, что придает им сходство с себорейной
экземой (1. е. seborrhoicus). К очень редким вариантам относится туберку-
лоидная красная волчанка (1. е. tuberculoides), по внешнему виду напо-
минающая! волчаночный туберкулез кожи. Геморрагический компонент
более свойственен системной форме, но кровоизлияния могут наблюдаться
и в очагах дискоидной красной волчанки (1. е. hemorrhagicus, рис. 57).
Так же преимущественно у больных с себорейной конституцией могут
наблюдаться фолликулярные (I. е. follicularis) и акнеиформные (1. е.
acneiformis) варианты. Диагностическое значение имеет наличие в глу-
бине ушных раковин сально-роговых пробок (симптом Г. X. Хачатурьяна),
после отторжения которых остается наперстковидная атрофия. И. И. Ле-
лис отмечает, что эти изменения долгое время могут быть единственным
проявлением дискоидной красной волчанки.
При папилломатозной форме (1. е. papillomotosus, s. verrucosus) очаги
приобретают бородавчатый характер, покрыты мощными роговыми наслое-
ниями, значительно возвышаются над окружающей кожей (рис. 58). Встре-
чается редко. Так, из 115 больных, наблюдавшихся А. А. Кройчнком
и М. М. Фуки в 1926—1934 гг., была только одна больная с гипертрофиче-
ской бородавчатой формой. Возникновение веррукозности авторы рассмат-
ривают как признак возможной злокачественной трансформации. Помимо
лица, волосистой части головы, веррукозные очаги могут располагаться
на руках, в том числе на кистях, имея большое сходство с бородавчатым
красным плоским лишаем или кератоакантомой (Uitto., 1978). Близка к
папилломатозной, а иногда и сочетается с ней, гиперкератотическая крас-
ная волчанка (1. е. hyperkeratoticus), когда поверхность выглядит (Маш-
киллейсон Л. Н., 1960) гипсовидной (1. е. gypseus рис. 59) или сходна с
кожным рогом (1. е. corneus). По мнению В. А. Рахманова и О. Л. Ивано-
ва (1964), развитие роговых изменений нередко является начальной ста-
дией рака.
При дисхромической красной волчанке (1. е. dyschromicus) централь-
ная зона белого цвета за счет исчезновения пигмента, периферическая—
6* 83
гиперпигментирована. Пигментация может занимать и весь очаг (1. е.
pigmentosus), который приобретает буроватые, коричневатые тона, что
особенно подчеркивается отсутствием резко выраженных гиперкератотиче-
ских изменений. Телеангиэктатическая красная волчанка (1. е. telangie-
ctaticus) характеризуется йаличием стойких сетчатых очагов, за счет боль-
шого количества телеангиэктазий. Относится к числу очень редких форм.
Так, В. Я- Арутюнов (1961) отмечает, что за многие годы работы удалось
наблюдать только один такой случай. И. И. Лелис (1970) рассматривает
ее как разновидность центробежной эритемы Иногда очаги поражения
приобретают кольцевидный характер, могут быть склеродермиформными.
Редко развиваются буллезные элементы, напоминающие многоформную
эксудативную эритему (Rowell et al., 1963; Lawrence et al., 1982). Rowell
и соавт. рассматривают это сочетание как особый синдром. Пузыри могут
быть как в очагах дискоидной красной волчанки (1. е. bullosus), иногда
с герпетиформным расположением элементов (1. е. herpetiformis), так и на
других участках кожного покрова. По мнению Cram и соавт. (1973), при
появлении буллезных высыпаний необходимо в первую очередь исключить
сочетание с порфириновой болезнью.
При опухолевидной красной волчанке (1. е. tumidus) гиперкератоз
выражен слабо, очаги отечны, значительно возвышаются, синюшно-
красйого цвета, покрыты множественными рубчиками. Они могут дости-
гать гигантских размеров.
Romero и соавт. (1977) описали 11 больных с клиническими, гистоло-
гическими и иммунофлюоресцентными признаками дискоидной красной
волчанки и красного плоского лишая. Очаги поражения располагались
в большинстве на дистальных частях конечностей в виде синюшно-крас-
ных или фиолетовых бляшек с атрофией, дисхромией, телеангиэктазиями,
в редких случаях с веррукозными, буллезными и язвенными изменения-
ми. Часты дистрофии ногтей. Наблюдалась также атрофия ногтевого ложа,
анонихия. На возможность сочетания признаков этих заболеваний указы-
вали также Copeman и соавт. (1970), Thorman (1977), Davies и соавт. (1977).
Одна из наиболее редких форм—глубокая красная волчанка (1. е.
profundus Kaposi—Yrgang). Проявляется плотными узловатостями в под-
кожной клетчатке, покрытыми нормальной или синюшно-красного цвета
кожей. Как правило, одновременно обнаруживают типичные очаги крас-
ной волчанки, чаще дискоидной (Izumi и Tagiguchi, 1983). После регрес-
сирования очагов остаются глубокие атрофические измененения. Возмож-
на кальцификация. Описан случай глубокой красной волчанки с гипертро-
фическими изменениями на поверхности (Otani, 1977).
Tufanelli (1971) наблюдал 6 больных с глубокой формой красной вол-
чанки . У 4 родственников 1 степени родства у одного из больных также бы-
ла красная волчанка. 2 пациентки были сестрами. У 4 больных одновре-
менно была дискоидная красная волчанка, 5—имели различные признаки
системности. Очаги поражения располагались глубоко под клинически
неизмененной кожей, были плотной консистенции, асимптомны, резко от-
граничены, от одного до нескольких сантиметров в диаметре. Большин-
84
Рис. 61. Люпус-карцинома.
Рис. 60. Люпус-карцинома.
Рис. 59. Гиперкератотическая
(гипсотидная) красная
волчанка.
Рис. 62. Люпус-карцинома.
ство элементов оставались без изменения годами. Излюбленной локали-
зацией были лоб, щеки, плечи и ягодицы. В областях плеч очаги пораже-
ния обнаруживались у всех больных. У 3 из них после регрессирования
остались участки атрофии, у 2—кожа стала эритематозной, атрофичной;
изъязвилась. У одного больного были и те и другие изменения. На сходное
распределение очагов глубокой красной волчанки указывают Zweiman и
соавт. (1975), наблюдавшие эту форму у больного тромбоцитопенической
пурпурой. Авторы отмечают большую склонность к системности при глу-
85
бокой по сравнению с дискоидной красной волчанкой. Thurston и Curtis
<1966) описали больного с множественными нодулярными очагами на лице,
туловище, верхних конечностях. У больной, наблюдавшейся Г. Г. Конд-
ратьевым (1961), глубокая форма на левой щеке развилась через 9 лет пос-
ле появления очага дискоидной красной волчанки на носу. Кожа над узло-
ватыми образованиями, размерами до 4 см в диаметре, была бледно-красно-
го цвета с синюшным оттенком, с отдельными мелкими беловатыми чешуй-
ками. Е. М. Каралицкий (1982) описал больную с типичными высыпания-
ми на лице и очагами глубокой красной волчанки на плече и ягодице. В
диагностике глубокой формы красной волчанки большое значение придает-
ся иммуноморфологическому исследованию (Izumi и Takiguchi, 1983).
В. А. Рахманов и соавт. (1964) отрицают целесообразность выделения как
особой формы глубокой красной волчанки. По их мнению, речь при этом
идет о сочетании дискоидной красной волчанки с другими заболеваниями,
чаще всего с глубокими саркоидами. Допускается, однако, что в отдельных
случаях может быть проявление дискоидной волчанки с глубокими гипо-
дермальными инфильтратами, свойственными красной волчанке, а не сар-
коидозу, хотя и указывается на возможность обнаружения при этом эле-
ментов туберкулоидного инфильтрата. Ранее такая точка зрения выска-
зывалась рядом авторов (Pautrier, 1953 и др ), но в настоящее время, как
следует из вышеприведенных материалов, большинством признается су-
ществование этой формы красной волчанки.
Большим своеобразием отличаются очаги красной волчанки на крас*
ной кайме губ, где они бывают, нередко длительное время, изолирован-
ными, и в полости рта. Чаще красная волчанка располагается на нижней
губе, может быть в типичной форме, протекать с выраженными воспалитель-
ными изменениями, отечностью, трещинами, обильными наслоениями чешуй-
кокорок, иногда с образованием эрозивно-язвенных очагов, редко—глубо-
ких узловатостей. Очаги нередко переходят на соседние участки кожи, а
также на слизистую рта. По наблюдениям Б. М. Пашкова и Е. Ф. Бе-
ляевой (1961), из 23 больных с изолированной локализацией красной вол-
чанки на красной кайме губ у 3 была типичная форма с гиперкератозом,
эритемой и атрофией; у 2—без явлений атрофии; у 10—эрозивно-язвенная;
у 7—в сочетании с выраженными эксудативными явлениями и образова-
нием пузырей, по типу эксфолиативного хейлита или экземы в стадии обо-
стрения; у одного больного—глубокая форма. Как и на коже, могут встре-
чаться очаги поражения с резко выраженными гиперкератотическими
изменениями, вплоть до развития кожного рога. К особенностям красной
волчанки красной каймы губ А. Л. Машкиллейсон (1984) относит разви-
тие вторичного гландулярного хейлита, встречающегося, по данным Т. Н.
Антоновой, у 25% больных, наиболее часто при эрозивно-язвенной форме.
В полости рта изолированные высыпания встречаются редко. Так, Shklar
и McCarthy (1978) наблюдали только один случай. Т. Н. Антонова (1965)
отмечает высокую частоту изолированного поражения красной каймы губ,
но из ПО больных с поражением губ и слизистой полости рта не было ни
одного больного, у которого бы очаги обнаруживались только в полости
86
рта. Очаги располагаются на щеках, преимущественно по линии смыкания
зубов, на небе, деснах, чаще в виде резко ограниченных, неправильных
очертаний очагов поражения, красного или красновато-синюшного цвета,
незначительно возвышающихся, больше по краю, с белесоватым слегка
атрофичным центром, ороговением, но могут проявляться в виде яркой
отечной эритемы, склонной к эрозированию или изъязвлению. Б. М. Паш-
ков и соавт. (1970) выделяют 3 формы красной волчанки в полости рта:
типичную, эксудативно-гиперемическую и эрозивно-язвенную. O’Neill и
соавт. (1977) описали болезненные изъязвления в полости рта и глотке
без других изменений на коже, как проявление системной красной вол-
чанки.
В редких случаях при красной волчанке поражается слизистая век,
чаще иижнего, что может приводить к деформации век за счет атрофиче-
ских изменений. Кератоконъюнктивит, как проявление красной волчанки,
наблюдал у 2 больных И. И. Лелис (1970).
В случаях диссеминированной красной волчанки (1. е. disseminatus)
очаги множественны, помимо кожи лица и волосистой части головы, рас-
полагаются на груди, в межлопаточной области, на кистях и стопах, реже
на других участках кожного покрова. В процесс может вовлекаться и сли-
зистая ануса. В перианальной области обнаруживаются эритематозные
бляшки с атрофией и изъязвлением (Roundtree et al., 1982).
Очень редко типичные очаги поражения могут быть на ладонях, где
они характеризуются выраженным гнперкератозом. Чаще, особенно при
диссеминированных и системных формах, обнаруживаются капилляриты в
области конечных фаланг, с телеангиэктазиями, нередко с атрофическими
изменениями, в том числе по типу белой атрофии.
К числу редких локализаций дискоидной красной волчанки отно-
сятся ногтевые пластинки. Клиническими признаками, указывающими на
возможную связь с красной волчанкой, по мнению Kint и van Кегре (1976),
являются синюшно-красная окраска, продольная исчерченность, повы-
шенная ломкость, подногтевой гиперкератоз. Диагностике помогает на-
личие типичных очагов красной волчанки или гистологические исследо-
вания. Большое клиническое сходство с дискоидной красной волчанкой
может иметь лимфоцитарная инфильтрация Jessner—Kanof, которую Bie-
licky и Trapl (1963) рассматривают как нерубцующийся вариант дискоид-
ной красной волчанки. По мнению же Cramer (1965), отнесение ее к крас-
ной волчанке является сомнительным прежде всего потому, что при этом
заболевании не обнаруживается гиперкератоз, атрофия мальпигиевою
слоя, дегенеративные изменения базальных клеток. Нет изменений ба-
зальной мембраны и соединительной ткани. Кроме того, не наблюдалось
развития типичных очагов красной волчанки, хотя заболевание и течет
длительно. Автор не исключает возмоййюсти, что лимфоцитарная инфильт-
рация Jessner—Kanof представляет собой стойкий вариант кольцевидной
эритемы. Неясно взаимоотношение ее и с лимфоцитомой, от которой ее от-
личает прежде всего течение, со спонтанными ремиссиями и рецидивами.
Клинически лимфоцитарная инфильтрация Jessner—Kanof проявляется
87
)
в виде небольших ограниченных эритемато-инфильтративных очагов, слег-
ка возвышающихся, красноватого, красновато-коричневатого или синюш-
ного цвета, в процессе периферического роста которых возможно образо-
вание кольцевидных элементов. Поверхность их гладкая, реже неровная,
но без фолликулярного гиперкератоза. Очаги могут быть единичными или
множественными, располагаются чаще на боковых поверхностях лица,
реже на туловище. Через несколько недель спонтанно исчезают, не остав-
ляя рубца. Рецидивируют. Гистологически обнаруживают лимфоцитарную
инфильтрацию с примесью гистиоцитов и плазматических клеток, с уси-
лением вокруг сосудов. По мнению Gottlieb и Winkelmann (1962), некото-
рые случаи лимфоцитарной инфильтрации могут рассматриваться как про-
явление полиморфного фотодерматоза. У 2 из 9 больных, у которых отме-
чалась повышенная светочувствительность, развилась дискоидная красная
волчанка. Кожные изменения при системной красной волчанке (1. е. syste-
micus) наблюдаются у большинства больных: у 85% больных по данным
А. И. Нестерова и Я- А. Сигидина (1966), 87,5%—И. И. Лелиса (1970);
практически у всех больных, по мнению В. А. Насоновой (1972); около
80%—по данным Sonnichsen, (1972). По мнению Е. М. Тареева (1965)
кожные изменения не являются доминирующими в клинической картине
системной красной волчанки, характеризуются полиморфизмом и распро-
страненностью по всему кожному покрову. В то же время указывается,
что они сохраняют важное диагностическое значение, особенно наиболее
типичные («бабочка», капилляриты на кончиках пальцев). Отмечая выра-
женное многообразие клинических проявлений, различный характер
течения, Jager (1976) указывает на трудность описания типичной клиниче-
ской картины. Г. X. Хачатурян (1958) указывает, что доминирующими
становятся эритема, эксудативные реакции вплоть до буллезных, гипер-
кератоз не носит того характера, как при дискоидной, атрофия выражена
в слабой степени. ЧЙсто отмечается геморрагический компонент, особенно
на кончиках пальцев. С учетом этого разнообразия автор выделяет не-
сколько вариантов: экзематоидный, эризипелоидный и форму, напоминаю-
щую многоформную эксудативную эритему. Могут быть и типичные оча-
ги дискоидной красной волчанки (обычно при трансформации последней в
системную), но чаще обнаруживаются изменения типа центробежной эри-
темы, яркая или ливидная, иногда рожеподобная краснота на лице (ery-
sipelas perstans faciei), симметричная, очаговая или диффузная, часто также
в виде бабочки, с отеком, иногда, при остром течении значительным, при-
водящим к сужению или закрытию глазной щели, рассеянные полиморф-
ные сыпи: эритематозные, эритемато-уртикарные, эритемато-сквамозные;
пятнисто-узелковые, в начальной стадии без склонности к гиперкератозу
и атрофии, скарлатиноформные, псориазиформные, напоминающие хрони-
ческую крапивницу, себореиды, токсидермии, часто с геморрагическим
компонентом, особенно на нижних конечностях, поверхностными изъязвле-
ниями; эритема и телеангиэктазии, кровоизлияния вокруг ногтей, ливедо.
Чаще высыпания располагаются на открытых участках тела, в области
суставов, в полости рта, на красной кайме губ (люпус-хейлит). На ладонях
88
и подошвах имеется диффузная эритема, характерным является наличие
на кончиках пальцев телеангиэктазий, капилляритов, следствием которых
могут быть атрофические изменения, некрозы, изъязвления» цианотиче-
ских, сходных с озноблениями пятен, в том числе с геморрагическим ком-
понентом, на тыле мелких суставов кистей. Возможны изменения типа
панникулита, узелковые высыпания вокруг мелких суставов, склеродер-
миформные изменения на лице и конечностях, феномен Рейно, ассоциа-
ция с синдромом Sjogren, ревматоидным артритом. Tufanelli и соавт, (1960)
наблюдали развитие дерматомиозита у дочери больной системной красной
волчанкой, а во втором случае—у одной из сестер из однояйцевой пары
близнецов—сосудистой пойкилодермии, у второй—системной красной вол-
чанки.
К числу сравнительно редких проявлений относятся буллезные высы-
пания чаще по типу многоформной эксудативной эритемы, в том числе с
геморрагическим содержимым, изъязвлением (Tromovitch и Hyman, 1961),
располагающиеся на конечностях, туловище, в полости рта, элементы типа
кольцевидной эритемы Дарье, периорбитальная эритема.
Буллезный атипический вариант системной красной волчанки (Penney
и Wiley, 1979; Pedros и Dahl, 1973; Krain и Rosenthal, 1973; Qrickx et al.,
1983) трудно дифференцировать от различных буллезных дерматозов, иног-
да ассоциированных, вследствие сходства аутоиммунных механизмов с
этой формой заболевания, от герпетиформного дерматита Дюринга (Мап-
cada, 1974; Davies, 1976; Meyer et al., 1976), пемфигоида (Kumar et al.,
1978; Binder et al., 1978; Clayton et al., 1982 и др.). Особенно затруднителен
может быть диагноз при относительно спокойном течении красной волчан-
ки с мало выраженной или нехарактерной висцеральной симптоматикой.
В этих случаях решающее значение в диагностике отводится обнаружению
антинуклеарных антител, аутоантител к нативной ДНК, LE-клеток. В
тех же случаях при диагностике буллезных высыпаний, когда трудно
решить вопрос на основании гистологических и иммуноморфологических
исследований, в частности при проведении дифференциального диагноза
с пемфигоидом, Olansus и соавт. (1983) рекомендуют применять иммуно-
электронную микроскопию. В пользу красной волчанки свидетельствует
отложение IgG главным образом ниже базальной мембраны, а не в зоне
lamina lucida.
Дистрофическим изменениям подвергаются волосы, ногти, может на-
блюдаться кальцификация в мышечной ткани, периартикулярно (Quismo-
rio, 1975). Как правило, нарушается общее состояние больных за счет ли-
хорадки, изменений внутренних органов и систем (волчаночный нефрит,
миозит, полисерозит, поражение лимфоузлов, печени и селезенки, суставов,
нервной системы, крови).
Дискоидная красная волчанка развивается чаще у женщин, преиму-
щественно у молодых, но может начаться в детском и преклонном возрасте.
В. Я- Арутюнов (1961) наблюдал красную волчанку у двухмесячного
ребенка. Описываются случаи развития высыпаний, сходных клинически
н гистологически с красной волчанкой, у новорожденных. Клинически об-
ед
наРуживаются ограниченные или сливающиеся эритематозные пятна на
лице, преимущественно вокруг глаз, часто с телеангиэктазиями, реже на
других участках кожного покрова (туловище, предплечья, спина). Матери
таких детей в большинстве случаев страдали системными заболеваниями
соединительной ткани, в том числе системной красной волчанкой. Lumpkin
et al. (1985) описали 3 больных детей, родившихся от клинически здоровых
матерей; но у тех и других обнаруживались антитела к Ro/SSA. Обычно в
конце первого года жизни, реже позднее эти высыпания исчезают, оставляя
дисхромию или атрофические изменения. В некоторых случаях наблюдается
вовлечение в процесс внутренних органов. Vonderheid и соавт. (1976) опи-
сали 4 девочек с подобной клинической картинкой, причем у матери одной
из них были высыпания сосудистого характера, напоминающие системную
красную волчанку, дед по отцу второй девочки страдал дискоидной крас-
ной волчанкой (отец не обследован). Мать еще одной больной отмечала
артралгии, особенно мелких суставов кистей и стоп, появление во время
беременности кратковременной зудящей эритемато-везикулезной сыпи на
конечностях. Fox и соавт. (1979) наблюдали развитие системной красной
волчанки у 19-летней женщины, перенесшей в период новорожденности
дискоидную красную волчанку, регрессировавшую в пятимесячном воз-
расте.
Обострения обычно наступают под влиянием внешнесредовых факто-
ров, в первую очередь от инсоляции, травм, а также от нервных перегру-
зок. С введением в терапию антималярийных и кортикостероидных препа-
ратов прогноз значительно улучшился, в том числе в отношении преду-
преждения обширных, уродующих изменений (1. е. mutilans).
Одним из тяжелых осложнений является развитие опухолей, чаще
спиналиомы (рис. 60,61, 62). По мнению Л. Н. Машкиллейсона (1960), чаще
подвергаются трансформации очаги на красной кайме нижней губы. Keith и
соавт. (1980) описали развитие рака кожи у 2 негров, в одном случае на го-
лове, в другом на верхней губе в зоне гипопигментированных атрофических
бляшек. Ertle и Hoting (1983) наблюдали развитие спиналиомы в очаге крас-
ной волчанки, существовавшем на голени 20 лет, с летальным исходом от
метастазов; И. Н. Винокуров и Г. Д. Хаймовский (1963)—на щеке; Р. М.
Ярушина (1974)—на ушной раковине; О. В. Лысенко и И. И. Ильин (1979)
—в полости рта. Основными факторами, способствующими раковому пере-
рождению, являются нерациональное лечение, особенно применение рент-
геновских лучей, гиперинсоляция. Сведения о частоте злокачественного
перерождения, приводимые в литературе, различны. Так, Л. Н. Машкил-
лейсон и соавт. наблюдали развитие рака у 5 из 1500 больных, И. И. Ле-
лис—у 2 из 518 человек. А. Л. Машкиллейсон (1970) нашел, что из 986
случаев рака губы в 1,2% ему предшествовала красная волчанка. По мне-
нию О. В. Лысенко и И. И. Ильина (1979), развитие опухоли в зоне крас-
ной волчанки является не исключением, а типичным осложнением, которое
они наблюдали у 4,2% больных, первично обратившихся по поводу дискоид-
ной красной волчанки. Системная красная волчанка развивается преиму-
щественно у женщин. Так, по данным В. А. Насоновой (1972), из 200 боль-
90
ных было только 3 мужчин. 79% жешцин заболели до 30 лет. Самой моло-
дой было 6 лет, самой старшей—54 года. У 1/3 больных было острое начало.
Прогноз ее тяжелый, особенно при остром течении. Важным является по-
этому своевременная диагностика и раннее полноценное лечение.
Гистопатология. Гистологически обнаруживают фолликулярный ги-
гиперкератоз, атрофию мальпигиевою слоя, вакуольную дегенерацию
клеток базального слоя, гиалинизацию и фибриноидные изменения кол-
лагена, лимфоцитарную инфильтрацию с примесью плазматических кле-
ток, гистиоцитов, больше выраженную вокруг придатков. В полости рта
характерными признаками являются паракератоз, вакуольная дегене-
рация базального слоя, дегенерация коллагена и лимфоцитарная пери-
васкулярная инфильтрация (Антонова Т. Н., 1965; Shklar и Me Carthy,
1978). При иммунофлюоресцентном исследовании в очагах дискоидной,
а при системной и в клинически неизменной коже выявляется отложение
иммуноглобулинов, преимущественно Ig G, реже Ig М, и С3 в зоне дермо-
эпидермальной границы. Для диагностики системной красной волчанки
особое значение имеет выявление с помощью реакции иммунофлюоресцен-
ции антинуклеарных антител, прежде всего против нативной ДНК. На-
блюдаются гомогенное, зернистое, перинуклеарное, периферическое и дру-
гие виды свечения, но наибольшую диагностическую значимость имеет
периферическое. /
Дифференциальный диагноз. Дифференцировать дискоидную красную
волчанку необходимо от розацеа, лимфоцитомы, эозинофильной грануле-
мы лица, себорейной экземы, псориаза, себорейной пузырчатки, лимфо-
цитарной инфильтрации Jessner—Kanof, на губах—от хейлита, в полости
рта—от красного плоского лишая, лейкоплакии; на волосистой части го-
ловы—от псевдопелады Брока, синдрома Лассюэра—Литтля, грибковых
заболеваний, пиодермии; системной—от дерматомиознта, многоформной
эксудативной эритемы, медикаментозных токсидермий, периодической
болезни.
Лечение. Антималярийные препараты (внутрь, обкалывание очагов,
кортикостероидные мази, фотозащитные средства, при системной красной
волчанке—кортикостероидные препараты, при угнетении клеточного им-
мунитета—иммуностимуляторы (Т-активин).
ЛИТЕРАТУРА
Гребенников В. А., Гавриленко В. Е. Особенности клиники хронической красной
волчанки в резко континентальном климате. Вестн. дерматол. и венерол. 1984, 5, 20—24.
Лысенко О. В., Ильин И. И. Lupus-carcinoma Вестн. дерма’тол. и венерол. 1979,
12, 35—40.
Машкиллейсон А. Л. Красная волчанка. В кн. «Заболевания слизистой оболочки
полости рта и губ.» Ред. Е. В. Боровский, А. Л. Машкиллейсон. М. Медицина, 1984, 204—
215.
Подымов В. К- Красная волчанка. Айастан, Ереван, 1981.
Скрипкин Ю. К- Коллагенозы кожные и венерические болезни. М. Мединина 1979. .
91
Ярушина Р. М. Плоскоклеточный рак, развившийся на очаге дискоидной красной
волчанки. Вести, дерматол. и венерол. 1974, 7, 85—88.
Agnello V., Gell J., Туе М. J. Partial genetic deficiency of the C4 component of
complement in discoid fupus erythematosus and urticaria (angioedema). J. A. Acad. Der-
matol., 1983, 9, 6, 894—898.
Binder W. L., SchotlandE., Beuther E. H. et al. Coexistence of bullous pemphigoid
and systemic fupus erythematosums. Arch, dermatof., 1978, 114, 8, 1187—1190.
Crickx B., Leoet R., Blechet-Butaye F. et al. Lupus erythemateu systesmiique
avec lesions bulfeuses. Ann. Dermatol. VenereoL, 1983, 110, 455—459.
Cubela V. Famifiare Faile von Lupus erythematodes. Ergebnisse systematischer Un-
tersuchungen. Derm. Mschr., 1983, 169, 191—193
Daoies M. G., Gorkiewicz A., Knight A. et al. Is there a reletionship between lupus
erythematosus and lichen planus? Brit. j. dermatoL, 1977, 96, 2, 145—154.
Douglas Af. C., Lamberg S. J., Lorincz A. L. et al. Lupus erythematosus—like
syndrome with a familial deficiency of C2. Arch, dermatof., 1976, 6, 112, 5, 671—672.
Ertle T., Hating E. Carcinoma spinocellulare in erythematodes. Z. Hautkr. 1983, 59,
9, 578—592.
Fox 1. R., Me Cuiston С. H., Schoch E. P. Systemic lupus erythematosus. Asso-
ciation with previous neonatal fupus erythematosus. Arch, dermatof., 1979, 115, 3, 340.
Inder A., Sterry W., Crunwald M. H. et al., Pfattenepithelkarzinome in discoiden
Lupus erythematodes-Herden. Z. Hautkr., 1983, 58, 18, 1284—1297.
Keith W. D., Kelly P- Squamous cell carcinoma arising in lesions of discoid fupus
erythematosus in black persons. Arch. dermatoL, 1980, 116, 3, 315—317.
Kint A., Van Негре L. Ungual anomalies in Lupus erythematosus discoides. Der-
matofogica, 1976, 153, 5/ 298—302
Lumpkin L. R., Hall J.,. Hogan J.D. et al.. Neonatal lupus erythematosus Arch
dermatof, 1985, 121, 3, 377—381
Millard L. G., Rowell N. R. Chilblain lupus erythematosus (Hutchinson), Brit. j.
dermatoL, 1978, 98, 5, 497—506
Millard L. G., Rowell N. R., Rajah S. M. Histocompattibifity antigens in discoid
and systemic lupus erythematosus. Brit. j. dermatoL, 1977, 96, 2, 139—144
Penneys N. S., Wiley И. E. Herpetiform blisters in systemic lupus erythematosus.
Arch. dermatoL, 1979, 115, 12, 1427—1428
Romero R. W., Nesbitt L. T., Reed R. J. Unusual variants of lupus erythematosus
0Г lichen planus. Arch. dermatoL, 1977, 113, 6, 741—748
Roundtree J., Weigand D., Burgdorf W. Lupus erythematosus with oral and peria-
nal mucous membrane involvement. Arch. dermatoL, 1982, 118, 55—56
UittoJ., Santa-Cruz D. J., Eisen A. Z. et al. Verrucous lesions in patients with
discoid lupus erythematosus. Brit. j. dermatoL, 1978, 98, 5, 507—520
Shklar G., Me Carthy P. L. Histopathology of oral lesion of discoid fupus erythema-
tosus. Arch. dermatoL, 1978, 114, 7, 1031—1035
Vonderheid E. C., Koblenzer P. J., Ming P. M. L. et al. Neonataf lupus erythema-
tosus. Arch. dermatoL, 1976, 112, 5, 698—705
Zweiman B.., Tomar R. ., Gross P. R. Lupus erythematosus profundus following
thrombocytopenic purpura. Arch, dermatof., 1975, III, 3, 347—351
92
ГАНГРЕНА ПОСЛЕОПЕРАЦИОННАЯ ПРОГРЕССИРУЮЩАЯ
(postoperative progressive Gangran).
В литературе считают, что заболевание описал Cullen в 1924 г. Однако
аналогичную клиническую картину в прошлом веке наблюдал Н. И. Пи-
рогов и называл ее поверхностной изъязвленной фагеденой.
Этиология и патогенез полностью не выяснены. Существует несколько
гипотез: бактериальная, вирусная, гематогенно-метастатическая (попа-
дание кишечных бактерий в кожу при язвенном колите), заболевание яв-
ляется следствием феномена Санарелли—Шварцмана, заболевание раз-
вивается вследствие гипогаммаглобулинемии.
Клиника. Изъязвление большей частью развивается после септических
операций, обычно через 6—14 дней. Характерны неудержимый центро-
бежный рост и в то же время ограничение распространения процесса в
глубину, удовлетворительное общее состояние, незначительное влияние
антибиотиков.
Участок поражения представлен язвой, окаймленной приподнятым
на 2—5 мм валикообразным сильно подрытым краем шириной в 1—3 мм
темно-ливидного цвета. Кнаружи от валика идет широкая зона яркой крас-
ноты, не четко отграниченная от здоровой кожи. Центральная часть яз-
вы имеет отчетливую тенденцию к образованию грануляций и эпителиза-
ции, по периферии же язва расширяется (рис 63).
Край язвы нередко пронизан множественными микроабсцессами, по-
этому при надавливании из краев и карманов под ними вытекает гной.
Края язвы крайне чувствительны и при перевязках причиняют резкие
боли.
Преимущественная локализация: живот, грудь, бедра, голени, ре-
же—руки, спина и как исключение—лицо и гениталии.
В качестве первичных эле-
ментов описываются пузырь с
серозным или серозно-геморра-
гическим содержимым, абсцес-
сы, фурункулы, иногда мно-
жественные пустулы, папулы.
Этн высыпания могут появиться
как в области операционной
раны, так и в непосредствен-
ной близости от нее. Как пра-
вило, повышается СОЭ, отме-
чается нормохромная анемия,
лейкоцитоз, иногда незначи-
тельная протеинурия. Часто
описывают гипогаммаглобу-
Рис. 63. Гангрена
послеоперационная прогрессирующая.
ад
линемию, наблюдается парапротеинемия, а иногда и клинически ма-
нифестная миелома. Может кооперироваться с язвенным колитом, мие-
ломой, полиартритом, пневмонией, холециститом, инфекцией мочевых пу-
тей. Но эти сочетания встречаются реже, чем при гангренозной пиодерме.
Прогноз большей частью благоприятный.
Гистопатология. Каких-либо специфических особенностей нет. В дер-
ме—явления острого воспаления, очаги некроза; в сосудах—отечность
стенок.
Дифференциальный диагноз. Проводят с язвенной формой пиодермии,
тяжелыми формами язвенного васкулита.
Лечение. Кортикостероиды в комбинации с антибиотиками, тран^г
фузии одногруппной крови и плазмы, витаминотерапия, анаболики.
ЛИТЕРАТУРА
Gilller R. Postoperative progressive Gangran und Dermatitis ulcerosa.— Hautarzt,
1968, 19, 408—422.
ГЕМАНГИОМА КАВЕРНОЗНАЯ (haemangioma cavemosum)
Этиология и патогенез. Гемангиома кавернозная представляет собой
доброкачественную опухоль из кровеносных сосудов, развивающуюся вслед-
ствие порока развития сосудистой системы. Кавернозная гемангиома встре-
чается значительно реже, чем плоская. Так, по данным Н. И. Кондрашки-
на (1963), из 8612 гемангиом кавернозных было 163. Опухоль у большинства
больных одиночна. При множественных гемангиомах кожи нередко опу-
холи обнаруживаются и во внутренних органах (Шанин Л. П., 1964).
Клиника. Ограниченная опухоль красного цвета с синюшным, фио-
летовым оттенком, реже (при глубоком расположении) цвета нормальной
кожи, обычно мягкой консистенции, чаще округлых очертаний, иногда
значительно возвышающаяся над кожей (рис. 64, 65). Поверхность глад-
кая, дольчатая, ио может быть гиперкератотичной и веррукозной (Imperi-
al и Helwig, 1967). При надавливании опухоль исчезает или уменьшается
в размерах, увеличивается в размерах, наливается кровью при крике ре-
бенка. Чаще всего располагается на голове и шее (веррукозная—на ногах).
Возникает обычно при рождении или в раннем детском возрасте, чаще у
девочек. В течение первых месяцев опухоль увеличивается в размерах,
иногда довольно быстро (Levine et al., 1960), занимая у ряда больных
обширные участки кожного покрова (например, половину лица, конечно-
сти и т. д.), затем рост прекращается. Течение длительное, обычно бессимп-
томное. При травме может наступить изъязвление, инфицирование, крово-
течение, иногда значительное, в редких случаях—тромбофлебит, эмболии,
сепсис. Неблагоприятно сказывается на течении гемангиомы беремен-
ность, в период которой может резко измениться рост опухоли.
Прогноз, как правило, благоприятный. Может иногда наблюдаться
спонтанное регрессирование до периода полового созревания. При лока-
лизации кавернозных гемангиом на конечностях, особенно при быстром
прогрессирующем росте вглубь, могут возникать застойные явления, тро-
94
фические расстройства, деструктивные изменения мягких тканей и костей
за счет прорастания в них опухоли, увеличение конечности с нарушением
функции. Гемангиомы, расположенные периорбитально, могут вызывать
нарушение зрения, поэтому, особенно при прогрессирующем увеличении
опухоли, не рекомендуется придерживаться выжидательной тактики (Gar-
cia и Dixon, 1984). Злокачественное перерождение если и бывает, то чрез-
вычайно редко. Тем не менее больные с кавернозной гемангиомой должны
быть тщательно обследованы для исключения гемангиоматоза висцераль-
ных органов, нервной системы, костей. Это особенно относится к случаям
неясного желудочно-кишечного кровотечения (Rice и Fischer, 1962). Опи-
сано сочетание кавернозной гемангиомы с тромбоцитопенией и пурпурой
(синдром Kasabach—Merritt). Этот синдром встречается редко. Так, Wil-
son и Haggard (1960) выявили его только у 2 из 310 больных с диагнозом
сосудистой опухоли. Сосудистые опухоли при этом могут быть доброкаче-
ственными и злокачественными. Вследствие этого, а также за счет гемор-
рагических осложнений, прогноз может быть серьезным. Lang и Dubin
(1975) обращают внимание на тщательное обследование таких больных для
исключения внутрисосудистой коагулопатии, прежде чем решать вопрос
о хирургическом лечении. Генерализованные кавернозные гемангиомы
кожи наблюдаются и при ряде других патологических состояний
(синдром Maffucci, blue rubber—bleb nevus и др.) как часть системного
ангиоматоза. Редким является сочетание кожных ангиоц с гемангиома-
тозом костей (болезнь Gorham), что может приводить к частичному или
полному остеолизу и замене костной ткани фиброзной (Frost и Caplan, 1965).
95
Гистопатология. Расположенные в дерме или гиподерме крупные по-
лости, заполненные кровью, стенка которых состоит из одного ряда эндо-
телиальных клеток, окруженных фиброзной тканью. Клеточная реакция
вокруг незначительна, обычно лимфоцитарного характера с примесью туч-
ных клеток.
Дифференциальный диагноз. Дифференцировать кавернозную геман-
гиому необходимо от гемангиоэндотелиомы, меланомы, саркомы Калоши,
синдрома Стюарта—Блюфарба и других сосудистых опухолей.
Лечение. Хирургическое, лучевое.
ЛИТЕРАТУРА
Frost J- F., Caplan R. М. Cutaneous hemangiomas and disappearing bones.— Arch.
Dermatol., 1965, v. 92 N 5, p.501—508.
Garcia R. L., Dixon S. L. Occlusion amblyopia secondary to a mixed capillary—ca-
vernous hemangioma. J. Am. Acad. DermatoL, 1984,10, 2(part 1), 263—267
Long P. G., Dubin H. V. Hamangioma-thrombocytopenia syndrome.— Arch. Der-
matoL, 1975, v. Ill, N I, p. 105—107.
ГЕМАНГИОП ЕРИ ЦИТОМА (haemangiopericytoma).
Синонимы: перицитома, перителиома, гем-, лимфангиоперицитома, пе-
риваскулярная гемангиоэндотелиома.
Редкое заболевание из группы злокачественных сосудистых опухо-
лей, которое впервые описали Stout и Murray в 1942 г. и предложили назва-
ние «гемангиоперицитома.» К настоящему времени опубликованы сообще-
ния о 700 больных.
Этиология и патогенез. Согласно данным литературы, источником
роста гемангиоперицитомы могут быть все элементы сосудистой стенки:
эндотелий, перициты, гломусные клетки, ангиобласты сосудистой мезен-
химы. Однако многие авторы (Stout и Murray, 1942; Kohot et al., 1961; Le-
ver и Schaumburg-Lever, 1975; O’Brien и et al. 1977; Enzinger и Smith,
1976; Andervall et al., 1978 и др.) придерживаются взгляда, что источником
роста гемангиоперицитомы являются перициты, описанные бернским ана-
I томом Zimmermann (1928), которые представляют модифицированные глад-
I комышечные клетки, прилегающие к стенкам капилляров и регулирующие
размеры капилляров. Некоторые авторы (Kay et al., 1951; Kauffman et
«1., 1960 и др.) рассматривают перициты как источники двух близких опу-
холей—гемангиоперицитомы и гломангиомы. Как показывают ультраст-
руктурные исследования (Murad et al., 1968; Kuh и Rosai, 1969 и др.), пе-
рициты обнаружены в стенках капилляров и венул. Они имеют длинные
L просты, окружающие сосуд и образующие прерывистый слой между эн-
дотелием и адвентициальной соединительной тканью. Обычно только ба-
зальная мембрана отделяет эндотелий от перицитов. Некоторые авторы
разделяют гемангиоперицитомы на доброкачественные и злокачественные.
Однако до сих пор не установлены точные границы между ними. Первые—
морфологически зрелые (дифференцированные), вторые—с признаками
анаплазии (недифференцированные). Большинство же авторов считают
9j
гемангиоперицитомы злокачественными или потенциально злокачественны-
ми, так как они рецидивируют и метастазирую- (Emanuel et al., 1974; An-
gervall et al., 1978; Henderson и Farrow, 1978; Wong и Jagoda, 1978; Калам-
карян А. А. и соавт., 1981 и др.). Отдаленные метастазы развиваются у
1/3 больных. Наиболее часто опухоль метастазирует в легкие, лимфатиче-
ские узлы и печень.
Клиника. Опухоль развивается у людей обоего пола, чаще у лиц сред-
него и пожилого возраста. Более 3/4 описанных больных были в возрасте
от 40 до 70 лет. Самому старому из зарегистрированных больных было 92
года. Описаны гемангиоперицитомы у детей (Попхристов, 1963; Enzinger
и Smith, 1967; Eiomoto, 1977). Stout и Murray наблюдали гемангиоперици-
тому у трех новорожденных. Среди 307 больных гемангиоперицитомой,
описанных в США, был 31 (10,1%) ребенок в возрасте от 2 месяцев до 15
лет (Kauffman и Stout, 1960). Отмечено, что гемангиоперицитома у детей
протекает агрессивно. Наиболее частая локализация гемангиоперицито-
мы—кожа, подкожная жировая клетчатка и скелетные мышцы. Но опухоль
может локализоваться всюду, где имеются капилляры, включая рото-
вую полость, медиастинум, ретроперитонеальные области (O’Brien и Bras-
field, 1965). Из 188 случаев гемангиоперицитомы, описанных в английской
литературе, у 103 (54,7%) больных очаги поражения локализовались в
коже и подкожной клетчатке и у 85—во внутренних органах (Spiro et al.,
1964). Suzuki et al. (1976) сообщили, что среди 88 опубликованных в Япо-
нии случаев гемангиоперицитомы в 52% опухоль поражала кожу и под-
кожные ткани, в 11%—забрюшинное пространство, в 10%—желудок.
Опухоли могут возникать на любом участке кожи, но чаще локализуются
на конечностях, особенно на нижних, несколько реже на туловище и голове.
Клинически гемангиоперицитома проявляется четко отграниченным буг-
ристым опухолевидным образованием, выступающим над уровнем окру-
жающей кожи. Опухоль имеет овальные или округлые очертания, состоит
из отдельных узлов плотноэластической консистенции, серовато-белого
или насыщенно-синюшного цвета (в зависимости от степени васкуляриза-
ции), безболезненная при пальпации; размеры ее от 0,5 до 5—8 см в диа-
метре. С течением времени опухоли могут изъязвляться и легко кровото-
чить. Обычно гемангиоперицитома—солитарная опухоль, хотя изредка
она может быть множественной, группируясь, как правило, на сравнитель-
но небольшом участке тела. В нашем наблюдении вокруг основного опухо-
левого поражения наблюдались сателлиты. Saunders и Fitzpatrick (1957)
описали больную 21 года с 15 очагами поражения на коже нижних и верх-
них конечностей. Следует подчеркнуть, что множественные опухоли, осо-
бенно синюшного цвета, могут представлять затруднения как для клини-
циста, так и для морфолога. Они могут быть легко приняты клиницистами
за саркому Капоши. Растут опухоли сравнительно медленно, хотя в части
случаев отмечается довольно быстрое увеличение их размеров. В одном
из наших наблюдений опухоль достигла диаметра 8 см за шестимесячный
период. Болезненность в части случаев развивается в результате сдавления
нервов.
7 Клиническая дерматология 97
Течение болезни, как правило, медленное (5—12 лет), но встречаются
случаи с быстрым смертельным исходом. Весьма часто опухоли рецидиви-
руют, иногда неоднократно. Рецидивы могут возникать через много лет
□осле удаления опухоли. Довольно часто наблюдаются метастазы. В лите-
ратуре процент метастазов при гемангиоперицитоме колеблется от 11,7%
(Stout et al., 1943) до 56,5% (O’Brien et al., 1965). От начала постановки
диагноза и до обнаружения метастазов проходило от 1 до 14 лет, в среднем
4,5 года (Enzinger н Smith, 1976). По данным ряда исследователей, леталь-
ный исход от гемангиоперицитомы колеблется от 23 до 47,8% в сроки от
3—5 до 15—20 лет, в среднем через 10 лет (Enzinger и Smith, 1976 и др.).
Недифференцированные формы гемангиоперицитомы дают более высокую
летальность.
Гистопатология. Опухоль состоит из большого количества капилляров
различной формы и размеров. Под эндотелием расположен слой удлинен-
ных клеток с многочисленными тонкими цитоплазматическими выростами.
Эти клетки, имеющие свойства типичных перицитов, окружены непрерыв-
ной базальной мембраной, кроме-зон непосредственного контакта с эндо-
телием. В некоторых случаях (преимущественно в недифференцированных
гемангиоперицитомах) в перицитах имеется много митотических фигур и
они проникают в просвет сосудов, поэтому такие опухоли следует рассмат-
ривать как злокачественные или потенциально злокачественные (Lever,
1975; Forrester и Houston, 1951; Fisher и соавт., 1952 и др.).
Дифференциальный диагноз. Его следует проводить с саркомой Ка-
лоши, ангиолейомиобластомой, фибросаркомой, ангиосаркомой, гломус-
ангиомой Барре-Массона, сосудистой леймиомой, саркомой Юинга.
Лечение. Более эффективно хирургическое лечение с последующим
облучением очагов поражения; применяют также цитостатики (проспи-
дин, винкристин, циклофосфамид, метотрексат и др.).
ЛИТЕРАТУРА
Каламкарян А. А., Сыч Л. И., Халилова В. И. Гемангиоперицитома кожи
Вести, дерматол. и венерол., 1981, 12, 58—62.
Anderoatl L., Kindblom L., Nielsen J., Soendsen P. Hemangiopericytoma a cjini
conathologis, angiographic and microangiographic study.— Cancer, 1978, 42, 5, 2412—2427'
' Enzinger F., Smith B. Hemangiopericytoma. An analysis of 106 cases.— Hum. Pat"
hoi., 1976, 7, I, 61—81.
ГЕМАНГИОЭНДОТЕЛИОМА КОЖИ (hemangioendothelioma cutis).
Синонимы: ангиосаркома, эндотелиома, лимфангиоэндотелиома, зло-
качественная ангиома, ангиобластическая ретикулосаркома.
Гемангиоэндотелиома, наименование которой было предложено Mal-
lory в 1908 году, относится к группе редко встречающихся опухолей. В
1964 г. Wilson Jones выделил из общей группы гемангиоэндотелиом опухоль
со своеобразной клинической картиной, которая характеризуется преиму-
щественно поражением кожи верхней половины лица и волосистой части
головы у лиц пожилого возраста и предложил для наименования этой опу-
98
холи термин ангиоэндотелиома кожи. В дальнейшем подобные описания
были опубликованы Л. И. Сыч и А. А. Каламкаряном (1979); Reed et. al.,
(1966); Bardwil et al. (1968); Girard et al. (1970); Haustein et al. (1974,
1983); Mehregan et al. (1976); Rosai et al. (1976); Thorman (1977); Martin
et al. (1980); Keahey (1982); Leon (1983), и др. В литературе приведены
описания не более 80 нааблюдений. • '
Этиология и патогенез. Гемангиоэндотелиома относится к группе опу-
холей, не поддающихся рациональной классификации,! так как гистогенез
ее в настоящее время остается спорным. Многие авторы считают источником
роста опухоли эндотелиальные клетки. По мнению Weidner и Braun-Falco
(1970), опухоль развивается из недифференцированных мезенхимальных
клеток. Albertini (1955) считает, что гемангиоэндотелиома является про-
изводным зародышевой ткани, способной к формированию сосудов, и назы-
вает эту опухоль ангиобластической ретикулосаркомой. Отдельные авторы
полагают, что источником роста гемангиоэндотелиом могут быть все эле-
менты сосудистой ткани: эндотелий, гломусные клетки, а также ангиобла-
сты сосудистой мезенхимы.
Клиника. Гемангиоэндотелиома кожи представляет собой чаще еди-
ничные, реже множественные резко отграниченные узлы-опухоли непра-
вильных очертаний, красновато-синюшного цвета размером от 1 до 10—15
см в диаметре. В части случаев опухоль представляет собой 2—3, а иногда
и больше тесно спаянных между собой узелков. Очаг поражения быстро
увеличивается по площади и в глубину, в подкожную клетчатку. В даль-
нейшем наступает распад опухолевой ткани с изъязвлением. Язва посте-
пенно увеличивается как в глубину, так и по площади. Дно ее неровное,
часто покрыто серозно-геморрагическими корками. По периферии опухо-
левого очага иногда наблюдаются отсевы. В центре опухоли нередко име-
ются гемангиомы, которые кровоточат при легкой травме. Часто отмеча-
ется чувствительность опухоли при ощупывании или надавливании; иногда
больные жалуются на довольно сильные боли, возникающие спонтанно в
форме кратковременных приступов. Гемангиоэндотелиома кожи может
развиваться на любом участке кожного покрова, однако излюбленная лока-
лизация ее (у 90—95% больных)—верхняя половина лица и волосистая
часть’Головы. Опухоль возникает преимущественно у людей старше 50—60
лет. Однако недавно Pasyk и Depowski (1978) опубликовали случай ангио-
эндотелиомы у 5-месячного ребенка. Часто фактором, предшествующим
развитию опухоли, является травма. Течение гемангиоэндотелиомы кожи,
как отмечают многие исследователи, характеризуется высокой степенью
злокачественности, агрессивным ростом, рецидивами и метастазированием
чаще всего в легкие, печень и лимфатические узлы. В большинстве слу-
чаев заканчивается смертью больных в течение нескольких месяцев.
Гистопатология. Гемангиоэндотелиома характеризуется наличием рас-
• ширенных, беспорядочно анастомозирующих тонкостенных сосудов, вы-
стланных одним или несколькими слоями атипичных эндотелиальных кле-
ток с крупными гиперхромными ядрами. Такие же клетки видны и в про-
свете некоторых сосудов. Много атипичных митозов. Встречаются гигант-
7* 99
ские многоядерные клетки, тяжи незрелых эндотелиальных клеток. Кол-
лагеновые волокна в виде обрывков: аргирофильные волокна окружают
эндотелиальные тяжи и сосуды по типу базальных мембран. Наблюдаются
кровоизлияния и отложения гемосидерина (Смольянников А. В., 1971;
Апатенко А. К., 1977; Weidner и Braun-Falco, 1970; Rosai et al., 1976;
и др.).
Дифференциальный диагноз. Проводят с гемангиомой, синдромом
Stewart—Treves, ангиолейомиосаркомой кожи.
Лечение. Хирургическое иссечение с последующим лучевым воздей-
ствием или применение химиотерапевтических цитостатических средств
(проспидин, циклофосфамид, дегранол, допан и др.).
ЛИТЕРАТУРА
Апатенко А. К. Мезенхимные и нейроэктодермальные опухоли и пороки разви-
тия кожи.— М.: Медицина, 1977.
Сыч Л. И., Каламкарян А. А. Гемангиоэндотелиома кожи.— Вести, дерматол. н
венерол., 1979, 2, 27—31.
Haustein U., Rytter М., Bethmann W. Hamangiosarcon der Kopfhaut.— Hautarzt,
1983, 34, 10, 515—517.
Keahey T., Bondi E. et al. Malignant angioendothefiom'atosis proliferans treated
with doxorubicin.— Arch. Dermatol., 1982, 118, 7, 512—514.
Leoni A., Cogo K-, Visona A. Angiosarcoma of the scafp'-fase.— Dermatofogica,
1983, 166, 5, 257—260.
Rosai J., Sumner H., Kostianoosky M., Perez—MesaC. Angiosarcoma of the skin.—
Hum. Pathol., 1976, 7, I, 83—106.
ГЕМОСИДЕРОЗ КОЖИ (haemosiderosis cutis).
Синонимы: пурпурозно-пигментные дерматозы; пурпурозный и пиг-
ментный капиллярит.
Изменения кожи, в основе которых лежит отложение гемосидерина в
дерме. Причиной является поражение капилляров.
В группу первичных гемосидерозов кожи входят: кольцевидная телеан-
гиэктатическая пурпура Майокки (purpura annularis teleangiectodes Majoc-
chi, 1896), прогрессирующий пигментный дерматоз Шамберга (dermatosis
pigmentosa progressiva Schamberg, 1901), пурпурозный и пигментный ли-
хеноидный ангиодермит Гужеро—Блюма (dermatitis liosachenoides purpura
et pigmentosa Gougerot—Blum, 1925). Сюда же относят дерматит цвета жел-
той охры (Favre-Chaix, 1924—1926), дугообразную телеангиэктатическую
ПУРПУРУ Touraine (1934, 1950), экзематидоподобную пурпуру Doucas—
Kapetanakis (1953) и зудящую пурпуру Loewenthal (1954) и др.
Этиология и патогенез. Некоторые авторы признают важную роль
аллергических изменений кожи (Скрипкин Ю. К. и соавт., 1969; Волкос-
лавская В. Н., 1971; Варшавский В. А., Иванов О. Л. и соавт., 1984,
Theisen, 1967; Zenner, 1970 и др ). В то же время В. И. Халилова (1971)
считает, что для этого иет достаточных оснований. Существует мнение, что
гемосидерозы кожи являются своеобразной реакцией кожи на различные
100 х
факторы—переутомление, охлаждение, инфекция, в том числе фокальная,
травмы, лекарственные препараты и др., которые способствуют повыше-
нию проницаемости и ломкости капилляров. Дискуссионным считается
вопрос, можно ли считать перечисленные выше основные разновидности
гемосцдерозов самостоятельными нозологическими единицами. За послед-
ние годы число разных клинических вариантов гемосидерозов увеличилось
за счет некоторых вновь описанных форм. Как показывает опыт кожной
клиники ЦКВИ и данные ряда авторов (Аствацатуров К- Р- и Казаков В. К.
1927; Машкиллейсон Л. Н. и Карасева 3. С., 1962; Bohnstedt, 1963; II-
lig и Kalkoff, 1970 и др.), некоторые из выделенных форм гемосидерозов ко-
жи по существу представляют собой лишь морфологические нюансы одного
и того же дерматоза. Значительное сходство в клинической и гистологиче-
ской картине различных форм первичных гемосидерозов кожи позволяет
прийти к выводу о целесообразности объединения этих заболеваний в одну
нозологическую группу—пигментно-пурпурозного дерматоза, сохранив КАК
подразделы основные клинические формы гемосидерозов кожи. Представ-
ляет интерес взаимоотношение лихеноидного, пурпурозного пигментного
ангиодерматита Гужеро—Блюма (Gougerot—Blum) с трехсимптомным синд-
ромом Гужеро—Дюперра (Gougerot—Duperrat). Есть основание предпо-
лагать, что ангиодерматит Гужеро—Блюма и синдром Гужеро—Дюперра
близки друг к другу и что между ними имеются переходные формы.
Клиника. При телеангиэктатической пурпуре Майокки очагам по-
ражения свойственна кольцевидная форма и наличие телеангиэктазий.
В сформировавшихся очагах поражений прогрессирующего пигментного
дерматита можно различать две зоны: центральную, представляющую пят-
но буровато-коричневого, ржавого цвета, иногда с легкой поверхностной
атрофией, и периферическую зону, образованную мелкими темно-красными
пятнышками, подобными зернышкам кайенского перца.
При ангиодерматите Гужеро—Блюма характерным симптомом является
группировка геморрагических пятен и узелков. У некоторых больных на-
ряду с группирующимися узелками может появляться и рассеянная
пятнисто-узелковая сыпь. Недавно возникшие элементы отличаются ярко-
красной, кирпично-красной окраской, более старые приобретают бурый
оттенок, на отдельных элементах можно отметить телеангиэктазии.
При гемосидерозе кожи обычно нет изменений ни в свертываемости,
ни в вязкости крови. Общее состояние пациентов остается удовлетвори-
тельным. Патологии со стороны внутренних органов не обнаруживается.
Субъективные ощущения в очагах поражений, как правило, отсутствуют.
Заболевание протекает хронически от нескольких месяцев до нескольких
лет, с тенденцией к. постепенному спонтанному разрешению.
Гистопатология. Гистопатологическая картина при разных формах
гемосидероза кожи однотипна. Поражение сосудов при гемосидерозе можно
охарактеризовать как продуктивно-инфильтративный капиллярит. Кро-
веносные капилляры в области сосочкового и подсосочкового слоев заметно
расширены, имеет место новообразование капилляров, их отечность и
расширение, диапедез, пролиферация эндотелия, отложение глыбок гемо-
101
сидерина в межуточном веществе, гистиоцитах и эндотелиальных клетках;
в окружности капилляров отмечается незначительная, лимфогистиоцнтар-
ная и плазмоклеточная инфильтрация. Фибриноидные изменения соеди-
нительной ткани отсутствуют.
Дифференциальный диагноз. Его следует проводить с атипичными
формами красного плоского лишая, особенно с пигментным красным пло-
ским лишаем, с псевдосаркомой Капоши, саркомой Капоши.
Лечение. Мероприятия, направленные на укрепление сосудистой
стенки, аскорбиновая кислота, рутин, препараты кальция, эскузан. При
наличии воспалительных явлений—мази с кортикостероидами.
ЛИТЕРАТУРА
Варшавский В. А., Иванов О. Л. и соавт. Иммуноморфологическая характеристика
васкулитов (ангиитов) кожи.— Вестн. дерматол. и-венерол., 1984, II, II—16.
Шапошников О. К., Деменкова И. В. Сосудистые поражения кожи.— Л., 1974.
Rook A., Wilkinson D., Ebling F. Textbook of Dermatology.— Blackwell, third
edition, Oxford, 1979, v. I, p. 981—992.
ГИДРОА ОСПЕННОВИДНАЯ (hydroa vacciniforme).
Синонимы: световая оспа; летняя гидроа; врожденный фотоактиниче-
ский эпидермолиз.
Впервые описана в 1892 г. французским дерматологом Bazin.
По-видимому, представляет собой неоднородную группу. Часть боль-
ных, у которых не выявляются нарушения порфиринового обмена, а
высыпания представлены только эритематозно-пузырьковыми элементами,
составляют группу истинной световой оспы. У других больных при тща-
тельном обследовании выявляют нарушения, специфичные для порфирий—
эритропоэтической протопорфирии, поздней кожной и неклассифициро-
ванной (Кривошеев Б. Н., 1981; Кузнецова Н. П. и соавт., 1981).
Этиология и патогенез. В основе заболевания лежит повышенная чув-
ствительность кожи к солнечным лучам, механизм развития которой не
ясен (Нерадов Л. А., 1964; Ashurst, 1969; Jaschke., 1975 и др.). Наследст-
венная Природа заболевания не доказана. Семейные случаи не выявлены.
Клиника. Заболевание чаще начинается‘у мальчиков в возрасте 2—5
лет, иногда в более раннем возрасте с первых же дней гГребывания на солнце.
Появлению сыпи обычно предшествуют продромальные симптомы общего
порядка. У ребенка, подвергшегося воздействию солнечного света, отме-
чаются явления возбуждения, потеря аппетита, тошнота, иногда рвота,
озноб, чувство жжения и зуда. Затем (обычно на следующий день после
облучения) появляется сыпь преимущественно на открытых частях тела—
на носу, щеках, ушных раковинах, тыле кистей в виде эритематозных пятен
размером с чечевицу или несколько крупнее и пузырьков шаровидной фор-
мы, наполненных прозрачным содержимым. Содержимое пузырьков быстро
мутнеет и пузырек западает в центре, напоминая пустулу при оспе. Обра-
зовавшийся в центре струп в виде черной корки приблизительно через
2—3 недели отпадает, оставив втянутый обесцвеченный рубчик, напоми-
102
нающий рубец от оспы. Пузырьки располагаются изолированно, но при
тесном расположении иногда сливаются. При тяжелых формах поражаются
также слизистые оболочки губ, глаз (светобоязнь, слезотечение, блефаро-
спазм, кератит), носа; наблюдается дистрофия ногтей и волос. Вспышки
болезни наступают весной и летом после более или менее длительного пре-
бывания на солнце. Интенсивность'вспышек уменьшается осенью, появле-
ние пузырьков прекращается полностью в зимнее время. У некоторых
больных к 15—30 годам заболевание может приобрести смягченное те-
чение и даже прекратиться. Ослабленная форма дерматоза (нерубцую-
щийся вариант)—hydroa aestivalis Hutchinson—чаще наблюдается у под-
ростков, не сопровождается некрозом и не оставляет рубцов.
Гистопатология. В эпидермисе пузыри, вначале многокамерные, а
затем, вследствие дегенерации эпидермальных клеток, однокамерные. Пу-
зыри выполнены фибрином, лейкоцитами и остатками дегенеративных
клеток мальпигиевой сети. В подсосочковом слое дермы на фоне тромбоза
сосудов очаги хронического воспаления с переходом в некроз и последую-
щим образованием рубца.'
Дифференциальный диагноз. Его следует проводить с поздней кожной
порфирией, летней почесухой, буллезным эпидермолизом, некротическими
угрями.
Лечение. Никотиновая кислота, препараты аминохинолинового ряда
(хлорохин, делагил, плаквенил), витамин В12, мази с фотозащитными сред-
ствами (парааминобензойная кислота, танин, хинин), кремы «Луч,» «Щит».
Больным рекомендуется избегать пребывания на солнце, носить фотоза-
щитные очки, шляпы с широкими полями, зонты.
ЛИТЕРАТУРА
Кривошеев Б. Н. Hydroa vacciniforme как клинический синдром кожных порфирий
и как самостоятельная нозологическая форма.— Вести, дерматол. и венерол., 1978,
4, 57 —61.
Кузнецова Н. П. и др. Порфирии.— М.: Медицина, 1981.
Jaschke Е., Reinken L., Frisch Н. Hydroa vacciniforme Bazin.— Hautarzt, 1975,
26, I, 11—17.
Lever W., Schaumburg-Lever G. Histopathology of the skin.— sth ed.— Philadelp-
hia, 1975, p. 193—197.
ГИПЕРКЕРАТОЗ СТОЙКИЙ ЛЕНТИКУЛЯРНЫЙ ФЛЕГЕЛЯ (hy-
perkeratosis lenticularis perstans—Flegel).
Этиология и патогенез. О возможной роли генетических факторов в
развитии дерматоза свидетельствуют семейные случаи (Dean, 1969; Frenk
и Tapernoux, 1976, и др.). В основе заболевания лежит нарушение керати-
низации неизвестной природы. Flegel (1958) полагал, что изменения на-
чинаются в дерме, в сосочковом слое, с последующим развитием гиперке-
ратоза. Frenk и Tapernoux (1976), напротив, считают, что формирование
гиперкератотических образований в зоне нормального эпидермиса—пер-
вичный процесс, все остальные изменения вторичны. Большое значение в
103
патогенезе заболевания авторы придают отсутствию телец Одланда и счи-
тают его аутосомно-доминантно наследуемым нарушением кератинизации,
вызванным небольшим клоном патологических кератиноцитов. Ikai и соавт.
(1977) обнаружили в гистиоцитах включения, природа которых неизвестна
(иммуноглобулины, патологические метаболиты или вирусы). В качестве
возможной причины развития дерматоза Krinitz и Schafer (1971) указывают
на длительный контакт с угольной пылью, Kocsard et al. (1968)—с дегтем.
Griineberg (1963) считал, что это заболевание является forme fruste фол-
ликулярного дискератоза Darier или редким вариантом верруциформного
акрокератоза Hopf. Muller и Schleicher (1968) рассматривают лентику-
лярный стойкий гиперкератоз Flegel как вариант болезни Kyrle. Frenk и
Tapemoux (1976) также обращают внимание на сходство этих дерматозов,
но считают стойкий лентикулярный гиперкератоз Flegel самостоятельным
дерматозом.
Клиника. Рассеянные мелкие (до 5 мм в диаметре) красновато-коричне-
ватые или желтовато-оранжевые роговые папулы, не сопровождающиеся
обычно субъективными расстройствами. После удаления роговых масс об-
наруживается углубление со слегка влажной поверхностью и феноменом
точечного кровотечения. Krinitz и Schafer (1971) наблюдали развитие в ос-
новании или вокруг папул желтовато-коричневатого или красновато-ко-
ричневатого валика. Высыпания располагаются преимущественно на ты-
ле стоп, голенях и бедрах. Значительно реже сыпь бывает и на руках
(Frenk и Tapernoux, 1976). У больного, описанного Kocsard и соавт. (1968),
были рассеянные высыпания и на туловище. Описан (Hunter и Donald,
1968) точечный кератоз на ладонях и подошвах в комбинации с рассеян-
ными роговыми папулами и мелкими псориазиформными бляшками. Koc-
sard и соавт. (1968) обнаружили маленькие округлые, величиной с острие
булавки вдавления, прерывающие кожный рисунок, на ладонях и в мень-
шей степени на стопах. Авторы полагают, что речь может идти о двух сход-
ных дерматозах: собственно стойком лентикулярном гиперкератозе Fle-
gel у пожилых мужчин с локализацией на конечностях и дискератотиче-
ском псориазиформном дерматозе, который может быть генерализованным;
встречается у мужчин и женщин с псориазиформными бляшечными про-
явлениями и очагами гиперкератоза. Общее состояние больных не стра-
дает.
Заболевание развивается у взрослых, несколько чаще у мужчин, с
пиком в интервале от 40 до 60 лет. Может протекать неопределенно долго,
без тенденции к регрессированию.
Гистопатология. Гиперкератоз, местами зоны истончения эпидермиса,
под участками гиперкератоза—акантоз, очаговый паракератоз, Kocsard
и соавт. (1968) подчеркивают, что утолщение рогового слоя папул носит
большей частью паракератотический, а не гиперкератотический характер.
В дерме—полосовидная инфильтрация из лимфоцитов и гистиоцитов. Miil-
1ег и Schleicher (1968) обнаружили преобладание в инфильтратах мелких
ретикулярных клеток, что дало им основание предположить возможность
ретикулярной гиперплазии неизвестного происхождения. Kuokkanen и
104
соавт. (1983) указывают на наличие в инфильтратах лимфоцитов с церебри-
формными ядрами. Raffle и Rogers (1969) обнаружили сосудистые измене-
ния в виде пролиферации эндотелия и утолщения стенок мелких сосудов.
Дифференциальный диагноз. Дифференцировать заболевание необхо-
димо от болезни Kyrle, псориаза, стуккокератоза, порокератоза Mibelli,
фолликулярного дискератоза Darier, бородавок, верруциформной эпидер-
модисплазии Левандовского—Лютца, ангиокератомы, парапсориаза, мышья-
кового кератоза.
Лечение. Удаление кюреткой наиболее крупных элементов, керато-
литические средства.
ЛИТЕРАТУРА
Frenk Е., Тарегпоих В. Hyperkeratosis lenticularis perstans (Flegel).— Dermatolo-
gica, 1976, 153, 4, 253—262.
Ikai K- Mural T., Takigawa M. et al. Intracytoplasmic inclusions in derma in-
filtrating cells of Hyperkeratosis lenticularis perstans.— Brit. J. Derm., 1977, 97, 4, 457—458.
Kuokkanen K-, Alaoaikko M., Pitranen R. Hyperkeratosis lenticularis perstans
(Flegel’s disease) Acta derm, venereol. (Stokh.), 1983, 63, 4, 357—360.
ГИПЕРКЕРАТОЗ ФОЛЛИКУЛЯРНЫЙ И ПАРАФОЛЛИКУЛЯРНЫЙ,
ПРОНИКАЮЩИЙ В КОЖУ (hyperkeratosis follicularis et parafollicularis
in cutem penetrans).
Синоним: болезнь Кирле.
Этиология и патогенез. Роль вирусной или бактериальной инфекции
и нарушение обмена витамина А, на что обращалось внимание, по мнению
Tappeiner и соавт. (1969), являются маловероятными. Наличие семейных
случаев свидетельствует о возможном участии наследственных факторов.
Наследование предположительно аутосомно-рецессивное. Tappeiner и соавт.
(1969) считают, что основное в патогенезе—это быстрое и преждевременное
ороговение клеток, которое может начинаться уже в нижних рядах ши-
повидного слоя. Тем самым граница между нормальными и ороговевшими
клетками смещается книзу, к базальному слою, клетки которого также
могут вовлекаться в этот процесс. В этот момент даже минимального меха-
нического воздействия достаточно, чтобы роговые массы проникли в дерму.
Клиника. Высыпания обычно множественны, состоят из гиперкера-
тотических папул с отчетливой склонностью к фолликулярному располо-
жению. Они сначала мелкие, с булавочную головку, постепенно увеличи-
ваются в размерах и достигают величины 1 см и более. Цвет папул серова-
тый, коричневато-красный или желтоватый. Окраска более интенсивна
по периферии папул. В центре их—гиперкератотическая пробка, размеры
которой увеличиваются по мере роста элементов (рис. 66) Если наслоения
роговых масс более или менее значительны, поверхность папул может ста-
новиться веррукозной. После удаления роговых пробок обнаруживается
слегка влажное или кровоточащее кратерообразное углубление. При слия-
нии высыпаний развиваются резко очерченные, нередко бородавчатые
бляшки неравномерных очертаний, иногда располагающиеся полосовидно,
большей частью в складках (подколенных, локтевых). Высыпания нахо-
' 105
Рис. 66. Гиперкератоэ фолликулярный и парафол-
ликулярный, проникающий в дерму (болезнь
Кирле).
дятся в разных стадиях раз-
вития: наряду с мелкими,
только что появившимися,
существуют и крупные очаги
поражения, частью уже ре-
грессирующие. Может на-
блюдаться феномен Кебнера.
Процесс может располагать-
ся на любом участке кожи,
быть распространенным, но
чаще локальный, с предпо-
чтительной локализацией в
области голеней, ягодиц,
предплечий. Высыпаний на
ладонях и подошвах не быва-
ет (Aram et al., 1969) или
это считается большой ред-
костью (Tappeiner et al..
' 1969). А. А. Антоньев (1953)
наблюдал высыпания в полости рта и на головке полового чле-
на. Общее состояние больных не страдает, субъективных расстройств
заболевание не вызывает. Имеются публикации о поражении кожи лица
(Abele и Dobson, 1961, и др.), но эта локализация, видимо, относится к
категории редких, как и волосистая часть головы (Gahlen и Grafendorf,
1980). Bjdrnberg (1959) указывает, что высыпания преимущественно рас-
полагаются на коже носа, где обнаруживаются насыщенно красные, ин-
фильтрированные очажки, покрытые слоистыми чешуйками, на внутрен-
ней поверхности которых обнаруживаются шипики.
Заболевание развивается обычно у взрослых, после 30 лет, но могут
болеть подростки и дети (Делис И. И. и Лилиене В. А., 1967). Протекает
длительно, отдельные высыпания могут регрессировать через 6—8 недель
с момента возникновения бесследно или оставляя пигментированные руб-
ЧИКИ, НО НЯ смену ИМ появляются новые. Самая большая продолжитель-
ность болезни—43 года (Bandmann и Schmid, 1968). Tappeiner и соавт. (1969), ,
проанализировавшие 28 опубликованных случаев болезни Кирле, пишут,
что полного выздоровления никто не наблюдал. Описаны сочетания с на-
личием у больных сахарного диабета, функциональных нарушений печени,
почек, удвоение почечной лоханки и мочеточника (Aram et al., 1969; Jacobi
и Engel, 1969; Petrozzi и Wartham, 1974). Tappeiner и соавт. (1969) реко-
мендуют обследовать всех больных для выявления латентного диабета.
Гистопатология. Она характерна и имеет диагностическую значимость
(Constantine и Carter, 1968); акантоз, резко выраженный фолликулярный
гиперкератоз, роговые пробки с паракератозом, с базофильным клеточным
детритом, проникновение в дерму роговых масс, гранулематозная реакция
вокруг них из лимфоцитов, эпителиоидных клеток, гигантских клеток
инородных тел.
106
Дифференциальнный диагноз. Проводят с бородавчатым плоским
лишаем, красным волосяным лишаем Девержи, фолликулярным дискера-
тозом Дарье, порокератозом Мибелли, стойким лентикулярным гиперке-
ратозом (Flegel), эластозом серпигинозным перфорирующим (Мишера—
Лютца), фолликулярным псориазом, ангиокератомой Мибелли.
Лечение. Витамин А, кератолитические и рассасывающие средства.
ЛИТЕРАТУРА
1
Gahlen W., Grussendorf Е.—J. Hyperkeratosis fofficularis et parafolficufaris im cu-
tem penetrans. In: Dermatologic in KTinik und Praxis. Hrsg G. W. Korting, Stuttgart, 1980,
Bd. 2, 2162—2164.
ГЛЮКАГОНОМА СИНДРОМ (glucagonomj syndromum)
Синоним: некротическая мигрирующая эритема.
Глюкагонома синдром—редкое клиническое состояние, характеризую-
щееся кожными высыпаниями, связанными со злокачественным новообра-
зованием поджелудочной железы. Becker и соавт. в 1942 г. впервые описали
связь кожных высыпаний с раковой опухолью панкреас. McGavran и соавт.
в 1966 указали на связь кожных изменений с карциномой глюкагонсекре-
тирующих а-клеток поджелудочной железы. Wilkinson (1971) опреде-
лил кожную симптоматику данного заболевания как некролитическую
мигрирующую эритему. Mallinson et al. (1974) ввели термин—Glucagono-
ma syndrome.
Sweet в 1974 г.’ наблюдал регрессию кожных поражений после хирур-
гического удаления опухоли а-клеток панкреас.
В настоящее время описано около 160 больных глюкагономой (Kahan
et al., 1977; Sweet, 1974; Freinkel et al., 1979; Lightman et al., 1974; Shu-
pack et al., 1978; Stacpoole et al., 1981 и др.). Мы не нашли в доступной
нам отечественной литературе описания данного синдрома.
Глюкагон является секретом а-клеток островков Лангерганса. Глю-
кагон и инсулин (гормон, продуцируемый Р-клетками) можно рассмат-
ривать как единую, бигормональную функциональную систему, которая
оказывает влияние на все виды обмена человеческого организма—липид-
ный, углеводный, белковый и минеральный. Глюкагон и инсулин облада-
ют диаметрально противоположным действием. Так, если глюкагон уве-
личивает уровень глюкозы в крови, что является одной из причин разви-
тия сахарного диабета у больных глюкагономой, то инсулин снижает уро-
вень глюкозы в крови. Глюкагонома секретирует значительное количест-
во глюкагона, причем его уровень в плазме крови может превышать нор-
мальный в 4—8 и более раз.
Этиология и патогенез. По мнению большинства современных авторов
(Wilkinson, 1973; Mallinson et al., 1974; Binnick et al., 1977; Pedersen и
Holst, 1976; Higgins et aL, 1979 и др.), гиперглюкагонемия может быть
следствием неопластического процесса в а-клетках островков Лангер--
ганса. Глюкагон в отличие от анаболического действия инсулина обла-
дает ярко выраженным катаболическим эффектом, что клинически про-
' 107
является различными дистрофическими процессами в коже, слизистых
оболочках, крови, нервной системе, потерей веса и др.
Клиника. Кожные поражения являются наиболее частыми и ранними
признаками синдрома, они нередко предшествуют клиническим симптомам
опухоли поджелудочной железы. Higgins et al. (1979), анализировавшие
клинические особенности случаев глюкагономы синдрома, опубликован-
ных в литературе до 1978 года, указывают, что только у 5 из 47 больных
не были обнаружены кожные поражения.
Возраст больных колеблется от 19 до 73 лет, однако около 85% описан-
ных больных были женщины в возрасте от 45 лет и старше, что дало осно-
вание некоторым авторам отнести этот синдром к болезням «после мено-
пауЬы».
На основании собственного наблюдения и анализа опубликованных
случаев можно систематизировать проявления синдрома: потеря веса (от
10 до 25 кг и более), слабость, плохой аппетит, анемия, глоссит, сахарный
диабет, изменения на коже.
Кожные высыпания начинаются в разных местах, обычно сразу ши-
роко распространяются, но наиболее тяжело поражается низ живота, бе-
дра, промежность, область между ягодицами. Кроме кожи, часто поража-
ются красная кайма губ, слизистая рта и влагалища. Поначалу появляются
эритематозные пятна, которые переходят в вялые пузыри. На месте пу-
зырей очень рано образуются корки, а на трущихся местах—мокнущие
поверхности. Высыпания имеют тенденцию к разрешению с центра с обра-
зованием распространяющейся кольцевидной корки красного цвета, что
дало всему кожному синдрому название некролитической эритемы (Wil-
kinson, 1973). Слияние отдельных очагов ведет к образованию крупных
бляшек, отграниченных от здоровой кожи фестончатым краем. Разрешение
обычно сопровождается гиперпигментацией. Клиническая картина некро-
литической мигрирующей эритемы отличается ярко выраженным поли-
морфизмом: рядом с гиперпигментированными разрешившимися участка-
ми отмечакУгся эритематозные пятна, вялые пузыри и корки. Обычная трав-
ма от обуви, серег и т. д. ведет к образованию новых высыпаний. Наблю-
дается дистрофия ногтей, диффузное поредение волос. Higgins (1979) у
1/3 больных отмечал стоматит или глоссит.
Кожные проявления глюкагономы нередко имеют атипичный псо-
риазоподобный вид, располагаются на нижних конечностях, на животе,
ягодицах, перианальной области (Freinkel et al., 1979; Stacpoole et al.,
1981; и др ). Мы (Каламкарян А. А. и Гордина А. А., 1984) наблюдали
в отделе дерматологии ЦКВИ пациентку 20 лет, у которой на фоне типич-
ной общей симптоматики глюкагономы имелись кожные высыпания, кото-
рые ошибочно воспринимались лечащими врачами как псориаз. Гистологи-
ческие иссследования исключали это заболевание. Отдельные высыпания
у нее начинались с мелких экземоподобного характера очажков с узелко-
вым компонентом. В дальнейшем очаги поражения покрывались корками
и чешуйками, приобретая псориазоподобный характер.
Прогноз большей частью неблагоприятный, течение медленное. За-
108
болевание, как правило, заканчивается метастазами через несколько лет
после развития опухоли. При своевременно проведенном оперативном
лечении прогноз несколько улучшается.
Гистопатология. Исследование свежих высыпаний показывает бурый
некролиз клеток поверхностных слоев эпидермиса и их отслоение в виде
корки или подкорнеального пузыря. Непостоянный акантоз СО СПОНГИО-
ЗОМ. Гистологически имеется сходство с токсическим эпидермальные не-
кролизом, но поражается только верхняя часть мальпигиевою СЛОЯ. От-
сутствие акантолиза, негативная реакция, иммунофлюоресценции.
Дифференциальный диагноз. Следует проводить с токсическим эпи-
дермальным некролизом, листовидной пузырчаткой, себорейной экземой,
обычным и пустулезным псориазом, герпетиформным импетиго Гебры,
доброкачественной семейной пузырчаткой, энтеропатическим акродерма-
титом, полиморфной эксудативной эритемой, герпетиформным дерматитом.
Лечение. Оперативное удаление опухоли, как правило, вызывает
регресс кожных высыпаний; рецидив высыпаний связан с развитием мета-
стазов В последнее время все больше внимания уделяется химиотерапии
стрептозотоцином Возможно применение а-аспарагиназы и цитостатиче-
ских химиотерапевтических средств. В целях компенсации диабета назна-
чают инсулинотерапию. Для наружного лечения мази с кортикостерои-
дами крем солкосерил и др. Необходимо систематическое диспансерное
наблюдение эндокринолога и дерматолога.
ЛИТЕРАТУРА
Abraira С., Debartolo М., Kaizen R. Disappearance of glucagonoma rach after sur-
gicaf resection, but not during dietary normalization of serum amino acids.— Amer. J. CTin.
Nutr., 1984, 39, 3, 351—355.
Binnick A. et al. Glucagonoma syndrome.— Arch, dermatof., 1977, 113, 749—754.
Freinkel R., Frienkel N. Dermatologic manifestations of endocrine disoders.— In:
Textbook of dermatofody. Oxford—London, 1979, v. 2, p. 1247—1265.
Higgins G., Recant L., Fischman A. The glucagonoma syndrome surgitally corabfe
diabetes.— Amer. J. Surg., 1979, 137, I, 142—148.
Kahan R. et al. Necrolytic. migratory erythema.— Arch. DermatoL, 1977, 113, 792—797.
Lightman S. et al. Cure of insulin-dependent diabetes mellitus by removal of a glu-
cagonoma.— Brit. Med. J., 1974, I, 367—374.
Lokich R. et al. Pancreatic alpha cell tumors.— Cancer, 1980, 45, 2675—2683.
Mallinson C. et al. A glucagonoma syndrome.— Lancet, 1974, v. 2, p. 1—5.
Pedersen N., Jonsson L., Holst J. Necrolytic migratory erytjiema and glucagon cell
tumor of the pancreas: the glucagonoma syndrome report of two cases.—Acta Dermato-vener.,
1976, 56, 391—395.
Riddle M. et al. Gfucanonoma syndrome in a 19-year-old woman.— Kest. J. Med.,
1978, 129, 68—72.
Scully R., McNeely B. Case records of the Massachusetts General Hospital.— N.
Engl. J. Med., 1975, 292, 1117—1123.
Shupack J. et al. The glucagonoma syndrome.— J. DemrmatoL Surg. Oncol., 1978,
4, 3—9.
109 *
Stacpoole P. et al. A familial glucagonoma syndrome. Genetic, clinical and biochemi-
cal features.— Amer. J. Med., 1981, 70, 5, 1017—1025.
Sweet R. Dermatosis specifically assciated with a tumour of pancreatic af[fha cells.—
Brit. J. Dermatol., 1974, 90, 301—308.
Swenson K-. Amon R.', Hani fin J. The glucagonoma syndrome.—Arch. Dermatol.,
1978, 114, 2, 224—228.
ГРАНУЛЕМА КОЛЬЦЕВИДНАЯ (granuloma anulare).
Этиология и патогенез. Наибольшее значение придается изменениям
в иммунной системе. Ряд авторов (Umbert и Winkelman, 1977; Kossard и
Winkelman, 1978 и др.) считает, что кольцевидная гранулема является
заболеванием, обусловленным гиперчувствительностью замедленного типа.
По их мнению, на это указывает обнаружение в очагах поражения фибри-
на, сходное с таковым при процессах, вызванных аллергическими реакция-
ми замедленного типа, мононуклеарный характер инфильтратов с нали-
.чием активированных макрофагов и фибробластов, иногда связь с сарко-
идозом, развитие болезни после туберкулиновых проб. Выявлено (Frid-
man-Birnbaum et al., 1983) нарушение клеточного иммунитета. Обнару-
жение в стенках мелких сосудов иммуноглобулина М и Q, фибриногена
послужило основанием для заключения, что кольцевидная гранулема яв-
ляется следствием васкулита (Dahl et al., 1977), тем самым изменения кол-
лагена рассматриваются как вторичные. Lever (1983) выражает сомнение
в наличии иммунокомплексного васкулита при этом заболевании, учитывая
разнородность данных иммуноморфологических исследований разных ав-
t< job, и допускает возможность очагового , повреждения коллагена как
первичного процесса. Такая точка зрения высказывалась Nishijama (1963),
Wolff и Maciejewski (1977). По мнению Л. Н. Машкиллейсона (I960),
кольцевидная гранулема нередко имеет туберкулезную природу, иногда
развивается после острых инфекций у людей, страдающих ревматизмом.
Наблюдалось развитие ее после травм, укусов клещей, насекомых, сол-
нечных ожогов и других неблагоприятных воздействий (Моуег, 1964;
Stankler et al., 1967; Dicken et al., 1969). Gross и Shelley (1971) обнаружили
связь генерализованной кольцевидной гранулемы с антитиреоидными ан-
тителами. Имеются сведения о возможной роли генетической предраспо-
ложенности (Rubin и Lynch, 1966, и др.). Что касается общих метаболиче-
ских нарушений, в частности углеводного обмена (Хазизов И. Е., 1984;
Кет и Schieff, 1960, Haim et al., 1973), то, наиболее вероятно, они имеют
большее значение при генерализованных формах. По мнению Haustein
(1976), имеется сходство ультраструктуры кольцевидной гранулемы и
диабетического липоидного некробиоза. На возможное различие в патоге-
незе разных вариантов кольцевидной гранулемы может указывать обна-
ружение (Friedman-Birnbaum и соавт. 1978) учащения при диссеминирован-
ной форме по сравнению с локальной антигена тканевой совместимости
В w 35.
Клиника. Наиболее часто встречающаяся форма имеет вид кольце-
видных очагов величиной 2—5 см и более (в редких случаях может дости-
гать гигантских размеров), овальных, округлых или г°же неправильных,
110
Рис 67. Кольцевидная
гранулема.
полициклических очертаний с несколько запавшей центральной частью,
состоящих из мелких узелковых элементов, плотноватой консистенции,
довольно глубоко залегающих в дерме с гладкой поверхностью, розовато-
сиреневатых или цвета нормальной кожи (рис. 67). Описаны проявления
в виде колец, вложенных друг в друга, как при многоформной эксудатив-
ной эритеме (Brown, 1972). Предпочтительной локализацией является
тыльная сторона кистей и стоп, реже высыпания располагаются в области
крупных суставов или других участков кожи. Так, из 18 больных, наблю-
давшихся Ю. К- Скрипкиным и О. Е. Хононовой (1963), у 3 больных они
располагались на голенях, у 2—на предплечьях, у 2—на локтевых суста-
вах, предплечьях и голенях. Лицо и волосистая часть головы поражаются
редко (Farlend et al., 1982). Из 6 случаев кольцевидной гранулемы на лице,
описанных Coskey (1979), у 3 высыпания располагались только на лице.
По наблюдениям авторов, они могут быть интрадермальными или подкож-
ными, одиночными или множественными. Глубокие, обычно единичные,
чаще располагаются периорбитально в области век. При локализации
на лице нодулярных элементов возможно сходство с лепрой (Leiker et al.,
1964). По наблюдениям В. И. Сяно (1967), у детей высыпания располага-
ются преимущественно на ногах, прежде всего на стопах, а у взрослых—
на руках, особенно на кистях. Описана локализация и в полости рта (Zan-
gel, 1963). У детей чаще, чем у взрослых, встречаются множественные очаги.
Высыпания асимптомны, как правило, не изъязвляются. Из других форм
следует в первую очередь отметить диссеминированную (генерализованную)
кольцевидную гранулему. В этих случаях высыпания множественны, рас-
сеянные или сливающиеся, что может придавать очагам сетчатый харак-
тер (Stankler и Leslie, 1967), но без значительной склонности к кольцевид-
ному расположению. Отмечается полиморфизм высыпаний (Kansky, 1975).
Dicken и соавт. (1969) наблюдали 26 больных этой формой. Гистологическая
111
картина была сходна' с таковой при типичной кольцевидной гранулеме,
но поражение было в основном у людей старше 40 лет. Отмечено большое
клиническое и гистологическое сходство с саркоидозом и лихеноидным
туберкулезом кожи. Мужчин было 8, женщин 18. Отмечались эритематоз-
ные, эритемато-папулезные, папулезные, кольцевидные и нодулярные
высыпания, цвета кожи или красноватые, желтоватые, коричневатые. У
большинства—асимптомные, зуд был только у 4 больных Не было ассо-
циации с какими-либо другими заболеваниями. В го же время другими
авторами обращается внимание на частое развитие диссеминированной фор-
мы у людей, страдающих нарушениями углеводного обмена По мнению
Romaine et al. (1968), атипичные формы кольцевидной гранулемы могут
рассматриваться как возможные проявления диабета Muller и Winkel-
mann (1966) считают липоидный некробиоз у диабетиков и кольцевидную
гранулему вариантами одного состояния на основании сходства гистоло-
гической картины. В то же время Meier-Ewert и Allnby (1971) не нашли
ассоциации между кольцевидной гранулемой и сахарным диабетом. Име-
ются наблюдения о связи генерализованной кольцевидной гранулемы с
антителами к тиреоидному глобулину (Goss и Shelley, 1971) Описано раз-
витие генерализованной гранулемы у годовалого ребенка (Takigawa и
Aushima, 1975). Dimitrowa и соавт. (1983) наблюдали лихеноидные фолли-
кулярные генерализованные высыпания у больного диабетом и саркои-
дозом, клинически сходные с лихеноидным туберкулезом, но гистологиче-
ски являющиеся кольцевидной гранулемой.
Более редкими из атипичных форм являются перфорирующая и под-
кожная (глубокая)—варианты кольцевидной гранулемы Перфорирующая
кольцевидная гранулема клинически имеет вид множественных мелких
папулезных элементов с западением в центре, располагающихся на дисталь-
ных частях конечностей, особенно на кистях (Owens и Freeman, 1971),
иногда на лице, шее и туловище (Izumi, 1973; Czarnecki и Hinter, 1979).
Может быть фолликулярной (1982). Регрессирование возможно
с рубцеванием. Подкожная кольцевидная гранулема развивается
почти исключительно в детском возрасте. Располагается глу-
боко в виде крупных плотноватых узелков, может быть спаяна с подле-
жащими тканями, встречается в основном на ногах, ягодицах, волосистой
части головы, на ладонях. Может быть изолированной, реже появляется
одновременно или предшествует типичным высыпаниям Обращается вни-
мание на трудности разграничения этой формы от ревматических узелков,
учитывая очень большое сходство гистологической картины при этих за-
болеваниях (Reinhard et al., 1971). Подчеркивается, что диагноз должен
ставиться с учетом всех клинических симптомов, выявленных у больных.
По мнению Lowney и Simons (1963), это в первую очередь отиоси'гря v изо-
лированным формам у взрослых, когда подкожные нодулярные элементы
могут сочетаться с другими признаками ревматоидного артрита или быть
связаны с системным сосудистым заболеванием. У детей—течение, сходное
с обычной формой кольцевидной гранулемы, если развитие подкожной
гранулемы не совпало с ревматизмом.
112
Описана эритематозная форма (Selmanowitz et al., 1966) с локализацией
на тыле кистей, предплечьях.
Pachinger и Kerb (1983) выделяют следующие атипичные варианты
кольцевидной гранулемы: мелкопапулезно-лихеноидная, эритематозно-ин-
фильтративная, полиморфно-бляшечная, туберозная, подкожная, пер-
форирующая.
Типичная форма кольцевидной гранулемы встречается преимущест-
венно у детей (в 70%—по данным Сяно В. И., 1967), из взрослых чаще
болеют женщины. Развивается медленно, течет неопределенно длительное
время, но в большинстве случаев регрессирует, в том числе спонтанно,
без рубцевания (может быть, как указывалось, при перфорирующей коль-
цевидной гранулеме) в течение 1—2 лет. Возможны рецидивы. Течение
генерализованных форм значительно более продолжительное, до 25 лет
(Stankler a. Leslie, 1967). Встречаются сочетания с разнообразными общими
и кожными болезнями: липоидным некробиозом (Schwartz, 1982), сахар-
ным диабетом (Romaine et al., 1968 и др.), ревматизмом (Eichmann, 1973),
туберкулезом, саркоидозом (Umbert a. Winkelmann, 1977).
Гистопатология. В эпидермисе особых изменений нет, за исключением
случаев перфорирующей формы; в дерме незначительные изменения в со-
судах (эндотелиальная пролиферация), околососудистые очаги дегенера-
ции коллагена с некрозом или некробиозом и отложением муцина, фиб-
рина, окруженные инфильтратами из фибробластов, лимфоидных клеток,
единичных гигантских клеток (туберкулоидная структура встречается
редко) Umbert и Winkelmann (1977) наблюдали преимущественно моно-
нуклеарную инфильтрацию в 72% случаев, палисадообразный—в 25%,
эпителиоидно-клеточный тип—в 3%. В поздних стадиях—фиброз. При
'перфорирующей форме—очаги дегенерации коллагена под эпидермисом с
проникновением некробиотических масс через эпидермис.
Лечение. Внутриочаговое введение кортикостероидов, кортикосте-
роидные мази под окклюзионную повязку, диметилсульфоксид, криоте-
рапия.
ЛИТЕРАТУРА
Хазиэов И. Е. Состояние углеводного обмена у больных кольцевидной гранулемой.
Вестн. дерматол. и венерол. 1984, 9, 53—57.
Bardach Н. G. Granuloma annulare with follicular perforation.— Derma tologica,
1982, 165, 47—53.
Coskey R. J. Facial granuloma annulare.— Arch, derm., 1979, 115, 7, 866—868.
Czarnecki N., Hinter H. Disseminiertes perforierendes Granuloma annulare.— Hau-
tarzt, 1979, 30, 6, 295—298.
Dahe M. V., Cherney K. J., Lindroos W. E. Elevated heparin-pricipitate fraction
of Plasma in granuloma anulare.— Arich. derm., 1979, 115, 9, 1059—1060.
Farland Me. J. P., Kanh Y. C. Luscombe H. A. Periorbital granuloma anulare.—
Arch, derm., 1985, 118, 3, 190—191.
Friedman-Birnbaum R., Haim S., Gideone O., Barzibei A. Histocompatibility
antigens in granuloma anulare.— Brit. J. derm., 1978, 98, 4, 425—427.
Friedman-Brirnbaum R., Gilhar A., Haim S., Golan D. T. Leucocyte inhibitory
8 Клиническая дерматология ИЗ
factor in granuloma annulare: a comparative study between the generalized and localized ty-
pes — Acta derm, venererol. (Stokh.), 1983, 63, 3, 242—243.
| Kansky A. Granuloma annulare disseminatum.— Hautarzt, 1975, 26, 9, 492—494.
Rossard S., Winkelmann R. K- Response of generalized granuloma anulare to al-
kyloting agents.— Arch, derm., 1978, 114, 2, 216—220.
Mensing H-, Janner M. Granuloma anulare giganteum.— Hautarzt, 1981, 32, 5,
25Л—257.
Pachinger W., Kerl H. Zur klinischen Vielfalt des Granuloma anulare.— Z. Haut-
fcrkh, 1983, 58, 21, 1559—1570.
Schwartz M. E. Necrobisis lipoidica and granuloma anulare.— Arch, derm., 1982,
118. 3. 192—193.
Stankler L., Leslie I. Generalized Granuloma anulare in a IS-rMonth-pld—Infant—
Dumatologica, 1976, 153, 3, 202.
Umbert P., Winkellmann R. K. Granuloma anulare and sarcoidosis. Brit. J. derm.,
1977, 97, 481—486.
ГРАНУЛЕМА ЛИЦА ГАН ГРЕН ЕСЦИРУЮЩАЯ (granuloma faciei
gangraenescens).
Синонимы: злокачественная срединная гранулема лица, прогресси-
рующая смертельная грануляционная язва носа.
В 1896 г. McBride впервые опубликовал наблюдение злокачественной
гранулемы носа. Из отечественных и зарубежных исследователей сообще-
ния о гангренесцирующей гранулеме содержатся в публикациях А. И.
Картамышева (1944), А. Ф. Фотина и соавт. (1961), А. Я- Прокопчука и
соавт. (1967, 1971), Э. А. Ладыженской (1979), Н. Е. Ярыгина и соавт.
(1980), Levan (1953), Alexander (1959), Altenburger и Pfeiffer (1962), Schle-
gel et al. (1979), Jshii и соавт. (1982) и др.).
Этиология и патогенез. Существуют инфекционная, опухолевая и
иммунная гипотезы. Некоторые авторы (Goldman и Churg, 1954; Попов Л.,
1961 и др.) рассматривают злокачественную или гангренесцирующую гра-
нулему лица и гранулематоз Вегенера как различные стадии одного забо-
левания. Однако А. И. Картамышев, (1944), А. Я- Прокопчук и соавт.
(1971), Е. П. Пирогова и В. С Шапиро, 1960; Н. Е. .Ярыгин и соавт. (1980),
Lever a. Schaumburg-Lever (1975), Simonis-Blumehfrucht и соавт. (1979).
и др. рассматривают гранулематоз Вегенера и гангренесцирующую гра-
нулему лица как самостоятельные нозологические формы, входящие в
группу системных аллергических васкулитов. По мнению Н. Е. Ярыгина
и соавт. (1980), в основе гангренесцирующей гранулемы лежит своеобраз-
ное иммунное воспаление слизистой оболочки и подлежащих тканей верх-
них отделов воздухоносных путей, сопровождающееся широким вовлече-
нием в процесс сосудов микроциркуляторного русла с последующей их
деструкцией.
Клиника. Мужчины болеют несколько чаще, чем женщины. Заболе-
вание встречается в любом возрасте, но чаще у лиц 30—50 лет. В течении
гангренесцирующей гранулемы различают три стадии. Первая стадия ха-
рактеризуется ринитом: серозно-водянистыми выделениями из носа, скоп-
114
Рис. 69. Ган*ренизирукщая
гранулема лица.
Рис. 68. Гангренивирующая
гранулема лица.
лением кровянисто-гнойных корок в носовых ходах, носовыми кровотече-
ниями. Во второй стадии (рис. 68, 69) идет прогрессирование язвенно-
некротических изменений, выделения из носа становятся гнойно-кровя-
нистыми, нарастает отек в гридаточных пазухах носа, в области носовых
раковин и перегородки образуются язвы, иногда с перфорацией. Мягкие
ткани лица и носа припухают. У отдельных больных наблюдаются дест-
рукция костно-хрящевой основы носа, костей верхней челюсти и влажная
гангрена мягких тканей лица (преддверие носа, верхняя губа, углы рта).
В тоегьей терминальной стадии болезни гнойно-некротические, некротиче-
ски-язвенные и гангренозные изменения в полости носа, околоносовых
пазухах, носоглотке, глотке, полости рта и воспалительно-деструктивные
процессы в костях носа и верхней челюсти достигают максимальной сте-
пени. Влажная гангрена с преддверия носа, верхней губы и углов рта рас-
пространяется на щеки, крылья носа и нижнюю губу. Происходит разру-
ление стенок гайморовых пазух, носовой перегородки, раковин и костей
стенки носа. Некроз распространяется на твердое небо, образуется рото-
нлглппй свищ Некротически-язвенные изменения изредка отмечаются так-
же в пищеводе, гортани, трахее и крупных бронхах. Резко ухудшается
общее состояние больных. Иногда воспалительные и деструктивные про-
цессы возникают в нижней челюсти, глазнице и внутреннем ухе. По свод-
ным данным Culter и Blatt (1956), поражение органа зрения встречается
в 42% случаев гангренесцирующей гранулемы. Основным симптомом па-
тологии глаз у таких больных является экзофтальм, возникающий в связи
с гранулематозной инфильтрацией глазных тканей и снижение зрения.
115
Прогноз неблагоприятный. Смерть наступает через 1—2 года. Боль-
ные погибают от сепсиса, нарастающей кахексии или массивного крово-
течения из эрозированных сосудов.
Гистопатология. Аллергические деструктивно-продуктивные васкулиты
микроциркуляторного русла, сопровождающиеся формированием изоли-
рованных полиморфноклеточных гранулем с последующим некрозом в
этих участках тканевого субстрата. Иногда наблюдается картина продук-
тивного или деструктивно-продуктивного артериолита с полным разруше-
нием сосудистых стенок.
Лечение. Эффективных средств лечения нет. Временный эффект на-
блюдается от комбинированного лечения массивными дозами кортикосте-
роидов в сочетании с цитостатиками (проспидин, циклофосфан), антибио-
тиками широкого спектра действия. В лечебный комплекс включают также
рентгеновское облучение.
ЛИТЕРАТУРА
Ярыгин Н. Е., Насонова В. А., Потехина Р. Н. Системные аллергические ва-
скулиты.— М.: Медицина, 1980.
Ishii et al. Nasal T-cell lymphoma as a type of so—called «lethal midline granuloma.»
— Cancer, 1982, 50, 11, 2336—2344.
McGuirt IT., Rose E. Lethal midline granuloma: a pathological spectrum.— J. La-
ryng., 1976, 90, 459—466.
Schlegel R., Krauft R. Midline Granuloma. Eine Literatiirubersicht mit besonderer
Beachtung von diagnostischen und therapeutischen Problemen, erganzt durch drei eigene FaL
Ibeobachtungen.— H. N. O., 1979, 27, 10, 334—344.
Simon-Blumefrucht A. et al. Le granulome malin centro-facial. A propos d’un cs.—
Dermatologica, 1979, 158, 3, 153—162.
ГРАНУЛЕМА ЛИЦА ОДОНТОГЕННАЯ ПОДКОЖНАЯ (granuloma
odontogenum subcutaneum).
Синоним: мигрирующая гранулема.
Хронические очаги продуктивного воспаления, возникающие в под-
кожной клетчатке около постоянных зубов, в нижней, реже верхней че-
люсти. Происхождение одонтогенной гранулемы находится в тесной связи
с той или иной формой заболевания зубов (Рабинович Л. М., 1955; Stoll
и Solomon, 1963; Caron и Sarcany, 1964; Pindborg, 1979; Scott, 1980 и др.).
Большинство подкожных гранулем стерильно. В ткани подкожной грану-
лемы не обнаружены специфические возбудители вирусной природы,
друзы актиномикоза. Весьма вероятно, что гранулемы вызываются про-
дуктами распада тканей, поступающими из пораженных зубов по лимфа-
тическим сосудам и межтканевым щелям. По мнению Т. Г. Робустовой
(1983), одонтогенная гранулема может быть вызвана возбудителем актино-
микоза—лучистым грибом.
Клиника. Одонтогенная подкожная гранулема лица наблюдается как
у мужчин, так и у женщин в возрасте от 10—15 до 50—60 лет; наиболее
поражаемый возраст 10—35 лет.
116
В области больного зуба, в подкожном слое, появляется безболезнен-
ное при пальпации опухолевидное воспалительное образование величиной
с горошину; кожа над ним не изменена. Со стороны полости рта прощупы-
вается плотный тяж, который тянется от альвеолы пораженного зуба. В
дальнейшем возможны две формы течения процесса: стационарная и пол-
зучая (Лукомский И. Г. ). Для стационарной формы характерно разви-
тие процесса лишь на ограниченном участке, над которым истонченная
кожа принимает синевато-багровый цвет. В дальнейшем обычно образуется
свищ, из которого выделяется кровянистая или кровянисто-гнойная жид-
кость. Такое состояние гранулемы остается стабильным в течение многих
месяцев и даже лет. Для ползучей формы заболевания характерно дальней-
шее распространение процесса поверхностно под кожей. В подкожной клет-
чатке образуются сплошные поля воспаленной грануляционной ткани.
Над очагом поражения кожа истончается, становится дряблой, вялой,
синеватой окраски, в различных местах вскрывается неглубокими свища-
ми, при зондировании которых обнаруживается слепой конец в мягких
тканях. Характерна способность процесса в одном участке стихать, а по
соседству обостряться с образованием новых очагов и свищей. Такое пол-
зучее воспаление захватывает иногда большие участки кожи, обезображи-
вая лицо. Излюбленным местом поражения на лице является тело нижней
челюсти (45,6%), щека (42,1%), подчелюстная область и реже область
угла нижней челюсти, шея и подбородочная область. Заболевание длится
от 3 месяцев до 2—3 лет, чаще от 3 до 6 месяцев. Рентгенологически у вер-
хушек корней пораженного зуба обнаруживается тень гранулемы, кисты,
ограниченные остеомиелитные очаги в челюсти. Hyman и Brownstein (1966)
наблюдали билатеральную одонтогенную гранулему.
Прогноз благоприятный, при современных методах лечения обычно
наступает выздоровление.
Гистопатолагия. Гистологически обычно находят < неспецифическую
хроническую гранулематозную ткань или хроническую дермальную во-
спалительную реакцию. Wende и Salomon (1942), Sakamoto и Stratigos
(1973) отмечают, что гистологические изменения, особенно фистул, напо-
минают эпителиому. Возможно, что эти находки представляют псевдо-
эпител иоматозную гиперплазию. В некоторых гранулемах микроскопиче-
ская картина указывает иа наличие внутриклеточных свободно располо-
женных капель жира (окраска Суданом III); по периферии таких жировых
включений встречаются многоядерные гигантские клетки, то единичные,
то в виде отдельных скоплений.
Дифференциальный диагноз. Его следует проводить с остеомиелитом
костей лица, с врожденной фистулой, пиодермиией щеки, гранулематозом
различного происхождения (актиномикоз, туберкулез кожи, гумма, микоти-
ческая и бактериальная инфекция).
Лечение. Удаление зуба, явившегося причиной заболевания, с обя-
зательным выскабливанием лунки после экстракции и назначение антибио-
тиков. В целях профилактики-необходимо своевременно удалять не под-
дающиеся, лечению зубы с обязательным выскабливанием лунки.
117
ЛИТЕРАТУРА
Робустова Т. Г. Актиномикоз челюстно-лицевой области. М., 1983.
Pindborg J. Disorders of the oral cavity and lips.— In: Textbook A. Rook, D. Kilkin-
son, F. Ebling. Oxford—London, 1979, v. 2, p. 1857—1932.
Scott. Cutaneous odontogenic sinus.— J. Amer. Acad. Dermatol., 1980, 2, 6, 521—
524.
ГРАНУЛЕМА ПИОГЕННАЯ (granuloma pyogenicum).
Синонимы: ботриомикома, телеангиэктатическая гранулема, добро-
качественная гранулема на ножке.
Термин «ботриомикоз» впервые предложил Боллингер в 1887 году
при описании поражения легких V лошадей, вызываемого, как тогда по-
лагали, Botryomyces equina.
Этиология и патогенез. В 1897 году Poncet и Dor на съезде француз-
ских хирургов сообщили о 4 случаях ботриомикоза у людей, полагая, что
заболевание вызвано возбудителем лошадиного ботриомикоза. Дальней-
шие исследования показали, что мнение о микотической природе пиоген-
ной гранулемы (ботриомикомы) оказалось вшибочным. В настоящее время
большинство авторов рассматривают это заболевание как своеобразную
форму пиодермии. Другие исследователи (Скрипник С. В., 1978; Nodi,
1955; Oehlschlangel и Milller, 1964 и др.) относят его к капиллярным ге-
мангиомам с вторичной гранулематозной реакцией. Zala в 1973 г. выска-
зал предположение о том, что в основе болезни лежит ангиобластома, к
которой присоединяется бактериальная инфекция. В патогенезе пиоген-
ной гранулемы большую роль играет травма—порез, укол, ожог и др.
(Никольский П. В., 1927; Пашков Б. М., 1964; Арутюнов В. fl., 1972;
Degos, 1971; Berlin, 1972, и др.).
Клиника. Через 2—5 месяцев на месте травмы появляется безболез-
ненное опухолевидное образование величиной с горошину или вишню
темно-красного цвета с гладкой или дольчатой поверхностью. Опухоль
имеет плотноэластическую консистенцию. Она легко кровоточит, произ-
водит впечатление сосудистой, в дальнейшем частично изъязвляется и
дает кровянисто-гнойное отделяемое. Опухоль сидит на ножке, что при-
дает ей сходство с грибом. Ножка может быть различной длины. Пиоген-
ная гранулема чаще локализуется на кистях (рис. 70), стопах, лице, но
может располагаться и на других участках кожного покрова, на генита-
лиях, по краю век, реже на губах, языке. Регионарные лимфоузлы, как
правило, в процесс не вовлекаются, за исключением редких случаев с
вторичным инфицированием. Очень редко наблюдаются гигантские пио-
генные гранулемы размерами до 3—4 см в диаметре (Макашева Р. К-, 1960;
Некачалов В. Я., 1963). Также редко встречаются множественные пио-
генные гранулемы (рис. 71) (Данилевская Е. Д-, 1968; Каламкарян А. А.
и соавт., 1980; Zala, 1973; Schukralan, 1974; Kaminsky, 1978 и др.).
Чаще они появляются после ожога.
Гистопатология. Разрастание грануляционной ткани с большим ко-
личеством расширенных сосудов с набухшим эндотелием, напоминающее
118
Рис. 71. Множественная
пиогенная гранулема.
ангиому. Инфильтрат состоит из полиморфных лейкоцитов, плазматиче-
эких клеток, лимфоцитов, тучных клеток. Часто обнаруживается значи-
тельное количество стафилококков, стрептококков и др. Гистологическое
строение скорее свидетельствует в пользу гемангиомы.
Дифференциальный диагноз. Его проводят с кавернозной ангиомой,
саркомой Капоши, ангиосаркомой, ангиолейомиосаркомой, вегетирующей
пиодермией.
Лечение. Хирургическое иссечение, электрокоагуляция, лазерное
облучение.
ЛИТЕРАТУРА
Каламкарян А. А., Суколин Г. И., Ракчеев А. П. Гигантские множественные
пиогенные гранулемы (ботриомикомы), леченные лучами лазера.— Вести, дерматол. н
венерол., 1981, I, 34—37.
Kaminsky D. MultipT disseminated pyogenic granuloma.— Brit.. J. Dermatol., 1978,
88, 4, 461—464.
- Zaynoun S., Juljlian H-, Kurban A. Pyogenic granuloma with multiple satellites —
Arch. Dermatol., 1974, 109, 5, 689—694.
119
ГРАНУЛЕМА ЭОЗИНОФИЛЬНАЯ (granuloma eosinophilicum).
Синоним: болезнь Таратынова.
Эозинофильная гранулема кожи представляет собой неоднородную
группу, в которую в настоящее время включают: эозинофильную грану-
лему лица, эозинофильную гранулему кожи других локализаций, эозино-
фильную гранулему кожи, как проявление эозинофильной гранулемы ко-
сти и симптоматическую эозинофильную гранулему (Pincus, 1950; Knoth,
1954; Lever и Schaumburg-Lever, 1975). В настоящее время большинство
авторов относят ее в группу гистиоцитоза X, обозначаемую также как про-
лиферативно-обменный ретикулогистиоцитоз. В типичных случаях характе-
ризуется изолированным или множественным поражением костей. Однако
наблюдаются и поражения других органов, в частности кожи. Эозинофиль-
ную гранулему впервые описал Н. Й. Таратынов в 1913 г. Эозинофильную
гранулему кости с кожными проявлениями описывали многие авторы—
Curtis и Cawley (1947), Kendrick и соавт. (1948), Lever и Leeper, (1950), Herz-
berg, (1961), Kleine-Natrop et al. (1970), Ulcan и Pais (1977), Burgess et al.,
1977; Mahzoon и Wood (1980), и др.
Этиология и патогенез. Высказываются мнения о неопластической,
инфекционной, вирусной, инфекционно-аллергической природе заболева-
ния. Существует точка зрения, рассматривающая эозинофильную грану-
лему как форму, стоящую на грани между гиперпластическими и опухо-
левыми процессами.
Клиника. Эозинофильная гранулема кости, по сравнению с другими
заболеваниями, объединенными в группу «гистиоцитоз X», отличается
сравнительно более доброкачественным течением. Однако иногда процесс
захватывает несколько костей или даже органов и тогда заболевание про-
текает неблагоприятно. Наиболее часто поражаются череп, ребра, таз,
длинные трубчатые кости. Рентгенограмма показывает характерные из-
менения, которые могут вызывать спонтанные переломы. Основная жалоба
большинства больных—ноющие боли постоянного характера, усиливаю-
щиеся при движении и ночью. Встречаются и внекостные локализации—
в лимфатических узлах, печени, почках, головном мозге, коже, легких,
желудке. Описаны случаи сочетанных поражений костей, внутренних ор-
ганов и кожи.
Эозинофильная гранулема кости встречается преимущественно в дет-
ском и юношеском возрасте; в более старшем возрасте она встречается лишь
В единичных случаях. Кожные проявления при эозинофильной гранулеме
кости отличаются выраженным полиморфизмом, имеют различную лока-
лизацию и трудны для диагностики. Очаги поражений могут • встречаться
всюду—на лице, на волосистой части головы, в подмышечных впадинах,
на спине, перианально и на других участках, а также на слизистых оболоч-
ках. Сыпь может быть ограниченной и генерализованной. Кожные пора-
жения при эозинофильной гранулеме кости можно разделить на три ос-
новные группы: 1) мелкие папулезные высыпания—желтоватого или ко-
ричневатого цвета с чешуйками или чешуйко-корками на поверхности.
Сливаясь, они образуют диффузные очаги, напоминаюшие себорейный
120
Рис. 73. Эозинофильная
гранулема слизистой
полости рта.
Рис 72. Эозинофильная
гранулема гениталии
I7'
дерматит; 2) пурпурозно-геморрагические высыпания, локализующиеся
преимущественно на лице, шее, верхней половине груди, а также на сли-
зистой оболочке полости рта; 3) гранулематозные (узловатые и опухоле-
видные) очаги.
В наших наблюдениях (рис. 72, 73) начальными высыпаниями при
эозинофильной гранулеме были пятнисто-пурпурозные элементы или эк-
судативно-шелушащиеся папулы Из указанных первичных элементов в
дальнейшем развивались инфильтративные бляшки и опухолевидные об-
разования. Рассеянные или сливающиеся инфильтративно-бляшечные оча-
ги поражения располагались на голове, лице, груди, животе, спине, яго-
дицах и в парагенитальной области. Образующиеся бляшки хорошо от-
граничены, имеют полушаровидную или "Чаще уплощенную форму; они
выстоят над общим уровнем кожи. Консистенция бляшек большей частью
мягковатая, иногда плотноэластическая. Узловато-опухолевидные очаги
достигают величины сливы и больше, плотны на ошупь, реже имеют плотно-
эластическую консистенцию, окраска их синюшно-багровая. Как узлы,
так и опухоли заложены в коже и подкожной клетчатке. Гранулематозные
очаги часто изъязвляются с образованием поверхностных или глубоких
язв, имеющих неправильные очертания, валикообразно утолщенные края;
они окрашены в насыщенный синюшно-багровый цвет. Язвы отличаются
вялым течением. Отмечается тенденция к локализации поражений в ро-
товой полости и в гениторектальной области. У многих больных увеличи-
ваются периферические лимфатические узлы. Наблюдаются также пора-
жения кожи в виде веррукозных и вегетирующих очагов в перианальной
и ректальной областях, на слизистой оболочке ануса. Domonkos (1971) ука-
зывает, что заболевание может начаться в коже и ограничиться только
121
кожей. Заболевание отличается вялым течением и рецидивами. При по-
явлении на коже генерализованных узловато-опухолевидных высыпаний
болезнь может быстро прогрессировать и больные погибают.
Гистопатология. Отмечается умеренный акантоз эпидермиса и поли-
морфноядерный гистиоцитарный инфильтрат в сосочковом слое дермы. В
глубине дермы выявляется массивный инфильтрат из гистиоцитов с силь-
но выраженным полиморфизмом (крупные многоядерные с уродливыми
ядрами клетки). Во многих гистиоцитах обнаруживаются интенсивно окра-
шенные базофильные различной величины ядра и слегка эозинофильная
протоплазма. Встречаются в небольшом количестве лимфоциты и эозино-
филы. Инфильтрат проникает в эпидермис, вызывая дистрофию эпидерми-
са и изъязвление.
Дифференциальный диагноз. Его следует проводить с туберкулезом
кожи, глубокими микозами, ретикулогемобластозами, хронической яз-
венно-вегетирующей пиодермией.
Лечение. Рентгенотерапия, кортикостероидные гормональные пре-
параты. Мы наблюдали прекрасный эффект в отношении как костных,
так и коденых проявлений от применения преднизолона (40 мг в сутки)
в сочетании с проспидином по 100—200 мг ежедневно (на курс 3—4 г).
ЛИТЕРАТУРА
Гребенюк В. Н. Эозинофильная гранулема и кожа.— Вестн. Дерматол. и венерол.,
1978, 12, 33—37.
Burgess G. et al. Eosinophilic ulcer of the tongue.— Arich. DemrmatoL, 1977, 113,
5, 644—645.
Mahzoon S., WoodM. Multifocal eosinophilic granuloma with skin ulceration. Histio-
cytosis X of the Hand—Schulfer—Christian type.— Arch. Dermatol., 1980, 116, 2, 218—220.
Rook A., Wilkinson D., Ebling F. Textbook of dermatology, Oxford—London— Mel-
bourne, 1979, 3rd, ed, p. 1513—1,518.
Stateoa S., Kiriakoo L., Mustakoo G. Kongenital Histiocytosis X. Papufonodose
nekrotische Form.— Hautarzt, 1980, 31, I, 26—29.
Vulcan P., Paris V. Asupra aspectefor efectronomicroscopice in feziunile cutane de
histiocitoza X. Dermato—Venerologia, 1977, 22, 81—96.
ГРАНУЛЕМА ЭОЗИНОФИЛЬНАЯ ЛИЦА (granuloma eosinophilictim
faciei).
Синонимы: гранулема лица, гранулема лица с эозинофилией.
Pinkus в- 1950—1952 годах из неоднородной группы эозинофильных
гранулем выделил гранулему лица с эозинофилией.
Этиология и патогенез. В литературе высказывается мнение об этио-
логической роли травмы (Nanta и Gadrat, 1937; Degos et al., 1960), травмы
и последующего иифицироваиия (Рабен А. С. и Васильева Н. Н., 1960).
Johnson и соавт. (1959) считают, что патогенез гранулемы лица базируется
на реакции антиген-антитело или реакции гиперчувствительности, в кото-
рой шоковой .тканью являются мелкие кровеносные сосуды и периваску-
122
Нис. /4. асзинофилъная
гранулема лица.
Рис. 76. Эозинофильная
гранулема лица.
лярная ткань верхней части дермы лица. Авторы считают, что причинную
роль могут играть очаговая инфекция, повышенная фоточувствительность,
травма, лекарства. Cream J. и соавт. (1975), String и соавт. (1967) указы-
вают, что при гранулеме лица нередко обнаруживаются явления васкулита
по типу феномена Артюса. Schroeter и соавт. (1971) методом прямой им-
мунофлюоресценции обнаружили в субэпидермальной зоне очагов эозино-
фильной гранулемы лица отложения IgG, IgM, IgA и С.
123
Клиника. Заболевание развивается чаще у мужчин, преимущественно
в зрелом возрасте. Среди пациентов Jonson и соавт. (1959) было 14 мужчин
и 1 женщина в возрасте от 25 до 65 лет. Мы наблюдали 11 мужчин и 3 жен-
щин в возрасте от 14 до 67 лет.
Заболевание начинается с появления на лице 1—2 очагов поражения,.
которые имеют характер пятен, реже бляшек, узелков или узлов (Dimi-
trescu et al., 1978). Число и размеры их могут медленно увеличиваться.
Величина высыпаний колеблется от нескольких миллиметров до 5—8 см
в диаметре. Цвет очагов коричневато-красный, разной ийтенсивности.
Поверхность гладкая, без шелушения и гиперкератоза, с расширенными
фолликулярными устьями, нередко с мелкими телеангиэктазиями, гра-
ницы отчетливые. Наиболее частая локазизация высыпаний—спинка носа,
щеки, лоб, реже предушные и височные области, ушные раковины (рис.74,
75, 76, 77).
У большинства пациентов субъективных ощущений они не вызывают,
отдельные лица указывают на небольшой зуд, жжение, чувствительность
в пораженных местах. Высыпания существуют неопределенно долго (мно-
гие годы). У некоторых больных очаги поражения становятся более вы-
раженными (усиливается окраска, появляется отечность) от воздействия
тепла, солнечных лучей. У 6 из 15 больных, наблюдавшихся Johnson и
соавт., в анамнезе отмечались по одному или нескольку приступов «аллер-
гической болезни» (сенная лихорадка, астма, синуситы, крапивница, ле-
карственная аллергия). Общее состояние больных не страдает, патологи-
ческих изменений в крови и костях нет.
Ряд авторов указывают, что эозинофильная гранулема лица является
наиболее частым проявлением эозинольной гранулемы кожи (Pinkus, Cra-
mer).
Rusin и соавт. (1976) сообщили о двух больных, у которых через 1 год
и 9 лет от начала заболевания высыпания распространились на плечи,
грудь и верхнюю часть спины. Мы наблюдали экстрафациальную локали-
зацию эозинофильной гранулемы у одной пациентки—пятнистоузелковые
высыпания в пахово-бедренных областях и на больших половых губах.
Гистопатология. Различают лейкоцитарную (раннюю) и фибробласти-
ческую (позднюю) стадии. Характерен полиморфноклеточный инфильтрат
в верхней половине дермы, отделенный от эпидермиса и придатков кожи
узкой неизмененной полосой. В ранней стадии сосуды расширены, в ин-
фильтрате преобладают эозинофилы, но также много нейтрофилов и гистио-
цитов. В верхней части дермы может присутствовать гемосидерин. В позд-
ней стадии в инфильтрате преобладают плазматические клетки, лимфоциты,
фибробласты, полосы коллагена пронизывают инфильтрат (W. Lever и
С. Schaumburg-Lever, 1975).
Диагноз основывается на сочетании характерных клинических и
гистопатологических данных.
Дифференциальный диагноз. Его следует проводить с саркоидозом,
красной волчанкой, лимфоцитарной инфильтрацией Jessner, фиксирован-
ным медикаментозным дерматитом, erythema elevatum et diutinum.
124
Лечение Заболевание отличается резистентностью к лечению, в том
числе к рентгенотерапии и системному применению кортикостероидов.
Описаны случаи успешного хирургического иссечения, в то же время сооб-
щается о нередких рецидивах после эксцизии. Мы наблюдали рецидив эози-
нофильной гранулемы лица рядом с операционным рубцом. У 5 больных
нами зафиксировано частичное и даже полное разрешение очагов пораже-
ния после длительного применения синалара под окклюзионную повязку.
Имеются сообщения о благоприятном действии дапсона (Anderson, 1975),
введения в очаг кортикостероидов, дермабразии.
Литература
Dimitrescu Al., Trifu Р., Trifu R., Chiotan F. Granufomuf eozinifific aT feteT.—
Dermato—Vener., 1978, 23, I, 57—64.
Leoer W. F., Schaumburg-Leuer C. Granuloma faciale.— In: Vascular Disease.—
In: Histopathology of the Skin. Philadelphia—Toronto, 1975, p. 166—167.
Rusin L., Dubin H., Taylor W. Disseminated granuloma faciale.— Arch. Derma-
tol., 1976, 112, II, 1575—1577. *
ГРАНУЛЕМАТОЗ ВЕГЕНЕРА (granulomatosis Wegener).
Синонимы: идиопатическая смертельная гранулема, злокачественная
гранулема, риногенная гранулема и др.
Системный гиперергический панваскулит, сочетающийся с развитием
в тканях некротизирующихся гранулем. Поражаются преимущественно
верхние дыхательные пути, легкие, почки. Относится к числу сравнитель-
но редких заболеваний.
Впервые заболевание описал в 1931 г. Klinger. Позднее Wegener (1936,
1939), на основании тщательного клинического и патологоанатомического
изучения трех случаев этой болезни, выделил ее в самостоятельную
нозологическую форму, назвав риногенной гранулемой. Godman и Churg
(1954), анализируя 7 собственных наблюдений и 22 случая, опубликован-
ных в литературе, описали характерные гистоморфологические признаки
заболевания, назвав его «гранулематозом Вегенера.»
Этиология и патогенез. До настоящего времени отсутствуют четкие,
представления об этиологии и патогенезе этого синдрома. Принадлежность
страдания к узелковому периартерииту признается многими клиницистами
и патоморфологами (Пирогова Е. П. и Шапиро В. С., 1960; Арутюнов
В. Я- и соавт., 1963; Kunze, 1973: Lever и Schaumburg-Lever, 1975 и др.).
Н. Е. Ярыгин и соавт. (1980) рассматривают гранулематоз Вегенера как
самостоятельное страдание, в возникновении которого, как и при других
формах системных аллергических васкулитов, первостепенное значение
имеют иммунные нарушения. Некоторые авторы (Fauci и Wolff, 1974; John-
son, 1971; Spira et al., 1974 и др.) полагают, что гранулематоз Вегенера
является аутоиммунным заболеванием. В основе поражения кожи, как и
заболевания в целом лежат многосторонние нарушения иммунитета (Меу-
rick et al. 1982).
125
Клиника. Заболевание характеризуется гранулематозно-некротиче-
ским поражением верхних дыхательных путей и легких, генерализован-
ным васкулитом, частым вовлечением в патологический процесс почек по
типу гломерулонефрита. Заболевание встречается в любом возрасте—от
2,5—3 месяцев (Биллер Т. Н. р соавт., 1968; Carrington и Liebow, 1966)
до 75 лет (Каралицкий Е. М., 1982, Walton, 1958), но чаще в возрасте 40—
50 лет. У мужчин гранулематоз Вегенера встречается чаще, чем у женщин
(соотношение 2:1). Как правило, заболевание развивается постепенно, но
возможно и острое его начало и быстрое прогрессирование. В зависимости
от локализации процесса начальный период гранулематоза Вегенера кли-
нически подразделяется на два варианта. При первом варианте более чем
у половины больных процесс начинается с поражения верхних дыхатель-
ных путей (носовой полости, околоносовых придаточных пазух и гортани)
и чаще всего носит некротически-язвенный характер. Реже первично по-
ражается слизистая оболочка рта, глотки. Второй вариант протекает с
преимущественным поражением трахеобронхиального дерева и легких,
клинически проявляется лихорадкой неправильного типа, болями в груд-
ной клетке, приступообразным кашлем с гнойно-кровянистой мокротой.
Симптомы первого варианта могут присоединяться в более* поздние перио-
ды заболевания. Более чем у 85% больных вовлекаются параназальные
синусы, чаще всего максиллярные и этмоидальные. Больные жалуются на
затрудненное дыхание, упорный, не поддающийся лечению насморк
с гнойным или гнойно-кровянистым отделяемым и боли в области
околоносовых пазух. Затем процесс переходит на носоглотку, мягкие тка-
ни носа и другие части лица. У 40% больных наблюдается снижение слуха
из-за развития среднего отита или закупорки носоглотки грануляциями.
У 25% в процесс вовлекаются ‘трахея и гортань. При прогрессировании
процесса небные, носовые ткани и альвеолярный отросток верхней челюсти
вместе с мягкими тканями подвергаются некрозу, разрушаются; на лице
образуются большие дефекты. Нередки тяжелые параназальные синуситы,
назофарингальные язвы с перфорацией перегородки носа, деформацией
носа. В результате разъедания сосудов наблюдаются кровотечения, кото-
рые могут быть смертельными. У 40% больных имеют место орбитальные
изменения, включающие гранулематозные изменения склеры, роговицы,
конъюнктивы. Одновременно появляются головные боли и повышение тем-
пературы тела до высоких цифр. По истечении нескольких недель или ме-
сяцев локальные явления первой стадии сменяются генерализацией (вто-
рая стадия) воспалительно-некротических изменений с вовлечением В
процесс многих органов и тканей. Распространение процесса на легочную
ткань клинически проявляется симптомами очаговой пневмонии—упор-
ный кашель, сопровождающийся гнойно-кровянистой мокротой, крово-
харканьем и даже смертельным легочным, кровотечением. Температура
носит обычно септический характер. Рентгенологически отмечается уси-
ленный бронхососудистый рисунок. с развитием участков инфильтрации
и образованием полостей. На высоте или в исходе заболевания у 80—85%
больных часто присоединяются поражения почек (некротический гломеру-
126
Рис. 79. Гранулематоз
Вегенера.
Рис. 78. Гранулематоз
Вегенера.
лит), проярляющиеся протеинурией, гематурией, цилиндрурией, а затем
азотемией. Выявляются признаки системного страдания: полиморфные
сыпи на теле и конечностях, геморрагический синдром, суставные и мышеч-
ные боли, полиартрит, изменения нервной системы, эндокринные наруше-
ния и др. Наблюдается анемия гипохромного типа, нейтрофильный лейко-
цитоз в начале заболевания при нарастании тяжести процесса сменяется
лейкопенией. Обнаруживается повышение СОЭ, С,, С4 компонентов комп-
лемента, гиперглобулинемия с повышением IgG и IgA; реакция бластной
трансформации лимфоцитов у многих больных меньше 30—40%. Пора-
жения кожи наблюдаются в ранних стадиях болезни у 1/4 больных, а в
поздних—у половины больных (Reed et al., 1963; Fauci и Wolff, 1973; Shel-
don et al., 1974; Hu Ch. H. et al., 1977; Meyrick и соавт., 1982; Fauci et al.,
1983. и др.). Поражения включают различные формы специфических и
неспецифических высыпаний, пурпуры, телеангиэктазии, геморрагии, пу-
зырьки, папулы, подкожные узлы и язвы. Типичными кожными пораже-
ниями при гранулематозе Вегенера являются узлы и язвы, обусловлен-
ные некротическим ангиитом дермальных сосудов с тромбозом и некрозом
(рис. 78, 79). Поражения слизистой оболочки полости рта встречаются поч-
ти у всех больных, они аналогичны поражениям респираторного тракта
и состоят из язв, особенно на нёбе, слизистой щек и десен.
Прогноз неблагоприятный, болезнь в большинстве случаев заканчи-
вается летально. В течение 2 лет умирает 90% больных. Средняя продол-
жительность жизни больных от начала заболевания до летального исхода
около 5 месяцев (от 3—4 недель до 3—4 лет). Смерть наступает чаше при
явлениях почечной или сердечно-легочной недостаточности, внезапного
массивного кровотечения.
Гистопатология. Наиболее характерным морфологическим проявле-
нием синдрома Вегенера является рассеянный панваскулит, сочетающийся
127
с наличием в пораженных органах и тканях некротизирующихся грану-
лем. В мелких и средних венах и артериях отмечается мукоидное набу-
хание стенок, фибриноидный некроз ц разрастание гранулематозной ткани
на небольших участках или на всем протяжении. ПрОСВСТЫ НЕКОТОРЫХ
сосудов тромбированы. Гранулемы состоят из эпителиоидных и лимфоид-
ных клеток, плазмоцитов, гистиоцитов, нейтрофильных и эозинофильных
лейкоцитов и гигантских клеток, имеющих сходство с клетками Штерн-
берга, Лангханса или с гигантскими клетками инородных тел.
Дифференциальный диагноз. Его следует проводить с риносклеромой,
обострением хронического тонзиллита, геморрагическим васкулитом, ото-
риногенным сепсисом, туберкулезом легких, актиномикозом верхней че-
люсти, ринитом, гайморитом, септической ангиной, сифилитической гум-
мой, узелковым периартериитом и др.
Лечение. Радикальных средств лечения нет. Антибиотики и кортико-
стероиды неэффективны или малоэффективны на ранних стадиях заболева-
ния. Цитостатики (проспидин, циклофосфамид, 6-меркаптопурин и др.)
в сочетании с массивными дозами кортикостероидов (преднизолон в дозе
60—100 мг в сутки) оказывают временный эффект (удлиняется жизнь боль-
ных на несколько месяцев).
ЛИТЕРАТУРА
Каралицкий Е. М. Злокачественный гранулематоз Вегенера в практике врача-дер-
матолога. Вести, дерматол. и венерол., 1982, II, 46—49.
Ярыгина Н. Е., Насонова В. А., Потехина Р. Н. Системные аллергические ва-
скулиты.— М.: Медицина, 1980.
Fauci A., Magnes В., Katz Р., Wolff S. Wegener’s granulomatosis. Prospective,
clinical and therapeutic experience with 85 pasients for 21 years.— Ann. intern, med., 1983,
98, 1, 76—85.
Hu Ch. H., O'Loughlin S., Winkelmann R. Cutaneous manifestations of .Wegener’s
granulomatosis.— Arch. Dermatol., 1977, 133, 2, 175—182.
Meyrick T., Rowland P., Martin B. Wegener’s granulomatosis presenting as pyoder-
ma gangrenosum. Clin, and exp. dermatol., 1982, 7, 5, 523—528.
Robbett W., Rubin J. Wegener’s granulomatosis: case report and review of the lite-
rature.— Bull. New York Acad. Men., 1980, 56, 7, 623—631.
Spioa D., George J., Sears D. Acure autoimmune hemolytic anemia elue to a tow
molecular weight IgM cold hemolysis associated with episodic limphoid granulomatous vascu-
litis.— Amer. J. Med., 1974, 56, 417—428.
ГРАНУЛЕМАТОЗ ХРОНИЧЕСКИЙ ПРОГРЕССИРУЮЩИЙ
ДИСКООБРАЗНЫЙ МИШЕРА—ЛЕДЕРА (granulomatosis disciformis
chronica et progressiva Miescher—Leder).
Синоним: туберкулоидный псевдосклеродермиформный симметрич-
ный гранулематоз.
Заболевание описано швейцарскими дерматологами Miescher и Leder
в 1948 г. В отечественной и зарубежной литературе имеются отдельные
128
публикации, посвященные данному дерматозу (Арутюнов В. Я. и Ино-
земцева А. Ф., 1958; Машкиллейсон Л. Н и соавт., 1964; Марьясис Е. Д.
и Павлик Л. В 1980; Tappeiner, 1952; Bazex et al., 1966; Chiale et al.,
1971; Noster et al., 1974; Epstein, 1977; Miura и Tanahashi, 1978; Hundei-
ker, 1979).
Этиология и патогенез. Некоторые исследователи связывают заболе-
вание с туберкулезной инфекцией (Miescher и Leder, 1948; Degos et al.,
1960), другие рассматривают как особую разновидность саркоидоза (Tap-
peiner’, 1952; Kogoj и Puretic, 1953; Марьясис Е. Д. и Павлик Л. В.,
1980 и др.). Chiale и соавт. (1971) расценивают его как заболевание иммуно-
аллергической природы.
Острые дискуссии вызывает вопрос об отношении гранулематоза Ми-
шера—Ледера к липоидному некробиозу. На большом клинико-гистоло-
гическом материале было установлено, что у больных липоидным некро-
биозом без диабета преобладает гранулематозный тип реакции, в то время
ка(к у больных диабетом преимущественно наблюдается некробиотический
тип (Арутюнов В Я , 1958; Каламкарян А. А. и Сыч Л И., 1963; Вла-
димиров В В , 1976; Miura и Tanahashi, 1978; Lever и Schaumburg-Lever,
1975; Vanbremeersch et al., 1966 и др.). По-видимому, наиболее правильно
рассматривать гранулематоз Мишера—Ледера не как самостоятельную но-
зологическую форму, а как своеобразный гистологический (гранулема-
тозный) вариант липоидного некробиоза Выраженность гранулематозного
или некробиотического компонента свидетельствует лишь о количествен-
ной разнице, но не дает убедительных данных для дифференциального
диагноза.
Клиника. Дискообразный гранулематоз характеризуется высыпания-
ми мелких узелков розового цвета, постепенно увеличивающихся в раз-
мерах и достигающих порой нескольких сантиметров в диаметре (до 3—5
см). Сформировавшиеся бляшки умеренно инфильтрированы, розовато-
красного цвета, часто имеют кольцевидные очертания; центр бляшек более
бледный с желтоватым оттенком, по периферии отчетливо выступает обо-
док красно-фиолетового цвета с мелким шелушением, остро-воспалительные
явления отсутствуют. При диаскопии бляшек изменения цвета нет. В
центральной части бляшек постепенно формируется рубцовая атрофия.
Зуда и других субъективных ощущений нет. Высыпания локализуются
обычно симметрично на передней и боковых поверхностях голеней, значи-
тельно реже на других участках—на лице, спине, груди, разгибательных
поверхностях рук и др. Помимо описанной типичной клинической картины,
встречаются описания своеобразного течения болезни в виде полиморфных
высыпаний, симулирующих липоидный некробиоз, красный плоский лишай,
красную волчанку. Окончательный диагноз устанавливается лишь после
гистологического исследования.
Прогноз благоприятный, хотя заболевание нередко продолжается
годами. Известны случаи самопроизвольного разрешения бляшек.
Гистопатология. Атрофия эпидермиса; в дерме на разной глубине
очаги некробиоза коллагена, окруженные инфильтратами из лимфоцитов,
9 Клиническая .дерматология 129
гистиоцитов, фибробластов, среди которых встречаются гигантские клетки
инородных тел. Значительно выражены сосудистые изменения, фиброз
стенок сосудов, пролиферация эндотелия, сужение просвета вплоть до
закрытия, иногда тромбозы мелких сосудов. Некоторые авторы подчерки-
вают, что некробиоз коллагена при наличии и отсутствии диабета обуслов-
ливается васкулярными изменениями (Knoth et al., 1955). При окраске
ia жир в очагах некробиоза коллагена липоидные отложения отсутствуют.
Дифференциальный диагноз. Следует проводить с липоидным некро-
биозом, кольцевидной гранулемой, саркоидами, индуративной эритемой,
склеродермией.
Лечение. Заболевание с трудом поддается лечению. Из средств общей
терапии рекомендуются синтетические противомалярийные препараты, в
частности резохин, делагил, витамины, особенно Е, ангиотрофин, ксмпла-
мин, противодиабетические препараты. Местно—кортикостероидные ма-
зи, содержащие антибиотики, криотерапия; мелкие очаги можно удалять
хирургическим путем.
ЛИТЕРАТУРА
Epstein W. Cutaneous granulomas.— Intern. J. Dermatol., 1977, 16, 7, 574—597.
Hundeiker M. Kfinik der «umschriebenen Granufomkrankheiten.— Z. Hautkr., 1979,
54, 11, 469—480.
Leoer W., Schaumburg-Lever G. Histopathology of the skin.— Ed. 5.— Philadelp-
hia: Lippincort C°, 1975.
Miura T., Tanahaihi Y. Granulomatosis disciformis chronica et progressiva with
Tymphadenopathy.— Arch. Dermatof., 1978, 114, 8, 1224—1225.
Nieboer C., Kalsbeek G. Direct immunofluorescence studies in granuloma annulare,
necrobiosis lipoidica and granulomatosis disciformis Miescher.— Dermatofogica, 1979, 159,
6, 427—432.
Noster U , Janer M., Schulz K- Zur Entitat der Necrobiosis maculosa. Granuloma-
tosis disciformis chronica et progressivn Miescher und ihre Beziehung zur Necrobiosis
lipoidica diabeticorum.— Hautarzt, 1974, 25, 4, 325—332.
ДЕРМАТИТ ГЕРПЕТИФОРМНЫЙ ДЮРЙНГА—БРОКА (dermatitis
herpetiformis Diihring—Brog).
Синонимы: болезнь Дюринга, болезнь Дюринга—Брока, полиморф-
ный болезненный дерматит Брока, пруригинозная пузырчатка Капоши и
ДР-
Заболевание описал Дюринг в 1884 г. Брок в 1888 г. дополнил описа-
нье клинической картины дерматоза. Герпетиформный дерматит встречается
относительно редко, составляя около 0,2% от других дерматозов (Eyster
и Kierland 1951).
Этнология и патогенез. Ряд авторов считают герпетиформный дерма-
тит полиэтиологическим синдромом (Коляденко В. Г. и соавт., 1980),
большинство же исследователей признают нозологическую самостоятель-
ность дерматоза. Существуют различные теории патогенеза дерматита Дю-
ринга. Одни из них не получили достаточного подтверждения (неврогенная,
130
эндокринная, инфекционная, вирусная), другие (токсико-аллергическая,
инфекционно-аллергическая) имеют своих приверженцев. Наиболее ши-
рокое признание получила аутоиммунная гипотеза. Начиная с 1966 г.
появилось много сообщений (Marks и др.) о наличии у больных герпети-
формным дерматитом изменений слизистой тонкого кишечника, схожих с
целиакией или глютеновой энтеропатией, в связи с чем получило распрост-
ранение мнение о близости этих патологий. В пользу такого мнения при-
водят исследования Gebnard и соавт. (1973), которые выявили антиген
HLA—А8 у 68% больных дерматитом Дюринга и‘у 84% при сочетании это-
го дерматоза с целиакией (в контрольной группе у 17:—30%), а также об-
наружение антиретикулиновых антител классов IgG и IgA как при герпе-
тиформном дерматите, так и при целиакии (Lancaster—Smith et al., 1975).
Аглютеновая диета, по мнению многих авторов, сама по себе улучшает
течение герпетиформного дер матита и усиливает эффект сульфоновых пре-
паратов. Упомянутые данные позволили Sneddon (1978) высказать пред-
положение, что в основе герпетиформного дерматита и целиакии лежат
одинаковые иммунные нарушения. Ряд авторов указывают, что в части
случаев герпетиформный дерматит можно рассматривать как паранеопла-
стическое заболевание (Штейнлухт Л. А. и соавт., 1979 и др.), в то же время
Jablonska и Chorzelski (1972) утверждают на основании иммунофлюорес-
центных исследований, что пузырные высыпания у больных неоплазмой
есть проявления буллезного пемфигоида.
Клиника. Заболевание встречается чаще у мужчин, развивается пре-
имущественно в молодом возрасте. Часть больных отмечает провоцирую-
щую роль нервных перегрузок, интеркуррентных заболеваний, менструа-
ций и др. Ощущения жжения, зуда, болезненности обычно за несколько
часов предшествуют появлению высыпаний, которые чаще бывают поли-
морфными. Однако в начале болезни или во время отдельных приступов
могут наблюдаться только уртикарные или только эритемато-папулезные
элементы. В дальнейшем на эритематозно-отечном фоне появляются сгруп-
пированные напряженные пузырьки диаметром 0,2—0,5 см с прозрачным
содержимым. Иногда, особенно у пожилых людей, наряду с мелкими могут
быть и крупные пузыри. На высоте приступа изменения кожи представляют
пеструю картину—уртикоподобные, узелковый, пузырьковые, иногда пу-
стулезные элементы, экскориации, гиперпигментации (рис. 80). Высыпа-
ния располагаются симметрично, имеют склонность к группировке и из-
любленную локализацию—локти, колени, плечи, ягодицы, область крестца.
Нередко поражается волосистая часть головы, шея, лицо. Высыпания
могут быть и на слизистой оболочке рта, гениталий, прямой кишки. Дан-
ные о частоте поражений слизистых оболочек колеблются от 0 (Sonnichsen,
1976) до 50% (Darier, 1930). Н. А. Торсуев и соавт. (1979) наблюдали их
у 9,6% больных; М. Н. Шеклакова (1980) у 4 из 21 больного типичной фор-
мой и у 3 из 6 больных с крупнопузырной формой дерматита Дюринга.
Высыпания сопровождаются сильным зудом, жжением, чувством напря-
жения. Характерна бурая пигментация после разрешившихся высыпаний.
Из-за сильного зуда и глубоких расчесов может присоединяться вторичная
9* 131
Рис. 80. Герпетиформный
дерматит
(пустулезная форма).
инфекция и остаются рубчики. Заболевание протекает длительно, иногда
десятилетиями. Частота, продолжительность и тяжесть приступов варьи-
руют у разных больных и в разные периоды болезни. Общее состояние
обычно не нарушено. Сезонности не отмечается.
Описаны атипичные формы герпетиформного дерматита—экзематоз-
ная, строфулоидная, трихофитоидная, пемфигоидная, вегетирующая, ло-
кализованная и др. Что касается herpes gestationis, то одни авторы (Маш-
киллейсон Л. Н., 1965) считают его герпетиформным дерматитом бере-
менных, другие (Bushkell et al., 1974 и др.) называют эту форму пем-
фигоидом беременных, основываясь на обнаружении при ней фиксиро-
ванных in vivo IgG в зоне базальной мембраны эпидермиса. Опубликованы
сообщения о сочетании герпетиформного дерматита с витилиго (Katz, 1978),
системной красной волчанкой (Aronson et al., 1979). В ЦКВИ наблюдалось
сочетание дерматита Дюринга с псориазом у двух больных, с красным пло-
ским лишаем—у одной и с системной красной волчанкой—у другой больной.
В последние годы появились описания смешанных форм герпетиформного
дерматита и буллезного пемфигоида (Barthelmes, 1977; Dabrowski et al.,
1978), подтвержденные иммуногистологическими исследованиями. Pape
и соавт. (1978) описали гломерулонефрит у больного герпетиформным дерма-
титом и установили отложения IgA и С3 в биоптатах почек и кожи, что
свидетельствует об общности их поражения. Для больных герпетиформ-
ным дерматитом характерна повышенная чувствительность к препаратам
йода (реже брома). На этом основана диагностическая проба с йодистым
калием (кожная и пероральная), предложенная Ядассоном. В жидкости
пузырей и в крови, как правило, имеется повышенное количество эозино-
филов. Szurgent и соавт. установили радиоиммунным методом повышенное
содержание простагландинов в пузырной жидкости (по сравнению с плаз-
мой и мочой). Они полагают, что простагландины играют роль медиатора
или модулятора в возникновении зуда.
132
Прогноз в отношении выздоровления часто неопределенный.
Гистопатология. Субэпидермальные пузыри, отек и периваскулярные
инфильтраты с примесью эозинофилов в верхней части дермы. Микроаб-
сцессы из нейтрофилов и эозинофилов на вершинах сосочков. ЛейкоЦито-
клазия. При электронно-микроскопическом исследовании установлено, что
основные изменения развиваются в верхних отделах дермы. Происходит
редукция и разрыв крепящих фибрилл, формируются участки лизиса кол-
лагеновых волокон и крепящих фибрилл (Шеклакова М. Н., 1981).
Иммунопатология. В срезах видимо интактной кожи по периферии
высыпаний выявляются отложения IgA. Они располагаются на вершинах
сосочков или линейно (van der Meer, 1969, и др.). Отложения иммуногло-
булина в очаге поражения захватываются лейкоцитами и потому не обна-
руживаются (Jablonska и Chorzelski, 1971). Циркулирующие анти IgA—
антитела при герпетиформном дерматите не выявляются, что, по мнению
некоторых авторов, говорит об умеренной выраженности аутоиммунных
процессов. Примерно у 20% больных обнаруживают антиретикулиновые
антитела, обычно наблюдаемые при целиакии (Holubar и Stingl, 1976). При
иммуноцитохимических исследованиях в электронном микроскопе уста-
новлено, что базальная мембрана свободна от отложений IgA. Они нахо-
дятся исключительно в субэпидермальных пучках микрофибрилл и фикси-
рованы в микрофибриллярной части эластических волокон (Honnigsmann
et al., 1977). В то же время Hideo и Stephen (1977) наблюдали у одного
больного связывание IgA-антител антигеном lam. lucida, а у другого—
фиксирующими волокнами суббазальной пластины.
Дифференциальный диагноз. Его следует проводить с истинной пу-
зырчаткой, буллезным пемфигоидом, буллезными токсикодермиями, суб-
кориеальным пустулезом.
Лечение. Основные препараты—сульфоны (ДДС, авлосульфон, диу-
цифои, димоцефон и др.). При недостаточной эффективности они комби-
нируются с кортикостероидами. Кроме того, применяются унитиол, транс-
фузии плазмы и крови, аутогемотерапия.
ЛИТЕРАТУРА
Доляденко В. Г., Головченко Д. Д. Шмыгло М. П. Сочетание герпетиформного
дерматита и псориаза у больных раком желудка.— Вестн. Дерматол. и венерол., 1980, 11,
58—60.
Торсуев Н. А., Шеклаков Н. Д., Романенко В. Н. Буллезные дерматозы.— М.:
Медицина, 1979 — с. 152—189.
Шеклакова М. Н. К вопросу о некоторых клинических особенностях герпетиформ-
ного дерматита и буллезного пемфигоида.— Вестн. дерматол. и венерол., 1980, 11, 39—41.
Штейнлухт Л. А., Придвижкин И. Г., Бурылева Д. Г. Пузырные дерматозы
как паранеопластические проявления.— Вестн. дерматол. и венерол., 1979, 3, 44—47.
Aronson A. J., Soltani К., Aronson I. Д., Ong Д. Т. Systemic lupus erythemato-
sus and dermatitis herpetiformis. Cc ncurence with Marfan’s syndrome.— Arch. Dermatol.,
1979, 115, 1, 68—70.
133
Barthelmes H. Koinzidenz von Parapemphigus und Dermatitis herpetiformis Duhring.
— Derm. Mschr., 1977, 163, 2, 152—153.
Bushkell L. L., Jordon R. E., Goltz R. W. Herpes Gestationis: new immunologic
findings.— Arch. Dermatol., 1974, 110, 1, 65—69.
Dabrowski J., Jablonska S., Chorzelski T.. P. et al'. Electron microscopic studies in
dermatitis herpetiformis in relation of the pattern of immune deposits in the skin.— Arch.
Dermatol. Res., 1977, 259, 3, 213—224.
Honigsmann H., Stingl G., Holubar K-, Wolff K- Efektronmikroskopische Loka-
fisation von Antikorpern und Immunkomplexen bei Immundermatosen.— Derm. Mschr.,
1977, 163, 3, 242—243.
Pape J., Mellbye O., Qystese B.,. Brodwal E. Glomerulonephritis in dermatitis her-
petiformis.—Acta Med. Scand., 1978, 203, 5, 445—448.
Sheddon J. B. Skin manifestations of immunological origin.— J. Roy Coll. Pkys.
London, 1978, 12, 2, 169—179.
Vaoita Hideo, Katz S. Circulatung IgA antibasement membrane zone antibodes in
dermatitis herpetiformis.— J. Invest. Dermatol., 1977, 69, 6, 558—560.
ДЕРМАТИТ ГОРШЕЧНЫЙ У ДЕТЕЙ (bedpan dermatitis in children).
Синоним: дерматит от пластмасс.
Контактный дерматит у маленьких детей, развивающийся при кон-
такте с пластмассовыми горшками.
Этиология п патогенез. Искусственные полимеры обладают выражен-
ными сенсибилизирующими кожу свойствами.
Заболевания кожи у лиц, имеющих контакт с различными пластмас-
сами и продуктами их переработки, еще недостаточно изучены, имеются
только отдельные сообщения (Антоньев А. А. и Танков Ю. П., 1971;
Багиова М. Д., 1984; Haustein et al., 1980 и др.).
Полагают, что профессиональные дерматит и экзема, развивающиеся
от контакта с синтетическими пластическими массами, зависят от сенсиби-
лизирующих свойств входящих в их состав синтетических смол и других
ингредиентов, в том числе формальдегида. Контакт с готовыми пласстмас-
совыми изделиями также в ряде случаев может приводить к развитию дер-
матита и экземы. z
Клиника. Воспалительные явления локализуются исключительно в
местах контакта со стенками горшка—на ягодицах, точно повторяя очер-
тания краев горшка (рис. 81). В некоторых случаях процесс на ягодицах
принимает более распространенный характер. Иногда поражение кожи
имеет вид не целого кольца, а как бы состоит из отдельных звеньев.
Высыпания располагаются большей частью симметрично в виде эри-
темного круга, с выраженной местной отечностью, зудом. На фоне отечно-
гиперемированиой кожи появляются везикулезные элементы; местами они
вскрываются и образуют эрозивные мокнущие участки, покрытые корками.
При контакте с пластмассовыми горшками у детей с аллергическим
фоном дерматит протекает особенно остро и может сопровождаться обо-
стрением экзематозного процесса (Коган А. И. и Богуш П. Г., 1981). В
отделе дерматологии ЦКВИ мы наблюдали ряд детей, у которых горшеч-
134 ’
Рис. 81. Дерматит
горшечный.
иый дерматт (а также пеленочный) протекал с
островоспалительными явлениями—с выражен-
ным отеком, везикулезными и нередко буллез-
ными высыпаниями. Общее состояние детей
при этом заметно не нарушается, хотя могут
иметь место нервозность, плаксивость, ухудше-
ние сна, сильный зуд.
Отечность, мокнутие через 3—4 дня умень-
шаются, корки начинают отторгаться, кожа
бледнеет. Как правило, воспалительные явле-
ния в течение последующих 2—3 дней исче-
зают, оставляя лишь слегка пигментированные
участки. При иррациональной терапии и не-
достаточном гигиеническом уходе за кожей
горшечный дерматит может трансформировать-
ся в экзему.
Дифференциальный диагноз. Диагностика
затруднений не вызываете отдельных случаях
следует дифференцировать от эксудативного диатеза, детского прур'иго,
детской экземы.
Лечение. Примочки, крем Унны пополам с кортикостероидными ма-
зями, 2—3% ихтиол-нафталановская паста.
ЛИТЕРАТУРА
Багнова М. Д. Профессиональные Дерматозы.— М.: Медицина, 1984.
Доган А. И., Богуш П. Г. Горшечный дерматит у детей.— Вестн. дерматол. и
венерол., 1981, № 9, с. 66—67.
ДЕРМАТИТ ОТ ЗОЛОТА (dermatitis of gold).
В отечественной и зарубежной литературе имеются лишь немного-
численные публикации, посвященные вопросу дерматитов и экзем, обу-
словленных золотом и его соединениями.
Этиология и патогенез Многие аспекты механизма развития «золотых»
дерматитов и экземы ие выяснены. Подчеркивается, что развитие заболе-
вания возможно лишь в результате повторного или непрерывного контакта
с препаратами золота, оказывающего преимущественно сенсибилизирую-
щее действие на кожу. Как полагают некоторые исследователи (Астапен-
ко М. Г. и соавт., 1961; Урбанюк К. Г., 1983; Comaish, 1969; Elgart, 1972;
Petros и Macmillan, 1973 и др.), в основе аллергических процессов от золота
лежит реакция антигена с антителом. Развитие аллергического процесса
от золота с соответствующей клиникой зависит от активности антигенного
воздействия и реактивности организма, а также от пути его поступления в
организм (прямой контакт, парентеральное введение).
Клиника. В отечественной и зарубежной дерматологической литерату-
ре классификации «золотых» дерматитов, насколько нам известно, нет.
135
Мы предлагаем (прелиминарно) все аллергические дерматиты от препа-
ратов золота по механизму развития и клиническим проявлениям разде-
лить на три основные группы: 1) профессиональные контактные аллерги-
ческие дерматиты, развивающиеся при работе с препаратами золота; 2) кон-
тактные аллергические дерматиты, обусловленные ношением ювелирных
украшений; 3) токсидермии, развивающиеся при лечении различных за-
болеваний препаратами золота (парентеральное введение). Такое деление,
разумеется, следует считать весьма условным, но полезным для практиче-
ских целей.
В литературе имеются немногочисленные сообщения о поражениях
кожи, развивающихся при производственном контакте с золотом и его
органическими и неорганическими соединениями. Б. А. Сомов и Г. Д.
Хаймовский (1964) наблюдали 37 больных профессиональным аллергиче-
ским дерматитом, имевших производственный контакт с раствором циани-
стого золота, в том числе у 24 рабочих цеха гальванического золочения
часового завода и у 13 работников других предприятий гальванической
промышленности. Пробы с 1% раствором цианистого золота оказались
положительными. Контакт с хлористым золотом может вызвать профес-
сиональный аллергический дерматит у фотографов. Контактный аллер-
гический дерматит от воздействия ауратцианамида калия наблюдали Raith
и соавт. (1980) у рабочих при изготовлении электронных аппаратов.
Клиническая картина дерматита, развившегося от контакта с золотом
(рис. 82, 83, 84, 85), характеризуется диффузной гиперемией без четких
границ, отеком, резким зудом, на фойе которых могут возникнуть зудя-
щие папулезные, везикулезные, а также буллезные высыпания. Если лока-
лизация на первых стадиях заболевания соответствует местам контакта
золота с кожей (кисти, предплечья, лицо), то затем может наступить рас-
пространение процесса с поражением туловища, гениталий и др. Положи-
тельные результаты компрессных кожных проб с солями золота (обычно
в 0,1%—0,5% концентрации) были получены у значительного большинства
больных «золотым» профессиональным дерматитом, у части больных одно-
временно наблюдается и очаговая реакция.
Течение профессиональных аллергических дерматитов от золота бла-
гоприятное; при переводе больных на другую работу рецидивов, как пра-
вило, не наблюдается, но специфическая чувствительность может сохра-
няться долго и поэтому возобновление контакта с этим аллергеном мо-
жет вызывать рецидив заболевания, вплоть до перехода его в профессио-
нальную экзему.
Токсидермии от золота (рис. 86) характеризуются гиперергической
реакцией кожи иа воздействие аллергена, проникающего в организм па-
рентерально. Препараты золота (кризанол, кризолган, аурофос) в недавнем
прошлом применяли для лечения красной волчанки, туберкулеза кожи,
легких и гортани, сифилиса и других болезней. Соли золота более 40—50
лет применяются для лечения ревматоидного артрита и инфекционного
неспецифического полиартрита.
Примерно у 25—30% больных, систематически лечившихся препара- «
136
Рис. 82. Дерматит
от золота.
Рис. 83. Дерматит
от золота.
Рис. 84. Дерматит
от золота.
Рис. 86. Таксидермия
от золота.
Рис. 85. Дерматит
от золота.
137
тами золота, наблюдаются токсико-аллергические осложнения, причем у
многих из них вовлекается кожа (Лебедев А. Я., 1930; Россиянская М. Л.,
1949; Астапенко М. Г. и соавт,, 1961; Берестен Т. И., 1979; Дермидонтова
Е. Н. и соавт., 1981; Урбанюк К. Г., 1983; Hartfall et al., 1937; McKenna,
1957; Boni et al., 1976; Petros Macmillan, 1973; Schneider et al., 1983,
и др4.).
Наиболее частым является лихеноидный дерматит («lichen aurique»)
который, как правило, носит распространненый характер и держится в
течение 1—2 недель после прекращения лечения препаратами золота.
Лихеноидные сыпи «золотого» происхождения обычно развиваются после
3—10 инъекций кризанола. Довольно часто заболевание проявляется урти-
карными элементами, геморрагической пурпурой, распространенными
эритематозными пятнами, иногда с отеком, везикулезными и буллезными
высыпаниями, В тяжело протекающих случаях может развиться универ-
сальная эритродермия с выраженным шелушением. Мы наблюдали боль-
ную в возрасте 37 лет, у которой после длительного применения кризанола
по поводу ревматоидного артрита развился генерализованный дерматит
(краснота, отек, инфильтрация, склонность к мокнутию, обильное шелу-
шение крупными чешуйками, сильный зуд) в сочетании с полутотальной
алопецией. После отмены кризанола и назначения комплексного лечения
(включая кортикостероиды) явления дерматита стихли, но высыпания на
некоторых участках кожного покрова держались несколько месяцев. К
более поздним изменениям можно отнести наблюдавшуюся нами своеобраз-
ную пигментацию серовато-ливидиого цвета, появившуюся через три ме?
сяца после лечения санокризином.
Очень редко наблюдаются контактные дерматиты от золота, вызван-
ные ношением золотых изделий (колец, браслетов, сережек, приколок и
др.
Cowan (1960), Shelly и Epstein (1963), Elgart (1972) и др. описали боль-
ных с аллергией к золотым предметам в виде экзематозной реакции уш-
ных раковин, пальцев рук и предплечий. В литературе описано 9 подобных
наблюдений. Кожные лоскутные пробы с солями золота оказались поло-
жительными.
Клиническая картина аллергического дерматита от контакта с золо-
тыми изделиями имеет некоторое своеобразие: отмечается резкий зуд и
узелковые высыпания («золотой лихеи»); нередко присоединяется везику-
ляция, мокнутие, образуются серозные корочки. Локализация дерматита
вначале точно соответствует местам контакта кожи с золотыми изделиями
(кольцом, браслетом и т. д.), а при повторных рецидивах процесс может
распространяться на лицо, верхние конечности. Мы наблюдали женщину
28 лет, у которой вскоре после прокола ушных мочек развилась аллергия
к золотым серьгам в виде лихеноидно-экзематозной реакции. Затем под
кольцом и браслетом появилась краснота, отек и везикуляция с мокнутием.
Однажды она отметила появление дерматита на волосистой части кожи го-
ловы (контакт с золотыми шпильками). После прекращения контакта с
золотыми изделиями воспалительный процесс стихал.
138
Как подчеркивают многие исследователи, для поражений кожи, раз-
вивающихся от контакта с золотыми изделиями, характерна сравнительно
быстрая трансформация дерматита в экзему.
Гистопатология. При контактном дерматите от золота гистологически
отмечается межклеточный отек (спонгиоз) в шиповидном слое эпидермиса
с образованием пузырьков Околососудистый инфильтрат в собственно
коже состоит из мононуклеаров, гистиоцитов и лимфоцитов; в жидкости
пузырьков преобладают мононуклеары.
Лечение. Обязательным условием лечения контактного аллергиче-
ского дерматита является полное устранение контакта с препаратами зо-
лота. В качестве средств неспецифической десенсибилизации применяют
тиосульфат натрия, хлорид кальция, антигистаминные препараты, инъек-
ции гистоглобул ина, большие дозы витамина С (до 1 г в сутки); наружное
симптоматическое лечение (примочки, пасты, мази, в том числе кортико-
стероидные). В тяжелых случаях токсидермии с генерализованным пора-
жением кожного покрова рекомендуется на короткое время назначение
кортикостероидов внутрь (преднизолон по 20—30 мг в сутки).
ЛИТЕРАТУРА
Берестен Т. И. Лечение ревматоидного артрита препаратами золота.— Тер. арх.,
1979, № 8, с. 118—123.
Дормидонтова Е. Н.,‘ Коршунов Н. И., Баранова Э. Я. Некоторые вопросы кризо-
терапии больных ревматоидным артритом.— Тер. арх., 1981, № 7, с. 78—80.
Урбанюк К. Г. и др. Применение кризанола при ревматоидном артрите.— Врач, де-
ло, 1983, № 1, с. 37—40.
Boni A., Fehr Д., Henninger Н. et al. —In: Pharmakotherape der rheurnatischen
Erkrankungen. Berlin, 1976, S. 69—85.
Schneider L. и соавт. Ashg. Dermatose — ein Fall mit spontaner involution der Hau-
ler scheinungen. Z. Hautkr., 199, 58, 2, 113—120.
ДЕРМАТИТ ПЕРИОРАЛЬНЫЙ (dermatitis perioralis).
• Синонимы: розацеаподобный дерматит, аллергический периоральный
дерматит, периоральный себорейный дерматит.
Своеобразный дерматоз лица с преимущественной локализацией во-
круг рта. Впервые описан в США Frames и Lewis в 1957 г. под названием
«светочувствительный себореид лица.» Mihan и Ayres (1964), подтвердив
эти наблюдения в отношении своеобразия клинической картины и локали-
зации процесса, преимущественного заболевания женщин, предложили
название «периоральный дерматит.» Steigleder (1970) предложил термин
«розацеаподобный дерматит». Обсуждается вопрос о нозологической само-
стоятельности периорального дерматита. Ряд авторов считают этот дерма-
тоз вариантом себорейного дерматита или атипичной формой розацеа (Сам-
цов В. И., 1977; Steigleder и Strempel, 1968; Dimitrescu, .1973; Gertler и
Tappeiner, 1973). Многие другие авторы указывают на существенные раз-
личия и считают периоральный дерматит самостоятельным, клинически
139
хорошо очерченным заболеванием, которое отличается от различных форм
розацеа, акие, себореи (Арутюнов В. Я.. 1977; Ариевич А. М. и соавт.,
1978; Hjorth et al., 1968; Petzold, 1971; Schubert и Rockl, 1972; Mellette
et al., 1976 и др.).
Этиология и патогенез. Основное значение придается действию на
кожу лица различных внешних факторов, в том числе активных моющих,
косметических средств, зубных паст (Бабаянц Р. С и соавт., 1973; 1983;
Steigleder, 1969; Baran и Civatte, 1971; Rocki и Schubert, 1971; Beetz et al.,
1974; Coskey, 1980, и др.). В. Я- Арутюнов (1977) считает, что в патогенезе
периорального дерматита доминирующую роль играют лекарственные и
другие формы аллергии. Ряд авторов (Зверькова Ф. А. и Литвинюк Н. В.,
1974; Boteng и Nolting, 1972; Weber, 1972; Saunders, 1975; Mellette et al.,
1976 и др.) указывают на связь заболевания с применением фторсодержа-
ЩИХ кортикостероидных мазей. Исследования Rockl и Schubert (1976) и
др. показали способность этих мазей изменять структуру коллагеновых
и эластических волокон, вызывать ломкость сосудов, что приводит к атро-
фии кожи, стойким эритемам, телеангиэктазиям. Имеются указания на
роль фузобактерий, demodex folliculorum, дисфункций нервной, эндокрин-
ной и пищеварительной систем в развитии периорального дерматита.
Клиника. Заболевание встречается у молодых женщин (25—35 лет),
очень редко у пожилых женщин и мужчин. Высыпания располагаются
более или менее симметрично, преимущественно на подбородке и в носо-
губных складках, на прилегающих к нйм участках щек, на коже верхней
и нижней губы, вокруг глаз. Единичные высыпания наблюдаются на ви-
сках, на переносице, на латеральных участках щек. Известны случаи пре-
имущественно периорбитальной локализации процесса. В то же время на-
блюдаются больные с диффузным поражением всего лица. В зоне пораже-
ния на фойе пятнистой или диффузной эритемы наблюдаются плоские,
конусовидные или Полусферические папулы, которые располагаются бес-
порядочно, иногда группируются, но не сливаются. Величина их от була-
вочной головки до 2—3 мм в диаметре. Окраска узелков меняется в зави-
симости от давности: от сочной красно-коричневой до синевато-красной в
ранней стадии и светло-коричневой в фазе регресса. Часто они превраща-
ются в папуло-пустулы или папуло-везикулы, которые через 2—3 недели
исчезают, оставляя буроватые пятна, в пределах которых вновь появля-
ются папулы. Считается типичным наличие свободной от высыпаний узкой
полоски кожи вокруг красной каймы губ. При периоральном дерматите
не бывает комедонов и keratitis rosacea, нет также и телеангиэктазий.
Заболевание течет с частыми обострениями. В периоды обострений больные
ощущают легкое жжение и зуд. Нередко отмечается усиление субъектив-
ных ощущений и гиперемии после приема горячей пищи, алкогольных на-
питков, смены температуры воздуха (холод, жара, ветер, солнце). Рези-
стентность этого косметически неприятного дерматоза к лечению часто
вызывает у больных раздражительность, подавленное настроение.
Прогноз благоприятный, продолжительность заболевания от 2—5
месяцев до 3—5 лет.
140
\
Гистопатология. Подавляющее большинство авторов подчеркивают
неспецифичность гистологических изменений при периоральном дермати-
те. В дерме—расширение сосудов, перифолликулярные и периваскуляр-
ные инфильтраты, состоящие из лимфоцитов, гистиоцитов, фибробластов
и плазматических клеток.
Дифференциальный диагноз. Его следует проводить с розовыми угря-
ми, себорейным дерматитом, контактным дерматитом, акнеподобными из-
менениями кожи вокруг рта у женщин во время менструаций.
Лечение. Большинство авторов в последнее время считают обязатель-
ным условием прекращение пользования фторсодержащими кортикосте-
роидными мазями. Мы наблюдали больных, у которых высыпания исчезли
в течение 2—3 месяцев после отмены этих мазей. Показано применение
витаминов группы В, антигистаминных препаратов и десенсибилизирую-
щих средств. У ряда больных хорошее действие оказывает трихопол (по
0,25x2 раза в день в течение 2—3 недель). В острой стадии дерматита ре-
комендуются холодные примочки из 1—2% водного раствора резорцина
или 2% раствора борной кислоты, в дальнейшем 2—5% серно-дегтярная
мазь «Ям» и др. Эффективным методом лечения является массаж жидким
азотом (В. Я- Арутюнов. 1982).
ЛИТЕРАТУРА
Ариевич А. М. Периоральный Дерматит.— Сов. мед., 1980, 3, 71—74.
В. Я- Арутюнов. Опыт лечения периорального дерматита жидким азотом. Вести,
дерматол. и венерол., 1982, 11, 35—37.
Coskey R. Dermatologic therapy: december, 1978, through november, 1979.— J. Amer.
Acad. Dermatol., 1980, 3, 2, 125—148.
Kalkoff K. Akne rosazea und perforate Dermatitis. Grundlagen der Differentialdiag-
nose und Therapie.— Mkrse arztt. Fortbitd., 1975, 25, 1, 19—23.
Kalkdff R., Buck A. Zur Pathogenese der perioraten Dermatitis.— Hautarzt, 1977,
28, 2, 7-! Г7.
Murks R., Wilkinson D. Rosacea and perioral dermatitis.— In.: Textbook of derma-
tology. Oxford, Lonnon, Mefburne, 1979, vol. 2, p. 1433—1440.
Rockl H., Schubert E. Die perioral Dermatitis.— Hautarzt, 1976, 27, 4, 147—152.
ДЕРМАТОЗ ОСТРЫЙ ЛИХОРАДОЧНЫЙ НЕЙТРОФИЛЬНЫЙ (acute
febrile neutrophilic dermatosis.).
. Синоним: синдром Свита.
Sweet в 1964 г. описал синдром, характеризующийся: 1) инфекцией
(наиболее часто верхних дыхательных путей) с лихорадкой; 2) болезнен-
ными дермальными инфильтратами на конечностях, лице и шее; 3) повы-
шением СОЭ и лейкоцитозом; 4) типичной гистологией (отек и инфильтра-
ция дермы, главным образом полинуклеарными лейкоцитами).
Этиология и патогенез. Неясны. У части больных с этим синдромом
наблюдались злокачественные новообразования и системные заболевания
крови (Matta et al., 1973; Raimer и Duncan, 1978; Pirard и Delannoy, 1977;
141
К’оск и Oken, 1976; Burton, 1980; Vole—Platzer, 1983, Storer и соавт.,
1983; Carot, 1983, и др.).
Клиника. В мировой литературе описано более 150 случаев этого синд-
рома. Возраст больных колеблется от 9 месяцев до 85 лет, составляя в
среднем 45—50 лет; в 80—90% случаев заболевание встречается у женщин.
К 1975 г. имелись сообщения о 14 заболевших мужчинах (Casado et al.,
1977), в том числе одно в отечественной печати (Г. Э. Шинский и соавт.,
1974). Более чем у половины больных развитию кожных изменений за одну-
две недели предшествуют респираторные заболевания. Клиническая кар-
тина характеризуется кожными высыпаниями в виде темно-красных бо-
лезненных папул, глубоких узлов и бляшек размером от 0,5 до 4—6 см в
диаметре на лице, волосистой части головы и конечностях, особенно в ди-
стальных отделах. Бляшки овальных и округлых очертаний с темно-крас-
ным центром и розово-отечным валиком иногда сопровождаются вези-
куляцией и образованием пузырей. У больных, как правило, наблюдается
высокая лихорадка (до 38—39,5°), лейкоцитоз (от 10000 до 20000, из них
75—80% сегментоядерные нейтрофилы). Появление новых элементов
сопровождается лихорадкой и нейтрофильным лейкоцитозом. Travers
(1978) наряду с кожными высыпаниями описал поражение слизистой обо-
лочки полости рта. Наблюдаются также артралгии (Каламкарян А. А. и
соавт., 1985)
Прогноз благоприятный, но возможны рецидивы.
Гистопатология. Эпидермис нормальный В дерме—отек, расширенные
капилляры, инфильтраты вокруг придатков кожи, состоящие в основном
из полинуклеарных нейтрофилов, а также лимфоцитов й гистиоцитов.
MacKie (1974) отрицает специфичность гистологических изменений при
этом заболевании.
Дифференциальный диагноз. Его следует проводить с многоформной
эксудативной эритемой, аллергическими васкулитами, erythema elevatum
diutinum, узловатой эритемой.
Лечение. Назначение кортикостероидных препаратов (20—30 мг пред-
низолона в сутки) приводит к быстрому клиническому выздоровлению.
Horio и соавт. (1980) наблюдали хороший эффект у 6 больных этим дер-
матозом от введения калий йодида. Suehisa и соавт. (1983) сообщили об
успешном лечении колхицином 3 больных с данным синдромом.
ЛИТЕРАТУРА
Каламкарян А. А., Суколин Г. И., Федоров С. М. Синдром Свита—острый лихо-
радочный нейтрофильный дерматоз. Вести, дерматол. и венерол., 1985, 9, стр. 32—36.
Шинский Г. Э., Зеленина О. И., Коробейникова Э. А. Острый лихорадочный
нейтрофильный дерматоз.— Вести, дерматол. и веиерол., 1974, 1, 76—79.
Burton J. Sweet’s syndrome, pyoderma and acute leukaemia.— Brit. J. DermatoL,
1980, 102, 2, 239—240.
Casalo Jimenez M., Solas J., Soto M lo J.-. RMo F. Syndrome Sweet.— Revista
de LeproroTogia, 1977, II, 3, 243—250.
1’42
Horio В. t al. Treatment of acute febrile neutrophilic, lermato s (Sweet’s syndrome)
with potassium iodide.— Dermatologica, 1980, 160, 5, 341—347.
Klock J., Oken R. Febrile neutrophilic dermatosis in acute myelogenous leukemia.—
Cancer, 1976, 37, 922—927.
Kostler E. Das Sweet-Syndrom (acute febrile neutrophile Dermatose).— Derm. Mschr.,
1979, 165, 8, 561—568.
Mac Kie R. Sweet’s syndrome. A further case of acute febrile neitrophific dermato-
sis.— Dermatologica, 1974, 149, 2, 69—73.
Pirard C., Delannoy A. Syndrome de Sweet et Teucemie myefoide.— Ann. Derm. Vene-
reof., 1974, 104, 2, 160.
Raimer S... Duncan W. Febrile neutrophylic dermatosis in acute leukemia.= Arch.
Dermatol., 1978, 114, 3, 413—414.
Storer J. и соавт. Sweets synofrome. —int. Z dermatol., 1983, 22, 1, 8—12.
Sweet R. Acute febrile neutriphilic dermatosis.— Brit. J. Dermatol., 1979, 100,
93—99.
Vole-Platzer B. Sweet’s syndrom. Zeitschrift fur Hautkrankheiten, 1983, 58, 3, 195—
196.
ДЕРМАТОЗЫ ЛИНЕЙНЫЕ (dermatitis linearis).
Rayer и Barensprung (Г8$3) описали случаи односторонне и линейно
расположенных невусов—Naevi unius lateralis. Besnier и Hallopeau, затем
Jadassohn (1895) предложили для невусов этого типа название «системных
невусов,» причем делили эту группу невусов на два вида: 1) линейно рас-
положенных и 2) диффузно распространенных на большей или меньшей
поверхности кожи. _ \
Barensprung (1863) полагал, что naevus lateralis развивается в резуль-
тате врожденного, уже во время утробной жизни наметизшегося заболе-
вания отдельных спинальных ганглий. Теория Barensprung была домини-
рующей в течение долгого времени и значительное количество случаев
линейно расположенных дерматозов, разнообразных по способу возник-
новения, клинической и гистологической картине и течению относились,
к невусам. Однако в дальнейшем, по мере того как были опубликованы
случзи линейного расположения определенных по своей клинической кар-
тине дерматозов (псориаз, экзема, нейродермит, красный плоский лишай,
склеродермия и др.), взгляд на принадлежность всех линейных дерматозов
к невусам многими авторами был оставлен.
В дерматологической литературе имеются немногочисленные публи-
кации, посвященные описанию унилатерального или линейного располо-
жения дерматоза—красного плоского лишая, склеродермии, псориаза,
нейродермита и др. (Полотебнов А. Г., 1883; Тыжненко А. М., 1921; Ганце
Ф. В. и Еременко М. Д., 1927; Пер М. И., 1929; Брауде Р. С. и Россиян-
ский Н. Л., 1935; Каламкарян А. А. и соавт., 1980; Strick и Hyman, 1961;
Diem и Fritsch, 1971; Farris и Carlewara, 1978 и др.).
Вопрос о причинах линейного расположения некоторых дерматозов
остается неясным. Большнинство авторов выдвигают предположение о
заболевании периферического нерва как причине линейного расположения
143
дерматоза. Эти дерматозы располагаются в определенном направлении:
на конечностях, на которых чаще всего наблюдаются линейные дерматозы,
они идут параллельно оси конечностей нередко во всю их длину; на туло-
вище идут чаще в виде горизонтальных, а также в виде вертикальных ли-
ний. При исследовании кожи констатирована как на туловище, так и на
конечностях гиперестезия и гипералгезия по направлению этих линий.
Рис. 87. Линейный
веррукозный невус.
Рис. 88. Линейный
дерматоз (комедо-невус).
Было обращено внимание, что в некоторых случаях направление линейных
дерматозов совпадает с направлением пограничных линий Voigt’а, т. е.
линий, ограничивающих области разветвления разных кожных нервов.
При линейно или в виде полос расположенных поражениях кожи, так же
как и при линейных невусах, нельзя отрицать возможности связи с нерв-
ными поражениями (Pecirko, 1893). Ряд публикаций посвящен описаниям
расположения дерматоза строго по ходу нервов—n. brachialis, ulnaris,
ischiadicus и др. (Якунер С. А., 1935; Маслов П. Е., 1941; Farris и Car-
leware, 1978 и др.).
Высыпания красного плоского лишая и псориаза по типу линейных
дерматозов могут возникнуть иа местах бывшего рентгеновского оГлучения
(Chamial и Damis, 1935), оспопрививания (Кузнецов А. В., 1954; Калам-
карян А. А., 1959) или heroes zoster (Bessone, 1940; Strik и Hyman, 1961).
144
Рис. 89. Линейный дерматоз
(комедоневус).
Рис. 91. Линейный
дерматоз (псориазиформный).
Рис. 90. Линейный дерматоз
(гиперкератотический)
Ю Клиническая дерматология
Рис. 92. Унилатеральный
дерматоз.
Унилатеральное расположение дерматозов объяснялось вегетативной,
биохимической и нервной асимметрией. При неврологическом обследова-
нии был обнаружен ряд патологических явлений двигательного, чувстви-
тельного, секреторного, сосудодвигательного и трофического характера.
А. А. Каламкарян' и соавт. (1980) наблюдали 9 больных в возрасте
от 12 до 55 лет. В двух случаях был диагностирован унилатеральный дер-
матоз, в семи—линейный дерматоз (красный плоский лишай и псориаз).
Эти наблюдения представлены на рисунках 87—92. На основании комп-
лекса электрофизиологических исследований (электромиография, ЭКГ,
( электросопротивление кожи, плетизмография и др.) авторы считают, что
локализация кожных высыпаний при псориазе и красном плоском лишае
по типу унилатеральных и линейных дерматозов обусловлена неврологиче-
скими нарушениями центрального или сегментарного характера. Во всех
наблюдавшихся нами случаях выявлялась асимметрия, отражающая
изменение в нервно-мышечном аппарате и вегетативной регуляции соответ-
ствующего сегмента.
Линейная локализация высыпаний при псориазе и красном плоском
лишае на местах нарушенной трофики, по их мнению, может быть своеоб-
разным проявлением феномена Кебнера.
Strick и Hyman (1961) сообщили о случае lichen planus linearis (фе-
номен Кебнера) на месте разрешившихся высыпаний опоясывающего
лишая.
По нашему мнению, не исключается возможность тесной зависимости
линейного расположения заболевания на коже от поражения центральной
нервной системы. Некоторые авторы (Solomon et al.;, 1968) подчерки-
вают сочетания линейного дерматоза с заболеванием головного мозга.
Клинический опыт отдела дерматологии ЦКВИ, а также анализ лите-
ратуры позволяет линейные заболевания кожи разделить на две группы:
1) линейные невусы: чаще всего веррукозные, сосудистые, пигментные,
папилломатозные и др.; 2) линейные дерматозы, которые по своей клиниче-
ской картине'укладываются в определенные дерматологические единицы.
Отдельные авторы выделяют третью группу линейных заболеваний
кожи, которые по своей клинической картине не укладываются в рамки
какого-либо из известных дерматозов. •
Все линейные дерматозы могут встречаться в качеству единственного
выражения болезни, в дальнейшем к линейному расположению могут при-
соединиться другие очаги того же дерматоза. Следует подчеркнуть, что
все линейные дерматозы, естественно, не могут рассматриваться как не-
вусы; в действительности наблюдаются, хотя и редко, несомненные случаи
красного плоского лишая, склеродермии, псориаза, нейродермита, экземы
и т. д., которые имеют топографию, идентичную с линейными невусами и
которые появляются в известном возрасте и уступают соответствующему
лечению.
Гистоморфологические проявления линейных дерматозов весьма раз-
нообразны и, как правило, укладываются в гистологическую картину из-
вестных дерматозов, располагающихся линейно.
146
ЛИТЕРАТУРА
I
Каламкарян, А. А. Фролов, Е. П. Акимов, В. Г. Красников. Ю. А. О так на-
зываемых унилатеральных и линейных дерматозах — Вестн. дерматол. и венерол., 1980,
10, 18—23.
Andreeo V: et al. Cutis marmorata teiangiectatica congenita in two sisters. — Brit.
J. Dermatol., 1979. v. 101, No 3, p. 345—350.
Cremer H. Cutis marmorata teiangiectatica congenita.— Hautarzt, 1982, B. 33, H. I,
S. 40—41. '
Farris A., Carleoaro E. Lichen lineare sello sciatico popliteo.— thron. Dermatol.,
1978, v. 2, p. 233—237. . /
ДЕРМАТОЗ ПОДРОГОВОЙ ПУСТУЛЕЗНЫЙ (dermatosis pustulosa
subcorneal is).
Синонимы: болезнь Снеддона—Вилькинсона, пустулез Снеддона—
Вилькинсона, субкорнеальный пустулез.
Заболевание описали в 1956 г. Sneddon и Wilkinson. Большинство
авторов (Самцов В. И., 1963. Машкиллейсон Л. Н., .1964; Скрипкин Ю. К.
и Хамаганова А. В., 1972; Торсуев Н. А. и соавт., 1979; Sneddon и Wil-
kinson, 1956, 1979; Duperrat, 1957; Verma et al., 1973; Kozminska, 1962
и др.) считают субкорнеальный пустулез самостоятельной нозологической
единицей. Мы присоединяемся к этому мнению. Однако не все исследова-
тели разделяют такую точку зрения. Reynaer (1958), Vilanova и Pinol (1958),
Musumeci (1958), Ito с соавт. (1960), Saul и Sulazar (1962;, Basombrio с
соавт. (1960) рассматривают этот дерматоз как своеобразную пустулезную
форму герпетиформного дерматита. Schwank и Trapl (1959) сближают его с
пустулезным псориазом; Cerutiti (1957) идентифицирует с хронической
доброкачественной семейной пузырчаткой; Ribuffo (1959)—с эритематоз-
ной пузырчаткой; Hellier (1956)—с пустулезным бактеридом.
Этиология и патогенез. Большую роль в развитии .болезни и обостре-
нии ее, по данным некоторых авторов, могут играть эндокринные расстрой-
ства, нарушения менструального цикла, психические травмы, аллергиче-
ские заболеванря и др. Так, М. Н. Бухарович и В. М. Ковалев (1979)
наблюдали субкорнеальный пустулезный дерматоз у двух сестер, страдав-
ших дисменореей; обострения болезни у них наступали после стрессовых
реакций, травм. Wolff (1971),Dahlouk с соавт. (1975), Cream (1977), Schnit-
zer с соавт. (1977) описали больных, длительно страдающих субкорнеаль-
ным пустулезным дерматозом, у которых отмечено повышение IgA, IgG_
Клиника. Подроговым (субкорнеальным) пустулезным дерматозом ча-
ще болеют женщины в возрасте 40—70 лет, хотя Beck и соавт. (1961), Zan-
са и Giebertoni (1974), Johnson и Cripps (1974) наблюдали детей, болевших
с первых недель жизни до пяти лет, a Fleger (1961)—женщину 86 лет,
Первичным элементом является пузырек с мутным содержимым, напоми-
нающий фликтену, или дряблая пустула, возникающая на нормальной
коже или чаще на участке предшествующей эритемы. После полного фор-
мирования пустулы гной скапливается в нижней половине, оставляя про-
10* ‘ . 147
Рис. 93. Синдром
Снеддона—Вилькинсона
(подроговой пустулезный
дерматоз).
зрачную жидкость в верхней по-
ловине. Величина первичных эле-
ментов колеблется от 2—3 мм в
диаметре до размера горошины;
очертания их округлые или оваль-
ные. Пузырьки и пустулы окру-
жены гиперемической каймой,
иногда кратковременной. Большей
частью они группируются, иног-
да сливаясь друг с другом, обра-
зуя причудливые фигуры в виде
концентрических колец, серпиги-
нирующих гирлянд, сопряженных
дуг и полудуг (рис. 93). Очаги
поражения постепенно увеличива-
ются в размерах за счет высыпа-
ния по периферии свежих элемен-
тов с заживлением в центре и
образованием тонких и поверхностных коричневатых корок. В период
разрешения высыпаний корки окружены по периферии бахромкой
отслаивающегося рогового слоя. После заживления остаются лишь гипер-
пигментированные пятна, на которых вновь могут появиться пустулы.
Высыпания преимущественно локализуются в подмышечных впадинах и
паховых складках, но они также часто возникают на груди, спине, верх-
них и нижних конечностях, на туловище, то есть на любых участках кож-
ного покрова, за исключением лица и волосистой части головы, кистей,
стоп. Поражения слизистых не наблюдается. Периодические «толчкообраз-
ные» обострения появляются обычно 2—3 раза в течение года. Они раз-
виваются быстро, продолжаются 1—2 недели, иногда дольше. Иногда бы-
вает незначительный зуд, чаще он отсутствует. Содержимое свежих вези-
куло-пустул обычно стерильное. В препаратах-отпечатках с поверхности
эрозий акантолитические клетки, как правило, не обнаруживаются; симп-
том Никольского отрицательный. Кожная проба с мазью, содержащей
50% йодида калия, или прием раствора йодида калия внутрь обострения
не вызывает. Прямая и непрямая реакция иммунофлюоресценции отри-
цательная. Общее состояние больных не нарушается, температура остается
нормальной, состав крови, СОЭ не изменяются.
Продолжительность дерматоза варьирует от нескольких месяцев до
многих лет (в наблюдениях Schwank иТгар!—29 лет). Характерно спонтан-
ное наступление ремиссий различной длительности (до нескольких лет).
148
Гистопатология. По мнению Sneddon и Wilkinson (1979), наиболее *
характерным гистологическим признаком субкорнеального пустулезного
дерматита является быстрая трансэпидермальная миграция нейтрофилов,
которые скапливакяся под роговым слоем. В мальпигиевом слое—отек и
спонгиоз. В папиллярном слое дермы—расширение капилляров и вокруг
них—инфильтрат, состоящий из нейтрофилов, небольшого количества
эозинофилов и мононуклеарных клеток. Акантолиза нет, но иногда встре-
чаются акантолитические клетки, возможно, обусловленные протеолитиче-
скими ферментами, имеющимися в пустулезных элементах (Burns и Fine,
1959). Электронно-микроскопические исследования Metz и Schropl (1970)
показали дегенеративные изменения в верхних слоях эпидермиса, особен-
но в зернистом слое. Расплавление плазматической мембраны и цитоплаз-
мы зернистых клеток вызывает образование субкорнеальных полостей.
Дифференциальный диагноз. Его следует проводить с герпетиформ-
ным импетиго Гебры, пустулезным псориазом, герпетиформным дерматитом
Дюринга, себорейным пемфигусом.
Лечение. Эффективного лечения нет. Рекомендуются кортикостерои-
ды, сульфоны, сульфоны в комбинации с кортикостероидами, сульфанила-
миды, рифампицин (бенёмицин); наружное лечение симптоматическое.
ЛИТЕРАТУРА ,
Бухарович М. Н., Ковалев В. М. Субкорнеальный пустулез Sneddon—Wilkinson
у двух сестер.— Вести. Дерматол. и венерол., 1979, 5, 37—40
Торсуев Н. А., Шеклаков Н. Д., Романенко В. Н. Буллезные дерматозы.— М.:-
Медицина., 1979.
Johnson S., Cripps D. Subcorneal pustular dermatosis in children.— Arch. Derma-
tol., 1974, 109,'I, 73—77.
Schnitzer L., Verret J., Schubert B. et al. Pustulose souscornee de Sneddon—Wil-
kinson avec acantholyse et myefome a IgA.— Ann. dermatol. Syph., 1977, 104, 107—112.
Sneddon J., Wilkinson D. Subcorneal pustular dermatosis.— Brit. J. Dermatol.,
1979, 100, I, 61—68.
ДЕРМАТОМИОЗИТ (dermatomyositis).
Синонимы: острый полимиозит, псевдотрихиноз.
Впервые описал Wagner в 1863 г. и отдельно Unverricht и Нерр в 1887 г.
Название дерматомиозит предложил Unverricht в 1891 г. Они наблюдали
острые формы заболевания. В 1903 г. Steiner предложил выделять ост-
рую, подострую и хроническую формы болезни. Изучение дерматомиозита
как системного заболевания началось в 40—50-х годах н. в.
Дерматомиозит—тяжелое общее страдание с преимущественным по-
ражением поперечнополосатой (меньше гладкой) мускулатуры и кожи,
соучастие которых в патологическом процессе может быть выражено в
разной степени. Встречается в любом возрасте. Данные о заболеваемости
мужчин и женщин разноречивы. Gertler (1970) говорит о том, что нет изби-
149
рательности в отношении пола, Sonnichsen (1975) чаще наблюдал заболева-
ние у мужчин, А. П. Соловьева (1980) и многие другие авторы—у жен-
щин.
Этиология п патогенез. Многие исследователи считают, что дермато-
миозит развивается в результате сенсибилизации организма к различным
антигенам—инфекционным, опухолевым и др. Имеются указания на раз-
витие дерматомиозита после сильной инсоляции, нервно-психических по-
трясений, контакта с эпоксидными смолами, фоторастворителями. Мы на-
блюдали развитие дерматомиозита после контакта с дезинсектицидами.
В литературе высказывается мнение об этиологической роли вирусов (Ha-
shimoto и Thompson, 1971; Klug и Sonnichsen, 1973; Секамова С. М. и соавт.,
1978). А. П. Соловьева (1980) полагает, что вирус или другой инфекцион-
ный фактор правильнее рассматривать в качестве пускового механизма
аутоаллергической реакции. В патогенезе дерматомиозита, по мнению
этого автора, больщая роль принанадлежит иммунным механизмам, гене-
тической предрасположенности.
Клиника. В настоящее время принято подразделять дерматомиозит
на первичный, идиопатический и вторичный, опухолевый, а по течению
на острый, подострый и хронический. Из-за некоторых особенностей вы-
деляют дерматомиозит у детей.
Изменения кожи, как правило, начинаются на лице в виде стойкого
отека и л иловатой эритемы, наиболее выраженных в области век (рис. 94).
Множественные телеангиэктазии делают окраску кожи более насыщенной.
Поражаются и другие участки кожи, преимущественно доступные инсоля-
ции—шея, открытая часть груди, спины, предплечья, кисти. Наряду
со стойкой эритемой могут наблюдаться узелки, геморрагии, эксудатив-
ные, пемфигоидные высыпания, пигментации. Над разгибательной поверх-
ностью суставов кистей, на предплечьях появляются склеродермоподобные
изменения. У ряда больных развивается пойкилодермия. В отдельных
случаях в течение какого-то времени кожные изменения могут быть доми-
нирующим или даже единственным симптомом болезни. Дерматомиозит
сопровождается трофическими нарушениями кожи—сухость, исчерчен-
ность, ломкость ногтей, выпадение войос. У части больных (у 20—83%)
наблюдаются изменения слизистых оболочек: конъюнктивит, стоматит,
эрозии, язвочки, пурпурозные элементы, высыпания, напоминающие крас-
ный плоский лишай, лейкоплакию.
Мышцы обычно поражаются уже в самом начале болезни—боли, отеч-
ность, слабость, затем—гипотония, атрофия, склерозирование, кальци-
ноз. В болезненный процесс вовлекаются или все мышцы скелета или их от-
дельные группы. Наиболее часто поражаются мышцы плечевого, а затем
тазового пояса. Характерно также поражение мышц пищевода, гортани
с явлениями дисфагии, дисфонии. Имеются указания на существование
дерматомиозита без поражения мышц (Rosner, 1976). Обострения'болезни
сопровождаются повышением температуры, артралгиями, похуданием,
астенизацией, ускоренной СОЭ, увеличением альфа—2 и гамма-глобу-
линов, повышением активности трансаминаз, появлением р моче креати-
150
на, изменениями электромиограммы. Висцериты
при дерматомиозите встречаются реже, чем при
системной красной волчанке и системной
склеродермии. У большинства больных так
называемым идиопатическим дерматомиозитом
рациональная терапия обеспечивает наступле-
ние ремиссии. При опухолевом дерматомиозите
прогноз сомнительный.
Гистопатология. В эпидермисе гипер- и па-
ракератоз. В дерме вначале отечность, позже
фиброз; расширенные сосуды окружены массив-
ными инфильтратами из лимфоцитов и плазма-
тических клеток. В мышцах—очаговое набуха-
ние; исчезновение псперечной исчерченности.
Гиалинизация. Очаговый распад мышечных
волокон. Позже—увеличение соединительной
ткани в эндо-и перимизиуме. Регенерацион-
Рас 94. Дерматомиозит
(вторичный).
ные процесса в сарколемме. При ультраструк-
турном анализе установлено, что основным
видом деструкции мышечных волокон является
миолиз. В сосудах гипертрофические и дистрофические изменения эндо-
телия и перицитов капилляров (Секамова С. М. и соавт., 1978).
Дифференциальный диагноз. Проводят с системной красной волчан-
кой и склеродермией.
Лечение. Основное место принадлежит кортикостероидным препара-
там. Их назначают также в комплексе с 6-меркаптопурином, азатиопрнном.
При хронической форме длительно применяют аминохинолиновые препа-
раты, салицилаты, индометацин, анаболики, витамины. Имеются указания
на эффективность лечения сульфадиазином, пириметамином и фолиевой
кислотой. При снижении активности болезни применяют массаж, чечебную
гимнастику и др. При опухолевом дерматомиозите наряду с лечением опу-
холевого процесса также показано применение кортикостероидов (Со-
ловьева А. П., 1979).
ЛИТЕРАТУРА
Исаева Л. А., Жвания М. А. Дерматомиозит у детей.— М.: Медицина, 1978.—
222 с.
Секамова С. М., Копьева Т. Н., Жвания М. А. Электронно-микроскопическое
исследование скелетных мышц при дерматомиозите у детей.— В кн.: Ультраструктурные
аспекты морфогенеза и регенерации в норме и патологии. М., 1976, с. 272—278.
Соловьева А. П. Дерматомиозит.•—М.: Медицина, 1980.— 176 с.
Barnes В. Е. Dermatomyositis and malignance.— Ann. Intern. Med., 1976, 84, I,
68—76.
Rosner E. Dermatomyositis ohne Myositis.— Hautarzt, 1976, 27, 11, 560.
151
ДЕРМАТОФИБРОСАРКОМА ВЫБУХАЮЩАЯ (dermatofibrosarcoma
protuberans)
Син. дерматофибросаркома прогрессирующая и рецидивирующая,
дерматофибросаркома выбухающая рецидивирующая Hoffman, болезнь
Darier—Ferrand.
Этиология и патогенез. Дерматофибросаркома выбухающая пред-
ставляет собой соединительнотканную опухоль. Несмотря на обычно доб-
рокачественное течение, она, согласно Гистологической классификации
опухолей кожи ВОЗ (1980), относится к группе злокачественных новообра-
зований фиброзной ткани. При этом указывается на случаи метастазиро-
вания. Fischer и Undeutsch (1979), рассматривающие дерматофибросаркому
выбухающую в группе злокачественных гистиоцитом, напротив, отмечают,
что метастазирование не является типичным для этой опухоли, и, если
имеется подозрение на таковое, то необходимо исключить фибросаркому,
или развитие новой опухоли, которая дала метастазы. Вопрос о происхож-
дении опухоли неясен. Ее рассматривают (Metz, 1979) как опухолевый
фиброматоз дизонтогенетического происхождения, как промежуточную
форму между дерматофибромой и фибросаркомой или как истинную сар-
кому кожи. Предполагалась возможность ее развития из эмбриональных
структур молочной железы, соединительной ткани сосудов. Данные элек-
тронно-микроскопических исследований свидетельствуют о вероятности ги-
стогенеза ^е из эндо- или периневральных клеток (Hashimoto et al., 1974)
или фибробластов (Ольховская И. Г. и Шубин А. С., 1977); Metz, 1979).
По мнению Taylor и Helwig (1962), дерматофибросаркома выбухающая
—опухоль соединительнотканного происхождения. Апатенко А. К. (1977)
считает ее злокачественным аналогом ангиофиброксантомы. и связывает
развитие с адвентициальными клетками. Fletcher и МсКое (1984) указывают,
что наиболее признанным является мнение о большей вероятности фибро-
гистиоцитарного происхождения опухоли.
Клиника. Дерматофибросаркома выбухающая возникает обычно у
взрослых, наиболее часто между 30 и 40 годами (Metz, 1979), чаще у муж-
чин (в 4 раза, по мнению From, 1979), но может быть и у детей, по данным
Metz, примерно в 10% случаев. По наблюдению этого автора, у ребенка
9 лет плоский очаг существовал с рождения, а с 3—4 лет начал приобре-
тать туморозную форму. Располагаться может на любом участке кожного
покрова, но чаще на туловище. По данным Groetschel и Cramer (1967),
из 344 случаев, опубликованных в литературе, в 45—опухоль располага-
лась в области плеч, в 63—на груди (из них 22 в области грудины), в 39—
на спине, в 42—на нижних конечностях, в 16—на голове, в 13—на животе,
в 23—в паховой области, значительно реже—на шее, щеках, ягодицах, в
люмбо-сакральной области и т. д. К числу очень редких локализаций от-
носятся щеки (Sauter и De Feo, 1971), нос (Petkov и Andreev, 1972), вульва
(Soltan, 1981). Клинически различают обычно 2 фазы: 1-я—плоский очаг
поражения, постепенно расширяющийся по периферии с гладкой или слег-
ка бугристой поверхностью, коричневатого или ливидного цвета; 2-я—опу-
холевая, наступающая через годы или даже десятилетия. Очаг как прави-
152 ' .
ло одиночен (рис. 95), хотя и
может состоять из нескольких
сливающихся склеродерме- или
келоидоподобных опухолевид-
ных узловатостей, достигает
нескольких сантиметров в ди-
аметре, значительно выступает
над уровнем кожи. Безболез-
нен, обычно медленно увели-
чивается в размерах, но описа-
ны случаи и с быстрым прогрес-
сированием (Sauter и De Feo,
1971). Опухоль расположена на
широком основании, вначале не
спаяна с подлежащими тка-
нями, но при рецидивах стано-
вится отчетливой склонность к
Рис. 95. Дерматофибросаркома
выбухающая.
инфильтрации окружающей кожи и подкожно-жировой клетчатки,
а иногда и мышц, фасций, костей (Groetschel и Cramer, 1967). Кожа в зоне
опухоли напряжена, истончена, пронизана телеангиэктазиями, в поздних
стадиях за счет усиленного роста может изъязвляться. Заболевание течет
медленно; метастазирование, если и бывает, то редко, при многолетнем
течении и после многочисленных рецидивов. Высокая частота рецидиви-
рования считается характерным признаком этой опухоли (Ольховская И. Г.
и Рабен А. С., 1976; Funk и Wagner, 1958; Groetschel и Cramer, 1967).
По данные И. Г. Ольховской и соавт. (1976), рецидивы после удаления
наблюдались у 16 из 26 больных. У 3 больных были метастазы в регионар-
ные лимфоузлы, у 5—рентгенологически выявлялись метастазы в легких,
у одного подтвержденные на аутопсии. Метастазы развились в сроки от
15 до 23 лет с момента развития опухоли, что, по мнению авторов, может
свидетельствовать об относительно благоприятном течении дерматофибро-
саркомы. Наличие отдаленных метастазов, по мнению Kutintschew и Dog-
ramadjeew (1965), вызывает сомнение в правильной диагностике. Рецидивы
возникают или за счет остатков опухоли в клинически неизмененной окру-
жающей коже или за счет врожденной готовности кожи больных к фибро-
матозным реакциям.
Kahn и соавт. (1978) наблюдали больную 27 лет с метастазами в лим-
фоузлы и в легкие, развившиеся после многократных хирургических опе-
раций, через 10 лет после первого удаления. Летальный исход наступил
через 16 лет с момента развития опухоли.
Гистопатология. Опухоль состоит из переплетающихся пучков соеди-
нительнотканных волокон; отмечается пролиферация молодых веретено-
образных клеток, встречаются уродливые ядра клеток, атипические ми-
тозы. В зависимости от степени дифференцировки может напоминать ити
фибросаркому, или дерматофиброму’ (Lever, 1975, Markowski и Zabel,
1978).
153
Rothlaender и Wolff (1982) описали наличие в очаге типичной дерма-
тофибросаркомы выбухающей отложений гемосидерина и липидов, что
считается более свойственным гистиоцитоме (Harstein и Weidner, 1979).
Авторы полагают, что в таких случаях приоритет при решении вопроса
о лечении и прогнозе должен быть отдан клинике.
Дифференциальный диагноз. Дифференцировать необходимо от опухо-
левых форм грибовидного микоза, ретикулосаркоматоза, гуммозного си-
филиса, фибросаркомы.
Лечение—хирургическое иссечение.
ЛИТЕРАТУРА
Апатенко А. К- Мезенхимные и нейроэктодермальные опухоли и пороки развития
кожи. М. Медицина, 1977
Бутов Ю. С., Борисенко К- К-, Савкина А. С., Попова И. С. Дерматофибросар-
кома Дарье—Феррана. ВестН. дерматол. и венерол., 1982, 42—44.
Ольховская И. Г., Пробатова Н. А., Казанцева И. А., О выбухающей дермато-
фибросаркоме. Арх. патол. 1976, 12, 17—24.
Ольховская И. Г., Рабен А. С. О клинико-морфологической характеристике Дер-
матофибросаркомы Дарье-—Феррана. Клин. мед. 1976, 6, 75—81.
Ольховская И. Г., Шубин А. С, О гистогенезе выбухающей дерматофибросарко-
мы.— Ар ..патол., 1977, 5, 33—37.
Fletcher С. D. М., McKee Р. Н. Sarcomas — a ch'nicopathological guide with par-
ticular reference to cutaneous manifestation Cfin. e p. dermatof. 1984, 9, 5, 451—465.
Hashimoto K-, Brownstein M. H., Jakobiec F. A. Dermatofibrosarkoma protuberans.
— Arch. Derm., 1974, 110, 5, 874—885.
Horstein О. B., Weidner F. Tumoren der Haut. In: Spezielle pathologische Anatomie
Hrsg. K- Doerr, G. Seifert, A. Uehfinger Bd. 7, (Histopathologie der. Haut) 1979, Berlin—
Heidelberg—N. Y., Springer—Verlag, 5, 252—254.
Lynn From, Neopfasmas of the deymis, in Dermatology in general Medicine Sec. ed.
by T. B. Fitzpatrick et af., 1979, 703—784.
Markowski R., Zabel I. Dermatofibrosarcoma protuberans Darier—Ferrand. Przegf.
Derm., 1978, 55, 2, 201—204.
Metz J. Dermatofibrosarcoma protuberans im Kinderalter. Hautarzt, 1978, 29, 8,
435—438.
Rothlaender J. P., Wolff H. H. Ungewohnlicher Dermatofibrosarcoma protuberans.—
Z. Hairtkrkh., 1982, 57, 22, 1664—1674.
Soltan M. Dermatofibrosarcoma protuberans of the vulva.— Brit. J. Derm., 1981,
88, 2, 203—205.
ДЕРМАТОФИБРОМА ЛЕНТИКУЛЯРНАЯ (dermatofibroma lenticula-
re)
Синонимы: котный узелок, простая фиброма, гистиоцитома.
Этиология и патогенез не выяснены. Полагают (Gertler, 1973), что
дерматофиброма лентикулярная—доброкачественная опухоль, развиваю-
154
щаяся из недифференцированных перителиальных ретикулярных клеток,
которые могут дифференцироваться в фиброциты, гистиоциты и эндоте-
лиальные клетки. По мнению Веаге и Cunliffe (1979), дерматофиброма
является не доброкачественной опухолью, а, вероятно, представляет собой
реактивную гиперплазию ретикулоэндотелиальных, сосудистых и фиброз-
ных клеточных элементов.
Развитию способствуют травмы, укусы насекомых (Thies и Hennies,
1968). Имеются указания на возможную роль вирусов (Newmann и Wal-
ter, 1973). Бывают семейные случаи (Roberts et al., 1981).
Клиника. Одиночные или реже множественные (у больной, наблюдав-
шейся Baraf и Schapiro, 1970, их было 61), внутридермальные опухоли
величиной в среднем около 1 см, округлых или овальных очертаний. Они
незначительно возвышаются над уровнем кожи, что более заметно в цент-
ральной части. Их опухолевидный характер отчетливо выявляется только
при пальпации. При этом возникает ощущение мелкой плотной пластинки,
заложенной в толще кожи. Поверхность гладкая, но может быть слегка
гиперкератотична, а в редких случаях и веррукозна. Дерматофиброма
подвижна, легко смещаема по отношению к подлежащим тканям. Окраска
может быть красноватой, бледно-серой, желтоватой, но чаще красновато-
коричневатая, более насыщенная в зоне старых элементов. Она. может
быть неравномерной, обычно более темная по краю. Субъективными рас-
стройствами, как правило, не сопровождается, лишь изредка некоторые
больные жалуются на зуд. Дерматофибромы лентикулярные могут рас-
полагаться на любом участке кожного покрова, но преимущественно—на
ногах, особенно в 'области голеней, на пояснице. Bedi и соавт. (1976) опи-
сали множественные дерматофибромы с локализацией только на ладонях
и подошвах, величиной от 0,5 до 1,5 см, округлых или овальных очертаний,
плотноватой консистенции.
Дерматофибромы преимущественно возникают в молодом возрасте,
чаще у женщин. Достигнув определенного размера, существуют без изме-
нения длительно. Общее состояние больных не страдает. Прогноз в боль-
шинстве случаев благоприятный. Изредка дерматофибромы лентикуляр-
ные могут исчезать самопроизвольно. После травмы может наступить изъяз-
вление, кровоточивость. Описано развитие злокачественной фиброзной
гистиоцитомы в легких у больного с множественными дерматофибромами
на коже (Roberts et al., 1981). Не исключалась возможность метастаза
из очага в коже, удаленного за 7 лет до возникновения процесса в легких,
хотя для этого предположения не было достаточных доказательств. Goette
и Helwig (1975), Bryant (1977) наблюдали развитие в зоне дерматофибромы
пигментной базальноклеточной карциономы. По данным Schoenfeld (1964),
атипическая пролиферация эпидермиса наблюдается в 4,6% случаев дер-
матофибром. Гистологически, как отмечает Uanowitz |и Goldstein (1964),
они могут быть неотличимы от базалиомы.
Halpryn и Allen (1959) нашли резко выраженные эпидермальные из-
менения в 25% случаев дерматофибром, причем в 8%—это была базалио-
ма, в 11%—себорейный кератоз, в 6%-^-кератоакантоз (гиперкератоз,
155
акантоз, папилломатоз). Arnold и Tilden (1960) выразили сомнение в столь
высокой частоте базалном, приведенной выше указанными авторами. Они
хотя и часто наблюдали гиперпластические процессы в зоне дерматофиб-
ром, но признаков базалиомы не отмечали.
Korom и Simon (1983) только в 15,2% из 255 случаев дерматофибромы
нашли неизмененный эпидермис, в 6,6%—он был атрофичным, в остальных
обнаруживалась гиперплазия (базалиомоподобная, фолликулярная, псев-
доэпителиоматозная, сходная с себорейным кератозом и др.), а в одном—
мультицентрическая базалиома. Причина столь частой гиперплазии эпи-
дермиса точно не установлена. Среди факторов, которые могут стимулиро-
вать пролиферацию эпидермиса, указываются накопления в дерме Р AS-
положительных и метахромазирующнх веществ, наличие самой дермато-
фибромы (Steigleder et al., 1962 и др.). Korom и Simon наблюдали измене-
ние эпидермиса прежде всего там, где дерматофиброма достигала дермо-
эпидермального соединения. Эти данные не согласуются с мнением Halp-
ryn и Allen, которые не нашли связи между степенью эпидермальных из-
менений и глубиной залегания опухоли.
Множественные дерматрфибромы лентикулярные могут сочетаться с
остеопойкилией (синдром Buschke—Ollendorf). Этот синдром наследуется
аутосомно-доминантно. В семьях наблюдается диссоциация признаков.
Это может касаться и характера кожных изменений. Так, Schorr с соавт.
(1972) среди 6 больных синдромом Buschke—Ollendorff наблюдали измене-
ния кожи только у 3, и они были представлены разными вариантами сое-
динительнотканного невуса. Дерматофибромы в этом случае имеют дис-
семинированный характер, сетевидное или полосовидное расположение с
концентрацией на бедрах, ягодицах, верхней части спины, шее, на пред-
плечьях. Они мелкие, обычно не более 1 см. Заболевание начинается как
правило в детстве.
Piette-Brion с соавт. (1984) сообщили о варианте синдрома Buschke—
Ollendorff с наличием, помимо дерматофибром, эластомы и двусторон-
ней глухоты. Tansch с соавт. (1984) наблюдали девочку, у которой гисто-
логически изменения в области высыпаний имели двоякий характер: соот-
ветствующие Nevuelasticus и лентикулярной дерматофиброме. Преиму-
щественное изменение эластических волокон (эластому) отметили в своем
наблюдении Cole и Вегг (1982). Поражение кожи появилось у женщины
63 лет в 58-летнем возрасте, расцененное вначале как псевдоксантома эла-
стическая.
Raque и Wood (1970) рассматривают диссеминированную лентикуляр-
ную дерматофиброму как вариант соединительнотканного невуса. По всей
вероятности, случай, который описан С. А. Глауберзоном и А. Я- Лебеде-
вым (1951) как множественная плоская дерматофиброма, был синдромом
Buschke—Ollendorff. Особенностью его было слияние отдельных высыпаний
в довольно крупные бляшки.
У пациентов с этим синдромом иногда отмечают склонность к образо-
ванию келоидов, полосовидную с анетодермией мелкоочаговую атрофию,
папилломы в области шеи, ихтиозиформную эритродермию, синдром Рендю—
156
Ослера, ладонно-подошвенные кератодермии, голубые склеры, дистрофии
зубов; может наблюдаться замедление роста. Помимо костной патологии
обнаруживают сахарный диабет, снижение интеллекта, инфантилизм,
катаракту, Schorr и соавт. наблюдали преждевременное половое созревание.
Гистопатология. Акантоз, гиперпигментация базального слоя, густые
сплетения новообразованного коллагена, пролиферация фибробластов,
новообразования сосудов, гистиоциты, содержащие гемосидерин, липиды.
Gertler (1973) указывает, что гистологически могут выявляться различные
морфологические структуры, изолированно или в разных комбинациях
в пределах одного и того же элемента,—структура плотной фибромы, ней-
рофибромы, гистиоцитомы и эндотелиомы. В чистом виде самой частой
является гистиоцитома, затем фиброма, редко—эндотелиома. По мнению
Lever (1983), напротив, фиброзный тип дерматофибромы встречается зна-
чительно чаще, чем клеточный (гистиоцитома). В обоих случаях выявля-
ются капилляры, иногда многочисленные, придающие опухоли ангиома-
тозный вид (склерозирующая гемангиома).
Дифференциальный диагноз. Дифференцировать дерматофиброму лен-
тикулярную необходимо с ксантомами, лейомиомой, гломусными опухо-
лями, невусами, меланомой, дерматофибросаркомой выбухающей.
Лечение. Обычно не проводится. При повторной травме—хирургиче-
ское.
ЛИТЕРАТУРА
Beare J. М., Cunliffe W. J. Histiocytome. In Textbook of dermatology Ed. Rook A.
et al.', Blackwell scientific publications, Oxford, 3-d ed., 1979, 2, 1522—1524.
GoetteD- K-, Helwig E. B. Basal cell carcinomas and basal cell carcinome—like chan-
ges overlying dermatofibromas. Arch. dermatoL, 1975, III, 5. 589—592.
Roberts J. T., Byrne E. H., Rosenthal D. Famifias variant of dermatofibroma with
malignancy in the proband.— Arch. dermatoL, 1981, 117, I, 12—15.
ДЖАНОТТИ—КРОСТИ СИНДРОМ (Gianotti—Crosti syndromum).
Синоним: папулезный акродерматит детей.
Этиология и патогенез. Предполагается вирусная природа заболева-
ния. Неясно, является ли этот синдром особой формой гепатита В или речь
идет о вирусиде (Milbradt, 1980). Castellano и соавт. (1978) не смогли обна-
ружить антиген в очагах поражения. Гистологически не было найдено дан-
ных за наличие иммунных комплексов. Тем не менее, авторы считают обя-
зательным во всех случаях тщательное обследование функции печени и
исключение гепатита типа В. Ф. А. Зверькова и И. Г. Придвижкин (1980)
различают 3 варианта этого синдрома: без поражения печени; в сочетании
с безжелтушным гепатитом; с тяжелым поражением печени, сопровождаю-
щемся желтухой. Milbradt и Nasemann (1975) с помощью электронной микро-
скопии у 2 больных в эндотелии мелких сосудов верхней части дермы обна-
ружили микротубулярные агрегаты размером около 200 А. Они не рассмат-
ривают их как вирусспецифические структуры, считая, что это может быть
157
микроморфологическим выражением иммунных процессов, например, в свя-
зи с антигенным воздействием. Gianotti (1976) подразделяет папулезные
акродерматиты детей на 2 формы: одна из них, собственно синдром Джа-
нетти—Крости, является малозаразным инфекционным заболеванием, обу-
словленным вирусом гепатита В; причина второй, более полиморфной, не-
известна. Автор предлагает до выяснения этиологии именовать ее акраль-
ным папуло-везикулезным синдромом.
Ramelet (1984) полагает, что, помимо этих 2 вариантов, существует
3-й, наблюдающийся у больных инфекционным мононуклеозом. Gengoux
и соавт. (1984) также наблюдали типичные для синдрома Джанотти—Кро-
сти высыпания у 18-месячного ребенка с гепатитом, но без антигена В,
а с антителами к вирусу Epstein—Barr. Robinson и Heath (1983) допускают
также широкое употребление диагноза синдрома Джанотти—Крости для
обозначения группы дерматозов, проявляющихся папулезными высыпа-
ниями на конечностях, пока неизвестна их причина, предлагая оставить
название болезнь Джанотти—Крости только для случаев папулезного ак-
родерматита детей, вызванных вирусом гепатита. В связи с этим особую
важность для постановки диагноза приобретает обнаружение антигена
вируса гепатита. Л. Боянов и соавт. (1963) выделяют 4 клинических при-
знака, характерных для этого синдрома: внезапное появление сыпи; сим-
метричная локализация на конечностях и лице; мономорфность высыпаний;
полиаденопатия.
Клиника. Высыпания развиваются внезапно, одномоментно. Мономорф-
ны, состоят из множественных густо расположенных, но не сливающихся,
отечных лентикулярных папул, не вызывающих субъективных расстройств.
Локализуются симметрично на конечностях, ягодицах, лице, которое по-
ражается в последнюю очередь (рис. 96, 97). Крупные складки обычно сво-
бодны от высыпаний (Gianotti, 1976). Цвет их розоватый или красный с
синюшным оттенком. На туловище высыпаний обычно не бывает, или они
слабо выражены и носят эритемато-сквамозный характер (Зверькова Ф. А.
и Придвижкин И. Г., 1980). Наблюдается нерезко выраженное увеличе-
ние периферических лимфатических узлов, особенно подмышечных и пахо-
вых, острый, как правило, безжелтушный вариант вирусного гепатита
(Gianotti, 1976). В крови—нерезко выраженная моноцитарная реакция,
повышение активности трансаминаз. Ринофарингит, тонзиллит, диспеп-
сия, умеренная лихорадка, общая слабость. Акральный папуло-везику-
лезный синдром, согласно описанию Gianotti (1976), является более частым
заболеванием, морфологически более вариабельным. Наряду с лентику-
лярными папулами, наблюдаются эритематозные и папуловезикулезные
высыпания, иногда с явлениями пурпуры, они могут сливаться, часто со-
провождаются зудом. Развивается в течение нескольких дней, часто в
несколько (Приступов. Высыпания располагаются симметрично на щеках,
ягодицах, ладонях и подошвах, в подмышечных впадинах, которые при
синдроме Джанотти—Крости, как правило, не поражаются. Может обна-
руживаться лимфаденит, но изменений в других системах не выявляется.
Выздоровление через 1—2 месяца, рецидивы редки. По-видимому, к этой
158
форме акродерматитов папулезных следует отнести наблюдение, описан-
ное Schwank (1966) как синдром Джанотти—Крости. Автор высказал мне-
ние о возможной роли аллергических реакций как причине высыпаний,
особенно если детей, больных инфекционными заболеваниями, лечат анти-
биотиками. Reich (1963) обратил внимание на выраженность геморрагиче-
ского компонента (пурпура Gianotti—Crosti). Начало с поражения кожи
лица, развитие пурпуры составили особенности и наблюдения Wiesner и
соавт. (1974). Winkelmann и Bourlond (1965) описали 18-месячную девочку
с папулезными и лихеноидными высыпаниями без какой-либо общей симп-
томатики и явлений пурпуры. Авторы считают, что вряд ли существует
принципиальная разница между двумя клиническими вариантами синдрома
Джанотти—Крости.
Заболевание развивается преимущественно у детей, в раннем детском
возрасте, протекает остро, регрессирует бесследно в течение 1—2 месяцев,
а иногда и раньше Исчезновению сыпи может предшествовать псориази-
формное отшелушивание. Рецидивов не отмечено. Гепатит существует зна-
чительно более длительно, до нескольких месяцев или лет. Faber и соавт.
(1983) наблюдали внутриутробное заражение вирусом гепатита В у 2-лет-
него больного с синдромом Джанотти—Крости. Мать этого ребенка в на-
чале беременности перенесла гепатит В с антигенами HBS и НВе. В крови
пуповины у новорожденного были обнаружены анти НВС—антитела клас-
са JgM. HBS—антиген не обнаруживался. При повторном обследовании,
159
через 1 неделю и 1 год, было выявлено нарастание титра антител к НВС.
В 2-летнем возрасте развился синдром Джанотти—Крости с положитель-
ным HBS—антигеном. Отсутствие его ранее, по мнению авторов, объяс-
няется необычно длительным инкубационным периодом.
Встречается обычно в виде спорадических случаев, но описаны и эпи-
демические формы (Ichimaru et al., 1976). Описано развитие заболевания
у взрослых (Clandy et al., 1977 и др.). Мы также наблюдали женщину с
этим синдромом.
Гистопатология. Изменения неспецифичны: очаговый гипер-и пара-
кератоз, акантоз, спонгиоз, расширение сосудов, умеренное набухание
эндотелия, лейкоцитоклазия, периваскулярные лимфогистиоцитарные ин-
фильтраты.
Дифференциальный диагноз. Дифференциррвать синдром Джанотти—
Крости необходимо от лихеноидных токсидермии, инфекционного моно-
нуклеоза, красного плоского лишая, болезни Abt—Letterer—Siwe.
Лечение симптоматическое. Обычно высыпания регрессируют само-
произвольно.
ЛИТЕРАТУРА
Зверькова Ф. А., Придвижкин И. Г. Детский папулезный акродерматит (синдром
Gianotti—Crosti). Вести, дерматол. н венерол. 1980, II, 34—37.
Gianotti F. Die infantilen papulosen Akrodermatitiden.— Hautarzt, .1976, 27, 10,
467—472.
Faber M., Berthold H., Remischoosky E. Gianotti—Crosti—Syndrom als Folge einer
pranatalen Hepatitis—В—Virusinfektion. Hautarzt, 1983, 34, 3, 123—125.
Castillano A., Schwertzer R., Tsang M. J., Omata M. Papular acrodermatitis of
childhood and hepatitis В infection.— Arch. Derm. 1978, 114,10, 1530—1532.
Milbradt R., Nasemann T. Uber die Entitat des Gianotti—Crosti Syndroms und seine
Beziehung zur Hepatitis—В—.Infection.— Hautarzt, 1975, 26, 9, 471—479.
Wiesner M., LinssG., SchwenkeW. Das Gianotti—Crosti—Syndrom:—Derm. Mschr.
1974, 160, 9, 758—761.
ДИСКЕРАТОЗ ФОЛЛИКУЛЯРНЫЙ ДАРЬЕ (diskeratosis follicularis
Darier).
Синонимы: псороспермоз, болезнь Дарье, вегетирующий кератоз.
В 1889 г. независимо друг от друга Darier во Франции и White в США
впервые описали это заболевание. Первоначально Дарье дал этому, забо-
леванию название фолликулярный вегетирующий псороспермоз (1899),
а затем фолликулярный дискератоз. Последний является наиболее при-
нятым синонимом болезни Дарье.
Этиология и патогенез неизвестны. По современным представлениям
фолликулярный дискератоз Дарье относится к генодерматозам с аутосом-
но-доминантным типом наследования (Gertler, 1970; Popchristov, 1971;
Lowe, 1978). Wedtofte с соавт. (1974) и др. полагают, что иммунологиче-
ский механизм является одним из звеньев патогенеза фолликулярного
дискератоза. Рееск и соавт. (1941) полагают, что фолликулярный диске-
160
ратоз является результатом дефицита витамина А. Однако с этим не соглас-
ны Carleton, Steven и многие другие исследователи. Существует мнение,
что буллезная форма болезни Дарье представляет собой своеобразную пе-
реходную стадию к доброкачественной семейной пузырчатке Gougerot—
Haily—Hayley (Touraine, 1954; Ellis, 1944; Canuldfield и Wilgram, 1963
и др.). Однако большинство других авторов (Смелов Н. С. и Трофимова
Л. Я-, 1956; Шеклаков Н. Д., 1961; Пер М. И., 1962; Jablonska et al.,
1959; Gertler, 1970; Lever, 1975 и др.) полагают, что болезнь Дарье яв-,
ляется самостоятельным заболеванием. Lachapelle с соавт. (1973) на
основании радиоаутографических исследований с использованием тими-
дина—6*—Н пришли к выводу, что это разные заболевания (отсутствие
акантолитических клеток и ранняя кератинизация при болезни Дарье).
Мы также полагаем, что, несмотря на большое сходство клинической и ги-
стологической картины, нет достаточных оснований для отождествления
болезни Дарье и доброкачественной семейной пузырчатки.
Клиника. Заболевание начинается часто в детском или в юношеском
возрасте. В литературе приводятся единичные наблюдения (Witten и Gel-
farb, 1963; Fishman,* 1975 и др.) внезапного развития болезни Дарье у
пожилых. Оба пола поражаются с одинаковой частотой. Болезнь может
встречаться среди членов одной семьи и даже в нескольких поколениях
(Hartwaguer, 1972 и др.). Высыпания симметричные, занимают обширные
участки, преимущественно в себорейных местах (рис. 98, 99). Излюблен-
ная локализация высыпаний волосистая часть головы, лицо (в носо-губ-
ных складках), область грудины, между лопатками, подмышечные впади-
ны, пахово-бедренные складки. В зависимости от характера морфологиче-
ских элементов различают узелково-роговую, узелково-везикулезную, пем-
фигоидную (буллезную) и вегетирующую формы. Основной морфологиче-
ский элемент—буровато-красная или бледно-розовая папула диаметром
0,2—0,5 см, располагается преимущественно в устье расширенных фолли-
кулов, но может быть и внефолликулярной. Поверхность ее покрыта плот-
носидящей роговой корочкой с клинообразным шипиком на нижней поверх-
ности (гиперкератотическая пробка); после удаления корочки обнажается
зияющее отверстие сально-волосяного фолликула. На тыльных поверх-
ностях кистей и стоп папулы напоминают плоские бородавки. Узелковые
элементы могут группироваться, сливаться, особенно на лице, волосистой
части головы, с образованием бляшек, покрытых жирными грязными
чешуйками или серозно-геморрагическими наслоениями, Несколько на-
поминающих очаги себорейной экземы; кожа в этих местах имеет сальный
блеск, волосяные фолликулы расширены. В части случаев при локализации
процесса в крупных складках тела очаги представляются в виде эритема-
тозно-мокнущих бляшек с щелевидными извилистыми-эрозиями-трещина-
ми на поверхности. Во время прогрессирования болезни хорошо заметен
воспалительный фликтенулезный ободок по краю очага. В области гени-
талий и вокруг ануса часто развиваются вегетации. При развитии вторич-
ной пиококковой или грибковой инфекции на первый план в клиниче-
ской картине фолликулярного дискератоза выступают проявления пиодер-
11 Клиническая дерматология 161
Рис. 98. Фолликулярный
дискератоэ Дарье.
Рис. 99. Фолликулярный
дискератоэ Дарье
(везикулезная форма).
мии, кандидамикоза и т. д. Н'. С. Смелов и Л. Я- Трофимова (1956), Pirard
и соавт. (1968) наблюдали больных, у которых одновременно с папуло-
корочковыми, папуло-пузырьковыми элементами на отдельных этапах
течения заболевания преобладали пузырьки; иногда последние являлись
основными морфологическими элементами. При этом клиническая картина
напоминала экзематозный процесс. Болезнь Дарье не является исключи-
тельно фолликулярным заболеванием, доказательство этому—поражение
ладоней и слизистых оболочек (Ellis, 1968). Ладонно-подошвенный гипер-
кератоз • наблюдается в 10—15% случаев. На ногтевых пластинках при
фолликулярном дискератозе могут образоваться борозды, углубления,
участки помутнения, подногтевой гиперкератоз. Zaias с соавт. (1973) у
7 из 37 пацие нтов обнаружили гистологически в подногтевом ложе пора-
женных ногтей специфические изменения (лакуны, акантолитические клет-
ки, многоядерные гигантские эпителиоидные клетки). Данные о частоте
поражения слизистой оболочки полости рта у больных фолликулярным
дискератозом весьма разноречивы и колеблются от 15 до 50% (Medak,
1971). Первое описание поражений слизистой оболочки полости рта бь ло
сделано Reenstiema в 1917 году. Как отмечают многие авторы (Shafer et
al., 1963; Getzler и Flint, 1966; Caulfield и Wilgram, 1963; Medak et al.,
1971; Weather et al., 1969; Krull et al., 1973; Rayne, 1970; Dellon, 1975;
Prindiville и Stern, 1976, и др.) высыпания во рту часто локализуются
на деснах, языке и слизистой щеки, но наиболее часто поражаются твердое
и мягкое небо. У отдельных больных поражается слизистая оболочка влага-
лища, гортани, глотки и пищевода (Brunauer, 1925; Dellon, 1975). Клиниче-
ская картина болезни Дарье на слизистых оболочках может быть различной:
в виде светло-белых узелков или белых линейных утолщений, стимулирую-
щих лейкоплакию. В некоторых случаях, как и на коже, отмечаются фи-
162
гурные высыпания, главным образом везикулезные. Основание полости
рта никогда не поражается. Язык поражается в тяжелых случаях и напо-
минает скротальный язык. Типичные высыпания дискератоза на слизи-
стых оболочках обычно не вызывают субъективных ощущений, боль и
жжение наблюдаются при эрозированных формах. Заболевание нередко
носит системный характер. Наряду с поражением кожи и слизистых обо-
лочек отмечаются нарушения психики (угнетенное состояние, слабость
памяти, умственная отсталость и др.), разнообразные эндокринопатии,
нарушения функции внутренних органов, дефекты развития костной ткани.
Общее состояние остается удовлетворительным.
Течение болезни хроническое с длительными спонтанными ремиссия-
ми.
Гистопатология. Наиболее характерные признаки— образование круг-
лых телец (corps ronds) и зерен (grain), внутри—эпидермальные щели (ла-
куны). Кроме того, отмечаются внутри-и внефолликулярный гиперкератоз,
акантоз и папилломатоз. В дерме лимфо-гистиоцитарный инфильтрат.
«Круглые тельца» локализуются в мальпигиевом слое, образуются в ре-
зультате преждевременной кератинизации клеток (доброкачественный ди-
скератоз). «Зерна» находятся в роговом слое. Во внутриэпидермальных
щелях (лакунах), расположенных чаще непосредственно над базальным
слоем, содержатся акантолитические клетки, лишенные межклеточных
мостиков, вследствие дегенеративных изменений. Электронно-микроскопи-
ческими исследованиями установлено, что в основе патологических измене-
ний лежат специфические процессы разрушения тонофиламентодесмосом-
ных комплексов. Вследствие этого клетки теряют десмосомный аппарат,
что обусловливает развитие акантолиза с последующим нарушением ке-
ратинизации (Forssmann и Holzmann, 1967; Caulfield и Wilgram, 1963;
Gottlieb и Lutzner, 1973). Изменения на слизистых аналогичны изменениям
на коже (круглые тельца, зерна и лакуны). Проведенные Довжанским С. И.
и Суворовым А. П. (1975) гистохимические исследования свидетельствуют
о глубоких изменениях (преимущественно в эпидермисе) метаболических
процессов, особенно белково-полисахаридного звена.
Дифференциальный диагноз. Следует проводить с вульгарной пузыр-
чаткой, себорейной экземой, верруциформной эпидермодисплазней Леван-
довского—Лютца, красным плоским лишаем, плоскими бородавками.
Лечение. Кортикостероиды (преднизолон по 15—30 мг в день), анти-
гистаминные препараты, витамин А, антибиотики, общие ванны с крах-
малом и др.; наружно—жидкость Кастеллани, кортикостероидные и са-
лициловые мази. В упорных случаях применяют Букки-или рентгено-
терапию. Имеются наблюдения (Orfanos, 1980; Lassus, 1980 и др.) о поло-
жительном действии ароматического ретиноида (Ro 10—9359).
ЛИТЕРАТУРА
Довжанский С. И.., Суворов А. Л. Фолликулярный дискератоз (Morbus Darier).—
Вести, дерматол. и венерол., 1975, 4, 40—45.
11* 163
Dellon A. L. Hypopharyngeal and laryngeal involvement withT Darier disease.—
Arch. Dermatol.,'1975, III, 6, 744—746.
Fishman H. Acute eruptive Darier disease (keratosis follicularis) occurence in an adult.
— Arch. Dermatol., 1975, Ilf, 2, 221—222.
Lassus A. Systemic treatment of psoriasis with an oral retinoic acid derivative (R°
10—9359).— Brit. J. Dermatol., 198x0, 102, 2, 195—202.
Orfanos C. Oral retinoid-present status.— Brit. J. Dermatol., 1980, 103, 5, 473—481.
Prindioille D. Oral manifestations of Darier disease.— J. Oral. Surg., 1976, 34, II,
1001—1006.
Sato A. Ultrastructure of dyskeratosis in morbus Darier.— J. Cuten Pathol., 1977,
4, 4, 173—184.
Weathers D., Driscoll R. Darier’s disease of the oral mucosa. Report of five cases.—
J. Oral. Surg. 1974, 37, 711—721.
Zachriae H. Dermabrasion in Darier's disease.— Acta Dermatovener., 1979, 59, 2
184—186.
ДИСПЛАЗИИ ЭКТОДЕРМАЛЬНЫЕ (displasie ectodermale).
Этиология и патогенез. Гетерогенная группа наследственных заболе-
ваний, обусловленных ненормальным развитием эктодермы. Различают
ангидротическую (dysplasia ectodermalis anhydrotica, s. hypotrichotica;
syndromum Christ—Siemens) и гидротическую (dysplasia ectodermalis hyd-
rotica, s. syndromum Clouston) формы, а также ряд синдромов, в которых
кожные дефекты ассоциированы с другими врожденными аномалиями
(синдром Marchall, синдром Helweg—Larsen и др.). Salomon и Кепег (1980)
считают, что термином «эктодермальная дисплазия» следует обозначать
такие состояния, которые являются врожденными, диффузными, не прог-
рессируют, затрагивают эпидермис и как минимум один из придатков кожи.
Ангидротическая форма без врожденных аномалий наследуется в большин-
стве случаев рецессивно-сцепленно с Х-хромосомой или аутосомно-ре-
цессивно (Альтшулер Б.'А. и Суворова К. Н., 1984). Имеются данные и о
вариантах, наследуемых аутосомно-доминантно (Witkowski и Prokop, 1974).
Гидротическая форма эктодермальной дисплазии представляет собой ауто-
сомно-доминантное заболевание (Williams и Fraser, 1967 и др.). Fried (1977)
указывает на возможность рецессивных форм. Биохимический дефект, по-
видимому, заключается в недостаточности дисульфидных связей во
фракции L-кератинового матрикса с высоким содержанием серы (Gold
и Scriver, 1972). Указывается на возможность развития дисплазий, по мень-
шей мере, по мнению Kuhlwein и Weiss (1982), спорадических форм, под
влиянием химических соединений, рентгеновых лучей, вирусных инфек-
ций и других вредностей.
Дисплазия эктодермальная ангидротическая (рис. 100, 101, 102, 103,
104, 105, 106).
, Клиника. Для ангидротической формы эктодермальной дисплазии
характерна следующая триада симптомов: гипо- или ангидроз, гипотрихоз,
гиподонтия. Кожа истончена, нежноскладчата, особенно вокруг глаз.
Потоотделение снижено или реже отсутствует по вс°му кожному покрову, ;
164
Рис. 100. Ангидротическая
эктодермальная дисплазия.
Рис. 101. Ангидротическая форма
эктодермальной дисплазии (гипотрихоз).
Рис. 102. Ангидротическая
эктодермальная дисплазия
(дистрофия зубов).
Рис. 103. Ангидротическая
эктодермальная дисплазия
(изменения ногтей).
165
за исключением мест локализации апокриновых желез, где оно в большей
или меньшей степени может сохраняться. Значительна сухость кожи, не-
сколько меньше в подмышечных впадинах, в области половых органов,
на лице и слизистых, что часто приводит к развитию конъюктивитов, ла-
рингитов, фарингитов, ринитов, стоматитов. У ряда больных наблюдается
ихтиозиформное шелушение, фолликулярный гиперкератоз, ладонно-по-
дошвенные кератодермии (Самцов В. И. и Граневский А. В., 1974, Маг-
tin-Pascnal, 1977). Kuhlwein и Weiss (1982) описали 12-летнего мальчика,
у которого, помимо типичной клинической картины, отмечался гиперке-
ратоз на дорзальной поверхности пальцев стоп. Резкое поредение волос
на голове или тотальная алопеция. Брови, а иногда и ресницы редкие,
скудное или отсутствует оволосение в подмышечных областях и на лобке.
Пушковые волосы обычно сохраняются. Зубы поздно прорезываются,
долго сохраняются на стадии молочных, могут быть не в полном количе-
стве, или даже отсутствовать йолностью, часто деформированы, с большими
промежутками между ними. За счет дистрофии зубов кожа вокруг рта мо-
жет иметь складки, напоминающие рубцы при врожденном сифилисе. Ха-
рактерен внешний вид больных: низкого роста, большой квадратный череп
с выступающей лобной частью, массивный подбородок, надбровные дуги,
высокие и широкие скуловые кости, впалые щеки, выпуклые губы, боль-
шие уши (уши сатира), седловидный нос. Reed и соавт. (1970), И. И. Лелис
(1978) описали сочетание с acanthosis nigricans. Из других состояний опи-
сана склонность к экзематозным реакциям (Korting и Salfeld, 1961), ато-
пический дерматит и астма (Reed et al., 1970), гипотиреоз Yapusan et al.,
1975), милиумподобные папулезные высыпания, обусловленные сально-
железистой гиперплазией, аплазия сосков (Martin-Pascual et al., 1977).
У женщин заболевание протекает в смягченной форме в виде нерезко вы-
раженных зубных аномалий, очаговых расстройств потоотделения, слабого
развития молочных желез (Суворова К- Н. и Антоньев А. А. 1977;
Witkowski и. Prokop, 1974).
Дисплазия эктодермальная гидротическая (рис. 107).
Наиболее характерными клиническими признаками являются дист-
рофия ногтей и гипотрихоз, в меньшей мере—ладонно-подошвенный ке-
ратоз, нарушение пигментации. Дистрофия ногтей является основным,
наиболее частым, а иногда и единственным признаком. Ногтевые пластинки
утолщены, хрупкие, легко обламываются, коричневатого цвета, деформи-
рованы, по краю как бы изъедены, расщеплены, продольно исчерчены.
Подноггевой «типеркератоз, часты паронихии. Ногти растут медленно.
Может наблюдаться атрофический тип ониходистрофии с почти полным
исчезновением ногтей. Этому также способствуют частые воспалительные
процессы, которые могут* приводить к рубцовым изменениям ногтевого ло-
жа. Кожа на кончиках пальцев, а также над суставами утолщена, склад-
чата. Rajagopalan и Chong Hai Тау (1977) обнаружили 3 типа ониходист-
рофии, описанных ранее Wiekey и Stevenson (1945). Наиболее частые и
самые тяжелые изменения заключались в том, что ногтевые пластинки
166
Рис. J04. Ангидротическая
эктодермальная дисплазия
(гиперкератотические изменения
на ладонях).
Рис. 105. Ангидротическая
эпидермальная дисплазия.
Рис. 106. Ангидротическая Рис. 107. Гидротическая
эпидермальная дисплазия. форма эктодермальной дисплазии
(гипотрихоз у отца и сына).
были уменьшены в размерах, конической или треугольной формы, туск-
лые, желтоватые. Вокруг них кожа была гипертрофирована, складчата.
, При втором типе изменений, который иногда встречается одновременно с
первым, ногти были нормального размера, но умеренно утолщены, их по-
верхность была дистрофически изменена: обнаруживались множественные
желтоватые и точечные углубления, продольные полосы, крошащаяся
ломкость по свободному краю и по бокам.
Третий тип ониходистрофии проявляется небольшим уменьшением
размера, уплощением пораженных ногтей, отделением дистальной части
167
ногтевых пластинок от ногтевого ложа, ломкостью. Выраженного утолще-
ния не наблюдалось.
Заболевание развивается вскоре после рождения или в раннем детском
возрасте, с одинаковой частотой у лиц обоего пола, чаще у блондинов.
Может быть значительной вариабельность степени выраженности и дис-
социация признаков в семьях. По наблюдениям Rajagopalan и Chong Hai
Тау (1977), изменения волос были более выражены у членов семьи женско-
го пола, а кератодермия—у мужчин. Полная клиническая картина обычно
формируется в период полового созревания. Изменения ногтей и ладонно-
подошвенный кератоз с возрастом прогрессируют. Волосы истончены, су-
хие, легко обламываются, поредевшие, иногда незначительно, иногда
до степени алопеции, в том числе тотальной (Me Nanghton et al., 1976).
Дистрофия волос обнаруживается и на других участках кожного покрова.
Брови и ресницы выпадают полностью или сохраняются только в медиаль-
ной части, Скудное оволосение наблюдается в подмышечных впадинах и
на лобке. Могут наблюдаться такие аномалии, как перекрученные волосы,
узловатая ломкость волос, расщепление волос. Обнаруживают утолще-
ния концевых фаланг, изменения зубов обычно отсутствуют, либо они
незначительны. Могут быть увеличены промежутки между зубами, ранний
кариес. Кожа нежная, истончена, суха, может быть ихтиозиформное ше-
лушение, кератоз на разгибательных поверхностях конечностей, иногда
с выраженной гиперпигментацией в области суставов, что может напоми-
нать acanthosis nigricans. Снижено может быть потоотделение, за исключе-
нием ладоней (Dethlefs и Tronnier, 1972), хотя есть указания, что у боль-
ных нередко наблюдается гипергидроз. Непостоянны и выражены в раз-
ной степени явления гиперкератоза на ладонях и подошвах. Утолщение
рогового слоя на ладонях и подошвах может быть очаговым, полосовидным
или сплошным, типа кератодермии Unna—Ihost (Theisen, 1963). Описано
также развитие доминантной формы Keratosis palmoplantaris transgrediens
(Landes, 1960). P. С. Диванян и А. Г. Мурадян (1980) описали сочетание
гидротической дисплазии с волчьей пастью. Могут быть болезненные тре-
щины, редко—пузыри. Описано (Wilkinson et al., 1977) развитие множе-
ственных мелких эккринных пором на кистях и стопах. Dethlefs и Tronnier
. (1972) обращают внимание на такие признаки, как повышенная чувствитель-
ность к холоду, функциональное изменение сосудов кожи по типу ангио-
патии, чему авторы придают большое значение в развитии поражения ног-
тей и волос. Указывается (Wiekey и Stevenson, 1945), что часто встречаются
глазные аномалии (страбизм, птеригиум, конъюнктивит, катаракта). В
редких случаях могут наблюдаться умственная отсталость, эпилепсия,
глухота, поли- и синдактилия, замедленный рост. Однако из-за малого
числа наблюдений трудно судить о том, насколько закономерны эти ас-
социации. Так, Rajagopalan и Chong Hai Тау (1977), обследовавшие боль-
шую китайскую семью в Малайзии, выявили 15 больных в 5 поколениях,
но ни у кого из них не было ни других кожных, ни общих заболеваний.
Таким образом, в отлитие от ангидротической формы, имеющей харак-
терный клинический признак—гипо- или ангидроз, в основе которого
168 >
лежит гипоплазия потовых и сальных желез кожи, гидротическая форма
имеет менее определенную и значительно более вариабельную симптома-
тику, что дало основание Jung (1980) высказать мнение о невозможности
четкого клинического отграничения этой формы эктодермальной диспла-
зии. К- Н. Суворова и А. А. Антоньев (1977) считают, что это заболевание
относится не к дисплазиям, а к группе наследственных нарушений кера-
тинизации.
Обе формы эктодермальной дисплазии развиваются в раннем детском
возрасте с формированием полной клинической картины к периоду полово-
го созревания, но ангидротическая дисплазия встречается во много раз
чаще у мальчиков, в то время как распределение гидротической формы не
связано с полом. Больные ангидротической формой страдают гипертер-
мией, что может иметь тяжелые последствия в нераспознанных случаях.
Иногда у них выявляются признаки снижения интеллекта, потеря слуха,
подверженность инфекции (Franken, 1966). Feftidi и соавт. (1964) наблю-
дали у 2 братьев выраженный нистагм и центральный синдром Horner.
Hazen и соавт. (1980) описали семью, в которой было 5 больных гидротиче-
ской эктодермальной дисплазией в 3 поколениях; 4 из них страдали дву-
сторонней преждевременной катарактой. Почти полная потеря зрения
наблюдалась у больного, данные о котором опубликовали И. Н. Ляшенко
и А. М. Соколов (1981).
Гистопатология. При ангидротической форме дисплазии отсутствие
эккринных потовых желез; апокриновые железы, волосяные фолликулы
и сальные железы могут быть изменены в разной степени, вплоть до от-
сутствия, но могут быть и нормальны. На аутопсии одного больного Reed
и соавт. (1976) обнаружили отсутствие слизистых желез в гортани, глотке,
трахее, бронхах и верхней части пищевода. При гидротической диспла-
зии—гипоплазия волос и сальных желез. По мнению Dominon и Ronisch
(1968), гистологический диагноз эктодермальной дисплазии может быть
поставлен только при сравнении идентичных участков здоровых людей.
Дифференциальный диагноз. Дифференцировать необходимо с про-
гериями, врожденным сифилисом, врожденной пахионихией.
Лечение симптоматическое. Необходимо предохранять больных с ан-
гидротической формой от перегревания.
ЛИТЕРАТУРА
Hazen Р. Y , Zamora I., Bruner W. Е., Muir W. A. Premature cataracts in a fa-
mily with hidrotic ectodermal Dysplasie. — Arch. Derm., 1980, 116, 12, 1385—1387.
Kuhlwein A., Weiss J. Anhidrotische ektodermale Dysplasie (Christ—Siemens—Tou-
raine syndrom) bei x-chromosomal—rezessiven Erbgang.— Derm. Mschr., 1982,168,1,34—43.'
Альтшулер Б. А., Суворова К- H. Изучение типа наследования ангидротической
эктодермальной Дисплазии. Весгн. дерматол. и венерол. 1984, 2, 15—18.
Р. С. Диванян, А. Г. Му рад ян. Синдром Клустона. Вести, дерматол. и венерол.
1980, 80, 46—48.
И. Н. Ляшенко, А. М. Соколов. Ангидротическая эктодермальная дисплазия у
больного 56 лет и ее осложнения. Весгн. Дерматол. и венерол. 1981, 5, 42—46.
169
И. И. Лелис. Аутосомно-рецессивная эктодермальная дисплазия — отдельная но-
зологическая единица. Вести- Дерматол. и венерол. 1978, 12, 56—59.
В. И. Самцов, А. В. Граневский. Ангидротическая эктодермальная дисплазия с
кератодермией. Вести, дерматол. и венерол. 1974,. 4, 64—67
К- Н. Суворова, А. А. Антоньев. Наследственные дерматозы. М. Медицина, 1974.
Altmeyer Р., Schindera I. Ein Beitrag zur hidrotischen ectodermafen Dyspfasie.—
Hautarzt, 1975, 26, 12, 631—637. ;
Gapusan /., Precup C., Porzolt A. Potidispfasia ectodermafe hidrotica ereditara.—
Dermato—venerofogica (Buc.), 1977, 20, 4, 289—297.
Jung E. Y. Ektodermaldysplasien In «Dermatologie in Klinik und Praxis.» Hrsg Y. K-
Korting, Stuttgart, 1980, Bd. 2, 20. 1—20. 3.
Martin-Pascual A., Unamuno P. D., Aparicio M., Hereros V. Anhidrotic (or hy-
pohidrotic) ectodermal dysplasi, Dermatologica, 1977, 154, 4, 235—243.
Me Naughton P. Z., Pierson D. L., Rodman O. Y. Hidrotic ectodermal dysplasia
in a black mother and daughter. — Arch. Derm., 1976, 112, 10, 1448—1450.
Raiagopalan K-, Chong Hai Tay. Hidrotic ectodermal dysplasia. Arch, dermatol.,
1977, 113, 4, 481—485.
Salomon L. M., Keuer E. J. The ectodermal dysplasias.— Arch. Derm., 1980, 116,
11, 1295—1299.
Wilkinson R. D., Schopflocher R., Rozenfeld M., Hidrotic ectodermal dysplasia with
diffuse eccrine poromatosis. Arch, dermatol., 1977, 113, 472—476.
Worret W. I., Burgdorf W. H. C., Kusch S. L. Anhidrotische ektodermale Dysp-
fasie.—Hautarzt, 1982, 135, 5, 289—290.
Witkowski R., Prokop 0. Genetik, Akademie—Verlag.— Berlin, 1974.
ДИСТРОФИЯ КОЖИ ПАПИЛЛЯРНО-ПИГМЕНТНАЯ (dystrophia cutis
papillaris et pigmentosa).
Синонимы: черный акаитоз, черный кератоз, сосочковая меланодер-
мия.
Описана в. 1890 г. Pollitzer и Janovsky. Различают три формы заболе-
вания: злокачественную, доброкачественную (ювенильную) и псевдоакан-
тоз.
Этиология и патогенез. Darier впервые отметил связь черного акан-
тоза с раком внутренних органов. В дальнейшем многие авторы подтвер-
дили это. По разным данным, рак обнаруживают у 50—100% больных с
черным акантозом. Именно эта форма является почти облигатным пара-
неопластическим процессом. Развитие паранеоплазии обусловливают слож-
ные обменные и иммунные расстройства организма и продукты катаболизма
опухолевых клеток (Бутов Ю. С. и Дмитриев В. И., 1979; Curt et al.,
1952, 1962; Mikhail et al., 1979; Rigel и Jacob, 1980; Gardama et al., 1980 и
др.). У больных мужчин чаще обнаруживается рак желудка, поджелудоч-
ной железы и кишечника, реже—легких, простаты, а у женщин—яични-
ков и молочной железы. В. С. Шапот (1981) относит папиллярно-пигмент-
ную дистрофию кожи в группу наследственной энзимопатии (нарушена
репарация поврежденной ДНК из-за отсутствия в отдельных случаях спе-
цифических эндонуклеаз).
170
Клиника. Изменения кожи характеризуются тремя основными при-
знаками: гиперпигментацией, папилломатозом и гиперкератозом. Они,
как правило, располагаются симметрично в местах излюбленной локализа-
ции: на шее, затылке, в области половы» органов, пупка, заднего прохода,
в подмышечных впадинах, паховых складках, локтевых и коленных сги-
бах, под грудными железами. Иногда очаги гиперпигментации бывают на
лице: вокруг рта, глаз, на подбородке. Пораженная кожа приобретает
буроватую или почти черную окраску, рисунок кожи усиливается. На
этом фоне возникают множественные ворсинчатые и бородавчатые разра-
стания аспидно-черного цвета величиной от просяного зерна до горошины.
Кожа имеет бархатистый вид из-за усиленного кожного рисунка и папил-
ломатозных разрастаний. Часто в очагах поражения встречаются мелкие
фибромы. В окружности очагов встречаются веснушки, пигментные невусы,
себорейные бородавки. Имеются описания пигментных и веррукозных из-
менений на слизистых оболочках полости рта, гортани, пищевода, влага-
лища и прямой кишки (Иванов Н. А. и соавт., 1969; Dornbush, 1973;
Schimeyer et al., 1974; Samitz, 1977 и др.). У некоторых больных заболе-
вание сопровождается алопецией, гиперкератозом ладоней и подошв, дис-
трофией ногтей. Субъективно отмечается умеренный зуд и покалывание.
У многих больных наблюдается нарушение общего состояния и постепен-
но нарастающая кахексия (рис. 108, 109).
1. Злокачественная форма сосочково-пигментной дистрофии кожи
встречается у взрослых (старше 25 лет). Поражения кожи ь 15—20% слу-
чаев на несколько лет предшествуют клиническим проявлениям рака;
у 60—65% они появляются одновременно с симптомами рака, у 20—22%—
после обнаружения злокачественного новообразования.
2. Доброкачественная (ювенильная) форма начинается в детском или
юношеском возрасте (рис. 110). Клинически и гистологически она анало-
гична злокачественной форме заболевания, отличаясь от нее лишь мень-
шей выраженностью пигментных изменений и сосочковой гипертрофии.
Общее состояние, как правило, не нарушается. Часто с наступлением по-
ловой зрелости высыпания регрессируют или остаются стационарными.
Эта форма в подавляющем большинстве случаев не сопровождается злока-
чественными новообразованиями внутренних органов (Никитина М. Н. и
Алехина, Г. М., 1976; Капкаев Р. А. и Аковбян В. А., 1980), хотя в единич-
ных случаях (Jacobi, 1959; Garrot, 1972 и др.) отмечено такое сочетание. Эту
форму, нередко сочетающуюся с различными аномалиями развития, врож-
денными уродствами, гипертрихозом, невусами и др., считают генетически
обусловленным заболеванием (Curth и Asdner, 1959; Senior и Gellis, 1964;
Lever и Schaumburg-Lever, 1975 и др.). Семейные случаи (Banuchi et al.,
1974; Marill et aL, 1973 и др.) указывают на наличие генетической пред-
расположенности. По мнению ряда авторов (Зенин А. С., 1928; Jadas-
sohn, 1948; Brown и Winkelmann, 1968 и др.), юношеская форма акантоза
развивается у молодых людей, страдающих эндокринными нарушениями
(диабет, ожирение, аменорея и др.).
171
Рис. 108. Акантоэис
нигриканс (злокачественный).
Рис. 109. Акантоэис
нигриканс
(злокачественный).
Псевдоакантоз (Curth, 1951). Встречается преимущественно у туч-
ных брюнеток. Имеет также сходную клиническую и гистологическую
картину с черным акантозом взрослых (рис. 111) Заболевание имеет добро-
качественный характер, появляется, когда больной прибавляет в весе, и
исчезает с похуданием больного.
Прогноз зависит от формы заболевания. При злокачественной форме
средняя продолжительность жизни больных после появления изменений
на коже составляет 1,5—2 года; пациенты погибают, несмотря на операцию.
Ввиду частого сочетания черного акантоза со злокачественными новообра-
172
зованиями внутренних органов, больные должны находиться под диспан-
серным наблюдением дерматолога и онколога.
Гистопатология. Выраженный гиперкератоз, папилломатоз, акантоз,
увеличение меланина в клетках базального слоя эпидермиса и наличие
в дерме большого числа хроматофоров. Воспалительные явления незначи-
тельны; в верхних слоях дермы—периваскулярный инфильтрат, состоя-
щий из лимфоцитов.
Дифференциальный диагноз. Следует проводить с аддисоновой болез-
нью, с фолликулярным дискератозом Дарье, мышьяковой меланодермией,
ихтиозом.
Лечение злокачественной формы болезни обычно безуспешно. В не-
которых случаях отмечается временное улучшение кожного процесса и
общего состояния после хирургического удаления злокачественного ново-
образования внутренних органов. При ювенильной форме показаны эндо-
кринные препараты, витамины, общеукрепляющие мероприятия, гигие-
нический уход за кожей, местно—смягчающие кремы. Р. А. Капкаев и
В. А. Аковбян (1980) успешно лечили псевдоакантоз аппликациями жид-
кого азота.
ЛИТЕРАТУРА
Бутов Ю. С., Дмитриев В. И. Злокачественная форма пигментнососочковой ди-
строфии кожи.— Вестн. Дерматол. и венерол., 1979, 7, 43—45.
Капкаев Р. А., Аковбян В. А. К вопросу о доброкачественных формах acanthosis
nigricans — Вестн. Дерматол. и венерол., 1980, 7, 50—52.
Никитина М. Н., Алехина Г. М. Случай Доброкачественной формы пигментно-
сосочковой дистрофин кожи.— Вестн. Дерматол. и венерол., 1976, 9, 66—69.
Шапот В. С. О некоторых генетических аспектах неоплазии.— Арх. патол., 1981,
2, 5—13.
Шупенько Н. М., Свирид А. А. Злокачественная форма папиллярно-пигментной
дистрофии кожи (acanthosis nigrigans malignum).— Вестн. дерматол. и венерол., 1980, 2.
37—40.
Curth Н. Classification of acanthosis nigricans.— Intern. J. Dermatol., 1976, 15,
592—603.
Gardama J., Gatt J., Stefani G-, Gatti C. Acanthosis nigricans asociado con adeno-
carcinoma de vejiga.— An. Bras. Dermatol., 1980, 55, 2, 91—94.
Iliescu O. Treatment chirurgial d’qn cas de Xeroderma pigmentosum.— In: Congres
national de dermatologie. Bucuresti, 1980, p. 240—241.
Liddell J., Jensen N. Malignant acanthosis nigricans and its unusial association with
carcinoma of the colon and carcinoma of the cervix.— Brit. J. Surg., 1976, 63, 248—250.
Mikhail G., D'Anne F., Drukker B. et al. Generalized malignant acanthosis nigri-
cans.—Arch. Dermatol., 1979, 115, 2, 201—203.
Rigel D., Jacobs M. Malignant acanthosis nigricans. A review J. Dermatol. Surg.
Oncol., 1980, 6, 11, 923—927.
173
ИМПЕТИГО ГЕРПЕТИФОРМНОЕ (impetigo herpetiformis).
Синонимы: импетиго беременных, герпес родильниц, пиемический
герпес
Впервые заболевание описал в 1872 г. F. НеЬга.
Этиология и патогенез. В первых описаниях НеЬга была установлена
тесная связь герпетиформного импетиго с беременностью и возникновение
заболевания рассматривалось как результат аутоинтоксикации. Kaposi
(1887) дополнил клиническую картину заболевания и указал на возможность
возникновения его у мужчин. Автор полагал, что герпетиформное импетиго
обусловлено пиемическими процессами при беременности и в послеродо-
вом периоде.
Ou F. и СоаЬг с соавт. (1982) наблюдали больную 26 лет. Высыпания
появились на 3-м месяце беременности. После рождения мертвого плода
кожные покровы полностью очистились от высыпаний.
Многие авторы считают, что дерматоз, по-видимому, связан с нару-
шениями функции желез внутренней секреции, в первую очередь паращи-
товидных желез и половых желез. Schardorn (1921), Leonhardi и Michel
(1958) считают, что гипокальциемия является важным признаком в диагно-
стике этого заболевания, хотя многие другие авторы,- и мы в том числе, не
полностью согласны с этим; во многих случаях уровень кальция в крови
нормальный. Можно полагать, что в основе развития заболевания лежат
сложные нейроэндокринные нарушения. В настоящее время нет единого
мнения о том, следует ли выделять герпетиформное импетиго в отдельную
нозологическую единицу. Некоторые авторы (Riecke, 1931; Lapiere, 1958;
Tielsch, 1962; Baker и Ryan, 1971; Macmillan и Champion, 1973; Oosterling
et al., 1978 и др.) видят в герпетиформном импетиго, хроническом акро-
дерматите Аллопо и пустулезном псориазе одно заболевание. Однако зна-
чительное большинство авторов (Машкиллейсон Л. Н., .1965; Калам-
карян А. А. и соавт., 1980; Зверькова Ф. А. и соавт., 1982; Scog, 1958;
Moslein, 1959; Lever, 1975; Mauric et al., 1960; Katzenellenbogen и
Fenerman, 1966; Bruckmann и Batli, 1978 и др.) рассматривают герпети-
формное импетиго как самостоятельное заболевание.
Клиника. Болеют преимущественно беременные женщины, но иногда
и небеременные, а также мужчины (Батунин М. П., 1929; Eberhartinger,
1958 и др.). Болеют чаще взрослые, но иногда заболевание начинается в
детском возрасте. Клинически на различных участках кожного покрова,
чаще в крупных складках (пахово-бедренных, подмышечных, под молоч-
ными железами, в области пупка), на воспалительном отечном фоне по-
являются герпетиформно расположенные мелкие пустулезные элементы.
Распространяясь центробежно, они образуют кольцевидные фигуры, рас-
полагающиеся беспорядочно. Пустулы содержат зеленовато-желтый гной,
после вскрытия они покрываются грязными гнойно-геморрагическими,
коричневатыми корками. Зуд обычно отсутствует. Содержимое свежих
пустул чаще стерильно, реже—содержит стафилококки или стрептококки.
В процесс часто вовлекаются слизисгые оболочки. В крови бывает гипо-
174
кальциемия (мы в наших шести наблюдениях гипокальциемии не
наблюдали).
Заболевание протекает хронически с тяжелыми обострениями, во вре-
мя которых появляются обильные распространенные (рассеянные и груп-
пирующиеся) болезненные высыпания, сопровождающиеся ознобами, подъе-
мами температуры тела (до 39°—40°), недомоганием, болями в суставах,
головной болью, повышенной СОЭ. С каждой последующей беременностью
высыпания становятся обильнее и общее состояние ухудшается. Беремен-
ность может заканчиваться выкидышем или рождением мертвого ребенка,
поэтому при тяжелом течении заболевания показано прерывание бере-
менности.
Ф. А. Зверькова и соавт. (1982) наблюдали больную 19 лет со вторич-
ным амилоидозом внутренних органов, развившимся на фоне длительного
тяжелого течения герпетиформного импетиго Гебры.
Прогноз заболевания серьезный, летальность значительная. Заболе-
вание протекает длительно.
Гистопатология. В верхних слоях мальпигиевой сети располагается
спонгиоформная пустула (Kogoj), представляющая собой, по-видимому,
гипертрофическую форму микроабсцесса Munro (Lapiere и др.).
Дифференциальный диагноз. Следует проводить с герпетиформным
дерматитом Дюринга, пустулезным псориазом, герпесом беременных и
акродерматитом Аллопо.
Лечение. Кортикостероиды, антибиотики, сульфаниламиды, перели-
вание крови, ПУВА-терапия, антисептические мази, жидкость Кастеллани.
Необходим соответствующий уход за больными.
ЛИТЕРАТУРА
!
Каламкарян А. А., Трофимова Л. fl., Акимов В. Г., Волошин Р. Н. К вопросу
о пустулезном псориазе, герпетиформном импетиго и акродерматите Галлопо.— Вести.
Дерматол. и венерол., 1980, 5, 40—43.
Зверькова Ф. А., Оловяшников О. В., Яцевич Л. Н. Герпетиформное импетиго и
амилоидоз.— Вести. Дерматол. и венерол., 1982, 11, 33—35.
Ou F., Krakowski A. et al. Impetigo herpetiformis with lowered serum level of vi-
tamin D.— Dermatologica, 1982, 164, 5, 360—365.
ЙОДОДЕРМА ТУБЕРОЗНАЯ (iododerma tuberosum).
Ricord в 1842 г. впервые обратил внимание на то, что в результате
применения йодистых препаратов могут возникать токсидермии «е только
типа акне, но и папулезно-гипертрофического типа с кровянисто-серозной
инфильтрацией соединительной ткани.
Впервые буллезные высыпания после приема йода (буллезная йодо-
дерма) описал Westmorelam (цит. по Миракянц Е. И., 1930) в 1855 го-
ду. Случаи туберозных и туберозно-буллезных йододермий в отечествен-
ной литературе описали А. И. Пбспелов (1890), А. И. Лянц (1911), ' В. А.
Поспелов (1912), М. Г. Мгебров (1916), А. А. Грин (1925), Е. И. Миракянц
(1930), М. Н. Егоров и А. Ф. Ястребова (1931), Л. Н. Машкиллейсон и
Е. Я- Герценберг (1938) и др.
175
Многие авторы (Farkas, 1979, Tester-Dalolerup, 1983 и др.) указыва-
ют, что медикаментозные высыпания, вызванные препаратами йода и про-
текающие по типу йодистых угрей, буллезной йододермы и туберозной
йододермы, могут возникнуть у больных, длительно получавших пре-
параты йода.
Этиология и патогенез. Существуют различные гипотезы, объясняю-
щие механизм и причины возникновения йодистых экзантем. И. Зеленев
еще в 1891 г. на заседании Русского сифилидологического и дерматологи-
ческого общества высказал мысль, что кожный йодизм имеет связь с бо-
лезнями почек и сердца. В дальнейшем многие авторы также подчеркивали
безусловную связь реакции кожи на йод с заболеваниями ряда внутренних
органов (хронический нефрит, гепатит, недостаточность кровообращения
и др.). Высокую аллергию к йоду некоторые авторы объясняют его сродст-
вом к альбумину. Rosenberg с соавт. (1972) описали больную 46 лет с веге-
тирующей йододермой, лимфоциты которой в культуре претерпевали блас-
тогенную трансформацию при воздействии йодированным сывороточным
альбумином. Эти данные согласуются с концепцией, что йод может дейст-
вовать как гаптен путем соединения с белком сыворотки, и указывают на
то, что аллергическая сверхчувствительность играет основную роль при
вегетирующей йододерме. Л. Н. Машкиллейсон и Е. Я- Герценберг (1938)
у больной с пузырчаткой и туберозной йододермой посмертно при гисто-
логическом исследовании обнаружили сосудистые изменения гиперергиче-
ского порядка.
Клиника, йодистые препараты могут вызывать побочные явления в
виде разнообразных кожных высыпаний—пурпуры, многоформной эксу-
дативной эритемы, узловатой эритемы, уртикарии, папул, пузырей, изъяз-
влений, вегетирующих узловатых образований.
Наиболее тяжелую форму йододермий представляют буллезная йодо-
дерма (йодистый пемфигус) и туберозная йододерма, часто осложняемые
вегетациями.
При буллезной йододерме высыпания большей частью начинаются с
пузырей величиной от небольшой горошины до лесного ореха, местами
сливающихся в крупные участки неправильной формы. 'Пузыри напря-
жены и часто наполнены геморрагическим содержимым. После вскрытия
пузырей обнажается дно, покрытое значительными вегетациями, достигаю-
щими высоты 0,5—1 см и симулирующими вегетирующую пузырчатку или
сифилиды. В других случаях вначале появляется узелковое высыпание,
которое затем в центре размягчается, образуется пузырь, довольно быстро
нагнаивающийся.
Туберозная йододерма чаще начинается с узелка, за короткий срок
превращающегося в пустулу, а затем в опухоль или инфильтративную бляш-
ку диаметром 3—5 см. По периферии бляшек и опухолей образуется вы-
пуклый край, состоящий из мелких пузырьков с жидким серозно-гиой-
ным содержимым. Консистенция йододермы почти пастозная, флюктуиру-
ющая, и только в редких случаях бляшка тверда на ощупь. Из многочислен-
ных межсосочковых лакун уже при легком надавливании выходит гной
176
часто с прЖесью крови. Пузыри и опухоли почти во всех случаях имеют
тенденцию к распаду и изъязвлению. При надавливании пальцем в не-
которых случаях больные жалуются на боль. Следует отметить, что иногда
туберозная йододерма возникает из йодистого пемфигуса или йодистых
угрей.
Очень редки описания йододермы на слизистых оболочках. В поло-
сти рта.йа языке и щеках образуются эрозии, затрудняющие прием пи-
щи: имеются- единичные описания распространения буллезного процесса
на миндалины, пищевод, двенадцатиперстную кишку и трахею. Продро-
мальные явления при йодистых токсидермиях выражаются в виде общей
слабости, лихорадки и рвоты.
Йододерма наблюдается чаще у мужчин (в 2/3 случаев), главным обра-
зом в пожилом и старческом возрасте. Излюбленная локализация йодо-
дермы: лицо (главным образом поражаются щеки, нос, лоб и реже уши),
шея, конечности, реже туловище и во'лосистая часть головы.
Сравнивая описания клинической картины туберозной йододермы и
бромодермы, С. Ф. Проскуряков (1910) обращает внимание на сходство
как отдельных элементов высыпи, так и общего ее вида. По-вцдимому,
химическое сходство этих лекарственных веществ ведет к однородной па-
тологии и почти одинаковому механизму возникновения. Патогистологи-
ческие исследования дают весьма близкие друг к другу картины.
Течение буллезной и туберозной йододермы различное: наряду с ча-
стыми доброкачественными случаями, оканчивающимися (после отмены
приема йода) в 3—4 недели выздоровлением, описаны весьма редкие слу-
чаи с печальным исходом. Следует отметить, что при возобновлении приема
препарата йода высыпания, как правило, рецидивируют.
Дифференциальный диагноз и лечение—как при бромодерме.
ЛИТЕРАТУРА
Farcas J. Jododerma tuberosum pri liecbe Solutsnom a jododerma bullosum po inge-
scii jodivanejsoli.— Cs. Dermatol., 1979, 54, 6, 351—355.
Тестер-Далдеруп К. (Tester-DalderupI). Лекарственные средства для лечения
бронхиальной астмы и противокашлевые средства. В кн.: Побочные действия лекарствен-
ных средств, пер. с английск, Москва, «Медицина.» 1983, 191—196.
ИХТИОЗ (ichthyosis).
Синонимы: рыбья чешуя, диффузная кератома, сауриаз.
В группу ихтиоза относятся различные заболевания, характеризую-
щиеся нарушением ороговения кожи. Общепринятой классификации нет.
В литературе описано много клинических форм ихтиоза на основании чисто
внешних проявлений болезни.
В 1964 г. Greiter предложил клинико-генетическую классификацию
ихтиоза, которая в 1965 г. была дополнена Wells и Кегг. Эти авторы вы-
делили аутосомно-доминантный тип (вульгарный ихтиоз, буллезная ихти-
озиформная эритродермия); аутосомно-рецессивный тип (ихтиозиформ-
ная эритродермия, ламелларный ихтиоз, синдром Рефсума, синдром Шег-
12 Клиническая дерматология 177
рена—Ларссена: рецессивный, сцепленный с Х-хромосомой. В отечест-
венной литературе имеются классификации, предложенные В. В. Кук-
линым (1974) и С. С. Кряжевой и соавт. (1977).
С. С. Кряжева и соавт. выделяют наследственный (истинный) ихти-
оз и ихтиозиформные изменения кожи (приобретенный ихтиоз). Наслед-
ственный ихтиоз они подразделяют не на 3, а на 4 группы, выделяя ау-
тосомно-рецессивно наследуемые синдромы, включающие ихтиоз как симп-
том: синдром Нетертона, синдром Юнга—Вогеля, синдром Шегрена—Ларс-
сена, синдром Герсума, синдром Руда. В группу приобретенного ихтиоза
они относят: 1—симптоматический (при гиповитаминозе А, болезнях крови,
злокачественных новообразованиях и др.); 2—сенильный; 3—pityriasis
circinata.
Этиология и патогенез. Ихтиоз—наследственное заболевание. Пато-
генез неясен. Имеются сообщения о нарушении у больных ихтиозом функ-
ции эндокринных желез, изменении в обмене аминокислот, ферментных
нарушениях, гиповитаминозе А, о снижении окислительных процессов
в клетках печени и снижении ассимиляции гликогена клетками эпидерми-
са (Потоцкий И. И. и Грабовская Л. А., 1976; Hofbauer и Schnyder, 1974
и др.)
Ихтиоз обыкновенный (ichtyosis vulgaris). Наиболее часто встре-
чающаяся форма с аутосомно-доминантным типом наследования. Разви-
вается обычно на 1—4 году жизни и прогрессирует до наступления периода
полового созревания. В дальнейшем интенсивность клинических проявле-
ний обычно уменьшается. В летнее время наступает улучшение, в легких
случаях—ремиссия. Заболевание проявляется сухостью, шелушением,
ощущением стянутости кожи,
иногда легким зудом. Окраска
кожи грязно-серая. Чешуйки име-
ют размеры от мельчайшего до 1
см в диаметре, цвет их от белого
до серого; большей частью они
тонкие, полупрозрачные, полиго-
нальной формы. В центре чешуй-
ка плотно прикреплена к коже, а
края отстают от нее. Характер
чешуек определяет клинические
варианты ихтиоза—ксеродерма,
блестящий или перламутровый,
белый и др. Наиболее выражен-
ные изменения наблюдаются над
коленными, локтевыми суставами,
на разгибательной поверхности
плеч, предплечий, голеней. Круп-
Рис. П2. Ихтиоз.
178
ные складки кожи—подмышечные, локтевые, подколенные, область поло-
вых органов, как правило, не поражаются. Ладони и подошвы сухие с под-
черкнутыми кожными линиями, складками, «сегментированные». Часто
на разгибательной поверхности плеч, на ягодицах имеется фолликулярный
кератоз. Волосы и ногти обычно не изменены; иногда они истончаются.
Сало- и потоотделение уменьшено. Имеются указания (Куклин В. Т. и
соавт., 1979) на повышенное ороговение слизистой оболочки щек и языка.
Кожа больных ихтиозом раздражима; от химических веществ, мыла часто
возникают дерматиты. С. С. Кряжева и соавт., наблюдавшие 138 больных
ихтиозом, у 48% установили аллергические реакции и аллергические за-
болевания кожи, у 1 больного—псориаз. Имеются указания на частоту
атопии у больных обыкновенным ихтиозом, склонность к развитию у них
пиококковой и грибковой инфекции. Teodosieva и Teodosiev (1979) у боль-
шинства обследованных ими больных вульгарным ихтиозом установили
врожденные почечные аномалии (двойные почки, изменения сосудов, на-
рушения клубочковой фильтрации, отклонения в выделении электроли-
тов). В. Т. Куклин и соавт. (1976) выявили нарушение гликогенной
функции печени. Braun-Falco и Landthaler (1978) описали больную, у
которой вульгарный ихтиоз сочетался с глухотой, аномалиями зубов и
Pili torti.
Гистопатология. Слоистый гиперкератоз; роговые пробки в устьях
волосяных фолликулов; зернистый, слой истончен или отсутствует. Осталь-
ные слои эпидермиса без изменений илн несколько истончены. В дерме
небольшая лимфоцитарная инфильтрация вокруг сосудов. Регрессивные
явления в волосяных фолликулах и сальных железах. При электронной
микроскопии—уменьшение зерен кератогиалина и нарушение их строения.
Дифференциальный диагноз. Проводится с ихтиозиформной эритро-
дермией, X-хромосомным рецессивным ихтиозом, с приобретенным их-
тиозом.
Ихтиоз Х-сцепленный рецессивный.
Синоним: черный ихтиоз.
Эта форма выделена Cockayne (1933) на основании генетических ис-
следований. Имеется описание больных Х-хромосомным рецессивным
ихтиозом у 6 братьев, мать которых страдала аутосомно-доминантным их-
тиозом (Mevorah и соавт., 1970). Встречается только у мужчин. Женщины
являются передатчиками мутантного гена.
Клиника. Заболевание протекает тяжелее, чем аутосомно-доминантный
ихтиоз. Начинается обычно на первом месяце жизни, прогрессирует до
периода полового созревания, в дальнейшем приобретает персистирующее
течение. В летнее время обычно наступает улучшение.. Кожа сухая, утол-
щенная, покрыта пластинчатыми чешуйками коричневатого, вплоть до чер-
ного цвета. Локализация такая же. как при вульгарном доминантном их-
тиозе, но чаще в процесс вовлекается кожа верхней части туловища, лица,
шеи. Примерно у 1/3 больных поражаются и крупные складки кожи. В
отличие от вульгарного ихтиоза нет фолликулярного кератоза и поражения
ладоней и подошв. Не отмечается атопия. У «“которых больных наблю-
\12* . 179.
дается гипогенитализм, катаракта, деменция, нарушения скелета, другие
пороки развития.
Гистопатология. Роговой слой толще, компактнее, чем при доминант-
ном вульгарном ихтиозе. Эпидермис с тенденцией к атрофии (Hofbauer и
Schnyder, 1974). С. С. Кряжева указывает на очаговый акантоз. Зернистый
слой сохранен или даже расширен. Регрессивные явления в волосяных
фолликулах и сальных железах. Потовые железы без изменений. При
электронной микроскопии—увеличение количества кератогиалиновых че-
рен, сохраняющих нормальную структуру.
Ихтиозиформная врожденная эритродермия Брока.
Синонимы: врожденный ихтиоз, тотальный ихтиозиформный гипер-
кератоз Дарье, красный врожденный кератоз Риля и др.
Заболевание описано Броком в 1902 г. Выделяют две формы—буллез-
ную и небуллезную.
Буллезная форма врожденной ихтиозиформной эритродермии
Синонимы: универсальный врожденный акантокератолиз Никольско-
го, врожденный ихтиозиформный эпидермолиз.
Наследуется аутосомно-доминантно.
Клиника. Сразу после рождения появляются краснота кожи, пузырь-
ки, крупнопластинчатое шелушение. Поражение чаще распространенное
и даже универсальное. При тяжелой форме, но описываются и ограничен-
ные формы, смерть может наступить вскоре после рождения. С возрастом
пузыри появляются все реже, но усиливаются явления кератоза. Гипер-
кератотические рыхлые наслоения преимущественно выражены в крупных
складках кожи, на шее. Отмечается гиперкератоз ладоней и подошв. Осталь-
ная кожа сухая, но достаточно эластичная. Потоотделение понижено.
На слизистой оболочке полости рта, глотки- бывают лейкоплакии. Общее
состояние обычно не страдает. Другие врожденные аномалии встречаются
редко.
Гистопатология. Гиперкератоз с дезорганизацией кератина. Очаги пара-
кератоза. Акантоз. Гранулез. Зернистая дегенерация клеток шиловидно-
го слоя с образованием полостей. Сосочки дермы гипертрофированы с пе-
риваскулярными преимущественно лимфоцитарными инфильтратами. Элект-
ронно-микроскопическими исследованиями показано, что в основе заболе-
вания лежит аномалия ороговения, в первую очередь нарушение синтеза
тонофибрилл (Jacobi и Heiderbach, 1977 и др.).
Дифференциальный диагноз. В раннем возрасте в период появления
пузырей следует дифференцировать с врожденным буллезным эпидермоли-
зом, пузырчаткой новорожденных, буллезным импетиго, доброкачествен-
ной семейной пузырчаткой Гужеро—Хейли—Хейл и, £ эксфолиативным дер-
матитом новорожденных. В более позднем возрасте при фёзко выраженном -
гиперкератозе дифференцируют с Ichthyosis hystrix, с сухой формой ихтио-
зиформной эритродермии. Golbus н соавт. (1980) использовали цитогенети-
ческий и биохимический анализ культивируемых клеток амниотической
жидкости, фетоскопию и биопсию кожи плода для пренатальной диагно-
180
стики врожденной буллезной ихтиозиформной эритродермии (отмечен риск
аборта в 5%).
НЕБУЛЛЕЗНАЯ (СУХАЯ) ФОРМА ВРОЖДЕННОЙ ИХТИОЗИФОРМ-
НОЙ ЭРИТРОДЕРМИИ.
Некоторые авторы отождествляют ее с ламелларным ихтиозом. Забо-
левание наследуется аутосомно-рецессивно. Этиология и патогенез не
выяснены. Имеются отдельные указания на энзимные нарушения—уве-
личение содержания в моче аргининово-сукцининовой кислоты (Wollshaut
и Muresan, 1974).
Клиника. Тяжесть заболевания может быть различной (рис. 113, 114,
115). При тяжелой форме ребенок обычно рождается недоношенным и
быстро погибает. В других случаях кожа родившегося ребенка покрыта
сухой .буроватой «коллодийной»
пленкой. У одних детей она затем
превращается в крупные чешуйки,
которые постепенно исчезают и
кожа приобретает нормальный
вид, у других эти чешуйки сохра-
няются в течение всей жизни. Ко-
жа под ними эритематозная. По-
ражение универсальное, в про-
цесс вовлечены и крупные склад-
ки. Обильное шелушение на во-
лосистой части головы. У ряда
больных эритема постепенно
уменьшается, но усиливается ги-
перкератоз. Отмечается усиленный
рост волос и ногтей, деформация
ногтей, подногтевой гиперкератоз.
Гиперкератоз ладоней и подошв.
Характерен выворот век. Значи-
тельно чаще, чем при других фор-
мах ихтиоза описываются больные
с рубцевидными очагами облысе-
ния, скрученными волосами, ке-
ратитом, глухотой, аномалиями
зубов, деменцией.
Гистопатология. Гиперкератоз.
Очаги паракератоза. Акантоз. Зер-
нистый слой утолщен. Керато-
гиалиновые гранулы увеличены.
Лимфоцитарные инфильтраты в рис пз Ихтиоэифо
верхней части дермы. 9ритродерМия.
181
Рис. 114. Ихтиозиформная Рис. 115. Ихтиозиформная
эритродермия (тот же больной). эритродермия (тот же больной).
।
Дифференциальный диагноз. Проводят с другими ихтиозиформными
генодерматозами.
Лечение. Витамин А внутрь и парентерально длительно, повторными
курсами. Препараты ретинойной кислоты. Витамины Вв, В12. Препараты
железа, фосфора. Введение плазмы крови. В тяжелых случаях ихтиози-
формной эритродермии—кортикостероидные препараты внутрь. Теплые
ванны с морской и поваренной солью, мази и кремы с витамином А, 10%
хлорида натрия, с мочевиной, молочной кислотой. Гелиоталассотерапия.
ЛИТЕРАТУРА
Кряжева С. С., Ведрова И. Н., Елецкий А. Ю. Клинико-генетические особенно-
сти различных форм ихтиоза.— Вестн. дерматол. и венерол., 1977, 9, 17—23.
Куклин В. Т., Ельцов В. К-, Белинская Р. Г., Величанская Т. Б. Гликогенная
функция печени у больных ихтиозом.— Вестн. Дерматол. и венерол., 1976, 2, 56—59.
Куклин В. Т., Шолохова А. И., Шумина Н. А. Изменения слизистой оболочки
полости рта у больных ихтиозом.— Вестн. дерматол. и венерол., 1979, 6, 37—40.
Потоцкий И. И'., ГрабовскаяЛ. А., Нарушения висцеральных органов и эндокрин-
ной системы при ихтиозе.— Вестн. дерматол. и венерол., 1976, 11, 52—55.
Суворова К- Н., Антоньев А. А. Нарушения кератинизации.— В кн.: Наследст-
венные Дерматозы. М., 1977, с. 60—84.
Braun—Falco О., Landihaler М. Ichthyosis vulgaris, Taubheit, Pili torti und Zahna-
nomalien.— Hautarzt, 1978, 29, 276—280.
Hofbauer M., Schnyder U. W. Zur Differentialdiagnose von autosomal—dominanter
Ichthyosis vulgaris und X—Chromosomaler Ichthyose.— Hautarzt, 1974, 26, 7, 319—325.
182
Jacobi H., Heiderbach U. Erythrodermia congenitalis ichthyosiformis bullosa.—
Derm. Mschr., 1977, 163, 10, 840—843.
Teodosieoa E. S., Teodosiev L. S. Nierenveranderungen bei Kranken mit ichthyosis
vulgaris.— Derm. Mschr., 1979, 165, 6, 407—411.
КАЛЬЦИНОЗ КОЖИ (calcinosis cutis).
Синонимы: метаболический кальциноз, липокальциногранулематоз,
болезнь Профише, липоидокальциноз, синдром Тейчлендера, идиопатиче-
ский кальциноз и т. д.
Первое описание универсального кальциноза принадлежит француз-
скому автору Profichet (1900).
Этиология и патогенез. Различают метастатический и метаболический
кальциноз. Первый обусловлен гиперкальциемией и крайне редко со-
провождается отложениями кальция в коже и подкожной клетчатке; обыч-
но обызвествление наблюдается во внутренних органах, в сосудах. Гипер-
кальциемию связывают с нарушением функции паращитовидных желез,
щитовидной железы, гипервитаминозом Д; он может наблюдаться при хро-
нических заболеваниях почек, костей, злокачественных опухолях и др.
Метаболический кальциноз развивается в результате местных нару-
шений обмена в тканях (Коляденко В. Г. и соавт., 1982; Хлебин К. И.,
Найденов Ю. Н., 1984; Абрикосов А. И. и Струков А. И., 1961; Lever
и Schaumburg-Lever, 1975). Эти нарушения объясняют нестойкостью бу-
ферных систем, развитием ацидоза, приводящих к снижению растворимо-
сти кальция и его отложениям в коже, подкожной клетчатке, мышцах, в
сухожильных влагалищах.
Клиника. Различают ограниченный и распространенный, универсаль-
ный метаболический кальциноз кожи. В случаях ограниченного кальци-
ноза кожи поражаются преимущественно верхние конечности, в первую
очередь кисти и область локтей, реже нижние конечности, ушные рако-
вины и др. При универсальном кальцинозе кожи возникают более или ме-
нее крупные узлы на различных участках тела, чаще на конечностях в
области крупных суставов, на спине rf ягодицах. Около 3/4 больных каль-
цинозом кожи—женщины. Ограниченный кальциноз кожи чаще встре-
чается в среднем и пожилом возрасте, универсальной формой заболева-
ния большей частью страдают молодые люди, а также дети и грудные мла-
денцы. И. И. Бухтоярова (1967) наблюдала врожденный диссеминирован-
ный кальциноз кожи у мальчика 3,5 месяцев, у которого сразу после рож-
дения появились множественные очаги уплотнения. А. М. Ариевич (1949)
демонстрировал на заседании Московского дермато-венерологического об-
щества кальциноз кожи у ребенка 3 лет, образовавшийся в трехнедельном
возрасте.
Множественные узелки и узлы невоспалительного характера распо-
лагаются на различных участках кожного покрова. Кальцинаты размера-
ми от горошины до лесного ореха, округлой или неправильной формы,
плотны на ощупь (каменной плотности), расположены в глубоких слоях
183
кожи или подкожной клетчатке, подвижны, безболезненны. Кожа спаяна
с ними, слегка втянута, напоминает лимонную корку, без признаков вос-
паления, слегка синюшно-красноватого цвета, с телеангиэктазиями. Поз-
же они размягчаются и вскрываются с образованием длительно существую-
щих язв и фистул, через которые выдавливается крошковатая масса с
примесью белых плотных крупинок извести. Течение таких «кальциевых
гумм» большей частью безболезненное, образующиеся фистулы, а иногда
и язвы протекают крайне вяло и рубцуются очень медленно, длительное
время отделяя известковые массы. Рентгенограммы соответствующих уча-
стков поражения дают четкую картину множественных отложений каль-
ция.
Кальциноз кожи, ограниченный и распространенный, нередко встре-
чается у больных дерматомиозитом, системной склеродермией (синдром
Т ибьержа—Вейссенбаха).
В литературе выделяют еще узловатую подкожную форму кальциноза
(Winer, 1952; Duperrat et al., 1968; Shanes и Wood, 1972; Kanitakis et al.,
1978; Van Brabandt, 1982 и др.), имеющую характер одиночных твердых
мелких узелков с гладкой поверхностью. Они выстоят куполообразно,
имеют желтовато-белый цвет. При травматизации из них может выделяться
творожистая масса. Локализация—голова, особенно щеки, ушные рако-
вины, реже—кисти и стопы. Заболевание обычно проявляется в первые
месяцы или годы жизни, редко—позже. Winer (1952) полагает, что это
обызвествленные гамартомы.
Помимо вышеописанных «первичных» форм кальциноза имеется «вто-
ричный» или дистрофический кальциноз, который развивается в резуль-
тате отложений кальция в воспалительных очагах, в кистах, рубцах, ново-
образованиях кожи (Van Brabandt, 1982; King et al., 1979; Swinehazt
и Golitz, 1982).
Прогноз неопределенный, лишь отдельные авторы сообщают о спон-
танном исчезновении небольших кальциевых отложений, чаще процесс
прогрессирует, появляются новые узлы, которые ведут к ограничению
подвижности или даже полному анкилозу суставов, к атрофии, кахексии
и даже смерти.
Дифференциальный диагноз. Следует проводить с саркоидозом Бенье—
Бека—Шаумана, подагрическими узлами, дерматофибромами, с коллик-
вативным туберкулезом кожи.
Гистопатология. Отложения кальция легко определяются в гисто-
логических препаратах, так как они окрашиваются гематоксилин-эозином
в насыщенно синий цвет и в черный цвет краской Косса. Отложения, со-
стоящие в основном из фосфорнокислого и в меньшей степени из углекис-
лого кальция, обнаруживаются в виде отдельных зерен или более или ме-
нее массивных скоплений между пучками соединительной ткани в собствен-
но коже и в подкожной жировой ткани. Между кальциевыми отложениями
имеются изменения фибросклеротического или гранулематозного харак-
тера, гигантские клетки инородных тел, а иногда и эпителиоидные клетки.
184
Лечение кальцинозов кожи, склонных к распаду, сводится к вскрытию
и опорожнению очагов хирургическим путем или электрокоагуляцией.
Крупные очаги обызвествления могут быть удалены хирургическим путем.
Назначаются йодистые щелочи, хлористый аммоний (до 2—3 г в сутки).
Местно—УВЧ, диатермия на инфильтрированные изменения при язвен-
ных поражениях. Рекомендуется избегать пищи, богатой кальцием и ви-
тамином Д.
ЛИТЕРАТУРА
Беренбейн Б. А., Лалаева А. М. Кальциноз кожи у больной ДисхонДроплазией._____
Вестн. дерматол. и венерол., 1976, 8, 67—71.
Коврижка И. М., Казимирчик 3. А. К вопросу о патологической анатомии лнпо-
кальциногранулематоза.— Вестн. Дерматол. и венерол., 1975, 9. 66—70.
Коляденко В. Г. и соавт. О распространенном кальцинозе кожи. Вестн. Дерматол.
И венерол., 1982, 3, 47—49.
Серов В. В., Шехтер А. Б. Соединительная ткань (функциональная морфология
и общая патология).— М. 1981.
Baurle G., Hornstein О. Generalized cutaneous calcinosis and mixed connective tis-
sue disease.— Dermatologica, 1979, 158, 4,
Hutchinson J., Abet B. Susskind W. Idiopathic calcinosis of the penis.— Brit. J.
Dermatol., 1980, 102, 3, 341—343.
KanitakisC., Tsoitis G. Calcinose nodulaire solitaire de la peau d’aspects clinique
et histologique particuliers. A propos de 2 cas.— Ann. Dermatol. Venereol., 1978, 105, 12,
1033—1040.
Kolten B., Pedersen J. Calcinosis cutis and renal failure.— Arch. Demrmatol., 1974,
110, 2, 256—257.
Swinehart J., Golitz L. Scrotal calcinosis. Distrophic calcification of epidermoid
cysts. Arch, dermatol., 1982, 118, 12, 985—988
Thomsen R. Subcutaneous fat necrosis of the newborn and idiopathic hypercalcemia.—
Arch. Dermatol., 1980, 116, 10, 1155—1158.
Yamada M. Ultrastructuraf and cytochemical studies on the calcification of the then-
don bonejoint.— Arch. Histol. Japan, 1976, 1976, 39, 347—378.
Van Brabandi S. Calcinosis circumscripta. Dermatologica (Basel), 1982, 165, 5, 514—
515.
КЕЛОИД (keloid).
Этиология и патогенез. Келоид представляет собой опухолевое раз-
растание и склероз соединительной ткани неизвестной природы. А. И.
Струков и А. Г. Бегларян (1963) относят келоиды к группе локализован-
ных первичных поражений соединительной ткани; в Гистологической клас-
сификации ВОЗ (1980) они рассматриваются среди опухолеподобных про-
цессов мягких тканей. М. В. Журавлева (1965) считает, что келоид разви-
вается в результате задержки созревания грануляционной ткани, обуслов-
ленной длительной активностью фибробластов и сохранением в межуточ-
ном веществе большого количества мукополисахаридов. Процесс со вре-
менем затихает, но полностью не прекращается, о чем свидетельствует
185
обнаружение в толще келоид*, в так называемых келоидных узелков (узел-
ков роста). Формирующиеся неполноценные коллагеновые волокна легко
подвергаются различным изменениям, вплоть до перестройки и дезоргани-
зации, гиалиноза. Болховитинова Л. А. и Павлова М. Н. (1977) также
указывают, что основным морфологическим признаком растущих келоидов
является незрелая соединительная ткань.
Различают спонтанные келоиды, развивающиеся на внешне неизмен-
ной коже, и вторичные, возникающие на фоне рубцов различного проис-
хождения (после ожогов, хирургических вмешательств, пиококковых про-
цессов, обычных угрей и др.). Это деление в значительной мере условно,
так как травматическое воздействие, на месте которого развился спонтан-
ный келоид, может быть минимальным и остаться без внимания. Так, по
данным Amar Inalsingh (1974), касающимся 501 больного, ни в одном слу-
чае не было спонтанных келоидов В качестве факторов, способствующих
разрастанию соединительной ткани, указываются гормональные наруше-
ния, прежде всего дисфункция половых желез, беременность, гипофункция
надпочечников, себорейная конституция (отсюда преимущественная лока-
лизация в так называемых себорейных местах), общие хронические инфек-
ции (туберкулез и др.) Отмечается роль вторичного инфицирования ране-
вой поверхности. О. Д. Кочура и соавт. (1975) наблюдали развитие мно-
жественных келоидов у больной, длительно страдавшей интенсивным зу-
дом, что, помимо постоянной травматизации, не исключает роли обменных
нарушений. Описано развитие келоидов на месте врожденных дефектов
волосистой части головы у двоюродных сибсов (Morschella, 1962), гигант-
ские келоиды у больного пахидермопериостозом (Hambrick и Carter, 1966).
Подтверждением роли обменных нарушений могут быть данные Kembl
и Brown (1976) о повышенной активности в келоидных рубцах кислой фос-
фатазы, NAD-диафоразы, а также Cohen и соавт. (1971), Craig (1973)—об
усилении активности коллагеназы.
Ford и соавт. (1983) указывают на патогенетическую роль локальной
гиперандрогении. Munzi и соавт. (1983) наблюдали развитие келоида после
введения гематопорфирина. Предполагается участие иммунных механиз-
мов с образованием антител к фибробластам, усилением их способности
синтезировать коллаген. Указывается и на значение механических свойств
кожи, в частности повышенного натяжения, различного в нормальных
условиях в разных зонах, чем объясняется преимущественная локализа-
ция в определенных частях тела (Ketchum et. al., 1974). При обсуждении
вопросов патогенеза келоидных рубцов Л. А. Болховитинова и М. Н.
Павлова (1977) обращают внимание на такие факторы, как ограниченное
количество тучных и плазматических клеток в растущих рубцах, тканевая
гипоксия.
Наличие семейных случаев может указывать на возможность генети-
ческой обусловленности системного поражения соединительной ткани.
Это, однако, в основном относится к множественным келоидам. Предпо-
лагается аутосомно-рецессивное наследование (Otto—Dare, 1975), но во
многих родословных оно более соответствует доминантному.
186
Рис. 116. Келоиды. Рис. 117. Келоиды.
Рис. 118. Келоидные Рис. 119. Келоидные
рубцы. рубцы.
187
Рис. 120. Келоидные
рубцы.
Laurentaci и Diognardi (1977) установили, что лица с антигенами тка-
невой совместимости В14 и BWle имеют повышенный риск развития кело-
идов. Об этом же может свидетельствовать большая частота келоидов среди
лиц с темным цветом кожи. Так, из группы больных (501 человек), наблю-
давшихся Amar Inalsingh (1974), европейцев было только 5, африканцев
—236.
Клиника. Спонтанные и вторичные келоиды сходны и представляют
собой плотные опухолевидные образования розового, красного или си-
нюшного цвета, различной формы, значительно больше в центральной
части, возвышающиеся над кожей, с блестящей гладкой, реже сморщен-
ной, дольчатой поверхностью, на которой часто видны множественные те-
леангиэктазии, иногда пигментация; кожа в зоне келоида напряжена, мо-
жет истончаться, при травме—воспаляться, изъязвляться с образованием
иногда трудно заживающих язв (рис. 116, 117, 118, 119, 120). Вторичные
келоиды соответствуют очертаниям рубцово измененной кожи (линейные,
округлые), но в отличие от гипертрофических рубцов выходят за пределы
первоначальной травмы. Спонтанные келоиды часто имеют неправильные
звездчатые очертания. Характерным является наличие клешневидных тя-
жей, проникающих в здоровую кожу, что отличает келоиды от гипертро-
фических рубцов.
Келоиды чаще развиваются у женщин. Но это может объясняться
чисто внешними причинами, например, косметическими проблемами. От
50 до 88% составляют лица до 30-летнего возоаста (Amar Inalsingh, 1974;
188
Ketchum et al., 1974), что объясняется большей подверженностью молоде-
жи травмам, особенностями структуры и функции кожи в этом возрасте
(повышенное натяжение, ускоренный синтез коллагена). По данным Л. А.
Болховитиновой и М. Н. Павловой (1977), келоиды на месте ожогов чаще
развивались после ожогов пламени, горячими жидкостями, химическими
веществами и паром. Вначале келоид имеет вид небольшого быстро расту-
щего образования. Затем, достигнув определенного размера (обычно в
течение 6—18 месяцев), годами остается в стационарном состоянии. После
оперативных вмешательств, по данным Amar Inalsingh (1974), большинство
келоидов развилось в первые 6 месяцев, около 17%—через год. Не было
ни одного случая келоида через две года после операции. Период между
заживлением ожога и ростом келоидов, возникших на месте ожогов II—
III степени, составлял от 1 до 2—3 месяцев (Болховитинова Л. А. и Пав-
лова, М. Н. 1977).
Келоиды, как правило, не исчезают в отличие от гипертрофических
рубцов, которые обычно регрессируют через несколько месяцев самопро-
извольно. Окраска их постепенно бледнеет, келоид становится белым или
приобретает пвет нормальной кожи. Субъективные расстройства непостоян-
ны, как правило, наиболее выражены в начальной фазе развития келоидов.
Может наблюдаться зуд, небольшой или мучительный, гиперестезия или
болезненность, самопроизвольная или при давлении. Боль может быть
значительной, особенно при развитии келоидного рубца на стопах после
нерационального удаления бородавок. Высока склонность к рецидивам
после оперативного вмешательства. При локализации келоидов на су-
ставах могут развиться контрактуры, на шее—ограничение подвижности.
Злокачественное перерождение бывает редко, при многолетнем сущест-
вовании многократных травмах преимущественно у пожилых. Так, М. В.
Журавлева наблюдала два случая рака, у мужчины 71 года и женщины
62 лет. Больше вероятность развития эпителиом в области келоидов, раз-
вившиеся на послеожоговых рубцах. Располагаться келоиды могут на
любом участке кожи, в том числе и в полости рта, но чаще бывают на гру-
ди, особенно в области грудины, на лице, ушных раковинах, шее, в межло-
паточной области. У ожоговых больных келоиды чаще наблюдались на
руках и ногах, лице, туловище, шее (Болховитинова Л. А. и Павлова,
М. Н. 1977). Случаи множественных келоидов не единичны. С. Ю. Зайцева
и И. Б. Трофимова (1982) описали множественные келоиды у брата и сестры
в сочетании с врожденными пороками сердца, эпилепсией, ревматизмом,
у сестры—с гирсутизмом, нарушением зрения (миопия), расширением
поверхностных вен ноги. Склонность к развитию келоидов наблюдается
при синдроме Noonan (Wyre, 1978). Своеобразная форма келоида описана
Edmundson (1951) на месте изъязвлений, вызванных хромом, в виде округ-
лых, сходных с пуговицей, слегка возвышающихся эритематозных опухо-
лей. v
Гистопатология. Разрастание коллагеновых волокон, в начальной
фазе с большим количеством фибробластов, тучных клеток, гликозамин-
гликанов, пролиферацией капилляров, в поздних—со скудной клеточной
, 189
реакцией, малым количеством сосудов, слабо выраженной эластической
сетью, гомогенизацией и гиалинизацией коллагеновых волокон.
Дифференциальный диагноз. Дифференцировать необходимо от дерма-
тофибросаркомы Дарье—Феррана, саркомы, липомы, фибромы, гипертро-
фических рубцов.
. Лечение. Лучи Букки, лазера, хирургическое иссечение, внутриочаго-
вое введение кортикостероидов, лидазы, пирогенные препараты, димексид,
лечебная физкультура, массаж.
ЛИТЕРАТУРА
Болховитинова Л. А., Павлова М. Н. Келоидные рубцы. М. Медицина, 1977.
Зайцева С. Ю., Трофимова И. Б. Два случая келоидного Диатеза в одной семье.
Вести, дерматол. и венерол., 1982, 4, 39—41.
Конура О. Д., Раппопорт Б. Н., Ремизова О. И. Множественный келоид, раз-
бившийся у пожилой больной, страдающей кожным зудом. Вести, дерматол., и венерол.,
1975, 8, 81—83.
Романенко В.'Н., Проценко Т. В. Пограничные лучи при лечении гипертрофиче-
ских рубцов. Вести, дерматол. и венерол., 1984, 9, 42—46.
Amar Inalsingh С. Н. Роа е perience in treating five hundred and one patients with
keloids. Johns Hopkins med., JI., 1974, 134—5, 284—290
Ford L. C,, KingD.F., Lagasse L. D. et al. Increased Androgen Binding in Kelo-
ids: A Preliminary Communication. J. Dermatol. Surg. Oncol., 1983, 9, 545—547.
Kemble J. V. H., Brown R. F. R. Enzyme activity in human scars and keloids. Brit.
J. dermatof., 1976. 94, 3, 301—305.
Ketchum L. D., Cohen I. K-, Masters F. W. Hypertrophic scars and keloids. A col-
lective review. Plast. Reconstr. Surg., 1974, 53, 140.
Laurentaci G., Diognardi D. HLA Antigens in Keloids and hypertrophic Scars. Arch.
dermatoL, 1977, 113, 12, 172 .
Nunzi E. Parodi A., Rebora A. Immunofluorescence findings in haematoporphy-
rin-induced keloid. Brit. J. dermatoL, 1983, 108, 3, 263—266
Otto-Dare P. Genetic studies on keloids. J. Nat. Med. Ass., 1975, 67, 428—432.
KEPATOAKAHTOMA (keratoacanthoma).
Синонимы: сальный моллюск; псевдокарциноматозный моллюск.
Бельгийский дерматолог A. Dupont в 1930 году сообщил о пациенте,
имевшем небольшую опухоль на лице, которую он расценил как добро-
качественное новообразование, исходящее из стенок сальной кисты. В
1936 г. Mac Cormac и Scauff сообщили о нескольких больных своеобразной
опухолью кожи, напоминающей спиноцеллюлярный рак, которую они
назвали <сальный моллюск». Rook и Whimster (1950) на основании клини-
ческого и гистоморфологического изучения 29 случаев предложили назы-
вать ее кератоакЗнтомой.
Этиология и патогенез. В настоящее время известное признание по-
лучила вирусная теория возникновения кератоакантомы (Прокопчук А. Я.
1969; Глазунов М. Ф., 1960; Ereauxet al., 1955; Bierwolf и Therman, 1965;
Nasemann, 1965; Kopf, 1976, и др). Однако никому еще не удалось выделить
190
Рис. 121. Кератоакантома.
Риг. 122. Кератоакантома.
Рис 124. Кератоакантома.
Рис. 123. Кератоакантома
гигантская.
вирус кератоакантомы и заразить животных. Montgomery (1967) указы-
вает на значение иммунных механизмов и генетических факторов в раз-
витии кератоакантомы. Некоторые авторы (Мас Согтас и Scarf, 1967; Bow-
man и Pincus, 1955 и др.) связывают ее развитие с длительным воздействием
на организм различных факторов: солнечного облучения, реытеновских
лучей, высокой температуры, эндокринных нарушений, воспалительных
изменений сальных желез, механических воздействий—царапины, уколы,
ожоги и др. Degos и соавт. (1967), Jacobson и соавт. (1974) сообщили о мно-
жественных кератоакантомах, развившихся у больных, получающих им-
мунодепрессивную терапию.
191
Клиника. Кератоакантома чаще развивается у мужчин в. среднем и
пожилом возрасте, но встречается и у молодых и даже у детей (Degliot и
Саго, 1976). Средний возраст, по данным Н. А. Торсуева (1959), составляет
61,5 года, по данным Rook и Whtmster (1979)—49 лет. Среди наблюдав-
шихся нами 15 больных он составил 52,6 года. Очаги поражения локали-
зуются преимущественно на открытых участках, большей частью на го-
лове (у 88,6%), кистях, предплечьях (у 10,9%), на красной кайме губ—у
5—7% и только у 2,2%—на закрытых участках тела (Ильин И. И. 1970;
Машкиллейсон А. Л., 1970; Беренбейн Б. А., 1980; Colombo et al., 1982
и др.). Имеются сообщения об атипичной локализации кератоакантомы
на слизистой оболочке полости рта, в перианальной области, на внутренней
поверхности препуциального мешка, в области подногтевого ложа, на конъ-
юнктиве и др.
Различают два основных типа кератоакантомы—одиночные (солитар-
ные) и множественные. Одиночные кератоакантомы встречаются чаще,
Обычно они возникают на внешне здоровой коже и им не предшествуют
какие-либо хронические дерматозы или пребластоматозные процессы. Лишь
немногие авторы видели образование кератоакантомы после травмы, у
больных экземой, красной волчанкой, пигментной ксеродермой, в пределах
линейного эпидермального невуса и т. п. (Исаева В. С. и соавт., 1966;
Ланнэ Т. К- и Мухина Л. Д., 1963; Randazzo и Fichera, 1963; Vulcan,
1964; Ciandi и Thivolet, 1975; Tennstedt et al., 1977; Ahmed, 1980; Want-
zin et al., 1980; Rosen, 1982; Temmerman и Clippele, 1982 и др.).
Заболевание начинается с появления безболезненного плотного по-
лушаровидного фолликулярного узелка цвета нормальной кожи, величиной
с булавочную головку, который очень быстро увеличивается (рис. 121, 122)
в размерах. Наиболее типичной формой является кратерообразная моллюс-
коподобная кератоакантома (рис. 123, 124), которая развивается более чем
в 90% случаев. Зрелая кератоакантома представляет собой плотное опухо-
левидное образование полушаровидной формы цвета нормальной кожи,
возвышающееся над уровнем кожи, диаметром от 0,5—1 см до 2—3 см.
Его краевая зона представляется в форме блестящего валика, а в центре
отмечается скопление роговых масс, выполняющих кратерообразное уг-
лубление (следствие своеобразного усиленного ороговения клеток кера-
тоакантомы). После удаления роговых масс обнажается кратерообразное
углубление. В дальнейшем опухоль регрессирует с оставлением слегка
вдавленного и пигментированного рубца.
Различают следующие фазы в развитии кератоакантомы (Buttner и
Klingmiiller, 1965): период роста (продолжительностью от 2 до 8 недель),
стационарную фазу (от 2 до 3—4 недель) и фазу обратного развития (от 2
до 8—10 недель). Полный цикл развития кератоакантомы—от возникно-
вения до формирования рубца—у значительного большинства больных
(по данным Rook и Whimster, 1979) длится 2—4 месяца. Однако бы-
вают случаи и более длительного речения (до 1—1,5 лет). Помимо описан-
ной в 10% случаев встречаются и другие клинические разновидности, кера-
тоакантомы кожи: по типу кожного рога, когда роговые массы не только
192
целиком заполняют центральный кратер, но в виде пробки выступают из
него, напоминая кожный рог; грибовидный или бляшковидный тип—отсут-
ствует центральный кратер, а роговые массы равномерно покрывают пло-
скую или выпуклую поверхность кератоакантомы; центробежный тип ха-
рактеризуется центробежным разрастанием опухоли, достигающей гигант-
ских размеров, с образованием изъязвлений и с выраженной воспалитель-
ной реакцией.
Большинство современных авторов убеждены в доброкачественности
кератоакантомы. Об этом свидетельствуют случаи самоизлечения (Задо-
рожный Б. А. и Шахова Ф. Б., 1962), а также отсутствие атипии и поли-
морфизма эпителиальных клеток. Наряду с этим высказывается мнение о
возможности озлокачествления кератоакантомы у глубоких стариков или
у лиц с «солнечной» атрофией кожи (Kestel и Blair, 1973; Rook и Whinster,
1979 и др ). Некоторые авторы наблюдали сочетание множественных и
солитарных кератоакантом с карциномами желудочно-кишечного тракта
(Tjon и Joe, 1971; Rulon и Helwig, 1973; Chapman Finn, 1974; Poleksic, 1974).
Гистопатология. Кратеровидное углубление заполнено роговыми мас-
сами; резко выраженный акантоз. Межсосочковые эпителиальные выросты
глубоко проникают в дерму, но атипического роста не наблюдается. В дер-
ме—очаговые инфильтраты из лимфоцитов, плазмоцитов, гистиоцитов; в
стадии разрешения—гигантские клетки инородных тел. Гистогенез кера-
тоакантомы до настоящего времени остается спорным. Наиболее распро-
страненным является представление о развитии кератоакантомы из покров-
ного эпителия (Глазунов М. Ф., Блинов Г. А., 1960; Апатенко А. К-,
1973; Ackerman, 1976 и др.).
Дифференциальный диагноз. Следует проводить с заразительным мол-
люском, старческой кератомой, базо- и спиноцеллюлярной эпителиомой.
Kalkoff (1960) и Hundeiker (1978) утверждают, что V5—V3 опухолей, диаг-
ностируемых как плоскоклеточный рак кожи, фактически является кера-
тоакантомой кожи.
Лечение. Хирургическое удаление, кюретаж, электрокоагуляция, цито-
статические препараты (метотрексат, блеомицин), лучевая терапия (Pul-
man et al., 1978). Рецидивы кератоакантом после хирургического уда-
ления составляют 4—6% (Rook et al., 1967; Poiares и Baptista, 1964; Ste-
vanovic, 1969 и др ). Sipollaro (1983) наблюдал хороший терапевтиче-
ский эффект при лечении подофиллином в виде аппликации 25 % раст-
вора один раз в неделю в течение нескольких недель.
ЛИТЕРАТУРА
Ackerman В. Histopathology of keratoacanthoma. in: Andrade eds Cancer of the skin.
Philadelphia, К. B. Saunders, 1976, pp.781—796.
Ahmed A. Muetiple keratoacanthoma.— Intern. J. Dermatol., 1980, 10, 9, 496—499.
Claudy A., Thivolet J. Multiple keratoacanthomas associatior with deficient cell
mediated immunity.— Brit. J. Dermatol., 1975, 93, 593—595.
13 Клиническая дерматология 193
Rook A., Whimster J. Keratoacacanthoma— a thirty year retrospect.— Brit. J.
DermatoL, 1979, 100, 1, 41—47.
Sipollaro V. The use of podophilin the treatmeum of keratoacanthoma. —intern. J.
dermafol., 1983, 22, 1, 436—440.
Tennstedt D., Lachapelle J. Keratoacanthomes et xeroderma pigmentosum.— Ann.
Dermatol. Venereol., 1977, 104, 2, 98—102.
Wantzin G., Agdal N., Soeigard E. Multiple kerato-acanthoma. A case report.—
Acta derma tovenereoL, 1980, 60, 5, 443—445.
КЕРАТОДЕРМИЯ КЛИМАКТЕРИЧЕСКАЯ (keratodermia climacteri-
cum).
Синонимы: постклимактерическая кератодермия, гипоэстрогенный ке-
ратодерматит ладоней и подошв, болезнь Хакстхаузена.
Brooke в 1891, Dubreuile в 1892 г. описали кожные изменения в климак-
терическом периоде, назвав их кератотической эритемой ладоней и подошв.
Haxthausen в 1934 г. привел подробное клиническое описание кожных из-
менений при климактерии у женщин и предложил название «Keratodermia
climacterium.» В СССР впервые этот синдром описали А. А. Каламкарян,
Е. Г. Федорова, Е. В. Бухарина (1984).
Этиология и патогенез. Анализ данных литературы и наши наблюде-
ния дают основание рассматривать климактерическую ладонно-подошвен-
ную кератодермию как часть климактерического синдрома,
встречающегося у 10—15% женшин. По мнению некоторых авторов (Сег-
vino et al., 1938; Lynch, 1943; Wolf, 1941; Gafbe, 1944; Samanta et al.,
1976 и др ), в основе гиперкератоза ладоней и подошв лежит гипофункция
яичников, щитовидной железы и др. П. В. Кожевников (1962) относит
климактерическую кератодермию в группу первичных приобретенных
кератозов, в подгруппу эндокринных кератозов. М. И. Пер (1959) считает
климактерическую кератодермию разновидностью гиперкератотической ла-
донно-подошвенной экземы.
Клиника. Заболевание встречается преимущественно у женщин, но
наблюдается и у мужчин (Barber, 1942; Ormsby и Montgomery, 1955; наши
наблюдения). Возраст больных женщин колеблется от 26 до 70 лет, но
средний возраст составляет 45—55; у мужчин подобные кератодермические
изменения на ладонях и подошвах наблюдаются между 50 и 60 годами.
Появление' климактерического синдрома, в том числе ладонно-подошвен-
ной кератодермии, особенно часто наблюдается ранней весной (февраль—
апрель). Клинически заболевание проявляется симметричным диф-
фузным или очаговым утолщением рогового слоя ладоней и подошв, обыч-
но в сочетании с различными соматогенными расстройствами; кожа стано-
вится сухой," появляются болезненные трещины. Везикуляция, мокнутие
и другие проявления экземы отсутствуют (рис. 125, 126, 127, 128, 129).
Гиперкератоз особенно выражен в местах давлений и трения (по краю по-
дошв). Зуд, нарастающий в ночное время, отмечается у многих больных.
194
Рис. 125. Кератодермия
климактерическая.
Рис. 126. Кератодермия
климактерическая.
Рис. 127. Кератодермия
климактерическая.
Рис. 128. Кератодермия
климактерическая.
У отдельных больных кератодермия ладоней и подошв наблюдается в со-
четании с псориазоподобными очаговыми высыпаниями на голенях, пред-
плечьях и других участках кожного покрова. Высыпания могут регрес-
сировать, уменьшаться, затем вновь обостряться. Как правило, после окон-
чания климактерического периода кожные высыпания полностью регрес-
13* 195
Рис. 129. Кератодермия
климактерическая.
сируют, однако у одной больной, наблюдавшейся Хастхаузеном, кожные
высыпания продолжались около 12 лет.
Гистопатология. Выраженный гиперкератоз и небольшой паракера-
тоз; акантоза и микроабсцессов нет. В дерме—инфильтрат из лимфоидных
клеток, расширение капилляров, дегенерация эластических и коллагено-
вых волокон.
Дифференциальный диагноз. Проводят с ладонно-подошвенным псо-
риазом, кератотической (роговой) экземой, веррукозным или гипертрофи-
ческим красным плоским лишаем, псориазиформным или кератодермиче-
ским сифилисом, нейродермитом.
Лечение. Эстрогены (в течение 2—3 недель), тиреоидные препараты,
теплые ванны для рук и ног, мази с 5—10% салициловой кислотой, пасты
и мази с нафталаном, дегтем, с кортикостероидами. Мы наблюдали благо-
приятный эффект от длительного применения витаминов А и Е (аевит).
ЛИТЕРАТУРА
Каламкарян А. А., Федорова Е. Г., Бухарина Е. В. Климактерическая керато-
дермня. Вести, дерматол. и венерол., 1984, 9, 4—8.
КЕРАТОДЕРМИЯ ЛАДОНЕЙ И ПОДОШВ НАСЛЕДСТВЕННАЯ
(keratodermia palmarum et plantarum hereditaria)
Этиология и патогенез. Гетерогенная группа генетически детермини-
рованных ладонно-подошвенных кератозов с неизвестным биохимическим
дефектом, которые могут быть изолированными или являются частью син-
196
дромов, сочетаясь с разнообразными аномалиями, прежде всего с эктодер-
мальными дисплазиями. Среди факторов, которые в той или иной мере
могут быть вовлечены в патогенез, Costello и Gibss (1967) указывают инто-
ксикации мышьяком и другими тяжелыми металлами, недостаточность
витамина А, ферментопатии, вирусную инфекцию, нарушения в иммун-
ной системе, а также гормональные дисфункции, в первую очередь поло-
вых желез. Riddick и соавт. (1971) наблюдали развитие кератодермии у
больного аденокарциномой легких. Anderson и Martt (1965)—после приме-
нения противохолестеринемического средства 20,25 диазахолестенола. Об-
щепризнанной классификации наследственных ладонно-подошвенных кера-
тодермий нет. Наиболее часто их классифицируют с учетом таких двух
критериев, как тип наследования и характер поражения (диффузный, оча-
говый, линейный и т. д.).
Zeligman (1983) рассматривает как наиболее важные следующие кри-
терии отнесения кератодермий ладоней и подошв в ту или иную группу:
факт наследования, «возраст» начала, диффузный или очаговый характер
кератоза, наличие очагов на других участках кожного покрова, наличие
ассоциации с другими патологическими процессами, эктодермальными или
системными. Автор высказывает некоторое сомнение в возможности раз-
граничения каждой клинической формы, так как в семьях могут быть
различные формы кератодермии. Наиболее удачной, с нашей точки зрения,
является классификация, предложенная Greither (1977), в которой учтены
морфологические особенности, тип наследования, наличие или отсутствие
ассоциированных симптомов. Автор различает диффузные (без и с ассо-
циированными симптомами), полосовидные и очаговые (без сопутствующих
симптомов) кератодермии, рассеянные папулезные формы (с или без дру-
гих признаков).
Клиника. Из диффузных кератодермий самой частой является kera-
tosis palmo-plantaris Unna—Thost. Характеризуется наличием массивного,
обычно сплошного ороговения ладоней И подошв, с гладкой или шерохо-
ватой, иногда веррукозной поверхностью, часто с трещинами, восковидно-
го, желтоватого или коричневатого цвета Многочисленные трещины могут
придавать очагам поражения мозаичный характер. Характерным призна-
ком является наличие на границе со здоровой кожей узкой полосы ливид-
ного цвета. В ранних стадиях эритема может быть отчетливой и на всей
поверхности ладоней и подошв, с возрастом же она ослабевает и в краевой
зоне. Гиперкератотические изменения в области свода стопы менее выра-
жены. У многих больных имеется гипергидроз, рост ногтей усилен, они
могут быть нормальными или утолщенными, деформированными, матового
цвета с гребешками на поверхности. Может наблюдаться онихогрифоз.
Заболевание развивается в большинстве случаев в течение первого года
жизни, но может существовать с рождения. С течением времени, обычно
до периода полового созревания, степень гиперкератотических изменений
нарастает. Наследуется аутосомно-домииантно. Могут быть значительные
меж- и внутрисемейные различия: изолированное поражение ладоней
или подошв, разная степень и характер кератоза и др. Субъективными
197
расстройствами обычно не сопровождается, иногда^небольшой зуд, жже-
ние. Трещины могут быть очень болезненны, особенно при воспалении
(рис. 130, 131, 132, 133, 134, 135).
В большинстве случаев у больных не выявляются другие кожные или
общие заболевания. Однако имеются описания сочетаний с глухотой (На-
tamochi et al., 1982)' аномалиями развития мягких тканей лица и лицево-
го скелета, гранулярным хейлитом (Егоров Н. А., 1978), с доминантно
наследуемым ихтиозом (Nielsen, 1983). Nielsen (1983) описал, как, вероятно,
особый вариант кератодермии Unna—Thost, кератодермию, развившуюся
в 2—3-летнем возрасте, с признаками мутиляции на пальцах, появившихся
в 16-летнем возрасте больного. Айнум в сочетании с диффузным кератозом
ладоней и подошв наблюдал Spencer (1942). Обнаруживались у больных
диффузной кератодермией и другие самые разнообразные изменения (гипо-
генитализм, дисфункции щитовидной железы, клинодактилия, утолщения
концевых фаланг в виде барабанных палочек, acanthosis nigricans, витилиго
и др ), однако для того, чтобы решить вопрос, закономерно ли это сочетание
или речь идет о разных по своей природе заболеваниях, необходимы допол-
нительные наблюдения, выяснение первичного дефекта.
Кератодермия, описанная Greither (keratosis palmo-plantaris progre-
diens et transgrediens), характеризуется следующими признаками: слабо
выраженный гиперкератоз подошв, гипергидроз ладоней, иногда с гипер-
кератотическими изменениями, наличие очагов гиперкератоза на других
участках кожи конечностей и туловища, сходных с наблюдаемыми при
эритрокератодермиях, прогрессирующее течение у детей, спонтанная ин-
волюция с возрастом. Клиническая картина сходна с наблюдаемой при
болезни острова Меледа (Hiibner и Ziegenbalg, 1969; Voiglander и Schnyder,
1980), но наследование—аутосомно-доминантное. Michalowski и Miturska
(1963) обращают внимание на наличие в семье больных кератодермией эпи-
лепсии, кератотические изменения в области сосков молочных желез, губ,
гипертрихоз. Авторы отмечают близость этой формы кератодермии к ихтио-
зу или ихтиозиформной эритродермии. Диффузные гиперкератотические
изменения могут наблюдаться при ряде синдромов. Описан (Shrank, 1966)
семейный случай «ограниченного кератоза», наследуемого аутосомно ре-
цессивно, клинически сходный с болезнью острова Меледа, проявляющийся
гиперкератотическими бляшками на тыле кистей и стоп, у 5 из 11 больных
—диффузной ладонно-подошвенной кератодермией. У одной больной были
очаги кератоза на лбу, щеках, подбородке. Волосы, зубы и глаза были
нормальны. На ногтях у некоторых больных—точечная истыканность. В
качестве отличительных признаков от болезни острова Меледа указывается
поражение туловища у всех больных, более раннее вовлечение в процесс
локтевых и коленных суставов, отсутствие более чем у половины больных
изменений на ладонях и подошвах.
При акрокератоэластоидозе Costa (acrokeratoelastoidosis Costa) кера-
тоз сочетается с дегенеративными изменениями эластических волокон.
Характеризуется наличием роговых папулезных элементов, сходных с
роговыми жемчужинами (Costa, 1954), по краю более или менее диффузного
198
Рис. 130. Кератодермия
подошв диффузная.
Рис. 131. Кератодермия
подошв диссеминированная
Рис. 134. Кератодермия Рис. 135. Кератодермия
наследственная. ограниченная.
утолщения рогового слоя ладоней и подошв. Изменения на ладонях наи-
более выражены в области тенара и гипотенара. Папулезные элементы
блестящие, плотные при пальпации, мелкие, 1—2 мм в диаметре, округ-
лые, овальные, или полигональные, беловато-желтоватого или сероватого
цвета, полупрозрачные, слегка возвышаются, частью как бы вдавленные
в кожу, что придает им некоторое сходство с ксантомами или с бородав-
ками. Могут сливаться в небольшие бляшки, располагаться кольцевидно,
линейно. Иногда в центре папулезных элементов может быть точечное вдав-
ление, плотно прилегающие чешуйки. Они могут занимать, кроме того,
нижние 2/3 голеней, область голеностопного сустава, на кистях—тыльную
поверхность пальцев, межпальцевых складок, лучезапястных суставов.
Кожные складки подчеркнуты, группы папулезных элементов разделяются
небольшими бороздками. Отмечается гипергидроз, волосы, ногти, слизи-
стые без изменений. Гистологически в наблюдениях Costa (1954) помимо
изменения количества и фрагментации эластических волокон обнаружива-
ется гомогенизация коллагена, однако гистологические и электронно-
микроскопические исследования Jung и соавт. (1974) не выявили изменений
коллагена. Заболевание развивается обычно в юношеском возрасте, рас-
сматривается как невоидная аномалия (Cocta, 1954). Jung и соавт наблю-
дали в одной семье 21 больного в трех поколениях. Наследование — ауто-
сомно-доминантное.
Из рецессивных ладонно-подошвенных кератодермий основными яв-
ляются болезнь острова Меледа (kerato; is palmo-plantaris progrediens et
200
transgrediens) и синдром Papillon—Lefevre. Reed и соавт. (1979) считают,
что наиболее характерными признаками болезни острова Меледа являются:
аутосомно-рецессивное наследование, начало с рождения или в раннем
детстве, гиперкератоз в виде перчаток или носков с резкими границами,
изредка кератотические изменения на локтях, коленях, лодыжках, оро-
говение в полости рта, резко выраженный гипергидроз, мацерация и не-
приятный запах, медленное прогрессирование без ремиссий. Кератодермия
диффузного или реже очагового характера, желтоватого, коричневатого
или грязно-серого цвета, по периферии—более отчетливая воспалительная
реакция в виде каймы красноватого цвета с ливидным оттенком. Глубокие
трещины, мацерация, иногда мокнутие, что отчасти придает изменениям
сходство с экзематозными. На тыле кистей обнаруживаются желтовато-
коричневатые лихеноидные роговые папулы, сливающиеся в небольшие
бляшки. Гиперкератотические очаги в области коленных и локтевых суста-
вов могут иметь псориазиформный характер. Вокруг рта наблюдается стой-
кая шелушащаяся эритема, небольшая инфильтрация. Кератодермии часто
сочетаются с дистрофией ногтей (койлонихия, подногтевой гиперкератоз,
поперечная и продольная исчерченность, вдавления, онихогрифоз), у
у ряда больных с умственным и физическим недоразвитием. У 3 из 11 боль-
ных Salomon (1981) наблюдал двойную окраску ногтевых пластинок: в
дистальной части—бледную, в проксимальной—розовую. Волосы и зубы
обычно нормальные. Лейкоплакия полости рта. Может развиться дерма-
тогенная контрактура. С возрастом интенсивность клинических проявле-
ний иногда становится меньше, особенно в случаях более позднего разви-
тия заболевания. Улучшение может наблюдаться во время беременности.
По мнению Salomon и соавт. (1982), болезнь острова Меледа является не
кератодермией, а эритрокератодермией с акральной локализацией. Изме-
нения кожи при синдроме Papillon—Lefevre сходны с болезнью острова
Меледа, по мнению Ohkawara и соавт. 1974), настолько, что эти состояния
очень трудно дифференцировать, пока не развился пародонтоз. Наблюда-
ется преимущественно диффузное эритемато-сквамозное утолщение кожи
ладоней и подошв, сочетающееся с гипергидрозом, аномалиями зубов,
кариесом, гингивитами, пародонтозом, выпадением зубов. Отмечаются ги-
перкератотические изменения в области ахилловых сухожилий, коленных
и локтевых суставов, которые могут иметь псориазиформный вид. На лице
эритемато-гиперкератотические изменения могут располагаться в виде
бабочки (Ziprakowski et al., 1963). Повышена склонность к инфекции (На.
neke et al., 1975; Bravo-Piris et al., 1983), может быть отставание в умствен,
ном и физическом развитии. Волосы обычно нормальные, но может быть
гипотрихоз, ногти дистрофичны. Отмечена кальцификация мозговых обо-
лочек (Gorlin et al., 1964; Brownstein и Skolnik, 1972), сочетание с врож-
денными бронхоэктазами (Bravo-Piris et al., 1976). Заболевание обычно
развивается в первые 4 года жизни, кожные и зубные изменения обычно
возникают почти одновременно, но иногда зубные аномалии развиваются
значительно раньше кератодермии (Brownstein и Skolnik, 1972) или на-
оборот (Ohkawara, et al., 1974). Молочные зубы прорезываются в обыч-
• 201
ные сроки, но как только появляется последний молочный зуб, развивается
гингивит, десны становятся отечными, кровоточащими, могут изъязвляться,
происходит разрушение круговой связки зубов с образованием абсцессов,
деструктивные изменения альвеолярных отростков челюстей, что приводит
к расшатыванию и выпадению зубов. Степень гиперкератотических измене-
ний совпадает по тяжести с наиболее выраженным поражением пародонта.
Молочные зубы обычно выпадают к 5 годам жизни, в этот же период стихают
воспалительные изменения, десны приобретают внешне нормальный вид,
уменьшается гиперкератоз. Рецидив, нередко в более интенсивной форме,
возникает после замены молочных зубов постоянными. Обычно к 16 годам
все зубы, за исключением троих моляров, выпадают, и патологический
процесс начинает ослабевать. Из других признаков у больных наблюдались
замедление физического развития, формирования скелета, остеопороз,
внутричерепная кальцификация, гипертония, гипергликемия, увеличение
щитовидной железы, микрофтальмия. Haim и Munk (1965), Verma и соавт.
(1979) описали арахнодактилию и атрофические изменения костной ткани
концевых фаланг пальцев с заострением и истончением. И. Ю. Голоусенко
и соавт. (1982) наблюдали у отца больного повышенную сухость кожи.
Из диффузных кератодермий следует отметить ладонно-подошвенную
кератодермию, сочетающуюся с карциномой пищевода. Развивается кера-
тоз обычно в 5—15 лет, рак—в 50-летнем возрасте. Наследование аутосом-
но-доминантное .
Voigtlander и Schnyder (1980) полагают, что термин keratosis palmo-
plantaris varians (Wachters) является наиболее подходящим для обозна-
чения ранее изолированно рассматриваемых полосовидных и очаговых
кератодермий, и считают вероятным, ссылаясь на работу Schnyder и Klun-
ker (1966), что такие формы как кератодермия Вгйпаиег—Fuchs и очаговый
кератоз ладоней и подошв Siemens являются только различными фенотипа-
ми указанного выше врожденного нарушения ороговения. Клинически
они занимают промежуточное положение между диффузными и точечными
кератодермиями. Имеются различные сочетания ограниченных бородав-
чатых или мозолеподобных с глубокими трещинами очагов ороговения и
полосовидных кератозов (чаще в области пальцев), иногда сочетающихся
с гиперкератотическими изменениями на тыле кистей и стоп, коленных и
локтевых суставах. Свод стопы, как правило, свободен от поражения. Ги-
пергидроза нет, могут быть дистрофические изменения ногтей, подногте-
вой гиперкератоз, иногда пузыри, сходные с наблюдающимися при буллез-
ном эпидермолизе. Поражение, как правило, симметричное, но может
быть унилатеральным. Развивается обычно в первые годы жизни, насле-
дуется аутосомно-доминантно. Ortega et al. (1983) описали аутосомно-
доминантную форму полосовидной кератодермий с поздним началом. Мо-
гут быть значительные меж-и внутрисемейные различия — от единичных
очажков гиперкератоза до диффузных кератодермий. Из других проявле-
ний наблюдались папилломатоз в полости рта и помутнение роговицы (Gray-
son, 1965); гипергидроз, расщепление ногтей, геликотрихия (Sutton-Wil-
liams, 1969); умственная отсталость, скелетные аномалии, фолликулярный
202
кератоз, гипертрихоз голеней (Salomon и Lazovic, 1962). Der Kaloustian
и Kurban (1979) представили описание ограниченной ладонно-подошвен-
ной кератодермии в области наибольшего давления на стопах, а также на
кончиках пальцев, гипотенаре, ассоциированной с умственной отсталостью,
лейкоплакией в полости рта, дистрофией ногтей, роговицы. Bologna (1966)
наблюдал семейную форму полосовидной кератодермии, которой страдали
преимущественно мужчины. Brehm (1956) отмечал у больной кератодер-
мией с полосовидным ладонно-подошвенным кератозом и бородавчатыми
изменениями на тыле кистей и стоп такие аномалии развития, как доба-
вочный сосок, многочисленные пигментные пятна цвета кофе с молоком,
систематизированный веррукозный невус. Описана (Bologna, 1962) семья,
в которой родители и все трое детей страдали кератодермией Briinauer.
Кератодермия Voerner по клинической картине сходна с кератодермией
Thost—Unna (Haneke, 1982 и др.). Диффузное утолщение рогового слоя
наиболее выражено на местах давления. Гистологическая особенность
заключается в наличии зернистой дегенерации (акантокератолиза), харак-
терного гистологического признака буллезной ихтиозиформной эритродер-
мии (Klaus et al., 1970; Lever, 1983). Wachters и соавт. (1983) указывают
на наличие зернистой дегенерации при монетовидной форме кератодермии,
которая клинически проявлялась ограниченными болезненными очагами
гиперкератоза на-стопах в местах наибольшего давления. Изменения на
ладонях незначительны, усиливались после физической работы. Кератоз
развивался в сроки от 1,1/5 до 15 лет, медленно прогрессировал. Наследо-
вание аутосомно-доминантное. Автор полагает, что эта форма кератодермии
идентична описанным ранее Roth и соавт. (1978) «наследственным болез-
ненным омозолелостям» и считает возможным выделить ее как самостоя-
тельную единицу из группы keratosis palmoplantaris varians. Насколько
подобное заключение правомерно, судить трудно без сравнительных кли-
нико-генетических и морфологических исследований различных форм ке-
ратодермий.
Из точечных кератодермий наиболее частыми являются keratodermia
maculosa symmetrica disseminata palmaris et plantaris Buschke—Fischer
и keratoma dissipatum palmare et plantare Brauer.
При кератодермии Buschke—Fischer, развивающейся обычно в 15—30-
летнем возрасте, имеются множественные плоские очажки ороговения,
округлые или овальные, величиной 2—10 мм, с центральным вдавлением,
чаще равномерно рассеянные на ладонях и подошвах, редко группирую-
щиеся, обычно не сливающиеся. При удалении роговых масс остается кра-
терообразное углубление.
Кератома Brauer появляется в первые годы жизни, характеризуется
мелкими множественными бородавчатыми очагами ороговения, округлых
или полигональных очертаний, полушаровидной формы, после удаления
которых остается блюдцеобразное углубление. Наследуется аутосомно-
доминантно. Voigtlander и Schnyder (1980), как и Greither. считают эти
формы идентичными и описывают их под названием keratosis palmoplan-
taris papulosa s. maculosa вместе с кератодермией Davies—Colley. Costel-
203
Io и Gibbs (1967) рассматривают их как синонимы keratosis punctata. Bra-
un-Falco и Marghescu (1967) описали больного с точечной ладонно-подош-
венной кератодермией, фолликулярным кератозом и распространенной,
с поражением туловища и конечностей, фолликулярной атрофодермией.
В наблюдении Cole (1976), помимо точечного кератоза, у 6 больных в трех
поколениях имелись генерализованные изолированные гипопигментные
пятна, занимающие почти все тело. Dupre и соавт. (1977) проанализировали
6 случаев (один—собственное наблюдение) точечного гиперкератоза ла-
донных линий и предполагают, что это, видимо, новый тип точечной кера-
тодермии. Клинически имелись мелкие роговые пробки, как бы вдавлен-
ные в складках ладонной поверхности пальцев. Гистологически обнаружи-
валось сходство с болезнью Кирле, но базальный слой оставался интакт-
ным. Точечная кератодермия может быть составной частью синдрома Aig-
ner вместе с остеопойкилозом.
Powell и соавт. (1983) описали точечную кератодермию и спастиче-
ские параличи в трех поколениях. Наряду с больными, имевшими и ту и
другую патологию, отмечалась диссоциация признаков. При синдроме
Richner—Hanhart наблюдаются ограниченные болезненные при на-
давливании кератозы, умеренное ороговение в области кончиков
пальцев с пониженной чувствительностью, олигофрения, дистрофия
роговицы. Обнаруживаются также аномалии пальцев, дистрофия
ногтей, гиперкератозы в области коленных и локтевых суставов, ночное
недержание мочи. Наследуется аутосомно-рецессивно. Связывают эту фор-
му кератодермии с тирозинемией (Goldsmith, 1975). Описываются и дру-
гие, самые разнообразные кератодермии, самостоятельные и как часть
синдромов (синдромы Volavsek—ладонно-подошвенный кератоз, ониходист-
рофии, барабанные палочки, симптомы сирингомиелии; Christ—Siemens—
Touraine (см. Дисплазии эктодермальные); Siemens (см. лишай шиповидный);
Spannlang—Tappeiner—очаговая кератодермия, гипергидроз, алопеция, ке-
ратит; Schafer—ладонно-подошвенная кератодермия, диссеминированный
фолликулярный кератоз, пахионихия, врожденная катаракта, нарушение
роста, аномалии черепа, олигофрения, половое недоразвитие: Hanhart—
акрокератоз, липомы, нижнечелюстной дизостоз с перомелией и микрогна-
тией, нарушение роста. Синдром Bureau—Barriere—Thomas—диффузная
ладонно-подошвенная кератодермия, барабанные палочки, ногтевые пла-
стинки в виде часовых стекол, гипертрофия длинных трубчатых костей,
гипергидроз. Наследование—аутосомно-доминантное. Синдром Naegeli—
Franceschetti—Jadassohn—умеренная ладонно-подошвенная кератодермия,
диффузная сетчатая пигментация, гипогидроз, фолликулярный кератоз,
дисплазия зубной эмали. Наследуется аутосомно-доминантно. Нозологи-
ческое место некоторых из них пока не определено. Jakac и Volf (1975)
описали семью, в которой у 4 из 5 детей на 2—6 году жизни развилась кера-
тодермия со значительной вариабельностью клинической картины у от-
дельных больных. Отмечались диффузные, очаговые изменения на ладонях
и подошвах без перехода на тыл, фолликулярный кератоз. Не было типич-
ным и течение с периодическими обострениями и ремиссиями. Обнаружи-
204
вались искривления костей кистей и стоп, дистрофия зубов, готическое
нёбо. Наследование аутосомно-рецессивное. Schiff и Hughes (1974) описали
необычное сочетание изолированных остроконечных точечных кератозов
ладоней и подошв величиной 1—3 мм бородавчато-гиперкератотического
характера с множественными папулезными высыпаниями размерами 3—
5 мм на лице коричневато-желтоватого цвета, при гистологическом Иссле-
довании которых выявлена сальножелезистая гиперплазия. Этих измене-
ний не наблюдалось у больных женщин. Наследуется точечная керато-
дермия аутосомно-доминантно (Buchanan, 1963, и др.). Спорадические
случаи точечной кератодермий Coupe и Usher (1963) рассматривают как
новую мутацию. Под названием «Hereditary papulotranslucent acrokerato-
derma» Onwukwe и соавт. (1973)- описали своеобразную семейную керато-
дермию, которая проявлялась мелкими желтовато-беловатыми полупроз-
рачными папулами и бляшками, сочетающуюся с тонкими волосами на
голове и аТопией. Гистологически выявлялись гиперкератоз, гипергра-
нулез, акантоз. Клинически эта форма имеет большое сходство с акроке-
ратоэластоидозом Costa Werni и соавт. (1983) описали больную 58 лет,
у которой в постпубертатном периоде на тыле пальцев кистей развились
многочисленные, постепенно увеличивающиеся в количестве мелкие верру-
козные узелки цвета нормальной кожи Кроме того, на боковых поверхно-
стях кистей с переходом на ладони имелись множественные диссеминиро-
ванные или сгруппированные, резко ограниченные, возвышающиеся узел-
ки, величиной с булавочную головку или чечевицу, с блестящей, гладкой,
лишь изредка веррукозной поверхностью. Авторы рассматривают эту фор-
му как идентичную акрокератодермии точечной наследственной, описан-
ной Lee и Nasemann (1974). От других точечных кератодермий ее отличает
локализация только на кистях, отсутствие характерного для них призна-
ка—точечного углубления после удаления роговых пробок. Наибольшим
было сходство с акрокератоэластоидозом Costa, но гистологически обна-
руживались только гиперкератоз, акантоз и гипергранулез без изменения
эластических волокон. Авторы не исключают возможности идентичности
этих заболеваний с учётом данных Matthews и Harman (1977) о случаях
акрокератоэластоидоза без изменения соединительнотканных волокон.
Описана (Tezuka, 1982) ограниченная, аутосомно-доминантно на-
следуемая форма ладонно-подощвенной кератодермий, гистологической
особенностью которой было отсутствие зернистого слоя.
Течение всех форм наследственной кератодермий ладоней и подошв
хроническое, прогноз для выздоровления неблагоприятный.
Гистопатология. Резко выраженный гиперкератоз, иногда акантоз,
нерезко выраженная воспалительная инфильтрация в дерме, более зна-
чительная при болезни острова Меледа. Brown (1971) описал при точеч-
ной кератодермий наличие паракератотической пробки, сходной с наблю-
даемой при порокератозе Mibelli. При точечной кератодермий Robenstein
и соавт. (1980) обнаружили с помощью электронного микроскопа внутри-
ядерные нарушения в виде множественных гипертрофированных ядрышек
205
в клетках базального и шиповидного слоев, которые, по мнению авторов,
способствуют развитию гиперкератоза.
Дифференциальный диагноз. Дифференцировать наследственную ке-
ратодермию ладоней и подошв следует от кератодермий дисменорейных,
климактерических, псориатических, травматических, профессиональных, от
ладонно-подошвенных сифилидов, кератозов при болезни Дарье, ихтиозе,
гонорее, диабете, тилотической экземе, мышьякового кератоза, бородавок,
мозолей.
Лечение. Длительный прием витамина А в больших дозах, аевит. от-
шелушивающие мази и пластыри. Имеются данные об эффективности лу-
чей лазера.
ЛИТЕРАТУРА
Braoo-Piris J., Villaron L. G., Martinez C., Garcia-Perez A. Papillon—Lefevre
syndrome: report of two familial cases.— Derma tofogica, 1976, 152, 3, 168—176.
Greither A. Erbliche Palmoplantarkeratosen.— Hautarzt, 1978, 28, 8, 395—403.
Goldsmith L. A. Tyrosinaemia—etiology of an inherited oculocutaneous syndrome.—
J. invest, derm., 1975, 64, 287.
Hatamochi A., Nakagawa S., Ueki H., Miyoshi K-, luchi I. Diffuse palmoplan-
tar keratoderma with deafness.— Arch, derm., 1982, 118, 8, 605—607.
Jakac D., Wolf A. Papilfomatos-verrukose Form der pafmoplantaren Keratodermie
kombiniert mit anderen Anomalien der Verhornung sowie dyspfastischen Zahnveranderun-
gen.— Hautarzt, 1975, 26, 1, 25—29.
Nielsen P. G. A curious Genetic coincidence found in a study of pafmopfantar kerato-
derma.— Dermaologica, 1983, 167, 310—313.
Nielsen P. G. Mutilating palmoplantar keratoderma.— Acta derm, vepereol. (Stokh.),
1983, 63, 4, 365—367.
. Ohkawara A., Miura I., Ishikawa J. Papillon—Lefevre syndrome.— Arch, derm.,
1974, 109, 5, 726—728.
Ortega M-, Quintana J., Camacho F. Keratosis palmoplantar striata. Acta derm,
venereol. ((Stokh.), 1983, 63, 3, 373—375
Reed M. L., Stanley J., Stengler F., Shupack J. L., Benjamin D. M. Mai de Me-
leda treated with 13-cis retinoic acid.— Arch. Dermatol., 1979, 115, 5, 605—608.
Robenstein D. G., Schwartz. R. A., Hansen R. C., Payne С. M. Punctate hyperke-
ratosis of the palms and soles.— J. Amer. Acad. Dermatol., 1980, 3, 1, 43—49.
Salomon T., Cezaravic B., Nardelli—Kooacici M., Schnyder U. M. Die Meleda—
Krankheit-eine Akroerythrokeratodermie.— Zeitschr. Hautkr., 1982, 57, 8, 580—586.
Schiff B. L., Hughes D. Palmoplantar keratosis acuminata with faciac, sebaceou
hyperplaria — Arch, dermatol., 1974, 109, 1, 86—87.
Tezuka T. Circumsribed palmoplantar keratoderma.—Dermatologica, 1982,165, 30—38.
Voigtlander V. Hereditare Verhornungsstorungen und Taubheit. Zeitschr. Hautkrkh.,
1977, 52, 1017—1025.
Voigtlander V.. Schnyder U. W. Palmoplantarkeratosen. In: Derm, in Pra is und
Klinik.— Hrsg. G. W. Korting, Stuttgart, 1980, Bd. 2, 2126—2136.
206
Wachters D. H. J., Frendorf E. L., Hausman R., van Dijk E. Keratosis palmoplan-
taris nummularis (hereditary painful callosities).— J. Acad. Dermatof., 1983, 9, 2, 204—209.
Werni R., Scharz Th., Gschnait F. Akrokeratoderma hereditarium punctatum.— Z
Hautkrkh., 1983, 59, 1, 37—40.
КЕРАТОДЕРМИЯ МУТИЛИРУЮЩАЯ ФОВИНКЕЛЯ (keratodermia
hereditarium mutilans Vohwinkel).
Синонимы: синдром Фовинкеля, наследственная мутилирующая кера-
тома
В 1929 году Vohwinkel впервые описал диффузный ладонно-подошвен-
ный гиперкератоз в сочетании с айнгумподобной перетяжкой пальцев, с
последующей ампутацией фаланг пальцев и тотальной алопецией у 24-
летней женщины Следует отметить, что до Vohwinkel описывались случаи
Рис. 136. Кератодермия
наследственная Фовинкеля.
Рис. 137. Кератодермия
наследственная Фовинкеля.
кератодермии ладоней и подошв в сочетании с перетяжками и ампутацией
концевых фаланг, однако Фовинкель дополнил этот симптомокомплекс то-
тальной алопецией, линейными гиперкератозами ладоней и подошв, гипер-
кератозом в области локтей и колен. Он же указал на семейность пораже-
ния и проследил доминантный путь наследования заболевания.
Клиника. В мировой литературе имеются лишь единичные описания
данного заболевания (Wizz, 1929; Gibbs и Frank, 1966; Callan, 1970; Sierra
et al., 1975; Cole et al., 1984).
Развитие кератодермии обычно начинается в раннем детстве, чаще
до года. Развитие перетяжек происходит в разные сроки, но чаще в воз-
расте от 4 до 10 лет.
207
Рис. 139. Гиперкератотические
изменения в области ахилловых
сухожилий у старшей сестры.
Рис. 138. Кератодермия ладоней
и кольцевидные перетяжки пальцев
у старшей сестры.
Рис. 140. Кератодермия
наследственная Фовинкеля.
Кератодермия, как правило, носит диффузный характер типа Унны-
Тоста с выраженными гиперкератотическими наслоениями толщиной до
1 см и более.
Поверхность ороговевших мест гладкая, бросается в глаза ее янтар-
но-желтый, желто-бурый цвет. Местами скопляются особенно мощные твер-
дые роговые наслоения.
По периферии очагов поражения отмечается красная или цианотиче-
ская полоса в 3—5 мм шириною, которая резко отграничивается от окру-
жающей нормальной кожи. Ее можно увидеть и на боковых поверхностях
пальцев рук, на месте перехода чрезмерно ороговевшего эпидермиса ла-
донной поверхности в нормальный тыльный. Здесь эта полоса значитель-
но уже.
208
Поражения с ладони переходят на прилежащие части внутренней по-
верхности предплечья, а с подошвы распространяются вдоль ахиллова
сухожилия. На коленях и локтях встречаются резко ограниченные буро-
красного цвета толстые бородавчатые бляшки. В ряде случаев это состоя-
ние усугубляется развитием глубоких трещин, присоединением вторичной
инфекции с последующим рубцеванием. Каких-либо субъективных ощуще-
ний, кроме болезненности на месте гиперкератоза, на месте образования
трещин не бывает.
Пальцы кистей и реже стоп в области концевых фаланг истончаются,
становятся коническими, желтоватого цвета и несколько напоминают паль-
цы при склеродактилии. Патологический процесс постепенно приводит к
формированию шнуровидных кольцевых перетяжек на пальцах, контрак-
тур и спонтанной ампутации пальцев. При рентгеновском исследовании
обнаруживаются на месте перетяжки резорбирование кости и ясные фо-
кусы рассасывания.
Диффузное выпадение волос наступает с 4—5 лет (Gibbs, 1966) без
признаков рубцовых изменений.
А. А. Каламкарян и Г. И. Суколин (1983) описали три случая раз-
личных наследственных видов кератодермий (наследственные поликера-
тозы Турена) в одной семье. У отца (пробанда) наблюдалась точечная кера-
тодермия, у младшей дочери—гиперкератотические явления на ногах и
тотальная алопеция, и, наконец, у старшей дочери—мутилирующий ва-
риант линейной кератодермий с тотальной алопецией. У последней боль-
ной наблюдались двойные перетяжки, одна из которых соответствовала
кожной складке между концевой и средней фалангами, а вторая соответ-
ствовала основанию ногтевой пластинки. Особенностью наблюдения яв-
ляется также наличие у последней больной ониходистрофии (ногтевые
пластинки типа часовых стекол), онихогрифоза и глубоких поперечных
перетяжек ногтевых пластинок, чего не отмечено в предыдущих сообще-
ниях (рис. 136, 137, 138, 139, 140).
Со стороны нервной системы, органов зрения и слуха, стоматологи-
ческого статуса, гениталий патологии у этих трех больных не выявле-
но. Грибы из очагов поражения не обнаружены, анализы крови, мочи, се-
рореакции на сифилис отрицательные.
Гистопатология. Мощный гиперкератоз, гранулез^ акантоз. В дерме
небольшие воспалительные инфильтраты из лимфоцйтов и гистиоцитов.
Дифференциальный диагноз. Следует проводить с другими врожден-
ными кератодермиями ладоней и подошв—Папийон—Лефевра, Унны—
Тоста, болезнью Меледа. Дифференцировать следует также с периорифи-
циальной кератодермией (Olmsted syndrome), которая, по мнению Poulin
и соавт. (1984), является самостоятельным заболеванием, отличным от му-
тил ирующей кератодермий Фовинкеля.
Лечение. Эффективных методов лечения нет. Состояние улучшается
под влиянием длительного приема витаминов А и Е, синтетических про-
изводных ароматических ретиноидов. Мы отмечали начало роста волос
14 Клиническая дерматология 209
под влиянием фотохимиотерапии. Для уменьшения риска развития ампу-
тации необходимо проводить терапию, направленную на улучшение трофи-
ки тканей (компламин, андеколин и др.).
ЛИТЕРАТУРА
Каламкарян А. А., Суколин Г. И. Мутилирующая кератодермия Фовинкеля.—
Вести. Дерматол. и венерол., 1985, 10, 32—37. х
Rook A. Hidrotic ectodermal dysplasia,— In: Textbook of dermatology. Oxford, 1979,
vol. 1, p. 116.
Sierra 0., Montero B. Syndrome de Vowinkel (etude de quatre cas).— Ann. Derma-
tol. Syphiligr., 1975, 102, 1, 41—45.
КИМУРЫ БОЛЕЗНЬ
Синоним: ангиолимфоидная гиперплазия с эозинофилией (morbus Ki-
mura).
Болезнь Кимуры впервыё описана в Японии в 1948 году (Kimura et
al.) как «необычный гранулематоз, сочетающийся с гиперпластическими
изменениями в лимфоидной ткани, с эозинофилией». С тех пор много слу-
чаев описано в странах Юго-Восточной Азии (Японии, Китае и др.). Позд-
нее появилось сообщение о подобном процессе, который авторы обозначили
как «ангиолимфоидная гиперплазия с эозинофилией» в английской, евро-
пейской литературе (Wells, Whimster, 1969). В последующих описаниях
одни исследователи (Mehregam, Shapiro, 1971; Reed et al., 1972; Caro,
Winkelmann, 1974; Lynn From, 1979; Hamrick et al., 1984; Flegel, 1984;
Kung Ignatus et al., 1984, и др.) считали эти заболевания разными стадия-
ми одной нозологической формы, другие—различными нозологическими
единицами (Bendl et al., 1977; Rosai et al., 1979; Nelson Dan, Michael,
1984, и др.). До 1981 г. описано 250 больных (Streinzer, 1985).
Этиология п патогенез. Большинство современных авторов рассматри-
вают заболевание как реактивный процесс с иммунными нарушениями (А1-
bertazzi et al., 1983, и др.). У некоторых больных обнаруживают гиперчувст-
вительность к ряду препаратов, астму или поллиноз в анамнезе, повышение
уровня иммуноглобулина Е в крови (Hamrick et al., 1984). Методом пря-
мой иммунофлюоресценции показано отложение иммуноглобулинов А, М
и Са в стенках пролиферирующих сосудов (Grimwood et al., 1979).
Клиника. Основными клиническими признаками заболевания явля-
ются одиночные или множественные кожные и подкожные узлы, увеличе-
ние лимфоузлов в сочетании с эозинофилией периферической крови. Опи-
сано вовлечение в процесс слюнных желез и подлежащих мышц. Общее
состояние больных остается хорошим, поражения внутренних органов ие
наблюдается. Заболевание встречается в любом возрасте (от 5 до 63 лет),
но чаще у лиц молодого возраста, преимущественно у женщин. Солитарные
или чаще множественные красно-бурые опухолевидные разрастания плот-
ноэластической консистенции располагаются внутрикожно или подкожно
на голове, шее, реже на других участках. Кожные узелки достигают раз-
меров до 1—1,5 см в диаметре, а подкожные узлы—3—7 см. Дерматологиче-
210
ские изменения могут быть весьма сходны с истинной опухолью, поэтому
иногда производят их радикальное удаление. После травмы или расчесов
образования иногда сильно кровоточат. Субъективные ощущения (зуд),
как правило, отсутствуют. Поражение кожи сопровождается, как пра-
вило, увеличением регионарных лимфатических узлов и сочетается с эози-
нофилией в крови. Заболевание характеризуется длительным течением и
отсутствием генерализации, но возможностью рецидива после удаления
очага поражения.
Прогноз благоприятный, течение заболевания длительное, общее со-
стояние больных не нарушается. У отдельных пациентов опухоли само-
стоятельно регрессируют.
Гистопатология. Узлы в коже и подкожной клетчатке представлены
пролиферирующими сосудами в основном капиллярного типа с утолщен-
ными стенками, воспалительным инфильтратом с большим количеством
эозинофилов, а также лимфоидными фолликулами со светлыми центрами.
В лимфатических узлах отмечается гиперплазия фолликулов, в паракор-
тикальных зонах и мозговом слое—очаги сосудистой гиперплазии и ин-
фильтрации эозинофилами.
Дифференциальную диагностику следует проводить с саркомой Капо-
ши, ангиосаркомой, гемангиоэндотелиомой и другими сосудистыми опухо-
лями, пиогенной и эозинофильной гранулемой, ретикулогистиоцитомой,
саркоидозом.
Лечение. Методы лечения ангиолимфоидной гиперплазии недостаточ-
но разработаны, положительный эффект у части больных наблюдается от
инъекции кортикостероидов в зону поражения кожи в сочетании с наруж-
ным применением кортикостероидных мазей.
В кожной клинике ЦКВИ мы (Каламкарян А. А., Вавилов А. М.,
Персина И. С. ) наблюдали больную 34 лет, у которой в правой заушной
области имелись множественные опухолевидные образования от 0,3 до
1 см в диаметре красновато-синюшного цвета, плотноэластической конси-
стенции, безболезненные. На поверхности отдельных узелков наблюдались
пупковидные вдавления с корочкой на поверхности. В полости пра-
вой ушной раковины имелось опухолевидное образование 1,5 см в диаметре,
по цвету и строению напоминавшее ягоду малины (состоит из отдельных
слившихся узелков). Увеличение шейных лимфатических узлов справа
до размеров грецкого ореха. В крови эозинофилы до 9%. Гистологически
эпидермис не изменен, в дерме ограниченное образование, состоящее из
множества сосудов капиллярного типа с расширенными просветами, выст-
ланное набухшим эндотелием без признаков атипии. Периваскулярные
воспалительные инфильтраты состоят из лимфоцитов, гистиоцитов, туч-
ных клеток и большого количества эозинофилов. Определяются скопле-
ния лимфоцитов, образующих структуры типа' лимфоидных фолликулов
со светлыми центрами, которые также инфильтрированы эозинофилами.
В нашем наблюдении имелись признаки, характерные как для ангио-
лимфоидной гиперплазии (пролиферация сосудов с образованием солид-
ных тяжей эндотелиальных клеток), так и для болезни Кимура (формиро-
14* 211
ванне лимфоидных фолликулов-в коже). Нам представляется более оправ-
данным рассматривать их как один патологический процесс. В нашей стра-
не это заболевание ранее не описано.
ЛИТЕРАТУРА
Takanaka Т., Okuda М. et al. Histological and immunological studies on eosinop-
hilic granuloma of soft tissue, so—called Kimura’s disease.— Clin, allergy, 1976, 6, 27—34.
Rosai J, et al. The hystiocytoid hemangiomas.— Hum. pathoL, 1979, 10, 707—710.
Baler G. Angiolymphoid hyperplasia with eosinophilia.— J. dermatol. Surg. oncol.,
1981, 7, 229—234.
Grimwood R. et al. Angiolymphoid hyperplasis with eosinophilia.— Arch, dermatol.,
1979, 115, 2, 205—207.
Inada S. et al. A case of eosinophilic lymph follicufosis of the skin (Kimura’s disea-
se).— J. dermatol., (Tokyo), 1977, 4, 207—214.
Castro C., Winkelmann R. Angiolymphoid hyperplasis with eosinophilia.— Cancer,
1974, 34, 1696—1705.
Flegel H. Angiolymphoid hyperplasie mit Eosinophilie.— Medizin aktuell, 1984, 10,
7, 316—317.
Rook A., Dawber R. Disease of the hair and scalp.— Blackwell scientific publica-
tions, Oxford—London, 1982, 5—534.
Hamrick H., Jennete J., LaForce C. Kimura’s disease: report of a peniatric case in
the United States.— J. Allergy and clinical immunology, 1984, 73, 5, 561—566.
Kawada A. Morbus Kimura: Darstellung der Erkrankung und ihre Differential Diag-
nose.— Hautazzt, 1976, 27, 309—316.
Nelson Dan, Michael. Angiolymphoid hyperplasia with eosinofilia.— Pediatr. derma-
toL, 1984, 1, 3, 210—214.
Kung Ignatus et al. Kimura’s disease. A clinico-pathological study of 21 cases and
its distinction from angio-Iymphoid hyperplasis with eosynophilia.— Pathology, 1984, 16, 1,
39—44.
Streinter W., Chott A., ZrunkM., Hartenau F. Angiolymphoide Hyperplasie mit
Eosinophilie (Kimura’s Diseuse). —Laryngel., Rhinol. etol., 1985,' 64, 7, 342—346.
КЛИППЕЛЯ—ТРЕНОНЕЯ СИНДРОМ (KHppel—Thenaunaysyndromum)
Синонимы: варикозный остеогипертрофический ангиоматозный невус
Клиппеля—Тренонея.
Klippel и Trenaunay в 1900 году описали врожденный дефект, который
характеризуется унилатеральным варикозным расширением вен, плоскими
или туберозными ангиомами, гипертрофией костной и мягких тканей с
удлинением пораженной конечности.
В литературе до 1973 г. описано всего 200 наблюдений этого заболе-
вания (Ayala, Donofrio, 1982)-
Этиология и патогенез. Согласно современной классификации отно-
сится к группе врожденных венозных дисплазий (Malan, Puglionisi, 1964).
Клиника. Как свидетельствуют литературные данные и как показы-
вают наши наблюдения, заболевание чаще начинается в 3—6-летнем воз-
212
расте с появления сосудистого родимого пятна. Развитие этого синдрома
с рождения встречается исключительно редко. Так, Weber (1907) наблю-
дал 12-недельную девочку с диффузным капиллярным невусом на боль-
шой поверхности тела с увеличением верхней и нижней конечности. В
дальнейшем отмечается одностороннее массивное варикозное расширение
вен, плоские или туберозные гемангиомы и гипертрофия костной и мягкой
тканей; пораженная конечность, чаще нижняя, по сравнению с нормаль-
ной заметно утолщена и удлинена (на несколько сантиметров).
Флебографическое исследование позволяет выявить ненормальное, раз-
витие венозных сосудов (рис. 141). Гипертрофия и удлинение конечности,
являющиеся характерным проявлением синдрома, связаны с влиянием
изменений гемодинамики на рост костей (Каламкарян А. А. и Суколин
Г. И., 1982).
Синдром Сервелла (syndromum Servell) является другой разновидно-
стью врожденной ангиодисплазии. Он характеризуется образованием на
нижних конечностях крупных варикозных узлов, свободных конкрементов
внутри сосудов, рентгенологически видимых как «конфетти», сильным остео-
порозом с пустотами в костях и укорочением пораженной конечности.
По характеру изменения костей этот синдром противоположен синдрому
Клиппеля—Тренонея. Флебография указывает на расширение вен, а ар-
териография изменений не обнаруживает.
Синдром Паркса Вебера (Parkes Weber syndromum). Представляется
важным разделять оригинальный синдром Клиппеля—Тренонея от синдро-
ма Паркса Вебера и других врожденных ангиодисплазий. В 1918 году Par-
kes Weber описал синдром Клиппеля—Тренонея, но с наличием артерио-
венозных фистул. Автор объединил эти две сосудистые аномалии (вари-
козные расширения вен, гипертрофия костной и мягкой тканей нижней
конечности, наличие кожных гемангиом, артерио-венозных анастомозов)
общим термином гемангиоэктатическая гипертрофия конечностей. Linde-
nauer (1965) предложил обозначить этот синдром как синдррм Клиппеля—
Тренонея—Вебера.
Врожденные артерио-венозные свищи (артерио-венозные фистулы)
могут долго клинически не проявляться. В большинстве случаев они вы-
являются в возрасте 10—20 лет, что связано с гемодинамической перестрой-
кой в связи с ростом организма; часто их появлению способствует чрез-
мерная физическая нагрузка и травма. Осложнением артерио-венозных
фистул (свищей) являются трофические нарушения в конечности, тромбо-
зы, эндоваскулиты, язвы, деструкция костей.
В некоторых случаях синдром Паркса Вебера приводит к таким кож-
ным проявлениям, которые напоминают саркому Капоши. Унилатераль-
ная локализация поражений, артериографические исследования позволя-
ют уточнить диагноз.
Лечение. Консервативное (тугая повязка, ношение эластических
чулок и др.). Хирургическое лечение (перевязка или удаление лишних
венозных сосудов) представляет определенную трудность.
213
ЛИТЕРАТУРА
Халамкарян А. А., Суколин Г. И. О синдроме Клиппеля—Тренонея.— В кн.: Па-
тогенез и терапия кожных и венерических заболеваний. Минск, 1982, с. 76—84.
Нуреев Г. Г., Вафина А. X. Синдром Клиппеля—Тренонея.— Вести, дерматол. и
венерол., 1975, 2, 56—57.
Ayala F., Donofrio Р. Sindrome di Klippel—Trennnannay—Wober.— G. Ital. Der-
matol., 1982, 117, 3, 187—189.
Parisch H. Klippel—Trenaunay—Weber syndrome.— Arch. DermatoL, 1974, 109, 4,
573—574.
КОЖА ВРОЖДЕННАЯ МРАМОРНАЯ ТЕЛЕАНГИЭКТАТИЧЕСКАЯ
(cutis marmorata teleangiectatica congenita)).
Первое описание и название болезни принадлежит Van Lohuizen (1922).
В США чаще употребляется термин «генерализованная кожная флебэкта-
зия». Встречается крайне редко, к настоящему времени в литературе на-
считывается не более 35 публикаций, посвященных этому заболеванию.
Этиология и патогенез. Allen, Barker и Henes относят заболевание к
диффузным системным гемангиомам, Neumark—к порокам развития, De-
gos (1967)—к врожденным доброкачественным дистрофиям кровеносной
системы; Zarilli (1959) основной причиной считает,изменение тонуса капил-
ляров в эмбриональном периоде. Arnold и Weber (1979), Hodr и Procharkova
(1979) описали семейные случаи этого заболевания.
Клиника. Заболевание наблюдается у лиц обоего пола, обнаружива-
ется уже при рождении (Hamphries, 1952; Moyer, 1966; Lapiere et al.,
1965; Laugier, 1966; Andreev, Pramatarov, 1979, и др.). Имеются единичные
наблюдения больных со схожей клинической картиной, но развивающейся
в молодом или даже зрелом возрасте. Типичными являются многочислен-
ные, располагающиеся на лице, шее, туловище и конечностях, включая
ладони и подошвы, пятна красновато-синюшного цвета, местами с фиолето-
вым оттенком, с отчетливо видными через тонкую кожу расширенными
венами. Кожа имеет подчеркнутый сетчатый рисунок, обусловленный рас-
ширенными кровеносными сосудами. Измененные участки чередуются с
отдельными участками видимо неизмененной кожи. Это придает коже вид
мраморности; отмечается выраженный акроцианоз. Расширенные сосуды
встречаются также на твердом и мягком нёбе, слизистой носовых ходов.
У отдельных больных в очагах поражения образуются длительно сущест-
вующие трофические язвы размером до 5—8 см в диаметре, с неровными,
местами подрытыми краями, вялыми грануляциями. Мы наблюдали боль-
ного мальчика с характерной картиной врожденного сосудистого пораже-
ния с трофическими нарушениями (множественные язвы на нижних ко-
нечностях). Нередко отмечается умеренно выраженное диффузное пореде-
ние волос на голове, заметное разрежение бровей и ресниц, замедленный
рост волос.
С возрастом отмечается ослабление мраморной пятнистости. Однако
нередко существенной динамики кожных изменений на протяжении многих
лет не отмечается.
214
Гистопатология. В дерме и гиподерме обширная сеть расширенных,
зияющих капилляров и мелких вен, переполненных кровью. Воспалитель-
ная инфильтрация отсутствует, эндотелий имеет нормальную структуру
(Degos et al., 1963; Lapiere, 1965; Lynch, 1967, и др.). Довольно выражен-
ная гипертрофия и дистомия сальных желез.
Дифференциальный диагноз. Проводят с рассеянными эссенциальными
телеангиэктазиями, плоскими ангиомами, сосудистой пойкилодермией и
сетчатым ливедо.
Лечение. Диатермокоагуляция сосудов может дать удовлетворитель-
ный в косметическом отношении результат.
ЛИТЕРАТУРА
Торсу ев Н. А., Бухарович М. Н. Своеобразный случай cutis marmorata teleangie-
ctatica.— Весгн. дерматол. и венерол., 1979, 1, 29—31.
Каламкарян А. А., Акимов В. Г., Вавилов А. М. Cutis marmorata tefeangiecta-
tica.— Весгн. дерматол. и венерол., 1981, 2, 12—16.
Andreev V., Pramatarov К- Cutis marmorata teleangiectatica congenita in two sisters.
Brit. J. dermatof., 1979, 101, 3, 345—350.
КОЖА ВЯЛАЯ (cutis laxa).
Синонимы: дерматохалазис, дерматолиз, генерализованный эластолиз.
Этиология и патогенез. Различают 2 формы: наследственную (идиопа-
тическую) и приобретенную. Наследственная форма подразделяется (Wit-
kowski и Prokop, 1974) на 3 варианта: 1) с началом в раннем детстве или
даже с рождения, протекающий с поражением легких, что может привести
к летальному исходу уже в первые годы жизни; 2) с развитием в период
полового созревания, прогрессирующим течением и вовлечением внутрен-
них органов; 3) ассоциированный с дистрофией костей. Развивается в
детском возрасте. Первые два варианта могут наследоваться аутосомно-
доминантно или рецессивно, третий—аутосомно-рецессивно. Приобретенная
cutis laxa развивается обычно после воспалительных процессов, реже в
зоне опухолей, в том числе нейрофибром при болезни Реклингаузена. Опи-
сано развитие cutis laxa после дерматитов, аллергических реакций на пе-
нициллин, ангионевротического отека системной красной волчанки, высы-
паний типа многоформной эксудативной эритемы, герпетиформного дер-
матита Duhring (Jabblonska, 1966, Reed et. al., 1971; Kell и Bury, 1975;
Scott et. al., 1976, Randle st. al., 1983). Так как клинически идиопатиче-
ская и приобретенная формы не различаются, может возникнуть, по мне-
нию Meigel (1980), вопрос, не является ли приобретенная форма поздним
проявлением наследственной, обусловленным воздействием внешних фак-
торов. Jablonska (1966) высказала мнение о возможной инфекционной
природе cutis laxa как идиопатической, так и приобретенной.Hashimoto
и Kanzaki (1975) предполагают, что развитие заболевания обусловлено
недостаточным образованием эластических волокон или повышением ката-
болических процессов в них, за счет повышенной активности эластазы и
215
Рис. 142. Вялая кожа.
недостаточной—ее ингибиторов. Scott и соавт. (1976) указывают на воз»
можную роль в патогенезе аутоиммунных реакций, гиперчувствительности,
аномалии обмена меди, так как ее дефицит ведет к нарушению структуры
и функции эластина.
Клиника. Кожа в пораженных местах лишена обычной эластичности,
дряблая, складчатая, свисает, имеет старческий вид. Это состояние может
развиться незаметно или ему предшествует стадия отека. При растягива-
нии кожа медленно возвращается к исходному состоянию. Наиболее вы-
ражены изменения на лице, но в процесс может вовлекаться кожа верхних
216
Рис. 143. Вялая кожа
у мальчика 12 лет.
конечностей, туловища (рис. 142, 143, 144). Поражение может быть гене-
рализованным '(Ledoux-Corbusier, 1983). По мнению Schreiber и Tilley
(1961), врожденные случаи обычно генерализованные, приобретенные—
локальные или ограниченные. Описано (Barsy et al., 1968) сочетание cutis
laxa с помутнением роговицы и умственной отсталостью, вероятно, пред-
ставляющее собой самостоятельный синдром (Me Kusick, 1976). К особым
формам cutis laxa относится блефарохалазис, протекающий с поражением
кожи век, и синдром Ascher, представляющий собой комбинацию блефаро-
халазиса, струмы и двойной губы (Klauder, 1959; Suter и Vakilzadeh, 1977;
Miihlendysr и Hundeiker, 1978). Klauder (1959) различает 4 типа блефаро-
халазиса (Fuchs, сенильный, наследственный и неопределённый), сходных
клинически, но неоднородных по своей природе (наследственность, сниже-
ние мышечного тонуса, вазомоторные расстройства, атрофические измене-
ния, отеки, дисфункции щитовидной железы, хронический нефрит, болезнь
Реклингаузена, cutis laxa и др.). Имеются наблюдения одностороннего
развития блефарохалазиса (Brazin et al., 1979).
Кроме изменений кожи обнаруживаются эмфизема легких, аневризма
аорты; дивертикулы желудочно-кишечного тракта, целиакия, грыжи, ги-
поплазия почек, пролапс слизистой прямой кишки (Goltz et al., 1965;
Reed et al., 1971; Kerl и Birg, 1975; Scott et al., 1976).
Ting и соавт. (1984) описали сочетание cutis laxa с множественной
миеломой, что, по их мнению, требует изучения вопроса о роли аутоим-
мунных нарушений в развитии приобретенной формы cutis laxa, на что
указывали ранее Nanko и соавт. (1979). хотя они и не обнаружили при
217
электронно-микроскопической прямой иммунофлюоресценции антител, свя-
занных с эластическими волокнами.
Заболевание встречается редко. Описаны врожденные случаи у сиб-
сов (Galbe et al., 1981). Прогноз при доминантных формах благоприят-
нее, чем при рецессивных, так как при них поражение внутренних органов
встречается реже. Могут быть тяжело протекающие осложнения, вызван-
ные эмфиземой и сердечной недостаточностью. В редких случаях процесс
со временем может улучшиться (Schreiber и Tilley, 1962).
Гистопатология. Фрагментация и зернистая дегенерация эластических
волокон1, местами их полное отсутствие. В то время как Hashimoto и Кап-
zaki (1975) не обнаружили изменений структуры коллагена, Klehr и соавт.
(1977) считали, что нри cutis laxa поражаются не только эластические, но
и коллагеновые волокна. Более того, высказано мнение, что в основе пато-
логического процесса лежит нарушение синтеза коллагена, что ведет к
внутриклеточному накоплению проколлагена. Byers и соавт. (1980) указы-
вают, что нарушение синтеза коллагена может быть обусловлено сниже-
нием активности лизилоксидазы.
Дифференциальный диагноз. Дифференцировать cutis laxa необходи-
мо с cutis hyperelastica, псевдоксантомой эластической.
Лечение. Симптоматическое, в частности хирургическое удаление из-
быточной кожи.
ЛИТЕРАТУРА
Маккьюсик В. А. Наследственные признаки человека. Пер. с англ. М. Медицина,
1976.
Brazin S. A., Stern L. J., Johnson W.'J. Unilateral blepharochalasis.— Arch. Derm.,
1979, 115, 4, 479—481.
Byers H., Siegel R. C. Holbrook et al. X—linked cutis laxa—defective cross—
link formation in collagen due to decreased lysyl oxydase activity.— N. Engl. J. Med., 1980,
.303, 61—65.
Galbe J., Marin M. C., Pastor I., Caloo A., Used M. M. Garciade dos casos en
una fratria.— Acta pediatr. esp., 1981, 455, 39, 331—334.
Hashimoto K., Kanzaki T. Cutis laxa. Ultrastructural and biochemical studies.—
Arch. Derm., 1975, 13, 7, 861—873.
Kerl H., Burg J. Erworbene (postinflammatorische) Dermatochalasis.— Hautarzt,
1975, 26, 4, 191—196.
Klerr N., Heine H., Schaeg G., Nasemann Th. Syntropie von Dermatochalasis und
Urticaria pigmentosa als Folge einer Germatosparoxis endoplasmaitica.— Hautarzt, 1977, 28,
10, 522—534.
Ledoux-Corbusier M. Cutis laxa. Congenital form with pulmonary emphysema: an
ultrast'uctural study.— J. Cutan. pathol., 1983, 10, 340—349.
Meigel W. N. Cutis laga In: Dermatologie in Klinik und Praxis Hrsg. G. W. Kor-
ting, Georg Thieme Verlag, Stuttgart, 1980, Bd. I'll, 2027—2028.
Muhlendyck H., Hundeiker M. Blepharochalasis (Fuchs) und Laffer—Ascher—Syn-
drom. Hautarzt., 1978, 29, 474, 477.
218
Randle H. W., Muller S. Generalized elastolysis associated with systemic lupus
erythematosus. Am. j. Acad. Dermatol., 1983, 8, 869—873.
Scott M. A., Rauh Y. C., Luscombe H. A. Acquired cutis laga associated with mul-
tiple myeloma.— Arch. dermatoL, 1976, 112, 6, 853—855.
Suter L., Vakiliaden F. Ascher—syndrom.— Hautarzt, 1977, 28, 5, 257—259,
КОЖА ГИПЕРЭЛАСТИЧЕСКАЯ (cutis hyperelaitica).
Синонимы: синдром A. H. Черногубова—Ehlers—Danlos, эластическая
фибродисплазия.
Этиология и патогенез. Гиперэластическая кожа представляет собой
гетерогенное наследственное заболевание. Выделяют восемь форм с раз-
личным типом наследования и, по-видимому, с неодинаковым первичным
биохимическим дефектом (Me Kusick, 1978). Первые три типа наследуются
аутосомно-доминантно; четвертая—гетерогенная (с аутосомно-рецессивным
и доминантным типом наследования); пятая наследуется рецессивно-сцеп-
ленно с Х-хромосомой; шестая и седьмая—аутосомно-рецессивно; восьмая—
аутосомно-доминантно. При некоторых формах выявлены первичный био-
химический дефект. Так (по данным Pinnel и Mr Kusick, 1979; Krieg et al.,
1981), при IV типе обнаружен недостаточный синтез III типа коллагена,
при V—дефект лизилоксидазы; при VI—недостаточность лизилгидрокси-
лазы; при VII—дефект проколлагенпептиазы. О. Е. Блинникова и соавт.
(1985) высказывают предположение о единой природе I и II типов синдро-
ма. Имеются указания, что у больных с подтипом IV типа синдрома Элерса-
Данлоса синтез III типа коллагена не отличается от нормы (Sulh et al.,
1984).
Следует подчеркнуть, что еще в 1954 году А. В. Русаков считал, что
в основе заболевания лежит врожденная недостаточность фибробластиче-
ских клеточных элементов, возможно отсутствие в них ферментов, контро-
лирующих нормальный синтез коллагена. С. А. Мельников и Ф. Е. Гор-
бачева (1965) видели причину заболевания в эмбриональной недостаточно-
сти трофической функции гипоталамо-гипофизарной области. Stevano-
vi£ и соавт. (1982) описали 12-летнего мальчика с синдромом Элерса—Данло-
са. Своеобразие этого наблюдения заключалось в наличии иммунодефи-
цита. Pommerening и Antonius (1960) не выявили у больных аномалий хро-
мосом.
Клиника. Приоритет первого детального клинического описания гипер-
эластической кожи принадлежит Н. А. Черногубову. В 1891 году он пред-
ставил на заседании Московского венерологического и дерматологического
общества больного 17 лет с диагнозом cutis laxa, нос симптоматикой, полно-
стью соответствующей заболеванию, названному позднее синдромом Элерса-
Данлоса. И. И. Лелис(1972) совершенно справедливо предлагает именовать
это заболевание синдромом Н. А. Черногубова—Элерса—Данлоса. Основны-
ми клиническими признаками заболевания являются: чрезмерная растяжи-
мость и гиперэластичность кожи, ее легкая ранимость, ломкость сосудов, с
развитием кровоподтеков, гематом, повышенная растяжимость связок, чрез-
мерная подвижность суставов («гуттаперчевый человек»), часто с подвыви-
219
хами. Кожа тонкая, нежная, бархатистая, особенно легко растяжима на
лице, шее, в области крупных суставов (рис. 145, 146). На кистях и сто-
пах, а в более позднем возрасте и в области локтей, может производить
впечатление избыточной. Ф. А. Зверькова и В. П.-Качанов (1976) наблю-
дали у одного из 7 детей развитие cutis laxa в области голеней, что проявля-
лось в виде «свободного чулка.» Часты признаки акроцианоза, может быть
синдром Рейно. Даже малейшая травма при небольшом кровотечении
вызывает обширные кровоподтеки, гематомы, плохо заживающие раны.
На месте травм, чаще в области крупных суставов и на кистях, могут раз-
виться моллюсковидные, ангиомоподобыые псевдоопухоли мягкой кон-
систенции, иногда кальцифицирующиеся, атрофические рубцы с гиперпиг-
ментациями' и телеангиэктазиями, напоминающие папиросную бумагу.
В редких случаях могут быть пузыри. Из-за большой растяжимости кожи
во время беременности не развиваются striae distensae. Гипермобильность
в межфаланговых суставах может быть выраженной в такой степени, что
пальцы легко растягиваются в длину на значительное расстояние, а также
в ульнарную и радиальную сторону (первый и пятый соответственно).
С. А. Мельников и Ф. Е. Горбачева (1965) считают, что в симптомокомп-
лекс синдрома Элерса—Данлоса может входить выраженная врожденная
гипомиотония. Ф. А. Зверькова и В П. Качанов (1976) отметили гипото-
нию мышц конечностей и туловища у 6 из наблюдаемых ими больных. Г. Н.
Нефедова (1968) наблюдала атрофию мышц дистальных отделов конечно-
стей. Помимо кожных и суставных, могут наблюдаться изменения в полости
рта (кровоточивость десен, аномалии зубов, периодонтит, гиперподвижность
языка, так что больные кончиком языка могут коснуться носа), глаз (птоз,
вывих хрусталика, ангиоидные полосы на глазном дне, голубые склеры,
отслойка сетчатки и др.), сердечно-сосудистой системы (расслаивающаяся
- аневризма аорты, спонтанный разрыв крупных сбсудов, внутричерепные
аневризмы с нарушением мозгового кровообращения, варикозное расшире-
ние вен, изменение клапанов сердца и др.), желудочно-кишечного тракта
(грыжи и дивертикулы пищевода, желудочно-кишечные дивертикулы, кро-
вотечения, прободение и разрыв кишечника и др.), легких (пневмо- реже
гемоторакс, медиастинальная и подкожная эмфизема и др.), мочевыделитель-
ной системы (поликистоз почек, дивертикул мочевого пузыря и др.). Опи-
сано (Kousseff, 1981) наличие в одной семье синдрома Элерса—Данлоса и
врожденного эпидермолиза. Hernandez и соавт. (1981) описали своеобраз-
ный вариант синдрома Элерса—Данлоса с признаками умственного недораз-
вития, аномалиями волос, ушных раковин, множественными пигментными
пятнами.
Заболевание развивается в детском возрасте, с примерно равной часто-
той у лиц обоего пола. С возрастом растяжимость кожи уменьшается. Из-
менения кожи могут быть распространенными, часто с множественной вис-
церальной патологией (Zalis и Roberts, 1967) или редко локальными (Eul-
len, 1979). Характер и степень проявлений различны в зависимости от фор-
мы заболевания (Pinnell и McKusick, 1979). При I типе синдрома Элерса—
Данлоса клинические проявления наиболее выражены; при II—они обна-
220
руживаются, но в смягченной
форме; при III—на первый план
выступает гипермобильность в
суставах; при IV—склонность к
поражению сосудов и кровотече-
ниям; при V—в основном обна-
руживаются изменения кожи;
при VI—доминирует глазная па-
тология; при VII типе отмечается
нарушение роста больных, силь-
ная гиперподвижность в суставах,
подвывихи, умеренное изменение
кожи; при VIII—кожно-сустав-
ные проявления ассоциируются с
аномалиями зубов и периодонто-
зом. Прогноз определяется выра-
женностью внутренней патологии.
Наиболее тяжело протекающими
осложнениями являются прободе-
ние кишечника, разрыв крупных
сосудов. Особенно высок риск
развития тяжелых последствий
болезни при беременности. Часты
преждевременные роды. Т. И. Тер-
нова и соавт. (1984) рекомендуют
Рис. 144. Гипермастическая
кожа.
Рис. 145. Гиперяластичсская
кожа.
обследовать детей для исключения данного синдрома при неясной эти-
ологии сердечно-сосудистых изменений.
Гистопатология. Уменьшение коллагеновых и относительное увеличе-
ние эластических волокон. По данным Sulica и соавт. (1979), только у
2 из 21 больного были отклонения от нормальной структуры коллагеновых
и эластических волокон. Так, у одного больного (IV тип) обнаруживались
в дерме скопления волокон, 'Напоминающих актинически поврежденные
эластические волокна. У второго больного (VI тип) отмечалось резкое истон-
чение волокон обоего типа. Pierard и соавт. (1983) указывают на морфо-
логические различия между I и II типами заболевания. При первом типе
коллагеновых пучков мало, они разрыхлены, истончены, эластические
волокна, напротив, многочисленны. При втором типе коллагеновые пучки
изменены мало, количество эластических волокон не увеличено, но они
неодинаковы по размерам, очертаниям, распределены неравномерно. Иног-
да, по мнению авторов, о'тличить от нормальной кожи может быть трудно.
StevanoviC и соавт. (1982) при электронно-микроскопическом исследо-
вании кожи у описанного ими больного с иммунодефицитом обнаружили
расщепление коллагеновых волокон на микрофибриллы, спирализацию
их, недостаточность эластина в эластических волокнах, наличие масс, на-
поминающих измененные эластические волокна.
Дифференциальный диагноз. Дифференцировать гиперэластическую
кожу необходимо от cutis laxa, псевдоксантомы эластической, синдромов
Марфана!, Ларсена, Тернера, Вернера.
Лечение. Симптоматическое.
ЛИТЕРАТУРА
Блинн"коваО. Е., Козлова С. И., Прытков А. Н. и соавт. Клинико-генетическая
характеристика синдрома Элерса—Дамлоса. Вести, дерматол. и венерол., 1985, 2, 45—48.
Зверькова Ф. А., Качанов В. П. О синдроме Элерса—Дамлоса. Вести, дерматол. и
венерол-, 1976, 2, 47—51.
Тернова Т. И., Бочкова Д. Н., Миримова Т. Д. и др. Изменения сердечно-сосу-
дистой системы при синдроме Элерса—Данлоса. Весгн. АМН СССР, 1984, 2, 65—68.
Kousseff В. G. Ehlers—Danlos syndrome and epidermolysis bullosa in the same fami-
li, Cutis, 1981, 27, 5, 519—521.
Krieg T., Ihme A., Weber L., Kirsch E., Mueller P. K- Molecular defects of col-
lagen metabolisms in the Ehlers —Danlos syndrome — Int. J. dermatof., 1981,
20, 6, 415—425
McKusick V. A. Mendefian Inheritance in Man Sth ed., Johns Hopkins Univ. Press,
1978.
Pinell S. R., McKusick V. A. Heritable disordes of connertive tissue skin changes.
In: Derm, in General Medicine Sec. Ed. Edby T. B. Fitzpatrick et al., 1979, 1144—1160.
Sulk H. M. B., Steinmann B., Rao V. H. et al. Ehlers —Danlos syndrome Type
IV D: An Autosomal recessive Disorder. Clin. Genet., 1984, 25, 278—287
222
SulicaV./., Cooper Р.П., PopeM., HombrickG. W., Gerson В. M., McKKusickV.A
Cutaneus histologic femtures in Ehlers—Danfos syndrome. Arch, dermatol., 1979, 115r. 1,
40—42.
КОЛЛОИД МИЛИУМ (colloid milium)
Сииоиимы: коллоидальный псевдомилиум, гиалома.
Этиология и патогенез. Причина заболевания неизвестна. Отмечается
роль актинического повреждения кожи (Guin и Seale, 1959; Hashimoto
et al., 1972; Briist, 1977). По данным Holzberger (1960), Помимо этого спо-
собствует развитию заболевания длительный контакт с продуктами нефти.
У больного, описанного Sullivan и Ellis (1961), помимо интенсивной ин-
соляции в детстве, имело место рентгеновское облучение лица в 15-летнем
возрасте по поводу угревой сыпи. Коллоидная дегенерация развилась при-
мерно в 40-летнем возрасте. Выделяется также редкая генетически детерми-
нированная форма, развивающаяся в детском возрасте, с предположительно
аутосомно-доминантным типом наследования. Miedzinski и соавт. (1960)
считают, что ювенильная форма имеет иной патогенез, чем коллоид милиум
взрослых на том основании, что ими не обнаружено при гистологическом
исследовании признаков дегенерации коллагена, отложений амилоида или
муцина. С точки зрения Sullivan и Ellis (1961), Brust (1977), изменения
кожи являются следствием дегенеративных изменений как коллагеновых,
так и эластических волокон, с последующим отложением вокруг повреж-
денных волокон материала сосудистого происхождения. По мнению Has-
himoto и соавт. (1972), накопление аморфного вещества в коже, лежащеё
в основе развития заболевания, происходит за счет продукции его фибро-
бластами и декомпозиции коллагеновых волокон. В более поздней работе
Hashimoto и соавт. (1975) показали, что основное количество коллоида
синтезируется активированными солнечными лучами фибробластами, а
лишь очень незначительная его часть является результатом дегенератив-
ных изменений коллагена. Этот вывод был сделан на том основании, что
коллоид не содержит характерных для коллагена гидроксипролина и гидро-
ксилизина, а фибробласты в культуре продуцируют сходное с коллоидом
вещество. Szymanski (1961) считает, что с общепатологической точки зре-
ния было бы более правильно называть это заболевание гиалиновой дегене-
рацией кожи, так как коллоид представляет собой гиалин. Zoon и соавт.
(1955), напротив, считали, что изменения могут развиваться первично в
сосудах с последующей диффузией белковых субстанций ме’Жду соединитель-
нотканными пучками.
Клиника. Множественные милиарные резко очерченные'восковидные,
просвечивающие узелки, желтоватого цвета, располагающиеся более или
менее симметрично, рассеянно или сгруппированно на коже лба, щек, на
шее, ушах, тыле кистей (рис. 147). По наблюдениям Guin и Seale (1959),
если имеется множественная локализация, наиболее выраженными явля-
ются изменения.на кистях. Holzberger наблюдал 28 больных мужчин с
локализацией процесса только на кистях. Они длительно (в среднем в те-
чение 17 лет) подвергались инсоляции, контактировали с бензином. При
223
Рис. 146. Коллоид милиум.
прокалывании элементов выделя-
ется прозрачная студенистая
жидкость- Если узелки сливают-
ся, кожа выглядит огрубевшей,
испещрена бороздами, разделяю-
щими отдельные группы высыпа-
ний. Если заболевание развива-
ется у детей, они выглядят старше
своего возраста (Wooldridge и Fre-
richs, 1960). Sullivan и Ellis
(1961) описали преимущественно
бляшечную форму коллоидной
дегенерации с размерами очагов
до 5 см, на части из которых
были множественные телеангиэк-
тазии. Телеангиэктазий не было
на других участках лица, так что
их развитие не связывалось авто-
рами с имевшей место в прошлом
рентгенотерапией. Brust (1977)
отметила наличие, помимо теле-
ангиэктазий, пигментаций, очаж-
ков атрофии в зоне поражения.
Субъективными расстройствами не
сопровождается.
Заболевание чаще возникает у мужчин среднего и пожилого возраста,
но может возникать и у детей до полового созревания (Warin, 1961). Раз-
вивается постепенно, в течение 2—3 лет, а затем существует стационарно
в течение всей жизни.
Гистопатология. Гиперкератоз, атрофия мальпигиевого слоя, в верх-
ней трети дермы зона базофильной дегенерации коллагена, отделенная от
эпидермиса узкой полосой неизмененного коллагена, отложение коллоида,
который окрашивается гематоксилин-эозином в розовый цвет. Природа
коллоида неизвестна. Основная часть его—аморфное вещество, по гистохи-
мическим реакциям сходное с амилоидом, но среди него встречаются фила-
менты, характеристика которых отличается от таковых при амилоидозе
(Hashimoto et al., 1972). По мнению Ebner и Gebhart (1978), коллоид при
ювенильной форме вероятнее всего эпидермального, а при заболевании,
развивающемся у взрослых,—дермального происхождения.
Дифференциальный диагноз. Дифференцировать коллоид милиум не-
обходимо с сенильным эластозом, белыми угрями, амилоидозом, эластои-
дозом кожи Favre—Racouchot, псевдоксантомой эластической.
Лечение. Фотозащитные кремы.
224
ЛИТЕРАТУРА
Brust В. Ober das Pseudomilium colloidale.— Derm. Mschr., 1977, 163, 6, 484 -489.
Hashimoto K-, Katzman R. L., Kang A. H. et al. Electron microscopical apa bio-
chemical analysis of colloid milium. Arch. dermatoL, 1975, 111, 1, 49—59
КОНДИЛОМЫ ОСТРОКОНЕЧНЫЕ ГИГАНТСКИЕ (condylomata acu-
minata giganticum)
Этиология и патогенез. Остроконечные кондиломы являются инфек-
ционным заболеванием, вызываемым вирусом папилломы человека. Пере-
дается при прямых контактах. Морфологически вирус неотличим от вируса
бородавок, но, как предполагается (Robinson и Heath, 1983), не идентичен
прежде всего в отношении иммунных реакций, вызываемых ими. По мне-
нию Oriel (1971), не получено данных и в отношении эпидемиологической
связи между обычными бородавками и остроконечными кондиломами, но
сочетание их, видимо, не является редкостью (Коган А. И. и Козлов А. И.,
1980). У больного, описанного Harvey и соавт. (1983), был выделен вирус
человеческой папилломы 6 ДНК (Gissman et al., 1982), что дало основание
авторам рассматривать гигантские кондиломы Buschke—Loewenstein как
вариант остроконечных кондилом. По мнению Harvey и соавт., требует
выяснения вопрос, существует ли особый субтип этого вируса, вызываю-
щий атипичные варианты кондилом, или они обусловлены неидентифици-
рованным вирусом папилломы. Gross (1984) в очагах экзофитно растущих
кондилом в перианальной области с помощью иммунофлуоресцентно-серо-
логических исследований получил данные о наличии 6-го и 1-го типа ви-
русов человеческой папилломы. Особенностью этого наблюдения было об-
наружение электронно-микроскопически повышенного количества вирус-
ных частиц, возможно, за счет сочетанной инфекции, дефекта Т-клеточного
иммунитету, вызванного наркоманией. Развитию гигантских форм способ-
ствуют трйвматизация, гипергидроз, беременность, иммунные нарушения,
наличие фимоза.
Клиника. Гигантские остроконечные кондиломы возникают вследст-
вие слияния отдельных кондилом, что приводит к формированию бородав-
чатых о'чагов розового или красного цвета, мягкой консистенции с доль-
чатой, влажной, мацерированной, нередко с кровоточащими трещинами,
поверхностью, сходной по внешнему виду с цветной капустой (рис. 148,
149). Эта форма остроконечных кондилом (карциномоподобные остроконеч-
ные кондиломы Buschke—Loewenstein) встречаются очень редко. Распола-
гается в основном на половых органах и вокруг ануса. Отмечена локали-
зация на плече (Greenberg и Wallace, 1963), множественные гигантские кон-
диломатозные очаги на руках и ногах у больной лимфедемой (Mulin et al.,
1983). Считается (Machacek и Weakley, 1960; Becker et al., 1969), что ги;
гантские кондиломы значительно чаще встречаются у мужчин, чем у жен-
щин.
Течение неопределенно долгое, иногда многолетнее. Прогноз обычно
благоприятный.
15 Клиническая дерматология 225
Towpic и Chroscicki (1979) при тщательном гистологическом исследо-
вании не обнаружили опухолевой метаплазии в очагах гигантских конди-
лом, существовавших в течение 25 лет. Возможна, чаще у лиц пожилого
возраста, трансформация в карциному, на что может указывать появление
болезненности, внезапный рост. Е. ЗабеЛь и соавт. (1960) описали развитие
бородавчатого рака через 16 лет после возникновения остроконечных кон-
дилом. Но длительность процесса не всегда имеет при этом особое значение.
Поэтому необходимо тщательное клиническое наблюдение и гистологиче-
ское исследование во' всех случаях гигантских остроконечных кондилом.
Lever (1975) обращает внимание на трудности раз1*раничения гистоло-
226
гически гигантских остроконечных кондилом и спиналиом и отмечает не-
обходимость повторных биопсий в сомнительных случаях, чтобы решить
вопрос о диагностике. По мнению Machacek и Weakley (1960), рецидиви-
рующий характер гигантских остроконечных кондилом мог бы служить
основанием для рассмотрения их как формы спиналиомы. Simmons (1983),
напротив, полагает, что гигантские кондиломы могут разрушать соседние
ткани, но гистологически остаются доброкачественными. Lever (1983) рас-
сматривает гигантские остроконечные кондиломы Buschke—Loewenstein не
как форму кондилом, а как веррукозную карциному.
Дифференциальный диагноз. Дифференцировать гигантские остроко-
нечные кондиломы необходимо от широких кондилом, веррукозных форм
кожного рака.
Гистопатология. Небольшое утолщение рогового слоя, паракератоз,
резко выраженный акантоз и папилломатоз, вакуолизация эпителиаль-
ных клеток. Немногочисленные митозы и отсутствие инвазий отличает
гигантские кондиломы от эпителиом.
Лечение. Хирургическое с гистологическим исследованием.
ЛИТЕРАТУРА
Забель Е., Люговска Д., Пибора Л., Карась 3., Бородавчатый спиноцеллюляр-
ный рак, развившийся на месте остроконечных кондилом. Вести, дерматол. ч венерол., -
1980, 12, 51—52.
Каган А. И., Козлов А. И., К вопросу об остроконечном кондиломатозе. Вестн.
дерматол. и венерол., 1980, 10, 53.
Robinson I. V. Е., Heath R. В. Virus diseases and the skin (hurchill Livingstone),
Edinburgh London, Melbourne, New York, 1983,
Simmons P. D. Genital warts.— Int. J. dermatol., 1983, 22, 7, 410—414.
КРАПИВНИЦА ПИГМЕНТНАЯ (urticaria pigmentosa)
Синоним: мастоцитоз кожи. °
Этиология и патогенез. По выражению Demis (1983), большинство
еще недавно существовавших теорий (инфекционная, метаболическая, то-
ксическая, воспалительная) зачахли в ожидании подкрепляющих данных.
Fischer (1962) рассматривает это заболевание как следствие доброкачест-
венной или злокачественной пролиферации ретикулоэндотелиальной систе-
мы. Bigelow (1961), Bear и Cunljffe (1979) проводят аналогию между масто-
цитозом и гистиоцитозами. Demis также указывает на сходство между пиг-
ментной крапивницей и гистиоцитозом X (пролиферативный характер, ин-
фильтраты из зрелых элементов, преимущественное развитие в детском
возрасте, возможность системного и лейкемического поражения и др.),
отмечая в то же время существенные различия между ними: худший прог-
ноз гистиоцитоза X при развитии у детей, наличие солитарных очагов на
коже при мастоцитозе, а в костях—при гистиоцитозе X и др. О возможной
роли генетических факторов свидетельствуют наблюдения семейных слу-
чаев (Gross и Hashimoto, 1964; James и Eady, 1981; Shaw, 1968; Jelinek,
1970 и др.), в том числе и у однояйцевых близнецов (Weber, 1973; Rockoff,
15* 227
1978.). Gay и соавт. (1970), найдя дискордантность у монозиготных близ-
нецов, усомнились, что пигментная крапивница представляет собой гене-
тическое расстройство, По-видимому, количество наблюдений слишком
мало, чтобы сделать определенное заключение, и вывод некоторых авторов
об аутосомно-доминантном типе наследования пигментной крапивницы
может быть верным только для отдельных родословных. По мнению Free-
man (1967), тучные клетки при пигментной крапивнице не отличаются по
структуре и процессу дегрануляции от имеющихся в непораженной коже.
Однако Naveh и соавт. (1975) обращают внимание на такие особенности
ультраструктуры как неравномерность очертаний, увеличение количества
длинных интердигитирующих микровиллей и склонности к агрегации туч-
ных клеток. Ludatscher и соавт. (1977) сходные изменения наблюдали лишь
при диффузных кожных и системных вариантах, в то время как при со-
литарной мастоцитоме структура тучных клеток была такой же, как и
в норме.
По мнению Selmanowitz и соавт. (1970), пигментную крапивницу сле-
дует рассматривать как состояние с мультифакториальным наследованием.
Затруднения при решении вопроса о роли наследственных факторов при
этом заболевании можно объяснить и трудностями получения надежной
информации о наличии (или отсутствии) поражения в семьях, учитывая,
что у многих процесс мбжет проявляться в виде немногочисленных, изо-
лированных элементов, исчезающих с возрастом, на которые больные,
а тем более их родственники, мбгли не обращать внимания.
♦ Клиника. Заболевание характеризуется большой вариабельностью
клинической картины. Обычно (Rinderman и Nasemann, 1973; Demis, 1983
и др.) различают доброкачественные (кожные и системные) и злокачествен-
ные (лейкемические) формы. Из доброкачественных форм наиболее частой
является мелкопятнистая и папулезная форма, которая чаще развивается
у детей, но может возникать и у взрослых. Проявляется в виде множествен-
ных небольших пятен и узелков в основном на туловище, в меньшей степе-
ни на конечностях, в том числе на ладонях и подошвах, на лице, редко в
полости рта, округлых или овальных очертаний, своеобразного краснова-
то-коричневатого цвета. Они незначительно возвышаются над уровнем
кожи, но при даже умеренном трении краснота и отечность усиливаются,
так что элементы принимают отчетливый волдырный характер, появляется'
или усиливается зуд, в обычных условиях незначительный или отсутствую-
щий. Пятна могут сливаться. Одновременно могут быть и узловатые образо-
вания, обычно немногочисленные, с более интенсивной пигментацией (риС.
150, 151, 152, 153). Может быть повышенной кровоточивость папуло-но-'
дозных элементов при травме. Так, Vilanova и соавт. (1961) наблюдали
9-месячного ребенка, которому пришлось проводить переливание крови
из-за сильного кровотечения после биопсии, вызванного циркулирующим
гепарино-подобным фактором. В редких случаях поверхность отдельных
узловатых элементов может быть веррукозной. Описываются случаи пиг-
ментной крапивницы, очень сходные с ксантомами, под прежним названием
пигментной крапивницы—ксантелазмоидеа. Так, Rasmussen (1978) наблю-
228
Рис. 149. Пигментная
крапивница пятнисто-узелковая.
Рис. 150. Пигментная
крапивница (телеан-
гивктатическая форма).
Рис. 151. Пигментная
крапивница (крупно-
пятнистая форма).
дал и а шее и туловище у 3-месячного ребенка Немногочисленные папулез-
ные высыпания овальных очертаний, с наибольшим диаметром около 1 см.
В зоне каждой папулы было 10—12 изолированно расположенных округ-
лых точечных пятен желтого цвета, размерами от 0,5—1 мм, что придавало
элементам сетчатый характер. При трении появлялась волдырная реак-
ция. Гистологически были обнаружены скопления тучных клеток. Буллез-
ная форма пигментной крапивницы развивается исключительно у детей
грудного или раннего детского возраста. Поданным Dewis и соавт. (1961),
69% всех опубликованных случаев развития пигментной крапивницы до
двухлетнего возраста проявлялись везико-буллезными высыпаниями. Cap-
lan приводит несколько меньшие цифры—57%. Они обычно встречаются
наряду с другими проявлениями, чаще с нодулярными (множественными
или изолированными), но могут им и предшествовать (Orkin et al., 1970).
Robinson и соавт. (1962) ни в одном из наблюдавшихся ими случаев не наб-
людали, чтобы развитие пузырей предшествовало пятнисто--узелковым вы-
сыпаниям. Буллезные высыпания существовали не более 3 лет с момента их
возникновения. Klaus и Winkelmann (1962) не рассматривают пузырные
проявления как особый морфологический вариант заболевания, если они
встречаются с другими изменениями, и не придают им прогностической
значимости. Эта форма может проявляться в Виде одиночных или множест-
венных пятнисто-папулезных или узловатых высыпаний, на поверхности
которых под влиянием трения или спонтанно появляются пузыри. Обра-
зование пузырей у детей объясняется большим, чем у взрослых, содержа-
нием в очагах тучных клеток и гистамина в их гранулах (Grijneberg et al.,
1967). Буллезные реакции могут развиваться на фоНе диффузной красно-
вато-коричневатой пигментации кожи, которая слегка инфильтрирована,
лихенифицирована, выглядит шагреневидной (Haensch и Ippen, 1968).
За счет инфильтрации может возникать повышенная складчатость в под-
мышечных и пахово-бедренных складках (Verbov, 1971). Пузыри могут
приобретать геморрагический характер (Broderman, 1978). Описано раз-
витие буллезных высыпаний у детей без других видимых признаков пиг-
ментной крапивницы, так называемый буллезный мастоцитоз (Robins,
1954; Miller и Shapiro, 1965, Plasmejer et al., 1972, и др.), в том числе в
сочетании с приступами бронхиальной астмы (Мс Кее et al., 1966). Они
могут начинаться с приступов крапивницы, Покраснения кожных покро-
вов, с последующим развитием буллезных элементов. По всей вероятности,
в случаях буллезного мастоцитоза, когда обширные буллезные высыпания
являются основным клиническим проявлением, речь идет о диффузном
варианте пигментной крапивницы, но со столь незначительной инфильт-
рацией кожи, что она выявляется преимущестйенно только при биопсии.
Диагностика в таких случаях затруднительна, так как поражение может
быть сходным с другими буллезными дерматозами, контактным дерматитом
(Welch et al., 1983). Солитарные мастоцитомы почти всегда развиваются
только у детей, составляя 10—15% всех случаев пигментной крапивницы
(Johnson и Helwig, 1961). Существуют с рождения или появляются в пер-
вые недели или месяцы жизни, редко позже (например Baraf и Shapiro,
230
1969. наблюдали развитие одиночной мастоцитомы у '16-летнего мальчика).
Описаны случаи мастоцитомы и у взрослых (Jacobs, 1958). Клинически
солитарная мастоцитома имеет вид небольшого опухолевидного образова-
ния или нескольких тесно расположенных узелков, при трении поверхности
которых отмечается положительный феномен Уины. У детей характерным
признаком является образование пузырей. Birt-и Nickerson (1959) наблю-
дали развитие пузыря после трения солитарной мастоцитомы у ребенка,
появление быстро угасающей генерализованной волдырной сыпи. После
травмы может быть покраснение лица или других участков кожного покро-
ва, сильный зуд. Располагается преимущественно на туловище и конечно-
стях. Обычно не связана с распространенными формами крапивницы и
поражением внутренних органов, но Lantis и Koblenzer (1969) сообщили
о ребенке, у которого на первом месяце жизни появился одиночный очаг
поражения с образованием пузырей, а в 6 месяцев развилась генерализо-
ванная пятнисто-папулезная сыпь. Могут наблюдаться и везикулезные
реакции. Barth и Haustein (1978) не наблюдали прогрессирования после
тотального удаления мастоцитомы. Редкой формой, наблюдающейся обыч-
но у взрослых, является телеангиэктатическая (teleangiectasia macularis
eruptiva perstans) с множественными высыпаниями красновато-коричне-
ватых мелких пятен с телеангиэктазиями на поверхности. Этот вариант
считается потенциально системным (Nickel, 1967, Allen, 1978). Диффузно-
эритродермический мастоцитоз у взрослых, в отличие от детей, протекает
обычно без пузырных реакций, но со значительно более выраженной ин-
фильтрацией. В зоне обширных очагов поражения могут возникать пиг-
ментации, телеангиэктазии, нодулярные, опухолевидные элементы, тре-
щины, эрозирование, очажки кровоизлияний, пурпура. К наиболее ред-
ким вариантам диффузно-инфильтрирующего мастоцитоза у взрослых отно-
сится пахидермический с резкой диффузной инфильтрацией кожи, которая
приобретает сходство с кожей крокодила. У больной этой формой пиг-
ментной крапивницы, описанной Meneghini и Angelini (1980), обнаружива-
лись, кроме того, многочисленные кожные опухоли, сходные с molluscum
fibrosum, большие комедоны, высокий уровень гистамина в крови, висце-
ральная патология (увеличение печени, тучноклеточная инфильтрация
костного мозга, желудочно-кишечные расстройства, спленомегалия с гемо-
литической анемией и лейкопенией, что вызвало необходимость спленэкто-
мии). Заболевание появилось в двухмесячном возрасте, постепенно про-
грессировало, но, несмотря на системность поражения, больная сохраняла
трудоспособность в 52-летнем возрасте. Диффузно-эритродермический ва-
риант с пахидермией описан и у детей (Потоцкий И. И., 1970). И. И. По-
тоцкий наблюдал опухолевидную форму мастоцитоза у пятилетней девочки,
у которой были обширные узловатости на лице, туловище и конечностях.
На лице они сливались, образуя facies leonina. Летальный исход наступил
от злокачественного мастоцитоза. К числу редких вариантов относится
форма пигментной крапивницы, высыпания при которой разрешались с
образованием атрофии типа анетодермии (Колпаков Ф. И. и соавт. 1984,
Thivolet et al., 1981 и др.).
231
Течение и прогноз различны, в основном в зависимости от сроков раз-
вития и типа заболевания: чем раньше развилось заболевание, тем про-
гноз лучше. По мнению Sochocky (1982), мастоцитоз, развившийся на
первом году жизни, в 70% случаев исчезает до периода полового созрева-
ния. Однако при диффузных буллезных формах считается (Orkin и соавт.,
1970 и др.), что прогноз при более позднем появлении у детей пузырей луч-
ше, чем в случае развития их вскоре после рождения. Подтверждением
этому может быть наблюдение Fromer и Jaffe (1973) ребенка, у которого
везикулезные, а затем пятнистые высыпания развились через 3 недели пос-
ле рождения, а в пять с половиной лет—признаки системности процесса
(гепатоспленомегалия, острая лимфобластная лейкемия). Описано (Ba-
kos et al., 1972) развитие системного мастоцитоза и в случаях врожден-
ной пятнистой крапивницы, до периода полового созревания протекавшей
спокойно, а затем стали появляться туморозные элементы на различных
участках кожного покрова, кровоточившие после травмирования, симпто-
мы, обусловленные повышенным выделением гистамина, поражение пече-
ни, лимфоузлов, костей. В среднем системные поражения наблюдаются
примерно у 10% больных, по Rohner и Rodefmund (1981),—значительно
чаще. По данным литературы (Caplan, 1963,) системные формы обычно
протекают доброкачественно, но у г/3 больных может развиться лейке-
мия, и эта вероятность выше при развитии заболевания у взрослых. При
наблюдении в среднем в течение 28 месяцев группы больных из 26 чело-
век болезнь закончилась летально в 6 случаях, в одном—из-за прогрес-
сирующего течения мастоцитоза, в одном—от первичного амилоидоза, в
одном—от лимфомы и в 3—от злокачественных опухолей эпителиального
происхождения (Webb et al., 1982). Прогноз солитарных мастоцитом и
диссеминированной пигментной крапивницы, возникшей у детей до полового
созревания, вполне хороший. Если генерализованная пигментная крапив-
ница возникла в период полового созревания, в 56% случаев она регрес-
сирует, в оставшихся—асимптомна. Нодулярные элементы регрессируют
раньше, чем пятнисто-папулезные (Klaus и Winkelmann, 1962). Считается
(Lantis и Koblenzer, 1969), что около 75% случаев развивается до периода
полового созревания, из них 1/3—до 6-месячного возраста, 25%—у взрос-
лых от 15 до 40 лет. Описаны врожденные случаи Davis и Weisman (1959)
и другими авторами. Caplan (1963) отметил развитие кожных изменений
до двулетнего возраста у 75% из 112 больных. На основании оценки дан-
ных больших выборок различных авторов Fine (1980) считает, что заболе-
вание развивается до этого возраста по меньшей мере у 50% больных. Из
139 больных, наблюдавшихся Demis, у 18 заболевание существовало с
рождения, у 49—развилось в течение первых 12 недель, у 20—с 3—12 ме-
сяцев. После 14-летнего возраста заболели остальные 20 человек.
Что касается отдельных форм, то. наиболее часты системные поражения
при диффузно-инфильтративных - вариантах, особенно при эритродерми-
ческом мастоцитозе у детей (Hoffbauer, 1974). У больных диссеминирован-
ным мастоцитозом, особенно под влиянием гистамин-высвобождающих
средств (аспирин, алкоголь, кодеин, полимиксин, витамин В, солнечные
232
ожоги, горячая вода и др.), могут быть проявления гистаминового шока
(покраснение лица, зуд, тахикардия, падение давления, головная боль,
желудочно-кишечные расстройства). Особенно это опасно у детей раннего
возраста.
Описываются ассоциации пигментной крапивницы с различными па-
тологическими состояниями: дефицитом антителообразования (Buchholz
et al., 1979), ювецильной ксантогранулемой (De Villezet al., 1975), скле-
родермией (Basler й Harrell, 1974), раком желудка (Lippert и Ketels-Har-
ken, 1974), анетодермией (Carr, 1971), рецидивирующим опоясывающим
лишаем и тяжело протекающим полирадикулоневритом (Kalz, 1975).
Одними из наиболее частых системных изменений являются пораже-
ния костей, возникающие вторично за счет проникновения тучных кле-
ток из костного мозга (Sostre и Handler, 1977). Эти изменения обычно на-
блюдаются у взрослых. Так, у 8 из 31 больного, у которых рентгенологи-
чески выявлялись зоны уплотнения и просветления костей (Rodermund
et al., 1978), был только один ребенок. Костные изменения, диффузные
или локальные, проявляются в виде очагов остеопороза или остеосклероза,
болей в костях, возможны патологические переломы. Demis (1983) считает,
что более вероятная частота костных изменений составляет 10—15%, а
не 30, как сообщалось в литературе. Из других систем поражается печень,
селезенка, желудочно-кишечный тракт, лимфоузлы (Rodermund et al.,
1978; Rohner и Rodermund, 1981 и др.). Изменения в желудочно-кишечном
тракте могут быть в виде тошноты, диареи, пептической язвы, болевого
синдрома. Гепатоспленомегалия, по данным Demis (1963), наблюдается в
10—15% случаев пигментной крапивницы, по мнению Sochocky (1982),—
в 48%. Могут наблюдаться изменения крови (анемия, эозинофилия, лейко-
пения, тромбоцитопения). Лейкемии, однако, встречаются редко (Parker и
Odland, 1979). Cooper и соавт. (1982) описали 6 больных пигментной кра-
пивницей, у которых развились злокачественные гематологические забо-
левания (лимфогранулематоз, лимфоцитарная лимфома). Причем, ни у
одного из них ранее не было признаков системного мастоцитоза. Высыпа-
ния были пятнисто-папулезные или бляшечные. У 2 больных—ювениль-
ная форма. Системные формы мастоцитоза могут протекать и без пораже-
ния кожи. Vam и соавт. (1980) указывают на значимость в этом случае
эозинофилии в крови и тканях и считают, что во всех случаях неясной эози-
нофилии необходимо исключение системного мастоцитоза (исследование
костного мозга, биопсия печени, селезенки, лимфоузлов).
Гистопатология. Повышенное содержание пигмента в эпидермисе,
диффузные или расположенные вокруг сосудов и придатков кожи инфильт-
раты из тучных клеток (Мельникова В. С. и Локунова Ж- К., 1977).
Дифференциальный диагноз. Дифференцировать пигментную крапив-
ницу необходимо с крапивницей с пигментацией, лекарственными сыпями,
ретикулезом кожи, гистиоцитомами, гистиоцитозом X, гемосидерозами,
пигментными невусами, ксантоматозом, пятнисто-папулезными сифили-
дами.
Лечение: антигистаминные средства, в малых дозах резерпин, сту-
герон, Пува-терапия (данные предварительные). 2зз
ЛИТЕРАТУРА
Колпаков Ф. И., Прохоренков В. И., Аникина Е. И. Семейная атрофическая пиг-
ментная крапивница. Вести, дерматол. и венерол., 1984, 2, 47—48.
Мельникова В. С., Л оку нова Ж- К- К вопросу о клинике, лечении и гистохимиче-
ской характеристике полисахаридного и белкового компонентов тучных клеток кожи боль-
ных мастоцитозом. Вести, дерматол. и венерол., 1977, 8, 27—30.
.Barth J., Haiistein U. F. BuIIoses Mastocytom dem Sauglings.— Derm. Monatschr.,
1978, 164, 114, 298—301.
Basler R. S. W., Harrell E. R. Urticaria Pigmentosa associated with scleroderma.—
Arch, dermatol., 1974, 109, 3, 393—394.
Beare J. M., Cunliffe W. J. Mastocytoses. In: Textbook of dermatology Ed. by A.
Rook et al, 3-ed., Blackwell secientific publtcutions, Oxford, 1979, v. r., 1587—1594.
Brodemann M. Mastocytosis cutanea diffusa generalisata mit grossfeldigen Sugilla-
tionen und hamorrhagischen Blasenschuben.— Dermatol.. Monatschr., 1978, 164, 7, 497—603.
Cooper A. J., Winkelmann R. K-, Wiltsie J. C. Hematologic malignancies occuring
in patients with urticaria pigmentosa.— J. American Acad. Derm., 1982, 7, 2, 215—220.
Hofbauer M. Uber eine Famifie mit Mastzelfenerythrodermie.— Hautarzt, 1974, 25,
5, 246—249.
James M. P., Eady R. A. J. Familial urticaria pigmentosa with giant mast cell gra-
nules.—1981, 117, 11, 713—718.
Katz F. Generalized mastocytosis Relapsing herpes zoster and polyradiculoneuritis.—
Dermatologies, 1975, 150, 6, 366—368.
Lippert H.D., Ketels—Harken H. Kasuistischer Beitrag zur Mastocytose beim Er-
wachsenen.^ Hautarzt, 1974, 25, 8, 398—403.
Meneghini C. L., Angelini G. Systemic mastocytosis with diffuse crocodile—like
pachydermic akin, pedunculated pseudofibromas and comedones.— Brit. J. Derm., 1980,
103, 5, 329—334.
Nickel W. R. Clinical spectrum of mastocytosis in man.— Arch, dermatol., 1976, 96,
4, 364—366.
Parker F., Odland G. F. The mastocytosis syndrome. In: «Dermatology in general Me-
dicine* sec. ed. Ed T. B. Fitzpatrick et af. Me Graw—Hil book company, N. Y., 1979, 772—
783.
RodermundO. S., Hauser W. Burkhardt R., Grips К-H., Stein G. Veranderungen
am Knochenmark bei Mastogytose.— Derm. Wschr., 1978, 164, 7, 493—496.
Rohner H. G., Rodermund О. E. Mastozytose und Gastrointestinaftrakt.— Hautarzt,
1981, 32, 12, 611—616.
КРАСНАЯ ЗЕРНИСТОСТЬ НОСА (granulosis rubra nasi)
Этиология и патогенез. Частые семейные случаи свидетельствуют о
генетической детерминации этого дерматоза. Предполагается аутосомно-
доминантный, реже аутосомно-рецессивный тип наследования со снижен-
ной пенетрантностью (Witkowski и Prokop, 1974). В развитии заболевания
придается значение вегетативной дисфункции сосудов (Gertler, 1970; Pastin-
szky и Racz, 1974).
И. А. Левин (1931) рассматривал красную зернистость носа как тубер-
кулид, однако, точка зрения о туберкулезнойагиологии красной зернисто-
234
' сти носа, по мнению С. А. Глауберзона и П. Г. Каминского (1950), Л. Н.
Машкиллейсона (1960), не имеет достаточных оснований^
Клиника. Вначале появляется розацеаподобная синюшная краснота,
в части случаев с телеангиэктазиями, гипергидроз в области носа, за-
тем высыпание мелких слегка остроконечных узелков от светло-розового
до темно-красного цвета, на поверхности которых иногда обнаруживаются
пузырьки. Комедоны отсутствуют, пустулы встречаются редко. Узелки
могут группироваться, но нб сливакугся. Характерным признаком явля-
ется наличие капелек потаи в виде росы. Изменения распространяются на
щеки, верхнюю губу, подбородок.
Болеют как правило ослабленные дети. Заболевание начинается обыч- .
но в первые годы жизни. В большинстве случаев к Моменту полового созре-
вания самостоятельно регрессирует, но может существовать неопределен-
но долго. Часто у больных обнаруживается гипергидроз, акроцианоз, иног-
да ладонно-подошвенный кератоз.
Гистопатология. Расширение выводных протоков потовых желез, со-
судов, небольшие инфильтраты, преимущественно из лимфоцитов вокруг
сосудов и потовых желез.
Дифференциальный диагноз. Необходимо проводить с мелкоузелковым
саркоидозом, розовыми угрями, себорейной экземой, бромистыми (иодисты-
ми) угрями.
Лечение. Обычно не требуется.
ЛИТЕРАТУРА
Pastinszky /., Racz 1. Hautveranderungen bei inneren Krankheiten. Verlag Volk und
Gesundheit. Berlin, 1974, Bd. 11.
Witkowski R., Prokop 0. Genetic erblicher Syndrome und Missbildungen. Akade-
mie—Ver lag, Berlin, 1974.
КРАУРОЗ ВУЛЬВЫ (kraurosis vulvae).
Синонимы: лейкоплакический вульвит, атрофическая лейкоплакия,
ограниченная склеродермия и др. (всего более двух десятков).
Breisky в 1885 г. впервые описал своеобразное заболевание слизисто-
кожных покровов наружных половых органов у женщин под названием
крауроз вульвы (греч. krauros—сухой).
Этиология и патогенез. До настоящего времени нет единого мнения
относительно причин и механизмов развития крауроза вульвы. Литератур-
ные данные свидетельствуют о многообразии этиологических факторов
крауроза. Большинство авторов защищает эндокринную гипотезу проис-
хождения крауроза вульвы (Декстер Л. И., 1965; Яковлева И. Я- и Штем-
бергМ. И., 1974; Скрипкин Ю. К- и Маркин И. Я-, 1974; Каламкарян А. А.
и Аковбян В. А., 1978; Bretland, 1957; Clare м Mailer, 1967; Pfister et
al., 1974 и др.). С этой теорией согласуется возникновение крауроза вуль-
вы у подавляющего большинства женщин во время климакса или мено-
паузы, а у части больных (у девочек и молодых женщин) после оператив-
ных вмешательств на половых органах. Замечено, что пересадка яичнико-
235
вой ткани положительно влияет на течение крауроза вульвы. Этиологиче-
ская роль воспалительных факторов (трихомоноз, кандидоз), по-видимому,
не так велика, как предполагали ранее (Декстер Л. И., 1965). По нашему
мнению, появление дрожжеподобных грибов на поверхности очагов крау-
роза можно объяснить ослаблением защитных свойств кожи. Нередко крау-
роз вульвы сочетается с лейкоплакией, причем если ранее лейкоплакию
рассматривали как стадию крауроза (Головин Д. И., 1975; Серебров А. М.
1961; Нудольская О. Е., 1962; Яковлева И. А. и Штёмберг М. Й., 1974),
то в настоящее время наблюдается тенденция рассматривать эти заболе-
вания как самостоятельные, которые могут сочетаться или следовать одно
за другим. Некоторые исследователи не исключают возможного участия
иммунопатологических механизмов в патогенезе крауроза вульвы. Суще-
ствует также мнение, согласно которому крауроз вульвы является одной
из форм склерозирующего и атрофического лишая с локализацией на ге-
ниталиях (Груздова А. И., 1969; Брайцев А. В. и Марзеева Г. И., 1973;
Laymon, 1951; Steigleder и Raab, 1961; Lutz, 1963; Lever и Schaumburg—
Lever, 1975, и др.). Л. Н. Машкиллейсон (1965) считает крауроз вульвы
первичной атрофией вульвы. Мы считаем, что, несмотря на гистологическую
схожесть между склероатрофическим лихеном и краурозом вульвы, их
следует рассматривать как раздельные самостоятельные заболевания.
Клиника. В начальном периоде заболевания слизисто-кожные покровы
больших и малых половых губ выглядят отечными, гиперемированными.
Весьма выражен зуд, принимающий порой нестерпимый характер. В даль-
нейшем (период атрофии) слизисто-кожные покровы наружных гениталий
становятся сухими, истончаются, теряют эластичность, приобретая блед-
»но-синеватый цвет или оттенок желтого воска. Кожа теряет блеск, стано-
вится шероховатой (вид сморщенной папиросной бумаги, смятого перга-
ментного листа), волосы на поверхности больших половых губ выпадают.
Наблюдается атрофия малых половых губ, клитора и частично больших
половых губ. Нередко в процесс вовлекается кожа промежности и пер ка-
нальной области. Период склероза характеризуется полной атрофией
наружных половых органов, развитием рубцового склероза всех частей
вульвы. Кожа становится не эластичной, плотной. Вход во влагалище рез-
ко суживается,' иногда до полной облитерации. Окраска—белесоватая
или пятнистая с пигментацией или телеангиэктазиями. На протяжении
всего развития болезни зуд и парестезии являются настолько
выраженными, что нередко приводят к развитию невротических состояний.
Прогноз для жизни большей частью благоприятный, но выздоровления
не наступает. Крауроз вульвы относят к так называемым предраковым
заболеваниям. Крауроз с лейкоплакией относят к облигатной форме пред-
рака, а крауроз без лейкоплакии—к факультативной форме. По данным
Яковлевой И. Я. и Штемберг М. И. (1,974), рак наблюдается у 19,2% боль-
ных со смешанной картиной болезни (крауроз в сочетании с лейкоплакией);
малигнизируются в основном очаги /лейкоплакии, а не крауроза.
Гистопатология. Сочетание атрбфии покровного эпителия с явлениями
гиперкератоза, характерна гидропическая дегенерация базального слоя
2зе
иногда с образованием субэпидермальных пузырей. В базальном слое мо-
жет наблюдаться накопление пигмента. В соединительной ткани располага-
ются инфильтраты, состоящие из лимфоцитов, тучных клеток и лейкоци-
тов. Эластическая ткань обнаруживается лишь в виде небольших обрыв-
ков и скрученных нитей. Коллагеновая ткань склерозирована. Электрон-
но-микроскопические исследования выявили выраженную вариабельность
толщины коллагеновых^ волокон, увеличение межволоконных промежут-
ков, фрагментацию волокон. В клетках эндотелия капилляров дермы—
мощная сеть микроканальцев, соединяющихся с пограничной мембраной.
Дифференциальный диагноз. Следует проводить с нейродермитом, гриб-
ковыми поражениями вульвы и влагалища, эссенциальным зудом вульвы,
склерозирующим и атрофическим лишаем, лейкоплакией.
Лечение. Малоэффективно. Эстрогены парентерально противопоказа-
ны, так как их применение может стимулировать переход крауроза в рак.
Полезны антигистаминные (супрастин, димедрол, диазолин), психотроп-
ные (седуксен, меллерил, аминазин и др.) препараты. Определенный тера-
певтический эффект может быть получен от спирт-новокаиновой блокады
(смесь этилового спирта 96° с 0,25% раствором новокаина в соотношении
1:4). Следует учесть, что кортикостероидные кремы, содержащие фтор,
нередко ведут к развитию атрофии кожи, поэтому их длительное применение
вряд ли целесообразно. Полезны кремы с синтетическими гормонами (те-
стостерон-пропионат в концентрации 0,2—0,3% или с метилтестостероном
0,25%) в комбинации с ваннами (хвойные и др.). В. С. Добронецкий и
В. А. Добрынин (1978) отметили высокий лечебный эффект от метилметио-
нинсульфония хлорида (витамин У).
ЛИТЕРАТУРА
Каламкарян А. А., Аковбян В. А. Современное состояние вопроса о краурозе вуль-
вы.— Вести, дерматол. и венерол., 1978, 12, 29—33.
Скрипкин Ю; К-, Маркин И. Я- К патогенезу и лечению больных краурозом вуль-
вы.— Сов. мед., 1974, 11, 148—149.
Яковлева И. А., Штемберг М. И. Лейкоплакия и рак вульвы.— Арх. патол.,
1974, 2, 59—63.
Pfister R. Therapie der Kraurosis vulvae.— Med. Klin., 1974, 69, 2, 74—75.
КРАУРОЗ ПОЛОВОГО ЧЛЕНА (kraurosis penis).
Синонимы: атрофический прогрессирующий баланит с краурозом.
В 1908 г. Delbanco описал трех больных с атрофией и сморщиванием
головки полового члена и внутреннего листка крайней плоти, назвав это
заболевание крауроз пениса и крайней плоти. Fournier еще в 1866 г. впер-
вые использовал термин «интерстициальный или глубокий баланит».
Этиология и патогенез. Наши наблюдения позволяют согласиться с
мнением тех исследователей, которые считают крауроз полового члена
полиэтиологическим заболеванием, обусловленным различными факторами:
хроническими воспалительными процессами, раздражением химическими,
механическими- воздействиями, рецидивирующим генитальным герпесом,
237
общим атеросклерозом, диабетом, старческой инволюцией и др. По-види-
I мому, у ряда больных крауроз является проявлением склероатрофического
лихена, но нельзя согласиться с мнением Ravits и Welsh (1957), Potter
(1959), Laymon (1961), Sonnichsen и Zabel (1976), Max (1975), Lever и
Schaumburg—Lever .(1975) о том, что крауроз полового члена следует рас-
сматривать только как форму склероатрофического лихена. По нашему
мнению, крауроз полового члена является самостоятельной нозологиче-
ской формой с первичной атрофией тканей головки полового члена. Fischer
и Nikolowski (1958) различают три разновидности крауроза полового чле-
на: 1) спонтанно возникающая, описанная Delbanco (1908); 2) послеопе-
рационная, описанная Stiihmer (1928); 3) после воспалительных прочее-1
сов, описанная Griitz (1937). Имеются все основания добавить еще одну,
четвертую (полиэтиологическую) разновидность—после воздействия мно-
гообразных факторов: вирусной герпетической инфекции, диабета, атеро-
склероза, мастурбации, кандидозной инфекции полового члена, старческой
инволюции и др.).
Клиника. Крауроз полового члена возникает преимущественно в воз-
расте 40—60 лет (Ломыскин А. И., 1976). Поражение локализуется на
головке полового члена, внутреннем листке крайней плоти и венечной бо-
розде. Во многих случаях заболевание начинается с уменьшения эластич-
ности, сухости внутреннего листка крайней плоти; последняя утолщена
и уплотнена до состояния невоспалительного фимоза с беловато-атрофиче-
скими блестящими участками. Головка полового члена поражается диффуз-
но (реже процесс носит очаговый характер), принимает серовато-белую
окраску, в некоторых случаях с голубоватым оттенком. В дальнейшем
появляются атрофия и сморщивание головки, мелкое шелушение, телеан-
гиэктазии; кожа головки на ощупь становится мягкой, тонкой, легко со-
бирается в мелкие складки. На препуции и уздечке часто развиваются
трещины. Мы наблюдали больно'го, у которого атрофические изменения
сопровождались геморрагическими пузырями. У отдельных больных могут
наблюдаться кольцевидный склероз в области бороздки и стеноз наружного
отверстия уретры, затрудняющий мочеиспускание. В части случаев раз-
витию крауроза предшествуют воспалительные процессы на головке и на.
внутреннем листке крайней плоти, после разрешения которых через раз-
личные промежутки времени развиваются склероатрофические измене-
ния. Крауроз полового члена сопровождается парестезией и зудом; у от-
дельных больных может протекать без зуда.
Течение заболевания длительное, лечение не всегда эффективно. Опи-
саны единичные наблюдения развития эпителиомы на фоне вторичной лей-
коплакии. .
Гистопатология. Некоторые исследователи считают, что гистологиче-
ские изменения при краурозе пениса такие же, как при краурозе вульвы и
при склероатрофическом лихене. В начальной стадии отмечается некото-
рое утолщение рогового и мальпигиева слоев эпидермиса с участками пара-
кератоза; в дальнейшем развивается атрофия. В дерме—воспалительно-
атрофические изменения: эластическая ткань обнаруживается лишь в
238
виде небольших обрывков и скрученных нитей; коллагеновая ткань ока-
зывается склерозированной. Придатки кожи (железы) отсутствуют.
, Дифференциальный диагноз. Следует проводить со склеротическим и
атрофическим лихеном, с красным плоским лишаем, первичным сифилити-
ческим баланитом Фольмана, эритроплазией Кейра, с хроническим ограни-
ченным доброкачественным плазмоцитарным баланопоститом.
Лечение. Из общей терапии рекомендуются мужские половые термо-
ны (инъекции тестостеронацетата 3 раза в неделю по 10 мг), концентрат
витамина Е (по 1 чайной ложке 2 раза в день), витамин А (внутрь по 100000
ЕД в сутки) в течение 1,5—2 месяцев. Назначают биогенные препараты:
лидаза (15 инъекций по 64 ЕД через день), стекловидное тело (20 инъекций
по 2 мл ежедневно). Спазмолитические средства: ангиотрофин, галидор,
компламин, андеколин, падутин. Из физиотерапевтических средств исполь-
зуют горячие ванночки для полового члена, местную и рефлекторную диа-
термию, электрофорез с лидазой и йодистчм калием. Кортикостероидные
и смягчающие мази.
ЛИТЕРАТУРА
Каламкарян А. А., Аковбян А. И. Современное состояние вопроса о краурозе
вульвы.— Вестн. дерматол. и венерол., 1978, 12, 29—33.
Ломыскин А. И. Склероатрофические дерматозы наружных гениталий у мужчин.—
Вестн. дерматол. и венерол., 1976, 7, 68—73.-
Харитонов В. А., Кудлой Л. К-, Григорьева Г. Н. Случай крауроза полового
члена.— Вестн. дерматол. и венерол., 1977, 1, 62—63.
Sonnichsen N., Zabel'R. Hautkrankheiten. Diagnostik und ТЬегарЦ.— Leipzig, 1976.
КРИОГЛОБУЛИНЕМИЯ (trioglobulinemia).
Термин «криоглобулины» был введен в 1947 году Lerner и Watson для
группы белков с общим свойством выпадать в осадок при охлаждении сы-
вороток ниже 37°. При подогревании сыворотки до 37° преципитат раство-
ряется, при охлаждении появляется вновь. Вследствие чувствительности
этой группы белков к холоду они были названы «крибглобулинами», а на-
личие криоглобулинов в крови «криоглобулинемией».
Этиология и патогенез. Полагают, что образование крибглобулина
может быть результатом инфекции, злокачественной опухоли, цирроза
печени и других причин. По мнению Вагг и соавт (1950), криоглобулины;
возможно, продуцируются плазматическими клетками. Высказывается так-
же мнение об иммунной природе проявлений криоглобулинемии (связы-
вают образование криоглобулинов с длительным антигенным раздражением).
В составе криоглобулинов были обнаружены компоненты комплемента.
Это позволяет предположить, что криоглобулины представляют собой им-
мунные комплексы (Cream, 1972; Bruet et al., 1974; Franklin, 1971 и др.).
Криоглобулинемия встречается или как первичная (идиопатическая) бо-
лезнь, или как вторичная (симптоматическая), связанная с опухолями—
множественной миеломой, хроническим лейкозом, лимфосаркомой и др.,
или воспалительными заболеваниями—хроническим гепатитом, нефри-
239
том, узелковым периартериитом, ревматоидным артритом, системной крас-
ной волчанкой, лепрой, сифилисом, васкулитами (Шинский Г. Э., 1963;
Stevanovic, 1974; Watanabe et al., 1974; Zinneman и Caperton, 1977; Hase-
gawa и Ke, 1970; Morrison et al., 1977; Lassus, 1969; Fountain, 1978; Jami-
son et al., 1978; Wallington и Reeves, 1978; Peirone, 1978; Martin, 1979 и
Thomas, 1982 и др.). Различают три степени криоглобулинемии: при пер-
вой степени—крио глобулины обнаруживаются в концентрации до 6 мг%;
при второй степени—от 6 до 25мг%; при третьей степени содержание крио-
глобулинов превышает 25 мг%. Криоглобулины могут состоять из одного
класса (простые криоглобулины), а чаще из двух или даже трех классов
иммуноглобулинов (смешанные криоглобулины типа IgG— IgM). Kovary
и соавт. (1980) у больного с пурпурой, неглубокими язвами и ливедо вы-
явили IgG3—каппакриоглобулинемию. Причиной некоторых проявлений
криоглобулинемии (пурпура, васкулиты) считают увеличенную вязкость
крови, содержащей криоглобулин или внутрисосудистое образование его
преципитата. Криопреципитация белков в капиллярах пальцев стоп
вызывает гипоксию тканей фаланг и тромбозы, что ведет к образованию
пузырей и в дальнейшем язв на коже. По сводным литературным данным
(Бережной Ю. 3., 1965; Гесс Л. В. и Костарева Н. И., 1974; Nicolau и
Badanoiu, 1964; Zarzycka—Stuchley, 1968; Garcia—Fountes et al., 1971;
Mayer, 1981; Levo, 1981 и др.), криоглобулинемия I—III степени обнаружи-
вается у 12—20% больных различными кожными заболеваниями. Так,
криоглобулины обнаружены у 40—50% больных системной красной вол-
чанкой, хронической крапивницей и васкулитами; у 1/4 больных склеро-
дермией, хронической дискоидной волчанкой, экземой; у 1/5 больных
псориазом и другими дерматозами. Smith и Arkin (1972) среди 36000 госпи-
тализированных не дерматологических больных у 3°/о обнаружили крио-
глобулинемию.
Клиника. Среди клинических проявлений криоглобулинемии пора-
жения кожи являются наиболее частыми и прогностически серьезными.
Одно из наиболее частых проявлений криоглобулинемии—пурпура, кото-
рая возникает, как правило, на голенях, бедрах, ягодицах, мошонке, ушах
(purpura cryoglobulinemica). Нередко отмечается обильная сыпь, состоя-
щая из петехий, экхимозов (до 1—2 см в диаметре); буллезных элементов,
изъязвлений, покрытых геморрагическими корочками (Harper et al., 1983).
(рис. 154). На участках подкожных кровоизлияний возможны припухлость,
сильные боли, жжение. Другим характерным проявлением криоглобули-
немии на коже являются васкулиты. Циркулирующие в крови крио-
глобулины отлагаются в мелких сосудах кожи и могут вызвать острые рас-
пространенные буллезно-некротические васкулиты с уртикарией, эрите-
мой, пурпурой. Нередко образуются синюшно-красного цвета, плотные
дермальные и гиподермальные узлы размером с горошину или мелкую сли-
ву. На верхушке узелков и узлов образуются пузыри и везикуло-пустулы,
которые подвергаются некрозу с последующим изъязвлением. На коже
туловища и конечностей могут появляться бугорки и отечные бляшки,
склонные к* некрозу, отек частей тела, подвергшихся охлаждению, в осо-
240
бенности ног, иногда в сочетании с ве-
зикуляцией, гангрена пальцев, буллез-
ная многоформная эритема. Общее со-
стояние больных чаще всего удовле-
творительное. Однако описаны боль-
ные, у которых отмечались лихорад-
ка, потеря веса, анорексия, нарушения
общего состояния вплоть до кахексии.
Под влиянием холодного воздуха или
воды (после купания в холодной воде)
у них могут развиться одышка, серд-
цебиение, отек гортани или охлажден-
ных участков кожи, не купирующиеся
антигистаминными препаратами. При
иммунофлюоресцентном исследовании
крови больных, страдающих холодовой
аллергией, было обнаружено отложе-
ние IgG и IgM в сосудах кожи, тогда
как в коже здоровых людей они отсут-
ствовали. Присутствие иммуноглобули-
нов тормозит СОЭ, препятствует прове-
рит. 153. Криоглобулинемия. дению электрофореза, мешает постанов-
ке осадочных реакций, в том числе на
сифилис. Все исследования в таких случаях нужно проводить в термостате.
Гистопатология. Картина капиллярита с умеренной круглоклеточной
инфильтрацией в окружности капилляров. Капилляры дермы расширены,
эндотелиальные клетки отечны, тромбов небольших сосудов с некротиче-
скими изменениями стенок; в крупных сосудах—изменения типа эндарте-
риита.
Лечение. Кортикостероиды (преднизолон 30—50 мг в сутки) раздельно
или в комбинации с цитостатиками. Фотрин, проспидин (наши наблюдения).
Теплая комната и теплая ванна успокаивают кожные проявления.
ЛИТЕРАТУРА
Гесс Л. В., Костарева Н. И. О криоглобулинемии у больных с некоторыми хрони-
ческими дерматозами.— Вести, дерматол. и венерол., 1974, 9, 68—71.
Gough W., Lightfoot R., Christian C. Cryoglobulins and complement in immune
complex disease.— Arthr. Rheum., 1974, 17, 497—502.
Harper J., Gray., Wilson—Janes E. Cryogloulinaemia and angiomatosis.— Brit. J.
Dermatol., 1983, 109, 4, 453—458.
Kovary P., Suter L., Steinkuhl H., Schmahl K- Clinical findings and immunoche-
mical studies in a patient with cpyoglobulinemia.— Arch. Demrmatol., Res., 1980, 268, 3,
289—296.
Levo У. Clinical, biological and biochemical aspects cryoglobulins.— Intern. J. Der-
matol., 1981, 20, 9, 590—591.
16 Клиническая дерматология 241
Martin S. Cryofibrinogenemia, monoclanal gammopathy, and purpura.— Arch. Der-
matol., 1979, 115, 2, 208—211.
Mayer F., Steyrer K., GschnaitF., Luger A. Hautnekrosen bei Kryoglobulinamie.—
Hautarzt, 1981, 32, 10, 535—537.
КРОНА БОЛЕЗНЬ (Crohn morbus).
Синонимы: гранулематозный энтерит, терминальный энтерит, регио-
нальный илеит, толстокишечная форма гранулематозного энтерита и т. д.
Болезнь Крона—хроническое гранулематозное воспаление кишечни-
ка, которому Crohn и соавт. в 1932 г. дали название региональный илеит.
В последние десятилетия установлено, что данная патология может пора-
жать любые отделы желудочно-кишечного тракта—от полости рта (Федо-
ров В. Д. и Левитан М. X., 1982; Schiller* et al., 1971; Kirsuer, 1978 и
др.) до анального отверстия; в тонкой и толстой кишках она наблюдается
наиболее часто (более чем у 50—60% больных). Основными симптомами
болезни Крона являются боли в животе, понос, лихорадка, анальные и
перианальные поражения, наружные и внутренние свищи, абсцессы, кро-
вотечения. По наблюдениям многих авторов (McCallum и Kinmont, 1968;
Dyer, 1970; Verbov, 1973; Greenstein и Sachar, 1976; Borgdorf et al., 1981;
Lebwohl et al., 1984, и др.), кожные поражения являются наиболее часты-
ми внекишечными проявлениями болезни Крона, они встречаются у 20—
48% больных.
Этиология и патогенез. Предлагаются различные теории. К числу
наиболее распространенных относятся инфекционная, психогенная, ал-
лергическая и аутоиммунная. Для болезни Крона, как и для других ауто-
иммунных расстройств, характерно длительное течение с наклонностью к
сезонн'ымобострениям, а также благоприятный эффект от принятия средств,
обладающих иммуносупрессивным действием.
Клиника. Болезнь Крона встречается во всех возрастных группах,
но преимущественно возникает у лиц молодого и среднего возраста (от 20
до 40 лет). Кожные проявления при болезни можно разделить на 3 группы.-
1. Перианальные поражения кожи. Они встречаются у 25—36% боль-
ных (Rankin et al., 1979; Lebwohl et al., 1984 и др.); чаще они развива-
ются после кишечных поражений, но могут предшествовать им.
К наиболее характерным поражениям болезни Крона относятся ишио-
ректальные абсцессы, фистулы, обширные изъязвления кожи перианаль-
ной области и промежности с линейными язвами, распространяющимися
на кожу половых органов и ягодиц. Язвы бывают множественными, отде-
ленными друг от друга участками здоровой кожи. Такие изменения наблю-
даются главным образом при обширном вовлечении в процесс толстой
кишки, включая прямую кишку.
К этой группе относятся также гранулематозные поражения полости
рта, наблюдавшиеся многими исследователями (Croft и Wilkinson, 1972;
Bazu и Asquit, 1980 и др.), а также язвенные поражения слизистой оболочки
полости рта.
2. Метастатические поражения кожи. Они развиваются вдали от же-
лудочно-кишечного тракта.
242
Рис. 154. Болезнь Крона.
Рис. 155. Болезнь
Крона.
В мировой литературе описано 10 случаев метастатической болезни
Крона на коже (Parks et al., 1965; Mountain, 1970; Wagner, 1971; Wit-
kowski et al., 1977; Burgdorf et al., 1981; Levine и Baugert, 1982; Lebwohl
et al., 1984 и др.). Анализ данных литературы показывает, что все боль-
ные с метастатической болезнью Крона на коже одновременно имели по-
ражения пищеварительного тракта.
Кожные метастатические поражения Могут располагаться на лице,
верхних и нижних конечностях, животе, груди и др. Они вариабельны по
характеру (рис. 155, 156). Это множественные лихеноидные буровато-
красного цвета папулы, изменения типа узловатой эритемы, гангренозной
пиодермии, хронической язвы, рожистого воспаления. Начальные кожные
гранулемы могут напоминать саркоидоз, гранулемы инородных тел, мико-
бактериальные и грибковые поражения. А. А. Каламкарян и С. М. Федо-
ров наблюдали больную 15 лет, которая была направлена в ЦКВИ-по по-
воду кожных высыпаний неясного генеза из Института проктологии с
диагнозом болезнь Крона. Диагноз был подтвержден эндоскопическим и
патогистологическим исследованием. Кожные изменения представлены
опухолевидной бляшкой в области ягодиц мягкоэластической консистен-
ции^ размерами 4X4 см, возвышающейся над поверхностью кожи на 0,5—
1 см. Цвет бляшки темно-малиновый с синюшным оттенком, при диаско-
пии—слабовыраженный симптом яблочного желе. В Институте проктоло-
гии была сделана операция резекции толстого кишечника, после которой
через несколько дней больная скончалась. При микроскопии кожного по-
ражения была обнаружена саркоидная реакция. Инфильтрат состоял из
гистиоцитов, лимфоцитов и единичных многоядерных гигантских клеток.
16* ' 243
3. Неспецифические (несаркоидные) поражения кожи, встречающиеся
при болезни Крона, характеризующиеся большим клиническим разнообра-
зием. Описаны кожные проявления по типу эксфолиативного дерматита,
синдрома Стивенса—Джонсона, псориаза, витилиго, крапивницы, опоясы-
вающего лишая, некротического васкулита, узловатой эритемы, гиперке-
ратоза, эритематозных пятен и др. Гистологически эти высыпания харак-
теризуются хроническими банальными (несаркоидными) воспалительными
изменениями.
Некоторые данные литературы (Bockus, 1976) показывают, что леталь-
ность при болезни Крона достигает 40%. К наиболее важным факторам,
определяющим прогноз при болезни Крона, относится возраст больных к
началу заболевания, протяженность поражения, длительность бокезни и
осложнения.
Гистопатология. Наиболее характерный гистологический признак— на-
личие в кожных поражениях гранулем саркоидного типа, состоящих из
многоядерных гигантских клеток. Морфологически они отличаются от
гранулемы при туберкулезе и саркоидозе.
Дифференциальный диагноз. Следует проводить с узловатой эритемой,
глубокой пиодермией, гангренозной пиодермией, глубокими микозами,
саркоидозом и др.
Лечение. Кортикостероиды, иммунодепрессанты, антибиотики, суль-
фасалазин. Отечественный препарат салазопиридазан (по 2 г в сутки в
течение 4—6 недель) оказывает хорошее действие на кишечные и кожные
проявления болезни Крона. Хирургическое вмешательство.
ЛИТЕРАТУРА
Федоров В. Д., Левитан М. X. Воспалительные заболевания толстой кишки.—
Ташкент, 1982.
Basu М., Asquith Р. Oral manifestations of inflammatory bowel disease.— Cfin Gast-
roenterol., 1980, 9, 2, 307—320.
Burgdorf W. Cutaneous manifestations of Crohn’s disease.— J. Amer. Acad. Derma-
tol., 1981’, 5, 6, 689—695.
Greenstein A., Sachar D. The extra—intestinal complication of Crohn’s disease and
ulcerative colitis: a study of 700 patienys.— Medicine, 1976, 55, 5, 401—412.
LebwohlM., Fleischmajer R., Present D., PrioleanPh. Metastatic Crohn’s disease.—
,J. Amer Acad. Dermatol., 1984, 10, 1, 33—38.
Levine W., Bangert J. Cutaneous granulomatosis in Crohn’s diseases.— Arch. Derma-
tol., 1982, 118, 1006—1009. .
McCallum D., Grag W. Metastatic Crohn’s disease.— Brit. J. Dermatof., 1979, 95, 6,
551—554.
КСАНТОМЫ ТУБЕРОЗНЫЕ (xanthoma tuberosum).
Синонимы: множественная ксантома, первичный гиперхолестеринеми-
ческий ксантоматоз.
Ксантоматоз кожи, в частности множественные туберозные (эруптив-
ные) ксантомы—одно из проявлений нарушенного липидного обмена. Све-
дения о ксантомах были впервые опубликованы французским дерматологом
244
Rayer в 1835 г. Термин «ксантомаж был введен в 1869 г. Smith, после чегЪ
в литературе появился ряд сообщений по этому вопросу (Ведров Н. С.,
1927; Олесов И. Н. и Селисский А. Б., 1928; Мамед-Заде А. С., 1938;
Каламкарян А. А. и Тимошин Г. Г., 1977; Montgomery и Osterberg, 1938;
Movittet al., 1951; Winkelmann и Welborn, 1969; Zemel et al., 1970; Rosen,
1978; Haller et al., 1979 и др.). Zollner и Wolfram (1970) наблюдали 78 боль-
ных гиперхолестеринемией, у 9 из них были различные формы ксантомы,
в том числе распространенные туберозные.
Этиология и патогенез; Гиперлипопротеинемии (гиперлипидемии) пред-
ставляют собой заболевания, при которых нарушения в образовании,
транспорте и расщеплении липопротеинов ведут к повышению уровня три-
глицеридов и холестерина в плазме крови. Повышение уровня липидов сы-
воротки бывает первичным и вторичнымХ Первичные гиперлипопротеине-
мии—когда без определенной причины уровень триглицеридов и холесте-
рина плазмы превышает принятые пограничные значения. Fredrickson и
соавт. (1965, 1967, 1971) предложили принцип дифференцирования- пяти
типов первичных гиперлипопротеинемий, который основывается на соот-
ношении липопротеиновых фракций плазмы и базируется прежде всего
на фенотипе электрофореза липидов. Разделение типов по Fredrickson и
соавт. (1967, 1971) рекомендовано к всеобщему применению Всемирной
организацией здравоохранения (Beaumont et al., 1970; Goldstein и Brown,
1975). Кожные поражения встречаются при всех пяти типах гиперлипо-
протеинемии. Они могут быть разделены на эруптивные ксантомы, тубе-
розные ксантомы, сухожильные ксантомы, ксантелазмы и плоские ксан-
томы (см. табл.).
Тип гиперлипопротеинемии Тип кожных проявлений (ксантом)
эруптив- ные тубе- розные сухо- жильные ксанте- лазма плоские
I тип— -гипер хиломикронемия, абдоминальные колики, гепатоспленомегалия. + — — — +
II тип— гипер ^-липопротеинемия (чаще развивается у детей) — + + + +
III тип— гипер-Э-липопротеинемия, гиперпре-Э-липопротеине- мия (повышенное содержание холестерина и триглицери- дов) -f- + ' "Ь + + ’
IV тип— гиперпре-Р-липопротеине- мия (повышено содержание холестерина и триглицеридов + + —W — —
V тип— является комбинацией I и IV типов + — — — —
245
Рис. 156. Ксантома туберозная.
Эруптивные ксантомы состоят из мелких мягких желтых папул с
преимущественной локализацией на ягодицах и бедрах. Туберозные ксан-
томы представлены крупными опухолями и- бляшками, локализующимися
преимущественно в области локтей, колен, ягодиц и пальцев. Сухожиль-
ные ксантомы наиболее часто поражают ахилловы сухожилия и сухожилия
разгибателей пальцев. Ксантелазма характеризуется слегка возвышающи-
мися, желтыми, мягкими бляшками в области век. Плоские ксантомы раз-
виваются в кожных складках, особенйо в складках ладоней.
Вторичные гиперлипопротеине-
мии (вторичный ксантоматоз) раз-
виваются на фоне других заболе-
ваний—сахарного диабета, нефрита,
нефротического синдрома, уремии,
панкреатита, системной красной
волчанки, лимфомы, билиарного
цирроза печени, алкоголизма и др.
На кожные поражения при сахар-
ном диабете, обусловленные сдвига-
ми в обмене веществ, указывают
многие авторы (Мартынова А. И. и
соавт., 1964; Хашимов Б. И., 1973;
Касько Ю. С. и соавт., 1980; Г. И.
Базуров, И. Е. Шахнес, 1983; Hunja
et al., 1974; Meinhof и Matzkies,
1977 и др.). Наблюдаются ли-
поидный некробиоз и ксантоматоз.
Ксантоматоз у больных диабетом
характеризуется появлением на коже туловища, конечностей и ягодиц
множественных образований в виде желто-коричневых узелков и опухолей,
обладающих склонностью к регрессу. Лечение сахарного диабета является
основным мероприятием в терапии больных диабетическим ксантоматозом,
что подтверждается быстротой регрессирования высыпаний на коже под
влиянием антидиабетических препаратов.
Клиника. Множественные симметрично расположенные узловатые, опу-
холевидные образования преимущественно в области крупных суставов—
коленных и локтевых, а также на ягодицах, плечах, тыльной поверхности
пальцев, на коже лица, волосистой части головы. Опухоли размерами от
крупной горошины до грецкого ореха, характерного желтого цвета с бу-
рым или фиолетовым оттенком, дольчатого строения, спаяны в конгломерат,
неподвижны, плотны. Некоторые из них как бы окружены красновато-
синюшной каймой. При диаскопии выявляется типичный желтоватый фон
очагов. Как правило, высыпания не сопровождаются, субъективными ощу-
щениями. Нередко туберозные ксантомы сочетаются с уплощенными ксан-
томами орбитальной области—ксантелазмами (xanthelasma palpebrarum)
и ксантоматозом сухожилий, особенно ахилловых (рис. 157). При тубе-
розном ксантоматозе нередко наблюдаются тяжелые атеросклеротические
246
расстройства коронарных сосудов сердца. А. А. Каламкарян и Г. Г. Тимо-
шин (1977) описали I тип ксантомы с множественными опухолевидными
высыпаниями на ладонях, на стопах, на тыльной и, особенно, на ладонной
поверхности пальцев кистей, над локтевыми и коленными суставами раз-
мерами до грецкого ореха. Уровень общих липидов в крови—1680 мг% (в
норме 400—700 мг%). Уровень холестерина в динамике в пределах 132—
164 мг%. Таким образом, наблюдение авторов не подтверждает существую-
щей концепции о взаимотесной связи туберозной ксантомы и гиперхоле-
стеринемии.
Базуров, Г. А. Шахнес И. Е. (1983) описали больного, у которого
имелся распространенный ксантоматоз с исходом в келоидные рубцы.
Больной, кроме того, страдал несахарным диабетом, что, по мнению авто-
ров, указывает на патогенетическую связь ксантоматоза с эндокринными
нарушениями.
Течение заболевания хроническое, прогноз для жизни определяется с
осторожностью, с учетом возможности атеросклеротических изменейий
коронарных сосудов и др.
Гистопатология. Эпидермис обычной толщины с неравномерно выра-
женными эпителиальными отростками, умеренный гиперкератоз. В толще
всего сетчатого слоя дермы—инфильтраты с наличием пенистых одно-
или многоядерных клеток, известных под названием гигантских клеток
Тутона (Touton). Одновременно обнаруживается воспалительная инфильт-
рация полинуклеарами и лимфоцитами с разрастанием капилляров и про-
лиферацией эндотелия. В старых очагах пенистые или ксантомные клетки
могут переходить в фибробласты и замещаться фиброзной тканью. Элект-
ронно-микроскопические исследования пенистых или ксантомных клеток
показывают, что они имеют неправильные очертания цитоплазмы, типичные
для макрофагов. Цитоплазма их содержит множество органелл, включая
лизосомы (Zemel et al., 1970; Wolf et al., 1970).
Лечение. Должно быть направлено на нормализацию нарушений ли-
пидного обмена и висцеральных расстройств. Применяют хирургическое
удаление крупных узлов и опухолей, диатермокоагуляцию мелких обра-
зований. Ограничение приема жиров, молочно-растительная пища. Sat-
macher (1967) получил хороший эффект у больного с туберозным ксанто-
матозом после 5-месячного лечения синтетическим правовращающим изо-
мером тироксина (а-тироксин) по 8—10 мг в день. Zollner и Wolfram (1970)
отметили положительное, но кратковременное действие р—пиридилкар-
бина.
।
ЛИТЕРАТУРА
Базуров Г. И., Шахнес И. Е. Множественная туберозная ксантома с исходом в ке-
лоидные рубцы. Вести, дерматол. и венерол., 1983, 5, 40—42.
Геллер Г., Ганефельд М., Ярове В. Нарушение липидного обмена. Диагностика,
Диагностика, клиника, терапия.— Пер. с нем.— М.: Медицина, 1979.
Каламкарян А. А., Тимошин Г. Г. Случай множественного туберозного ксанто-
матоза.— Вестн. Дерматол. и венерол., 1977, 5, 41—44.
Касько Ю. С., Трутяк Л. Н., Квятковская Г. В- Случай диабетического ксанто-
матоза.— Вестн. Дерматол. и венерол., 1980, 1, 50—51.
Meinof W., Matzkies F. Rflckbildung von Xanthoma bei endogener Hypertriglyze-
ridamie unter Kohlenhydratarmer Ernahrung.— Hautarzt, 1977, 28, 12, 648—652.
Rosen T. Dystrophic xanthomatosis in mycosis fungoides.— Arch. Dermatol., 1978,
114, 1, 102—103.
КСЕРОДЕРМА ПИГМЕНТНАЯ (xeroderma pigmentosum).
Синонимы: пигментная атрофическая аденома, прогрессирующий лен-
тикулярный меланоз, пигментная атрофодермии, злокачественное лен-
тиго.
Довольно редко встречающееся заболевание, впервые описанное в
1870 г. Kaposi. В дальнейшем о единичных подобных наблюдениях сооб-
щили отечественные и зарубежные авторы: Кудиш В. М. (1906, 1910),
Пер М. И. (1925), Жирмунская. К- М. и Минц М. М. (1934), Кривояз М. Л.
(1940), Тер-Григоряи Г. Д. (1957), Бабаянц Р. С. (1958), Михельсон В. М.
и Э. В. Айказян (1972), Абдуллаев, А. X. Белова, Л. В. 1983). Cleaver
и Carter (1973), Guerrier с соавт. (1973), Diaconnu и Popescu (1980) и др.
Этиология и патогенез. Пигментная ксеродерма—генетически детер-
минированное заболевание, передается аутосомным геном. Большей ча-
стью носит семейный характер (Тер-Григорян Г. Д. 1957). Lynch с соавт.
(1967) обследовали одну семью, в которой у 5 из 7 братьев и сестер имелись
проявления пигментной ксеродермы. Болезнь встречается чаще при близ-
кородственных браках и вообще в изолятах—географических,' националь-
ных, религиозных и т. д. (Кудиш В. М., 1906; Тер-Григорян Г. Д., 1957;
Venkatesan, 1969, Misra et al., 1971; Lever и Schaumburg-Lever, 1975 и др.).
Механизм возникновения'своеобразной сенсибилизации к ультрафиолето-
вым лучам при пигментной ксеродерме пока не ясен. Мнение об увеличе-
нии фотодинамических субстанций в организме больных пигментной ксеро-
дермой большинством авторов не поддерживается и порфирина у боль-
шинства больных не находили (Rook и Lever, 1945 и др.). По мнению ряда
исследователей (Cleaver, 1968; 1972; 1973; Robbins и Craermer, 1972; Ba-
den et al., 1972; Stich et al., 1972; Setlov et al., 1976 и др.), в основе пигмент-
ной ксеродермы лежит отсутствие или недостаточность в клетках (фибро-
бластах) больных фермента УФ-эндонуклеазы, участвующего в репарации
ДНК, поврежденной ультрафиолетовыми лучами. По данным Михельсо-
на В. М. и Айказяиа Э. В. (1975) у некоторых больных пигментной ксеро-
дермой дефектен фермент ДНК—полимераза—1, необходимый для зас-
траивания брешей в нити ДНК-
Клиника. Заболевание поражает оба пола, хотя некоторые авторы счи-
тают, что среди девочек оно встречается чаще. Первые симптомы более
чем у 75% больных появляются между 6 месяцами и тремя годами, но мо-
гут наблюдаться и в более старшем (14—35 лет) и даже в пожилом возрасте
(на 65 году). С раннего детства наблюдается светобоязнь и повышенная
248
I
Рис. 157. Ксеродерма
пигментная.
фоточувствительность с образованием на от-
крытых участках кожи после непродолжитель-
ной инсоляции красноватых пятен, на кото-
рых впоследствии и откладывается пигмент.
На коже лица, шеи, кистей, разгибательных
поверхностей предплечий появляются обиль-
ные пигментные пятна (в виде веснушек)
темно- и светло-коричневого цвета, неправи-
льных очертаний, диаметром 0,1—0,5 см, а
также мелкие плоские папулы желтоватого
цвета, едва выступающие над поверхностью
кожи (рис. 158, 159). Затем кожа начинает
шелушиться, пигментированные места смор-
щиваются и становятся атрофичными (вторая
стадия заболевания). На коже носа, губ, на
красной кайме губ—многочисленные телеанги-
эктазии. Наряду с пигментированными пятна-
ми встречаются атрофические рубцы беловатого
цвета и небольшие гиперкератотические очаги. Развивается микростомия
за счет атрофии и рубцовых изменений нижней губы, красная кайма кото-
рой истончена; на губах—трещины. В общем пораженная кожа сухая,
истонченная, местами напряжена, натянута, не собирается в складки,
имеет клиническое сходство с кожей при хроническом радиодермите. Атро-
фические изменения нередко ведут к различным обезображиваниям: энтро-
пиону, истончению ушей и кончика носа, атрезии отверстий рта и носа. Те-
леангиэктазии и ангиомы, помимо кожи, наблюдаются на языке, слизистой
ротовой полости. Помимо кожи, у 80—85% больных поражаются глаза—
конъюнктивит, кератит, поражение радужной оболочки (гиперпигмента-
ция, атрофия края зрачка и стромы) и роговицы глаз, что приводит к сни-
жению зрения (El-Hefnaw и Mordata, 1965). Volcker и Nauman (1975)
указывают, что при пигментной ксеродерме почти всегда поражается кожа
век. На веках могут быть дисхромии, телеангиэктазии, гиперкератозы и
различные опухоли. В поздней, третьей, стадии заболевания кожа пред-
ставляет картину преждевременной старческой дистрофии с возникнове-
нием бородавчатых разращений, которые зачастую раково перерождаются.
Тер-Григорян Г. Д. (1957), Schimpf (1979), Fischer и соавт. (1982) относят
лигментную ксеродерму в группу облигатных преканкрозов. Развитие
злокачественных новообразований на местах пигментных и атрофических
пятен происходит иногда уже в первые годы жизни, особенно же с 8—10-
летнего возраста. Очень быстро опухоли распадаются, дают метастазы во
внутренние органы и приводят к смерти. Появляющиеся опухоли имеют
различный характер. Чаще это базалиомы и спиналиомы, реже меланомы,
саркомы, перителиомы, трихоэпителиомы, эндотелиомы, ангиосаркомы.
Кератоакантома является необычным осложнением пигментной ксеродер-
мы. В литературе описано 15 подобных случаев (Tenstedt и Lachapelle,
1977, Braun—Falco et al., 1982). Нередко у больных пигментной ксеро-
249
Рис. 158. Ксеродерма
пигментная.
дермой наблюдаются общие дистрофические изменения органов и тканей:
синдактилия II—IV пальцев на стопах, дистрофия зубов, рост отстает от
возрастной нормы на 10—25 см, тотальное отсутствие волос (Diem и Fritsch,
1973) Интеллект больных обычно не страдает, но описана особая форма
пигментной ксеродермы—синдром де Санктиса—Какионе (De Sanetis и Сас-
chione, 1932), характеризующаяся умственной отсталостью и другими нев-
рологическими симптомами (Суворова К. Н. и Сазонова Н. С., 1970;
Тег-Koloustian et al., 1974).
Б. А. Беренбейн и соавт. (1983) наблюдали двух родных братьев с
пигментным ксероидом Юнга (Junge, 1980), для которого характерно бо-
лее легкое течение, позднее начало заболевания, менее выраженные кожные
проявления, благоприятный прогноз.
Прогноз заболевания неудовлетворительный и тем хуже, чем раньше
началась болезнь. В большинстве случаев болезнь прогрессирует и ведет
к смерти в возрасте 10—15 лет. Имеются единичные описания, где боль-
ные доживали до 60—70 лет (Нерадов Л. А., 1964, Kaposi, 1870; Herzheimer,
1922; Rielle, 1924).
Гистопатология. В ранней стадии заболевания: гиперкератоз, истон-
чение мальпигиевой сети с атрофией части эпидермальных отростков, отек
250 /
верхней части дермы, хронический воспалительный инфильтрат (преиму-
щественно вокруг сосудов) в верхней части 'дермы, пятнистая меланиновая
пигментация базального слоя с наличием меланофоров в верхней части дер-
мы. Во второй стадии гиперкератоз и гиперпигментация усиливаются. Эпи-
дермис в некоторых местах атрофичен, в других—акантотичен. Ядра не-
которых эпителиальных клеток атипичны, сильнее окрашены, как при
старческом кератозе. В верхней части дермы дегенеративные изменения
коллагена и эластических волокон такого же характера, как при старче-
ской дегенерации кожи. В третьей (опухолевой) стадии—гистологическая
картина рака. Электронно-микроскопические исследования (Guerrier et
aL, 1973; Caputo и Celifano, 1971; Tsuji, 1975) показывают уплотнение,
уменьшение кератиноцитов и их ядер, атипию ядрышек. Меланоциты также
атипичны, с полиморфными меланосомами, крупные (гигантские) пигмент-
ные зерна, гранулы в меланоцитах или кератиноцитах.
Лечение. Симптоматическое. Опухоли удаляются хирургически, жид-
ким азотом, лучами лазера. Horkay и соавт. (1974) получили хороший эф-
фект от применения 5-фторурацила. Больным рекомендуется избегать
воздействия инсоляции, носить красные и желтые вуали, шляпы с широ-
кими полями, смазывать кожу фотозащитными кремами и мазями (крем
«Щит», мази, содержащие 5—10% хинина или 15—20% парааминобензой-
ной кислоты и др.). Имеются сообщения (Braun-Falco et al., 1982;) о
благоприятном терапевтическом эффекте у больных пигментной ксеро-
дермой от длительного (до года) приема ароматического ретиноида ((Ro
10-9359).
ЛИТЕРАТУРА
Михельсон В. М., Смирнов В. С. О генетической неоднородности пигментной ксе-
родермы. — Вести, дерматол. и венерол., 1975, 3, 70—75.
Braun-Falco О. и соавт. Tumorprophyfaxe bei Xeroderma pigmentosum, mit aroma-
tischen Retinoid (R° 10—9359). Hautarzt, 1982, 33, 8, 445—448.
Diaconnu J., Popescu S. Quelques considerations sur le jeroderma pigmentosum.—
[n: Congres national de dermatologie. Bucuresti, 1980, p. 47—48.
Leclaire M., Benoit M., Desmans F. Xeroderma pigmentosum chez deux soeurs. Etude
du caryotype.— Soc. Franc. Derm. Syphilogr., 1974, 81, 1, 18—20.
Schipf A. Das Ptattenepithelkarzinom der Haut und Halbschleimhaute. Prakanzero-
sen.— In: Nicht emtzflndlische Dermatosen. 11 IB. Gottron und Korting. Berlin: Springer—
Verlag, 1979, 1, 130—196.
Tennstedt D., Lachapelle J. Keratoacanthomes et xeroderma pigmentosum.— Ann.
Dermatcf. VenereoL, 1977, 104, 2, 98—102.
Ter-Kaloustian et al. The genetic defect in the de Sanctis—Cacchione syndrome.—
J. invest. Dermatol., 1974, 63, 5, 392—396.
Tsuji T. Electron microscopic studies of xeroderma pigmentosum: unusual chanes in
the keratinocyte.— Brit. J. Dermatol., 1974, 91, 6, 657—666.
Volcker H., Naumann G. Conjunctivale und corneale Augenveranderungen bei Xero-
derma pigmentosum.— Hautarzt, 1974, 15, 11, 561—565.
251
ЛАССЮЭРА—ЛИТТЛ Я СИНДРОМ (Lassueur—Little syndromum).
Синонимы: плоский волосяной лишай, красный фолликулярный руб-
цующийся лишай.
Редкое заболевание, самостоятельность которого остается спорной.
В 1915 г. Little описал больного с очаговой рубцовой алопецией волосистой
части головы, нерубцовой алопецией подмышечной области и лобка и фол-
ликулярными шиповидными высыпаниями на туловище и назвал это за-
болевание lichen spinulosus et folliculitis decalvans. Beatty (1915) предло-
жил назвать указанный дерматоз синдромом Лассюэра—Литтля. В связи
с тем, что сочетание подобных клинических симптомов еще в 1914 г. описы-
вал Picardi, в ряде стран принято название: синдром Picardi—Lassaueur—
Little. Мы присоединяемся к мнению большинства авторов о том, что синд-
ром Лассюэра—Литтля представляет собой атрофический вариант крас-
ного плоского лишая с преимущественным поражением фолликулярного
аппарата кожи (Машкиллейсон Л. Н., 1965; Брайцев А. В. и соавт.,
1968; Довжанский С. И. и Суворов А. П., 1976; Silver et al., 1953; Altman
и Perry, 1961; Domonkos, 1971; Gertler, 1972; Horn et al., 1982 и др.).
Клиника. В настоящее время установлено, что у большинства больных
с синдромом Лассюэра—Литтля одновременно обнаруживаются типичные
высыпания красного плоского лишая, остроконечные высыпания типа
шиповидного лихена на коже туловища и конечностей и, участки псевдо-
пелады на волосистой части головы, причем во всех поражениях гистоло-
гически обнаруживается картина красного плоского лишая. Начальные
признаки заболевания заключаются в появлении зуда и высыпаний лихе-
ноидных элементов почти одновременно на коже волосистой части головы,
туловища и конечностей. У части больных узелки вначале локализуются
только на туловище и лишь через несколько лет на волосистой части го-
ловы появляются участки рубцового облысения. На коже преимущественно
разгибательных поверхностей плеч, в области локтевых суставов, запя-
стий и бедер имеется множество мелких фолликулярных, остроконечных
узелков телесного цвета с небольшими роговыми шипиками на верхушке,
без признаков воспаления. При поглаживании кожа представляется сухой,
жесткой, шероховатой и создает впечатление терки. Наряду с этим на коже
груди, внутренних поверхностей предплечий, поясницы, голеней имеются
типичные для красного плоского лишая розовато-фиолетовые, блестящие,
полигональные папулы с пупковидным вдавлением в центре. Субъектив-
ные ощущения, как правило, отсутствуют, но иногда отмечается
сильный зуд. На волосистой части головы, обычно в области те-
мени, затылка и висков, образуются небольшой величины плешин-
ки круглой или овальной формы. Постепенно (крайне медленно)
количество и размеры их увеличиваются. Группируясь на ограни-
ченном участке, плешинки сливаютс^ и образуют участки значи-
тельной величины (3x4—5X7 см) с фестончатыми очертаниями (рис. 161,
162, 163). У части больных по периферии основного облысевшего уча-
стка образуются мелкие плешинки, й поражение мо*жет постепенно рас-
пространяться по всей волосистой части головы. Кожа в пределах очагов
252
Рис. 159. Синдром
Лассюэра—Литтля
Рис. 160. Синдром
Лассюэра—Литтля
Рис. 161. Лассюэра—Литтля
синдром.
облысения натянута, блестит, атрофична и как бы вдавлена, окраска ее
чаще нормальная, реже она имеет розовато-синюшный цвет. Могут быть
серозно-геморрагические корки и следы расчесов; на больших плешинках
нередко отмечаются островки нормальных волос. По периферии очагов
алопеции нередко располагаются фолликулярные конусовидные узелки.
Волосы в подмышечных ямках и на лобке разрежены, часть их обломана и
истончена (нерубцовые участки облысения). У некоторых больных описан-
ные симптомы сочетаются с проявлениями типичного красного плоского
лишая на слизистых оболочках в виде опаловых папул. Иногда при дан-
ном синдроме отмечаются дистрофические изменения ногтевых пластинок.
Со стороны внутренних органов патологических изменений не выявлено.
Клинические и биохимические анализы без особенностей. Как показыва-
ют наши наблюдения, при синдроме Лассюэра-Литтля всех признаков триа-
ды может не быть. Наиболее часто встречаются явления рубцовой алопеции
и лихеноидные высыпания. В связи с этим можно предположить, что псев-
допелада не самостоятельное заболевание, а проявление этого синдрома.
Заболевание относится к наиболее резистентным формам красного
плоского лишая. Длительность течения дерматоза может достигать 15—
20 лет.
Гистопатология. Очаговый гиперкератоз, неравномерный гиперграну-
лез, отек в шиповидном слое, вакуольная дистрофия клеток базального
слоя, полосовидные или периваскулярные инфильтраты в верхних слоях
дермы.
Дифференциальный диагноз. Следует проводить с дискоидной крас-
ной волчанкой, декальвирующим фолликулитом, рубцующейся эритемой
лица Унны—Тенцёра, болезнью Дарье, волосяным кератозом.
Лечение. Терапия представляет значительные трудности. Рекомен-
дуются синтетические антималярийные препараты (делагил, плаквеиил),
пресоцил, инъекции витамина А, аевита, гистоглобулина, кремы и мази
с кортикостероидами. Мы наблюдали хороший эффект от фотохимиотера-
пии.
ЛИТЕРАТУРА
Базыка Д. А. Сравнительные результаты лечения красного плоского лишая раз-
личными методами.— ВестН. дерматол. и венерол., 1977, 2, 54—57.
Довжанский С. И., Суворов А. П. Синдром Little—Lassueur.— Вести, дерматол. и
венерол., 1976, 7, 73—76.
Жаворонкова Т. С., Шахнес И. Е. О синдроме Литтля—Лассюэра.— Вести. Дерма-
тол. и венерол., 1980, 9, 44—47.
Рупишпейн Л. Г., Ройтбурд М.Ф., Фадеева В. И. К вопросу о синдроме Лассю-
эра—Литтля.— Вести, дерматол. и венерол., 1977, 4, 58—60.
Разнатдвский И. М-, Родионов А. Н. О синдроме Piccardi—Lassueur—Little —
Вести, дерматол. и венерол., 1978, 4, 54—57.
Штейнлухт Л. А., Придвижкин И. Г., Котина Р. И., Павлова В. И. Пресоцил
254
if терапии больных красным плоским лишаем,— Вестн. Дерматол. и венерол., 1978, 1, 74—
76.
Coltoin A., Mateescu D. Sindrom Graham—Little—Lassueur.— Derm.— Vener.,
1974, 19, 137—140.
Horn R. Goette D. и соавт. immuno-fluorescent finding and clinical overlap in two
cases of follicular lichen planus. — J. Amer. Acad, dermatol., 1982, 7, 2, 203—207.
Samman P. Lichen planus and lichenoid eruptions.— In: Textbook of dermatology.
Philadelphia, 1979, 3 rd ed., p. 1483—1502.
ЛЕЙКОЗЫ КОЖИ (leucosis cutis)
Синонимы: лейкемиды кожи, гемодермия, злокачественная лимфо-
дермия, лимфаденическое пруриго.
Обычно возникают при лейкозах на фоне четких изменений в гемо-
граммах и развернутой клинической симптоматики. Одиако кожные по-
ражения могут быть начальными симптомами лейкоза, когда другие кли-
нические признаки (изменения крови, лимфоузлов и костного мозга) еще
отсутствуют.
Этиология и патогенез. Природа лейкозов остается неясной. Роль
генетических факторов ограничена формированием предрасположенности
к развитию лейкоза, которая реализуется под влиянием различных воз-
действий: радиации, химических веществ, вирусов. взкцинаций и др.
Большинство современных отечественных и зарубежных исследова-
телей предполагают, что в основе лейкозов лежит гиперпластический опу-
холевый процесс в кроветворной ткани. Кожные поражения при лейкозах
разделяются на специфические лейкемиды, обусловленные пролиферацией
клеток, свойственных данной форме лейкоза, и неспецифичеекие лейкеми-
ды, гистологическое строение которых не имеет характерной картины.
Лейкемиды кожи описаны многими отечественными и зарубежными авто-
рами (Даштаянц Г. А., 1953; Потоцкий И. И., 1958; Хачатурян Г. X.,
1961; Арутюнов В. Я., 1961; Каламкарян А. А., 1961; 1975; Ашмарин
Ю. Я., 1976; Biesiadecky, 1876; Kaposi, 1885; Nicolau, 1904; Bluefarb,
1959). Полиморфизм кожных высыпаний при лейкозах очень велик и не
всегда удается по характеру кожных изменений дифференцировать раз-
личные формы лейкозов (Потекаев Н. С., Иванов О. Л., 1983).
Клиника. Кожные поражения являются частыми проявлениями ост-
рого (миелобластного, лимфобластного, монобластного и недифференциро-
ванного) лейкоза, они встречаются у 50—60% больных.
Неспецифические лейкемиды кожи при остром лейкозе характеризу-
ются полиморфизмом: геморрагии, пруригоподобные высыпания, уртика-
рия, иногда они напоминают многоформную эксудативную эритему. Наи-
большую группу составляют больные с геморрагическим синдромом, кото-
рый встречается у 65—80% больных острым лейкозом. Геморрагические
явления чаще развиваются остро, внезапно в виде багрово-синюшных пят-
нистых сыпей на коже, кровоточивости десен, реже кровотечений из но-
са и гениталий. Высыпания не исчезают при надавливании, обычно носят
255
распространенный характер,, располагаются на туловище, конечностях и
реже на лице.
Специфические лейкемиды при остром лейкозе обычно рассеяны по
всему кожному покрову в виде плотноватых, хорошо очерченных, слегка
болезненных узлов, величиной от горошины до грецкого ореха. Они воз-
вышаются над уровнем кожи в виде синюшно-багровых полушаровидных
образований. Путем периферического роста и слияния соседних очагов
возникают более крупные инфильтраты неправильных очертаний с ладонь
взрослого и крупнее. Опухоли при остром лейкозе, в о’тличие от специфи-
ческих лейкемидов при лимфо-и миелолейкозе, довольно часто распада-
ются с образованием глубоких, резко болезненных язв. У отдельных боль-
ных острым лейкозом лейкемическая инфильтрация кожи может протекать
под видом язвенно-некротических изменений. Лейкозная инфильтрация
слизистой оболочки у таких больных имеет характер гипертрофического
гингивита и язвенного стоматита.
Кожные поражения при хронических лейкозах встречаются при лим-
фатическом лейкозе чаще, чем при других формах лейкозов. Среди наблю-
давшихся нами больных различными формами лейкозов с кожными пора-
жениями у 61,3% был лимфолейкоз, у 14,5%—миелолейкоз, у 11,3%—ост-
рый лейкоз, у 3,2%—миеломная болезнь, у 9,7%—эритремия.
Специфические узловато-опухолевидные образования при хрониче-
ском лимфолейкозе (рис. 163) имеют вид ограниченных опухолей, величи-
ной от горошины до куриного яйца и больше, округлых, овальных или
неправильных очертаний, форма их обычно полушаровидная, иногда они
имеют вид массивных плоских дисков. Кожа над ними гладкая, блестящая,
часто с телеангиэктазиями. Консистенция опухолей большей частью мяг-
коватая, плотно-эластическая, но встречаются и плотные опухоли. Цвет
элементов вначале насыщенно красный, сравнительно скоро становится
синюшно-багровым, ливидным. Постепенно увеличиваясь в числе и сли-
ваясь, очаги поражения в отдельных случаях достигают значительной
величины и занимают большие участки кожи. Опухоли располагаются на
любом участке кожного покрова, но чаще на лице, создавая клиническое
сходство с facies leonina при лепре. Они медленно подвергаются изъязвле-
нию. Неповрежденные опухоли большей частью безболезненные, но со-
провождаются зудом. Значительно реже специфические лейкемиды при
хроническом лимфолейкозе проявляются в виде универсальных эритро-
дермий. Кожа сплошь представляется отечной, интенсивно красной с си-
нюшно-багровым оттенком, инфильтрированной, с глубокими трещинами;
как правило, отмечается крупнопластинчатое шелушение; мучительный
непрерывный зуд, дистрофические изменения придатков кожи, увеличен-
ные периферические лимфоузлы.
Диагностика лейкемической эритродермии нередко (особенно у боль-
ных сублейкемическим и алейкемическим лимфолейкозом) затруднительна.
Решающее значение для определения специфичности поражения кожи на-
ряду с исследованиями гемограммы, миелограммы, аденограммы и клиниче-
скими данными имеют результаты гистологического исследования.
256
Неспецифические Кожине изменения значительно чаще наблюдаются
при лимфолейкозе, чем при других формах лейкоза. Клинически
неспецифические высыпания (лейкемиды) характеризуются уртико-пру-
ригинозными элементами, геморрагическими, эксудативно-пемфигоидны-
ми высыпаниями, пигментно-трофическими изменениями кожи и ее
придатков, экзематозными изменениями. Нередко у больных лимфолейко-
зом встречаются пиодермии, опоясывающий лишай. Наши многолетние
клинико-лабораторные наблюдения позволяют высказать предположение,
что они, по-видимому, являются результатом действия аутотоксических
и иммунных факторов. Очевидно, существуют и другие факторы, обуслов-
ливающие развитие неспецифических лейкемидов, но роль их изучена не-
достаточно.
Специфические кожные изменения при хроническом миелолейкозе
клинически имеют большое сходство с таковыми при лимфолейкозе и
характеризуются развитием преимущественно на туловище, реже на ко-
нечностях и лице опухолевидно-узловатых образований размерами от мел-
кой горошины до куриного яйца, синюшно-красного цвета с самыми раз-
нообразными оттенками. Консистенция плотноэластическая. В части слу-
гаев опухоли и инфильтративные бляшки сливаются в объемные бугристые
образования с причудливыми очертаниями. В большинстве случаев они
ie сопровождаются зудом, но нередко бывают болезненны при пальпации.
Исключительно редко наблюдаются очаги поражения в виде синюшно-
красных опухолей величиной от горошины до лесного ореха на слизистой
ютовой полости, склонные к изъязвлению с образованием глубоких резко
олезненных язв.
7 Клиническая дерматология 257
Неспецифические лейкемиды при миелолейкозе лишены характерных
клинико-морфологических черт и отличаются большим полиморфизмом:
уртико-пруригинозные элементы, геморрагические эксудативно-пемфиго-
вдные высыпания и др.
Прогноз неблагоприятный, появление и резкое распространение кож-
ных поражений (особенно специфических) у больных хроническим миело-
и лпмфолейкозом, как правило, свидетельствует об ухудшении основного
процесса, наступлении терминального‘«бластного» криза и сравнительно
быстром летальном исходе.
Гистологическое строение специфических инфильтратов кожи при
хроническом лимфолейкозе характеризуется периваскулярной и периглан-
дулярной, преимущественно мономорфной, пролиферацией в сосочковом и
подсосочковом слоях дермы, состоящей из малых и средних лимфоцитов,
пролимфоцитов. Среди них часто встречаются клетки, достигающие боль-
ших размеров, с выраженной базофилией цитоплазмы и нежной сетчатой
структурой ядра-лимфобласты.
Гистопатология. Гистологическая картина специфических опухоле-
вых поражений кожи при хроническом миелолейкозе характеризуется лей-
козной инфильтрацией кожи преимущественно молодыми клетками мие-
лоидного ряда.
Лечение. При относительно благоприятном течении лейкоза с неспе-
цифическими кожными высыпаниями рекомендуется ограничиться назна-
чением противозудных средств, антигистаминных препаратов, транквили-
заторов и различных местных лекарственных средств (кортикостероидные
кремы и мази, ментол в спирту (1—3%), мази с анестезином (3—5%) и
др.). При очень распространенных высыпаниях, сопровождающихся силь-
ным зудом, рекомендуются кортикостероиды (преднизолон по 15—30 мг в
сутки). Больным со специфическими кожными поражениями следует при-
менять соответствующие (в зависимости от формы лейкоза) методы тера-
пии: цитостатики, рентген, кортикостероиды и др.
ЛИТЕРАТУРА
Потекаев Н. С., Иванов О. Л. Гемобластозы кожи. В кн. Дифференциальная диаг-
ностика кожных болезней. М. 1983, 305—331.
Потоцкий И. И. Заболевания кожи при лейкозах.— Киев: Здоров’я, 1978.
ЛЕЙОМИОМА КОЖИ (leiomyoma cutis)
Синонимы: ангиолейомирма.
Первое описание лейомиомы принадлежит R. Virchow (1854), подверг-
шему гистологическому исследованию опухоли, возникшие на коже груди
у 32-летнего мужчины. Позднее Besnier (1873, 1880) и Babes (1884) пред-
ложили классификацию лейомиом кожи, выделив при этом сосудистую
лейомиому. В настоящее время, соответственно гистогенезу, различают
3 типа лейомиом кожи, характеризующихся свойственными каждому из
них клиническими и гистоморфологическими чертами. I тип—множест-
258
венные лейомиомы, развивающиеся из гладких мышц, поднимающих волос
или диагональных мышц, (Богров С. Л., 1922; Каламкарян А. А. и соавт.,
1978; Ormsby и Montgomery, 1955; Macetela-Ruiz, 1963; Runne et al., 1980
и др.). II тип—дартоидные (генитальные) солитарные лейомиомы, разви-
вающиеся из tunica dartos мошонки и гладких мышц грудных сосков (Со-
колов А. А., 1873; Lever и Schaumburg—Lever, 1975). Ill тип—солитарные
ангиолейомиомы, развивающиеся из мышечных стенок замыкающих ар-
терий и гладкомышечных элементов стенок мелких сосудов (Разумихин
В. С., 1928; Апатенко А. К., 1977; MacDonald и Sanderson, 1974; Harald
и Herwig, 1975). Некоторые авторы (Добронравов В. Н., 1960; Dahing и
Ayer, 1959) считают, что лейомиома и ангиолейомиома скорее порок раз-
вития, чем неоплазма в полном смысле этого слова. Встречаются единичные
описания семейной лейомиомы, что позволяет считать это заболевание
генетически обусловленным.
Клиника. Множественные лейомиомы кожи встречаются у мужчин
почти в два раза чаще, чем у женщин; ангиолейомиомы, напротив, у жен-
щин регистрируются чаще. Лейомиомы из мышц, поднимающих волос,
как правило, характеризуются мультицентрическим ростом, то есть яв-
ляются множественными. Это плотные приподнятые узелки правильных
очертаний, застойно-красного, коричневатого, синевато-красноватого цве-
та, величиной от булавочной головки до чечевицы, крупной фасоли и
больше. Поверхность узелков гладкая, блестящая. Локализуются на
туловище и конечностях, чаще располагаются группами (рис. 164, 165).
Характерной чертой лейомиом кожи является появление или усиление
болезненности под влиянием механического раздражения (трение одеждой.
Рис. 163. Лейомиома
кожи.
17*
Рис. 164. Лейомиома кожи генерализованная.
почесывание, давление или прикосновение) и охлаждения, а также спон-
танные приступы жестоких невыносимых болей длительностью в несколько
минут, нередко сопровождающихся расширением зрачков, снижением ар-
териального давления, побледнением кожи. Сдавление нервных клеток
лейомиомой может обусловить болевой синдром. Некоторые авторы отме-
чают медленное червеобразное сокращение узелков на холоде. Мы наблю-
дали нескольких больных со строго односторонним расположением множе-
ственных высыпных элементов, что не исключает возможности участия
периферической нервной системы в патогенезе.
Ангиолейомиома, по мнению многих авторов (Апатенко А. К-, 1963;
Ormsby и Montgomery, 1955; Gomez et al., 1983; и др.), развивается из
мышечных элементов мелких сосудов кожи. Клинически это солитарные,
но нередко диффузно-распространенные или локально-множественные об-
разования в виде плотных узелков-опухолей, слегка возвышающихся над
поверхностью кожи. Опухоль болезненна при надавливании, но менее чем
при лейомиомах. Часто ангиолейомиома располагается около крупных
суставов. Возраст больных—средний и пожилой.
Генитальная лейомиома (лейомиома tunica dartos) встречается редко.
Runne и соавт. (1980) обнаружили в литературе данные о 26 лейомиомах
мошонки. Локализуется чаще на мошонке, больших половых губах, реже—
на сосках и характеризуется одиночными плотными опухолевидными об-
разованиями буровато-красного цвета, величиной до 1,5—3 см в диаметре.
260
При воздействии холода или при трении может червеобразно сокращаться.
Лейомиома мошонки может развиваться из изолированно лежащих от-
дельных мышечных клеток дермы, напоминающих гистологически миофиб-
робласты. За редким исключением все эти опухоли доброкачественные.
Гистопатология. Лейомиома состоит из переплетающихся пучков глад-
комышечных волокон с умеренными соединительно-тканными прослойками-
в них, как правило, мало сосудов, но много нервных волокон (Торсуев
Н. А., 1940; Stout, 1953 и др.).
Ангиолейомиома располагается в глубине дермы и в подкожной клет-
чатке; состоит из переплетающихся тонких и коротких мышечных волокон;
ядра мышечных клеток овальные, веретенообразные, гиперхромные. От-
мечается обилие сосудов среди мышечных волокон (Апатенко А. К-, 1963)
Дифференциальный диагноз. Следует проводить с фибромами, ангио-
мами, фибросаркомами, нейрофибромами, ангиоретикулезом (саркомой)
Капоши, эпителиомами, лейомиосаркомами кожи и другими опухолями.
Лечение. Хирургическое иссечение, криотерапия, внутривенные или
внутримышечные вливания проспидина на курс 2—2,5 г.
ЛИТЕРАТУРА
Апатенко А. К- Мезенхимные и нейроэктодермальные опухоли и пороки развития
кожи.— Москва, 1977.
Каламкарян А. А., Самсонов В. А., Акимов В. Г. К вопросу о лейомиомах.—
Вестн. Дерматол. и венерол., 1978 4, 46—50.
Gomez М., Cabrera Н., Costa J. Lieiomiomas solitaries de la carai dos observaciones.
— Rev. Argeut. dermatol., 1983, 64, 2, 203—208.
Harald B., Hdriidig E. Das Angioleiomyoma der Haut.— Hautarzt, 1975, 26, 12, 638—
642.
Lever W., Schaumburg—Lewer G. Histopathology of the skin.— Sth edition.— Phi-
ladelphia, Lippincott, 1975.
MacDonald D., Sanderson K- Angioleiomyoma of the skin.— Brit. J. Dermatol., 1974,
91, 2, 161—168.
Runnel)., AntzH., PullmannH. Verrucoses Leiomyom des Skrotums unter dem BiL
de Spitzer Kondylome.— Z. Hautkr., 1980, 55, 10, 652—660.
ЛЕЙОМИОСАРКОМА КОЖИ (leiomyosarcoma cutis).
Синонимы: ангиолейомиосаркома, злокачественная лейомиома, лейо-
миобластома кожи, лейомиосаркома кожи и подкожной клетчатки.
Редко встречающаяся опухоль кожи гладкомышечного происхожде-
ния, представляет собой злокачественный аналог лейомиомы (ангиолейомио-
мы). Лейомиосаркома возникает либо первично из мышц поднимающих
волос и из мышц стенок сосудов, либо вторично в результате саркоматоз-
ного озлокачествления лейомиомы (Tappeiner и Wodniansky, 1961; Wevin-
ser и Newman, 1977 и др.).
Этиология и патогенез. Неизвестны.
261
Клиника. Лейомиосаркома может встречаться в любом возрасте. Опи-
сана она у 2-и 5-месячных детей (Heieck и Organ, 1970; Stout и Hill, 1958;
Gannopoulos и Stout, 1962), у 6-летнего мальчика (Glucker et al., 1972),
однако наиболее часто она регистрируется у лиц среднего и пожилого воз-
раста (старше 50—60 лет) одинаково часто у мужчин и женщин. Лейомио-
саркома—крупная опухоль с четкими контурами, плотноэластической
консистенции, располагающаяся в глубоких слоях дермы, с гладкой по-
верхностью, розоватого, розовато-синюшного цвета, размерами от 1,5 до
5—6 см и более в диаметре. Редко наблюдаются участки изъязвления и
геморрагий в центре опухолей. Иногда опухоль выбухает над поверхностью,
кожи, что придает ей сходство с дерматофибросаркомой. Наиболее часто
(у 50—60%) лейомиосаркомы локализуются на нижних конечностях; у
V4 больных леймиосаркома локализуется на голове и шее (Templeton,
1976; Dahl и Angervall, 1974; Kantor и Csato, 1979; Orellana-Diaz et al.,
1983 и др.). Из редких локализаций лейомиосарком следует назвать гени-
тальную (кожа полового члена и больших половых губ). В литературе мы
нашли лишь 13 случаев подобной локализации (Levy, 1932; Fagundes et
al., 1962; Hutcheson et al., 1969; Pandhl et al., 1975 и др.). Обычно лейо-
миосаркомы представлены солитарными опухолями, но встречаются и
множественные (Каламкарян А. А. и соавт., 1982).
Последние авторы приводят наблюдения больного 62 лет первичной
ангиолейомиосаркомой кожи и подкожной клетчатки с необычайной гене-
рализацией опухолевого процесса в коже в сочетании с метастазами в лег-
ких. Dahl и Andervall (1974) наблюдали лишь 7 таких случаев из 47 лейо-
миосарком кожи и подкожной клетчатки. Лейомиосаркома чаще не дает
субъективных ощущений, однако в терминальном периоде она может со-
провождаться сильными болями (Каламкарян А. А. и соавт., 1982; Haim
и Gellei, 1970; Fields и Helwig, 1981 и др.).
Лейомиосаркомы отличаются выраженной злокачественностью и обыч-
но приводят к летальному исходу в сроки от нескольких месяцев до 3—
6 лет. Рецидивы и метастазы после радикального хирургического иссече-
ния наблюдаются у 70—80% больных. Метастазирование при лейомио-
саркоме обычно гематогенное—в легкие, печень, селезенку, костный мозг,
.сердце, позвоночник и др.; лимфогенное распространение лейомиосаркомы
встречается значительно реже (Haim и Gellei, 1970 и др.).
Гистопатология. Разрастание атипичных уродливых гладкомышечных
элементов с обилием фигур патологических митозов. Обращает внимание
выраженный ядерный и клеточный полиморфизм. Почти в каждой опухо-
ли можно обнаружить известную склонность к расположению клеток в
виде частокола.
Дифференциальный диагноз. Следует проводить с карциномой, сар-
комой, ангиоретикулезом Капоши, дерматофибросаркомой, фибросарко-
мой, лейомиомой, ангиосаркомой и др.
Лечение. Радикальное иссечение с последующей лучевой или химио-
терапией.
262
ЛИТЕРАТУРА
Каламкарян А. А., Самсонов В. А., Акимов В. Г. К вопросу о лейомиомах кожи.
Вести, дерматол. и венерол., 1978, 4, 46—50.
Каламкарян А. А., Даниелян Э. Е. и соавт. Аигиолейомиосаркома кожи и под-
кожной клетчатки. Вести- Дерматол. и венерол., 1983, 3, 31—35.
Dahl J., Angewall L. Cutaneous and subcutaneous leiomyosarcoma. A clinico-patho-
iogic study of 47 patients.— Path. Europ., 1974, 4, 307—314.
Harnal P. Leiomyosarcoma of the penis. Caise report and review of the literature.—
Brit. J. Urol., 1975, 47, 3, 319—324.
Kantor E., Csato J. Leiomiosarcom.— Dermaro—venrrologia, 1979, 1, 55—58.
Pandhi R. Leiomyosarcoma of labium majus with extensive metastases.— Dermatolo-
gies, 1975, 150. 2, 70—74.
Templeton A. Superficial vascular tumours in Ugandan Africans.— E. Afr. Med. J.,
1976, 53, 3, 130—135.
Wang P., Hornstein O., Schricker K- Kutaneus Leiomyosarcom und osteomedullares
Plazmozytom mit Nachweis von IgA Kappa Paraprotein in Serum Und Hauttumor. — Haut-
arzt, 1976, 27, 9, 441—448.
Fields J., Helwig E. — Leiomyosarcoma of the skin and subcutaneous tissue. Cancer,
1981, 47, 1, 156—169.
ЛЕЙШМАНИОЗ КОЖИ (leishmaniosis cutis)
Синоним: Боровского болезнь
Этиология и патогенез. Возбудитель лейшманиоза был открыт и опи-
сан Боровским П. Ф. в 1898 году. Основным источником инфекции яв-
ляются грызуны (суслики, песчанки), реже больные люди; переносчики—
москиты, обычно Phlebptomus papatasii. Является эндемичным заболева-
нием. В нашей стране встречается преимущественно в Средней Азии, Азер-
байджанской ССР.
Клиника. Кожевников П. В. (1941) выделил две формы болезни Бо-
ровского: сельский, или остро некротизирующийся тип, возбудителем
которого является Leishmania tropica major, и городской, или поздно изъ-
язвляющийся тип, возбудителем которого является Leishmania tropica.
Сельский тип клинически проявляется возникновением на месте уку-
сов москитов фурункулоподобных узлов, регрессирующих в течение 3—
8 месяцев, оставляющих после себя иммунитет к этому возбудителю. Го-
родской тип протекает менее остро, характеризуется возникновением на
открытых участках бугорковых элементов, существующих длительно,
поздно изъязвляющихся (через 5—6 месяцев) и медленно рубцующихся
(в среднем в течение года). В редких случаях возникает туберкулоидный
(люпоидный) вариант болезни Боровского в зоне (реже вне ее) регресси-
ровавших ранее очагов поражения, на рубцах или вокруг них (рис. 166).
Реактивации оставшихся в тканях возбудителей способствуют общие
или локальные иммунные нарушения (Kurban, 1973; Farah, 1979; Добр-
жанская Р. С., 1969; Родякин Н. Ф, 1982). Таким образом, туберкуло-
идный тип болезни Боровского представляет собой затяжную или рециди-
263
вирующую форму (leishmaniasis
cutis recidivans) этого заболе-
вания. Она может развиться во
время или после окончания
рубцевания (иногда через не-
сколько лет) обычно протекав-
шего поздно изъязвляющегося
типа болезни Боровского (До-
бржанская Р. С., 1979). По
наблюдениям Родякина Н. Ф.
(1982), не было случаев тубер-
кулоидного лейшманиоза у лиц,
не болевших кожным лейшма-
ниозом, равным образом как и
у больных зоонозным типом
этого заболевания. Эту форму
впервые описал Christopherson
в 1923 году (цит. по Strick et
al., 1983). В нашей стране ее
выделил И. И. Гительзон (1932)
под названием металейшманиоз.
В тех случаях, когда процесс
развивается на некотором рас-
Рис. 165. Бугорковый лейшманиоз. стоянии от первоначального
очага, предполагается возмож-
ность реинфекции (Harman, 1979). Это подтверждается развитием тубер-
кулоидной реакции на месте введения больным L. tropica. У здоровых
на месте инъекции возникает гистиоцитарная пролиферация.
Появляются мелкие бугорки, располагающиеся сгруппированно в виде
кольцевидных, полициклических фигур,' полудуг. Цвет их желтовато-
буроватый, при диаскопии—отчетлив феномен яблочного желе. Реже встре-
чаются гипертрофические, напоминающие келоиды, папилломатозные, вер-
рукозные изменения. Описано развитие деструктивно-мутилирующих форм
(Kochs, 1958), псориазиформных очагов (Barsky et al., 1978). Добржан-
ская Р. С. (1964,1979) выделяет следующие разновидности: наиболее частую
нодулярную, затем диффузно-инфильтративную, язвенно-деформирующую,
эритематозную, опухолевидную и смешанную. Наиболее частная локали-
зация—лицо (до 80%, по данным Кожевникова П. В., 1961). Указывается
на возможность распространенных очагов, когда обширные участки тела
были покрыты псориазиформными бляшками (Strick et al., 1983).
Заболевание развивается чаще в молодом возрасте. По данным Родя-
кина Н. Ф. (1982), 2/3 больных—лица в возрасте от 2 до 18 лет. В 6,8% слу-
чаев развитие заболевания отмечено у пожилых. Может продолжаться
неопределенно долго, годами. Если очаги достигают больших размеров,
в их зоне возможно изъязвление.
264
Гистопатология. Туберкулоидные инфильтраты из гистиоцитов, лим-
фоцитов, гигантских и эпителиоидных клеток. Плазматических клеток и
полинуклеаров мало (Kurban et al., 1960). Возбудитель обнаруживают
редко; он менее вирулентен, чем выделенный от больных с поздно изъязв-
ляющимся лейшманиозом.
Дифференциальный диагноз. Дифференцировать заболевание необхо-
димо с туберкулезом кожи, саркоидозом, бугорковым сифилидом.
Лечение: солюсурмин, мономицин, криотерапия.
ЛИТЕРАТУРА
Добржанская Р. С. Клинико-серологические особенности и терапия кожного лейш-
маниоза. Дисс. докт., Ашхабад, 1979.
Baraky S., Storino W., Salgea K-, Knapp P. Cutaneous leishmaniasia.— Arch.
Derm., 1978, 114, 9, 1354—1355.
Farah F. S. Protozoan and helminth infections. In: «Dermatology in general Medicine»
Ed. Th. B. Fitzpatrick et al., 1979, 2-d ed., 1638—1656.
Harman R. R. Cutaneous leishmaniasis In «Textbook of Dermatology. Ed. by Rook
A. et al. Blackwell scientific publications, 3-d ed., Oxford, 1979, 1, 902—904.
Strick R. A., Borok M., Gasiorowski'H. C- Recurrent cutaneous leishmaniasis. J.
Am. Acad. Dermatol., 1983, 9, 3, 437—443
ЛЕЙШМАНИОЗ АМЕРИКАНСКИЙ КОЖНЫЙ (leishmaniosis cutis
americana).
Синонимы: бразильский кожный лейшманиоз, южноамериканский кож-
ный лейшманиоз, кожно-слизистый лейшманиоз, эспундия.
Распространенное эндемическое заболевание тропических и субтро-
пических стран; встречается преимущественно в Южной и Центральной
Америке. Спорадические случаи отмечены в других странах, в частности
в США, у лиц прибывших из эндемических зон.
Эпидемиология. Возбудителем американского лейшманиоза является
L. braziliensis, морфологически неотличимый от L. tropica, но отличаю-
щийся по антигенным свойствам (Koerber et al., 1978). Согласно исследо-
ваниям Walton et al., 1977; Price и Silvers, 197^; Kerdel-Vegas, 1982, и
др.), природными резервуарами лейшманиозной инфекции являются глав-
ным образом мелкие лесные грызуны. Переносчиками инфекции являются
кровососущие насекомые из рода Phlebotomus. Остается нерешенным воп-
рос о возможности прямого заражения от больного человека.
Клиника. Встречается у лиц обоего пола и любого возраста. Инкуба-
ционный период, как правило, длится от 2 недель до 3 месяцев; однако
известны случаи с более длительным инкубационным периодом. Заболева-
ние начинается с появления на коже открытых участков тела и особенно
на лице единичных и реже множественных мелких папул красновато-бу-
роватого цвета, имеющих на поверхности едва заметную тонкую чешуйку.
Через несколько недель папулы самостоятельно регрессируют, или, по-
степенно увеличиваясь, превращаются в узлы с веррукозными разраста-
265
Рис. 166. Лейшманиоз американский кожный.
ниями на поверхности. Узлы имеют мягкую тестоватую консистенцию.
В результате небольшой эксудации в углублениях и щелях боро-
давчатых разрастаний образуются плотные корки. В дальнейшем
в центре узла образуется некроз, а затем глубокая язва с плотноватыми,
неровными, изъеденными краями. Дно язвы чаще неровное, покрытое
серозно-гнойными корками и остатками некротического распада (рис. 166).
Язва может увеличиваться до значительных размеров, при этом во-
круг нее сохраняется довольно широкий вал нераспавшегося инфильтра-
та. Очень характерны регионарные лимфангоиты и лимфадениты, часто
уже на ранних стадиях заболевания. По ходу лимфатических сосудов не-
редко удается определить четкообразные утолщения, слегка болезненные
при пальпации. В некоторых случаях возникают не отдельные папулы или
узлы, а плоскостные инфильтраты, на поверхности которых в дальнейшем
образуются язвочки. У части больных в этой стадии могут быть общая
слабость, недомогание, головные и мышечные боли и др.
Для американского лейшманиоза в отличие от пендинской язвы или
кожного лейшманиоза характерно более частое поражение слизистых обо-
266
лочек с деструкцией тканей носа, полости рта, глотки и гортани. Аме-
риканская лейшмания не дает приобретенного иммунитета и поэтому воз-
можно гематогенное или лимфогенное распространение болезни на слизи-
стые оболочки после полного регрессирования кожных очагов пораже-
ния. Это вторичное вовлечение, известное как espundia, свидетельствует
о низком уровне иммунологических показателей, как это наблюдается
при иммунодепрессивной терапии (Moriarty, 1978).
По даннымHeimgariner и соавт. (1974), Kerdel-Vegas (1982), Faran(1983)
и др., слизистые оболочки поражаются у 30—80% больных.
В большинстве случаев через много лет (от 3 до 25 лет) после первых
кожных поражений, после так называемого скрытого периода, развиваются
поздние проявления заболевания. Они имеют вид обширных гранулема-
тозно-деструктивных и язвенных поражений, располагаются не только
на коже, но и на слизистой оболочке полости рта и носоглотки. Почти
всегда в начале поражается слизистая носа (хронический насморк, эксу-
дат, мелкие кровоизлияния, перфорация перегородки, деформация носа),
потом патологический процесс может перейти на верхнюю губу, мягкое'
небо, зев, миндалины, глотку, голосовые связки, гортань, трахею, бронхи
и др. Довольно рано вовлекаются хрящи носа, которые вскоре полностью
разрушаются, в результате чего нос деформируется (загибается книзу),
а верхняя губа утолщается и выступает вперед и вверх («нос тапира»).
Довольно часто у больных появляется кашель, охриплость голоса, дис-
фагия. Следует подчеркнуть, что поражаются в основном хрящи; даже в
самых тяжелых случаях кости лица, а также мышцы щек и языка оста-
ются непораженными. Общее состояние больных страдает мало. Иногда
отмечается умеренный лейкоцитоз, увеличение СОЭ. Следует отметить,
что обнаружить лейшмании в крови не удается.
Диагноз заболевания основывается на учете типичной клинической
картины и нахождения возбудителя в патологическом материале (микро-
скопически и бактериологически). Применяют и некоторые иммунологиче-
ские реакции (непрямой иммунофлюоресцентный тест), тест Монтенегро-
интрадермальное введение 0,1 мл взвеси жгутиковых лептомонад (лейш-
манина); тест положителен в 97—98% случаев (Mezzarda и Lazzaro, 1973;
Walton et al., 1974; Aikawa et al., 1982; Faran, 1983, и др.).
Прогноз благоприятный, хотя в некоторых злокачественных случаях
возможны значительные разрушения и распад мягких тканей лица, носо-
глотки, дыхательных путей, приводящие к смертельному исходу в ре-
зультате присоединения вторичной пиококковой инфекции или острой
респираторной инфекции (Faran, 1983).
Гистопатология. В зависимости от клинической картины и стадии за-
болевания (давность заболевания) в гистопатологии могут превалировать
иди явления хронического неспецифического воспаления, или туберку-
лоидные структуры. В свежих высыпаниях в-дерме имеется гранулема-
тозный инфильтрат, состоящий преимущественно из гистиоцитов и плаз-
матических клеток, макрофагов, а также небольшого числа нейтрофилов.
В старых высыпаниях инфильтрат приобретает туберкулоидное строение.
267
Язвенные поражения слизистых характеризуются преимущественно не-
специфическим воспалительным инфильтратом. В эпидермисе могут быть
выражены акантоз, псевдокарциноматозная гиперплазия.
Дифференциальный диагноз. Следует проводить с фрамбезией, пиодер-
мией, споротрихозом, эпителиомой, хромобластомикозом, карциномой,
лепрой, риносклеромой, гистиоцитозом, сифилисом.
Лечение. В настоящее время нет специфического средства для ле-
чения американского лейшманиоза. Выбор способа лечения во многом за-
висит от характера и локализации заболевания и от его длительности.
Назначают препараты сурьмы; так показано применение солюсурьмина
(препарат пятивалентной сурьмы), который вводят внутривенно ежед-
невно, начиная с 3 мл, постепенно увеличивая дозу до 8—10 мл. Полезно
также применение солюстибозана внутримышечно, на курс 60—80 мл.
Многие авторы сообщают об успешном применении глюкантима. Препарат
вводится внутривенно или внутримышечно ежедневно по 20 мл в течение
15—80 дней; после двухнедельного перерыва курс лечения повторяют.
Применяют также амфотерицин В, который вводят по 30 мг, курсовая до-
за 400—1800 мг.
При пиококковых осложнениях показано применение антибиотиков.
Местная противовоспалительная и дезинфицирующая терапия в стадии
изъязвления.
В отделе дерматологии ЦКВИ мы (Каламкарян А. А., Суколин Г. И.,
Федоров С. М., Зиновьев Г. А., 1985) наблюдали первый случай амери-
канского (слизисто-кожного) лейшманиоза в СССР у больного 60-ти лет с
длительно незаживающими язвами, отеком, эксудацией верхней губы,
перфорацией хрящевой перегородки носа, искривлением и деформацией
кончика носа (искривлением его книзу).
Больной с 1930 по 1957 г. проживал в Парагвае. Заболевание носа
началось после возвращения в СССР в 1961 г) Клинический диагноз кожно-
слизистого лейшманиоза подтвержден лабораторными исследованиями: по-
сев материала из области язвы на среду дал рост лейшманий Braziliensis.
Реакция непрямой иммунофлюоресценции с лейшманиозным антигеном
оказалась положительной в титрах Г.320. После курса лечения глюканти-
мом (по 20 мл ежедневно № 15) наступила полная эпитализация язвы верх-
ней губы. Больной выписан из клиники в состоянии клинической ремис-
сии.
ЛИТЕРАТУРА
Aikawa М., Hendricks L. et al. Interactions between macrophagelike cells and leish-
mania braziliensis in vitro.— Amer. J. Pathol., 1982, 108, 1, 50—59.
Castro R. Tratamento da leishmanioze tegumentar americane.— Ann. Bras. Dermatol.,
1980, 55, 2, 87—89.
Faran. F. Parasitic disease.— In: Clinical dermatology. Philadelphia, 1983, 4, 18, 2,
1—21.
268
Koerber W. et al. Treatment of cutaneous leishmaniasis with antimoni sodium gluco-
nate.— Arch. Dermatol., 1978, 114, 1226—1229.
Moriarty P. Diagnosis and prognosis of New World Leishmaniasis.— Arch- Dermatol.,
1978, 114, 962—963.
Price S., Silvers D. New Korld leishmaniasis.— Arch. DermatoL, 1977, 113—, 1415
1416.
Shaw J., Lainson R. Leishmaniasis in Brazil: some observations on intradermal rea-
ctions to different trypanosomatid antigens of patients suffering from cutaneuosus and muco-
cutaneous leishmaniasis.— Trans. R. Soc. Prop. Med. Hyg., 1975, 69, 323—335.
Walton B. et al. American cutaneous leishmaniasis.— JAMA, 1974, 228, 1256—1258.
Walton B. et al. Differences in biological characteristics of three leishmania isolates
from patients with espundia.— Amer. J. Trop. Med. Hyg., 1977, 76, 850—855.
ЛЕНТИГИНОЗ ПЕРИОРИФИЦИАЛЬНЫЙ (lentiginosis periorificial is)
Синоним: синдром Пейтца—Егерса—Турена.
Этиология и патогенез. Заболевание рассматривается как нейро-ме-
зенхимальная дисплазия, вызванная генной мутацией, передающаяся
аутосомно-доминантно с плейоотропным эффектом. Eberle и Klostermann
(1968) не обнаружили изменений в структуре и количестве хромосом, ха-
рактерных для этого синдрома. Klostermann (1963), наблюдавший в семье
15 больных, относит этот синдром к факоматозам. Из 31 случая, проанали-
зированных Touraine и Coder (1946), в 22 было несколько больных в се-
мье в 3—4 поколениях. Может быть значительная вариабельность и дис-
социация признаков среди родственников. Например, у больной, наблю-
давшейся Farmer и соавт. (1963), был полный синдром, у ее брата—
только пигментации, у отца—полипоз. По поводу малосимптомных слу-
чаев Klostermann замечает, что при оценке этого явления должны учиты-
ваться даже минимальные изменения, недостаточные для постановки диаг-
ноза, но могущие быть связанными со специфическим эффектом г'ена, а
также вариабельность временного интервала, в котором более характерные
признаки появятся или, наоборот, исчезнут, как, например, пигментация.
Кроме того, отсутствие клинически и рентгенологически полипоза еще
не исключает изменений в слизистой, которые могли проявляться в микро-
скопически малых папилломах. Подтверждением этому могут служить дан-
ные Р. В. Савиной и соавт. (1983), обнаруживших гистологически гамартом-
ные кисты в зоне макроскопически неизмененных участков слизистой обо-
лочки желудка у больной с синдромом Пейтца—Егерса 18 лет, погибшей
от рака желудка. Спорадические случаи считаются проявлением новых
мутаций (Reddy et al., 1976).
Клиника. Множественные мелкие пигментные пятна от светло-коричне-
вого до черного цвета овальных или округлых очертаний, густо располо-
женные вокруг и в полости рта, на губах, особенно нижней, периназально,
периорбитально, реже на конечностях (на ладонях и подошвах, тыльной
поверхности пальцев) —рисунки 168, 169, 170. Описаны случаи с ограни-
ченными высыпаниями в полости рта и на красной кайме губ (Hamack,
1960). В полости рта могут быть и папилломатозные изменения (Lowe,
269
Рис. 168. Лентигиноз
периорифициальный
(синдром Пейтц а—Егерса—Турена).
Рис. 167. Лентигиноз
периорифициальный
(синдрсм Пейтца—Егерса—Турена).
1975). А. В. Брайцев и Г. М.
Большакова (1960) описали ге-
нерализованные лентигинозные
высыпания. По. данным Bart-
nolomew (1957), красная кайма
губ поражается у 96% больных,
слизистая щек—у 83%, кожа
вокруг естественных отверстий
лица—у 36%, конечностей—у
32%. Изменения могут быть и
на роговице (Degos, 1981).
Заболевание ограничивает-
ся только кожными изменени-
ями лишь у единичных боль-
ных, у большинства наблюда-
ется сочетание с множествен-
Рис. 169.' Синдром
Пейтца—Егерса—Турена.
270
ными полипами желудочно-кишечного тракта, особенно тонкого кишеч-
ника (Klostermann, 1966). Полипы могут быть и другой локализации:
мочевой пузырь, почечные лоханки, мочеточник, бронхи, нос (Me Kusick,
1976). На основании анализа литературы Klostermann пришел к выводу,
что в 33% случаев проявления полипоза развиваются в первом десятилетии
жизни, 71 %—к концу второго и 31 %—третьего. Но эти сроки, по наблю-
дениям автора, могут быть значительно более поздними, с появлением ки-
шечной симптоматики в 38- и даже 50-летнем возрасте. Bandler (1950)
описал семью, в которой у одного из членов семьи имелся множественный
лентигиноз с локализацией на губах, периорально, на слизистой щек, а
также на тыльной поверхности кистей и пальцев, в сочетании с множест-
венными гемангиомами тонкого кишечника, но без полипов. У нескольких
других родственников был множественный гемангиоматоз кишечника без
лентигиноза. Автор‘ставит вопрос о взаимодействии генов, ответственных
за развитие пигментации, кишечных полипов и гемангиом. У больных,
описанных Торсуевым Н. А. (1973), наблюдалось сочетание с множествен-
ными пигментными, в том числе гигантскими, невусами. Заварова Р. Ф. и
соавт. (1979) наблюдали больного с аденоидами и ахилией.
Заболевание развивается в первые годы жизни, но может существо-
вать с рождения. В редких случаях периорифициальный лентигиноз воз-
никает у взрослых (Hofer et al., 1967). С возрастом интенсивность пигмен-
тации на коже может изменяться, в полости рта она обычно остается без
изменений (Me Allester et al., 1967). Начиная с подросткового возраста
или немного позже, начинает клинически проявляться полипоз желудка и
кишечника: боли, рвота, кровотечение, симптомы непроходимости, инваги-
нации, вторичной анемии. Зачастую синдром выявляется из-за развившихся
осложнений после хирургического вмешательства по поводу непроходи-
мости или развития опухоли (Payson и Мои mgis, 1967; Griffits и Bisset, 1980).
Иногда больные подвергаются многократным операциям. Наиболее неблаго-
приятным считается повышенный риск злокачественного перерождения
полипов, преимущественно желудка, 12-перстной кишки, толстого кишеч-
ника. Данные о частоте развития карцином неоднородны. В частности,
имеется мнение (Belisario, 1971), что полипы при этом синдроме представ-
ляют собой гамартомы и поэтому протекают доброкачественно. Castelman
и Kruickstein (1962) нашли в 33 случаях из 57, диагностированных как
карцинома на фоне полипов, аденоматозные полипы с очаговой атипией.
Newbold (1970), приводя данные Achord и Proctor (1963) о том, что висце-
ральный канцер развивается примерн э у 20% всех больных лентигинозом
периорифициальным, отмечает трудность истинной оценки частоты, так
как неясно, все ли случаи, исследованные гистологически, являются зло-
качественными опухолями, а не гамартомами. Кроме того, в эту серию
могли быть включены другие формы наследственных полипозов кишечника
с большей потенциальностью к раковому перерождению. Более вероятными
автор считает данные Asman и Pierse (1970) о 10%-ной или меньшей частоте
развития рака в сравнении с 60% или более—при семейном полипозе.
По мнению Reid (1974), при синдроме Пейтца—Егерса—Турена имеется
271
повышенный риск развития рака кишечника, составляющий 2—3%. Опу-
холи чаще развиваются в зоне 12-перстной кишки. Неоплазма может воз-
никнуть или на нормальной слизистой или из полипов, развившихся при
этом синдроме. Что касается полипов толстого кишечника, то здесь может
быть сочетание двух типов—гамартомных и аденоматозных, часто пере-
ходящих в карциному. Автор поддерживает точку зрения Olansky и Achord
(1969) о том, что превентивное хирургическое вмешательство не показано
и не выполнимо, но настороженность—обязательна. Harnack (1960) пола-
гает, что оперативное вмешательство в основном показано как средство
профилактики инвагинаций. Nishiyama и соавт. (1965), описывая диффе-
ренциальную диагностику между синдромом Peutz—Jeghers и другими
состояниями, протекающими с изменениями кожи и полипозом кишечника,
отмечают в качестве одного из признаков, отличающих этот синдром, боль-
шую опасность возникновения инвагинаций, чем злокачественной опухоли.
Указывается, что злокачественное перерождение с метастазами наблюда-
ется чрезвычайно редко. К таким достоверным случаям автор относит толь-
ко наблюдения Achord и Proctor (1963) и Нот и соавт. (1963). По мнению
Degos (1981), злокачественное перерождение наблюдается в 2—5% случаев.
На основании анализа литературы Farmer и соавт. (1963) отмечают, что
наиболее частым является боль в животе, вызванная инвагинацией поли-
пов (около 85%), затем кровотечение, анемия, общая слабость. Хирургиче-
ское вмешательство рекомендуется только при непроходимости или крово-
течении, но не как мера профилактики, а радикальные или обширные хи-
рургические вмешательства считаются нецелесообразными. С точки зрения
автора, необходимо проводить тщательный дифференциальный диагноз
между почти совершенно доброкачественным синдромам Peutz—Jeghers и
предзлокачественным семейным полипозом толстого кишечника, что бы-
вает трудно, если нет пигментаций или имеется выраженный полипоз тол-
стого кишечника. Но даже, если при синдроме Peutz—Jeghers обнаружи-
вается эта локализация полипов, это не является показанием для тоталь-
ной колонэктомии, как при семейном • полипозе или синдроме Gardner.
Авторы считают, что полипы при этом синдроме являются гамартомами.
Newbold заключает: чем тяжелее полипозное поражение кишечника, тем
больше частота изменений на коже и больше вероятность развития раковой
опухоли. Burdick и Prior (1982) указывают на повышенный риск развития
у больных также опухолей мочеполовых органов, легких, яичников, мо-
лочной железы.
Гистопатология. Увеличение пигмента, количества меланоцитов в ба-
зальном слое, макрофагов, содержащих пигмент, в дерме. В некоторых зо-
нах меланоциты могут выявляться в виде гнезд в зоне дермо-эпидермаль-
ного соединения.
Дифференциальный диагноз. Дифференцировать лентигиноз периори-
фициальный необходимо от веснушек, различных форм генерализованного
лентиго (Leopard—синдром и др.), центрофационального лентигиноза, га-
строинтестинального полипоза (Cronkite—Canada синдром), болезни Ад-
дисона, синдрома Gardner.
272
Лечение. Хирургическое удаление полипов. Диспансерное наблюде-
ние за больными с обследованием родственников I степени родства.
ЛИТЕРАТУРА
Заварова Т. Ф., Шупенько Н. М., Белобровая Д'. М. Синдром Пейтца—Турена—
Егерса. Вести, дерматол. и венерол. 1979, 6, 34—36.
Савина Т. В., Поляков В. С., Будаев К- Д- Кисты тела желез слизистой оболочки
желудка у больной с синдромом Пейтца—Егерса и раком желудка. Арх. патол. 1983, 1,
65—68.
Degos R. Derma tologie, Paris 1981, 3, 754 d-754 p.
Griff its C. D. M., Bisset W. H. Peutz—Jeghezs syndrome.— Arch. dis. childhood,
1980, 55, 11, 866—869.
Lowe N. J. Peutz—Jeghers syndrome with pigmented oral papillomas.— Arch, der-
matol., 1975, 111, 4, 503—505.
(McRusik V. A. у MakkbiocuK. B. A. — Наследственные признаки человека. Пер. с
англ., М., Медицина, 1976,
Reddy В. S. N., Singh G. Chandra S., Rao R. Peutz—Jeghers syndrome.— Qerma-
tologica, 1976, 152, 6, 367—371.
Reid J. D. Intestinal carcinoma in the Peutz—Jeghers syndrome.— JAMA, 1974, 229,
7, 833—834.
ЛЕНТИГИНОЗ ЭНДОБУКАЛЬНЫЙ (lentiginose endobuccale).
Синонимы: идиопатическая лентикулярная пигментация слизистой
щек, болезнь Ложье.
В 1970 г. Laugier и Hunziker описали случай идиопатической ленти-
кулярной, меланотической пигментации слизистой щек и губ. Тщательно
проведенное гастро-энтерологическое исследование позволило авторам иск-
лючить наличие у больного злокачественного периорифициального лен-
тигиноза (синдрома Пейтца—Егерса—Турена) и описываемые изменения
рассматривать как самостоятельное заболевание. Pellerat (1971) предло-
жил называть данное заболевание идиопатической меланотической пигмен-
тацией Ложье. На заседании Французского дерматологического общества
12—13 марта 1976 г. в Лилле был представлен аналогичный случай под
названием болезни Ложье, или «эндобукального лентигиноза». В сообщени-
ях Graciansky и соавт. (1975), Grupper (1976), Konrad и Wolff (1973), Sarto-
ris и соавт. (1975) и др. указывается на исключительную локализацию пиг-
ментаций на слизистой рта и красной кайме губ и отсутствие периорифи-
циальных высыпаний. Ни в одном случае не выявлено желудочно-кишеч-
ной симптоматики периорифициального лентигиноза (висцерального по-
липоза).
Этиология и патогенез. Они до настоящего времени остаются невыяс-
ненными; возможно, что это врожденная локальная аномалия пигменто-
образования. Не исключена возможность и наследственного характера.
Клиника. Заболевание встречается одинаково часто у мужчин и жен-
щин в возрасте от 8 до 87 лет. Время начала заболевания неизвестно, так
как высыпания не сопровождаются субъективными ощущениями и выяв-
18 Клиническая дерматология 273
ляются случайно. Клиническая картина довольно четкая -наличие тем-
но-бурых пятен на слизистой рта, иногда с захватом красной каймы губ
(но никогда периорифициально). Основным отличием от синдрома Пейтца—
Егерса—Турена является отсутствие висцерального полипоза. Отсюда
важность гастро-энтерологического обследования больных меланотически-
ми высыпаниями на слизистых.
В отделе дерматологии ЦКВИ (Каламкарян А. А. и Суколин Г. И.,
1985) наблюдали больного 42 лет, у которого на слизистой щек и красной
кайме губ в течение 15—20 лет имеются буроватые, темно-коричневые
с фиолетовым оттенком пятнистые элементы, не дающие субъективных ощу-
щений. Цвет пятен при надавливании не изменяется. Ректоромано-эзофа-
. гогастродуоденоскопия патологии не выявила. Все вышеизложенное по-
зволило установить диагноз идиопатической лентикулярной пигментации
слизистой щек (болезнь Ложье).
Гистопатология. В пятнистых очагах выраженная гиперпигментация
базальных клеток, других изменений нет. Базальная мембрана интактна,
меланоциты в дерме не обнаруживаются. Электронно-микроскопически
выявляется большое количество меланосом различного размера.
Дифференциальный диагноз. Следует проводить с синдромом Пейтца-
Егерса—Турена, пигментными высыпаниями медикаментозной природы
(например, фиксированная лекарственная эритема).
Лечение. С косметической целью, при локализации пигментаций на
красной кайме губ можно применить дерматоэксцизию в пределах эпидер-
миса.
ЛИТЕРАТУРА
Cordonnier V., Beer Р et al. Maladie de Laugier (lentiginose endobuccale). Communi-
cation presentee a la Soc. Franc. Derm., Reunion de Lille, 12—13 mars, 1976.
Graciansky P., de Beer F., Dassonoille M. Pigmentation melanique acquise de fa
(evre inferieure.— Bull. Soc. RFranc. Derm. Syph., 1975, 82, 258—263.
Grupper Ch. Pigmentation de la muqueuse buccale et labiate.— Soc. Frans. Derm,,
rnai. 1976.
Sartoris S., Pippione M, De Paoli M., Ceroetti 0. Pigmentazione melanica idiopa-
tica.— G. Min. Derm., 1975, 110, 382—385.
ЛИМФОЦИТОМА КОЖИ (lymphocytoma cutis).
Синонимы: доброкачественный лимфоденоз Бефферштедта, саркоид
Шпйглера—Фендта, лимфоплазия кожи, доброкачественная лимфоцитома
кожи.
До настоящего времени еще нет всеми признанного определения лим-
фоцитомы кожи. В литературе наиболее часто употребляется термин лим-
фоцитома, предложенный Kaufmann-Wolf в 1921 г. Однако первое опи-
сание принадлежит Spiegler (1894) и Fendt (1900), которые описали подоб-
ное заболевание под названием саркоматоз (позднее как саркоид Шпиг-
лера—Фендта). В 1943 г. Bafverstedt объединил доброкачественные опу-
холи кожи с поражением лимфоретикулярной ткани под названием добро-
274
качественного лимфоденоза кожи. Среди отечественных авторов лимфо-
цитому наблюдали Поспелов А. И. (1895—1896), Жорно Я. Ф. и Василье-
ва Л. И. (1952), Шинский Г. Э. и соавт. (1965), Потекаев Н. С. и соавт.,
(1978, 1980).
Этиология и патогенез. О сущности лимфоцитомы высказаны разно-
образные точки зрения. Pillsbury и соавт. (1956) локализованные формы
рассматривают как фолликулярную лимфому, а диссеминированные формы
относят к ретикулярноклеточной лимфоме. Piorini (1959) также относит
лнмфоцитому к доброкачественным фолликулярным лимфомам, противо-
поставляя их злокачественным лимфомам. Некоторые авторы (Gottron,
1960; Rimbaud et al., 1962; Wentholt et al., 1953 и др.) относят лимфо-
цитому к преходящим воспалительно-реактивным ретикулезам, т. е.
к ретикулярной гиперплазии в ответ на раздражение. В 1965 г. Mach, обоб-
щив данные литературы и 107 собственных наблюдений, предложил новый
термин—доброкачественные лимфоплазии кожи, основанный на концепции
реактивной гиперплазии предшествующей лимфоидной ткани. Саго и Hel-
wig (1969) среди кожной лимфоидной гиперплазии выделяют лимфоцитомы,
лимфоцитарную инфильтрацию Jessner—Kanof и гранулематозные реакции
на укус насекомых. Cramer (1962), Mach (1966) лимфоцитарную инфильт-
рацию Jessner—Kanof относят к лимфоцитоме кожи, что, по нашему мне-
нию, является недостаточно обоснованным. В последнее время Потекаев
Н. С. и соавт. (1978, 1980), Lever и Schaumburg—Lever (1975), Berg и
соавт. (1978) относят лнмфоцитому в группу псевдолимфом кожи. Выска-
зывается мнение, что лимфоцитома кожи вызывается различными раздра-
жителями (укусы насекомых, травмы, прививочные рубцы, инфекции),
она может сопровождать фотодерматозы, опухолевые процессы, хрониче-
ский атрофирующий акросклероз, хроническую мигриующую эритему Аф-
целиуса—Липшютца. Мы, как и большинство авторов, полагаем, что лим-
фоцитому следует отнести к доброкачественной реактивной лимфоретику-
лярной пролиферации.
Клиника. Лимфоцитома встречается во всех возрастных группах, пре-
имущественно в молодом и зрелом возрасте, несколько чаще у женщин.
Клинические проявления лимфоцитомы кожи отличаются большим поли-
морфизмом, что приводит к частым диагностическим ошибкам и поздней
диагностике. Среди ошибочных диагнозов наиболее часто встречаются
саркоид, лимфосаркома, ретикулосаркома, дискоидная красная волчан-
ка, узловатая почесуха, красный плоский лишай.
Встречаются два типа этого заболевания: локализованный (солитарный)
и диссеминированный. Больные с одиночными опухолями составляют 2/3
случаев, с множественными—1/3 случаев. После 20—30 лет увеличивается
число больных с множественными опухолями. При обеих формах вы-
сыпания представлены узелками, поверхностными плоскими инфильтра-
тами и опухолями. Узелки величиной с горошину или крупнее четко вы-
деляются на фоне окружающей кожи, они имеют мягковатую или плотно-
эластическую консистенцию (рис. 171, 1'72). Цвет узелков синюшно-фио-
летовый или буро-коричневый. Нередко они сопровождаются нежной кож-
18* 275
кожи.
Рис. 171. Лимфоцитами
кожи (после пиявок).
ной атрофией, по-видимому, остающейся после спонтанно регрессирующей
лимфоцитомы. При диаскопии узелков видна желтая окраска, однородная
или с просвечивающими зернами (в результате мелких отложений гемо-
сидерина), напоминающими люпомы. Субъективных ощущений обычно Нет.
Узелковые высыпания растут медленно и редко рецидивируют после лече-
ния рентгеновскими лучами.
Поверхностно-инфильтративная форма клинически характеризуется
дискообразными слегка возвышенными инфильтратами буровато-коричне-
вого или розовато-синюшного цвета с гладкой поверхностью, иногда с
мелкими узелками в окружности. Наблюдающееся в редких случаях незна-
чительное шелушение создает сходство с дискоидной красной волчанкой.
Этот вариант клинически может также напоминать лимфоцитарную ин-
фильтрацию Иесснера—Канофа.
При опухолевой форме—опухоли величиной от фасоли до вишни и
больше чаще имеют плотно-эластическую консистенцию, возвышаются на
1—2 см над уровнем кожи. Цвет их вначале розовато-красный, позже при-
обретает синюшно-розоватую окраску. Опухоли, как правило, заложены
в коже и подкожной клетчатке, тесно спаяны с кожей, но подвижны в от-
ношении подлежащих тканей.
Течение лимфоцитомы хроническое (от нескольких месяцев до мно-
гих лет), волнообразное; спонтанные ремиссии сменяются частыми реци-
дивами после любого метода лечения. Лимфоцитома кожи, как правило,
не сопровождается увеличением периферических лимфатических узлов,
изменения миелограммы не существенны, гематологические показатели
'у многих больных остаются нормальными. Лишь у части больных в пери-
ферической крови выявляется относительный лимфоцитоз (ст 30 до 40%).
276
В биохимических показателях сыворотки крови (уровень сахара, били-
рубина, холестерина, остаточного азота, протромбина и белковых фракций)
не выявлены особенности, характерные для этого заболевания.
Прогноз благоприятный, лимфоцитома кожи в основном представляет
собой доброкачественный процесс. Потенциальная злокачественность лим-
фоцитомы очень мала, однако вопрос о возможной трансформации ее в
истинную лимфому пока остается открытым.
Гистопатология. В дерме и нередко гиподерме отмечается массивный
инфильтрат, состоящий из лимфоидно-ретикулярных и гистиоцитарных
клеток, обычно отграниченный от эпидермиса узкой зоной нормального
коллагена. При этом часто обнаруживается фолликулярное строение, т. е.
имеются структуры, похожие на фолликулы (центры размножения) лим-
фоузлов. В составе инфильтрата встречаются также эозинофильные и
нейтрофильные лейкоциты, плазматические клетки. Эпидермис не изменен.
Mach (1965) выделяет пять гистологических типов лимфоцитомы: лимфати-
ческий, ретикулярно-клеточный, гранулематозный, гигантофолликуляр-
ный и плазмоклеточный, подчеркивая, что каждый из указанных типов
может переходить в любой другой.
Электронно-микроскопическое исследование лимфоцитомы показало,
что она почти исключительно состоит из лимфоидных и ретикулярных кле-
ток в различных стадиях развития (Mach, 1966). Иммунологические и
энзимоцитохимические исследования обнаруживают в дерме инфильтраты,
состоящие преимущественно из малых лимфоцитов и больших ретикуляр-
ных клеток. В-лимфоциты преобладают над Т-лимфоцитами. Большие
ретикулярные клетки показывают высокую активность кислой фосфатазы
и неспецифнческих эстераз (Braun-Falco и Burg, 1975)
Дифференциальный диагноз. В зависимости от клинических проявле-
ний лимфоцитомы его следует проводить с эозинофильной гранулемой, сар-
коидом, туберкулезной волчанкой, красной волчанкой, лимфоцитарной
инфильтрациией Иесснера-Канофа, со специфическими кожными инфильт-
ратами при лейкозах и др.
Лечение: антибиотики, рентгенотерапия (высокая чувствительность к
рентгеновским лучам), хирургическое удаление с последующим облучением
умеренными дозами рентгеновских лучей.
ЛИТЕРАТУРА
Потекаев И. С., Иванов О. Л., Сергеев Ю. В. Доброкачественная лимфоплазия
кожи.— Вестн. дерматол. и венерол., 1980, 6, 47—52.
Braun-Falco О. L., Burg G. Lymphoretikulare Proliferationen in der Haut. Cyto-
chemische und immuncytologische Untersuchungen bei Lymphadenosis benigna cutis.— Hau-
tarzt, 1975, 26, 124—132.
Lever W. F., Schaumburg-Lever G. Histopathology of the skin.— Philadelphiaa,
1975.
277
ЛИПОДИСТРОФИЯ ПРОГРЕССИРУЮЩАЯ (lipodystrophia progressi-
va).
Синонимы: липоатрофия, болезнь Барракера-Симона.
Относительно редкое заболевание, характеризующееся атрофией под-
кожной жировой клетчатки лица, шеи, плечевого пояса, грудной клетки
при нормальном или избыточном отложении жира в нижней половине тела.
В отечественной и зарубежной дерматологической литературе имеются
единичные описания этого заболевания (Каламкарян А. А., 1958; Бухаро-
вич М. Н и соавт., 1981; Schirren, 1962; Amroliwalla, 1977).
Этиология и патогенез. Кожевников П В. (1964), Degos (1961) рас-
сматривают прогрессирующую липодистрофию как аномалию развития.
В литературе придается значение нарушениям мезэнцефалона, гипофиза,
щитовидной железы, яичников. Иногда развитию липодистрофии пред-
шествуют общие инфекции (корь, скарлатина, коклюш, энцефалит), трав-
ма черепа и др.
Рис. 172. Липодистрофия
прогрессирующая.
278
Клиника. Заболевание чаще встречается у девочек и начинается в
6—8-летнем возрасте (Dupperrat, 1959; Попов Л., 1963; Danidson, 1977 и
др.). Иногда заболевание носит семейный характер. Процесс атрофии под-
кожной жировой ткани развивается медленно, начинаясь с головы и по-
степенно симметрично распространяясь вниз на грудь и верхние конечно-
сти. Достигнув максимального распространения, процесс как бы заканчи-
вается, и клиническая картина остается стабильной до конца жизни. В
отдельных случаях атрофия подкожного жира ограничивается только ли-
цом и дальше не распространяется. Подкожная жировая клетчатка в ниж-
ней части живота, на лобке, бедрах сохраняется. Из-за атрофии подкожной
клетчатки в верхней части туловища создается впечатление, что живот,
ягодицы, бедра, чрезмерно крупные. Довольно часто при липодистрофии
отмечаются трофические изменения кожи, ногтей, волос. В очагах
поражения кожа несколько истончена, сероватого цвета, легко собирается в
складку (рис. 173) Общее состояние пациентов обычно удовлетворитель-
ное. У некоторых из них при физической нагрузке отмечается повышенная
утомляемость, мышечная слабость. Со стороны внутренних органов каких-
либо типичных изменений не определяется. Лабораторные исследования
не выявляют заметных отклонений в процессах обмена, в составе крови
и мочи. В части случаев заболевание может сочетаться с диабетом, пора-
жением почек, нарушением менструального цикла, гиперхолестеринемией,
психической лабильнос"ью (Rook et al., 1979; Peters, 1973 и др.). Течение
заболевания может быть различным. Наряду со стабильными на протяже-
нии ряда лет формами наблюдаются случаи быстрого прогрессирования
атрофии подкожной жировой ткани.
Гистопатология. Почти полное отсутствие жира в подкожной клетчатке.
Дифференциальный диагноз. Следует проводить с прогрессирующей
гемиатрофией лица Ромберга, с локальной липоатрофией на месте введе-
ния инсулина у больных сахарным диабетом.
Лечение. Лечебные мероприятия следует направлять на устранение
сопутствующих нарушений эндокринных желез и нервной системы. Вита-
мины комплекса В, бромиды и другие укрепляющие нервную систему сред-
ства.
ЛИТЕРАТУРА
Бухарович М. Н., Остропалец С. С., Баграчева А. И. Случай симметричной про-
грессирующей липодистрофии.— Вести, дерматол. и венерол., 1981, 1, 37—40.
Amroliwalla F. Vaccination scar with solf—tissue atrophy restored by local insulin
treatment.— Brit. med. J'., 1977, 1, 1389—1390.
ЛИХЕН СКЛЕРОЗИРУЮЩИЙ И АТРОФИЧЕСКИЙ (lichen sclerosus
et atrophicans).
Синонимы: белый лишай Цумбуша, болезнь белых пятен.
Впервые описал Hallopeau в 1889 г. как склерозный тип плоского ли-
шая. Однако в дальнейшем он различал две формы склерозирующего ли-
279
хена: первично склерозирующий лихен и вторичный, развивающийся из
обычного красного плоского лишая.
Этиология и патогенез. Патогенетическую роль могут играть различ-
ные факторы, в том числе эндокринные, инфекционные, нейрогенные и др.
Не решен вопрос о самостоятельности первичного склерозирующего ли-
хена. Ряд авторов (Miescher, 1948; Montgomery и Hill, 1940; Kogoj, 1934;
Bedi и Arunthathi, 1975 и др.) выделяют его в самостоятельную клиниче-
скую форму, отличающуюся от локализованной склеродермии и от атрофи-
ческой фоРмы красного плоского лишая. В то же время Unna и соавт.
(1931), Машкиллейсон А. Л. (1958), Кожевников П. В. (1964), Вербенко
Е. В. и соавт., 1979, Каламкарян А. А. и Федорова Е. Г. (1980), Никити-
на М. Н. (1980), Трофимова Л. Я. и соавт. (1983), Чистякова И. А., 1984
и др. считают его разновидностью склеродермии, указывая, что нередко
у одного и того же больного одновременно можно видеть склероатрофиче-
ский лихен и типичные очаги бляшечной склеродермии. Не так редко встре-
чаются случаи изолированного склероатрофического лихена головки и
препуциального листка полового члена (Post и Janner, 1975). Lever и Scha-
umburg-Lever (1975) полагают, что крауроз вульвы и облитерирующий
ксеротический баланит, рассматривавшиеся в прошлом как самостоятель-
ные дерматозы, по-видимому, идентичны склеротическому и атрофическому
лихену. Некоторые современные авторы (Каламкарян А. А. и Аковбян
Рис. 173. Склероатрофический
лихен.
Рис. 174. Склероатрофический
лихен.
В. А., 1978; Montgomery и Laymon, 1955) указывают, что склероатрофиче-
ский лихен гениталий и крауроз вульвы являются самостоятельными забо-
леваниями, подчеркивая при этом разницу в течении и терапии этих забо-
леваний.
Клиника. Болеют преимущественно взрослые, чаще женщины, очень
редко дети (Bedi и Arunthathi, 1975 и др.). Появляются мелкие узелки
(3—6 мм в диаметре) округлых или реже неправильных очертаний, раз-
бросанные или сгруппированные, иногда сливающиеся в небольшие бляш-
ки. Цвет узелков алебастровый, асбестовый с перламутровым блеском.
При локализации процесса на нижних конечностях окраска как отдель-
280
Рис. 175. Буллезная
форма склероатрофи-
ческого лихена.
ных высыпаний, так и бляшек имеет ливидный оттенок. Кожный рисунок
на узелках сглажен, они производят впечатление несколько запавшйх,
пергаментных (рис. 174, 175, 176). Субъективных жалоб не вызывают. В
некоторых случаях периферическая часть этих пятен-узелков лилового
цвета, напоминает сиреневое кольцо, наблюдающееся при бляшечной
склеродермии. В очагах поражения иногда возникают пузыри. Буллез-
ную или пемфигоидную форму болезни белых пятен наблюдали Георгиев
(1960), Машкиллейсон А. Л. (1978), Каламкарян А. А. и Федорова Е. Г.
(1982), Голоусенко И. Ю. (1982), Garb и Sims (1959), Eppard и Sneddon
(1975), Silverio и Serri (1975) и др. Локализуются высыпания большей ча-
стью на шее, верхней части груди, плечах, на спине, животе, бедрах и дру-
гих местах. Склеротический и атрофический лихен наблюдается не только
на коже, но и в области вульвы и на головке полового члена, где пораже-
ние может быть изолированным. Течение болезни хроническое. При вторич-
ном склерозирующем и атрофическом лихене часто удается обнаружить
остатки типичных высыпаний красного плоского лишая. Кроме того, боль-
ные обычно жалуются на зуд.
Hadlich, Linse (1983) описали у двух больных гиперкератотическую
и телеангиэктатическую форму склероатрофического лихена.
Гистопатология. Гиперкератоз с роговыми пробками, атрофия маль-
пигиевой сети, выраженный отек и гомогенизация коллагена в верхней час-
ти дермы и воспалительный инфильтрат в средней части дермы.
Дифференциальный диагноз. Следует проводить с красным плоским
лишаем, кра^розом вульвы и полового члена.
Лечение. Витамины А, Е, Д2, антибиотики (пенициллин), ферментные
препараты (лидаза, ронидаза, трипсин и др.), физиотерапия (местная диа-
термия, ультразвук) и др. August и Milwar (1980) рекомендуют криохирур-
гию склероатрофического лихена вульвы. Из-за возможности злокачествен-
ного перерождения рекомендуют во всех случаях производить обрезание с
гистологическим исследованием удаленной части препуциального мешка.
281
ЛИТЕРАТУРА
Голоусенко И. Ю. Буллезная форма склеротического и атрофического лихена Гал-
лопо—Дарье.— Веста, дерматол. и венерол., 1982, 8, 60—62.
Каламкарян А. А., Федорова Е. Г. Склероатрофический лихен и его взаимосвязь
. со склеродермией?— Вести, дерматол. и венерол., 1980, 12, 4—8.
Никитина М. Н. и др. Очаговая склеродермия наружных половых органов у де-
тей.— Веста, дерматол. и венерол., 1980, 11, 49—52.
Трофимова Л. Я-, Олисова М. О., Авербах Е. В. Некоторые аспекты взаимоотно-
шения склероатрофического лихена, склеродермии и витилиго.— Вести, дерматол. и вене-
рол., 1983, 2, 52—54.
Чистякова И. А. Склероатрофический лихен с необычной клинической картиной.—
Веста, дерматол. и венерол., 1984, 4, 54—55.
August Р., Milward Т. Cryosurgery in the treatment of lichen sclerosus et atrophicus
of the vulva.— Brit. J. Dermatol., 1980, 103, 6, 667—670.
Bedi B., Arunthathi S. Lichen sclerosus et atrophicus_Indian J. Derm. Vener., 1975,
41, 2, 49—53.
Hadlich J., Linse R. Zur klinisch-histologischen Variationsbreite des Lichen sclero
sus et atrophicus.— Hautapzt, 1983, 34, 9, 553—455.
Post B., Janner M. Lichen sclerosus et atrophicus.— Z. Hautkr. 1975, 50, 6, 655—
681.
ЛИШАЙ БЛЕСТЯЩИЙ (lichen nitidus)
Синоним: гранулема блестящая.
Этиология и патогенез. Лишай блестящий является редким дермато-
зом, вопрос о нозологической принадлежности которого является спорным.
Высказывается точка зрения о самостоятельности этого дерматоза (Pincus,
1973 и др.), но наиболее часто лишай блестящий рассматривается как ва-
риант красного плоского лишая (Samman, 1979; Steigleder, 1975; Korzing
и Denk, 1974 и др.). Так, Wilson и Bett (1961) считают, что у 25—3G% боль-
ных красным плоским лишаем могут быть найдены высыпания, неотличи-
мые от наблюдаемых при блестящем лишае. Авторы полагают, что термин
lichen nitidus должен быть сохранен только для тех случаев, когда лихе-
ноидные папулы мономорфны, мелкие, группируются, хотя не исключено,
что и в этих случаях это может быть милиарная форма красного плоского
лишая. Но эта точка зрения разделяется не всеми (Gertler et al., 1970;
Weiss и Cohen, 1971 и др.). Так, Pincus (1973), Weiss и Cohen (1971) обра-
щают внимание на такую особенность гистологической картины, как нали-
чие массивной паракератотической пробки в центре папулы, что, по их
мнению, никогда'не встречается при красном плоском лишае.
По мнению Л. Н. Машкиллейсона (1965), в пользу той точки зрения,
что блестящий лишай является самостоятельным заболеванием, свидетель-
ствуют особенности гистологической картины, отличной от таковой при
красном плоском лишае, а также отсутствие у значительной части больных
туберкулоидной структуры. Kellett и Beck (1984) в качестве аргумента
против единства лишая блестящего и красного плоского указывают на
разный характер изменения при этих заболеваниях ногтевых пластинок.
282 ' s
Рис. 176. Лишай
блестящий.
Waisman и соавт. (1973) при иммунофлюоресцеитном исследовании не об-
наружили отложения иммуноглобулинов при блестящем лишае, в то вре-
мя как они выявлялись у 95% больных красным плоским лишаем. Ger-
tler и соавт., считая, что нет достоверной связи этого дерматоза ни с крас-
ным плоским лишаем, ни с туберкулезом, предполагают возможность со-
судистого процесса (аллергического) как причины блестящего лишая. На
основании анализа литературы Nasemann (1980) высказывает мнение, что
это или самостоятельный дерматоз или милиарная форма кольцевидной
гранулемы. Как полагает автор, сходство гистологической картины при
блестящем лишае и саркоидозе может служить основанием и для вопроса,
не является ли это заболевание аллергидом при саркоидозе.
Клиника. Высыпания множественных симметрично расположенных
папул величиной с булавочную головку, плоских или слегка полушаровид-
ных, с блестящей не шелушащейся поверхностью, иногда с небольшим
вдавлением в центре, цвета нормальной кожи или бледно-розовых. Очер-
тания папул округлые, реже полигональные (рис. 177). Сыпь густо рас-
283
положена, группируется, могут быть кольцевидные фигуры. Обычно высы-
пания не сливаются, но это может быть при локализации папул в области
коленных и локтевых суставов, тогда возникает псориазиформное или пити-
риазиформное шелушение (Samman, 1979), или на ладонях и подошвах с
развитием гиперекератотических, шелушащихся сухих очагов с трещинами,
имеющих сходство с экзематозными (Gertler et al., 1970). Сыпь может рас-
полагаться на любом участке кожного покрова, но наиболее часто—на
коже полового члена. В редких случаях бывает генерализованной (granu-
loma nitidum generalisatum), развивается приступообразно (Schumachers,
1966). Изредка могут наблюдаться изменения слизистых, ногтевых пласти-
нок (утолщения, истыканность, исчерченность). Описаны геморрагические
и везикулярные формы (Jetton et al., 1972).
Banse—Kupin и соавт. (1983) наблюдали перфорирующий блестящий
лишай. У больного одновременно были типичные высыпания красного
плоского лишая. Может быть положительным феномен Кебнера. Субъек-
тивных расстройств обычно не бывает, иногда незначительный зуд. Раз-
вивается преимущественно у детей. Описан семейный случай заболевания
у брата и сестры с месячным интервалом в его развитии (Guiuducer, 1962).
Течение длительное, но прогноз благоприятный, так как высыпания спон-
танно регрессируют.
Дифференциальный диагноз. Дифференцировать необходимо с фол-
ликулярным муцинозом, лихеноидным туберкулезом кожи, папулезными
сифилидами, красным плоским лишаем, аллергидами различного происхож-
дения. '
ч Гистопатология. Расположенные субэпидермально периваскулярные
гранулемы из лимфо- и гистиоцитарных элементов, эпителиоидных кле-
ток, немногочисленных гигантских, плазматических, тучных клеток.
Лечение. Симптоматические средства.
ЛИТЕРАТУРА
Banse-Kupin L. Morales A., Kleinsmith A. Perforating lichen nitidus. J. Am. Acad.
Dermatol., 1983, 9, 452—456.
Kallett J. K-, Beck M. H. Lichen nitinus associated with distinc*ive nail changes.—
Clin. exp. dermatol., 1984, 9, 2, 201—204.
Korting G. W., Denk D. Dermatologische Differentia Idiagnose— Stuttgart—N. Y.,
1974.
Nasemann Th. Papulose Krankheiten der Haut. In: «Dermatologie in Praxis und КП-
nik.»— Hrsg. G. W. Korting, Stuttgart, 1980, Bd. 2, 171—17, 23
Samman P. D. Lichen planus and lichenolid eruptions. In: «Textbock of Dermatology»,
2-ed ed., by A. Rooketal. —Oxford, 1979, v. 2, 1438—1502.
ЛИШАЙ КРАСНЫЙ ОТРУБЕВИДНЫЙ ВОЛОСЯНОЙ ДЕВЕРЖИ
(pityriasis rubra pilaris Devergie).
Синонимы: красный остроконечный лишай, универсальный многоформ-
ный кератоз, ограниченный кератоз.
284
Заболевание было описано Claudius Tarral в 1829 г., а затем Р. Rayer
(1835) под названием общего псориаза (psoriasis universalis). В 1857 г. Де-
вержи дал подробное описание заболевания и предложил называть его
волосяной лишай.Besnier (1889) предложил название красный волосяной
лишай, которое удерживается по настоящее время.
Красный отрубевидный волосяной лишай—заболевание, характери-
зующееся фолликулярным гиперкератозом, отрубевидным шелушением и
эритемой. Дерматоз встречается редко и мало изучен. По данным Griffi-
ths (1976), один случай болезни Девержи приходится на 3500 кожных боль-
ных, т. е. частота среди больных дерматозами составляет 0,03%, по дан-
ным Cozza (1965)—0,25%, Lawahrg (1964)—1,3%. Красный волосяной ли-
шай описан у лиц различной этнической принадлежности и цвета кожи.
Этиология и патогенез. В прошлом имела место теория туберкулез-,
ной этиологии. Павлов Т. П., Никольский П. В-, Иордан А. П. и др. придава-
ли известное значение нервно-эндокринным нарушениям. Brunstig и She-
ard (1941), Colomb (1959), Gertler (1960), Lamar и Gaethe (1964), Waldorf
и соавт. (1965), Binnick (1978) и др. усматривают этиологическую и пато-
генетическую связь данного дерматоза с дефицитом витамина А. Однако
наши наблюдения и литературные данные (Л. Н. Машкиллейсон, 1965;
Schuppener, 1957 и др.) показывают, что лечение витамином А больных
болезнью Devergie часто недостаточно эффективно. Кроме того, исследо-
вания Niemi и соавт. (1976) и др. установили, что уровень витамина А в
сыворотке крови у подавляющего большинства больных остается в пре-
делах нормы. Некоторыми авторами высказывалось предположение об
инфекционной этиологии. Duperrat рассматривает красный волосяной ли-
шай как полиэтиологический синдром. Описано около 30 семейных случа-
ев красного волосяного лишая (Griffiths, 1975), наличие которых не исклю-
чает наследственного характера заболевания.
Клиника. Болезнь встречается чаще у мужчин в возрасте 20—50 лет,
хотя наблюдаются случаи ее в различные возрастное периоды, в том числе
у детей, подростков и стариков.
Некоторые авторы различают две формы красного отрубевидного во-
лосяного лишая: детскую, проявляющуюся впервые в детском или юно-
шеском возрасте, иногда семейную, что предполагает участие наследствен-
ного фактора по типу аутосомальной, домин анты, и другую, взрослую—
начинающуюся в среднем возрасте, не семейную и не наследственную.
Основным клиническим проявлением красного волосяного лишая яв-
ляется фолликулярный гиперкератоз. Высыпания представляют собой мел-
кую плотную фолликулярную коническую папулу размером с просяное
зерно, с роговой верхушкой или же покрытую белесоватой чешуйкой. В
центре такого уэелка всегда можно найти волос, часто обломанный, атрофи-
рованный или скрученный. Цвет папулы варьирует от телесного до желто-
вато-розового и темнее. Затем, тесно скучиваясь, узелки сливаются в одно-
образные псориазоподобные бляшки, сильно инфильтрированные, с су-
хой, шелушащейся поверхностью, перекрещенной резко обозначенными
кожными бороздами. Увеличиваясь в числе и размерах, эти бляшки сли-
235
ваются в широкие поля и могут занять всю поверхность кожи. При паль-
пации тесно расположенных папул создается впечатление терки. В связи
с наличием гипер- и паракератоза бляшки имеют иногда весьма значитель-
ное сходство с псориатическими, особенно на локтях и коленях. Разви-
тие процесса медленное, но нередко (у 60—70% взрослых) заболевание
начинается остро и за 2—3 недели может образоваться универсальная эрит-
родермия с островками нормальной кожи. Высыпания располагаются
симметрично, чаще на разгибательных поверхностях конечностей, в области
локтевых и коленных суставов, на лице и шее (рис. 178, 179, 180, 181, 182,
183, 184). Типичны мелкие остроконечные фолликулярные роговые папулы
на тыльных поверхностях первых и вторых фаланг пальцев рук («конусы
Бенье>). У 90—95% больных кожа ладоней и подошв диффузно утолщается
и пронизывается глубокими бороздами, из-за чего больные вынуждены
держать кисти в полусогнутом положении. Ногги часто утолщены, про-
дольно или поперечно исчерчены, ломки, тусклы, желтой окраски;
иногда на них отмечаются псориазиформные вдавления. Волосы иногда
истончены и разрежены.
При обширных поражениях кожи может встречаться лимфаденопатия.
Aguilar и соав”’. (1973) высказывают мнение, что отрубевидный волосяной
лишай не является только заболеванием кожи, что возможны генерали-
зованные случаи с вовлечением других систем. Изменения мышц при отру-
бевидном волосяном лишае—миопатия, дистрофия, неспецифический мио-
зит и др.—наблюдали Kierland и Kulwin (1950), Orbaneja и соавт. (1959),
Kaldore и Hambrick (1965). В очень редких слу чаях в процесс вовлекается
слизистая оболочка полости рта; при этом чаще на твердом небе отмечаются
Рис. 17Р Кр^ный
волосяной лишай Дсжржи.
Рис. 177. Красный
волосяной лишай Девержи.
286
Рис. 180. Красный
вол сякой лишай Девержи.
Рис. 179. Красный
волосяной лишай Девержи.
Рис. 182. Красный
волосяной лишай Девержи.
Рис. 181. Красный
волосяной лишай Девержи.
287
Рис. 183. Красный
волосяной лишай
Девержи.
отдельные мелкие остроконечные узелки и диффузные белесоватые пятна.
Поражения глаз также встречаются, однако трудно сказать, являются
ли они первичными или же возникают при поражении краев век глаза
(Franceschetti, 1961). Больные ощущают чувство напряжения и стягивания
кожи, иногда небольшой зуд. Хотя высыпания всюду почти одинаковы,
топография поражения откладывает определенный отпечаток на их клини-
ческую характеристику. Так, например, на туловище и конечностях име-
ются главным образом фолликулярные узелки и покраснение, на ладонях
и подошвах—диффузная кератодермия с глубокими бороздками и трещина-
ми, на лице покраснение и обильное шелушение (кожа лица гиперемиро-
вана, напряжена, натянута, блестящая; может образоваться выворот
нижних век), а на волосистой части головы имеется только обильное ше-
лушение, производящее впечатление перхоти. Течение красного волося-
ного лишая может ухудшаться во время беременности и в период менструа-
ции. Тяжесть течения болезни имеет весьма большой диапозон: в одних
случаях узелки носят строго локализованный характер, а в других—пре-
обладает наклонность к быстрому распространению по всему телу.
Гистопатология. Фолликулярный гиперкератоз с мелкими гнездами
паракератоза над сосочками. Неравномерный акантоз. Вакуольная дегене-
рация базального слоя. Незначительный инфильтрат дермы, состоящий
из лимфоцитов, нейтрофилов и немногочисленных тучных и плазматиче-
ских клеток, располагается преимущественно вокруг умеренно расширен-
ных сосудов и волосяных фолликулов.
Дифференциальный диагноз. Следует проводить с псориазом, фол-
ликулярной экземой, лекарственными высыпаниями, фолликулярным их-
тиозом, симметричной прогрессирующей эритрокератодермией, красным
плоским лишаем. В некоторых случаях клиническое -сходство болезни Де-
вержи с чешуйчатым лишаем бывает настолько большим, что приходится
288
говорить о псориазоподобной форме красного отрубевцдного волосяного
лишая. Наличие по краям бляшек или обширных шелушащихся очагов
отдельных фолликулярных узелков значительно облегчает диагностику.
Braun-Falco и соавт. (1983) на основании результатов патогистоло-
гического, гистохимического, авторадиографического и электронно-микро-
скопического исследования биоптатов кожи, полученных от 5 больных
болезнью Девержи, пришли к выводу, что болезнь Девержи—самостояте-
льное заболевание, отличное от псориаза.
Прогноз для жизни благоприятный, в отношении излечения неопреде-
ленный. Длительность заболевания разная, иногда значительная (до 10—
25 лет). Вслед за кратковременной неполной ремиссией наступает обост-
рение.
Лечение. Рекомендуют длительный прием витамина А внутрь и парен-
терально повторными курсами. Имеются сообщения о благоприятном дей-
ствии кортикостероидов, метотрексата, азатиоприна. В острых случаях
показано применение тиосульфата натрия, антигистаминных препаратов.
Полезны теплые ванны, кератолитические, смягчающие мази, кремы, мази
с кортикостероидами.
А. А. Каламкарян, Г. Ц. Марзеева, Е. В. Авербах, М. О. Олисова
(1984) наблюдали хороший терапевтический эффект (клиническая ремис-
сия и значительное улучшение) у четырех больных красным волосяным
лишаем Девержи от фотохимиотерапии. De Bast и соавт. (1982) отметили
хороший эффект от применения кортикостероидов (50 мг в день) в сочета-
нии с тигазоном (75—50 мг в день) в течение нескольких месяцев.
ЛИТЕРАТУРА
А. А. Каламкарян, Г. И. Марзеева, Е. В. Авербах, М. О. Олисова. Опыт при-
менения фотохимиотерапии при красном волосяном лишае Девержи. Вести. Дерматол. и
венерол., 1984, 2, 4—8.
С. И. Полищук, В. В. Гончаров. Красный отрубевидный волосяной лишай Де-
вержи.— Вести. Дерматол. и венерол., 1982, 1, 61—63.
Binnick S. Pityriasis rubra pilaris responding to aminonicotinamide.— Arch. Derma-
tol., 1978, 114, 9, 1348—1349.
Braun—Falco О. и соавт. Pityriasis rubra pilarisi A clinico—pathological and thera-
peutic stuly with special reference to histochemistry, autoradiography and elestron micros-
copy.— Arch, dermatol. les., 1983, 275, 5, 287—298.
Griffiths W. Pityriasis rubra pilaris.— Clin. Exp. Dermatol., 1980, 5, 1, 105—112.
Griffiths W. Pityriasis rubra pilaris — an historical approach. Clinical features.—
Clin. Dermatol., 1976, 1, 1, 37—50.
Niemi K-, Kousa M., Stargards K-, Karoonen J. Pityriasis rubra pilaris: a clinico-
pathological study with a special reference to autoradiography and histocompatibility anti-
gens.— Dermatoologica, 1976, 152, 2, 109—118.
19 Клиническая дерматология 289
ЛИШАЙ КРАСНЫЙ ПЛОСКИЙ (lichen ruber planus—атипичные
формы)
Заболевание впервые описал английский дерматолог Е. Wilson в 1869
году. В отечественной литературе обстоятельно вопросы клиники и мор-
фологии дерматоза изучил В. В. Иванов в 1902 году.
Как отмечают многие авторы, в течение последних двух десятилетий
безусловно наблюдается увеличение заболеваемости красным плоским ли-
шаем. Частота красного плоского лишая варьирует от 0,78 до 2,4% по от-
ношению ко всем кожным заболеваниям (Тураноь Н. М., Каламкарян А. А. и
соавт., 1976; Sehgal et al., 1973; Sedano, 1978; Hornstein et al., 1980; Arndt,
1979 и др.). Этот дерматоз весьма распространен на африканском конти-
ненте, составляя 5% первичных обращений в Сенегале и 6% в Нигерии
(Soyinka, 1973). При обследовании на рак и предраковые заболевания
сельского населения округа Эрнакулам в штате Керала (Южная Индия)
у 1,5% обследованных (из '639) зарегистрированы высыпания плоского
лишая на слизистой оболочкё полости рта (Pindborg et al., 1972).
Причины этого большинство исследователей связывают с более широ-
ким применением различных видов лекарственной терапии и реальным
увеличением лекарственной патологии, увеличением контакта с различны-
ми химическими аллергенами в быту, в профессиональной среде и т. д.
Клиническая картина обычной формы красного плоского лишая ха-
рактеризуется высыпаниями, состоящими из мелких плоских, блестящих,
особенно при боковом освещении, полигональных узелков темно-красного
и фиолетового цвета с центральным пупковидным вдавлением. Высыпания,
как правило, сопровождаются зудом разной интенсивности. Слизистая
оболочка полости рта поражается в среднем у 30—40% больных красным
Рис. 184. Красный плоский лишай
(изолированное поражение).
290
Рис 185. Гиперкератотический
красный плоский лишай.
Рис. 186. Гиперкерртотический
красный плоский лишай.
Рис. 187. Зостериформный
красный плоский лишай.
Рис. 188. Зостериформный
красный плоский лишай.
плоским лишаем. Помимо типич-
I ной формы красного плоского ли-
шая существует ряд атипичных
вариантов этого заболевания, име-
ющих определенную гистологиче-
скую характеристику (рис. 185,
186, 187, 188, 189).
КОРАЛЛОВИДНЫЙ КРАС-
НЫЙ ПЛОСКИЙ ЛИШАЙ (lichen
ruber planus moniliformis).
Впервые эту очень редкую разновидность красного плоского лишая
описал Капоши в 1886 г. Среди более чем 300 больных, наблюдавшихся
в ЦКВИ, эту разновидность мы установили лишь у одной больной с локали-
зацией процесса на шее, груди и плечах. Клинически заболевание харак-
теризуется образованием крупных, величиной до вишневой косточки, упло-
щенных папул, расположенных в виде четок. Узелки располагаются друг
около друга, образуя полосы, напоминающие нитки с кораллами (от ла-
19* 291
тинского monile—ожерелье). Около полос разбросаны типичные папулы крас-
ного плоского лишая и коричневые пятнышки. Высыпания локализуются
на шее, в области плечевого пояса, локтей, коленей, нижней части живота.
Язвенный красный плоский лишай (lichen ruber ulcerosus). Встреча-
ется крайне редко. Male и Azambuja (1975) отметили, что к 1975 году в
литературе описано только 24 случая. За последние 15 лет в ЦКВИ обра-
тилось только 2 больных с язвенной разновидностью заболевания среди
более чем 300 больных с обычной формой. Эрозивно-язвенная форма крас-
ного лишая слизистой оболочки полости рта наблюдается чаще. Так, Е. И.
Абрамова (1965) обнаружила ее у 45 (15%) из 294 больных красным плоским
лишаем. Язвенные поражения обычно локализуются на коже нижних ко-
нечностей (Потекаев Н. С. й соавт., 1964; Гребенюк В. Н. и Розыева
А. А., 1980; Ариевич А. М., 1983; Weidner и Ummenhofer, 1979; Thorman,
1974; Babtista et al., 1980). Очаг поражения представляет собой язву
Рис. 189. Инфильтративно-язвенная форма красного
плоского лишая.
чаще с фестончатыми очертаниями размером от 1 до 4—5 и более см. Края
язвы плотные, розовато-синюшной окраски, возвышаются над уровнем
окружающей здоровой кожи. Дно язвы покрыто вялыми зернистыми
грануляциями с некротическим налетом. Язвы сопровождаются болезнен-
ностью, усиливающейся при ходьбе (рис. 190, 191). Типичные для' крас-
ного плоского лишая высыпания одновременно располагаются на других
участках кожного покрова и в полости рта. Гистологически в биоптатах
292
Рис. 190. Инфильтративно-язвенная форма красного
плоского лишая.
язвы обнаруживаются изменения, характерные для красного плоского
лишая: гиперкератоз, акантоз, гранулез.
Заболевание характеризуется упорным течением, трудно поддается
лечению; единственным эффективным методом лечения является Хирургиче-
ская аутопластика кожи.
При длительном существовании язвенные поражения приобретают
тенденцию к злокачественному перерождению.
Cram и соавт. (1966), Degos и Schnitzler (1967) представили обзор ли-
тературы и описали необычный синдром плоского лишая, характеризу-
ющийся язвами стоп и больших пальцев, непрерывным отторжением ног-
тей и рубцовой алопецией головы. Всего было опубликовано 25 наблюдений.
Thorman (1974) приводит наблюдения с болезненными язвами на подош-
вах и на больших пальцах. На слизистой щек отмечали белые папулы в
виде сетки; типичные фиолетовые папулы плоского лишая были рассеяны
на бедрах и на туловище. На больших пальцах имелись резко ограничен-
ные язвы.
Вегетирующий роговой красный плоский лишай (lichen ruber planus
comeus vegetans).
Эта форма является исключительно редкой разновидностью. На по-
верхности очагов поражения образуются папилломатозные разрастания.
Мы наблюдали 48-летнего мужчину, у которого на пальцах кистей и стоп,
а также на половом члене имелись распространенные роговые и бородав-
чатые бляшки, покрытые обильным количеством папилломатозных. раз-
растаний, окруженные широким синюшно-ливидным венчиком. У этого
293
Рис. 191. Гипертрофический
красный' плоский лишай.
Рис. 192. Гипертрофический
красный плоский лишай.
же больного на других участках кожи, а также на слизистой оболочке по-
лости рта имелись типичные высыпания красного плоского лишая (рис. 192,
193).
Гистологически—картина красного плоского лишая с резко выражен-
ным акантозом и гиперкератозом, отчетливым утолщением зернистого слоя
(гипергранулез).
Бородавчатый (гипертрофический) красный плоский лишай (lichen ru-
ber planus verrucosus s. hypertrophicus).
Эта форма встречается нечасто, ее наблюдали Н. Н. Кушуева (1926),
В. Н. Космадис (1930), Г. И. Уманский (1963), А. А. Каламкарян и А. А.
Розыева (1983), Cram и Muller (1966), Domonkos (1971), Gunter (1975) и др.
Клиническая картина (рис. 194, 195) характеризуется образованием
на передне - боковых поверхностях голеней, реже на кистях, мошонке и
других участках кожного покрова плоских, грубых бородавчатых, резко
очерченных разрастаний, образующих бляшки размерами 4—7 см в диа-
метре, чаще неправильно округлых или овальных очертаний. Цвет бляшек
ливидный с фиолетовым оттенком. В большинстве случаев появляется
один или несколько очагов поражения, которые у отдельных больных,
сливаясь, образуют сплошные участки поражения голеней с переходом
на стопы. Поверхность бляшек неровная, бугристая, испещрена бородав-
чатыми выступами с множеством углублений, ноздреватая с ячеистой по-
верхностью, напоминающей сито; оии довольно значительно выступают
над окружающей кожей По периферии основных очагов поражения у
многих больных обнаруживаются мелкие фиолетово-красноватые узелки,
характерные для типичной формы красного плоского лишая.
294
J
Рис. 193. Веррукозный
красный плоский лишай.
Рис. 194. Веррукозный
красный плоский ,мишй.
У некоторых больных наблюдаются на поверхности бляшек вегети-
рующие сосочково-роговые разрастания.
Отличительные особенности этой разновидности—мучительный зуд и
необычайное упорство к терапевтическим мероприятиям.
Пемфигоидный красный плоский лишай (lichen ruber planus pemphi-
goides).
Среди атипических форм красного плоского лишая одной из наибо-
лее редких является пемфигоидная, везико-буллезная форма. В 1881 г.
Baker-London впервые описал разновидность красного плоского лишая,
протекающую с образованием пузырей. Kaposi в 1891 г. подобным случа-
ям дал наименование пемфигоидного красного лишая. Эту разновидность
наблюдали ряд авторов (Иордан А. П., 1927; Коляда Ф. и Ровинский Я.,
1931; Глауберзон С. А., 1937; Лаптев В. А. и Каламкарян А. А., 1954;
Sarkany et al., 1964; Fenerman и Sandbank, 1971; Kavli, 1979; Wozel et al.,
1983 и др ). Причина образования пузырей пока остается невыясненной.
Мы склонны присоединиться к точке зрения тех авторов, которые рассмат-
ривают буллезные высыпания как следствие особенно бурных воспалитель-
ных явлений, причел., несомненно, внешние и внутренние раздражающие
факторы благоприятствуют их появлению.
Клинически заболевание характеризуется образованием пузырьков
или пузырей на бляшках и папулах красного лишая или на эритематозных
участках и на неповрежденной коже. Высыпания имеют различную величи-
ну— от чечевицы до вишни, голубиного яйца и более Как правило, пузыри
бывают не в большом числе (рис. 196). Толстая напряженная покрышка
в дальнейшем становится дряблой, морщинистой. Содержимое пузырей
прозрачное, слегка опалесцирующее с желтоватым оттенком, местами с
295
примесью крови (геморрагическое).
Излюбленной локализации пузыри
не имеют, хотя несколько чаще на-
блюдаются на конечностях. Мы сов-
местно с А. А. Розыевой наблюдали
типичный пемфигоидный лишай у
больного с локализованным процес-
сом в об тасти голеней. Спустя не-
сколько дней пузырные эффлорес-
ценции подсыхают и на их местах
образуется легкое шелушение. Забо-
левание в этой форме обычно соп-
ровождается сильным зудом.
Гистологически пемфигоида ая
форма характеризуется образова-
нием подэпителиального пузыря, ко-
торый образуется в результате дис-
трофических изменений клеток ба-
Рис. 195. Пемфигоидный зального слоя (Sobel et al., 1976;
красный плоский лиишй. Saurat et а1;> 1977> Методом прямой
и непрямой иммунофлюоресценции
обнаружены отложения Ig(j в области эпидермальной базальной мембраны.
У некоторых больных, страдающих типичным красным лишаем кожи,
пузыри развиваются й на слизистой оболочке полости рта, причем эти вы-
сыпания быстро превращаются в эрозивные или язвенные (Shklar, 1968).
Диагноз подобных поражений во рту может быть затруднен ввиду сходства
их с пемфигусом, пемфигоидом, эксудативной эритемой.
Субтропический красный плоский лишай (lichen planus subtropicus).
В 1919 г. Fordyce впервые описал клиническую картину этой разно-
видности заболевания, однако он рассматривал ее как кольцевидный пиг-
ментный тип красного плоского лишая. В 1941 г. Niles опубликовал по-
добный случай В 1949 г. Dostrovsky и Sagher высказались в пользу само-
стоятельности этой формы и дали подробное клиническое описание, от-
метив при этом некоторые клинико-эпидемиологические особенности в
отличие от классической формы красного лишая. El-Zawarry (1965) опи-
сал несколько случаев этой формы заболевания. Dilaimy (1976) назвал
эту разновидность „lichen planus subtropicus4*, так как она встречается
преимущественно в странах Среднего Востока В отечественных дермато-
логических руководствах эта форма не приводится, хотя она может встре-
чаться в субтропических зонах Узбекистана, Туркменистана, Таджикиста-
на и Кавказа, кроме того, информация об этой форме полезна для врачей,
которые обслуживают иностранцев, приехавших на учебу или работу в
СССР
Клиника. Эта форма чаще встречается в странах Среднего Востока -
Египте, Палестине, Ираке и др. (где она составляет 30—40% от общего чис-
ла больных красным плоским лишаем). Возраст больных от 10 до 60 лет, но
296
чаще 20—30 лет. У молодых пациентов эта форма встречается намного
чаще, чем классическая форма красного лишая. Женщины чаще болеют,
чем мужчины. Первые высыпания обычно появляются и рецидивируют
весной и летом; локализуются высыпания преимущественно на открытых
участках кожного покрова (лицо, шея, тыльные поверхности кистей,
предплечья); реже поражаются туловище, гениталии и слизистая полости
рта. Ногти не вовлекаются.
Заболевание начинается с появления пятен буровато-синюшного цве-
та, без чешуек, округлых или овальных, с хорошо отграниченными краями,
состоящими из мелких узелков размерами от 0,2 до 3 см в диаметре. Пят-
на и узелки имеют тенденцию к слиянию с соседними высыпаниями, образуя
при этом бляшки и кольцевидные фигуры, которые весьма характерны для
клинической картины заболевания. При этом образуются кольцевидные
фигуры с синюшно-красного цвета валиком и коричневым центром, узелки
выступают не так четко, как при типичном красном плоском лишае. Недав-
но появившиеся пятна и узелки почти не шелушатся, регрессирующие
высыпания бывают покрыты тонкими серовато-белыми чешуйками. Зуд,
если он есть, минимальный и наблюдается только летом.
Гистопатология. Изменения характеризуются незначительным ги-
перкератозом, акантозом, неравномерным гипергранулезом. В дерме—
небольшой лимфо - и гистиоцитарный инфильтрат с поредением эластиче-
ских волокон..
Дифференциальный диагноз. Следует проводить с пигментной базаль-
но-клеточной карциномой, дискоидной красной волчанкой, порокератозом
Мибелли, кольцевидной гранулемой. Течение длительное, ремиссия зи-
мой, обострения летом.
Лечение. Антималярийные препараты (хлорохин, резохин, делагил)
в сочетании с кортикостероидными препаратами.
Пигментный красный плоский лишай (lichen ruber planus pigmentosus).
Впервые эту разновидность описал Pirila в 1918 году. Остро на зна-
чительной части кожного покрова, на туловище, лице и конечностях воз-
никают множественные буроватые пятнистее высыпания, которые сравни-
тельно быстро сливаются в диффузные очаги поражения. При этом обна-
ружить отдельные узелки, типичные для плоского лишая, не удается,
пока не уменьшится общая пигментация кожи. Интересно, что при появле-
нии пятен зуда, как правило, не бывает, последний возникает на третьей-
четвертой неделе заболевания.
Очень редко эта разновидность сочетается с типичными проявлениями
красного плоского лишая слизистой оболочки полости рта.
Случаи пигментного красного лишая описаны Л. А. Ишув (1960),
А. А. Каламкаряном и соавт. (1983), Bhutani et al. (1974, 1979) и др.
Принадлежность этой формы к красному плоскому лишаю, помимо клини-
ческих данных, должна подтверждаться гистологическими исследования-
ми.
297
В сосочковом слое кожи обнаруживается множество крупных клеток,
содержащих бурый пигмент, не дающий реакцию на железо. Murti и соавт.
(1979) обнаружили заметное повышение ДОПА-положительных мелано-
цитов. Дифференцировать следует с токсической лихеноидной меланодер-
мией и меланодермией.
Приплюснутый красный плоский лишай ((lichen ruber obtusus).
При этой форме преимущественно на голенях располагаются ограни-
ченные очаги, состоящие из несколько возвышенных и приплюснутых
узелков, плотно-эластической консистенции с гладкой поверхностью или
покрытых небольшим количеством чешуек. Цвет высыпаний ливидно-
коричневый, синевато-красный. Очаги поражения могут развиваться не-
зависимо или в сочетании с обычной формой красного плоского лишая. В
редких случаях подобные очаги поражения могут появляться и на других
участках кожного покрова. Гистологические изменения отличаются вы-
раженным акантозом и более мощным инфильтратом в дерме.
Атрофический красный плоский лишай (lichen ruber planus atrophicus).
Эта разновидность впервые описана Hallopeau в 1887 г., ее особенно-
стью является исход высыпных элементов в атрофию. Высыпания чаще
локализуются на голове, туловище, подмышечных впадинах и половых
органах. Они характеризуются немногочисленными очагами поражения,
состоящими из отдельных типичных для красного плоского лишая узел-
ковых высыпаний и атрофических пятен с характерной лиловатой и жел-
товато-буроватой окраской. При слиянии образуются синевато-буроватые
атрофические бляшки диаметром от 1 до 2—3 см. Могут образоваться
кольцевидные очаги поражения (lichen ruber planus annularis atrophicus) c
едва заметным буровато-синюшного цвета валиком и коричневатым цент-,
ром. Особенным упорством течения отличаются кольцевидные атрофиче-
ские поражения.
Гистологическая картина характеризуется исчезновением межсосочко-
вых выростов эпителия, почти полным отсутствием сосочков и отсутствием
эластических волокон в области инфильтрата.
Кроме описанных выше разновидностей красного плоского лишая,
следует упомянуть о линейной форме красного лишая (lichen ruber planus
linearis), при которой высыпные элементы располагаются в виде непрерыв-
ных или прерывистых полос и чаще встречаются у детей; иногда высыпания
группируются и располагаются исключительно зостериформно (ЦсЬеп ru-
ber planus zosteriformis).
Красный плоский лишай не относится к дерматозам, потенциально
злокачественным, однако, как отмечают некоторые исследователи (Grin-
span, 1966; Koberg et al., 1965; Arndt, 1979; Samman, 1979 и др.), при
длительном существовании, особенно при локализации на слизистой, а
также при веррукозно-гипертрофическом и буллезно-язвенном типе воз-
можно злокачественное перерождение. Раздражающая местная терапия и
особенно лечение Х-лучами могут способствовать неопластическим из-
менениям. А. М. Беженко (1961) сообщил о перерождении веррукозных
298
бляшек красного плоского лишая иа голени в плоскоклеточный орогове-
вающий рак у 2 больных через 21 и 33 года от начала заболевания.
ЛИТЕРАТУРА
А. М. Ариевич. Буллезно-язвенная форма красного плоского лишая стоп и кистей.
Вестн. дерматол. и венерол., 1983, 6, с, 45—48.
Гребенюк В. Н., Розыева А. А. Эрозивно-язвенная форма красного плоского ли-
шая.— Вестн. Дерматол. и венерол., 1980, № 8, с. 57—60.
Каламкарян А. А., Сыч Л. И., Розыева А. А. Клинико-морфологические особен-
ности различных форм красногр плоского лишая в процессе лечения.— Вестн. дерматол. и
венерол., 1983, № 9, с. 4—8.
Розыева А. А. Клинико-морфологические особенности красного плоского лишая и
новые методы лечения. Автор, дис. канД. мед. наук (14.00.11).— М., 1981.
Arndt К- Lichen planus.— In: Fitzpatric, T., Eisen A., Freedberg J., Austen K. Der-
matology in general medicine. New York, 1979, v. 14, p. 655—-661.
Dilaimy M. Lichen planus subtropical countries.— Arch. Dermatol., 1976, 112, No 9,
p. 1251—1253.
Sobel S., Miller R. Lichen planus pemphigoides: immunofluorescence finding.— Arch.
Dermatol., 1976, v. 112, No 9, p. 1280—1283.
Wozel G. и соавт. Lichen ruber planus pemphigoides durch Gofddehandlung bei Rheu-
matoid — Arthritis. — Dermatol. Msdr.,’ 1983, 169, 2, 125—129.
ЛИШАЙ ОПОЯСЫВАЮЩИЙ ОБЛАСТИ ГЛАЗ (herpes zoster ophthal-
micus)
Среди различных форм опоясывающего лишая особое место по ча-
стоте, тяжести и многообразию клинических проявлений занимает опоясы-
вающий лишай области глаз, что определяется анатомическими особенно-
стями глазной ветви тройничного нерва. . .
Этиология и патогенез. В настоящее время большинством авторов
(Глоба Э. А. и Нугманова М. Л., 1972; Данилов Л, Н., 1977; Крисон
Е. Д., 1973; Шишов А. С. и Уманский К- Г., 1981; Hope-Simpson, 1965;
Gold, 1966; Chatak et al., 1973; Hosegawa, 1971; Ishikawa и Hondo, 1974;
Hata, 1975; Daniel, 1976; Scovby, Sullivan, 1982, и др.) установлено, что
опоясывающий лишай и ветряная оспа вызываются одним и тем же ней-
ротропным фильтрующимся вирусом—varicella—zoster virus. Патогенез
до конца не выяснен. Одни авторы считают, что вирус первично поражает
кожу и затем по нервным стволам проникает в ганглии. Другие авторы
высказывают мнение, что кожный процесс является нейротрофическим,
вторичным, обусловленным поражением в ганглио - радикулярном отделе
периферической нервной системы. Наиболее распространенной клиниче-
ской формой герпетического поражения нервной системы являются ганг-
лионевриты. Работами последних лет установлено, что вирусы ветряной
оспы—опоясывающего лишая способны длительное время персистиро-
вать в организме человека и, в частности, в клетках чувствительных ганг-
лиев (спинальные ганглии и ганглии черепных нервов). Возможно длитель-
299
ное персистирование вируса в клетках кожи и слизистых оболочек без
проникновения в нервную систему.
Клиника. Herpes zoster ophthalmicus, по данным А. А. Каламкаряна
и В. Д. Кочеткова (1964, 1973), составляет почти 1/3 всех случаев опоясы-
вающего лишая. Эта форма встречается в зрелом и в пожилом возрасте,
чаще между 50—60 и 70—80 годами, преимущественно у лиц с тяжелым
иммунологическим дефектом (Nasemann et al., 1973; Runn, 1978, и др.).
[Чаще наблюдается осенью и зимой; у многих больных заболеванию пред-
шествует переохлаждение. Как отмечают многие авторы (Каламкарян А. А.
и Кочетков В. Д., 1964, 1966, 1973; Гыцу и соавт., 1976; Мальцева Н. Н.
и соавт., 1978; Piebenga и Laibson, 1973; Thomas и Howard, 1972; Nord et
al., 1977), в основе herpes zoster ophthalmicus лежит поражение гассерова
узла и первой ветви тройничного нерва. У 3/4 больных поражается область
иннервации первой ветви, а у 1/4—второй ветви тройничного нерва. В боль-
шинстве случаев герпетическим высыпаниям за 1—5 дней предшествуют
невралгические явления в виде дергающих, колющих, жгучих болей в
пределах расположения будущего высыпания; нередко они сочетаются
с парестезиями и гиперестезиями. Пузырьковые высыпания на лице и
голове (кожа лба, верхнее веко, спинка носа, теменная область) характе-
ризуются односторонностью поражений без перехода за среднюю линию,
Бесцветное вначале содержимое пузырьков постепенно (через 12—24 ча-
са) становится у большинства больных геморрагическим (herpes zoster
haemorrhagicus), а сами пузыри укрупняются порой до размеров голуби-
ного яйца (herpes zoster bullosus). Возникающее в дальнейшем западение
центральной части пузырьковых элементов придает им большое сходство с
варицеллоподобными высыпаниями. У ослабленных пожилых больных,
страдающих тяжелыми сопутствующими заболеваниями (лейкозы, грибо-
видный микоз, саркома Капоши и др.), наблюдается наиболее тяжелая
разновидность заболевания herpes zoster ophthalmicus gangrenosus, кото-
рая клинически проявляется образованием в очагах поражения долго
не заживающих глубоких изъязвлений и гангренозных участков с мелко-
фестончатыми очертаниями. По выздоровлении на их месте остаются слегка
вдавленные рубцы. При поражении второй ветви тройничного нерва (и.
maxi Haris) очаги поражения захватывают кожу височной области и щек,
боковой поверхности носа, нижнего века и верхней губы.
Герпетические ганглиониты гассерова узла с синдромом невралгии
тройничного нерва наблюдаются у 15—20% больных опоясывающим ли-
шаем. У многих больных средняя продолжительность постгерпетических
невралгий не превышает 1,5—6 месяцев. Однако у лиц пожилого возраста
боли эти могут продолжаться до 8—12 месяцев и более. Примерно у 8—10%
наблюдаются односторонние высыпания на слизистой оболочке ротовой
полости, глотки, зева, твердого и мягкого неба в виде отдельных групп
пузырьков со светлым содержимым, окруженных воспалительной гипере-
мией. Покрышка пузырьков быстро разрывается, обнажая резко болез-
ненные эрозии с микрофестончатыми краями. Одновременно с герпетиче-
ским поражением кожи наблюдаются пузырьковые высыпания на конъюк-
300
тиве и роговице глаз с исходом в мельчайшие рубцовые изменения, иногда
могущие приводить к полной потере зрения. Менингоэнцефалиты и ме-
нингиты встречаются при опоясывающем лишае редко, но протекают тя-
жело. У подавляющего большинства больных (70—75%) в остром периоде
опоясывающего лишая отмечаются умеренно выраженные общемозговые
симптомы в виде нарушения сознания, сонливости, адинамии, диффузных
головных болей, головокружения, тошноты, рвоты. У многих больных
наблюдаются выраженные вегетативные нарушения с пигментацией кожи,
нарушением потоотделения и др. Наши наблюдения свидетельствуют, что
herpes zoster ophthalmicus имеет все симптомы острого общеинфекционного
заболевания (повышение температуры нередко до 38—39,5°, выраженные
признаки общей интоксикации). У отдельных больных при поражении
коленчатого узла (ganglion geniculi) развивается синдром Ханта—одно-
временное поражение лицевого, слухового и тройничного нерва с герпети-
ческими высыпаниями в ротовой полости и наружном слуховом проходе.
Гистопатология. Характерна баллонируклцая дегенерация клеток
шиповидного слоя с последующим образованием пузырей и воспалительной
инфильтрацией в дерме преимущественно лимфоцитами и в меньшей сте-
пени нейтрофилами. В ядрах дегенерированных эпителиальных клеток
отмечаются своеобразные включения—тельца Липшютца.
Дифференциальный диагноз. Следует проводить с рожей, особенно
когда еще недостаточно отчетливо выражен пузырьковый характер сыпи,
с острой экземой, с поверхностной стрептодермией.
Прогноз в подавляющем большинстве случаев благоприятный. Ре-
цидивы herpes zoster ophthalmicus исключительно редки.
Лечение. Комплексное лечение включает: антибиотики (в целях про-
филактики осложнений), салицилаты, витамины группы B.Bj, В12), индук-
тор интерферона (вводится внутримышечно по 1 мл через 3 дня), гидро-
хлорид эметина (1 % раствор 2 мл ежедневно внутримышечно), У ФО, диа-
динамические токи, диадинамоэлектрофорез с анальгезирующей смесью—
100 мл 0,5% раствора новокаина+1 мл 0,1% раствора адреналина, моче-
гонные средства, вливание 40% раствора уротропина. При очень сильных
невралгических болях показано применение небольших доз кортикостеро-
идных препаратов (преднизолон 15—20 мг). У части больных, получающих
преднизолон, удается предупредить развитие постгерпетической неврал-
гии (Каламкарян А. А., 1973; Pearce, 1973; Keczkes и Basher, 1980). Мази с
антибиотиками и кортикостероидами, аппликации димексида (ДМСО).
ЛИТЕРАТУРА
Шишов А. С., Уманский К- Г. Опоясывающий герпес и его генерализованные
формы.— Клин, мед., 1981, 9, 66—72.
Fania D., Soltz—Szotz. Interferon und Interferonstimulierung. Neue Ansatze in der
Therapie von Virusinfektioncn.— Hautarzt, 1974, 25, 4, 313—318.
Keszkes %., Basheer A- Do corticosteroids prevent post-herpetic"neuralgia?— Brit. J-
Dermatof., 1980, 102, 5, 551—555.
Miller L. Herpes zoster in elderly.— Cutis, 1976, 18, 427—432.
301
NordE., Weinberger D., Benjamin D., PinkhasJ. Motor paralisis complicating her-
pes zoster.— Dermatologica, 1977, 154, 5, 301—304.
Runne U. Sehr generalisiert Zosterverlaufe durch zellulare Immundefekte. Bedeutung
einer absoluten oder relativen T—Zellmangels.— Hautarzt, 1978, 29, 3, 147—152.
Skooby F.,' Sullioan M. Herpes zosber and varicella in children with Hadgkins disease.
— Acta pediatr. Scand., 1982, 71, 2, 269—273.
ЛИШАЙ ШИЛОВИДНЫЙ (lichen spinulosus).
Синонимы: шиповидный фолликулярный кератоз, шиповидный кератоз.
Этиология и патогенез. В большинстве генетически детерминирован-
ный дерматоз, наследуемый аутосомно-доминаитно (Meyer et al., 1965).
Ebling и Rook (1979) рассматривают лишай шиповидный как вариант ke-
ratosis pilaris. Gahlen и Grussendorf (1980) под этим названием описывают
3 формы фолликулярного гиперкератоза: как варианты keratosis pilaris,
красного плоского лишая (синдром Little—Lassueur), лихеноидной ал-
лергической реакции при инфекционных заболеваниях (lichen scrofuloso-
rum; s. trichophyticus; s. syhhiliticus; сальварсанная токсидермии). Ли-
хеноидные роговые высыпания могут наблюдаться и при недостаточности
некоторых витаминов (в первую очередь А и С), развиваться под влиянием
экзогенных факторов (контакт с продуктами каменноугольного дегтя и
др ), быть симптомом многих наследственных заболеваний (кератодермии,
эктодермальные дисплазии, врожденная пахионихия, монилетрикс и др.).
Клиника. Наиболее часто встречается форма заболевания, которая
обозначается как собственно лишай шиповидный Crocker—Adamson. Об-
наруживаются мелкие, чаще фолликулярные, в большинстве шиповатые
роговые папулы, группирующиеся в виде очагов самых разноообразных
очертаний, кожа в зоне которых имеет розовато-красноватую окраску с
синюшным оттенком, обычно нерезко выраженную. Может быть обильное
муковидное шелушение, из-за чего очаг становится белым, как бы посыпан-
ным пудрой. За счет наличия шипиков на папулах при проведении ла-
донью по поверхности очага создается впечатление прикосновения к терке.
После удаления папулы обнаруживается небольшое углубление, в нем
остатки обломанных волос (рис. 197). Высыпания наиболее часто распола-
гаются симметрично на задней поверхности шеи, разгибательной поверх-
ности плеч, в области спины, живота, ягодиц, бедер. Нередко сочетаются с
keratosis pilaris. Субъективных расстройств они обычно не вызывают, лишь
некоторые больные отмечают небольшой зуд.
Болеют преимущественно дети, чаще мальчики. Прогноз для выздо-
ровления благоприятный, с возрастом наступает самопроизвольная ремис-
сия. Более Тяжело (за счет ассоциированной симптоматики) протекающи-
ми формами являются keratosis follicularis spinulosa decalvans (Siemens) и
keratosis follicularis congenita Siemens. В первом случае распространенные
высыпания фолликулярных роговых папул, располагающихся на лице,
шее и других участках кожного покрова, сочетаются с рубцовой алопецией
и изменениями глаз (выпадение бровей, ресниц, блефарит, воспаление
конъюнктивы, роговицы, фотобоязнь, выворот век и др.). Franceschetti et
302
i
Рис. 196. Лишай
шиловидный.
al. (1956) считают, что глазные изменения не являются следствием механи-
ческого повреждения, вызванного заменой ресниц роговыми шиповатыми
папулами, как полагал Siemens, а что дегенеративные изменения роговицы
представляют собой первичный процесс. Кроме того, могут наблюдаться
атрофодермии, изменения ногтей, ладонно-подошвенные кератодермий (Ки-
okkanen, 1971; Gahlen и Grussendorf, 1980). Заболевание развивается в пер-
вые недели или месяцы жизни, клинические проявления более выражены
у лиц мужского пола. У женщин могут быть forme fruste заболевания (Fran-
ceschett et al., 1957). Britton и соавт. (1978) наблюдали также отставание
в росте, глухоту, транзиторную гепатоспленомегалию, подверженность ин-
фекции без выявленных признаков иммунного дефекта, Adler и Nyhan
(1969) — врожденную глаукому, катаракту, умственную отсталость, ар-
ахнодактилию и аминоацидурию.
Цысыпания гиперкератотических папул при keratosis follicularis
congenita Siemens располагаются преимущественно на разгибательной
поверхности конечностей, ягодицах, периорально, имеют некоторое сход-
ство с комедонами и угревой сыпью, сочетаются с ограниченным керато-
зом ладоней и подошв, гипергидрозом, пахионихией, лейкоплакическими
изменениями в полости рта, иногда пузырями, чаще на конечностях, умст-
венной отсталостью.
Гистопатология. Умеренно выраженный гиперкератоз, роговые проб-
ки в устьях расширенных фолликулов, слабо выраженная лимфоцитарная
инфильтрация в дерме вокруг волосяных фолликулов.
Дифференциальный диагноз. Необходимо проводить с лихеноидным
туберкулезом кожи, красным волосяным лишаем Девержи, фолликуляр-
ным муцинозом, волосяным лишаем.
Лечение. Отшелушивающие средства, витамин А.
зоз
ЛИТЕРАТУРА
Britton И., Lustig J., Thompson В. J., Meyer S., Esterly N. B. Keratosis follicu-
laris spinulosa decailvans.— Arch. Derm., 1978, 114, 5, 761—764.
Ebling F. J., Rook A. Disorders of Keratinization In «Textbook of dermatologie.»
Oxford, 1979, 2, 1290—1290.
Gohlen W., Grussendorf E. I. Follicular Keratosen und benigne Diskeratosaen. In:
«Dermatologie im Praxis und Klinik.» Hrsg G. W. Korting, Stuttgart, 1980, Bd. 2, 2137—2171.
ЛУИ-БАР СИНДРОМ (Louis-Bar syndrome).
Синонимы: атаксия-телеангиэктазия, цефало-окуоло-кутанная теле-
ангиэктазия.
В 1941 г. Луи-Бар описала синдром, характеризующийся мозжечко-
вой атаксией и кожно-конъюнктивальной телеангиэктазией. В 1958 г.
Boder и Sedgwick предложили название атаксия-телеангиэктазия и доба-
вили третий важный компонент этого синдрома—рецидивирующую тяже-
лую синопульмональную инфекцию.
Этиология и патогенез. Этот синдром в различных классификациях
рассматривается как спинно-мозжечковая дегенерация (Toller и Millichap,
1961) или как факоматоз (Boder и Sedgwick, 1958; Centerwall и Miller, 1958).
Термин факоматоз (phakomatosrls) предложил van der Hoeve в 1923 г. для
заболеваний с комбинированным поражением нервной системы и кожи (врож-
денные нейро-экто-мезодермальные дисплазии). Boder и Sedgwick (1964)
в 15Q случаях синдрома Луи-Бар обнаружили дегенеративные явления в
ЦНС, дегенерацию пирамидальных клеток, расширение менингеальных
сосудов и др.
Значительная часть описанных наблюдений относится к семейно-
наследственной патологии, тип наследования аутосомно-рецессивный (Боч-
ков Н. П. и соаЬт., 1974; Антимоний Р. Г. и Тимченко А. В., 1983;
Tadjoedin и Fraser, 1965; Lampert, 1969; Paterson, 1976; McKusick, 1976
и др.).
Многие современные авторы синдром Луи-Бар относят к сочетанным
иммунодефицитным состояниям. В основе лежат гипоплазия тимуса и де-
фицит IgA и IgE, т. е. нарушение функции клеточного и гуморального
звеньев иммунитета, чем и объясняют частые рецидивирующие инфекци-
онные заболевания органов дыхания, пищеварительного тракта, кожи и
др. Помимо гипоплазии вилочковой железы отмечается гипо-, атрофия
лимфатических узлов, селезенки и лимфатического аппарата, пищевари-
тельного канала (Куликов В. Б., 1974; Ивановская Т. Е., 1975; Коваль-
чук Л. В., 1979; Вельтищев Ю. Е., 1984; Gutman и Lemli, 1963; Fireman
et al., 1964; Haerer et al., 1969; Griscelli et aL, 1973, и др.). По мнению
указанных авторов, снижение иммунитета приводит к ранней гибели боль-
ных от инфекционных заболеваний и усиливает тенденцию к развитию
злокачественных новообразований лимфатической системы.
Клиника. Заболевание встречается редко. Оно включает в себя пора-
жения нервной системы, кожно-конъюнктивальные телеангиэктазии и
304
инфекционные заболевания. Среди неврологических манифестаций этого
синдрома атаксия мозжечкового характера является первым симптомом
у всех больных; начинается с раннего детства, становится особенно замет-
ной, когда ребенок начинает ходить. Вследствие прогрессирования моз-
жечковых расстройств больные часто полностью лишены возможности
ходить. Отмечается атаксия позы и конечностей, гипотония мышц, замед-
ленная скандированная невнятная речь по типу мозжечковой дизартрии,
нистагм, тремор, нарушения иннервации движений глазных яблок. Сухо-
жильные рефлексы низкие или отсутствуют. Окуломоторная апрексия
встречается в 80% случаев, затем развивается офтальмоплегия.
Телеангиэктазии обычно появляются после атаксии, в возрасте 4—6
лет, в некоторых случаях позже, а иногда на первом месяце жизни (Thi-
effry et al., 1961). Телеангиэктазии вначале развиваются на глазных
яблоках (бульбарная конъюнктива), затем на веках и коже лица.
Инфекционные заболевания главным образом органов дыхания обна-
руживаются у 80% больных. Они проявляются рецидивирующими рино-
фарингитами, хроническими бронхитами, бронхопневмониями, бронхо-
эктазиями. Описываются гнойные отиты. Патогенетически их связывают с
иммунным дефицитом.
Дерматологическая картина синдрома Луи-Бар (рис. 198) характе-
ризуется наличием кожных телеангиэктазий у 100% больных. Другие
кожные проявления (резкая сухость кожи и волос, фолликулярный ке-
ратоз на предплечьях и голенях, пигментации цвета «кофе с молоком» и
сетчатые дисхроматически-атрофические поражения на коже лица) наблю-
даются у 50—70% больных.
20 Клиническая дерматология 305
Кожные изменения не являются специфическими для атаксии—теле-
ангиэктазии, однако диагностическая важность этих изменений делает не-
обходимым их раннее выявление в целях более успешного лечения; часто
диагноз ставится на основании дерматологической картины.
Вначале телеангиэктазии появляются на бульбарной конъюнктиве,
затем распространяются на кожу вёк, носа, ушных раковин, шеи; появля-
ются на коже локтевых сгибов, предплечий, подколенных областей, тыль-
ной поверхности кистей, стоп. Телеангиэктазии особенно выражены на
участках кожи, которые подвергаются солнечному облучению. На лице,
в области щек, они принимают вид «пучков» и расширения сосудистой сети.
Веки часто становятся неэластичными, кожа лица плотной, напоминающей
склеродермию.
Иногда телеангиэктазии также наблюдаются на мягком и твердом
нёбе. Из других кожных проявлений болезни, имеющих важное значение
для диагноза, следует отметить гиперпигментацию и гипопигментацию с
атрофией и телеангиэктазиями, напоминающие пойкилодермию.
Луи-Бар в оригинальном описании указывала на наличие пятен
цвета кофе с молоком, размерами от чечевицы и больше, напоминающих
веснушки на коже носа и щек, подбородка, груди и межлопаточной области.
Кроме того, у многих больных наблюдаются фолликулярный кератоз и
сухость кожи. У части больных наблюдаются экзематозные поражения,
преждевременное поседение волос, гипертрихоз, псориазоподобные высы-
пания, акнеиформные элементы на щеках и др.
Мы (Каламкарян А. А. и Самсонов В. А., 1984) наблюдали девочку
4 лет 8 месяцев, у которой на лице имелись множественные телеангиэктазии
и пигментные пятна цвета кофе с молоком. С раннего детства ребенок от-
ставал в физическом развитии; часто отмечались бронхиты и пневмонии
затяжного характера с повышением температуры тела до 38—39°. К кон-
цу первого года жизни девочка стала подниматься, но ходить не могла—
падала. Общее состояние постепенно ухудшалось. С двухлетнего возраста
передвигается с посторонней помощью; походка неустойчивая, танцую-
щая. На третьем году жизни появилось нарушение речи, она стала скан-
дированной с носовым оттенком. Имеются телеангиэктазии бульбарной
конъюнктивы в области глазных щелей. Сухожильные рефлексы резко
снижены, выраженная атаксия при коленно-пяточной пробе.
Прогноз неблагоприятный, более половины больных погибают через
5—10 лет после появления первых симптомов болезни от воспаления лег-
ких или лимфоретикулярных злокачественных опухолей.
Гистопатология. В участках кожи с гиперпигментацией и телеанги-
эктазиями нерезко выраженный гиперкератоз и порокератоз, отложение
меланина в эпидермисе. В дерме—воспалительная реакция, расширение
капилляров с периваскулярной инфильтрацией из нейтрофилов и лим-
фоцитов.
При патологоанатомическом исследовании лимфатической системы
выявляются гипоплазия тимуса, гипо - и атрофия лимфатического аппа-
рата желудочно-кишечного тракта. В мозжечке обнаруживается дегене-
306
рация мозжечковых клеток, включая клетки Пупкинье, зернистых кле-
ток и отросгчатых клеток.
Дифференциальный диагноз. Его следует проводить с атаксией Фрид-
рейха, болезнью Пьера Мари, болезнью Рандю—Ослера. Синдром Луи-Бар в
отношении дерматологической симптоматики следует отличать от стертых
и атипичных форм разного вида кожно-глазо-мозговых ангиоматозов, таких,
как синдромы Гиппеля—Линдау, Штурге—Вебера—Краббе и других, при
которых наблюдаются сосудистые изменения со стороны глаз.
Лечение. В связи с частыми инфекционными заболеваниями легких
необходимо назначение антибиотиков, сульфаниламидов и других про-
тивовоспалительных препаратов. Больные подлежат диспансерному на-
блюдению у невропатолога, педиатра и дерматолога.
ЛИТЕРАТУРА
Велыпищев Ю. Е.' Иммунодефицитные состояни.— В кн.: Прикладная иммунология.
Киев, 1984, с. 76—105.
Егоров Е. А. Офтальмологические проявления синдрома Луи-Бар.— Вести- оф-
гальмол., 1977, № 3, с. 76—78.
Синявская О. А., Зерницкая М. В., Захарова С. Б. Сищфом Луи-Бар у ребенка
6 лет.— Вопр. охраны материнства и детства, 1983, № 2, с. 67—69.
Adams R. Neurocutaneous disease.— In: Dermatology in general medicine. Fitzpat-
rick et al. McGram—hill book company, 1979, p. 1206—1247.
Armata J. et al. Lymphoblastic leukemia in a boy with ataxia telangiectasia.— Pedi-
Jtr. Pol., 1983, 58, 6, 555—557.
Champion R. Disorders ffecting small blood vessels erythema and teleangiectasia.—
In: Rook, Kilkinson, Ebling, Blackwell scientific publications, Thied ediation, Oxford—
London, 1979, v. 1, p. 955—970.
‘ , Skoglund R. Ataxia telangiectasisa.— Arch. Neurol., 1984, 41, 2, 137—146.
Waldmann T. et al. Disorders of В cell and helper T cell in the pathogenesis of the
immunoglobulin deficiency of patients with ataxia-telangiectasis.— J. Invest., 1983, 71, 2,
282—285.
МЕЛАНОЗ ПРЕДРАКОВЫЙ ОГРАНИЧЕННЫЙ ДЮБРЕЯ (Melanosis
circumscripta praecancerosa Dubreuilh)
Синоним: злокачественное лентиго Hutchinson.
Этиология и патогенез. Согласно гистологической классификации
опухолей кожи ВОЗ (1980), заболевание относится к группе предраковых
состояний. Существуют и другие взгляды. Так, по аналогии с болезнью
Боуэна, меланоз предраковый рассматривается как melanoma in situ, так
как наиболее существенным признаком является пролиферация анапла-
стических меланоцитических клеток в эпидермисе и волосяных фоллику-
лах (Braun-Falco et al., 1975; Kalkoff и Kiihnl-Petzoldt, 1973). Стадия
интраэпидермальной пролиферации представляет собой, таким образом,
очень раннюю фазу злокачественной меланомы (Dawber и Wilkinson, 1979).
Переход опухолевых клеток через базальную мембрану (стадия вертикаль-
ного распространения) свидетельствует о развитии собственно злокаче-
20* 307
ственной меланомы (Clark и Mihm 1969; Kalkoff и Kiihnl-Petzoldt, 1973).
По мнению Schreus (1965), ограниченный преканцерозный меланоз пер-
вично не является меланомой, а представляет собой пролиферативный про-
цесс, захватывающий клетки пограничного слоя эпидермиса, который мо-
жет существовать годами без прогрессирования и, даже без лечения, иног-
да спонтанно регрессировать. При дальнейшем развитии может возникнуть
картина псевдоспиналиомы или псевдомеланомы—состояний, которые го-
дами могут не терять своей доброкачественности. С точки зрения Schreus,
вопрос о возможности и сроках злокачественного перерождения требует
дальнейшего изучения, более правилен термин Melanosis extensive et pro-
liferative Hutchinson.
Cramer (1967) считает, что длительное течение (7—10 и даже 20 лет)
меланоза пребластоматозного несовместимо с обозначением его как ме-
ланоцитобластомы. В то же время автор не исключает, что иногда мелано-
цитобластома какое-то время может ограничиваться чисто эпидермальным
расстройством, т. е. быть по сути melanoma in situ. По мнению Gertler и
Lartmann (1957), частота перехода в бластому ограниченного преканцероза
завышена и он должен рассматриваться только как факультативный пре-
меланомный дерматоз. К своеобразным невусам относили преканцерозный
ограниченный меланоз Дюбрея А. А. Левков и В. В. Зданович (1967).
Следует подчеркнуть, однако, что уже в ранней стадии с помощью элект-
ронной микроскопии (Anton—Lamprecht et al., 1971, Lupulescu et al.,
1973) выявляется в базальном слое пролиферация патологических мела-
ноцитов.
Клиника. Заболевание проявляется в виде одиночного медленно рас-
тущего очага, с неправильными полициклическими очертаниями, без уп-
лотнения в основании, вначале светло-коричневого, при длительном суще-
ствовании—темно-коричневого или черного цвета. Характерна неравномер-
ность окраски: с одной стороны наличие на фоне менее интенсивно окра-
шенных участков отдельных резко пигментированных точек, с другой—
наряду с резко пигментированными участками черного цвета могут быть
зоны с менее интенсивной окраской, просветлениями и даже депигмента-
цией, что наблюдается обычно при спонтанном регрессировании в какой-
то части очага поражения при продолжающемся распространении в дру-
гих (рис. 199). Описаны и непигментированные варианты. Например, Cra-
mer (1967) наблюдал у 70-летней женщины поражение на коже голени,
клинически очень сходное с болезнью Bowen. При гистологическом исследо-
вании был установлен диагноз непигментированного пребластоматозного
меланоза. Меланоциты были выявлены с помощью Dopa—реакции по Bloch
и окраски на пигмент по Mason. Амеланотическая форма преканцерозного
меланоза может развиться и как рецидив на месте неполностью удаленного
типичного очага (Su и Bradby, 1980). Элиминацию пигмента авторы склон-
ны объяснить как следствие воспалительной реакции типа инородного
тела на рубец. Поверхность очага гладкая, хотя и может наблюдаться неж-
ное шелушение. Размеры очага обычно 2—6 см в диаметре, но при много-
летнем существовании могут достигать величины 8—10 см и более. Субъек-
308
Рис. 198. Меланоз предраковый
ограниченный Дюбрея.
тивных расстройств заболевание
не вызывает. Наиболее часто ог-
раниченный предраковый меланоз
располагается на открытых, под-
вергающихся наиболее интенсив-
ной инсоляции, участках тела: на
лице, тыле кистей, реже на пред-
плечьях.
Из 88 больных, наблюдавших-
ся Arma, Szlachcic и соавт. (1970),
у 83% очаги поражения были на
лице. По данным Jung и Kolzsch
(1968), из 17 случаев в 31,6% на
лице, в 41,2%—на конечностях,
в 27,2%—на туловище. Не исклю-
чено, что среди этих лиц были и
больные с поверхностно распро-
страняющейся меланомой, кото-
рая, в отличие от преканцероз-
ного ограниченного меланоза, мо-
жет располагаться на любом
участке кожи. Kalkoff и Kiihnl-
Petzoldt (1973) ставят под сомне-
ние возможность локализации ог-
раниченного преканцероза на туловище. Не скрывается ли, по их
мнению, в этих случаях часто, если не всегда, поверхностно распрост-
раняющаяся меланома, сходная с ограниченным преканцерозом. Имеются
описания меланоза Дюбрея конъюнктивы (Reese; 1955, Klauder, 1959),
в полости рта (Costello et al., 1959), на кончике пальцев (Lupulescu et al.,
1973). Длительное время поражение находится на уровне кожи или едва,
равномерно возвышается над ней, не претерпевая особых изменений, за
исключением усиления интенсивности пигментации и увеличения в раз-
мерах-. Однако рано или поздно какая-то из зон приобретает узловатый,
опухолевидный характер, сначала незначительно, а со временем все более
возвышаясь над уровнем остального очага поражения. Поверхность в этом
месте изменяется, становится папилломатозной, усиливается шелушение,
может наступить эрозирование. Вокруг пятна появляется эритема. Это
является признаками развития злокачественной меланомы.
Заболевание развивается примерно одинаково часто у мужчин и женщин.
Так, из. 17 больных, наблюдаемых Jung и Kolzsch (1968), было 8 мужчин и
9 женщин. У мужчин возрастной пик—50—60 лет, у женщин процесс раз-
вивался с примерно равной частотой по десятилетиям от 50 до 80 лет. В
то же время имеются сведения о более частом развитии заболевания у
женщин. Так, все 7 больных, наблюдавшихся Н. Н. Трапезниковым и
соавт. (1976), были женщины старше 45 лет. Прогноз тем лучше, чем рань-
309
ше поставлен диагноз. Без лечения у значительного числа больных (до
40%, по данным И. С. Дерижановой и соавт., 1980; у большинства—по
мнению К- В. Даниель-Бек и А. А. Колобяковой, 1979) развивается ме-
ланома (лентиго-меланома), что обычно происходит через 10—15 лет (по
данным Costello et al., 1959, этот срок колеблется от 21 месяца до 30 лет)
с момента появления ограниченного предракового меланоза. Michalic и
соавт. (1983) описали 2 больных с глубокой инвазией уже через 2 и 4 года
с момента развития заболевания. Считается, что прогноз при меланоме
этого типа более благоприятный, чем при других формах (Иконописов Р.
и соавт., 1977; Kalkoff и Kuhnl-Petzold, 1973, и др.). Так, Waste и Helwig
(1968) наблюдали развитие метастазов только у 4 больных из 45. Может
наблюдаться спонтанное регрессирование (Schreus, 1965). При локализа-
ции на конъюктиве опасность перерождения выше (Rees, 1955).
Гистопатология. Акантоз, повышенное содержание пигмента в базаль-
ном слое, концентрация, и гнезда меланоцитов в зоне дермо-эпидермального
соединения. В дерме—повышенное содержание меланоцитов, воспалитель-
ный инфильтрат.
Дифференциальный диагноз. Следует проводить с сенильным керато-
зом, пигментной базалиомой, поверхностно распространяющейся формой
меланомы.
Лечение. Хирургическое, близкофокусная рентгенотерапия.
ЛИТЕРАТУРА
Дерижанова И. С. Антоньев А. А., Парщикова С. М. Меланоз. БМЭ, 3-е изд. М.«
1980, т. 14, 494—496.
Даниель-Бек К. В., Колобяков А. А., Злокачественные опухолй кожи н мягких
тканей. М., Медицина, 1979.
Иконописов Р-, РайчевР., Киров С., Черноземам И. Пигментные опухоли. Меди-
цина и физкультура., София, 1977.
И. Н. Трапезникова, А. С. Равен, В. В. Дворовский, Г. Б. Титенер. Пигмент-
ные невусы и новообразования кожи. М., Медицина, 1976.
Braun-Falco О., Lucacs S., Schoefinins И. Н. Zur Behandlung der Melansis circum
sczipta praecancerosa Dubreuilh — Hautarzt, 1975, 26, 4, 307, 10.
Dawber R. P. R., Wilkinson J. D. Melanotic freckle of Hutchinson treatment ofmacu-
far aijd nodubarphases with cryotherapy.— Brit. J. derm., 1979, 101, 1, 47—49.
Michalik E. E., Fitzpatrick T. B., Sober A. J. Rapid progression of lentigo maligna
to deeply invasive lentigo maoigna meofanoma.— Arch, dermatol., 1983,119, 10,831—835.
МЕЛАНОМА ЗЛОКАЧЕСТВЕННАЯ КОЖИ (melanoma malignum cu-
tis)
Синонимы: мёланокарцинома; невокарцинома.
Меланома представляет собой злокачественную невоклеточную опу-
холь. Встречается относительно редко: по данным Gertler (1970), Callen и
соавт. (1978), Hunter и соавт, (1979), Orfanos и Doering (1983)—4—5 слу-
чаев на 100000 населения.
310
Этиология и патогенез. Наиболее вероятно, что меланома представ-
ляет собой мультифакториальное заболевание, в развитии которого при-
нимают участие как средовые (доминирующие), так и генетические факторы.
На роль последних прежде всего указывают публикации семейных слу-
чаев. меланомы, в том числе у близнецов (Katzellenbogen и Sandbank, 1966;
Andrews, 1978; Clark et al., 1978; White и Mac Donald, 1981; Wassilev et
al., 1981), а также высокий риск развития семейной меланомы из наследу-
емых невоклеточных невусов (синдром В—Kmole). Из средовых факто-
ров, способствующих развитию меланомы, указываются, в первую очередь,
солнечные лучи, травмы невусов, особенно пограничных. О влиянии солн-
ца свидетельствуют данные эпидемиологических исследований о неодина-
ковом географическом распределении меланомы. В то же’ время Wiske-
man (1974) не нашел, за исключением щек, связи между локализацией
меланомы и местом наиболее выраженной поглощаемости лучевой энергии.
На основании этого сделан вывод, что свет действует не как локальный
инициатор трансформации меланоцитов, а как канцероген. Отмечается
роль негативного воздействия солнечных лучей на клетки Лангенгарса
(Mackie, 1982). Особенно часто (до 90%) располагается меланома на откры-
тых участках тела у женщин (Schneller, 1980). Что касается ассоциации
меланомы с такими признаками как цвет глаз, волос, кожи, то данные по
этому вопросу неоднородны, хотя и имеются сведения о меньшей поражае-
мое™ брюнетов (Gellin et al., 1969) и наибольшем риске лиц с рыжими
волосами и со светлой кожей (Beral et al., 1983). Определенное значение в
развитии меланомы придается нарушениям в иммунной системе. Возник-
новению меланомы из пигментных невусов способствует травмирование
их, не обязательно многократное, солнечные ожоги, прижигания и другие
раздражающие вмешательства. Признаками, указывающими на развитие
меланомы, могут быть внезапное усиление роста, изменение поверхности,
уплотнение, обычно неравномерное, ослабление или чаще усиление окраски,
выпадение волос в зоне невуса, появление вокруг невуса эритематозной
каймы, точечных пигментаций, кровоточивость, появление зуда, жжения,
болезненности.
Клиника. Различают следующие варианты (ВОЗ, 1980): 1) злокаче-
ственная меланома; 2) злокачественная меланома, возникающая в пред-
раковом меланозе, включая меланотическое пятно Гетчинсона; 3) злокаче-
ственная меланома, возникающая из голубого невуса; 4) злокачественная
меланома, возникающая из гигантского пигментированного невуса. По
данным Hunter (1979), гистогенетически меланома в 14% случаев связана
с ограниченным преканцерозным меланозом Дюбрея, в 74%—является
поверхностно распространяющейся меланомой, в 37%—нодулярной. Пер-
вая форма злокачественной меланомы является наиболее частой из пере-
численных и наиболее злокачественной. Соответствует термину нодуляр-
ная меланома, обычно встречающемуся в литературе (Clark et al., 1969;
Sober et al., 1979, и др.). Может развиться на видимо здоровой коже,
но чаще ей предшествуют те или иные изменения, в первую очередь пиг-
ментные невусы, а из них—пограничные. Но меланома может развиться и
311
Рис. 199. Меланома кожи.
из дермального невуса без микроскопических признаков пограничной ак-
тивности (Okun и Banman 1965; Okun et. al., 1974). Очень высока частота
развития нодулярной меланомы у больных пигментной ксеродермой (Jung,
1980). А. П. Шанин (1964) не наблюдал развития меланомы из пигмент-
ных волосатых невусов. Описано развитие меланомы в зоне псориатиче-
ского очага, подвергавшегося воздействию рентгеновских лучей, препа-
ратов дегтя, УФО (Proctor et al., 1980) при длительном лечении мето-
трексатом (Wemmer, 1983). Клинически имеет вид опухолевидного образо-
вания или пигментированного пятна, быстро увеличивающегося в объе-
ме, неправильных очертаний, насыщенного желтоватого или темно-корич-
невого цвета (рис. 200). Интенсивная пигментация в зоне опухоли, однако,
наблюдается не всегда. Встречаются полностью или частично ахроматиче-
ские меланбмы (Borkovic и Schwarta, 1983, и др.). Клинически они могут
проявляться в виде опухолевидных узловатостей красного цвета, без или
с изъязвлением. При подозрении на беспигментную меланому Morishima
и соавт. (1983) рекомендуют применять иммунофлюоресцентный метод с
использованием формальдегида, предложенный Falck и соавт. (1962), для
выявления катехоламинов, 5-гидрокситриптамина, ДОФА и 5-гидроксит-
риптофана.
Наблюдаются случаи так называемой воспалительной меланомы, про-
текающей с резко выраженными краснотой, отеком, инфильтрацией, бо-
лезненностью (Haupt et al., 1984), клинически сходной с метастатической
рожеподобной карциномой. По мере роста опухоли, иногда достигающей
очень больших размеров, поверхность ее, вначале гладкая, становится
неровной, гиперкератотичной, покрытой корками, бугристой, неравно-
мерно возвышающейся над кожей, легко травмируемой, кровоточивой.
Могут быть пузыри (Knoth, 1960). Усиливается пигментация, окраска
становится почти черной с голубовато-синеватыми оттенками Может на-
312
ступить изъязвление, распад опухоли. Вокруг нее появляются мелкие
пигментированные опухолевидные элементы, увеличиваются и становятся
плотными регионарные лимфоузлы. Метастатические очаги на коже могут
быть пятнисто-папулезными, пигментированными или нет, нодулярными,
фунгозными с изъязвлением, редко—везико-буллезными (Goldman, 1966).
В более поздние сроки возникают регионарные метастазы в лимфоузлы и от-
даленные, гематогенные, наиболее часто в легкие, печень, головной мозг,
кости (Weidner, 1981). Редким метастатическим феноменом является диф-
фузный меланоз. Konrad и Wolff (1974) рассматривают диффузный меланоз
как уникальную форму метастазирования отдельными клетками. В редких
случаях первым признаком развившейся меланомы являются метастазы
в лимфоузлы, что может наблюдаться в случаях амеланотической мела-
номы, спонтанно регрессирующей первичной опухоли, а также при очень
малых размерах меланомы.
Hula (1975) наблюдал феномен спонтанного регрессирования первич-
ной меланобластомы у 8,1% больных меланомой. Прогноз тот же, что
и у больных без спонтанного исчезновения опухоли. Аналогичного мнения
придерживаются 3. Б. Гольберт и соавт. (1977). Honigsman (1974) допу-
скает возможность развития меланомы первично в лимфоузлах из невус-
ных клеток, что, по их мнению, могло бы быть объяснением тех случаев
метастатической меланомы, в которых не обнаружен первичный очаг.
Редко первичные меланомы могут быть множественными (Beard more и
Davis, 1975; Remy и Bocnendahe, 1975). По данным Beardmore и Davis
(1975), только у 3,9% из 1444 больных меланомой было более одной опухо-
ли. В этих случаях может возникать вопрос, не являются ли они метаста-
тическими? В известной мере помогают решению этого вопроса гистологиче-
ские исследования, в частности отсутствие активности в зоне дермо-эпи-
дермального соединения может свидетельствовать в пользу метастаза (Ког-
berg et al., 1978). Tritsch (1970) обращает внимание на отсутствие или не-
значительную выраженность воспалительной реакции при метастатической
меланоме. Однако это не абсолютные критерии, тем более что некоторые
авторы, в частности Gans и Steigleler (1957), не находят принципиальных
различий в морфологическом строении первичных и метастатических опу-
холей. Особенно большими затруднениями могут быть при метастазе ме-
ланомы в эпидермис, когда, по мнению Balus и соавт. (1968), эти два состоя-
ния практически разграничить невозможно. Очень сходна по клинической
картине и гистологической структуре с меланомой, развившейся на фоне
ограниченного преканцерозного меланоза Дюбрея, так называемая поверх-
ностно распространяющаяся меланома. Отличия между ними, по данным
Kalkoff и Kiihnl-Petzoldt (1973), заключаются в том, что поверхностно
распространяющаяся меланома может возникнуть на любом участке кожи
(а не на открытых предпочтительно, как ограниченный преканцерозный
меланоз Дюбрея), в том числе и на слизистых; растет быстрее, вертикаль-
ный рост начинается при значительно меньших размерах (1,5—2 см) и это
может произойти уже в течение первого года; клинически уже в ранней
стадии она более инфильтрирована, а гистологически в- этот же период
313
выявляется инвазия меланоцитов в дерму. Может наблюдаться спонтан-
ное частичное регрессирование (Happle et al., 1975). По-видимому, к бы-
стро прогрессирующим вариантам поверхностно распространяющейся
меланомы относится выделяемая как особая форма акральная лентиги-
нозная меланома, встречающаяся чаще у лиц с черной и желтой кожей
на подошве и в области подногтевого ложа, характеризующаяся особенно
плохим прогнозом (Vazguez et al., 1984 и др.). Меланома наиболее часто
располагается на конечностях, особенно на ногах, туловище, затем в об-
ласти головы, шеи, но может развиться на любом участке кожи, в том числе
на слизистых. По данным Kiihne и Sochor (1978), при примерно равной
частоте локализации меланомы на голове у мужчин и женщин, выявлено
значительное преобладание поражаемости туловища у мужчин, а нижних
конечностей—у женщин. Описано сочетание меланомы кожи и глаза (Kor-
ting et al., 1983), причем поверхностно распространяющаяся меланома
на коже развилась спустя 6 лет после удаления глаза по поводу веретено-
клеточной меланомы радужки. Сведения о частоте развития меланомы у
людей с гигантским пигментированным невусом неоднородны. Так, Krinitz
и Wozniak (1972) наблюдали развитие меланомы у 11,8% больных. По
материалам ВОЗ (1980), злокачественная меланома возникает примерно
у 1/3 больных гигантским пигментированным невусом. Этот вид меланомы
возникает в более раннем возрасте, чем другие формы. Считается, что в
большинстве случаев это происходит до 10-летнего возраста (Sober et al.,
1979). У ряда больных протекает с поражением центральной нервной си-
стемы (Braun-Falco и Schoefinius, 1973). Описано развитие меланомы на
фоне гигантского церебриформного невуса (Gross и Carter, 1967). В зоне
голубого невуса меланома развивается редко (Koch, 1959 и др.). Gartmann
и Lischka (1972) отмечают, что без изъязвления и (или) метастазов невоз-
можно отличить злокачественно перерожденный невус от доброкачествен-
ного. Трудна и гистологическая дифференциальная диагностика. Kittken
и Negri (1966) описали развитие меланомы из голубого невуса у негритянки
с метастазами и гибелью больной спустя 14,5 лет после появления голубого
невуса и 7,5 лет после его неполного удаления. Значительно более быстрое
течение заболевания описал Hernandez (1'973). Больной 35 лет погиб от
метастазов через год после удаления опухоли и через 17 месяцев с момента,
как невус был обнаружен. Своеобразна клиническая картина меланомы
на ногтевом ложе (меланотический панариций). Вначале опухоль просве-
чивает через ногтевую пластинку в виде коричневатого пятна, которое,
увеличиваясь в размерах и объеме, приподнимает ноготь, постепенно де-
формирует его и разрушает. Описана метастатическая меланома в области
ногтевого ложа в виде пигментированных полос (Petras и Samman, 1983).
По наблюдениям Wojnerowiz и соавт. (1976), подногтевая меланома
составила 2,3% всех случаев меланом. Авторы считают, что при появле-
нии любой опухоли, повреждающей ноготь, необходимо исключить мела-
ному. Меланома бывает сходной с бородавкой, папилломой, фибромой (Sha-
piro и Bodian, 1969). Иногда, особенно если имеется изъязвление и кро-
воточивость, меланома может напоминать пиогенную гранулему (Niken
314
и Lubin, 1975). К числу редких локализаций относится меланома полового
члена, чаще всего встречающаяся на головке. По данным Tober и Hart-
mann (1977), в литературе, до их наблюдения двух больных, с 1859 года
опубликован 41 случай. К моменту постановки диагноза у 50% больных
уже имелись метастазы. Редко встречается первичная меланома полости
рта. На основании анализа литературы Chaudhry и соавт. (1958) указы-
вают, что за 100 лет (1859—1957) опубликован только 91 случай с изолиро-
ванной меланомой-этой локализации. В большинстве случаев она распо-
лагалась на твердом небе, альвеолярных отростках верхней челюсти.
Средняя продолжительность до постановки диагноза составила 16,7 ме-
сяцев, что, возможно, объясняется отсутствием боли. Отмечается частое
предшествование меланоме гиперпигментации слизистой полости рта. Мо-
жет развиваться изъязвление, значительны разрушения окружающих
тканей, раннее метастазирование. Так, в момент постановки диагноза они
наблюдались у половины больных, в том числе у 25%—отдаленные.
Было только 3 больных, переживших 5-летний период.
Меланома злокачественная может развиться в любом возрасте, но ред-
ко бывает в предпубертатном периоде. Allen (1960) изучил 28 случаев зло-
качественной меланомы у детей. В 15 наступил летальный исход, в 7—с
метастазами. Характер течения болезни у других больных неизвестен.
Ни / одного из больных не было данных за ювенильную меланому. Гисто-
логически опухоль была более злокачественна чем у взрослых. В то.же
время имеются данные о том, что меланома у детей протекает более благо-
приятно (Misgeld, 1973). У мужчи меланома встречается несколько реже.
По данным Hempel и Remmele (1 '73), основанным на анализе статистики
с 1927 по 1971 г., касающемся 13 19 случаев меланомы, соотношение жен-
щин и мужчин составляет 1:0,84. Имеются указания на частое развитие
у больных меланомой витилигоподобной лейкодермы, которая рассматри-
вается как благоприятный прогностический признак (Nordlung et al.,
1983; Koh et dl.,). Прогноз для жизни больных меланомой зависит от
типа меланомы, локализации опухоли, ее величины, гистологической струк-
туры (степень плеоморфизма и проникновения в дерму опухолевых клеток,
наличие их в сосудах, частота митотических фигур, интенсивность воспа-
лительной реакции стромы), сроков начала лечения. Friedman и соавт.
(1983) указывают, что течение меланомы более благоприятное, если в клет-
ках опухоли обнаруживаются клетки приобретенного меланотического
невуса. Прогноз меланомы, развившейся из голубого невуса, более бла-
гоприятен, чем других форм (Трапезников Н. Н. и соавт., 1976; ВОЗ,
1980 и др.). По мнению Renman и Stringer (1971), 20% меланом, развив-
шихся в зоне врожденного гигантского невуса, заканчиваете^ фатально.
Наименее благоприятен прогноз при нодулярной меланоме. Прогноз ме-
ланомы у женщин на открытых участках лучше, чем на закрытых, у муж-
чин он не зависит от локализации (Schneller, 1980).
Степень инфильтративного роста оценивается гистологически соглас-
но критериям Clare и соавт. (1969) или, что точнее, по измерению толщины
опухоли. Первая стадия, по Clare и соавт.—это эпидермальная локали-
315
зация опухоли; II—опухолевые клетки проникают в сосочковый слой дер-
мы; III—атипичные меланоциты инфильтрируют сосочковый слой вплоть
до сетчатого; IV—пролиферация опухолевых клеток в сетчатом слое; V—
проникновение их в подкожную клетчатку. Прогноз более благоприятен
в первых двух стадиях. Kiihnl-Petzoldt и соавт. (1982) приводят следую-
щие оценки прогноза, основанные на данных измерения толщины опухоли:
хороший—92% больных живут более 8 лет: это женщины, без изъязвления
опухоли и толщиной ее менее 1,5 мм; средний—78% переживших 8-летний
период—это мужчины, без изъязвления опухоли и при толщине ее менее
1,5мм; плохой (4'6%)—меланома толще 1,5мм с изъязвлением. Для оценки
прогноза имеет значение степень выведения с мочой меланогенов (Schwartz
et al., 1969). Диффузный меланоз кожи и мел анурия—свидетельство
терминальной стадии (Silberberg et. al., 1968). По данным Storck (1977),
основанным на материалах литературы, 5-летний период переживает 51%
больных при наличии опухоли без клинически выявляемого увеличения
лимфоузлов (1 стадия), 12,6% при вовлечении в процесс регионарных лим-
фоузлов (II стадия); все больные с лимфо-или гематогенной диссемина-
цией (III стадия) погибают за этот период. Прогноз более благоприятен,
если размеры опухоли не превышают 2 см в диаметре. К такому же выводу
пришел ранее Buchanan (1961). По мнению Mishima (1968), прогноз более
благоприятен при так называемой меланоцитической меланоме, развив-
шейся на фоне сенильного лентиго, ограниченного пребластоматозного
меланоза Дюбрея, из клеток меланобластического ряда, чем при невоцити-
ческой меланоме, возникающей из обычного лентиго, пограничного невуса,
из клеток невобластического (шванновского) ряда. Неблагрприятен про-
гноз при локализации меланомы на слизистых. Ч*го касается данных об
ухудшении течения меланомы во время беременности, то, как считает Zaun
(1983), это мнение эпидемиологически недостаточно обосновано.
Гистопатология. Опухоль состоит из кубических, полигональных и в
меньшей степени веретенообразных меланоцитов, проникающих в дерму.
Могут встречаться гигантские клетки. Степень пигментации неоднородна
так же, как и частота митотических фигур. Опухолевые клетки окружены
воспалительным инфильтратом, преимущественно лимфо-гистиоцитарным,
интенсивность которого уменьшается по мере прогрессирования опухоли.
М. А. Корницкий (1983) описал сочетание в опухоли атипичных мелано-
цитов и кератиноцитов, считая такие опухоли истинными меланокарци-
номами.
Дифференциальный диагноз. Дифференцировать меланому необходи-
мо от пигментированной базалиомы, .болезни Боуэна, гемангиоперицитомы,
фибросаркомы, псевдомеланомы, пиогенной гранулемы.
Лечение. Хирургическое, лучевое, комбинированное (включая им-
мунотерапию).
316
ЛИТЕРАТУРА
ГольбертЗ. В., Романова О. А., Червонная Л. В. О спонтанной регрессии злокаче-
ственной меланомы. Арх. патол. 1977, 6, 36—42.
Корницкий М. А. О гистогенезе меланокарциномы кожи. Вести, дерматол. и вене-
рол., 1983, 11, 40—41.
Albert D. М., Todes-Taylor N. , Wagoner М., Nordlung J. J. , Lerner A. B. Viti-
ligo or Halo nevi occuring in two patients with choroidal melanoma Arch, dermatol., 1982,
118, 1, 34—36.
Beadmore G. L., Daois N. C. Multiple primary cutaneous melanoms.— Arch, derma-
tol., 1975, 3, 5, 603—609.
Bork K-, Korting G. W., Rumplet H. J. Diffuse Melanos bei malignen Mefanom.—
Hautarzt, 1977, 28, 9, 463—468.
Borkooic S. P., Schwartz R. A. Amelanotic lentigo maligna melannoma manifesting
as a dermatitis-like plaque.— Arch, dermatol., 1983, 119, 5, 423-^425.
Callen J. P., Chanda J. J., Stawiski M. A. Malignant melanoma.— Arch, dermatol.,
1978, 114, 3, 369—370.
Clark W. H., Reimer R. R., Greene M., Ainsworth A. M., Mastrangelo M. J. Ori-
gin of familial malignant melanomas from heritable meoanocytic lesions.— Arch, dermatol.,
1978, 114, 5, 732—738.
Friedman R. J., Riegel D. S., Kopf A. etal. Favorable prognosis for malignant me-
lanomas associated with acquired melanocytic nevi. Arch, dermatol., 1983, 119, 614 55—462.
Haupt H. M., Hood A. F., Cohen M. H. Inflammatory melanoma. J. Am. Acad,
deni. 1984, 10, 1, 52—55.
Happle R., Schotola /., Macher E. Spontanregression und Leukoderma beini malig-
nen Melanom.— Hautarzt, 1975, 26, 3, 120—123.
Hula M. Spontanni regrese primarniho melanoblastomu.— Cs. dermatologie, 1975,
50, 3, 156—168.
Hunter J. A. A., Pondest S., White H., Me Intyre M. A. The incidence and prog-
nosis of cutaneous malignant melanoma in south-east Scotland Brit. J. derm., 1979,101, suppl.
17, 9.
Jung E. G. Xeroderma pigmentosum In: «Dermatologie in Praxis und Klinik Hrsg.
W. Korting Stuttgart, 1980, Bd. 2. 1584—1587.
Kokoschka E. M. Immuntherapeutische Moglichkeiten der Behandlung des malignen
Melanoms.— Hautarzt, 1979, 30, 12, 656—661.
Nioen J., Lubin J. Peducufated malignant melanoma.— Arch, derm., 1975, 3, 6,
755—756.
Nordlung J. J., Kirkwood J. M., Forget В. M. et al. Vitiligo in patients with me-
tastatic melanoma: A good prognostic sign.— J. Am. Acad. Derm., 1983, 9, 5, 689—696.
Orfanos С. E., Doring Ch. MaTignes Melanom: Eine aktuelle Bestandsaufnahme Z.
Hautkrkh., 1983, 12, 15, 881—900.
Proctor M. S., Cox A. J. Grais L. S. Lentigo jnaligna melanoma in a treated psoria-
tic plaque.— Arch, dermatol., 1981, 117, 3, 149—150.
Remy W., Bockendahe H. Gleichzeitiges Vorkommen zweier primaren Melanoma bei
einem Patienten.— Hautarzt, 1975, 26, 6, 327—329.
Sober A., Mihm M. C., Fitzpatrick T. B., Clare H. H. Maligant melanoma of the
317
skin and beningen neoplasm and hyperplasias of melanocytes in the skin: In «Dermatology
in general Medicine,» Ed by T. B. Fitzpatrick et al., Sec. ed. Me Graw—Hill book company,
N. Y., 1979, 629—654.
,kck H. Klinik, Statistik und Risikofaktoren des malignen MeTanoms. Dermatolo-
gies, 1977, 155, 3, 129—142.
Taber H., Hartmann H. H. Das maligne Melanom des Penis.— Hautarzt, 1977, 28,
115, 239—240.
Su W. P. D„ Bradley R. R. Amelanotic lentigo maligna.— Arch, dermatol., 1980,
116, 1, 82—83.
Wassilew S., Fanner M., Schaeg G. FamiTiare multiple naevogene Melanoma.— Z.
Haufkrh., 1981, 56, 14, 931—941.
Weidner F. Metastasierung maligner Hautmelanome Klinische und histologische As-
pekte.— Aktuel. Dermatol., 1981, 7, 2, 31—33.
МЕЛАНОМА ЮНОШЕСКАЯ (melanoma juvenile)
Синоним: невус Шпитца. ,
Этиология и патогенез. Согласно гистологической классификации
опухолей кожи ВОЗ (1980), юношеская меланома представляет собой
эпителиоидный и(или) веретеноклеточный невус. Schreus (1969) считает,
что невоклеточные невусы принадлежат к органоидным новообразованиям,
состоящим из меланоцитов и шванновских клеток; Несмотря на то, что
комбинированные невусы, к которым относится и юношеская меланома,
редко предшествуют злокачественной меланоме, Р. Иконописов и соавт.
(1977) рассматривают это новообразование как пигментный предрак.
Клиника. Обычно асимптомное опухолевидное (выбухающее) обра-
зование плоской или полушаровидной формы, небольших размеров (около
1 см), с четкими границами, плотной консистенции, цвет которого колеб-
лется от светло-красного до темно-коричневого или реже черного. По наб-
людениям Rupee и соавт. (1979), только в одном случае из 80 опухоль была
множественной.
Burket (1979) наблюдал 2 больных, у которых было от 20 до 50 элемен-
тов, причем у одной из них они были распространенными. Вначале опухоль
растет быстро, а затем остается годами в стационарном состоянии. По пе-
риферии могут располагаться телеангиэктазии. Поверхность гладкая,
лишена волос, реже гиперкератотичная, бородавчатая. В редких случаях
возможно изъязвление (Paniago-Pereira et al., 1978). Локализация—
преимущественно на лице, конечностях, реже на шее, туловище. По дан-
ным Rupee и соавт. (1979), на голове располагается опухоль в 54% случаев,
в остальном на конечностях и только у 3 из 80 человек—на туловище. Име-
ются сведения о локализации на конъюнктиве (Gartman и Thufm, 1960),
в области ягодиц (Thorman, 1976). По мнению Alien (1960), при локализации
опухоли на конъюнктиве, в первую очередь, надо думать о возможности
злокачественной, а не ювенильной меланомы. Frenk и соавт. (1975) в своей
работе приводят классификацию Duperrat и Dufourmental, различающих 4
клинических варианта: 1) малопигментированная опухоль, мягко-эластиче-
ской консистенции, розового или светло-коричневого цвета, исчезающего
318
при диаскопии; 2) малопигментированная опухоль плотной консистенции
(фиброматозный тип,' иногда с телеангиэктазиями); 3) пигментированное
новообразование с гладкой, редко с шелушащейся поверхностью; 4) мно-
жественные элементы красновато-коричневатого цвета на фоне пятен типа
кофе с молоком. Кроме того, авторы отмечают такие редкие варианты, как
развившиеся на фоне гигантского невуса с депигментацией вокруг, мно-
жественные—на здоровой коже.
Anrade (1968) различает следующие клинические варианты ювениль-
ной меланомы: бляшечную и мультинодулярную, множественную сгруппи-
рованную в зоне пятен кофе с молоком; ангиоматозную, или сходную с пно-
генной гранулемой; фиброматозную, плотную, с выраженной пигментацией.
Prose и соавт. (1983) на основании данных литературы выделяют 4 типа
множественных ювенильных меланом: диссеминированные меланомы, сгруп-
пированные на здоровой, гипо - или гиперпигментированной коже.
Заболевание развивается преимущественно у детей, с частотой, не-
зависимой от пола. Имеются сведения о семейных случаях (Anrade, 1968).
Frenk и соавт. (1975) считают, что в большинстве случаев развивается в
возрасте 3—15 лет; 15%—в период полового созревания и у взрослых, но
может существовать с рождения или появиться у лиц старше 60 лет. Зло-
качественное перерождение наблюдается редко (Allen, 1960; Saber et al.,
1979). По мнению Allen, возможность развития злокачественной меланомы
не выше, чем на фоне любого смешанного невуса. Как считает автор, не
опубликовано ни одного случая с метастазами, в котором бы гистологиче-
ская картина соответствовала критериям ювенильной меланомы.
Гистопатология. В поверхностных слоях дермы имеются в разных
соотношениях, в том числе с преобладанием того или иного вида, веретено-
образные клетки, часто располагающиеся перпендикулярно к поверхности
эпидермиса» эпителиоидные клетки, в том числе многоядерные, некоторые
из них, из-за расположения ядер по периферии клетки, сходны с клетками
Тутона. Гистологически юношеская меланома может иметь сходство со
злокачественнной (Okun, 1979 и др.). Как считает А. К. Апатенко (1977),
ювенильную меланому отличает от злокачественной поверхностный харак-
тер расположения клеток, небольшое количество пигмента, меньший ати-
пизм клеток, преобладание веретенообразных клеточных элементов, нали-
чие многоядерных гигантских клеток. По мнению Sprafke (1964), сходст-
во может достигать такой степени, что только по данным морфологии нель-
зя с достаточной достоверностью решить вопрос, идет ли речь о злокаче-
ственной меланоме или безвредной юношеской. По данным Paniago-Perei-
га (1978), в 9,5% обнаруживается пограничный тип невуса, в 66%—сме-
шанный, в 24,5%—интрадермальный, преимущественно веретеноклеточ-
ный—в 54,5% эпителиоидноклеточный—в 21%, комбинированный—в 24,5%
Дифференциальный диагноз. Дифференцировать юношескую меланому
необходимо с меланомой злокачественной, гистиоцитомой, келоидами, бо-
родавками, ангиомой, туберкулезной волчанкой.
Лечение. Хирургическое иссечение.
319
ЛИТЕРАТУРА
Апатенко А. К., Мезенхимные и нейроэктодермальные опухоли и пороки развития
кожи- М., Медицина, 1977. Гистологическая классификация опухолей кожи. ВОЗ, Женева,
1980.
Burket J. М. Multiple benign juvenile melanoma.— Arch, dermatol., 1979, 115, 2,
229.
Frenk R., Meoorah B., Delacretaz J. Seltenere Melanozyten und NaevuszeTlnaevi.—
Hautarzt, 1975, 26, 12, 619—624.
Okun M. R. Melanoma resembling spindle and epithelioid cell nevus.— Arch. Derm.,
1979, Ц5, 12, 1416—*420.'
Paniago—Pereira C., Maize J. C., Ackermann A. B. Nevus of large spindleand epit-
helioid cells (Spitz’s nevus).— Arch. Derm., 1978, 114, 12, 1811—1823.
Prose N. S., Heilman E., FelmanY. M. etal. Multiple benign juvenile melanoma.—
J. Am. Acad. Dermatol., 1983, 9, 2, 236—242.
Rupee M., Hint A., Horm W. Das juvenile Melanom.— Hautarit, 1979, 30, 11, 581—
584.
Sober A. J., Mihm M. C., Fitzpatrick T. B., Clark W. H. Ma ignant melanoma of
the skin and benign neoplasms and hyperplasias of melanocytes in the akin. In: «Dermatolo-
gy in genera! Medicine.» Ed. T. B. Fitzpatrick et al., 2—d 1979, 629—654.
Thorman Th. Benignes juveniles Melanom Spitz in dermalen Navuszellnavus bei einem
Erwachsehen.— Derm. Machr., 1976, 162, 8, 686—689.
МИКОЗ ГРИБОВИДНЫЙ (mycosis fungoides).
Синонимы: грибовидная гранулема, генерализованный саркоматоз,
кожная лимфадения и др.'
Грибовидный микоз впервые описал французский дерматолог Алибер
(J. Alibert) в 1806 г. Он же в 1832 году предложил название грибовидный
микоз, подчеркнув характерные клинические проявления болезни—круп-
ные грибовидные опухоли. В 1879 г. другой французский дерматолог Ba-
zin подробно описал клинику так называемой классической формы грибо-
видного микоза и с тех пор заболевание носит имя Алибера—Базена. Vi-
dal и Brocq в 1885 г. описали вторую форму—mycosis fungoides a’tumeurs
d’emblee. Hallopeau и Besnier в 1892 г. описали третью—эритродермиче-
скую форму. Первое описание грибовидного микоза в России сделал А. М.
Стуковенков в 1889 году.
Этиология и патогенез. До сих пор нет единой точки зрения на этио-
логию и патогенез грибовидного микоза. Не решен вопрос о характере
заболевания—опухоль или первично-воспалительный процесс, поддержи-
ваемый иммунными нарушениями. Не получены достоверные данные об
этиологической роли инфекции, в том числе вирусной По представлениям
многих современных авторов в основе грибовидного микоза лежит опухоле-
вое поражение ретикуло-гистиоцитарной (лимфоидно-ретикулярной) си-
стемы кожи (Каламкарян А. А., 1968, 1977; 1983; Ашмарин Ю. Я., 1972;
Пр’оскурнина В. С., 1975; Degos, 1963; Montgomery и Winkelmann, 1968;
Beer et al., 1970 и др ). Американские и английские авторы (Gall и Mallery’
1942; Rappaport, 1976; Lever, 1975; Lukes, 1978; Carr et al., 1980, и др-j
320
относят грибовидный микоз в группу злокачественных лимфом. Rappa-
port (1978), Steigleder (1978), Edelson (1978), Buchner et al. (1979) полага-
ют, что грибовидный микоз является Т-клеточной лимфомой, которая на-
чинается во многих участках с лимфоидно-ретикулярного инфильтрата
преимущественно в коже. Согласно классификации ВОЗ (1976), грибовид-
ный микоз отнесен в группу опухолей кроветворной и лимфатической тка-
ней. Однако представление о решающем значении изменений гемопоэтиче-
ских органов в механизме развития грибовидного микоза недостаточно
обосновано. Изложенные материалы позволяют считать, что проблема
грибовидного микоза не имеет абсолютно четких контуров, и перспектива
ее исследований еще очень широка.
Клиника. Грибовидный микоз в 70—80% случаев начинается в возра-
сте от 40 до 60. лет. Мужчины болеют чаще, чем женщины. Мы считаем целе-
сообразным придерживаться традиционной классификации, разделяющей
грибовидный микоз на три основные формы: классическую Алибера—Базена,
обезглавленную Брока и Видаля и эритродермическую Галлопо—Бенье.
Классическая форма грибовидного микоза ‘ разделяется на три ста-
дии: 1) экзематоидно-эритродермическую, 2) бляшечную и 3) опухоле-
вую (рис. 201, 202, 203).
* В экзематоидно-эритродермической стадии высыпания могут напоми-
нать парапсориаз, экзему, псориаз, нейродермит или же имеется картина
генерализованной эритродермии. Для диагностики этой стадии извест-
ное значение имеют длительность и упорство течения, резкость границ
очагов поражения, увеличение периферических лимфатических узлов,
непрекращающийся зуд, резистентность к противовспалительной и десен-
сибилизирующей терапии.
Инфильтративно-бляшечная стадия характеризуется резко ограничен-
ными, несколько возвышающимися плоскими инфильтративными бляшками
с грубой шагреневой поверхностью, размерами от фасоли до ладони и
больше. Бляшки отличаются насыщенным темно-красным или синюшным
цветом с буроватым оттенком.
Опухолевая стадия характеризуется появлением опухолевых по-
ражений. Только опухолевые поражения встречаются у небольшого чи-
сла больных; у подавляющего же большинства обнаруживаются проявле-
ния всех периодов заболевания, т. е. наряду с опухолями встречаются
экзематоидные и инфильтративно-бляшечные изменения. Насыщенно-крас-
ные с синюшно-багровым оттенком опухоли достигают величины от вишни
до куриного яйца и больше, плотны на ощупь, реже имеют плотно эластиче-
скую консистенцию, возвышаются на 1—4 см над уровнем кожи. Характер-
ны быстрый рост и распад опухолей (несколько недель), в результате чего
образуются глубокие язвы с валикообразно-утолщенными краями, кровя-
нисто-гнойным отделяемым и остатками гангренозно распавшихся тканей.
У многих больных в этот период появляется слабость, недомогание, теря-
ется аппетит, повышается температура тела и т. д.
Поражения слизистых оболочек при грибовидном микозе встречаются
сравнительно редко. Во рту опухолевые образования располагаются глав-
21 Клиническая дерматология 321
Рис. 201. Грибовидный микоз.
Рис. 200. Грибовидный
микоз (опухолевая стадия)
ным образом на твердом и мягком
нёбе, реже на слизистой оболочке
щек. Они склонны к быстрому рас-
паду с образованием обширных язв.
Мучительный зуд и генерализованное
поражение периферических лимфати-
ческих узлов являются частыми и
ранними симптомами грибовидного
микоза (у 90—95 % больных). Раз-
нообразные нарушения трофики об-
наруживаются почти у 2/3 больных
(пигментация кожного покрова, раз-
витие пойкилодермических измене-
ний, дистрофия волос и ногтей, на-
рушения потоотделения и др.).
Вторая форма—обезглавленный
грибовидный микоз—в настоящее
время диагностируется редко, по-ви-
Рис. 202. Грибовидный микоз. димому, в связи с тем, что ее от-
носят в группу ретикулосаркоматоза
кожи (ретикулосаркомы, лимфосаркомы). Для этой формы является типич-
ным развитие опухолей на туловище, конечностях и лице без предшест-
вующих экзематоидной и бляшечной стадии.
Третья—эритродермическая—форма грибовидного микоза значитель-
но отличается от классической формы. У большинства больных эритро-
дермия развивается сразу, без стадии предшествующих высыпаний, рас-
пространяется по всему кожному покрову, сопровождается отеком ко-
жи, выраженной инфильтрацией, обильным шелушением, плохим общим
322
состоянием, высокой общей температурной реакцией. Ярко-красная окра-
ска на лице и туловище постепенно темнеет, приобретает в дальнейшем
синюшно-фиолетовый оттенок.
Количественные и качественные изменения периферической крови в
различных стадиях грибовидного микоза бывают неодинаковыми. На'ран-
них стадиях наиболее часто отмечается лейкоцитоз, нейтрофилез с палоч-
ко-ядерным сдвигом, лимфоцитоз, а в терминальном периоде и при эритро-
дермической форме—лейкопения, лимфопения и увеличение СОЭ; эозино-
филия регистрируется почти у 70% больных во всех стадиях заболевания.
У многих больных (95—97%) грибовидный микоз протекает без сущест-
венных признаков нарушения костно-мозгового кроветворения.
Прогноз при грибовидном микозе тяжелый; болезнь в большинстве
случаев смертельна. Течение, как правило, злокачественное, неуклонно
прогрессирующее, средняя продолжительность жизни больных грибовид-
ным микозом 5—8 лет. Однако описаны случаи, когда заболевание длилось
15—20 лет и более. При эритродермической форме средняя продолжитель-
ность жизни составляет 2,8 года.
Гистопатология. В первой стадии—акантоз, отек и экзоцитоз в нижних
рядах клеток шиповидного слоя эпидермиса, расширение кровеносных и
лимфатических сосудов дёрмы. Основную мйссу полосовидного пролифе-
рата в дерме составляют лимфоциты, фибробласты, плазматические клетки,
различное количество эозинофилов, тучных клеток и нейтрофильных гра-
нулоцитов. Во второй, инфильтративно-бляшечной стадии, отмечается резко
выраженный акантоз с образованием небольших гнезд лимфоцитов и гисти-
оцитов (микроабсцессы Pautrier), которым придается большое диагностиче-
ское значение. В третьей, опухолевой стадии, пролиферат занимает всю
толщину дермы, распространяясь вглубь, иногда захватывая подкожную
клетчатку и подлежащие ткани. Среди пролиферирующих элементов зна-
чительно чаще наблюдаются лимфоидно-ретикулярные и ретикулярные
клетки в разных стадиях развития, что создает картину, сходную с опухо-
левым процессом. Наблюдается много фигур митозов. Так называемые
микозные клетки, представляющие собой атипичные дермальные гистио-
циты, тканевые лимфоциты или первичные лимфоидно-ретикулярные (мо-
нонуклеарные) клетки с крупным мозговидным или почкообразным ядром,
имеют диагностическое значение только в комплексе с другими данными
микро' конической картины.
Грибовидный микоз первично и преимущественно поражает кожу,
однако в терминальной стадии нередко поражаются и внутренние органы.
Висцеральная патология на аутопсии обнаруживается у 20—60% больных
(Беляева Е. Ф. и соавт., 1955; Borok et al., 1980). Инфильтрат во внут-
ренних органах обычно аналогичен инфильтрату в поражениях кожи:
отмечается характерная для грибовидного микоза множественность кле-
точных типов и полиморфизм лимфоидно-ретикулярных клеток.
Лечение. Кортикостероидные препараты, химиотерапевтические цито-
статические средства (проспидин, фосфазин, дипин, циклофосфамид), лучи
Рентгена. Указанные средства применяется раздельно и в различных ком-
21* 323
бинациях. Nisce и соавт. (1978) рекомендуют схему фракционного облуче-
ния электронными лучами всей кожи. На курс лечения 3400 рад.
ЛИТЕРАТУРА
Каламкарян А. А. Некоторые актуальные вопросы клиники грибовидного микоза.—
Вести, дерматол. и венерол., 1977, 12, 3—8.
А. А. Каламкарян. Клиника и терапия ретикулезов кожи. Ереван, Айастан, 1983.
Проскурина В. С. Материалы к изучению патогенеза грибовидного микоза. Автореф.
дисс. докт. мед. наук.— М., 1975,
Bounet Н., Farrer-Brown G., Henry К-, Jelliff A. Letters to the editor. Classifica.
tion of non Hodgkin’s lymphomas.— Lancet, 1974, , 405—406-
Bucher S. Eezymzytochemische und immunzytologische Differepzierung von Infilt-
ratzelle in der Haut Mycosis fungoides.— Dermatofogica, 1979, 159, 5, 386—392.
Burg G., Braun-Falco O. Quantitative enzymecytochemical studies in mycosis fun-
goides.— In: Al III Cortgres National de Dermatologie. Bucuresti, 24—26 Oktombrie, 1974,
s. 53—54.
Edelson R. Recent advances in the cutaneous T—cell lymphomas.— Bull, du cancer,
1977, 2, 209—224.
Hagedorn M., BornW., RunzF., RiedeU. Further similaritis between Sezary synd-
rome and mycosis fungoides.— Arch. Dermatol. Res., 1978, 261, 3, 253—258.
Heilman D., Ackerman A. A histological atlas of pseudomalignant and malignant
fymphoreticufar disorders of the skin.— J. Dermatol. Surg. Oncol., 1980, 6, 8, 646—651.
Macklin R., FouldsJ., Nelson H. Histological changes observed in the skin of patients
with mycosis fungoides receiving photochemotherapy.— CTin. Exp. Dermatof., 1980, 5, 4,
405—413.
Molin L., Thomsen K-, Volden G., Groth О. Photochemotherapy (PUVA) in the pre-
tumour stage of mycosis fungoides.— Acta DermatovenereoL, 1981, 61, 1, 57—51.
МИКСЕДЕМА ОГРАНИЧЕННАЯ ПРЕТИБИАЛЬНАЯ (myxoedema cir-
cumscriptum praetibiale).
Микседема или слизистый отек—заболевание, обусловленное выпа-
дением или понижением функции щитовидной железы. Микседема кожи
проявляется в трех формах: 1) микседема при гипотиреозе—генерали-
зованная туберозная и диффузная (myxoedema generalisatum tuberosum
et diffisum); 2) микседема при гипертиреозе—ограниченная претибиальная
микседема (myxoedema circumscriptum prae'tibiale); 3) микседема при нор-
мальной функции щитовидной железы (myxoedema papulatum lichenoides и
sclero myxoede ma).
Ограниченную претибиальную микседему впервые описали Richter
(1927) и Keining (1928). По этому вопросу имеются описания лишь еди-
ничных наблюдений (Прокопчук А. Я. и соавт., 1968; Штульман Д. Р.,
1961; Ширков Л. и Ялемов М., 1969; Петрова И. Л. и Зайцева Л. И., 1984;
Trotter и Eden, 1942; Watson, 1946; Diamond, 1959; Wodniansky, 1966;
Lynch, 1973; Kind и Homstein, 1976; Cheung et al., 1978; Dandon et al.,
1979; Goette, 1980. Feinsiber, 1983).-
324
Рис. 204. Претибиальная
микседема-
Рис. 203. Претибиальная
микседема.
Рис. 205. Претибиальная
микседема.
Рис. 206. Претибиальная
микседема
Этиология и патогенез. В основе заболевания, по-видимому, лежит
дистиреоз, т. е. нарушения функции щитовидной железы как в сторону
повышения, так и в сторону снижения. Претибиальная микседема воз-
никает при гипертиреозе, что послужило поводом для создания тиреотокси-
325
ческой теории, после струмэктомии, после лечения тиреотоксикоза рент-
геновскими лучами, метилурацилом и другими антитиреоидными препара-
тами. Кожным изменениям либо предшествуют, либо сопутствуют явления
тиреотоксикоза. Струмэктомия, как правило, не влияет на динамику кож-
ных изменений. Гипофизарная (нейроэндокринная) теория (Хавин И. Б.
1967; Cortis et al., 1949; Diamond, 1959; Kind и Hornstein, 1976; Goette,
1980) о связи претибиальной микседемы с избыточной секрецией гипофи-
зом тиреотропного гормона не может объяснить избирательного пораже-
ния кожи голеней. Аутоиммунный патогенез претибиальной микседемы
предложили Kind и Hornstein (1976) после обнаружения ими у двух боль-
ных антител против тиреоглобулина и микросомальной ткани щитовидной
железы. Следует однако отметить, что ни гипофизарная, ни тиреотокси-
ческая, ни аутоиммунная теории не исчерпывают патогенеза претибиаль-
ной микседемы. Korting и соавт. (1968) на основании проведенных элект-
ронно-микроскопических исследований высказывают гипотезу о возмож-
ности непосредственной секреции хроматропного вещества (муцина) сое-
динительнотканными клетками кожи.
Клиника. Претибиальная микседема наблюдается преимущественно
у женщин старше 40—50 лет. Заболевание характеризуется симметричным
поражением кожи передней и передне-боковых поверхностей голеней в
форме возвышающихся (на 10—15 мм) полушаровидных подушкообразных
узловатостей или плоских опухолевидных образований различных разме-
ров, изолированных или сливающихся. Кожа на пораженных участках
уплотнена до хрящевидной плотности, не собирается в складку. При надав-
ливании пальцем ямки в противоположность другим видам отека не оста-
ется. Цвет кожи в начальных стадиях напоминает цвет апельсиновой кор-
ки, позднее кожа становится восковидной, желтовато-серой или слегка
цианотичной. Фолликулярный аппарат резко выражен, волосяные фолли-
кулы втянуты, кожа напряжена, в ряде случаев напоминает апельсиновую
корку. В очагах поражения иногда наблюдается усиление пигментации,
гипертрихоз. При длительно и неблагоприятно текущих формах очаги
поражения, сливаясь, могут занимать всю поверхность голеней с развитием
слоновости голеней и стоп (так называемые элефантиастические формы
претибиальной микседемы, рис. 204, 205, 206, 207).
У большинства больных после сформирования болезни она не обна-
руживает тенденции к прогрессированию, а в некоторых случаях наблю-
дается спонтанный регресс через несколько лет.
Гистопатология. В эпидермисе гиперкератоз. Эпителиальные отростки
часто уплощены. В дерме, особенно в нижней ее части, массивные отложе-
ния муцина, вызывающие значительное отделение коллагеновых волокон
друг от друга. Сосуды в верхней части дермы расширены и окружены слабо
выраженным воспалительным инфильтратом. Отек кожи связан с накоп-
лением в коже слизеподобного муцинозного вещества, содержащего гиа-
луроновую и хондроитинсерную кислоты (протеин полисахаридный комп-
лекс).
Дифференциальный диагноз. .Следует проводить со слоновостью.
326
Лечение—тиреоидин, рентгеновское облучение области гипофиза, впры-
скивание гиалуронидазы в очаги поражений.
• ЛИТЕРАТУРА
Z
Петрова И. Л., Зайцева Л. И. Претибиальная микседема.— Вестн. дерматол. и
венерол., 1984, 4, 47—50.
Dandona Р et al. Secsussful treatment egophthalijiosand pretibia' myxoedema with
plasmopheresis.— Brit. Med. J. 1979, 6160, 374—376.
Feinsiber D. Mixedema cirecumscripto pretebia i: Varienad verugosa.— Rev. Argent
Dermato., 198 , 4, 2, 142—148.
GoetteD. Thyroid acropachy.— Arch. Dermatol., 1980, 116, 2, 205—g06.
Kind R., Hornstein O. Klinik des praetibialen Myxodema uns neue Aspekte zur Diag-
nostik und Pathogenese.— Hautarzt, 1976, 27, 8, 375—381.
Lever W., Schaumburg-Leoer G. Histopathology of the disease skin.— 5th ed.— Phi-
ladelphia—Toronto: J. B. Lippincott C°, 1975. '
Sonnichsen N., Zabel R. Hautkrankheiten. Diagnostik und Therapie.— Leipzig, 1976.
МОНИЛЕТРИКС (monilethrix).
Синоним: аплазия волос перемежающаяся, аплазия четкообразная.
Этиология и патогенез. Заболевание представляет собой генетически,
детерминированную патологию волос, наследуемую аутосомно-доминантно
с высокой пенетрантностью, но со значительно вариабельной экспрес-
сивностью гена даже в пределах одной и той же семьи. Об этом свидетель-
ствуют и наши наблюдения, и данные литературы (Heydt, 1964, и др.). Этим
объясняется наличие, помимо больных с резко выраженным процессом,
достигающим практически тотальной алопеции, лиц с едва заметными про-
явлениями, обнаруживаемыми лишь при целенаправленном обследовании
родственников. Zimmerman (1983) отметила снижение экспрессивности у
больных в нисходящих 5 поколениях. Schnyder и Salomon (1962) указы-
вали на возможность рецессивного типа наследования, Hamm и сотр.
(1984) ее отрицают. Первичный дефект неизвестен, и, как отмечает Bent-
ley-Phillips (1979), до сих пор не удалось решить даже вопрос, является
. ли монилетрикс следствием функциональных или структурных нарушений.
Salomon и Schnyder (1962) указывали на значимость в патогенезе функцио-
нальных нарушений, вероятно, центральной нервной системы.
В основе истончения лежит, по мнению Luboch и Triantos (1979), перио-
дическое торможение синтеза кератина, причина которого не ясна. Пред-
положительная роль нарушения обмена аминокислот не подтвердилась
(Heydt, 1964, Mader и Rose, 1969).
Клиника. Основными симптомами являются поредение волос на голове
или на других участках кожи, покрытых волосами, разная степень облы-
сения, вплоть до тотального, наличие мелких красноватых фолликулярных
гиперкератотических папул, пронизанных обломками волос. Heydt (1964)
относит к основным проявлениям также плато- и койлонихию. Волосы
327
Рис. 207. Моналетрикс. . . Рис. 208. Монилетрикс.
328
короткие, обломаны на уровне 1—2 см, сухие, тусклые, деформирован-
ные, имеют веретенообразные вздутия, чередующиеся с перетяжками,
примерно с равными интервалами, примерно в 1 мм. Поражение
волосистой части головы может быть изолированным, чаще наиболее вы-
раженным в затылочной области, или диффузным, но и в этом случае можно
обнаружить участки с нормально растущими воЛосами. Описаны случаи,
когда изменения наблюдались только на других участках тела (Hamm et
al., 1984). Различной может быть и выраженность фолликулярного керато-
за. Он может развиться до или после появления патологии волос, а то и
вообще отсутствовать. Фолликулярный кератоз может обнаруживаться и
на разгибательной поверхности конечностей, хотя и в меньшей степени,
чем на волосистой части головы и задней поверхности шеи (рис.208,
209, 210).
Заболевание может существовать с рождения, но чаще развивается
на первом году жизни (когда на смену пушковым волосам нормальные не
отрастают, а появляются обломанные), в некоторых случаях в более
позднем детском возрасте, очень редко—у взрослых. Постепенно прогрес-
сирует до пубертатного периода, затем может существовать без изменений
в течение всей жизни, несколько улучшаясь в летнее время. При длитель-
ном течении—возможны атрофические изменения. Отмечено благоприят-
ное влияние беременности (Salomon и Green 1963), в редких случаях с воз-
растом возможно самопроизвольное излечение (Heydt, 1964). Считалось,
что монилетрикс чаще развивается у лиц с темными волосами, но все наблю-
давшиеся Heydt (1964) больные имели голубые глаза и светлые волосы.
Общее состояние больных обычно не страдает, но Salomon и Schnyder (1962)
полагают, что это заболевание чаще, чем считается, ассоциируется с изме-
нениями ногтей, зубов, нейропсихическими нарушениями. Из других при-
знаков описывались преждевременная катаракта (Thiel, 1959), синдакти-
лия (Salomon и Green, 1963), фолликулярный кератоз и дебильность (Ma-
der и Rose, 1969), нейродегенеративные болезни (Jfray, 1965), бронхиаль-
ная астма, диффузный нейродермит (Heydt, 1964). Roock и Dawbe (1982)
полагают, что при такого рода ассоциациях, вероятно, идет речь не о доми-
нантной, а о рецессивной форме заболевания. Необходимы, однако, даль-
нейшие исследования, чтобы решить вопрос о закономерности подобных
сочетаний, о том, являются ли они следствием плейотропного действия гена
или простым совпадением.
Гистопатология. Зоны сужения и расширения обычно без существен-
ных изменений в структуре волосяных фолликулов. Диагностическую зна-
чимость имеет обнаружение на стержне волос веретенообразных утолще-
ний.
Дифференциальный диагноз. С трихотилломанией, pili torti, грибко-
выми болезнями, узловатой ломкостью волос, псевдомонилетриксом.
Лечение. Эффективных методов лечения нет. .
329
ЛИТЕРАТУРА
Bentley-Phillips В. Monilethrix und Pseudomonilethrix. In «Haar und Haarkrank-
eiten.» Hrsg. С. E. Orfanos. Gustav Fischer Verlang, Stuttgart NY, 1979, 447—465.
Hamm H., Echternacht-Happle R. , Happle R. Monilethrix: ausschlisslicher Befall
der Korperbehaarung. Z. Hautkr., 1984, 59, 17, 1177—1178.
Lubach D., Triantos N. Untersuchung uber die Monilethrix. Hautarzt, 1979, 30, 5,
253—256.
Rook A., Dawber R. Disesses of the hair and scalp. Blackwell Scientific Publications.
Oxford London Edinburgh Boston (Melbourne., 1982, 180—186.
Zimmermann R. Zur Monilethrix. Derm. Mschr., 1983, 169, 10, 638—645.
МУЦИНОЗ ФОЛЛИКУЛЯРНЫЙ (mucinosis follicularis).
Сущность заболевания составляют дегенеративные изменения воло-
сяного фолликула и сальных желез с разрушением их структуры и муци-
нозные отложения. Н. Pinkus в 1957 г. выделил как нозологическую форму
и предложил название «alopecia mucinosa». В том же году Braun-Falco
описал аналогичные изменения под названием «mucophanerosis intrafol-
licularis et seboglandularis». Больных с подобными изменениями описывали
ранее Kreibich в 1926 г., Gougerot и Blum в 1932 г., Szodoray и Lehner в
1939 г. Так как не всегда это заболевание сопровождается алопецией, Jab-
lonska и соавт. (1959) предложили термин фолликулярный муциноз (mu-
cinosis follicularis)
Этиология и патогенез. Браун-Фалько (1957) произвел гистохимиче-
ские исследования и уточнил гликопротеидный характер вещества, напол-
няющего дегенерированные фолликулы. Он считает, что появление муци-
нозного вещества в волосяных фолликулах и сальных железах происходит
из-за разрушения клеток, наступающего под влиянием инфекционных фак-
торов, высказал предположение о вирусном происхождении заболевания.
В настоящее время многие исследователи рассматривают заболевание как
дегенеративный муциноз волосяных фолликулов и сальных желез, этиоло-
гия которого еще неизвестна.
Муцинозы в настоящее время подразделяют на три основные группы:
1. Муцинозы, связанные с дисглобулинемией и плазмоцитозом костного
мозга, куда входит большинство папулезных муцинозов (микседематозный
лйхен, склеромикседема); 2. Муцинозы, наблюдаемые на фоне дистиреоза
(гипо-или гипертиреоидизма), включающие диффузную микседему и пре-
тибиальную микседему; 3. Муцинозы, связанные с обменными нарушения-
ми, приводящими к изменениям основного вещества соединительной ткани
парциального характера (миксомы, мукоидные кисты, фокальные муцинозы)
и распространенные формы (муциноз папулезный, папуло-туберозный му-
циноз). Причины нарушений химизма и структуры основного вещества
соединительной ткани неясны. В случаях, связанных с дисглобулинемией,
можно предполагать отложение в дерме сывороточных глобулинов и му-
цина, при парциальных формах возможно локальное нарушение синтеза
фибробластами основного вещества соединительной ткани и коллагена с
330 •
образованием муцина. Различают первичный (идиопатический) муциноз,
который развивается без предшествующих кожных заболеваний, и вторич-
ный (симптоматический) муциноз, который наблюдается в сочетании с
грибовидным микозом, ретикулезами кожи, болезнью Ходжкина, лейко-
зами кожи и, как исключение, при хронических воспалительных дерма-
тозах (нейродермит, красный плоский лишай, красная волчанка и др.).
По нашим наблюдениям и данным других авторов (Каламкарян А. А.,
1980; Johnson et al., 1959; Kim и Winkelmann, 1962; Bransting, 1962; Hy-
man et al., 1962; Degos et al., 1966; Emmerson, 1969; Stankler et al., 1975;
Wolff et al., 1978; Binnick et aL, 1978; Gammon et al., 1978; Guilhon et
al., 1980. и др.), сочетание фолликулярного муциноза с ретикулогемоблас-
тозами встречается в 10—25% случаев. По мнению Hagedom (1979), ча-
стое сочетание фолликулярного муциноза с ретикулогемобластозами (зло-
качественными лимфомами кожи) позволят рассматривать этот дерматоз
как паранеоплазию.
Не всегда легко дифференцировать первичный и вторичный муцино-
зы. Мы наблюдали больных первичным фолликулярным муцинозом, клини-
ческая картина у которых симулировала грибовидный микоз. Дегенера-
тивные изменения фолликулов при этих обеих формах аналогичны, и раз-
личие заключается в том, чю при симптоматическом варианте проявля-
ются также другие х оакирные гистологические признаки основного за-
болевания.
! ic 210. Фолликулярный
муциноз.
Рис . 1. Ф 1Л.. уля, -их.,
муцинчз.
331
Клиника. Заболевание встречается в любом возрасте, но чаще в период
от 15—20 до 40^50 лет. Мужчины болеют в три раза чаще, чем женщины.
Клинически можно различать две стадии в развитии заболевания; началь-
ная—фолликулярно-папулезная стадия и следующая—опухолевидно-бля-
шечная стадия. Фолликулярно-папулезная форма чаще локализуется на
лице и волосистой части головы; нередко процесс принимает диссеминиро-
ванный характер с поражением туловища и конечностей. Высыпания на-
поминают гусиную кожу, состоят из многочисленных мелких (размером
2—3 миллиметра) фолликулярных узелков розовато-синюшного цвета
плотной консистенции, часто с выраженным кератозом. Имеются воспали-
тельные явления, особенно в начальной стадии; при дальнейшем течении
они уменьшаются (рис. 211, 212). Большей частью сильный, реже небольшой
зуд. Бляшечные очаги развиваются почти у 40—50% больных и тогда кли-
ническая картина напоминает грибовидный микоз или ретикулосаркомй-
тоз кожи. Эта клиническая форма характеризуется появлением'на лице, во-
лосистой части головы, затылке, реже на конечностях и других местах
бляшек величиной от 1—2 до 4—5 см в диаметре, инфильтрированных,
преимущественно плоских, резко отграниченных. Бляшки всзвышаются
над окружающей кожей, большей частью покрыты мелкими чешуйками,
нередко видны расширенные, заполненные роговыми массами отверстия
волосяных фолликулов. Консистенция очагов различная, но обычно до-
вольно плотная. Зуд сильный. Наряду; с бляшками имеются и фолликуляр-
но-узелковые элементы. У отдельных больных опухолевидно-бляшечные
очаги поражения подвергаются некроти’ческому распаду с образованием
более или менее глубоких болезненных язв. Поверхностная рубцовая
алопеция наблюдается у 50—60% больных. Алопеция обычно ограничен-
ная, но описываются случаи распространенной алопеции (Degos et al.,
1961; Kim и Winkelmann, 1962, и др.). Мы наблюдали двух больных с пер-
вичным фолликулярным муцинозом, у которых была поражена вся воло-
систая часть головы, в результате развилась тотальная алопеция.
Вторичный (симптоматический, злокачественный) муциноз клиниче-
ски более напоминает основное заболевание (грибовидный микоз, рети-
кулосаркоматоз кожи и др.), чем фолликулярный муциноз.
Прогноз плохой у больных с первичным муцинозом при наличии опу-
холевидно-бляшечных высыпаний и при вторичном (злокачественном) му-
цинозе, связанном с ретикулогемобластозами.
Гистопатология. Дегенеративные изменения волосяных фолликулов и
сальных желез с формированием кистозных полостей, заполненных гомо-
генными массами (муцин), окрашивающимися метахроматично толуиди-
новым синим. Иногда муцин не обнаруживается. В дерме—инфильтрат,
состоящий из лимфоцитов н гистиоцитов, иногда с наличием эозинофилов,
тучных и гигантских клеток. Электронно-микроскопически (Bonerand i et
al., 1980) в дерме определяется аморфная электроннопрозрачная субстан-
ция, в ней наблюдаются микрофибриллы от 3 до 4 нм в диаметре. Кол-
лагеновые фибриллы сохраняют нормальную периодическую структуру
с диаметром 87—121,5 нм. Среди кистозных элементов обнаруживаются
332
фибробласты с хорошо развитой эндоплазматической сетзю, лимфоциты и
гистиоциты с лакунами в цитоплазме. При злокачественном (вторичном)
муцинозе, связанном с грибовидным микозом, воспалительный инфильтрат
состоит из клеток, свойственных грибовидному микозу с микроабсцессами
Потрие в эпидермисе. Взгляды на сущность муцинозных изменений по
мере накопления новых фактов менялись. Некоторые исследователи счи-
тают, что муцин эпителиального происхождения, но механизм его образо-
вания неясен. В него входят главным образом кислые мукополисахариды и
в меньшей степени нейтральные.
Дифференциальный диагноз. Его проводят с парапсориазом (особенно
с бляшечным), нумулярной экземой, грибовидным микозом, ретикуле-
зами кожи, волосяным кератозом, красным волосяным лишаем Девержи,
саркоидозом, себорейной экземой, синдромом Лассуэра—Литтла.. Течение
заболевания длительное, рецидивирующее. Начало в большинстве случаев
острое; обычно отдельные высыпания держатся несколько месяцев и исче-
зают, не оставляя после себя следов, в то же время могут появляться
новые высыпания; так что процесс может продолжаться много лет.
Лечение. В первой стадии идиопатического муциноза рекомендуется
витамин А, кортикостероидные мази и кремы. В опухолевидно-бляшеч-
ной стадии—кортикостероиды (преднизолон 20—40 мг в сутки) в сочетании
с цитостатиками (проспидин, циклофосфамид, винкристин, винбластин),
лучи Букки или Рентгена.
i
ЛИТЕРАТУРА
Binnick A., Wax F., Clendenning W. Alopecia mucinosa of the face associated with
nycosis fungoides.— Arch. Dermatol., 1978, 114, 5, "91—792.
Bonerandi J., Andrac L., Follana J. et al. Muyinose cutanee juvenile spontanement
resolutive.— Ann. Dermatol. Venereol., 1980, 107, 1—2, 51—57.
Gammon W., Caro J., Leng J., Wheeler C. Secondary cutaneous mucinosis with sy-
s'emic lupus erythematodes.— Arch. Dermatol., 1978, 3, 432—435.
Hagedorn M. Mucinosis follicularis: Eine paraneoplastische oder praeblastomatose
Dermatose?— Z. Hautkr., 1979, 54, 15, 692—699.
Harman R., English M., Halford M. et al. Botryomycosis: a complication of exten-
sive follicular mucinosis.— Brit. J. Dermatol., 1980, 102, 2, 215—222.
Steoanooic D. Mucinosis in erythematous plaques of Steigleder and Perry. A histo-
chemical and electrnomicroscopic study.— Derm. Mschr., 1980, 166, 11, 736—746.
Wolff H., Kinney J., Ackerman A. Angiolymphoid hyperplasia with follicular mu-
cinosis.— Arch. Dermatol., 114, 2, 229—232.
НЕВУС OTA (nevus Ota).
Синонимы: серо-синюшный глазоверхнечелюстный невус.
Этиология и патогенез. Неясно, является ли невус Ота гаматомой
или невоидным образованием (Lever, 1975). Некоторыми авторами рассмат-
ривается как вариант голубого невуса (Г. В. Фалилеев и А. Ф. Бровкина,
333
Рис. 213. Невус Ота.
Рис. 212. Невус Ота.
1981; Steigleder, 1975). Семейные случаи редки. Так, из 210 больных, наб-
людавшихся Ikeda и соавт. (1967), было только 2 семейные.
Клиника. Как правило, односторонее изменение окраски кожи в зоне
иннервации тройничного нерва (I—II ветви), главным образом на лбу, пе-
риорбитально, в области висков, щек, носа, уха. Часто на конъюнктиве,
роговице, радужной оболочке глаза, К числу менее частых локализаций
относится зона губ, полости рта (мягкое нёбо, глотка), слизистая носа.
Двустороннее расположение невуса наблюдается лишь у немногих боль-
ных (О. М. Гурдус и О. С. Бочарникова, 1983; Иконописов Р. и соавт.,
1977; Albertazi et al., 1981; Pfrunder и Eichmann, 1984). Цвет кожи в. зоне
поражения колеблется от светло-коричневого до серо-черного с голубова-
тым оттенком. Склеры обычно окрашиваются в голубой цвет, конъюнк-
тива—в коричневый. Пигментация может быть диффузной или очаговой,
сетчатой, как правило границы ее неправильные, постепенно переходя-
щие в здоровую кожу. Поверхность невуса гладкая, обычно находится на
уровне кожи, без уплотнения. Kopf и Weidman (1962) описали в зоне пиг-
ментации небольшие узелковые элементы, сходные с голубыми невусами
(рис. 213, 214).
Невус Ота может существовать с рождения или появляется в раннем
детском или подростковом возрасте; Из 240 больных, по наблюдениям Ike-
da и соавт. (1967), у 119 заболевание развилось до года, у 121—после 2 лет
Два пика—один в раннем детском возрасте, второй—в период полового
созревания. Возникает примерно в 5 раз чаще у женщин. Случаи возникно-
вения его у взрослых редки. Наиболее распространен среди японцев, но
встречается у людей и других национальностей. Из 3 больных, описан-
ных А. С. Рабеном и Г. В. Титинером (1976), были 2 женщины (армянка
и калмычка) и один мужчина, отец которого—бурят, мать—русская. Обе
больные, которые наблюдались А. М. Вавиловым и Л. М. Розентул (1980),
—русские. Самое позднее развитие отмечено в 26-летнем возрасте (Ikeda,
334
1967). С течением времени зона пигментации может расширяться, по ин-
тенсивности окраска ослабевает. Перерождение в меланому наблюдается
редко. Р. Иконописов и соавт. (1977) нашли 3 сообщения о развитии ме-
ланомы в зоне невуса Ота. О. М. Гурдус и О. О. Бочарникова (1983) по-
лагают, что из-за возможности развития меланомы нецелесообразно про-
изводить какие-либо травмирующие манипуляции.
Зрение обычно не страдает, но описаны случаи с врожденной катарак-
той (Gewiztzman и Rasmussen, 1976), глаукомой, пигментированным
ангиоматозом (Reinke et aL, 1974). Имеются наблюдения о сочетании
невусов Ota и Ito (пигментация боковой части шеи, верхней части груди
и плеча—Hidano et al., 1965).
Гистопатология. Характерно наличие дендритических меланоцитов
между пучками коллагеновых волокон.
Дифференциальный диагноз. С другими пигментными невусами.
Лечение. Не требуется. Необходимо диспансерное наблюдение у дер-
матолога и окулиста.
ЛИТЕРАТУРА
Вавилов А. М., Розентул Л. М. Два случая невуса Ота. Вести, дерматол. и вене-
рол., 1980, 4, 35—37.
Иконописов Р., Райчев Р., Киров С., Черноземски И. Пигментные опухоли. Меди-
цина и физкультура. Сйфия, 1977.
Рабен А. С., Титенер Г. Б. О так называемом невусе Ота. Вестн. дерматол. и ве-
нерол. 1976, 12, 54-*-56.
Фалил?ев Г. Ф., Бровкина А. Ф. Невус, БМЭ, 3-е изд. М., 1981, 16, 261—264.
Гурдус О. М., Б&арникова О. С. Синдром Ота. Вестн. дерматол. и венерол. 1983
10, 63—65.
Albertazi F., Strarii G. F., Laoarino A., Offino R. Annunziata M. Un caso di nevo
di Ota bilaterale.— Chron. dermatol., 1981, 12, 1, 47—52.
Lewitzman G. B., Rasmussen I. E. Nevus of Ota Zith ipsilateral congenital cataract. —
Arch, dermatof., 1976, 112, 9, 1284—1285.
Pfunder H. Eichmann A. Bifateraler Naevus Ota. Akt. Dermatof., 1984, 1 , 76—78.
Reinke R. T., Haber K-, Josselson A. Ota nevus, multip e hemangiomas and Takaya-
su arteritis.— Arch, dermato ., 1974, 110, 3, 447—450.
Steigleder G. K- Dermato ogie und.Venero ogie, Stuttgart, 1975.
Даудярис И. П., Даудярене В. И. Синдром Марторелла (гипертонические язвы
голени).— Вестн. дерматол. и венерол., 1978, 3, 54—58.
Измайлов Г. А., Морозов В. Г. Синдром Марторелла. Клинич. мед., 1984, 9, 118—
120.
Орликов Г. А. Синдром Марторелла.— Клинич. мед. 1976, 2, 134—135.
Скрипкин Ю- К- Случай синдрома Марторелла. Вестн. дерматол. и венерол., 1982,
3, 36—36.
Ryan Т., Wilkinson D.— In: Textbook of dermatology. Oxford-London—Edinburg—
Mefbourn, 1979, 3rd ed., v. 1, p. 993—1058.
335
НЕДЕРЖАНИЕ ПИГМЕНТА (Incontinentia pigmenti)
Синонимы: синдром Блоха—Сульцбергера, системный меланобластоз
кожи, дегенеративный меланоз кожи.
Первая демонстрация больного с клинической картиной недержания
пигмента принадлежит Garrod и Adamson (1905). Bloch в 1925 г. описал
клинику болезни и предложил название недержание пигмента.
Ассистент Блоха Sulzberger в 1928 г. подробно разработал случай
Блоха.
Этиология и патогенез. Блох и Сульцбергер указали, что заболевание
является врожденным, при нем нарушается функция пигментообразующих
клеток базального слоя эпидермиса и пигмент проникает в дерму. Заболе-
вание носит системный характер с вовлечением многих экто- и мезодер-
мальных тканевых структур (Barth et al., 1980). Причину развития болез-
ни одни авторы видят во внутриутробной аллергизации плода (Wodnian-
ski, 1955; Sprafke, 1963), другие—в нарушениях нейрогуморального, со-
судистого характера (Jadasshon и Franceschetti, 1933). С. И. Довжанский
и И. И. Павлова (1974) придают значение травмам и инфекции матери во
время беременности; Г. Б. Беленький и Г. М. Большакова (1960), Kita-
mura (1969) и др.—вирусной инфекции матери, приводящей к пренатальным
поражениям плода. Имеются указания на хромосомные аномалии у боль-
ных недержанием пигмента и их родственников (Vissian etal., 1978). Пред-
полагается наследование, сцепленное с полом, или аутосомно-доминант-
ное наследование с летальным эффектом мутантного гена для лиц муж-
ского пола (Суворова К- Н. 1977). Fellner и Weinstein (1978) наблюдали
мальчика с недержанием пигмента; они полагают, что в данном случае
речь шла о спонтанной мутации. Гипотеза об аутосомно-доминантной пе-
редаче заболевания по женской линии, по мнению Carney (1976), не может
объяснить всех опубликованных в литературе наблюдений.
Клиника. Выделяют три формы недержания пигмента: 1. Синдром
Bloch—Sulzberger; 2. Синдром Naegeli—Franceschetti—Jadasshon; 3. Синд-
ром Ito—бесцветное недержание пигмента.
Синдром Блоха—Сульцбергера проходит в своем развитии 4 стадии:
везикулезно-буллезную, папулезно-веррукозную, пигментную и стадию
склероза, атрофии и депигментации (Sprafke, 1963).
Кожные проявления развиваются большей частью в первые дни или
недели жизни, когда на туловище, конечностях появляется пятнистая
эритема, местами с шелушением, местами с пузырьковыми высыпаниями.
В дальнейшем—папулы, покрытые корочками. Часть элементов приобрета-
ет веррукозный характер. Везикулезные и папулезные элементы появля-
ются приступообразно—однократно или повторно. Иногда первоначаль-
ные высыпания напоминают поверхностную ангиому, крапивницу. Имеется
тенденция к полосовидному расположению сыпи. В дальнейшем воспали-
тельные явления разрешаются и появляется своеобразная пигментация в
виде брызг, завихрений. Они располагаются преимущественно на боковых
поверхностях туловища, в подмышечных областях, на плечах и бедрах,
редко на местах прежних высыпаний. С годами пигментации бледнеют,
336
Рис. 214. HeilepxcaHue
пае цента.
Рш 215. Недержание
пиг цента (та же бальная).
22 Клиническая дерматология
постепенно исчезают, иногда оставляя легкую атрофию. В ранней (воспа-
лительной) стадии болезни наблюдается эозинофилия крови. По данным
разных авторов, она колеблется от 2—4 до 50%. Наряду с кожными симп-
томами в 75—80% случаев регистрируются различные эктомезодермальные
дефекты—костной системы, зубов, ногтей, глаз, нервно-психической сфе-
ры, которые могут выявляться на более поздних этапах развития болезни.
Клиническая картина заболевания широко варьирует в зависимости от
присутствия тех или иных симптомов и их комбинации. Описываются от-
дельные случаи без неправильностей развития у лиц с хорошими, даже
недюжинными способностями (рис. 215, 216, 217, 218).
Недержание пигмента встречается почти исключительно у женщин.
При статистическом анализе 653 достоверных случаев, проведенном Car-
ney (1976), было 593 женщины, 16 мужчин (в 44 случаях пол не указан).
К этой форме недержания пигмента Н. М. Савиных и Б. Н. Кривошеев
(1978) относят буллезный кератогенный и пигментный дерматит или син-
дром Asboe— Hansen, рассматривая его как раннюю стадию заболевания.
II. Сетчатый пигментный дерматоз Naegeli или синдром Franceschet-
ti—Jadasshon. В возрасте около двух лет появляются сетевидные пигмен-
тации, которые комбинируются с ладонно-подошвенными кератозами, умень-
шением потоотделения, желтыми пятнами на зубах. Поражаются оба
полд. Наследование по доминантному типу. Заболевание относят к хро-
матофорным невусам.
III. Бесцветная форма недержания пигмента, или синдром Ito. Ito
в 1951 г. описал первый случай дисхромии у японской женщины, у которой
с детства существовали депигментированные пятна в виде полос, завитков;
брызг. В дальнейших публикациях японских авторов сообщалось о со-
четании описанных кожных изменений с различными аномалиями, соот-
ветствующими таковым при синдроме Блоха—Сульцбергера. Grosshans и со-
авт. (1971) наблюдали 4 случая синдрома Ито в одной семье (мать и три
дочери). Они же упоминают о синдроме Ито у матери и старшей дочери,
тогда как у младшей дочери был классический тип недержания пигмента.
Badanoiu и соавт. (1970) указывают, что не все описанные в литературе
наблюдения могут быть отнесены к вышеуказанным трем типам недержа-
ния пигмента—речь идет об отдельных случаях несемейного характера,
преимущественно у мужчин. Они их рассматривают как переходные формы.
Гистопатология. В ранней стадии болезни (везикулезной)—спонгиоз,
густо расположенные внутриэпидермальные пузырьки, содержащие эози-
нофилы. Остальной эпидермис пронизан эозинофилами, исходящими из
периваскулярных областей верхней части дермы, где имеется неспецйфи-
ческая воспалительная инфильтрация. В стадии гиперкератотических
изменений гистологическая картина может быть схожей с псевдоэпителиог
матозной гиперплазией. В стадии пигментации—часть клеток базального
слоя вакуолизирована; количество пигмента в базальных клетках нормаль-
но или уменьшено. В верхней части дермы—значительные отложения ме-
ланина, преимущественно внутри меланофоров. Ультраструктурные ис-
следования в стадии пигментации, проведенные Badanoiu и соавт. (1979),
338
выявили нарушения непрерывности базальной мембраны, создающие
проход для меланинсодержащих отростков цитоплазмы меланоцитов в на-
правлении к дерме, где они захватываются меланофагами.
Дифференциальный диагноз. В ранней стадии может представлять
трудности. Следует дифференцировать с герпетиформным дерматитом Дю-
ринга, врожденным эпидермолизом.
Лечение. Симптоматическое.
ЛИТЕРАТУРА
Белостоцкая Е. С. и соавт. О «недержании пигмента» у сестер. — Вестн. Дерматол.
и венерол., 1983, 10, 57—58.
Довжанский С. И., Павлова И. И. Недержание пигмента (синдром Bloch—Sulzber-
ger).— Вестн. дерматол. и венерол., 1974, 5, 65—68.
Савиных Н. М., Кривошеев Б. Н. Синдром Asboe—Hansen как ранняя стадия не-
держания пигмента.— Вестн. Дерматол. и венерол., 1978, 3, 47—49.
Суворова К- И., Антоньев А. А. Наследственные дерматозы.— М.: Медицина,
1977.— с. 57—60.
Badanoiu A., Vulcan V., Pais V. Bemerkungen zur Intermediaform der Incontinen-
tia pigmenti.— Derm. Mschr., 1976, 162, 1, 17—23.
Fellner M., Weinstein L. H. Incontinentia pigmenti in a boy. Intern. J. Dermatol.,
1978, 17, 1, 67—68.
VissianL., Emeriti., Leoy A., Vaillaudl.—C. Incontinentia pigmenti. Etude chro-
mosomique d’une famille.— Ann. Dermatol. Venereol., 1978, 105, 2,-119—121.
НЕКРОБИОЗ ЛИПОИДНЫЙ (necrobiosis lipoidica)
Синонимы: липоидный диабетический некробиоз (Urbach); атрофиче-
ский пятнистый липоидный диабетический дерматит (Oppenheim); псевдо-
склеродермиформный симметричный хронический гранулематоз (Gottron);
хронический прогрессирующий дисковидный гранулематоз (Miescher et
Leder).
Представляет собой сравнительно редкое страдание. Brzowski (1975)
сообщил, что липоидный некробиоз кожи установлен у 4% больных сахар-
ной болезнью.
Этиология и патогенез. Большинство имеющихся в настоящее время
данных (Каламкарян А. А. и Сыч Л. И., 1963; Владимиров В. В., 1971;
Машкиллейсон Н. А- и Зайцева С. Ю., 1981; Поляков В. А., 1983; Ка-
ламкарян А. А. и Абрамова Е. А., 1984; Шапошников О. К- и Хазизов
Е. И. , 1985; Lucaryk et al., 1978; Ullman и Dahl, 1977; Kobayasi et al.,
1974; Scheider et al., 1980, и др.) свидетельствуют о том, что диабетиче-
ская ангиопатия лежит в основе липоидного некробиоза; причиной некро-
биоза коллагена в очагах поражения являются сосудистые поражения,
вызванные возможно не только циркулирующими в крови токсинами при
сахарном диабете (Urbach,. 1932), но и различными нейроэндокринными
нарушениями, иммунологическими сдвигами (наличие в сыворотке больных
циркулирующих иммунных комплексов) и др. Дезорганизация соедин'и-
22* ЗЗУ
тельной ткани и липоидные отложения в коже, по-видимому, носят вторич-
ный характер и обычно происходят в местах дистрофии или некробиоза
коллагена. Ведущую роль в патогенезе липоидного некробиоза играют
поражения сосудистой стенки, причем в первую очередь микроциркуляр-
ного русла. Связь липоидного некробиоза кожи с аллергией, инфекцией,
гипертонической болезнью, длительным куреньем не доказана. У части
больных возникновению липоидного некробиоза предшествовали травмы
(ушибы, укусы насекомых, царапины и др.).
Клиника. Заболевание может возникнуть в любом возрасте, чаще в
20—40 лёт, несколько реже в старческом и детском. Большинство авторов
подчеркивает более частое заболевание женщин (70—75%) по сравнению
с мужчинами (25—30%). У 18—20% больных кожные изменения появля-
ются задолго (1—ГО лет) до развития сахарного диабета. У 25—32% они
развиваются одновременно с ним, у 55—60% больных Сахарный диабет
предшествует поражению кожи. Нельзя отметить прямой зависимости
между тяжестью диабета, выраженностью и прогрессированием липоидного
некробиоза.
Мы выделяем следующие основные формы липоидного некробиоза:
склеродермоподобная, поверхностно-бляшечная, типа кольцевидной гра-
нулемы и узелковая. Склеродермоподобная форма встречается наиболее
часто. Она характеризуется наличием на коже единичных, реже множе-
ственных бляшек. При склеродермоподобной форме липоидного некробио-
за можно выделить 3 стадии развития. Первая стадия характеризуется
появлением розовато-красных узелков конусовидной, чаще полусфериче-
ской формы с гладкой поверхностью и перламутровым блеском. Вторая
стадия характеризуется образованием инфильтрированных бляшек бурова-
то-красного цвета с резко очерченными границами, размерами от 10-копееч-
ной монеты до детской ладони. Весь очаг поражения возвышается над уров-
нем окружающей кожи; уже в этой фазе узелки, расположенные по краю
очага, приобретают фиолетовые оттенки и возвышаются над уровнем его
центральной части. Наконец, после длительного существования (от не-
скольких месяцев до 2—3 лет), заболевание переходит в третью заключи-
тельную стадию, которая характеризуется образованием округлых бляшек,
чаще неправильных очертаний с блестящей поверхностью, с запавшим
желтовато-буроватым центром и слегка возвышающимся краем фиолетово-
красного цвета. При пальпации очагов поражения определяется склеро-
дермоподобное уплотнение, более выраженное в центральной части. Про-
цесс обычно заканчивается образованием рубцевидной атрофии. Весьма
часто на поверхности пораженных участков отмечаются многочисленные
телеангиэктазии. Таким образом, сформировавшиеся очаги поражения
состоят из двух зон: в центре—кожа цвета слоновой кости блестящая, глад-
кая, ясно выявляется рубцевидная атрофия, поверхность плотная на ощупь,
напоминающая бляшечную склеродермию. По периферии отмечается слег-
ка возвышающийся узкий валик розовато-синюшного цвета, состоящий из
слившихся мелких блестящих узелков; волосяные фолликулы отсутствуют
(рис. 219, 220, 221).
340
некробиоз (оиабетический).
Рис. 219. Липоидный
некробиоз (диабетический).
Рис. 220. Липоидный
некробидз (недиабети-
ческий, напоминающий
кольцевидную гранулему).
У подавляющего большинства больных очаги поражения множествен-
ные, локализуются на передне-боковых поверхностях голеней и голено-
стопных суставов; значительно реже на кистях, туловище й волосистой
части головы. Субъективные явления обычно отсутствуют, лишь при изъ-
язвлении появляется болезненность. Примерно в 1/4 всех случаев липо-
идного некробиоза очаги поражения сопровождаются изъязвлениями.
При этом язвы ча!це встречаются у больных диабетом, чем без диабета.
Язвы поверхностные, как правило, неправильных очертаний, края нх за-
341
частую мягкие, слегка подрыты. Отделяемое язв серозно-геморрагическое,
при Подсыхании образуются темно-коричневые корки.
Липоидный некробиоз типа кольцевидной гранулемы характеризуется
наличием на различных участках кожного покрова (кисти, стопы и т. д.)
очагов поражений округлых очертаний, диаметром в 3—4 см и более.
Очаги поражений по периферии окружены валиком сииюшно-красного
или розовато-синюшного цвета с желтоватым оттенком, шириной 2—5 мм,
состоящим из отдельных папулезных элементов. В центре очага кожа на
вид слегка атрофична или не изменена.
Поверхностно-бляшечная разновидность липоидного некробиоза ха-
рактеризуется образованием на предплечьях и плечах, тыле кистей, жи-
воте, груди, спине очагов поражения округлых или неправильных фестон-
чатых очертаний размерами от небольшой монеты до ладони взрослого и
больше, розовато-желтоватого цвета, окаймленных фиолетово-краснова-
тым или сиреневато-розовым ободком шириной до 5—10 мм. Края бляшек
резко очерчены, поверхность их гладкая, в центре очагов отмечается не-
большое западение, уплотнение в основании бляшек отсутствует.
Прогноз для жизни благоприятный. В отношении выздоровления сом-
нительный.
Гистопатология. Изменения сосудов/некробиоз коллагена, отложение
липоидов, инфильтрат, состоящий из гистиоцитов, лимфоцитов, фибробла-
стов, эпителиоидных и гигантских клеток чужеродных тел. В кровеносных
сосудах обнаруживаются характерные признаки диабетической микро-
ангиопатии (фиброз и гиалинов стенок, пролиферация эндотелия и тром-
боз малых сосудов). Со стороны эпидермиса—гиперкератоз, изредка атро-
фия и вакуольная дистрофия базального слоя. Установлено (Miiller,
1966, и др.), что у больных без диабета преобладает гранулематозный тип,
в то время как у больных диабетом—некробиотический тип. При электрон-
но-микроскопическом исследовании эпидермиса и подлежащих слоев дер-
мы обнаружены изменения ультраструктуры клеток, которые можно рас-
ценивать как дистрофические и атрофические (Kobayasi et al., 1974; Lu-
karyk et al., 1978). По мнению многих авторов (Ormsby и Montgomery,
1955; Lever и Schaumburg-Lever, 1975; Nosier et al., 1974; Wells и Smft,
1979; Mobacken et al., 1970; Gotz, 1983, и др.) хронический прогрессирую-
щий дискообразный гранулематоз Мишера, по-видимому, представляет
собой липоидный некробиоз без липоидных отложений. При кольцевидной
гранулеме значительно менее выражены сосудистые изменения, отсутствуют
липоидные отложения, почти не встречаются гигантские клетки.
Лечение. Диета бедная углеводами и жирами, внутриочаговые инъек-
ции кортикостероидов и инсулина, витамин Е, гепарин, липокаин, амид
липоевой кислоты, ангиотрофин, метионин, компламин, ультразвук, лучи
лазера, длительные втирания кортикостероидных мазей и др.
342
ЛИТЕРАТУРА
Каламкарян А. А.. Абрамов А. А. Клинические варианты липоидного некробиоза-
В кн. Современные методы диагностики и терапии дерматозов. Сб. научных трудов 4 глав-
ного упр. МЗСССР. М., 1984, 41—44.
Поляков В. А. Мелкоузелковая форма липоидного некробиоза.—Вестн. дерматол. и
венерол., 1983, 6, 62—63.
Шапошников О. К., Хазизов И. Е. Современные аспекты патогенеза и терапии кольце-
видной гранулемы и липоидного некробиоза.—Вестн. дерматол. и венерол., 1985, 5, 4—8.
Kobayasi Т., Danielsen L., Asbpe-Hansen G. Uftratructure of necrobiosis lipoidica
diabeticorum.— Acta Dermatovener., 1974, 54, 6, 427—431.
Mackey J. Necrobiosis lipoidica diabeticorum involving calp and face.— Brit. J. Der-
matof., 1975, 93, 6, 729—730.
Schneider H., Thormann Th. Generalisierte diabetische Derma ngiopathie.— Z. Ha-
utkr., 1980, 55, 7, 441—447.
Ullman S.r Dahl M. Necrobiesis lipoidica.— Arch. Dermatol., 1977, 113, 1671—1673.
Gotz H. Uber die Necrobiosis lipoidica (diabeticorum) sowie zur Frage der Zugehorig-
keit der Granulomatosis disciformis chrohic et progressive Mischer. —Hautarzt, 1983, 34, 7,
322—325.
НЕКРОЛИЗ ТОКСИЧЕСКИЙ ЭПИДЕРМАЛЬНЫЙ (toxic epidermal
necrolysis).
Синонимы: острый токсико-аллергический ожоговидный некроэпидер-
молиз, острый эпидермальный некролиз, синдром Лайелла, токсический
эпидермальный некролиз.
В 1954 г. английский дерматолог Lyell описал заболевание под назва-
нием «toxic epidermal necrolysis.» Синдром Лайелла—остро возникающее
лихорадочное заболевание, сопровождающееся генерализованными бул-
лезными высыпаниями на коже и слизистых оболочках полости рта, глаз
и половых органов.
Этиология и патогенез. Обсуждаются следующие гипотезы—инфекцион-
ная (стафилококковая, вирусная), токсико-аллергическая, в том числе
лекарственная (после приема пенициллина, хлортетрациклина, аспирина,
пирамидона, сульфаниламидов, аминазина, люминала, кодеина и многих
других лекарственных средств). Поиски специфического вируса были без-
результатны. Собственные клинические наблюдения, а также анализ мно-
гочисленных литературных данных позволяют нам рассматривать эпи-
дермальный некролиз как своеобразный токсико-аллергический синдром,
являющийся выражением гиперергической реакции организма на различ-
ные инфекционные и неинфекцнонные агенты (медикаменты, инфекции,
нервно-эндокринные и нервно-психические расстройства, характер пита-
ния и т. д.). Примерно 10% человеческой популяции несет генетическую
предрасположенность к аллергической сенсибилизации организма (Кува-
ева И. Б. и Максимова Н. Г., 1978). Многие работы показывают, что судь-
ба лекарств в организме в значительной мере определяется генетическими
свойствами организма и его изменениями в процессе болезни. Характерно
343
возникновение извращенной реакции на определенные лекарственные сред-
ства в связи с наличием биохимических (ферментативных) продуктов (Боч-
ков Н. П., 1976; Вартанян М. Е., 1978). Сравнение клинических и гисто-
морфологических данных при синдроме Стивенса—Джонсона и синдроме
Лайелла свидетельствует о том, что качественно это один и тот же процесс,
который, однако, у разных больных имеет различную количественную сте-
пень проявления в силу разной реактивности (Пашков Б. М., 1966; Гра-
чева Н., 1978; Самцов В. И. и Подвысоцкая И. И., 1979, 1980; Каламка-
рян А. А. и Самсонов В. А., 1980; Бухарович М. Н., 1980; Ohlensche-
ger, 1966; Samuels, 1967, и др.).
Клиника. Болеют большей частью молодые люди и дети, чаще в весен-
нее и осеннее время. Не во всех случаях можно отметить связь заболевания
с предшествующим приемом лекарств. Наиболее характерным и домини-
рующим в клинической картине синдрома Лайелла, как и при синдро-
ме Стивенса—Дженсона, является поражение кожи и слизистых оболочек
на фоне тяжелого общего состояния. На коже туловища, разгибательных
поверхностей верхних и нижних конечностей, а также на лице и в области
половых органов внезапно появляются распространенные, эритематозно-
отечные пятна багрово-красного цвета, которые, сливаясь, образуют круп-
ные бляшки. Исключительно быстрое развитие воспалительных явлений,
буквально в течение нескольких часов, приводит к образованию плоских,
дряблых пузырей с фестончатыми очертаниями, величиной до ладони взрос-
лого человека и больше, наполненных серозной и серозно-кровянистой
жидкостью. Покрышка пузырей морщинистая, вялая, легко отделяется,
обнажая обширные мокнущие, резко болезненные эрозивные поверхности
с обвисшими кусками эпидермиса по краям. Симптом Никольского резко
положительный: при малейшем прикосновении эпидермис легко отделя-
ется на обширном протяжении («симптом простыни»). На ладонях он отде-
ляется обширными пластинами, наподобие перчатки. На тыльных поверх-
ностях кистей и стоп, на разгибательных поверхностях предплечий и
голеней имеются пятнистые высыпания, напоминающие элементы эксу-
дативной полиморфной эритемы. По клинической картине поражения на
спине, животе, ягодицах, бедрах напоминают кожу при ожоге II степени.
На половом чЛене, мошонке, малых и больших половых губах, в окологе-
нитальной области и промежности локализуются эритематозно-отечные
пятна с синюшным оттенком и крупные дряблые пузыри, после вскрытия
которых образуются эрозии мясо-красного цвета. Поражения слизистых
оболочек рта, носа характеризуются гиперемией, отеком с последующим
развитием крупных пузырей, которые, быстро вскрываясь, образуют край-
не болезненные обширные эрозии и изъязвления с остатками покрышек
пузырей. Губы отекают, покрываются кровянистыми и грязными корками
и глубокими трещинами. Из-за резкой болезненности затруднен прием
даже жидкой пищи. В отдельных случаях воспалительный ппоцесс распро-
страняется на слизистую оболочку глотки, желудочно-кишечного тракта,
гортани, трахеи и бронхов, вызывает тяжелые приступы одышки. Наблю-
дается застойно-синюшная гиперемия и отек кожи, конъюктивы йек с об-
344
разованием пузырей и эрозий, покрытых серозно-кровянистыми корками.
Заболевание протекает тяжело, с высокой температурой, часто сопро-
вождается дистрофическими изменениями внутренних органов. Во мно-
гих случаях выявляется умеренный лейкоцитоз со сдвигом влево. Посевы
крови и содержимое невскрывшихся пузырей, как правило, оказываются
стерильными. Примерно 25—30% больных относительно быстро погибают.
У взрослых поражение больше 70—80% поверхности кожи является
опасным для жизни.
Гистопатология. Некроз поверхностных слоев эпидермиса. Клетки
мальпигиева слоя отечные, межклеточная и эпидермо-дермальная связь
нарушена (отслаивание эпидермиса от дермы), в результате этого на коже
и слизистых оболочках образуются пузыри, которые располагаются внутри
и подэпителиально.
Дифференциальный диагноз. Следует проводить с острым пемфигусом,
эксфолиативным дерматитом новорожденных (болезнь Риттера).
Лечение. Раннее применение больших суточных доз кортикостероидов
(50—100 мг и более преднизолона в сутки), антибиотики, витамины. Дето-
ксицирующие мероприятия—введение плазмы, кровезаменителей, жидко-
стей, электролитов; стерильное белье.
ЛИТЕРАТУРА
Браиловский А. Я- По поводу статьи А. А. Каламкаряна и В. А. Самсонова «О
многоформной эксу дативной эритеме, синдроме Стивенса—Джонсона, токсическом эпи-
дермальном некролизе (синдроме Лайелла) и их взаимоотношении.»— Вести, дерматол.
и венерол., 1980, 11, 29—31.
Бухарович М. Н. Некоторые аспекты взаимоотношения многоформной эксудатнв-
ной эритемы, синдромов Стивенса—Джонсона и Лайелла.— Веста, дерматол. и венерол.,
1980, 9, 33—34.
Каламкарян А. А., Самсонов В. А. О многоформной эксудативной эритеме, синд-
роме Стивенса—Джонсона, токсическом эпидермальном некролизе (синдроме Лайелла) и
их взаимоотношении.— Веста, дерматол. венерол., 1980, 3, 36—42.
Куваева И. Б., Максимова Н. Г- Аллергические реакции на пищевые антигены и
нх связь с функциональным состоянием желудочно-кишечного тракта и иммунной системы
организма.— Вести. АМН СССР, 1978, 3, 82—90.
Самцов В. И., Подвысоцкиая И. И., Лисовская Н. Д. Замечания по поводу острого
эпидермального некролиза.— Веста. Дерматол. и венерол., 1980, 11, 31—34.
Lyell A. Texic epidermal necrolysis (the scalded skin syndrome): a reappraisal.— Brit.
J. Dermatol., 1979. 100, 1, 69—86.
НЕКРОТИЧЕСКИЕ УГРИ (acne necrotica).
Синонимы: acne varioliformis; acne necrotica miliaris.
Некротические акне Бека (Boeck) соответствуют вариолиформным акне
Гебры (НеЬга).
Форма acne necrotica miliaris, описанная R. Sabouraud в 1928 г., весьма
дискутабельна и в настоящее время связывается с себорейным дерматитом.
По мнению некоторых авторов (Lane, 1933; Ormsby, 1934; Montgomery М.,
345
1937; Dillaha et al., 1983), милиарные некротические акне являются малой
(абортивной) формой некротических акне Гебры.
Этиология, патогенез. Акне-некротика редкое заболевание, в настоящее
время этот диагноз ставится исключительно редко, хотя его раньше широко
ставили.
Этиология этой формы акне неизвестна. Можно думать о значении на-
рушений желудочно-кишечного тракта, эндокринных органов, нервной си-
стемы. Бактериальная инфекция, действующая снаружи, может иметь пре-
имущественное значение. Возможно, что заболевание вызывается золотистым
стафилококком с высокой вирулентностью. Из пустул выделены золотистые
стафилококки (Tach'ibanna et al., 1964; Ormsby, 1934, и др.). Сложность
патогенеза пиодермических заболеваний заставляет нас не переоценивать
роли чисто этиологического агента, т. е. пиококков. Gertler (1972) рассматри-
вает это заболевание как одну из разновидностей угрей.
Клиника. Заболевание наблюдается у мужчин и женщин зрелого возра-
ста. Наиболее поражаемый возраст от 15 до 40 лет, но в отдельных случаях
высыпания возникают даже после 50—60 лет. Клиническая картина характе-
ризуется высыпанием розовых или багрово-красных фолликулярных и не-
фолликулярных узелков величиной от булавочной головки до чечевицы, в
центре которых образуется похожий (симулируя пузырьки-пустулы) на пу-
стулу очаг некроза, постепенно подсыхающий в кровянисто-черную корку.
При этом комедоны большей частью отсутствуют. После отхождения корки
остается вдавленный, не исчезающий вариолиформный, вначале розоватый,
затем белый рубец. Таким больным обычно ставится диагноз папулонекроти-
ческого туберкулеза кожи, либо третичного сифилиса.
Анализ крови, мочи, биохимические исследования без отклонений. Ко-
личество высыпаний весьма различно—от единичных до очень большого числа.
Характерно для клинической картины дерматоза длительное приступообраз-
ное, рецидивирующее течение заболевания. Новые высыпания возникают как
бы толчками, приступами, течение процесса хроническое.
Высыпания локализуются на лбу (на границе роста волос), на висках,
в ушных раковинах, иногда на носу и в носогубных складках и лишь очень
редко занимает туловище (середину спины и груди).
У больной 49 лет, находившейся под нашим наблюдением, высыпа, ‘я
имелись на лбу, висках, ушных раковинах и на носу.
У отдельных больных заболевание сопровождается непостоянным сл.
бым зудом.
Гистологическая картина соответствует сухому некрозу в зоне волосяно-
го фолликула и тромбозу сосудов в воспалительной периферической зоне.
Инфильтрат перифокальный состоит из нейтрофилов и тучных клеток. Обра-
зование некроза обусловлено не только гипервирулентностью бактерий, но
и состоянием кожной сенсибилизации к стафилококкам.
Дифференциальный диагноз следует проводить с папулонекротическим
туберкулезом кожи и бугорковым сифилисом.
Почти исключительная локализация некротических угрей на волосистой
части головы и на лбу, типичные вариолиформные рубцы, а также более жи-
346
Рис. 2216. Acne necrotica. Оспенновидные
рубцы на ушных раковинах.
Рис. 221\ Acne necrotica. Типичные
вариолиформные рубцы на лбу.
Рис. 221в. Acne necrotica. Типичные
оспенновидные рубцы на носу.
ные. Рубцы при некротическом
вые воспалительные явления по окру-
жности высыпаний позволяют уже
клинически исключить папулонекро-
тический туберкулез. Локализуется
папулонекротический туберкулез пре-
имущественно на конечностях; чаще
на разгибательных поверхностях. Вто-
рое место занимает область ягодиц;
гораздо реже бывают поражения во-
лосистой части головы. Гистологи-
чески здесь всегда обнаруживается
классическое бугорковое строение
инфильтрата. В отличие от некроти-
ческих акне сифилитические бугорки
всегда плотны. Остающиеся после
бугоркового сифилиса рубцы имеют
фестончатые очертания, они неров-
акне гораздо более заметны, чем
рубцы бугоркового сифилиса, которые постепенно в значительной степени
сглаживаются. Кроме того, правильному диагнозу могут помочь и другие
моменты: наличие других признаков сифилиса, результаты серологических
исследований и др.
Лечение. Заболевание очень трудно поддается лечению. Лечение, как
правило, должно быть комплексным: антибиотиками (тетрациклин,
вибромицин, террамицин, стрептомицин, пенициллин, линкомицин, фузидин
и др.) раздельно или в комплексе с кортикостероидными гормонами (предни-
золон 20—30 мг в сутки). При некротическом акне антибиотикотерапия яв-
ляется одной из составных частей сложного комплексного лечения. Исполь-
347
зуют инъекции стафилококковой вакцины или стафилококкового анатокси-
на в комбинации с антибиотиками, сульфаниламидами, аутогемотерапией и
др. Положительное влияние оказывают антисеборейный витамин биотин (Ras-
set., 1959). Knigge (1977) рекомендует введение пенициллина в очаг пораже-
ния. Возможно положительное действие пресоцила внутрь по 2 таблетки 3 раза
в день в течение 3—4 недель. Назначаются витамины А, С, витамины ком-
плекса В и др. При этой форме пиодермии целесообразно наружно при-
менять мази с антибиотиками.
ЛИТЕРАТУРА
Degos R. Dermatologie.—Paris, 1981.—Vol. I.—P.492—493.
Gertler W. Systematische Dermatologie.—1972.—S. 1191—1192.
ffnigge H. Zur Therapie der Acne necrotica. Zeitschrift Haut Geschlechtskr.—1957.—
Bd. 23, H.12.—S.357—358.
Montgomery H. Acne necrotica miliaris of the scalp. Arch. Dermatol. Syph.—1937.—
Vol. 36, N 1.—P.40—44.
Muller H. XBeitrag zur Therapie der Akne nekroticans (Brock). Dermatol. Wschr.—
1964.—Bd. 149, H.19.—P.495—500.
Ormsby 0. Practical treatise on diseases of the skin for the use of students ad pra-
ctitioners. —4 ed.—Philadelphia, 1934.—P. 1210—1211.
-Stritzler C., Friedman R., Looeman A. Acne necrotica. Arch. Dermatol. Syph.—
1951.— Vol.64, N4.—P.464—469.
Tachibana K- et. al. A study of adne necrotica miliaris. Acta Dermatologica Kyoto
(Jap..).—1964.— Vol.59, N 4.—P.253—259.
ОПУХОЛЬ ГЛОМУСНАЯ (tumor glomus).
Синонимы: болезнь Barre—Masson, ангионейромиома, артериальная
ангиомионейрома, гломус-ангиома.
Впервые о гломусной опухоли доложил на Страссбургском конгрессе
психиатров е 1920 году французский невропатолог Barre, а в 1922 году
он впервые описал клиническую симптоматику опухоли. Первое опи-
сание гломусной опухоли в отечественной литературе принадлежит Г. И.
Маркелову (1934). Гистологии^ опухоли впервые изучил Masson (1924).
Единой номенклатуры и классификации гломусных опухолей нет. В оте-
чественной литературе их обычно включают в группу сосудистых новообра-
зований (Чернеховская Н. Е. и соавт., 1978).
Этиология и патогенез. Большинство авторов (Самотокин Б. А., 1952;
Апатенко А. К- и Турусов В. С., 1962; Masson, 1924; Ekestrom, 1950;
Horton, 1955; Schumacher, 1955 и др.) считают, что опухоли Барре—Массона
развиваются из гломусов (артериовенозных анастомозов) кожи, при этом
подчеркивается их происхождение из функциональной части анастомоза—
канала Суке—Гойера (Sucquet—Hojer), выстланного эндотелием и окру-
женного эпителиоидными (гломусными) клетками. Эти клетки рассматри-
ваются как видоизмененные гладкомышечные клетки, способные сокра-
щаться (Weidman и Wise, 1937) или набухать, изменяя просвет анастомо-
348
зов. Гломусы богато иннервированы. Некоторые авторы указывают на
связь гломусной опухоли с травмой. Однако случаи с множественными
опухолями и с разнообразной локализацией говорят против этого.
Клиника. Различают одиночные и множественные гломусные
опухоли. Одиночный гломус-тумор представляет , собой небольшое
округлое опухолевидное образование (узелок), локализующееся пре-
имущественно в области ногтевого ложа или концевых фаланг паль-
цев кистей. Он пурпурного или синевато-красного цвета, мягкой
консистенции, глубоко расположен в дерме. Величина опухолей на
пальцах колеблется от 0,1X0,2 до 0,5x0,6 см. Опухоли внутренних
органов (желудка, легких, печени, кишечника) достигают размеров
10x15 см (Черкес В. Л. и Белоус Т. А., 1973). Если опухоль лежит
под ногтем, она представляется в виде округлого пятна диаметром в не-
сколько миллиметров, красновато-синего цвета. Типичной локализацией
гломусных опухолей раньше считали ногтевое ложе (около 90% гломус-
ангиом располагаются на пальцах). Но так Как артерио-венозные анасто-
мозы гломусного типа найдены почти во всех органах и тканях (Долго-
Сабуров Б. А.), то эти опухоли могут возникнуть в коже самых различных
областей тела, а также в желудочно-кишечном тракте, легких, печени и
других органах. Опухоли характеризуются резкими, часто приступообраз-
ными болями, особенно усиливающимися при различных раздражениях
(механических, температурных и др.). Нередко приступы болей сопровож-
даются вегетативными реакциями: ощущением страха и жара, покрасне-
нием лица, шеи, «замиранием» сердца, потливостью, стенокардией и др.
Множественные гломусные опухоли, описанные многими авторами
(Цимеринов А. А. и Литвиненко Д. И., 1938; Апатенко А. К- и Турусов
В. О., 1962; Альбицкий Б. А. и Журавлева Н. И., 1*975; Косачев Н. Д.
и РитнюК Р. И., 1975; Johnson, 1976 и др.), клинически и гистологически
отличаются от солитарных. Они исключительно редко располагаются под
ногтем, чаще на других участках кожного покрова, часто безболезненны,
появляются обычно в детском возрасте. Нередко выявляется их наслед-
ственный характер с аутосомно-доминантным типом наследования (Мор-
довцев В. Н. и Сыч Л. И., 1974; Einzinger и Smith, 1976, и др.).
Течение заболевания хрс- шческое, опухоль растет медленно, как пра-
вило, не метастазирует, не подвергается злокачественному перерождению
(Kohaut и Stout, 1962).
Лечение. Оперативное (хирургическое). Гломус-тумор нечувствителен
к лучевой терапии, а после электрокоагуляции часто рецидивирует. В опу-
холи постоянно выявляется (i ри импрегнации серебром) богатая сеть нерв-
ных волокон. Kohout и Stout (1961) на основании культуры тканей и мор-
фологии полагают, что гломусные клетки—это перициты. Однако недавние
электронно-микроскопические исследования (Marad et al., 1968; Venkata-
chalm и Greally, 1969; Tamowski и Hashimoto, 1969 и др.) показали бли-
зость этих клеток к гладкомышечным клеткам. В зависимости от преобла-
дания тех или иных составных частей (артерий, нервных, мышечных эле-
ментов) различают три формы гломусных опухолей: эпителиоидную, невро-
349
матозную, ангиоматозную (Masson). Множественные опухоли по гистоло-
гическому строению напоминают кавернозную ангиому. Они содержат
мало эпителиоидных клеток. Эти опухоли расценивают как порок развития.
Дифференциальный диагноз. Следует проводить с ангиомой, мелано-
мой, невриномой, голубым невусом, дерматофибромой, лейомиомой.
Гистопатология. Патоморфологически гломусная опухоль воспроиз-
водит структурные элементы артерио-венозных анастомозов. Она состоит
из большого количества беспорядочно переплетающихся между собой со-
судов, окруженных эпителиоидными (гломусными) клетками.
ЛИТЕРАТУРА
Альбицкий Б. А., Журавлев Н. И. Опухоль Барре—Массона.— В кн.: Некоторые
вопросы клиническо-эк'спериментальной онкологии. Томск, 1975, с. 83—86.
Косачев И. Д., Ритнюк Р. И. Клубочковые опухоли артерио-венозных анастомо-
зов.— Вестн. хирургии, 1975, 114, 6, 73—76.
Мордовцев В. И., Сыч Л. И. Случай семейных множественных гломусных опухо-
лей.— Вестн. Дерматол. и венерол., 1974, 17, 82—85.
Потекаев Н. С., Иванов О. Л., Сыч Л. И. Одиночная гломусная опухоль кожи.—
Вестн. дерматол.-и венерол., 1979, 10, 48—51.
Чернеховская Н. Е., Чечулина А. И., Анучкова Э. Л. Лапароскопическая диагно-
стика гломусных опухолей.— Вести, хирургии, 1978, 6, 90—92.
Carapeto F., Garcia A., Winkelmann R. Acral arteriovenous tumor.— Acta Dermato-
vener., 1977, 57, 2, 155—158.
Rook A., Winkinson D.. Ebling F. In: Textbook of dermatology. Third ed. Oxford,
1979, v. 11, p. 2220—2221.
ПАПИЛЛОМАТОЗ КОЖИ КАРЦИНОИДНЫЙ ГОТТРОНА (papillo-
matosis cutis carcinoides Gottron).
В 1932—1936 гг. Gottron описал Двух больных, у которых имелись
опухолевидные и папилломатозные разрастания кожи голеней и подошвы,
гистологически характеризующиеся инвазивным ростом эпидермиса, ги-
перкератозом, но не обладающие другими признаками злокачественности.
Автор выделил это заболевание под названием папилломатоз кожи. Nico-
lowski и Eisenlohr (1960), учитывая клиническое и гистологическое сходство
очагов поражения при папилломатозе кожи с плоскоклеточным раком дали
ему название карциноидный папилломатоз кожи Готтрона. К 1980 г. в
мировой литературе описано около 40 наблюдений (Христин Л- И. и соавт.,
1963; Фомин К- Ф. и соавт., 1971; Беренбейн Б. А., 1980; Каламкарян
А. А., 1980; Nicolowsky и Eisenborn, 1950; Mischer, 1950; Frank, 1959;
Wodniansky, 1960; Schimpf и Seller, 1963; Fischer^ 1975; Riboldi и Pozzo,
1976; Quednow et al., 1983, и др.). Civat'te (1967) и другие авторы относят
карциноидный папилломатоз кожи к хронической вегетирующей пиодермии
Азу a (Azua, 1904).
Этиология и патогенез. Nicolowsky и Eisenlohr (1950), Woldniansky
(1960), Dorin (1961), Б. А. Беренбейн и соавт. (1983) и др. придают важное
значение в развитии.заболевания, предшествующему воспалению. Schimpf
350
Рис. 222. Карциноидный
папилломатоз кожи Готтрона.
и Seller (1975) связывают его развитие
с нарушением кровообращения в облас-
ти нижних конечностей.
- Клиника. Мужчины болеют немно-
го чаще, чем женщины. Возраст боль-
ных колеблется от 40 до 80 и более лет.
Очаги поражения асимметричны и мо-
гут возникнуть на любом участке ко-
жи, но чаще локализуются на конеч-
ностях, особенно нижних (иа голенях,
стопах), несколько реже- -на туловище,
половых органах и на слизистой обо-
лочке полости рта. Заболевание разви-
вается на фоне какого-либо длительно
существующего дерматоза (красный
плоский лишай, экзема, псориаз, ва-
рикозный симптомокомплекс, туберку-
лез кожи и др.), на местах бывших
травм и на рубцах, где постепенно
появляются папилломатозные бородав-
чатые разрастания (рис. 222) Эти
разрастания чаще образуют одиноч-
ную опухоль или уплощенную бляшку
плотной консистенции размером с детскую ладонь и больше, рез-
ко отграниченную от здоровой кожи и выступающую над ее уровнем
на 1—2 см. Папилломатозные и веррукозные разрастания придают очагам
сходство с цветной капустой. В углублениях между сосочковыми разра-
станиями скапливаются чешуйки и корки, по удалении которых обнаружи-
вается мацерация и изъязвления. При сдавливании очага с боков из глу-
бины выделяется сливкообразный гной в виде небольших капель. На по-
верхности местами имеются кровянисто-гнойные корки.
Течение длительное, доброкачественное. Однако имеется сообщение
Schuppener и Steinke (1960) о злокачественном перерождении очага папил-
ломатоза кожи у больного, ежедневно применявшего в течение 9 месяцев
примочки из раствора азотнокислого серебра (химическое раздражение)
Гистопатология. Резко выраженные разрастания всех слоев эпидермиса
с образованием причудливых эпителиальных тяжей, глубоко проникающих
в дерму. Атипии клеток эпидермиса нет, базальная мембрана сохранена.
В сосочковом слое дермы массивный инфильтрат, состоящий из лимфоцитов
с примесью эпителиоидных клеток, гистиоцитов и фибробластов.
Дифференциальный диагноз. Следует проводить с хронической вегети-
рующей пиодермией (псевдоэпителиомой Azua), плоскоклеточным раком,
глубокими микозами, бородавчатым туберкулезом кожи.
Лечение. Рентгенотерапия, цитостатики, 0,5% колхаминовая мазь,
20—50% проспидиновая мазь.
351
ЛИТЕРАТУРА
Беренбейн Б. А. Псевдорак кожи.— М.: Медицина, 1980-
Quednow Ch., Motsch Н., Jahuke G. Zur Froge malignen Entartung der Papilloma-
tosis lutis carcinoicles Gottron bei vorbestehendem Lichen sclerosus et atrophicans genitalis.
Dermatol. Monatssche, 1983, 169, 3, 179—184.
ПАПУЛЕЗ ЗЛОКАЧЕСТВЕННЫЙ АТРОФИЧЕСКИЙ (papulosis
atrophicans maligna (Degos)).
Синонимы: смертельный кожно-кишечный синдром, синдром Дегоса.
В 1942 г. французские дерматологи Degos, Delort и Tricot сообщили
об этом заболевании, назвав его атрофическим папулезно-сквамозным дерма-
титом. В 1952 г. Дегос предложил назвать его злокачественным атрофиче-
ским папулезом или смертельным кожно-кишечным синдромом. Синдром
Дегоса (правильнее синдром Дегоса—Делора—Трико)—редко встречающий-
ся симптомокомплекс неясной этиологии, характеризующийся поражением
кожи и внутренних органов, главным образом желудочно-кишечного тракта.
В настоящее время в литературе описано около 80 случаев злокачествен-
ного атрофического папулеза Дегоса (Иванов О. Л. и Потекаев Н. С.,
1966; Аравийский А. Н. и соавт., 1969; Сосновский А. Т., 1980; Калам-
карян А. А., Суколин Г. И., Трофимова Л. Я., 1982; Black et al., 1973;
Boddaert et al.,'1973; Enjolras et al., 1974; Olmos и Laugier, 1977; Degos,
1979 и др.).
Этиология и патогенез. Nishida и Howard (1968) при помощи электрон-
но-микроскопических исследований нашли внутриклеточные включения в
эндотелии сосудов размером 25—30 нм, что не исключает вирусы. КоЫ-
meier, Lansecker, Winiarsgki рассматривают эту болезнь как кожно-кишеч-
ную форму облитерирующего тромбангиита. Высказываются предположе-
ния об аутоиммунном характере поражения.
Клиника. Заболевание развивается преимущественно в молодом воз-
расте. Было описано 2 случая у детей (Henkind и Clark, 1966; Cabre et al.,
1974) в возрасте нескольких недель и 7 случаев у лиц старше 55 лет. Обычно
преобладают мужчины, однако в последнее время описано много случаев
среди женщин. Анализ литературных данных и наши собственные наблю-
дения позволяют в развитии клинической картины выделить две стадии.
Первая—продолжается в среднем от нескольких месяцев до двух-трех
лет, характеризуется преимущественным поражением кожи. Как отмечают
Black и Wilcon Jones (1971) из 45 больных, описанных к этому времени, у
И кожные поражения длительное время (до 14 лет) были единственным
проявлением болезни. Изменения кожи начинаются с появления округ-
лых полусферических отечных бледно-розовых папул диаметром от 2—3 до
5—10 мм. Центральная часть их постепенно западает и становится фаян-
сово-белой. Развитой высыпной элемент состоит из двух зон: центральной—
запавшей, атрофичной, фаянсово-белой, как бы гофрированной, с плотно
,прилегающими чешуйко-корками и периферической—валикообразной, яр-
352
корозовой с синюшным оттенком, с телеангиэктазиями. Кожные поражения
вначале единичные, а затем обычно становятся множественными, от 10
до нескольких десятков. Они рассеяны по всей кожной поверхности, пре-
имущественно на туловище и конечностях. Высыпные элементы, кйк
правило, располагаются изолированно и лишь изредка имеют наклонность
к слиянию. Высыпания обычно появляются толчкообразно, цикл развития
отдельного элемента продолжается много месяцев и даже лет. На месте
регрессирующих папул остаются оспенноподобные «штампованные» руб-
чики. Одновременно на коже больного можно видеть элементы, находящие-
ся на разных этапах эволюции (эволюционный полиморфизм). Общее со-
стояние больного не изменяется, субъективные ощущения, как правило,
отсутствуют. Описаны атипичные проявления—волдыри, пустулы, изъяз-
вления. Слизистые .оболочки поражаются в редких случаях.
Мы наблюдали больную 33 лет, у которой заболевание характеризо-
валось длительным существованием кожного процесса (5 лет), необыкно-
венной множественностью и распространенностью высыпаний (1776 элемен-
тов), локализацией высыпаний на лице и веках, которые, как правило,
не вовлекаются в процесс, не описанным ранее поражением голосовых
связок.
Вторая стадия, длительность которой составляет 1—3 недели, харак-
теризуется внезапным развитием симптомов поражения желудочно-
кишечного тракта. Появляются множественные перфорации, преимущест-
венно тонкого кишечника, первым признаком которых являются резкЛ
приступообразные боли в животе, тошнота, рвота, развивается картина
острого живота. Olmos и Laugier (1977) нашли у 61% больных поражения
кишечника. Рано или поздно кишечные поражения развиваются у всех
больных. Как уже отмечалось, кишечные поражения могут появляться
через 2—3, а иногда и 14 лет после появления первых кожных высыпаний.
При вскрытии брюшной полости, при лапароскопии видны на кишечнике
белые, желтоватые, бледно-розовые пятна в подслизистой. Они определя-
ются как белый инфаркт, варьирующий в размерах от миллиметра до не-
скольких сантиметров в диаметре, часто гораздо большего размера, чем
кожные пятна. Поражение не всегда локализуется в тонком кишечнике;
описано много случаев с вовлечением желудка, толстого кишечника, 12-
перстной кишки, пищевода и прямой кишки. Кровь обычно нормальна.
Ряд авторов указывал на увеличение фибринолитической активности, на
отложения IgA, IgG и С3 в дерме и в периваскулярных областях. Иногда
находили гиперглобулинемию, с наибольшим увеличением аз-глобулинов.
По данным Olmos и Laugier (1977), которые анализировали 70 Случаев,
описанных в литературе, у 17% больных были симптомы вовлечения в
процесс центральной и периферической нервной системы. Описаны случаи
с преимущественным поражением центральной нервной системы и характер-
ной кожной сыпью, но без выраженных кишечных симптомов. Эти больные
погибали от тромбоза сосудов мозга (Huriez et al., 1975). А. Н. Аравийский
и соавт. (1970) наблюдали кожно-глазнично-нервный вариант болезни Де-
госа. Неврологическая симптоматика очень разнообразна: гемипарезы с
23 Клиническая дерматология - 353
афазией или без нее, вовлечейие черепно-мозговых нервов, моноплегии,
расстройства чувствительности и др. Нейро-офтальмологическая симпто-
матика включает птозы, неврит зрительного нерва, диплопию, нарушение
полей зрения, отек дисков зрительного нерва. Миокардит, фиброзный пери-
кардит на аутопсии обнаруживали Nomland и Layton (1960), Naylor и
соавт. (1960), Strole и соавт. (1967), Cabre и соавт. (1974).
Заболевание в подавляющем большинстве случаев заканчивается ле-
тальным исходом. Olmos и Laugier (1977) отметили 38 летальных исходов
из 70, но у 32 оставшихся в живых от начала заболевания прошло не более
двух лет. Чем больше длительность заболевания, тем больше возможность
летального исхода. Смерть чаще всего наступает после кишечной перфора-
ции и перитонита, иногда от поражения головного мозга, в части случаев—
легких, легочного абсцесса, инфарктов почек.
Гистопатология. Изменения мелких артерий (эндангиит и облитери-
рующий тромбангиит) с последующим образованием очагов инфаркта в
различных органах, главным образом в коже и желудочно-кишечном трак-
те. В эпидермисе отмечается небольшой гиперкератоз с паракератозом и
заметная атрофия мальпигиевой сети. В дерме—облитерация и тромбоз
мелких сосудов дермы. Обширные очаги некроза с гиалиновой дегенера-
цией коллагеновых пучков. Воспалительная инфильтрация выражена сла-
бо. Электронно-микроскопические исследования показывают дегенерацию
эндотелиальных клеток (везикуляцию цитоплазмы).
• Дифференциальный диагноз. Следует проводить с васкулитами, па-
пуло-некротическим туберкулезом кожи, вариолиформным парапсориа-
зом, анетодермией и склеродермией.
Лечение. Не эффективно
ЛИТЕРАТУРА
Довжанскйй С. И., Никифорова Н. Е., Соколов Е. Д., Гаврилов В. А. Злокачест-
венный атрофирующий папулез Дегоса.— Вести, дерматол. и венерол., 1979, 10, 43—46.
Каламкарян А. А., Суколин Г. И., Трофимова. Злокачественный атрофирующий
папулез Дегоса. Вести, дерматол. и венерол., 1982, 3, 36—40.
Degos R. Ma ignant atrophic papuiosis.— Brit. J. Derma to ., 1979, 100, 1, 21—35.
Olmos L., Laugier P. U trastructure de la maladie de Degos.— Ann. Dermatol. Vene-
reol., 1977, 104, 280—291.
ПАРАНЕОПЛАЗИЯ (paraneoplasia)
Синонимы: паранеопластические заболевания кожи.
В дерматологии под паранеопластическими заболеваниями подразуме-
ваются такие кожные симптомы, которые сочетаются со злокачественными
опухолями внутренних органов, но не являются их метастазами.
Патогенез. Предложен ряд теорий для объяснения механизмов воз-
никновения паранеоплазий—энзимная, токсико-аллергическая, иммуно-
логическая и др. Критериями паранеопластических заболеваний принято
считать: сосуществование с онкологическим заболеванием, их парал-
354
лельное развитие, исчезновение после хирургического удаления опухоли,
а также после эффективного химиотерапевтического или лучевого воздей-
ствия, возобновление паранеопластического синдрома при рецидиве опу-
холи, статистически достоверная корреляция обоих процессов.
Кожные изменения у онкологических больных составляют основную
группу паранеоплазий. В настоящее время описано более 60 паранеопла-
стических кожных синдромов. Частота их у онкологических больных ко-
леблется от 3,7% (Noster, 1966) до 61,5% (Мизонова Т. П., 1974; Калам-
карян А. А., Шадыев X. К-, 1984). Большинство ученых считает, что
никакой закономерной зависимости между характером, локализацией
опухоли и типом кожного поражения не существует. Паранеопластиче-
ские изменения кожи могут предшествовать клиническому проявлению
опухолевого заболевания, появляться одновременно с ним или позже.
Термин паранеопластические поражения кожи обозначает как раз-
личные дерматозы, так и высыпания, которые нельзя отнести к опреде-
ленной нозологической форме (пигментация, гиперкератозы, эритемато-
сквамозные и другие сыпи) и которые с различной частотой сочетаются с
новообразованиями.
Сосочково-пигментная дистрофия кожи (acanthosis nigricans).
Облигатный паранеопластический дерматоз. Развитие его у лиц стар-
ше 40 лет в 80—92% случаев указывает на рак внутренних органов, чаще
на аденокарциному желудка или кишечника (Darier, 1896; Curth, 1950;
Bariere, 1975; Storch, 1976 и др.). В области естественных складок тела
появляются гиперпигментация, затем кожа здесь утолщается, покры-
вается грязно-серыми папилломатозными разрастаниями. Примерно у
половины больных папилломатозные разрастания появляются на слизи-
стой оболочке рта, половых губ. Наряду с описанной формой существует
доброкачественный юношеский черный акантоз и симптоматический, наб-
людающийся у тучных женщин.
Дерматомиозит (dermatomyositis).
От 14 до 23% случаев дерматомиозита составляет вторичный паранео-
пластический дерматомиозит (Соловьева А. П., 1980). У больных старше
40 лет частота епГувеличивается до 50—69% (Федоровская Р. Ф. и Рут-
штейн Л. Г., 1982., Wong, 1969 Niebauer, 1974). По клиническим проявле-
ниям ои не отличается от обычного идиопатического дерматомиозита, на-
блюдается при опухолях различной локализации—яичники, молочная
железа, матка, желудок, почки, органы дыхания и др.
Везико-буллезные дерматозы.
Эритематозная и эксфолиативная пузырчатка, буллезный пемфигоид,
герпетиформный дерматит Дюринга, субкорнеальный пустулезный дерма-
тоз встречаются у 2—18% онкологических больных, в основном пожилого
возраста. Приблизительно у половины больных их появление предшествует
диагностике злокачественной опухоли.
Гиперкецатотические дерматозы.
Паранеопластический псориазиформный акрокератоз или синдром
Базекса (Bazex). Характеризуется появлением на кончиках пальцев, на
23* 355
тыле межфаланговых суставов рук и ног, на кончике носа, по краю уш-
ных раковин псориазоподобных, пятнисто-чешуйчатых зудящих очагов.
Заболевание в основном встречается у мужчин при раке гортани, глотки,
легких, пищевода.
Приобретенный ихтиоз.
Относительно редкий паранеопластический синдром. Встречается у
1—2% больных раком внутренних органов.
Эритемы.
Круговидная, внезапно возникающая эритема Гаммеля (Erythema gy-
ratum repens Gammel) и центробежная кольцевидная эритема Дарье (eryt-
hema anulare centrifugum Darier) чаще чем другие формы эритем наблю-
даются при злокачественных процессах. Эритема Гаммеля обычно появля-
ется за несколько месяцев до выявления признаков злокачественной
опухоли, иногда же одновременно с ними. Располагается на шее, груди,
на других участках туловища, реже на лице и конечностях, оставляет
свободными кисти и стопы. Элементы растут по периферии, существуют
сп. йко, регрессируют только после удаления опухоли, а также после до-
стижения эффекта лучевой йли химиотерапии.
Кожный зуд. Возникновение беспричинного кожного зуда, плохо под-
дающегося лечению, у больных старше 50 лет требует тщательного онколо-
гического обследования. По данным ряда современных авторов, у 5—20%
больных кожным зудом последний носит паранеопластический характер.
Зуд чаще появляется внезапно и бывает различной интенсивности. Он
может быть универсальным или локализованным. При раке легких, же-
лудка, предстательнсй железы описаны случаи возникновения генерализо-
ванного зуда за несколько лет до первых клинических симптомов опухоли.
При системной склеродермии, согласно данным различных авторов
(Андреев В., 1973; Kreysel et al., 1976 и др.), рак внутренних органов
встречается у 1,7—6,5% больных. При ограниченней форме склеродер-
мии связь с раком внутренних органов не установлена.
Приобретенный гипертрихоз пушковых волос или приобретенная пуш-
ковая волосатость (hypertrichosis lanuginosa acquisita). Впервые описана в
1951 г. Lycle. По наблюдениям Wysocki (1971) и Storck (1976) в 65—75%
случаев приобретенный пушковый гипертрихоз возникает на почве опухо-
левого процесса. Поэтому относят его к облигатным паранеоплазиям. Вне-
запно кожа лица, туловища и конечностей покрывается длинными пушко-
выми волосами, что придает больному обезьяноподобный вид. Приобретен-
ная пушковая волосат сть наблюдается при раке желчного пузыря, мо-
чевого пузыря, легкого, толстого кишечника.
Гиперпигментация кожи чаще пп< является в виде дугообразной пиг-
ментации кожи лба (linea fusca). Наблюдается при опухолях мозга.
Каламкарян А. А. и Шадыев X. К. (1984) среди 1212 больных раз-
личными паранес пластическими заболеваниями ксжи у 185 (15,396) наблю-
дали гиперпигментированные пятна, не поддающиеся классификации.
Причем в 178 (96,2%) случаях гиперпигментация опережала онкологиче-
ское заболевание. Авторы считают, что гиперпигментированные пятна у
356
мужчин в возрасте от 51 года до 60 лет и у женщин в возрасте от 61 года до
70 лет должны вызывать повышенную онкологическую настороженность.
ЛИТЕРАТУРА
Машкиллейсон А. Л. и др. Об изменениях кожи при висцеральном раке.— Клин,
мед., 1975, 6; 42—44.
Мизонова Т. П. К вопросу об изменениях кожи у больных злокачественными ново-
образованиями.— Вести, дерматол. и венерол., 1974, 11, 46—49.
Каламкарян А. А., Шадыев X. К- Паранеопластическая гиперпигментация кожи.—
Вести, дерматол. и венерол., 1984, 6, 4—7.
Федоровская Р. Ф. и Рутиипейн Л. Г. К вопросу о паранеопластических пораже-
ниях кожи. Вести. Дерматол. и венерол., 1982, 11, 43—45.
Bazex A. Paraneoplastische Akrokeratose.— Hautarzt, 1979, 30, 3, 119—123.
Lausecker H. Die Hautmetastasen der bosartigen Geschwulste. In: Nicht netzundliche
dermatosen IIIB Herausgegeben H. A. Gottron und G. K- Korting. Berlin, 1979, Bd. 11,
S. 328—343.
Niebauer G. Die paraneaplastischen Dermatosen.— Wien. Med. Kschr., 1974, 124,
683—688.
Prioat Y. Maladie du Duhring—Broq et cervicite dysplasique (forme paraneoplasique
ou association fortuite?).— Bull. Soc. Er. Derm. Syph., 1974, 81, 1, 44—49.
Thomas C. Das paraneoplastische Syndrome.— Med. Klin., 1975, 70, 2053—2065.
ПАРАПСОРИАЗ (parapsoriasis).
Синонимы: болезнь Брока, бляшечная, питириазиформная диссеми-
нированная эритродермия, стойкая ксантоэритродермия и др.
Французский дерматолог Brodq в 1902 г. объединил в одну группу
под названием парапсориаз три дерматоза, описанные ранее под различными
названиями, имеющие некоторые общие черты: хроническое течение, по-
верхностный характер кожных высыпаний, отсутствие субъективных ощу-
щений, резистентность к терапии. В настоящее время различают 4 клини-
ческие формы парапсориаза: каплевидную, лихеноидную, бляшечную и
острую вариолиформную. Брок допускал существование переходных форм.
Большинство отечественных дерматологов (Богров С. Л., 1910; Маш-
киллейсон Л. Н., 1965; Кузнец М. М., 1960; Касумбейли X. Г., 1967)
поддерживают концепцию Брока о родстве отдельных разновидностей пара-
псориаза. Разнатовский И. М. (1976, 1977) рассматривает бляшечную, кап-
левидную и лихеноидную форму парапсориаза как самостоятельные забо-
левания, не имеющие-ничего общего между собой.
Наши клинические, гистологические, гистохимические и энзимологи-
ческие исследования подкрепляют представления о нозологическом род-
стве разных форм парапсориаза.
Этиология и патогенез. Высказываются мнения о возможности этиоло-
гической роли инфекции (грипп, ангина, тонзиллит, стрептококковая ин-
фекция и др.), нарушений функции эндокринной системы, изменений ал-
лергической реактивности больных.
357
Рис. 223. Парапсориаз.
Клиника. Парапсориаз у мужчин встречается чаще, чем у женщин.
Возраст больных колеблется в широких пределах—от 6 до 80 лет, однако
в группе больных бляшечным парапсориазом около 80% составляют лица
в возрасте от 40 до 70 лет. Среди больных каплевидным и вариолиформным
парапсориазом преобладают лица молодого и среднего возраста—от 20 до
40 лет. Имеются отдельные наблюдения смешанного парапсориаза—капле-
видного и бляшечного, капле-
видного и лихеноидного.
Бляшечный парапсориаз
(parapsoriasis en plaques).
Клиническая картина ха-
рактеризуется появлением пя-
тен или слабо инфильтрирован-
ных бляшек желтовато-розового,
желтовато-красного цвета; ве-
личина их от трехкопеечной
монеты до ладони взрослого и
больше, очертания овальные,
округлые или неправильные.
.Овальные, вытянутые, с заос-
тренными концами, полосовид-
ные бляшки чаще располагают-
ся на коже грудной клетки, па-
раллельно ребрам, на конеч-
ностях—параллельно длинной
оси. Очаги поражения, как пра-
вило, плоские, не возвышаются
над уровнем окружающей нор-
мальной кожи, сколько-нибудь
выраженной инфильтрации
обычно не наблюдается (рис.
223). Довольно часто имеется
мелкопластинчатое или отрубе-
видное шелушение серовато-беловатыми чешуйками. У части больных
поверхность очагов поражения бывает морщинистой, шероховатой, го-
фрированной и напоминает смятую папиросную бумагу. Субъективные
ощущения отсутствуют; иногда наблюдается небольшой непостоянный
зуд. Увеличение периферических лимфатических узлов наблюдается лишь
у единичных больных.
2. Каплевидный парапсориаз (parapsoriasis guttata).
Заболевание характеризуется появлением на коже туловища, верхних
и нижних конечностей, а также на лице и кистях многочисленных округ-
лых узелков величиной до чечевицы, светло-розового или светло-коричне-
вого цвета, покрытых в центре коричневато-сероватой чешуйкой, напоми-
нающей пленку коллодия. Описывают три характерных феномена: феномен
облатки (при осторожном снятии чешуйки она отделяется целиком), фе-
358
номен скрытого шелушения (при поскабливании поверхности высыпаний
удается вызвать отрубевидное шелушение) и симптом пурпуры (при интен-
сивном поскабливании высыпаний появляются точечные кровоизлияния).
Высыпания регрессируют, не оставляя следа. У отдельных больных на
месте разрешившихся высыпаний остаются лейкодермические пятна, ко-
торые, по нашему мнению, образуются под действием инсоляции. Субъектив-
ные ощущения отсутствуют.
3. Лихеноидный парапсориаз (parapsoriasis lichenoides, parakeratosis va-
riegata, lichen variegatus).
Встречается редко. На туловище и конечностях появляются множест-
венные, распространенные высыпания конусовидных узелков величиной
с просяное зерно, красновато-желтоватого цвета, слегка блестящих. Зуд
отсутствует. Течение затяжное, иногда с ремиссиями и обострениями.
Отличается особенно большой резистентностью к лечению. Существование
лихеноидного парапсориаза представляется сомнительным. Мы присоеди-
няемся к мнению. С. Т. Павлова (I960) и считаем, что лихеноидный пара-
псориаз представляет собой начальную форму сосудистой атрофической
пойкилодермии Jacobi.
4. Острый парапсориаз (parapsoriasis acuta, morbus Mucha-Habermann;
pityriasis lichenoides et varioliformis acuta).
Эту форму описали Mucha (1916) и Habermann (1925). В отечественной
литературе острый парапсориаз наблюдали Б. Н. Зильберман и Н. Л.
Россиянский (1927), Е. Д. Ашурков и И. Г. Зон (1938), X. Г. Касумбейли
(1959), А. А. Каламкарян и соавт. (1980) и др. Клиническая картина харак-
теризуется полиморфизмом—имеются одновременно пузырьковые, пусту-
лезные, варицеллоподобные элементы, геморрагические узелки, некротиче-
ские корочки. Кроме того, имеются высыпания, типичные для каплевидного
парапсориаза. У единичных больных увеличиваются периферические лим-
фатические узлы. На месте разрешившихся высыпаний остаются мелкие,
слегка вдавленные осповидные атрофические рубчики. У отдельных боль-
ных высыпания распространяются на слизистые оболочки полости рта и
половых органов. Заболевание обычно возникает внезапно с быстрой гене-
рализацией. Иногда наблюдаются продромальные явления в виде головной
боли, общей слабости и недомогания, субфебрильной температуры. Сыпь
рассеяна, симметрична, не группируется, в отличие от других форм пара-
псориаза может распространяться на кожные покровы волосистой части
головы, лица, кистей, стоп (Каламкарян А. А. и соавт., 1980; Касумбейли
X. Г., 1967; Burke et al., 1969; Ellsworth et al., 1982 и др.).
По нашим наблюдениям, острый парапсориаз либо полностью проходит,
либо переходит в хроническую каплевидную форму. Ряд авторов (Калам-
карян А. А. и соавт., 1980) рассматривают вариолиформный парапсориаз
как вариант каплевидной формы с острым течением. Некоторые исследо-
ватели (Шапошников О. К. и Деменкова Н. В., 1974; Разнатсвский И. М.,
1977; Bloc et al., 1976; Ritzenfeld, 1963; Winkelmann, 1960 и др.) объеди-
няют вариолиформный и каплевидный парапсориаз и включают их в груп-
359
пу аллергических васкулитов. В периферической крови больных парапсо-
риазом изменений обычно не наблюдается.
Прогноз для жизни благоприятный, в отношении выздоровления сом-
нительный. Возможна трансформация в грибовидный микоз бляшечного и
лихеноидного парапсориаза (Каламкарян А. А., 1964, 1966, 1967; Сыч
Л. И. и Касумбейли X. Г., 1968; Потекаев Н. С., Константинов А. В.
и соавт., 1975; Lapiere, 1953, 1962; Hewitt et al., 1971; Samman, 1972,
1975; Chiergato et al., 1973; Danda et al., 1975; Heid et al., 1977; Bonva-
let et al., 1977 и др.).
Гистопатология. Все формы парапсориаза не имеют патогномоничной
гистологической картины. Им свойственны изменения типа хронического
дерматита: акантоз, слабовыраженный паракератоз. При каплевидном
парапсориазе—межклеточный и внутриклеточный отек мальпигиевою слоя.
В дерме расширены сосуды, вокруг них инфильтрат из полиморфноядерных
лейкоцитов, лимфоцит в и гистиоцитов. При остром парапсориазе—гипер-
кератоз, акантоз, вакуольное перерождение клеток мальпигиевой сети с
образованием в некоторых случаях внутриэпидермальных пузырьков; в соб-
ственно коже—диффузный и околососудистый инфильтрат с преобладанием
лимфоцитов; участки гибели ткани с образованием некротических корок.
Данные гистохимических и гистоэнзимслогических исслед ваьий (Калам-
карян А. А., Сыч Л. И., Оразымбетова Ж- А., 1980) свидетельствуют о
невысокой функциональной активности клеток инфильтрата при всех фор-
мах парапсориаза.
Дифференциальный диагноз проводят с псориазом, вторичным па-
пулезным сифилисом, красным плоским лишаем и пойкилодермией типа
Jaci bi. Бляшечный парапсориаз дифференцируют с грибовидным микозом,
себорейной экземой, хронической трихофитией кожи. Острый парапсориаз—
с ветряной оспой.
Лечение. Никотиновая кислота, витамины В,, В2, аскорбиновая кис-
лота. Иногда вызывают улучшение синтетические противомалярийные
препараты. При остром парапсориазе—кортикг стероидные препараты (пред-
низолон 20—30 мг в сутки) в сочетании с антибиотиками широкого спектра
действия. Компламин в сочетании с метионином дает определенный эффект
при всех формах парапсориаза, особенно при каплевидном варианте. Сле-
дует воздерживаться от длительных солнечных ванн и ультрафиолетовых
облучений.
ЛИТЕРАТУРА
Каламкарян А. А., Сыч Л. И., Оразымбетова Ж. А. Клинические, гистоморфо-
логические и терапевтические аспекты парапсориаза.— Вестн. дерматол. и венерол., 1980,
6, 4—10.
Раэнатовский И. М. Эволюция взглядов на группу парапсориазов Брока.— Вестн.
дерматол. и венерол., 197b, 2, 41—47.
Bonoalet A., Colan-John К., BelaichS., Degos R. Le diferent formes die parapsoria-
sis en plaques.— Ann. Dermatol. Syph., 1977, 104, 1, 18—25.
360
Danton J. , Maly E., Vanukooa J., Vosmic F. Problematics prechoda parapsoriasis
en plaques mycosis fungoides.— Cesk. Derm., 1975, 49, 5, 116—131.
Ellsworth J. et al. Mucha—Habermann disease in children. The association with rheu-
matic diseases. — J. Rheumatol., 1982, 9, 2, 319—324.
HeidE., Desoaux J., Brandie J., Grosshans E. Der Verlauf der Papapsoriasis en pla-
ques (Brocq’sche Kraukheit).— Z. Hautkr., 1977, 52, 12, 658—662.
Samtnan P. D. Cutaneous reticulosis.—' Trans. St. John’s Hosp. Derm. Soc., 1975,
61, 1—2, 11—15.
ПАТОМИМИЯ (pathomimia).
Синонимы: синдром Мюнхгаузена, симуляция.
Впервые заболевание, принадлежащее к этой группе—экскориирован-
ные акне молодых женщин было описано в 1898 г. М. Brocg.
На основании данных литературы (Минскер О. Б., 1969; Adamson,
1915; Pusey и Senear, 1920; Halprin, 1966; Moriame, 1967; Janus, 1972; Пэу-
неску-Подяну, 1976; Fruensgaard et aL, 1979) и собственных наблюде-
ний мы относим к патомимии всю группу повреждений кожи, вызываемых
и умышленно или подсознательно поддерживаемых самими больными.
Патомимии подразделяются на следующие группы:
1> Дерматомаиии: трихотилломания (выдергивание волос), онихофа-
гия (привычка грызть ногти), невротические экскориации. Дерматомании
рассматриваются как самсповреждения психопатического происхождения,
навязчивые состояния или привычки.
II. Членовредительство (артефакт):
а) симуляция, аггравация—повреждения вызываются на интактной
коже с целью извлечения выгоды из мошеннических побуждений (освобож-
дение от работы по болезни, уклонение от военной службы и т. д.). По-
будительной причиной нанесения себе повреждений служат определенная
материальная или моральная выгода, социальные преимущества или укло-
нение от определенных обязательств. Такие больные, хотя они и симу-
лянты, страдают психическими расстройствами (Пэунеску-Подяну,
1976).
б) патомимии (в узком смысле слова)—форма самоповреждений встре-
чается у психопатов, которые прибегают к самоистязанию с целью полу-
чить удовольствие или удовлетворение.
III. Психопатии—само повреждения кожи у психических больных (при-
кладывание к коже различных раздражающих веществ или нагретых пред-
метов, введение под кожу разнообразных веществ и др.).
Экскориированные акне молодых женщин (по Броку) встречаются
не только в возрасте 16—20 лет, но и у женщин в возрасте около 40 лет.
Поэтому целесообразно исключить определение «молодые» и называть за-
болевание—экскориированные акне женщин, хотя невротические экскориа-
ции наблюдаются и у мужчин.
Эти больные подолгу занимаются своей внешностью, сидят перед зер-
калом, вскрывают папулы и пустулы иглой, ногтями, выдавливая мнимые
или имеющиеся вульгарные акне и комедоны, повреждая кожу. Даже при
361
отсутствии зуда больные не могут удержаться, чтобы постоянно не чесать
и ковырять кожу. \
Эти поражения обычно свидетельствуют о фиксированном неврозе
или психозе; больные критически относятся к своему состоянию, но не
отказываются от самоповреждения и связывают его с неудержимым жела-
нием экскориировать кожу, сдирать кожу (невроз навязчивых действий).
'Некоторые больные производят указанные экскориации преимущественно
в дневное время, другие—в ночИое или вечернее время (перед сном).
Патомимия (синдром Мюнхгаузена)—это особое психическое состоя-
ние, при котором больной, вызывая у себя повреждения кожи (путем ме-
. ханического, термического или химического раздражения), упорно отри-
цает сам факт их нанесения.
Клинически эти поражения очень разнообразны, необычны, не уклады-
ваются в картину определенного дерматоза и часто принимаются за атипич-
ное проявление различных дерматозов.
Локализация артефактов различна. У части больных отмечается от-
носительная ограниченность патологического процесса только на лице
(лоб, нос, щеки, подбородок); у значительно большей части больных наб-
людаются различные варианты расположения поражения (например: лицо,
грудь и плечи; предплечья и живот и др., рис. 224, 225, 226, 227).
Доказательством диагноза патомимии служит признание уловок или
разоблачение больного, обнаружение орудий самоповреждения, изъятие
которых приводит к быстрому исчезновению патологических симптомов.
При психиатрическом обследовании у больных нередко обнаруживают
психопатические черты личности со склонностью к истерии и аггравации.
В большинстве случаев больные производят манипуляции (выдавлива-
ние, трение, царапание, выскабливание) как бы бессознательно. Эти мани-
пуляции приносят удовлетворение, доставляют удовольствие, снижают
нервное напряжение.
Дерматозойный бред (синдром тактильного галлюциноза). Представ-
ляет собой форму психического заболевания. Первое описание дермато-
зойного бреда и сам этот термин принадлежит шведскому дерматологу Экбо-
му (1938).
Этот синдром может встречаться при эндогенных психозах, при ипо-
хондрической форме шизофрении, в депрессивной фазе маниакально-депрес-
сивного психоза (Морозов В. М. , 1963; Архангельская Е. И. и Архангель-
ский А. Е., 1977; Каламкарян А. А., В. Н. Гребенюк, Е. А. Брюн, 1978,
1979), а также носи! внушенный психогенно обусловленный характер
(Столяров Г. Л., 1966; Schott et al., 1973).
Клиническую картину этого синдрома характеризуют многочислен-
ные тактильные галлюцинации при отсутствии зрительных и слуховых
обманов.
Больные ощущают на коже ползанье насекомых, их укусы, сильный
зуд, насекомые вгрызаются в мясо. Галлюцинаторные ощущения носят
или генерализованный, или ограниченный характер (кожа головы,. поло-
вые органы, подмышечные впадины).
362
Рис. 225. Патомимия.
Рис. 226. Патомимия.
Рис. 227. Трихотиломания.
363
Во время наплыва галлюцинаций больные говорят, что они находятся
как в муравейнике и множество насекомых бегает по их телу. Ощущения
иногда вызывают чувство ужаса, больные вскакивают, осматривают свое
тело и одежду, хотя и знают, что ничего не найдут. Резко меняется поведе-
ние больных: они постояннр заняты стиркой и кипячением белья, без кон-
ца моются, натирают тело керосином или используют изуверские методы
самолечения. Почти всегда высказывают бредовые суждения часто в форме
простой констатации наличия насекомых.
Такой сформированный образ присутствия на коже постороннего объек-
та без наличия действительной причины ощущений позволяет трактовать
его как галлюцинаторный образ (тактильный галлюциноз).
Течение патомимии как дерматологического симптомокомплекса за-
висит от того или иного психопатологического синдрома. В связи с этим
диагностика данного расстройства должна основываться на выявлении пси-
хопатологического синдрома с указанием той или иной формы патоми-
мии.
Лечение. Комбинация психотропных средств (меллерил, амитрипти-
лин и др.)^ психотерапия, беседы об опасности нанесения себе повреждений;
наложение фиксированной (лейкопластырной) повязки. Больные должны
наблюдаться и лечиться одновременно у психиатра и дерматолога.
ЛИТЕРАТУРА
Архангельская Е. И., Архангельский А- Е. Синдром дерматозойного бреда в кли-
нической практике.— Вестн. дерматол. и‘венерол., 1977, 11, 69—72.
Каламкарян А. А., Брюн Е. А., Гребенюк В. Н. Дерматологические и психопато-
логические аспекты патомимии.— Вестн. Дерматол. и венерол., 1979, 4—9.
Каламкарян А. А., Гребенюк В. Н. К вопросу о патомимии.— Вестн. дерматол.
и венерол., 1978, 11, 3—6.
Каламкарян А. А., Брюн Е. А-, Гребенюк В. Н. Кожный зуд, протекающий по
типу тактильного галлюциноза.— Вестн. дерматол. и венерол., 1978, 8, 90—92.
Fruensgaard К- et al. Neurotic excoriations.— Intern. J. Dermatol., 1979, 17, 10,
761—767.
ПАХИДЕРМОПЕРИОСТОЗ (pachydermoperiostosis).
Синоним: синдром Турена—Соланта—Голе.
Этиология и патогенез. По-видимому, гетерогенное заболевание, про-
текающее с изменением кожи, обычно по типу пахидермии, утолщением
концевых фаланг пальцев рук в виде барабанных палочек, утолщением и
окостенением периоста, преимущественно дистальных частей длинных труб-
чатых костей. Различают идиопатические (семейные) и симптоматические
(вторичные) формы, клинически сходные, но изменения при вторичном
пахидермопериостозе чаще ограничиваются конечностями (Stevanovic,
1968). Вторичная форма наиболее часто развивается при тяжело протекаю-
щих хронических заболеваниях легких, плевры, сердца (Stevanovic, 1968).
Schneider (1974) предложил следующую классификацию пахидермо-
периостоза.
364
I. Пахидермопериостоз семейный (синоним: идиопатлческая гипертро- '
фическая остеоартропатия, синдром Friedreich—Erb—Arnold, синдром
Touraine—Solente—Gole'): а) моносимптомные и стертые формы; б) семей--
ное утолщение пальцев в виде барабанных палочек; в) форма с полным
симптомокомплексом.
II. Пахидермопериостоз симптоматический (синоним: остеоартропа-
тия гипертрофическая легочная, синдром Bamberger—Pierre—Marie: г) па-
хидермопериостоз злокачественный; б) пахидермопериостоз с пневмопа-
тией; в) пахидермопериостоз, связанный с интра - и экстраторакальным
заболеванием/*
III. Комбинация пахидермопериостоза с Acanthosis nigricans: а) зло-
качественные формы этих заболеваний; б) доброкачественные формы этих
заболеваний;
Пахидермопериостоз, наблюдающийся у больных карциномой легких,
рассматривается как паранеопластический синдром. Указывается на ча-
стое сочетание с Acanthosis nigricans (Marmelzat, 1964; Stevanovic, 1968;
Schneider, 1974).
Наследуется аутосомно-доминантно, реже аутосомно-рецессивно. Пер-
вичный дефект неизвестен. Среди возможных факторов указываются гор-
мональные (гипофиз, щитовидная железа, надпочечники) и нейроцирку-
ляторные нарушения (Hambrick и Carter, 1966), но пока нет достаточно
убедительных доказательств их причинной роли. По мнению Salfeld и Spal-
ckhaver (1966), временной гормональной дисрегуляцией могло бы быть
объяснено нарушение роста в пубертатный период, в частности за счет
преходящего увеличения секреции соматотропного гормона. Против
длительно протекающих органических изменений за счет гормомальных
нарушений, как при акромегалии, свидетельствует тот факт, что спустя
какой-то период времени заболевание становится стацйонарным.
Клиника. Изменения кожи заключаются в значительном утолщении
ее, развитии складок, разделенных глубокими бороздами, на волосистой
части головы и на лбу (cutis verticis et frontitis gyrata), в меньшей степени
на щеках, тыле кистей и стоп. При вовлечении в процесс век глазная щель
резко суживается. Б. Н. Кривошеев и соавт. (1978) описали больную 24
лет с резко выраженной гипертрофией молочных желез, развившейся в
18-летнем возрасте в период беременности, потребовавшей прерывания
беременности и ампутации молочных желез, вес каждой достигал 8 кг..
Другие симптомы поражения кожи непостоянны; отмечены себорея, угре-
вая сыпь, акроцианоз, гипергидроз, скудное оволосение, линейный ладон-
но-подошвенный кератоз, узелковый муциноз, келоиды (Schneider, 1974;
Hambrick и Carter, 1966; Salfeld и Spalckhaver, 1966). За счет костных из-
менений голени и предплечья цилиндрически утолщены, кисти и стопы
увеличены в размерах и напоминают по форме лопату, концевые фаланги
утолщены в виде барабанных палочек, ногти приобретают вид часовых сте-
кол. Описано развитие тотальной лейконихии при вторичном (злокачест-
венном) пахидермопериостозе (Marmelzat, 1964).
365
Rosenthal и Kloepfer (1962) отметили сочетание костных изменений и
помутнения роговицы, развившихся в раннем детском возрасте, со склад-
чатой кожей лица и головы, возникшей после 30 лет.
Заболевание развивается в период полового созревания или вскоре
после него. В течение 5—10 лет развивается полная клиническая картина
и как правило становится стационарной в 20—30-летнем возрасте (Ham-
trick и Carter, 1966; Schneider, 1974). Болеют почти исключительно муж-
чины. Так, по данным Salfeld и Spalckhaver (1966), взятым из литературы,
из 91 больного было только 2 женщины.
Заболевание может протекать с полным симптомокомплексом или с
частичными проявлениями. По данным Salfeld и Spalckhaver (1966), ос-
нованных на анализе 91 случая, опубликованного в литературе, изменения
костей наблюдались у 85 больных, пахидермия в области головы—у 57,
конечностей—у 17, утолщение концевых фаланг—у 75. Ollendorf—Curth
et al. (1961) описали семейный случай утолщения концевых фаланг паль-
цев в виде барабанных палочек, не связанный с висцеральными заболева-
ниями, с признаками гипертрофической остеоартропатии только у членов
семьи с резко выраженными изменениями пальцев. Из общих симптомов
наблюдаются усталость, угнетенность, ослабление памяти, сниженная ра-
ботоспособность, половые расстройства, тяжесть в ногах, артралгии, ле-
тучие опухания коленных суставов (Salfeld и Spalckhaver, 1966; Hambrick
и Carter, 1966; Schneider, 1974). Больные приобретенной формой должны
быть обследованы для исключения рака, в первую очередь, легких. Опи-
сано развитие вторичного пахидермопериостоза и при опухоли матки (Ste-
vanovic, 1968).
Гистопатология. Утолщение дермы, увеличение количества коллаге-
новых волокон, основного вещества, гиперплазия сальных желез, незна-
чительные периваскулярные инфильтраты.
Дифференциальный диагноз. Проводится с акромегалией, тиреоидной
акропатцей.
Лечение. Симптоматическое.
ЛИТЕРАТУРА
Кривошеев Б. Н., Богатырев А. В., Тонышев Р. И., Ершов В. Н. Пахидермо-
периостоз (синдром Tonraine—Sofente—Gole) Вести, дерматол. и венерол., 1978, 2, 57—60.
Schneider Н. G. Zur Klinik und KTassification der Pachydermopariostose.— Derm.
Machr., 1974, 160, 10, 818—824.
ПАХИОНИХИЯ ВРОЖДЕННАЯ (pachyonychia congenita)
Синонимы: синдром Ядассона—Левандовского, врожденный полике-
ратоз, миогоформный идиопатический кератоз Сименса, врожденный много-
формный кератоз.
Этиология и патогенез. Генодерматоз, наследуемый аутосомно-доми-
нантно. Первичный дефект неизвестен. Неясен вопрос о самостоятельности
этого заболевания. По-видимому, оно является гетерогенным, так же как
366
и вся группа ониходистрофий вообще, и изменения нитей, характерные
для него, могут быть одним из симптомов различных с генетической точки
зрения дисплазий. К таким заболеваниям, отличающимся клинически
по ассоциированным проявлениям, по мнению Voigtlander и Schnyder
(1980), могут в первую очередь относиться синдромы Fischer; Spannlang и
Tappeiner; Briinaiuer; Schafer.
Vogt и соавт. (1971) описали больного 42 лет с комбинацией признаков
синдрома Ядассона—Левандовского и Schafer—пахионихия (отсутствую-
щая в описании Schafer), ладонно-подошвенная кератодермия, гипотрихоз,
низкий рост, гипогонадизм, и-полагают, что оба синдрома идентичны. Кроме
того, у больного обнаружены микрофтальмия с уменьшенным размером
роговицы, катаракта.
Клиника. Клиническая картина полиморфна, но основным является
врожденное поражение ногтевых пластинок, с которым могут быть с раз-
ной частотой ассоциированы другие изменения. Ногтевые пластинки зна-
чительно утолщены (до 1 см), очень уплотнены, теряют естественный блеск,
становятся помутневшими, цвет их колеблется от желтовато-коричневатого
до серо-черного, на поверхности ьх—множественные продольные полосы.
Имеется резко выраженный подногтевой гиперкератоз, деформация ногте-
вых пластинок, они могут напоминать часовые стекла, быть искривлены,
онихогрифоз, часты паронихии. Moldenhauer (1968). проанализировал дан-
ные о 93 больных, опубликованные в литературе. У 67 больных имелся
ладонно-подошвенный кератоз (очаговый, реже диффузный или полосо-
видный, омозолелости); у 55—фолликулярный гиперкератоз с локализа-
цией на разгибательных поверхностях конечностей, особенно в области
крупных суставов, на слизистых—лейкокератоз, лейкоплакия; образование
пузырей наблюдалось у 36, чаще на стопах; у 22—гипергидроз; у 11—из-
менение роста (чаще замедленное) или формы волос; у 11 человек—анома-
лии зубов (иногда единичные зубы имеются уже при рождении, раннее вы-
падение); у 9 больных—снижение интеллекта; у 6—изменение костной
системы (избыточная подвижность в суставах, удлинение конечностей,
нарушение окостенения); у 3 больных обнаруживалась катаракта, помут-
нение роговицы; у 3—замедленное развитие половых органов. Комбинации
симптомов различны, а у большинства больных отмечалось наличие 3—5
признаков. Диссоциация признаков может наблюдаться у членов одной
родословной (Joseph, 1964). Формы, проявляющиеся 1—2 симптомами,
могут вызывать значительные диагностические трудности и чаще, по-ви-
димому, рассматриваются как варианты ониходистрофий. Korting и Holz-
mann (1969) считают важным тестом для выявления латентных форм опре-
деление гидроксипролин'а и мукополисахаридов, содержание которых в
сыворотке крови у больных повышено. Moldenhauer и Seidel (1973) обра-
щают внимание на частое обнаружение у лиц с пахионихией себоциётома-
тоза. Vineyard и Scott (1961) наблюдали подобное сочетание у 8 родствен-
ников в 4 поколениях одной родословной. Banscher (1973) наблюдал у
больного частичную атрофию слезных протоков, резкое сужение носовых
отверстий; Soderquist и Reed (1968)—эпидермальные кисты у 3 членов од-
• 367
Рис. 229. Пахионихия
врожденная.
Рис. 228. Пахионихия
врожденная.
Рис. 231. Пахионихия
врожденная.
368
Рис. 230. Пахионихия
врожденная.
ной семьи, клинически сходные с себоцистоматозом, но гистологически
отличные от него. Кроме этого, у больных отмечался гиперкератоз на сто-
пах с образованием пузырей, сухие петлеобразные волосы, наличие с рож-
дения зубов. Н. А. Егоров и соавт. (1978) описали сочетание пахионихии,
кератотических изменений с врожденными аномалиями сердца, легких,
яичек, слабоумием, костной патологией.
Заболевание встречается чаще у мальчиков; примерное соотношение
мужчин и женщин 71:41 (Moldenhauer, 1968). Обычно с рождения сущест-
вуют изменения ногтей, другие признаки появляются позже (рис. 228, 229,
230, 231). Прогноз для выздоровления неблагоприятный. Под влиянием
травм легко развиваются паронихии. Возможно злокачественное перерож-
дение очагов лейкокератоза и лейкоплакий в полости рта, глухота (Voigt-
langer и Schnyder, 1980). Микроцефалию, отставание в умственном разви-
тии, отсутствие вторичных половых признаков в 16-летнем возрасте наблю-
дали у одного из больных В. А. Гребенников и Р. А. Чалимова (1978).
Обращает на себя внимание в этом наблюдении повышенная ранимость
кожи с образованием пузырей после травмы на лице, туловище, конечно-
стях. Н. Д. Шеклаков и соавт. (1970) описали, помимо фолликулярного
и ладонно-подошвенного гиперкератоза, наличие крупных, до 1,5 см, боро-
давчатых высыпаний в области ягодиц, внутренней поверхности бедер,
пояса и локтевых суставов. Степень выраженности ладонно-подошвенного
кератоза может быть различной. У больных, наблюдавшихся Р. С. Дива-
нян и А. Г. Мурадян (1984), были умеренно выраженные очаги гиперке-
ратоза, в то время как Р. А. Капкаев и соавт. (1984) наблюдали больных
с резко выраженным кератозом в области стоп, распространенными гипер-
кератотическими изменениями. Michalowski (1983) описал больного с па-
хионихией I и V пальцев стоп в сочетании с гиперкератозом на ладонях,
резко очерченными эритемато-гиперкератотическими очагами, часто вер-
рукозными, расположенными преимущественно вокруг естественных от-
верстий тела. Нозологическая принадлежность этого наблюдения не ясна.
Автор считает, что отсутствие в полости рта лейкоплакии не позволяет
рассматривать его как синдром Ядассона—Левандовского. Учитывая пред-
почтительную локализацию и характер кожных изменений, предложено
название «Erythrokeratodermia periorificialis.»
Дифференциальный диагноз. Проводят с ниходистрофией, пахиони-
хией профессиональной.
Лечение. Назначают длительный прием витамина А, рибофлавин,
фолиевую кислоту, желатин, общеукрепляющие средства, по показа-
ниям—гормональные препараты, горячие содовые ванночки, 2—10% сали-
циловые мази, уреапласт, мази с включением 5—10% салициловой, мо-
лочной кислоты, резорцина для удаления гиперкератотических наслоений
и последующим применением смягчающих мазей.
24 Клиническая дерматология
369
ЛИТЕРАТУРА
Диванян Р. С., Мурадян А. Г., Семейный случай синдрома Ядассона—Левдидов-
ского. Вестн. дерматол. и венерол. '
Гребенников А. А., Чалимова Р. А. К вопросу о наследственной пахионихии. Вести,
дерматол. и венерол., 1978, 7, 71—75.
Егоров Н. А., Григорьева Г. Н., Бобрик В. В. Врожденный поликератоз Ядассо-
на—Левандовского. Вестн. Дерматол. и венерол., 1978, 2, 61—63.
Michalowski R. Etythrokeratodermia periorificialis mit Akrenbeteiligung. Hautarzt,
1983, 34, 9, 465—467.
Vaigtlander V., Schnyder U. W. Pachyonychia congenita-syndrom In: «Dermatologie
in Klinik und Praxis.» Hrsg G. W. Korting, Stuttgart, 1980, Bd. 2, 2130—2131.
ПЕДЖЕТА БОЛЕЗНЬ (morbus Paget)
Этиология и патогенез. Традиционно болезнь Педжета относят к груп-
пе облигатных преканцерозов, но отводят ей особое место (Венкеи и Шугар,
1962; Schimpf, 1979 и др.) Это объясняется (Schimpf) наличием как экстра-
маммарных первично внутриэпидермальных опухолей, которые в большин-
стве случаев не связаны с аденокарциномой и собственно поэтому являю-
щихся преканцерозом, и форм, располагающихся в области молочных же-
лез и представляющих собой, по мнению большинства, метастаз per coiitinui-
tatem рака молочной железы. Указывается на роль травмы в качестве про-
воцирующего фактора, рубцовых изменений, воздействия неизвестного
экзо-или эндогенного онкогенного фактора. Мс Кее и Hertogs (1980) счи-
тают, что экстрамаммарные формы обычно связаны с карциномой потовых
желез. Авторы описали болезнь Paget у больной раком шейки матки и
расценивают ее как метастаз.
Клиника. Заболевание развивается как правило у лиц старше 40 лет,
преимущественно у женщин. Обычно располагается в области молочных
желез и значительно реже на других участках кожи (половые органы, про-
межность, живот, подмышечные впадины). Если поражение локализуется
на молочных железах, то оно как правило одностороннее. На соске молоч-
ной железы или вокруг него вначале возникает едва заметный небольшой
шелушащийся очаг поражения красного цвета, несколько напоминающий
экзематозный, тем более что могут быть выражены эксудативные явления
вплоть до легкого мокнутия, зуд и небольшое жжение. Однако в отличие
от экземы уже с самого начала границы очага резкие, а по периферии его
имеется едва возвышающееся уплотнение. Очертания очага поражения
чаще неправильные, полициклические, зона его очень медленно расширяется
выходит за пределы околососкового кружка. В течение месяцев или лет
усиливается мацерация, уплотнение становится более отчетливым, особен-
но по краям, очаг эрозируется, покрывается серозно-кровянистыми кор-
ками, после снятия которых обнаруживается влажная зернистая за счет
вегетаций, слегка кровоточащая поверхность. В центре может наблюдаться
частичное рубцевание. Сосок втягивается, хотя и не всегда, в молочной
370L
железе может пальпироваться в. этот период опухолевидное уплотнение,
увеличиваются регионарные лимфатические узлы. Наиболее частой экстра-
маммарной локализацией болезни Педжета является аногенитальная
область (Mohs и Blanchard, 1979). Очаги могут быть как одиночными, так и
многочисленными. Клинически может быть больший полиморфизм, чем
при локализации в области молочных желез. Существует длительно, по-
степенно распространяясь на близлежащие участки кожи. Описаны веге-
тирующие, пигментированные проявления (Teller, 1959). В своем обзоре
Schimpf отмечает, что не всеми признается существование экстрамаммар-
ной формы болезни Педжета. Полагают, что это могут быть нераспознанные
другие опухоли, в первую очередь педжетоидные варианты поверхностной
меланокарциномы. Becker et al. (1960) описали 81-летне;го больного с на-
личием болезни Педжета в области лобка, половых органов, перианальной
зоне, существующей 24 года. При гистологическом исследовании плотного
гранулематозного очага на мошонке в эпидермисе были обнаружены много-
численные клетки Педжета, а в дерме—большие дендритические клетки,
содержащие мало пигмента. Больной погиб через год после этого от мета-
стазов меланомы. Сходство с амеланотической меланомой при гистологиче-
ском» исследовании было и у второй больной с локализацией болезни Пед-
жета в области вульвы. Для дифференциальной диагностики этих состоя-
ний авторы считают важным окраску на тирозин нефиксированных препа-
ратов, на муцин и восстановленное серебро—фиксированных (меланома
не содержит муцина). На значительные диагностические трудности при
экстрамаммарных формах болезни Педжета, особенно у мужчин, обращают
внимание Sonnichsen и Вагшапп (1973). Обычно они сочетаются с карци-
номами различной локализации, в том числе молочной железы. Чаще—
это аденокарциномы потовых желез, но могут быть следствием метастази-
рования опухолей других локализаций (Sanderson и МасЮе, 1979, Gunn
и Fox, 1971). Lever (1975) различает 3 формы экстрамаммарной болезни
Педжета: 2 из них располагаются в подмышечной и генито-перианальной
областях и связаны с карциномой апо - или эккринных потовых желез,
3-я—в перианальной зоне и представляет собой метастаз рака прямой киш-
ки. Вопрос о возможности развития болезни Педжета не только из апокри-
новых, но и из эккринных потовых желез, по мнению Maciejewski et al.
(1979), остается дискутабельным. Используя иммуноцитогистохимический
метод выявления особого глико протеин а-макер а эпителия апокриновых
потовых желез, Merot et al. (1985) высказали предположение, что периа-
нальная болезнь Педжета может иметь апокриновую дифференцировку,
как и другие экстрамаммарные локализации опухоли, а также может про-
исходить из неапокриновых желез промежности. Авторы указали на необ-
ходимость дальнейших исследований. Kawatsu и Miki (1971) описали одно-
временное развитие болезни Педжета в обеих подмышечных впадинах и на
половых органах у 77-летнего мужчины. Одновременное развитие болезни
Педжета на 3 участках, по мнению Ueki и Kohda (1979), может свидетель-
ствовать об общем воздействии какого-то неизвестного фактора или о тем,
что такие зоны у некоторых людей имеют генетически детерминированную
24* 371
склонность к формированию педжетоидных клеток. О. К. Шапошников
и соавт. (1976), Yoelle и Price (1960) рекомендуют производить гистологи-
ческое исследование для исключения болезни Педжета во всех случаях
стойких очагов поражения в подмышечных впадинах и генито-перианаль-
ной области..
Течение заболевания длительное, многолетнее. Прогноз неблагоприят-
ный, так как спонтанного выздоровления не наступает, при отсутствии лече-
ния возникают метастазы, несколько позднее при экстрамаммарных фор-
мах.
Murrell и Me Mullan (1962) наблюдали больного, у которого распрост-
раненные метастазы привели к летальному исходу через 25 лет после раз-
вития экстрамаммарной болезни Педжета половых органов. Опухоль рас-
пространялась медленно, постепенно вовлекая в процесс кожу мошонки,
полового члена, лобка.
Гистопатология. Акантоз, папилломатоз, полиморфизм шиповидных
клеток, наличие педжетовых клеток (большие, округлые или овальные,
светлые вакуолизированные клетки с большим округлым или почковид-
ным бледно-окрашиваемым или гиперхромным ядром). Клетки лишены меж-
клеточных связей. В дерме наблюдается воспалительная реакция, пре-
имущественно из лимфоцитов, плазматических клеток, немногочисленных
тучных клеток. В отношении происхождения педжетовых клеток нет еди-
ного мнения. Допускается трансформация in situ кератиноцитов в педже-
товы клетки под влиянием онкогенного фактора или прямое проникновение
опухолевых клеток в эпидермис (Greenwood и Minkowitz, 1971). Medenica
и Sahihi (1979) полагают, что трансформация кератиноцитов in situ воз-
можна как результат реакции эпидермальных клеток на наличие в нем
опухолевых клеток.
Дифференциальный диагноз. Следует проводить с микробной экземой,
чесоткой, первичным сифилисом, хронической пиодермией, болезнью Боуэ-
на, поверхностным кандидозом, базалиомой, грибовидным микозом, ней-
родермитом, герпетической инфекцией, меланомой.
Лечение. Хирургическое и/или лучевое.
ЛИТЕРАТУРА
Шапошников О. К-, Разнатовский И. М., Родинов А. Н. Экстрамаммарная болезнь
Педжета.— Вести, дерматол. и венерол., 1976, 11, 56—58.
Maciejewski W., Bandmann Н.—J., Schmid М. A. Extramammarer Morbus Paget.—
Hautarzt, 1979, 5, 271—272.
McKee P. H., Hertogs К- T. Endocervical adenocarcinoma and vulval Paget's disea-
se: a significayt assocition.— Brit. J. Derm., 1980, 103, 4, 443—447.
Merot У., MazoujianG., Pincus G. et al. Extra mammary Pagets disease of the Peria-
nal and perineal regioni Arch, dermatol., 1985, 121, 6, 750—752.
Mohs F. E., Blanchard L. Microscopilly controlled surgery for extramammary Paget’s
disease.— Arch. Derm., 1979, 115, 6, 706—708.
Sqnderson К- V., Mackie R. Tumors of the skin In: «Textbook of Derm.» Ed. A. Rook,
372
D. S. Kiikinson, F. J. G., Ebling, Oxford, London, Edinburgh, Melbourne, 1979, 3-d Ed.
v. 2, 2129—2232. v
Schimpf A. Das PTattenepithelkarzinoma der Haut und Halbschleimhaute In: Hand-
buch Haut—und Geschlechskrankheiten. J. Jadassohn, Erganzungswerk, Nicht entzundliche
Dermatosen III B., Bosartige Geschwulste, -Leukamie, Hrsgh. Gottron, G. M. Korting, Ber-
lin, Heidelberg, New—York, 1979, 130—327.
ПЕЛЛАГРА (pellagra)
Этиология и патогенез. В основе заболевания лежит нарушение об-
мена веществ, вызванное дефицитом комплекса витамина В, особенно ни-
котиновой кислоты, и (или) триптофана эндогенного или экзогенного про-
исхождения: длительное голодание или неполноценное (с избытком угле-
водов, недостатком белков, овощей, витаминов) питание, алкоголизм, хро-
нические заболевания желудочно-кишечного тракта (нарушение всасыва-
ния, холецистит, панкреатит, язвенный колит и др.), длительное примене-
ние медикаментов, в первую очередь являющихся антагонистами витами-
нов РР и Ве, обладающих гепатотоксическим и повреждающим слизистую
оболочку кишечника действием. Указывается (Cohen et al., 1974; May-
rick Tomas et al., 1981 и др.) на значимость для развития заболевания за-
медленной инактивации этих веществ. Нарушение обмена триптофана мо-
жет быть генетически детерминированным. Примером является рецессив-
но-наследуемый синдром Hartnup, основные симптомы которого—аминоа-
цидурия, пеллагроидные изменения кожи у лиц раннего детского возра-
ста, стоматит, глоссит, диарея, мозжечковая атаксия, реже глазная па-
тология (нистагм, диплопия и др.), психические нарушения. Stadler et al.
(1982) описали развитие пеллагры у больной, годами принимавшей аналь-
гетики, антидепрессанты. Из других средств наиболее часто указываются
противотуберкулезные препараты, салициламид, цитостатики. Пеллагро-
идные изменения кожи могут быть одним из проявлений карциноидного
синдрома, наряду с приступообразно развивающимися «приливами», скле-
родермиморфными изменениями (Fleischmajer и Hyman, 1961; Castiello
и Lynch, 1972 и др). Так как у больных пеллагрой выявляется ряд неблаго-
приятных факторов, как экзо-, так и эндогенных, видимо, деление пеллаг-
ры на первичную и вторичную вряд ли целесообразно, тем более, что па-
тогенез их сходен.
Клиника. Основные клинические проявления—встречающиеся в раз-
личных сочетаниях изменения кожи, желудочно-кишечные нарушения,
неврологическая симптоматика, чем и определяется характерная триа-
да—дерматит, диарея, деменция.
Stadler и соавт. (1982) отмечают, что кожные изменения наблюдаются
более чем в 80% случаев пеллагры. А. С. Лебедев и Т. А. Лебедева (1934)
й другие авторы наблюдали их у всех больных. Видимо, это объясняется
обследованием больных различными авторами в разное время года. По мне-
нию Н. Г. Хорошавина (1934), кожная симптоматика наиболее часто встре-
чается в июне—июле, а случаи pellagra sine pelle agra—в зимне-весеннее
(январь—март). Обычно изменения кожи являются наиболее ранним симп-
373
Рис. 232. Пеллагра.
Рис. 233. Пеллагра.
томом, на который обращают больные внимание. Однако им часто пред-'
шествуют общие признаки заболевания (общая слабость, снижение аппети-
та, нарушение сна, ослабление памяти, неустойчивое настроение, расстрой-
ство пищеварения), которые, как правило, остаются незамеченными, осо-
бенно если слабо выражены. М. А. Розентул и И. 3. Талалов (1934) отме-
тили, что изменения кожи были первыми симптомами, на которые обратили
внимание больные, только в 17,5% случаев пеллагры. У 10,8% больных
они появились одновременно с другими симптомами, у 8,1% поражение
кожи развилось в течение первой недели после изменений нервной системы
или кишечника. Обращает на себя внимание высокая частота позднего
вовлечения кожи в патологический процесс: в сроки от 1 до 3 недель у
24,3% больных, от 1 до 5 месяцев—у 39,3%. По мнению авторов, это за-
висит от сроков появления первых признаков заболевания: чем раньше
весной оно развилось, тем позднее возникает пеллагрозная кожа. Отсюда
значимость настороженности в отношении этого заболевания, которое,
видимо, встречается чаще, чем принято считать, а утверждения некоторых
авторов о том, что в нашей стране нет пеллагры, не соответствует истинному
положению вещей. Об этом свидетельствуют и наши наблюдения, и публи-
кации в литературе последних лет (Сосновский А. Т. и соавт., 1980; Чи-
стякова И. А. и соавт., 1985). Действительно, случаи пеллагры единичны,
она перестала быть массовой, как это наблюдалось много десятилетий
тому назад, однако, не исключено, что вследствие стертой клинической
симптоматики в ряде случаев заболевание просто не диагностируется.
374
Изменения кожи располагаются преимущественно на открытых и
подвергающихся травматизации участках тела. Наиболее часто—это лицо,
шея, тыл кистей и стоп, нижняя треть предплечий и голеней (рис. 232,
233). Проявляются в виде резко очерченной фоточувствительной эритемы,
вначале яркой, типа солнечного ожога, затем быстро приобретающей крас-
новато-коричневатые, грязновато-коричневатые оттенки. Гиперпигмен-
тация иногда настолько выражена, что, особенно при наличии общей сла-
бости, пониженного артериального давления, может приближать пеллагру
к клинике недостаточности функции надпочечников (Иоссель Б. М., 1934).
Встречаются хлоазмоподобные пигментации. Менее часто вовлекаются в
процесс закрытые участки тела (область суставов, промежность, половые
органы и др.). Поражение, как правило, симметрично, характерным яв-
ляется расположение на кистях и предплечьях в виде «пеллагроидных
перчаток», на стопах и голенях—«пеллагроидных сапожек». С боковых
поверхностей шеи очаги переходят в виде полосы различной ширины на
на грудь («ожерелье», «воротник» Казаля), иногда эта полоса принимает
V-образные очертания. Кожа в очаге поражения суха, слегка инфильтри-
рована, гиперкератотична, особенно по краю, покрыта мелкоотрубевидны-
ми чешуйками. Могут быть болезненные трещины, поверхностные изъязв-
ления (Kingreen и Breger, 1983), резко выраженный фолликулярный гипер-
кератоз (Сосновский А. Т. и соавт., 1980 и др.). Степень выраженности
воспаления может быть различной. При более или менее остром течении
отчетлив эксудативный компонент, достигающий везико-буллезных реак-
ций (pemphigus pellagrosus). Пузыри с прозрачным, реже геморрагиче-
ским или гнойным при инфицировании, содержимым. Очаги поражения
могут иметь экзематоидный вид. Пузыри чаще появляются на ногах, чем
на руках—в 70 и 28,7%, соответственно, по наблюдениям М. А. Розентула
и 3. И. Талалова (1934). Могут быть и на подошвах. На лице высыпания
могут располагаться в форме бабочки, быть сходными с себорейным дерма-
титом, а при наличии помимо эритемато-сквамозных изменений, пузырей,
корок, пигментации лицо становится маскообразным («пеллагрозная ма-
ска»).
Изменения на лице чаще встречаются у женщин и.детей, они менее
выражены, чем на других участках кожи. Характерным считается нали-
чие узкой полосы неизмененной Кожи по границе с волосистой частью го-
ловы и вокруг рта (Me Laren, 1979; Samitz, 1981 и др.). Но это не является
правилом. Gertler (1962) отметил у больной наличие на лице, преимуществен-
но на коже носа, носогубных складок, периорально, а также в верхней
части множественных фолликулярных остроконечных, в носогубных склад-
ках нитевидных, гиперкератотических элементов. Углы рта были гипер-
кератотически изменены, с трещинами. Comaish et al. (1976) наблюдали
множественный комедоноподсбный фолликулярный кератоз в области
бровей, щек, носа, что в совокупности с обильным шелушением придавало
сходство очагам поражения с красной волчанкой. На веках могут быть
эритематозные полулунные диски с геморрагическим компонентом, на-
поминающие свежие кровоподтеки (Мещерский Г. И., 1933), вокруг глаз-
375
резко выраженная гиперпигментация в виде «пеллагрозных очков» (Розен-
тул М. А. и Талалов 3. И., 1934). Пальцы утолщаются, надсуставные
складки сглаживаются. Ладони приобретают желтоватый оттенок, шелу-
шатся, иногда наблюдается мозолеподобный гиперкератоз (Gertler, 1960).
М. А. Розентул и 3. И. Талалов наблюдали переход очагов поражения
на ладони в виде широких красных полос и эритематозных пятен, с выра-
женным отшелушиванием при регрессировании. На ногтях отмечаются
широкие поперечные белые полосы (Donald et al., 1962), деформация ног-
тевых пластинок (часовые стекла и др.). На подошвах описано развитие
кератоза, диффузного или на местах давления (Costello и Gibbs, 1967).
Травма может быть провоцирующим фактором, способствующим появле-
нию поражения кожи и на других участках кожного покрова. Над круп-
ными суставами обнаруживаются слегка инфильтрированные гиперкера-
тотически бляшки. В области голеностопных суставов пеллагрозная эри-
тема может располагаться кольцеобразно в виде полного или частичного
браслета (Мещерский Г. И., 1933.). М. А. Розентул и И. 3. Талалов на-
блюдали «эрозивный браслет» в нижней трети предплечий. В складках
поражение иногда напоминает acanthosis nigricans. Вокруг ануса могут
возникать вегетирующие очаги поражения, сходные с сифилитическими
(Мещерский Г. И., 1933). Высыпания сопровождаются зудом, жжением,
но могут быть асимптомными. С течением времени постепенно в централь-
ной части очагов поражения воспалительные изменения стихают, пр.' ис-
ходит отшелушивание, гиперкератоз сохраняется на границе со здоровой
кожей; при обострении, обычно после инсоляции, эритема появляется
вновь. При этом с каждым обострением, особенно в начале болезни, усили-
вается гиперпигментация, инфильтрация. При длительном течении воз-
можны атрофические изменения, в том числе на ладонях и подошвах. Кожа
становится вялой, сморщенной. Как следствие выраженных трофических
нарушений возможно развитие гангренозных изменений в области паль-
цев стоп, выпадение волос. Наряду с типичными пеллагрозными изменения-
ми наблюдается сухость, сероватая окраска всего кожного покрова, эри-
тематозные пятна с растрескиванием эпидермиса, фолликулярный гипер-
кератоз («гусиная кожа»), иногда с геморрагическими изменениями в зоне
роговых папул, чаще расположенных на голенях. Довольно часты измене-
ния слизистых оболочек, особенно при более остром течении. Наблюда-
ется отечность губ, хейлит, стоматит, в том числе афтозный, глоссит. Язык
становится отечным, малиново-красного цвета, блестящим, увеличенным
в размерах, нередко с трещинами на поверхности, участками атрофии и
гипертрофии сосочков, вдавлениями от зубов на боковых поверхностях.
Нередки вульвиты, баланопоститы. Поражение желудочно-кишечного трак-
та проявляется слюнотечением, снижением аппетита, рвотой, диареей,
болью в животе, потерей веса. Неврологическая симптоматика разнообраз-
на: астенический синдром, слабость, адинамия, бессонница, раздражитель-
ность, депрессия, ослабление памяти, делирий, психозы, энцефалопатия,
полиневропатия и др. Пеллагра может быть провоцирующим фактгргм
для шизофрении. Как указывалось, желудочно-кишечные расстройства
376
и неврологические нарушения могут развиваться до, одновременно или
позже кожных изменений. - Описано развитие пеллагроидной энцефало-
патии без других симптомов пеллагры (кожных сыпей, диареи) у алкого-
ликов (Serdaru et al., 1981).
Ishii и Mishihara (1981) указывают на необходимость исключения пел-
лагры во всех случаях психо-неврологической или желудочно-кишечной
симптоматики у алкоголиков, так как она часто может протекать без кож-
ных изменений. Так, на секции у 20 из 74 хронических алкоголиков были
найдены невропатологические признаки пеллагры. У большинства не было
кожных изменений.
Заболевание встречается преимущественно у взрослых. У детей про-
явления несколько менее выражены. Протекает чаще хронически, с пол-
ной симптоматикой или олигосимптомно. Ухудшение наступает в весенне*
летний период. Прогноз при своевременном распознавании и лечении бла-
гоприятный. Но пеллагра может внезапно закончиться летальным исхо-
дом. Это может наблюдаться при остром (тифоидном) течении, а также быть
при сравнительно небольших клинических проявлениях заболевания.
Опасны для жизни тяжело текущие хронические формы с кахексией, вы-
раженной нервно-психической симптоматикой. Психические расстройства
и неврологические изменения могут длительно сохраняться и после выздо-
ровления. Freundlich et al. (1981) описали семейный случай пеллагроид-
ных высыпаний в комбинации с неврологическими нарушениями (атаксия,
дизартрия, нистагм, кома). У пробанда от кровнородственного брака вы-
сыпания на коже впервые появились в 13-месячном возрасте, а невроло-
гические нарушения—в 14 лет, хотя рецидивы пеллагры с гистологическим
подтверждением диагноза и регрессированием после препаратов никоти-
новой кислоты наблюдались за этот период несколько раз. В связи с тем,
что у больного не наблюдалось аминоацидурии, нарушения всасывания
триптофана, свойственных синдрому Hartnup, автор сделал вывод о нали-
чии у больного генетически-детерминированного заболевания с возможным
биохимическим дефектом, заключающимся в блоке обмена триптофана.
Гистопатология. Изменения неспецифичны. Основными являются ги-
перкератоз, очаговый паракератоз, акантоз, в поздних стадиях истонче-
ние мальпигиевою слоя, спонгиоз, образование пузырьков, повышенное
содержание пигмента, отек сосочкового слоя дермы, сосудов, преимущест-
венно лимфоцитарная инфильтрация, иногда дегенеративные изменения
волокнистых субстанций дермы.
Дифференциальный диагноз. Дифференцировать пеллагру необходимо
от фотодерматозов, экземы, нейродермита, порфирии, псориаза, много-
формной эксудативной эритемы, кератодермий, микозов стоп.
Лечение: комплекс витаминов В (РР, В5, Вв, Ва).
ЛИТЕРАТУРА
Сосновский А. Т., ^нушевскии О. С., Корсун В. Ф. и др. О спорадической пеллагре.
Вестн. дерматол. и венерол., 1980, 8, 60—62.
377
Freundlich E., Stotter M., Jatzio S. Familial pellagra-like skin rach with neurologi-
cal manifestations. Arch. dis. chiin., 1981, 56, 2, 146.,
fshii N., Nishihara Y. Pelfargra among alocoholics: clinical and pathological study
of 20 necropsy cases. J. Neurol. Neupsurg. Psychiatr., 1981, 44, 3, 209—215.
Kingreen J. Ch., Breger G. Pellagra bei «Morazon»—Abusus. Z. Hautker., 1983, 59,
9, 573—577
Me Laren D. S. Cutaneous changes in nutritional deficiencies. In «Dermatology in
general Medicine.» Ed. T. Fitzpatrick et al. In «Dermatology in general Medicine.» Ed. T.
Fitzpatrick et al., Sec. ed., Me Graw Hill Book Company, N. Y. 1979, 1026—1027
Mayrick Thomas R. H., Rowland Payne С. M. E., Black M. M. Isoniazidinnucen
peliagra. Brit. men. j., 1981, 283, 6286, 287—288
Samitz M. H. Cutaneous disorders of the lower exrtremities. Ses. ed., J. B. Lippincott
Company, Philadelphia Toronto. 1979.
Serdaru M., Escourolle P., C. de, Baecque et al.. L'encephalopathi pseudo-pellagreuse
chez I’alcoofique. Nouv. Pres. Medicall., 1981, 10, 45, 3705—3707.
Stadler R. , Orfanos E. , /monel C. Medicamentos induzierte Pellagra. Hautarzt,
1982, 33, 276—280.
ПЕМФИГОИД БУЛЛЕЗНЫЙ ЛЕВЕРА (pemphigoid bullosa Lever)
Синонимы: пемфигоид (Roov и Waddington), парапемфигус (Prakken
и Woerdeman), неакантолитическая пузырчатка (Шеклаков Н. Д.), гер-
петиформный буллезный дерматит.
Lever в 1951 г. выделил это заболевание из группы пемфигуса и обозна-
чил вначале как доброкачественный хронический пемфигус, но затем дал
название буллезный пемфигоид.
Этиология и патогенез. Предложена иммунологическая гипотеза па-
тогенеза. Она основана на обнаружении в крови больных антител к базаль-
ной мембране эпидермиса (Машкиллейсон А. Л. и Голоусенко И. Ю.,
1984, Jordon, Saffls, Beutner, 1969 и др.). Использование в реакции имму-
нофлюоресценции анти С3-конъюгата и свечение при этом базальной
мембраны позволило говорить о комплементсвязывающей активности пем-
фигоидных антител. Антитела к базальной мембране эпидермиса, способные
связывать комплемент, были обнаружены и в жидкости пузырей, что сви-
детельствует о формировании в них иммунного комплекса.
Гипотеза патогенеза буллезного пемфигоида схематично представля-
ется следующим образом. Антитела реагируют с гомологичным антигеном
в зоне базальной мембраны, при этом активизируется система комплемен-
та. Вместе с фиксацией 3, 5, 6 и 7 компонентов комплемента освобождаются
хемотаксические факторы, привлекающие полиморфноядерные лейкоциты.
Эти лейкоциты высвобождают лизосомальные энзимы, которые вызывают
деструкцию базальной мембраны.
Клиника. Заболевание встречается в любом возрасте, но чаще начина-
ется после 55—60 лет. Средний возраст больных, наблюдавшихся Левером,
был 66 лет, М. Н. Шеклаковой—70 лет. Женщины болеют несколько чаще,
чем мужчины. Пемфигоид характеризуется появлением напряженных пузы-
378
7 Рис. 234. Буллезный
пемфигоид.
Рис. 235. Буллезный
пемфигоид.
рей диаметром 0,5—1 см и более. Пузыри появляются как на видимо неизме-
ненной коже, так и на эритематозно-уртикарном фоне, располагаются изо-
лированно или группируются. Преимущественная локализация—живот,
паховые складки, конечности. Однако высыпания могут быть на любом
участке тела. Наблюдаются распространенные и локализованные формы
(рис. 234, 235).
Содержимое пузырей прозрачное, иногда геморрагическое. Образо-
вавшиеся на их месте эрозии не имеют тенденции к периферическому ро-
сту, в отличие от истинной пузырчатки довольно быстро эпителизируются.
Симптом Никольского отрицательный. Может наблюдаться небольшая от-
слойка эпидермиса при потягивании за покрышку пузыря. Слизистые обо-
лочки вовлекаются примерно в половине случаев—чаще рот, нос, гортань,
пищевод, реже—наружные гениталии, прямая кишка.
Поражения слизистых обычно хорошо поддаются терапии, но у от-
дельных больных они протекают очень упорно, несмотря на активную те-
рапию. Н. Д. Шеклаков (1967) выделяет четыре клинических разновидности
пемфигоида: слизистых оболочек, кожи с мономорфными и полиморфными
высыпаниями и универсальную с поражением кожи и слизистых. Bean
и соавт. (1976) наблюдали у 7 больных везикулярный вариант пемфигоида.
Неприятных субъективных ощущений обычно нет. Зуд и жжение бывают у
отдельных больных с полиморфными высыпаниями. М. Н. Шеклакова
(1980) наблюдала умеренный зуд у 12 из 21 больного. Пемфигоид протекает
379
длительно, иногда годами, приступообразно, реже непрерывно. Акантоли-
тические клетки не обнаруживаются. Нередко наблюдается эозинофилия в
пузырной жидкости и в крови. Пробы с иодистым калием обычно отрица-
тельные, у части больных могут быть положительными. Методы иммуно-
флюоресценции у 70—80% больных пемфигоидом выявляют циркулирую-
щие и фиксированные в коже антитела класса IgG, редко IgA к базальной
мембране эпидермиса. Титр их в отличие от пузырчатки часто не корре-
лирует с активностью заболевания.
Ряд авторов считает, что буллезный пемфигоид может быть паранео-
пластическим заболеванием (Андреев В., 1973; Машкиллейсон А. Л. и
соавт., 1975 и др.). В то же время Stork и соавт. (1973) полагают, что буллез-
ные дерматозы, в том числе пемфигоид, редко являются паранеопластиче-
скими реакциями. Имеется ряд сообщений о сочетании пемфигоида с
псориазом (Koerber et al., 1978 и др.), красной волчанкой (Jordon et. al.,
1969), истинной пузырчаткой (Chorzelski et al., 1974; Harrington и Sned-
don, 1979), витилиго и билиарным циррозом (Hamilton и Me Kenzie, 1978),
герпетиформным дерматитом (Barthelmes, 1977).
Гистопатология. Под эпидермисом располагаются большие пузыри,
содержащие много эозинофилов. В верхней части дермы вокруг расширен-
ных сосудов—отек, лимфоцитарные инфильтраты с наличием эозинофилов.
Акантолиза нет. Старые пузыри, вследствие регенерации, могут находиться
интраэпителиально.
Электронно-микроскопически установлено, что пузырь располагается
между базальной мембраной и цитоплазматической мембраной клеток ба-
зального слоя (Caputo et al., 1973; Торсуев Н. А. и соавт., 1979), антитела
локализуются преимущественно в lamina lucida базальной мембраны, они
ассоциируются с полудесмосомами (Honigsmann et al., 1977).
Дифференциальный диагноз. Проводят с истинной пузырчаткой, гер-
петиформным дерматитом.
Лечение. Кортикостероиды, суточную дозу которых определяет тя-
жесть заболевания. При недостаточной эффективности их комбинируют с
цитостатиками (метотрексат, азатиоприн). Трансфузии плазмы, крови.
ЛИТЕРАТУРА
Машкиллейсон А. Л., Голоусенко И. Б. Система комплемента при буллезном и
рубцующем пемфигоидах. Вестн. дерматол. и венерол., 1984,.1, 14—17.
Об изменениях кожи при висцеральном раке (А. Л. Машкиллейсон, С. А. Кутин,
Л. Г. Рутштейн, М. Ф. Ройтбург.— Клин. мед. 1975, 6, 42.
Шеклакова М. Н. К вопросу о некоторых клинических особенностях герпетиформ-
ного дерматита и буллезного пемфигоида. Вестн. дерматол. и венерол., 1980, 11, 39—41.
Beah S. F., Michel В., Furey N. et al. Vesicular Pemphigoid.— Arch. Dermatol.,
1976, 112, 1402—1404.
Chorzelski T. P., Maciejowski E., Jablonska S. et al. Coeristence ol pemphigus and
bullous pemphigoid.— Arch. Dermatol., 41974, 109, 6, 849—853.
Honigsmann H., Singl G., Holubar K-, Wolff K- Elektronenmikroskopische Loka-
380
hsation von Antikorpern und ImmunkompTexen bei Immundermatosen.— Dermatol., Mschr.,
1977, 163, 3, 242—243.
Koerber W. A., Price N. M., Watson W. Coexistent psoriasis and bullous pemphi-
goid.— Arch. QDermatoT., 1978, 114, 1643—1646.
ПИОДЕРМИЯ ГАНГРЕНОЗНАЯ (pyodermia gangrenosum)
Синонимы: язвенный дерматит, язвенная серпигинирующая пио-
дермия.
Термин pyodermia (ecthyma) gangrenosum предложен Brunsting, Go-
eckerman и O’Leary в 1930 году.
Этиология и патогенез. Заболевание длительное время рассматрива-
лось как редкая разновидность пиодермии. Некоторые авторы придержи-
ваются и сейчас этого мнения. В последние годы многие исследователи счи-
тают, что в основе гангренозной пиодермии лежит аллергический васку-
лит, а различным микроорганизмам отводят роль сенсибилизирующего
фактора (Ашмарин Ю. Я-, 1967; Королев К). Ф. и Ходюков Э. Я-, 1974;
Thompson et al., 1973 и др.). Во многих работах указывают на связь ганг-
ренозной пиодермии с иммунными нарушениями, изменением спектра сы-
вороточных протеинов, с нарушением функции иммунных клеток (Rockl
et al., 1964; Branchtel и Lemmel, 1976; Zabel, 1976; Norris et al., 1978;
Савова и Десев, 1980; Lozzy et al., 1982; Greenberg et al., 19Й2; Check et
al., 1983). Описывают сочетание гангренозной пиодермии с парапротеине-
мией, криоглобулинемией, с ревматоидным артритом, язвенным колитом,
различными формами лейкемий (Perry и Brunstiiig, 1957; Lazarus et al.
1972; Perry и Winkelmann, 1972; W aldenstrom et al., 1978; Pioihozka et
al., 1981 и др.).
Клиника. Заболевание встречается обычно у взрослых, дети болеют
редко. Изменения кожи начинаются с появления пузырька, пустулы, иног-
да с узловатого или плоского инфильтрата, которые быстро некротизиру-
ются. Изъязвление быстро увеличивается в размерах. Края язвы си-
нюшно-красные, подрытые, они приподняты в виде валика шириной
1—1,5 см, окруженного зоной гиперемии. В пределах валикообразной
инфильтрации имеются множественные мелкие пустулы и очаги некроза.
Дно язвы неровное, покрыто легко кровоточащими грануляциями, отде-
ляемое обильное, кровянисто-гнойное (рис. 236, 237). Очаги серпигинируют
во всех или в одном каком-либо направлении, одновременно рубцуясь в
другом. Очаги имеют склонность располагаться на конечностях, преиму-
щественно на нижних, реже на туловище, сопровождаются значительной
болезненностью. Содержимое начальных пустул может быть стерильным.
В отделяемом язв обнаруживают разнообразную кокковую и бактериаль-
ную флору. Общее состояние страдает относительно мало. Может быть
преходящая лихорадка.
Прогноз не всегда благоприятный, определяется в значительной мере
часто имеющимся одновременно системным заболеванием (лейкоз, хрониче-
ский полиартрит, язвенный колит и др.). Характерна склонность к реци-
дивам.
381
Рис. 236. Пиодермия
ангренозная.
Рис. 237. Гангренозная
пиодермия.
Гистопатология. Отечность стенок сосудов вплоть до закрытия про-
света, тромбоз сосудов в верхней части дермы. Гранулематозные инфильт-
раты во всей толще дермы, состоящие из лимфоцитов, нейтрофильных
лейкоцитов, плазматических клеток, фибробластов. Очаги деструкции.
Дифференциальный диагноз. Следует проводить с хронической язвен-
но-вегетирующей пиодермией, гранулематозом Вегенера.
Лечение. Комплексное применение кортикостероидов, азатиоприна,
антибиотиков.
ЛИТЕРАТУРА
Королев Ю. Ф., Ходюков Э. Я- К вопросу об этиологии, патогенезе и лечении ганг-
ренозной пиодермии.— Вести, дерматол. венерол., 1974, 11, 11—14.
Савова И., Десев Д. Случай на pyoderma gangrenosum в съчетание с криоглобули-
немия.— Дерматол. и венерол., 1980, XIX, 1, 37—39.
Brachtel R., Lemmel Е. М. Chronisch vegetirende Piodermie bei zellularen Immunde-
fekt.—Hautarzt, 1976, 27, 488—491.
Greenbery S. и соавт. Pyoderma gangrenosum.— Arch, dermatol., 1982, 7, 118, 502—
507.
Larry M. et al. Pyoderma gangrenosum. Jmmukologic findings.— Arch, dermatol.,
1982, 118, 10, 769—773.
Norris D. A., Weston W. R., Thorne E. G., Hum bert J. R. Pyoderma Gangreno-
sum. Abnormal Monocyte Funktion corrected in vitro with Hydrocortisone.— Arch. Derma-
tol., 1978, 114, 906—911.
Zabel M. Pyoderma gangrenosum mit IgG—Paraproteinamie bei Plasmozytom.—
Hautarzt, 1976, 27, 603—605.
382
ПИОДЕРМИЯ ШАН КРИФОРМНАЯ (pyodermia chancriformis).
Термин шанкриформная пиодермия впервые использовал Hoffmann в
1934 г. из-за сходства поражения с сифилитическим твердым шанкром.
Относится к очень редким атипичным пиодермитам, вызываемым стафило-
кокком, реже стрептококком. Важное практическое значение заболевания
заключается в его чрезвычайно большом клиническом сходстве с первич-
ной сифиломой, могущим послужить поводом для ошибочного диагноза. В
отечественной и зарубежной литературе опубликовано небольшое число
работ с описанием случаев шанкриформной пиодермии (Каплан М. Я.,
1940; Торсуев Н. А., 1941; Выгодский Д. С., 1941; Аствацатуров К- Р.
и Розентул М. А., 1951; Шахтмейстер И. Я. и Карпушкин В. П., 1964;
Абрамова Е. И. и Ремизов С. М., 1970; Gougerot и Besnier, 1936; Branom
et al., 1963; Laymon et al., 1954; Lejman et al., 1973; Gianadda et al., 1982
и др.).
Клиника. Шанкриформная
пиодермия может развиваться
как у взрослых, так и у детей,
независимо от поча. Локализу-
ется в области гениталий и
экстраген итально (красная кай-
ма верхней и нижней губы),
слизистые оболочки полости
рта и щек, язык, углы рта,
подбородок, спинка носа, веки,
брови и др.). У большинства
больных процесс располагается
в области половых органов: у
мужчин на головке полового
члена, в заголовочной борозде
и крайней плоти; у женщин на
больших и малых половых гу-
бах и на слизистой влагалища.
Заболевание обычно начинается
Рис. 238. Пиодермия
шанкриформная.
с появления пузырька, после
вскрытия которого остается
эрозия, весьма напоминающая
твердый шанкр, или язва округлых или овальных очертаний, основа-
ние которых всегда уплотнено в различной степени; края покатые, не круто
обрезанные, зачастую приподнятые. Отделяемое не обильное, серозное.
В некоторых случаях по периферии имеется узкий воспалительный ободок.
Изъязвления могут достигать 1—2 см в диаметре. Дно почти всегда ровное,
розовато-красное, иногда покрыто незначительным гнойным распадом (рис.
238). Субъективных ощущений поражение не вызывает. Регионарные лим-
фатические узлы плотны, безболезненны, не спаяны между собой и с под-
лежащими тканями. При шанкриформной пиодермии поражение чаще все-
го одиночное, но может быть множественным. Следует отметить, что не-
383
редко у больных, помимо основного шанкриформного элемента, могут быть
другие высыпания пиодермии. В отделяемом эрозий и язв обычно обнару-
живают стафилококки и стрептококки. Для исключения сифилиса необхо-
димы тщательные исследования отделяемого язвы на бледную трепонему
и серологические исследования.
Гистопатология. Она достаточно характерна и позволяет уверенно
исключить первичную сифилому. Выраженный акантоз, отек, разрастание
лимфатических и кровеносных сосудов. Периваскулярный и диффузный
инфильтрат, состоящий из полиморфноядерных нейтрофилов, эозинофилов,
лимфоидных элементов и гистиоцитов, в противоположность первичной
сифиломе, которая является в гистологическом отношении плазмомой.
Дифференциальный диагноз. Следует проводить в первую очередь с
твердым шанкром. Ни в одном случае, как бы клиническая картина не
походила на первичную сифилому, до нахождения бледных трепонем в
язве или положительных серологических реакций на сифилис приступать
к специфическому лечению нельзя:
Лечение. Антибиотики (пенициллин, метациклин, олететрин и др.),
сульфаниламиды, местно анилиновые краски, 5—10% линимент синтоми-
цина, 4% гелиомициновая мазь и др.
ЛИТЕРАТУРА
Gianadda В., Gianadda Е., Fazio М. Pyodermie chancriforme. — Dermatologica
(Basel), 1982, 164, 3, 195—200.
ПЛАЗМОЦИТОМА КОЖИ (plasmacytoma cutis).
Синонимы: миеломная болезнь, плазмоклеточный парапротеинемиче-
ский ретикулез, болезкз Рустицкого—Калера.
Этиология и патогенез. Плазмоцитому, наряду с макроглобулинемией
Вальденстрема и болезнью тяжелых цепей, большинство современных
исследователей относят в группу парапротеинемических гемобластозов,
объединяющих опухолевые процессы в системе иммунокомпетентных кле-
ток, осуществляющих функцию гуморального иммунитета. И. А. Кассир-
ский и Г. А. Алексеев (1970) и Н. Е. Андреева (1974) рассматривают мие-
ломную болезнь как бластомную моноклоновую, реже диклоновую имму-
ноглобулинопатию. Особенностью парапротеинемических гемобластозов яв-
ляется способность миеломных клеток синтезировать иммуноглобулины
(Ig) или их фрагменты—парапротеины (патологические белковые продук-
ты). В зависимости от типа иммуноглобулина различают IgM—миелому,
IgG—миелому, IgA—миелому, миелому Бенс—Джонса.
Клиника. Миеломная болезнь развивается преимущественно в возра-
сте 45—65 лет, одинаково часто у мужчин и женщин. Заболевание пред-
ставляет собой сложный симптомокомплекс, складывающийся из следу-
ющих основных синдромов: медуллярного (оссальгического, костного),
экстрамедуллярного (почечного, анемического, гепатоспленомегалическо-
го, кожного, нервного, амилоидного) и смешанного (медуллярного-экстра-
медуллярного). Различают: солитарную плазмоцитому (костную и вне-
384
костную) и генерализованную
плазмоцитому (множественную
миелому). Кожные изменения
подразделяются на специфиче-
ские и неспецифические. Встре-
чаются два вида специфических
поражений кожи: первичные,
обнаруживаемые преимущест-
венно в коже, и вторичные, раз-
вивающиеся вследствие рас-
пространения процесса с пора-
женных костей или внутренних
органов. Клинически они мало
отличаются друг от друга. Пер-
вичные (солитарные) плазмо-
цитомы кожи исключительно
Рис. 239. Миеломная болезнь с кожными
проявлениями (инфильтративно-гемор-
рагические изменения).
редки: в литературе описано
всего 11 случаев (Hedinger, 1911;
Krutsay, 1936; Dolin и Dewar,
1956; Johnson и Taylor, 1970;
Mikhail и Spindler, 1970; La
Perriere et al., 1973; Canlas
et al., Nini et al., 1983 и др.).
Специфические миеломные по-
ражения кожи проявляются
развитием плотных опухолей и
бляшек величиной от вишни до
грецкого ореха и больше. Цвет
их синевато-красный или буро-
вато-синюшный различной на-
сыщенности. Опухоли чаще рас-
полагаются изолированно, реже
сливаются в объемные бугрис-
тые образования. Субъектив-
ные расстройства отсутствуют.
Рис. 240. Миеломная болезнь
(геморрагические изменения).
Редко наблюдается изъязвление
опухолей с образованием резко
болезненных глубоких язв. Плазмоцитомы кожи часто локализуются
на волосистой части головы, лице, верхней половине груди и на
руках. В некоторых случаях первичная кожная плазмоцитома мета-
стазирует в регионарные лимфатические узлы (Mikhail et al., 1970)
или в висцеральные органы (Johnson и Taylor, 1970). Вторичные специфи-
ческие поражения кожи встречаются примерно у 7—10% больных миелом-
ной болезнью. Bluefarb (1955) обнаружил их у 9 из 88 больных множествен-
ной миеломой костей. Клинически вторичные специфические плазмоцитомы
кожи характеризуются появлением на конечностях и туловище опухолеввд-
25 Клиническая дерматология
385
Рис 241. Миеломная
болезнь (милиум-подобные
изменения в подмышечной
впадине).
Рис. 244. Плазмоцитома кожи —миеломная болезнь
(изменения кожи по типу шиповидного лихена).
386
них образований размерами от горошины до голубиного яйца. Они отли*
чаются мягковатой или плотноэластической консистенцией, малой болез*
ценностью, имеют синюшно-красный цвет с буроватым оттенком. При не-
кротическом распаде опухолевых элементов образуется язва с кровянисто-
гнойным отделяемым (рис. 239, 240, 241, 242, 243, 244). Диагностическими
критериями генерализованной миеломы является наличие патологических
иммуноглобулинов в сыворотке крови, белка Бенс—Джонса в моче
(Вепсе—Jones протеинурия), остеолитические очаги или диффузный остео-
пороз, повышенное содержание атипичных плазматических клеток в кост-
ном мозге (более 10%), наличие плазмоклеточных опухолей мягких тканей
(Wilmanus, 1975).
Неспецифические кожные высыпания встречаются значительно чаще,
чем специфические. Они могут быть результатом нарушения белкового
обмена, интоксикации организма продуктами метаболизма. Одной из при-
чин неспецифических поражений кожи при миеломной болезни являются
отложения амилоида в коже, которые, по данным Н. Е. Андреевой (1976);
встречаются у 15—20% больных и могут сопровождаться макроглоссией,
Клинически они проявляются узелками и бляшками, нередко с некротиче-
скими и язвенными изменениями.
А. А. Каламкарян (1966), Hodl, Turek, Kerl (1982) наблюдали боль-
ных JgA—плазмоцитомой с системным амилоидозом с тяжелыми язвенно-
некротическими поражениями кожи и нервной системы.
Токсические кожные проявления характеризуются алопецией, эри-
темато-отечными пятнами, пигментацией кожи, себорейноподобными из-
менениями на лице и др. Изменения крови (лейкопения, анемия, тром-
боцитопения) могут быть причиной кожных кровоизлияний, пурпуры,
экхимозов. При миеломатозном поражении почек нередко развивается
отек кожи, сопровождающийся зудом, а при локализации миеломы в нерв-
ных ганглиях часто развивается опоясывающий лишай. Мы наблюдали
женщину 30 лет с генерализованной диффузной формой плазмоцитомы с
распространенными, обширными гангренозно-некротическими поражениями
кожи, с кахексией, лихорадкой, анемией, ускоренной СОЭ, общей слабо-
стью, поражениями костной системы (оссалгия), симптомами поражения
нервной системы и психики: резкая головная боль, падение зрения, ослаб-
ленная память, бредовые высказывания, галлюцинации, гемиплегия. В
литературе описано около 20 случаев комбинации миеломной болезни с
ксантоматозом кожи (Алексеев Г. А., 1970; Moscholla, 1970 и др.).
Прогноз неблагоприятный, смерть наступает при явлениях общей
кахексии, анемии, геморрагического диатеза, уремии, пневмоний и др.
Гистопатология. При специфических поражениях кожи выявляются
массивные инфильтраты преимущественно из атипичных плазматических
клеток. При цитологическом исследовании пунктата опухоли также обна-
руживают характерные миеломные клетки.
Дифференциальный диагноз. Кожные изменения дифференцируют с гри-
бовидным микозом, ретикулосаркоматозом кожи, дерматофибросаркомой,
гистиоцитомами.
25*
387
Лечение. Цитостатические средства (сарколизин, проспвдин, мельфа-
лан, циклофосфан, натулан и др.), лучевая терапия, кортикостероиды,
анаболические гормоны.
ЛИТЕРАТУРА
Андреева Н. Е. В кн.: Патогенез и терапия лейкозов. М., 1976, с. 277—293.
Bataille R., Rosenberg F. Myelome multiple des os.— Med. actuelle, 1978, 5, 9, 323—
327.
Canlas M., Dillon M., Loughrin J. Primary cutaneous plasmacytoma.— Arch. Der-
matol., 1979; 115, 6, 722—724.
Hodl S., Turek T., Kerl H. Plasmozyfom—assoziirte bullos—hamorrhagische Amy-
loiolose der Haut.—Hautarzt, 1982, 33, 10, 556—588. ;
mburg—Leoer G. Histopathology of the skin. V ed.— Philadelphia:-. , . 1
J. P. Lippincott company, 1975.
Stankler L., DaoidsonJ- Multiple extra-medullary plasmocytomas of ’he skin.— Brit
J. Dermatol.? 1974, 90, 2, 217—221. ; / ;
ПОДУШЕЧКИ ПАЛЬЦЕВ ОКОЛОСУСТАВНЫЕ (pulvillus digit!).
Синонимы: периартикулярный эпидермо-гипертрофический невус.
Околосуставные подушечки пальцев впервые флли описаны Garrod
(1893). Gougerot и Yoannon описали это заболевание в 1926 г. под назва-
нием эпидермогипертрофические невусы кистей и стоп. Исключительная
локализация процесса в области суставов позволила Л. Н. Машкиллей-
сону (1928) добавить слово околосуставные к этому названию.
Этиология и патогенез. Многие авторы (Машкиллейсон Л. Н., 1928;
Gougerot et al., 1926; Allison, 1966 и др.) рассматривают заболевание как
невоидный дерматоз; по нашим наблюдениям, провоцирующим фактором
может быть травма, в частности, профессионального характера. Возможны
семейные случаи (Мордовцев В. Н., 1979; Hermann, 1959).
Клиника. Несколько возвышенные безболезненные подушкообразные
утолщения отмечаются на тыле межфаланговых суставов пальцев; диа-
метр — 1—2 см, цвет коричневато-красный или не отличается от цвета нор-
мальной кожи. Они подвижны, не спаяны с подлежащими тканями, плотно-
эластической или плотноватой консистенции. Поверхность грубая, шеро-
ховатая, рисунок кожи заметно подчеркнут. Воспалительных явлений по
периферии нет. Указанные изменения носят стойкий характер, чаще наб-
людаются на руках, но могут быть и на пальцах ног. Заболевание встре-
чается преимущественно у детей и подростков, реже у взрослых.
Течение заболевания длительное, в редких случаях подушечки спон-
танно регрессируют.
Гистопатология. Утолщение всех слоев эпидермиса, гиперкератоз, вы-
раженный акантоз и гипергранулез; в дерме—слабый периваскулярный
инфильтрат без расширения сосудов.
Дифференциальный диагноз. Следует проводить с гистиоцитомой, рев-
матическими и подагрическими узелками, кольцевидной гранулемой, фиб-
роматозными невусами, синовиальной кистой.
388
Лечение малоуспешное. Применяют лучи Букки, лучи Рентгена, корти-
костероидные мази. Е. И. Шахнес и Л. В. Таранова (1980) сообщили о
хорошем клиническом эффекте от назначения витамина А внутрь в соче-
тании с кремом Rettin А в виде компрессов.
ЛИТЕРАТУРА
Шахнес И. Е., Таранова Л. В. Околосуставные подушечки пальцев.— Вестн. дер-
матол. и венерол., 1980, 12, 43—44.
Rook A., Wilianson D., Ebling F. In: Textbook of derma)tolory. Oxford—London,
1979, p. 1633—1634.
ПОЙКИЛОДЕРМИЯ АТРОФИЧЕСКАЯ СОСУДИСТАЯ ЯКОБИ (poi-
kilodemiia atrophicans vascularis Jacobi).
Синонимы: эритематозная сетчатая атрофодермия.
В 1906 г. Е. Jacobi описал заболевание, характеризующееся телеанги-
эктазиями, пигментацией, капиллярными геморрагиями и атрофией, кото-
рому он затем в 1907 году дал название сосудистая атрофическая пойкило-
дермия, В. И. Теребинский (1916), А. А. Халипский (1928), Т. М. Юринов
(1946), Wolf и SeimanoWitz (1970), Murray и Zimmerman (1971), Hlavaty
и Lukadova (1975) и др., как и Якоби, выделяют идиопатическую атрофию
кожи как самостоятельную форму и относят ее к классу атрофий. Большин-
ство других исследователей (Павлов С. 1., 1924; Машкиллейсон Л. Н. и
Позин Л. С., 1933; Шапошников О. К-, 1969; Кгеп, 1926; Gougerot и
Burnier, 1934; Rollier, 1951; Griineberg, 1958; Dougherty, 1971; Gertler,
1972 и др.) рассматривают пойкилодермию Якоби не как самостоятельное
заболевание, а как исход некоторых дерматозов—дерматомиозита, скле-
родермии, красной волчанки, грибовидного микоза, лимфогранулематоза,
красного плоского лишая, парапсориаза и др. Наконец, третья группа
авторов (Каламкарян А. А., 1969; Разнатовский И. М., 1976; Keil, 1938;
Linzer, 1951; Waddington, 1953; Palmer, 1962 и др.) высказывает взгляд
на. пойкилодермию, как на симптомокомплекс, тесно связанный с опухоле-
вым процессом, в частности являющийся фазой предопухолевого периода
грибовидного микоза или ретикулосаркоматоза кожи или развивающегося
у больных висцеральным (печень) раком (Сопи и Gogonea, 1973).
Этиология и патогенез. Механизмы развития заболевания не изучены.
Клиника. Заболевание характеризуется пигментными пятнами, атрофи-
ческими участками и телеангиэктазиями. Иногда болезнь начинается остро-
воспалительными явлениями в коже, отеком и т. д. или она с сацого на-
чала носит хронический характер. Наиболее характерен для этого заболе-
вания пестрый рисунок кожных высыпаний. Поражение кожи распро-
страненное, занимает лицо, шею, туловище, плечи, предплечья, бедра,
голени и характеризуется наличием пятен бурого цвета и атрофических
участков, которые, располагаясь рядом, образуют полосы и мелкопятни-
стую сеть, причудливо сочетаются и образуют разнообразные фигуры:
кольца, дуги, овалы и т. п. Атрофические линии окаймляют то гипер-
пигментированные, то с виду нормальные участки кожи. Телеангиэктазии
389
видны то в виде отдельных тончайших сосудов, пробегающих в коже, то
они сгруппированы, имеют вид кружевоподобной пигментной сетки, в
которую зкраплены светлые участки; при надавливании стеклом они ис-
чезают. Поверхность пойкилодермического очага сухая, слегка шелу-
шится с наличием капиллярных геморрагий и красновато-коричневых фол-
ликулярных узелков величиной с булавочную головку. В отдельных слу-
чаях отмечено поражение слизистой оболочки полости рта. Иногда отме-
чается общее недомогание и нерезкий зуд. Таким образом, изменения кожи
клинически весьма напоминают так называемую рентгеновскую кожу, но
без предшествующего облучения рентгеновскими лучами. Заболевание
характеризуется хроническим течением и стойкостью очагов поражения.
Гистопатология. На ранних стадиях заболевания отмечается истон-
чение эпидермиса и сглаживание эпидермально-дермальной линии. В ба-
зальном слое—выраженная вакуольная дегенерация и повышенное содер-
жание меланина. В верхней части дермы капилляры несколько расширены,
«округ них полосовидно располагается инфильтрат, состоящий в основном
из лимфоидных клеток и гистиоцитов. В поздних стадиях—атрофия эпи-
дермиса усиливается, в дерме воспалительный инфильтрат уменьшается;
эластические волокна разрушены или резко фрагментированы; гомогени-
зация и склероз коллагена.
Дифференциальный диагноз. Следует проводить с красной волчанкой,
атрофическим красным плоским лишаем, прогрессирующей идиопатической
атрофией кожи, дерматомиозитом, бляшечным и лихеноидным парапсориа-
зом, грибовидным микозом.
Лечение такое же, как при атрофиях кожи. При вторичных пойкило-
дермиях важно своеовременное рациональное лечение первичных заболе-
ваний (дерматомиозита, красной волчанки, парапсориаза, грибовидного
микоза и др.). Больные должны находиться под диспансерным наблюдением
для своевременного обнаружения и лечения опухолевых процессов.
' ЛИТЕРАТУРА
РазнатовскиД И. М. Дифференциальная диагностика между болезнью Брока и
грибовидным микозом.— Вести, дерматол. и венерол., 1976, 8, 29—34.
Hlavaiy Р., Lukacova Е. Poiikiloderma atrophicans vascularia Jacobi.— Cs. Derm.,
1975, 50, 1, 60—63.
ПОЙКИЛОДЕРМИЯ ВРОЖДЕННАЯ ТОМСОНА (poikilodermia conge-
nitale Thomson).
Синонимы: синдром Томсона, врожденная пойкилодермия.
В 1923 г. Thomson описал сетчатые кожные изменения на лице
и конечностях у двух сестер. По классификации, предложенной Bazex
и Dupre (1958), синдром Томсона, как и синдром Ротмунда, относится
к ранним формам врожденных пойкилодермий. До сего времени остается
спорным вопрос о взаимоотношении этих синдромов. Braun и Unger (1965),
Diem (1975), Kristensen (1975) и др. их идентифицируют, указывая, что
они отличаются только наличием или отсутствием катаракты. Marghescu
390
Рис. 245. Пойкилодермия
врожденная Томсона.
Рис. 246. Врожденная
пойкилодермия (та же больная).
и Braun-Falco (1965) считают, что в группе врожденных пойкилсдермий
определенную самостоятельность имеют шесть синдромов и среди них
синдром Томсона. Rodermund и Hausmann (1977) также высказываются
за нозологическую обособленность этого синдрома. Они собрали в литера-
туре описание 40 достоверных случаев и опубликовали одно собственное
н 1блюдение. Соотношение лиц мужского и женского пола среди этих паци-
ентов было 19:22.
Этиология и патогенез. Имеются лишь общие указания на эмбриональ-
ное повреждение мезо- и эктодермы. Синдром Томсона относят к числу
врожденных заболеваний с аутосомно-рецессивным типом наследования.
Хромосомные аномалии не описаны. Нередки семейные случаи заболевания,
чаще у братьев и сестер. Родители больных, как правило, не являются
кровными родственниками.
Клиника. Кожные изменения начинаются большей частью на первом
году жизни, редко в 2—3 года. На щеках, на лбу, подбородке, ушных ра-
ковинах появляются эритема и отечность, постепенно распространяющиеся
на разгибательную поверхность рук, ног, на ягодицы. В дальнейшем раз-
виваются сетчатая, пятнистая атрофия, телеангиэктазии, гипер - и де-
пигментации (рис. 245,246). Через 2—3 года эти изменения перестают нара-
стать и принимают персистирующий характер. На туловище высыпания
встречаются редко. Описаны гиперкератозы, фолликулярные и лихеноид-
ные папулы, сухость, шелушение кожи. Случаи с бородавчатыми гипер-
кератозами выделяются как веррукозный тип синдрома Томсона. У части
391
больных наблюдается повышенная чувствительность к солнечным лучам,
проявляющаяся в усилении кожных изменений, зуде или в форме буллез-
ного дерматита. С возрастом выраженность телеангиэктатических изменений
уменьшается (Thomson, 1936; Feldreich, 1955). При синдроме Томсона встре-
чаются различные костные аномалии: треугольное лицо с высоким широким
лбом и узким подбородком, «дырчатый череп», фронтальные гиперостозы,
готическое нёбо, нарушение оссификации длинных костей, гипо-или ап-
лазия фаланг и др. Часто наблюдается гипотрихоз, аномалии зубов. Боль-
шинство авторов сообщает о маленьком росте пациентов и нормальном ин-
теллекте.
Прогноз для жизни благоприятный.
. Гистопатология. Атрофия эпидермиса, отечная дегенерация клеток
базального слоя. Сглаженность соединительнотканевых сосочков, расши-
рение капилляров в верхней части дермы, разрежение эластики.
Дифференциальный диагноз проводят с синдромом Ротмунда, синдро-
мом Блюма. *
Лечение. Симптоматическое.
ЛИТЕРАТУРА
Diem Е. Rothmund—Thomson—Syndrom Kasuistischer Beitrang.— Hautarzt, 1975,
26, 8, 425—429.
Kristensen K. Poikiloderma congenitale en early case of Rothmund—Thomson’s Synd-
rome.— Acta Dermato-vener., 1975, 55, 4, 316—3188.
Rodermund O.—E., Hausmann D. Typus verrucosus des Thomsonsyndroms. Ein Beit-
rag zu den kongenitalen Poikifodermien.— Hautarzt, 1977, 28, 308—313.
ПОРОКЕРАТОЗ (porokeratosis)
Этиология и патогенез. Порокератоз рассматривается как наследст-
венное заболевание, передающееся аутосомно-доминантно. Reed и Leone
(1970) относят порокератоз Мибелли к группе эпителиальных невусов,
обусловленных патологическим клоном клеток, более чувствительных к
ультрафиолетовым лучам чем нормальные клетки эпидермиса. Тем са-
мым отмечается сходство с актиническим кератозом. По мнению Braun-
Falco и Balsa (1969), существенным моментом в патогенезе порокератоза
является нарушение процессов дифференцировки в ограниченных зонах
эпидермиса, ведущее к преждевременному и патологическому ороговению,
с образованием паракератотической пробки. По мнению М. С. Ихтейман
(1931), более подходящее название этого заболевания не порокератоз, а
keratosis centrifuga atrophicans, предложенный Respighi, описавшим этот
дерматоз в один год с Mibelli, в связи с тем, что патологические очаги не
ограничиваются устьями потовых желез, располагаются на слизистых.
Клиника. Высыпания представляют собой мелкие роговые папулы,
красновато-коричневатого или серого цвета, имеющие кратероформное
углубление в центре, заполненное роговой пробкой, с медленным эксцент-
рическим ростом и слиянием в округлые или полициклические фигурные
очаги поражения до нескольких см в диаметре. Центральная часть их вы-
392
Рис. 247. Порокератоз
Мибелли.
Рис. 248. Порокератоз
Мибелли линеарный-
глядит запавшей, часто слегка атрофичной, гипер - или депигментирована,
может быть с явлениями гиперкератоза, бородавчата (Chernosky, 1979,
Beer и Smith, 1984). В ней могут появляться новые папулезные элементы,
что приводит к образованию кокардовидных очагов поражения. Потоотде-
ление снижено или отсутствует. Периферическая зона приподнята в виде
рогового валика, на поверхности которого имеется продольная бороздка
(рис. 247, 248). Субъективные расстройства обычно отсутствуют, при диссе-
минированных формах может быть нерезко выраженный зуд. Локализация
— конечности, особенно стопы и кисти, лицо, шея, половые органы. Высы-
пания могут быть на слизистой полости рта, в виде округлых или полицик-
лических опалесцирующих бляшек, окруженных белесоватым валиком. В
редких случаях очаги могут располагаться на волосистой части головы,
вызывая рубцовую алопецию.
Высыпания обычно немногочисленны, за исключением случаев, опи-
санных как поверхностный диссеминированный актинический порокератоз
(Chernosky, 1967), когда высыпания множественны, располагаются на от-
крытых участках тела, меньшего размера и менее гиперкератотичны, чем
при обычной форме порокератоза. При типичной форме порокератоза Ми-
белли высыпания на подошвах и ладонях бывают нечасто, обычно в виде
гиперкератотических папул (Michail и Wertheimer, 1968).
Guss и соавт. (1971) описали porokeratosis plantaris, palmaris et dis-
393
seminata как особую форму заболевания. Клинически проявляется в виде
точечных гиперкератотических папул или небольших очагов (около 1 см
в диаметре) поражения, претерпевающих ту же динамику, что и типичный
порокератоз Мибелли, но в отличие от него, высыпания более многочислен-
ны, с наличием элементов не только на ладонях и подошвах, но и на дру-
гих участках тела, в том числе и на слизистых, без склонности располагать-
ся на открытых участках тела, как при поверхностном актиническом поро-
кератозе. В связи с этой публикацией Michail и Wertheimer (1972) указы-
вают на неоправданного» выделения описанной клинической картины как
особой единицы, так как она может укладываться в известные клинические
варианты, которых, по мнению авторов, 6: классический порокератоз (Ми-
белли); поверхностный диссеминированный эруптивный (Респиги); диссе-
минированный поверхностный актинический (Черноски и Фримаиа), ко-
торый, вероятно, является вариантом порокератоза Респиги; гиперкера-
тотический веррукозный порокератоз; гиперпластический и «forma minima»
Фрейнда. Деление на различные клинические формы, однако, в значитель-
ной мере условно, так как у одного и того же больного могут быть проявле-
ния, рассматриваемые как особые варианты. Так, на протяжении 69-лет-
ней продолжительности заболевания (Shatin, 1969) у больного 79 лет, не
было только веррукозных очагов вокруг и под ногтевыми пластинками,
на слизистых и алопеции, упоминаемых в литературе. Описан линейный
порокератоз, который по клинической картине напоминает линейный вер-
рукозный невус (Goldnes, 1971; Rahbari и соавт., 1974). Развивается пре-
имущественно в детском возрасте. Локализуется предпочтительно на ди-
стальных частях конечностей, где обнаруживаются мелкие кольцевидные,
веррукозные и гиперкератотические очаги, лихеноидные высыпания. В
зоне некоторых из них выявляется центральный участок атрофии и корич-
неватая пигментация. Schramm и Bork (1982) наблюдали линейный неви-
формный порокератоз, простиравшийся от области пупка на внутреннюю
поверхность правого4 бедра. Линейный унилатеральный порокератоз на-
блюдали Strani и соавт. (1983).
Описано сочетание линейного порокератоза и диссеминированного
поверхностного актинического порокератоза (Dover et al., 1986).
Порокератоз может развиться в любом возрасте, но чаще в детском,
считается, что реже у женщин, но все больные актиническим порокерато-
зом, описанные Каралицким Е. М. (1984), были женщины, из 8 больных,
наблюдавшихся Б. Т. Глухеньким и соавт. (1984)—5 было женщин. Тече-
ние длительное. У больных, описанных Reed и Leone (1970), очаги пора-
жения в большинстве существе вали от 3 месяцев до 15 лет, а у одного боль-
ного—в течение 53 лет. Возможно спонтанное исчезновение. Описано раз-
витие спиналиомы на фоне порокератоза Мибелли (Ehlers и Rothe, 1971),
в том числе множественных в сочетании с гломерулонефритом (Feu Ids и
Slater, 1983). Borowski и Strckowska (1983) описали больного с ладонно-
подошвенным и диссеминированным порокератозом, у которого после 40-
летнего существования дерматоза в зоне очагов развился множественный
спиноцеллюлярный рак. Это дало авторам основание рассматривать эту
394
форму порокератоза как предраковый дерматоз. Walther (1966) наблюдал
больного порокератозом Мибелли, который одновременно страдал болез-
нью Дауна, аномалиями глаз (птеригиум, эпикантус, кератоконус, ка-
таракта).
М. С. Ихтейман (1931) помимо одностороннего поражения кожи было
обнаружено spina bifida, низкий рост, замедление полового созревания,
что дало основание автору рассматривать порокератоз Мибелли как врож-
денную кожную дистрофию типа дискератоза. .
Гистопатология. В зоне рогового гребешка—резко отграниченный
акантоз и порокератотическое утолщение рогового слоя, отсутствие зер-
нистого слоя под ним, истончение мальпигиевою, в дерме—расширение
сосудов, периваскулярные лимфогистиоцитарные инфильтраты. При акти-
ническом, ладонно-подошвенном вариантах эти изменения менее выражены.
Дифференциальный диагноз. Порокератоз Мибелли необходимо от-
личать от эластоза перфорирующего серпигинирующего Мишера—Лютца,
эпидермальных невусов, веррукозного туберкулеза кожи, псориаза, крас-
ного плоского лишая, стойкого лентикулярного гиперкератоза Флегеля,
бородавок, базалиомы, солнечного кератоза, кольцевидной гранулемы,
себорейной экземы, дискоидной красной волчанки. -
Лечение. Кератолитические средства, внутриочаговое введение корти-
костероидов, криотерапия.
ЛИТЕРАТУРА
Глухенький Б. Т., Козий Л. М., Калюжная Л. Д. К диагностике порокератоза
Мибелли. Вести, дерматол. и венерол., 1984, 5, 45—48.
Каралицкий Е. М. Актинический порокератоз. Вести. Дерматол. и венеорол., 1984,
9, 59—61.
Beer W. Е., Smith N. Р. Hyperkeratotic porokeratosis (Mibelli) with psoriasis, res-
ponse to an aromatic retinoid.— Clinl exp. derm., 1984, 9, 5, 509—513.
Chernosky M. D. Porokeratosis. In: «Dermatology in general Medicine* Sd. T. Fitz-
patrick et al. Sec ed., 1979, Me Gran—Hill book company N—I, 279—282.
Dooer J. S., Miller J. A., Leoene G. M. Lcnear porokeratosis of Mibelli and DSAP.
Clin. exp. Dermatol., 1986, 11, 1, 79—83.
Foulds I. S„ Slater D. N. Porokeratosis of Mibelli and immune complex glomerulo-
nephritis.— Cliin. exp. Dermatol., 1983, 8, 1, 69—75.
Rahbari H., Cerdero A. A., Mehregan A. H. Lineal porokeratosis Arch. Dermatol.,
1974, 109, 4, 526—528.
Schramm P., Bork ,K. Naeviforme Poroceratosis—kein distinktes Krankheitsbild,
sondern mophologiscbe Variante der Porokeratosis Mibelf.— Zeit. Hautkr., 1982, 57, 13,
963—970.
Strani G. F., Sartoris S., Messineeo A. et al. Un caso di Porocheratosi di Mibelli. A
disposizione zoniforme. Chron. derm., 1983, XIV, 4, 525—527.
ПОРОМА ЭККРИННАЯ (eccrine poroma)
Этиология и патогенез. Порома эккринная является редко встречаю-
щейся доброкачественной опухолью кожи, гистологически связанной с
395
элементами внутриэпидермального отдела выводного протока эккринных
потовых желез (Pincus et al., 1956; Головин Д. И., 1958; Апатенко А. К.,
1968 и др.). Гистохимические и электронно-микроскопические ис-
следования позволили Lever и Hashimoto (1966) прийти к заключению, что
клетки эккринной поромы представляют собой незрелые эпидермальные
клетки, которые могут развиваться в направлении клеток выводных про-
токов эккринных потовых желез и образовывать просветы. Наличие в них
большого количества гликогена делает их сходными с эмбриональными
эккринными потовыми железами. По мнению Krinitz (1967), в развитии
опухоли может принимать участие и интрадермальная часть выводного
протока потовых желез.
Клиника. Небольших размеров, в среднем не более 1—2 см в диаметре,
опухоль, розоватого, темно-красного или красновато-коричневатого цвета,
от мягкой до плотно-эластической консистенции, иногда значительно воз-
вышающаяся над кожей. В зоне опухоли могут обнаруживаться теле-
ангиэктазии. Наблюдаются опухоли, расположенные на ножке. Опухоль
как правило одиночна, в редких случаях множественна, как это было в
описании Soldner (1970), когда у больной в области ладоней и подошв было
более 100 опухолей. Поверхность опухоли гладкая или слегка папилло-
матозна, гиперкератотична. Может быть дольчатой (Yasuda et al., 1964).
Безболезненна. Локализуется наиболее часто на подошвах, затем на ладо-
нях, реже на других участках кожного покрова—голове, шее, спине, груди,
предплечьях (Okun et al., 1963 и др.). Описано развитие выраженной
пигментации (Krinitz, 1967). Диагноз во всех случаях требует гистологи-
ческого подтверждения.
Заболевание развивается обычно в среднем и пожилом возрасте, оди-
наково часто у мужчин и женщин. 14 из 16 больных, наблюдавшихся А. К.
Апатенко (1968), были старше 40 лет. Только у 2 человек эккринная порома
развилась в 20 и 28 лет. Опухоль растет медленно, может изъязвляться,
что наблюдается обычно после травмы. Возможно злокачественное пере-
рождение, на что может указывать кровоточивость, боль, зуд.
Гистопатология. Разрастания клеток, сходных с кератиноцитами, но
меньших размеров, кубической формы, с округлым базофильным ядром, в
виде тяжей, проникающих в дерму, кистозные полости, наполненные гомо-
генным содержимым, лимфогистиоцитарная реакция стромы (Апатенко.
А. К-, 1966; German, 1964). Knox и Spiller (1958) обращают внимание на
повышенную васкуляризацию опухоли, что придает опухоли большое
сходство с пиогенной гранулемой. Характерным считается наличие гли-
когена в клетках опухоли, что подтверждено и электронно-микроскопиче-
скими данными (Lever и Hashimoto, 1966). Krinitz (1967) обнаружила у
2 из 4 больных явления гиалиноза вокруг сосудов в зоне опухоли. Pyly-
ser и соавт. (1983) выделяют 3 гистологических типа: доброкачественный,
предзлокачественный, или диспластический и инвазивный, злокачествен-
ный.
Darnall и Моррег (1966) обнаружили в опухоли на коже пальца пра-
вой стопы, клинически напоминающей больше пиогенную гранулему, но
396
сходную и с меланомой или спиналиомой, признаки эккринной поромы и
болезни Боуэна. Клинически и морфологически сходную картину описали
Winkelmann и Me Leod (1966), но с дермальной локализацией патологиче-
ского процесса (атипическая, дермальная порома).
Дифференциальный диагноз. Дифференцировать порому эккринную
необходимо от базалиомы, фибромы, кератоакантомы, меланомы, пиоген--
ной гранулемы, себорейных бородавок.
Лечение. Хирургическое.
ЛИТЕРАТУРА
Lever W.F., Hashimoto К- Die Histogenese einiger Hautanhangstumoren. — Hautarzt,
1976, 17, 4, 161—173.
Pylyser K-, De Wolf-Peters C., Marten К. The histology of eccrine poromas: a stiidy
of 14 cases.— Dermatologica, 1983, 167, 243—249.
ПОРФИРИЯ КОЖИ ПОЗДНЯЯ (porphyria cutanea tarda).
Этиология и патогенез. Поздняя порфирия кожи представляет собой
обычно ненаследственную форму порфириновой болезни, главным образом
вызванную латентным или манифестным повреждением печени с последую-
щим нарушением обмена порфиринов. Наиболее убедительным примером
генеза поздней порфирии такого типа являются порфирии от производст-
венных факторов (Bleiberg et al., 1964), а также при опухолях печени и осо-
бенно массовые случаи заболевания в Турции (porp'hyria turcica), вызван-
ные употреблением в пищу хлеба из пшеницы, обработанной фунгицидом
гексахлорбензолом. При обследовании, спустя 20 лет, 32 больных со сред-
ним возрастом 29 лет, Cripps и соавт. (1980) обнаружили разнообразную
патологию: гиперпигментации, гирсутизм, рубцы, нарушение роста, без-
болезненные артриты, увеличение содержания в сыворотке крови железа.
Уровень порфиринов в моче был повышен. Среди обычно встречающихся
экзогенных факторов, которые могут служить порфириногенами, по дан-
ным Ippen ((1967), основанным на анализе наблюдений 116 больных, наи-
большее значение имеют свинец, алкоголь, производственные химические
соединения, медикаменты (тяжелые металлы, мышьяк, противотубер-
кулезные препараты, седативные средства, эстрогены, железо и др.), раз-
личные хронические заболевания (малярия, туберкулез, сифилис и др.).
Чаще, по-видимому, имеет место комбинированное воздействие неблаго-
приятных факторов. Так, по наблюдениям Bielicky (1966), 73% больных,
соприкасавшихся со свинцом, были алкоголиками, а у 42%—был в анам-
незе сифилис. Все 13 больных, которых обследовали М. М. Левин и соавт.
(1973), злоупотребляли алкоголем, 6 человек из них имели длительный
контакт с нефтепродуктам!!, 4—с ядохимикатами, 1—со свинцом; у боль-
шинства работа была связана с длительным пребыванием под открытым
небом. Имеются сообщения о развитии поздней кожной порфирии у больных
почечной недостаточностью, леченных гемодиализом (Lichtenstein et al.,
1981), гепатомой (Keczkes и Barker, 1976), после длительного приема эстро-
генов (Becker, 1965; Barth и Kipping, 1971) или тетрациклина (Epstein
» 397
et al., 1976), антидиабетических сульфаниламидных препаратов (Malina
и Bartos, 1975), метотрексата (Malina et al., 1983) Порфириногенная ак-
тивность некоторых химических соединений и медикаментов (гексахло-
ран, барбитураты) подтверждается и экспериментальными исследованиями
(Панков Б. С., 1968, Смирнов В. С. и Колесников Ю. Г., 1974). К фак-
торам, которые могут способствовать развитию поздней порфирии кожи,
М. С.' Бидрат (1982) относит нарушения в иммунной системе, М. А. Штейн-
берг (I960)—дефицит витамина РР. По данным Thivolet (1979), клиниче-
ски очень сходное с поздней порфирией поражение развивается примерно
у 16% больных, леченных диализом. Предполагается, что в основе лежит
воздействие фотосенсибилизирующих веществ, не порфириногенных. При
иммуноморфологическом исследовании выявлено отложение иммуногло-
булинов в сосудах кожи и в области базальной мембраны. По данным ряда
авторов (Капралов И. К-, 1967; Welishler, 1972 и др.), для развития па-
тологии обмена порфиринов имеет значение повышенное содержание же-
леза, влияние которого может проявиться в повышении активности син-
тетазы аминолевуленовой кислоты и снижении активности уропорфири-
ноген-декарбоксилазы, играющих важную роль в биосинтезе порфири-
нов. Что касается роли наследственных факторов в развитии поздней пор-
фирий кожи, то, в отличие от других форм порфирии, она, по-видимому,
незначительна. Являются редкостью семейные случаи, незначительна ча-
стота поздней кожной порфирии среди родственников больных первой сте-
пени родства, что указывает на доминирующее значение в развитии ее сре-
довых факторов. Возможно и существует какая-то форма, которая обуслов-
лена генетическими факторами, наследуемыми аутосомно-доминантно, о
чем свидетельствует публикация некоторых родословных (Vibrans, 1973;
Goerz, 1979). Однако не исключено, что семейные случаи поздней порфи-
риновой болезни являются, по меньшей мере частично, вариантами porp-
hyria variegata, учитывая их возможное сходство (Tori et al., 1977, Bickers
et al., 1979). Вместе с тем, нельзя не учитывать данных о некотором сход-
стве нарушений порфиринового обмена у больных и их родственников
(Идельсон’Л. И., 1981, Magnin, 1982) и особенно того факта, что для раз-
вития эффекта повреждающих факторов в виде поздней порфирии кожи
важным является снижение активности уропорфириноген-декарбокси-
лазы в эритроцитах и печени (Kushner et al., 1976, Hammer и Golz, 1982).
Следовательно, речь может Идти о поздней порфириновой болезни как о
мультифактор^альном заболевании (Simon et al., 1978), с преимуществен-
ным значением средовой компоненты. Этот вопрос требует дальнейшего
изучения. По мнению Doutre и соавт. (1981), целесообразно разделение позд-
ней порфирии кожи на 2 формы: спорадическую, обусловленную экзоген-
ными токсическими факторами, при которой недостаточность уропорфи-
риноген-декарбоксилазы обнаруживается только в печени, и наследствен-
ную, аутосомно-доминантную, с дефектом этого фермента в эритроцитах.
Поданным Der Kaloustian и Kurban (1979), самая высокая частота кожной
порфирии в популяции Bantu в Южной Африке.
Клиника. Заболевание проявляется развитием после инсоляции или
398
Рис. 249. Поздняя кожная порфирия
(пойкилодермические и склеродермопо-
добные изменения).
Рис. 250. Поздняя кожная порфирия
(пойкилодермические и склеродермоподобные
изменения).
травмы пузырей на неизмененной или слегка покрасневшей и отечной коже
открытых участках тела, в первую очередь на лице, ушных раковинах,
тыле кистей. Они редко бывают крупными, обычно напряженные, покрышка
легко разрывается, содержимое серозное, в редких случаях серозно-гемор-
рагическое. Кожа легко ранима. В непосредственной близости к пузырям
может быть положительным симптом Никольского. Вскрывшиеся пузыри
быстро покрываются серозно-геморрагическими корочками, после оттор-
жения которых часто остаются поверхностные рубчики, милиумподобные
кисты; наличие участков гиперпигментации и участков витилигоподобной
депигментации придает коже больных пестрый вид (рис. 249, 250). В редких
случаях пигментация является доминирующим признаком (меланодерми-
ческая форма), может предшествовать развитию типичных клинических
проявлений (Мирзоева М. Г., 1964; Bielicky, 1966). При локализации
пузырей на волосистой части головы возможно очаговое облысение. У 6
из 42 больных, наблюдавшихся Р. Г. Акимочкиной (1965), имелись атрофи-
ческие изменения мягких тканей пальцев рук. Может наблюдаться ливид-
ное поражение кожи лица, изменения типа пеллагроидного дерматита,
хейлит, гипертрихоз, изменения ногтей, склеродермиформные поражения,
которые чаще ограничиваются кожей открытых участков тела (Bielicky,
1966), но могут быть диффузными. Развитие последних связывается с на-
рушением синтеза коллагена (Kint и De Weert, 1978). При этом обращается
399
внимание на сходство изменений коллагена при склеродермиформном типе
кожнбй порфирии и очаговой склеродермии (Para et al., 1979). В то же
время Redeker и Bronow (1962) у больных акросклерозом не нашли наруше-
ний порфиринового обмена за исключением 1 из 8, у которого предполага-
ется диагноз поздней кожной порфирии. К сходному выводу пришли Д. П.
Кочетов и Б. Н. Кривошеев (1976). По наблюдениям Е. М. Каралицкого
и Е Ф. Сербиной (1983), большинство больных склеровитилигинозной
формой поздней кожной порфирией—мужчины старше 50 лет, длительно
страдающие этим заболеванием, злоупотребляющие алкоголем, подвер-
гающиеся длительной инсоляции. Но вряд ли склеродермоподобные
изменения у больных порфирией следует считать склеродермией, сопутст-
вующей порфирии, как рекомендует М. Г. Мирзоева (1969). При длитель-
ном течении болезни появляются признаки преждевременного старения
кожи, в первую очередь повышенная складчатость ее, особенно на лбу.
При наличии комедонов, кистозных образований в области висков, у на-
ружного угла глаз клиническая картина может быть сходной с болезнью
Фавра—Ракушо. Описано (Гребенюк В. Н., 1977) сочетание склеродерми-
ческих и склеровитилигинозных проявлений поздней порфирии с пора-
жением глаз. Указывается на частое сочетание поздней порфириии с сахар-
ным диабетом. У больных сахарным диабетом, по мнению Malina и Bartos
(1975), порфирия выявляется с частотой, равной 0,21%. Bielicky (1966)
совместно с Berman обследовали 3000 больных диабетом, мужчин и женщин
поровну. Нарушение порфиринового обмена обнаружили у 14 мужчин, у
7 из которых были Ложные проявления. На основании этих данных реко-
мендуется обследовать больных диабетом, особенно мужчин, для выявле-1
ния скрытой порфирии. Выявляется разнообразная глазная патология
(Кузнецова Н. П. и Наделяева В. М., 1972; Berth et al., 1972): конъюнк-
тивиты, пигментации конъюнктивы, расширение сосудов конъюнктивы и
склеры, нарушение цветного зрения, изменение глазного дна, пузырьки
на роговице с последующим помутнением и др. Что касается сочетаний
поздней кожной порфирии и красной волчанки, то данные в этом отношении
неоднородны (Тумаркин Б. М., Головинов Э. Д., 1972; Cram et al., 1973;
Haneke и Schwartze, 1971; Clemmensen и Thomsen, 1982). Моча больных
окрашивается в розовый цвет, характерна красная флюоресценция в ультра-
фиолетовых лучах. В ней повышено содержание уропорфиринов (I и III),
а также, но в значительно меньшей степени, копропорфиринов (III). В
сыворотке крови повышено содержание железа, сочетающееся с недостаточ-
ностью трансферина (Epstein и Pinski, 1965). Указывается на диагностиче-
скую значимость у нелеченных больных повышенного содержания в сы-
воротке 7-глютамилтрансферазы (Adjarov и Ivanov, 1980).
Заболевание развивается преимущественно у мужчин среднего возра-
ста, чаще у злоупотребляющих'алкоголем, особенно работающих на откры-
том воздухе и связанных с нефтепродуктами. По данным, опубликован-
ным Н. П. Кузнецовой и соавт. (1981), более 1/3 больных—шоферы, трак-
тористы, автомеханики. В последние годы имеются сведения об учаще-
нии поздней кожной порфирии у женщин, что связывается с распрост-
400
ранением гормональных препаратов, в первую очередь как средств конт-
рацепции (Becker, 1965; Baer,. 1979; Fiedler et al., 1981). Если поздняя
порфирия развивается у женщин, по мнению Korting и Denk (1974), в пер-
вую очередь следует думать о возможности паранеопластической или ме-
дикаментозной порфирии. Нельзя забывать, естественно, и о таких факто-
рах, как профессиональные вредности, алкоголь. По данным Н. П. Куз-
нецовой (1968), поздняя кожная порфирия у женщин чаще, чем у мужчин,
протекает с общей симптоматикой. У детей поздняя кожная порфирия
бывает редко. Заболевание течет длительно, рецидивирует весной, про-
гноз зависит от степени повреждения печени, устранения факторов, выз-
вавших его. В. С. Смирнов (1981) отмечает высокую частоту развития ра-
ка печени у бальных поздней кожной порфирией. Из других осложнений
наблюдается гемохроматоз.
Гистопатология. Подэпидермальный пузырь (лишь старые пузыри
могут частично или полностью располагаться в эпидермисе); небольшая
преимущественно лимфоцитарная периваскулярная инфильтрация, мел-
кие эпидермальные кисты, может наблюдаться сенильный эластоз, исчез-
новение эластики. Определяется расширение, гомогенное утолщение сте-
нок сосудов, отложение в них PAS-положительных веществ, резистент-
ных к диастазе, выявляются с помощью реакции иммунофлюоресценции
иммуноглобулины (прежде всего IgG); электронно-микроскопически ре-
дупликация сосудистой базальной мембраны, вокруг сосудов массы нежно-
фибриллярного материала. Epstein и соавт. (1973) на основании этих данных
пришли к заключению, что первичные изменения развиваются в сосудах
капиллярного слоя дермы. /
Дифференциальный диагноз. С врожденным эпидермолизом, истинной
пузырчаткой, многоформной эксудативной эритемой, фотодерматитами,
световой оспой, пеллагрой, склеродермией.
Лечение. Запрещение лекарственных средств-порфириногенов, алко-
голя, диета, рекомендуемая для больных хроническим гепатитом, метио-
нин, пиридоксальфосфат, никотиновая кислота, унитиол, натрия бикар-
бонат. Имеются сообщения об эффективности малых доз антималярийцых
препаратов.
ЛИТЕРАТУРА
Гребенюк В. И. Поздняя кожная порфирия со склеродермическими и склеровитили-
гииозными проявлениями. Вестн. дерматол. и венерол., 1977, 11, 62—655.
Идельсон Л. И. Порфирии. М., 1981.
Каралицкий Е. М., Сербина Е. Ф. О склеровитилигинозной форме поздней кожной
порфирии. Вестн. дерматол. и венерол., 1983, 9, 42—45.
Кочетов Д. П., Кривошеев Б. Н. Склеродермия и поздняя кожная порфирия с
клиническими проявлениями склеродермии. Вестн. дерматол. и венерол. 1976, 1, 64—68.
Кузнецова Н. П., Панков Б. С., Чубарова А. С., Кривошеев Б. Н., Капралов И. К.
Порфирии. М. Медицина, 1981.
Baer R. L. Porphyrien: Newe klinische und therapeutische Aspekte In: «Fortschritte
26 Клиническая дерматология 401
der practischen Dermatologie und Venerologie,» Hrsg. O. Braun—Falco, H. H. Kolff, Sprin-
rer—Verlag, 1979, Bd. 9, 183—188.
Clemmensen O., Thomsen K- Porphyria cutanea trarda and systemic lupus erythema-
tosus.— Arch, derm., 1982, 118, 3, 150—162.
Cripps D. J., Gocmen A., Peters H. A. Porphyria tuuurcica.— Arch, dermatol., 1980,
116, 1, 46—50.
Dourte M. S., Beylot C., Bionlac P., Nordmann Y. Porphyrie cutanea dite tardive
de I’enfant. A propos d’un cas avec etude enzymatique Familiale.— Ann. dermatol., venerol.,
1981, 10, 751—757.
Epstein J. H., Tuffanelli D. L., Seibert J. S., Epstein W. L. Porphyria like cutane-
ous changes induced bei tetracycline hydrochloride photosensitizasion.— Arch, derm., 1976,
112, 5, 661—666.
Fiedler H., Zaumsell R. P., Wienmeister H. Porphyria cutanea tarda bei Frauen un-
ter besonderer Berucksichtigung der hormonellen Kontrazeption.— Derm. Mschr., 1981, 167,
8, 481—485.
Joerz G. Porphyria cutanea tarda In: Dermatologie in Klinik und Praxis. Hrsg. G W.
Korting, Stuttgart, 1979, Bd. 3, 3852—3860.
Hammer G., Goerz G. Pathogenetische Aspekte der Porphyria cutanea tarda.— Z. Hau-
utkrh., 1982, 57, 8, 605.
Kushner J. P., Barbuto A. J., Lee J. R. An inheritel enzyme defekt in porphyria
cutanea tar da:decreased uroporphyrinogen decarboxylase activity.— J. clin. invest, 1976, 5
8, 1089—1097.
Malina L., Chlumsky I., Chlumska A., Krtek V. Porphyria cutanea tarda (PCT) als
Folge der Methotrexat—und Buthioptirin—Therapie.— Z. Hautkrkrkh., 1983, 58, 4, 241—
243.
Parra C. A., Pizzi N. de Parra, Dimeter of the collagen fibrils in the sclerodermatosus
Parra C. A., Pizzi N. de Parra, Dimeter of the collagen fibrils in
skin porphyria cutanea tarda.— Brit. j. derm., 1979, 9, 5, 573—578.
Simon N., Hunyadi J., Szorenyi A., Dobozy A. Porphyria cutanea tarda—eine mul-
tifaktoriell vererbliche Erkrankung.— Hautarzt, 1978, 29, 7, 378—382.
Thioolet J. Pseudporphyria cutanea tarda bei Hamodlalysepatiemten.— Hautarzt»
Thioolet -J. Pseudporphyria cutanea tarda bei Hamodialysepatiemten.— Hautarzt,
1979, 30, 3, 154, 157.
. Tori I., D. Alessandro Landolfo. Inheritance of porphyria cutanea tarda.— Bnt. J.
Derm., 1977, 97, 6, 617—627. \
ПОРФИРИЯ ВРОЖДЕННАЯ ЭРИТРОПОЭТИЧЕСКАЯ (porphyria eiyfli-
ropoetica congenita)
Этиология и патогенез. Редкое аутосомно:рецессивное заболевание. У
больных обнаруживается повышение активности синтазы 8-аминолевулено-
вой кислоты и снижение активности уропорфириноген а III косинтазы. Уро-
порфирин I накапливается в костном мозге, в эритроцитах, плазме, в коже,
зубах, что приводит к развитию гемолитической анемии и фотосенсибилиза-
цни кожи, эритродонтии. В моче обнаруживают большое количество уро-
порфирина I, в кале—копропорфирина I. Определение косинтазы уро-
порфириногена III в эритроцитах применяется для. выявления гетерози-
402
Рис. 251. Эритропоэтическая порфирия у двух братьев.
Рис. 252. Эритропоэтическая Рис. 253. Эритропоэтическая
порфирия—поражение кистей •' порфирия—поражение кистей
у тех же больных. у тех же больных.
готных носителей гена. Среди механизмов, обусловливающих кожные из-
менения, предполагается участие лизосомальных гидролитических фермен-
тов, высвобождающихся под действием света биологически активных ве-
ществ, таких, как гистамин или гистаминоподобные вещества, брадикинин
(цит. по Gellis и Feingold, 1975; Der Kaloustian и Kurban, 1979).
Клиника. Основными кожными симптомами являются фоточувствн-
тельная эритема, повышенная ранимость кожи,, пузырьково-пузырные
26* 403
высыпания, мастью геморрагические, на открытых участках тела, вскры-
вающиеся с образованием покрытых корками, трудно заживающих, легко
инфицирующихся эрозий и язв. Кожа приобретает коричневатую окраску,
как при загаре, на этом фоне обнаруживаются очаги атрофии, депигмента-
ций, телеангиэктазии, милиумподобные кисты. За счет рубцовых измене-
ний’ склерозирования могут быть дерматогенные контрактуры. Наблюда-
ются линейные рубцы вокруг рта, мутилирующие деформации пальцев,
носа глаз. Выворот век, конъюнктивит, кератит, возможно развитие сле-
поты. Волосы и ногти дистрофичны. Может быть гипертрихоз, на открытых
участках тела илй генерализованный (Maleville et al., 1982), иногда очаги
рубцовой алопеции на голове, анонихия. Зубы темнеют, приобретают крас-
новатый оттенок, особенно отчетливый в ультрафиолетовых лучах. Раз-
витие склеродермоподобных изменений на лице делает его маскообразным.
Повышена чувствительность к свету: ожоговые реакции даже при неболь-
шой экспозиции (рис. 251, 252, 253). Для диагностики важное значение
имеет определение повышенного содержания уропорфирина I в моче, эрит-
роцитах и плазме, копропорфирина—в кале.
Развивается обычно в первые 5 лет жизни, часто в первые недели или
месяцы с примерно равной частотой у мальчиков и девочек. Описано и
позднее (в 15-летнем возрасте) начало заболевания (Duterque et al., 1983).
Протекает тяжело. У больных выявляются гемолитическая анемий, сплено-
мегалия, повышена частота переломов костей. Ухудшение наблюдается
летом. Прогноз неблагоприятный. Больные плохо растут, подвержены
инфекции, обычно погибают в молодом возрасте. Первые признаки забо-
левания могут быть уже при рождении в виде красного окрашивания пеле-
нок из-за наличия в моче большого количества порфиринов.
На заседании Московского научного общества дермато-венерологов
им. А. И. Поспелова (июнь 1984) старший научный сотрудник ЦКВИ М3
СССР Г. И. Суколшгпредставил двух братьев, страдающих врожденной эри-
тропоэтической порфирией. Заболевание у них развилось на первом году
жизни и с течением времени привело к резко выраженным деформациям в
области головы, кистей и стоп. Наступило почти полное рассасывание кон-
цевых фаланг, хрящевой части носа, в значительной мере—ушных раковин.
Обращало на себя внимание резкое повышение содержания в моче уро-
порфиринов (о&оло 1000 мг/сутки).
Гистопатология. Субэпидермальные пузыри, небольшая воспалитель-
ная реакция в дерме.
Дифференциальный диагноз. Дифференцировать необходимо от фото-
дерматозов, световой оспы, буллезного эпидермолиза, пигментной ксеро-
дермы.
Лечение. Спленэктомия, каротиноиды, переливание эритроцитарной
массы, фотозащитные средства.
ЛИТЕРАТУРА
Duterque М., Cioatte J., Jeanmougin М., et at. Porphyrie Erythropoietique de Gun-
ter de Revelation tardive. Ann. Dermatol. Venereoll., 1983, 110, 709—710.
404
Gallis S. S., Feingpld M. Congenital erythropoetic porphyria. Am. J. Dis. Child.,
1975, 129, 6, 702.
Idriss Z. H., Nadj jar S. S., Der Kaloustjan V. M. et al. Congenital erythropoetic
porphyria. Am. J. Dis. Child., 1975, 129, 701—702
Kaloustian Der V. M., Kurban A. K- Genetic diseases of the skin. Springer—Verlag,
Berlin—Heidelberg—N. Y., 1979
Maleoille J., Babin J—P., Mallard Z. et al. Porphyrie erythropoie—tique congeni-
tale ue Gunter et carotinoidesu. Ann. Dermatol. Venereol., 1982, 109, 883—887.
КРОВАВЫЙ ПОТ (haematidrosis)
Синоним: синдром кровавого пота.
Давно известная, но очень редкая патология. К 1980 году опублико-
вано не более 20 наблюдений. Упоминание о кровавом поте имеется в
евангелии от Луки (80—90 гг. и. э.), где сказано, что Иисус страдает так
жестоко, что вместо пота у него стекают капли крови.
Этиология и патогенез. Кровавый пот образуется благодаря приме-
шиванию крови путем эритродиаледеза из капилляров, окружающих по-
товые железы. Изменения стенок сосудов, приводящие к повышению их
проницаемости, могут быть вызваны диэнцефальными нарушениями, раз-
вившимися под воздействием инфекции, физических и психических травм
и др. (Сперанский А. Д., 1936; Вайсфельд И. Л. и Кассиль Г. Н., 1956;
Гращенков Н. И., 1956; Роговер А. Б. и Вейн А. М., 1957).
Имеются сообщения о появлении кровавого пота у лиц, страдающих
истерией, тяжелыми психическими заболеваниями. Bianchi (1926) сообщает
о больной 28 лет с классической картиной истерии, у которой во время мо-
литвы появлялся кровавый пот, выделение которого усиливалось под влия-
нием религиозного экстаза. Vincento (1926) опубликовал случай кровавого
пота у религиозных стигматов; кровь выделялась у больной каплями из
нескольких мест лба, количество ее увеличивалось в период экстаза.
Г. К. Лавский (1932) описал больную девушку 24 лет с наличием ши-
зофренических, истерических и психастенических реакций. Период при-
ступа истерии сопровождался выделением кровяного пота на носу, левом
предплечье, в области ногтевых фаланг, а также на левой ноге вблизи на-
ружного мыщелка. На месте выделения кровянистой липкой жидкости
нарушения целости кожи не обнаружено, что указывает на выделение кро-
ви путем эритродиапедеза.
А. Б..Роговер и А. М. Вейн (1957) наблюдали больную 11 лет с кро-
вавым потом. В возрасте 8 лет у больной через несколько дней после тя-
желой психической травмы появился под глазами кровавый пот. В даль-
нейшем неоднократно появлялась кровь на лице в виде мазков на лбу,
веках, щеках, носу. Иногда кровь стекала каплями. Кожа всегда остава-
лась неповрежденной. Несколько раз возникали кровотечения из носа.
Мы наблюдали женщину 28 лет, у которой во время обострения мани-
акально-депрессивного психоза появлялся кровавый пот в подмышечных
и пахово-бедренных областях.
Имеются указания на возможность носового кровотечения, кровавого
405
пота, повышения температуры тела под влиянием внушения. Приведен-
ные факты говорят о том, что резкие, часто повторяющиеся эмоциональ-
ные напряжения могут приводить к усиленной продукции нейрогормонов,
вызывающих вазодилатацию и повышенную сосудистую проницаемость,
тем самым обусловливать потоотделение с примесью крови.
Клиника. Кровавый пот чаще наблюдается в области лица. Он может
выделяться каплями на нескольких участках лба, носа. Одновременно
может появляться и в других местах (предплечья, ногтевые фаланги,
ноги). Нарушения целости кожи в этих участках нет. При сильных психо-
эмоциональных напряжениях (например, во время религиозного экстаза)
количество кровавого пота увеличивается.
Дифференциальный диагноз. Следует проводить с окрашенным по-
том.
Лечение. Гипноз, психотерапия.
ЛИТЕРАТУРА
Вайсфельд И. Л., Кассиль Г. Н. Функциональное состояние капилляров кожи при
некоторых заболеваниях нервной системы.— Доклады АН СССР, 1956, 109, 5, 1057—1060.
Косидожкий 3. Сказания евангелистов.—Пер. с польск.— М. 5 Политиздат, 1979.
Роговер А. Б., Вейн А. М. Синдром «кровавого пота» н его патогенез.— Жури,
невропатол. и психиатрии, 1957, 57, 7, 849—851.
ПСЕВДО КСАНТОМА ЭЛАСТИЧЕСКАЯ (pseudoxanthoma elasticum)
Синонимы: эластическая ксантома Бальзера, эластический невус
Гутмана, системный эласторексис Турена.
Впервые заболевание было описано Balzer в 1884 г. под названием
эластической ксантомы. Изучив гистологическую картину пораженной
кожи, Darier в 1896 г. дал этому заболеванию название эластической псев-
доксантомы. В 1889 г. Doyne описал поражение глазного дна, которому в
1892 г. Knapp дал название ангиоидные полосы (цит. по Danielsen et al.,
1970). В 1929 г. Groenblad и Strandberg сообщили о случаях сочетания псев-
доксантомы с ангиоцдными полосами в сетчатке глаз (болезнь пигментных
полос сетчатки). Впоследствии такое сочетание офтальмологи стали назы-
вать синдромом Греиблада—Страндберга. Однако наши наблюдения и
данные других авторов свидетельствуют о том, что развернутая картина
эластической псевдоксантомы не укладывается в рамки этого синдрома,
так как помимо кожи и глаз в процесс вовлекаются и внутренние органы,
т. е. речь идет о системном поражении эластической ткани.
Этиология и патогенез. Большинство авторов считает эластическую
псевдоксантому врожденным системным заболеванием, клиническому вы-
явлению которого могут способствовать некоторые эндогенные влияния
(беременность, эндокринные нарушения, диабет, половое созревание), а
также травмы, различные инфекционные болезни и др. Л. И. Степанов и
соавт. (1980) указывают на связь заболевания с токсоплазмозом. К настоя-
щему времени утвердилось мнение о наследственной передаче эластической
псевдоксантомы по аутосомно-рецессивному или аутосомно-доминантному
406
типу (Popl, 1974; Harvey et al., 1975). Довольно часто заболевание носит
семейный характер (Pautrier и Renard, 1947; Worikger и Gerhardt, 1957;
Gadehold, 1960).
Клиника. Женщины, по-вцдимому, болеют несколько чаще мужчин.
Заболевание развивается преимущественно в возрасте до 20 лет, нередко
у детей. Ф. А. Зверькова (1979) наблюдала эластическую псевдоксантому
у детей в возрасте от 3 до 12 лет.
Клиническая картина эластической псевдоксантомы складывается
из поражения кожи, глаз и внутренних органов. Изменение кожи харак-
теризуется образованием ксантомоподобных, желтоватого цвета или цвета
слоновой кости, слегка возвышающихся плоских узелковых элементов,
округлых или овальных очертаний, величиной от просяного зерна до че-
чевицы. Они часто располагаются линейно или же сливаются в ограничен-
ные и диффузные бляшки. Тесно расположенные папулы как бы вкраплены
в кожу, создавая сетчатость рисунка. Пораженная кожа лимонно-желтого
цвета, несколько утолщена и одновременно с этим дряблая, легко со-
бирается в складки. Высыпания обычно располагаются симметрично на
коже шеи, чаще на боковых ее поверхностях, сгибательных поверхностях
верхних и нижних конечностей, в подмышечных впадинах, в области локте-
вых, подколенных сгибов, паховых складок; реже—йа других участках
туловища. Почти всегда остаются свободными лицо, ладони и подошвы.
Узелки желтоватого цвета могут наблюдаться на слизистой рта, чаще на
мягком небе, нижней губе, а также на слизистой влагалища и прямой кишки.
Поверхность языка иногда имеет вид географической карты. Субъектив-
ные явления отсутствуют. Поражения глаз, так называемые ангиоидные
полосы в сетчатке, встречаются у 25—80% больных эластической псевдо-
ксантомой (Архангельский В. Н., 1960; Головченко Д. и соавт., 1984;
Benedict и Montgomery, 1935; Krill и Bertnol, 1973; Hogan и Heaton, 1973;
Miyoshi et al., 1980; Sing et al., 1984 и др.).
В течении процесса на глазном дне окулистами отмечается 3 стадии
по Вивальди: 1—бессимптомная, когда на глазном дне в области эласти-
ческой мембраны сетчатки (мембрана Бруха) обнаруживаются ангиоидные
полосы; II стадия характеризуется резким падением зрения, вследствие
геморрагий сетчатки; в III стадии происходит рубцовое перерождение оча-
га, развитие скотомы и необратимой потери зрения. Ангиоидные полосы,
по мнению большинства офтальмологов, представляют собой трещины и
надрывы в эластичном слое мембраны Бруха (Елисеев Э. Г. и соавт., 1978
и др.). Гипертония, стенокардия, перемежающаяся хромота, желудочно-
кишечные кровотечения и др. являются следствием поражений сосудистой
системы.
Течение болезни хроническое; начавшись в молодом возрасте, оно тя-
нется десятки лет.
Гистопатология. В глубоких слоях дермы и в подкожной клетчатке
отмечается увеличение эластической ткани, имеются ограниченные очаги
дегенерации и распада, эластические волокна скручены, разорваны, места-
^ми утолщены в виде комков (эласторексис или эластоклазис Дарье); резко
407
изменяется их способность окрашиваться—они становятся базофильными.
Эпидермис не изменен или на некоторых участках отмечается умеренный
акантоз. Электронно-микроскопические исследования кожи и слизистых
оболочек полости рта указывают на первичный характер поражения эла-
стических волокон (Danielson и Kobayasi, 1974). Установлено, что эласти-
ческая ткань изменена не только на участках кожного поражения, но и на
клинически здоровых участках кожи, а также в стенках кровеносных со-
судов. В других пораженных органах обнаруживаются дегенерация эла-
стической ткани и кальцификация периферических артерий. Микроскопиче-
ские и ультраструктурные эндомиокардиальные исследования (Przybojew-
ski et al., 1981) при помощи кардиальной катетеризации со взятием биоп-
сии показали, что причиной сердечных жалоб была вторичная диффузная
артериопатия, связанная с гиперэластозом, поражающим мелкие интра-
муральные коронарные сосуды.
Лечение. Ликвидация фокальной инфекции, лечение эндокринных
расстройств. Длительное применение витаминов Е и А, аскорбиновой кис-
лоты, рутина. При наличии глазных симптомов—фотокоагуляция (Offret
и соавт., 1970), которая приостанавливает процесс путем рубцевания со-
судоподобных трещин мембраны Бруха. Аргонлазеркоагуляция (Елисеева
Э. Г. и соавт.! 1978).
ЛИТЕРАТУРА
Головченко Д. Д. и соавт. О синдроме Гренблад—Страндберга. — Вести, дерматол.
и венерол., 1984, 5, 52—54.
Елисеева Э. Г., Димитрова В. Г., Подгорная Н. Н., Переверзина О.'К. Флюорес-
центная ангиография и лазерокоагуляция при синдроме Гренблад—Страндберга.— Вести,
офтальмологии, 1978, 3, 30—35.
Зверькова Ф. А. Об эластической псевдоксантоме у детей.— Вести, дерматол. и
венерол., 1979, 10, 39—42.
Степанов Л. И., Маслюк Н. И., Есенина К. П. Эластическая псевдоксантома
Дарье.— Вести, дерматол. и венерол., 1980, 9, 48—50.
Danielsen L., Kobayasi Т. Pseudoxanthoma elasticum.— Acta Dermatovener., 1974,
54, 121—127.
MiyoshiN., NishiT., NonakaS. Two cases of pseudoxanthoma elasticum.— Nishini-
hon J. Dermatol., 1980, 32, 5, 811—818.
Przybojewski J., Moritz F., Tiedi F., Van der Walt J. Pseudoxanthoma elasticum
with cardiac involvement. A case report and review of the literature.— South Afr. Med. J.,
1981, 59, 8, 268—275.
Sing R„ Kumar B., Kaur S. Pseudoxanthoma elasticum. A report of six cases.— J.
Ass Physic. India, 1982, 30, 11, 845—847.
ПСЕВДОСАРКОМА КАПОШИ (pseudosarcoma Kaposi).
Синонимы: псевдоангиосаркоматоз Капоши, акроангйодерматит.
Псевдосаркома Капоши—поражение кожи преимущественно нижних
конечностей, клинически чрезвычайно напоминающее истинную болезнь
Капоши.
408
Этиология и патогенез. В настоящее время различают два варианта
псевдосаркомы Капоши: 1. особая форма ангиодермита, вызванная хро-
нической венозной недостаточностью и описанная в 1965 году Mali и соавт.
Позже этому вопросу посвятили свои работы также Laugier (1977), Rosen
и Martin (1979); 2. Врожденная артериовенозная недостаточность с мно-
жественными артериовенозными фистулами (Каламкарян А. А. и соавт.,
1982; Bruefarb и Adams, 1967; Stewart, 1967; Waterson et al., 1969; Ear-
hart et al., 1974; Amon, 1975; Allesi et al., 1975; Laugier, 1977; Bedbur
et al., 1983 и др.). Псевдосаркома Капоши—это отдельная нозологическая
форма, являющаяся результатом порока эмбрионального развития со-
судистой системы. Своевременное выявление ложного ангиосаркоматоза
Капоши (Stewart, 1967) имеет особую практическую значимость, так как
избавляет больных от терапии цитостатиками и рентгеновскими лучами.
Клиника. Псевдосарксма Капоши типа Мали характеризуется появле-
нием обычно у мужчин в возрасте 40—50 лет на фоне хронической веноз-
ной недостаточности множественных пурпурозного цвета пятен и крупных
бляшек синюшно-коричневого цвета на коже голеней и стоп. Очаги пораже-
ния размером до ладони представляют собой плоские инфильтраты слегка
приподнятые, которые отличаются выраженной болезненностью, плохо
поддаются терапии, часто изъязвляются. Часто у больных наблюдается
сетчатая пигментация кожи в области лодыжек, распространяющаяся на
голень и пальцы. Здесь же у ряда больных имеются мелкие очаги атрофии.
Для второго типа псевдосаркомы Капоши (Bluefarb—Stewart, 1967) ха-
рактерны кожные проявления, клинически схожие с болезнью Капоши,
множественные врожденные артерио-
венозные фистулы, строго односто-
ронние кожные и сосудистые прояв-
ления. Заболевание, как правило,
проявляется в молодом возрасте
(20—30 лет), в то время как саркома
Капоши обычно развивается у пожи-
лых людей. На тыльных поверхностях
1, 2 и 3 пальцев стопы, в области
щиколоток и нижней половины одной
из голеней появляются пятнистые,
узелковые и бляшечные высыпания
с четкими границами, синюшно-ко-
ричневого и ливидного цвета. Очаги
поражения варьируют от мелких
пятен на большом пальце стопы до
бляшки величиной с ладонь в облас-
ти задней поверхности щиколотки и
голени с явлениями центрального
изъязвления (рис. 254, 255, 256, 257,
258). Иногда эти поражения возвы-
шаются над поверхностью здоровой
Рис 254 Псевдосаркома
Капоим (тип Маля).
409
Рис. 255. Псевдосаркома Рис. 25С. Псевдосаркома
Капоши (тип Мали). Капоши (тип Мали).
Рис. 257. Псевдосаркома
Капоши.
Рис. 258. Псевдосаркома
Капоши (тип Блюфарба—Стюарта).
кожи. Могут появляться папилломатозные разрастания. Довольно часто
отмечается отек щиколоток или даже всей нижней конечности. Нередко
псевдоангиосаркоматоз сочетается с варикозиь’м расширением вен, увели-
чением объема и удлинением всей конечности. Больные жалуются на рез-
кую болезненность. При артерио- и флебографии выявляется врожденная
410
ангиодисплазия с множественными мелкими артериовенозными фистула-
ми (шунтами). Артериовенозные фистулы могут рассматриваться как со-
судистая патология, при которой имеет место прямая коммуникация меж-
ду артериями и венами без посредничества капиллярного русла. Эта
патология может иметь травматический или чаще врожденный характер.
Как правило, они унилатеральные, множественные и более часто отме-
чаются на нижних, чем на верхних конечностях.
Гистопатология. Изменения, схожие с саркомой Капоши: пролифера-
ция мелких сосудов, фибробластов, экстравазаты эритроцитов.
Дифференциальный диагноз. Проводят с истинной саркомой Капоши,
красным плоским лишаем, ангиокератомой. Показано применение под-
кожной артериографии, при которой определяется сосудистая патология
врожденного характера (артериовенозные фистулы).
Лечение. При псевдосаркоме (тип Mali) лечение такое же, как и при хро-
нический венозной недостаточности. При псевдосаркоме Капоши (тип Ste-
wart—Bluefarb) множественные артериовенозные фистулы лучше перво-
начально лечить консервативно-тугим бинтованием. При недостаточной
эффективности ц в случаях мучительных болей и множественных некро-.
зов лечение хирургическое—ушивание артериовенозных фистул или ам-
путация конечности.
ЛИТЕРАТУРА
I Каламкарян А. А., Суколин Г. И., Мордовцев В. Н. Псевдосаркома Капоши:
особеннрсти клинических проявлений, взаимосвязь с врожденными сосудистыми диспла-
зиями.— Вестн. дерматол. и венерол., 1983, 1, 10—14.
А топ R. Arteriovenous malformation resemblung Kaposi sarcoma.— Arch. Derma-
tol., 1975, 3, 12, 1656—1558.
Bedbur M., Loose D., Hofmann N., GoerzG. Pseudo—Kaposi—Serkoma. Z. Hautkr.,
1983, 58, 11, 834—842.
Laugier P. Fausse angiosarcomatose de Kaposi.— Arch. 1'Uhion Med. Balk., 1977,
15, 3—4, 445—446.
Rosen Th., Martin S. Radionuclide scanning in pseudo—Kaposfa sarcoma.— Arch.
Dermatol., 1979, 115, 6, 747—748.
ПСОРИАЗ ВЕРРУКОЗНЫЙ (psoriasis verrucosa)
Этиология и патогенез. Причины развития этой очень редкой формы
псориаза неясны. Ф. В. Гинце н Я- А. Мериин (1929), Д. Г. Дудина
(1948), Г. И. Уманский и В. П. Малевич (1959) полагали, что в основе,
видимо, лежит изменение реактивности организма под влиянием различ-
ных внешних факторов (мышьяк, пиококковая инфекция и др.). М. М.
Кузнец (1948) наблюдал у 6 из 190 военнослужащих, страдающих псориа-
зом, бородавчатый псориаз и объясняет такую высокую частоту его раз-
вития длительным охлаждением, повышенной влажностью и другими не-
благоприятными фронтовыми условиями. Из описанных нами (Трофимо-
ва Л. Я. и Мордовцев В. Н., 1975) 7 больных мы отметили только в одном
случае длительный прием мышьяка, но обратили внимание на возможность
411
развития веррукозных элементов под влиянием нерациональной наружной
терапии. Rotchford и Hyman (1961) описали гиперкератотический, частью
веррукозный псориаз у девочки, страдающей болезнью Дауна.
Клиника. Анализ собственных наблюдений и данных литературы поз-
воляет выделить 2 формы веррукозного псориаза. Одна из них, развиваю-
щаяся на фоне обычной формы псориаза, не вызывает особых диагностиче-
ских трудностей, принадлежность же второй, проявляющейся обычно в
виде линейных веррукозных изменений, в комбинации с типичными псо-
риатическими высыпаниями или без них, к псориазу или к линейному не-
вусу с воспалением остается неясной. В связи с этим мы рассмотрим эти
варианты раздельно. В первом случае в зоне некоторых из типичных высы-
паний псориаза обнаруживается более значительная, чем обычно, инфильт-
рация, очаговые веррукозные разрастания с отчетливым эксудативным
компонентом, покрытые массивными наслоениями чешуек и корок. Обычно
они располагаются на конечностях, чаще на ногах. Развивается преиму-
щественно у мужчин. Особенностью является значительная резистент-
ность к лечению (рис. 259, 260, 261). При гистологическом исследовании
выявляется картина, типичная для псориаза. При втором варианте обычно
имеются односторонние веррукозные очаги поражения, резко очерченные,
значительно возвышающиеся над окружающей кожей, красноватого цвета,
покрытые наслоениями множественных чешуек, а иногда и корок, с отчет-
ливой тенденцией к линейному расположению. Наряду с этим часто обна-
руживаются типичные псориатические высыпания, обычно более много-
численные на той стороне, где имеются веррукозные высыпания. Они мо-
гут появляться одновременно, но чаще раньше, иногда значительно, воз-
никают бородавчатые проявления. Так, у наблюдавшегося Bennet и соавт.
(1973) больного распространенный эпидермальный невус существовал с
детства, а типичный псориаз развился в 63-летнем возрасте, в том числе
веррукозного характера в зоне эпидермального невуса н прилегающих уча-
стков кожи. Авторы высказали предположение, что псориазиформные вос-
палительные изменения в области эпидермального линейного невуса, от-
сутствие которых до развития типичного псориаза было доказано гисто-.
логическим исследованием, представляют собой феномен Кебнера, а сам
эпидермальный невус является псориазогенным очагом у лиц с предрас-
положением к псориазу. К сходному выводу ранее пришли Sugai и соавт.
(1970). На возможность обнаружения в зоне некоторых эпидермальных
невусов гистологической картины псориаза обратил внимание в 1896 году
Унна. Для эпидермальных невусов, протекающих с воспалительными из-
менениями, Altman и Mehregan (1971) предложили название «inflammatory
linear verrucose epidermal nevus». По их мнению, в большинстве случаев
линейный псориаз представляет собой клинически и гистологически ва-
риант этого невуса. Ни у одного из 25 наблюдавшихся ими больных не было
типичных псориатических высыпаний на других участках кожного покрова.
Изменения были сходны с наблюдаемыми при спориазе или нейродермите.
Не обнаружили псориаза ни у больных с воспалительным линейным эпи-
дермальным невусом, ни у их родственников Thormann и соавт. (1978).
412
Рис. 259. Веррукозный
псориаз.
Рис. 260. Веррукозный
псориаз.
Рис. 261. Веррукозный
псориаз.
Против причинной связи с псориазом подобного типа изменений высказы-
ваются Haustein и Suss (1978). Они считают типичными для линейного во-
спалительного веррукозного эпидермального невуса следующие клинико-
морфологические признаки: раннее начало, поражение чаще женщин, ло-
кализация на левой ноге, зуд, воспалительная псориазиформная гистоло-
гическая картина, резистентность к лечению. Hodge и соавт. (1978) склонны
413
рассматривать это заболевание как особую единицу, а именно псориази-
формный линейный невус, а не псориаз, так как, по их данным, гистологи-
ческая картина, хотя и псориазиформная, но не типична для псориаза.
Dupre (1977) считает это заболевание особой нозологической единицей со
специфическим нарушением кератинизации и формированием мозаических
структур в виде чередования участков гипер- и паракератотического утол-
щения рогового слоя и соответственно увеличением или отсутствием зер-
нистого слоя.
Большинство же авторов, описывающих псориазиформные эпидермаль-
ные невусы, отмечают, как это упоминалось уже выше, что рано или позд-
но у этих больных развивается типичный псориаз. Иллюстрацией этому
может служить, в частности, и наблюдение Gethner (1961), обнаружившего
типичную картину как в очаге линейного псориазиформного эпидермального
невуса, так и в развившихся позже изменениях обычной характеристики.
Geerts и соавт. (1976) наблюдали больного с линейным веррукозным не-
вусом с воспалением, развившемся в 18-летнем возрасте после нервно-пси-
хической травмы. Отец и его дядя страдали псориазом. Помимо линейно
расположенных веррукозных очагов на правой половине тела, имелись
высыпания на лице и волосистой части головы. Гистологически обнаруже-
ны спонгиоформные пустулы. В целом морфологическая картина была
очень похожа на псориаз, что дало основание авторам заключить, что гра-
ницу между псориазом и линейным воспалительным эпидермальным не-
вусом провести сложно.
Ogino и Higuchi (1976) наблюдали девочку 9 лет, страдавшую пусту-
лезным генерализованным псориазом с 6 лет. В 9-летнем возрасте было
впервые замечено наличие унилатеральных линейных веррукозных измене-
ний с воспалением, гистологически сходных с псориазом. Под влиянием
лечения метотрексатом пустулезные Высыпания регрессировали, равным
образом как и пустулы, появлявшиеся в зоне линейных очагов поражения,
последние же оставались без изменения.
Не исключено, что противоречие во взглядах на возможность развития
линейного веррукозного псориаза объясняется тем, что основное внимание
было уделено морфологическим проявлениям, которые могут быть сходны,
несмотря на существенные различия в причине, их вызвавшей. По нашему
мнению, воспалительные линейные веррукозные изменения могут прини-
мать ту или иную морфологическую картину в зависимости от предраспо-
ложенности, в том числе и к псориазу. Этим еще раз доказывается, что псо-
риаз с точки зрения патогенеза (структуры предрасположенности) явля-
ется неоднородным заболеванием. Линейность же распределения высыпа-
ний во многом обусловлена неврологическими нарушениями центрального
или сегментарного характера (Каламкарян А. А. и соавт., 1980). Возмож-
но, что этим объясняется и своеобразие клинических проявлений, которые
могут быть одними из фенотипических проявлений псориатического гено-
типа.
Дифференциальный диагноз., Дифференцировать веррукозный псориаз
414
/
необходимо от бородавчатого невуса, красного плоского лишая, нейро-
дермита, вегетирующего дискератоза Дарье.
Лечение. Псориазин, антипсориатикум, витамин В12 в больших дозах
(500—1000), при резком прогрессировании и опасности развития злока-
чественных новообразований—иссечение, рентгеновские лучи.
ЛИТЕРАТУРА
Каламкарян А. Л., Фролов Е. П., Акимов В. Г., Красников Ю. А. О так называемых
унилатеральных и линейных Дерматозах. — Вести, дерматол. и венерол. 1980,10,18—23.
Dupre A. Inflammatory linear verrucose epidermal nevus.— Arch, dermatol., 1977,
113, 6, 767—769.
Geerts M. L., KM A., De Vleeschonwer L. Inflammatory verrucose epidermal naevus
or linear psoriasis.— Dermatologica, 1976, 153, 5, 284—289.
Haustein U.—F.,, Suss E. Inflammatoriecher linearer verrukoser epildermaler Navus.—
Derm. Machr., 1978, 164, 2, 120—129.
Hodge S. J., Barr J. M., Owen L. G. Inflammatory linear verrucose epidermal ne-
vus.’—Arch. dermatol., 1978, 114, 3, 436—438.
Ogino A., Higuchi T. Generalized pustular psoriasis and pustulisation on the linear
epidermal nevus.— Dermatologica, 1976, 152, 6, 376—384.
Thormann Th., Sonnichsen N., Klug H. Zabel R. Klinische und morphologische Un-
tersuchungen zum Krankheitsbild des entzundlichen linearen verrucosen epidermalen Navus.
Derm. Mschr., 1978, 164, 4, 241—254.
ПСОРИАЗ ПУСТУЛЕЗНЫЙ (psoriasis pustulosa).
Впервые пустулезный псориаз описал Zumbusch в 1910 году.
Этиология и патогенез. Большинство современных авторов полагают,
что в патогенезе псориаза могут играть роль различные неблагоприятные
эндогенные и экзогенные факторы, в первую очередь нервно-психические и
инфекционные (вирусные). Следует отличать от пустулезного псориаза
«псориаз с пустулизацией», при котором пустулезные высыпания возни-
кают от вторичной инфекции или раздражающей медикаментозной терапии
(Moslein, 1959). Riecke (1931), Andrews (1939), Ingram (1958), Macmillan и
Champion (1973), Oosterling и соавт. (1978) отождествляют пустулезный
псориаз с хроническим акродерматитом Hallopeau и герпетиформным
импетиго НеЬга. Значительное число дерматологов (Машкиллейсон Л. Н.,
1965; Каламкарян А. А. и соавт., 1980; Ашмарин Ю. Я., 1981; Капкаев
Р. А. и Аковбян В. А., 1981; Антоньев А. А. и Суворова К- Н., 1981;
Kogoj, 1937; Scod, 1958; Moslein, 1959; Katzenellenbogen и Fanerman, 1966;
Lever и Schaumburg—Lever, 1975; Bruckman и Batli, 1978 и др.) их раз-
граничивают.
Клиника. Различают две формы пустулезного псориаза: генерализо-
ванный (Zumbusch, 1910) и пустулезный псориаз ладоней и подошв (Bar-
ber, 1930). Мужчины, видимо, заболевают несколько чаще, чем женщины.
В подавляющем большинстве случаев пустулезный псориаз начинается в
возрасте между 15 и 35 годами. На гиперемированных участках кожи воз-
никают поверхностные пустулы величиной с булавочную головку. Пусту-
415
лы окаймлены ярко-красным, резко очерченным венчиком. Некоторые из
них сливаются, образуя сгнойные озера.» Не вскрываясь или иногда вскры-
ваясь, пустулы ссыхаются с образованием коричневато-желтоватых корок.
Содержимое пустул всегда стерильно. Локализация пустулезного псориа-
за—грудь, спина, боковые поверхности туловигца, разгибательные поверх-
ности верхних и нижних конечностей. Высыпания, распространяясь, вре-
менами занимают весь кожный покров. Некоторые авторы отмечают, что
наряду с распространенными пустулезными высыпаниями у больных могут
быть проявления обычного псориаза. По нашим наблюдениям, это спра-
ведливо в отношении лишь пустулезного псориаза Barber. У больных с
распространенными, универсальными поражениями заболевание протекает
тяжело, сопровождаясь, высокой лихорадкой, поражением суставов, сла-
бостью, общей дистрофией, часто нефропатией и резкой болезненностью
кожи. Обычно отмечается лейкоцитоз в крови. Слизистые оболочки, по-
видимому, не поражаются или поражаются исключительно редко. Нередко
наблюдается поражение ногтевых пластинок.
В литературе нет единого мнения и о взаимоотношении пустулезного
псориаза ладоней и подошв с пустулезным бактеридом Эндрюса, нозоло-
гическая самостоятельность которого, на наш взгляд, сомнительна (Ка-
ламкарян А. А., Ашмарин Ю. Я-, Трофимова Л. Я- 1982).
Гистопатология. Умеренный акантоз, порокератоз. Крупные микро-
абсцессы (Munro), занимающие большую часть мальпигиева слоя, проника-
ющие в роговой слой. Воспалительные явления в дерме выражены сильнее,
чем при акродерматите и импетиго Гебры. В верхней части собственно
кожи отмечается расширение капилляров и более или менее выраженный
воспалительный инфильтрат из лимфоцитов и гистиоцитов.
Дифференциальный диагноз. Проводят с герпетиформным импетиго
Гебры, стойким акродерматитом Галлопо, пустулезным бактеридом Энд-
рюса.
Лечение. Кортикостероиды, антибиотики, цитостатические препараты,
фотохимиотерапия, витамины (А, Вт, В12 и др.), общие ванны, мази с корти-
костероидами, жидкость Кастеллани и др.
ЛИТЕРАТУРА
Ашмарин Ю. Я- По поводу статьи А. А. Каламкаряиа, Л. Я- Трофимовой, Р. Н.
Волошина, В. Г. Акимова <К вопросу о пустулезном псориазе, герпетиформном импетиго
и акродерматите Галлопо.»— Вестн. Дерматол. и венерол., 1981, 2, 39—40.
Антоньев А. А., Суворова К- Н. К дискуссии о некоторых Дерматозах с пустулез-
ными высыпаниями.— Вестн. дерматол. и венерол., 1981, 8, 43—45.
Каламкарян- А. А., Трофимова Л. Я-, Акимов В. Г., Волошин Р. Н. К вопросу
о пустулезном псориазе, герпетиформном импетиго и акродерматите Галлопо.— Вестн.
Дерматол. и венерол., 1980, 5, 40—43.
Каламкарян А. А., Ашмарин Ю. Я-, Трофимова Л. Я- Об атипичных формах
псориаза.— Вести, дерматол. и венерол., 1982, 8, 8—13.
Капкаев Р. А., Аковбян В. А. Нозологические варианты некоторых пустулезных
дерматозов.— Вестн. дерматол. и венерол., 1981, 7, 50—53
416
ПУЗЫРЧАТКА (pemphigus).
Этиология и патогенез. Пузырчатка рассматривается как аутоиммун-
ное заболевание, патогенетическое значение в развитии которого придается
антителам преимущественно IgG класса к цементирующей межклеточной
субстанции эпидермиса (Beutner et al., 1973 и др.). Это подтверждено мно-
гочисленными клиническими наблюдениями, включая и данные (Jab lon-
ska et al., 1970; Maize et al., 1975; Krain, 1974; Kavli, 1978; Wilhelm et al.,
1982 и др.) об ассоциации пузырчатки с другими аутоиммунными заболева-
ниями (например, с myasthenia gravis), а также экспериментальными ис-
следованиями'(Binder et al., 1978). Van Joost (1976) предполагает 3 меха-
низма иммунных нарушений, которые могут вести к развитию заболевания:
центральное, возможно генетически детерминированное, повреждение им-
мунной системы, в развитии которого определенная роль принадлежит ти-
мусу, чем и объясняется связь пузырчатки с другими иммунными болез-
нями и опухолей лимфоидной ткани. Развивающаяся при этом пузырчат-
ка рассматривается как идиопатическое аутоиммунное заболевание; цент-
ральйое ослабление иммунной системы, обусловленное экзогенными фак-
торами, с развитием пузырчатки как вторичной аутоиммунной болезни;
развитие в эпидермисе патологического процесса с.накоплением в межкле-
точном пространстве антигена, ведущего к образованию аутоантител. На
возможную роль генетических факторов указывают семейные случаи, уча-
щение у больных пемфигусом некоторых антигенов тканевой совместимости,
в частности, HLA—10 (Krain et al., 1973).
Имеются сообщения (Alinovi et al., 1982; Ruocco и Pisani, 1982; Chri-
steler et al., 1982; Tinozzi, 1982 и др.) о развитии буллезных дерматозов,
сходных клинически, морфологически и иммунологически с пузырчаткой
(листовидной, эритематозной), под влиянием самых разнообразных внеш-
них факторов, прежде всего медикаментов (D-пеницилламин, тиопронин,
саптоприл, рифампицин и др), применяющихся для лечения ревматоид-
ного артрита, склеродермии, гипертонической и других болезней. В одних
случаях высыпания регрессировали вскоре после отмены препаратов, в
других—имели длительное течение подобно истинной пузырчатке.
Клиника. Различают 4 клинических формы истинной (акантолитиче-
ской) пузырчатки: обычная, вегетирующая, листовидная и эритематозная.
Общим для них является развитие пузырей, положительный симптом Ни-
кольского, наличие акантолитических клеток, аутоантител класса I'gG к
межклеточной субстанции эпидермиса, гистологически—интраэпидермаль-
ных пузырей за счет акантолиза. Из перечисленных форм наименьшие диаг-
ностические трудности при локализации пузырей на К( же представляют
высыпания обычной пузырчатки. Что касается изолированного поражения
полости рта, а оно в начальном периоде болезни бывает часто (у 60,6%
больных, по наблюдениям Н. Д. Шеклакова, 1961; у 67 из 107 больных
Rosenberg и соавт., 1971; около 60% по сведениям Lorenzen, 1983, осно-
ванным на обзоре данных литературы), то процент диагностических ошибок
довольно высок. Например, Bean (1974) отмечает, что ни в одном случае
пемфигуса у детей с локализацией пузырей в полости рта этот диагноз не
27 Клиническая дерматология *417
предполагался. Наиболее часто ставится диагноз: стоматит, многоформ-
ная экссудативная эритема. Облегчают распознавание пузырчатки в поло-
сти рта такие признаки, как эфемерность пузырей, болезненность эрозий,
наличие обрывков эпителия по их краю, фибринозного налета, торпидность,
наиболее частое расположение на слизистой щек. На мысль о возможности
пузырчатки и необходимости соответствующего обследования должны на-
водить также обычно средний или пожилой возраст больных (следует в то
же время помнить, что обычная пузырчатка у детей так же, как правило,
начинается в полости рта), безуспешность традиционной, для стоматита
терапии.
Изложенное, однако, не означает, что при постановке диагноза обыч-
ной пузырчатки на коже вообще не возникает проблем. Они бывают, и не
так уж редко, особенно на ранних стадиях болезни, которые нередко про-
текают атипично, и, о чем уже упоминалось, у детей, когда ранняя поста-
новка диагноза чрезвычайно важна, ибо только своевременное, массивное
(а не средними дозами) лечение кортикостероидами улучшает прогноз за-
болевания (Jordon et al., 1969; Bean, 1974; Levine et al., 1983 и др.). При-
мером могут служить следующие клинические наблюдения. Известны слу-
чаи, когда единственным проявлением пузырчатки длительное время может
быть изолированный очаг (так называемый примераффект). Он может на-
поминать импетиго, себорейный дерматит, осложнившийся вторичной ин-
фекцией (Торсуев Н. А. и соавт., 1974), быть в виде резко ограниченной
ссадины ярко-красного цвета со слегка неровной бархатистой поверх-
ностью, частично покрытой корками, на пояснице (Павлов С. Т., 1961)
или в виде экссудативного хейлита на красной кайме нижней губы (Левин
М. М. и Лосева В. А., 1969). В последнем наблюдении в одном случае
высыпания пузырей на коже и в полости рта появились через 11 месяцев,
во вторам—через 2 года. Из-за диагностической ошибки обе больные по-
лучили близкофокусную R-терапию.
Мы наблюдали больную в молодом возрасте, когда клинические про-
явления длительное время были сходны с высыпаниями розового лишая.
По данным Senear (1952), в начальной фазе высыпания могут напоминать
буллезную экссудативную эритему. Hashimoto и соавт. (1983) описали 79-
летнюю больную, у которой была зудящая кольцевидная эритема без пу-
зырей. При гистологическом исследовании был выявлен эозинофильный
спонгиоз, при прямой иммунофлюоресценции—отложение IgG и С3 в
межклеточном пространстве эпидермиса. И хотя у больной не было об-
наружено признаков акантолиза, образования пузырей, был отрицатель-
ным симптом Никольского, положительным эффект от DDS, авторы рас-
ценили это наблюдение как атипичный вариант пузырчатки. Начальные
проявления обычной пузырчатки на волосистой части головы могут иметь
сходство с пиодермией или себорейной экземой.
Tappeiner и Holubar (1970) описали начальное изолированное пораже-
ние стоп в виде дисгидрозиформных везикуло-буллезных элементов, отли-
чавшихся торпидным течением, с развитием через несколько месяцев, сна-
чала сходных с герпетиформным дерматитом Дюринга, а затем типичных
418
Рис. 262. Себорейная Рис. 263. Себорейная
пузырчатка. пузырчатка.
буллезных высыпаний на туловище. Диагноз был подтвержден гистологи-
ческими и иммунологическими исследованиями, в том числе в биоптатах с
очагов на стопах. Авторы отождествляют это наблюдение с Pemphigus
serpiginosus, описанным НеЬга. Атипично может протекать обычная пу-
зырчатка у больных опухолями. Мс Кее и соавт. (1978) описали сочетание
обычной пузырчатки с ретроперитонеальной параганглиомой. Изменения
кожи появились через год после появления клинических признаков опу-
холи в виде зудящих эритемато-узелковых высыпаний, частью с неболь-
шими пузырями на поверхности. Вокруг рта были изменения, напоминаю-
щие герпетические, в полости рта—поверхностные язвы на языке и в обла-
сти глотки. Диагноз пузырчатки был подтвержден гистологическим и им-
муноморфологическим исследованием. Известные затруднения в правиль-
ной диагностике представляют случаи смешанных буллезных дерматозов,
когда, например, клинические проявления более свойственны пузырчатке,
а гистологически выявляются признаки герпетиформного дерматита Дюрин-
га или наоборот. Такие случаи описываются под названиями: акантолити-
ческий герпетиформный дерматит, сульфон чувствительная пузырчатка
(Winkelmann и Roth, 1960; De Mento и Grover, 1973), смешанный буллезный
27* 419
Рис. 265. Пузырчатка—смешанная
форма (та же больная).
Рис. 264. Пузырчатка — смешанная
форма с герпетиформным дерматитом.
дерматоз (Ваггапсо и Tulsa, 1974), герпетиформный пемфигус (Jablonska
et al., 1975; Chorzelski и Jablonska, 1976). Решающее значение в диагно-
стике придается иммунным и иммуноморфологическим нарушениям.
Б. А. Пасечник и Л. А. Орлова (1981) наблюдали начальные прояв-
ления атипичной пузырчатки, сходные с герпетиформным дерматитом Дю-
ринга, у 13 из 48 больных. Л. Я- Трофимова и В. И. Хапилова (1981),
В. И. Хапилова (1981) наблюдалй больных с атипичным началом пузыр-
чатки, сходным с герпетиформным дерматитом Дюринга. В отдельные
периоды течения болезни признаки обоих заболеваний существовали Од-
новременно. Аналогичная ситуация может быть и между пузырчаткой и
пемфигоидом (Tappeiner и Holubar, 1970). В наблюдении Guggenberger и
соавт. (1979) у больной 36 лет были клинические признаки пемфигоида
(большие напряженные пузыри на неизмененной коже) и герпетиформного
дерматита Дюринга (зуд, герпетиформность высыпаний). Симптом Николь-
ского был отрицательным. Диагноз пузырчатки поставлен на основании
гистологических и иммунофлюоресцентных критериев. К смешанным фор-
мам относят и случаи сосуществования пемфигуса и буллезного пемфигои-
да, когда имеется образование акантолитических пузырей и аутоантител
к межклеточной субстанции и базальной мембране эпидермиса. Chorzelski
и соавт.. (1974) наблюдали 3 больных с клиническими и иммунологически-
420
Рис. 266. Пузырчатка
вульгарная (изолированное
поражение полового члена).
ми признаками пузырчатки и пемфигоида. Причем у одного
из них смешанная форма развилась с самого начала, у других—
пемфигус возник раньше. Авторы считают, что в осгсве смешанных форм
лежат иммунные нарушения, которые могут давать различные виды ауто-
иммунных реакций. Характерным для герпетиформного пемфигуса морфо-
логическим признаком является эозинофильный спонгиоз (Emmerson и
Wilson-Jones, 1968), которому придается диагностическое значение (Bau-
er et al., 1983 и др.), хотя и не абсолютное (Lorenzen, 1983).
К числу редких локализаций обыч зй пузырчатки относится конъ-
юнктива. Bean и соавт. (1975) опубликовали данные о 2 больных с пузыр-
чаткой глаза. В одном случае были изменения и на коже, в полости рта, в
другом—в полости рта, на половых органах. С помощью реакции иммуно-
флюоресценции было обнаружено межклеточное свечение конъюнктивы.
По наблюдениям Rosenberg и соавт. (1976), касающимся 107 больных раз-
личными формами пузырчатки, конъюнктива была вовлечена в процесс у 7
человек, никогда первично, и только при обычной пузырчатке.
При обычной и эксфолиативной пузырчатке наблюдается поражение
ладоней и подошв в виде отдельных или сливающихся везикуло-буллезных
элементов, после исчез» вения которых возможно резко выраженное пла-
стинчатое шелушение, развитие гиперкератотических изменений. Costello
и Gibbs (1967) указывают на возможность гиперкератоза и без предшест-
вующих пузырей. Может наблюдаться поражение околоногтевых тканей,
как начальный процесс (Торсуев Н. А. и соавт., 1979).
421
Своеобразие вегетирующей пузырчатки заключается в том, что пузыри
часто располагаются в полости рта, периорифициально, в крупных склад-
ках, они быстро вскрываются и эрозии покрываются папилломатозно-вер-
рукозными разрастаниями, нередко инфицирующимися, что придает Им
сходство с вегетирующей пиодермией. Некоторые авторы (Nelson et al.,
1977 и др.) выделяют как особую форму вегетирующий пемфигус Hallopeau,
отличающийся от вегетирующей пузырчатки Neumann более доброкачест-
венным течением и отсутствием буллезных элементов. Процесс начинается
с высыпания пустул, быстро вскрывающихся и трансформирующихся в
папйлломатозные, вегетирующие очаги с преимущественной локализацией
в складках. Могут быть эрозивные очаги в полости рта. Для дифференци-
ровки от вегетирующей пиодермии наиболее важной считается реакция
иммунофлюоресценции (Leroy, 1982 и др.). Вегетирующие, гиперкератоти-
ческие очаги могут развиваться и при обычной пузырчатке в процессе им-
муносупрессивной терапии, в том числе при отсутствии пузырей (Faber
et al., 1983).
Трудности в диагностике эксфолиативной пузырчатки обусловлены
тем, что пузыри за счет поверхностного расположения часто мало замет-
ны, они быстро покрываются чешуйко-корочками, что придает, особенно
при генерализованном процессе, очагам поражения характер эксфолиа-
тивной эритродермии. Это сходство усиливается тем, что пузыри возникают
на эритематозном фоне. Слизистая оболочка полости рта обычно остается
не вовлеченной в процесс, что резко отличает эту форму пузырчатки от
других разновидностей (Пашков Б. М. и соавт., 1970). Perry (1961) под-
черкивает трудности диагностики этой формы заболевания и указывает,
что больным эксфолиативной пузырчаткой ставились такие диагнозы, как
псориаз, себорейный дерматит, розовый лишай, опоясывающий лишай,
грибковая инфекция, экзема, семейный доб)рокачественный пемфигус Хей-
ли-Хейли. Bruckner и соавт. (1980) наблюдали острое развитие эксфолиа-
тивной пузырчатки, причем высыпания • были представлены множеством
ограниченных гйперпигментированных гиперкератотических очагов, клини-
чески напоминавших себорейный кератоз.
Из публикации Winkelmann и Roth (1960) следует, что высыпания,
в зоне которых гистологически выявляется картина листовидной пузыр-
чатки, клинически могут быть сходны с герпетиформным дерматитом Дю-
ринга, тем более что был получен положительный эффект от сульфонов.
Сходные случаи наблюдали Chorzelski и Jablonska (1970) у 2 больных с
клинической картиной, сходной с герпетиформным дерматитом Дюринга.
Гистологически выявлялся наряду с акантолизом эозинофильный спон-
гиоз. С помощью РИФ обнаружены циркулирующие и связанные аутоан-
титела. Но, в отличие от случая Winkelmann и Roth, без эффекта от суль-
фонов. Описано у 2 больных 15 и 20 лет, страдающих эксфолиативной пу-
зырчаткой, наличие на коже туловища толстых гиперкератотических че-
шуек с кератотическими шипами, входящих в волосяные фолликулы, у
одной из них—ладонно-подошвенный гиперкератоз (Торсуев Н. А. и с* авт.,
1979).
422
Эритематозная пузырчатка, или синдром Senear—Usher в настоящее
время рассматривается как ограниченный вариант листовидной, хотя еще
сравнительно недавно некоторые авторы считали ее буллезной формой
красной волчанки (Арутюнов В. Я-, 1961 и др.), с которой она имеет наи-
большее клиническое сходство, которое может усиливаться за счет раз-
вивающейся иногда рубцевидной атрофии, а также положительным эффек-
том только от наружного применения кортикостероидов (Туе et al., 1964).
Не исключено сочетание эритематозной пузырчатки и красной волчанки,
как клиническое, так и особенно иммунологическое (Maize et al., 1982
и др.).
Поэтому, во всех подозрительных на сочетанную патологию случаях
необходимо проводить тщательное морфологическое и иммунологическое
обследование. Так, Van Yoost и соавт. (1984) обнаружили в одном случае
наряду с признаками пузырчатки (акантолиз, отложение в межклеточных
пространствах IgG и С3) ЛЕ-клетки и антитела к ДНК; в другом—гисто-
логические и иммуноморфологические данные, характерные для красной
волчанки, а с помощью электронной микроскопии—акантолиз. При обиль-
ном наслоении чешуйко-корок очаги могут быть сходны с себореидами,
импетиго, экссудативным псориазом.
Tappeiner и Holubar (1970) наблюдали 66-летнюю пациентку, которая
страдала повышенной ранимостью кожи головы при расчесывании волос.
Клиническая картина 2-х монетовидных очагов напоминала пиодермию,
актинический кератоз, поверхностную эпителиому или дискоидную крас-
ную волчанку. Только после трехкратной биопсии был поставлен диагноз
эритематозной пузырчатки. У одной из больных, описанных Л. Г. Рут-
штейи и соавт. (1980), в начале заболевания были стойкие высыпания на
лице и волосистой части головы, расцениваемые как красная волчанка.
Типичные для эритематозной пузырчатки высыпания появились только
через год.
Атипичность пузырчатки у детей заключается уже в самом факте ее
развития в этом возрасте, необычной клинической картине, сходной с мно-
гоформной экссудативной эритемой, стоматитом, пиодермией, токсиче-
ским буллезным дерматитом. Поэтому во всех случаях необъяснимо затя-
нувшегося стоматита, пиодермии необходимо проводить обследование для
исключения пузырчатки. Описаны случаи развития обычной пузырчатки в
3 1/2 года (Berger et al., 1973), 6 1/2 лет (Хамаганова А. В. и соавт., 1977),
у 13—14-летних детей (Jordon et al., 1969; Levine et al., 1982; Kock и
Lobach, 1983). Kahn и Lewis (1971) наблюдали эксфолиативную пузырчат-
ку у 18-месячного ребенка. Schroeter и соавт. описали эксфолиативную
пузырчатку у 3-летнего мальчика, которая вначале располагалась на во-
лосистой части головы и была сходна с импетиго или себореей. В отличие
от обычной пузырчатки, начинающейся у детей обычно в полости рта, при
эксфолиативной поражение локализуется только на коже (Bean, 1974).
Течение может быть относительно спокойным. Так, в наблюдении Ber-
ger и соавт. (1973) генерализованные высыпания появились только через
8 лет после развития поражения в полости рта. Но чаще оно агрессивное,
< 423
на что указывают как летальные исходы, которые могут наступить уже в
течение первого года болезни (Jordon et al., 1969), так и необходимость
применения для купирования болезни высоких, даже для взрослых, доз
кортикостероидных препаратов.
Судя по публикациям, складывается впечатление, что у детей эксфо-
лиативная пузырчатка встречается чаще по отношению к обычной, чем у
взрослых. Клинической особенностью обычной пузырчатки у детей может
быть развитие вегетирующих очагов. Рис. 262, 263, 264, 265, 266, 267.
Гистопатология. Интраэпидермальные пузыри, возникшие за счет
акантолиза, при обычной пузырчатке в нижней части шиповидного слоя,
при листовидной—в зернистом или в поверхностных слоях шиповидного.
При вегетирующей пузырчатке обнаруживаются помимо пузырей папил-
ломатоз, акантоз, внутриэпидермальные, преимущественно эозинофильные
абсцессы.
Дифференциальный диагноз. Дифференцировать пузырчатку необхо-
димо от пемфигоида, герпетиформного дерматита Дюринга, многоформной
экссудативной эритемы, буллезных медикаментозных реакций.
Лечение. Большие дозы кортикостероидов при начальной дозе 60—80
и более мг преднизолона, иммунодепрессанты (азатиоприн, метотрексат),
полноценное питание, анаболические гормоны, ограничение поваренной
соли.
ЛИТЕРАТУРА
Пасечник Б. А., Орлова'Л. Н. К вопросу об атипичном течении вульгарной пузыр-
чатки. Вестн. дерматол. и венерол., 1981, 2, 44—47. •
Торсуев Н. А., Шеклаков Н. Д., Романенко В. Н. Буллезные Дерматозы. М. Ме-
дицина, 1979.
Трофимова Л. Я-, Халилова В. И- К вопросу о клинике и терапии пузырчатки,
Вестн. дерматол. и венерол., 1981, 12, 17—19.
Халилова В. И. Случай атипичной вульгарной пузырчатки. Вестн. дерматол. и
венерол. 1981, 3, 56—57.
Хамаганова А. В., Ведрова И. И., Лукьянова 3. П. Вульгарная пузырчатка у
мальчика 6г/2 лет. Вестн. дерматол. и венерол., 1977, 2, 57—58.
Alinovi A., Benoldi D., Manganelli Р. Pemphigus erythematosus induced by Thiop-
ronine. Acta dermatoverier. (Stockh.), 1982, 62, 453—454.
Bean S. F. Chronic nonhereditary blistering disease in children. Arch, dermatol., 1974,
110, 6, 941—944
Bean S. F., Holubar K., Gillet R. B. Pemphigus involving the eyes. Arch, dermatol.,
1975, 3, 14, 1484—1486.
Binder W. L., Beutner E. H. Chorzelski T. P. In vitro effects of pemphigus antibodies
on skin. Brit. J. Dermatol., 1978, 99, 1, 39—*2
- Bruckner N., Katz R. A., Hood 1. Г Pemphigus foliaceus resembling eruptive sebor-
rheic keratoses. Arch, dermatol., 1980, 136. 7, 815—817
Chorzelski T. P., Maciejewska E., Jahlonska S. et al. Coexistence of pemphigus and
bullous pemphigoid. Arch, dermatol., 1974 109, 849—853
424
Guggenbergeg K-, Maciejewski W., Scherer R. Eine klinisch ungewonliche Erscheinun-
gsform des Pemphigus vulgaris. Hautartz, 1979, 30, 1, 33—35.
Hashimoto T., Sugiura M., Kurihara S. et al. Atypical pemphigus showing eosinop-
hilic spongiosis. Cl. Exp. Dermatol., 1983, 8, 1, 37—40
JablonskaS., ChorzelskiT. P., Beutner E. H. etal. Herpetiform pemphigus, a variab-
le patterns of pemphigus. Int. J. dermatol., 1975, 14, 353—359.
Jeanmougin M., BonoaletD., Bertail M. A. et al. Pemphigus avec aspect cliniq d’e-
rytheme annulaire centrifuge. Ann. Derm. Venereol., 1982, 109, 9, 775—776.
Van Joost T. Pemphiguskrankheiten. Hautarzt, 1976, 27, 6, 253—260.
Kock B.—W., Lubach D. Ober den kindlichen Pemphigus vulgaris. Hautarzt, 1
34, 9, 456—458.
Krain L. S. The association of pemphigus with thymoma or malignapcy. A critical
review. Brit. j. dermatol., 1974, 90, 397—405.
Leoine L., Bernstein J. E., Soltani K- et al. Coexistence childhood pemphigus folia-
ceus and Grave’s disease. Arch, dermatol., 1982, 118, 8, 602—604.
Leroy D. Pemphigus vegetant a type clinique de dermatite pustuleuse chronique de
Hallopeau. Ann. Dermatol. Venereol., 1982, 109, 6—7, 549—555
Maize J. C., Green D-, Prooost T. T. Pemphigus foliaceus: A case with serologic fea-
tures of Senear—Usher syndrome and other autoimmune abnormalities. J. Am. Acad. Derma-
tol., 1982, 7, 736—741
Me Кее P.H., Me Clelland M.,' Sandford J.G. Coexistence of pemphigus, anti-
skeletal muscle antibody and a retroperitoneal paraganglioma. Brit. j. dermatol., 1978, 99, 4,
441—445.
Nelson C. G., Apisarnthanarax P., Bean S. et cd. Pemphigus vegetans of Hallopeau.
Arch, dermatol., 1977, 113, 7, 942—94
Ruocco V. Pisani M. Induced pemphigus. Arch, dermatol. Res., 1982, 274, 123—140.
Wilhelm T., Metz J., Deuchert K- Pemphigus erythematosus und malignes Thymom.
Hautarzt, 1982, 33, 3, 156—158
ПУЗЫРЧАТКА ХРОНИЧЕСКАЯ ДОБРОКАЧЕСТВЕННАЯ СЕМЕЙ-
НАЯ ГУЖЕРО—ХЕЙЛИ—ХЕЙЛИ (pemphigus benignus chronicus familia-
ris Gougeret—Hailey—Hailey). '
Синонимы: семейный буллезный дискератоз и др.
Первое описание заболевания принадлежит Gougerot и Allee (1933),
которые наблюдали распространенные формы болезни, начинающиеся в
детском возрасте. Howard и Hugh Hailey в 1939 г. сообщили о двух бра-
тьях, у которых заболевание развилось в возрасте 26 и 28 лет, и высыпания
ограничивались крупными складками кожи. Они включили в свое сообще-
ние описание еще двух братьев, наблюдавшихся доктором Allison с анало-
гичной клинической картиной и поздним началом болезни.
'Этиология и патогенез. Еще нет единого мнения о нозологическом
месте доброкачественной семейной пузырчатки. Многие современные ав-
торы считают ее самостоятельным доминантно наследуемым генодерматозом
с непостоянной пенетрантностью и относят в группу акантолитических за-
болеваний (Jablonska et al., 1959; Marsch и Stuttgen, 1978; Beno Michel,
425
1982 и др.)* Другие исследователи (Finnerud и Szymanski, 1950; Ellis, 1950
и др-) рассматривают как буллезную разновидность фолликулярного ди-
скератоза (болезни Дарье).
Colomb и соавт. (1968) при исследовании хромосом больных добро-
качественной семейной пузырчаткой обнаружили поломки хромосом в
50 случаях митозов.
Механизм акантолиза, лежащего в основе заболевания, еще не раскрыт.
Gottlib и Lutzner (1970) в результате исследований на ультраструктурном
уровне пришли к заключению, что при болезни Гужеро—Хейли—Хейли не-
нормальное функционирование генов нарушает сцепление эпидермальных
клеток друг с другом по линии микроворсин ки-тонофиламенты-цельные
десмосомы. Особое значение авторы придают существованию причудливых
микроворсинок на поверхности кератиноцитов и патологической конфигу-
рации тонофиламентов, считая эти признаки патогномоничными для бо-
лезни Гужеро—Хейли—Хейли.
Четко установлено, что бактерии, дрожжи, травма, ультрафиолетовая
радиация провоцируют появление заболевания у генетически предраспо-
ложенных людей. Однако не ясны пути, по которым эти факторы вызывают
акантолиз. Остаются не раскрытыми природа изменений функций генов,
биохимические процессы в кератиноцитах при акантолизе (Muchel, 1982).
Клиника. Заболевание чаще начинается в детстве, реже в возрасте
20—30 лет. Примерно в 1/3- -2/3 случаев оно имеет семейный характер,
иногда прослеживается в 2—3 поколениях.
Излюбленная локализация высыпаний—затылок, задне-боковые по-
верхности шеи, подмышечные, паховые складки, область пупка. В ред-
ких случаях высыпания могут быть генерализованными (Birnbaum et
al., 1980). Типичные изменения образуются преимущественно в склад-
ках, где трение и влажность активизируют микробную флору. Первичный
элемент—фликтенулезный пузырек на слегка воспаленном основании. Пу-
тем слияния их образуются очаги округлых, овальных, полициклических
очертаний, величиной от 10-копеечной монеты до ладони взрослого человека.
Центральная часть очагов красна, отечна, мокнет, покрыта извилистыми
эрозиями-трещинами, между которыми имеются вегетации в виде низких
гребешков. Очаги окаймлены отечным фликтенулезным венчиком, за счет
которого они могут увеличиваться по площади. В фазе регрессирования
приступа болезни очаги в центре эпителизируются, принимают буровато-
синюшный оттенок, па краю же еще могут возникать новые фликтены, ко-
торые ссыхаются в тонкие корочки. В результате присоединения вторич-
ной инфекции корочки становятся рыхлыми буровато-желтыми. В не-
посредственной близости от очагов может быть положительный симптом
Никольского. В препаратах-отпечатках со дна эрозий обнаруживают акан-
толитические клетки.
Заболевание чаще обостряется в весенне-летний период, затухает
зимой. Имеются указания на зависимость обострений от фазы менструаль-
ного цикла (Грабчак К. А., 1977). Общее состояние больных, как правило,
не страдает.
426
В редких случаях высыпания могут располагаться на слизистых обо-
лочках рта, пищевода и др. (Fischer и Nikolowski, 1961; Schneider et al.,
1966; Witkowski и Parish, 1973).
Heinze (1979) описал больную с поражением слизистой оболочки рта,
гениталий и конъюнктивы. Он указывает, что диагноз доброкачественной
семейной пузырчатки на слизистых не может быть установлен без гисто-
логического исследования. Постановке диагноза также помогает обследо-
вание других членов семьи, которое следует проводить летом. Имеются
сообщения о сочетании доброкачественной семейной пузырчатки с псориа-
зом (Heaphy и Winkelmann, 1976).
Заболевание длится неопределенно долго. Обострения сменяются ре-
миссиями различной продолжительности.
Гистопатология. В свежих элементах над базальным слоем эпидермиса
обнаруживаются мелкие щели. В полностью сформировавшихся элемен-
тах—надбазальные пузыри, которые содержат акантолитические клетки,
часть которых находится в состоянии ороговения. Могут быть признаки
дискератоЗа, когда некоторые акантолитические клетки сморщиваются,
приобретая вид зерен.
Удлиненные дермальные сосочки проникают в полость пузырей; они
обычно покрыты одним слоем базальных клеток.
Дифференциальный диагноз. Проводят с вульгарным пемфигусом,
везикулезной формой болезни Дарье, интертригинозной экземой. При по-
ражении слизистых—с афтозным стоматитом, красным плоским лишаем.
Лечение. Как правило оно недостаточно успешно. При оценке его эффек-
тивности следует принимать во внимание волнообразность течения болез-
ни, склонность к спонтанному улучшению в зимнее время.
Применяют антибиотики, сульфоны, витамин А. При тяжелых обо-
стрениях с обширным поражением кожи—кортикостероиды (20—30 мг
преднизолона в сутки) в течение непродолжительного времени (2—4 не-
дели). Наружно—жидкость Кастеллани, спиртовой раствор бриллиантово-
го зеленого, мази с антисептиками и кортикостероидами. Часто отмечается
плохая переносимость мазевых основ.
ЛИТЕРАТУРА
Грабчак К- А. К наследственному происхождению хронической семейной добро-
качественной пузырчатки.— Вестн. дерматол. и венерол., 1977, № 5, с. 53—56.
Beno Michel. Commentary: Hailey—Hailey Disease. Familial benign chronic pemphi-
gus.— Arch. Dermatol., 1982, v. 118, № 10, p. 781—783.
Heinze R. Pemphigus chronicus benignus familiaris (Gougerot—Hailey—Hailey) mit
Schleimhautbeteiligung bei einer Diabetikerin.— Derm. Mschr., 1979, v. 165, N 12, p. 862—
867.
Kahn D., Hutchinson E. Esophageal involvement in familial benign chronic pemp-
higus.— Arch. Dermatol., 1974, v. 109, p. 718—719.
ПУСТУЛЕЗНЫЙ И АТРОФИЧЕСКИЙ ДЕРМАТИТ ГОЛЕНЕЙ (der-
matitis cruris pustulosa et atrophicans)
427
Синонимы: сикоз голеней, пустулезный и атрофический фолликулит
голеней.
Впервые термин «dermatitis cruris pustulosa et atrophicans» применил
G. Clarke в 1952 году, описав это заболевание в Лагосе. Следующая раоота
принадлежит R. Harman, который в 1968 году подробно охарактеризовал
клинические черты этого дерматоза. Частота нозологии в разных географи-
ческих зонах вариабельна. В Нигерии и Лагосе (Clarke G., 1952; Shrank А.
и Harman R., 1968; Poster J., 1963 и др.) она составляет от 0,5 до 4,8%
среди всех дерматозов, но почти не встречается' в Восточной и Южной
Африке.
В иностранной европейской и отечественной дерматологической ли-
тературе сообщений о атрофическом пустулезе голеней мы не встретили.
Лишь совсем недавно А. А. Каламкарян и М. Балтабаев наблюдали в
ЦКВИ больного 31 года с характерными данными этого заболевания. Кли-
нически заболевание характеризовалось наличием множества фоллику-
лярных пустул с исходом в атрофию. Высыпания локализовались на коже
голеней и четко были отграничены от здоровой кожи на голеностопных
суставах.
Этиология и патогенез. При бактериологическом исследовании по-
чти постоянно выделяется золотистый, реже белый, стафилококк (Suge-
than Р. и Zacarian J., 1973). Имеет-
ся также точка зрения о возможной
роли стрептококка, бактерий. В па-
тогенезе пустулезного дерматита су-
щественное значение имеет наличие
на коже так называемых ворот ин-
фекции, т. е. микротравм.
Клиника. Клиническая характе-
ристика пустулезного дерматита го-
леней неполная и требует дополни-
тельного уточнения и накопления
материала.
Оба пола поражаются одинаково
часто, возраст больных колеблется
от 14 до 32 лет. Процесс начинается
чаще всего с появления на голенях
множественных сикозиформных фол-
ликулярных узелков, пустул, окру-
женных узким воспалительным вен-
чиком. Просуществовав несколько
дней (а иногда несколько недель), эти
высыпания вскрываются и покрыва-
ются корками (рис. 268). Пустулы
проявляют явную тенденцию к слия-
Рис. 268. Пустулезный нию, с образованием отека и неболь-
и атрофический дерматит голеней- ШОГО зуда. В процессе дальнейшего
428
I
тг.ения, через 3—4 недели на месте фолликулитов формируются атрофи-
ческие рубцы. Давая общую характеристику этой формы пиодермии, можно
прежде всего указать на сравнительно слабую клиническую выраженность
островоспалительных явлений. Как правило, на тыле стоп высыпаний
нет, но могут быть единичные пустулы на разгибательной поверхности
предплечий. Общее состояние удовлетворительное. В ряде случаев появ-
ляются все новые и новые элементы, которые располагаются как изолиро-
ванно, так и группами, захватывая обширные поверхности голеней. В та-
ких случаях заболевание длится месяцы, даже годы, может сопровождать-
ся небольшим повышением температуры, недомоганием, общей сла-
бостью, головными болями.
Прогноз обычно благоприятный, хотя в отдельных случаях возмож-
но развитие элефантиаза. Длительность заболевания от 3 месяцев до 4 лет,
у большинства больных 12—20 месяцев.
Гистопатология. В эпидермисе акантоз, паракератоз, внутриклеточ-
ный отек. Отек сосочков дермы, инфильтрация лимфоцитами, плазмати-
ческими клетками, эозинофилами, полиморфноядерными лейкоцитами,
гистиоцитами и иногда гигантскими клетками. Волосяные фолликулы со-
держат пустулы. Встречается атрофия эпидермиса и волосяных фолли-
кулов, фиброз дермы.
Дифференциальный диагноз. Диагноз устанавливается на основании
наличия хронических фолликулярных пустул, атрофических изменений
на переднебоковых поверхностях голеней. Дифференцировать следует
с декальвирующим фолликулитом.
Лечение. Эта форма пиодермии отличается значительной резистент-
ностью к лечению. W. Jacyk (1976) в Нигерии получил хороший терапев-
тический эффект у 30 больных пустулезным дерматитом голеней от септ-
рина; препарат оказался эффективным у 75—80% больных. В некоторых
случаях хороший эффект наблюдался от длительного приема тетрацикли-
нов (Harman R., 1968).
А. А. Каламкарян и М. Балтабаев (1986) наблюдали хороший резуль-
тат от комплексной терапии—проспидином (по 100 мг ежедневно, общая
курсовая доза—2300 мг) в сочетании с кортикостероидами (преднизолон
по 20 мг ежедневно) и антибиотиками (вибрамицин по 0,1 в течение 2—3
недель).
ЛИТЕРАТУРА
Clarke G. A note on dermatitis cruris pustulosa ef atrophicans Trans. R. Soc. Trop.
Med. Hyg.— 1952.— Vol. 46, N 5.— P. 558—559.
Harman R. Dermatitis cruris pustulosa et atrophicans. The Nigerian skin disease. Brit.
J. Dermatol.— 1968.— Vol. 80, N 2.— P. 97—107.
Jacyk W. Septrin in the treatment of dermatitis cruris pustulosa et atrophicans Brit. J.
Dermatol.— 1976,—Vol. 95, N 1.— P. 71—73.
Poster J. Dermatitis cruris pustulosa et atrophicans. Clinical Dermatology.— Phila-
delphia, 1983.—Vol. I.—P. 1—46.
429
Shrank A., Harman R. The incidence of skin disease in a Nigerian teaching hospital
dermatological clinic Brit. J. Dermatol.— 1966.— Vol. 78, N 3.— P. 235—239.
Sugethan P., Zacarian J., Jog M. Folliculitis cruris pustulosa et atrophicans. Indian
J. Dermatol. Venereol.— 1973.—Vol. 39, N 1.—P. 35—38.
РАК МЕТАСТАТИЧЕСКИЙ КОЖИ (carcinoma cutis metastatica).
Этиология и патогенез. По данным литературы (Ehlers и Krause, 1970;
Rosen, 1980), частота метастазов рака внутренних органов в кожу колеб-
лется от 0,29 до 3,3%. По мнению Ehlers и Krause, требуют ответа такие
вопросы, как: почему метастазирование в кожу бывает редко, склонность
отдельных опухолей метастазировать неодинакова, предпочтительное по-
ражение определенных зон кожи; какова связь между видом опухоли и
локализацией метастаза. Несмотря на то, что метастатический процесс
может располагаться на любом участке кожного покрова, чаще метастазы
•обнаруживаются на груди, животе, в подмышечных впадинах и в пери-
генитальной области (Lausecker et al., 1979; Debois, 1982 и др.). По мне-
нию Popchristow и соавт. (1960), редкость кожных метастазов объясняется
прежде всего тем, что кожа обладает хорошими защитными свойствами, в
результате чего в большинстве случаев опухолевые клетки, достигшие
кожного покрова, элиминируются. Beerman (1969) также придает ответу
организма на развивающуюся опухоль первостепенное значение. Недиф-
ференцированные опухоли дают метастазы в кожу чаще, чем зрелые. По
мнению Ordonez и Smith (1983), вероятность метастазирования в кожу выше
у тех опухолей, которые чаще дают метастазы в другие органы. Метастази-
рование происходит гемато-или лимфогенно, реже per contituitatem или
другим путем (Lausecker et al., 1979). На основании .изучения данных о
о 724 больных, у которых доказан первичный источник метастаза в кожу,
Browstein и Helwig (1972) пришли к выводу, что у мужчин наиболее часто
кожные метастазы имели происхождение из опухолей легких (24%), да-
лее—толстого кишечника (19%), меланомы (13%); у женщин—молочной
железы (69%), толстого кишечника (9%), матки (5%) и карциномы яични-
ков (4%). Debois (1982) считает, что наиболее часто метастазирует рак молоч-
ной железы, желудка, поджелудочной железы, бронхов, яичников. По
мнению Freeman и соавт. (1982), метастазы шейки матки бывают очень ред-
ко, даже в терминальной стадии. Редко наблюдаются и метастазы опухо-
лей печени и желчного пузыря (Reingold и Smith, 1978, Padilla et al.,
1982), предстательной железы (Rockl и Albert, 1962).
Клиника. Изменение кожи на месте метастазов характеризуется боль-
шим полиморфизмом. Наиболее часто встречаются узелковоподрбные и узло-
ватые метастазы, величина которых колеблется от нескольких миллиметров
до 2—5 см и более, количество—от единиц до сотен (рис. 269). Они распола-
гаются интрадермально, и в таких случаях смещаемы или инфильтрируют
подкожную клетчатку. Кожа над глубоко расположенными элементами
на вид не изменена, или напряжена, с нерезко выраженным розоватым или
синюшным оттенком, слегка блестит, над более поверхностными—окраска
ее более интенсивна, преобладают застойные тона. Характерной особен-
ностью метастазов является плотная консистенция, быстрый рост, склон-
430
ность к изъязвлению и распаду, неэффективность консервативной тера-
пии. Обычно субъективных расстройств не бывает или они незначительны,
хотя'описана и резкая болезненность в очагах поражения (Rudner et al.,
1965). Они располагаются рассеянно, иногда линейно, зостериформно.
Hodge и соавт. (1979) наблюдали метастазы карциномы легких в кожу рук
и груди, клинически очень сходную с herpes zoster. Описана экзантемати-
ческая форма метастаза рака молочной железы (Ehlers и Krause, 1970). К
своеобразным вариантам относятся эризипелоидная (carcinoma erysipeloi-
des) и склеродермиформная (carcinoma sclerodermiformes) разновидности
кожных метастазов. Эризипелоидная форма встречается преимущественно
при раке молочной железы, а также легких (Haselrigg, 1977), проявляется
в виде внезапно развивающейся насыщенной эритемы с синюшным оттен-
ком, быстро и неравномерно распространяющейся по периферии. При паль-
пации выявляется отчетливо выраженное уплотнение. С течением времени
интенсивность окраски слабеет, доминируют желтовато-коричневатые тона,
но уплотнение усиливается. На поверхности, на фоне застойной эритемы
и инфильтрации, выделяются полосовидные участки более значительного
уплотнения, множественные мелкие опухолевидные элементы (carcinoma
lenticulare), часто с телеангиэктазиями на поверхности (carcinoma telean?
giectoides) и пузырьковидные высыпания, иногда с геморрагическим ком-
понентом, имеющие сходство с гем-лимфан гномами (Lymphangiosis carcino-
matosa), Bemecker и Bachmann (1979) наблюдали Erythema anulare-
подобную разновидность эризипелоидной карциномы при метастазе рака
яичников.
Эризипелоидная форма наблюдается и при раке других локализаций,
в частности при опухолях мочевого пузыря, толстого кишечника, а также
при раке простаты и желудка (Ebner и El-Mansy, 1971).
Склеродермоподобный метастатический рак кожи обычно проявля-
ется в виде глубоких очагов постепенно распространяющегося уплотне-
ния, кожа в зоне которого не изменена или приобретает ливидный отте-
нок. Может быть ограниченным или занимать обширные участки тела,
например, при раке молочной железы. Одним из вариантов этого типа мета-
статической карциномы является Cancer en cuirasse в виде доскообразной
плотности сплошных очагов, охватывающих в виде панциря грудную клет-
ку. Описан и при метастатической меланоме (Day, 1957), раке слезной же-
лезы (Klehr, 1979). Мы наблюдали подобную форму при метастазе в кожу
рака щитовидной железы. Метастазы, располагающиеся на голове, могут
иметь сходство с сальножелезистой кистой, как это имело в случае мета-
статической карциномы простаты (Peison, 1971). В зоне опухоли может
развиться alopecia neoplastica (Cohen et al., 1961; Nelson, 1972, Baum et
al., 1981). К редким клиническим вариантам относится elephantiasis carci-
nomatosa нижних конечностей, который может развиться при метастазе
в кожу рака половых органов (Littis и Zuehlke, 1979). Своеобразной была
клиническая картина метастаза рака желудка, описанного Szodoray (1959).
На коже верхней части спины и на шее были обширные веррукозные очаги,
очень сходные с микседематозными.
431
Рис. 269. Рак кожа
метастатический.
Wiesner и соавт. (1984) наблюдали метастаз рака желудка в виде об-
ширных конгломератов на шее, состоящих из кистовидных опухолей.
На прилегающих участках груди элементы были сходными с высыпаниями
хронической крапивницы.
Несмотря на то, что специфичность тех или иных кожных изменений
для определенного вида метастазйрующего рака отрицается, указывается,
что метастатический рак молочной железы чаще проявляется в виде лен-
тикулярной, эризипелоидной и склеродермиформной карциномы, включая
Cancer en cuirasse (Lausecker et al., 1979; Haselrigg и Rudolph, 1977). Осо-
бым вариантом является лимфангиосаркома (синдром Stewart—Treves).
Метастазы органов брюшной полости имеют повышенную склонность рас-
полагаться на животе, особенно в области пупка, преимущественно в виде
нодулярных образований, единичных или множественных (Zeligman и
Schwilm, 1974, Samitz, 1975). В область пупка может метастазировать и
карцинома яичников, но чаще метастазы определяются в зоне послеопера-
ционного рубца (Lausecker et al., 1979). Волосистая часть головы счита-
ется редкой локализацией метастазов (Bleeman, 1969). Но в этой области
могут выявляться метастазы любой опухоли. Чаще—это рак молочной
железы, бронхов, почек (Подлящук Е. Л. и Устинова В. Ф., 1979; Ro-
senthal и Lever, 1957; Lausecker et al., 1979). Опухоли бронхов метаста-
зируют в кожу головы, груди, живота (Ehlers и Krause, 1970); почек—в
область лица (Ehlers и Krause, 1970; Braun-Falco и Lukacs, 1971). Hodl
(1980) считает метастаз в концевые фаланги характерным признаком рака
бронхов.
Довольно часто метастаз в кожу представляет собой первый признак
наличия в организме опухоли. Так, из 84 больных, наблюдавшихся Post
и Janner (1974), у 30—это был первый признак карциномы. Это относится
в основном к метастазам, распространяющимся гематогенно, особенно к
432
опухолям легких и почек (Browstein и Helwig, Ehlers и Krause). Как пра-
вило, это неблагоприятный в прогностическом смысле признак, так как
свидетельствует о прогрессирующем течении или рецидиве (Stolp и Fiige-
mann, .1976; Post и Janner, 1974; Price и Kopf, 1974 и др.). Так, у 42 из 84
больных, наблюдавшихся Post и Janner (1974), летальный исход наступил
в течение года. Наиболее продолжительным считается течение cancer еп
cuirasse. Продолжительность жизни больных с метастазами в кожу, рака
молочной железы, по данным И. Э. Теплицкого (1976), составляла от 1
месяца до 16 лет.
Гистопатология. Общим гистологическим признаком метастатического
рака в коже является отсутствие воспалительного инфильтрата, в отличие
от первичных опухолей, когда почти всегда имеется выраженное в той или
иной степени воспаление (Lausecker et al., 1979). Гистологическое иссле-
дование во многих случаях не дает возможности поставить диагноз первич-
-ной опухоли (Cawby и Weary, 1964, Ehlers и Krause, 1970, LeVer, 1975;
Lausecker et al., 1979). Считается, что наиболее часто совпадает струк-
тура метастаза и первичного очага при таких опухолях, как гипернефрома,
рак печени, щитовидной железы, желудка, хорионэпителиома. Количество
опухолевых клеток различно, меньше всего их при длительном течении can-
cer en cuirasse.
Дифференциальный диагноз. Дифференцировать метастатический рак
кожи необходимо от саркомы кожи, ретикулосаркоматоза кожи, грибо-
видного микоза (опухолевая стадия), липоматоза, ксантоматоза, ретикуло-
. гистиоцитомы.
Лечение. Лучевое, хирургическое, цитостатики.
ЛИТЕРАТУРА
Подлящук. Е. Л., Устинов В. Ф. Об изолированных метастазах рака в кожу головы.
Вестн- дерматол. и венерол. 1979, 9, 45—47.
Теплицкий И. Э. О метастазировании в кожу рака молочной железы. Клин, мед.,
1976, 6, 73—74.
Altmeyer Р. Kutane Metastasen eines Bronchialkarcinomas unter dem histologischen
Bild des ekkrinen Porokarzinoms.— Hautarzt, 1977, 28, 12, 661—663.
Baum E. M., Omura E. F., Payne R. R. Little LW. P. Alopecia neoplastica.— a
rare form of cutaneous metastasis.— J. Am. Acad. Der., 1981, 4, 6, 688—694.
Bernecker H. A., Bachmann W. Hautmetastasen eines Ovarialkarzinomas unter
dem Bild eines Erythema annulare.— Hautarzt, 1979, 30, 3, 164—165.
Debois J. M. Endometrial adenocarcinoma metastatic to the scalp.— Arch, dermatol.,
1982,* 118, 1, 42—43.
Haitschuh W., Bersch A. Hautmetastasen eines Magenkarzinoms unter dem Bild eines
beriignen mesenchymalen Tumors.— Hautarzt, 1981, 32, 9, 476—478.
HaselriggD. E., Rudolph A. H. Inflammatory metastatic carcinoma.— Arch. Derm.,
1977, 113, 1, 69—70.
Hode S. Fingermetastasen bei Brronchuscarcinom. Aktuel Derm. 1980, 6, 5, 248—254.
Freeman C. R.. Rosenfeld M., Schopflocher C. Cutaneous metastases from carcinoma
of the cervix.— Arch. Derm., 19Й2, 1982, 118, 1, 40—41.
28 Клиническая дерматология 433
Klehr N., Sehaeg G., Nasemann T. Cancer en cuirasse induziert durch einen maligns
Tranendrusenmischtumor.— Hautarzt, 1979, 30, 5, 299—304.
Lausecker H.t Steppert A., Gschnait F. Hautmetastasen der bosartigen Geschwulste.
In: Nicbt Entzundliche Dermatosen III B. Bdsartige Geschwulste. Leukamie. Hrsg. H. A.
Gottron, G. K. Korting, Springer—Verlag, Berlin. 1979, 328—417.
Littis P. J., Zuehlke R. L. Cutaneous metastatic carcinoma and elephantiasis sympto-
matica.— Arch. Derm., 1979, 1115, 1, 83—84.
Ordonez N. Y., Smith J. L. Peritoneal malignant mesothelioma with multiple distant
skin metastases.— Arch, derm., 1983, 119, 10, 827—830.
Padilla R. S., Sarmillo M.., Dao A. et al. Cutaneous metastatic adenocarcinoma of
gallbladder origin.— Arch, derm., 1982, 118, 7, 515—517.
Post B., Fanner M. Zur Haufigkeit und Lokalization des sekundaren Hautcarcino-
mas.— Hautarzt, 1974, 25, 1, 17—20.
Price N. M., Kopf A. W. Metastases to skin from occult malignant neoplasms.— Arch.
Derm., 1974, 109, 4, 547—550.
Rosen T. Cutaneous metastases.— Med. clin. North. Am., 1980, 64, 885—900.
Samitz M. H. Umbilical metastasis from carcinoma of the stomach.— Arch. Derm.,
1975, 111, 11, 1478—1479).
Stolp A., FUgemann S. Hautmetastasen eines hypernephroiden Karzinoms Derm.
Mschr., 1976, 162, 5, 426—433.
Zeligmarm L, Schwilm A. Umbilical metastasis from carcinoma of the colon.— Arch.
Derm., 1974, 110, 6, 911—912.
Wiesner M., Stolp A., Pambor M. etal. Ungewonliche Hautmetastasen eines Nagen-
karzinoms.— Derm. Mschr., 1984, 170, 5, 336—341.
РЕКЛИНГХАУЗЕНА БОЛЕЗНЬ (morbus Reclinghausen)
Синоним: нейрофиброматоз. -
Этиология и патогенез. Болезнь Реклингхаузена является аутосомно-
доминантным заболеванием из группы факоматозов. Значительная часть
случаев обусловлена спонтанными мутациями, частота которых, по мнению
А. С. Сергеева (1973), колеблется от 4,4 до 4,9х10~4. Около одного слу-
чая этого заболевания приходится на 3000 новорожденных (Christophers,
1979). Считается (Венкеи и Шугар, 1962), что нейрофибромы могут возни-
кать как из шванновских клеток, так и из соединительной ткани, окружаю-
щей нерв. По мнению А. К. Апатенко (1977), нейрофиброма развивается из
эндо-и периневральных соединительнотканных элементов. Не исключается
в то же время участие шванновских клеток.
Клиника. Клиническая картина многообразна. Ведущими являются
нейрокутанные изменения, но часто выявляются признаки поражения
внутренних органов, костной ткани, желез внутренней секреции, глаз.
На коже в большинстве случаев обнаруживаются пигментные пятна и
нейрофибромы, кожные и подкожные. Как правило наиболее ранним симп-
томом являются пигментные пятна, которые появляются вскоре после
рождения, обычно множественные, с гладкой поверхностью, характерным
желтовато-коричневатым цветом (цвет «кофе с молоком»). Величина их
обычно небольшая (0,5—1 см), но большую диагностическую значимость
434
Рис. 271. Болезнь
Реклингхаузена.
Рис. 270. Болезнь
Реклингхаузена (нейрофиброматоз).
имеют пятна, размер которых превышает 1,5—2 см, особенно у взрослых,
когда наличие более чем 6 подобного типа пятен делает диагноз нейрофиб-
роматоза вероятным (рис. 270, 271, 272, 273). Степень этой вероятности воз-
растает, если одновременно обнаруживают в подмышечных впадинах мно-
жественные мелкие (типа веснушек), скученно расположенные пятна. We-
sterhof и Konrad (1982) обращают внимание на диагностическую значимость
2 феноменов: голубовато-красноватые и псевдоатрофические пятна. Голубо-
вато-красноватые пятна могут возникнуть до или в период полового созре-
вания, имеют размер в среднем около 3 см, чаще располагаются на туло-
вище, могут слегка возвышаться над кожей, исчезают при диаскопии.
Псевдоатрофические пятна немногочисленны, размером от 5 до 10 см, рас-
полагаются на бедре, пояснице, животе. Могут существовать с рождения
или появляться в раннем возрасте. По данным Е. А. Абаммасовой и Р. Н.
Бунятова (1979), пигментные пятна наблюдались у 79,5% больных из 219.
В 82,5% случаев только наличия пигментаций на коЯсе было достаточно
для постановки диагноза. Нейрофибромы появляются обычно позже, чем
пигментные пятна, в позднем детском или подростковом возрасте. По дан-
ным Р. С. Бабаянца и А. С. Сергеева (1973), около 65% больных отметили
появление кожных опухолей в возрасте до 16 лет. У 4—они были с рожде-
ния, у 6 человек—нейрофибромы стали появляться позже 25 лет. Они мо-
гут иметь вид мягких фибром, цвета нормальной кожи или слегка пигмен-
тированных, расположенных преимущественно на туловище, небольших
размеров, нередко на ножке, мягкой консистенции, или плотноватых опу-
холевидных новообразований, расположенных в подкожной клетчатке. В
28* 435
этом случае они могут располагаться по ходу периферических нервов (обыч-
но в области тройничного и верхнего шейного). Над глубоко лежащими
нейрофибромами у ряда больных обнаруживаются «пустые» грыжевидные
выпячивания, в зоне которых кожа слегка цианотична. Количество опу-
холей различно—от нескольких десятков до сотен и даже тысяч (Шекла-
ковН. Д., 1950; Шелыгина Н. М. и Зарецкий М. М., 1974; Бахадыров
К. Б. и соавт. 1983). Вначале их обычно немного, но с течением времени
количество их нарастает, особенно в период полового созревания и бере-
менности. Величина их у разных больных, да и у одного и того же больного
различна, иногда достигает размера кулака взрослого человека и более
(Mukherji, 1974). В ряде случаев (по данным Crowe et al., 1956, у 16% боль-
ных) развивается диффузный нейрофиброматоз с избыточным разраста-
436
нием соединительной ткани кожи и подковной клетчатки с возникновением
опухолевидных образований гигантских размеров (elephantiasis neuro-
fibromatosa), Henington и Caros (1959) наблюдали обширные свисающие с
лица и шеи, плеч и верхней части груди в виде крупных складок фибро-
матозные разрастания (fibroma pendulum) у больной нейрофиброматозом
69 лет. Первые признаки заболевания появились в 16 лет, когда разви-
лись множественные мелкие опухолевидные узелки (нейрофибромы). На
шее и лице слева они стали постепенно увеличиваться в размерах и до
30-летнего возраста больной достигли почти уровня пупка. В дальнейшем
рост замедлился и в момент обследования разрастания были на уровне
лобка. Наряду с этим обнаруживались сотни нейрофибром, немногочис-
ленные пятна цвета кофе с молоком.
В зоне нейрофибром могут быть нарушения различных видов чувст-
вительности, иногда болезненность. Из других изменений кожи описыва-
ются невусы, пигментные и сосудистые, cutis laxa, синдром Ehlers—Dan-
los, аденома сальных желез, ихтиоз (Зарбаева С. Ф., 1971; .Куклин В. Т.
и соавт., 1976 и др.). Нейрофибромы в полости рта обнаруживаются у 1—
5% больных (Freedus и Doyle, 1975). В наблюдении И. Ю. Голоусенко
(1982), у больной помимо множественных изменений на коже обнаружива-
лось утолщение десен, множественные опухолевидные образования в по-
лости рта. Клинические проявления изменений нервной системы зависят от
локализации и характера поражения (ограниченное или диффузное). Они
наблюдаются у 3—8% больных (Crowe et al., 1956). Наиболее часто в про-
цесс вовлекаются глазной, слуховой и тройничный нерв, в результате чего
могут развиваться головные боли, головокружения, невралгии, снижение
слуха, зрения, экзофтальм. Больные часто апатичны, страдают депрессией,
у них снижен интеллект. Развитие внутричерепных опухолей может вызы-
вать эпилепсию. Изменения костной системы обнаруживаются у многих
больных и характеризуются остеодистрофией, кифосколиозом, гипоплазией
ребер, возникновением субпериостальных кист, деформацией твердого не-
ба, челюстей, наличием spina bifida.
Е. А. Абаммасова и Р. Н. Бунятов (1979) обнаружили деформацию
позвоночника (кифоз, сколиоз) у 121 из 219 больных. М. Е. Липец и соавт.
(1982) указывают на возможность развития костных изменений в виде саб-
левидных голеней. По данным М. О. Злотникова (1930), не является ред-
костью фиброматоз, полипоз внутренних органов. Имеются сообщения о
различных изменениях сердечно-сосудистой системы у больных нейрофиб-
роматозом (врожденные пороки сердца, гипертоническая болезнь и др.),
частота которых, по мнению Person и Perry (1978), большая, чем принято
считать. Нет ясности и в отношении эндокринных нарушений при нейро-
фиброматозе. Наблюдаются акромегалия, диабет, гипогонадизм, недоста-
точность щитовидной и паращитовидных желез, но самым частым считается
феохромоцитома (Crowe et al., 1956). От 10 до 20% взрослых имеют болезни
легких (Davis и Kaplan, 1978). Возникновение неврином в желудке и ки-
шечнике наблюдается примерно у 10% больных (Hochberg et al., 1974),
что может сопровождаться кровотечением, симптомами непроходимости
437
(Sivak et al., 1975). В зависимости от комбинации тех или иных признаков
кожных и висцеральных изменений Riccardi (1982) выделяет 8 клинических
вариантов.
Прогноз для выздоровления неблагоприятный. Появившись, ней-
рофибромы существуют в течение всей жизни. Субъективных расстройств
они обычно не вызывают. Злокачественное перерождение, по мнению Un-
dentsch (1957), наблюдается редко, по данным же других авторов (Crowe
et al., D’Agostino et al., 1963) это происходит в 2—3,1% случаев, преиму-
щественно в области гигантских нейрофибром. Злокачественные опухоли в
центральной нервной системе—это главным образом астроцитомы. Him-
stedt и соавт’. (1982) полагают, что частота саркоматозного перерождения
ниже 3%. Khight и соавт. (1973) считают, что злокачественное перерожде-
ние нейрофибром происходит в 3—15% случаев, но, по их мнению, точная
оценка не представляется возможной из-за отсутствия адекватного наблю-
дения за больными, диагноз которым поставлен в детстве. Описано разви-
тие меланомы у больного нейрофиброматозом (Khight et al., 1973; Mastran-
gelo et al., 1979), системного мастоцитоза (Merot et al., 1984). Riccardi (1982)
описывает 11 типов опухоли при болезни Реклингхаузена (кожные и висце-
ральные нейрофибромы, глиомы зрительного нерва, невромы слухового
нерва, менингомы и т. д.). Больные могут погибать от нарушения ликво-
обращения при локализации нейрофибром в головном мозгу (Челноков
В. С. и Игошин Ю. М., 1980). Braun (1958) указывает на повышенную склон-
ность больных к развитию злокачественных новообразований. В то же вре-
мя могут встречаться, чаще у родственников больного типичным нейро-
фиброматозом, малосимптомные формы болезни Реклингхаузена, проявляю-
щиеся наличием немногочисленных пигментных пятен цвета кофе с молоком.
Гистопатология. Строма опухоли состоит из полосовидно располо-
женных волнистых фиброзных волокон; множественны сосуды, встречаются
тучные клетки, тонкие нервные волокна (Lever, 1975). В пигментных пят-
нах и клинически неизмененной коже могут обнаруживаться гигантские
пигментные гранулы в эпидермальных клетках и меланоцитах, они не об-
наруживаются в норме, но встречаются при синдроме Albright (Rook, 1979),
лентиго и naevus spilus (Morris et al., 1982). По данным Weber и Braun-
Falco (1972), основными клеточными элементами нейрофибром являются
шванновские клетки. А. К- Апатенко (1977) выделяет 3 морфологических
варианта: фиброзный, с преобладанием клеточных структур и смешанный.
Дифференциальный диагноз. Следует проводить с фибромами, липо-
мами, невусами, синдромом эпидермального невуса (Stein et al., 1972).
Лечение. Отдельные наиболее крупные нейрофибромы могут быть
удалены хирургически. Однако хирургическое вмешательство обычно по-
казано при локализации опухолей в других органах и системах. -
ЛИТЕРАТУРА
Апатенко А. К- Мезенхимные и нейроэктодермальные опухоли и пороки развития
кожи. М. Медицина, <977. !
438
Бабаянц Р. С., Сергеев А. С. К вопросу о диагностике болезни Реклингхаузена
(медико-генетический аспект). Вестн. дерматол. и венерол., 1973, 8, 10—16.
Шелыгина Н. М., Зарецкий М. М., Редкий случай множественного нейрофибро-
матоза. Вестн. дерматол. и венерол., 1974, 3, 82—83.
Куклин В. Т., Теплоухова Г. И., Астахова К- Т. О сочетании ихтиоза с болезнью
Реклингхаузена. Вестн. дерматол. и венерол. 1976, 11, 65—67
Абаммасова Е. А., Б унятое Р. Н. Роль кожной пигментации в диагностике сколио-
за при нейрофиброматозе (болезнь Реклингхаузена) Вести, дерматол. и венерол., 1979,
10, 52—55.
Челноков В. С., Игошин Ю. М. Два случая необычного исхода болезни Реклинг-
хаузена. Вестн. Дерматол. и венерол. 1980, 80, 62—64.
Липец М. Е., Прокофьева Н. М-, Шибаева Л. Н. К костной симптоматике при бо-
лезни Реклингхаузена. Вестн. дерматол. и венерол. 1982, 11, 52—55.
Crowe F. V., Schull W. J., Neel J. V. Clinical pathological and genetic study of
multiple neurofibromatosis.— Springsfeld, Charles, C. Thomas 1956, cit. J. R. Person, H. O.
Perry Recent aedvances In the phakomatoses.— Int. J. Dermatol., 1978, 17, 1, 11—13.
Daois S., Kaplan R. L. Neurofibromatosis and interstitial lung disease.— Arch, der*
mat., 1978, 114, 9, 1368—1369.
Freedus M. S-, Doyle P. K- Multiple neurofibromatosis with oral manifestations.—
J. oral, surg., 1975, 33, 360.
Hlmsiedt P. D.f Markowitz A. G., Muller H. Sarcomatose Entartung bei Neurofibro-
matosae — Recklinghausen. Hautarzt, 1982, 33, 529—532.
Hochberg F. H.t Dasiloa A. B., Galdabini J., Richardson E. P. Gastrointestinal
involvement in von Reclinghausen’s neurofibromatosis. Neurology, 1974, 24, 1144.
Mastrangelo M. J., Goepp С. E., Patel Y. A. Clark N. H. Cutaneous melanoma in a
patient with neurofibromatosis.— Arch, derm., 1979, 115, 7, 864—865.
Merot Y., Banquis C., Saurai J. H. Neurofibromatoss de von Recklinghausen et
mastocytose systemique.— Dermatologica, 1984, 168, 25—30.
Morris P. J., Johnson W. G., Silvers D. N. Giant pigment granules in biopsy speci-
mes from cafe au lait spors in neurofibromatosis.— Arch, derm., 1982, 6, 385—388.
Rook A. Geneticsin dermatology In: Textbook of dermatology. Ed. Rook A. Wilkin-
son D. S. F. J. G. Ebling, Oxford, London, Edinburgh, Melbourne, 1979, 1, 97—140.
Sivak M. V., Sullivan В. H., Farmer R. G. Neurogenic tumors of the small
intenstine.— Gastroenterology, 1975, 68, 374.
Westerhof W., Konrad K- Blue-red macules and pseudoatrophic macules. Arch, derm.,
1982, 118, 8, 577—581. t
РЕТИКУЛОГИСТИОЦИТОМА КОЖИ (reticulohistiocytoma cutis).
Синонимы: кожная гигантоклеточная опухоль, ретикулогистиоцитар-
ная гранулема, гигантоклеточная гранулема, множественная ретикуло-
гистиоцитома.
Ретикулогистиоцитома относится к доброкачественным фиброгистио-
цитарным опухолям кожи. Первое клинико-морфологическое описание фи-
бромы в русской литературе принадлежит видному московскому дермато-
439
логу Е. С. Главче (1902). К 1973 г. в мировой литературе приведены дан-
ное о 95 больных ретинулогистиоцитомой.
Этиология и патогенез. Сущность и гистогенез ретикулогистиоцитомы
неясны. Некоторые авторы (Саго и Senesr, 1952; Dupout и Vandaele, 1957;
Montgomery et al., 1958 и др.) считают ее реактивно-воспалительным гра-
нулематозом. Popchristov (1963), Gordon (1964) полагают, что ретикуло-.
гистиоцитома близка к ангиофиброксантомам, гистиоцитомам и ксантомам,
хотя, возможно, и представляет собой особую онко-нозологическую форму.
Ю. В. Постнов (1962) рассматривает ретикулогистиоцитому не как грану-
лему или истинную опухоль, а как своеобразную очаговую дисплазию рети-
кулярной ткани кожи. А. К- Апатенко (1973, 1977) считает, что есть осно-
вания связать гистогенетически ретикулогистиоцитому с полипотентными
адвентициальными клетками сосудов. Он относит ретикулогистиоцитому'
к особой разновидности ангиофиброксантомы, где основным компонентом
опухоли являются гистиоцитарно-макрофагальные элементы. Мы рас-
сматриваем ретикулогистиоцитому кожи как самостоятельную нозологи-
ческую опухолевую форму. Кроме солитарных ретикулогистиоцитом в ли-
тературе описаны множественные ретикулогистиоцитомы (Суворова К. Н.,
1968; Каламкарян А. А. и Гребенюк В. Н., 1980; Каламкарян А. А. и
соавт., 1985; Weber и Freudenthal, 1937; Warin и Senear, 1957; Barrow и
Holubar, 1967 и др.), характеризующиеся множественными опухолевидно-
узловатыми поражениями кожи. Множественную ретикулогистиоцитому
следует отличать от множественного ретикулогистиоцитоза, который пред-
ставляет собой системное заболевание (в процесс вовлекаются многие ор-
ганы—печень, почки, сердце, суставы, сосуды, нервная система и др.) с
кожными проявлениями. По-видимому, они близки, но не идентичны.
Catterall и White (1978) считают, что множественный ретикулогистиоцитоз,
подобно дерматомиозиту, должен учитываться как редкое проявление ма-
лигнитета. Из 41 случая множественного ретикулогистиоцитоза, описан-
ного в литературе, у 11 (27%) был рак внутренних органов. А. А. Калам-
карян и В. Н. Гребенюк (1980) на основании собственных исследований
допускают существование промежуточных форм заболевания между рети-
кулогистиоцитомой и множественным ретикулогистиоцитозом. Согласно
Международной классификации опухолей (Женева, ВОЗ, 1974), ретикуло-
гистиоцитома относится к опухолям из соединительной ткани.
Клиника. Ретикулогистноцитома встречается в любом возрасте, оди-
наково часто у мужчин и женщин; множественная ретикулогистноцитома—
чаще у женщин старше 30 лет. Появляются шаровидные или уплощенные
узлы и опухоли величиной от горошины до лесного ореха и даже до голу-
биного яйца. Очаги поражения безболезненны, слегка зудят, возвышаются
над уровнем окружающей кожи на 0,5—2 см; некоторые мелкие узелки
вначале не выдаются над общим уровнем кожи и заметны лишь вследствие
своей буроватой окраски (рис. 274, 275, 276). Консистенция опухолей и
бляшек плотно-эластическая; окраска—коричневатая с розовато-синюш-
ным оттенком. Отдельные опухоли могут сливаться в крупный очаг с буг-
ристой поверхностью. Локализация—различные участки кожного покрова
440
Рис. 274. Ретикулогистиоцитома
множ ес таенная
Рис. 275. Ретикулогистиоцитома
множественная (та же больная).
Рис. 276. Ретикулогистиоцитома кожи.
цитоплазмой. Вокруг них—мелкок
(лицо, нижние и верхние конеч-
ности, туловище, волосистая часть
головы, гениталии). Иногда узел-
ки и узлы рассеяны по всей коже;
в других случаях они группиру-
ются в одной области, распола-
гаясь близко друг к другу. Очаги
поражения растут медленно, в
течение многих лет увеличиваются
в размере, изредка подвергаются
инволюции. На месте разрешив-
шихся узлов и опухолей остается
выраженная пигментация или ат-
рофически-пигментные изменения.
Гистопатология. В дерме об-
наруживаются многоядерные ги-
гантские клетки с базофильной
ная инфильтрация. В цитоплазме
некоторых гигантских клеток определяются липиды и гемосидерин.
Дифференциальный диагноз. Проводят с ретикулосаркоматозом ко-
жи, дерматофибросаркомой, множественным (системным) ретикулогистио-
цитозом, ангиофиброксантомой, дерматофибромой, фибросаркомой.
Лечение. Достаточно эффективной терапии ретикулогистиоцитомы
нет. Мы наблюдали хороший эффект от комплексной терапии цитостати-
ком проспидином (на курс 3—3,5 г) в сочетании с кортикостероидами (пред-
низолон по 20—30 мг в сутки).
ЛИТЕРАТУРА
Апатенко А. К., Потекаев Н. С. О так называемом ретикулогистиоцитозе (кожно-
артропатический гигантоклеточный ретикулогистиоцитоз).— Арх. патол., 1976, 6, 61—64.
Каламкарян А. А., Гребенюк В. Н. Множественная ретикулогистиоцитома. —
Вести, дерматол. н венерол., 1980, 9. 28—32.
441
Каламкарян Л. А.,- Рутштейн Л. Г., Ройтбурд М- Ф.« Забамова В. Е. Множест'
венная ретикулогистноцитома, успешно леченная проспиДином и преднизолоном.— Вести-
дерматол. н венерол., 1985, 6.
Caiterall М., White J. Multicentric reticulohistiocytosis and malignan disease.—
Brit. J. Dermatol., 1978, 98, 2, 221—224.
Cheozant-Breton J. La reticulo-histiocytose multicentrique revue de la litterature
recente.— Ann. Dermatol. Venereol., 1977, 104, 745—753.
РЕТИКУЛОСАРКОМАТОЗ КОЖИ (reticulosarcomatosis cutis).
Синонимы: злокачественный ретикулосаркоматоз кожи, лимфобласто-
ма, злокачественная лимфома.
В 1950 году Gottron ввел понятие «так называемый ретикулосарко-
матоз кожи», определив это заболевание как переходное между ретику-
лезом и ретотельсаркомой.
Этиология и патогенез. Большинство современных авторов и среди
них Н. С. Смелов и Е. Ф. Беляева (1958), А. А. Каламкарян (1969, 1976,
1983), Ю. Я. Ашмарин (1972), Н. С. Потекаев (1974), В. С. Проскурнина
(1975), Р. Д. Штерн (1976), Knoth и Sandritter (1965) и др. считают рети-
кулосаркоматоз кожи злокачественным пролиферативным процессом в лим-
фоидно-ретикулярной системе кожи. Lukes и Collins (1973), Lessana-Lei-
bovitch (1975), Mathe и соавт. (1975), Lever и Schaumburg-Lever (1975)
полагают, что речь идет о злокачественной лимфоме, о кожных проявле-
ниях лимфобластоматоза.
Клиника. Мужчины болеют в 1,5—2 раза чаще, чем женщины; забо-
левание встречается преимущественно в среднем и пожилом возрасте.
Обосновано выделение острой и подострой форм, характеризующихся
быстротой и тяжестью течения. При острой форме заболевание начина-
ется внезапно высыпанием на лице, груди, спине и конечностях розово-
синюшного цвета пятен и мелких фолликулярных узелков, из которых в
течение 1,5—3 недель формируются инфильтративные бляшки уплощенной
формы, хорошо отграниченные, мягковатой и плотно-эластической кон-
систенции и опухолевидные образования. Опухоли достигают величины
вишни, куриного яйца, плотны на оЩупь, имеют синюшно-багровый цвет.
Поверхность гладкая с наличием множественных телеангиэктазий и ге-
моррагий. Характерен быстрый распад с образованием глубоких язв. Ча-
сто отмечается увеличение периферических лимфатических узлов.
При подострой форме—опухоли, инфильтративные бляшки, величиной
от мелкого ореха до детского кулака, резко отграничены, умеренно плот-
ны на ощупь, возвышаются на 1—4 см над уровнем кожи и имеют насы-
щенно красный с синюшно-багровым оттенком цвет. Опухоли спаяны с
кожей, но подвижны по отношению к подлежащим тканям. Поверхность
их гладкая или бугристая, блестящая с подчеркнутым фолликулярным
рисунком, напоминающим апельсиновую корку. На поверхности видны
телеангиэктазии и точечные геморрагии. Опухоли чаще располагаются
изолированно, реже сливаются в крупные бугристые образования. Субъ-
ективные ощущения отсутствуют, опухоли безболезненны при пальпации.
442
Рис. 277.
Ретикулосаркоматоз
кожи.
Рис. 278.
Ретикулосаркоматоз
кожи.
I
Рис. 280.
Ретикулосаркоматоз
кожи.
Рис. 279.
Ретикулосаркоматоз
кожи.
443
Рис. 282. Ретикулосаркоматоз
кожи
Рис. 283. Ретикулосаркоматоз
ножи
Рис. 284. Ретикулосаркоматоз
коки.
Рис. 281. Ретикулосаркоматоз
кожи.
444
Рис. 285.
Ретикулосаркоматоз
кожи.
Рис. 286.
Ретикулосаркоматоз
кожи.
Рис. 287.
Ретикулосаркоматоз
кожи.
Рис. 288.
Ретикулосаркоматоз
кожи.
445
Рис. 289. Сифилис,
клинически напоминающий
ретикулосаркоматоз
Рис. 290. Ретикулосаркоматоз кожи
(сифилис, симулирующий
ретикулосаркоматоз кожи).
Рис. 291. Сифилис,
напоминающий
ретикулосаркоматоз.
Рис. 292. Сифилис,
симулирующий
ретикулосаркоматоз.
446
В результате распада опухолей образуются глубокие болезненные язвы с
резко очерченными, валикообразно утолщенными краями, окрашенными
в синюшно-багровый цвет с гнойно-кровянистым отделяемым и остат-
ками гангренозно распавшихся тканей (рис. 277—292)
Bourgeois, Spinasse и соавт. (1970), Hadida и соавт. (1970) описали
опухолевидно-буллезные поражения при злокачественном ретикулезе кожи.
Нередко поражается слизистая оболочка полости рта с образованием
массивных, хорошо отграниченных опухолей и инфильтратов багрово-
синеватого цвета, через 1—2 недели подвергающихся некротическому рас-
паду с образованием резко болезненных, глубоких язв неправильных очер-
таний с вывороченными краями и с грязно-серым распадом в центре. До-
вольно часто отмечается генерализованное увеличение периферических
лимфатических узлов. Характерны трофические нарушения: очаги облы-
сения на волосистой части головы, выпадение бровей, нарушение потоот-
деления. При подостро текущих формах, несмотря на тяжесть и обшир-
ность поражения кожного покрова, длительное время, иногда вплоть до
терминального периода сохраняется удовлетворительное общее состояние,
отсутствует лихорадка.
Изменения гемограммы и миелограммы в большинстве случаев не от-
ражают тяжести процесса и могут иметь диагностическое и прогностиче-
ское значение только в совокупности со всей клинической картиной забо-
левания. В терминальном периоде заболевания часто наблюдается уве-
личение СОЭ, тромбоцитопения, лейкопения, моноцитоз, анемия. У по-
давляющего большинства больных в пунктате лимфатических узлов обна-
руживается ретикулярно-лимфоидная типерплазия; дермограммы (пре-
параты-отпечатки биопсированной кожи) характеризуются выраженной
ретикулярной и ретикулярно-лимфоидной гиперплазией, что свидетельст-
вует о высокой степени злокачественности. Поражения внутренних орга-
нов появляются лишь у части больных в терминальной стадии.
Течение болезни злокачественное, прогрессирующее. Длительность
жизни при остром ретикулосаркоматозе 3—12 месяцев, при подостром—
не более 1,5—3 лет.
Гистопатология. Эпидермис без специфических изменений, часто об-
наруживается атрофия и сглаживание эпителиальных отростков. В дерме
и подкожной клетчатке выявляются диффузные пролифераты из круп-
ных ретикулярных и лимфоидно-ретикулярных клеток с гиперхромными
ядрами и многочисленными митозами (Каламкарян А. А., 1969; Пдте-
каев Н. С., 1974; Проскурнина В. С., 1975; Burg и Braun-Falco, 1975 и
др.). Располагаются пролифераты преимущественно вокруг сосудов и при-
датков кожи. Под эпидермисом сохраняется полоса неизмененной соедини-
тельной ткани; в отдельных случаях специфический пролиферат проника-
ет в эпидермис. В области пролиферата разрушаются коллагеновые и эла-
стические волокна. В лимфатических узлах наблюдается диффузная про-
лиферация ретикулярных и лимфоидно-ретикулярных клеток. Многими
исследователями подчеркивается высокая метаболическая и пролифера-
тивная активность клеток у больных ретикулОсаркоматозом кожи.
447
. Лечение. Цитостатики (проспидин, винкристин, винбластин, мето-
трексат, 6-меркапторурин, циклофосфан, фосфазин), кортикостероиды (пред-
низолон, дексаметазон, триамцинолон), рентгеновские лучи. Наиболее
целесообразно их комплексное применение.
ЛИТЕРАТУРА
/
Каламкарян А. А. Некоторые новые аспекты классификации ретикулогемобласто-
зов.— Вестн. дерматол. и венерол., 1976, 9, 3—8.
Каламкарян А. А. Клиника и терапия ретикулезов кожи. «Айастан,» Ереван, 1983.
Шапошников О. Кч Геншер Е. И., Родионов А. Н. Функциональное состояние
лимфоцитов из лимфатических узлов у больных злокачественными лимфомами кожи.—
Вестн. дерматол. и венерол., 1982, 8, 4—7.
Потекаев И. С. Первичный ретикулез кожи: автореф. дисс. докт. мед. наук.— М., 1974.
Проскурнина В. С. Материалы к изучению патогенеза грибовидного микоза: автореф.
докт. мед. наук.— М., 1975.
BouoaletD. Histopathologic les hematodermies. Dermatologica, 1978,157, 5, 257—268.
Civaite J., Blanchet P. Les hematodermies. Actuual. hematol. Fr., 1975, 9, 153—161.
Lessana-Leibooitch M., Hewitt J. Nosologie des reticuloses cutanees.— Ann. Med.
Interne Fr., 1975, 126, 3, 183—177.
Lever IF., Schaumburg-Lever G- Histopathology of the skin.— 5th ed.— Philadep-
hia, 1975.
Mathe G., Belpomme D„ Dantcheo D. Hematosarcomas non hodgkinienns.— Revi
sion actueolle de la nomenclature et de la classification.— Nouv. Presse med., 1975, 4, 4, 241—
245.
Steigleder G. Bengn and malignant proliferative response. Dermal infiltration, origin
and importance.— Acta Derma to venereol., 1976, 56, 1, 33—41.
Walk S. Primary malignant lymphoma cutis. — Can. Med. Ass. J., 1977, 117, 7, 750—
752.
РИНОФИМА (rhinophyma)
Этиология и патогенез. Рассматривается как осложнение (Marx и Wil-
kinson, 1979) или как форма розовых угрей: железистый гиперпластиче-
ский тип (Lever, 1975). Kleine-Natrop и соавт. (1981) считают сочетание
ринофимы с розовыми угрями обычным явлением. По мнению Korting и
Denk (1974), ринофима часто сочетается с розацеа или выраженной се-
бореей, но это не является обязательным. В группе розацеа описывает ри-
нофиму и Steigleder (1975) как ограниченное разрастание сальных желез,
хотя и подчеркивается, что ринофима может наблюдаться и без других
симптомов розацеа. По данным Acker и Helwig (1967), acne rosacea как
ранние проявления ринофимы наблюдались только у 30% больных. Е. И.
Рыжкова (1974) рассматривает ринофиму как инфильтративно-продуктив-
ную стадию розацеа. Предрасполагающими факторами могут быть хро-
нические заболевания желудочно-кишечного тракта, неблагоприятные
средовые воздействия, в первую очередь актиническое повреждение кожи,
переохлаждение, злоупотребление алкоголем. Следует подчеркнуть од-
нако, что Marx и Wilkinson (1979), отмечая традиционную связь развития
448
Рис. 293. Ринофима.
Рис. 294. Начинающаяся ринофима,
лимфостаз у больного розовыми
угрями.
ринофимы с алкоголизмом, считают, что для этого нет достаточных дока-
зательств.
Клиника. Резкое увеличение размеров носа за счет развития бугри-
стых дольчатых узловатостей, разделенных бороздками. Консистенция их
обычно мягкая, цвет застойный, на поверхности—множественные телеан-
гиэктазии, часто возникают пиогенные элементы. Фолликулы расширены,
при надавливании из них выделяется салообразная масса. Узловатости
могут сливаться, достигать гигантских размеров, что приводит к резкой
деформации носа и обезображиванию лица. В редких случаях поражаются
другие участки лица (лоб, щеки, подбородок, уши), при этом могут раз-
виваться лепроподобные изменения. Хрящ, кости носа в процесс не вовле-
каются (Konz, 1975). Рисунки 293, 294.
Заболевание возникает преимущественно у мужчин среднего и особен-
но пожилого возраста, развивается медленно. У больных часты признаки
розацеа, могут наблюдаться заболевания глаз (конъюнктивит, блефарит,
кератит), явления эластоидоза, иногда развивается базалиома. По мне-
нию Acker и Helwig (1967), базалиомы при ринофиме бывают, вероятно,
чаще, чем на нормальной коже носа.
Гистопатология. Разрастание фиброзной ткани, гиперплазия ''альных
желез, атрофия волосяных фолликулов, гиперваскуляризация, микро-
циркуляторные нарушения, воспалительная инфильтрация с примесью
ней рофилов (Цветкова Г. М. и соавт., 1977).
29 Клиническая дерматология 449
Дифференциальный диагноз. Дифференцировать ринофиму необходимо
от бромодермы, лепры, ретикулосаркоматоза, грибовидного микоза.
Лечение. Хирургическое.
ЛИТЕРАТУРА
Рыжкова Е. И- Клинико-морфологические особенности, патогенез и лечение роза-
цеа. Дисс. докт. М. 1976.
Acker W., Helwig Е. В. Phinophyma with carcinoma..— Arch. Derm., 1977, 95, 3,
250—254.
Konz B. Zur operativen Behandlung des Rhinophymes.— Hautarzt, 1975, 26, 4, 211—
214.
Korting Y. W., Denk R. Dermatologische Differentiqldiagnose. Stuttgart, N—Y,
1974.
Kleine-Natrop H. E., Sebastian G., Norn K- Uber Regeln der Rhinophym—Chirur-
gie.— Derm. Mschr., 1981, 167. 6, 370—376.
Marx R., Wilkinson D. S. Rosacea and perioral dermatitis In: «Textbook of dermato-
logy Ed. byl. -Rook et al. Oxford, 1979, 3 ed, 1433—1440.
Steigleder G. K- Dermatologie und Venerologie, Stuttgart, 1975, 476—481.
РОГ КОЖНЫЙ (cornu cutaneum)
Этиология и патогенез. Кожный рог представляет собой новообразо-
вание из роговых масс. Вопрос о его самостоятельности спорный. По мне-
нию Jakolza и соавт. (1976),—это, как правило, собирательное понятие,
которое по клиническим проявлениям сходно, но по патогенезу и течению
различно. Преимущественно возникает на фоне преканцеррзной пролифе-
рации эпидермиса, особенно часто на фоне сенильного кератоза. Korting
и Denk (1974) считают, что кожный рог в большинстве случаев представляет
собой вариант сенильной кератомы и так как может маскировать керато-
акантому, спиналиому или болезнь Боуэна, во всех случаях необходимо
гистологическое исследование. По мнению Д. И. Головина (1982), кожный
рог представляет собой собирательное понятие, а не отдельную онконозоло-
гическую единицу. Он может оказаться папилломой, раком in situ, в том
числе типа Боуэна, и кератоакантомой.
С точки зрения Junge и соавт. (1973), понятие «кожный рог» может
служить лишь вспомогательным средством для обозначения клинических
особенностей, и сохранение его как нозологической единицы не оправдано.
Базу для развития кожного рога составляет ряд морфологически и этиоло-
гически гетерогенных изменений кожи, из которых наиболее частыми, по
наблюдениям авторов, являются солнечный кератоз, спиналиома, бородав-
ки обыкновенные и кератоакантома. И по мнению Cramer и Kahlert (1964),
обозначение «кожный рог» должно служить только как предварительный
клинический диагноз, постановка которого должна предполагать во всех
случаях гистологическое исследование. Как полагает Higoumenakis (1967),
для развития кожного рога необходима предрасположенность, которая
реализуется при хроническом локальном раздражении самого различного
характера. Среди провоцирующих факторов отмечаются микротравматиза-
450
ция, имеющая значение в случае
развития кожного рога на здоровой
на вид коже, актинические повреж-
дения эпидермиса, вирусная инфек-
ция и др. (Higoumenakis, 1967; Rit-
chie и Mullins, 1968; Alkiewicz, 1975
и др).
Клиника. Ограниченные разрас-
тания роговых масс, напоминающие
рога животных, большей частью ко-
нической формы, обычно прямые,
желтовато-коричневатого или темно-
го цвета, плотноватой или плотной
консистенции (рис. 295). Поверх-
ность гладкая или неровная с мно-
жественными бороздами. Воспали-
тельные явления выявляются только
в непосредственной близости к осно-
ванию рога в виде узкого эритема-
тозного венчика. Роговые новообра-
зования могут достигать очень боль-
ших размеров (до 30 см), реже рост
в длину небольшой, разрастания
занимают более обширные участки
по поверхности, но и в этих случаях
Рис. 295. Кожный рог.
размеры верхушки значительно уже основания. Высота кожного рога, по
мнению Cramer и Kahlert (1964), может служить в известной мере прогнос-
тическим признаком. Так, кожный рог, размеры которого не превышали
1 см, обычно развивается на фоне базалиомы и сенильной кератомы. В ос-
новании кожного рога большего размера гистологически выявлялись
себорейные бородавки, кератоакантома, ороговевающая папиллома. На
красной кайме губ высота кожного рога обычно не превышает 0,5—1 см
(Машкиллейсон А. Л., 1970). Поражается значительно чаще нижняя губа,
нередко фоном служат различные патологические процессы (красная и
туберкулезная волчанка, лейкоплакия и др.).
Кожный рог обычно одиночный, множественные новообразования
являются редкостью (Desar и Stiittgen, 1968). Описанный Л. Н. Химки-
ной и Э. И. Радченко (1983) случай множественного кожного рога, по всей
вероятности, представляет собой паранеопластическое поражение у боль-
ного раком желудка.
Развивается несколько чаще у женщин, особенно старших возрастов,
располагается преимущественно на лице (уши, щеки) и волосистой части
головы. По наблюдениям Cramer и Kahlert (1964) кожный рог, располагав-
шийся на других, кроме головы, участках кожного покрова, был доброка-
чественным. Редко кожный рог располагается на слизистых и полуслизи-
стых. Течение и прогноз зависят от дерматоза, на фоне которого развился
29*
451
кожный рог. Растет обычно медленно. Наиболее часто выявляется рак в
случаях кожного рога, развившегося на фоне сенильного кератоза, не
считая тех случаев, когда он возник в зоне опухолей.
Гистопатология. Резко выраженный гиперкератоз, папилломатоз; в
основании, как указывалось, могут быть самые разнообразные процессы—
преканцерозные, злокачественные и доброкачественные опухолевые, ин-
фекционные, обусловленные травмой и др. (Junge et al., 1973; Alkiewicz,
1975; Gibbs и Hyman, 1968; Sandbank, 1971 и др.).
Дифференциальный диагноз. Дифференцировать кожный рог необхо-
димо от бородавок, мозолей, фибром, ангиокератомы ограниченной невифор-
мной, бородавчатых невусов, веррукозного псориаза.
Лечение хирургическое.
ЛИТЕРАТУРА
Головин Д. И. Ошибки и трудности гистологической диагностики опухолей. М.
Медицина, 1982.
Alkiewicz J., Zur Histologie und Klinik des Cornu cutaneum) — Hautarzt, 1975, 26,
3, 155—157.
JakolzaD. Lehnert N., Thormann T. B. Cornu cutaneum gigantum auf hypertrophi-
schem Keratoma senile. Derm. Kschr., 1976, 162, 7, 591—593.
Korting G. W., Denk R. Dermatologische Differentialdiagnose, Stuttgart, 1974.
РОССОЛИМО—МЕЛЬКЕРССОНА—РОЗЕНТАЛЯ СИНДРОМ (Rossoli-
mo—Melkersson—'Rosenthal syndromum)
Синонимы: синдром Мелькерссона—Розенталя.
В 1928 г. Melkersson описал больного с рецидивирующим парезом ли-
цевого нерва и стойким отеком губ. В 1931 г. Rosenthal отметил третий
симптом этого заболевания—складчатый или скротальный язык. Lucher
(1919) предложил заболевание, характеризующееся невритом лицевого
нерва, отеком лица и складчатым языком именовать синдромом Мелькерс-
сона—Розенталя. На триаду симптомов, типичных для данного синдрома,
как указывают французские авторы Mouly и Vaillont (1962), еще в 1901 г.
указал русский невропатолог Г. И. Россолимо. Поэтому многие авторы
(Смелов Н. С., 1964; Явчуновская М. А. и Сажина И. А. , 1966; Стоянов
Б. Г., 1973; Mouly и Vaillont, 1962; Rauch, 1967 и др.) предлагают назва-
ние <синдром Россолимо—Мелькерссона—Розенталя».
Гранулематозный хейлит, описанный Miescher в 1945 г., многие сов-
ременные авторы (Смелов Н. С и соавт., 1960; Шапошников О. К., 1960;
Пашков Б. М. 1970; Ангелюк Н. Н. и Падалко П. И., 1971; Schimpf,
1959; Hornstein и Schuermann, 1962; Grinspan et al., 1968; Worozowski
и Swieboda, 1969; Malaesku et al., 1978 и д.) относят к синдрому Мелькерс-
сона—Розенталя, полагая, что в ряде случаев он является моносимптом-
ным проявлением этого синдрома.
Этиология и патогенез. Единого взгляда на сущность заболевания в
настоящее время нет. Некоторые авторы (Evans, 1965; Wilkinson, 1969;
Grinspan et al., 1968) подчеркивают тесную связь с саркоидозом. Против
452
Рис. 296. Синдром Россолимо—Мель-
керссона—Розенталя.
Рис. 297. Синдром
Россолимо—Мель-
керссона—Розенталя.
этого высказываются сторонники инфекционно-аллергической природы
этого заболевания (Нещадим Н. Н. и соавт., 1972; Стоянов Б. Г., 1973;
Filipowicz и Michalowiez, 1959; Berger, 1956; Gottwald, 1967; Mazurkiewicz,
1968 и др.). К факторам, предшествующим началу болезни и ее рецидивам,
относятся грипп, простой пузырьковый лишай, ангины, травмы или тре-
щины красной каймы губ, охлаждение, прием лекарств и др. (Коляденко
В. Г. и соавт., 1959 и др.). Согласно другим представлениям, развитие
этого синдрома обусловлено нарушением функции вегетативных волокон
лицевого нерва, в частности парасимпатических. Сообщается о функцио-
нальных изменениях различных периферических нервов и центральной
нервной системы (Россолимо Г. И., 1901; Melkersson, 1928; Rosenthal,
1931; Hornstein, 1961; Bodechtel, 1963; Gorrwaald, 1967 и др.).
Клиника. Характерная триада—стойкая припухлость лица, чаще
губ, рецидивирующий неврит лицевого нерва и отечно-складчатый язык—
встречается не более чем у 20—30% больных; основную группу (70—80%)
составляют больные с моно - и двусимптомными проявлениями (макро-
хейлия или макрохейлия в сочетании с невритом лицевого нерва или склад-
чатым языком). Заболевание может развиться у лиц обоего пола и в любом
возрасте, чаще в периоде от 16—17 до 35—55 лет (75—80% больных.). Ве-
дущим симптомом является макрохейлия или отек губ, который наблю-
дается у 95% больных. Губы, особенно верхняя, увеличиваются в разме-
рах (превышая обычные размеры в 3—4 раза), приобретают плотно-эласти-
ческую консистенцию и синюшно-розовую ркраску; при надавливании на
453
них ямка не остается. Припухлость наподобие хоботка выворачивает крас-
ную кайму губ наружу. Слизистая губ, примыкающая к деснам, обычно
слегка гиперемирована. Отмечается чувство напряжения (распирания),
неловкость при разговоре, еде, сухость красной каймы. Часто макрохейлия
сочетается с отечностью щек (пареит): вздутие занимает всю щеку, а иногда
распространяется на другие участки мягких тканей лица (рис. 296, 297).
Болезненность й воспалительные явления в области припухлости отсутст-
вуют, В редких случаях наблюдается отечность всего лица, создающая
впечатление «львиной маски». Гранулематозный блефарит наблюдается в
единичных случаях (утолщение кожной части века, сужение глазной щели
и др.). Вторым основным симптомом (наблюдается у 35—40% больных) яв-
ляется неврит лицевого нерва, который у большинства больных проявля-
ется в виде пареза и в отдельных случаях паралича мимической мускула-
туры. Нередко на стороне отека сглажена носогубная складка. У многих
больных имеются мигренеобразные головные боли, чаще на стороне отека
или неврита лицевого нерва. Третьим основным симптомом является отеч-
но-складчатый язык—особая форма макроглоссии—гранулематозный глос-
сит; он встречается у 39—52% больных (Савкина Г. Д. и Ангелюк Н. Н.,
1972; Стоянов Б. Г., 1973; Hornstein и Shuermann, 1962 и др.). Для него
характерна отечность языка, выраженная в разной степени. Поверхность
языка бугристая, разделена на отдельные «холмики» и «дольки», перемежаю-
щиеся с глубокими бороздками. Язык постепенно сильно увеличивается в
размерах, становится малоподвижным, порой он заполняет всю ширину
ротовой щели. По боковой поверхности языка обнаруживаются отпечатки
зубов.
Некоторые авторы (Altmeyer и Schimpt, 1975; Larsson и Westermark,
1978) подчеркивают возможность развития гранулематозных очагов вне
лица—в области вульвы, на ягодицах и др. Принадлежность этих очагов
поражения к синдрому была доказана на основании данных гистологиче-
ского исследования.
Гистопатология. Характерны клеточные реакции узелкового типа,
обозначаемые как гранулемы. Они локализуются преимущественно вокруг
мелких сосудов, чаще вокруг капилляров, посткапиллярных венул и лим-
фатических сосудов. Различают несколько основных типов морфологиче-,
ских реакций: туберкулоидный, неспецифический, лимфонодулярно-плаз-
моцитарный и саркоидный. Чаще встречается туберкулоидный тип скопле-
ния эпителиоидных клеток, окруженных небольшим количеством лимфо-
идных и ретикулярных элементов. Некроз не встречается. Саркоидный
тип реакции характеризуется резко отграниченными скоплениями эпите-
лиоидноклеточных или гигантоклеточных гранулем, окруженных незна-
чительным количеством лимфоцитов. Для лимфонодулярно-плазмоцитар-
ного типа характерен густой инфильтрат в дерме или собственном слое
слизистой, состоящий из лимфатических клеток с примесью ретикулярных
и плазматических клеток. Неспецифический тип тканевой реакции прояв-
ляется массивным отеком со слабо выраженной клеточной реакцией.
Полагают, что характер гистологических изменений зависит от давности
454
проявлений заболевания. Заболевание протекает длительно—от несколь-
ких лет до нескольких десятков лет. Оно отличается приступообразным
характером течения.
Дифференциальный диагноз. Следует проводить с острым ангионевро-
тическим ограниченным отеком кожи, рожистым воспалением лица, слоно-
востью губ, кавернозной гемангиомой, лимфангиомой, туберкулезной вол-
чанкой, третичным сифилисом, лепрой.
Лечение. Комплексная терапия, включающая кортикостероиды (пред-
низолон по 25—30 мг в сутки), антибиотики широкого спектра действия
(окситетрациклин по 100—200 тыс. ЕД 4—5 раз в сутки; метациклин по
300 тыс. ЕД 2 раза в сутки и др.), препараты аминохинолинового ряда (де-
лагил, хлорохин-дифосфат, плаквенил по 0,25 г 2 раза в день, 20—25 г на
курс); антигистаминные препараты; витамины (аскорбиновая кислота и
др.). К. П. Фомберштейн (1979) с успехом применил акупунктуру в со-
четании с сероводородными ваннами.
ЛИТЕРАТУРА
I
Коляденко В. Г., Шупенько Н. М., Свирид А. А. О синдроме Россолимо-Мелькерс-
сона—Розенталя.— Вестн. дерматол. и венерол., 1979, 9, 22—24.
Altmeyer P., Schimpt A. Aborfives'Melke sEvn^Rosenthal Syndrom mit Cheilitis
et Pareitis granulomatosa, Bucca lobata und Hedraitis granulomatosa.— Akt. Dermatol.,
1975, 1, 3, 125—130.
РОТМУНДА СИНДРОМ (Rothmund syndromum).
Впервые его описал мюнхенский офтальмолог August Rothmund млад^
ший в 1868 году как катаракту со своеобразной дегенерацией кожи. Боль-
шинство исследователей относит синдром Ротмунда в группу врожденных
пойкилодермий. Одни авторы признают его известную самостоятельность
в этой группе (Marghescu и Braun-Falco, 1965; Rodermund и Hausmann,
1977 и др.), другие авторы отождествляют с синдромом Томсона (Carleton,
1943; Sexton, 1954; Schirren и Nasemann, 1962; Wozniak и Bohm, 1972;
Kristensen, 1975 г. и др.) и называют синдромом Ротмунда—Томсона.
Wodniansky (1957) считает целесообразным для этих синдромов единое
обозначение—врожденная пойкилодермия.
Этиология и патогенез. Синдром Ротмунда—врожденное заболевание,
наследуемое по указаниям одних авторов аутосомно-рецессивно, по дру-
гим—доминантно с неполной пенетрантностью (Wodniansky, 1957). Gertler
(1970) указывает, что в его основе лежит эмбриональное повреждение мезо-
и эктодермы с реализацией на пятой неделе жизни плода. В большинстве
опубликованных сообщений отмечается кровное родство родителей и семей-
ные случаи болезни.
Клиника. Заболевание встречается редко, по-видимому, чаще у жен-
щин. Rodermund и Hausmann (1977) собрали литературные описания 41
больного, среди них 18 мужчин и 23 женщины. В Центральном кожно-ве-
нерологическом институте Минздрава СССР за последние 35 лет наблюдали
лишь одного пациента с синдромом Ротмунда.
455
Заболевание начинается на первом году, реже в первые годы жизни.
Появляется покраснение кожи лица, в первую очередь щек, лба, подбо-
родка, ушных раковин, которое затем распространяется на разгибатель-
ные поверхности рук, ног, на ягодицы. Кожа туловища поражается как
исключение. Вслед за покраснением кожи развивается пойкилодермия.
Обычно к концу первого года жизни картина кожных изменений полно-
стью сформирована и дальше не прогрессирует. На пораженных участ-
ках отмечается мраморность кожи за счет сетевидных тонких красных по-
лосок, которые слегка шелушатся; на их месте постепенно развиваются
атрофические полосы. Чередование красных атрофических и пигментных
полос придает коже пестрый вид. Вся остальная кожа нежная, тонкая,
прозрачная. Часто отмечают сухость кожи, иногда гиперкератозы. Весьма
характерны парциальный ййи общий гипотрихоз (брови, ресницы, волосы
на голове), акромикрия (маленькие руки и ноги с короткими неуклюжими
пальцами), седловидный нос, приземистая фигура, гипогенитализм, гипо-
тиреоз, аномалии зубов и ногтей. В то же время Marghescu и Braun-Falco
(1965) указывают, что аномалии зубов и ногтей едва ли бывают при синдро-
ме РотмундЗ, они обращают внимание на частую гипо - или аплазию пото-
вых и сальных желез. Почти у всех больных между 3 и 7 годами жизни раз-
вивается двусторонняя катаракта; которая быстро прогрессирует и при-
водит к слепоте. Почти-все авторы отмечают нормальные психику и ин-
теллект пациентов^-Синдромом Ротмунда и отсутствие у них хромосомных
. аномалий^-'"'''-
.— Прогноз в отношений продолжительности жизни благоприятный. По-
следняя пациентка Ротмунда умерла в 1955 году в возрасте 92 лет.
Гистопатология. Атрофия эпидермиса, нередко гиперкератоз, сгла-
женные сосочки кожи, большей частью лимфо- и гистиоцитарный пери-
васкулярный инфильтрат, дегенерация коллагеновой ткани, разрежение
и отчасти фрагментация эластических волокон, атрофия или отсутствие
сальных и потовых желез, уменьшение волосяных фолликулов.
Дифференциальный диагноз. Проводят с синдромом Томсона, синдромом
Вернера.
Лечение. Симптоматическое.
ЛИТЕРАТУРА
Kristensen К- Poikiloderma congenitale — ап early case of Rothmund—Thomson’
syndrome.— Acta Dermatovenereol., 1975, 55, 4, 316—318.
Rodermund О. E., Hausmann D. Das.Rothmund—Syndrom. Ein Beitrag zu der kon-
genitalen Poikilodermien.— Z. Hautkr., 1977, 52, 4, 129—141.
Wozniack K- D-, Bohm W. Zum Problem der Poikilodermia congenita Thomson.—
Z Haut u. Geschl.— Kr., 1972, 47, 625—632.
САРКОИДОЗ КОЖИ (sarcoidosis cutis).
Синонимы: болезнь Бенье—Бека—Шаумана, доброкачественный лим-
фогранулематоз Шаумана, хронический эпителиоидноклеточный ретику-
лоэндотелиоз, болезнь Бека, доброкачественный милиарный люпоид и др.
456
Саркоидоз—системное заболевание, поражающее самые различные ор-
ганы и ткани: легкие, медиастинальные лимфатические узлы, кожу, гла-
за, кости и др. Саркоиды являются различными кожными проявлениями
саркоидоза.
Различают следующие клинические формы саркоидов: саркоид Бе-
ка в трех его основных разновидностях (рассеянная мелкоузелковая, круп-
ноузловатая и диффузно-инфильтративная); ознобленная волчанка Бенье-
Теннесона и ангиолюпоид Брока—Потрие, подкожные саркоиды Дарье—
Русси.
Этиология и патогенез. Длительное время господствовал взгляд на
саркоидоз, как на своеобразное ослабленное проявление туберкулезной
инфекции. Однако, как указывают многие исследователи, в настоящее
время нет для этого оснований. Отдельные авторы описывают семейный
саркоидоз, что указывает на роль генетических факторов. James (1979) на-
блюдал 16 семей, у 33 членов которых имелся саркоидоз. Некоторые авторы
рассматривают саркоидоз как полиэтиологический синдром. Многие со-
временные авторы рассматривают саркоидоз как своеобразную форму эпи-
телиоидноклеточного ретикулоэндотелиоза.
Саркоидоз часто встречается в странах северной Европы и в США.
В США он в 10 раз чаще встречается у черных, чем у лиц белой популяции,
в Южной Африке в 10 раз чаще среди племени Банту, чем у белых (James,
1976).
Частота поражения кожи при саркоидозе колеблется от 15% до 30—
40%, James (1979) на основании анализа 3676 больных/описанных в ми-
ровой литературе, установил, что женщины болеют чаще (57%), чем мужчи-
ны (43%). Чаще болеют взрослые в возрасте до 40 лет (рис. 298,299,30U,3Ol)^
Клиника. 1. Саркоид Бека. Эту клиническую разновидность саркоидо-
за впервые описал норвежский дерматолог Бек (1899) под названием «мно-
жественные саркоиды кожи.» Более чем у 3/4 больных саркоидом Бека
выявляются саркоидные поражения и других органов (легких, медиа-
стинальных лимфатических узлов, глаз, костей—кистоидный остит Мо-
розова—Юнглинга).
Клиника саркоида Бека разнообразна. Мелкоузелковая разновид-
ность характеризуется высыпанием узелков размерами от булавочной го-
ловки до горошины, полушаровидных, возвышающихся над окружающей
кожей, плотно-эластической консистенции, буровато-синюшного цвета.
Почти при всех формах саркоидов при диаскопии выявляются мельчайшие
желтовато-буроватые пятнышки («пылинки»), сходные с феноменом «яб-
лочного желе» при туберкулезной волчанке. В процессе обратного разви-
тия узелки уплощаются и на поверхности их появляются тонкие чешуйки.
При полном регрессе узелков остается гиперпигментация или поверхност-
ный рубчик с пигментацией по периферии. Крупноузловатая форма про-
является единичными или множественными резко ограниченными плоски-
ми очагами поражения (бляшками) синюшно-буроватого цвета, размером
от 10 до 20-копеечной монеты и более. В остальном клиническая характе-
ристика узлов такая же, как и мелко-узелковых элементов.
457
458
Диффузно-инфильтративная форма встречается реже двух предыду-
щих и характеризуется образованием не резко отграниченных инфиль-
тративных очагов поражения плотноватой консистенции, буровато-синего
цвета, величиной до ладони взрослого и больше. Субъективных ощущений,
как и при предыдущих формах, нет.
При всех формах саркоидоза кожи туберкулиновые реакции большей
частью отрицательны; внутрикожная проба с суспензией из саркоидной
ткани (реакция Nickerson, 1939 и Kveim, 1941) у большинства больных
(70—80%) положительная.
2. Ангиолюпоид описан французскими дерматологами Броком и По-
трие в 1913 году. Многие современные исследователи рассматривают ангио-
люпоид как клинический телеангиэктатический вариант саркоида Бека.
Заболевание характеризуется появлением на лбу, боковых поверхностях
верхней части носа одиночных очагов поражения до 1,5—2 см или несколь-
ко более в диаметре, синеватого цвета с буроватым оттенком, мягкой кон-
систенции с многочисленными телеангиэктазиями на поверхности бляшек.
Субъективных ощущений нет. При диаскопии обнаруживается диффузная
желтовато-коричневая окраска.
3. Ознобленная волчанка Бенье—Теннесона, описанная Бенье (1888)
и Теннесоном (1892), многими современными исследователями рассматри-
вается как клиническая разновидность саркоида Бека. На коже щек,
носа, ушных раковин, подбородка и лба возникают фиолетово-красные
плоские или слегка выпуклые очаги поражения с довольно четкими гра-
ницами. Шелушение отсутствует, атрофии или рубцов не остается. У ча-
сти больных обнаруживаются поражения легких, костей, суставов, уве-
личение лимфатических узлов. Отмечается ухудшение процессе в холод-
ное время года. При диаскопии выявляются желтоватые точки—пятнышки.-^
Гистопатологические изменения такие уе, как при саркоиде Бека.
Дифференцировать следует с лейкемией кожи, туберкулезной вол-
чанкой, лепрой.
4. Подкожные саркоиды Дарье—Русси, описанные этими дерматолога-
ми в 1904—1906 гг., встречаются редко. До сих пор дискутируется вопрос
об их принадлежности к болезни Бенье—Бека—Шаумана. Многие авторы
считают саркоиды Дарье—Русси разновидностью уплотненной эритемы Ба-
зена. Наблюдения кожной клиники ЦКВИ позволяют согласиться с
мнением этих исследователей.
Клинически заболевание характеризуется образованием подкожных
узлов размерами от горошины до грецкого ореха и больше, как правило,
немногочисленных, безболезненных. При слиянии образуются бляшечные
.инфильтраты больших размеров. Кожа, покрывающая очаги, тускло-ро-
зового цвета. Локализуются узлы преимущественно на боковых поверх-
ностях туловища, животе и бедрах. Увеличиваясь в размерах, они спаи-
ваются с кожей, но почти никогда не изъязвляются в противоположность
узлам индуративного туберкулеза кожи.
Гистопатологическая картина более соответствует таковой при уплот-
ненной эритеме Базена.
459
Дифференцировать следует с индуративной и узловатой эритемой,
инфекционно-аллергическими васкулитами, гуммозным сифилисом.
Кроме перечисленных типичных поражений кожи, при саркоидозе вы-
деляют различные клинически атипичные формы, отнесение которых к
саркоидозу возможно лишь на основании гистологических исследований.
Среди клинически атипичных форм поражения кожи при саркоидозе сле-
дует упомянуть: лихеноидную, напоминающую красный плоский лишай
(Hallopeau и Еск, 19'02; Berg, 1963); веррукозную и папилломатозную фор-
мы (Irgang, 1950; Pallet et al., 1983); ихтиозиформную (Kaub et al.,
1978); анулярную и плоские цирцинарные формы, клинически напоми-
нающие базалиому и красную волчанку (Hanuksele, 1971), грибовидный
микоз (McFarland et al., 1978); форму, клинически не отличимую от папу-
ло-некротического туберкулеза кожи. Среди атипичных форм саркоидов
кожи нередко описывается язвенная разновидность (Павлов П. А., 1903;
Мещерский Г. И., 1904; Nathan et al., 1974; Hopf и Krebs 1974; Metz и
Hartmann, 1977). Язвы образуются в основном в пределах первичного мел-
ко-и крупноузловатого очага. Почти всегда отмечается выраженная
болезненность. При гистологическом исследовании изъязвившихся сар-
коидных папул обнаруживают поверхностно расположенный неспецифи-
ческий воспалительный инфильтрат и типичные эпителиоидноклеточные
гранулемы в глубоких слоях дермы.
В патогенезе язвенной формы саркоидоза кожи придают значение ох-
лаждению, давлению, венозному стазу, гипертонии, сахарному диабету;
однако эти факторы, по-видимому, играют второстепенную роль.
Прогноз, как правило, благоприятный, течение длительное. Исклю-
чительно редко (в 1—2% случаев) саркоидоз кожи с вовлечением в процесс
внутренних органов ведет к смерти.
Гистопатология. В средних и глубоких отделах дермы обнаружива-
ются эпителиоидноклеточные гранулемы с примесью лимфоидных и гистио-
цитарных элементов, с единичными гигантскими клетками типа Лангханса
или инородных тел. В отлнчие от туберкулеза казеозный, творожистый
некроз, как правило, отсутствует (Bjerke et al., 1981; Bower, 1981).
Дифференциальный диагноз. Следует проводить с туберкулезной вол-
чанкой (с плоской формой волчанки), ретикулезами кожи, бугорковым и
папуло-пустулезным сифилисом, эозинофильной гранулемой лица/ крас-
ным плоским лишаем, папуло-некротическим туберкулезом, бугорковым
лейшманиозом кожи.
Лечение. Наиболее эффективным методом лечения является длитель-
ное (в течение нескольких месяцев) назначение кортикостероидных пре-
паратов (преднизолон по 20—30 мг в сутки) раздельно или в сочетании с
цитостатиками (проспидин, циклофосфамид). Полезно комбинировать кор-
тикостероидные и противотуберкулезные препараты. Целесообразно на-
правление больных на южные курорты (Крым, Кавказ).
460
ЛИТЕРАТУРА
Bower J. Pulmonary evaluation of patients presenting with dermatological manifesta
tionof sarcoidosis.— Intern. J. Dermatol., 1981, v. 20, No 5, p. 385—389.
Bjerke J., Keogh H., Matre R. In situ identification of mononuclear cells in cutaneous
infiltrates in discoid lupus erythematosus, sarcoidosis and secondary syphilis. Acta dermato-
venerol. 1981, v. 61, No 5, 371—380.
Hopf B., Krebs .4. «Ulcera cruris» als soltone form einer sar koi dose.— Dermatologies,
1974, v. 149, No 1, p. 55—62.
James D. et al. A worldwide review of sarcoidosis.— Ann. N. Y. Acad. Sci., 1976, v.
v. 278, p. 321—333.
James M. Sarcoidosis of the skin.— In: Fitzpatrick et al. Dermatology in general me-
dicine. 2nd ed. New York, 1979, p. 1336—1343.
KauhJ., Googy H., Luscombe H. Ichthyosiform sarcoidosis.— Arch. Dermatol., 1978,
v. 114, No 1, p. 100—102.
McFarland J., KauhJ., Luscombe H. Sarcoidosis associated with mycosis fungoides.—
Arch. Dermatol., 1978, v. 114, No 6, p. 912—915.
Nathan M., Pinsker R., Chase P., Elguezabel. Sarcoidosis presenting ?s lymphede-
ma.— Arch. Dermatol., 1974, v. 109, No 4, p. 543—544.
Paller A., Surek Ch. et al. Cutaneous sarcoidosis associated with sarcoidosis of the up-
per airway.— Arch. Dermatol., 1983, v. 119, No 7, p. 592—596.
САРКОМА МНОЖЕСТВЕННАЯ ИДИОПАТИЧЕСКАЯ
ГЕМОРРАГИЧЕСКАЯ КАПОШИ. (sarcoma idiopaticum haemorrhagicum
multiplex Kaposi).
Синонимы: ангиоретикулез Капоши, ангиосаркома Капоши, саркома
Капоши, ангиоэндотелиома кожи и др.
Заболевание впервые описал Kaposi в 1872 году.
Этиология и патогенез. Большинство современных авторов рассмат-
ривают ее как болезнь неизвестной этиологии, имеющую черты опухолевых
заболеваний околососудистой ретикулогистиоцитарной ткани (Каламка-
рян, А. А., 1967, 1978; Dorffel, 1932; Tedeschi, 1958; Murray и Lothe, 1962;
Templeton и Dhru, 1975).
Клиника. Заболевание встречается у представителей всех континентов,
однако распространено неравномерно. В Азии оно представляет казуисти-
ческую редкость. Велика заболеваемость в Африке, где она составляет от
0,04% в Тунисе (Chadli, 1960) до 9,1 % в Конго (Thijs, 1957; Farrat, 1982), по
отношению ко всем злокачественным опухолям. Довольно часто сар-
кому Капоши наблюдают в Европе—в СССР, Италии, Венгрии, ГДР,
Франции и др. По длительности и особенно по тяжести течения выделяют
три формы заболевания. При хронической форме прогрессирование мед-
ленное и больные живут в среднем 6—8 лет, иногда дольше (до 15—20 лет).
При подострой форме заболевание быстро прогрессирует и в течение 2—3
лет приводит к гибели. Острая форма характеризуется ранней генерали-
зацией процесса, неуклонным прогрессированием и летальным исходом
через несколько месяцев от начала болезни. Мужчины заболевают чаще,
461
Рис. 302. Саркома Капоши.
Рис. 303 Саркома Капоши.
Рис. 304. Сапкима. Капоши.
Рис. 305. Саркома Капоши.
462
Рис. 311. Саркома
Капоши.
Рис. 313. Саркома
Капоши
Рис. 312. Саркома Капоши (п»ра-
женив слизистой полости рта).
Рис. 314. Саркома
Капоши.
s
464
чем женщины, соотношение между ними колеблется от 3:1 до 32:1, состав-
ляя в среднем 8:1 (Каламкарян А. А., 1978; Акимов В. Г., 1978; Lothe, 1963;
Pisanty и Garfunkel, 1970 и Др.). В Европе ангиоретикулез чаще поражает
людей в возрасте от 40 до 70 лет, в Африке—от 20 до 40 лет. Заболевание
часто начинается с появления красновато-синюшных пятен величиной
от чечевицы до 10-копеечной монеты. Постепенно увеличиваясь в размерах,
они достигают в диаметре 2—5 см. Пятна хорошо отграничены, поверх-
ность их гладкая. Другое частое раннее проявление ангиоретикулеза—
узелок сферической, реже плоской формы величиной от просяного зерна
до мелкой горошины розовато-синюшного цвета, мягкой консистенции.
Располагаются узелки изолированно, часто сгруппированно. В дальней-
шем образуются опухоли и узлы полушаровидной формы, размерами от
крупной горошины до лесного ореха, отчетливо возвышающиеся над нор-
мальной кожей. Консистенция опухолей плотно-эластическая; поверх-
ность их блестящая, неровная, с мелкими углублениями, подобно апель-
синовой корке, реже гладкая. Располагаются они или изолированно, или
сливаются в крупные бугристые образования. Цвет, очагов поражения в
начале заболевания красновато-синюшный, позднее интенсивность окраски
усиливается до синюшно-буроватой. В результате распада опухолей и
узлов, который наблюдается примерно у 15—20% больных, образуются
глубокие, резко болезненные («стреляющие» боли) язвы неправильных
очертаний, с несколько вывороченными краями, окрашенными в насыщен-
ный синюшно-багровый цвет (рис. 302—322). Дно язв бугристое, покрыто
кровянисто-гангренозным отделяемым. У многих больных отмечаются ве-
гетации грануляционной ткани и веррукозные разрастания. Редко наб-
людаются множественные буллезные и везикулезные высыпания, распола-
гающиеся на отечных голенях и стопах и клинически напоминающие лим-
фангиому. Многоочаговость, распространенность и симметричность высы-
паний являются важной особенностью ангиоретикулеза Капоши. Чаще
всего (почти у 80—90% больных) процесс носит распространенный харак-
тер с преимущественной локализацией на стопах, голенях и тыле кистей.
Из более редких локализаций первичных высыпаний следует упомянуть
ушные раковины, щеки, нос, спину, живот, половой член. Поражение
слизистой оболочки полости рта встречается у 2—12% больных ангиорети-
кулезом. Чаще высыпания располагаются на мягком, твердом небе, затем
на щеках, в глотке, гортани, на языке и дужках. У больных со значитель-
ным распространением очагов поражения они почти всегда сопровожда-
ются интенсивными, мучительными болями. Примерно у 25—40% больных
наблюдаются отеки пораженных областей, главным образом стоп, голе-
ней и кистей, нередко развивается выраженный элефантиаз с множест-
венными папилломатозными и гиперкератотическими разрастаниями. Уве-
личение периферических лимфатических узлов не является ведущим симп-
томом при ангиоретикулезе, оно встречается примерно у 20—25% больных,
чаще при подострой форме заболевания. Гемограмма в большинстве слу-
чаев остается нормальной. У 2/3 больных обнаруживается лимфоцитоз и
моноцитоз. Поражение костей, характеризующееся остеопорозом (чаще
30 Клиническая дерматология 465
Рис. 315. Саркома
Калоши.
Рис. 316. Саркома
Калоши на коже
верхнего века.
Рис. 318. Саркома
Калоши
Рис. 317. Саркома
Калоши на коже
верхнего °ека.
466
Рис. 319. Саркома Капоши
(локализация на носу).
Рис. 320. Саркома Капоши
(локализация на половом члене).
Рис. 321. Саркома
Капоши.
Рис. 322 Саркома .Капоши
(поражение уха).
30*
467
стоп) и периостозом, атрофией суставных концов фаланг, обнаруживают
рентгенологически примерно у 1/3 больных ангиоретикулезом Капоши,
главным образом с распространенными опухолевидно-узловатыми высы-
паниями.
Гистопатология. В дерме многочисленные сосудистые просветы, окру-
женные эндотелиальными и перителиальными клетками; выраженная про-
лиферация клеток веретенообразной формы—молодых фибробластов; не-
которые из них атипичны. Встречаются участки геморрагий и отложений
гемосидерина. Обнаруживаются митозы клеток.
Дифференциальный диагноз. Следует проводить со злокачественной
меланомой, фиброматозом, дерматофибросаркомой, красным плоским ли-
шаем, ангиомой, болезнью Рейно, тромбофлебитом, ретикулезами кожи,
грибовидным микозом, псевдосаркомой Капоши и др.
Лечение. Пенициллин в начальных стадиях заболевания до 20—30 млн.
на курс; цитостатики (проспидин, фосфазин, метотрексат, 6-меркаптопу-
рин) рекомендуются больным с распространенными узелковыми и опухо-
левидно-узловатыми образованиями. Рентгенотерапия показана при огра-
ниченных опухолевидных очагах поражения. Хирургическое удаление
крупных опухолей целесообразно сочетать с до - или послеоперационной
химиотерапией.
ЛИТЕРАТУРА
Акимов В. Г. Клинические особенности ангиоретикулеза Капоши у африканцев
народности Банту:—Вести, дерматол. и венерол., 1978, 1, 62—65.
Каламкарян А. А. Клинические аспекты ангиоретикулеза Капоши (sarcoma Kapo-
si).— Вести- дерматол. и веиерол., 1978, 1, 3—8-
Farfat Р. Kaposis sarcoma in childhood. South Afr. Med. J., 1982, 61, 17, 636—637.
Pisanty S., Garfunkel A. Kaposi's sarcoma.— J. Oral. Med., 1970, 25, 3, 89—92.
Templeton A., Dhru D. Kaposi’s sarcoma in holf-brothers.— Trop. reogr. med., 1975,
‘27, 3, 324—325.
САРКОМА КАПОШИ—как проявление СПИД
В последнее время установлена связь саркомы Капоши с нарушением
иммунного статуса. Впервые это было отмечено у больных, .подвергшихся
пересадке почки и получавших иммунодепрессивную терапию.
Первое сообщение о таком сочетании принадлежит Siegel и соавт.
(1969). Вслед за ним последовало большое количество аналогичных публи-
каций (Farman et al., 1975; Hardy et al., 1976; Harwood et al.,
1979; Hood et al., 1982; Каламкарян А. А. и соавт., 1984 и др.).
Известно, что саркома Капоши может сочетаться с другими заболева-
ниями, которые требуют длительных курсов лечения кортикостероидами
или цитостатиками—с красной волчанкой, пузырчаткой, дерматомиозитом,
бронхиальной астмой, хроническим миелолейкозом, грибовидным мико-
зом и другими (Шапошников О. К. и Родионов А. Н., 1980; Каламкарян
А. А. и соавт., 1984; Ulbrigth, 1981; Snyder и Schwartz, 1982; Weis и Seru-
chan, 1982 и др.). Важным доказательством иммунозависимости саркомы
468
Капоши у этих пациентов служит факт ремиссии опухоли при полной отме-
не или снижении доз иммунодепрессантов.
Таким образом, клинические наблюдения позволяют выделить са-
мостоятельную иммунозависимую форму саркомы Капоши, которая ха-
рактеризуется чрезвычайно злокачественным течением, возникновением
на фоне массивной терапии иммунодепрессантами ранее существующих
заболеваний.
В последние годы (с 1981 г.) в зарубежной литературе широко обсуж-
дается связь ряда заболеваний, включая саркому Капоши, у лиц с приоб-
ретенной недостаточностью иммунитета (AIDS—Aquired immune deficien-
cy syndrome, СПИД—синдром приобретенного иммунодефицита).
Синдром начал регистрироваться в США с лета 1979 г., причем 3/4 па-
циентов жили в Нью-Йорке, Лос-Анджелесе и Сан-Франциско—городах
с высоким уровнем мужского гомосексуализма, а 75% больных являлись
гомосексуалистами.
Первые сообщения были сделаны в 1981 году (Friedman-Kein, Ну-
mes и др., 1981), хотя, как упоминалось выше, первые такие случаи реги-
стрировались уже в 1979 году (Ioffe et al., 1983). К осени 1984 гоАа в
США было зарегистрировано уже около 3000 случаев СПИД со смертно-
стью в 38—43%. Согласно статистическим данным Центра инфекционных
заболеваний, летальность среди американцев, страдающих СПИД, дости-
гает 95%, если наблюдение за ними продолжается в течение 2 лет.
С 1981 г. стали появляться первые сообщения о саркоме Капоши,
связанной со СПИД, у европейцев—испанцев, французов, немцев, датчан
(Vilsseca et al., 1982; Gorin et al., 1982; Gerstoft et al., 1982; Dotz
и Berman, 1983; Orfanos et al., 1983 и др.). В Европе к концу 1984 года
было зарегистрировано около 300 случаев СПИД, более всего во Франции.
В США эта форма саркомы Капоши рассматривается как одно из про-
явлений СПИД (Gottlieb et al., 1983; Sonnabend et al., 1983; Abema-
уог и Colaterra, 1983 и др.). Помимо саркомы Капоши в этот синдром вхо-
дят различные инфекционные заболевания, такие как пневмоцистная пнев-
мония, инфекционный мононуклеоз, генерализованный простой герпес,
менингиты и энцефалиты, обусловленные аспергилезом, системный кан-
дидоз, В-клеточные лимфомы и другие.
Наиболее часто при СПИД встречается пневмоцистная пневмония
—в 52%, реже саркома Капоши—в 28%, а их сочетание наблюдается в
9% (Gottlieb et al., 1983; Sonnichsen и Dorte Ruff, 1984 и др.).
Обобщив многочисленные исследования, имеющиеся в литературе
(Wallace et al., 1982; Rasmussen et al., 1982; Friedman-Kein et al.,
1982; Stahl et al., 1982; Schroff et al., 1983; Ammann et al., 1983;
Rogers et al., 1983 и др.), можно полагать, что решающим для СПИД
критерием изменения иммунной системы является недостаточность клеточ-
ного иммунитета, причем на первый план выходят: отчетливая лимфопения
в периферической крови, отсутствие кожных реакций на тесты с различны-
ми антигенами, снижение сооти^"><-иия Т-хелперов к Т-супрессорам
469
(менее 1). Природа такого иммунодефицита остается неясной. Наиболее
популярной является инфекционная гипотеза; предполагается, что име-
ется какой-то инфекционный агент, возможно, вирус, который способен
передаваться половым путем, подобно вирусу гепатита В и, обладая лимфо-
цитотропностью, вызывать состояние иммунодефицита. Эта гипотеза ос-
нована на обнаружении у подавляющего большинства больных высоких
титров антител к вирусу цитомегалии и вирусу Эпштейн—Барр.
Несмотря на кажущуюся убедительность приведенных фактов, ин-
фекционная теория, по нашему мнению, не в состоянии объяснить этиопа-
тогенез саркомы Капоши.
На конец 1984 года в США было известно около 600 случаев саркомы
Капоши у мужчин-гомосексуалистов, но ни разу не сообщалось о заболе-
ваемости среди членов их семей, или, например, у профессиональных про-
ституток.
Эпидемиология человеческих болезней не знает примеров передачи
половым путем инфекционного агента только лицу того же пола. Мы раз-
деляем мнение тех исследователей, которые основным звеном патогенеза
саркомы Капоши у гомосексуалистов считают нарушение иммунитета, обу-
словленное разными факторами, в том числе и вирусами (Stahl et al., 1982;
Geursen, 1983; Frennan, 1982 и др.).
Клиника. От обычной саркомы Капоши иммунодепрессивную форму
отличают: молодой возраст, внезапное появление генерализованных форм
узелковых и опухолевидных образований, редкость или отсутствие пятни-
стых высыпаний, атипичное расположение кожных элементов—голова,
шея, ротовая полость, туловище, стремительное прогрессирование процес-
са, более частое поражение висцеральных органов и лимфатических узлов,
резистентность к проводимой терапии, плохой прогноз, 50%—60% больных
умирают в сроки от 3 месяцев до 2 лет.
Идентичность так называемого агрессивного варианта саркомы Капоши
у гомосексуалистов острой форме этого заболевания очевидна и не диску-
тируется.
Лечение. Эффективных методов терапии нет.
ЛИТЕРАТУРА
Dotz W., Berman В. Kaposi’ ssarcoma, chronic ulcerative herpes simplex, and acqui-
red immunodeficiency.— Arch. Dermatol., 1983, 119, 1, 93—94.
Friedman-Kein A. E. Disseminated Kaposi’s sarcoma: An epidemic in homosexual
men.— J. Amer. Acad. Dermatol., 1983, 8, 5, 737—738.
Geursen R. G. AIDS syndrome der erworeenen Immundefiriend.— Die gelen Heffte,
1983, 23, 1—7.
Modlin R., Crissey J., Rea T. Kaposi’ ssarcoma.— Interm. J. Dermatol.. 1983, 22, 8,
443—448.
OrfanosC., Vossman D., Bauer R. Disseminiertes Kaposi—Sarkom bei jungen homo-
sexullen Mannern mit erworbener Immundefiens in der Bundesrepublik Eingatz von Inter-
feron.— Akt. Dermatol., 1983, 9, 6, 199—206.
470
Snyder R., Schwartz R. Telangiectatic Kaposi’s sarcoma.— Arch. Dermatol., 1982,
118, 12, 1020—1021.
СЕБОЦИСТОМАТОЗ (sebecystomatos)
Синонимы: стеатоцистоматоз, множественные стеатоцистомы, врожден-
ные сальные кисты.
Анализ литературы показывает, что клинико-анатомическая характе-
ристика кистозных образований недостаточно четкая, а их терминология
разнообразна. Гистогенез этих опухолей до настоящего времени остается
предметом споров. Bosellini (1898), которому принадлежит первое описа-
ние себоцистоматоза, считал, что кисты образуются в результате избыточ-
ной кератинизации, ведущей к задержке секрета сальных желез. Многие
ранние авторы (Ormsby и Finnernd, 1930 и др.) также расценивали очаги
поражения, как жировые или ретенционные кисты. Большинство же со-
временных авторов (Sachs, 1938; Vyneyard и Scott, 1961; Kligman, 1964;
Gorlin и Jeusen, 1975; Cole, 1976; Woznack и Slukova, 1976; Nodes и Norins,
1977; Bode и Piewig, 1980 и др.) отрицают концепцию «ретенционных кист»
и считают, что эти образования имеют невоидный характер (генодерматоз)
и передаются по аутосомно-доминантному типу. К кистозным образованиям
невусного происхождения относятся милиум, дермоид, эпидермальные кисты
и сальные кисты. В клинико-морфологическом отношении они являются
доброкачественными опухолями. Некоторые эпидермальные кисты рассмат-
риваются как пороки развития, их возникновение объясняют отшнуров-
кой эпидермиса в эмбриогенезе (Головин Д. И., 1958); они сочетаются с
другими пороками развития. Гистологические исследования Kligman и
Kirschbaum (1964) показали, что стеатоцистоматоз является разновидно-
стью дермоидных опухолей. Кисты, подобные множественным стеатоцисто-
мам, были описаны при других заболеваниях—врожденной пахионихии,
верруциформном акрокератозе Гопфа, гипертрофическом красном плоском
лишае, койлонихии и др. (Ronchese, 1944; Conters и Costello, 1957;
Vinegard и Scott, 1961; Verbov, 1972; Cole, 1976 и др.).
Клиника. Заболевание начинается в раннем детстве; мужчины и жен-
щины болеют одинаково часто. Нередко наблюдаются семейные случаи в
нескольких поколениях. Высыпания обычно очень распространены, множе-
ственны, располагаются на лице, волосистой части головы, плечах, тулови-
ще, спине, бедрах, мошонке; грудь является наиболее частой локализацией.
На указанных участках кожного покрова разбросаны множественные круг-
лые опухолевидные образования, покрытые нормальной кожей, мягко
эластической консистенции, величиной от чечевицы до крупной горошины
и более (рис. 323, 324, 325, 326). Большинство высыпаний желтоватого
цвета, а некоторые цвета нормальной кожи. Опухоли спаяны с верхним
слоем кожи и хорошо подвижны вместе с ней. При вскрытии выделяется
прозрачная маслянистая жидкость молочного или желтого цвета, без за-
паха или плотная сырообразная масса, которая по своему химическому
составу имеет сходство с липидами крови. Клинические анализы крови и
мочи без патологии; содержание холестерина в крови нормальное. Посев
содержимого кист роста пиококков не дает. Заболевание обычно не соче-
471
Рис. 323 Себоцистоматоз.
Рис. 325. Себоцистоматоз.
Рис. 326. Себоцистоматоз.
472
тается с вульгарными угрями; комедоны очень редко встречаются при мно-
жественном себоцистоматозе.
Течение заболевания хроническое; в редких случаях наблюдалось
перерождение в базальноклеточную эпителиому.
Гистопатология. Оболочка кист состоит из двух слоев: внутреннего
эпителиального и наружного соединительнотканного. В сальных железах
изменений нет, не обнаруживается закупорка фолликула или протока
сальной железы. Кисты открываются внутрь волосяного ф'лликула, они
связаны с эпидермисом только короткими тяжами недифференцированных
клеток. Электронная микроскопия показывает, что стенки кист идентичны
эпидермису—имеется базальная мембрана, базально-клеточный слой, про-
межуточный и роговой слой (Hashimoto et al., 1964).
Дифференциальный диагноз. Проводится с milium, эпидермальными
кистами, жировыми дермоидными кистами и кистами при юношеских уг-
рях.
Лечение. Хирургическое иссечение крупных опухолей, электрокоагу-
ляция.
ЛИТЕРАТУРА
Bode U., Piewig G. Klassifikation follikulare Zysten: Epidermalzysten einschliesslich
Sebocystomatpsis Gunther, Steatocystoma multiplex und Trichilemmalzysten.— Hautarzt,
1980, 31, 1, 1—9.
Cole A. Steatocystoma multiplex.— Arch. Dermatol., 1976, 10, 1437—1439.
Sanderson R., Mackie R. Tumors of the skin.— In: Textbook of dermatology. Ed.
Rook, Wilkinson, Ebling. Third edition. Oxford, 1979, p. 2129—2231.
СЕЗАРИ СИНДРОМ (Sezary syndrom)
Синонимы: доброкачественный, гиперпластический кожный ретикуло-
гистиоцитоз, эритродермический ретикулез с ретикулемией.
Этиология и патогенез. Французский дерматолог Sezary (1₽38, 1949)
описал 59-летнюю женщину с генерализованной эритродермией, лимфаде-
нопатией, интенсивным зудом, с наличием в периферической крови «чу-
довищных», «уродливых» моноцитоидных клеток. Так как густой инфильт-
рат из подобных клеток присутствовал и в дерме, было заключено, что она
является их источником. По мнению Сезари, эти клетки имеют мезен-
химное, ретикулярное и гистиоцитарное происхождение и аналогичны
клеткам, наблюдаемым при грибовидном микозе. Большинство современ-
ных авторов (Каламкарян А. А., 1967; 1968; Андреев В. и соавт., 1978;
Tedeschi и Lansinger, 1965; Prunieras, 1974; Brehmer-Andersson, 1976;
Cledeuning et al., 1964, 1977; Saxe et al., 1977; Cooperrider et al.,
1978; Price и Microvich, 1978; Higedorn et al., 1978; Engel и Klug, 1978
и др.) на основании клинических, гистологических, цитологических, гемато-
логических, цитохимических и электронно-микроскопических исследова-
ний склоняются к мнению, что синдром Сезари является не самостоятель-
ным заболеванием, а клинической равновидностью грибовидного микоза.
Как свидетельствуют исследования вышеуказанных авторов, «необычные»,
«уродливые», «монструозные» клетки в коже больных грибовидным микозом
473
и в коже и крови больных синдромом Сезари не являются специфическим
маркером этих заболеваний. Эти клетки находят также в воспалительных
инфильтратах при доброкачественных дерматозах—при псориазе, парап-
сориазе, красном плоском лишае, дискоидной красной волчанке, васкулите.
От 10 до 15% лимфоцитов здоровых людей имеют «мозговидные», «скрючен-
ные» ядра (Сезариподобные клетки). По мнению Jeckley и соавт. (1975),
Saglier-Guedon и соавт. (1977), клетки с церебриформными ядрами пред-
ставляют собой реактивные (стимулированные) лимфоциты. Engel и Klug
(1978), Duncan и Winkelmann (1978), Edelson и соавт. (1974) Thomas (1976)
и др. считают, что синдром Сезари является, по-видимому, лейкемическим
вариантом грибовидного микоза. При обоих заболеваниях речь идет о
кожных лимфомах из Т-клеток. Kato и соавт. (1974) при исследовании
аутопсийного материала выявили пролиферацию ретикуло-гистиоцитарных
клеток. Braun-Falco и соавт. (1977) также отмечают, что заметная ретику-
лярная гиперплазия наблюдается в кожных поражениях грибовидного
микоза и синдрома Сезари. Значение клеток Сезари и «микозных» клеток
остается неясным.
Клиника. Синдром Сезари, как и грибовидный микоз, встречается в
основном у лиц старше 50 лет; чаще болеют мужчины. Высыпания на коже
первоначально обычно расценивают как экзематозные, парапсориазиформ-
ные, как нейродермит. Сравнительно быстро происходит генерализация
процесса; в течение 6—12 месяцев процесс принимает универсальный ха-
рактер. В период сформировавшейся эритродермии кожа сплошь резко
гиперемировав а, отечна, инфильтрирована, с обильным средне- и круп-
нопластинчатым шелушением. Волосистая часть головы сплошь покрыта
сероватыми жирными'чешуйками. Наиболее сильно-шелушение выражено
на голенях, бедрах и предплечьях. Почти как правило, наблюдается кера-
тодермия ладоней и подошв, ониходистрофия—ногтевые пластинки стано-
вятся деформированными, крошатся, отпадают. На голове волосы заметно
редеют. Интенсивная ярко-красная окраска на туловище и лице, на конеч-
ностях постепенно приобретает синюшный оттенок; местами имеются очаги
с неравномерной бурой пигментацией. Периферические лимфатические
узлы, особенно паховые, бедренные и подмышечные, увеличены до разме-
ров лесного и грецкого ореха, плотноэластической консистенции, не спая-
ны с окружающими тканями, безболезненные. Нередко нарушается общее
состояние, повышается температура, больных беспокоит мучительный уни-
версальный зуд, появляются участки мокнутия, трещины, покрытые сероз-
но-кровянистыми корочками. Часто беспокоит постоянное ощущение оз-
ноба. По свидетельству многих авторов и по нашим наблюдениям, у боль-
ных с синдромом Сезари нет существенных изменений со стороны перифе-
рической крови и миелограммы. Лишь в терминальной фазе изменения пери-
ферической крови приобретают черты сублейкемическото лейкоза. Нозоло-
гическая классификация лейкемических изменений, появляющихся при
синдроме Сезари (грибовидном микозе), все еще затруднительна. Учиты-
вая отсутствие изменений в гемограмме и миелограмме, по нашему мнению,
логично допустить, что «наводнение» крови до 10—12% моноцитными или
474
лимфоидными клетками (клетки Сезари) происходит не из костного мозга,
а из кожных очагов поражения.
Длительность заболевания, как правило, от 1 года до 6 лет.
Гистопатология. Обильное разрастание клеток в дерме и в подкожной
клетчатке. Клеточная структура пролифератов отличается полиморфизмом.
Среди разных клеток должны быть отмечены лимфоциты, плазмоциты,
ретикулярные клетки, фибробласты, значительное количество гистиоцитов,
полйнуклеарных нейтрофилов и эозинофилов. Встречаются так называе-
мые «микозные клетки»—атипичные ретикулярные, лимфоидные или гистио-
цитарные клетки с крупными, неправильной формы, хорошо окрашиваю-
щимися ядрами. В мальпигиевом слое иногда можно обнаружить так назы-
ваемые микроабсцессы Потрие, состоящие из небольших групп клеток—
преимущественно лимфоцитов и гистиоцитов. В эпидермисе имеется вы-
раженный акантоз с увеличением эпидермальных отростков.
Дифференциальный диагноз. Следует проводить с первичным ретику-
лезом и ретикулосаркоматозом кожи, эритродермиями при лейкозах, бо-
лезни Ходжкина, парапсориазе, а также с экзематозной, псориатической
и другими эритродермиями.
Лечение. Кортикостероиды, цитостатики и рентгеновские лучи раз-
дельно или комплексно. Из цитостатических препаратов более эффективны
проспидин, циклофосфамид, винкристин, винбластин, дипин, фосфазин,
фотрин и фопурин. Эффективность их заметно усиливается при сочетанном
применении нескольких препаратов с разным механизмом действия (поли-
химиотерапия). Гелио- и климатотерапия с очень умеренным использо-
ванием солнечных лучей могут быть рекомендованы как дополнительный
лечебный метод. Между курсами полихимиотерапии рекомендуется назна-
чение поддерживающей терапии кортикостероидами и 6-меркаптопури-
ном.
ЛИТЕРАТУРА
Каламкарян А. А. Некоторые актуальные вопросы клиники грибовидного микоза.—
Вести, дерматол. и венерол., 1977, 12, 3—9.
Brehmer-Andersson Е. Mycqsis fungoiles and its relation to Sezary’s syndrome.— Acta
dermatoveneor., 1976, 56, 142—153.
Bondi E., Leyden J., RossTh.. Sezary cell syndrome ismall cell variant).— Arch. Der-
matol., 1978, 114, 12, 1864—1865.
Duncan S-, Winkelmann R. Circulating Sezary cells in hospitalized dermatology pa-
tients.— Brit. J. Dermatol., 1978, 99, 3, 171—178.
Engel S., Klug H. Klinischer Beitrag zur Problematik von Mycosis fungoides und
Sezary-Syndrome.— Derm. Mschr., 1978, 164, 2, 130—137.
Haustein U. Mycosis fungoides — Bewahrtes und Neues.— Derm. Mschr., 1977, 163,
11, 865—885.
Loffler H., Meyhofer W., Lange R. Sezary—Syndrome eine leukamische Variante
der Mycosis fungoides.— Dtsch. Med. Mschr., 1974, 99, 10, 429—434.
Prunieras M. Le syndrome et la cellule de Sezary.— Dermatologica, 1978, 157, 6,
371—376.
475
СИРИНГОМА (syringoma).
Синонимы: сирингоцистоаденома (Тогок, 1899), множественная тубе-
розная лимфангиома (Kaposi, 1872), распространенная гидраденома (Jac-
quet-Darier, 1887) и др.
Этиология и патогенез. Сирингома является доброкачественной опу-
холью потовых желез. Одни считают, что она развивается из интраэпидер-
мальной части выводных протоков эккринных потовых желез (Lever и
Hashimoto, 1966; Hashimoto et al., 1966; 1967, 1980), другие (Головин
Д. И., 1958; Haensch, 1971); что при сирингоме имеет место апокринная диф-
ференцировка клеток. По мнению Hacusch и соавт. (1971), о развитии си-
рингомы из апокринных желез свидетельствуют такие данные, как локализа-
ция, сочетание с другими опухолями первичного эпителиального ростка,
такими, как трихоэпителиома или adenoma sebaceum, связь с наружным
корневым влагалищем, а не с эккринными потовыми железами. С точки
зрения авторов можно было бы допустить возможность развития сирин-
гомы из 2 различных производных эктодермы, в частности, располагающих-
ся на веках и грудной клетке, но считают это маловероятным и недоказан-
ным. Высказывалась точка зрения (Winkelmann и Miiller, 1964), основан-
ная на гистохимических исследованиях, о возможной связи сирингомы с
секреторной частью эккринных потовых желез. Hashimoto et al. (1985)
с помощью моноклональных антител подтвердили, что эруптивная сирин-
гома и сирингома, располагающаяся в области век, является одной и той
же опухолью, развивающейся из протока эккринных потовых желез. По
мнению А. К. Апатенко (1973), сирингому правильнее рассматривать не
как истинную доброкачественную опухоль, а как порок развития потовых
желез, гистогенетически связанный с выводными протоками эккринных
потовых желез. Описаны семейные случаи (Yesudian и Thambiah, 1975 и
др.). Предполагают возможность эндокринной предрасположенности, на-
следуемой по аутосомно-доминантному типу.
Клиника. Высыпания чаще множественны, располагаются симметрич-
но на лице, особенно периорбитально, шее, в области плечевого пояса»
грудной клетки, половых органов. Реже—на животе, разгибательной по-
верхности конечностей (Hugues и Apisarnthanarax, 1977). Но они могут
быть немногочисленными и локализоваться только на лице (периорбиталь-
но) или на шее. По данным Allington и соавт. (1968), сирингома является
самой частой опухолью век. Это мелкие, едва возвышающиеся над уровнем
кожи, резко ограниченные, плотноватые милиум-подобные опухолевидные
элементы, слегка блестящие, как бы просвечивающие, темно-розовые, жел-
товато-коричневатые или чаще цвета нормальной кожи. Слияние отдель-
ных элементов наблюдается в очень редких случаях (Рукавишникова В. М.,
1976). Диссеминированные формы не часты. И. А. Кауфман и Л. А. Пет-
ренко (1966) наблюдали больную с множественными высыпаниями, распо-
лагающимися по всему кожному покрову, со слиянием мелких изолирован-
ных элементов в подмышечных впадинах, паховых складках, промежности.
При желтовато-коричневатой окраске высыпаний может быть клиническое
сходство с пигментной крапивницей (Haneke и Gutschmidt, 1978). Описаны
476
изолированные сирингомы, а также располагающиеся на тыле пальцев
кистей—акральная сирингома (Hugnes и Apisarnthanarax, 1977). В редких
случаях сирингома может располагаться только на больших половых гу-
бах (Carneiro et al., 1971), на половом члене (Zalla и Perry, 1971). Субъ-
ективных расстройств не бывает, если не считатьнеболыпого зуда у неко-
торых больных с диссеминированными формами.
Заболевание развивается, как правило, в период полового созревания,
примерно в 2 раза чаще у женщин. Эруптивные формы развиваются раньше
(Zalla и Perry, 1971). Раз возникнув, существует неопределенно долго, лишь
в редких случаях наблюдается частичное регрессирование, преимуществен-
но при распространенных формах. Прогноз для жизни благоприятный.
В. М. Рукавишникова (1976) обнаружила лишь одно описание злокачествен-
ного перерождения сирингомы. У больных нередко отмечаются и другие
доброкачественные опухоли, прежде всего трихоэпителиомы. Указывается
(Butterworth et al., 1964; Lechner и Dotzer, 1981 и др.) на частое развитие
сирингомы, обычно изолированной, располагающейся периорбитально у
лиц, страдающих болезнью Дауна.
Dupre и Bonafe (1977) описали сочетание сирингомы, болезни Дауна,
синдрома Марфана и синдрома Элерса—Данлоса. Обращено внимание на
своеобразие сирингомы, которая образовывала большие эритематозные очаги
причудливых очертаний, расположенных на ихтиозиформной коже.
Гистопатология. В верхней части дермы обнаруживаются тяжи из
клеток, напоминающих клетки базального слоя эпидермиса, маленькие
кисты, связанные с выводными протоками потовых желез, окруженные
двумя рядами эпителиальных клеток. Кисты содержат коллоидоподобные
просвечивающие массы. Описаны варианты, в которых гистологически до-
минировали светлые клетки (Headington et al., 1972).
Дифференциальный диагноз. С узелковыми ксантомами, трихоэпи-
телиомой, базалиомой.
Лечение. Отдельные крупные элементы могут быть удалены диатер-
мокоагуляцией.
ЛИТЕРАТУРА
Рукавишникова В. М. Случай сирингомы. Вести, дерматол. и венерол., 1976, 11,
60—63.
Haneke Е., Gutschmidt Е. General isierte syringome.— Hautarzt, 1978, 29, 4, 222—223.
Hashimoto K-, BlumD., FukayaT. et al. Familial syringoma Arch, dermatol., 1985,
121, 6, 75k—760.
Hugues P. S. H. Apisarnthanarax P. Acral syringoma.— Arch, dermatol., 1977, 113,
10, 1435—1436.
Lechner W., Dotter V. Multiple syringome bei Trisomie 21.— Z. Hautkrkh., 1981,
56, 22, 1467—1470.
СКЛЕРЕДЕМА ВЗРОСЛЫХ БУШКЕ (scleroedema adultorum Buschke).
Синонимы: отечная склеродермия, доброкачественное прогрессирую-
щее уплотнение подкожной клетчатки, склерема взрослых.
Buschke (1900) впервые описал клинические признаки заболевания
Термин «взрослых» является недостаточно верным, так как заболевание
нередко встречается и у детей. Janet и соавт. (1935) различают доброкачест-
венную «детскую» форму склередемы, регрессирующую без лечения, и
«взрослую» форму, характеризующуюся различной длительностью. Rei-
chenberger (1964) указывает на две вершины подъема этой болезни: в воз-
расте от одного года до двадцати лет и от сорока одного года до пятидесяти
лет. Женщины болеют в два раза чаще, чем мужчины.
Этиология и патогенез. Возникновение склередемы после острых ин-
фекций заставляет думать об инфекционно-аллергическом или инфекционно-
токсическом происхождении.
Клиника. Склередема развивается внезапно, почти всегда после ин-
фекционного заболевания (ангины, тонзиллита, фарингита, скарлатины,
гриппа, пневмонии, рожи и др.). Появляется уплотнение кожи лица и
шеи, неловкость и затруднение при повороте головы. Уплотнение очень
быстро распространяется на плечи, грудь, верхнюю часть опины, иногда
на нижние конечности, оставляя свободными кисти рук. Мимика лица
уменьшается, кожные складки и морщины сглаживаются. Явлений атро-
фии не наблюдается. Пораженная кожа натянута, бледная или слегка
синюшна; нередко отмечается выраженная гиперемия. Больные жалуются
на скованность при движениях шеи, затруднении движения в плечевом
поясе, чувство стягивания. Примерно у 10—12% больных вовлекается
в процесс язык (макроглоссия), в противоположность микроглоссии
при системной склеродермии. Волосы не выпадают. Хотя нетронутость
кистей и стоп является дифференциально-диагностическим признаком
этого заболевания, однако Л. Н. Машкиллейсон (1965), Roy и соавт. (1972)
допускают, что в 8—10% они вовлекаются в процесс. В настоящее время
многие авторы обнаруживают у больных склередемой изменения внутрен-
них и эндокринных органов, вегетативной нервной системы, белковых фрак-
ций сыворотки крови и т. д. (Кузнец М. М. и Кальянова М. И., 1948;
Марьясис Е. Д. и Митихина 3. И., 1966; Полуэктов Ю. А. и Арский
С. А., 1968; O’Leary, 1940; Breinin, 1953; Greenberg et al., 1963; Curtis
и Chulak, 1965; Holubar и Mach, 1967; Jogaman и Echeverria, 1974; Meyer
и Quantoc, 1974 и др.). Заболевание в большинстве руководств описыва-
ется как сравнительно быстро протекающее и самостоятельно проходящее.
По нашим наблюдениям (Каламкарян А. А. и Чистякова И. А., 1977;)
склередема часто имеет хроническое течение. В среднем длительность бо-
лезни составляет 1—3 года; в некоторых случаях значительно больше—
до 12—19 лет. В последние годы некоторые авторы сталй выделять особую
форму склередемы у больных сахарным диабетом, характеризующуюся
упорным многолетним течением. Изменения кожи и подкожной клетчатки
при этой форме чаще локализуются в области затылка, задней поверхно-
сти шеи и верхней части спины.
478
Гистопатология. В глубоких слоях дермы—набухшие расщепленные
коллагеновые волокна раздвинуты так, что между ними остаются про-
странства, заполненные метахроматически окрашивающимся веществом
(гиалуроновая кислота). Гистохимические исследования выявляют, что
муциноподобное вещество есть мукополисахарид (Cohn et al., 1970).
Эпидермис нормален, иногда с незначительным гиперкератозом.
Дифференциальный диагноз. Проводят с диффузной склеродермией,
, дерматомиозитом, микседемой кожи.
Лечение. Антибиотики, кортикостероидные препараты (преднизолон
в средних дозах), сульфаниламиды, бальнео- и физиотерапия. А. А. Ду-
бинский и П. П. Гуида (1976) сообщили об успешном применении уни-
тиола.
ЛИТЕРАТУРА
Дубинский А. А., Гуйда П. П. Случай склередемы, успешно леченной унитиолом.—
Вестн. дерматол. и венерол., 1978, 8, 49—51.
Каламкарян А. А., Чистякова И. А. О склередеме взрослых Бушке.— Вестн. дер-
матол. и венерол., 1977, 9, 6—10.
Meyer R., Quantock О. Chronic scleroedema.— S. African. Med. J., 1974, 48, 5, Г64—
166.
Yogman M., Echeverria P. Schleredema and carditis.— Pediatrics, 1974, 54, 1, 108—
III.
СКЛЕРОМИКСЕДЕМА АРНДТА—ГОТТРОНА (scleromyxoedema Arndt-
Gottron).
Синонимы: папулезная лихеноидная микседема, микседематозный ли-
хен.
Этиология и патогенез. Winand (1972), Fowlkes и соавт. (1967) выска-
зывают предположение об аутоиммунной природе заболевания. Другие
исследователи относят склеромикседему к заболеваниям с парапротеине-
мией типа каппа или лямбда (Colomb et al., 1966; James et al., 1967; Huth
et al., 1972; Proppe, 1969; Krebs и Muller, 1980). Действительно napanpo-
теинемия при склеромикседеме и микседематозном лихене наблюдается
нередко. Кожа приобретает антигенные свойства,, в ответ на которые плаз-
матические клетки костного мозга вырабатывают особые антитела-пара-
протеины, относяещиеся к IgG с легкими цепями каппа или лямбда. Это
подтверждается обнаружением в сгустке крови больных парапротеина
(Каламкарян А. А. и соавт., 1976 и др.). По мнению большинства авторов
(Смелов Н. С., 1964; Шапошников О. К-, 1960 Каламкарян А. А. и со-
авт., 1976; Braun-Falco, 1970; Hollman, 1970 и др.), склеромикседема
относится к группе муцинозов, не связанных с нарушением функций щи-
товидной железы, хотя в описании отдельных наблюдений имеются указа-
ния на связь с гипотиреозом. Нарушения белково-синтетической функции
печени, по мнению Montgomery и Underwood, лежат в основе образования
патологических белковых фракций и муцина, откладывающегося в коже.
Опираясь на электронно-микроскопические исследования Carli-Basset
и соавт. (1979), Granowska и ссавт. (1981) считают, что при этом заболева-
479
Рис. 327. Склеромикседема
А рндта—Готтрона.
нии муцин вырабатывается непосредственно в дерме фибробластами. Слу-
чаи склеромикседемы наблюдали А. А. Каламкарян, В. А. Самсонов и
соавт., 1976; Б. А. Беренбейн и Т. С. Жаворонкова, 1977; А. С. Лемперт и
соавт., 1978; Е. М. Каралицкий, 1980; Grubendorf и Gahlen, 1973; Lai и
Fat, 1973; Bridgen, 1974; Hollman et al., 1970; Feldman et. al., 1969; Liden
et al., 1971 и др.
Клиника. Заболевание встречается преимущественно у женщин старше
40—50 лет, характеризуется наличием многочисленных мелких узелков
диаметром 3—5 мм, полукруглых очертаний, блестящих (особенно при
боковом освещении), располагающихся группами. Узелки имеют тенден-
цию к слиянию, образуя очаги поражения с утолщенной, плотной (скле-
родермиформная плотность), не собирающейся в складки кожей. Отмеча-
ется своеобразный распространенный отек кожи, который в противопо-
ложность другим видам отека не оставляет ямки при надавливании. Ха-
рактерен темно-розовый с восковым блеском цвет возвышающихся скла-
док и массивных инфильтратов на туловище, кистях, особенно на лице
(рис. 327). Поражение кожи, как правило, распространенное, высыпания
располагаются на лице, туловище, на верхних конечностях, особенно на
предплечьях и кистях, на ягодицах, реже на ногах. Волосяные фоллику-
лы расширены, втянуты, что придает коже сходство с апельсиновой кор-
кой. Обращает на себя внимание общий вид больных: маскообразное лицо,
узкие глазные щели из-за отечности век, утолщенные губы и нос. Деформа-
ция лица придает ему сходство с facies leonina. Кисти приобретают Хапо-
480
образный вид, сгибание в пястно-фаланговых и межфаланговых суставах
затруднено, объем движений в них ограничен. Отмечается выраженный
гиперкератоз, особенно в области ладоней и подошв. Секреция сальных и
потовых желез уменьшена. Волосы сухие, отмечается диффузное их по-
редение на голове, наружной части бровей и в подмышечных впадинах.
Больные часто жалуются на общую слабость, снижение работоспособности,
скованность в движениях. Часты нарушения сердечно-сосудистой деятель-
ности, острые психические расстройства, дизартрия и др.
Прогноз при своевременно начатом лечении благоприятный.
Гистопатология. Отложение муцина, расщепление коллагеновых фиб-
рилл, главным образом в ьерхней трети дермы, увеличение числа фиброблас-
тов. При использовании специальных красителей в поверхностных слоях дер- ,
мы определяются скопления муцина между пучками коллагеновых волокон,
а также звездчатой конфигурации фибробласты, которым, как? уже отмеча-
лось, некоторые исследователи приписывают роль производителей муцина.
Существует прямая связь между характером этих изменений и степенью
выраженности клинических проявлений заболевания (Hadida е) al.,\1969;
Sourmond и Furth, 1968 и др.). При электронно-микроскопическом иссле-
довании обнаруживается большое количество фибробластов.
Дифференциальный диагноз. Следует проводить с диффузной склеро-
дермией.
Лечение. Кортикостероиды (20—30 мг в сутки в пересчете на «предни-
золон), дийодтирозин (по 0,05 г 2 раза в день трехнедельными циклами с
10-дневными интервалами между циклами), тиреоидин (по 0,05 г 2—3 ра-
за в день раздельно или в комбинации с кортикостероидными препаратами)*
Наблюдение дерматолога и эндокринолога.
ЛИТЕРАТУРА
Беренбейн Б. А., Жаворонкова Т. С. Склеромикседема-— Вести, дерматол. и ве-
нерол., 1977, 12, 36—40.
Каламкарян А. А., Самсонов В. А., Завадская Ю. Н. Склеромикседема.— Вести,
дерматол. и веиерол., 1976, 1, 53—56.
Каралицкий Е. М. К вопросу о склеромикседеме АриДта—Готтрона.— Вести, дер-
матол. и веиерол., 1980, 12, 59—62.
Лемперт А. С., Панков Б. С., Белова Г. С. Склеромикседема Арндта—Готтрона.
— Вести. Дерматол. и венерол., 1978, 7, 65—68.
Carli-Basset С., Loret G., Alison G. et al. Apparition retardee d’une globulin monoc-
lonale au cours d’une mucinose papuleuse.— Ann. Dermatol. Venereol., 1979, 106, 175—179.
Granowska et al. Electron microskopy findings in scleromyxoedema.— In: XIII natio-
nal Congress of the Hungerian Society of dermatology. Budapest, March 90— April. 1981,
p. 106—107.
Krebs A., Muller A. Lichen mxyoedermatosus in multiples Myelom vom Typ IgG/Kap-
pa.— Hautarzt, 1980, 31, 12, 649—653.
31 Клиническая дерматология
481
СЛИЗИСТО-КОЖНЫЙ ЛИМФАТИЧЕСКИЙ СИНДРОМ (mucocutaneo-
us lymph node syndromum).
Синонимы: болезнь Кавасаки, узелковоподобный артериит.
Болезнь впервые описана в Японии в 1967 г. Т. Kawasaki на основе
наблюдения за 50 детьми. Начиная с 1973 г., появились сообщения об этой
болезни в других странах мира—в США, Корее, Франции, Греции, Канаде
и др. (Kim J. et al., 1973; Radford et al., 1976; Fanconi, 1978; Gorin et al.,
1978; Berel et al., 1980 и др.). В отечественной дерматологической литера-
туре описания этого заболевания мы не встречали.
Этиология и патогенез. Причина болезни окончательно не выяснена.
Дискутируется возможность вирусной этиологии. При электронной микро-
скопии (Hamashima et al., 1973; Fatterman и Hashida, 1974) биоптатов
кожи и лимфатических узлов лиц с болезнью Кавасаки обнаруживаются
риккетсиоподобные тельца.
При болезни Кавасаки имеется системное поражение мелких и сред-
них артерий, морфологически подобное изменениям при узелковом пери-
артериите (Gossege et al., 1979; Keren et al., 1979; Nagington и Rook, 1979.
Rossignol et al., 1980). Характерная особенность—пораженное сердце, вы-
являвшееся во всех патологоанатомических наблюдениях (Tanaka et
al., 1976). Помимо сердечной патологии, нередко обнаруживают пораже-
ние сосудов почек, гипоплазию вилочковой железы и очаговые воспали-
тельно-некротические изменения в лимфатических узлах.
Клиника. Болезнь Кавасаки встречается у детей в возрасте от 3 меся-
цев до 10 лет, чаще у мальчиков в первые 2 года жизни. Развивается пре-
имущественно летом.
Заболевание начинается с повышения температуры до 38—40°, не
поддающегося воздействию антибиотиков. Спустя несколько дней присое-
диняется конъюнктивит, диффузная гипеоемия слизистой оболочки рта и
и глотки, заложенность носа, гиперемия зева и распространенные пятни-
стые полиморфные эритематозные высыпания (без везикуляции) на коже
лица, туловища и конечностей. Отмечается отек сосочков языка (по типу
«малинового» языка), острое увеличение шейных лимфатических узлов,
резкая гиперемия и плоский отек ладоней и подошв. Кроме того, могут
наблюдаться нарушения коронарного кровообращения, диарее умерен-
ный лейкоцитоз, повышение СОЭ, анемия, нередко повышение IgE.
Продержавшись 2—3 недели, температура нормализуется, гиперемия
слизистой рта, зева, конъюнктивы исчезает, увеличение шейных лимфатиче-
ских узлов проходит, плотный отек на ладонях и подошвах рассасывается,.
отмечается поверхностная десквамация эпидермиса вначале на кончиках
пальцев, а затем на ладонях. В отделе дерматологии ЦКВИ мы (Каламка-
рян А. А., Гребенюк В. Н., Федоров С. М., 1985) наблюдали ребенка
2,5 лет с типичными поражениями кожи, слизистых оболочек и лимфатиче-
ских узлов на фоне высокой температуры, диспептических расстройств,
лейкоцитоза с нейтрофилезом (78%) и сдвигом влево, увеличением СОЭ
(52 мм час). Терапия пенициллином была не эффективной. Преднизолон
в небольших дозах оказал благоприятное воздействие. Отек кистей и стоп,
482
эритема ладоней, подошв, слизистой ротовой полости, конъюнктивит, лим-
фаденопатия и кожные высыпания на туловище уменьшились, а затем ис-
чезли вслед за прекращением лихорадки.
Приводим частоту отдельных симптомов при болезни Кавасаки, по
данным японских авторов: лихорадка (38—40°, не поддающаяся антибио-
тикотерапии)—у 95% больных, полиморфные высыпания на коже лица и
туловища—у 92%, диффузная гиперемия слизистой рта и зева—у 90%,
гиперемия и отек ладоней и стоп—у 88—94%, конъюнктивит—у 88%, уве-
личение шейных лимфатических узлов—у 75—80%.
Прогноз чаще благоприятный, но 1,7—2% больных погибают от сер-
дечной патологии. Непосредственной причиной смерти являются разрыв
аневризмы, тромбоз коронарных сосудов, инфаркт миокарда (Kawasaki
et al., 1974; Kato et al., 1975; Radford et al., 1976 и др.).
Гистопатология. Изменения неспецифичны (Kawasaki et al., 1974).
Так, по данным Riley (1976), исследовавшего биоптаты кожи у 7 больных,
гистологические изменения не имели отличительных особенностей. Автор
обнаружил лишь отек дермы, расширение капилляров и умеренную пери-
васкулярную инфильтрацию лимфоцитами, гистиоцитами и моноцитами.
В биоптатах лимфоузлов обнаруживается неспецифическая гиперплазия
ретикулярных и лимфоидных клеток.
Дифференциальный диагноз. Следует проводить с ревматоидным арт-
ритом, узелковым периартериитом, синдромом Стивенса—Джонсона, много-
формной экссудативной эритемой, инфекционным мононуклеозом, лекар-
ственной болезнью, паратонзиллитом, флегмоной дна полости рта.
Лечение. Антибиотики малоэффективны. В тяжело протекающих слу-
чаях назначают кортикостероиды (чаще преднизолон) в максимальной
суточной дозе 1,5—2 мг/кг массы тела с последующим постепенным сниже-
нием дозы до полной отмены через несколько месяцев. Симптоматические
средства. На кожные очаги поражения рекомендуются кремы и мази, со-
держащие кортикостероиды.
ЛИТЕРАТУРА
Aterman К-, Dische М., Franke et al. Aneurysms of the coronary arteries in infants
and children. A review and report of six cases.— Arch, path, Anat., 1979, 373, 27—44.
BellD-. Brink E. et al. Kawasaki's syndrome.— N. Engl. J. Med., 1981, 304, 1568—
1575.
Fanconi A., Bodmer P., Egloff B. Akutes mukokutaneos Lymphknotensyndrom (Ka-
wasaki syndrom) mit letaler Periarteriitis nodosa der krouzarterien.— Helv. paediat. Acts,
1978, 33, 2, 135—140.
Kawasaki T. et al. A new infantil febrile mucocutaneous lymph node syndorme prevai-
ling in Japan.— Pediatrics, 1974, 54, 271—276.
Kegel S. et al. Cardiac death in mucocutaneous lymph node syndrome.— Amer. J.
Cardiol., 1977, 40, 2, 282—286.
Kusakawa S.. Heiner D. Elevated levels of immunoglobulin E in the acute febrile
mucocutaneous lymph node syndrome.— Pediatr. Res., 1976, 10, 108—111.
31* 483
N'agington J., Rook A. Virus and related infection.— In: Tevtbook of dermatology
(Ed. A. Rook, D. Wilkinson, F.) Ebling. Oxford—London, 1979, v. 1, p. 607—676.
Riley H. Mucocutaneous lumph node syndrome (Kawasaki disease).— J. Infect. Dis.,
1976, 134, 3, 302—305.
Rossignol A., Bost M., Pasquier D. Thrombose mortelle d’un anevrysme coronarien
chez use enfant. Maladie de Kawasaki on periarterite nodeuse? (letter).— Nouv. Presse Med.,
1980, 9, 6, 386—393.
Spector R. Mucocutaneous lymph node syndrome.— In: Clinical dermatolody. Philadel-
phia, 1983, 2, 7, 44.
Weston W., Huff J. The mucocutaneous lymph node syndrome: A critical re-exami-
nation.— Clin. Exp. Dermatol., 1981, 6, 167—178-
СТИВЕНСА—ДЖОНСОНА СИНДРОМ (syndromun Stevens—Johnson).
Синонимы: эктодермоз эрозивный плюриорифициальный Фиссенже—
Рандю, дерматостоматит Баадера, пемфигоидная многоформная эритема.
Синдром описан в 1922 г. американскими педиатрами Стивенсом и
Джонсоном. По мнению многих исследователей, является тяжелой формой
многоформной экссудативной эритемы (Каламкарян А. А., 1959; Паш-
ков Б. М., 1964; Каламкарян А. А. и Самсонов В. А., 1980; Самцов В. И.,
1979; Королев Ю. Ф., 1980; Браиловский А. Я., 1980; Бухарович М. Н.,
1980; Callahan, 1976; Wermut et al., 1975; Kauppinen, 1972 и др.).
Этнология и -патогенез. Инфекционная, в частности вирусная, гипо-
теза не получили убедительных доказательств. Большинство современных
авторов относит синдром Стивенса—Джонсона к токсико-аллергическим
заболеваниям. Работами многих исследователей показано, что повышен-
ная чувствительность больных при этом синдроме характеризуется возник-
новением необычной, извращенной реакции на введение очень малых ко-
личеств лекарственных средств (антибиотики, сульфаниламидные препара-
ты, анальгетики, барбитураты, жаропонижающие и многие другие). Выска-
зывается обоснованное мнение о близости синдрома Стивенса—Джонсона
и синдрома Лайелла, по существу менаду ними имеется лишь количествен-
ное, а не качественное различие.
Клиника. Заболевание характеризуется бурным началом с выражен-
ными общими расстройствами, высокой лихорадкой и преобладанием во
всей клинической картине тяжелых буллезных поражений слизистых обо-
лочек и кожных покровов. На туловище, верхних и нижних конечностях,
а также на лице и в области половых органов появляются эритематозно-
отечные пятна и багрово-красные бляшки с синюшным оттенком, размером
от одно-до пятикопеечной монеты и больше. Затем очень быстро (в течение
иепкольких часов) на этих местах формируются плоские пузыри с фестон-
чатыми или округлыми очертаниями размером до ладони взрослого и боль-
ше; пузыри, сливаясь, могут достигать огромных размеров. На некоторых
покрасневших участках кожного покрова эпидермис «сползает» под влия-
нием легкого надавливания или прикосновения (резко положительный
симптом Никольского). Пузыри тонкостенные, вялые, содержимое их
большей частью серозное или серозно-кровянистое, в случае присоедине-
ния вторичной пиококковой инфекции оно становится гнойным. На месте
484
вскрывшихся пузырей обнажаются обширные резко болезненные мокну-
щие поверхности ярко-красного цвета с остатками покрышек по периферии
(«эпидермальный воротник»). В тяжелых случаях отмечается выпадение
волос, паронихии с последующим отторжением ногтевых пластинок. На
тыле кистей, предплечий и голеней часто появляются элементы с запавшим
синюшным центром, окруженным ярким венчиком гиперемии, клинически
весьма напоминающие высыпания многоформной экссудативной эритемы.
Часто наблюдаются тяжелые поражения слизистых оболочек. На слизи-
стой оболочке рта и носа появляются гиперемия, отек, вялые пузыри,
которые, вскрываясь, образуют крайне болезненные обширные эрозии и
изъязвления с остатками покрышек пузырей. Губы резко отечны, гйпере-
мированы, покрыты корками, кровоточащими эрозиями, трещинами и
пузырями. Нередко наблюдается блефароконъюнктивит с серозно-гнойным
отделяемым из глазной щели, иридоциклит, иногда даже панофтальмит с
полной потерей зрения (Байкова Р. А. и Фишман Г. А., 1975; Elias et al.,
1977; LJbaidy и Nally, 1976; Trevison et al., 1982 и др.). Описаны случаи
баланита, вульвовагинита, вовлечения уретры и мочевого пузыря, а также
перехода процесса на трахеобронхиальное дерево, желудочно-кишечный
тракт, почйи и другие висцеральные органы. Заболевание сопровождается
общими тяжелыми явлениями: высокой температурой (38—40°), расстрой-
ством нервной системы (головные боли, бессонница, раздражительность,
коматозное состояние и др.), поражением пищеварительных органов (диспеп-
тические явления, гепатит, гастрит), дыхательных органов (бронхит,
плеврит, пневмония). •
Поражение 70—75% поверхности кожи рассматривается как опасное
для жизни. Летальный исход наступает у 18—20%, а по некоторым данным
у 40—45% больных.
Гистопатология. Некроз поверхностных слоев эпидермиса, наруше-
ние эпидермо-дермальной связи.
Лечение. Антибиотики, кортикостероиды (дозировка и длительность
назначения последних определяется тяжестью процесса—в среднем 30—
50 мг в сутки преднизолона), антигистаминные, десенсибилизирующие
средства, внутривенное капельное введение водно-электролитных раст-
воров, 5% раствора глюкозы с небольшими дозами инсулина, гемодеза.
ЛИТЕРАТУРА
Бочков Н. П. Генетика человека. Наследственность и патология.— М.: Медицина
1978.
Каламкарян А. А., Самсонов В. А. О миогоформной экссудативной эритеме, сннд-.
роме Стивенса—Джонсона, токсическом эпидермальном некролизе (синдроме Лайелла) и
их взаимоотношении.— Вести, дерматол. и венерол., 1980, 3, 36—42.
Киселев И. В. Синдром Стивенса—Джонсона н токсико-аллергический гепатит у
больных туберкулезным эксудативным плевритом.— Клин, мед., 1978, 2, 113—114.
Мельник Н. К особенностям клиники синдрома Стивенса—Джонсона.— Клин,
мед., 1978, 8, 130—133.
485
Самцов В. И. Подвысоцкая И. И. Токсический эпидермальный некролиз.— Вестн.
дерматол. и венерол., 1979, 12, 16—23.
Assad D. Toxic epidermal necrolisis in Stevens—Johnson syndrom.— Can. Med. Ass.,
1978, 118, 2, 154—156.
Elias P., Fritsch P., Epstein E. Staphylococcal scalded skin syndrome.— Arch. Der-
matol., 1977, 113, 4, 207—212.
Lyell A. Toxic epidermal necrolysis (the scalded skin syndrome): a reppraisol.— Brit.
J. Dermatol., 1979, 100, 1, 69—86.
Trevison G. et al. Syndrome d: Stevens—Johnson. G. ital. dermaetsl., 1982, 117,
3, 183—185.
СТЮАРТА—ТРИВСА СИНДРОМ (syndromum Stewart—Treves).
Синонимы: постмастэктомическая лимфангиосаркома, злокачествен-
ная лимфостатйческая эндотелиома.
В 1948 г. Stewart и Treves впервые описали шесть случаев лимфангио-
саркомы. В литературе к настоящему времени опубликовано 226 случаев.
Этиология и патогенез. В основе синдрома Стюарта—Тривса лежит
развитие злокачественной сосудистой опухоли на фоне хронической лим-
федемы и элефантиаза руки той стороны, где производилась в прошлому
радикальная мастэктомия по поводу рака молочной железы. В большинстве'
случаев радикальная мастэктомия сочеталась с удалением лимфоузлов
или рентгеновским облучением. У женщин, перенесших мастэктомию и
подвергшихся облучению, хроническая лимфедема встречается в 10 раз
чаще, чем у женщин, лечившихся без рентгенооблучения. Данные о часто-
те отека и элефантиаза у оперированных женщин колеблются между 20—
30% и 70—80% (Woodward et al., 1972; Kleemann и Stihl, 1972; Adolfsson,
1973; Foldi, 1977; Sandmark, 1977; Eggert et al., 1977; Duran et al., 1978
и др.). Лимфангиосаркома развивается в 0,45—0,8% случаев. В генезе
отека после мастэктомии главное значение имеют циркуляторные сосуди-
стые расстройства верхней конечности в результате тяжелых анатомических
и функциональных нарушений в системе крово-и лимфообращения на
стороне операции (блок лимфооттока, интерстициальный фиброз, образо-
вание рубцов и др.). Лимфо-и гемангиоэндотелиомы могут развиться рано,
через год (Bacman et al., 1979). и поздно, через 27—49 лет (Schaffer et al.,
1979), но в большинстве случаев они развиваются спустя 8—10 лет после
мастэктомии (Woodward et al., 1972; Winkler, 1975; Duran et al., 1978;
Kostler et al., 1978; Pizzi et al., 1983; Hoberle, 1983 и др.). В литературе
нет единой точки зрения на гистогенез этой опухоли. Одни авторы (Stewart
и Treves, 1948; Негу et al., 1972; Wolf, 1963; McConnel и Haslam, 1959;
Silverberg et al., 1974; Glass, 1976 и др.) полагают, что опухоль является
заболеванием sui generis, развивающимся из лимфатическиих сосудов, т. е.
не связана с карциномой грудной железы. Другие (Delaure, 1962; Martin
и Vilain, 1960; Salm, 1963 и др.) считают, что ангиосаркома развивается из
предшествующего рака грудной железы. На основании гистохимических и
электронно-микроскопических методов исследования Friederici и Roberts,
1967; Kermarek и Varini, 1975; Laugier и Olmos, 1973; Silverberg и соавт.,
1971 и др. утверждают, что синдром Стюарта—Тривса представляет собою
486
-I
сосудистую саркому (лимфангиосаркому), развивающуюся на фоне пред-
шествующего хронического лимфостаза.
Клиника. Синдром встречается почти только у женщин преимуществен-
но пожилого возраста (средний возраст больных 62—68 лет—Sanderson
и MacKie, 1979; Woodward, 1972), хотя иногда встречается и у молодых
женщин 25—30 лет (Ryan et al., 1974). Известны отдельные случаи и у
мужчин (Oettle et al., 1963; Ramsey et al., 1968). Поражение имеет одно-
стороннюю локализацию—в коже и подкожной клетчатке плеча и пред-
плечья, где появляются узелки синюшно-красного или багрово-циано-
тичного цвета, размером до горошины, часто растущие экзофитно; вокруг
них образуются сателлиты, петехиальные кровоизлияния и телеангиэкта-
зии. Узелки, сливаясь, образуют массивные узлы, опухоли синевато-чер-
ного цвета размером до сливы и больше, возвышающиеся над уровнем кожи
на 2—3 см. Затем, как правило, наступает изъязвление кожи над опухолью
и распад опухолевой ткани; эти явления быстро прогрессируют. Язва
увеличивается как по протяженности, так и в глубину. Дно ее неровное,
часто покрыто серозно-геморрагическими и некротическими корками,
отмечается обильное выделение кровянисто-серозной жидкости.
Болезнь характеризуется высокой злокачественностью, агрессивным
ростом с метастазированием в легкие, печень, надпочечники, яичники и
другие органы. Заболевание всегда заканчивается смертью больных в сред-
нем через 15—20 месяцев после развития ангиосаркомы (Woodward et al.,
1972; Winkler, 1975; Eby et al., 1967; Dimitrescu и Vulcan, 1980 и др.).
Гистопатология. Обилие расширенных, беспорядочно анастомозирую-
щих тонкостенных сосудов, выстланных несколькими слоями атипичных
полиморфных эндотелиальных клеток с большим количеством митозов и
скопление клеток с крупными ядрами.
Электронная микроскопия. Сосуды выстланы эндотелиальными клет-
ками, содержащими несколько пузырьков пиноцитоза и микрофиламенты,
четко выраженная базальная мембрана с «пеницитными» клетками (Pizzi
et al., 1982, 1983).
Дифференциальный диагноз. Следует проводить с ангиолейомиосар-
комой, фибросаркомой, гемангиоэндотелиомой, саркомой Капоши, мелано-
мой, гемангиоперицитомой и другими злокачественными сосудистыми ново-
образованиями.
Лечение рентгеновыми лучами, цитостатиками и др. малоэффективно.
ЛИТЕРАТУРА
Dimitrescu A., Vulcan Р. Sindrom Stewart—Treves; Studiu anatomoclinic in legatura
cu un caz.— Dermato—Vener., 1980, 25, 1, 35—42.
Kermarek J., Varini J. Etude microscopique et ultrastructurale d’upe tumeur de Ste-
wart—Treves.— Arch. Anat. Pathol., 1975, 23, 193—198.
Kestrel E., Roitzsch E., Kuntze J. Zum Stewart—Treves—syndrom (angioplasti-
scher Sarcom bei chronischen Lymphodem).— Derm. Mschr., 1978, 164, 12, 882—888.
Olmos L., Laugier P. Stewart—Treves syndrome the histopathological evolution of
epithelial metastases.— J. Dermatol. Surg. Oncol., 1977, 3, 3, 295—299. .
487
Pizzi и соавт. Syndrome de Stewart—Treves. Deux cas avec etade — ultrastructurale.
Sem. hop. Paris., 1983, 59, 10, 687—694.
Sanderson K-, MacKie R. Tumors of the skin.— In: Textbook of dermatology. Ed
Rook Wilkinson. Therd edition. Oxford, 1979, 2199—2232.
Sandsmark M. Angiosarcoma in postmastectomy lymphoedema. A report of one case
of Stewart—Treves Syndrome.— Ann. Chir. Gynecol., 1977, 66, 5, 251—253.A.
Winkler K. Das Stewart—Treves syndrom.— Z. Hautkr., 1975, 50, 14, 593—600.
ТРИХОЭПИТЕЛИОМА (trichoepithelioma)
Синонимы: кистозная железистая эпителиома (Brook, 1892), кистоз-
ная трихобазалиома, множественная папулезная трихобазалиома (Jarisch,
1894).
Этиология и патогенез. Трихоэпителиома представляет собой добро-
качественную опухоль с дифференцировкой в сторону волосяных структур.
Часты семейные случаи с аутосомно-доминантным типом наследования.
Считается, что одиночная опухоль не наследуется. По мнению Lever (1975),
трихоэпителиома развивается из незрелых клеток с тенденцией к диффе-
ренцировке в первичные эпителиальные зародышевые структуры. Д. И.
Головин (1958) считает, что при трихоэпителиоме имеет место эпидермоид-
ная и аденоидная дифференцировка, но наиболее характерными являются
пилоидные образования—уродливые волосы, роговые и гиалиновые массы,
заключенные в тяжи и гнезда темных клеток. Автор придерживается диз-
эмбриогенетической теории развития этой опухоли. Bandmann и Bosse
(1968) различают 2 группы трихоэпителиомы: I—трихоэпителиомы, не свя-
занные с опухолями другого типа или дефектами—а) солитарные; б) мно-
жественные мономорфные; в) множественные полиморфные, П—трихоэпи-
телиомы, ассоциированные с другими дефектами—а) только с опухолями
первичного эпителиального ростка, например, с сирингомой, цилиндромой;
б) в рамках факоматозов. По мнению авторов, трихоэпителиома разви-
вается из ductus pilosebaceus.
Клицика. Высыпания обычно множественные, имеют вид мелких опу-
холевидных элементов, полушаровидных, округлых очертаний, плотно-
ватых, по цвету не отличающихся от нормальной кожи или светло-розо-
вых. Размеры их на лице обычно не превышают 0,2—0,5 см, на волосистой
части головы они могут быть крупнее, до 2—3 см. Поверхность их гладкая,
на более крупных элементах часто обнаруживаются телеангиэктазии.
Располагаются преимущественно на лице (носогубные складки, веки, верх-
няя губа, лоб), реже на волосистой части головы, шее, верхней половине
туловища. Имеются сообщения о диффузных и солитарных формах три-
хоэпителиомы (Вавилов А. М. и соавт. 1981). Zeligman (1980), описывая
4 случая солитарной трихоэпителиомы, отмечает, что этот вариант может
развиться в детском возрасте и у взрослых, располагается преимуществен-
но на лице, в области щек и подбородка, имеет небольшие размеры, не более
2 см в диаметре, овальных очертаний в виде одиночного или чаще несколь-
ких опухолевидных элементов, сгруппированных в виде кольца. Клиниче-
ское предположение возможной солитарной трихоэпителиомы всегда тре-
бует гистологического подтверждения. Czernobilsky (1972) указывает, что
488
в отличие от множественных трихоэпителиом изолированная может рас-
полагаться и на конечностях. Nasemann и соавт. (1973) описали атипич-
ную трихоэпителиому на коже подбородка в виде крупного бляшковидного
плотного очага. Подчеркиваются большие затруднения в диагностике.
Так, Steigleder (1972) приводит мнение Helwig и Gray о том, что клинически
при солитарной трихоэпителиоме правильный диагноз ставится только в
8% случаев, гистологически—только в 50%. Для диагностики, как счи-
тает Steigleder, лучше удалять весь элемент. Считается (Zeligman, 1960),
что диагноз солитарной трихоэпителиомы должен ставиться в тех случаях,
когда имеются множественные роговые кисты с элементами рудиментар-
ных волос и очень мало очагов, характерных для базалиомы. Одиночные
очаги, в которых доминирует картина, сходная с наблюдаемой при база-
лиоме, Hashimoto и Lever (1979) считают более правильным рассматри-
вать как кератотическую базалиому. У больных могут быть и другие доб-
рокачественные опухоли, такие как сирингома, цилиндрома (Rasmussen,
1975). Л. Н. Машкиллейсон и Н. С. Смелов (1931) рассматривали трихо-
эпителиому и цилиндрому как варианты или стадии одной и той же опухо-
ли. У больного, описанного Г. И. Рогайлиным и Е. В. Вербенко (1966),
были множественные милиумподобные высыпания и комедоны на клиниче-
ски неизмененной коже и на поверхности трихоэпителиом. Описано со-
четание с наследственной дистрофией ногтей (Cramers, 1981), гломеруло-
нефритом, множественными кистами почек и Легких, эпилепсией, шизофре-
нией (Grag и Helwig, 1963), одновременное развитие с карциномой молоч-
ной железы (Sandbank и Bashan 1978).
Заболевание встречается чаще у женщин. В выборке Gray и Helwig
(1963) мужчин было почти в 3 раза больше, чем женщин, но это объясняется
особенностями выборки больных, которые формировались из континген-
та военнослужащих. Развивается в период полового созревания, в юноше-
ском возрасте, реже—у взрослых, иногда существует с рождения. С тече-
нием времени количество опухолей постепенно увеличивается, становятся
несколько большими и их размеры. Самопроизвольного регрессирования
не наблюдается. Иногда возможно изъязвление, превращение в базалиому
типа ulcus rod ens. Но это признается не всеми (Lever, 1975). Мы наблюдали
изолированную трихоэпителиому на коже носа со значительной деструкцией
тканей.
Гистопатология. Роговые кисты в дерме, окруженные слоем базаль-
ных клеток, базальноклеточные островки и тяжи, нередко связанные с
волосами, иногда рудиментарные волосы. В зависимости от особенностей
гистологического строения А. К. Апатенко (1093) выделяет следующие
варианты опухоли: кистозные, светлоклеточные, солидные и сложного
строения.
Дифференциальный диагноз. С сирингомой, базалиомой.
Лечение. Хирургическое удаление при подозрении на развитие кар-
циномы.
489
ЛИТЕРАТУРА
Cramers М. Trichoepithelioma multiplex and dystrophia unguium congenita a new
syndrome? Acta derm, venerol. (Stockholm), 1981, 61, 4, 364—365.
Hashimoto K-, Lever W. F. Tumors of skin appendages. In: Dermatology in general
Medicine. Ed. by T. B. Fitzpatrick et al. Me Graw-Hill book Company Sec. ed. 1979, 4, 485—
506.
Lever W. F., Schaumburg-Leper G. Histopatology of the skin J. B. Lippincott com-
pany, 1975, 515—518.
ТРОФЕДЕМА НОННЕ—МИЛЬРОЯ—МЕЙЖА (trophoedema Nonne—
Milroy—Meige).
Синонимы: наследственная (семейная) лимфедема.
Virchow в 1863 г. описал лимфатический отек с пороками развития.
Первое описание «Elephantiasis congenita hereditaria» принадлежит Nonne
(1890). Milroy (1892) описал врожденный плотный безболезненный отек
ног у 22 больных в 6 поколениях одной семьи, Meige в 1898, 1901 и 1933
годах описал под названием трофедемы наследственный отек, развиваю-
щийся к моменту полового созревания. Наследственный характер трофе-
демы, описанной Nonne, Milroy и Meige, порок развития лимфатической
системы, лежащий в основе этих форм, позволяет многим авторам (Lau-
secker, 1949; Schnyder, 1966; Brunner, 1969; Kostler, 1975 и др.) объединить
эти формы под названием наследственной (семейной) трофедемы, подраз-
деляющейся на врожденную форму Nonne—Milroy и позднюю форму Meige.
Этиология и патогенез. Наследственное заболевание с аутосомно-
доминантным типом наследования. Описано также Х-хромосомно-доминант-
ное наследование. Пенетрантность гена около 85% (Schnyder, 1966). Име-
ется сообщение (Szarmach, 1961) о врожденной трофедеме у двух братьев,
у которых не было аналогичного заболевания в предшествующих поколе-
ниях. Patterson и соавт. (1968) доказали доминантное наследование врож-
денной лимфедемы у собак. Причину лимфедемы они видят в генетически
детерминированном дефекте развития периферической лимфатической си-
стемы. Этиология не известна. В литературе высказывается мнение о зна-
чении инфекционных, механических, эндокринных, гуморальных, нервных,
метаболических факторов.
Клиника. Заболевание встречается редко, у женщин несколько чаще,
чем у мужчин. По данным Schroder и соавт. (1950), соотношение женщин и
мужчин при врожденной наследственной трофедеме 1,29:1, а при поздней
форме—1,58:1. Заболевание существует с рождения или появляется в пер-
вые месяцы жизни, во многих случаях развивается в пубертатный период.
У большинства больных поражены обе ноги, степень их поражения может
быть разной. Отек не переходит за паховую складку, он может ограничи-
ваться только голенями, которые тогда приобретают цилиндрическую фор-
му. В начале заболевания при надавливании пальцем остается ямка’, позже
ямка не образуется. Поверхность кожи гладкая, бледная или ливидная,
редко' шишковато-папилломатозная с гиперкератозами. Изъязвления опи-
сываются очень редко. Появлению наследственной трофедемы не пред-
490
шествует воспаление. Исключение может быть при поздней форме, когда
воспаление может играть роль провоцирующего фактора (Hausten и Meseg,
1970). Болезненности, как правило, нет. Взрослые пациенты отмечают тя-
жесть в ногах, усиление отечности по вечерам, после длительного стояния
или сидения. Заболевание держится в течение всей жизни. Осложнения—
рожистое воспаление, экзема, пиодермия, язвы; злокачественные ново-
образования наблюдаются редко. Заболевание может сочетаться с дру-
гими пороками развития (spina bifida, анэнцефалия, гипер-и гипоплазия
эндокринных желез, шейные ребра, полидактилия, врожденные пороки
сердца и др.). Dhar и Hajini (1976) описали трех членов одной семьи, стра-
дающих болезнью Мильроя, у двух из которых развился нефротический
синдром. К- С. Чеботарев и X. X. Хейчетян (1979) наблюдали больного с
трофическим отеком Мильроя—Мейжа, аномалией зубов и врожденным
фимозом.
Гистопатология. В дерме, подкожной жировой клетчатке—резко вы-
раженный интерстициальный отек, лимфоэктазы. Увеличение количества
коллагена. Имеются указания на отсутствие лимфатических сосудов. Pfle-
ger (1965) установил признаки облитерирующей лимфангиопатии—расши-
рение интимы, происходящее без воспаления, приводящее к сужению и
постепенной облитерации просвета. При проведении теста красящими
веществами эвакуация краски отсутствует или появляются мелкие окра-
шенные пятнышки, что говорит об аплазии или гипоплазии лимфатических
сосудов (Marghescu и Hettier, 1964).
Дифференциальный диагноз. Проводят с другими формами лимфедемы,
микседемой, прогрессирующей липодистрофией, отеками, вследствие ве-
нозной, почечной, сердечной недостаточности, диспротеинемией и др.
Лечение. Эффективных методов лечения нет. Применяют диуретики,
препараты гиалуронидазы, инфильтрацию кортикостероидами, бинтование
эластичным бинтом, последовательное иссечение участков подкожной тка-
ни и др.
ЛИТЕРАТУРА
Чеботарев К- С., Хейчетян X. X. Случай трофического отека Мильроя—Мейжа.—
Вестн. дерматол. и венерол., 1979, 8, 49—52.
Dhar S. N., Hajini G. Н. Milroys disease with nephrotic syndrom.— Indian J. Der-
matol., Venereol., 1976, 42, 2, 60—82.
Kostler E. Das Trophodem (Nonne—Milroy—Meige) Karzinombildung als seltene
Komplikation.— Derm. Mschr., 1976, 169, 6, 465—477.
ТУБЕРКУЛЕЗ КОЖИ ВОЛЧАНОЧНЫЙ (tuberculosis cutis luposa).
Этиология и патогенез. Туберкулезная волчанка является одной из
наиболее частых форм туберкулеза кожи. Так, по данным В. А. Рахманова
и соавт. (1966), касающимся наблюдений клиники за 70-летний период
(1895—1964), волчаночный туберкулез кожи составил 70,2% всех форм ту-
беркулеза кожй.
Из 437 больных, наблюдавшихся М. И. Шаповал (1980), 116 болели
туберкулезной волчанкой. Чаще регистрировалась только уплотненная
491
Рис. 328. Туберкулез
кожи волчаночный.
Рис. 330. Туберкулезная волчанка.
492
Рис. 329. Туберкулезная волчанка
(после прокалывания ушей).
эритема—у 238 человек. Некоторое
изменение в соотношении частоты
этих форм, видимо, является след-
ствием учащения в последние годы
диссеминированных форм туберкуле-
за кожи. Возбудителем является ту-
беркулезная палочка, которая про-
никает в кожу преимущественно
лимфо- или гематогенно из других
очагов туберкулезной инфекции в
организме (легких, лимфоузлов и
др.) и вызывает специфическое вос-
паление с формированием бугорка за
счет ограниченной пролиферации
клеток (эпителиоидных, гигантских,
лимфоидных) и их частичного рас-
пада (творожистого некроза). У
больных туберкулезной волчанкой
довольно часто встречаются туберку-
лезные поражения легких, но они
как правило находятся в неактивном
состоянии. Так, М. И. Шаповал
(1980) при рентгенотомографическом обследовании обнаружила у 17 %
больных туберкулезной волчанкой следы перенесенного туберкулеза лег-
ких (кальцинаты в лимфатических узлах средостения и легких, плев-
ральные сращения и др.). В патогенезе важная роль принадлежит ал-
лергии и снижению иммунитета, а также воздействию разнообразных не-
специфических факторов, в том числе снижение естественной резистент-
ности, острые инфекции, травмы, нейро-эндокринные, вегетативно-т;ю-
фические расстройства, недостаточное питание, гиповитаминозы, болезни
крови, беременность, кортикостероидная терапия и др. (Россиянский
Н. Л. и Смелов Н. С., 1947; Агапкин И. Н. и Багаева М. И., 1959; Маш-
киллейсон Л. Н., 1960; Нерадов Л. А., 1961; Вейнеров И. Б., 1963;
Авербах М.М., 1976 и др.). Несомненное значение в развитии заболева-
ния имеют предрасположенность к туберкулезу, климато-географические
особенности, эпидемиологическая обстановка по туберкулезу вообще.
Клиника. Наиболее частой формой волчаночного туберкулеза кожи
является плоская волчанка (lupus vulgaris plantis). Так, у 15 из 18 боль-
ных, леченных И. Н. Агапкиным (1975), была плоская волчанка, у 2—опу-
холевидная, язвенная—у одного. Она характеризуется высыпанием не-
больших бугорков, незначительно возвышающихся над уровнем кожи,
сливающихся за счет периферического роста в сплошные очаги различных
очертаний и величины, иногда значительной (1. v. diffusus). Цвет их желто-
вато-красный или красновато-коричневатый, меняющийся при диаскопии
на желтовато-буроватый (симптом «яблочного желе»). Бугорки мягкие,
легко разрываются при надавливании зондом (симптом «зонда»), при этом
возникает выраженная болезненность, кровотечение (рис. 328, 329, 330).
Вокруг основного очага. более крупного, могут возникать множественные
мелкие (1. v. corymbiformis). Поверхность очага поражения обычно глад-
кая, шелушение отсутствует или небольшое, лишь в редких случаях оно
значительное, отрубевидное или мелкопластинчатое (1. v. squamosus, •
pityriasiformis; s. psoriasiformis; s. exfoliativus). Иногда появляются боро-
давчатые, папилломатозные разрастания (1. v. verrucosus; s. papillomato-
sus). При резко выраженном ороговении может возникать картина, сход-
ная с кожным рогом (1. v. cornutus). Во всех случаях необходимо тщатель-
ное наблюдение, так как это может быть признаком злокачественного пере-
рождения. Довольно часто наблюдается изъязвление (lupus vulgaris exul-
cerans), особенно при травматизации, с возникновением поверхностных
язвочек с мягкими краями, зернистым, легко кровоточащим дном, окру4
женных ободком инфильтрата, в зоне которого отчетливо выявляются
бугорки. Иногда выражены явления экссудации (1. v. exudativus). Чаще
это бывает при вторичном инфицировании (что может наблюдаться и при
отсутствии видимых дефектов кожи). В этих случаях воспалительные яв-
ления становятся более выраженными, очаг покрывается корками, при
инфицировании—гнойными (1. v. crustosus; s. impetiginosus). Может раз-
виться рожистое воспаление, иногда со вторичным элефантиазом при ре-
цидивах (1. V. elephantiaticus), что чаще наблюдается на ногах и в области
верхней губы. По данным А. А. Штейна (1935), наиболее часто рожистое
493
воспаление наблюдается при плоской и изъязвленной туберкулезной вол-
чанке. Причем, как правило, локализация рожистого воспаления и лю-
позного процесса совпадают. Наиболее часто язвенные формы встречаются
на слизистых или на участках кожи, граничащих с ними. Обычно язвенные
поражения не склонны к распространению, но, если это наблюдается, то
может происходить как по периферии с регрессированием в одних участках
и появлением новых элементов в других (1. v. serpiginosus), так, что осо-
бенно неблагоприятно, и вглубь, что может привести к разрушению под-
кожной клетчатки, хрящевой части носа, ушей, значительному обезображи-
ванию, отторжению фаланг пальцев (1. v. excedens; 1. v. vorax; 1. v. muti-
lans). Особой формой язвенной волчанки является «ранняя» инфильтра-
тивная волчанка (Подвысоцкая О. Н., Каплан Я. Б., 1935), которая
протекает с развитием островоспалительной инфильтрациии в области кон-
чика носа, а также хрящевой его части, увеличением носа в размерах,
быстрым распадом, формированием обезображивающих рубцов. Реже по-
ражение встречается на губах, щеках, на мягком небе и других участках.
Нехарактерны для обычной клинической картины довольно яркая (иногда
рожеподобная) эритема с размытыми границами. Помогает диагностике из-
менение окраски на бурую при диаскопии, наличие поражения на слизис-
той носа, нередко с перфорацией носовой перегородки, увеличение перифе-
рических лимфоузлов, отсутствие или незначительная выраженность боли,
даже при больших разрушениях ткани, положительная реакция на ту-
беркулин, гистология, наличие признаков перенесенной туберкулезной
инфекции других органов, чаще легких. В редких случаях особенностью
туберкулезной волчанки является вертикальный рост с развитием очагов,
значительно возвышающихся над кожей (lupus vulgaris tuberosus; 1. v.
tumidus). Эта форма имеет вид опухолевидных образований желто-бурого
цвета, чаще расположенных на кончике носа, ушной раковине, нередко
сочетается с элементами плоской волчанки на других участках кожи (Агап-
кин И. Н. и Багаева М. И., 1959). К редким вариантам относится дис-
семинированная туберкулезная волчанка (1. v. disseminatus), когда высы-
пания обнаруживаются одновременно на разных участках кожного покрова,
и эритематозная (1. v. erythematoides), сходная с красной волчанкой. В
большинстве случаев туберкулезная волчанка возникает в детском или
юношеском возрасте, чаще у лиц женского пола. В последнее десятилетие
отмечается значительное уменьшение числа больных туберкулезом кожи,
в том числе детей (Рахманов В. А. и соавт., 1966; Литовченко О. В. и
Шаповал М. И., 1976). Особенностью современного течения волчанки
является и скудность клинических проявлений, что приводит к позднему
ее выявлению (по данным М. И. Шаповал, 1980, у 44,8% больных через
5 и более лет), а также развитие заболевания у взрослых. Располагается
чаще всего на лице, особенно на коже носа, затем на щеках. Часто пора-
жается слизистая носа (нередко с разрушением хрящевой части носовой
перегородки с деформацией кончика носа) и полости рта. По данным И. Г.
Лукомского (1945), Б. М. Пашкова и соавт. (1970), очаги туберкулезной
волчанки в полости рта локализуются преимущественно на деснах, твердом
494
и мягком небе, затем на губах, щеках, языке и язычке. Может в процесс
вовлекаться конъюнктива с развитием эктропиона. По данным И. Б. Вей-
нерова (1963), при туберкулезной волчанке кожи часто наблюдается по-
ражение ушей с гнойными и катаральными отитами и рубцеванием бара-
банной перепонки. Следует отметить, что у 7 из 16 больных туберкулезной
волчанкой наружного уха, наблюдавшихся А. М. Ольховским (1975), пер-
вичные высыпания возникли на ушной раковине. В последние годы не так
уж редко встречаются обширные очаги поражения на закрытых участках
тела, особенно на ягодицах.
Течение волчаночного туберкулеза кожи торпидное, многолетнее. На
месте регрессировавших очагов остается рубцовая атрофия, по внешнему
виду напоминающая папиросную бумагу, или рубцы, иногда келоидоподоТ)-
ные (lupus vulgaris keloidiformis; s. sclerosus; s. fibromatosus). В зоне руб-
цовых изменений, а также вокруг них характерно наличие люпом. Общее
состояние больных, как правило, не страдает, чем, видимо, и объясняется
то обстоятельство, что больные иногда десятилетиями не обращают внима-
ния на изменения кожи, особенно на закрытых участках тела,
Прогноз при лечении благоприятный. У части больных (4,1% по дан-
ным В. А. Рахманова и соавт., 1966), туберкулезная волчанка сочетается
с другими формами туберкулеза кожи. Наиболее тяжелым осложнением
туберкулезной волчанки, развивающимся в большинстве случаев в ре-
зультате нерационального лечения (особенно лучевого) или раздражающих
воздействий, в том числе метеорологических, является lupus-carcinoma,
которая протекает более тяжело, чем обычная спиналиома (Агапкин И. Н.,
Багаева М. И., 1959). Обычно возникает спиналиома, реже базалиома и
редко другие формы опухолей. В. А. Рахманов и соавт. (1966) наблюдали
развитие люпус-карциномы у 1,9% больных туберкулезной волчанкой.
Ни один из них не получал лучевой терапии, но все больные длительно
работали на открытом воздухе, многократно лечились раздражающими
средствами и УФО. Частота люпус-карциномы, по данным О. В. Санта-
ловой, составила 1,5% от числа больных, лечившихся в Ленинградском
люпозории в 1947—1958 гг. Чаще рак развивался на фоне язвенной вол-
чанки или изъязвившейся плоской. Однако имеются Сведения и о более
высокой частоте злокачественного перерождения волчаночного туберкуле-
за кожи. Так, М. А. Зеликман (1978) наблюдал развитие люпус-карци-
номы у 12,5% больных туберкулезной волчанкой, несколько чаще у муж-
чин, преимущественно в возрасте 50— 70 лет с давностью процесса свыше
10 лет. Обращается автором внимание на роль позднего начала лечения,
рецидивов и гиперкератотических изменений в зоне рубцов. Описано раз-
витие люпус-карцином и на фоне других форм туберкулеза кожи, в част-
ности скрофулодермы (Кардашенко Б. Я- и соавт., 1981). Располагается
люпус-карцинома чаще на лице, но может быть и на других участках
кожного покрова (Лалаева А. М. и соавт., 1981).
Гистопатология. Бугорки из эпителиоидных клеток, окруженных
лимфоцитами, гигантских клеток Лангханса, как правило, казеозный не-
кроз в центре бугорка различной степени выраженности. Могут выявлять-
495
ся туберкулезные палочки. Для диагностики, кроме того, используется
заражение морских свинок, получение культуры возбудителя, реакция
Пирке и Манту (положительные у большинства больных), а также тера-
пия ex juvantibus противотуберкулезными препаратами. М. И. Шаповал
(1980) предложила гистаминопексический туберкулиновый тест, заклю-
чающийся в том, что после внутрикожного введения туберкулина наб-
людается снижение гистаминопексии сыворотки у больных туберкулезом
кожи более чем на 5%.
Дифференциальный диагноз. С третичным (бугорковым) сифилисом,
саркоидозом, лимфоцитомой, красной волчанкой, туберкулоидной лепрой,
туберкулоидной формой лейшманиоза (металейшманиоз), с люпоидным
сикозом, спиналиомой, с хронической язвенной пиодермией, актиномико-
зом, споротрихозом, хромомикозом.
Лечение. Стрептомицин, производные гидразида изоникотиновой кис-
лоты, ПАСК.
ЛИТЕРАТУРА
Авербах М. М. Иммунология и иммунопатология туберкулеза. М. Медицина. 1976.
Зеликман М. А. К вопросу об озлокачествлении туберкулезной волчанки. Вести,
дерматол. и венерол., 1981, 7, 53—55.
Кардашенко Б. Я., Шахтмейстер И. Я-, Анименко М. М. Люпус-карцинома.
Вести, дерматол. и венерол., 1981, 2, 53—54.
Литовченко О. В., Шаповал М. И. Особенности туберкулеза кожи у детей в настоя-
щее время. В ки. «Туберкулез у детей», М., 1976, 133—141.
Агапкин И. Н. Лечение больных туберкулезом кожи препаратами ГИНК в амбу-
латорных условиях. Веста, дерматол. и венерол. J975, 7, 71—75.
Лалаева А. М., Левинтова Г. И., Деменкова Н. В. Люпус-карцинома. Вести, дер-
матол. и венерол. 1981, 7, 53—55.
Ольховский А. М. Туберкулезная волчанка наружного уха. Веста, дерматол. и ве-
нерол. 1975, 12, 60—61.
Шаповал М. И. Ранняя диагностика туберкулеза кожи. Канд. дисс. М. 1980.
Шевляков Л. В., Владимиров М. М. Эритематозная разновидность туберкулезной
волчанки. Веста, дерматол; и венерол., 1977, 10, 81—82.
УЗЕЛКИ ДОИЛЬЩИЦ (tuberkulum mulgentium).
Термин принадлежит Winternitz (1899), который впервые установил
заражение доярок от больных коров. Заболевание описывалось немецкими
авторами под названием «узелки доильщиц», а французскими—«красная
вакциния». В СССР впервые узелки доильщиц описаны в 1929 г. Ф. А.
Коганом и Л. А. Нерядовым. Это своеобразное профессиональное заболе-
вание работников, ухаживающих за коровами,—доилыодц, ветеринарного
персонала и др. Большинство авторов (Долгов А. П. и Морозов М. А.,
1933; Залкан П. М. и Пазникова М. Ф., 1932; Штарк А. М., 1940; По-
тоцкий И. И., 1965; Lipeschutz, 1915; Zumbusch, 1926; Braun, 1975 и др.)
придерживаются мнения, что узелки доильщиц вызываются вирусом пара-
вакцины—strongyloplasma paravaccina. В цитоплазме и в ядрах пора-
496
женных вирусом эпителиальных клеток человека находили характерные
округлые включения («элементарные тельца»), весьма сходные с включе-
ниями при оспе коров, овец и других хламидозоонозах. Вирус обнаружи-
вали в электронном микроскопе (Davis и Musil, 1968), размеры вируса от
140 до 300 нм (Evins et al., 1971). Паравакциния (paravaccina), своеобраз-
ная оспоподобная болезнь коров, характеризующаяся специфической
сыпью на вымени и сосках и способная передаваться людям (зооноза).
Клиника. Все больные доильщицы—молодые женщины в возрасте от
17 до 40 лет. Заболевание начинается с появления небольшого розового
пятна, на месте которого затем развиваются узелки. Обычно появляется
один узелок и'затем спустя 3—5 дней возникают еще несколько узелков
или же несколько узелков появляются одновременно. Узелки обычно округ-
лых или овальных очертаний величиной от чечевицы до средней горошины,
значительно возвышаются над поверхностью кожи, имеют резко очерчен-
ные границы и не сопровождаются реактивными воспалительными явле-
ниями со стороны окружающей кожи. Цвет узелков варьирует от ярко-
красного до сине-красного, в зависимости от времени существования узел-
ков. Узелки плотны на ощупь и слегка болезненны при дотрагивании.
Начиная с 5—7 дня центральная часть опускается, образуя пупковидное
вдавление (очаг поражения по внешнему виду несколько напоминает высы-
пания при оспе). Просуществовав в среднем 6—12 недель, узелки расса-
сываются, оставляя после себя пигментацию. Количество высыпных эле-
ментов от 1 до 5. Общих явлений обычно не наблюдается, но иногда раз-
вивается лимфангоит и регионарный лимфаденит (Ormsby и Montgomery,
1955). Тыл пальцев и кистей является самой частой локализацией кожного
поражения. Реже описывается появление узелковых высыпаний на дру-
гих частях кожного покрова: на предплечьях, носу, щеках, в углах рта,
в межпальцевых складках рук.
Гистопатология. В эпидермисе—акантоз, гиперкератоз и паракера-
тоз. В дерме—густой неспецифический хронический воспалительный ин-
фильтрат, состоящий из полинуклеаров, эозинофильных, эпителиоидных и
лимфоцитарных клеток. Много расширенных капилляров, окруженных
густым клеточным инфильтратом.
Дифференциальный диагноз. Следует проводить с бородавками, с истин-
ной коровьей оспой, папуло-некротическим туберкулезом, пиогенной гра-
нулемой.
Лечение. Спиртовые анилиновые краски, йодная настойка, жидкость
Кастеллани, 5—10% линимент синтомицина.
ЛИТЕРАТУРТА
Braun W. Dermatologie, Ein Lehrbuch fur Studenten.— Berlin, 1975.
УЛЬЭРИТЕМА НАДБРОВНАЯ (ulerythema ophryogenes).
Синонимы: надбровная рубцующаяся эритема, волосяной кератоз ли-
ца, красный атрофический волосяной кератоз лица.
Впервые описана Taenzer (1889) и детально изучена Unna. Фоллику-
32 Клиническая дерматология 497
лярный кератоз преимущественно бровей, сопровождающийся эритемой
и заканчивающийся стойким выпадением волос, появлением атрофических
рубчиков, придающих пораженной коже вид нежной сетки. Многие совре-
менные дерматологи относят надбровную эритему в группу кератозов,
куда входят также разъедающая червеобразная атрофодермия (atropho-
dermia vermicularis), девальвирующий фолликулярный кератоз (kerato-
sis pilaris decalvans).
Этиология и патогенез. Некоторые исследователи придают значение в
возникновении болезни эндокринно-вегетативным расстройствам (Розен-
тул М. А., 1937), а также хронической инфекции (туберкулез и др.). Се-
мейный характер ряда случаев дает основание предполагать наследствен-
ную природу заболевания.
Клиника. Начало преимущественно в детском и юношеском возрасте.
Считается, что оба пола поражаются одинаково часто; по нашим наблюде-
ниям, дерматоз чаще встречается у девушек. Начинаясь на наружной части
бровей, процесс может распространяться на лоб, виски, щеки, предушные
области. Отмечается покраснение симметричных участков кожи, на кото-
рых располагаются не сливающиеся мелкие фолликулярные узелки, соз-
дающие впечатление зернистости: при поглаживании кожа шероховатая.
Ороговение в устьях фолликулов. Нередко заметно мелкое отрубевидное
шелушение. Волосы на пораженных местах часто выпадают, волосяной
мешок атрофируется и в конце процесс приводит к рубцевидной фоллику-
лярной атрофии. Брови редеют. Иногда отчетливо заметен нежный сете-
видный характер рубцовых изменений. Субъективно иногда жжение и зуд.
Нередко заболевание сочетается с красным волосяным лишаем (keratosis
follicularis rubra), поражения при котором располагаются на шее, раз-
гибательных поверхностях плеч и туловище.
С годамц процесс самопроизвольно перестает прогрессировать и на
коже остаются едва заметные участки сетчатой атрофии.
Гистопатология. Резко выраженный глубокий фолликулярный ги-
перкератоз, местами небольшой акантоз, резкое истончение эпидермиса
на отдельных участках; в собственно коже в окружности фолликулов, саль-
ных и потовых желез—инфильтрат из лимфоцитов и плазматических клеток.
Сосуды кожи расширены, имеется незначительный отек.
Дифференциальный диагноз. Следует проводить с красной волчанкой,
фолликулярным дискератозом Дарье, сикозиформной и акнеиформной
ульэритемой.
Лечение. Повторные курсы витамина А внутрь или парентерально.
Мази с 2—3% салициловой кислотой, витамином А.
ЛИТЕРАТУРА
Csermely Е., de Panfilis G. L’uleritema ofriogeno.— G. Min. Dermatol., 1974, 109, 5,
292—293.
Eiling F., Rook A. Disorders of keratinization — In: Textbook of dermatology. Ox-
ford-London, 1979, 3rd., p. 1253—1314.
498
ФАСЦИИТ ЭОЗИНОФИЛЬНЫЙ (fasciitis eosinophilicum).
Синонимы; синдром Шульмана, диффузный фасциит.
Американский ревматолог L. Shulman в 1974 г. впервые описал двух
мужчин, у которых после резкой физической нагрузки появился отек, а
затем стойкое уплотнение кожи различных участков конечностей, сопровож-
давшиеся эозинофилией периферической крови и гипергаммаглобулинёмией.
При глубокой биопсии обнаружено утолщение подкожной фасции, лимфо-
цитарная и плазматическая инфильтрация ее. Он предложил наименование
болезни «диффузный фасциит». В связи с наличием эозинофилии в инфильт-
ратах Rodnan и соавт. (1975) предложили термин «эозинофильный фасциит».
Caperton и Hathaway (1975) предложили название—синдром Шульмана.
К настоящему времени в мировой литературе имеются описания 30 боль-
ных эозинофильным фасциитом, сделанные в основном ревматологами (На-
сонова В. А. и соавт., 1978; Caperton и Hathaway, 1975; Gray и Рорро,
1977; Krayser и Fathill, 1977; Fleisihmojer et al., 1978; Lupton и Goette,
1979; Solomon et al., 1982 и др.). В отечественной дерматологической ли-
тературе первое описание синдрома Шульмана принадлежит А. А. Калам-
каряну и соавт. (1981).
Этиология и патогенез. Shulman (1974, 1975) считает, что синдром
является самостоятельным заболеванием, поскольку клинические и ла-
бораторные данные отличаются от таковых при склеродермии. Некоторые
авторы (Caperton и Hathaway, 1975; Bennet и Feinstein, 1977) склонны рас-
сматривать его как разновидность системной склеродермии. Atherton (1976)
высказал мнение, что возможно склередема Бушке и эозинофильный фас-
циит являются вариантами одного заболевания. Мы полагаем, что эозино-
фильный фасциит и системная склеродермия могут быть близки по своей
этиологии, но клинически, гистологически и по течению—это, по-видимо-
му, два различных заболевания. По мнению Boisen и соавт. (1983), наличие
антител против ДНК и гистологические изменения сближают данное за-
болевание с дерматомиозитом.
Клиника. Признаками эозинофильного фасциита являются: 1) частое
совпадение начала заболевания со значительной физической перегрузкой.
2) болезненность и уплотнение кожи, обычно дистальных участков конеч-
ностей; 3) сгибательные контрактуры; 4) отсутствие синдрома Рейно и
висцеральной патологии; 5) эозинофилия крови, увеличение уровня сы-
вороточных иммуноглобулинов; 6) утолщение фасции (подкожной и меж-
мышечных) вследствие развития фиброза и периваскулярных инфильт-
ратов, состоящих из лимфоидных, плазматических клеток и гистиоцитов с
наличием эозинофилов; 7) отсутствие выраженных изменений кожи и мышц;
8) благоприятное действие кортикостероидной терапии.
Болеют оба пола, несколько чаще мужчины. В большинстве случаев
это люди среднего возраста (39—45лет), но иногда дерматоз наблюдается
у пожилых. У многих больных после резкого физического перенапряжения
появляются боли, отек и уплотнение кожи кистей, стоп, бедер и предпле-
чий. В течение последующих двух-трех месяцев уплотнение кожи, подкож-
ной клетчатки и мышц распространяется на лицо, грудь, живот, развива-
32* 499
4
ются явления сгибательной контрактуры пальцев кистей, ограничение дви-
жений в лучезапястных, локтевых, плечевых суставах. Больные не могут
поднять руки, не могут раздеться без посторонней помощи. Кисти и стопы
холодные на ощупь, но синдром Рейно отсутствует. Кожа в складку не со-
бирается. Несмотря на проводимое интенсивное лечение (пенициллин, де-
лагил и др.), заболевание продолжает прогрессировать. Кожные покровы
вне очагов поражения бледно-розового цвета с выраженным буроватым
оттенком. Висцеральные поражения, свойственные системной склеродер-
мии, не наблюдаются. У 3/4 больных отмечается эозинофилия в перифери-
ческой крови (от 5—8 до 12—18% и более); у многих больных обнаружива-
ется гипергаммаглобулинемия, повышение СОЭ.
Гистопатология. Barnes и соавт. (1979), анализировавшие гистологи-
ческую картину 20 случаев эозинофильного фасциита, опубликованных в
литературе, указывают, что наиболее выраженные изменения обнаружи-
ваются в подкожных и межмышечных фасциях при отсутствии или слабой
выраженности изменений в скелетных мышцах и коже. Фасция представ-
ляется резко отечной, утолщенной в 2—15 раз по сравнению с нормой, скле-
розированной, с очаговыми и диффузными инфильтратами, состоящими в
основном из лимфоидных, гистиоцитарных и плазматических клеток с при-
месью лейкоцитов (эозинофилов). Ни в одном из описанных случаев выра-
женного утолщения дермы, характерного для системной склеродермии, не
было отмечено. Тканевая эозинофилия может быть очаговой или диффуз-
ной и обычно наблюдается в фасции или подкожной клетчатке. Отложение
иммуноглобулинов (IgG) обнаружено в 5 из 8 биоптатов при прямой им-
мунофлюоресценции, что позволяет подагать, что иммунологические фак-
торы могут стимулировать начало заболевания.
Дифференциальный диагноз. Следует проводить с системной склеро-
дермией, дерматомиозитом и склередемой Бушке.
Лечение. Отмечается хороший терапевтический эффект от применения
кортикостероидных препаратов (преднизолон по 30—50 мг в сутки до по-
лучения клинической ремиссии). Рекомендуются витамины А, Е, Д; физио-
терапия (местная диатермия); теплые ванны, гимнастика.
ЛИТЕРАТУРА
Каламкарян А. А., Большакова Г. М., Цветкова Г. М., Сыч Л. И. Эозинофиль-
ный фасциит (синдром Шульмана). Вестн. дерматол. и венерол., 1981, 4, 4—7.
Насонова В. А., Иванова М. М., Ахназарова В. Д , Оскилко Т. Г. Диффузный
эозинофильный фасциит.— Тер. архив, 1978, 5, 7—13.
Barnes L., Roduan В., Medsger Т., Short D. Eosinophilic fasciitis.— Amer. J. Pat-
hol., 1979, 96, 2, 493—518.
Boisen M., Keiding L., Thomsen K- Eosinophilic fasciitis. — Dermatologica (Basel),
1983, 167, 3, 142—144.
Chonda J., Callen Y., Taylor W., Diffuse faciitis with eosinophilia.— Arch. Der-
matol., 1978, 114, 10, 1522—1524.
Gray R., Poppo M. Eosinophilic fasciitisi a sclerodermalike illness.— JAMA, 1977,
237, 529—530.
500
Krauser R. Eosinophilic fasciitis.— Arch. Dermatol., 1977, 113, 8, 1092—1093.
Lupton G. Goette D. Localized eosinophilic fasciitis.— Arch. Dermatol.. 1979, 115, 1,
85—87.
Schulman L. Diffuse fasciitis with hypergammaglobinemia and eosinophilia: a new
syndrome abstracted.— J. Rheumatol., 1974, 1, (suppl.), 46—48.
Weinstein D., Schwartz R. Eosinophilic fasciitis.— Arch. Dermatol., 1978, 114, 7,
1047—1049.
ФЛЕБОТОДЕРМИЯ (phlebotodermia). z
Синонимы: москитный дерматоз.
Имеются лишь единичные работы (Казаков В. М., 1934; Фурманов
С. И., 1941; Павлов С. Т. и соавт., 1943; Dostrowsky, 1925; Holman,
1926 и др.), посвященные этому вопросу.
Этиология и патогенез. Термин phlebotodermia определяет анатоми-
ческую локализацию патологического процесса и его этиологию. В патоге-
незе флеботодермии играют роль токсические или токсико-аллергические
свойства слюнного секрета москита. Возможность хронического течения
дерматоза, а также появления высыпаний на участках, не подвергшихся
укусам москитов, свидетельствует о развитии сенсибилизации к продуктам
слюнных желез насекомых, попадающим в кожу человека во время укуса.
Клиника. Кусают флеботомусы как ночью, так и днем, но особенно с
наступлением темноты. Преимущественно жертвами укусов москитов яв-
ляются люди с нежной, кожей. По характеру течения флеботодермии де-
лятся на острые и хронические. Появлению кожных элементов после
укусов москитов предшествует период от нескольких минут до нескольких
часов. Сыпь появляется чаще всего на открытых местах тела: на лице, шее,
тыле кистей, стоп, разгибательных поверхностях предплечий и голеней,
реже на туловище. На месте укуса москита развивается уртикарно-эрите-
матозное пятно, которое быстро увеличивается, достигая размеров 10—15-
копеечной монеты. Спустя некоторое время (от нескольких часов до суток
и больше) пятно начинает бледнеть и исчезает, иногда оставляя в центре
точечную геморрагию. У некоторых больных заболевание протекает по
типу отека Квинке или буллезных высыпаний. Буллезная форма флебото-
дермии наблюдается у людей с повышенной чувствительностью кожи на
укусы москитов, т. е. у людей, предрасположенных к аллергическим забо-
леваниям,—крапивнице, экземе и др. Вначале на месте укуса москитов
появляются на коже эритематозно-уртикарные элементы, вслед за которыми
формируются везикулы и крупные пузыри, достигающие размеров голу-
биного яйца и больше. Вокруг буллезных высыпаний почти всегда имеется
эритематозный венчик шириной в 3—5 см. Довольно часто наблюдаются
общие явления—головная боль, слабость, понижение аппетита и др. Пу-
зыри регрессируют в течение нескольких дней. Смешанная форма флебото-
дермии характеризуется полиморфизмом сыпи с наличием волдырей, пру-
ригинозных, папуло-везикулезных и буллезных элементов, кровянистых
корочек. В подавляющем большинстве случаев высыпания исчезают после
прекращения укусов москитов. Однако у небольшой части лиц высыпания
501
продолжаются месяцами и годами, принимая характер хронической флебо-
тодермии с клиническими проявлениями узловатой почесухи (prurigo no-
dularis). Заболевание проявляется высыпанием полушаровидных узлов
от 5 до 15 мм в диаметре с преимущественной локализацией на разгибатель-
ных поверхностях конечностей, спине, пояснице и ягодицах. Цвет узелков
и узлов коричневато-серый, поверхность покрыта плотно сидящими чешуй-
ками и корочками. Высыпные элементы располагаются всегда изолирован-
но и наблюдаются в сравнительно небольшом количестве. Больные обыч-
но расчесывают высыпания до крови и тогда зуд прекращается, поэтому
на многих элементах сыпи наблюдаются кровянистые корочки, а иногда и
пустулизация, вследствие присоединения пиококковой инфекции. Нередко
развивается лихенификация со множеством экскориаций и линейных рас-
чесов. Кожа становится утолщенной, грубой, пигментированной. У боль-
ных из-за сильного зуда возникают резко выраженные невротические рас-
стройства, бессонница, потеря аппетита и др. Сыпь локализуется не толь-
ко на открытых частях тела, но и на закрытых местах -
Прогноз обычно благоприятный, хотя иногда заболевание может про-
должаться в течение нескольких лет и упорно противостоит различным
методам лечения. Временами наблюдаются неполные и непродолжитель-
ные ремиссии.
Гистопатология. При острой флеботодермии в дерме обнаруживают
явления острого экссудативно-инфильтративного воспаления (разрыхление
и отслойка эпителия с образованием внутриэпителиальных пузырей; в
поверхностной части дермы—гнездные скопления негустого инфильтрата,
состоящего из лимфоцитов и единичных плазмоцитов. При хронической
флеботодермии—в эпидермисе резко выраженный акантоз и явления не-
кробиоза, в дерме—диффузный инфильтрат вокруг некротической зоны,
состоящий в основном из нейтрофилов, плазмоцитов и гистиоцитов.
Дифференциальный диагноз. Следует проводить с высыпаниями, выз-
ванными укусами клешей, клопов, блох, вшей и пр., с крапивницей и пру-
риго различной этиологии, герпетиформным дерматитом Дюринга и др.
зудящими дерматозами.
Лечение. Антигистаминные средства, аминазин (наши наблюдения),
гелиотерапия, морские купания, сероводородные и радоновые ванны, ап-
пликации озокерита, мази, содержащие серу (5—10%), деготь (3—5%),
нафталан, кортикостероиды, лазеротерапия.
ЛИТЕРАТУРА
Казаков В. М. Опыт изучения реакции кожи и крови под влиянием укусов флебото*
мусов.— Сов. вести, веиерол. и дерматол., 1934, 5, 447—450.
Фурманов С. И. Клиника москитных дерматозов.— Вести, веиерол- и дерматол.,
1941, 9—10, 19—25.
ФОКСА—ФОРДАЙСА БОЛЕЗНЬ (morbus Fox—Fordyce).
Этиология, и патогенез. Причина заболевания неизвестна. Точка зре-
ния о том, что это заболевание из группы нейродермита, в настоящее время
не разделяется. Основное значение в развитии придается дисфункции апо-
502
криновых желез (Shelly et al., 1956; Heite и Zaun, 1961; Hurtby, 1979).
По данным Shelly и соавт. (1956), Montes и соавт. (1959), основные
нарушения, ведущие к развитию в последующем клинических проявле-
ний, закрытие верхней части протока апокриновых желез кератотически-
ми массами, разрыв интраэпидермальной части протока, образование микро-
везикул с последующей воспалительной реакцией, дегенеративными изме-
нениями, отложением муцина. Преимущественное развитие болёзни Fox-
Fordyce у женщин, частое обнаружение дисфункции яичников (дис-и аме-
норея и др.), улучшение во время беременности, особенно в 3-м триместре,
когда содержание эстрогенов наиболее высокое, усиление зуда в пременст-
руальном периоде послужило основанием для гипотезы о причинной связи
заболевания с овариальной недостаточностью. Montes и соавт. (1959, 1961)
наблюдали улучшение от эстрогенных препаратов, отметив корреляцию
между улучшением течения заболевания и подавлением фолликулостиму-
лирующего гормона. Угнетающим действием на фолликулостимулирующий
гормон Kronthal и соавт. (1965) считают возможным объяснить положитель-
ный эффект контрацептивов при этом заболевании. Turner (1976), однако,
не обнаружил отклонения от нормы у больных андрогенов и эстрогенов.
Leyh (1973) придает большое значение в развитии этого процесса нару-
шению нейрогуморальной регуляции менструального цикла, проявляю-
щемуся в относительном избытке эстрогенов и недостаточности желтого
тела, замечая, однако, что редкость болезни Фокса—Фордайса при часто
встречающейся дисфункции половых желез и нарушении менструального
цикла свидетельствует о наличиии индивидуальной предрасположенности
к этому дерматозу. Heite и Zaun (1961) обращают внимание на такие фак-
торы, как предшествующие развитию заболевания абсцессы потовых желез,
повышенную нервную возбудимость. На основании анализа данных лите-
ратуры авторы выделили следующие 3 группы наиболее частых заболеваний,
предшествующих или сопровождающих болезнь Фокса—Фордайса: гидра-
денит, фурункулез и другие виды пиодермии подмышечных впадин, лобка
и других мест излюбленной локализации болезни Фокса—Фордайса; на-
рушение функции половых желез, нарушение щитовидной железы. Graham
и соавт. (1960) полагают, что наличие болезни у однояйцевых близнецов
могло бы быть свидетельством невоидной природы заболевания или нали-
чия предрасположенности к его развитию, вероятно, заключающейся в
неизвестном эндокринном расстройстве. В то же время не исключается
возможность развития заболевания вследствие метаболических наруше-
ний, в первую очередь накопления кислых и нейтральных мукополисахари-
дов. Winkelmann и Montgomery (1956) придавали этиологическое значение
муцинозной дегенерации, обнаружив муцинозный материал в просветах
апокриновых желез и свободно в дерме.
Клиника. Поражаются преимущественно места локализации апо-
криновых потовых желез, в первую очередь подмышечные впадины, область
лобка, промежность, в редких случаях высыпания могут быть и на других
участках тела. Сыпь мелкопапулезная, величина узелков уменьшается к
503
периферии очага, чаще форма их
полушаровидная, редко кониче-
ская, очертания круглые, отчетлива
склонность к фолликулярному или
парафолликулярному расположе-
нию. Папулы располагаются гус-
то, как правило, не сливаются
(рис. 331). Поверхность часто ги-
перкератотична, цвет их может не
отличаться от цвета нормальной
кожи, но чаще красноватый с си-
нюшным оттенком. Кожа вокруг
узелков без воспалительных изме-
нений, суха, волосы разрежены,
часто обломаны. Сопровождается
сильным зудом, интенсивность ко-
торого становится большей перед
Рис. 331. Фокса—Фордайса болезнь. менструациями, после нервных
перенапряжений. В редких слу-
чаях зуд может отсутствовать. Heite и Zaun (1961) обнаружили 13 таких
случаев из 304 опубликованных и проанализированных ими.
Заболевание развивается во много раз чаще у женщин, чем у мужчин.
Winkelmann и соавт. (1956) полагают, что соотношение мужчин и женщин
составляет 1:10. Описан (Graham et al., 1960) редкий случай болезни Фо-
кса-Фордайса у однояйцевых близнецов мужского пола. Обычно возни-
кает вскоре после полового созревания у детей (Heite и Zaun, 1961), в
период менопаузы (Montes et al., 1959). Течение хроническое, может
длиться десятилетиями, вплоть до менопаузы, но возможно развитие бо-
лезни и в климактерическом периоде. Улучшение наблюдается во время
беременности.
Гистопатология. Гиперкератоз, акантоз, расширение и дегенератив-
ные изменения потовых желез, разрыв протоков их, лимфоцито-гистиэци-
тарные инфильтраты в дерме вокруг волосяных фолликулов, потовых же-
лез, сосудов, скопление муцина.
Дифференциальный диагноз. Следует проводить с ограниченным ней-
родермитом, красным плоским лишаем, хронической экземой, псевдоксаи-
томой эластической, сир ингомой.
Лечение. УФО, внутриочаговое введение кортикостероидов, кортико-
стероидные мази под пленку, контрацептивы, половые гормоны (после
обследования).
ЛИТЕРАТУРА
Hurtby Н. J. Fox—Fordyce disease. In: Dermatology in general medicine. Sec fcd.Ed.
by T. B. Fitzpatrick et. al., 1979, Me Graw—Hill Blook company, 477—480.
Leyh F. Morbus Fox—Fordyce. Uberlegungen zur Pathogenese. Hautarzt, 1973, 24, 11,
482—485.
504
ХЕНДА—ШЮЛЛЕРА— КРИСЧЕНА СИНДРОМ (Hand—Schuller-
Christian synnromum).
Синонимы: болезнь Хенда—Шюллера—Крисчена, липогранулематоз,
гистиоцитоз X и др.
В 1953 г. Lichtenstein предложил объединить синдром Хенда—Шюл-
лера— Крисчена, синдром Абта—Леттерера—Сиве и эозинофильную гра-
нулему костей (болезнь Таратынова)—в одну группу, для обозначения
которой предложил термин гистиоцитоз X. Он исходил из того, что гистио-
цит является характерным клеточным компонентом при всех трех формах.
Буква «X» указывает на неизвестную этиологию, заболевания.
Синдром Хенда—'Шюллера—Крисчена представляет собой хроническую
диссеминированную разновидность гистиоцитоза X, синдром Абта—Леттере-
ра—Сиве—острую диссеминированную, а эозинофильная гранулема—лока-
лизованную (Whitehead, 1972).
Этиология и патогенез. Заболевание рассматривается как хрониче-
ская форма ретикулогистиоцитоза со вторичным нарушением холестери-
нового обмена (Серов В. В., 1980; Кайлаков А. М., 1976 и др.). К- А.
Москачева и соавт. (1967) и др. полагают, что болезнь Хенда—Шюллера—
Крисчена развивается в результате изменений Неопластической природы
в системе мононуклеарных фагоцитов с последующим нарушением холе-
стеринового метаболизма.
Электронно-микроскопическое исследование, проведенное А. А. Ка-
ламкаряном и соавт., свидетельствует о том, что в основе болезни лежит
пролиферация клеток Лангерганса.
Kragballe и соавт. (1981) на основании иммунологических исследований
у 6 детей, страдающих гистиоцитозом X, пришли к выводу о существовании
функционального дефекта в мононуклеарно-фагоцитарной системе больных
гистиоцитозом X, о частичном сохранении клеточного иммунитета за счет
неизмененных нейтрофилов.
Заболеванию нередко предшествует травма, вирусные инфекции (грипп,
корь, ветряная оспа). Описаны случаи развития болезни у нескольких
членов семьи, что может свидетельствовать о роли наследственных факторов.
Клиника. Заболевание протекает хронически, характеризуется изме-
нением костной системы, особенно костей черепа («череп типа географиче-
ской карты»), несахарным мочеизнурением, экзофтальмом, разнообразными
поражениями кожи.
Болезнь поражает главным образом детей (в возрасте до 4—5 лет),
но ее можно встретить и у взрослых (Каламкарян А. А. и соавт., 1985;
Nethercott et al., 1983). Описано в литературе 10 случаев врожденного
гистиоцитоза X—у всех пациентов имелись генерализованные высыпания
на коже. Fitzpatrick и соавт. (1981) сообщили о проявлениях болезни Хен-
да—Шюллера—Крисчена на коже у 76% пациентов.
На лице, туловище, в подмышечных и перианальной областях появля-
ются многочисленные желтовато-коричневого цвета папулезно-пустулез-
иые, пустулезно-корочковые и эрозивно-язвенные элементы, вызванные
пропитыванием кожи ксантомными клетками. Иногда возникают болезнен-
505
ные экссудативно-инфильтративные очаги поражения красно-бурого цвета,
размерами до 3—5 см; в центральной части некоторых элементов образу-
ются некротические изъязвления, покрытые темно-бурыми корками.
Наблюдаются поражения слизистой оболочки полости рта, которые
характеризуются болью, опуханием десен, изъязвлением, а также расша-
тыванием зубов и их выпадением. Нередко отмечается диффузное пореде-
ние волос на голове. Гепато-спленомегалия, лимфаденопатия развиваются
у 20—25% пациентов, экзофтальм—у 35—40% больных.
Деструкция костей вследствие развития гранулем встречается у
всех больных в виде множественных мелких и крупных дефектов причудли-
вых очертаний. Несахарное мочеизнурение встречается у 50—60% боль-
ных. Оно обусловлено поражением гипоталамуса и гипофиза гранулема-
тозной тканью. (
В крови часто отмечаются анемия, лейкоцитоз, эозинофилия, повы-
шенная СОЭ (30—50 мм в час). В сыворотке крови—повышение уровня хо-
лестерина до 300 мг% и выше и С-липопротеидов (800—900 мг%).
Прогноз большей частью неблагоприятный. Летальный исход насту-
пает у 30—70% больных (Fitzpatrick et al., 1981). Прогноз болезни
улучшился после введения в терапию цитостатиков и кортикостероидов.
Гистопатология. В эпидермисе, участки акантоза и атрофии, экзоцитоз,
иногда разрушение базального слоя клетками инфильтрата. В дерме—гу-
стые инфильтраты, состоящие из скопления гистиоцитов, лимфоцитов,
эозинофилов. Просветы кровеносных и лимфатических сосудов расширены,
эндотелий набухший. При окраске Суданом—III зйачительная часть ги-
стиоцитов дает положительную реакцию на жир. При электронно-микро-
скопическом исследовании (Каламкарян А. А. и соавт., 1985; Carrington
et al., 1972; Andrade et al., 1978; Tarnowski и Hashimoto, 1967 и др.)
кожи из очага поражения обнаруживаются инфильтраты, состоящие пре-
имущественно из гистиоцитов, в цитоплазме которых имеются хорошо раз-
витые органоиды: пластинчатый комплекс, цистерны эндоплазматического
ретикулума, митохондрии, лизосомы, рибосомы и полисомы. В некоторых
из этих клеток определяются гранулы Лангерганса, что позволяет иден-
тифицировать их как клетки Лангерганса.
Дифференциальный диагноз. Следует проводить с ксантоматозом, врож-
денным сифилисом, лимфогранулематозом, болезнью Абта—Леттерера—Сиве.
Лечение. При локальных поражениях положительных результатов
удается достигнуть применением малых доз лучей Рентгена. Кортикосте-
роидные гормоны (преднизолон 30—40 мг ежедневно) способствуют ино-
гда обратному развитию кожных поражений. При распространенном кож-
ном процессе используют также химиотерапию. Методом выбора является
применение преднизолона в сочетании с проспидином (по 100 мг внутри-
мышечно ежедневно, курсовая доза 2—2,5 г). Диета с ограничением жи-
ров, богатая белками.
506
ЛИТЕРАТУРА
Каламкарян А. А., Персина И. С., Зотова И. Н., Парастаев С. А. Два случая
синдрома Хенда—Шюллера—Крисчена. Клинико-морфологическое исследование.— Вести,
дерматол. и венерол., 1985, № 6, с. 4—9.
Ляшенко И. Н., Соколов А. М. Болезнь Хенда—Шюллера—Крисчена в практике
дерматолога.— Вести, дерматол. и венерол., 1984, № 3, с. 48—50.
Favara В., McCarthy R. et al. Histiocytosis X.— Hum. Pathol., 1983, 14, 8, 663—
676.
Fitzpatric R., Rappoport M., Siloa D. Histiocytosis X.— Arch. Dermatol., 1981,
117, 3, 253—257.
Kragballe K., Zacharie H., Herlin T. Histiocytosis X — an immune deficiency disea-
se? Studies on antibody — dependent monocytemediated cytotoxicity.— Brit. J. Dermatol.,
1981, 105, 1, 13—18.
Neihercott J., Murray A., Chaloardjan A. Histiocytosis X in two aduldas.— Arch.
Dermatol., 1983, 119, 2, 157—16’
Stateva S., Kiriakow J., Mustakov G. Kongenitale Histocytosis X. Papulonodose
necrotische Form.— Hautarzt, 1980, 31, 1, 26—29.
Zachariae H. Histocytosis X in two infants-treated. with topical nitrogen mustard.—
Brit. J. Dermatol., 1979, 100, 4, 433—438.
ХОНДРОДЕРМАТИТ УЗЕЛКОВЫЙ УШНОЙ РАКОВИНЫ (chondro-
dermatitis nodularis chronica helicis).
Этиология и патогенез. Развивается обычно после травм, отмороже-
ний и других экзогенных воздействий, вызывающих нарушение кровооб-
ращения ушных раковин и приводящих к дегенеративным изменениям
перихондрия и/или соединительной ткани (Goette, 1980). Процесс в эпи-
дермисе рассматривается как вторичный, сходный с реакцией эпидермиса
иа выведение кальция при кальцинозе кожи или некробиотической ткани
при кольцевидной гранулеме. Определенное значение придается анатомиче-
ским особенностям ушных раковин, а также возрастным факторам: истон-
чение кожи, потеря эластичности ткани с возрастом, дегенеративные изме-
нения сосудов и соединительной ткани.
Клиника. Обычно по верхнему краю завитка ушной раковины, как
правило, единичный плотный узелок, полушаровидной или овальной фор-
мы, величиной 0,5—2 см. Цвет его не отличается от нормальной кожи или
желтовато-красноватый. На его поверхности часто обнаруживается плот-
но прилегающая чешуйка или корочка. Узелок спаяй с подлежащей хря-
щевой тканью, воспалительные изменения в его основании минимальны
или отсутствуют. Важным симптомом является болезненность, возникаю-
щая на холоде и особенно при надавливании, так что больные не могут
спать на стороне поражения. На других участках ушной раковины хондро-
дерматит встречается редко (Lester, 1977). Поражается чаще всего правое
ухо. По данным Elste (1965), у 50 из 80 больных процесс локализовался
на правом ухе, у 25—на левом, у 5—на обоих. Только у 3 человек узелок
располагался в зоне antihelix. По мнению Newcomer и соавт. (1953), эта
локализация более свойственна заболеванию у женщин. По наблюдениям
507
Yaffee (1963), у женщин хоидродерматит может с одинаковой частотой по-
ражать как правое, так и левое ухо, с преимущественной локализацией
узелка в области козелка и противозавитка.
Заболевание развивается преимущественно у мужчин старше 50 лет,
наиболее часто в 60—70-летнем возрасте. Wilkinson (1979) наблюдал боль-
ного 20 лет, отец и дядя которого также страдали этим заболеванием. Жен-
щины болеют значительно реже. Так, из 80 больных, наблюдавшихся Elste
(1965), женщин было только 10. Процесс без лечения существует длительно,
самопроизвольное регрессирование наблюдается редко (Arndt, 1979).
Гистопатология. Гиперкератоз с признаками псевдоэпителиоматоз-
ной гиперплазии, акантоз, нередко дефект эпидермиса в центре узелка, в
виде канала проникающего в дерму, заполненного некротическими массами
и клетками воспалительного инфильтрата. Вокруг него фибриноидный не-
кроз с нейтрофильной инфильтрацией, гранулемы из лимфоцитов, плазма-
тических клеток, гистиоцитов. Диагностическое значение имеет воспаление
перихондрия, часто с дегенерацией подлежащего хряща (Goette, 1980).
Дифференциальный диагноз. С базалиомой, сенильной кератомой, коль-
цевидной гранулемой.
Лечение. Хирургическое, внутриочаговое введение кортикостероидов.
ЛИТЕРАТУРА
Arndt К. A. Inflammatory disorders of cartilage. In: Dermatology in general Medi-
cine Ed. by T B. Fitzpatrick et al., Sec ed. New-York, 1977, 692—694.
Goette D. K- Chondrodermatias nodularis chronica chelicis-A trapepithelial elinina-
tion disorder.— Dermatologica, I960, 161, 2, 101—111.
Wilkinson D. S. Diseases of the external ear. In: Textbook of Dermatology Ed by A.
Rook et al., Oxford, 1979, 3-d ed., v. 2, 1909—1922.
Lester A. J. Chondrodermatitis nodularis chronica chelicis.— Arch, dermatol., 1977,
113, 525.
'ЦИЛИНДРОМА (cylindroma)
Синонимы: гиалинизированная трихобазалиома.
Этиология и патогенез. Цилиндрома представляет собой редко встре-
чающуюся доброкачественную опухоль. Происхождение ее неясно. В гисто-
логической классификации опухолей ВОЗ (1980) цилиндрома рассматри-
вается как эккринная опухоль, но указывается иа наличие мнений о раз-
витии из апокриновых желез, а также из волосяных структур. Lever и Has-
himoto (1966) относят цилиндрому к опухолям из незрелых структур апо-
криновых желез.
Точки зрения об эккринном происхождении цилиндромы придержи-
ваются Crain и Helwig (1961), Munger и соавт. (1962), Urbach и соавт. (1963).
Л. Н. Машкиллейсон и Н. С. Смелов (1931) считали, что гистогенетически
цилиндрома связана с волосяными фолликулами, и назвали ее гиалинизи-
рованной трихоэпителиомой. По мнению А. К. Апатенко (1972), наличие
в разных опухолях железистой и пилоидной структур свидетельствует о.
гетерогенности цилиндромы, которая может быть связана как с потовыми
508
(эккринными) железами, так и с волосяными фолликулами. Сходство кле-
ток цилиндромы с клетками волосяных фолликулов и потовых желез эмбрио-
на дало основание А. К- Апатенко думать о дисэмбригенетической природе
этой опухоли. Анализ семейных случаев свидетельствует о возможности
аутосомно-доминантного типа наследования (Korting et al., 1970). Соли-
тарные цилиндромы—спорадические (Crain, Helwig, 1961). Описано раз-
витие их после рентгеновской эпиляции (Black и Wilson, 1971).
Клиника. Узловатые опухоли, внешне сходные с помидорами, кашта
нами или гроздьями винограда, мягкой или слегка плотноватой консис-
тенции, обычно безболезненные. Количество опухолей различно, чаще
они более или менее многочисленны, солитарные встречаются редко (Ja-
nicka и Stepien, 1978). Поверхность их гладкая, часто усеяна множествен-
ными телеангиэктазиями. Кожа в их зоне лишена волос, синюшного, розо-
ватого или красного цвета, напряжена, может эрозироваться. Величина
может быть различной, но редко превышает 4—6 см. Опухоли локализу-
ются преимущественно на волосистой части головы и лице, редко на дру-
гих участках кожи, изолированно, но из-за тесного расположения имеют
вид конгломератов, значительно возвышающихся над кожей, иногда зани-
мающих значительные участки кожи, например, покрывающих волосистую
часть головы, наподобие тюрбана, иногда закрывая и часть лба (отсюда на-
звание—тюрбанная опухоль). Обычно развиваемся в молодом возрасте,
509
чаще у женщин (рис. 332, 333). Т. А. Главинская и Г. А. Петров (1983)
описали больную, у которой первые признаки цилиндромы развились в
37-летнем возрасте. Но может существовать и с детства. В этих случаях
оии обычно множественны, часто наследуемы. С возрастом размеры и
количество опухолей увеличиваются. Достигнув определенного размера,
длительно остаются в стационарном состоянии. Злокачественное перерож-
дение наблюдается редко; о нем может свидетельствовать изъязвление, с
метастазами (Korting et al., 1970). Возможность развития злокачественных
опухолей, по мнению А. К. Апатенко (1973), сомнительна. У больных мно-
жественной цилиндромой могут быть также трихоэпителиомы (Rasmussen,
1975).
Гистопатология. В дерме множественные полосы и гнезда из клеток,
сходных с клетками базального слоя, содержащих гиалин, и окруженных
гиалиновой оболочкой. В пределах опухолевых островков имеются клетки
2 видов: расположенные в центре имеют светлоокрашенное ядро овальной
формы, по периферии—клетки расположены в виде палисада, меньших
размеров, имеют темноокрашенные ядра. А. К- Апатенко (1973) разли-
чает 4 морфологических варианта: недифференцированный, гидраденома-
тозный, трихоэпителиоматозный и смешанный.
Дифференциальный диагноз. Необходимо дифференцировать с липо-
матозом, атероматозом, себоцистоматозом.
Лечение. Отдельные наиболее крупные опухоли могут быть удалены
хирургически.
ЛИТЕРАТУРА
Rasmussen J. Е. A syndrome of trichoepitheliomas milia and cylindromas.— Arch,
dermatol., 1975, 111, 5, 610—614.
ЧЕСОТКА НОРВЕЖСКАЯ (norvegica scabies).
Впервые описана норвежскими учеными Boeck и Dantelssen в 1847 го-
ду у больных проказой. Если в прежние годы заболевание встречалось ред-
ко, то в последние годы стали чаще публиковаться случаи «норвежской»
или «корковой» чесотки (Михеев Г. Н., 1984; Leroy et al., 1980; Ward,
1981; Haydon и Kaplan, 1981; Reimer, 1983; Itani, 1960; Leibowitz et al.,
1980; Pezzarossa et al., 1983).
Этиология и патогенез. Норвежская чесотка вызывается тем же чесо-
точным клещом, что и обычная чесотка. Несостоятельным оказалось предпо-
ложение, что норвежская чесотка вызывается особой формой человеческого
зудня или зуднями животных. Об идентичности возбудителя обычной и
норвежской чесотки свидетельствует возникновение типичной формы забо-
левания у медицинского персонала, членов семьи и других лиц, бывших в
контакте с больными норвежской чесоткой.
В патогенезе норвежской чесотки ведущими, по-видимому, являются
иммунные механизмы. Этот своеобразный и тяжелый вариант обычной
чесотки человека чаще встречается у лиц, ослабленных тяжелыми забо-
леваниями (лейкемия, туберкулез, красная волчанка, склеродермия, си-
510
Рис. 334. Норвежская
чесотка.
Рис. 335- Норвежская чесотка
(тот же больной).
Рис. 336. Норвежская чесотка
(тот же больной).
рингомиелия, лепра и пр.), умствен-
но отсталых людей (болезнь Дауна,
сенильная деменция и др.), у лиц с
дефицитом иммунитета, при длитель-
ном лечении цитостатиками и корти-
костероидными гормонами.
Количество клещей на больном
норвежской чесоткой велико; в кор-
ках, в многочисленнцх переплетаю-
щихся и пересекающихся многоярус-
ных ходах в огромном количестве
находят клещей на разных стадиях
развития, в связи с чем заболевание
отличается высокой контагиозностью
и возникновением локальных эпиде-
мий (Reimer, 1983; Jagavcar, 1980).
Itani и соавт. (1979) наблюдали боль-
ную с болезнью Дауна, у которой в
течение 25.лет было кожное забо-
левание неясного характера, расце-
нивавшееся как пиодермия или хро-
ническая экзема. При гистологиче-
ском й микроскопическом исследова-
511
ниях обнаружены чесоточные клещи. За время пребывания в клинике
больная заразила чесоткой 11 стационарных больных и 8 медицинских
сестер. У всех зараженных заболевание протекало с клинической карти-
ной обычной чесотки.
Haydon и Kaplan (1971) наблюдали вспышку чесотки в дерматологи-
ческой больнице в США у 54 больных. Инфекцию занес больной 57 лет,
страдавший в течение многих лет недиагностированной норвежской че-
соткой. Правильный диагноз был поставлен только спустя 19 лет. Его за-
болевание никогда полностью не регрессировало от «соответствующей» те-
рапии. После лечения 25% кремом с бензил-бензоатом у всех больных
наступило выздоровление. В отделе дерматологии ЦКВИ недавно наблю-
дался 37-летний больной норвежской чесоткой с массивными гиперкера-
тотическими наслоениями по всему кожному покрову. В корках были об-
наружены множественные ходы с клещами в различных стадиях развития,
ходы в корках были расположены в несколько этажей. После трехднев-
ного, лечения 20% водно-мыльной эмульсией бензил-бензоата больной
был выписан из стационара здоровым. Для размягчения корок перед каж-
дым втиранием препарата делалась ванна со слабым раствором перман-
ганата калия.
Клиника. Клинической особенностью норвежской чесотки является
массивное наслоение гиперкератических масс, корок на различных участ-
ках кожного покрова, что создает сходство с гиперкератотической фор-
мой псориаза. Корки желтовато-грязного, буро-черного цвета, плотные,
с гладкой или бороздчатой поверхностью толщиной от нескольких мил-
лиметров до 2—3 см располагаются в несколько слоев (5—7). Между этими
слоями и под всей коркой обнаруживается большое число чесоточных кле-
щей; на нижней поверхности корковых слоев видны извилистые углубления,
соответствующие чесоточным ходам. При снятии корковых наслоений об-
наруживаются обширные мокнущие эрозивные поверхности. Обращает
на себя внимание почти полное, а в некоторых случаях и полное отсутствие
зуда, что, по-видимому, следует поставить в связь с иммунодефицитным
состоянием организма (рис. 334, 335, 336).
В. И. Фельдман и М. И. Пер (1928) и др. описали случаи одновремен-
ного заболевания норвежской чесоткой и распространенной трихофитией.
Для указанной формы заболевания характерна типичная для чесотки ло-
кализация высыпаний (сгибательные поверхности конечностей, межпаль-
цевые складки кистей, внутренняя поверхность бедер, жнвот н др.). В
отличие от обычной чесотки процесс может поражать кожу лица, шеи, во-
лосистой части головы, нередко он имеет генерализованный характер. Бо-
лезнь часто осложняется вторичной пиодермией, лимфаденитом, сопро-
вождается эозинофилией, лейкоцитозом и ускоренной СОЭ. Норвежская
чесотка течет хронически и без антипаразитарного лечения может длиться
годами, при своевременно начатом адекватном лечении заболевание быстро
регрессирует.
Гистопатология. В утолщенном роговом слое большое количество
чесоточных ходов, расположенных в ряд этажей; зернистый слой на этих
512
участках отсутствует/Акантоз, паракератоз. В сосочковом сЛое дермы не-
большие воспалительные явления.
Дифференциальный диагноз следует проводить с пиодермией, экзе-
мой, осложненной пиодермией, гиперкератотнческой формой псориаза.
Лечение. По мнению многих отечественных и зарубежных авторов
(Гумаров В. 3., 1968; 1969; Смелов Н. С. и Большакова Г. М., 1968; Скрип-
кин Ю. К- и соавт., 1970; Nowak Z. и Alkiewicz J., 1970; Paterson W. et
al., 1973; Pezzarossa E. et al., 1983) лучшим средством лечения чесотки
(в том числе норвежской) является водно-мыльная эмульсия бензил-
бензоата (20—25% для взрослых и 10% для детей.). При недостаточном
эффекте можно назначить лечение по методу Демьяновича (последователь-
ное втирание в кожу 60% раствора гипосульфита натрия и 6% раствора
соляной кислоты). Не потеряли значение мазь Вилькинсона, 33% серная
мазь.
По мнению некоторых зарубежных авторов (Leibowitz et al. 1980;
Jagavar, 1980; Reimer, 1983 и др.), наиболее эффективным акарицидным
средством является линдановая мазь (для лечения детей грудного и ран-
него возраста рекомендуют 0,3% линдан, а для взрослых 1% мазь в тече-
ние 2—3 дней). Лечение начинают .после ванны (для размягчения эпидер-
миса), а затем втирают мазь в течение 2—3 дней.
ЛИТЕРАТУРА
Михеев Г. Н. Возникновение норвежской чесотки у 2 больных красной волчанкой.,
— Веста, дерматол. и венерол., 1984, 4, 57—61.
Jagmcar С. Scabies.— Quaterly Med. Rev., 1980, 31, 3, 1—31.
Leibowitz M. et al. Keratotic scabies (Norvegian. Scabies).— S. Afr. Med., J. 1980,
57, 10, 363—366.
Orfils G. et al. Sarna nourvega asociacion con colagenosis (sclerodermia generalizada)
у con nocardiosis.— Rev. argent. Dermatol., 1983, 64, 3, 299—303.
Pezzarossa E., BenoldiD., Alinovi A. Scabbia norverese.— G. Ital. Dermatol., 1983,
118, 1, 27—29.
Reimer G., Schofer H., AUmeyer R. Zum Krankheitsild der Scabies Norvegica.—
Akt. Dermatol., 1983, 9, 3, 81—83.
Ting H., Wang E. Scabies and systemic lupus erythematodes.— Inteern. J. Derma-
tol., 1983, 22, 8, 473—476.
ШЕГРЕНА СИНДРОМ (Sjogren syndromum).
Синонимы: сухой кератоконъюнктивит, сухой синдром, ксеродерма-
тоз и др.
Заболевание описал шведский окулист Sjogren в 1933 г. Ранее (в
1931 г.) аналогичную клиническую картину описал отечественный клини-
цист С. А. Спектор. В дерматологической литературе первое описание при-
надлежит А. А. Каламкаряну и В. Г. Стоянову (1966).
Этиология и патогенез. В развитии заболевания придается значение
хронической инфекций (Sjogren), эндокринным нарушениям—климакс, аме-
норея, овариальная недостаточность и др. (Спектор С. А., 1931; Ромачева
33 Клиническая дерматология 513
И. Ф. и Саксонова Е: А., 1959). Е. М. Тареев, Morgen’ и др. относят су-
хой синдром к коллагенозам, другие считают его вариантом системной
красной волчанки. Ряд авторов (Машкиллейсон Л. Н., 1964; Каламкарян
А. А. и Стоянов Б. Г., 1966; Touraine, 1961) усматривают связь заболева-
ния с дефицитом витамина А экзогенного и эндогенного характера. Moat-
sopoulos и соавт. (1980) полагают, что синдром Шегрена развивается в ре-
зультате лимфоцитарной инфильтрации и закупорки эккринных желез, при-
водящих к уменьшению или подавлению секреции. Повышенная концент-
рация лизоцима в слюне околоушных желез, по их мнению, является спе-
цифическим признаком синдрома Шегрена. Tapper-Jones и Alfred (1980)
установили заметное повышение секреции слезных желез в ответ на дачу
бромгексина и отсутствие повышения секреции слюнных желез.
Клиника. Большинство авторов отмечают, что «сухой синдром» наблю-
дается в среднем возрасте, преимущественно у женщин в климактерическом
периоде. Клиническая картина характеризуется четко выраженным со-
четанием поражения слизистых оболочек, кожи и суставов. Со стороны
глаз отмечается ксерофтальмия, гиперемия, конъюнктивы переходных скла-
док и склер. Конъюнктива сухая, больные жалуются на постоянное ощу-
щение сухости в'глазах: «глаза как будто песком засыпаны». Влажность
конъюнктивальных мешков очень мала: «плачут без слез». Слизистая обо-
лочка ротовой полости отличается резкой сухостью (ксеростомия), нередко
беспокоит хрипота, сухой кашель, больные не могут проглотить пищу, не
запивая водой. Слизистая рта лакированная, гладкая, нередко покрыта
трещинами, особенно иа языке и в углах рта. При далеко .зашедшем про-
цессе сухость распространяется на глотку, гортань, трахею, а также вла-
галище, уретру и прямую кишку. Нередко понижается секреция желудка
и кишечника, что приводит к гастриту, колиту (Тальковский С. И., Ма-
каренко И. И., 1963). У многих больных увеличиваются околоушные слюн-
ные железы. Примерно в 50—60% случаев отмечается поражение суста-
вов типа ревматоидного артрита (Спектор, С. А., 1931; Sjogren, 1948; Sten-
stam, 1961 и др.): боли, деформации суставов, мышечная атрофия. Отдель-
ные авторы (Каламкарян А. А. и Стоянов Б. Г., 1966) сообщают о нару-
шениях психики в терминальном периоде заболевания, выражающихся в
депрессии и чувстве страха (тревожно-депрессивное состояние). Снижение
секреции потовых и сальных желез обусловливает сухость кожи. Кожа те-
ряет свой обычный блеск, тускнеет, шелушится, нередко отмечается фол-
ликулярный гиперкератоз, сильный зуд.’ Волосы становятся тусклыми,
хрупкими, наблюдается их выпадение—диффузное или частичное. Часто
поражаются ногти, они теряют прозрачность, тускнеют, подчас значитель-
но утолщаются за счет подногтевого гйперкератоза. Заболевание имеет
хроническое (от нескольких месяцев до многих лет) волнообразное тече-
ние; ремиссии, как правило, бывают кратковременными.
Гистопатология. Атрофия эпидермиса, сглаживание сосочкового слоя,
отсутствие базальных клеток. В дерме—лимфоцитарная инфильтрация.
Придатки атрофированы.
Дифференциальный диагноз. Следует проводить с системной красной
514
волчанкой, неспецифйческим инфекционным артритом, ревматоидным арт-
ритом, распространенной себорейной экземой, саркоидозом.
Лечение. Комбинированная терапия кортикостероидными препара-
тами в обычных терапевтических дозах с витаминами (А, комплекс В, С и
Е) и другими лекарственными средствами (резохин, анальгин, параамино-
салицилова'я кислота и др.); половые гормоны, тиреоидин и др. Местно—
смягчающие кремы и мази.
ЛИТЕРАТУРА
Moutsopoulos Н., Chased Т., Mann D., Klippel J. et al. Sjogren’s syndrome (sicca
sindrome).— Ann. Int. Med., 1980, 92, 2, 212—216.
Tapper-Jones M., Alfred M. Sjogren's syndrome treated with bromhexine.— Brit.
Med. J., 1980, 62, 28, 1356—1357.
ЭКЗЕМА ГЕРПЕТИЧЕСКАЯ КАПОШИ (eczema herpeticus Kaposi).
Синонимы: варицеллеформная сыпь, острый варицеллеформный пусту-
лез, острый вакциниформный пустулез.
Название «герпетическая экзема» более оправдано, так как указывает
на связь заболевания с экземой и на эпидемиологические особенности дер-
матоза. Герпетическая экзема, описанная впервые Капоши в 1893 г.,
- сравнительно редкое и довольно тяжелое заболевание, представляющее
определенные трудности для диагностики и лечения.
Этиология и патогенез. Seidenberg (1941) впервые выделил из пузырь-
ков герпетической экземы вирус простого герпеса. Новые исследования
(Nasemann и Herzberg, 1974; Lever и Schaumburg-Lever, 1975; Suter et al.,
1977; Segal и Watsom, 1978) подтвердили этиологическую роль вируса гер-
песа. В очагах поражения при исследовании в электронном микроскопе
выявлены частицы вируса, морфологически соответствующие вирусам груп-
пы простого герпеса. Это было подтверждено культуральным методом и
реакцией связывания комплемента. Иногда герпетическая экзема является
результатом случайной инокуляции в кожу вируса вакцин ии (eczema vac-
cinatum). Герпетическая экзема большей частью возникает при зараже-
нии от больных простым герпесом (Каламкарян А. А., 1964; Фомин К. Ф.,
Федорова В. А., 1971; Hitselberger и Burns, 1961; Scully et al., 1975 и
др.), а вакцинальная—при контакте с привитыми против оспы лицами
или в связи с противооспенной вакцинацией самого больного. Высказы-
вается мнение, что герпетическая и вакцинальная экземы представляют
одно заболевание. Существует и другой взгляд: несмотря на сходство кли-
нических проявлений, эти заболевания являются самостоятельными ви-
русными осложнениями детской экземы.
Герпетическая (варицеллеформная) экзема, как правило, развива-
ется у больных с предшествующими хроническими дерматозами, атопиче-
ским дерматитом, особенно у детей, и диффузным нейродермитом у взрослых.
Иногда высыпания развиваются и при других кожных заболеваниях—при
себорейном дерматите, пузырчатке, фолликулярном дискератозе Дарье,
юношеских угрях, грибовидном микозе и др. (Hitselberger и Burus, 1961;
33* - 515
Weiss et al., 1965; Jank и Soltz-Szots, 1966; Silverstein и Buruett, 1967;
Doeglas nMoolhuysen, 1969; Schully et al., 1975; Hazen и Eppes, 1977 и др.)
Полагают, что развитие варицеллеформной экземы является результатом
нарушения клеточного (Miiller et al., 1972) или гуморального (Deresinski
и Sjteveus, 1974) иммунитета под влиянием избыточного солнечного облу-
чения, инфекции, терапии большими дозами кортикостероидов, проводи-
мой по поводу основного заболевания, ПУВА-терапии. Наши наблюде-
ния, а также данные литературы (Марьясис Е. Д. и Павлик Л. В., 1980
и др.) позволяют считать, что возникновение герпетической экземы нередко
связано с переохлаждением, которое приводит к активации дремлющей
герпетической инфекции.
Клиника. Клиническая картина герпетической экземы весьма харак-
терна и описана в литературе многими отечественными и зарубежными
авторами (Голосовкер С. Я. и Зверькова Ф. А., 1955; Розентул М. А. и
Белякова А. Г., 1969; Фомин К- Ф. и Федорова В. А., 1971; Студницин
А. А. и соавт., 1971; Марьясис Е. Д., Павлик Л. В., 1980; Estavoger et
al., 1976; Nagington et al., 1979 и др.). Болезнь поражает детей, но также и
взрослых, которые больны или недавно болели экземой, нейродермитом
и др. Заболевание начинается внезапно, сопровождается высокой темпера-
турой (до 39—40°), резко выраженным токсикозом и тяжелым общим со-
стоянием. На фоне отека и гиперемии кожи появляются более или менее
обильные, расположенные группами пузырьки, величиной от просяного
зерна до чечевицы, быстро переходящие в пустулы с характерным пупко-
видным вдавлеиием в центре. При слиянии пустул образуются большие с
' микроциклическими очертаниями бляшки. Свежие прозрачные пузырьки с
пупковидным вдавлением в центре производят впечатление ветряной
оспы. Сыпь располагается на измененных экземой, нейродермитом, фолли-
кулярным дискератозом, а также на других участках кожи (чаще на ли-
це, шее, груди, кистях, предплечьях и др.). Отдельные или сгруппирован-
ные по 4—5 мелкие пустулы встречаются и на непораженной коже. В про-
цессе обратного развития высыпания подсыхают, на месте пустул остаются
кровянистые корочки. После отпадения корочек на месте высыпаний оста-
ются розовые пятна или вторичная пигментация, исключительно редко
поверхностные рубчики. Герпетическая экзема нередко протекает по типу
токсико-септического заболевания с явлениями менингизма, осложня-
ется пневмонией, отитом. В крови нередко наблюдается анэозинофилия.
Возможно одновременное герпетическое поражение слизистой оболочки
рта в виде афт, а также конъюнктивы и роговицы (светобоязнь и резкое ,
припухание век).
Раньше смертность от герпетической экземы достигала 16—25% (Ber-
ten и Brusting, 1944; Copeman и Brusting, 1964;) с введением в практику
сульфаниламидных препаратов, антибиотиков и кортикостероидов она
снизилась до 2—3%.
Гистопатология. Везикуло-пустулы в эпидермисе, баллонирующая
дегенерация эпителиальных клеток, скопление нейтрофильных лейкоцитов.
Обнаруживаются внутриклеточные включения.
516
Дифференциальный диагноз. Следует проводить с. ветряной оспой,
вакцинией, пиодермией. Трудно, а порой и невозможно провести дифферент
циальную диагностику между варицеллиформным пустулезом (герпетиче-
ской экземой) и вакцинальной экземой. Некоторые авторы не разделяют
обе эти формы, считая их одним и тем же заболеванием.
Лечение. Антибиотики, сульфаниламиды, кортикостероиды, интерфе-
рон (парентерально), антигистаминные препараты, общеукрепляющие сред-
ства, витамины. Наружно—жидкость Кастеллани, мази с антибиотиками
(гелиомицин, линкомицин и др.). Больные должны быть изолированы. С
целью профилактики вирусных осложнений дети, страдающие зудящими
дерматозами, не должны контактировать с лицами, больными герпесом,
подвергшимися противооспенной вакцинации.
v ЛИТЕРАТУРА
Марыкис Е. Д., Павлик Л. В. О герпетиформной экземе Капоши—Юлиусберга
у взрослых.— Вести, дерматол. и венерол., 1980, 9, 50—52.
Estaooger J., Saimont С,, Menget С. et al. Pustulose varioliforme de Kaposi—Juli-
usberg chez une fillette de 11 ans.'— Pediatrie, 1976, 31, 3, 295—303.
Nagington J., Rook A. Virus and related infection.— In: Textbook of dermatology.
Ed.: Rook, Wilkinson, Ebling. Oxfort, 1979, 3d ed., p. 607—676.
Segal R., Watson W. Kaposi’s varicellform eruption in mycosis fungoides.— Arch.
Dermatol., 1978, 114, 7, 1067—1069. X,
Schully R., Galdamini J., McNeely B. Case records of Massachusetts general hospi-
tal.— N. Engl. J. Med., 1975, 293, 598—603.
Suter L., Bilek t., Vakilzadeh F. Eczema herpeticatum Kaposi bei Pemphigus folia-
ceum.—Z. Hautkr., 1977, 52, 5, 151—154.
ЭЛАСТОЗ ПЕРФОРИРУЮЩИЙ СЕРПИГИНИРУЮЩИЙ (elastosis .
perforans serpiginosa)
Этиология и патогенез. Заболевание относится, по-видимому, к группе
дерматозов, характерной чертой которых является трансэпителиальная
элиминация материала, ставшего чужеродным. В качестве одной из воз-
можных причин указывается на нарушение созревания эластических во-
локон (Pass et al., 1973). По мнению Hitch и соавт. (1959), первичные из-
менения заключаются в своеобразной пролиферации грубых волокон, имею-
щих характеристику эластических. Это наблюдается в основном в верхней
части сетчатого, в субсосочковом слое, в ^еньшей мере—в зоне сосочков.
Woerdemann и Scott (1959) на основании гистологических исследований по-
лагают, что речь идет об эластоидной дегенерации коллагена, а не гипер-
плазии эластической ткани. Авторы предположили, что фолликулярность
расположения зависит от вовлечения в процесс сальных желез.
Некротически измененные эластические волокна вызывают реакцию
инородного тела, в результате чего элиминируются через эпидермис. Опи-
сание семейных форм (Woerdemann et al., 1965), отмечаемая (Reed и Pidgeon,
1964; Korting, 1966; Mehregan, 1968) связь эластоза перфорирующего серпи-
гинирующего с другими генетическими заболеваниями (синдром Ehlers Dan-
517
los, osteogenesis imperfecta, болезнь Дауна и др.) свидетельствует о возмож-
ной роли генетических факторов в развитии дефекта эластической ткани.
Описано развитие эластоза серпигинирующего после длительного введения
Д-пеницилламина (Pass et al., 1973; Gloor и Bersch, 1982).
Клиника. Фигурные, линейные, полициклические, овальные, кольце-
видные очаги поражения из гиперкератотических папул, иногда кониче-
ских, веррукозных, размерами до 5 мм, цвета нормальной кожи, розовато-
красноватых или коричневатых, имеющих в центре небольшое углубление,
заполненное роговой пробкой. Очаги обладают периферическим, серпиги-
нирующим ростом, при этом в центральной части высыпания регрессируют,
оставляя нерезко выраженную атрофию и депигментацию. Высыпания
обычно немногочисленны; диссеминированные формы описаны при монго-
лоидизме (Rasmussen, 1972). Излюбленной локализацией являются заты-
лок, шея. Реже очаги поражения располагаются на руках, щеках, бедрах.
Имеется описание эластоза перфорирующего серпигинирующего на ушных
раковинах (Weidmann и Allyn, 1971), в области лба (Hauss, 1963), диссе-
минированной формы (Pedro и Garcia, 1974). У 2 из 19 больных, описанных
Woerdemann и Scott (1959), очаги развились в области коленных и локтевых
суставов при отсутствии их на шее. Комбинации поражений локтей и ко-
леней с другими локализациями наблюдались еще у 5 больных.
Заболевание развивается у детей или лиц молодого возраста, чаще у
мужчин. Существует неопределенно долго; может спонтанно регрессиро-
вать, оставляя рубцевидную атрофию.
Гистопатология. Акантоз, гиперкератоз, наличие базофильно окраши-
ваемого детрита в эпидермисе, проникающего из дермы, по периферии кото-
рого выявляется гранулематозная реакция; количество эластических воло-
кон увеличено, они огрубевшие, гомогенизированы.
Дифференциальный диагноз следует проводить с болезнью Кирле, коль-
цевидной гранулемой, липоидным некробиозом, порокератозом Мибелли.
Лечение. Криотерапия, кортикостероиды наружно и внутриочагово,
к^р это литические мази.
ЛИТЕРАТУРА
Gloor М., Bersch A. Elastoma intrapapillare perforans verruciforme (Lutz—Miescher)
als Folge einer Langzeittherapie mit D-penizillamine.— Hautarzt, 1982, 33, 5, 219—293.
ЭЛАСТОИДОЗ КОЖИ УЗЕЛКОВЫЙ С КИСТАМИ И КОМЕДОНАМИ
(elastoidosis cutis nodularis cystica et comedonica).
Синоним: болезнь Фавра—Ракушо.
Сравнительно редкое заболевание кожи дистрофического характера.
Впервые описано Favre-Racouchot в 1931—1937 годах.
Этиология и патогенез. Многие авторы (Kunz, 1961; Korting, 1959;
Dressiler, 1958; English, 1971 и др.) указывают, что длительное воздействие
инсоляции и других атмосферных факторов (ветра, непогоды и др.) пред-
располагают к развитию заболевания. Это подтверждается относительной
частотой его у сельскохозяйственных рабочих и локализацией на коже от-
518
крытых участков, в первую очередь на лице и шее. Pierard и Fontaine (1953),
Rood (1972) указывают на врожденную предрасположенность к данному
заболеванию. Fuga (1959) и Maragnam (1964) считают эластоидоз разно-
видностью старческой атрофии. Огшеа и Depaoli (1965) на основании иден-
тичных данных, полученных при гистологическом и гистохимическом ис-
следовании кожи при болезни Фавра—Ракушо и при ромбовидной коже,
рассматривают их как варианты старческих изменений кожи.
Клиника. Заболевание встречается преимущественно у мужчин стар-
ше 50 лет. Как правило, очаги поражения локализуются на коже лица,
особенно вокруг глаз и в височных областях. Иногда захватываются
ушные раковины и задняя поверхность шеи. Degos и соавт. (1954), Л. И.
Глебова и соавт. (1980) описали больных с распространенными поражениями
кожи верхних и нижних конечностей, туловища. Кожа лба, шеи, за ушными
раковинами утолщена, покрыта морщинами, неровная, плотная, желто-
вато-красного цвета, напоминает «булыжную мостовую.» На этих участках
имеются множественные глубокие узловатые элементы размером до круп-
ной горошины, вишни, полупросвечивающиеся кисты беловато-желтоватого
цвета диаметром 1—5 мм и комедоны коричневато-черного цвета, располага-
ющиеся в центральной части большинства узелков и кистозных образова-
ний. При сдавливании комедонов из них выходит кремоподобная масса
белого цвета. Наиболее сильные дистрофические изменения происходят в
коже шеи и лица. В коже закрытых участков они также обнаруживаются,
хотя и в меньшей степени (Кожевников П. В., 1964).
Гистопатология. В дерме множество кист различных размеров. Стенки
кист состоят из нескольких слоев эпидермальных клеток, содержащих
кератин. Отмечается базофильная дегенерация соединительной ткани верх-
них слоев дермы, отделенной от эпидермиса слоем нормального коллагена.
Базофильная дегенерация волокнистой ткани, видимо, очень характерна
для этого заболевания, так как отмечена почти всеми авторами. Сальные
железы атрофированы, потовые железы уменьшены в количестве и разме-
рах. Сосуды верхнего слоя собственно кожи расширены, стенки их утол-
щены. Вокруг сосудов располагается инфильтрат, состоящий преимуще-
ственно из гистиоцитов и лимфоцитов. Для гистохимической картины эла-
стоидоза характерно отсутствие кислых мукополисахаридов (Niebauer и
Stockinger, 1964). Возможно, что эпидермальные кисты развиваются как
результат нарушения кератинизации сально-волосяного фолликула.
Дифференциальный диагноз. Следует проводить с диффузной эластомой
Дюбрейля, коллоид-милиумом, акне-келоидом, ромбовидной кожей, си-
рингомой, трихоэпителиомой.
Лечение. Положительный терапевтический эффект наблюдается от
дермабразии.
ЛИТЕРАТУРА
Глебова Л. И., Машкиллейсон Н. А., Таумин М- Р-, Абрамов В. М. Случай рас-
пространенного elastoidosis cutis nodularis cystica et comedonica (болезнь Favre—Racouchot).
— Вести, дерматол. и венерол., 1980, 7, 53—55.
519
Lever IF., Schaumburg-Lever G. Nodular elastosis with cysts and comedones (Favre
and Racouchot).— In: Histopathology of the skin. Philadelphia, 1975, p. 251.
ЭНДОМЕТРИОЗ КОЖНЫЙ (endometriosis cutis)
Синонимы: эндометриальная гетеротопия; менструирующая опухоль,
аденофиброз и др., всего 33 синонима.
Под эндометриозом следует понимать разрастание тканей в виде со-
литарных или множественных узлов, по морфологическому строению и
функциональным свойствам напоминающим эндометрий. .
Эта патология была описана МйПег (1854) и Rokitansky (1860), а пер-
вые случаи кожного эндометриоза опубликовали Recklinghausen в 1885 г.,
а затем Sampson в 1925 г.
Этиология и патогенез. Имеется много теорий происхождения эндо-
метриоза, однако ни одна из них не решает проблемы патогенеза этого за-
болевания и не раскрывает причины многообразия его локализации. В ос-
новном эта патология встречается в возрасте от 30 до 55 лет у 8—15 % мен-
струирующих женщин. Кожный эндометриоз, по различным авторам (Ба-
скаков В. П., 1979; Палка В. С. и Минельман С. И., 1970; Жук А. Г.,
1976; Field et al., 1962; Williams et al., 1976), составляет от 0,42 до
. 4,0% по отношению ко всем эндометриозам. Существуют три различных тео-
рии развития эндометриоза: эмбриональная (Recklinghausen, 1886), мета-
пластическая (Иванов Н. С., 1897) и теория эндометриального происхож-
дения (Sampson, 1921—1922). Сторонники последней связывают возникно-
вение эндометриозов с ретроградным поступлением клеток эндометрия
вместе с менструальной кровью через маточные трубы в брюшную полость
и органы малого таза при нарушении естественного оттока. Сторонники
гормональной теории считают, что нарушение в организме содержания
гормонов (абсолютная или относительная гиперэстрогения) создает благо-
приятный фон для развития эндометриоза. Гормональный генез эндомет-
риоза подтверждается циклическим характером заболевания и регрессом
его во время беременности и менопаузы. Ряд авторов (Rodшапп и Jones,
1962; Scott, 1962 и др.) описали эндометриоз легких, предполагая гемато-
генный и, возможно, лимфогенный занос клеток маточного эндометрия. Раз-
витие эндометриоза в коже, наружных гениталиях также объясняется, в
основном, распространением по сосудам (Палка В. С. и Минельман С. И.,
1970; Жук А. Г., 1976; Адамян Л. В., 1977; Nora et al., 1956). Кроме
сосудистой трансплантации маточного эндометрия возможен механический
перенос, механическая имплантация во время различных оперативных
вмешательств—кесарева сечения и сальпингэктомии, аппендектомии, лапа-
ротомии, грыжесечения. Современные клиницисты и практические врачи
пользуются следующей классификацией эндометриозов (Баскаков В. П.,
1966; Мажбиц А. М., 1971): 1. Генитальный эндометриоз (внутренний,
наружный, комбинированный) составляет 92—94% всего эндометриоза.
2. Экстрагенитальный эндометриоз (эндометриоз слепой кишки, мочевого
пузыря, почек, пупка, легких, послеоперационных рубцов кожи и др.)
составляет 6—8%. Наиболее частыми локализациями кожного эндометрио-
520
Рис. 337. Эндометриоз
кожный-
за являются пупочная область и послеоперационные абдоминальные руб-
цы. Описаны также локализации на конечностях, в паху, на вульве и вок-
руг ануса.
Клиника. Эндометриоз кожи представлен дольчатым опухолевидным
образованием буроватого цвета, размером до вишни или несколько больше,
не имеющим капсулы, состоящим
из отдельных узлов плотно-элас-
тической консистенции (рис. 337).
В клинической картине кожного
эндометриоза прослеживается цик-
лическое течение, связанное с та-
кого же рода изменениями слизис-
той матки (Лопатина Т. В., 1971;
Азимова Д. А., 1975; Адамян Л.
В., 1977; Duperrat и Ficheux, 1970;
Movers, 1971; Popoff et al., 1962;
Цвелев Ю. В., 1978 и др.). За
3—5 дней до менструации в кож-
ной опухоли появляются сильные
боли, сна увеличивается в объеме
и плотности. Во время менструа-
ции эти явления резко усиливают-
ся и на поверхности опухоли
появляются кровянистые выделе-
ния. По окойчании менструации опухоль уменьшается в размере, боли
и кровотечения прекращаются.
Метаплазия в злокачественную опухоль наблюдается крайне редко,
обычно при генитальном эндометриозе Сообщений о малигнизации кожного
эндометриоза мы не встретили (Каламкарян А. А. и соавт., 1981).
Гистопатология.' Опухоль имеет ячеистое строение, заполнена кровя-
нистой жидкостью; в дерме и эпидермисе—кисты и канальцевые железы
различных размеров и формы; имеются признаки секреции кист.
Дифференциальный диагноз. Проводят с аденокарциномой, пиоген-
ной гранулемой, папилломами, дерматофибромой, варикозным расширением
пупочных вен, ангиомами.
Лечение. Хирургическое иссечение эндометриозных участков в пре-
делах здоровых тканей с последующей гормональной терапией (эстроген-
гестагенные препараты и гестагены).
ЛИТЕРАТУРА
Адамян Л. В. Особенности клинического течения редких локализация эндометри-
оза.— В кн.: Актуальные вопросы современной хирургии. Сб. тр. Ин-та хирургии им.
Вишневского. М-, 1977, с. 129—133.
Азимова Д. А. Функциональное состояние гипоталамо-гипофкзарно-яичниковой
системы у больных наружным эндометриозом.— Сов. мед., 1975, 6, 26—30.
Баскаков В. И. Клиника и лечение эндометриоза.— Л., 1979.
521
Жук А. Г. Об эндометриозе пупка.— В ки.: Актуальные вопросы онкологии. Кеме-
рово, 1976, вып- III, с. 223—224.
Каламкарян А. А., Стрижаков А. Н. Адамян Л. В. и др. Кожный эндометриоз
(клиника, диагностика и лечение).— Вести, дерматол. и венерол., 1981, 2, 4—9.
Williams Н., BarskyS., StorinoW. Umbilical endometrioma. Arch. Dermatol., 1976,
112, 10, 1435—1436.
ЭПИДЕРМОДИСПЛАЗИЯ ВЕРРУЦИФОРМНАЯ ЛЕВАНДОВСКОГО—
ЛЮТЦА (epidermodysplasia verruciformis Lewandowski—Lutz).
Синонимы: генерализованная бородавчатость, врожденные дискера-
тотические бородавки.
Описана в 1922 г. Левандовским и Лютцем.
Этиология и патогенез. Одни авторы (Тыжненко А. И., 1931; Шах-
нес И. Е. н соавт., 1977; Jablonska и Formas, 1966; Rook и Wilkinson,
1979 и др.) полагают, что это генерализованная форма плоских бородавок;
другие (Ведрова И. Н., 1949; Машкиллейсон Л. Н., 1960; Диванян Р. С.
и Мокроусов М. С., 1977; Tsuji et al., 1970; Daroczy et al., 1972 и др.)
рассматривают верруциформную эпидермодисплазию как самостоятельное
заболевание, относящееся к прекарциноматозным невоидным дерматозам.
Мы, как и ряд других авторов (Каламкарян А. А. и Самсонов В. А., 1977;
Каралицкий Е. М. и соавт., 1982; Gorda et al., 1969; Depaoli, 1971; Gra-
nispan et al., 1972; Pieyre, 1974; Claudy et al., 1982; Haustein, 1982, Ber-
tani et al., 1983, и др.) рассматриваем верруциформную эпидермодиспла-
зию Левандовского—Лютца как генодерматоз, вероятно, вирусной этио-
логии (человеческим папилловирусом типа 3,5) с относительно высокой
потенцией к бластоматозной трансформации (от 12—15 до 30—75%). Из-
вестны семейные случаи, иногда в нескольких поколениях. (Шмелев К. А.,
1949; Gorda, 1969 и др.). Gianotti и соавт. (1969), Tsuji и соавт. (1970), Grup-
рег и соавт. (1971), Depaoli (1971) на основании положительной ауто-и ге-
тероинокуляции, нахождения в ядрах клеток рогового и зернистого слоев
эпидермиса элементарных телец, высказывают мнение о вирусной природе
дерматоза. Многие другие авторы не смогли подтвердить этого. Исследо-
вания, проведенные в последние годы у небольшого числа больных вер-
руциформной эпидермодисплазией, обнаружили некоторые общие повреж-
дения клеточного иммунитета (Jablonska et al., 1976; Pyrhonen et al., 1978;
' Bundino et al., 1980 и др.). Полученные при этом данные отражают неко-
торые наследственные отклонения иммунных реакций или угнетение им-
мунных функций, вызванные вирусной инфекцией.
Клиника. Болезнь характеризуется, как правило, множественными
высыпаниями, сходными но внешнему виду с плоскими и вульгарными
бородавками. Узелковые элементы размером от просяного зерна до чече-
вицы и больше локализуются в значительном количестве на тыле кистей,
стоп, предплечьях, голенях, бедрах, на лбу, щеках, боковых поверхностях
шеи и в меньшем количестве—на спине, животе (рис. 338, 339), ягодицах.
Границы элементов четкие, цвет коричневатый, по консистенции элемен-
ты несколько плотнее окружающей, видимо, здоровой кожи. На отдельных
522
Рис. 339. Верруциформная
зпидермодисплазия Леван-
довского—Лютца (тот ясе больной).
Рис. 338. Верруциформная
зпидермодисплазия Леван-
доеского—Лютца.
участках кожного покрова (бедра, голени) на месте травматизации опре-
деляются бородавчатые линейно расположенные элементы (изоморфная
реакция). На лице и шее высыпания имеют вид плоских бородавчатых папул
диаметром от 2 до 6 мм. На туловище и конечностях они обычно более круп-
ные и более плотные, возвышаются над уровнем кожи, имеют четкие гра-
ницы и напоминают вульгарные бородавки. На тыльных поверхностях
кистей бородавчатые образования сливаются между собой, образуя буро-
вато-синюшного оттенка бляшки. Субъективнее ощущения отсутствуют.
Болезнь проявляется чаще всего в детском возрасте, реже у взрослых и
пожилых людей.
Дерматоз стойкий, длится всю жизнь, нередко бородавчатые образо-
вания озлокачествляются.
Гистопатология. Гиперкератоз, паракераФоз, акантоз и вакуолизация
клеток утолщенного зернистого и шиповидного слоев. Характерны так
называемые «пустые» клетки, подвергшиеся вакуольной дегенерации. Ва-
куолизированные клетки мальпигиевою слоя отличают сморщенные пикно-
тические ядра.
Дифференциальный диагноз. Следует проводить с верруциформным
акрокератозом Hopf, бородавчатым туберкулезом кожи, бородавчатым
плоским лишаем.
Лечение. Теплые ванны, кератолитические средства, лучи Букки,
поливитамины.
ЛИТЕРАТУРА
Абдуллаев А. X. Верруциформная зпидермодисплазия Левандовского—Лютца.—
Вестн. дерматол. и венерол., 1980, 12, 40—43.
Диванян Р. С.. Мокроусов М. С. Своеобразный случай верруциформной эпидермо-
дисплазии Левандовского—Лютца.— Вестн. дерматол. и венерол., 1977, 1, 64—5Ь.
523
Каламкарян Л. А., Самсонов В, А. К вопросу о малигнизации верруциформной
эпидермодисплазии ЛеванДовского—Лютца.— Вести, дерматол и венерол., 1977, 1, 48—51.
Карлицкий Е. М. и др. К вопросу о верруциформной эпидермодисплазии.— Вести,
дерматол. и венерол., 1982, 1, 55—58.
Шахнес И. Е., Кауфман И. А., Лунева О. И. Случай генерализованного верруко-
за.— Вести, дерматол. и венерол., 1977, 1, 51—54.
Bundino S., Zina A., Bernengo М. Dyskeratosis congenita with epidermidysplasis
verruciformis of Lewandowsky and Lutz.— Dermatologica, 1978, 156, 2, 15—22.
Cloudy A., Touraine J., Mitanne D. Epidermodysplasia verruciformis induced by a
new human papillomavirus (HPV—8).— Arch. Dermatol., 1982, 27, 3—4, 213—219.
Haustein U. Epidermodysplasia verruciformis Lewandowsky—Lutz mit multiplen
Plattenepithel — und Bowenkarzinomen.— Dermatol. Mschr., 1982, 168, 12, 821—828.
Jablonska S. et al. Regression of the lesions of epidermodysplasia verruciformis.—
Brit. J. Dermatol., 1982, 107, 1, 109—116.
Pyrhonen S-, Jablonska S., Obalek S., Kuismanen E. Immune reactions in epider-
modysplasia verruciformis.— Brit. J. Dermatol., 1980, 102, 3, 247—253.
ЭПИДЕРМОЛИЗ БУЛЛЕЗНЫЙ (epidermolysis bullosa hereditaria).
Синонимы: врожденная пузырчатка, наследственный травматический
буллезный дерматоз (Hallopeau).
Клиническую картину болезни первым описал Hutchinson в 1875 г.-
как врожденное заболевание первым описал Goldscheider в 1882 г. Kobner
в 1886 г. предложил название врожденный буллезный эпидермолиз.
Hallopeau в 1896 г. выделил две формы эпидермолиза—простую и ди-
строфическую. Siemens в 1923 г. указал, что простой эпидермолиз насле-
дуется аутосомно-доминантно, а дистрофический—аутосомно-рецессивно.
Он допускал, что эти формы не относятся к единой клинической группе.
Частота составляет 1:60000 (Breit, 1979).
Этиология и патогенез. Получены данные, говорящие о том, что в ос-
нове буллезного эпидермолиза лежит врожденный дефект синергизма тка-
невых энзимов и ингибиторов (Barth и Duck, 1972; Ratzenhofer et aL, 1977).
Некоторые исследователи указывают на роль нарушений механизма свер-
тывания крови, другие отрицают это (Schnyder et al., 1964; Gedde-Dahl
et al., 1966).
Gordon и соавт. (1979) сообщили о повышенном содержании а-фе-
топротеина в амниотической жидкости и сыворотке крови матери, кото-
рая родила ребенка с проявлениями буллезного эпидермолиза. Полагают,
что указанный тест можно использовать для исследования женщин, имею-
щих одного ребенка с буллезным эпидермолизом. Описаны дети с дистро-
фическим эпидермолизом, родители которых были кровными родственника-
ми (Groot et al., 1978 и др.). Хромосомные аномалии при врожденном эпи-
дермолизе не установлены.
ПРОСТОЙ ИЛИ ДОБРОКАЧЕСТВЕННЫЙ БУЛЛЕЗНЫЙ
ЭПИДЕРМОЛИЗ (epidermolysis bullosa hereditaria simplex).
Заболевание начинается в раннем детстве или с рождения. Склон-
ность к образованию пузырей особенно четко выявляется, когда ребенок
524
Рис. 340. Эпидермолиз буллезный дистрофический-
Рис. 341. Буллезный эпидермолиз
(форма Вебера—Кокаина).
начинает ползать, хо-
дить. На локтях, ко-
ленях, кистях, стопах,
пояснице, т. е. на мес-
тах давления, частой
травматнзации, образу-
ются прозрачные, редко
геморрагические пузы-
ри, которые быстро за-
живают, не оставляя
следа. Слизистые обо-
лочки поражаются очень
редко, ногти в процесс
не вовлекаются. Общее
состояние детей не стра-
дает. Помимо кожных,
других патологических
изменений у них нет.
Нередко к пубертатному
периоду проявления
заболевания ослабевают
или даже проходят. У
взрослых пузыри обра-
зуются чаще летом, пос-
ле длительной ходьбы.
Выделяют особую клиничес^ю разновидность простого буллезного
эпидермолиза —позднюю (epidermolysis bullosa hereditaria tarda Weber
— Cockayne или летний
буллезный эпидермолиз
Л. Н. Машкиллейсона).
Эта форма протекает более
мягко, наблюдается у взро-
слых, пузыри появляются
на ногах (редко на руках)
преимущественно в летнее
время или после горячей
ванны. Часто сочетается с
гипергидрозом, а иногда с
кератодермией подошв
(Штамова Л. В. и Прорвич
Л. В., 1949). Маджаров и
соавт (1977) считают эпи-
дермолиз Вебера—Кокайн а
абортивной сезонной фор-
мой простого эпидермо-
лиза.
525
ДИСТРОФИЧЕСКИЙ БУЛЛЕЗНЫЙ ЭПИДЕРМОЛИЗ (epidermolysis
bullosa dystrophica).
Встречается чаще, чем простая форма. Начинается с детства, неред-
ко с рождения. Продолжается многие годы. Часто наблюдаются семейные
случаи. Тяжесть заболевания у членов семьи может быть разной. Пузыри
заживают с образованием рубцов, застойно-бурых., гиперпигментирован-
ных атрофических участков, милиумподобных эпидермальных кист. Пу-
зыри возникают на любом участке кожного покрова, но чаще на местах,
подвергающихся давлению (рис. 340, 341).
Рубцевание очагов поражения на кистях, стопах может привести к
образованию контрактур. Характерны изменения ногтей, вплоть до ано-
нихии; нередко дистрофии волос, зубов, акроцианоз, ладонно-подошвен-
ный гипергидроз.
Часто в процесс вовлекаются слизистые оболочки рта, гортани, пище-
вода, носа, кишечника, гениталий. Дети, страдающие дистрофическим бул-
лезным эпидермолизом, нередко отстают в физическом развитии, но умст-
венно обычно развиты нормально. Ряд авторов (Graciansky et al., 1974 и
др.) указывают, что врожденный дистрофический эпидермолиз нередко
осложняется амилоидозом внутренних органов, особенно почек.
В настоящее время принято выделять несколько разновидностей дист-
рофического буллезного эпидермолиза:
1. Epidermolysis bullosa hereditaria letalis Herlitz.
При этой аутосомно-рецессивной форме пузыри на коже и слизистых обо-
лочках возникают с момента рождения. Тяжелые повреждения образу-
ются при сосании, в результате чего прием пищи становится невозможным.
Смерть обычно наступает в первые месяцы жизни.
2. Epidermolysis bullosa hyperplastica (Cockayne)
Наследуется доминантно. Наряду с необильными пузырями появляются
к злоидоподобные образования, бородавчатые гиперкератозы, онихогри-
фозы. Поражаются слизистые. Возможен вторичный стеноз пищевода, по-
мутнение роговицы, искривление позвоночника.
3. Epidermolysis albopapuloides (Pasini)
Заболевание начинается в детстве и’ протекает как дистрофическая фор-
ма, но после 10-летнего возраста на туловище появляются округлые бе-
лые выстоящие над уровнем окружающей кожи плотные уртикоподобиые
/ рубцы величиной с чечевицу. Они образуются как на местах предшествую-
щих пузырей, так и на до того не измененной коже. Наследуется аутосомно-
рецессивно.
4. Epidermolysis bullosa dystrophyca ulcero-vegetans (Nicolas, Moutot).
Очень редкая форма, начинающаяся с рождения. Сопровождается мно-
жественными дистрофиями. На месте пузырей—язвенный распад, вегета-
ции. Развивается вторичный амилоидоз, гепато- и спленомегалия. Насле-
дуется аутосомно-рецессивно.
5. Epidermolysis bullosa distrophica inverse (Gedde-Dahl, 1971; Hashi-
moto et al., 1976; Breit, 1979).
Высыпания преимущественно или исключительно располагаются в
626
паховых, подмышечных, анальной складках, в промежности. Пузырные,
пиодермоподобные высыпания начинаются в период новорожденное™,
сопровождаются рецидивирующими кератитами, паронихиями. В конце
детства появляются рубцы, атрофии кожи, дистрофии ногтей, дефекты
зубной эмали, позже—стриктуры в пищеводе. Наследуется аутосомно-
рецессивно.
Эти симптомы могут существовать не одновременно, а выявляться в
различных комбинациях. Описаны и другие очень редкие формы дистро-
фического эпидермолиза, которые, по-видимому, являются следствием раз-
личной выраженности и разного сочетания многочисленных поражений,
встречающихся при этом заболевании.
Прогноз для жизни большей частью благоприятный. В редких описа-
ниях летального исхода болезни в качестве причин смерти приводят сеп-
тические процессы, пневмонии, туберкулез, нефрит, карциному и саркому,
стеноз пищевода, кахексию, амилоидоз (Schubert и Hochheim, 1975). Опи-
саны случаи развйтия рака кожи и слизистых на очагах дистрофического
эпидермолиза (Kohl, 1956; Schiller, 1960).
Дифференциальный диагноз. Следует проводить с пузырными заболе-
ваниями, наблюдающимися в период новорожденное™: пузырчатка ново-
рожденных, сифилитическая пузырчатка, hydroa vacciniformia, с ранней
фазой недержания пигмента, кожной порфирией, токсическими экзанте-
мами.
Гистопатология. При простом эпидермолизе пузыри располагаются
всегда над базальной мембраной, т. е. интраэпидермально, эластика не
страдает.
При дистрофической форме эпидермолиза пузыри располагаются под
базальной мембраной; при типе Herlitz расщепление происходит в La-
mina lucida, т. е. между плазматической мембраной базальных клеток и
базальной мембраной. Эластика разрушена.
Имеются данные о недостаточности эластической ткани в клиниче-
ски неизмененной коже у больных дистрофическим эпидермолизом.
При альбопапулоидной форме эпидермолиза наблюдаются пучки со-
единительнотканных волокон, в области которых исчезли эластические
волокна. В области уртикоподобных рубцов, образовавшихся как будто
бы без предшествующих пузырей, между эпидермисом и дермой обнаружи-
вают клинически невидимые микропузыри.
Лечение. Трансфузии цельной крови, плазмы; при выраженных во-
спалительных явлениях—глюкокортикоиды, при осложнениях вторичной
инфекцией—антибиотики; препараты железа, фосфора, витамины. При
стенозе пищевода—бужирование. Важное значение имеет правильное тру-
доустройство больных, достигших совершеннолетия (Штейнлухт Л. А.,
1974).
ЛИТЕРАТУРА
Лелис И. И., Лелиене В. А. Наследственныйбуллезный эпидермолиз и его вариан-
ты.—Вести, дерматол. и венерол., 1977, 6, 63—66
527
Студницин А. А., Никитина М. Н., Алехина Г. М., Белякова А. Г. Клиника и
течение буллезного эпидермолиза у детей.— Вестн. дерматол. и венерол., 1974, 4, 3—7.
Штейнлухт J1. А. О клинике и лечении врожденного эпидермолиза.— Вестн. дер-
матол., 1974, 5, П—15.
Маджаров И., Атанасов В., Минков Д. и др. Върху клиниката и фамилния харак-
тер на Epidermolisis bullosa simplex тип Keder—Cockayne.— Берматол. и венерол., 1977,
16, 2, 103—106.
Gordon Y., Kirby J. D., Kitau M. J. et al. Materna serum and amniotic fluid concent-
rations of alphafetoprotein in Epidermolysis bullosa simplex.— Brit. Med., 1979, 1, 307—
308.
Graciansky P-, Larreque M., Blocher G., Oliver-Martin D. Epidermolyse bulleuse
conjenitale dystrophique transmise en recessivite avec syndrome nephrotique terminal.—
Bui,. Soc. franc. Derm. Syph., 1974, 81, 2, 186—190.
Groot W. G. D., Postuma R., Hunter A. G. W. Familial piloric atresis associated
with epidermolysis bullosa.— J. Pediat., 1978, 92, 429—431.
Hashimoto J., Anton-Lamprecht J., Hofbauer M. Epidermolysis bullosa dystrop-
hies inverse.— Hautarzt, 1976, 27, 532—537.
Ratzenhofer E , Lubec G. Blaseninhalt bei Epidermolysis hereditaria bullosa dystrop-
hies spalted Alpha— I—Antitrypsin.— Hautarzt, 1977, 28, 481—482.
Schubert H., Hochheim A. Epidermolysis bullosa hereditaria dystrophica polydysp-
lastica mit todlichem Ausgang bei sekundarer Amyloidose.— Derm. Mschr., 1975, 161, 399—
401.
ЭПИТЕЛИОМА ОБЫЗВЕСТВЛЕННАЯ (epithelioma calcificans)
Синоним: пиломатриксома.
Этиология и патогенез. Считается (Forbis и Helnig, 1961; Lever и Hashi-
moto, 1966 и др.), что эпителиома обызвествленная развивается из первич-
ного эпителиального ростка с дифференцировкой в направлении волося-
ных структур. Базофильные клетки, составляющие основу опухоли в на-
чале развития, рассматриваются как эквивалент клеток волосяного мат-
рикса. А. К. Апатенко (1973), напротив, отмечает, что при некротизирую-
щейся эпителиоме не бывает четкой дифференцировки в направлении воло-
сяных фолликулов, и полагает, что опухоль гистогенетически связана со
стенкой эпидермальной кисты. Трансформацию стенки эпидермальной кисты
в обызвествленную эпителиому описали Kanitakis и соавт. (1984). Дина-
мика патологического процесса представляется как постепенный переход
от активных базофильных до теневых клеток с накоплением в последних
мелкогранулярных отложений и развитием оссификации преимущественно
за счет остеобластической реакции стромы (Wiederberg, 1971). По мнению
Д. И. Головина (1958), эпителиому следует называть не обызвествленной,
а некротизирующейся, так как основным признаком ее является не отло-
жение кальция, а некроз темноклеточной паренхимы. С такой точкой зре-
ния согласен А. К. Апатенко (1973). Развитию способствуют травмы. Опи-
саны семейные случаи (Geiser, I960).
Клиника. Как правило, опухоль одиночна. Из 228 больных, наблюдав-
шихся Forbis и Helwig (1961), у 7 человек было по 2 опухоли, у одного—3
528
и еще у одного—4. Wong и соавт. (1972) наблюдали множественную пило-
матриксому. У девочки на протяжении четырехлетнего периода, с 6 до
10 Лет, появились на ногах и руках 5 опухолевидных образований. Пило-
матриксома обычно шаровидной или овальной формы. Расположена в глу-
боких слоях кожи, вначале из-за небольших размеров почти не выступает
над уровнем кожи. При длительном многолетнем течении увеличивается
до нескольких см в диаметре. Характерной особенностью ее является вы-
раженная плотность. Опухоль не спаяна, подвижна, в большинстве слу-
чаев покрыта неизмененной или реже слегка покрасневшей кожей. Встре-
чаются пигментные формы. Casers и соавт. (1974) наблюдали их у 3 из 37
больных, Forbis и Helwig (1961)—у 19 из 240 больных. Субъективных рас-
стройств, как правило, не вызывает. При надавливании опухоль может
быть болезненной, иногда больные отмечают зуд или жжение. Располагается
чаще на лице, волосистой части головы, шее, бедрах, реже на плечах, ту-
ловище. Forbis и Helwig (1961) не нашли ни одного случая локализации
опухоли на ладонях и подошвах. Диагноз во всех случаях требует гистоло-
гического подтверждения, так как эпителиома обызвествленная зачастую
рассматривается как атерома, фиброма, сальножелезистая киста и другие
новообразования. Только в одном из 50 случаев эпителиомы, по наблюде-
ниям Wiedersberg (1971), был поставлен правильный клинический диагноз.
В. Г. Рощин (1966) сообщает о 4 больных с локализацией опухоли в обла-
сти век, клинически расцененной как атерома. И по наблюдениям Hauss
(1962), атерома является одним из самых частых заболеваний, диагностиро-
ванных клинически вместо обызвествленной эпителиомы. Иногда ошибочно
ставится диагноз спиналиомы (Вот и Рагга, 1964), даже после гистологиче-
ского исследования.
Развивается преимущественно в детском возрасте, несколько чаще у
женщин (40,6 и 59,4% у мужчин и женщин, соответственно, по данным
Moehlenbeck, 1974, касающимся материалов литературы и наблюдений
автора, всего 99.4 больных). 36% больных, наблюдавшихся Forbis и Helwig
(1961), были моложе 20 лет, 43%—от 20 до 30 лет. По данным Moehlenbeck
(1973), 40% опухолей развивается до 10 лет и более 60%—до 20-летнего
возраста.
Родионова Н. А., Цыркунов Л. П. (1983) описали больного с разви-
тием опухоли в 55-летнем возрасте. Течение длительное (до 50 лет, по на-
блюдениям Wiederberg, 1971), опухоль растет медленно; Swerlick и соавт.
(1982) описали 6 случаев быстро растущей пиломатриксомы, что является
необычным. Изъязвляется редко. Инвазивная опухоль рассматривается
как исключение (Forbis и Helwig, 1961), после удаления может
рецидивировать, при этом приобретать черты базалномы (Lopansri et al.,
1980). А. П. Прандецкий и А. К. Юзвинкевич. (1969) описали развитие
спиналиомы в зоне пиломатриксомы, Gould и соавт. (1984)—метастаза в
легких, возникшего через 4 года после первого удаления рецидивирующей
пиломатриксомы спины. Мы также наблюдали случай метастазирующей
обызвествленной эпителиомы. Общее состояние больных обычно не страдает.
Runne и соавт. (1982) описали женщину 42 лет с дистрофической миото-
34 Клиническая дерматология 529
нией Curschmann—Steinert, у которой были множественные обызвествлен-
ные эпителиомы. Особенностями эпителиом, ассоциирующихся с миото-
нией дистрофической, считается также частая семейная аггрегация, раз-
витие в более позднем возрасте.
Гистопатология. Опухоль окружена в большинстве случаев оболочкой,
состоит из 2 типов клеток: по периферии из базофильных, представляющих
собой небольшие клетки со скудной цитоплазмой, нечеткими границами,
резко базофильным ядром, и в центре из теневых, имеющих более Четкие
границы, чем базофильные, и неокрашенное ядро. В длительно существую-
щих очагах базофильных клеток становится мало; кальцификация и осси-
фикация, напротив—более выраженными (Peterson и Hult, 1964). Очаги
ороговения, некроза, часто обызвествленные, иногда окостенение. Могут
встречаться структуры, напоминающие незрелые волосы. При пигментных
формах обнаруживается меланин в теневых клетках и клетках стромы, а
иногда в дендритических меланоцитах (Cazers et al., 1974).
Дифференциальный диагноз. Следует проводить с фибромами, цилинд-
ромой, атеромой, сальножелезистыми и эпидермоидными кистами.
Лечение. Хирургическое. Может рецидивировать.
ЛИТЕРАТУРА
Casers J. S., Okun М. R., Pearson S. Н. Pigmented calcifing epithelioma.— Arch.
Derm., 1974, 110, 5, 773—774.
Runne U., Chief G. H., Zentner J. Multiple Pilomatrixome als symptom der Myotonia
dystrophica Curschmann — Steinert.— Hautarzt, 1982, 33, 5, 271—275.
Surrlick R. A., Cooper P. H., Mackel S. E. Rapid enlargement of piloma trixoma.—
J. Am. Acad. Derm., 1982, 7, 1.
ЭПИТЕЛИОМА СПИНОЦЕЛЛЮЛЯРНАЯ (epithelioma spinocellulare).
Синонимы: спиноцеллюлярная карцинома, планецеллюляриая эпи-
телиома, спиналиома.
Этиология и патогенез. Спиналиома представляет собой злокачествен-
ную опухоль (карциному), развивающуюся в эпидермисе или придатках
кожи (Головин Д. И., 1982), обладающую инвазивным ростом, склон-
ностью к раннему метастазированию. Клетки ее имеют тенденцию к орого-.
вению, неодинаковую у разных больных. Различают ороговевающий и не-
ороговевающий плоскоклеточный рак кожи. Менее дифференцированные
формы обычно являются более агрессивными (Фалилеев Г. В., 1979; Lund,
1965; Даниель-Бек К- В. и Колобяков А. А., 1979). Провоцирующими
факторами могут служить воздействие канцерогенных веществ (каменно-
угольная смола, пек, мышьяк и др.), интенсивная инсоляция, лучевая
энергия, травмы, ожоги и т. д.. Среди факторов, способствующих разви-
тию рака полового члена, основное значение придается фимозу (Айвазян
А. В., 1967), рака нижней губы—курению (Тазуев А. Ш., 1976). Опухоль
может развиться на клинически неизмененной коже, но чаще возникает на
фоне самых разнообразных патологических соостояний: лучевые поврежде-
ния кожи (Tempel et al., 1970; Козбагаров С. Г. и Фелькер А. Я.', 1973),
530
болезнь Боуэна, эритроплазия Kefipa(Schoefinius et al., 1974), туберкулез-
ная волчанка, язвенная или изъязвляющаяся плоская форма, особенно
после облучения (Wagner и Pilchowski,. 1959; Санталова О. В., 1960; Да-
даева А. М. и соавт., 1981), скрофулодерма (Кардашенко Б. Я. и соавт.-,
1981), актинические изменения кожи (Szabo, 1975), пигментная ксеродерма
(Moss et al., 1965), остроконечные кондиломы (Е. Забель и соавт., 1980),
буллезный эпидермолиз (Carapeto et al., 1982), порокератоз Мибелли (Eh-
lers и Rothe, 1971; Oberste-Lehn и Moll, 1968), рубцы после вакцинации
(Marmelzat, 1968), профессиональные изменения кожи (Friederich и Schnel-
len, 1965), флоридный папилломатоз полости рта (Капее, 1969), красный
плоский лишай, в том числе в полости рта (Kronenberg et al., 1971; Fulling,
1973; Dobleleer et al., 1982), хронический кандидоз полости рта (Eyre и
Nally, 1971), споротрихоз (Gottron и Nikolowsky, 1957), гидраденит (Donsky
и Mendelson, 1964), крауроз вульвы и полового члена, кератоакантома, вер-
руциформная эпидермодисплазия (Schimpf, 1979), идиопатическая атрофия
кожи (Swanbeck и Hillstrom, 1969), красная волчанка (Винокуров И. Н. и
Хаймовский Г. О., 1963; И. В. Старцев и соавт., 1969; Лысенко О. В. и
Ильин И. И., 1979; Profirov, 1964); хронические язвы голеней (Gertler,
1964), псориаз (Коновалов Р. В. и Смирнов Ю. П., 1978; Завадский В. Н.
и Есенин А. А., 1982) и другие самые разнообразные хронические воспа-
лительные, процессы (Бабаянц Р. С. и соавт., 1973; Кадыров Ф. А. и Ланин
А. А., 1967; Баран Л. А. и соавт., 1972).
Verdich (1979) описал развитие спиналиомы у 2 больных грибовидным
микозом, которым в течение 3 лет проводилось лечение метоксаленом и
длинноволновыми ультрафиолетовыми лучами. Опухоль развилась на здо-
ровой коже. Одному из больных до назначения Пува-терапии применяли
кортикостероиды и тио-теф (местно), рентгеновские лучи; второму—толь-
ко кортикостероидные мази. Имеются сообщения о развитии плоскокле-
точного рака кожи после Пува-терапии больных псориазом (Tam et al.,
1979). О значимости как фактора риска длительной иммуносупрессивной
терапин свидетельствуют случаи спиналиом, развившихся у больных пос-
ле пересадки почки (Westbury и Stone, 1973). Lovell и соавт. (1980) наблю-
дали развитие опухоли, гистологически имеющей черты спиналиомы и ба-
залиомы, в очагах эрозивного пустулезного дерматоза на волосистой части
головы, возникшего после травмы, по поводу которого в течение 50 лет
применялась мазь, содержащая ртуть. Vitaliano и Urbach (1980) обращают
особое внимание на такие факторы риска, как способность к загару и воз-
раст. rto их мнению, способность загорать лучше предупреждает развитие
опухоли, чем черный цвет кожи. По мнению Swanbeck (1971), пока не
получено доказательств роли вирусов при плоскоклеточном раке кожи.
Клиника. Наиболее частой формой спиналиомы является нодулярная
или поверхностная, при которой возникает вначале небольших размеров,
плотной консистенции безболезненное опухолевидное образование, по цве-
ту не отличающееся от нормальной кожи или розоватой окраски. Поверх-
ность гладкая или слегка гиперкератотична, иногда папилломатозна,
может быть покрыта чешуйками и корочками. Характерен довольно быст-
34* 531
Рис. 342. Эпителиома спиноцеллюлярная.
Рис. 343. Эпителиома
спиноцеллюлярная.
Рис. 344. Спиноцел-
люлярная эпителиома
хадони.
\
Рис. 345. Спиноцел-
люлярная эпите.мома
нижней губы.
532
Рис. 346. Спиноцеллюлярная
эпителиома мошонки.
Рис. 347. Спиноцеллюлярная
эпителиома полового члена.
Рис. 348. Веррукозная
эпителиома.
Рис. 349. Эпителиома
у больного псориазом.
533
Рис. 350. Люпус-карцинома.
Рис. 351. Эпителиома спиноцеллюлярная
у больного красным плоским лишаем.
Рис. 352. Солитарная
трихоэпителиома.
рый рост опухоли, усиление окраски, приобретающей насыщенные ярко-
красные или красновато-коричневатые тона, инфильтрирующий рост в
глубину, распад с образованием глубокой, болезненной язвы, частично
покрытой корками (язвенно-ннфильтрирующая карцинома). Края язвы
плотные, развороченные, дно неровное, зернистое, иногда папилломатоз-
ное, легко кровоточит (рнс. 342—356). Опухоль становится неподвижной,
может вызывать обширные разрушения подлежащих тканей, включая ко-
сти, сосуда', жизненно важные органы, что, наряду с метастазированием,
представляет собой проявление малигнитета (Schimpf, 1979). Реже ветре-
534
Рис. 353. Трихоэпителиома
у больной с цилиндромой.
Рис. 354. Трихоэпителиома.
чается глубокая форма спиналиомы,
располагающаяся в нижележащих сло-
ях кожи, инфильтрирующая подкожную
клетчатку и изъязвляющаяся с обра-
зованием язвенного ракового пораже-
ния. Эта форма может протекать с
выраженными воспалительными изме-
нениями, что придает сходство с пио-
генными процессами (Мордовцев В. Н.,
1970). Иногда плоскоклеточный рак
кожи имеет вид желтовато- или красно-
вато-коричневатого очага, поверхность
которого бугриста, с гиперкератозом,
бородавчатыми разрастаниями, покрыта
корками (веррукозная карцинома).
Веррукозная форма растет медленно,
по мнению Dimitrowa и соавт. (1983),
редко метастазирует. Папилломатозная,
фунгоидная форма спиналиомы хотя и PliC^355_ Эпителиома спи ноцеллюляр
близка по клинической картине к вер- на пом>вим члене
рукозной, но отличается большими
размерами, вегетирующими разрастаниями типа цветной капусты, быстрым
ростом, большой склонностью к распаду и метастазированию. Спиналиома
может иметь форму кожного рога. Вариантом веррукозной спиналиомы.
535
по всей вероятности, является epithelioma cuniculatum, располагающаяся
на стопах в виде бородавчатой, экзофитно растущей опухоли (Brown и
Freeman, 1976 и др.). В ее развитии придается значение хронической трав-
ме, воспалению, вирусу человеческой папилломы, препаратам мышьяка
(Wilkinson et al., 1981; Owen et al-, 1978; Seehafer et al., 1979; Klima и
соавт., 1980; Levi et al., 1983). В межпальцевых складках стоп спиналиома
может быть в виде трещин, язвенных, язвенно-вегетирующих поражений
(Baptista и Freitas, 1976).
На слизистой полости рта спиналиома встречается в виде язвенной
формы, папиллярных и плотных узловатых образований (Пашков Б. М
и соавт., 1970). В 78% случаев спиналиома располагается на красной кай-
ме нижней губы, в 9,6%—на языке, в 4,4%— на слизистой щек, в 4%—
на дне полости рта, в 2%—на нёбе, в 2%—в области альвеолярных отро-
стков челюстей, на красной кайме верхней губы и в других областях. При
локализации на языке он становится плотным, малоподвижным, болезнен-
ным. По мнению Mahele (1968), прогноз при раке, располагающемся во
рту, значительно хуже, чем при локализации его на нижней губе, и прибли-
жается к таковому при меланоме. На половых органах спиналиома чаще
развивается на фоне лейкоплакии или крауроза, проявляется в виде быстро
изъязвляющихся бугристых разрастаний или плоских гиперкератотиче-
ских, инфильтрированных очагов. Спиналиома располагается преимущест-
венно на коже лица, особенно на нижней губе (51% всех опухолей этого
типа, располагающихся в области головы, по данным Waissmann и Konz,
1979), полуслизистых и слизистых оболочках, в области половых органов,
ануса, на тыле кистей. Значительно реже—на ногах и особенно на руках
(исключая кисти) и туловище. На ладонях (Weinrauch et al., 1983) спи-
налиома бывает редко.
По данным Б. И. Зак и Т. И. Бакулиной (1975), первичный рак мо-
шонки составляет 0,18% всех новообразований мочеполовой сферы, из
13 случаев 11 были спиналиомой, 2—базалиомой.
Частота плоскоклеточного рака нарастает с возрастом (Becker и Bren-
nan, 1961). Обычно он встречается у лиц пожилого возраста, чаще у муж-
чин. Из 751 больного спиноцеллюлярной эпителиомой, данные о которых
представлены Jung и соавт. (1968), было 509 мужчин и 242 женщины. Мета-
стазирование может наступить уже в ранние сроки. Обычно—в регионар-
ные лимфоузлы, редко в кости и внутренние органы (Фалилеев Г. В.,
1979). Согласно данным Moller и соавт. (1979), у больных спиналиомой не
выявлено учащения рака внутренних органов, хотя, как указывают авторы,
об этом имелись сведения в литературе, особенно в американской. По Lund
(1965), плоскоклеточный рак кожи, развившийся на актинически изменен-
ной коже, метастазирует реже, чем возникший на рубцах, ожогах, хрони-
ческих язвах, мышьяковом кератозе, рентгеновском повреждении кожи.
Moller и соавт. отмечают, что наиболее опасны в смысле метастазирования
спиналиомы, развившиеся на фоне воспалительных и дегенеративных из-
менений кожи, затем располагающиеся на полуслизистых. Реже всего
метастазы, по. их мнению, наблюдаются при первично кожном раке.
536
Carcinoma caniculatum обладает преимущественно локальным инва-
зивным ростом, но метастазы в регионарные лимфоузлы не исключаются
(Мс Кее et al., 1981; Neill et al., 1984). (
Указывается (Turner и Callen, 1981) на резкое усиление агрессивности
спиналиомы у больных лимфомами, что связывается как с иммунными на-
рушениями, присущими этой патологии, так и с иммунодепрессивной те-
рапией. При раке губы соотношение мужчин и женщин, по данным Wagner
(1974), колеблется от 4:1 до 20:1. Из 241 больного с раком нижней губы,
в выборке Jung и соавт. (1968), было только 8 женщин. По данным М. П.
Федюшина (1955), из больных раком губы, с гистологически установленным
диагнозом, было 95% мужчин и 4,9% женщин. Большей склонностью к
рецидивам после хирургического удаления, по данным Dzubow и соавт.
' (1982), обладают опухоли у лиц мужского пола, развившиеся в более мо-
лодом возрасте, большего размера, располагающиеся на нижних конеч-
ностях.
Гистопатология. Глубоко проникающие в дерму пролифераты раковых
клеток с эозинофильной протоплазмой, напоминающих клетки шиповид-
ного слоя; отсутствие межклеточных связей, гиперплазия и гиперхрома-
тоз ядер, полиморфизм, увеличение числа митозов, множественные ати-
пические митотические фигуры, ороговение отдельных клеток с образова-
нием роговых «жемчужин», реакция стромы в виде скопления лимфоцитов,
плазматических клеток, гистиоцитов, полинуклеаров. Интенсивность реак-
ции стромы зависит от стадии процесса, в далеко зашедших случаях—она
незначительна и почти полностью вытесняется опухолевыми клетками.
Дифференциальный диагноз. С сенильным кератозом, базалиомой,
хронической пиодермией, туберкулезной волчанкой, первичным сифили-
сом, кератоакантомой, псевдоэпителиоматозной гиперплазией.
Лечение. Хирургическое, лучевое.
ЛИТЕРАТУРА
Кардашенко Б. %., ШахтмеОстер И. Я-, Алименко М. М. Люпус-карцинома.
Веста, дерматол. и венерол. 1981. 2, 53—54.
Коновалов Р. В., Смирнов Ю. П. Развитие множественного рака кожи у больных
псориазом. Вести, дерматол. и венерол., 1978, 9, 47—49
Лалаева А. М., Левинтова Г. И., Деменкова Н. В. Люпус-карцинома. Вести, дер-
матол. и венерол., 1981, 7, 53—54.
Даниель-Бек К- В., Колобяков А. А. Злокачественные опухоли кожи и мягких
тканей. М. Медицина, 1979.
Забель Е., Люгавская Д., ПшибораЛ., Карась 3. Бородавчатый спиноцеллюляр-
ный рак, развившийся на месте остроконечных кондилом. Веста, дерматол. и венерол.,
1980, 12, 51—52.
Завадский В. Н., Есенин А. А. Множественный плоскоклеточный рак кожи у боль-
ного псориатической эритродермией. Вести, дерматол. и венерол., 1982, 2, 65—66.
Зоек Б. И., Бакунина Т. И. Рак кожи мошонки. Вести, дерматол. и венерол., .1975,
12, 54—57.
537
Тазуев А. Ш. К патогенезу рака нижней губы. Вести, дерматол. и веиерол., 1976,
10, 68—70.
Baptista А. Р., Freitas L. С. Epitheliomas spino-cellulaires interdigitaux des piens.—
Ann. Derm. Syph., 1976, 102, 5—6, 401—408.
Brown S., Freeman G. Epithelioma cuniculatum.— Arch, dermatol., 1976, 112, 9,
1295—1296.
Carapeto F. Y., Raster J. A., Martin J., Agurrura J. Recessive dystrophic epider-
molysis bullosa and multiple squamous cell cercinomas.— Dermatologica, 1982, 165, 39—46.
Dimitrowa J., Obreschkowa E., Balabanowa M-, Zankow W. Carcinoma verrucosum
Z. Hautkrkh., 1983, 58, 1, 39—47.
DobbeleerG., Brasseur T., Ledoux M. Epithelioma spipocebulaire du talon se develop-
pant sur un' lichen plan ersif — Dermatologica, 1982, 165, 5,/479—479.
Dzubow L. M-, Rigel D. S., Robins P. Risk factors for local recurrence of primary
cutaneous squamous cell carcimomas: treatment by microscopically controlled excision.—
Arch, dermatol., 1982, 118, 11, 900—902. /
Klima M., Kurtis B., Jordan P. H. Verrucous carcinoma of skin. J. cutan. pathol.,
1980, 7, 88—98.
, Led L., Bratina G., Sala G. P. et al. Considerazioni su di un caso di epjtelioma cuni-
culatum.— Giorn. it. derm., vener. 1983, 118, 159—162.
Mahrle Y. Das Lippenkarzinom und seine operative Behandlung.— Hautarzt, 1978,
29, 5, 251—258.
Me Кее P. H., Wilkinson J. D., Corbett et al. Carcinoma cuniculatum: a case meta-
stasizing to skin and lymph node.— Clin. egp. derm., 1981, 6, 6. 613—618.
Moller R., Reymann F. Hou-Gensen K. Metastases in dermatological patients with
squamous cell carcinoma.— Arch, dermatol., 1979, 115, Q 703—705.
Moller R., Nielsen A., Reyman F., Hou-Gensen K- Squamous cell carcinoma of
the and internal malignant neoplasms.— Arch, dermatol. 1979, 115, 3, 304—305.
Neill S. M-, Calnan C. D., Rahim G. F. Carcinoma cuniculatum Clin: exp. derm., '
1984, 9, 3, 309—311. /
Seehafer J. R., Muller S. A., Dicken C.iH. et al. Bilateral verrucous carcinoma of
the feet.—Arch, dermatol. 1979, 115, 10, 12224-1223.
Szabo P. Klinik und Rontgentherapie voh Lippencarcinomen.— Hautarzt, 1975, 26,
10, 524—528.
Tam D. W., van Scott E. J., Urbach F. Bowen's disease and squamous cell, carci-
noma.— Arch, dermatol., 1979, 115, 2, 203—204.
Turner J. E., Callen J. P. Aggressive behavior of squamous cell carcinoma in a patient
with proceeding lymphocytic lymphoma.— J. Am. Acad. Derm., 1981, 4, 4, 446—450.
Verdich J. Squamous cell carcinoma.— Arch, dermatol.. 1979. 115, 11, 1338—1339- '
Vitaliano P. P., Urbach F. The relative importance of risk factors in nonmelanoma
carcinoma.— Arch, dermatol., 1980, 116, 4, 454—457.
Wagner G. Epidemiologie der Malignome der Mundschleimhaut und des Rachens.—
Hautarzt, 1977, 25, 3, 110—118.
Weinrauch L., Peled J., Goldberg et al. Squamous cell carcinoma of the palm.— Der
matologica, 1983, 166, 89—91.
Weissmann J., Konz B. Dermatologische Behandlungsergebnisse bei spinocellularen
538
Karzinomen. In: Operative Dermatologie. Springer, Berlin—Heidelberg, New York, 1979,
207—213.
Wilkinson J. D., Me Кее P. H., Black M. M. et al. A case of carcinoma cunicula-
tum with co-existent viral plantar wart.— Clin. exp. dermatol., 1981, 6, 6, 619—623.
ЭРИЗИПЕЛОИД (erysipeloid).
Синонимы: свиная рожа, псевдорожа, ползучая эритема.
Зоонозное заболевание диких и сельскохозяйственных животных (пре-
имущественно свиней), а также человека. Первые сведения об эризипелои-
де в СССР принадлежат В. К- Стефанскому и А. А. Гринфельду (1930) Они
наблюдали в Одессе вспышку заболевания у рабочих рыбоконсервных фаб-
рик, домохозяек и поваров.
‘ Этиология. Возбудитель эризипелоида erysipelothrix rhusiopathiae
впервые выделил у человека A. Rosenbach в 1884 г.; он предложил термин
эризипелоид для отграничения этого заболевания от рожистого воспале-
ния стрептоккоковой этиологии. Возбудитель эризипелоида—грамполо-
'жительная палочка, может сапрофитировать на различных сортах мяса и
рыбы. Бактерионосительство при эризипелоиде имеет широкое распрост-
ранение в природе среди домашних животных и птиц; исследование клини-
чески здоровых свиней показало, что носительство эризипелотриксов со-,
ставляет от 1J jio 35—50% (Wood, -J974). Эризипелоид имеет наибольшее
распространение среди мясников, работников боен, рыбаков, продавцов
мяса и рыбы, поваров, охотников и других, имеющих дело с продуктами
животного происхождения (Токарева К. Н., 1969; Антоньев А. А. и
соавт., 1971; Errlich, 1944; Nelson, 1955; Barnett J., Estes S. etaL, 1983 и др.)
Клиника. У 4/5 больных возникновению профессионального эризипе-
лоида предшествует производственная микро- или макротравма. А. А.
Антоньев и соавт. (1974) выделяют кожный эризипелоид (ограниченный и
распространенный), острый и хронический рецидивирующий артрит, анги-
нозную, кишечную и септическую формы. При ограниченном кожном эри-
зипелоиде через 2—3 суток (инкубационный период) после попадания в ко-
жу возбудителя на месте повреждения эпидермиса появляется краснота и
—припухлость (редко пузырь), сопровождающиеся зудом, жжением, иногда
дергающими болями. Очаг поражения представляет собой багрово-красную
инфильтрированную бляшку округлых или овальных очертаний, резко
отграниченную от здоровой кожи. Бляшка эксцентрично растет на 1—3 см
в сутки, достигая величины от 3—4 до 5—10 см в диаметре в течение 3—5
дней. По мере роста бляшки центр ее бледнеет; приобретает синюшный
оттенок, в нем уменьшается отечность и инфильтрация. Край очага пора-
жения имеет вид яркого отечного валика, приподнятого над окружающей
здоровой кожей. В дальнейшем края очага постепенно сглаживаются.
Локализация очагов поражения на пальцах рук (чаще указательного или
большого) и кистях отмечается в 96—98% случаев. Значительно реже эри-
зипелоид встречается в области запястий и предплечий. Как правило, по-
ражение возникает на одной конечности. Возможна гематогенная и лимфи-
генная диссеминацня процесса. При распространенном кожном эризипе-
539
лоиде высыпания захватывают предплечья, плечи, туловище и нижнйе
конечности.
Эризипелоидный артрит возникает обычно через 1—2 суток после по-
явления очагов на коже и наблюдается у 70—85% больных. Поражаются
преимущественно межфаланговые суставы. Отмечается резкий отек, сги-
бательная контрактура, движения в суставах становятся резко болезнен-
ными, паЛец приобретает веретенообразную форму.
Редко встречающаяся злокачественная форма (септическая) эризи-
пелоида характеризуется хроническим течением, тяжелыми общими явле-
ниями, поражением внутренних органов, температурой гектического ха-
рактера, может заканчиваться летально.
Ангинозная форма эризипелоида начинается остро, с ознобом, лихо-
радкой, болями в горле, общей слабостью. Отмечается яркая гиперемия
нёба, язычка, дужек и миндалин. На коже туловища и верхних конечно-
стей часто появляются крупные эритематозные пятна. При кишечной
форме, которая начинается также остро, с ознобом, лихорадкой, рвотой,
поносом, болями в животе, на коже появляется распространенная круп-
нопятнистая розовая сыпь. Заболевание имеет некоторую сезонность пре-
имущественного развития в летние и осенние месяцы. Возможны рецидивы
и повторные заражения, так как после перенесенного эризипелоида им-
мунитета нет.
Прогноз, как правило, благоприятный. Продолжительность заболе-
вания от 2 до 6—8 Недель.
Гистопатология. Серозно-инфильтративное воспаление кожи с отеком
всех слоев и периваскулярной круглоклеточной (преимущественно лимфоид-
ной) инфильтрацией. В эпидермисе—акантоз и отек.
Дифференциальный диагноз. Следует проводить с рожистым воспале-
нием, паронихией, панарицием, многоформной экссудативной эритемой,
мигрирующей эритемой.
Лечение. Антибиотики—пенициллин, тетрациклины, левомицетин. На-
ружно—линимент синтомицина, гелиомициновая, неомициновая, ихтиоло-
вая мази. Ультрафиолетовые облучения.
Профилактика. Своевременная обработка мелких травм у лиц, занятых
обработкой мяса, рыбы.
ЛИТЕРАТУРА /
Антоньее А. А., Чайковский В. Т., Сосонкин И. Е. К вопросу об эризипелоиде.—
Вести. Дерматол. и венерол., 1974, 6, 24—30.
Ковалев Г. К- Эризипелоид (эпизоотология, эпидемиология, клиника и профилакти-
ка).— Сов. мед., 1979, 12, 66—70.
Barnet J-, Estes S. et al. Erysipeloid. J. Amer. acad. dermatol., 1983, 9, 1, 116—123.
Braun W. Dermatologie. Ein Lehrbuch fur Studenten.— Berlin, 1975. z
ЭРИТЕМА ВОЗВЫШАЮЩАЯСЯ ДЛИТЕЛЬНО ПРОТЕКАЮЩАЯ
(erytnema elevatum diutinum).
Впервые заболевание описали Hutchinson (1888) и Bury (1889). Однако
540
дали подробное клиническое описание и предложили~название erythema
elevatum diutinum Williams и Crocker (1894, 1902).
Этиология и патогенез. Описанные в литературе 2 типа возвышающей-
ся эритемы являются по существу различными стадиями единого процес-
са: 1) тип Hutchinson (ранний период заболевания) гистологически, по-
видимому, близок к васкулиту (периваскулярный инфильтрат, состоящий
в основном из нейтрофилов и лимфоцитов); 2) тип Bury (более старые по-
ражения гистологически напоминает кольцевидную гранулему с развитием
фиброза.
Учитывая изменения, обнаруженные в артериолах, венулах и капил-
лярах, возвышающуюся длительно протекающую эритему многие авторы
(Каралицкий Е. М., 1966; Haber, 1955; Marshall, 1960; Laymon, 1962;
Mraz et al., 1967; Cream et al., 1971; Stanson и Wilson, 1973; Sonnichsen и
Zabel, 1976; Kovary et al., 1977; Wolff et al., Statham et al., 1978, и др.)
относят к доброкачественным формам аллергических васкулитов. Некото-
рые авторы (Селисский А. Б., Попхристов, 1973; Nicolau и Badanoiu, 1960;
Little и др.) рассматривают это заболевание как разновидность кольце-
видной гранулемы, с этим не согласны Garuier (1952), Haber (1955), Lugt
(1959), Laymon (1962) и др. Наш клинический опыт позволяет рассматривать
возвышающуюся эритему как самостоятельное заболевание, важную роль
в патогенезе которой могут играть лекарственная аллергия и токсико-ин-
фекционные воздействия (грипп, ангина, ревматизм и др.). Имеются отдель-
ные сообщения (Duperrat и Rappaport, 1959; Wolf, 1971; Stevanovic, 1971)
о сочетании стойкой возвышающейся эритемы с парапротеинемией. Mor-
rison (1977) обнаружил у больных криоглобулины (IgM— IgG).
Wolff, Scherer, Braun-Falco (1978) на основании иммуно-электронно-
микроскопических исследований поддерживают гипотезу, согласно которой
в патогенезе заболевания имеет значение реакция типа Артюса, вызванная
бактериальным антигеном.
Клиника. Заболевание чаще встречается у мужчин; возраст больных
колеблется от 5 до 75 лет, но в большинстве случаев заболевание развива-
ется между 30 и 60 годами.
Клинически заболевание проявляется узелками величиной с чечевицу,
которые, сливаясь, образуют бляшки размером до 5—10 см. Очаги поражения
неправильной конфигурации, иногда цирцинарные, красновато-синюшного,
а в дальнейшем буровато-красного цвета, плотноэластические, гладкие,
поблескивающие, с четко очерченными краями значительно возвышаются
над окружающей кожей; субъективные «ощущения, как правило, отсутст-
вуют (рис. 356, 357, 358). Высыпания располагаются более или менее сим-
метрично, распределяются беспорядочно и локализуются на разгиба-
тельных поверхностях конечностей, на локтях и коленях, на тыле кистей и
разгибательной поверхности пальцев рук, реже—на спине, шее, ягодицах,
животе и ушных раковинах. Редко на поверхности очагов поражения фор-
мируются буллезные геморрагические высыпания с последующим изъязвле-
нием. Различные показатели «рутинных» лабораторных исследований нор-
мальные. Общее состояние больных не нарушается. Выраженных измене-
541
Рис. 358. Эрат ча возвышающаяся
длительно протекающая.
Рис. 357. Эритема возвышающаяся дли-
т?..ьно протекающая.
ний со стороны внутренних органов
нет. Заболевание протекает годами.
Stonton и Wilson (1973) описали
“ужчину 55 лет, у которого заболе-
вание длилось 25 лет с частыми ре-
миссиями и рецидивами. На месте
разрешившихся высыпаний остается
бурая пигментация.
Гистопатология. В ранних ста-
диях заболевания определяется кар-
тина аллергического васкулита. Со-
суды в дерме резко расширены, эндо-
телий отечный, сосудистая стенка
утолщена, гиалинизирована и окру-
жена периваскулярным инфильтра-
том, состоящим из нейтрофилов, лим-
фоцитов, гистиоцитов, иногда с при-
месью эозинофилов и плазматических
клеток. Коллагеновые волокна гиа-
лииизированы. Фибриноидная деге-
нерация сосудистых стенок. В более
поздних стадиях наблюдаются явле-
ния фиброза и отложение (вторич-
542
ное) в дерме липидов (экстрацеллюлярный холестероз). В эпидермисе
отмечается небольшой акантоз и гиперкератоз. ДУ о Iff и соавт. (1978) на ос-
новании электронной микроскопии, гистологии и иммунофлюоресценции
обнаружили лейкоцитокластический васкулит у больной с типичной
клинической картиной стойкой возвышающейся эритемы.
Дифференциальный диагноз. Следует проводить с кольцевидной центро-
бежной эритемой Дарье, саркоидозом, красным плоским лишаем, лепрой,
хронической папулезной крапивницей, липоидным некробиозом, кольце-
видной гранулемой, герпетиформным дерматитом Дюринга.
Лечение. Общеукрепляющие средства, витамины, аутогемотерапия,
кортикостероидные мази под окклюзионную повязку, антигистаминные
препараты. В части случаев эффективны кортикостероидные препараты
внутрь (преднизолон 10—20 мг в сутки). Есть указания на хороший ре-
зультат при применении дапсона (4,4’-диаминодифенилсульфон) (Tanzer
и Lynfield, 1978; Katz et al., 1977).
ЛИТЕРАТУРА
Barriert H., Burean В., LeRoux P. Erythema elevatum diutinum.— Ann. Dermatol.
Venereol., 1977, 194, 1, 75—76.
Fort S., Rodman 0. Erythema elevatum diutinum. Response to dapsone.— Arch,
dermatol., 1977, 113, 9, 819—822.
Losonczy Z. Erythema elevatum et diutinum.— In: XIII national Congress Hungerian
Soc. Dermatology. Budapest, March—April., 1981, p. 237—238.
Statham B., Greenwood R., Cotterill J. Erythema elevatum diutinum—a reportof
two unisual. pafieuts.— Clin. egp. dermatol., 1983, 8, 5, 549—552.
Wolff H., Maciejewski W., Scherer R. Erythema elevatum diutinum.— Arch. Der-
matol. Res., 1978, 261, 1, 7—16.
Wolff H., Scherer R., Braun-Falco 0- Erythema elevatum diutinum. Immunelektro-
nenmikroskopische Untersuchung der leukocytoklastischen Vasculitis in einer Intrakutanreak-
tion mit Streptokokkenantigen.— Arch. Dermatol. Res., 1978, 261, 1, 17—26.
ЭРИТЕМА ДИСХРОМИЧЕСКАЯ СТОЙКАЯ (erythema dyschromicum
perstans).
Синонимы: пепельно-серый дерматоз, хроническая фигурная мелано-
дермическая эритема, идиопатическая пятнистая множественная пиг-
ментация, стойкая пятнистая пигментация.
Это заболевание впервые было описано Рамиресом (Ramirez) в 1957 г.
в Эль-Салвадоре под названием «пепельно-серый дерматоз» (ashy der-
matosis, cenicienta). -Впоследствии Convit и Bodriguez (1961) описали
5 больных из Венесуэлы. Они расценивали это заболевание как новый ва-
риант эритемы perstans, назвав его «erythema chronicum figuratum melano-
dermicum». Sulzberger обратил внимание на вариабельность окраски, кото-
рая колебалась от пепельно-черной, голубовато-серой до коричнево-серой и
красной. В некоторых случаях он видел гипопигментацию и предложил
название «erythema dyschromicum perstans». В дальнейшем это название
было использовано Convit (1961). Заболевание наиболее распространено в
543
Центральной Америке, имелись сообщения также из США, Японии, Цент-
ральной Европы (Mashima, 1961; Stevenson и Miura, 1966; Knox ё! al.,
1968; Gellin и Hilger, 1974; Peachey, 1976; Badanoiu et al., 1979; Moller-
Veitheer и Goos, 1977; Bleehen и Ebling, 1979; Person и Rogers, 1981; Kos-
tler, 1983 и др.). К настоящему времени в литературе описано более 100
случаев стойкой дисхромической эритемы.
Этиология и патогенез. Наличие асимптомных пигментных пятен, ба-
зально-клеточная дегенерация, воспалительный инфильтрат, недержание
пигмента весьма напоминают pigmentatio macularis perstans японских
авторов. Persob и Rogers (1981) полагают, что «пепельный дерматоз» явля-
ется следствием послевоспалительной гиперпигментации, имеющей в ос-
нове базально-клеточный апоптоз. Гистологическое изучение во всех 9 ими
наблюдавшихся случаях показало вакуолизацию базальных клеток эпи-
дермиса. Прямая иммунофлюоресценция, произведенная в 4 случаях, по-
казала отложение IgM в дермо-эпидермальной пограничной зоне.
В последние годы появились сообщения (Bhutani et al., 1974; Kark и
Litt, 1980; Naidorf и Cohen, 1982 и др.), в которых на основании гистоло-
гических исследований стойкая дисхромическая эритема рассматривается
как клинический вариант красного плоского лишая.
Preissl и Faff (1978) считают, что речь идет об аллергическом процессе,
вызванном пищевым аллергеном, гельминтами, фотозащитными и космети-
ческими средствами, лекарственными препаратами.
Reachey (1976) описал «пепельный» дерматоз у девушки 16 лет, у кото-
рой после иммунизации против краснухи развилась распространенная
пигментация на шее, туловище и проксимальных отделах конечностей.
Предложен термин «лихеноидный меланодерматит» как название груп-
пы различных дерматозов (пигментный лихен, меланодерматит Гофмана и
Габермана, меланоз Риля, пойкилодермия Сиватта, летние солнечные лихе-
ноидные высыпания, эритема дисхромическая стойкая, лихеноидная ме-
дикаментозная реакция, блестящий лишай), характеризующихся лихеноид-
ной тканевой реакцией (Pincus, 1973; Weedon, 1982). Лихеноидная ткане-
вая реакция диагностируется на основании морфологически специфичного
вида повреждение базальных клеток эпидермиса (апоптоза), которое при-
нимает форму вакуольной дистрофии и в своей основе отличается от некроза.
По мнению Kostler (1983), лихеноидная тканевая реакция лежит в основе
этого полиэтиологического заболевания.
В кожной клинике ЦКВИ мы наблюдали 3 бЬльных с дисхромической
эритемой, у которых клинически имелись высыпания, напоминающие ле-
карственные высыпания или пигментную форму красного плоского лишая
(lichen demblee). У двух больных начало пигментации совпало с приемом
лекарств. По нашему мнению, эритема дисхромическая стойкая представ-
ляет заболевание, при котором иммунные (аллергические) механизмы игра-
ют значительную роль в патогенезе.
Клиника. Основным клиническим признаком заболевания являются
множественные пепельно-серые пигментные пятна. Заболевание встреча-
ется в любом возрасте (от 4,5 до 75 лет), одинаково часто у мужчин и жен-
544
Рис. 359. Стойкая дисхромическая
эритема (пепельный дерматоз).
Рис. 360. Стойкая дисхромическая
эритема (тот ясе больной).
щин. Поражаются все национальности, однако большинство случаев опи-
сано в Латинской Америке.
Заболевание может возникнуть внезапно и проявляться множественны-
ми высыпаниями или развиваться медленно из одного или нескольких эле-
ментов.
Без каких-либо предшествующих элементов на кожном покрове по-
являются округлые или овальные пятна диаметром от нескольких мм до
детской ладони, не сливающиеся между собой, аспидной или грязно-бу-
рой окраски, иногда с некоторой голубизной или с легким синюшным от-
тенком, не зудящие, не шелушащиеся, не изменяющиеся при трении (рис.
359, 360).
В ранней стадии заболевания обнаруживается нерезко выраженная
краевая эритема (активный край). Высыпания локализуются на шее, пле-
чах, спине, реже—на передней поверхности туловища и бедрах. Слизи-
стые оболочки не поражаются. Обще$ состояние не нарушается. Патоло-
гия со стороны внутренних органов не выявляется. Субъективные жало-
бы отсутствуют. Пигментация или гипопигментация могут присутствовать
одновременно на одних и тех же пятнах. Течение заболевания длительное,
однако в некоторых случаях оно разрешается самопроизвольно в срок от
2—3 месяцев до 1,5—2 лет.
35 Клиническая дерматология 545
Гистопатология. Наблюдается гидропически-вакуольная дегенерация
базальных кератиноцитов и уменьшение количества меланоцито.в в базаль-
ной области. В верхних слоях дермы присутствие множественных макро-
фагов, нагруженных большим количеством меланиновых зерен таким обра-
зом, что эти зерна полностью маскируют клеточные ядра. В дерме—уме-
ренно выраженные периваскулярные лимфогистиоцитарные инфильтраты.
Электронная микроскопия выявляет поражение кератиноцитов, меж-
клеточный отек, западение (втягивание) десмосом, разрыв базальной мем-
браны и дермальный меланофагоцитоз, в дерме много меланофагов, состоя-
щих из скопления меланосом, заключенных в лизосомальных мембранах
(Stoter et al., 1968; Tschen et al., 1980).
Наличие вакуолизации базальных клеток и недержание пигмента при
ashy dermatosis показывает, что первичный процесс заболевания разви-
вается в районе базальной мембраны (Tschen et al., 1980). Эти авторы по-
лагают, что «пепельный дерматоз» представляет лихеноидную реакцию.
Дифференциальный диагноз. Прежде всего следует проводить с фикси-
рованной лекарственной сыпью, используя данные анамнеза, клиниче-
ского течения заболевания и гистологической картины. Следует также
дифференцировать с пятнистой меланодермией, некоторыми формами крас-
ного плоского лишая, недержанием пигмента, лепрой, пятнистым мастоци-
тозом.
Лечение. Методы лечения дисхромической эритемы не разработаны,
все виды терапин мало эффективны.
ЛИТЕРАТУРА
Badanoiu A., Popesku В., Stocescu / Erythema dyschromicum perstans.— Dermato-
Vener., 1979, 24, 3, 215—223.
Bhutani L., Bedi T., Pandhi R., Wajak W. Lichen planus pigmentosus.— Dermato-
logica, 1974, 149, 1, 43—50. \
Kark E., Litte S. Ashy dermatosis.— a variant of lichen planus? Cutis, 1980, 25, 5,
k 631—633___
Rostler E. Erythema dyschromicum perstans.— Derm. Zschr., 1983, 169, 5, 322—327.
Moller-Veitheer M., Goos M. Erythema dyschromicum perstans.— Hautarzt, 1977,
28, 10, 539—541.
Naidorf K., Cohen S. Erythema dyschromicum perstans and lichen planus.— Arch.
Dermatol., 1982, 118, 9, 683—686.
Keachey R. Ashy dermatosis.— Brit. J. Dermatol., 1976, 94, 2, 227—228.
Tschen J. et al. Erythema dyschromicum perstans.— J- Amer. Acad. Dermatol., 1980,
2, 295—302.
ЭРИТЕМА КОЛЬЦЕВИДНАЯ ЦЕНТРОБЕЖНАЯ ДАРЬЕ (erythema
annulare centrifugum Darier).
Синонимы: стойкая эритема, длительно протекающая фигурная и
кольцевидная эритема.
ош/сая Дзрье в )9)Б г.
Этиология и патогенез. Развитие кольцевидной эритемы патогенетиче-
546
Рис. 363. Центробежная Рис. 364. Центробежная
эритема Дарье. эритема Дарье (гигантская).
35* 547
ски связывают с воздействием токсических, инфекционных, аллергических
факторов, с иммунными и аутоиммунными расстройствами (Shelley, I960;
Aschurst, 1967; Wite и Perry, 1969; Rekant и Becker, 1973; Cream, 1975;
Sonnichsen и Zabel, 1976 и др.). В ряде случаев центробежная эритема яв-
ляется паранеопластическим дерматозом у больных раком внутренних
органов (Belisario, 1973 и др.), лейкозом (Kimmig et al., 1967).
Клиника. Болеют оба пола, несколько чаще мужчины. Заболевание
наблюдается обычно у лиц среднего возраста, но встречается и у детей.
Rekant и Becker (1973), Gianotti и Ermacora (1975) описали эритему Darier
у новорожденных, a Hammar и Ronnerfalt (1977) описали три случая коль-
цевидной эритемы, развившейся у детей в первые несколько недель после
рождения. Заболевание начинается появлением красного цвета пятен, бы-
стро превращающихся в уртикоподобные папулы или бляшки, величиною
с десятикопеечную монету и больше. Дальнейшая эволюция этих очагов
такова: центр их уплощается, розово-красный цвет сменяется ливидным,
затем буроватым; по периферии же продолжается рост элемента в виде
кольцевидного возвышенного валика; таким образом образуются кольца с
большим (15—20 см) диаметром. При слиянии соседних колец появляются
очаги в виде гирлянд, а от частичного разрешения колец—дугооборазные
элементы. Описывают и крупные уртикарно-геморрагические пятна раз-
личной формы и величины, сливающиеся между собой и образующие при-
чудливые узоры на большой поверхности кожного покрова. Просущест-
вовав 2—3 недели, они исчезают, нередко оставляя после себя застойно-
бурую пигментацию. По соседству же появляются новые высыпания,
которые сливаются, образуя причудливые полициклические фигуры. Не-
которые кольца распадаются на фрагменты, каждый из которых продол-
жает свой цикл развития (рис. 361, 362, 363, 364).
В всыпания эритемы Дарье не имеют излюбленной локализации. Они
могут располагаться на лице, шее, груди, спине, животе, пояснице, пле-
чах, предплечьях, стопах и голенях. Зуд выражен в различной степени,
редко бывает очень сильным (наши наблюдения). Заболевание течет хро-
нически, дает частые рецидивы, затягиваясь обычно на много месяцев или
даже лет.
Среди клинических разновидностей центробежной эритемы, наблюдав-
шихся рядом авторов (Штамова Л. В., 1927; МгебровМ. Г. иСинаниМ. Ф.,
1929; Никитина А. Ф. и Большакова Г. М., 1956; Harnack, 1960; Kind,
1975; Sonnichsen и Zabel, 1976 и др.), заслуживают внимания «шелушащая-
ся эритема», сопровождающаяся шелушением наружного края очагов no-z
ражения и «везикулезная («везико-буллезная») центробежная эритема с
наличием быстропреходящих везикулезных или буллезных элементов
по краям высыпаний. Везикулезную эритему Дарье в ряде случаев, по-
видимому, можно рассматривать как форму переходную к герпетиформному
дерматиту Дюринга (Машкиллейсон Л. Н., 1964). Мы наблюдали несколько
больных, у которых начало герпетиформного дерматита протекало под
маской центробежной эритемы Дарье.
Прогноз для жизни благоприятный, за исключением случаев, когда
центробежная эритема является паранеопластический дерматозом. Прог-
ноз в отношении выздоровления часто неопределенный.
Гистопатология. В мальпигиевом слое эпидермиса небольшой межкле-
точный и внутриклеточный отек. В дерме умеренный отек, расширение
капилляров, небольшие периваскулярные инфильтраты из лимфоцитов и
гистиоцитов, иногда с примесью эозинофилов и нейтрофилов.
Дифференциальный диагноз. Следует проводить с крапивницей, герпе-
тиформным дерматитом Дюринга, септической эритемой, многоформной
экссудативной эритемой, ретикулогемобластозами, саркоидозом, кольце-
видной гранулемой, туберкулоидной лепрой, себорейной экземой.
Лечение. Аутогемотерапия, антигистаминные препараты, поливитами-
ны. При наличии очагов инфекции, заболеваний желудочно-кишечного
тракта—антибиотики. В особенно упорных случаях—кортикостероиды.
ЛИТЕРАТУРА
Champion R. Disorders affecting smoll bloon vessels: erythema and teleangiectasia,)—
In: Tegtbook Rook, Zilkinson, Ebling. Oxford—London, 1979, vol. 1, p. 955—980.
Gianotti F., Ermacora E. Erythema gyratum atrophicans trausien neonatale.— Arch.
Dermatol., 1975, 111, 5, 615—616.
Hammar H., Ronnerfalt L. Annular erythemas in infants associated with autoimmune
disorders in their mother.— Dermatologies, .1977, 154, 2, 115—127.
Kind R. Bui lose Variante des Erythema annulare centifugum Darier bei Candida-
albicans-Infektion.— Hautarzt, 1975, 26, 9, 466—470.
Soltani K-, Pacernick L., Lorincz A. Lupus erythematodeslike lesions in newborn
infants. —Arch. Dermatol., 1974, 110, 4, 435—437.
Sonnichsen N., Zabel R. Hautkrankheiten. Diagnostik und Therapie.— Leipzig. 1976.
ЭРИТЕМА МНОГОФОРМНАЯ ЭКССУДАТИВНАЯ (БУЛЛЕЗНАЯ
РАЗНОВИДНОСТЬ) (erythema exudativum multiforme bullosum).
Заболевание впервые в 1860 г. описал F. Hebra под названием много-
формной экссудативной эритемы. Буллезная разцовидностьявляется ред-
кой атипичной формой экссудативной эритемы. Ее наблюдали А. А. Калам-
карян (1959), Б. М. Пашков (1966), А. Я. Браиловский и Э. Н. Солошенко
(1975), Н. С. Шашель (1975), Prutkin и Fellner (1971), Azulay и соавт. (1975),
Kirby и Darby (1978), Vinke-Gengoux и Delbrouck-Poot (1979), Dogliotti
(1980) и др.
Этиология и патогенез. Большинство авторов придерживаются токси-
ко-аллергической полиэтиологической концепции развития заболевания,
указывая на роль медикаментозных, аллергических, инфекционных фак-
торов, эндокринных, желудочно-кишечных, нервно-психических нарушений
и т. д. Tonnesen и Soter (1979) с патогенетической точки зрения рассматри-
вают многоформную эритему как реакцию сверхчувствительности к целому
ряду факторов (инфекции, вирусы, медикаментозные средства и др.). Ука-
зывается на связь экссудативной эритемы с вирусами (Королев Ю. Ф.,
1975; Monnet, 1976; Ubaidy и Nally, 1976), однако настойчивые поиски
549
Рис. 366. Эритема многоформная
экссудативная (буллезная, гемор-
рагическая форма).
Рис. 365. Многоформная экссудативная
эритема (буллезная).
специфического вируса остаются безрезультатными. Высказывается мне-
ние об иммунном механизме пока неизвестного типа.
Многие современные авторы выделяют идиопатическую (при которой
не удается установить причинный фактор) и симптоматическую (при ко-
торой известен этиологический фактор) форму. По мнению А. А. Каламка-
ряна и В. А. Самсонова (198Q), в формировании патологического процесса,
вероятно, играет роль генетически обусловленная «готовность» организма,
которая реализуется под влиянием различных факторов (эмоциональный
стресс, простуда, инфекция, медикаментозная интоксикация и др.), спо-
собствующих аллергической перестройке, и проявляющейся по типу ги-
перергической реакции немедленно-замедленного типа. Мы в течение не-
скольких лет наблюдали болпного с тяжелой формой экссудативной эрите-
мы, возникающей каждый раз после тяжелых нервно-психических пере-
живаний, причем вначале появлялась генерализованная пятнисто-папу-
лезная и затем буллезная сыпь преимущественно на верхних и нижних
конечностях.
Особо следует остановиться на вопросе о взаимоотношении буллезной
многоформной эритемы с синдромами Стивенса—Джонсона и Лайелла.
Многие исследователи (Каламкарян А. А., 1959; Пашков Б. М., 1963; Ка-
ламкарян А. А. и Самсонов В. А., 1980; Бухарович М. Н., 1980; Браи-
ловский А. Я., 1980; Самцов В. И., 1980; Civatte, 1967; Gertler, 1970;
Kano, 1974; Sonnichsen и Zabel, 1976; Champion, 1979 и др.) считают, что
Б50
синдромы Стивенса—Джонсона и Лайелла являются в сущности синони-
мами тяжелых форм буллезной многоформной эритемы, а их клинические
различия зависят от степени выраженности реакции организма. Мнение о
единстве этих патологических форм не противоречит целесообразности
сохранения дифференцированного подхода к указанным заболеваниям,
т. к. он предопределяет прогноз и терапевтическую тактику.
Клиника. Наиболее часто болеют лица в возрасте от 15—20 до 30—40
лет. Многоформная эритема, как и синдромы Стивенса—Джонсона и Лайел-
ла, описаны у людей обоего пола, тяжелые формы в 2—3 раза чаще регист-
рируются у мужчин, чем у женщин. Заболевание начинается внезапно
подъемом температурь^ до 38—40°, резким ухудшением самочувствия, го-
ловными болями; могут наблюдаться тошнота, рвота, боли в мышцах, рес-
пираторные симптомы и т. д.
Характерными клиническими морфологическими признаками на ко-
же на различных этапах развития многоформной экссудативной эритемы,
токсического эпидермального некролиза и синдрома Стивенса—Джонсона в
большинстве случаев являются эритематозные, эритематозноотечные с
багрово-синюшным оттенком, типичным эксцентричным ростом симметрич-
ные высыпания различных размеров, с преимущественной локализацией
на разгибательных, дистальных участках Конечностей, на лице—вплоть
до распространенных очагов поражения, захватывающих значительные
участки кожного покрова с характерным симптомом так называемой ми-
шени, радужной оболочки, бычьего глаза и т. д. (рис. 365, 366).
Пузыри имеют различные размеры и очертания, а в некоторых слу-
чаях, чаще при синдромах Лайелла и Стивенса—Джонсона, они могут до-
стигать гигантских размеров. Содержимое пузырей серозное или серозно-
гнойное. Покрышки пузырей тонки, сравнительно легко разрушаются,
образуя обширные весьма болезненные, кровоточащие при дотрагивании
ярко-красные эрозированные поверхности, окаймленные бахромкой об-
рывков покрышек пузырей—«эпидермальный воротник». У части больных
наблюдаются геморрагические и петехиальные изменения. В тяжелых
случаях отмечается выпадение волос, паронихии с последующим отторже-
нием ногтевых пластинок.
Вовлечение в процесс слизистых оболочек области естественных от-
верстий, особенно при тяжелой буллезной форме полиморфной эритемы и
синдроме Стивенса—Джонсона может быть довольно значительным, но
наиболее выраженные изменения чаще регистрируются в полости рта,
гортани, носа, области глаз. Пузыри быстро вскрываются, образуя резко
болезненные, кровоточащие эрозии. Губы и десна красные, опухшие с
кровянистыми корками, что делает прием пищи чрезвычайно затрудни-
тельным. При поражении глаз (у 20—25% больных) наблюдается блефаро-
конъюнктивит с серозно-гнойным отделяемым из глазной щели, иридо-
циклит, кератит, помутнение роговицы и потеря зрения. К особенностям
этих заболеваний следует отнести вовлечение в процесс внутренних органов
по типу острой токсико-аллергической миокардиодистрофии, острого брон-
хита, гепатита и др.
551
Для многоформной экссудативной эритемы характерно хроническое
рецидивирующее течение, нередко отмечается сезонность заболевания,
преимущественно в осенне-весеннее время.
Буллезные формы чаще встречаются в холодное время—зимой, ран-
ней весной и осенью, значительно реже летом.
Исход заболевания при обычной форме, как правило, благоприятный,
при буллезных формах во многом зависит от общего состояния, возраста
больных, распространенности и тяжести процесса, своевременности диаг-
ностики, причинного фактора.
Гистопатология. В эпидермисе определяется внутри- и межклеточный
отек, пикноз ядер, гидропическая дегенерация базальных клеток в ком-
бинации с очаговым некрозом эпидермиса, что может вести к формированию
пузырей. При буллезных высыпаниях пузыри в зависимости от интенсив-
ности и остроты поражения локализуются под эпидермисом и только в
старых высыпаниях они иногда располагаются внутриэпидермально
(Ackerman, 1971; Braun-Falco, 1970). В дерме наблюдается отек сосочково-
го слоя, расширение сосудов, периваскулярные инфильтраты из лимфоцитов,
нейтрофилов с примесью эозинофилов.
При электронно-микроскопическом исследовании (Lever и Schaum-
burg-Lever, 1975; Orf ados et al., 1974) в базальных клетках эпидермиса
выявляются интрацитоплазматические повреждения с потерей органелл.
В средних отделах эпидермиса могут определяться электронно-плотные
дискератотические тельца. Поврежденные эпидермальные клетки часто не
содержат органелл.
Дифференциальный диагноз. Следует проводить с токсикодермией,
герпетиформным дерматитом Дюринга, пузырчаткой, афтозным стоматитом,
узловатой эритемой, «большим афтозом» Турена.
Лечение. При легком течении многоформной эритемы достаточно на-
значения антигистаминных и десенсибилизирующих средств. В более тя-
желых случаях и распространенных буллезных формах присоединяют ан-
тибиотики и кортикостероиды (30—50 мг преднизолона в сутки).
Можно рекомендовать капельное внутривенное вливание 2 раза в день
смеси 5% раствора глюкозы (500—1000 мл), 5% аскорбиновой кислоты
(1 мл), преднизолона (30 мг), инсулина (8 ЕД).
В связи с бурным освобождением лизосомальных ферментов, обуслов-
ливающих эпидермальный некролиз (Misgeld, 1974), с успехом применяют
ингибиторы протеолитических ферментов (трасилол и др.). Местное лече-
ние—растворы анилиновых красок, жидкость Кастеллани, дезинфицирую-
щие и кортикостероидные мази.
ЛИТЕРАТУРА
Браиловский А. По поводу статьи А. А. Каламкаряна и В. А. Самсонова.—
Вестн. дерматол. и венерол., 1980, № И, с. 29—31.
Бухарович М. Н. Некоторые аспекты взаимоотношения многоформнрй экссудатив-
ной эритемы, синдромов Стивенса—Джононса и Лайелла.— Вести, дерматол. нвенерол.,
1980, № 9, с. 33—34.
552
Каламкарян А. А., Самсонов В. А. О многоформной экссудативной эритеме, синд-
роме Стивенса—Джонсона, токсическом эпидермальном некролизе (синдроме Лайелла)
их взаимоотношении.— Вести, дерматол. и веиерол., 1980, № 3, с. 36—42.
Самцов В. И., Подвысоцкая И. И., Лисовская Н. Д. Замечания по поводу острого
эпидермального некролиза.—Вести. Дерматол. н венерол., 1980, Ns 11, с. 31—34.
Azulay Д., Queoedo L., Gifueura R-, Manhaes L. Erythema multiforme in fanttnn
atrophicans.— Int. J. Dermatol., 1975, v. 14, No 1, p. 56—58.
Degos R. Dermatologie.— Paris, 1976,
Dogkiotti M. Erythema multiforme an unusual reaction to BC9 vaccination.— S. Afr.
Med. J., 1980, v. 57, No 9, p.332—334.
Kirly J-, Darby C. Erythema multiform associated with a contact dermatitis to ter-
penes.— Cont. Dermatitis, 1978, v. 4, No 4, p. 238—239.
OrfadosC., Germany W., LeoerG., LeoerW. Dermal and epidermal types of erythema
multiforme. A histopathologie study of 24 cases.— Arch. Dermatol., 1974, v. 109, No 5, p
p. 682—688.
Sonnichsen N., Zabel R. Hautkrankheiten. Diagnostik und Therapie.— Leipzig, 1976.
Tonnesen M-, Soter N. Erythema multiforme.— J. Amer. Acad. QDermatol., 1979.
v. 1, No 4, p. 357—364.
Vincke-Gengoux P., Delbrouck-Poot F. Erytheme polymorphe bulleux chronique.—
Dermatologica, 1979, v. 158, No 3, p. 207—213.
ЭРИТРОКЕРАТОДЕРМИЯ ФИГУРНАЯ ВАРИАБЕЛЬНАЯ (erythro-
keratodermia figurata variabilis)
Этиология и патогенез. Заболевание относится к группе генодермато-
зов. По-видимому, является гетерогенным, наследуется преимущественно
аутосомно-доминантно. Gewirtzman и соавт. (1978) описали семью, в ко-
торой было 12 больных в 5 поколениях. Указывается на возможную роль
в патогенезе нарушения обмена витамина A (Wulf et al., 1960), отмечается
роль хронической травмы (Brown и Kierland, 1966; Borza, 1963), снижение
в эпидермисе количества клеток Лангерганса (Van der Schoeff et al., 1982).
Многие (Rinsema, 1928, Popoff, 1963, Gans и Steigleder, 1957) считают эрит-
рокератодермию фигурную вариабельную одной из форм врожденной их-
тиозиформной эритродермии. Однако Miescher и Staheli (1949), Galen (1960),
Schneider и соавт. (1962), Brown и Kierland (1966) обращали внимание на
ряд различий в клинических проявлениях и течении этих заболеваний
Brown и Kierland (1966) дают сравнительную характеристику их в виде
таблицы. ,
Vandeesteen и Miiller (1971) на основании гистохимических и электрон-
номикроскопических исследований пришли к выводу, что вариабельная
эритрокератодермия—заболевание sui generis, не связанное с врожденной
ихтиозиформной эритродермией. Оно представляет собой первичное рас-
стройство кератинизации эпидермального происхождения, существенным
компонентом которого является значительное уменьшение количества ке-
ратиносом. Обнаружено также увеличение количества немиелинизирован-
ных нервов в сосочковом слое дермы, гидролитических ферментов—в эпи-
дермисе. На различие между этими заболеваниями указывает также отсут-
553
Пок зателн Эритрокератодермии вариабельная Ихтиозиформная эритродермия
Начало Наследование Эритродермия Гиперкератоз Волосистая часть Волосы, иогти Пузыри Г нстопатология В раннем детстве Аутосомно-доминантное Ограниченная, фигурная изменяющаяся Ограниченный Без изменений Не изменены Не бывает Неспецифична, может наблюдаться при любом из эпидермальных невусов или при ихтиозиформной эритродермии. С рождения Рецессивное? Генерализоьанная Стабильная Генерализованный Себорейное шелушение Ускоренный рост Могут, быть Может быть специфической: баллонирующая дегенерация в средней части эпидермиса и в зернистом слое с частым образованием микровезикул и пузырей.
ствие повышенной пролиферативной активности эпидермоцитов (Schellan-
der и Fritsch, 1969).
Gans и Koch (1951) рассматривали вариабельную эритрокератодермию
как вариант сосудистой дисплазии и полагали, что в основе нарушения
кератинизации лежит патологическое расширение сосудов. Сходной точки
зрения придерживался Gertler (1957), приводя в качестве примера, под-
тверждающего ее, развитие гиперкератоза при ангиокератоме. Капааг
(1965) усматривал в эритрокератодермии вариабельной^ вариант псо-
риаза. Коляденко В. Г. и соавт. (1983) обращают внимание на возмож-
ное клиническое сходство с пустулезным субкорнеальным дерматозом Вил-
кинсона.
В группу вариабельных эритрокератодермий Galen (1960), Fleger и
соавт. (1966) относят ichthyosis linearis circumflexa, описанный в 1949 г.
Cornel (Friihwald полагает, что этот дерматоз был описан в 1922 г. Rille).
По мнению Schnyder и Wiegand (1968), ichthyosis linearis circumflexa за-
нимает промежуточное положение между эритрокератодермиями и врож-
денным ихтиозом. Другие же авторы (Литвинюк Н. В. и Тузова 3. М.,
1966; Коляденко В. Г. и соавт., 1983; Franck, 1956; Малое, 1958; Steva-
novic и Pavic, 1958; Dimitrowa и Georgiewa, 1961; Vinejard et al., 1961;
Ruzicka et al., 1963; Stankler и Cochrane, 1967) относят его к врожденной
ихтиозиформцой эритродермии. .
Sedlacek и Кгепаг (1971) наблюдали больную с ichthyosis linearis cir-
cumflexa Cornel, у которой были массивные папилломатозные разрастания
в промежности, резко усилившиеся во время беременности, несмотря на
то, что изменения на остальной коже практически регрессировали. К цир-
цинарным вариабельным эритемато-сквамозным генодерматозам Gianotti
1969) относит и синдром Netherton, проявляющийся изменениями на
554
Рис. 367. Вариабельная
эритрокератодермия.
Рис. 368. Вариабельная
зритрокератодермия.
Рис. 369. Вариабельная
зритрокератодермия.
555
коже в виде врожденной ихтиозиформной эритродермии; аномалией волос—
множественные утолщения по их длине, сходные с бамбуковой палочкой
(trichorrexis invaginata, по определению Wilkinson), реже—piltorti; ато-
пией.
Schnyder и Weigand (1968) наблюдали подобные изменения волос и при
ichthyosis linearis circumflexa; Julius и Keeran (1971')—кроме того, в соче-
тании с умственным недоразвитием и аномалиями почек. Из 15 больных с
trichorrhexis invaginata, наблюдавшихся Hurwitz и соавт. (19711, у 12 кож-
ные проявления были в виде ichthyosis linearis circumflexa, у остальныхЗ—-
других форм ихтиоза. У 3 больных с ichthyosis linearis circumflexa име-
лись другие аномалии волос (ломкость, pili torti, узловатая ломкость во-
лос), у 6 человек—волосы были нормальны. По мнению Altman и Stroud
(1969), синдром Netherton и ichthyosis linearis circumflexa представляют
собой одну и ту же нозологическую единицу, в основе которой лежит на-
рушение кератинизации, захватывающее волосы и кожу. Авторы относят это
состояние к атипическим формам ихтиоза. Учитывая, что характерной осо-
бенностью его является псориазиформная гистологическая картина оча-
гов поражения, они считают возможным предположить существование осо-
бой группы псориазиформного ихтиоза, к которой следовало бы относить
все случаи, когда при гистологическом исследовании обнаруживается кар-
тина, сходная с ихтиозом. Имеются описания и более редких вариантов
эритрокератодермии: erythrokeratodeermia cum nanismo, olygophrenia et
osteoporosi (Popoff, 1963), erythemes circines variables, erythemes periorifi-
ciels et hypotrichose congenitaux (Barez и Dupre, 1956); Erythrokeratoder-
mia progressiva symmetrica (Gottron, 1922), Erythrokeratodermia en cocar-
des (Degos et al., 1947); Erythrokeratodermia progressiva (Schnyder et al.,
1964). По мнению H. С. Смелова и соавт. (1971), Б. Т. Глухенького и со-
556
авт. (1983), Cram (1970), описанные выше формы по существу представ-
ляют собой варианты одного заболевания. Marghescu и соавт. (1982) опи-
сывают больную с «атипичной эритрокератодермией*, у которой с 6-месяч-
ного возраста наступила двусторонняя глухота, больная осталась глухо-
немой. Эта разновидность эритрокератодермии была впервые описана Schny-
der в 1963 году; в литературе сообщают о 8 подобных наблюдениях.
Клиника. Фигурные эритематосквамозные очаги, границы которых
меняются в течение нескольких часов или дней, реже недель. В одних
местах процесс разрешается, нередко оставляя гиперпигментацию, в
других—появляются новые очаги, постепенно увеличивающиеся в раз-
мерах за счет эксцентрического роста. В центральной части интенсивность
воспаления при этом уменьшается. В редких случаях могут наблюдаться
явления экзематизации (Н. С. Смелов и соавт., 1971). Высыпания у боль-
шинства больных распространенные с наибольшей выраженностью на ли-
це, шее, плечах, туловище. Иногда процесс занимает весь кожный покров,
(рис. 367,368, 369, 370).
Может быть ладонно-подошвенный гиперкератоз. Особенностью вариа-
бельной эритрокератодермии, описанной Schellander и Fritsch (1969), было
универсальное поражение кожи с наличием фигурных эритемато-сквамоз-
ных изменений и массивными очагами гиперкератоза, достигающими круп-
ных размеров, особенно на ягодицах и ногах, существующих с детства.
Ладонно-подошвенная кератодермия, гипертрихоз, особенно в зоне гипер-
кератотических бляшек. Сходную клиническую форму наблюдали Wulf
и соавт. (1960). Субъективные расстройства непостоянны, в виде нерезко
выраженного зуда, стягивания кожи, иногда болезненности. Эритема
усиливается под влиянием эмоциональных факторов, холода, ветра, жары.
Заболевание существует с рождения или чаще развивается в раннем
детском возрасте. Как правило, не исчезает, хотя с возрастом интенсивность
воспалительных явлений уменьшается и у взрослых обычно доминируют
кератотические изменения. Giroux и Barbeau (1972) наблюдали сочетание
эритрокератодермии с атаксией. Gertler (1970) опубликовал случай, когда
заболевание протекало с псориазиформными проявлениями на волосистой
части головы, изменениями типа acanthosis nigricans в подмышечных впа-
динах и на внутренней поверхности бедер, околосуставными подушечками,
ладонно-подошвенным кератозом. Hurwitz и соавт. (1971) приводят следую-
щую таблицу проявлений ichthyosis linearis circumflexa и эритрокерато-
дермии вариабельной.
Cram (1970) относит к важным дифференциально-диагностическим при-
знакам постоянно наблюдающуюся лихенификацию складок и развитие
в некоторых случаях вялых субкорнеальных пузырьков. Faninger (1956)
наблюдал своеобразную форму семейной ограниченной эритрокерато-
дермии с локализацией на руках, главным образом на кистях.
Гистопатология. Выраженный гиперкератоз, паракератоз, иногда спон-
гиоз, расширение мальпигиевой сети. В верхней трети дермы небольшой
отек, лимфоцитарные инфильтраты. Глухенький Б. Т. и соавт. (1983) вы-
557
Покалателн Ichthyosis linearis circumflexa Эритрокератодермия вариабельная
Начало Наследование Локализация С рождения Рецессивное Туловище и проксимальные части конечностей В раннем детстве Доминантное Конечности и лицо
Лицо Эритродермия Течение Гистопатология диффузная краснота и шелушение Мигрирующие полицикличе- ские очаги с удвоенным шелушащимся краем Частичная ремиссия Акантоз и паракератоз Фиксированная эритема, особенно вокруг естественных отверстий Полуциклические бляшки гипер- кератоза (часто с центральным шелушением) Персистенция в течение жизни Гиперкератоз и акантоз
явили дистрофические изменения в эпидермисе и дерме, одной из вероятных
причин которых, по мнению авторов, является гипоксия за счет измене-
ния сосудов, возможно генетического происхождения. Van der Schroeff и
соавт. (1982) сделали предположение о возможной роли снижения количе/
ства эпидермальных клеток Лангерганса в развитии патологического оро-
говения.
Дифференциальный диагноз. С себорейной экземой, псориазом, ихтио-
зом, красным волосяным лишаем. /
Лечение. Витамин А, аевит, рибофлавин, никотиновая кислота, вита-
мин В12, слабые салициловые и серно-салициловые мази.
ЛИТЕРАТУРА
Gewirtzman J. В., Winkler N. N., Dobson R. L. Erythrokeratodermia variabilis.—
Arch. Derm., 1978, 114, 2, 259—261.
Marghescu S., Wolff H., Braun-Falco 0. Kongenita'le Erythrokeratodermie mit Taub-
heit Schnyder Hautarzt. 1982, 33, 8, 416—419.
Van der Schroeff J. G., Rutter D. J., Bots G. T. A. M. Epidermal Langergans cell
|П Erythrokeratodermia variabilis.— Arch, derm., Res., 1982, 274, 339—348.
ЭРИТРОПЛАЗИЯ КЕЙРА (erythroplasia Queyrat)
Синоним: эпителиома бархатистая.
Этиология и патогенез. В большинстве случаев эритроплазия Кейра
рассматривается как вариант болезни Боуэна слизистых и полуслизистых
(Hyman и Leider, 1961; Schimpf, 1979). Steigleder (1964) считает сомнитель-
ным, что эти заболевания во всех случаях идентичны и полагает, что эрит-
роплазия Кейра с большим основанием, чем болезнь Боуэна, может быть
558
отнесена в группу карцином in situ. По материалам ВОЗ (1980), разделение
этих заболеваний оправд но тем, что при эритроплазии Кейра большая
вероятность инфильтративного роста и нет склонности к ассоциации с ви-
сцеральными формами рака. Goette (1974) среди факторов, которые могут
способствовать развитию болезни, отмечает хроническое раздражение
смегмой, так как болезнь практически не развивается у лиц с удаленной
крайней плотью.
Клиника. Обычно одиночный очаг поражения, с неравномерными,
резко очерченными границами, овальных, округлых или неправильных
очертаний, нередко фестончатых, слегка отечный и вначале мало инфильт-
рированный, в связи с чем едва возвышающийся. Характерна его поверх-
ность—насыщенно-красного цвета, часто с коричневатыми оттенками, влаж-
ная, блестящая, лоснящаяся, как бы бархатистая (рис. 371,372). По мере раз-
вития процесса усиливается инфильтрация, поверхность может покрывать-
ся корочками, легко кровоточит, иногда становится вегетирующей, эрозиро-
ванной, что может служить признаком развития эпителиомы. Очаг, как
правило, располагается на половых органах (у мужчин на головке поло-
вого члена, внутренней поверхности крайней плоти, у женщин—в области
вульвы, особенно на внутренней поверхности малых половых губ). Имеются
сообщения (Vulcan et al., 1976) об одновременном наличии болезни Боуэ-
на в области лобка и эритрсплазин Кейра на головке полового члена. Опи-
саны также поражения полости рта. И. И. Ильин и О. Г. Карповская
(1968) сообщают о локализации эритроплазии на нижней губе При лока-
лизации в области вульвы англо-американские авторы ставят диагноз бо-
лезни Боуэна (Sanderson, 1979). По мнению Веикеи и Шугар (1962), при
экстрагенитальной локализации поражения более вероятно, что это не
эритроплазия Кейра, а болезнь Боуэна.
559
Эритроплазия Кейра возникает значительно чаще у мужчин, как пра-
вило, старше 50 лет. Как и при болезни Боуэна, может длительное время
(до десятилетий) оставаться чисто эпидермальным процессом без инвазии
глубжележащих тканей и метастазов. Но прогноз эритроплазии Кейра
считается неблагоприятным (Steigleder, 1964). Переход в карциному от-
мечен у 20—40% больных (Schimpf, 1979). По мнению Венкеи и Шугар
(1962), это наблюдается во всех случаях. В. Г. Коляденко и соавт. (1979)
допускают возможность течения одних вариантов по типу болезни Боуэна
с большей вероятностью злокачественного перерождения, других—с бо-
лее доброкачественной динамикой.
Гистопатология. Неравномерный акантоз, очаговый гипер- и пара-
кератоз, нарушение созревания клеток с образованием атипических кле-
ток, в том числе многоядерных, со светлой вакуолизированной цитопла-
зией, гиперхромными ядрами, дискератозом, многочисленными митозами.
В дерме расширение сосудов, лимфоцитарные инфильтраты с примесью
плазматических клеток.
Дифференциальный диагноз. Дифференцировать эритроплазию Кейра
необходимо от баланопостита (вульвита), крауроза, плазмрцеллюлярного
Ограниченного баланопостита Zoon, ограниченного псориаза, экземы, фи-
ксированной эритемы, красного плоского лишая, спиналиомы, педжетоид-
ной эпителиомы, болезни Педжета, сифилиса.
Лечение. Блеомицин, лучевое, хирургическое, цитостатические мази.
ЛИТЕРАТУРА
В. Г. Коляденко, Н. М. Левковский, Н. М. Шупенько, Д. Д. Головченко. Эрит-
роплазня Кейра. Вестн. дерматол. н венерол., 1979, 12, 32—35.
Duncan Р., Vulkan V., Dumitriu Е. Maladia Bowen cutanata (Pubiata si a mucoasei
peniene (eritroplazie Queyrat).— Derm. Vener., 1976, 21, 2, 151—153.
Goette D. K- Erythroplasia of Queyrat. Arch. Derm., 1974, 110, 2, 271—273.
Sanderson К- V. Premalignant and malignant lesions of the penis and scrotum. In:
«Tegtbook of dermatology Ed.» by Rook A. et al. Blackwell scientific publications, Oxford,
1979, 2, 1963—1964.
Schiphf. Erythroplasia Queyrat. In: <Nicht entzundlieche Dermatosen III B. Bosartige
Geschwulste, Leukeamie.» Hrmsg H. A. Gottron und G. K- Korting Springer—Verlag, 1979,
145—150.
ЯЗВЫ ГОЛЕНИ ГИПЕРТОНИЧЕСКИЕ (ulcus cruris hypertonicum).
Синонимы: гипертензионрые ишемические язвы, синдром Марторелла.
В 1945 г. испанский ангиолог Martorell впервые описал больных
гипертонической болезнью, у которых на голенях развились резко бо-
лезненные и длительно незаживающие язвы, выделив эту патологию в
самостоятельную единицу. В дальнейшем появились описания подобных
наблюдений, подтверждающие существование гипертонических язв, полу-
чивших название синдрома Марторелла (Farber и Schmidt, 1950; Castermans-
Ellias, 1959; Schmitz, 1955; Mozes et al., 1962; Kluge, 1965; Schnier et al.,
1966; Fairbaitn et al., 1972 й др.). В отечественной литературе по этому
560
Рис. 373. Синдром
Марторелла (язвы
гипертонические).
вопросу опубликованы работы О. К- Шапошникова и М. И. Крупно (1963),
Н. С. Потекаева и А. В. Константинова (1966), А. П. Михеевой (1972),
Г. А. Орликова (1976), И. П. Даудярис и Б. И. Даудярене (1978), Ю. К.
Скрипкина и соавт. (1982), Г. А. Азиавлова, В. Г. Морозова, 1984).
Этиология и патогенез. Расстройства трофики и язвы являются след-
ствием облитерации мелких артерий и артериол кожи голеней на ограничен-
ном участке. Однако не все авторы признают ишемию как патогенетический
фактор, ибо не всегда при гистологическом исследовании язвы и окружаю-
щих ее тканей находили гиалиноз или эндартериальную пролиферацию.
В отделе дерматологии мы наблюдали двух больных, у которых четко
прослеживалась взаимосвязь между тяжестью клинического течения син-
дрома Марторелла и проявлениями гипертонической болезни, что позволяет
считать этот синдром одним из осложнений гипертонической болезни.
Клиника. Заболевание начинается с появления небольшого болезнен-
ного красного пятна в средней или нижней трети голени. Наиболее часто
пятно локализуется на передне-или задненаружной поверхности голени и
довольно быстро становится синюшно-красным, буроватым или пурпурным.
Процесс может начинаться и с возникновения небольших папул, иногда
имеющих геморрагический компонент. После легкой травмы или без види-
мой причины на местах первичных высыпаний появляется некротическая
корочка, под которой обнаруживается изъязвление (рис. 373). Гипертони-
ческие язвы голеней бывают большей частью множественными, полицикли-
ческими, поверхностными, размерами от 2 до 5 см в диаметре, с фестонча-
тыми, слегка подрытыми краями, некротическим дном и незначительным
серозно-гнойным отделяемым. Как правило, они располагаются симмет-
рично на обеих голенях. Язвы отличаются резкой болезненностью и иск-
36 Клиническая дерматология 561
лючительной чувствительностью даже при самом легком прикосновении.
Для болей характерно постоянство, они не зависят от времени суток и поло-
жения пораженной конечности.
У больных почти всегда имеются характерные изменения глазного
дна не только в виде гипертонической ангиоретинопатии, но и мелких кро-
воизлияний в сетчатку ?Чаще болеют женщины в возрасте 40—60 лет, т. е.
в период угасания половой гормональной активности. Наблюдающиеся у
них изменения со стороны сердечно-сосудистой системы и почек свидетель-
ствуют о тяжелой форме гипертонической болезни. Варикозного расши-
рения вен и проявлений хронического тромбофлебита обычно не отмеча-
ется. Осциллографические исследования на нижних конечностях ука-
зывают на проходимость магистральных сосудов. Признаков значительных
расстройств периферического кровообращения не наблюдается.
Язвы существуют длительно (месяцы, годы) и трудно поддаются кон-
сервативной терапии.
Гистопатология. Просвет мелких периферических сосудов сужен,
имеются значительно выраженные явления эндартериальной пролифера-
ции и субэндотелиального гиалиноза, т. е. деструктивные изменения, кото-
рые при гипертонической болезни находят в различных органах и систе-
мах. Это дает основание рассматривать синдром Марторелла как тяжелое
осложнение гипертонической болезни.
Дифференциальный диагноз. Следует проводить с варикозным симпто-
мокомплексом, изъязвлением при постфлебическом синдроме, с облитери-
рующим эндартериитом и- облитерирующим артериосклерозом нейротро-
фическими и гуммозными язвами.
Лечение. Гипотензивные средства; мази, содержащие антибиотики,
антисептики, анальгетики; УВЧ, местное ультрафиолетовое облучение,
новокаиновые блокады. Эффект наступает только при стойком снижении
артериального давления.
ЛИТЕРАТУРА
Скрипкин Ю. К., Уджуху В. Ю., Короткий И. Г. и др. Случай синдрома Марторелла.
Вести, дерматол. и венерол., 1983, 3, 34—36. ,
Nitzschner И. et al. Zur Problematic des Ulcus Martorell. Z. Haut.—u. Geschl.
Krankh., 1966, 40, 6, 188—193
содержание
Введение ................................................................... 3
Аденома сальных желез симметричная........................................... 5
Акне фульминанс ............................................................. 6
Акротерия семейная . .............................................. 9
Акродерматит стойкий пустулезный адлопо • .......................10
Акродерматит энтеропатический .............................................. 13
Акрокератоз верруциформный Гопфа ........................................... 17
Акропатия псевдосирингомиелитическая ..................................... 19
Амилоидоз кожи ..............................................................21
Ангиокератома.............................................................. 26
Ангиокератома Мибелли ...................................................... 26
Ангиокератома туловища диффузная Фабри ..................................... 27
Ангиокератома мошонки (вульвы)............................................ 29
Ангиокератома туловища ограниченная невиформная........................... 30
Ангиокератома солитарная папулезная ........................................ 33
Ангиоматоз наследственный семейный геморрагический........................ 34
Анетодермия ................................................................ 35
Атрофия кожи ............................................................. 37
Базалиома .................................................................. 45
Базальноклеточного невуса синдром ................................ 53
Баланит ксеротический облитерирующий ....................................... 56
Бехчета синдром ............................................................ 58
Боуэна болезнь ............................................................. 61
Блума синдром .......................................................... 65
Бромодерма ............................................................... 68
Васкулит кожи аллергический ................................................ 70
Вернера синдром .......................................................... 73
Вискотта—Олдрича синдром ................................•.................. 74
Волосатик ................................................................ 77
Волчанка красная ...................................................... . 79
Гангрена послеоперационная прогрессирующая ................................. 93
Гемангиома кавернозная ..................................................... 94
Гемангиоперицитома ......................................'.................. 96
Гемангиоэндотелиома кожи .................................................. 98
ГемосиДероз кожи .......................................................... ’00
Гидроа оспенновидная ...................................................... ,О-
Гиперкератоз стойкий лентикулярный Флегеля..............................'• 1°3
36* 563
Гиперкератоз фолликулярный и парафолликулярный, проникающий в кожу . 105
Глкжаганома синдром......................................................Ю7
Гранулема кольцевидная...................................................ИО
Гранулема лица гангренесцнрующая............................................И4
Гранулема лица одонтогенная подкожная..............................116
Гранулема пиогенная.........................................................И8
Гранулема эозинофильная....................................................120
Гранулема эозинофильная лица...............................................122
Гранулематоз Вегенера....................................................125
Гранулематоз хронический прогрессирующий дискообразный Мишера-Ледера . 128
Дерматит герпетиформный Дюринга..........................................130
Дерматит горшечный у детей...............................................134
Дерматит от золота................................ • . • • • • • 1^5
Дерматит периоральиый....................................................139
Дерматоз острый лихорадочный нейтрофильный............................141
Дерматозы линейные . . . . .............................7 . 143
Дерматоз подроговой пустулезный............................................147
Дерматомиозит ......................................................... • 149
Дерматофибросаркома выбухающая.......................................... .152
Дерматофиброма лентикулярная .......................................г .154
Джоноттн—Крости синдром..................................................157
Дискератоз фолликулярный Дарье...............................'. . • 160
Дисплазии эктодермальные...................................................164
Дистрофия кожи папиллярно-пигментная.......................................170
Импетиго герпетиформное . ................................174
Иододерма туберозная ......................................................175
Небуллезная (сухая) форма врожденной ихтиознформной эритродермии . . 181
Кальциноз кожи.............................................................183
Келоид...............................................................* 185
Кератоакантома.............................................................190
Кератодермия климактерическая .............................................194
Кератодермия ладоней и подошв наследственная...............................196
Кератодермия мутилирующая Фовинкеля........................................207
Кимуры болезнь.............................................................210
Клиппеля—Треноне синдром ..................................................212
Кожа врожденная мраморная телеангиэктатическая.........................214
Кожа вялая.................................................................215
Кожа гиперэластнческая ....................................................219
Коллонд мнлиум ........................................................... 223
Кондиломы остроконечные гигантские.........................................225
Крапивница пигментная ...... .................... 227
Красная зернистость носа...................................................234
Крауроз вульвы.............................................................235
Крауроз полового члена................................................. . 237
Криоглобулинемия ..........................................................239
Крона болезнь .............................................................242
564
Ксантомы туберозные ....к.............................................. 244
Ксеродерма пигментная ................................................. 248
Лассюэра—Литтля синдром ............................................. 252
Лейкозы кожи ....................................................... 255
Лейомиома кожи . ... ~..............~. .................. 258
Лейомиосаркома кожи ................................................... 261
Лейшманнбз кожи ..................................................... 263
Лейшманиоз американский кожный ........................................ Jgg
Лентигиноз периорифициальный .......................................... 269
Лентигиноз эндсбукальный .............................................. 278
Лимфоцитома кожи ...................................................... 274
Липодистрофия прогрессирующая ................................... 278
Лихен склерозирующий и атрофический.................................. 279
Лншай блестящий ..................................................... 282
Лишай красный отрубевидный волосяной Девержи .......................... 284
Лншай красный плоский ................................................. 291
Лишай опоясывающий области глаза '.................................. 299
Лишай шиловидный ...................................................... 302
Лун—Бар синдром ....................................................... 304
Меланоз предраковый ограниченный Дюбрея ............................... 307
Меланома злокачественная кожи .......................................... ЗЮ
Меланома юношеская .................................................... 318
Микоз грибовидный 320
Микседема ограниченная претибиальная .................................. 324
Монилетрикс ...»....................................................... 327
Муциноз фолликулярный ................................................. 330
Невус Ота ............................................................ 333
Недержание пигмента .......................;........................... 336
Некробиоз липоидный ................................................... 339
Некролиз токсический эпидермальный .................................... 343
Некротические угрн ..............................•..................... 345
Опухоль гломусная ..................................................... 348
Папилломатоз кожи карциноидный Готтрона ............................... 350
Папулез злокачественный атрофический .................................. 352
Паранеоплазня ......................................................... 354
Парапсориаз ...............................;........................... 357
Патомимия ............................................................. 361
Пахндермопериостоз .................................................... 364
Пахионнхня врожденная ................................................. 367
Педжета болезнь ....................................................... 370
Пеллагра ............................................................. 373
Пемфигоид буллезный Левера ............................................ 378
Пиодермия гангренозная ................................................ 381
Пиодермия шанкриформная .............................................. 383
Плазмони % ма кожи .................................................... 384
Подушечьи дальцев околосуставные ...................................... 388
565
ПоййилоДермня атрофическая сосудистая Якоби t ........................... 389
Пойкялодермия врожденная Томсона ........................................ 390
Порокератоз ............................................................. 392
Порома эккринная ........................................................ 395
Порфирия кожи поздняя ................................................... 397
Порфирия врожденная эритропоэтическая .................................. 402
Кровавый пот ........................................................ 405
Псевдоксантома эластическая ............................................ 406
Псевдосаркома Капоши .................................................... 408
Псориаз веррукозный...................................................... 411
Псориаз тпустулезный ............................................z . . . 415
Пузырчатка ............................................................ 417
Пузырчатка хроническая доброкачественная семейная ....................... 425
Пустулезный и атрофический дерматит голеней ............................. 427
Рак метастатический кожи ................................................ 430
Реклингаузена болезнь ................................................... 434
Ретикулогистноцитома кожи ............................................. 439
Ретикулосаркоматоз кожи ' 442
Ринофима .............................................................. 448
Рог кожный .......................................................... 450
Россолимо—Мелькерссона—Розенталя синдром ................................ 452
Ротмунда синдром ........................................................ 455
Саркоидоз кожи .......................................................... 456
Саркома множественная идиопатическая геморрагическая Капоши.............. 461
Саркома Капоши — как проявление СПИД .................................. 468
Себоцистоматоз ....................... . . . t........................ 471
Сезари синдром .......................................................... 473
Снрннгома ...............................................'............... 476
Склередема взрослых Бушке ............................................... 478
Склеромикседема Арндта—Готтрона ....................................... 479
Слнзисто-кожный лимфатический синдром.................................... 482
Стивенса—Джонсона синдром ............................................. 484
Стюарта—Тревса синдром .................................................. 486
Трихоэпителиома ......................................................... 488
Трофедема Ноние—Мильроя—Мейжа ........................................... 490
Туберкулез кожи волчаночный ............................................. 491
Узелки доильщиц ......................................................... 496
Ульэритема надбровная ................................................... 497
Фасциит эозинофильный ................................................. 499
ФлеботоДермня ........................................................... 501
Фокса—Фордайса болезнь .................................................. 502
Хенда—Шюллера—Крисчена синдром .......................; . . . 505
Хондродерматит узелковый ушной раковины.................................. 507
Цилиндрома .............................................................. 508
Чесотка норвежская ...................................................... 510
566
Шегрена синдром ........................................................... 513
Экзема герпетическая Капоши ...............................................'515
Эластоз перфорирующий серпегинирующий ..................................... 517
Эластоидоз кожи узелковый ................................................. 518
Эндометриоз кожиый ........................................................ 520
Эпидермсдисплазия верруциформная Левандовского—Лютца ...................... 522
Эпидермолиз буллезный ..................................................... 524
Эпителиома обызвествленная ................................................ 528
Эпителиома спнибцеллюлярная .............................................. 530
Эризипелоид ..............................'................................ 539
Эритема возвышающаяся длительно протекающая ............................... 540
Эритема дисхромическая стойкая ............................................ 543
Эритема кольцевидная центробежная Дарье ................................... 546
Эритема многоформная экссудативная (буллезная разновидность)............... 549
Эритрокератодермии фигурная вариабельная ................................. 553
Эритроплазня Кейра ......................................................; 558
Язвы голенн гипертонические ............................................... 560
Амаяк Артемович Каламкарян
Владимир Николаевич Мордовцев
Лидия Яновна Трофимова
КЛИНИЧЕСКАЯ ДЕРМАТОЛОГИЯ
(редкие и атипичные дерматозы)
Спец, редактор Э. Е. Даниелян
Редактор издательства Н. И. Колесникова
Художник Г. К. Мнацаканян I
Художественный редактор Д. Папян
Технический редактор К. Г. Саркисян
Корректор А. А. Азарян
ИБ
Сдано в набор 08. 04. 1987 г. Подписано в печать 19. 04. 1989 г. ВФ 01303. Формат
70 X90V16. Бумага офсетная. Гарнитура <Литературная». Печать офсетная. 41,53 усл.
печ. л. 166,12 усл. кр.-отт. 41,46 уч. изд. л. Тираж 5000. Заказ 2462. Цена 8 р. 60 коп.
Издательство «Айастан», Ереван-9, ул. А. Исаакяна, 28.
Полнграфкомбннат им. Акопа Мегапарта Госкомитета Арм. ССР, по делам изда-
тельств, полиграфии и книжной торговли. Ереван-9, ул. Теряна, 91.