/


















































































































































































































































































































Текст
В,Н Кулезнев
СМЕСИ
ПОЛИМЕРОВ
УДК 678.13 + 678.19
Кулезнев В. Н.
Смеси полимеров.— М.: Химия, 1980.— 304.,
ил.
В книге систематизированы результаты исследования од-
нофазных, двухфазных и многофазных смесей полимеров. Из-
ложены современные термодинамические и структурно-физи-
ческие теории смесей полимеров, модельные представления о
свойствах смесей полимеров Описаны структура и свойства
растворов смесей полимеров. Особое внимание уделено зако-
номерностям смешения полимеров друг с другом и различ-
ными ингредиентами. Приведены примеры применения кон-
кретных смесей полимеров в различных областях техники.
Книга рассчитана иа научных сотрудников, инженерно-
технических работников, занимающихся получением и пере-
работкой полимерных материалов, а также для аспирантов
и студентов старших курсов химико-технологических вузов,
специализирующихся в области физикохимии, механики и пе-
реработки полимерных материалов.
304 с., 28 табл., 160 рис., список литературы 592 ссылки.
Рецензент: докт. физ.-мат. наук, проф Л. Я. МАЛКИН.
3MIU-UOI
К 050(01 f80 51 80-2H03090t,t,U-
С Издательство «Химия», 1980 г.
Смес
<J
э
Смет
в
в
I
I
1
Г
В
Валерий Николаевич Кулезнев
I
I
СМЕСИ ПОЛИМЕРОВ
1
Смо.
Смо.
Сов*
Совг
Сов?
Соп<
г
1
<
Соп]
1
Ста»
Сте1
Сте!
Стр?
Редакторы Рогайлина А. А., Ромм Р. С.
Художник Киреев В. В.
Художественный редактор Носов Н. В.
Технический редактор Косачева Г. И.
Корректоры Васина Т. С., Лазуткина Л. В.
ИБ № 290
Сдано в набор 20.03.80. Поди, в псч. 6.06.80. Т-11139. Формат бумаги
60X90*/ie. Бумага тип. Ns 1. Гари, литературная. Печать высокая. Уел.
печ. л. 19. Уч.-изд. л. 22,12. Тираж 4200 экз. Заказ Кв 244. Цена 2 р. 80 к.
Изд. № 1225.
Ордена «Знак Почета» издательство «Химия». 107076, Москва,
Стромынка, 13.
Московская типография JV° 11 Союзполиграфпрсма при Государст-
венном комитете Совета Министров СССР по делам издательств, по-
лиграфии и книжной торговли. 113105, Москва, Нагатинская ул., д. 1.
TiK
Так
Тем
Теп
Тер
Тер
Тер
Тер
ТсГ
Тег
301
СОДЕРЖАНИЕ
Предисловие......................................................... 5
Глава I. Термодинамика растворения полимера в полимере 7
Литература .............................35
Глава II. Истинные и коллоидные растворы смесей полимеров 3d
§ 1 Истинные растворы смесей полимеров...............................38
§ 2. Расслаивание растворов смесей полимеров и свойства получающихся
эмульсий .............................................................52
§ 3. Межфазное натяжение и самопроизвольное эмульгирование в раство-
рах смесей полимеров..................................................68
§ 4. Растворы трехкомпоиентных смесей полимеров.....................72
§ 5. Влияние растворителя на структуру пленок смесей полимеров, полу-
чаемых из растворов . ... .75
Литература . . . .... 78
Глава III. Механизм смешения полимеров 42
§ 1. Эмульсия ньютоновских жидкостей................................. 83
§ 2. Эмульсия ньютоновских и неиьютсновских жидкостей .... 91
§ 3. Эмульсии вязкоупругих жидкостей, образованные растворами и рас-
плавами полимеров . ................... 94
Литература . 123
Глава IV. Коллоиднохимическая структура смесей полимеров 125
§ I. Смесь полимеров как коллоидная система . ... 125
§ 2. Размер и форма частиц .... . . 136
§ 3 Обращение фаз . ... ... 146
§ 4 Межфазный слой . 152
Литература . . . . 172
Глава V Особенности физико-механических свойств смесей полимеров 177
§ 1. Физические свойства ............................................177
§ 2. Механизм деформации ... ..........................190
§ 3. Реолш ическис свойства .... - 194
3
§ 4. Релаксация напряжений и полз'честь ... ... 225
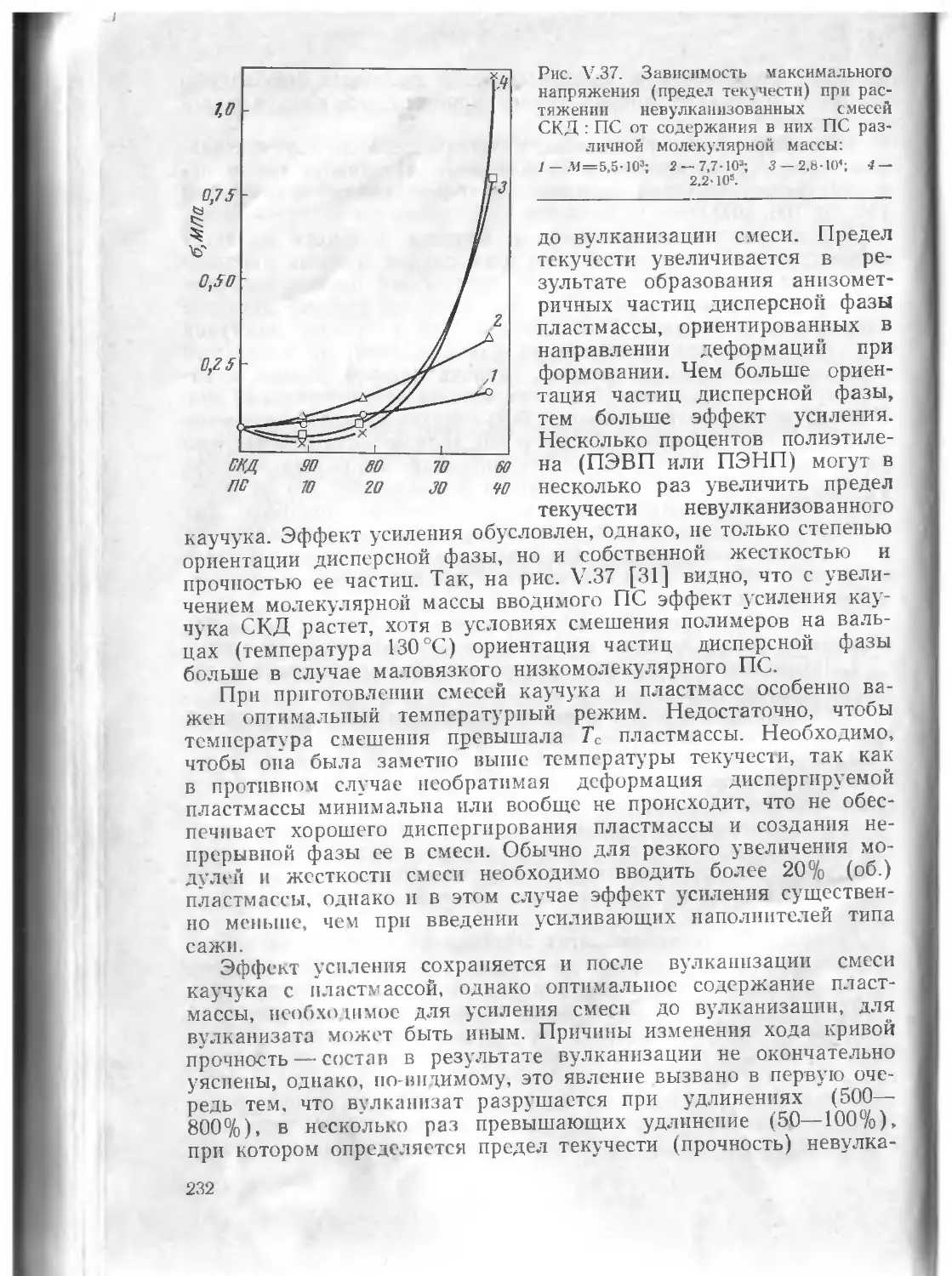
§ 5. Прочность и разрушение .... .................. 230
Литература . . ............. 250
Глава VI. Многокомпонентные системы на основе смесей полимеров 254
§ 1. Трехкимпонентные смеси полимеров...............................255
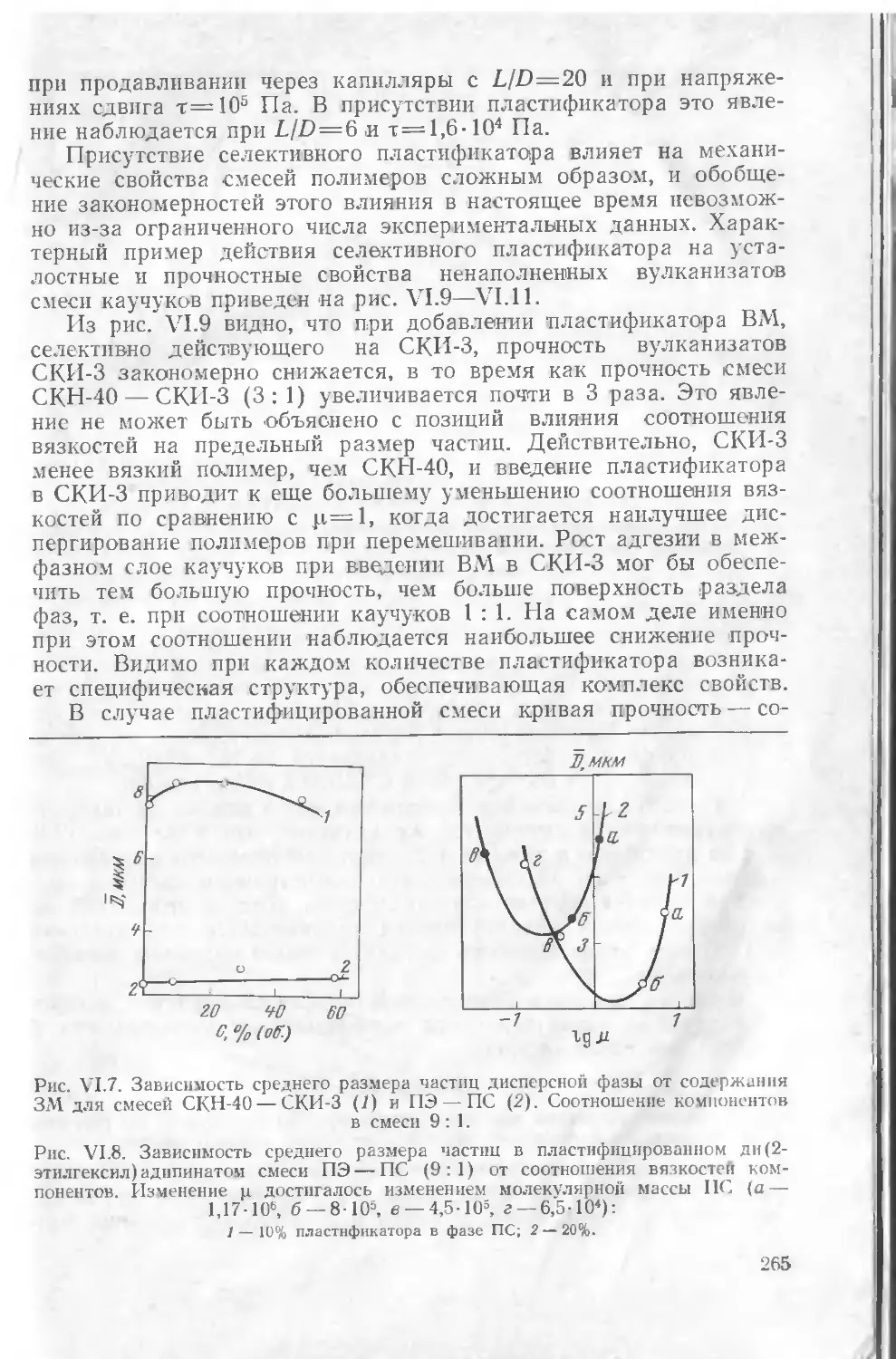
§ 2. Пластификация смесей полимеров 261
3. Наполнение смесей полимеров...................................268
§ 4. Вулканизация смесей полимеров................................ 272
Литература ... 280
Глава VII. Критические явления в смесях полимеров 282
Литература 295
Предметный указатель................................................298
ПРЕДИСЛОВИЕ
Полимерные системы, в состав которых входят два полимера,
начали применяться давно для получения материалов, сочетающих
свойства обоих смешиваемых полимеров. Так, еще в начале этого
столетия хрупкость полистирола пытались уменьшить добавлением
каучука. Это привело в конечном итоге к созданию широкой гам-
мы ударопрочных пластмасс, хрупкость которых удалось резко по-
низить благодаря наличию микрофазы эластомера*, В 30-х и осо-
бенно в 40-х годах с освоением промышленного синтеза большого
числа новых полимеров (в том числе каучуков) расширилось и
применение их смесей. Существовавшее среди ученых в 50-х —
начале 60-х годов критическое отношение, а иногда и просто недо-
верие, к смесям полимеров (вследствие обнаруженной «всеобщей
несовместимости» полимеров) позднее изменилось, так как стали
очевидны преимущества свойств микронеоднородвых систем поли-
меров. В настоящее время общепринятым является мнение, что
различные материалы можно получать, используя любые сочета-
ния полимеров, не опасаясь каких-либо нежелательных последст-
вий при применении смеси «несовместимых» полимеров. Несмотря
на то, что применение смесей быстро растет, они все еще исполь-
зуются недостаточно, особенно в промышленности переработки
пластмасс. Сопоставление этих смесей со сплавами (смесями) ме-
таллов позволяет предположить, что смеси полимеров со временем
будут широко применяться для создания новых ценных конструк-
ционных материалов.
Очевидно, следует относить к смесям полимеров системы, по-
лученные смешением двух или более полимеров в условиях, при
которых смешиваемые компоненты могут необратимо деформиро-
ваться Эти условия включают смешение полимеров при темпера-
турах выше температуры стеклования или температуры плавления
(не обязательно выше температуры текучести), смешение раство-
ров с последующим высушиванием или осаждением, смешение по-
лимеров с олигомерами с последующим отверждением, смешение
латексов или водных дисперсий с последующими коагуляцией и
перемешиванием.
Смеси полимеров являются одним из видов полимер-полимер-
ных систем, включающих смеси с полимерным наполнителем, взаи-
мопроникающие сетки, привитые полимеры и т. п. Фазовая струк-
тура этих систем задается не только (и не столько) условиями сме-
5
шения, а в первую очередь дисперсностью полимерного наполни-
теля, сохраняющего форму частиц при смешении, степенью набу-
хания полимеров в мономере, кинетикой прилитой полимеризации
и т. п.
Полчмер-полимерные системы в свою очередь являются состав-
ной частью композиционных материалов, в полимерной матрице
которых наряду с другой (или другими) полимерной фазой могут
присутствовать частицы минеральных и вообще неполимерных
фаз.
Клаоючфпкация композиционных материалов сейчас только
складывается и не является окончательной^ На основе вышеска-
занного с учетом личного опыта работы в области смесей полиме-
ров автор ограничил рамки книги собственно смесями полимеров.
Таким образом, привитые и блок-сополимеры, взаимопроникающие
сетки и т. п. привлекаются для сопоставления и и ^люстрации и не
рассматриваются систематически.
Автор выражает благодарность своим коллегам В. Д. Клыко-
вой, Л С. Крохиной, Ю. П. Мирошникову за помощь, которую они
оказали з работе над рукописью, а также всем ученикам и сотруд-
никам, участие которых в экспериментах и в их обсуждении поз-
волило сформулировать многие современные принципы создания
смесей.
Автор признателен А. С. Левашовой за помощь при оформле-
нии рукописи
Представленная книга является первой попыткой обобщить
разносторонний опыт науки и практики в создании смесей полиме-
ров; она, естественно, не могла полностью охватить накопленный
в этой области материал, поэтому все критические замечания чи-
тателей будут приняты автором с благодарностью.
Глава I
ТЕРМОДИНАМИКА РАСТВОРЕНИИ ПОЛИМЕРА
В ПОЛИМЕРЕ
Полимеры смешивают друг с другом чаше всего при температу-
ре нише их температуры текучести. При этом они находятся в вяз-
koi екучем состоянии, независимо от того, были полимеры до на-
I рсиания кристаллическими или аморфными стеклообразными. Та-
ким образом, процесс смешения полимеров в большинстве случаев
является процессом взаимоднспергирования и последующего взаи-
морлствореиня двух или более вязкоупругих жидкостей Термо-
шнамика растворения полимера в полимере — это термодинамика
изанморастворения жидких фаз, особенностью которых является
участие в процессе растворения гибких молекул достаточно боль-
шой длины.
( мешение жидкостей протекает самопроизвольно вплоть до об-
разования истинного раствора (однофазной системы), если процесс
сопровождается уменьшением изобарно-изотермического потенциа-
ла, г. е. при условии AGm<;0. Известно, что
AGm = \Нт ТASm (1)
не ДЯт— изменение энтальпии; ASm — изменение энтропии.
Если при смешении происходит выделение теплоты (АЯт<0)
и увеличение энтропии, то Gm уменьшается. Уменьшение Gm мо-
жет произойти и при поглощении тепла (эндотермический про-
цесс), если соблюдается условие |A//m| < | TASm|.
При образовании идеальной смеси молекул жидкостей 1 и 2
изменение энтропии ASm определяется следующим уравнением:
л, л, •
= — knx In — — Лл2 In - , д- (2)
где Hi и ла—число молекул смешиваемых полимеров в единице объема; k — кон-
станта Больцмана.
При переходе к полимерам, т. е. с увеличением молекулярной
массы, уменьшается число молекул в единице объема (П| и п2),
чю обусловливает малую величину ASm в соответствии с уравне-
нием (2). Так, изменение энтропии 7"ASm при образовании идеаль-
ной смеси 1 см3 одной низкомолекулярной жидкости с 1 см3 дру-
гой низкомолекулярной жидкости равно 34,8 Дж/2 см3, при смеше-
нии 1 см’ полимера, имеющего молекулярную массу 10 , с 1 см3
низкомолекулярной жидкости 7’ASm = 18,9 Дж/2 см3, а при смеше-
7
нии тех же количеств двух полимеров друг с другом 7'\5т=
= 0,0348 Дж/2 см3 [1]. -
Из этих расчетов следует важный вывод о том, что изменение
Gm при образовании идеальной смеси полимеров определяется
практически только величиной &Нт в виду малости T&Sm.
Три основные термодинамические теории смешения полимеров
были развиты Скоттом [2], Томпа [3] и Мароном [4] Эти работы
основывались на теории растворов полимеров Флори [5] и Хаггин
са [6] с учетом изменения энтропии, соответствующего идеально-
му процессу смешения. Теории Скотта, Томпа и Марона в основе
своей одинаковы, поэтому ниже излагается только теория Скотта
или, как ее часто называют, теория Флори — Скотта, что отражает
тесную связь теории [2] с теорией растворов полимеров Флори.
Теоретически термодинамический потенциал смешения полиме-
ров определяется следующим образом [2];
= р-— 'п fi + 1° 4'2 Т /isflfrj (3)
где V—общий объем смеси; V, — мольный объем мономерного звена; qr и <ы —
объемные доли полимеров 1 и 2 в смеси; xt и хз— степени полимеризации моно-
меров; Х12 — параметр взаимодействия полимеров, который определяется только
энтальпией взаимодействия мономерных звеньев цепных молекул.
При значительном увеличении молекулярной массы полимеров,
когда —ъоо, уравнение (3) упрощается:
AG,„ = (4)
Двухфазные смеси полимеров получаются при составах и тем-
пературах, соответствующих области фазовой диаграммы, ограни
ченной бннодалью (рис. 1.1). Это область неполной взаимораство-
римости полимеров при AGm>0. Внешняя область фазовой диаг-
раммы— область неограниченной взачморастворимости, т. е. об-
ласть составов и температур, в которой ДСт<0. Если вторая и
Рис. 1.1. Фазовая диаграмма бинарной системы жидкость — жидкость с нижней
(а) и верхней (б) критической температурой смешения:
1 — бннодаль; 3 — спннпцаль.
ipcii.H производная от AGm по <р обращаются в нуль, т. е.
J =d3AG>^<9q ?=0, то смесь оказывается в критической точ-
ке (вершина бинотали) Это условие позволяет рассчитать значе-
пп< параметра взаимодействия полимеров в критической точке:
* I / 1 1 V
/.икр— 2 I xVi + хЧг ) (-5’
Ih уравнения (5) видно, что чем больше молекулярная масса
(чем больше Xi и *г), тем меньше Хиг т- Это значит, что даже не-
iii.i'in le.ibiioe поглощение тепла при смешении может привести к
расслаиванию полимеров.
На рис 1.1 показана фазовая диаграмма бинарной системы
жидкость—жидкость, ( иинодаль —линия, ограничивающая об-
ои ж абсолюпю нестабильных смесей. Между бинодалыо и спи^,_________
по ia 1ыо заключена область метастабильного состояния, характе-
ри нющегося наличием особенно крупных флуктуаций состава, ког-
да система устойчива лишь по отношению к малым изменениям
сосьтва. Критическая точка является общей для бинодали и спи-
нотали. Лля любой точки спинодалн характерен относительный
максимум отрицательного значения AGm, определяемый условием
=0, в соответствии с которым из уравнения (3) можно
получить уравнение спинодалн:
1 / 1 1 \
Х12.Р= 2 х1Ф1ф+ х2ф2,Р;
Бинодаль является совокупностью точек диаграммы, в которых
химические потенциалы полимеров 1 и 2 в сосуществующих фазах
ранни. Это условие позволяет записать систему уравнений, опре-
1е.1яющую па диа1рамме положение бинодали:
In <Ti' + (1 ~ ^ ’) Ф» + *1X12 (<й)2 = In 4- (1 —) Фз + «го» (<Й)2 (?)
|П <Р2 + (1 — ) Ч>1 + *2/12 (<₽|')= = 1П <Й + (1 — j + *27.1» (ч?)2 (8)
В этих уравнениях буквы со штрихами относятся к соответству-
ющей фазе. Уравнения бинодали (7), (8) более сложны, чем урав-
нения спинодалн (6), в то же время спннодаль, как правило, рас-
положена близко к бинодали, поэтому для расчетов фазовой диаг-
раммы часто пользуются уравнением спинодалн.
В присутствии растворителя выражение для термодинамическо-
го потенциала имеет вид:
_ RTV / <г. ф,
—'чч1п<р5-|-— 1пф!+— 1п<р2 +
+ /лгФтФз + 7.is4Ws + 7»1ФгФ1) (9)
Hie V. моЛьиыП обз.ем растворителя. Хи 11 Хз» — параметры взапмодеиствин
полимеров 1 и 2 с р.п творителем; — объемная доля растворителя.
9
Координаты точки, лежащей на бннодали, для системы поли-
мер— полимер — растворитель определяются из условия
/ d=AG,„ \ / дЧвт х ( f>2.\Glrl \
V /I ^Т? а
Само уравнение бинодали можно записать, исходя из уравне-
ний (9) и (10), приняв одновременно для упрощения, что парамет-
ры взаимодействия каждого полимера с растворителем равны меж-
ду собой, а полимеры имеют одинаковую степень полимеризации:
InSi + x/nfl-lJ^)2 1п»а + х/12(1-ф,)(«;)а (Н)
где = Ч>!/(Ч! + ф2) н ₽2 = Ча (ЧI + Ч'г).
Важно отметить, что уравнение (11) выглядит аналогично урав-
нению (7) для полимеров с равной степенью полимеризации, за
исключением того, что вместо параметра взаимодействия полиме-
ров xi2 в уравнении (7) в уравнение (И) входит величина Х12 (1—
<р5). Эта последняя может рассматриваться в качестве параметра
взаимодействия в системе полимер — полимер — растворитель. Ес-
ли учесть, что в уравнение (11) не входят величины /и и Хг» (они
приняты равными и поэтому сократились в процессе преобразова-
ний), то из теории Флори — Скотта сразу следует важный вывод:
растворитель служит только для того, чтобы уменьшить эффектив-
ный параметр взаимодействия х<2(1—<Г»). Действительно, чем
меньше концентрация полимера (1—<j\). тем меньше эффективный
параметр взаимодействия и при достаточно большом 4s система
неизбежно становится однофазной. По существу аналогичные вы-
воды следуют из теории Марона [4].
По аналогии с уравнением (5) для раствора двух полимеров в
общем растворителе величина параметра взаимодействия в крити-
ческой точке определяется следующим образом
Из уравнения (12) видно, что параметр взаимодействия xi2 в
критической точке достигается для данной пары полимеров неза-
висимо от природы растворителя при условии равенства хы —Хг»-
Экспериментально показано, что в разбавленных растворах все
полимеры образуют однофазные смеси. Это подтверждает теорию
Флори — Скотта. Однако эксперимент ц/ю же время показывает
(см. гл. II), что природа растворителя существенно влияет на ве-
личину концентрации, при которой данная пара полимеров рас-
слаивается в растворе. Вообще строгая проверка уравнения (12)
невозможна, потому что невозможно подобрать растворитель с
одинаковым сродством к обоим полимерам.
Теория Флори — Скотта предсказывает и возможность расслаи-
вания сополимеров, полученных из одних и тех же мономеров при
различном соотношении мономеров в макромолекулах. Экспери-
10
мент подтвердил, что сополимеры разного химического состава мо-
I ут расслаиваться Условие расслаивания сополимера определяет-
> я уравнением [7]
Ч Ч ( RTP V7* /141
До о.о I ——I (1о)
\ 2MW J
д< р—- П.1О1 ность полимера; Дб— разность параметров взаимодействия макро-
м<и|<-н\.| сополимеров.
Если макромолекулы сополимера ие однородны по составу, а
имеют гауссово распределение параметров взаимодействия 6, и
90% сополимерных макромолекул удовлетворяют условию (13),
ю сополимер является однофазной системой. Если А6 превышает
величину, определяемую условием (13), то в смеси сополимерных
макромолекул произойдет расслаивание. Впоследствии этот вы-
нь i теории был с успехом подтвержден (качественно) рядом ав-
торов [8—10], которые показали, что при достижении высокой
конверсии в процессе сополимеризации в системе происходит рас-
слаивание.
Устойчивость смеси сополимеров можно оценить по параметру
растворимости сополимера 6Г. если известен параметр раствори-
мости соответствующих гомополимеров [11]:
бс=26‘Тт <14>
где <р<—объемная доля мономера с параметром растворимости бь
В литературе имеется сравнительно немного диаграмм взаим-
ной растворимости в системе полимер — полимер — растворитель
и еще меньше диаграмм растворимости полимер — полимер. В ра-
боте [12] исследовано равновесие в смеси олигомеров полиизо-
бутилен—полндиметнлсилоксан разной молекулярной массы, в
работе [13] равновесие в смеси олигомеров полистирол — поли-
июпрен, а в [14] смеси полимеров ПВХ — ПММА (рис. 1.2). Каче-
ственно эти работы подтверждают теорию Флори—Скотта, однако
ля того, чтобы дать новые идеи для развития теории данных
слишком мало.
В существующем виде теория дает возможность оценить весьма
приближенно, а в случае одинаковых
молекулярных масс и близкой химиче-
ской природы полимеров довольно точ-
но, их способность к взаимному раство-
рению. Действительно, в соответствии с
Pi 12 Фяаоняя диаграмма бинарной системы
НИХ — IIMMA, полученная методом электронно-
«III юпгн о мт роаналнза. Молекулярная масса
ПММ . 8Q-10*. ПВХ 110-103.
№0
140
J00
0,2 0,8 а)„вя
11
теорией Гильдебранда для регулярных растворов можно на-
писать:
у
'1.11 ~ RT (^1 (Г')
где 6| и йг — параметры растворимости смешиваемых полимеров.
Если /12^X12 кр» то полимеры при смешении способны образо-
вать однофазную смесь, и по уравнению (5) можно рассчитать
критическое значение xi2- Аналогичным образом можно поступить
при расчете взаимной растворимости полимеров при определен-
ном их соотношении. При этом х<2. рассчитанное по (15), сравни-
вают с тем же параметром, рассчитанным по (6) или по (12) для
случая растворов смеси полимеров. Параметр растворимости по-
лимеров можно найти либо в справочных таблицах, либо рассчи-
тать пи правилу Смолла [15—17].
Ориентировочная оценка взаимной растворимости полимеров
расчетным путем имеет преимущества, главным образом, в тех
случаях, когда составляются композиции для применения в виде
растворов (клеев, лаков, красок и т. п.), а также при составлении
смесей малоизученных полимеров или сополимеров, эксперимен-
тальные данные по взаимной растворимости которых отсутствуют.
Большинство обычных полимеров растворяются друг в друге
в количестве долей процента или немногих процентов, а критиче-
ская температура смешения, например, высокомолекулярных по-
листирола и полпизопрепа. рассчитанная, исходя из теории Фло-
ри—Скотта, составляет более тысячи градусов. Очевидно, что при
температурах эксплуатации и переработки полимеров на диаграм-
ме фазового состояния их смесей (см. рис. 1.1) в основном остает-
ся область двухфазных систем: бинодаль практически примыкает
к оси ординат.
Наиболее исчерпывающие данные по качественной оценке спо-
собности полимеров к взаиморастворению («совместимость — не-
совместимость») приведены в обзоре [18].
Теория Флори — Скотта позволяет сделать в целом правильные
заключения о том, что при значительной степени полимеризации
(большой молекулярной массе) параметр Х12 очень мал [урав-
нение (6)], т. е. взаимная растворимость полимеров мала. Теория,
однако, предсказывает наличие лишь верхней критической темпе-
ратуры смешения (ВКТС), а эксперимент обнаруживает для неко-
торых систем и нижние критические температуры смешения
(НКТС).
Исходя из теории Флори — Скотта и учитывая малое значение
энтропии смешения, предложены другие простые методы оценки
сродства полимеров друг к другу. Так, для оценки совместимости
полимеров можно рассчитывать параметр ₽= (61—6г)2 и считать,
что полимеры «совместимы» при р<0,0697 Дж/см3 [15]. Предло-
жено [19] рассчитывать АНт из эмпирического уравнения
Д//т = ^(61-вг)2(«-<Г1)211/1 (16)
12
I la основании сравнения параметров 6 для пар совместимых и не-
совместимых полимеров с литературными данными можно пред-
положить, что при Х//т<4.2-10 2 Дж,'см3 полимеры взакмнорас-
коримы (совместимы). Оба эти подхода могут служить для ха-
p.iKh puciiihii сродства полимеров, но не для определения их вза-
имной р;и гворимости. Например, смесь нпс-полибутадиена и цис-
н <11П1 ширена является взанмнонерастворимой [20], но в то же
время оба полимера, входящие в ее состав, практически вообще
не различаются по параметрам растворимости. Все это ограничи-
вает применимость теории Флори — Скотта для оценки взаимной
растворимости полимеров.
Для установления роли энтальпийного члена при оценке взаим-
ной растворимости полимеров были определены теплоты смешения
15 пар полимеров [21]. Оказалось, что большинство полимеров
несовместимо (совместимость оценивалась по расслаиванию смеси
в растворе в общем растворителе) и теплота при их смешении
поглощается, только в двух парах теплота выделялась и полиме-
ры действительно не расслаивались в растворе. Однако три пары
(СКБ—ПС, СКВ—СКС-30 и НЦ-АЦ) образовывали при смешении
в растворе двухфазные системы при положительном тепловом эф-
фекте смешения. Эти пары вообще не могут образовывать при ком-
натной температуре однофазные смеси ни в растворе, ни в блоке.
Полученные данные нельзя объяснить без предположения о замет-
ном вкладе энтропийной составляющей в термодинамику сме-
шения. ,
Расчет изменения энтропии смешения по уравнению (2) дает
пппь величину комбпнагориальиой составляющей энтропии, опре-
(еляемой клк изменение числа перестановок разнородны молекул
(комбинаторика процесса) при образовании смеси. Некомбинато-
рнальная составляющая обусловлена определенным взаимодейст-
вием между молекулами. Поскольку взаимодействие молекул по-
лимера 1 (взаимодействие 1—1) обычно не равно взаимодействию
2 2 (полимер 2), а это, в свою очередь, не равно взаимодействию
1—2, то при смешении может происходить изменение ближнего
порядка в системе (как увеличение, так и уменьшение), что дает
большую величину некомбинаторнальной составляющей того или
трутого знака.
Необходимость учета изменения энтропии при смешении поли-
хиров привела к попыткам «усовершенствования> теории Фло-
ри Скотта. В уравнении (3) для изменения термодинамического
поюнциала смешения параметр X12 можно выразить в зависимости
не только от энтальпии, но и от полной энтропии смешения, учет
которой в величине AGm должен способствовать лучшему совпа-
iciniio теоретических и экспериментальных результатов. Поэтому
можно пре (положить наличие концентрационной зависимости х>2
и ни те ря ja [22]:
7 7i + 7гФ + 7зФ2 +.................... I17)
13
Тогда уравнение спинодалн запишется в виде:
1/(1 — ф«) 4- (ф*«)’1'- 2£о — 2₽i (1 — 3Ф<) — 6g2<₽j (1 — 2<р{) - . (1а)
где U+1 = <*+!)(£* — £*+1): *=1.2. . .
Аналогично рассчитывают и бинодаль. Сравнение результатов
расчета с экспериментальными данными [12], при котором рас-
четные кривые строят подбирая Xi 11 %2 позволяет, естественно, хо-
рошо описать экспериментальные фазовые диаграммы. Однако
предсказать форму бинодали и спинодалн при этом невозможно,
так как коэффициенты ряда в уравнении (17) находят эмпиричес-
ким путем.
Некомбпнаторнальное изменение энтропии при смешении поли-
меров учитывалась в теории Флори [23—24], которая основана
на представлениях Пригожина о связи термодинамического пове-
дения смесей с локальной структурой (ближним порядком) сме-
шиваемых компонентов. Термодинамический эффект обусловлен
изменением объема при смешении, происходящим вследствие раз-
личий в свободных объемах компонентов. Понятие сегмента в этой
теории аналогично понятию сегмента в теории Флори — Скотта, но
различие состоит в том, что здесь сегменты имеют определенную
поверхность, по которой и осуществляется взаимодействие типа
1 — 1, 2—2 и 1—2. Величина взаимодействия по поверхностям сег-
ментов оценивается параметром контактных взаимодействий х)2.
Зависимость термодинамических функций от структуры смеси
требует введения в соответствующие уравнения параметров состоя-
ния компонентов (приведенные параметры):
''Нт = V [<р,(с^1 — сг1) 4- <г„Р£ (Vi1 — tr’) + Ф^Х^гг1] (19)
где v* — характеристически» удельный объем 1 г смеси; ? — приведенный объем
компонентов и их смеси; Р*, Т*/—характеристическое давление и температура;
0«— поверхностная доля «-того компонента, рассчитанная из соотношения по-
верхностей сегментов смешиваемого полимеров; Х12 — параметр, характеризую
щнй изменение энергии при контакте полимера 1 с полимером 2 (измеряется в
Дж/см3 или Дж/г).
Экспериментально трудно определить характеристические и
приведенные параметры, необходимые для расчета \Нт и по
уравнениям (19) и (20), поэтому их рассчитывают ^коэффициен-
там теплового расширения и изотермической сжимаемости. Одна-
ко в целом расчеты в рамках новой теории Флори достаточно
сложны, что и обусловило попытки модернизировать теорию Фло-
ри— Скотта [22].
Исходя из предложенной теории, Флори [24] рассчитал изме-
нение энтальпии и энтропии смешения полиизобутилена и линей-
ного полиэтилена в жидком состоянии (табл. 1.1). Необходимые
параметры состояния полимеров были рассчитаны из прежних ра-
14
Таблица 1.1 Термодинамические функции смешения полиизобутилена
и полиэтилена при одинаковом числе сегментов каждого полимера в смеси
t. *с ДН,П-101, Дж/г 7Д5Л1101. Длг ДСт-101. Дж/г
•л 1,1658 12.56 —23,45 35,59
ПО 1 2051 8,37 —26,38 34,33
1 .0 1,2306 10.47 —22,61 33,08
6<и автора по изучению термодинамики смешения нормальных
алканов.
Для расчета величин, приведенных в табл. 1.1, использовали
параметр Xi2 = 0,8-l Дж/г, определенный также на основе изучения
нормальных алканов. Из таблицы видно, что энтропия смешения
отрицательна, т. е. уменьшается при смешении. Это в свою оче-
редь указывает на увеличение ближнего порядка в смеси, т. е. на
го, что некомбинаториальная составляющая энтропии перекрыва-
ет '-омбинаториальную. Энтропийный вклад превосходит энталь-
пийный, и в целом AGm>0, при этом \Gm мало зависит от темпе-
ратуры. Флори отмечает, что величина \Gm находится вне преде-
лов погрешностей теории и ошибок в оценю; параметров, входя-
щих в уравнения. То обстоятельство, что рассчитанное значение
химических потенциалов положительно по отношению к исходным
жи 1ким компонентам во всем интервале составов, указывает на
взаимную несовместимость этих полимеров в условиях равнове-
сия [21|. В гл. II приведены данные, ука 1ывающне на увеличение
степени ассоциации каждого полимера в присутствии другого, что
хорошо объясняет уменьшения энтропии при смешении (см.
табл. 1.1).
Изменение XGm определяется молекулярной массой смешивае-
мых полимеров. На рис. 1.3 показаны результаты расчета AGm
при смешении при 77°C полиоксиэтилена с полииксипропиленом
разной длины цепи. Видно, что AGm<0 (полимеры смешиваются),
если пень одного из полимеров состоит из четырех мономерных
е шпиц. Полимеры плохи смешиваются (XGm«sO), если в цепь
входит 17 мономерных единиц (мол. масса 800—1000). Если в
цсш. входит 50 мономерных еди
15
Рис. 1.4. Влияние молекулярной массы Л1 и термического коэффициента расши-
рения а на вид фазовой диаграммы состояния бинарной системы полимер — по-
лимер (а, =0.580-10-3 К-1, Х1а=0)
№ кривой МХ=М, а» 103 (а2—<Х|) 100, ой
1 Б 10» 0.640 10.3
2 5-10» 0.630 8.6
3 1-105 0.630 8.6
4 1-105 0,620 6.9
5 1-103 0.615 6,0
6 2 105 0.610 Б. 2
7 2105 0,605 4.3
Рис 1.5. Влияние параметра взаимодействия Х|2 на вид фазовой диаграммы би-
нарной системы полимер — полимер (под № 5 на рис. 1.4):
1 — Хи---0,0063; 2 — 0; 3 — 0,0022; 4 — 0,0042; 5 — 0,00515; 5 — 0.0063; 7 — 0,022 Дж/см’.
рамм смесей полимер — полимер [25J. Получены сложные уравне-
ния бинодали и спинодали, в которые входит большое число пара-
метров состояния смешиваемых полимеров.
Рассчитаны две смеси ПС и ПЭ с одинаковыми и с различными
молекулярными массами полимеров. ПС и ПЭ были выбраны по-
тому, что для них в литературе опубликовано много данных по
параметрам состояния. Молекулярные массы подобрались так,
чтобы критическая температура смешения попала в область от
—100 до +300°C. Во всех случаях объем смеси и Sm уменьшались
при смешении. Параметры состояния ПС и ПЭ были близки к
параметрам многих других полимеров. Кроме тога в расчетах учи-
тывались вариации параметров в широких предежпх, поэтому полу-
ченные результаты для указанной системы можно считать типич-
ными и для других систем полимер — полимер.
16
Оказалось, что для системы полимер — полимер характерна
НКТС, а не ВКТС. Уменьшение взаимной растворимости с повы-
шением температуры наблюдалось всегда при Х|2^0 (рис. I 4 и
1.5) [25]. С увеличением молекулярной массы полимеров НКТС
сдвигается в область более низких температур. Влияние Х|2 пока-
зано на рис 1.5. При Х|2=#=0 область растворимости расширяется,
а при Xi2<0, соответствующем взаимодействию полярных полиме-
ров, диаграмма качественно не меняется, лишь КТС сдвигается в
область более высоких температур. Уже небольшие положитель-
ные значения Х)2 сильно изменяют картину. Бинодали и спинодалн
принимают форму песочных часов (кривые 5—7), характерную
для систем с двумя КТС. В этом случае взаимная растворимость
увеличивается при нагревании от —100°С. В области от —50 до
0°C верхняя и нижняя КТС сливаются и при положительных тем-
пературах растворимость уменьшается при дальнейшем нагрева-
нии. На рис. 1.5 не приведена нижняя ветвь бинодалей и спинода-
лей <3 и 4, так как для кривой 4 ВКТС соответствует— 131,6°C.
Наличие обеих КТС очень редкое явление для смесей полиме-
ров. Области значений Х12, при которых возникают две КТС, очень
узка, а температуры, при которых полимеры смешиваются, часто
лежат ниже нуля.
Из параметров состояния наиболее существенное влияние на
вид диаграммы оказывает термический коэффициент расшире-
ния а. Так, для смеси с All = Af2 = 200 000 различие между а обоих
полимеров в 4% уже достаточно, чтобы оказать влияние на взаим
ную растворимость полимеров. Различие в 20% приводит к рас-
слаиванию смеси полимеров, если молекулярная масса одного из
них составляет 4000. Молекулярно-массовое распределение (ММР)
практически не изменяет положения критической точки, если оно
меняется одинаково в обоих полимерах. Если ММР меняется нео-
динаково, то критическая точка сдвигается из области средних со-
ставов смеси в область крайних составов. Большое различие в
ММР может привести к потере взаимной растворимости. Общая
тенденция состоит также в увеличении НКТС с повышением внеш
него давления, при этом большое различие в а может привести к
начальному снижению НКТС с последующим ее ростом. С повы-
шением давления уменьшается область двухфазных систем также
благодаря сближению ветвей бинодали. Экспериментально это бы-
ло продемонстрировано на примере смеси ПММА с ПВА, которая
под давлением 1000 МПа переходила в состояние однофазной
(один максимум механических потерь) и снова расслаивалась при
нагревании выше Тс при атмосферном давлении [26].
Наиболее важным результатом, полученным в [25], является
предсказание существования НКТС для смесей полимеров. Сравне-
ние теоретических и экспериментально полученных бинодалей в
этой работе не проводилось. Построенные автором по точкам по-
мутнения фазовые диаграммы двух смесей полимеров (рис. 1.6)
имели выраженные НКТС, однако теоретическая бинодаль по по-
2—244
17
Рис. I Б. Фазовые диаграммы бинарных систем
полистирол — поливннилметиловый эфир (/) [25]
и сополимер стирола с 28% акрилонитрила —
поликаприлактон (2) [25] (темные и светлые
точки соответствуют началу и завершению помут-
нения смеси)
ПС ПВМЭ Сополимер пкл
£ £ X 3 23.6101 3.2 1,332-101 1.73 22.3-101 2.06 3.5-10» 15.6
лученнь ы параметрам не строилась. Сопоставление значений
<Г«р, рассчитанных в работе [25], со значениями, полученными по
теории Флори — Скотта, показало совпадение в пределах ±10%,
которое было тем полнее, чем ближе были значения параметров
состояния. Проведенное аналогичным образом сопоставление зна-
чении Х12кр показало расхождение в пределах 4% уже при малом
различии в параметрах состояния полимеров. Сложность расчетов
по теории Флори и по равнениям, приведенным в [25], обеспечи-
вает широкую применимость теории Флори — Скотта для оценки
состояния взаимной растворимости смесей полимеров, в том чис-
ле и в присутствии растворителя.
(^Экспериментальные данные по определению взаимной раство-
римости полимеров сравнительно немногочисленны и крайне про-
тиворечивы. Это объясняется трудностью достижения равновесия
вследствие высокой вязкости, а также тем обстоятельством, что
разные способы определения фазового состава дают разное число
фаз в смеси (см. гл. IV).
Имеющиеся данные по взаимной растворимости, или по «тер-
модинамической совместимости полимеров», можно разделить на
следующие группы.
Смешивают равпоконцетрпрованные растворы двух полимеров
в общем растворителе и проводят эксперимент при максимальной
общей концентрации смеси в растворе. Отсутствие расслаивания
принимают за указание на взаимную растворимость полимеров.
Вывод этот, как правило, неверен, поскольку исследуемый интер-
вал концентраций не превышает 10—15%, а иногда еше меньше.
Расслаивание же при удалении растворителя наступает и при бо-
лее высоких концентрациях, что приводит к получению мутных
пленок из прозрачных растворов.
Из раствора получают прозрачные пленки однофазной структу-
ры. Вывод о взаимной растворимости полимеров может быть оши-
бочным, поскольку структура пленки зависит от природы выбран-
ного растворителя, поэтому пленки, полученные из разных раство-
рителей, могут быть как мутными, так и прозрачными. Равновесие
же в пленке в отсутствие растворителя практически не достигает-
ся, и оценить фазовый состав в условиях равновесия, без учета
влияния растворителя, не удается.
Смешивают полимеры на вальцах или в другом смесительном
оборудовании в отсутствие растворителя. Получаемые смеси оче-
видно неравновесны, поскольку в вязкой смеси даже при переме-
шивании процесс взаиморастворения может не завершиться, или
интенсивное перемешивание, сопровождающееся механохимически-
ми реакциями, может привести к получению однофазной смеси
несовместимых полимеров вследствие механодеструкции и после-
дующей прививки^
Очень небольшое число смесей можно надежно отнести к числу
взаимнорастворимых независимо от соотношения полимеров в сме-
си. Это, тЪ-первых, смесь НЦ и ПВА, однофазность которой при
получении из разных растворителей была подтверждена разными
методами [18, 27, 28]; полимеры во всей области составов смеши-
вались экзотермически [28], при этом во всей области составов
\Gm<0.
Однофазна смесь цис-полипзопрена и разветвленного полибу-
тадиена СКВ [20]. В зависимости от условий получаются одно-
фазные смеси ПС с полиметилвнниловым эфиром [29, 30] и одно-
фазные смеси ПВХ и СКН-40 [31].
Полные данные о взаимной растворимости полимеров данной
молекулярной массы могут быть получены только в результате ис-
следования температурной зависимости растворимости. Это осо-
бенно важно тля полимеров, поскольку при их смешении уменьше-
ние энтропии видимо типичное явление, а при ASm<0 фазовая
диаграмма характеризуется наличием НКТС. Диаграмма смеше-
ния олигомер — олигомер приведена в [13], а полимер — по-
Рнс. 1.7. Зависимость растворимости полистирола и полиметнлетнрола в полннзо-
прене (/) и иолиметилметакрилате (2) от молекулярной массы полимеров в
обычных (б) и логарифмических координатах (о). Молекулярная масса ПС:
2660. 2780, 5620, 9,8-103—550-103; ПМС: 870, 6670 [36].
2*
19
лимер— на рис. 1.6. Если молекулярные массы ПС увеличить до
2- 10s, а ПВМЭ до 51,5-103, то НКТС смеси снижается со 120 до
80 °C.
Сравнение данных показывает, что, по-видимиму, для олиго-
меров вероятнее существование диаграмм с ВКТС, а для поли-
меров— диаграмм с НКТС. Например, уменьшение взаимной рас-
творимости полимеров с повышением температуры наблюдали
для смесей стиролакрилонитрильного сополимера с ПММА [32],
пол и дифенилового эфира с ПС [33] и бутилкаучука с этилен-про-
пиленовым сополимером [34].
Взаимная растворимость полимеров зависит от молекулярной
массы компонентов (рис. I 7). Видно, что при больших молекуляр-
ных массах (более 104) растворимость с изменением молекулярной
массы не изменяется. Растворение одного полимера в другом (ког-
да пределы растворимости малы) можно рассматривать как рас-
творение в плохом растворителе. Зависимость мольной доли поли-
мера V в растворе в плохом растворителе от молекулярной мас-
сы полимера [35] описывается формулой
N-=e~A”
где А — константа для данной пары полимер — растворитель.
Эта формула хорошо описывает также растворимость полиме-
ра в полимере; электрополяцня к 100%-ной концентрации позво-
ляет оценить молекулярную массу ПС, соответствующую неограни-
ченной смешиваемости ПС с ПИП и ПММА, равную соответствен-
но 500 и 1600.
Данные по растворимости полимеров в полимере приведены в
табл. 1.2.
Приведенные данные не всегда являются количественно стро-
гими. Так, в таблице приведена растворимость ПС в ПММА по
данным маллуглового рассеяния. Величина растворимости суще-
ственно ниже величины, полученной по данным оптической плот-
ности [36] Одной из причин является то, что разные методы мо-
гут давать разные результаты (даже при одинаков^ условиях
приготовления образцов). Так, в методе оптической плотности по-
явление мутности пленки наблюдается по достижении частицами
размера 700—900 А, поэтому момент выделения второй фазы фик-
сируется с запозданием (при более высокой концентрации раство-
ряемого полимера); в методе малоуглового рассеяния могут быть
зафиксированы ассоциаты макромолекул, не являющиеся еще ча-
стицами второй фазы, т. е. предел растворимости оказывается за-
ниженным Известно, например, что в смеси сополимеров метилмет-
акрилата и бутилметакрилата при большом различии в химиче-
ском составе макромолекул наблюдается гетерогенность в пленке,
полученной из раствора. В электронном микроскопе гетероген-
ность заметна при различии в содержании ММА в смешанных со-
20
Таблица 1.2. Растворимость полимера 1 в полимере 2
Полимер 1 Afj Полимер 2 М2 Раствори- мость С, % (масс ) Литературный источник, методы
ПС ПММА 0.9 [36]. оптическая плот-
(370 10s) («7-10’) 0,14 ность, электронная мик- роскопия
ПС (370 10’) ПИП (1-10») То же
СКН-18* СКД 3,0 [37], оптическая плот- ность
СКД* CKH-18 0,8 То же
< :кс-зоаркм* HK 5.0 >
НК* CKC-30APKM 1,0 »
Олигомер БА (2400) ПММА (1.65-10s) 5,0 [38], оптическая плот- ность, вязкость
Олигомер этилгексил- акрилата (2200) 1,0 То же
Олигомер ЭА 40,0 То же
Полихлоропрен Полнтрихлорбута- 13.6 [39], оптическая плот-
(1610’) диен ность
(25-10’) 12.7 То же
(60 10s) 9,1 »
(90 10‘) 7,5 >
(1,44- 10s) 7.2 >
(2.20 10s) 6.4 >
(3.63 10s) 6,3 >
(7.95- 10s) 6.2 »
(1.1-10») 6,2 [40]. оптическая плот-
СКИ ПММА 0,8
9.0 10s ПММА (0.9 10s) (0.9-10') СКИ ность, физико-механиче- ские показатели
CK I (9.0-10s) ПС 1 1 То же
(2.5 10s) (3.7-10’) 0,5 >
ПС (3.7 10s) СКД (2.5-10s) 0 4 >
ПЭ* ПОД)* 2,0 [41], деструкция в плаз- ме, ВЧ-разряда, плот- ность
11OM ПЭ* 3,0 То же
IIOM* ПЭ* [42], рентгеноструктур- ный метод
C.KII* СКД* 1,0 [20], термомеханический и РТЛ метод
< кд* СКИ* 1,0 То же
IIC* ПММА* 0,008 [43], малоугловое рас- сеяние
• Молекулярные массы не указаны, технические полимеры не подвергались дополни-
тельной очистке.
полимерах 10%. мутность в плеш е появляется при различии в
13%, а второй максимум на температурной зависимости механиче-
ских потерь возникает при 20% различия в содержании ММА [10].
Характерно, что в случае сополимеров ММА и БМА взаимная
растворимость компонентов не увеличивается при добавлении
третьего компонента, порознь совместимого с каждым полимером
смеси, как это наблюдалось для системы стирол — акрилонитрил
[44]. В смесях образцов с широким ММР увеличенная взаимная
растворимость (см. табл. 1.2) может возникнуть в результате пе-
рехода из фазы в фазу низкомолекулярных фракций.
Способностью неограниченно смешиваться обладают ПС и
атактический ПП с молекулярными массами 330—630 [45]; П.ММА
с молекулярной массой около 1500 является тета-растворителем
для высокомолекулярного ПС [46].
Из табл. 1.2 и рис. 1.7 видно, что предел молекулярной массы,
ниже которого начинается увеличение растворимости с уменьше-
нием степени полимеризации зависит от природы полимеров. Так,
при растворении полнхлоропрена в политрнхлорбутадиене этот
предел заметно выше, чем в парах ПС — ПММА и ПС— ППП.
Значительная взаимная растворимость наблюдается для следу-
ющих полимеров в смесях: ПВХ — ПВА [47], ПВХ — сополимер
бутадиена и метилстирола [48], поливинилиденфторид и ПММА
[49], ацетобутпрат целлюлозы и СКН-40 [50], бутадиенстироль-
ный каучук СК^ЗО t сополимер СКС-85 [51].
Растворимость полимеров др) г в друге повышается при их свя-
зывании химическими связями в молекулы блок-сополимеров. Так,
блок-сополимеры стирола н а-метилегирола однофазны, даже если
молекулярные массы блоков превышают 200 000; при этих молеку-
лярных массах механическая смесь указанных гимополимеров оп-
ределенно двухфазна [52]. Аналогичное явление продемонстриро-
вано на примере блок-сополимеров бутадиена н стирола [53]:
миьрорассланвание в растворе блок-сополимера происходит при
более высокой концентрации, чем в смеси растворов ПБ и ПС той
же молекулярной массы н при том же соотношении компонен-
тов.
Кристаллические полимеры могут образовывать однофазные
смеси только при изоморфном строении макромолекул. Изомор-
физм крайне редкое явление, без которого невозможно построение
смешанных кристаллов.
Наиболее полная характеристика взаимной растворимости в
системе жидкость — жидкость дается зависимостью термодинами-
ческого потенциала Gm от состава. Попытки экспериментального
определения этой величины для смесей полимеров оказывались
обычно безуспешными из-за высокой вязкости и трудности дости-
жения равновесия. В настоящее время получены некоторые экспе-
риментальные данные в этой области.
Метод определения давления пара растворителя над набухши-
ми полимерами или их смесью позволяет рассчитать AGm [54].
22
Действительно, раствор смеси полимеров в общем растворителе
может быть получен двумя путями:
т, полимера 1 + тг полимера 2 раствор 3 — AGm
раствор 3 + растворитель = раствор 4 — АОщ
или
т, полимера 1 + растворитель — раствср 1 — AGj
гп„ пслимера 2 + растворитель = раствор 2 — ДСц
раствор 1 + раствор 2 = раствор 4 — AGjy
Отсюда, в соответствии с законом Гесса, следует
— AGm— AGjn= —AGj — AGji — AGiv (21)
Поскольку для очень разбавленных растворов AGp—Н). свобод-
ная энергия смещения полимеров мпжет быть вычислена фактиче-
ски по принципу аддитивности:
- AG„, = Д0ш - (AG, + Д0п) (22)
где AGi. AGn, AGm— свободные энергии смешения полимеров 1 н 2 и их смеси
с общим растворителем в расчете на 1 г полимера.
Если массовые доли полимеров смеси ic'i и и>2, то свободная
энергия смешения на 1 г смеси вычисляется как разность между
экспериментально определенной величиной взаимодействия компо-
нентов в смеси и средневзвешенной величиной взаимодействья
макромолекул в смешиваемых полимерах:
= AG]j| — (wt AG] -|- WjAGji) (23)
Можно опре делить свободную энергию смешения полимеров
н< 1аппы\ по обращенной газовой хроматографии [55,56]. Поэто-
му методу на носитель в хроматографической колонке наносят тон-
кий слой смеси полимеров (смешанная фаза) и определяют удержи-
ваемый мольный объем растворителя Ут. Если среднюю молеку-
лярную массу смешанной фазы обозначить через Af, а мольную-
юлю (-того компонента смеси — через х-, то свободную энергию
смешения компонентов AG,„ можно определить по уравнению
1п И„, = 2 Xl In Vmi - (24)
I
Этими двумя методами были определены значения AGm сме-
сей олигомеров [56] и смесей полимеров [54].
Hi рис. 1.8 видно, что в смеси сополимеров термодинамические
параметры смешения отрицательны. Так, JAScO, что указывает
на увеличение ближнего порядка в смеси по сравнению с исходны-
ми компонентами и на большой вклад некомбинаториальной энт-
ропии смешения в общую величину энтропии.
В табл. 1.3 приведены значения &gm для разных пар полиме-
ров. образованных взаимнонерастворимыми полимерами, которые
полому двухфазны. Несмотря на двухфазную структуру и, следо-
23
Рис. 18. Зависимость термодинамических
функций (Дж/г смеси) смешения ПВЛ и
нитрата целлюлозы от состава смеси. Неза-
висимыми методами доказана однофазиость
смесей, полученных испарением растворите-
ля из смеси растворов [57]:
2-Лйт: 3-7X3,,,.
вательно, наличие свободной поверхностной энергии, некоторые
значения AGm отрицательны, что может указывать на значитель-
ное сегментальное растворение в межфазном слое [57]. Следует
отметить, однако, что в двухфазных смесях избыточная энергия
«смешения» есть свободная поверхностная энергия. Если срав-
нить [58] приведенные в табл. 1.3 значения с величинами, рассчи-
танными, исходя из фактической удельной поверхности раздела
фаз в смеси, и из величины экспериментально определенного меж-
фазного натяжения, то рассчитанные величины \Gm, конечно, по-
ложительны и на два-три порядка ниже табличных. Это различие
пока не нашло объяснения, особенно в связи с результатами рас-
чета AGm из рис. 1.3, показывающего, что вся кривая AGm—<р
смещается в область положительных значений с ростом степени
полимеризации смешиваемых компонентов.
Таблица 13. Свободная энергия смешения в некоторых
смесях полимеров [57]
Соотношение полимеров Лдт. Дж/г Смесь получена
АЦ:ПВА = 7:3 ЛИ НЦ=7:3 ПММА: ПБМА=2 :8 ПВХ ПММА=7:3 ПВХ :СКН-26=1 : 1 ПВХ :СКБ=1 : 1 —3,5 —5,0 0 2 —4,0 —0,7 + 1.1 +0,6 Из раствора То же » » Прессованием смеси порошков Смешешгем расплавов То же
Фазовый состав смесей
полимер (1) — полимер (2) — растворитель
Как уже отмечалось [см. уравнение (11)], в уравнение бино-
дали раствора смеси полимеров вместо параметра взаимодействия
полимеров входит величина xiz(l—<F»). которая рассматривается
как характеристика взаимодействия в системе полимер — поли-
мер— растворитель. Теория Флори — Скотта, таким образом, рас-
сматривает растворитель как простой разбавитель, снижающий
взаимодействие взаимнонерастворимых полимеров, что при малой
24
Рис. I 9. Фазовая диаграмма трехкомпонентной системы полимер — по 1нмер —
растворитель при Х| = х2=«1000. Хо=Хг« 11 Х«=20 (°)- 8 (б) и 4 (в) [2]
Рис. 1.10. Бинодали фазовой диаграммы полимер — полимер — растворитель для
Xt=4000. х2=250 и х=0.02 npi х>» 11 Х21. равных соответственно 0.2 и 0.3 (с),
0,3 и 0.45 (б). 0,45 и 0,3 (в) [59].
концентрации раствора может сделать систему однофазной. С уве-
личением параметра Х12 (рис. 1.9) уменьшается область взаимной
растворимости полимеров в растворе, описываемая симметричной
бннодалью, критическая точка на которой соответствует соотно-
шению полимеров 1:1.
Расчеты бинодалей для растворов смесей полимеров в большом
объеме приведены в работе [3]. Несимметричные фазовые диаг-
раммы были получ< ны автором для системы, в которой молекуляр-
ные массы полимеров различались в 10 раз, при этом критиче-
ская точка смещалась по оси составов в сторону полимера с мень-
шей молекулярной массой. Оказалась несимметричной и фазовая
диаграмма для случая xt—х3. когда взаимодействие одного из по-
лимеров с растворителем было равно hj.tio (xi«=0). а взаимодей-
ствие другого равно взаимодействию полимеров (x2«=Xi2)- Во всех
проделанных расчетах влияние природы растворителя на взаимную
растворимость полимеров практически не исследовалось, хотя по-
следний случаи, рассмотренный в [3], jназывает на ошибочность
подхода к растворителю как к простому разбавителю смеси поли-
меров. снижающему параметр взаимодействия в соответствии с
формулой на xi2< 1—Ф«).
В более поздних работах, например [59], на основе теории
Флори — Скотта были рассчитаны бинодали для тройных систем с
применением современной вычислительной техники, в том числе и
1ля случаев, рассмотренных ранее Скоттом и Томпа. Для систем
<• одинаковой степенью полимеризации и одинаковым взаимодейст-
вием обоих полимеров с растворителем бинодаль симметрична,
при Xi^=x2 симметрия бинодали нарушается, а при увеличении
параметра в 1аимодействия полимеров (возрастание Х’г) умень-
1п ц*тся область взаимной растворимости, что было в принципе по-
25
казано и ранее. Основная ценность работы [59] состоит в том, что
в ней была проанализирована расчетным путем роль природы рас-
творителя в системе (рис. 1.10). На рис. 1.10 показаны бинодали
при условии А‘15^Х2 при ПОСТОЯННОМ отношении Х1в/Х2« (кривые а
и б) или при постоянной разности (хы—х»») (кривые б и в). Из
рисунка видно, что чем больше различие во взаимодействии поли-
мера с растворителем, т. е. чем больше разность (хы—Хг«) 1,лп от-
ношение хы/%2«, тем меньше область взаимной растворимости поли-
меров в растворе; при этом значительно нарушается симметрия
диаграммы фазового состояния. При 'постоянном отношении х «//2«
растворимость полимеров уменьшается с ухудшением абсолютно-
го значения качества растворителя по отношению к обоим полиме-
рам (рис. 1.10, кривые а и б) и с приближением х<« к критической
величине 0,5. Расчет показал, однако (па рис. 1.10 не приводится),
что при одинаковом взаимодействии полимеров с растворителем
(например, х>«—Х2« = 0) положение бинодали не зависит от абсо-
лютного значения Xi«* а определяется лишь взаимодействием са-
мих полимеров Х12-
Несколько ранее расчет спинодалей различных тройных систем
был проведен в работе [60]. Закономерности изменения вида спи-
нодалей аналогичны изменению вида бинодали при изменении
взаимодействия компонентов системы, однако особенность расчета
[60] состояла в том, что он был выполнен для полимеров (xi2—0),
помешенных в растворители, сильно различающиеся по взаимо-
действию со смешанными полимерами. Оказалось, что под влия-
нием растворителя может произойти расслаивание полимеров
с образованием фазовой диаграммы в визе петли, размер кото-
рой зависит от природы растворителя (рис. 1.11, кривые г и г').
Расчетным путем показано также, что синодаль в виде замкнутой
петли получается для систем, в которых взаимодействие полиме-
ров (Х12) близко к критическому хыкр. з растворитель неодинаков
по отношению к обоим полимерам, например Х'2/Х1з=1.2—1,5.
Из работ [59, 60] следует вывод о том, что на взаимную рас-
творимость полимеров в растворе влияет не только собственно вза-
имодействие полимеров, но и природа растворителя, которая в оп-
ределенных условиях может обеспечить образование однофазно-
го раствора смеси несовместимых полимеров или двухфазного рас-
твора смеси совместимых полимеров. Отсюда следует, что метод
оценки совместимости полимеров по расслаиванию их растворов
не может дать однозначных результатов, даже на качественном
уровне («совместимы» — «не совместимы») и даже при условии ис-
следования фазового состава смеси растворов в широком интерва-
ле концентраций смеси полимеров.
Рассмотренные выше теории растворов смесей полимеров и рас-
считанные бинодали могут быть сопоставлены с рядом экспери-
ментальных данных. Экспериментальные данные по определению
спинодалей в растворах смесей полимеров практически отсутствуют.
Первые экспериментально определенные бинодали тройных
26
Рис. Ill. Спинодалн фазовой диаграммы смеси полимеров в растворе при Х]=
=х2=1000 для Xi2=0,025 (а, б, в. в') и Xi2=° (г. г'). Параметры х>« и x2i рав-
ны соответственно:
а — 0,40 и 0.40; б — 0.40 и 0.45; в — 0,40 н 0,50; е' — 0,40 и 0,30; г — 0,40 и 0,50; г' — 0,40
и 0,30 [601.
Рис. 112. Фазовая диаграмма системы ацетат целлюлозы — поливинилацеталь —
ацетон. Молекулярная масса полимеров:
a— «,=9.7-10' и «,=5.6-10'; б — 9.7 -10* и 1,6-10*. в — 3,9 10* и 5.6 10*; г — 3.910' н 1,5 13*.
систем полимер — полимер — растворитель были получены [61]
еще до появления теории Флори — Скотта. В работе [61] впервые
были систематически изучены диаграммы растворимости или про-
сто растворимость полимеров при неизменном их соотношении и
переменной концентрации смеси в растворе на примере 35 систем.
Были получены треугольные диаграммы для систем ПС—НЦ в ме-
тнлэтнлкетонс, ПВА — поливинилацеталь в метилэтнлкетоне,
ПВА—ПС в хлороформе и метилэтнлкетоне, НЦ — поливинилаце-
таль в метилэтнлкетоне и ацетоне, НК — в бензоле и по четыре
системы, различающиеся молекулярными массами в смесях ПС —
поливинилацеталя в хлороформе и ацетата целлюлозы — поливи-
нилацеталя в ацетоне.
На рис. 1.12 приведена диаграмма состояния для смесей ацета-
та целлюлозы различных молекулярных масс с поливинилацета-
лем в ацетоне. Из рисунка видно, что диаграмма несимметрична
вследствие разной длины смешиваемых макромолекул и что основ-
ную часть диаграммы занимает область нестабильных расслаиваю-
щихся растворов. Эта область увеличивается с возрастанием моле-
кулярной массы. В большинстве систем, рассмотренных в [61],
расслаивались уже 2—5%-ные растворы. Характер бинодалей ка-
чественно согласуется с основными выводами теории Флори —
< котта.
Экспериментально установленное уменьшение взаимной раство-
римости полимеров в растворе с увеличением их молекулярной
массы согласуется как с предсказаниями теории Флори — Скотта,
। iK и с результатами последующих работ [62—64, 46, 65]. В ука-
27
занных работах исследовали, как правило, фракции полимеров в
одном или нескольких растворителях, ограничиваясь изменением
концентрации смеси в растворе при постоянном соотношении поли-
меров.
Количественное сравнение бинодалей, полученных эксперимен-
тально и рассчитанных по теории Флори — Скотта для полимеров
с разными молекулярными массами, приведено на рис. 1.13 и 1 14.
[63, 66]. Смешиваемые полимеры имели близкие молекуляр-
ные массы, а выбранный растворитель (судя по значениям /и и
/2я) был хорошим по отношению к обоим полимерам (рис. 1.14).
В работе [66] (рис. 1.13) параметры взаимодействия полимеров с
растворителем не приведены. Для расчета бинодалей необходим
также параметр взаимодействия /|2. Автор работы [66] экспери-
ментально не определял его, а подбирал таким образом, чтобы
рассчитанная бинодаль проходила вблизи экспериментальной. На
рис. 1.13 экспериментальные точки сравниваются с рассчитанными
бииодалями 1 и 2, полученными при выбранных величинах /|2,
равных соответственно 0,038 и 0,025. Бинодаль, рассчитанная по
Скотту для /12=0,038 (кривая 7), совпадает с экспериментальной
вблизи вершины. С увеличением концентрации кривые расходятся,
и в этой области составов с экспериментальными точками совпа-
дает кривая, полученная для /12=0,025 (кривая 2).
На рис. 1.14 бинодаль рассчитывалась при /12=0,62, экспери-
ментально определенным по данным состава сосуществующих фаз
расслоившихся растворов полимеров [67]. Совпадение экспери-
ментальных данных наблюдалось в достаточно широкой области
составов, однако из анализа рис. 1.13 и I 14 можно заключить, что
полного совпадения теории и эксперимента во всей области со-
ставов не наблюдается.
Рис. 1.13. Фазовая диаграмма систем НК — ПММА—бензол (а) и НК—
ПММА— и-бутилацетат (б):
а — М3=3-I05: бинодали рассчитаны для Хи 0,038 (/> и 0.025 (2), светлые кружки
экспериментальные точки; aMi=3IO5 н ЛЬ==1.6-10в: бинодаль (3). темные кружки — экспе-
риментальные точки; б — !—.M|=Mj==3-105; 2 — Л11 = 3-105 и Ма=10"-
28
Рис. 1.14. Фазовая диаграмма системы ПС—ПП — толуол при 30°C [63):
а — бпнодаль, рассчнтакн я по Скотту для Л1,=5.64 10* н М2=6,010* при <Хц=0,62. а1$ =
=4,28 и а =4,40 (моль/л) при Vs =61.41 л/моль; б — экпернментальная бпнодаль для
нсфрикционных полимеров с Л1| — 27.5-10* и Л(а—10,5-10* (а-=х/1')_
Рис. 1.15. Фазовая диаграмма смеси полиизобутилен—полистирол—растворитель:
Молекулярные массы олигомеров; 0—1100 и 2700 (толуол) •—1100 н 2720 (бензол); А —
1670 и 2720 (толуол); А — 1670 н 2720 (четыреххлорисгыП углерод) |70).
При смешении полимеров с небольшой молекулярной массой
растворы остаются однофазными вплоть до довольно высоких кон-
центраций, и значительная часть диаграммы является областью
взаимной растворимости, как это было показано на примере сме-
си олигомеров ПС и полиизобутилена (рис. 1.15) [68].
Экспериментально установлено как увеличение [69], так и
уменьшение [65, 69, 70] концентрации расслаивания полимеров в
растворе с повышением температуры. Так, отмечено [69], что
смесь ПС и ПММА в о-ксилоле, расслаивающаяся при 25°C, ста-
новится однофазной при 85 °C, и в то же время эта смесь в меток-
сибензоле гомогенна при 25 °C и расслаивается при 85 °C. Сниже-
ние концентрации расслаивания при повышении температуры на-
блюдали в растворах ПС и ПБ в тетралине [65] и стироле [70] и
в стирольных растворах смесей ПС с бутадиен-стирольными сопо-
шмерами [70]. Совершенно ясно, что рост концентрации расслаи-
вания полимеров в растворе с повышением температуры не согла-
суется с теорией Флори — Скотта и может быть объяснен только,
исходя из теории Флори и основанных на ней расчетах [25].
В соответствии с теорией Флори — Скотта растворитель рас-
• матривается как простой разбавитель, а согласно теории Флори,
р.нвитой в [59, 60], взаимная растворимость полимеров зависит
>1 природы применяемого растворителя. Экспериментально пока-
то, что критические концентрации расслаивания различны в раз-
ных растворителях [61, 69]. Наибольшая смешиваемость полиме-
рии наблюдалась в растворителях, параметры взаимодействия ko-
i' 'рых 6 были близки к 6 полимеров. В плохом растворителе рас-
шивание пронсхо шт при более низких концентрациях (ср. взаим-
29
пую растворимость каучука и ПС в смеси их растворов в бензоле
и бутилацетате, рис. 1.13). Влияние природы растворителя на сме-
шиваемость полимеров видно и из данных рис. 1.15. На примере
[71] смеси ПС — ПММА и ПММА — поли-1,2-винилпиридина, ког-
да сродство полимера к растворителю оценивали по величине кон-
станты а в уравнении Куна — Марка — Хаувипка сделай вывод о
том, что с увеличением разности au—etas взаимная растворимость
уменьшается. Это качественно подтверждает вывод работ [59, 60],
показавших, что взаимная растворимость полимеров уменьшается
с возрастанием разности %ie—%2Я. В работе [63], в которой иссле-
довались смеси атактического ПС с ПП в девяти растворителях
отмечено влияние растворителя на взаимную растворимость поли-
меров, но определенной закономерности такого влияния не обна-
ружено. В работе [36] показано, что с ухудшением качества рас-
творителя в растворе смеси ПС — ПММА, оцениваемого по харак-
теристической вязкости растворов полимеров, бинодаль сдвигалась
в область более низких концентраций полимеров.
Указанные работы подтверждают влияние природы растворите-
ля на диаграмму фазового состава, поскольку состояние системы,
ее фазовый состав, естественно, определяется взаимодействиями
полимер — полимер, полимер (1)—растворитель и полимер (2) —
растворитель. Эти работы, однако, не отвечают на вопрос о том,
остается ли собственно взаимодействие полимеров, определяемое
величиной %12, неизменным при изменении природы растворителя,
что чрезвычайно важно при определении величины Х12 методом из-
мерения состава сосуществующих слоев, по второму вириальному
коэффициенту и другим методам, связанным с исследованием рас-
творов смесей полимеров.
В настоящее время известны экспериментальные факты, пока-
зывающие, что взаимодействие самих полимеров в растворе зави-
сит от природы растворителя. Меняя природу растворителя, взаи-
модействие полимеров можно изменить настолько, что из данной
пары полимеров можно формовать либо однофазные, либо двух-
фазные пленки. Это было показано на примере смеси ПС с поли-
метнлвнниловым эфиром [72], хлоркаучука с сополимером этилена
с винилацетатом [73] и смесей некоторых полярных полимеров
[27], что свидетельствует о зависимости самого эффекта совмести-
мости или несовместимости от природы растворителя.
Имеется и прямое указание па изменение взаимодействия поли-
меров в растворе в зависимости от природы растворителя [74],
которое основывается на следующем эксперименте. Хлорирован-
ный полихлоропреп (63% хлора) с молекулярной массой 20-103 и
метилвинилпиридиповыи сополимер (СКМВР 10) с молекулярной
массой 5-103 могут реагировать друг с другом с образованием ио-
на пиридиния, определяемого по отношению интенсивности полос
поглощения 1570 и 1450 см *. Оказалось, что интенсивность погло-
щения, т. е. количество ионов пиридиния в растворе и в пленке,
полученной из растворов в толуоле, меньше, чем в растворе в
30
I’„с. 1.16. Содержание иона пиридиния, опре-
деляемого по относительной оптической плот-
ности Z>i57o/£>u5o в растворе (/, 3) и в прогре-
тых пленках {2, 4).
хлороформе (рис. 1.16). Независимы-
ми опытами было определено качест-
во растворителей по отношению к
обоим полимерам. По данным харак-
теристической вязкости, по измерению
тепловых эффектов конденсации па-
ров растворителя из раствора разной
концентрации и по количеству осади-
теля, необходимого для осаждения
полимера из 7%-ного раствора, оказалось, что толуол является с
термодинамической точки зрения более хорошим растворителем,
чем хлороформ (характеристическая вязкость растворов полиме-
ров в толуоле выше, чем в хлороформе). Рис. 1.16, таким образом,
показывает, что в хорошем растворителе взаимодействие полиме-
ров менее интенсивно, чем в плохом. Видимо, при уменьшении
взаимодействий типа 1—S и 2—S увеличивается взаимодействие
полимеров 1—2. Повышение взаимодействия полимеров по поляр-
ным группам следует также и из уменьшенного содержания по-
лярных групп на поверхности пленки, полученной из плохого рас-
творителя: угол смачивания пленок водой меньше для образцов,
полученных из плохого растворителя.
Вывод о зависимости параметра х12 от природы растворителя
следует и из экспериментов со смесью 11(3(1) и ПММА(2) в рас-
творе в бензоле, хлороформе и этилацетате [75]. Молекулярные
массы полимеров колебались в пределах от 4-Ю4 до 4-106, пара-
метры взаимодействия обоих полимеров с каждым растворителем
в отдельности были практически одинаковы. Так, xi2 для ПС и
ПММА в бензоле равнялись 0,446 и 0,437, в этилацетате соответст-
венно 0,486 и 0,475, а в хлороформе 0,380 и 0,402. В каждом из
указанных растворителей для четырех смесей, различающихся мо-
лекулярными массами полимеров, по точкам помутнения были по-
строены диаграммы фазовых состояний. Параметр взаимодействия
определяли по формуле Скотта, справедливой для случая, когда
параметры взаимодействия каждого полимера с выбранным рас-
творителем равны между собой:
«1гкр = = 0.5 (Vf0-5 + V-0'5)2(1-фжр)-* (25)
me ,,р — объемная доля растворителя в критической точке; Vt и Г2— мольные
объемы полимеров 1 и 2; V—мольный объем мономерной группировки полиме-
ров. Обычно принимают V= Vs.
Точность определения критической концентрации не ниже 5%.
Результаты определения приведены на рис. 1.17. Видно, что пара-
метр взаимодействия полимер — полимер аи кр возрастает с умень-
31
Рис. 1.17. Зависимость параметра взаимодействия ПС и НММЛ в критических
точках расслаивания от молекулярной массы полимеров и природы растворителя:
1 — 3.2- 10’ и 4,0-10’; 2 — 3.2-10’ и 2,610е; 3—3.2-10® и 9,8-10*; 4-4,0-10* и 9,8-10*. Раствори-
тели: этилацетат xls =0,48; метилэтилкетои xls =0,47 и бензол xls =0,44.
шением молекулярной массы полимеров и с ухудшением термоди-
намического качества растворителя. Эта же закономерность сох-
раняется и для расслоившихся растворов [76].
Полученная линейная зависимость параметра ецгкр для разных
фракций полимеров позволяет упростить способ определения 712.
Достаточно для пары полимеров с любыми молекулярными масса-
ми определить сигкр и полученную точку в координатах <П2кр—Скр
соединить с нулем. Положение (угол наклона) прямой определяет-
ся природой полимеров и природой растворителя. При этом опре-
деление ai2 кр должно быть проведено при соблюдении условия
ob=«2s. Если определение проводилось для разных пар полиме-
ров, например в хороших растворителях, то угол наклона прямой
зависел в основном от природы контактирующих полимеров. Этот
метод позволил оценить параметры сигкр для разных полимеров и
расположить эти пары по мере убывания взаимной растворимости
(совместимости) в следующий ряд: ПС—ПММА, ПС—СКИ,
СКТ—СКИ, СКТ—ПС, ПС ННБ, скэпт—скс-зо, скэпт—
СКН-40.
Значения авкр можно найти по Гильдебранду: ui2kP=(6i—
—b’}?IRT. Тогда указанные полимеры по мере роста «12Кр (ухуд-
шению взаимной растворимости) расположатся в следующий ряд:
ПС—ПММА, СКЭПТ—СКС-ЗО, СКТ—СКИ, СКТ—ПС, ПС—СКИ.
ПС—НИВ, СКЭПТ—СКН-40.
Различие относительной оценки совместимости, очевидное из
сравнения этих рядов, определяется, видимо, гем, что параметр
взаимодействия по Гильдебранду отражает только энтальпийную
составляющую взаимодействия, тогда как экспериментально опре-
деленный параметр а12 Кр включает и энтропийную составляющую
и является наиболее надежным способом оценки взаимодействия
полимер — полимер. Несоответствие указанных рядов еще раз под-
тверждает принципиальную ошибочность оценки совместимости
полимеров на основании сопоставления их плотностей энергии
когезии.
32
Из теорий растворов смесей полимеров наименее эксперимен-
тально подтвержденной [68, 77] является теория Марона 14]. Со-
поставление значений xi2. рассчитанных по этой теории, со значе-
ниями, рассчитанными по другим теориям, приводит к противоре-
чивым результатам. Так, для олигомеров [68] величина xi2 по тео-
рии Марона на порядок превышала величины, рассчитанные по
теории Флори — Скотта [67] В то же время результаты, получен-
ные в работе [77], указывают на согласование расчетов по тео-
рии [4] с расчетами по другим теориям.
Таким образом, достоинства и недостатки теории Марона в
должной мере еще не выявлены.
Основные методы определения
термодинамических параметров взаимодействия
полимеров Х12 и ui2 в системах полимер (1) -
полимер (2) и растворитель (S) -полимер (1) -полимер (2)
Параметры Х12 и aJ2 имеют одинаковый физический смысл, по-
скольку а2з=%2з/П [где V— мольный объем мономерной (повто-
ряющейся) группировки в макромолекуле полимера, выраженный
и м'/моль и принимаемый обычно равным около 10-4 м3/моль].
Летод Скотта [2] применим к тройным системам, в которых
рпгпюритель практически одинаково взаимодействует с каждым
in полимеров^ при соблюдении условия |хы—X2s| <C1/VS; размеры
макромолекул смешиваемых полимеров, выраженные через моль-
ные .1б|дмы макромолекул, должны удовлетворять условию V°5<
Г* В 1ПКИХ системах (па илвасмых иногда «системами Скот-
1Я») iiapaMi ip и laiiMi'Teni гния полимеров в критической точке
нинынасия уравнением (25).
'1ля расчета «12 кР строят диаграмму состояния тройной систе-
мы у щвлетворяющей указанным условиям, и, определив критиче-
кую концентрацию, при которой наступает расслаивание, рассчи-
।ын.нот <pSKp. Метод Скотта применяли для определения парамет-
ра ..заимодействия, например, в системах бензол — ПС—ПММА
]78|, бензол — каучук—ПММА [66], толуол — полипропилен
Iаыктический)—ПС [45, 63], а также в смесях ПС—ПММА в
о< и юле, метилэтнлкетоне, и этилапетате [76].
Метод определения «12 по составу контактирующих слоев. Ав-
юры работы [67] предложили следующее уравнение:
„ (l/^l) 1П (ф1/ф!) + (1/У2) 1П (Ф2/Ф2) + («is — «2.0 (ф» — Фе)
12 = (<Й-Ф2) + (Ф1-Ф1) l26)
1 ie И и Vz — мольные объемы полимеров 1 и 2; <pi, <j>2, <ps — объемные доли по-
шмеров 1 и 2 и растворителя в одной из фаз (параметры другой фазы выделе-
ны штрихом); Иц и ais — термодинамические параметры взаимодействия полиме-
ров с растворителем, определенные независимым методом в бинарной системе по-
шмер — растворитель.
Уравнение (26) получено из условия равенства химических по-
|<‘нциалов компонентов в равновесных сосуществующих фазах.
.4 -214
33
Расчет по уравнению (26) можно проводить, если известен состав
верхнего и нижнего слоя в расслоившейся системе после достиже-
ния равновесия. Состав фаз может быть проанализирован любым
методом. Метод Аллена применялся для расчетов, например в ра-
ботах [45, 63, 67, 76].
Метод определения хы по значениям второго вириального ко-
эффициента. Второй вириальный коэффициент получен из данных
ио осмотическому давлению [77] или по светорассеянию [78].
В работе [78] Х12 был рассчитан по данным светорассеяния для
смеси ПС и ПММА в бутаноне, из зависимости избыточной мут-
ности НС!х" от общей концентрации полимеров в растворе с —
= ci + c2. При этом второй вириальный коэффициент связан с тер-
модинамическими параметрами следующим образом:
В12 = (1 + 7.12 — Xis — 7.2s)/2l sPiPa (27)
где pi и [>2 — плотность каждого полимера.
Параметры хм и хг« были определены для соответствующих
растворов каждого полимера в данном растворителе. Величина хы
была рассчитана [77] из данных по осмотическому давлению на
основе усовершенствованной авторами теории Марона.
Метод обращенной газовой хроматографии [55]. Газовая хро-
матография позволяет определить взаимодействие полимеров xi2,
если в качестве неподвижной фазы применяется смесь двух поли-
меров, нанесенная на носитель, а в газовом потоке находится до-
зированное количество паров жидкости, обычно не являющейся
хорошим растворителем для данной пары полимеров. Измеряя ко-
личество паров, сорбированных полимерами и их смесью, т. е. оп-
ределяя удерживаемый объем сорбата Vg, можно рассчитать тер-
модинамический параметр взаимодействия сорбат — смесь:
I / 7.1 S \ . / 7.2.Ч \ . / /12 \ , ] ,,
7.s(i,2)—Н I ЧТ +1 р ) Фа — I pi H'i'IalH
273/? (10,41+^) 7 Vs \ Л Л /л ю
— n PsV Vg — ( — Vj IФг — ( — v2 ) 'f2 — [pp (Bs Vs) (28)
где Xi» и X»«— параметр взаимодействия каждого полимера смеси с растворите-
лем; <р — объемные доли компонентов; Vs, V] и V?— мольные объемы сорбата и
полимеров соответственно; Ps — давление паров сорбата; щ и п2— удельные
объемы полимеров; w, и w%—их массовые доли.
Второй вириальный коэффициент сорбата Bs можно рассчитать
из критических условий существования растворителя:
Н.РкР 9
^=Т2Н1-6<Г*>/7^ <29)
Далее определяют параметр взаимодействия сорбат—смесь х«а,2)
и отдельно параметры для каждого полимера xis ИХ2«, после этого
рассчитывают величину хы по уравнению (28). Метод применялся
для оценки взаимодействия в смесях некоторых полимеров и в
смесях олигомер — олигомер [56].
34
Метод расчета по плотности энергии когезии. По параметрам
растворимости fii и 62 можно рассчитать параметр взаимодейст-
вия:
Vs 01 ~^)2
RT
(30>
Если Vs~10-4 м3/моль и Т^300 К, то «)2=(б1—62)2/6. Метод
применялся, например, в работах [75, 67, 18J, в этих же работах
рассчитанное значение си 2 сопоставлялось с данными, полученны-
ми экспериментально.
ЛИТЕРАТУРА
1. С ее G. — Quart. Rev. Chem. Soc., 1947, v. 1, p. 265—283.
2. Scott R L. — J. Chem. Phys., 1949, v. 17, p. 279—287.
3. Tompa H. Polymer Solutions. London, Butterworths, 1956. 320 p.
4. Maron S. H., Nakajima N. — J. Polymer Sci., 1957, v. 40, p. 59; 1959, v. 42,
p. 327—340; 1960, v. 47, p. 157—168: 1961, v. 54, p. 587—598.
5. Flory P. J. Principles of Polymer Chemistry. N. Y., Cornell Univ. Press. Ithaca.
1953. 410 p.
6. Huggins Al. L. — J. Chem. Phys., 1941, v. 9, p. 440—449; Ann. N. Y. Acad. Sci.,
1942, v. 43, p. 1—19.
7. Scott R. L. — J. Polymer Sci., 1952, v. 9, p. 423—432.
8. Chandler L. A., Collins E. A.— ACS Polymer Prepr., 1968, v. 9, p. 1416—
1420.
9. Breners W., Hild W., Wolff H. e. a. — Plaste u. Kautschuk, 1954, Bd. 1,
S. 170—180.
10. Коллинский Ф., Маркерт Дж. — В кн.: Многокомпонентные полимерные
системы. Пер. с англ./Под ред. М. Я. Малкина и В. Н. Кулезнева. М., Химия,
1974, с. 72—82.
11. Krause S., Smith A. L.-t Duden AL G.—J. Chem. Phys., 1965, v. 43, p. 2144—
2145.
12. Allen G., Gee G., Nicholson J. P.— Polymer, 1961, v. 2, p. 8—14.
13. McIntyre D., Rounds N„ Campos-Lopez E. — ACS Polymer Prepr., 1969, v. 10,
p. 531—537.
14. Чалых A. E., Сапожникова И. H., Алиев А. Д.—ДАН СССР, 1978, т. 238,
№ 5, с. 1144—1146.
15. Шварц А. Г., Динзбург Б. Н. — Совмещение каучуков с пластиками и син-
тетическими смолами. М., Химия, 1972. 224 с.
16. Small Р. А. — J. Appl. Chem., 1953, v. 3, р. 71—74.
17. Ноу К. L. — J. Paint Techno!., 1970, v. 42, р. 76—79.
18. Krause S. — J. Macromol. Sci., 1973, pt. C, v. 7, № 2, p. 251—314.
19. Schneier B. — J. Appl. Polymer Sci., 1973, v. 17, p. 175—179.
20. Мельникова О. Л., Кулезнев В. Н., Аулов В. А., Клыкова В. Д. — Высоко-
мол. соед., 1976, сер. Б., т. 19, № 12, с. 903—906.
21. Слонимский Г. Л., Стриминский Г. В.—ЖФХ, 1956, т. 30, № 9, с. 1941—
1945; № 10, с. 2144—2149.
22. Koningsveld R., Kleintens L. А. — In: Macromolecular Chemistry Plenary and
Main Lecture, Intern. Svmp. Macromol., Helsinki, 1972. London, 1973, V. 8,
p. 197—230.
23. Flory P. J. — J. Am. Chem. Soc., 1965, v. 87, p. 1833.
24. Flory P. J., Eichinger В. E., Orwoll R. A. — Macromolecules, 1968, v. 1, № 3,
p. 287—288.
25. McMaster L. P.—Macromolecules, 1973, v. 6, p. 760—773.
26. Miyata S., Hata T. Rep. Progr. Polymer Phvs. Japan, 1969, v. 11. p. 313—316.
27. Frise Д’. —Plaste u. Kautschuk, 1968, Bd. 15, S. 646—650.
28. Tager A. A., Scholochovitch T. I., Bessonov Ju. B. — Europ. Polymer J., 1975,
v. 11, № 4, p. 321—326.
3'
35
29. Bank M., Leffingwell J., Thies C. — J. Polymer Sci., 1972, pt. A-2, v. 10,
p. 1097—1110.
30. Kwei T. K., Nishi T., Roberts R. F. — Macromolecules, 1974, v. 7, p. 667—
671.
31. Оганесов Ю. Г., Кулезнев В. Н„ Воюцкий С. С. — Высокомол. соед., 1970,
сер. Б, т. 12, № 19, с. 691—693.
32. .McMaster L. Р. — ACS Polymer Prepr., 1974, v. 15, p. 254—259.
33. Shaw M. T. — J. Appl. Polymer Sci., 1974, v. 18, p. 449—472.
34. Koningsveld R., Kleintjens L. A., Schoffeleers H. M. — Pure Appl. Chem.,
1974, v. 39, p. 1—31.
35. Моравец Г. Макромолекулы в растворе. М., Мир, 1967. 398 с.
36. Цулезнев В. Н., Крохина Л. С., Оганесов Ю. Г., Злацен Л. М. — Коллоид,
ж., 1971, т. 33, № 1, с. 98—104.
37. Кулезнев В. Н., Догадкин Б. А., Клыкова В. Д. — Коллоид, ж., 1968, т. 30,
с. 255—257.
38. Радбиль Т. И., Разинская И. Н., Штаркман Б. П. — Высокомол. соед., 1967,
сер. Б, т. 9, с. 692—695.
39. Сафаров А. Ш., Геворкян А. В., Овсепян М. Е. — Учен. зап. Ереван, гос.
ин-та, 1973, вып. 3, с. 78—80.
40. Кулезнев В. Н., Клыкова В. Д., Чернин Е. И., Евреинов Ю. В. — Коллоид,
ж., 1975, т. 37, с. 267—272.
41. Лебедев Е. В.. Липатов Ю. С., Привалко В. П. — Высокомол. соед., 1975,
сер. А, т. 17, с. 148—153.
42. Шилов В. В., Безрук Л. И., Комото Т.. Ковач Т., Липатов Ю. С. — Высоко-
мол. соед., 1976, сер. А, т. 18, с. 2793—2800.
43. Yeun Н. К-, Kinsinger I. В. — Macromolecules, 1974, v. 7, р. 329—336.
44. Molau G. Е. — J. Polymer Sci., 1965, p. B, v. 3, p. 1007—1015.
45. Berek D., Bohmer B., Lath D.—Plaste u. Kautschuk, 1967, Bd. 14, S. 556—
563.
46. Kuhn R., Cantow H. I., Burchard W. — Angew. makromol. Chem., 1968, Bd. 2,
S. 146—151.
47. Vasile C., Asand N., Schneider J. — Cell. Chem. a. Technol., 1971, v. 5, p. 597—
606.
48. Feldman D., Rusu M. — J. Polymer Sci., Polymer Symp., 1973, № 42, pt 2,
p. 239—246.
49. Paul P. R., Altamirano J. O. — ACS Polymer Prepr., 1974, v. 15, p. 409—
411.
50. Асимова P. M„ Козлов П. В., Каргин В. А., Вторыган С. М. — Высокомол.
соед., 1962, т. 4, с. 554—559.
51. Смирнова Н. В.. Айвазов А. Б., Динзбург Б. If., Алексеенко В. И. — Высо-
комол. соед., 1975, сер. Б, т. 17, с. 405—409.
52. Robeson L. М., Matzner М., Fetters L. J., McGrath I. Е. — ACS Polymer
Prepr., 1973, v. 14, № 2, p. 1063—1068.
53. Hoffman H., Pampus G., Matwede G. — Kautschuk u. Gummi, 1969, Bd. 22,
S. 691—706.
54. Тагер А. А., Шолохович T. И., Цилипоткина M. B. — Высокомол. соед., 1972,
сер. А, т. 14, с. 1423—1424; Тагер А. А. — Высокомол. соед., 1972, сер. А,
т. 14, с. 2690 2706.
55. Deshpande I). D., Patterson D., Schreider H. P., Hsu C. S. — Macromolecules,
1974, v. 7, p. 530-537.
56. Нестеров A. E., Липатов К). С. — Обращенная газовая хроматтрафия в тер-
модинамике полимеров. Киев, Наукова думка, 1976. 128 с.
57. Шолохович Т. И. Канд. дне. Свердловск, Уральск, ун-т, 1975.
58. Кулезнев В. Н. — Коллоид, ж., 1977, т. 39, № 2, с 407—408.
59. Hsu С. С., Prauznitz Л М. — Macromolecules, 1974, v. 7, р. 320—324.
60. Zeman L., Patterson D. — Macromolecules, 1972, v. 5, p. 513—516.
61. Dobry A., Boyer-Kawenoki F.— ,1. Polymer Sci., 1947, v. 2, p. 90—100.
62 Burkhard F., Majer H., Kuhn W. — Helv. chim. acta, 1960, v. 43, p. 1192—
1199.
36
i. i Berek D., Lath D . Durdovic V. — J Polymer Sci., 1967, pt. C,'.pt. 2, p. 659—
667.
«.I Кулезнев В. H„ Крохина Л. С. — Усп. хим., 1973, т. 42, с. 1278—1319.
Welugan D. С., Burns С. М. — J. Appl. Polvmer Sci., 1974, v. 18, р. 521—
529.
*•<>. Bristow G. M. — J. Appl. Polymer Sci., 1959, v. 2, p. 120—124.
ii7. Allen G., Gee G., Nicholson J. P. — Polymer, 1960, v. 1, p. 56—62.
OH. Paxton Th. R. — J. Appl. Polymer Sci., 1963, v. 7, p. 1499—1508.
69. Kern R. L.—J. Polymer Sci., 1956, v. 21, p. 19—25.
70. White J. L., Patel R. D. — J. Appl. Polymer Sci., 1975, v. 19, p. 1775—
1782.
71. Hugelin C., Dondos A. —Makromol. Chem., 1969, Bd. 126, S. 206—214.
72. Bank M., Lelfingwell J., Thies C. — Macromolecules, 1971, v. 4, p. 43—48.
73. Purcell A., Thies C.— ACS Polymer Prepr., 1968, v. 9, p. 115—122.
74. Крохина Л. С.. Кулезнев В. H„ Люсова Л. Р., Глаголев В. А. Высокомол.
соед., 1976, сер. А, т. 18, № 3 ,с. 663—668.
73. Крохина Л. С., Кулезнев В. Н. — В кн.: Термодинамика органических соеди-
нений. Межвуз. сб. Горький, 1976, № 5, с. 68—73.
76. Крохина Л. С., Кулезнев В. И.— Высокомол. соед., 1978, сер А, с 20 № 9
с. 1981—1987.
77. Cerny L. С., Newman Е. И., Staciw D. .14., Visally W.—J. Macromol. Sci,
1973, pt. В, 7, p. 19—26.
78. Stockmayer W. H., Stanley H. E — J. Chem. Phys., 1950, v. 18, p. 153—154.
Глава II
ИСТИННЫЕ И КОЛЛОИДНЫЕ РАСТВОРЫ
СМЕСЕЙ ПОЛИМЕРОВ
Раствор смеси полимеров представляет собой систему поли-
мер— полимер — растворитель, в которой растворитель является
самостоятельным компонентом и не может рассматриваться как
некий простой разбавитель. Поэтому данные, получаемые при изу-
чении поведения смеси двух полимеров в общем растворителе не
могут однозначно характеризовать взаимодействие (тем более по-
ведение при переработке) смеси этих двух полимеров в отсутствие
растворителя. В присутствии растворителя изменяются не только
характерные времена релаксации системы, но также и термодина-
мика взаимодействия полимерных компонентов. Вопрос о взаимной
растворимости или нерастворимости полимеров (их совместимо-
сти) может быть диаметрально противоположным для смеси в
присутствии и отсутствие растворителя. Несмотря на это, кажу-
щаяся легкость работы с раствором как с объектом научного ис-
следования привела к тому, что в очень многих работах на осно-
вании экспериментов с растворами смеси полимеров делаются ма-
лообоснованные выводы о «совместимости» самих полимеров в
отсутствие растворителя.
В этой главе суммируются результаты исследования истинных
(однофазных) и коллоидных растворов смесей полимеров, а также
факторов, влияющих па один из важнейших параметров системы
полимер—полимер — растворитель на концентрацию, при которой
происходит расслаивание смеси полимеров в растворе.
§ 1. ИСТИННЫЕ РАСТВОРЫ СМЕСЕЙ ПОЛИМЕРОВ
Известно, что в растворах неполярных полимеров при очень ма-
лой концентрации макромолекулы разобщены растворителем на-
столько, что находятся в виде отдельных клубков. С возрастанием
концентрации сферы действия молекулярных клубков с учетом их
диффузной, размытой границы перекрываются. Концентрация, при
которой происходит контактирование клубков, определяется по пе-
регибу на кривой свойство — состав [1, 2|. В частности, в [2] бы-
ло показано, что при достижении этой концентрации наблюдается
перегиб на кривой С для толуольного раствора полистиро-
ла (ПС), где /?эо°—интенсивность рассеяния под углом 90°С, а
С — концентрация раствора. Для ПС с молекулярной массой
38
'• К)6 точка перегиба соответствовала концентрации 0,3% раствора
IIC в толуоле [2].
Очевидно, что подобное изучение смесей полимеров в растворе
не является достаточно надежным, поскольку молекулярная масса
каждого полимера различна и размер клубков макромолекул так-
же неодинаков. Поэтому перегиб на кривой может быть результа-
там достижения критической концентрации одним из компонентов
или смесью в целом.
В 1964 г. в работе [3] был предложен метод исследования
структурных изменений полимера, растворенного в полимерном
растворителе. Суть метода состоит в том, что смесь полимеров
помешают в растворитель, показатель преломления которого ра-
нен показателю преломления одного из полимеров (этот полимер
называется «балластным» [4]). В такой системе инкремент пока-
зателя преломления балластного полимера приближается к нулю,
п поэтому его избыточное светорассеяние /?<ю° (избыточное по срав-
нению с рассеянием растворителя) также близко к нулю незави-
симо от молекулярной массы балластного полимера и концентрации
раствора. Таким образом, в смеси исследуемого «рассеивающего»
полимера с балластным рассеивает один исследуемый полимер.
Если общая концентрация раствора велика, а концентрация иссле-
дуемого (небалластного) полимера в нем мала, то к такому рас-
твору можно применить теорию рассеяния света разбавленных рас-
творов и проследить за конформационными изменениями исследуе-
мого полимера, применяя растворы с разной концентрацией бал-
ластного полимера. В частности, с увеличением концентрации рас-
твора можно определить точку перегиба на кривой — концент-
рация н установить момент начала перекрывания сферы действия
Рис. II.1. Зависимость /?%<> (а) и KdR'w (б) растворов ПС (/) и смеси ПС и
ПИБ (2) в толуоле от концентрации ПС. Молекулярная масса ПС 5,5-105, ПИБ
3,0 10е (5].
39
клубков и переход от раствора с изолированными молекулами к
системе, в которой начинается ассоциация макромолекул.
Из рис. 11.1 видно, чю при малых концентрациях раствора кри-
вые концентрационной зависимости светорассеяния раствора ПС в
толуоле и аналогичной зависимости светорассеяния смеси раство-
ров ПС и ПИБ в толуоле (ПИБ— балластный полимер) совпада-
ют, а начиная с некоторой концентрации, резко расходятся [5].
Выбор толуола в качестве растворителя обусловлен тем, что пока-
затель преломления его практически равен показателю преломле-
ния ПИБ. Поэтому в системе ПС—ПИБ — толуол основной вклад
в рассеяние дает ПС, так как избыточное рассеяние ПИБ в толуо-
ле близко к нулю.
Из данных, приведенных на рис. 11.1, можно заключить, что,
начиная с некоторой концентрации, макромолекулы ПС в смеси с
ПИБ рассеивают свет гораздо интенсивнее, чем в толуоле [5]. Это
можно объяснить лишь тем, что при данной концентрации начина-
ется ассоциация макромолекул ПС. Перегиб на кривой рассеяния
света в растворе смеси полимеров наблюдается при концентрации
меньшей, чем в растворе полимера в обычном растворителе, как
это видно из того же рис. II. 1.
Концентрация, при которой отмечается несовпадение кривых
для раствора ПС в толуоле и в «полимерном» растворителе, назы-
вается концентрацией начала ассоциации и обозначается G. На
рис. 11.1,6 те же экспериментальные данные приведены в коорди-
натах KCIRmf—С, где К — константа в уравнении светорассеяния
KCIR"^=1 IMw+2A2C. Совершенно очевидно, что концентрацию
Ci удобнее определять в координатах, представленных на
рис. II.1,6. Значения Сь найденные указанным способом, приведе-
ны в табл. II.1.
Из табл. II. 1 видно, что природа растворителя также влияет на
концентрацию начала ассоциации С\. В качестве «хорошего» рас-
творителя по отношению к ПИБ был взят СС1.|. Для ПИБ с моле-
кулярной массой 30,0-105 характеристическая вязкость в толуоле
составляет 3,7 дл/г, а в СС14 — 8,2 дл/г. Для ПС оба растворителя
Таблица II.1. Концентрация, при которой начинается ассоциация Ci
и концентрация, при которой происходит расслаивание С»(г/дл)
в смесях растворов ПС и ПИБ (7:3) в толуоле и четыреххлористом углероде
Mw.10S Толуол Четиргххлористый углерод
ПС пиь С1 С, Cl са
5,5 30,0 0,09 0,9 0,25
3,7 30,0 — 1,1 0,36 1,5
2,2 30,0 0,20 1,3 0,42 —
1,2 30,0 0,24 1,7 0,65 —
0,65 30,0 0,41 2,4 0,79 —
40
Рис. П.2. Зависимость второго вириального коэффициента
/12 (по светорассеянию) от концентрации ПИБ в полимер-
ном растворителе. Нулевая концентрация ПИБ соответст-
вует раствору ПС в толуоле. Молекулярная масса ПС
1-Ю6, ПИБ 1,2-106 [3].
являются «хорошими». Концентрация начала ас-
социации увеличивается с уменьшением молеку-
лярной массы и уменьшается с понижением
сродства полимера к растворителю, что согла-
суется с общими представлениями о закономер-
ностях ассоциации полимеров в растворе.
Из рис. II. 1 видно, что значение Ci в поли-
мерном растворителе ниже, чем в растворе в то-
луоле, что подтверждается также и сравнением
данных рис. II.1 с данными работы [2]. Это яв-
ление объясняется следующим образом. При на-
личии второго полимера, сродство которого к
первому невелико (полимеры «несовместимы»),
уменьшается число контактов сегментов разнородных макромоле-
кул, что в результате приводит к уменьшению объема, в котором
могут располагаться молекулы обоих полимеров. Уменьшение та-
кого «разрешенного» объема приводит к тому, что ассоциация од-
нородных макромолекул начинается при меньших концентрациях,
чем в растворе одного полимера, следовательно, Ci должна быть
меньше в растворе полимера в «полимерном» растворителе по
сравнению с обычным раствором.
Это нужно учитывать при определении молекулярной массы и
размерив молекул в смеси полимеров или в блок-сополимерах, в
которых область концентраций, соответствующих предельно раз-
бавленным растворам, может оказаться существенно меньше
области концентраций, в которых можно определять молекуляр-
ные параметры полимеров, растворенных в обычном раствори-
теле.
Приведенные рассуждения позволяют провести аналогию меж-
ду действием одного полимера па другой в растворе и действием
на полимер обычного, ни зкомолскулярного («плохого») раствори-
теля. Эта аналогия подтверждается тем, что при переходе от обыч-
ного растворителя к «полимерному» уменьшается величина второ-
го вириального коэффициента Л2 (рис. П.2, табл. II.2). Действи-
тельно, как это видно из рис. II.2, второй вириальный коэффици-
ент для ПС в растворе в толуоле резко уменьшается при замене
толуола раствором ПИБ в толуоле и может даже стать отрица-
тельным.
Метод «полимерного» растворителя позволяет не только уста-
новить наличие избыточной ассоциации в растворе смеси полиме-
ров, но и проследить за конформационными превращениями мак-
ромолекулярных клубков в области концентраций, непосредственно
41
Таблица II.2. Результаты определения Л2 и размера макромолекул ПС
в толуоле и в полимерном растворителе [5]
Образец Концентрация ПИБ, г/дл V ю*. СМЗДГ2-МОЛЬ) Радиус инер- с цин R. А (Л2)'/г. Л
1 (мол. масса 6,45-103) 0 4,55 434 960
0,13 0.19 434 —
0,225 0,16 495 —
2 (мел-масса 9,55-10“) 0 3,3 620 1520
о,1 0,9 620 —
0,2 0,9 720 —
0,3 —0,3 848 —
предшествующей началу ассоциации. Полученные результаты при-
ведены в табл. II.2.
Если, например, раствор смеси ПС:ПИ=1 : 1 с обшей концент-
рацией 0,26 г/дл и парциальной концентрацией ПС 0,13 г/дл после
определения угловой (8) зависимости избы точного светорассеяния
разбавить раствором смеси растворов ПИБ в толуоле с концент-
рацией 0,13 г/дл, то парциальная концентрация ПС уменьшится, а
концентрация ПИБ в растворе останется постоянной. Зависимость
KCIRI от парциальной концентрации ПС дает возможность опре-
делить молекулярную массу ПС, а угловая зависимость светорас-
сеяния позволяет с помощью диаграммы Зимма определить раз-
мер молекул ПС. Таким образом можно найти зависимость разме-
ра клубка и молекулярной массы молекул ПС от концентрации
балластного полимера, в данном случае ПИБ, которая и представ-
лена в табл. П.2.
Диаграммы Зимма искривлены в области больших концентра-
ций, однако при малых концентрациях и малых углах можно про-
вести экстраполяцию к нулевому углу и нулевой концентрации.
С ростом концентрации полимерного растворителя ассоциация
макромолекул ПС не происходит, на что указывает постоянство
молекулярной массы ПС. Это означает, что исследованная область
концентраций несущественно превышает Сь При малых концентра-
циях ПИБ не происходит также и изменения размеров клубка (ра-
диуса инерции) ПС. Очевидно, при концентрации раствора мень-
ше Ci клубки достаточно удалены друг от друга и не оказывают
взаимного влияния в той мере, в какой было бы необходимо для
увеличения степени ассоциации и для изменения формы клубка.
Если концентрация ПИБ в полимерном растворителе становится
такой, что парциальная концентрация ПС оказывается несколько
выше Ci или равной ей, то размер клубка ПС увеличивается, хотя
роста молекулярной массы в этих условиях еще не наблюдается.
Приведенные экспериментальные результаты можно объяснить
следующим образом.^ При концентрации полимера выше Ct рас-
твор смеси полимеров однофазен, т. с. граница раздела отсутству-
42
< i. Рыхлые полимерные клубки со степенью заполнения полиме-
ром не более 1—2%, контактируя друг с другом, могут проникать
п объемы, занятые клубками других макромолекул. Таким обра-
ти, сегменты, например ПИБ, могут находиться как внутри клуб-
ков ПС, так и вне их. Предпочтительность контактов сегментов ти-
па ПС—ПС и ПИБ—ПИБ в отличие от контактов ПС—ПИБ
обусловливает возникновение сил отталкивания разнородных сег-
ментов. и благодаря наличию сегментов ПИБ как внутри клубка
ИС, так и вне его последний может либо сжиматься, либо развер-
Iываться.
Предложенный метод «полимерного» растворителя [3] впослед-
ствии неоднократно применялся для исследования блок-сополи-
меров и смесей полимеров [4—7]. Полученные результаты неодно-
шачны: в работах [4, 6] наблюдали уменьшение размеров молеку-
лярного клубка с ростом концентрации полимерного растворителя,
в то же время отмечалась нсидентичность низкомолекулярного и
полимерного 6-растворителя. В работе [6.J прямо указано на то,
что размер макромолекулярного клубка в полимерном растворите-
ле больше, чем в низкомолекулярном 6-растворителе. В этих рабо-
iax концентрация полимеров в растворе представляется много
меньше Ci, хотя авторы специально этот вопрос не рассматривали
и величину Ci не определяли. Эти данные не согласуются с резуль-
татами работ [7] и [5], авторы которых показали, что размер
клубка ПС не зависит от концентрации полимерного растворителя
(ПИБ + толуол), что объясняли [7] практически нулевым парамет-
ром взаимодействия полимеров. Неоднозначность полученных ре-
зультатов проявляется и в том, что, по данным [6], размер клуб-
ков ПС в полимерном растворителе (ПММА в бензоле) увеличи-
вается с возрастанием молекулярной массы ПММА.
Видимо, в системе ПС—ПММА — растворитель характер меж-
молекулярных взаимодействий таков, что сжатие клубков более
аметно, чем свертывание. Так, в работе [100] были получены
пленки дейтерированного ПММА, содержащего 1 % поли-а-метил-
стирола с молекулярной массой около 2,5-105. Дифракция нейтро-
нов показала, что ПС. нахо щтся в ПММА в агрегированном со-
стоянии с числом молекул в агрегате около 16. При этом молеку-
лярные клубки агрегатов сжаты по сравнению с их предполагаемым
объемом в низкомолекулярном 6-растворителе: при указан-
ной выше молекулярной массе поли-а-метилстирола размер клуб-
ков составляет около 170 А. Однако клубки ассоциатов не являют-
ся предельно сжатыми, что, естественно, предполагает взаимопро-
никновение клубков поли-а-метилстирола и ПММА. Кроме того,
наблюдается очень широкое распределение клубков по размерам.
Таким образом, в больших клубках взаимопроникновение полиме-
ров может быть еще более значительным.
Возможность изменения размеров молекулярных клубков поли-
меров с изменением концентрации раствора в низкомолекулярном
растворителе признается многими исследователями. Так, в работе
43
Френкеля с сотр. [8, с. 102] высказывается следующее соображе-
ние: «При малых концентрациях в результате увеличения доли по-
лимера в растворе уменьшаются размеры макромолекулярных
клубков вследствие уменьшения свободного объема, но по мере
роста концентрации полимера ограничение подвижности сегментов
в результате помех, создаваемых соседями, может привести к ча-
стичному распрямлению (разворачиванию) клубков, т. е. к увели-
чению размеров макромолекул».
Если раствор одного полимера разбавлять полимерным раство-
рителем довольно высокой концентрации так, чтобы общая кон-
центрация раствора стала близка к пределу расслаивания смеси,
то, как показано в [9], первый полимер в растворе не перейдет в
систему с изолированными макромолекулами, а в полимерном
растворителе будут существовать клубки со степенью ассоциации
около 55.
Увеличение размеров клубков полиизопрена (натурального кау-
чука) в присутствии ПС в растворе в бензоле наблюдали по кос-
венным данным [10]. Так, при вулканизации разбавленных рас-
творов ПК в бензоле при комнатной температуре единой структу-
ры геля не образуется при концентрации НК менее 0,28 г/дл.
В присутствии ПС в растворе гель возникает даже при концентра-
ции НК 0,25 г/дл, при этом макромолекулы ПС в состав простран-
ственной сетки геля не входят. Это означает, что при указанной
концентрации, которая близка к Сь увеличение размеров клубков
вулканизующегося полимера позволяет создать единую простран-
ственную структуру в условиях, в которых при обычных размерах
клубков она не возникает.
Конформационные изменения макромолекулярных клубков в
присутствии второго полимера можно изучать путем измерения
вязкости и седиментации разбавленных растворов смесей полиме-
ров в полимерном растворителе.
В работе [11] измеряли характеристическую вязкость разных
полимеров в полимерных растворителях. Было обнаружено умень-
шение размеров клубков полимеров, о чем можно было судить по
снижению характеристической вязкости но сравнению с расчетным
значением. Сообщается [12] о результатах исследования гидроди-
намических свойств систем с полимерным растворителем:
ПММА ПС — толуол, ПММА — полиэтиленгликоль — бензол, по-
ливинилпирролидон (ПВП) —полиметилакрилат (ПМА)—дихлор-
этан и другие. Молекулярные массы основного полимера, измерен-
ные по светорассеянию и седиментации, остаются неизменными, а
характеристическая вязкость основного полимера уменьшается с
возрастанием концентрации и молекулярной массы балластного
полимера. Константа а в уравнении Марка—Куна — Хувинка, ви-
димо, не меняется при переходе к полимерному растворителю
(рис. П.З).
Исследование [13] седиментации ПС, помещенного в раствор
ПИБ в толуоле, показало (рис. П.4), что с ростом концентрации
44
Рис. П.З. Зависимость вязкости от молекулярной массы пилим< тилметакрилата
в бензоле и полимерных растворителях:
1- бензол или 0,3%-иый бензольный раствор полиэтилен! ликоля с молекулярной массой
1,5-10*; 2, 3 —0,2%-ный и 0,3%-ный бензольные растворы полиэтиленгликоля и молекулярной
массой 1,5-10® [12].
Рис. II.4. Зависимость коэффициента седиментации полистирола Sue от концент-
рации полимерного растворителя (раствор ПИЬ в толуоле). Парциальная кон-
центрация ПС:
1 — 0; 2 — 0,1; 3 — 0,2; 4 — 0,3; 5 — 0,4; 6 — 0,5 г/дл [13].
полимерного растворителя коэффициент седиментации ПС умень-
шается (увеличивается гидродинамическое сопротивление среды),
а начиная о некоторой концентрации, наблюдается даже возраста-
ние коэффициента седиментации, вероятно, вследствие ассоциации
макромолекул и увеличения массы седиментирующих частиц.
Применение метода полимерного растворителя оказывается
плодотворным и при исследовании зависимости свойство — состав
|5]. На рис. 11.5 приведена зависимость избыточного светорассея-
ния в системе ПС — ПИБ — толуол от соотношения полимеров при
их постоянной концентрации в растворе.
Экспериментально полученная зависимость свойство — состав
может служить источником информации об изменениях, происходя-
щих в структуре при изменении соотношения компонентов лишь в
том случае, если теория предсказывает, как должно изменяться с
составом указанное свойство при отсутствии специфических взаи-
модействий в системе. Простейшей теоретической зависимостью
свойство — состав является, как известно, линейная зависимость
явления пара над идеальными смесями (растворами) от состава,
выражаемая законом Рауля, которая выражает аддитивность
свойств. Подавляющее большинство работ по смесям полимеров, в
которых была получена экспериментальная кривая свойство —
состав, постулировало аддитивное изменение исследуемого свойст-
ва с составом. Вместе с тем очевидно, что очень многие свойства
вовсе не являются аддитивными. Нет априори никаких оснований
г читать вязкость, прочность, светорассеяние, модуль и т. п. адди-
45
Рис. II.5. Зависимость /?££ от соотноше-
ния полимеров для растворов смесей ПС
и ПИБ (1—4) при постоянной концент-
рации раствора смеси полимеров
0,5 г/дл. Молекулярная масса ПИБ
3,0-10е, ПС: 5,5-105 (/), 2,2-105 (2),
1,2-105 (3), 0,65-105 (4); 1'—4' — соот-
ветствующие экспериментальные зависи-
мости AJyo от концентрации ПС в то-
луоле в отсутствие ПИБ [51.
тивными величинами. Поэтому
нельзя делать какие-либо выво-
ды о структуре смеси и взаимо-
действии компонентов в ней пу-
тем сравнения эксперименталь-
ной и аддитивной кривых свой-
ство — состав (для большинства
свойств).
В системе, приведенной на рис. II.5, рассеивает свет, т. е. «-ви-
дим* только один полимер — полистирол. Пунктирные линии на
рис. 11.5 представляют собой экспериментальные значения избы-
точного светорассеяния в толуоле при концентрации, равной пар-
циальной концентрации ПС в смеси. Они показывают, таким обра-
зом, как рассеивал бы свет ПС в отсутствие второго полимера.
Экспериментальные кривые сильно отличаются от теоретических и
свидетельствуют о значительном увеличении светорассеяния ПС в
присутствии ПИБ. Избыточное рассеяние в смеси даже превышает
рассеяние раствора ПС в толуоле при концентрации, равной пре-
дельной концентрации ПС в данном растворе. Экспериментальная
кривая имеет максимум; это значит, что в точке максимума (около
70% ПС в смеси с ПИБ) рассеяние выше, чем рассеяние раствора
ПС в толуоле, концентрация которого равна концентрации смеси
полимеров. Разность между рассеянием ИС в смеси и рассеянием
его в толуоле обусловлена, очевидно, избыточной ассоциа-
цией П( в присутствии ПИБ по сравнению с ассоциацией ПС в
толуоле при одинаковых концентрациях ПС в обоих случаях.
Теория не позволяет определить размер ассоциатов в растворе
конечной концентрации по данным светорассеяния, поэтому по из-
быточному рассеянию можно лиг.ь качественно судить о размере
ассоциатов. Степень ассоциации ПС при этом тем больше, чем
больше интенсивность светорассеяния ПС в полимерном раствори-
теле превышает соответствующее значение светорассеяния ПС в
толуоле при одинаковых концентрациях ПС.
Если принять в качестве меры избыточной ассоциации величи-
ну АА*90° при соотношении полимеров в расторе ПС :ППБ = 7:3,
то из графика зависимости от концентрации раствора смеси
полимеров (рис.11.6) видно, что избыточное рассеяние света уве-
личивается с возрастанием как концентрации смеси полимеров в
растворе, так и молекулярной массы ПС. Зависимость Д/?90° от
46
। шцснтрации раствора выражается прямой линией, пересекаю-
щей ось абсцисс при C = Ci (значение Ci определено независимым
методом). Это еще раз подчеркивает то обстоятельство, что избы-
шчпая ассоциация ПС, обусловленная присутствием ПИБ, начина-
юн при C = Ci и далее увеличивается с концентрацией вплоть до
< , т. е. вплоть до концентрации, при которой происходит расслаи-
нание, когда ассоциаты ПС достигают такого размера и плотно-
। in, что между разнородными ассоциатами образуется граница
раздела и раствор разделяется в микрообъемах на две фазы.
Разные полимеры в зависимости от их химической природы, мо-
лекулярной массы, типа растворителя и других факторов имеют
весьма различные значения Ci и С2, и увеличение размера и плот-
ности ассоциатов с возрастанием концентрации происходит, по-ви-
шмому, тем интенсивнее, чем меньше интервал концентраций
С'1—С2. Можно предположить, что в смеси полимеров существуют
некоторые соответственные (эквивалентные) состояния ассоциации
н области однофазных растворов. Если концентрация раствора
имеет некоторое значение, которое находится как раз посередине
интервала Ct—С2, то другая пара полимеров в растворе при дру-
|ой концентрации, но также лежащей посередине интервала
Е’1—С2, должна характеризоваться эквивалентной степенью ассо-
циации. Это положение пока еще не удалось проверить на различ-
ных парах полимеров, так как избыточное рассеяние в них зависит
от инкремента показателя преломления полимеров и от вклада
каждого полимера в рассеяние, а не только от степени избыточной
Рис. 11.6. Концентрационная зависимость избыточного рассеяния A/?[%<. ПС в сме-
< и с ПИБ (ПС: ПИБ=7 :3) Д/?оо° рассчитывали по разности между рассеянием
IIC в растворе полиизобутилена в толуоле, и рассеянием ПС в толуоле. Молеку-
ырная масса ПС10“5 1 (1, 5); 2,2 (2); 1,2 (3); 0,65 (4); 1—4— растворы в
толуоле; 5 — раствор в СС14.
Рис П.7. Зависимость избыточного рассеяния ПС от приведенной концент-
рации ПС С* = (С—С1)/(С2—Ci). Обозначения см. на рис. П.6 [5].
47
ассоциации. Однако это можно провести на одной и той же паре
полимеров с различными молекулярными массами.
Принцип соответственных состояний ассоциации иллюстрирует-
ся рис. 11.7, на котором показана зависимость избыточного рассея-
ния Л#эо° (по данным рис. II.6) от приведенной концентрации
С*=(С—Ci)/(C2—Ci). Приведенная концентрация является мерой
соответственного состояния ассоциации смеси полимеров в раство-
ре. Растворы с разными массовыми концентрациями, но с одинако-
выми приведенными концентрациями должны иметь близкую или
даже одинаковую степень ассоциации макромолекул. Рис. II.7
подтверждает это положение: независимо от молекулярной массы
ПС, т. е. независимо от Ci и С2, зависимость избыточного рассея-
ния от приведенной концентрации выражается одной прямой.
Можно полагать, таким образом, что одинаковое избыточное рас-
сеяние вблизи точки расслаивания для всех образцов ПС независи-
мо от молекулярной массы соответствует одинаковому значению
степени ассоциации и плотности полимера в ассоциатах, необходи-
мых для того, чтобы образовалась поверхность раздела между ас-
социатами разных полимерных молекул и чтобы произошло разде-
ление раствора на две фазы.
Для сравнения на рис. 11.6 показана зависимость избыточного
рассеяния от концентрации для раствора смеси ПС и ПИБ в CCU,
для которых избыточное рассеяние выше вследствие большого ин-
кремента показателя преломления ПС в ССП, а также вследствие
большего вклада ПИБ в общее рассеяние смеси полимеров.
Результаты, полученные методом светорассеяния, подтвержда-
ются данным по вязкости. Так, на рис. II.8 показана зависимость
относительной вязкости смеси полимеров от состава в полулога-
рифмических координатах. В смесях жидкостей вязкость смеси
подчиняется достаточно надежно правилу логарифмической адди-
тивности (пунктирная линия). Отклонение экспериментального
значения вязкости от значения, лежащего на пунктирной прямой,
возрастает с увеличением концентрации раствора, и прямая
AlgrjoTH—С пересекает ось абсцисс в точке, соответствующей кон-
центрации раствора 0,2 г/дл. Это значение представляет собой кон-
центрацию С|, полученную по данным вязкости. Она превышает
величину Ci = 0,09 г/дл (см. табл. П.1), полученную по данным све-
торассеяния.
Завышенное значение концентрации, при которой начинается
ассоциация (по данным вязкости), можно объяснить распадом ас-
социатов при сдвиговых деформациях (при течении), обнаружен- I
ным в опытах по влиянию напряжения сдвига на предел расслаи- j
вания, когда увеличение напряжения сдвига при течении раствора |
смеси полимеров приводит к росту предела расслаивания [14]. |
Приведенные данные свидетельствуют о повышенной степени
ассоциации в растворах смесей полимеров. Закономерности ассо-
циации, полученные на примере смеси ПС и ПИБ, очевидно, не
являются специфическими только для этой пары, а характерны для
48
Рис. II.8. Зависимость
относительной вязкости
раствора (IgTH™) от со-
отношения ПС (А/а,=
=5,5-Ю5) и ПИБ (Mw=
= 3-106) в толуольных
растворах с общей кон-
центрацией полимера 0,8
(1), 0,6 (2), 0,4 (3), 0,2
(4) г/дл (а) и от кон-
центрации раствора сме-
си ПС и ПИБ (б); пунк-
тир — аддитивная зави-
симость [51.
। меси полимеров, макромолекулы которых не имеют сильнополяр-
ны.х функциональных групп, взаимодействие которых может изме-
нить установленные выше закономерности.
Гаким образом, для смеси полимеров в растворе можно выде-
лив три типа структур (рис. П.9). При концентрациях меньше
раствор состоит из смеси изолированных макромолекул обоих
полимеров, при концентрации выше С2 раствор двухфазен и пред-
1авляет собой эмульсию, а в интервале Ci—С? система однофаз-
11.1, однако является смесью не изолированных молекул, а их ас-
• ।пшатов. На наличие ассоциатов в смеси растворов полимеров
\ называл еще Хаггинс: «если притяжение между однородными мо-
к’кулами больше, чем между разнородными, о чем свидетельству-
। расслоение растворов при высокой концентрации, то молекулы
растворенного вещества образуют агрегаты при концентрации не-
колько ниже той, при которой происходит образование второй
фазы» [15].
Влияние одного полимера на степень ассоциации или, иначе го-
воря, па ближний порядок макромолекул другого полимера может
привести к изменению структуры каждой фазы полимера в смеси
и свойств фазы. Это сказывается, например, на закономерностях
кристаллизации смесей полимеров (как это было показано на сме-
II ПЭ и ПП), когда интервал между температурами кристаллиза-
ции и плавления каждой фазы в смеси сужался, что свидетельству-
। об упорядоченности структуры расплава каждой фазы [16]. По-
। 214
49
1,0 2.0 3,0 fO 5,ОМ,, -w
ПС
Рис. II.9. Концентрационные пре-
делы существования растворов
смесей ПС и ПИБ (7:3) в зави-
симости от молекулярной массы
ПС.
вышепием степени ближне-
го порядка можно объяс-
нить и смещение температу-
ры стеклования в смесях,
которое наблюдалось в ра-
боте [17].
Влияние второго полиме-
ра на степень ближнего по-
рядка и Гс поливинилхло-
рида в смеси с ПММА изу-
чалось в работе [18]. Имен-
но это влияние приводит к
неаддитивному изменению плотности смеси полимеров с составом,
о чем указывалось ранее [19]. Повышение степени ассоциации
(ближнего порядка) в смеси полимеров объясняет уменьшение
энтропии ври смешении полимеров, которое было обнаружено экс-
периментально [20] и показано путем расчета [21].
Для расчета кривой свойство — состав можно исходить из адди-
тивности молекулярных масс смешиваемых полимеров или фрак-
ций полимеров с последующим расчетом кривой свойство — состав
на основе известных зависимостей свойства от молекулярной мас-
сы. Такой подход был впервые предложен и применен конкретно
к анализу данных по светорассеянию в работе [22].
При смешении полимеров средневзвешенную молекулярную
массу смеси можно рассчитать по уравнению
/Исм = MjUl, + Л12Щ2
(1)
где W, и w%—массовые доли полимера 1 и 2, а ДЦ и .П2—их молекулярные
массы.
Зависимость избыточного светорассеяния от молекулярной мас-
сы выражается формулой
feVC __ 1
^9J° Mw
2AZC
(2)
где A'=2n* 2n2/AW: v — инкремент показателя преломления; п— показатель пре-
ломления среды; No—число Лвагадро; X— длина волны падающего света.
Отсюда после соответствующей подстановки и упрощения по-
лучено следующее выражение:
см /?2шайсм/))2 (3)
где
% = k'v-C — 2,4аС7?"0 .
Полученное уравнение показывает, что избыточное рассеяние
не аддитивно массовой доле, как это предполагалось в работе
50
I'm 11.10. Зависимость избыточного светорассея-
ния А/?э0 раствора смеси ПС и ПИБ от соотно-
шения полимеров при общей концентрации рас-
iiiopa 1,8%. Молекулярная масса ПС 2,9-105,
ПИБ 1,2-106:
I экспериментальная кривая; 2 — кривая, рассчитанная
уравнению (3); пунктир- аддитивная зависимость [22].
|23]. Известно, что мутность (или свето-
рассеяние) смеси пропорциональна со-
ставу для бинарной смеси низкомолеку-
лярных жидкостей, если смесь по струк-
iype близка к идеальной [24]. В систе-
ме полимер — полимер — растворитель
интенсивность рассеяния зависит не толь-
ко от молекулярной массы полимера, но
и от его сродства к растворителю (зна-
чение А2) и от квадрата инкремента по-
казателя преломления (v2), а также от
ругих физических констант, входяших
!. величину k'.
На рис. 11.10 сравниваются теоретическое уравнение (3) и экс-
периментальные данные для смеси полимеров. Рассчитанная по
\ равнению (3) кривая оказывается выпуклой в направлении оси
бсцисс. Это означает, что избыточное светорассеяние, рассчитан-
ное из условия аддитивности, ниже, чем разность между экспери-
ментальным и теоретическим значением, рассчитанным по уравне-
нию (3). Разумеется, предложенный метод расчета не является
ipojini для области высоких копненiраций, так как уравнение (2)
ираведливо для разбавленных растворов. Таким образом, полу-
ченные данные свидетельствуют об относительной близости адди-
। ивной и теоретической кривой (см. рис. II. 10), а также показыва-
ioi направление отклонения расчетной кривой от аддитивной за-
висимости.
Аналогичный метод расчета применим и для анализа данных
но вязкости раствора смеси полимеров [25, 26].
Было предпринято несколько попыток теоретического описания
1.ШЧСИМОСТИ удельной вязкости от состава смеси растворов полиме-
ров с учетом параметров, позволяющих оценить взаимодействие
полимеров в смеси. Так, в работе [27] предложена формула:
9 уд см Т- 1'912 6*2 -J- ^цС’1 -| 2 у Т' />226*2
(4)
" />12=1 ЬцЬц, Ьц и Ь22— вискозиметрические параметры взаимодействия между
породными макромолекулами; bi2— параметр взаимодействия между разнород-
ими макромолекулами.
Аналогично уравнение для т)удсм, отличающееся от (4) лишь
особом выражения параметра взаимодействия bi2, предложено
г
51
в работе [28]:
Иуд см = I'lJiC'i + + ЬцС1 + (Ьц 4 Ь22) бгС2 4~ Ь22С2 (5)
где b12= (6U +b22)/2.
Получено [29] также теоретическое выражение для констан-
ты Хаггинса k' бинарной смеси полимеров в растворе и выраже-
ние для вязкости смеси:
т‘удсм = [qljCj 4- [1]]аСа 4- k' 4- fea МгСа + 2 1 kjkl [т]]; Мг^^г /6y
С Q4-C2
Если принять параметр взаимодействия й=А'[ц]2, то зависимость
(6) окажется аналогичной уравнению (4). Авторы [27, 28] пред-
лагают различие между экспериментальным и теоретическим зна-
чением параметра Дй12 = й12 эксп — Ь12теор принять за меру сродства
полимеров в растворе. Считают [30], что при подстановке в оба
уравнения среднеарифметического значения Ьц= (bu + bi2)/2 полу-
чается лучшее совпадение теории с экспериментом. В работах [31,
32] параметр ЛЬ12 использовали для оценки совместимости поли-
меров в растворе. Авторы [31] нашли, что совместимость, оцени-
ваемая по величине отклонения \bi2 от теории, совпадает с оцен-
кой, проведенной по расслаиванию. В другой работе [32] показано,
что с помощью параметра &bi2 можно установить область сов-
местимости полимеров, однако авторы считают возможности мето-
да ограниченными.
Все указанные выше работы позволяют оценить взаимодействие
полимер — полимер только в относительно разбавленных раство-
рах. В работе [33] была даже установлена корреляционная связь
между параметром Ьг2 и параметром взаимодействия Флори 712,
определенным из соотношения объемов расслоившихся растворов
по уравнению Скотта.
Во многих случаях вполне допустимо сравнение эксперимен-
тальных значений относительной, абсолютной или удельной вяз-
кости растворов с логарифмическими аддитивными значениями
(как это было сделано на примере рис. II.8).
В некоторых работах по вязкости растворов смесей полимеров
в качестве «аддитивных» использовались различные зависимости,
в том числе и полученные в результате сложения парциальных
значений свойств или построенные по принципу логарифмической
или арифметической аддитивности. В большинстве случаев экспе-
риментальные значения вязкости не совпадают с «аддитивностью»,
принятой авторами [34—39]. Совпадение наблюдается лишь в
разбавленных растворах или при повышении температуры.
§ 2. РАССЛАИВАНИЕ РАСТВОРОВ СМЕСЕЙ ПОЛИМЕРОВ
И СВОЙСТВА получаемых эмульсий
При повышении концентрации раствора смеси полимеров обыч-
но наступает момент, когда однофазный истинный раствор перехо
дит в двухфазный, мутный. При его хранении могут образоватьс»
52
ша слоя, различающихся по химическому составу и концентрации
полимера в слоях. Чем выше концентрация раствора смеси полиме-
ров, по достижении которой он расслаивается, тем шире область
• уществования однофазных, термодинамически устойчивых систем.
Если смесь полимеров в отсутствие растворителя устойчива прак-
тчески неограниченное время и расслаивание в ней не происходит
вследствие высокой вязкости компонентов, то вязкость растворов
обычно достаточно низка, чтобы расслаивание произошло сравни-
1сльно быстро. Это позволяет измерить многие важные парамет-
ры, такие, как время расслаивания, состав и объем слоев равно-
весной расслоившейся системы или же просто установить сам факт
наличия или отсутствия расслаивания, что не всегда просто сде-
1ать в смеси полимеров без растворителя. Поэтому изучение за-
висимости концентрационного предела расслаивания от различных
внутренних и внешних факторов является одним из косвенных пу-
тей оценки влияния тех же факторов на совместимость полимеров
в блоке (в отсутствие растворителя), где такая оценка сильно за-
|руднена. Подобного рода исследование имеет и самостоятельное
научное значение в связи с применением смесей полимеров в среде
общего растворителя для последующей переработки.
На предел расслаивания раствора смеси полимеров влияют сле-
(ующие факторы: природа и структура макромолекул смешивае-
мых полимеров; соотношение полимеров в смеси; молекулярная
масса и молекулярно-массовое распределение; химическая приро-
ы растворителя, его сродство к полимерам в смеси; температура;
напряжение сдвига (если раствор деформируется — течет или пе-
ремешивается); малые добавки других веществ.
Природа и структура смешиваемых полимеров. Концентрация
раствора, при которой полимеры расслаиваются, оказывается том
чипе, чем ближе полимеры по химической природе. Если разли-
чи- в химической природе велико, то расслаивание может проп-
|>|йти при концентрации менее 1%. Сильное различие в химической
природе, обусловленное наличием полярных функциональных
рупп, может, наоборот, привести к образованию смесей, не рас-
। .швающихся в растворе. Так, сополимеры, содержащие в макро-
кискулах некоторое количество карбоксильных групп (карбокси-
|цные каучуки с 7—15% карбоксильных групп), совместимы в
рп-творе с сополимерами винилпиридипа (винилпиридиповые кау-
|\кп с 5—15% пиридиновых групп). Близкие по природе полимеры
।пут иметь столь близкие физические константы, что микрорас-
|.швание в растворе не переходит в макрорасслаивание (это мо-
। привести к ошибочному выводу об однофазности смеси). Так,
чесь ipzc-полиизопрена и ф/с-полибутадиена в бензольном рас-
ni'tpe микрорасслаивается при довольно высокой концентрации,
пако полимеры имеют практически одинаковую плотность и мак-
р|'расслаивание не происходит, а из-за близких показателей пре-
'"млеиия помутнение смеси в результате микрорасслаивания не-
I MCI но. *
53
Наиболее полный анализ пар полимеров с точки зрения их
совместимости в растворе и в блоке дан в обзоре [40]. Фундамен-
тальными экспериментальными работами по расслаиванию поли-
меров в растворе являются работы [41 и 42].
В работе [41] были исследованы 35 пар полимеров в разных
растворителях. Все пары расслаивались, за исключением пар нит-
рат целлюлозы — поливинилацетат, нитрат целлюлозы-—полиме-
тилметакрилат, бензилцеллюлоза — полистирол. Впоследствии под-
тверждено, что нитрат целлюлозы и поливинилацетат действитель-
но образуют однофазную смесь [20]; надежные подтверждения
однофазности для других двух пар пока отсутствуют. Выло отме-
чено, что смесь нитрата целлюлозы и бензилцеллюлозы расслаи-
вается в растворе. Таким образом, нитрат целлюлозы «совместим»
с поливинилацетатом и несовместим с бензилцеллюлозой, к кото-
рой он ближе по химической природе. Различие в полярностях
ничего не объясняет, поскольку несовместимые полибутадиен и
полиизопрен не различаются по полярности, если в качестве ее ме-
ры использовать плотность энергии когезии этих полимеров. В ра-
боте [42] из 30 исследованных пар полимеров только три не рас-
слаивались в растворе: полистирол — поли-о-метилстирол, поливи-
нилацетат — полиметилакрилат, поли-л1-метилстирол — поли-п-ме-
тилстирол. Имеется, однако, информация, что полимеры второй
пары расслаиваются [41]. Расслаиваются даже такие пары, как
полиметил- и полиэтилакрилаты, поли-о- и ноли n-метилстиролы,
полиметилметакрилат и полиметилакрилат [42]. Обнаружена вза-
имная нерастворимость натурального каучука и гуттаперчи, кото-
рые являются цис- и трпнс-изомерами полиизопрена. Таким обра-
зом, даже небольшие различия в структуре макромолекул смеши-
ваемых полимеров могут привести к расслаиванию смеси. Это
обстоятельство полезно сопоставить с трудностью или даже невоз-
можностью образования совместных кристаллов в смеси кристал-
лических полимеров. Видимо, требования к максимально плотной
упаковке особенно высоки в случае полимеров, склонных к обра-
зованию надмолекулярных структур (ассоциатов) в аморфном со-
стоянии и в растворах.
Трудности исследования влияния структуры макромолекул по-
лимеров па предел их смешиваемости в растворах определяются
в значи 1елыюй мере сложностью получения полимеров с одина-
ковой молекулярной массой и разной структурой. Эта трудность
не была преодолена и в работе [43], в которой изучалось влияние
структуры полиметакрилатов и полиакрилатов на предел расслаи-
вания. Были определены пределы расслаивания в бензоле раство-j
ров смесей полиакрилатов (метил-, этил- и н-бутил) друг с дру-
гом. смесей полимегакрилатов с различной длиной эфирной боко-1
вой цепи (метил-, изо- и н-бутил, гексил-), а также смесей поли-1
акрилатов с полиметакрилатами. Полученные результаты сопостав-1
лились с числом углеродных атомов и мономерных звеньях сме|
шиваемых полимеров. Молекулярные массы смешиваемых полиме-1
54
ров не были одинаковыми и даже близкими, но тем не менее
прослеживалась некоторая общая тенденция, состоящая в том, что
различие в один углеродный атом (в основной или боковой цепи)
(остаточно, чтобы полимеры становились несовместимыми. Так, в
растворе расслаивались пары полиметилакрилат — полнэтилакрн-
лат и полиметилакрилат — полиметилметакрилат. Расслаивались
(аже растворы смесей полиизобутнлметакрилата с поли-н-бутил-
метакрилатом. При увеличении различии в числе углеродных ато-
мов в мономерных звеньях смешиваемых макромолекул увеличи-
вались различия в структуре макромолекул и уменьшался предел
расслаивания.
Влияние разветвленности на предел раслаивання изучалось в
работе [44]. В работе [45] исследовался предельный случай смеси
линейного и разветвленного полимеров на примере раствора смеси
линейного ПС с молекулярной массой (2—5) 10s и микрогеля ПС.
Микрогель ПС был получен эмульсионной полимеризацией стиро-
ла в присутствии 0,3% дивинилбензола. При помещении в толуол
микрогель ПС образовывал устойчивые растворы, однако после
смешения с толуольным раствором линейного ПС система расслаи-
валась, так, что в нижнем слое концентрировался микрогель, а в
верхнем ПС.
В этой связи интересно обратить внимание па представления,
высказанные в работе [41] о механизме расслаивания полимеров.
Именно в этой работе впервые было обращено внимание на роль
фуктурного фактора при оценке совместимости полимеров. Авто-
ры [41] считают, что каждая макромолекула в растворе окружена
молекулами растворителя, которые образуют сольватный слой оп-
ределенной шрукгуры, близкой к структуре тех элементов молоку-
ил, которые они окружают. Таким образом возникает некоторая
норядоченность расположения молекул растворителя и всякая
юлекула другой формы, имеющая иные «квазикристаллические
|ементы структуры», подойдя вплотную к первой, нарушит упо-
г-1 (оченпый слой растворителя. Это соответствует началу выделе-
ны полимера в виде второй фазы. Если же увеличить концентра-
цию одного какого-либо полимера выше той, при которой прояс-
нит расслаивание смеси, то в отсутствие другого полимера
|Ц1(еления твердой фазы не произойдет. Отсюда делается вывод,
•но при сближении одинаковых молекул упорядоченные (сольват-
ные) слои растворителя не мешают друг другу, а взаимно достра-
нот один другой так, что образуется ассоциат молекул с оди-
....вымн квазнкристаллическими элементами структуры.
(>риснтация молекул растворителя, находящегося в непосред-
ик-ипом контакте с макромолекулами полимеров (в том числе
юполярпых), доказана в работах [46].
Такое объяснение подтверждает предложенную в этой главе
1 выше) аналогию между образованием совместных кристалли-
ки х структур в смесях кристаллических полимеров и ассоциа-
• н макромолекул аморфных полимеров. Расслаивание растворов
55
смесей полимеров, видимо, является косвенным указанием на до-
вольно высокую степень ближнего порядка, т. е. высокую надмоле-
кулярную организацию в аморфных полимерах и их растворах,
при которой образование смешанных ассоциатов макромолекул
может произойти только при очень близкой структуре макромоле-
кул смешиваемых полимеров. Именно близкая конфигурация (кон-
формация) макромолекул смешиваемых полимеров, необходимая
для возникновения смешанных ассоциатов (что редко выполняет-
ся практически), приводит к раздельной ассоциации макромоле-
кул смешиваемых полимеров, определяющей значительные измене-
ния энтропии смешения.
Соотношение полимеров в смеси. Способность полимеров к сов-
мещению оценивают иногда по способности к расслаиванию смеси
их растворов. Расслаивание растворов смеси полимеров зависит не
только от концентрации, но и от соотношения полимеров в смеси.
Эта зависимость следует из вида бинодали (см. гл. I). При увели-
чении содержания компонента, находящегося в смеси в относитель-
но малом количестве, предел расслаивания обычно уменьшается.
Области с малым содержанием одного из полимерных компонен-
тов как правило исследованы мало. Более того, не раз отмечалось
[37, 39, 47], что при наличии в смеси менее 10% одного из поли-
меров расслаивания не происходит. Этот вывод, конечно, зависит
и от методики, с помощью которой изучали расслаивание раство-
ров. Обычно смешивают в различных пропорциях растворы одина-
ковой концентрации, заведомо большей предела расслаивания при
соотношении полимеров 1 : 1, и эта концентрация может оказаться
недостаточной для расслаивания растворов с иным соотношением
полимеров.
Молекулярные массы и ММР полимеров. Экспериментально
уменьшение предела расслаивания с возрастанием молекулярной
массы было установлено еще в работе [11]. В гл. I было показа-
но, что как в ранних, так и в современных теориях растворов смесей
полимеров указывается, что взаимная растворимость полимеров
стремится к нулю при стремлении молекулярной массы к бесконеч-
ности. Теория, однако, не всегда применима к конкретным поли-
мерным системам, иногда из-за отсутствия надежных значений для
расчета фи шческих параметров, иногда из-за неадекватности тео-
рии. В лой связи было предпринято немало попыток установить
эмпирические зависимости предела расслаивания от молекулярной
массы.
В работе [IS] установлена следующая эмпирическая зависи-
мость критической концентрации С2 от молекулярной массы поли-
меров, подтвержденная впоследствии в [49]:
С3 = А (Л1+)-’/« + (7)
где Л7+= (Лй-М,)0-6 — среднегеометрическая молекулярная масса; Mi, Mj — моле-
кулярные массы смешиваемых полимеров, Л и С™ — константы для данной па-
ры полимеров.
56
Это уравнение справедливо для полимеров как с узким, так и
Юстаточно широким распределением по молекулярным массам.
\вторы [48] считают, что только в том случае, если исследуется
комбинация полимеров, молекулярные массы которых очень силь-
но различаются и для которых не выполняются условия ('/ц,—
Z2s)<l и х°^_<х2<х*, значения С2 не ложатся на прямую зависи-
мости С 2—
Уравнение (7) подобно уравнению, предложенному в работе
[50]. По данным другой работы [51], зависимость С2 от молеку-
лярной массы для системы ПС — ПММА — бензол (менялась мо-
лекулярная масса ПС) хорошо описывается уравнением
l/lnlnc ~ а1 ’П ^2^) + f (4Упс
где v — средний удельный объем полимера; Ci и f — константы.
Аналогичное уравнение можно записать для зависимости С2 от
молекулярной массы ПММА, но с другими константами.
При необходимости многократных определений молекулярной
массы полимеров одинаковой химической природы и при относи-
тельно постоянном значении ММР не следует игнорировать воз-
можность определения ее путем «титрования» растворителем рас-
твора смеси полимера с другим, эталонным полимером и определе-
ния концентрации, при которой происходит просветление смеси.
Наличие калибровочной кривой для данной пары полимеров и от-
носительно постоянная растворимость одного полимера в другом
при ограниченном изменении молекулярной! массы исследуемого
полимера позволяет быстро получить падежные значения молеку-
лярной массы исследуемого полимера.
Влияние молекулярной массы на предел расслаивания в раз-
личных растворителях было изучено в работе [52]. Интенсивность
взаимодействия полимера с растворителем оценивалась по вязко-
сти 1%-ных растворов полимеров в данном растворителе. На осно-
вании большого количества экспериментальных данных по опреде-
лению предела расслаивания установлена следующая эмпиричес-
кая зависимость:
Oh /Л + 1]г/В2) b=kC2 (9)
где Ц| и 1)2 — удельные вязкости растворов полимеров при концентрации 1 г/дл;
Pi и Р2 — степени полимеризации компонентов; b и k — константы.
Влияние молекулярной массы на совместимость полимеров в
растворе может проявиться и в «чистом виде» при полной идентич-
ности химической природы и структуры макромолекул смешивае-
мых полимеров. Так, расслаивание в растворе смеси фракций по-
лиэтилена, молекулярные массы которых различаются более чем
в 10 раз, наблюдалось авторами работы [53]. Правда, расслаива-
ние в этой системе наблюдалось в сравнительно узком темпера-
турном интервале, что, однако, непосредственно следует из теории
растворов Томпа. Имеется ряд данных, показывающих, что смеси
57
фракций одного и того же полимера, значительно различающиеся
по молекулярной массе, обнаруживают признаки двухфазной
структуры и в отсутствие растворителя.
Несколько меньше влияние молекулярной массы на предел
расслаивания проявляется для блок-сополимеров, в которых мак-
ромолекулярные блоки разнородных полимеров связаны химиче-
ской связью, что предотвращает расслаивание; поэтому оно на-
ступает при значительно больших молекулярных массах полимер-
ных компонентов. В работе [54] экспериментально показано, что
блок-сополимер стирола и а-метилстирола типа Л—Б остается од-
нофазным (одна Тс) даже по достижении молекулярной массы бло-
ков в 2-105, т. е. в условиях, в которых смесь гомополимеров име-
ет ярко выраженную двухфазную структуру.
Природа растворителя. Отсутствие точного критерия при опен-
ке сродства растворителя к полимеру привело к тому, что при ис-
следовании влияния природы растворителя па совместимость каж-
дый автор применял свой критерий оценки качества растворителя.
В работе [55] о качестве растворителя судили по величине па-
раметра растворимости б. Сравнивая параметры растворимости
различных растворителей с параметрами смешиваемых полимеров,
определяли наибольшую совместимость полимеров в растворе в
тех растворителях, 6 которых были близки к б полимеров.
Качество растворителя оценивали и по величине удельной вяз-
кости растворов при концентрации 1 г/дл [52]. Вязкость рассмат-
ривалась как мера сольватирующей способности растворителем
данного полимера. Эмпирическая зависимость предела расслаива-
ния от природы растворителя и молекулярной массы полимера
описывается уравнением (9). Из этого уравнения следует, что чем
хуже растворитель (меньше вязкость раствора), тем при более
низкой концентрации растворы расслаиваются. Этот вывод был
подтвержден для смесей других полимеров, состав которых изме-
нялся в широких пределах [56]. Качество растворителя в работе
[56] более корректно оценивали по характеристической вязкости
раствора [ц].
Предлагалось оценивать сродство полимера к растворителю
по константе а в уравнении Куна—Марка—Хаувинка [ц]=КА1“,
которая характеризует степень набухания полимерного клубка в
данном растворителе [57]. Предел расслаивания увеличивался с
уменьшением разности констант для смешиваемых полимеров. Это
означает, что совместимость полимеров в растворе ухудшается,
если растворитель оказывается плохим по отношению к одному из
полимеров. Однако в работе [57] не исследовались смеси полиме-
ров в растворителе, являющемся плохим по отношению к обоим
полимерам: в этом случае разность констант может оказаться
очень небольшой и предел расслаивания также довольно низким.
В настоящее время на основании многих экспериментальных
результатов (см., например, [52, 56, 58]) можно однозначно за-
58
ключить, что предел расслаивания данной пары полимеров в рас-
творе в общем растворителе уменьшается с ухудшением качества
растворителя. Эю обстоятельство приводит к тому, что концент-
рационная область существования однофазных смесей полимеров
в растворе уменьшается при переходе к «плохим» раствори-
телям.
Температура. Характер влияния температуры на фазовый со-
став системы полимер — полимер — растворитель определяется
тем, какое положение на фазовой диаграмме занимает эта система
при комнатной температуре, а также определяется видом фазо-
вой диаграммы (см. гл. I).
Различия в молекулярной массе и во взаимодействии выбран-
ного растворителя с полимерными компонентами смеси вызывают
резкую асимметрию бинодали, и критическая температура (верх-
няя или нижняя точка диаграммы для диаграмм соответственно
с ВКТС и НКТС) может оказаться в области соотношений поли-
меров со значительным преобладанием одного из компонентов.
В этом случае соотношение полимеров 1 : 1 в выбранном интерва-
ле температур может соответствовать крайней части восходящей
или нисходящей ветви бинодали. Фазовые диаграммы смесей поли-
меров в растворе и в отсутствие растворителя получены для огра-
ниченного числа систем, поэтому часто приходится пользоваться
результатами изучения температурной зависимости предела рас-
слаивания для ограниченной области составов и температур. Эти
результаты подтверждают высказанные общие соображения о
влиянии температуры на «совместимость» полимеров в растворе.
Так, ряд авторов утверждает, что температура практически не
влияет [6, 9] или влияет очень незначительно [41, 59] на предел
расслаивания растворов.
В других работах [55, 60] отмечается, наоборот, важная роль
температуры, влияющей на гибкость макромолекул и на их соль-
ватацию растворителем, а следовательно, и на предел расслаива-
ния. Например, смесь ПС с ПММА в о-ксилоле, расслаивающаяся
при 25 °C, становится однофазной при 85 °C [55]. В то же время
в л-оксибензоле эта смесь была гомогенной при 25 °C и расслаива-
лась при 85 °C. Смесь хлоркаучука с сополимером этиленвинилаце-
тата образовывала однофазную систему при 25 °C, которая рас-
слаивалась при повышении температуры [60].
Систематические исследования влияния температуры на предел
расслаивания не потеряли актуальности с появлением термодина-
мических теорий взаиморастворимости полимеров. Такие исследо-
вания могли бы помочь сформулировать некое эмпирическое пра-
вило, облегчающее поиск растворителей для данной пары поли-
меров, в которых бы обеспечивался наибольший температурный
интервал существования однофазных, а следовательно, устойчивых
во времени растворов смесей полимеров.
Напряжение сдвига. Внешние деформирующие усилия влияют
на фазовые переходы различным образом. Впервые влияние на-
59
пряжения сдвига на расслаивание растворов полимеров было об-
наружено в работе [61, 62].
Эксперименты проводили на примере смеси бензольных раство-
ров ПС и этилцеллюлозы, используемых в соотношении бензол:
ПС : ЭЦ=97.1 : 1,1 : 1,8. При этом относительный объем фаз рас-
твора ПС и ЭЦ составлял соответственно 47 и 53% при 21,7 °C,
а критическая температура, при которой система становилась одно-
фазной, равнялась 31,7 °C.
Если в такой системе создать градиент скорости, например
при сдвиге жидкости в зазоре между коаксиальными цилиндра-
ми, то при градиенте в 200 с-1 система «просветляется», т. е. ее оп-
тическая плотность становится равной оптической плотности рас-
твора ПС и ЭЦ при эквивалентной концентрации. Авторы отмеча-
ют, что это наблюдается при удалении от Ткр по температурной
шкале на 10°. Таким образом, эффект, вызываемый сдвигом с гра-
диентом скорости 200 с-1, аналогичен нагреванию на 10° этого
раствора, или, как это экспериментально показано, разбавлению
системы растворителем в 1,15 раза.
Интересно объяснение, данное авторами наблюдаемому эффек-
ту. Само по себе дробление не может привести к уменьшению ка-
пель до размера, при котором система становится оптически про-
зрачной. Соотношение градиента скорости и межфазного натяже-
ния определяет размер капель только до достижения каплями диа-
метра около 10-5 м. Далее для достижения оптической однород-
ности системы за счет простого дробления частоту вращения рото-
ра следовало бы увеличить в 1000 раз по сравнению с экспери-
ментальной.
По-видимому, отмечают авторы, межфазное натяжение на гра-
нице растворов полимеров, составляющее при данной концентра-
ции около 10 6 Н/м, уменьшается при дальнейшем дроблении, что
облегчает диспергирование. Оптическая прозрачность достигается
тогда, когда размер капель становится соизмеримым с размерами
межфазного слоя. Это и есть момент полного исчезновения призна-
1 Ков фазового расслаивания.
Более подробное исследование влияния напряжения сдвига на
предел расслаивания проведено в работах [14, 63] на примере
растворов смесей ПС—ПИП в толуоле (хороший растворитель,
С2 = 2,85%) и в декалине (плохой растворитель, С2 = 1,5%) и сме-
сей ПС- ПММА в толуоле (С2 = 10,5%) и в ксилоле (С2=5.0%).
На рис. II II показана зависимость оптической плотности рас-
творов от напряжения сдвига для растворов в хорошем и плохом
растворителе. Видно, что при достижении определенной величины
напряжения сдвига раствор становится более прозрачным и, на-
конец, оптическая плотность его становится равной оптической
плотности, характерной 1ля однофазного раствора ПС или ПИП
при данной концентрации (пунктир на рисунках). Из приведен-
ных данных следуют некоторые важные выводы. Видно, что чем
меньше концентрация рас шора, т. е. чем ближе она к пределу
60
Рис. II.11. Зависимость оптической плотности растворов смесей ПС и ПИБ (I : 1)
в декалине (а) и в толуоле (6) от напряжения сдвига. Цифры у кривых- кон-
центрация полимеров в г/дл.
расслаивания, тем меньшее напряжение требуется для перевода
системы в однофазную; при большом удалении от предела расслаи-
вания оптическая плотность под влиянием напряжения сдвига хо-
тя и снижается, но не достигает значений оптической плотности
однофазных растворов; и, наконец, очевидно, что разрушение ка-
пель в хорошем растворителе происходит легче, чем в плохом.
Переход от двухфазного раствора к однофазному под действи-
ем сдвига означает, что расслаивание с возрастанием напряжения
сдвига происходит при большей концентрации раствора. Действи-
тельно, по данным рис. 11.11 можно построить зависимость преде-
ла расслаивания от напряжения сдвига па примере растворов сме-
си ПС и ПИП (рис. 11.12). Аналогичные результаты получены и
для растворов смесей ПС и ПММА.
На рис. 11.13 показана зависимость растворимости одного рас-
твора в другом от напряжения сдвига. Одновременное воздействие
температуры и напряжения сдвига на растворы показало, что уве-
личение взаимной растворимости полимеров (возрастание Сг),
вызванное нагреванием, может быть усилено действием сдвига.
Повышение температуры оказывает большее воздействие на рас-
творы смесей полимеров в плохих растворителях, а увеличение на-
пряжения сдвига на растворы в хороших растворителях.
Влияние напряжения сдвига на фазовый состав смеси полиме-
ров может проявиться при перемешивании, взбалтывании, перека-
чивании растворов; может при перемешивании на механическом
оборудовании, особенно при повышенной температуре, может про-
исходить увеличение взаимной растворимости полимеров и в отсут-
ствие растворителя.
Малые добавки других веществ. Малые добавки веществ, вво-
димые в количествах, не меняющих качество растворителя в це-
лом, иногда приводят к изменению предела расслаивания. Так, в
работе [35] действие спирта и ацетона на смесь растворов ПВХ и
бутадиен-стирольного сополимера объяснялось возможным блоки-
рованием этими веществами полярных групп ПВХ. Однако анало-
61
Рис. П.12. Зависимость С2 от напряжения сдвига для растворов смеси ПС и
ПИП (1:1) в толуоле (7) и в декалине (2).
Рис. 11.13. Зависимость растворимости раствора ПИП в растворе ПС (/) в де-
калине и раствора ПС в растворе ПММА (2) в декалине от напряжения сдвига.
гичный эффект был вызван введением метанола в смесь растворов
натурального и бута диен-стирольного каучуков, в которых поляр-
ные группы отсутствовали [39]. Предотвращение расслаивания
растворов ПС и ПВА при введении в раствор ацетона объясняли
[64] агрегированием молекул и изменением их формы. В то же
время в работе [65], в которой исследовалось влияние ряда низ-
комолекулярных веществ на систему ПВХ—-нитрат целлюлозы —
ацетон, расслаивание задерживалось веществами с более высоким
поверхностным натяжением, чем у ацетона. Количественной взаи-
мосвязи между концентрацией добавки, устраняющей фазовое рас-
слаивание, и ее поверхностным натяжением нс установлено.
В работе [47] исследовано действие 23 ра «личных добавок на
смесь ПС—ПММЛ в бензоле. В качестве добавок использовали
спирты, кетоны, органические кислоты, сложные эфиры, амины
(анилин, диметиланилин) и такие вещества, как дихлорэтан, хлор-
бензол, этиленхлоргидрин, пиридин, тиофен. Установлено, что не-
которые вещества очень эффективно предотвращают расслаивание
растворов или замедляют его, некоторые, наоборот, ускоряют рас-
слаивание. Па основании полученных экспериментальных данных
не удалось найти непосредственной связи между эффектом дейст-
вия добавки и ее поверхностным натяжением, дипольным момен-
том, диэлектрической проницаемостью или теплотой испарения.
Специфичность действия добавки и ее эффективность для дан-
ной системы полимер — полимер— растворитель исследовались в
работе [66]. В системе ПС—ПИП—бензол [66] проверяли дейст-
вие добавок, которые были эффективны в смесях, содержащих по-
лярные полимеры [35, 47, 64]. Ни одно из применяемых веществ
не оказалось эффективным. Было обнаружено, однако, что хинон,
вводимый в раствор смеси ПС—ПИП в виде бензольного раство-
62
а с
Рис. II 14. Зависимость предела расслаивания С2 бензольного раствора смеси ПС
и ПШ1 (1:1) (а) и относительной вязкости Т]|)1Н бензольных растворов ПИП
(1) и ПС (2) (б) от количества введенного хинона.
ра, увеличивает концентрацию расслаивания (рис. 11.14); при этом
собственно эффект повышения С2 связан, видимо, с изменениями
конформации макромолекул ПИП (рис. 11.14, б). Механизм дейст-
вия хинона в данном случае не вполне ясен. На рис. 11.15 показа-
но, что эффект этот развивается во времени и не проявляется в
растворе полибутадиена и его сополимеров. При 3%-ной концент-
рации раствора смеси полимеров и содержании хинона макси-
мально 10% (в пересчете на сухой остаток) концентрация хинона
в растворе не превышает 0,3% и, следовательно, не изменяет ка-
чества растворителя но отношению к полимеру. В то же время
доказано, что химического взаимодействия хинона с полимерами
не происходит.
По-видимому, хинон специфиче-
ски взаимодействует с макромоле-
кулами полиизопрена, как бы «сор-
бируясь» на них, приводя к разру-
шению ассоциатов макромолекул,
что затрудняет образование ассо-
циатов достаточно больших для воз-
никновения поверхности раздела
Природа этого явления, видимо, та-
кая же, как и разрушение «по сорб-
ционному механизму» узлов флук-
Рис. II. 15. Кинетика изменения относитель-
ной вязкости бензольных растворов ПИП
(/), НК (2), СКС-ЗО АКР (3) и СКД (4)
при введении 10% (масс.) хинона (в рас-
чете на полимер).
63
ту анионной сетки (а ими могут также быть ассоциаты!)
[67].
Привитые и блок-сополимеры, добавляемые к смеси растворов
гомополимеров такого же химического состава, как и блоки сопо-
лимеров, не образуют однофазную систему и не изменяют предел
расслаивания. Если размер блоков блок-сополимера достаточно
велик, чтобы он мог образовывать домены дисперсной фазы в от-
сутствие растворителя, то это является важным условием стабили-
зирующего эффекта блок-сополимера в растворе смеси гомополи-
меров. Такой блок-сополимер в отсутствие гомополимеров в рас-
творе при некоторой концентрации образует двухфазную систему,
и по достижении этой концентрации раствор блок-сополимера сам
по себе становится мутным. При меньшей концентрации блок-сопо-
лимер образует в растворе мицеллы и гомополимеры ассоциируют
с одинаковыми по химическому составу блоками сополимера. Воз-
никает сначала однофазная (концентрация меньше предела рас-
слаивания), а затем с ростом концентрации двухфазная система,
которая, однако, может быть очень стабильной во времени [68,
69]. Таким образом, эффективность действия блок-сополимеров
проявляется в увеличении стабильности эмульсии смеси растворов
полимеров и регулировании размера частиц эмульсии, а не в уве-
личении концентрации расслаивания в этой системе [58].
Объем и состав слоев расслоившейся смеси растворов. При
смешении растворов полимеров с концентрацией выше предела
расслаивания получается эмульсия, обычно не являющаяся агрега-
тивно устойчивой и поэтому со временем расслаивающаяся на два
слоя. Расслаивание завершается образованием четкой границы
раздела между слоями, каждый из которых содержит преимущест-
венно один полимер, а расположение слоев соответствует значени-
ям плотности полимеров. Обычно объемы слоев не равны объемам
исходных растворов полимеров, использованных для смешения.
Это связано с перемещением как макромолекул полимеров, так и
растворителя из одного слоя в другой. Измерение объема, концент-
рации растворов в слоях и состава слоев дает полезную информа-
цию о сродстве полимеров друг к другу и о влиянии температуры,
природы растворителя и других факторов на взаимодействие по-
лимер — полимер.
Возникновение границы раздела между слоями происходит
сравнительно быстро, но объемы слоев со временем несколько из-
меняются. Например, в смеси 3%-ных толуольных растворов ПС
(мол. масса 3,7-10’) и ПИБ (мол. масса 3,0-10') при комнатной
температуре граница раздела возникает через 2 ч, а ее перемеще-
ние прекращается через 5 сут. Время установления равновесия
зависит от общей высоты столбика расслаивающегося раствора и
колеблется от недели при высоте столбика 10 см до 2 мес, при вы-
соте 50 см. Сначала после установления границы раздела имеется
перепад концентрации в слоях — она увеличивается сверху вниз
в каждом слое, однако по достижении равновесия концентрация
64
становится постоянной по высоте одного слоя. При значительной
вязкости растворов и малой разности плотностей полимеров рас-
слаивание почти не ускоряется при центрифугировании.
Скорость расслаивания уменьшается с возрастанием концент-
рации и увеличивается с повышением температуры, что связано с
изменением вязкости [70]. Отрицательное влияние температуры
на устойчивость растворов было обнаружено впервые в рабо-
те [41].
В работе [41] было высказано также предположение, что рас-
творитель перераспределяется между слоями растворов пропор-
ционально осмотическому давлению в каждом слое. Это дает воз-
можность рассматривать границу раздела как полупроницаемую
перегородку, через которую диффундирует (после установления
равновесия) только растворитель, и положение которой фиксиру-
ется в момент достижения равенства осмотических давлений в
слоях.
Осмотическое давление в растворе с концентрацией С равно
n/C = R7’f-=l- + H2c') (10)
\ Мп /
При условии равенства
тих растворов получается:
осмотических давлений контактирую-
___1 /41g -р 42С
С2 ~ 1/М^А'^
(П)
(12)
При полном или почти полном разделении полимеров по сло-
ям концентрацию в каждом слое можно выразить через объем
раствора: C=CoVo/V (где Со и Vo — исходные концентрация и
объем). Обычно исходные растворы имеют равные концентрации
при соотношении 1:1; отсюда окончательно получается [63]:
Уг _ l/Mj+A"2Ct
Vi " 1/^+4^
Уравнение (12) показывает, что если молекулярные массы рав-
ны, то больший объем займет тот слой, в котором больше второй
вириальиый коэффициент А2 взаимодействия полимер — раство-
ритель. Это хорошо подтверждается тем, что для одной и той же
пары полимеров объем данного слоя в зависимости от природы
растворителя тем больше, чем больше характеристическая вяз-
кость полимера в этом растворителе. Если А'2 =А ", то объем сло-
ев не находится в пропорциональной зависимости от молекуляр-
ной массы. Если значение А2С велико, то даже значительные из-
менения молекулярной массы не приведут к существенным
изменениям объема слоев. Только в 0-растворителе при условии
^2 =7)? =0 соблюдается следующая простая зависимость [41, 34]:
Е, _ g2
V2 М,
(13)
5-244
65
что, правда, не получило экспериментального подтверждения, по-
скольку авторы не учли влияния природы растворителя на объем
слоев. Экспериментально показано, что при расслаивании, напри-
мер, смеси ПС и ПИБ в толуоле с исходной концентрацией поли-
меров 2,9 г/дл объемы слоев, хотя и не равны исходным, но и не
изменяются при изменении молекулярной массы ПС в интервале
(0,65—8,3) -105 при сохранении постоянной молекулярной массы
ПИБ 3-106.
Соотношение (13) является приближенным, поскольку при зна-
чительных концентрациях уравнение (10) не корректно, и при кон-
центрациях, близких к пределу расслаивания, может происходить
значительный переход макромолекул одного полимера в другой.
На системе ПС — атактический ПП — толуол установлена кон-
центрационная зависимость содержания ПП в слое ПС, начиная
с концентрации, лишь на десятые доли процента превышающей
критическую [59]. Вблизи критической концентрации в слое ПС
содержалось до 50% общего количества ПП. При увеличении кон-
центрации содержание ПП в слое резко падало до 4—5% и затем
мало менялось при дальнейшем изменении концентрации. Расчет
параметра взаимодействия по уравнению Скотта показал, что с
увеличением концентрации Х12 также уменьшается, асимптотически
приближаясь к практически постоянной величине.
Это последнее наблюдение согласуется с зависимостями объема
слоев от общей концентрации раствора смеси полимеров и от тем-
пературы (рис. 11.16), из которых видно, что при удалении от кри-
тической концентрации или при удалении от Ткр объемы слоев из-
меняются сначало сильно, а затем очень мало.
Экспериментальное определение зависимости объема слоев от
соотношения полимеров позволяет оценить взаимную раствори-
мость полимеров в растворе данной концентрации, а построение
кривых при нескольких температурах дает во )можность опреде-
лить влияние температуры на взаимную растворимость (рис. 11.17
и II.18). Если кривые выходят не из начала координат, то отсе-
каемый ими отрезок на оси составов соответствует значению вза-
Рис. 11.16. Зависимость объема слоя ПС в pin слоившейся смеси ПС и ПИП
(1:1) от концентрации («) и температуры (о) Цифры у кривых — температура
в "С (п) и концентрация в % (б).
66
Рис. 11.17. Изменение объема верхнего слоя в зависимости от состава расслоив-
шихся 10%-ных бензольных растворов ПС и ПММА при 25 (/) и 35 3С (2). Мо-
лекулярная масса ПС 3-105, ПММА 8,5-10s.
Рис. П.18. Изменение объема нижн<го слоя расслоившихся 2,8%-ных хлорофор-
менных растворов смеси ПС — ПИБ пси 25 (/) и 35°C (2). Молекулярная мас-
са ПС 3-105, ПИБ 1,2-10е.
имной растворимости полимеров. Из рис. 11.18 видно, что ПИБ с
большой молекулярной массой не растворяется в растворе ПС в
заметных количествах, поскольку кривая выходит из начала коор-
динат и объем слоя меняется пропорционально исходным объемам
растворов. ПС лучше растворяется в растворе ПИБ, поскольку
объем слоя ПС при преобладании ПИБ оказывается меньше, чем
объем раствора ПС, взятого для смешения. С повышением темпе-
ратуры объем слоев в смеси ПС—ПММА приближается к аддитив-
ной прямой, а в смеси ПС—ПИБ — удаляется. Это означает, что
взаимная растворимость полимеров ухудшается с повышением тем-
пературы в первом случае (система с НКТС) и улучшается во
втором случае (система с ВКТС).
Кривая свойство — состав раствора смеси полимеров при кон-
центрации выше предела расслаивания. Выше предела расслаива-
ния исследование растворов осложняется высокой вязкостью, зна-
чительной ее аномалией, делающей практически необходимым сня-
тие кривой течения, а также относительной неустойчивостью рас-
творов во времени вследствие их двухфазности. Эти причины обус-
ловили весьма ограниченное число работ по вязкости концентри-
рованных растворов смесей полимеров.
В работе [70], одной из первых в этой области, на примере
смеси диацетата целлюлозы с полярными полиакрилонитрилом
или хлорированным ПВХ показано, что удельная вязкость раство-
ров в ацетоне не является аддитивной функцией состава (аддитив-
ность— прямая линия в координатах удельная вязкость — состав),
5'
67
а значительно ниже аддитивной прямой. С увеличением общей кон-
центрации полимеров в растворе это отклонение увеличивается.
В обзоре [71] приводятся данные по весьма большому числу
систем и указывается, что для концентрированных растворов сме-
сей полимеров вязкость чаще всего ниже аддитивной, однако в
смесях полярных полимеров с сильным межмолекулярным взаимо-
действием часто наблюдаются отклонения от этого правила.
Реологическим свойствам концентрированных растворов смеси
производных целлюлозы с другими полярными полимерами посвя-
щен ряд работ [72—74], в которых также обращается внимание на
то, что вязкость растворов смеси полимеров ниже аддитивной и
сильно зависит от напряжения и скорости сдвига (сильная анома-
лия вязкости).
Интересные экспериментальные результаты по вязкости кон-
центрированных растворов смесей полимеров получены в работах
[75—78] на примере смесей полярных полимеров с полярными и
с неполярными полимерами при изучении волокнообразования и
пленкообразования из этих растворов.
По существу единой теорией растворов смесей полимеров при
концентрации выше предела расслаивания могла бы явиться тео-
рия, учитывающая течение эмульсии с переменным сечением ка-
пель, меняющих форму в процессе течения под влиянием напря-
жения сдвига. Основным уравнением, описывающим течение
эмульсий, является формула Тэйлора, которая, однако, не учиты-
вает изменения формы капель при течении, приводящего даже к
волокнообразованию в потоке двухфазной жидкой системы, как
это показано в работах [61, 62].
Изменение формы капель эмульсии в процессе течения являет-
ся основой теории смешения полимеров в расплаве и растворе и
рассматривается в главе III, в то время как явление обращения
фаз в растворах смеси полимеров имеет значение для построения
зависимости свойство — состав не только растворов, но и распла-
вов смесей полимеров и рассматривается в главе V.
« 3. МЕЖФАЗНОЕ НАТЯЖЕНИЕ И САМОПРОИЗВОЛЬНОЕ
ЭМУЛЬГИРОВАНИЕ В РАСТВОРАХ СМЕСЕЙ ПОЛИМЕРОВ
В работах |61, 62] было указано, что межфазное натяжение на
границе раздела растворов полимеров в общем растворителе рав-
няется примерно 10 6 П/м, однако конкретные экспериментальные
данные не приводились и достоверность полученного значения не
обсуждалась. Позднее были выполнены две независимые работы
по определению межфа шого натяжения в растворах смесей поли-
меров [79, 80]. Выяснилось, что межфазное натяжение о действи-
тельно мало и колеблется в пределах сотых и тысячных долей
П/м в зависимости от концентрации раствора. Малое значение о
приводило даже к диспергированию капли одного раствора в дру-
68
Рис. 11.19. Зависимость межфазного натяжения
от концентрации расслоившейся смеси раство- '
ров ПС и ПИБ в толуоле при 20 СС. Молоку- '
лярная масса ПС: 8,3-105 (/), 2,9-105 (2),
3,7- 10й (3), 1,24 10s (-#), 0,65-10s (5); 3 — не-
фракциопированиый блочный ПС [63, 65].
гом при определении межфазного на-
тяжения методом выдавливания кап-
ли на горизонтальную подложку. z-°
На рис. 11.19 показана зависи-
мость <т от концентрации раствора и
молекулярной массы полимеров [81].
Несмотря на сильную зависимость о
от концентрации раствора, межфазное
натяжение в исследованных пределах
концентраций остается в пределах ты- 10 г,о j,v
сячных долей Н/м. Пересечение оси с.г/ЮОмя
концентраций в точках С2 при продол-
жении кривых подтверждает известную закономерность стремле-
ния к нулю межфазного натяжения в критической точке. Изотерма
межфазного натяжения спрямляется в координатах Igo—lg(C—
С2)3/2. Методом экстраполяции этой зависимости к 100%-ной кон-
центрации раствора получено значение о па границе раздела ПС—
ПИБ, равное 10-3 Н/м. Эти данные хорошо согласуются с данны-
ми, появившимися впоследствии в результате прямого измерения,
экстраполяции или расчета (гл. IV).
Межфазное натяжение как известно (гл. IV), связано с разме-
ром межфазного слоя, уменьшение п сопровождается увеличением
толщины слоя, распространяющегося на весь объем фазы по до-
стижении системой критической точки. Уменьшение о с уменьше-
нием концентрации раствора свидетельствует о большой протя-
женности межфазных слоев на границе раздела растворов смесей
полимеров. Характерно, что данные рис. 11.19 указывают на сла-
бую зависимость ст от молекулярной массы. Затухающий характер
соответствующей кривой [81] и стремление к пределу является
еще одним косвенным доказательством того, что межфазное натя-
жение в высокомолекулярных системах определяется сегменталь-
ной растворимостью и поэтому мало зависит от молекулярной
массы контактирующих полимеров. В области относительно не-
больших значений молекулярной массы поверхностное натяжение
описывается формулой а = Ооо— KIM*1*, где К—константа, а —
поверхностное натяжение при бесконечно большой молекулярной
массе [82].
В работе [83] рассчитано значение межфазного натяжения для
растворов полимеров. Расчет формально подобен расчетам по
рассеянию света полимерными растворами вблизи критической
температуры (по теории Дебая). Автор [83] утверждает, что и
расслаивание смеси растворов несовместимых полимеров, и рас-
69
спаивание раствора полимера при критической температуре — по-
добны. Полученное в [83] уравнение справедливо лишь для сим-
метричной системы для полимеров с одинаковыми молекулярными
массами. Рассчитанная величина по порядку согласуется с экспе-
риментом.
Малое межфазное натяжение и уменьшение его при уменьше-
нии концентрации раствора или повышении температуры может
приводить к самопроизвольному диспергированию! раствора одно-
го полимера в среде другого, теоретическое обоснование возмож-
ности которого в коллоидных системах дапо в работах [84, 851
При повышении температуры граница раздела растворов, плос-
кая при комнатной температуре, становится выпуклой в сторону
менее плотного раствора, как это можно наблюдать для смеси
ПС—ПИП в декалине с молекулярными массами 2,9 -105 и 2,5-
•105. При нагревании ее до 80- 100 °C граница вообще исчезает.
Диспергирование одного раствора в другом приводит иногда к
образованию очень стабильной эмульсии, способной к опалесцен-
ции (см. главу VII).
Устойчивые эмульсии, возникающие в одном из слоев после
перемешивания и расслаивания растворов полимеров,— довольно
типичное явление, которое наблюдалось неоднократно. В литера-
туре были упоминания о том, что один из слоев иногда оказыва-
ется мутным, но никакого объяснения этому не давалось. Так, в
смеси 3%-иых растворов ПИЬ (3- 10г) и ПС с различными молеку-
лярными массами (см. рис. 19) мутным всегда является верхний
слой, содержащий преимущественно ПИБ. Раствор содержит кап-
ли раствора ПС размером около 10 мк. Во всех случаях более
вязким раствором был раствор ПИБ. Таким образом само по себе
изменение мол. массы пе является достаточным условием для об-
разования эмульсии в противоположном слое. В толуольном рас-
творе смеси ПИБ (мол. масса 1,08-105) и ПС (9.25-105) после
расслаивания более вязким оказывается1 нижний слой с преобла-
данием ПС, и именно этот слой оказывается мутным. В мутном
слое со держится 24% ПИБ, а в прозрачном слое ПИБ лишь 3%
ПС.
Аналогичный вывод можно сделать из рассмотрения данных
табл. П.З, согласно которым в нижнем слое всегда находится
ПММА, а в порхнем ПС. Независимо от природы растворителя и
полимера мутным, т. е. содержащим эмульсию другого полимера,
оказываете я ют слой, в котором вязкость выше В му гном слое со-
держится 20 26% другого полимера [81]
Полученные данные можно объяснить с точки зрения влияния
вязкости дисперсионной среды па кинетическую устойчивость
эмульсий. Очевидно, что с увеличением вя«кости ’дисперсионной
среды уменьшается интенсивность броуновского движения капель
эмульсии.) Поскольку капельки не защищены поверхностно-актив-
ными веществами, каждое их столкновение может привести к коа-
лесценции. Естественно поэтому, что уменьшение скорости броу-
70
Таблица 11.3. Влияние вязкости растворов на характер расслаивания
в системе полимер — полимер — растворитель
Молекулярная масса М-105 Растворитель Характеристика исходных растворов Мутный слой
ПС ПММ1 Конц, с, г/дл ПС Т[, Па с ПММА Г), Па-с
3,7 0,87 30,9 Бензол Хлороформ Бутилацетат Бензол Хлороформ Бутилацетат 20,0 7,0 4,0 60,4 53,7 49,5 3,1 3,4 0,5 3,7 3,3 2,7 997,8 2296.2 500,8 Верхний > Нижний » >
Примечание. Вязкость определяли на вискозиметре Хепплера с падающим ша-
риком.
невского движения вследствие роста вязкости среды (увеличение
молекулярной массы или понижение температуры) должно эффек-
тивно влиять на устойчивость эмульсин. Видимо эмульсии стано-
вятся устойчивыми лишь при достижении определенного значения
вязкости среды. При значениях вязкости, приведенных в табл. П.З,
устойчивость эмульсии настолько велика, что она не разрушается
даже при центрифугировании с частотой вращения 7000 об/мин.
Эмульсии, возникающие после перемешивания растворов и об-
ладающие высокой стабильностью, могут быть устойчивыми кине-
тически, но не термодинамически. Доказательством термодинами-
ческой устойчивости системы является ее самопроизвольное обра-
зование, т. е. самопроизвольное диспергирование одной жидкости
в другой. Такой процесс самопроизвольного диспергирования одно-
го раствора полимера в другом наблюдается, если один из раство-
ров (легкий) наслаивается на другой и система термостатируется.
Измерение оптической плотности во времени показывает, что в
одном из слоев образуется устойчивая эмульсия с размером ка-
пель 0,5—3,0 мкм. Помутнение слоя проявляется в появлении лег-
кой опалесценции и сопровождается значительным увеличением со-
держания в этом слое тругого полимера (табл. И.4).
Данные в табл. II.4 указывают на то, что в случае самопроиз-
вольного эмульгирования стабильная эмульсия возникает в слое
полимера с большей вязкостью раствора (большая молекулярная
масса).
Известно, что при искривлении поверхности раздела фаз по-
верхностное натяжение может увеличиваться или уменьшаться в
зависимости от того, в какую сторону искривляется поверхность.
Известно также, что поверхностное натяжение зависит от кривиз-
ны поверхности. При эмульгировании жидкостей должна образо-
вываться та эмульсия, для которой поверхностное натяжение ни-
же [86, с. (127—128)]. В низкомолекулярных жидкостях, напри-
мер в системе гексан — метанол, более плотная жидкость эмульги-
ровалась в менее плотной. Очевидно, что в приведенных полимер-
71
Таблица П.4. Самопроизвольное эмульгирование растворов полимеров [81]
Полимер Al^-10— Б С, г/дл Содержание полимеров Характеристика слоя
в верхнем слое в нижнем слое
ПС блочный ПИБ ПС эмульсион- ный ПИБ ПС блочный ПИБ 3,7 1,08 9,25 1,08 3,7 30,0 10,0 3,0 6,5 93,5 2.7 97,3 5,0 95,0 76,0 24,0 76,0 24,0 98,0 2,0 Слегка мутный нижний слой Мутный нижний слой Слегка мутный верхний слой
пых системах плотность раствора не меняется с изменением моле-
кулярной массы и плотность раствора ПС всегда больше плот-
ности раствора ПИБ независимо от молекулярной массы. Видимо,
вязкость при постоянной температуре определяет скорость броу-
новского движения и кинетическая устойчивость возрастает с уве-
личением вязкости подобно росту ее с понижением температуры.
Вязкость определяет поэтому нс направление процесса эмульгиро-
вания, а устойчивость эмульсии: более устойчива эмульсия в бо-
лее вязкой среде.
§ 4. РАСТВОРЫ TPEXliOMIlOIlfll 1П1.ТХ СМЕСЕЙ ПОЛИМЕРОВ
Исследование фазового состава и свойств в системе полимер —
полимер — полимер — растворитель представляет интерес в связи
с информацией о состоянии взаимной растворимости полимеров в
таких системах (что практически недостижимо в смесях расплавов
трех полимеров), а также в связи с возможностью косвенного
суждения о структуре тройных полимерных смесей, Практическое
значение тройных смесей очевидно.
На рис. 11.20 показаны примеры фазовых диаграмм трехкомпо-
нентных растворов. Видно, что взаимная растворимость компонен-
тов в бинарных смесях незначительна, и что с ростом общей кон-
центрации раствора область взаимной растворимости полимеров
уменьшается.
Растворимость полимера в бинарной смеси двух других поли-
меров выше, чем в каждом полимере бинарной смеси. Так, в сме-
си полибут» пипа (ПБ) с ПИБ и ПС в 4%-иом хлороформенном
растворе растворимости ПИБ в ПБ и ПС составляет 1,4%, а рас-
творимость ПБ в ПС и ПИБ соответственно 9,9 и 24%. В то же
время растворимость ПИБ в смеси ПБ:ПС«=75:25 равна 13%.
Аналогичная картина наблюдается и для системы ПИБ —
ПИП—ПС (рис. II 20,а). Видно также, что уменьшение концент-
рации растворов с 2,5 до 2,25% неодинаково влияет на раствори-
мость полимеров друг в друге. Например, растворимость ПИБ в
ПИП и ПС увеличилась незначительно, тогда как растворимость
72
Рис. 11.20. Фазовые диаграммы трой-
ных смесей полимеров в растворе в
общем растворителе при 20 °C и по-
стоянной общей концентрации поли-
меров:
о — смесь ПИП, ПИБ, ПС в хлороформе
с концентрацией 2,25% (2, 3) и 2,5% (/, 4);
б — то же, но с концентрацией 4%; в —
смесь СКТВ, ПИП, ПС в толуоле с общей
концентрацией 2%.
ПИП и ПС в ПИБ заметно повысилась. Так, из рис. 1120, и сле-
дует, что при уменьшении концентрации системы с 2,5 до 2,25%
растворимость ПИБ в ПИП увеличилась в 1,17 раза, тогда как
растворимость ПИБ в смеси ПИП:ПС=1:1 увеличилась в
2,3 раза.
Диаграммы рис. П.20 показывают, что в зависимости от кон-
центрации раствора и природы полимеров можно получать фазо-
вые диаграммы различных типов [87].
Повышение взаимной растворимости полимеров в растворе
тройной смеси аналогично случаю жидких систем, в которых три
жидкости попарно расслаивающиеся, могут образовать однофаз-
ную систему при составлении трехкомпонентиой системы [88].
Можно следующим образом объяснить увеличение раствори-
мости полимера в смеси по сравнению с растворимостью его в
каждом компоненте. Ассоциация полимеров бипарной смеси уве-
личена по сравнению с растворами отдельных полимеров (при той
же концентрации) и расслаивание наступает по достижении кри
тического размера ассоциатов, когда происходит астабилизация
73
однородных ассоциатов с образованием отдельных фаз. Введение
в бинарную смесь третьего полимера вызывает уменьшение пар-
циальной концентрации каждого компонента в растворе, вследст-
вие чего уменьшается размер ассоциатов данного полимера, что
приводит к увеличению смешиваемости полимеров, расширению
области существования однофазных систем.
Логично предположить дальнейшее увеличение взаимной сме-
шиваемости (взаимной растворимости, совместимости) полимеров
при увеличении числа полимерных компонентов.
В этой главе уже указывалось па полезную информацию, по-
лучаемую при исследовании светорассеяния в растворах смесей
полимеров. На рис. П.21 показана зависимость избыточного свето-
рассеяния от соотношения полимеров в растворе, а па сторонах
треугольной диаграммы даны зависимости светорассеяния от соот-
ношения полимеров в соответствующих бинарных смесях в том же
растворителе. Диаграмма построена методом планирования экспе-
римента по Шеффе [87, 89], видно отсутствие максимумов на трой-
ной диаграмме (по сравнению с уровнем светорассеяния в би-
нарной смеси). Многочисленные эксперименты с различными по-
лимерами показывают, что такие диаграммы (см. рис. 11.21) ти-
пичны для зависимостей свойство-—состав.
В тройной смеси не происходит дополнительной, избыточной
ассоциации по сравнению с ассоциацией в бинарных смесях. Этот
вывод согласуется с повышенной взаимной растворимостью поли-
меров в тройной смеси.
Отсутствие принципиальных изменений в фазовой структуре
тройной смеси по сравнению с бинарной обусловливает исключи-
тельную пологую форму поверхности свойство — состав в приме-
Рпс. 11.21. Избыточное светорассеяние см-1) растворов смесей полиме-
ров в хлороформе при С—1,8 (а) и 1,5 г/дл (б).
74
Рис. 11.22. Эффективная вязкость (П)
растворов смесей ПБ — ПММА — ПС
в бензоле при концентрации раствора
3 г/дл и температуре 20 °C.
нении к вязкости тройных смесей
(рис. 11.22). Плоский, нерельефный
характер поверхности существует,
несмотря па заметное уменьшение
вязкости бинарных смесей по срав-
нению с аддитивными значениями,
которые вообще типичны для би-
нарных смесей малополярных по-
лимеров.
§ 5. ВЛИЯНИЕ РАСТВОРИТЕЛЯ ПА СТРУКТУРУ ПЛЕНОК
СМЕСИ ПОЛИМЕРОВ, ПОЛУЧАЕМЫХ ИЗ РАСТВОР V
При получении пленки из раствора смеси полимеров.путем вы-
паривания растворителя система проходит интервал концентра-
ции от исходной концентрации раствора до 100%-пой концентра-
ции. Если исходный раствор однофазен и прозрачен, то при уда-
лении растворителя растущая концентрация в определенный мо-
мент становится равной пределу расслаивания и далее процесс
удаления растворителя сопровождается расслаиванием системы.
Чем медленнее удаляется растворитель и чем меньше вязкость в
момент достижения предела расслаивания, тем легче и полнее
проходит разделение раствора па фазы. При медленном удалении
растворителя из раствора многих смесей полимеров происходит
образование в пленке частиц дисперсной фазы одного из полиме-
ров размером в сотни микрон. При очень медленном удалении рас-
творителя из смеси несовместимых полимеров можно получить
прозрачную пленку, благодаря тому, что раствор успевает рассло-
иться на два слоя, и пленка в этом случае также получается двух-
слойной и прозрачной, поскольку прозрачен каждый ее слой.
В работе 190] экспериментально показано увеличение гомоген-
ности пленок смесей ПИП- ПИБ и бутадиен — стирольный сопо-
лимер — ПС при ускорении выпаривания толуола из раствора.
В этой же работе пленки с малым (0,2—0,6 мкм) размером ча-
стиц дисперсной фазы I [ИБ в смеси с ПИП получены выпариванием
толуола из раствора при комнатной температуре при содержании
дисперсной фазы (ПИБ), равном 3%. При этом получаются ча-
стицы круглой формы и с узким распределением по размерам.
Малое содержание полимера дисперсной фазы в смеси не яв-
ляется достаточным условием получения сферических частиц ма-
лого размера. Так, в работе [90] показано, что если в смеси ПИП—
ПИБ содержится всего 3% ПИП, то последний тем не менее обра-
зует непрерывную фазу в виде топкой фазовой сетки в матрице
ПИБ.
75
Рис. П.23. Э.1скт1юнно-М11кроскопические фотографии пленок ПВХ и ПММХ,
полученных in 1%-ного раствора в тетрагндрофуране с содержанием ПММА
в смеси i (и) К) (б}. 20 (в), 50 (г), 60 (д), 80 (е)", 90 (ж) и 98 (з) [99J.
Известно, что расслаивание может происходить по разным ме-
ханизмам— путем образования зародышей с последующим их уве-
личением в размерах и по механизму спиподальпого расслаивания,
при котором начальной формой фазовой структуры является тон -
кая сетка второй фазы, возникающая спонтанно по достижении
критических условий выделения второй фазы. Видимо, механизм
расслаивания, наряду со скоростью удаления растворителя и вяз-
76
Рис. 11.24. Разрушающее напряжение при
растяжении вулканизатов смеси СКБ с
20% ПС, полученных методом сублимации
бензола из растворов разной концентрации.
Пунктир — предел расслаивания.
костью системы, оказывает сильное
влияние на фазовую структуру
пленки из смесей полимеров.
На рис. 11.23 [91] показано из-
менение фазовой структуры пленок
из смесей ПВХ — ПММА в широ-
ком диапазоне составов. Эта кар-
для пленок, полученных из
тина является, по-видимому, типичной
раствора.
При получении пленок из растворов методом сублимации рас-
творителя структура исходного раствора сохраняется в большей
степени. Так, в пленке смеси бутадиенового каучука СКБ с 20%
ПС, полученной методом сублимации бензола из 0,5%-ного рас-
твора, частицы ПС имеют размер 1 мкм и невидимы в световом
микроскопе, тогда как при исходной концентрации раствора 6%
(выше предела расслаивания) система оказывается мутной, и раз-
мер частиц составляет несколько микрон. Изменение размера ча-
стиц в зависимости от исходной концентрации раствора настолько
велико, чго проявляется в изменении разрушающего напряжения
при растяжении вулканизатов (рис. П.24), значение которого
уменьшается с увеличением размера частиц.
Условия (главным образом скорость) удаления растворителя
при получении пленок из раствора смеси полимеров влияют па
структуру и свойства пленок, что, в свою очередь, маскирует роль
природы растворителя, которая на самом деле может быть весьма
значительной. Так, в работах [92, 93] показана возможность по-
лучения как однофазных, так и двухфазных пленок смесей поли-
меров в зависимости от природы растворителя. Пленки смесей ПС
и полиметилвипилового эфира, получаемые при комнатной темпе-
ратуре, оказываются однофазными (прозрачными), если получены
из растворов в бензоле или толуоле, но двухфазными (мутными),
если получены из раствора в трихлорэтилене или хлороформе.
Фазовый состав контролировали (помимо контроля оптических
свойств) по температуре стеклования, определенной методом ска-
нирующей калориметрии и по частотной зависимости диэлектриче-
ских потерь. Однофазные пленки имели одну Тс и широкий пик
на термограмме, тогда как двухфазные пленки характеризовались
наличием двух Тс, совпадающих с Тс исходных компонентов. Ха
рактерно, что зависимость Тс от состава смеси в однофазных плен-
ках не совпадала с рассчитанной зависимостью для статистических
сополимеров этих же мономеров. Это означает, что смесь нс явля-
ется столь же гомогенной, как и статистический сополимер такого
77
же состава, однако микронеоднородности (ассоциаты макромоле-
кул) в однофазной смеси слишком малы, чтобы обнаруживать
признаки наличия второй фазы.
Аналогичное явление наблюдается [93] для смеси хлорирован-
ного каучука (63% связанного С1) и этиленвинилацетатного сопо-
лимера [около 40% (масс.) винилацетата]. Смесь однофазна и
прозрачна, если опа получена из раствора в толуоле, ксилоле или
тетрахлорэтилене, но мутна и двухфазна, если опа получена из
растворов в хлороформе или метнленхлориде.
Как пленки из смеси ПС — ПВМЭ, так и пленки смеси ХК—
ЭВА, будучи однофазными, расслаиваются, выделяя частички мик-
рофазы одного из полимеров при нагревании. Первая смесь рас-
слаивается (появляется опалесценция) при 125 °C, а вторая в ин-
тервале 180—225 °C.
Прямые экспериментальные подтверждения влияния раствори-
теля на характер взаимодействия полимеров в растворе были по-
лучены в работе [94]. В смеси хлорированного ПХП п метилви-
нилпиридинового каучука наблюдается большее взаимодействие
полярных групп (оцененное спектрофотометрическп), если смесь
находится в растворе в плохом растворителе (хлороформе) по
сравнению с той же смесью, растворе иной в хорошем растворите-
ле (толуоле). Повышенное взаимодействие полимеров характерно
и для пленок, полученных из растворов в плохом растворителе. По-
вышенное взаимодействие полимеров в пленках, полученных из
хлороформа, обусловливает и повышенное значение разрушающего
напряжения при растяжении (30 МПа по сравнению с 24,5 МПа
для пленок из толуола).
В то же время повышенное взаимодействие полимеров в плен-
ке приводит к более интенсивному расходованию полярных групп
и снижению прочности связи с металлом пленок, полученных из
хлороформа (4,3 МПа) по сравнению с пленками, полученными
из раствора в толуоле (7,5 МПа). Меньшая прочность связи плен-
ки с металлом обусловлена в конечном счете тем, что в пленках,
полученных из плохого растворителя, меньшее количество поляр-
ных групп выходит на поверхность, па что указывает пониженное
значение угла смачивания пленки водой (70° против 65,5" для
пленки из толуольного раствора).
Заметное а иног ца и очень сильное влияние природы раствори-
теля и исходной концентрации раствора на свойства пленок изве-
стно и для пленок индивидуальных поли леров [95 99].
ЛИТЕРАТУР\
1. Boyer R. F., Spencer R. S.—J. Polymer Sci., 1950, v. 5, p. 375—376.
2. Doty P., Steiner R.— Ibid., p. 383 384.
3. Кулезнев В. H., Крохина Л. С., Лякин Ю. И., Догадкин Б. А. — Коллоид,
ж., 1964, т 26, № 4. с. 475 180.
4. Эскин В. Е„ Нестеров А / Коллоид, ж., 1966, т. 28, с. 904—906.
78
5, Кулезнев В. И., Крохина Л. С. — Высокомол. соед., 1973, сер. А, т. 15, № 4,
с. 906—916.
6. Kuhn R., Cantow Н J. — Makromol. Chem 1969, Bd. 122, S. 65--81.
7 Hude H. J., Tanner A. G. — J. Coll. Interface Sci., 1968, v. 28, p. 179—
186.
8. Френкель С. Я., Е.и яшевич Г. K-, Панов Ю. Н. — В кн.: Успехи химии и
физики пслимеров. М., Химия, 1970, с. 87—138.
9. Cantow Н. J., Kuhn R., Burchard W.— In: International Symposium on Mac-
romolecular Chemistry, Toronto, 1968, Prepr. Sci. Papers, pt. A, v. 1, p. 14.
10. Кулезнев В. И., Крохина Л. С., Догадкин Б. А.— Коллоид, ж., 1965, т. 27,
№ 5, с. 715—719.
11. Hugelin Ch., Dondos А.— Makromol. Chem., 1969, Bd. 126, S. 206—214.
12. Бектуров E. А. Тройные полимерные системы в растворах. Алма-Ата, Наска,
1975. 251 с.
13. Sundelot L. (J. — Chem. zvesti, 1971, v. 25. p. 203—208.
14. Кулезнев В. H Кандырин Л. Б., Крохина Л. С., Буканова Е. Ф. — Колло-
ид. ж., 1971, т. 33, с. 539—545.
15. Huggins М. L. — J. Am. Chem. Soc., 1942, v. 64, р. 2716—2727.
16. Виноградов Г. В., Яновский 10. Г., Кулезнев В Н., Иваненко Т. А. — Кол-
лоид. ж., 1966 , т. 28, с. 640—644.
17. Manale S., Murakami R., Takayanagi M.— Intern. J. Polvmer. Mater., 1971,
v. 1, № 1, p. 47—73.
18. Разинскаа И. H., Штаркман Б. П. — Высокомол. соед., 1977, сер. Б, т. 19,
№ 10, с. 765—767.
19. Кулезнев В. Н., Игошева К- М.— Высокомол. соед., 1962, т. 4. с. 1858—1861.
20 Тагер А. А., Шолохович Т. И., Цилипоткина М. Б. — Высокомол. соед., 1у72
сер. А, т. 11, с. 1423—1424,
21. Flory D., Eichinger В. Е., Orwoll R. А. — Macromolecules. 1968. v. 1, р. 287—-
"93.
22. Кулезнев В. Н„ Андреева В. М —Высокомол. соед., 1962, т. 4, № 12,
с. 1851—1857.
23. Bushuk !¥'.• Benoit Н. — Canad J. Chem., 1958, v. 36, p. 1617—1623.
24. Рощина Г П. Термодинамика и строение растворов. М„ 11тд-во АН СССР,
1959. 233 с.
25. Гуль В. Е., Ленская Е. А., Кулезнев В. Н. — Коллоид, ж., 1965, т. 27, № 3,
q 341—345.
26. Гуль В. Е. Ленская Е. А.. Кулезнев В. Н., Арутюнова С. Г. — ДАН СССР,
1965. т. 160. № 1, с. 154—157.
27. Krigbaum U" R., Wall Т. F.— J. Polymer Sci., 1950, v. 5, p. 505—514.
28. Catsiff E. H., Heweff IT A. — J. Appl. Polymer Sci., 1962, v. 6, № 23,
p. 530- 532.
29. C’-ai'g I. H , Bigelow С. C. — J. Polymer Sci.. 1955. v. 16, p. 177—191.
30. Williamson G. R., If rigid B. — J. Polymer Sci.. 1965. pt. A. v. 3, p. 3885—-
3891.
31. Bohmer B., Berek D., Florian S. — Europ. Polvmer J.. 1970. v. 6, p. 471—
478.
32. Vasile C., Sihneider J. A. — Makromol. Chem., 1971, Bd. 141, S. 127—144.
33. Berek Г)., Bohmer B. — In: International Symposium Macromolecular Che-
mistry, Toronto, 1968, Prepr. Sci. Papers, pt. A, v. 1, p. 15—17.
34. Липатов E. M., Липатова Г. M.—-Коллоид, ж., 1959, т. 21, с. 517—521.
35. В<~1юцкий С. С., Зайончковский А. Д„ Резникова Г А. — Коллоид, ж., 1956,
т. 18, с. 515--520.
36. Слонимский Г. Л., Комская Н. Ф. — ЖФХ, 1956, т. 30, с. 1749—1754
37. Михайлов Н. В., Зеликман С. Г. — Коллоид, ж., 1957, т. 19, с. 35—41.
38. Алексеенко В. И., Мишистин И. У. — Высокомол. соед., 1959, т 1, с. 1593—
1598.
39. Догадкин Б. А.. Килезнев В. Н., Пряхина С. Ф. — Коллоид, ж., 1959 т. 21,
№ 2, с. 174—180.
40. Krause S. — J. Macromol. Sci., 1972, pt. C, v. 7, № 2, p. 251—314
41. Dobry A., Boyer-Kawenoki F.—J. Polymer Sci., 1947, v. 2, p. 90—100.
79
42. Kern R. J., Slocomble R. J.—J. Polymer Sci., 1955, v. 15, p. 183—192.
43. Fuchs O. — Angew. makromol. Chem., 1969, Bd. 6, S. 79—85.
44. Mladenov I., Nikolinsky P., Gul V., Petrov A.-Compt. rend, beige, 1961,
v. 14, p. 615—619.
45. Sieglaff C. L.—J. Polymer Sci., 1959, v 41, № 138, p. 319—326.
46. Фрисман Э. В. и др. — Высокомол. соед., 1962, т. 4, с. 1559—1563; 1964,
1. 6, с. 341—345; 1966, т. 8, с. 1359—1364.
47. Кулезнев В. Н., Игошева К. М.— Коллоид, ж., 1962, т. 24, № 3, с. 306—308.
48 Berek D., Bohmcr В., Lath D. — Plaste u. Kautschuk, 1967, Bd. 14, S. 556—
561.
49. Gyogyi-Edelenyi J., Wolfram E. — Ann, Univ. Sci. Budepest, sect, chim.,
1969, v. 11, p. 125—128.
50. Burchardt F., Majer H., Kuhn W. — Helv. chim. acta, 1960, v. 43, p. 1192—
1198.
51. Kuhn W., Cantow H. J., Burchard №.— Angew. makromol. Chem., 1968,
Bd. 2, S. 146—151
52. Fuchs O.— Makromol. Chem., 1966, Bd. 90, S. 293—297.
53. Koningsveld R„ Staverman A. J. — Koll.-Z. u. Z. Polymere. 1966, Bd. 210,
S. 151—163; 1967, Bd. 220, S. 31-42.
54. Roberson L. M„ Matzner M., Fetters L. J.. McCrath J. E. — ACS Polymer
Prepr., 1973, v. 14, № 2, p. 1063—1068.
55. Kern R. L. — J. Polymer Sci., 1956, v. 21, p. 19—25.
56. Кулезнев В. H., Крохина Л. С., Оганесов Ю. Г., Злацен Л. М. — Коллоид,
ж., 1971, т. 33, с. 98- 104.
57. Hugelin Ch., Rondos А. — Macromol Chem., 1969, Bd. 126, S. 206—214.
58. Кулезнев В. И., Крохина Л. С. — Усп хим., 1973, т. 42, № 7, с. 1278—1309.
59. Berek D., Lath D., Durdovic V.— J. Polymer Sci., 1967, pt. C, № 16, pt 2,
p. 659—667.
60. Pursell A., Thies C. — ACS Polymer Prepr., 1968, v. 9, № 1, p. 115—122.
61. Silberberg A., Kuhn №. — Nature, 1952, v. 170, p. 450 460.
62. Silberberg A., Kuhn №.—J. Polymer Sci., 1954, v. 13, p. 21—42.
63. Кандырин Л. Б. Канд. дис. M., МИТХТ им. М. В. Ломоносова, 1971.
64. Липатов С. М. — Коллоид, ж., 1960, т. 22, с. 639—640.
65. Hunyar A.. Weisner Е. — J. Polymer Sci., 1958, v. 30, р. 645—650.
66. Крохина Л. С.. Килезнев В. Н., Б у канава Е. Ф. — Высокомол. соед., 1974,
сер. А, т. 16, № 7, с. 1576—1580.
67 Ассонова Т. А., Трапезников А. А. — Коллоид, ж., 1964, т. 26, с. 640 —644.
68 Molau G. — J. Polvmer Sci., 1965, pt. A, v. 3, p. 1267—1278, p. 4235—4242;
Koll.-Z. u. Z. Polymere, 1970, Bd. 238, S. 493-497.
69 Авербух M. 3., Никанорова H. И., Розиноер Я. M.— Коллоид, ж., 1976,
т. 38, № 3, с. 419—424.
70. Михайлов И. В. и др. — Хим. волокна, 1959, № 3, с. 18—22; 1961, № 1.
с 24 29.
71. Friese К —Plaste u. Kautschuk, 1968. Bd. 15, S. 646—650.
72 Каналюнас Р И., Лившиц Р. М., Ложкин В. Е.. Роговин 3. А. — Высокомол.
соед., 1967, сер. А, т. 9, с. 2597—2603.
73. Мигранян Т. III., Пенькова М. П., Конкин А. А. — Хим. волокна, 1970, № 2,
с. 25-27.
74. Киселева Г. Ф„ Пенькова М. П., Конкин А. А. — Хим. волокна, 1970, № 1,
с. 13—16.
75. Соломон 3. Г., Кудрявцев Г. И. — Высокомол. соед., 1970, сер. А, т. 12,
с. 2582—2587.
76. Feldman D., Rusu М. —Europ. Polymer J., 1971, v. 7, p. 215—222.
77. Геллер Б. Э. и др.— Хим. волокна, 1970, № 3, с. 34—35; № 4, с. 38—39;
№ 5, с. 36—39.
78. Тульчук 3. Д., Милевская Р. А., Тараканов О. Г. — Хим. волокна, 1970,
№ 6, с. 57—60.
79. Langhummer G., Kes tier L. — Makromol. Chem., 1965, Bd. 88, S. HO-
IS?.
80
80. Кулезнев В. Н., Крохина Л. С., Догадкин Б. А. — Коллоид, ж., 1967, т. 29,
№ 1, с. 170 1 Т.
81. Кулезнев В. Н., Крохина Л. С., Догадкин Б. А. — Коллоид, ж., 1969, т. 31,
№ 6, с. 853 859; в кн.: Труды симпозиума по поверхностным явлениям в
жидкостях и жидких растворах. Л., Изд-во ЛГУ, 1971, с. 111—119.
82. Липатов Ю. С. Физическая химия наполненных полимеров. М., Химия, 1977.
303 с.
83. Vrij A. — J. Polymer Sci., 1968, pt. A, v. 6, р. 1919—1932.
84. Volmer M. — Z. phys. Chem., 1927, Bd. 125, S. 151—162; 1931, Bd. 155,
S. 281—298; 1957, Bd. 206, S. 181—184; Bd. 207, S. 307—316.
85. Щукин E. Д., Ребиндер П. A. — Коллоид, ж., 1958, т. 20, с. 645—654.
86. Русанов А. И. Термодинамика поверхностных явлений. Л., Изд-во ЛГУ,
1960. 154 с.
87. Мирошников Ю. П. Канд. дис. М., МИТХТ им. М. В. Ломоносова, 1971.
88. Hughes L. J., Britt G. Е. — J. Appl. Polymer Sci., 1961, v. 5, № 15, p. 337—
348.
89. Scheffe H.— J. Roy. Statist. Soc., 1958, pt. B, v. 20. № 2, p. 344—358.
90. Бакеев H. Ф., Берестнева 3. Я., Жарикова 3. Ф., Каждан М. В. — Высоко-
мол. соед., 1973, сер. А, т. 15, № 9, с. 2128—2130.
91. Батуева Л. И. Канд. дис. М.. МГУ им. М. В. Ломоносова. 1976.
92. Bank М., Leffingw ell J., Thies C.—Macromolecules, 1971, v. 4, № 1, p. 43—
46.
93. Leffingwell J., Thies C., Gertzman H. — ACS Polymer Prepr., 1973, v. 14,
№ 1, p. 596—600.
94. Крохина Л. С., Кулезнев В. H., Люсова Л. Р., Глаголев В. А. — Высокомол.
соед., 1976, сер. А, т. 18, № 3, с. 663—668.
95. Дорохина Т. В., Новиков А. С., Зубов П. И. — Высокомол. соед., 1959, т. 1,
с. 36—45.
96. Кулезнев В. Н., Клыкова В. Д., Евреинов Ю. В., Догадкин Б. А. — В кн.:
Механизмы процессов пленкообразования из полимерных растворов и дис-
персий. М., Наука, 1966, с. 143—146.
97. Ботвинник Г. О., Тагер А. А., Древаль В. Е. — В кн.: Химия и технология
производных целлюлозы. Владимир, ВНИИСС, 1971, с. 146—148.
98. Зверев М. П., Зубов П. И., Бар А. Н., Никанорова Л. П., Иванова Л. В.—
Высокомол. соед., 1974, сер. А, т. 16, с. 511- -518.
99. Андрианова Г. П., Нарожная Е. Л. — Высокомол. соед., 1975, сер. А, т. 17,
с. 923—928.
100. Kruse W. A., Kirste R. G., Haas J., Schmitt В. J., Stein D. J. — Makromol.
Chem., 1976, Bd. 177, S. 1145—1160.
6—244
Глава III
МЕХАНИЗМ СМЕШЕНИИ ПОЛИМЕРОВ
Теория смешения основывается па представлениях об идеаль-
ном процессе, при котором перемешиваемые среды находятся под
действием однородного простого сдвига и одна из поверхностей,
ограничивающих систему, движется с определенной скоростью от-
носительно другой, неподвижной поверхности. В этом случае сдвиг
вызывает изменение формы частиц смешиваемых материалов, на-
ходящихся в зазоре между пластинами. Изменение формы частиц
выражается в переходе от относительно сферических к вытянутым,
сильно ализометричным формам. Уменьшение одного из размеров
«полосы» диспергируемого материала («утончение полос») и рас-
сматривается как процесс смешения, соировож чающийся увеличе-
нием поверхности раздела фаз [1, с. 210 214].
Представление об идеальном процессе смешения недостаточно
для понимания закономерностей формирования фазовой структу-
ры смеси полимеров, поскольку увеличение поверхности раздела
фаз может приводить к формированию разных структур, в том чис-
ле различающихся типом непрерывной фазы. Сам процесс обра-
щения фаз при смешении полимеров, который может вообще не
соировож чаться заметным изменением поверхности раздела, при
водит в то же время к ЬрииципиальнИм и членениям свойств смеси,
чего не учитывают представления об идеальном процессе смеше-
ния II наконец, идеальный процесс смешения пе предполагает
дробления жидких капель на более мелкие при сдвиге, хотя этот
процесс и является в первую очередь ответственным за формиро-
вание (разовой структуры смеси полимеров.
Очевидно, таким образом, что представления о процессе сме-
шения полимеров, основанные на закономерностях деформации
сплошных срс i, недостаточны для понимания процесса смешения
полимеров. Закономерности смешения полимеров (формирование
фазовых структур) могут быть сформулированы на основе рассмот-
рения микро лехапики деформации двухфазных смесей полимеров,
т. е. механики деформации и разрушения жидких капель одной
среды, помещенных в другую жидкую среду
В этой главе рассматривается дефор пирование капель жидких
эмульсий, из которых простейшими являются эмульсии двух не-
смешивающихся ньютоновских жидкостей. Такие эмульсии пе мо-
гут полностью моделировать смеси полимеров. Смесь полимеров
является эмульсией двух высокомолекулярных жидкостей, как
82
правило, вязкоупругих, закономерности течения которых зависят не
просто от соотношения вязкоупругости фаз, но и от условий обра-
зования смешанной флуктуационной сетки в межфазном слое. По-
лимеры, строго говоря, не могут рассматриваться как абсолютно
несмешивающиеся. Чем выше температура смешения, тем меньше
упругость, эластичность расплавов и тем ближе они по поведению
напоминают ньютоновские жидкости. В то же время с повыше-
нием температуры может сильно изменяться (в том числе увели-
чиваться) взаимная растворимость полимеров, что снова обусловит
отличия смеси полимеров от модели двух песмешивающихся нью-
тоновских жидкостей.
Современные представления о микромеханике деформации
жидких эмульсий позволяют объяснить некоторые закономерности
смешения полимеров и формирования в них фазовых структур.
§ 1. ЭМУЛЬСИЯ НЬЮТОНОВСКИХ ЖИДКОСТЕЙ
Наибольшее число работ по исследованию поведения капли
жидкости, помещенной в жидкую среду, относится к куэттовскому
потоку (рис. III.1,а), хотя основные закономерности поведения
капель характерны и для пуазейлевского и других типов потоков
[2].
Капля, помещенная на осевую линию потока, где существует
нулевой градиент скорости, не перемещается вдоль потока, однако
вокруг нее возникают возмущения в текущей матрице и искривле-
ния линий тока, что приводит к возникновению внутренних напря-
жений вблизи капли и последующей деформации капли. При ма-
лом градиенте скорости у (рис. III. 1,6) капля принимает форму
эллипсоида вращения. Для оценки величины деформации необхо-
димо рассчитать поле скоростей в матрице и в капле.
6*
Рис. III.1. Куэттовский поток в зазоре между параллельными поверхностями,
движущимися в противоположных направлениях:
а — схема действующих напряжений; 6 — параметры капли, деформированной в потоке.
83
Впервые эту задачу решил Тэйлор в 1934 г., распространивший
теорию Эйнштейна вязкости суспензий на случай разбавленных
эмульсий (см., например [3]). До настоящего времени теория эта
является основой расчетов вязкости эмульсий и поведения капель
в потоке. Исходные условия, принятые Тэйлором, сводятся к тому,
что радиус капли г мал, градиент скорости небольшой, а межфаз-
ное натяжение <г велико, так что можно поддерживать каплю по
форме, близкой к сферической. Касательные напряжения на гра-
нице раздела фаз непрерывны, а это эквивалентно отсутствию про-
скальзывания на границе между матрицей и каплей.
Под действием касательных напряжений капля вращается в
потоке (как это показано стрелкой на рис. III.1, а) с периодом
вращения / = 4л/у. Если вязкость капли меньше вязкости матрицы,
касательные напряжения в матрице вызывают циркуляционные
потоки внутри капли как реакцию на внешнее напряжение. На ос-
новании теории Тэйлора можно рассчитать траектории линий то-
ков внутри капли [4] по уравнению (см. рис. II 1.1,б):
[(x®/r2) cos 2Ч -| |* 4 1 ]31'х2/г=1 - 11= = К (1)
где г — радиус сферической капли: |i = )|i/i]o — соотношение вязкостей капли-и
матрицы соответственно: К — константа для данной линии тока, изменяющаяся
непрерывно от 0 на поверхности до (ц+ 1 )' в центре капли.
Из уравнения (1) следует, чго линии тока в капле имеют эл-
липтическую форму (рис. III.2) с эксцентриситетом, увеличиваю-
щимся с приближением к центру. При малой вязкости капли (pi<
<0,5) в ней возникают два циркуляционных контура с застойной
зоной между ними (рис. III.2, б). На основании уравнения (1)
можно рассчитать величину угловой скорости в разных точках
капли (при разных К). Оказывается, что при угле ориентации <р =
Рис. III.2. Циркуляция кнутри жидкой капли, находящейся в куэттовском пото-
ке. Линии токов рассчитаны по уравнению (/), значение К возрастает с увели-
чением номера кривой. Видны два объема жидкости, циркулирующей вблизи
центра капли:
a — J1—1; б-ц—0,25.
Я4
= 45° жидкость на всех линиях тока имеет одинаковую скорость
(о=у/2. На поверхности капли (К=0) минимальное и максималь-
ное значения <в реализуются соответственно при ф = 90° и <р=0°. а
для линий тока внутри капли картина меняется: наибольшая уг-
ловая скорость наблюдается при <j = 90°.
Нормальные напряжения в отличие от касательных не являют-
ся непрерывными на межфазной поверхности. Разность давлений
внутри и снаружи капли, создаваемая ими, равна
• { 19р+ 16 \
Д₽у = ( 16ц+16-J sin2?’ <2>
Кроме величины \Р-. создаваемой потоком и стремящейся дефор-
мировать каплю, на нее действует лапласовская сила, создаваемая
межфазным натяжением о, которая для сферической капли равна
ЬРО = 2а/г (3>
Из уравнения (2) видно, что ДР* становится максимально по-
ложительным или отрицательным соответственно при <р = л/4 и
<р=Зл/2 (на рис. III. 1,а обозначено стрелками). Под действием
ДР* капля деформируется до тех пор, пока силы ДР- и ДРП не-
уравновесятся. В соответствии с теорией Тэйлора это произойдет
при деформации D=(L—B)/(L-\-B), где L и В — соответственно
длина большой и малой полуосей капли-эллипсоида в экватори-
альной плоскости:
<т
19|i 16
16|л+ 16
(4>
где k и f(n) —безразмерные параметры, характеризующие деформируемость кап-
ли в потоке.
Теория Тэйлора справедлива лишь при малых скоростях сдви-
га, при которых LIB =1,5, и деформация капли фактически отлича-
ется от рассчитанной по уравнению (6). Угол ориентации, который
по теории Тэйлора может быть предельно равен 45°, в действитель-
ности иногда превышает это значение. Уточнение теории [5, 6]
привело к следующему уравнению для угла ориентации:
я / 3 4- 2и \
Чт= 4 ~Т ( г; ) D (5)’
Теория Тэйлора получила дальнейшее развитие в работах Кок-
са [7] также в предположении малых деформаций и отсутствия
инерционных эффектов. Оказалось возможным рассчитать кинети-
ку деформации D=f(t) сферической капли по уравнению
D = Dm [1—2 exp ( — 2Щ’//19Лр) cos yt Д- exp ( — 4Оу</19йц)]1/2 (бу
где
D =____________5(19р+ 16)__________
т 4(1 + ji)/(19|1)S-|-(20*-1)2
8>
•является величиной равновесной деформации. Изменение угла ори-
ентации деформированной капли <р вплоть до предельного угла
равно
л 1
<г-= — —— tg-1
4 2
19ц [ехр (—207?/19£ц) cos yt—1] -|- 20&-1 exp (—20yf,'19fyt) sin yi
—20k-1 [exp (- -20yt/ 19fyi) cos yi—1 ] + 19ц exp (—20yZ/lQfep) sin yt
/ 19Лц \
\ 20 /
(7)
л 1
<Fm = -4-4-
Анализ уравнений (6) и (7) показывает, что деформирование
капли, фактически являющееся пульсирующим, может рассматри-
ваться как затухающий колебательный процесс со временем ре-
лаксации т, которое приближенно определяется следующим обра-
зом:
т= pk/y = /-iii/o
(8)
Из теории следует также, что если капля помещена в куэттов-
ском потоке несимметрично относительно оси потока, то возника-
ющие в потоке возмущения приводят к миграции капли по направ-
лению от стенок к центру и смещение описывается следующим
уравнением:
Зп,т=гЧ
*з=хз+—/'ос
(9)
33(54ц2+ 102р+54)( 19ц+16)
где/'(ц) =------44Ч0(ц41)8-----’ х° и х— соответственно исходное и прой-
денное за время наблюдения t расстояние, измеряемое от стенки до центра капли.
Интересно, что теория не предсказывает миграцию жестких
сфер в отсутствие инерционных эффектов ни в куэттовском, ни в
пуазейлевском потоках [8].
В соответствии с теорией Тэйлора (см. [2]) при значительном
увеличении деформации капли она должна разрушаться, что на-
блюдалось и экспериментально. Условие разрушения можно за-
писать в виде:
B’lof (н) > 2ст/г (Ю)
Отсюда величина критической деформации в момент разруше-
ния капли в куэттовском потоке равна
ОКР = Л .f(p)=4 (in
Критический ря щус способной к разрушению капли определя-
ется по формуле
2а 1
скр = ~------- (12)
ТПо /(10
«6
Экспериментальная проверка показала [2], что теория Тэйло-
ра качественно описывает деформацию капель вплоть до 7)<С0,2
Из уравнения (4) следует, что для вязких капель при большом ц
выражение упрощается, переходя в
О = е/4ц. <рт=--л/2 (13>
Этот вывод также подтвержден экспериментально — в систе-
мах с большим р капля ориентировалась вдоль потока. Теория
Кокса 17] хорошо описывает зависимость деформации от скорости
сдвига вплоть до деформации 0=С7)=С0,4. Зависимость угла ори-
ентации от вязкости хорошо следует теории Кокса для систем с
ц>1 и теории Серфа fjравнение (5)] для р<1.
В работе [4] впервые были экспериментально обнаружены за-
тухающие колебания формы и угла ориентации капли при внезап-
ном инициировании потока. Полученные при этом зависимости де-
формации и угла ориентации капли от времени качественно сов
падали с теорией Кокса [7].
Экспериментально подтверждена и миграция капель в потоке-
(см., например [2]), которая как в куэттовском, так и в пуазейлев-
ском потоке направлена к осевой липни, т. е. в область с пулевым
градиентом скорости, где возмущения потока, связанные с конеч-
ным расстоянием капли от противоположных стенок, компенсиру-
ют друг друга. Скорость миграции возрастает с уменьшением за-
зора между деформирующими поверхностями, увеличением разме-
ра капли, скорости сдвига и отношения В пуазейлевском
потоке скорость миграции капли тем больше, чем больше ее на-
чальное расстояние от центра потока. .Миграция капель удовлет-
ворительно описывается уравнением (9).
При значительной деформации капли зависимость ее деформа-
ции от скорости сдвига не укладывается в рамки существующих
теорий, при дальнейшем увеличении деформации капля разруша-
ется. Мэсон [2] выделил четыре типа разрушения капель (рис.
Ш.З).
Первый тип р<0,2. Пи достиже-
нии критического градиента скорости
капля принимает S-образную форму и
от ее заостренных концов ((рт>45°)
отрываются капельки размером около
50 мкм.
Второй тип ц>0,2. Центральная
часть капли внезапно начинает вытя-
гиваться в шейку, что приводит к раз-
Рис Ш.З. Поведение капель в средах с раз-
личными р в неровновесных условиях дефор-
мирования:
а — ц<0,2; б — р>0,2; в — р<2; г — ц>2.
87
рушению частицы на две дочерние (большие) капли и три капли-
спутника.
Третий тип ц <2. Капля выдерживает большие деформации и
по достижении определенной деформации мгновенно распадается
надмножество мелких капель.
- Четвертый тип ц>2. Капля пе разрушается даже при высоких
скоростях сдвига (до 40 с-1). При больших у опа ориентируется
вдоль потока и деформация ее становится равновесной.
Интересно, что при очень медленном возрастании скорости
сдвига (около 0,01 с-1/мин), когда капля в каждый момент вре-
мени характеризуется равновесным в данных условиях значением
деформации, все системы независимо от у распадаются по второ-
му типу.
Легче всего разрушение капель происходит в системах с 0,3^
^у^0,6. Это соответствует теоретическим расчетам [9], согласно
которым максимальное рассеяние энергии в капле происходит при
11 = 0,632.
Разрушение происходит также при остановке потока. В мо-
мент остановки потока капля, стабильная при заданном градиенте
скорости, распадается на множество мелких капель. '
Наблюдаемые экспериментально закономерности деформации
и разрушения капель справедливы обычно как для куэттовского,
так и для пуазейлевского потоков.
Введение поверхностно-активных веществ в эмульсию с дос-
таточно большим межфазным натяжением обеспечивает возникно-
вение прочного адсорбционного слоя молекул ПАВ на границе
раздела фаз капля — среда. Наличие на поверхности капли диа-
метром 1 мм всего 6-10“8 г ПАВ достаточно для прекращения
внутренней циркуляции жидкости, что происходит, по-видимому,
вследствие затрат энергии потока на деформацию упругой оболоч-
ки ПАВ [4]. Деформация оболочки всего на 1,25% достаточна
для подавления циркуляции жидкости в капле, что в свою очередь
существенно увеличивает деформируемость капли до момента ее
распада.
Возникновение упругости оболочки капли в результате адсорб-
ции ПАВ снижает скорость миграции капли в направлении пуле-
вого градиента скорости.
Устойчивое и. жи дкого цилиндра. Капля ньютоновской жидко-
сти может быть деформирована в среде другой ньютоновской жид-
кости как путем увеличения скорости сдвига среды, так и путем
деформации капли в покоящейся среде. И в потоке, и в покоящей-
ся жидкости деформирование капли приводит к образованию (в
пределе) цилиндра определенных критических размеров, по дости-
жении которых цилиндр внезапно разрушается на множество мел-
ких капель. Закономерности разрушения, достаточно сложные и
разнообразные, как это было показано выше, упрощаются при
разрушении цилиндра в покоящейся жидкости.
«8
Рис. III.4. Схема образования стоячей
волны длиной X и амплитудой а0 на по-
верхности жидкого цилиндра радиу-
сом R.
Еще Плато и позднее Рэлей показали, что причиной разруше-
ния жидкого цилиндра в покоящейся жидкости является форми-
рование на его поверхности возмущений в виде симметричных сто-
ячих воли (рис. Ш.4), уравнение которых можно записать в виде
(14)
где а — отклонение линии поверхности от исходного положения в данной точ-
ке; х'— расстояние по оси цилиндра.
Если Х<;2л7?, то колебания затухают, если Х>2лР, то коле-
бания прорастают со скоростью
а = аоехр(^/) (15)
где I] — коэффициент, характеризующий степень неустойчивости цилиндра.
Из всех волн, возникающих на поверхности жидкого цилиндра,
разрушение вызывает та, амплитуда которой растет с наибольшей
скоростью.
Из рис. III.4 видно, что возникшая на поверхности цилиндра
стоячая волна приводит к образованию в самом цилиндре череду-
ющихся «варикозных» утолщений и утоныненин, которые сразу
увеличивают поверхность цилиндра и, следовательно, его поверх-
ностную энергию. Как только диаметр цилиндра в местах перетя-
жек становится достаточно малым, стремление системы к умень-
шению свободной поверхностной энергии делает предпочтительным
не выравнивание поверхности цилиндра, а дальнейшее уменьше-
ние диаметра перетяжки и разрыв цилиндра с образованием сфе-
рических капель, что и приводит к экспоненциальной зависимости
амплитуды волны от времени (15).
На основе динамической теории жидкого цилиндра Рэлея и
экспериментальных данных Тэйлора, Томотика разработал теорию
устойчивости жидкого цилиндра [10], нейтрально взвешенного в
другой несмешивающейся жидкости. В соответствии с данными
Томотики выражение для коэффициента нестабильности q при от-
сутствии инерционных эффектов записывается в виде
а а
q = ~ 1 = 2т)07? F I1)
(16)
где x=2nR!k и f(x, ц) —сложная функция параметров х и ц.
Из (16) следует, что жидкая пить устойчива (колебания затуха-
ют) при х>1 и д<0 и распадается на капли при х^1 и q^O.
89
Рис. HI.5. Зависимость волнового числа хт=2лЯ/Ат от
соотношения вязкостей фаз, рассчитанная по теории
Томотики.
r3P
Для определения длины волны, амплитуда
которой растет с максимальной скоростью и
которая приводит к разрушению жидкого ци-
линдра Ат, строят тависимость функции
f (х, р) от х при p = const. Но значению хт со-
ответствующему положению максимума на
этой кривой, находят длину наиболее опасной
ВОЛНЫ Ат = 2л/?/Л'т.
Рассчитав Zm для разных значений р, То-
мотика получил зависимость безразмерного
параметра хт от р, которая приведена на
рис. III.5. Кривая на рис. III.5 показывает,
что для любой системы с заданным значени-
ем р характерна определенная длина волны, при возникновении
которой жидкий цилиндр максимально нестабилен. Максимум кри-
вой соответствует р=0,28, откуда Alu = 5,33-2/?; сильное уменьшение
или увеличение р сопровождается увеличением 7.т.
В данной жидкости предельно стабильны жидкие цилиндры как
с очень большой, так и с очень малой вязкостью.
Из (15) следует, что время жизни жидкого цилиндра выража-
ется уравнением
(17)
При этом, полагая равенство объема цилиндра и суммы объемов
капель, можно рассчитать радиус капли:
(18)
который при заданном R зависит, как это следует из (18), только
от длины разрушающей волны. Из (18) следует важный вывод о
том, что кривая па рис. III.5 характеризует также зависимость
размера капель, образовавшихся из жидкого цилиндра, от соотно-
шения вязкое гей цилиндра и среды. Этот вывод представляет ос-
нову понимания механизма диспергирования полимера в полиме-
ре при сдвиге, экструзии и т. п.
Экспериментальная проверка теории Томотнки [И] в целом
подтвердила се основные выводы. Наблюдаемое иногда [11] от-
клонение поведения жидкого цилиндра от предсказываемого тео-
рией не может быть в настоящее время свидетельством несовер-
шенства теории, а может просто объясняться наличием примесей,
способных адсорбироваться на поверхности раздела фаз.
Устойчивость жи iKoi о цилиндра в потоке существенно выше
его устойчивости в покоящейся жидкости [2J. Скорость роста амп-
«0
литуды разрушающей волны увеличивается па 12 десятичных по-
рядков при остановке потока [10]. Замедление роста амплитуды
волны при наличии потока окружающей цилиндр жидкости мож-
но объяснить (наряду с другими причинами) наблюдавшейся экс-
периментально неравномерностью роста амплитуды [12]. Если в
покоящейся системе амплитуда волны растет со временем экспо-
ненциально. то при инициировании потока развитие амплитуды
волны сначала подавляется, затем увеличивается, а после снова
подавляется.
При заданном yt скорость роста амплитуды волны М проходит
через минимум при определенном значении Хо(х=Хо при /=0),
обозначаемом как (x0)oj>t. Величина Л4макс повышается при уве-
личении у/ и тем быстрее, чем меньше р. Можно рассчитать зави-
симостьхор4^(Хо)ор/Схр(—3/zyt), т. е. зависимость волнового числа
для волны с максимальной аплитудой от величины yt для раз-
ных значений ц. Оказывается, что при у/ = 0 и <j/t]ofy=^—>оо зна-
чения хор1 приближаются к величинам, рассчитанным Томотикой
для покоящейся системы. При увеличении yt, xopt уменьшается и
* 2rv
приближается к некоторой асимптоте. При yt—>оо, хор<=-— при-
\)pf
нимает постоянной значение у, пе зависящее от k, но сильно зави-
сящее от ц. Теперь можно рассчитать зависимость у от ц. Оказы-
вается, что максимум соответствует координатам ^„>=0,185 и ц=
0,14 в отличие от покоящейся системы, где хт — 0,589 и ц =
= 0,28. Таким образом, зависимость у от ц качественно аналогична
кривой на рис. III.5, отличаясь координатами максимума.
В соответствии с данными работы [12] можно также сделать
вывод, что и в стационарном потоке зависимость размера капель.
возникших при разрушении жидкого цилиндра, от соотношения
вязкостей р описывается кривой с минимумом. Экспериментальная
проверка теории в той же работе [12] была проведена на двух си-
стемах с ц=1,46 и (1 = 0.148. Выяснилось прежде всего (в соответ-
ствии с приведенной выше классификацией механизма распада
цилиндра), что в потоке цилиндр распадается не одновременно по
всей длине, образующиеся капли имеют неодинаковый размер и
расстояние между ними неодинаковое. В целом же теория качест-
венно подтверждается экспериментом.
§ 2. ЭМУЛЬСИЯ НЬЮТОНОВСКИХ И ИЕПЬЮТОНОВСКИХ ЖИДКОСТЕЙ
Теория, описывающая деформацию капель в несмешивающей-
ся жидкости в случае, когда одна из жидкостей является неньюто-
новской, отсутствует. Поэтому поведение подобного рода систем
оценивается путем сравнения с поведением эмульсии, в которой
обе фазы являются непьютоновскими жидкостями. Системы такого
рода (смешанные) представляют интерес и в том смысле, что по
91
закономерностям микромеханики они оказываются промежуточны-
ми между модельными ньютоновскими жидкостями и смесями рас-
плавов или растворов полимеров, в которых обе фазы не подчи-
няются закону Ньютона. Системы этого промежуточного типа мо-
гут быть эмульсиями ньютоновской жидкости в неньютоновской
либо неньютоповской жидкости в ньютоновской среде.
Ньютоновские капли в неньютоновской среде. В работах [8,
13] было установлено, что поведение ньютоновских капель в по-
токе псевдопластичпой или вязкоупругой жидкости характеризует-
ся всеми внешними признаками поведения ньютоновских систем.
Количественная аналогия отсутствует. Так, деформация капель в
смешанных системах увеличивается с ростом скорости сдвига бы-
стрее, чем в ньютоновских, что в случае псевдопластичных сред
можно объяснить снижением вязкости среды в процессе сдвига и
соответственным увеличением р, приводящим в соответствии с тео-
рией к росту D [см. (4)]. Ориентация капли в псевдопластичной
жидкости укладывается в рамки теорий Серфа и Кокса [5, 7].
При замене псевдопластичной среды (раствор карбоксивипило-
вого полимера) па вязкоупругую жидкость (4%-ный раствор поли-
акриламида) деформация и ориентация капли не подчинялись
предсказаниям теории.
Полученные результаты справедливы как для куэттовского, так
и для пуазейлевского потоков.
В отличие от закономерностей деформации н ориентации зако-
номерности процесса радиальной миграции капель в смешанной
системе не совпадают с таковыми для ньютоновской эмульсии.
Отличие состоит в том, что ньютоновская капля в куэттовском
потоке вязкоупругой или псевдопластичной жидкости, мигрируя
к центру потока, занимает равновесное положение, не совпадаю-
щее с центром потока. Капля находится ближе к внутреннему
цилин 1ру в случае псевдопластичной и ближе к внешнему в слу-
чае вязкоупругой среды. Положение равновесия зависит от вели-
чины у и отношения радиуса капли к толщине кольцевого зазора
r/(Rit—Ri) между коаксиальными цилиндрами прибора Куэтта.
Скорость перемещения капли возрастает с ростом у, увеличением
отношения радиуса капли к толщине зазора между цилиндрами
r/(Rlt- Ri), ростом удаленности капли от положения равновесия,
что качественно совпадает с закономерностями для ньютоновских
систем.
В пуазейлсвом потоке вязкоупругой жидкости капля смещает-
ся к оси трубы, скорость смещения возрастает с ростом у и пре-
вышает значения скорости, рассчитанные из уравнения (9).
В псевдопластичной жидкости капли мигрируют по-разному: те,
что первоначально были ближе к оси трубы, перемещаются к
стенкам, а капли от стенок — к оси трубы [13].
Жесткие сферы (при ц—>-0) в ньютоновской среде вообще не
мигрируют, а в вя «коунругой жидкости смещаются к стенке
92
Таблица Ш.1. Поведение изолированных капель в потоке
Ньютоновская среда Вязкоупругая среда Псевдопластичиая среда
Деформация и разруше- ние в потоке Ньютоновская капля Вязкоупругая капля Псевдопластичиая капля Ориентация в потоке Ньютоновская капля При малых D соответствует теоретическим значениям Разрушается по первому второму и третьему типу Экспериментальные значе- ния D меньше теоретиче- ских. Разрушается по вто- рому типу При малых D подчиняется теории. Разрушается по вто- рому типу При малых ц описывается уравнением (5), при боль- ших— уравнением (7) Нс совпадает с теоретическим значе- нием Разрушается по первому, второму и третьему типу Не исследовано Не исследовано Не соответствует теории Не совпадает с теоретиче- ским значением. Разрушается по первому и второму типу Не исследовано Не исследовано При малых ц описывается уравнением (5), при боль- ших — ие соответствует тео-
Вязкоупругая или псев- допластичная капля Радиальная миграция в куэттовском потоке При малых ц описывается уравнением (5) Не исследовано Не исследовано
Твердые сферы Ньютоновские капли Не мигрируют От стенок к центру зазора В направлении к поверхности внеш- него цилиндра От стенок к равновесному положе- нию ближе к поверхности внешнего цилиндра _ г — Ri При >0,75 или <0,75 соответственно к по- верхности внутреннего или внешнего цилиндра От ограничивающих поверх- ностей к равновесному по- ложению ближе к поверхио- сти внутреннего цилиндра
внешнего цилиндра аппарата Куэтта независимо от того, какой
из цилиндров оставался неподвижным и в какую сторону вращал-
ся [8]. Миграция жестких сфер в вязкоупругой среде обуслов-
лена, по-видимому, совместным действием нормальных напряже-
ний и скорости сдвига, которые максимальны на внутренней и
минимальны на внешней стенках аппарата Куэтта. В пуазейлев-
ском потоке твердые частицы мигрируют к стенке, по медленнее,
чем жидкие капли.
Неньююновские капли в ньютоновской жидкости. Эта система
также исследована в работе [13] в условиях куэттовского и пуа-
зейлевского потоков. Вязкость систем подобрана так, чтобы урав-
нения Кокса и Тэйлора были идентичными const, k—>0).
Оказалось, что деформация псевдопластичных капель в ньютонов-
ской среде хорошо описывалась теорией, развитой для ньютонов-
ских систем. Для вязкоупругих капель деформация, оставаясь ли-
нейной функцией скорости сдвига уг, оказывалась меньше, чем
следует из теории. Снижение концентрации раствора полимера,
составляющего каплю, способствовало уменьшению расхождения
теории и эксперимента. Угол ориентации псевдопластичных ка-
пель хорошо следует существующей теории, предполагающей-'ве-
личину угла, равную л/4.
Ориентация вязкоупругих капель укладывается в рамки тео-
рии при малых ц и отличается от теоретических значений при
р>2,8.
Многие закономерности поведения вязкоупругих капель в нью-
тоновских жидкостях исследованы на примере красных кровяных
телец в плазме крови [2].
В табл. III.1 приводятся обобщенные данные по микромеха-
нике эмульсий, частично табулированные в [13].
§ 3. ЭМУЛЬСИН ВЯЗКОУПРУГИХ ЖИДКОСТЕЙ,
ОНРХЛОПАННЫЕ РАСТВОРАМИ И РАСПЛАВАМИ ПОЛИМЕРОВ
В эмульсиях, образованных двумя растворами или расплава-
ми полимеров, наблюдаются все основные явления, известные для
эмульсий ньютоновских жидкостей. Так же как и в ньютоновских
эмульсиях, в этих реологически сложных системах наблюдается
вытягивание капель и распад жидких цилиндров па мелкие кап-
ли в зависимости от соотношения скорости сдвига и межфазного
натяжения. Количественного согласия наблюдаемых закономер-
ностей с теоретически вычисленными для ньютоновских систем
здесь ожидать не приходится, учитывая опыт исследования сме-
шанных систем. Поэтому первостепенное значение приобретает
здесь накопление экспериментальных фактов и установление ка-
чественного совпадения или различий в поведении полимерных
эмульсий по сравнению с эмульсиями, рассмотренными в преды-
дущих разделах этой главы.
94
Эмульсии па основе растворов
несовместимых полимеров
Закономерности разрушения жидких цилиндров, образованных
вязкоупругим раствором полимера (растворы ПС разной моле-
кулярной массы) в растворе несовместимого полимера (полиме-
тилвиннлсилоксан), изучены в работе [14]. Цилиндрик вязкоуп-
ругой жидкости вытягивался в среде другой вязкоупругой жидко-
сти с разной скоростью и разрушался, образуя цепочку одина-
ковых по размеру мелких капель. Вязкоупругость системы прояв-
лялась в характерной зависимости приведенного времени жизни
tb=tb!Ry от скорости вытягивания (рис. III.6). Такая зависимость
обусловлена, видимо, ростом внутренних напряжений в сформи-
рованном цилиндре с увеличением скорости вытягивания, что ус-
коряет распад. Вообще же сама по себе вязкоупругость приводит
к замедлению роста разрушающей волны, как это показано в
опытах Гордона с сотр. [15]. В работе [15] показано, что струя
водного раствора полимера (0,05—0,1 %-пая концентрация рас-
твора) на воздухе постепенно астабилизуется, что сопровождает-
ся появлением на ее поверхности варикозных утолщений, и рост
вязкоупругости раствора сопровождается торможением скорости
формирования варикозпости.
Образование нитей раствора полимера при сдвиге эмульсии
двух 15%-ных растворов полиуретана и полиакрилонитрила, один
из которых был подкрашен небольшим количеством сажи, наблю-
дали в капилляре и в вискозиметре типа конус — плоскость [16].
Возникновение волокнистых структур в эмульсиях поливини-
лового спирта и некоторых биополимеров в условиях простого
сдвига между стеклянными пластинками наблюдали в работе
[17]. Отмечено, что после прекращения сдвига волокна быстро
распадались на мелкие капли. В концентрированных эмульсиях
Рис. III.6. Зависимость приведенного времени жизни вязкоупругого жидкого
цилиндра (ц=0,87) от скорости вытягивания.
Рис. Ш.7. Зависимость степени асимметрии (Л) и угла ориентации (<рт) капель
дисперсной фазы от параметра ф=ут]Г1/з/о).Состав эмульсии: равные по объ-
ему бензольные растворы ПС и этилцеллюлозы, ц»1.
95
исследование затруднено невозможностью наблюдать отдельные
нити, а также частыми актами коалесценций капель и жидкиа
нитей.
Способность капли к деформации авторы работы [18] оцени-
вали по величине параметра 0, аналогичного по смыслу парамет-
ру k в теории Тэйлора [см. уравнение (4)]:
р = ^у’/з/а (19)
где 1]=Ч1=По; V—объем капли.
Оказалось, что вытянутая капля обладает неограниченной ус-
тойчивостью при условии 0<0^0,74 и распадается при 0>О,74.
На рис. III.7 показана зависимость полученной экспериментально
степени анизометричности капли (отношение длин главных полу-
осей капли-эллипсоида) и угла ориентации от величины парамет-
ра 0. Из рис. Ш.7 видно, что при Л>8 угол ориентации стремит-
ся к 90°, а зависимость А от 0 оказывается практически линейной
и может быть представлена уравнением А = 3,0 00-88.
Время жизни деформированных капель в потоке в этих же
условиях можно рассчитать по формуле
При высокой концентрации эмульсии (равное соотношение
объемов каждой фазы) процесс ориентации и деформации капель
Рис. 1П.8. Зависимость степени анизометричности капель дисперсной фазы от
скорости сдвига (а), соотношения вязкостей фаз (б) и исходного диаметра ка-
пель (я) при 32,5 °C. Объемная доля дисперсной файл <f = 0,4. Состав эмульсий
на основе водных растворов декстрана (Д), желатина (Ж) и ЛВС различной
концентрации:
а- Г — ПВО <5%) - Ж (10%); 2-Ж (10%) - ПВО (5%). З-Д (8%) - Ж (10%), ц=0,Г.
4-Д (10%)-ПВС (5%), ц-=0.05; 5 - ПВС (5%)-Д (10%). (1=20; 6 -Ж (10%) - Д (8%),
ц=10;
б — скорость сдвига у=3.9 с-1, различные по составу эмульсии с разным р;
е: Г-ПВС (5%) — Ж (10%); 2-Д (8%)-Ж (10%), т=15 с-1.
I
4.6 fZ 1,S 5.0 5.1 pH
Рис. Ш.9. Зависимость деформируемости капель (Л) от pH среды в системе Д
(8%)—Ж (10%)—вода при у=3,9 с-1.
Рис. Ш.10. Зависимость Л от р для эмульсий на основе водных растворов:
I - ПВС (Б%)-Ж (10%); 2- ж (10%)-ПВС (5%); З-Д (8%) — Ж (10%); <-Д (10%) —
ПВС (5%). Объемная доля дисперсной фазы <р=0,4.
в потоке обязательно сопровождается коалесценцией. Коалесцен-
ция сразу увеличивает размер капли и, следовательно, увеличива-
ет параметр р, что в свою очередь приводит к быстрой деформа-
ции и разрушению капли. Поэтому в концентрированной эмуль-
сии в процессе сдвига возникает значительная полидисперспость
капель при заданном значении у.
Метод малоуглового рассеяния света был применен в работе
[17] для исследования концентрированных эмульсий па основе
водных растворов декстрана и желатина. На рис. Ш.8 приведена
зависимость степени анизометричности капель в этих системах от
скорости сдвига, соотношения вязкостей и исходного диаметра ка-
пель. Видно, что деформируемость капель при заданном значе-
нии у возрастает с ростом исходного диаметра капель и с приб-
лижением соотношения вязкостей к единице. В этой же работе
приведена зависимость деформируемости капель эмульсии от pH
среды (рис. Ш.9). При изменении pH среды за пределами 4,5-—
5,1 система становится однофазной, и измерение деформируемости
капель не проводится. Внутри указанного интервала pH система
двухфазна, и деформируемость резко снижается от границ интер-
вала к его середине. Учитывая, чго при приближении к точке
фазового перехода межфазное натяжение в любой системе стре-
мится к нулю, можно с уверенностью рассматривать зависимость,
приведенную на рис. Ш.8, как зависимость деформируемости
капель от величины межфазного натяжения: деформируемость
резко возрастает при уменьшении межфазного натяжения.
Исследована также зависимость деформируемости А от пара-
метра р (рис. Ш.10). Видно, что в исследованном интервале ско-
ростей сдвига и размеров капель наблюдается линейная зависи-
мость анизометричности от р, которая может быть выражена
уравнением X = const-pu’fi3. Оказалось, что для разных систем, по-
казанных на рис. III. 10, значение показателя степени при р оди-
7—244 97
каково, но меняется при изменении объемной доли дисперсной фа-
зы. т. е., по-видимому, показатель степени отражает вклад процес-
сов коалесценции в общую ориентацию капель эмульсии.
Растворы смесей полимеров в известной мере моделируют рас-
плавы смесей, являясь относительно более простыми тля иссле-
дования системами, поскольку в них всле (сгвие малой вязкости
равновесное состояние деформации капель может быть достигну-
то за время опыта. Раьновесное состояние деформации капель
эмульсии в расплавах смесей полимеров устанавливается не
всегда.
Формирование фазовой структуры
в смесях полимеров в отсутствие растворителя
Перемешивание низкомолекулярных, маловязких жидкостей
друг с другом с образованием эмульсии заключается в образова-
нии струй, разрывающихся под действием касательных напряже-
ний или в результате обычной в таких случаях сильной турбули-
зации потока. В расплаве смеси полимеров при перемешивании
турбулизация в обычном смысле слова никогда не возникает. Из-
менение направления потока расплава может приводить к непо-
средственному разрыву возникающих струй дисперсной фазы.
Однако в настоящее время такой механизм нельзя считать един-
ственным, учитывая изложенные выше экспериментальные факты
и теории, указывающие на возможность самопроизвольного раз-
рушения деформированных капель эмульсии. Очевидно, что если
диспергирование происходит только в результате непосредствен-
ного разрушения жидких нитей путем изменения направления
движения расплава, то изменения степени дисперсности даже
очень грубодисперсной смеси полимеров, помещенной в зазор с
однородным сдвигом (например, прибор типа конус— плоскость),
вообще пе должно происходить. На самом деле диспергирование
полимера в полимере происходит и и результате однородного
с щига, что может быть, по-видимому, объяснено только распадом
жидгого цилиндра по механизму Рэлея — Тэйлора — Томотики.
В какой мере каждый механизм диспергирования определяет про-
цесс смешения полимеров в применяемых на практике смеситель-
ных аппаратах, неизвестно.
В этом разделе приводятся экспериментальные цпшые, отра-
жающие состояние исследований в области изучения механизма
диспергирования полимеров друг в друге при деформировании
смеси, пахо ппцспся в вязкотекучем состоянии
Деформация и разрушение капель. Экспериментально показа-
но, что сдвиговое течение полимерной эмульсии в отсутствие рас-
творителя соировож дается в общем случае деформацией капель
дисперсной фазы и их ориентацией в потоке [19]. На рис. III.11
показана мвдрофотогряфия среза экструдата смеси ПММА:ПС=
= 80:20. Из рисунка видно, что при за апной скорости сдвига
98
Рис. III.11. Микрофотография среза смеси
ПММА: ПС (80:20), полученной при про-
давливании через капилляр при 180 °C и
у=0,5 с-1; р — 0,055.
(у = 0,5 с-1) деформируются только
крупные частицы, в то время как
мелкие остаются практически неде-
формированными. Увеличение ско-
рости сдвига приводит к деформа-
ции все более мелких частиц.
Первые наблюдения, указываю-
щие на самопроизвольное разрушение нитевидных частиц дисперс-
ной фазы непосредственно в потоке вязкоупругой смеси распла-
вов полимеров, сделаны в работе [14]. В смесях, исследованных
в этой работе, можно было встретить и варикозные волокна дис-
персной фазы, в которых распад начался, но не успел завершить-
ся образованием капель.
В дальнейшем было проведено систематическое исследование
устойчивости жидкого цилиндра [20] на примере полиметилсило-
ксановых олигомеров различной молекулярной массы, помещен-
ных в матрицу олигомера полибутадиена. Соотношение вязкостей
фаз (полиметилсилоксан — полибутадиен) изменяли в широком
интервале (табл. Ш.2). Жидкий цилиндр получали путем дефор-
мации жидкой капли, помещенной в аппарат, схема которого по-
казана на рис. 111.12. Капля, помещенная в центр пространства
между деформирующими роторами, вытягивалась в тонкую нить
вдоль оси х. После остановки роторов па поверхности покоящей-
ся жидкой нити появлялись утоныпения и утолщения, которые
увеличивались со временем, приводя цилиндр к разрушению па
цепочку капель. На рис. III.13 показаны результаты фотосъемки
жидкой нити через определенные промежутки времени вплоть до
разрушения нити.
Полученные результаты позволили измерить амплитуду вол-
ны разрушения и построить
У
зависимость амплитуды от времени
1ля капли, образованной низкомо-
лекулярным и относительно высоко-
молекулярным образцом ПМС
(рис. 111.14). Теория Рэлея — Тэй-
лора — Томотики (РТТ) предска-
зывает линейную зависимость при-
веденной амплитуды возмущения
(см. рис. Ш.4) от времени. Из
рис. III. 14 видно, что на начальных
Рис. 111.12. Схема потока жидкости, ини-
циируемого четырьмя роторами. Стрелками
показано направление вращения роторов и
направлений линий тока.
7’
99
Рис. 111.13. Микрофотографии
процесса разрушения жидкой
нити для системы № 6 (см.
табл. III.2) [20]. Масштаб
0,4 мм,
стадиях разрушения для
полимерного жидкого ци-
линдра действительно иа-
блю тается линейная за-
в ней м ос Iь н р иведенпой
амплитуды от времени.
На завершающих стадиях
разрушения рост ампли-
туды существенно замед-
ляется, что хорошо видно
на примере высокомоле-
кулярного образца ПМС.
Анализ микрофотографий
показал, что начало от-
клонения кривой In(а/
//?)—t от прямолинейной
зависимости совпадает с
возникновением тонких
перетяжек между почти
сформированными капля-
ми. Очевидно, что меха-
низм указанных отклоне-
ний обусловлен высоко-
молекулярной природой
жидкого цилиндра: при большой скорости нарастания амплитуды
на завершающих стадиях (логарифмический такой изменения ам-
плитуды со временем) в перетяжках возникают большие эласти-
ческие теформапии, приводящие также к ориентации макромоле-
кул в перетяжке и, следовательно, к резкому повышению про-
дольной вязкости жидкости и увеличению ее сопротивления разру-
шению.
В полимерных системах с меньшей вязкостью (рис. III.14,
кривые /, 2) эластичность пе проявляется, и линейный характер
зависимости In(ti/R)—t сохраняется на всех стадиях деформа-
ции, по-видимому, вследствие того, что большая скорость релак-
сации в такой системе оказывается выше скорости роста ампли-
туды волны, и шметпых эластических деформаций (и ориентации
молекул) в перетяжке пе возникает. Поэтому разрушение проте-
кает по классическому механизму для низкомолекулярных систем.
Из микрофотографии на рис. III.13 видно, что разрушение пе-
ретяжек между сформировавшимися каплями приводит к обра-
зованию капель-сиу । пиков со значительно меньшими размерами,
чем основные капли. В системе с высокомолекулярным цилиндром
образуется, как правило, три видимых капли-спутника (папример,
100
Таблица III.Z Свойства систем ПМС—ПБ при 20 СС
(свойства ПБ; Л7г.=0,21 • 10\ ЛД./Л?„= 1,9, (3 = 892 кг/м3,
ц.„=36,6 Па-с)
Номер систе- мы <? 1 О С 1 ПМС Л1„ р20.10-3 ПМС, кг/м3 ПМС, Па-с О ё ^ПБ с -103, н/м Ik С1 | Оо 2лД Д.,10-5, С/М </76105, м/с а
'""В j=: *~в 1^
1 0,5/0,4 1,8 0,961 0,11 0,003 5,4 0,26 0,44 0,17 7,03 1,05
2 1 1/0,4 1,2 0,970 0,46 0,012 5,4 0,10 0,38 0,27 4,10 0,67
3 2,0/0,4 1,5 0,970 0,95 0,26 5,3 0,41 0.36 0,38 2,65 0,40
4 6,0/0,4 — 0,975 19,9 0,55 / ( 0,51 0,31 1,28 0,91 0,094
5 8,0/0,4 -— 0,970 39,3 1, 07 6,0 0,47 0,34 2.00 0,48 0,065
6 11,5/0,4 —. 0,952 117,5 3,21 6,8 0,40 0,36 2,68 0,97 0,043
7 18,5 /0,4 — 0,969 414,6 11,32 5,4 0,25 0,40 3,10 0.13 0,019
системы 4 -7 в табл.; 1II.2), а в системах с меньшей молекуляр-
ной массой разрушение перетяжек сопровождается образованием
множества очень мелких, с трудом различимых в микроскопе ка-
пель.
Полученные данные позволили рассчитать коэффициент неста-
бильности q в уравнении Рэлея [15] по наклону кривых на рис.
111.14. Из данных табл. III.2 видно, что qR резко увеличивается
при уменьшении соотношения вязкостей фаз, т. е. при этом увели-
чивается нестабильность цилиндра. Найденная путем экстраполя-
ции начальная амплитуда волны о0 («о=<» при /=0) составляет
величину порядка 10 5—10 6 см, т. е. совпадает с соответствующи-
ми значениями для низкомолекулярных систем.
Таким образом, механизм астабилизации и распада жидкого
цилиндра в вязкоупругих полимерных системах качественно не
отличается от механизма для
чественпые отклонения могут
нием вязкости полимерной сре-
ды, в которой находится жид-
кий цилиндр даже при таком
же как и в сравниваемой низ-
комолекулярной системе зна-
чении ц. Специфика разруше-
низкомолекулярных систем. 1\оли-
быть обусловлены высоким значе-
Рис. III. 14. Экспериментальная зави-
симость амплитуды возмущения от
времени для систем ПМС (ци-
линдр) — ПБ (среда) при различных
значениях ц:
I. 2 — полимеры № 1 (табл. Ш.2); 3, 4 —
полимеры № 6 (табл. II 1.2).
о w во но 7во гоо zwtt
101
-3,0 -г,о -ю о ioig/j.
Рис. Ш.15. Теоретическая кривая Томотики (/) и экспериментальная зависи-
мость (2) волнового числа 2n/?/Zm от соотношения вязкостей фаз в системе № 6
(табл. Ш.2).
Рис. Ш.16. Экспериментальная (1} и теоретическая (2) зависимость радиуса
капель а0, разорвавшихся в результате распада жидкого цилиндра в системе № 6
(табл. Ш.2), от ц.
иия, обусловленная способностью полимерной жидкости к ориента-
ции и упрочнению, приводит к возникновению полидисперсности
образующихся капель.
На рис. Ш.15 показана теоретическая и экспериментальная
зависимости длины разрушающей волны от соотношения вязко-
стей фаз. Положение максимума на теоретической (р,=0,28) и
экспериментальной! кривой ((.1=0,25) хорошо совпадают, однако
экспериментальная кривая лежит ниже теоретической, что озна-
чает превышение длины волны разрушения цилиндра полимерной
жидкости по сравнению с рассчитанными значениями. Длина раз-
рушающей волны определяет и размер образующихся капель, по-
скольку объем жидкости, израсходованной на образование капель-
спутников в целом невелик. Поэтому кривая на рис. Ш.15 доста-
точно хорошо отражает фактический размер частиц (рис. III.16),
в особенности это касается положения экстремальных точек по
отношению оси соотношения вязкостей.
Для характеристики процесса смешения полимеров большую
роль играет время жизни жидкого цилиндра, который при боль-
шой устойчивости может и не успеть разрушиться за время сме-
шения. Экспериментальная зависимость времени жизни (приве-
денного к ра гнусу цилиндра) от соотношения вязкостей и теоре-
тическая кривая, рассчитанная по (17), приведены на рис. III.17.
Видно, что стабильность цилиндра монотонно увеличивается с ро-
стом р. Это может свидетельствовать о различной роли астаби-
лизации жидкого цилиндра для процесса смешения полимеров
при разных соотношениях вязкостей смешиваемых полимеров.
102
Рис. III.17. Зависимость приведенного
времени жизни жидкого цилиндра от р,:
/ -экспериментальная кривая; 2 — кривая,
рассчитанная по уравнению (17).
В работе [19] исследовали
формирование цилиндров в рас-
плаве смеси полимеров и прове-
зи анализ размеров образующих-
ся частиц с точки зрения аста-
билизации цилиндров по меха-
низму РТТ. Исследования подоб-
ного рода трудоемки, поскольку
требуют анализа большого числа микрофотографий для объек-
тивной оценки размеров частиц. Результаты исследования могут
к тому же искажаться возможной коалесценцией капель при сдви-
говом деформировании системы в целом.
Было проведено исследование смеси ПММА : ПС=80 : 20, в ко-
торой матрицей являлся ПММА. Соотношение вязкостей изменя-
ли варьированием молекулярной массы ПММА (матрицы). Пред-
варительно полимеры смешивали на вальцах при 190 °C и в по-
лученной смеси (эмульсии одного полимера в другом) формиро-
вали жидкие цилиндры продавливанием через капилляр вискози-
метра (//(/ = 5) при нескольких фиксированных скоростях сдвига
в интервале 0,02—1,4 с *. Фиксация волокнистой структуры до-
стигалась продавливанием расплава в воду со льдом. Структуру
полученных волокнистых систем изучали с помощью микроско-
па (рис. III. 18,о, б).
Рис. Ш.18. Электронно-микроскопические фотографии срезов смеси ПММА : ПС
(80:20) после выхода из капилляра вискозиметра (а) (180°С. у=0,5 с р —
= 0.23) и после прогрева этой смеси 5 мин при 170 °C (б)
103
-15^0-0,5 О 0,5 lOlffji
Рис. III.19. Экспериментальная зависимость волнового числа (а) и приведенного
времени жизни цилиндра ПС (б) от ц для смесей ПММА: ПС (80:20). Темпе-
ратура. при которой происходит распад цилиндра 170 °C; а — волокна получены
при у=0,5 с-1 (/) и 1,4 с-1 (2).
Исследование микроструктуры волокнистых образцов, прогре-
тых в состоянии расплава в течение различного времени при
170 °C, показало, что при прогреве происходит распад цилиндров
па цепочки капель (рис. III.18. б). Было определено время до рас-
пада цилиндров и длина волны разрушения в смесях с разным р.
Полученные результаты приведены на рис. III.19. Видно, что ка-
чественно зависимость волнового числа от соотношения вязкостей
фаз хорошо коррелирует с кривой Томотоки, приведенной па рис.
Ш.15. Таким образом, распад деформированных частиц дисперс-
ной фазы в смесях полимеров действительно происходит по меха-
низму астабилизации жидкого цилиндра. Зависимость приведен-
ного времени жизни цилиндра от соотношения вязкостей дана па
рис. III.19, б, из которого видно, что эта зависимость обратна при-
веденной на рис. Ш.17. Это указывает на то, что время жизни
зависит в сильной степени не. только от соотношения вязкостей
фаз, по и от абсолютной величины вязкости среды. В опытах, ил-
люстрируемых рис. III. 17. изменение р достигалось изменением
вязкости фазы при постоянной вязкости матрицы, тогда как в
экспериментах, описываемых рис. III.19, б, изменение р сопровож-
далось сильным изменением вязкости матрицы, рост которой (пе-
реход к малым р) приводил к сильному увеличению времени
жизни цилиндра.
Полученные выше закономерности имеют прямое отношение к
механизму диспергирования полимеров при их смешении меха-
ническим способом с той, однако, оговоркой, что смешение и,
соответственно, распад цилиндра происходит здесь непосредствен-
но в потоке Прямые экспериментальные данные по устойчивости
полимерных цилиндров в движущейся матрице в настоящее вре-
мя отсутствуют.
104
Информацию о механизме диспергирования полимера в поли-
мере в реальных условиях смешения можно получить, изучая за-
висимость предельного размера частиц дисперсной фазы от про-
должительности смешения. Оказалось, что размер частиц с уве-
личением времени смешения уменьшается до некоторого предела,
после которого даже длительное перемешивание оказывается не-
эффективным [21] Предельный размер частиц зависит от интен-
сивности перемешивания, от соотношения компонентов, от их при-
роды. Характер этих зависимостей установлен не во всех слу-
чаях.
Естественно предположить, что при увеличении содержания
диспергируемого компонента размер частиц должен возрастать,
поскольку с повышением концентрации частиц растет вероятность
их коалесценции. Уменьшение степени дисперсности частиц в сме-
си полимеров в области равных соотношений компонентов было
установлено количественно (рис. Ш.20) [22--28].
В ряде работ [28—32] отмечалось уменьшение размера частиц
с уменьшением вязкости полимера, находящегося в виде дисперс-
ной фазы Эти результаты, однако, не были полученье при изме-
нении соотношения вязкостей в широком интервале. В работах
[24, 33, 34] было обнаружено, что при перемешивании на обыч-
ном смесительном оборудовании, когда разрушение цилиндров
дисперсной фазы может происходить в условиях непрерывного
сдвига в переменных направлениях, зависимость предельного раз-
мера частиц дисперсной фазы от соотношения вязкостей фаз так-
же выражается кривой, аналогичной кривой, характерной для
Рис. Ш.20. Влияние соотношения компонентов в смеси ПММА: ПВХ на вели-
чину абсолютной (/) и относительной (2) удельной поверхности частиц дисперс-
ной фазы [22].
Рис. Ш.21. Зависимость среднего диаметра частиц дисперсной фазы в смесях
ПММА : ПС (80:20). полученных в камере пластографа Врабендера при 190 °C
и 30 об/мин:
1 — после 10 мин смешения; 2— после 50 мии смешения (предельный размер частиц).
105
разрушения покоящегося цилиндра. Так. в работе [33] исследо-
вали смешение в пластографе Брабендера смеси ПММА:ПС=
= 80:20 при 190°С. Из рис. III.21 видно, что минимум кривой со-
ответствует соотношению вязкостей фаз р = 0,4—0,5 и форма кри-
вой аналогична форме кривой, полученной при определении раз-
меров капель, образующихся в результате распада покоящегося
жидкого цилиндра (см. рис. III. 16).
Кривая а=[(р) с минимумом и области p«l получена так-
же и в работе [24] для смеси IIK СКЭ1 IT = 80 : 20
Влияние нормальны1, напряжений (упругости) на предельный
размер частиц систематически не изучалось. Представляется ве-
роятным, что с увеличением эластичности жидких цилиндров
возрастает их сопротивление деформированию и повышается ста-
бильность по отношению к разрушению (как это было показано
выше на примере покоящегося цилиндра). Экспериментально ус-
тановлено, что при получении в сопоставимых реологических ус-
ловиях смесей пластмасс и смесей каучуков предельный размер
частиц в последних оказывается почти па порядок выше при зна-
чительно большей эластичности каучуков в условиях смешения.
Таким образом, увеличение эластичности расплава приводит к
увеличению предельного размера частиц, что подтверждается и
данными работ [29, 30, 35].
Изменение скорости сдвига при смешении не оказывает силь-
ного влияния на предельный размер частиц, как это было показа-
но также на примере смеси ПС : ПММА с различным соотноше-
нием компонентов [33, 34]. Это может быть связано как с малым
интервалом изменения скоростей сдвига (изменение частоты вра-
щения ротора в смесителе Брабепдера в интервале 20—
140 об/мин), так и с тем обстоятельством, что увеличение интен-
сивности перемешивания сопровождается увеличением доли эла-
стических (упругих, обратимых) деформаций, которое, наоборот»
должно приводить к укрупнению частиц.
Температура влияет па предельный рашер частиц, по-видимо-
му, нс непосредственно, а через изменение соотношения вязкостей
фаз. Температурный коэффициент вязкости для разных полимеров
может сильно различаться, что приводит к неодинаковой скорости
и 1менения вязкости в разных полимерах с температурой и обус-
ловливает изменение соотношения вязкостей. Отмечено, что фор-
ма кривых зависимости среднего' размера частиц от температуры
смешения совпадает с формой кривых зависимости р от темпера-
туры [34].
Феноменологические теории смешения
При рассмотрении процесса смешения с феноменологической
точки зрения в качестве исходных предпосылок принимают сле-
дующее.
106
Рис. III.22. Схема процесса ламинарного смешения в куэттовском аппарате при
различном положении смешиваемого (темного) компонента в зазоре; п—число
оборотов (от начала процесса; внутреннего цилиндра.
1. Смешение полимеров (независимо от того смешивается ли
полимер с полимером или полимер с наполнителем) производит-
ся реально при низких числах Рейнольдса вследствие высокой
вязкости системы. Это означает, что поток полимера в смеситель-
ном аппарате является ламинарным, и смешение в таких услови-
ях называют ламинарным или идеальным процессом смешения.
2. Эффективность смешения оценивают по величине (или соот-
ветственно скорости) прироста поверхности раздела' фаз. Это на-
глядно представлено на рис. Ш.22, а, из которого видно, что по-
лоска добавдепного вещества в процессе смешения деформирует-
ся и вытягивается в тонкую линию. Поскольку объем дисперсной
фазы сохраняется постоянным, то эффективность смешения мож-
но оценивать по уменьшению толщины полосы, которое в свою
очередь является мерой увеличения поверхности раздела фаз.
Рассмотрим кратко теорию ламинарного смешения, следуя
подходу, принятому в [1].
Пусть объем вещества, подлежащего диспергированию, поме-
щен в матрицу, в которой он должен быть диспергирован; и по-
верхность раздела фаз в пространственных координатах х, у, z
определяется зависимостью вида
F(x.y, г) = о (21)
Направляющие косинусы нормали к этой поверхности определят-
ся выражением
I ^(dF/dx^
В результате деформирования образуется большая поверхность
раздела (диспергирование на отдельные частицы еще, не произо-
107
шло) и эта новая поверхность описывается соотношением
F' (х’, у' г') = 0 (23)
Если предположить, что поверхности раздела остаются при де-
формации плоскими, то можно определить объем материала меж-
ду поверхностями раздела:
V =- S'r/2 (24)
где S' — площадь поверхности раздела после деформации
Из (24) определяются граничные условия для площади по-
верхности раздела
S'r/2 V1-,S'r/2>V2 (25)
где И и Vs — объемы смешиваемых компонентов.
Предлагаемая теория решает задачу определения площади
поверхности раздела после деформации, если известен механизм
деформации и первоначальная плота ц> поверхности раздела.
Для характеристиги деформации следует принять вектор пе-
ремещения и(х, у. Л), который определяется во всем объеме не-
деформированиого материала. Проекции этого вектора на оси х,
у, z обозначаются соответственно через их, иу и иг. Выразим по-
верхность раздела после деформации через ис одну ю поверхность:
F' (х', у’, г') - F (х, у, г) 0 (26)
где Xi=x'i—Ui{x, у, z); (х, у, г) и (х', у', г') —координаты точки в исходном со-
стояЯ,Ш1*Тг после деформации.
С помошью уравнения (26) выразим косинусы нормали к по-
верхности Е'(0) в точке (х', у’, г') через косинусы норма чп к ис-
ходной поверхности Е(0) в соответственной точке (х, у, z)
/ Vi ct)S
2j
cos =—-——---------------------------------------- (27>
1 -2 22 szcos “гeos + X 2 hz cos2
I i i / 4 3 >
Поверхность раздела после деформации можно определить
следующим образом
(28)
Полученное выражение можно проинтегрировать по площади
Stj проекции поверхности S' на плоскость ij.
В случае идеального ламинарного смешения, когда в смеси-
тельном аппарате (ем. рис. III.22) осуществляется чистый сдвиг,
с помошью уравнений (27) и (28) можно определить S. так как
проекция поверхности раздела па плоскость, перпендикулярную
108
Рис. 111.23. Изменение толщины полос смешиваемого материала:
а — до сдвига; б — после сдвига.
вектору перемещения, остается при деформации неизменной. По-
следнее справедливо, если радиусы цилиндров в аппарате Куэтта
(см. рис. III.22) велики, и осуществляемый сдвиг можно аппрок-
симировать сдвигом между плоскостями в направлении осн х, как
это показано на рис. III.23. В этом слхчае векторы смещения в
направлении у и z равны пулю, и площадь поверхности раздела
определяется по уравнению
_ С С Г dux I 0их \2 1 di/dx
JН1-2 ) cos2a*jT^ <29>
Если сама поверхность раздела представляет собой плоскость,
как например, па рис. 111.23, то косинусы нормали пе зависят от
координат точек поверхности и выражение (29) принимает вид:
[дих [ дих \2 Т¥е
1—2 cos ах cos atJ + I ——I cos2 ax (30)
Влияние начальной ориентации поверхности учитывается углами
Ох и аг
Экспериментально показано, что поверхность раздела увеличи-
вается максимально, если вектор смещения (деформации) пер-
пеидикулярен поверхности раздела (а.х = 0). Увеличение поверх-
ности раздела минимально, если вектор деформации расположен
под углом л/2 к поверхности раздела (см. рис. III.22, в).
У равнение (30) позволяет определить необходимую деформа-
цию сдвига, если и >вестпы начальная гс и конечная г ширина по-
лос (см. рис. III.23):
tn
г — ---г------------------тг; где у = дих'ду (31)
(1 — 2у cos tzA- cos ау |- у2 cos2 а2)1/2
Уравнение (30) определяет также влияние первоначальной
ориентации диспергируемого вещества по отношению к направле-
нию сдвига, что качественно продемонстрировано на рис. III.22.
Наиболее интенсивное смешение происходит при первоначальной
ориентации диспергируемого вещества перпендикулярно направ-
лению сдвига. При ориентации вдоль сдвига перемешивание пол-
ностью отсутствует.
В рамках указанной теории легко обосновать влияние реоло-
гических характеристик матрицы на интенсивность смешения.
109
Действительно, напряжения сдвига от деформирующей поверхно-
сти передаются непосредственно только на матрицу, тогда как
дисперсная фаза воспринимает их в свою очередь от материала
матрицы. Если представить себе два слоя полимеров, помещенных
горизонтально один на другой и находящихся между горизонталь-
ными плоскопараллельными плоскостями, io при движении одной
из плоскостей начнут деформироваться и слои полимеров. В ре-
жиме установившегося течения напряжения сдвига во всех слоях
равны, т. е. нет скачка напряжений пи па границе, слоев, пи внут-
ри их. Напряжение с гвига равно:
Q = П1Т1 = 'П2Т2 (32)
1де т]1 и т)2 — вязкости первого и второго полимера (матрицы и дисперсной фа-
зы); у, и у’2 — соответственно их скорости сдвига в установившемся режиме де-
формации.
Видно, что чем больше вязкость слоя, тем меньше скорость
его деформации Понятно, что при диспергировании высоковяз-
кого полимера в среде маловязкого деформируется, главным об-
разом, среда, и возникающие в ней напряжения при данной ско-
рости деформации окажутся недостаточными для деформации и
диспергирования высоковязкой диспергируемой фазы.
Из (30) и (32) можно получить следующее выражение для
скорости сдвига диспергируемой фазы (см. обозначения на рис.
Ш.23):
Г / H — 2h 2Л \-i
Тг= Н\Н + Н (33)
Из (33) видно, что при (.1=3)2/111 стремящемся к бесконечности,
скорость сдвига дисперсной фазы стремится к пулю, если же
ц—И), то предельное (максимальное) значение скорости сдвига
в диспергируемой фазе равно:
V
т2- H — 2h (34)
В реальных условиях смешения компоненты подвергаются не
только сдвигу, по и растяжению; в последнем случае увеличение
поверхности раздела определяется по формуле
(cos2 ах cos2 аи cos2 а, \’/а
и 4- (35)
xi у- z2 /
где х, у. z — отношение расстояний Между точками до деформации к расстоянию
между теми же точками шнле деформации, определенному в направлении соот-
ветствующих координатных осей х = дих'дх, y=duJ9y, z — uu /дг.
Практически при смешении полимеров схема деформирующих
усилий и деформаций намного сложнее представленной выше.
Для расчета прибегают к аппроксимации, разбивая пехотную не-
плоскую поверхность диспергируемой фазы па ряд плоских, а
НО
сложную деформацию на ряд этапов, в каждом из которых ее
можно свести либо к чистому сдвигу, либо к одноосному растя-
жению. Результаты по отдельным этапам суммируются.
В соответствии с положениями развитой выше феноменологи-
ческой теории смешения интенсивность и качество смешения оп-
ределяются толщиной полос (волокон) диспергируемого материа-
ла после определенной деформации. Чем меньше толщина полос,
тем равномернее распределен диспергируемый материал по объ-
ему. Экспериментально равномерность смешения можно опреде-
лить не только по толщине полос, по, что более практически удоб-
но, по степени равномерности распределения дисперсной фазы по
объему. Равномерность распределения определяют по содержанию
диспергируемого полимера в достаточно большом числе проб из
разных объемов смеси. Для этой цели можно применять любой
метод, например газовой хроматографии, ПК-спектроскопии, хи-
мические методы, пригодный для конкретных случаев определе-
ния состава смеси полимеров. Если обозначить относительное
содержание дисперсной фазы в объеме через q, а, число частиц
дисперсной фазы в пробе — через N, то в идеальном случае пре-
дельно перемешанной смеси ее равномерность характеризуется
статистическим критерием о2, называемым генеральной диспер-
сией:
<t2 = q(1 — q)/N (36)
Практически содержание частиц дисперсной фазы в каждой про-
бе может быть неодинаковым (недостаточно равномерное смеше-
ние) и тогда фактическая (экспериментально определяемая) рав-
номерность смешения определяется величиной выборочной дис-
персии:
1 'м
= д/__j У (*i — Ч2 (37)
1=1
где М — число проб; х< — содержание частиц диспергируемой фазы в каждой
пробе; х — среднее значение доли частиц диспергируемой фазы в смеси для М
м
проб, равное х=(1/Л1) У*;-
Отбирая достаточное (не менее десяти) число проб из раз-
ных мест смеси, получаем предельную о2 и фактическую S2 ха-
рактеристики однородности, которые позволяют рассчитать индекс
смешения:
Z=o2/S2 (38)
При /—Ч) система не перемешана, при I—>-1 система идеально
перемешана.
В соответствии с [36] за однократный проход через зону сме-
сительного действия эффективность смешения, как мера прироста
111
поверхности раздела фаз может быть рассчитана по формуле
S/S0 = y/3 (39)
где у — общая величина деформации сдвига.
Еще один подход к оценке эффективности смешения состоит
в том, что для очень грубой смеси, проходящей через зону смеси-
тельного действия, толщина полос уменьшается (с учетом соотно-
шения вязкостей) по уравнению
г=^1‘ (40)
где Го — первоначальная толщина полос (в соответствии с рис. 111.23).
Для расчета деформации сдвига, обеспечивающей заданную
эффективность смешения, нужно зпать индекс смешения I, при
котором смесь обладает удовлетворительным комплексом свойств,
начальную толщину полос г0 и масштаб разрешения при оценке
однородности (минимальный объем пробы). Конечная толщина
полос (размер частиц) и индекс смешения святапы уравнением
С учетом (39) можно рассчитать минимальную деформацию
сдвига, необходимую для заданной степени смешения, которая
характеризуется заданным значением индекса смешения:
Изложенная выше теория смешения позволяет сделать следу-
ющие выводы [1, с. 218]:
1. Смесительный аппарат осуществляет смесительное действие
за счет большой деформации сдвига, п его конструкция предпо-
лагает и вменение направлений сдвига так, чтобы сдвиг был всег-
да направлен перпендикулярно (или близко к перпендикулярно-
му) направлению ориентации диспергируемого вещества.
2. Чем крупнее, начальные агрегаты и чем меньше конечные
заданные ра шеры частиц (толщина полос), тем больше должна
быть деформация сдвига (больше интенсивность смешения).
3. Чем меньше относительная объемная концентрация щепер-
гируемого компошита, тем больше должна быть интенсивность
смешения. Значительно труднее смешать малое количество мате-
риала с большим, чем приготовить смесь, в которой соотношение
компонентов близко к 1 : 1.
Очевидно, что пе все выводы теории ламинарного смешения
справедливы в применении к смешению полимеров. Так, сопо-
ставление третьего вывода и цанных рис. 111.20 показывает оче-
видное противоречие: коалесценция частиц диспергируемого по-
лимера с ростом его объемной доли в смеси приводит к получе-
112
нию наиболее грубодисперсной смеси как раз в соотношении 1:1.
т. е. третий вывод справедлив лишь для твердых частиц наполни-
теля, но не для системы полимер — полимер. Второй вывод не
учитывает время стабильности жидких цилиндров (нитей) и веро-
ятности самопроизвольного их распада в процессе смешения.
Отмеченные ограничения применимости теории ламинарного
смешения к системе полимер — полимер указывают на необходи-
мость новых подходов к анализу процесса. Одним из таких под-
ходов является теория, рассматривающая процесс смешения как
процесс вытягивания частиц дисперсной фазы с последующими
множественными механическими разрывами нитей на отдельные
частицы, например при перемене направления деформации мат-
рицы [24]. Существенно в теории [24] то, что в ней процесс сме-
шения рассматривается как динамическое равновесие между
дроблением (микроразрывы) и коалесценцией капель дисперсной
фазы.
Константа скорости процесса дробления частиц записывается
следующим образом:
/д = 4V7(“ + Зсг/г) (43)
где 1] — эффективная вязкость смеси; у — скорость сдвига; и — энергия разруше-
ния частиц дисперсной фазы; о — межфазное натяжение; г — радиус исходных
сферических частиц.
Видно, что константа скорости дробления уменьшается при
увеличении энергии разрушения и уменьшении размера частиц.
Константа скорости процесса коалесценции выражается урав-
нением
4
k«- = ~ W <44}
где 1Г=0—1 вероятность удачных столкновений частиц, приводящих к коалес-
ценции; <р — объемная доля частиц дисперсной фазы.
В момент установления динамического равновесия соблюдает-
ся условие ki=k2, откуда можно получить выражение для предель-
ного радиуса частиц в процессе смешения:
(12/л) (Готр
гео = ;—(45)
чу — — ir<(.u
Для того чтобы произошло дробление, напряжение сдвига т]у
должно превышать плотность энергии разрушения частиц. С уче-
том этого условия уравнение (45) можно преобразовать:
12 (Гасс / 4Ц7ф« \
гею ——г*- /1 4-----т— ] (46)
лцу у лцу /
Из уравнений (45) и (46) следует, что предельный размер ча-
стиц уменьшается при увеличении вязкости матрицы и уменыпе-
8—244
113
пни и; о и де это качественно находит подтверждение в практике
переработки смесей полимеров. Количественная проверка теории
[24] оказалась невозможной, поскольку в уравнения входят не-
известные значения К и и. Из уравнения (46) следует увеличение
предо. 1ыюго размера частиц с возрастанием упругости дисперги-
руемого полимера, поскольку рост упругости неизбежно приводит
к повышению энергии разрушения пшеничных капель дисперсной
фазы.
Теория [241 предсказывает увеличение размера частиц при
уменьшении вязкости смеси (например, при повышении темпера-
туры смешения), однако в действительности зависимость оказы-
вается обратной, т. е. снижается с повышением температуры.
Автор [24] объясняет это снижением межфазного натяжения при
повышении температуры. На самом деле причина снижения раз-
мера частиц с повышением температуры не вполне ясна, если
учесть, что межфазное натяжение на границе раздела полимер—
полимер невелико и нс очень сильно меняется с температурой.
Теория смешения в смесителях закрытого типа со сложпыде
профилем скоростей только начинает складываться и не позво-
ляет прогнозировать параметры структуры формируемой смеси.
Типы фазовых структур в смесях полимеров
Высокая вязкость полимеров приводит к тому, что равновес-
ная форма частиц дисперсной фазы не возникает при сколь угод-
но длительном хранении или даже в результате специально про-
веденного «отжига» смеси. Равновесная форма частиц тем более
пе может возникнуть в системах, в которых один или оба компо-
нента при охлаждении после извлечения из смесительного аппара-
та стеклуются или кристаллизуются., Это обусловливает большое
разнообразие форм фазовых структур, существующих в смесях
полимеров. На рис. 111.24 приведены основные типы структур. Не-
равновссность фазовых структур вызывает возникновение допол-
нительных внутренних напряжений, которые могут приводить (как
отменллось в гл. V) к повышенному разбуханию струи полимера
на выходе из формующей) отверстия или к повышенной усадке
с меси в процессе отжига.
Возникновение той или иной фазовой структуры обусловлено
как свойствами смешиваемых компонентов, так и условиями сме-
шения. Опыт переработки полимеров показывает, что легче всего
непрерывную фату образует полимер с мепыпей вязкостью. Усло-
вий возникновения более сложных типов структур нс могут быть
предсказаны часто нс только количественно, по и качественно.
Первой попыткой определить количественно условия возник-
новения разных типов фазовых структур является! теория, разви-
тая Ваноэном [29]. В основе теории лежит представление о «ди-
намическом межфазном натяжении». Действительно, межфазное
натяжение является, как известно, тензорной величиной, и в про-
цессе деформации системы имеют заметную величину такие, в
114
Рис. 111.24. Типы фазовой структуры в смесях полимеров:
с — обычная дисперсная (форма частиц близка к сферической); б — волокнистая: в — сло-
истая (дисперсная фаза в виде кольцевых слоев, в предельном случае в виде коаксиальных
цилиндров); г — микроэмульсия (в частицах дисперсной фазы); д — матричная структура
(«матрица в матрице»), обе фазы непрерывны.
частности, компоненты тензора, как нормальные напряжения па
границе раздела фаз. Поэтому динамическое межфазное натяже-
ние может оказаться даже равным пулю при соответствующем
соотношении компонентов тензора. При этом не следует ожидать
взаимодиспсргирования или взаиморастворения фаз, поскольку
статическое межфазное натяжение, определяемое природой кон-
тактирующих фаз, остается в процессе деформации неизменным.
Поведение межфазной границы, например ее искривление в сторо-
ну той пли иной фазы, определяется в процессе деформации имен-
но динамическим межфазным натяжением.
Так по Вапоэну, тип возникающей структуры в смеси полиме-
ров А и В в потоке Пуазейля определяется радиусом частиц г,
главной1 разностью нормальных напряжений полимеров Р2 и ста-
тическим межфазным натяжением одв. Для эмульсий полимера А
в матрице В или для эмульсий полимера В в матрице А могут
быть соответственно написаны уравнения для динамического
межфазного натя/кения:
пдв = <т°4в + -i- гА _ (Рв)в] (4")
ОВА = n-w _ -L Гв [(р2)л _ (Р2;в] (48)
Теперь можно сформулировать пути количественного подхода к
оценке закономерностей формирования типов структур рис. III 24.
Обычная дисперсия полимера А в полимере В возникает наи-
более часто, в особенности при явном количественном преобла-
дании полимера матрицы в смеси и в условиях хорошего смеше-
ния. Равновесие сил на межфазной поверхности, определяющее
формирование дискретной частицы, соответствует условию <т, ^0.
Таким образом, уравнение (47) имеет смысл в двух случаях
(Аг)л — (Л>)в > 0 (4!1)
°лв > + 4" [(^)а - (A>)bJ f-0)
8
115
Из этих условий следует, что при (Р1)х~>СРч)ъ фаза А всегда
распределяется в матрице В в виде частиц сферической или- вы-
тянутой фирмы. Образование дискретных частиц компонента В в
матрице Л возможно лишь (согласно [50]) при очень малом ис-
ходном размере частиц, т. е. при условии
'в < 6сцв/[(А,).\ — (Р2)В]
(51>
При достаточно высоких скоростях сдвига нормальные напря-
жения в расплавах полимеров представляют собой величину того
же порядка, что и касательные напряжения, т. с. 10 2—10 1 МПа
[20, 29], а величина о"в Для. большинства полимерных систем в
среднем равна около 0,5 Па (ем. гл. IV). Из этих данных сле-
дует, что условие (51) выполняется при гс^1—0,1 мкм, что
хорошо согласуется с экспериментом [29]. Сформулированные
условия образования обычных дисперсий справедливы и для во-
локнистых структур (рис. 111.24). Однако практически волокнис-
тые структуры, как правило, не возникают при малом содержании
дисперсной фазы и при смешении в аппаратах со сложными пере-
крещивающимися линиями потоков. Волокнистые структуры пе
образуются, если вязкость и вязкоупругость диспергируемого по-
лимера существенно выше этих показателей для полимера мат-
рицы.
Образованию волокнистых структур благоприятствует дефор-
мация смеси в потоке с параллельными линиями токов (пуазей-
левский, куэттовский, гиперболический), когда начальный' размер
частиц достаточно велик или частицы могут коалесцировать на
начальной стадии деформации, а также при достаточно высокой
скорости и напряжении сдвига при малом диаметре капилляра
(для пуазейлевского потока) и невысокой по сравнению со средой
вязкостью и вязкоупругостью частиц дисперсной фаты [16, 19, 34,
37 45]. Образование волокнистых структур в смесях каучуков
наблющли в |38|. Тип потока также определяет возможность
формирования волокнистой структуры. Так, в гиперболическом
поток! можно получить сильно деформированную каплю с вязко
стью более чем в 10 раз превышающую вязкость среды [12, 20],
тогда как в куэттовском и пуазейлевском потоке такие частицы
почти не цформируются.
Слоиная структура (второй тип на рис. 111.21) возникает в
тех случаях, когда ра «мер частиц велик. Это укладывается в рам-
ки теории Вапозпа, поскольку при соблюдении условия (49) из
уравнения (18) сл< тует, что при размере частиц более 1 0,1 мкм
динамическое межфазное натяжение может стать очень малым.
Таким образом появляется возможность формирования очень
больших поверхпосий, необходимых для образования слоистой
структуры. Во пшкновенис слоистой структуры или кольцевых
слоев в пуазейлевском потоке можно представить упрощенно как
расположение большой частицы дисперсной фазы в слоях с су-
щественно различной скоростью движения вследствие наличия
116
градиента скорости. Поэтому разные участки частицы начинают
двигаться с разной скоростью, что приводит к деформации ча-
стицы, направленной к тому, чтобы всю частицу перевести в слой
с минимальным градиентом, т. е. в весьма тонкий кольцевой слой
в пуазейлевском потоке, ориентированный в направлении течения.
Кольцевые слои наблюдали экспериментально [29, 44—47].
Опп располагаются, как правило, ближе к стенкам капилляра,
центральную часть потока занимают волокна и вырожденные сло-
истые образования типа лент. Кольцевые слои в форме коаксиаль-
ных цилиндров, как предельный случай кольцевых структур воз-
никают при больших напряжениях сдвига (т>10г’ Н/м2), сред-
них значениях отношения длины капилляра к диаметру и при
наличии очень крупных частиц дисперсной фазы. Кольцевые слои
наблюдали и в смесях каучуков [48].
Микроэмульсия полимера матрицы в частицах дисперсной фа-
зы возникает, как правило, при получении ударопрочного поли-
стирола в момент обращения фаз в процессе полимеризации сти-
рола. Микрочастицы показаны схематически на рис. III.24, чет-
вертый тип. Ваноэн теоретически и экспериментально показал,
что это явление характерно и для смесей полимеров, в которых
эластичность (нормальные напряжения) меньше эластичности
дисперсной среды. При этом менее эластичный компонент как бы
обволакивает частицы более упругой фазы. Таким образом, воз-
никновение микроэмульсии в процессе получения ударопрочного
ПС не является следствием протекания полимеризациопных про-
цессов. хотя последние и могут способствовать этому. В работе
[29] приведены микрофотографии срезов смеси ПММА : ПС с мо-
лекулярными массами соответственно 1,3- 105 и 2,75-105, из кото-
рых видно, что менее эластичные частицы ПММА в смеси с ПС
содержат частицы более эластичной полистиролыюй матрицы.
Ваноэн показал также, что образование микроэмульсии согласу-
ется с требованием минимизации общей свободной поверхностной
энергии системы. Если дисперсная фаза образована более элас-
тичным компонентом, то микроэмульсия не возникает.
В связи с этим пеобхо щмо упомянуть работу Уайта с сотр.
[46], в которой исследовали влияние вязкости и нормальных на-
пряжений (первая и вторая разность) па форму границы раздела
расплавов в процессе совместного течения двуслойной массы ПЭ
и ПС в трубе. Независимо от нормальных напряжений менее вяз-
кий компонент всегда имел вогнутую границу раздела и даже мог
капсюлировать (обволакивать) более вязкий полимер, причем
скорость процесса возрастала с увеличением разности в вязкости
обоих слоев.
Магричная структура возникает с увеличением содержания
дисперсной фазы и с приближением соотношения полимеров к со-
отношению 1:1, При этом обе фазы непрерывны, что вообще ха-
рактерно только для высокомолекулярных эмульсий с большой
вязкостью системы. Матричная структура образуется при тем
117
меньшем содержании дисперсной фазы, чем меньше вязкость дис-
персной фазы и ниже ее эластичность. Иногда матричные струк-
туры относят к категории взаимопроникающих сеток, что вряд
ди оправданно или, по крайней мерс, вряд ли полезно, поскольку
под взаимопроникающими сетками первоначально понимали мо-
лекулярные, а пе фазовые сетки. Для полимерных систем суще-
ствование матричных структур можно наблюдать в широком ин-
тервале составов смеси в пределах от 3: 7 до 7:3 (см. гл. IV), а
иногда и в более широком интервале.
Матричные системы возникают час го при смешении пеполи-
мерных компонентов с полимерами. Примером может служить
•смесь солей (мыла) жирных кислот и металлов, которые уже в
небольшом количестве образуют непрерывную структуру в каучу-
ке и сильно изменяют его свойства, в частности стойкость к рас-
творителям.
Обращение фаз
Обращение фаз имеет особое значение для формирования
свойств смесей полимеров. Оно может наблюдаться не обязатель-
но при равных соотношениях компонентов, но и при явном преоб-
ладании одного из компонентов, причем возможно образование
непрерывной фазы компонентом, находящимся в меньшем коли-
честве. При изменении соотношения полимеров в смеси область
составов, в которой непрерывная фаза образована компонентом
А, отделена от области, в которой непрерывная фаза состоит из
компонента В, промежуточной областью, соответствующей обра-
зованию матричной системы, когда обе фазы непрерывны.
Возможность обращения фаз, г. е. перевод дисперсной фазы в
непрерывную, зависит как от вязкоупругости компонентов, так и
ют условий смешений и, в первую очередь, от напряжения и скоро-
сти сдвига и от температуры. Эффективность действия внешних
условий (температура, интенсивность смешения) определяется
различием их влияния па проявление вязкоупругости [35, 49].
На рис. III.25 приведена зависимость условной вязкости от
скорости сдвига”)! от температуры. Ви iho, что как аномалия вяз-
кости, так и ее температурный коэффициент сильно различаются.
Если смешикаlb, например, бутилкаучук с ПММА при 130°C, то
(при соотношении г эмпонентов 1:1) бутилкаучук как менее вяз-
кий компонент, легче перейдет в непрерывную фазу. При 180 иС,
наоборот, ПММА становится менее вязким и теперь именно
ПММА легче, образует непрерывную фазу. Уже из приведенного
примера становятся понятными причины возможного обращения
фаз при изменении температуры и скорости смешения полимеров.
В табл. III.3 представлены данные, свидетельствующие об об-
ращении фаз в некоторых смесях полимеров В таблице подобра-
ны пары полимеров, содержащие каучук и пластик. Это облегча-
ет идентификацию возникших структур, поскольку модули ком-
118
Таблица ITI.3. Свойства смесей полимеров, полученных при различных температурах и скоростях сдвига
Смесь полимеров Состав смеси U -П*/П2 о, МПа Ен, МПа ?, % .4, отн. ед.
‘макс “макс ^макс ’мни 0)макс ^макс Чмакс ’'макс ^макс ’"макс “макс
*мии “мин “иип ^мии "‘мни ’мИН “мин ’'мин “мин
ПММА : БК 20:80 — 1,1 0,8 — 1,2 1,0 — 600 540 — —
^мин = 140 °C 30:70 0,5 2,2 0,9 1,8 1,4 0.7 1,4 1,0 3,0 1,4 3,0 1.4 600 300 150 500 78 86 —
Ыиакс=140 Об/мин 40:60 1,6 0,8 2,3 1,0 3,3 1,6 5,5 4,0 50 40 50 100 176 80 176 103
45:55 — 1,6 1,2 — 7,8 5,6 70 100 — 116 109
ПИБ:ПЭНД 10:9 — 4,5 3,0 — 0,6 0.5 — 800 850 — —
/и„к=120°С 20:80 0,7 2,3 0,7 1,2 10 2,0 80 6,0 1,2 0,4 0,9 0,8 600 1000 750 800 263 39 77 92
«И1КС = 120 об/мин 25:75 11 8,0 11 9,0 1,4 0,9 1,2 1,0 550 700 700 750 — 139 116
Продолжение
Смесь полимеров Состав смеси О, МПа £и, МПа е, % А, отн. ед.
*М£КС ю макс *макс “макс ^макс ® макс ?мак: “макс ^макс ммакс
MUJ “мин ^мпн “мин ^мин “мин ^мин “мин ^ыии “мин
0.6 1,2 2,1 1,5 20 70 118
ПС: БК 40:60 1,8 1,5 Й8 1,2 25 100 101 —
fB„=130°C
Ь»маке = 140 Об/мин
0,5 0,5 1200
СКЭПТ:БК 25:75 — 0,4 — 0,3 — 1800 — — —
0,8
<иин=130°С 1,4
0,6 0,8 800
40:60 0,5 — 0,5 — 1300 — —
ХПЭ:ПИБ 10:90 1,4 0,7 1200
1,2 0,5 1000
0,7 3,0 1,0 1400
£мин= 110 °C 20.80 1,4 — 2,0 0,7 — 1100 — —
6,3 1,3 1750
30:70 5,7 — 1,0 — 1900 — — •—
* Значения и о> во всех случаях равны соотвеютвенно 180 СС и 20 об/мин.
макс МИ"
Рис. III.25. Зависимость условной вязкости (крутящий момент/частота враще-
ния роторов, об/мин) полимеров от температуры (а) и частоты вращения ро-
торов (б):
1 - БК; 2 — СКЭПТ; 3 — ПС; 4 — ПИБ; 5 — ПММА; 6 — ХПЭ; 7 — ПЭНД.
попентов различаются значительно, и обращение фаз приводит к
резкому изменению свойств смеси.
Другой пример приведен на рис. III.26. Кривые нагрузка — де-
формация построены для смесей ПММА: БК, полученных в раз-
ных условиях смешения. С повышением температуры или скоро-
сти сдвига ПММА стремится перейти в непрерывную фазу, по-
скольку при 180°C, он имеет вязкость меньше, чем БК (см. рис.
Ш.25). Об этом свидетельствует большая площадь под кривой
(кривая 1) и более высокие модули смеси. При 140 °C менее вяз-
ким становится БК, но различие в вязкостях не слишком велико.
И форма кривых ст—е„ и прямые исследования микроструктуры
указывают на то, что в такой смеси возникает матричная структу-
ра, и обе фазы оказываются непрерывными.
Из рис. III.26 хорошо видно и влияние частоты вращения ро-
торов смесителя на свойства получаемой композиции. Кривые /
и 2 свидетельствуют о том, что при одной и той же температуре
смешения (150 °C) при частоте вращения роторов 140 об/мин об-
разуется смесь с пределом текучести вдвое большим, чем предел
текучести смеси, полученной при частоте вращения роторов
20 об/мин.
Еще более наглядно иллюстрируют явление обращения фаз
результаты исследования смеси полиэтилена высокого давления
и полиизобутилена (рис. III.27). При 180 °C полиэтилен, как ме-
нее вязкий компонент, образует непрерывный каркас в смеси, что
обусловливает увеличение модуля смеси и повышение напряже-
ния при заданном значении удлинения. Снижение температуры до
120 °C приводит к тому, что изменяется соотношение вязкостей фаз
и, в соответствии с изложенной выше теорией, непрерывной фа-
зой становится ПИБ, смесь делается мягкой, каучукоподобной.
В процессе деформации разрушение каркаса полиэтилена проис-
ходит в большей степени для смеси, полученной при 180 °C, что
снова указывает на непрерывность фазы ПЭ в этом случае.
121
Рис. Ш.26. Зависимость напряжение—деформация смссец 1ГЧ.М \ : БК (40 : 60),
полученных пои различных условиях смешения:
J —180 °C, 40 об/мин: 2—140 'С, 40 об/мин; /'—150 °C, 140 об/мин; 2'— 150 °C, 20 об'мин.
Рис III 27. Зависимость о — ч смесей ПЭ: ПИБ (20:80), полученных при 120 °C
(1 1') и 180 °C (2,2'):
1,2 — первый цикл деформации; Г, 2' — второй цикл деформации.
Непрерывность фазы того или иного полимера в смеси можно
показать надежно в опытах по селективному растворению. Если
на смесь, в которой непрерывной фазой предполагается данный
полимер, подействовать селективным растворителем, растворяю-
щим только полимер непрерывной фазы, то образец потеряет це-
лостность и в микроскопе можно будет наблюдать от тельные ча-
стицы дисперсной фазы после растворения матрицы. Если же этот
полимер находится в дисперсной фазе, то (епствие селективного
растворителя не нарушит целостности обра ща, а вызовет лишь
некоторое набухание смеси в целом. Опыты эти служат более па-
дежным подтверждением непрерывности той или иной фазы, чем
исследование микроструктуры смеси, поскольку при достаточно
высокой копцепграции дисперсной фазы и неправильной форме
ее частиц, оказывается весьма затруднительным установить дейст-
вительное слияние частиц и момент возникновения матричной
структуры.
Приведенные выше данные свидетельствуют о большом влия-
нии как температуры, так и скорости сдвига на структуру смеси.
Не только конечный размер частиц, но и природа матрицы зависят
от скорости смешения, а не только от обшей величины деформа-
ции, как это предполагается в теории ламинарного смешения.
Свойства смеси полимеров значительно сильнее зависят от преды-
стории их приготовления, чем свойства индивидуальных полиме-
ров, что хорошо видно и 1 и «ложенпого материала. Видимо, интен-
122
сификацпя процессов смешения, как главная тенденция в совре-
менной промышленности переработки полимеров, может дать
разные результаты в применении к различным смесям полиме-
ров, а это требует индивидуального подхода к оценке эффектив-
ности смешения в случае конкретных смесей. Воспроизводимость
показателей свойств материалов на основе смеси полимеров от
одной партии к другой может быть обеспечена лишь с учетом на-
личия двухфазной легко изменяемой структуры смеси и тщатель-
ного контроля температурно-временных условий получения смеси
и ее последующего формирования, термообработки готового изде-
лия и т. п. Неравновеспость двухфазных систем на основе смесей
полимеров может приводить к изменению свойств в процессе
термообработки и термореактивных отвержденных систем.
ЛИТЕРАТУРА
1. Торнер Р. В. Теоретические основы переработки полимеров (Механика про-
цессов). М., Химия, 1977. 462 с.
2. Goldsmith И. L„ Mason. S. G. — In: Rheology: Theory and Applications/Ed. by
F. R. Eirich. N. Y., Acad. Press, 1967, V. 4, Ch. 2, p. 85—250.
3. Рейнер M. Реология. Пер. с англ. М., Наука, 1965. 223 с.
4. Rumscheidt F. D., Mason S. G. — J. Coll. Sci., 1961, v. 16, p. 210—237,
238—261.
5. Cerf R. — J. chim. Phys., 1951, v. 48, p. 59— 73.
6. Chaff ey C. F., Frenner H. — J. Coll. Interface Sci., 1967, v. 24, p. 258—269.
7. Cox R. G. — J. Fluid Meeh., 1969, v. 37, p. 601—620.
Karnis A., Mason S. G.— J. Coll. Interface Sci., 1967. v. 24, p. 164—169.
v. Chaffey С. E., Mason S. G.-—J. Coll. Interface Sci., 1966, v. 21, p. 254—
278.
10. Tomotika S. — Proc. Rov. Soc. (London), 1935, pt. A, v. 150, p. 322—347;
1936, pt. A, v 153, p. 302-318.
11. Rumscheidt F. D., Mason S. G. — J. Coll. Sci., 1962, v. 17, p. 260—282.
12. Mikami T., Cox R. G., Mason S. G. — Intern. J. Multiphase Flow, 1975, v. 2,
p. 113—118.
13. Gauthier F., Goldsmith H. L.. Mason S. G. — Rheol. Acta, 1971, Bd. 10, S. 344;
Trans. Soc. Rheol., 1971. v. 15, p. 297—330.
14. Мирошников Ю. П., Петросян M. K-, Кулезнев В. II.— Коллоид, ж 1976,
т. 38, с. 279—285.
15. Gordon М., Yerushalmi J., Shinner R. —Trans. Soc. Rheol., 1973, v. 17, p. 303—
319.
16. Лопперт Г., Овердип В. — В кн.: Многокомпонентные полимерные системы.
Пер. с англ./Под ред. А. Я. Малкина и В. II. Кулезнева. М., Химия, 1974,
с. 61—71.
17. Г улов В. Я. Канд. дис. М., ИНЭОС АН СССР, 1973.
18. Silberberg A., Kuhn IF.— J. Polymer Sci., 1954, v. 13, p. 21—42.
19. Мирошников К). П„ Гольман А. М„ Кулезнев В. Н. — Коллоид, ж., 1979
т. 41, с. 1120—1125.
20 Мирошников Ю. П„ Каминский М., Кулезнев В. Н. — Там же, с 1112—
1119.
21. Rehner I. jr., Wei Р. Е. — Rubb. Chem. Technol., 1969, v. 42, p. 985.
22. Разинская И. И., Батуева Л. И., Штаркман Б. П. — Коллоид, ж, 1974 т. 36
с. 291 297.
23. Heikens D„ Barentsen W. — Polymer, 1977, v. 18, р. 69—72.
24. Tokita .V. Reprints (№ 26) ACS Rubb. Div. Meeting, San Francisko, Oct. 6,
1976.
25. Благов С. С., Печковская К. А., Лыкин А. С., Симановская С. А., Шмигель-
ский В. К- — Каучук и резина, 1959, № 3, с. 12-—14.
123
26. Andrews E. H. Rubb. Chem. Technol., 1967, v. 40, p. 635—651.
27. Gardiner J. B. — Rubb. Chem. Technol., 1970, v. 43, p. 370—-384.
28. Marsh P. A., Voet A., Price L. D., Millins T. J. — Rubb. Chem. Technol., 1968,
v. 41, p. 344 363.
29. Vanoene H. — J. Coll. Internface Sci., 1972, v. 40, p. 448—467.
30. Starita J. Л4. — Trans Soc. Rheol., 1972, v. 16, p. 339—361.
31. Walters Al. H., Kejte D. N. — Trans. Proc. Inst. Rubb. Ind., 1962, v. 38, № 9,
p. 40—52.
32. Hess IP. AT, Scott С. E„ Callan I. E.— Rubb. Cfiem. Technol., 1967, v. 40,
p. 371—390.
33. Кулезнев В. И., Грачев А. В., Мирошников Ю. П. — Коллоид, ж., 1976, т. 38,
с. 263-270.
34. Мирошников Ю. П., Белоусова Е., Смирнов Ю., Кулезнев В. II. — В кн.: Ма-
шины и технология переработки каучуков, полимеров и резиновых смесей.
Межвуз. сб. науч, трудов, 1978, вып. 2, с. 35—37.
35. Кулезнев В. И., Клыкова В. Д„ Мирошников Ю. П. — В кн.: Международная
конференция но каучуку и резине, Киев, 1978. Секция А. Препринты.
36. Mohr W. D„ Saxton R. L., Jepson С. H. — Ind. Eng. Chem.. 1957, v. 49, № 11.
p. 1855- 1863.
37. Heitmiller R. F., Naur R. Z., Zabusky H. H. — J. Appl. Polymer Sci., 1964,
v. 8, p. 873 882.
38. Rovatti W., Bobalek E. G. — J. Appl. Polvmer Sci.. 1963. v. 7, p. 2269—
2281.
39. Козлов П. В.—ЖВХО им. Д. И. Менделеева, 1964, т. 9, с. 660—665.
40. Кандырин Л. Б. Канд. дне. М., M1ITXT им. М. В. Ломоносова, 1971.
41. Барамбойм Н. К, Ракитянский В. Ф. — Коллоид, ж., 1974, т. 36, с. 129—132.
42. Цебренко М. В., Юдин А. В., Кучинка ЛЕ Ю„ Виноградов Г. В., Зубо-
вич К А. — Высокомол. соед., 1973. сер. Б, т. 15, с. 566 571.
43. Цебренко ЛЕ В., Аблазова Т. И., Юдин А. В., Виноградов Г. В. — Коллоид,
ж., 1976, т. 38, с. 204—208.
44. Vinogradov G. V., Yarlykov В. V., Tscbrenko ЛЕ V., Yudin А. V., Ablazo-
va Т. I. — Polvmer, 1975, v. 16, p. 609 614.
45. Walczak Z. A.’—J. Appl. Polymer Sci., 1973, v. 17, p. 169—182.
46. White J. L., Ufford R. C.. Dharod К R-> Price R. L. — J. Appl. Polymer Sci.,
1972, v. 16, p. 1313—1322.
47. Han C. D., Yu T. C.—J. Appl. Polymer Sci., 1971, v. 15, p. 1163—1178.
48. Kiyek H., Schoon Th. С. J. — Koll.-Z. u. Z. Polymere, 1966, Bd. 208, S. 22—
41.'
49. Avgeropoulos G. N., Weissert Г. C., Biddison P. IE, Bohm G. G. — Rubb.
Chem. Technol., 1976, v. 49, p. 93 104.
Глава IV
КОЛЛОИДТЮХИМИЧЕСКАЯ СТРУКТУРА
СМЕСЕЙ ПОЛИМЕРОВ
§ 1. СМЕСЬ ПОЛИМЕРОВ КАК КОЛЛОИДНАЯ СИСТЕМА
Большинство смесей полимеров можно рассматривать как осо-
бый класс коллоидных систем типа «полимер в полимере». Такое
выделение смесей полимеров в особый класс коллоидных систем
подчеркивает их специфику: двухфазную структуру, несмотря на
близкую или практически одинаковую полярность компонентов,
высокую агрегативную устойчивость благодаря огромной вязко-
сти и малому межфазному натяжению, развитый межфазный слой
и другие особенности. 'Термин «полимер в полимере» кажется бо-
лее удачным, чем предлагаемый иногда для смесей полимеров
термин «масло в масле», поскольку он лучше отражает указан-
ную специфику этих коллоидных систем.
Для характеристики свойств систем с коллоиднодисверены ми
пли грубодисперсными частицами требуется применение основных
законов коллоидной химии и физико-химической механики, а т.ак-
же коллоиднохимический подход к анализу структуры смесей.[ Вза-
имодействие полимеров в двухфазных смесях реализуется по гра-
ницам раздела частиц и матрицы. Поэтому термодинамическое
сродство полимеров является определяющим для оценки интенсив-
ности взаимодействия па границе раздела фаз, но не является оп-
ределяющим (единственным) для формирования комплекса фи-
зических свойств смеси. /
В соответствии с основными представлениями коллоидной хи-
мии механические свойства двухфазной смеси полимеров зависят
от следующих внутренних параметров.
1. От соотношения объемов дисперсной фазы и дисперсионной
среды (матрицы).
2. От природы дисперсной фазы и матрицы, поскольку данные
полимерные компоненты могут быть перемешаны в различных ус-
ловиях, и при заданном соотношении объемов фаз либо один по-
лимер, либо другой может образовать непрерывную среду, меха-
нические свойства которой в наибольшей степени определяют
свойства системы в целом.
3. От размера и формы частиц дисперсной фазы.
4. От интенсивности взаимодействия полимеров на границе
раздела фаз (прочность связи, способность к образованию меж-
фазных слоев и их структура).
Важно с самого начала выяснить, сохраняется ли полимерная
фаза неизменной по физическим и химическим свойствам после
125
Рис. IV. 1. Интервал AT между температу-
рами плавления и кристаллизации ПЭ (/) и
ПП (2) в их смесях (АТ — соответствует
ширине гистерезисных петель температурной
зависимости модуля G' при нагревании и
охлаждении).
диспергирования ее в полимерной
матрице другого химического строе-
ния Прежде всего вспомним, что
Методы получения смеси полимеров,
как это было показано в предыду-
щей главе, в большинстве сводятся
к смешению расплавов или раство-
ров полимеров. Если один из поли-
меров является кристаллическим, его кристаллизация происходит
после формования пленки, т. е. после удаления растворителя или
охлаждения расплава, а следовательно, кристаллизация идет в
вязкой среде, что оказывает влияние на ее кинетику и на структу-
ру кристаллической фазы. Кроме того, возможно проникновение
одного полимера в среду другого в виде истинного раствора (хотя
истинное растворение может составлять лишь незначительный про-
цент) или путем частичного взаимопроникновения сегментов мак-
ромолекул в переходном межфазном слое./ Как было показано в
гл. II, один полимер является плохим растворителем для другого.
Поэтому в расплаве (а, особенно, в растворе) при образовании
смеси степень ассоциации каждого полимера вследствие их взаим-
ного влияния может меняться так же, как это происходит в рас-
творе полимера в «плохом» низкомолекулярном растворителе На-
личие возможного взаиморастворения, сегментальное проникнове-
ние в межфазном слое, значительная вязкость матрицы наклады-
вают отпечаток на закономерности кристаллизации и структуру
кристаллической фазы. 1
Впервые влияние одного полимера в смеси па кристаллизацию
другого было отмечено в работе [1], где исследовались смеси ПЭ
и IIП, полученные смешением расплавов полимеров в ротационном
вискозиметре при 190°С. Температурную зависимость комплекс-
ного модуля 6*. модуля упругости G' и потерь G" снимали в ча-
стотном реометре при ничтожной общей деформации, не возму-
щающей внутренней структуры расплава. Скорость нагревания не
превышала 3 5 грач/ч. Поскольку скорости кристаллизации и
плавления указанных полимеров различны, кривые G'—T при на-
гревании и охлаждении не совпадали, т. е. возникала петля «ги-
стерезиса кристаллизации». Ширина петли характеризует разность
между темпера।\ рами плавления и кристаллизации АТ, совпада-
ющую с этой же величиной, определяемой но исчезновению и по-
явлению кристаллических структур при наблюдении в микроско-
пе. На рис. IV.1 [1| показано, что в смеси ПП—ПЭ АТ для каж-
дой фазы (например, для фазы ПЭ) не равно АТ для исходного-
126
компонента (например, для того же ПЭ) и зависит от содержа-
ния дисперсной фазы, т. е. от поверхности раздела. Видимо, вслед-
ствие малого сродства полимеров в случае их несовместимости
количество контактов полимер 1 — полимер 2 стремится к мини-
муму. Поэтому в смеси несовместимых компонентов свободный
объем в межфазном слое, где могут располагаться макромолекулы
обоих компонентов, существенно увеличивается. Увеличение плот-
ности укладки компонентов в каждой фазе может приводить к
повышению плотности смеси в целом [2], по-видимому, благодаря
росту степени ассоциации однородных сегментов и увеличению
степени ближнего порядка. Это в свою очередь приводит к росту
числа зародышей кристаллизации и уменьшению интервала между
температурами плавления и кристаллизации в смеси (см. рис.
IV.1).
В смеси ПП с 10% ПБ при размере частиц дисперсной фазы
1—7 мкм уменьшается индукционный период кристаллизации и
вдвое увеличивается скорость кристаллизации. Это явление нельзя
объяснить только наличием частиц дисперсной фазы, которые са-
ми по себе могут служить зародышами кристаллизации: ведь в
момент кристаллизации полипропилена полнбутадиен остается
в жидком (вязкотекучем) состоянии [3]. Присутствие второго
полимера, близкого по химическому строению, замедляет кристал-
лизацию диспергированного в нем кристаллического полимера,
видимо, вследствие истинного взаиморастворения или диффузии в
межфазном слое, что было показано на примере смесей ПЭ, ПП,
ПЭТФ [4] и кристаллизующихся каучуков с другими полимера-
ми. В то же время присутствие полимера, далекого по химическо-
му строению («носовметающегося»), может вообще не влиять на
скорость кристаллизации. Примером служит натуральный каучук,
скорость кристаллизации которого снижается в смеси с бутадиен-
стирольным и почти не меняется в смеси с полпхлоропрепом.
Иногда второй полимер влияет на температуру плавления.
Так, по данным дериватографического исследования добавление
к ПЭНП на вальцах при 130—135 °C бутадиен-стирольного блок-
сополимера ДСТ-30 в количестве 70% приводит к снижению т. пл.
ПЭНП на 5 °C [5].
Конечный эффект присутствия второго полимера в смеси с кри-
сталлическим не однозначен и зависит не только от природы до-
бавленного полимера, по и от условий получения смеси, ее струк-
туры.
Изменение свойств фазы в смеси некристаллизующихся поли-
меров во многих случаях также не получило надежного объясне-
ния. Одной из важных причин изменения свойств фазы, напри-
мер Тс, может быть уменьшение размера частиц ниже_опре делен
ной величины. При уменьшении размера частицы отношение массы
вещества в объеме к его массе на поверхности частицы в меж-
фазном слое непрерывно уменьшается и при малом размере ча-
стицы большая часть ее вещества оказывается непосредственно
127
Рис. IV.2. Зависимость снижения температуры
стеклования A7'c в блок-сополимерах стирола
и бутадиена (СБС) от соотношения S/V.
прилегающей к поверхности и нахо-
дится под влиянием поверхностных
сил и вещества матрицы.
Понижение Тс полистирольной фа-
зы с уменьшением размера частиц бы-
ло показано па примере термоэласто-
пластов. Так, на основе обработки
многочисленных, в том числе и лите-
ратурных данных, установлено [6],
что удельная поверхность частицы 3
и ее объем V линейно связаны с величиной снижения Тс (рис.
IV.2), хотя разброс значений велик. Много работ, в том числе и
цитированных в [6], подтверждают зависимость Тс от размера
частиц дисперсной фазы. Здесь пе следует, видимо, делать разли-
чий между смесями и блок-сополимерами, поскольку число хими-
ческих связей на границе раздела фаз в блок-сополимерах отно-
сительно невелико и не может изменить принципиально энергию
межмолекулярного взаимодействия в межфазном слое. Смеси же
полимеров никогда не могут быть столь высокодисперсными, как
блок-сополимеры, и потому в них меньше проявляется влияние
размера частиц. Указывают, однако [7], что в смесях ПС—ПИБ
фаза ПС имеет Тс на 4 °C ниже, чем у исходного ПС. В смеси
бутадиен-стирольного сополимера СКМС-ЗОРП со смолой СКС-85
температура стеклования сополимера увеличивается на 8 °C по ме-
ре повышения температуры смешения с 40 до 100 °C [8]. Правда,
нет убежденности в том, что два последних случая не являются
следствием частичного растворения одного полимера в другом.
В то же время, чтобы обеспечить указанное снижение Гс, количе-
ство полимера, перешедшего в ipyryio фазу, должно быть выше
предела его растворимости.
Пшеиение свойств полимерных фаз при смешении — еще од-
па причина относиться с осторожностью к оценке сродства поли-
меров по кривым свойство—состав, где на осях ординат в точках,
соответствующих индивидуальным полимерам, обычно откладыва-
ют пока ннелн свойств исходных полимеров, а пе, как это следо-
вало бы, их свойств в полимерной дисперсной фазе и в матрице.
/Изменение свойств вещества с увеличением степени дисперс-
ности— хорошо известный факт в области коллоидной химии.
Однако непосредственная зависимость физических свойств ве-
ществ от размеров частиц часто ускользай! из внимания потому,
что редки попытки связать свойства коллоидных систем со свой-
ствами дисперсной фазы. Так, с уменьшением размера капель во-
ды увеличивается давление пара над каплями, и при достижении
размера капель 10 нм р/р0= 1,108 (р0 — давление' пара над плос-
128
кой поверхностью воды при данной температуре, р — давление
пара над каплей). Очевидно, что на диаграмме состояния воды
повышенному давлению пара будет соответствовать пониженная
температура замерзания (плавления) капель воды. Гипотетичес-
кий композиционный материал, в котором при Т<;0оС мы предпо-
лагали наличие дисперсной твердой фазы в виде льда, на самом
деле содержит жидкую дисперсную фазу, если частицы фазы до-
статочно малы!
расчет показывает [9, с. 7—8], что доля «особенных» молекул
(расположенных близко к поверхности и на поверхности), нахо-
дящихся в энергетическом состоянии, отличающемся от состояния
молекул в объеме, меняется в зависимости от размера частиц по
кривой с максимумом, соответствующим размеру несколько более
10~9 м. Доля «особенных» молекул становится пренебрежимо ма-
лой (менее 1%) при размере частиц 10 7 м и более, когда свой-
ства вещества дисперсной фазы перестают (в пределах точности
эксперимента) зависеть от объема фазы. Скачкообразный качест-
венный переход от двухфазных к однофазным системам происхо-
дит в области размеров частиц в пределах 10~9—10~7 м, в кото-
рой свойства вещества зависят от размера частиц. При большом
размере молекул полимеров указанный интервал размеров частиц
может быть шире.
Известные сегодня механические модели, характеризующие
свойства двухфазных систем, основаны на представлении о неиз-
менности свойств вещества при диспергировании. Трудно пред-
ставить себе возможность создания механических моделей, учиты-
вающих изменение свойств дисперсной фазы при изменении раз-
мера частиц. __
Изменение механических свойств при увеличении отношения
поверхности к объему вещества экспериментально показано при
исследовании тонких дублированных пленок и покрытий [10]. Эти
изменения могут быть столь значительными, что полимер, не спо-
собный к вынужденноэластической деформации в виде свободной
пленки, может значительно деформироваться при растяжении ад-
гезионного соединения полимер — подложка.
Другой причиной изменения свойств фазы полимера в смеси
по сравнению со свойствами исходного полимера является раз-
личие термических коэффициентов расширения разных фаз, что
создает при охлаждении смеси внутренние напряжения, приводя-
щие. в частности, к сдвигу температуры стеклования. Впервые та-
кие представления были высказаны в работе [11], когда было об-
наружено увеличение Тс ПС в смесях с полибутадиеновым, бута-
диен-стирольным и другими каучуками, а также с ПММА, ПВХ и
др. Была развита теория двухфазных систем с межфазным слоем,
которому приписывались некоторые свойства фазы, и рассчитан-
ные значения изменения Тс оказались совпадающими с экспери-
ментальными. Так, при значениях термических коэффициентов
расширения ПС (а=5,55-10-4 град-1) и ПБ (а=7,90-10“'* град-1)
9—244
129
при охлаждении смеси в матрице ПС (80% от массы смеси) со-
здаются внутренние напряжения, которые и приводят, по-видимо-
му, к увеличению Гс ПС на 9,7°, тогда как рассчитанное значение
ЛГС в зависимости от выбранной модели составляет 8—12 °C. Для
всех смесей наблюдали совпадение расчетных и эксперименталь-
ных Тс. Различие рассчитанных и экспериментальных значений
Тс ПС, находящегося в матрице ПММА, авторы объясняют «мо-
лекулярным взаимодействием», которое не учитывалось в расче-
те», т. е. неучтенным взаимодействием в межфазном слое.
Аналогичное явление наблюдали на примере смолы АБС
[12], когда Тс фазы ПБ снижалась па 1,5—4 °C. Зная разность
термических коэффициентов расширения и зависимость Тс от
давления, рассчитали, что для согласования экспериментальных
и расчетных данных необходимо предположить наличие межфаз-
ного слоя толщиной 18 нм на частицах среднего размера
0,2 мкм.
Интересно, что в этой работе отмечен рост ударной вязкости
с ростом толщины межфазного слоя.
Возникновение напряженного состояния наблюдалось непосред-
ственно [13], когда частицы каучука помещали в эпоксидную смо-
лу, моделирующую матрицу ПС, и отверждали смолу.
Можно полагать, что смещение температур стеклования или
плавления отдельных фаз в смесях наблюдали многие исследова-
тели, по далеко не все они могли быть уверены в том, что это
смещение выходит за рамки точности измерения Тс. Заметим, од-
нако, что, несмотря на различие причин, приводящих к изменению
свойств каждой фазы (в частности, Тс), наблюдаемое иногда уве-
личение Тс пластмассы и уменьшение Тс каучука [11, 12] пе мо-
жет быть объяснено взаимным растворением компонентов, так
как опо должно было бы привести к обратному эффекту. Наблю-
даемое явление не противоречит, однако, представлению о воз-
можном изменении степени ассоциации каждого компонента в сме-
си полимеров, т. е. об изменении ближнего порядка в микрообъ-
емах фаз, непосредственно примыкающих к межфазным слоям.
Для оценки коллоиднохпмической структуры смеси полимеров
решающее шачепие приобретает установление ее фазового соста-
ва. Это особенно важно для смесей, состав которых не очень силь-
но отличается от предела растворимости и где не может быть ап-
риорной уверенности в том, что число полимерных компонентов
соответствует числу фаз, а свойства фаз, например Тс, идентичны
' свойствам исходных полимеров. .
Число фаз устанавливают гю .числу температур плавления для
смесей кристаллических полимеров или по числу релаксационных
переходов (температур стеклования), определенных любым ме-
тодом, обеспечивающим необходимую точность.
Впервые динамический механический метод был применен к
смеси полимеров в работе [14], а затем в работе [15] В этих
работах было обращено внимание на то, что, если смесь образо-
130
Рис. IV.3. Динамические механические по-
тер!1 смеси ПС и БСК.
вана полимерами, считающимися
несовместимыми, кривая зависимо-
сти механических потерь от темпе-
ратуры имеет число максимумов,
равное числу полимерных компонен-
тов Пример термомеханической
кривой смеси несовместимых поли-
меров приведен на рис. IV.3 [14],
где температурная зависимость мо-
дуля упругости и логарифмическо-
го декремента затухания (эксисрп
мент проводился на маятниковом
приборе) представляет собой на-
ложение кривых, характерных для каждого компонента смеси. По-
ложения максимумов соответствуют температурам стеклования
исходных полимеров, а высота максимума или площадь, ограни-
ченная им, определяется содержанием полимеров в смеси Такой
вид кривых указывает на типично двухфазную структуру смеси,
где свойства фаз остались неизменными при смешении,'а заметного
взаимодействия полимеров в межфазном слое не происходит.
Взаиморастворимые компоненты в смеси дают один максимум
[15], расположенный между максимумами смешиваемых компо-
нентов (см. также [14]). Во всех случаях исследователи отмечали
большее или меньшее (в зависимости от природы смешиваемых
полимеров) расширение максимума потерь, что объясняли опре-
деленным взаимодействием полимеров в межфазном слое, не при-
водящим, однако, к сдвигу основного максимума по температур-
ной шкале или появлению дополнительного, промежуточного мак-
симум аЛ>
Метод определения числа фаз в смеси полимеров по кривой
зависимости механических или диэлектрических потерь от темпе-
ратуры имеет наибольшее распространение как по причине относи-
тельной доступности приборов, так и потому, что наличие макси-
мума позволяет обычно более точно фиксировать температуру пе-
рехода по сравнению с зависимостями, имеющими при Тс лишь
перегиб. На рис. IV.4 приведены типы кривых для разных смесей.
Смесь а — типично двухфазная смесь, подобная приведенной на
рис. IV.3. Кривая б имеет сдвиг максимумов в сторону сближе-
ния, что может_быть результатом определенной пластификации
полимера с высокой Тс и «ужесточения»'“полимера с малой Тс.
Это объясняется либо истинным частичным взаиморастворснием
полимеров, либо их взаиморастворснием на границе раздела, ли-
бо изменением структуры каждой фазы в области, прилегающей
к межфазному слою, в результате межмолекулярного взаимодей-
ствия полимеров и изменения степени ближнего порядка. Величи-
9'
131
Рис. IV.4. Схематическое изображение типов тем-
пературной зависимости tg б для бинарных смесей
полимеров различной структуры (пунктир — поло-
жения максимумов исходных полимеров).
на сдвига максимумов типа, показанного
на рис. 1V.4, б зависит от размера частиц.
Кривая в указывает на возникновение
термоусадочных напряжений в двухфаз-
ной системе и изменении Тс по механиз-
му Такаянаги — Поля [11, 12]. Из кри-
вой г можно заключить, что число фаз в
смеси больше, чем число исходных ком-
понентов, и в системе имеется развитый
межфазный слой, обладающий свойства-
ми фазы (об этом подробнее будет ска-
зано позднее). Наконец, кривая д указы-
вает па полное взаиморастворение поли-
меров с образованием однофазной смеси.
Здесь имеется один промежуточный мак-
симум, который при изменении соотно-
шения полимеров перемещается по шкале температур по закону,
близкому к тому, что описывает зависимость температуры стекло-
вания от состава для статистических сополимеров.
Реальная картина потерь в смеси полимеров может быть слож-
нее, включая смещение одного из максимумов, смещение с появ-
лением промежуточного максимума и т. п. Принципиальным пре-
пятствием применению метода, является близость температур пе-
реходов и возможное перекрывание отдельных максимумов. Это
следует особенно иметь в виду, поскольку ширина максимума для
смеси обычно заметно больше, чем ширина максимума для исход-
ных полимеров.
Температуры переходов в смесях полимеров можно определять
по диэлектрическим потерям [16], по максимуму эластичности
[17], по модулю упругости [18], методом ЯМР [19], трибометри-
чески [2()|, но термическому коэффициенту линейного [21] или
объемного расширения [22] и другими методами.
Отметим особо два метода, получающие большое распростра-
нение. Метод дифференциального термического анализа (ДТА)
пригоден для определения температуры фазовых переходов, ког-
да выделяемое тепло фиксируется в виде четкого пика на термо-
грамме. Этим методом можно фиксировать также переходы типа
стеклования, когда теплоемкость с температурой изменяется скач-
кообразно, что приводит к дополнительному поглощению тепла и,
следовательно, поянлению температурного пика в точке перехода,
позволяющему определить Тс. Метод ДТА очень перспективен
[23, 24], поскольку для снятия термограммы в современном ска-
нирующем калориметре можно применять очень тонкие (десятки
132
мкм) образцы, помещаемые между дисками нагревателя и диском
термопары, что позволяет снимать термограмму со скоростью на-
гревания в десятки градусов в минуту. Применяя этот метод, так-
же нужно быть уверенным в достаточно большом расстоянии меж-
ду главными релаксационными переходами по температурной
шкале.-Иногда при работе в области низких температур может
выделиться теплота кристаллизации одного из компонентов и за-
маскировать релаксационный переход. Видимо это обстоятельство
привело авторов [25] к ошибочному отнесению смеси ПС и ПБ
к числу однофазных.
Метод радиотермолюминесценции (РТЛ), впервые применен-
ный для исследования полимеров в нашей стране [26], состоит в
облучении полимера ионизирующей радиацией при температуре
много ниже Тс (обычно при температуре жидкого азота). Возник-
шие активные центры практически не аннигилируют со временем
и оказываются стойкими при нагревании вплоть до температуры,
близкой к Тс- При достижении Тс активные центры реагируют и
запасенная ими энергия превращается в световую. Зависимость
свечения от температуры выражается кривой с максимумом в точ-
ке стеклования. Для многих полимеров максимум оказывается
очень отчетливым и TQ может определяться с большой точностью,
причем высота максимума в ряде случаев столь высока, что по-
зволяет определить присутствие данного полимера в смеси, когда
его содержание ие превышает 1—2% [27].
Широко распространено изучение смесей полимеров с помощью
микроскопии (см. Кулезнев в сб. [28], с. 10—60). Электронная
микроскопия смесей полимеров стала быстро развиваться после
того, как для контрастирования объектов была применена четырех-
окись осмия или травление в низкотемпературной плазме кисло-
рода (см. ссылки в обзоре [28], с. 10—60). В случае световой мик-
роскопии можно применять весьма простые методы контрастиро-
вания. Так, подвергая смесь полимеров набуханию в парах
селективного растворителя [29], можно вызвать выдавливание
частиц набухающего полимера из ненабухающей матрицы. Воз-
никший рельеф хорошо просматривается в микроскопе. Метод
прост и дает хорошие результаты, даже если полимеры имеют
близкие показатели преломления. Так, при набухании смеси НК
и полиизобутилена в парах циклогексанона непосредственно на
предметном столике микроскопа удалось снять кривую распреде-
ления частиц набухшего НК в полиизобутилене. При этом макси-
мальное набухание НК не превышало 3*0%, что соответствовало
искажению линейных размеров не более чем на 10% [30].
Здесь пе даются исчерпывающие ссылки на работы по элект-
ронной и световой микроскопии смесей полимеров. Многие работы
цитируются в связи с изложенном данных по структуре смесей.
Нужно отметить, что вопросы препарирования смесей полимеров
очень сложны и пе решены окончательно, поэтому непременным
условием получения надежных результатов является многократ-
133
пос исследование одной и той же смеси разными методами для
получения уверенности в объективности снимков [25].
В связи с методами исследования микроструктуры смесей по-
лимеров следует сделать одно принципиальное замечание. Метод
исследования микроструктуры смесей полимеров с помощью мик-
роскопа не дает однозначного ответа на вопрос о фазовом соста-
ве смеси. Это значит, что наличие микроиеодпородностей в поле
зрения микроскопа не является падежным указанием на наличие
второй фазы в смеси полимеров. Этот вывод прямо следует из
классического определения фазы [31, с. 232]: «фаза — совокуп-
ность всех гомогенных частей системы, одинаковых по составу и
по всем химическим и физическим свойствам (не зависящим от
количества вещества) и отграниченных от других частей системы
некоторой поверхностью (поверхность раздела)». Указание на не-
зависимость химических и физических свойств от количества ве-
щества наиболее важно для понимания того, что такое фаза в
смеси полимеров.(Если один полимер полностью растворен в дру-
гом, то растворенные макромолекулы, размер клубков которых
составляет десятки им, могут быть видны как микроиеодпородно-
сти, а смесь фактически является однофазной. В истинных раство-
рах низкомолекулярных и полимерных веществ, ^как известно,
существует ассоциация однородных молекул. Естественно, что ас-
социаты макромолекул могут быть столь большими, что даже в
истинном растворе полимера в полимере они могут быть ошибоч-
но приняты за частицы второй фазы. В этом отношении характе-
рен опыт Мацуо [32], показавшего, что в смеси ПВХ и СКН-40
(100:15) имеется один максимум модуля механических потерь,
а па микрофотографиях отчетливо просматриваются частицы раз-
мером около 10 нм, т. е. микронеоднородности существуют и в
однофазной смеси полимеров?
г Еще одним оптическим методом исследования структуры смеси
полимеров является рассеяние света или просто определение опти-
ческой плотности. Зависимость оптической плотности от концент-
рации второго полимера позволяет установить предел раствори-
мости полимера в полимере, как это было сделано, например, для
смесей НС с ПММА и полиизопреном [33]. Высокомолекулярный
ПС растворялся в ПММА в количестве около 0,1%.
При изучении рассеяния вертикально поляризованного лазер-
ного пучка под разными углами в растворе ПС в ПММА было
найдено [31], что при концентрации ПС 0,0001—0,005% имеются
флуктуации концентрации, которые могут рассматриваться как
гомофазные, а при содержании ПС около 0,01% он, по-видимому,
выделяется в виде частиц повой фазы. Это означает, что резуль-
таты определения числа фаз в смеси полимеров могут зависеть от
применяемого метода. Положение аналогично нахождению раз-
ной степени кристалличности в зависимости от того, применен ли
рентгеноструктурный или щлатометрический метод.
Ниже следуют примеры зависимости результатов определения
134
фазового состава смеси полимеров от применяемого метода иссле-
дования.
Смесь ПВХ (Л1 = 1,59-105) и ПММА (М= 1,02-105), получен-
ная совместным осаждением воднометанольной смесью из раство-
ра в тетрагидрофуране, по данным термомеханических исследова-
ний и ДТА имеет одну Гс [35]. Величина Тс меняется с изменением
состава смеси, что указывает на однофазность системы. Опти-
ческая плотность пленок из этих смесей резко повышена, что ука-
зывает па наличие крупных микропеодпородностей. В электрон-
ном микроскопе можно установить появление микронеоднородно-
стей при введении 5% ПММА в ПВХ и 2% ПВХ в ПММА. К со-
жалению, образцы для микроскопии и определения оптической
плотности получены в этой работе выпариванием растворителя,
что могло привести к большему расслаиванию системы, чем при
осаждении смеси из раствора. Однако очевидно, что следует ос-
торожно подходить к оценке фазового состава, в том числе и к
выбору’ метода получения смеси, учитывая, что достижение равно-
весия в смесях полимеров, ввиду их огромной вязкости, часто бы-
вает совершенно невозможно.
Коллинскпй и Маркерт (см. сб. «Многокомпонентные полимер-
ные системы» [24, с. 72—83]) синтезировали сополимеры ММА—
БА с разным содержанием мономеров в макромолекулах. Пленки
смесей сополимеров получали из внешне гомогенных растворов в
этплацетате выпариванием па подложке. Если разница в содер-
жании ММА в смешиваемых сополимерах составляла 13% (мол.)
[определено с точностью до 1% (мол.)], то пленки получались
мутными. В то же время электронная микроскопия показывает на-
личие микронеоднородностей уже при различии в содержании
ММА 10% (мол.). Во всех этих пленках имеется одип максимум
механических потерь (маятниковый прибор), и максимум этот
разделяется на два лишь при разнице в составе сополимеров в
20% (мол.). Четкий второй максимум возникает при 25% (мол.)
различия в составе сополимеров. Поскольку образцы были полу-
чены одним и тем же методом, можно считать, что при различии
составов в 20% появляются частицы второй фазы, тогда как при
меньшем различии составов имеются только гомофазные флуктуа-
ции концентрации, хотя и приводящие к помутнению пленки.
Мак-Найт, Стелдипг и Караж [28, с. 129—140] получали смеси
ПС с поли-2,6-диметил-1,4-оксифепилеиом в разных соотношениях
литьем под давлением при 280 СС, а затем переплавляли и прессо-
вали для получения образцов для испытаний. Смеси прозрачны
и методом дифференциальной сканирующей калориметрии в них
обнаруживается одна 7С в промежутке между температурами
стеклования ПС (90°C) и ПОФ (209°C). При измерении темпе-
ратурной зависимости модуля потерь методом сдвиговых вынуж-
денных колебаний обнаруживается два максимума, указывающих
на наличие двух фаз, из которых каждая содержит также другой
полимер. При измерении диэлектрических потерь при той же ча-
135
стоте, что и механических, обнаруживается одна Тс, которая, од-
нако. меняется с составом смеси по S-образной кривой, что ука-
зывает на неоднородное распределение компонентов в объеме.
Можно предположить, что при некоторой степени дисперсности
частиц одного полимера в другом, структура аморфной смеси на-
поминает структуру жидкой эвтектики с микрообластями твердых
растворов и размытых границ между ними В этом случае, в за-
висимости от применяемого метода, можно зарегистрировать одну
температуру перехода подобно одной температуре плавления в
низкомолекулярной эвтектике.
Вследствие перавновеспости большинства смесей (высокая
вязкость системы) и двойственной природы полимеров (цепные
молекулы очень больших размеров, состоящие из сравнительно
небольших кинетических единиц — сегментов) определение фазо-
вого состава в смесях затруднительно и для получения надежных
результатов требуется применение нескольких методов. Всегда
необходимо учитывать принципиальное различие между мнкроне-
одпородностыо и многофазностыо смеси.
§ 2. РАЗМЕР И ФОРМА ЧАСТИЦ
Количественная характеристика микроструктуры включает оп-
ределение размера частиц, распределения по размерам, формы
частиц (например, как отношение полуосей эллипсов, если части-
цы анизометричны), степени ориентации апизометричных частиц,
если ориентация их пе вполне произвольна. Иногда, если известна
объемная доля дисперсной фазы и размер частиц, для характе-
ристики системы рассчитывают среднее расстояние между части-
цами. Методы усреднения размеров обычны и пе имеют специфи-
ки в применении к смесям полимеров.
Количественная оценка размеров и формы частиц проста в
случае правильной формы частиц и четких границ раздела фаз.
Объективность оценки снижается, если форма становится произ-
вольной и неправильной, как это показано на рис. IV 5 [36]. Из-
мерение размеров частиц обычными способами становится невоз-
можным при большом их содержании и значительной коалесцен-
ции дисперсной фазы, ког да структура смеси приближается к
структуре двух непрерывных фаз (дисперсная фаза фактически
становится непрерывной). В таких случаях бывает полезным рас-
считывать не средний размер частиц, который при неправильной
их форме не имеет исходного физического смысла, а удельную по-
верхность раздела фаз в смеси [37].
Для определения количественных параметров фазовой струк-
туры дисперсных систем, в том числе и смесей полимеров, суще-
ствует несколько методов.
Визуальное измерение. Микрофотографию, увеличенную до соответствующих
размеров, анализируют таким образом, что измеряют не менее 100 частиц. Если
136
Рис. IV.5. Микрофотографии смесей СКИ—ПИБ и СКС-ЗО—ПС с различным со-
отношением компонентов:
а— смесь СКИ — ПИБ, 50% СКИ; 6 — то же, 80% СКИ; в — то же, 97% СКИ: г — смесь
СКС-ЗО—ПС. 50%ПС; д — то же; 20% ПС; е — то же. 90% ПС.
----------------Sj-----------------------•
♦
частицы анизометрпчны, то измеряют больший и меньший размер каждой части-
цы и вычисляют среднее по большему и меньшему размерам. В этом случае
одновременно получают меру анизометричности частиц. Отношение среднеарифме-
тических или среднеквадратичных размеров частиц по большему или меньшему
диаметру даст среднее значение анизотропии. Если имеется негативный отпеча-
ток на пленке, его проектируют н# экран и все измерения проводят на экране
при удобном увеличении. Полезно проводить проектирование на экран с нане-
сенной на него измерительной сеткой.
Метод взвешивания. Негатив проектируют на экран из бумаги (в том числе
из кальки, при проектировании негатива с обратной стороны экрана), изображе-
ния частиц оконтуривают карандашом и затем вырезают оконтуренные участки.
Зная толщину и плотность бумаги и массу вырезанных «частиц», можно рассчи-
тать среднюю площадь «частицы» и ее средний размер, если форму частицы
аппроксимировать сферой. Метод вполне объективен, если граница раздела фаз,
спроектированная на экран, достаточно четка.
Методы формальной стереометрии. Эти методы широко применяемые в ме-
таллографии и биологии [38], были применены для исследования смесей поли-
меров [37].
137
Рис. IV.6. Схема определения объемной доли дисперсной фазы линейным («) и
точечным методом (б).
Объемное содержание дисперсной фазы определяют методами, основанными
на принципе Кавальсри — Акера, который может быть сформулирован следую-
щим образом: доля данной фазы в объеме смеси, на площади сечения смеси и
на линии, проходящей через площадь сечения, одинакова. На этом принципе ос-
нованы два наиболее распространенных метода: линейный метод Розиваля, когда
объемная доля фазы оценивается по суммарной длине отрезков секущих, прохо-
дящих через все измеряемые частицы данной фазы (/) (рис. IV.6, а), и точечный
метод Глаголева, когда считают число перекрестий сетки экрана (х), попавших
во все частицы дисперсной фазы (рис. IV.6, б). Зная в первом методе суммар-
ную для всех секущих, проходящих через поле микрофотографии (L), а во втс -
ром методе — общее число перекрестий экрана (А')> определяют объемную долю
дисперсной фазы как отношение 1/L или х/Х.
В линейном методе секущие могут быть непараллельны и их направление
может быть произвольным, как это предполагает метод Хэйна [38], Статистика
показывает, что средняя длина произвольно направленных секущих внутри кон-
тура частицы I связана со средним значением диаметра d соотношением d=1.16I,
откуда определяют диаметр частиц. Если частицы вытянутой формы ориентиро-
ваны преимущественно в одном направлении, то сетку секущих (см. рис. IV, а)
можно нанести не произвольно, а направленно: сначала в направлении ориента-
ции, а затем после подсчета — в направлении, перпендикулярном ориентации.
В этом случае можно получить средние значения большого и малого диаметра
частиц, что позволит оцепить их анизометричность.
Метод машинного счета. Существуют приборы [38], в котовых производится
сканирование микрофотографий или негативов Узкий пучок ^н'та, проходя че-
рез исследуемую микрофотографию нли негатив, отмечает точки изменения опти-
ческой плотности исследуемого объекта и с помощью ЭВМ, входящей в состав
комплекта, дает характеристики среднего размера, распределения по размерам
или удельной поверхности частиц. Возможно параллельное изображение на теле-
визионный экран, что позволяет следить за правильностью сканирования. Прибо-
ры такого типа, видимо, будут основой количественного исследования смесей
полимеров, no lofiiio их применению уже сейчас к биологическим объектам и
сплава м
Результаты определения размера частиц можно суммировать
следующим обра «ом
Дисперсное i ь полимеров в смеси велика, если полимеры близки
по вязкости п влас.тнчности (см. гл. III), а также имеют близкое
химическое строение, что обусловливает хорошее взаимодействие
на границе раздела фа«, большую интенсивность сдвига и в конеч-
ном счете приводит к лучшему диспергированию. Размер частиц
в хорошо перемешанной смеси каучуков колеблется в пределах
138
0,5—5 мкхц если полимеры сильно различаются пс вя ш-эстп и хи-
мическому строению, размер частиц увеличиваетсяСдо 50—70 мкм~.
Отпако было показано, что изменение размера частиц в пределах
2—80 мкм (табл. IV.1) мало влияет на механические свойства сме-
си после вулканизации [39, 40, 41].
Влияние размера частиц стеклообразного или кристаллического
полимера на свойства каучуков не выяснено. Здесь, видимо, долж-
на быть закономерность, сходная с влиянием размера частиц не-
полимерного наполнителя на механические свойства полимеров.
Было, например, установлено, что при уменьшении размера частиц
минеральных наполнителей в полимерах до 0,2 мкм прочность ра-
стет и относительное удлинение падает линейно с ростом обратной
величины диаметра частиц наполнителя [42].
< Наиболее отчетливые результаты по выяснению влияния разме-
ра'частиц полимера на свойства смеси были выполнены Берком
(цитировано по [43]) па примере модельной смеси бутадиен-сти-
рольного каучука, содержащего в качестве наполнителя сополимер
стирола, дивинилбензола и акрилонитрила (75:20:5). Сополимер
получали полимеризацией в латексе с разными количествами
эмульгатора, причем крупные частицы были синтезированы двух-
стадийной полимеризацией. Латексы отличались узким распреде-
лением частиц по размерам. Были получены сополимеры с разме-
рами частиц: 0,025; 0,03; 0,04; 0,05; 0,25; 0,45; 0,55; 0,65 мкм. Проч-
ность модельной смеси возрастает, достигая максимума при
размере частиц_0,05 мкм, эффект усиления исчезает и прочность
снижается при вв<' гении сополимера с размером частиц 0,25 мкм
и выше. Относительное удлинение и модуль мало зависят от разме-
ра частиц.
Наиболее подробно исследовано влияние размера частиц каучу-
ка на ударрпрочность пластмасс. Большинство авторов считает,
что оптимальным размером частиц каучука следует считать 0,1—
10 мкм [44—^]. Обычно ударопрочные пластмассы, получаемые
Таблица IV.1. Влияние размера частиц НК в смесях с БСК и СКД
на механические свойства ненаполненных вулканизатов
(соотношение каучуков 1:1)
Смесь Способ получения смеси Размер частиц, мкм Эластич- ность, о/ /0 Разрушающее напряжение при растяже- нии, МПа Модуль 300%, МПа
НК-БСК Из раствора 100 81,5 8.4 1,3
Плохо перемешана 10 81,5 5,9 1,6
Нормально перемешана 2 81,7 9.0 1,5
Сильно перемешана 2 81.6 8,2 1.1
НК-СКД Из раствора 80 89,7 5,5 1,4
Плохо перемешана 5 88.6 5,5 1.2
Нормально перемешана 2 87.9 7,8 1 2
Сильно перемешана 2 88,3 7,5 1,3
139
полимеризацией мономера, в котором растворен каучук, содержат
частицы каучука, в свою очередь содержащие окклюдированные
микрочастицы пластмассы [48]. Это характерно и для ударопроч-
ного ПС, и для смол АБС [45, 46, 48]. Если полимеризация ведется
в латексе прививкой на частицы каучука, то включений микроча-
стиц пластмассы в частицы каучука не происходит (один из типов
смол АБС) и оптимальный размер частиц каучука оказывается
равным 0,1—0,5 мкм, т. е. на порядок меньше [44, 47]. Полимери-
зация может проходить в массе, т. е. в растворе каучука в моно-
мере (стироле) или, например, в растворе каучука в смеси стирола
и акрилонитрила при получении смол АБС. При этом всегда обра-
зуются окклюзии пластмассы в частицах каучука, и оптимальный
размер частиц достигает 1—6 мкм [47, 48]. Здесь можно заметить,
что количество окклюзий довольно велико, поскольку при содер-
жании в исходной смеси 5—10% (об.) каучука реальное содержа-
ние дисперсной фазы каучука (вместе с окклюзиями) достигает
15—20% (об.). Характерно, что модуль сдвига в ударопрочных
пластмассах зависит не от количества введенного каучука, а от со-
держания дисперсной фазы, т. с. от общего количества каучука
вместе с окклюдированным пластиком [49, 50].
Минимальный размер частиц дисперсной фазы каучука в мат-
рице стеклообразного полимера, видимо, определяется радиусом
кривизны растущей трещины; эффект повышения стойкости к у да-
ру исчезает, когда диаметр частицы становится меньше радиуса
кривизны растущей трещины, равного приблизительно 0,1 мкм
[44]. Дополнительная информация о связи структуры ударопрочных
полимеров с их механическими свойствами приводится в обзоре
Брагау (см. статью в сб. [28]).
Оптимальный размер частиц иногда находится в довольно уз-
ких пределах. Так, в работе [51] было установлено, что в системе
типа смолы АБС наибольшей удельной ударной вязкбетью облада-
ют образцы с частицами размером 0.18 мкм, при ^сличении раз-
мера до 0,34 мкм удельная ударная вязкость несколько снижается,
а при уменьшении частиц до 0,02 мкм — падает более чем в 2 раза.
Для дисперсий каучука в каучуке наличие оптимума в размере
частиц подтверждается лишь косвенно. В работе [521 в резиновые
смеси на основе /^гс-полибутадиенового каучука СКД с вязкостью
по Муни 50 ед. вводили измельченной каучук СКД с вязкостью
по Муни 110 ед. Последний являлся высокомолекулярным каучу-
ком, но разветвленность обоих была примерно одинаковой. Каж-
дый каучук содержал необходимые вулканизующие агенты. Жест-
кий каучук был ра । дроблен на молотковой дробилке при низкой
температуре и рассеян па фракции с размерами частиц менее 1 мм,
1—2 мм, 2—3 мм и более 3 мм. Измельченную смесь на основе
жесткого каучука вводили в мягкую смесь на шпрнц-машине. Ко-
нечно, каждая фракция в процессе введения дробилась на более
мелкие частицы, однако, поскольку время смешения и условия
были одинаковыми, размер частиц в смеси был, вероятно, пример-
140
но пропорциональным исходному размеру частиц вводимого порош-
ка каучука. Оказалось, что в соответствии с теорией взаимоусиле-
пия каучуков [53] при введении жесткого каучука в мягкий со-
противление утомлению вулканизатов увеличивается, причем
максимум сопротивления утомлению достигается при введении
фракции жесткого СКД с исходным размером частиц 1—2 мм. Та-
ким образом доказано наличие оптимального размера частиц, хотя
истинный оптимальный размер частиц дисперсной фазы в вулка-
низате неизвестен. Интересно, чго, если смесь с оптимальным раз-
мером частиц пропустить еще 4 раза через шприц-машину, то в ре-
зультате дополнительного диспергирования частиц дисперсной фа-
зы сопротивление утомлению снижается.
Таким образом, чрезмерное дробление частиц дисперсной фазы
нельзя считать полезным. Это подтверждается и экспериментами
со смесями ПВХ и СКН-40, когда при предельном диспергировании
с переходом к однофазной смеси прочность при растяжении снижа-
лась по сравнению с двухфазной, т. е. по сравнению с более грубо-
дисперсной смесью [54, 55]. Исследование микроструктуры смеси
указанных полимеров в электронном микроскопе (с помощью
двухступенчатых реплик со сколов) определенно указывает на на-
личие «умеренной» степени диспергирования каучука в ПВХ в сме-
сях с максимальной стойкостью к удару.
В табл. IV.2 приведены размеры частиц в смесях эластомеров,
полученных на лабораторных вальцах при температурах, обычно
принятых для смешения указанных эластомеров.
Указанные в таблице размеры являются ориентировочными, по-
скольку они зависят (см. гл. III) не только от природы смешивае-
мых полимеров, но в большей степени от соотношения вязкости и
эластичности компонентов.
Размер частиц в смеси полимеров, полученной из раствора мед-
ленным выпариванием растворителя, составляет сотни мкм. Если
небольшое количество раствора нанести па большую поверхность,
Таблица IV.2. Средний размер зон
в смесях разных каучуков (Н—полимер в непрерывной фазе)
Компоненты Размер частиц, мкм
25 : 75 50:50 75 : 25
БСК:НК 0,3:Н 5.0:Н НО, 7
БСК:СКН 1,3:Н 2,0:Н Н:2,0
СКД:СКН 0,2:Н 0,3:Н Н:О,7
СКН* :СКН 30,0:Н 25,0:Н 11:20,0
СКН* :БСК 6,0:Н — 11:4.0
ПХП : НК 2,5:Н 4.0:Н 8,0:11
ПХП:СКД 0,3:Н 11:1,0
* СКН — бутадиен-ннтрнльный сополимер с содержанием
акрилонитрила 40%.
141
инн‘б/я.
Рис. IV.7. Изменение относительной мутности т/<р
(ф—объемная доля частиц) с изменением отно-
шения т=п!по (п и По показатели преломления
частицы и среды). Расчет проведен для показа-
теля преломления среды по=1,60 и Х=0,5893 мкм.
Цифры у кривых — размер частиц в мкм.
то в результате быстрого испарения рас-
творителя размер частиц может оказать-
ся менее 1 мкм, т. е. таким же, как и
при смешении на вальцах или в закры-
тых смесителях [36].
Размер частиц оказывает большое
влияние на оптические свойства смесей,
их прозрачность, что особенно важно для
ударопрочных пластмасс, часто исполь-
зуемых в светотехнике. По расчетам
[56], максимальная мутность смеси при отношении показателей
преломления частицы и среды (zn) от 0,93 до 1,20 характеризует
смеси с размером частиц соответственно от 5 до 1 мкм. На рис.
IV.7 показана сильная зависимость мутности системы от величи-
ны т. Если размер частиц меньше 50—70 нм, то они очень мало
рассеивают свет, и поэтому блок-сополимеры (в том числе термо-
эластопласты) прозрачны, несмотря па большую разность в пока-
зателях преломления. Обычно процесс полимеризации для получе-
ния ударопрочных пластмасс проводят таким образом, что показа-
тели преломления практически равны. Так, в ударопрочном
материале С1А [57], представляющем собой смесь (с включением
привитого сополимера) бутадиен-метилметакрилатного каучука и
сополимера стирола, метилметакрилата и акрилонитрила, размер
частиц каучука 0,1- 0,8 мкм, но равенство показателей преломле-
ния частиц и среды обеспечивает полную прозрачность материала.
Показатель преломления в каучуке (как и удельный объем) па-
дает с температурой быстрее, чем в стеклообразной матрице, и по-
этому т непрерывно меняется. Для того, чтобы сохранить прозрач-
ность материала в широком интервале температур, необходимо
добиться высокой дисперсности частиц (рис. IV.8), что не всегда
позволяет сохранить хорошую стойкость к удару.
В смесях полимеров частицы дисперсной фазы распределены
статистически, что предполагает распределение пе только по раз-
мерам частиц, но и распределение расстояний между ними. В блок-
сополимерах в случае практически монодисперсного распределения
блоков, особенно каучуковых, по молекулярным массам и получе-
ния пленки из расIвора при медленном удалении растворителя до-
стигается идеально равномерное распределение частиц дисперсной
фазы в матрице основного полимера (рис. IV.9) [57]. Если в объем
бутадиен-стирольного блок-сополимера с идеально равномерным
распределением частиц добавить оба гомополимера в тех же соот-
142
Рис. 1V.8. Зависимость прозрачности ///о от тем-
пературы для смеси полиизопрена и полиакрило-
нитрила (Л = 0,5893 мкм, толщина образна около 0,6
3 мм): в
1 — размер частиц 0,1 мкм; 2 — размер частиц 1,0 мкм. \
---------------------------------------- 0,4
ношениях, то первоначальная структура
блок-сополимера остается практически °
неизменной, но размер доменов ПС уве-
личится 158]. Детальные исследования
структуры и свойств смесей двух гомополимерив с блок-сополиме-
ром соответствующего состава проведены в работе [59].
Была сделана попытка рассчитать для блок-сополимеров раз-
мер доменов полистирола при условии сохранения среднестатисти-
ческой конформации блоков и минимума избыточной поверхност-
ной энергии. При этом учитывали, что между фазами имеются хи-
мические связи и полное разделение системы на две фазы (дости-
жение подлинного минимума свободной эпер1ии) невозможно [60].
Ввиду относительно низкой концентрации химических связей на
межфазной границе (примерно одна связь ПС--ПБ на 10 нм2)
можно считать, что многие закономерности струкгурообразованпя
в блок-сополимерах справедливы и для смесей полимеров с разме-
ром частиц, близким к таковым в блок-сополимерах. Из рис. IV. 10
[60] видно, что для блок-сополимера с молекулярной массой бло-
ков ПС 14-10е минимуму свободной энергии отвечает радиус ча-
стиц около 9 нм при толщине переходного слоя 4 нм. Расчет по-
казал, что сохранение среднестатистических конформаций невоз-
можно без образования переходного слоя (см § 3). Очевидно, что
рост молекулярной массы блоков ПС должен привести к увеличе-
нию размеров их статистических клубков н к росту размеров до-
менов.
Релаксационные процессы в полимерах протекают медленно пз-
за высокой вязкости среды. Поэтому фор”а частиц в смесях поли-
меров, полученных перемешиванием расплавов, является не строго
Рис IV.9. Микрофотография среза блок-сополимера бутадиена и стирола (32 : 68)
(а) и светооптическая диаграмма рассеяния (б). Масштаб на (а) 100 нм.
143
Рис IV 10. Зависимость свободной энергии фа-
зового расслаивания от радиуса R частиц дис-
персной фазы — доменов ПС в матрице ПБ
(А/? — толщина переходного слоя при 7=
=373 К; разность параметров растворимости
бпс-6пб=0,74; молекулярные массы блоков:
14-10*—70-103—14-103). На оси абсцисс от-
ложены R 101, нм, на кривых — AR-102 нм.
сферической, а ани.зомстричной. Иног-
да анизометричность формы становит-
ся столь значительной, что в струе рас-
плава, выходящей из капилляра вис-
козиметра, |могут возникнуть волокна
тисп-ерсной фазы, имеющие очень
большую длину.
В смеси ПВХ и бутадиен-нитри ль-
пого каучука волокнистая структура характерна для системы с
наилучшими механическими свойствам^ |61].
Волокна дисперсной фазы в матрице полимера, образующего
непрерывную фазу, были обнаружены при непосредственном наб-
людении в электронном и световом микроскопе в смеси ПС и
ПММА. В работе [62] был сделан вывод, что «частицы дисперсной
фазы при течении... приобретают вид волокон, ориентированных в
направлении течения». Результаты этой работы изложены также в
статье [63]. Волокна были обнаружены в смесях ПС с полиолефи-
нами [64], а также в смесях полиамидов с полиолефинами [65].
В работах [64, 65] струю, вышедшую из капиллярного вискозимет-
ра, обрабатывали селективным растворителем, растворявшим по-
лимерную матрицу. В результате выделяли волокна дисперсной
фазы, длину и диаметр которых можно было непосредственно оп-
ределить. Подробное исследование явления волокпообразования
на примере смесей полиоксиметилена и сополиамида было прове-
дено в ряде работ, например [66, 67].
Если диаметр капель велик, то вместо волокон образуются
пленки или даже коаксиальные цилиндры в результате перехода
капель при течении в тонкий кольцевой слой. При возникновении
пленочной структуры или структуры концентрических слоев реа-
лизуется механизм слойного течения, когда наиболее интенсивная
деформация происходит в маловязком слое (см. Г. Допперт и
В. Овердии в сб. |28]). Это приводит к сильному снижению вязко-
сти смеси при сравнительно малом содержании маловязкого по-
лимера дисперсной фазы. Если такую смесь дополнительно переме-
шать для увеличения дисперсности частиц, то слойное течение не
реализуется и сдвиговые деформации более равномерно распреде-
ляются в потоке, в том числе и в более вязкой среде полимера,
содержание которого преоблатает. Вальцевание смеси в этом слу-
чае приводит к росту вя «кости расплава почти на целый десятич-
ный порядок [63, 68].
144
Рис. IV.11. Анизотропия прочности в смесях ПЭ—
бутилкаучука:
О — без прогрева, сразу после выхода из экструдера;
,Д — после прогр=ва при ЬО °C □ — после прогрева при
120 °C.
Анизометричность частиц дисперсной
фазы, крайним выражением которой яв-
ляется образование волокнистой с грук-
туры, ориентированной в направлении
деформирующих усилий, приводит к ани-
зотропии механических свойств. Так, было показано [69, 70], что
относительный прирост прочности вдоль оси ориентации по срав-
нению с прочностью в поперечном направлении в вулканизатах
смесей СКД -СКН-18 составляет 20%, а в вулканизатах отдельно
взятых СКД или СКН-18— всего 4—6%. В еще большей степени
эффект анизотропии прочности проявляется для смесей с участием
кристаллических или стеклообразных полимеров, когда анизомет-
ричные частицы дисперсной фазы при выходе из экструдера фик
сируются благодаря быстрой кристаллизации. Так, смесь ПЭ—БК
(1:1) имеет прочность вдоль направления экструзии (без вытяж-
ки), более чем в 2 раза превышающую прочность в перпендику-
лярном направлении. В тех же условиях экструзии пленки из са-
мих ПЭ или БК практически не обладают анизотропией прочности
(рис. IV.11, [70]).
Анизометричность частиц дисперсной фазы в смесях полимеров
является важной особенностью этого класса систем наряду со спо-
собностью частиц менять форму при переработке и занимать пре-
имущественную ориентацию в направлении завершающих дефор-
маций при экструзии, литье, вальцевании и пр. Для ударопрочных
полимеров характерны [71] понижение стойкости к удару и повы-
шение хрупкости при получении образцов литьем под давлением
при пониженных температурах. В этих условиях, когда усилия
сдвига ьелики и деформация частиц каучука также велика, а ре-
лаксация формы частиц затруднена, повышается ориентация ча-
стиц в направлении литья и облегчается рост трещин в перпенди-
кулярном направлении. Повышение температуры литья приводит
к меньшей деформации частиц в направлении сдвига и создает ус-
ловия для их последующей усадки и дезориентации, что обеспе-
чивает повышение стойкости к удару образца в целом. По этой
причине данные испытания образцов вдоль направления ориента-
ции лучше отражают истинные свойства материала.
Присутствие в смеси привитого или блок-сополимера обеспечи-
вает достаточно сферическую фирму частицам, которые в уелови
ях смещения в отсутствие сополимера были неправильной формы
[72] Наличие привитых и блок-сополимеров в ударопрочных пласт-
массах сводит к минимуму нежелательные изменения свойств,
вызванные ориентацией частиц каучука в процессе переработ-
ки [71].
10—244
145
Во многих случаях волокнообразовапие в дисперсной фазе сме-
си полимеров является положительным явлением, вызывая эффект
армирования [65—67] моноволокна и упрочнение смеси даже без
преимущественной ориентации волокон дисперсной фазы. Эго по-
казано па примере смеси ПВХ и бутадиен-нитрильного сополиме-
ра [73] и на других примерах [28]. Волокнообразовапие частицами
дисперсной фазы приводит к возможности создания напряженных
конструкций. Так, если порошок ПЭ (15 20%) ввести на холоду
в резиновую смесь, завулкапизовать, а затем растянуть в нагретом
состоянии и охладить, то ориентированные при деформации нагре-
той смеси волокна ПЭ при охлаждении армируют резину и она
останется растянутой при снятии нагрузки [74]. Трубки из такой
резины, надетые на проволоку при нагревании, сжимаются и плот-
но охватывают проволоку, служа надежной изоляцией.
Частицы неправильной формы могут возникать и при получе-
нии пленок из растворов [36].
§ 3. ОБРАЩЕНИЕ ФАЗ
В смесях полимеров, как и во всякой коллоидной системе, при
изменении соотношения компонентов происходит обращение фаз.
При добавлении малого количества одного полимера к другому об-
разуется дисперсия добавленного полимера в матрице основного,
преобладающего компонента. Если же количество добавляемого
компонента существенно больше половины объема, он образует
матрицу, а исходный компонент становится непрерывной фазой.
Такие явления очень характерны для нестабилизированных эмуль-
сий низкомолекулярных веществ типа «вода в масле», когда при
достижении концентрации воды 50% (об.) происходит переход
эмульсии в систему «масло в воде», на что указывает резкое уве-
личение электропроводности системы [75].
В смесях полимеров при высокой вязкости системы и низком
.межфазном натяжении анизометричные частицы не претерпевают
быстрой коалесценции, что приводит к возникновению твердооб-
разиых эмульсий подобно структурообразованию в суспензиях кол-
лоидных частиц неправильной формы [76]. По терминологии
П. А. Ребиндера структуры, возникающие прй коагуляции суспен-
зий, когда частицы коагулюма разделены прослойками жидкой
дисперсионной среды, называются коагуляционными, а при нали-
чии непосредственных связей между дисперсными частицами в
коагулюме — конденсационными (или кристаллизационными).
С точки зрения механических свойств переход от коагуляционной
к конденсационной структуре сопровождается возникновением яр-
ковыраженной хрупкости системы. Рассмотрим явления такого
типа на примере смесей полистирола с каучуками.
ПС с молекулярной массой 3-105 смешивали с разными каучу-
ками при 130°C, а затем прессовали пластинки при 150°C. Опре-
деляли механические свойства смесей и скорость их набухания
146
Рис. IV. 12. Зависимость коэффициента усиления
(по модулю упругости) смесей СКД — ПС от со-
держания ПС:
О — смесь получена вальцеванием при 25 °C с последую-
щим прессованием прн 25 °C; А — то же, прессование при §
150 °C; □ — вальцевание при 130 °C, прессование при &
150 МС. у
в диметилформамиде, в котором набуха- §
от только ПС. На рис. IV. 12 показана
зависимость коэффициента усиления сме-
СИ ПО модулю упругости (К = £см/£скд) I
от содержания ПС. Модуль определяли
по величине предельной деформации, раз-
вивающейся под действием заданной на-
грузки во времени. Видно, что уже при
содержании 20% (об.) ПС начинается
нарастание модуля, что указывает на сближение концов анизомет-
ричных частиц ПС и образование каркаса пластмассы в матрице
каучука. Характерно, что (тот же рисунок) при введении ПС на
холодных вальцах с последующим холодным илй горячим прес-
сованием усиления практически не происходит и некоторый эф-
фект замечен только при большом содержании ПС. Это свиде-
тельствует о большом значении анизометричной формы частиц ПС
для обеспечения эффекта усиления, поскольку частицы ПС в ис-
ходном состоянии и 'В смеси, полученной на холоду, были сфери-
ческими. В то же время смеси даже с содержанием ПС до 40%
обладали некоторой эластичностью, обеспечивающей удлинение
при разрыве до 80 -100%. Это указывает па существование в сме-
си коагуляционных структур, в которых частицы ПС разделены
прослойками каучука. Прослойки хорошо видны на микрофотогра-
фии и имеют толщину до 0,1 мкм (рис. IV.13).
Переход к конденсационным структурам происходит при содер-
жании ПС 50% и более. На рис. IV. 14 показана зависимость ско-
рости набухания смеси в ДМФ от содержания ПС. Видно, что ско-
рость набухания достигает предела, равного скорости набухания
чистого ПС, только при содержании ПС 50% • Разрывное удлине
ние при этом падает до нескольких процентов. Все это свидетель-
ствует об образовании единого и непрерывного каркаса ПС, когда
его содержание в смеси составля-
ет 50%. Исследование микро-
структуры смесей, содержащих
20—50% ПС, при умеренном
увеличении, когда прослойки кау-
чука между частицами нс видны,
а виден полистирольнын каркас
Рис. IV.13. Электронно-микроскопичв
скан фотография смеси 11С — СКД
(4:6), показывающий наличие прослоек
СКД между частицами ПС.
10'
147
Рис. IV.14. Зависимость скорости набу-
хания смеси ПС — СКД в диметилформ-
амиде от содержания ПС (обозначения
см. на рис. IV.12):
а — угол наклона кинетической кривой набу-
хания Q—HF Л, где Q — количество погло-
щенного ДМФ. %; t — время, с).
в целом, приводит к ошибочному
выводу о том, что обе фазы не-
прерывны.
Приведенный здесь пример ти-
пичен и указывает та необходимость применения нескольких мето-
дов контроля |непреры.ьности каждой фазы, чтобы судить о том,
произошло ли обращение фаз в смеси. Возникноьение коагуля-
ционного каркаса дисперсной фазы, приводящее к тому, что на
микрофотографии обе фазы представляются непрерывными, в со-
четании со значительным измененном механических свойств в за-
висимости от состава смеси, наблюдается в интервале 30—70% со-
держания каждого полимера. Если 'вязкость расплава одного из
полимеров значительно меньше .вязкости другого, то маловязкий
полимер может образовать коагуляционный или конденсационный
каркас уже при содержании свыше 10% [28, с. 46, 47].
Фазовая структура смеси полимеров, которая выглядела на
микрофотографии как переплетение двух непрерывных фаз, наблю-
далась неоднократно. По-видимому, Уолтерс и Кейт первые свя-
зали возникновение такой структуры с равенством вязкостей сме-
шиваемых полимеров [40]. Вывод о наличии двух непрерывных фаз
в смеси (па примере смеси ПЭ—ПП) был сделан на основании
изучения температурной зависимости модуля упругости в работе
[1]. Впоследствии непрерывные фазы в смесях полимеров наблю-
дались неоднократно на основании изучения микроструктуры сме-
сей. Естественно, что заключения подобного рода достаточно субъ-
ективны, когда дело касается микрофотографий смеси с сильно1
анизомегричными частицами неправильной формы при соотноше-
нии полимеров от 3 : 7 до 7 : 3.
Обращение фаз в смесях полимеров возможно при изменении
условий перемешивания. Вопрос этот в настоящее время совер-
шенно не изучен Важность этого вопроса можно пояснить, напом-
нив, что смесь каучук — пластик обладает комплексом свойств
каучука или пластика в зависимости от того, образована ли непре-
рывная фаза соответственно каучуком или пластиком.
Более полярный полимер обнаруживает более сильную зависи-
мость вязкости от температуры, чем полимер неполярный. Такая
зависимость типична в том смысле, что энергия активации вязкого
течения, определяющая температурную зависимость вязкости, ра-
стет с ростом полярное!и полимера. Можно подобрать пару поли-
меров ПММА и ПИБ так, чтобы ПММА при 160°C был более
вязким, чем ПИБ, а при повышении температуры до 180 °C оказал-
148
ся менее вязким, чем ППБ. Это приводит к тому, что при темпе-
ратуре смешения 180 °C непрерывной фазой оказывается ПММА,
а при 160 °C - ПИБ. Подтверждением такого заключения является
наличие смесей, для которых кривые нагрузка — удлинение и гис-
терезисные кривые существенно различаются, если смесь одного и
того же состава получать при разных температурах (см. гл. III)
Если смесь ПММА—ПИБ. полученную при 180°C, когда, судя
по свойствам, непрерывной фазой является ПММА, поместить в
растворитель, растворяющий селективно только ПММА (напри
мер, в метилэтилкетон), то вся смесь растворяется и остается
только порошок дисперсной фазы ПИБ. В то же время такая же
смесь, полученная при 160 °C, где непрерывной фазой является
ПИБ, сохраняет целостность в метилэтилкетоне и из нее вымыва-
ется только дисперсная фаза ПММА.
На микрофотографиях смесей, полученных при разных темпера-
турах, видно, что для одной и той же пары полимеров и при не-
изменном их соотношении дисперсная фаза состоит из частиц
полимера с большей в данных условиях смешения вязкостью. Та
ким образом, наблюдаемый эффект может служить как основой
для выбора значений вязкости смешиваемых компонентов, так
и для объяснения кажущихся «загадочными» результатов смеше-
ния полимеров, когда небольшое изменение условий перемешива-
ния, главным образом температуры, оказывает решающее влияние
на механические свойства композиций.
Недостаточно освещена роль вязкоупругости в обращении фаз.
Имеется одна работа [77], в которой свойства полимеров, от ко-
торых зависит фазовая структура смеси, предлагали оценивать по
величине отношения G”/G'. Если G''IG'^>\, то полимеры смеши-
ваются хорошо (мала доля упругой деформации в процессе сдви-
га), если G"/G' = 0,4 и меньше, полимеры смешиваются плохо.
Если свойства полимеров различаются, то полимер, у которого
G"!G'^>\, легче образует непрерывную фазу, чем полимер с ма-
лым G"IG'.
Очевидно также, что легче всего обращение фаз можно на-
блюдать для полимеров, сильно различающихся по энергии акти-
вации вязкого течения. Это не всегда предполагает различие в
«полярности», не определенной количественно. Так, в той же рабо-
те [77] явление обращения фаз наблюдали на примере смеси поли-
бутадиена и этилен-пропиленового тройного сополимера.
Скорость смешения (число оборотов ротора смесителя) влияет
менее заметно на обращение фаз, поскольку практически она из-
меняется в небольших пределах. Большее влияние температуры на
обращение фаз, чем скорости перемешивания, непосредственно сле-
дует из принципа температурно-временной эквивалентности, когда
эффект влияния температуры эквивалентен эффекту, вызванному
изменением логарифма скорости деформации. Заметим, что в тех
случаях, когда скорость перемешивания может изменяться в широ-
ких пределах, это изменение определенно вызывает обращение
149
фаз. Так, при получении ударопрочного ПС методом полимериза-
ции стирола в стирольном растворе каучука, сначала образуется
дисперсия ПС в матрице каучука (более вязкого на начальных
стадиях полимеризации, когда обращение фаз возможно), а затем
дисперсия каучука в ПС при интенсивном перемешивании, когда
в результате интенсивных сдвиговых деформаций происходит пе-
реход более вязкого каучука в дисперсную фазу [71].
Комплекс физико-механических и химических свойств смеси по-
лимеров зависит в первую очередь от природы непрерывной фазы,
в связи с чем определение природы непрерывной фазы представ-
ляется важнейшим этапом в анализе фазовой структуры смеси.
Основой для такого анализа может служить любое свойство, за-
метно зависящее от природы непрерывной фазы.
Модуль упругости меняется с составом смеси по S-образной кривой, если
в смеси наблюдается обращение фаз (рис. IV.15). Видно, что экспериментальные
точки отклоняются от кривых, рассчитанных по уравнению Такаянаги [78] для
системы «капли полимера 1 в матрице полимера 2» (кривая /) и для системы
«капли полимера 2 в матрице полимера 1> (кривая г). Область отклонений от
кривых 1 и Г можно считать областью составов, в которой происходит обраще-
ние фаз, имея в виду, что наряду с полным контактом частиц дисперсной фазы
возможен и контакт через прослойки полимерной матрицы. Экспериментальные
точки хорошо совпадают с кривыми, построенными по уравнениям Пакулы —- Кры-
шевского [79] и Кернера [80], отражающим обращение фаз в системе и учиты-
вающим изменения модуля контактированием частиц непосредственно и через про-
слойки (особенно хорошо совпадает кривая Пакулы—Крышевского). Как пока-
зывает пример, приведенный на рис IV. 15, хороший результат можно получить и
для системы каучук—каучук, если температура, при которой определяют мо-
дуль, лежит в промежутке между температурами стеклования каучуков, что
обеспечивает сильное изменение модуля с составом и уверенное определение об-
ласти обращения фаз.
СКС-ЗОЛРК 20 00 60 80 100
Рис. IV. 15. Сравнение экспериментальных (точки) и расчетных значений (кри-
вые) модуля упругости смеси ПБ— ПИП с узким ММР (температура опыта —
ГС(11ИП)= 68°С, Тс(ПБ)»—ЮО’С):
Л /'-—расчет по Такиянлгн; 2 — по Пакуле — Крышевскому; 3—по уравнению полиагрега-
тивной модели Кернера.
Рис. IV.16. Сопротивление отслаиванию (Р) пленки каучука СКЭПТ-50 (кри-
вая /) и каучука СКС ЗОАРК (кривая 2) от пленок их смесей с разными соот-
ношениями каучуков (пункт ир— сопротивление отслаиванию при соотношении
каучуков на поверхности смеси, равном их соотношению в объеме).
150
Вязьость расплава или раствора зависит от состава по S-образпой кривой,
если полимеры монодисперсны и не проявляют аномалий вязкости, а также в
случае определения начальной ньютоновской вязкости (подробнее см. гл. V).
Прочностные характеристики (предел текучести, прочность при растяжении,
удлинение при разрыве) меньше реагируют на обращение фаз, поскольку разру-
шение связано обычно с большими деформациями, приводящими к изменению
структуры смеси в такой степени, что это маскирует эффект обращения фаз.
Относительное удлинение при разрыве и предел текучести более пригодны для
определения природы непрерывной фазы, чем прочность при растяжении.
Адгезия и смачивание. В низкомолекулярных эмульсиях природу непрерыв-
ной фазы определяют по растеканию капли эмульсии на подложке из парафина.
Этот прием не пригоден для смесей полимеров. Вместо этого на поверхность
пленки из смеси полимеров можно наносить каплю жидкости, угол смачивания
которой для полимеров, входящих в состав смеси, различен. Тогда, зная угол
смачивания капли на пленке смеси и сравнивая его с углами смачивания на каж-
дом полимере, можно определить природу непрерывной фазы, как это было сде-
лано на примере ПЭТФ с ПП или с ПС в работе [81].
Можно исследовать адгезию смеси к полимерам, входящим в ее состав. Ад-
гезия смеси полимеров Л+В к субстрату из полимера Л велика и практически
равна аутогезии полимера Л, если полимер Л является непрерывной фазой. Ад-
гезия же смеси А-1-В к субстрату из полимера В должна быть низкой, как это
часто бывает в случае адгезии полимеров, обладающих малым сродством друг
к другу. На рис. IV. 16 [821 показана зависимость адгезии смеси бутадиеи-сти-
рольпого каучука СКС-ЗОАРК к этилен-пропиленовому тройному сополимеру
СКЭПТ-50 от соотношения полимеров в смеси (адгезия осуществлялась пооче-
редно к каучуку СКС-ЗОАРК и к СКЭПТ). Полученные данные показывают, что
при введении уже небольших количеств СКЭПТ последний образует непрерыв-
ную среду в смеси, благодаря чему адгезия смеси к субстрату из СКЭПТ прак-
тически равна аутогезии самого СКЭПТ.
Метод адгезии имеет ограничения. Он неприменим при очень малой адгезии
и аутогезии исследуемых полимеров, а также при малой когезии полимеров, ког-
да распространение трещины при расслаивании идет не по границе раздела по-
лимеров, а по фазе одного из полимеров в непосредственной близости от грани-
цы раздела фаз. Преимуществом метода является возможность контроля полу-
ченных данных: область обращения фаз должна быть одинаковой в эксперимен-
те по расслаиванию склеек смеси как с одним полимером, так и с другим. Если
результаты определения адгезии к одному из полимеров не согласуются с ре-
зультатами определения адгезии к другому полимеру, это указывает на несовпа-
дение поверхности расслаивания с поверхностью раздела.
Электропроводность. Для низкомолекулярных эмульсий типа «масло — вода*
или «вода — масло» измерение электропроводности является классическим мето-
дом определения непрерывной фазы. Метод применим и для смесей полимеров
при условии большого различия электропроводности смешиваемых компонентов.
Чем меньше точность измерения электропроводности, тем больше должно быть
различие величин электропроводности исходных полимеров, чтобы по получае-
мой S-образной кривой электропроводность — состав можно было надежно оп-
ределить область обращения фаз.
Диффузия и проницаемость. По этим параметрам также можно определить
природу непрерывной фазы. Пример такого определения был приведен выше для
случая набухания смеси каучуков с ПС в парах метилэтилкетона. Скорость на-
бухания смеси становится равной скорости набухания ПС в тот момент, когда
последний образовал непрерывную фазовую сетку в смеси. В литературе приво-
дятся и другие данные по исследованию фазовой структуры смеси полимеров
методом диффузии и проницаемости (подробнее см. гл. V).
Другие методы определения природы непрерывной фазы могут быть приме-
нены для тех или иных конкретных систем, исходя из изложенных общих прин-
ципов. Так, например, способность полимеров к воспламенению зависит сущест-
венно от того, образовал ли защитный полимер непрерывную фазу в смеси.
Внедрение ПВХ в смолы АБС в количестве около 50% (об.) приводит к тому,
151
что смесь становится негорючей; аналогичный эффект достигается при введении
хлорированного ПЭ в обычный ПЭ для снижения горючести кабельной изоля-
ции. Наибольшая эффективность защитного действия достигается тогда, когда
негорючий полимер образовал непрерывную фазу в смеси.
Насыщенный полимер, вводимый в ненасыщенный, повышает озоностойкость
смеси, если в ней возникает непрерывная фаза ненасыщенного полимера [82].
§ 4. МЕЖФЛЗПЫП СЛОИ
Силы, действующие па молекулу, находящуюся на поверхности
раздела фаз, различны в направлении каждой фазы. Это простое
следствие различия свойств контактирующих фаз. По этой при-
чине возникают специфические поверхностные силы, которые вме-
сте с тепловым движением частиц, образующих систему, приводят
к нарушению геометрической правильности границы раздела. Гра-
ница раздела фаз представляет собой не плоскость и не поверх-
ность, а поверхностный слой определенной толщины б, зависящей
от свойств контактирующих фаз. Учение о поверхностных слоях,
изучение их структуры и свойств является центральным вопросом
коллоидной химии |9|.
Понятие поверхностного (или межфазного) слоя ввел впервые
Гиббс, который называл поверхностный слой «поверхностью раз-
рыва», имея в виду разрыв непрерывности изменения термодина-
мических функций в этом слое. По Гиббсу, «поверхность разры-
ва — неоднородный тонкий слой, разделяющий объемные фазы и
обладающий, следовательно, конечной толщиной и объемом». Учи-
тывая конечную толщину и определенный объем поверхностного
слоя, Гуггенгейм ввел понятие «поверхностная фаза». Границы
поверхностной фазы определить точно нельзя, поскольку она пе-
реходит в объемную фазу непрерывно и, строго говоря, свойства
поверхностного слоя идентичны свойствам фазы лишь на бесконеч-
ном удалении от границы раздела. Поэтому толщина слоя 5 от-
считывается как расстояние, на котором отклонение свойств или
состава слоя от свойств или состава объемной фазы становится
незначительным, меньше определенной заданной величины. При
этом следует учитывать, что отдельные свойства могут изменяться
при удалении от середины слоя самым различным образом, по
разным законам, и по этой причине величина б зависит от того, по
изменению какого свойства она была определена.
Если толщина слоя меньше радиуса кривизны поверхности
раздела, то его состояние наряду с энергетическими параметрами
задается поверхностью раздела фаз:
du’ = T'dSx + ods Н X (1}
где и — внутренняя энергия; S — энтропия; Т — температура; р. — химический по-
тенциал; п — число компонентов; о — поверхностное натяжение; s — удельная по-
верхность (индекс s относит данную величину к поверхностному слою).
152
Это уравнение аналогично уравнению состояния для объемной
фазы а:
dua = Пза - ПУ0 + <-2)
i
где вместо величины удельной поверхности фигурирует объем
фазы V. Внутренняя энергия системы равна сумме иаи us.
Можно определить зависимость между поверхностным натяже-
нием системы и толщиной поверхностного слоя: увеличение по-
верхностного натяжения (уменьшение термодинамического сродст-
ва фаз) приводит к уменьшению толщины поверхностного слоя.
Надежные экспериментальные данные по определению толщи-
ны поверхностных слоев немногочисленны и относятся к границам
раздела фаз, находящихся в разных агрегатных состояниях. Так,
на границе раздела раствор соли воздух в жидкой фазе имеется
поверхностный слой толщиной 0,5 нм [83], на границе раздела
кварц — полидиметилсилоксановый олигомер — слой олигомера
толщиной 10 нм [84]. Определение усилия, необходимого для
сближения полированных кварцевых пластин с жидкой прослой-
кой фурфуролацетонового мономера между ними, показало нали-
чие слоя с повышенной вязкостью толщиной в 0,2_мкм J85] _Эти
данные приведены в качестве примера существования поверхност-
ных слоев иногда огромной протяженности в низкомолекулярных
системах.
Межфазцый слой на границе раздела полимер — полимер име-
ет особенности строения, обусловленные высокомолекулярной при-
родой контактирующих фаз.
В гл. I (см. рис. 1.7) было показано, что растворимость поли-
мера в полимере ничтожно мала и возрастает с уменьшением мо-
лекулярной массы, лишь начиная с некоторого предела. Этот пре-
дел для таких полимерных пар, как ПС—ПИП или ПС—ПММА
совпадает с областью перехода от полимера к олигомеру. Когда
молекулярная масса одного из указанных полимеров становится
меньше этого предела и приближается к молекулярной массе ки-
нетического сегмента, растворимость таких малых молекул во вто-
ром полимере становится неограниченной. Так, ПС неограниченно
растворяется в ПИП и ПММА при достижении молекулярной мас-
сы соответственно 500 и 1600. Отсюда понятно, почему мономеры
подавляющего большинства полимеров смешиваются неограничен-
но, в то время как соответствующие полимеры несовместимы. Паи
более характерный пример — полиизопрен и полибутадиен. Гид-
рированные мономеры этих полимеров, которые могли бы моде-
лировать элементарный отрезок цепи полимера являются жидко-
стями, смеси которых по свойствам близки к идеальным, подчи-
няющимся закону Рауля. Соответствующие полимеры с молеку-
лярной массой в несколько сотен тысяч смешиваются друг с
другом в количестве менее 1%, т. е. практически взаимопераство-
римы [27].
153
Особенности теплового движения в полимерах связаны, как
известно, с наличием кинетических сегментов, перемещающихся
как единое целое в элементарном акте теплового движения. Пере-
ход макромолекул из слоя одного полимера в слой другого невоз-
можен ввиду практической взаимонерастворимости полимеров. Пе-
реход же части сегментов данной макромолекулы возможен, по-
скольку молекулярная масса сегментов соответствует области
значительной их растворимости в другом полимере. Таким обра-
зом, большинство полимерных пар, компоненты которых практиче-
ски взаимонерастворимы должны быть способны к взаимораство-
рению сегментов в межфазном слое. Это дало возможность выска-
зать гипотезу о сегментальной растворимости полимеров [86, 87,
28], согласно которой на границе раздела полимер — полимер об-
разуется межфазный слой, представляющий собой своеобразный
«раствор» сегментов одного полимера в другом.
Следует подчеркнуть, что явление сегментальной растворимо-
сти характерно именно для поверхности раздела. Глубина взаимо-
проникновения сегментов, т. е. толщина слоя, должна зависеть от
изменения термодинамических параметров поверхностного слоя
контактирующих полимеров в соответствии с общим уравнением
Гиббса [1], примененным к полимерным объектам. Растворение
сегментов одного полимера в другом при малом сродстве компо-
нентов друг к другу может осуществляться, видимо, главным об-
разом при увеличении поверхностной энтропии вследствие перехо-
да конформаций поверхностных молекулярных клубков от менее
вероятных к более вероятным. Схематически этот переход показан
на рис. IV.17. Поверхностный характер явления сегментальной
растворимости указывает на относительность и качественный ха-
рактер сопоставления с растворимостью олигомеров в полимерах.
При растворении олигомера в полимере процесс определяется из-
менением энтропии системы в целом, растущей вследствие перено-
са большого числа малых молекул олигомера в полимер, а в меж-
фазном слое процесс определяется соотношением сродства поли-
Рис. IV.17. Схема более вероятного расположения поверхностных клубков мак-
ромолекул несовместимых полимеров за счет сегментального растворения и об-
разования межфазного слоя
154
мер—-полимер и ростом энтропии при данном изменении формы
макромолекулярных клубков в межфазном слое.
Изменение энергетического состояния одного сегмента мало
влияет на состояние макромолекулы в целом. Так, изменение эн-
тропии макромолекулы при кристаллизации в расчете на сегмент
при вхождении в кристалл одного сегмента незначительно и резко
возрастает при участии последующих сегментов в построении кри-
сталлической решетки. Эта аналогия наглядно показывает, что на-
чало сегментального растворения полимеров в области их контак-
та качественно аналогично растворению соответствующих пар оли-
гомер — полимер. Очевидно, что если к переходу в другую фазу
сегмента, то процесс сегментального растворения в межфазном
способны отрезки макромолекулы размером меньше кинетического
слое не сможет осуществиться. Если сегментальная диффузия воз-
можна, то ее предел определяется сродством полимеров и измене-
нием энтропии приповерхностных клубков в результате изменения
их конформации.
Представления о сегментальной растворимости дали возмож-
ность объяснить многочисленные явления, наблюдаемые при кон-
такте полимер — полимер, которые раньше объясняли диффузией
макромолекул, возрастающей с увеличением сродства полимеров.
Очевидно, что при взаимной растворимости полимеров менее 1 %
трудно ожидать взаи.модиффузии макромолекул при тех значениях
вязкости системы, которые наблюдаются на опыте. Времена ре-
лаксации сегментов невелики [28] и это приводит к достаточно
быстрому возникновению слоя сегментальной растворимости после
установления контакта полимер — полимер.
В отличие от низкомолекулярных систем, где поверхностный
слой образован в результате диффузии молекул, атомов или ионов,
в системах полимер — полимер межфазный слой образован в ре-
зультате взаимоднффузии сегментов, которая обусловлена возмож-
ностью сегментальной растворимости полимеров.
Выше отмечалось, что в системах с развитым межфазным сло-
ем наблюдается малое межфазное натяжение. Зависимость меж-
фазного натяжения а от толщины слоя 6 может быть получена
формально, исходя из представлений Гиббса о поверхностном слое,
как это было сделано например в работе [88]:
б — (°2 — *2°° / 3)
RTra/r^f
где п и г2 — степени полимеризации каждого полимера; Г|о=—мольный объем
сегмента; с2— поверхностное натяжение одного из полимеров; о12— межфазное
натяжение; <pi — объемная доля компонента 1 в межфазном слое.
Чем меньше о, тем больше б. Таким образом, толщину меж-
фазного слоя можно оценить, измерив межфазное натяжение на
границе раздела полимер — полимер.
155
Межфазное натяжение определяется следующим выражением:
+ —?ФЙ—2<] р
(4)
где Пи — межфазное натяжение между фазами 1 и 2; щ и а2— соответственно
поверхностные натяжения фаз 1 и 2; Ф'! и ‘1>г' выражают вклад дисперсионного
и диполь-дипольного взаимодействия на границе раздела фаз.
Предложено и экспериментально обосновано [89] выражение
для U>d и Фр:
201Оз
ф4 =
(5)
где и а?—компоненты поверхностного натяжения каждой фазы, обусловлен-
ные дисперсионным и диполь-дипольным взаимодействием.
Отсюда выражение для oi2:
. d d
, 4OJO2
Giz — Oj + as — d
"Г O2
(6)
°? F°a
Уравнение (6) показывает, что чем ближе значения полярно-
стей фаз, тем меньше межфазное натяжение. Оно позволяет также
оценить вклад полярного взаимодействия, если измерено межфаз-
ное натяжение на границе раздела полярного полимера с неполяр-
ным, в котором о*—«-0. Экспериментальные значения межфазного
натяжения на границе раздела расплавов полимеров и результат
расчета щг для температур ниже температуры стеклования или
плавления приведены в табл. IV.3 и IV.4.
В литературе имеются указания, что значения межфазного на-
тяжения на границе ПС—ПИП 0,83-10-3 Н/м [92] и на границе
ПС—ПБ — около 10-3 Н/м [93] удовлетворительно согласуются с
экспериментально найденными размерами доменов ПС в триблок-
сополимерах соответствующего состава.
В табл. IV.3 приведены результаты экспериментального опреде-
ления межфазного натяжения, в ряде случаев экстраполированные
к 150 °C. Температурный коэффициент межфазного натяжения ра-
вен для большинства систем 0,01-10 3 Н/м-град, однако для си-
стемы ПЭВА—ПВА приводится значение — 0,022-10-3 Н/м-град.
Положительный температурный коэффициент а был обнаружен
толы о для системы ПЭ—ПДМС, но здесь не исключена ошибка
эксперимента [94].
Значения поверхностного натяжения расплавов полимеров на
границе с во «духом [89] приведены в табл. IV.5.
Представления о сегментальной растворимости полимеров в
межфазном слое фактически явились основой развитой Гельфан-
дом [95—97] теории, учитывающей также прежние работы по изу-
чению структуры макромолекул на поверхности раздела [98—
101]. Эта теория основывается на представлениях об идеальной эн-
тропии смешения полимеров, т. е. она не учитывает наличия ближ-
него порядка. Конформационная энтропия рассчитывается на ос-
нове гауссовой статистики, а энергетическое взаимодействие поли-
156
Таблица IV.3. Межфазное натяжение на границе
раздела полимер — полимер при 150 °C
Контактирующие полимеры СГ-108, Н/м Контактирующие полимеры а-103, Н/м
ПЭ—ПП 1,1 ПС—ПВА 3.7
ПЭ—ПС 5.0 ПС—ПЭВА (38,7% винил- 5,6
ацетата)
ПЭВП—ПС 5,7 ПС ПММА 1,6
НЭНП-ПХП 3,6 ПС—ПДМС 6,0
ПЭ—ПВА 10,5 ПХП—П-н-БМА 1,6
ПЭВП—ПВА 11,0 ПХП ПДМС 6.5
ПЭНП—ПВА 9,8 ПВА—ПЭВА (25% винил- 5,8
ацетата)
ПЭ—ПЭВА (38.7% ви- 3,5 ИВА П-н-БМА 2,8
нилацетата)
ПЭ—ПЭВА (30,9% ви- 3,3 ПВА ПГМГ 4,5
нилацетата)
ПЭ—ПЭВА (25.0% ви- 1,3 ПВА—ПДМС 7,4
нилацетата)
ПЭВП- ПММЛ 9,5 ПММА—П-н-БМА 1,8
ПЭВП —П-н-БМА 5,2 ПММА—П-грет-БМА 2,3
ПЭНП- 11-изо-БМА 4,2
ПЭНП—П-трет-БМА 3,7
ПЭНП—Полиэтиленгли- 9,5 ПЭГ—ЭВА (25% винилаце- 5.7
КОЛЬ тата)
пэнп—птмг 4,1 ПЭГ—ПТМГ 3,8
ПЭНП—пдме 5,4 ПЭГ- ПДМС 9,8
ПЭНП—пдме 5,1 ПТМГ—ЭВА (25% винил- 1,2
• ацетата)
ПП—ПС 5,1 ПГМГ ПДМС 6,3
ПП—пдме 3,0 ПДМС- П н-БМА 3.8
ПИБ—ПВА 7,4 ПДМС—II трет-БУХХ 3,3
ПИБ ПДМС 4,3
ПС—ПХП 0,5
меров оценивается параметром взаимодействия Флори — Хаггин-
сах- В результате может быть рассчитана толщина межфазного
слоя, профиль изменения концентрации сегментов и изменение
плотности полимерной массы на границе раздела, а также поверх-
ностная свободная энергия, хорошее совпадение которой со зна-
чениями межфазного натяжения служит подтверждением правиль-
ности предложенной теории.
Гельфанд „использовал три основных подхода для достижения
указанных выше результатов: а) на основе предположения о сим-
метрии и тождественности энергетических полей молекулярных
сил каждого полимера; б) с учетом несимметричности (неодина-
ковости) полей молекулярных сил со среднегеометрическим их
усреднением и в) на основе решетчатой модели Флори, развитой
для случая гетерофазной системы.
Выводы этих трех направлений исследования качественно рав
неценны, поэтому ниже излагается в основном теория, развитая в
157
Таблица IV.4. Межфазное натяжение на границе
раздела полимер — полимер при разных температурах
Контактирующие полимеры в-10», Н/М Метод определения
при 20 °C при 140 °C при 180 °C
ПХП--ПЭНП 4,6 3,7 3,4 Метод лежащей кап-
ПВА—ПЭВП 14,5 п.з 10,2 ли [89] То же
ПММА—ПЭВП 11,9 9,7 9,0 »
П-н-БМА—ПЭВП 7,1 5,3 4,7
П-кзо-БМА—ПЭНП 5,5 4,3 3,9
П-трет-БМА—ПЭНП 5,9 3,8 3,4 »
ПС—ПЭВП 8,3 5,9 5,1
ПТГФ—ПЭНП (политетра- 5,0 4,2 3,9
гидрофуран) ПДМС—ПЭНП 5,3 5,1 5,0
ПВА—ПДМС 8,4 7,4 7,1 »
ПХП—ПДМС 7,1 6,5 6,3 >
ПТГФ—ПДМС 6,4 6,3 6,2 »
П-н-БМА—ПДМС 4,2 3,8 3,6 »
П-трег-БМЛ—ПДМС 3,6 3,3 3,2 »
ПХП—П-н-БМА -— 1,6 — »
ПХП—ПС — 0,5 —
ПВА—ПС 4,2 3,7 3,5 »
ПВА—П-н-БМА 4,2 2,9 2,5
ПММА—П-н-БМА 3,4 1.9 1,5 »
ПММА—П-трет-БМА 3,0 2,3 2,2
ПММА—ПС 3,1 1,7 1,2
ПС—пип 1,0 — — Измерение с после-
ПА’-ПС 5,2 дующей экстраполя- цией к 100%-ной кон- центрации [90] Измерение длины вол-
НА”- ПС 5,9 иы возмущения в струе расплава при 266--283 °C [91] То же
ПА”’—ПС — — 7,5
ПА* ПЭНП — — 6,4 >
ПЭТФ -ПЭНП — — 15 >
* Полиамид -с вязкостью расплава 0,076-103 Па-с.
*♦ Полиамид с вязкостью расплава 0,11-103 Па-с.
*** Полиамид, наполненный сажей, с вязкостью расплава 0,8-108 Па-с.
предположении симметрии силовых полей взаимодействующих по-
лимеров.
Пусть полимеры Л и В состоят из статистических клубков мак-
ромолекул со степенью полимеризации z и длиной сегмента в
(средпеквадратичеекос расстояние между концами цепи равно
zb2), находящихся в ноле молекулярного притяжения с интенсив-
ностью и(г) в расчете на сегмент. Статистической мерой вероятно-
сти состояния служит zt, где t масштабный параметр, меняющийся
от 0 до 1 при начальном значении г=го. Ищем функцию q(r0,t),
158
Таблица IV.5. Поверхностное натяжение полимеров
Полимер Поверхностное натяжение, а -103, Н/м Полимер Поверхностное натяже ние, o-ЮЗ, Н/М
при 20 °C при 140 °C при 180 °C при 20 °C при 140 °C при 180 °C
пхп 13,6 33,2 29,8 П-трет-БМА 30,5 23,3 21,0
ПС 40,7 32,1 29,2 ПТГФ 31,9 24,6 22,2
ПММА ПВА 41,1 36,5 32,0 28,6 28,9 25,9 ПДМС 19,8 14,1 12,2
П-н-БМЛ 31,2 24,1 21,7 ПЭВП 35,7 28,8 26,5
П-изо-ЬМА 30,9 23,7 21,3 ПЭНП 35,3 27,3 24,6
являющуюся отношением функции распределения в пределах поля
и (г) к той же функции вне пределов поля:
<7 РЪ О = j drx. . .drMP (r0, rlt. . ., rM)X
X exp £ — ! fry' | । | dr±... dr^P (r0,..., гм) (7)
где M=zt, n — радиус-вектор, определяющий положение конца i-того сегмента.
Значение Р определяется гауссовой статистикой:
• Р(гп---rM) drM (3/2лР)ЗМ'2 х
х exp |- 3/26"- (rl+1 - r{)*] (8)
Отношение плотностей вероятности при данном г к аналогич-
ному значению в отсутствие поля дается выражением q(r, t). Если
(by и)2 мало, то q(r, t) может быть выражено несколько изменен-
ным уравнением диффузии
2-1 [d<? (r,t)/dt] = 4- b\*q (г, t) - [u (r)/kT\ q (r, t) (9)
при начальном условии q(r. 0) = 1.
При наличии границы раздела фаз полимеров А и В сегмент
молекулы А находится под действием силы
^ХРв(г)/р0 (Ю)
возникающей благодаря меньшему взаимодействию сегментов типа
А—А по сравнению с А—В и направленной внутрь фазы Л, и под
действием силы
[(1/р?*) - (kT/zpB)] [рА (г) + Рв (Г) - PoJ (11)
возникающей вследствие тенденции перемещения сегмента А в
зону с плотностью (рд+рв)<ро, и выталкивается из зоны с плот-
159
ностью (ра+ре)>Ро, где рл и рв — парциальная плотность сегмен-
тов А и В, а и— сжимаемость, равная для полимеров в вязкотеку-
чем состоянии около 5-10-6 м2/Н.
Силы дальнодействия, важные для низкомолекулярных соеди-
нений, могут быть опущены ввиду их малости по сравнению с си-
лами дальнодействия, возникающими из-за связанности сегментов
в единую макромолекулу, как это видно в уравнении (9) из сла-
гаемых, содержащих производные. Отсюда можно записать два
симметричных выражения для функции распре целения по г сег-
ментов А и В:
d?A(r,0 1 (
—dt— = "б" 6 v 9А~ И
Рв (Л)
Ро
(12)
Рв(<) .11 , Л
-7---1 ?аМ
Ро JJ
! ддъ (г, t)
dt ~
1 , .. ( Ра (г) ,
~6-*-V2<7bM)-(7.~^-4
Г Ра (г) Рв(') _
+ Ч Ро + Ро
• ?в (г, О
(13)
де
\ Р<Л7\ I г '
Начальные условия процесса образования межфазного слоя:
<7А (г, 0) = дв (г, 0) = 1 (14)
Если принять, что разделяющая фазы поверхность (начало кон-
такта) проходит в точке х=0 и положительное направление обога-
щено сегментами А, а отрицательное—-сегментами В, тогда гра-
ничные условия определятся следующим образом:
9a(°°.<) 9в( —0 = 1 (15)
Va( —~>0 = 9в(ос.0 = 0 (16)
Очевидно, что кривые q_\ и (соответственно рА и рв) асимп-
тотически приближаются к соответствующим значениям в чистых
фазах А и В, учитывая малую взаимную растворимость полиме-
ров, к тому же стремящуюся к нулю с ростом молекулярной мас-
сы (см. гл. I).
Изменение плотности, например полимера А, с расстоянием
перпендикулярно поверхности раздела может быть выражено урав-
нением
Ра W _
Ро
1
J dtq^(r, \ (г, t)
0
(17)
и если считать сжимаемость полимера ничтожной, т. е. %/£->-0, а
молекулярную массу большой, т. е. долю кольцевых сегментов ма-
160
Рис. IV.18. Профиль концентрации сегмен-
тов полимера А в межфазном слое рд/ро
на границе с полимером В, рассчитанный
для x/g=O и %/g=0.1. Верхняя кривая —
изменение общей плотности (Рд+рв)/Ро по
толщине слоя. Ось ординат соответствует
положению первоначальной (до сегменталь-
ного растворения) поверхности раздела фаз.
лой, то из уравнений (12) и (13) можно получить выражения для
профиля концентрации сегментов А и В:
Ра W/p„ =(;/(!-«) (18)
PbW/Po=1/(1 + 1;2) (1Q)
где ? — exp [(6-/J1-'2 x/i].
Изменение профилей концентраций в соответствии с (18) и
(19) приведено на рис. IV.18.
Толщина межфазного слоя определяется выражением:
Ро | (dPAjdx) | л=0 = 2б/(бД1/2 (20)
Если принять Ь—0,7 нм, а %=0,03, то толщина слоя 6=3,3 нм.(
Теория дает возможность расчета межфазного натяжения. Ко-
нечное выражение может быть записано следующим образом
' " (х/б)’7» pobkT (21)
Оно хорошо подтверждается экспериментальными данными по
межфазному натяжению пар полимеров, мало различающихся по
полярности. Так, в парах ПС—ПММА, ПММА—П-н-БМА, П-н-
БМА—ПВА при 150 °C экспериментально найденные значения о
равны 1,5-10-3; 1,8- 10 -3; 1,9-10—3 Н/м, а рассчитанные — соответ-
ственно 1.0- 1(Н; 2,0-10-3; 1,9- 10-3 Н/м [89].
Для систем, образованных полимерами, сильно различающими-
ся по полярности, применима формула, полученная с учетом не-
симметричности поля межмолекулярного взаимодействия:
ЛТа,/2
’ Ра + Рв
2
1 (Р2а- Рв)2]
6 Ра + Рв j
(22)
где ₽л2 = -g-poA&A2 И Рв2=-|ров&в2; Роа И ров — плотности фаз А и В
(если А и В разнополярны, то и плотности их существенно различ-
ны); а определяют по уравнению Гильдебранда:
а= (1/£7) (6д —бв)2 (23)
где бд и бв — плотности энергии когезии фаз А и В.
11—244
161
Таблица IV.6. Сравнение экспериментальных и расчетных значений
межфазного натяжения на границе раздела полимер — полимер
Контактирующие полимеры Межфазное натяжение и-10* s, Н/м 6-102, НМ а- Ю-з м—3
эксп. (при 140 °C) расч. (22) расч. (24)
ПЭ—ПС 5,9 4,6 4,7 150 1,8
ПЭ—ПВА 11,3 7,1 7,4 ПО 3,7
ПЭ—ПММА 9,7 4,9 5,1 140 2,1
ПЭ—П-н-БМА 5,3 2,7 2,8 210 0,8
ПС—ПХП 0,5 0,7 0,7 880 0,05
ПС—ПВА 3,7 1 9 1,9 310 0,4
ПС—ПММА* 1,7 0,3 0.3 1600 0,01
ПДМС—ПВА 7.4 7.2 7,8 80 5,1
ПДМС- ПХП 6,5 4,7 4 ° 140 2,1
ПДМС—П-н-БМА 3,8 3.2 3,3 130 1,5
ПДМС- ПТГФ 6,3 4,3 4.4 170 1,5
П-н-БМА—ПВА 2,9 3,0 3,1 160 1.1
П-н БМА—ПХП 1,6 0,7 0,7 820 0,05
П-н-БМА—ПММА 1 9 1,5 1,5 290 0,3
* Система ПС — ПММА одна из немногих, для которых а определено непосредственно
Штокыайером и Стэнли: а— (0,15±0,1)Х1°5 м-3, что соответствует а—0,11-103 м~3 при 140 "С
если предположить, что а обратно пропорционально температуре. Расчет о12 с учетом этих
данных приводит к значению 10-3 Н/м.
С учетом эффекта дальнодействия межмолекулярных сил фор-
мула (22) запишется в виде разложения в ряд:
о = Л7а1/2
Ра + Рв
1 „ { 2
+ 18 Ра + Рв
, 1 iPa-Eb)2) L
+ 6 ₽д-₽Б ) +
2 (Ра-Рв)2 \ ,
5 (Ра + Рв)3 / +
(24)
где у — мера расстояния, на котором проявляется дальнодействие; у2 — второй
момент фАНкции корреляции.
Результаты расчета межфазного натяжения по формулам (21)
и (22) приведены в табл. IV.6, (где сравниваются с эксперимен-
тальными данными табл. IV.4).
Из табл. IV.6 видно, что различие между значениями межфаз-
ного натяжения, рассчитанными по формулам (22) и (24), неве-
лико.
Развитая Гельфандом теория позволяет сделать следующие вы-
воды, часшчно отмеченные выше.
1. Нт границе раздела смесей полимеров, пе обладающих спо-
собностью к" в нтиморастворению, существует межфазный слощ
возникающий в результате взаиморастворепия сегментов полиме-
ров, причем толщина этого слоя больше, чем в системах из низко-
молекулярных жидкостей. Для полимеров близких по химической
природе, таких как ПС ПММА толщина слоя может достигать
16 нм (см. табл. IV.G).
162
2. Состав слоя меняется монотонно, асимптотически приближа-
ясь к состав}' соответствующей фазы (см. рис. IV.18).
3. Плотность межфазного слоя понижена по сравнению с адди-
тивным значением плотностей каждой фазы (см. рис. 1V.18) и так-
же асимптотически приближается к плотностям соответствующих
фаз.
4. Рассчитанные значения межфазного натяжения совпадают с
экспериментально полученными, подтверждая правильность раз-
витой теории.
5. Низкомолекулярный компонент (растворитель) преимущест-
венно сорбируется в межфазном слое, благодаря чему слой обо-
гащается растворителем, а взаимопроникновение сегментов фаз А
и В в межфазном слое увеличивается.
Следует остановиться на работах, посвященных изучению меж-
фазных слоев в блок-сополимерах. Размер и форма доменов в
блок-сополимерах определяются несколько ипыни факторами, чем
в смесях полимеров, причем важнейшую роль играет соотношение
размера домена и размера тех блоков сополимера, которые обра-
зуют домены. Требования сохранения сплошности домена (при-
мерно равной плотности по его диаметру), приблизительного ра-
венства среднеквадратичных расстояний между концами блока в
домене и в изолированном блоке, а также необходимость разме-
щения химических связей между блоками па границе раздела фаз
(или в межфазном слое) являются теми принципами, на которых
базируются расчеты размера и формы доменов, например ПС в
блок-сополимерах стирола с изопреном или с бутадиеном [102,
103, 104].
Факторы, определяющие образование межфазного слоя в
блок-сополимерах, видимо, аналогичны таковым для смесей по-
лимеров. Свидетельством этого является относительно малая кон-
центрация химических «межблочных» связей на границе доменов,
составляющая для сополимеров стирол — изопрен — стирол около
одной связи на 10 им2 поверхности раздела баз [105], а также
то обстоятельство, что межфазное натяжение, определенное для
системы ПС—ПИП (см. табл. IV.4), хорошо согласуется со зна-
чением его для блок-сополимеров, обеспечивающим практически
наблюдаемый размер и форму доменов [104]. Поэтому интересно
рассмотреть модельные представления и экспериментальные дан-
ные по установлению наличия межфазного слоя в блок-сополи-
мерах.
Теория межфазного слоя в блок-сополимерах развита преиму-
щественно в работе [106], а также [107].
В работе 1106] ь качестве рабочей модели двухфазной структу-
ры блок-сополимера типа С—Б—С приняли модель сферических
доменов радиусом 7?, окруженных слоем, состоящим из смеси сег-
ментов толщиной А/?, находящихся в таком же соотношении, как
в блок-сополимере. Домены диспергированы в матрице полибута-
диена. Учитывая малое число контактов разнородных молекул по
11* 163
границе раздела фаз, энтальпийную составляющую свободной
энергии образования домена находят из теории регулярных рас-
творов:
ДЯ = - Vm<pc<PB (6с - 6в)2 (1 - /) (25
где Vm — мольный объем полимера; <р — обьемая доля; 6 — параметр раствори-
мости; f — объемная доля межфазного слоя.
Энтропийная составляющая распадается на три части. Первая
обусловлена необходимостью расположения межблочных химиче-
ских связей в межфазном слое Д/?:
bS^Nklnpf + P) (26)
где N — число макромолекул блок-сополимера в единице объема.
Вторая обусловлена необходимостью расположения блоков по-
листирола внутри домена и может быть записана в соответствии
с [102]:
( Г 9L2 I 9L2
= 2Nk j In РС - 3 [- 1 ~ In (27)
где пс—число статистических сегментов полистирола; /с — их длина; L — фак-
тический радиус домена, т е. АН АЛ?; множитель 2 отражает наличие двух бло-
ков полистирола в каждой молекуле сополимера; Рс — вероятность нахождения
всех сегментов в пределах радиуса L.
Третья составляющая Д5В обусловлена необходимостью поме-
щения блоков полибутадиена вне доменов и помещения второго
«межблочного» узла снова в интервале ДУ? какого-либо домена.
Вероятность нахождения цепи из пв сегментов (блок полибутадие-
на длиной /в) в таком положении, что сегменты не соприкасаются
с поверхностью домена, а второй «межблочный» узел находится в
интервале Д/?, выразится следующим образом (ось х перпендику-
лярна поверхности домена и блок полибутадиена начинается в
точке х=0, х = Д7?/2):
где d— расстояние между доменами.
Интегрирование (27) от г=0 до г=оо дает выражение для
W(x)dx. Учи. иная, что конец блока полибу гадиена может входить
в любой слой толщиной получим выражение искомой вероят-
ности для цепи полибутадиена:
ад с
/>и . J W(x)dx+ J W(x)dx (29)
0 d-AR
164
п необходимое значение энтропии:
Д5в = МНпРь (30)
Полученные выражения позволяют найти Дб образования до-
мена:
ДО = АЯ — 7 (ASt + ASC + ASB) (31)
Задаваясь разными значениями R и Д7? и составом сополимера,
можно с помощью ЭВМ найти условия минимума Дб и соответст-
вующие этому минимуму значения параметров структуры. Резуль-
таты приведены на рис. IV.10 (см. с. 144). Наиболее глубокий ми-
нимум Дб соответствует толщине межфазного слоя 4 нм, что
дает размер домена (диаметр) от 18 до 26 нм в полном согла-
сии с экспериментальными данными [108].
Автором [107] приводятся следующие данные по фазовой
структуре блок-сополимера С—Б С типа «Кратон 101», рассчи-
танные в соответствии с моделью, аналогичной предыдущей. Мо-
лекулярные массы блоков соответственно П-103—54• 103—11 103,
и при 300 К минимизация свободной энергии приводит к необходи-
мости существования на границе раздела межфазного слоя, пред-
ставляющего собой смесь сегментов с молекулярной массой (в
среднем) для ПС 3950 и для ПБ 1030. При этом сегмент каждого
полимера содержит одинаковое число атомов углерода в главной
цепи — 76. Рассчитанное соотношение сегментов в слое по объему
ПС:ПБ = 77:23. Плотность межфазного слоя рассчитывается по
принципу аддитивности, и тогда объемные доли фаз полибутадие-
на, полистирола и межфазного слоя составляют соответственно
0,71; 0,17 и 0,12 при толщине слоя 1,15 нм и диаметре частиц вме-
сте со слоем 17,3 нм.
Таким образом, исходя из аналогий, общих для коллоидной хи-
мии и применимых как к низкомолекулярным, так и к высокомо-
лекулярным системам, а также основываясь на расчетах, проде-
ланных разными авторами специально для дисперсных систем типа
«полимер в полимере», необходимо допустить существование меж-
фазного слоя на границе раздела полимер — полимер. Межфазный
слой, исходя из этих соображений, является раствором сегментов
одного полимера в другом, и протяженность слоя в направлении,
перпендикулярном границе раздела, составляет десятки или не-
многие сотни нм. Приведенные выше расчеты показывают, что об-
разование межфазного слоя сегментальной растворимости — явле-
ние равновесное, обусловленное не кинетическими, а термодинами-
ческими закономерностями.
Рассмотрим экспериментальные данные, подтверждающие сде-
ланные выше выводы.
Наиболее непосредственным доказательством сегментальной
растворимости полимеров в межфазном слое является зависи-
мость адгезии полимеров от их химической природы. Это явление
широко и обоснованно использовалось многими исследователями
для подтверждения диффузионной теории адгезии. Речь идет о
165
равновесных значениях адгезии, т. е. и таких величинах, которые
являются предельными во времени при данной температуре кон-
такта и давлении при дублировании образцов. Таким образом,
усилие расслаивания, принимаемое за меру адгезионного взаимо-
действия, является равновесной в данных условиях величиной, за-
висящей от глубины взаимодиффузии на границе раздела, которая
в свою очередь определяется сродством полимеров, грубо прибли-
женно определяющимся разностью параметров растворимости кон-
тактирующих полимеров. Увеличение адгезии полимеров с умень-
шением разницы параметров растворимости объясняли увеличени-
ем «совместимости» полимеров. Поэтому адгезию полимеров пред-
лагали использовать как критерий их совместимости [109], или,
зная «совместимость» полимеров (точнее, соотношение полярно-
стей), судить о предельно достижимой величине их адгезии [НО].
Учитывая малую взаимную растворимость полимеров, когда даже
такие близкие по химической природе полимеры, как ПБ и ПИП,
взаиморастворимы не более чем на 1%, можно считать, что обще-
известная зависимость адгезии от соотношения параметров рас-
творимости является подтверждением сегментальной растворимо-
сти в межфазном слое, которая заметно зависит от соотношения
параметров растворимости (см. табл. IV.6 и данные по раствори-
мости ПС в ПИП и ПММА в гл. I). Из формул (22) и (24) видно,
что межфазное натяжение пропорционально разности плотностей
энергии когезии контактирующих полимеров [ср. формулу (23)],
а это значит, что чем больше указанная разность, тем больше
Рис. IV.19. Зависимость удельной энергии когезии от соотношения каучуков:
/ — НК —СКН-40; 2 — HK-CKI118; 3 - НК — СКВ; 4 - СКВ —СКН-40; 5 — СКВ — СКН-18;
S — СКВ — СКС-ЗО.
166
межфазное натяжение и тем меньше слой сегментальной рас-
творимости.
Смесь полимеров обладает пониженной энергией когезии fill,
I 12]. Энергию когезии определяли из опытов по набуханию дефор-
мированных вулканизатов полимеров и их смесей. Полученные
данные приведены на рис. IV.19 [111], из которого видно, что
энергия когезии меняется непропорционально составу, располага-
ясь ниже аддитивной прямой. Если бы взаиморастворення поли-
меров па границе раздела не происходило и смесь оставалась бы
строго гетерогенной, энергия когезии должна была бы меняться
пропорционально составу. Пониженное взаимодействие полимеров
в переходном слое и некоторое снижение плотности в нем, на
что указывают расчеты [95- 97] и экспериментальные данные по
плотности смесей [113] приводит к уменьшению энергии когезии.
Чем ближе полимеры по полярности, тем больше отклонение, ко-
торое практически отсутствует в случае смеси бутадиенового
каучука (СКД) с полярным СКН-40, когда межфазный слой, по-
видимому, минимален.
В пользу существования межфазного слоя говорят и опыты по
создаМю взаимопроникающих сеток ВПС [108, 114]. В [108] ука-
зывается, в частности, на один из способов получения ВПС, когда
латексы двух полимеров, содержащие вулканизующие вещества,
строго селективные для каждого полимера, смешивались с после-
дующим отливом пленки и вулканизацией. Вулканизующие аген-
ты подобраны таким образом, чтобы исключить химические реак-
ции между разнородными полимерами. Исследования полученных
ВПС динамическим механическим методом, электронной микро-
скопией, сканирующей колориметрией и определение густоты про-
странственной сетки убедительно показали наличие химических
связей между полимерами на границе раздела фаз, возникнове-
ние которых невозможно без сегментальной растворимости.
Проводились прямые опыты по определению сегментальной
растворимости на границе раздела полимеров методов флуорес-
центной метки и парамагнитного зонда. В работе [115] методом
флуоресцентной метки показано, что в привитых сополимерах сти-
рола и акрилонитрила на полибута диене и в привитых сополиме-
рах винилхлорида на ПБ блоки указанных полимеров могут на-
ходиться как на границе раздела, так и в противоположной фазе
полимера. При исследовании смеси tpc-ПБ и бутадиен-стироль-
ного каучука СКС-50 методом парамагнитного зонда [116] ста-
бильный радикал химически прививали к макромолекулам ПБ.
Частота вращения радикала-зонда уменьшалась во времени после
приведения в контакт пленок ПБ и СКС-50. Это свидетельствует
о сегментальном растворении полимеров в граничном слое. Авто-
ры отмечают, однако, что образующийся слой имеет незначитель-
ную толщину.
При прямом электронно-микроскопическом определении толщи-
ны межфазного слоя [117] было найдено, что для системы ПВХ—
167
ПММА она составляет 100—1300 нм при 160—170°С и до 300 нм
при 210—220 °C, а для пары ПС—ПММА — 30 нм, т. е. намного
превышает величины, получаемые расчетным^ путем. Толщины,
превышающие среднеквадратичное расстояние между концами
макромолекул, сомнительны, так как для их достижения должна
происходить диффузия целых макромолекуч, а не части образую-
щих их сегментов, что является признаком заметной взаимной
растворимости компонентов. Следует указать, что в этих опытах
не удаляли способной к диффузии низкомолекулярной фракции
полимеров. Для пары ПС—ПММА глубина слоя в 30 нм по поряд-
ку величины неплохо совпадает с вычисленным 6=16 нм (см. табл.
IV.6).
Многие исследователи, изучая микроструктуру смесей полиме-
ров, обращали внимание на то, что размытость границы раздела
тем больше, чем ближе по химической природе смешиваемые по-
лимеры. Так, Мацуо [118] отмечает,, что граница частиц бутадпен-
нитрилыюго каучука, .диспергированных х ПВХ, более размыта,
чем частиц ПБ в том же ПВХ, причем размытость границ растёт
с увеличением содержания в сополимере акрилонитрила, т. е. с
увеличением сродства ПВХ и сополимера. Количественное опре-
деление толщины межфазных слоев обычными методами элек-
тронной микроскопии, видимо, затруднительно, а во многих случа-
ях просто невозможно, если эти толщины согласно табл. IV.6 не
превышают немногие хотни нм. Качественное же электронно-мик-
роскопическое подтверждение наличия взаимоднффузии полиме-
ров в результате сегментальной растворимости на границе разде-
ла полимеров не вызывает сомнений.
Если толщина слоя достигала бы 100 нм, го при размере ча-
стиц дисперсной фазы 1 мкм количество полимера в межфаз
ном слое составляло бы около 27% от общего объема диспергиро-
ванных частиц (119]. ОднакЯ при толщине слоя 3—5 ня ко-
личество полимера в межфазном слое становится заметным лишь
при развитой удельной поверхности, что характерно для блок-
сонолимеров, у которых размер частиц составляет немногие десят-
ки нм. Именно для блок-сополимеров обнаруживаются проме-
жуточные максимумы на температурной зависимости механиче-
ских потерь, объясняемые релаксационными явлениями в межфаз-
ном слое. Так, в работах [120—122] наблюдали промежуточный
(третий) максимум механических потерь для образцов триблбк-
сополимеров бутадиена и стирола, полученных из разных раство-
рителей [120, 121] или на вальцах с последующим прессованием
[122]. Пример из работы [122] приведен на рис. IV.20. Промежу-
точные максимумы для указанных образцов были обнаружены и
на термограммах методом ДТА [120, 121]. Во всех случаях авторы
объясняли возникновение максимума образованием «промежуточ-
ного слоя» на границе раздела фаз. Условия получения образцов
блок-сополимеров в приведенных выше опытах можно считать
близкими к равновесным, особенно при получении образцов из
168
Рис. IV.20. Температурная зависимость тангенса угла механических потерь блок-
сополимеров бутадиена и стирола:
1 молекулярные массы блоков бутадиена 1,68-10®, стирола 0,58-10ь; 2— бутадиена 9.56*10*,
стирола 6,64-10*.
растворителей, когда матрица полибутадиена в блок-сополимере
оставалась при температуре гораздо выше Тс. Поэтому и образо-
вание промежуточного максимума указывает на равновесный ха-
рактер возникающего межфазного слоя. Однако для многих си-
стем промежуточный максимум никогда не наблюдался, что может
быть связано с малой дисперсностью системы (малая удельная
поверхность), малой толщиной слоя, большим расстоянием между
•кспсриментальными точками по температурной шкале (когда мак-
симум мог <не попасп>» в область измерения), низкой точностью
замера потерь, не позволяющей зафиксировать небольшой по вы
соте максимум, или, наконец, близостью основных релаксацион-
ных максимумов, внутренние «плечи» которых, перекрываясь, не
иошоляют выявить потери в межфазном слое.
Отмечено, что максимум потерь данного полимерного компо-
нент в смеси шире максимума индивидуального полимера. Если
бы компоненты смеси взаимодействовали только по поверхности
рлыела (без образования межфазного слоя), то при размере ча-
сти в несколько микрон нм это не должно сказаться на их релак-
сационных свойствах и площадь максимума должна быть пропор-
циональна объемной доле данного полимера в смеси. Образование
межфазного слоя приводит к появлению новых релаксирующих^
клементов в смеси, и релаксационный переход происходит Тюлее
плавно: максимум потерь расширяется. Расширение максимума
in ыектрических и механических потерь в смесях полимеров было
впервые систематически исследовано в работах [123, 124]. Как
ни то из рис. IV.21 [124], для смеси СКД—СКН-18 в отношении
I 1 ширина максимума, измеренная на половине его высоты, уве-
тчилась на 50% по сравнению с максимумом самого СКН-18.
Расширение максимума отмечалось и в работах [125,126]. В рабо-
|г [126] обращалось внимание на то, что в ударопроч-
169
Рис. IV.21. Температурная зависимость тан-
генса угла механических потерь смесей
СКД— СКН-18, измеренная на торсионном
маятнике:
/ — 100% СКД; 2 — 75% СКД; 3-50% СКД; 4 —
25% СКД; 5 — 100% СКН-18.
ном ПС площадь максимума по-
терь пропорциональна содержанию
каучука только для серии полиме-
ров, полученных в одинаковых ус-
ловиях. Изменение условии синте-
за или переработки наполненного
каучуком полистирола ведет к на-
'ее Температура. ° с 20 ™ РуШР11ИЮ пропорциональности. Эго
указывает на переход части каучу-
ка в межфазный слой, вызывающий изменение ширины макси-
мума потерь.
В пользу существования межфазного слоя свидетельствует и
ряд других экспериментальных данных. Обнаружено [127] сме-
щение спектра времен релаксации для смеси 40% ПММА и 60%
ПС по сравнению с рассчитанным, исходя из предположения об
аддитивности, и не обнаружено смещения спектра для смеси ПС—
ПЭВП, где компоненты сильно различаются по химической при-
роде и образование глубокого межфазного слоя маловероятно
(см. также [128]). Зависимость фактора приведения ат от темпе-
ратуры для блок-сополимеров типа С—Б—С описывается урав-
нением ВЛФ лишь в областях ниже —50 °C и выше +80 °C, что
авторы [12У] объясняют наличием межфазного слоя. Многие ме-
ханические, и в особенности прочностные, и релаксационные свой-
ства блок-сополимеров пе мотут быть объяснены без предполо-
жения о наличии межфазного слоя [130].
Образование слоя сегментальной совместимости специфично
для смесей полимеров, в связи с чем его образованию и зако-
номерностям уделено в этой главе большое внимание. Однако
слой cel ментальной совместимости лишь часть общего гранич-
ного слоя в смеси полимеров, протяженность которого может
быть много больше и достигать десятки или сотни нм. Возникнове-
ние протяженных граничных слоев в смесях полимеров является
следствием гетерофазной структуры таких систем. Структура гра-
ничных слоев в смесях полимеров по многим принципиальным
чертам аналогична структуре граничных слоев в наполненных по-
лимерах. Современные представления о граничных слоях в напол-
ненных полим<рах (в том числе с полимерным наполнителем)
изложены в книге [131].
Полимер, благодаря большому размеру макромолекул, спо-
собен образовывать протяженные граничные слои. Если поверх-
ность частицы наполнителя ограничивает подвижность близлежа-
щих макромолекул, то изменяется их конформация (в сторону
170
Рис. IV.22. Связь полимерного наполнителя
с полимерной матрицей:
Л с слой сегментальной растворимости; £>к —
граничный слой с измененной надмолекулярной
структурой матрицы н с частицами микрофазы
контактирующего полимера.
отклонения от наиболее вероятной),
что в свою очередь приводит к из-
менению надмолекулярной струк-
туры, образованной граничными
макромолекулами. Однако в непосредственной близости от поверх-
ности раздела надмолекулярные структуры включают и часть сег-
ментов более удаленных макромолекул, в связи с чем изменение
конформаций макромолекул и изменение надмолекулярных струк-
тур проникает (с ослаблением) и далее в объем матрицы. Если
полимер кристалличен, то уже небольшое количество твердых ча-
стиц (иногда всего доли % по объему) может изменить кристал-
лическую структуру всего полимера, что указывает на очень силь-
ное «дальнодействие» поверхности по «эстафетному» механизму.
Возникновение граничных слоев в смеси полимеров может
происходить по обе стороны от границы раздела, т. е. обе фазы
могут содержать протяженные граничные слои, возникшие вслед-
ствие «эстафетного» изменения конформаций макромолекул и
надмолекулярных структур.
Изменение структуры граничных слоев всегда равносильно оп-
ределенной деформации макромолекул и поэтому сопровождает-
ся возникновением внутренних напряжений. Если деформации
имеют предпочтительное направление, то и внутренние напряже-
ния могут действовать направленно на те или иные микрообъемы
граничных слоев. При этом в соответствии с [131, с. 207] «...на-
личие напряжений в контактной зоне может приводить к дис-
локации отдельных элементов и их агрегатов. Следовательно, в
контактной зоне возможны локальное искажение фазовых границ
вследствие их неоднородности и перенос значительных объемов
одного полимера в межструктурныс области другого. При этом,
разумеется, не исключается взаимная диффузия вследствие сег-
ментальной подвижности».
Малое межфазное натяжение на границе раздела полимер —
полимер приводит, как это было показано в гл. II, к самопроиз-
вольному эмульгированию полимера в полимере. В расплаве сме-
си полимеров образование микроэмульсии возможно непосредст-
венно вблизи фазовой границы, что также вносит новые элемен-
ты в структуру граничного слоя.
Таким образом, наряду со слоем сегментальной растворимо-
сти в смеси полимеров может существовать протяженный гра-
ничный слой, состоящий из полимера с измененной надмолеку-
лярной структурой, в том числе содержащий частицы полимера
другой фазы. Схема такого слоя приведена на рис. IV.22 [131]
171
Представления о структуре граничных слоев непрерывно ме-
няются главным образом ввиду очень скудного фактического ма-
териала в этой области, который можно было бы считать досто-
верным.
ЛИТЕРАТУРА
1. Виноградов Г. В., Яновский Ю. Г., Кулезнев В. Н., Иваненко Т. А.— Кол-
лоид. ж., 1966, т. 28, № 5, с. 640 644.
2. Кулезнев В. Н., Игошева К- М. — Высокомол. соед., 1962, т. 4, № 12,
с. 1858—1862.
3. Kakutani Т. — Japan Plast. Age, 1964, v. 2, № 9, p. 52—53.
4. Parrini P., Corrieri G. — Makromol. Chem., 1965, Bd. 86, As 6, S. 271—
279.
5. Кокоулина И. Г., Дорофеева Л. Г., Ильин С. Н., Январева Т. С. — Пласт,
массы, 1972, № 4, с. 46—47.
6. Bares J. — Macromolecules, 1975, v. 8, Xs 2, p. 244—246.
7. Boyer R. F., Spencer R. 5, —J. Appl. Phys., 1944, v. 15, p. 398—449.
8. Смирнова H. В., Динзбург Б. И., Никольский В. Г. — Каучук и резина. 1972,
№ 11, с. 21—22.
9. Фридрихсберг Д. А. Курс коллоидной химии. Л., Химия. 1974. 351 с.
10. Гуль В. Е., Кулезнев В. Н. Структура и механические свойства полимеров.
М., Высшая школа, 1979. 351 с.; Басин В. Е.—Мех. полимер., 1971, As 4,
с. 731—733.
11. Manabe S., Murakami R., Takayanagi М.— Mem. Fac. Eng. Kyushu Univ.,
1969, v. 28, p. 295—309; Intern. J. Polymer. Mater., 1971, v. 1, p. 47—73.
12. Pohl G. Plaste u. Kautschuk. 1973, Bd. 20, Xs 5, S. 340—342.
13. Schmitt I. A. — J. Polymer Sci., 1970, pt. C. Xs 30, p. 437—445.
14. Buchdahl R., Nielsen L. E.—J. Appl. Phys., 1950, v. 21, p. 482; Нильсен Л.
Механические свойства полимеров и полимерных композиций. М., Химия,
1978. 310 с.
15. Brener W., Hilil W., Wolf H. — Plaste u. Kautschuk, 1054, Bd. 1, S. 170—
185; 1957, Bd. 4, S. 244—259.
16. Михайлов Г. П.—Усп. хим., 1955, т. 24, с. 127—149, 875—898; Кулез-
нев В. Н., Клыкова В. Д. — Коллоид, ж., 1968, т. 30, Xs 1, с. 44—48.
17. Mullins L. R. — Rubb. Chem. Technol., 1947, v. 20, p. 998—1021; Марей А. И..
Сидорович E. A. — Мех. полимер., 1965, Xs 5, c. 85—87.
18. Meltzer M., Dermody IF. /.. Tobolsky A. V.— J. Appl. Polymer Sci., 1964,
v. 8, p. 765—778.
19. Айвазов А. Б., Миндиаров X. Г., Зеленев Ю. В., Оганесов Ю. Г., Раев-
ский В. Г. — Высокомол. соед., 1970, сер. Б, т. 12, Xs 1, с. 10—14.
20. Френкин Э. И., Яновский Ю. Г., Виноградов Г. В.—Мех. полимер., 1966,
№ 6. с. 895—899.
21. Floyd б I Brit. J. Appl. Phys., 1952, Xs 3, p. 373—388.
22. Бартенев Г. M., Конгаров Г. С. — Высокомол. соед., 1960, т. 2, с. 1962—
1967.
23. Sato Т., Takahashi М. — J. Appl. Polymer Sci., 1969, v. 13, p. 2665'—
2676.
24. Ke B. — J. Polymer Sci., 1962, v. 61, p. 47—62.
25. Marsh P. A., Voet A., Price L. D. — Rubb. Chem. Technol., 1968, v. 41, Xs 2,
p. 344—355.
26 Бубен H. Я- и ар — ДАН СССР, 1965, т. 162, с. 370—373; Высокомол. соед.,
1967, сер. А, т 9, с. 2275 - 2286; ДАН СССР, 1968, т. 178. с. 383—384.
27. Мельникова О. Л., Кулезнев В. Н., Аулов В. А., Клыкова В. Д. — Высоко-
мол. соед., 1976, сер. Б, т. 19, Xs 12, с. 903—906.
28. Многокомпонентные полимерные системы. Пер. с англ./Под ред. А. Я. Мал-
кина и В. Н. Кулезнева. М., Химия, 1974. 328 с.
172
29. Младенов И., Николинский II., Васильева С. — Докл. Ьълг. ЛИ. 19ь.1, i HI,
с. 837—843.
30. Кулезнев В. Н., Гуль В. Е., Евреинов Ю. В. — В кн.: Механизм процессов
пленкообразования из полимерных растворов и дисперсий. М„ Наука, 1966,
с. 177—179.
31. Киреев В. А. Курс физической химии. М., Госхимиздат, 1955. 832 с.
32. Matsuo М., Nozaki G., lyo У.— Polymer Eng. a. Sci., 1969, v. 9, № 3,
p. 197—205.
33. Кулезнев В. И., Крохина Л. С., Оганесов Ю. Г., Злацен Л. М. — Коллоид,
ж., 1971, т. 33, с. 98—105.
34. Yhen Н. К-, Kinsinger I. В.—Macromolecules, 1974, v. 7, № 3, р. 329—
336.
35. Разинская И. Н., Видяйкина Л. И., Радбиль Т. И., Штаркман Б. П. — Высо-
комол. соед., 1972, сер. А, т. 14, № 4, с. 968—973.
36. Бакеев Н. Ф., Берестнева 3. Я-, Жарикова 3. Ф., Каждан М. В. — Высоко-
мол. соед., 1973, сер. А, т. 15, № 9, с. 2128—2130.
37. Разинская И. Н., Батуева Л. И., Штаркман Б. П,-—Коллоид, ж., 1974, т. 36,
№ 2, с. 291—296.
38. Салтыков С. А. Стереометрическая металлография. М., Металлургия, 1970.
163 с.; Современные проблемы машинного анализа биологических структур.
М., Наука, 1970. 144 с.
39. Marsh Р. A., Voet A., Price L. D. — Rubb. Chem. Technok, 1967, v. 40, № 2,
p. 359—370; 1968, v. 41, № 2, p. 344—355.
40. Walters Л1. H., Keyte D. H.— Trans. Proc. Ins. Rubb. Ind., 1962, v. 38,
№ 2, p. 40—55.
41. Благов С. С., Печковская К- А., Лыкин А. С. и dp. — Каучук и резина, 1959,
№ 3, с. 12—14.
42. Alter Н. — J. Appl. Polymer Sci., 1965, v. 9, № 4, p. 1525—1539.
43. Усиление эластомеров. Пер. с англ. М., Химия, 1968. 483 с.
44. Davenport N. Е., Hubbard L. W., Pettit М. R.— Brit. Plast, 1959, v. 32,
№ 12, p. 549—564.
45. Bender B. W. — J. Polymer Sci., 1965, v. 9, № 8, p. 2887—2899.
46. Баллова Г. Д. и др. — Пласт, массы, 1972, № 7, с. 52—54; 1974, № 2,
с. 13—14.
47. Rohl G.. Gruber К., Taeger F. — Plaste u. Kautschuk, 1972, Bd. 19, S. 255—
264.
48. Kato K. — Koll.-Z. u. Z. Polymere, 1967, Bd. 220, S. 24—31.
49. Cigna G. — J. Appl. Polymer Sci., 1970, v. 14, p. 1781—1792.
50. Baer M. — J. Appl. Polymer Sci., 1972, v. 16, p. 1109—1121.
51. Коршак В. В., Еремина Е. И., Вылегжанина К. А. — Пласт, массы, 1972,
№ 7, с. 56—58.
52. Захаров Н. Д.. Бабюк В. Н., Кулезнев В. И., Захаркин О. А. — Коллоид,
ж., 1974, т. 36, № 2, с. 252—257; Кулезнев В. Н. — В кн.: Композиционные
полимерные материалы. Киев, Паукова думка, 1975, с. 93—109.
53. Кулезнев В. Н„ Клыкова В. Д., Догадкин Б. А. — Коллоид, ж., 1968, т. 30,
№ 5, с. 707—712.
54. Оганесов Ю. Г., Кулезнев В. И., Воюцкий С. С. — Высокомол. соед., 1970,
сер. Б, т. 12, № 9, с. 691—693.
55. Кулезнев В. Н. — В кн.: Композиционные полимерные материалы. Киев,
Наукова думка, 1975, с. 93—109.
56. Conaghan В. F., Rosen S. L. — Polymer Eng. a. Sci., 1972, v. 12, № 2,
p. 134—153.
57. Kampf G., Hoffman R., Kromer H. — Ber. Bunsenges phys. Chem., 1970,
Bd. 74, № 8/9, S. 851—859.
58. McIntyre D., Campos-Lopez E. — In: Blok Polymers/Ed. by S. L. Aggarwal.
N. Y., Plenum Press, 1970, p. 19—46.
59. Kohler I., Riess I., Baunderet A. — Europ. Polymer J., 1968, v. 4, № 1, p. 173—
205.
60. Leary D. F.. Williams M. C. — J. Polymer Sci., Polymer Phys. Ed, 1973,
v. 11, p. 345—459.
173
61. Rovaiti W., Bobalek E. — J. Appl. Polymer Sci., 1963, v. 7, № 10, p. 2269—
2292.
'62 . Кандырин Л. Б. Автореф. канд. дис. М., МИТХТ им. М. В. Ломоносова, 1971.
63. Кандырин Л. Б., Кулезнев В. Н. — Коллоид, ж., 1974, т. 36, № 3, с. 473—479.
64. White J. L., Ufford R. С., Dhurod К R-, Price R. L.—J. Appl. Polymer Sci.,
1972, v. 16, № 6, p. 1313—1330.
65. Барамбойм H. К., Ракитянский В. Ф.—Коллоид, ж., 1974, т. 36, № 1
с. 129—132.
66. Цебренко М. В., Юдин А. В., Кучинка М. Ю., Виноградов Г. В., Зубо-
вич К- А.— Высокомол. соед., 1973, сер. Б, л. 15, № 8, с. 556—567.
67. Vinogradov G. V., Yarlykov В. V., Tsebrenko М. V. Yudin А. V., Ablazo-
va Т. I. — Polymer, 1975, v. 16, № 8, p. 609 614; Сизевич T. И. Автореф.
канд. дис. Киев, Ин-т легкой пром., 1974.
68. Кулезнев В. II, Мельникова О. Л., Клыкова В. Д., Скворцов В. П., Глу-
хсвский В. С. — Коллоид, ж., 1975, т. 37, № 2, с. 273—279.
69. Клыкова В. Д. — Канд. дис. М., МИТХТ им. М. В. Ломоносова, 1969.
70. Кулезнев В. И.. Евреинов Ю. В.. Клыкова В. Д., Шапошникова М. И. —
Коллоид, ж., 1973, т. 35, с. 281—285.
71. Keskkulu Н.— Appl. Polymer Symp., 1970, № 15. р. 51—78.
72. Dinges К-, Schuster Н. — Makromol. Chem., 1967, Bd. 101, S. 200—213;
Ide F., Hasegawa A. — J. Appl. Polymer Sci., 1974, v. 18, № 4, p. 963—
974.
73. Rovatti IV’., Bobalek E. — J. Appl. Polymer Sci., 1963, v. ., № 10, p. 2269—
2292.
74. Пат. 3326869, 1967 г. (США).
75. Воюцкий С. С. Курс коллоидной химии. М., Химия, 1964. 574 с.
V 76. Ребиндер П. А. — Физико-химическая механика. М., Знание, 1958. 64 с.;
Фридрихсберг Д. А. Курс коллоидной химии. Л., Химия, 1974. 351 с.
77. Avgeropulos G. N., Weissert F. С., Biddison Р. Н., Bohm G. G. A. — Rubb.
Chem. Technol., 197b, Bd. 49, S. 93- 104.
78. Vemura S., Takayatiagi M. — J. Appl. Polymer Sci., 1966, v. 10, p. 113—
139.
79. Pakula T., Kryszewski M, Grebowicz I., Galrsky A. — Polymer J., 1974, v. 6,
p. 94—112.
80. Kerner E. H. — Proc. Phys. Soc., 1956, Ser. B, v. 69, p. 802—808.
81. Власов С. В., Сагалаев Г. В., Кулезнев В. Н., Марков Н. Г. — Пласт, массы,
1975, № 9, с. 37—38.
82. Малошук Ю. С., Кулезнев В. Н., Ханин С. Е. — Коллоид, ж., 1973, т 35,
№ 2; с. 408—410; Ханин С. Е„ Ангерт Л. Г., Кулезнев В. Н., Мало-
шук Ю. С. — Коллоид, ж., 1975, т. 37, № 1, с. 99—104.
83. Русанов А. И. Фазовые равновесия и поверхностные явления. Л., Химия,
1970. 188 с.; Фактор Э. А. Канд. дис. Л., ЛГУ, 1972.
84. Дерягин Б. В. — В кн.: Успехи коллоидной химии. М., Наука, 1973, с. 30—38.
85. Чернин Е. И. Автореф. канд. дис. М., МИТХТ им. М. В. Ломоносова, 1977.
86. Кулезнев В. Н., Крохина Л. С., Догадкин Б. А. — Коллоид, ж., 1967, т. 29,
с. 170—171; Кулезнев В. И., Догадкин Б. А., Клыкова В. Д. — Коллоид, ж.,
1968, т. 30, № 2, с. 255—257.
87. Кулезнев В. Н. Совместимость полимеров. — Энциклопедия полимеров. М.,
Советская энциклопедия, 1977. Т. 3, с. 433—439.
88. Kammer Н. W.— Wiss. Z. Techn. Univ. Dresden, 1975, Bd. 24, Ns 1, S. 35—
89. IVu S. — J. Polymer Sci., 1974, pt. C, № 34, p. 19—30.
90. Кулезнев В. H., Крохина Л. С., Догадкин Б. А. — Коллоид, ж., 1969, т. 31,
№ 6, с. 853- -859.
91. Chappelear D. С. — ACS Polymer Prepr., 1964, v. 5, № 22, p. 363—371.
92. Krigbaum W. R., Yazgan S., Tolbert W. R.— J. Polymer Sci., Polymer Phys.
Ed., 1973, v. 11, Ns 3, p. 511—527.
93. Campoz-Lopez E., McIntyre D., Fetters L. J. — Macromolecules, 1973, v. 6,
Ns 3, p. 415—423.
94. Gaines G. L. — Polymer Eng. a. Sci., 1972, v. 12, Ns 1, p. 1—11.
174
95. Helfand Е., Tagami Y.— J. Polymer Sci., 1971. pt. B, v. 9, № 10, p. 711 74t>;
J. Chem. Phys., 1972, v. 56, № 7, p. 3592—3601.
96. Helfand E., Sapse A. M. — J. Chem. Phys., 1975, v. 62, № 4, p. 1327—
1331.
97. Helfand E.—J. Chem. Phys., 1975, v. 63. № 5, p. 2192—2198.
98. Pouchly J.—Coll. Czech. Chem. Commun., 1963, v. 28, p. 1804—1812.
99. Di Marzio E. A.-—J. Chem. Phys., 1965, v. 42, p. 2101—2121.
100. Vrij A. •—J. Polymer Sci., 1968, pt A-2, v. 6, p. 1919—1927.
101. Edwards S. F. — Proc. Phys. Soc.. 1965, Ser. B, v. 85, p. 613—641.
102. Meier D. J. — J. Polymer Sci., 1969, pt C, v. 26, p. 81—102; ACS Polymer
Prepr., 1970, v. 11, p. 400—405.
103. Krause S. — J. Polymer Sci., 1969, pt. A-2, v. 7, p. 249; Macromolecules,
1970, v. 3, p. 84—99.
104. Krigbaum W. R., Yazgan S„ Tolbert W. R.— J. Polymer Sci., Polymer Phys.
Ed., 1973, v. 11, № 3, p. 511—527.
105. Echida T„ Soen T., Inoue T., Kawai H. — J. Polymer Sci., 1970, pt. A-2. v. 8.
p. 101—124.
106. Leary D. F., Williams M. C. — J. Polymer Sci., Polymer Lett. Ed., 1970, v. 8,
p. 335—340; J. Polymer Sci., Polymer Phys. Ed., 1973, v. 11, p. 345—459.
107. Kaelble D. H. — Trans. Soc. Rheol., 1971, v. 15, p. 235 247: Kaelble D. H„
Cirlin E. H.—J. Polymer Sci., Polymer Symp. Ed., 1973, №43, p. 131—
148.
108. Manson J., Sperling L. Polymer Biens and Composites. N. Y., Plenum Press,
1977. 513 p.
109. Воюцкий С. C. — Легкая пром., 1953, № 1, с. 42; Воюцкий С. С., Ваку-
ла В. Л.—Усп. хим., 1964, т. 33, № 2, с. 205—232.
НО. Дерягин Б. В., Жеребков С. К, Медведева В. Н. — Коллоид, ж., 1956, т. 18,
№ 4, с. 404—409.
111. Орехов С. В., Захаров Н. Д., Кулезнев В. Н„ Догадкин Б. А. — Коллоид,
ж., 1970, т 32, № 2, с. 245—250.
112. Кулезнев В. Н., Шварц А. Г., Клыкова В. Д„ Догадкин Б. А.— Коллоид,
ж., 1965, т. 27, № 2, с. 211—215.
ИЗ. Кулезнев В. Н., Игошева К. М. — Высокомол. соед., 1962, т. 4, № 12,
с. 1858—1862.
114. Липатов Ю. С., Сергеева Л. М. Взаимопроникающие полимерные сетки. Ки-
ев, Наукова думка, 1977. 157 с.
115. Riess G. — In: 23rd International Congress Pure a. Applied Chemistry, Bos-
ton, 1971, Macromolecular Prepr. V. 1, Ser. 1, p. 485—491.
116. Коварский А. Л., Аркина С. H., Вассерман A. M. — Высокомол. соед., 1970,
сер Б, т. 12, с. 38—43; Коварский А. Л., Буркова С. Г., Вассерман А. М„
Морозов Ю. Л, —ДАН СССР, 1971, т. 196, с. 383-384.
117. Каменский А. Н., Фодиман Н. М., Воюцкий С. С. — Высокомол. соед., 1969,
сер. А, т. 11, № 2, с. 394—399.
118. Maisuo М„ Nozaki С., Jyo У.— Polymer Eng. a. Sci., 1969, v. 9, № 3, р. 197—
205.
119. Кулезнев В. Н„ Догадкин Б. А., Клыкова В. Д. — Коллоид, ж., 1968, т. 30,
с. 255—257.
120. Marker L. —ACS Polymer Prepr., 1969, v. 10, № 2, p. 524—529.
121. Miyamoto T„ Kodama M„ Shibayama K. Reports on Progress of Polymer
Physics in Japan, Tokyo, 1969. V. 12, p. 325—328.
122. Оськин В. H„ Янковский Ю. Г., Малкин А. Я., Кулезнев В. Н„ Альтзи-
цер В. С., Туторский И. А.—Высокомол. соед., 1972, сер. А, т. 14, № 10,
с. 2120—2123.
123. Кулезнев В. Н. Докт. дис. М., МИТХТ им. М. В. Ломоносова. 1973.
124. Кулезнев В. Н., Клыкова В. Д. — Коллоид, ж., 1968, т. 30, № 1, с. 44—48.
125. Зеленев Ю. В., Никифорова А. В. — Высокомол. соед., 1969, т. II. № 2,
с. 138—144.
126. Keskkula Н., Turley S. G., Boyer R. F. — J. Appl. Polymer Sci., 1971, v. 15,
№ 2. c. 351—367.
175
127. Hill A. S., Maxwell B.— Poljmer Eng. a. Sci., 1970, v. 10, № 5 p 289—
292. H
128. Полистирол/Малкин А. Я., Вольфсон С. А., Кулезнев В. H., Файдель Г. И.
М., Химия, 1975. 288 с.
129. Shen В.. Kaelble D. Н. — J. Polymer Sci., 1970, pt. В, v. 8, № 3, p. 149—
159.
130. Morton M. — J. Elastoplastics, 1971, v. 3, № 4, p 112—125.
131. Липатов Ю. С. Физическая химия наполненных полимеров М.. Химия 1977
304 с.
Глава V
ОСОБЕННОСТИ ФИЗИКО-МЕХАНИЧЕСКИХ СВОЙСТВ
СМЕСЕЙ ПОЛИМЕРОВ
§ 1. ФИЗИЧЕСКИЕ СВОЙСТВА
Стеклование
Стеклование в смесях полимеров имеет ряд характерных осо-
бенностей, связанных со структурой смесей. Стеклование может
происходить раздельно в каждой фазе, если полимеры не взаи-
модействуют друг с другом. В другом крайнем случае стеклова-
ние в смесях является полностью кооперативным процессом, в ко-
тором участвуют сегменты макромолекул смешанных полимеров.
В связи с этим соответствие числа главных релаксационных пе-
реходов (температур стеклования) числу полимерных компонен-
тов в смеси дает представление о ее фазовом составе. Рассмот-
рим стеклование бинарных смесей полимеров.
Обычно бинарные смеси полимеров двухфазны и в них обна-
руживаются две температуры стеклования, соответствующие в
пределах ошибки эксперимента' значениям Тс исходных полиме-
ров. В 1947 г. впервые было отмечено наличие двух минимумов
эластичности по отскоку па температурной зависимости для сме-
си натурального и бутадиен-нитрилыюго каучуков [1], что соот-
ветствовало двум независимым релаксационным переходам и
указывало на наличие двух температур стеклования. Впоследст-
вии [2] при исследовании смесей полимеров и сополимеров меха-
ническим динамическим методом было показано, что для смесей
ПС с каучуками обнаруживается две температуры стеклования,
соответствуюшпе Тс исходных компонентов. В настоящее время
этот метод наиболее широко применяется для исследований сме-
сей полимеров наряду с методом ДТА. Как показано па много-
численных смесях, в случае несовместимых полимеров число ре
лаксационных переходов соответствует числу компонентов, тогда
как в смесях совместимых полимеров наблюдается один релакса-
ционный переход (одна температура стеклования), положение ко-
торого на температурной шкале зависит от состава смеси [3]
На рис. V.1 показан типичный пример стеклования однофаз
пых и двухфазных смесей [4]. При перемешивании ПВХ с сопо
лимером СКН-18 на вальцах при 165—170 °C смесь двухфазна и
при средних соотношениях компонентов обнаруживаются две тем
пературы стеклования, близкие к Тс соответственно ПВХ и
СКН-18. Смесь ПВХ с СКН-26, полученная в тех же условиях,
что и предыдущая, однофазна и имеет одну Тс, монотонно измс
няющуюся с составом.
12—244
177
Рис. V.l. Температуры стеклования смесей
каучуков с ПВХ (Те определяли термоме-
ханическим методом):
/ — смесь ПВХ—СКН-18; 2 — смесь ПВХ —
СКН-26.
Закономерности изменения Тс с
составом смеси не одинаковы для
разных смесей. Логично предполо-
жить, что для однофазной смеси, в
которой полимерные компоненты
равномерно распределены друг в
друге, зависимость Тс от состава
должна быть такой же, как в ста-
тистических сополимерах. Для со-
полимеров зависимость Тс от состава описывается уравнениями
Фокса [б] или Гордона—Тэйлора [6]. Согласно Фоксу:
1
Тс1,2
(1)
где Тс1.2 — температура стеклования сополимера, содержащего массовую долю
Wi одного и W2 — другого мономера; Tci и Тсг — температуры стеклования гомо-
полпмеров 1 и 2.
Уравнение Гордона—Тэйлора имеет следующий вид:
Tci 2 — 7С1 Л- fao2 (Тс2 — ^a2^'wi
(2)
где k — отношение разностей термических коэффициентов расширения в эласти-
ческом и стеклообразном состоянии для гомопилимеров 1 и 2.
На рис. V.2 показано [7] удовлетворительное совпадение экс-
периментальных значений Тг для смеси поли-е-капролактопа и
ПВХ с рассчитанными по уравнению Гордона — Тэйлора. Поли-
капролактон кристаллический полимер, и наилучшее совпаде-
ние теории с экспериментом наблюдается, если для него прини-
мают экстраполированное значение Тс. Хорошее совпадение на-
блюдается и с данными, рассчитанными по уравнению Ф<жса.
Такое совладение указывает на большую однородность смеси, что
подтверждается также отсутствием кристаллизации полпкапро-
лактона в смеси (молекулы ПВХ, равномерно распределенные в
поликапролактонс, препятствуют кристаллизации).
Если смесь однофазна, но не вполне однородна (возможное
наличие ассоциатов, флуктуаций концентрации однородных сег-
ментов), то экспериментальные данные отличаются от расчетных
(рис. V.3) [8].
При интенсивном взаимодействии полимерных компонентов,
ограничивающем подвижность сегментов, температура стеклова-
ния смеси может оказаться существенно выше Tf исходных поли-
меров. Тогда Тс смеси изменяется с составом по кривой с мак-
симумом. Такая кривая характерна, например, для смеси поли-
178
винилнитрата и поливинилацетата, полученной смешением рас-
творов в общем растворителе (Тс определена дилатометрическим
методом) [9]. Максимум на кривой Тс — состав смеси указывает
на весьма значительные отклонения от уравнений (1) и (2).
Имеется указание, что снижение Тс может быть пропорцио-
нальным объемной доле второго полимера, как это показано [10]
па примерах смеси натурального каучука с разветвленным поли-
бутадиеном (каучук СКВ), и смеси бутадиен-иитрильных сопо-
лимеров СКН-18 и СКН-40.
В двухфазных смесях часто наблюдается отличие Тс одной
или обеих фаз от Тс исходных полимеров. Точность определения
Тс во многих работах невелика, и смещение Тс не может быть
уверенно измерено. Для случаев, когда смещение Тс велико и на-
дежно измеримо, можно сформулировать следующие причины
этого явления.
К изменению Тс каждой фазы может приводить ограниченная
взаимная растворимость полимеров. В этом случае смесь поли-
меров представляет собой дисперсию растворов полимеров друг
в друге. В каждой фазе Тс может изменяться в результате рас-
творения в ней другого полимера, как это показано выше для
однофазных смесей. Растворимость полимеров друг в друге, в
особенности при наличии широкого спектра ММР, может дости-
гать нескольких процентов [11, 12]. В работе [3], по-видимому,
впервые отмечено повышение Тс бутадиен-стирольного сополиме-
ра в смеси с ПС, когда содержание ПС достигало 20—40%. Этот
эффект автор объяснил возможным растворением ПС в фазе со-
Рис. V.2. Зависимость Тс смесь поли-е-капролактон — ПВХ от ее состава (точ-
ки— данные эксперимента, кривые рассчитаны по уравнению Гордона Тейлора)
/ — при й=0,519, Гг1 =202 К; 2—при 6=0,420, =213 К.
Рис. V.3. Зависимость Тс смеси ПС — полиметилвинпловый эфир от содержа-
ния ПС:
/—экспериментальная кривая; 2—расчет по Гордону — Тэйлору при fc—0,25; 3 — то же,
прн fe=0,5; 4 — расчет по Фоксу.
12*
179
вания каждой фазы может
Рис. V.4. Зависимость Гс ПММЛ от содер-
жания ПВХ в смеси:
1 — по термомеханическнм кривым; 2 — по мето-
ду ДТА.
полимера. Однако нет работ, в ко-
торых бы измерение Тс каждой фа-
зы сопровождалось бы независи-
мым определением взаимной рас-
створимости полимерных компонен-
тов.
Изменение температуры стекло-
происходпть также вследствие изме-
нения надмолекулярной структуры полимеров, поскольку формо-
вание частиц дисперсной фазы в растворе или расплаве смеси по-
лимеров физически подобно формированию ассоциатов (с после-
дующим возможным фазовым разделением) в растворе в «пло-
хом» растворителе (см. гл. IV). Изменение свойств полимера в
зависимости от природы растворителя, из которого он выделен,
хорошо известно. Увеличение степени ассоциации каждого поли-
мера в смеси и соответствующее увеличение плотности смеси под-
тверждено экспериментально [13, 14]. На рис. V.4 [15] показана
зависимость Тс ПММА от содержания ПВХ в смеси, полученной
смешением в расплаве при 150—160°C (Тс определяли методом
ДТА и по термомеханическим кривым). При этом Тс ПВХ не из-
меняется. Очевидно, что растворение ПВХ в фазе ПММА приво-
дило бы к снижению Тс ПММА, что не подтверждается результа-
тами опыта рис. V.4).
Изменение компонентов в смеси возможно в результате
различий коэффициентов теплового расширения смешиваемых по-
лимеров в области выше и ниже Тс.
Эти различия вызывают возникнове-
ние значительных «усадочных» напря-
жений при охлаждении расплава по-
лученной смеси, которые могут при-
вести к изменению Тс. Количественное
сопоставление разности термических
коэффициентов расширения с измене-
нием Д проведено в работе [16]. Ис-
Рис. V.5. Зависимость Тг ПС от его содержа-
ния в смеси с разными полимерами (смеси по-
лучали смешением латексов или полимерных
растворов с последующим осаждением раство-
ром СаС1г или выливанием растворов полиме-
ров в нерастворитель; образцы формовали в
прессе):
1 — смесь ПС с цис-полибут адиеном; 2 — то же, с бу-
таднен-стнрольным сополимером; 3 — с бутадиен-нит-
рильным сополимером; 4 — с сополимером винилаце-
тата и винилхлорида; 5 — с поливинилацетатом; 6 —
с поливинилхлоридом.
180
№ кривой
Содержание стирола,
% (масс.)
Мол. масса блока ПС
ЛЫО-б
Мол. масса сополиме-
ра Л1-10—5
J
2
3
4
28
31
46
75
0,207
0,584
0.694
1,95
0,90
2.16
1,65
2,60
следуя различные смеси, содержащие ПС, автор обнаружил, что
повышение Тс ПС зависит от природы второго полимера (рис. V.5).
При этом Тс всех добавляемых полимеров были ниже, чем у ПС
(Тс определяли дилатометрически). Основываясь на предложен-
ной модели смеси, авторы [ 16] рассчитали изменение Тс для слу-
чая, когда сжатие ПС за счет разности коэффициентов расшире-
ния полимеров в смеси реализуется исключительно путем умень-
шения свободного объема. В этим случае Тс, естественно, повыша-
ется. Наблюдалась качественная корреляция между повышением
Тс фазы ПС и ростом разности термических коэффициентов рас-
ширения двух полимеров (см. рис. V.5) при температуре выше
значений Тс обоих компонентов.
В работе [16] отмечается, однако, что различием термических
коэффициентов расширения нельзя объяснить для всех смесей на-
блюдаемое увеличение Тс, которое может быть частично обуслов-
лено «молекулярным взаимодействием контактирующих иолиме
ров». Это особенно характерно для смеси ПС—ПММЛ, для кото-
рой ДТС совершенно не укладывается в рамки развитой теории
181
Такое несоответствие, видимо, можно сопоставить с большой тол-
щиной межфазного слоя на границе раздела ПС—ПММА, как
это следует из данных электронно-микроскопических исследова-
ний и из расчетов по теории Гельфанда (см. гл. IV).
Концепция свободного объема была применена к процессам
стеклования смесей полимеров на основе теории Симхи— Бойера
в работе [17], где была показана возможная локализация свобод-
ного объема в межфазном слое и уплотнение полимеров каждой
фазы при нагревании смеси выше Тс.
Особенностью процесса стеклования в двухфазных смесях по-
лимеров является появление релаксационных переходов, зани-
мающих на температурной шкале промежуточное положение меж-
ду главными релаксационными переходами компонентов смеси.
Такие промежуточные переходы, обусловленные наличием меж-
фазного слоя, легче всего обнаруживаются в блок-сополимерах,
где размер частиц минимален и межфазная поверхность чрезвы-
чайно развита. На рис. V.6 [18] наличие промежуточного макси-
мума механических потерь показано для разных образцов термо-
эластопластов — трпблок-сополимеров бутадиена и стирола типа
СБС. Наличие промежуточного максимума и его величина зави-
сят от природы растворителя, из раствора в котором выделены
образцы для исследования [19, 20].
Наличие промежуточного релаксационного перехода, соответ-
ствующего стеклованию смеси полимеров в межфазном слое, по-
казано и для смесей ПВХ с бутадиен-нитрильными сополимерами
СКН-18 и СКН-26 [21].
Плавление и кристаллизация
Образование совместных кристаллов в смеси полимеров, кото-
рое может наблюдаться при наличии изоморфизма смешиваемых
полимеров,— явление крайне редкое.
Достоверные данные об образовании совместных кристаллов
отсутствуют.
В смесях несовместимых полимеров взаимное влияние компо-
нентов осуществляется через фазовую границу и при кристалли-
зации вызывает явления, в принципе аналогичные явлениям при
стекловании. Вследствие ограниченной растворимости взаимное
проникновение полимеров из фазы в фазу может вызвать замед-
ление кристаллизации. Упорядочение надмолекулярной структу-
ры в каждой фазе под влиянием второго полимера как нераство-
рителя может привести к упорядочению кристаллической струк-
туры. Известны случаи такой раздельной кристаллизации полиме-
ров в смеси, когда параметры кристаллической структуры или
кинетические параметры кристаллизации каждого полимера оста-
ются неизменными в фазах полимеров в смеси. Следующие при-
меры иллюстрируют сказанное.
Смеси натурального каучука и циклизованного натурального
каучука обладают повышенными прочностными показателями,
182
хорошим еопро। ив.'к, пнем не i ирпнию и пониженным уилингшн u.
что свидетельствует об эффекте усиления при преобладании в
смеси либо одного, либо другого полимера. При средних соогно
тениях полимеров от 4:6 до 6:4 прочность понижена, а относи
1ельное удлинение повышено. Рентгенограммы растянутых образ-
цов показывают, что в области средних составов смесей кристал-
лизация НК при растяжении не происходит [22].
Исследуя смеси НК с бутадиен-стирольным каучуком и с по-
лихлоропреном методом крутильного маятника, авторы [23] по-
казали, что БСК замедляет кристаллизацию НК больше, чем это
следует из простого эффекта разбавления, тогда как присутствие
ПХП не сказывается на кристаллизации НК. Это, по-видимому,
указывает на определенную растворимость БСК в НК, в то время
как растворимость ПХП в НК незначительна. По данным [24]
БСК в количестве 30 масс. ч. тормозит кристаллизацию ПХП и
снижает количество кристаллической фазы в нем. Во всех этих
случаях один полимер проникал в другой в процессе смешения
па вальцах при высокой температуре и больших сдвиговых уси-
лиях, так что содержание одного полимера в среде другого могло
быть выше равновесного значения.
^wc-Полибутадиен и Zfwc-полиизопрен (СКД и СКИ) практи-
чески нерастворимы друг в друге [25], а их взаимное влияние
благодаря близости химического строения невелико. Поэтому для
смесей этих каучуков во всей области составов имеется два мак-
симума скорости кристаллизации, положение которых точно со-
ответствует положению соответствующих максимумов для исход-
ных полимеров. Однако при увеличении содержания СКИ в сме-
си отмечена затрудненность роста сферолитов СКД [26].
Механическим динамическим методом вынужденных колеба-
ний исследовали смеси ПЭ—ПП в разных соотношениях. Смеси
получали перемешиванием в расплаве [27]. Температурная зави-
симость модуля упругости каждого из этих полимеров, снятая
при нагревании и при охлаждении, выражается кривой, образую-
щей характерную петлю, указывающую на несовпадение темпера-
тур плавления и кристаллизации. В случае смеси полимеров воз-
никают две петли, что позволяет определить интервал между тем-
пературами плавления и кристаллизации в каждой фазе раздель-
но. На рис. IV.1 с. 126 показана зависимость А7’=7’Пл—Др от
соотношения полимеров в смеси. Как видно из рисунка, при всех
соотношениях полимеров в смеси АТ меньше, чем у индивидуаль-
ных полимеров, что указывает на большее упорядочение струк-
туры расплава в смеси, чем в исходных ПЭ и ПП.
Кристаллизация полимеров в смесях изучена недостаточно.
Вместе с тем представляется важным изучение закономерностей
кристаллизации полимеров в вязкой среде другого полимера, спо-
собного оказывать влияние на структуру расплава и кристалличе-
скую структуру первого полимера, а также установление влияния
кристаллизации одного из компонентов смеси на ее механические
183
свойства. Так, в резиновом производстве широкое практическое
применение нашло добавление нескольких процентов ПЭ в кау-
чук для улучшения его реологических и прочностных свойств до
стадии вулканизации. Известно, что кристаллизация некоторых
органических веществ в среде полимера приводит к резкому из-
менению его механических свойств. Например, кристаллизация
всего 5% фенил-.р-нафтиламина в среде каучука приводит к тако-
му же повышению модуля, как введение 30—40% сажи, являю-
щейся усиливающим наполнителем [28].
Плотность
При смешении полимеров аддитивность плотности означает
пропорциональное изменение ее с изменением объемной доли ком-
понентов. Отклонение от аддитивности указывает на изменение
свободного объема в смеси. Существовало мнение, что «совмести-
мость» компонентов, т. е. способность образовывать однофазную
систему при смешении, обусловливает повышение плотности сме-
си по сравнению с аддитивными значениями; «несовместимость»,
т. е. образование двухфазной смеси — приводит к понижению
плотности. Мнение это в общем виде неверно, поскольку, как
уже отмечено выше, образование однофазной смеси (в равновес-
ном пли «принудительном» процессе смешения) может приводить
к разрыхлению системы, поскольку полимеры в большинстве сво-
ем не изоморфны, а это сказывается на плотности упаковки не
только в кристаллическом, но и в аморфном состоянии. Таким
образом^' плотность смеси должна зависеть от способа ее получе-
ния и не может являться критерием «совместимости» компонен-
тов. Сказанное видно на примере рис. V.7 [13], где плотность сме-
* сей ПС и ПММА оказывается
выше или равна аддитивной ве-
личине в зависимости от способа
получения смеси. Характерно,
что если те же смеси получать
смешением расплавов на валь-
цах, то при всех соотношениях
компонентов плотность сущест-
венно ниже аддитивной [13].
Примеры смесей несовместимых
полимеров с плотностью выше
аддитивной приведены в работе
Рис. V.7. Зависимость плотности пленок
смеси ПС —ПММА от состава при раз-
ных условиях формования образцов
(пунктир — аддитивная зависимость):
I — пленка получена из раствора в толуоле
при 20 °C; 2 — то же, при 80 °C; 3 — то же,
при 100 °C; 4 — то же, при 20 °C с последую-
щим прогревом 150 ч при 130 °C.
184
[29]. Плотность таким образом является показателем структуры
смеси, но характер ее изменения при смешении не отвечает на
вопрос о «совместимости» полимеров.
Диффузия газов и жидкостей
Диффузия газов и жидкостей через пленку (мембрану) из сме-
си полимеров определяется закономерностями диффузии через
многофазные (например, наполненные) системы при условии, что
частицы диспергированного полимера не участвуют в процессе
переноса диффузанта. Однако и в этом случае увеличение содер-
жания дисперсной фазы может привести к обращению фаз, что
резко проявляется, например, в характере зависимости коэффи-
циента диффузии от состава смеси полимеров. Если оба компо-
нента двухфазной системы участвуют в переносе вещества (и,
как правило, с разными коэффициентами диффузии), то в зако-
номерностях диффузии появляются дополнительные особенности.
Введение полимера с пониженной газопроницаемостью умень-
шает газопроницаемость основного полимера, и поэтому путем
смешения полимеров удается регулировать показатели газопро-
ницаемости. Изменение газопроницаемости происходит по более
сложным закономерностям, чем эффект разбавления одного поли-
мера другим. Фазовая структура смеси, по-видимому, играет здесь
важную роль [30].
Законно предположить, что скорость диффузии (или набуха-
ния) смеси полимеров в жидкости, в которой способен набухать
только один компонент, должна определяться тем. какой полимер
образует непрерывную фазу в смеси. На примере смеси ПС с
ПБ, набухающей в диметилформамиде, было показано [31] уве-
личение скорости набухания с ростом содержания ПС в смеси.
Только при содержании ПС, равном 50%, скорость набухания
становится равной скорости набухания самого ПС, что указывает
на образование действительно непрерывной фазовой структуры
ПС в смеси (см. также гл. IV). Р этот момент между частицами
дисперсной фазы действительно возникают физические контакты,
обусловливающие частичную коалесценцию частиц.
Зависимость коэффициентов диффузии в смеси фенолофор-
мальдегидная смола — поливинилбутираль была изучена для двух
типов диффузантов — раствора азотной кислоты и раствора ще
лочи [32]. Кислота диффундирует через ПВБ и плохо переносит-
ся фазой смолы; щелочь, наоборот, диффундирует преимуществен
но через фазу смолы. Зависимость коэффициентов диффузии обо
их указанных селективных диффузантов от состава смеси спиде
тельствовала об обращении фаз в системе с изменением состава.
Наиболее подробное исследование газопроницаемости смесей
полимеров на примере смеси СКЭПТ — бутилкаучук было прове-
дено в работе [33]. В смесях каучуков с практически одинаковой
вязкостью, обеспечивающей наилучшее смешение и наибольшую
185
XgP-tgPgK
1,0
0,6
0,6
0,0
0,2
Рис. V.8. Зависимость среднечислового размера дисперсных фаз (а) и ширина
распределения (б) от состава смеси БК — СКЭПТ:
О — невулканизованные смеси; ф — вулканизаты (на осях ординат отложены ^пэф-102, нм).
Рис. V.9. Обобщенная зависимость коэффициентов диффузии и проницаемости
азота для смесей полимеров:
/ — БК — СКЭПТ при 150 °C; 2— то же, при 100 °C; 3 — то же, при 80 °C; 4 — то же, при
60 °C; 5 —НК—ПИБ; 6 — НК — ПВХ.
дисперсность частиц, и в вулканизатах этих смесей размеры ча-
стиц были равны и закономерности диффузии были одинаковы-
ми. Размер частиц каждого полимера увеличивался с увеличе-
нием его содержания в смеси (рис. V.8), что было установлено
электронно-микроскопически. Точки па осях ординат на рис. V.8
характеризуют размер структурных (не фазовых!) микронеодно-
родпостей в смешиваемых каучуках. Диффузию и проницаемость
образцов по азоту исследовали газохроматографическим методом
[30]. Диффузию паров бензола исследовали сорбционным мето-
дом на весах Мак-Бена.
На рис. V.9 показана зависимость эффективного коэффициен-
та диффузии D и проницаемости азота Р в приведенных коорди-
натах для смеси БК—СКЭПТ при разных температурах [33] и
для смесей других каучуков по литературным данным ([30],
с. 256). Единая кривая показывает, что для данных пар полиме-
ров закономерности структурообразования в смесях, а следова-
тельно, и закономерности проницаемости одинаковы.
Для смесей БК—СКЭПТ, в которых дисперсионная среда
СКЭПТ, изменение коэффициента диффузии и проницаемости с
составом описывается следующей зависимостью:
В Р<₽2
In D - In Пскэпт — -д' fi + Рф2 (3)
186
Для смеси, где дисперсионная среда
некие 'Ь
1пО = 1пРбк + 7Г Л+к
(4)
Здесь fi и f? — доли свободного объема СКЭПТ и БК; ₽ =
=/2—/г, В — постоянная.
Очевидно, что уравнения (3) и (4) применимы к любой двух-
фазной системе, где непрерывной средой является полимер с
большей (3) или меньшей (4) проницаемостью.
Аналогичные закономерности изменения газопроницаемости
получены для смесей ПВХ — этиленвинилацетатный сополимер
(ЭВА) при содержании винилацетата в составе сополимера 45%
[34]. При этом двухфазная структура смеси была подтверждена
S-образиой зависимостью оптической плотности от состава смеси.
По данным оптической плотности и проницаемости обращение
фаз происходит уже при содержании ЭВА более 7,5%. Характер-
но, что смеси ПВХ с ЭВА, содержащим 65% винилацетата, явля-
ются однофазными, на что указывает практически линейная за-
висимость коэффициента диффузии от состава. Однако энергия
активации процесса диффузии меняется с составом по кривой с
максимумом.
Обычные методы определения коэффициента диффузии для
двухфазной системы, в которой и среда и дисперсная фаза участ-
вуют в процессах сорбции и переноса, не учитывают двухфазно-
стн, и расчет; проводится так, как если бы система была одно-
фазной. В действительности для таких систем целесообразно раз-
личать коэффициенты диффузии для дисперсной фазы Е)ц (ло-
кальный коэффициент диффузии) и для непрерывной фазы —
(макроскопический коэффициент диффузии). Изменение Dj и Е)ц
с составом смеси БК и СКЭПТ представлено на рис. V.10 [33].
Естественно, что Dj уменьшается по мере увеличения содержания
бутилкаучука в среде СКЭПТ, поскольку увеличение концентра-
ции частиц дисперсной фазы в единице объема приводит к удли-
нению траектории диффундирующих молекул газов (или раство-
рителей) вследствие эффекта огибания дисперсных частиц. Ло-^
кальиый коэффициент диффузии Он
является функцией свободного объема _3
материала дисперсных частиц, т. е.
функцией плотности упаковки макро- ё? Г/
молекул в частицах дисперсной фазы.
Его постоянство независимо от соста- с
----------------------------------- S’
Рис. V.10. Зависимость эффективного (1, 2), _Е
макроскопического (.3) и локальных (4, 5) ко-
эффициентов диффузии паров бензола в смеси
БК — СКЭПТ при 20 °C. Кривые 1, 2 рассчи-
таны для двух различных размеров частиц
дисперсной фазы БК (1—30 нм; 2—80 им).
187
ва смеси указывает на то, что структура полимера в частицах
дисперсной фазы тождественна структуре исходного полимера.
Изменение Di с составом системы описывается следующим
уравнением [33]:
( в <ь» \
^ = ^ехр(-7Гта^) (5)
где £>i — коэффициент диффузии в полимере 1; £>i — макроскопический коэффи-
циент диффузии (миграция в дисперсной среде).
Уравнение (5) написано для случая, когда компонент 1 явля-
ется непрерывной фазой.
Коэффициент диффузии смеси полимеров связан с коэффици-
ентами Di и Du следующим образом [35]:
/2 (1 + Г) Ч?! Т £>ц
где Г—постоянная Генри для суммарной изотермы сорбции; R — радиус частиц
дисперсной фазы; I — толщина образца.
Подставив в (6) выражение (5), получим уравнение, описы-
вающее изменение коэффициента диффузии в двухфазной си-
стеме:
/ з ф, \
exp I — —с , 1D2
\ 11 1 Ч 2 /
/ В ф2 \
— (l+D-^expf- --г^у + Р2
(7)
Расчет по этому уравнению коэффициента диффузии для раз-
личных по размеру частиц дисперсной фазы показан на рис. V.10,
который демонстрирует хорошее согласие теории и эксперимен-
тальных данных. Важность уравнения (7) еще и в том, что оно
показывает зависимость диффузионных характеристик смеси по-
лимеров от размеров частиц: при одном и том же составе смеси
изменение размеров частиц в указанных пределах приводит к из-
менению коэффициента диффузии на 20%.
Определяя зависимость коэффициентов диффузии или прони-
цаемости смесей полимеров от состава, следует обращать вни-
мание на постоянство структуры смеси во время опыта, что осо-
бенно важно для смесей, содержащих стеклообразные или кри-
сталлические полимеры. Так, имеются указания, что в процессе
сорбции жидкого или парообразного гексана пленками смеси
ПС — полифенилепоксид происходит растрескивание матрицы ПС
и быстрая кристаллизация дисперсной фазы полифениленоксида.
Такие изменения структуры смеси заметно влияют на характер
зависимости диффузионных характеристик от состава смеси.
188
Оптические свойства
Оптические свойства смеси полимеров зависят от прозрачно-
сти, цвета и показателей преломления смешиваемых компонентов,
от размера частиц дисперсной фазы и присутствия других ингре-
диентов, в особенности стабилизаторов и наполнителей. Необхо-
димым условием высокой прозрачности смеси является прозрач-
ность смешиваемых полимеров, применение нетемнеющего на све-
ту стабилизатора и отсутствие наполнителей. При соблюдении
этих условий прозрачность смеси определяется размером частиц
дисперсной фазы и соотношением показателей преломления сме-
шиваемых компонентов.
Если показатели преломления компонентов различаются, их
можно сблизить прививкой или сополимеризацией, вводя в одну
или обе фазы смеси тот или иной мономер. Не исследована воз-
можность сближения показателей преломления фаз введением
селективного пластификатора, концентрирующегося в одной из
фаз и меняющего ее показатель преломления в нужном направ-
лении.
Примером смеси, имеющей выраженную двухфазную структу-
ру при полной прозрачности благодаря равенству показателей
преломления, является смесь цис-полиизопрена и цис-полибута-
диена.
Рассеяние света и потеря прозрачности смеси полимеров про-
исходит вследствие наличия поверхности раздела фаз, которая
увеличивается с уменьшением размера частиц. Поэтому прозрач-
ность должна монотонно возрастать с увеличением размера ча-
стиц. В действительности прозрачность начинает расти, когда
размер частиц становится сопоставим с длиной волны падающего
света.
В области размеров частиц, обеспечивающих дифракцию све-
товых волн, последние рассеиваются под разными углами в за-
висимости от длины волны. Это явление, обычное для коллоид-
ных систем, приводит к опалесценции дисперсной системы, кажу-
щейся более голубой в отраженном и более оранжевой в прохо-
дящем белом свете.
При широком распределении частиц по размерам, и в особен-
ности, когда внутри частиц дисперсной фазы присутствует микро-
фаза матричного полимера, смесь не только опалесцирует, но и
имеет цветовой оттенок (например, коричневый для дисперсии
3% ПС в матрице ПММА). В этой смеси наряду с крупными,
присутствуют частицы размером 0,075—0,125 мкм [36].
Максимум мутности системы при соотношении показателей
преломления частицы и среды 0,93 и 1,20 соответствуют 5 и 1 мкм
соответственно (см. гл. IV). Если размер частиц снижается до
50—70 нм, то частицы рассеивают очень мало и система про-
зрачна. Очевидно, что оптимум оптических свойств не совпадает
с оптимумом механических свойств, а потому оптические свойства
189
смесей нельзя регулировать только путем изменения размеров ча-
стиц дисперсной фазы.
Поскольку полимеры в высокоэластическом состоянии харак-
теризуются более сильной температурной зависимостью удельного
объема, а следовательно, и показателей преломления, чем в стек-
лообразном состоянии, а также вследствие различия температур-
ных коэффициентов показателя преломления разных полимеров,
смесь, прозрачная при комнатной температуре, может помутнеть
при нагревании или охлаждении.
Горючесть
Термическая устойчивость и горючесть смесей полимеров за-
висят от того, может ли легко деструктирующпйся при нагрева-
нии полимер инициировать термодеструкцию другого, более ста-
бильного полимера. Если такого взаимного влияния не наблюда-
ется, то термодеструкция и особенно воспламеняемость смеси
снижаются при добавлении термостабильного или трудновоспла-
меняющегося (негорючего) полимера.
Эффект защиты горючего полимера негорючим максимален
тогда, когда негорючий компонент образует непрерывную фазу
в смеси. Так, смесь легко горючего АБС и негорючего ПВХ при-
ближается по стойкости к воспламенению к ПВХ, когда содержа-
ние последнего в смеси составляет 50% и он образует непрерыв-
ную фазу. Аналогичный эффект достигается введением негорю-
чего хлорированного ПЭ в обычный ПЭ для создания негорючей
кабельной изоляции.
Фазовая структура смеси влияет на горючесть двухфазных
систем больше, чем соотношение полимеров.
§ 2. МЕХАНИЗМ ДЕФОРМАЦИИ
Однофазные смеси полимеров деформируются как однород-
ные системы, если в процессе растяжения нс происходит расслаи-
вание. Двухфазные системы редко деформируются однородно:
если модули фаз сильно различаются, то перенапряжения на гра-
нице раздела фаз могут привести к отслаиванию матрицы от ча-
стиц дисперсной фазы. Механизм разрастания трещины в про-
цессе разрушения рассматривается ниже.
Имеются косвенные указания, что 'при больших деформациях
в однофазных (пли близких к однофазным) системах [37, 38] за-
трудняется кристаллизация и ориентация при растяжении. При-
чиной является то, что в таких смесях с соотношением компонен-
тов, близким к 1 : 1, отсутствует совокупность макромолекул оди-
накового строения, способных образовать кристаллическую
структуру в растянутом состоя ни!?) Это проявляется как в специ-
ально приготовленных однородных смесях НК и БСК [37], так и
в однофазных смесях ПВХ с бутадиен-нитрильным каучуком [38].
190
Рис. V.11. Микрофотография структуры пленки (а) и схема взаимодействия мат-
рицы (ПЭТФ) и наполнителя (ПП) (б). Микрофотография получена с пленки
растянутой в одном направлении при 90 °C со скоростью 2’50%/мин.
Прямого определения структурных превращений в указанных ра-
ботах не делалось.
В смесях полимеров с близкими модулями диспергированных
частиц и матрицы величины деформации обеих фаз довольно
близки. Отслаивание по поверхности раздела фаз наблюдается
лишь при больших деформациях, J
Проведенные модельные опыты с глобулой полимера, поме-
щенной изолированно или рядом с другой такой же глобулой в
матрицу другого полимера, показали, что закономерности дефор-
мации совокупности двух или более частиц отличаются от дефор-
мации одной частицы. Деформация частицы, например ПВА в
матрице ППБ, уменьшается при нанесении эмульгатора на по-
верхность частицы (при Т>Т, поливинилацетата) [39]. Таким
образом, даже при относительно близких значениях модулей фаз,
когда оба полимера находятся в одинаковом физическом состоя-
нии, деформация частиц и матрицы не является одинаковой.
Различие в величине деформации фаз особенно заметно, когда
полимеры находятся в разных физических состояниях [40].
На рис. V. 11 показана микрофотография отслоения матрицы
ПЭТФ от наполнителя ПП при 90 °C и скорости одноосной вы-
тяжки пленки 250 %/мин. Возникающий ореол ограничивает об-
ласть отслаивания. На том же рисунке приведена схема действия
сил на частицы в расслоившейся системе, из которой следует, что
d
P" = P'~D W
iде P'— растягивающее усилие; Р"— сила сжатия частицы; d — диаметр части-
цы; D — диаметр полости, возникшей при отслоении.
Таким образом, в начале растяжения, когда расслаивание еще
не произошло, частицы деформируются под действием сил растя-
жения, непосредственно приложенных к межфазной поверхности,
и сил сжатия, вызванных уменьшением поперечного сечения мат-
рицы при деформации. После отслаивания деформация частицы
может быть обеспечена лишь силой сжатия (см. рис. V.11), кото-
рая уменьшается с ростом диаметра полости отслаивания [41].
191
Таким образом, отслаивание часто приводит к прекращению де-
формации частиц.
Частицы пластмассы в матрице каучука деформируются не-
значительно и главным образом при наличии химических связей
па границе раздела фаз (например, в блок-сополимерах), а ча-
стицы каучука в матрице пластмассы деформируются вместе с
матрицей. В смеси же твердых в условиях деформации полимеров
(стеклообразных или кристаллических) способность частицы со-
противляться деформации определяется условием [41]
Л = °Г,(СЖ) > 1 (9)
IY
где Cv(ok) — предел текучести материала частицы при сжатии (в расчете на
истинное сечение); />—предел текучести материала матрицы в условиях растя-
жения.
Значения пределов текучести разных полимеров в условиях
растяжения известны, и поэтому измерение предела текучести
при сжатии может дать ответ на вопрос о поведении частицы при
деформации смеси после возникновения отслоений.
Неоднородность структуры смеси полимеров, как и всякой
композиционной системы, приводит к неоднородности деформации
в микрообъемах. Это можно продемонстрировать на примере сме-
си ПЭТФ—ПС, в которой при растяжении со скоростью 200—
4000%/мин деформируется практически лишь матрица ПЭТФ,
а средние размеры частиц ПС остаются неизменными. Расслаи-
ваний не наблюдается [42].
Пусть образец в целом деформируется на величину е, а мат-
рица на величину ем, причем ем>е, поскольку часть образца заня-
та недеформируемым полимерным наполнителем. Если диаметр
частиц d, а расстояние между центрами частиц наполнителя до
деформации /, то после деформации расстояние между центрами
частиц равно /(е+1), а кратчайшее расстояние между поверхно-
стями частиц Z(e+1)—d. Первоначальная (до деформации) дли-
на участка матрицы между поверхностями частиц I—d, поэтому
удлинение матрицы равно
ем = ------yTTd-------- (1 °)
Можно показать, что отношение d/l равно объемной доле на-
полнителя в смеси. Поэтому
®M = -T=V (")
Уравнение такого же вида можно получить и другим спосо-
бом [43].
Уравнение (11) позволяет рассчитать действительную дефор-
мацию полимера матрицы. Так, из экспериментальных данных
рис. V.12 можно найти для смеси ПЭТФ с 20% ПС напряжение
192
Экспериментально опре-
Рис. V.12. Зависимость напряжения от дефор-
мации при растяжении пленок ПЭТФ и его
смесей с ПС со скоростью 1000%/мин при 90 °C:
1— ПЭТФ; 2 — ПЭТФ+10% ПС; 3— ПЭТФ+20% ПС; ы
4 — ПЭТФ+30% ПС. С;
____ ________________________________ 5
V?
7,2 МПа при е = 100%, а по уравнению
(11) и,, = 129%. Для иенаполненного
П.41Ф при том же о величина е=Ем
сгя ыпляет 150%- Несовпадение этих
ш iii'iiiii свидетельствует о том, что
II »1Ф в присутствии ПС становится
б"'нт жестким (менее деформируемым).
jii юппая плотность полимера-матрицы (ПТЭФ) оказалась выше
п । ниш ди исходного ПТЭФ, и это превышение плотности сохраня-
ла I. при деформации. Таким образом, уменьшенной деформируе-
ма >г in матрицы соответствует повышенная плотность.
Прямые экспериментальные данные свидетельствуют о нали-
чии межфазных слоев в наполненных полимерах и в мономерах
to их отверждения в присутствии наполнителя [44]. Можно пред-
iio.io кп 1Ь поэтому, что увеличение плотности матрицы характер-
in in слоев, примыкающих к наполнителю.
Приняв, что размер недеформированных частиц наполнителя
и пбьемс матрицы увеличивается за счет межфазного слоя, а ос-
Твиш.тяся часть ПЭТФ деформируется так же, как ПЭТФ в He-
ir по i пенной системе, можно рассчитать по уравнению (11) кажу-
ппг<я шачение объемной доли наполнителя фк. Рассчитанные
1П1Г111П1Я <рк приведены в табл. V.I.
II । табл. V. 1 видно, что <рк больше, чем <р, но по мере увеличе-
I и п происходит их сближение. Зная qK, .можно рассчитать сум-
Mnpiivio толщину межфазного слоя с измененной плотностью, по-
(t ) и.ку средний размер частиц наполнителя (3 мкм) определен
и нк риментально. Уменьшение <рк с ростом напряжения при де-
формации указывает на уменьшение толщины межфазного слоя,
Которое можно объяснить тем, что свойства этого слоя постепен-
но меняются с удалением от поверхности частицы ПС. С ростом
напряжения в матрице ПЭТФ в процесс деформации вовлекают-
ся нее новые внешние участки межфазного слоя.
Г л блица V.I. Значения кажущейся объемной доли <рк частиц ПС
в смеси ПС—ПЭТФ (90 °C, скорость растяжения 1000%/мин)
при разных значениях напряжения вытягивания с
( "Д< ржание I ]< (б. доли а, МПа
3,4 5,2 7,1 11,0 17,0 23,6 28,0
0,12 0,240 0,200 0,180 0,151 0,132 0,121 0,118
0.23 0,460 0,400 0,334 0.292 0,260 0,244 0,231
0,34 0,680 0,580 0,506 0,450 0,378 0,358 0,343
13—244
193
Рис. V.13. Изменение предела текучести
ПЭТФ в его смесях с ПС по толщине межфаз-
ного слоя при различных скоростях деформа-
ции:
О-ПЭТФ+10% ПС; X —ПЭТФ+20% ПС; Д —
ПЭТФ+30% ПС; 1 — скорость деформации 200%/мин;
2— 1000%/мии; 3— 4000%/мин.
Зная толщину неразрушенной части межфазного слоя при
данном напряжении, можно рассчитать зависимость предела теку-
чести полимера в межфазном слое от расстояния до поверхности
частицы, т. е. проследить изменение механических свойств поли-
мера по толщине 6 межфазного слоя (рис. V.13). Как видно из
рис. V.13, кривые для разного содержания наполнителя совпада-
ют, что указывает на независимость свойств слоя от содержания
наполнителя. В то же время температура и скорость деформации
влияют на зависимость <тг=[(6), что указывает па релаксацион-
ный характер деформации межфазного слоя.
§ 3. РЕОЛОГИЧЕСКИЕ СВОЙСТВА
Количественная оценка зависимости вязкоупругости смесей
полимеров от их состава проводилась применительно к началь-
ному модулю упругости и начальной ньютоновской вязкости. За-
висимость модуля от состава смеси при больших деформациях
и зависимость вязкости от состава при наличии значительной
аномалии являются способом оценки свойств смеси, но не ее
структуры, поскольку сравнение экспериментальных данных с
расчетом на основе адекватной модели невозможно.
Однофазные смеси
Впервые эмпирическую формулу для описания зависимости
вязкости от состава жидких смесей предложил Аррениус [45,
с. 483]:
1п Г] = А\ In Дх + V2 In Г], (12)
где и Лг2 —мольные доли компонентов; тц и т]2 — вязкости жидких компонен-
тов смеси.
Формула Аррениуса означает логарифмическую аддитивность
вязкости, когда состав смеси выражен в мольных долях.
Экспериментально установлено, что для начальной ньютонов-
ской вязкости т]<| смесей фракций полимеров (на примере фрак-
ций полистирола) характерна аддитивная зависимость логариф-
мов вязкости от массовых долей Wi компонентов смеси [16]:
1g По “= ®i 1g Пл + ЧЬ 1g Т)о2 (13)
194
• 1бе эти формулы выражают логарифмическую аддитивность
11кости, что является частным случаем более общего закона ад-
III।пкности изменений вязкости в зависимости от температуры,
in к-кулярной массы, состава и других факторов [47].
Согласно [45], выражение типа (12) справедливо лишь для
|\чая, когда добавление второго компонента не изменяет энер-
iiiio активации вязкого течения, т. е. является лишь грубым при-
Л шженнем в качестве обобщенного закона течения жидких сме-
||ц Если взаимодействие между разнородными компонентами
fni.ii.nie, чем между однородными, то смесь образуется с умень-
шу пнем объема и кривая вязкость — состав проходит выше аддп-
пцшой [см. уравнение (12)]. В случае же, когда взаимодействие
и смеси меньше, чем в смешиваемых однородных жидкостях,
смесь образуется с увеличением объема и вязкость меняется по
кривой ниже аддитивной. В однофазных жидких смесях вязкость
выжна меняться с составом по уравнению типа [45, с. 483]:
In т) = -у- Nf In Т)! + -у- A'i In Т]2 + N±N2 In ц1>2 (14}
। в 41.2—величина, отражающая взаимодействие молекул разного типа.
Оценка взаимодействия разнородных молекул и соответствую-
щий расчет вклада этого взаимодействия в величину вязкости за-
ipv щительна для низкомолекулярных смесей и в особенности для
пи.inмсров. По этой причине более удобно описание зависимо-
||| вязкостй от состава смеси на основе какой-либо зависимости,
шляющейся бесспорной. В качестве такой можно выбрать прин-
цип аддитивности молекулярной массы смеси полимеров [48]:
Ми) = М: W± т- МаЕ>2 (15)
Зная зависимость вязкости от молекулярной массы для каждо-
Н1 полимера, можно рассчитать вязкость смеси как расплавов,
i.iK и растворов полимеров. Так, для расплава полимера:
Т)0=^ 116)
и i|n начальная ньютоновская вязкость расплава; К. и а — константы.
I ели смесь образована фракциями одного и того же полимера,
hi в этом случае в широком интервале молекулярных масс Л'1 =
А • К и 01 = 02 = а. Тогда можно написать:
i)o(c«) = (^ПоГ + а’2 ПюТ X17)
Ч"им>, Hoi и т)о2 — начальная ньютоновская вязкость смеси и смешиваемых
1МЧ1ИН нтов; »i и W2 — массовые доли компонентов.
Уравнение (17) описывает вязкость смеси макромолекул pai
пип । 1 ины, которые образуют «неаддитивную» сетку зацеплений,
и ыи'орой число узлов с увеличением длины молекул растет очень
Гии ipo Очевидно, что при переходе к области молекулярных
I । 195
масс меньше критической, когда сетка зацеплений отсутствует и
а= 1, уравнение (17) переходит в (13).
Уравнение (17) применимо для расчета вязкости однофазной
смеси взаиморастворимых полимеров с одинаковой гибкостью
макромолекул, когда значения Л’ и а смешиваемых полимеров и
их смеси одинаковы. Последнее означает, что узлы флуктуацион-
ной сетки зацеплений в исходных полимерах и в их смеси релак-
сируют с одинаковой скоростью. Приведенный выше подход впер-
вые был применен для расчета светорассеяния в разбавленных
растворах смеси полимеров [49] (см. также гл. II). Впоследствии
он был использован для расчета вязкости расплавов [48] и рас-
творов [51] смесей полимеров, а также вязкоупругих характери-
стик смесей фракций [50], в том числе и их зависимости от ча-
стоты ы. Так, обобщенная формула для расчета динамической
вязкости г]' имеет вид [50]:
Ч' (со) = t®, [т)1 + ^21»12 (о>/М1,/а1“ (18)
где >,; = — параметр, отражающий смещение спектральных функций
компонентов в их релаксационном спектре при получении смеси; — средне-
массовая молекулярная масса смеси; k - - эмпирическая константа, сравнительно
мало меняющаяся при переходе от одного гомологического ряда полимеров к
другому.
Уравнение (18) переходит в (17) при со->-0.
Из (18) можно получить формулу для модуля потерь
б"И= )щ1[бП<оА1)л1]1''“ + щ2[02(«А2)Л2]1'а)а (19)
а затем, используя любой численный метод, найти выражение
для G'(w).
В ряде работ (см., например [52]) было предложено уравнение
для вязкости смеси фракций полимеров, справедливое в том чис-
ле и при значительных различиях в молекулярной массе компо-
нентов:
Чем — д,ф2 (20)
где Чем, i]i и пг — вязкость смеси и фракций; <pi и — объемные доли фракций;
D и У2,1 вспомогательные безразмерные критерии (Z) = ZjtlX!j2и A’a.i = Хя'Пг/М'П!);
и ).2 — константы, определяющие относительное сопротивление трению макро-
молекул фракций полимера в смеси.
В координатах 1g г)— состав это уравнение дает кривые, ле-
жащие ниже аддитивных зависимостей.
Сопоставление экспериментальных и расчетных данных по вяз-
кости или модулям смесей полимеров или их фракций, имеющих
однофазную структуру, не позволяет отдать во всех случаях пред-
почтение какому-либо из предложенных уравнений. Так, в рабо-
те [53] на примере полибутадиенов с узким ММР, и поэтому не
имеющих аномалии вязкости, показано, что зависимость вязко-
сти от состава смесей полибутадиенов с разной молекулярной
196
массой хорошо описывается уравнением (17), т. е. кривой, лежа-
щей несколько выше аддитивной зависимости. В той же работе
чстановлено, что в однофазной смеси ^нс-полиизопрена и развет-
вленного полибутадиена (каучук СКВ) вязкость в логарифмиче-
ских координатах меняется линейно с составом. В то же время
I 1я смеси фракций ПС зависимость вязкости от состава является
а щитивний [46], а для смесей фракций ПВА и ПИБ (ссылки
см. в [52]) описывается уравнением (20), т. е. вязкость меняется
по кривой, лежащей ниже аддитивной. В этой связи заметим, что
при большой вязкости полимеров не удается уверенно получать
равновесные смеси, в том числе смеси взаиморастворимых поли-
меров и фракций одного и того же полимера. Смешение в отсут-
ствие растворителя может быть недостаточным и привести даже
к возникновению фазовой неоднородности в смеси взаиыораство
рпмых полимеров [53]. При получении же смеси из растворов
свойства ее могут зависеть от качества растворителя. Должна
учитываться также возможность фазового расслаивания в смеси
фракций одного и тою же полимера, сильно различающихся по
молекулярной массе. Известно, во всяком случае, что вязкость
смеси фракций ПЭ, полученной в отсутствие растворителя, увели-
чивается с улучшением качества смешения [54].
Зависимость модуля потерь от состава смеси фракций доста-
1ОЧП0 хорошо описывается уравнением (19), в том числе и для
мееей, в которых на частотной зависимости модуля упругости
имеется перегиб, соответствующий релаксационному переходу вто-
рой фракции [50]. Для смесей взаиморастворимых полимеров, та-
ких как ПИП—СКВ [53], модуль, определенный по углу наклона
швейного участка кривой напряжение — деформация, меняется
пшейно с составом.
Двухфазные смеси
( д шествующие теории вязкоупругости смесей полимеров по-
шо.шют приближенно рассчитать модуль или вязкость двухфаз-
iKiii смеси взаимонерастворимых полимеров в случае, если значе-
ния рассчитываемых параметров (вязкость, модуль) смешиваемых
компонентов сильно различаются. Практически .расчет и экспери
мсп гильпая проверка расчета модулей применимы только к сме-
<ям каучуков с пластмассами. Для смесей двух пластмасс дли
»цу\ каучуков, модули которых различаются незначительно, рас-
ям модуля нецелесообразен ввиду значительного расхождения
ич перпмснта и теории, не учитывающей многих факторов, ха рак
кртмощих структуру смеси. При этом относительная ошибка
рпсчета тем больше, чем меньше разница модулей смешиваемых
компонентов.
I лк им’образом, сравнение теории с экспериментом можно про-
iiKiiiii. на смесях полимеров, эквивалентных наполненным поли-
M.piM (каучук в пластмассе или, наоборот, пластмасса в каучу-
1у7
ке). По этой причине необходим анализ уравнений, полученных
как в применении собственно к смесям полимеров, так и перво-
начально выведенных для наполненных систем. Количество урав-
нений, предложенных для описания свойств двухфазных компо-
зиционных материалов, велико, и они приведены в соответствую-
щей литературе [55, 56], поэтому в данном разделе рассматрива-
ются основные расчетные формулы и некоторые эксперименталь-
ные данные, свидетельствующие о степени их достоверности.
Зная влияние полимера-наполиителя на вязкость смеси, можно
рассчитать его влияние и на модуль. При этом надо иметь в виду,
что относительный прирост модуля и вязкости с введением дан-
ного количества наполнителя одинаков, если при деформации не
происходит изменения объема материала, т. е. коэффициенты Пуас-
сона фазы v/ и матрицы vm одинаковы и равны 0,5. При этом
— = -£- (21)
flm Grn
где I] и G — вязкость и модуль смеси; Gm и i]m — модуль и вязкость матрицы.
Если частицы дисперсной фазы сферические, а модуль дис-
персной фазы существенно больше модуля матрицы, то с учетом
изменения объема системы при деформации:
Таким образом, для наполненных систем при vrn=v^=v=0,5
нижеследующие (23—27) уравнения для вязкости эквивалентны
уравнениям для модулей, и наоборот.
В разбавленных суспензиях частицы вносят возмущения в
плавное течение жидкой матрицы, что приводит к повышению
вязкости системы. Эйнштейн, проанализировав эту проблему для
случая частиц, малых по сравнению с размером сосуда и боль-
ших по сравнению с молекулами жидкости, предложил формулу
-’1 = । + (23)
Чт
где ср/ — объемная доля дисперсной фазы; кв — коэффициент Эйнштейна.
Коэффйциент kE, равный 2,5 для сферических частиц, увели-
чивается для анизометричных частиц при малой скорости сдвига
и уменьшается до
1.15/
kE~ л In 2/ (24)
для вытянутых частиц, ориентирующихся в потоке при большой
скорости сдвига. Здесь / — отношение большой и малой осей ани-
зометричной частицы.
Для разбавленной эмульсии, когда необходимо учитывать вяз-
кость среды и вязкость капель, уравнение, аналогичное (23),
198
лы.п) получено Тэйлором [57]:
Ч___, , „ г 2Пге + 5?у
Чт -*+2’ь 5ф„ + 5щ
(25)
IJ этом уравнении было принято во внимание наличие меж-
i|i.i того натяжения, но не учтено скольжение по межфазной гра-
нице.
Уравнение (23) экспериментально хорошо подтверждается до
объемной концентрации дисперсной фазы не более 6%. С ростом
содержания дисперсной фазы предпочтительнее уравнение Гута
|58|:
-^- = 1+2,507+14.0^+... (26)
। и- коэффициент при третьем члене ряда может быть меньше
и колебаться для многих систем в пределах 7—10. Для сфериче-
< ких частиц достаточно большого размера при отсутствии замет-
ного эффекта оседания уравнение (26) справедливо до 30% (об.)
«одержания дисперсной фазы [59].
Принципиально новым подходом к расчету вязкости (соответ-
।пенно модулей) композиционных материалов явилось уравнение
Муни
Ч \ ЬеЧГ /1У7.
I Чти ) 1—4>1/4>макс (27)
t ни гывающсё то обстоятельство, что для эффекта повышения вяз-
кости (модуля) существенна не сама величина tpf, а доля этой
пел и чины от предельной концентрации фмакс. при которой возни-
।зет плотная упаковка частиц и выше которой наполнение невоз-
можно. Отношение ф//фмакс является поэтому величиной, характе-
рп »ующей степень заполнения матрицы частицами дисперсной
<|> । пи в виде доли от предельно плотной упаковки.
Величину фмаис удобно определить экспериментально [60] по
объему пустот в известном объеме сосуда, заполненного частица-
ми наполнителя. Можно воспользоваться значениями истинной
и ни пости вещества наполнителя и его насыпной плотности. Отно-
шение <pf/(pмакс> где задается в данном опыте, а фмакс определя-
ки экспериментально, является мерой заполнения объема в до-
О1ч от максимального. При этом зависимость модуля сдвига G от
ч '|«п,с выражается для данного типа полимера-матрицы единой
। itiiiciiMOCTHO для всех наполнителей в широком интервале раз-
м-рои частиц [60]. В смеси полимеров, полученной механическим
in pi мгшиванием расплавов, предельное заполнение невозможно
онрг юлить экспериментально. Оно может быть оценено прибли-
।«lino по моменту образования непрерывной структуры дисперс-
ной фазы.
< >бразование агломератов наполнителем высокой дисперсности
рнно шт не только к заметному изменению фмакс, но и к увеличе-
199
нию коэффициента Эйнштейна [55]:
2,5(VS+VL)
kE =----------- (28)
где Vs — фактический объем твердых частиц в агломерате; Vl — объем вещест-
ва матрицы в эффективном объеме агломерата.
Проблема определения максимального заполнения объема ча-
стицами дисперсной фазы в смесях полимеров часто не находит
решения в связи со сложной формой частиц и образованием ими
непрерывной фазы в результате коалесценции при смешении.
В связи с этим приведенные выше уравнения для вязкости и мо-
дулей применимы к смесям полимеров в области содержания
дисперсной фазы до 15—20% (об.), а в случае определения моду-
лей — также и при условии Vf=Vm=v=0,5.
Системы, характеризующиеся значениями коэффициентов Пуас-
сона меньше 0,5, могут быть проанализированы на основе уравне-
ния Кернера [61]:
_________<ffGf________ Ут
(7 — 5vm)Grn -f- (8 — lOvm)Gf ~г 15(1— vm)
G = Gm------------г-----------------~------ (29)
(Pm
(( — 5vm) Grn + (8 — 10v/n) Gf 15(1 vm)
где индексы tn и f относятся к матрице и фазе, а символы G, v и ф означают
модуль, коэффициент Пуассона и объемную долю дисперсной фазы.
В первой статье Кернера [61, с. 802—808] рассмотрены ди-
электрические свойства дисперсных систем. Уравнение (29) опи-
сывает зависимость свойств от состава смеси (или дисперсной сис-
темы вообще) без учета неизбежного обращения фаз в смеси
или возникновения предельных структур, например плотной упа-
ковки частиц твердой дисперсной фазы при объемной доле час-
тиц, отличающейся от дд=1. В соответствии с уравнением (29)
могут быть получены две кривые зависимости свойств от состава,
отражающие формальное предположение, что либо одна, либо
другая фаза остается непрерывной во всей области составов
смеси.
Такаянаги получил аналогичную формулу [62], которая так-
же графически выражается двумя кривыми, соответствующими
предельным случаям, когда либо одна, либо другая фаза остает-
ся непрерывной во всей области составов:
„ _ , (7 — 5ут) Gm -р (8 — ЮУ'т) Gf — (7 — 5vm) (Gm Gf) tpf
G (7 —5vm) Gm 4- (8 - 10v,„) Gf + (8 - 10vm) (Gm - Gf) <pf
В уравнениях (29) и (30) значения модуля упругости могут
быть заменены значениями комплексного модуля G*, вязкости ц
пли комплексной вязкости ц* [62].
200
Нильсен показал, что наиболее общим уравнением, фактиче-
ски обобщающим уравнения (23), (27), (29), (30) для системы
« пп-рдой дисперсной фазой в мягкой матрице, является следую-
щее [55]:
G 1 + AB<f!
Gm ~
(31)
I ie G— модуль (сдвига, растяжения или объемный); А— константа, характерн-
ее кнцая форму частиц дисперсной фазы и зависящая от коэффициента Пуассона
торицы (если vm=0,5, А=йе—1, где kE— обобщенный коэффициент Эйнштей-
И.1)
Для сферических частиц можно проследить простую зависимость
А = (7 —5v„,)/(8-10v„,) (32)
Константа В зависит от соотношения модулей дисперсной фа-
ы и матрицы и от величины А:
_ GfiGm —1
Gf'Gm + А
(33)
Коэффициент определяется соотношением q>f и <рМакс:
, / 1 фмакс \
\ Фмакс /
4'f
(34)
Произведение Zxpf является приведенной объемной долей дис-
персной фазы, причем £=1 при фМакс=1.
Значения коэффициента Эйнштейна уменьшаются с уменьше-
нием коэффициента Пуассона приблизительно следующим обра-
и>м:
v 0,50 0,40 0,35 0,30 0,20
kE . . . . . 2,50 2,25 2,16 2,10 2,00
Приведенные выше уравнения показывают, что чем больше
р.ииица между модулями фазы и матрицы, т. е. чем больше от-
ношение GjlGm. тем больше константа В и тем выше модуль сме-
। и при данном соотношении компонентов. При очень большом
pni.iipiiiH модулей можно упростить уравнение Кернера, положив
д 1я стеклообразной фазы в эластичной или вязкой матрице мо-
ц и. фазы Gf = °° или для систем типа поропластов (дисперсная
<|>.i bi —.воздух) — модуль Gf=O.
Довольно большое распространение для расчетов модулей или
пн 1КОСТИ смесей полимеров получили модельные представления
1.1каянаги — Каваи [63, 64]. На рис. V.14 показаны две модели,
и которых буквой т обозначена непрерывная фаза (матрица),
I внутренняя (дисперсная) фаза. Модель 1 является двухфаз-
ной моделью с последовательным соединением элементов, а mo-
di и. 2—с параллельным соединением элементов. Значения X
и i| характеризуют структуру смеси, характер распределения
компонентов в смеси. Так, например, если внутренняя фаза твер-
201
Рис. V.14. Механические модели, отражающие ме-
ханические свойства двухфазных смесей полиме-
ров:
т — непрерывная фаза — матрица; f — дисперсная фаза.
дая, то при ее распределении в виде аг-
регатов или вытянутых частиц образу-
ется структура типа коагуляционной,
когда твердые частицы разделены отно-
сительно небольшими прослойками эла-
стичной матрицы. В этом случае свойст-
ва смеси и в первую очередь модуль бу-
дут приближаться к свойствам дисперс-
ной фазы, и этой структуре будет соот-
ветствовать модель 1 или 2 с большим
значением ф и малым X. При соответст-
вующем выборе параметров ф и X обе
модели эквиваленты и дают одинаковые
значения модуля упругости. Таким обра-
зом, в принципе характер изменения ф
и X с изменением объемной доли дис-
персной фазы отражает изменение фазо-
вой структуры смеси.
Уравнения для модулей смеси в соответствии с моделями
рис. V.14 удобно сравнивать с уравнением Кернера (29), если за-
писать его в следующем виде:
G* (1 - <pf) G^ +(<х + Фй g;
Gm - — “Ф/) Grn + “ (X — ф?) g;
(35)
где а = 2(4 —5vm)/(7 —5vm).
Для модели с последовательным соединением элементов (мо-
дель 1) выражение для комплексного модуля (и аналогично для
модуля сдвига) запишется в виде
[ф. 1 — ф, I-1
X^ + d-X^G^ + ~~G^~ ] (36)
где ХДд = ф/.
Параметры ф1 и Xi можно выразить через а, входящее в урав-
нение (35):
4’1 = ф (1 + «V(a + Ф)
Xi = (а + ф)/(1 +а)
При этом уравнение (35) становится идентичным уравнению
(29). Это означает, что при определенном выборе параметров ф1
и Xi, т. е. при определенной фазовой структуре смеси уравнение
Кернера дает то же значение модуля упругости, что и уравнение
202
ппхфазной модели. Индекс 1 в приведенных выше формулах оз-
il.... принадлежность к модели 1.
Аналогично для модели с параллельным соединением элемен-
и>|| выражение для комплексного модуля:
G* = Х21 -gj- + Z}» 1 + (1 — Х2) Е*т
L f т J
(I ie Ф/=Х2фг) переходит в уравнение (29), если соблюдаются
пни ношения
Ф2 = (1 + «<Pf)/(l + «)
Х2 = ср (1 + сх),'{1 + а<р)
Уравнения (36) и (37) в свою очередь становятся идентичны-
ми, если соблюдаются следующие соотношения:
= 1 - ср/ ф2» = Х2Ф2 ~ ф/
Комплексный модуль растяжения связан с комплексным моду-
дем сдвига следующим образом:
£* = 2 (1 + V) G’
। u- v* = v'-H‘v"— коэффициент Пуассона для вязкоупругого тела.
Если v*=v'=v, уравнение (29) для £* запишется в виде
£*_________________(!+ +
Ет V (1 + “Фр Ет + «₽ (1 — Фр £/
«
। де
₽ = (1 + vm)/(l + v;)
y = (l + v)/(l +vm)
В случае равенства коэффициентов Пуассона матрицы и фазы
уравнение (38) аналогично уравнениям (36) и (37). При этом
можно пользоваться и соответствующими равенствами для ф
и X, приведенными выше. Выражения для Е' и Е", а также для
1g Л=£"/Е'обсуждаются в [65].
Исходя нз двухфазной модели Такаянаги — Каваи, можно рас-
щипать фотоупругую константу смеси полимеров, блок-сополнме-
р.ч и других двухфазных систем [66].
Модельные представления о свойствах смесей полимеров бы-
||| развиты в работе [66], где предложена аналогичная
рис. V.15 модель, являющаяся объемной. Расположение и конфи-
i\рання объемных элементов таковы, что при изменении направ-
ц-пня действия силы на модель результаты расчета не изменяют-
<ч При расчете модулей Е' и Е" модель не дает лучшего согла-
। ин с опытом, чем модель рис. V.15, и несколько лучше
шлагуется с экспериментом при расчете фотоупругой константы.
Приведенные выше модели и уравнения не учитывают обра-
щения фаз при изменении соотношения компонентов в системе.
203
Рис. V.15. Схема, поясняющая возможное опи-
сание эффекта обращения фаз в смесях с по-
мощью уравнений (31) и (41).
Для данной смеси, предполагая по
очереди, что то один, то другой ком-
понент является непрерывной фазой,
можно получить две кривые, подоб-
ные изображенным на рис. V.17 (см.
с. 206), тогда как в действительности
при обращении фаз в системе зависи-
мость свойство -— состав в принципе
должна выражаться S-ибразной кривой. Кернер учитывал это об-
стоятельство [61] и предложил более общее уравнение для мно-
гофазной системы
Vi------------------------------- л (391
L> (7 — 5v) G + (8 — 1 Ov) Cl ’ u '
и соответственно для двухфазной смеси полимеров при v=0,5
(G-Gm)4m (G-Cf)4f
4,5G+3G,;i +4,5G-f-3Gz —
(40)
где v — коэффициент Пуассона смеси; индекс i — относится к одной из фаз си-
стемы; индексы т и / — обозначают одну из фаз системы, причем фаза т при
малом ее содержании в действительности оказывается дисперсной (внутренней).
Уравнение (40) дает S-образную кривую в координатах мо-
дуль— состав, которая отражает обращение фаз, если принять,
что при плотной упаковке частиц дисперсной фазы модуль смеси
становится равен модулю дисперсной фазы еще до того, как <р/ =
= 1. Этот подход к учету обращения фаз аналогичен в принципе
тому, как в уравнении Муни и Нильсена учитывается возникнове-
ние плотной упаковки частиц дисперсной фазы при увеличении ее
содержания в системе.
Нильсен предложил [55] применить уравнение (31) для слу-
чая обращения фаз в смеси. При этом уравнение, описывающее
систему, в которой более твердая дисперсная фаза стала непре-
рывной, записывается следующим образом:
Gm 1 +
G 1 — Bitty
где константы А и В равны соответственно:
8 - !0vm _ _G,„/<?— 1
l~ 7 —5vm ’ Gm/Gf + A
Индекс т по-прежнему обозначает непрерывную фазу, но в от-
личие от уравнения (31) теперь эта фаза более жесткая (высоко-
(41)
204
модульная), чем дисперсная фаза. Коэффициент £ рассчитывает-
я так же, как это показано выше, но вместо фмакс подставляют
Чмнкс — объемную долю «мягких» частиц дисперсной фазы.
На рис. V.15 приведена схема, поясняющая применимость
равнений (31) и (41) к обращению фаз. Верхняя кривая соот-
ветствует дисперсии мягких частиц в твердой матрице [уравне-
ние (41)], а нижняя — дисперсии твердых частиц в мягкой матри-
це [уравнение (31)]. Модуль первой системы стремится к моду-
лю мягкой матрицы еще до того, как объемная доля матрицы ста-
ла равной 1, т. е. предел этот определяется величиной фмакс, если
отсчитывать справа, и (1—фмакс) при отсчете от начала коорди-
нат. Предел концентрации твердой фазы определяется, естествен-
но, величиной фмакс, как показано на рис. V.15. Очевидно, что ес-
ли область обращения фаз известна, то можно задать такие
значения фмакс и фмакс, что обе кривые сольются, образуя S-образ-
ную кривую, как показано на рис. V.15. Практически удобно
[35] в области составов между точками пересечения верхней
и нижней кривых на рис. V.15 найти значения модулей смеси,
воспользовавшись правилом логарифмической аддитивности. По-
лученная зависимость модуль — состав действительно приводит
к хорошему совпадению расчета с экспериментом, как это будет
показано далее на рис. V.19.
Для частиц правильной (или известной) формы можно рас-
считать фмакс ориентировочно или определить экспериментально
|(>0]. Нахождение фмакс затруднительно, если смешение полиме-
ров производилось на механическом оборудовании. В этих случа-
ях частицы дисперсной фазы анизометричны, форма их произ-
вольна и меняется с изменением соотношения компонентов. По-
>юму найти величины фмакс и фмакс можно лишь для данной па-
ры полимеров, полученных в выбранных условиях смешения.
В гл. IV было показано, что интервал обращения фаз может
простираться от соотношений 3:7 до 7:3. Поэтому всякое урав-
нение зависимости свойств смеси от состава, основанное на мо-
ц'льных представлениях, не имеет предсказательной силы и совпа-
l'itc V.16. Схематическое изображение комплексной двухмерной модели.
905
дает с экспериментальными данными лишь при соответствующем
подборе констант для конкретной смеси полимеров.
Предложена комплексная модель [67], в которой обращение
фаз учитывается самой конструкцией модели, как это ясно из
рис. V.16, а. При соотношении 1 : 1 обе фазы являются непрерыв-
ными, что и соответствует 'моменту обращения фаз. Расчет прово-
дится в соответствии со схемой рис. V.16, б. На этой схеме части
I и III имеют одинаковые, свойства — свойства матрицы. Части
IV и II имеют также одинаковые свойства, которые можно выра-
зить с помощью одной из двухфазных моделей Такаянаги — Ка-
ван. Таким образом можно рассчитать комплексные модули Е*
частей модели, а затем комплексные податливости частей I и III
(CJ и С[) и частей II и IV (С}1 и СР), где
СТ=1/£’ и C2 = -vm/f
Комплексный модуль смеси рассчитывается по формуле
р._____(W)a-(4+c^ ________________'
- 2 jCy [(C]7-(C^J +CJ [(Cjr-OT (
Зависимость модуля от состава смеси выражается S-образной
кривой.
Если частицы дисперсной фазы сильно анизометричны (волок-
ноподобны) и ориентированы в одном направлении, то можно
вычислить модуль смеси по следующему уравнению:
G (Gf/Gm) + х [ 1 + (Gf/Gm - 1) <г71
Gm ~ Gf/Gm — 4f (Gf/Gm — 1) + x
где G — модуль сдвига системы в направлении, параллельном или перпендику-
лярном ориентации волокон дисперсной фазы, или модуль растяжения в направ-
лении, перпендикулярном ориентации; х — константа, зависящая от геометрии
дисперсных частиц и вида деформации.
Экспериментальное исследование модулей упругости смесей
полимеров. В области содержания второй фазы до 10—15% (об.)
наблюдается иногда хорошее совпадение теоретических и экспе-
риментальных значений модуля [55, 62]. Уравнения (30, 36, 37)
лучше описывают системы с мягкой дисперсной фазой, а уравне-
ние (29) лучше подходит для опи-
сания смесей с твердой дисперс-
ной фазой или с твердым минераль-
ным наполнителем. Полная кривая
зависимости модуля от соотноше-
ния компонентов во всем интервале
Рис. V.17. Зависимость начального модуля
упругости при сжатии от состава смеси
ПБ —ПИП при — У0°С:
1 И /' — кривые, рассчитанные по уравнениям (36)
и (37); 2 — по уравнению (42); 3 — по уравнению
(40); точки — экспериментальные данные
206
Рис. V.18. Зависимость начального модуля
сжатия от состава грубодисперсиой смеси СКВ
(/с=-47°С) и ПИП (Ус =—64 °C), получен-
ной вальцеванием в течение 3 мин (кривая /)
и однофазной смеси, полученной испарением
растворителя из смеси растворов указанных
полимеров (кривые 2. 3. 4). Модули определя-
ли при температурах —58 °C (1, 2) и при
52 ДЗ (3); на кривой 4 каждая точка получе-
на при температуре на 14 °C выше температу-
ры стеклования смеси данного состава.
соотношений лишь приближенно опи-
сывает экспериментальные данные
[53], как это видно из рис. V.17. Ко-
нечно, особенно плохо описывается
область обращения фаз.
В однофазных смесях взаиморас-
(воримых полимеров модуль линейно
швисит от состава при температурах
выше или ниже температур стеклова-
ния полимеров. В области температур,
промежуточной между температурами
полимеров, имеются две линейные зависимости, пересекающиеся
в точке, соответствующей смеси с Тс, равной температуре измере-
ния модуля. Поэтому при разных температурах измерения модуля
наклон линейных участков разный (рис. V.18). Из рис. V.18 видно,
чго при недостаточном перемешивании полимеров на вальцах да-
же в принципе совместимые компоненты могут образовать двух-
фазную композицию. При этом в начальной области составов мо-
дуль практически не зависит от количества дисперсного «мягкого»
наполнителя и полностью определяется модулем твердой матрицы.
Несовпадение теории и эксперимента при определении моду-
лей упругости двухфазных смесей полимеров обусловлено двумя
|.тавными причинами.
Первой причиной является неучет существующими теориями
и 1мепения структуры и свойств полимера при переходе его в дис-
персное и особенно в коллоиднодисперсное состояние. Задача уче-
i.i такого изменения необычайно сложна, как и сложен учет из-
менения свойств материалов при деформации (нелинейная вязко-
хпругость по сравнению с линейной вязкоупругостью, когда изме-
нениями в структуре материала можно пренебречь).
Изменение структуры и свойств полимера при переходе в дис-
персное состояние в процессе смешения определяется в основном
|ремя факторами: частичной взаимной растворимостью компонен-
|ов, внутренними напряжениями как в матрице, так и в дисперс-
ной фазе, и возникновением протяженного граничного слоя. Рас-
। чпнрим кратко механизм влияния указанных факторов па изме-
нение структуры и свойств полимеров при смешении.
207
ло. близко к
Риг. V.19. Зависимость модуля Юнга от состава
блок-сополимера СБС (точки — эксперим< нталь-
ные данные):
1 — кривая, рассчитанная по уравнению (31) при А .=0,86,
<₽макс“°’8&; 2 —то же при Д=3.0. <Рмаке =0,80; 3 — тео-
ретическая кривая, полученная усреднением кривых 1
и 2 по правилу логарифмической аддитивности.
Взаимная растворимость полимеров
обычно ничтожно мала (см. гл. IV). При
аддитивном изменении свойств поли 4 ер а
при растворении в нем другого внесение
нескольких процентов второго полимера
не приведет к заметному изменению
свойств. Так. при смешении полимеров.
Тс которых различаются на 50 °C (это
обычно довольно большая разница), вве-
дение 2% второго полимера приведет к
тзмененню Тс на 1 °C. Точность измере-
ния Тс обычно мала, и изменение в 1 °C,
___ х , ошибке опыта. То же можно сказать и о
изменении модуля, если учесть, что при взаимном растворении
полимеров модуль линейно зависит от состава смеси (см.
рис. V.19). Известно, однако, что если в процессе смешения обра-
зуется насыщенный раствор одного полимера в другом, то при
охлаждении такого раствора часть добавленного полимера может
выделиться в виде мелкодисперсной коллоидной эмульсии. Такие
эмульсии чрезвычайно стабильны вследствие малого межфазного
натяжения и высокой вязкости системы и характеризуются экстре-
мальным изменением свойств (см. гл. VII). В частности, при до-
бавлении, например, долей процента ПММА к ПС происходит
снижение в несколько раз вязкости расплава ПС и заметный рост
модуля в твердом состоянии [68, 69]. Если при смешении поли-
меров создались условия для возникновения структуры, близкой
к критической, достаточно перехода долей процента одного поли-
мера в среду другого (в матрицу или в частицы дисперсной фазы),
чтобы ре."ко изменить свойства этой фазы.
Внутренние напряжения, как было показано в § I этой главы,
могут быть столь велики, что приведут к заметному изменению
Тс, что указывает па сильное изменение структуры фазы. При
этом может и пе происходить изменения надмолекулярной струк-
туры, а только изменение межчастичных расстояний и соответст-
вующее изменение межмолекулярного взаимодействия. Однако
естественно, что возникновение внутреннего напряжения сопро-
вождается деформацией, которая приводит к соответствующему
изменению структуры, а также и свойств дисперсной фазы или
матрицы. В полимерах, содержащих твердый минеральный напол-
нитель, внутренние напряжения, конечно, не меняют структуры
пли свойств наполнителя, и поэтому все изменения сводятся к об-
208
разованию напряженных граничных слоев полимера вокруг твер-
дой частины наполнителя. Естественно, что в смеси полимеров
благодаря относительной близости модулей дисперсной фазы
и матрицы, глубокие изменения структуры и свойств могут про-
исходить в каждой фазе.
Внутренние напряжения можно приближенно определить по
уравнению [55]
Ьвн КЕто (Рт — (Л> 7) (44)
где К — константа, Слизкая к единице; £т0— модуль матрицы, в которой воз-
никли внутренние напряжения; ат — термический коэффициент расширения мат-
рицы; щ — термический коэффициент расширения частицы фазы; Т — температу-
ра измерения модуля; То— температура, при которой начали возникать внутрен-
ние напряжения (чаще всего это температура стеклования).
Внутренние напряжения за счет разности теплового расшире-
ния приводят к снижению модуля матрицы вблизи частицы. Чем
ниже температура, т< м больше внутренние напряжения, и отно-
шение модуля смеси к модулю матрицы может составлять всего
0,5 от ожидаемого по теории.
Изменение структуры полимера при переходе в дисперсное со-
стояние может происходить под непосредственным влиянием по-
верхности раздела (поверхностных сил). Радиус действия поверх-
ностных сил в воде или других полярных жидкостях либо в силь-
но аиизометрнчных (также полярных) жидкостях типа жидких
кристаллов составляет не менее 0,1 мкм, а в неполярных жидко-
стях— около 10 нм (эти данные получены на основе прямых из-
мерений толщины граничных слоев жидкостей па твердых по-
верхностях). Очевидно, что если размер частицы близок или ра-
вен протяженности действия межфазных сил, то структура веще-
ства частицы изменится. Для полимеров характерно еще большее
дальнодействие сил, поскольку изменение структуры полимера
в прилегающем к границе раздела слое ведет к изменению струк-
туры в более удаленных слоях благодаря «передаче информации)»
через цепочку химических связей.
Изменение механических свойств при диспергировании может
происходить вследствие адсорбционного понижения прочности или
твердости, «эффекту Ребиндера». Адсорбция одного полимера на
поверхности другого, заполнение мнкропустот и микротрещин ве-
ществом матрицы могут привести к значительному изменению мо-
дуля, вязкости и других свойств (не только предельных свойств,
таких как прочность или удлинение при разрыве). На модельных
системах показано, что при дублировании двух слоев, например
полимера и металла, изменяются свойства обоих слоев, и в хруп-
ком полимере в результате дублирования может появиться пре-
дел текучести, т. е устраняется хрупкость [70].
Заметное изменение свойств вещества дисперсной фазы проис-
ходит, как правило, в тех случаях, когда размер частиц дисперс-
14—244
209
ной фазы становится меньше 104 нм, т. е. когда вещество перехо-
дит в коллоиднодисперсное состояние. Диспергируемое вещество
при переходе в коллоиднодисперсное состояние теряет один из
двух основных признаков фазы (независимость свойств от объе-
ма). С точки зрения механики дисперсных систем и, в частности,
в применении к смесям полимеров, смеси с размером частиц ме-
нее 104 им являются в механическом смысле сложными дисперс-
ными системами, рассчитать механические свойства которых на
основе обычных двухфазных моделей в большинстве случаев
в принципе невозможно. Более грубодисперсные системы могут
быть моделированы простыми двухфазными моделями, если для
данной системы показана независимость свойств фазы от степени
ее диспергирования в матрице, а также при относительно незна-
чительной протяженности межфазного слоя.
Вторая причина возможного несовпадения результатов расче-
та механических свойств двухфазной смеси полимеров с экспери-
ментальными данными состоит в невозможности надежного прог-
нозирования формы частиц дисперсной фазы и связанного с фор-
мой частиц момента обращения фаз или интервала составов сме-
си, в котором оно происходит.
Форма частиц дисперсной фазы зависит от соотношения вяз-
костей фаз при температуре смешения, а для вязкоупругих си-
стем — от соотношения вязкостей и упругостей фаз при темпера-
туре и напряжении сдвига в процессе смешения^ Количественная
связь между механическими свойствами фаз и их структурообра-
зованием в заданных условиях смешения неизвестна. Известно,
однако, что довольно малые количества дисперсной фазы могут
образовать непрерывную структуру. Поэтому в интервале соотно-
шений полимеров от 3 : 7 до 7 : 3 надежное количественное прог-
нозирование механических свойств смеси с учетом только свойств
смешиваемых полимеров в настоящее время невозможно.
Непрерывная фаза, как известно, выходит на поверхность об-
разца [71], и поэтому химические и механические свойства по-
верхностного слоя определяются природой непрерывной фазы [55,
72]. Образец, сформованный из двухфазной системы, неизбежно
оказывается покрыт «корой» из полимера непрерывной фазы.
По этой же причине адгезионные свойства поверхности смеси по-
лимеров, сформованной в прессе, существенно отличаются от
свойств поверхности, образованной путем среза [73]. Эффект
«коры» можно учесть в виде следующего уравнения [55]:
GDS
G GD3
Ga ~ (G-Gm)(d~D)3 + GmD3
(45)
где G — истинный модуль сдвига двухфазной системы (то же для модуля растя-
жения или сжатия); Ga — экспериментально найденный модуль, зависящий от
толщины «коры», которая в свою очередь определяется диаметром d частиц,
дисперсной фазы; D — толщина образца прямоугольного сечения.
210
Уравнение (45) получено и проверено для полимеров с мине-
ральным наполнителем и для пен. Для тонких образцов влияние
коры» приводит к изменению модуля па 10—20%.
В некоторых случаях возможно, если не прогнозирование, то
достаточно точное описание экспериментальных кривых свойст-
во — состав с помощью приведенных выше уравнений или моде-
лей. Так, на рис. V.19 показано хорошее совпадение эксперимеп-
глльных данных по модулям блок-сополимеров бутадиена и сти-
рола с расчетными значениями, подученными по формуле (31).
<иачения констант здесь подобраны произвольно так, чтобы при-
близить расчетную кривую к экспериментальным данным. Харак-
к-рно, что константы «прямого» и «обратного» уравнений несим-
метричны. это объясняется несимметричным интервалом обраще-
ния фаз, происходящим в интервале объемных долей 0,15—0,80.
Если для аналогичного типа блок-сополимера (Кратон 101)
исходить из значений модулей исходных полимеров, то разница
между экспериментальным значением модуля (1,13-109 Па) и рас-
четным (1,85-106 Па) окажется огромной [74].
Если смесь полимеров получена совместной полимеризацией
двух не взаимодействующих химически мономеров или полимери-
|;щией мономера в присутствии полимера иной химической при-
роды, то возникает мелкодисперсная система, структура которой
близка к однофазной. На границе раздела развит межфазный
слой, и область обращения фаз растянута практически на всю об-
1'и V.20. Модули сдвига смесей ПУ—ПАН (точки — экспериментальные
данные):
кривая для системы е обращением фаз; 2 — расчет по уравнению
где а=1/5.
Рис. V.21. Модули смесей ПВХ с хлорированным ПЭ при 27 °C:
кривые, рассчитанные по уравнению (29) в предположении, что либо ПВХ (/), либо
II 1 (.’) являются непрерывной фазой; 3 — по уравнению (40). Точки — зкенсримен г ильные
in светлые — для смесей ХПЭ с высокомолекулярным (26-10’) ПВХ; темные - для
смесей ХПЭ с низкомолекулярным (0,40-105) ПВХ
I Г
211
ласть составов смеси. Зависимость модуля от состава для такой
системы может быть описана уравнением, аналогичным уравне-
нию для вязкости (17) (см. с. 195) с показателем степени a=1/s
[75], как это показано на рис. V.20. В таких системах роль меж-
фазного слоя особенно велика, что подтверждается данными по
релаксации напряжения и ползучести, приведенными в работе [75].
На рис. V.21 [76] показано влияние молекулярной массы од-
ного из полимеров на величину модуля смеси. Чем меньше моле-
кулярная масса поливинилхлорида, тем меньше вязкость его рас-
плава и тем легче (при меньшем его содержании) он образует
непрерывную фазу (темные точки). Модули сдвига смесей опре-
деляли при температуре 27°C. Видно поэтому, что они выше для
смесей с менее вязким поливинилхлоридом. Модуль самого поли-
винилхлорида при температуре 27 СС выше модуля хлорированно-
го полиэтилена и практически не зависит от молекулярной массы.
Комплексный модуль и его компоненты зависят от температу-
ры смеси полимеров, и зависимость эта часто используется для
определения числа фаз в смеси (см. гл. IV). Наиболее характер-
ны следующие типы температурной зависимости модуля потерь
или тангенса утла потерь tg6: 1) два максимума потерь, положе-
ние которых на шкале температур точно соответствует положению
максимумов для исходных полимеров; 2) максимумы потерь сме-
си смещены относительно температур стеклования исходных по-
Рис. V.22. Температурная зависимость модуля упругости С смесей двух бута-
диен-стирольных сополимеров с содержанием стирола 16 и 50%. Содержание вы-
сокостирольного компонента в смеси указано цифрами на кривых.
212
Гис. V.23. Температурная зависимость
/' и tg 6 блок-сополи «еров СБС с 30%
шрола (мол. масса 15,1-Ю-’ для каждо-
блока), полученных в виде пленок из
разных растворителей:
- метилэтшкетои; 2 — этилацетат; 3 — толу-
ол; 4 — СС14.
шмеров; 3) наряду со смещен-
ными или неизмененными по
шмпературе максимумами появ-
ляется третий, промежуточный
максимум; 4) вместо двух исход-
ных в смеси появляется один
промежуточный максимум. При-
чины возникновения той или
иной зависимости модуля или
ыигенса угла потерь от состава
билли изложены ранее. Отметим
г< ьтько, что в смеси полимеров максимумы всегда расширены по
сравнению с максимумами исходных полимеров, даже если их по-
южение по шкале температур не изменилось. Расширение макси-
мумов всегда указывает на образование граничного слоя или на
танмное влияние контактирующих фаз на их внутреннюю струк-
i\py, что и приводит к расширению главной области релаксации
in данной фазы.
На рис. V.22 [55] показан ход кривых G'—Т для типично
шухфазной системы. Из рисунка V.22 видно, что температуры
н-кловаиия в смесях и в исходных полимерах практически одп
и 1ковы, и после размягчения полимер переходит в высокоэласти-
nichoe состояние, характеризуемое наличием плато высокоэластич-
цпеги. Горизонтальные участки кривых в области между темпера-
прами стеклования компонентов расположены тем выше, чем
Польше в смеси полимера с более высокой температурой стекле-
ния.
1 ипичная зависимость динамических свойств от температуры
। in такой двухфазной системы, как блок-сополимер бутадиена
и । пцюла, приведена на рис. V 23 [55]. Характерно, что при ис-
пиленном массовом содержании каждого компонента для пленок,
ш> i\ченных из разных растворителей, высота пика tgrt разная.
11< м лучше растворитель данного блока, тем больший объем эти
|<п\п занимают в смеси, и высота пика растет с увеличением
iii.emioii доли (реальной объемной доли!) ПС в пленке. Для НС
1мы л «хорошим» растворителем является метилэтилкетон, и пик
IK в пленке из этого растворителя самый высокий. Для каучу-
iiioif фазы максимум потерь самый высокий в пленке, полечен-
ш и четыреххлористого углерода, который является «хорошим»
, ншоритслем для каучуковой фазы. Кривые температурной за-
213
Рис. V.24. Зависимость вязкости от со-
става расплавов (1) и 10%-ных бензоль-
ных растворов (2) смеси полибутадис-
нов с узким ММР [Mw/Mn= 1,1) и мо-
лекулярными массами 1,0-105 и 2.6-105.
висимости G' не меняют положе-
ния при перемене растворителя и
в этом смысле они менее инфор-
мативны, чем кривые tgfi—Т или
G"— Т.
Обширное исследование при-
менимости разных моделей и
уравнений типа приведенных вы-
ше к двухфазным полимерным
системам показало необходи-
мость корректировки параметров
уравнений для обеспечения наи-
лучшего совпадения с экспери-
ментом [76]. Корректировка сво-
дилась к подстановке в уравнения значений объемной доли с уче-
том взаимопроникновения полимеров из фазы в фазу, а также с
учетом окклюзии полимера, т. е. наличия в частицах дисперсной
фазы мелких частиц полимера матрицы. В последнем случае сна-
чала рассчитывали свойства дисперсной фазы по одному из при-
веденных выше уравнений, а затем свойства композиции в целом.
В согласии с рис. V.24 оказалось, что модуль упругости зависит
главным образом от объемного содержания дисперсной фазы (дис-
персная фаза ПС на рис. V.24 имеет наибольший объем в пленке,
полученной из метилэтилкетона, и ее модуль упругости выше).
В то же время модуль потерь или тангенс угла потерь зависят от
морфологии и от концентрации дисперсной фазы. Расчетные дан-
ные [76] подтверждаются как собственными данными автора, по-
лученными на примере смеси ПММА и пространственно сшитого
сополимера бутилакрилата с 1,3-бутилендиметакрилатом. так и
данными рис. V.24, где высота максимума для каучука зависит от
структуры дисперсной фазы ПС.
Таким образом, и расчет, и экспериментальные данные пока-
зывают, что вид кривых зависимости компонентов комплексного
модуля от температуры может дать качественную информацию
о структуре смеси, хотя информация эта не всегда является од-
нозначной, поскольку потери зависят от многих факторов. Боль-
шое количество данных по сравнению экспериментальных и рас-
четных кривых зависимости модулей упругости и потерь от со-
става для разных смесей приведено в работах [77, 78].
Смеси кристаллических полимеров также описываются в рам-
ках двухфа шых моделей. Это можно видеть на примере смеси по-
лиэтиленов высокой и низкой плотности [79]. В этом случае па-
раметры двухфазной модели, обеспечивающие совпадение теории
214
г экспериментом, заметно зависят от условий кристаллизации
( меси.
Экспериментальное исследование вязкости смесей полимеров.
Наиболее простой моделью однофазной смеси полимеров являет-
ся смесь фракций одного и того же полимера. Зависимость вяз-
кости от состава для такой смеси описывается уравнением (17),
которое в согласии с экспериментальными данными предполага-
ет изменение вязкости по кривой выше аддитивной (рис. V.24)
|55]. Такой же характер кривой наблюдается и для однофазной
смеси ПС и полиэтиленоксида [80]. В то же время зависимость
вязкости от состава строго аддитивна для смеси ПИП — СКВ
[53] и для смеси фракций атактического ПС [46]. Во всех указан-
ных случаях смеси были составлены из полимеров, не содержа-
щих таких функциональных групп, которые могли бы сильно
взаимодействовать друг с другом. Примером пары с сильным
взаимодействием компонентов является смесь сополимера бута-
шена с 10% мстплвинилпиридина и хлорированного полихлоро-
прена [81]. Взаимодействие между функциональными группами
приводит к отклонению характеристической вязкости растворов
смеси полимеров от аддитивной в сторону увеличения.
Характер кривой вязкость — состав в однофазной смеси поли-
меров с узким распределением зависит от структуры смеси, кото-
рая определяет изменение свободного объема с составом и его
распределение в смеси. Если свободный объем смеси складыва-
ется аддитивно, то эффективный коэффициент внутреннего тре-
пня сегментов меняется тоже аддитивно, а при примерно равных
шачениях свободного объема и вовсе не зависит от состава [80].
При этом, однако, число узлов флуктуационной сетки в смеси не
। 11ИТИВИО, что приводит к росту вязкости выше аддитивной и так-
ие к росту модуля упругости в области плато, как это показано
it гой же работе [80].
Можно полагать, что в зависимости от структуры смеси, в зна-
чительной мере определяемой изоморфизмом смешиваемых мак-
ромолекул, свободный объем может меняться различным образом.
>го и предопределяет вид кривой 1?т]о — состав. Очевидно, что
। иитивность вязкости однофазных смесей является частным слу-
чаем, связанным с некоторым увеличением свободного объема
и смеси по сравнению с аддитивным его значением. В однофазных
месях наличие широкого ММР не влияет на характер зависпмо-
(III от состава смеси, как это показано на примере упомянутой
выше смеси ПИП — СКВ [53]. В двухфазных же смесях переход
io полимеров с узким ММР к полимерам с широким ММР при-
1'0 шт к принципиальным изменениям формы кривой зависимости
ффективной вязкости от состава.
В полимерах с узким ММР отсутствует аномалия вязкости
поэтому аномалия здесь не влияет на форму кривой вязкость —•
•став. Как и в случае модулей, форма кривой вязкость — состав
|ражает происходящее с изменением состава обращение фаз
215
Рис. V.25. Зависимость вязкости от соста-
ва для смесей ПБ — ПИП с узким ММР,
полученных на вальцах (/), испарением
растворителя (2, 5) и совместным осажде-
нием из раствора (3); 4 — смесь полиме-
ров с широким ММР, полученная испаре-
нием растворителя. В скобках указаны тем-
пературы, при которых определяли вяз-
кость.
(рис. V.25). На рис. V.25 видно, что
при любом из исследованных мето-
дов получения смеси ПБ—ПИП она
остается двухфазной и это обуслов-
ливает S-образную форму кривой
вязкость — состав (кривые 1, 2, 3).
Поскольку определение вязкости
неизбежно сопровождается силь-
ной деформацией смеси и, следова-
тельно, искажением структуры, су-
ществовавшей в недеформированном состоянии, область обраще-
ния фаз сильно растянута. Область обращения фаз фиксируется
тем четче, чем больше компоненты различаются по вязкости (ср.
кривые 1 и 3). При малой разнице вязкостей обращение фаз во-
обще не фиксируется и зависимость оказывается прямолинейной
(см. кривую 5). Разумеется, фактически обращение фаз происхо-
дит независимо от соотношения вязкостей и, если в смеси поли-
меров с равными вязкостями исследовать изменение модуля в ин-
тервале температур T'C<Z Т<Т”С> промежуточном между темпера-
турами стеклования компонентов, то S-образное изменение модуля
обнаруживается отчетливо.
Для полимеров с широким ММР также характерна S-образ-
ная кривая зависимости начальной ньютоновской вязкости от со-
става. Природу (непрерывной фазы и область обращения фаз мож-
но быстро оценить, сравнивая экспериментальные данные с рас-
считанными по эмпирической формуле [82]
1g’] = <(; Ig’lz+C1 —Чп
(46)
По формуле (46) можно рассчитать, как менялась бы вяз-
кость смеси, если бы один из компонентов оставался дисперсной
фазой во всей области составов. Полагая в качестве дисперсной
фазы (соответственно и матрицы) то один, то другой компонент,
можно построить ^предельные» кривые, аналогичные кривым, рас-
считанным по более сложным уравнениям (29) и (30).
Если частицы дисперсной фазы очень крупны, то при дефор-
мации сдвига они вытягиваются не только в тонкие волокна,
ориентированные в направлении сдвига, как это было показано
выше (см. также [82]), но и образуют тонкие пленки, распола-
216
Рис. V.26. Модель чередующихся кольцевых слоев.
। ающиеся в слое с одинаковым, возможно,
наименьшим градиентом скорости сдвига.
При течении в капилляре это приводит к
образованию незамкнутых кольцевых сло-
ев, моделью которых, как это показано в
работе [83], является система замкнутых
чередующихся кольцевых слоев в попереч-
ном сечении потока. В каждом слое течет
одна из несмеыиьающихся жидкостей
Стенка
(рис. V.26).
Приведенная объемная скорость течения любого цилиндриче-
ского слоя равна:
?j = Qj<Q = (p°)2-(p"+i)2 (47)
где р=т/тш— приведенный радиус кривизны слоя; т — напряжение сдвига;
т^. — напряжение сдвига у стенки капилляра; ц=тЕ,/у— эффективная вязкость;
у=41'о/Т? — эффективное значение скорости сдвига у стенки капилляра радиу-
са R, которое имела бы ньютоновская жидкость.
Реологическое уравнение состояния запишем в ьиде
J_=_L (1+^2) (48)
где Цо и k — константы данного материала.
С четом того, что эффективная вязкость смеси жидкостей
ц =/?тц;-/4Ро, можно написать уравнения линейной скорости 'сдви-
га в слоях:
Vi+l = V]+- р]+1 + *4 (- р/+1)] <49>
АР-+1 + Р2+1 - ЕРр , + С = О (СО)
.де р;=Р2; л = b = ^jJ- + 2P7-;
с=4- *4 р]+^'jPj ~^~+Рр
У стенки капилляра (/=1): щ=0; Р/=1.
На оси капилляра (j=‘2N): P2n=0.
Здесь A'=q7?/d— число концентрических слоев толщиной d на
входе в капилляр для жидкости, объемная доля которой равна q.
Выбирая произвольные значения т), можно рассчитать каждое
течение Pj и V, по уравнениям (47, 49, 50) при заданном напря-
жении сдвига у стенки. Методом последовательных приближений
можно рассчитать истинное значение т], отвечающее условию
Р2Л = 0.
В работе [83] было исследовано течение эмульсии из смеси
растворов полиакрилонитрила (ПАН) и полиуретана (ПУ) в ка-
пиллярном вискозиметре. Средний размер капель составлял
5 мкм. Расчелы проведены для случаев, когда у стенки капилля-
217
ра находится либо раствор ПУ, либо раствор ПАН. Эксперимен-
тальные данные в широкой области составов хорошо совпадают
с кривой, средней между этими двумя вариантами. Только при
преобладании в системе более вязкого ПАН эксперимент лучше
совпадает с вариантом расчета, где у стенки капилляра предпо-
лагается наличие слоя раствора ПАН (рис. V.27).
Расчет был проведен для значений:
Раствор ПУ Раствор ПАН
По................. 12,6 222
k.................. О 2,5-10-8
Из рис. V.27 видно, что в системе, структура которой близка
к структуре концентрических чередующихся слоев, вязкость меня-
ется по кривой, расположенной ниже аддитивной, а эффект обра-
щения фаз, естественно, не проявляется. Как только возникает
слой жидкости с меньшей вязкостью, так сразу именно в нем раз-
вивается наибольшая скорость сдвига при заданном напряжении
сдвига. В неньютоновских жидкостях это приводит к немедленно-
му дополнительному снижению вязкости менее вязкой жидкости
с ростом напряжения сдвига.
Эффект, аналогичный показанному на рис. V.27, виден и на
рис. V.28 [82], где приведены данные по вязкости смесей ПС
и ПММА с разной степенью дисперсности. Видно, что в грубодис-
персных смесях вязкость меняется по кривой, аналогичной полу-
чаемой расчетом в предположении системы концентрических сло-
ев. После вальцевания смеси, приводящего к увеличению степе-
Рис. V.27. Зависимость вязкости эмульсии растворов ПАН и ПУ от состава по
данным капиллярной вискозиметрии:
/ у стенки раствор ПЛИ; 2 — у стенки раствор ПУ; 3— кривая для усредненной вязкости.
Рис. V.28. Зависимость эффективной вязкости (lgT=5,25 [Па]) смеси ПС —
ПММА при 170°C от состава (точки — экспериментальные данные):
о — грубодиспереиые смеси, полученные прессованием порошков; б — высокодисперсиые сме-
си после вальцевания.
218
ни диспергирования компонента с большей вязкостью, этот более
низкий компонент становится непрерывной фазой в смесях с пре-
обладающим его содержанием. Это в свою очередь обусловлива-
I парадоксальный на первый взгляд эффект: вязкость в резуль-
iare вальцевания возрастает почти на целый десятичный порядок.
Хналогичный эффект наблюдается при сравнении смесей, Полу-
ниных на вальцах и из раствора: если при получении пленки из
раствора смесь сильно расслаивается и образуются крупные ча-
стицы, то при преобладании более вязкого компонента вязкость
меси оказывается существенно ниже, чем у смеси такою же со-
1ава, полученной на вальцах в условиях хорошего диспергиро-
вания компонентов [53].
В отличие от с\ есей полимеров с узким ММР, в смесях с ши-
роким ММР зависимость вязкости от состава, как правило, опи-
ывается кривой, лежащей ниже аддитивной. Это можно показать
ия смесей концентрированных растворов НК и БСК [84], сме-
еей ПЭ с ПП [48], ПБ с СКН-18 [85]. ПС с ПП и ПЭ [86, 87]
и для многих других. Причины различий в поведении смесей с уз-
ким и широким ММР не вполне ясны и будут рассмотрены ниже.
В случаях, когда между компонентами возникают специфические
взаимодействия, обусловленные наличием функциональных групп,
как, например, в смеси полиамида и полиоксиметилена, вязкость
расплава смеси оказывается выше аддитивной и кривая даже мо-
нет иметь значительный максимум [88].
При течении двухфазной смеси возникает анизотропия свойств,
которая является следствием анизотропии фазовой структуры.
Наряду с релаксацией молекулярных клубков, ориентированных
и потоке, в двухфазной системе происходит релаксация фор-
мы самих частиц, что и приводит к повышенной анизотропии
меси, выходящей из капилляра или экструдера или снятой
валвцев [89]. Следствием всего этого является возникновение
и «Пыточных нормальных напряжений в смеси полимеров в про-
цессе сдвига и повышение ее эластичности по сравнению с адди-
И1ВПЫМИ значениями. Как известно, эластичности смесей поли
н-ров повышена и при матых деформациях [80]. В работах [86,
М'. | величина нормальных напряжений в потоке была определена
непосредственно с помощью капиллярного вискозиметра с ще-
к-вым .или кольцеобразным зазором, позволяющим измерять
I.тление в струе на выходе из капилляра. На рис. V.29 [86] по-
нана для смеси ПС — ПП типичная зависимость вязкости
и нормальных напряжений от состава. Можно лишь замети гь,
р.пшивая данные рис. 29, а с данными других работ подобного
|H>ia, что резкое снижение вязкости более вязкого компонента
при добавлении 10—20% менее вязкого компонента является
ip.шилом для смесей неполярных полимеров. Снижение вязкости
< нее вязкого компонента при добавлении небольшою количест-
|| | более вязкою (левая часть кривых на рис. 29, а) менее ти-
пично.
219
Рис. V.29. Зависимость вязкости (а) и первой разности нормальных напряжений
(б) для смеси ПС—ПП от состава (Т=200°С):
/ — =0,6 10’ Па; 2 — ти)=0,7105 Па.
Условия деформации, как уже было отмечено выше, сильно
влияют на структуру смеси. Сравнительно малые различия фазо-
вой структуры приводят к большим изменениям характера кри-
вой свойство — состав. Поэтому механические параметры ано-
мальных жидких смесей важно определять в соответственных
состояниях. По-видимому, наиболее правильно сопоставлять кри-
вые свойство — состав, полученные при одинаковых значениях
напряжения, а не скорости сдвига, так как скорость сдвига ме-
няется скачкообразно от фазы к фазе, тогда как напряжение
сдвига в установившемся потоке меняется непрерывно, не пре-
терпевая разрыва и на межфазной границе. Это следует из тех
простых соображений, что избыточное напряжение в любом мик-
рообъеме потока привело бы к избыточной деформации, при кото-
рой перенапряжение могло бы релаксировать. Действительно,
кривые свойство — состав, каждая точка которых получена при
одной и той же скорости сдвига, имеют разную форму для дан-
ной смеси при одной и той же температуре в зависимости от на-
пряжения, тогда как при одинаковых напряжениях сдвига фор-
ма кривых не зависит от их величины (меняется лишь сама .вели-
чина вязкости или первой разности нормальных напряжений)
[86].
По указанным причинам зависимость вязкости от состава сме-
си может быть принципиально различной при измерении на раз-
ных приборах, особенно когда смесь обладает значительной элас-
тичностью, что характерно для смесей каучуков [90]. На рис.
V.30 показана зависимость различных вязкостных показателей от
состава смеси ПК—ПБ при температурах 100°С (по Муни), 94°C
(пластограф Брабендера) и 126°C (капиллярный вискозиметр).
Возможно лишь качественное сопоставление показателей в пре-
делах формы кривой свойство—состав.* Однако из рисунка вид-
* О количественной взаимосвязи вязкости по Муни, индекса расплава и
эффективной вязкости см. [91].
220
но, что вязкость по Муни меняется по кривой ниже аддитивной,
ак же как и усилие продавливания через капилляр при малых
скоростях продавливания. В то же время кривые, полученные
в пластографе Брабендера, располагаются существенно .выше ад-
щтивной зависимости, так же как и кривые усилия продавлива-
ния, полученные при больших скоростях. Очень хорошо видно,
что форма кривых усилие продавливания—состав принципиаль-
но .различна при разных скоростях течения. Это связано с тем,
что скорости сдвига при определении по Муни и в пластографе
Брабендера различны и близки к скоростям, при которых в ка-
пиллярном вискозиметре обеспечивается качественное совпаде-
ние формы кривых либо с данными по Муни, либо с данными,
полученными в Брабендере.
Нет информации о величине потерь на входе в капилляр для
приведенной группы данных. Потери определяются величиной эла-
стичности и, очевидно, пе могут меняться пропорционально соста-
ву смеси или пропорционально напряжению сдвига. Потери эти
отсутствуют в пластометре Муни и заметны .в пластографе Бра-
1‘пс V.30. Зависимость вязкости по Муни, вращающего момента на пластографе
!•!> Шеидера и усилия продавливания через капилляр от состава смеси НК—ПЬ:
низкость по Муни; б — вращающий момент (/— время перемешивания 2 мин; 2 — 4 мин;
10 мин); в — продавливание через капилляр (/ — скорость 0,3 с-1, 2 -0.7 с-1, .3— 1.6 г >.
II) с-1); г— то же (5 —скорость 7,5 с-1, 6—15 с-1, 7 — 30 с-1); д -тоже (в — .К10 с-1,
у — зооо с-‘).
221
Рис. V.31. Зависимость индекса течения
п от состава смесей ПБ—ПС и ПБ—
ПИП:
1 — ПБ — ПС (полимеры с узким ММР, смесь
получена смешением 20%-ных растворов); 2—
то же, смесь получена испарением растворите-
ля; 3 — ПБ — ПИП (полимеры с узким ММР.
смесь получена испарением растворителя); 4 —
то же ПИП с широким ММР: (Afai/Aln= 1,4).
бендера, где смесь движется в камере с постоянно меняющимся
сечением. Они могут составлять более половины усилия продав
ливания в капиллярном вискозиметре с малым отношением дли-
ны капилляра к его диаметру.
Вопросы, связанные с аномалией вязкости в двухфазных по-
лимерных смесях, мало изучены. Очевидно, что даже эмульсия,
составленная из ньютоновских жидкостей, должна в принципе
являться реологически аномальной системой, поскольку в процес-
се течения меняется ее структура (капли становятся анизометрич-
ными), а следовательно, вязкость должна зависеть от напряжения
сдвит а.
Величину аномалии вязкости удобно оценивать по отклонению
от единицы индекса течения п в степенном законе течения:
у — Стп (51)
На рис. V.31 приведена зависимость п от состава для разных
смесей. Из этих данных видно, что при отсутствии аномалии вяз-
кости в смешиваемых полимерах аномалия в смесях может и не
наблюдаться (кривая 3). Следсватетьно, сама по себе двухфаз-
ность системы не является достаточным условием для возникно-
вения аномалии. Это подтверждается тем, что в растворах смеси
ПБ и ПС гри концентрациях раствора в интервале между преде-
лом расслаивания смеси С2 и концентрацией, при которой возни-
кает сетка зацепления, аномалия отсутствует, хотя система двух-
фазна [53, 92].
Таким образом, аномалия вязкости в смеси полимеров явля-
ется следствием возникновения флуктуационной сетки зацепле-
ний, образованной одинаковыми по химической природе макромо-
лекулами в пределах каждой фазы и разнородными молекулами
в межфазном слое. Решающее значение приобретает различие
в гибкости макромолекул, образующих сетку зацеплений. Так,
гибкость макромолекул ПБ и ПИП практически одинакова, по-
скольку отношение радиусов инерции клубков в 0-растворителе
и рассчитанного из модели свободного вращения (Ае)1/«/(Л*)1/»
для ПИП равно 1,5, а для ПБ— 1,6. В такой системе узлы, обра-
зованные макромолекулами ПИП — ПИП и ПБ — ПБ, мехачиче-
222
кн эквивалентны узлам ПИП—ПБ, поэтому аномалия пя.мо-
ги не возникает. Отношение радиусов инерции для ПС .равно
2,2—2,4, т. е. гибкость макромолекул ПС и ПБ резко различна,
потому в межфазном слое возникают узлы флуктуационной сет-
ки, существенно различающиеся по релаксационным свойствам,
чго и приводит к возникновению аномалии вязкости в таких сме-
сях (см. рис. V.31, кривые 1, 2). Поскольку сетка зацеплений
в растворе выбранных образцов ПБ и ПС возникает при концент-
рации около 18%, аномалия возникает лишь после того, как воз-
никла сетка, т. е. при концентрации раствора 20% и более.
Если хотя бы один из полимеров обладает аномалией вязко-
< ги, то аномалия в смеси существенно увеличивается (см. рис.
V.31, кривая 4). Если аномалией обладают оба полимера, то ано-
малия смеси увеличивается значительно, как это отмечено на
примере расплава смеси полимолекулярных образцов ПЭ и ППр
|93].
По-видимому, именно рост аномалии вязкости в смеси поли-
меров маскирует эффект обращения фаз по данным вязкости
и приводит к тому, что кривая вязкость—состав отклоняется от
аддитивной в сторону уменьшения вязкости во всей области со-
ставов. Весьма вероятно, что увеличение аномалии вязкости в сме-
си связано со структурой флуктуационной сетки, образованной
разнородными макромолекулами, поскольку даже при одинако-
вых значениях свободного объема смешиваемых полимеров и в
о щофазной системе число узлов сетки меняется неаддитивно со-
таьу смеси [80].
Энергия активации вязкого течения измеряется аналогично ее
тмерению в индивидуальных полимерах. Зависимость In т]—1/7
1ля смесей полимеров линейна в достаточно широком интервале
юмператур для смесей, компоненты которых также характеризу-
ются линейной зависимостью. Зависимость энергии активации от
состава смеси •определяется структурой смеси, и если один из
компонентов образует непрерывную фазу, энергия активации сме-
си близка к энергии активации этого компонента. Изменение фа-
Ю.В0Й структуры смеси под действием внешних условий приводит
к тому, что энершя активации ДЯТ (определенная при постоян-
ном значении напряжения сдвига т—const) зависит от величины
п.1 пряжения сдвига в тех случаях, когда для индивидуальных по-
шмеров такой зависимости не наблюдается. Так, в смеси ПС—
ПММА [82] в интервале температур 150—170°C зависимость
I 1 •]—1/7 прямолинейна, а Д/7-r .возрастаете ростом напряжения
шига. Отмеченное изменение \НХ касается не всех смесей,
। только тех, которые содержат избыток высоковязкого компо
пента.
Эффект увеличения теплоты ак гивацил вязкого течения с уве-
шчением напряжения сдвига отмечался и ранее, например для
шцентрированных растворов ПС в «плохих» растворителях, об
। тающих весьма прочной флуктуационной сеткой ассоциатов
223
макромолекул [94]. Явление это объяснялось тем, что при воз-
действии достаточно интенсивного сдвигового напряжения, струк-
тура, имеющаяся в растворе, не разрушается, а, наоборот, упроч-
няется в процессе ориентации. Аналогично этому в расплаве ука-
занной смеси можно непосредственно наблюдать образование вы-
тянутых волокон дисперсной фазы.
В смеси ПС — ПП при сравнительно высоких температурах,
например в интервале 180—250°С, Л//х слабо зависит от напря-
жения сдвига при 180сС и заметно снижается при 250°С. Это
означает, что общие закономерности изменения ХНх с напря-
жением сдвига сформулировать не представляется возможным,
за исключением того, что такая зависимость может быть резко
выражена для смесей полимеров в отличие от ДНГ —т индивиду-
альных компонентов смеси. Такой вывод имеет аналогию с рас-
творами полимеров в разных растворителях [94].
Повышенная эластичность смесей полимеров, обусловленная
анизотропией формы частиц дисперсной фазы, приводит к повы-
шенному разбуханию струи смеси на выходе из формующего при-
способления. Это наиболее ярко выражено для несформировав-
шегося потока при малых отношениях длины капилляра к диа-
метру lid. Если Z/d>20, то напряжение .в струе успевает отрелак-
сировать, по-видимому, как вследствие распада струй дисперсной
фазы >на капли по механизму Томотики (ем. гл. III), так и вслед-
ствие релаксации напряжений на границе раздела фаз. Это мо-
жет привести к понижению эластичности (разбухания) струи сме-
си полимеров [95]. Обычно же разбухание струи смеси полимеров
оказывается выше аддитивных значений или даже изменяется
в зависимости от состава по кривой с максимумом. Если частицы
дисперсной фазы образованы пространственно сшитым полимером
(микрогелем), то разбухание струи, наоборот,снижается [96, 97].
Напряжение сдвига, при котором происходит срыв струи в ка-
пилляре, меняется с составом смеси близким к аддитивному зна-
чению [98], тогда как соответствующая критическая скорость
сдвига изменяется более значительно. Эластичная турбулентность
(шероховатость) струи в смесях может иногда резко снижаться.
Причины этого .не всегда ясны. Это, однако, облегчает перера-
ботку. Вводя всего несколько процентов полиэтилена в ПС, мож-
но увеличить напряжение появления шероховатости в струе, да-
же если ПЭ имеет большую вязкость, чем ПС [99].
Работ, количественно связывающих параметры фазовой струк-
туры .в потоке с реологическими свойствами смесей, не имеется.
Качественная связь прослеживается лишь в том смысле, что свой-
ства смеси определяются в основном свойствами полимера, обра-
зующего .непрерывную фазу. Определить какой компонент обра-
зует непрерывную фазу не всегда просто. Как правило, можно
быть уверенным лишь в том, что непрерывную фазу образует
полимер с малой вязкостью, и притом преобладающий по количе-
ству .в смеси. Это хорошо прослеживается на примере смесей
224
Рис V.32. Зависимость эффективной вяз-
кости смесей СКИ-3 ПС от молоку--
лирной массы ПС (температура опыта
130°C, lgr=5,5 Па):
1 - 10% ПС; 2 - 20% ПС; 3 - 40% ПС.
с разной молекулярной массой
одного из компонентов. Так, на
рис. V.32 [96] показана зависи-
мость вязкости от молекулярной
массы ПС в смеси его с СКИ-3.
Для индивидуального ПС в та-
ких координатах наблюдается
прямая линия, а в смеси
СКИ-3—ПС с увеличением моле-
кулярной массы ПС происходит
обращение фаз. При малых мо-
лекулярных массах полистирол
образует непрерывную фазу, и вязкость смеси сильно снижается
с увеличением содержания в ней ПС. Высокомолекулярный ПС
образует дисперсную фазу в матрице СКИ-3, и вязкость смеси не-
значительно увеличивается с ростом молекулярной массы ПС, по-
скольку вязкость матрицы (СКИ-3) остается постоянной.
Аналогично вязкости с изменением молекулярной массы одно-
го из компонентов меняется энергия активации вязкого течения
и индекс течения п, которые также определяются главным обра-
разом природой непрерывной фазы [96].
§ 4. РЕЛАКСАЦИЯ НАПРЯЖЕНИЯ И ПОЛЗУЧЕСТЬ
Систематические исследования релаксации напряжения
и ползучести смесей полимеров в широком интервале составов
практически отсутствуют. На основании имеющихся эксперимен-
тальных данных и общих соображений о механизме деформации
смесей можно тредстаьить 'следующие закономерности процесса.
? В однофазных смесях скорость релаксации напряжений или
ползучести должна увеличиваться приблизительно пропорциональ-
но содержанию более податливого полимера. Это справедливо
только для таких систем, в которых нет сильного межмолекуляр-
ного и иного взаимодействия между .разнородными макромолеку-
лами. Если же такое взаимодействие реализуется, напоимер для
смеси эластичного сополимера бутадиена и 'Винилпиридина с со-
полимером бутадиена и акриловой кислоты, система теряет рас-
творимость в органических .растворителях вследствие реакции
функциональных групп друг с другом. Такая смесь приобретает
свойства пространственно сшитого полимера и ползучесть ее
ограничена сеткой прочных межмолекулярных связей.
В двухфазных смесях релаксация .напряжения и ползу честь
пирс шляются главным образом свойствами полимера, образую-
225
I > Н
щего матрицу, и скорость релаксационных /процессов претерпева-
ет сильные изменения в области составов, где происходит обра-
щение фаз. Зависимость скорости релаксационного процесса от
состава смеси должна при этом выражаться S-образной кривой
во всей области составов, о чем подробно говорилось в предыду-
щем разделе на примере вязкости и модулей смесей. В этом слу-
чае невозможно предполагать даже приближение к аддитивности
в зависимости скооо^ти релаксации напряжения или ползучести
от состава смеси. /Цдпако в ограниченной области составов мо-
жет наблюдаться известная пропорциональность в изменении ре-
лаксационных свойств: возможно увеличение скорости релаксации
и ползучести при добавлении каучукоподобиого полимера к стек-
лообразному или снижение этих показателей при добавлении
твердого полимера к .каучукоподобному (как это происходило бы
при введении минеральных наполнителей).
Существенное ограничение указанных закономерностей обу-
словлено возможностью расслаивания смеси полимеров при боль-
ших деформациях или напряжениях [100]. При отслаивании мат-
рицы от частиц дисперсной фазы ползучесть резко возрастает. Это
явление кажется довольно очевидным, однако экспериментально
подтверждено только для полимеров, содержащих минеральный
наполнитель.
Очень приближенно и только для малых деформаций и мало-
го содержания дисперсной фазы в отсутствие отслаивания на
межфазной границе можно пользоваться соотношением
8 (0 Gm
(0 б
где е(О и 6m (О—ползучесть смеси и полимера, образующего матрицу; G и
Gm — релаксационные модули [55].
Закономерности ползучести могут резко изменяться при боль-
ших деформациях, приводящих к образованию волокнистой
структуры (когда частицы дисперсной фазы превращаются
в волокна). При определенном соотношении модулей упругости
фаз такое волокнообразование может привести к «усилению» сме-
си и снижению ползучести. Ориентационное упрочнение особенно
характерно для смесей полимеров, содержащих кристаллический
(кристаллизующийся) компонент в качестве дисперсной фазы.
При близких значениях модуля дисперсной фазы и матрицы оп-
ределенное значение приобретает деформация в межфазном слое.
Она приводит к ускорению ползучести, поскольку энергия коге-
зии в межфазном слое, .как правило, ниже аддитивного значения.
Смеси полимеров являются реологически сложными объекта-
ми, и по этой причине применение к ним принципа суперпозиции
ограничено. Если в смеси преобладает один из полимеров, то
он образует матрицу и тогда именно преобладающий полимер оп-
ределяет релаксационные свойства системы. Принцип темпера-
турно-временной эквивалентности применим в этом случае к сме-
(52)
226
I'i V.33. Релаксационные модели
..-и ПВА —ГШМАсш (1:1). Циф-
i.i у кривых — температура опыта.
тг го3 то4
ftp см л, с
< и !ак же, как к полимеру
м.нрпцы, содержащему напол-
ни! ель [56]. Приведение осу-
ществляется не только по тем-
пературе и времени действия
(илы, по и по концентрации
наполнителя. Можно думать,
ио эластичный наполнитель в
пюрдой матрице в этом смыс-
ле не является исключением.
В отношении смесей со
средним соотношением компо-
нентов решение вопроса о при-
менимости принципа темпера-
пр по-временной эквивалент-
ное гн не столь однозначно.
Экспериментальные данные го-
нор ят о том, что приведение
но (можно для тех частей обоб-
щенной кривой, которые ле-
। 11 непосредственно в окрестностях температур стеклования
। к того компонента. В промежуточной области температур,
ни да механическое поведение компонентов крайне различно,
приведение невозможно. Заметим, что промежуточная область
и мператур — это та область, в которой особенно четко проявля-
г.>|ся релаксационные свойства межфазного слоя, что иногда обус-
вшливает появление промежуточного максимума потерь, который
ишечно «не предусматривается» классической теорией приведе-
ния.
< амо по себе наличие перегибов на кривой G'—Т, соответст-
вующих релаксационным переходам каждого компонента, не яв-
чк-н я принципиальным препятствием для применения принципа
•юшналснтности. Так, для смесей полистиролов, в которых моле-
(ярпые массы фракций сильно различаются, обнаружено два
нгрегнба на термомеханической кривой. Однако указанные два
перегиба могут быть получены чисто теоретически, если релакса-
ционные спектры двух фракций ПС принять идеализированными
(прямоугольными) и если эти спектры занимают разное положе-
ние и а оси времен. Два перегиба получаются в результате рас-
। I, исходя из принципа аддитивного сложения спектров, без
npi [положений о фазовой неоднородности смеси [101]. Таким
Htp । (ом, аддитивность релаксационных свойств (аддитивность.
iiiBhi шого объема) может обеспечить применимость принципа
•юшн.1 iciiTiiocTH даже для реологически сложных систем.
227
Рис. V.34. Обобщенные кривые для го-
мополимеров ИВА и ПММЛСШ и для
их смеси (1:1) при 112 °C по данным
рис. V.33.
Примером применения прин-
ципа эквивалентности к смесям
полимеров является получение
обобщенной кривой для смеси
ПВА с ПММА, частицы которого
диаметром около 100 мкм были
пространственно сшиты в процес-
се эмульсионной полимеризации
путем добавления 0,5% аллилметакрилата [102]. Эксперименталь-
ные данные по временной зависимости релаксационного модуля
приведены на рис. V.33 [102]. Значения Тс для ПВА около 35 °C,
для ПММА около ПО °C. Приведенные данные типичны в том
смысле, что здесь отчетливо видны два релаксационных перехода,
соответствующих каждой фазе полимера в смеси. Можно приме-
нить для описания полученных результатов модель Такаяпаги
[103], но для лучшего совпадения теории и эксперимента, в соот-
ветствии со сказанным выше, параметры модели а и ф следует
взять разными в разных областях температур. В данном случае
такими областями температур оказались найденные эмпирически
области: Г<40°С; 40оС=СГ< 110°C; ТД>Л10оС. Теперь можно по-
строить обобщенную кривую для гомополимеров и их смеси
(рис. V.34), исходя из известных свойств гомополимеров (элемен-
тов модели) и параметров модели, т. е. способа сочетания свойств
гомополимеров в смеси [103]. Поскольку выбор элементов модели
не обоснован физически, построение обобщенной кривой также
проведено по существу эмпирически. Преимущества выбранной
системы в том, что в ней природа непрерывной фазы определяется
не столько соотношением компонентов, сколько наличием про-
странственной сшивки в ПММА и, следовательно, его неспо-
собностью образовать матрицу при соотношении компонентов
1:1.
На рис. V.35 [103] показана зависимость фактора смещения
1g а.т от температуры для двух времен действия силы, которые
фактически являются границами экспериментального интервала
измерения релаксационного модуля (см. рис. V.33). Из рис. V.35
видно, что указанная зависимость является функцией времени,
при котором производился отсчет релаксационного модуля. Это
одно из основных отличий поведения смеси от поведения .индиви-
дуальных полимеров. Если построить обобщенные кривые или
кривые 1g ат—Т при разных температурах приведения (например,
112 и 40,8°C), то наклон кривых окажется разным, что также ха-
рактерно именно для смесей и полимеров, поскольку для индиви-
дуальных полимеров изменение температуры приведения может
228
привести только к смещению обобщенной кривой без изменения
ее наклона. Указанные отличия поведения смеси полимеров от
поведения индивидуальных полимеров наиболее заметны в обла-
сти температур, промежуточной между Тс взятых компонентов,
чем и вызвано нарушение непрерывности кривой 1g ат—Т в этой
области, которое особенно отчетливо видно на примере кривой
для ударопрочного ПС на рис. V.36 [104]. Для областей темпе-
ратур или .времен действия силы вблизи Тс компонентов можно
пользоваться значениями ат соответствующих индивидуальных
полимеров. Поскольку модель двухфазной системы применима
для описания релаксационных свойств смеси в широком интерва-
ле температур и времен действия силы только при произвольном
выборе параметров, величина фактора смещения для смеси поли-
меров в конкретных областях температур и времен может быть
определена только экспериментально.
Аналогичные результаты получены на примере ударопрочного
ПС и его смеси с полибутадиеном, для которой авторы [104]
обнаружили промежуточный максимум при температуре около
—40 °C, возникший вследствие «перепутывания разнородных мак-
ромолекул». Зависимость фактора приведения от температуры
для ударопрочного ПС показана на рис. V.36. Релаксационные
переходы проявляются при —80 и +85 °C, т. е. в области стекло-
вания индивидуальных полимеров данной структуры и молеку-
лярной массы. При температуре ниже —80 °C релаксирует глав-
ным образом'только ПБ и применима формула ВЛФ:
. R С^Т — Т„)
lg аг с2+(Т — Tv)
Рис. V.35. Температурная зависимость 1g ат для смесей ПВЛ: ПММА (1:1)
при температуре приведения 112 °C для времени действия силы 100 и 3000 с
Рис. V.36. Фактор приведения для ударопрочного ПС при температуре приве-
дения 87°C (расчетные и экспериментальные данные).
229
где а^— фактор приведения каучуковой фазы; С! = 11,2 и С2=60,5 для ПБ; тем-
пература приведения То=—87 °C.
Данные при высокой температуре (выше О °C) определяются
релаксацией фазы ПС. Для этой области оказалось хорошо при-
менимым уравнение ВЛФ, измененное в работе [105], с учетом
температурного коэффициента свободного объема Да/:
с —|7’ (T)/&a.f — То|
lg = Ci + [Т + wf {T}!\af — То| т> Т2 I54)
—Ci [Т2 - Wf (T')/&a.f — 7Д
lg = Ci + |7’2 + w/(n/Aa/-7’0] т ^т2 (55)
где индекс G означает стеклообразную фазу; Wf(T)—доля неравновесного сво-
бодного объема; Ci=J2,4; С2=41,0; 7'2=59°С; температура приведения Ти=
= 100 °C; Да,=4,5-10 4 град-1.
Данные по Wf(T) взяты из работы [105]. Расчет по уравне-
ниям (53), (54), (55) дает кривую, приведенную на рис. V.36.
Кривая совпадает с экспериментальными данными во всей обла-
сти температур, кроме промежуточных между Тс индивидуальных
компонентов.
§ 5. ПРОЧНОСТЬ И РАЗРУШЕНИЕ
Закономерности прочности и разрушения смесей полимеров
изучены мало, а обобщенное представление об этих закономер-
ностях начинает складываться только для ударопрочных пласт-
масс. Поэтому накопленный экспериментальный материал целе-
сообразно рассматривать с позиций прочности наполненных поли-
меров, которые исследовались давно, и степень обобщения экспе-
риментальных данных здесь относительно велика. В этом пара-
графе закономерности прочности полимеров, содержащих непо-
лимерный наполнитель, не будут рассматриваться подробно.
Для состояния теории прочности наполненных систем, как
и вообще для теории прочности композиционных материалов,
характерно обилие представлений, исходных предпосылок и ги-
потез, выдвигаемых для обоснования механизма разрушения
двухфазных систем с выраженной границей раздела. Такое поло-
жение обусловлено тем, что для разных композиционных мате-
риалов и в разных условиях испытания (определения прочности)
основные причины, ответственные за получаемое значение проч-
ности, различны. Различны и механизмы разрушенияТРДля пони-
мания механизма разрушения композиционного материала нель-
зя рассматривать его подобно однофазной полимерной системе
как сплошную среду. Действительно, все закономерности прочно-
сти, отличающие композиционный материал от материала одно-
фазного (в том числе однофазного, но микрогетерогенного, как
все полимерные материалы), связаны с наличием выраженной
поверхности раздела. Поэтому для понимания закономерностей
230
процесса разрушения и закономерностей прочности необходимо
глубокое понимание микромеханики процесса зарождения и ро-
ста трещин.
Нет необходимости подробно излагать здесь эксперименталь-
ный материал по прочности наполненных материалов ввиду’ на-
1ичия соответствующих недавно опубликованных монографий
[55,56,106,107].'
\ Всякая микронеоднородность в матрице полимера является
концентратором напряжений, и в этом смысле частица наполни-
теля не является исключением. На сферической частице возника-
ют перенапряжения, в три раза превышающие среднее значение
напряжения в матрице. При наличии острых углов, выступов
и т. п. концентрация напряжения увеличивается: на возникшей
в растягиваемой резине трещине, глубина которой 30 мкм, а ра-
диус кривизны в вершине 10 нм, напряжение повышается в 10 раз.
Поэтому реально наблюдаемое в большинстве случаев увеличение
прочности при введении наполнителей должно быть обусловлено
рядом причин, которые бы «нейтрализовали» отрицательный эф-
фект наполнителя как концентратора напряжений. Эго не отно-
сится к системам наполнитель — кристаллический полимер, где
частицы наполнителя могут явиться зародышами кристаллизации
и так сильно изменить характер кристаллической структуры поли-
мера, что одно это может привести к существенному изменению
механических свойств.
Увеличение прочности (эффект усиления) в разных типах на-
полненных систем вызвано следующими основными причинами:
1. Изменение направления роста трещины при встрече с ча-
стицей наполнителя.
2. Ориентация и упрочнение полимера в межфазном слое на
границе раздела полимер — наполнитель.
3. Возникновение собственной структуры наполнителя в ре-
зультате нехаотчческого (в том числе цепочечною) расположе-
ния его частиц.
4. Разрыхление и ослабление полимера в межфазном слое,
где перенапряжения, в том числе в вершине растущей трещины,
при деформации релаксируют; изменяется направление роста тре-
щины; увеличивается область пластической деформации в вер-
шине трещины.
Рассмотрим проявление этих механизмов усиления полимера
при введении в него другого полимера, т. е. при получении сме-
си. Однифазные смеси полимеров при этом не рассматриваются.
Усиление полимера более твердым полимером. При введении
пластиков в каучуки иногда большую роль играет даже не увели-
чение прочности при растяжении, а увеличение предела текуче-
сти при растяжении невулканизовапной смеси. Этот показатель,
называемый в резиновой технологии «прочностью невулканизо-
ванной смеци» или «каркасностью», определяет способность сме-
си сохранясь форму, приданную ей в процессе формования, вплоть
231
Рис. V.37. Зависимость максимального
напряжения (предел текучести) при рас-
тяжении невулкаиизованных смесей
СКД : ПС от содержания в них ПС раз-
личной молекулярной массы:
7 —,М=5,5-103; 2 —7.7-103; 3 — 2,8-10*; 4 —
2.2-10’.
до вулканизации смеси. Предел
текучести увеличивается в ре-
зультате образования анпзомет-
ричных частиц дисперсной фазы
пластмассы, ориентированных в
направлении деформаций при
формовании. Чем больше ориен-
тация частиц дисперсной фазы,
тем больше эффект усиления.
Несколько процентов полиэтиле-
на (ПЭВП или ПЭНП) могут в
несколько раз увеличить предел
текучести невулканизованного
каучука. Эффект усиления обусловлен, однако, не только степенью
ориентации дисперсной фазы, но и собственной жесткостью и
прочностью ее частиц. Так, на рис. V.37 [31] видно, что с увели-
чением молекулярной массы вводимого ПС эффект усиления кау-
чука СКД растет, хотя в условиях смешения полимеров на валь-
цах (температура 130°C) ориентация частиц дисперсной фазы
больше в случае маловязкого низкомолекулярного ПС.
При приготовлении смесей каучука и пластмасс особенно ва-
жен оптимальный температурный режим. Недостаточно, чтобы
температура смешения превышала Тс пластмассы. Необходимо,
чтобы она была заметно выше температуры текучести, так как
в противном случае необратимая деформация диспергируемой
пластмассы минимальна или вообще не происходит, что не обес-
печивает хорошего диспергирования пластмассы и создания не-
прерывной фазы ее в смеси. Обычно для резкого увеличения мо-
дулей и жесткости смеси необходимо вводить более 20% (об.)
пластмассы, однако и в этом случае эффект усиления существен-
но меньше, чем при введении усиливающих наполнителей типа
сажи.
Эффект усиления сохраняется и после вулканизации смеси
каучука с пластмассой, однако оптимальное содержание пласт-
массы, необходимое для усиления смеси до вулканизации, для
вулканизата может быть иным. Причины изменения хода кривой
прочность — состав в результате вулканизации не окончательно
уяснены, однако, по-видимому, это явление вызвано в первую оче-
редь тем, что вулканизат разрушается при удлинениях (500—
800%), в несколько раз превышающих удлинение (50—100%),
при котором определяется предел текучести (прочность) невулка-
232
низованной смеси. При разрушении вулканизата, естественно,
разрушается каркас, образованный фазой пластмассы, и конечная
прочность вулканизата зависит в значительной мере от способно-
сти пространственно сшитого каучука к ориентации и от того,
препятствует разрушение каркаса пластмассы этой ориентации
или, наоборот, способствует. Для вулканизатов характерно также
увеличение прочности при растяжении с увеличением твердости
(модуля) дисперсной фазы. Это видно из данных табл. V.2. Проч-
ность при растяжении растет, а относительное удлинение падает
при переходе от бутадиен-стирольного сополимера с 65% стирола
(СКС-65) к ПС в качестве наполнителя [108, с. 38].
Некоторые аспекты структуры и прочностных свойств каучу-
ков, наполненных пластмассой, подробно рассматриваются в мо-
нографии [56].
К числу каучуков, наполненных пластмассой, относятся и блок-
сополимеры, например блок-сополимеры бутадиена и стирола
(термоэластопласты). Блок-сополимеры бутадиена и стирола име-
ют прочность свыше 30 МПа (см. например, [74, 109]) и двух-
фазную структуру, представляющую собой дисперсию доменов
ПС в матрице ПБ (см. гл. IV). Наличие редких химических свя-
зей па границе раздела фаз предотвращает расслаивание при
больших деформациях, тогда как в обычных смесях каучуков
с пластмассами отслаивание наблюдается так же, как в смеси
двух пластмасс (см. § 2). Наличие химических связей между до-
менами ПС и матрицей ПБ вряд ли является главной причиной
повышенной прочности блок-сополимеров и уж во всяком случае
это не единственная причина. Дело в том, что смешением полиме-
ров, например ПБ и ПС, невозможно получить смесь с регулярным
расположением частиц дисперсной фазы размером порядка 180 нм,
как это требуется для оптимальных прочностных свойств в ударо-
прочном полимере [НО], и тем более с размером частиц в 15—
Таблица V.2. Свойства вулканизатов СКС-ЗОАРК,
наполненного высокостиротьными сополимерами (смолы и ПС введены
в количестве, обеспечивающем содержание стирола в смеси 45%)
Показатель Полимер
СКС-65 СКС-75 СКС-85 СКС-95 ПС
Разрушающее напряжение при растяжении, МПа 7,17 8,92 9,28 11,2 11,14
Относительное удлинение при разрыве, % Остаточное удлинение, % 511 398 313 304 206
16 17 49 55 24
Твердость по ТМ-2 75 84 90 91 88
Температура хрупкости, СС —23 —24 —27 —31 —35
Эластичность по отскоку, % 6 11 16 19
233
25 нм как в термоэластопласте. Поэтому нельзя проверить весьма
простое и логичное предположение, что большая прочность тер-
моэластопластов может быть обусловлена малым размером, а
следовательно, и большой удельной поверхностью раздела фаз,
которая определяет эффект усиления (см. гл. IV). Возник-
новение смешанной флуктуационной сетки в межфазном слое
каучук — пластмасса может обеспечить при малом размере час-
тиц достаточную связанность системы в целом. Роль регулярности
расположения доменов ПС подтверждается экспериментальными
данными [111]. Из этих данных следует, что расширение ММР
блоков ПС (Мк-/Мп) от 1,05 до 2,5 приводит к снижению прочно-
сти термоэластопласта с 16,5 до 13,5 МПа. Расширение же ММР
полибутадиеновых блоков (ММР полибутадиена определяет рав-
номерность расположения доменов ПС и возникновение внутрен-
них напряжений при наличии широкого ММР) с 1,05 до 2,6 при-
водит к снижению прочности с 16,5 до 6.0 МПа, т. е. до величины,
характерной для простой смеси каучука с ПС (см. рис. V.37). За-
метим также, что само по себе наличие химических связей в меж-
фазном слое не всегда приводит к росту прочности смеси поли-
меров, а иногда даже вызывает ее снижение. Так, эффект усиле-
ния вулканизата бутадиен-стирольного каучука полистиролом
больше, чем сополимером стирола с 10% бутадиена (СКС-90),
хотя при вулканизации между частицами сополимера СКС-90
и матрицей каучука возникают химические связи. Если снижение
прочности в сополимере СКС-90 обусловлено некоторым умень-
шением его модуля, следовательно эффект химических связей
в межфазном слое во всяком случае недостаточен, чтобы ком-
пенсировать это влияние [И2].
Механизм усиления каучука пластмассой определяется следу-
ющими факторами из числа перечисленных на с. 231.
1. При наличии в качестве дисперсной фазы частиц твердого
полимера перенапряжения в вершине растущей трещины недоста-
точны для того, чтобы разрушить такую частицу. Поэтому трещи-
на огибает частицы или меняет направление (роста, что приводит
к увеличению поверхности разрушения, а значит и к повышению
прочности, которая по Гриффитсу определяется суммарной вели-
чиной энергии образования новой поверхности.
2. Анизометричные частицы дисперсной фазы, соединяясь не-
посредственно в результате коалесценции (конденсационная
структура) или через тонкие прослойки дисперсионной среды
(коагуляционная структура), образуют каркас. Агломерация час-
тиц дисперсной фазы может происходить в результате плохого
смачивания матрицей всей частицы или ее части, покрытой тем
или иным адсорбционным слоем, или, наоборот, вследствие хоро-
шего смачивания модифицированной части и плохого смачивания
непокрытого полимера. Возникновение анизометричных частиц
дисперсной фазы, и тем более слияние их в единый каркас, при-
водит к увеличению вероятности встречи трещины с частицей
234
и к наиболее эффективному увеличению энергии новообразования
поверхности. Увеличение дисперсности частиц (уменьшение их
диаметра) также приводит к удлинению пути растущей трещины,
поскольку увеличивается число частиц в единице объема при од-
ной и той же объемной доле полимера-наполнителя.
3. Межфазный слой на границе раздела полимер — полимер
является ослабленным элементом системы, где энергия когезии
понижена (см. гл. IV). В этий ослабленной зоне происходит
быстрая релаксация напряжений в вершинах трещин и они ста-
билизируются или распространяются преимущественно по меж-
фазным слоям, т. е. снова многократно меняется направление ро-
ста трещин. Деформация матрицы в устье трещины переходит
в деформацию межфазного слоя, а иногда и в деформацию час-
тицы диспергированного полимера. Это хорошо показано на при-
мере термоэластопластов. где в матрице развиваются большие
напряжения, не сопровождающиеся отслоением в межфазном
слое, и где деформацию доменов ПС наблюдали непосредственно
[74].
Механизм усиления, отмеченный под № 2 в приведенном
выше общем списке возможных причин усиления, в применении
к смесям полимеров, видимо, не проявляется в значительной
степени, поскольку межфазное натяжение на границе расплавов
смесей полимеров невелико, а следовательно, невелика и адсорб-
ция одного полимера на поверхности другого. Однако некоторое
уплотнение полимеров в межфазных слоях нельзя исключать
[56].
Усиление твердого полимера мягким низкомодульным полиме-
ром происходит при введении каучука в пластмассу, т. е. в пер-
вую очередь при получении ударопрочных полимеров. Дисперс-
ная фаза каучука, однако, обеспечивает не только увеличение
ударной вязкости, но и повышение прочности при одноосном рас-
тяжении и при других видах деформации, что особенно эффек-
тивно проявляется при наполнении каучуками термореактивных
полимеров типа эпоксидных, полиэфирных и т. п. В этих случаях
разрушение материала происходит по механизму хрупкого раз-
рушения, на что указывают малые значения разрывного удлине-
ния. При этом, как в случае удара, так и в случае одноосного
растяжения трещина распространяется с большой скоростью. Это
обстоятельство сразу выдвигает требование к повышенной элас-
тичности каучука — его температура стеклования должна сильно
отличаться от температуры испытания, иначе в условиях быстро-
го прорастания трещины частицы каучука могут потерять элас-
тичность и вести себя фактически как стеклообразные включения
в стеклообразной матрице. Ударными нагрузками называют
обычно механические воздействия на материал со скоростью 5--
25 м/мин.
Нет никаких обобщений разрозненных экспериментальных дан-
ных о том, как каучуковая фаза влияет на прочность стеклооб-
235
разных или кристаллических полимеров в условиях больших,
в том числе вынужденноэластических деформаций. Однако уста-
новлено, что при введении каучука в пластмассу прочность ее
при обычных скоростях деформации и при нехрупком механизме
разрушения, как правило, снижается. Что касается предела те-
кучести, то он снижается при введении каучука до величины,
приблизительно равной произведению предела текучести полиме-
ра матрицы на объемную долю каучука [ИЗ].
Интенсивная диссипация энергии и перенапряжений в верши-
не растущей трещины происходит тогда, когда велики механиче-
ские потери при деформации, т. е. в температурно-временных ус-
ловиях, соответствующих максимуму на кривой зависимости тан-
генса угла механических потерь от скорости действия силы или
от температуры. Если tgfi$c0,02, полимер является хрупким, если
tgft>O,l, полимер обладает большой стойкостью к удару.
Закономерности прочности хрупких полимеров, содержащих
каучуки, наиболее широко исследованы на примере полистирола.
Полистирол чрезвычайно хрупкий полимер. Для него не харак-
терны p-релаксациоиные переходы, которые в таких полимерах,
как полиметилметакрилат, поликарбонат и др., обеспечивают дис-
сипацию механической энергии в растущей трещине и повышение
сопротивляемости разрушению при ударе. Поэтому в ПС вводят
каучуки, способные рассеивать энергию удара при температурах
от —40 °C и выше, поскольку в этой области температур они
обычно обладают большим tgfi. Для введения каучука обычно
используют метод прививки в латексе или в растворе. Простое
смешение на механическом оборудовании применяется гораздо
реже, так как обеспечивает менее эффективную защиту от удара.
Как правило, при получении ударопрочных полимеров (ударо-
прочного ПС, смол АБС) каучуковая фаза должна быть слегка
структурированной, что достигается либо введением структури-
рующих агентов в процессе полимеризации в растворе, либо пред-
варительным структурированием каучука в латексе.
На рис. V.38 показано влияние содержания микрогеля (сши-
тых частиц) бутадиен-стирольного каучука на характер кривых
нагрузка — удлинение смесей ПС — БСК. Из рисунка видно, что
с повышением содержания каучука
полистирол перестает разрушаться
хрупко, и площадь пот кривой увели-
чивается, т. е. возрастает энергия раз-
рушения пластмассы. Еще более ин-
формативна табл. V.3 [113], из кото-
Рис. V.38. Кривые нагрузка — удлинение при
скорости 3,4 м/мин механических смесей ПС и
пространственно сшитого бутадиен-стирольно-
го сополимера. Цифры на кривых — содержа-
ние сополимера в %.
236
Таблица V.3. Механические свойства различных ударопрочных композиций
на основе ПС в сравнении со свойствами исходного ПС
Материал Скорость растяжения, М/МИН Модуль упругости £-10-2, МПа Разрушающее напряжение при растяже- нии, МПа Удлинение при разры- ве, % Энергия, затрачиваемая для достиже- ния предела текучести, 17-10-ь, Нм/мЗ Энергия, затрачиваемая на разрушение образца £7-10—5, Нм, м3
л* 1,25-IO-3 2,22 36,0 25,3 2,08 82,0
2,510-2 2,28 40,5 19,6 3,60 72,5
2,5 2,52 52,8 8,15 5,65 36,5
7,3-10 3,28 70,7 7,7 7,61 45,5
в** 1,25-10-з 2,56 26,9 20,5 1,05 47,8
2,5-IO-2 2,55 32,2 16,4 2,04 47,4
1,85 3,32 45,4 6,1 3,36 23,5
6,3 4,30 60,0 6,0 4,18 30.3
ПС 1,25-10-з 3,50 45,5 1,55 2,97 4.05
4,5-10 4,50 76,0 2,2 6,45 10,0
• Предел текучести
** Предел текучести
30,3 МПа.
23,3 МПа.
рой видно, что ^ведение каучука приводит к снижению модуля
упругости, предела текучести и прочности материала по сравне-
нию с этими показателями у исходного ПС, но значительно увели
чивает величину работы, необходимой для достижения предела те-
кучести и для разрушения образца во всем исследованном интер-
вале скоростей растяжения. Из таблицы видно, что при повышении
скорости деформации модуль упругости и прочность при растяже-
нии как для наполненного, так и для ненаполненного ПС возра-
стают, но энергия, требуемая для разрушения наполненного ПС,
уменьшается.
Многие исследователи пытались выяснить механизм повыше-
ния ударной вязкости хрупких пластмасс при введении каучука,
однако, полной картины этого механизма пока нет.
Предполагалось, что образующаяся в пластмассе трещина,
контактируя с частицей каучука, деформирует ее. Перенапряже-
ния в вершине трещины расходуются на деформацию этой ча-
стицы, диссипируясь и превращаясь в конечном итоге в теплоту
внутреннего трения каучука [114]. Как показано, в микротрещи-
нах, возникающих при ударе, действительно имеются тяжи ори-
ентированного каучука. Когда размер частиц каучука становится
соизмеримым с радиусом кривизны вершины трещины, т. е.
меньше 10 им, эффект усиления практически исчезает [115].
Отмечено [116], что в ударопрочном ПС всегда возникает
большое количество микротрещин, заполненных тяжами ориенти-
рованного полимера, в отличие от макротрещин, в которых ори-
ентированного материала нет или он уже разрушился. Обилие
микрот рещин свидетельствует об очень большой поверхности
237
разрушения, которая и поглотает энергию удара. В настоящее
время, пожалуй, установилось единое мнение о том, что именно
сильно развитая поверхность разрушения, создаваемая разветв-
ленной сеткой микротрещин, является причиной повышения
ударной вязкости хрупких пластмасс при введении в них дис-
персии каучуков. Авторы [116] рассматривали частицы каучука
как концентраторы напряжений, которые и инициируют развитие
микротрещин. Оказывается, существенную роль играет число час-
тиц дисперсной фазы в единице объема [117]. Так, при расстоя-
нии между частицами не более, чем в 1,5 раза превышающем их
диаметр, рост микротрещины начинается при небольших напря-
жениях и, как показано на примере ударопрочного ПС, трещины
растут по направлению от частицы к частице, а не возникают
исключительно в направлении, перпендикулярном действию силы.
Практически далеко не все частицы каучука генерируют микро-
трещины: например, около частиц размером более 4 мкм трещи-
ны не возникают. Существенно, что введение частиц твердого
наполнителя или пузырьков воздуха, также являющихся концент-
раторами напряжения, не приводит к 'повышению стойкости
к удару.
Считается, что при деформации в присутствии каучука увели-
чение механических потерь может привести к возникновению мест-
ных очагов теплообразования и вызвать повышение температуры,
досгаточное для возникновения вынужденноэластической дефор-
мации в микрообъемах ПС или другого стеклообразованного
хрупкого полимера. Было высказано предположение [118], что
каучук вызывает релаксационные f-переходы в ПС, которые так-
же способствуют диссипации энергии и росту микротрещин, за-
полненных ориентированным полимером матрицы. Это подтверж-
дается практически одинаковым значением энергии активации
смещения максимума потерь в каучуке, f-перехода и процесса
образования микротрещин. Если частицы каучуковой фазы спо-
собствуют рассеянию энергии растущей трещины благодаря де-
формации каучука, то стойкость к удару полимера, наполненного
каучуком, при данных условиях деформации должна меняться
с изменением величины механических потерь именно в каучуке.
В действительности, однако, пропорциональность между величи-
ной механических потерь в каучуковой фазе и ударной вязкостью
наблюдается лишь для систем, полученных одинаковым способом
[119], что было показано в частности на примере смол АБС
[109].
Высокая ударная вязкость материала при относительно малом
содержании каучука может быть достигнута только при подвул-
канизации каучука и наличии хотя бы редких химических связей
между каучуком и матрицей при достаточно большом химическом
сродстве каучука и пластмассы, приводящем к возникновению
межфазного слоя в результате сегментального растворения ком-
понентов. Хорошее взаимодействие полимеров в межфазном слое
238
и прочная связь фаз необходимы в первую очередь для предот-
вращения отслаивания полимеров из-за различий коэффициентов
их теплового расширения. Так, термические коэффициенты ПС
и каучука различаются более чем в 4 раза (6-10~5 и 25-10
1/°С.). Наличие химических поперечных связей в каучуке и на
межфазной границе обусловливает пространственнонапряженное
состояние в каучуке и в прилегающем слое пластмассы [120].
В таком состоянии отслоение каучука от матрицы происходит
лишь при ударе, а это в свою очередь вызывает возникновение
радиальных микротрещин вокруг частиц. Если каучук не вулка-
низован и со средой не связан, то различие коэффициентов тер-
мического расширения приводит к возникновению пустот либо
в фазе каучука, либо на межфазной границе, что сразу уменьша-
ет эффект усиления хрупкого полимера.
Каучук и стеклообразный полимер (например, ПС) имеют раз-
ные коэффициенты Пуассона (0,49 и 0,33). Поэтому при дефор-
мации возникает гидростатическое давление в матрице, приводя-
щее к увеличению свободного объема и появлению способности
НС к вынужденноэластической деформации [121]. Этим, однако,
нельзя объяснить, почему при введении в ПС твердых частиц,
например двуокиси титана, также приводящих к возникновению
трехмернонапряженного состояния, ударопрочность ПС не увели-
чивается.
Новые представления о механизме усиления пластмасс каучу-
ками развиты в теории, предложенной Брагау [122]. В своей
работе Брагау анализирует теории упрочнения и отмечает их не-
достатки и неточности, не согласующиеся с экспериментальными
данными. Так, теория, основывающаяся на представлениях о по-
глощении энергии при прорастании трещины через частицу кау-
чука, будучи соответственно развита, предсказывает, что только
0,1 часть энергии удара может быть поглощена самими частица-
ми каучука. С помощью этой теории нельзя объяснить более вы-
сокие удлинения при разрушении ударопрочных полимеров по
сравнению с полимером матрицы, а также сильную зависимость
ударной вязкости от свойств матрицы при одних и тех же струк-
турномеханических параметрах частиц каучукового наполнителя.
Кроме того, толщина микротрещин в ряде случаев сравнима
с размером усиливающих частиц каучука, и поэтому энергия, по-
глощаемая при прорастании трещины в матрице (более высоко-
модульной, чем каучук), существенно больше, чем при прораста-
нии в относительно немногочисленных частицах каучука.
Теория, основывающаяся на представлениях об увеличении
свободного объема при наполнении хрупкой матрицы, также при
ближайшем рассмотрении в определенной степени нелогична.
Так, увеличение свободного объема за счет разности термических
коэффициентов расширения может быть еще больше, если вместо
каучука взягь твердые частицы, по размерам и концентрации
точно соответствующие частицам эластичного наполнителя. Ци-
239
тируемые в [122] данные показывают, что наполнение стирол-
акрилонитрильных сополимеров диоксидом титана приводит
к увеличению хрупкости и снижению ударной вязкости по Изоду,
т. е. эффект оказывается обратным по сравнению с наполнением
каучуком. При этом расчет показывает, что расширение матрицы,
обусловленное присутствием частиц каучука (при известных ко-
эффициентах объемного расширения), эквивалентно расширению,
вызванному нагреванием на 12 °C. Такой незначительный эффект
не может объяснить увеличения стойкости к удару при комнат-
ной температуре или тем более при —40 °C, что характерно для
-смол АБС. Данная теория не предсказывает зависимость эффек-
та упрочнения от размера частиц каучука, от величины адгезии
на межфазной границе.
Теория, рассматривающая частицы каучука как концентрато-
ры напряжений, недостаточна потому, что рассчитанный по из-
вестной в механике формуле Гудьира эффект концентрации на-
пряжений в зависимости от модуля частиц оказывается выше для
твердых или газообразных частиц в матрице хрупкого полимера,
чем для частиц каучука.
Теория Брагау основывается на том, что поскольку распреде-
ление упругой энергии в вершине макротрещины и микротрешины
одинаково, на процесс роста микротрещин можно распростра-
нить динамическую теорию развития трещин Гриффита и в осо-
бенности теорию Иоффе. По Гриффису, развитие трещины в од-
нородном хрупком теле ускоряется с ростом длины трещины по-
сле достижения некоторой критической длины, и в определенный
момент скорость достигает своего предельного значения, равного
приблизительно половине скорости упругопоперечной волны
в этом же теле. По достижении этой скорости трещина начинает
разветвляться. Это явление было объяснено Иоффе, показавшим,
что при большой скорости роста трещины максимальные напря-
жения концентрируются в двух разных плоскостях, не совпадаю-
щих с первоначальной плоскостью роста трещины. Поэтому тре-
щина может или изменить направление или разветвиться. Если
эти представления перенести на систему пластмасса — каучук,
то окажется, что критическая скорость роста трещины в матрице
составляет ~620 м/с, а в каучуке ~29 м/с. Поэтому, когда тре-
щина из матрицы перейдет в фазу каучука, там должно произой-
ти ее разветвление. Чем чаще встречается трещина с частицами
каучука, тем больше она ветвится, тем больше поглощение энер-
гии и тем вероятнее полная диссипация энергии удара и прекра-
щение роста трещины.
Теория критических скоростей ветвления подтверждается кор-
реляцией изменения скорости звука и ударной прочности с тем-
пературой [122]. Согласно теории критических скоростей ветвле-
ния, трещина ветвится в частицах каучука, имеющих определен-
ную протяженность, при которой частица приобретает все свойства
фазы каучука (см. гл. IV), а это соответствует размеру не менее
240
0,1 мкм. Поскольку в смолах АБС толщина микротрещины со-
ставляет 0,5—0,6 мкм, а в ударопрочном ПС около 2 мкм, то
этим объясняется, почему оптимальный размер частиц каучука
в ударопрочном ПС больше, чем в смоле АБС. Если частица
меньше толщины трещины, ветвления не происходит. Чем больше
размер частиц каучука, тем меньше их число (обратно пропорцио-
нально третьей степени диаметра), поэтому вероятность встречи
микротрещины с частицами быстро уменьшается и, соответствен-
но, снижается стойкость образца к удару. Если необходимо уве-
личить ударную вязкость без общего понижения модуля образца,
надо получить дисперсию каучука в матрице с размером частиц,
близким к оптимальному, и с узким распределением по размерам.
Понятно также, что при малой прочности связи частицы с матри-
цей микротрещина не переходит в фазу каучука, а просто огибает
частицу, как пустоту.
Довольно подробно эффект усиления каучуком эпоксидной
смолы исследован в условиях деформации образца с постоянной
заданной скоростью, в том числе на образцах, находящихся меж-
ду металлическими поверхностями, когда фактически наполненная
каучуком эпоксидная смола играла роль адгезива [123]. Отмече-
но, что переход от «пластмассы, усиленной каучуком, к смеси по-
лимеров» происходит при содержании каучука 15—20%, когда
модуль материала резко снижается и меняются все закономерно-
сти механических свойств. Почти все образцы разрушаются та-
ким образом, что трещина растет рывками, т. е. периодически ско-
рость роста трещины становится больше скорости деформации,
а затем уменьшается, напряжение снижается, и рост трещины
временно прекращается. Энергия разрушения, необходимая для
инициирования роста трещины после ее «остановки», в присутст-
вии оптимального количества каучука увеличивается в 30 раз,
тогда как энергия, •соответствующая спаду скорости роста трещи-
ны, в наполненной эпоксидной смоле лишь немного выше, чем
в отсутствие наполнителя. Область начинающегося разрушения
всегда может быть выявлена по интенсивному побелению вслед-
ствие развития микротрещин участка, где отмечены значительные
деформации как матрицы, так и каучука. В работе [123] сдела-
ны следующие важные выводы. В условиях деформации одноос-
ного растяжения частицы каучука ведут себя так, как если бы
они находились в трехмернонапряженном состоянии. При боль-
шой скорости роста трещины она разветвляется, и поверхность
разрыва оказывается очень неровной. Рассчитанный на основа-
нии экспериментальных данных диаметр зоны пластической де-
формации в вершине трещины прямо пропорционален энергии
разрыва. Оказалось, что с введением частиц каучука сильно уве-
личивается размер зоны пластической деформации в вершине
трещины. Высказано предположение, что увеличение зоны пла-
стичности может быть обусловлено двумя причинами: пластифи-
кацией матрицы за счет некоторой доли каучука, находящегося
16—244
241
Рис. V.39. Сопротивление утомлению вулка-
низатов смесей СКД и СКН-18:
1 — знакопеременный (симметричный) изгиб с кру-
чением; 2 — многократное растяжение от 8,=0 до
е2=150%; 3 — коэффициент утомляемости Р (по
данным многократного растяжения).
в матрице в состоянии истинного
раствора (или вообще в молеку-
ляриодисперспом состоянии), а
также наличием значительного диф-
фузного слоя на границе каучук —
матрица. Большие размеры диф-
фузного слоя обусловлены тем, что
система возникла в условиях, близ-
ких к критическим условиям расслаивания (в процессе роста мо-
лекулярной массы смолы при ее отверждении).
Если разрушение происходит в материале, работающем как
адгезив, то в тонких слоях адгезива толщина диффузного слоя
может совпадать с размером зоны пластичности в вершине тре-
щины, что и ведет к сильному искривлению пути трещины и со-
ответственному росту энергии разрушения. Подсчеты авторов да-
ют основание предполагать увеличение диаметра пластической
зоны с 0,001 до 0,035 см в результате введения частиц каучука
в условиях, близких к критическим, когда частицы возникают в
процессе выделения каучука как новой фазы (см. также с. 292).
Полученные в [123] экспериментальные результаты указыва-
ют на общность закономерностей разрушения наполненных кау-
чуком пластмасс как при ударе, так и при растяжении, близком
к одноосному, а также подтверждают большое значение разви-
того межфазного слоя в повышении стойкости к удару.
Таким образом, причины усиления пластмасс каучуками ясны
лишь в самом общем виде. Из перечисленных выше четырех воз-
можных причин усиления в данном случае имеют значение, види-
мо, первая и четвертая. Действительно, при наличии эффекта уси-
ления (например, роста ударопрочности) трещина многократно
меняет направление роста, а наличие разрыхленного межфазного
слоя обусловливает расширение зоны пластической деформации
в вершине трещины.
Взаимоусиление полимеров, близких по значениям модулей,
сформулированное как общий в таких системах эффект, было
обнаружено в работах автора [124, 85, 11, 125, 96] на примерах
увеличения сопротивления утомлению для смесей каучуков нату-
рального и бутадиен-стирольного, дщополибутадиенового и бута-
диен-нитрильного (СКН-18), полиизопренового и бутадиен-нит-
рильного (СКН-40). Типичные результаты для вулканизатов сме-
си СКД и СКН-18 приведены на рис. V.39, из которого видно, что
динамическая выносливость смесей в разных режимах утомления
изменяется по кривой с максимумом.
242
Наиболее надежной характеристикой усталостной выносливо-
сти является скорость уменьшения прочности резины в процессе
утомления. При этом число циклов до разрушения определяется
следующим образом:
ЛГ=(ог,/оЛ)₽ (56)
где Пр —прочность резины при однократном растяжении образца до разрыва;
сЛ-— механическое напряжение, вызывающее разрыв после N циклов; р — коэф-
фициент утомляемости.
Коэффициент р характеризует способность резины сохранять
свои механические свойства при динамическом нагружении, т. е.
является наиболее объективной и не зависящей от метода испы-
таний характеристикой усталостных свойств резины. Как видно
из рис. V.39. значение р также превышает соответствующие зна-
чения для индивидуальных каучуков, т. е. вулканизаты из смесей
каучуков лучше сопротивляются многократным деформациям,
чем вулканизаты индивидуальных каучуков.
Повышенное сопротивление утомлению любого полимерного
материала обеспечивается высокой статической прочностью, низ-
ким уровнем механических потерь, высоким сопротивлением старе-
нию. Для рассматриваемой пары полимеров все указанные пока-
затели свойств изменяются с составом смеси по кривой, близкой,
к аддитивной. Видимо имеется и другая причина повышенного соп-
ротивления утомлению, связанная непосредственно с двухфазной
структурой смеси.
Смешиваемые каучуки обычно различаются по значениям мо-
дуля (податливости), т. е. возникающая при их совмещении двух-
фазная система характеризуется различием модулей дисперсной
и непрерывной фазы (матрицы). Это различие часто еще более
увеличивается в результате вулканизации. Разрушение образца
при многократных деформациях начинается с роста микротрещи-
ны. Если микротрещина зарождается в матрице с меньшим моду-
лем, то при встрече с частицей высокомодулыюго каучука рост
трещины затормозится, а то и вовсе прекратится, что создает усло-
вия для релаксации перенапряжений в вершине трешины. Если
микротрещина растет в матрице с более высоким модулем (мо-
дуль непрерывной среды выше, чем модуль дисперсной фазы), то
перенапряжения в вершине растущей трещины сразу релаксируют
при встрече с частицей дисперсной фазы, имеющей большую по-
датливость. Все это вместе создает условия для большей динами-
ческой долговечности. Таким образом, эффект усиления проявля-
ется нс только при добавлении пластмассы к каучуку или каучука
к пластмассе, но и при совмещении полимеров, находящихся в од-
ном и том же физическом состоянии. Поскольку смешиваемые по-
лимеры, как правило, неодинаковы по величине модуля, прочности
и т. п., это создает ту оптимально неоднородную (микронеодпород-
ную) систему, которая лучше сопротивляется разрушению под дей-
ствием внутренних или внешних механических сил.
16
243
Рис. V.40. Число циклов N многократного изгиба,
необходимое для роста надреза в ченаполненных
вулканизатах смеси СКД— СКН-18:
1 — рост надреза в интервале 2—4 мм; 2— то же, в ин-
тервале 2—12 мм.
Предлагаемое объяснение подтверж-
дается прямыми измерениями скорости
роста трещины в процессе многократно-
го изгиба пластинки вулканизата с на-
скн-18 25 55 75 wo песенным проколом (рис. V.40), а также
тем, что повг рхность разрушения вулка-
низатов индивидуальных каучуков вы-
глядит более гладкой по сравнению с поверхностью разрушения
вулканизатов из смесей полимеров, особенно по сравнению с об-
разцами с максимальной динамической выносливостью (рис.У.41).
Может быть принято и другое объяснение эффекта взаимоуси-
ления. Как известно, энергия когезии в слое сегментальной рас-
творимости на границе раздела фаз ниже ее среднего значения
для смеси, что приводит к большей скорости релаксационных про-
цессов в этом слое. Перенапряжения в вершине трещины релакси-
руют при встрече с межфазным слоем, следствием чего является
либо прекращение роста трещины, либо распространение ее преи-
мущественно по границе раздела фаз. В работе [126] было пока-
зано, чти в грубидисперсной смеси каучуков развивающаяся тре-
щина разрушает частицы, не огибая их. При увеличении дисперс-
ности частиц трещина иногда проходит через частицы, а иногда
огибает их по поверхности раздела. Очевидно, что чем меньше
скорость роста трещины, тем более вероятно ее распространение
по наиболее ослабленным участкам системы, которыми и являют-
ся межфазные слои в смеси полимеров. При наличии двух непре-
рывных фаз в смеси кау чу ков при средних их соотношениях тре-
Рис. V.41. Внешний пт поверхности разрушения вулканизатов смеси СКД_____________
СКН 18 после многократного изгиба:
Г — СКД: 2-25% СКН 18; 3-50% СКН-18; 4 75% СКН-18; 5 - СКН-1.8.
244
Минимальнпя деформация, %
Рис. V.42. Зависимость сопротивления
утомлению наполненных вулканизатов
смесей НК — БСК от режима испытания
(для испытания изготовлены образцы с 7
одинаковыми модулями):
1 — БСК; 2 — 25% НК; 3 — 50% НК; -4 — 75% g
НК; 5 —НК. 5
__________________________________ а
а-
щина может распространяться Р
как по межфазным слоям, так и а
неизбежно разрушать частицы
дисперсной фазы.
Явления взаимоусиления ха-
рактерно не только для смесей
каучуков, но и для смесей кри-
сталлических или стеклообраз-
ных полимеров. Так, устойчивость к многократному изгибу воло-
кон из смеси ПЭ—ПП (1:1) в 3—5 раз выше, чем у волокон ПЭ
или ПП. Эффект сохраняется и в наполненных смесях каучуков
(см. обзор [125]). В одном цикле растяжения эффект вьаимоуси-
ления наблюдается реже, и прочность смеси полимеров не всегда
меняется по кривой с максимумом в зависимости от состава сме-
си. Несколько примеров роста прочности и появления резкого мак-
симума на кривой прочность — состав приводится в работе [127]
для пленок из смесей ПММА и хлорированного ПВХ, ПММА и
сополимера винилхлорида с винилацетатом, ПММА и ПВА.
Разное проявление закономерностей взаимоусиления в динами-
ческих и статических испытаниях указывает на зависимость эф-
фекта от режима испытания. Характерен в этом отношении рис.
V. 42 [128], где приведены данные об испытаниях на машине, поз-
воляющей осуществлять растяжение и сжатие в динамическом ре-
жиме. Переход от сжатия к растяжению во всех режимах мог
проводиться в одном цикле с общей величиной в» 75 %. На оси
абсцисс отложено значение минимальной деформации, так, что
—75% означает режим многократного сжатия с амплитудой дефор-
мации от 75 до 0%,—37,5% означает знакопеременное сжатие
(—37,5%) —растяжение (+37,5%), 0% означает растяжение от О
до 75%, а +50%—растяжение от 50 до 125%. Из рис. V. 42 вид-
но, что максимум сопротивления утомлению определяется нс толь-
ко соотношением ка\чуков, но и режимом утомления. Эффект
взаимоусиления наиболее отчетливо проявляется в наиболее жест-
ком режиме испытания (от 50 до 125%), что было отмечено и ра
псе [129]. Такой эффект характерен для смесей НК и с другими
каучуками [128]. Динамическую выносливость данного каучука в
заданном режиме утомления можно улучшить подбором другого
каучука для получения смеси.
Необходимо остановиться отдельно на применении основных
концепций кинетической теории прочности к композиционным ма-
териалам, к числу которых относятся смеси полимеров. Исследр-
245
вання в этой области немногочисленны. Они обобщены в обзоре
[130], из которого следует, что применимость основного уравнения
кинетической теории прочности к композиционным материалам
ограничена непостоянством констант уравнения. Согласно кинети-
ческой теории прочности, время до разрушения данного материа-
ла, находящегося под нагрузкой, определяющей возникновение в
материале напряжения тр, описывается уравнением
U — у а
Тр = т0 ехр
(57)
где То — имеющий размерность времени коэффициент, определяющий координату
полюса точки пересечения семейства кривых 1g тг—о при разных температурах;
— константа, имеющая смысл энергии активации процесса разрушения при
напряжении, стремящемся к нулю; у — структурночувствительный коэффициент;
Т — температура опыта.
В принципе уравнение (57) применимо к композиционным ма-
териалам, причем в широком интервале изменения температуры и
напряжения константы Uo, у, То остаются для данного материала
постоянными. При этом получается нормальный «'веер» прямых
для разных напряжений или температур, что было продемонстри-
ровано на примере ориентированных армированных композитов
[130]. Экспериментальные данные позволяют сделать следующие
(хотя и неокончательные) выводы о закономерностях и величинах
коэффициентов уравнения (57).
Коэффициент тр (в секундах) и по .величине, и по физическому
смыслу не отличается от такового для гомогенных материалов.
Он структурнонечувствителен и не меняется при изменении при-
роды и свойств компонентов композиционного материала.
Коэффициент Uo (в Дж/моль) для волокнонаполпенных ориен-
тированных композитов типа эпоксидной смолы, наполненной стек-
ловолокном, также оказывается структурнонечувствитсльным и по
величине близок или совпадает с Uo той фазы, которая находится
в композите в избытке. Переход от Uo для эпоксидной смолы к
С'о стекловолокон при увеличении объемной доли волокна в ком-
позите происходит довольно резко и в узком интервале составов.
Это аналогично тому, как энергия активации вязкого течения сме-
сей полимеров существенно определяется полимером, находящим-
ся в виде непрерывной фазы (см. § 3 этой главы). Разумеется,
указанный вывод может быть сделан с точностью до предположе-
ния о неизменности свойств материала фазы в смеси и в исходном
(до смешения) состоянии и поэтому не может рассматриваться
как универсальный.
Коэффициент -у, имеющий размерность объема, часто приводит-
ся, однако, в кДж-м2/( моль-Н) или в ккал-мм2/(моль-кг). Он за-
висит не только от структурных особенностей дисперсной фазы и
матрицы, но и от характера межфазных слоев, от анизомет-рично-
сти и ориентации частиц дисперсной фазы и от других параметров
структуры композита [130]. Согласно кинетической теории проч-
246
ности коэффициент у является произведением усредненного коэф-
фициента перенапряжений на активационный объем. В компози-
те, где помимо перенапряжений в местах случайных дефектов
структуры возникают перенапряжения вследствие фазовой неод-
нородности структуры материала, применение понятия усреднен-
ного коэффициента перенапряжений может быть некорректным.
Разумеется все, что известно об изменении коэффициента у, отно-
сится к испытаниям малодеформируемых материалов при напряже-
ниях ниже предела текучести. Структура композиционного материа-
ла, а особенно смеси полимеров, может в процессе деформации
меняться столь значительно, что применение уравнения (57) вооб-
ще окажется невозможным.
Так, в работе [131] показано, что для смесей ПВХ с ПММА
в зависимости от их состава и фазовой структуры возможно как
смещение полюса, так и аномальное расположение веера прямых
долговременной прочности. В частности, для смеси ПВХ : ПММА=
= 1:1 кривые зависимости 1g тР—1/Т не сходятся к полюсу, а рас-
ходятся по направлению к нему, тогда как та же смесь при соот-
ношении компонентов 80:20 характеризуется «нормальным» рас-
положением веера прямых. В работе указывается, что при созда-
нии пространственной сетки химических связей в фазе ПММА
отмеченные аномалии устраняются.
Исследование долговременной прочности композиционных ма-
териалов, а также изучение аномальных случаев их поведения
под действием нагрузки важно не только вследствие значения
композитов кйк конструкционных материалов. «Задача исследова-
ний таких случаев для композитов особенно интересна потому,
что причины наблюдаемых усложнений для композитов часто
выяснить легче, чем для гомогенных тел» [130].
При определении прочности смесей полимеров необходимо учи-
тывать значительную анизотропию прочности, возникающую
вследствие ориентации частиц дисперсной фазы при формовании
образца [89]. В гл. IV (см. рис. IV. 11) уже приводились данные
по анизотропии смесей ПЭ и бутилкаучука. 14з рисунка видно,
что прочность смеси в направлении ориентации (направление съе-
ма с вальнев) выше, чем .в перпендикулярном направлении, и пре-
вышение это для смесей больше, чем для смешиваемых исходных
ПЭ и БК. Наличие анизотропии прочности было также показано
на примере других смесей каучуков с пластмассами, каучуков с
каучуками (вулканизаты) и смесей двух пластмасс [125].
Заканчивая рассмотрение прочностных свойств смесей полиме-
ров как представителей класса композиционных материалов, необ-
ходимо еще раз подчеркнуть, что преимущества, присущие двух-
фазным смесям полимеров, возникают именно благодаря опреде-
ленной степени микронеоднородности системы, а не вопреки ей. При
коллоиднохимическом подходе к интерпретации закономерностей
прочности реальных полимерных тел вообще, можно отметить об-
щую закономерность связи структуры со свойствами, характер-
247
ную и для микронеоднородных (однофазных) полимерных мате-
риалов с их надмолекулярной структурой, и, особенно, для двух-
фазных смесей полимеров. Такой особенностью является сочета-
ние в структуре реального тела областей с более прочными и об-
ластей с менее прочными химическими и межмолекулярными свя-
зями. Процесс деформации или даже (в пределе) разрушение
системы происходит поэтому неравномерно, не по классическому"
механизму разрастания единой, однонаправленной 'магистральной
трещины или однородной деформации всего тела по принципам
механики сплошной среды. Именно эта неоднородность свойств
тела, доведенная или нс доведенная до уровня двухфазности при
формовании тела, приводит к улучшению свойств, в том числе и
прочностных. Пути проявления этой закономерности весьма раз-
личны. Вот некоторые примеры.
В работах Б. А. Догадкина с сотр. (см. ссылки в [129]) было
установлено повышение усталостной выносливости и прочности
при растяжении вулканизатов в тех случаях, когда в процессе
вулканизации в системе соз щвалась комбинация «прочных и сла-
бых лабильных вулканизационных связей». Имея в виду, что «свя-
зи» фактически являются частицами коллоидных размеров, можно
полагать, что комбинацию прочных и слабых связей правильнее
рассматривать как совокупность чередующихся в пространстве
более прочных и более слабых «микрообъемов» вулканизата. При
наложении нагрузки в областях с меньшим 'взаимодействием про-
исходит ускоренная релаксация напряжений, что делает поле на-
пряжений более однородным и, следовательно, всю систему —
лучше сопротивляющейся нагрузкам.
В смеси любых полимеров на границе раздела фаз образуется
переходный слой. Этот слой формируется как следствие сегмен-
тальной растворимости на границе раздела фаз. По-существу слой
сегментальной растворимости является микрообъемом, в котором
межмолекулярное взаимодействие ослаблено. Ослабление межмо-
лекулярного взаимодействия между полимерами в слое сегмен-
тальной совместимости по сравнению со среднеаддитивиым значе
нием следует из малого термодинамического сродства полимеров,
приводящего к их несовместимости. Ослабление это показано так-
же и экспериментально [125], как понижение энергии когезии
смеси каучуков по сравнению с аддитивными значениями. Ослаб-
ленное взаимодействие полимеров может наблюдаться не только
в слое сегментальной растворимости, но и в других областях пе-
реходного слоя, где структура каждого полимера изменена по
сравнению с его структурой в объеме, что приводит к пониженной
плотности вещества в переходном слое [13, 56]. Тзким образом,
чередование «более прочных и менее прочных» об частей в смеси
полимеров наблюдается даже в тех случаях, когда прочность, мо-
ду 1и, вязкость или другой показатель свойств одной фазы близок
к такому же показателю другой фазы.
В работах П. И. Зубова с сотр. (см. ссылки в [129]) было по-
24Я
казано, что при формировании покрытий из полимера, когда он
растворен в «плохом» растворителе, процесс пленкообразования
проходит через стадию гелеобразования. Это способствует возник-
новению структуры, где уплотненные (более прочные) микрообъ-
емы, являющиеся узлами пространственной структуры геля, чере-
дуются с ослабленными микрообластями. Такая микрогетероген-
ная (но безусловно однофазная) структура сохраняется в пленке
и после удаления растворителя. Чередование прочных и ослаблен-
ных областей приводит как полагают авторы, к ускорению релак-
сации напряжений, по-видимому, вследствие смещения более проч-
ных микриблоков за счет ослабленного взаимодействия между
ними. Это и ведет к резкому снижению внутренних напряжений
по сравнению с имеющей более однородную структуру пленкой,
сформированную из «хорошего» растворителя.
Приведенные здесь примеры показывают, что чередование мик-
рообластей с более прочным и более ослабленным взаимодейст-
вием характерно для широкого круга систем, но что это качество
является наиболее общим и наиболее принципиальным именно для
смесей полимеров. Таким образом, специфика коллоиднохшмичес-
кого подхода к изучению структуры и свойств смесей полимеров
определяется тем, что эти смеси являются двухфазными система-
ми с чередующимися микрообъемами с повышенным и ослаблен-
ным химическим или межмолекулярным взаимодействием. Под
микрообъемами можно, видимо, понимать как частицы дисперс-
ной фазы, так и граничные слои, имеющие характер «полуколло-
идных» образований, т. е. не обладающие характерными призна-
ками отдельной фазы.
Таким образом, прочность является свойством, менее других
механических свойств подчиняющимся «правилу аддитивности»
или «правилу смесей». Строго говоря, аддитивными являются не-
которые (не все) свойства идеальных смесей, идеальных раство-
ров. Типичной кривой свойство — состав в двухфазной смеси, как
это показано на примере вязкости в этой главе, является S-образ-
ная кривая, отражающая обращение фаз. Частицы второй фазы
являются концентраторами напряжений, и поэтому формально
прочность двухфазных систем должна быть ниже, чем однофаз-
ных однородных смесей. В действительности ряд факторов и в
числе их, в первую очередь, изменение структуры полимера ,в сме-
си (в граничных слоях или во всем объеме частицы фазы) приво-
дят к возникновению неоднородности системы. Неоднородность
выражается в чередовании более прочных и менее прочных (соот-
ветственно более или менее жестких, плотных и т. д.) микрообъ-
емов. Такая неоднородность приводит к облегчению релаксации
напряжений в процессе деформации смеси или при ее формирова-
нии.
Простая двухфазная модель смеси полимеров, пригодная в
первом приближении для расчета модулей или податливости сме-
си, не годится для расчета прочности, поскольку в основе модели
249
лежит предположение о неизменности структуры и свойств фаз
при переходе от отдельно взятых полимеров к их смеси. Уже ка-
чественные представления о механизме повышения удельной удар-
ной вязкости пластмасс при введении каучуков предполагают на-
личие определенной, достаточно' большой адгезии на границе раз-
дела фаз, в то время как при расчете модуля в двухфазной моде-
ли величина адгезии не учитывается.
ЛИТЕРАТУРА
1. Mullins L. R.— Rubb. Chem. Technol., 1947, v. 20, p. 998—1021.
2. Buchdahl R., Nielsen L. E.—J. Appl. Phys., 1950, v. 21, p. 482—499; J. Po-
lymer Sci., 1955, v. 15, № 1;Нильсен Л. Механические свойства полимеров
и полимерных композиций. М., Химия, 1978. 310 с.
3. Brener й"., Hild W., Wolf И. — Plaste u. Kautschuk, 1954, Bd. 1, S. 170;
Wolf H. — Plaste u. Kautschuk, 1957, Bd. 4, S. 244.
4. Белякова Л. К., Гузеев В. В., Малинский Ю. М. — Пласт, массы, 1973, № 12,
с. 39—41.
5. Fox Т. G. — Bull. Am. Phys. Soc., 1956, v. 2, p. 123.
6. Gurdon M., Taylor J. S. — J. Appl. Chem., 1952, v. 2, p. 493.
7. Koleske J. V., Lundberg R. D.—J. Polymer Sci., 1969, pt. A-2, v. 7, p. 795—
807.
8. Bank M., Leffingwell I., Tides C. — Macromolecules, 1971, v. 4, № 1, p. 43—
46.
9. Akiyama S. — Bull. Chem. Soc. Japan, 1972, v. 45, p. 1381—1384.
10. Бартенев Г. M., К ангаров Г. С. — Высокомол. соед., 1960, т. 2, № 11,
с. 1692—1697.
11. Кулезнев В. Н. Совместимость полимеров. — Энциклопедия полимеров. М.,
Советская энциклопедия, 1977. Т. 3, с. 433—439.
12. Килезнев В. FL, Клыкова В. Д., ДогаОкин Б. А.— Коллоид, ж., 1968, т. 30,
, № 5, с. 707—712.
13. Кулезнев В. И., Игошева К- М. —Высокомол. соед., 1962, т. 4, с. 1858—1862.
14. Кулезнев В. И., Крохина Л. С. — Высокомол. соед., 1973, сер. А, т. 15,
с. 906—916.
15. Разинская И. И., Штаркман Б. П. — Высокомол. соед., 1977, сер. Б, т. 19,
№ 10, с. 765—767.
16. Manabe S., Murakami R.. Takayanagi М., Uemura S. — Intern. J. Polymer.
Mater.. 1971, v. 1, № 1, p. 47—73.
17. Липатов Ю. С., Виленский В. A.— Высокомол. соед., 1975, сер. А, т. 17,
№ 9, с. 2069—2075.
18. Оськин В. И., Яновский Ю. Г. Малкин А. Я.. Кулезнев В. EL, Альзит-
цер В. С, Татарский И. А. — Высокомол. соед., 1972, сер. А, т. 14, № 10.
с. 2120—2123.
19. Becher J Е., Marker L., Bradford R. D., Aggarwal S. L.—J. Polymer Sci.,
1969, pt. C. № 26. p. 117—131.
20. Miyamoto T.. Kodama K-, Shibayama K- — J. Polymer Sci., 1970, pt. A-2, v. 8,
p. 2095 -2108.
21. Айвазов А. Б., Миндиаров X. Г., Зеленев Ю. В., Оганесов Ю. Г., Раев-
ский В. Г. — Высокомол. соед., 1970, сер. Б, т. 12, № 1, с. 10—14.
22. Thies И. R. — Ind. Eng. Chem., 1941, v. 33, p. 389—393.
23. Beatty J. R._ Davies J. M.—J. Appl. Phys., 1949, v. 20, p. 533—545.
24. Kell P. M. — Rubb. World., 1958, v. 138, № 4, p. 586—595.
25. Мельникова О. Л., Кулезнев В. И., Аулов В. А., Клыкова В. Д. — Высоко-
мол. соед., 1976, сер. Б, т. 19, № 12, с. 903—906.
26. Бухина М Ф. Кристаллизация каучука и резины. М„ Химия, 1973. 239 с.
27. Виноградов Г. В., Яновский Ю. Г., Кулезнев В. FL, Иваненко Т. А. — Кол-
лоид. ж„ 1966, т. 28, № 5, с. 640—644.
250
28. Linnig F. J., Parks E. J., Stiehler R. D. — Rubb. Chem. Technol., 1966, v. 39,
p. 1041—1058; Payul Д. R.— Rubb. Chem. Technol., 1968, v. 41, p. 1203—
1225.
29. Слонимский Г. Л., Мусаэлян И. Н., Казанцева В. И. — Высокомол. соед.,
1962, т. 4, с. 219—225, 815- 819, 823-830.
30. Рейтлингер С. Д. Проницаемость полимерных материалов. М., Химия, 1974.
269 с.
31. Евреинов Ю. В. Канд. дис. М., МИ1ХТ им М. В. Ломоносова, 1973.
32. Тросгянская Е. Б., Бельник Д. Р., Чалых А. Е., Черникова О. Д. — Высоко-
мол. соед., 1974, сер. Б т. 16, № 7, с. 48р—488.
33. Никишин Е. Л., Чалых А. Е., Авгонов А. Е., Кулизнев В. И., Морозо-
ва А. В., Неверов А. Н. — Коллоид, ж., 1978, т. 40, № 4, с. 789—793.
34. Ranby В. С. — J. Polymer Sci., Polymer Symp. Ed., 1975, № 51, p. 89—104.
35. Чалых A. E., Улин В И., Золотарев П. П., Авгонов А. Е., Алиев А. Д. —
Изв. АН СССР. Сер. хим., 1976, 4, с. 735—738.
36. Uemura У., Takeyanagy А4.— J. Appl. Polymer Sci., 1975, v. 11, № 4, p. 1029—
1035.
37. Кулезнев В. H„ Догадкин Б. А. — Коллоид, ж., 1962, т. 24, № 5, с. 632—633.
38. Оганесов Ю. Г., Кулезнев В. Н., Воюцкий С. С. — Высокомол. соед., 1970,
сер. Б, т. 12, Л" 9, с. 691—693.
39. Топуридзе Н. С. Канд. дис. М., НИФХИ им. Л. Я. Карпова, 1973.
40. Inoue Т . Ishihara Н., Kawai Н., Ио У., Kato К- — In: Proceeding of Interna-
tional Conference on Mechanic Behavior of Materials, Kyoto, 1971. V. 3.
41. Марков H. Г., Власов С. В., Сагалаев Г. В., Кулезнев В. Н., Марков А. В.—
Коллоид, ж., 1976, т. 38, № 4, с. 698—703.
42. Марков Н. Г., Власов С. В., Сагалаев Г. В., Кулезнев В. Н., Марков А. В.—
Коллоид, ж., 1977, т. 39, № 3, с. 467—471.
43. Ziegel К. D., Frensdorf Н. К., Fogiel A. W.— J. Appl. Polymer Sci., 1969,
v. 13, р. 867—869.
44. Чернин Е. И. Автореф. канд. дис. М.. МИТХТ им. М. В. Ломоносова, 1977.
45. Френкель Я. И. Кинетическая теория жидкостей. Л., Наука, 1975. 592 с.
46. Horio М., Fuji/ Т., Onogy S.—J. Phys. Chem., 1964, v. 68, p. 778—791.
47. Бартенев Г. Al, Вишницкая Л. А. — Высокомол. соед., 1964, т. 6, № 4,
с. 751—757.
48. Кулезнев В. Н., Конюх И. В., Виноградов Г. В., Дмитриева И. П. — Колло-
ид. ж., 1965, т. 21, № 4, с. 540—545.
49. Кулезнев В. Н., Андреева В. М. — Высокомол. соед., 1962, т. 4, № 12,
с. 1851—1857.
50. Малкин А. Я., Сабсай О. Ю., Жанге рее ва Г. Ж-, Забушина Л1. 77., Вино-
градов Г. В. — Мех. полимер., 1977, № 3, с. 519—523.
51. Гуль В. Е., Пенская Е. А., Кулезнев В. Н. — Коллоид, ж., 1965, т. 27, № 3,
с 341______345
52. Ninomiya К., Ferry J. — J. Macromol. Sci., 1969, pt. В, v. 3, № 2, p. 237—
25c.
53. Kuleznev V. N., Melnikova O. L., Klykova V. D. — Europ. Polymer J., 1978,
v. 14, p. 455—464.
54. Daring G.. Leugerung H. J.-—Kunststoffe, 1963, Bd. 53, S. 11—28.
55. Нильсен Л. Механические свойства полимеров и полимерных кондиций. Пер.
с англ. М.. Химия, 1978. 309 с.
56. Липатов Ю. С. Физическая химия наполненных полимеров. М., Химия, 1977.
304 с.
57. Taylor G. I.— Proc. Roy. Soc. (London), 1932, Sec. A, v 138, p. 41—48.
58. Guth E„ Simha R. — Koll.-Z., 1936, Bd. 74, S. 276—285.
59. Sweeny К- H., Geckler R. D. — J. Appl. Phys., 1954, v. 25, p. 1135—1141.
60. Кандырин Л. Б., Кулезнев В. H., Чернин Е. И., Фрейдин А. С., Грин-
берг С. М.— Коллоид, ж., 1977, т. 39, № 5, с. 966—969.
61. Kerner Е. Н. — Proc. Phys. Soc., 1956, Ser. В, v. 69, p. 802—807, 808—813.
62. Uemura S., Takayanagi M.—J. Appl. Polymer Sci., 1966, v. 10, p. 113—
115.
251
63. Takayanagi M.— In: Proceedings of the 4th International Congress of Reo-
logy. New York, Interscience, 1965. Pt 1, 161 p.
€4. Fujino K-, Ogawa Y., Kawai H.— J. Appl. Polymer Sci., 1964, v. 8, p. 2147—
2161.
65. Dickie B. A. — J. Appl. Polymer Sci.. 1973, v. 17, p. 45—63.
66. Краус Г., Роллман К- В. — В кн.: Многокомпонентные пвлимерные системы.
Пер. с ан1л./Под ред А. Я. Малкина и В. Н. Кулезнева. М., Химия, 1974,
328 с.
67. Pakula Т., Kn/szewski М., Grebowicz 1., Galrski А. — Polymer J., 1974, v. 6,
№ 2, p. 94—102.
68. Кулезнев В. И., Клыкова В. Д., Чсрнин Е. И., Евреинов Ю. В — Коллоид
ж., 1975, т. 37, № 2. с. 267- 272.
69. Кулезнев В. И., Кандырин Л. Б., Клыкова В. Д.— Коллоид, ж., 1972, т. 34,
с. 231—234.
70. Басин В. Е. — Лакокрас. материалы и их прямей., 1976, № 1, с. 41—44; Мех.
полимер., 1971, № 4, с. 731—733.
71. Малощук Ю. С., Кулезнев В. Н„ Ханин С. Е. — Коллоид, ж., 1973, т. 35,
№ 2, с. 408—410.
72. Ханин С. Е„ Ангерт Л. Г., Кулезнев В. Н., Шашков А. С. — Каучук и рези-
на. 1974, № 1, с. 30—32.
73. Левит Е. 3., Кулезнев В. Н., Евреинов Д. Б., Богуславский Д. Б. — Каучук
и резина, 1978, № 9, с. 28—31.
74. Aggurwal S. L.-—Polymer, 1976, v. 17, р 938—956.
75. Allen G., Bowden M I., Tadd S. At, Blundell D. ]., Jeffs G. M., Da-
vies W. E. — Polymer, 1974, v. 15, p. 28—32.
76. Dickie R. A., Mo-Fung Cheung. — J. Appl. Polymer Sci., 1973, v. 17, p. 79—
94.
77. Zitek P., Zelinger J. — J. Polymer Sci., 1968, pt. A-l, v. 6, № 3, p. 467—
474.
78. Ziegel K-, Romanov A.— J. Appl. Polymer Sci., 1973, v. 17, p. 1133—1139.
79. Okamoto T., Takayanagi M.—J. Polymer Sci., 1968, pt. C, № 23, p. 597—
606.
80. Prest W. M., Porter R S. — J. Polymer Sci., 1972, pt. A-2, v. 10, p. 1639—
1655.
81. Крохина Л Gt, Кулезнев В. H., Люсова Л. Р., Глаголев В А. — Высокомол.
соед., 1976, сер. А, т. 18, № 3, с. 663 -668.
82 Кандырин Л. Б., Кулезнев В. Н. — Коллоид, ж., 1974, т. 36, с. 473—477.
83. Допперт Г., Овердип В. — В кн.: Многокомпонентные полимерные системы.
Пер. с англ./Под ред. А. Я. Малкина и В. Н. Кулезнева. М., Химия, 1974,
с. 61—71.
84 Догадкин Б. А., Килезнев В. Н., Пряхина С. Ф. — Коллоид, ж., 1959, т. 21,
№ 2, с. 174—180.
85. Кулезнев В. И., Шварц А. Г., Клыкова В. Д., Догадкин Б. А. — Коллоид,
ж., 1965, т. 27. № 2, с. 211—215.
86. Han С. D., Kim Y. W., Chen S. J.—J. Appl. Polymer Sci., 1975, v. 19, № 10,
p. 2831 -2843.
87. Kim Y. W„ Han C. D. — J. Appl. Polymer Sci., 1976, v. 20, № 10, p. 2905—
2912.
88. Цебренко M. В., Юдин А. В., Виноградов Г. В. — Высокомол. соед., 1973,
сер. А, т. 15, с. 1790—1797.
89. Кулезнев В. IE, Евреинов Ю. В., Клыкова В. Д., Шпошникова М. И. — Кол-
лоид. ж., 1973, т. 35, № 2, с. 281—285.
90. Folt V. L., Smith R. W.— Rubb. Chem. Technol., 1973, v. 46, № 5, p. 1113—
1209.
91. Кандырин Л. Б., Анфимов Б. H., Кулезнев В. И., Альзитцер В. С. — Каучук
и резина, 1976, № 11, с. 20—22.
92. Мельникова О. Л. Канд. дис. М., МИТХТ им. М. В. Ломоносова, 1975.
93. Plochocki А. — J. Appl. Polymer Sci., 1972, v. 16, p. 987—1003.
94. Древаль В. E., Тагер A. A , Сычева Э. И., Взведская 3. П.—Мех. полимер.,
1970, Ns 5, с. 920—923.
252
95. Han C. D., Yu T. C. — Polymer Eng. a. Sci., 1972, v. 12, № 2, p. 81—90.
96. Кулезнев В. H. Докт. дис. ЛЕ, МИТХТ им. М. В. Ломоносова, 1973.
97. Кулезнев В. Н., Элькина И. А., Банькова Л. И., Догадкин Б. А. — Коллоид,
ж., 1970, т. 32, № 3, с. 381—387.
98. Кулезнев В. Н„ Кандырин Л. Б., Альтзицер В. С., Анфимов Б. И. — Каучук
и резина, 1976, № 9, с. 19--22.
99. Пап С. D., Lamonte R. R. — Polymer Eng. a. Sci., 1972, v. 12, № 2, p. 77—
80.
100. Марков И. Г., Власов С. В., Сагалаев Г. В., Кулезнев В. И., Марков А. В.—
Коллоид. ж., 1976, т. 38, Кв 4, с. 698—703.
101. Murakami К-, Оно К. — J. Polvmer Sci., 1972, pt. В, v. 10, № 8, р. 593—
599.
102. Horino Т. — J. Appl. Polymer Sci., 1965, v. 9, p. 2261—2272.
103. Kaplan D., Tschoegl N. E. — Polymer Eng. a. Sci., 1974, v. 14, № 1, p. 43—
49.
104. Choi C., Kaya A., Shen M Polymer Eng. a. Sci., 1973, v. 13, № 3, p. 231—
235.
105. Rusch К. C. — J. Macromol. Sci., 1968, pt. B, v. 2, p. 179—187.
106. Усиление эластомеров. Пер. с англ. М., Химия, 1968. 483 с.
107. Гуль В. Е. Структура и прочность полимеров. М., Химия, 1978. 327 с.
108. Шварц А. Г., Динзбург Б. Н. Совмещение каучуков с пластиками и синте-
тическими смолами. М., Химия, 1972. 224 с.
109. Полистирол. Физико-химические основы получения и переработки/Мал-
кин А. Я.. Вольфсон С. А., Кулезнев В. II., Файдель И. И. М., Химия,
1975. 288 с.
ПО. Кормак В. В., Еремина Е. Н.. Вылегжанина К. Л., Матусевич Е. С., Усма-
нова И. Ф., Кудрявцева Г. В. — Пласт, массы, 1972, Кв 7, с. 56—57.
111. Эренбург Е. Г., Еремина М. А.. Пискарева Е. П.. Толстопятов Г. М., Под-
дубный И. А. — Высокомол. соед., 1972, сер. А, т. 14, Кв 12, с. 2627—2632.
112. Мортон М. — В кн.: Многокомпонентные полимерные системы. Пер. с англ./
Под ред. А. Я. Малкина и В. Н. Кулезнева. М., Химия, 1974, с. 97—114.
113. Strella S. — Г} кн.: Конструкционные свойства пластмасс. Пер. с англ./Под
ред. Г. В. Виноградова. М., Химия, 1967, 463 с.
114. Merz Е. И., Claver G. С., Baer М. — J. Polymer Sci., 1956, v. 22, р. 325.
115. Metsuo М. — Polymer Eng. a. Sci., 1969, v. 9, № 3, p. 197—212.
116. Bucknall С. B., Smith R P. — Polymer, 1965, v. 6, p. 437—450.
117. Matsuo M., Wang T. T., Kwei T. K. — ACS Polymer Prepr., 1971, v 12,
Кв 1, p. 676—683.
118. Boyer R. F. — J. Polymer Sci., 1968, pt. A, v. 6, p. 161—180.
119. Keskkula H. — Appl. Polymer Symp., 1970, Ks 15, p. 51—78.
120. Schmitt J. A. —J. Polymer Sci., 1970, pt. C, Ks 30, p. 437—445.
121. Newman S., Strella S. — J. Appl. Polymer Sci., 1965, v. 9, p. 2297—2309.
122. Брагау С. Д. — В кн.: Многокомпонентные полимерные системы. Пер. с
англ./Под ред. А. Я. Малкина и В. 11. Кулезнева. М„ Химия, 1974, с. 123.
123. Bascom W. D., Cottington R. L., Jones R L., Peyser P. — J. Appl. Polymer
Sci., 1975, v. 19, Ks 9, p. 2545—2562.
124. Кулезнев В. H. Канд. дис. М., МИТХТ им. М. В. Ломоносова, 1959; Догад-
кин Б. А., Кулезнев В. И., Тарасова 3. Н. — Коллоид, ж., 1958, т. 20, Кв 1,
с. 43—51.
125. Кулезнев В. И. — В кн.: Многокомпонентные полимерные системы. Пер. с
англ./Под ред. А. Я. Малкина и В. Н. Кулезнева. М„ Химия, 1974, с. 10—60.
126. Wallers М. Н.. Keyte D. N.—Trans. Proc. Tnst. Rubb. Ind., 1962, v. 38, Kb 9,
p. 40—54.
127. Friese K. — Plaste u. Kautschuk, 1968, Bd. 15, № 9, S. 646—650.
128. Beatty J. R. — In: International Rubber Conference, ACS, Rubber Division,
San Francisco, Oct. 1976. Preprints Ks 37.
129. Кулезнев В. H., Шершнев В. А.— Каучук и резина, 1977, Кв 11, с. 56—63.
130. Рсгель В. Р. — Высокомол. соед., 1977, сер. А, т. 19, Кв 9, с. 1915—1923.
131. Пичугина С. В., Разинская И. Н-, Слуцкер А. И. — Мех. полимер., 1977, Кв 3,
с. 558.
253
Глава VI
МНОГОКОМПОНЕНТНЫЕ СИСТЕМЫ
НА ОСНОВЕ СМЕСЕЙ ПОЛИМЕРОВ
В большинстве случаев практического применения смеси по-
лимеров содержат дополнительные ингредиенты. В смесях каучу-
ков наличие ингредиентов абсолютно необходимо для проведения
вулканизации. Вообще же в полимеры и их смеси вводят, как
правило, стабилизатор (противостарнтель), а в ряде случаев—-
также пластификаторы и наполнители.
Подобрать стабилизатор, наполнитель, пластификатор, вул-
канизующий агент или третий 'полимер, которые имели бы одина-
ковое сродство к каждому из двух полимеров, входящих в состав
смеси, невозможно. Это усугубляется еще и тем, что смеси по-
лимеров составляют, как правило, из полимеров, сильно разли-
чающихся по химической природе, поскольку именно в этом слу-
чае можно ожидать получения материала с необычным и желатель-
ным комплексом свойств. Чем больше различаются полимерные
компоненты смеси по химической природе, тем больше различие
их сродства к третьему полимеру или к неполимерным ингредиен-
там композиции. Поэтому вводимые в смесь ингредиенты не рас-
пределяются равномерно в каждой фазе, а концентрируются преи-
мущественно либо в дисперсной фазе, либо в матрице в зависи-
мости от их сродства к данному полимерному веществу. Свойства
каждой фазы при этом изменяются и могут сильно отличаться как
от свойств исходных полимеров, так и от свойств полимеров, содер-
жащих среднее количество ингредиента, которое имелось бы в
каждой фазе при равномерном распределении ингредиента в сме-
си. Это приводит к неконтролируемому изменению свойств самой
смеси, и требуется очень большое число опытов для эмпирическо-
го нахождения оптимальной дозировки ингредиента.
Можно ввести ингредиенты в заданном количестве в один
из полимеров или в оба, а затем перемешать полимеры. Диффузия
ингредиентов из фазы в фазу в процессе формования изделия или
его эксплуатации приведет снова к их неравномерному распреде-
лению. Если полимер, подверженный старению в процессе пере-
работки или эксплуатации, заправить противостарителем и сме-
шать с полимером, устойчивым к процессу старения и не содер-
жащим или содержащим мало противостарителя, то со време-
нем противостарнтель может мигрировать из фазы первого поли-
мера в фазу второго. В результате полимер, чувствительный к
254
старению, может быстро разрушиться, и смесь в целом быстро
потеряет эксплуатационные свойства.
Большая протяженность межфазных слоев в смесях полимеров
и пониженная энергия когезии в этих слоях может привести к
концентрированию в них ингредиентов, что также может карди-
нально изменить свойства смеси в целом.
В настоящее время имеются лишь отрывочные данные о рас-
творимости ингредиентов в полимерах или о их сродстве к поли-
мерам. Неизвестно, например, сколько пластификатора из ряда
фталатов содержится в фазе ПВХ и сколько в фазе бутадиен-ннт-
рнльного сополимера СКН-18, если в смесь этих полимеров вве-
дено известное количество указанного пластификатора. Это за-
трудняет рациональное построение рецептур композиций даже на
основе смесей хорошо изученных и широко применяемых поли-
меров.
В этой главе будут рассмотрены закономерности структуры
и свойств двухкомпонентных смесей полимеров, содержащих тре-
тий полимер, смесей, содержащих наполнитель, пластификатор
или вулканизующий агент. Работ, посвященных исследованию
стабилизации смесей полимеров в связи с возможным перераспре-
делением стабилизатора между фазами, не имеется, несмотря на
принципиальное значение этого вопроса.
§ 1. ТРЕХКОМПОНЕПТНЫЕ СМЕСИ ПО ДИМЕРОВ
Малые количества третьего полимера, .введенные в смесь пер-
вых двух, могут перейти в состояние истинного раствора в сме-
си, поскольку взаимная растворимость полимеров .в трехкомпо-
нентной системе выше, чем в бинарной (см. гл. II). Если количест-
во третьего полимера выше предела его растворимости «в смеси,
но продолжает оставаться малым, этот полимер может .распреде-
литься главным образом либо в частицах одной из фаз (по-'види-
мому, которая обладает наименьшей упругостью), либо на
границах раздела фаз. Если же три полимера смешаны в соизме-
римых количествах, каждый полимер образует свою фазу, протя
женность которой соответствует его объемной доле. Для понима-
ния того, как формируется комплекс свойств смеси полимеров,
принципиально важно, что, переходя от индивидуального полиме-
ра к бинарной смеси, мы кардинально меняем структуру от одно-
фазной до двухфазной, при переходе же от бинарной смеси к
тройной (и вообще к многокомпонентным смесям) меняется лишь
число tba_>. а система по-прежнему остается гетерофазной., Работ
по изучению структуры тройных смесей каучуков опубликовано
мало. В работе [1] на примере тройных смесей каучуков хлорбу-
тилкаучук (ХБК) — этиленпропилендиеновый сополимер
(СКЭПТ)—натуральный каучук (НК) и ХБК—СКЭПТ—поли-
бутадиен (ПБ) в разных соотношениях показано, что при введе-
нии третьего каучука гомогенность смесей не улучшается, а смесь,
255
Рис. VI.1. Свойства невулканизованиых трехкомпонентных смесей СКД —
СКМС-ЗОАРК — СКН-18:
с—предел текучести оу-10, МПа; б — эффективная вязкость lgТ] 10 [Па с] при напряже-
нии сдвига г=6,4-10!, Н/м2; в —усадка в % через сутки после продавливания через капил-
ляр; г — напряжение, при котором появляется нерегулярность струи при выходе из капил-
ляра (т-10’, Н,м2).
скорее, делается даже менее однородной. Отмечается, что один
из каучуков концентрируется преимущественно в межфазных слоях
первых двух. В работе [2] на примере смесей НК—хлорирован-
ный полибутадиен (ХПБ)— (СКЭПТ) (50:30:20) исследована
структура тройных смесей и показано, что в ненаполненных сме-
сях фазы НК и ХПБ обе являются непрерывными, причем толщи-
на фазовой сетки ХПБ значительно меньше и составляет примерно
3 мкм, а частицы СКЭПТ диаметром от 0,2 до нескольких мкм
равномерно распределены в матрице, составляющей основную не-
прерывную фазу.
Наглядной формой представления результатов исследования
тройных смесей полимеров является треугольная диаграмма (тре-
угольник Гиббса). На рис. VI. 1 и VI.2 представлены в качестве
256
Рис. VI .2. Свойства вулканизатов трехкомпонентных смесей СКД —
СКМС-ЗОАРК — СКН-18:
а — разрушающее напряжение при растяжении ар • 10 МПа; б — относительное удлинение
при разрыве, %; в — сопротивление раздиру (сг-10, МПа); г — сопротивление утомлению (на
рисунке указано число циклов М-ll—4 до разрушения) при растяжении образца от 100
ди 250%.
примера диаграммы свойство — состав невулканизованиых смесей
и вулканизатов смесей СКД—СКМС-30АРК--СКН-18. Вулкани-
заты были получены при введении на холодных вальцах 100 масс. ч.
каучука 10 масс. ч. фенолоформальдегидной смолы «фенофор
Б» и 3 масс. ч. стеарата пинка с последующей вулканизацией
60 мин при 163 °C. Построение треугольников, подобных приведен-
ным па рис. VI I и VI.2, весьма трудоемкая задача. При иссле-
довании смесей через 10% по составу построение диаграммы свой-
ство— состав для бинарной смеси потребует исследования девяти
смесей, а для трехкомпонентной смеси — исследования 66 смесей.
Для построения таких диаграмм целесообразно применение
методов планирования эксперимента. Так, при применении метода
планирования эксперимента на симплексе [3], впервые использо-
ванного для построения диаграмм тройных смесей полимеров в
работе [4], можно ограничиться испытанием 15 смесей разного
состава (включая три исходных полимера и бинарные смеси) да-
же для построения полинома четвертого порядка. Диаграммы рис.
17—244
257
VI. 1 и VI.2 получены указанным способом. Часто кривизна изо-
линий свойства невелика, и тогда можно ограничиться примене-
нием полинома второго 'или третьего порядка, для решения кото-
рых необходимо испытать соответственно 6 или 10 смесей, вклю-
чая индивидуальные полимеры.
Как видно из данных рис. VI.1 и VI.2, большинство диаграмм
свойство — состав представлено достаточно сложными поверхнос-
тями (изолинии здесь подобны изолиниям рельефа на топографи-
ческой карте) с впадинами, вершинами, седловинами, что свиде- .
тсльствует о значительных изменениях структуры при изменении
состава смесей. Сложный рельеф поверхности свойство — состав
для тройной смеси полимеров в большей мере характерен для со-
ставов, в которых один из компонентов преобладает, а два других
составляют меньшую часть. Таким образом, основные изменения
свойств смесей происходят в области небольших добавок других
полимеров к первому, основному. В пределах треугольника соста-
вов (см. рис. VI. 1 и VI.2) не наблюдается таких областей, в ко-
торых бы происходило значительное ухудшение или значительное
улучшение свойств по сравнению с бинарными композициями.
Исключение составляет сопротивление утомлению тройной смеси
(см. рис. VI.2,г), для которого наблюдается относительный мак-
симум. Видимо, в тройной смеси, где поверхность раздела фаз на-
иболее развита, возможно значительное искривление пути роста
трещины, что обусловливает диссипацию энергии и повышение
усталостной выносливости. Таким образом, эффект взаимоусиле-
ния, характерный для бинарных смесей полимеров и, в частности,
для бинарных смесей каучуков, установленный в 1958 г. н в пос-
ледующих работах автора с сотр. [5], проявляется и в тройных
смесях. Эффект этот велик, что особенно хорошо видно при по-
строении сечений треугольной диаграммы .в разных направлениях.
Если провести сечение, соответствующее добавлению, например,
каучука СКН-18 к смеси СКД—СКМС-30 в отношении 80:20, то
можно видеть, что уже небольшого количества (10—-15%)
СКН-18 достаточно для повышения усталостной выносливости вул-
канизата в 7- 10 раз. Повышение прочностных свойств и устало-'
стнби выносливости резин на основе НК с quc-полибутадиеном
при введении бутадиен-стирольного каучука отмечено в работе1
[6]. Улучшение комплекса свойств, в том числе технологических
и усталостных, а также рост сопротивления истиранию наблюда-
ется в тройных композициях каучуков НК—полибутадиен—поли-
изопрен и НК — полибутадиен — бутадиен-стирольный каучук,
предназначенных для протекторов большегрузных и средних гру-
зовых автомашин [7, 8]. Хорошие эксплуатационные качества
имеет смесь паирита, СКМС-ЗОАРКМ-15 и СКД [9]. Улучшение
озоностойкости в сочетании с ростом динамической выносливости,
морозостойкости, износостойкости и других свойств наблюдается
при введении в резины из диеновых каучуков совместно этилен-
пропиленовых и галогенсодержащих полимеров [10].
258
Рис. VI.3. Схема расчета свойств трехком-
понентной смеси.
Ввиду незначительного измене-
ния свойств тройных смесей с со-
ставом в средней части треугольни-
ка есть возможность рассчитать
свойства тройной смеси, исходя из
свойств бинарных смесей. Это хоро-
шо видно на примере, приведенном
на рис. VI.3. Здесь показана точка
О в треугольнике составов, соответ-
ствующая смеси, образованной либо совмещением полимеров 1, 2
и 3 в соотношении : <р2: <рз, либо совмещением бинарных смесей,
состав которых определен точками на сторонах треугольника в
соотношении <12 : <pi3-<р2з. Если известны уровни свойств полиме-
ров z/i, у2, уя и их бинарных смесей у^, yts, у2з, то можно написать
уравнение плоскости, проходящей через вершины треугольника:
Е — J/14P1 + У2Ч>2 + f/зФз
(1>
или через точки на его сторонах, выражающие состав и уровень
свойств соответствующих бинарных смесей:
1
у = “Г С'/1гФ1Я + /йзДТз + г/ззЧзз) (2)
Уравнение (1) предполагает аддитивность свойств трехкомпо-
нентной смеси по отношению к свойствам индивидуальных поли-
меров. Это уравнение при прогнозировании свойств тройных сме-
сей не соблюдается, т. е. аддитивность свойств тройной смеси, так
же как и бинарной, по отношению к свойствам смешиваемых ин-
дивидуальных полимеров отсутствует.
Уравнение (2) отражает аддитивность по отношению к свойст-
вам г/12, У1з, 1/23 таких пар полимеров, смешав которые в количест-
вах <ji2, <13, <2з, можно получить данную тройную смесь. Из рис.
VI.3 видно, что для нахождения значения свойства в любой точке
треугольника (например, в точке 0) следует провести прямые из
каждой вершины через выбранную точку, найти уровень свойств
(У12, //1з, Уъз) бинарных смесей, состав которых определяется пере-
сечением прямых со сторонами треугольника, и определить объем-
ную долю каждой бинарной смеси (<pi2, <тз, Фгз)- Затем надо по
уравнению (2) рассчитать свойство тройной смеси, которое и вы-
ражается точкой 0 на данной поверхности свойство — состав.
Таким образом, можно сформулировать принцип аддитивности
бинарных смесей по отношению к свойствам тройных трехфазных
систем, согласно которому показатель свойства в данной точке
треугольника составов аппроксимируется плоскостью, проходящей
через точки на сторонах треугольника, соответствующие бинар-
ным смесям, из которых образована тройная композиция.
17
253
Принцип «парной» или «бинарной» аддитивности [11] пока-
зывает, что аддитивность свойств смесей трех полимеров может
соблюдаться по отношению к бинарным двухфазным смесям, ког-
да структура трехфазной смеси сравнивается со структурой двух-
фазной, т. е. меняется число фаз, а в принципе гетерофазность со-
храняется.
Иногда свойства бинарных смесей, из которых получена трой-
ная смесь данного состава, оказываются близкими по уровню.
Тогда плоскость составов тройных смесей практически горизон-
тальна, и свойство тройной смеси может быть найдено как среднее
арифметическое свойств бинарных композиций:
1 = з (Д12 + У is "Т У из)
Достаточно точное (до 10%) прогнозирование свойств тройных
смесей по уравнениям (1) и (2) возможно, когда компоненты пе
обладают заметной взаимной растворимостью, не содержат функ-
циональных групп, способных к сильному межмолекулярному
взаимодействию, а область составов достаточно удалена от сторон
треугольника. Однако даже для смеси СКД—ПВХ—СКН-40, где
последние два полимера обладают значительной взаимной раство-
римостью, возможно качественное прогнозирование свойств трой-
ных смесей с ошибкой в пределах 10—40%.
При исследовании свойств тройных смесей с помощью метода
планирования эксперимента и построения тройных диаграмм свой-
ство— состав возможно .применение общих для таких диаграмм то-
пологических принципов. Так, меняя способ получения смеси, ко-
личество добавленного пластификатора, наполнителя и т. п., мож-
но установить закономерности движения пи треугольнику интере-
сующей исследователя характеристической точки (седловины, вер-
шины, впадины и т. п.) и предсказать условия смешения или
количество и характер ингредиентов, необходимых для перемеще-
ния характеристической точки в заданную область составов.
Примером может быть максимум на диаграмме сопротивления
утомлению вулканизатов (см. VI.2,г), который смещается по тре-
угольнику составов в зависимости от режима утомления, что по-
зволяет произвести направленный выбор состава смеси для дан-
ных условий эксплуатации материала.
Смеси с числом полимеров более трех не могут быть охарак-
теризованы наглядной диаграммой состав — свойство. Если диаг-
рамма для бинарной смеси линейна (кривая), для тройной — дву-
мерна (поверхность), то для четырехкомпонентной смеси она трех-
мерна. Интересующие нас экстремальные точки на трехмерной
диаграмме состав — свойство можно найти либо построением
множества секущих плоскостей, либо с помощью ЭВМ, которая по
заданной программе может отыскать, какому составу смеси соот-
ветствует максимум (например, максимум сопротивления утомле-
260
пню) или другой экстремум в системе. Такой же принцип может
быть положен в основу отыскания состава, обладающего если не
максимальным, то просто заданным уровнем свойств.
§ 2. ПЛАСТИФИКАЦИЯ СМЕСЕЙ ПОЛИМЕРОВ
Смеси полимеров применяются давно, и в ряде случаев в них
вводят пластификаторы. К числу пластифицированных смесей по-
лимеров относятся некоторые крупнотоннажные смеси, такие как
смесь ПВХ и бутадиен-нитрильных сополимеров, а также смеси
каучуков для производства шин и резиновых технических изде-
лий. Обычно пластификаторы в указанных смесях пластифициру-
ют оба полимера, причем выбор пластификатора обусловлен при-
родой полимера, находящегося в смеси в преобладающем коли-
честве. Закономерности пластификации смеси ПВХ с бутадиен-
нитрильными сополимерами приведены в [12] пластификация
смеси НК—СКС-30 продуктом пиролиза рё5ины изучена в работе
[13]. Технологический опыт пластификации других смесей каучу-
ков до настоящего времени не обобщен. Имеющиеся публикации
позволяют сделать вывод, что закономерности пластификации
смесей полимеров общими пластификаторами .по крайней мере
качественно аналогичны закономерностям пластификации инди-
видуального полимера. В этом разделе внимание уделяется имен-
но системам полимер — полимер, содержащим длретификатецщее-
лективного действия, пластифицирующий эффект которого отли-
чается от действия общего для обоих полимеров пластифика-
тора.
Присутствие пластификатора в смеси полимеров меняет взаи-
модействие полимеров в межфазном слое и адгезию полимеров
друг к другу. Неодинаковое сродство пластификатора к разным
полимерам ведет к его концентрированию в одной из фаз и, сле-
довательно, меняет соотношение вязкостей. В гл. III было пока-
зано, что взаимодействие жидких фаз на границе раздела и ве-
личина соотношения вязкостей фаз влияют на деформируемость
жидких капель дисперсной фазы и на предельный размер частиц.
Изменение же предельного размера частиц должно привести к из-
менению свойств смеси. Таким образом, введение пластификатора,
имеющего обычно, .неодинаковое сродство к компонентам смеси,
приведет к изменению размера частиц и их способности к ориен-
тации в процессе деформации при переработке и эксплуатации.
Это обусловит изменение свойств смеси.
Влияние природы пластификатора и его сродства к полимерам
на их адгезию друг к другу исследовано в работе [14] на приме-
ре каучуков СКИ-3 и СКН-40, в которые вводили медицинское ва-
зелиновое масло (ВМ), диметилфталат (ДМФ) и продукт пиро-
лиза тяжелых фракций нефти, содержащий более 80% тяжелых
ароматических соединений, имеющих техническое название зеле-
ное масло (ЗМ). Каучук СКИ-3 набухал неограниченно в ВМ
261
Рис. VI.4. Зависимость прочности связи дублированных слоев СКИ-3 и С КН-40
jot содержания пластификатора в масс. ч. на 100 масс. ч. полимера (если пла-
стификатор содержится в обоих полимерах, то количества его одинаковы):
с — в СКИ-3 введено ЕМ. в СКН-40 введены 1 — ВМ, 2— ДМФ, 3 — ЗМ, 4 — без пластифи-
катора; 6 — в СКИ-3 введено ЗМ, в СКН-40 введены 1 — ЗМ. 2 — ДМФ. 3 — без пластифи-
катора; б — в СКИ-3 введен ДМФ, СКН-40 без пластификатора; г — СКИ-3 без пластифи-
катора, в СКН-40 введены 1 — ВМ, 2 — ДМФ, 3 — ЗМ.
и не более чем на 5% в ДМФ; СКН-40, наоборот, 'неограниченно
набухал в ДМФ и не более 5% в ВМ; оба полимера неограничен-
но набухали в ЗМ; ВМ и ДМФ неограниченно смешивались
с ЗМ и не смешивались друг с другом. Из обобщенных данных,
приведенных на рис. VI.4, видно, что введение пластификатора
в СКН-40 пе влияет на величину адгезии — она остается такой же,
как и при контакте каучуков, не содержащих пластификатора.
При введении же любого пластификатора в СКИ-3 адгезия уве-
личивается независимо от того, какой пластификатор содержится
в слое СКН-40. Из этого правила для данной пары полимеров нет
исключений, и если адгезия не меняется при добавлении ВМ в оба
каучука в равных количествах (см. рис. VI.4, а, кривая 1), то это
объясняется тем, что в СКН-40 нельзя ввести более 4% ВМ,
а этого количества недостаточно для увеличения адгезии (см.
кривые 2—4). Другой особенностью является наличие экстре-
мальных точек на зависимости адгезии от содержания пластифи-
катора, когда СКИ-3, содержащий ЗМ, дублировали с СКН-40,
не содержащим пластификатора (см. рис. VI.4, б, кривая 1). Да-
же когда полимеры, находящиеся в контакте, содержали пласти-
фикаторы, не совместимые друг с другом (кривая 2 на рис.
VI.4, а), это тоже приводило к росту адгезии по сравнению с си-
стемой без пластификатора.
Указанные эффекты не могут иметь лишь кинетическую при-
роду (снижение вязкости), поскольку значения адгезии, приве-
денные на рис. VI.4, были получены в условиях достижения пре-
дельной величины во времени. Немонотонное изменение адгезии
с ростом содержания пластификатора наблюдалось и в случае
контакта полимеров с металлами [15]. Немонотонный характер
зависимости адгезии от содержания пластификаторов в контак-
тирующих слоях свидетельствует о принципиальной возможности
направленного регулирования прочности связи полимеров в гра-
262
ничком слое. Следует, однако, иметь ,в виду, что переход к высо-
ким температурам, при которых проводится смешение полимеров,
может изменить характер зависимости адгезии от концентрации
и природы пластификатора.
Влияние пластификатора на взапмодиспергирование полиме-
ров и, в частности, на предельный размер частиц изучено в [16]
на примере смесей ПЭНП с ПС в соотношениях 9:1 и 7 : 3. Дис-
персная фаза ПС селективно пластифицировалась добавлением
ди (2-этилгексил) адипината и дпбутилфталата. Исследовалась
также смесь СКИ-3 — СКН-40 в соотношении 9:1, в которой
СКП-40 пластифицировался диметилфталатом.
Введение селективного пластификатора в дисперсною фазу
приводит к изменению соотношения вязкостей фазы и матрицы
(ц=|)1/г]2) Зависимость размера частиц от соотношения вязко-
стей, дисперсной фазы и матрицы (см. гл. III) должна иметь вид
кривой с минимумом при ц=1. Если изменение ц вызвано введе-
нием пластификатора, зависимость приобретает вид кривой
с максимумом (рис. VI.5) [16]. Видимо при высоком содержании
пластификатора в фазе ПС происходит его «выдавливание» на
поверхность раздела ПС — ПЭ. Такое концентрирование пласти-
фикатора в межфазном слое вызывает уменьшение прочности
связи полимеров и «проскальзывание» при деформации. Соответ-
ственно уменьшается сдвиговое усилие, передаваемое матрицей
полиэтилена на частицу ПС. Рост размера частиц ПС с увели-
чением содержания пластификатора в нем происходит до тех пор,
пока вязкость‘фазы ПС не станет достаточно малой (ц^1), что-
бы дробление частиц фазы происходило даже при условии кон-
центрирования пластификатора в межфазном слое.
Характерно различие в положении левых ветвей кривых на
рис. VI.5. В этой области соотношений вязкостей механизм диспер-
гирования маловязких частиц идентичен, по-видимому, механизму
диспергирования жидких капель, рассмотренному в гл. III Увели-
чение содержания дисперсной фазы с 10 до 30% приводит, как
и следовало ожидать, к росту размера частиц (кривые /и 2).
Введение дибутилфталата, в присутствии которого фаза ПС обла-
дает меньшей эластичностью, чем в присутствии того же количе-
ства (по объему) адипинового эфира, приводит к уменьшению
размера частиц. Возможной причиной уменьшения размера ча-
стиц при переходе от адипината к фталату при одинаковом ц
может быть и увеличение адгезии в межфазном слое в присутст-
вии фталата, которое, однако, экспериментально пе исследовали.
На рис. VI.6 и VI.7 показано изменение размера частиц при
смешении каучуков СКН-40 и СКИ-3 в присутствии селективного
(ДМФ) и общего пластификатора (ЗМ), а также зависимость
размера частиц от содержания ЗМ в смеси ПЭ—ПС. Видно, что
увеличение размера частиц при уменьшении ц (добавление се-
лективного пластификатора), наблюдается и в этом случае. Как
видно из рис. VI.7, размер частиц не зависит от дозировки пласти-
263
Рнс. VI.5. Зависимость предельного размера частиц фазы ПС от соотношения
вязкостей фаз ПС и ПЭ при введении в ПС пластификаторов (смешение на
вальцах при 150°C):
1 — ди(2-этилгексил)адипииат, ПЭ : ПС=9 : 1; 2— то же. ПЭ : ПС=7 : 3; 3 — днбугилфта-
лат, ПЭ : ПС=9 : 1»
Рис. VI.6. Зависимость среднего размера частиц фазы СКН-40 от соотношения
вязкостей СКИ-3 и СК.Н-40 при введении в СКН-40 ДМФ.
фикатора, если последний является обшим для обоих полимеров
и не меняет заметным образом ц, а также равномерно распреде-
ляется в двухфазной смеси, не концентрируясь, по-видимому,
в межфазных слоях.
Если соотношение вязкостей варьируют путем изменения мо-
лекулярной массы полимера дисперсной фазы, как это делают
обычно в таких исследованиях, то присутствие селективного пла-
стификатора не меняет типичного характера кривой зависимости
предельного размера частиц от соотношения вязкостей: эта зави-
симость по-прежнему выражается кривой с минимумом (рис. VI.8)
[16]-
Введение селективного пластификатора приводит к изменению
способности дисперсной фазы ориентироваться в процессе сдви-
га в капилляре [17]. Это было показано на примере смеси поли-
оксиметилена (ПОМ) с сополиамидом (СПА), в который вводили
селективный пластификатор — триэтиленгликоль. Оказалось, что
для тройной системы характерна ярко выраженная концентриче-
ская структура экструдатов, которая в отличие от смеси без
пластификатора образуется при значительно более низких напря-
жениях сдвига и в более коротких капиллярах. Так, для смеси
ПОМ—СПА «телескопичносгь» слоев, распределяющихся в виде
концентрических колец различной ширины, наблюдается только
264
при продавливании через капилляры с LID=‘2.0 и при напряже-
ниях сдвига х—105 Па. В присутствии пластификатора это явле-
ние наблюдается при LID=§ ,и т=1,6-104 Па.
Присутствие селективного пластификатора влияет на механи-
ческие свойства смесей полимеров сложным образом, и обобще-
ние закономерностей этого влияния в настоящее время невозмож-
но из-за ограниченного числа экспериментальных данных. Харак-
терный пример действия селективного пластификатора на уста-
лостные и прочностные свойства ненаполненных вулканизатов
смеси каучуков приведен на рис. VI.9—VI.11.
Из рис. VI.9 видно, что при добавлении пластификатора ВМ,
селективно действующего на СКИ-3, прочность вулканизатов
СКИ-3 закономерно снижается, в то время как прочность смеси
СКН-40 — СКИ-3 (3: 1) увеличивается почти в 3 раза. Это явле-
ние не может быть объяснено с позиций влияния соотношения
вязкостей на предельный размер частиц. Действительно, СКИ-3
менее вязкий полимер, чем СКН-40, и введение пластификатора
в СКИ-3 приводит к еще большему уменьшению соотношения вяз-
костей по сравнению с р— 1, когда достигается наилучшее дис-
пергирование полимеров при перемешивании. Рост адгезии в меж-
фазном слое каучуков при введении ВМ в СКИ-3 мог бы обеспе-
чить тем большую прочность, чем больше поверхность раздела
фаз, т. е. при соотношении каучуков 1:1. На самом деле именно
при этом соотношении наблюдается наибольшее снижение проч-
ности. Видимо при каждом количестве пластификатора возника-
ет специфическая структура, обеспечивающая комплекс свойств.
В случае пластифицированной смеси кривая прочность — со-
Рис. VI.7. Зависимость среднего размера частиц дисперсной фазы от содержания
ЗМ для смесей СКН-40 — СКИ-3 (?) и ПЭ — ПС (2). Соотношение компонентов
в смеси 9:1.
Рис. VI.8. Зависимость среднего размера частиц в пластифицированном ди(2-
этилгексил) адипинатом смеси ПЭ — ПС (9:1) от соотношения вязкостей ком-
понентов. Изменение ц достигалось изменением молекулярной массы ПС (а —
1,17-106, б — 8-10% в — 4,5-105, г — 6,5-104):
1~ 10% пластификатора в фазе ПС; 2 — 20%.
265
Рис. VI.9. Влияние селективного пластификатора (вазелиновое масло) на зави-
симость прочности от состава смеси СКН-40 — СКИ-3:
1 — без пластификатора; 2— в СКИ 3 введено 15 масс. ч. ВМ.
Рис. VI. 10. Влияние сел< ктивного пластификатора на сопротивление утомлению
при знакопеременном изгибе и постоянном значении напряжения Л'п для смесей,
приведенных на рис. VI.9.
став даже для таких различных полимеров, как СКИ-3 и СКН- 40,
располагается выше аддитивной. Этим еще раз подтверждается,
что деление полимеров на совместимые .и несовместимые в смыс-
ле качества получаемых из .них смесей не только условно, но и не-
верно: можно получить удовлетворительные материалы на основе
смесей полимеров, бесспорно являющихся несовместимыми, при
правильном выборе состава смеси и условий ее получения.
На рис 1.10 показана зависимость числа циклов до разруше-
ния вулканизатов смесей тех же каучуков, что и на рис. VI.9,
откуда видно, что в режиме постоянно действующего напряжения
при многократном знакопеременном симметричном изгибе в сме-
си наблюдается эффект взаимоусиления. хотя и небольшой по
величине. Эффект взаимоусиления увеличивается при введении
ВМ, причем это увеличение составляет около половины десятич-
ного порядка.
Известно, что усталостные свойства смесей каучуков наилуч-
шим образом характеризуются коэффициентом утомляемости р
в уравнении Рсзниковского:
А'=(ор/ол0₽ (3)
где (тр — прочность резины при однократном растяжении образца до разрыва:
о л — механическое напряжение, обусловившее разрыв после N циклов.
Оказалось, что коэффициент р сильно изменяется при введе-
нии селективных пластификаторов; он увеличивается, когда се-
лективный пластификатор вводится в полимер, образующий мат-
266
рицу. Таким образом, для повышения прочности или усталостной
выносливости необходимо пластифицировать дисперсную фазу,
а для увеличения коэффициента утомляемости нужно пластифи-
цировать непрерывную фазу (матрицу) [18]. Общность этих вы-
водов для других пар полимеров подлежит экспериментальной
проверке.
Интересно, что введение пластификаторов в смесь несовмести-
мых полимеров приводит не к снижению, а даже к увеличению
динамической выносливости двухфазных смесей, которые, как это
можно было бы предполагать, должны расслаиваться с ухудше-
нием свойств ввиду их термодинамической несовместимости и не-
равновесности. На рис. VI. 11 приведена зависимость прочности
и коэффициента утомляемости в относительных единицах так,
чтобы легко можно было проследить общую тенденцию измене-
ния свойств при введении пласгифика горов. Из этих результатов,
а также из данных, приведенных в [19]. следует, что эффект не-
монотонности изменения свойств («антипластификации») [20]
наблюдается при действии селективного пластификатора на смесь
каучуков гораздо чаще, чем при пластификации индивидуальных
полимеров. Действие совместных пластификаторов на смесь по-
лимеров по общим закономерностям не отличается от действия
тех же пластификаторов на индивидуальные полимеры, как это
видно из опыта пластификации смеси ПВХ — СКН-40 нефтяными
пластификаторами типа 354 [21].
Исследования селективной пластификации смеси пластмасс на
примере смеси ПС — ПММА показали [22], что при введении ди-
изобутилазелаината или бутоксиэтилстеарата в матрицу ПС мож-
но получить эластичные композиции, наполненные стеклообраз-
ным ПММА, а при пластификации фазы ПММА полиоксипропи-
0 5 10 15 5 10 75
ВМ, масс. ч. ОМ, масс, ч
Рис. VI.И. Зависимость относительных значений прочности при растяжении (а)
и коэффициента утомляемости (б) вулканизатов смесей СКИ-3— СКН-40 от ко-
личества пластификатора ВМ, введенного в СКИ-3:
; —СКИ-3; 2 — СКИ-3 ; СКИ-40=3 : 1; 3 — СКИ-3 : СКН-40= 1 : 3.
267
ленгликолем в стеклообразной матрице ПС можно получить уда-
ропрочный ПС, если количество пластификатора достаточно для
снижения Тс ПММА ниже комнатной.
Приведенные экспериментальные данные показывают еще од-
ну важную возможность «конструирования» фазовых структур за-
данного типа в смесях полимеров и соответствующего направлен-
ного изменения свойств композиционных материалов.
§ 3. НАПОЛНЕНИЕ СМЕСЕЙ ПОЛИМЕРОВ
Наполнитель, как и пластификатор, может неравномерно рас-
пределяться между фазами полимеров в гетерофазной смеси.
Принципиальное различие распределения наполнителей в смеси
состоит в том, что они не могут диффундировать в покоящейся
или перемешиваемой смеси. Поэтому самопроизвольное перерас-
пределение наполнителей между фазами смеси невозможно в от-
личие от пластификаторов, способных перераспределяться между
фазами в соответствии с их термодинамическим сродством к по-
лимерным компонентам смесиН
Перенос наполнителей из фазы в фазу и переход их из свобод-
нодисперсного состояния в состояние дисперсии в вязком полиме-
ре происходит в процессе механического перемешивания, вслед-
ствие чего условия процесса смешения и порядок введения напол-
нителя оказывают решающее влияние на его распределение меж-
ду фазами. Это влияние иногда более существенно, чем влияние
различий в сродстве полимеров смеси к поверхности данного на-
полнителя.
На распределение наполнителя в смеси полимеров оказывают
влияние следующие факторы.
1. Порядок введения наполнителя в смесь или в компоненты
смеси. Имеется в виду возможность введения наполнителя либо
в один полимер, либо в другой, либо в оба полимера в заданных
количествах с последующим перемешиванием. Возможно введение
наполнителя в заранее приготовленную смесь, а также другие ва-
рианты режима смешения, обеспечивающие резко различное со-
держание наполнителя в фазах полимеров д смеси.
2. Различие сродства смешиваемых полимеров к поверхности
наполнителя, обусловливающее различное смачивание поверхно-
сти разными полимерами и поэтому преобладание на поверхности
частиц наполнителя сегментов того полимера, который лучше
смачивает эту поверхность.
3. Различие вязкости смешиваемых полимеров, что приводит
к разной скорости деформации каждой фазы при заданном на-
пряжении сдвига в смесительном оборудовании. Наполнитель
преимущественно переходит в быстро деформирующуюся фазу.
Рассмотрим механизм проявления указанных "факторов более
подробно.
Различие вязкости смешиваемых полимеров влияет на перерас-
пределение наполнителя между полимерами в том смысле, что
268
поток быстро деформирующегося полимера увлекает частицу на-
полнителя, которая обволакивается этим полимером быстрее, чем
более вязким полимером, оказываясь таким образом вовлеченной
в фазу менее вязкого, т. е. деформируемого полимера.
Наибольшее значение наполнение как процесс модификации
полимеров имеет в технологии переработки эластомеров, где наи-
более распространено наполнение сажей. Поэтому принципы вве-
дения наполнителей в смесь полимеров рассмотрены в литерату-
ре главным образом на примере смеси эластомеров, наполненной
сажей [23—26].
Влияние вязкости полимеров на распределение сажи реализу-
ется по принципу саморегулирования, когда наполнитель входит
преимущественно в маловязкую фазу, повышая ее вязкость до тех
пор, пока вязкости фаз не уравняются, после чего распределение
наполнителя происходит главным образом в соответствии с соот-
ношением сродства наполнителя к полимерам смеси [23].
Характерным примером является введение НК или СКД
в этилен-пропиленовый тройной сополимер (СКЭПТ), содержа-
щий сажу. Если вводимый каучук (НК или СКД) имеет малую
молекулярную массу и, следовательно, низкую вязкость, то при
длительном перемешивании сажа переходит в него. Если же в са-
женаполненный СКЭПТ вводить высокомолекулярный и высоко-
вязкий НК или СКД, то сажа остается в основном в фа-
зе СКЭПТ. Причина того, что сажа остается в фазе СКЭПТ,
сродство которого к саже меньше, чем у СКД или НК
является кинетической: в процессе смешения малодеформируемый
высоковязкий НК или СКД не успевают заместить СКЭПТ на по-
верхности частицы сажи, тогда как низковязкий быстродеформи-
руемый СКД или НК успевают за время перемешивания занять
поверхность сажи. Аналогичный пример можно привести и для
смеси СКД и НК- Каучук СКД имеет большее сродство к саже,
и она при смешении оказывается в основном в фазе СКД. Одна-
ко при длительном перемешивании молекулярная масса НК быст-
ро снижается в результате механодеструкции, что приводит в ко-
нечном счете к переходу сажи в фазу НК-
Во всех случаях термин «переход наполнителя» следует по-
нимать условно, поскольку частицы наполнителя могут переме-
щаться только под влиянием сдвиговых усилий, но не в результа-
те наличия градиента концентраций наполнителя в полимере.
Различие в сродстве полимеров к поверхности наполнителя мо-
жет оказаться очень значительным. Очевидно, что при введении
наполнителя с полярной поверхностью в смесь полярного и непо-
лярного полимеров произойдет переход наполнителя в фазу по-
лярного полимера ввиду его высокого сродства к полярной по-
верхности наполнителя. Поэтому иногда возникает необходимость
модификации (олеофилизации, гидрофобизации) полярного напол-
нителя (например, минерального) при введении его в неполяр-
ный полимер. Закономерности сродства полимеров одной какой-
269
либо группы, например карбоцспных (относительно неполярных),
к такому распространенному наполнителю как сажа, окончатель-
но не установлены. Наибольшим сродством к саже обладают дие-
новые эластомеры и в наибольшей степени полибутадиеп. Поэто-
му, если сажа вводится в смесь СКД с другими каучуками, то
опа преимущественно накапливается в фазе СКД- Увеличение
полярности каучука должно приводить к пониженной адсорбируе-
мости его на саже, однако практически часто наблюдают доволь-
но высокое содержание сажи в фазе бутадисн-нитрильного каучу-
ка даже в его смесях с СКД, НК и др. [26, 27]. Видимо, наличие
в макромолекулах каучука даже небольшого количества двойных
связей приводит к повышению его сродства к саже. Так, этилен-
пропиленовый тройной сополимер СКЭПТ с небольшим количест-
вом диеновых звеньев в макромолекуле обладает высоким сродст-
вом к саже, а этилен-пропиленовый двойной сополимер СКЭП
так же, как бутилкаучук или хлорбутилкаучук, — весьма низким
сродством.
Аномальное поведение сажи наблюдалось также в с меси НК
и ПХП [2, 27, 28]. Сажа остается в фазе ПХП при введении НК
или БСК в сажевую маточпую смесь на основе ПХП. Даже при
введении сажи в смесь ПХП с НК (или с БСК) она распределя-
ется неравномерно, оставаясь преимущественно в фазе ПХП.
И, наконец, замечено, что разбавление маточной смеси сажи с НК
путем введения полихлиропрена приводит к тому, что сажа кон-
центрируется на границе раздела каучуков, а часть ее переходит
в фазу ПХП. Аналогичный эффект высокого сродства ПХП к са-
же обнаруживается и для смесей других эластомерэь
Сродство полимера к наполнителю определяется в значитель-
ной мере и природой поверхности наполнителя. Это хорошо вид-
но на примере эластомеров, содержащих сажу пли аэросил. Пред-
варительно окисленная сажа или модифицированный аэроспл об-
наруживают большее сродство к НК в его смеси с СКД, или
большее сродство к бутилкаучуку в его смесях с НК или СКЭПТ
[24, 26].
Порядок введения наполнителя в смесь полимеров может ре-
шающим образом изменить характер pacnpt целения наполнителя
между фазами смеси. При любом соотношении сродства пли вяз-
кости полимеров можно «принудительно» увеличить содержание
наполнителя в компоненте, имеющем меньшее сродство или более
высокую вязкость, если предварительно ввести большую часть
или весь наполнитель в этот компонент с последующим смешени-
ем его с другим полимером. Если смешение с наполнителем не
предполагается длительным, то для большей равномерности рас-
пределения наполнителя можно смешивать заранее приготовлен-
ные маточные смеси. При смешении полимеров в закрытом смеси-
теле, где интенсивность смешения существенным образом зависит
от степени заполнения камеры смесителя, раздельное введение
ингредиентов может привести к удлинению цикла смешения за
270
Таблица VI.1. Механические свойства вулканизатов смесей каучуков (1:1)
с 40 масс. ч. сажи ДГ-100
Каучук Порядок введения сажи* Напряжение при 300% удлинения, МПа Разрушающее напряжен не при растяже- нии, МПа Относительное удлинение, %
СКД+СКС-30-1 I 6,2 10,0 495
II 6,9 18,7 530
III 7,2 20,0 540
IV 5,4 8,0 480
СКД+СКН-40 I 12,3 24,4 485
II 10,2 21,3 490
III 16,2 21,6 402
IV 9,3 9,8 321
•Способы введения сажи: I — (каучук А+сажа) +(каучук В+сажа); II—(каучук А+
+каучук Б)4-сажа; III — (каучук Л+сажа)4-каучук Б; IV — (каучук Б4-сажа)4-каучук А.
счет того, что в начальный период времени, когда в один из кау-
чуков вводится наполнитель или предварительно смешиваются
два каучука без наполнителя, камера нс полностью загружена. Ин-
тенсивный провесе смешения в этом случае начнется лишь после
того, как в камеру смесителя будут помещены все ингредиенты,
т. е. оба каучука и наполнитель.
Необходимо специально подчеркнуть, что вследствие двухфаз-
ной структуры смеси полимеров порядок введения наполнителей,
как и других ингредиентов, играет гораздо большую роль, чем
в случае композиции иа основе одного полимера. Оптимальный
комплекс механических свойств наполненной смеси полимеров
обеспечивается при оптимальном распределении наполнителя
между фазами. Оптимальное распределение не всегда означает
равномерное распределение. Направленное распределение напол-
нителя между фазами создает ту степень микрогетерогенности
в системе, которая обеспечивает и наилучшие условия для релак-
сации перенапряжений и наилучшим образом препятствует разра-
станию разрушающих трещин, что в совокупности ведет к улуч-
шению прочностных и усталостных свойств. Это видно из табл.
VI. 1 [25] на примере смесей СКД с бутадиен-нитрильным
(СКН-40) и карбоксилатным каучуком СКС-30-1, содержащим
в макромолекулах 1 % (мол.) звеньев акриловой кислоты.
На распределение наполнителя в смеси полимеров влияет
и общее количество введенного наполнителя, о чем подробнее см.
в [26, 28].
Еще более сложный случай распределения ингредиентов в сме-
си полимеров представляет система двух каучуков с одновремен-
ным введением наполнителя и пластификатора. При смешении
в открытом смесительном оборудовании (типа вальцев) порядок
смешения может быть весьма произвольным в зависимости от це-
лесообразности увеличения или уменьшения вязкости каждой
271
фазы, тогда как в закрытом смесителе периодического действия
(типа смесителя Бенбери) эффект смешения часто максимален
при одновременной загрузке ингредиентов [29].
Введением пластификатора можно уменьшить, а введением
наполнителя увеличить вязкость той или иной фазы полимера
в смеси. Таким образом, введение наполнителя также позволяет
регулировать соотношение вязкостей фаз и размер частиц дис-
пергируемого полимера. При сильной адсорбции обоих полимеров
на наполнителе интенсифицируется процесс смешения полимеров,
поскольку частицы наполнителя как бы соединяют воедино мак-
ромолекулы разнородных полимеров. Так, введение сажи в смесь
каучуков в определенных случаях предотвращает расслаивание
раствора, что практически важно при приготовлении клеев и ла-
ков из смесей полимеров.
§ 4. ВУЛКАНИЗАЦИЯ СМЕСКИ ПОЛИМЕРОВ
Вулканизующие агенты, входящие в состав вулканизующей
группы (при химической вулканизации), различаются по раство-
римости в полимерах, образующих смесь. Кроме того, среди вул-
канизующих агентов могут быть вещества, вовсе нерастворимые
в полимерах (например, полностью нерастворим оксид цинка,
применяемый в качестве активатора серной вулканизации). Неко-
торые агенты вулканизации вводятся в количестве, большем пре-
дела растворимости, и тогда основная масса их также присутст-
вует в виде твердых частиц. Ингредиенты вулканизующей группы,
растворимые в полимерах в условиях перемешивания или вулка-
низации, распределяются в соответствии с закономерностями, по-
казанными для пластификаторов, а нерастворимые ингредиенты
(в том числе нерастворимые лишь при температуре смешения)
распределяются главным образом по механизму, известному для
наполнителей. Очевидно, что неравномерное распределение вулка-
низующей группы между фазами полимеров в смеси должно быть
типичным явлением при вулканизации смесей полимеров и опре-
делять ее специфику.
Вторым важным фактором, определяющим специфику вулка-
низации смесей полимеров, является совулканизация в межфаз-
ном слое, оказывающая влияние па свойства вулканизата. Для
каждого полимера смеси можно подобрать такие вулканизующие
системы, которые обеспечат раздельную вулканизацию каждой
фазы каучука без образования поперечных связей между разно-
родными макромолекулами.
Хранение смеси полимеров, содержащей вулканизующие аген-
ты, и нагревание се перед началом пространственного сшивания
при вулканизации приводят к изменению параметров фазовой
структуры. В результате последующей вулканизации (образова-
ния пространственной сетки химических связей) изменение фазо-
вой структуры становится необратимым, т. е. такие параметры
272
Таблица VI.2. Коэффициенты распределения некоторых вулканизующих
агентов в смесях полимеров при 153 °C
Смесь полимеров А—Б К = са/сб
Сера МВТ Ди-о-ТГ 1 тмтд
СКЭПТ—Хлорбутилкаучук 1,25 1,6 0,76 1,52
НК—Хлорбутилкаучук 1,56 2.95 1,7 4,8
БСК-1502—Хлорбутил- 1,84 4,25 3,14 10
каучук
ПБ—Хлорбутилкаучук 2,0 2,7 1,47 10
Неопрен—Хлорбутилкаучук 2,5 6 3,6 10
НК—скэпт 1.25 1,85 2,22 3,17
БСК-1502—НК 1.18 1,44 1,86 2
ЦБ - БСК-1-502 1,09 0,64 0,46 —
ПБ—НК 1,26 0,92 0,85 —
фазовой структуры невулканизованпой смеси, как размер и форма
частиц, величина межфазного слоя и даже число фаз в смеси, мо-
гут отличаться от параметров вулканизата. Это третий фактор,
влияющий на свойства смесей полимеров, и специфичный именно
для них.
В этом параграфе будут разобраны указанные факторы, оп-
ределяющие специфику вулканизации гетерофазных смесей поли-
меров. Закономерности вулканизации смешиваемых полимеров
рассмотрены в ряде монографий и обзоров [30—33].
Распределение растворимых вулканизующих агентов между
фазами полимеров в смесях происходит в результате диффузии
в соответствии с растворимостью агента в данном полимере. Ко-
эффициенты диффузии большинства растворимых ингредиентов
составляют 10~7—10-8 см2/с, а размер частиц дисперсной фазы
колеблется в пределах нескольких мкм. Поэтому концентрацион-
ное равновесие в фазах полимеров в смеси может быть достигну-
то за очень короткое время, за несколько секунд [1, 34] В табл.
VI.2 приведены коэффициенты распределения некоторых вулкани-
зующих агентов в смесях полимеров при температуре вулканиза-
ции [35].
Температурная зависимость растворимости ингредиентов в
разных каучуках различна (рис. VI.12). Как видно из рисунка,
при повышении (или понижении) температуры может измениться
направление диффузии данного ингредиента, который при одной
температуре преимущественно растворяется в одном каучуке, а
при другой температуре 'больше растворяется в другом каучуке.
Концентрация вулканизующего агента в каждой фазе меняет-
ся в процессе вулканизации вследствие убыли свободного агента
в одной из фаз в результате его присоединения к полимеру. В сме-
си насыщенного полимера с ненасыщенным вулканизующие аген-
ты быстрее расходуются в фазе ненасыщенного полимера, где
273.
18—244
Сумасе, ч.
Рис. VI.12. Растворимость серы в каучуках:
/ — в бутилкаучуке; 2 — в хлорбутилкаучуке; 3 — в
этилен-пропиленовом каучуке; 4 — в НК; 5 — в бута-
диен-стирольном каучуке; 6 — в цис-полибутадиене;
7 — в полихлоропрене.
суммарная скорость вулканизации
много больше, чем в фазе насыщен-
ного полимера (или полимера с ма-
лой степенью ненасыщенности). Поэ-
тому, если даже в результате подбора
соответствующих агентов вулканиза-
ции или применения подходящего ме-
тода распределения ингредиентов уда-
лось сосредоточить необходимое ко-
личество вулканизующего агента в фазе полимера с малой нена-
сыщенностью, то в процессе вулканизации все равно большая
часть вулканизующего агента перейдет в фазу более ненасыщен-
ного полимера, который может оказаться перевулканнзованным.
Неравномерное распределение ингредиентов в смеси приводит к
неаддитивному изменению кинетических параметров вулканиза-
ционного процесса с изменением соотношения полимеров в смеси
(рис. VI.13) [26] Вследствие этого возникает необходимость
корректировки на практике состава вулканизующей группы эм-
пирическим путем. Такая корректировка может фактически пре-
вратиться в поиск вулканизующей группы совершенно другой хи-
мической природы, когда необходимо устранить отклонения от
а ддитивности. Так, замена тиурама на сантокюр в смесях НК—
СКН-40 приводит к устранению минимума на зависимости скоро-
сти вулканизации смесей от их состава (кривые 1 и 2). Разумеет-
ся, приведенные на рис. VI.13 отклонения от аддитивности при-
сущи смесям полимеров, в которых компоненты не различаются
слишком сильно по степени ненасыщенности. Если различие ве-
лико, го отклонения от аддитивности кинетических характеристик
вулканизационного процесса становятся неизбежными в подав-
ляющем большинстве случаев.
Распределение агентов вулканизации в смеси полимеров мож-
но регулировать, в ряде случаев обеспечивая хорошее совпадение
скоростей вулканизации в разных фазах. Одн им из таких методов
является введение всех или части вулканизующих агентов в один
из полимеров смеси или введение их в один из полимеров в пре-
обладающем количестве. Кинетика вулканизации и свойства резин
могут при этом изменяться в довольно широких пределах [1,26,
36, 37]. Другой метод заключается в такой модификации полиме-
ров, чтобы агенты вулканизации были химически связаны с ними
или чтобы полимеры содержали функциональные группы, быст-
рее реагирующие с введенными агентами вулканизации. Так, для
обеспечения равной степени вулканизации в смеси ненасыщенных
274
и насыщенных (СКЭПТ, БК) полимеров, последние предваритель-
но модифицируют в растворе [38,39] или в массе прививкой
ускорителей или галогенированием [40]. Галогеносодержашпе на-
сыщенные полимеры, такие как хлор- и бромбутилкаучук, хлор-
сульфополиэтилен, галогенированный СКЭПТ и т. п., ’ обнаружи-
вают значительно большую скорость вулканизации, а также спо-
собность вулканизоваться специфическими вулканизующими аген-
тами, которыми исходные каучуки не вулканизуются.
Еще одним путем регулирования распределения вулканизую-
щей группы в полимерах смеси является изменение размеров не-
активной части органических молекул, входящих в состав вулка-
низующей группы. Обычно неактивной частью является алкильная
скдню is so г5 о скд юо is 50 г5 о
СКС-Я-Щ 15 50 75 100 CKC-JO-1 0 15 50 75 100
Рис. VI.13. Изменение кинетических характеристик вулканизации ненаполнеппых
(сплошные линии) и наполненных (пунктир) смесей каучуков в зависимости от
типа вулканизующей системы:
/ — сера 2,0+тиурам 0,3; 2 — сера 2,0+сантокюр 0,8; 3 — сера 0,5+тиурам 3,0: 4 — сера 2.0+
+каптакс 0,75.
18'
275-
группа в органических ускорителях вулканизации при серной
вулканизации или углеводородная цепочка смол при смоляной
вулканизации. Изменение размеров углеводородной цепи влияет в
известной степени на активность функциональных групп, участ-
вующих непосредственно в образовании пространственных сшивок,
однако в то же время меняется растворимость органического
соединения в полимере. Обычные вулканизующие агенты (в осо-
бенности ускорители вулканизации) более полярны, чем каучуки
(т. е. имеют больший, чем каучук, параметр растворимости), по-
этому удлинение углеводородной цепи молекул ускорителей при-
водит к повышению их растворимости в каучуках, в особенности
в неполярных. Это может резко изменить равновесную концент-
рацию вулканизующей группы в фазах смеси полимеров. Пе ис-
следован вопрос о применении смесей нескольких ускорителей,
подобранных в соответствии с их растворимостью в фазах смеси
полимеров так, чтобы в фазе каждого полимера концентрировался
преимущественно один из ускорителей, что обеспечивало бы не-
обходимую скорость вулканизации и заданный тип химических
вулканизационных связей. Наличие больших алкильных радика-
лов влияет не только на равновесную растворимость ингредиентов,
по и на скорость их диффузии, уменьшая ее, что приводит к со-
хранению в процессе вулканизации того распределения ингредиен-
тов, которое задано было при смешении [41, 42].
Снижение скорости перераспределения ингредиентов в процес-
се вулканизации может быть достигнуто путем применения ин-
гредиентов, образующих нерастворимые соединения с ускорителя-
ми и вулканизующими агентами, или замедляющими их диффу-
зию. Как отмечается [42], замена оксида цинка на оксид свинца
в обычных сероускорительных системах позволяет получать ка-
чественные вулканизаты на основе смесей разнородных по хими-
ческой природе полимеров (таких, как СКЭПТ и СКН). Это до-
стигается благодаря тому, что свинцовая соль ускорителей в от-
личие от цинковой не растворяется в полимерах, чем предотвра-
щается перераспределение ускорителей. Улучшение свойств ре-
зин на основе смесей диеновых полимеров с БК может быть до-
стигнуто при серной вулканизации в присутствии аминосодержа-
щих ускорителей, которые снижают диффузию вулканизующих
агентов из БК в другой полимер [1].
Наконец, можно использовать несерные вулканизующие си-
стемы (перекисные, смоляные) или системы с галогенсэдержащи-
ми вулканизующими агентами, растворимость которых в обеих
фазах примерно одинакова [33,43—45].
Полагают, что равенство скоростей вулканизации полимерных
компонентов смеси является важнейшим фактором, обеспечиваю-
щим получение резины с оптимальным комплексом свойств [43—
45]. Это является тем эмпирически установленным правилом, ко-
торое позволяет подбирать рецептуры вулканизации смесей по-
лимеров.
276
По-видимому, равенство скоростей вулканизации достигается
при условии, что диффузия из фазы в фазу при прочих равных
условиях минимальна. Это принципиально важно, так как в про-
тивоположном предельном случае одна из фаз может оказаться
вообще невулканизовапной. При радиационной вулканизации до-
зы, требуемые для гелеобразования полимеров смеси различны, и,
следовательно, нарастание частоты вулканизационной сетки в фа-
зах не синхронизировано. Тем не менее при радиационной вулка-
низации чаще, чем при химической, обеспечивается аддитивность
изменения механических свойств вулканизатов с составом. Именно
в случае радиационной вулканизации ввиду отсутствия вулкани-
зующих агентов исключены диффузионные процессы.
Синхронизация поперечного сшивания в фазах имеет тем мень-
шее значение, чем больше плато вулканизации.
Совулканизация (процесс образования химических связей
между разнородными макромолекулами в смеси полимеров) в
случае двухфазной смеси осуществляется только в межфазном
слое—слое сегментальной растворимости. При большом разли-
чии в химической природе полимеров, когда слой сегментальной
растворимости оказывается минимальным и недостаточным для
образования флуктуационной сетки из разнородных макромоле-
кул, не происходит в достаточной степени и совулканизация. Со-
вулканизацию полибутадиена с бутаднен-нитрильными сополиме-
рами обеспечить тем легче, чем меньше нитрильных групп содер-
жится в макромолекуле сополимера, хотя ненасыщенность бута-
диен-нитрильнмх сополимеров всегда достаточно высока, чтобы
обеспечить принципиальную возможность совулканизации их с
диеновыми гомополимерами.
Прямые доказательства совулканизации полимеров в смесях
практически отсутствуют. Они базируются в основном на совул-
канизации в слоях полимеров, соединенных друг с другом в виде
адгезионного соединения (дублированные слои). Характер изме-
нения механических свойств с составом смеси в общем случае ни
прямо, ни косвенно не является мерой совулканизации полимеров
в смеси.
Предложен метод оценки степени совулканизации, основанный
на набухании вулканизата смеси полимеров в селективных рас-
творителях, т. е. в таких, в которых набухает только один поли-
мер [46]. Метод основан на расчетах Крауса интенсивности взаи-
модействия полимер — наполнитель при набухании. Чем больше
взаимодействие полимер — наполнитель или чем больше число
химических или сильных межмолекулярных связей между поли-
мером и наполнителем, тем меньше набухает собственно каучуко-
вая фаза. Уменьшение степени набухания полимерной фазы при
увеличении содержания наполнителя и есть мера взаимодействия
полимер — наполнитель. При аналогичном подходе к вулканизату
смеси полимеров ошибка в оценке межфазных связей (степени
совулканизации полимеров) возрастает вследствие того, что вул-
277
канизованный .полимер дисперсной фазы также набухает в опре-
деленной степени в.любом выбранном растворителе, а миграция
вулканизующих агентов из фазы в фазу может изменить степень
вулканизации непрерывной (набухающей) фазы даже больше, чем
ее изменяет совулканизация в межфазном слое. Поэтому предло-
женный метод [46] в общем случае также дает лишь весьма при-
ближенную оценку степени совулканизации.
Наличие химических связей между разнородными полимерами
при двухфазной структуре смесей доказано для случая смеси по-
лимеров с функциональными группами, способными реагировать
друг с другом. К числу таких смесей относится смесь бутадиен-
метилвинилпиридинового сополимера (СКМВП) с карбоксилат-
ным каучуком (сополимер диенового мономера с акриловой кис-
лотой), простой прогрев которой приводит к потере растворимости
и образованию вулканизационной структуры, а также смесь
СКМВП с хлорсульфополиэтиленом или полихлоропреном, бута-
диен-нитрильного сополимера с хлорсульфополиэтиленом или по-
лихлоропреном и т. п. [26, 47, 48].
При хранении невулканизованной смеси полимеров в ней мо-
гут происходить релаксационные процессы, приводящие к измене-
нию фазовой морфологии. На примере смеси гщс-полибутадиена и
СКЭПТ [49] показано, что наиболее высокая степень дисперсно-
сти полимеров в смеси наблюдается сразу после смешения для
системы, в которой вязкости компонентов близки между собой.
Затем в процессе вылежки смеси образуются более крупные ча-
стицы каждой фазы, которые и фиксируются в процессе вулкани-
зации, вследствие чего фазовая структура смеси сразу после сме-
шения и после вулканизации неидентична. Причины такого явления
неясны. Наиболее реально предположить (авторы этого не выска-
зывают), что релаксация формы частиц, вытянутых при смеше-
нии и переходящих в сферические при вылежке смеси, приводит
к увеличению среднего диаметра частиц, измеряемого на срезах.
При прогреве смеси в процессе вулканизации наблюдается и
уменьшение размеров частиц, и переход от двухфазной смеси к
однофазной. Это можно видеть на примере смеси СКД и БСК.
для которой возникновение промежуточного максимума на кривой
радиотермолюмннесценции (РТЛ) при вулканизации считалось
доказательством совулканизации [50]. В действительности обра-
зование однофазной смеси, в которой вместо двух максимумов
РТЛ образуется один промежуточный, происходит при нагрева-
нии и без вулканизации. Этот промежуточный максимум можно
зафиксировать, если смесь после прогрева сразу охладить в жид-
ком азоте, чтобы однофазная смесь не успела расслоиться при
охлаждении. Эта смесь имеет верхнюю критическую температуру
смешения: она становится однофазной при нагревании до 150 °C
за несколько минут, а расслаивается в течение недели (появляют-
ся два максимума) при быстром охлаждении до комнатной тем-
пературы [51].
278
Таким образом, экспериментальные данные, имеющиеся в на-
стоящее время, показывают, чго в двухфазной системе вулканиза-
ция фиксирует ту фазовую структуру, которая имелась в системе
к моменту возникновения пространственной сетки химических
связей. Эта фазовая структура может отличаться от заданной в
процессе смешения. Изменения фазовой структуры могут быть
направлены как в сторону укрупнения частиц, так и в сторону
уменьшения их размеров, вплоть до перехода к однофазной сис-
теме в результате достижения предела растворимости полимеров
при нагревании смеси до начала вулканизации.
Принципиальное улучшение механических или физико-хими-
ческих свойств достигается, конечно, при правильном подборе по-
лимеров для смешения. Дальнейшее, часто очень значительное
изменение свойств данной смеси, может быть достигнуто путем
корректировки рецептуры и режима введения ингредиентов. Силь-
ная зависимость свойств вулканизата от выбора ингредиентов и
способа их введения затрудняет установление общих закономер-
ностей изменения свойств вулканизата в зависимости от соотно-
шения полимеров и их химической природы. Даже качественный
характер изменения свойств (выше или ниже аддитивной зависи-
мости) может зависеть от выбора ингредиентов или способа их
введения.
Видимо, вулканизаты смесей полимеров не являются менее
однородными, чем вулканизаты отдельно взятых полимеров, как
это следует из данных по статистической обработке результатов
механических.испытаний [52]. Для вулканизатов смесей полиме-
ров характерна повышенная динамическая выносливость в режиме
многократных деформаций [41], которая не всегда сочетается с
повышенной прочностью при однократном растяжении или с по-
вышенным сопротивлением раздиру [53]. Механизм взаимоусиле-
ния связывают с облегченной релаксацией напряжения в вершине
растущей трещины при встрече с межфазным слоем, в котором
межмолекулярное взаимодействие, по-видимому, ослаблено, что
следует из данных по определению энергии когезии смеси поли-
меров и прочности связи дублированных слоев разнородных по-
лимеров в динамическом режиме [54]. Подтверждением этого яв-
ляется также наблюдаемая пониженная величина константы С2
в уравнении Муни — Ривлина [44].
Возможность миграции вулканизующих агентов из фазы в
фазу в смеси полимеров в процессах смешения и вулканизации
затрудняет прогнозирование свойств вулканизатов, открывая, од-
нако, возможности для направленного воздействия на структуру
и механические свойства смесей, руководствуясь принципами, об-
щими для всех гетерофазных систем (см. гл. V).
Обычно принятым путем построения таблиц или графиков
трудно проследить за изменением даже одного свойства при из-
менении содержания нескольких ингредиентов и при изменении
порядка их введения. Если же требуется учет изменения многих
279
свойств вулканизата смеси полимеров, то оптимизация качества
становится возможной только с помощью ЭВVI Создание специ-
альных алгоритмов для подсчета критерия качества материала
дает возможность объективно оценить эффективность создания
рецептур в таких сложных системах, как вулканизаты на основе
смеси (полимеров, содержащей наполнители, пластификаторы и
другие ингредиенты. Гетерофазная структура смесей приводит к
усилению влияния состава и условий получения вулканизата на
критерии его качества [55].
ЛИТЕРАТУРА
1. Gardiner I. В. — Rubb. Chem. Technol., 1970, v. 43, p. 370—379.
2. Marsh P. A., Mullens T. J.. Price L. D. — Rubb. Age, 1969, v. 101, № 9,
p. 77—84; Rubb. Chem. Technol., 1970, v. 43. p. 400—412.
3. Scheffe H., Roy J. —J. Statist. b< c., 1958, ut B, v. 20, p. 344—355.
4. Кулезнев В. И.. Мирошников Ю. П., Догадкин Б. А.— Коллоид, ж., 1971,
т. 33, № 3, с. 390—395.
5. Кулезнев В. Н., Шварц А. Г., Клыкова В. Д„ Догадкин Б. А. — Коллоид. ж.,
1965, т. 27, № 2, с. 211—215; Догадкин Б. А., Кулезнев В. Н., Тарасо-
ва 3. Н. — Коллоид, ж., 1958, т. 20, № 1, с. 43—51.
6. Vohwinkel R. — Kautschuk u. Gummi, Kunststoffe, 1968, Bd. 21, № 8, S. 436—
444.
7. Krol L. H. — Rubb. Chem. Technol., 1°66, v. 39, p. 452—463.
8. Mekel A. L., Bierman H. — Rubb. J., 1967, v. 149, p. 54—60.
9. Нейечкирхен Ю. H., Захаров H. Д„ Орехов С. B. — Каучук и резина, 1972,
№ 11, с. 10—12.
10. Speranzini А. Н., Drost S. J.— Rubb. Chem. Technol., 1970, v. 43, p. 482—
499.
11 Мирошников Ю. П., Кулезнев В. H. — Коллоид. ж., 1972, т. 34, № 6, с. 884—
888.
12. Тиниус К. Пластификаторы. М—Л , Химия, 1964. 916 с.
13. Младенов И., Николински П„ Грозлеков П. — Кожи, обувки, каучук, пласт-
маси (София), 1961, т. 2, № 4, с. 6—9.
14. Кулезнев В. И., Малошук Ю. С., Григорьян Г. И., Догадкин Б. А. — Высоко-
му п соед., 1971, сер. А, т. 13, № 1, с. 55—58; Григорьян Г. И., Кулезнев В. И.,
Малошук Ю. С., Догаокин Б. А. — Учен зап. МИТХТ им. ЛА. В. Ломоносова,
1970, т. 1, Ns 2, с. 111—116.
15. Грибкова Н. Я., Козлов П. В., Якубович С. В. — Высокомол. соед. 1965, т. 7,
с. 751—756.
16. Кулезнев В. Н., Понизивская Н. В., Мирошников Ю. П.—Межвузовск. тем.
сб., сер. Химия и технол. органич. произв. М., 1978, т. 8, с. 102—-106.
17. Цебренко М. В., Аблазова Т. И.. Юдин А. В., Виноградов Г. В. — Коллоид,
ж, 1976, т. 38, Nt 1. с. 204—209.
18. Кулезнев В. Н., Бродский Г. И., Догадкин Б. А., Григорьян Г. И. — Каучук и
резина, 1972. № 7, с. 24—26.
19. Григорьян Г. И. Канд. дис. М., МИТХТ им. М. В. Ломоносова, 1972.
20. Гуль В. Е„ Федюкин Д. Л., Догадкин Б. А. — Коллоид, ж., 1953, т. 13, Ns 1,
с. 11 — 15; 1957, т. 19, № 3, с. 287—292.
21. Эстрин В. Н., Шевелев Б. П., Кулезнев В. И., Эстрина А. А., Мирошни-
ков Ю. П. — Транспорт и хранение нефти и нефтепродуктов, 1971, Ns 7,
с. 13—16.
22. Periard J., Banderet A.. Riess G. — Angew. macromol. Chem., 1971, Bd. 15.
S. 37—46, 55—64.
23. Walters M. H., Keyte D. N. — Trans. IRI. 1962, v. 38, p. 40—52; Rubb. Chem.
Technol., 1965, v. 38, p. 62—71.
260
24. Hess W., Scott С. E., Callan J. E. — Rubb. Age, 1966, v. 98, p. 68—75; Rubb.
Chem. Technol., 1967, v. 40, p. 371—379.
25. Чиркова И. В. Канд. дис. Ярославль, Ярославский политехи, ин-т, 1971.
26. Чиркова Н. В.. Орехов С. В., Захаров Н. Д.— Резиновые смеси на основе
комбинации каучуков. М., ЦНИИ информ, и технико-эконом. исслед. нефте-
перераб. и нефтехим. пром., 1974. 48 с.
27. Marsh Р. A., Voet A., Price L. D., Mullens Т. J. — Rubb. Chem. Technol.,
1968, v. 41, p. 344—352.
28. Corish P. J., Powell B. D. — Rubb. Chem. Technol., 1974, v. 47, p. 481—510.
29. Никишин E. А. Канд. дис. M„ НИИ шинной пром., 1979.
30. Догадкин Б. А. Химия эластомеров. М., Химия, 1972. 391 с.
31. Донцов А. А., Тарасова 3. Н., Шершнев В. А. — Коллоид, ж., 1973, т. 35,
№ 2, с. 211—225.
32. Донцов А. А. Процессы структурирования эластомеров. М., Химия, 1978.
287 с.
33. Блох Г. А. — Органические ускорители вулканизации и вулканизующие си-
стемы для эластомеров. Л., Химия, 1978. 240 с.
34. Гршиин Б. С., Туторский И. А., Юровская И. С. — Высокомол. соед., 1970,
сер. Б, т. 12, № 5, с. 387—389.
35. Gardiner 1. В. — Rubb. Chem. Technol., 1968, v. 41, p. 1312—1323.
36. Pehner Wei P. E. — Rubb. Chem. Technol., 1969, v. 42, p. 985—994.
37. Mitchell 1. ,M. — J. Elastom. a. Plast., 1977, v. 9, № 3, p. 329—340.
38. Hopper R. J. — Rubb. Chem. Technol., 1976, v. 49, p. 341—352.
39. Baranwal К. C., Son P. H. — Rubb. Chem. Technol., 1974, v. 47, p. 88—89.
40. Morrissey R. 7". — Rubb. Chem. Technol., 1976, v. 49, p. 353—366.
41. Mastromatteo R. P.. Mitchell J. M., Brett T. I. — Rubb. Chem. Technol., 1971,
v. 44, p. 1065—1079.
42. Woods M. E., Davidson I. A. — Rubb. Chem. Technol., 1976, v. 49, p. 112—
117.
43. Кошелев Ф. Ф., Корнев A. E., Буканов A. M. Общая технология резины. M.,
Химия, 1978. 527 с.
44. Кулезнев В. H.f Шершнев В. А. — Каучук и резина, 1977, № И, с. 56—63.
45. Шварц А. Г., Динзбург Б. Н. Совмещение каучуков с пластиками и синтети-
ческими смолами. М., Химия, 1972. 224 с.
46. Запп Р. Л. — В кн.: Многокомпонентные полимерные системы. Пер. с англ./
Под ред. Л. Я. Малкина и В. Н. К>лезнева. М., Химия, 1974, с. 114—129.
47. Sherchnev V. A., Kuleznev V. М, Anfimov В. Н. — Rubb. Age, 1976, v. 108,
№ 9, р. 65.
48 Захаров Н. Д. Хлоропреновые каучуки и резины на их основе. М., Химия,
1978. 271 с.
49. Bohm G. G. — Rubb. Age., 1976, v. 108, № 9, p. 29—38.
50. Златкевич Л. Ю., Каплунов Л1. Я., Борисов В. А., Никольский В. Г. — Каучук
и резина, 1970, № 9, с. 45—47.
51. Пестов С. С., Кулезнев В. Н., Шершнев В. А. — Коллоид, ж., 1978, т. 40. № 4,
с. 705—711.
52. Клыкова В. Д., Кулезнев В. Н., Догадкин Б. А.—Коллоид, ж., 1971, т. 33,
№ 3, с. 464—465.
53. Пестов С. С. Канд. дис. М., МИТХТ им. М. В. Ломоносова, 1979.
54. Орехов С. В., Захаров Н. Д., Кулезнев В. Н., Догадкин Б. А. — Коллоид, ж.,
т. 32, № 2, с. 245—250.
55. Шершнев В. А., Пестов С. С. — Каучук и резина, 1979, № 9, с. 11—19.
Глава VII
КРИТИЧЕСКИЕ ЯВЛЕНИЯ В СМЕСЯХ ПОЛИМЕРОВ
Под критическими явлениями понимают обычно явления, на-
блюдаемые вблизи критической температуры (7’кр), критического
давления (Ркр), критического объема (VK₽) или критического
состава (Скр). Они характерны для переходов жидкость—пар,
жидкость — жидкость (например, переход гелия из одной моди-
фикации в другую, тоже жидкую модификацию), при расслаива-
нии жидкой системы и т. п. Для таких явлений характерна отно-
сительная постепенность изменения удельного объема и внутрен-
ней энергии системы.
Как известно (см. гл I), в системах полимер — растворитель,
полимер — полимер и полимер 1 — полимер 2 —- растворитель мо-
гут существовать как верхние, так и нижние критические темпе-
ратуры смешения, а иногда и те и другие одновременно.
Границей между однофазной и двухфазной системами на фа-
зовой диаграмме является бинодаль, которую еще Гиббс назвал
границей абсолютной устойчивости фазы. За этой границей начи-
нается область, в которой система устойчива по отношению к ма-
лым ^непрерывным) изменениям термодинамических параметров,
но не устойчива при возникновении в ней зародышей второй фа-
зы. Такое состояние системы простирается вплоть до спинодали,
которая (также по определению Гиббса) является границей су-
щественной неустойчивости, разделяющей область фазовой диаг-
раммы на термодинамически метастабильную и нестабильною.
Фактически вблизи перехода в двухфазную область возникаю!
крупномасштабные флуктуации плотности или концентрации, яв-
ляющиеся статистическими образованиями, которые отличаются
от обычных флуктуаций в первую очередь своими размерами [1].
Размеры флуктуаций могут характеризоваться коэффициента-
ми устойчивости — величинами по смыслу' обратно пропорциональ-
ными размерам флуктуаций [2]. Коэффициенты устойчивости для
простой системы, находящейся в равновесии в условиях, соответ-
ствующих бинодали, удовлетворяют условиям—(ДР/<Д')Г>0 (ме-
ханический коэффициент устойчивости) и (dl'ldS)p=TICp>Q (тер-
мический коэффициент устойчивости). Поскольку критическая
точка принадлежит и бинодали и спинодали, а для последней
(dT/dS)p = T/CP=0 и —(dP/dV)T= fV₽r)-1=0, то в критической
точке и Ср—^оо. Поэтому и выше критической точки су-
ществует область пониженной термодинамической устойчивости,
282
характеризуемая сильным развитием крупномасштабных флуктуа-
ций, создающих во всей системе зернистую микроструктуру с со-
хранением внешней сплошности и однородности. Такая структура
обусловливает явление критической опалесценции, правильное объ-
яснение которому дал впервые Смолуховский и которое в настоя-
щее время описано количественно [3].
Исследования критической опалесценции в низкомолекуляр-
ных системах были проведены главным образом на примере сме-
сей органических жидкостей [4—6], как более простом для ис-
следования объекте. Примеры изучения критической опалесцен-
ции в точке перехода жидкость — пар приведены в работах [7, 8].
В критической области существования системы жидкость —
жидкость наблюдается ряд интересных явлений, связанных с по-
ниженной скоростью диффузии. Так, в системе метанол — н-гек-
сан, имеющей верхнюю критическую температуру смешения, ме-
тодом микрофотосъемки показано, что при переходе из однофаз-
ного в двухфазное состояние (понижение температуры) в крити-
ческой точке образуется мелкодисперсная критическая микро-
эмульсия, в которой развиваются струйные потоки вверх и вниз
[9]. Создается как бы нитевидная, волокнистая структура систе-
мы. В этой же системе при нагревании до Т=Ткр образуется про-
межуточная (третья) фаза в виде сильно опалесцирующего вяз-
кого и прочного слоя, который не разрушается при прохождении
через него мешалки. Остатки этого слоя наблюдаются в течение
2,5 сут при 7'=7’Кр+1,5°С. Это значит, что возникшая в растворе
особая критическая структура очень медленно распадается при
повышении температуры (медленная «структурная» диффузия).
И на других примерах можно показать, что в растворах вблизи
критического состояния проявляется ряд особенностей, которые
не могут быть истолкованы, исходя из представлений об обычной
диффузии [10, 11]. Диффузия протекает в два этапа: на первом
процесс крайне замедлен, затем наступает резкое ускорение диф-
фузии, проходящей со скоростью и по законам, характерным для
некритических систем аналогичного состава. Высказывается мысль
о том, что двухстадийная диффузия является характерным свой-
ством систем вблизи критической области [11].
Даже в низкомолекулярных системах вблизи критического со-
стояния показано сильное увеличение толщины межфазного слоя.
В системе метанол — циклогексан по данным отражения све-
та от границ раздела фаз обнаружено, что при 7'=7'КР+О,38°С
толщина слоя 6«И00 нм по сравнению с 6 = 1 нм (вдали от 7кр
[12]).
Возникновение вблизи критической точки крупномасштабных
флуктуаций, являющихся фактически зародышами новой фазы,
приводит к появлению экстремумов в изменении свойств. Зафик-
сировано изменение сжимаемости, теплового расширения и тепло-
емкости [13,14]. В то же время результаты разных авторов по
теплопроводности [15,16] и вязкости [17—21] вблизи критической
283
точки различаются не только количественно, но иногда не сов-
падают даже качественно. Это снова указывает на аномалии диф-
фузионных процессов в околокритической области и влияние
этих аномалий на свойства, обусловленные тепло- и массоперено-
сом. Усредненный коэффициент диффузии в околокритической об-
ласти иногда не превышает 10-8 см2-с-1 [3]. .
Времена релаксации в системах с участием полимеров велики,
и поэтому аномалии свойств, характерные для критических состо-
яний в низкомолекулярных системах, должны быть здесь еще бо-
лее заметными.
Критические явления в системе полимер — растворитель наи-
более широко исследовались по данным критической опалесцен-
ции [22—33]. Теория рассеяния света в однокомпонентных систе-
мах была развита в работах [34], а в двухкомпоненгных — в ра-
ботах [30], в том числе и применительно к растворам полимеров.
Был обнаружен максимум асимметрии рассеяния света в бинар-
ной системе при концентрации несколько ниже критической точки
(см., например, [22] и [31]). Установлено, что аномалии рассея-
ния света обусловлены сжатием полимерных клубков в однофаз-
ной области вблизи критической точки. Так, в работе [33] мето-
дом рассеяния света и диффузного рассеяния рентгеновских лу-
чей в растворе поли-а-метилстирола в циклогексане показано, что
размеры клубков в предкритической области растут с увеличени-
ем молекулярной массы медленнее, чем обычно, т. е. непропор-
ционально М1!*. Отношение средних размеров клубков вблизи
7кр к размерам невозмущенных высокомолекулярных образцов
составляет около 0,4, что указывает на сильное сжатие клубков
и переход их в состояние, близкое к глобулизации. Аналогичные
результаты были получены ранее для других полимеров (см.,
например, [35]), что дало возможность отметить две общие особен-
ности структуры раствора полимера в критических условиях (Скр
или 7’кр). Первая особенность состоит в сильном сжатии клубков,
так что указанное отношение критических размеров к размерам в
О-точке составляет для разных полимеров 0,14—0,65. Вторая осо-
бенность в том, что в определенном интервале температур вблизи
критической эти размеры с температурой не меняются.
Интервал температур, в котором клубки не меняют разме-
ры, может быть достаточно протяженным и достигать 12 °C, вклю-
чая 0-температуру. Чем больше сжатие клубка в критических
условиях, тем шире интервал температур, в котором кубок не ме-
няет размеры.
В предкритической области в растворах конечной концентра-
ции наблюдается ассоциация макромолекул. В гл. II это было
показано на примере смесей растворов полимеров, а в работе
[36] показано спектроскопически на примере растворов полиокси-
пропилендиола в н-гексане.
Изменение структуры раствора приводит к увеличению вре-
мени релаксации и энергии активации релаксационных процессов,
284
что было показано на примере дипольной поляризации [37]. Ас-
социация приводит к появлению двух сильно различных групп
времени релаксации: малых времен, отражающих релаксацию
неассоциириванных молекул, и больших времен, характеризую-
щих релаксационные процессы, связанные с перемещением круп-
ных ассоциатов молекул.
Особенностью растворов полимеров при достижении критичес-
кой температуры является характерная температурная зависи-
мость вязкости [31,38]. Так, для раствора ПС в циклогексане
наблюдается монотонная (обычная) зависимость температурного
, . 1 ch „„
коэффициента вязкости—•от концентрации ПС, если тем-
пература на 2 °C и более отличается от критической. На той же
зависимости появляется максимум, если температура лишь на
0,2 пли 0.02 СС отличается от критической. Чем меньше отклоне-
ние температуры от критической, тем больше величина максиму-
ма. Для системы ПС — о-триметилформамид — диэтиловый эфир
с нижней критической температурой смешения (НКТС) был об-
наружен рост вязкости непосредственно перед расслаиванием
[39].
В околокритической области возникающие частицы новой
фазы несут малый запас избыточной поверхностной энергии. При
удалении от критических условий быстро завершается формиро-
вание межфазного слоя предельной структуры, и избыточная меж-
фазная энергия возрастает. В системах с малым предельным
межфазным натяжением область концентраций, давлений, темпе-
ратур и т. п., в которой происходит формирование межфазного
слоя, оказывается протяженной. В этой протяженной области
межфазное натяжение особенно низкое, что обусловливает воз-
никновение термодинамически устойчивых коллоидных систем.
Гиббс предложил считать работу образования критического
зародыша мерой устойчивости метастабильной фазы. В таком
направлении мысль Гиббса получила развитие в работах [40], а
затем [41]. Если межфазное натяжение мало (менее 10-4—10~5
Н/м), то увеличение энтропии, достигаемое участием возникших
капелек в броуновском движении, превосходит увеличение сво-
бодной энергии системы за счет образования новой поверхности.
Свободная энергия системы в целом меняется при этом с изме-
нением размера частиц по кривой с минимумом, что и обусловли-
вает термодинамическую устойчивость эмульсии и самопроиз-
вольность ее образования.
Схематически область возникновения термодинамически ус-
тойчивых эмульсий [45] в качестве одного из возможных вариан-
тов показана на рис. VII. 1. Примеры возникновения термодина-
мически устойчивых эмульсий в системах полимер — жидкость и
жидкость — жидкость приведены в работах [40—45].
Таким образом, в условиях расслаивания и выделения новой
фазы в системе возникает граница раздела фаз, которая вблизи
285
1
Рис. VII.1. Области существования на фазовой ди-
аграмме однофазных стабильных (/), метастабиль
пых (2), лабильных (3) систем и термодинамически
стабильных дисперсных систем (4).
критической области характеризуется чрез-
вычайно низким межфазным натяжением.
При этом расчет [461. подтвержденный
экспериментом (см. также гл. IV), показы-
вает, что в полимер-полимерных системах
межфазное натяжение в момент выделения
новой фазы вблизи критической области
составляет 10-7 Н/м. Это в 100 раз меньше
межфазного натяжения, измеренного в со-
ответствующих условиях расслаивания низкомолекулярных жид-
костей, ив 104 раз меньше межфазного натяжения в полимер-
полимерных системах в условиях, далеких от расслаивания.
Возникающий в условиях расслаивания межфазный слой
включает большой объем вещества обеих фаз потому, что толщи-
на слоя и поверхность раздела фаз, обусловленная малым разме-
ром частиц, велики. .Малое межмолекулярное взаимодействие
несовместимых полимеров в межфазном слое приводит к пониже-
нию плотности системы [47], снижению плотности энергии коге-
зии смеси полимеров в целом [48], что эквивалентно «концентри-
рованию» свободного объема в межфазном слое [49]. Возникает
своеобразная система зернистой (или слоистой) структуры, в ко-
торой механические свойства определяются чередованием более
прочных (зерна, слои) и менее прочных (межслоевые, межфазные
промежутки) микрообъемов вещества [50]. Подобная структура
облегчает релаксацию внутренних напряжений или напряжений в
вершине растущей трещины при встрече с межфазным слоем.
Структура такой системы обусловливает:
а) наилучшие условия для релаксации внутренних напряже-
ний или перенапряжений ввиду большой протяженности межфаз-
ного слоя;
б) наиболее статистически равномерное расположение колло-
идных частиц выделяющейся фазы, а следовательно, наименьший
средний путь трещины при данной концентрации частиц в объе-
ме;
в) наибольшее количество частиц при данной объемной доле
вещества выделяющейся фазы ввиду их малого размера и узкого
распределения по размерам, т. е. снова наименьший средний путь
трещины в системе;
г) наименьшее количество вещества выделяющейся фазы для
достижения такой величины поверхности раздела и количества
частиц в единице объема, которые бы обеспечили наиболее рав-
номерное распределение напряжений в системе и быструю релак-
сацию перенапряжений.
286
Рис. VII.2. Концентрационная зависимость
оптической плотности (/), вязкости (2) и
объема нижнего слоя (5) бензольного рас-
твора смеси ПС — ПИП (7:3).
Если после критической точки
система переходит в состояние тер-
модинамически устойчивой эмуль-
сии, то относительная протяжен-
ность интервала составов или тем-
ператур, в которых такая эмульсия
существует, позволяет исследовать
ее механические свойства. Впервые
механические свойства термодинамически устойчивых эмульсий
были исследованы на примере смесей полимеров и растворов сме-
сей полимеров, где при переходе от однофазной системы к двух-
фазной (расслаивание) пе присходит такого резкого изменения
механических свойств, как при расслаивании раствора одного по-
лимера, и поэтому аномалии изменения механических свойств,
особенно заметны. Сам комплекс механических свойств является
здесь важной характеристикой системы в противоположность
смеси низкомолекулярных жидкостей [51—55]. В этих работах
изменение механических свойств фиксировали одновременно с из-
мерением структурных характеристик (например, оптической плот-
ности), что дало возможность разделить наблюдаемые закономер-
ности свойств в. предкритической и в закритической областях, т. е.
выделить область существования термодинамически устойчивых
эмульсий.
На рис. VII. 2 приведена концентрационная зависимость опти-
ческой плотности, вязкости и объема нижнего слоя бензольного
раствора смеси ПС с полиизопреном (ПС—ПИП 7:3). Оптическая
плотность практически не меняется в области прозрачных пред-
критических растворов, несколько увеличивается и сохраняется
постоянной для области существования устойчивой эмульсии, а
затем снова возрастает, когда эмульсия начинает быстро рассла-
иваться. Второй слой (макрорасслаивание) появляется тогда, ког-
да на кривой оптической плотности появляется второй подъем,
т. е. при переходе к состоянию быстро расслаивающейся эмуль-
сии. Вязкость несколько возрастает в области начала формирова-
ния устойчивой эмульсии (фактически этот рост происходит в
предкритической области). Формирование термодинамически ус-
тойчивой эмульсии сопровождается сильным падением вязкости, а
после перехода системы в состояние быстро расслаивающейся
эмульсии вязкость снова возрастает. Формирование термодинами-
чески устойчивой эмульсии сопровождается падением вязкости
системы в 2—10 и более раз для разных систем.
На рис. VII. 3 показано, как раствор смеси полимеров дважды
проходит точку расслаивания и оба раза наблюдается минимум
287
W 20 W 60 10 100
ПИП BO 60 40 20 0
------1------1_____сю_____I_____
ПО 20 00 60 60 '100
ПИР BO 60 00 20 0
Рис. VII.3. Зависимость теплоты актива-
ции вязкого течения (а), логарифма вяз-
кости (б) и концентрации, соответствую-
щей расслаиванию (в), от содержания
ПИП в смеси с ПС. Между кривыми на
рис. в расположена серповидная об-
ласть существования устойчивых эмуль-
сий (ср. с областью 4 па рис. VII.1).
вязкости. В предкритической об-
ласти, так же как и во многих
низкомолекулярных смесях, че-
рез максимум проходит теплота
активации вязкого течения. Ми-
нимум вязкости совпадает с пе-
реходом термодинамически ус-
тойчивой эмульсии в область
двухфазных систем. На рис. VII.3
хорошо видна серповидная об-
ласть существования термодина-
мически устойчивых, самопроиз-
вольно возникающих эмульсий
(ср. с рис. VII.1). Падение вяз-
кости наблюдается как при до-
бавлении менее вязкого раствора
к более вязкому, так и наобо-
рот. Это еще раз подтверждает,
что указанный эффект ничего общего не имеет с простым разбав-
лением системы.
Увеличение напряжения сдвига, при котором измеряется вяз-
кость, приводит к гомогенизации системы, и переход от однофаз-
ных растворов к двухфазным становится более плавным, что со-
провождается уменьшением глубины минимума, а в пределе —
его полным исчезновением (рис. VII. 4) [53]. Аналогичный эф-
фект наблюдается и при повышении температуры, которая также
приводит к уменьшению, а затем и исчезновению минимума на
концентрационной зависимости вязкости раствора смеси полиме-
ров [51].
В гл. I отмечалась сильная зависимость взаимной растворимо-
сти полимеров от молекулярной массы. В полимере с широким
ММР разные фракции выделяются в виде второй фазы и в раз-
ных условиях и фазовый переход оказывается размытым. Поэто-
му реологические эффекты в области расслаивания различаются
в зависимости от ширины ММР полимеров в смеси, как это видно
из рис. VII. 5. Данные, приведенные на рис. VII. 5, показывают
наличие значительного по величине максимума вязкости (модуль
потерь) в предкритической области в полимерах с узким ММР.
На рис. VII. 5, а кривая 1 построена по данным непосредственно-
го измерения свойств раствора смеси, полученной обычным сме-
гревания — охлаждения раствор в крити-
ческой области гомогенизировался при нагревании и расслаивал-
ся при охлаждении, приближаясь к термодинамическому равнове-
сию, что обусловило появление минимума вязкости. Смеси поли-
меров с широким ММР (рис. VII.5, б) быстрее образуют термоди-
намически устойчивую эмульсию, и минимум вязкости наблюдается
в смеси сразу после ее перемешивания.
При образовании термодинамически устойчивых эмульсий
важную роль играет временной эффект, что естественно при воз-
никновении новой фазы в вязких системах, какими являются по-
лимеры или их растворы. Это особенно легко можно наблюдать
на кривых температурной зависимости модуля упругости или мо-
дуля потерь растворов смесей полимеров в интервале, включаю-
щем температуру расслаивания. При медленном охлаждении
структура раствора успевает перестроиться и можно наблюдать
минимум вязкости, тогда как при быстром охлаждении система
«проскакивает» критическую область, и минимума вязкости об-
наружить не удается. Замедленность релаксационных процессов
в растворах смесей полимеров обусловливает и повышенный «ги-
стерезис» в них: несовпадение кривых изменения вязкости с тем-
пературой при нагревании и охлаждении.
Рис. VII.5. Зависимость модуля потерь раствора ПИП в присутствии малых
добавок раствора ПС (а — полимеры с узким ММР, б — с широким ММР):
1 — при 20 °C; 2 — при 20 °C после цикла; нагревание до 80 °C и последующее охлаждение
до 20 °C.
19—244
289
Критические явления и возникновение термодинамически ус-
тойчивых эмульсий наблюдается и в расплавах смесей полимеров.
Впервые падение вязкости в области расслаивания было отме-
чено в работе [56]. В этой работе смесь ПЭВП и ПП (90:10)
перемешивали в ротационном вискозиметре при 190 °C, т. е. в
условиях, когда смесь достигала состояния раствора, близкого к
насыщенному. При последующем охлаждении до 155 °C ПП ча-
стично выпадал в виде отдельной фазы, что сопровождалось зна-
чительным ростом модуля упругости системы. Как можно пред-
положить, структура раствора была при этом близка к структуре
термодинамически устойчивой эмульсии, что вызывало значитель-
ное (в 2,5 раза) падение вязкости, определенной при напряжении
сдвига, стремящегося к нулю. В указанной работе было отмечено
лишь качественное совпадение предела расслаивания и минимума
вязкости смеси.
В последующих работах [51—55] экстремальное изменение
вязкости было количественно сопоставлено с пределом расслаи-
вания и с существованием термодинамически устойчивых эмуль-
сий одного полимера в другом (или их растворов в общем рас-
творителе). Обнаруженный эффект снижения вязкости в области
расслаивания полимеров является, по-видимому, общим, так как
в смесях полимеров, сильно различающихся по структуре, он на-
блюдается и в отсутствие растворителя. Так, при концентрации
ПИП в смеси с ПММА, отвечающей пределу его растворимости,
наблюдается резкое уменьшение вязкости (около десятичного
порядка), как это видно из рис. VII. 6 [53]. Снижение вязкости
наблюдается и в смесях двух пластиков. Так, на том же рисунке
видно снижение вязкости как при добавлении расплава ПС к
расплаву ПММА, так и наоборот. При этом характерно, что при
содержании ПММА в смеси с ПС менее 0,2%, когда наблюдается
минимум вязкости, струя расплава после выхода из канала ка-
пиллярного вискозиметра становится опалесцирующей, а при
увеличении содержания ПММА расплав становится после охлаж-
дения молочно-белым и этот переход соответствует прохождению
вязкости через минимум и возвращению к прежним значениям.
Соответствие минимума вязкости расплава смеси ацетобути-
рата целлюлозы и полиоксиметилена пределу расслаивания поли-
меров в смеси показано в работе [57]. Здесь предел расслаива-
ния определяли по изменению параметра Флори xi2, найденного
по данным обращенной газовой хроматографии. Минимуму вяз-
кости расплава смеси ацетобутират целлюлозы — ПОМ при 10%
ПОМ соответствует максимум аномалии вязкости и минимум па-
раметра Флори.
В работе [54] было показано, что экстремальное изменение
свойств смесей в области термодинамически устойчивых эмульсий
характерно не только для расплавов, но и для смесей в твердом
состоянии, о чем свидетельствуют максимумы прочности и пре-
дела текучести и минимумы твердости и модуля.
290
Рис. VII.6. Зависи-
мость вязкости
расплава смеси
ПММА — ПС от
концентрации ПС
(а), ПММА—ПИП
от концентрации
ПИП (б) и ПС—
ПММА от концен-
трации ПММА (в).
Измерение» оптической плотности показывает (см. рис. VII.2)
область концентрации системы, в которой существует стабильная
эмульсия. Определение размера частиц в этой эмульсии методом
спектра мутности показало, что в растворах смеси полимеров
размер частиц устойчивой эмульсии составлял не более 100 нм
и оставался неизменным в течение 6 мес., причем сохранялась
узкая кривая распределения по размерам частиц. Если концент-
рация или состав раствора превышает предел стабильности
эмульсии, размер частиц быстро возрастает до 600—1000 им и
более и система немедленно расслаивается на два слоя. Этот про-
цесс в самом начале сопровождается расширением кривой распре-
деления капель эмульсии по размерам. Переход к быстро расслаи-
вающейся эмульсии приводит к скачкообразному уменьшению
значения поверхности раздела почти на три десятичных порядка
при ничтожном изменении состава или температуры си-
стемы.
В ряде случаев при получении полимер-полимерных смесей
возникают системы, близкие по структуре к термодинамически
устойчивым эмульсиям, но не являющиеся в действительности
равновесными. К числу таких систем относятся композиции, по-
лученные полимеризацией мономера или олигомера в матрице
полимера. Можно создать условия, в которых исходный олигомер
(или мономер) будет полностью растворим в полимере. Однако
19'
291
Рис. VII 7. Зависимость ар вулканизатов СКД от
концентрации олигоэфиракрплата:
1 — СКД+а.<ь-метакрил(бис-гриэтилепгликоль)фталат; 2 —
а-метакрил-со-триметакрнлпеитаэритрит - ди(диметакрил-
пентаэритрит)адипинат. Стрелками показан предел рас-
творимости олигоэфира при 150 °C (сплошные линии) и
при 20 °C (пунктир).
растворимость будет уменьшаться по мере полимеризации добавки,
и после достижения предела растворимости дальнейшее наращи-
вание молекулярной массы приведет к выделению частиц дис-
персной фазы, обладающих, как правило, малыми размерами и
сильно развитой поверхностью раздела, а также очень узким рас-
пределением по размерам. Такая система высокооднородна, не-
смотря на наличие двухфазной структуры. В работе [58] было
впервые проведено сопоставление растворимости олигоэфиракри-
латов в каучуке и показано (рис. VII. 7), что максимум прочно-
сти вулканизатов после полимеризации олигоэфира совпадает с
пределом растворимости последнего в каучуке. Исследование
микроструктуры вулканизатов показало, что ниже предела рас-
творимости олигоэфира его полимер распределен равномерно в
виде частиц одинакового размера. Такая система возникает, ког-
да в процессе полимеризации частицы полиэфира выделяются в
виде отдельной фазы из раствора в каучуке.
При хорошем сродстве полимера и полимеризующегося олиго-
мера последний выпадает в виде чрезвычайно мелкодисперсных
частиц. Так, при полимеризации в среде полиднметилсилоксачо-
вого каучука олигоорганогидридсилоксана с молекулярной массой
1000, содержащего 0,5—0,6% HSi-групп, в исходной однофазной
системе возникают частицы размером около 10 нм, о чем сви-
детельствуют данные рентгеновского рассеяния под малыми угла-
ми. В этой системе при содержании олигомера около 30% проч-
ность проходит через максимум [59].
Таким образом, потеря растворимости полимера в полимере в
результате изменения температуры, состава системы (концентра-
ция раствора, соотношение полимеров) или вследствие роста мо-
лекулярной массы одного или обоих компонентов является осно-
вой различных методов создания термодинамически устойчивых
систем.
Введением малых количеств одного полимера в расплав дру-
гого на вальцах или другом смесительном оборудовании можно
в ряде случаев обеспечить высокую дисперсность образующейся
эмульсии. Это достигается подбором таких компонентов для
смешения, которые бы имели достаточно большое термодинами-
ческое сродство и такие условия смешения, при которых обеспе-
чивается хорошая дисперсность и достаточная взаимодиффузия
на границе раздела, и такой концентрации добавки (менее 10%),
при которой коалесценция частиц незначительна (см. гл. IV).
292
Рис. VII.8. Зависимость разрушающего напря-
жения при изгибе (сплошные линии) и растя-
жении (пунктир) ПВХ от содержания в нем
«легирующих добавок»:
/, /'—ПАС-22 (олигоэфир); 2, 2'— ПДЭА (олиго-
эфир); 3, S'— ППА-4 (олигоэфир): 4, 4'— СКН мар-
ки «Бреон», латекс; 5, 5'— СКС ЗОАРК марки «Бу-
лекс», латекс.
Высокая дисперсность частиц и рав-
номерность распределения по разме-
рам в отсутствие коалесценции, а так-
же значительный межфазный слой
обеспечивают получение систем, в
которых структура близка к структу-
ре термодинамически устойчивых
эмульсий. Конечно, эмульсин эти мо-
гут быть неравновесными, и эффект
немонотонного изменения свойств
обусловлен замедленностью диффузии
о 2 в в ю
Содержание, (маос.)
макромолекул, препятствующей разделению системы на два слоя,
что характерно для смесей полимеров вообще. Возможно при этом
и возникновение действительно равновесных эмульсий. В тех слу-
чаях, когда отсутствуют данные о равновесии таких систем и нет
данных о взаимной растворимости компонентов, системы указан-
ного выше типа называют «системами с малыми добавками» или
«системами с легирующими добавками».
В качестве'примера можно привести работы [60—64], в кото-
рых исследовано действие олигомеров и эластомеров па свойства
твердых пластмасс. Наиболее характерные результаты приведены
на рис. VII. 8 [64], где видно немонотонное изменение механиче-
ских свойств ПВХ при добавлении легирующих добавок. Авторы
[64] отмечают, что «улучшение прочностных, реологических и де-
формационных свойств модифицированных ПВХ-материалов, по-
видимому, происходит за счет проникания легирующих веществ в
дефектные зоны полимера, возникающие при формовании образ-
цов. При этом возрастает подвижность всей системы и ускоряются
процессы релаксации, которые протекают достаточно полно, что
в свою очередь снижает остаточные микронапряжения». Таким
образом, налицо своеобразный эффект антипластификации — не-
монотонного изменения свойств при введении высокомолекулярно-
го (или олигомерного) пластификатора в твердый полимер. Внеш-
нее проявление этого эффекта аналогично изменению свойств при
возникновении термодинамически устойчивых эмульсий.
Заметим, что эффект межструктурной пластификации при до-
бавлении в полимеры малых количеств низкомолекулярных пласти-
фикаторов, может быть связан также с выделением некоторого
количества пластификатора в виде частичек микрофазы, вызван-
ным ограниченной растворимостью пластификатора в полимере,
на что указывалось в работе [52].
293
Неравновесные системы со структурой, близкой к структуре
термодинамически равновесных эмульсий, могут возникать не
только в расплавах смесей полимеров, но и в металлических
сплавах. В современном металловедении они получили название
«дисперсноупрочненные материалы», для создания которых «высо-
кодисперсную смесь фаз получают путем закалки сплава на пе-
ресыщенный твердый раствор с последующим упрочняющим от-
пуском или старением» [65, с. 7]. В металловедении в результате
многолетнего практического применения дисперсноупрочпенпых
материалов эмпирически установлено, что размер упрочняющих
частиц не должен превышать 0,01—0,05 мкм, оптимальное сред-
нее расстояние между частицами составляет 0,1—0,5 мкм при
равномерном их распределении в матрице, и таким образом, эф-
фективное упрочнение обеспечивается при содержании упрочня-
ющей фазы не более 5—10% (об.). Одним из основных способов
получения дисперсноупрочненных материалов является выделение
тугоплавкого металла из его раствора в легкоплавким при пони-
жении температуры с последующей фиксацией выделившейся
новой фазы, например путем закалки системы в целом. Представ-
ления о механизме упрочнения главным образом основываются
на предположении о локализации и перестройке дислокаций кри-
сталлической решетки матрицы при встрече с дисперсными ча-
стицами, что в конечном счете также связано с более равномер-
ным распределением напряжений в системе и релаксацией пере-
напряжений.
В заключение заметим, что изменения структуры в растворах
смеси полимеров могут происходить и в разбавленных системах
вдали от концентрации расслаивания. Речь идет о концентрации
С] (см. гл. II), когда разнородные клубки макромолекул в рас-
творе смеси полимеров с концентрацией около 0,1 г/100 мл начи-
нают контактировать. Происходящее изменение структуры рас-
твора состоит, видимо, в сжатии клубков, т. е. происходит свое-
образная подготовка к выделению новой фазы. Однако парци-
альная концентрация данного полимера слишком мала, чтобы
несколько однородных молекул могли образовать частицу новой
фазы, и поэтому дальнейший рост концентрации раствора ведет
уже не к сжатию клубков, а к их расширению благодаря проник-
новению сегментов другого полимера внутрь клубка (см. данные
по светорассеянию в гл. II). Момент сжатия клубка под влиянием
окружающих его макромолекул другого полимера сопровожда-
ется снижением вязкости раствора до минимального значения с
последующим ее повышением до прежних значений с ростом
концентрации и начавшимся взаимопроникновением клубков. Это
было продемонстрировано в работе [66] на примере смеси ПС—
ПММА—бензол, причем бензольный раствор ПММА являлся для
ПС «полимерным растворителем». В этом опыте концентрацию
ПММА сохраняли постоянной, а концентрацию ПС меняли, что
позволяло определить изменение вязкости собственно ПС, раство-
294
ренного в бензольном растворе ПММА. Если построить зависимость
ГпЗпсОт концентрации ПММА, то минимум вязкости наблюдается
при концентрации 0,08 г/100 мл.
Минимум вязкости на кривой вязкость — температура обна-
ружен и для растворов триблок-сополимера ПММА—ПС—ПММА
[67]. Минимум соответствует переходу с понижением температу-
ры от структуры десегрегированных блоков к структуре сегреги-
рованных в пределах макромолекулярного клубка. Это по су-
ществу переход от относительно гомогенной структуры к более
гетерогенной структуре раствора, поэтому и наблюдаемый эффект
аналогичен отмеченному выше эффекту снижения вязкости в рас-
творе смеси ПС—ПММА.
Приведенные в этой главе данные показывают, что эффект не-
монотонного изменения свойств термодинамически устойчивых
эмульсий с участием полимеров является общим явлением. Ха-
рактер изменения свойств при этом отличается от закономерно-
стей, известных для предкритических систем. Явление имеет ана-
логии в разбавленных растворах смесей полимеров, когда с ро-
стом концентрации раствора возникает своеобразная (безусловно
термодинамически устойчивая) «эмульсия» изолированных макро-
молекул одного полимера в среде другого полимера. Внешне
аналогичный эффект изменения свойств наблюдается и в таких
полимер-полимерных системах или в системах полимер — олиго-
мер, когда структура фактически близка к структуре термодина-
мически устойчивых эмульсий, но сами эти системы неравновес-
ны, хотя достаточно устойчивы во времени. Большая поверхность
раздела, протяженный межфазный слой и регулярность фазовой
структуры определяют основные свойства таких дисперсных сис-
тем, которые можно назвать системами с «идеальной» коллоидной
структурой.
ЛИТЕРАТУРА
1. Френкель Я- И. Кинетическая теория жидк< стей. Л., Наука, 1975. 591 с.
2. Семенченко В. К. Избранные главы теоретической физики. М.. Учпедгиз,
1960. 324 с.; Семенченко В. К.. Мартынюк М. М. — Коллоид, ж., 1962, т. 24,
№ 5, с. 611—613.
3. Скрипов В. П. Метастабильная жидкость. М., Наука, 1972. 184 с.
4. Кукс Л1. Ф., Лиснянский Л. Л —В кн.: Критические явления и флуктуации
в растворах. М. Изд-во АН СССР 1960, с. 27.
5. Беридзе Д. К., Шахпаронов М. И. — Там же, с. 21.
6. Chu В. - - J. Am Chem. Soc., 1964, v. 86, р. 3557—3558.
7. Blosser L. G., Drickammer H. G. — J. Chem. Phys., 1951, v. 19, p. 1244 -
1252.
8. Колпаков Ю. Д., Скрипов В. П.— Оптика и спектроскопия, 1965, т. 19,
с. 616—618.
9. Смирнов Б. А. — В кн Всесоюзное совещание по тепло- и массообмену,
Минск, 5—9 мая, 1964.
10. Кричевский И. Р., Хазанова М. Е., Лесневская Л. С. — ИФЖ, 1962. № 2,
с. 14—16.
11. Хазанова М. Е., Лесневская Л. С. — Ж ФХ, 1966, т. 40, с. 76—79.
295
12. Gilmar G. H., Gilmer W., Huang J., Webb W. — Phys. Rev., Letters, 1965,
v. 14, p. 491—493.
13. Шейндлин A. E.—Теплоэнергетика, 1954, № 1, c. 11—14.
14. Амерханов X. И., Керимов A. M. — ДАН СССР, 1957, т. 113, с. 368—370.
15. Боровик Е. Ж- — Эксп. теор. физ., 1949, т. 19, с. 561—569. Амирханов X. И.,
Адамов А. 77. — Теплоэнергетика, 1963, № 7, с. 77—81.
16. Bailey В. I.. Kelner Я. —Brit. J. Appl. Phys., 1967, v. 18, p. 1645—1655.
17. Rothmund V. — Z. phys. Chem., 1908, Bd. 63, S. 54—71.
18. Naldrett S. N„ Mass O. — Canad. J. Res., 1940, pt. B., v. 18, p. 322—327.
19. Michels A., Bofzen A., Schuzman A. — Physica, 1954, Bd. 20, S. 1141—1148.
20. Голубев И. Ф. Вязкость газов и газовых смесей. М., Физматгиз, 1959. 245 с.
21. Kestin J., Whitelaw J. Н., Zien Т. F. — Physica, 1964, Bd. 30, S. 161—171.
22. Кулезнев В. H., Андреева В. М. — Высокомол. соед., 1962, т. 4, № 12,
с. 1851—1857.
23. Кулезнев В. Н., Крохина Л. С., Лякин Ю. Н., Догадкин Б. А. — Коллоид, ж.,
1964, т. 26, № 4, с. 475—480.
24. McIntyre D., Winis A., Green М. S. — J. Chem. Phys., 1962. v. 37, p. 3019—
3025.
25. Цветков В. И., Эскин В. Е., Сказка В. С. — Укр. физ. ж., 1962, т. 7, с. 923—
927.
26. Эскин В. Е., Нестеров А. Е. — Высокомол. соед., 1966, т. 8, с. 1051—1054.
27. Chu В. — J. Chem. Phys., 1965, v. 42, р. 426—431.
28. Тагер А. А., Аникеева А. А., Андреева В. М., Чумарова Т. Я., Черноскуто-
ва Л. А. — Высокомол. соед., 1968, сер. А, т. 10, с. 1661—1667.
29. Borchard И'. — Вег. Bunsenges. Phys. Chem., 1972, Bd. 76, S. 224—228.
30. Debye P. — In: Electromagnetic Scattering/Ed. by M. Keller. N. Y., 1963.
485 p.
31. Borchard W., Rehage G. — In: Multicomponent Polymer Systems. Am. Chem.
Soc. Washington, 1971, p. 42.
32. Chu B. — J. Chem. Phys., 1965, v. 42, p. 2293—2301.
33. Волкова Л. А., Андреева H. А., Подольский А. Ф., Таран А. А., Эскин В. E.—
Высокомол. соед., 1977, сер. А, т. 19, № 3, с. 475—479.
34. Ornstein L. S., Zernicke F. 1. — Z. Phys., 1926, Bd. 27, S. 761—795.
35. Debye P., Woerzman. D., Chu B.—J. Chem. Phvs., 1962. v. 36, p. 851, 1803—
1817.
36. Лирова Б. И., Смоленский А. Л., Савченко Т. Л., Тагер А. А. — Высокомол.
соед., 1972, сер. Б, т. 14, № 4, с. 265—271.
37. Бурштейн Л. Л. и др. — Высокомол. соед., 1974, сер. Б, т. 16, № 5, с. 16,
326—329; 1975, сер. А, т. 17, с. 883—887: 1976, сер. А, т. 18, Ns 2, с. 291—295.
38. Debye Р. — J. Chem. Phys., 1959, v. 31, р. 680—696.
39. Rigler J. R., Wolf B. A., Briteinbach L. W. — Angew. makromol. Chem., 1977.
Bd. 57, № 854, S. 15—21.
40. Volmer M. — Z. Phys. Chem., 1927, Bd. 125, S. 151; 1931, Bd. 155, S. 281—
292; 1957, Bd. 206, S. 181—194; Bd. 207, S. 307—309.
41. Щукин E. Д., Ребиндер П. А.— Коллоид, ж., 1958, т. 20, с. 645—649.
42. Русанов А. И., Щукин Е. Д., Ребиндср П. А.—Коллоид, ж., 1968, т. 30,
с. 573—578.
43. Кочанова Л. А., Федосеева Н. П., Кичумова В. М., Перцов А. В., Ребин-
дер П. А. — Коллоид, ж., 1973, т. 35, с. 838—843, 843—848.
44. Абросенкова В. Ф., Влодавец И. Н. — Коллоид, ж., 1975, т. 37, № 5, с. 926—
929.
45. Федосеева Н. П., Кучумова В. М., Кочанова Л. А., Щукин Е. Д. Коллоид, ж.,
1978, т. 40, 946—949.
46. Joanny 1. F., Leiblier L. — J. Phys., 1978, v. 39, № 9, p. 951.
47. Кулезнев В. H., Игошева К- М. — Высокомол. соед., 1962, т. 4, № 12, с. 1858—
18b2.
48. Кулезнев В. И., Догадкин Б. А., Клыкова В. Д. — Коллоид, ж., 1968, т. 30,
№ 1, с. 255—257; Орехов С. В., Захаров Н. Д., Кулезнев В. Н„ Догад-
кин Б. А. — Коллоид, ж., 1970, т. 32, № 2, с. 245—250.
296
49. Липатов Ю. С. Физическая химия наполненных полимеров. 2-е изд. М., Хи-
мия, 1977. 303 с.
50. Кулезнев В. Н. — В кн.: Смеси и сплавы полимеров. Киев, Паукова думка,
1978, с. 24—38; Кулезнев В. Н., Шершнев В. А. — Каучук и резина, 1977,
№ 11, с. 56—63.
51. Кулезнев В. Н., Кандырин Л. Б., Крохина Л. С., Куканова Е. Ф.— Коллоид,
ж., 1971, т. 33, с. 539.
52. Кулезнев А. И., Кандырин Л. Б., Клыкова В. Д. — Коллоид, ж., 1972, т. 34,
№ 2, с. 231—234.
53. Кулезнев В. Н. — В кн.: Многокомпонентные полимерные системы. Пер. с
англ./Под ред. А. Я. Малкина и В. Н. Кулезнева. М., Химия, 1974, с. 10—60.
54. Кулезнев В. Н., Клыкова В. Д., Чернин Е. М., Евреинов Ю. В. — Коллоид,
ж., 1975. т. 37, № 2, с. 267—272.
55. Kuleznev V. N.. KUkova V. D., Dogadkin В. A. — In: IV Conference Europenne
des Plastiques et des Coautchoucs. Resumes des Communications, Paris, 4—
7 VI, 1974, p. 35.
56. Виноградов Г. В., Малкин А. Я., Кулезнев В. Н., Ларионов В. Ф. — Коллоид,
ж., 1966, т. 28, № 6, с. 809—813; Кулезнев В. Н. и др. Коллоид, ж. 1965,
т 27 с 540-^—545
57. Липатов Ю. С., Шумский В. Ф., Лебедев Е. Ф. — ДАН СССР. 1979, т. 244,
№ 1, с. 148—151.
58. Кулезнев В. И., Клыкова В. Д„ Васильченко Е. И., Берлин А. А., Межиков-
ский С. М. — Коллоид, ж., 1976, т. 38, № 1, с. 176—180; Plaste u. Kautschuk,
1977, Bd. 24, № 2, S. 96—97.
59. Ушанов С. М., Плавник Г. М., Нанушьян С. Р. — Высокомол. соед., 1979,
сер. Б, г. 31, № 10, с. 735—738.
60. Натов М. А., Джагарова Е. X. — Высокомол. соед., 1966, т. 8, с. 1835—1840.
61. Акутин М. С. — Пласт, массы, 1971, 0 1, с. 36—37.
62. Кербер М. Л. — Пласт, массы, 1971, № 5, с. 59—66.
63. Соголова Т. И., Акутин М. С., Цванкин Д. Я., Кербер М. Л. — Высокомол.
соед., 1975, сер. А, т. 17, № 11, с. 2505—2511.
64. Атанасова Ш К, Кербер М. Л., Акутин М. С.—Пласт, массы, 1977, № 8,
с. 13—14.
65. Портной К- И., Бабич Б. Н. Дисперсноупрочненные материалы. М., Метал-
лургия, 1974. 199 с.
66. Cragg L. Н., Bigelow С. С.—J. Polymer Sci., 1957, v. 24, р. 429—438.
67. Dondos A., Rempp Р., Benoit Я. — Polymer, 1972, v. 13, p. 97—105.
предметный указатель
Адгезия 151, 165, 262 сл.
«Антипластнфикация» 267
Аррениуса уравнение 194
Ассоциация макромолекул 40 сл., 48
Бенз ил целлюлоз а 54
Блок-сополимеры 22, 41, 58, 127, 142 сл.,
170, 181, 192, 211, 233
и межфазный слой 163
фазовая структура 165
Брагау теория 239
Бутилметакрилат 20
Вальцевание 144, 218
Баноэна теория 114
Верхняя критическая температура смеше-
ния 12, 17 сл., 59, 67
Взаимодиспергирование 115
Взанмодиффузия 292
Взанморастворимость 7 сл., 20, 61, 66, 74,
115, 131, 179, 288
диаграммы 11 сл.
Взв нм ©усиление 242, 258, 266 сл.
Волновое число 90, 102, 104
Волокнистые структуры 95, 103 сл., 116,
144, 146. 226
Волокнообразование 146
Воспламеняемость 190
Вулканизаты 77, 233 сл., 242. 248, 257, 280,
292
механические свойства 271
нсиаполнениые 139
прочность 265
утомление 141, 260
Вулканизация 272 сл.
радиационная 277 сл.
серная 276
Вулканизующие агенты 254, 273
Вынужденноэластическое состояние 236 сл.
Высокоэластическое состояние 190, 213
Вязкость 151, 268, 287
динамическая 195
комплексная 200
и молекулярная масса 45, 195
и напряжение сдвига 222
ньютоновская 194
однофазной смеси 196
относительная 63
расплавов 195 сл.
растворов 49, 67
смесей полимеров 48, 215 сл., 268 сл.
и состав смеси 51. 195
и температура 285, 295 сл.
ударная 130, 140, 238, 240
удельная 58, 67
фаз 101 сл.
и характер расслаивания 71
характеристическая 40, 44, 58
эффективная 75, ИЗ
Вязкоупругие среды 92, 93. 101
Вязкоупругость 95, 118, 194, 207
Гелеобразование 249
Гельфанда теория 156. 162
Гесса закон 23
Гиббса треугольник 256
Гиббса энергия 15
Гильдебранда теория 11, 32 сл.
Гильдебранда уравнение 161
Гистерезис 126, 149, 289
Глаголева метод 138
Гордона— Гэйлора уравнение 178
Горючесть смеси полимеров 190 сл.
Гриффита теория 240
Гута уравнение 199
Гуттаперча 54
Двухфазные системы 8 сл., 125 сл.
Деоая теория 69
Декстран 96, 97
Деформация 111
выпуждснноэластическая 12У,
238 сл.
кинетика 85
критическая 86
в межфазном слое 194, 226
механизм 190 сл.
и предел текучести 192
сдвига 112
смесей 122
236,
сплошных сред 82
эмульсий 82 сл., 98 сл.
Диаграмма (ы)
Зимма 42
треугольная 256
фазовые 286
Дибутилфталат 263
Диметилфтал ат 263
Динамическая выносливость 258
Диспергирование 70 сл., 90. 98, 104
Дисперсность 128, 138
Днсперсноупрочиенные материалы 294
Дифракция 43
Дифференциальный термический анализ
132
Диффузия 151, 283
газов 185 сл.
жидкостей 185 сл.
макромолекул 155
Долговечность полимера 243
Домены полистирола 144. 156. 164, 234
Дробление частиц 141
Дублированные слои 277
298
Желатин 96, 97
Жидкости ньютоновские 82 сл.
Зимма диаграммы 42
Идеальные смеси 7
Износостойкость 258
Изобарно-изотермический потенциал 7
Изоморфизм 22
Индекс
смешения 111 сл.
течения расплава 222
Иоффе теория 240
Истинные растворы 38 сл.
Кавельери — Акера принцип 138
Капсюлироваиие 117
Каркасность 231
Каучук(и) 19 сл., 32 сл., 53, 63, 106, 118 сл.,
127 сл., 133 сл., 144 сл., 167 сл.,
177 сл., 190 сл., 214 сл., 225, 232,
245, 254 сл.
натуральный 21, 27 сл., 44, 54, 63, 106,
127 133
Кернера уравнение 200, 202, 205
Коагуляционные структуры 146
Коалесценция 96, 103, 112, 200, 234, 292
Кокса теория 87
Коллоиднодисперсное состояние 210
Коллоидные системы 38 сл., 125 сл.
Комплексный модуль 126
Конденсационные структуры 146
Концентраторы напряжений 231, 249
Коэффициент
вязкости 106, 2«5
диффузии 185 сл., 273, 284
изотермической сжимаемости 14
межфазного натяжения 156
нестабильности 89, 1U1
проницаемости 186 сл.
Пуассона Ж, 201, 239
седиментации 45
теплового расширения 14, 239
усиления 147
устойчивости 282
утомляемости 266
Эйнштейна 200, 201
Кратон 101, 211
Кристаллизация 49, 126, 145, 182
Кристаллические полимеры 188, 226
Кина — Марка — Хаувинка уравнение 58
Куэтта аппарат 107
Киэттовский поток 83, 86 сл., 92
Латексы 139
Макрорасслаивапие 287
Макротрещины 237 сл.
Марона теория 8
Матричная структура 117
Межблочные химические связи 104
Межмолекулярные связи 248, 277
Мс?жфазиое натяжение 60, 68 сл., 84 сл.,
s 97, 114 сл., 156 сл., 162, 199, 235»
285
и концентрация смесн 69
и межфазный слой 69
и эмульгирование 68 сл.
Межфазный слой 83, 126, 152, 182, 193, 235,
285 сл.
в блок-сополимерах 163
Межфазный слой
деформация 194, 226
и межфазное натяжение 155 сл.
и пластификатор 263
плотность 165
и растворитель 163
релаксационные свойства 227
состав 163
толщина 161, 167 сл., 193, 283
и химические связи 164, 234
и энергия межмолекулярного взаимо-
действия 128
Метод
взвешивания 137
визуального измерения параметров 136
дифференциального термического ана-
лиза 132, 177
исследования структурных изменений
полимера 39
малоуглового рассеяния 97
машинного счета 138
обращенной газовой хроматографии 34
определения параметров по составу
контактирующих фаз 33 сл.
определения термодинамических па-
раметров взаимодействия полиме-
ров 33 сл.
планирования эксперимента 257 сл.
полимерного растворителя 41, 43, 45
радиотермолюмииесценции 133
расчета по плотности энергии когезии
35
световой микроскопии 133
Скотта 33
формальной стереометрии 137
электронной микроскопии 133
Микрогель 55, 224, 236
Микронеодиородности 78, 134, 186, 231 сл.»
247
Микроразрывы 113
Микрорассланваиис 22, 53
Микроструктура 134
Микротрещнны 237 сл., 243
Микроэмульсии 117, 171, 283
Многокомпонентные системы 254 сл.
Модуль
потерь 196, 288
растяжения 203
релаксационный 227 сл.
сдвига 197 сл., 199, 203
упругости 121, 126, 148 сл., 194, 200 сл.,
212, 289 сл.
Молекулярная масса 19. 56 сл., 71
и вязкость 195
и концентрация 56
и коэффициент расширения 16
Молекулярно-массовое распределение 17,
56 сл., 196, 214 сл., 219, 234, 288 сл.
Морозостойкость 258
Муни пластометр 220 сл.
Муни уравнение 199. 204
Мутность 51. 142, 189
Набухание 58, 133, 147, 185. 277 сл.
Наполнение 268
вязкость смешиваемых полимеров 268
порядок введения наполнителя 270
распределение наполнителя 271
сродство полимеров к поверхности на-
полнителя 269 сл.
Наполненные полимеры 170. 185. 193. 230
Наполнители 171. 184, 189. 193. 199. 226,
254. 268
Напряженис(я)
внутренние 209
при изгибе 293
при растяжении 77, 139, 193
релаксация 225, 248
сдвига 59, 62, ПО, 118, 223
299
Натяжение межфазное 60, 68 сл., 84 ел.,
97, 199, 235, 285
Искриста ллизующиеся полимеры 127
Ненаполнсиные вулканизаты 139
Нсиаполиеиные полимеры 193, 244
Неоднородности системы 249
Несовместимые полимеры 41, 54, 95 сл.,
154, 182, 266
Нижняя критическая температура смеше-
ния 12, 17 сл., 59, 67, 285
Нильсена уравнение 201, 204
Ньютоновские жидкости 82 сл.
Ньютоновские капли
деформация в потоке 93
миграция в потоке 93
в ньютоновской среде 92 сл.
ориентация в потоке тока 93
разрушение в потоке 93
Обращение фаз 118, 146 сл., 185. 204, 211.
215 СЛ., 223, 249
и адгезия 151
и вязкость 151
и вязкоупругость 149
и диффузия 151
и модуль упругости 150
и проницаемость 151
и смачивание 1Б1
и электропроводность 151
Однофазные смеси 197 сл.
вязкость 215 сл.
модуль упругости 206 сл.
Озоностойкость 152, 258
Окклюзия пластмассы 140
полимера 214
Олеофилизация 269
Олигомеры 34, 99, 153
растворимость 21
смешение 19
Опалесценция 189, 283 сл.
эмульсий 70
Оптическая плотность 31, 60, 287
Оптические свойства полимеров 189 сл.
Осмотическое давление 65
Отслаивание 191, 233, 239
Параметр взаимодействия Флори — Хаг-
гинса 157
Плавление смеси полимеров 182 сл.
Планирование эксперимента 257 сл.
Пластификаторы 189, 254, 261 сл., 293
Пластификация 261 сл.
Пластмассы 239, 293 сл.
ударопрочные 139
хрупкие 237
Пластограф Брабендера 220 сл.
Пластометр Муни 220 сл.
Пленки 18, 20. 30. 43, 126 сл., 129. 144. 184.
193. 213, 216. 249
Плотность смеси полимеров 184, 286
Поверхностная фаза 152
’Поверхностно-активные вещества 88
Поверхностное натяжение 69, 71, 153, 159
Поверхностный слой 152 сл.
Поверхность раздела фаз 108, 134, 152
Показатель посломления 142
Ползучесть 225 сл.
Полиакриламид 92
Полиакрилаты 54
Полиакрилонитрил 67, 95, 143, 211, 217 сл.
Полиамиды 144. 219
Полибмтадиен 19. 54. 72, 99, 153, 163 сл.,
197, 234, 255. 270. 277
мыс-Полнбутадиен 13. 53, 180, 183, 189
Полибутилен 29. 101 сл., 127 сл., 133.
' 166 с л., 185. 206 сл.
Поли-н-бутилметакрилат 65
Поливинилацеталь 27
Поливинилацетат 17, 19, 22 сл., 54, 61 сл.,
159, 179 сл., 191, 227
Поливинилбутираль 185
Поливинилиденфторид 22
Поливинил метиловый эфир 18
Поливинилиитрат 179
Поливиниловый спирт 95 сл.
Поливиннлнирролидон 44
Поливинилхлорид 11, 19, 22, 24, 50, 61, 67,
75, 129, 134 сл., 141 сл., 151, 177 сл.,
190, 211 сл., 245 сл., 255, 267
Поли-2,6-диметнл-1,4-окснфенилен 135
Полнднметилсилоксаи 11
Полидисперсность 102
Поли дифениловый эфир 19
Полиизобутилен 11, 14, 29, 32, 39 сл., 46 сл..
64 сл., 121, 128, 133, 118 сл., 191
Полиизобутилметакрилат 55
Полиизопрен И сл., 19, 44, 53 сл., 63. 287
^ыс-Полиизопрен 13, 19, 53, 183, 189, 197
Полиизопропилен 20 сл., 60, 63. 72, 134,
143, 153, 166 сл., 206 сл., 214
Поликапролактон 18, 178
Поликарбонаты 236
Полимеризация 140
Полиметакрилаты 54
Полиметилакрнлаты 44, 54
Полиметилвиннловый эфир 19, 77, 179
Полиметил метакрилат 11, 17, 19 сл., 28 сл.,
43 сл., 50 сл., 59 сл.. 67, 71, 103.
129, 134, 144, 148 сл., 159 сл., 16о сл.,
180, 184, 214 сл., 223 сл., 236, 245 сл.»
267 сл., 290
Полиметилсилоксан 99 сл.
Поли метилстирол 285
Поли-а-метилстнрол 43
Поли-ж-метилстирол 54
Поли-о-метилстирол 54
Поли-я-мстилстирол 54
Полиолефины 144
Полиоксиметилен 144, 219, 290
Полиоксипропилен 15
Полиоксипропилеигликоль 267
Полиоксипропилеидиол 284
Полиоксиэтилен 15
Полипропилен 22, 29, 33, 49, 66 сл., 126 сл.,
148 сл., 183 сл., 191, 219. 245. 290
Полистирол 11 сл., 18 сл.. 27 сл., 38 сл..
46 сл., 54 сл., 59 сл.. 95, 103, 117,
128 сл., 133 сл., 146, 162 сл., 177 сл.,
184, 192. 213 сл., 227 сл., 263 сл.,
285
и молекулярная масса 42
и размер макромолекулы 42
степень ассоциации 46
ударопрочный 140, 149. 170, 229 сл.,
236 сл., 268
Политрихлорбутадиен 22
Полиуретаны 95. 211, 217 сл.
Полифенилсноксид 188
Полихлоропрен 21, 22, 30, 127, 159, 2/0,
278
Полиэтилакрилат 54
Полиэтилен 14 сл., 21, 49, 117, 121, 126 сл.,
145 сл., 183 сл., 197. 212 сл., 224.
245 сл., 263 сл.
Полиэтиленгликоль 44
Полнэтиленоксид 215
Полиэтилентепефталат 191 сл.. 127, 151
Поропласты 201
Предел текучести 192, 194, 231, 256
Привитые сополимеры 64, 145, 167
Пригожина теория 14
Принцип
«бинарной» аддитивности 260
суперпозиции 226
температуро-временной эквивалентно-
сти 226
300
Проницаемость 151
Пространс I веннонапряженнос состояние
239
Противостарителн 254
Прочность 249
вулканизатов 265
невулканизованной смесн 231
смеси полимеров 230 сл.
и состав смесн 266
Псевдопластнчные среды 92, 93
Пуазейлсвский поток 83, 86 сл., 92
Пуассона коэффициент 200, 201, 239
Радиотермолюминесценция 133, 278
Разветвленность макромолекул 55
Разрушение
смеси полимеров 230 сл.
время 246
механизм 230
энергия 241, 242
Расплавы полимеров 94 сл., 103, 180, 214,
290
индекс течения 222
смешение 184
турбулизация 98
Рассеяние света 134, 189, 284 сл.
Расслаивание 9 сл., 18, 38 сл.. 47, 52 сл.»
166, 191, 197, 22b, 233, 242, 278,
285 сл.
время 53
и добавки 61 сл.
и концентрация 38 сл.. 53, 67
механизм 55, 76
и молекулярная масса 56 сл.
и молекулярно-массовое распределение
56 сл.
напряжение сдвига 59
и объем слоев 64 сл.
предел 55, 56. 63
и природа полимеров 53 сл.. 58 сл.
и соотношение полимеров 56
сополимеров 10 сл.
и состав слоев 64, 67
и структура полимеров 53
и температура 59 сл.
Растворение
вязкоупругих жидкостей 7
полимера в полимере 7 сл., 21
селективное 122
Растворимость 21
Растворители 10 сл., 26, 38 сл., 144, 213
ацетон 61, 67
балластный 39 сл.
бензол 44 сл., 53, 62 сл., 71, 187
бути л ацетат 71
н-гсксан 285
декалин 60 сл.. 70
дихлорэтан 44
диэтиловый эфир 285
ксилол 60
о-ксилол 59
метил эти л кетон 149, 214
jw-okch бензол 59
полимерный 39 сл.
природа 58 сл., 180
толуол 38 сл., 60 сл.
хлороформ 40 сл., 47, 71
циклогексан 284
Растворы полимеров 94 сл.
вязкость 49
диспергирование 70
истинные 38 сл.
коллоидные 38 сл.
разбавленные 39
и свойства 67
трехкомпонентные 72 сл.
эмульгирование 72
Растяжение ПО, 241
Рауля закон 45, 153
Ребиндера эффект 209
Резниковского уравнение 266 ~
Релаксационные переходы 177, 182, 228
Релаксация напряжений 225 сл., 248. 271,
286
время 86, 155, 284
скорость 225
и состав смеси 226
Розиваля метод 138
Рэлея теория 89
Сажа 269 сл.
Сантокюр 274
Световая микроскопия 133
Светорассеяние 39, 45 сл., 74
и концентрационная зависимость 39,
40 сл.
и молекулярная масса 44, 50
в разбавленных растворах 39
уравнение 40
Седиментация 44
Сера 273
Серфа теория 87
Симхи — Бойера теория 182
Скорость
ветвления критическая 24U
диффузии 185
дробления частиц 113
кристаллизации 126
набухания 147, 185
плавления 126
сдвига 110, 118
Скотта системы 33
Скотта теория 8
Скотта уравнение 31
Слоистая структура 116, 217
Смачивание 151
Смеси полимеров
вулканизация 272 сл.
вязкость см. Вязкость
горючесть 190 сл.
дисперсии 115
диффузия газов и жидкостей 185 сл.
истинные растворы 38 сл.
коллоиднодисперсное состояние 210
коллонднохнмичсская структура 125 сл.
коллоидные растворы 38 сл.
кристаллизация 49, 182 сл.
критические явления 282 сл.
межфазное натяжение 68 ст.
механизм деформации 190 сл.
микроструктура 134.
многокомпонентные 254 сл.
модели 201 сл.
модуль упругости 206 сл.
и молекулярная масса 39 сл.
наполнение 268 сл.
однофазные 197 сл.
оптические свойства 189 сл.
плавление 182 сл.
плотность 184
ползучесть 225 сл.
прочностные свойства 177 сл , 230 сл.
разрушение 230 сл.
расплав 98 сл., 180, 290 сл.
расслаивание см. Расслаивание смесн
полимеров
релаксация напряжения 225 сл.
свойства 119 сл.
стеклование 177 сл.
структура см. Структура смеси поли-
меров
термическая устойчивость 190 сл.
термодинамические параметры 33 сл.
тип структуры 49 сл.
трехкомпоиентиые 72 сл.
фазовая структура 82
фазовые диаграммы 59
фазовые структуры 114 с л.
301
Смеси полимеров
фнзнко-механнческие свойства 177 сл.
физические свойства 177 сл.
эмульгирование 68 сл.
эмульсии см. Эмульсии полимеров
Смешение 7 сл.» 180, 207, 271
верхняя критическая температура 12,
17 сл.
время 102, 105
идеальное 82, 107
изобарно-изотермический потенциал 7
индекс 111
интенсивность 112
критическая температура 8, 12
ламинарное 107, 112
механизм 82 сл.
нижняя критическая температура 12,
17 сл.
олигомеров 19
равномерность 111
расплавов 184
в смесителях 113
теории 8 сл., 106 сл., 113
теплота 7, 13
термодинамические параметры 8, 24,
33 сл.
эффективность 107
Смилла правило 12
Смолы 128, 130, 140. 151
АБС 240
фен о лоформ альдегидная 185, 257
эпоксидная 241
Совместимость полимеров 12, 184
Совместимые полимеры 54, 59, 266
Совулкаинзация 272 сл.
Сополимеры 20, 22, 29 сл., 79, 180, 277
и параметр растворимости 11
расслаивание 10 сл.
стирола 18
Сопротивление
раздиру 257, 279
утомлению 258 сл.
Стабилизаторы 189, 254
Стеклование 177 сл.
Стеклообразные полимеры 188
Структура смесей полимеров 39, 82 сл.,
125 сл.
волокнистая 95, 144, 244, 283
зернистая 286
коагуляционная 234
конденсационная 234
размер частиц 136 сл.
слоистая 286
тнп 49
форма частиц 136 сл.
Тзкаянаги уравнение 200
Такаянаги — Каваи двухфазная модель 203
Температура
кристаллизации 126, 183
плавления 126, 183
расслаивания смесн 59
смешения 8, 12, 17 сл., 59, 67, 232, 278,
283
стеклования 50, 77, 127 сл.. 135, 177 сл.,
207, 213, 228, 232, 268
текучести 232
Теплота
активации 223, 288
смешения 7, 13
Гермодеструкция 190
Термодинамика растворения полимера в
полимере 7 сл.
Термодинамическая совместимость поли-
меров 18
Термодинамические параметры взаимодей-
ствия полимеров 33 сл.
Термодинамический потенциал смешения 8
Термореактивные полимеры 235
Тсрмоэластопласты 128, 142, 182, 233
Тиурам 274
Томотики теория 89, 224
Томпа теория 8, 57
Трехкомпонентные смеси
вязкость 268 сл.
наполнение 268 сл.
иевулканнзованные 256
пластификация 261 сл.
прочность 266 сл.
свойства вулканизатов 257
сопротивление утомлению 258
фазовые диаграммы 72
Трехмернонапряженное состояние 241
Трещины 190 сл., 237, 240
Трнблок-сополнмеры 168, 182
Тэйлора теория 84 сл., 96
Тэйлора уравнение 199
Ударопрочные пластмассы 139, 230
Ударопрочные полимеры 140, 233, 235, 236
Удлинение при разрыве 147, 232» 257 сл.
Уравнение
Аррениуса 194
ВЛФ 229
времени до разрушения полимера 246
Гильдебранда 161
Гордона — Тэйлора 178
Гута 199
деформации полимера 192
диффузии 159
изменения энтропии
Кернера 200, 202, 204
критической концентрации 56 сл.
Куна — Марка — Хаувннка 30, 44. 58
Муки 199, 205
Нильсена 201, 204
растворимости полимера в полимере
20
Резинковского 266
светорассеяния 40
Скотта 31
Такаянаги 200
Тэйлора 199
условий расслаивания полимеров II
Фокса 178
Эйнштейна 198
Усиление полимеров 147, 183 сл., 226, 231.
242 сл.
механизм 239
мягким низкомодульным полимером
235 сл.
твердым полимером 231 сл.
Усталостная выносливость 243, 267
Утомление вулканизатов 141, 242. 260
тройной смеси 258
Фазовые диаграммы 8 сл., 11, 16, 25, 28 сл.»
59, 72, 286 сл.
Фазовые (ая) структуры (а) 82
блок-сополимеров 165
волокнистая 116
и деформация капель 98 сл.
количественные параметры 136 сл.
кольцевые 117
матричная 117
методы определения параметров 136 сл.
микроэмульсия 117
обычная дисперсия 116
и разрушение капель 98 сл.
слоистая 116
типы 114 сл.
формирование 98 сл.
Фазовый переход 288
Фенофор Б 257
Флори параметр 290
Флори теория 8 сл., 14
Флори — Скотта теория 8, 14, 24, 25, 33
302
«3, 215 сл., 222, 234,
Флуктациониая сетка
277
Фокса уравнение 178
Хаггинса теория 8
Химические связи 247
Хлоркаучук 30
Хлорсульфополиэтилен
Хрупкие полимеры 238
Хрупкость 146
сл.» 277
278
Целлюлоза
ацетат 24, 27
ацетобутнрат 22, 290
ди ацетат 67
нитрат 19, 24, 27, 54
Эйнштейна коэффициент 200, 201
Эйнштейна уравнение 198
Электронная микроскопия 133
Электропроводность 151
Эмульгирование 68 сл., 72
Эмульсин 49, 64, 68, 82 сл., 198, 218, 285 сл.
вязкоупругих жидкостей 94 сл.
механические свойства 287
неиыотоиовских жидкостей 91 сл.
несовместимых полимеров 95 сл.
Эмульсии
'нестабилизнроваиныс 146
ньютоновских жидкостей 83 сл.. 88 сл.
опалесценция 70
на основе водных растворов 97
свойства 52 сл.
твердообразные 146
Эмульсионная полимеризация 55
Энергия
активации 149, 195, 223, 246, 284 сл.
когезии 32, 54. 166 сл., 226, 244 , 255,
285 сл.
межмолекулярного взаимодействия 128
разрушения 113, 241 сл.
свободная 24, 143, 164
смешения 15, 23, 24
Энтальпия 13, 14
Энтропия 7, 12 сл., 50, 154 сл., 285
Этилцеллюлоза 60, 95
Эффект
антипластификацнн 293
взаимоусилення полимеров 244 сл., 258,
266’сл.
межструктурной пластификации 293
немонотонности изменения свойств 267
обращения фаз 204
огибания дисперсных частиц 187
Ребиндера 209
смешения, тепловой 13
усиления полимера 183, 231, 243