/





















































































































































































































































































































































































































































































Текст
Э. ЛЕМ/ЮВ
3. АЕШСВ
МЕЖОИЗНЫЙ
К4И/1ИЗ
Перевод с английского
канд. хим. наук С. С. Юфита
под редакцией
д-ра хим. наук Л. А. Яновской
Москва «Мир» 1987
Содержание
Предисловие редактора перевода 5
Предисловие авторов ко второму изданию 7
Предисловие авторов к первому изданию . 9
Наиболее часто встречающиеся обозначения 11
Глава 1. Ионные пары и экстракция ионных пар 12
1.1. Введение. Природа межфазного катализа 12
1.2. Ионные пары в органической фазе 16
1.3. Экстракция ионных пар из водных растворов 20
1.3.1. Принципы ,. 20
1.3.2. Влияние растворителя 24
1.3.3. Влияние ониевого катиона 27
1.3.4. Влияние аниона 30
1.4. Краун-эфиры, криптанды и.другие соединения, образующие хе-
латы,. как экстрагеиты 37
1.5. Анионный обмен, между твердой и жидкой фазами , 41
Глава 2. Механизм межфазиого катализа 44
2.1. Изучение'механизма реакций в различных условиях ... 44
2.1.1. Механизм реакций в нейтральных средах .... 44
2.1.2. Механизмы реакций, .проходящих в присутствии гидрок-
сидов щелочных металлов . 54
2.1.3. Механизм реакций в присутствии других оснований . . 66
2.2. Эмпирическая оценка эффективности катализаторов , . .67
2.3. Необычные катализаторы и катализаторы на полимерных
носителях ...".., 77
Глава 3. Практическое использование межфазного катализа 80
3.1. Общие экспериментальные методы 81
3.1.1. Обычные катализаторы , . 81
3.1.2. Условия проведения реакций 88
3.1.3. Общие замечания о проведении реакций в присутствии
щелочей 95
3.1.4. Катализаторы, связанные с полимерами .... 97
3.1.5. Хиральные катализаторы 102
3.2. Получение галогенсодержащих соединений . .... 109
3.2.1. Обмен галогенов 109
3.2.2. Обмен фторидов ....-.- 114
3.2.3. Галогениды из спиртов, эфиров и диазометанов . . 116
3.3. Получение нитрилов 119
3.4. Получение сложных эфиров 124
3.4.1. Эфиры карбоновых кислот 124
; 3.4.2. Эфиры других кислот 134
3.5. Различные реакции замещения 137
3.6. Тнолы и сульфиды 142
3.7. Получение простых эфнров 148
3.8. N-алкилирование 160
3.9. С-алкилнрование активированных связей С—Н .... 172
3.9.1. Алкилярование арнлацетоннтрилов 173
3.9.2. Алкилнрование других активированных цнанатов и изо-
цианатов 181
3.9.3. Алкилнрование малонового зфнра 183
3.9.4. Алкилирование бензилкетонов, арилуксусных эфнров н
амидов 185
3.9.5. Алкнлирование других соединений, содержащих, дважды
• активированные СН-связи 190
3.9.6. Алкилнрование простых карбонильных и карбоксильных
производных . . . - . ■. . ■ -. 192
319.7. Алкилирование. углеводородов - . - " 895
3.10. Алкйлнрованне амбндентных анионов 196
3.11. Изомеризация аНД}-обмен .,, - . , ... ..,.-..,,-.". . 214
3.12. Присоединение по непредельным- связим С—С . . .' . 217
3.12.1. Присоединение к. ацетиленам .• .-.'". - .-1* .£.* 217
3!12j2. Присоединение по Михаэлю . . ,. . ... . .' * 219
3.12.3. Присоединения к неактив ировашшм, двойным связям ' 226
3.13. Присоединение по связям.С=.О « C=N . . . ... . ;.. I . 228
3". 13. i. Бензоиновая конденсация ■.. ... . ;■• . .' .' " . 228
ЗЛ3.2. Конденсации, а ль дольного типа .■ . ■;. •. '*.'.:■ . 228
ЗЛЗ.З. Другие типы реакций i .."..".". 232
3.14. в,3я^и)»н}к>ва.ииелг;:. ^ , .:v -.,-. . п . ■■■■:".' ' ■: .".." Д! 237
3.15. Реакции гидролиза. .. ^ .. .. .-.■;•.■. .•■-... 244
3I15.L Различные реакции гидролиза .;.--. ;" . . . "244
3.15.2. Омыление сложных эфнров 246
3.16. Генерирование и превращение фосфониевых в сульфонневых
' илидов' . .' 250
3.16Л. Реашдая Виттига 250
3.16.2. Получение олефтюв ■ по Хорнеру (активированная
связь РО) ...... . 257
3.16.3. Реакции .сульфонилидов и оксосульфонилйдов . ... 259
3.17. Нуклеофильное ароматическое замещение . . ... , . 265
3.18. Различные реакции, проходящие в присутствии оснований, ие
расЬмотренные в других разделах 272
3.18.1. Перенос- диазогруппы ." .' .'.■.'. . . . 272
3.18.2. Y-Элиминирование , - ... 272
3.18.3. Получение ангидридов и фторангидридов кислот . . 273
3.18.4. Перегруппировки .... -. . . . ,. ' 274
3-.18.5. Радикальные реакции . . . .... . . . 277
3.18.6. Реакции с -Гетрагалогеаидами углерода .... 277
3.18.7. Некоторые реакции соединений серы . . ■ ■. '. . 278
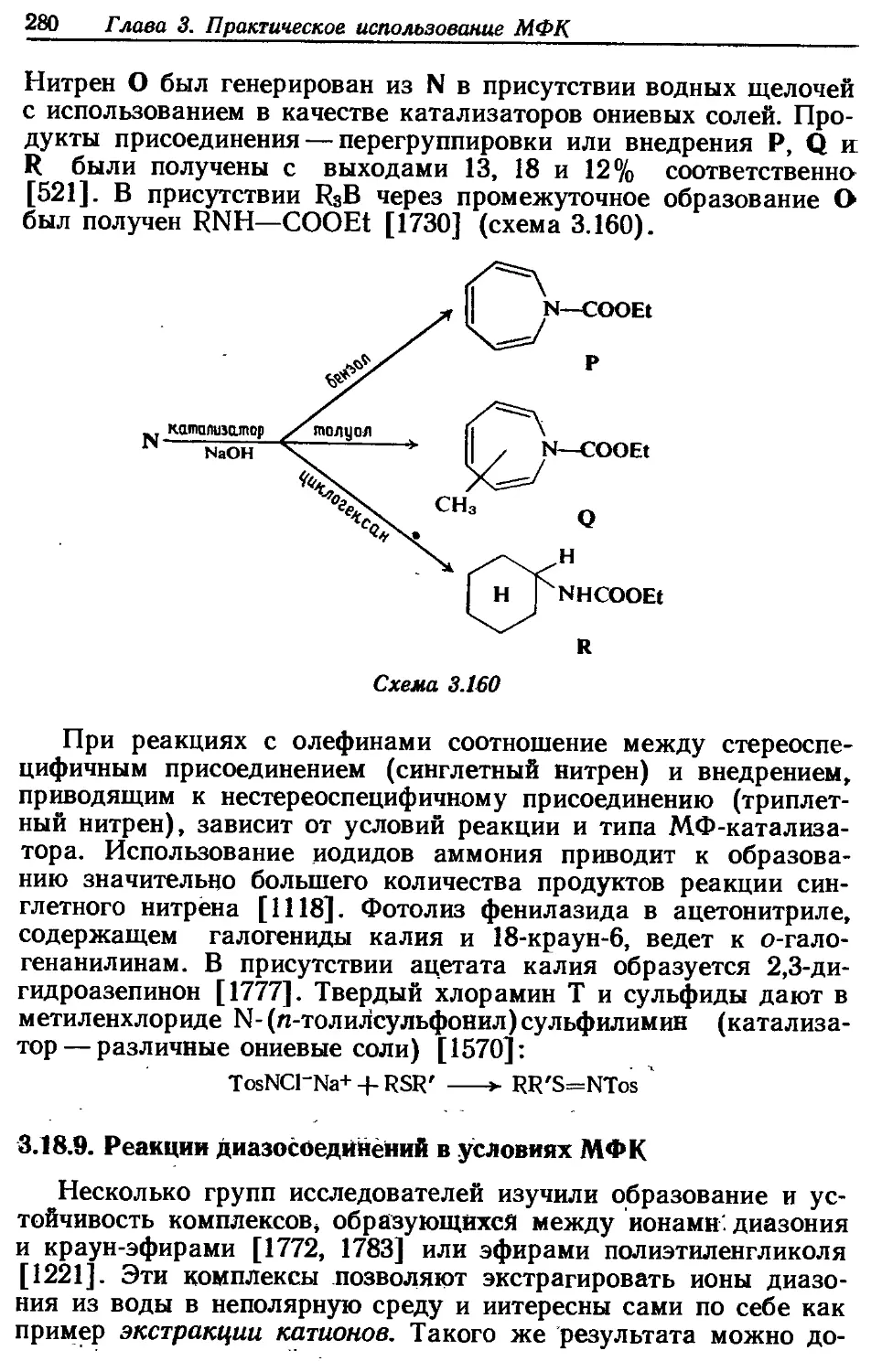
3.18.8. Генерирование нитренов . . ....... 279
3:18.9. Реакции диазосоедйнений в условиях МФК • . • 280
3.18.10. Получение гидразонов и использование нх в МФК 282
3.18.11. Электрохимия ; 283
З.Ю. Применение МФК в химии металлоорганических соединений 283
. 3.20. а-Элимннир'ование . . ■ ... 289
3.20.1. Реакции образования и присоединения дихлоркарбена .. 289
3.20.2. Другие реакции дихлоркарбена и трихлбрметильного
аннона - . ' ■ 320
3.20.3. Дибромкарбеи . . •. . ....... 334
3.20.4. Прочие днгалокарбены. ,. . ... . •. . 349
3.20.5. Другие смешанные карбены 357
Содержание 485
3.21. Реакции восстановления 365
3.21.1. Комплексные гидриды и днборан 365
3.21.2. Другие восстанавливающие реагенты ..... 374
3.22. Реакции окисления 378
3.22.1. Окисление перманганатом 379
3.22.2. Роль пероксида водорода в межфазных реакциях 387
3.22.3. Реакции с супероксндом калия 390
3.22.4. Хроматы 399
3.22.5. Гипохлорнт 400
3.22.6. Калийгексацнаноферрат(Ш) 403
3.22.7. Пернодат 404
3.22.8. Надуксусная кислота 406
3.22.9. Кислород как дкислитель 407
3.22.10. Другие реагенты-окислители' 409
Литература . . . . . 411
Работы, опубликованные на русском языке . . . . . . . 462
Предметный указатель •. '-. . . . . . . . . . . . 467
Наиболее часто встречающиеся обозначения
Ец «тбутил
Hex «-гексил
Hep и-гептил
Oct и-октил
Pent w-пентил
Prop (Рг) к-пропил
МФК межфазный катализ
Q+ четвертичный аммониевый катион
ТЭБА бензилтриэтиламмонийхлорид
ТЭБА-бромид бензилтриэтиламмонийбромид
адоген 464 "1 метилтриоктиламмонийхлорид
аликват 336 J (технический)
Глава 1
Ионные пары
и экстракция ионных пар
1.1. Введение. Природа межфазного катализа
Межфазным катализом (МФК) называют ускорение реакций
между химическими соединениями, находящимися в различных
фазах. Как правило, это реакции между солями,
растворенными в воде или присутствующими в твердом состоянии, с одной
стороны, и веществами, растворенными в органической
фазе, — с другой. В отсутствие катализатора такие реакции
обычно протекают медленно и неэффективны или не происходят
вообще. Традиционная методика проведения реакций включает
растворение реагентов в гомогенной среде. Если используется
гидроксилсодержащий растворитель, реакция может
замедляться из-за сильной сольватации аниона. Побочные реакции с
растворителем иногда снижают скорость еще больше. Часто
превосходные результаты дает применение полярных апротон-
ных растворителей. Но они обычно дороги, трудно отделяются
после реакции и могут вести к возникновению экологических
проблем при широкомасштабном использовании. Кроме того,
в некоторых случаях, например при О- или С-алкилировании
амбидентных анионов, полярные апротонные растворители
могут в результате преобладающего образования нежелательных
продуктов в заметной степени подавлять, а не промотировать
реакцию.
Межфазный катализ делает возможным или ускоряет
реакции в малополярных растворителях между ионными
соединениями и органическими (не растворимыми в воде)
веществами. Наиболее часто используемыми катализаторами являются
ониевые соли или комплексообразователи, которые могут
связывать ионы щелочных металлов и таким образом переводить
их в раствор. Основная функция катализатора состоит в
переносе анионов реагирующей соли в органическую фазу в форме
ионных пар. В апротонных растворителях они фактически не
сольватированы и ничем не экранированы (за исключением,
может быть, их противоионов) и, следовательно, обладают
высокой реакционной способностью.
/./. Введение. Природа межфазного катализа 13
Из этого, ясно, что межфазный катализ имеет значительные
преимущества по сравнению с общепринятыми методами:
— не требуются дорогостоящие безводные или апротонные
растворители;
— обеспечиваются более высокие скорости реакций и/или
более низкие температуры реакций;
— во многих случаях можно использовать более простое
оборудование;
— вместо алкбголятов, амида натрия, гидрида натрия или
металлического натрия можно использовать водные растворы
тидроксидов щелочных металлов.
Кроме того, имеются еще и особые преимущества,
например:
— возможность осуществления реакций, которые не идут в
других условиях;
— изменение селективности;
— изменение соотношения продуктов (например, О- и С-
алкилирования);
— более высокие выходы в результате подавления побочных
реакций.
Хотя большинство реакций, рассмотренных в этой главе,
включает экстракцию анионов в органические растворители
под действием катиона-катализатора, общее понятие
«межфазный катализ» шире. Оно, кроме того, включает в себя
экстракцию катионов и даже нейтральных молекул из одной
фазы в другую с помощью катализатора.
Межфазный катализ — относительно новая область химии,
возникшая на основе исследовательской работы трех
независимых групп. Начало было положено в середине и конце 60-х
годов Макошей, Старксом и Брендстрёмом.
Реакции, включающие межфазный перенос, конечно,
проводились и ранее, и, несомненно, значительное количество таких
реакций «похоронено» в старой литературе [1—3] и особенно
в патентах |[4], причем самый старый, из известных в
настоящее время, датирован 1913 г. Некоторые из первых авторов
вступили в эту область более или менее случайно и,
по-видимому, не интересовались механизмами таких каталитических
реакций. И главное — никто из них не осознавал
потенциальных возможностей и широты нового метода.
Известные в настоящее время методики межфазного
катализа появились в работах Макоши и сотр. [5] в 1965 г. Эти
авторы начали систематическое исследование алкилирования
и других реакций в двухфазных системах, главным образом в
таких, которые содержали концентрированные водные
растворы гидроксидов щелочных металлов. В описании они использо-
14 Глава 1. Ионные пары и экстракция ионных пар
вали термины «каталитические двухфазные реакции»,
«каталитическое алкилирование анионов», «каталитическое
генерирование карбенов» и др. Во многих случаях даже сейчас при
обсуждении механизмов реакций Макоша предпочитает эти
термины. Его работы стали более известны после публикации в
«Tetrahedron Letters» в 1969 г. i[6] сообщения об открытии
генерирования дихлоркарбена. Первые работы Врендстрёма
имели более физикохимический и аналитический характер. Его
первые статьи, относящиеся к МФК, появились в 1969 г. /[7],
затем в 1970 г. последовал ранний обзор |[8], названный им
«Экстракция ионных пар в препаративной органической химии».
Брендстрём использует термин «экстрактивное алкилирование»
для реакций алкилирования в двухфазной системе в
присутствии молярных количеств катализатора.
Термин «межфазный катализ» («phase transfer catalysis»)
был введен Старксом и впервые использован в патентах в
1968 г. [9]. Новый метод проведения реакций и термин
«межфазный катализ» получили признание, по-видимому, после
публикации статьи Старкса в 1971 г. в «Journal of American
Chemical Society» [10]. Впервые были ясно очерчены границы
метода, а сам метод распространен за пределы первоначального
применения, т. е. алкилирования и генерирования карбенов.
Более того, была предложена единая механистическая
концепция для всех этих реакций, что дало сильнейший толчок
развитию работ в этой области. С тех пор появился целый поток
статей, в которых межфазный катализ был использован для
проведения реакций новых типов, таких, как реакции между
жидкой и твердой фазами, реакции под влиянием катализаторов,
«пришитых» к полимерам, а также разработаны новые
каталитические реагенты, например краун-эфиры. Они будут
рассмотрены в следующих разделах.
Термин «межфазный катализ» в настоящее время наиболее
употребителен*. Он используется как в учебниках,
справочниках, рефератах и энциклопедиях, так и в сборниках
органических синтезов. Для того чтобы применять вышеупомянутый
общий механизм к различным реакциям межфазного катализа»
необходимо модифицировать этот механизм**. Макоша
предпочитает исключать некоторые реакции из числа «истинных»
межфазных каталитических реакций и называет их «так
называемыми межфазными каталитическими реакциями» или
«каталитическими двухфазными реакциями». Однако в настоящем
* В отечественной литературе используются также термины, являющиеся
дословным переводом с английского: «катализ фазового переноса» (КФП) или
«катализ межфазного переноса» (КМФП). — Прим. перев.
** Например, депротонирование часто происходит на границе раздела
двухфазной системы.
1.1. Введение. Природа межфазного катализа 15
обзоре авторы предпочитают объединить под названием
«межфазный катализ» все двухфазные реакции между твердыми или
растворенными в воде солями, кислотами, основаниями или
даже нейтральными молекулами и веществами, растворенными в
органических растворителях, которые катализируются ониевы-
ми солями, комплексами краун-эфир/соли щелочных металлов
или сходными комплексными солями. Это позволяет объединять
реакции с одинаковыми каталитическими эффектами или с
одинаковыми катализаторами независимо от того, известен ли их
механизм или нетД Такое название является практически
удобным феноменологическим определением: оно ие учитывает
специфического действия катализатора, а лишь указывает на
наличие двух фаз и катализатора. Реакции, протекающие
исключительно или преимущественно на межфазной границе,
также включены в рассмотрение при условии, что они
ускоряются катализатором. Таких реакций известно очень много.
Может возникнуть вопрос: следует ли включать в
монографию по межфазному катализу мицеллярный катализ? Где
нужно провести границу? Мицеллярным реакциям посвящено много
обзоров '[29—31]. Такие реакции обычно проводятся в
гомогенных или псевдогомогенных фазах. Поэтому они не подходят под
наше определение реакций МФК и, следовательно, здесь, как
правило, не рассматриваются. Однако реакции МФК по
определению, принятому в данной книге, могут происходить с
механистической точки зрения в мицеллах или инвертированных
мицеллах. Хотя это и не является типичной реакцией МФК и
не существует достоверных примеров подобных реакций,
некоторые указания на возможность протекания такого процесса
имеются.
Ранние обзоры по МФК находятся в работе Брендстрёма
«Препаративная экстракция ионных пар» 1[11], в статьях
одного из авторов данной книги '[12, 13], в двух докладах на
конференциях и статьях Макоши [14, 15, 18, 114] ив обзоре Док-
са, включающем рассмотрение других областей применения
четвертичных аммониевых соединений ![16]. В литературе
имеются также более краткие обзоры, вводящие читателя в эту
область, например i[17, 19, 115]. Брендстрём недавно
опубликовал главу о принципах МФК в журнале «Advances in Physical
Organic Chemistry» [112]. Наконец, многие области
применения МФК описаны в недавно появившихся книгах*
«Межфазный катализ в органической химии» Вебера и Гокеля [113]
и «Межфазный катализ, принципы и методики» Старкса и Ли-
отты [116]
* По межфазному катализу см. также кннгн: Яновская Л. А., Юфит С. С.
Органический синтез в двухфазных системах. — М.: Химия, 1982; Юфит С. С.
Механизм мефазного катализа.—М.: Наука, 1984. — Прим. перев.
16 Глава 1. Ионные пары и экстракция ионных пар
у
1.2. Ионные пары в органической фазе
Во многих случаях МФК состоит в экстракций ионных
молекул органическим растворителем или их растворении в нем.
В связи с этим полезно иметь необходимые данные о структуре
и свойствах таких растворов. Полный обзор /этого предмета
выходит за рамки настоящей книги. Однако в данном разделе
будет представлено его краткое качественное изложение. Для
более глубокого ознакомления с физико-химическими
концепциями, методами и полученными результатами читатель может
воспользоваться учебниками по физической, химии, физической
органической химии (например, [21]) или последними
монографиями |[22, 23, 39]. Структура и реакционная способность
карбанионов в ионных парах и карбанионоидных металлоорга-
нических соединениях рассмотрены в обзоре [40] и
специальных монографиях s[41—-43].
Обычные неорганические соли натрия и калия не
растворимы в неполярных органических растворителях. Это верно и
для солей неорганических анионов с небольшими
органическими катионами, например для тетраметиламмония. Подобные
аммонийные соли часто способны, однако, растворяться в ди-
хлорметане и хлороформе. Более того, использование
относительно больших органических анионов может обеспечивать
растворимость солей щелочных металлов в таких
растворителях, как бензол. Например, диэтил-«-бутилмалонат натрия
дает 0,14 М раствор в бензоле, для которого понижение точки
замерзания неизмеримо мало, что говорит о высокой степени
ассоциации. Подобным образом большие ониевые катионы
(например, тетра-«-гексиламмония) делают растворимыми соли
даже небольших органофобных анионов (например, гидроксид-
ионов) в углеводородах. Ионофоры, т. е. молекулы, состоящие
из ионов в кристаллической решетке, диссоциируют (полностью
или частично) на сольватированные катионы и анионы в
растворителях с высокими диэлектрическими проницаемостями.
Подобные растворы в воде являются хорошими проводниками.
В менее полярных растворителях даже сильные электролиты
могут растворяться с образованием растворов с низкой
электропроводностью; это означает, что только часть растворенной
соли диссоциирована на свободные ионы. Чтобы обьяснить такое
поведение растворов, Бьеррум выдвинул в 1926 г. гипотезу
ионных пар. Впоследствии его гипотеза была
усовершенствована Фуоссом '[38] и рядом других исследователей. Ионные пары
представляют собой ассоциаты противоположно заряженных
ионов и являются нейтральными частицами. Стабильность
ионных пар обеспечивается в основном кулоновскими силами,
но иногда этому способствует и сильное взаимодействие с ок-
1.2. Ионные пары в органической фазе
ружающей средой. Ионные пары — это термодинамически
индивидуальные частицы, находящиеся в равновесии со
свободными ионами:
Главное различие между свободными ионами и ионными
парами состоит в там, что растворы, содержащие только ионные
пары, не проводит электрический ток. Таким образом,
измерение проводимости\ позволяет определить содержание свободных
ионов. Что касается криоскопии и измерения давления паров,,
то в этих случаях ионные пары ведут себя как отдельные
частицы. Константы диссоциации ионных пар известны для
многих растворителей. Как правило, при низких концентрациях в-
растворителях с диэлектрической проницаемостью больше 40
находятся главным образом диссоциированные ионы. В
растворителях с диэлектрической проницаемостью ниже 10—15 даже
при высоком разбавлении свободные ионы почти полностью
отсутствуют.
В любом растворителе чем больше ионы, тем больше
степень диссоциации. Например, константа равновесия
образования ионной пары К в нитробензоле равна 80 для хлорида тет-
раэтиламмония, 62 для бромида, слишком мала, чтобы
определить ее для пикрата тетра-«-бутиламмония, но равна 7 для.
пикрата тетраэтиламмония i[26].
Поведение и структура ионных пар и более сложных
комплексов широко изучались такими методами, как кондуктомет-
рия, спектроскопия комбинационного рассеяния, спектроскопия
в УФ-, видимой и ИК-областях, а также методами электронного
и ядерного магнитного резонанса. Эти методы и полученные
результаты описаны в обзоре/[22].
Растворители представляют собой однородные
структурированные субстанции. При контакте между молекулами
растворителя и растворенного вещества имеют место ион-дипольные
взаимодействия. Степень сольватации указывает на
количество таких взаимодействий. Взаимодействие тем больше, чем-
ближе контакт между растворимым веществом и
растворителем. Дипольные, дисперсионные и индукционные
взаимодействия, а также водородные связи действуют совместно с кулонов-
скими силами, и все вместе определяют стабильность и
свойства ионных пар. Поэтому большое значение имеет природа1
как растворенного вещества, так и растворителя. Сольватная.
оболочка уменьшает подвижность и коэффициенты диффузии
как ионов, так и ионных пар. Способность апротонного
растворителя к сольватированию не зависит от диэлектрической
проницаемости, но в значительной степени определяется его элект-
ронодонорными или электроноакцепторными свойствами. Рол»
18 Глава 1. Ионные пары и экстракция ионных пар
растворителей в органической химии посвящен обширный
обзор [24].
Как упоминалось выше, большое влияние на /физические и
химические свойства ионных пар оказывает взаимодействие их
с растворителем. В этом отношении растворители можно
подразделить на три группы.
1. Полярные протонные растворители легко сольватируют
как анионы, так и катионы. Неорганические /катионы
взаимодействуют со свободными электронными парами, тогда как
анионы сольватируются путем образования водородных связей.
Крупные четвертичные аммониевые ионы не
сольватируются '[37] или по крайней мере сольватируются не специфично,
т. е. сильного непосредственного взаимодействия с
растворителем не существует. В этих растворителях имеет место
высокая степень диссоциации на свободные сольватированные
ионы. Однако многие анионы обладают относительно низкой
реакционноспособностью (нуклеофильностью) из-за сильного
экранирования сольватной оболочкой.
2. Полярные апротонные растворители (например, ДМСО,
ДМФА) легко сольватируют катионы. Однако анионы
сольватируются слабо, так как положительный конец диполя
растворителя пространственно затруднен, и поэтому скорости
реакций в таких растворителях высоки. Соли в них сильно
диссоциированы.
3. Реакции МФК легко протекают в малополярных апротон-
ных растворителях. Их диэлектрические проницаемости
изменяются от 8,9 (дихлорметан), 4,7 (хлороформ) и 4,2 (диэтиловый
эфир) до 2,3 (бензол) и 1,9 (гексан). Хотя растворимость
обычных неорганических солей в этих растворителях
пренебрежимо мала, органические четвертичные аммониевые, фосфоние-
вые и другие ониевые соли, так же как и «замаскированные»
органической оболочкой соли щелочных металлов, часто
достаточно растворимы, особенно в дихлорметане и хлороформе.
В этих растворителях концентрация свободных ионов
незначительна и доминируют ионные пары. Вследствие слабого
взаимодействия между ионными парами и молекулами
растворителя реакция с электрофилами в органической фазе идет
быстро, и некоторые обычно слабые нуклеофилы (например,
ацетат) оказываются сильными. Так, например, в гомогенных
растворах в ацетонитриле относительная нуклеофильность
солей тетраэтиламмония в реакции замещения с различными
анионами от азида до фторида различается всего в 80 раз,
причем фторид является наиболее сильным нуклеофилом среди га-
логенидов ![127]. Различия в реакционной способности ионов
в таких растворителях по сравнению с «нормальным
поведением» в некоторых случаях бывают просто поразительными, и та-
1.2. Ионные пары в органической фазе 19
кие ионные, пары иногда называют «голыми» или
«обнаженными» ионами) Однако это название может ввести в заблуждение,
так как ионнЪге пары присутствуют и в менее полярных
растворителях. Кроме того, влияние растворителя на относительные и
абсолютные скорости реакций очень значительно даже в «не-
сольватирующеЙ» среде.
Самоассоциация между ионными парами ведет к
образованию агрегатов, например димеров, тримеров или квадруплетов.
Такая ассоциация энергетически выгодна и часто наблюдается
в неполярной среде, если растворы не бесконечно разбавлены.
Ассоциация становится измеримой уже при таких низких
концентрациях, как 0,001 моль/л. Например, криоскопическая
степень ассоциации (отношение экспериментально найденной
молекулярной массы к формульной) для тиоцианата тетра-к-бу-
тиламмония в бензоле составляет 2,5 при концентрации
0,0013 моля на 1000 г растворителя, увеличивается до 31,9 при
0,281 моля на 1000 г растворителя и снова несколько снижается
при более высоких концентрациях (22,7 при 0,753 моля на
1000 г растворителя) '[25]. Такая ассоциация ионных пар
оказывает очень сильное влияние на экстракцию солей из водной
фазы в органическую (разд. 1.3.1). Степень ассоциации
зависит от катиона, аниона, растворителя и концентрации. Тримеры
одновалентных ионов являются заряженными частицами и
проводят электрический ток таким же образом, как и ионные пары,
содержащие многовалентные ионы.
Исходя из физических и кинетических доказательств,
предполагают, что существует два типа ионных пар, для которых
предложены различные названия:
1) свободные (рыхлые), внешние или разделенные
растворителем ионные пары;
2) связанные (тесные), внутренние, близкие или контактные
ионные пары.
Сольватированный свободный ион может беспрепятственно
приближаться к своему противоиону до соприкосновения двух
сольватных оболочек с образованием свободной (разделенной)
ионной пары. Если два иона подошли еще ближе друг к другу
и выдавили разделяющие их молекулы растворителя, то
образуется контактная (тесная) ионная пара. В зависимости от
природы и концентрации катиона, аниона и растворителя и в
пределах определенных интервалов температур оба типа
ионных пар могут сосуществовать как термодинамически
различные частицы в динамическом равновесии. В эфирных
растворителях образование разделенных растворителем ионных пар
обычно экзотермично. Если эти два типа ионных пар
сосуществуют, их диссоциация на свободные ионы описывается тремя
взаимосвязанными равновесиями. Положение этих равновесий
20 Глава 1. Ионные пары и экстракция ионных пар ,
сильно зависит от стерических эффектов, связанных с
катионом. Для контактных ионных пар стереоспецифи«ность более
вероятна; это проявляется, например, в реакциях^ H/D-обмена
{28]. Известно, что краун-эфиры превращают шюгие (но не
все; см., например, [17]) контактные ионные цары катионов
щелочных металлов в разделенные растворителем ионные
пары. Последние реагируют менее специфично /[28]. Влияние
различных эфирных растворителей (например, эфиров поли-
этиленгликоля или добавленных краун-эфиров) на структуру
ионных пар рассмотрено в обзоре {32].
Рассмотрение роли ионных пар в нуклеофильном
алифатическом замещении к данному случаю не имеет прямого
отношения, однако сведения, приведенные в работах [33—36],
могут оказаться полезными.
1.3. Экстракция ионных пар из водных растворов
1.3.1. Принципы
Экстракция растворителем ионных соединений из водной
фазы в органическую — область, хорошо известная химикам-
аналитикам и практикам. Экстракция металлов и кислот
также детально изучена i[71—74].
Даже наиболее простая двухфазная (вода/органическая
фаза) реакция замещения между анионом соли и органическим
веществом включает ряд равновесных процессов:
а) общую реакцию
[Q+X-]
где индексы ВФ и ОФ обозначают водную и органическую
фазы соответственно, Q+ — катион катализатора, a I[Q+X~] —
ионные пары. Общее уравнение можно разбить на два:
б) химическую реакцию в органической фазе
в) экстракционное равновесие
[<Э+Х-]оф + Na+вФ + У-Вф ч=* [Q+Y-Ьф + Na+B<t> + Х~Вф
Информацию относительно факторов, определяющих
положение равновесия (в), можно получить, рассматривая следую-
лцие более простые равновесия экстракции:
г) <2+вф + Х"ВФ < * 1<3+Х~]оф
д) Q+вф + Y-вф ч=* [Q+Y-Ьф
^ 1.3. Экстракция ионных пар из водных растворов 21
Лоследнее\уравнение существенно для понимания всего МФК.
К счастью, Экспериментальные методы исследования этих
экстракций разработаны, и для использования в аналитических
целях получены\ многие количественные данные (литературу см.
в таблицах).
Шилл и Модин ([44, 45] называют величину EQX,
получаемую из уравнения (г), стехиометрической константой
экстракции:
£Qx- [<2+]ВФ[Х-]ВФ
Следует отметить, что EQX не тождественна более известной
константе распределения. Так как знаменатель этого
выражения включает концентрацию продуктов реакции, то
экстракционное равновесие зависит от концентрации как катионов, так и
анионов в водной фазе. Для точного определения константы
экстракции в не слишком разбавленных растворах необходимо
вместо концентраций использовать активности*. В связи с тем
что на экстракционную систему обычно кроме равновесия,
представленного в уравнении (г), влияет еще ряд факторов,
ситуация еще более усложняется. Среди этих факторов
следующие:
1) ассоциация или диссоциация ионных пар в органической
фазе, которые снижают концентрацию i[Q+X~]o<i> и тем самым
способствуют экстракции;
2) ассоциативные эффекты в водной среде, ведущие к
уменьшению экстракции;
3) зависимость равновесия в водной фазе от рН, что влияет
на эффективную концентрацию аниона:
X" + Н3О+ т—*- НХ + Н2О
или эффективную концентрацию катиона, если Q+ — первичный,
вторичный или третичный аммониевый ион НВ+:
НВ+ + Н2О -<—у В + Н2О+
4) экстракция незаряженных частиц, таких, как НХ и В, в
органический слой и еще к тому же возможное образование
ионных ассоциатов [HQ+X2-] и [Q+X~, 2HX] в органической
среде.
* При использовании активности получают термодинамическую
константу экстракции, которая непосредствеиио измеряется редко. Для установления
относительных величии свободных энергий переноса аниона из воды в 1,2-ди-
хлорэтан использовали экстраполяцию до полной диссоциации соли в обеих
фазах Г И 9].
22 Глава 1. Ионные пары, и экстракция ионных пар
Шилл и Модин ([44, 45] считают равновесие (г/ основным
процессом, а все остальные (1—4) —«побочными процессами».
Они определяют условную константу экстракции $*<зх, которая
включает все конкурирующие «побочные реакции» и связана
со стехиометрической константой экстракции, следующим
образом:
«Q.X
где a — поправочные коэффициенты. Они отклоняются от
единицы с усилением влияния «побочных процессов» и равны
единице, когда влияние этих процессов пренебрежимо мало.
Хотя константы диссоциации четвертичных аммониевых
соединений в дихлорметане и хлороформе имеют порядок
10~4—10~5 их влиянием в часто используемых разбавленных
растворах нельзя пренебрегать. Желательно, чтобы в
органической фазе происходила ассоциация ионных пар, так как этот
процесс способствует экстракции. Поэтому более
концентрированные растворы обладают преимуществом. Если анион
вводится в систему частично в виде неорганической соли NaX, то
высокая концентрация и избыток NaX в водной фазе
увеличивают экстракцию i[Q+X~] в органическую фазу. В то же
время возможная ассоциация ионов неорганической соли в
водной фазе в большинстве случаев не оказывает
неблагоприятного действия на процесс в целом.
Факторы 3 и 4 достаточно распространены и там, где это
необходимо, должны приниматься во внимание. При
определенных условиях важность одного или нескольких «побочных
процессов» может повышаться. В принципе можно измерить
все процессы этого типа и рассчитать их влияние. Брендстрём
[11, 112] приводит все уравнения, необходимые для таких
расчетов с использованием логарифмических экстракционных
диаграмм, включающих многие очень сложные равновесия.
Результаты этих расчетов помогают ответить как на аналитические,
так и на практические вопросы типа:
Как провести селективную экстракцию?
Как выбрать оптимальные условия для количественной экст-,
ракции (рН, концентрацию)?
Каковы идеальные условия для количественного разделения,
смеси?
Однако в большинстве случаев МФК достаточно
качественного представления о возможности экстракции. Практически
происходит конкурентная экстракция двух или более анионов.
В случае реакции замещения (а), необходимо рассматривать
экстракционное равновесие (в). Используя основное уравнение1
1.3. Экстракция ионных пар из водных растворов 23
экстракции для [Q+X~] и '.[Q+Y-], получаем следующее
уравнение:
EQX ~ [Q+X-Ьф ' [Y-Ьф
Если EQx и Eqy известны, легко рассчитать, может ли
экстракция аниона Y~ в органическую фазу конкурировать с
экстракцией уходящей группы Х~ для любой данной концентрации
X", Y~ или катиона катализатора на любой стадии реакции.
-С этой целью полагают, что вместо активностей достаточно
использовать концентрации и что «побочные процессы»,
описанные выше, не мешают или по крайней мере их влияние можно
оценить. Кроме того, необходимо допустить, что катион Q+ до
такой степени липофилен, что присутствует фактически только
в органической фазе. В этих условиях появляется возможность
рассчитать, какая часть катиона катализатора находится в
паре с Х~ и какая с Y~ i[13], и предсказать результат общей
реакции. В большинстве случаев это будет грубая оценка, но
она достаточна для препаративной работы. Для оптимизации
исследования экстракции и для аналитической работы
необходимо более длительное и дорогостоящее изучение «побочных
процессов» 1[ 11, 112].
Необходимо подчеркнуть, что «побочными процессами»
нельзя пренебрегать и что простая оценка условных констант
экстракции может ввести в заблуждение. Брендстрём подробно
описывает распределение B114NCI между водой и дихлормета-
ном при 25°С в широком диапазоне температур 1[112]. В этом
случае коэффициенты активности в водном слое известны,
коэффициенты активности в органическом слое можно
рассчитать, а константы диссоциации и димеризации в органической
фазе — измерить. Получив эти данные, сравнивают расчетные
и экспериментальные абсолютные концентрации в обеих
фазах, предполагая возможность самопроизвольной экстракции,
диссоциации и димеризации. В изученном интервале
концентраций [112] условная константа экстракции сначала
уменьшается от 3,54 до 1,51, а затем повышается до 7,54 с
увеличением разбавления.
Величина стехиометрической константы экстракции зависит
не только от органического растворителя, но и от размеров и
структуры аниона и катиона. Эти факторы рассматриваются в
следующих разделах. Нужно отметить, что экстракция
возможна при очень большом разнообразии величин констант
экстракции благодаря правильному выбору ионов: даже
наиболее гидрофильные анионы (например, ОН~)
экстрагируются при использовании очень липофильных катионов, а
наиболее гидрофильные катионы, такие, как Me4N+, переносятся в
24 Глава 1. Ионные пары и экстракция ионных пар
органическую фазу очень липофильными анионами. Из многих
катионов, способных экстрагировать анионы (замещенные
аммониевые, фосфониевые, сульфониевые, арсониевые ионы и
т. п., а также комплексы краун-эфиров с катионами щелочных
металлов), систематически исследовались лишь аммониевые
ионы. Поэтому наше обсуждение сконцентрировано на них,,
однако предполагается, что структурные эффекты при
экстракции других катионов будут такими же.
1.3.2. Влияние растворителя
Брендстрём 1[46, 112] определил большое число кажущихся
констант экстракции между водой и различными растворителяг
ми для стандартной четвертичной аммониевой соли — бромида
тетра -м-бутиламмония (табл. 1.1). Растворитель, используемый
в работе по МФК, должен быть не смешивающимся с водой,,
так как в противном случае будут образовываться сильно гид-
ратированные «экранированные» ионные пары с низкой
реакционной способностью. Чтобы избежать образования водородных
связей с анионами ионных пар, растворитель, кроме того,
должен быть апротонным. Приведенные в табл. 1.1 данные
показывают, что величины констант экстракции очень сильно
изменяются. Растворители из последней колонки таблицы в
целом не подходят для МФК: некоторые из них частично
смешиваются с водой, другие слишком активны и могут мешать мжь
гим процессам. Однако для рассматриваемой стандартной соли^
которая обладает средней липофильностью, все эти растворит
тели являются хорошими или отличными экстрагентами.
Родственные по структуре, несколько более полярные соединения
(например, гомологи) должны иметь сходную способность к
экстрагированию ионных пар. Это позволяет сделать важный,
вывод: если в качестве реагентов в реакциях в условиях МФК»
например в алкилировании, используются соединения типа
приведенных в последней колонке табл. 1.1, то органический
растворитель не требуется, так как экстракция ионных пар в
чистую органическую фазу будет вполне удовлетворительной.
Из данных первой и второй колонок табл. 1.1 следует, что
имеет место большое различие в экстракционной способности
растворителей при, казалось бы, малых структурных изменена
ях (сравните цис- и гра«с-1,2-дихлорэтилен, 1,1,2-трихлорэтан,
1,1,2,2-тетрахлорэтан и пентахлорэтан). Специфические
взаимодействия между растворителем и растворенным веществом
должны играть определенную роль даже в таких
предположительно «несольватирующих» растворителях. С практической
точки зрения еще более важно, что низкокипящие
хлорированные углеводороды (хлороформ, дихлорметан и в меньшей сте-
Таблица 1,1. Кажущиеся константы экстракции £ви4ГСВг=[Ви4МВг]оф/[Ви4^]вф*[В1—]вФ, рассчитанные
для распределения 0,1 М бромида тетра-я-бутиламмония между водой и приведенными растворителями [46]
Растворитель
CHjCl2
CHCls
CDCb
ecu
CH3CHC12
С1СН2-СН2С1
С1СН2—CHClj
С12СН-СНС12
С12СН-СС13
я-С8Н7С1
С1-(СН2)3-С1
С1-(СН2)4С1
BujNBr Растворитель
35 CHg-CHCl—CHjCl
47 С1СН2-СНС1-СН2С1
41 C6HSC1
<0,1 о-С12С6Н4
0,5 СН2=СС12
6,1 туюяс-С1СН=СНС1
8,6 ч«с-С1СН=СНС1
145 С1СН=СС12
<0,1 С2Н5ОС2Н5
<0,1 С1СН2СН2—О—СН2СН2С1
2,9 СН3СООСгН5
0,3 С2Н5СОС2Н3
К-С3Н7СОСН3
£Bu4NBr
0,5
6,1
<0,1
<0,1
<0,1
<0,1
33
0,2
<0,1
2,8
0,2
1,1
1,7
Растворитель
C2HSCOCH3
CH3NO2
«-C3H7NO2
С1СН2СООС2Н5
NCCH2—COOCH3
н-С4Н9ОН
СНзСНОН—С2Н5
C1CH2CN
ClsC-CN
CH2=CHCN
C1CH2CH2CN
h-C3H7CN
CH2=CH--CH2CN
CH3O—CH2CN
CH3O—CH2—CH2CN
C2Hs0-CH2-CH2CN
£Bu4NBr
14
168
9,0
1,4
54
69
23
17000
2,3
130
940
13,7
67
84
91
38
26 Глава 1. Ионные пары и экстракция ионных пар
пени 1,2-дихлорэтан), как оказалось, являются лучшими
растворителями. Они не только обладают высокой экстракционной
способностью для стандартной соли (данные для других солей
см. в разд. 1.3.3 и 1.3.4), но также дешевы и легко удаляются.
Недостаток таких растворителей состоит в том, что они могут
вызывать побочные реакции, однако это не очень опасно,
поскольку большинство реакций МФК протекает очень быстро.
Следует отметить, что, как и можно было ожидать, диэти-
ловый эфир и этилацетат имеют низкую экстракционную
способность. Менее понятны плохие характеристики хлорбензола;
и о-дихлорбензола. Эти растворители часто используют в тех-
случаях, когда существует опасность побочных реакций с хло-.
роформом или дихлорметаном. Вовсе не обязательно исключать-
растворитель с низкой константой экстракции как
неподходящий, однако его применение означает, что в любой данный:
момент во время реакции лишь небольшая часть теоретически"
возможного количества ионных пар присутствует в
органической фазе, и, следовательно, реакция будет идти в таких
растворителях медленнее. Впрочем при использовании более
крупных, более липофильных катионов (см. следующий раздел),,
этот эффект в некоторой степени нейтрализуется.
Эффекты высаливания. Величина константы экстракции
зависит не только от системы растворителей, но и от
присутствия других солей. Большинство констант экстракции,
приведенных в литературе, было определено при постоянной ионной'
силе. Однако в этих условиях может иметь место очень
сильный эффект высаливания. Брендстрём 1[П2] изучал
относительные константы экстракции Bu4NCl и Bu4NBr в системе во-
да/дихлорметан в присутствии карбоната калия. Было
обнаружено, что величины коистант изменяются одинаково и
находятся в линейной зависимости от моляльности К2СО3.
Конкурентная экстракция карбоната или гидрокарбоната не
наблюдается, поэтому происходит истинное высаливание.
Добавление 2 молей КгСОз на 1 л приводило к увеличению константы
экстракции почти в 1000 раз. Этот эффект высаливания имеет
важное значение для МФК, особенно в случае реакций с
концентрированным (50%-ным) водным раствором гидроксид*
натрия. В такой среде почти все соли четвертичного аммония
малорастворимы и легко экстрагируются. Кроме того, он
действует как осушитель органической фазы. В ряде случаев
можно использовать растворители, смешивающиеся с водой, на-,
пример ацетонитрил или ТГФ, если только добавление другой-
соли обеспечивает образование двухфазной системы. Таким
образом, действие соли может быть двояким: она отделяет
органический растворитель и высаливает ионные пары.
Недавно было исследовано распределение хлорида метил-
трибутиламмония между водой или 0,1 М водным раствором
1.3. Экстракция ионных пар из водных растворов 27
NaCl и следующими растворителями: гексаном, ксилолом, тет-
рахлоридом углерода, бензолом, хлороформом, дихлорметаном,
бутил ацетатом, 4-метилпентаноном-2 и З-метилбутанолом-1
[121].
1.3.3. Влияние ониевого катиона
Хорошо известно, что высокомолекулярные амины могут
экстрагироваться в виде ионных пар аммониевых солей с
различными противоионами из водных растворов в среду,
подобную хлороформу. Недавно селективная экстракция такого
типа была положена в основу ряда аналитических методов {44,
47—51, 54, 58] и способов разделения <[7, 52, 53]. Как уже
упоминалось в разд. 1.3.1 и хорошо описано в обзорах Бренд
стрёма [11, 112], могут существовать чрезвычайно сложные
равновесные системы с несколькими константами, которые зависят
от структуры аниона, катиона и растворителя, а также от рН,
ионной силы и концентраций. В результате физико-химических
и аналитических исследований подобного равновесия
установлено, что существует взаимосвязь между размером катиона и
константой экстракции. Этот факт очень важен для МФК.
Ясно, что увеличение числа атомов С, окружающих
центральный атом N аммониевых катионов, будет увеличивать их
липофильность, повышая тем самым константу экстракции EQx.
Густави '[53] наблюдал линейную зависимость между
igEox и п (числом атомов С в аммониевых ионах). Он
экстрагировал пикраты в метиленхлорид, используя первичные
амины, а также симметричные вторичные и третичные амины и
симметричные четвертичные аммониевые соли. Были найдены
следующие зависимости для аммониевых ионов из
первичных аминов: lgEQnKKP&T=—2,40+0,63п
вторичных аминов: lgEQvKKpaT=—1,35+0,61 п
третичных аминов: lgEQnHKpaT=0,10+0,54n
четвертичных аммониевых солей: lg£QmiKpaT=—2,00+0,54 п
Для целей МФК особый интерес представляют
четвертичные аммониевые ионы, так как их участие в основной реакции
менее вероятно. В связи с этим важно проверить, являются ли
данные соотношения справедливыми для других растворителей
и противоионов или они относятся только к симметричным
катионам. Константы экстракции пикратов для некоторых
растворителей приведены в табл. 1.2 [49, 53], из которой ясно
видно увеличение величины lgEQx в среднем на 0,54 с
добавлением каждого атома С. И снова метиленхлорид и хлороформ
являются лучшими растворителями для экстракции, хотя их
28 Глава 1. Ионные пары и экстракция ионных пар
Таблица 1.2. Константы экстракции £B4NnintpaT=[R4NnHKpaT]/[R4N+]B<i>.
•[Пикрат-]Вф четвертичных аммониевых пикратов, экстрагируемых из
водного раствора в органический растворитель [53, 49]
р пинрат Для следующих катионов:
Растворителе
(CHs)4№- I (C2H6)4N+ I (H-C3H7)4N+ I <K-C4He)4N+
(K-C5H,,)4N+
CH2C12
CHCls
C6H6
ССЦ
1.5
0,22
220
21
0,22
2
4
35
.9-
,4-
10*
10s
4.8-
8.1.
3,9-
87
10»
10»
10»
2,45-
7,9-
2,9-
108
105
10*
относительное положение в таблице противоположно
положению их в табл. 1.1.
Было исследовано и влияние размера катиона на
экстракцию других анионов. В первом приближении («побочные
процессы» и другие экспериментальные ошибки исключить
непросто) снова было найдено то же увеличение примерно на 0,54
единицы на дополнительный атом С. Ниже приводятся анионы»,
экстракция которых симметричными четвертичными ионами а
хлороформ подчиняется этому правилу*: ацетат, фенил ацетат,
бензоат, салицилат, 3-гидроксибензоат |[55], л-толуолсульфонат,.
р-нафталинсульфонат, тринитробензолсульфонат, 2,4-динитро-
ct-нафтол, дипикриламин, сульфонатная группа в азокрасите-
лях [35], нитрит, перхлорат, хлорид, бромид, иодид ([58].
Результаты Цапкевич-Тутай и Цапкевича [59], относящиеся к
экстракции бромида четвертичного аммония в 1,2-дихлорэтан,
представлены в графической форме. Значения igiiQx,
рассчитанные на основании этих результатов, представлены в табл. 1.3-
Хотя ошибка, конечно, велика из-за ограниченности
имеющихся данных, эти сведения, несомненно, полезны.
Из последней таблицы видно, что даже для
несимметричных катионов совсем в другом растворителе 'lg^Qx
увеличивается на 0,54—0,61 единицы на атом С, если одна из цепей
удлиняется. Однако, как и ожидалось, число атомов С не
является единственным фактором, контролирующим константу
экстракции; поэтому расчет неизвестных констант экстракции
окажется надежным только для симметричных ионов, если R —
гомологи, или при изменении длины лишь одной из четырех
углеродных цепей. Например, бензильные группы значительно*
менее липофильны, чем н-гептильные, и их вклад в экстракци-
* Например, для RjN+ при переходе от R=Memn к R=amn число
атомов С увеличивается на четыре.
1.3. Экстракция ионных пар из водных растворов
Таблица 1.3. Логарифмы кажущихся констант экстракции,
экспериментально определенные дли бромидов аммония
(вода/1,2-дихлорэтан) [117} или вычисленные с помощью диаграмм
из работы [59]
Соль
Общее
число атомов
углерода
''QX
,) (CH3)3NBr
(H-C,2H25)(CH3)3NBr
(H-C14H2S)(C2H6)3NBr
(H-Ci2H25)(C2H5)3NBr
(k-Ci0H21) (w-C3H7)3NBr
(K-CsH19)(«-C3H7)3NBr
(H-C10H21)Bu3NBr
Bu4NBr
(C2H5)Bu3NBr
(CH3)Bu3NBr
(H-C5H,,)4NBr
(H-C6H13)4NBr
19
18
17
15
20
18
16
19
18
22
16
14
13
20
24
3,88
3,27
2,67
1,52
3,7&
2,55-
1,36
2,87
2.2S
4,49
0,82
—0,14
—0,41
3,03
5,22
онную способность лежит где-то между вкладами бутильной
и пропильной групп. Так, хлорид бензилтриэтиламмония
(обычно называемый ТЭБА) находится главным образом в
водной фазе, если отсутствует «эффект высаливания», который
возникает под действием концентрированного гидроксида
натрия.
Следовательно, в целом логарифмы констант экстракции
гомологических серий четвертичных аммониевых солей
увеличиваются приблизительно на одну и ту же величину — около
0,54 на дополнительный атом С — независимо от типа
неполярного апротонного растворителя и аниона.
Аналогичные общие тенденции наблюдаются для алкилтри-
фенилфосфониевых и алкилтрифениларсониевых ионов. Арсо-
ниевые ионы кажутся более липофильными. Аллильная
группа делает фосфониевый ион более гидрофильным по сравнению
с н-пропильным аналогом [112]. Установлено .[20], что ионы по-
липофильности располагаются в следующий ряд: Hex4N+>
> Pent4N+> Ph4As+> (ызо-С5Нп) 4N+> Bu4 P+> Bu4N+.
Глава 1. Ионные пары и экстракция ионных пар
1.3.4. Влияние аниона
Огромное влияние аниона на экстракцию может быть
показано в лекционных демонстрациях с катионоидными
красителями, находящимися в форме солей HSO4~, С1~, Вг~ и 1~ |[129].
Частичный или полный обмен аниона неоднократно
проводился путем эквилибрирования органического раствора
четвертичной аммониевой соли [Q+X~] с водным раствором NaY. На
основе этих исследований можно построить шкалу липофиль-
ности. Клиффорд и Ирвинг ([63] установили следующий
порядок экстракционной способности ионов, начиная с липофиль-
ного иона С1О4~ и кончая гидрофильным ионом РО43~, для
системы хлороформ/вода:
сю4- » сю„- > no3- > сг » hso4- > он- > so4*- > ссу- > ро4*-
Для других систем получены следующие результаты:
вода/СН2С12 или ClCHj—СН2С1 [118]: ClO4->SCN->I->ClO,->NO8->
>Вг->ВгО3->С1-
вода/ксилол [121]: ClO4->SCN->I->NOs->Br->Cl-
вода/нитробензол [76]: 1->Вг->С1~
водные растворы кислот/СС14 [64]: Br->N0s->Cl->HS04->CHsCO2->
F
ClO4->I->
>f->oh-
вода/толуол или вода/СН2С1а [65] : ClO4->I->NO3->C6H5COj->Br->
>ci->hso4->hco,->ch3co2-foh
Наконец, Макоша и сотр. {66] включили в этот ряд цианид:
I->Br->CN->Cl->OH->F->SO42-. В их экспериментах
органической фазой служил бензол, хлорбензол или о-дихлорбен-
зол, а водной фазой — концентрированный гидроксид натрия.
Другой ряд анионов, которые можно обменять на хлорид,
включает ВН4~, SCN-, [Fe(CN)6]3-, MnO4~ NQr, N3~ [66].
Для катионов тетрафенилфосфония также были
исследованы некоторые другие анионы [75]: МпО4~>С1О4~>1~>
> HCrO4-> NO3-> Br~> BrO3-> NO2- С1~> Ю4-> Ю3~>
>s2o38->s2->so42-.
Ясно, что один и тот же порядок проявляется в различных
апротонных растворителях. Это естественно для разбавленных
растворов, где различия в энергии сольватации для анионов
Х~иУ- в органических растворителях и воде являются
главными факторами, определяющими экстракционную способность.
Однако для некоторых использованных солей (поскольку
применялись различные катионы) границы растворимости в любой
фазе, а также способность к гидратации, диссоциации и
ассоциации могут изменить порядок экстракционной способности.
Один из этих факторов, вероятно, обусловливает, неожиданное
положение бензоата в приведенной выше серии (ср. с табл.
Таблица 1.4. Константы экстракции £qx тетра-к-бутиламмонийных ионных пар в системе вода/хлороформ
Анион
С1-
Вг~
I-
сю4-
NO3-
сн3со2-
CeHs—CHj-COj-
CeH5CO2-
Салицилат
8 В скобках приведены
%с8
0,78 (0.35)
19,5 (17)
1023 (2188)
3020 (4,37-10*)
24,5 (79)
7,6.10"»
1,86
2,45
263
Литере- | -
тура |
53, 58
53, 58
53, 58
53, 58
53, 58
55
55
55
55
значения констант экстракция в системе
' Анион
З-Гидроксибензоат
Фениксид
Пикрат
я-Толуолсульфонат
Нафталин-2-сульфонат
Антрацен-2-сульфонат
Триннтробензолсульфонат
2,4- Динитро-1 -нафтоксид
вода/метиленхлорид.
0,03
0,93
8,1.10* (4,8.10е)
214
2818
1,3-10*
2,9-10*
2.8-10*
Литература
55
55
49, 53
45
45
62
45
45
32 Глава 1. Ионные пары и экстракция ионных пар
1.4). Положение гидроксида и фторида также кажется
сомнительным. Комбинируя эти значения с данными, полученными
позднее (табл. 1.4), авторы данной книги пришли к
следующему ряду:
пикрат>МпО4->С1О4->5СМ->1->СЮз-=толуолсульфонат>
> NO3- > Br- > CN-=ВгО3-=бензоат > NO2-=
=Cl->HSO4->HCO3->aueTaT>F-,OH->SO42->CO32->P(V-
Такой же ряд липофильности обычно наблюдается для
различных алкилтрифенилфосфониевых катионов [112], а также тет-
рафенилфосфониевых, тетрафениларсониевых и трифенил-
•сульфониевых катионов '[75]. Необходимо подчеркнуть один
очень важный вывод, вытекающий из этих данных. При
использовании малых катионов типа Bu4N+ анионы двух- или
трехосновных кислот экстрагируются значительно труднее, чем
гидро- или дигидроанионы соответствующих кислот, т. е.
HS04->SO*-; H2PO4->H
НСгО4- > СгО4з- • HS-
НО„С—(Ш„)4—СО2" > -О2С—(СН^—СО2~
Однако с увеличением Q+ экстрагируемость Q2SO4,
например, растет быстрее, чем экстрагируемость QHSO4. В случае
Hex4N+ необходимы сильнокислые растворы для
предотвращения экстракции (Hex4N)2SO4 вместе с Hex+NHSO4 ;[11].
Практический вывод из этих фактов состоит в том, что для
катионов средних размеров гидросульфаты являются не только
хорошим исходным материалом для приготовления многих
ониевых солей, но также и очень полезными МФ-катализато-
рами. Добавление 1 мол. экв. гидроксида натрия превратит
анион гидросульфата в нейтральный сульфат, который не
может мешать, так как он экстрагируется труднее почти всех
других органических и неорганических анионов. Кроме
гидросульфата удобно использовать хлориды. Более гидрофильные
четвертичные аммониевые соли с такими анионами, как
ацетат, фторид или гидроксид, трудно приготовить: они сильно
гигроскопичны и/или нестабильны.
Обратимся теперь к количественным аспектам МФК-
Определены многие константы экстракции* для системы
вода/хлороформ или дихлорметан [45, 53, 55, 60—62]. Соответствующие
данные для МФК суммированы в табл. 1.4 и 1.5. Во многих
случаях константы для родственных симметричных катионов
• Коэффициенты распределения некоторых анионов и ионных пар были
рассчитаны из свободных энергий сольватации и гидратации для системы
вода/различные апротоиные растворители [130].
1.3. Экстракция ионных пар из водных растворов 33
Таблица 1.5. Константы экстракции £qx различных солей
в системе вода/1,2-дихлорэтан [20]
Катионы
Bu«N+
Bu«P+
(C5Hn)«N+
(u3o-C5Hu)4N+
(C6H13)«N+
Ph«As+
Ph«P+
(C12H25)NEt3+
EQX для следующих анионов:
CI-
0,13
0,24в
0,57
38,2
9,95
7,56
7,2
13,2В
e я-Толуолсульфонат.
" л-Нитрофедолят.
"Данные работы [120].
Bi-
I-
CH3OSO3-
ToiH1
6,15 830 18,1 180
6,58В 528В
28.4 3730
970
1,3-106
349 4,3-10* 820 1,Ы0*
357В
n-NP-6
247
610
6,4-10*
1,6-10*
~10*
1,9-10*
1,9-10*
также известны. Было бы весьма желательным определение
констант и для других анионов.
Примечателен широкий диапазон величин констант
экстракции в ряду галогенидов. Очевидно, иодид, так же как и
перхлорат, экстрагируется предпочтительнее. Отсюда
непосредственно следует, что, если присутствуют небольшие количества
катионов, они будут экстрагироваться в виде ионных пар в
основном с очень липофильными анионами. Этот эффект,
вероятно, будет мешать желаемой реакции МФК, особенно если в
процессе реакции образуются иодид-ионы. Поэтому иодиды
(и в меньшей степени бромиды), как правило, не следует
использовать в качестве катализаторов МФК.
Среди органических анионов наиболее гидрофильными
являются ацетат и формиат. При сравнении констант экстракции
салицилата (внутренняя водородная связь!) и 3-гидроксибензо-
ата необходимо учитывать сильные структурные отличия этих
соединений. В принципе влияние структуры установлено и для
гомологических рядов анионов. Каждая дополнительная СН2-
группа делает анион более липофильным. Кроме алкильных
групп сильно увеличивают константы экстракции другие липо-
фильные заместители, такие, как нитрогруппа, хлор, бром
и т. д.
Фактически в реакциях МФК имеет место конкурентная
экстракция двух или более анионов. Селективность экстракции
34 Глава I. Ионные пары и экстракция ионных пар
в принципе можно рассчитать из известных значений £qx, как
показано на с. 23. Для грубой оценки можно считать, что
относительные константы экстракции для различных катионов и
неполярных растворителей очень близки между собой. Это
справедливо лишь в редких случаях только как первое
приближение и является слишком большим упрощением в других
случаях. Часто реагент или одна из ионных пар, участвующих
в истинной реакции, присутствуют в концентрации, близкой к
насыщению. Тогда следует ожидать отклонений от идеального
поведения. Более того, полярность и способность растворителя
к образованию водородных связей по-разному влияет на
различные анионы. Известны «константы селективности» Кх >-y
для конкурентной экстракции хлорида по отношению к
бромиду, иодиду и перхлорату из воды в 11 растворителях i[121] и
для хлорида относительно цианида в 8 растворителях <[122].
Как ожидалось, Ка—»-сы изменяется незначительно, причем
максимальный интервал изменения от 0,9
(вода/цыс-1,2-дихлорэтан) до 3,1 (вода/бензонитрил). Специфичное влияние
растворителя более ярко выражено для серий анионов, сильно
различающихся по липофильности i[121]. Следует особо
отметить, что гидроксилсодержащие растворители выравнивают
различия:
Растворитель
А"" следующих
анионов а-
Вг~
СКГ
Гексан
Бензол
Дихлорметан
Тетрахлорид углерода
Нитробензол
З-Метилбутанол-1
Хлороформ
8,1
24,5
39,8
43,7
44,7
3,0
23,4
а Катион — метилтриоктиламмоиий.
525
3800
3090
8510
7585
15,8
891
17 000
250000
60250
43 650
1,15-106
28,2
17,38
Нужно упомянуть, между прочим, возможность экстракций
кислот НХ в форме ионных пар Q+X----HX. Известны факты
практического применения этой «экстракции водородными свя-'
зями» для HF2-, НС12- и Х- ... НООН (гл. 3).
Так как многие реакции МФК проводятся в присутствии
сильных щелочей — гидроксидов щелочных металлов, особый
1.3. Экстракция ионных пар из водных растворов 35
интерес представляет относительный порядок изменения
констант экстракции четвертичных аммониевых оснований. Хотя
в литературе имеется несколько ссылок на константы
экстракции ОН", большинство из них ненадежны. Некоторые
определены в растворителях, частично смешивающихся с водой, и,
следовательно, не представляют для нас интереса. Другие
приведены для хлороформа или дихлорметана как органической
фазы [55, 56], но можно показать, что эти фазы быстро
реагируют с гидроксид-ионом [67]. В то же время, четвертичные
аммониевые основания совершенно не растворимы в
неполярных растворителях, если отсутствуют следовые количества
протонных растворителей. Например, продажный «гидроксид тет-
рабутиламмония в толуоле» часто используется как реагент.
Он готовится путем разбавления промышленного метанольного
раствора толуолом и частичной (но не полной) отгонки
метанола. Гидроксид-ион часто сильно гидратирован и всегда
сосуществует с некоторым количеством воды или других гидрок-
силсодержащих растворителей (см. [96]). Таким образом, в
силу этих причин возможность определения констант экстракции
путем измерения распределения четвертичных аммониевых
оснований между водой и неполярными растворителями
затруднена. Кроме того, растворы R4N+OH~ относительно
нестабильны (разд. 3.1.2). При разложении образуется R3N, который
также может титроваться кислотой, что приводит к неправильным
заключениям относительно концентрации гидроксид-иона в
любой фазе.
В случае метиленхлорида можно показать, что Bu4N+OH~
может экстрагироваться в некоторой степени из
свежеприготовленного водного раствора. Однако титр раствора быстро
уменьшается вследствие реакции с растворителем »[67]. Если
раствор бисульфата тетрабутиламмония находится в
равновесии с концентрированным гидроксидом натрия, то этим
растворителем экстрагируется заметное, но быстро уменьшающееся
количество Bu4N+OH~. Это согласуется с порядком
экстракционной способности OH~>SO42~. Уже через 10 мин из гидрокси-
да аммония получается 67% хлорида в результате
взаимодействия с растворителем [67]:
2ОН" + CHsjCIs! >■ СН2О + НгО + 2С1-
В противоположность этому в 0,13 М растворе Pent^+OH- в
Дихлорметане концентрация ОН-групп в течение 60 мин при
20°С снижается только на 5% i[123]. Этот раствор содержит
10—И молей Н2О на 1 моль ОН~, а стабилизирующее действие
гидроксилсодержащих растворителей на четвертичные
аммониевые основания известно [124]. Есть основания предполагать
36 Глава 1. Ионные пары и экстракция ионных пар
[125], что упомянутый выше раствор Bu4N+OH- содержал во^
ды меньше, так как был в контакте с сильным осушителем —
концентрированным раствором гидроксида натрия. Даже
относительно стабильный PenUN+OH"- 10H2O в СНгС12 через 24 ч
разделяется на две фазы — маленькую водную и большую
органическую [123]. Дальнейшие исследования в лаборатории
авторов этой книги i[ 131] подтвердили эти предположения:
растворы R4'N+OH~ (R = Bu до Oct) с концентрацией от 0,05 до
0,1 М имеют период полураспада в метиленхлориде от 5 до
10 ч. В хлорбензоле концентрация снижается лишь на 10% в
течение 100 ч. Однако если присутствует 50%-ный водный NaOH,
по объему одинаковый с хлорбензолом, то период
полураспада уменьшается до 2 ч. Таким образом, присутствие
концентрированного гидроксида натрия сильно ускоряет распад.
Согласно грубым оценкам, константа экстракции
гидроксида тетра-н-гептиламмония в бензол из воды равна 1, что в
Ю4 раз меньше константы для хлорида >[67]. В типичном
эксперименте по МФК с гидроксидом натрия в качестве реагента
различия в константах экстракции частично компенсируются
использованием большого молярного избытка NaOH в
присутствии малых, каталитических количеств хлоридов аммония.
При равновесии между бензольными растворами очень липо-
фильных катионов (например, тетра-я-гексиламмония или
тетра-н-гептиламмония), введенных вначале в виде хлоридов, и
большим избытком гидроксида натрия только несколько
процентов* ионов аммония в органической фазе будут связаны
с гидроксидом, остальные — с хлоридом. С менее липофильны-
ми катионами (например, тетра-я-бутиламмония или бензил-
триэтиламмония) как растворимость гидроксидов, так и
экстракционная способность ионных пар малы. Поэтому лишь
небольшая часть ионов аммония — главным образом в виде
хлоридов (96—99,5%) —будет в органической фазе|[67].
В заключение необходимо отметить, что многие другие
анионы экстрагируются в органическую фазу вместе с
определенным количеством сольватирующей воды. Степень гидратации
ионных пар зависит от аниона, катиона, растворителя, а
также условий реакции. Проведены различные исследования этих
эффектов (например, [68—70]).
Макоша и сотр. ;[66] разработали препаративный метод
получения безводных солей бензилтрибутиламмония со многими
анионами, исходя из хлоридов, путем обмена анионов в
присутствии концентрированного раствора гидроксида натрия.
* Более высокое значение, приведенное в работе [67], оказалось
ошибочным [1311.
1.4. Краун-эфиры, криптанды и другие соединения
37
1.4. Краун-эфиры, криптанды и другие соединения,
образующие хелаты, как экстрагенты
Краунами называют макрогетероциклы, обычно содержащие
в качестве основной единицы (—Y—СН2—СН2)П, где Y — это
О,S или N
н
н
■N-"
о.
5
Схема 1.1
Особенно большой интерес к крауи-эфирам возник в последнее
Десятилетие. Были опубликованы работы, посвященные
различным аспектам их получения и исследованию химических
свойств [77—82]. Так как систематическая номенклатура этих
соединений очень неудобна, обычно используются тривиальные
названия. Примером служит 18-краун-6 (1), в котором 18
означает число атомов в кольце, краун — класс (в данном слу-
38 Глава 1. Ионные пары и экстракция ионных пар
чае это краун-эфир), а 6 — число атомов кислорода в
положениях /, 4, 7 и т. д. Другими общеизвестными, имеющимися в
продаже краун-эфирами являются дибензо-18-краун-6 (2), ди-
циклогексано-18-краун-6 (3) (часто неправильно называемый
«дициклогексил-18-краун-6») и 15-краун-5 (4).
Имеются сообщения о новых краун-эфирах, их азо-аналогах,
полиокса-полиаза-микроциклах, аналогах, содержащих аннели-
рованные гетероцнклы, и о би- и полициклических аналогах.
Один из них привлек особое внимание. Соединение 5,
названное Леном и сотр. [83] криптат[2.2.2]*, продается под
названием «криптофикс '[222]». Общей чертой всех краунов и
родственных веществ является наличие центральной «дыры» или
полости. В результате хелатирования внутри этой «дыры»**
могут образовываться комплексы с другими частицами,
различные по стабильности в зависимости от их радиуса и
электронной конфигурации. Это могут быть катионы, анионы,
нейтральные (нульвалентные) металлы и нейтральные молекулы,
такие, как нитрилы ,[108].
Для МФК представляют интерес катионные комплексы,
образованные с катионами натрия и калия. Наиболее стабильные
калиевые комплексы образуются с 18-членными кольцами
(соединения 1, 2, 3 или 5), тогда как натрий образует комплексы
преимущественно с соединением 4 и другими 15-членными крау-
нами. Среди других катионов, образующих комплексы,
имеются ион гидроксония Н3О+ [106], ионы аммония [84] и ионы ди-
азония [91, 111]. Крам и сотр. ([84] показали, что, когда такое
комплексообразоваиие типа «хозяин — гость» проводят с хи-
ральными краун-эфирами и замещенными рацемическими
первичными аммониевыми солями, такие комплексы можно
использовать для разделения оптических изомеров. Они
использовали оптически активные бинафтильные единицы, однако
известны многие другие диастереомерные крауны,
потенциально или в действительности оптически активные '[85]. Во многих
случаях физическими методами, включая рентгеновский
анализ, были точно установлены структуры комплексов.
Хелаты типа ион щелочного металла/краун представляют
особый интерес при обсуждении МФК. Другие родственные
явления, например введение анионов в криптаты [88],
растворение щелочных металлов в различных растворителях с помощью
краунов |[89], выделение устойчивой кристаллической соли
Ыа~/криптат iNa+ ![90] и образование анион-радикалов из аро-
* Цифры обозначают три мостика из двух кислородных атомов. Согласно
номенклатуре ИЮПАК, это соединение следует называть криптандом. — Прим.)
ред.
** Существуют также комплексы, в которых ион металла расположен вне
плоскости крауна.
1.4. Краун-эфиры, криптанды и другие соединения 39
матических углеводородов, краунов и щелочных металлов
[95], могут найти применение в будущих работах по МФК-
Вернемся к комплексообразованию между ионами щелочных
металлов и краунами или криптандами, которое ведет к
разнообразным следствиям:
а) «Органическая оболочка» щелочного металла
обеспечивает образование, по существу, «ониевого иона», который
может экстрагироваться или растворяться вместе с анионами
точно так же, как и сами ониевые соли, в неполярных
органических растворителях*. Несмотря на меньшую устойчивость,
полиэфиры с открытыми цепями и различные полиамины
образуют сходные комплексы и также могут применяться для
экстракции солей ![97]. Комплексы краун-эфиров можно, кроме
того, использовать в качестве моделей для транспорта и
разделения ионов через жидкие мембраны i[86, 98], а в случае
фоточувствительных краунов можно даже добиться «транспорта
активных ионов» |[87].
б) Анионная часть ионной пары, присутствующая в
органическом растворителе, «активирована», т. е. более реакцион-
носпособна, чем без комплексующего катионного агента, даже
в тех случаях, когда солюбилизирующего действия краун-эфи-
ра не требуется, так как сама неорганическая соль
растворима. В полярных апротонных растворителях, где имеет место
высокая степень диссоциации, этот эффект наиболее заметен.
Такие системы называют системами, включающими «реакцию
голых анионов». Однако это образное название ошибочно, так
как сильные взаимодействия между растворителем и
растворенным веществом протекают даже в «слабо сольватирующей»
и «несольватирующей» среде![99].
«Активация аниона» посредством 18-крауна-6 в ацетонитри-
ле (диэлектрическая проницаемость 39) была изучена в
работе '[99], где показано, что при этом происходит выравнивание
нуклеофильности. Константы скоростей замещения в бензил-
тозилате на N3"~, Ас~, CN~, F~, Cl~, Вг~ и 1~ отличались
меньше чем на порядок величины. Ацетат и фторид проявляли
значительно более высокую реакционную способность по
сравнению с «нормальными» реакциями в гидроксилсодержащих
растворителях. Хотя этот эффект «активации аниона» часто
использовался в гомогенной среде, мы приведем только один
поразительный пример. Меррифилд и сотр. [100] селективно от-
Щепляли защищенные аминокислоты и пептиды от оксиациль-
ных смол, используя цианид калия в ДМФ, N-метилпирролидо-
Известны коэффициенты распределения комплексов 18-крауиа-6 со щелоч-
? металлами и семью неорганическими противоионами в CH2CI2/H2O
40 Глава 1. Ионные пары и экстракция ионных пар
не или ацетонитриле, при комнатной температуре. В
присутствии дициклогексано-18-крауна-6 (3) выход 90—97%
наблюдался через 8—16 ч, тогда как в отсутствие краун-эфира реакции
протекала лишь на 5—6% за 72 ч.
R'
I
Вое—NH—СН—С
R'
I
Вое—NH—СН—СОа© + NC—СН—С
Схема 12
i
Когда такие факторы, как природа субстрата, нуклеофила и
уходящей группы, постоянны, «активация аниона» зависит от
растворителя, а также от природы и концентрации лиганда.
Бициклические криптанды, такие, как 5, оказывают более
сильное влияние, так как они в большей степени охватывают
катион, образуя тем самым более стабильные комплексы*. В
полярных апротонных растворителях крауны обусловливают
усиление диссоциации. В других системах (например, грег-бутоксид
натрия в ДМСО) ионные агрегаты разрушаются в результате
комплексообразования с краунами, что приводит к
увеличению основности алкоксида, измеряемой скоростью отщепления
протона /[101]. В менее полярной среде, такой, как ТГФ или
диоксан, доминирующими частицами являются ионные пары.
В этом случае краун-эфиры могут благоприятствовать
образованию разделенных растворителем более свободных (рыхлых)
ионных пар >[38, 81] с более высокой реакционной
способностью |[102]. Даже в гидроксилсодержащих растворителях при
добавлении краунов наблюдаются удивительные эффекты, так
как изменяются структура и состав сольватной оболочки вокруг
ионной пары и ионные агрегаты частично разрушаются.
Например, сильно изменяется соотношение син/анти-изомеров при
элиминировании, катализируемом основаниями i[ 103].
Кнёхель и сотр. i[93, 104, 105, 107] провели обширные
исследования активации анионов в полярных апротонных средах как
в гомогенных, так и в условиях МФК. В абсолютном
ацетонитриле гомогенные реакции между бензилхлоридом и ацетат-
ионом, генерированном из нескольких щелочных ацетатов и
• Для целей МФК, по-видимому, можно принять следующий ряд
активности при «активации аниона»: краун<криптанд« аммониевый катион [129].
/.5. Анионный обмен между твердой и жидкой фазами 41
различных серий лигандов, ускорялись циклическими аминопо-
лиэфирами значительно сильнее, чем краунами. На основании
данных 'Н-ЯМР-спектроскопии такое поведение было
соотнесено со степенью разделения ионных пар 1[104]. Если
использовать каталитические количества комплексанта меньше стехио-
метрических, та же реакция проходит, как в МФК-системе
жидкость/твердая фаза. В этих условиях процесс активации
анионов связан с другими факторами: растворением и комп-
лексообразованием соли, анионным обменом и процессами
переноса. Было проведено сравнение действия большого числа
лигандов в стандартной двухфазной реакции |[93, 107]. Как и
ожидалось, простой зависимости между активацией аниона,
растворением и общей скоростью реакции не существует.
В заключение необходимо подчеркнуть, что активация
анионов легко подавляется присутствующей в среде в следовых
количествах водой в результате сольватации ионной пары или
анионов и соответствующего снижения их нуклеофильности
[107, 109, ПО]. Кроме того, крауны имеют склонность к
переносу воды даже в такие неполярные растворители, как
хлороформ i[l 10].
1.5. Анионный обмен между твердой и жидкой
фазами
Первоначально МФК проводился исключительно с
использованием водной и органической фаз. Эту методику некоторые
авторы называют «МФК в системе жидкость/жидкость». Как
упоминалось в предыдущих разделах, часто происходит соэкст-
ракция некоторого количества гидратной воды, которая может
мешать желаемой реакции, подавляя ее и/или изменяя ее
направление. Поэтому можно было предполагать, что в таких
случаях следовало бы отказаться от использования воды и
проводить МФК с твердыми солями. Подходящим примером
является генерирование дихлоркарбена из трихлорацетата натрия —
реакция, проводящаяся обычно в абсолютированном диметокси-
этане (стоимость которого высока) г
Na+ -О2ССС1„ *■ NaCI + СО2 + [СС12] >- дальнейшие реакции
Трихлорацетат натрия разлагается водой по реакции
Na+-O2CCCls + H2O >■ NaHCOs + HCCIs
и, хотя ионы трихлорацетата могут экстрагироваться с
помощью четвертичных аммониевых солей из водных растворов
в хлороформ, содержащий олефин в качестве акцептора карбе-
42 Глава 1. Ионные пары и экстракция ионных пар
на, совместная экстракция гидратной воды подавляет реакцию
карбена. Однако если твердый трихлорацетат натрия,
каталитические количества четвертичной аммониевой соли, хлороформ
и олефин взбалтывать при 25—80 °С, то дихлорциклопропаны
получаются с весьма удовлетворительными выходами [92].
Такой вариант реакции получил название «МФК в системе
твердая фаза/жидкость», и в недавнем обзоре [81] предполагали
даже, что краун-эфиры являются единственными подходящими
катализаторами для этой цели. Это предположение
основывалось на том, что краун ведет себя как двумерная система с
несколькими полярными центрами, что позволяет ему подойти
к кристаллической решетке соли настолько близко, что путь
катиона из решетки к лиганду становится очень коротким. В
противоположность этому у ониевых катионов положительный
ключевой атом пространственно экранирован, и поэтому такие
катионы не могут подойти к решетке близко. Более того,
предполагают, что эффективность краунов как катализаторов может
быть частично объяснена колебательным выделением из
комплексных продуктов свободных (несвязанных в комплекс) ли-
гандов; в результате этого процесса комплексы могут сначала
осаждаться, а затем «освобождать» краун для дальнейшего
комплексообразования с реагирующей солью.
На самом же деле во многих случаях ониевые соли также
очень эффективные катализаторы в системах твердая фаза/
жидкость, и даже имеются случаи, когда крауны менее
эффективны. Это является следствием того, что первоначальное комп-
лексообразование катион/лиганд может проходить быстро
только, в гомогенном растворе в гидроксилсодержащих
растворителях. Если комплекс образуется таким путем, то нежелательный
растворитель необходимо заменить на неполярный
растворитель.: Последний, однако, несомненно, образует более или
менее существенную сольватную оболочку ионных пар. Поскольку
комплексы краунов со щелочными катионами достаточно
устойчивы, то вопрос об «освобождении» крауна остается открытым.
Впрочем, каталитические процессы очень близки как для
комплексов ониевых ионов, так и для краунов: растворенная ионная
пара в том и другом случае просто обменивается анионами с
поверхностью кристаллической решетки твердой фазы. Вполне
возможно, что имеется еще какой-то неизвестный пока фактор,
который определяет ход реакций в таких системах.
Такими факторами могут быть скорость перемешивания,
размер частиц реагирующей соли, возможность отложения
соли, образующейся в результате реакции, на поверхности
растворяющейся соли. Проведению процесса МФК может
способствовать перемешивание с помощью эффективной скребковой
мешалки или использование шаровой мельницы.
1.5. Анионный обмен между твердой и жидкой фазами 49
Недавно было показано [126], что растворенный комплекс
КС1 с криптатом |[2.2.2] обменивает анионы с твердым
ацетатом натрия. Более того, растворенные комплексы 22Na- или
42К-криптат [2.2.2] обменивались радиоактивной меткой с
твердыми солями в ходе некоторых реакций замещения в системе
твердая фаза/жидкость i[126]. Таким образом, по крайней
мере в некоторых реакциях МФК происходит, по-видимому,
разрушение комплексов, возможно связанное с типом
растворителя, аниона и катиона:
Равновесие обмена анионов между ониевыми хлоридами,
растворенными в 1,2-дихлорэтане, и твердым л-нитрофенолятом
натрия устанавливалось за 10—30 мин, т. е. медленнее, чем
при обмене между жидкими фазами. По скорости обмена
(Х~оф замещается нитрофенолятом) катионы и анионы
растворенными в 1,2-дихлорэтане, и твердым п-нитрофенолятом
полагаются в следующий ряд [20]:
[2.2.1]-Na+>Ph4As+>[15-KpayH-5] - Na+>Hex^N+>Bu4N+
Cl~ > n-толуол сульфонат>- Вг
Имеются указания на то, что следовые количества воды
могут оказывать существенное влияние на скорости обмена
между жидкой и твердой фазами при использовании как ониевых
солей, так и краунов, но это нуждается в подтверждении в
будущих работах.
Кроме краун-эфиров, криптатов и ониевых солей для
катализа реакций между твердыми солями Щелочных металлов и
алкилирующими агентами могут быть использованы
полиэфиры с открытой цепью, диамины и полиамины ([93, 94]. '
В заключение необходимо снова подчеркнуть преимущества
метода МФК в системе твердая фаза/жидкость. «Активация1
аниона» происходит в полярных растворителях, которые
смешиваются с водой, например в ацетонитриле. Поскольку
растворенная соль в таком растворителе до некоторой степени
диссоциирует и анионы сольватируются непрочно^ реакции
протекают быстро. Это справедливо даже для анионов,
считающихся слабыми нуклеофилами в гидроксилсодержащих
растворителях (например, для ацетатов, фторидов).
Глава 2
Механизм
межфазного катализа
2.1. Изучение механизма реакций
в различных условиях*
В разд. 1.1 межфазный катализ был определен как
двухфазная реакция между солями (в твердой форме или в виде
водных растворов), кислотами или основаниями и субстратами,
находящимися в органических растворителях, протекающая в
присутствии так называемых межфазных катализаторов.
Типичными представителями таких катализаторов являются оние-
вые соли или вещества, образующие комплексы с катионами
щелочных металлов, такие, как краун-эфиры, криптанды или
их аналоги с открытой цепью. Как уже указывалось в разд. 1.1,
определение МФК основано скорее на наблюдаемых эффектах,
а не на каком-либо едином механизме. Впрочем, широкие
исследования этих эффектов привели к выяснению механизма
многих реакций МФК.
Все реакции можно разделить на те, которые проходят в
нейтральных или кислотных условиях, и те, которые идут в
присутствии высококонцентрированных растворов
неорганических щелочей. Вначале рассмотрим первый тип реакций. К
нему относятся многочисленные реакции замещения, окисления
и восстановления, протекающие в нейтральных или кислых
средах.
2.1.1. Механизм реакций в нейтральных средах
Первоначальный механизм МФК, предложенный Старксом
[2], для реакций замещения приведен ниже
RX + [Q®Ye] > RY + [Q®Xe] (органическая фаза)
» 3 гЕ5СЛ1 (поверхность
1 г ф раздела (раз)
Na® Ye Q® Xе (войная фаза)
Схема 2.1
• Подробное обсуждение физико-хнмнческих аспектов МФК проведено
в работах [1, 68].
2.1. Изучение механизма реакций в различных условиях 45
Ионная пара i[Q+Y~], возникающая при экстракции аниона Y~
в органическую фазу с помощью ониевого катиона Q+, быстро
вступает в реакцию обмена с RX. Образующаяся при этом
новая соль i[Q+X~] возвращается в водную фазу, где Q+ снова
захватывает ион Y~ и начинается следующий цикл.
Предполагаемый механизм очень логичен, поскольку
известно, что ионные пары легко экстрагируются в неполярный орга*
нический растворитель, а ониевые соли в таких гомогенных
растворах обладают высокой реакционной способностью
(гл. 1), однако тщательное исследование его все-таки
необходимо.
Поскольку МФ-катализатор ониевого типа может понижать
поверхностное натяжение или обладать свойствами
поверхностно активного вещества, необходимо вначале установить,
действительно ли реакция проходит в органической среде, а
не на поверхности раздела фаз или в водной фазе.
Некоторые авторы изучили влияние скорости
перемешивания на кинетику реакции. Известно, что скорость типичной
реакции, идущей через поверхность раздела фаз,
пропорциональна скорости перемешивания при изменении последней от 600
до 1700 об/мин i[6]. В то же время реакции МФК не
чувствительны к изменению скорости перемешивания после
достижения определенного минимума, необходимого для разрушения
градиента концентрации по обе стороны от поверхности раздела
фаз. Установлено, что при использовании стандартного лабора^
торного оборудования (магнитные мешалки или механические
мешалки с плоскими лопастями в круглодонных колбах)
скорости реакций с нейтральными реагентами не изменялись
после достижения скорости перемешивания больше 250 [2],
200 t[4], 350 [5] об/мин. В присутствии концентрированного
раствора гидроксида натрия для получения воспроизводимых
результатов необходимы большие скорости перемешивания.
Так, при синтезе эфиров из спиртов и алкилхлоридов в
присутствии разбавленного водного раствора NaOH и катализатора
скорость перемешивания составляла всего 80 об/мин i[7], в то
же время для проведения реакции между спиртами и диметил*-
сульфатом в системе NaOH/катализатор требовалось
существенно более интенсивное перемешивание [8]. При синтезе ди-
галокарбенов из галоформа, 50%-ного водного NaOH и ТЭБА
для получения воспроизводимых результатов необходимо
перемешивание со скоростью 750—800 об/мин [9]. Такого рода
различия могут быть объяснены разным механизмом
протекания реакции (см. ниже), но очевидно, что в нейтральных
условиях реакция не протекает на поверхности раздела фаз.
Тот факт, что й органический субстрат, и липофильный
катализатор в большей или меньшей степени нерастворимы в вод-
46 Глава 2. Механизм межфазного катализа
ной фазе, подтверждает высказанное предположение о
протекании реакции либо в органической фазе, либо (что также
возможно) в мицеллярной фазе. Правильность этих выводов
была показана в ходе дальнейших исследований.
Предположение о быстром установлении экстракционного равновесия
(см. схему 2.1) подтверждено при изучении обмена анионов с
помощью ион-селективных электродов. Из этих данных
следует, что стадия, определяющая скорость, должна проходить в
органической фазе. Константы скорости второго порядка для ал-
килирования нитрофенолята тетрапентиламмония были почти
одинаковы для гомогенной реакции в дихлорметане и для
реакции в системе CH2CI2/H2O {72]. Влияние различных
катализаторов на ход реакции 1-бромоктана с ионом тиофенолята
изучено в системе бензол/вода \[3, 4].
График в координатах 1п(,[1фг]оф/[СбН58-]вф) —время
представляет собой прямую линию, что указывает на обычную
кинетику реакции второго порядка, которую можно было
ожидать, полагая, что никакой границы раздела фаз не
существует. К тому же константа скорости линейно зависела от
концентрации катализатора даже при 20-кратном изменении последней.
Это позволило сопоставить константы скорости, полученные
для реакций в присутствии различных катализаторов.
Аналогичным образом были определены константы скоростей
реакции «-октилметансульфоната в хлорбензоле при большом
избытке хлорида, бромида или иодида в водном растворе. В
настоящей работе в качестве катализаторов использовали гало-
гениды трибутилгексадецилфосфония.
Реакция имела псевдопервый порядок по субстрату:
Скорость = V[ROSOaCHsl
а наблюдаемая константа kK линейно зависела от,
концентрации катализатора [5].
Суммарная реакция
МФК
ИХоф + Y-цф >* RYO* + Х"вф (1)
может быть разбита в соответствии с постулатами МФК на
две стадии — стадию экстракции (2) и собственно химическую
реакцию (3):
[Q+X-Ъф + Y-вф ^ [Q+Y-юф + Х"вф (2>
[Q+Y-](^ + RX -^[Q+x-]<*>+RY (3)
было показано в разд. 1.3.1,
[Q+X-Ьф-твФ
2.1. Изучение механизма реакций в различных условиях 4?
Если обозначить исходную концентрацию катализатора через
4о» то*
{<2+У-]оф-[Х-]Вф = *■(*- IQ+Y-fo*) [¥-]ВФ (5)
ф _
Пвф (6)
Выражение для скорости реакции в органической фазе будет
иметь вид
d [Rxydt = MQ+Y-]<*HRX] (7)
или после интегрирования
£o(Q*) — CO(RX) c0(QY) IRXj
где cO(qy) и cO(RX) — начальные концентрации соответственно
Q+Y~ и RX в органической фазе. Подстановка уравнения (6) в
уравнение (8) дает
n Co(QY) ([
Или после упрощений
COnst/ = COIlst-ln /[V-L 1 y,^i TrovT (1°)
([X Ьф + а[х ]вф)1кХ] v '
Если
1Х-]вф + К РПвф = const (11)
то график в координатах время — 1п(|[У-]Вф/|[КХ]) [или
обратная величина (см. [4])] дает прямую линию.
Равенство (11) выполняется строго только при К=1, как
это бывает в большинстве обычных случаев изотопного обмена.
В тех случаях, когда К не сильно отличается от единицы,
отклонения от прямой могут быть не очень велики.
В работе i[ 10] были рассчитаны профили скоростей второго
порядка для фиксированных условий при варьировании
величины К от 1000 до 0,001. Если 1СЗ>1, то это означает
неправдоподобно большую концентрацию Y~ в органической фазе, что
приводит к кривой, сходной с кривой, получаемой при авто-
катализе. При К<.0,1 реакция останавливается. Это и есть
нежелательный «эффект отравления катализатора», уже
обсуждавшийся в гл. 1: ониевый катион в ходе реакции все больше
и больше ассоциируется с уходящим анионом, что н приводит
к постепенному понижению концентрации катализатора. Отрав-
* В уравнениях типа закона действия масс квадратные скобки означают
концентрацию, в остальных случаях таким способом выделяются ионные пары.
48 Глава 2. Механизм межфазного катализа
ления такого типа можно избежать, если использовать в
реакции молярные количества катализатора. При /С=0,01
увеличение количества катализатора от 1 до 20 мол. % достаточно,
чтобы реакция не останавливалась [10].
Кинетика псевдопервого порядка для процессов при МФК
была изучена неоднократно (см. i[5]). Однако процесс,
подчиняющийся закономерностям реакций первого порядка,
можно ожидать только в том случае, если совместное влияние
величины К и избытка [Y~]b<i> сделают '[Q+Y~]oo
действительно постоянной {уравнение (6)]. В табл. 2.1 приведены вы-
Та блица 2.1. Вычисленные
минимальные молярные соотношения
[Y-]/[RX], достаточные
для наблюдения кинетики
псевдопервого порядка
К
1
1.33
2
10
100
1000
Минимальное- соотношение
[Y-J/IRXJ
10
8
4
2
0.8
0,4
численные минимальные соотношения '[Y~]b*/[RX],
достаточные для наблюдения условий кинетики псевдопервого порядка.
В том случае, когда К очень велико, для наблюдения
кинетики псевдопервого порядка необходимо поддерживать
концентрацию Y~~ ниже, чем эквивалентная, по крайней мере до
тех пор, пока источник ионов Y~ не иссякнет и реакция не
остановится i[10].
Необходимо подчеркнуть, что на величину констант EQX и
Eqy (и соответственно К) оказывают влияние не только
активность ионов, но также и многие другие, рассмотренные в разд.
1.3 факторы, объединенные общим названием «побочные
процессы». Так, например, величины К. >[2] и Jt2 i[2, 4] резко
возрастают, если водная фаза насыщена реагирующей или
образующейся солью.
Кинетика псевдопервого порядка по алкилгалогениду,
растворенному в октане, наблюдалась при его реакции с водным
раствором цианида натрия; катализатором служил трибутил-
гексадецилфосфонийбромид [2]. При этом наблюдалась ли-
2.1. Изучение механизма реакций в различных условиях 49
нейная зависимость между константой скорости и
концентрацией катализатора, величина которой изменялась в 20 раз ;[2].
Однако если в качестве катализатора использовать тетрабутил-
фосфонийбромид, то связь между скоростью и концентрацией
катализатора оказывается не такой простой. Реакция
начинается медленно, и скорость ее увеличивается с течением
времени. Авторы отмечают, что в двухфазной системе бромоктан/во-
да почти весь трибутилгексадецилфосфонийбромид находится в
органической фазе, в то время как большая часть тетрабутил-
фосфониевой соли остается в воде. В ходе реакции происходит
накопление продукта реакции — октилцианида (нонанитрила),
являющегося экстрагентом для тетрабутилфосфониевых солей.
В результате скорость реакции постепенно увеличивается. Для
того чтобы проверить, действительно ли концентрация тетрабу-
тилфосфониевой соли в органической фазе возрастает по мере
увеличения конверсии 1-бромоктана, был использован
ВщР+Вг", содержавший 14С. В ходе реакции проводили анализ
проб, взятых из органической фазы, определяя в них
количество исходного вещества, продукта реакции и 14С. Из
полученных данных были вычислены константы скорости первого
порядка, определена концентрация Bu4P+Bi— и построены
соответствующие графики. Оказалось, что в двойных
логарифмических координатах между ними существует линейная
зависимость, причем прямая имеет наклон, равный единице.
Из этих данных однозначно следует, что
а) реакция проходит в органической фазе;
б) скорость ее пропорциональна концентрации катиона
катализатора в органической фазе.
Эти результаты исключают участие в реакции мицелл.
Установив, что реакция происходит в органической фазе, Старке и
Оуэне i[2] выдвинули другое предположение — о возможности
протекания реакции в инвертных мицеллах. Для разрешения
этого вопроса были изучены агрегация и гидратация ониевых
солей в органических растворителях. Установлено, что при
концентрации солей от 0,04 до 0,25 моль/л трибутилгексадецил-
фосфонийбромид, трибутилгексадециламмонийбромид и триде-
цилметиламмонийхлорид в бензоле и 1-бромпропане образуют
агрегаты с числом молекул от 1,2 до 5,4. Подобные же
значения величин агрегирования получены и для длинноцепочечиых
аммониевых солей жирных кислот в неполярных
растворителях, что и позволило сделать предположение о существовании:
«инвертных мицелл» ([11].
Однако в границах указанных выше концентраций,
несмотря на значительные изменения в степени агрегации (особенна
в бензоле), линейная зависимость скорости реакции от
концентрации катализатора сохраняется. Кроме того, необходим
90 Глава 2. Механизм межфазного катализа
мым условием механизма с участием инвертных мицелл
является перенос этими мицеллами в органическую фазу цианида
натрия. Однако доказать этого не удалось: концентрация
цианида натрия в органическом слое в системе 1-хлороктан/вод-
ный раствор NaCN и 0,01—0,1 моль/л четвертичной соли была
меньше 10~5 моль/л.
Исследованию ассоциации ониевых солей уделялось
внимание и в последующих работах [66, 67]. Показано, что степень,
ассоциации возрастает при уменьшении полярности раствори-;
теля. Так, при концентрации 0,5—5-10~2 М степень агрегации
хлорида и бромида гексадецилтрибутиламмония изменяется
от 1—2 в хлорбензоле до 4—6 в циклогексане '[67].
В этих неполярных растворителях в гомогенных условиях
соблюдается тот же порядок активности групп при замещении,
что и в диполярных апротонных растворителях: СМ~>Ыз~>
>C1~>-Br~>-I->SCN-. Константы скорости в циклогексане
в 5—7 раз больше, чем в хлорбензоле, но еще большие
различия наблюдаются при переходе от циклогексана к ДМСО и
особенно при переходе от цнклогексана к метанолу [67]:
*цг/*дмсо = 10-30; Ацг/*сн,(ОН = 20-1600(0
Степень гидратации отдельных ионов была измерена путем
встряхивания раствора (k-Ci6H33)BuP+X~ в толуоле или в
1-цианоктане с тритированной водой [2]. Средняя величина
гидратации для X~=NO3~, Сг~ и CN~ была равна
соответственно 0,4, 4 и 5. Количество экстрагированной воды в
хлорбензол при X~=Q-, Вг~, 1~ составляет 3,4, 2,1 и 1,1 молекулы
Н2О на 1 анион (катион был тот же).
Впоследствии было проведено сравнение скоростей реакций
яуклеофильного замещения между к-октилметансульфонатом и
ионами галогенидов, измеренных как в гомогенных условиях,
так и в условиях МФК*-
В безводных гомогенных условиях абсолютные скорости
реакций более высокие. Относительные величины скоростей для
С1~:Вг~:1~ равны 6,5:2,5:1,0. В условиях МФК из-за влияния
гидратации изменялись и абсолютные величины скоростей и их
соотношение: для С1-:Вг~:1~ оно стало 0,6:1,1:1,0.
Если в хлорбензол добавить столько же воды, сколько ее
переносят с собой фосфониевые галогениды в двухфазной
системе, и провести реакцию замещения в гомогенных условиях,
то воспроизводятся и абсолютные, и относительные величины
скоростей, характерные для МФК [5]. Таким образом, на
общую скорость реакции влияет гидратная оболочка ионной па-
* 1 моль к-октилметансульфоната экстрагирует в органический слой
0,15 моля Н2О.
2.1. Изучение механизма реакций в различных условиях 51
ры, включающая некоторое ограниченное число молекул воды.
В ходе дальнейших исследований реакции к-октилсульфона-
та с ионами Cl~, N$~, Вг~ и 1~ в системе хлорбензол/вода
использовали эти же катализаторы — СюНззВщР+Вг-, ВщР+Вг-
и ВщЫ+Вг-
Изменение концентрации неорганической соли в водном
слое вплоть до насыщения (~6М) не влияло на степень
гидратации и тем самым на реакционную способность
экстрагируемых анионов. Однако в присутствии 60%-ного КОН или
50%-ного NaOH скорости становились такими же, как и в
гомогенной реакции в безводных условиях, а сами скорости
увеличивались в 13, 4, 2,6 и 1,4 раза для С1-, Ns", Вг~ и I-
соответственно. Этот эффект не наблюдался при использовании
менее концентрированных растворов щелочи или насыщенного
раствора NaF i[98].
При изучении реакции л-нитрофенолята с метилиодидом в
различных растворителях было найдено [68], что скорость
реакции в гомогенных условиях в «сухих* растворителях выше в
5—34 раза, чем скорость реакции в растворителях,
насыщенных водой. Однако если в водный слой добавить 30—50%,
NaOH, то из-за его осушающего действия скорость реакции
возрастает, достигая lU (в присутствии 30% щелочи) н */г
(для 50%) от величины скорости в сухом растворителе.
В статье, посвященной кинетике МФК. Гордон и Кутина
[10] подробно обсуждают возможность совместного
использования соотношений, описывающих процесс экстракции и
химическую реакцию [уравнения (1) и (2)]. Некоторые из их
расчетов и графиков пригодны для особых целей. С их помощью
можно выбрать такую систему для измерения скоростей
реакций, которая будет вести себя в согласии с простыми
кинетическими закономерностями. При этом особое внимание
следует проявлять при сравнении катализаторов в сходных
экспериментах с использованием различных концентраций ониевых
ионов и/или ионов щелочных металлов.
Однако еще большее значение имеют те случаи, когда
суммарная реакция является обратимой:
RY Х-ВФ (1а)
Составные части этого равновесия можно представить в
форме уравнения (2) (неизмененное) и уравнения (За):
(2)
ОФ
[Q+Y-Ьф + КХ ч=* [Q*X-]o* + RY (За)
52 Глава 2. Механизм межфазного катализа
Из них следует, что
(12)
Однако более тщательный расчет показал, что равновесие,
описываемое уравнением (12), в общем случае может быть
достигнуто только при К, близком к единице, или же в
присутствии молярного количества катализатора. Если К сильно
отличается от единицы (например, 1000 или 0,001), то истинное
положение равновесия (по крайней мере в практически
доступном интервале времени) будет определяться количеством исг
пользованного катализатора. Рассмотрим реакцию, в которой
использовали 1% катализатора (0,01 моля), /Соф = 10000, К=
=0,001, а концентрация и RX и Y"~ равны 1 моль/л. По мере
образования Х~ он постепенно «отравляет» катализатор и прямая
реакция (1а) идет все медленнее и медленнее. В это же время
обратная реакция (1а) ускоряется. Используя уравнение (4),
можно вычислить* число молей катализатора z, входящих в
ионную пару с Y~, при конверсии 10%:
*' ==°-001 и г = 8-
(р,О-«)-О.Э
Таким образом, уже при 10%-ной конверсии 99,1%
катализатора находится в форме ионной пары с Х~, а не с Y~, и прямая
реакция фактически останавливается. Найденное положение
равновесия соответствует приблизительно 10%-ной конверсии,
и это несмотря на то, что максимальное значение К* может
■быть равно 10! Другие примеры такого рода расчетов
приведены в работе ;[10].
На общей схеме МФК (с. 44) показано, что катион
катализатора мигрирует вместе с различными противоионами из
одной фазы в другую. Возможность такого механизма
исключается при замене стадии (1) или (1а) на стадию (2) и (3)
или (За) соответственно. Тем не менее остается вопрос:
необходимо ли вообще физическое перемещение катиона между
двумя фазами?
Количество катализатора, находящегося в органической или
в водной фазе, зависит от типа растворителя и от липофильно-
•сти используемой ониевой соли. Независимо друг от друга
Брендстрём и Монтанари показали, что для проявления МФК
нет необходимости в миграции катиона катализатора**.
* При этом предполагается, как это было принято в разд. 1.3, что
побочные реакции отсутствуют и вместо активностей компонентов используются для
■расчетов их концентрации.
** Недавно тот же вывод был сделан на основе изучения сцинтилляции
жидкостей [97].
2.1. Изучение механизма реакций в различных условиях 53
В узкий цилиндр, наполненный водным раствором 4-(2,4г
динитрофенилазо)феноксида натрия, ниже верхнего уровня
.жидкости вводили по каплям раствор бромида четвертичного
аммония в дихлорметане [12]. В воде этот индикатор имеет
красный цвет, соответствующий его анионной форме, а в
дихлорметане — голубой, характерный для его ионной пары с
Q+. При падении капли в цилиндре она становится голубой,
что указывает на обмен анионами. Если бы одновременно в
водную фазу экстрагировался и Q+, то он не смог бы
вернуться обратно в покинутую им быстро падающую каплю. В
другом эксперименте использовали U-образную трубку,
содержащую две независимые органические фазы, разделенные
водным раствором неорганической соли [13]. Применяемые
катализаторы сильно различались по липофильности — от очень
липофильных, которые на 100% находились в органической
фазе, до таких, которые частично растворялись в водном слое.
В одну из органических фаз прибавляли «-октилметансульфо-
лат, а в другую — один из катализаторов.
При использовании менее липофильного катализатора
спустя некоторое время, необходимое для того, чтобы прошла его
диффузия через среднюю фазу, начиналась реакция, скорость
которой постепенно возрастала от нуля до некоторой
постоянной величины. Однако при введении высоколипофильных катаг
лизаторов Od^N+Bi— и (С^Нзз) ВизР+Вг~ никакой межфазной
реакции в другой органической фазе не наблюдалось. Но
реакция начиналась тотчас же при добавлении катализаторов в
органическую фазу, содержащую субстрат.
Из этих экспериментов ясно, что схема 2.1 должна быть
несколько изменена. На схеме 2.2 с учетом этих данных
изображен реальный процесс МФК с применением липофильных
катализаторов.
(органическая
RX + [QeY6] > RY + [QeX6] qjcaa)
I 1 _ (поверхность
у " " ~ i развела <раз)
Na®Ye Xе (вовная фаза)
Схема 2.2
Для понимания механизма МФК важно ответить еще и на
другой вопрос: осуществляется ли реакция в органической
среде через ионные пары или же через свободные анионы?
В гл. 1 было показано, что константа диссоциации ионных пар
в неполярных средах, типичных для МФК, имеет порядок
10~5. Таким образом, концентрация свободных анионов очень
мала и реакция с ними крайне маловероятна. Тем не менее
Брендстрём для ответа на этот вопрос исследовал кинетику
64 Глава 2. Механизм межфазного катализа
обмена галогенов в условиях МФК 1[14]. Если реакция
проходит через стадию образования свободных анионов, то ее
скорость должна быть пропорциональна корню квадратному
из концентрации -[Q+X"] в органической фазе. Однако
поскольку на самом деле скорость прямо пропорциональна
концентрации '[Q+X~], то это означает, что доминантными нуклео-
филами в пределах исследованных концентраций являются
ионные пары. В другом обширном исследовании по введению
алкильных групп в р-дикетоны методом экстрактивного а л кил и-
рования (МФК) i[15] было найдено, что в качестве нуклеофила
могут в принципе выступать как свободные анионы, так и
ионные пары с тетрабутиламмониевым противоионом. Но при
проведении препаративных синтезов в растворах дихлормета-
на все же только ионные пары являются действительно
активными нуклеофилами.
Образование сложных эфиров при реакции бензилхлорида
с твердыми солями в толуоле, катализируемой третичными
аминами или четвертичными аммониевыми солями, также
проходит в органической фазе и имеет первый порядок по субстрату
и катализатору '[94]. Кинетика замещения i[81—83] и этери-
фикации с использованием краун-эфиров в качестве МФК в
системах жидкость/жидкость [55, 81—83] и твердая
фаза/жидкость [73] также подтверждает общую схему механизма МФК.
Реакцию между твердым фенолятом калия и алкилгалогенида-
ми в толуоле могут катализировать даже линейные полиэфиры,
связанные с полимерным носителем, и кинетика реакции
оказывается точно такой же, как и с растворенным катализатором.
Эти наблюдения указывают на возможность тесного контакта
между смолой-носителем катализатора и твердой солью ;[74].
2.1.2. Механизмы реакций, проходящих в присутствии
гидроксидов щелочных металлов
Выше мы уже показали, что механизм МФК в нейтральных
условиях был полностью изучен и разъяснен. Все наблюдаемые-
эффекты могли быть удовлетворительно объяснены схемой 2.1
или 2.2 и соответствующими уравнениями, описывающими
равновесные и кинетические стадии. Теперь давайте рассмотрим
другой тип МФК, включающий реакции, которые идут в
присутствии оснований. Мы увидим, что в этом случае картина не
является достаточно четкой.
В присутствии сильных оснований проводят реакции
различного типа, такие, как С-, О-, N-алкилйроваййе,
изомеризация, H/D-обмен, присоединение, а- и р-элиминирование,
гидролиз и многие другие.
2.1. Изучение механизма реакций в различных условиях 55
Примеры практического использования МФК для
проведения этих, по-видимому, наиболее многочисленных и интересных
-с препаративной точки зрения реакций рассмотрены в гл. 3.
Начнем наше рассмотрение с реакции алкилирования,
которая должна характеризоваться сильной зависимостью от рКа
соответствующей С—Н-, N—Н- или О—Н-кислоты.
Относительно сильные кислоты, такие, как, например, ацетилацетон,
растворяются в гидроксиде натрия. Соответственно работа
катализатора межфазного переноса состоит в реэкстракции аниона
в форме ионной пары обратно в органическую фазу, где и
проходит С- или О-алкилирование (в разд. 3.10 см. о направлении
алкилирования амбидентных анионов). Другими словами, в
этом случае действует механизм, представленный на схеме 2.2.
Однако легко показать, что при алкилировании слабых
кислот, например алифатических спиртов (р/Св«18), в
органическую среду экстрагируются одновременно незаряженный
субстрат и его анион. Основными равновесными стадиями будут
NaOH + ROH ч—*- Na+OIT-f Н2О (водная фаза)
ИОНвф ■?—*- ШНОФ (обе фазы)
[Q+X"b* + Na+OR-вф =«=* (Q+OR-fofl, + №+Х-Вф (обе фазы)
Кроме того, важное значение могут иметь следующие
равновесия:
[Q+X-foo + Na+OH-вф =<=* [Q+OH-Ьф + Na+Х'вФ (обе фазы)
[Q+OH-]O<I> -f- ROHq* s=fc [Q+OR-Ьф + НЯО (органическая фаза)
и наконец, необратимая стадия образования эфира:
[Q+OR-Ьф + R'X »■ R'OR + [Q+ХТоф (органическая фаза)
В разд. 1.3.4 уже было показано, что по сравнению с ионом
галогена очень гидрофильный гидроксил-ион экстрагируется с
трудом. Поскольку OR- более липофилен, чем ОН-1, то оба
равновесия, включающие [Q+OH~], реализуются, вероятно,
только крайне редко. Тем не менее очевидно, что описание
взаимодействия остальных равновесных стадии существенно
сложнее, чем для простых реакций замещения.
Фридман и Дюбуа [7] получили несимметричный эфир из
первичного алкилхлорида и спирта встряхиванием их с
пятикратным избытком (по отношению к спирту) 50%-ного водного
раствора едкого натра при 25—70 °С. В качестве
катализатора использовали 3—5 мол. % B114NHSO4, а растворителем
служил сам алкилгалогенид. Механизм этой реакции был
подробно изучен. При использовании одного эквивалента бензилхло-
рида в ТГФ, избытка м-бутанола и раствора едкого натра,
насыщенного поваренной солью, реакция имела первый порядок
>6 Глава 2. Механизм межфазного катализа
по катализатору и по бензилхлориду. При
использовании эквимольных количеств реактантов наблюдался ясно-
выраженный второй порядок вплоть до 80% превращения.
Энергия и энтропия активации были 13,9±0,5 ккал/моль и
—25,5±1,6 энтр. ед. соответственно. Как и следовало
ожидать, исходный противоион в катализаторе оказывал замет^
ное влияние на скорость реакции вследствие конкурентной
экстракции. Самая большая скорость наблюдалась с HSO4~*^
очень малая — с 1~ и еще меньшая — с CICU".
Эти результаты полностью соответствуют обычной схеме-
МФК, при этом экстракция иона RO~ и, возможно, его
образование в органической фазе (хотя и в меньшей степени)
являются быстрыми процессами. Были получены следующие
данные о сравнительной экстрагируемости ОН~: в равновесных
условиях из водной фазы, содержащей 10~2 М BU4NHSO4, 50%
NaOH и насыщенной NaCl, в ТГФ переходит 86% катиона. Иа
них только 10,7% связаны с ОН~, а остальные — с хлоридом-
Если в ТГФ растворен 1 моль/л бутанола, то весь ВщЫ+
находится в органической фазе и только 5,4% его связаны с С1,
а остальные катионы образуют [C4H9O~Bu4N+] (последующие
работы, посвященные кинетике образования эфиров, см. в
[71]).
Когда реакция прошла на 80% и более, а содержание
исходного ROH в реакционной смеси снизилось, становится
заметен гидролиз избытка алкилгалогенида до спирта, затем этот
новый спирт образует симметричный эфир {7]:
R'Cl »- R'OH >- R'O—R'
Этот тип реакции впервые был открыт Херриотом и Пик-
кером |[3, 23]. Эти же авторы исследовали реакцию между
вторичными алкилбромидами, водным NaOH и межфазными
катализаторами. Основная часть субстрата (81—94%)
подвергается элиминированию, а остаток гидролизуется. Такое
соотношение характерно для реакций в органических
растворителях в отличие от реакций в водных растворах.
Рассмотренные выше результаты Фридмана и Дюбуа [7]
были подвергнуты Гордоном и Кутиной [10] подробному
математическому анализу. Предполагая, что приведенные выше
равновесия имеют место, последние установили, что Фридман
и Дюбуа работали в границах экспериментальных условий,
обеспечивающих линейность кинетических данных в
координатах графика для реакций второго порядка. Фридман и Дюбуа
использовали довольно необычную систему (ТГФ/конц. NaOH,
* В NaOH иои HSO4- превращается в не способный к экстракции ион
SO*2- (см. гл. П.
2.1. Изучение механизма реакций в различных условиях 57
насыщенный NaCl) для того, чтобы установить факт
экстракции аниона слабой кислоты — к-бутанола. В работе [25] была
выбрана система, более обычная для МФК: 0,01 моля бензил-
триэтиламмонийхлорида в 100 мл бензилцианида находились
в равновесии со 100 мл концентрированного раствора NaOH*.
В органической фазе было обнаружено 63% основания от
теоретического количества. Следовательно, в этих условиях
только треть катализатора находится в форме хлорида
и может осуществляться нормальная реакция МФК-
Основность в органической фазе обусловлена ионной парой
rPhCHj*N+Et3-CH(CN)Ph]. Конкурирующая с ней ионная пара
{PhCHaN+Et3OH-] либо быстро вступает в кислотно-основное
взаимодействие с избытком фенилацетонитрила, либо остается
в водной фазе, поскольку ее липофильность недостаточна для
экстракции в органическую фазу в сколько-нибудь заметном
количестве. Было показано, что при равновесии между
бензольным раствором ifBtuN+Q-]** и раствором NaOH около 50%
катиона находится в органическом слое и только 4% этого
количества связаны с ОН~ '[30].
Хотя растворимость i[R4N+OH-] в средах типа
фенилацетонитрила лучше» чем в бензоле, однако различие в константах
экстракции ОН~ и С1~ препятствует присутствию в
органической фазе заметного количества гидроксида (разд. 1.3). Кроме
того, как уже отмечалось выше, это незначительное количество
ОН~ будет реагировать с раствором фенилацетонитрила.
Теперь рассмотрим существенно более слабые С—Н- и
N—Н-кислоты. Было показано, что в присутствии
концентрированной водной натриевой щелочи, выступающей в качестве
депротонирующего агента, может проходить МФК-алкилирова*
ние даже таких слабых кислот, рКа которых около 22—25
(в зависимости от выбранной шкалы значений рКа). Это тем
более удивительно, что промежуточный анион на много
порядков величины основнее, чем вода.
В ранних работах по МФК реакций в присутствии сильных
неорганических оснований предполагалось, что механизм этих
реакций включает экстракцию [Q+OHh] в органическую среду,
где и происходит депротонирование и алкилирование
субстрата:
[Q+X-Ьф + НО-ВФ =?=5= [Q+OH-Ьф + Х-Вф (обе фазы)
[О+ОН-Ьф + HSub ^—*■ [Q+Sub-Ьф + Н2О (органическая фаза)
[<2+8иЬ-]Оф + RX >- [<2+Х-]ОФ + R—Sub (органическая фаза)
* Хотя точное значение рКа бензилцианида в водных средах неизвестно,
но очевидно, что это приблизительно 15—16. Фенилуксусный эфир имеет
рКа=17 Г24].
** ТЭБА еще более гидрофилен и практически не растворим в бензоле.
58 Глава 2. Механизм межфазного катализа
Однако против этой схемы было выдвинуто много
возражений. Мы уже видели, что гидроксид по своей природе
экстрагируется с трудом. Это связано как с его низкой
растворимостью, так и с малой константой экстракции. Эти трудности:
увеличиваются еще больше, если в конкуренцию за
каталитическое количество имеющегося четвертичного аммониевого
катиона вступает какой-либо более липофильный анион,
например галоген. По мере образования иона галогена в ходе
реакции экстракция гидроксида будет все больше и больше ингиби-
роваться. Правда, это рассуждение не имеет общего характера;,
например, оно, по-видимому, не пригодно для хлорид-иона.
Кроме того, равновесие депротонирования между Н—Sub ir
гидроксидом четвертичного аммония в органической фазе
сильно сдвинуто влево из-за большой разницы в кислотности
(субстрата и воды).
В более поздней гипотезе, предложенной Макошей {26, 27],.
было высказано предположение, что депротонирование
субстрата происходит на поверхности раздела фаз. Если
катализатор в системе отсутствует, то на поверхности раздела фаз
образуется как бы двухслойная структура, включающая со
стороны водной фазы катион щелочного металла, а со стороны
органической фазы депротонированный анион субстрата. Из-за
взаимной нерастворимости в противоположных фазах ионы
иммобилизуются и в значительной степени дезактивируются. Эта
ситуация похожа на обычную адсорбцию на поверхности.
Макоша показал, что некоторые реакции алкилирования;
проходят в двухфазных системах в отсутствие катализатора
межфазного переноса. Из этого следует, что местом реакции
является поверхность раздела фаз '[26, 27, 31]. Так, например»
при встряхивании фенилацетонитрила, и-бутилиодида и 50% •
ного водного едкого натра при 80 °С в течение 7 ч алкилирова-
ние проходит с выходом 70% i[31]. Установлено, что
концентрация нитрила в водной фазе в этих условиях составляет
менее 2 млн.-1, а концентрация натриевой соли субстрата в
органической фазе — около 5 млн.-1. Таким образом, реакция
возможна только на поверхности раздела фаз \[31]. АЛкилброми-
ды R'Br в этих условиях не вступают в реакцию, однако если
использовать смесь R/Br/C4H9I, то образуются два продукта
алкилирования: Ph—CH(R')— CN и Ph—CH(C4H9)—CN.
Очевидно, что реакция проходит через образование R'l.
Поскольку в отдельном эксперименте было обнаружено, что между
водным раствором иодида натрия и R'Br обмена галогенидами не
происходит, то этот обмен осуществляется'на поверхности
раздела фаз со стороны органической фазы между иодидом,
освобождающимся из С4Н91 при алкилировании, и R'Br i[31].
Результаты исследования межфазных реакций в этой и других
2.1. Изучение механизма реакций в различных условиях 59
работах [26, 27] подчеркнули важную роль поверхности
раздела фаз. Эта точка зрения подтверждается также
необходимостью для получения стабильных и воспроизводимых
результатов более сильного перемешивания при проведении
межфазных реакций с участием водных растворов едкого натра (см.
выше), хотя в этом случае положение, несомненно, еще
осложняется более высокой вязкостью концентрированных растворов
NaOH.
В случае обычных реакций МФК также было показано, что
депротонирование идет на поверхности раздела фаз. Действие
катализатора в этом случае может быть двояким:
а) Катализатор способствует депротонированию. Это может
быть следствием того, что положительный заряд катиона,
противостоящий реакционному центру, облегчает уход протона.
б) Катализатор способствует освобождению аниона
субстрата, «закрепленного» на поверхности раздела фаз* (ПРФ).
Этот отрыв депротонированного субстрата от поверхности
раздела фаз имеет более важное значение, чем а), поскольку
сопровождается одновременным переходом исходного противоио-
на катализатора в водную фазу.
Это равновесие в целом сдвинуто в нужную сторону,
поскольку типичные анионы органических субстратов более липо-
фильны, чем ионы галогенидов.
Однако время от времени появляются работы, в которых
описывают «отравление катализатора» липофильными
неорганическими анионами (например, перхлоратом, пикратом**,
иодидом). Это происходит в тех случаях, когда образующийся
на промежуточной стадии органический анион умеренно-липо-
филен.
Таким образом, мы получаем следующую, вполне
удовлетворительную схему механизма алкилирования слабых кислот
в условиях МФК (ПРФ — поверхность раздела фаз, на
которой закреплены соответствующие ионы):
Ыа+ВфОН~вф + HSuboo < * Na+прф + Sub~np« + Н4О (межфазно)
Sub-прф + [Q+X-fo* :*=* [Q+Sub-foo + Х'вф (обе фазы)
[Q+Sub-ЬФ + RX *■ R—Sub + [Q+Х'Ьф (органическая фаза)
* Это выражение, конечно, не означает, что одни и тот же анион
закреплен на поверхности в течение длительного времени, скорее всего имеет место
динамический процесс обмена.
** Первоначальные данные, полученные в лаборатории авторов [28] об
одинаково быстром алкилировании бутилциаиида как с помощью пикрата, так
и с помощью хлорида тетрабутиламмоиия, оказались ошибочными. Пикрат
работает очень плохо. Однако при генерировании дигалокарбенов пикраты
являются хорошими катализаторами. Оказалось, что в этой реакции иои пикрата
исчезает, заменяясь в конце концов на хлорид.
60 Глава 2. Механизм межфазного катализа
В отличие от рассмотренной ранее экстракции i[Q+X~]oo из.
нейтральной водной фазы в органическую ионная пара
[Q+Sub~]o* не может быть гидратирована в сколько-нибудь-
заметной степени. Это происходит вследствие действия двух
факторов: связывания воды концентрированным NaOH,
который является сильным осушителем, и разрушения ионных пар
водой, представляющей собой в этих условиях более сильную-
кислоту.
Эксперименты в отсутствие катализатора позволяют сделать
предположение, что в ходе реакции i[Q+Sub~] образуется
только в малой концентрации и что заключительная стадия
реакции происходит на поверхности раздела фаз. Однако на
самом деле это оказалось не так.
Если использовать концентрированный раствор гидроксида
натрия и катализатор, то можно получить устойчивый раствор
ионных пар, содержащих анион даже такой слабой кислоты,,
как флуорен*. В серии экспериментов [69] раствор 0,1 М
Нех4&Вг в бензоле был эквилиброван с равным объемом
концентрированного водного раствора NaOH (~ 200-кратный
молярный избыток). Титрование органической фазы показало, что
в ней содержится только 1,5% Q+, связанного с анионом ОН~,
а 98,5% соли находится в форме бромида! Если органическая
фаза содержит еще дополнительно и флуорен, то количество
щелочи в ней возрастает до 21%. Значительное увеличение
щелочи, определяемой титриметрически в присутствии флуорена,
окраска органической фазы и положение равновесия для гидр-
оксидов аммония (см. выше) показывают, что «щелочность» в
этом случае следует отнести к ионной паре |[Нех4№С1зНэ~]оФ.
Почднее были тщательно изучены различные параметры
реакции этилирования фенилацетонитрила этилбромидом в
присутствии 50%-ного NaOH и Bu4NBr '[70]. В этом случае
скорость перемешивания в пределах 35—1950 об/мин сильно
влияет на начальную скорость исчезновения реактаитов при
55 °С, и еще большее значение скорости реакции было
установлено при проведении реакции в реакторе высокоэффективного
перемешивания. Наибольшее возрастание скорости (в 20 раз)
наблюдалось при увеличении числа оборотов в минуту от 110
до 200, что сопровождалось исчезновением первоначально
раздельных слоев с образованием однородной смеси. По графику
Аррениуса была вычислена энергия активации, составившая
приблизительно 20 ккал/моль. В двойных логарифмических
координатах график зависимости скорости реакции от
концентрации катализатора представляет собой прямую линию с на-
• В зависимости от способа определения и типа шкалы кислотности
приблизительное значение рКа флуорена равно 20,5—22,8.
2.1. Изучение механизма реакций в различных условиях 61
клоном 0,6 (от 0,0125 до 0,4 М Bu4NBr). Из этого следует, что
при нормальном механизме МФК не следует ожидать простой
линейной зависимости от концентрации катализатора.
Изменения в скорости реакции, полученные при варьировании
концентрации гидроксида натрия в водной фазе и добавления в
нее бромида, также не согласовывались с предположением о
том, что эти изменения связаны с простой экстракцией
гидроксида четвертичного аммония в органическую фазу. Хотя общее
уравнение кинетики реакции вывести не удалось, полученные
данные лучше всего соответствуют механизму, рассмотренному
выше.
Теперь, разобравшись с механизмом алкилирования в
условиях МФК, перейдем к рассмотрению механизма генерирования
дигалокарбенов. Мы тщательно изучим все факты,
относящиеся к генерированию дихлоркарбена, однако полученные
выводы равным образом будут применимы ко всем карбенам,
образующимся при межфазных реакциях. Проведение
конкурентных реакций показало, что дихлоркарбен, генерируемый при
МФК, идентичен дихлоркарбену, получаемому другими
методами i[2, 29], и не является карбеноидом. Кроме того, можно
показать, что в условиях МФК карбен :CXY может, обменивая
галогены, превращаться в :СХ2 и :СУг*. Надо добавить, что в
отличие от всех других методов генерирования
дигалокарбенов при МФК реакция проходит при комнатной температуре
как необратимый быстрый одностадийный процесс. В то время
как смесь трет-бутилата калия с хлороформом реагирует при
—20 °С независимо от присутствия или отсутствия субстрата, а
ЫСС1з распадается обратимо даже при такой низкой
температуре, как —72 °С, реакционная смесь, используемая в МФК —
хлороформ/конц. NaOH/катализатор, — в том случае, когда
отсутствует реактант, взаимодействующий с карбеном, сохраняет
свою способность давать :СС12 даже при комнатной
температуре в течение нескольких дней. Поскольку между хлороформом*
и концентрированным раствором NaOD/D2O наблюдается очень,
быстрый H/D-обмен, который происходит и без всякого
катализатора, то первой стадией должно быть депротонирование на
границе раздела фаз. Предположительно при этом образуется
двойной слой того же типа, что и обсуждавшийся выше:
НСС13 б<ыст > СС13~закреплен (граница раздела фаз)
В отсутствие катализатора гидролиз идет очень медленно;
добавление четвертичных аммониевых солей несколько увеличи-
* Подробное обсуждение см. в разд. 3.20.1.
62 Глава 2. Механизм межфазного катализа
вает скорость гидролиза (см. табл. 3.18). При введении в смесь
олефина скорость расхода хлороформа возрастает, а
конкурирующая реакция гидролиза подавляется. Величина этого
эффекта зависит от нуклеофильности олефина. Другими словами,
имеется конкуренция между олефином и водой за :ССЬ, в
которой олефин побеждает, так как он находится в той же фазе,
что и предшественник карбена. Аналогично тому, как это было
сделано выше при обсуждении алкилирования слабых СН- и
NH-кислот и флуорена, можно предположить, что катализатор
снимает ССЦ~ с поверхности раздела фаз в форме
[R4N+CCls~] оф- Для того чтобы проверить это предположение,
была приготовлена смесь 0,1 М раствора [R^N+CCb"] в
хлороформе и концентрированном NaOH. После достижения
равновесия смесь разделяли, органическую фазу высушивали,
отбирали одну аликвотную часть фазы и встряхивали ее с
олефином. Другую аликвоту оттитровывали НС1, а третью осторожно
упаривали. Никаких доказательств существования :[R4N+CCl3~]
или ее соответствующих производных получено не было <[9, 29,
32].
Впоследствии было показано, что ![R4N+CCl3~] неустойчива
при комнатной температуре. Твердый трихлорацетат натрия
можно частично перевести в хлороформ с помощью '[R4N+C1~],
который дает i[R4N+~O2CCCl3]. Эта соль медленно декарбокси-
лируется при комнатной температуре, давая дихлоркарбен,
который можно уловить олефином. Если «ловушки> нет, то
выделить [R4N+CCl3~] не удается вследствие быстрого осмоления
(33]. Хотя i[R4N+CCl3~] и не устойчива в растворе, она
должна быть интермедиатом при генерировании :СС12. Механизм
карбеновых реакций в условиях МФК, который включает все
доступные доказательства, приведен ниже. Первой стадией
является депротонирование на поверхности раздела фаз и
образование «закрепленной» тригалометилидного иона, НаО
остается в водной фазе:
НСОаоф H-Na+вф + ОН"вф =♦=*= СС13-ПРФ + Na+пРф + Н„О (межфазно)
Затем следует стадия «снятия» аниона. Однако равновесие
на этой стадии сильно сдвинуто влево, что стабилизирует все
последующие стадии:
ОС18-Прф + Р*4М+Х-]оф =*==* [R4N+CCl8-]o« + Х"ВФ (обе фазы)
Известно, что образование карбена обратимо:
[R4N+CCl8-]c№ *=± Р^+С1-]ОФ + [СС12]Оф (органическая
2.1. Изучение механизма реакций в различных условиях 63
Л образование аддукта необратимо:
С1С1
[СС]3]О<Р + ^>=<^ *. -/ \ (органическая фаза)
Схема 2.3
Эти реакции конкурируют с необратимой реакцией гидролиза,
которая протекает на границе фаз и потому идет медленно:
[СС13]П_ + ОН.фв > [НОСвС12] —^
^__^.формиат + СО (обе фазы)
[СС13]оф + Н2ОВ<Р > [НОСНСУ -""^
Схема 2.4
Кроме того, могут проходить реакции обмена:
[СС13]Оф+[NR,*Xe]0(p ;p=^ [NR<*CXCl2e]o<p (органическая (раза)
[М^*СХС13е]0(р+ Xetf ^=± CXCIf прф + [NR^xe]^№.6е фазы)
НСХС13
Схема 2.5
Другие медленные побочные процессы включают
взаимодействие СС1Э- и СС12 с НСС13 (разд. 3.20.1).
Еще раз следует подчеркнуть, что важной особенностыо
предлагаемого механизма является стабилизация
предшественника карбена, «динамически связанного» в форме тригало-
метилидиого аниона на границе раздела фаз. Кинетика таких
реакций и реакций алкилирования слабых кислот не
исследована. Их изучение осложняется гетерогенностью системы,
конкурентными реакциями, сложными равновесиями, а также
общими ограничениями, связанными с получением линейных
зависимостей для констант скоростей второго порядка (см. [10]).
Однако, несмотря на все эти трудности, известные факты, по-
видимому, согласуются с рассмотренным выше механизмом.
Мы уже показали, что в случае МФК алкилирование и
генерирование карбенов в присутствии гидроксида натрия могут
проходить по трем мало отличающимся механизмам, однако
ясно, что МФК включает целый ряд и других механизмов.
<64 Глава 2. Механизм межфазного катализа
Наиболее простой механизм МФК в присутствии сильных
щелочей (например, механизмы H/D-обмена и изомеризации),
по всей видимости, включают экстракцию гидроксида. Многие
другие механизмы глубоко не изучены. В случае МФК
механизмы могут сильно изменяться в зависимости от характера
субстрата и условий реакции. Так, например, ^-элиминирование
может проходить межфазно, если катализатор облегчает
стадию депротонирования. В то же время, если в органической
фазе присутствуют малые количества ионов гидроксида
четвертичного аммония, то и депротонирование будет осуществляться
в этой же фазе. Однако известен еще и третий механизм. Он
наблюдается в отсутствие оснований при повышенных
температурах. В неполярных средах относительно несольватированные
ионы галогенидов ведут себя как основания (см. гл. 1); на-
лример, пентахлорэтан дегидрохлорируется галогенидами
аммония в условиях запатентованного промышленного процесса:
R4NX
С13С-СНС12 -j^ CJ2C=CCJ2 + HCl(ra3)
полагать, что и некоторые другие реакции
элиминирования включают образование соли /[R4N+HX2~], которая затем
диффундирует к границе раздела фаз, где и происходит
нейтрализация.
Другие реакции МФК требуют, очевидно, экстракции гидр-
■оксид-иона. Среди них отметим реакции гидролиза и омыления.
Как было показано при использовании в качестве модельного
-субстрата дихлорметана и Bu4NHSO4 как катализатора,
гидролиз проходит довольно быстро, поскольку в отсутствие более
липофильного, чем ОН~, аниона [Bu4N+OH~] может
экстрагироваться в органическую фазу ![30]. Скорость реакции
постепенно снижается до очень низкой из-за того, что образующийся
хлорид-ион дает ионную пару с катализатором в органической
фазе.
Реакция омыления в условиях МФК чрезвычайно
чувствительна ко многим факторам — растворителю, катализатору и
структуре субстрата [34], в результате чего механизм реакции
может быть различным. В том случае, когда образующаяся
новая кислота очень гидрофильна (например, низкомолекулярная
дикарбоновая кислота), МФК очень эффективно ускоряет
омыление. Напротив, МФК-омыление эфиров липофильных кислот
протекает вяло. При варьировании исходного аниона
катализатора (катион Вщ№")при омылении диэтилового эфира адипи-
новой кислоты в одинаковых условиях был установлен
следующий порядок скоростей реакции: HSO4~»Cl->Br->Г~>
>С1О4~. При реакции в Bu4N+HSO4~ аммониевая соль в
органической фазе находилась в форме ,[Bu4N+HOOC(CH2)4COO~]
2.1. Изучение механизма реакций в различных условиях 65
[34]. Этот анион, по-видимому, легко обменивается через
поверхность раздела фаз с ОН~. При наличии других, более ли-
пофильных неорганических ионов экстракция ОН~, а также
экстракция и транспорт НООС(СН2)4СОО~ частично ингибиру-
ются.
При омылении диэтиладипата нейтральные
поверхностно-активные вещества типа
QeHs3(O-CH2CH2)n—R(R = OH, ОСОСН3, ОСН3, С1)
были активны в той же степени, что и ониевые соли [35].
Очень эффективен был также стеарат натрия, что указывает на
возможность поверхностных реакций (см. табл. 2.7). Хотя
полиэфиры могут давать комплексы с гидроксидом натрия того
же типа, что и краун-эфиры, их влияние часто объясняется
поверхностно-активными свойствами или образованием мицелл.
Однако многие детали механизма еще требуют выяснения. На
основании изучения скорости диффузии были определены в
широких пределах скорости гидролиза эфиров в водно-толуольных
смесях без катализатора; интенсивное перемешивание
приводило к огромному увеличению скорости. Однако прибавление
анионного ПАВ — додецилсульфата натрия — несколько тормозило
реакцию '[41].
Предварительные эксперименты показали, что по крайней
мере в типичных условиях МФК каталитическая активность
(рниевых солей не изменяется параллельно их влиянию на по-
лерхностное натяжение i[35]. В то же время считается, что
многие реакции гидролиза проходят в мицеллах и обратных
мицеллах. Некоторые реакции иного типа, рассмотренные в
этой книге, также могут проходить в условиях образования
мицелл [11].
Надо заметить, что возможность протекания реакции между
алкилгалогенидами и основаниями внутри мицелл в качестве
реакционной среды исключается на том основании, что
эффективные МФ-катализаторы, как правило, представляют собой
липофильные ониевые соли с объемистыми, большей частью
симметричными заместителями [23]. Типичные мицеллообразу-
ющие агенты имеют небольшую полярную группу [например,
(СНз)з№—] и длинный липофильный хвост. Хотя некоторые
симметричные тетраалкиламмониевые соли могут до некоторой
степени агрегироваться в воде [36], вопрос о том, является ли
этот процесс следствием мицеллообразования, остается
открытым [37]. Кроме того, симметричные ониевые ионы имеют
более низкую степень агрегирования, чем типичные мицеллообра-
зующие агенты, и хуже растворяют органические субстраты
i[38]. Однако еще более важен тот факт, что типичные мицел-
лярные реакции проводят в «гомогенных» водных или органи-
66 Глава 2. Механизм межфазного катализа
ческих растворах. В случае МФК субстрат и объемистый
катализатор находятся обычно в органической фазе.
Если субстрат, продукты и органический растворитель
будут путешествовать в водную фазу мицеллы и обратно, то это
сильно запутает все транспортные и диффузионные процессы.
Во всяком случае для некоторых реакций МФК следует
исключить из числа участников процесса как мицеллы, так и инверт-
ные мицеллы. Действительно, вопрос о том, следует ли считать
мицеллами такие агрегаты, у которых степень ассоциации в
органическом растворителе меньше чем 10, остается предметом
обсуждения.
Однако в некоторых случаях мицеллярный катализ может
наблюдаться. Например, «аликват 336» (метилтриоктиламмо-
нийхлорид) является очень эффективным липофильным МФ-
катализатором (см. ниже). Сам по себе он мицеллы не
образует. В водных растворах в отсутствие органических
растворителей он существует в виде масляной суспензии. Однако, если
добавить в смесь какой-либо неионный мицеллообразующий
агент (например, полиоксиэтиленгликоль), аликват уходит
внутрь или на поверхность неионной мицеллы. Образующийся
таким способом катализатор оказывается очень эффективным
во многих процессах [39]. В воде при очень низких
концентрациях (10~4—10~5 М) аликват 336 образует самоассоциаты.
И хотя они существенно меньше, чем обычные глобулярные
мицеллы, они катализируют нуклеофильный гидролиз и реакции
декарбоксилироваиия {40]. Совершенно ясно, что механизм
гидролиза нуждается в дальнейшем тщательном изучении.
2.1.3. Механизм реакций в присутствии других оснований
Наряду с гидроксидами щелочных металлов в МФК
используют также и другие основания: твердые фториды щелочных
металлов, бикарбонаты и карбонаты, гидриды и амиды.
Вопросы о механизме участия в МФК первых двух анионов не
представляют особого труда, так как эти анионы могут
экстрагироваться в органические растворители при обычном
проведении МФК в системе жидкая фаза/твердая фаза (о солюби-
лизации НСО3~ см. в [75]). Однако что касается остальных
анионов, то в противоречии с предположениями, высказанными
во многих статьях, оказалось, что они экстрагируются в
неполярные среды достаточно трудно как с помощью ониевых
солей, так и с помощью краун-эфиров.
Карбонаты. Было показано, что никакой солюбилизации
карбонатного иона из твердой КгСО3 ни в толуол, ни в ацето-
нитрил не происходит по крайней мере при использовании
следующих катализаторов: BmNBr, Hex4NBr, 18-краун-6 i[69, 76].
2.2. Эмпирическая оценка эффективности катализаторов 67
Несмотря на это, МФК-алкилирование в присутствии К2СО3
проходит, так как Н—субстрат образует ионную пару [Q+суб-
страт-]. Это может происходить в некоторых случаях из-за
присутствия следовых количеств бикарбоната, который легко
экстрагируется (см., например, [77]). Однако чаще акт депрото-
нирования происходит на поверхности твердой соли, а действие
катализатора состоит в снятии депротонированного субстрата.
Гидриды и амиды щелочных металлов. Твердые амиды и
гидриды также не растворяются при добавлении таких комп-
лексообразующих агентов, как 18-краун-6 [69] или криптанд
[2.2.2] {78]. Сообщения и предположения об их солюбилизации,
появлявшиеся в литературе, ошибочны. Поэтому все реакции
алкилирования в (присутствии МФ-катализаторов должны
начинаться со стадии депротонирования на поверхности кристалла.
В работе [79] утверждается, что в присутствии чрезвычайно
большого избытка 18-краун-6 происходит частичное растворение
КН в ТГФ. Полученный при фильтровании в инертной
атмосфере раствор был способен депротонировать углеводороды с рКа
ниже 36,3. До этого было уже известно, что депротонирование
такого типа проходит даже в толуоле под действием трет-аыи-
лата натрия в присутствии .криптанда [2.2.2] {80]. Очень похоже,
что гидроксилсодержащие примеси (например, открытый аналог
краун-эфира — гексаэтиленгликоль), которые могут быть в
использованном для реакции большом количестве 18-краун-6,
привели к появлению растворимых алкоксидов, которые в свою
очередь и депротонировали углеводороды. Следует отметить, что
в системе КН/НСРЬз может образовываться ионная пара
[(К"-18-краун-6)+СРЬз~], которая затем в зависимости от
растворителя и примененного комплексанта может либо
раствориться, либо дать тонкую суспензию {59].
2.2. Эмпирическая оценка эффективности
катализаторов
В этом разделе будет сделан обзор имеющихся в литературе
сравнительных данных об эффективности .катализаторов.
Вначале охарактеризуем различные классы ониевых солей и краун-
эфиров — катализаторов, дающих комплекс с катионом. Затем
обсудим селективность реакций, которая наблюдается при
использовании некоторых типов катализаторов.
Основные факторы, которые влияют на поведение
катализаторов, уже обсуждались в разд. 1.3.1—1.3.4 и 2.1. Катион
катализатора должен быть достаточно липофильным, чтобы
обеспечить как достаточную растворимость экстрагируемой ионной
пары, так и высокую степень экстрагируемости ее в органиче-
63г,; Глава 2. Механизм межфазного катализа
скую фазу. Для получения благоприятной константы экстракции
необходимо, чтобы взятый первоначально анион катализатора
и анион, образующийся в ходе реакции, были как можно 'более
гидрофильны. Однако на практике необходимость отделения
использованного катализатора от продуктов реакции может
потребовать применения менее липофильных .катализаторов,
которые переходили бы в водный слой при промывке реакционной
смеси. В результате при выборе ониевой соли приходится
принимать компромиссное решение (разд. 3.1).
Однако следует помнить, что при работе в неполярных
средах даже с самыми лвпофильными катионами их растворимость
и способность к экстракции являются только предварительно
необходимым условием успешного проведения химической
реакции. Как было показано в гл. 1, при сравнении различных
катализаторов видно, что прямая связь между их растворимостью,
экстрактивными свойствами и «анионной активностью» или
скоростью реакции отсутствует. Следует учитывать наряду с
равновесиями, предшествующими реакции, также и другие факторы.
Особенно важными среди них являются взаимодействие аниона
и катиона в ионной паре и количество гидратной воды,
переносимое в органический слой. Поэтому неудивительно, что в
гомологическом ряду, например для симметричных тетраалкиламмо-
нийных солей, при переходе от очень гидрофильных к лшюфиль-
ным ионам активность возрастает чрезвычайно резко, а затем,
когда начинают сказываться эти «другие факторы», она
медленно уменьшается.
Хотя многие авторы проводят сравнение характеристик
нескольких катализаторов, использованных ими в реакции, эти
оценки не пригодны для обобщений, так .как они получены
в различных условиях. Необходимо подчеркнуть, что даже в
параллельных экспериментах сравнение результатов 1может быть
ошибочным, если меняются концентрации катиона
катализатора и неорганического катиона [10].
Ниже мы будем обсуждать работу, где было проведено
такое сравнение при весьма постоянных условиях проведения
реакции. Как правило, рассчитывают только выход реакции, но
в некоторых случаях определяют также и константы скорости.
Поскольку использование гидрофильных катализаторов в
реакциях, идущих в присутствии щелочей, основано на
высаливающем эффекте щелочей, то мы будем рассматривать их
отдельно, как это было сделано в предыдущем разделе.
В табл. 2.2 приведены константы скорости реакции между
тиофеноксидом и 1-бромоктаном в системе бензол/вода [4]. Как
и можно было ожидать, максимальной эффективностью
обладали большие, более липофильные соединения, а эффективность
2.2. Эмпирическая оценка эффективности катализаторов
Таблица 2.2. Константы скорости второго порядка для
реакции тиофеноксида с 1-бромоктаном в системе
бензол/вода [4]
Катализатор
Константа
скорости8 k, 1<Н> М-'-с-1
Me4NBr
Prop4NBr
Bu4NBr
Bu4NI
MeOctjNCl (аликват 336)
(PhCH2)Et3NBr (ТЭБА-бромид)
Пиридил—BuBr
Пиридил—HepBr
ПирИДИЛ—С12Н25ВГ
HexEt3NBr
OctEtsNBr
(C,0H21)Et3NBr
(C12H25)Et3NBr
(C16H33)Et3NBr
(C,eH33)Me3NBr
Ph4PBr
Ph4PCl
PhsMePBr
Bu4PCl
Oct3EtPBr
(C16H33)Et3PBr
Ph«AsCl
Дициклогексано-18-крауи-б
<0,0016
0,0056
5,2
7,4
31
<0,0016
<0,0016
0,023
0,092
0,015
0,16
0,24
0,28
0,48
0,15
2.5
2,7
1,7
37
37
1,8
1,4
41
a Наблюдаемые константы скорости экстраполированы до
содержания катализатора 0,00137 моля.
более симметричных ионов выше, чем у тех, которые имеют
одну длинную цепочку. Замещенные фосфониевые ионы иногда
более эффективны, чем аналогично замещенные аммониевые
ионы. Как уже было показано в гл. 1, бензильные и арильные
группы по каталитическим свойствам не эквивалентны С7- и
Сб-алкильным группам. В гептане скорость в 100 раз 'Медленнее,
а в более полярном растворителе о-дихлорбензоле она
возрастает (особенно при использовании слабых катализаторов) в 5—
300 раз. Максимальная величина константы скорости в МФК-
70 Глава 2. Механизм межфазного катализа
реакции в 10 раз больше, чем в гомогенной реакции в этаноле,
но почти в 300 раз меньше, чем в абсолютном ДМФА [4].
Следует, конечно, помнить, что истинная скорость в
экспериментальных условиях проведения МФК-реакции может регулироваться
количеством использованного катализатора.
Были определены выходы и время реакции замещения брома
на иод в м-октилбромиде при использовании в качестве
катализаторов различных краун-эфиров и трибутил-м-гексадециламмо-
нийбромида (см. табл. 3.4). Лучше ониевой соли оказались
только некоторые .криптанды, имеющие боковую цепь.
Недавно были определены кинетические параметры для ряда
реакций замещения, в которых использовали следующие
катализаторы: 2-тетрадецил[2.2.2] '[82], лергидротрибензо[2.2.2] [93],
дициклогексано-18-краун-6 [81], 2-децил[2.2.2], 18-краун-6,
различные алкилзамещенные дибензо-18-крауны-6 {83] и гексаде-
цилтрибутилфосфониевые соли [81]. Основной вывод из этих
работ следующий: обычные краун-эфиры достаточно липофиль-
ны сами по себе и добавочные боковые цепи не вносят
существенного вклада в их свойства. Действительно, активность МФК
снижается по мере удлинения боковой цепи [83]. В общем
в реакциях МФК-замещения использование краун-эфиров не
имеет преимуществ [81, 83]. Они работают особенно плохо в тех
случаях, когда в качестве нуклеофилов используют С1~ и CN~
[81]. Липофильные криптанды ведут реакцию в 2—5 раз
быстрее, чем ониевые соли. В системе Н2О/хлорбензол анионы по
своей активности располагаются в следующий ряд [82]: ЭДз~>
>iGN->Br->J->Cl->SCN-. Сравнение изменений в гидро-
фильности, влияющей на экстракцию и каталитическую
активность в зависимости от липофильных факторов в молекуле
катализатора, было исследовано только в японской работе {84].
Экстрактивная сила катализатора больше всего проявляется
в реакциях окисления перманганатом. Было показано [42], что
умеренный избыток аммониевой соли почти количественно
экстрагирует МпО4~ (табл. 2.3).
При восстановлении боргидридами самая высокая скорость
у реакции, идущей в присутствии четвертичного аммониевого
катализатора, который имеет гидроксильную группу в р-поло-
жении к атому азота, например с (—)-Ы-додецил-Ы-метилэфед-
ринийбромидом, чем с аликватом 336 или трибутилгексадецил-
фосфонийбромидом [43]. Однако в других МФК-реакциях этот
«особый» катализатор оказался менее эффективным, чем
обычные. Одним из возможных объяснений такой уникальной
эффективности именно в реакции восстановления боргидридами
является предположение об активировании карбонильной
группы к атаке ионом ВН4~ благодаря предварительному
образованию водородной связи. Некоторые авторы считают, что опти-
2.2. Эмпирическая оценка эффективности катализаторов 71
Таблица 2.3. Экстракция 1,0 миоля МпО«~ из 0,02 М
водного раствора в бензол с использованием четвертичных
солей [42]
Катализатор
Количество
катализатора,
ммсщь
Количество
экстрагированных
ионов МпО<-
в бензоле,
множь
ТЭБА
Bu«NBr
Bu4PCl
(CI6H33)Me3NBr
Аликват 336
1,51
1,63
1,54
1,54
1,53
1,52
0,0
0,74
0,95
0,96
0,86
0,93
чески активные катализаторы, несущие ОН-грулпу у хирально-
го центра, проявляют слабую асимметрическую индукцию
в МФК-'боргидридном восстановлении (разд. 3.1.5).
В работе [65] было лроведено сравнение каталитических
характеристик типичной для МФК четвертичной аммониевой
соли, аликвата 336, 18-крауна-6 и тетраметилэтилендиаминов
в системе твердая фаза/жидкая фаза. Аммониевый катализатор
показал одинаковые результаты или даже превосходил другие
в реакциях замещения ацетатных, фторидных и аденильных
анионов, однако в случае цианидного аниона реакция с краун-
эфиром протекала ло крайней мере в 100 раз быстрее, чем
с аликватом 336 (разд. 1.6).
Рассмотрим теперь реакцию алкилироваиия в присутствии
концентрированного раствора МаОН. Макоша и сотр. [44]
сравнили ряд катализаторов в стандартных условиях на примере
реакции этилирования бензилцианида этилхлоридом.
Полученные результаты приведены в табл. 2.4.
Эти ранние результаты показывают, что более или менее
оптимальным катализатором является ТЭБА. Другие
катализаторы, близкие по своей активности к ТЭБА, или гигроскопичны,
или дороги. Интересно отметить, что при использовании
последующих гомологов ТЭБА выход был более низкий. Это,
по-видимому, связано со спецификой взаимодействия между анионом
и катионом внутри ионной пары [R4N+PhCH-—GN]. В этой
и других реакциях вновь наблюдались постепенное увеличение
выхода вплоть до максимального и затем' его медленное
снижение по мере роста липофильности катиона катализатора
(табл. 2.6—2.9).
Макоша нашел также, что многие неорганические анионы,
добавленные в водную фазу, конкурентно подавляют алкилиро-
72 Глава 2. Механизм межфазного катализа
Таблица 2.4. Вммвие типа-катализатора на выход в реакции
этилирования бензнлцнаннда этнлхлорядом [44]
Катализатор
.Выход,
Катализатор
Выход, %
(PhCH2)MesNCl
(PhCH2)Me2EtNCl
(PhCH2)MeEt2NCl
(PhCH2)Et3NCl (ТЭБА)
(PhCH2)Et2PropNCl
(PhCH2)EtProp2NCl
(PhCH2)MeEtPropNCl
(PhCH2)Et2BuNCl
<PhCH2)PropsNCl
(PhCH2)2Et2NCl
32
40
45
50
47
44
43
45
43
15
n-НзСО—РЬСНг—NEt3Cl
л-Cl—PhCHsHNEtsCl
=CH2)NC1
М-Аллил-М-бензилпипери-
динийхлорид
H2C=CH—СНг—NEtsCl
Н2С=СН—CHj—NProp3Cl
(PhCH2)Et3NBr
Et4NCl
54
38
Q
О
5
23
6
35
51
Ьание. Наиболее сильнодействующими являются 1~ и СЮ4 .
Меньшим эффектом обладают ионы Br~, NO2", СОз2~, CN~.
Фторид, хлорид и сульфат-ионы не влияют на ход реакции [44].
МФл-реакция бензилирования бензилцианида была изучена
в таких условиях, когда в системе до начала реакции было
только незначительное количество посторонних ионов, которые могли
Таблица 2.5. Влияние аниона тетрабутиламмонийвых
катализаторов на выход в реакции бензилирования
бензилцианида бензилхлорндома [45, 46]
Анион катализатора
Степень моно-
бензилчрова-
н»я, %■
Степень дибея-
зилировання, %
hso4-
С1-
Вг-
I-
сю4-
J5-H афталннсульфонат
Бензоат
л-Нитробензоат
Пикрат
а Условия: 0,05 моля PhCHjCN+0,05
лнзатора, 0,1 моля крнц. NaOH; 1 ч, 0 "С.
42,8
40,6
35,1
8,6
24,1
40,2
45,3
44,1
15,1
моля PhCH3CI, 0,001
11,8
9,3
7,4
0,5
3.5
12,8
12,2
12,2
3,7
ноля ката-
2.2. Эмпирическая оценка эффективности катализаторов 73
бы распределиться по фазам с катионом катализатора. Даже
в этих условиях наблюдалась очень сильная зависимость
(табл. 2.5) выхода продукта от типа аниона* катализатора [45].
Хотя в этом случае имеются трудно объяснимые отклонения, но
в целом тенденция влияния типа аниона на выход
соответствовала тенденции в изменении их коэффициентов экстракции;
иными словами, очень липофильные анионы трудно межфазно
обмениваются с анионом депротонированного субстрата.
При сравнении влияния даже очень широкой серии
различных катионов катализаторов и краун-эфиров в той же самой
реакции никаких ясных выводов сделать не удалось {45].
Доке {47] исследовал влияние длины заместителя в молекуле
катализатора на образование стирола при действии
концентрированного NaOH на фенилэтилбромид (табл. 2.6).
Таблица 2.6, Влияние типа
катализатора, на выход, стирола,
полученного из фенил эти лбромида
под действием концентрированного
гидрокенда натрия [47]
Катализатор
Et4NBr
PropEt3NBr
BuEt»NBr
PentEtsNBr
HexEt3NBr
НерЕЩВг
OctEtsNBr
(CbHuJEtsNBr
(CiaHMjEtsNBr
Выход, %
3
7
12
50
53
45,5
44
42
38
Было проведено также сравнейие различных катализаторов
в реакции омыления (34, 35]. Однако эти результаты относятся
только к омылению сложных эфиров, анион кислоты в которых
очень гидрофилен (таол. 2.7). Сильное влияние на эту реакцию
стеарата натрия указывает на возможность реализации
различных механизмов (разд. 2.1). Примечательно, что в этой реакции
полиэфиры с открытой цепью были столь же активны, как и
бисульфат тетрабутиламмония, а краун-эфиры оказались
неактивными. При сравнении констант скоростей гидролиз?
п-нитрофенйлацетата, катализируемого Ph4P+, BU4N4", PheGe^
Ph3Pb+, Ph3Sn+, R3Sn+ и Bu2Cl2Sn, оказалось, что при использо-
74 Глава 2. Механизм межфазного катализа
Таблица 2.7. Влияние типа катализатора на выход при омылении
днэтиладипината под действием концентрированного
гндроксида натрия" [34]
Катализатор
Выход, „ Выход,
% |
Катализатор I %
Отсутствует
B114NHSO4
Bu4NCl
Bu4NBr
Bu4NI
BU4NCIO4
Bu«N-6eH3oaT
Аликват 336
Oc^NBr
(Ci6H33)Me3NBr
ТЭБА
Цетилпиридиний—Cl
15-Краун-5
18-Краун-6
11
93
32
18
15
14
32
50
40
39
18
20
23
11
Дибензо-18-краун-6
Bu4NBr
Pent4NBr
Hex4NBr
Hep4NBr
Oct4NBr
Стеарат натрия
Et4N+C8F17SO3-
Ci6H33(O—CHs—CH2)29OCOCH3
Ci6H33 (O—СНг^СН2) зо—OCH3
Ci6H33 (O—CH2—CH2) зо—Cl
C,6H33(O—CHs—CH2)30—OH
12
18
20
45
46
39
60
39
95
84
76
74
a Условия: 0,01 моля субстрата, 0,05 ноля 50%-иого NaOH, 5 мл петролейното
эфира, 0,0001 ноля катализатора; 1 ч, комнатная температура.
вании BtuNBr скорость по крайней мере в 2,5 раза выше, чем
во всех остальных случаях [87].
Сравнение активности катализаторов при получении
дихлоркарбена впервые было 'проведено химиками на производстве
[48]. Более современные данные по получению дихлоркарбена
приведены в табл. 2.8, а для дибромкарбена — в табл. 2.9.
Изучение данных, представленных в этих таблицах,
выявляет ряд интересных эффектов. Характеристики катализаторов
при получении дихлоркарбена не совладают с их
характеристиками при генерировании дибромкарбена. Отрицательное
влияние липофильности добавленного аниона гораздо сильнее
проявляется в первом случае. Этот факт находится в прекрасном
соответствии с механизмом, предложенным в разд. 2.1.
Действительно, поскольку СВг3~ намного .более липофилен, чем СС13~,
то уход его с поверхности раздела фаз задержать не так просто.
'Соответственно при получении :СВг2 влияние изменений катио-
ща катализатора сказывается не так сильно, как в случае :СС1г.
Поразительно, однако, что третичные амины, которые в ходе
реакции превращаются в настоящие катализаторы R3NCHX2+X~
((см. разд. 3.20.3), являются лучшими катализаторами при
2.2. Эмпирическая оценка эффективности катализаторов
Таблица 2.8. Влияние типа катализатора при получении
дихлоркарбена» (выход дихлорноркарана из циклогексена) [9, 29]
Катализатор
Выход,
%
Катализатор
Me4NI
Et4NBr
Prop4NCl
Prop4NBr
Bu4NHSO4
Bu4NCl
Bu4NBr
Bu4NI
Bu4NCl04
BujN—бензоат
BiuN—и-нитробензоат
Bu#N—нафталинсульфонат
Bu4N—пикрат6
Pent4NBr
Hex4NBr
Hep4NBr
Oct4NBr
Аликват 336
ТЭБА
ТЭБ А-бромид
(PhCH2)Prop3NCl
(PhCH2)Bu3NCl
(C16H33)Me3NBr
(C16H33)Et3NBr
1
44,2
34,2
26,1
45,7
38,8
29,1
22,6
18,6
24,5
16,3
8,6
36,6
32,4
34,8
28,4
23,4
41,9
51,0
35,2
48,3
48.8
42,6
39,7
(Ci6H33)Prop3NBr
(C16H33)Bu3NBr
(C,6H33)Me2(PhCH2)NCl
Et4N+CsH17SO3-
Prop4PCl
Bu4PCl
(Ci6H33)Bu3PBr
Ph3 фурфурил РВг
Ph3(PhCH2)PCl
Ph4PCl
Ph4AsCl
Ci6H33—пиридиний—Cl
18-Краун-6
15-Краун-5
Дициклогексано- 18-краун-6
Дибензо-18-крауи-6
Prop3N
Ci6H33(OCH2—СН2)зоОН
С,6Н33 (О—CHs—СН2)29ОСОСН3
С,6Н33(О—СН2—СН2)40ОСН3
С^Нзз (О—CHg—СН2) 40CI
C,2H26NO(Me)2
C16H33NO(Me)2
a Условия: 0,1 ноля циклогексена, 0,4 моля СНСЬ, 0,2 моля 50%-ного
0,001 моля катализатора; 4 ч, комнатная температура.
Пнкрат-ион разрушается i
з условиях реакции.
39,0
35.5
48,2
4,0
35,4
38,3
39,6
1
1
1
62,0
1
54,9
55,0
41,7
18,2
74.0
7,9
7,0
7,6
6,3
8,5
8,3
NaOH,
получении :СС12. Получение :CBr2 в их присутствии идет
существенно хуже.
Фенилзамещенные фосфониевые соли оказались
катализаторами с очень низкой активностью, что резко отличает их от
обладающего превосходными характеристика-ми тетрафенилар-
сонийхлорида. Это объясняется тем, что первые разрушаются
щелочами, давая R3PO. И наконец, в согласии с развитыми
76 Глава 2. Механизм межфазного катализа
Таблица 2.9. Влияние типа катализатора при получении
дибромкарбенаа (выход дибромноркарана из циклогексена) [49]
Катализатор
Выход.
Катализатор
Выход,
%
ТЭБА
Аликват 336
Prop3N
BU3N
Prop4NBr
Bu4NBr
PeruNBr
Hex4NBr
Hep4NBr
Oct4NBr
Ph4PCl
Ph4AsCl
<CieH33)Me3NBr
<C,eHs3)Bu3NBr
44,8
52,6
33.0
31.4
40,0
48.7
48,0
55,0
50,2
43,0
0,7
41,0
48,3
51,5
(CieH33)M€2(PhCH2>NCl
CieHs3—пиридиний—Cl
18-Краун-6
Дибензо-18-краун-6
Bu4NHSO4
Bu4NCl
Bu4NBr
Bu4NI
Bu4NC104
Bu4N—бензоат
BiuN—я-нитробензоат
Bu^—пикрат"
BU4N—р-нафталинсульфонат
49.0
38,5
47.5
48,0
44,56
38,26
39,06
38,06
32,56
35,56
29.76
37,06
44.06
Условия: 0,1 моля циклогексена, 0,4 ноля 50%-ного NaOH, 0,4 моля СНВг3,
Ю.001 моля катализатора; 4 ч, комнатная температура.
В реакционной смеси только 0,2, моля NaOH.
Пикрат-ион разрушается в условиях реакции.
выше представлениями о механизме этой реакции очень
плохими катализаторами являются нейтральные ПАВ, оксиды ами-
Нбв, полиэфиры с открытой цепью, а также анионные ПАВ.
'"■^Для того чтобы сравнить катализаторы, время реакции и
другие переменные параметры поддерживают практически
постоянными. Поэтому на самом деле большая часть
рассмотренных выше катализаторов может использоваться в других
условиях для получения карбенов.
Японские исследователи утверждают, : что дихлоркарбен,
генерированный с использованием разных катализаторов, может
отличаться по своей «селективности», определяемой по соотно1
лююда mohq-. и .бисаддуктов, которые образуются при
взаимодействии его с полиолефинами. В то время как р-гидроксиалкил^
■п;риалкиламмониевый катализатор дает, как это описано, [50,
Ed1]., 4почти исключительно моноаддукт, цетилтриметиламмоний
приводит к полиаддуктам. Там же сообщалось, что при
использовании оптически активного гидроксилсодержащего катализа-
«ора-{Образуются.- оптически активные: продукты присоединения
2.3. Катализаторы на полимерных носителях 77
дихлоркарбена [51]. Это сообщение не подтвердилось [52].
Попытки воспроизвести опыты по селективному присоединению
также не удались [53].
Недавно были опубликованы {54] результаты изучения влия
иия катализаторов на селективность .присоединения к одной шп.
к двум двойным связям в сопряженных или несопряженных
диенах и триенах. Были определены константы скорости
псевдопервого порядка для моно- и бисприсоединения дихлоркарбена
к изопрену. Их отношение быстро растет в ряду: цетилтриме-
тиламмоний > хинуклидин > трибутиламин > 1,4-диазаби-
цикло[2.2.2]октан. Другими словами, количество бисаддукта
уменьшается при переходе от цетилтриметиламмония к 1,4-ди-
азабицикло[2.2.2]октану. В присутствии первого в подходящих
условиях выход достигает 88% (соотношение моно:бис = 2: 1),
а в присутствии второго с выходом 23% образуется чистый
моноаддукт. Эти результаты трудно сопоставить с другой
имеющейся информацией о поведении катализаторов и о реакциях
дихлоркарбена. Из данных табл. 2.8 следует, что третичные
амины — одни из наиболее эффективных катализаторов, хотя
авторы работы [54] установили, что амины намного менее
активны, чем четвертичные аммониевые соли. Однако условия
проведения реакции у этих авторов были несколько иными (менее
концентрированный NaOH!). Было сделано предположение, что
атом азота действует как переносчик карбена, однако
впоследствии оно было опровергнуто Макошей (разд. 3.20.1).
Очевидно, и утверждение о «селективности» требует независимого
подтверждения. Некоторая новая информация, относящаяся к
этому вопросу, обсуждается в разд. 3.20.1.
2.3. Необычные катализаторы и катализаторы
на полимерных носителях
В качестве катализаторов кроме ониевых солей и краун-эфи-
ров можно использовать и другие соединения. Это и зфиры по-
лиэтиленгликоля с открытой цепью, и такие молекулы, которые
имеют несколько цепей такого же типа, прикрепленных к
центральному ароматическому ядру (так называемые «полиподы» —
многоножки). Их каталитические свойства и механизм действия
аналогичны действию краун-эфиров. Хотя они иногда являются
менее эффективными экстрагентами, этот недостаток часто
можно устранить, используя их в более высоких концентрациях
(гл. 3).
Подробно описаны растворяющая способность различных
солей, комплексообразующие свойства и .каталитическая
характеристика полиэтиленгликолей и их эфиров [89—92].
78 Глава 2. Механизм межфазного катализа
Иногда в качестве катализаторов используются самые
неожиданные вещества. Реакции обмена проводили в присутствии
10—15 >мол.% фосфортриамида (А). Радикал R представляет
собой какую-либо липофильную группу (но не метальную) [16].
Реакции замещения S#2 катализировали гегракыс-(алкилсуль-
финилметил) метаном [95] или метил-2-пиридилсульфоксидом
[96]. Соединения В—F также использовали при проведении
алкилирования бензилметилкетона алкилгалогеиидами в
присутствии водного раствора гидроксида натрия при комнатной
температуре (17].
[(CH3)2N]2PON(CH3)R (RO)2PO-CH2-SOR'
А В
(RO)2PO—CR2'—SCXR" (EtO)2PO—CH2SO-CH2—PO(OEt)2
С D
PhSO—CH2—SOPh (EtO)2PO—CH2—PO(OEt)2
E F
Макоша в реакциях алкилирования часто использует диме-
тилсульфоксид (ДМСО) в качестве «сокатализатора» или в
некоторых случаях в качестве собственно катализатора (примеры
см. в гл. 3). Можно полагать, что каталитическое действие
соединений А—F так же, как и ДМСО, состоит в солюбилизации
солей или NaOH в органическом слое благодаря специфической
сольватации и/или хелатообразованию.
Пиридиниевые соли обычно мало пригодны в качестве
катализаторов в МФК-реакциях, однако недавно было обнаружено,
что соль 2-диалкиламинопиридиния (G) можно использовать
с этой целью при проведении алкилирования в присутствии
концентрированного раствора NaOH (18].
(R1 = Н, СН3; Ra = Me, Et; R3 = блинная цепь)
«J—R3
Bt\° w | 1
R2 Et G
Схема 2.6
В качестве катализаторов для генерирования дихлоркарбена
были предложены оксиды аминов [85]. Их же использовали при
получении бензилацетата из бензилхлорида [86]. В том и в
другом случае настоящим катализатором была, очевидно,
четвертичная аммониевая соль, образовавшаяся in situ. В случае
реакции этерификации предложена следующая последовательность
стадий:
R3NO > RSN+—CH2Ph > R3N+OCH—Ph
RSN >■ RSN+—CH2PhCr"
2.3. Катализаторы на полимерных носителях 79
Сообщается также о моделях МФК-реакций для систем
жидкость/жидкость с использованием в качестве катализаторов
необычных циклических фосфониевых и арсониевых солей [59],
а для системы твердая фаза/жидкая фаза — особой октопус-мо-
декулы («осьминога») [60], сложных то структуре {88] и простых
зфиров зтиленгликоля [61], лолиэтиленаминов {62], тетраметил-
этилендиамина {63] и замещенных р-аминофосфамидов [64].
Ряд авторов описали МФ-катализаторы, фиксированные на
полимерных подложках. Такие катализаторы представляют
большой интерес для промышленного применения, поскольку их
легко отделять после окончания реакции и, .кроме того, можно
использовать в непрерывных процессах. Этот метод МФК
подучил название «трехфазный катализ» {19, 21, 22]. Реакция
замещения с 1-бромоктаном при использовании закрепленной
аммониевой соли имеет первый .порядок ло субстрату. Если
полистирол содержит 1—21% групп —CH2NR3+ у фенильных
колец, то активность таких смол прямо пропорциональна числу
этих групп. Увеличение количества фенильных колец, имеющих
группы —СН2—NMe3+, в микропорах полистирола до 46—76%
приводит к резкому снижению каталитической активности.
Продажные анионообменные смолы обычно мало подходят в
качестве МФ-катализаторов [19]. Результаты изучения действия
иммобилизованных ониевых солей, краун-эфиров и криптандов {20]
показали, что в основном механизм реакций с этими
катализаторами сходен с «нормальным» механизмом МФК-реакций.
Недавно было показано что при гидролизе 1-бромадаманта-
на в системе толуол/вода в присутствии полистирола с
полиэфирными боковыми цепями образуется 1-гидроксиадамантан.
Это явление получило название «эффект твердофазного сораст-
ворителя» («solid-phase cosolvent») и впоследствии было
изучено с помощью спиновых меток {22]. В качестве МФ-катализа-
торов были использованы также триамиды фосфорной кислоты
на полимерных носителях [57]. В общем можно сказать, что
катализаторы на полимерных носителях, по-видимому, менее
эффективны, чем те же катализаторы сами по себе. Однако при
длительном использовании они могут быть удобнее, особенно
если цена катализаторов на полимерных подложках станет
доступной. Возможности этой новой области катализа еще
ожидают своего развития. Практические аспекты использования
изученных до настоящего времени катализаторов, связанных
с полимерными носителями, приведены в гл. 3.
Необходимо отметить, что трехфазные катализаторы типа
твердофазных сорастворителей ведут реакцию феноксида с
гомологами алкилбромидов избирательно, что не наблюдалось
при обычной МФК-реакции. Так, превращение 1-бромбутана
идет намного быстрее, чем 1-бромоктана [58]. Обзор по
трехфазным катализаторам дан в работе [56].
Глава 3
Практическое использование
межфазного катализа
3.1. Общие экспериментальные методы
3.1.1. Обычные катализаторы
В патентах в качестве катализаторов предлагается множество
четвертичных аммониевых (N), фосфониевых (Р), арсониевых
(As), антимониевых (Sb), висмутониевых (Bi) и третичных
сульфониевых (S) солей. Однако на практике широко
используют только небольшое число аммониевых и фосфониевых
солей, которые вырабатываются промышленностью и поэтому
доступны. Их стоимость не превышает стоимости типичных
промышленных .продуктов тонкого органического синтеза.
Стоимость многих ониевых солей 10—60 долл. за 100 г, а некоторых
даже выше 150 долл. за 100 г, а краун-эфиры ещё дороже.
В то же время промышленное получение этих соединений не
представляет особых трудностей, так что можно ожидать, что
по мере возрастания, спроса и увеличения производства цены
на них будут снижаться. В 1977 г. наиболее дешевым из
доступных катализаторов был «трикаприлметиламмонийхлорид»,
который представляет собой технический продукт, содержавший
соли со смешанными алкильными радикалами Се—Сю. Торговое
название этой смеси солей Aliquat 336 (аликват 336)* или Ado-
gen 464 (адоген 464)**. Соединения типа бензилтриэтиламмо-
нийхлорид (ТЭБА) или бензилтриэтиламмонийбромид (ТЭБА-
бромид), бензилтриметиламмонийхлорид, бензилтриметиламмо-
нийбромид или бензилтриметиламмонийгидроксид (тритон В
в метаноле или в воде), тетра-и-бутиламмонийхлорид, или тет-
ра-н-бутилам.монийбромид, тетра-«-бутиламмонийиодид или тет-
ра-и-бутиламмонийгидроксид (в виде раствора в воде, в
метаноле или в смеси метанол/толуол) и цетилтриметиламмонийхло-
рид или цетилтриметиламмонийбромид поставляются всеми
основными химическими фирмами, производящими реактивы.
Поскольку МФК становится модным, то некоторые фирмы вы-
* Фирмы: Fluka AG, Buchs, Switzerland; General Mills Co., Kankasee,
Illinois, USA.
** Фирмы: Aldrich Chemical Сотр., Milwaukee, Wisconsin, USA; Ashland
Chemical Co., USA.
3.1. Общие экспериментальные методы 8J
деляют в каталогах МФ-катализаторы в отдельный раздел.
По заказам можно получить и другие соли, такие, как бензил-
трибутил-, тетра-и-пентил-, тетра-н-гексил-, триоктилпропил-
аммонийхлориды или -бромиды, а также многие другие.
Стоимость бромидов обычно выше. В последнее время различные
фирмы стали предлагать кислые сульфаты (бисульфаты) тет-
ра-н-бутил- и тетра-и-гексиламмония. Из фоофониевых солей
легко .можно получить трибутилгексадецилфосфонийбромид,
этилтрифенилфоофонийбромид, тетрафенилфосфонийхлорид-
Легко доступны следующие краун-эфиры и криптанды: 15-кра-
ун-5, 18-краун-6, дибензо-18-краун-6, дициклогексано-18-краун-б,
4,7,13,16,21 -пентаокса-1,10-диазабицикло[8.8.5]трикозав (Krypto-
fix 221—криптофикс 221), 4,7,13,18-тетраокса-1,10-диазабицик-
ло[8.5.5]эйкозан (Kryptofix 211 — криптофикс 211) и наиболее
часта используемый «[2.2.2]» (Kryptofix 222 — криптофикс 222) —
4,7,13,16,21,24-гексаокса-1,Ю-диазабицикло[8.8.8]гексакозан и его
бензопроизводное (Kryptofix 222 В —криптофикс 222 В).
Большую часть этих катализаторов легко приготовить в
лаборатории. Поскольку кватернизация третичных аминов
особенно <быстро идет в ацетоиитриле, то в последние годы этот
растворитель широко используется. Некоторые примеры приведены
ниже. Бензилтрибутиламмонийхлорид получают с выходом 85%
при кипячении в течение 1 нед смеси три-и-бутиламина и бен-
зилхлорида в ацетонитриле [1]. Бензилтриэтиламмонийхлорид
образуется с количественным выходом из триэтиламина и бен-
зилхлорида в толуоле при кипячении смеси в течение 2—4 сут
[2]. Если хороший выход не требуется, то реагенты растворяют
в ацетонитриле [2] или ацетоне {3] и оставляют на ночь при
комнатной температуре. При нагревании до 65 °С в течение
3 сут 1-бромгексадекана и трибутилфосфина образуется три-
бутилгексадецилфосфонийхлорид [4]. Тетрагексиламмонийбро-
мид получают с выходом 59% при кипячении соответствующего
амина и бромида в ацетонитриле в течение 48 ч. Для получения
три-и-бутилэтилам'монийбисульфата кипятят 5 ч трибутиламин
и диэтилсульфат в ацетонитриле, удаляют растворитель,
к остатку добавляют разбавленный водный раствор серной
кислоты и кипятят еще 48 ч. Это необходимо для гидролиза
образующегося при реакции этилсульфата в кислый сульфат {5].
Из приведенных выше примеров видно, что кватернизация
проводится главным образом с использованием алкилбромидов,
алкилиодидов и диалкилсульфатов. В случае хлоридов этот ме*
тод применим только для метил-, бензил- и аллилхлорида.
Хлориды с другими радикалами реагируют с третичными аминами
слишком медленно, чтобы их можно было использовать для
практических целей. При кватернизации с диалкилсульфатами
образуются тетраалкиламмонийалкилсульфаты R4N+ "tOSC^OR
82 Глава 3. Практическое использование МФК
Как показано выше, их можно гидролизовать до кислых
сульфатов.
Эти соли полезно использовать в качестве МФ-катализатора
в тех случаях, когда анион катализатора должен переходить
в органическую фазу намного хуже, чем реагирующий анион
(по терминологии Брендстрёма такой процесс^ ^называется
препаративная экстракция ионных пар). Изо всех обычных анионов
наиболее подходящими являются бисульфат и хлорид. Во
многих случаях можно использовать бромиды, однако применение
иодидов часто вызывает трудности, особенно в тех случаях,
когда в реакцию вводят алкилиодиды, что вызывает образование
в ходе реакции дополнительных количеств иодид-ионов. При
этом наблюдается «отравление» катализатора, которое состоит
в том, что весь катализатор экстрагируется в форме иодида
в органическую фазу и реакция останавливается. Так же как и
в случае гомогенных реакций с предварительно полученной
аммониевой солью, в системах с иодидами большую роль может
играть ионный обмен. Следует подчеркнуть, что такой обмен
в большинстве типичных МФК-реакций не является
необходимым. Однако в некоторых реакциях в присутствии
катализаторов добавление небольших количеств иодида ускоряет процесс:
иодид обменивается с галогенидом в алкилирующем агенте,
делая его более активным (RX+I~—HRI+X~). Таким способом
можно влиять на соотношение С/О-изомеров, образующихся при
алкилировании амбидентных анионов (ом., например, [1716]).
Метод экстракции часто может заменить применявшиеся
ранее методы обмена анионов, описанные в книге Губен-Вайля
[6]. С этой целью используют следующие методики:
а) Реакцию Р^Ы+^галогенида с серебряной солью желаемого
аниона (очень старый дорогой метод).
б) Пропускание водного или водно-метанольного раствора оние-
вой соли через ионнообменную смолу, содержащую желаемый
анион. Прочность связывания анионов смолой изменяется
в ряду SO42->I->NO3->Br->Cl->OH->F- [7].
Подробное описание получения четвертичного аммонийгидрокси-
да в метанольном растворе с использованием сильноосновной
лолистирольной смолы, содержащей алкилированный
четвертичный аммонийгидроксид, приведено в «Органических
синтезах» {8].
в) Двойное использование соли щелочного металла. Для этого
подбирают такой растворитель, в котором желаемая
четвертичная аммониевая или фосфониевая соль растворялась бы,
а щелочная соль с ненужным анионом выпадала в осадок.
г) Реакция четвертичных аммониевых иодидов с кислотой НХ
в п исутствии эпоксидов {6]
3.1. Общие экспериментальные методы 83
NR4®I«> + НаС—СН—R + НХ ► NR4eXe + IHaC—CH(OH)R
О
Схема 3.1
д) Взаимодействие четвертичных аммониевых иодидов и
бромидов с диметилсульфатом в высококипящем растворителе
(например, в хлорбензоле). Образующиеся при реакции ме-
тилиодид или .метилбромид отгоняются, а метилсульфат гид-
рол изуется [9]. Использование этого метода позволяет
регенерировать дорогостоящий СН31:
R4N+X~ + (CHS)2SO4 *■ ReN+'OSOgOCHj + CHSX
е) Окисление иодидов пероксидом водорода в присутствии
кислоты НХ. При этом трииодид четвертичного аммония,
который не растворим в воде, выпадает в осадок {6, 33]:
3R4N+I" + НаО2 + 2НХ >• R4N+IS" + 2R4N+X~ + H2O
ж) Обмен анионов с использованием экстракции
растворителями. Часто этот метод оказывается самым простым.
Если вводимый анион более липофилен, чем тот, который
з&мещается (например, иодид обменивается с хлоридом,
хлорид— с бисульфатом, азид — с нейтральным сульфатом,
цианид— с хлоридом*), то никаких проблем не возникает.
Исходную соль растворяют в метиленхлориде и встряхивают
с избытком концентрированного** водного раствора соли
щелочного металла, содержащей соответствующий необходимый
анион. Эту операцию повторяют два или три раза, используя
каждый раз свежий водный раствор. Часто удобнее
использовать в качестве исходной соли кислый сульфат тетрабутиламмо-
ния, так как образующийся после нейтрализации сульфат не
растворим в органической фазе. Таким методом в
кристаллическом виде получены различные галогениды, цианиды, азиды,
нитрилы, бензоаты, феноляты и еноляты р-дикетонов, р-циано-
эфиров, ip-кетосульфонов и диметилбензоилмалоната.
Трудности возникают в тех случаях, когда образующиеся
четвертичные ониевые соли гидрофильны и в заметной степени
остаются в водной фазе или когда оба аниона имеют близкие по
величине константы экстракции. Некоторые из этих трудностей
удалось преодолеть Брендстрёму (10]. Другим возможным
решением проблемы является понижение растворимости соли
R4N+ в воде в результате использования концентрированных
растворов гидроксида натрия в качестве водной фазы. Ионный
* Более полный ряд «экстракции анионов» см в разд. 1.3.4.
** Наилучшие результаты получаются при использовании возможно более
концентрированного раствора.
Глава. 3. Практическое использование МФК
обмен в системах с концентрированным водным раствором
.NaOH лучше проводить, используя по рекомендации Макоши
[11] бензол, о-дихлорбензол или их смеси с ацетонитрилом в
качестве органической фазы. При этом не обязательно добиваться
лолного растворения в двухфазной системе исходной натриевой
я четвертичной аммониевой соли, так как при ионном обмене
•обычно образуются более растворимые соединения. Если
исходить из хлоридов, то таким методом можно получить
следующие соли: бромиды, иодиды, тетрагидробораты, родадады,
перхлораты, тексацианоферраты(Ш), цианиды, перманганаты,
нитриты, нитраты и азиды. Этот метод, если брать в качестве
исходных солей сульфаты или фториды, пригоден также и для
лолучения гидроксидов, хотя их растворимость в органических
•средах очень мала (даже в тех случаях, когда используют
объемистые катионы) (11, 12].
Согласно другой методике, галогениды четвертичного аммо*
лия растворяют в метаноле в присутствии кислоты, анион
.которой необходимо ввести в соль взамен галогенид-иона. Для ко№*
центрирования соли в работе [1017] предлагается удалять исходг
иый анион в форме метилгалогенида, хотя широкую
применимость этого метода еще следует доказать. Другие авторы вводя1
в реакцию исходную аммониевую соль с эфиратом трехфторя^
«того бора, в результате чего образуется тетрафторборат. ЭтсйГ
анион легко обменивается на другие анионы [1018].
Особую проблему представляет получение фторидов, яОг
■скольку этот анион очень тидрофилеи и поэтому трудно
экстрагируется. Кроме того, его трудно отделить от гидратной воды.
Тетрэбутиламмонийфторид получают нейтрализацией тетрафуг
тиламмонийгидроксида плавиковой кислотой-. Он выделяется
в форме кристаллического клатрата (Bu4N+F~-32,8 H2O) пщ
€—10 °С. Дегидратация клатрата при 30—50°С/15 мм рт. с?».
м высушивания образующейся пасты при 30—40 °С/0,5 мм рт. ст>
над пентоксидом фосфора в течение 15—20 ч дает безводную,
чрезвычайно гигроскопичную соль {24—26}. 'При выпаривании
водного раствора тетраэтиламмонийфторида образуются
кристаллы Et*N*F--2H2O. При высушивании они частично распаг
даются (36]:, ■
1 (C2H5)4N+F-
Очень удобную для применения в качестве катализатора форму
tyxoro iRi'N^F- получают следующим образом: выпаривают
водный раствор соли в присутствии трехкратного избытка силикаг
геля, затем добавляют метанол и снова упаривают, после этого
остаток высушивают при 100 °С. Полученный продукт
негигроскопичен и сохраняет свою высокую каталитическую активность
даже после долгого пребывания на воздухе {1019].
.3.1. Общие экспериментальные методы 85
Даже простые краун-эфиры весьма дороги, хотя их получение
в лабораторных условиях не так уж и трудно*. 1,4,7,10,13,16-Гек-
саоксациклооктадекан (18-краун-6) (1) может быть получен из
триэтилеигликоля и его дитозилата несколькими способами:
с грет-бутоксидом калия в бензоле (выход 33%) {13], в ТГФ
(выход 60%) [14] или же из того же самого гликоля и
продажного дихлорида (1,8-дихлор-3,6-диоксаоктана) с гидроксидом
калия в водном ТГФ (выход 40%) [15]. Наконец, обработка
бис- (2-хлорэтилового) эфира и тетраэтиленгликоля гидроксидом
калия в ТГФ дает 18-краун-6 с выходом 30% (20]. Полученный
сырой .продукт очищается через комплекс с ацетонитрилом.
(Методы синтеза см. в (1006], другие способы очистки — в (1881],
методы .получения гидрокс»метил-18-.крауна-6 — в [1380, 1745],
а 2,6-диметил-18-крауна-6 — в [1707]; недавние синтезы.
различных оптически активных краун-эфиров описаны в [1618, 1741,
1773, 1882], обзор дан в {1891].)
По методу, описанному в работе [16], можно получать дибен-
зо-18-краун-6 (2); несколько модифицированная методика, при
которой достигаются лучшие выходы, приводится в [17]. Ката-
Схема 3.2
* Обзор [1394]. Новые препаративные варианты синтезов см. -в [1316,
1421, 1466]. " ' ,! :' ' ■' ^ • .- - '■-. \ •:■...■.. ..
86 Глава 3. Практическое использование
литическое гидрирование (2) над рутениевым катализатором,
нанесенным на окись алюминия, дает дицнклогексано-18-краун-!&
(3) (16].
Эфир (3) может существовать в виде пяти диастереомеров,
поэтому в результате гидрирования 2 образуется смесь,
состоящая из цис-син-цис- и цис-анти-цис-продукто&'-Были
разработаны специальные методы, позволяющие получать
транс-антитранс- и транс-син-транс-изамеры [18J, а также оптически
активные (+)-гра«с-акгы'гранс-соединения {19].
При кипячении в диоксане в течение 24 ч смеси 0,5 моля
триэтиленгликоля с 1,25 моля бис- (2-хлорэтилового) эфира и
2,3 моля гидроксида натрия с выходом 38% образуется
1,4,7,10,13-пентаоксациклопентадекан (15-краун-5) (4) '{20].
Если исходить из 1,8-дихлор-3,6-диоксаоктана и диэтиленгли-
коля, то выход 4 составляет всего 14% {21]>
Схема 3.3
4
Схема 3.4
Селективная олигомеризация этиленоксида в присутствии
тетрафторборатов лития, меди или цинка приводит ж
комплексам 15-крауна-5 в качестве основного продукта. Однако детали
этого очень простого препаративного метода до сих пор еще
не опубликованы* {22].
Другие методы получения см. в [1316, 1421, 1466].
3.1. Общие экспериментальные методы 87
Крнптанд [2.2.2] (криптофикс [222], 1,10-диаза-4,7,13,16,21,24-
гексаоксабицикло[8.8.8]гексакозан) (5) был получен Леном
и сотр. [23] в две стадии, как показано на схеме 3.5, с
использованием техники высокого разбавления. Другие методы см.
в работе [1303], гидроксиметил[2.2.2] ом. в [1320]. Описан также
синтез криптанда [1.1.1] с выходом 40% {1770].
Схема 3.5
В литературе приведены методы получения множества
других комплексующих агентов, аналогичных по строению, однако
большая часть из них не нашла практического применения.
Некоторые из лигандов такого рода рассмотрены в гл. 1
(разд. 1.4).
88 Глава 3. Практическое использование МФК
Катализаторы для' экстракции катионов. Все до сих лор
рассмотренные катализаторы используются для переноса
анионов в неполяряую среду. Обратный процесс — перенос
отдельных катионов в эти фазы — используется не очень часто, хотя;
он легко осуществим: так, для переноса солей щелочных
металлов необходимо использовать лююфильные анионы, например
иодид {1398], липофильные сульфонаты [1257, 1737], длинноцепо-
чечные карбоксилаты или тетраарилбораты {1795].
3.1.2. Условия проведения реакций
Типичные условия проведения реакций (хотя они и не во
всех случаях оптимальны) описаны в последующих разделах.
Так, до сих пор нет ясного понимания того, как влияет
структура катализатора на скорость и выход реакция {27, 28]. Влияние
растворителя, катализатора и перемешивания обсуждалось
в гл. 1 и 2, поэтому здесь мы дадим только некоторые
практические рекомендации.
3.1.2.1. Растворитель
Часто в качестве органической фазы применяется сам
исходный жидкий субстрат. В принципе для этой цели можно
использовать многие органические растворители. Однако они не должны
даже частично смешиваться с водой, чтобы избежать
сильной гидратации ионных пар. Следует иметь в' виду,' что в
малополярных растворителях таких, как гептан или бензол, ионные
пары из водной фазы в органическую переходят лишь в
незначительной степени, если только сочетание аниона с катионом не
является очень липофильным. Так, например, ТЭБА весьма
неэффективен как катализатор в системе бензол/вода [28] и даже
в такой системе, как дихлорметан/вода [2]. При использовании
этих растворителей рекомендуют соли тетрабутиламмония или
соли даже с еще большими катионами, такими, как тетра-н-пен-
тиламмоний, тетра-н-гексиламмоний или аликват 336.
В общем можно сказать, что ионные пары охотнее переходят
в органическую фазу и соответственно скорости реакций
увеличиваются в таких растворителях, как дихлорметан,
1,2-дихлорэтан и хлороформ.
Хотя алкилирование, как правило, идет намного быстрее,
чем реакции с карбенами, все же в тех случаях, когда можно
опасаться этой побочной реакции, не следует в присутствии
водных растворов щелочей использовать в качестве
органической фазы хлороформ. Выбор катализаторов при применении
хлорированных растворителей проще. Однакр сильные нуклео-
филы могут реагировать с ними. Реакции такого типа особенно
. 3.1. Общие экспериментальные методы 89
хорошо изучены для дихлорметановых растворов хороших нук-
леофилов в присутствии менее реакциониоспособных алкили-
рующих агентов. В этих случаях нежелательные побочные
продукты могут образовываться в заметных количествах [29, 30]:
2R4N+X- + CH2C12 >■ X—СНа—X+2R4N+C1-
Х = О—арил, О—алкил, SR, О2С—R, СН (акцептор)2
При подборе соответствующих условий проведения реакции эти
соединения можно получать в качестве основного продукта.
Если все же необходимо использовать хлорированный
растворитель, но в то же время желательно избежать реакций
внедрения, то в качестве такого инертного хлорированного
растворителя можно взять о-дихлорбензол, несмотря на то что он
является довольно плохим акстрагентом ионных пар.
3.1.2.2. Перемешивание
Для проведения большинства лабораторных МФК-синтезов
можно использовать магнитную мешалку. Однако следует иметь
в виду, что иногда результаты не воспроизводятся, особенно
в тех случаях, когда для реакции используют 50%-ные растворы
гидроксида натрия или калия, которые из-за их вязкости
перемешиваются слишком медленно. Рекомендуются следующие
скорости перемешивания: для МФК-реакций в нейтральных
условиях в системе вода/органическая фаза более 200 об/мин [27],
для реакций в присутствии гидроксида натрия и в системах
твердая фаза/жидкость 750—800 об/мин {31, 32]. В некоторых
случаях в системах твердая фаза/жидкость приходится
использовать высокоэффективные скребковые мешалки..
3.1.2.3. Количество катализатора
По имеющимся в литературе данным количество
катализатора в зависимости от типа системы варьируется от нескольких
молярных процентов до нескольких молей.
Поскольку скорость реакции зависит от концентрации
катализатора (в органическом слое), то очень маленькие
концентрации его следует использовать только в сильноэкзотермических
процессах или в тех случаях, .когда стоимость катализатора
слишком высока. В большинстве случаев нормальным
количеством считается 1—3 мол.%.
Молярные количества катализатора желательно
использовать в следующих случаях:
а) если в ходе реакции образуется иодид-ион, который может
связывать ониевый катион в органической фазе;
б) если используется малоактивный алкилирующий агент;
90 Глава S. Практическое использование МФК
в) если алкилирующий агент склонен к побочным реакциям
(например, к гидролизу под влиянием водного раствора
щелочи) ;
г) если желательно провести реакцию с полифункциональной
молекулой селективно.
3.1.2.4. Стабильность катализатора
Обычно катализаторы выполняют свои функции очень
хорошо, однако следует иметь в виду, что аммониевые и фосфоние-
вые соли могут подвергаться деструкции в ходе реакции.
Известно, например, что четвертичные гидроксиды претерпевают при
повышенных температурах гофмановское расщепление
(элиминирование), однако в зависимости от структуры эта реакция
может идти и при комнатной температуре. Кроме того, при
наличии подходящих заместителей возможен распад:
RSNR'+OH- >■ RsN-f R'OH
По легкости отщепления от гидроксида четвертичного
аммония радикалы можно расположить в следующий ряд (34]: (J-фе-
нилэтил > аллил > бензил > этил > пропил > циклогек-
сил > метил > изобутил > фенил. Препаративно
элиминирование по Гофману можно провести, нагревая аммониевую соль
с КОН в полиэтиленгликоле (1275].
Четвертичные аммониевые соли с бензильными
заместителями в присутствии сильных нуклеофилов, таких, как феноляты
или тиоляты, могут иногда образовывать >бензиловые эфиры или
соответственно сульфиды [211, 226, 231, 933]. Описано много и
других примеров деалкилирования. Недавно эта реакция была
использована в препаративных целях для удаления бензильной
или метильной группы в очень мягких условиях (например,
с применением тиолятов или других нуклеофилов в ГМФТ при
комнатной температуре [848—850]). Сообщалось также, что
в условиях МФК под действием тиолятов может происходить
декватернизация гетероциклов [1020, 1021]. Внутримолекулярное
деалкилирование в триалкилсульфонийгалогенидах в
малополярных средах происходит очень быстро; при 50 °С время
полупревращения в хлорбензоле при концентрации раствора
10~2 моль/л изменяется от 10 до 40 мин в зависимости от типа
противоиона: 1~>Вг~>С1~ [1641].
Однако все это не страшно, так как в подавляющем
большинстве методик МФК применяются довольно мягкие условия,
а используемые обычно катализаторы достаточно устойчивы
в этих условиях и не распадаются при комнатной температуре
в течение многих дней.
3.1. Общие экспериментальные методы 91
Повышение температуры способствует распаду. Так, в
присутствии концентрированной натриевой щелочи тетрабутилам-
мониевая соль за 7 сут распадается до трибутиламина на 52%
при 60 СС и на 97% лри 100 °С. ТЭБА с высоким выходом дает
диэтилбензиламин, а бензилтриметиламмонийхлорид — равные
количества дибензилового эфира и диметилбензиламина [12J.
Эфир образуется в несколько стадий по реакции, сходной с
реакциями фенолятов:
CeH5CHa—NMe8+.OH- *■ СвН6СН,,—ОН + MesN
С„НвСН—NMea+ -f "О—СН2СвНб *■ СвН6СНа—О—CH2CeH5 + MesN
Аналогично при действии NaOH распадается бензилгексадецил-
днметиламмонийбромид, давая диметилгексадециламин, диме-
тилбензиламин, дибензиловый эфир и гексадецен-1 112].
Распад этих катализаторов проходит при (повышенных
температурах, а бензйлдиметилфениламмонийгидроксид, как
сообщается в работе 1225], разлагается при упаривании
спиртового раствора при комнатной температуре. Этоксид, пропоксид
и гидроксид а-аллилбензилметилфениламмо.ния распадаются
при упаривании их спиртовых или водных растворов или
высушивании {1007]. (Другие данные о ста0ильности R4NOH при
комнатной температуре см. в разд. 1.3.4, последняя часть,
и в работах {12, 1229, 1475].)
Фосфониевые соли, используемые .в качестве катализаторов,
при контакте с концентрированными водными щелочами более
стабильны, чем соответствующие аммониевые соли. Однако они
вступают в реакции типа реакции Виттига и мЪгут, если
содержат фенильные заместители, распадаться с образованием
оксида трифенилфосфина {1027]. Так, трифенилалкилфосфониевые
соли претерпевают МФК-реакцию Виттига при комнатной
температуре, в то же время трибутилгексадецилфосфонийбромид не
изменялся в этих условиях в течение 16 ч при 100 °С (12]. Тетра-
фенилфосфонийхлорид распадается быстрее, чем бромид, и
намного быстрее, чем иодид. Эта реакция ускоряется BiuNBr.
В присутствии концентрированного водного раствора NaOH за
15 ч при 20 °С распад проходит на 83% в дихлорметане и на
17% в бензоле или в гексане [1242].
Катализаторы, содержащие бензильную группу, .могут быть
особенно чувствительны к действию окислителей [2].
Фосфониевые соли в отсутствие водного раствора гидроксида натрия,
по-видимому, более стабильны, чем соответствующие соли ам-
моиия, примерно до 200 °С {4]. Однако при получении тиоэфиров
было отмечено, что некоторые фосфониевые ионы распадаются
быстрее аммониевых даже в мягких условиях {27].
Процессы разложения катализаторов, рассмотренные выше,
могут приводить к появлению в ходе реакции побочных продук-
92 Глава 3. Практическое использование МФК
тов. Однако изменения каталитической активности часто не
наблюдается, так как присутствующие в реакционной смеси алки-
лирующие агенты реагируют с образующимися аминами и фос-
финами, давая вновь четвертичные ионы.
3.1.2.5. Выбор катализатора*
Хороший катализатор для проведения реакций в
нейтральных средах должен иметь 15 или больше атомов углерода. При
предварительном изучении новых МФК-реакций в нейтральных
или кислых средах лучше всего использовать тетрабутиламмо-
ниевые соли, в частности бисульфат или аликват 336. В при-;
сутствии концентрированных водных щелочей прежде всего
следует испытать Bu4NX, ТЭБА или аликват 336. Однако, как было
показано в других разделах, эффективными могут оказаться
и многие другие .катализаторы — ониевые соли, краун-эфиры и
криптанды. В ранних работах высказывалось предположение,
что для МФК в системах жидкость/твердая фаза лучше всего
использовать краун-эфиры, однако оказалось, что в этих систе-'
мах ониевые соли столь же эффективны [624, 953]. В некоторых
случаях хорошие результаты были получены с необычными для
МФК катализаторами: полиамидами [117, 118], полиэфирами-
с открытой цепью [47], 2-диалкиламинопиримидиновыми солями'
[312], полифосфорамидами [851] и др. [46]. Весьма удобными
катализаторами являются соли бензилтрибутиламмония, посколь-1
ку их легко приготовить в лабораторных условиях, а иопользо-'
вание их очень часто дает хорошие результаты.
3.1.2.6. Выделение и регенерация катализатора
Отделение катализатора от продуктов реакции после синтеза,,
как правило, не ■представляет никаких трудностей, так как
в большинстве случаев только сам катализатор растворим
в воде. Иногда достаточно несколько раз промыть реакционную:
смесь водой. В других случаях упаривают использованный
органический растворитель, остаток обрабатывают водой и водный
раствор экстрагируют подходящим растворителем, например
эфиром. Для того чтобы регенерировать катализатор, водный
раствор ониевой соли насыщают NaOH или неорганической
солью и экстрагируют дихлорметаном [1719]. Такая методика
высаливания может быть использована для получения
различных водных фаз. В 1%-ном растворе NaOH растворяется 27%
Bu4NBr, а в 15%-ном NaOH —только 0,07% [1716]. Тетрабутил-
* Небольшой обзор см. в [1460].
S.I. Общие-экспериментальные методы
аммониевые соли из различных источников можно собирать
вместе и растворять в дихлорметане. После встряхивания с
избытком концентрированного водного раствора иодида натрия
при рН ниже 3 и удаления растворителя получается иодид,
.который можно перекристаллизовать из бутанона {33]. Эту соль
можно превратить в бисульфат тетрабутиламмония по одной
из трех приведенных ниже методик [33]:
толуол
1) Bu4N+r + H2SO4 + (k-C8H17)3N —->- Bu4N+HSO4- + (h-C8H17)8NH+I-
/ вода
2) Bu4N+I" + (CH3)2SO4 » Bu4N+ -OSO2OCH3 + CH3I
H+
Bu4N+-OSO2OCHj + H2O » Bu4N+HSO4" + CH3OH
с12с=сна
3) 3Bu4N+I" + 2H2SO4 + H2O2 »-
' вода
>■ Bu4N+I3- + 2Bu4N+HSO4-
Для выделения 18-крауна-6 часто используют следующую
процедуру: реакционную смесь несколько раз промывают
подкисленным насыщенным раствором хлорида калия. Водные
экстракты от нескольких серий опытов смешивают и упаривают
на роторном испарителе. Полученный твердый остаток
несколько раз экстрагируют метиленхлоридом. Экстракты сушат
сульфатом магния, фильтруют и упаривают. Остающийся после
упаривания твердый остаток содержит хлорид калия; для очистки'
его можно сублимировать при 130—140сС и давлении
0,05 мм рт. ст. или перекристаллизовать из ацетонитрила. После
этого получается лродукт, достаточно чистый, и, хотя он и
содержит следы хлорида калия, его можно использовать для
проведения большинства реакций [35].
Выделение липофильных катализаторов (например, аликва-
та 336 или С1бН3зРВи3Вг) из неполярных растворителей не
всегда проходит легко (см. также [1801]). Часто продукт реакции
можно отогнать или отделить перекристаллизацией. По новой"
методике реакционную смесь после завершения реакции
встряхивают с ионообменной смолой (Н-форма) при комнатной
температуре. Смола захватывает катион, который может быть
регенерирован промывкой водно-этанольным раствором НС1 [1499].
В некоторых случаях распределение R4NX между водной к
органической фазами сильно зависит от температуры. Известен
по крайней мере один случай, когда для переведения всего
катализатора в водный слой достаточно простого охлаждения
[1733], поскольку коэффициенты распределения D0°c и D25°c
различаются приблизительно в 100 раз.
94 Глава 3. Практическое использование МФК
3.1.2.7. Дополнения
Для того чтобы облегчить экстракцию ассоциированных
ионных пар в органическую фазу в водно-органических двухфазных
системах, водная фаза должна быть по возможности
концентрированной. Хотя среди МФК-реакций очень мало экзотермичных,
все же разумно при проведении неизвестных реакций соблюдать
предосторожности — иметь наготове баню со льдом или
добавлять реагент небольшими порциями. В присутствии
концентрированных растворов едкого натра часто образуются устойчивые
эмульсии. В некоторых случаях разрушению эмульсии помогает
нейтрализация или центрифугирование, однако, чтобы убрать:
избыток щелочи, часто проще промыть смесь несколько раз
водой. Такая промывка способствует разделению фаз. Следует
помнить, что при использовании R4N+HSO4" для нейтрализации
необходимо добавлять больше чем один эквивалент щелочи. .,
Иногда желательно знать, где находится большая часть ка-"
тализатора (ониевого катиона): в начале реакции, в ходе ее
или в конце — перед началом обработки. Для определения
аммонийных ионов иногда используют титрование лаурилсульфа-
том натрия в двухфазной системе хлороформ/водный буфер;
в присутствии метилового желтого в качестве индикатора [1797],
Брендстрём [1791] рекомендует титрование 0,1 М раствором
калиевой соли 3,5-ди-грег-бутил-2 гидроксибензолсульфоната (6)
в фосфатном буфере (0,5 М Н3РО4/0,5 М К.Н2РО4) /дихлорметан.
Для установления конечной точки титрования он использует
2-10~4 М раствор диэтиламиноиндонафтола синего (7), цвет
которого меняется от густо-синего до почти бесцветного.
Переход можно сделать еще более четким, если добавить в смесь
в качестве контрастного красного цвета следы Судана III. Такое
титрование с успехом используется для определения Bu4N+ или
еще более липофильных катионов. Более простая, хотя и менее
чувствительная, методика основана на использовании в качестве
индикатора Fe3+. В точке эквивалентности происходит хелато-
образование и водный слой меняет желтую окраску на
зеленовато-голубую [1716].
он
Схема 3.6
3.1. Общие экспериментальные методы 95
3.1.3. Общие замечания о проведении реакций
в присутствии щелочей
Хотя лимитирующая стадия для большей части реакций
обмена, катализируемых основаниями, и для реакций замещения
одинакова, однако в случае реакций со слабыми кислотами
необходима предварительная стадия депротонирования. Таким
образом, эти реакции включают по крайней мере еще одно
равновесие.
Наиболее часто используемые неорганические основания
представляют собой твердые гидроксиды щелочных металлов:
или их концентрированные водные растворы. Перемешивание
таких систем может представлять проблему, так как растворы
становятся вязкими и склонны давать эмульсии. Выходы и
скорости в случае, если не удается добиться идентичных условий
перемешивания, невоспроизводимы. Как правило, для
перемешивания таких систем лучше использовать не магнитную,,
а механическую мешалку или вибромешалку.
В общем 'можно считать, что система водный раствор гидрок-
сида натрия/аммониевый катализатор способна депротонировать
субстраты с рКа ниже 20—25. Сведения о рКа использованных
субстратов можно найти в {212, 213]. В этих условиях флуорен
алкилируется, а ацетонитрил не реагирует {214]. Однако в
присутствии порошкообразного гидроксида калия и 18-крауна-6
ацетонитрил вступает в альдольную конденсацию {215].
Аналогично алкины-1 не алкилируются, но .могут реагировать с
карбонильной группой. Недавно была описана изомеризация 3-фенил-
пропена-1 (рКа 34) в 1-фенилпропен-1 .под действием 50%-ного
NaOH/R^NX {1647]. Потенциальные возможности системы
твердый порошкообразный КОН/катализатор (иногда при
добавлении инертного высушивающего агента) пока еще используются
не полностью.
Сильными основаниями в системах МФК являются фториды
[1235, 1297, 1361, 1605, 1638] и твердые карбонаты щелочных
металлов {128, 1048], хотя они и несколько слабее, чем водный
NaOH [2]. При проведении реакций в условиях МФК с
карбонатами в .качестве оснований смесь нагревают обычно до 80—
100 °С или даже выше (см. также с. 67). Дитрих и Леен [359J
показали, что комбинация криптофикс[2.2.2](5) /ЫаЫН2/ТГФ-
способна депротонировать дифенил- и трифенилметан.
Многообещающими являются, по-видимому, такие
комбинации, как твердый трет-бутоксид калия/краун-эфир/лодходящий
растворитель. Рассмотрим вначале некоторые лучше изученные
гомогенные системы. Известно, что скорость депротонирования
грег-бутоксидом калия зависит как от его концентрации, так
и от типа растворителя. Сложность этой системы объясняется
96 Глава 3. Практическое использование МФК
не только тем, что в ней имеется целая серия равновесных
реакций между .мономерами и агрегатами ионных пар грег-бутокси-
да калия, но также и тем, что он может давать комплексы
с растворителем [216, 217]. Каталитической активностью в
реакциях изомеризации, рацемизации и Н/Ю-обмена обладает только
мономерная форма основания. Естественно, что относительная
концентрация мономерных молекул будет больше всего при
яизкой .концентрации щелочи. Было локазано, что при
одинаковой концентрации (10~3 М) в rper-бутаноле грег-бутоксид бен-
зилтриметиламмония по крайней мере в 1000 раз более сильное
основание, чем rper-бутоксид калия [218]. Добавление полярных
растворителей, например ДМСО, к раствору КОС(СН3)3 в трет-
бутаноле увеличивает основность по нескольким причинам:
такой растворитель увеличивает диэлектрическую
проницаемость раствора, специфически сольватирует катион К+ (что
соответственно сдвигает равновесие между агрегатами и
мономерами в сторону мономеров) и устанавливается новое
равновесие с его сопряженным основанием {218]. Поэтому в чистом
ДМСО грег-бутоксид калия должен быть намного более
сильным основанием, чем в грег-бутаноле. Действительно, было
установлено, что H/D-обмен в ДМСО идет в 105 раз быстрее,
чем в грег-бутаноле {216]. Но все-таки даже в таком
растворителе основность увеличивается при добавлении .краун-эфира.
При изменении .концентрации комплекса КОС(|СН3)з/краун-
зфир от 0,01 до 0,33 М наблюдается линейная зависимость
изменения скорости изомеризации. Это означает, что добавление
краун-эфира действительно эффективно лодавляет ассоциацию
ионных пар {219]. Недавно -было показано, что в тетрагидрофу-
ране или в бензоле краун-эфир увеличивает нуклеофильность
грег-бутоксида калия сильнее, чем его основность: бензилхлорид
дает с выходом 77—83% грег-бутоксибензиловый эфир и только
очень небольшое количество стильбена. Карбен не образуется.
Дифенилметан частично депротонируется [931].
В результате превращения при действии краун-эфира
агрегатов ионных пар или контактных ионных пар в сольватнораз-
деленные ионные лары всегда образуется более реакционноспо-
собный анион. Этот эффект был использован во многих
случаях. Некоторые из них, относящиеся к МФК, будут
рассмотрены в других разделах этой книги. Что же касается реакций,
катализируемых основаниями, то следует помнить, что по
сравнению с гомогенными реакциями применение краун^иров
в МФК изменяет не только скорость реакции {434, 918, 1284], но
даже отношение сым-изомер/амгы-изомер и региоселективность
элиминирования НХ [220—222].
Хотя 18-краун-6 и не способен солюбилизировать гидрид
калия в ТГФ [1397], такая комбинация, как было показано [430],
3.1. Общие экспериментальные методы 97
может металлировать трифенилметан (рКа 31,5), дифенилметан
(рКа 33,1), ди-п-толилметан (рКа 35,1) и даже до некоторой
степени ди-2,4-ксилилметан {рКа 36,3). Применение {2.2.2]
чрезвычайно усиливает такого рода металлирование слабых кислот
и енолизацию кетонов, что позволяет проводить их алкилирова-
ние {1309, 1310, 1379, 1600]. Реакция проходит, по-видимому, на
поверхности кристаллов NaH или КН.
12-Краун-4 солюбилизирует натрий, образуя ионную пару
[комплекс—cNa+][Na~]. Добавление хлористого лития осаждает
NaCl, и в .конце концов можно приготовить раствор, который
содержит 0,1 М Na~. Такой раствор действует как чрезвычайно
сильное основание, которое может депротонировать
трифенилметан и аммиак {1247], однако данных о его использовании пока
не имеется. Раствор rper-бутоксида бензилтриметиламмония
в rper-бутаноле можно приготовить простым смешиванием трет-
бутоксида калия с галогенидом аммония в грег-бутаноле и
отделением выпадающего осадка КХ центрифугированием {218].
Ценность использования этого реагента и других ониевых солей
применительно к другим растворителям и субстратам не
очевидна, поскольку они претерпевают глубокий распад {1060].
Опыт показывает, что лучшие результаты получаются при
комбинировании краун-эфиров с различными сильными
основаниями в твердом состоянии. Использование дорогого 'Краун-зфира
при проведении различных реакций оправданно в тех случаях,
когда в результате этого можно избежать применения дорогих
безводных растворителей или металлоорганических соединений.
В заключение этих общих замечаний о проведении реакций,
катализируемых щелочами, необходимо указать, что известно
довольно большое число реакций, катализируемых тритоном В
(бензилтриметиламмонийгидроксидом) в гомогенных растворах.
Сюда относятся: присоединение по Михаэлю, альдольная
конденсация и самоокисление карбанионов {223]. По-видимому,
многие из них могут быть осуществлены в двухфазных системах
с использованием в качестве МФ-катализатора той же самой
или иной аммониевой соли. Однако до сих пор описано
сравнительно небольшое число таких реакций (см. ниже).
3.1.4. Катализаторы, связанные с полимерами
Хотя межфазные катализаторы обычно не регенерируются,
это, конечно, необходимо при применении их в больших
количествах или при использовании в непрерывных процессах.
В этих случаях нерастворимые, связанные с полимерами
катализаторы («трехфазные катализаторы») обладают широкими
потенциальными возможностями применения. Как будет
подробно показано в других разделах, такими катализаторами мо-
7—751
93 Глава 3. Практическое использование МФК
гут быть четвертичные амминиевые соли [62, 64, 68, 775, 860,
902], фосфониевые соли [775, 858], триамиды фосфорной кислоты
[1025], краун-эфиры [775, 858, 1023, 1024], полиэфиры [316,
1022] и криптанды (775], связанные с полистирольной матрицей.
Иногда для МФК-реакций используют технические анионооб-
менные смолы {299, 616], но они мало подходят для этой цели
из-за высокого содержания —NR3+-rpynn в полимере '[64].
Кинетическая активность специально полученных катализаторов на
полимерах может быть столь же велика, как и у растворимых
МФ-катализаторов. Недавно был описан {1008] легкий и
экономичный синтез крупнопористых смол и катализаторов на их
основе. Однако, несмотря на очевидные преимущества, эти
катализаторы еще не получили широкого распространения в
промышленности. Во многих случаях наблюдается небольшая
потеря активности, даже если катализатор работает всего несколько
циклов {1670—1673], и, по-видимому, никто не может
предложить тест, который лозволил бы сказать, сколько циклов будет
работать катализатор или как долго можно с ним вести
процесс. Одной из причин снижения активности является слишком
низкая механическая прочность в условиях сильного перемешич
вания. Еще одна, более важная причина состоит в том, что
фосфониевые и аммониевые трехфазные катализаторы подвергав
ются деалкилированию (см. разд. 3.1.2.4 и [933]). Это лозволяет
предположить, что в будущем получат распространение трех*
фазные катализаторы на основе связанных с полимерным
носителем краун-эфиров, криптандов и эфиров полиэтиленгликоля^
Однако появление лростых методов выделения и регенерации
обычных .катализаторов [1499] «ожет сделать их более
предпочтительными, чем трехфазные катализаторы. Были
предложены '[1801] 'катализаторы на основе глины, которая способна
адсорбировать одновременно и водную фазу, и ониевую соль*
образуя необычный трехфазный катализатор. В другом
примере [1807] рекомендуется использовать в качестве катализатора
для превращения iCH2X2 в CH2(OR)2 монтмориллонит (торговое
название Tixogel VP, фирма Sudchemie, Munchen), 'который
является ионообменником для четвертичных аммониевых ионов.
Тщательное изучение показало, что реакции с
катализаторами, связанными с полимерами, имеют отчетливо выраженную
кинетику лсевдопервого порядка в тех случаях, когда
каталитическая часть связана с матрицей длинной несущей цепью —
«спейсером» — такой, чтобы до матрицы было 30—40 атомов.
Наиболее ярко параллельный рост .каталитической активности
и длины цепи наблюдали при использовании в качестве
растворителя м-гептана для аммониевых [860], фосфониевых [8581
и краун-эфирных {858] каталитических групп. Эти данные
позволяют предположить, что каталитическая активность зависит
Таблица 3.1. Приготовление и сравнение катализаторов,
оптимизации препаративных методик при трехфазном катализе
Основа: ПС — хлорметилированный сшитые полистирол; Ст — стекло или
кремнезем; АОС — продажная анионообменная смола; ПМА — полиметил-
акрилат.
Активные группы: А — RsN+; В — R$P+; С — краун-эфир; D — криптанд; Е —
полиэтиленгликоль (эфир); F — амид фосфорной кислоты; G — эфир фосфо-
ниевой кислоты; Н — аминоксид; I — азакраун-эфир; К — AsO3H2; L —
N(O)R2
Тип реакции
Катализатор
активная
группа
Литература
R—X—»-R—Y (обмен галогена)
ROSO2CH3—>-RCI
RX
ПС
Ст
ПС
ПС
ПС
ПС
ПС
ПС
ПС
ПС
А, В, С, D
А, В
D
Е
С
А
В
F
А, В, Е, F
А, В
(Модифицированный
декстран-ани-
онный обменник)
(Специальный по-
лиамидэфир)
ПС
ПС
ПС
ПС
ПС
ПС
ПС
ПС
ПС
ПС
ПС
ПС
ПС
ПС
(Moj
G
В
А, В, С, D
А, В, С, F
F
А
А, В, С, Е, F
В
С
Н
I
А, В
D
L
ниФицирован-
755,
859,
1678
1684,
1681,
1023,
62
1008,
1025
1675
1683
1673
1695
1413,
1687
775,
1670
1025
858
1026,
, 1748
, 1769
, 1689
, 1745,
, 1670,
1440
1745,
62, 64, 68,
1672,
1008,
1803,
1690
1686
1683
1684
1674
1673
1675
1612,
1804
1672,
1769
1671
1769
1802
1802
ный декстран-ани-
онный обменник)
(Бензокрауны, со- 1734
полимеризованные
с фенолами и
формальдегидом)
(Полиэфир, сши- 1682
тый с полидиаце-
тиленом)
(Модифицирован- 1677
ный привитой эти-
лениминстироль-
ный сополимер)
100 Глава 3. Практическое использование
МФК
Продолжение табл. 3.1
Тин^реакции
Катализатор
основа
активная
группа
Литература
RX—>JR—О—CO-iR'
RBr—»-ROAr и
RBr—>-ROR'
RBr—»-R—SR'
RX
PhCH2CN—vPhCHRCN
(конц. NaOH)
PhCH2COCH3—vPhCHRCOCH3
(конц. NaOH)
RBr—*ROH
ПС А, В, Е, F
ПС А
ПС в
ПС А, В
Ст С
АОС
ПМА С
(Модифицированный привитой эти-
лениминсти
рольный сополимер)
(Полиэфир,
сшитый с полидиаце-
тиленом)
ПС Е
1675
1321
1670
1672
1226
1321
1685
1677
1682
316, 1022, 1691
ПС А, В
ПС C,D
ПС А, В, Е, F
ПС А
(Этиленстирол-
оксидный
блок-сополимер)
Ст В
ПС В
(Полиакриламиды)
(Полимеры винил-
краун-эфиров)
(Бензокрауны, со-
полимеризованные
с фенолами и
формальдегидом)
ПС В
ПС А, В
ПС C,D
ПС А, В
ПС В
ПС А
АОС
ПС А, В
ПС в
ПС C.D
ПС Е. F
1672
1745,
1693
62
1688
1678
1670
1781
1024
1734
1008,
1672,
1769
1672
1670
902,
299,
1683
1008
1769
313,
1769
1670
1683
1805
958
1025, 1694
3.1. Общие экспериментальные методы
Продолжение табл. З.Г
Тип реакции
Катализатор
основа
активная
группа
(Полиэфир,
сшитый с полидиаце-
тиленом)
ПС А
АОС
(Модифицированный привитой эти-
лениминстироль-
ный сополимер)
АОС
ПС А
Ст В
ПС F
ПС C.D
(Модифицированный декстранани-
онный обменник)
ПС А
АОС
Литература
1682
62, 68, 902
616
1677
616
62
859, 167&
1025
1769
1673
62
1806
RBr—«-JROR+ROH
: ССЬ-присоединение
(НСС13, конц. NaOH)
: СВгг-присоединение
RCHBr—CHBrR'—vRCH=CHR'
(Nal, Na2S2O3)
RCOR'—-.-RCHOHR'
(NaBH4)
RCHOHR' - RCOR'
(OCle)
RCH=CH—CO—R' -►
RCH—CHCO—R' (H2O2)
чо7
RCOR'
ПС А
RCH—CHR' -► RCH—CHR'
ПС C,D
Ароматическое нуклеофильное заме- Ст в
щение
1676
1281
1282
1726
1679
1678
102 Глава 3. Практическое использование МФК
от того, насколько каталитический центр выступает над
поверхностью носителя, как >бы «растворяясь» в органическом
растворителе. Влияние структуры носителя было тщательно изучено
[1800] (см. обзор {56] в гл. 2).
Подобного же типа катализаторы, включающие
четвертичный фосфониевый катализатор, связанный с силикагелем,
оказались активными в реакциях обмена, алкилирования и
восстановления боргидридом. Соединение А было более активным,
чем В.
*~°\ Ч \ *
О—Si(CH2)3PBu3Bre ^ —О—Si(CH2)3NHCO(CH2)10PBu3Bre
</ j-c/
А В
Схема 3.7
Активными катализаторами были также ониевые соли,
связанные с аэрозолем-200 или с аминоалкилированным стеклом
марки Corning [1026]. Катализатором в реакциях замещения
и окисления может быть даже продажный глинозем (оксид
алюминия) [1680].
В табл. 3.1 приведен обзор методов получения и
использования трехфазных катализаторов, которые здесь подробно не
рассматриваются.
3.1.5. Хиральиые катализаторы
Межфазный катализ включает образование ионных пар,
в которых анион и катион довольно тесно связаны. Возможно,
поэтому ассиметричное влияние хирального катиона
катализатора на реакции анионов приводит к частичному разделению
рацематов, т. е. к оптической индукции. Необходимым условием
такого эффекта является достаточно тесное взаимодействие
аниона и катиона и только в одном из нескольких возможных
положений и конформаций. Высокая подвижность аниона по
отношению « катиону препятствует этому эффекту.
Использование с этой целью четвертичных аммониевых солей с хираль-
ным центром в углеродном скелете, по-видимому,
малоперспективно, если только анион-катионное взаимодействие не
усиливается дополнительной полярной группой (например,
группой ОН, способной образовывать водородную связь). «Лучшими
катализаторами могут быть соединения с хиральным
аммонийным азотом, который с трех сторон стерически экранирован
[1173, 1601].
Большая часть экспериментов по оптической индукции про-
3.1. Общие экспериментальные методы 103"
ведена с катализаторами на основе эфедрина (А):
Me Me Rl R2
I I , R' e H
H — С — N <,r2 г ^ Mee-jl a Me CH2Ph
H — С — OH H % b Me Me
= Ph
Ph А В с Me Et
MeO.
OH
R1 R2
d Et Et
с Me C12 H2.
N-<:H2Ph
D
Схема 3.8
Имеются две возможные причины, которые могут привести
к ошибочному истолкованию такого рода экспериментов.
1. Распад этого катализатора в условиях МФК проходит
довольно легко {384]. При этом образуется оптически активный
оксиран (В) с очень высоким удельным вращением ([o]d
+ 117,6°, 'без растворителя) {544].
2. В процессе кватернизации почти всегда образуются
небольшие количества гидрогалогенида третичного амина.
Свободный амин регенерируется при обработке реакционной смеси
после проведения МФК-реакции. Примесь веществ,
образующихся в результате процессов 1 или 2, или же продуктов,
образовавшихся из этих примесей, может симулировать оптическую
индукцию, несмотря на тщательные меры предосторожности.
Хлориды бензилхинина или хинидина (С), так же как и А,
могут распадаться, давая диастереомерные элоксиды (D) с
удельным вращением {a]D22 —53,2° или +118,2° соответственно (1697]."
В табл. 3.2 приведены данные о скоростях распада С. У
более липофильных солей распад происходит медленнее.
Алкилирование
Файд [383] сообщил, что если в качестве .катализатора
использовать Аа в хлороформе, то наблюдается асимметричное
алкилирование соединений Е (схема 3.9). Максимальная
асимметрическая индукция составляла 6%, с катализатором АЬ
выход был ниже, в растворе гексана оптической активности
совсем не наблюдалось.
104 Глава 3. Практическое использование МФК
Таблица 3.2. Разложение бензилхининийхлорида (1)
(бензилхинидинийхлорид разлагается с такой же скоростью),
1Ч-додецил-1Ч-метилэфедринийбромида (2) и
1Ч-бензил-1Ч-метилэфедринийхлорида (3) в различных условиях [1697]
-Соединение
Водная фаза
Температура, °С
Время,
ч
Степень
рааложе-
ния, %
0,6 М NaBH4a
30%-ный H2O2/2MNaOH
Водный тиофенолят
10%-ный водн. NaOH
30%-ный водн. NaOH
50%-ный водн. NaOH
Водный KCN
0,6 М NaBH4a
10%-ный водн.
30%-ный водн.
50%-ный водн.
0,6 М NaBH4a
10%-ный водн.
30%-ный водн.
50%-ный водн.
NaOH
NaOH
NaOH
NaOH
NaOH
NaOH
Комнатная
50
Комнатная
50
Комнатная
21
50
80
21
50
80
21
50
•80
80
0
Комнатная
Комнатная
Комнатная
0
Комнатная
Комнатная
Комнаиная
12
72
24
5
24
72
1,
0,
18
1
0,
о о о ел
24
1
10
12
6
1
15
10
10
5
75
5
75
5
0
50
18
40
0
80
70
65
85
85
80
60
80
80
87
25
0
5
15
50
0
10
25
65
арН, равный вначале 9, медленно возрастает до 10,2.
При воспроизведении данных Файда оказалось, что
наблюдавшаяся удивительно высокая оптическая индукция для
необработанного продукта (по крайней мере на 98%, т. е.
практически полностью) обусловлена наличием примесей (843, 949, 1697].
о „о
-С—Z
+ RBr
3.1. Общие экспериментальные методы 105
Если водный раствор натриевой соли DL-миндальной или
DL-2-фенилмасляной кислоты обработать этилбромидом в мети-
ленхлориде в присутствии 0,5 моля оптически активного
Ы.Ы.Н-триэтилметиламмонийбромида, то образуются
соответствующие этиловые эфиры с оптической чистотой 3,2 и 8,6% [949].
В этом случае распад катализатора проходит с трудом, а
другие примеси были исключены. В .последующих работах
описано энантиоселективное алкилирование, в котором также
использовали р-гидроксиаммониевые катализаторы. Для большинства
продуктов была получена низкая оптическая чистота. Часто
трудно решить, насколько тщательно были очищены продукты
и насколько реальны приведенные данные о выходе оптически
активного продукта. Однако максимальная оптическая чистота
19% точно установлена на основе кинетики разделения
рацемата в системе фталимид/катализатор С и этил-2-бромпропионат
[1469, 1661]. Помимо этих реакций были изучены алкилирование
соединений типа Е и других веществ с активной кислой ме-
тиленовой группой в присутствии хирального бензилдиме-
тил- (£{«с-2-гидроксиметил) циклогексиламмонийбромида [1333],
превращения арилацетонитрилов с различными катализаторами.
[1333, 1469, 1661], тиофенолятов, фенолятов и бензоатов с
.катализаторами типа А и С [1469, 1661] и присоединение
хлороформа к карбонильной группе бензальдегида [1648].
Присоединение дихлоркарбена
В работе {622] сообщалось, что катализатор Ас наводит
небольшую асимметрическую индукцию в продуктах
присоединения дихлоркарбена к олефинам в системе НСС13/конц. NaOH.
Тщательное исследование аналогичной реакции с оптически
активным катализатором Ad показало [384], что оптически
активное вещество содержится только в неочищенном продукте;
это оптически активное вещество оказалось эпоксидом В,
образовавшемся из катализатора. По-видимому, механизм реакции
также не благоприятствует образованию оптически активных,
аддуктов дихлоркарбена. Неоднократно было показано, что.
интермедиатом в таких реакциях является свободный карбен.
н н
R—С—R'
1
1
Bu— N—Ви
F
R—С—R'
|
Ви—Ne—Ви
СС12е G
Схема 3.10
106 Глава 3. Практическое использование МФК
Поэтому другие сообщения [623] о некоторой небольшой
оптической индукции при присоединении :СС12 в присутствии
оптически активных аминов F при R=Et, Ph и R'=Me, Et
(схема 3.10) были встречены сдержанно. Несмотря на то что
Макоша рассмотрел предположения о появлении интермедиата
типа G, который представляет собой илид, образованный при
взаимодействии карбена с атомом азота, и локазал {433], что
его образование невозможно, данные этой работы игнорируются
в некоторых более поздних исследованиях, и такие структуры
все еще используются {623] для объяснения хода реакции. Для
того чтобы твердо установить, возможна ли оптическая
индукция подобного типа, .была проведена реакция с хлороформом
и концентрированным раствором гидроксида натрия в
присутствии (5)-(+)-М,1М-диметилфенилэтиламина. Перегнанный
продукт реакции действительно обладал небольшим оптическим
вращением, которое, однако, исчезало при тщательной очистке
[843, 1697].
Восстановление боргидридом
Имеются по крайней мере 12 работ по боргидридиому
восстановлению кетонов с хиральными катализаторами большей
частью типа А или С, в которых сообщается [543], что при
восстановлении ацетофенона в системе 1,2-дихлорэтан/2 н.
NaOH/NaBH4 с катализатором Ае оптическая чистота продукта
достигает 39%. Однако данные о столь высоком вращении не
смогли воспроизвести другие авторы [542, 773, 949, 1014].
Контрольный эксперимент, в котором использовали катализатор Ае
и заранее полученный неактивный 1-фенилэтанол в тех же
самых условиях восстановления, но без добавления NaBH4,
показал, что у выделенного из реакционной смеси 1-фенилэтанола
наблюдается небольшое положительное вращение [545].
Анализ методом ГЖХ показал {949], что даже после хрома-
тографического разделения и фракционной перегонки в
продукте содержится эпоксид В. Этот результат не означает, .конечно,
что все результаты с такими катализаторами ошибочны,
поскольку в нейтральных условиях восстановления боргидридом
катализатор распадается 1медленно (табл. 3.2). Продажный
NaBH4 часто содержит небольшие примеси NaH; это приводит
к тому, что иногда его водные растворы становятся
сильнощелочными ;(см. [1697]).
Авторы последующих работ по боргидридному
восстановлению с катализаторами типа А или С [542, 773, 1170—1172, 1519,
1661] считают, что некоторая степень оптической индукции
может быть получена при использовании кетонов с большими
заместителями (например, пивалофенон и С дают продукт
3.1. Общие экспериментальные методы 107
28,5%-ной оптической чистоты [1661]. Однако необходимо вновь
подчеркнуть, что оценить тщательность работы авторов трудно.
Диалогичные катализаторы, но без ОН-группы не давали
оптически активных продуктов [542, 773, 905, 1519]. Авторы
объясняли это тем, что взаимодействие ОН-группы катализатора с
карбонильной группой субстрата благоприятствует образованию
в переходном состоянии одного из двух возможных диастерео-
меров. Но ведь такие катализаторы без р-гидроксила и не
распадаются так легко!
В присутствии специальных катализаторов, содержащих
фосфониевые соли (1519], несколько гидроксильных групп [905,
1519], две аммонийных группы [905], частично защищенные
моносахариды [1201] и хиральные краун-эфиры (максимальный
оптический выход 8%) [1344] также могут образовывать
оптически активные продукты.
Другие реакции
Небольшое вращение было обнаружено при галогенирова-
нии олефинов, образовании циангидринов [1292], при бисгидр-
оксилировании с помощью перманганата [1761] и в реакции Дар-
зана [923, 951, 1566] в присутствии Ае, С и аналогичных
катализаторов. Утверждения о проведении энантиоселективного
синтеза фенилоксиранов из триметилсульфонийиодида и бенз-
альдегида в присутствии 50%-ного водного NaOH и
катализатора А {500, 555] оказались, как было показано [501] позже,
ошибочными; оптическая активность в этих опытах была
обусловлена наличием В. Однако в присутствии белка водный
раствор iNaIO4 окисляет сульфиды в сульфоксиды с оптической
индукцией до 41 % [1272]. Эта реакция протекает в водной фазе и,
по-видимому, имеет мицеллярный характер.
Для энантиоселективного синтеза сложных эфиров
использовали оптически активные полиамины (полученные из
производных аминокислот). Продукты имели очень низкую
оптическую чистоту [1722]. Еще в одной группе опытов была
поставлена цель получить сложные эфиры DL-2-фенилмасляной и
DL-миндальной кислот при использовании серии хиральных
катализаторов с асимметрическим углеродным скелетом с гидр-
оксильными группами и без них. Только в присутствии бромида
(1R) - (4^-изопропил)-(1г-метил)-(Зс-триэтиламмоний)циклогек-
сана был достигнут небольшой оптический выход [843, 949].
Оксим сополимера 4-винилпиридина и (5)-5-метилгептен-1-
она-3 показал очень умеренное хиральное различие при
гидролизе эфира (ОЬ)-п-нитрофенил-З-метилпентановой кислоты
Таблица 3.3. Присоединений по МйХаэлю и §п6ксиДйрбваниё t испбльзоваиием хирйлЬнЫ* катализатброй
Тип компонентой
Катализаторы
Наибольший
оптический
выход, %
Растворитель, условия реакции
Литера,
тура
балкой, нитрометаи
Халкои, нитрометан
Бутенои, иитросоединения
Бутенон, ip-кетоэфир
Ненасыщенный кетон, тиол
Ненасыщенный сульфоксимид и
нитрометан, тиолы.р-кетоэфнры
Халкон, тиолы, нитросоедине-
ння
Бутенон, р-кетоэфиры
Циклогексеноны, тиолы
Халкон, Н2О2, ОН-
Дипнон, Н2О2, ОН-
Халкон, водн. NaOCl
Циклогексеион,
rper-BuOOH/NaOH
Нафтохнноны,
rper-BuOOH/NaOH
Аа, Ае
Ае, С в F-'форме
Хинин
С
Хиральный краун/КОтрет-Ви
С
Алкалоиды хинной коры
Хинин илн С
Эфедрин, Аа, Ае
N-Метилхининийгидрокснд
Хинин
Хининиевые солн
С
Поли-(5)-аланин
С
С
С
с
26
23
56
18
99
81
75
13
23
25
76
36
48
93
26
25
20
78
Толуол, твердый KF
Толуол, гомогенные
Толуол, гомогенные
Гомогенные, неполярный
растворитель
Толуол
Гомогенные
Гомогенные
СН2С12 илн CH2C12/H2O/KF
Гомогенные или двухфазные
Гомогенные
ССЦ
Толуол, твердый KF
Толуол
Толуол
СН2С12
Толуол
Толуол
Толуол
1660
924
424
1011
1814
1011
554, 1701
1614
1211
1039
1660
603, 1011
1589
1250
95b
1369
1813
1 3.2. Получение галогенсодержащих соединений 109
Присоединение по Михаэлю
и эпоксидирование ненасыщенных кетонов
Стереоселективное присоединение по Михаэлю
представляет собой выдающийся пример МФК-реакции с использованием
оптически активных .катализаторов, которая устояла под
ударами критики [ш97]. Первыми химиками, добившимися высоких
оптических выходов в этой реакции и в реакции эпоксидирова-
ния, были Уайнберг с соавторами (обзор {1810]).
В табл. 3.3 приведены результаты их работы. Общий вывод
таков: наибольший энантиомерный избыток достигается в
неполярных растворителях, в этаноле он равен нулю. В качестве
катализаторов присоединения по Михаэлю использовались
также алкалоиды цинхоны, связанные с полимерным носителем
[892, 1811], и хиральные ониевые соли, являющиеся
производными L-метионина [1812]. С этими катализаторами общий выход
был высоким, но оптический выход разочаровывал.
3.2. Получение галогенсодержащих соединений
Методы МФК были использованы для проведения реакций
замещения следующих типов:
Алкил(арил, ацил)галогенид *■ Различные галогениды
(реакции обмена галогенов)
Алкилсульфонат *■ Алкилгалогенид
Первичный спирт -f-HCl »■ Алкилхлорид
. Диарилдиазометан *■ Диарилметилфторид
(нуклеофильное
ароматическое замещение)
3.2.1. Обмен галогенов
Препаративно наиболее важными являются синтезы иоди-
дов и фторидов. Однако техника МФК может быть
использована также и для получения хлоридов, бромидов и иодидов,
содержащих изотопную метку. Старке [4] нашел, что полное
равновесие 35С1/36С1 между 1-хлороктаном и Na36Cl в присутствии
четвертичной соли в качестве катализатора достигается при
кипении смеси за 5 ч. Аналогичный обмен иод — радиоактивный
иод при 100 °С проходит полностью за 5 мин [4]. При обмене
химически неэквивалентных групп Х~ и Y~~ превращение могут
лимитировать как равновесие экстракции двух ионных пар
Q+X~ и Q+Y~, так и химическое равновесие RX+Y~ +± RY+X~.
Например, если необходимо превратить алкилбромид в
алкилхлорид, то используют избыток хлорида натрия в водном рас-
ПО Глава 3. Практическое использование МФК
творе. После того как МФК-равновесие достигнуто/ водную
фазу заменяют свежим раствором NaCl. Такую обработку
можно повторить несколько раз {4] (см. гл. 2). Самым легким
методом даже для простого обмена I—>-С1 является поэтому
смешивание эквимолярных количеств алкилиодида и безводного
Bu4NGl (который легко приготовить) и отгонка/алкилхлорида
по мере его образования [899].
RX+Nal —-* RI+NaX
При получении первичных алкилиодидов. из хлоридов или
бромидов обменной реакцией с водным раствором Nal
приходится нагревать реакционную смесь до 10&°С в присутствий
четвертичной аммониевой соли (несколько молярных
процентов) в течение 5 или 2 ч соответственно {38, 39]. Реакция идет
намного быстрее с 'Молярным количеством катализатора;
Брендстрём превратил хлорацето-2,6-ксилидид в
соответствующее иодпроизводное при кипячении смеси в течение всего
90 мин (выход 92%) {899]. Вторичные бромиды в этих условиях
дают главным образом олефины. Триметилсилилметилхлорид
превращается в иодид за 2,5 ч кипячения (водный Nal, ВщМ);
однако превращение в 'бромид требует уже 11-сут кипячения
[1210]. Многочисленные попытки превратить хлортриметилсилан
в иод- или бромтриметилсилан не открыли легких путей
достижения этой цели. В тоже время фтортриметилсилан таким
способом получить 'можно (разд. 3.2.2) [1897]. Радиоактивный
иодид обменивается с неактивным иодидом при 10-мин
кипячении с НШ1 в смеси вода/гептан в присутствии гексадецилтрибу-
тиламмонийбромида в .качестве катализатора {1642]. ТЭБА был
использован для превращения
С1—СН2СН2—ОН »- I—СН2СН2—ОН
в системе толуол/вода [1760].
Наряду с четвертичными ониевыми солями в качестве
катализаторов используются и краун-эфиры {43—45, 48]. В табл. 3.4
представлены данные об эффективности различных
катализаторов при реакции 5-молярного избытка водного насыщенного
раствора иодида щелочного металла с н-октилбромидом в
присутствии 0,05 мол. экв. соответствующего катализатора.
Как видно из этих результатов, ониевые соли являются
лучшими .катализаторами, чем большая часть «раун-эфиров,
за исключением только модифицированных аза-макробицикли-
ческих полиэфиров 6а и 6Ь, .которые, однако, не являются
доступными продажными препаратами*. Скорости, полученные
* Еще более высокие скорости были получены при использовании трнцнк-
логексано [2.2.2] (пергидротрибензогексаоксадиаза [8.8.8] эйкозан) [992].
ч 3.2. Получение галогенсодержащих соединений 111
Таблица 3.4. Эффективность различных катализаторов в реакции
К(№)1+н^октилбромид
5-молярный избыток водного насыщенного раствора соли щелочного
металла, 0,09\мол. экв. катализатора
Соль
Катализатор
Температура, "С
Время,
ч
Выход,
Литература
KI
К1
KI
KI
К1
Nal
KI
KI
Отсутствуем
Дибензо-18-иоаун-6 (2)
Дициклогекса»И8-краун-6 (3)
{2.2.2] (5)
к-С1бН3зРВи3+Вг-
Бензо-15-краун-5
6а
6Ь
80
80
80
60
60
80
60
60
24
40
3
14
1
21
0,2
0.5
<4
80
100
90
93
80
100
92
44
44
44
48
48
44
48
48
при применении аммониевых, фосфониевых, краун-эфирных
и криптатных катализаторов, привитых к нерастворимым
полимерам [775, 1023, 1026], а также краун-эфиров с липофильными
боковыми цепями [776], не выходили из обычных границ. Эти
трехфазные катализаторы легко отделяются от реакционной
смеси, но их необходимо готовить специально (см. также
разд. 3.1.4).
R
О
О.
N
Р
О.
Р \ N
Р
a: R = н-СцН2в
Ol R = И-СцГ123
Схема 3.11
Вместо дорогих краун-эфиров предлагают использовать в
качестве катализаторов так называемые полиподные молекулы
и технические смеси полиэфиров с открытыми цепями [46, 47].
Наиболее эффективным из испытанных полиподных молекул
был замещенный триазен 7. По своим каталитическим
свойствам он был близок ,к трибутилгексадецилфосфонийбромиду
и только несколько хуже алкилзамещенного аза-макробицикли-
112 Глава 3. Практическое использование МФК
ческого полиэфира 6. Так, в реакции н-октилбромщн—>-иодид
(5-кратный избыток КД) применение 1 мол.% 7 привело к
выходу иодида 85% за 3 ч при 60 °С [46], а при использовании в той
же реакции 2 мол.% технического диметилового вфира поли-
этиленгликоля (п—8,39) (8) в качестве катализатора через 3 ч
при 100 °С выход иодида был 44% [47]. В реакцииУФинкельштей-
на испытаны также циклические фосфониевые/ и арсониевые
соли [1029J, специальные краун-эфиры и циклические амино-
эфиры [1023, 1028} и гексаалкилфосфамиды [851].
17—(О—СН2—CH2)4]aN N N[(CH2—СН2О)4—н-СвН17]2
N[(CH2—СН2—О)4—нСвН17]2
7
НзС—О(СН2—СН2—О)п—СНз
8
Схема 3.12
Тундо [1364, 1365] предложил использовать в МФК-реакциях
газообразные алкилгалогениды. Так, реакция шла при
пропускании RX через колонку, заполненную KI и расплавленным или
нанесенным на полимер (в некоторых случаях на инертный
носитель) МФ-катализатором при 150—160 °С. Аналогично
проходил обмен галогенами .между дихлорметаном и этилбромидом
при пропускании их через нагретую колонку с А12О3, которая
пропитана фоофониевой солью [1593].
Nat
Метиленхлорид превращается в два приведенных выше
продукта обмена в присутствии трибутилгексадецилфосфонийбро-
мида [40] или некоторых других катализаторов (41]. Реакцию
проводят в качающемся автоклаве при 100—110°С от 10 до
20 ч и молярном соотношении CH2C12/Nal, равном 2,4. Хлориод-
метан и дииодметан были получены с выходом 67 и 14%
соответственно.
При 150 °С этот обмен проходит и без катализатора (1030].
С хлороформом и бромоформом такой обмен не происходит,
вероятно, из-за стерических препятствий подходу нуклеофила.
Но все же обмен можно провести другим, хотя препаративно
и не очень удобным методом с использованием карбенного
механизма в присутствии водной щелочи и МФ-катализатора {42].
3.2. Получение галогенсодержащих соединений WS-
МФКЧреакция замещения протекает намного быстрее, если
в качеств» уходящей использовать метансульфонатную группу.
Для реакции
растворителя
реакционная способность ионов галогенов изменяется в ряду
X=I~>Br~>-Ci~^F~* [44]. Наиболее быстрая реакция
заканчивается за 7 мин при 100 °С, а самая медленная проходит при
115°С,на 65% за 42 ч. Соединение 9, которое легко гидролизует-
ся, было получено по методу, приведенному ниже (49],
продукт 10 синтезирован из соответствующего тозилата в
гомогенном растворе ацетонитрила при кипячении с безводным
Bu4N+F~ в течение 6 ч [26].
Хотя, как было показано выше, вторичные бромиды в
условиях МФК-замещения дают главным образом алкены, более
активные мезилаты превращаются во вторичные галогениды
с относительно хорошими выходами. Из оптически активного
2-октилмезилата были получены оптически активные хлорид
(выход 83%, оптическая чистота 89%) и бромид (выход 78%,
оптическая чистота 82%). Реакцию проводили в присутствии
5 мол.% аликвата 336 или трибутилгексадецилам.монийброми-
да при 100 °С в течение 1,5 или 0,5 ч соответственно. Для
уменьшения рацемизации в результате повторного обмена при
получении фторида, который реагирует слишком медленно,
использовали эквимолярное количество неорганической соли.
ОСНз РСНз
НзС
СН2—OSO2—СНз
СНз
бензол, войн. К.С1
аликват 336,
комн. темп., гомин
FHaC
к
Т
1
_/°
OBz
Ql
т
СНз
9
\ н
НИ
чсн„а
н
Схема 3.13
н о-
10
-С—СНз
СНз
* При использовании в этой же реакции в качестве катализатора соли
три-я-бутилгексадецилфосфония в системе хлорбензол/вода соотношение
активностей следующее: I- : Вг~ : С1~=1 : 1,1 :0,60 [50].
S14 Глава 3. Практическое использование МФК
^
В аналогичных условиях иодид получался в рацемической
•форме {51]. По-видимому, количество катализатора влияет
на оптический выход, однако этот вопрос остался неизученным.
Очень хорошие выходы для различных обменных реакций
были достигнуты при использовании «трехфазных/
.катализаторов при кипячении за время от 24 до 100 ч [62] (см. табл. 3.1).
Обычные анионообменные смолы для этих целей, как правило,
не пригодны. В качестве альтернативы было предложено
«заряжать» анионообменную смолу требуемым ионбм и затем
кипятить ее с субстратом в инертном растворителе [777]. Этот метод,
ло-видимому, значительно .менее привлекателен, чем МФК!
3.2.2. Обмен фторидов
Возвращаясь теперь к обсуждению методов получения
фторидов, которые имеют свои особенности, вспомним, что
сравнительно мало сольватированные фторидные ионные пары в МФК-
реакциях выступают и как нуклеофилы, и как основания. Это
приводит к тому, что возрастает конкуренция между
замещением, гидролизом и элиминированием. Монтанари и сотр. (52]
проводили реакцию следующим образом: первичный или
вторичный алкилбромид, хлорид или мезилат встряхивали при
100—160 °С с насыщенным раствором KF и каталитическим
количеством трибутилгексадецилфосфонийбромида в течение
7—16 ч. Первичные алкилбромиды и бензильные соединения
превращались во фториды с выходами от 70 до 90%; из
вторичных алкилбромидов выход был намного хуже. Циклогексилхло-
рид и 2-октилбромид давали только олефин и спирт. Однако
2-октилмезилат превращался во фторид с выходом 54%.
Использование в качестве катализаторов краун-эфиров в
двухфазной системе вода/органический растворитель не дало
никаких преимуществ {44, 45], однако Лиотта и сотр. [53] предлагают
использовать 18-краун-6 для МФК в системе твердая
фаза/жидкая фаза, лредставляющей собой избыток твердого KF в
бензоле или ацетонитриле. Для большинства первичных алкилгало-
генидов и бензильных галогенидов реакция проходила за то же
время, что и в системах жидкость/жидкость с ониевыми солями
в качестве катализаторов (от 24 до 120 ч). Однако для обмена
C1/F в арилхлорметилсульфидах в ацетонитриле с KF/краун
требовалось кипячение в течение 4—5 сут {895]. В отличие от
рассмотренных выше данных о реакции в системе
вода/органическая фаза твердофазная система позволяет превратить
2-октилбромид во фторид (выход 32%). Однако элиминирование
преобладает и в этом случае. 2,4-Динитрохлорбензол и ацетил-
хлорид также были превращены в соответствующие фториды
в этой системе {53]. Для некоторых из таких процессов с твер-
, 3.2. Получение галогенсодержащих соединений 115
дофазнйй системой наилучшим катализатором из испытанных
краун-эфшюв оказался крипотофико[2.2.2] {60]. Следует снова
отметить, Уго в этих реакциях можно использовать также более
дешевые производные полиэтиленгликолей с открытой цепью
типа 8. Эти катализаторы менее активны, чем жраун-эфиры,
однако этот Недостаток компенсируется тем, что можно
применять в более высокой концентрации дешевый технический
продукт. Для проведения реакций рекомендуют в качестве
растворителя бензол, ацетонитрил или сам эфир полиэтиленгликоля
[61, 1015]. Система KF/CH3CN/18-KpayH-6 позволяет провести
замену хлора, находящегося в а-положении к карбонильной
группе, как это показано на схеме 3.14:
Для обмена C1/F в сульфонилхлоридах {396],
жарбомоилхлоридах и хлорформиатах лучше всего использовать твердый
KF/18-краун-б без растворителя или в ацетонитриле при
комнатной температуре. При использовании системы iKjF/18-кра-
ун-6/ацетонитрил наблюдается реакция элиминирования — п^ч-
соединения {1510]:
PhSe—СНЯ—CFaCl »- PhSe—CH=CF2 »- PhSe—CH2—CF8
Трифторметильные гетероароматические соединения
медленно обменивают изотопы 19F/18F при 100 °С в присутствии краун-
эфира {1424]. Известны также и другие примеры замещения
X/F в бензильном положении в ароматических и
гетероциклических соединениях {1436, 1446]. При нагревании хлортриметил-
силана с системой KF/18-краун-б в ацетонитриле образуется
фтортрвметилсилан, который конденсируется в охлаждаемой
116 Глава 3. Практическое использование МФК
7
ловушке (1897]. Некоторые 'примеры обмена C1/F предъявлены
да схеме 3.15: '
Cl COOR F COOR
C__q niB.KF, оичиклогексано-18-крпун-Б V / /
COOR » * •
a
о Ъ о
-а
о
F
[1541]
17% 52% 23% 8%
[1429]
V
Схема 3.15
Различные аспекты использования фторидов
(элиминирование, присоединение) обсуждаются в других разделах.
3.2.3. Галогениды из спиртов, эфиров и диазометанов
%0Н катализатор ~* %Cl
Экстракция галогеноводородов не является характерным лро'
цессом для МФК. Например, при установлении равновесий
в системе метиленхлорид/конц. соляная кислота в органическуй
фазу переходит только небольшое количество кислоты. Однако
в присутствии тетрабутиламмонийхлорида (~0,1 М в СН2С1г)
в метиленхлориде оттитровывается почти эквимолярное количе*
«тво соляной кислоты (57]. Можно предположить, что
водородная связь между хлорид-ионом и гидрохлоридом достаточна
сильна, чтобы могла осуществиться экстракция ВМ+НСЪ*
* Существование таких комплексов, как R$N»2HC1, предполагается в
бензольных растворах [879]; соединение EtsNH+HCl2- известно как в твердом cti
стоянии, так и в растворе [880]. !
л 3.2. Получение галогенсодержащих соединений 117
\
Более лрпофильные катализаторы (например, Hep4NCl)
позволяют экстрагировать соляную кислоту даже в бензол.
Аналогично из концентрированного водного KHF2 экстрагируется
Bu4N+HF2~. По-видимому, некоторое количество хлорид-ионов
остается ассоциированным с четвертичным .катионом, однако
титрование кислоты в органическом слое постоянно дает
завышенные результаты по сравнению с теорией. В то же время
из системы HBr/Bu4N+Br~ экстрагируется меньше эквивалента
НВг [57].
До сих пор только в нескольких работах использовался
перенос галогеноводородов в неполярный органический
растворитель. Ландини и сотр. [58] превращали н-алканолы в
соответствующие хлориды нагреванием спиртов с 5 молями НС1 и
0,1 моля трибутилгексадецилфосфонийбромида при 105 °С
за 30—45 ч. В случае спиртов* С8—Ci6 выходы были
больше 90%-
Если спирты частично смешивались с водой, то и скорость
реакции, и выходы были ниже. При получении бромидов
кипячением спиртов с концентрированной бромистоводородной
кислотой присутствие катализатора очень слабо ускоряет их
образование [2].
Однако при расщеплении диалкил- и алкилариловых эфиров
кипячением с 47% -ной НВг добавление трибутилгексадецилам-
монийбромида сильно ускоряет реакцию [1032]. Кроме того,
превращение 2-ацетилбутиролактона в 5-бромпентанон-2 в
присутствии ТЭБА происходит при .комнатной температуре [1727].
Ph2CHN2 —» Ph2CHF
: Взаимодействие дифенилдиазометана в .метиленхлориде
с водным раствором дифторида калия KHF2 в присутствии пер-
клората тетрабутиламмония за 48 ч в темноте дало 50% дифе-
нилметилфторида, 10% бензгидрола и 35 % полимера. Аналогично
ведут себя и другие диарилдиазометаны, однако 9-диазо-
флуорен оказался более стабильным в этой реакции и
образовалось только 14% фторида [59]. Другим методом превращения
спиртов в «лориды в щелочной среде является реакция с ди-
хлоркарбеном, ставшая доступной благодаря МФК. Этот метод
будет обсужден в разд. 3.20.2.
Различные патентные данные, относящиеся к этому разделу,
можно найти в табл. 3.5.
Некоторые типичные методики приводятся ниже.
"* В работе Парнес н сотрудников было показано, что высшие спирты сами
способны переносить НС1 из водной фазы (Коломникова Г. Д., Калин-
кин М. И., Парнес 3. Н., Курсанов Д. Н. — Докл. АН СССР, 1982, т. 265, №2,
с. 352). — Прим. ред.
Таблица 3.5. Дополнительные литературные данные по материалам разд. 3.2, 3.3 и 3.S
Тип реакции
Условия
KF/HjO, аммониевые соли
KF/HjO, аммониевые соли
KF/HjO, аммониевые соли
NaCl/HjO, фосфониевые соли
Ониевые соли
HCl/HjO, аммониевые соли
NaCN/НгО, аммониевые соли
(K)NaCN/H2O, ониевые соли
NaNOs, ониевые соли
NaSCN/HjO, ониевые соли
металл + -OsSAr, ониевые соли
-ОАг, ониевые соли
-SR, ониевые соли
NajSOs/HzO, R4NX
NazSO»/HzO, BU4NX
NaNs/H2O, аликват 336, R4NX
Литература
130
131
132
133
134
135
136, 137, 138, 1455
139, 141, 142, 1452
141
142
143
144
1033
145
146, 147, 946
ArCOCl—»ArCOF
ArSO2CI — ArSO2F
(Me2N)2POCl — (Me2N)2POF
BrCH2—CH2Br — C1CH2—CH2Ct
ROH + HC1 -» RCI
HO(CH2)4OH — Cl—(CH2)»—Cl
C1(CH2)3—Br -» CI(CH2)3—CN
RX -* RCN
RX -* RNO2
CH2Br2 -* CH2(SCN)2
RX -* RSCN
Me3Sn(CH2)2X -* Me3Sn(CH2)2—SO2—Ar
-* Me3Sn(CH2)2—OAr
— Me3Sn(CH2)2—SR
RCI -♦ RSO3Na
Полимер— COj—CHa—НС—CHa »■
ЧО7 Полимер-С02—СН2—СН(ОН)—СИ,—SO39
R—СО—С1 — R—CO—Na — R—N=C=O
R—
о
—R
RCI + KOCN
к
СН2=СН—CH2CI + EtOH + KOCN ■
RCI + KOCN — RN=C=O
(K)NaOCN сухой, кетонный растворитель, 148, 149
ониеваи соль
■ СН2=СН—СНг—NH2—COOEt CH8CN, фосфониевые соли
KOCN, следы Н2О, аммониевые соли
150
151
3.3. Получение нитрилов 119
1-Фтороктан {52]: 0,1 моля 1-хлороктана, 0,5 моля KF-2H2O,
30 мл воды и 0,01 моля трибутилгексадецилфосфонийбромида
перемешивают в течение 7 ч в автоклаве при 160 °С. После
охлаждения отделяют органический слой, промывают его водой,
конц. H2SO4, снова водой и сушат СаС12. Перегонка дает 77%
(10,2 г) желаемого продукта, т. ,кип. 142—144 °С.
(+)-(5)-2-хлороктан [51]: 0,05 моля свежеприготовленного
оптически активного (/?)-2-октилметансульфоната, 0,05 моля
КС1, 0,0025 моля аликвата 336 и 5 мл воды перемешивают
90 мин при 100 °С. Органический слой отделяют, водный слой
экстрагируют лентаном. Органический слой и вытяжку
объединяют, промывают водой, концентрированной серной кислотой,
снова водой, а затем сушат СаС12. После отгонки растворителя
остаток перегоняют в вакууме, получают 74% (5,5 г) продукта,
т. кип. 69—70°С/20 мм рт. ст., [a]D20+33,4° (без растворителя,
/=1), оптическая чистота 89%.
3.3. Получение нитрилов
Среди наиболее тщательных работ по МФК-реакциям самые
ранние, датированные 50-ми и началом 60-х годов, посвящены
замещению R—X—*R—CN (69, 73]. Эту же реакцию
использовали для фундаментального изучения механизма МФК [4, 63] и
кинетики, «трехфазного» катализа i[64]. Было изучено множество
катализаторов самой разнообразной структуры, и общая
разработка этой темы в литературе сопоставима с обсуждением
вопросов, связанных с обменом галогенов.
Использование обычных, четвертичных аммониевых или фос-
фониевых солей {4, 38, 39], краун-эфиров {44, 77] или полипод-
ных молекул типа 7 {46] не привело к существенному
улучшению процесса*, за исключением тех случаев, когда вследствие
чувствительности субстрата к воде в реакцию вводился твердый
RCN (1035]. Например, в случае аллильных и бензильных гало-
генидов при применении водных растворов цианидов гидролиз
может подавлять реакцию замещения. Тем не менее в
литературе можно найти ^методики проведения таких реакций с
использованием водного KCN [70, 71, 82, 897]. Если желательно
избежать присутствия воды, то для получения замещенных бен-
зилцианидов можно с успехом применять твердофазную МФК-
систему с 18-краун-6 в качестве катализатора. Аналогично
получаются триметилсилилцианид [65] и цианформиат (R=фенил,
* Хотя в гомогенных условиях скорость реакции возрастала, особенно в
ГМФА [978].
120 Глава 3. Практическое использование МФК
бензил, алкил; выход 62—94%) 166]:
(HoQaSi-CI '°'*PCn > (H3C)3SiCN
О
KCN ' — C—CN
Схема 3.16
Цианид трибутилолова — новый цианирующий агент — также
■может быть синтезирован в этих условиях {1404]. Однако
реагенты, находящиеся в органической фазе, достаточно хорошо
защищены от гидролиза, например даже хлорфррмиат можно
превратить в цианформиат в системе, содержащей воду (Н2О/то-
луол, NaCN, Bu4NBr; 0—40 °С, 3—30 мин; выход 31—87%)
[1573].
'В другом синтезе триметилсилилцианида в твердофазной
МФК-системе вместо краун-эфира в качестве -катализатора
с успехом были использованы ониевые соли (растворитель N-ме-
тилпирролидон, NaCN; 36 ч, 100 °С; выход 60—70%) {1277].
Однако Симхен и Коблер [67] считают, что при синтезе
чувствительных к гидролизу соединений лучше использовать
предварительно полученный и выделенный цианид четвертичного
аммония в апротонных растворителях, таких, как ДМСО, ацетонит-
рил или метиленхлорид {67]. Описано также применение анио-
нообменных смол в GN-форме [1507]. В обычном МФК-процессе
вместо краун-эфира можно использовать более дешевый
катализатор — эфир полиэтиленгликоля 8, хотя он и несколько
менее активен [47, 61]. В более поздних работах рекомендуют
применять «трехфазный» .катализ [62, 64, 68, 775, 860]. Как уже
указывалось в разд. 3.1.4, эта техника в принципе очень
привлекательна. Так, выдан патент на получение адипонитрила из
1,4-дихлорбутана с использованием в качестве катализатора
ионообменной смолы амберлит ЩА-400 [69]. Однако недавно
было показано, что каталитическая активность «трехфазного»
катализатора на основе полистирола с поперечными связями
зависит от числа имеющихся групп R4N+. Высокая степень
замещения в кольцах, .как это характерно для продажных
ионообменных смол, снижает возможность их использования в МФК-
реакциях {64].
И наконец, в .качестве катализаторов при синтезе цианидов
можно использовать даже первичные, вторичные или третичные
амины, если только они не слишком низкомолекулярны и в силу
этого хорошо растворимы в воде (70, 72]. По-видимому, амины
алкилируются in situ, давая четвертичные аммониевые соли —
истинные катализаторы.
3.3. Получение нитрилов 121
Получение цианидов, катализируемое ониевыми солями,
проходит намного быстрее с первичными (2—8 г при 100 °С; выход
95%), чем со вторичными, субстратами. Циклогексилгалогениды
и третичные алкилгалогениды дают главным образом продукты
элиминирования. 2-Хлороктан на 85—90% превращается в
цианид и на 10—15% элиминируется. При реакции замещения
в оптически активном 2-октилметансульфонате образуется
инвертированный продукт, рацемизация достигает 30% 14].
Субстраты, содержащие бензильную группу, реагируют уже при
комнатной температуре (17 ч). Скорость реакции зависит от
величины уходящих групп, т. е. OMes>Br>Cl.
Реакция с иодидом и п-толуолсульфонатом спустя короткое
время останавливается вследствие того, что катион
катализатора образует ионную пару с этими анионами [4]. Этот недостаток
можно преодолеть двумя способами: либо используя большее
количество катализатора*, либо заменяя в ходе реакции
несколько раз водную фазу на Свежий раствор цианида. Наряду
с простыми алкил- и бензилцианидами были получены и более
сложные [74, 75, 967, 1268, 1324, 1441]. Из этих синтезов
наиболее интересно получение ароилцианидов {76]:
if
Ar-i-CN
CN
~60% ~30%
Схема 3.17
Труднодоступные винилнитрилы .могут быть получены сте-
реоспецифически с прекрасным выходом (90%) путем
обработки винилгалогенидов при 75 °С 2 экв. KCN в бензоле в
присутствии небольших количеств РсЦРЬзРК и 18-крауна-6 [893]:
R1 R3 Rl R3
С-С + KCN Pd(Ph^- '»*№«, V_/
С—С + KCN *g^j С—С
R2 X R2 CN
Схема 3.18
* В случае мезилатов или бромидов добавление слишком большого
количества катализатора приводит к тому, что реакция протекает очень бурно к
становится неконтролируемой [4].
122 Глава 3. Практическое использование МФК
а-Иминонитрил (имидоилцианид) получают, перемешивая ими-
доилхлорид в хлороформе с водным цианидом калия в
присутствии гексадецилтриметиламмонийхлорида или ТЭБА при
комнатной температуре {1036]:
Ar—C(C1)=N—CeH6 >- Ar—C(CN)=N-CeH6
Замещение на цианид в условиях МФК используют также для
модификации некоторых типов полимеров: хлорметилированно-
го лолистирола {1217, 1259], продукта присоединения алкилсуль-
фенилхлоридов к цис- 1,4-полибутадиену [1444] и поливинилхлор-
формиату {1557]. Во всех рассмотренных до сих лор реакциях
замещения ионом CN~ образуются только цианиды, но не изо-
цианаты. Однако оказалось, что в гомогенных условиях
активированные галогениды с тетраметиламмонийдицианатом серебра
в ацетонитриле образуют только изонитрилы {78]:
2RX + Me4N+Ag(CN)2- >■ 2R—NC + Me4N+AgX2-
С трифенилметил- и бензгидрилгалогенидами реакция протекает
при комнатной температуре за 2—15 ч с выходами около 90%.
4-Нитрозамещенные соединения требуют кипячения в течение
нескольких часов. Метилбромид реагирует очень медленно,
метилиодид необходимо кипятить с реагентом около 30 мин,
фенилиодид и бензоилхлорид в реакцию не вступают {78].
Получение изонитрилов МФК-реакцией с карбаминами обсуждается
в разд. 3.20.2.
Другим, менее распространенным методом получения
цианидов является реакция спиртов с трибутилфосфином в системе
тетрахлорид углерода/твердый KCN и 18-краун-6 в
ацетонитриле при комнатной или более высокой температуре (1607].
Меченый цианид калия в присутствии 18-крауна-6 обменивается
с нитрильной грулпой в ацетонитриле при кипячении за 25 .мин.
Вероятно, эта реакция не является простым замещением, в
работе {1378] предложен альтернативный механизм.
Наконец, следует упомянуть об окислительном цианировании
и фотоцианировании, хотя их только условно можно отнести
к МФК-реакциям.
В системе метиленхлорид/водный NaCN с тетрабутиламмо-
нийсульфатом или аликватом 336 в качестве электролита и
переносчика было осуществлено анодное цианирование
нафталина, стильбена, анизола и других ароматических эфиров и
аминов {79, 863, 864, 1034]. В этих условиях 9,10-диалкилантрацен
присоединяет в положения 9 и 10 как нитрильную, так и изо-
нитрильную группу {95]. Подобным же образом осуществляется
3.3. Получение нитрилов 123
и анодное ацетоксилирование {862]:
CN
оТо] * ГоТо
Схема 3.19
Фотоцианирование ароматических углеводородов было
проведено в безводном ацетонитриле или дихлорметане с
использованием систем KCN/18-краун-б [883], Bu4NCN >[916] или водного
KCN/Bu4NCN/CH2Cl2 {916].
Дополнительные ссылки на патенты, имеющие отношение
к проблемам, рассмотренным в этом разделе, приведены
в табл. 3.5.
Ниже приведены типичные экспериментальные методики.
Катализ краун-эфирами
Хриметилсилилцианид [65]: 0,1 моля сухого KCN, 0,11 моля
триметилсилилхлорида и 0,4 моля 18-крауна-6 кипятят без
доступа кислорода в течение 24 ч. Смесь перегоняют прямо из
реакционного сосуда, применяя .колонку Вигре. Выход 36%.
т. кип. 114—117 °С.
Катализ ониевыми солями
1-Цианооктан [4]: 0,67 моля 1-хлороктана, 2 моля NaCN,
25 мл воды и 0,01 моля трибутилгексадецилфосфонийбромида
кипятят 2 ч при 105 °С. Выход продукта после обработки
органической фазы 94%, чистота 97%.
124 Глава 3. Практическое использование МФК
3.4. Получение сложных эфиров
3.4.1. Эфиры карбоновых кислот
В обычных условиях замещения в протонных растворителях
карбоксилат-анион является одним из наиболее слабых нуклео-
филов. Главный фактор, снижающий нуклеофильность, —
сильная сольватация аниона. Ионные пары в неполярных апротон-
ных растворителях (ситуация, характерная для МФК) должны
Таблица 3.6. Дополнительные литературные данные о получении
сложных эфиров.
Водные растворы солей (или твердые соли), ониевые катализаторы
Тип реакции
Литература
CH2=C(R)—СО2еКв + Н2С—СН—СН2С1
о
fan г4 I* r*n Prt ?
VeO2C—R—СО2е)металлг
R—СО2еметаллф
г*+ С1СН2-СН=СН-СН2С1
) металл2*
(еО2С—R—СО2е)металлг+ + Cl-полимер -*сшитьш полимер
<еО2С—R—СО2е)металп2+ R,x
C»F2n+,—СН2СН2Х + КвеО2С—R
сн3(сн=сн)2—со2екф + а—сн2—сн2—о—сн=сн2
CH2=C(R)—СО2еметаллф + Cl—CH2—SR'
CH2=C(R)—СО2еМетоллф + Cl—CH2—COOEt
ХСН2—CH(OCOR)—СН2Х + MemQJ/PeO2CR/ -
Циануротя кислота,Ыа-сояь+ С1СН2—НС—СН2
V
БисфенолА,Ыа-соль+ С1СН2—НС—СН2
+ С12СО
+ асо—r—coci
+ C1O2S—R—SO2CI
+ a2cs
152—161
162—168,
276
169—173
174, 175
176
177—180
181
182
183
184
185
186
186
187—189,
277,917,1711
190—194,
1582, 1867
195,196,1523
1580, 1868
196
3.4.
Тип реакции
Получение
СЛОЖНЫХ
эфиров
Продолжение табл.
125-
3.6
Литература
О V-он + cico—свн4—
coci
CO2eNae
BrCHj—C6H4—CHjBr-f-KjCOs (краун-эфир) —«-поликарбонат
+ CICO2Et
RX+Na+-SCO—NR2
Эстрадиол- 17-Р+карбамоилхлорид
919
197—199'
200
839, 172В
926
1037,
1562
1362
1717
проявлять и действительно проявляют повышенную
реакционную способность.
Ранние данные об использовании ониевых солей при
получении эфиров находятся главным образом в патентах.
Действительно, имеется множество запатентованных процессов
получения полимеров из солей щелочных металлов моно- или дикар-
боновых кислот взаимодействием их с дигалогеналканами или:
с эпихлоргидрином, а также из солей щелочных металлов
фенолов с хлоридами дикарбоновых кислот, хлоридами дисульфоно-
вых кислот, фосгеном и тиофосфгеном, катализируемые в пастах
или в смесях органических растворителей с водой при
повышенных температурах. В числе возможных катализаторов,
упоминают четвертичные аммониевые, фосфониевые, арсоние-
вые и третичные сульфониевые соли. Однако наиболее часто-
в описанных примерах используют бензилтриэтил- и бензилтри-
летилам'мониевые соли. Эти данные суммированы в табл. 3.6.
Ониевые соли часто также применяют для проведения реакции:
между кислотами и эпоксидами. Однако большая часть этих:
126 Глава 3. Практическое использование МФК
запатентованных процессов не является МФК-реакциями
(см. (9651).
Истинная этерификация в условиях МФК была
использована для модификации поливинилхлорида, хлорметилированного
полистирола и других полимеров (1227, 1259, 1444, 1503, 1739J.
Обратимся теперь к соединениям низкой молекулярной
массы. Для них имеется несколько вариантов проведения реакции
получения эфиров взаимодействием солей карбоновых кислот
и алкилирующих агентов в растворителях и без них [96, 97].
Иногда используют свободные кислоты и третичные амины.
В этом случае не ясно, то ли кислая триалкиламмониевая соль
солюбилизирует карбоксилат-ион, то ли вначале алкилирующий
агент быстро образует тетраалкиламмониевую соль, которая
и действует далее как истинный катализатор {98—100].
Например, в патенте описан синтез ди-м-октил- и дибензилфталатов
из фталата натрия (оба с выходом 91%) [101]. Дри этом
использовали в качестве катализаторов триметилоктиламмонийбромид
или бензилтриметиламмонийхлорид; реакционная омесь
вода/органический слой содержала избыток алкилирующего агента.
В ходе реакции проводили азеотропную отгонку (отгонялась
смесь вода/алкилирующий агент), а в конце смесь кипятили
при 150—160 °С. Эти условия, вероятно, не являются
оптимальными, так как использованные катализаторы не очеяь липо-
фильны. Так, при применении метилтриоктиламмонийхлорида
(аликвата 336) для получения ацетата из 1-бромоктана берут
8-кратный избыток водного ацетата натрия при нагревании
смеси до 105 °С. Реакция проходит практически полностью за
1 ч (38, 39].
h-CioH2i—Вг:—»- н-СюНц—ОАс
Примеры образования эфиров по такому типу, приведенные
в литературе, включают многочисленные случаи взаимодействия
реада-антов, имеющих несколько функциональных групп {1238,
1435, 1468, 1567]. Менее интересны, по-видимому, методики,
в которых используются анионообменные смолы (в' карбокси-
латной форме), которые нагревают с RX {1321]. Данные о
трехфазном катализе при .получении сложных эфиров рассмотрены
в разд. 3.1.4. ч«с-3,5-Дибромциклопентен-1 (А) может быть
лревращен в ^ыс-диацетоксипродукт В также при
использовании аликвата 336 в системе четыреххлористый углерод/водный
ацетат .калия {102] (ср. [427]). За 5 ч при 60 °С .конверсия
достигает 68%, а через 9 ч при 42 °С она бывает и 75%. В
продукте реакции содержится только 3—5% продукта аллильной
перегруппировки — 3,4-диацетоксициклопентеяа-1. Тем же
способом циклогексенон превращается в 4-ацетоксициклогек-
3.4. Получение сложных афиров 127
сен-2-он-1 лри бромировании N-бромсукцинимидом и
следующим за этим в условиях МФК замещением (7 ч при 33 °С)_
ОАс
Ионные пары, переходящие в органическую фазу из водной,,
всегда несут с собой некоторое количество молекул воды, и
поэтому их реакционная способность меньше, чем в полностью"
безводных условиях. Поэтому четвертичную аммониевую соль,
иногда лучше сначала выделить и высушить, а потом уже
вводить в реакцию алкилирования {104]. Последняя стадия часто-
хорошо идет в метиленхлориде. В этом растворителе, за
исключением некоторых медленно реагирующих алкилхлоридов,
можно применять любые алкилирующие агенты, так как сам по себе
растворитель является очень слабым реагентом. При кипячении
сухого бензоата в дихлорметане в течение 24 ч в присутствии
ГЭБА образовалось только 15% дибензоилоксиметана [103]-
Различные (в том числе и стерически затрудненные) диэфиры
метилена были получены при кипячении выделенных тетрабу-
тиламмонийкарбоксилатов в метиленхлориде в течение 2—3 суг
[104].
2RCOO-—+RCOO—CH2—OOCR
Стандартная методика Брендстрёма [105] для получения эфи-
ров состоит в нейтрализации эквивалентных количеств кислоты
и бисульфата тетрабутиламмония 2 н. раствором гидроксида
натрия, прибавлении избытка алкилирующего агента в
дихлорметане и кипячении около 30 мин. Выделение аммониевой соли:
в случае иодидов достигается выпариванием метиленхлорида
и экстрагированием находящегося в остатке продукта этерифи-
кации эфиром. Если при реакции образуется тетрабутиламмо-
нийбромид, то его можно удалить промывкой эфирного
раствора водой. В тех случаях, когда в качестве алкилирующего
агента используется диметилсульфат, следует брать только катали-
128 Глава 3. Практическое использование МФК
тгическое количество бисульфата тетрабутиламмония, поскольку
образующийся метилсульфатный анион переходит главным об«
разом в водную фазу и в таком количестве, которое не
препятствует экстракции карбоксилатного иона.
Используя этот метод Брендстрём смог получить (с выходом
до 90%) зфиры даже таких стерически затрудненных кислот,
как о,о'-диметил- или диметоксизамещенных бензойных кислот.
Получение эфиров дикарбоновых кислот в большинстве случаев
.проходит без каких-либо затруднений. Только очень лштофиль-
ные кислоты дают низкий выход (например, выход эфиров
винной кислоты 40%). Однако аминокислоты этим методом этери-
фицировать нельзя. В то же время N-замещенные аминокислоты
легко дают разнообразные эфиры: растворяют N-производное
-аминокислоты в насыщенном водном растворе бикарбоната
натрия и добавляют смесь молярного количества адогена 464 и
небольшого избытка алкилгалогенида. Смесь выдерживают при
комнатной температуре в течение 3—24 ч {1225].
Гидроксибензойная кислота может быть проалкилирована
•селективно по кислотной группе, если использовать только
1 моль тетрабутиламмониевой соли {106]. Аналогично при ком-
латной температуре этерифицируется р-гидроксидитиокоричная
кислота С [107]. МФК-реакция имидоилхлорида с фенолами
(тиофенолами) приводит к арилимидатам (арилтиоимидатам),
.в то время как с карбоксилатами образуются диароиламиды
J1332].
- о с, (S)
\S Pfr-C-NR . Ph-C=NR . Ph-C=N-R
ЮН''' д I
С Ar—C=O
Схема 3.21
Эрссон [108] использовал этот *метод для газохроматографи-
■ческого определения карбоновых кислот и фенолов. Метод
включает экстракцию кислоты в форме ионной пары в метилен-
хлорид и получение производного с пентафторбензилбромидом.
Скорость реакции увеличивается в зависимости от структуры
противоиона и при увеличении его концентрации. Для
повышения скорости реакции гораздо лучше использовать вместо тет-
3>абутиламмониевых солей более липофильные соли тетра-м-пен-
тиламмония. Имеется обзор, посвященный применению
экстрактивного алкилирования для анализа фармацевтических
препаратов [1052], а недавно описана микромодификация этого
метода с твердофазной системой МФК и использованием в
качестве щелочи карбоната натрия [1053].
ЗА. Получение сложных эфиров 129
Подробно были изучены солюбилизация, экстракция и
«активация анионов» карбоксилатов щелочных металлов различными
комплексантами. Кнёхель сравнил активность в качестве МФ-
катализаторов для реакции между твердым ацетатом калия
и бензилхлоридом в ацетонитриле многих лигандов (краун-эфи-
ров, аминополиэфиров, нонактина, полиподов и полиэфиров
с открытой цепью) {109]. Результаты исследования показали,
что между степенью солюбилизации и скоростью реакции нет
простой корреляции.
Наибольшее значение имеют следующие факторы:
1) стабильность образующегося комплекса;
2) липофильность лиганда;
3) жесткость молекулы лиганда.
Наиболее активными катализаторами оказались дициклогек-
сано-18-краун-6 и криптофикс 221 (11); из других работ
следует, что в твердофазной МФК системе аликват 336 был таким же
хорошим катализатором, как и краун-эфиры [1041]:
11
Схема 3.22
Лиотта и сотр. (110] показали, что можно достигнуть высокой
концентрации комплекса 18-крауна-6 с ацетатом калия как
в бензоле (до 0,8 моль/л), так и в ацетонитриле. При 'этом
по крайней мере 80% краун-эфира находятся в форме
комплекса. Однако 'более экономично использовать только 3—10 мол.%
катализатора вместе с избытком твердого ацетата калия в
бензоле или в ацетонитриле, где реакция идет быстрее. Такой
ацетатный реагент Лиотта назвал «голым» ацетатом, однако,
как уже было показано в разд. 1.4, этот термин может вводить
в заблуждение. В этих условиях ацетат действительно является
мощным нуклеофилом, но очень слабым основанием. Для
окончания реакции с первичными алкилгалогенидами необходимо
при комнатной температуре несколько суток, а при кипении
смеси — несколько часов. Активированные бромиды (например,
бензилбромид) требуют только нескольких часов при комнатной
температуре. Первичные алкилгалогениды совершенно не
образуют продуктов элиминирования, что еще раз свидетельствует
130 Глава 3. Практическое использование МФК
о низкой основности «голого» ацетата. Соотношение скоростей
элиминирования для различных уходящих групп в реакции
с 1-замещенными гексанами составляло приблизительно Вг:
: OTs : С1 = 4 : 2 : 1. Два изученных вторичных алкилгалогенида
дали только по 10% олефина — продукта элиминирования.
о
II
СОСН2Вг ^\ /СО—СН2—О—С—R
Аналогично были получены я-бромфенациловые эфиры при
кипячении калиевых солей карбоновых .кислот с я-бромфенацил-
бромидом в ацетонитриле или в бензоле (где реакция идет
медленнее) в течение 15 мин (иногда до 30 мин) в присутствии
5 мол. % дициклогексано-18-крауна-6 или 18-крауна-6 [111].
Выходы продуктов после выделения составили 90—100% даже
для таких стерически затрудненных карбоксилатов, ,как 2-иод-
бензойная кислота или мезитойная кислота. Последнюю
реакцию можно проводить и без кипячения, перемешивая смесь
в течение нескольких часов при комнатной температуре.
Утверждают [111], что этот метод по выходам и легкости проведения
превосходит все до сих пор известные синтезы фенациловых
эфиров.
Столь же хорошие выходы (85—99%) можно получить
при кипячении в бензоле в течение 3 ч калиевых солей
карбоновых кислот, алкилбромидов и небольшого количества крипто-
фикса {222] (5) (112]. Получение метилтиометиловых эфиров
было предложено в качестве защиты карбоксильной группы.
Эфиры образуются яри кипячении карбоксилата калия с хлор-
метилметилсульфидом и каталитическим количеством иодида
натрия и 18-крауиа-6 в бензоле. Удаление защитной группы
происходит лри последовательной обработке HgCb в кипящей
смеси ацетонитрил/вода и сероводородом [1042]:
R—COOH + C1CH2SCH3
Этерификация, катализируемая краун-зфирами,
используется также в аналитических целях 1113, 114, 1038]. Предельно
малое количество фенациловых эфиров, которое можно было
определить, используя УФ-детектор в жидкостной хроматографии,
1 и 60 нг соответственно для производных С2- и С2о-кислот [114].
Ниже приведен 'более сложный случай использования метода
МФК [115]:
3.4. Получение сложных эфиров 131
сно
CH5-C-CeH5
.6>
(тверйып) ^г
гн сно сн2—свн5
СвН, | |
с=о
Схема 3.24
Первой стадией пептидного синтеза Меррифилда является
сшивание аминокислот (с защищенной азотной функцией) с хлор-
метилированным полистиролом путем образования сложноэфир-
ной группы. Эту стадию можно ускорить, используя калиевую
соль Вое-аминокислоты и молярное количество 18-крауна-6
в ДМФА {972].
Кроме краун-эфиров были рекомендованы и другие комплек-
сообразующие лиганды: технические эфиры полиэтиленгликоля
(8, и«8) [61, 1004], различные замещенные триамиды
фосфорной кислоты [117] и диамины [117], среди которых самым
простым является Ы,Ы'-тетраметилэтилендиамин (12) {117, 118].
Однако позднее было показано (1013], что в ходе реакции с мо-
нобензилхлорвдом 12 образует соль, превращаясь в настоящий
катализатор [1013]:
Н3СО-(СН2-СН2—O)n-CHS
8
Me2N—CH2—CH2—NMe2
12
В тех случаях, когда реакцию проводят с твердой солью-,
в .качестве алкилирующих агентов применяют бензилбромид
или бензилхлорид, а катализатор растворяют в бензоле (для 8)
или в ацетонитриле (для 12); выходы конечных продуктов, как
правило, достигают почти количественных даже при
использовании вторичных алкилирующих агентов и стерически
затрудненных .кислот [118]. В присутствии 12, триэтилендиамина или
18-крауна-б в бензоле или ацетонитриле (80 °С, от 30 .мин до
24 ч) бромгидрин вступает в реакцию с твердым ацетатом
[1534]. Реакция между триметилсилилхлоридом и твердым
ацетатом натрия в тетрахлорметане проходит при кипячении с
катализаторами Bu4'NCl или ВщМ в течение 4 или 6 сут
соответственно (1210].
В качестве основания при твердофазном алкилировании
кислот в условиях МФК, в первую очередь для определения их
132 Глава 3. Практическое использование МФК
методом газовой хроматографии, можно использовать
бикарбонат натрия. Этот метод особенно пригоден для соединений,
чувствительных к гидролизу. Bu4N+HCO3~ (образующийся из
Bu4NHSO4 и ЫаНСОз) экстрагируется в органическую фазу, где
и происходит депротонирование кислоты и соответственно ее
алкилирование. В этом случае полезно в качестве
растворителей использовать кетоны, в которых BuN+HCO3~ устойчив даже
при повышенных температурах. Наличие следовых количеств
воды и этанола чрезвычайно сильно сказывается на скорости
алкилирования {1212, 1475, 1476].
Получение карбонатов методом МФК представляет собой
особую проблему, поскольку карбонатный ион очень трудно
экстрагировать в органический растворитель из водной фазы
или из твердых солей. Лиссель и Демлов [1560] показали, что
эфиры угольной кислоты не образуются со сколько-нибудь
заметным выходом при нагревании твердого, сухого карбоната
щелочного металла с алкилгалогенидом и небольшим
количеством аликвата 336. Однако если использовать твердый, сухой
бикарбонат, то образуются симметричные карбонаты (толуол,
100°С, 8—15 ч, выход 70—90%): экстрагирующийся вначале
R4N+HGO3- превращается в полуэфир RO—СО—ОН, который
депротонируется в таких условиях быстрее, чем декарбоксили-
руется. При этом образуется 'ROCO2~R4N+, который затем тоже
алкилируется [1560]. По мнению авторов, запатентованный
процесс получения поликарбонатов методом МФК (табл. 3.6)
работает только потому, что используемый карбонат содержит
некоторое количество бикарбоната.
Недавно был предложен не совсем обычный способ алкили-
рования с использованием легкодоступных алкилдифенилсульфо-
ниевых солей; при этом этерифицируемую кислоту, сульфоние-
вую соль и твердый сухой карбонат калия перемешивают
в метиленхлориде {1594]:
ЯХХ + Ph2S v I^SPVX-
R*SPh2+X- + R2CO2-K+ v R2CO—OR1 + KX + Ph-jS
Карбонаты, тиокарбонаты и карбаматы енолов можно
синтезировать соответственно из енольных триметилсилильных эфи-
ров и фторформиатов, фтортиоформиатов или карбомоилфтори-
дов, нагревая их с каталитическим количеством системы
KF/18-краун-б или с раствором R4NF в ТГФ {1300, 1511]:
R2C=CR— OSiMe3 >- R2C=CR—СГ >- R2C=CR—О—СО—X
(X = OR, NR2, SR)
Ацетаты коричных спиртов (Е- или Z-изомеры) были
получены при стереоспецифическом раскрытии кольца в 2-арилцик-
3.4. Получение сложных зфирое 133
лопропилбромидах в гетерогенной системе ацетат калия/18-кра-
ун-6, содержащей ДМСО в .качестве растворителя [1706].
Рассмотрим теперь получение эфиров из хлор ангидридов
кислот и спиртов. В ранних экспериментах были сделаны попытки
использовать в реакции Шоттена — Баумана бензоилхлорид и
различные спирты в присутствии МФ-катализаторов и водного
раствора гидроксида натрия. Однако стандартных условий для
простых субстратов подобрать не удалось. Хотя реакция и шла
быстрее по сравнению с некаталитическим процессом, но часть
хлорида гидролизовалась, а скорость зависела от липофильно-
сти спирта {882]. Впоследствии этот тип реакции был изучен
более детально. При прибавлении ангидридов или хлорангидридов
кислот к смеси 30%-ного водного раствора гидроксида натрия
или калия и спирта с 10 мол.% Bu4NCl в инертном
растворителе активные первичные и некоторые вторичные спирты уже
за 10 мин при 0°С давали соответствующие эфиры с выходом
65—91 % {1355]. Применение менее активных третичных спиртов
в этих условиях приводило к образованию ангидридов, поэтому
в случае третичных спиртов для реакции используют
твердофазный МФК-процесс с твердыми соединениями щелочных
металлов в качестве оснований. Подобным способом в присутствии
40%-ного водного гидроксида натрия и Bu4NCl при комнатной
температуре в бензоле или метиленхлориде были ацетилирова-
ны, бензоилированы и тозилированы частично защищенные
углеводы. Показано [1357, 1586, 1587], что в этой реакции более
липофильный B114NI не проявляет каталитических свойств. Эте-
рификация может проходить селективно: гидроксильные группы,
склонные 'К образованию внутримолекулярной водородной связи,
при этерификации в условиях МФК проявляют меньшую
реакционную способность, что нельзя объяснить только стерически-
ми препятствиями. Так, иногда вторичные ОН-группы в сахарах
ацетилируются легче, чем первичные [1696].
Для быстрого определения меченного 14С канцерогена —
Ы-гидрокси-]М-ацетил-2-аминофлуорена — его ацилированный
гидроксиламин был О-ацилирован уксусным ангидридом
в присутствии ТЭБА в системе СНгСЬ/разбавленный раствор
гидроксида натрия [1445].
Твердофазный МФК-процесс позволяет провести ацилиро-
вание даже сильно затрудненных стерически фенолов; для этого
к хорошо перемешиваемой смеси фенола и порошкообразного
твердого КОН или NaOH и Bu4NHSO4 в бензоле, дихлорметане
или диоксане при комнатной температуре прибавляют по
каплям ацилхлорид [1278, 1450].
Синтез Ы,Ы-диалкиламиносульфенилкарбаматных
инсектицидов был проведен с выходом 38—75% из карбомоилфторидов
и спиртов в системе водный NaOH/толуол (катализатор
134 Глава 3. Практическое использование МФК
Bu4NHSO4) или NaOH/дихлорметан (катализатор ТЭБА)
[1040]:
R^N—S—N(CH3)—COF -» R'R2N—S—N(CH3)—COOR3
R2N^COC1 +
Схема 3.25
Сходным образом, N-замещенный карбомоилхлорид реагиру*
ет со стероидным фенольным гидроксилом в присутствии
различных ониевых солей и краун-эфиров в системе хлорированный
углеводород/водный раствор гидроксида натрия {123].
3.4.2. Эфиры других кислот
При .комнатной температуре в системе 30%-ный водный
NaOH/ТЭБА/бензол из алканолов и сульфохлоридов образуют^
ся эфиры сульфокислот [1394]. Описано монотозилирование дио*
лов в частично защищенных сахарах тозилхлоридом с
использованием системы дихлорметан/водный раствор гидроксида
натрия и избытка тетрабутиламмонийбисульфата (30 мин,
комнатная температура) (119]. В зависимости от стереохимии исходных
Сахаров образуются различные количества производных; так,
например, .метил-4,6-0-бензилиден^-0-глюшпиранозид дает
55% 2-тозилата, 31% 3-тозилата и 7% 2,3-дитозилата, а .метил?
4,6-О-бензилиден-та-Е)-маннопиранозид образует 95% 2-тозилата.
Другие примеры ом. в работе {1383].
В системе бензол/водный гидроксид натрия с 10 мол.%
ТЭБА в качестве катализатора (1,5 ч при 60 °С) были получены
диалкилсульфаминаты стероидных фенолов (D). Вторая гид-
роксильиая группа при этом не реагирует [ПО, 121; ср. 976]:
,OSO2Ph
NH2
CH—SO,—OR'
R
Схема 3.26
3.4. Получение сложных эфиров 135
Подобным же способом можно приготовить алкансудьфона-
ты (F) {121, 122]. Изучение механизма реакции этерификации
показало, что основной маршрут включает образование в
качестве промежуточных продуктов фенилоульфенов [122]. Без
МФ-катализаторов аминофенолы дают смесь сульфоновых
эфиров и амидов. Очень хорошую селективность при получении Е
можно достигнуть при проведении реакции в смеси
вода/хлорбензол с R4iNX в качестве катализатора и поддержании
рассчитанной величины рН. Расчет проводят по формуле рКа—
_рН = +0,5-=—1,5 {1405] (см. схему 3.26).
Сообщают [1547], что синтез алкилдифенилдитиофосфинатов
в МФК-системе можно осуществить с выходом 47—95%.
Первой стадией реакции, приведенной на схеме 3.27, является
нейтрализация моноалкилфосфата раствором гидроксида тетраме-
тиламмония в метаноле. Растворитель затем удаляют, а остаток
кипятят с 1 молем алкилгалогенида [124]. Очевидно, что
превращение может быть проведено в двухфазной системе с
использованием достаточно липофильного катализатора.
о
RiO—РО3ае ► R'O—P—ORa R'O—POaHe
ОН
Схема 3.27
Аналогично, экстрагируя тетрабутиламмониевую соль моно-
алкилфоофита в CH2CI2 и алкилируя ее затем в ацетонитриле
при 50 °С, получают смешанные диалкилфосфиты {957] (схема
3.27). Последние были использованы для получения смешанных
триалкилфосфатов {1044] и алкилфосфинатов [1448].
Ароматические тюлифосфонаты синтезировали, исходя из PhPOCfe, бис-
фенола А и KOH/R4NX [1592].
Из алкилгалогенидов или алкилмезилатов и О-алкилдитиокар-
бонатов .калия или непосредственно из спиртов, CS2 и NaOH
с различными катализаторами в некоторых растворителях или
без них были получены разнообразные О,8-диалкилдитиокарбо-
наты [925, 1009, 1043, 1245, 1474, 1538, 1662]. Простой синтез
несимметричных сульфидов можно провести без выделения
промежуточных продуктов («в одном горшке» — one-pot synthesis),
нагревая необработанную реакционную смесь с гидроксидом
калия
RxX-f K+-S2C—OR2 ► RXS—CS—OR* »- R*SR*
RS—CS—OC(CHS)S ► RSH
при 80 °C (выход 25—85% в зависимости от структуры) [1043].
В том случае, когда заместителем в дитиокарбонате является
136 Глава 3. Практическое использование МФК
трет-бутил, нагревание реакционной смеси со слабыми
щелочами приводит тс алкантиолам с выходами 60—91% {915].
При нагревании до 100 °С в присутствии аликвата 336 O.S-
диалкилдитиокарбонаты перегруппировываются в S.S-диалкил-
производные [1245, 1662].
Интересный пример сокатализа был недавно изучен
Фридманом и сотр. {125]. При проведении реакции между фенолятом
в воде и диалкилтиофосфорилхлоридом, растворенном в мети-
ленхлориде, в отсутствие катализатора образуются главным
образом продукты гидролиза:
ArO-Na++(MeO)2P(S)Cl »- АгО—PS(OMe)a
Активирование фенолята с помощью МФ-катализатора,
например бисульфата тетрабутиламмония, сильно ускоряет
реакцию, и выход желаемого продукта возрастает; в то же
время при катализе N-метилимидазолом, который является нуклео-
фильным катализатором, активирующим ацилирующий агент,
скорость реакции возрастает еще больше, но одновременно
увеличивается и гидролиз. Использование этих двух
катализаторов вместе приводит ,к удивительному эффекту: скорость
реакции превосходит суммарное увеличение скорости реакции от
отдельных катализаторов, а гидролиз фосфорилхлорида идет
только на 60%.
Цвиржак и сотр. [126—129] предложили метод фосфорили-
рования в условиях МФК аминов, гидроксиламинов, гидразина
и спиртов. Используя методику Атертона и Тодда,
промежуточные галогенангидриды диалкилфосфорной кислоты (Н) можно
получить in situ из диалкилфосфитов (G) и тетрагалометанов:
R1O
о
H
"R'O
VI
NHRaR"
.R'O
I
20%-КЫп NaOH, ТЭБА'
C1J
50%-нып КОН
H2NOH, CCU
ТЭБА
R'V°
R'c/ NNHOH
50%-нып NaOH
ССЦ, ТЭБА
R2OH
К
R'O/ NH—NH2
L
Схема 3.28
M
3.5. Различные реакции замещения 137
При проведении реакций .«в одном горшке» с достаточно
хорошими выходами можно толучить соединения I {126], К (127],
L .[128] или М {129].
Ниже приводятся некоторые типичные .методики.
Катализ краун-эфирами
л-Бромфенацилацетат [111]: 1 ммоль КОАс, 1 ммоль ю,л-ди-
бромацетофенона и 15 мг дициклогексано-18-крауна-6
суспендируют в 10 ,мл ащетонитрила. После 15-мин кипячения
растворитель удаляют под вакуумом, а остаток растворяют в бензоле и
пропускают этот раствор через короткую колонку с сухим сили-
кагелем для того, чтобы удалить краун-эфир. После уларивания
получают 252 мг (98%) желаемого продукта, т. пл. 85—86 СС.
Катализ малыми количествами четвертичных аммониевых солей
1-Децилацетат (38]: 0,25 моля 1-бромдекана, 2 моля трйгид-
рата ацетата натрия и 10 г аликвата 336 перемешивают 2 ч при
105 СС. Добавляют воду и экстрагируют смесь органическим
растворителем. После обычной обработки и перегонки получают
продукт реакции. Выход почти количественный.
Катализ с использованием молярных количеств
тетрабутиламмониевого иона
Метиловый эфир .карбоновой кислоты (общая методика)
[105]: 0,1 моля карбоновой кислоты и 0,1 моля B114NHSO4
растворяют в 100 мл 2 М NaOH, к смеси прибавляют 0,13 моля
СН31 в 100 мл дихлорметана и перемешивают при температуре
кипения в течение 15 мин. После разделения фаз отделяют
органический слой и удаляют растворитель.' Остаток
обрабатывают 100 мл эфира, при этом ВщШ остается в осадке.- Его
отфильтровывают (выход 100%), а эфирную вытяжку
упаривают. Остаток перекристаллизовывают или перегоняют. Выходы
обычно не ниже 90%; так, метиловый эфир 2,4,6-триметилбен-
зойной кислоты образуется с выходом 94%.
3.5. Различные реакции замещения
Наряду с реакциями замещения, приводящими к получению
галогенидов, нитрилов и сложных эфиров, известно и много
других реакций замещения, для проведения которых используют
МФК. Условия их проведения и применяемые .катализаторы не
отличаются от тех, что уже описаны в предыдущих разделах.
138 Глава 3. Практическое использование МФК
Следует помнить, что в относительно неполярных, апротонных
растворителях ионные пары имеют другой порядок изменения
реакционной способности, чем в обычных условиях проведения
реакций в водно-этанольных или подобных средах. Так,
некоторые «слабые» нуклеофилы оказываются очень сильными
в МФК-реакциях.
R—X водн- SCN~ -^ R—SCN
Эта реакция с первичными и вторичными субстратами
проходит за 1—24 ч при кипячении, выходы до 90% [4, 38, 39, 73,
82, 1045]. В качестве катализаторов были испытаны как
четвертичные аммониевые соли, так и краун-эфиры, а также многие
первичные, вторичные и третичные амины. Последние в реакци:
онной смеси превращаются в четвертичную соль (кватернизуют-
ся), что иногда 'Приводит к увеличению времени реакции [82];
Комплекс тиоцианата калия с 18->краунам-6 является сравни-;
тельно слабым нуклеофилом в гомогенном ацетонитрильно*|!
растворе; так, он реагирует с бензилтозилатом в 32 раза мед*
леннее, чем ацетат калия (83]. В гексахлорциклотрифосфазенё
можно заменить все шесть атомов хлора на группы SCN {9841,
Растворимый, частично хлорметилированный полистирол был
модифицирован путем обработки тиоцианатом натрия в
присутствии криптанда{2.2.2] [1217]. Другие реакции замещения на
тиоцианатную группу проводят с твердыми солями щелочных
металлов в присутствии 18-крауна-6 [1534] и под действием
анионообменной смолы амберлит А26 (в тиоцианатной форме)
в кипящем толуоле [1507].
R—X ,—* R—
В работе [4] сообщалось, что в водно-органических смесях
в присутствии МФ-катализаторов может проходить замещение
на нитритную группу, однако детали условий реакции не
приводятся. Этот гароцесс, по-видимому, не очень эффективен (65].
Твердый нитрит в ацетонитриле в присутствии 18-крауна-6 при
25—40 °С дает главным образом нитропроизводные; нитриты
при этом являются побочными продуктами. Выходы продуктов
в этой реакции несколько меньше, но близки, как и соотношение
продуктов к результатам, полученным лри проведении реакции
в ДМСО {65]: 50—70% нитропроизводных при реакции с
первичными галогенидами (см., однако, [1703]). Циклогексилбро-
мид дает только продукты элиминирования. В гомогенных
условиях можно использовать предварительно полученную соль
R4NNO2; при этом нитропроизводные образуются с выходом
3.5. Различные реакции замещения 139
40—77%. а отношение групп нитро/нитрит в продуктах
приблизительно равно 7 : 3 '[1323].
Для полноты картины рассмотрим еще одну методику, хотя,
строго говоря, она и не относится .к истинным МФК-процессам.
Сначала продажную сильноосновную анионообменную смолу
(например, амберлит IRA 900) превращают в нитритную форму,
промывают водой, этанолом и 'бензолом и сушат в вакууме.
Затем для получения нитросоединений двойное количество
смолы перемешивают и нагревают с бромэфирами карбоновых
кислот или с алкилбромидами. Таким способом, например, были
получены 1-нитропропан (25 °С, 15 ч, 47%), фенилнитрометан
(25 °С, 4 ч, 87%), этил-2-метил-2-нитропропионат (50 °С, 24 ч,
60%) [116]. Известны также и другие реакции с анионообмен-
ными смолами, включающими специфические, необходимые для
реакции ионы (см. {116] и другие работы, рассмотренные выше).
Я—Л г-*гЯ—ONO2
Реакцию замещения на нитратную группу в условиях МФК
еще не проводили. Однако было показано, что в гомогенных
условиях в присутствии агентов, дающих комплекс с катионом
соли, нитрат-ион является очень сильным нуклеофилом. В
зависимости от типа растворителя ацетобромглкжоза и система
нитрат серебра/криптофикс {222] дают смесь продуктов сольволи-
за А и нитратных эфиров В. Соотношение этих (продуктов
изменяется от А : В=98 : 1 в метаноле до 0 : 100 в диглиме {84].
мо2
сн2
,—ОАс
ОАс
ОАс
AgNO3, ROH
В.2.2]
СН2—ОАс
* |^ ОАс
Is*—г
4OR
ОАс ОАс
А
Схема 3.29
СН2—ОАс
ОАс
ОАс
В
При реакции н-ал кил бромидов с 25%-ным водным раствором
NaN3 в присутствии аликвата 336 при 100 °С уже за 6 ч
образуются азиды с почти количественным выходом [85, 1635].
Хлориды реагируют несколько медленнее, а иодиды — быстрее. Цикло-
гексилгалогениды дают 25% циклогексена.
140 Глава 3. Практическое использование МФК
По приведенной ниже схеме можно получить с
удовлетворительными выходами аминокислоты, исходя из ct-бромэфиров, при
этом в качестве катализаторов используют Bu4NBr (в воде) или
18-краун-6 (в .бензоле). При реакции с «-бром-р-гидроксипро-
пионатом (R=HOCH2—) глицидат не образуется в качестве
промежуточного продукта, так как в противном случае
раскрытие оксидного кольца приводило бы к продуктам с другим
расположением групп [865]:
А,
^ ^ R-CH-COOH
катализатор •
NH
R = Н, Me, Et, HOCH2, MeOCH2
. N3—СН2—СН—COOR
нштишатор |
ОН
Схема ЗЖ>
В недавних работах было показано, что при реакциях эритро-
и грео-метил-р-гидрокси(и {}-метокси-)-р-фенил-«-бромпропиона~
тов на регио- и стереохимию влияют как тип использованного
катализатора, так и относительная конфигурация реактанта
[1046]. Исследование этой же реакции в системе твердая
фаза/жидкость в 'присутствии краун-эфиров проведено в работе
[1534].
При использовании в качестве катализатора технического
диметилового эфира полиэтиленгликоля (8) реакция между
твердым азидом калия и бензилбромидом при проведении ее
в бензоле или в ацетонитриле заканчивалась за 8 или 2 ч
соответственно [61].
Исследовано {88, 89] замещение хлора на азидную группу
в поливинилхлориде (ПВХ). Реакцию проводили в гетерогенной
смеси порошкообразного ПВХ и водного раствора азида натрия
в присутствии четвертичных аммониевых солей. Тетрабутилам-
монийхлорид и тетрабутиламмонийбромид как катализаторы
были лучше, чем более обычные поверхностноактивные вещества
[88]. Тетрабутиламмонийхлорид был также наилучшим
катализатором и при проведении реакции в растворе ТГФ, в .котором
ПВХ растворим, а натриевая соль нерастворима [88].
Авторы обсуждают наблюдаемые в этих реакциях
кинетические особенности при использовании длинноцепочечных поверх^
ностноактивных веществ, считая, что система аммонийная
соль/катализатор адсорбируется на молекулах ПВХ как на
твердых поверхностях, так и в растворе ПВХ {88, 89].
3.5. Различные реакции замещения 141
RCO—X r-^RCO—N3 —+ R—N=C=O
Азид тетрабутиламмония можно приготовить в виде чистых
кристаллов путем прямой экстракции из водной смеси
бисульфата тетрабутиламмония, азида натрия и избытка гидроксида
натрия (86]. Его, в свою очередь, можно превратить в ацилазид
при взаимодействии с ацилхлоридом в толуоле при 25 °С. При
более высоких температурах (50—90 °С) происходит
перегруппировка Курциуса и образуются с выходом 52—89% различные
изоцианаты {86]. Используя метод МФК, можно провести эту
реакцию путем прямого взаимодействия водного раствора азида
натрия с ацилхлоридом в присутствии четвертичного аммоний-
хлорида и последующим 'пиролизом. Эта методика была
запатентована, так как она эффективна даже при реакции с
малорастворимыми хлорангидридами кислот [87].
R—X+OCN- —* R—N= C= О
Возможно также провести прямое замещение, дающее изо-
цианат. В 1958 г. была запатентована реакция бензилхлорида
с цианатом натрия в присутствии различных растворителей
в ДМФА или в отсутствие растворителей, катализируемая тетра-
этиламмонийиодидом. Она протекает очень быстро при 170 °G
[90]. Этот же катализатор использовали для получения силиль-
ных соединений в ДМФА (145°С, 30 мин, выход 89%) {91]. Этот
факт указывает на возможность замещения хлорид-иона
в исходном соединении в качестве первой стадии реакции под
действием аммонийиодида. Такое предположение
подтверждается тем, что KI также катализирует эту реакцию, хотя и менее
эффективно {91].
RsSi(CH2)nCl >■ RsSi(CH2)n— N=C=O
(С1(СН2)„—SiRR')2O >■ (O=C=N—(СН2)„—SiRR')2O [91]
RBr+KOCN(H2O) »■ R—NH—CO—NH—R [38,39]
' \ i г аликват 336 l
Если замещение проводить в водной среде, то, естественно,
образуются производные мочевины [38, 39]. В присутствии краун-
эфиров в N-метилпирролидоне или в о-хлорбензоле твердый
цианат калия реагирует с аллилхлоридом и алкилбромидом [60].
Как и в рассмотренном выше случае, на ход реакции может
влиять димеризация и тримеризация изоцианатов. В разд. 3.18
приведены случаи образования изоцианатов в результате гоф-
мановского расщепления в условиях МФК.
Тетрабутиламмоний-я-толуолсульфинат был выделен
методом экстракции ионных пар в СНгСЬ из смеси и-толуолсульфи-
ната натрия и Bu4N+iCb. Этот реагент гладко взаимодействует
142 Глава 3. Практическое использование МФК
в ТГФ с первичными алкил-, бензил- и аллилгалогенидами яри
температурах 10—40 °С, давая соответствующие сульфоны с
довольно высокими выходами [92]. Среди других соединений,
которые могут быть превращены в арилсульфоны, отмечены
изшропилбромид, хлорацетонитрил, этилбромацетат, фенацил-
хлорид, 1,3-дихлорацетон (продукты моно- или бисзамещения
образуются в зависимости от условий реакции), метиленбромид
(монопродукт). С 1,2-дибромэтаном помимо замещения
проходит частичное дегидробромирование [92].
RX + NBu4eOaes—( { ) У-СНЭ > R—SOa
Схема 3.31
В других случаях используют твердофазный вариант МФК,
арилсульфонат натрия суспендируют в ДМЭ и нагревают с RX
и небольшим количеством Bu^NBr. Выходы превосходные [1368].
В табл. 3.5 лриведена сводка литературных данных,
относящихся к реакциям замещения.
3.6. Тиолы и сульфиды
Как известно, сульфидные и тиолатные анионы являются
сильными нуклеофилами. Кроме того, они легко переходят с
четвертичным ониевым противоионом из водной фазы в
органическую. Таким образом, они должны быть идеальными субстратами
в МФК-реэкциях. Действительно, в фундаментальных
работах Херриота и Пиккера [28, 201], посвященных изучению
механизма МФК и влиянию структуры катализаторов, была
использована система тиофенол/алкилирующий агент.
При комнатной температуре из системы KHS/бензилбромид
в бензоле за 30 мин был получен бензилтиол; в качестве комп-
лексанта-катализатора использовали диметшювый эфир поли-
этиленгликоля (8) {61]:
PhCH2Br + KHS 1- PhCH2SH
При нагревании первичных алкилбромидов или хлоридов
с водным раствором сульфида натрия в присутствии трибутил-
гексадецилфосфонийбромида или тетрабутиламмонийхлорида
при сильном перемешивании в течение 40 мин при 70 СС были
получены симметричные сульфиды с почти количественным
выходом [202, 1010]:
RX + NajjS > R—S-R
3.6. Тиолы и сульфиды 143
При использовании бензилхлорида для этого потребовалось
меньше 10 мин. Вторичные алкилгалогениды реагируют
медленнее— требуется нагревание в течение 2 ч при 110°С. Стериче-
ски затрудненный неопентилбромид дает без перегруппировки
динеолентилсульфид за 500 мин при 70 °С [202]. В качестве
катализатора можно использовать также и дилаурилдиметиламмо-
нийхлорид [203], хотя при этом для реакции требуется больше
времени \(в случае н-октилхлорида 2 ч яри 105 °iC). В этих же
условиях с аликватом 336 я-нитрохлорбензол образует с
выходом 42% ди-п-нитрофенилсульфид [204]. Использование дицик-
логексано-18-крауна-6 совместно с K2S не дает, по-видимому,
каких-либо экспериментальных преимуществ [45]. МФК
использовали также при получении диалкилполисульфидов [203].
RS-Na++R'X—*/?—S—/?'
Одной из самых первых реакций такого рода было алкили-
рование тиофенола и этантиола замещенными аллилхлоридами
в присутствии 10 н. КОН и каталитического количества
катализаторов типа дибензилдиметиламмонийхлорида [211]. В тех же
условиях, которые используются для получения симметричных
аналогов, только за (более короткое время или при более
низкой температуре, можно синтезировать и несимметричные
сульфиды. Ландини и Ролла [202] использовали трибутилгексадецил-
фосфонийбромид без какого-либо растворителя. В этих условиях
даже неопентилбромид реагирует с тиофенолятом натрия, давая
несимметричный сульфид, выход 78—85% 1856]. Херриот и Пик-
кер (205] предпочитают систему бензол/аликват 336. При
двукратном избытке алкилирующего агента арилтиоляты
реагируют при комнатной температуре даже со вторичными галогенида-
ми, образуя меньше чем за 1 ч продукт реакции с выходом
более 90% (205]. Проведение реакции с а л кил меркаптанами
требует кипячения (202, 203]. При проведении этой реакции, так же
как и в случае других реакций замещения, в качестве
катализаторов испытывались полиподные лиганды. Они не были
достаточно эффективны при обычном использовании [46]. В реакции
n-метилтиофенолята натрия с {R)-(—)-2-хлороктаном
наблюдалась 98%-ная инверсия с образованием (5)-( + )-2-октил-и-
толилсульфида [202]. В последнее время метод МФК был
использован при синтезе лантионина и его диастереомеров (1656],
а также при получении ряда полифункциональных сульфидных
комплексов (1529, 1532, 1546].
144 Глава 3. Практическое использование МФК
>- (RS)2CH
При проведении некоторых реакций в дихлорметане в
качестве побочных продуктов образуются симметричные дитиоаце-
тали формальдегида. Если отсутствуют другие алкилирующие
агенты, то реакция идет с выходом более 90% за 15 .мин при
комнатной температуре (катализатор — аликват 336) [205].
Дитиоацеталь легко алкилируется по центральному атому
углерода, давая после гидролиза соответствующий альдегид [205].
Соединения типа Аг—S—СН2С1 были получены из СН2ВгС1
[994], а соединения типа (RS)zCH—COOEt — из этилдихлораце-
тата в системе твердый КгСО3/аликват 336 [1826]. При
обработке CBr2F2 и тиофенола системой К.ОН/ТЭБА образуется 70%
PhS—CF2Br и 30% PhS—CF2H. В этом случае интермедиатом
является дифторкарбен {1828]. При действии водного раствора
диазониевой соли в присутствии адогена 464 (который есть
то же самое, что и аликват 336) из метиленбромида при
кипячении в течение 9,5 ч образуются замещенные 1,3-бензоксатио-
лы (А) {206]. Продукт А {К;=5-(1-этоксиэтокси)] был получен
из соответствующего 1,3-бензоксатиол-2-она при кипячении его
в дибромметане с системой водный ЫаНСОз/аликват 336. Эта
реакция представляет собой одновременное омыление и алкили-
рование {1244]. Из CD2C12 и 1,2-бензендитиола в присутствии
системы NaOH/аликват 336 был получен 2,2-дидейтеробен-
зо-1,3-дитиолан с выходом 86% [1749].
OeNae
SeNae
R
Схема 3.32
Восстановление соединения В с помощью тетрабутиламмо-
нийтетрагидроборана в СН2С12 при —30 °С странным образом
привело к D [207]. Его образование можно объяснить алкилиро-
ванием избытком В первичного продукта восстановления С:
RSO2 RSO2 ^Se RSO2 S—С—SR'
R S К S R S HI
BCD
Схема 3.22a
Для алкилирования амбидентных тиосоединений используют
метод «экстрактивного алкилирования», описанный в разд. 3.10.
3.6. Тиолы и сульфиды 145
Однако депротонированные соединения Е и G ведут себя
с химической точки зрения как истинные тиолятные анионы*
поэтому их алкилирование обсуждается в этом разделе. Про-
паргилбромид в метиленхлориде в присутствии 2 молей водного
гидроксида натрия и 1 .моля тетрабутиламмонийбисульфата*
реагирует с Е, давая за 18 ч при .комнатной температуре алле-
нилсульфид F [208].
Соединение G (R=CH3, C6H5) было проалкшшровано
в СН2С1г или в СНС1з аллил-, кротил- и пропаргилбромидами
при комнатной температуре за время от 1,5 до 5 ч {209].
R .COOEt
R'S COOEt R'S H
(Z)-H (£)-H
Схема 3.33
В качестве экстрактанта был использован бисульфат тетра-
бутиламмония. В том случае, когда R = CH3, образовывались
Z- и £-изомеры соединения Н, а при R=C6H5 был получен
только Z-изомер. В случае 2-меркаптокоричной кислоты (G-кис-
лота при R=C6H5) при действии молярного количества алкили-
рующего агента вначале происходит S-алкилирование, после
чего можно провести еще одно алкилирование — с образованием
сложного эфира [907].
Дициклогексано-18-краун-6 .катализирует образование
сульфидов из тиолов и хлор-2,4-динитробензола в ацетонитриле
даже в тех случаях, когда в системе нет щелочи и тиолы не
находятся в тиолятной форме £210].
Однако при этом реакция включает образование НС1 и идет
существенно медленнее, чем с тиолятами.
Необычный луть получения некоторых сульфидов,
совершенно отличный по механизму от реакций с тиолятами, состоит
в реакции алифатических или ароматических тиоцианатов с
соединениями, содержащими кислую группу С—Н. Эта реакция
проходит под влиянием системы концентрированный
146 Глава 3. Практическое использование МФК
NaOH/ТЭБА [300, 1420]:
R—S—CN + НСС13 "*"">■ R—S—СС1з + NaCN
I эЪА,
R—S—CN + Ph—CH—CN
Из фенилацетилена и алкилтиоцианатов под действием
концентрированного NaOH/ТЭБА образуются фенилэтинилсульфи-
ды [769]:
NaOH
Ph— C=CH + RSCN -—*■ Ph—C=C—SR + CN~
ТЭБА
При действии системы концентрированный
NaOH/ТЭБА/следы ДМСО может проходить тиоалкилирование арилацетонит-
рилов; в качестве побочных продуктов неясного происхождения
образуются аюлисульфиды [1047]:
PhCHfR1)—CN + S + R2X —v PhCR^SR^-CN
R2X + S ► R2-(S)n—Ra
Превращения 2-хлорциклогексанона при взаимодействии его
с водным раствором сульфида натрия в присутствии Bu4NCl
относятся к МФК-реакциям в нейтральных средах и приводят
в зависимости от условий к одному из указанных ниже
продуктов [995]:
Схема 3.35
С помощью высокоэффективного МФК-процесса, показанного
на схеме 3.36, -были синтезированы серусодержащие гетероцик-
лы. При использовании 25%-ного водного NaOH, ВщЫВг
и дибромметана или 1,2-дихлорэтана были получены 1,3-дитио-
лан (I) и 1,4-дитиан (J) с выходом 47 и 87% соответственно.
Благодаря продуманному выбору оптимальных условий для
проведения реакций было подавлено образование полимерных
продуктов {1488].
3.6. Тиолы и сульфиды 147"
50-«0% [1559, 1826];
||
H V_K I J [1488],
Схема 3.36
Из соответствующих а-галогенпроизводных были получены
при комнатной температуре с прекрасным выходом а-алкилмер*
каптокетоны, а-алкилмеркаптоэфиры и а-алкилмеракаптолакто-
ны; для этого использовали систему сухой КгСОз/аликват 336
[1827]. Присоединение сульфида к ацетонитрилу с образованием
быс-(цианоэтил) сульфида происходит в результате
экзотермической МФК-реакции. Этот процесс можно использовать для
удаления следов ацетонитрила из газов (1829].
При использовании хиральных катализаторов для получения
сульфидов с помощью алкилирования {1469] и присоединений
к активированной двойной связи в некоторых случаях
наблюдался высокий выход оптически активных продуктов (разд. 3.1.5).
Высушенный с помощью вымораживания фторид калия
является очень активным основанием, .которое позволяет проводить
алкилирование тиолов даже в отсутствие МФ-катализаторов
[1605].
В области полимерной химии описано применение методов
МФК для модификации хлорметилированных полистиролов
путем замещения атома хлора на серусодержащие остатки {1227,
1259, 1739, 1830] и для получения полимеров из дигалогенов
и дитиолов [1279] или Na2S (1458].
148 Глава 3. Практическое использование МФК
3.7. Получение простых эфиров
Метод получения эфиров с помощью МФК был открыт
случайно. В 1951 г. Жаррусс нашел, что вторичные спирты и р-хлор-
этилдиэтиламин в присутствии водного раствора гидроксида
натрия дают простые эфиры:
СНз /СНз
ROH + ClCHa-CHa-N(
eoaKN,oH x
СНз СНз
Схема 3.37
Он сделал предположение, что в ходе реакции образуются
следовые количества Ы,Ы,Ы',Ы'-тетраметилпиперазинийдихлори-
да, который действует как катализатор {224]. Впоследствии он
показал, что под действием водного раствора гидроксида натрия
и ТЭБА из йензилхлорида и циклогексанола образуется смесь
простых эфиров. Однако работа Жаррусса не привлекла
внимания химиков. Точно так же и ранние работы по МФК-алки-
дированию фенола и бензилового спирта замещенными аллил-
хлоридами в присутствии системы КОН/четвертичные
аммониевые, хлориды остались погребенными в литературе [211, 225, 226].
Примерно в то же самое время в патентной литературе были
описаны некоторые реакции, которые в широком понимании
можно считать МФК-процессами, например получение
эпоксидных смол из дифенолов [186, 228] или из циануровой кислоты
[186] и зпихлоргидрина в присутствии щелочей и аммониевых
солей.
2RX __^L^ R0R
водн. NaOH
Только в 1972 г. Херриот и Пиккер сообщили, что 1-бром-
«ктан в присутствии 2 н. NaOH и четвертичной аммониевой соли
дает эфир и, кроме того, 20% октанола-1 и 5% октена в
качестве побочных продуктов. Добавление в реакционную смесь
спирта увеличивало как выход, так и скорость образования эфира.
Из этого следовало, что первой стадией реакции был гидролиз
.алкилгалогенида. В таком случае остается неясным, почему
же образуется эфир, а не спирт, который является продуктом
гидролиза. В присутствии концентрированного раствора гидр-
хжсида натрия имеет место равновесная реакция депротониро-
вания спиртов. Независимо от того, является ли спирт
гидрофильным и вследствие этого находится главным образом в
водной фазе или же он с самого начала был в органической фазе,
3.7. Получение простых эфиров 149
при экстракции в органическую фазу будет переходить
преимущественно алкоксидный анион, так как он заведомо более липо-
*филен, чем гидроксид-ион. Поскольку алкоксид-ионы являются
-также более сильными нуклеофилами, то в результате
происходит образование эфира.
Другие примеры получения симметричных простых эфиров
из галогенидов или сульфатов с использованием аликвата 336
(как самого лучшего катализатора) находятся в патентах [229].
При использовании (С^НгвЬ^СНгСОгСНз Вг~ в качестве
катализатора удалось превратить 1,4-дихлорбутан в тетрагидро-
.фуран [230].
RX+R'OH МФК > R—O—R'
водн. NaOH
Если в этой реакции использовать 2 моля первичного
спирта на 1 моль первичного 'бромида, то можно получить около
■60% смешанного простого эфира {28].
Однако почти в таком же количестве образуются и
симметричные эфиры. Во многих случаях их смесь разделить трудно.
Простые вторичные алкилгалогениды не дают в этих условиях
-эфиров, поскольку подвергаются элиминированию (с
образованием олефинов) и гидролизу.
В работах |232, 1051] сообщается, что оптимальными
условиями получения несимметричных эфиров методом МФК явля»
ются: 5-кратный избыток 50%-ного водного NaOH по
отношению к спирту, избыток алкилхлорида (который желательно
использовать также и как растворитель), 3—5 мол. % тетрабутил-
аммонийбиоульфата. Эту смесь перемешивают при 25—70 °С.
Первичные спирты образуют эфиры с выходом 80—95% за 3—
4 ч, а для образования вторичных спиртов требуется более
длительное время или большее количество катализатора. При
изучении этой реакции в стандартных условиях наблюдалось четко
выраженное влияние противоиона первоначально взятого
катализатора: бисульфат (превращающийся в сульфат в ходе
реакции) был эффективнее, чем иодид, который в свою очередь был
лучше, чем перхлорат {232]. Симметричные эфиры образуются
в количестве менее 10% главным образом в конце реакции,
т. е. когда соотношение концентраций реагентов становится
неблагоприятным. Эти трудности отсутствуют при получении
метиловых эфиров с помощью диметилсульфата. В присутствии
тетрабутиламмонийиодида [233] выходы были почти
количественными, однако с успехом были испытаны катализаторы и
с более гидрофильными анионами.
Оказалось, что реакция может протекать как в самом спирте,
так и в различных растворителях, таких, как петролейный эфир,
150 Глава 3. Практическое использование МФК
бензол, эфир или дихлорметан. Реакция одинаково гладко
проходит как с насыщенными первичными спиртами, так и с
«активированными» первичными, вторичными и третичными спирта'
ми, которые содержат непредельные или электроноакцепторные
группы. Насыщенные вторичные спирты реагируют медленнее,
а третичные не реагируют совсем. Стерические затруднения,
по-видимому, не влияют на ход реакции. Однако
водорастворимые соединения (например, углеводы) в реакцию не вступают,
так как их ионные пары не переходят в органическую фазу.
С помощью этой реакции 1,4-дибромбутан может быть
превращен в 1-бензокси-4-бромбутан с выходом 88% [874]. При
использовании в синтезе эфиров твердого гидроксида натрия
в качестве основания подавляются побочные реакции
(образование олефинов, спиртов, симметричных эфиров) [1406]. Ниже
приведен необычайный синтез смешанных диалкиловых или ал-
килариловых эфиров с использованием алкилдифенилсульфоние-
вых солей (.как алкилирующих агентов) в системе тверда*
фаза/жидкость с карбонатом калия в качестве основания [1596]
R*X + Ph2S »- I^SPVX"
X" + K+ "OR2 » I^OR* + K+X" + Ph2S
В работе [1258] изучены некоторые группы, которые способ
ны давать эфиры гидроксиламина и оксимов. Оксим и дихлор*^
метан [система водный NaOH/R4NX или твердый КгСОз/18-кра-
ун-6 или КОа/комплекс Pd(II)] образуют соединений
R2C=N—О—СНа—О—N=CR2 [1274, 1296, 1467, 1863].
Бутилбромид, взаимодействуя с сык-бензальдоксимом, дае*
58% эфира син-оксима и следы нитрона, в то время как анти-
бензальдоксим в качестве главного продукта образует нитрой
[1467]:
a«mu-PhCH=NOH »- PhCH=N(O)—С4Н„ (48%) +
+ a«mu-PhCH=N—О—С4Н„ (12%)
При нагревании до 80 °С спирта, силилхлорида, сухого
карбоната калия и каталитического количества аликвата 336
образуются триметилсилильные и трет-бутилдиметилсилильные
эфиры.
Силилированию подвергаются первичные, вторичные и
третичные спирты. Используемые реагенты доступны и недороги,
а растворитель (петролейный эфир или толуол) необходим
только для реакций с твердыми спиртами [1627].
Аддукт, образующийся при присоединении хлороформа к
двойной связи карбонильного соединения А, можно О-метили-
ровать в тех же условиях, хотя в зависимости от изменения
условий проведения реакция может идти по различным
альтернативным направлениям (разд. 3.20.2). Мерц и Томахо [235],
3.7. Получение простых эфиров 151
проводя реакцию 1 моля карбонильного соединения, 1,2 моля
диметилсульфата, 1 мол.% ТЭБА в 3 молях хлороформа в
присутствии 2,4 моля 5%-иого водного гидроксида натрия три
30—35 °С, не смогли выделить промежуточное соединение А
(схема 3.38). Другие авторы [236] использовали в этой реакции
метилиодид с тетрабутиламмонийиодидом в качестве
катализатора в бензоле/12 М NaOH. При этом достигался выход
порядка 80—95%.
А
Схема 3.38
Более трудным случаем этерификации является реакция
между ендиолятом бензоина и быс-тозилатом диэтиленгликоля
[428; см. также I860]:
PhCH(OH)—CO-Ph + Tos—О—(СН2)2—О—(СН2)2—OTos
OUiN Br, 14ч , 80 С
74%
При перемешивании в течение нескольких часов при 100 °С
с избытком системы бензилхлорид/50°/о-ный водный NaOH
проходит бензилирование 1,2-О-изопропилиденглицерина; в
качестве катализатора используют бензйл-три-к-бутиламмонийбромид
[399].
Аналогичным образом было проведено монобензилирование
моносахаридов с двумя ОН-группами, таких, как метил-4,6-0-
бензилиден-а-О-глюкопиранозид (В) или метил-2,3-ди-О-бензил-
o-D-глюкопиранозид (С), кипячением их в течение 48 ч в ме-
тиленхлориде с 1,7 моля бензилбромида, 20 мол.% Bu4N+HSO4~
н 50%-ным раствором гидроксида натрия [239].. Общий выход
составил —80%. Из В образовывалось 6% 2,3-дибензилового
эфира, 54% 2-бензилового эфира и 20% 3-бензилового эфира.
152 Глава 3. Практическое использование МФК
Из С—18% метил-2,3,4-трибензил-а-О-глюкопиранозида и
68% 2,3,6-трибензилового эфира. Описано получение в условиях
МФК 1,2,5,6-тетраоктадецил-Е)-маннитола через его липофиль-
ные 3,4-производные [1005]. О-Алкилирование изопропилиден-
и бензилиденпроизводных моносахаридов может быть
осуществлено при комнатной температуре метилбромидом, бензилхлори-
дом или 1,2-дихлорэтаном в системе бензол/50%-ный водный
гидроксид натрия [240, 1049]. Алкилирование метилиодидом.
Me ХУ-
Me О"
я-
>h—СН
V
СН
-сн2
/-
is
2ОН
А
^/оснз
1
он
Me О^
носн2
/
с
3.40
~СН2ОСН2—Ph
О
,/оСНз
OBz
идет медленнее, так как выделяющийся при реакции иодид-ион
отравляет катализатор. Из двух катализаторов (ТЭБА и
ВщЫВг) последний был намного более эффективным. При
использовании дихлорметана одновременно в качестве алкили-
рующего агента и в качестве растворителя наблюдалось
образование ацеталей формальдегида (см. ниже) [240].
Метилирование и бензоилирование моносахаридов с разной степенью-
замещения описано в работах [1416, 1463, 1597, 1861].Региосе-
лективность реакций таких полифункциональных субстратов
определяется, по-видимому, относительной кислотностью гидр-
оксильных групп [239, 1416]. Путем конденсации производных
Сахаров, имеющих две рядом стоящие свободные гидроксиль-
ные группы с бис- (2-хлорэтиловым) эфиром, который
использовался и как растворитель, и как реагент, в присутствии конц.
NaOH/R4NX и последующей циклизации продукта конденсации
(под влиянием системы катехин/сухой NaOH в бутаноле-1)
выла получена серия хиральных краун-эфиров [1249].
Использование системы NaOH/аликват 336 в дихлорметане
позволяет получать также и «-(аллилокси)ацетонитрилы (Е)
из циангидринов альдегидов (D) и аллилбромидов [237].
Анион циангидрина в сильнощелочной среде частично распадается,
давая альдегид и цианид-ион. Этот ион конкурирует с аллил-
галогенидом, что приводит к образованию побочных соединений
3.7. Получение простых зфиров 153
типа F. Аналогичным способом было получено и соединение Н
[238]. Хотя выходы соединений типа Е не слишком велики (24—
46%), они являются полезными промежуточными продуктами,
которые можно превратить в Р/у-ненасыщенные кетоны типа G,
действуя на них при —78 °С диизопропиламидом лития в ТГФ,
при этом происходит [2, 3]-сигматропная перегруппировка,
сопровождающаяся элиминированием LiCN, что и приводит к G.
он R-<»° — ^
МаОН \\ CN CN
МФК
R-CHO + CNS > )c=CH-CH2-CN
н
Схема 3.41
С помощью МФК можно получить эфирные производные
хлорметилированного полистирола [1050, 1217, 1227, 1259].
Подобной модификации можно подвергнуть и другие полимеры,
содержащие галоген [1444].
Фенолятный анион в условиях МФК подвергается О-алкили-
рованию еще легче, чем алифатические спирты.
Кинетику этой реакции в гомогенных условиях (в диоксане)
исследовали Угелстед и сотр. [249]; оказалось, что ионная
пара тетрабутиламмонийфенолята реагирует с бутилбромидом в
3-Ю4 раз быстрее, чем простая калиевая соль. Комплекс
калиевой соли с дициклогексано-18-крауном-6 реагирует быстрее
простой соли в 2—3 раза [249] с достаточно хорошими
выходами (большей частью 80—95%). Применение МФК особенно
выгодно в тех случаях, когда кроме основной реакции может
проходить С-алкилирование [29, 278]. В условиях МФК С-алки-
лирование идет всегда с выходом менее 5% [29, 380]. Были
испытаны [29, 966] следующие алкилирующие агенты:
первичные и вторичные алкилгалогениды, эпихлоргидрин и диалкил-
154 Глава 3. Практическое использование МФК
сульфаты. В качестве катализаторов применяли бензил-три-м-
бутиламмониевые соли, а реакцию проводили в смеси метилен-
хлорид/водный гидроксид натрия при интенсивном
перемешивании (вибромиксер) при комнатной температуре. Стерические-
препятствия не мешали метилированию. Так, даже 2,4,6-три-
rper-бутилфенол с диметилсульфатом давал продукт реакции с
выходом 93%. В то же время без катализатора реакция не шла.
Однако при использовании в качестве алкилирующих агентов
этил- или изопропилиодидов реакция с сильно затрудненными
фенолами не проходила. В этом случае, как и во многих других
МФК-реакциях, при использовании различных алкилирующих.
агентов успех был достигнут введением в реакцию
каталитических количеств четвертичной аммониевой соли. Однако такая:
методика не пригодна при применении алкилиодидов. Иодид-
ионы экстрагируются в органическую фазу лучше, нем
феноляты, что приводит к отравлению катализатора. В то же время
бензилхлорид настолько активен, что он гидролизуется прежде,,
чем имеющееся в системе небольшое количество катализатора
успевает перенести весь фенолят-ион в органическую фазу. По-
этим причинам* для реакций с алкилиодидами и бензнлгало-
генидами рекомендуют использовать стехеометрическое
соотношение фенол/катализатор.
При применении избытка реакционноспособного алкнлнрую-
щего агента О-алкилирование 2/-гидроксихалконов проходит с
превосходными выходами [875]. В качестве побочной реакции:
происходит замыкание кольца с образованием флавононов-
(схема 3.42). Синтез замещенных 3,3-дигидро-2Н-1,4-бензокса-
зинов был осуществлен под действием системы твердый
гидроксид натрия/аликват 336 в растворе ацетонйтрила или дихлор-
метана [1241}:
н
Схема 3.42
* В работе [291 приведено другое объяснение этим фактам, основанное на
том, что бензилхлорид в условиях МФК имеет очень малую реакционную
способность. ' '-■',■■■■''
3.7. Получение простых эфиров 155
При МФК-алкилировании 2-амино-З-гидроксипиридинов
образуется только О-производное (выход 20—87%) [1862].
Довольно необычное перемещение алкильного радикала от
третичного амина к фенолятному иону проходит (с выходами
28—56%) при перемешивании амина с хлороформом и
бензолом. Предлагается следующий механизм [1371]:
НСС18 ?► CC1S R3N -f CC12 » R3N+—~СС12
R3N+—-CCI2-f-HCCl3 » R3N+—CHCI2-CC13
R3N+—СНС12-СС13 + Аг0Н > R3N+—CHCljfOAr + HCC18
R3N+—СНС12"ОАг » R—OAr + R2N—CHC12
При отсутствии алкилирующего агента дихлорметан,
являющийся растворителем, начинает взаимодействовать с фенолят-
ным анионом, образуя в качестве побочного продукта диарил-
оксиметан [29]. Небольшие изменения в условиях проведения
этой реакции позволяют получать этот продукт с очень
высоким выходом [234]. С этой целью твердый порошкообразный
гидроксид калия и фенол перемешивают в метиленхлориде в
присутствии 5—10 мол.% аликвата 336 в течение 8—16 ч при
комнатной температуре. При использовании водного раствора
гидроксида щелочного металла или менее липофильного
катализатора— ТЭБА скорость реакции резко снижается.
Метиловые эфиры катехинов получают, вводя в реакцию метиленбро-
мид, водный гидроксид натрия и адоген 464 в качестве
катализатора [244, 245]. В этой реакции можно использовать
также метиленхлорид [873]:
АгОН + СН2С12 >- ArO—OV-OAr
R—ОН +СН2С12 v RO—CH2—OR
Поскольку алкоксилатные ионы менее нуклеофильны, чем
фенолятные, то для получения ацеталей формальдегида с
выходом 65—80% при использовании твердого КОН приходится
вести процесс около 15 ч [234]. Этим методом можно провести
метиленированне различных 1,2-диолов, включая и углеводы
[909,954].
Для завершения более сложной реакции, приведенной на
схеме 3.43, лотребовалось перемешивание при комнатной
температуре в течение суток [429]. Аналогичное превращение
наблюдалось при взаимодействии тетра-О-бензил-а-О-глюкопирано-
зилбромида с различными фенолятами в системе вода/СН2С1г/
/ТЭБА. Образование арил-р-Б-производных происходило сте-
реоспецифически [1228]. Подобный же результат был получен и
при ацетилировании глюко- и маннопиранозидов [1865].
Для получения эфиров фенолов очень часто используют в
качестве катализатора бисульфат тетрабутиламмония [243].
При реакции с гетероциклическим аналогом I применялся
156 Глава 3. Практическое использование МФК
ТЭБА [247] (схема 3.44), но иногда рекомендуется криптанд.
[2.2.2]. Синтезы Вильямсоиа с фенолятами или алкоксилатами,.
генерируемыми КН или NaH в бензоле или в ТГФ, могут
проходить при комнатной температуре [1224, 1379].
СООМе
АсО
+ н
но
Bu4NHSO. ?
NaOH/CHaCfe
СООМе
АсО
сна
ОАс
Схема 3.43
Cl
+ C1CH2—СООМе
X°jC
Cl " О—СН2—СООМ*
I
Схема 3.44
В результате кипячения смеси 2-хлорциклогексанона с фенолом
в бензоле, водного гидроксида натрия и небольшого количества
аликвата 336 Пиккер [246] выделил 58% 2-феноксициклогекса-
нона К. При этом обычного продукта реакции Фаворского
обнаружено не было (схема 3.45). В соединения типа L и М
можно ввести метоксиметильную защитную группу, если провести
реакцию калиевой соли соответствующего фенола с хлордиме-
тиловым эфиром в ацетонитриле в присутствии 10 мол.%
18-крауна-6 при комнатной температуре [241].
По другой (улучшенной) методике реакцию проводят
следующим образом: к перемешиваемой смеси раствора фенола
в СНгСЬ, небольшого количества адогена 464 и водного
раствора гидроксида натрия медленно, по каплям прибавляют трех-,,
пятикратный избыток хлорэфира (выход конечного продукта
79—81%) [950]. В недавних публикациях сообщается об алки-
лировании в условиях МФК о-ацилфенолов [1056] и реакциях
между фенолятами и эпихлоргидринами [1057] или хлорангид*
ридом циануровой кислоты [1058].
Для синтеза фенольных эфиров и с меньшим успехом для
синтеза алифатических эфиров используют так называемый трех-
3.7. Получение простых эфиров 157
фазный катализ. Подробности, относящиеся к такого рода
реакциям, включая описание реакций с твердофазными сорас-
творителями («solid-phase cosolvents») (неионные боковые
группы в ионообменных смолах), см. в разд. 3.1.4.
освн-,
ОСН2—ОСНз
м
Схема 3.45
Экстрактивное алкилирование (метод Брендстрёма) было
использовано химиками-аналитиками для количественного газо-
хроматографического определения фенолсодержащих
анальгетиков— пентазоцина (N) и соединения О. При рН 10,5
приготовляют бензольный экстракт человеческой плазмы, затем его
упаривают, остаток вновь экстрагируют разбавленным водным
раствором кислоты и, наконец, добавляют водный раствор гидр-
оксида натрия. После этой обработки проводят реакцию
образования производных, перемешивая полученный продукт е
пентафторбензилбромидом и тетрабутиламмонийбисульфатом в
дихлорметане при комнатной температуре [250].
ОТнс-сн
N—СН2—СН=С
|
но
но
N
с—с,н5
N—СНз
Схема 3.46
Другим примером аналитического применения МФК
является определение с помощью газожидкостной хроматографии
соединений типа эстрогенов. Для этого их экстрагируют в мети-
ленхлорид в форме ионных пар с тетрагексиламмониевым ио-
158 Глава 3. Практическое использование МФК
ном и перед хроматографированием алкилируют метилиодидом.
Высоколипофильиый катион обеспечивает быструю экстракцию
и метилирование при комнатной температуре [242]. Обзор
работ, посвященных применению экстрактивного алкилирования
для аналитических целей, дан в [1052], другие примеры
использования этого метода см. в [1054, 1487]. При алкилировании в
двухфазных системах феноляты реагируют с пентафторбензил-
бромидом и другими бензилгалогенидами и в отсутствие МФ-ка-
тализатора, в то же время алкилирование карбоксилатов без
катализатора не идет; это позволяет легко отличать их друг от
друга [1055, 1583]. Катализатор не требуется также и при
синтезе некоторых эфиров с использованием в качестве основания
лиофильно высушенного KF [1605]. Библиографические ссылки
на другие работы, охватывающие все типы реакций получения
эфиров, приведены в табл. 3.7.
Образование енольных эфиров кетонов, О- или С-алкилиро-
вание р-дикетонов, р-кетоэфиров и гетероциклов будет
обсуждаться в разд. 3.10. Получение эпоксидов по реакции Дарзана
списано в разд. 3.13, а реакции образования эпоксидов через
илиды сульфония можно найти в разд. 3.16. И наконец,
разд. 3.17 содержит описание синтезов эфиров при нуклеофиль-
ном ароматическом замещении.
Ниже приводятся типичные экспериментальные методики.
Получение метиловых эфиров (см. [233])
Интенсивно перемешивая в течение 15—30 мин, достигают
равновесия в двухфазной системе, содержащей раствор спирта
(0,5 моля) и 1 г тетрабутиламмонийгалогенида в 200 мл петро-
дейного эфира (т. кип. 50—70 °С) и 50%-ный водный раствор
гидроксида натрия (1,3 моля). При этом наблюдается слабое
разогревание. К смеси в течение 1 ч при охлаждении
добавляют по каплям 0,6—0,75 моля диметилсульфата с такой
скоростью, чтобы температура не поднималась выше 45 °С.
Перемешивают еще 2—3 ч и затем при комнатной температуре
прибавляют 10 мл конц. NH4OH в течение 30 мин. Наконец, смесь
выливают в воду и отделяют органическую фазу, промывают
водой, сушат Na2SO4 и обрабатывают затем обычными методами.
Выход в определенной степени зависит от скорости
перемешивания. Для полумикромодификации при использовании
магнитной мешалки рекомендуют брать 1,5-кратный избыток
(CH3)2SO4.
Получение фенольных эфиров [29]
Смесь 50 мл СНгСЬ, '50 мл Н2О, 10 ммолей фенола, 15 ммо-
лей NaOH, 20—30 ммолей алкилирующего агента и 0,1 —
3.7. Получение простых эфиров 159
Таблица 3.7. Дополнительные литературные данные
об образовании простых афиров в условиях межфазиого катализа
Тип реакции, продукты реакции, замечания
Литература
Замещенные полнфениленоксиды 1530, 1531,
* 1603
Ароматические полиэфиры 1253, 1289,
1478, 1576,
1864
Полимеры цианурхлорида, бромировашюго бисфенола А и 1372
2,4,6-трибромфенола
Полимеры циклических простых эфиров, карбоксилатов и 1628
карбонатов
Бензилирование целлюлозы: наиболее эффективный катали- 1388
затор Me4NX (!)
ROH+C1CH2—CH(OMe)2 1602
Аминофенольные эфиры 1645
о-Метилаллилоксифенол из катехина 1550
Днбензиловые эфиры 1454
Бензилглицидиловый эфир 1505
ClsCH (ОН) 2+этиленоксид—>~2,7-бис- (трихлорметил) - 1322
1,3,6,8-тетраоксадекан
Алкилирование бензгидрола, изучение влияния изменений ус- -1229, 1475
ловий реакции
Моноалкилирование эфиров олигоэтиленовых гликолей, не- 1264
значительное ускорение при использовании R4NX
Алкилирование феноксидов в присутствии краунов и их по- 1581
лимеров
Бензил-к-бутиловый эфир (методика для студентов) 1709
Синтез простых эфиров при наличии а-кетогруппы 1710
Модификация полихлорметилстирола путем образования про- 1227
стых эфиров
Модификация поливинилбромида путем образования простых 1739
эфиров
О-Алкилнрование каменного угля 1757
О-Алкилирование N-замещенных тирозинов 1775
О-Алкилирование 1-гидроксибензтриазола 1774
Образование бензодиоксанов и бензодиоксоланов 1751
Гваяцилметилхлорид — новый реагент для защиты спиртов в 1753
условиях межфазного катализа
160 Глава 3. Практическое использование МФК
1 ммоль бензил-три-н-бутиламмонийбромида* перемешивают
вибромешалкой при комнатной температуре в течение 2—12 ч.
Органический слой отделяют, а водный слой экстрагируют
двумя порциями СН2С12. Объединенный с экстрактом органический
•слой упаривают, к остатку добавляют воду и вновь
экстрагируют эфиром или пентаном. Органическую фазу дважды
промывают 2 н. NH3 (для удаления диметилсульфата) или, если это
необходимо, обрабатывают метанольным раствором ЫНз (для
разрушения диэтилсульфата) и наконец промывают
насыщенным раствором NaCl. После высушивания над Na2SO4 раствор
упаривают и оставшийся эфир очищают перегонкой или
перекристаллизацией.
3.8. N-алкилирование
N-алкилирование в классическом варианте проводится в
двухфазной системе, содержащей карбонат натрия или щелочь.
Для того чтобы вторичные или третичные аммониевые соли
могли образоваться из аминов, последние должны подойти к
поверхности раздела фаз. Скорость реакции будет
определяться нуклеофильностью амина; из этого следует, что МФ-катали-
затор не должен оказывать заметного влияния на реакции
нормальных аминов. Водный раствор натриевой щелочи не
является достаточно сильным основанием для депротонирования
неактивированных аминов. Однако депротонирование становится
возможным, если кислотность NH-группы повышается под
влиянием соседних электроноакцепторных групп:
рКа NH8 34—36, ArCONH2 ~ 25, PhSO8NH2 ~ 10
Таким образом, при получении очень нуклеофильных амид-
ных анионов катализатор может действовать одним из двух
способов: либо 1) переносить гидроксид-ион, осуществляющий
депротонирование, в органическую фазу, либо 2) убирать де-
протонировэнную молекулу с поверхности раздела фаз. Эти
предположения находят подтверждение в большинстве
исследований, выполненных в данной области. Действительно, в
литературе имеется только несколько публикаций, в которых
сообщается об алкилировании неактивированной NH-связи в
присутствии четвертичных аммониевых катализаторов. В
присутствии водных растворов гидроксидов калия или натрия были
проалкилированы 1,3-дихлорбутеном-2 и бензилхлоридом раз-
* При использовании в качестве алкилирующих агентов СН31 или бензил-
хлорида лучшие результаты получаются со стехиометрическими количествами
катализатора.
3.8. N-алкилирование 161
личные ароматические амины. При использовании в качестве
катализаторов специальных четвертичных аммониевых
хлоридов или ТЭБА выходы были в 2—3 раза выше [225, 226, 251,
1548]. Также успешно было проведено циклическое быс-алкили-
рование первичных аминов A [R=CH3, (СН3)гСН, трет-бутил]
соединением В в смеси растворителей хлороформ/этанол/вода
в присутствии бензилтриаммонийметоксида. Выходы достигали
65—90% [252]. И наконец, в условиях МФК происходит
ускорение реакции антранилонитрила с 1-хлор-триацетил-Ь-рибопи-
ранозой (выход 10%) [1059] и образования азиридинов [1870].
Под действием системы NaOH/ТЭБА происходит алкилирование
9-аминоакридина, при этом с выходом 40—50% образуется
9-алкиламинопроизводные [1496].
В отличие от водного гидроксида гидрид калия плюскрип-
танд[2.2.2] количественно превращает анилин в анилид в
бензоле или ТГФ при комнатной температуре. Алкилирование
заканчивается за.З мин. [1379]. Другим необычным основанием,
которое предлагается для N-алкилирования анилина, является
лиофильно высушенный фторид калия без каких-либо добавок
[1605].
Вг
R—N
i
Вг
Схема 3.47
В гидр азобензоле связь N—Н существенно более кислая,
поэтому небольшая равновесная концентрация аниона может
образоваться в присутствии карбоната калия или водной
щелочи. G K2CO3 в этаноле или ДМФА моноалкилирование идет
плохо. Применение техники экстрактивного алкилирования с
метиленхлоридом в качестве растворителя в присутствии 0,5-^
1 моля тетрабутиламмония гидроксида и- 0,5—1 моля водного
гидроксида натрия нри кипячении в течение 1—24 ч до
нейтральной реакции позволяет значительно увеличить выход. Пер^
вичные иодиды, бензилбромид я аллилбромид дают хорошие
выходы, вторичные иодиды — хуже [253] (схема 3.48; см.
также [1548]). Аналогично можно проалкилировать фенилгидра-
зон С; при этом образуется D с выходом 43—98%, Условия
реакции: перемешивание с 50%-ным водным раствором гидрокси-
162 Глава 3. Практическое использование МФК
да натрия, избытком алкилирующего агента и 5 мол.% тетрабу-
тиламмонийхлорида в течение 5 ч при 30—60 °С [254].
к
I
СвН5—NH—NH—СвН5 > СвН5—NH—N—CeHs
Схема 3.48
Гидролиз разбавленной серной кислотой приводит к 1-ал-
кил-1-фенилгидразону Е. Соединения типа F алкилируются в
системе дихлорметан/15%-ный водный раствор гидроксида
натрия с бензилтриметиламмониевым катализатором (8 ч,
комнатная температура) [273]. Эти соединения распадаются под
действием конц. NaOH/BtuNCl в отсутствие алкилирующего агента
и дают диазосоединения [308]. В условиях МФК можно также
проалкилировать диазоаминосоединения (1,3-диарилтриазены
[1062]).
C=N—N-
Схема 3.49
Брендстрём [255а] показал, что ацетанилид можно проме-
тилйровать в СНгСЬ в присутствии 1 моля тетрабутиламмоний-
бисульфата, 2 молей водного NaOH и избытка метилиодида.
В этих условиях легко алкилируются также сульфонамиды,
изатин и фталимид [255Ь]. Однако бензамид алкилируется с
трудом, поскольку в нем связь N—Н недостаточно кислая [256].
В таких случаях для депротонирования связи N—Н в амидах
используют гидриды щелочных металлов в инертных
растворителях и проводят алкилирование в присутствии 18-крауна-6
[941] или аликвата 336 [942, 1398]. Однако с препаративной
точки зрения более привлекательна возможность использовать
для проведения таких реакций конц. NaOH/Bu4NHSO4 в
кипящем бензоле (моноалкилирование бензамида) или
порошкообразные твердые NaOH/K2CO3/Bu4NHSO4 при кипячении с
обратным холодильником (бисалкилирование бензамида, моноал-
3.8. N-алкилирование 163
килирование карбоксамидов, бисалкилирование сульфонамидов)
[1754].
о о
II II
NH—С—Rl R\ /\ /N—С—R1
R3X/NaOH
ТЭБА
G
О О
II II
Лн ^N.aOH> С NR
OU-
Схема 3.50
Алкилирование в присутствии NaOH можно проводить и с
1—10 мол. % катализатора; время и температуру подбирают
в зависимости от структуры применяемого агента. Например,
для соединений типа G: в случае (CH3)2SO4 реакция проходит
за 45 мин при комнатной температуре, а в случае к-С4Нэ1— за
24 ч при 80 °С. Выходы порядка 80—95%. Без катализатора,
если К1 = алкил, реакция идет очень медленно [257].
Превосходные выходы макроциклических N-тозилазациклоалканов были
получены при диалкилировании тозиламида дибромидами
(реакцию проводят при медленном добавлении алкилирующего
агента или в очень разбавленных растворах с применением Bu4NI
и разбавленного NaOH) [1787]. Однако форманилид реагирует
в двухфазной системе в отсутствие ТЭБА [257]. Скорость
конкурирующей реакции — гидролиза амида — во всех случаях
была очень мала. Циклические амиды, такие, как пирролидон,
е и и-капролактам, также можно алкилировать при 40—50 СС
в присутствии системы концентрированный NaOH/бензол/ТЭБА
[847]. Другие члены этого ряда с большими циклами реагируют
медленнее. Были проалкилированы [1063, 1545, 1869] даже
Р-лактамы, которые образуются в этих условиях в результате
внутримолекулярной циклизации [1064]. И та и другая
реакция идут с высокими выходами. В одной из препаративных
методик используют твердый KOH/Bu4NBr в ТГФ [1553].
Как уже упоминалось, очень эффективной щелочной
системой для алкилирования амидов является смесь твердых
порошкообразных NaOH/КгСОз в бензоле в присутствии Bu4NHSO4
[1261, 1374]. Арилкарбаматы также можно проалкилировать
по азоту [1291].
Твердый фталимид калия можно алкилировать в толуоле в
присутствии 10 мол.% трибутилгексадецилфосфонийбромида
164 Глава 3. Практическое использование МФК
йри 100 °С бензилхлоридом (20 мин), циклогексил- или неопен-
тилбромидом (40 ч); выходы 85—100%. Оптически чистый ал1
килметансульфонат дает продукты с инверсией около 90%. Для
некоторых вторичных субстратов и неопентилбромида в
качестве побочной реакции наблюдается элиминирование. Скорость
реакций намного ниже при использовании в качестве
растворителя амилового спирта. Это наблюдение согласуется с
предположением о том, что для протекания быстрой МФК-реакции
необходимо, чтобы ионные пары были несольватированы [258,.
1524]. (О реакциях, идущих в присутствии краун-эфиров см.
[1108, 1379, 1534].) При алкилировании 2-бромалканоатами в-
системе твердая фаза/жидкая фаза с хиральным катализатором
были получены оптически активные 2-фталимидные эфиры с
низкими или умеренными оптическими выходами [940, 1469];
см. разд. 3.1.5.
о
(твердый)
Виз(и-С16Н33)РВг>
толуол . RX - '
Схема 3.51
N—R
Алкилирование цианамидов избытком RX в присутствий
50%-ного водного гидроксида натрия и ониевой соли дает толь-!
ко продукты диалкилирорания. Они легко расщепляются до
вторичных аминов [1065, 1621]. Связи N—Н можно
активировать, вводя остаток фосфорной кислоты. Цвиржак и сотр.
[272] превращали первлчные амины во вторичные, получая про?
межуточно диэтилфосфорамиды, которые затем алкилировали и
подвергали расщеплению. Стадии 1 и 2 представляют собой
МФК-реакции.
о
о
Стадия 1 (R1O)ZPH + СХ4 + R2NHZ ™""> (R'O)ZP—NH—Ra + HCXa [1261
ТлБА
О
II
Слтайия 2 (R'OfcP-NH—Rz
О R3
II I HC1
СтаЭия 3 (R1O)2P—N—Rz -Ш++ HN—Rz
Схема 3.62
о к3
(R'O)i-N-R»
[2721
R3
' 3.& N-aAKUAupoeanue 165
В первой реакции интермедиатом является диэтилхлорфос-
фит (см. разд. 3.4 и 3.18.6). На стадии 1 амин и диалйАлфое»
фит в метиленхлориде прибавляют к двухфазной системе,
состоящей из дихлорметана и тетрахлорида или тетрабррмида
углерода, 20%-ного водного раствора гидроксида натрия .и ТЭБА
при комнатной или более" низкой температуре. Выходы
желаемых продуктов достигали обычно приблизительно 80—90%...Ал-
килирование на стадии 2 проводят в системе толуол/50%-ный
водный „NaOH/тетрабутиламмонийбисульфат (кипячение в течение
4 ч). Выходы продуктов большей частью колеблются между 6? и
98%. Конечная стадия 3 —гидролиз — протекает при растворении
амида в ТГФ, насыщенном газообразным НС1, и выдерживании
этой смеси при комнатной температуре в течение 12 ч [272].
В то же время доступные дифенилфосфиновыё амиды PttePONHa
легко дают продукты моно- и биеалкилирования в системе бен-
зол/конц. NaOH при использовании в качестве катализатора
Bu4NHSQ4. После гидролиза получают первичные или
вторичные амины [885]. Гидр азиды дифеншгфосфина были проалки-
лированы в условиях МФК с использованием в качестве
комбинированного основания твердых NaOH и Na2CO3 [I061]. При
алкилировании амида или анилида дифенилфосфина указан- -
ным выше способом вторичные галогениды даюТ главным обра-"
зом продукты элиминирования. Частично это направление
реакции можно подавить, используя систему твердый ЫаОН/КгСОз/
BU4NHSO4 в бензоле при 80 °С [1480]. При этом протекает
только моноалкилирование. Другая модификация метода позволяет
проводить все стадии «в одном горшке»: •'■:■ , )
—NH2 + CH3SO2C1 + ROH >■ Ph2PO—NHR
В этом случае в качестве основания также используюттвер*
дый NaOH/КгСОз [1663]. Если учесть легкость гидролиза
продукта и доступность исходных материалов, то этот метод,
несомненно, явится привлекательной альтернативой
классическому синтезу Габриэля.
Me3Si—N{Na)—PO(OEt)2 представляет собой замещенный
амид натрия, растворимый в бензоле, однако алкгогарование
этого производного в бензоле не происходит. Добавление
Bu4NBr позволяет провести эту реакцию при температуре
кипения. По-видимому, натрий в этом случае связан крвалентно и
потому нереакционноспособен; добавление ониевой соли
приводит к образованию ионной пары, которая и Подвергается алки-
лированию [1756].
Дифенилфосфониевые гидразиды также относятся к соед^
нениям, которые можно алкилировать, используя систему
твердый NaOH/K2CO3 [1061]. В ацетилированном продукте-вторая
связь N-H становится активной и способна к последующему-ал»
166 Глава 3. Практическое использование МФК
килированию. Гидролиз приводит к 1,2-диалкилгидразинам
[1755]:
PbaPO—NH—NH2 >- PhsjPO—NR1—NHa >-
PhaTO—NRi—NH—COCHg >■
Ph,PO—NR1—NR*—COCH3 >■ R*NH—NHR*
Ионообменная смола дуолит А-109, ТЭБА или BtuNI в
системе 60%-ный водный NaOH/CH2Cl2 служили катализаторами
для следующих реакций циклизации:
1) внутримолекулярной конденсации сс-галокарбоксамидов
R'CHX—СО—NHR2 с образованием дикетопиперазинов
(выходы 46—88%);
2) внутримолекулярного алкилирования бромкарбоксамидов
Br—(CH2)n—CR'Br—CO—NHR2 с образованием а-бромлак-
тамов (выходы 63—95%);
3) синтеза «в одном горшке» р-лактамов из а-метилча,р-дибром-
пропионилхлорида и фенилаланина (схема 3.53).
СН3 СООН СН3 о .
I I Br-J f СООН
ВгСНд—С-СО—a + H2N-CH—СН2—Ph > III
I • N CH-CH,-Ph (45-65%)
Нг *
Схема 3.53
Отклонимся ненадолго от предмета нашего изложения,
чтобы рассмотреть некоторые реакции N-ацилирования, в
которых реагент генерируется в ходе процесса МФК. Если к смеси
водного раствора солей щелочных металлов органических
кислот, амина и быс-(о-нитрофенил)фенилфосфоната в СНгС12
добавить при перемешивании небольшое количество BtuNX, то
экстрагирующиеся карбоксилат-ионы превращаются в
смешанный ангидрид, являющийся промежуточным продуктом. Он
взаимодействует с амином, образуя амид [1762]. Аналогично
можно получить р-лактамы из N-бензил-р-аминокислот, проводя
дегидратацию через промежуточное образование смешанных
ангидридов с метансульфонилхлоридами в системе водный
КНСОз/хлороформ/15 мол.% BtuNX. В этом случае выходы
продуктов достигают 60—80% без применения техники
высокого разбавления [1340] (схема 3.54).
При реакции амидов с дикетеном в толуоле в присутствии
четвертичных аммониевых солей с прекрасным выходом
образуются N-ацил-З-оксобутирамиды [1899]. Как показано на
схеме 3.54, раскрытие кольца лактона вызывается, по-видимому,
галогенидом. Образующиеся N-ацил-З-оксобутирамиды могут
3.8. N-алкилирование 167
затем реагировать с дикетеном в присутствии аликвата 336,
давая с высоким выходом Ы-ацил-3-ацетил-4-гидрокси-6-метилпи-
ридоны [1899]. Эти реакции проводят в гомогенном растворе.
Н
-CHS— CO2eNR*e R'-CH—CHS—CO-0-OS02CH3 R'
N
RCO-NH-CO-CH2-CO-CH3
R2/
CH3
Схема 3.54
Возвратимся теперь вновь к N-алкшшрованию в условиях
МФК. Являясь слабыми основаниями и нуклеофилами азириди-
ны с трудом алкилируются в обычных условиях, но легко
претерпевают перегруппировку, превращаясь в продукты с
открытой цепью типа Н. Если эти продукты перемешивать с бензил-
хлоридом или к-бутилхлоридом и ТЭБА в 30%-ном водном
NaOH в течение 6 ч при комнатной температуре или
соответственно 8 сут при 45 °С, то с количественным выходом
образуется I [259].
RNH—СН2—СНа
н
кшпализатор\11Х, NaOH
Схема 3.55
Инверсия у атома азота в азиридинах в растворе ускоряется
следами воды. Поскольку в системе азиридин/СНгСЬ/ТЭБА/
/конц. водный NaOH никакой инверсии не наблюдается, то,
следовательно, органическая фаза должна быть безводной [943].
3,5-Дициано-1,4-дигидропиридины К (R', R2, И8=алкил или
арил) можно проалкилировать с выходами 62—86% алкилио-
168 Глава 3. Практическое использование МФК
дидамй или бензилбромидом в толуоле или метиленхлориде, ис-
иользуя бензилдодецивдиметиламмонийбромид [260]. Без
катализатора такие реакции идут только в гомогенных растворах
КОН/ДМСО.
н
к
CN
R1
R«X. NaOH
катализатор
I
R*
Схема 3.56
Об алкилировании индола имеется много публикаций [261—•
264, 934, 935]. При работе без растворителя [261, 262] или в
бензоле [263] с концентрированным раствором гидроксида
натрия в качестве основания реакцию ведут при комнатной темпе-
ратуре-в; течение 6—22 ч или при 50 °С примерно 1 ч.
6TVCQ
N
Схема 3.57
Выходы L составляют 80—90%. В качестве катализаторов
использовались ТЭБА [261], аликват 336 [262] или Bu4NHSO4
[263]. Более активные галогениды и алкилсульфаты требуют
добавок только нескольких молярных процентов катализатора,
а вторичные бромиды, метилиодид и л-нитрофенилбромид —
молярных количеств катализатора. При использовании аллил- и
бензилгалогенидов доминирует образование побочных
продуктов Ми N. Более тщательное исследование обнаружило, что даже
при применении молярных количеств катализатора эти реакци-
онноспособные реагенты все же образуют небольшие
количества М и N [262]. Напротив, соединения типа L можно получить
и в. отсутствие катализатора при использовании двухфазной
системы; содержащей умеренное количество ДМСО или ГМФТ.
В этом случае реакция проходит более энергично и образуются
меньшие количества М и N [261]. Описаны также N-сульфо-
нирование [936] и N-ацилирование [1107] в условиях МФК.
Система КР/18-краун^ была использована для введения Z-за:
.. ;• -, . 3.8. Ы-ал^илироваяие 169
щитной группы в триптофан [921];. Тем же способом можно
провести N-алкилирование 2-амино?4.--хлор|5ензофенанов [1789]
и дифениламина [261]. Тетрабутиламмонийбромид и концент^-
рированный. гидроксид натрия оказались наилучшей.
комбинацией при алкилировании незамещенного имидазола (Р) и пираг
зола (Q) [274]. Максимальный выход при алкилировании Р
в растворе бензола (3 ч, кипячение) достигал 71%
при.использовании 1-бромбутана. и 76% в случае >бенаилхлоряда. В "к&г
честве алкилирующего агента был применен .также бромцикло:
пентан. Интересно отметить, что в начале реакции как. Р, так
и Q находятся почти количественно в концентрированном
растворе гидроксида натрия, однако в ходе реакции катализатор
экстрагирует небольшие количества депротонированного
субстрата для алкилирования.
В быстро растущем потоке1 литературы по N-алкилированию^
гетероциклов содержатся новые или дополнительные сведения;
о пирролах [261, 938, 1336, 15371, имйдазолах [1537, 1544], пи-
разолах [1251, 1255, 1387, 1537], бензимидазолах [1317, 1491,
1875], бензотриазолах [937, 1317, 1537], карбазолах [26!, 1537,
1877], индолах [1336, 1537], триптофане [1537], акридонах
[1262, 1876, 1877], аденине [1718, 1872], теобромине [1873],
пиримидинонах [1735,1874], пиридазиноиах [1879], 2-хлорфенотиа-
зинах [275, 279], 3,7-диметилксантине [1794], индено[1,2-<1]-
азепинонах [1708], 1,3-дигидро{2Н)-бензо[2,3-Ь]диазешшонах-
1,5 [1786], (5Н)-дибенз[Ь,1]азепине и его дигидропроизводных
[1655], 1,2,3,4-тетрагидро-2,4 диоксохиназолине [1270], хинок-
салино[2,3-Ь][1,4]бензотиазине '[1880] и фуранопирроле [1434].
Некоторые четвертичные соли изохинолин-4-карбоксамида
конденсируются в щелочных условиях МФВйг-давая
[1407].
Из замещенных 7Н'Пирроло[2,3^]шримдйинов ..и.
2,3,5-трм-0-бензил-В-арабинофуранозы или- О-рибез
МФК с 50%-ным водным NaOH. <5ыла. синт£3|фр?$ш«ршщ
нуклеозиды и аналоги нуклеозадов [1342^ 1386, :lj63(i ,Ш$, \Щ,
1792, 1S78]. Интересно отметить, дао яа «ooTH0W№8e-ifr
а-анойеров-могут влиять как тип и количество.МФ^атйди^т
ра, так и концентрация гидроксида натрия. Разбавление;^f
приводит к уменьшению общего выхода и возрастание доли
р-продукта [1871]. Изменение количества катализатора влщет,
по-видимому, как на скорость i$:«) -аномеризацгии, так и на
хкорость распада исходных соединений.
Необходимо упомянуть хотя бы вкратце об одном необычг
ном случае N-алкйлирования. Алкильные группы сухой алкилт
дифенилсульфониевой соли переносятся на амии в системе тверг
дая фаза/жидкая фаза [1596]
- + HNR2' + К2СО8 —* RNR2' + SPh2 +. Ш + КНСО,
170 Глава 3. Практическое использование МФК
В некоторых случаях внутримолекулярное * алкилирование
идет так легко, что реакция протекает без катализатора на
поверхности раздела фаз. Так, образование азиридина из R и S
наблюдалось при простом смешивании бензольного раствора
этих соединений с 50%-ным водным раствором гидроксида
натрия [265]. Такая методика представляет препаративный
интерес, так как в гомогенных метанольных растворах пиридина или
гидроксида натрия проходит элиминирование, приводящее к
образованию двойной связи или к последующему нежелательному
расщеплению азиридинового кольца.
Г J>N—СООМе
Химики-аналитики использовали метод экстрактивного ал-
килирования для метилирования микроколичеств соединений,
выделенных из биологических объектов, для газохроматографи-
ческого или масс-спектроскопического изучения, например нит-
разепама (Т) [266], хлорталидона (U) [267], барбитуратов
(V) фенобарбитоиа и пентобарбитона [268] и замещенных
5-арил-5-фенил-гидантоинов (W) [269]. При использовании в
качестве алкилирующего агента метилиодида и тетраалкилам-
монийгидроксида в качестве экстрагирующего агента и проти-
воиона образуется смесь от моно- до полиалкилированных
производных.
Наибольшее влияние на скорость реакции оказывает липо-
фильность аниона и катиона. Поскольку скорость стадии алки-
лирования, протекающей в органической фазе, очень велика, то
общая скорость реакции зависит главным образом от
равновесия процесса экстракции ионных пар. В случае фенобарбитона
(V, R=CeH5) при проведении реакции в системе фосфатный
буфер (рН 10)/CS2 и 25 °С с тетрабутиламмонием в качестве ката-
3.8. N-алкилироваяие 171
Vnh
A I
^J У=О Ar—SO»—NHR
W
Схема 3.69
лизатора реакция проходит на 10% за 60 мин, с
тетрапентиламмонием за то же время реакция проходит полностью, а при
использовании тетрагексиламмония время, необходимое для
получения количественного выхода в этих же условиях,
составляет всего 5 мин. При алкилировании более липофильного иона
пентабарбитона [V, К=СН(СНз)—СН2—C2H3] для достижения
100%-ного выхода за 15 мин достаточно взять тетрапентиламмо-
ний [268]. Аналогичная техника экстракции ионных пар была
использована для аналитического определения сульфоиами-
дов X [270] и теофиллина Y [271] в форме пентафторбензиль-
ных производных. Другие примеры экстрактивного алкилирова-
ния лекарственных веществ см. в [939, 983, 1052].
Реакции, которые формально можно рассматривать как «ал-
килирование» вторичных и первичных аминов, а также
гидразина дихлоркарбеном, приводят к формамидам, изоиитрилам и
диазометану соответственно. Оаи обсуждаются в разд. 3.20.2,
a N-, О- ила S-алкилирование—в разд. 3.10.
Типичные экспериментальные методики приводятся ниже*
Алкилирование щетанилида
А. Молярное количество катализатора: 0,1 ноля тетрабутил-
аммонийбисульфата растворяют в 100 мл 2 М NaOH. К этому
раствору добавляют 0,1 моля ацетанилида и 0,18 моля метилио-
дида в 100 мл СН2С12. Смесь кипятят 30 мин, после разделения
двух фаз органический слой упаривают и к остатку
прибавляют 100 мл эфира, что приводит к выпадению в осадок тетрабу»
тиламмоиийиодида, который затем отфильтровывают. После
172 Глава-3. Практическое использование МФК
удаления растворителя получают К-ацетил-М-метиланилин с
выходом 90% [255а].
Б. Небольшое количество катализатора: смесь 0,02 моля
ацетанилида, 0,001 моля ТЭБА, 50 мл бензола, 0,1 моля NaOH
и 4 мл воды энергично перемешивают при комнатной
температуре до образования пасты. После этого нагревают до 60—
70 °С, -прибавляют по каплям 0,04 моля и-пропилбромида и
смесь перемешивают 200 мин. За это время паста растворяется.
Органическую фазу отделяют, промывают 2 н. НС1, водой и
высушивают, после удаления растворителя вещество перекристал-
лизовывают; т. пл. 45—48 °С, выход 82% [257].
Алкилирование простых амидов: щелочная система NaOH/KzCOz
Раствор 0,075 моля алкилгалогенида в бензоле добавляют
по каплям при перемешивании к кипящей смеси 0,05 моля
N-моноз вмещенного карбоксамида, 7 г тонкоизмельченного
гидроксида натрия, 14 г карбоната калия и 0,005 моля тетра-и-
бутиламмонийбисульфата в 50 мл бензола. После кипячения в
течение 4 ч смесь.обрабатывают водой. Выход 73—96% {1374].
3.9. С-алкилирование активированных связей С—Н
Алййлирование карбанионов представляет большой интерес
для {органического синтеза, поэтому среди МФК-реакций Оал-
килнроВанйе Наиболее изучено. В 1951 г. Жаррусс [224]
обнаружил,- чТ<Х фйшлацетонитрил можно проалкилировать этил- и
бензилЭшорядами в; присутствии концентрированных водных
щелочей; й^ТЭБ А, Макоша расширил область применения этой
реакции, оптимизировал; условия, а.также дал ее общее обоснова-
ниег БШда показано,- что метод МФК имеет- много преимуществ
шу сра!*нёйвд© i более обычными методиками, которые требуют
не ТоЖк©*'дорогих" основавши (амида натрия, трег-бутоксида
ка&йя, ййрдцов эдетадлев, -трифенилметидов и др.)., но также
и безводных растворителей (абсолютного эфира, бензола, ди-
ме*йлсулШфбксида,лдйЖетйлформамида и др.): Во многих,
случаях МФК проще, чем другие методики. При использовании
МФК реакции идут с высокрй селектярностью и приводят к
получению более чистых продуктов .с хорошими выходами. Для
этих реакций в качестве катализатора* таще всего яслользует-
ся.ТЗБА.
* В -качестве катализаторов можно использовать- и другие ониевые соли.
Jtfio широко. изучено г[9971 также влияние "изм^нениаГуслЦвий. реакции. При
^пользовании обычных анионообменных смой дЛй1 тр'ехфязного катализа
реакции-шли существенно медленнее [958]
3.9. С-йлкпЛирование активированных связей С—Я 173
3.9.1. Алкилирование арилацетонитрилов
Алкилирование фенилацетонитрила в системе этилбромид/
/концентрированный водный гидроксид натрия/1 мол.% ТЭБА
•описан в «Органических синтезах» [280].
Реакция проходит слабоэкзотермично и при 28—35 °С
заканчивается за 3—5 ч, выход продукта алкилирования
достигает 78—84%. С целью облегчить отделение продукта реакции
от непрореагировавшего исходного субстрата продукт
превращают в а-фенилциннамонитрил, добавляя бензальдегид в
реакционную смесь после того, как реакция в основном закончится
[280]. По этой общей методике было получено
большое--количество различных замещенных арилацетонитрилов (см. табл. 3.8
и схему 3.60). Механизм этой реакции был подробно изучен.
При использовании обычных методик из соединений с
активной метиленовой группой обычно образуется смесь продуктов
моно- и диалкилирования. Однако в условиях МФК
концентрация ионных пар карбанионов в органической фазе мала,
поскольку мала концентрация катализатора. В тех случаях, когда
PhCH2CN является более сильной кислотой, чем Ph—CH(R) —
—CN (как чаще всего бывает), диалкилирование не идет и
стадия моноалкилйрования проходит селективно. Однако из
экспериментальных данных следует, что низкомолекулярные и реак-
ционноспособные алкилирующие агенты (например, аллил-- и
бензилгалогениды) имеют тенденцию образовывать
относительно больше продуктов диалкилирования. В тех случаях, когда
желательно именно это направление реакции, ее рекомендуют
проводить при более высоких температурах [283]. При О^С
бензилирование бензилцианида в присутствии различных
катализаторов дает моно- и биспродукты в соотношении от 3 до. 6
(1 ч, конверсия 40—60%) [852]. При метилирований о-, м-,
л-метилфенилацетонитрила метилиодидом м- и л-изомеры
претерпевают диалкилирование и лишь о-соединение даегг только
продукт моноалкилйрования [1732]. Макоша и сотр. [13J2J
разработали легкий метод для очистки моноалкилфецилацето-
нитрилов: эти вещества вводят в реакцию с винил ацетатом в
условиях МФК и получают соединения типа PhCR(CN)-^-
—СН(СНз)—О—СОСН3. Гидролиз и расщепление этих
соединений карбонатом натрия в водном этаноле (которое проводится
после отделения их перегонкой от PI1CR2CN) приводят к
чистому PhCHRCN.
При использовании большого избытка концентриршанногр
раствора гидроксида натрия алкилбромиды, как правило,
являются болеае эффективными алкилирующими агентами, few
хлориды [281, 282, 285]. Иодиды и в некоторой степени бромй1
ды (при меньшем избытке NaOH) отрицательно влияют на ал*
Ta6JfHS»3.8.
2
Субстрат
АяснЯВртющЖГ artHf
Продукт
Выход.
Литература
Ar©H(R)-CI*
№ » Й, алкия>
RX
X—(CH,)»— X
CHi)»—CH—Ph
€3N
CN
Щ
CN
(СНЙ.-Х
78^-84
в, б
>90
93
280
281-283, 285, 308, 314,
ЗГ5, 1343, Г54Э
284
286
286
Ph—CH(R)—CN
Cl- (CH,)»CN
PhCH(R)—CN
PhCH,CN
PhCH(R)—CN
PhCH,CN
PhCHaCN
PhCH(R)CN
Ph-CR-CN
(СНЛ.--СЫ.
HN
70-90
288, 774
XCH,
NO,
XCH,—CR=CR'R'
CICH(R')—OR"
CICH(R')—OR"
X—C««— COOR'f
X—CH,—COOR.'1"
CH,—CR-CR'R"
Ph—CR—CN
R—CH—OR"
Ph—CH—CH—CH—Ph
CN R' CN
CHaCOOR'
Ph—CH^CN + Ph—C—CN
Sfr-98
25-60
7-95»
68-88
I
CH,—COOR1
Ph—CiH-CN
)R' C!
Hr-COOR*
26-95»
290
291
291
294
292
Продолжение табл. 3.8
Субстрат
Алкилирующий агент
Продукт
Выход, %
Литература
Ph—CH—(CHa),NR3 R'X
CN
PhCH2CN
PhCH(R)CN
Ph—CH—CN
,NCOPh
H' "CN
Ph—CR—(CH2)nNRa
CN
Ph CN
R—N(CH2CH,Cl)a
(R =алк.ил,тозил)
•-N-
I
R
Вг(СН2)аС(СН3)^О2 Ph—CR—CN
(CHS)2C(CH3)2-NO3
R
RX
RX
Ph—CR—CN
i-C-OR'
CH3 R"
_N—COPh
R' CN
34—88a
30—68
20—80
a, б
76—82
293
297, 317
309
301
295-320
310, 1068
PhCH(R)CN
PhCH(R)CN
Ar—CH(R>—CN
R'SCN
Ph—CR—CN
SR'
Ph—C(R)—CN
45-77
300
58—67
(7-11).
303—307, 513
330
I Зависит от заместителей и условий.
'Обычно высокий.
Для R-H, л-1, 2.
т~Я'-трет- или втор-алкил.
в
178 Глава 3. Практическое использование МФК
килирование [281], поскольку иодид экстрагируется в
органическую фазу лучше, чем анион фенилацетонитрила. Поэтому
применение иодидов не рекомендуется в тех случаях, когда
используются каталитические количества ТЭБА. Чтобы избежать
этого, Брендстрём и сотр. [321] при алкилировании алкилио-
дидами вводят молярные количества тетрабутиламмониевых
солей. Как и ожидалось, в обычных реакциях с применением
ТЭБА вторичные галогениды реагируют медленнее и дают более
низкие выходы, чем первичные галогениды [283, 285].
Растворители тормозят реакцию; спирты более или менее ингибируют
ее, а если гидроксид натрия заменить на гидроксид калия,
реакция вообще не идет [281]. Хотя некоторые реакции проводятся
при 60—80 °С, гидролиз нитрильных и эфирных групп почти не
происходит, и только при температурах около 90е он становится
заметным.
Ph—CH—CN
I
R—СН
Ph—CH—CN
Ph—CH—CN
(СНа)„—CN
-CN
(СНа)»—CN
I / С1СНа—COOR \х-(СНЛ.—CN
(СНа)^
Ph-CH-CN Ph-CH^CN
(СНа)„ H.C-COOR j
+ (СНа)„—CN
НаС—COOR
I
Ph—С—CN
I
НаС—COOR
Схема 3.60
Недавно были осуществлены алкилирование и ацилирование
в бензольном растворе в присутствии системы твердый К2СО3/
/дибензо-18-краун-6 [1048].
На предыдущей схеме показаны различные типы
соединений, пригодные для МФК. Во многих реакциях были использо-
3.9. С-алкилирование активированных связей С—Н 17&
ваны простые алкил-, аллил- и бензилгалогениды [280—285,
290], а также фенилацетонитрил, алкилфенил- или дифенилаце-
тонитрилы. Можно использовать также циклы, имеющее фе-
нильный заместитель [301, 314, 315, 318]. При реакциях с дига-
логенпроизводными соединениями могут быть получены гомо-
циклические [286] и гетероциклические [297, 317] кольца. Ал-
килирование я-нитробензилхлоридом в присутствии
относительно разбавленного раствора гидроксида натрия и четвертичного
аммониевого катализатора проходит (по крайней мере
частично) по радикальному механизму, так как было доказано
существование промежуточно образующихся радикалов [319].
При использовании концентрированного раствора гидроксида
натрия и ТЭБА замещение является основным процессом для
о-, м- и я-нитробензилхлоридов, хотя наблюдается также
образование динитростильбенов — типичных продуктов рекомбинации
радикалов [289, 308]. В качестве алкилирующих агентов были
использованы также галонитрилы [287, 288], трет- или втор-
алкилгалоацетаты* [292, 294], многие другие замещенные га-
лоалканы [297, 309], 9-хлорфлуорен [1066] и даже та-хлорэфи-
ры [291].
Последние дают простые продукты замещения с алкилфе-
нилацетонитрилами, замещенными глутаронитрилами и с фенил-
ацетонитрилами [291]. Аналогично реагируют а-диалкиламино-
[293] и сс-алкоксифенилацетонитрилы [295, 320], реакция
облегчается при добавлении ДМСО.
PhCHO
R1
О—С—СН,
OR»
R3XT3BA> Ph-C-R3
NaOH
N—COPh
I
CN
Метиловые я этиловые эфиры слишком быстро гидролизуются.
ft80 Глава:S. Практическое использование МФК. ' ." :*,
Поскольку исходные вещества можно легко, получить из
циангидрина бензальдегида, то приведенная выше на схеме щ>-
следовательность реакций представляет собой, йо. существу,
синтез арилкетонов из. бензальдегида [295, 320] или из других
ароматических альдегидов.
Показано, что в двухфазной системе соединения Рейссерта
реагируют с выделением тепла с алкилбромидами, бензилхло-
ридом [310, 1068] и с 9-хлоракридином [870]; образующиеся в
результате аддукты затем превращают в изохинолины..
Поскольку карбанионы являются мягкими основаниями, тиоциана-
ты. предпочтительно атакуют их атомом серы. Именно поэтому
тиоалкилирование фенил ацетонитр ил ов алкилтиоцианатами
действительно наблюдается (хотя это и удивительно на первый
взгляд) в условиях межфазного катализа [300]:
«1—CH(R)—CN + R SCN
Ph—CH2CN 4- NCS—CH2—CH2—SCN
Схема S:62
Обычным катализатором, применяемым в этой реакции,
является ТЭБА. Следует отметить, что можно использовать также
дибензо-18-краун-6 [302]. Хотя во многих случаях он не имеет
явных преимуществ, однако он оказался более селективным
катализатором при синтезе Ph2C(CN)—СН2—СН(СН3)'—NMe2.
Это соединение — важный промежуточный продукт Метадона —
обычно образуется В—-форме смеси изомеров Pli2C(CN)—
—СН(СН3)—СН2—NMe2 в соотношении 1:1. Если же в
качестве алкилирующего агента используется С1СН(СН3)—СН2—
—NMe2, то соотношение изменяется до 72:28. Для нормального
алкилирования проведено сравнение ряда катализаторов и
установлено [299], что ТЭБА и тетрабутиламмонийбромид
являются лучшими катализаторами, чём гексадецилтриметиламмо-
вийбромид, трибутилгексадецилфосфонийбромид или смола
дауэкс 1 (трехфазный катализ). Эпизодически используют
специально приготовленные твердые.чкатализаторы, нанесенные на
полимеры. Недавно было найдено [312], что 2-диалкиламино-
пиридинийтетрафенилбораты (А) являются эффективными
катализаторами. Даже триаякилам^ины могут быть катализатора-
g.P. С-алкилирование активированных, связей С-^И 3BS
мп при диалкилировании «в одйом горшке», поскольку избыток
RX превращает их в истинные катализаторы £1069].
А
Схема S.63
Для понимания механизма очень важным является тот факт,
что очень активные алкилирующие агенты (например, бензил-
хлорид) реагируют с фенилацетонитрилом даже в отсутствие
катализатора, хотя реакция идет и намного медленнее, чем в
условиях МФК- При повышенных температурах (80" °>С) алкилио-
диды также реагируют довольно быстро без катализаторов
[298]. Эти наблюдения, как и результаты конкурентного алки-
лирования, указывают на важную роль поверхности раздела
фаз при алкилировании. [298J. Работы по энантйоселективному
алкилированию фенилацетонитрилов с хиральными
катализаторами рассмотрены в разд. 3.1.5. Применение
фенилацетонитрилов для нуклеофильного аррмэтического замещения описано в
разд. 3.17. *'"' '•'" J
3.9.2. Алкилирование других активированных цианатов
„. и изоцианатов
Алифатические" нитрилы, которые не имеют активирующих
rpynrt, не алкйлйруются в условиях МФК. Однако-фенилтие^-
группы в соёдмениях типа нитрилов $>-фенилти6гликоля В
"{296]t,\ аналогичных селенопроизводных [876] или в дитйо-
карбаматах типа С [322] достаточно активны, что позволяет ал-
килировать йк нри- коА1натной температуре. Соединения С
являются промежуточными в новом синтезе кетонов [322], а селе-
новйе производные можно превратить в о^р-ненасыщенные
нитрилы обработкой" N-хлорсукцинимидом во влажном ацетонит^
риле илиН2О2в ТГФ/вода [876]. ■
- Еще более активными являются нитрилы, 'несущие фосфор-
ноэфйрные (Щ или фосфорноамидные (4Е) заместители.
Каталитическое алкилирование этих соединений с иодйдами в
качестве алкилирующих агентов было проведено в дихлорметане при
45 °С (1 ч, выходы 30—80%) в присутствии молярных количеств
тетра-«-бутиламмонийбромида [323—325]; в случае алкилбро-
мидов используют 5—10 мол.% ТЭБА [325]. В цианоуксусный
эфир можно ввести нитроарильную группу (ТЭБА, п-хлорнит-
182 Глава S. Практическое использование МФК
NC—CHa—SPh * NC—CH—SPh > NC—C—SPh
i-
s s
NC—CHa—Cl + NaeS©—C—NMe, * NC—CHa—S—C—NMe2
С
R1 S R1 S »
I II I II «a n он \
С »■ H—С—S—С—NMea >- R2—C—S—C—NMe» *; J.go > C=O
CN CN
CHa—R1
NC—CH2—SePh > >■ NC—C—SePh
Ra Ra
COOMe
(RO)—P—CHa—CN (RaN)a—P—CHaCN NBu«eeCH^
D E CN
F
Схема 3.64
роарильное соединение) [303]. Брендстрём выделил тетрабутил-
аммониевую соль метилцианоацетата (F) экстракцией
растворителем и затем проалкилировал ее в хлороформе. Эти реакции
заканчиваются в течение 10—120 мин при комнатной
температуре. В отличие от приведенных выше примеров в данном
случае была выделена смесь исходного вещества и продуктов мо-
но- и диалкилирования [326]. Малодннитрил, по-видимому, дает
исключительно продукты диалкилирования даже с
каталитическими количествами аммониевых солей [4, 38]. Из тозилацето-
нитрила -в условиях МФК получают смесь моно- и диалкилиро-
ванных продуктов. В реакции с 1,8-диазабицикло [5.4.0] ундеце-
ном в гомогенном бензольном растворе был обнаружен только
моноаддукт [888]. Алкилирование 1,2-дибромэтаном как мало-
динитрила, так и цианоацетата в концентрированном растворе
NaOH с использованием молярного количества ТЭ.БА дает циа-
ноциклопропан-1-карбоновую кислоту [329]:
•* < NC—CHa—COOR
СООН
Схема 3.65
3.9. С-алкилирование активированных связей С—Н 183
Легкодоступные шиффовы основания Ph2C=N—CH2COOR
и РЬгС=Ы—CH2CN алкилируются по простой методике МФК
(10%-ный NaOH, BU4NHSO4 для эфира; 50%-ный NaOH, ТЭБА
.для нитрила) после гидролиза получают аминокислоты [1070].
Обычный синтез u-меркаптоалкановых кислот включает МФК-
алкилирование S-цианометилтиокарбаматов или дитиокарбама-
тов (NC—CH2S—СХ—NR2, X=O, S) и последующий гидролиз
[1071].
Химическая модификация хлорметилированных и цианоме-
тилированных полистирольных смол была проведена реакцией
соответственно с малодинитрилом или хлорацетонитрилом по
МФК-методике в присутствии щелочи [1072].
Только недавно было изучено алкилирование в «-положение
к изонитрильной группе. Для этого необходимо наличие в
молекуле активирующей группы. В системе ТЭБА/конц. NaOH с
G реагируют только аллил- и ' бензилгалогениды. Последний
дает дибензильный продукт [327]).
C=N— CH2— COO—mpem -Bu
НзС
—n=c.
н
Схема 3.66
В то же время более активированный субстрат Н реагирует
даже со вторичными хлоридами при использовании в общем-то
неподходящего катализатора — тетра-я-бутиламмонийиодида
при б°С [328]. При более высоких температурах начинается
диалкилирование. По другой методике Н, СБг, 10%-ный NaOH
и молярное количество тетрабутиламмонийбромида образуют
соль, структура которой показана ниже. Эту соль можно затем
проалкилировать и получить с прекрасными выходами б-алкил-
тио-4-тозил-1,3-тиазолы [431]:
CS3, HCCI3, NaOH ?
Вц^Вг
н
Tos—С—N=C
NBu«e
L s4
Tos
Tos
Схема 3.67
3.9.3. Алкилирование малонового эфира
Еще в 1954 г. Бабаян и сотр. [340] проалкилировали этил-
малонат замещенными алкилхлоридами в присутствии 10%
четвертичных аммониевых соединений и 40%-ного водного гид-
роксида калия.
184 Глава '3. Практическое использование МФК
Потенциальные возможности, заложенные в этом.
эксперименте/оставались скрытыми многие годы. Позднее Брендстрём
использовал свой метод экстрактивного алкилирования (мо>
лярные количества или небольшой избыток тетра-к-бутиламмо-.
нийбисульфата, 2 моля 2 М NaOH, СН2С1г) для алкилирований1
малонового эфира алкилиодидами [321]. Обычно (за исключен
нием алкилирования метилиодидом) наблюдается только моно?
алкилирование. При использовании метода Макоши (ТЭБА|
/конц. NaOH) реакция между третбутилмалонатом и этилбро-
мидом, бензил- и аллилхлоридом идет экзотермически [338]т
При. использовании эквимолярных количеств реактантов образу*
ется смесь моно- и дизамещенных продуктов. Менее активный
алкилирующие агенты, например бутилбромид, дают более
высокие выходы, если в реакционную смесь добавить ДМСО1
Применение избытка реагента позволяет проверти двойное ал*
килирование. С а,<в-дибромалканами. образуются как трех-г таги
и пятичленные циклы с выходом.90 и 75% соответственно [338}l
Этилмалонат и 1,2-дибромэтан дают циклопропан-1,1-дикарбо-
новую кислоту. В присутствии молярного количества ТЭБА
идут одновременно двойное алкилирование и гидролиз [329].
Аналогично реакции с фенилацетонитрилом, рассмотренной
выше, происходит нитроарилирование малонового эфира
замещенными л-нитрохлорбензолами (ТЭБА/конц. NaOH) [303].
В последних публикациях предлагается использовать твер»
р,ые карбонаты щелочных металлов с ониевыми солями в ка^
чёстве катализаторов [1048] и применение метода экстрактна?
ного алкилирования для получения комплексов с малонов*'
Эфиром [1073]. Опубликованы сообщения о синтезе амино*
лот с помощью, алкилирования ациламиномалоновых э<~
(твердый К2СО3, 18-краун-6 или R4NX) [1585], о химическр!
модификации сшитого хлорметилированного полистирола npi
реакции с анионами цианоуксусного эфира, малодинитрила йщ
малонового эфира [1259] и о получении О, S-диэтилбромметйл^
тиомалоната посредством двухкратного применения—метода
экстрактивного алкилирования сначала метилиодидом, а затем
дибромметаном [1699]. Тозил- и 2,4-динитрофенилгидразонШ
подвергаются а-бромированию фенилтриметиламмонийтриброт
мидом в ТГФ и затем взаимодействию с системой
порошкообразный NaOH/Bu4NHSO4/MaflOH0Bbm эфир, давая соединения
указанного ниже типа. Обе стадии реакции проводятся «в одном
горшке» [1231]:
RXCH2—C(R2)=N—NHX >■ RiCHBr—C(R2)=N—NHX »-
: —* (H6C2OOC)2CH—CHR1—C(R*)=N—NHX
. Алкилирование ацетоуксусных эфиров и родственных соеди-*
нений рассмотрено в разд. 3.10.
3.9. С-йАкиЛирование активированных связей С—Н 185
3.9.4. Алкилирование бензилкетонов, арилуксусных эфиров
и амидов
Все реакции, рассмотренные в этом разделе, сведены в
табл. 3.9. Хотя енолятные анионы могут в принципе
подвергаться как О-, так и С-алкилированию, обычно протекав* только
С-алкилирование. Так, при использовании метода Брендстрёма
(1 моль катализатора) фенилацетон реагирует с метилиодидом,
давая 92% моно-С-продукта [321, 1075]. При использований
метода Макоши с ТЭБА и различными алкил-, аллил- и бензил-,
галогенидами высокие выходы монопродукта получают только
в том случае, если алкилирующий агент берут с небольшим из'т
бытком. Применение большего избытка (2,5—3 моля) приводи^
к дизамещению. Для изучения механизма реакции интересен
тот факт, что с алкил- и бензилгалогенидами медленная
реакция проходит даже в отсутствие ТЭБА [331]. Это является
серьезным доводом в пользу протекания реакции на прверхно-
сти раздела фаз. Кроме того, было найдено [432], что многие
первичные, вторичные и третичные амины катализируют
алкилирование фенилацетона бутилбромидом и другими алкилгало-
генидами в присутствии 50 %-ного водного гидроксида калия или
твердого КОН. Это возможно благодаря тому, что в условия^
реакции in situ может происходить образование четвертичных
аминониевых солей, которые и являются истинными катализа^
торами [432]. Некоторые амины не проявляют каталитической
активности. К ним относятся амины, у которых атом азота сте*
рически затруднен, или амины со структурой анилина! или Щг
ридина. То, что трибутиламин не катализирует бутилирование
фенилацетонитрила [433], объясняется чрезвычайно |мягкими
условиями проведения реакции. Кватернизация, приводящей к
тетрабутиламмонийбромиду — «активному» катализатору,в этих
условиях не протекает. Однако при 100 °С реакция с т|)ибутйл-
амином идет [432]. 1,2-Дибромэтан и 1,4-дибромбутан: дают в
качестве основных продуктов 1 или К и L соответствен^). Одра-
ко с 1,3-дибромпропаном образуется смесь продуктов,
Доказанная ниже [332] (схема 3.68). ? ;
Соединение М является продуктом О-алкилирования. При
использовании изопропилбромида и дезоксибёнзоина также
образуются продукты С- и О-алкилирования — соединения N и Р
в соотношении 2,3: 1 [333]. Аналогично изопропилирование т|ет-
ралона дает почти равные количества О- и С-продуктрв [335],
а из дифенилацетальдегида образуются только О-производные
[1048]. В катализируемой ТЭБА реакции между бензилкетона-
ми и а-хлорэфирами О-алкилирование является основным
процессом, а продукты С-алкилирования выделить не ; удалось
[337]. трет-Бутиларилацетаты менее реакционноспособны, чем
Таблица Э.9. Алкилирование бензнлкетонов, аридукеусвых эфиров и амидов в условиях МФК
Субстрат
Алкнлврующий агат
Продукт
Выход,
Литература
Ph—СНа—СОСН» СН,1
Н
Ph—СНа—COR
Ph—CHr—COR'
Ph—CH2—COR
RX (1 моль)
RX (избыток.)
х-(сна).-х
я - 1-4
СНэ
Ph—СИ—СОСНз
Ph—CH—COR
Ph—CH—COR'
Ph—CHa—COR
Ph—CH—COR
I
(CHa).
Ph—CH—COR
Ph—CH—COR
(CH2),-X
Ph—CR—COR
I
Ph-CHR-COR СМсН*)а-СООСН(СН,)г (CH?), COOCH(CH3)a
92
44—72
77-94
a
68—92
321
330
331, 333, 334
332-334, 903
333
RX
Ph—CH,—COR CICH,OR'
Ph—CH—CO—mpta-Bu R'X
Ph—CH=C—R
I
O—CHaOR'
R' О
Ph—С—СО —/я/нзд-Bii
SO (R-алкил)
80—90 (R-бензил,
аллил)
25—60
45-706
50-89
334, 335
334, 336
337
338
339
1740
OR О
OR О
Продолжение табл. 3.9
Субстрат
Алкилярующвй агент
Продукт
Выход, %
Литература
N
R
PhCHjX
СООМе R'X
R'X
CHj-CH—СН2Вг
т
R
R
Q V-С—СООМе
R'
xf-
54-72
73
1ООВ
100е
69—96
339
341, 930
1492
Выходы зависят от условий.
6 Для получения хороших выходов необходимо добавление небольших количеств ДМСОк реакционной смеси.
в Комплекс разлагается солью церия.
3.9. С-алкиларование активированных связей С—Н
PhCH—<СН2)«—СНР>
COR COR
PhCH—COR Ph—CH—COR Ph—CH-<CHS)3—CH—Ph
(CHa)3Br H2C—CH=CHa COR .COR \ - /'
M
Ph—СНа—COPh
Ph—CH—COPh Ph—CH=C—Ph
I + I
HC(CH3)a OCH(CH3)2
N
* Схема 3.68
О
соответствующие арилацетонитрилы. Для получения хороших
выходов необходимо добавлять небольшие количества ДМСО
[338]. Чтобы избежать гидролиза сложных эфиров, следует
использовать объемистые трег-бутильные группы. Метиловые эфи-
ры можно применять в том случае, если кислотные свойства
активной метиленовой группы усилены. Для этого, например,
подходит образование л-комплексов типа арен-Сг(СО)3, в которых
атом металла является электроноакцептором [341, 930]. Эти
комплексы получают при кипячении с гексакарбонилом хрома.
После почти количественного алкилирования (СН2С1г или
СбНб/цетилтриметиламмонийбромид/50%-ный NaOH, 1,5—3 ч
при комнатной температуре) комплексы можно легко разрушить
солями церия [341, 390].
Для синтеза 3-алкил-, 3,3-диалкил- или - нитрофенилсоедине-
ннй из Ы^замещенных оксиндолов были использованы ТЭБА
и ДМСО в качестве сокатализатора [339, 1074]. Алкилировач
кие. в условиях МФК комплекса гетероар ил-уксусный эфир
является одной из стадий нового элегантного метода. синтеза
алкалоидов [1076]. .''?--»
190 Глава 3. Практическое использование МФК
Во всех реакциях, которые мы до сих пор обсуждали, в
качестве катализаторов использовали аммониевые соли — чаще
всего ТЭБА. Недавно было показано, что 2-диалкиламинопири-
диниевые соли являются также активными катализаторами,
хотя они и не превосходят ТЭБА. Для катализа были
использованы также краун-эфиры [45] и замещенные азамакробнцикличе-
ские полиэфиры [48а]. Однако их преимущества сводятся на нет
вследствие их высокой стоимости.
-3.9.5. Алкилирование других соединений,
содержащих дважды активированные СН-связи
р-Кетосульфоны, формула которых приведена ниже, легко
экстрагируются в форме ионной пары с различными противоио-
нами, например с тетрабутиламмонием. Из этих ионных пар
•были получены продукты моноалкилирования двумя
способами: 1) ионные пары выделяли и заменяли дихлорметан,
служивший экстрагирующим растворителем, на этилацетат; 2)
проводили непосредственно экстрактивное алкилирование (метод
Брендстрёма). Выходы большей частью превышали 70% [355].
АгСО—СН2—SOjjR RCH2—CO—СНг—SO2Ph
В трехстадийном синтезе 1,4-дикетонов промежуточно
образующиеся р-кетосульфоны алкилировали замещенными фена-
дилбромидами, как это показано на нижеследующей схеме
[342]:
О
R»COOR + Н3С—SO,—СНэ *■ R1—С—CH,—SO,—CH, ►
V
О СНа-С-Аг О О
Ar—CO—CH,—Br
RS—СНа—С—Ph
р
Схема 3.69
Ионные пары, образованные р-кетосульфоновым анионом и
тетрабутиламмониевым катионом, экстрагировали в хлороформ
и перед реакцией алкилирования высушивали. Однако
необходимость этой меры предосторожности в данном случае не
очевидна [342]. Выходы продуктов алкилироваиия составляли 63—
«0%.
3.9. С-алкилирование активированных связей С—Н 191
Другим классом соединений, которые можно легко алкили-
ровать в присутствии ТЭБА, являются со-сульфиды ацетофено-
на (Р) [343]. В зависимости от типа алкилирующего агента и
радикала К изменяется выход продуктов О-алкилирования. С
менее реакционноспособными алкилгалогенидами образуются
сложные смеси. В последнем случае сокатализатором являлся
ДМСО [343].
Аналогичным образом, используя систему ТЭБА/конц. NaOH,
можно проалкилировать галогенметиларилсульфоны (Q),
а также соединения типа R [323, 344, 1077]:
Ar—SO2—CHSX (EtO)2PO—CH2—COOEt
(Х = С1, Br)
Q R
Препаративный интерес представляет алкилирование
этилового эфира 3-метил-4-бензосульфонилбутен-2-овой кислоты с
помощью 1-бром-3-метилбутена-2 или 1-бром-3,7-диметилокта-
диена-2,6 в системе CH2Cl2/NaOH/T3BA [906].
Восстановительное десульфурирование продуктов алкилирования является
удобным способом получения терпенов:
СНз
PhSOa—СН2—С=СН—COOEt »• PhSOa—CH—C==CH—COOEt
СН,
R—CHa—C=CH—COOEt
Схема 3.70
Метод МФК позволяет алкилировать р-фенилсульфоиилэфи-
ры почти количественно [1078].
Алкилирование хлорметансульфонамидов идет с выходами
55—88%, если наряду с Bu4NBn и 50%-ным NaOH
присутствуют небольшие количества ГМФТА [1087]. Связи СН в
активированных «- и "у-метильных группах пиридиниевых солей также
могут быть проалкилированы с помощью МФК- Примером
может служить превращение 1,2,4,6-тетраметилпиридинийиодида
в 4-71ре7'-бутил-2,6-диизопропил-1-метилпиридинийиодид при
действии метилиодида (выход 20%) [1979].
Алкилирование аллилсульфонов, бензилсульфонов и амидов
бензилсульфоновых кислот в системе RBr/Bu4Br/50%-HHfi NaOH
идет медленно. Этот процесс сильно ускоряется в присутствии
небольших количеств ГМФТА и при 35—60 °С за 2—5 ч прохо-
192 Глава 3. Практическое использование МФК
дит с выходом 56—90% [1266, 1840]:
С„Н6—CHR»—SO2X —
(X = Rs или NR2s) .
PhCH2NH2 > PhCH2—N=CHPh >■
PhCHR—N=CHPh v PhCHR—NH2
Описаны две группы методов ia-алкилирования бензиламина,
'6н превращается в бензилиденовое производное, которое можно
депротонировать и алкилировать в МФК-системе (выходы до
■95%). Последующий гидролиз дает алкилированный продукт.
Можно также провести присоединение в условиях МФК к акри-
лонитрилу или стиролу с образованием пирролидинов [1588,
1715].
Классическим методом с NaH в качестве основания был
проалкилирован этиловый эфир 1,3-дитиан-2-карбоновой
кислоты— реагент, рекомендуемый для получения а-кетоэфиров.
Это вещество было получено в одной из первых МФК-реакций
улучшенным способом из пропандитиола-1,3 и дихлорацетата
(К2СО3, аликват 336, толуол, комнатная температура, выход
"57%) и алкилирован в той же самой системе при 60 °С (выход
37—81%) [1559].
3.9.6. Алкилирование простых карбонильных
и карбоксильных производных ■-
Реакции этого типа .в условиях межфазного катализа
провести намного труднее,"чем- реакции, рассмотренные до сих пор:
во-первых, в этих соединениях СН-связи существенно менее
активны, вследствие чего необходимо проводить процесс при
более высоких температурах или увеличивать время реакции;
во-вторых, конкурирующая реакция альдольного типа может
•стать основной. Поэтому алкилирование простых алифатических
альдегидов и кетонов не используется для синтеза, хотя
некоторые примеры этих реакций известны [334]. В патентной
литературе описано алкилирование ацетона в присутствии гидрокси-
да натрия и фосфониевых и аммониевых солей в качестве
катализаторов очень реакционноспособными замещенными аллил-
длоридами с хорошими выходами [345—348, 1081]:
СН„—СО—СН3 + С1—СН2—СН=С(СН3)2 *-
-—»■ СН3—СО—СН2—СН2—СН=С(СН3)2
Алкилирование циклогексанона, 2-метилциклогексанона,
3-метилциклогексанона и циклопентанона тем же пренилхлори-
дом (конц. NaOH/ТЭБА) дает выходы в области 20—50%, хотя
образуется много побочных продуктов [352]. Интересно
отметить, что очень похожие результаты были получены и при ис-
3.9. С-алкилирование активированных связей С—Н 193
пользовании твердого гидроксида калия в качестве
конденсирующего агента без какого-либо катализатора. Результаты
изучения направления и скорости реакции пренилирования в
условиях МФК различных карбонильных соединений были недавно
опубликованы советскими авторами [1373, 1502, 1590].
В случае ацетофенонов, однако, для того же типа реакций
более эффективной, чем твердый КОН, была комбинация
водного гидроксида натрия с ТЭБА (50 °С, 3 ч). Выходы продуктов
также были не очень хорошими [352, 1477]. При использовании
техники экстрактивного алкилирования по Брендстрёму была
получена смесь моно- и диметилированных ацетофенонов
[356]. Редкое использование этих реакций для синтеза можно
объяснить тем, что арилкетоны обладают малой кислотностью.
Ранние кинетические исследования показали, что в эфире
ионные пары с четвертичным аммониевым катионом образуются
быстро и сразу алкилируются [353].
R1N / R4 \
R3—С—СНО + С=СН—OR3
R-' \ */ I
Схема 3.71
Альдегиды, содержащие только один а-водородный атом
(например, изомасляный альдегид), алкилируются активными
реагентами, такими, как метилиодид, аллил- и бензилгалогенид,
в системе бензол/концентрированный гидроксид натрия с тетра-
бутиламмониевыми солями в качестве катализатора [349, 350,
1080, 1230, 1563, 1716]. Для того чтобы свести к минимуму
самоконденсацию, алкилирующий агент и альдегид прибавляют
по каплям к перемешиваемой реакционной смеси при
температурах 20—70 °С, однако, несмотря на это, выходы продуктов
часто бывают очень низкими. Изомасляный альдегид и 2-этил*
гексаналь дают смесь продуктов С- и О-алкилирования [349,
1716]. Как и следовало ожидать, изопропилбромид приводит
только к продуктам О-алкилирования с выходом 21%; хлорук-
сусные эфиры также реагируют с низкими выходами [351].
XOCHaCOOR3
Схема 3.72
Более подробно изучена реакция между изомасляным
альдегидом и бензилхлоридом в системе 50%-ный водный NaOH/
/толуол при 70 °С в течение 4 ч. Установлено [1716], что с раз-.
194 Глава 3. Практическое использование МФК
личными катализаторами соотношение продуктов С- и О-алки-
лирования было следующим: Bu4NI 7,6; Bu4NHSO4 4,3;
Bu4NBr 3,7; Prop4NI 10,9; аликват 336 5,8 Bu4NCl 4,3.
Удивительно, что обычный эффект «отравления
катализатора» иодидами в этой реакции не наблюдается, более того,
реакция с иодидами проходит быстрее всего. Было высказано
предположение, что перед алкилированием бензилхлорид
превращается в иодид, который имеет более легко уходящую группу, что
ускоряет С-алкилирование (см. разд. 3.10).
Дибензилирование фенилацетальдегида проходит с выходом
19—29% [1230]. При попытке провести Y-алкилирование юур-не-
предельных альдегидов О- или 7"алкилиРование вообще не
наблюдалось, а шло исключительно а-алкилирование [1230,
1502, 1604]:
RlCH2—CH=CR«—CHO + R*X »- RlCH=CH—CR2RS—CHO (34—65%)
Несколько более низкие выходы наблюдаются в том случае,
когда альдольная конденсация и бензилирование проводятся
одновременно, «в одном горшке» [1604]:
r)—CHO
Удивительно, что при попытке бензилирования в условиях
МФК N-пропилиденанилина образовался Ы-бензил-1,2-дигидро-
хинолин (выход 15%) [1230]. В то же время платиновый
комплекс (S) алкилируется диметилсульфатом или бензил хлоридом
в положение, соседнее с карбоксильной группой с
превосходными выходами (ВщШБСУЫаОН/НгО/ацетон) [354].
РГ
е-0
Bu,p" XO
-: : ' S
Схема 3.73
Следует упомянуть две работы о применении оснований более
сильных, чем гидроксид натрия; в одной из них описано
получение растворимых литиевых, натриевых и калиевых енолятов
циклогексанона при действии твердых LiH, NaH и КН, которое
становится возможным или ускоряется в присутствии криптан-
дов. Полученные активированные еноляты способны отрывать
протоны даже от эфиров, служащих растворителем [1309].
В другой работе отмечено, что бутиллитий не реагирует с
карбонильными соединениями или карбоксилатами в присутствии
криптанда [2,1.1]; вместо этого идет депротонирование в а-поло-
жение [1482].
^ 3.9. С-алкиЛирование активированных связей С—Н 195
3.9.7. Алкилирование углеводородов
Система водный гидроксид натрия/ТЭБА достаточно основ-
на, чтобы депротонировать инден и флуорен. Различные
производные индена были получены с удовлетворительными
выходами 50—73% при действии первичных алкил-, бензил- и аллил-
галогенидов [358].
R
Схема 3.74
Флуорен алкилируется несколько труднее, поскольку он
является гораздо более слабой кислотой. Как и в случае других
слабых кислот, для получения хороших результатов
необходимо добавлять к реакционной смеси, включающей насыщенный
алкилбромид, небольшое количество ДМСО. В этих условиях
при 80—100 °С образуется смесь моно- и диалкилированных
продуктов [357]. Алкилирование самого циклопентадиена
должно проходить легко, и оно описано в литературе, но без
экспериментальных подробностей [214, 360]. Однако можно
предположить, что при этом образуются сложные смеси. Катализ
краун-эфирами также был использован при алкилировании
индена [45]. Следует подчеркнуть, что комплексующие агенты
можно использовать <с большим эффектом, чем ониевые соли,
в очень основных средах в отсутствие воды, поскольку ониевые
соли в этих условиях распадаются слишком быстро. Дитрих и
Леен [359], используя азамакробициклический полиэфир крип-
тофикс[2.2.2] (5) и твердый гидроксид калия/ТГФ или амид
натрия/криптофикс[2.2.2]/ТГФ, провели депротонирование
соединений, имеющих очень высокие рКа [359]. В последней
системе были генерированы окрашенные анионы трифенилметана
н дифенилметана и получены продукты их бензилирования
196 Глава 3. Практическое использование МФК
[359]. Металлирование дифенилметана (р/Со 33,4) и трифенил-
метана (рКа 31,1) гидридами натрия или калия в присутствии
[2.2.1] или 5 соответственно может быть осуществлено как в
ТГФ, так и в бензоле. Этот процесс включает депротонирование
на поверхности твердых нерастворимых гидридов [1310].
Система гидрид лития/криптанд[2.1.1] для проведения этих
реакций не пригодна.
Ниже приводятся типичные экспериментальные методики.
Алкилирование алкилацетонитрила,
каталитическое количество ТЭБА
4-Бром-2,2-дифенилбутиронитрил: при смешивании 0,025 моля
дифенилацетонитрила, 0,06 моля 1,2-дибромэтана, 0,1 г ТЭБА
и 10 мл 50%-ного водного гидроксида натрия развивается
слабая экзотермическая реакция. Спустя 15 ч фазы разделяются и
избыток 1,2-дибромэтана удаляют перегонкой с паром. Остаток
экстрагируют и перекристаллизовывают из лигроина, получают
продукт с т. пл. 68—69 °С, выход 94% [287, 360].
Алкилирование трет-бутилфенилацетата,
каталитическое количество ТЭБА+ДМСО
трет-Бутил-2-фенилгексаноат: 0,025 моля трег-бутилфенил-
ацетата, 0,038 моля 1-бромбутана, 12 мл 50%-ного водного
гидроксида натрия, 0,1 г ТЭБА и 5 мл ДМСО перемешивают
при 60 °С в течение 3 ч. Смесь разбавляют водой и экстрагируют
бензолом. После высушивания и удаления растворителя
продукт очищают вакуумной перегонкой, т. кип. 129—131 qC
(8 мм рт. ст.), выход 45% [338].
Алкилирование малонового эфира,
: молярное количество катализатора
Диэтилэтилмалонат: 0,24 моля NaOH в 50 мл воды
прибавляют к 0,12 моля тетрабутиламмонийбисульфата в 50 мл воды.
Сюда же добавляют раствор 0,1 моля диэтилмалоната и
0,2 моля этилиодида в 75 мл дйхлорметана. Смесь
перемешивают и кипятят в течение 20 мин. Фазы разделяют и удаляют
органический растворитель на роторном испарителе. К остатку
добавляют 200 мл эфира и отфильтровывают нерастворивший-
ся Bu4NI (выход 100%). После того как растворитель удаляют,
выделяют продукт с почти количественным выходом [321].
ЗЛО. Алкилирование амбидентных анионов
В разд. 3.9 были рассмотрены реакции, приводящие к
продуктам С-алкилирования. Однако и при изучении простых
карбонильных енолятов наблюдаются некоторые тенденции к О-ал-
3.10. Алкилирование амбидентных анионов 197
килированию. В этом разделе мы рассмотрим реакции {$-дикар-
бонильных и сходных с ними соединений, для которых вопрос о
соотношении продуктов С- и гетероатом-алкилирования
является достаточно сложным. Из всей суммы имеющих значение
факторов мы рассмотрим прежде всего влияние структуры аниона
на направление алкилирования и/или ацилирования. В этой
области применение МФК нацелено на решение вопроса о
способе изменения направления алкилирования в нужную сторону
(например, С- или О-), при этом сравниваются
экспериментальные достижения метода МФК с более старыми методиками.
Соответственно мы обсудим процессы МФК наряду с
классическими методами и методиками, занимающими промежуточное
положение, т. е. гомогенными реакциями с четвертичными
аммониевыми противоионами и краун-эфирами. Было показано, что
направление С- или (гетероатом-алкилирования зависит от
многих факторов (структура субстрата и алкилирующего агента,
растворитель, концентрация, природа катиона, наличие краун-
эфира, температура и т. п.) и что (полного количественного
описания взаимодействия всех этих факторов пока еще не
достигнуто. Подробное обсуждение этих вопросов можно найти в
работах [361, 368, 896]. Нежесткий §-дикарбоиильный аниои в
принципе может существовать в трех конформациях—U, W
hS [361]:
w-форма
{ЕЕ)
Схема 3.75
S- форма
(EZ)
Обычно в растворе устанавливается равновесие между
этими тремя формами, положение которого зависит от различных
факторов (см. ниже), однако наличие в молекуле жесткого
скелета может привести к тому, что будет существовать только
одна форма. Свойства этих трех форм и особенно кислотность
и способность к образованию ионных пар и к их диссоциации
весьма различаются. Было показано [362], что в неполярных
растворителях еноляты щелочных металлов ациклических р-ке-
тосоединений находятся главным образом в U-форме и между
анионом и катионом существует сильная «ассоциативная» связь.
Это взаимодействие остается сильным даже в водных
растворах [362].
198 Глава 3. Практическое использование МФК
рЛГиаО) 5,2
Схема 3.76
В одинаковых условиях еноляты в W-форме
диссоциированы сильнее [362]. Это связано с двумя факторами: а)
ассоциированные U-еиоляты по сравнению с ассоциированными W-ено-
лятами стабилизованы в результате хелатообразования с ионом
металла; б) диссоциированные U-еноляты по сравнению с
диссоциированной W-формой енолята дестабилизированы
сильнее вследствие электростатического отталкивания час-
тичнозаряженных атомов кислорода. -Эти факторы играют
более важную роль в апротониых растворителях, поскольку в
протонных растворителях водородные связи уменьшают
внутримолекулярные электростатические взаимодействия [361].
Следует добавить, что в неполярных растворителях
ассоциированные U-образные соли склонны к более сильному
агрегированию. В таких растворителях в зависимости от их типа и от типа
противоиона в растворе могут одновременно присутствовать как
контактные, так и сольватноразделенные ионные пары, а
также диссоциированные ионы.
Недавние физико-химические исследования (дальняя ИК-,
ЯМР-спектроскопия, кондуктометрические измерения) в ТГФ и
ДМСО подтвердили, что основным типом енолятов является
ионная пара с анионом в U-форме. Особенно поражает тот факт,
что соли тетрабутиламмония ведут себя так же, как и соли
щелочных металлов. Это указывает на ионность связи в этих ено-
лятах и, что еще более важно, на отсутствие жестких требований
к положению катиона по отношению к узкой области
локализации заряда аниона. В то время как небольшой ион
щелочного металла может располагаться на плоскости между О-ато-
мами (истинный хелат), ион аммония вынужден находиться
выше плоскости U-образного аниона [363].
Рентгенографическое изучение кристаллической структуры
комплекса 18-крауна-6 и калиевой соли этилацетоацетата
показало, что К+ координирован с." шестью атомами кислорода
краун-эфира и с двумя О-атомами плоского U-енолята [364].
Раньше многие авторы считали, что реально существуют
только U- и W-еноляты, однако недавно было показано, что в
3.10. Алкилирование амбидентных анионов 199
метанольном растворе симметричная форма W на самом деле
находится в стерически менее затрудненной форме S. Второй
метильный сигнал в ЯМР-спектре ацетилацетоиата (W-форма)
в действительности перекрывался другим сигналом [365, 366].
Изучение ЯМР-спектров при низкой температуре в CD8OD
показало, что в U-форме находится 23% хелата, в то время как
большинство анионов имеет нехелатированную S-форму.. В
метаноле молекулы растворителя выступают в качестве лигандов
по отношению к натрию, поэтому хелатирование идет в
меньшей степени '[365]. Даже в пиридине, если имеется избыток
18-крауна-6, преобладает U-форма [366]. Увеличение концент-.
рации краун-эфира приводит к уменьшению концентрации
U-формы. В результате тщательного изучения енолятов в
метаноле был сделан вывод, что еноляты натрия, находящиеся в
U-форме, образуют хелаты и хелатную ионную пару, в то
время как S-анионы в значительной степени диссоциированы.
Литиевые еноляты ацетилацетоната лрактически недиссоциирова-
ны, а соли калия диссоциированы сильнее, чем соли натрия,
причем анионы находятся в S-форме [366J.
Алкилирование и ацилирование р-дикарбонильных
соединений может приводить к трем первичным продуктам: С-, Оцис-
(Z) и Охране- (Е). Гелин и сотр. [367], взяв в качестве примера
ацетоуксусный эфир, составили обзорную схему этих
направлений реакции:
металл
металл'
О?
Н3С OEt
U-форма
RCOCI
5Et
W-форма
RCOCI
Н3С—С—СН—С—OEt
С—OEt
С- ацил
Схема 3.77
200 Глава S. Практическое использование МФК
На приведенной выше схеме видно, что U-форма приводит к
смеси Оцис- и С-продуктов, а W-форма дает только ОГракс-про-
дукты. Хотя такого рода рассуждения и распространены в
литературе, однако на самом деле различия в структуре енолятов
сказываются на структуре продуктов только в том случае, если
алкилирование идет быстрее, чем установление равновесия
между различными формами енолятов. Хотя этот вывод
подразумевался многими авторами, но только в одном случае было
показано [889], что нельзя делать предположения о структуре
продуктов, исходя из данных о структуре енолятов. При алки-
лировании симметричного р-дикетона S-форма енолята, ролью
которой часто пренебрегают [364], может приводить к смеси
Оцис- и Огракс-продуктов, а W-форма енолята дает только
Огракс-продукт алкилирования. Возможно, что имеющиеся в
литературе разногласия вызваны тем, что не учитывается роль
S-формы енолятов. Еще большие трудности возникают в тех
случаях, когда нет уверенности, определяется ли ход реакции
кинетическим или термодинамическим контролем. Нобль [368]
сформулировал правила, определяющие направление
алкилирования амбидентных анионов: «чем свободнее анион, тем больше
тенденция к алкилированию по наиболее
электроотрицательному положению». ,
Следовательно, О-алкилированию будут способствовать:
1) полярный, апротонный растворитель;
2) большой противоиои;
3) низкая концентрация енолят-аниона;
4) наличие «жесткой» уходящей группы в алкилирующем
агенте или такой реагент, который малоактивен в реакциях типа
SN2;
5) стерически затрудненный алкилирующий агент.
Классический метод алкилирования в этаноле с
использованием в качестве реактантов алкилгалогенидов приводит к тому,
что атаке подвергается реакционный центр с наименьшей элект-
роотрицательностью, следствием чего является преобладание
С-алкилирования. Водородные связи с растворителем
стабилизируют U-образный енолят и экранируют О-атомы от атаки
реагентом. В общем образование ионных пар уменьшает вклад
алкилирования по более электроотрицательному реакционному
центру. Так, реакция по центру с наименьшей
электроотрицательностью, т. е. по С-атому, наблюдается в апротонных
растворителях с низкой полярностью (ТГФ, эфиры, СНС13, СНгС12,
СбН6). Напротив, в диполярных апротонных растворителях
(ДМСО, ГМФТА, ДМФА) преобладает О-алкилирование [361,
367]. В очень разбавленных растворах тетрабутиламмониевая
соль этилацетоацетата существует главным образом в диссоции-
ЗЛО, Алкилирование. амбидентных анионов 201
рованной W-форме; следствием этого является преобладание
О-алкилирования. Однако увеличение доли .С-алкилирования в
менее полярных растворителях достаточно четко выражено:
соотношение продуктов О- и С-алкилирования равно 7,7 в ДМФА
и 3,9 в диметоксиэтане [361].
Брендстрём и Юнггрен [369] изучили изопропилирование
ацетилацетоната тетрабутиламмония в различных
растворителях. Следует, однако, учитывать, что в этом случае существуют
стерические факторы, которые весьма способствуют О-алкили-
рованию. Как можно видеть из табл. 3.10, соотношение
продукта б л и ц а 3.10. Алкилирование BiuN-ацётилацетоната
(0,05 моля соли в 50 мл растворителя и 0,1 моля изопропилиодида)
Растворитель
Параметр
Тафта Р
Диэлектрическая
проницаемость £
Содержание
продуктов, %
С-алкилирования
О-ал^или-
рования
С/О
дмсо
Ацетон
Ацетонитрил
Хлороформ
Диоксан
Толуол
2,60
1,95
2,30
2,30
1,25
0,55
48,9
20.5
37,5
4,8
2,2
2,4
, 42
42
48 ■
51
63
69
.58:
58
52
49
33
5
,0,72
0,72
0,92
1,04
1,91
13,8
тов С- и О-алкилирования возрастает с уменьшением
диэлектрической проницаемости е и параметра Тафта Р. (который
является мерой сольватационных свойств растворителя).
По-видимому, единственным исключением является ацетон. Таким
образом, можно сделать вывод, что «амым лучшим растворителем
для получения максимального выхода продуктов
С-алкилирования в условиях МФК служит толуол.
Следует отметить, что наиболее часто используемый в МФК
растворитель — хлороформ — не оказывает заметного влияния
на направление этой сложной реакции. Так, было найдено, что
растворитель (ДМФА или СНС13) не оказывает никакого
влияния на алкилирование диалкилоксалацетатов и алкил-2-диано-2*
фенилпируватов [370]. .
Важную проблему представляет также образование Оцис-
или Ограяс-продуктов при ацилировании или -алкйлировании.
Для того чтобы продемонстрировать слбжность влияния различ-
202 Глава 3. Практическое использование МФК
ных факторов, рассмотрим реакцию между натриевой солью
этилацетоацетата и О,О-диэтилфосфорхлоридтиоата [371]:
s
Na« ||
О НзС (ЕЮ)аР-О
/Л
COOEt НзС COOEt
Схема 3.78
Хлорангидрид кислоты взаимодействует с
электроотрицательным атомом кислорода, а полярные растворители
благоприятствуют образованию диссоциированной W-формы еноля-
та. Поэтому в таком растворителе образуется почти
исключительно ОТранс (Е) -продукт, в то время как в, бензоле основным
является O4UC(Z)-продукт (образующийся из недиссоциирован-
ной U-формы енолята).
Колинд-Андерсен и Лавессон [373] использовали эту
реакцию для получения многих производных типа А. Они сравнили
соотношение продуктов Z и Е при реакции натриевой соли в
кипящем этаноле и (предварительно выделенной) тетрабутил-
аммониевой соли в дихлорметане.
R—CO—СН—COR' *■ R—С=СН—COR'
O-=-P(SXOEt)2
Z и Е
К = СН3, РЬ
R' = ОМе. OEt. СНз. Ph
: Схема 3.79
Как и ожидалось, натриевая соль в этаноле (U-форма
енолята) дала преимущественно Z-продукт, а тетрабутиламмоние-
вая соль в дихлорметане (диссоциированная W-форма
енолята) — только Е (Отроке) -продукт.
Влияние катиона было изучено в реакции алкилирования
этилацетоацетата этилтозилатом в диметоксиэтане [372].
с/о
L£+
20
2.2
Nai+
6
6
K+
0,9
4.7
Bu4N+
Следы
0,26
Оцис
3.10.. Алкилирование амбидентных анионов 203
Уменьшение размера катиона и «жесткости» уходящей
группы благоприятствует О-алкилированию. Большая величина
катионов и W-форма енолята способствуют образованию Отранс-
продуктов. Использование BiuN-соли вместе с различными
другими алкилирующими агентами приводит в тех же условиях к
увеличению количества продуктов С-алкилирования [372]
(другие примеры см. в работе [382]).
Условия: 0,05 М Bu4N+CHsCOCH-—COOEt, ДМЭ
Алкилирующий агент 0,1 М EtI 0,3 М EtBr . пом ЕЮТ
С/О 8,4 2,9 0,26
Однако выводы следует делать с осторожностью, поскольку,
как уже было показано, низкая концентрация в принципе
благоприятствует О-алкилированию (особенно Отранс) [368, 3/fllJ,.
В то же время известны случаи, когда изменение концентрации
не влияет на соотношение продуктов [370, 1083]. Как было
установлено в ряде случаев, соотношение О/С-изомеров в продуктах
алкилирования не зависит от типа уходящей группы в алкили-
рующем агенте (бензилхлорид, бензилбромид, бензилтозилат)
[380]. Спорным является также вопрос о корреляции междуре-:
акционной способностью алкилирующих агентов в Б^-реакци-
ях и соотношением О/С-изомеров [370]. Ясно только, что
различные факторы очень сложно влияют на ход реакции. Недавно,
было обращено внимание еще на один из таких фактов:
оказалось, что на соотношение С/О-изомеров сильно влияет
содержание воды в апротонных растворителях, причем..наличие
воды способствует С-алкилированию. Это явление наблюдается
независимо от присутствия или отсутствия краун-эфиров 5 даже
при концентрациях воды порядка миллимолей (например, в так
называемом сухом ТГФ) [1345]. Аналогично соотношение
продуктов О- и С-алкилирования дезоксибензоина диметилсульфа-
том зависит от количества воды, экстрадируемрй в ходе МФК-
реакции. Концентрация воды в: органической фазе зависит от
концентрации водного гидроксида натрия. Соотношение О/С
колеблется от 1,73 (твердый NaOH, Hex4NBr, бензол) до 1,46
(30%-ный водный NaOH, условия те же) [1746].
Камбиллау и др. [377] изучили алкилирование этилтозила-
том и этилиодидом натриевых енолятов этилацетоацетата в
ТГФ как в присутствии, так и в отсутствие 18-крауна-6 и крип-
тата [222]. Скорость реакции увеличивалась в присутствии кра-
ун-эфира и еще больше повышалась в присутствии криптата.
В общем, процент О-алкшгарования'(особенно ОтРа«с-алкилиро-
вания) также повышался. Комплексообразование с крауи-эфи-;
ром разрушает агрегированные ионные пары, что и приводит
соответственно к увеличению Скорости 'реакции. . Кроме того,,
204 Глава 3. Практическое использование МФК
положение равновесия смещается в сторону образования
диссоциированной W-формы енолята. Другие обширные
исследования с использованием различных краун-эфиров щелочных
катионов и тетрафениларсониевых ионов в различных
растворителях позволили качественно сделать те же выводы [378, 379,
1082, 1609]. Влияние краун-эфира на сдвиг соотношения
продуктов С- или О-алкилирования в ту или иную сторону
максимально сильно проявляется в наименее полярных
растворителях [379]. Влияние краун-эфира и криптата [222] на
соотношение С- и О-алкилирования метоксикарбонилциклогексанона
было исследовано в смеси ДМСО/СН3ОН, чистом ДМСО и
ДМЭ, при этом наблюдалась та же тенденция [382, 1508].
Рассмотрим теперь несколько практических аспектов. Можно ис-
нользовать две методики МФК, включающих:
а) выделение четвертичной аммониевой соли енолята с
помощью экстракции растворителем и последующей реакцией
в том же или в другом растворителе;
б) проведение «истинной» МФК-реакции в двухфазной системе.
Оба метода широко используются.
В некоторых случаях использование четвертичных
аммониевых солей амбидентных анионов является единственным
возможным методом алкилирования. Например, диалкилацилмало-
наты — довольно сильные кислоты, а соответствующие им
анионы—слабые нуклеофилы. Вследствие этого обычные методы
алкилирования их оказываются непригодными, так как после
алкилирования соединение может очень легко отщеплять ациль-
ную группу. Брендстрём [374], используя избыток алкилиодида,
изучил алкилирование тетрабутиламмониевых солей диэтилбен-
зоилмалоната в дихлорметане и получил следующие результаты:
Содержание продуктов, %
СН31
С2Н51
«30-C3H7I
С-алклли-
рования
100
54
47
14
Очалкили-
роваиня
0
46
53
86
■ Как было показано выше, при использовании толуола
может увеличиваться процент С-алкилирования [369].
' Рассмотрим еще раз реакции с ацетилацетоном. При
проведении реакций, с этим соединением в обычных условиях в про-
3.10. Алкилирование амбидентных анионов 20$
тонных растворителях возникают проблемы, поскольку этот ди-
кетон алкшщруется намного медленнее, чем этилацетоацетат
или диэтилмалонат. Тетрабутиламмониевые соли ацетилацето-
на (предварительно выделенные с помощью экстракции) были
проалкилированы в хлороформе (10—30 мин, кипячение).
Результаты приведши ниже [375] (см. также [1083]):
Алкилнодид
СН31
С2Н51
ызо-С3Н71
H-C4HflI
Содержание продуктов, %
моно-С
72
50,5
87
ди-С
98,5
16
_
—
О
1,5
12
49,5
13
Джонес и сотр. [837] изучили ацилирование ацетилацетона
и этилацетоацетата в системе дихлорметан/2н. NaOH в
присутствии молярных количеств тетрабутиламмонийбисульфата. Как
и в рассмотренном выше примере с О.О-диэтилфосфорхлорид-
тиоатом, ацилирование этих соединении ацетил- и бензоилхло-
ридом прошло только по кислороду. При реакции с пентанди-
оном, так же как и в случае ацетоуксусного эфира, доля
образующегося £-изомера выше, чем Z-формы (93—96% Е и 7—
4% Z). В статье Джонеса и сотр. [837] формулы продуктов
напечатаны с ошибкой, но смысл ясен из текста.
О
О О
' II II
НзС—С—СН2—С—R
(R = СН3, ОСаН5)
B^WX/NaOH
НзС Н
НаС
C—R
О
P-R
Н
R'—С=О
Z
Схема 3.80
О R'—С=О
(главные провукты) Е
При использовании только каталитических количеств
четвертичных аммониевых солей реакция этилирования
ацетилацетона проходит очень медленно [854]. Соотношение продуктов С- и
О-алкилирования не зависит от размеров катиона (в
изученных автором пределах). Если реакцию (проводят в метиленхло-
риде с предварительно выделенным тетра-м-пентиламмонийаце'-
206 Глава 3. Практическое использование МФК
тилацетонатом (20 °С, 15 мин), то соотношение О/С/бледующее:
С2Н51 1:5,7; ОДД- 1:2; (CSHSO)2SO^2:1
Изучено [380] также бензилирование ацетилацетона в вод-
ноорганической среде с использованием молярного количества
бромида тетрабутиламмония в качестве катализатора.
Соотношение 0/С-изомеров варьировало от 1:3 до Li 1 при изменении
типа уходящей группы и условий реакции. 1#стинное алкилиро-
вание в условиях МФК этилацетоацетата и ацетилацетона
впервые было проведено с использованием бензилхлорида и 1,3-ди-
хлорбутена-2, водного гидроксида калия и дибензилдиметилам-
монийхлорида или ди-(3-хлорбутен-2-ил)диметиламмонийхлори-
да, взятых в эквимолярных количествах [226, 340].
Впоследствии алкилирование ацетоуксусного эфира 1-хлор-З-метилбу-
теном-2 было подробно изучено с целью оптимизации условий
для промышленных делей. При использовании ТЭБА и КгСО3
(твердый или 50%-ный водный раствор) образовывался моно-
пренилацетоуксусный эфир, а с концентрированным водным
раствором КОН — продукт диалкилирования и/или метилгелтенон
[1495, 1533].
Метилацетоацетат был проалкилирован в системе
хлороформ/водный гидроксид натрия в присутствии молярного
количества тетрабутиламмонийбисульфата [376]. С метил-, этил- и
бутилиодидами проходило только С-алкилирование (в основном
образовывался продукт моноалкилирования и немного
диалкилирования). Соотношение C/0-изомеров при алкилировании изо-
пропилиодидом было равно 3:1. В тех же самых условиях при
добавлении 2,4 моля СН31 бензилацетоацетат дал 66% моно-С
и 33% ди-С-продукта [398].
Hlo другой методике растворенный в бензоле ацетоуксусный
эфир обрабатывают вначале-гидридом натрия [397], после
этого прибавляют алкилирующий агент, 10 мол.% аликвата 336
и кипятят реакционную смесь 8 ч. С аллил- и бензилгалогени--;
дами получают очень хороший выход моно-С-продукта.
Простые алифатические галогениды требуют увеличения времени
реакции и, кроме того, дают некоторое количество О-продуктов,
что препятствует широкому использованию этих галогенидов в
МФК [397].
Кларк и Миллер [381] предложили необычный метод
получения продуктов моно-С-алкилирования алкилацетоацетатов и
1,3-дикетонов. При смешивании водного раствора тетраэтилам-
монийфторида и р-дикетона в ТГФ образуется моносольватный
комплекс с сильной водородной связью. После удаления
растворителя прибавляют при комнатной температуре избыток алкил-
йодида в хлороформе. В результате получают моно-С-продукт
с прекрасным выходом. Улучшенный вариант этой методики со-
ЗЛО. Алкилирование амбидентных анионов 207
стоит в перемешивании р\-дикарбонильного соединения с
твердым дигидратом фторида калия и Bu^NCl в ацетонитриле при
комнатной тевшературе в течение 9 ч [1235]. Очень тонко
раздробленный и «цательно высушенный с помощью глубокого
охлаждения KF можно использовать в этой реакции без всякого
катализатора [1605].
в»
К.оГ
А В
Схема 3.81
Данные об алкилировании А и сходных с ним соединений в
присутствии хиральных катализаторов см. в разд. 3.1.5.
Соединение А можно проалкилировать в две стадии — сначала
провести реакцию А с гидроксидом натрия, а затем алкилировать
в присутствии четвертичной аммониевой соли, при этом
образуется 84% С-алкилированного продукта В [385]. При
алкилировании ацетоуксусного эфира 2-октилтозилатом или 2-октилиоди-
дом наблюдаются инверсия и частичная рацемизация, при этом
энантиомерная чистота продукта О-алкилирования была выше,
чем у продукта С-алкилирования [1418]. В других работах
сообщается о С-алкилировании анилида ацетоуксусной кислоты
[1561] и об образовании пятичленного цикла при реакции
между ацетоуксусным эфиром и 1,2,4,5-тетрабромметилбензолом в
условиях МФК [1442].
Недавно был предложен другой метод алкилирования в
условиях МФК [116]: анионообменную смолу (например, амбер-
лит ЩА 900) в ОН-форме обрабатывают этанольным
раствором циклического fj-дикарбонильного соединения. Затем р-ди-
кетонатную форму смолы встряхивают в этаноле с алкялирую-
щим агентом (при этом проходит только С-алкилирование) или
в толуоле (в этом случае кроме С-продукта образуется и
некоторое количество О-продукта).
Алкилирование различных бензоинов и дезоксибензоинов
было осуществлено с использованием в качестве катализатора
Bu4NBr [900]. Диметилсульфат всегда дает смесь {E/Z)
-изомеров а,(р-диметоксистильбенов с небольшим избытком £-изомера.
В то же время аллильные и бензильные производные
образуют главным образом и-аллил- или бензилбензоины и
аллильные или бензильные эфиры соответственно. Эти данные
согласуются с гипотезой о том, что мягкие алкилирующие агенты дают
главным образом С-продукты. При этилировании в бензоле
208 Глава 3. Практическое использование МФК
лри .60 СС субстратов с различными уходящими
получены следующие результаты:
Ph
I' ■
0=0
I
снон
Ph
NaOH, EtX
Bu4NBr, бензол
Ph
I
С—OEt
Ph
I
C=O
C—QEt Et—C-OEt
I I
Ph (E + Z) Ph
ами были
Относительный выхой,%
EtOSO3
Mes-O-
Tos-O
Br
I
92
90
90
30
21
2
48
8
10
10
68
30
. ' , . Схема 3.82
Соотношение продуктов О/С-алкилирования при этилирован
нии дезоксибензоина с различной уходящей группой изменялось
следующим образом: OS02OC2H5 2,22; OMes 3,0; OTos 2,7;
Br 0,39; I 0,03 [900]. Алкилирование различными алкилиоди-r
дами р-кетосульфоксидов проходит главным образом по С-атог
му; исключением является только изопропилиодид [1866]. Ал*
копирование 9-бензоилфлуорена (NaOH, 18-краун-6, бензол)
протекает очень селективно, давая С-продукт [1207], а 3-хрома-
ион образует смесь 4,4-диалкил-З-хроманона и З-алкокси-4,4-
диалкил-4Н-хромена [1451}. Алкилирование в условиях МФК
rper-бутилхлоридом приводит к 9-грег-бутил-9-бензоилфлуорену
[1207]! .
- . Соединения типа; D, имеющие активированную .метиленовую
ipynnyj можно превратить в анионы и, добавив тетрабутйламмо-
ниевый катион» экстрагировать в органическую среду. Реакция
е дисульфидом углерода приводит к смеси таутомерных
анионов Ё'и F дйтйокислот и геж-меркаптотиолатам соответственно.
Последние в свою очередь могут быть, депротонированы
щелочью с образованием дианиона С;
Примерами соединений типа D, которые реагируют таким же
Образом, могут служить1 диэтилмалонат [391], метилцианоаце-
тат [39Ц, малодинитрил [391], ацетилацетон [30], бензоил-
ацетон [30], дибензоилметан [30], метил- и этилацетоацетат
(392], этилбензоилацетат [392] и цианоацетон [392].
Некоторые соли E/F были выделены в кристаллическом виде. При neper
мешйвании их в хлороформе при комнатной температуре в
течение "18 ч с алкйЛйруюЩйм агентом получают дитиоэфиры Н
н^йокётали кетеййЛ. Поскольку основность-'аниона F близка к
\ ".З.Ж Алкилирование амбидёнтных анионов
основности\аниона, который образуется из тиоэфиров Н, то де-
лротонирова\ше соединений Н частично проходит даже в том
случае, когда\вначале присутствуют только моносоли. В
результате всегда наблюдается в какой-то степени образование
продуктов диалкилЬрования кетена I [30, 391, 392]. Так, например,
если X—Y=COGEt, а И=зтил или аллил, то Н и I образуются
в соотношении 2 5.1. Диалкилирование метиленхлорида дает К.
Описаны также различные перегруппировки этих серусодержа-
щих соединений и^их внутримолекулярные конденсации [30г
391,392, 107,209].
NR«® NR«e
; С—С »■ Х- "
/' ч\ основание
Y' e 4S Y
Е F
COOR, CN, COPh, COR)
С=С ЪН3
У SR Y' ^SR Y/ NS
Н I К
Схема 3.83
В системе бензол/водный NaOH/Bu4NOH взаимодействие
между 2-нитропропаном и n-нитробензилхлоридом дает только
продукты С-алкилирования, которое проходит как цепной
анион-радикальный процесс. Эта же реакция с бензилхлоридом
идет как реакция Sn2 и дает продукты О-алкилирования,
превращающиеся в конечном итоге в соответствующие альдегиды
[838]:
п-NOg—CjHj—CHaCl >■ n-NO2—CeH4—CHj—CMe2—NOj
ОД,—CH2C1 »» CeH4—CH2—O—N(O)=CMe2 -*-^- CeH5CHO
Как уже было сказано раньше, при С-алкилировании фенолов
обычно не возникает каких-либо трудностей. Однако если
2,4,6-тригидроксифенилкетоны в условиях МФК дают с пренил-
бромидом продукты только С-алкилирования [1084], то антрон
и акридон образуют О-метильные производные [1085].
Подробно были изучены и родственные гетероциклические
таутомерные системы:
хк
н,с
Схема 3.84
н
Na0HCHCI»
Реагент
Относительны!! выхоЭ, %
СН31
(CH,ObSOa
12
90
86
4
Схема 3.85
н
-сС ,Л~
■Рейгент
Относительный 8ыхоЭ,%
О
S
СН,1
(CH,O)aSOj
СИэ1
2
54
35
98
46
65
8.М> Алкилирование амбидентных анионов 211;
Экстрактивное алкилирование 2-гвдроксифуранов избытком
йодистого метила с молярным количеством бисульфата тетрабу*
тиламмония дает только продукт алкилирования в положение 3
с низким выходом вследствие протекающего одновременно
раскрытия цикла [387]. Несколько продуктов образуются также
и при алкилнровании 2-гидрокситиофенона и «го таутомеров
[386] (схема 3.85). В зависимости от типа уходящей группы в
алкилирующем агенте наблюдается, как и можно было
ожидать, региоселективвое алкилирование; при наличии «лучшей»
уходящей группы —Ьетоксисульфонильной — продукт О-алки-
лирования образуется-с наиболее высоким выходом. В этом
случае соотношение продуктов О- и С-алкилирования близко к
соотношению, полученному при использовании таллиевых солей.
Однако метод экстракции ионных пар более прост и безопасен,
поскольку реакция проводится при комнатной температуре.
Сходным образом реагировали и многие другие 2-гидрокситио-
фены и 2-гидроксиселенофены с заместителями в разных
положениях. Наблюдаемые закономерности были аналогичны тем.
которые описаны для примеров, приведенных выше [386—390].
Изучены [904] также 3-гидроксифураны, 3-гидрокситиофены и
3-гидроксиселенофены (схема 3.86). В условиях МФК протекает
только О-(или S) -алкилирование в реакциях: 4-гндроксифурано-
на с диалкилсульфатами [1428], тиофенона с а-бромйзобутира-
том и 1-меркаптотиофена с ю-бромизобутиратом [1473].
Бензо[Ь]тиофен-2(ЗН)он (L) в виде тетрабутиламмониевой
соли был экстрагирован в хлороформ, а после высушивания
раствора пррметилирован. При этом образовалась смесь про-
212 Глава 3. Практическое использование МФК
дуктов, показанная на схеме 3.87. Реакции с натриевыми или
таллиевыми солями также дали сложные смеси продуктов,
которые труднее разделялись, чем смесь, полученная по методу
экстракции растворителем [395].
В двухфазной системе бензол/вода в присутствии всего
6 мол.% тетрабутиламмонийбромида (6 ч, 60,"С) были лроал-
килированы различными реагентами 2-тиоксо.-£,3-дигидроимида-
зол и его 1-метилпроизводные [393]. Последние подвергаются
только S-алкилированию. В незамещенных соединениях
протекает лишь N.S-диалкилирование (схема 3.88). При диалкилиро-
вании 2-тиоксоимидазолидина образуются N.S-продукты [1516]-*.
R
н I
f>SR
В аналогичных условиях 5,5-диметилйзоксазолидон дает
62—85% продуктов N-алкилирования и 38—15% О-алкилйрова-
ния (в зависимости от RX) [962]. Д4-Тиазолинтионы-2 и
родственные им соединения подвергаются только S-алкилированию
[1086]. Опубликована также работа об О- или N-алкилирова-
нии гидроксипиридинов в системе бензол/мода при 60 °С [394].
Как и следовало ожидать, 3-гидроксипиридин алкилируется
только по кислороду, однако 4-пиридон дает смесь O/N-продук-^
гов в соотношении 11ь'-Чъ, почти независящем от типа алкили-
рующего агента (к-бутил-, изобутил- и а лл ил бром иды). 2-Пи-
Схема &Я9
3.10. Алкилирование амбидентных анионов 21&
ридон дае* только 15—25% продукта О-алкилирования [394],
а акридон образует смесь продуктов О- и N-алкилирования
в соотношений, изменяющемся от 0:100 до 35:65 в зависимости
ют действующего реагента [1262]. Амбидентный дианион 1,2,3,4-
тетрагидро-2,4-диоксохиназолина образует N.N-дипродукт с
выходом 84—95% -[1270]. Было изучено также алкилирование 4-
гидроксихинолинсю в условиях МФК [871]. В этих же условиях
5-пиридин-, 2-пиримидин- и бензоксазолтионы [932] и тиоакри-
доны [1514] давали только S-алкильные производные.
Ниже приводятся типичные экспериментальные методики.
Промежуточное выделение соли енолята
Алкилирование ацетилацетона. К холодному раствору 1,1
моля NaOH в 500 мл Н2О добавляют 0,5 моля бисульфата тетра-
бутиламмония. После прибавления 0,5 моля ацетилацетона
проводят экстракцию раствора 500 мл хлороформа. Экстракт
высушивают и упаривают; в остатке остается соль -г- тетрабутил-
аммонийацетилацетонат, т. пл. 155 °С (после
перекристаллизации из ацетона), выход 70%. Эту соль можно проалкилировать
в различных растворителях [375].
Экстрактивное алкилирование по Брендстрёму
(использование молярных количеств катализатора)
Алкилирование метилацетоацетата. К раствору 0,1 моля ме-
тилацетоацетата и 0,2 моля алкилиодида в 75 мл хлороформа
прибавляют при перемешивании раствор 0,1 моля бисульфата
тетрабутиламмония и 0,2 моля NaOH в 75 мл воды. Проходит
экзотермическая реакция. Спустя ~15 мин после окончания
нейтрализации слои разделяют, хлороформ упаривают, а тет-
рабутиламмонийиодид осаждают, добавляя эфир. После
фильтрования растворитель удаляют в вакууме [376].
Алкилирование при использовании малых количеств
МФ-катализаторов
S-Алкилирование 1-метил-2-тиоксо-2,3-дигидроимидазола:
0,05 моля субстрата, 0,05 моля алкилгалогенида и 0,003 моля
тетрабутиламмонийбромида растворяют в 150 мл бензола.
Прибавляют 15 мл 40%-ного водного раствора г.идроксида натрия
и смесь перемешивают 6 ч при 60 °С. После этого органический
слой отделяют и высушивают, а растворитель удаляют.
Остаток перегоняют в вакууме или перекристаллизовывают из эта»
иола [393].
214 Глава 3. Практическое использование МФК
3.11. Изомеризация и H/D-обмен
Хотя эти процессы являются самыми простым^ из
возможных реакций, протекающих в присутствии конщнггрированных
водных растворов гидроксидов щелочных металлов, механизм
этих превращений долгое время был непонятен. Депротоинро-
вание может протекать на поверхности раздев фаз, в
органической фазе под действием экстрагированною гидроксида
аммония или же внутри инвертной мицеллы. В настоящее время
известно, что этот процесс протекает в результате экстракции
QOH. Поскольку при этом не образуется липофильного галоге-
нид-аниона, то «отравления катализатора» не происходит. Про-
тонирование или дейтерирование промежуточно
образовавшегося карбаниона протекает под действием небольших количеств
Н2О или D2O, которые экстрагируются вместе с анионом.
Если активированные (например, бензильной группой) аце-,
тилены перемешивать в дихлорметане с твердым гидроксидом
калия в присутствии ТЭБА, то изомеризация проходит с почти
количественным выходом (А-*-В, R=H, CH3, C6Hs). Смесь бен-
зилацетилена и фенилаллена, образующегося при реакции про-
паргилбромида с фенилмагнийбромидом, количественно
превращается в 1-фенил пропин-1 [400].
CeH5—CH—G=C—CeH5 > CeHs—С=С=СН—СвН„
I I
R R
А В
C,HS—СНа—С=СН
t CeHe—С=С—СН3
НС==С—СНа—S—C=CH—COOEt
R
Схема 3S0
Все эти реакции идут значительно быстрее в присутствии
катализатора. Менее кислые соединения не изомеризуются.
Например, метилпропаргиловый эфир не превращается в этих
условиях в более стабильный метоксиаллен [400]. Однако тио-
эфир С (R=CH3, С6Нб) можно превратить в алленовое
производное под действием системы 2 М NaOH/тетрабутиламмоний-
бйсульфат [209]. Изомеризация аллилбензола в смесь
цис/транс* 1 -фенил- 1-пропенов под действием 50%-ного водного
гидроксида натрия с различными катализаторами
проходит со следующими временами полупревращения (ti/t):
Pent4NBr 82 мин, Hex4NBr 66 мин, адоген 464 12 мин [1647].
3.11. Изомеризация и Н/Р-обмен 215
Для изомеризации менее активных алкенов требуется
применение более сильных лцелочей; в этих случаях используют смесь
трег-бутилата калия и 18-крауна-6. Так, например, в этой
системе октин-1 при нагревании до 150 ^С превращается в октин-2 с
выходом 94%, а гептадиен-1,2 при 70°С дает смесь 93% гепти-
на-2, 2% гептина-1 и 5% гептадиена-1,2 [1105].
Хлорметилсульфид и пропаргилсульфид в присутствии NaOH
и аликвата 336 претерпевают ряд удивительных превращений
1971]:
PhSCH2Cl + СНз—С=С—СНа—SPh
Ph
СНэ
PhS CH==CH2
-X
PhS CH3
Схема 3.91
Показано [413], что в отсутствие воды тетрабутиламмоний-
цианид вызывает изомеризацию бутен-3-нитрила в кротонони-
трил (который склонен димеризоваться и полимеризоваться),
а гексен-3-динитрила в сопряженный изомер. В использованном
для проведения этих реакций растворителе — ацетонйтриле —
цианид-ион ведет себя как основание, так как не образует
водородных связей. '
- NC—СН=зСНг-СН8
>- NC—CH=CH—(CH2)a—CN
NC—СНа—СН=СН—СНа—CN
Старке [4] показал, что если перемешивать октанон-2 с
50%-ным раствором NaOD в D2O в присутствии 5%
четвертичной аммониевой соли в течение 30 мин при комнатной
температуре, то происходит полный H/D-обмен атомов водорода и у С1
и у С3. Без катализатора (аликват 336?) за 3 ч обмен водорода
на дейтерий проходит только на б% [4]. Трехкратное
повторение этой процедуры с 1-ацетилциклогексаном и 1-ацетилцикло-
пентаном дает продукты, содержащие более 99% D4 [401]. Этот
метод был использован также для дейтерирования более
сложного соединения D, в котором дейтерий входил в положения,
указанные на схеме 3.92 [402};:. -.,
Мб Глава 3. Практическое использование МФК
И-CnHan + » S
I
Xе
.СНз
СНз
Другие авторы [975] работали с системой Кгз/г/
кват 336. В системе CH2Cl2/NaOD/D2O/T3BA с выходом 95%
проходили H/D-обмен и изомеризация 1-алкилиндеиов в З-алкиль*
ные производные [986]. Примеры включают также обмен в
производных стероидов [1423, 1471], дитиоацеталях
[1826],хлороформе [1765], у первого атома мостика в бицикло- [5.3.1 ]-ун-
деканоне-11 [1447], диазометане [1088], норборнаноне [1089],
а также обмен в С-метильной группе ArNMe—CS—СН3.
[1438]. В F2CH--O—CF2—CHCliF обмен проходит быстро в
хлорфторметильной группе, но медленно в CHF2 [1824].
Кислотный обмен с DC1 в D2O не ускоряется в присутствии МФ-ка-
тализаторов. Что касается механизма щелочного обмена, то
предполагают, что экстрагированный R4N+OD~-"(D2O)*
действует и как основание, и как переносчик D2O [853]. Исследование
различных катализаторов H/D-обмена в системе 5% NaOD в
D2O показало, что наиболее эффективны максимально липо-
фильные катализаторы: очень эффективен (C7Hi5)4N+, a BiuNt
значительно менее эффективен, так как эта соль
распределяется между водной и органической фазами. В этом случае краун-
эфиры как катализаторы совсем неэффективны [853, 1825]'.
Быстрее всего реакция шла без растворителя; углеводородные
растворители проявляли большую эффективность, чем хлориро1'
ванные углеводороды [853, 1825]. Изучена [406] скорость
H/D-обмена под влиянием щелочей в метильных группах суль-
-
V/
Н3С
VV~*
меЭленно
медленнее
Схема 3.93 ,
3.12. Присоединение по непредельным связям С—С 21?
фонийгалогенидов Е в гомогенной системе D2O.. Оказалось, что
скорость обмена в длинноцепочечных алкилах (и=10, 12) была
намного выше, чем в соединениях с короткими цепями (и=
= 1—8). При использовании лаурилдиметилсульфониевых солей
(Е, и=12) скорость обмена возрастала при изменении аниона X
в следующей последовательности: КВг<С1. Наблюдаемые
закономерности объясняют мицеллярными эффектами.
Очень подробно с изменением всех возможных параметров
был изучен H/D-обмен в гетероциклических соединениях [403—
405, 1825]. Некоторые направления обмена в гетероциклах
показаны на схеме 3.93.
3.12. Присоединение по непредельным связям С—С
3.12.1. Присоединение к ацетиленам
^^, (Ph—CH=CH)aS
^~~~* рь—сн=сн—sr
Схема 3.94
Присоединение сульфида натрия или этилтиолата к фенил-
ацетилену в водном растворе катализируется дибензо-18-крау-
ном-6 или (менее эффективно) ТЭБА [407]. С помощью
Et3NH+HCl2~ [880] без растворителя или в апротоииых
растворителях удается присоединить к тройной связи НС1 [881]. В
реакцию присоединения вступают различные соединения, такие,
как 1-фенилалкины, диметиловый эфир ацетилендикарбоновой
кислоты и 3-хлорпропин. Было показано, что такого рода
процессы могут осуществляться в условиях межфазного катализа
при использовании системы конц. НС1/аликват 336 путем
экстракции R^N+HCb" [882]. В другой работе [1513]
присоединение НВг к алкинам действием R4N+HBr2~~ проводилось в СН2С1а.
Авторы получали реагенты, насыщая R4N+Br~ газообразным
НВг, однако эта методика существенно менее привлекательна,
чем межфазный катализ.
Макоша и сотр. [408] описали присоединение 2-фенилбути-
ронитрила (А) к ацетилену и фенилацетилену. Реакция
проходит экзотермически при использовании порошкообразного
твердого едкого кали вместе с ТЭБА в ДМСО (выход около 60%);
в тех же условиях алкилфенилацетонитрилы (А)
присоединяются к фенилацетилену [409], алкилтиоацетилену [410] и эток-
сиацетилену [410] при использовании иногда вместо КОН гид-
218 Глава 3. Практическое использование МФК
роксида натрия. Выходы колеблются обычно в пределах 80—-
95%.
CN CN ":CN H
I III
Ph—С— Н + НС=С—R' »■ Ph—С—С=С—R' + Ph—С—С=С—R*
R R Н Н : R Н
А В (R'= Н, Ph, S—R) С D
CN
А + НС=С—ОСаН. > Ph—С—С=СНЯ
R ОСаН.
Е
Схема 3.95
Установлено, что присоединение к фенилацетилеяу и алкал-
тиоацетилену идет в одном направлении с образованием
продуктов типа С и D, причем цис-изомер С образуется обычно в
больших количествах, чем трамс-изомер. Однако эпоксиацетилен
дает соединение типа Е.
АгМН2
r"^"*OCH3 цис/траНе R *OCU3
М = As, P
Bu Bu
нс сн ЧУ
— V
трет-Ъи цис/mpaw трет-аа трет-аа
М = As, P
t' н н В%/Ви
(м) ^_ \/ — (*)
>Si^ Si ^Si^
Jx R R. R. ■ _ Ix. i\.
M = As, P R = СНз, Ph
Схема 8.96
3.12. Присоединение по непредельным связям С—С 219
Циклоприсоединение арилфосфинов и арсинов к пентади-
инам-1,4 и их аналогам с гетероатомом в положении 3
катализируется небольшими количествами твердого гидроксида калия
и 18-крауна-6 в бензоле [914]. Реакция часто протекает
экзотермически и приводит к продуктам с выходом около 90%.
Против ожидания даже гидрид ди-к-бутилолова
присоединяется в этих условиях с вполне удовлетворительным выходом.
Однако такого рода реакции циклоприсоединения проходят
только с терминальными ацетиленами в отличие от реакций
присоединения в жидком аммиаке в присутствии амида лития
(схема 3.96) [914].
Наконец, описано [1090] весьма необычное
внутримолекулярное присоединение в условиях межфазного катализа амида
азота, полученного in situ, к алленовому эфиру.
3.12.2. Присоединение по Михаэлю
Тритон В (бензилтриметиламмонийгидроксид) давно
используется как катализатор в реакциях присоединения,
преимущественно таких, которые осуществляются в гомогенной среде.
В литературе можно найти много примеров такого типа реакций
(соответствующую библиографию см. в [411а, 412]).
Менее изучена гомогенная реакция, катализируемая цианид-
и фторид-ионами. Несмотря на то что некоторые из них были
уже давно известны (ср. [411Ь]), только в последнее время они
привлекли к себе особое внимание. Например, тетрабутиламмо-
нийцианид в ТГФ или ацетонитриле вызывает присоединение
нитроалканов, спиртов и хлороформа к а, ^-ненасыщенным кето-
нам и сложным эфирам ![413]. В этих растворителях ионные
пары нитрил/четвертичный аммоний не защищены водородными
связями и ведут себя как основания. Кротонитрил димеризуется,
акрилонитрил полимеризуется [413].
Реакции присоединения тетраалкиламмонийфторида и
реакции, катализируемые им в гомогенных растворах, представлены
на следующей схеме:
2Н3С—СН=СН—CN BuNCN > НзС—СН=С—СН—СНа—CN
CNCH3
Схема 3.97
Использование фторид-иона как «основания» в этих
конденсациях весьма выгодно, потому что низшие нитроолефины
крайне чувствительны к обычным основаниям. В некоторых
приведенных ниже примерах по имеющимся данным нельзя
судить о степени растворения фторида калия в кипящем кси-
220 Глава 3. Практическое использование МФК
лоле. Поэтому в последних реакциях (см. схему 3.98)' вместе с
фторидом калия использованы небольшие количества 18-крау-
на-6 для того, чтобы увеличить растворимость катализатора в-
таких растворителях, как бензол, дихлорметан и ацетонитрил*
[416]. Присоединение метиленовых групп, активированных нит-
ро- и цианзаместителями, также происходит с высоким выходом
[416, 1092]. Полезной альтернативной системой катализатор/'
основание является система ВщМС1/твердый KF в толуоле или
ацетонитриле [1211, 1235]. Использование фторида как основа*
йия рассмотрено в обзоре [1664]. :
C1FC=CF2 E^F * CIFHC—CF3 [36]
F2C=CF—CF3 >■ F3C—CHF—CF3 [36]
H2C—CN
V,,./ HaCCN.ffiTTn. \l /
NNO2 / 4NO2
' .RSH, ТПР I I И
>=<II ^g^ «S-C-C-i- ВД
c- ' н
О О
.ксилол I у >^
учение I I /
CH3
O+H^C=< -^Tl I >-CH. [4.51
NO2 ПХ/^О
СНз
I
CH2—CH—NO2
° CH3 <ГНз
CH2—C=O
[415 J
H
Схема 8.98
3.12. Присоединение по непредельным связям С—С 221
Как показано на схеме 3.99, система 18-краун-6/сухой цианид
калия в ацетонитриле использована для присоединения CN~ к
метакрилонитрилу [77], Интересно отметить, что ацетонциангид-
рин может служить в качестве источника HCN. Система KCN/
/18-краун-6/ацетонциангидрин является простым, эффективным
и стереоселективным реагентом гидроцианирования [417].
Раствор субстрата и краун-эфира (моль на моль) в подходящем
растворителе (например, в бензоле или ацетонитриле)
добавляют к избытку сухого цианида калия. Затем вводят яцетон-
циангидрин (1,2 моля) и полученную двухфазную систему либо
интенсивно перемешивают, либо кипятят. Выходы достигают
85%. Если возможно образование двух стереоизомеров,
преобладает более термодинамически устойчивый продукт [417].
Н2С=С—CN KCN-'g-"4»DH-6, NC—
I
СНз
KCN, 18-кроун-6
(CH3)SC(OH)CN. СН.
CN CN
1:4— 1:10
Наибольшее распространение получили межфазные реакции,,
проводимые с использованием 50%-ного NaOH как основания
и ониевых солей или краунов как катализаторов [1492, 1539,.
1601]. Иногда применяют смесь твердых ЫаОН/КгСОз в
присутствии ТЭБА и ацетонитрила как растворителя [1715]. Варьируя
концентрацию гидроксида натрия, можно изменять
направление самоконденсации кротонового альдегида (СНгСЫвода,.
Bu4NI, 0°C): при использовании 50%-ного водного NaOH
образуется 6-метилциклогексадиен-1,3-карбоксальдегид (49%), а*
применение 20%-ного водного NaOH ведет только к цис, транс-
5,6-дигидро-2,6-диметил-2Н-пиран-3-карбоксальдегиду (30—
40%) [1539]. В присутствии четвертичного аммониевого
фторида в ТГФ образуется другой димер — 2-этилиден-З-метилглута-
ровый альдегид.
ОАс1
O2N-
о
OaN
ОСН3
ОСН3
(X - COOR, COR)
Н
ОАс4
ОСН3
н
ОСЯэ
осн$
О
НзС—с—н
о
осн3
^ 3.12. Присоединение по непредельным связям С—С 22$
Присоединение малонового и ацетоуксусного эфиров к а, р-
непредельным альдегидам в условиях межфазного катализа в-
присутствии конц. NaOH и ТЭБА приводит к глубокому осмоле-
нию исходных соединений. Однако проведение реакции с
твердым поташем или карбонатом натрия и ТЭБА в бензоле
позволяет получать приемлемые выходы продуктов [1093, 1301, 1837]^
При использовании методики межфазного катализа для
присоединения по Михаэлю диэтилмалоната, ацетилацетона и ди-
бензоилметана к З-нитро-2-енпиранозиду G или его
предшественнику F образовывались только термодинамически менее
устойчивые манно-изомеры [418] (схема 3.100). Однако малоди-
нитрил давал смесь обоих гладко- и манно-изомеров. Как было-
показано, манно-изомер изомеризовался в глюко-изомер в
условиях реакции. Эти реакции проводили в системе бензол/0,2 н.
NaOH при комнатной температуре в присутствии трибутилгек-;
садецилфосфоиийбромнда. Ту же методику использовали для;
другой серии р-глюкозидйых нитросахаров [419] и аналогична
получили предшественники для Сахаров с разветвленными
цепями {1091]. При самой мёдяеяной реакции с использованием;
трибутилгёксиламмонийбромида в течение 4 сут при комнатной*
температуре из Н в системе бензол/водный раствор нитрата
натрия получалась смесь 20% I, 40% К и 20% исходного
продукта [425]. Хотя выход I невелик, метод оказался удачным,,
так как К может быть превращен в Н. В другом примере
использования метода межфазного катализа для реакции Михаэля?
ацетат спирта I был превращен в L с выходом 7Г% [425].
В этом случае в качестве СН-кислоты был использован ацетил-
ацетон, первичный продукт расщеплялся в условиях реакции.
Путем последовательных реакций присоединения по
Михаэлю, внутримолекулярной альдольной конденсации и
дегидратации без выделения промежуточных продуктов (процесс «в
одном горшке») можно синтезировать 5-тиациклогексенкарбокс-
альдегид. Типичным примером является реакция между М и-
кротоновым альдегидом, приводящая к N с выходом 84%'
(СН2С12/50%-ный водн. NaOH/Bu4N+I-) [426]. Из М и акро-
леина получают более сложную смесь продуктов, поскольку-
протекает еще одна — конкурентная —реакция частичного от-1
щепления тйоацетата с образованием кротонового альдегида.1
Взаимодействие а-меркаптокарбонильных соединений с 2-хлор-
акрилоиитрилом в аналогичных условиях приводит к дигидро-
тиофенэпоксидам (О) с превосходными выходами [426]. ;
В присутствии 50%-ного водного едкого натра и ТЭБА фе-
нилвинилсульфон вступает в реакцию с а-хлорпропионнтри-
лом. В течение 1 ч при комнатной температуре образуются сое-'
динеиие Р и следовые количества стереойзомерных
циклопропанов Q и R [421]. В более жестких условиях (40 °С, 2 ч, несколь-
£24 Глава 3. Практическое использование МФК
СНО СНО ^^ХНО
М N .:
^SAc
ст
Г/'т
о
Схема 3.101
ко миллилитров ДМСО в качестве сокатализатора) образуются
только Q и R. Циклизация промежуточного карбаниона,
образующегося при реакции с rper-бутилакрилатом или акрилонит-
рилом, выступающими в качестве активированных олефинов,
происходит очень быстро в мягких условиях, в результате чего
получаются только циклопропановые продукты [421]:
СН3 СНз
I I
i»h—SOa— CH=CHa + НС—CN > Ph—SOa—СН2—СН2—С—CN *■
ci a
р
PhSO2, СНз PhSO2 СХ
*CN + нЛГЛ'СНз
Q R
Схема 3.102
rper-Бутоксид калия в толуоле, ТГФ или в ДМФА и бис-
«(триметилсилил) амид лития в ТГФ катализируют реакцию
соединений типа S [R = H, Ph; R^rper-бутил, (—)-ментил] с трет-
•бутилакрилатом лития в ТГФ [422]. Для проведения этой
реакции могут быть использованы также и условия? межфазного
катализа (Bu4NBr/50%-Hbifi водный NaOH) [422]. Выходы
продуктов при использовании межфазного катализа сравнимы с
выходами, получаемыми при применении других методов. При
использовании межфазных катализаторов или грег-бутоксида
калия из двух возможных изомеров — Z и Е — образуется
преимущественно £-изомер, тогда как при проведении реакции в
других системах основание/растворитель преобладает Z-изомер
J422].
3.12. Присоединение по непредельным связям С-^С ,225
H2C=CH—COO-^№Bu+ R—CHCI—COOR' »
S
R H
R'OOC \ / COO-v^
z
Схема 3.103
Cl
1
- R—C—CH2—CH2
COOR'
/ \
/ X
R
imU R'OOC A /
V
E
—COO—mpem$>
СОО—трт-Ы
H
Такой тип реакции присоединения по Михаэлю с
последующим замыканием циклопропанового кольца наблюдается в
реакциях между ненасыщенными нитрилами, кетонами,
альдегидами, сложными эфирами, фосфонатами и фосфониевыми
солями с а-хлорнитроалканами, галогенмалоновыми эфирами, а-хлор-
нитрилами и а-хлорэфирами [1094, 1095, 1096, 1214, 1215, 1427].
Как правило, всегда необходимо использовать
концентрированный гидроксид натрия и ониевую соль, однако по крайней мере
в некоторых случаях медленная реакция возможна и в
отсутствие катализатора.
Что касается селективности, то она меняется в зависимости
от варианта применяемой каталитической реакции [1096] (см.
подобные эффекты в реакции Дарзана, разд. 3.13.3). В ряде
работ, в которых применялись твердые порошкообразные NaOH
или КгСО3, было показано, что соотношение f/Z-изомеров
может определяться в какой-то степени выбором используемого
катализатора [1214, 1215].
Обращение стереоселективности наблюдается также в
реакции циклизации Т, которую проводят при —40 °С в хлорбензоле
с использованием каталитических количеств грег-бутоксида
калия в присутствии 18-крауна-6 или без него [423]. В первом
случае соотношение U/V равно 62:38, во втором — 38:62. На
последней строчке схемы 3.104 показан новый синтез [1504]
замещенных пиридонов, осуществленный путем экзотермической
реакции (конц. NaOH, ТЭБА, выход 85%), которая включает
присоединение по Михаэлю и присоединение к нитрильной
группе.
Хотя винилацетат и не является акцептором в реакции
Михаэля [1167], он реагирует с фенилацетонитрилом,
соединениями Рейссерта и хлороформом при перемешивании в бензоле в
присутствии концентрированного гидроксида натрия и ТЭБА
[420]. Реакция с хлороформом будет рассмотрена позже вместе
с реакциями присоединения дихлоркарбена.
226 Глава 3. Практическое использование МФК
j
I COOR
о
н
COOR
Н
-COOR
Ph
T
ЕЮ2СЧ 4
С
1
с
OEt
1
£.—Н
Л
IT '
/
■ сн2
Ph
и
>
III
N
ЕЮ2С
Схема 3.104
Ph
V
Н
.CN
О
Ph
CN
+онв
N—COPh + H2C=CH—OAc I
H CN
Схема 3.105
О
CH—ОС—Ph
I
СНз
При использовании оптически активных межфазных
катализаторов возможно стереоселективное присоединение по реакции
Михаэля, при этом могут быть получены высокие оптические
выходы; подробности см. в разд. 3.1.5.
3.12.3. Присоединения к неактивированным двойным связям
При кипячении алкенов с концентрированными водными
растворами НХ и гексадецилтрибутилфосфонийгалогенидов в
течение от 15 мин до 50 ч происходит присоединение НС1, НВг
или HF к олефинам в соответствии с правилом Марковникова.
В этом случае экстрагируется комплекс с водородной связью
[R4P+X--HX-] [1634], Если для генерирования хлора
использовать НС1 и Н2Ог в присутствии хирального катализатора, то
3.12. Присоединение по непредельным связям С—С 227
хлорирование алкенов происходит с частичной асимметрической
индукцией [1292] (см. также разд. 3.1.5).
В то время как ^ыс-гидроксилирование описано в разд. 3.22.1,
вицинальное ^ыс-гидроксиаминирование ненасыщенных
углеводородов следует обсудить в данном разделе. Реагентом для
такого типа реакций служат хлорамин T/OsO4 в присутствии или
в отсутствие межфазного катализатора, однако в этих двух
случаях наблюдается весьма значительное различие в
соотношении диастереомеров [1254].
Присоединение иодтиоцианата к алкенам ускоряется при
использовании двухфазной системы и адогена 464 в качестве
межфазного катализатора [556]. Так, 1,25 ммоля циклогексана,
3 ммоля иода, 6,25 ммоля тиоцианата калия реагируют в
двухфазной системе 10 мл хлороформа/1 мл воды в присутствии
0,96 ммоля адогена 464, давая смесь W и X в соотношении 2:1.
Другие субстраты, в том числе 5а-андростен-2, дают смесь
четырех региоизомеров. Этот метод, не требующий использования
ни хлорида иода, ни сульфолана, может быть, распространен
на синтез вицинальных иодазидов. Реакция проходит с выходом
88% за 48 ч при 20 °С. В качестве катализатора может быть
использован также 18-краун-6.
На последней строчке схемы 3.106 показан необычный метод
синтеза 4,5-дигидро-1Н-1,2-бенздиазепинов. Реакция проходит
при 25—40 °С в системе бензол/вода в присутствии гексадецил-
трибутилфосфонийбромида. В качестве аниона X могут быть:
N3", CN~ или ОАс~; выходы достигают 85% [1526].
Л
Ь, NaN3
Н«О, СНСЬ
аЗоген464
ОН
X R1
Схема 3.106
228 Глава 3. Практическое использование МФК
Нитромеркурирование двойных связей в цыс-полибута диене
действием HgCl2/NaNO2 ускоряется при использовании
двухфазной системы вода/дихлорметан в присутствии BU4NHSO4;
соотношение нитрито- и нитросоединений в конечном продукте
составляет от 0,2 до 0,7 [1606].
3.13. Присоединение по связям С=О и C=N
3.13.1. Бензоиновая конденсация
При перемешивании бензальдегида с 0,13 моля тетрабутил-
аммонийцианида в воде при комнатной температуре проходит
бензоиновая конденсация с выходом 70% [435]. Проведение
реакции в ТГФ или ацетонитриле при комнатной температуре
требует присутствия только 0,02 моля четвертичного
аммониевого цианида [413]. В этом состоит существенное отличие от
общепринятой методики (кипячение в этаноле или метаноле),
в которой применяется 0,2—0,4 моля цианида щелочного
металла на 1 моль бензальдегида. Очень гигроскопичные тетра-
алкиламмониевые цианиды приготовляют из бромидов в
абсолютном метаноле путем ионного обмена на колонке со смолой
ЩА-400 (CN-форма) [436]. Если использовать водный раствор
KCN и аликват 336 [437], то образуются лишь следы бензоина,
вероятно, потому, что хлорид и цианид имеют близкие
константы экстракции. Бензоиновая конденсация осуществляется также
в присутствии 18-крауна-6 или дибензо-18-крауна-6 в качестве
катализаторов при 25—60 °С либо в системе водный цианид
калия/ароматический альдегид без растворителя, либо в системе
твердый KCN/альдегид, растворенный в бензоле или
ацетонитриле [437].
7—47% бензоина получается в результате реакции,
известной как ацильная перегруппировка Виттига (см. [1836]), когда
бензальдегид взаимодействует с гидридом калия и 18-крауном-6.
3.13.2. Конденсации альдольного типа
Альдольные конденсации под действием гидроксида натрия
ускоряются в условиях межфазного катализа. Например,
масляный альдегид в присутствии аликвата 336 дает (после
дегидратации) 2-этилгексен-2-аль с выходом 90%, а в отсутствие
межфазного катализатора выход продукта составляет лишь
14% [1714]. В присутствии ТЭБА порядок реакции конденсации
ацетона меняется со второго на третий (относительно ацетона),
и в результате образуется диацетоновый спирт, который далее
превращается в окись мезитила и форон [1547].
3.13. Присоединение по связям С=О и C=N 229
В ряде патентов и статей описаны: поликонденсация
алифатических и циклических кетонов в системе водный NaOH/алик-
ват 336 [1456], взаимодействие оксида цитраля с ацетоуксус-
ным эфиром (водн. NaOH, ТЭБА) [1594], реакция типа кондеет-
сации Кнёвенагеля для цитраля (твердый Na2CO3, ТЭБА,
бензол; выход 30—61%) [1301]. В этих же условиях другие а,р-не-
предельные альдегиды дают продукты присоединения по
Михаэлю.
При взаимодействии циклопентадиена с кетонами при
комнатной температуре в течение 2 ч в ТГФ в присутствии
порошкообразного гидроксида калия и каталитических количеств
18-крауна-6 получают фульвены с выходом 17—54% [1515].
В нескольких сообщениях [1222] описана конденсация пиперо-
наля с пиперидидом кротоновой кислоты в ДМСО при
использовании 50%-ного гидроксида калия и ТЭБА. При дальнейшем
изучении установлено, что реакции, приведенные на схеме 3.107>
можно осуществить с очень высоким выходом (до 97%), если
проводить их в кипящем толуоле в присутствии безводного
карбоната калия и аликвата 336. В этих случаях R должен быть
ароматическим остатком или третичным ал кил ом [1613].
Схема 3.107
1-Алкины не могут быть проалкилированы в системе водный!
раствор основания/межфазный катализатор, несмотря на то чта
депротонирование в этом случае происходит. Однако
присоединение 1-алкинов к карбонильным соединениям сильно
ускоряется при проведении реакции в системе толуол/13 %-ный водный
NaOH в присутствии аликвата 336 [1838].
При использовании системы 50%-ный NaOH/ТЭБА
ароматические альдегиды конденсируются с N-бензилиденбензиламином
при комнатной температуре с образованием 2-амино-1-арил-Ы-
бензилиден-2-фенилэтанолов, которые можно далее гидролизо-
вать до замещенных аминоэтанолов [1097]:
ArCHO + PhCH=N—CH2Ph »- АгСН(ОН)—CH(Ph)—N=CHPh
230 Глава 3. Практическое использование МФК
В тех же условиях из арилацетонитрилов и 3- или 4-заме-
1ценных нитрозобензолов получают с высокими выходами а-фе-
ниламинофенилацетонитрилы и/или 1-анилино-2-циан-1,2-диарил-
этены в зависимости от имеющихся заместителей [1098]:
AriCH2CN+ON—Аг* * Ar*—C(CN)=N—Ar*
Ari—C(CN)=N—Ar2 + AriCH2CN >- Ar1C(NHAri')=qCN)Ari
.Реакция Кнёвенагеля между циануксусным или малоновым
эфиром и а, р-ненасыщенными альдегидами протекает с
приемлемыми выходами только при использовании системы твердый
К2СО3/ТЭБА/бензол [1093]:
Z—CH2COOR + R2C=CH—CHO ■ >- R2C=CH—CH=C(Z)—COOR
Ранее упоминалось несколько случаев внутримолекулярной
альдольной конденсации [115, 426]. 2-Метилбензоксазол и 2-ме-
тилбензтиазол при использовании системы 50%-ный водный гид-
роксид натрия/ТЭБА реагируют с ароматическими альдегидами
в отсутствие растворителя при комнатной температуре в
течение 1—24 ч [438]. В некоторых случаях вместо стирольных
производных или наряду с ними были выделены спирты:
СНз
V-CH=CH—Аг
X
Схема 3.108
R1—SO2—CH2—R2
А
R3CHO >- R1—SO2—C=CHR3
I
Ra
r
H3C—GN + R—CO—R'
Схема 3.109
Сульфоны A (RJ=Ph или Me2N; R2=H, Ph, Me и Ме2С=СН)
реагируют с ароматическими альдегидами при комнатной
температуре в течение 2—6 ч при использовании системы метиленхло-
3.13. Присоединение по связям С—О и C=N 231
рид/50%-ный водный раствор гидроксида натрия/ТЭБА [439].
В этих условиях ароматические кетоны оказались нереакцион-
носпособными, в то время как алифатические альдегиды давали
продукты самоконденсации. В тех же самых условиях 3,5-дифе-
нилтиадиоксан-З.Б-диоксид (В) может быть получен из А
(R' = CH3; R2 = H) и бензальдегида [440]. Однако при замене
ТЭБА на 18-краун-6 течение реакции усложняется из-за
появления конкурентной реакции Канниццаро. Подробное изучение
факторов, влияющих на этот процесс, показало, что наилучшие
выходы в реакции Канниццаро достигаются в отсутствие каких-
либо катализаторов. Это находится в хорошем согласии с
принятым для реакции Канниццаро шестичленным циклическим
переходным состоянием. Комплексообразование с краун-эфиром
подавляет участие металла в координации переходного
состояния [440].
Непосредственное превращение кетонов в а,р-непредельные
кетоны С при действии таблетированного твердого КОН в
ацетонитриле сопровождается в ряде случаев образованием р.у-не-
насыщенных соединений [215, 857, 1248]. При этом р-гидрокси-
нитрилы не образуются. При проведении реакции в
присутствии такого сорастворителя, как бензол, необходимо
использовать 18-краун-6 в качестве катализатора; при проведении
реакции в ацетонитриле катализатор не нужен. С наименее
удовлетворительными выходами реагируют альдегиды, метилкетоны и
сильноенолировэнные кетоны. Ароматические альдегиды дают
ароматические ненасыщенные нитрилы с выходом 70% при
использовании системы 50%-ный NaOH/толуол/полиэтиленгликоль
(в каталитических количествах) [1831].
Последовательные реакции альдольной конденсации и
элиминирования, осуществляемые при 40 °С в течение 20 мин,
приводят к предшественнику простагландина с хорошим выходом
н sch3 Л
/ * 6 войн. LiOH *" \ if
онс—' V II
но'
Схема 3.110
Первой стадией всех альдольных конденсаций,
рассмотренных до сих пор, является депротонирование под действием гид-
роксидов или карбонатов. Используя в качестве основания
гомогенный раствор Ei4NF-2H2O в ацетонитриле, Рожков и др.
[441] осуществили следующие реакции:
Ph—С=СН + Н3С—СО—СН3 »- Ph—С=С—С(СН3)2ОН
Ph—CO—R+CHS—CN —»- Ph—CR(OH)—CH2CN
232 Глава 3. Практическое использование МФК
TR4NF в ТГФ вызывает также альдолизацию масляного
альдегида [1539]. При использовании системы KF/18-краун-б в
кипящем ксилоле был получен с небольшим выходом продукт
аннелирования циклопептенона, в то время как циклизация с
образованием 2,2-дизамещенных циклопентандиона-1,3 не
наблюдалась [1498].
о
=о
Схема 3.111
реакция
*не иоет
О
3.13.3. Другие типы реакций
Родственными альдольной конденсации и очень удобными
в препаративном отношении, хотя и не всегда
осуществляемыми в межфазных условиях, являются реакции, в которых триме-
тилсилильные соединения атакуются присутствующими в
каталитических количествах тетраалкиламмонийфторидами или
цианидами, что приводит к образованию промежуточных ацети-
лидов, енолятов или алкоксилатов, которые немедленно
присоединяются по карбонильной группе:
OSiMe3
CHO
tPh-C=ceNBu4*]
НО Н
\ .OSiMe3
Y
/
[442]
Me3Si—CH2—COOR
[NBu
—COOR]
С=С—Ph
[24]
R OSiMe3
[443]
CH2—COOR
R OSiMe3
;v X
[444,445]
R OSiMe3
RX 4N3
Схема 3.112
[444]
3.13. Присоединение по связям С=0 и C=N 233
Впоследствии было показано, что для снятия защиты в аце-
тонитриле '[1235]' или дихлорметане [1273] можно
использовать систему твердый KiF/катализатор (ВщС1 или краун-эфир).
Так, первую реакцию, приведенную на схеме 3.112, можно
осуществить при простом нагревании компонентов в метиленхлори-
де в присутствии системы KiF/18-краун-б [1273]. Аналогично
реагируют бензил- и циннамоилтриметилсилан [1832], а в
«Organic Syntheses» описывается присоединение триметилсилил-
цианида к бензохинону (18-краун-6, KCN, тетрахлорид углерода
[1833].
С использованием системы водный KCN/бензол/ТЭБА из
соответствующей кетозы был получен циангидрин (1,2:5,6-ди-О-
циклогексилиден-З-С-циано-а-О-аллофураноза) с выходом 86%
[1099].
В присутствии ониевых солей или краун-эфиров
ароматические альдегиды реагируют с цианидом натрия и ацилхлоридом
с образованием эфиров циангидринов [887, 982, 1292, 1584].
CN
сно о J
+ NaCN + Cl-C-R1"""""'"/
Схема 3.113
Реакции подобного типа наблюдаются и для алифатических
альдегидов [963]. Кроме того, в присутствии аллил- или бен-
зилбромидов, KCN и ТЭБА эфиры циангидринов получаются
с выходом 44—70% [963, 1490]:
RCHO -f KCN + Вг—СН2—СН=СН—R1 v
»- R—CH(CN)—О—СН2—СН=СН—R1
В присутствии межфазных катализаторов ускоряется также
образование бисульфитных производных ароматических
альдегидов [1729]. Более необычным является опубликованный
недавно трехфазный метод, который осуществляется в условиях
кислотного катализа на полистиролсульфокислотной смоле:
растворенные в бензоле ароматические кетоны конденсируются с
формальдегидом (водным), давая 4-арил-1,3-диоксаны с почти
количественным выходом [1652]. При комнатной температуре
и перемешивании в течение 30 мин был осуществлен синтез гли-
цидных нитрилов D с выходом 55—80% из ароматических или
алифатических альдегидов и кетонов и хлорацетонитрила в.
стандартной системе концентрированный раствор гидроксида
натрия/катализатор [448, 1492, 1759]. При этом несимметрична
234 Глава 3. Практическое использование МФК
замещенные кетоны образуют смесь стереоизомеров D. Вместо
ТЭБА можно использовать дибензо-18-краун-6 [302]. Стереосе-
лективность реакции Дарзана изучали путем сравнения
взаимодействия бензальдегида с rper-бутилхлорацетатом, грет-бутил-
2-хлорпропионатом или фенилхлорацетонитрилом в трех
различных экспериментальных условиях [503].
Ph
О
COO-mpem-Ы ph COO—mpem-Ъп Ph CM
ц A 7v л Л
H\/H H\/CH3 H\/Ph
О О О
Е F G
Схема 3.114
В условиях межфазного катализа (молярные количества
BmNBr и бензальдегида, избыток хлорзамещенного соединения
и 50%-ный NaOH) и при использовании гидрида натрия или
грет-бутоксида натрия в ГМФТА наблюдалось одинаковое
соотношение стереоизомеров Е, F и G, что подтверждало
одинаковую степень ассоциации и «плотность» первоначально
образующегося промежуточного продукта. Другое соотношение
стереоизомеров было получено при проведении реакции в ТГФ в
присутствии КаОС(СНз)з [503]. Если же реакцию между а-хлор-
фенилацетонитрилом и бензальдегидом проводить в бензоле в
присутствии 50%-ного водного гидроксида натрия, то стереохи-
мический результат реакции сильно зависит от того, имеется ли
в реакционной смеси ТЭБА или нет [952, ср. также 1834, 1835]:
PhCHO + Ph—CH(C1)CN у \ /И» V/ CN
О О
В присутствии ТЭБА: 92 8
В отсутствие ТЭБА: 32 68
Схема 3.115
Стадия циклизации должна быть медленной в сравнении со
стадией обратимого первоначального присоединения, и (что
доказано) образование эпоксида в условиях межфазного катализа
осуществляется из ионных пар в органической фазе, где нор-
3.13. Присоединение по связям С=0 и C=N 235
мальные сферические эффекты благоприятствуют образованию
того промежуточного продукта, который ведет к грскс-соедине-
нию. В отсутствие катализатора процесс должен
осуществляться на границе раздела фаз, и тогда, как предположили Макоша
и сотр. [952], ион-ионное и ион-дипольное взаимодействия
приводят к образованию предшественника и/или переходного
состояния, наиболее благоприятного для возникновения цис-изомера:
цис- продукт
Схема 3.116
Позднее в условиях межфазного катализа при
использовании Na2CO3 или КгСО3 как твердых оснований в межфазной
реакции Дарзана (см. [1048]) были получены эпоксиды из
карбонильных соединений и 9-хлорфлуорена [1066] и хлорметан-
сульфонамидов [1087].
К оптически активным эпоксидам привела конденсация п-
хлорбензальдегида с фенацилхлоридом в системе толуол/
/10%-ный NaOH в присутствии бензилхининийхлорида при
перемешивании в течение 3 ч [951]:
Я-С1—СвН4—СНО + CIGHaCO—СеН5
Схема 3.117
Аналогично в системе водный цианид натрия/толуол из
бромизобутилфенола получается оптически активный эпоксид.с
оптической чистотой, неизвестной ранее для катализаторов
такого типа* [951]:
О
о
II
С—CR2Br
NaCN
катализатор
Схема 3.118
* Подробную дискуссию об этих и других реакциях с хиральными
катализаторами см. в разд. 3.1.5. • -
236 Глава 3. Практическое использование МФК
Присоединение дикетена к карбонильным соед*
луоле катализируется R«NX в присутствии твец
Предполагается следующий механизм этой pea»
о
нсГ
ииям в
того K2CO3.
[1766]:
о
Схема 3.119
Соединения Рейссерта Н—К были получены с более
высокими выходами, чем позволяют давно разработанные методики.
Реакцию проводят, добавляя постепенно при комнатной
температуре ацетилхлорид к перемешиваемой смеси хинолина или
изохинолина, дихлорметана, водного раствора цианида калия и
ТЗБА [449].
Схема 3.120
Недавно в межфазных условиях на основе фталазина [993]
и 1,6-нафтиридина [1556] были также получены соединения
Рейссерта. Соединения типа I (R=Rh или СН3) реагируют как
■с алифатическими, так и с ароматическими альдегидами илн
жетонами, давая продукты конденсации L и побочно спирт М с
общим выходом, часто превышающим 90% [311, 886]. Эти
превращения осуществляются в бензоле или ацетоиитриле в
присутствии 50%-ного водного раствора гидроксида натрия. Ре-
|—Н
Схема 3.121
3.14. ^-Элиминирование 237
акция проводит даже в отсутствие катализатора, и этот факт
является одним из основных указаний на то, что межфазные
реакции могут происходить в действительности на границе
раздела фаз. Добавки ТЭБА улучшают выходы в случае кетонов,
однако для альдегидов этого не наблюдается [311].
Сероводород присоединяется к тройной связи нитрилов с
образованием тиоамидов при перемешивании бензольного
раствора нитрила с избытком водного раствора сульфида натрия при
70 °С в присутствии четвертичной аммониевой соли и под
давлением от 1 до 2 ат4 [1100, 1472]:
RCN + H2S—^R—CS—NH2
Присоединение хлороформа к СО-группе будет рассмотрено
в разд. 3.20.2.
Ниже приводятся типичные экспериментальные методики:
Конденсация альдольного типа, основание — карбонат калия.
Смесь 0,1 моля анисового альдегида, 0,1 моля пиперидида сор-
45иновой кислоты, 1,0 г аликвата 336 и 4,0 г сухого КгСО3 в 30 мл
толуола нагревают при 90 °С в течение 10 ч. Реакционную смесь
обрабатывают 50 мл воды, слои разделяют, водный слой
дополнительно экстрагируют дихлорметаном. Объединенный
органический экстракт сушат и концентрируют на роторном
испарителе. Остаток обрабатывают 10—20 мл эфира и затем осторожно
добавляют петролейный эфир и оставляют кристаллизоваться
на холоду. Аликват остается в растворе. Пиперидид 7-(4-меток-
сифенил)гептатриен-2,4,6-овой кислоты (т. пл. 133 °С)
получается с выходом 63% [1613].
Присоединение по Михаэлю, основание — фторид калия.
1 ммоль «донора Михаэля» добавляют при комнатной
температуре к смеси 1 ммоля «акцептора Михаэля», 1,5 моля
безводного KF и 0,025 ммоля гексадецилтрибутилфосфонийбромида в
10 мл метиленхлорида. Реакционную смесь перемешивают 2 ч
и затем выливают в воду. Слои разделяют и органический слой
промывают разбавленной НС1, сушат, упаривают и при
необходимости хроматографируют. Выход около 100% [1614].
3.14 ^-Элиминирование
Реакция элиминирования в условиях МФК всегда
сопровождается конкурирующей с ней реакцией замещения, которая в
этом случае является нежелательным побочным процессом (см.
разд. 3.2 и 3.4). Вторичные субстраты (особенно циклогексиль-
ные производные) образуют главным образом продукты
элиминирования, в частности при действии нуклеофилов типа
фторидов илн ацетатов. Третичные соединения дают олефины в каче-
238 Глава 3. Практическое использование МФК
стве единственного продукта. Подробное изучение покйзало, что
под действием тетраэтиламмонийфторида в ацетонитоиле
реакция идёт быстрее, а продуктов отщепления по Зайц/ву в общей
смеси продуктов реакции значительно больше, чем/ри действии-
rper-бутоксида в г/эег-бутаноле либо ДМСО или этоксида в
этаноле [453]. В препаративных целях такое элиминирование
проводят большей частью, как это показано ниж<^ в гомогенных
растворах, но иногда используют систему твеодый KF/катали-
затор [450, 1510]. Твердый KF, высушенный сыюмощью
глубокого охлаждения, является, по-видимому, очень активным реа-
агентом даже в отсутствие катализатора [1606]. Имеется обзор,
посвященный использованию фторидов в качестве оснований
[1664].
:><
СН
R
-CHa—
чиОи^ктиксоно-М-
H3CCN, 25 «С
СНз
CHz—С—OSO2CH3
СНз
краан-6) ДгС_
Bu<,NBr,TT4'>
C-R
СНз
R V
[450]
-^-СН
а—СН-
хсн3
[45Ц
Rl SiPh3
R3—С—С=СН2 "4КР-ДМСО> ;С=С=СН3 [452]
I £ч, кони. темп. /
>
I
С!
D2
Схема 3.122
Сходные реагенты и условия реакции были с успехом
использованы для проведения элиминирования в более сложных
случаях [55, 520], например
NC
аД:
МеООС
СООМе
KCI/крааи.А, мс—С=С—СООМе
хсооме чльЧ1Олан
СООМе
b-ci
н
tOOMe МеООС
Схема 3.123
гСООМе
-
С1
При дегидрогалогенировании при температурах до 250 °С в
лрисутствии четвертичных аммониевых или фосфониевых солей
образуется газообразный НС1 и алкен. Ниже приведены типич-
3.14. ^-Элиминирование . 239
ные примеру такого элиминирования:
снснДсн2ci н3с-сн=с—ch2ci + н-,с-с=сн—сн2а
с, а а а
+ Н3С—СН—С=СН2 [4631
а а
О—СН2-СН-СН-)|Н2С1 RaNX a > Н2С=С-СН=СН2 [464]
CI
С -СН2—СН2 CN -^|. H2C=CH—CN [465]
Схема 3.124
Используя систему твердый KF/18-краун-б, можно получать при
элиминировании нестабильные продукты, показанные на
схеме 3.125.
[1102]
[ПОЗ]
Схема 3.125
Другим типом двухфазной реакции элиминирования
является легко протекающее дегалогенирование вицинальных дибром-
алканов (А) под влиянием иона иодида.
Rl—CH—CH—R2 »°8"N''-N"S'°33 R._CH=CH_^i.
I I катализатор
Вг Вг
А
Схема 3.126
Раствор субстрата в толуоле перемешивают при 90 °С с
водным раствором иодида натрия (10 мол. %) и избытком
тиосульфата натрия в присутствии 10 мол.% трибутилгексадецилфосфо-
нийбромида [456]. Без катализатора под влиянием одного
240 Глава 3. Практическое использование МФК
сульфата натрия реакция тоже идет, но скорость ее намного
меньше. Соединение А в мезо- или эритро-формш реагирует
быстрее, давая только транс-олефин, а ^./-диастеоеомеры или
rpeo-соединения превращаются в смесь цис- и rpJwc-олефинов.
Эта реакция может быть проведена и как «трехфазный»
каталитический процесс со специально приготовленн/нм
катализатором, связанным с сшитым полистиролом [62].
Для элиминирования элементов воды от бен/альдоксима под
влиянием KCN или KF и катализатора в двухфазной системе
был предложен механизм основного катализа /929]:
AiCH=NOH >- ArCN *
Л
С такими основаниями, как rper-бутоксид калия, реакции
проводят большей частью в полярных апротонных
растворителях, однако иногда используют и бензол, в котором такие
основания растворяются довольно плохо. В том и другом случае при-!
бавление краун-эфира не только изменяет растворимость, но,
кроме того, оказывает сильное влияние на ассоциацию ионов.
Это приводит, как уже указывалось выше, к радикальному
изменению скоростей реакций, ориентации и стереохимии р-элими-
нирования [454, обзор: 455]. Гладко и в мягких условиях
проходит дегидрогалогенирование хлор- и бромалканов при нагре-
вании их с твердым грег-бутоксидом калия и 1 мол. % 18-крау-
на-6 в петролейном эфире при температуре более низкой, чем
температура кипения образующегося алкена. В этих условиях бор-
нилхлорид, например, за 6 ч при 120°С образует 92% борнена
без примеси камфена и трициклена [1104]. В сходных условиях
из 1,2- и 1,1-дигалогенидов можно получить 1-алкины. Геминаль-
ные дихлориды (полученные из кетонов и PCls) с прекрасным
выходом дают замещенные алкины. Изомеризация этих алки-
нов в аллены или сдвиг тройной связи в другое положение
протекает существенно медленнее, чем обычный процесс
элиминирования. £-Галогеналкены подвергаются с«м-элиминированик>
под действием системы грег-ВиОК/краун, давая алкины с
хорошим выходом [1105].
Обратимся теперь к рассмотрению реакций, протекающих
под действием оснований, наиболее часто используемых в
МФК.
Такими основаниями являются гидроксиды щелочных
металлов. Дегидрогалогенирование в их присутствии проходит только
в тех случаях, когда в соединении имеется протон,
активированный электроноакцепторным заместителем или находящийся в
бензильном или винилогичном к бензильному положению. Такие
МФК-реакции очень похожи на обсуждавшиеся выше реакции
в гомогенных условиях, при которых выделяется газообразный
НС1, и в том и в другом случае используются одинаковые ката-
3.14. ^-Элиминирование 241
лизаторы\ Однако в общем при применении водного раствора
гидроксида натрия требуются значительно более низкие
температуры [13М, 1591, 1713]
—CH(<5j)—СН=СН2
С1ХН—СНС1
PhO—CFjppCHCl
ArO—CHa—CH,— Cl
ониевая соль
вода. NaOH
аликват 336
СН2=СС1—СН=СН2 [457—4611
вода. NaOH
тритон Б
*■
водн. КОН
Bu4NHSO4
вода. NaOH
>■ С12С=СН2 [462]
PhO—CF=CC12 J472]
ArO—CH=CH2 [1319]
Элиминирование НВг от фенетилбромида с 50%-ным NaOH
при использовании в качестве катализатора н-бутилтриэтилам-
монийхлорида при 90 °С полностью заканчивается за 2 ч. Беа
катализатора в этих условиях образуется только 1% стирола
[466, 467]. Соединение В дает при комнатной температуре 60%.
замещенного бутадиена за 4 ч [466, 467], а для
элиминирования С с выходом 83% необходимо всего 3 ч [468].
Ph—Ша—СНаВг
-*■ P.hCH=CH2
(n-F—С6Н4)2С=СН—СН2-СН2—С1
NaOH/ТЭБА
(n-F—СвН4)С=СН—СН=СН2)
в
Ph—СН(Вг)—CHgBr
с
бензол, аликват 336
водн. КОН
Ph—(Br)C=CHa
Изучено влияние длины цепи (R) катализатора на выход
стирола в стандартных условиях (60 °С, конц. NaOH) [467]:
Катализатор REt^N+Br~
Выход, % 3
R
Выход, %
45,5
44
к-С4Н9
12
42
к-С5Нц
50
53
38
Возрастание выхода в начале серии не является
неожиданным, учитывая изменение в растворимости солей. Однако
последующее уменьшение выхода объяснить труднее. Подобное
явление было отмечено и в случае реакций с дигалокарбенами (см.
ниже). Другие примеры элиминирования НХ от соединений с
активированным водородом в системе водн. NaOH/катализатор
можно найти в работах [1815—1823].
542 Глава 3. Практическое использование МФК
В работах [469, 470] сообщалось о получении ацетиленов
при дегидрогалогенировании в присутствии тритонаJB (бензил-
триметиламмонийгидроксида). Доступный продажный
продукт — раствор этой щелочи в метаноле — был Превращен в
толуольные, бензольные или пиридиновые раствсшы путем
разбавления метанольного раствора избытком нового растворителя
и отгонки большей части метанола. Тритон В/не всегда
полностью растворяется в бензоле или в толуоле/однако в
большинстве случаев это не имеет значения. Для Работы с
нестойкими кетонами или сложными эфирами (которые не
подвергаются омылению) лучше использовать пиридин. Температура и
зремя реакции лежат в пределах от 70°С/$0 мин до (—10) —
(—30) °С/1—5 мин. Некоторые типичные примеры реакций,
идущих с выходом 40—85%. приведены ниже:
п-Х—Свн4—СС12—СН3 v
n-X-CeH4— CsCH
n-X—CeH4—CC1=CH2 *
R_€H=CBr2 » R—C=C—Br [R = Ph, CH(OEt)2, CC1=CCI2]
n_X—C6H4—CH=CBr—COOiMe »» n-X—Cgfy—CsC-COOiMe
CI2C=CC1—CH=CX—COiMe >- CISC=CC1—C==C—COiMe
Недавно эти же реакции были проведены с высокими
выходами (от 70 до 87%) в истинных условиях МФК [471].
Субстраты, растворенные в пентане, перемешивали с 50%-ным вод-
яшм раствором гидроксида натрия и с бисульфатом тетрабутил-
аммония (более 2 молей). Некоторые из этих реакций
элиминирования проходили экзотермично. Для понимания механизма
имеет значение тот факт, что в этих мягких условиях при за-
змене бисульфата тетрабутиламмония на его бромид или иодид
никакой реакции не происходит; не протекает реакция и в
присутствии ТЭБА или гексадецилтриметиламмонийбромида [471].
Метод был проверен и опубликован в «Органических
синтезах» [1306]. Однако он имеет серьезный недостаток, поскольку
требует применения большого количества дорогого BiuNHSAt-
ГНедавно Лиссель нашел, что такого рода элиминирование
проходит и с каталитическим количеством аммониевой соли, если
«она высоколипофильна, например с тетраоктиламмонийброми-
дом [1611] (см. ниже экспериментальную методику).
Простой метод проведения элиминирования по Гофману
состоит в нагревании аммониевых солей с гидроксидом калия в
-метиловом эфире полиэтиленгликоля [1275].
Рассмотрим теперь более подробно механизм МФК-элимини-
■рования в присутствии NaOH. Совершенно очевидно, что
существуют по крайней мере два маршрута реакции.
\ 3.14. ^-Элиминирование 24i£
Маршрут а: Экстракция 0Н~ в органическую фазу с
последующим элиминированием требует молярного количества
катализатора в iiape с первоначальным противоионом, более
гидрофильным, чек! гидроксил. Эта реакция легко проходит при
низких температурах. Не очень подходит для этой цели молярное
количество тритона В, не полностью растворенного в толуоле
или в аналогичном растворителе, поскольку образующийся га-
логенид более растворим, чем гидроксид.
Маршрут б: Элиминирование, проходящее с участием ионной
пары R4N+X-: ъ
\ Ан + NR4®Xe
X
Схема 3.127
Дополнительным подтверждением возможности такого
маршрута является тот факт, что элиминирование может протекать
и без щелочи с выделением газообразного НС1. Реальность
существования R4N+HX2~ в органических средах обсуждалась в
разд. 3.2. В присутствии оснований R4N+HX2~ нейтрализуется
на поверхности раздела фаз, что сдвигает равновесие реакции
элиминирования — присоединения и увеличивает общее время
реакции. Ниже приведены типичные экспериментальные
методики.
Щелочная система трет-бутоксид калия/краун-эфир
2-Этилгексен-1: Раствор 20 ммолей 1-хлор-2-этилгексана в.
20 мл петролейного эфира (т. кип. 160—180 °С) перемешивают
с 2,5 г трет-ВиОК и 0,2 ммоля 18-крауна-6 в течение 7 ч при
130 °С. Затем смесь выливают в воду. После отделения
органическую фазу промывают водой, а водную фазу несколько раз-,
экстрагируют петролейным эфиром. Все органические
экстракты объединяют, сушат и перегоняют. Выход олефина 93%, т. кип..
119 °С. Если необходимо получить высококипящие олефины, то-
в качестве растворителя используют низкокипящий петролей-
ный эфир [11041. Аналогичным образом можно получать и алкн-
ны [1105].
Щелочная система порошкообразный гидроксид калия/OcUNBr
Гексадецин-1: Раствор 0,1 моля 1,2-дибромгексадекана и
1 ммоля тетраоктиламмонийбромида в 100 мл петролейного
эфира (т. кип. 85—100 °С) прибавляют к 14 г сухого
порошкообразного КОН и перемешивают 6 ч при 90 °С. После этого смесь
фильтруют и перегоняют, т. кип. 95°С/0,1 мм рт. ст., выход
16*
244 Глава 3. Практическое использование МФК
Если необходимо получить низкокипящие алкины, то в качестве
растворителя используют высококипящий петролеТшый эфир
[1611].
Щелочная система 50%-ный водный гидроксид
натрия!ButNHSOt и активированные субстраты
Арилвиниловые эфиры: 5 ммолей арил-2-хло~рэтилового
эфира в 50 мл бензола и 5 мл 50%-ного водного раствора NaOH
перемешивают с 5 ммолями BU4NHSO4 в течение 1 ч при
комнатной температуре. После обычной обработки Получают продукт
реакции с выходом 90—96% [1319].
3.15. Реакция гидролиза
Многие реакции гидролиза, как полагают, проходят в
мицеллах [473] или в инвертных мицеллах [473, 474]. Однако
непосредственное сравнение скоростей реакций и других физико-
химических данных, полученных при проведении этих реакций
в разбавленных «гомогенных» растворах, с препаративным
омылением и другими двухфазными МФК-реакциями гидролиза,
невозможно.
■3.15.1. Различные реакции гидролиза
При нагревании пропиленоксида с водой и метилтрибутил-
■фосфонийиодидом в автоклаве в атмосфере СО2 образуется про-
ландиол-1,2 почти без примесей полигликолей [1527]. Гидролиз
лростых и сложных силильных эфиров (включая защитные
$- (триметилсилил) этилоксикарбониламиногруппы) происходит
лри перемешивании с Bu4NCl и дигидратом фторида калия в
ацетонитриле или ТГФ [1235]. С помощью МФК-гидролиза
хлоридов были получены также стерически затрудненные триал-
коксисиланолы [1619].
Гидролиз алкилгалогенидов для получения спиртов в
условиях МФК невыгоден, поскольку при этом в качестве главных
продуктов образуются простые эфиры, иногда с довольно
хорошими выходами (разд. 3.7). Только в некоторых особых случаях
эта реакция гидролиза проходит успешно. Например, гидролиз
Н3С—С(С1) = СН—СНгС1 водными растворами гидроксидов
щелочных металлов ускоряется аммониевыми солями [225]. При
кипячении в бензоле в течение 48 ч соединения А (Х=Вг или
С1) с системой водный NaOH/аликват 336 образуется
соединение В с выходом 50 или 64% соответственно [246]. Довольно
удивительно, что при этом не. происходит образования
нормальных продуктов реакции Фаворского. В водном ЫаНСОз с
3.15. Реакции гидролиза 245
гексадецилтрибутилфосфонийбромидом при 100 °С и под
давлением СОг Происходит гидролиз 1,2-дихлорпропана с
образованием диолаЧпОб]. Галогениды гидролизуются также при
кипячении в бензоле в течение 4 ч с амберлитом А26 в карбонатной
форме [1858].
ОН ^\ ХС13 /\ /СООН
с
Схема 3.128
Гидролиз чистого а,а,а-трихлортолуола (С) до бензойной
кислоты в 20%-ном водном растворе гидроксида натрия при
80 °С сильно ускоряется добавлением 0,01 М гексадецилтриме-
тиламмонийбромида или в меньшей степени 0,006 М
нейтрального ПАВ — брий 35*. С 0,02 М BmN+Br- эта реакция шла
хуже [475]. Разбавление С бензолом при использовании катион-
ного ПАВ увеличивает время реакции в 11 раз. Авторы
интерпретируют эти данные как косвенное, хотя и не точное,
доказательство эмульсионного или мицеллярного катализа, а не
истинного МФК-процесса.
Продолжительное кипячение 2-нитропропана с водным тетра-
этиламмонийфторидом приводит к гидролизу нитрогруппы через
промежуточное образование сч«-таутомера; в результате
гидролиза образуется ацетон [1092].
PhPO(OBu)F быстро гидролизуется в щелочном растворе
пероксида водорода. В этом случае наиболее эффективны
катализаторы, содержащие ОН-группу [1859].
Гидролиз сульфурилхлоридов в двухфазной системе
существенно ускоряется четвертичными аммониевыми солями. На
основании ранних кинетических исследований [476] гидролиза
SO2Cl2 в тетрахлоруглероде с катализаторами типа
детергентов был сделан вывод, что реакция проходит межфазно, а
детергент ускоряет реакцию, уменьшая поверхностное натяжение.
Старке [4] показал, что гидролиз чистых или растворенных
длинноцепочечных алкансульфонилхлоридов водным раствором
гидроксида натрия проходит очень медленно. Однако
добавление небольшого количества четвертичного аммониевого
катализатора приводит к тому, что реакция протекает быстро и с
разогревом [4, 38, 39]. Предварительные результаты по гидролизу
тозилфторида гидроксидом натрия указывают на сочетание
• Брий 35 —это C,2H2s(O—CHs—СН2)2з0Н.
246 Глава 3. Практическое использование МФК
МФК с катализом на поверхности [1720]. Частичный/гидролиз
нитрилов до амидов был осуществлен в системах пиоидин/вода/
/гидроксид калия/ВщЫВг [1501] или дихлорметан/|года/гидрок-
сид натрия/30 %-ный H2O2/B114NHSO4 [1232].
Очень мягкий метод окислительного гидролиза тиокеталей
и тиоацеталей состоит в перемешивании субстрата в водно-
дихлорметановой смеси с 1 мол. экв. гидробромрда пиридиний-
пербромида, 1 мол. экв. пиридина и 0,1 мол. экв. Bi^NBr при
комнатной температуре [1758].
3.15.2. Омыление сложных эфиров
Для омыления стерически затрудненных сложных эфиров,
которые трудно гидролизуются в обычных условиях, оказалось
очень полезным использование краун-эфиров и криптатов. Пе-
дерсен и сотр. [504, 505] обнаружили, что дициклогексано-18-
краун-6 способствует растворению гидроксидов калия и натрия
в бензоле. При этом можно приготовить растворы
концентрацией до 1 моль/л. В большинстве случаев этот комплекс
сначала получают в метаноле,' который затем замещают на бензол»
однако около 1% СН3ОН удалить не удается. Таким образом,,
хотя в конечном растворе находятся как ионы ОН~~, так и
СН8О~ [43, 504], этого вполне достаточно для гидролиза эфиров
2,4,6-триметилбензойной кислоты. Бензольный раствор метил-
тетрадеканоата гидролизуется при комнатной температуре при
перемешивании с порошкообразным гидроксидом
калия/каталитическое количество 18-крауна-б в течение 12 ч [969].
Реакции гидролиза и декарбоксилирование сложного эфира,
содержащего а-активирующую группу, были проведены «в
одном горшке» [35].
х
R1—С—COOEt
Ra
КОН/18-к.раци-б^
комн. темп.
X
R1—С—СО2еКв
R2
~1ОО°С
X = —COOEt, —COR, —CN R1—С—Н
R2
Схема S.129
В этом случае стадия омыления проходит в системе
гидроксид калия/эквимолярное количество 18-крауна-6 в смеси около
3.15. Реакции гидролиза 247
6 об.% этгкнола/бензол при комнатной температуре. Если
неспособные к еиолизации субстраты гидролизуются очень быстро,
то для гидр&шза кислых субстратов, таких, как диэтилфенил-
малонат, требуется при комнатной температуре почти 20 ч.
Стадия декарбоксилирования проходит при кипячении в течение
2—20 ч. Если'йарбоксилат декарбоксилируется с трудом
(например, моноэтилацетамидомалонат), то в качестве
растворителя применяют диоксан. Эта методика получения «в одном
горшке» не пригодна для декарбоксилирования очень кислых
субстратов (р/Са<Ю), так как реакция тормозится образованием
нереакционноспособной енолятной формы. Известны и другие
реакции, в которых краун-эфир оказывает заметное влияние на
скорость декарбоксилирования [506, 507, 1101].
Рассмотренные выше эксперименты проводили в
гомогенных растворах. Дитрих и Леен [359] сравнили эффективность
системы гидроксид калия/дициклогексано-18-краун-6 в
гомогенном растворе толуол/1 % метанол с эффективностью смеси
твердого порошкообразного гидроксида калия/криптофикс [222]
(5 на схеме 3.5). Результаты омыления метилмезитоата
приведены в табл. 3.11.
Таблица 3.11. Гидролиз метилмезитоатов
Катализатор
Отсутствует
Дициклогексаио-18-кра-
ун-6
Криптофикс [222]
Криптофикс [222]
Растворитель
Пропанол-1
(гомогенные условия)
Толуол
(гомогенные условия)
Толуол
(гетерогенные условия)
ДМСО
(гомогенные условия)
Время,
ч
5
31
12
2 мии
Температура, «С
75
74
25
25
Выход,
%
0
58
80
50
Из этих результатов ясно, что мягкий МФК'процесс с крип-
тофиксом [222] в качестве катализатора очень удобен для сте-
рически затрудненных соединений, хотя стоимость реагента
может препятствовать его широкому использованию. Следует
подчеркнуть, что по отношению к сложному эфиру необходимо
использовать эквивалентные количества краун-эфира или крип-
танда.
Показано, что дешевые ониевые соли пригодны как
катализаторы и в простых реакциях омыления. Так, гидролиз диметил-
адипата 50%-ным водным гидроксидом натрия в отсутствие
катализатора при комнатной температуре проходит медленно, но
248 Глава 3. Практическое использование МФК
Таблица 3.12. МФК-омыление сложных эфиров с использованием
в качестве катализатора аликвата 336 в стандартных условиях (10 ~2 моля
сложного эфира, 6 мл легкого петролейного вфира, 3 мл Б0%«ного водного-
NaOH, 10~4 моля аликвата; 1 ч, комнатная температура) [1109]
Эфир
Диэтиладипат
Диэтилфталат
Этилдекаиоат
Этилстеарат
я-Октилстеарат
Этил-2-фенилбутират
Этил-3-фенилпропиоиат
Этил-я-нитробеизоат
к-Бутилбензоат
Изопропвлбензоат
Бензил бензоат
Этилбензоат
Метилбензоат
Этил-2,4,6-триметил бензоат
Метилсалицилат
Этилфенил ацетат
«-Октил ацетат
Циклогексилацетат
Этилпропионат
Этил-2,2-диметилпропвонат
Изопропил-2,2-диметилпропионат
Этил-3,3-диметилбутнрат
Этил-3-метилбутират
Циклогексил-3-метилбутират
Этил-З-метилбутеН'2-оат
Этил-и-толуолсульфонат
к-Октил-п-толуолсульфонат
Выход, %
(а) без
катализатора
11
14
7
13
5
3
20
10
27
15
40
5
12
8
95
20
6
13
37
27
54
19
24
20
20
9
5
(б) с
катализатором
50
46
7
20
13
8
32
47
33
19
56
24
34
9
100
98
42
44
85
41
72
36
27
22
26
12
6
Ускорение
(бЖа)
4,5
3,2
1
1,5
2.6
2.7
1,6
4,7
1.2
1,3
1.4
4,8
2.8
1,1
1.05
4.9
7,0
3.4
2,3
1.5
1.3
1.9
1.1
1.1
1,3
1.3
1.2
при добавлении аликвата 336 происходит быстр"ая
экзотермическая реакция [4, 38, 39]. Однако, по-видимому, в тех случаях,
когда карбоксильная часть сложного эфира имеет длинную
цепь, карбоксилат образует ассоциаты с катионом
катализатора и каталитический эффект пропадает [4],
3.15. Реакции гидролиза 249
Омыление в условиях МФК было подробно изучено [477,
1109] (табл. 3.12). Растворители влияли на реакцию омыления
диэтиладипйта в системе 50%-ный водный гидроксид натрия/
/аликват 336 следующим образом: петролейный эфир лучше
бензола; бензол лучше эфира; эфир лучше метиленхлорида.
Эти результаты на первый взгляд удивительны, так как в
«нормальных» МФК-реакциях наблюдается обратный порядок
влияния растворителей, когда наименее полярный растворитель
является наилучшим. Это может быть истолковано как еще одно
доказательство того, что ОН~ не переносится в органическую
фазу в отличие от реакций с краун-эфирами и криптатами,
рассмотренных выше. В стандартных условиях (0,01 моля
эфира, 0,05 моля концентрированного гидроксида натрия, 10~* моля
катализатора, 1 ч; комнатная температура) эфиры
гидрофильных гидроксикарбоновых и дикарбоновых кислот гидролизуются
намного быстрее, чем простые длинноцепочечные аналоги. В
реакциях с последними каталитический эффект аммониевых солей
не наблюдается или же проявляется очень слабо. Омыление
стерически затрудненных метилмезитоатов слабо ускоряется
тетрабутиламмониевым катализатором при 100 СС. При
варьировании аниона катализатора в реакции омыления диэтиладипа-
та в петролейном эфире эффективность тетрабутиламмониевых
солей возрастала в ряду: иодид<бромид<хлорид<бисуль-
фат<гидроадипат.
При замене симметричных катионов в стандартных условиях
(0,01 моля диэтиладипата, 5 мл петролейного эфира, 0,05 моля
концентрированного гидроксида натрия, 10~* моля
катализатора; 1 ч, комнатная температура) были получены следующие
результаты:
Катализатор (C4H9)4NBr (CsH,,),,NBr (QH^hNBr (C7H15)4NBr (C8HI7)«NBr
Выход, % 18 20 45 46 39
Наилучшим катализатором из имеющихся в продаже
аммониевых солей является бисульфат тетрабутиламмония. Столь же
эффективен, однако, и неионный ПАВ (D). Таким же хорошим
оказалось и соединение Е (R = H, CH3) см. [477]).
Н88С1в-О(СЦ8-СН,-О)м-О-С0-СН8
D Е
Хотя анионный поверхностноактивный стеарат натрия
считается довольно хорошим катализатором, в этих реакционных
условиях краун-эфиры были не очень эффективными. Более
подробное обсуждение различных факторов, влияющих на эту
реакцию, см. в разд. 2.2.
На приведенной ниже схеме представлен простой метод
превращения альдегидов в а,§-ненасыщенные соединения. Послед-
250 Глава 3. Практическое использование МФК
няя стадия проводится в водном NaOH в присутствии Bu4NHSO4
[1439]:
NtBC
RCH2—CH2—CHO v R—СНа—СН=СН—ОАс ——■>
: »- R—СНВг—СН=СН—ОАс > R—СН=СН—СНО
В заключение можно сказать, что проведение омыления в
условиях МФК синтетически выгодно в случае стерически затруд-^
ненных эфиров. При этом следует использовать систему
твердый- гидроксид калия/толуол и краун-эфиры или криптанды в:
качестве катализаторов. Кроме того, скорость гидролиза
простых эфиров карбоновых кислот концентрированным водным
раствором гидроксида натрия значительно выше для
гидрофильных карбоксилатов. Хорошими катализаторами являются
четвертичные аммониевые соли, особенно Bu4NHSO4 и некоторые
анионные и неионные ПАВ. Это указывает на то, что может
осуществляться любой из трех возможных механизмов: реакции
на поверхности, мицеллярный катализ или «истинная» МФК-
реакция. В зависимости от условий может реализоваться
каждый из этих механизмов. Как было показано раньше, при МФК
возможна экстракция кислот в форме ионной пары R4N+X---HY
[57]. Ранние работы, в которых рассматривалось кислотное
МФК-омыление, оказались ошибочными [1202, 1348]. Однако
недавно было описано мягкое и селективное расщепление трет-
бутиловых эфиров, которое происходит при перемешивании с
10 М H2SO4, 9 М НВг или 12 М НС1 при комнатной
температуре в присутствии С^НззРВизВг. В отличие от щелочного
гидролиза этот метод не лимитируется гидрофильностью кислот
[1857].
3.16. Генерирование и превращение фосфониевых
и сульфониевых илидов
3.16.1. Реакция Виттига
Первой стадией нормальной реакции Виттига является де-
протонирование фосфониевой (в большинстве случаев трифенил-
фосфониевой) соли [478, 480]:
илиЭ илен
Схема 3.130
3.16. Генерирование и превращение илидов 251
Кислотно-основной характер системы определяется типом
заместителей R3 и R4: электроноакцепторные группы усиливают
кислотность соли или основность соответствующего илида.
В этих случаях для отрыва а-протона пригодны слабые
основания, например карбонат калия. В более общем случае, когда
заместителей, сильно повышающих кислотность, мало или они
отсутствуют, используют, как правило, сильные щелочи: литий-
органические соединения, амид натрия в жидком аммиаке, ал-
коксиды щелочных металлов в гидроксильных растворителях
или в диметилсульфоксиде либо димсильный анион в ДМСО.
Стабилизованные (наличием групп R=COOR, CN и др.) илиды
можно выделить. В то же время хорошо известно, что обычные
фосфониевые илиды чувствительны и к воде, и к кислороду,
поэтому стандартная методика требует применения тщательно
высушенных растворителей и инертной атмосферы. Под
действием воды происходит необратимый распад с образованием ал-
килдифенилфосфина и бензола. На воздухе протекают
следующие реакции:
HPh
O=PPh3 + R2C=O RaC
Схема 3.131
Скорость распада фосфониевых солей под действием гидрокси-
да натрия зависит от растворителя (СНгС12>«-бутанол^>бензол,
гексан>эфир >без растворителя) и, по-видимому,
катализируется тетрабутиламмонийгалогенидами С1~>Вг~>1"-) [1242].
Эти факторы действуют и при проведении реакции Виттига
в обычных условиях. На второй стадии илид и карбонильное
соединение взаимодействуют, образуя бетаин в форме трео- или
эрытро-стереоизомеров. Они в свою очередь превращаются
соответственно в Е- или Z-олефины. Взаимопревращение
изомеров бетаинов возможно только при его распаде до исходных
соединений. Поскольку этот процесс идет очень медленно или
вообще не идет, то влиять на соотношение Z/E трудно.
Резонансно стабилизованные илйды (например, R3=COOEt) дают
главным образом £-продукт, а вестабилизованные илиды
(например, R3=aлкил) дают в основном Z-олефины. Небольших
изменений в соотношении Z/f-изомеров можно достигнуть
подбором растворителя и добавлением солей лития [480, 481, 482].
252 Глава 3. Практическое использование МФК
Таким образом, на основе того, что нам известно о стандартной
реакции Виттига, на первый взгляд перспектива улучшения ее
проведения в МФК-условиях маловероятна. Однако в 1973 г.
Меркль и Мерц [483] показали, что для проведения реакции
Виттига даже с неактивированными фосфониевыми солями
можно использовать систему концентрированный раствор гид-
роксида натрия/органический растворитель. С тех пор эту
препаративно очень простую методику. широко используют [483].
Вопросы, связанные с механизмом реакции, все еще остаются
не совсем ясными. Некоторые авторы использовали в качестве
катализаторов аммониевые соли или краун-эфиры, другие
обходились без катализаторов, аргументируя это тем, что, как
известно, фосфониевые соли сами являются межфазными
катализаторами. Однако во многих случаях при использовании
водного гидроксида натрия первая стадия депротонирования
проходит, по-видимому, межфазно. Образующийся илен является
нейтральной частицей, и поэтому для облегчения его диффузии
в глубь органического слоя катализатор не нужен. Это
приводит к тому, что конкурирующая реакция с водой не
происходит. В других случаях в качестве щелочей использовали
твердый грег-бутоксид калия или карбонат калия [484], твердый
фторид калия [1297] или твердый гидроксид натрия [1782].
Реакция между альдегидами и н-бутилтрифенилфосфоний-
бромидом в диоксане с твердым К2СО3 приводит к
одинаковому выходу продуктов в присутствии как дициклогексано-18-
крауна-6, так и небольшого количества воды [1599], в то
время как в абсолютном растворителе олефин почти не
образуется.
Предположение о том, что депротонирование во всех
случаях проходит на поверхности раздела фаз, причем присутствие 1
катализатора не является необходимым, находится в
противоречии с некоторыми данными, имеющимися в литературе. Для
выяснения этого вопроса типичная МФК-реакция Виттига была
проведена в следующих системах основание/растворитель:
твердый КгСОз/бензол, трег-бутоксид калия/бензол, 50%-ный
водный NaOH/дихлорметан, твердый NaOH/бензол или дихлорме-
тан, твердый KF/бензол или дихлорметан. Во всех случаях
выходы в присутствии катализатора и без него были очень близки
.'[1632]. Таким образом, по крайней мере в большинстве
случаев влияние МФ-катализаторов в реакции Виттига очень
невелико. С точки зрения механизма реакцию Виттига не следовало
бы включать в число МФК-реакций. Но, как будет показано
ниже, весьма близкая к ней реакция Хорнера — Эммонса,
несомненно, относится к МФК-реакциям!
В различных растворителях (ТГФ, бензоле, метиленхлориде)
соли типа [Ph3P—СН2—R3]+X~ (где К3 = арил) с различными
3.16. Генерирование и превращение илидов 253.
альдегидами образуют олефины с выходом 60—80% как с
твердыми, так и с водными основаниями как в присутствии
катализаторов, так и без них [483—486]. Однако если R3 = CH3, то в-
системе концентрированный гидроксид натрия/метиленхлорид
без катализатора выходы составляют только 20—30% [483].
В работе [484] сообщалось, что при реакции с твердым трет-
бутоксидом калия или карбонтом калия и 18-крауном-6 в ТГФ
или метиленхлориде некоторые субстраты дают очень высокие
выходы. Однако эти данные в препаративных условиях
воспроизвести не удалось [1634].
Реакция Виттига между глиоксалем и арилметилфосфоние-
выми солями по обеим альдегидным группам в присутствии
водного раствора гидроксида натрия ведет к образованию
только около 20% диарилбутадиена [487].
При использовании в качестве органической фазы бензола:
выход олефина зависит от концентрации гидроксида натрия:
при повышении концентрации сначала наблюдается увеличение
выхода до максимума; дальнейшее повышение концентрации:
приводит к уменьшению выхода. Оптимальные условия
определяются не только анионом фосфониевой соли, но также и R3"
[485]:
Оптимальная концентрация NaOH для получения
максимального выхода в реакции
R»
С6НВ
н
н
СН2=СН
X
С1
I
С1
С1
NaOH. моль/л
6
6
1
1
Выход, %■
80
80
70
60
Исследование влияния характера оснований и условий
проведения реакций, описанное выше, не привело к созданию
единой методики, пригодной для препаративных работ.
Оптимальные условия, по-видимому, необходимо подбирать для каждого-
отдельного субстрата. Поэтому для сравнения влияния
различных оснований и растворителей была использована серия
реакций олефинирования по Виттигу (см. [1632]). Из полученных-
данных следует, что применение карбоната и фторида калия
целесообразно только в случае более кислых фосфониевых
солей. Наилучшей изо всех испытанных комбинаций была систе-
Таблица 3.13. Реакции Виттнга н Хорнера в условных межфазного каталйзй
Реакция
Основание
Растворитель
Катализатор
Выход, %
Литера-
Тура
R'CHO+Ph3P+CHR»R«X-
№'=арнл; Х=С1, Вг, I;
R», R4=H, CH,, C6H5)
R^HO+R2—CH2PPh3+X-
(R1, Рг=алкил, фенил)
R'COR*+PhsP+CHR3R«X-
(RVR**" арил, Н или алкил,
включая ненасыщ.; R3, R*»*apM, Н
или алкил)
ArCOCF3+Ph3P+CH3ArCl-
Защищенный углевод-СНО+
-1-Ph.P+rHoRX-
Ферроценальдегид+Р11зР+СНЛ1-*
Полимерсвязанный-РЬРРЬ2СН2Р'+
+АгСНО (R'=бензил, алкил)
ArCHO+Ph,P+CH2R
Аг—CH2PPh3+X-+CH2O
2Ar-CH2PPh3+X-+OCH-CHO
Ph3P= NCR!R*R*+ Ph2C *= С=0
(RS R*. R»=H, CH,, QHs)
Ar—COR+
PPh,+-CH = CH—NHArCl-
RCHO+R1S-CH(RS)—PPhs+Cl-
Конц. NaOH
Твердый трет-ВиОК.
или КгСО,
5 и. NaOH
Твердый К2СО3
г/jer-BuOK
KF
Разбавленный NaOH
25%-ный NaOH
Конц. NaOH
Твердый NaOH
Конц. NaOH
Конц. NaOH
Твердый КОН
NaOH
NaOH -
Н2О/СН2С12
ТГФ илн СН2С12
Н2О/С6Н6
Диоксан
Бензол
CHsCN
Н2О/С6Н6 нлн СН2С12
Н2О/С6Н6
Н2О/СН2С12
Бензол, диоксан, ТГФ
или СН2С1г(
—
Н2О/СН2С12
Эфир
Н2О/СН2С12
Н2О/СН2С12
Отсутствует
Bu4N+I-
18-Краун-6
Отсутствует
Дициклогексано-
18-краун-6 или следы
Н2О
Отсутствует
Дибензо-18-краун-6
ТЭБА или
отсутствует -
R4N+X-
Отсутствует
Отсутствует
Отсутствует
Отсутствует
Отсутствует
20—90
82-97
60—99
<98
Различный
<93
88—100
45-85
79—87
45-97
80-90
13—23
65-85
низкий
~90
483,
1110,1457
484
ср. 1634
485
1599
1632
1297
1343,
1375
1484
1239
1782
486
487
488
1111
1112
Ar-CHO+ (EtOhPO-CH2-X
(X=CN, COOR, COPh, S-Ph,
SO—R, SO2R)
Аг—СНО+ (EtO)2POCH2—X
(X=-SR, -SO2R, -SOR)
Ar—CH = CH—CHO+ )
+ (EtO)2POCH2Ar I
ArCHO+ . I
(EtO)2POCH2—CH=CH—Ar '
RlRKO+ (EtO)2POCH2-X
(R1, R2=H, алкнл, арил; X=CN,
COOR, гетероароматический)
CH9
1
Аг—СНО+ (EtO)2PO—CH—CN
Ферроценальдегид+
+ (EtO)2POCH2—Ph
CH3COCH2PO(OEt)2+CsHnCHO
X—CH2—PO (OEt) 2+ PhCHO
(X=€N, COOR)
ArCH2PO(OMe)2+R'R'CO
(R'*=apM; R2=H, CH3 или арил)
R'CHj-PO (ОСзН7-изо) 2+R8R3CO
(К'=Арил, кротил, циннамоил;
К2,Р3=алкил, Н, алкенил, арил)
ArSeCH2PO(OEt)2+ArCHO
PhCHCl—POfOEtJ^R'RaCO
Конц. NaOH
Конц. NaOH
Конц. NaOH
Конц. NaOH
Порошок КОН
Разбавленный NaOH
Разбавленный NaOH
50%-ный NaOH
Разбавленный NaOH
KF-2H2O
NaH
Порошок КОН
NaOH
50%-ный NaOH
Н2О/СН2С12
Н2О/СН2С12
Н2О/С6Н6
Н2О/СН2С12
ТГФ
Н2О/СН2С12
НгО/различные
растворители
Н2О/СН2С12
н2о/сн2сь
ДМФ илн CH3CN
ТГФ или диглнм
Бензол
Н2О/СН2С12
Отсутствует, Bu«N+I~
Различные R4N+X-,
краун-эфиры
Bu4N+I-
BtuN+I-
Отсутствует
BtuN+Br-
Разлнчные ониевые
соли
Bu4N+I-
BU4N+I-
Bu«N+I- или 18-кра-
уи-6
15-Краун-5
18-Краун-6
ТЭБА
Bu4NHSO4
55—95
55—87
72-81
30-77
76—98
80
Высокий
81-98
82
39—83
86-99
16-77
63
44-82
492, 1256
493
494, 1457
495, 1114
1360
323
496
1484
1542
1361
1631,
1658
1633
1808
1809
■256 Глава 3. Практическое использование МФК
ма трет-бутоксид калия/бензол, т. е. именно та, которая ближе
всего напоминает «классические» условия проведения реакции
Виттига, особенно тем, что это основание частично растворимо
в бензоле. В присутствии этой системы протекают такие
реакции, которые не проходят в других условиях МФК, например
реакции кетонов и алифатических альдегидов с
неактивированными фосфониевыми соединениями. Хотя в этих случаях реак-
дии идут только с умеренными выходами, простота этих
условий может сделать эту методику привлекательной [1113, 1632].
Соотношение Z/f-изомеров в МФК-реакции Виттига не
отличается от такового при использовании алкоксидного метода.
Ph3P+—CH2—Я3 и R'CHO дают Z/£«l/l с R»-фенил или ж-,
л-замещенный фенил. Если R3=C6H5—СН=СН или 9-антраце-
.нил, то количество Z уменьшается до 36 и 0% соответственно
[483]. При использовании системы 18-краун-6/карбонат калия
или грет-бутоксид калия было отмечено влияние растворителя
ла стереохимию образующихся олефинов. В реакции R'CHO-f-
+R3CH2—РРЬз+Х- с R^QHs или C2HS, R3 = C6HS как в ТГФ,
так и в СН2СЬ в основном образовывался гранс-изомер, причем
эв последнем растворителе его было несколько больше.
Напротив, при R3 = CH3, R1 = C6H5 соотношение Z/E было 85/15 в ТГФ
и 22/78 в СН2С12 [484]. Использование твердого NaOH в
аналогичном исследовании [1782] привело к увеличению соотношения
Z\E в ТГФ, бензоле и диоксане приблизительно до 85/15, в то
время как в СН2С12 это соотношение было равно 53/47. В этих
экспериментах катализатор не использовали.
Введение NH-связи в положение, соседнее с фосфониевым
центром, увеличивает кислотность настолько, что для депрото-
нирования можно использовать твердый гидроксид калия.
Отделение образующихся при этом фосфиналкилиминов от щелочи
и последующее введение их в реакцию с дифенилкетеном дает
кетенимины. Таким образом можно получить их в оптически
-активной форме без заметной рацемизации у асимметрического
.центра, непосредственно связанного с азотом [488]:
R1 R1
Ph3PBr2 > Ph3P—NHC—R2 J^U- Ph3P=NC—R2 ""C°C==°>
Bre 4R3 XR3
R1
Ph2C=C=NC—R»
4
Схема 3.132
В табл. 3.13 суммированы результаты опубликованных
^работ.
3.16. Генерирование и превращение илидов 257
3.16.2. Получение олефинов по Хорнеру
(активированная связь РО)
В этой реакции, родственной реакции Виттига, вместо
илидов фосфора используются фосфонатные карбанионы [489]. Эта
реакция обладает следующими преимуществами: во-первых,
фосфонатный карбанион более нуклеофилен и реагирует в
мягких условиях с самыми разнообразными альдегидами и кетона-
ми; во-вторых, растворимость фосфонатов в воде облегчает
выделение продуктов реакции из реакционной смеси при
обработке; в-третьих, фосфонаты, которые получают по реакции
Арбузова, дешевле и более доступны. Обычные фосфонаты, с
успехом используемые в реакции Хорнера, включают заместитель
R3, резонансно стабилизирующий карбанион. Если R3=H или
алкил, то олефины образуются с низким выходом. С точдси
зрения стереохимии образованию трамс-олефинов
благоприятствуют небольшие заместители у а-углерода фосфоната. Стериче-
ские затруднения как в фосфонате, так и в карбонильном реак-
танте способствуют промежуточному образованию бетаина, что
приводит к цис-олефинам [490, 491].
(Etoy + RSffiaX ► R3CH2-P(OEt)s+X-
-н+
>■ R3CH2—PO(OEt)2 >■ R3CH--PO(OEt)2
(R3 = COR, COOR, CN, арил и др.)
Недавно реакция Хорнера была осуществлена в условиях
МФК. Как и в случае реакции Виттига, некоторые авторы
обходятся без катализаторов, а другие используют аммониевые
соли или краун-эфиры. Все эксперименты, проведенные в
лаборатории авторов книги, показали, что в присутствии
катализаторов выходы выше [1633]. Это совершенно естественно,
поскольку при депротонировании фосфоната образуется анион,
который не может передвигаться свободно без противоиона. Ббль-
шая часть результатов указывает, по-видимому, на
преимущественное образование гранс-олефина. В системе СН2С12/50%-ный
водный NaOH без катализатора из альдегидов и (ЕЮ)2РОСН2Х
(X = COOR, SO2CH3, PO(OEt)2) образуется 100% чистого
£-изомера [492]. Реакция
(ЕЮЬРО—СН2—S— R3 + R'CHO -> R1—CH=CH—S—R3
ff». (и = 0.1.2) (О).
Схема 3.133
в системе СН2С12/водный NaOH сТЭБА специфична для
ароматических альдегидов. Для кетонов и алифатических альдегидов
258 Глава 3. Практическое использование МФК
эта реакция не пригодна. При я=0 или 1 образуется смесь
цис/транс-изоыеров, а при п=2 получают чистые
транс-соединения. Миколайчик и др. [493] обнаружили, что при п=0
катализатор удивительным образом влияет на стереохимию олефинов.
В случае четвертичного аммонийхлорида образуются
эквивалентные количества Z- и £-алкенов, в то время как
соответствующие бромиды, иодиды и краун-эфиры приводят к смеси, где
Z<C.E. По-видимому, эти соли влияют на относительную
стабильность промежуточно образующихся бетаинов. В другой
работе соединения с п=2 (схема 3.133) сначала были проалки-
лированы в условиях МФК (СН2С12/0,5М NaOH/Bu4NBr) и
затем введены в реакцию с альдегидами (CH2CI2, 50%-ный
водный NaOH, Bu4NBr). Полученные в результате олефи-
ны имели в основном Z-конфигурацию [1256]. Типичная для
МФК щелочь — гидроксид натрия — может быть
использована только для таких фосфонатов, которые включают
повышающую кислотность группу в р-положении. К
соединениям, которые можно ввести в эту реакцию, относятся
диэтилбензил- или диметилбензил-, циннамил- и кротил-
фосфонаты. Серьезным ограничением реакции является
омыление эфиров. Применение изопропилфосфонатов приводит к
заметному увеличению выхода. Поиски оптимальных условий
показали, что наилучшей является система порошкообразный
гидроксид калия/бензол и 18-краун-6 в качестве катализатора.
Циннамил- и кротилфосфонаты в этой системе даже с
алифатическими кетонами и ненасыщенными альдегидами дают не очень
большие выходы [1633]. Другие варианты проведения реакций
в системе гидрид натрия/15-краун-5 в ТГФ или диглиме см. в
работах [1631, 1658].
Была изучена также стереохимия реакции, приведенной на
следующей схеме [496]:
о
II
PhCHO + (EtO)aP—CH—CN
СН3
Схема 3.134
При варьировании растворителя и катализатора наблюдалось
слабое, но достоверное изменение соотношения Z/E. Однако эта
соотношение очень сильно отличалось от величины, полученной
в ГМФТА. Реакцию Хорнера в условиях МФК успешно
используют также для синтеза сопряженных диенов и полиенов. В
системе водный гидроксид натрия и BiuNI эти соединения могут
3.16. Генерирование и превращение илидов 259
быть синтезированы двумя способами [494, 1457]:
о о
II II
(EtO)2P—CH2Ar + Ph—CH=CH—CHO (EtObP— CH2—CH=CHPh + АгСНО
' ArCH=CH—CH=Ch—Ph
Схема 3.135
В табл. 3.13 приведены соответствующие литературные
данные.
Оптически активный 3-замещенный дигидроизокарбостирил
(А) реагирует с диэтилцианометилфосфонатом и 50%-ным NaOH
при комнатной температуре, давая диастереомеры В и С. Диа-
стереомерный избыток В зависит от растворителя. Без
катализатора реакция идет значительно медленнее, но несколько более
селективно [975].
ОН
Bi^NBr, CH^Cl^
Bu^NBr, Зиоксан
Без катализатора СН^1
70,5
75,5
С1г 77
Схема 3.136
29.5
£4,5
гъ
3.16.3. Реакции сульфонилидов и оксосульфонилидов
Подобно Р-илидам, S-илиды также могут реагировать с
карбонильными соединениями. Однако известно, ч/го диметилсуль-
фонийметилиды дают со всеми карбонильными соединениями
оксираны. Напротив, диметилоксосульфонийметилиды образуют
оксйраны только из обычных карбонильных и циклопропилза-
мещенных соединений, полученных из сопряженных продуктов
о
♦ (H3Q2S=CHa
RCHO
(H3C)3SO«>ieoc__^ (H3C),S=CHa RHC.CH_eOft,
Схема 3.137
COR'
260 Глава 3. Практическое использование МФК
Сульфониевые соли получают при реакции органических
сульфидов с алкилгалогенидами. Они в свою очередь депротони-
руются безводными основаниями в согласии с более старой
препаративной методикой. В некоторых случаях можно работать
по МФК-методике и без катализатора. Мерц и Меркль [498]
показали, что триалкиламмониевые соли с небольшими алкиль-
ными группами не вступают в реакцию без катализатора в
системе метиленхлорид/50%-ный водный гидроксид натрия. Бенз-
альдегид и коричный альдегид (50 °С, 48 ч) превращаются с
выходом 90% в присутствии 1—5 мол. % ВщШ. С кетонами
выход ниже: за 72 ч ацетофенон образует 36% оксирана, а
бензофенон — только 18%. Причина каталитического эффекта
при добавлении аммониевой соли и их влияние на механизм
реакции не понятны. Среди всех галогенидов иодид менее всего
пригоден для переноса гидроксила. Мерц и Меркль
предполагают, что триметилсульфонийиодид сам по себе не проявляет
каталитической активности вследствие того, что имеет
небольшие алкильные группы. Однако депротонирование на
поверхности раздела фаз приводит к образованию нейтральных частиц
(см. выше обсуждение реакции Виттига). Решение этих
вопросов требует новых работ по изучению механизма реакции.
Группа швейцарских химиков провела реакцию {3-ионона с
триметилсульфонийгалогенидами в присутствии ТЭБА и без
него, при этом был получен (£)-2-метил-4-(2',6',6'-триметил-1'-
циклогексенил)-1,2-эпоксибутен-3 [1640]. ТЭБА увеличивает
скорость реакции вдвое; бромид, использованный в качестве
противоиона для МезЭ4", приводит к торможению реакции, иодид
также оказался неактивным, а фторид реагировал
экзотермически.
Яно и др. [497] изучили реакцию между бензальдегидом
и триметилсульфонийиодидом (А) и лаурилдиметилсульфоний-
иодидом (В) без катализатора в двухфазной системе бензол/
водный гидроксид натрия, варьируя его концентрацию. В 5 н.
NaOH А с PhCHO оксиран не давал; по-видимому, А находился
в водной фазе. При 15 н. концентрации NaOH было получено
83% фенилоксирана. Единственное отличие этих опытов от
описанных ранее неудачных попыток провести эту реакцию [498],,
по-видимому, состояло в использовании бензола вместо мети-
ленхлорида.
(CHS)3S+I- C12H25S+(CHS)2I-
А В
При использовании соединения В, которое может действовать
как типичный детергент и мицеллообразователь, даже с 5 н.
NaOH было получено 23% фенилоксирана, ас 15 н. NaOH
выход вырос до 81%. Реакция между хлоридом или иодидом три-
3.16. Генерирование и превращение илидов 261
метилсульфония и кетоном идет с высоким выходом при
перемешивании с 15 н. NaOH в течение 10 ч. В присутствии
хлорида выход выше, чем с иодидом [497] (табл. 3.14).
Таблица 3.14. Выходы оксираиов в реакции
R1.
\
C=O+(CH3)3S+X-
Карбонильное соединение
с6н5—сно
Циклогексаион
Циклогексанон
Ацетофенон
Камфора
X
С1
С1
I
С1
С1
Выход,
86
88
27
85
40
Оксираны были получены из бензальдегида, формальдегида
и ацетальдегида с использованием ряда сульфониевых илидов
(табл. 3.15). При этом была использована следующая методи-
Таблица 3.15. Выходы оксиранов R—СН—СН—R' [499]
О
Сульфониевая соль
Выход, %
С.Н5—СНО
СН2О
СВДСНО
Me3S+Cl-
Et3S+Cl-
СН3—СН=СН—SMe2+Cl-
С6Н5—CH2SMe2+Cl-
68
80
70
85
—
—
8
87
—
—
—
48
ка: избыток 50%-ного водного гидроксида натрия прибавляли к
теплой смеси водного раствора сульфониевой соли,
карбонильного соединения и (обычно) несмешивающегося растворителя.
В качестве растворителей применяли бензол, толуол, к-гексан,
метанол, изопропанол, а также их смеси. В продуктах реакции
преобладали гранс-оксираны. В случае алифатических
альдегидов синтез осложнялся реакцией Канниццаро и альдольной
конденсацией. С аллилдиметилсульфонийхлоридом также про-
262 Глава 3. Практическое использование МФК
ходила нежелательная побочная реакция с образованием 2-ме-
тилоксирана:
НзС—СН=СН—S(CH3)2eCle °"f> Н3С—СН—СН2—S(CH3);'
Н2О |
он
оне НзС—СН—СН2 + S(CH3)2
6
Н2С=СН—CH=S(CH3)2 R,CHO> Н2С=СН—HC-CHR'
Схема 3.138
Сообщалось [500, 555] также об энантиоселективном
синтезе фенилоксиранов из бензальдегида и триметилсульфонийиоди-
да под действием 50%-ного водного гидроксида натрия с
использованием в качестве хирального катализатора
производного эфедрина. Однако эти данные об оптической индукции
оказались ошибочными [501]. Подробное обсуждение см. в
разд. 3.1.5.
Триметилоксосульфонийгалогениды являются также
предшественниками диметилоксосуЛьфонййметйЛидов, которые в свою
очередь могут реагировать с карбонильными соединениями,
давая оксираны. Брендстрём отметил, что в этой системе имеют
преимущество хлориды, поскольку они растворимы в ТГФ.
В обычном методе синтеза получают иодиды. До последнего
времени для превращения иодидов в хлориды необходимо было
.использовать элементарный хлор. Применение методики
экстракции ионных пар Брендстрём а позволяет провести это
превращение очень легко. Иодид и эквимолярное количество бензилтри-
бутиламмонийхлорида распределяются между метиленхлоридом
и водой. В воде находится триметилоксосульфонийхлорид, а в
органической фазе — С6Н5—CH2NBu3+I~ [1].
Мерц и Меркль [498] обнаружили, что образование фенил-
оксирана в указанных выше условиях сопровождается реакцией
альдольного типа:
о
с.н.-сно ^^^ нА
с6н5
Схема 3.139
3.16. Генерирование и превращение илидов 263
а,р-Ненасыщенные ароматические кетоны образуют смесь
27.Е-Циклопропилкетонов с выходами 70—80%:
Аг—СН=СН—С—QH5 з
О
Схема
► Аг— СН
<
3.140
СН—С—С6Н5
снГ 1
1,5-Дифенилпентадиен-1,4-он-3 дает смесь продуктов моно-
и бисциклопропанирования [498]. В табл. 3.16 представлены
реакции сульфонийилидов.
Ниже приводятся типичные экспериментальные методики.
Олефины (реакция Виттига): Гетерогенная смесь 3 ммолей
альдегида, например бензальдегида или коричного альдегида,
5 ммолей алкилтрифенилфосфонийгалогенида, 5 мл бензола и
10 мл водного гидроксида натрия перемешивают при комнатной
температуре от нескольких минут до нескольких суток. Затем
смесь обрабатывается обычным образом.
цис/транс-Стильбены: 10 мл 50%-ного водного гидроксида
натрия прибавляют к хорошо перемешиваемой смеси 20 ммолей
бензальдегида и 20 ммолей бензилтрифенилфосфонийхлорида в
20 мл дихлорэтана. Реакция заканчивается за 10 мин.
Обработка дает почти равные количества цш> и гранс-продукта [483].
Рекомендуемая методика: 10 ммолей фосфониевой соли и
20 ммолей трет-ВиОК перемешивают в 40 мл бензола в
атмосфере азота при 0—4°С. После появления красно-оранжевой
окраски прибавляют по каплям 10 ммолей карбонильного
соединения, растворенного в 30 мл бензола. Смесь перемешивают на
холоду в течение 2 ч, а затем при комнатной температуре
до 72 ч. Окончание реакции контролируется ТСХ. Дальнейшую
обработку проводят обычным способом [1632].
Олефины (реакция Хорнера). Раствор 20 ммолей фосфоиата
и 20 ммолей альдегида в 5 мл бензола прибавляют при
комнатной температуре к двухфазной системе 20 мл бензола/20 мл
50%-ного водного гидроксида натрия, содержащей 1 ммоль
BtuNI в качестве катализатора. Смесь энергично перемешивают
и кипятят в течение 30 мин и затем обрабатывают [494].
Рекомендуемая методика: 10 ммолей дийзопропилового
эфира фосфониевой кислоты и 10 ммолей карбонильного соединения
в 10 мл бензола добавляют по каплям в кипящую смесь 26
ммолей порошкообразного КОН и 1,6 ммоля 18-крауна-6 в 30 мл
бензола в атмосфере азота. Реакционную смесь кипятят 20 ч и
затем обрабатывают обычным способом [1633].
Таблица 3.16. Реакции сульфониевых и оксосульфониевых илидов с карбонильными соединениями
в условиях межфазиого катализа
Реакция
Основание
Растворитель
Катализатор
Выход,
%
Литература,
Конц. NaOH
Н2О/СН2С12
[я—0,1; R1, R2=H, Ph, Ph—CH = CH,
(R-=CH3, ОСН.)]
5, 10, 15 н. NaOH Н2О/С„Нв
(Rs-Me, лаурил; Х=С1, I; R',R2=
=Н, Me, Ph, циклогексанон,
камфора)
Конц. NaOH
(R' = H, Me, Ph; R3=R5=Me, Et;
RA=Me, Et, аллнл, бензил)
PhHCO+Me3S+I-
Конц. NaOH
ArCOR+полистнрол—S(Me2)+X- Конц. NaOH
[-S(Et2)+X~]
Н2О/бензол
Н2О/толуол
Н2О/метанол
Н2О/этанол
Н2О/изопропанол
Н2О/я-гексан
Н2О/СНгС12
Н2О/СН2С12
Bu4N+I- и Me3S+I- 20—90 498
Отсутствует
Отсутствует
(—) N,N-Днметил-
эфедринийбромид
Bu4N+I-
30—90 497
0-90 499 .
67 500, 555
86—99 1115
3.17. Нуклеофильное ароматическое замещение 265
2-Фенилоксиран: 100 мл 50%-ного водного гидроксида
натрия прибавляют при перемешивании к смеси 0,1 моля бензаль-
дегида, 0,1 моля диметилсульфонийиодида и 1 ммоля BiuNI в
100 мл метиленхлорида. Реакционную смесь энергично
перемешивают при 50 °С в течение 48 ч. Обработка дает 2-фенилокси-
ран с выходом 92% [498].
3.17. Нуклеофильное ароматическое замещение
Было проведено огромное количество реакций в условиях
МФК, в которых галогены в активированных ароматических и
гетероциклических соединениях замещались на фтор, эфирную,
а также эфирную или аминную группу, тиоциан- или сульфо-
группу (табл. 3.17). Макоша и сотрудники подробно изучили
реакции анионов бензилцианидов. ■;
МФК, по-видимому, является наиболее подходящим методом
нитроарилирования алкилфенил- и дифенилацетонитрилов; при
этом используют о- и n-галогеннитробензолы, их производные
[303—307, 313], а также хлорнитропиридины [1067]. Сам фе-
нилацетонитрил в реакцию не вступает, так как уже следы
продукта нитроарилирования отравляют катализатор. X может
представлять собой галоген, CN, COPh, фенил, алкил, NO2 или
rper-BuOOC
Ph—CH(R)—CN +
4
NO2
NO2
Ph—CH(R)—CN +
Схема 3.141
В 2,4-дихлорнитробензоле замещается главным образом атом
хлора в положении 4 [304, 306]. Если нитрогруппа расположена
в орто- или пара-положении к сильному электроноакцепторно*
Таблица 3,17. Нуклеофильное ароматическое замещение (реакции, ие упомянутые в тексте или на схемах)
Тип реакции
Растворитель, катализатор, условия
Литература
X
Д
No2 (х-а,вг)
SCN
О
г
NO2
SO3H
NO1
NO2
F
NO2
г
NO2
► Замещение
К-фталимидом,
Na-сахаридом,
Na-ацетилацетоиатом
o-(O2N)—С(Н4—С1—*Эфиры, тиоэфиры
Сульфирование активированных сополимеров
Активированный АгС1—»-Ar—NH5
НаО/толуол, R4NX, 1—2 ч, 90 СС
Н2О/СН4С12, KsSA, RsHNX, 3 ч, 0°С
CH3QN, тв. I(F, J8 краун-6, I ч, 83 °С
CHgCN, твердые соли, крауны
Bu«NBr
Вода» NH,, R4N7 R«PX, Ю ч, 150 "С
1525
1692
53
1380
1798
1799
1677
Ol > — IО 1 >
CHaCN, тв. KF, дибензо-18-краун-6, 43 ч, 82 "С 1031
ОАг
. растворитель, R4NCI, 15 ч
1704
Т.в. KF, аликват 336
1363
82 "С
i та./KsGQs, Д8?краун-6, от 6 ч до 2 сут,
(X-CUF, NO,)
Продолжение табл. 3.17
Тип реакции
Растворитель, катализатор, условия
Литера,
тура
C(CFj),—COOEt
F,
(X - NO,, Cl; Nu - SCN, OR,
PhCR—CN)
Cl
Nu
Я-
(X - Cl, F; Nu = <рталимиЭ,сахаринат )
ГбТ- fo^CN
'N
OR
.Cl
О J^N + CHj(COOR)2
S
ДМФА, KF, 18-краун-6
CH3GN, сухой KF, [2.2.2], комн. темп,
или 18 краун-6, 83 °С
CH3CN, KNu, 18-краун-6
Толуол, вода, NaOH, алнкват 336
нлн краун
NaOH, Bu4NBr
1437
QHe, Н2О, NaOH, ТЭБА нлн аликват 336 1415,
1595
56, 1425
1380
1399
1555
I
NH,
3.17. Нуклеофильное ароматическое замещение 269
му заместителю, то она также может замещаться ,[305, 3Q7,
313]:
[313J
Ph—С
Схема 3.142
Аналогичным образом в системе, содержащей бензол/коиц.
NaOH и небольшое количество ДМСО, в присутствии BiuNCl
при 50 °С было проведено нуклеофильное замещение хлора в
9-хлоракридине производными феиилацетонитрила [870].
Схема 3.143
Было высказано предположение [307], что в
сильнощелочных средах, используемых в МФК, дифенилацетонитрил и сте-
рически затрудненные алкилфенилацетонитрилы могут
участвовать в различных редокс-процессах с ароматическими нитросое-
динениями. В некоторых случаях эти процессы становятся
главными.
Недавно предложены варианты синтезов, включая
нормальное замещение енолятами бензилкетонов [1285] и замещение
арилацетонитрильными анионами в N-оксидах пиридина [1287],
2-хлорхинолинах, 1-хлоризохинолинах [1286] и в N-оксидах
4-хлорхинолинов [1269]. Более необычную реакцию Макоша
назвал «вице-замещением». Суть вице-замещения состоит в том,
что при взаимодействии ароматического соединения, не
имеющего уходящей группы, с анионом а-хлорацетонитрила роль от-
270 Глава 3. Практическое использование МФК
сутствующей уходящей группы выполняет атом хлора. Эта
реакция проходит без катализатора под действием NaOH в ДМСО
[131]
RCHCl—CN +
NO2
CHR— CN
Замещение галогена в ароматическом кольце в случае хлор-
2,4-динитробензола и перхлорпиридина обсуждалось ранее.
Используя сильные электроноакцепторные свойства диазоние-
вой группы, Гокель и сотр. [80] провели реакцию в метиленхло-
риде между 0,1 М 4-бромбензолдиазонийтетрафторборатом и
5-молярным избытком четвертичного аммонийхлорида (ТЭБА
или тетрабутиламмонийхлорид). Хотя сам тетрафторборат и не
растворим, однако при двукратной обработке хлоридом
аммония (реакция обмена противоионами) арилдиазониевые ионы
переходят в раствор [81]. При 30 °С спустя 24 ч около 70%
брома замещается на хлор. Последующее восстановление с
помощью Н3РОг в присутствии каталитических количеств оксида
меди приводит к смеси продуктов [80]:
Вг
Схема 3.145
3.17. Нуклеофильное ароматическое замещение 271
Если использовать 5-молярный избыток хлорида и более
разбавленные растворы, то выходы повышаются. Интересно
отметить, что добавление 18-краун-6 замедляет реакцию обмена,
поскольку положительный заряд в комплексе крауна с диазо-
ниевой группой делокализован, что ослабляет способность к
нуклеофильному замещению. Описаны также реакции между
4-бромпиридином и циклическими вторичными аминами
(50%-ный водный NaOH, ТЭБА-Вг; 100°С, 5 ч) [1328]. Если
принять во внимание величину кислотности NH-связи, то
отнесение этой реакции к МФК-процессам вызывает сомнение (см.
разд. 3.8).
Разнообразные по типу механизма реакции замещения в
условиях МФК можно осуществить, используя комплексы
тяжелых металлов:
Аг—X
Процесс включает взаимодействие ароматического галогенида
в системе бензол/вода в присутствии каталитического
количества никель-трифенилфосфониевого комплекса [М[РРп3]з или
транс-хлорнафтил-быс- (трифенилфосфин) никель] - трифенилфос-
фина и аликвата 336. Раствор цианида натрия медленно
добавляют при 55 °С. Выходы продуктов замещения 37—98% [1236].
Замещение в азокрасителях атома галогена в активированном
кольце на группу CN или SO2CH3 проходит при нагревании с
натриевыми солями (NaCN или ЫаОгЗСНз) в системе анизол/
/вода с каталитическим количеством соли Cu(I) и R4NX
(схема 3.146). При этом, вероятно, происходит экстракция
комплекса меди [1280, 1329]. Соединения BtuN+CuX2- (Х=С1, Вг, I,
CN) можно приготовить в чистом виде [1892].
N=N—/ Q )-NEt2 . O,N
У-"
NO2 NHAc
Х=гологеи; Y - CN, SOjCH3
Схема 3.146
(Различные по механизму реакции замещения обсуждаются на
с. 392 и 395.)
272 Глава 3. Практическое использование МФК
3.18. Различные реакции, проходящие в присутствии
оснований, не рассмотренные в других разделах
3.18.1. Перенос диазогруппы
Y основание
(X, Y = COOR, Ph, COR, H)
ийяоятвяюр
R_NH2 + Tos
. Схема 3.147
В МФК-системе бензол, пентан или дихлорметан/водный
карбонат или ЮМ NaOH/аликват 336 или тетрабутиламмонийбро-
мид проходит быстрая реакция с образованием а-диазокарбо-
нильных соединений, не содержащих примеси непрореагировав-
шего. тозилазида [508, 1327]. Смесь перемешивают обычно 15 ч
при комнатной температуре; выходы продуктов в пределах 77—
100%. Применение 2,4,6-триизопропилфенилсульфонилазида
позволяет аналогичным способом получить циклические а-диазоке-
тоны [1543]. При использовании 10—50-молярного избытка
амина по отношению к тозилазиду в системе 50%-ный водный
NaOH/ТЭБА можно получить арилазиды с выходами до 94%.
В некоторых случаях выходы повышаются при использовании
тетрагексиламмонийбромида. Алифатические амины, как
правило, реагируют хуже [509], и только очень немногие
гетероциклические амины вступают в эту реакцию [1494].
Монозамещенные гидразины можно восстановить до
углеводородов кипячением в течение 2—10 ч с тозилазидом в системе
бензол/водный NaOH/тетраэтиламмонийбромид [1116]:
RNHNH2 + TosNs v RH + 2N2 + TosNH2
3.18.2. ^-Элиминирование
Некоторые реакции у-элиминирования с последующим
присоединением по Михаэлю описаны в разд. 3.12.
С(СНз)гС1
Аг—CR=CH—СН—СНг—COOR X—(СН2)3—CN
Ar—CR=СН-СН—СН—COOR
\/
/ V
сн3 сн3
Схема 3.148
^ 3.18. Реакции, не рассмотренные в других разделах 27S
Реакции образования циклопропанов, представленные на
схеме 3.148, описаны в патентной литературе [513, 1764] (условия:
концентрированные растворы гидроксидов щелочных металлов,.
R4NX). При нагревании хлоргидринов с 20 %-ным водным гидрок-
сидом натрия в присутствии гидррксида тетрабутиламмония
образуются эпоксиды [1117] (см. также разд. 3.13.3).
3.18.3. Получение ангидридов и фторангидридов кислот
В работе [514] описан метод получения алифатических и
ароматических фторангидридов в условиях МФК из
соответствующих кислот, карбоната калия и димера гексафторпропена,.
перфтор-2-метилпентена-2 (А) или его тримеров в дихлормета-
не. Исходная реакционная смесь состоит из трех фаз: А, соль
калия и дихлорметан. Добавки 18-крауна-6, аликвата 336 или
глима с четырьмя или более этиленоксидными звеньями
ускоряют реакцию настолько, что она заканчивается за 2—24 ч при
комнатной температуре.
JF II рС
RCOOH + K2CO3+ >=< капал-*™» R_^F + 4CH-C-C2FS
СН—С—F
О—С—R |
II
О
Схема 3.149
Японские авторы [1422] усовершенствовали первоначальную
МФК-методику, включив промежуточную стадию — экстракцию
в водную фазу (схема 3.150). Для получения смешанного
ангидрида бензойной и уксусной кислот требуется но крайней
мере 0,3 моля катализатора
РЬСОСЦорг.) +
СН, СН, С1е
S-CO-Ph ftofe) + NaOAcfrta) ► №С1(«йн.) + PhC0-O-Ac(opi.) + N
I СН3
СИ,
Схема 3.150
274 Глава 3. Практическое использование МФК
3.18.4. Перегруппировки
При перемешивании сульфонамидов типа В в дихлорметане
с 18-крауном-6 и твердым гидроксидом калия они претерпевают
перегруппировку Смайлса с образованием соединений С. В
отсутствие краун-эфира выделить С из водного раствора щелочи
не удается, так как он подвергается дальнейшим
перегруппировкам с образованием D [515].
2_CH2—N—SO2e > O2N-/ О )-OCH2—CH,—NH
O3N
O3N-
-N—СНа—СН2—ОН
(R = Me, Et, Ph)
П
Схема 3.151
Перегруппировка Фрича — Буттенберга — Вихелля проходит
в условиях, близких к МФК при комнатной температуре в
системе толуол/20 %-ный ДМСО1 с 3 экв. грег-бутоксида калия и
Д экв. криптанда [2.2.2] [552].
Аг—С=С—Аг
Схема 3.152
Новая модификация синтеза индолов по Фишеру,
катализируемого щелочами, осуществляется нагреванием реактантов в
сульфолане с твердым карбонатом калия и молярным
количеством катализатора. Если в качестве МФ-катализаторов
используют 18-краун-6, то источником R1 является ониевая соль
RR'N+B или алкилгалогенид:
сн3 .
Схема 3.153
-РЬ
3.18. Реакции, не рассмотренные в других разделах 275
Гофмановское расщепление
R—CONHa
R—N=C=O
вторичных алкилзамещенных карбоксамидов дает изоцианаты
с выходом 70—80%. Для достижения высоких выходов
реакцию проводят в дихлорметане с системой бром/0,5 М NaOH/
/BU4NHSO4 при комнатной температуре, тщательно
контролируя процесс, чтобы избежать дальнейшего гидролиза изоциана-
тов с образованием мочевины. Однако в случае н-алкилкарбокс-
амидов избежать гидролиза не удается [1354].
Перегруппировка Курциуса
R—CO—Q >- R—NHS
является еще одним современным приложением МФК- Ее
проводят кипячением хлорангидрида кислоты с трнметилсилилази-
дом в бензоле в присутствии азида калия и 18-крауна-6; выходы
продуктов до 88% [1367].
Реакция Рамберга — Беклунда
ЯЧ*»СН—SO8—СН(Х)—R* >-
может быть проведена в условиях МФК в системе 10%-ный
водный NaOH/аликват 336/дихлорметан; выход 75—90% [1893, см.
также 1883]. На схеме 3.154 показаны еще четыре реакции:
1)
R—СО—СНС1—R' + R—CO—CHF— R'
Схема 3.154
O2N
CH2CI
Ri,NeOH9
CeH.NOj/CCI,*
L
O2N
NO2
NO2 O3N
NOj
PaN
СНз
R<,NeOHe
ecu *
ссп
O2N
CHC12
H
oTTlg) + @Г~*Т9.
62N NO2 O2N NOS
I К
O2N
CC1
Схема 3.155
3.18. Реакции, не рассмотренные в других разделах 277
1) изомеризация эпоксидов в кетоны, проходящая при —70 СС
под действием MePBu3F [1368]', 2) расширение цикла,
происходящее под влиянием KF/18-крауна-б в дихлорметане [1569];
3) МФК-перегруппировка Виттига (твердый КОН, краун-эфир
или ониевая соль) [1204]; наконец, 4) декарбоксилирование
6-нитробензизоксазол-З-карбоновой кислоты. Эта реакция
обычно ускоряется в органическом слое двухслойной мембраны.
Такой мембраной может быть используемый в качестве
катализатора аликват 336 в слабо агрегированной форме [1657].
3.18.5. Радикальные реакции
n-Нитробензилхлорид и бензилтриэтиламмонийгидроксид в
смеси четыреххлористого углерода и нитробензола (1:2) дают
до 98% Е. В качестве побочных продуктов образуются F и G
[516]. В отсутствие нитробензола Е образуется только с
выходом 25%, а в отсутствие четыреххлористого углерода Е вообще
не образуется. В аналогичных условиях из п-нитротолуола или
нитробензола и четыреххлористого углерода наряду с
хлороформом и гексахлорэтаном можно получить также Е.
Попытки доказать возможное образование карбена в качестве
промежуточного продукта при реакции Н в условиях МФК
оказались безуспешными. В результате были получены стереоизо-
меры Е, а также соединения F, I, К [517]. Реакция между нит-
робензил хлоридам и и замещенными фенилацетонитрилами в
системе концентрированный гидроксид натрия/катализатор [308,
319] приводит главным образом к продуктам замещения
наряду с некоторым количеством продуктов взаимодействия
радикалов (более подробно см, в разд. 3.9.1).
ЗЛ8.6. Реакции с тетрагалогенидами углерода
Макоша и сотр: [522] нашли, что в системе
концентрированный гидроксид натрия/ТЭБА с высокими выходами проходит
реакция между арилацетонитрилами и тетрахлоридом углерода
(схема 3.156), ,
По-видимому, С1+ отрывается от CCU и присоединяется к
промежуточно образующемуся карбаниону в ходе сложной
серии последовательных стадий. Анионы хлорфенилацетонитрила
можно уловить в виде продуктов реакции с альдегидами и аце-
тонитрилом [1120]. Аналогичный перенос положительно
заряженного иона хлора должен происходить и в процессе синтеза
ди-трет-бутилфосфоргалогенидов, которые образуются с
выходом 90% (схема 3.157) [523]. Диэфиры
моногалогенидфосфорной кислоты были получены раньше в недостаточно чистом
виде реакцией с'М-галйгенсукцийимйдЬм. "-:
278 Глава 3. Практическое использование МФК
PhCHa—CN + CCI4 T?EA/N"OH> \:*=C/
NC Ph
Ph Ph
PhaCH—CN + CCI4 > ,C—C(
Ph I I Ph
NC CN
PhCH2--CN--Ph-C-CN
I 1 н
a
К „ у—^см
H,C=CH-CN NC \ /
Схема 3.156
(CH3)3COV V (CH3)3CC> V
p-h + гх и>У«аи«он/теБА M
y ib, 2ft25 c
(СН3)зСО (СН3)3СО
x == a, Br
Схема 3.157
Эту реакцию без промежуточного выделения диалкоксифосфор-
галогенидов использовали для получения смешанных сложных
эфиров [129], диэфиров моноамидов [126, 272], диэфиров гид-
разидов [128] и диэфиров алкоксиамидов [127] фосфорной
кислоты (см[. разд. 3.4 и 3.8).
Наконец, фенилацетилен был превращен в феиилхлорацети-
лен при добавлении 50%-ного водного гидроксида натрия к
энергично перемешиваемой смеси алкинов, СОЦ и ТЭБА при
35°С [769]:
Pb-CasCH
Этим же методом был хлорирован и ряд других соединений
[1120, 1883]; например, грег-бутилфенилацетат дал 75% Ди-
хлорпроизводного, а дибензилсульфон количественно
превращался в стильбен. '
3.18.7. Некоторые реакции соединений серы
При реакции N-бензилиминосульфодифторида (L) и димети-
лового эфира ацетилендикарбоновой кислоты (12 ч, 132 °С) в
хлорбензоле с 2 экв. NaF и 0,1 экв. 18-крауна-6 образуется 65%
аддукта М [518].
Возможно осуществить прямое превращение в 2-алкенонит-
рилы^ алифатических карбонильных соединений перемешива-
3.18. Реакции, не рассмотренные в других разделах 279
Ph—CH»—N=SFa
NaF
lS-крауи-б
[Ph—C=N*—Se]
Схема 3.158
МеООС,
МеООС
М
нием их с О-этил-5-цианометилдитиокарбонатом или S-цианоме-
тилдиэтилфосфортиоатом в системе 50%-ный водный NaOH/
/аликват 336 в ацетонитриле (выходы 38—79%) [1359].
При взаимодействии тиоцианатов (схема 3.159) с 5—15 М
NaOH и этилтриметиламмонийбромидом в СН2С12 можно
наблюдать необычную реакцию. Те же продукты образуются при
реакции соответствующих бромидов с водным NaSCN/NaOH [1370]:
EtOCS—S—CHR1—CN
или
(EtO),PO—&-CHR1—CN
R\
R* ,R»
Яэ' CN
Ph2CH—SCN —
PhCOCHj—SCN
PhCH2—SCN —
Ph2C=S(90%)
H H
(45%)
-» PhCH2—S—S—CH2Ph (30%)
(96%)
Схема 3.159
3.18.8. Генерирование нитреиов
Использование фторидов для проведения а-элиминирования
с образованием нестабильных нитреиов было впервые описано
применительно к гетерогенным и гомогенным растворам:
KF, кипящий
Ph—CO—NHCI ; »- Pb—N=C=O »- вторичные продукты (519]
—COOEt
продукты внедрения (520]
n-OsN—С„Н4—SOS—NH—COOEt
N
280 Глава 3. Практическое использование МФК
Нитрен О был генерирован из N в присутствии водных щелочей
с использованием в качестве катализаторов ониевых солей.
Продукты присоединения — перегруппировки или внедрения Р, Q и
R были получены с выходами 13, 18 и 12% соответственно-
[521]. В присутствии R3B через промежуточное образование О
был получен RNH—COOEt [1730] (схема 3.160).
Схема 3.160
При реакциях с олефинами соотношение между стереоспе-
цифичным присоединением (синглётный нитрен) и внедрением,
приводящим к нестереоспецифичному присоединению (триплет-
ный нитрен), зависит от условий реакции и типа МФ-катализа-
тора. Использование иодидов аммония приводит к
образованию значительно большего количества продуктов реакции син-
глетного нитрена [1118]. Фотолиз фенилазида в ацетонитриле,
содержащем галогениды калия и 18-краун-6, ведет к о-гало-
генанилинам. В присутствии ацетата калия образуется 2,3-ди-
гидроазепинон [1777]. Твердый хлорамин Т и сульфиды дают в
метиленхлориде К[-(га-толилсульфонил)сульфилимин
(катализатор— различные ониевые соли) [1570]:
TosNCl-Na+ + RSR' >■ RR'S=NTos "
3.18.9. Реакции диазосоединений в условиях МФК
Несколько групп исследователей изучили образование и
устойчивость комплексов, образующихся между ионами', диазония
и краун-эфирами [1772, 1783] или эфирами полиэтиленгликоля
[1221]. Эти комплексы позволяют экстрагировать ионы
диазония из воды в неполярную среду и интересны сами по себе как
пример экстракции катионов. Такого же результата можно до-
3.18. Реакции, не рассмотренные в других разделах 281
стигнуть, естественно, при обмене противоиона на очень липо-
фильный анион. Эти комплексы довольно устойчивы к
термическому или фотохимическому разложению с выделением азота.
Так, арендиазонийтетрафторбораты обычно нерастворимы в
хлороформе, однако в присутствии дициклогексано-18-крауна-6
можно провести в этом растворителе диазосочетание с пиррола-
ми. Другие методики проведения этой реакции намного сложнее
[1119]. В свою очередь, введение R^NCl в систему позволяет
получить в органической фазе растворимые хлориды диазония
[81]. Для сильного ускорения азосочетания в системе твердая
фаза/жидкая фаза или твердая фаза/твердая фаза
рекомендуется использовать n-додецилбензолсульфонат натрия [1257] или
тетракис- [3,5-ди(перфторметил) фенил] борат [1795].
4-бромфе„ил-Н2*
4-6poM<peHit/i-D 1,4-бибромбензол 4-бромбисренил
(100%) (50%) (81%)
ArH KOAc/l8-Kptt!<H-6> Of) y_Ar 55-81%
Схема 3.161
При обработке и-бромбензолдиазонийтетрафторбората
ацетатом калия в присутствии 18-крауна-6 при комнатной
температуре |[81] проходит реакция, альтернативная реакции Занд-
майера. Предполагают, что в этом случае солюбилизированный
краун-эфиром ацетат калия вступает в реакцию обмена проти-
воионами с нерастворимой исходной диазониевой солью. Можно
представить себе, что нестабильный диазоацетат превращается
в диазоангидрид, который соответственно дает арильный
радикал, а последний в свою очередь атакует растворитель [81, 227].
Как было показано выше, этот метод может быть использован
для синтеза смешанных диарилов из стабильных, легко
получаемых тетрафторборатов или гексафторфосфатов арилдиазония
[227]. В качестве катализаторов наряду с краун-эфирами
используются липофильные анионы, ониевые соли или глимы
[1506]. В этих случаях выходы намного выше по сравнению
с обычными методиками. Если нагревать соединение,
содержащее диазогруппу в метиленхлориде с каталитическим
количеством дициклогексано-18-крауиа-6 и следами порошка меди, то
282 Глава 3. Практическое использование МФК
можно провести восстановление диазогруппы, т. е. формально
замещение на водород (0,5—4 ч, 40 СС [93]). По другой
методике диазосоединение перемешивают с гипофосфорной кислотой
в хлороформе в присутствии небольшого количества оксида
меди и (если необходимо) 18-крауна-6 [94]. На основе получения
краун-катионных комплексов и последующем генерировании
арилрадикалов были разработаны идущие с высокими
выходами методы синтеза арилбромидов и арилиодидов [855]. Галоге-
нирование проводится в хлороформе с использованием
стабильных и безопасных тетрафенилборатов арилдиазония в
присутствии каталитического количества 18-крауна-6 и либо небольшого
избытка бромтрихлорметана для получения бромидов, либо
подметана или молекулярного иода для получения иодидов.
В ходе реакции образуется некоторое количество продуктов
восстановления и хлорирования (0—8%). Если растворителем
является бромхлорметан, то в качестве побочного продукта
образуется гексахлорэтан.
Из тетрафторборатов диазония в системе твердый цианид
калия/18-краун-6 в дихлорметане были получены арендиазо-
цианиды, п-Х—Аг—N2—CN. Эти соединения при гидролизе
образуют Аг—N=N—CONH2; кроме того, их можно использовать
в качестве 2л-компонентов в реакции [4+2]-циклоприсоединения
[1206].
3.18.10. Получение гидразоиов и использование их в МФК
Гидразины экстрагируют в неполярную органическую среду
как путем образования их комплексов с краун-эфирами [1391],
так и с помощью ониевых солей, с которыми они образуют
водородную связь [R4N+X"~-"H2N—NH2] [1778]. Описано также
последующее получение гидразонов [1391]. Достаточно
кипячения в течение 3 ч для того, чтобы прошло восстановление гидра-
зона бензофенона по Вольфу — Кижнеру (толуол, КОН, дибен-
зо-18-краун-6) [1453]. Было проведено также разложение то-
зилгидразонов в условиях МФК действием 50%-ного водного
гидроксида натрия [908, 1771]. Производные бензофенона
давали 63% дифенилдиазометана, а производные флуорена — 92%
9-диазофлуорена [908]. Тозилгидразоны циклопентанона и ди-
бензилкетона были превращены в соответствующие алкены [908,
1771]. «,{}-Ненасыщеиные тозилгидразоны с ароматическими
заместителями давали пиразолы, а при использовании
гидразонов с алифатическими заместителями никаких определенных
продуктов получено не было. При реакции с системой боргидрид
натрия/BtuNBr происходит восстановление тозилгидразона ди-
бензилкетона до 1,3-дифенилпропана [1771].
3.19. МФК в химии металлоорганических соединений 283
3.18.11. Электрохимия
В заключение обзора различных типов реакций,
проведенного в разд. 3.18, необходимо указать, что четвертичные
аммониевые соли помимо хорошо известного использования их в
качестве фоновых электролитов могут найти и другое применение
в электрохимии. Установлено [524], что действие постоянного
тока на неактивную редокс-систему Си2+/[У(СО)б]~,
представляющую собой гетерогенную систему жидкость/жидкость,
вызывает выпадение слоя меди на границе раздела фаз i[524].
На платиновом аноде было проведено также окисление
системы, содержащей 3 М водный NaCN, нафталин или анизол в
метиленхлориде в присутствии МФ-катализатора [79]. При
этом были получены с выходами до 70% моноцианопроизвод-
ные. Эта методика пригодна также для проведения ацилокси:
лироваьия.
3.19. Применение МФК в химии
металлоорганических соединений*
орго-Металлированцые комплексы S с донорными лиганда-
ми, содержащими серу, были легко получены [510] при
комнатной температуре с помощью реакции:
R R
=S + Fe3(CO)ia
бензол
(R = NMe2, OMe, H, Me)
Схема 3.162
Предполагается, что частицей, которая переходит в
органическую фазу, является HFes(CO)ir. Источником бензильного
* Опубликован специальный обзор по использованию МФК в химви
переходных металлов Г1395].
284 Глава 3. Практическое использование МФК
водорода в продукте реакции служит орго-положение исходной
молекулы. Те же самые тиобензофеноны при реакции с ди-
мером дикарбонил (цикл опентадиенил) железа при обработке
50%-ным водным гидроксидом натрия и МФ-катализатором в
бензоле при комнатной температуре дают фульвены с
превосходными выходами. По-видимому, промежуточным продуктом в
этом случае является ионная пара Q4i[Fe(CsH5) (СО)2]~ [1122].
Восстановительные свойства дикарбонил (циклопентадие-
нил) железа были использованы в новом методе превращения
1,1-дихлорциклопропанов в 1-хлорциклопропаны. При попытке
упростить методику с помощью МФК было обнаружено
образование димеров винилиденжелеза с мостиковой связью:
грамс-1,1-дихлор-2,3-дифенилциклопропан и б«с-([дикарбо-
нил (циклопентадиенил) железо] в системе 50%-ный водный
NaOH/TI^/BiuNHSC^ дают цис- и гранс-изомеры ц- (бензил-
фенил винил идеи) - ц - карбонил - бис- [карбонил( цикл
опентадиенил) железа]. В качестве промежуточного продукта
образовывался 1-хлор-2-фенилциклопропен [1654]:
a a
R
Схема 3.163
В системе твердая фаза/жидкая фаза метод МФК
позволяет провести окислительный обмен лигандами в пентакарбониле
железа при комнатной температуре в метиленхлориде или ТГФ
[1890]:
NaNO2 + QC1 + Fe(CO)B » Q+[Fe(CO)sNO]- + NaCl + CO + CO2
В тех же условиях можно восстановить октакарбонилдико-
бальт )[1890]:
NaNO2 + QC1 + Со2(СО)8 >■
>- Q+[Co(CO)4]"" + продукты диспропорционирования
Нитрат кобальта (II) превращается в кластер
(С1С)Соз(СО)э, если его раствор (H2O/NH4OH, бензол)
обработать дитионитом натрия и оксидом углерода в присутствии
C,oH2,NMe3Br [1889].
3.19. МФК в химии металлоорганических соединений 285-
При реакции аллилброыида с октакарбонилдикобальтом в
бензольном растворе с 5 н. NaOH и ТЭБА при комнатной
температуре был получен трикарбоннльный комплекс (Т) я-аллил-
кобальта с выходом 70—80% ;[511]. В этом случае
МФК-методика также превосходит старые методы синтеза по выходамг
скорости реакции, простоте и мягкости условий.
Экстрагируемым анионом является, по-видимому, Со(СО)4~.
RCXj + Co,(COV , Sll NmOH таКА /1 \
(R - а Вг. РЬ, CO2»Bu, S"-fa^J^- iCOhCo _J->Co(CO),
CH2OH; X - О, Вг) отимл \ I /
Со
(СО)?
и
Схема 3.164
В сходных условиях из геж-тригалогенпроизводных [511J
и циклопентадиенилаллилкарбонильных комплексов
(C5H5)Met(CO)«(n-C3Hs), где Met=Mn, Fe, Mo) с хорошими
выходами были получены кластеры U. Исходные продукты
получают из металлкарбонил(циклопентадиенил)галогенидов и
аллилгалогенидов [1265, 1884].
Катализируемое палладием карбонилирование бензил-,
арил-, винил- или гетерилгалогенидов также ускоряется в
условиях МФК ![512]. Методика состоит в следующем: к смеси
30%-ного водного гидроксида натрия, ксилола, тетрабутилам-
монийиодида, Pdi[P(C6H5)3]2Cl2 и небольшого количества три-
фенилфосфина (чтобы избежать выпадения металлического
Pd) при 95 °С в атмосфере оксида углерода при давлении 5 бар
медленно по каплям прибавляют органический галогенид.
Функция катализатора состоит в освобождении
карбоксильного аниона из промежуточного Pd-комплекса и переноса его в
водный слой.
30%-ный NaOH, Bu4NI
RX + СО -f- 2NaOH *■ RCO2"Na+ + NaX + Н.О
Pd-комплекс, ксилол 2 ' *
Реакции такого же типа проходят с арил- и винилгалогенида-
ми при облучении их в присутствии Bu4NBr и Со2(СО)8 в
бензольном растворе [1885].
286 Глава 3. Практическое использование МФК
Замещение в условиях МФК наблюдалось [551] также и в
карбонилах металлов группы VI. В качестве одного из
примеров можно привести следующую реакцию:
бензол, 80 "С, 2 ч
—»- Mo(CO)4[Ph2P—(CH2)2—PPhJ
Аналогичные реакции проведены с карбонилами хрома,
молибдена и вольфрама; в качестве лигандов использовались АэРпз,
PPh3 и дипиридин. Реакции в условиях МФК протекали
гораздо быстрее, чем реакции, проводимые как по старым
методикам, так и по более новым гомогенным каталитическим
методам с гидроксидом натрия в водном метаноле.
Предполагается, что гидроксид натрия действует следующим образом:
вначале вытесняет лабильную частицу из [М(СО)5СООН]-, а
затем сам вытесняется новым лигандом [551]. Эта схема
подтверждается включением 18О из Н218О в карбонилы металлов
группы 6Б в условиях МФК [928, 1464]. К 11,3 М раствору
Na18OH добавляют смесь карбонила металла и Bu^NI в
бензоле и при сильном перемешивании нагревают до 75 °С [928].
Использование этой методики позволяет проводить обмен в
следующих комплексах: M(CO)5L (M=Cr, Mo, W; L=CO,
PPh3, PBu3) и Fe(CO)4PPh. В результате обмена СО на изо-
нитрил образуется Met (СО) б (CNR) (Met = Cr, Mo, W). Реакция
идет в системе 50%-ный водный раствор NaOH/ТЭБА [1578].
Хлороплатиновыё(П) комплексы четвертичных фосфинов
растворимы в бензоле или дихлорметане, но нерастворимы в
воде. В присутствии 18-крауна-6 или дициклогексано-18-крау-
на-6 был проведен обмен лигандов на гидроксид- или цианид-
ионы действием водного раствора гидроксид а калия или
твердого цианида калия соответственно [944]:
l2(OH)2L2] + [K(18-KpayH-6)][Pta,PEt3]
PMe3Ph, PEt2Ph)
(РШ2(циклооктадиен)] *■ [K(18-KpayH-6)]2[Pt(CN)2R2]
(R = 2-THemui, бензофуранил-2, 4-метоксифенйл, метил)
4«c-[PtMe2(PEts)2] »- [К(18-краун-6
Длинноцепочечные аммониевые иоиы вели себя так же, как и
МФ-катализаторы обмена галогенных лигандов в реакциях
типа [1123]:
»- [OsFCIs]2-
При облучении гексакарбонилов металлов в системе метиленхло-
рид/ТГФ в присутствии крауи-эфира и гидроксида калия или
фторида калия в течение 2 ч ртутной кварцевой лампой вы-
8.19. МФК в химии металле/органических соединений 287
сокого давления были получены комплексные анионы
[W(CO)SOH]- '[Cr(CO)sOH]- [W(CO)5F]- и [Cr(CO)gF]-
с противоионом [К(Дибензо-18-краун-6)]+ [922]. Бензилброми-
ды можно превратить в арилуксусные кислоты с помощью кар-
бонилирования в условиях МФК в присутствии 2 мол. % окта-
карбонилдикобальта, 5 н. NaOH и R*NX (комнатная
температура, 10—12 ч в атмосфере СО) [920, 996, 1246, 1497, 1572].
По-видимому, проходит бензилирование промежуточно
образующейся ионной пары '[R4N+[Co(CO)4]-]:
ArCH2Br + R4N+[Co(CO)4]- >■ ArCH2—Co(CO)4 + R4N+Br"
а затем взаимодействие с СО и последующий гидролиз
АгСН2—Со(СО)4 + СО *- Аг—СН2—СО—Со(СО)4
АгСН2—СО—Со(СО)4 + 2ОН~ »- АгСН2—СОа" + [Со(СО)4]~ + Н2О
Сходным образом о-дигалометиларильные соединения, октакар-
бонилдикобальт и краун-эфир дают комплекс с КОН и
превращаются в циклические кетоны [1124]. Если присутствует сте-
хиометрическое количество ТЭБА, то его б"ензильная группа
входит в состав фенилуксусной кислоты [1401]. Наконец, при
реакции фенилацетилена с метилиодидом в атмосфере оксида
углерода (1 бар) в присутствии системы 5М NaOH/бензол и
небольших количеств октакарбонилдикобальта и цетилтриметил-
аммонийбромида образуется с выходом 44% 4-гидрокси-4-ме-
тил-2-фенилбутен-2-олид [1125]. В этих же условиях бензилга-
логениды в присутствии ТЭБА/Со2 (СО) 8 дают арилзамещенные
пировиноградные кислоты [446].
При действии тетракарбонила никеля в системе 30-ный
водный NaOH/R4NX в атмосфере оксида углерода проходит
карбонилирование аллилхлоридов с образованием бутеновых
кислот. Это свидетельствует, по-видимому, об образовании
промежуточного соединения—[Ni5(CO)i2]2~ и '['Ni6(CO)i2]2~.
Молярное соотношение аллилгалогенида и карбонила никеля
должно быть равно (12—25) : 1 [1400]. Региослецифическое
взаимодействие диена, СО и метилиодида ведет к гра«с-дие-
нилметилкетону с выходом 43—86%, Эту реакцию проводят в
бензоле с 5 М NaOH, небольшим количеством Co2(CO)r
и R4NX [1125, 1209]; например,
СН2=С(СН3)—СН=СН2 >■ СН2=С(СН3)—СН=СН—СО—СН„
транс
Описано образование кетонов RCOR из алкилгалогенидов с
использованием Fe(CO)5', реакцию проводят с 33%-иым
водным гидроксидом натрия и тетрабутиламмонийбромидом в бен-,
золе при комнатной температуре. Бензильные бромиды и иод-,
октан реагировали с выходом выше 90% [1295]. Несимметрич-
288 Глава 3. Практическое использование МФК
ные этилкетоны образуются с более низкими выходами. Для их
получения сначала менее реакционноспособные галогениды
(как правило, бромиды) обрабатывают смесью карбонил же-
леза/№ОН/катализатор, а затем прибавляют этилиодид.
Недавно система Со2(СО)8/СО/водный гидроксид натрия/
/ТЭБА и метилиодид была использована для получения
1-ацилфульвенов из фульвенов [1796] и ненасыщенных гидрок-
сикетонов и диенонов из ал ленов:
R2iC=C=CHR* >- R2iC(OH)-C(COCH3)=CHR2+[R*CH=C(CR2OH)]2CO
Последние соединения образуются также в отсутствие метил-
иодида и МФ-катализатора [1784]. При реакции хлорида
Fe(II) с циклопентадиеном в присутствии твердого гидроксида
калия и 18-крауна-6 в ТГФ при комнатной температуре можно
получать ферроцены [1335].
Образование фенолкарбонатов проходит при
взаимодействии фенолов, СО, гидроксида натрия в присутствии
МФ-катализатора, окислителя и катализатора, содержащего элементы
группы 8Б [1552].. В двух исследованиях описана
изомеризация следующего типа, протекающая в присутствии родиевого
катализатора:
СН2=СН—СНОН—R »- С2Н5—СО—R
СН=СН—СН2—Y >■ СН3—СН=СН—Y
(Y = Аг или MeNAr)
В одном случае RhCl3 экстрагировался в форме I^N+RhCU"
[1886], другие же авторы [1461, 1887] применяли комплекс
R[h(CO)2Cl]2 вместе с 8М NaOH и R4NX.
Использование димера дихлордикарбонилродия(1) было
предложено для мягкого дегидрирования бензильных спиртов
в карбонильные соединения; реакция идет в системе 8 М NaOH/
/бензол/ТЭБА.
Ph—СНОН—R »- Ph—CO—R
Существуют доказательства того, что эта реакция протекает
через образование орго-металлированного комплекса [1643].
Палладиевый комплекс Р<1С12(РРпз)2 был использован в
системе Си1/ТЭБА/10%-ный водный гидроксид натрия/бензол для
проведения реакции сочетания (выход 32—90%) [1888]:
R1—CseCH + RaCX=CHRs >- R1—С==С-CR2=CHRS
В присутствии каталитических количеств Bu4NCl и PdCl2 в
системе дихлорметан/3 н. НС1 из а,(Р-ненасыщенных кетонов и
хлорида фенилртути или тетрафенилолова были получены
р-фенилкетоны. В этом случае активной экстрагируемой
частицей, по-видимому, является BtuN+PdCl3~ ![1276]:
RCH=CH—CO—R' —->■ RCH(Ph)—CH2—CO—R'
3.20. а-Элиминирование 289
Среди других методик MOJK с использованием металлооргани-
ческих соединений можно отметить удобный препаративный
способ алкилирования арилуксусных кислот в комплексе с
Сг(СО)з (разд. 3.9.4), восстановление нитросоединений с
помощью Ре3(СО)12/катализатор (разд. 3.21.2) и дегалогенирова-
ние «-бромкетонов в системе Со2 (СО) g/NaOH/ТЭБА (разд.
3.21.2).
3.20. «-Элиминирование
Концентрированные неорганические основания могут часто
вызывать «-элиминирование. В самом деле, катализируемое
основаниями образование дигалокарбенов является одной из
наиболее широко применяемых МФК-реакций. Первая стадия
этой реакции — депротонирование, как правило, идет намного
быстрее, чем последующее отщепление галогенид-иона. Таким
образом, в присутствии подходящего акцептора реакции
первоначально образовавшихся анионов могут конкурировать или
даже преобладать над реакциями карбенов. В данном разделе
эти родственные реакции рассматриваются совместно. Уделено
внимание также описанию современных методик получения
дигалокарбенов в условиях МФК в отсутствие оснований, хотя,
строго говоря, эти методики и не относятся к настоящему
разделу. Наконец, будут рассмотрены реакции других карбенов.
3.20.1. Реакции образования
и присоединения дихлоркарбена
Долгое время считалось, что образование дихлоркарбена
из хлороформа под действием основания представляет собой
реакцию; чувствительную к влаге, поскольку хорошо известен
гидролиз хлороформа с образованием оксида углерода и фор-
миат-иона -(см. обзор литературы по химии дигалокарбенов до
1970 г. [605]). Хотя классическая реакция Реймера — Тимана
проводится в водном растворе гйдроксида натрия, тем не менее
Дёринг и Хофман '.[606] в одной из пионерских работ, по химии
СС12 в 1954 г. обнаружили лишь 0,5% дихлорнокарана после
проведения реакции циклогексена с хлороформом в водном
растворе гйдроксида калия. Вслед за этим были высказаны
осторожные предположения, что СХ2 может быть не так сильно
чувствителен к воде, как считалось. Например, реакция олефи-
нов с хлороформом и гидроксидом натрия была проведена в
тетраглиме при 95 СС [607] или в смеси СНСЦ/плавленый
85%-ный КОН при 110°С [840]. Однако лишь после опубли*
290 Глава 3. Практическое использование МФК
кования в 1969 г. ставшей теперь знаменитой работы Макощи
и Вавжиневича [608] получение дигалокарбенов в присутствии
водных растворов сильных оснований и катализатора (обычно
ТЭБА) стало общепринятым методом*. С тех пор вследствие
простоты и эффективности этот метод нашел широчайшее при-:
менение. Метод Макоши предпочитают теперь для
большинства реакций присоединения дихлоркарбена. Даже в тех случаях,
когда обычные дихлоркарбеновые реагенты не реагируют, т. е.
для стерически затрудненных или электронодезактивированных
алкенов, он приводит к удовлетворительным или очень высоким
выходам. Известно лишь очень небольшое число примеров,
когда реагент Зейферта (PhHgCBrClg) или декарбоксилирование
трихлорацетата натрия приводит к результатам лучшим, чем в
методе Макоши. Это обычно относится к субстратам,
чувствительным к основанию. При этом необходимо отметить, что
реагент Зейферта сам может быть получен реакцией в условиях
МФК и что декарбоксилирование трихлорацетата натрия
также успешно протекает в МФК-процессе, например при
пониженных температурах. Тщательное изучение селективности
показало, что дихлоркарбен, генерированный в различных
системах, всегда является свободным дихлоркарбеном, а не кар-
беиоидом [609—611]. Это также относится к СС12, полученному
в условиях МФК [4]. Недавно Мосс и сотр. [612] сравнили
селективность реакций ряда карбенов/карбеноидов как в
присутствии, так и в отсутствие крауи^эфиров. В отличие от
других систем селективность в случае СС12 не изменялась, что
свидетельствовало в пользу того, что он действительно
существует в свободном виде. Чем же можно объяснить то, что система
МФК/СС12 часто дает более высокие выходы или является
последним шансом даже в случае безнадежно
дезактивированных субстратов?
Это отличие объясняется рядом факторов. Во-первых,
дешевле и легче использовать для генерации дихлоркарбена
огромный избыток хлороформа и гидроксида натрия. Во-вторых,
в отличие от других методов получения СС12 в МФК-реакции
он образуется обратимо и разлагается постепенно. Таким
образом, нежелательные побочные реакции (атака на растворитель
или исходный реагент, димеризация, полимеризаци) не прояв-
* Следует отметить, что недавно в качестве эффективной системы для
получения СС12 была предложена смесь измельченного КОН (предпочтительно)
или NaOH/ТЭБА и CHC1S [868, 1738]. Выход продукта такой же, как и в
оригинальном, методе Макоши, но с твердым основанием реакция идет немного
быстрее. Для медленно реагирующих олефииов это является недостатком, так
как ССЬ может гидролизоваться, вместо того етобы сохраняться и
расходоваться по мере необходимости.
3.20. а-Элиминирование 291
ляются в заметной степени. Наконец, хлороформ как
растворитель довольно инертен по отношению к СС12.
Преимущество данного метода по сравнению с остальными
проявляется наиболее четко при тщательном рассмотрении
второго фактора.
а) Разложение LiCCl3 происходит при температуре выше
—73°С'[610].
б) Реакция между хлороформом и грег-бутилатом калия
при температурах —20 °С и выше проходит мгновенно.
в) Трихлорацетат натрия отщепляет СО2 в глиме при 80 °С.
В МФК-реакциях в неполярных растворителях сухой
трихлорацетат аммония разлагается медленно даже при комнатной
температуре. При этом не было обнаружено [31] стабильного
интермедиата Ё4№"СС1з~.
г) Реагент Зейферта (бромдихлорметилидфенилртуть)
разлагается около 80 °С на PhHgBr и СС12. В присутствии иодид-
иона при пониженных температурах эта реакция частично
обратима [611].
Время жизии образовавшегося дихлоркарбена всегда
крайне мало. В отсутствие реагирующего олефина он «выдыхается».
Например, хорошо изучены многостадийные и сложные реак-
ци с дихлоркарбеном, полученным из трихлорацетата натрия
[614]. Однако в случае реакции Макоши весь дихлоркарбен не
образуется одновременно. Побочные реакции и гидролиз идут
медленно, и система остается реакционноспособной в течение
длительного времени даже в отсутствие хорошего акцептора
карбена. Таким образом, находящийся в равновесии с
исходным реагентом СС12 может «ждать» субстрат, и поэтому
становится возможной реакция даже с очень дезактивированными
субстратами. На практике применяют 50%-ный
(концентрированный) водный раствор гидроксида натрия в присутствии
ТЭБА как катализатора и хлороформа в качестве
растворителя. Общие тенденции к образованию, присоединению и
гидролизу СС12 приведены в табл. 3.18. В отсутствие олефина
медленный гидролиз хлороформа ускоряется примерно в 6 раз под
действием ТЭБА. Добавление олефина приводит к повышению
расхода хлороформа, величина ускорения зависит от природы
олефина. Гораздо большее значение имеет то, что соотношение
Скоростей присоединения карбена и гидролиза хлороформа
зависит от нуклеофильности олефина и может изменяться в
очень широких пределах [384]. Поэтому малореакционноспо-
собные субстраты следует перемешивать с большим избытком
основания и хлороформа длительное время. Из данных,
приведенных в табл. 3.18, видно, что условий, оптимальных для
всех олефинов, ие существует. Тем не менее была проделана
большая и успешная работа по оптимизации условий реакции
292 Глава 3. Практическое использование МФК
водного NaOH и
ТЭБА при 23 °С
Олефин
0,1 моля
[384]
олефина в
присутствии 1
Расход
НССЬ, ммоль
ММОЛЯ
Выход продукта8
гидролиза HCCIs
1ММОЛ|>
i%
лрасоедвве-
ния СС12.
/ммошь
Циклогексен без ТЭБА
Без олефина, но с
ТЭБА
3,3-Диметилбутен-1
Гексен-1
Циклогексен
2-Метилбутен-2
а Через 4 ч после начала
3,4
20,8
25,0
53,6
67,3
80,4
реакции.
3,4
20,8
14,5
8,4.'
11,85
3,3
100
58
15
17
4
,7
,6
,1
0
12
45
55
п
,0
,2
,45
,1
для олефинов со средней реакционной способностью. Следует
отметить следующие основные факторы, влияющие на ход
реакции.
Скорость перемешивания
Перемешивание должно быть энергичным. При скорости
перемешивания 800 об/мин и более получают воспроизводимые
результаты. Не имеет значения, используется магнитная или
механическая мешалка. Виброперемешивание приводит к
несколько большей скорости реакции, что обусловлено лучшим
теплопереносом, а не лучшим смешением [32].
Температура и время реакции
Повышение температуры увеличивает скорость реакции, и
часто при 50 °С более высокий выход продукта достигается за
более короткое время. Однако надо помнить, что гидролиз
конкурирует £ присоединением, и влияние температуры на эти
процессы различно. Иногда реакция Макоши проходит бурно и
невоспроизводимо. Наблюдались случаи, когда из двух,
по-видимому, идентичных реакций одна неожиданно шла с разогре^
вом и становилась неконтролируемой, в то время как другая
проходила нормально. Поэтому обычно принимают какую-либо
из двух мер предосторожности. Либо смешивают реагенты при
5°С и добавляют обычные реакционноспособные субстраты
при охлаждении на ледяной бане, а затем при комнатной
температуре; через несколько часов смесь нагревают в течение 1 ч
3.20. а-Элиминирование 293
лри 50 °С [613]. Либо перемешивают .субстрат и катализатор в
хлорофордое при повышенной температуре: в в течение
нескольких часов прибавляют по каплям концентрированный л раствор
гидроксида натрия. Это основано на наблюдении, что выходы в
стандартной реакции (0,1 моля циклогексена, 0,4 моля СНСЬ,
0,2 моля гидроксида натрия, 1 ммоля ТЭБА, 23 °С)
увеличиваются со временем: за 1 ч реакция протекает на 20%, за 4 ч —
на 46%, за 24 ч.— на 74%, за 72 ч —на 86%- При
использовании 0,4 моля гидроксида натрия при 50 °С выход в той же pej
акции за 2 ч составляет более 90%. Аналогичные эффекты на^
блюдалисьдля гексена-1, З-метилбутена-2 и 3,3-диметилбутена-1
[613]. В случае инертных, термически нестабильных или
чувствительных к гидролизу субстратов смесь обычно
перемешивают очень продолжительное время (вплоть до 100 ч) при ком-г
натной температуре.
Количество хлороформа
При проведении реакций в небольшом масштабе (1—20 ммс
лей) избыток хлороформа используют как растворитель я как
реагент. При работе с 0,1—'1,0 молями олефина общий объем
смеси приобретает большее значение. Установлено, что в
условиях, указанных в табл. 3.18, 3—4-кратный избыток
хлороформа по отношению к алкену дает наилучшие результаты.. В
реакциях Макоши выпадение осадка коричневых нерастворимых
побочных продуктов (полимерный GCb?) наряду с образовав
нием стойких эмульсий может затруднить перемешивание,
особенно при медленных реакциях. В таких случаях полезно
применение сорастворителя. Некоторые авторы применяли бензол,
но использование дихлорметана [1856] предпочтительнее, так
как он, по-видимому, слегка ускоряет основную реакцию. По^
добный эффект еще более ярко выражен для других дигало-
карбенов.
Количество основания
Большинство исследователей используют
концентрированный (50%-ный) раствор гидроксида натрия. Однако .иногда
применяют концентрированный раствор гидроксида калия и
Ва(ОН)2. В стандартных условиях (4 ч, 4 мол? СНС13 на
1 моль алкена) выходы резко растут вплоть до -соотношения
NaOH и олефина 4 : 1. Дальнейшее увеличение выхода
невелико [613].
Количество катализатора
Обычно используют 1 мол. % катализатора по отношению
к олефину. На выход 7,7-дихлорноркарана -в стандартных
условиях практически не влияет изменение концентрации ТЭБА в
294 Глава 3. Практическое использование МФК
интервале 0,5—20 мол. %. При меньших концентрациях
катализатора выход резко падает, по всей видимости, вследствие
того, что в этом случае катализатор находится в основном в
водной фазе [613].
Тип катализатора
Подробное обсуждение влияния катиона и аниона
катализатора на скорость реакции приведено в гл. 2. На практике
успешно применяют многие соли аммония и небольшие
различия в их влиянии на скорость сглаживались при обычном
времени реакции (несколько часов). В качестве катализаторов
предпочтительнее использовать хлориды, -но ни в коем случае
не иодиды. Наиболее широко используемой солью аммония
является ТЭБА, однако аналогичное действие оказывает алик-
ват 336, цетилтриметиламмонийбромид и многие другие соли,
включая некоторые соли фосфония и арсония [32, 613, 616].
Иногда используют сокатализатор — небольшое количество
этанола: 1 мл на 1 ммоль ТЭБА (впрочем, это лишь очень
слабо увеличивает скорость реакции в случае ССЬ) и, как
упоминалось ранее, дихлорметан в качестве сорастворителя.
Другие типы катализаторов
В качестве таковых используют специально полученные,
связанные с полистиролом соли аммония [62, 68, 902], поли-
этиленоксиды на полимерной подложке [1121] и
промышленную ионообменную смолу амберлит ЩА-400 [616]. В одном
патенте [1000] применяли оксид амина, но наш опыт
показывает, что такие катализаторы не обладают преимуществами- по
сравнению с ониевыми солями. В другом патенте [620]
использовали соль сульфония и в качестве сокатализатора неионное
ПАВ. Было также найдено, что третичные (но не первичные
или вторичные) амины также являются очень эффективными
катализаторами [617]. Макоша и сотр. [433] объяснили этот
довольно поразительный результат, показав, что в реакции
участвует ряд нестойких промежуточных продуктов (см. разд.
3.20.2). Наиболее активными из всех катализаторов оказались
три-к-пропиламии и три-н-бутиламин [613] (см. табл. 2.8).
Поэтому удивительно, что некоторые третичные амины были
рекомендованы как селективные катализаторы
моноприсоединения СС12 к полиенам [866]. Недавно Ривз и сотр. [432]
показали, что можно использовать даже первичные или
вторичные амины, если присутствует алкилирующий агент (например,
этилбромид), генерирующий действующий катализатор.
Ряд исследователей вводили в качестве катализаторов
краун-эфнры, йо они, по всей вероятности, не дают каких-либо
реальных преимуществ [45, 302, 618]. Однако недавно было
ЗЛО. а-Элиминирование 295
обружено [619], что они сильно влияют на селективность
присоединения дихлоркарбена. Показано [621, 622] также, что
селективными катализаторами являются ионы триалкюьр-окси-
этиламмония. Было установлено, что катализатор А приводит
к образованию из соответствующих олефинов моноаддуктов В,
С, D и Е, в то время как в случае цетилтриметиламмонийбро-
мида образуются бис- или грис-аддукты. Однако попытки
подтвердить этот эффект привели к неоднозначным результатам
[619]. Позже было показано, что образование моно-, ди- или
триаддуктов присоединения СС12 к этим алкенам зависит как
от природы катализаторов (липофильные катализаторы дают
больший процент продуктов полиприсоединения), так и от их
относительной концентрации. Поразительно, что в случае более
низких концентраций полиаддукты образуются с большими
выходами. Наличие гидроксильных групп в различных
положениях катализатора не влияет на селективность [1898].
О якобы наблюдавшейся оптической индукции при
присоединении дихлоркарбена и опровержение этого наблюдения
см. разд. 3.1.5.
а
-а
Ph—CH2—N(Me)2—СН2—СН2ОН
в
ОН®
А
а а
.он
в
а
а
о
Схема 3.165
Недавно был обнаружен интересный и иеобъясненный до
настоящего времени факт [ 1299]: смесь твердого карбоната
калия и 1 мол. % твердого фторида^ калия является очень
эффективной синергической щелочной системой при условии, что обе
используемые соли тщательно высушены, а в качестве
катализатора применяют BU4NHSO4; реакционную смесь кипятят
1 сут. Любая из этих солей может действовать самостоятельно
как основание, но при этом в идентичных условиях выходы
заметно снижаются. При использовании концентрированных
водных растворов КОН или NaOH добавление KF снижает выход
аддукта дихлоркарбена.
296 Глава 3. Практическое- использование МФК
Типичная методика Макоши [624, 625]
В смеси 0,4 моля хлороформа и 1 мл этанола растворяют
0,1 моля субстрата и, 1 ммол'ь ТЭБА (или три-к-пропиламина).
К раствору добавляют 0,4 моля охлажденного льдом
свежеприготовленного 50%-ного раствора NaOH и перемешивают
сначала на ледяной бане, затем при комнатной температуре в
течение 1—2 ч. Затем реакционную смесь нагревают и
перемешивают еще 3—5 ч при 50 °С, после чего реакционную массу
выливают в большое количество воды. Органическую фазу
многократно обрабатывают свежими порциями воды или
разбавленной соляной кислоты для разрушения стойких эмульсий;
эти операции не требуются в тех случаях, когда фазы легко
разделяются и происходит выпадение осадка полимеров
коричневого цвета. Растворитель после высушивания отгоняют.
Продукты реакции, если это возможно, выделяют
перегонкой. Неполярные твердые или высококипящие продукты
отделяют от коричневых побочных продуктов пропусканием их
раствора в смеси пётролейный эфир/серный эфир или в толуоле
через короткую колонку, заполненную силикагелем.
Получение дихлоркарбена из трихлорацетата натрия
NaO2CCCls »- NaCl + CO2 + СС1г
Иногда применение сильных оснований для получения
дихлоркарбена нежелательно. Здесь альтернативой может быть
хорошо известная реакция декарбоксилирования
трихлорацетата натрия.
В классическом варианте этот метод требует
использования дорогого безводного диметоксиэтана в качестве
растворителя и температуры около 80 °С. Была разработана [675, 31]
улучшенная модификация этого метода в условиях МФК,
которая исключает дорогой растворитель и позволяет вести
реакцию при пониженных температурах; получение термически
нестойких соединений, таких, как К, возможно даже при
комнатной температуре. Однако в последнем случае реакция идет
несколько суток. 11ри проведении реакции в кипящем растворе
хлороформа с 1—2 мол. % ТЭБА или аликвата 336 и 2
молями трихлорацетата натрия на одну двойную связь достигают
выхода 65—88%.
3.20. а-Элиминирование ,297
Декарбоксилирования не происходит, если при реакции в
условиях. МФК используют концентрированный водный' раствор
трихлорацетата натрия, хотя (гидратированный) анион'
экстрагируется. В данном случае основную роль играет количество
катализатора. Когда катализатора слишком много, в растворе
находится и разлагается за единицу времени относительно
большое количество трихлорацетата натрия; это ведёт к
.заметному развитию хорошо известных побочных реакций [6Й]
(атака СС12 или СС1з~ на трихлорацетат и осмоление),,
и СС12 «выдыхается». В отличие от метода Макоши в
данном случае олефин не влияет на скорость расходования
источника карбена [675]. Было изучено [676] также влияние
катионов (K+>Na+>Li+) как в присутствии, так и в отсутствие
краун-эфиров на декарбоксилирование и присоединение
дихлоркарбена. Выводы были аналогичными: при быстром дё^-
карбоксилировании выход продукта относительно низок.
Внедрение в Si—Н-связь дихлоркарбена, полученного по
МФК-реакции из NaO2CCQ3, идет с замедлением: [1855].
Типичная методика трихлорацетатного. способа] [675]
В 50 мл <£НС1з растворяют 1 ммоль катализатора и 0^05 моля
олефина. К раствору прибавляют 0,1 моля твердого, тонко
измельченного трихлорацетата натрия (промышленный, г^рбициД,
содержащий 96,7% основного вещества, без дальнейшей
очистки), смесь нагревают и энергично перемешивают при £0°С JI
прекращения выделения СО2 (5—6 ч). В хБде реакции1*
становится черной или коричневой. После добавления Щ
воды фазы разделяют. Нерастворимый хлбпьевидйый ос
отделяют. Органическую фазу сушат и продукт выделяют пе
регонкой или кристаллизацией.
Получение дихлоркарбена из (^рихлорметил)триметилсилана,
Me8SiCCl8 + KF (краун) >■ MegSiF + KC1 + СС1г
Описан новый мягкий метод получения дихлоркарбена (по
вышеприведенной реакции. Олефин и кремнийорганйчеркое fcoj-
единение перемешивают 1 сут в диглиме при температуре 25;°G
с твердым KF и 18-крауном-6 [1459]. В случае дибром)карбёна
используют тот же метод; типичные выходы находятся в Ш4г
тервале 55—80%. Реагенты могут быть :получены декарбокеиг
лированием триметилсилилтригалогенацетата [1785]
Получение и присоединение дихлоркарбена
по методу Макоши
Вернемся теперь к наиоолее часто применяемой системе
Макоши. Среди аналогичных реакций Макоши с ССЬ, описан1
ных в литературе, общие реакции приведены в таблицах : и
лишь отдельные примеры обсуждаются детальна
осадок
Таблица 3.19. Реакции йондпрИсоедийения СС& к/о^финам • условиях межфаэного
Субстрат
Примечания
Литература
Различный алифатические и арсша- Обычна опей» "высокий При наличии стоических лрепятст- 4, 31, 32, 433» 608-»
тические алкены
-'Н, алкил, арил)
Ферродензамевденные алкены
Стероиды
Ph
(R = Н, Me, Ph)
вйя выходы лизкие
Обычно- очень высоки* Тетрафешштклея не реагирует
4?
Обычно очей* высокий
Обычно очень вьйокня Малоустойчивые аддукты:
Обычно очень
Боковая цепь
Циклы
Обыаяа о<1ечь': высокий
6Г7, 636, 641-643
675, ?36, 1298, 1384
1408; 1594, 1790
626-633, 732
735
627", 634, 1484
31, 635
801
639,, 1*33
640г 644
646
647
40
648
(R » Me, Ac)
92
85,75
81, 5
Четыре стереоизомерных моноаддук-
та
Конкурирующее внедрение по С—Н 649
947
955
956
'ОАс
Продолжение табл. 3.19
Субстрат
Выход,, %
Примечания
Литература
R'CH^CH2—SiMe3
Другие полициклические соединения
СН2 = СН—СН2Вг
С1Н2С—СН = СН— СНаС1
Енйны
R—C = C—SiMe3
so.
45
60
55-72
80
45
26
71
Многочисленные перегруппировки
Присоединение по двойной связи
Гидролиз. до. 1-алкилциклопропенона
Присоединение по незатрудненной
двойной связи
693
693
650
1267, 1430, 1389
1352
652, 1131
1326
1750
695, 968
653, 1126
' Различные замещенные циклогексён'ы'
R2C = C-COOR2
R1
(R'¥=H)
R2C = C-CONH2.
R1
H2C.=C^CN
CH3
О
it
R-C-CR' = CR2R3
Циклопентадиеноны
До 85
До 92
42
До 90
= CH—CHR1—OH
= C(R)— CR2OH
Циклоалкен-2-олы
R2C = CR—CH?—OCH (OR1) CH3
R'HC = CH—CR2(OR3)2
2 Кеталь циклогексенона
75^83
60-92
70—90
654, 655, 1462, Г512,
1615
616, 671—673, 677,
872
Побочный продукт: 4,4,4-трихлор-2-
метйлбут'иронитрил
Простые кетоны склонны к осмоле-
нию
5- и 6-членные циклы: главным
образом , .сын- по отношению к ОН-
группе; 7- и 8-членные — только ан-
ти-.
38—8*2
71
616
671
674
1493; 1650, 1788
694
616, 621
502
1471
663
660-662, 674
'■* 8Х
Субстрвт
Выход, %
Продолжение табл. 3.19
Примечания
Литература
CD
Енольные эфиры различных типов
X'
(X - Н. OR)
Еиольиые ацетаты кетонов
Енамииы
Имины Ar—CH«=N—Аг'
(R)
78
До 97
До 95
До 70
До 86
До 90
Часто низкие выходы
75
Имины (R,Ar)~CH=NR
Днгидропираны
Ненасыщенные производные Сахаров Высокие выходы
Винилсульфиды До 56
1 -Силациклогексаднены-2,4
Атака только в {^-положение
651
656, 722, 1127, 1392,
1431, 1705
657—659
420, 686
664-666, 1623
Некоторые аддукты легко
перегруппировываются в условиях реакции
Некоторые ,аддукты легко перегруп- 667, 669, 670, 964,
пйровываются в условиях реакции " "
1128
Иногда нестабильные аддукты
Место атаки зависит от заместителя
667,
987, 1130,1485
1132
1637, 1705
1313
. ■■'■ . &Ж а-Элиминирование 303
В табл. \ 19 приведены реакции СС1а с соединениями
с.одной двойнойЧсвязыо. С. простыми олефинами легко
достигаются выходы *5—95%, с сильно стерически затрудненными ал-
кенами (например, 3,3-диметилбутеном-1) выход даже в
благоприятных условиях достигает только 40%. Тетрафенилэтилен
в отличие от трифенилэтилена не реагирует.
Границы применимости метода обнаруживаются.для
некоторых олефинов с элёктроноакцепторными заместителями. В
случае ди-, три- и тетрахлорэтиленов реагент Макоши не вступает
в реакцию, но с первыми двумя субстратами применим три-
хлорацетатный метод. Как указывалось выше, в процессе
Макоши дихлоркарбен находится в равновееии с трихлорметиль-
ным анионом:
-н+
CHGL ■ »- СО," zj—» СС1- + С1-
3 быстро а * '
В реакцию вступают или электрофил СС1г, или нуклеофил
СС13~, или одновременно обе частицы в зависимости от
природы олефина (электронообогаеденный, элёкт-ронодефици'Гныи или
«нейтральный») в реакционной смеси. Таким образом,-
небольшие изменения структуры олефина будут определять изменение
направления реакции в сторону образования дихлорциклопрр-
пана (с) или присоединения хлороформа но двойной связи
(б). ■ • ■'•- к ■■ -:•
Cl Cl
Схема 3,167
Как правило, когда R1«=R2*«=R3=H, то наблюдается только
направление (б). Но при R3—алкил или фенил и Rl, R2=*H
или алкил реакция идет по направлению (а) [420, 671, 674,
677, 686Г 768]. Ситуация становится более сложной, если один,
или оба радикала Д1^8==фенил или алкил и R3=H: образу?
ются смеси аддуктов и продуктов вторичных реакций, рассмот-.
ренные в разд. 3.20.2. Аллильный гидрокснл до некоторой сте«,
пени дезактивирует двойную связь по отношению к атаке CClj
[502]. Было проведено обширное исследование [990] влияния
соседнего циклопропанового кольца на реакционную
способность двойной связи по отношению к генерированному в МФК-
реакции GC12. Оказалось, что циклопропилалкены реагируют
примерно в 10 раз быстрее, чем изопропилалкены, и примерно
так же, как соответствующие метил ал кены.
304 Глава 3. Практическое использование МФК
-. Ацетилены также являются плохими нуклеофйлами, и их
реакции с дихлоркарбеном обычно проходят медленно. Реагент
Макоши не является исключением. Кроме топ>, .первичные
продукты реакции — дихлорциклопропены — в ходе реакции
частично гидролизуются с образованием с^р-ацетиЛеновых кетонов
и циклопропенонов, ,в которых в свою очередь может
раскрываться цикл:
ci а
—СгС—
Схема 3.168
Таким образом, реакция ацетиленов с реагентом Макоши не
приводит к удовлетворительным выходам, однако вследствие
своей простоты используется для синтеза фенил-, грег-бутил-,
грег-бутокси- и циклопропилзамещенных циклопропенонов
[626, 678, 891]. Реакция алкинов с трихлорацетатом натрия
дает в качестве основного продукта дихлорциклобутенон [679].
о.
га
—с==с—
/
Схема 3.169
Продукты присоединения к полициклическим мостиковым
алкенам при условии соблюдения стерических требований
быстро перегруппировываются. В этих случаях перегруппировки
могут проходить во время реакции, и образующиеся продукты
способны к присоединению второй молекулы карбена, что
приводит к сложным смесям продуктов. Механизм реакции
характеризуется тем, что сын-атом хлора временно отщепляется в
виде хлорид-иона и дисротаторное раскрытие трехчленного
цикла ведет к образованию аллильного катиона, который снова
присоединяет хлорид-анион с той же самой стороны молекулы:
Cl* a
Схема 3.170
3.20.. а-Элиминирование 305
Примерькприведены ниже:
С1
н
С1 [696,697,11291
[7061
С1
Скема 3.171
306 Глава 3. Практическое использование МФК
Менее напряженные циклопропаны в этих условиях ие
изменяются:
С1
1695J
[700, 955]
R = Н, CI, ОМе, ОАс
Схема 3.172
Наиболее интересной из всех является циклическая система
бицикло [2.2.1] гептадиена, где три конкурирующих первичных
присоединения—1,2-экзо-, эндо- и 1,4-гомоприсоединение —
ведут к получению большого числа продуктов [687, 703—705,
1665], особенно при использовании замещенных соединений.
Аналогично ведет себя квадрициклан [1288], однако в этом
случае преобладает третий продукт, структура которого
приведена на схеме 3.173. Реакция с квадрицикланом
свидетельствует, что генерируемый в условиях МФК СС1г атакует даже
напряженные циклопропаны. Другим примером является
трехчленный цикл в 1,3-дегидроадамантане [1349]:
+ биспройукт
Схема 3.173
Системы с двумя и более двойными связями реагируют с
обычными дихлоркарбеновыми реагентами только по одной связи,
{666]
[502]
COOR
[68Ц
CI CI
CI2C=CH—CH==CH—CH(OR)2 *■ C!2C=CH-
-CH(OR)2 [682J
CI CI
CH3 CH3
ArCH=CH—CH=CH2
СНз СНз
CI CI
ArCH=CH
[683]
CI CI
[684]
CI CI
R1 R2
н —~
(+6ucnpo8yn.m)
Ph
I
CH2=C—CH=CH2 >
R1 R2
■-=-= [737]
OR
CI CI 1 .
Схема 3.174
CI CI
CH=CH2 [685]
OR
—C— =
[738]
CI CI
Таблица 3.20. Многократное присоединение СС/2 в условиях межфазного катализа
Реакция
Примечания
Литература
г
а
о
Се
С\2
си
_/
R.R* R'R2
(R\ R2 = Н.алкм.арил)
>=С-(СНЛ-С=с(
К- R:> R3
(R1, R2, R3 = Н.аппилдрил ; л = О, 1, 2)
=СНГ-(СН,Ь—С=СН—CH(OEt)2
СНз
R1 R2
(СН2)„—ft-
cu
С12
Смеси стереоизо- 684, 685, 687, 688
меров
Смеси стереоизо- 626, 689, 1146,
меров 1147, 1252, 1302
СН2)2—С=СН—CH(OEt)2 ■
77%
758
(CH,)a-
СНз
си
62%, два изомера 683
СН3 СНз
СНз СНз
CIa
R / \ R1
c=c=c
R R1 R' R' R'
(R = Н,алкил,арил; R' = Н,алкил)
R1
Cl2
Ar2C=C=C=CAr2
Ar2
CI»
CIa
Ar2 + Ar2
=CAr2
cia
ci2
^C=C=C=C
R' 4R'
(R = Ph; R' =алкил ufluR = R' =
CH2=CH-O—CH = CH2 >
R' R'
42%, два- изомера
692
Смеси стереоизо- 707, 708, 1144,
меров 1145, 1325, 1382
626
7.09; 710
CIa
-о-сн=сна
R'
С1а
1а СЬ
•о-
ёэо
Продолжение табл. 8.20
Примечания
Литературе
Только один изо- 687
мер
Только один
изомер
685, 699
Только один
изомер
Только один
изомер
Два стереоизомера 31
Реакция
R
(R - Н, Ph)
ч
сь
711, 713
626, 712, 713
622
730, 693
Продолжение табл. 3.20
— ■
Примечания
Литература
968
693
691
948-
1148—1150, 1432,
1493
Реакция
Другие полициклические соединения
ЗЛО. а-Элиминирование 313
поскольку электроотрицательные" атомы хлора дезактивируют
систему по отношению к дальнейшей атаке. Однако в случае
реакций в условиях МФК часто наблюдается присоединение по
нескольким связям, и иногда процесс трудно остановить на
стадии моноприсоединения. Если имеется несколько
изолированных кратных связей, то в первую очередь будет атакована
наиболее электронообогащенная. У сопряженных систем
наиболее активированная часть может оказаться единственной
реагирующей, как показано на схеме 3.174.
Однако обычно, множественные присоединения преобладают,
хотя выходы высших аддуктов снижаются. Известные примеры
множественных присоединений СС12, генерированного в
условиях МФК, приведены в табл. 3.20.
Иногда наблюдаются неожиданные перегруппировки
первичных аддуктов. Известна также довольно уникальная реакция
фенилаллена [707, 708]:
PhCH=C=CH2
С1
н
Схема 3.175
Были предложены механизмы этих превращений:
о
N. Н
Чс=с/
/ \
Н3С COOEt
_N
J>>—C=C-i-COOEt
C1<T ' ■"
ЧУ [779]
Схема 3.176
Таблица 3.21. Присоединение дихлоркарбена к ароматическим и гетероциклическим системам
в условиях межфазного катализа
СО
Реакция
Примечания
Литература
Низкий выход 708
Трудности привое- 1900
произведении ~
Выход 79%"
636, 637,
638, 733
I
Главным образом 730
моиопродукт
Низкий выход 719
(+изомеры )
R - Н, Cl, OtjH,
Алкилированиые ароматические соединения—>-спиро[2,6]нонатриены
О Me
ОМе
ОМе
Другие примеры
Низкий выход 719
718, 719
720
Низкий выход 723
I
723, 734
S2
1ся
Продолжение табл. 3.21
со
■Ре
Реакция
Примечания
Литература
ОМс
CI
С1
4 J +
2%
2,3-Диметоксипронзводное—>-семь продуктов с очень низким выходом
721
724, 1622
HN
C12CH
R.
Cl
О
I
CHCI2
// \
и
.Cl
,CHC12
с
70 и
19%
37 и '44%
(ниже для менее
замещенных)
39%'
1346
1522
728
731
728
I
H
-сн
N
N
-N
Of»
CHC!2
C!
La
X = 0,S '
Cl
Л-
MeO О ОМс МеО О °Ме
3 н 63%
1 и 33%
13-25%
15%
731, 987
728
987
1522
718, 731
987
1
Продолжение табл. 3.21
Реакция
Примечания
Литература
rooc—:
61—78%
30—70%
25%
713, 714
«в
Й)
645, 1136
728, 729,
987
I
731, 979,
987, 1347
718
о 6
Берберин, оксиберберин—►аддукты
о
3%
987
Низкий выход
82%
50—55%
72%
83%
730
1137,
1481
1483
987
987
■320 Глава 3. Практическое использование МФК
Реакции генерированного в условиях МФК дихлоркарбена
с ароматическими и гетероциклическими соединениями
приведены в табл. 3.21.
Обычно циклоприсоединение СС12 по двойной связи,
входящей в ароматическую систему, не идет; такие реакции
возможны только при условии, что двойная связь сохраняет некоторую
долю олефинового характера (например, в фенантрене) или
имеет высокую электронную плотность и частично
локализована (например, в 2-метоксинафталине). В большинстве случаев
выходы низкие или даже очень низкие. Необычные
превращения алкилированных ароматических углеводородов приводят к
образованию смесей спирононатриенов [718—720], как
показано на примере 2-метилнафталина:
.СНз
С1
Схема 3.177
Исследовались реакции многих других соединений, при этом
часто наблюдали образование сложных смесей и низкие
выходы продуктов (литературу см. в табл. 3.21).
Побочные реакции с хлороформом. Как указывалось выше,
система NaOH/СНСЛз/катализатор остается пригодной для
проведения реакции в течение длительного времени. Тем не менее
в отсутствие хороших акцепторов карбена идут медленные
побочные реакции СС12 с хлороформом. Образуются небольшие
количества СН2С12, CC'U, C2C14, С2НС15 и полимеров.
Предложены [ 1143] механизмы этих реакций.
3.20.2. Другие реакции дихлоркарбена
и трихлорметильногр аниона
|
Кроме присоединения по двойным С=С-связям равновесная
система ССЬ/ССЦ" обладает другими богатыми
синтетическими возможностями, которые будут рассмотрены в этом разделе.
3.20. а-Эламинирование 321
Реакции внедрения
Реакции внедрения генерированных обычным способом ди-
хлоркарбенов по С—Н-связи довольно редки и идут с низкими
выходами, даже если используют хлороформ и трет-бутилат
или метилат натрия и трихлоруксусный эфир. Несколько
лучшие результаты получаются при использовании раствора три-
хлорацетата натрия в глиме. Еще более высокие выходы дает
применение бромдихлорметилфенилртути при несколько
повышенных температурах. Поэтому предполагалось, что для
Таблица 3.22. Внедрение дихлоркарбена, генерированного
в условиях МФК, по связи С—Н
Соединение
Положение
внедрения
Выход, %
Литература
Кумол
Этнлбензол
Тетралин
Изопентан
Метшщиклогексан
quc-Декалнн
транс-Декалин
Адамаятан
Диамантан
трис-Гомобаррелан
трис- Гомобульвален
Метиловый эфир бензгидрола
п-Хлорбензилметнловый эфнр
Дибензиловый эфир
Диэтнловый эфнр
Метоксицнклогексан
Тетрагндрофуран
Диизопропиловый эфнр
1 -Метил-1,2-дигидрохинолин
2-Алкил-1,3-Дноксоланы
2-Алквл-1,3-дноксавы
2,5-Дналкилтетрагидрофураны
трет-Н
СН»
1
трет-Н
трет-Н
9
9
1
U
1
1
трет-Н
СНЙ
СНа
СН»
трет-Н
2
трет-Н
2- н 4-
СН
СН
СН
24
2
21
12
5
29
<з
54
63
37
64
59
17
18
26
8
5
18
43
30
30—90
5—18
87
739, 741, 742
741 _
741
742
739, 742
739, 742
739
740
747
744, 745
745
742
742
742
742
739
741, 742
739, 742, 743
746
980, 1133—
1135, 1353
980, 1133,
1134
1351
322 Глава 3. Практическое использование МФК
внедрения карбен нуждается в избытке термической энергии.
Опыты, проведенные с СС12, генерированным в условиях МФК,
при 0°С опровергли это предположение [739]. Однако даже
при использовании этого метода препаративные выходы
достигаются только тогда, когда внедрение проходит в голову
мостика или в .«-положение к активированной группе. Чем менее
стерически затруднена голова мостика, тем выше выход.
В случае изомерных декалинов обнаруживается поразительное
различие: реагирует только цыс-изомер с относительно
«свободным» третичным атомом водорода. Методики проведения
реакции аналогичны методикам присоединения СС12. В табл. 3.22
указаны известные выходы.
Образование нитрилов дегидратацией
при помощи МФК-дихлоркарбена
Сараи и сотр. [748] и независимо от них Хёфли [749]
обнаружили, что алифатические и ароматические амиды, тиоамиды
и альдоксимы и некоторые мочевины (R2N—CONH2 —>•
—ь R2N—CN) при взаимодействии с реагентом Макоши могут
быть превращены в нитрилы. Субстрат растворяют или
диспергируют в хлороформе, добавляют ТЭБА и теплый
40%-ный раствор NaOH и кипятят смесь 30 мин. Для липо-
фильных соединений выходы приближаются к 95%, низшие
алифатические амиды дают выходы 10—20%. В последнем
случае, как и для некоторых диамидов, преобладает гидролиз.
Один из возможных механизмов приведен на схеме 3.178:
* NaOH,CHCla
R—CsN + Na®O®—CHO + 2NaCl
Схема 3.178
Поскольку реакции дегидратации этого типа обычно
проводят в кислой среде, может иногда оказаться полезным и
процесс с участием основания. Грэфе [750] вводил в реакцию ами-
дииы (выходы 61—92%):
Схема 8.179
320. а-Элиминирование 323
Аналогично из N-алкилмочевин образуются карбодиимиды
с очень низким выходом [1700]:
RNH—CO—NH2 v RNH—C=N
RNH—CO—NHR' v R—N=C=N—R'
При помощи СС12, генерированного в условиях МФК, та-оксике-
токсимы могут быть расщеплены на нитрилы и кетоны [1139]:
RSC(OH)—C(R') = NOH v RaCO+R'CN
Реакции с аминами
В давно и хорошо известной карбиламинной реакции
НСС13
R-N=C:
обычно получают умеренные, но плохо воспроизводимые
выходы продуктов. Эти недостатки преодолеваются ее
МФК-модификацией. При использовании ТЭБА как катализатора выходы
повышаются до 20—76% [751—754]. Известна методика этой
реакции, опубликованная в «Органических реакциях» [754], в
которой раствор 1,93 моля амина, 0,98 моля хлороформа и 2 г
ТЭБА в метиленхлориде прибавляют по каплям к
концентрированному раствору гидроксида натрия при 45 °С.
SOCI2 CHC13, тв. КОН
R—NH2 *- R—N=SO *■ R—N=C:
краун-эфир
Считается, что альтернативный синтез изонитрилов дает
лучшие результаты: по этой методике амины сначала
превращают в N-сульфиниламины взаимодействием с тионилхлоридом,
а последние в свою очередь вводят во взаимодействие с
хлороформом, твердым гидроксидом калия и дибензо-18-крауном-6
(или еще лучше с дициклогексано-18-крауном-6) в циклогекса-
не или бензоле [755]. В этом случае МФК-процесс с использо^
ванием системы ТЭБА/водный гидроксид натрия не обладает
преимуществами и даже приводит к образованию менее чистых
продуктов.
Образование диазометана из гидразина, хлороформа и
гидроксида натрия было впервые обнаружено Штаудингером в
1912 г.:
НСС13/КОН
N2H4 »- CH2N2
катализатор
В МФК-модификации эта реакция дает выход 48% при
использовании системы гидроксид калия/18-краун-6 или прибли-
324 Глава 3. Практическое использование МФК
зительно 35%, если применяют систему гидроксид
натрия/четвертичный аммониевый катализатор [756]. Полученные
растворы диазометана в эфире или метиленхлориде содержат
некоторое количество гидразина, захваченного с диазометаном при
азеотропной перегонке. Из растворов в метиленхлориде
гидразин можно удалить экстракцией водой [756]. В настоящее
время, когда N-нитрозосоединения непопулярны вследствие
канцерогенной опасности, этот метод может найти применение.
СНС13/ТЭБА
R—CO—NH—NH2 — »- R—CO—NH—NH—ОС—R + CH2N2
Ацилгидразиды (И=арил, бензил) при использовании СС12,
полученного в условиях МФК, превращаются в диацилгндрази-
ны (выход 40—50%) и диазометан (27%) [757]. Механизм
этой реакции все еще неясен.
При взаимодействии с СС12, генерированным в условиях
МФК, вторичные амины превращаются в формамиды [759, 760,
.1216] (схема 3.180). Выход — в интервале 39-^-91%. Иногда в
качестве сорастворителя используют метиленхлорид. Атака по
двойной связи идет гораздо медленнее, чем атака на неподе-
ленную электронную пару атома азота. Так, из диаллиламина
образуется NyN-диаллилформамид:
Схема 3.180
В то же время 2,2,6,6-тетраметил-4-пиперидон подвергается
атаке по СО-группе, а не по экранированному звену NH, и в
результате перегруппировок образуются соединения,
показанные на схеме 3.181 [1305, 1311]. Недавно были обнаружены
другие сложные реакции экранированных аминов с СС12 и ке-
тонами (схема 3.181), которые позволяют получать с
большими выходами высокозамещенные амиды. Считают, что в данном
случае промежуточными продуктами являются 2,2-дихлорокси-
раны.
В реакции дихлоркарбена с третичными аминами на первой
стадий должен образовываться илид аммония (А). Об этом
говорилось в предыдущем разделе в связи со способностью
третичных аминов катализировать образование дихлоркарбена в
3.20. а-Элиминирование 325
H
[1305, 1311]
R1RVNHi i
+ R4
Н
Ri_NH—R2 + R3—CO—R4 + R5—NH—R6
P1 О R3 R5
\ I I /
N—С—С—N [1520)
R2 R4 R6
3.181
условиях МФК [617, 433] и предположительном образовании
оптически активных аддуктов карбена {623]:
Показано [617], что при взаимодействии третичного амина,
олефина и хлороформа (2 ч, 50°С) в отсутствие гидроксида
натрия не образуется аддуктов дихлоркарбена. Далее, не
наблюдалось также какой-либо реакции при продолжительном
кипячении три-к-пропиламина и хлороформа [447], Кимура и
сотр. [623] считают, что А переносит дихлоркарбен к олефину.
Однако Макоша [433] показал, что образование А не является
обратимым: при взаимодействии эквимолярных количеств
третичного амина и хлороформа со стиролом и NaOH не
образуется аддукт дихлоркарбена со стиролом. Макоша считает, что
при нормальных условиях (при избытке хлороформа) А де-
протонирует хлороформ с образованием ионной пары В,
которая затем может переносить СС1г с образованием С. Затем С
разлагается на хлороформ и амин, и цикл может начаться
снова [433].
RSN
-RsN+—CHC12C1~ + CC12
С
RSN+—CHC12CC1S- *
В
»- продукты присоединения
Однако недавно было показано, что R3N, хлороформ и гидрок-
326 Глава 3. Практическое использование МФК
сид натрия образуют темноватую сиропообразную массу,
содержащую хлорид четвертичного аммония и растворимую в
воде. Эта масса способна действовать как МФ-катализатор в
новой реакции присоединения СС12. При перемешивании раствора
этого продукта в метиленхлориде со смесью циклогексен/гидр-
оксид натрия дихлорноркаран не образуется '[447]. Поэтому
представляется возможным, что данный продукт содержит С
и что катализ третичными аминами в реакциях дихлоркарбена
должен включать образование С (истинного МФ-катализато-
ра). Это объяснение несколько отличается от первоначального
предположения Макоши, поскольку С не разлагается вновь, а
действует скорее как обычный катализатор. Реакция D с МФК-
системой включает атаку двух молекул СС12 и распад кольца с
образованием Е [716].
I'll
D
1 cci3
^/ ВОвн. NaOH
Схежа
Г
3.182
—7N—CHO
^N—CHO
*h
Б
Реакции с другими соединениями
Атака на соединение F идет скорее по атому серы, чем по
двойной связи. Эндрюс и Эванс [715] наблюдали [2,3]-сигма-
тропную перегруппировку
SPh
SPh
CCIa
смесь
стереоизомерое
Схема 3.183
Прочие аллилсульфиды в результате внедрения СС12 по
связи S—С образуют 1-хлор-1-меркаптобутадиены '[1138], а сульф-
оксиды могут быть восстановлены до сульфидов при действии
СС12, генерированным в МФК-системе, с выходом 80—96%
[1142, 1649]:
RaSO + CClg »- R2S + СОС18
3.20. а-Элиминирование 327
Эта реакция особенно эффективна для объемистых сульфок-
сидов, где, по всей видимости, идет ускорение из-за стериче-
ских факторов. Менее затрудненные соединения (например,
грамс-тиадекалин) претерпевают обратную реакцию, т. е.
формальное окисление с выходом 5—30%. Очевидно, интермедиат
R2S=CC12 гидролизуется, и окисление и восстановление идут
одновременно '[1646].
Реакция Реймера — Тимана
Реакция обычно проходит без катализатора. Для высоколи-
пофильных субстратов оказалось, однако, полезным проводить
взаимодействие в присутствии катализатора — соли цетилтри-
метиламмония [761]. Действительно, «аномальная» реакция
Реймера — Тимана в условиях МФК идет при комнатной
температуре с 10%-ным раствором NaOH, в то время как в
отсутствие катализатора требуются более высокие температуры и
более концентрированное основание [841].
он о
СНэ
Н3С СНС1а
78%
н
НзС
НзС
СНз
Схема 3.184
Дальнейшие эксперименты показали, что алкильные группы
от катионов четвертичного аммония или третичных аминов
могут переноситься с образованием эфиров фенолов [1338, 1371].
Третичные амины — более эффективные катализаторы в
реакции Реймера — Тимана, чем R4NX.
В то время как в присутствии R»N выход салицилового
альдегида сильно возрастает, выход л-оксибензальдегида
практически не изменяется. Может протекать следующая реакция
[1338]:
RSN > RSN—CHCl£Cl- »- RSN—CHC1JO-—Ph »-
o-Cl2CH—CeH4—ОН »- o-OCH—CeH4—ОН
328 Глава 3. Практическое использование МФК
Реакции со спиртами
Данная реакция между низшими спиртами, избытком
хлороформа и 50%-ным водным раствором гидроксида натрия в
присутствии ТЭБА приводит к сложным смесям продуктов.
Препаративный интерес представляют только реакции с
этанолом и 2,2,2-трифторэтанолом, которые приводят к образованию
ортоформиатов с выходами 36 и 33% [762]. В случае этанола
побочными продуктами являлись этилхлорид, карбонат и окса-
лат.
. д«- ?
R—О—С—Н
\
RCI
Схема 3.185
По аналогии со вторичными аминами можно ожидать, что в
реакции с высшими спиртами основными продуктами будут
формиаты. Однако они образуются лишь в небольшой степени.
Табуши и сотр. [763] обнаружили, что вместо этого с высоким
выходом образуются алкилхлориды. За исключением тех
случаев, когда реакция является экзотермичной, перемешивание
продолжают в течение 5 ч при комнатной температуре. Этот
метод применяли [3, 644] для реакций ряда стероидов и
терпенов, и как доказательство SNi-механизма наблюдалось
сохранение конфигурации. Однако в некоторых случаях наблюдались
перегруппировки и инверсии конфигурации [763], и детали
механизма требуют дальнейшего исследования. Тем не менее этот
метод представляет препаративный интерес, поскольку он
позволяет проводить превращение спирт — алкилхлорид в
основных условиях при комнатной температуре. Соединения с
двойными связями обычно подвергаются циклопропанированию
с сохранением гидроксильной группы. Фенолы, включая
стероиды, превращаются в соответствующие хлориды [3]. Эта
реакция может быть распространена и на получение алкилбромидов
[764].
^^^ ,он
CHCb. N«OH^
катализатор
R'
Схема 3.186
3.20. а-Элиминироваяие 329
Сложные реакции имеют место, если гликоли реагируют с
двухфазной дихлоркарбеновой системой. Среди получаемых
продуктов есть кетоны, эпоксиды, олефины и дихлорциклопро-
паны; предложены [765] механизмы, объясняющие их
образование. Дихлоркарбеновая система способна деоксигенировать
эпоксиды. Образовавшиеся олефины превращаются главным
образом в дихлорциклопропаны, но общие выходы не очень
велики [766]. Обе реакции — деоксигенирования и присоединения
ССЬ — стереоспецифичны.
R R' R R' „ „, R R'
HCCI,, NaOH
катализатор
R R' R R' *■ * R R'
>=<
Реакции трихлорметильных анионов
В разд. 3.20.1 упоминалось, что в двухфазной системе
NaOH/СНОз/катализатор образуются и СС12, и СС1з~ и что в
зависимости от субстрата в реакцию вступает та или другая
частица. К очень электронодефицитным олефинам, таким, как
винилацетат, акрилонитрил или акриловые эфиры, идет только
присоединение хлороформа по двойной связи; но иногда
наличие «-заместителя может оказаться достаточным для сдвига
реакции в сторону образования циклопропана. В метакрялонит-
риле обе возможности реализуются одновременно (схема 3.188).
Присоединение к винил ацетату (схема 3.188) идет через
образование небольшого количества свободного ацетальдегида
[1167]:
СН8=СН—ОАс >■ СН8=СН—О" >■ СНа—СНО »-
>- СН,—СН(СС1„)—О-
СН,—CH(CC1S)—О- + СН2=СН—ОАс »- CHS—СН(СС1„)ОАс + СН,=СН—О"
Устаиовлено, что в этих условиях а,р-непредельные эфиры
типа G превращаются в спиропентаиы Н, если по крайней ме-
330 Глава 3. Практическое использование МФК
Н2С=СНОАс >
Н2С=СН—COOR -
Н2С=С—COOR
СНз
Н2С=С—CN
СНз
НзС—СН—ОАс
СС.з ^ ^^
[420,671, " f420*686!
677, 768]
»- С1зС—СН2—СН2—CQOR
[671]
COOR
" \7сн3
С12
[616,671-673, 677]
CN
-> \ А + С13С—СН2—СН—CN
V СНэ сн
С12 СНз
[671]
Схема 3.138
ре один из заместителей R1 и R2 является алкилом или
фенилом:
ci2 ci2
R" H CHCb,NaQH у Г67. ggQ,
R2 COOR
G Н
Схема 3.189
Тетрахлор- и тетрабромспиросоедннения получаются из
некоторых непредельных стероидных кетонов [644], однако более
простые непредельные карбонильные соединения дают в
основном продукты осмоления. Механизм образования Н
первоначально включает нормальное присоединение дихлоркарбена
[680]. Затем в сильноосновной среде полученные аддукты (I)
дегидрохлорируются с образованием К. Известно, что эфиры
циклопропенкарбоновых кислот, подобные К, мгновенно
присоединяют любой доступный нуклеофил. Так, присоединение
гидроксил-иона ведет к образованию побочного продукта (L) —
полуэфира замещенной янтарной кислоты. Альтернативное
присоединение трихлорметильного аниона приводит к образова-
3.20. а-Элимшшрование 331
нию М, из которого в результате последовательных реакций
элиминирования — присоединения образуется N. Последний в
свою очередь подвергается атаке другого аниона СС13~,
который отрывает положительный атом хлора с образованием
побочного продукта — тетрахлорида углерода и спиропентана Н.
:oor
OR
\ соон
-CHjCOOR
CI СС13
н
COOR
м
ссь ссь
А»
COOR
N
Схема S.190
COOR
н
Способность МФК-системы дегалогенировать была
обнаружена также в следующей последовательности реакций [680]:
С1зС^-СС12-СС13
С12С=СС1—СС13 -
(главный продукт)
[С12С=С=СС1а]
С1Я
С1а
(побочный
продукт)
Схема 3.191
Реакция между алкилтиоцианатами и хлороформом в
присутствии системы NaOH/ТЭБА умеренно экзотермична и (в
результате замещения цианид-иона) приводит к образованию
332 Глава 3. Практическое использование МФК
трихлорметилсульфидов с прекрасным выходом [300, .1420]:
NaOH
RS—CN + CHCL —-*- RS—CCl.+ NaCN
ТЭБА
Аналогичным образом ведут себя селеноцианаты [1486].
В качестве альтернативных источников дигалокарбена для
присоединения к затрудненным субстратам в нейтральных
условиях используют соединения тригалогенметилфенилртути
(реагенты Зейферта).
PhHgCl >■ PhHgCXj,
Синтез этих реагентов обычно довольно сложен. Однако Ма-
кошей был найден легкий способ их получения [767]. Раствор
20 г гидроксида натрия и 37 г фторида калия в 100 г воды
медленно прибавляют по каплям к суспензии 0,05 моля
хлорида фенилртути и 0,4 г ТЭБА в 70 мл хлороформа и
перемешивают 2 ч при комнатной температуре. Продукт получают с
выходом 72%. Аналогичным образом могут быть получены
другие соединения тригалогенметилфенилртути (X3=BrCl2, Вгз).
По всей вероятности, фторид действует как дополнительное
сильное основание.
Под действием хлороформа и твердого Ва(ОН)2-8НгО
четвертичные никотинамиды О превращаются в трихлорметильные
производные; некоторые из< них -могут в свою очередь
перегруппировываться в циклопропаны [725]. Изучали [726, 727]
также дальнейшие превращения этих систем в условиях МФК:
rONH2
Схема 3.192
3.20. а-Элиминировшше 333
МФЮреакция дихлоркарбена с дегидролюциферином (Р)
дает показанный на схеме альдегид вместо ожидаемого
циклопропана [7t8]:
МеО
N—Me
Схема 3.193
Большой препаративный интерес представляют реакции
карбонильных соединений с .системой СС18~/СС12. Альдегиды и
реакционноспособные алифатические кетоны при быстром
прибавлении 50%-ного раствора NaOH при 0°С с выходами до
80% превращаются в та-трихлорметилкарбинолы (Q) в системе
хлороформ/1—5 мол. % ТЭБА [235]. В присутствии диметил-
сульфата образуются соответствующие эфиры.
При повышении температуры выход продуктов Q быстро
снижается вследствие ретрораспада, проходящего в водной
фазе.
\
R'
С=О
NaOH. CHCI3
ТЭБА. 0°С
он
(CH3)aSO4
\о3
cci3
Q
Схема 3.194
Конкурирующая реакция с ароматическими альдегидами — это
реакция Канннццаро. Однако в узком температурном интервале
56±2°С ароматические альдегиды с выходом 75-^83%.
образуют миндальные кислоты [235, 770, 772, 1564, 1648]. Иногда
получают 2-арилакриловые кислоты [1554]. Из ацетона и али-
циклических кетонов образуются смеси а-гидрокси- (R),
а-хлор- (S) и а,р-непредельных кислот (Т) (схема 3.195) [235,
771].
Здесь, по всей видимости, температура не играет очень
большой роли [771]. При температуре 20°С практически
единственными продуктами являются хлоркислрты S, при 56 °С
преобладают соединения R (выход 13—55%). Далее соединение
Q [R—И'^СНгЫ может быть превращено в S [R—R'=
= (СН2)4] при 20 С {771] j но при более высокой температуре
334 Глава 3. Практическое использование МФК
R
Схема 3.195
превращение 2,2,2-трихлор-1-фенилэтанола в миндальную
кислоту невозможно [235, 770]. Обсуждался [1267] механизм,
предполагающий образование в качестве интермедиата дихлор-
оксирана (схема 3.196), и в некоторых случаях были выделены
промежуточные а-хлорацилхлориды [1267].
Схема 3.196
В системе, содержащей водную фазу КОН/ЫС1/ЫНз,
органическую фазу СНС1з/СН2С12 и катализатор ТЭБА,
ароматические альдегиды могут быть превращены с выходом 29—81% в
а-аминоарилуксусные кислоты [1141].
3.20.3. Дибромкарбен
Аддукты дибромкарбена часто предпочитают хлорным
аналогам, поскольку их гораздо легче модифицировать в
последующих реакциях. МФК-химия дибромкарбена в основном
аналогична химии СС12. Рассмотрев данные по селективности
реакций в системе бромоформ/трет-бутилат калия, Мосс и сотр.
[780] пришли к выводу, что в данной системе «кинетически
эффективное участие карбеноида минимально». Однако дибром-
320. а-Элиминирование 335
карбен \должен быть более редкционноспособным, чем дихлор-
карбен. уго предположение подтверждается опытами по
конкурирующем реакциям: равные количества хлороформа и бро-
моформа, стирола, ТЭБА и концентрированного раствора NaOH
при реакции в качестве основного продукта дают 1,1-дибром-
2-фенилциклопропан [384]. Более раннее сообщение [781], в
котором в качестве основного продукта описывалось дихлор-
производное, было впоследствии опровергнуто (см. примечание
16 в работе [384]). Поскольку СВг2 более реакционноспособен,
чем СС12, можно предположить, что он более чувствителен к
гидролизу. Действительно, Хайн [782] нашел, что
относительная скорость гидролиза СНВг3 в гомогенной водной среде в
6 раз выше скорости гидролиза СНС1з. В условиях МФК
скорости гидролиза в отсутствие олефина различаются и при
комнатной температуре [1140]. В присутствии акцепторов карбена,
отличных от воды, наблюдается заметное различие в
температурной чувствительности реакций присоединения СС12 и СВг2.
Скорости реакций и выходы продуктов присоединения ди-
хлоркарбена в большинстве случаев возрастают при
повышении температуры от комнатной до 40—60 °С. В аналогичных
реакциях дибромкарбена повышение температуры увеличивает
скорость гидролиза сильнее, чем скорость присоединения, что
зависит от нуклеофильности олефина. При проведении опытов
с различными олефинами было найдено [783], что при
повышении температуры выходы продуктов иногда возрастают,
иногда снижаются, а иногда остаются постоянными. Отсюда
вытекает, что подбор оптимальных условий присоединения
СВг2 гораздо сложнее, чем в случае СС12, и единого набора
оптимальных условий не существует.
Из ранних публикаций по реакциям дибромкарбена в
условиях МФК известно, что выход аддуктов СВг2 всегда ниже,
чем в реакциях присоединения СС12. В литературе имеется ряд
сообщений по оптимизации условий реакции, но, поскольку
изменялись многие параметры, часто трудно сравнивать
результаты. Скаттеболом и сотр. [784] установлено, что лучше всего
проводить реакцию при комнатной температуре в течение
длительного времени (до 96 ч). Необходимо эффективное
перемешивание, и в случае менее реакционноспособных олефинов
требуется избыток бромоформа вплоть до четырехкратного.
Стирол оказался более реакционноспособным, чем 2-метилбутен-2,
так как реакция с ним проходила при меньшем избытке
бромоформа и за более короткое время. Макоша и Федорыньски
[785] наблюдали, что добавление небольших количеств
этанола (0,8 мл при загрузке реагентов порядка 0,2 моля) к
реакционной массе повышает выход на 10—30%. Они рекомендовали
проводить реакцию со 100%-ным избытком СНВгз в течение
336 Глава 3. Практическое использование МФК
3 ч при 40—45 °С. Влияние этанола не совсем понятно,
предполагается, что может иметь место специфическая сольватация
ионной пары R^N+CBr3~, которая стабилизирует ее илюзволяет
проникнуть дальше в глубь органического слоя, зайиедляя
таким образом гидролиз. Другое возможное объяснение состоит
в том, что этилат-ионы экстрагируются в органическую фазу
гораздо легче, чем гидроксид-ионы, поэтому кинетика реакции
до некоторой степени изменяется. Были иайдёны [786] еще
лучшие условия реакции для малореакционноспособных оле-
финов. Они аналогичны вышеприведенным, но в качестве
катализатора применяют три-н-бутиламин и как сорастворитель
метиленхлорид (в объеме, равном объему бромоформа).
Большинство авторов применяют не менее 2 молей СНВг3 на 1 моль
олефина. Однако для некоторых а-замещенных а^р-непредель-
ных кетонов хорошие выходы достигались при использовании
лишь 1,25 моля галоформа на 1 моль субстрата [674].
Изучено также влияние многих других факторов [783, 787,
1140], из которых следует упомянуть два наиболее важных.
1. Бромоформ — высококипящий растворитель, и часто
бывает трудно очистить продукты реакции с близкими
температурами кипения от его следов; поэтому некоторые приведенные
в литературе выходы могут оказаться неточными.
2. Крайне необходимым является энергичное
перемешивание. При использовании магнитной мешалки скорость
перемешивания должна быть не ниже 600—800 об/мин.
Ниже приведены данные о влиянии некоторых из
исследованных факторов.
Количество гидроксида натрия. Наилучшие результаты
достигаются при использовании 5—10 экв. концентрированного
раствора гидроксида натрия на 1 моль СНВг3.
Количество системы СНВг3/сорастворитель. В большинстве
случаев достаточно применения 2 молей СНВг3 на 1 моль
двойной связи. Для облегчения перемешивания можно добавить
равный объем метиленхлорида, хотя его влияние на выход не
очень велико.
Температура и время реакции. Рекомендуется проводить
реакцию при комнатной температуре. Обычное время реакции
для стирола и циклогексена составляет 4—6 ч. Для более сте-
рически затрудненных олефинов (3,3-диметилбутен) или элект-
ронодефицитных дезактивированных систем (полнприсоедине-
ние к циклооктатетраену) реакцию ведут гораздо дольше (от 2
до 6 сут). Хотя в этих случаях выход день ото дня возрастает,
для экономии времени лучше через 1—2 сут разделить
реакционную смесь и сырой продукт снова ввести в реакцию.
Количество катализатора. Обычно достаточно 1 мол. %
катализатора по отношению к олефину.
N
3.20. а-Эламинирование 337
Тип катализатора и сокатализатора. Хорошие результаты
достигаются при использовании многих катализаторов,
включая симметричные тетраалкиламмонийгалогениды, аликват 336,
ТЭБА, три-н-бутиламин и три-н-пропиламин, а также краун-
эфиры. Добавление этанола при использовании солей
четвертичного аммония оказывает ярко выраженный
благоприятный эффект.
Ниже приведены типичные методики реакций присоединения
СВг2.
Метод Макоши и Федорыньского [786]
Смесь 0,1 моля алкена, 0,2 моля бромоформа, 25 мл мети-
ленхлорида, 0,4 мл три-н-бутиламина и 0,5 моля (40 мл);
50%-ного водного раствора гидроксида натрия энергично
перемешивают 4 ч при температуре 40—45 °С. Реакционную массу
разбавляют водой и отделяют метиленхлорид. Водный слой
дважды экстрагируют метиленхлоридом, объединенные
органические слои промывают разбавленной соляной кислотой, во-;
дой, сушат и выделяют продукт.
Метод Демлова [783, 787]
Смесь 0,1 моля алкена, 0,2 моля бромоформа, 20 мл мети-
ленхлорида, 1 мл этанола, 250 мг ТЭБА и 1—2 моля 50%-ного
водного раствора гидроксида натрия энергично перемешивают
при комнатной температуре от 4—6 ч до 2—6 сут, затем
реакционную массу обрабатывают, как указано выше.
В первой части табл. 3.23 указаны реакции в частично
оптимизированных условиях, а затем приведены другие реакции
дибромкарбена.
Из результатов, представленных в табл. 3.23, можно
сделать ряд выводов. Наличие в олефине свободной гидроксиль*
ной группы ведет, вероятно, к образованию только одного
изомера с транс-положением дибромциклопропанового кольца по
отношению к ОН-группе :[502, 788—790]. Гидроксильная
группа в аллильном положении дезактивирует олефин по
сравнению с незамещенными углеводородами [502]. В то же времй
реакцией с СВг2 насыщенные спирты могут быть переведены
в бромиды даже в тех случаях, когда обычные реагенты
(SOBr2 и Ph3PBr2) не действуют [764]. То же самое
наблюдалось и для СС12.
Внедрение СВг2 по СН-связям широко не изучалось;
выходы часто ниже, чем при действии СС12 [742]. В реакции с
2,5-дигидрофураном соотношение продуктов присоединения и
внедрения при 0 °С составляет 65 :35 и при комнатной
температуре— 40:60 [791, 987]. В реакции 2,3-дигидро-1,4-диокса-
Таблица 3.23. Присоединение днбромкарбена в условиях межфазногб катализа
Олефин
а) Оптимизированные условия реакций
С« 1-Хлор-2-метилпропен-1
С8 2-Метнлбутен-2
1-Хлор-3-метилбутен-2
С6 Гексен-1
Гексен-2
Циклогексен
н-Бутнлвиниловый вфир
3,3-Диметилбутен-1
Cj Гептен-1
Тетраметялаллен
Этил-З-метилбутен-2-оат
Температура,
45
Комн.
45
45
45
45
Комн.
Комн.
Комн.
45
45
45
40
45
Комн.
45
Коми.
45
Молярное
соотношение
CHBrg/ал-
кен
2
4
2
2
2
2
3,5
2
2
2
2
4
2,5
2
4
2
6?
2
Катализатор/условия
Bu3N/CH2Cls
ТЭБА
ТЭБА/следы СгН6ОН
ТЭБА/следы CjHsOH
Bu3N/CH2Cl2
BU3/CH2CI2
ТЭБА
ТЭБА/следы С2НВОН
ТЭБА/следы С2НВОН
ТЭБА/следы С2НвОН
Bu3N/CH2Cl2
Ме4Р1/СН2С12
Дибензо-18-краун-в
ТЭБА/следы С2НВОН
ТЭБА/следы С2Н6ОН,
10 молей NaOH
Bu3N/CHaCl2
ТЭБА
ТЭБА/следы С2НВОН
Время,
ч
4
72
3
3
4
4
96
20
4
3
4
2
3
24
4
72?
3
Выход,
%
65
73
81
89
61
67
72
86
73,5
92
76
-100
74
*50
30
67
10
89
Литература
786
784
785
785
786, 797
786
784
787
787
797
786
616
618
785
792, 797
786
784
785
Се Стирол
-i
Октен-1
Октен-2
Циклооктен
Октадиен-1,5 (биспродукт)
я-Бутилметакрнлат
С9 а-Метилстирол
Изопропенилбензол
СюДецен-1
о-Днвннилбензол (бнспродукт)
Си Додецен-1
б) Неоптимизнрованные условия реакций
Ph2C=CH2
Стильбен
1-Фенилпропен-1
Цнклооктатетраен
Коми.
45
45
45
45
45
Коми.
45
45
45
43
45
Комн.
45
Коми.
Коми.
40
Кони.
Комн.
Комн.
2
2
2
2
S
2
3
4
2
2
2
2
6?
2
7
7
2,5
2,5
2,5
4
ТЭБА
ТЭБА/следы С2Н5ОН
Bu3N/CH2Cl2
ТЭБА/следы С2Н8ОН+
+СН»С1г+ 10-кратное
молярное количество NaOH
Bu3N/CH2Cl2
Bu3N/CH2Cl2
ТЭБА
BU3N/CH2C12
ТЭБА/следы С2Н6ОН
ТЭБА/следы С2Н5ОН
BUsN/CHjClj
Bu3N/CH2Cl2
ТЭБА
ТЭБА/следы С2Н5ОН
ТЭБА
ТЭБА
Днбензо-18-краун-6
ТЭБА/СН»С12/вибропере-
мешивание
ТЭБА
ТЭБА/следы СгНвОН/СНгС1г
24
3
4
4
4
4
48
4
3
3
4
4
<24
3
15
18
9
4
16
240
66
78
88
95
75
75
70
60
76
80
89
77
50
78
34
32
35
80
23
38 (1
25 (2
1 h
784
785, 797
786
783
786
786
784
786, 626
785
785
786
786
784
798
626
626
618
1409
626
а 626,
800,
730
0,3 (4)
Продолжение табл. 3.23
Олефин
Темпе-
ратура,
Молярное
соотношение
СНВг8/ал-
кен
Катализатор/условия
Время,
Выход,
%
Литература
Вга
(R = Н, Me.Ph)
Комн.
5-10
Комн.
(5исаЭоук.т)
45'
ТЭБА/следы CsHsOH/CHjClj 240 42 (l)a 730
10 (2)
1,2 (3)
ТЭБА/бензол ? 711
ТЭБА/бензол
ТЭБА
Комн. 2 ТЭБА/следы С2Н6ОН
4 ТЭБА/следы С2Н8ОН
54 648
59 794
3,5 22-60 647
799
50 ? Цетилтрнметиламмоннй- 2,5 80—90 704
хлорид
Комн. 11 ТЭБА
Комн. 7—10 ТЭБА
Комн. 10 ТЭБА/следы С2Н8ОН
Комн. 10 ТЭБА
94 94 700
91; 72 48—94 700, 955,
1153
18 64 802
Комн. 10 ТЭБА/следы C2HSOH 18 57 €02
48 55 695, 968
I
Продолжение табл. 3.23
Олефин
Температура,
Молярное
соотношение
СНВга/ап-
кен
Катализатор/условия
Время,
Выход,
%
Литература
,СН
(СНа).
он
он
(бисаЭ9ук.т)
OEt
О
Комн.
Цетилтриметиламмоний-
бромнд
Комн. 5 ТЭБА
40 2 ТЭБА
Коми. 2,5 ТЭБА
0 1 Ci4H«NMe3Cl
45 4 ТЭБА/следы СгН5ОН
12 25-63 789, 790
53 788
75
24 39
722
658
7 17 791
30 40 948
ВТ:
=C"CN J
R4 СГ
R" R4 О'
-О
О
Me' ^f Me
О
R2CH=C—COOR
I
Rl
Комн. 1,25 ТЭБА
Комн. 1,25 ТЭБА
Комн. 1,25 ТЭБА
2-24 20-90 674, 796,
1151
2? 87 796
2 30—60 674, 1152
Коми. 6,9 ТЭБА/следы С2Н5ОН 18 69 793, 1414
45 5,5 ТЭБА/следы С2Н5ОН/СН2С12 4 57 691
ТЭБА, цетрнмид
4 до 90 795, 672,
1419
Продолжение табл. 3.23
Олефин
Темпе-
рат^ра,
Молярное
соотношение
СНВгз/ал-
кеи
Катализатор/условия
Время,
Выход
%
Литература
2— CR3R4OH Комн.
(СН,)2С=СН-<СН2)2-С(ОН)(СН,)-СН=СН2
Вг2
(СНз)
сн3
(CHS)2—С—СН=СН2 Комн.
он
в) Другие примеры
Аллилгалогениды
Аллнльные эфиры и спирты
ТЭБА
ТЭБА
10,7 Bu4NBr
16 34—88 502
16 93
90
502
867
1131,
1154,
1426
1155,
1156,
1403,
1426,
1449
Сопряженные и несопряженные бута- 457,
диены, включая циклические н поли- 1290,
циклические 1313,
1377,
1412
4-Вннилциклогексеи 1159
Бульвален 730
Различные гетероциклы, включая заме- 987,
щеиные гидроароматические соеднне- 1568,
ния 1636
Виниловые эфнры, виниловые тиоэфиры 1392,
1728'
Ar2C=CHs, R2C=CH2, включая поли- 1390,
циклические структуры 1408,
14oU
Высоко замещенные гидроароматиче- 1615
ские соединения
Замещенные енины 1326 в
Замещенные циклопентадиеноны 1788
Цифры в скобках указывают двойную связь, по которой идет присоединение
CBrj, — Прим. перев.
346 Глава 3. Практическое использование МФК
на продукт внедрения, очевидно, образуется в результате
термического распада аддукта дибромкарбена [658]. Продукты
внедрения по активированным СН-связям 2-алкил-1,3-диоксо-
ланов дают соединения А (выход 60—90%) ;[980, 1134, 1353].
Внедрение по СН-связям диенов, входящих в комплексы с три-
карбонилом железа, приводит к соединениям В, которые могут
быть гидролизованы и окислены в несопряженные альдегиды
[1208]:
СНВг2 Fe(CO)3 В
Схема 3.197
В случае очень мало реакционноспособных субстратов
могут идти необычные побочные реакции: трибромметильный
анион атакует бромоформ с образованием тетрабромида углерода1
СВг8~ -}- НСВг8 >- СВг4 -|- НСВга~~ >- ЫаСВга
В присутствии воздуха и на свету СВг4 и СНВгз могут
присоединяться к олефину по радикальному механизму. Так, при
взаимодействии с 3,3-диметилбутеном образуются три
продукта, соотношение которых зависит от условий реакции ![1143]:
Вг2
>рСН=СН2 > А/ \ + >рСН-СНа-СВгэ + >рСН-СН2-СНВгг
Н Вг Вг
Схема 3.198
Радикальные реакции можно исключить, проводя
взаимодействие в темноте в атмосфере азота. Другая необычная реакция,
по всей вероятности, зависит от применяемого катализатора:
когда реакция с аллилбромидами ведется в присутствии
бромида цетилтриметиламмония, то со средним выходом образуются
продукты нормального присоединения CBrg, а при
использовании ТЭБА или бромида тетрабутил аммония реакция идет по
пути замещения брома на трибромметильную группу [1154,
1896, см. 1131]:
r2c=C(R')—енавг —»-
3.20. а-Элимширование 347
Эта реакция недавно была распространена на бензилброми-
ды ,'[1205].
Рассматривая новые неожиданные реакции СВгг, следует
учитывать возможность равновесия триплетного дибромкарбена
с синглетным, поскольку разница энергий между этими двумя
формами, очевидно, очень мала [1158].
В отличие от OF2, CFC1 и СС12 реакция дибромкарбена с
нор борнадиеном дает четыре продукта перегруппировки, ни
один из которых не является продуктом линейной хелетропной
реакции. Очевидно, что стерические требования к 1,4-гомопри-
соединению слишком жестки ![704]:
КО— mpem-Bu/CHBr3
МФК-мет сВ 51%
26%
22%
25%
19,5%
13%
7,5%
Схема 8.199
Множественное присоединение СВгг к соединениям с
несколькими двойными связями возможно, как и в реакциях с
ССЬ. Иногда электронные и стерические факторы затрудняют
эти процессы. Так, циклооктатетраен в прямой реакции дает
очень малые выходы три- и тетрааддуктов [730]. Для
получения заметных количеств этих соединений надо повторно
вводить в реакцию выделенные моно- и диаддукты. После
удаления двух атомов брома диаддукта выход продуктов нового
присоединения СВгг резко увеличивается [792] (схема 3.200).
Электронодефицитные, замещенные электроноакцепторными
группами олефины, как и при использовании системы
хлороформ/основание, присоединяют СВгз~ вместо СВгг. Так, винил-
ацетат, акрилонитрил и эфиры акриловой кислоты образуют
приведенные ниже продукты [384, 768]:
RCH=CH—О—СО—R' *■ R-£Ha—СН—О—СО—R'
СВг8
НаС=СН—X *■ Вг8С—СНа—СН,—X (Xc=CN, COOR)
Соединения с «-метальными группами дают циклопропаны
'[616, 672, 674, 691, 795, 796]. Из метакрилонитрила также об-
н
Схема 3.200
3.20. а-Элимннирование 349
разуется только продукт присоединения дибромкарбена [384]:
С12 Вг,
CJ,C—CHa-CH-CN
' i
Схема 3.201
В этом состоит отличие данного процесса от обычной реакции
с хлороформом, в которой образуются и циклические продукты,
и продукты с открытой цепью. При использовании
соответствующих исходных 'реагентов, могут получаться тетрабромспиро-
пентаны, реакция получения которых обсуждалась ранее
(в связи с реакциями хлороформа) [644].
Совершенно отличный метод проведения МФК-реакций для
получения аддуктов дибромкарбена заключается в разложении
трибромацетата натрия в присутствии соли четвертичного
аммония в неполярном растворителе. Как подробно,было
изложено в разделе, посвященном1 ОСЬ, эйот метод позвййя^т избе*
жать применения основания, обязательного! в методе Макоши.
Из циклогексена образуется дибромиоркаран с выходом 78%
[31].
3.20.4. Прочие дигалокарбены
В МФК-процессе могут быть получены все возможные дига-
логенные сочетания СХ£ Однако для подучения хороших
результатов условия проведения реакций с GClj нельзя
использовать без изменений. Во-первых, многие из галоформов
довольно дороги и не могут применяться в большом избытке, и,
во-вторых, реакционная способность других карбенов
отличается от реакционной способности СС12. Порядок реакционной
способности хорошо установлен (с учетом нед'айних
определений и уточнений принципа реакционная способность —
селективность ' [815, 1160]):
СН„ CBi-jj CCIa CFC1 CFa
увеличение электрофнльности
увеличение селективности
Здесь приводятся некоторые положения из ставшей теперь
классической пионерской работы Хайна по а-элнминированик>
в присутствии оснований (см. обзоры [782, ШЗ]):
1. Скорости образования карбаннонов ^CHXYZ г—**CXYZ~)
350 Глава 3. Практическое использование МФК
изменяются в следующем порядке: I«Br>Cl>'F. Эта стадия
протекает гораздо быстрее, чем последующие. Величины р/Са
равны: СНВгз 9, СНС13 15, CHFa 26,5 :[1161].
2. Относительная легкость отщепления галогенид-аниона от
промежуточного тригалогенкарбаниона (CXYZ~ —>- CXY+Z-)
изменяется следующим образом: Br^sI>Cl»F.
3. Способность атома галогена к стабилизации CXY
изменяется в следующем ряду: F»Cl>Br>I. Следовательно,
наиболее стабильный карбен является и наиболее селективным.
4. Возможен также обмен галогена с другими галогенид-
ионами.
НСХэ
±н«>
СХз
+хе
СХ2
последующие
реакции
+н*
HCX2Y ^=± CX2Y®
Схема 3.202
Бромхлоркарбен, бромиодкарбен и процессы обмена галогена
Выше упоминалась возможность обмена галогена.
Теоретически для смешанных галоформов возможны процессы,
приведенные на схеме 3.203.
HCXsY
CX2Ye
конечные
продукты
' с открытой
цепью
x2- циклопропаны
XY-циклолропаны
HCXY2
Ye + :CXY
Xе + :CY8
Схема 3.203
Если эти реакции идут быстро, то в реакцию обмена может
вступать даже посторонний неорганический галогенид-анион
(например, вносимый с катализатором). Эти предположения
частично подтвердились экспериментами: при использовании
водного раствора гидроксида натрия, насыщенного хлоридом
иатрия, и ТЭБА в реакции присоединения дибромкарбена к
циклогексену продукты реакции содержали 3% бромхлорнорка-
-рана (В), образовавшегося в реакции обмена. Обратный про-
3.20. а-Элиминирование 351
цесс с раствором гидроксида натрия, насыщенным бромидом
натрия, с дихлоркарбеном не идет. Как упоминалось ранее,,
бромид-ион отщепляется гораздо легче, чем хлорид, и из
небольшого количества аниона СВгСЬ", возможно, снова
образуется СС12. Локальная концентрация постороннего галогенид-
иона должна быть очень мала, поскольку концентрация ТЭБА-
невелика. Контрольный опыт показал, что в отсутствие
гидроксида натрия обмен нуклеофильного галогенида с МФ-катали-
затором не идет [384]. Для смешанных галоформов можно-
ожидать более высокие локальные концентрации различных га-
логенидов и, возможно, благоприятные условия для процессов,
обмена. Действительно, несмотря на то что бромид-ион
является лучшей уходящей группой, в реакциях СНВгСЬ и СНВг2С1'
с циклогексеном и ТЭБА образуются все три возможные
комбинации соединений А—С со следующими выходами [384]:
С1
Вг
CHBrClz
CHBr*CI
A
78
22
В
19
69
Схема 3.204
С
3
9
В то же время из дихлорфторметана образуются только-
продукты присоединения хлорфторкарбена. В случае CHBr2Cf
степень обмена и суммарный выход сильно зависят от нуклео-
фильности и стерической доступности двойной связи (табл. 3.24)
[989].
Таблица 3.24. Состав продуктов реакции СНВг2С1 с различными
алкевами в стандартных условиях (катализатор ТЭБА) [989]
Алкен
Выход аддуктов, %
суммарный
СС12
свга
СВг2
3,3-Диметилбутен-1
Гексен-1
Чыс-Бутен-2
Изобутен
Циклогексен
2-Метилбутен-2
8,1
10,5
27,1
54,3
54,6
62,6
4.9
3,1
8,9
17.9
13,1
25,7
2,8
5,7
15,7
33,7
32,2
33,2
0,4
1,7
2,4
2,7
9,2
3,8
352 Глава 3. Практическое использование МФК.
Внедрение
в присутствии
Кумол
Адамантан
по СН-связям кэрбенов, полученных из СНВг2С1
ТЭБА, дало следующие результаты [989]:
Выход продуктов внедрения, %
суммарный | CCUs
* CBrCl
7,0 2,0 1,3
15,6 3,7 7,2
СВг2
1,3
4,7
С препаративной точки зрения получение бромхлорцикло-
пропанов в нормальных условиях МФК является неудобным
процессом, поскольку образующиеся смеси аддуктов часто
трудно разделить. Однако Федорыньски установил, что
результат реакции в системе алкен/СНВг2С1/НаОН в присутствии кра-
ун-эфира как катализатора зависит от типа крауна. При
использовании 18-краун-6 или 15-краун-5 обычно получают смесь
всех трех дигалогенциклопропанов, хотя селективность обычно
выше, чем при использовании в качестве катализаторов
четвертичных солей аммония. В присутствии дибензо-18-крауна-6
образуются только бромхлорциклопропаны [835]: при
смешении эквимольных количеств алкена и СНВг2С1, 2 мол. % ди-
бензо-18-крауна-6 и 50%-ного раствора NaOH и
перемешивании в течение 4 ч при 45 °С образуются продукты с выходом
43—76% |[835]. Однако контрольные опыты показали, что:
а) дибензо-18-краун-6 в отсутствие алкенов катализирует га-
логенообмен между галоформами; б) даже в присутствии ди-
бензо-18-крауна-6 чистые аддукты CBrCl образуются только в
очень узком интервале условий опыта; в) присутствии
алкенов относительные скорости обмена и присоединения зависят
от природы олефина. Иногда образуются чистые аддукты
CBrCl, а в других случаях получаются разнообразные смеси
галогенидов [989, 1012].
Причины специфического действия дибензо-18-крауна-6 все
еще не ясны. Более понятен тот факт, что система трет-бути-
лат калия/CBrCI дает только продукты присоединения CBrCl
(см., например, [842]), поскольку в этих условиях образование
карбена практически необратимо. То же самое верно для
синтеза бромхлорциклопропана из реагента Зейферта — дибром-
хлорметилида фенилртути PhHgCBr2CI [804].
Обменные процессы в условиях МФК идут даже при
конкурентном образовании карбенов из равных молярных
количеств бромоформа и хлороформа; например, реакция со
стиролом в специфических условиях в присутствии системы гидр-
оксид натрия/ТЭБА приводит к образованию 78% аддукта
СВг2, 15% аддукта СС12 и 7% аддукта CBrCl i[384]. В отсут-
3.20. а-Элиминированце. 353
етвие акцептора карбена э.та реакция обмена может быть
использована для получения смешанных галоформов из
хлороформа и бромформа. При соответствующих условиях степень
гидролиза не превышает 16%. Реакцией 2 молей СНС1з и
СНВгз, 3 г ТЭБА и 200 мл 50%-ного .водного раствора NaOH
при 35 °С в течение 1—2 ч (вначале, необходимо охлаждение) и
последующей перегонкой .. на эффективной колонке получают
0,66 моля СНВгСЬ и 1,08 моля СНВг2С1, а также исходные
соединения и остаток [836]. При реакции в системах бромоформ/
/NaOH, насыщенный NaBr/ТЭБА ъ при 150 °С „в ..течение 24 ч
получают СНВггС1 и СНВгС12 с выходом только 10% каждого
[843].
По обычным методикам эти превращения проводят с
использованием в качестве катализатора хлорида алюминия.
Недавно с этой же целью в реакции использовали систему эти-
леноксид/галогенид тетраалкиламмония [844].
Аналогичные процессы обмена галогена могут идти в МФК-
реакциях получения карбенов из СНВг21 или CHBrI2.
Большинство образующихся бромиодциклопропанов и дииодцикло-
пропанов довольно неустойчиво. Если используются алкены с
низкой электронной плотностью (например, метакрилонитрил),
то из СНВг21 и СНВг12 получаются почти равные количества
аддуктов CBr2, CBrI и С12. Однако, когда используются более
реакционноспособные акцепторы (например, 1,1-дифенилэти-
лен), из СНВг21 в качестве основного продукта получают ди-
бромциклопропан, а из СНВг12 — аддукт бромиодкарбена
[1839]. Таким образом, порядок уходящих групп в условиях
МФК следующий: 1>Вг отличается от порядка, найденного
Хайном при гомогенном гидролизе (см. выше).
Фторсодержащие карбены: CF2, CCIF, CBrF, CFI
Попытки генерировать CF2 в условиях МФК в системах
СНСМУводный раствор NaOH/катализатор [808, 845] или
№О2ССР3/катализатор <[31] оказались тщетными. Однако
недавно при использовании систем твердый К2СО3 или твердый
NaOH/катализатор были получены низкие (около 10%)
выходы аддуктов GF2 [988]. В противоположность CF2 карбен
CC1F легко генерируется. Его аддукты являются ценными
промежуточными продуктами для получения фторалкенов, фтор-
диенов и фторалкилспиртов. Шлоссер и сотр. [805—807, 834]
использовали в качестве основания концентрированный
раствор гидроксида калия и ТЭБА (или дициклогексано-18-кра-
ун-6 либо 18-краун-6) как катализаторы. В качестве
растворителя применяли избыток CHCkF и реакции проводили в
течение 0,5—2 ч при температуре 0°С (обратный холодильник
охлаждали до —30 °С), используя внбромешалку. Исследовано
354 Глава 3. Практическое использование МФК
множество различных олефинов (см. [1162, 1163, 1213]). В
реакцию вступают даже соединения с «неудобными»
заместителями (аллильный атом галогена, сложноэфирная группа, аце-
таль, гидроксиальная группа), и выходы колеблются от 30 до
70%. Вейершталь и сотр. [808] рекомендуют перемешивать при
температурах 0—20 °С в течение 2—3 сут. В качестве
катализатора они рекомендуют ТЭБА-бромид. При использовании
этого катализатора в растворе гидроксида натрия из соединения А
образуются (в зависимости от избытка CHC12F) только моно-
аддукты В и С или через 2 сут диаддукт D [666].
Cl F
С1СНа
Cl
АсО—(СН,)3
Cl F
СН—СНа—СН2
•—о
Cl F
Cl F
НОСН,
15%
24%
= 4:1)
ОАс
ОАс
Схема 8.205
3.20. а-Элиминирование 355
В то время как из 2,5-дигидрофурана и дигидропирана
образуются стабильные аддукты CC1F, один из аддуктов 2,3-ди-
гидрофурана и оба аддукта 2,3-диметилиндола претерпевают
дальнейшие превращения [987]:
(ОЧСНЬ
немного)
Полученные реакцией с CBrF 1-бром-1-фторциклопропаны
являются интересными промежуточными продуктами для
дальнейших селективных превращений. В реакциях Фриделя —
Крафтса отщепляется атом фтора [801, 811], в результате нук-
леофильной атаки получаются фторсодержащие соединения с
открытой цепью, и обмен брома дает фторзамещенные
циклопропаны [812]. Реакцию проводят с избытком CHBrgF или,
что лучше, с метиленхлоридом в качестве сорастворителя и
ТЭБА как катализатором. Избыток концентрированного гидр-
оксида натрия прибавляют с такой скоростью, чтобы смесь
медленно закипала. Затем реакционную массу кипятят 3 ч и
перемешивают еще 1,5 ч. Выходы колеблются от 51% (3,3-ди-
метилбутен-1) до 88% («-метилстирол) [808, 811, 812, 1164,
1790]. В качестве катализатора также используют дибензо-18-
краун-6 [959]. Соотношение с««/а«г«-аддуктов в случае арил-
этиленов меняется в широком интервале в зависимости от
заместителей в ароматическом ядре [1164].
При перемешивании смеси конц. NaOH ТЭБА/раствор
CHFI2 в метиленхлориде в течение 4 ч при 20 °С образуются
аддукты CFI. Галоформ был получен реакцией CHBr2F и
иодида натрия в ацетоне (7 сут, 100 СС). Поскольку CHBr^F —
наиболее трудно доступный реагент, то олефины берут в
избытке. Со стиролом достигается 60%-ная конверсия (в расчете
356 Глава 3. Практическое использование МФК
на CHFI2), но при использовании а-метилстирола, циклогексе-
на, 2-метилбутена-1, 3,3-диметилбутена-1 [809] и дифенилэти-
лена [810] выходы аддуктов были не столь высокими.
Дииодкарбен и хлориодкарбен
Циклопропановые производные этих карбенов могут быть
получены из йодоформа или хлордииодметана, ТЭБА и
концентрированного водного раствора гидроксида натрия с
использованием метиленхлорида в качестве сорастворителя при
температуре 60 °С (перемешивание в течение 3—5 ч) [810,
813]. Сырые продукты довольно неустойчивы, и некоторые из
них при попытке перегонки взрывались. Эта неустойчивость,
вероятно, вызвана наличием очень чувствительных побочных
продуктов, образующихся в результате окисления в щелочной
среде и, возможно, включающих иодаты. Считают, что именно
они инициируют разложение. Даже «очищенные» алкилдииод-
циклопропаны, полученные в условиях МФК, разлагаются
довольно быстро как в чистом виде, так и в растворе. Арильные
производные и хлориодциклопропаны несколько более
стабильны [810]. Следует упомянуть, что те же самые соединения,
полученные альтернативным Методом без использования
гидроксида натрия, гораздо устойчивее [814]. Выходы дииодцикло-
пропанов обычно невелики: (2—20%), за исключением л-хлор-
фенильного производного, которое было получено с выходом,
59% [813]. Однородные хлориодциклопропаны или смеси]
сым/амгы-изомеров были получены с выходом 50—65%- Даже
из 3,3-диметилбутена-1 образуется смесь сым/амгы-изомеров
[810]. Вейершталь [809, 810, 813] неожиданно обнаружил ка-]
талитический эффект ТЭБА при получении CFI, CC1I и С12 в
условиях МФК- Это удивительно, поскольку известно, что в
некоторых реакциях замещения и алкилирования в условиях-
МФК иодид-ион вызывает «отравление катализатора». Авторы
считают, что использование сорастворителя — метиленхлорида'
позволяет преодолеть это затруднение. Отсутствие отравления
можно объяснить также различиями в липофильности пары
тригалогенметильный анион/иодид-анион. Было показано, что
образование СС1й замедляется прибавлением иодид-иона [32],
т. е. анионный обмен ;
I" ч * фиксированный CClg~
проводимый катионом RtN+ на границе раздела фаз, не так
благоприятен, как обмен
С1- < > фиксированный CClg"
Однако иодсодержащие метильные анионы гораздо более ли-»
ЗЛО, а-Элиминирование 357
пофильны, чем СС13~, и должны легко обмениваться даже с
иодид-ионом.
Недавно было описано получение других 1-иод-1-галоген-
циклопропанов, их ЯМР-спектры и реакции [1318].
3.20.5. Другие смешанные карбены
Моногалокарбены
Этот тип карбенов не может быть получен в условиях МФК
даже при повышенных температурах. Система
концентрированный гидроксид натрия/катализатор вызывает лишь медленный
гидролиз метилендигалогенидов. Аналогично оказались
тщетными попытки уловить арилгалокарбены и стирилгалокарбены
при попытках генерации их из сходных продуктов А и В [816].
Во всех этих случаях скорости депротонирования не могут
конкурировать со скоростями гидролиза.
Аг—СНС12 С6Н6—СН=СН—СНС12 CeH6SCHCl2
А В С
CeH6—S—СН2С1 СН,—S—CHC12 RS(C1)C=C(C1)SR
D E F
Хлормеркаптокарбены
Опыты генерирования и улавливания хлорфенилтиокарбена
в экзотермической реакции в системе олефин/С в смеси с
50%-ным водным раствором гидроксида натрия и
каталитическими количествами ТЭБА оказались более успешными.
Выходы продуктов колебались от 20% (гексен-1) до 63% (стирол);.
Побочно образовывалось около 10% продукта димеризации F
(R=C6H5) :[8Г7]. Аналогично из D и олефина в метиленхлори-
де в системе ТЭБА/гидроксид натрия с использованием вибро-
Таблица 3.25. Получение фенилтиоциклопропанов
из QH5—S—СН2С1 и олефинов в метиленхлориде
в присутствии ТЭБА/NaOH
Субстрат
транс-Стильбеи
Стирол
Циклогексеи
цис-Бутен-2
граяс-Бутен-2
Винилэтиловый эфир
Выход О, %
78
70
67
65
79
60
Соотношение
экзо/эндо ил»
цисЦтранс
9.0
ь&
5.3
—
5,5
358 Глава 3. Практическое использование МФК
мешалки в течение 4 ч получают фенилтиоциклопропаны (G)
(табл. 3.25) [818]:
c,h5s н
СеНв—S—СНВС1
D G
Схема 3.207
Можно ожидать, что хлорметилтиокарбен менее электро-
филен, чем хлорфенилтиокарбен. Мосс и Пилькевич [819] улав*
ливали эти карбены различными олефинами при реакции с Б
и грег-бутилатом калия в качестве основания и получили
продукты реакции с выходом от низкого до умеренного.
Нежелательным побочным продуктом, образующимся с высоким
выходом, является формальный димер карбена F (R=CH3) (см.
формулу на с. 357). В единичном эксперименте с тетраметил-
этиленом было показано [819], что в условиях МФК можно
получить более высокий выход аддукта и меньший выход F
(система конц. NaOH/ТЭБА, З ч при 50 °С) [819]. Показано [846]
также, что хлорметнлтиокарбен — это свободный карбен, а не
карбеноид.
Алкилиденкарбены
Ньюмзн и сотр. [820, 821] широко изучили щелочное
разложение N-нитрозооксазолидонов (Н). В присутствии олефи-
нов были получены продукты I. Они, вероятно, образуются из
алкилиденкарбенов. Однако кроме этих продуктов в
присутствии метанола образуются соединения типа J и К. Позднее
было найдено, что аддукты непредельных карбенов образуются
с более высоким выходом из М-[2-йндроксиалкил]~:Ы-нитрозо-
ацетамидов L с открытой цепью при использовании аликвата
336 и водного гидроксида натрия в качестве основания [821,
822]. Эта реакция благоприятствует образованию соединений I
по сравнению с продуктами конкурирующей
перегруппировки J. Исключением являются L (R=C6H5, 1?'=цикло-СзН5),
которые образуют только J (R=C6H6, К'=цикло-СзН7) [821].
В опубликованной позднее работе изопропилиденкарбен
получали из диметилвинилтрифторметансульфоната и грег-бути-
лата калия как в присутствии 18-крауна-6, так и без него.
Электрофильность изопропилиденкарбена была определена
методом конкурирующей реакции с замещенными стиролами.
Корреляцией по уравнению Гаммета вычислено р=—0,75
независимо от того, проводили реакцию с краун-эфиром или без
N—NO
H
CteC—R' + C=CHOCH3
NO
CHS—N—COCHt
I
360 Глава Д. Практическое.использование МФК
него. Это свидетельствовало в пользу промежуточного
образования свободного карбена [823].
Аналогичные опыты с фенилгалокарбенами показали, что
карбен и карбеноид обладают различной селективностью и их
можно различить при помощи краун-эфиров [612]. Величина
параметра р для изопропилиденкарбена, полученного из окса-
золидона Н, равна —3,4 [820], что указывает на то, что он не
является свободным карбеном. Формы, полученные из L в
условиях МФК, непосредственно не сравнивали между собой, но,
ориентируясь по величинам р, можно предположить, что это
«свободные» карбены. В условиях МФК приведенные ниже
соединения были получены с выходами 35—80% [824].
NO pi R
R
ОН
ЧС-СН,—N—СОСНз +
г» '
R*O
\ NaOH
р=СН, аликв(тззб-
R1
L R/R' = (CH2)S; (сн«)4; chs> сн3
R> = CHS, Ph, OC2HS, H
R2 = CHS, C2H5
Схема 8.209
Раскрытие цикла при помощи ацетата ртути и обработка
сероводородом или Kh .превращает эти продукты с высоким
выходом в |р,т>-йепредель'ные эфиры или их 1|3-иодпроизводные.
Циклогексилиденкарбен генерируется при температурах от —10
до —5°С из L (R—R'= (СН2)5) (см. схему 3.209 или 3.211) в
растворе олефина в присутствии аликвата 336 при медленном
прибавлении водного раствора гидроксида натрия. Он
присоединяется к 1,4-циклогексадйену и норборнадиену с
образованием М и N. Однако в реакции с циклопентадиеном вместо
ожидаемого О с выходом 76% образуется Р [825].
ОО /V0
м
N
Схема 3210
3.20. а-ЭлиминирОвание • , 361
Перегруппировка идет даже при 30 °С. В тех же условиях МФК
из L [R-~R/:= (СНгЬ] в присутствии нуклеафилов образуются
соединения Q с выходом 40—60%. Аналогично из альдегидов
RCHO или кетонов R—СО—СН2—R' и соединений L
образуются продукты S или Т соответственно. Однако ие было
установлено, образуются соединения Q, S и Т через карбен или нет
[826]. .::
ООН
' NaOH, Хв
U-N-COCH3 ^
NO
L (R, R' = (CH2)5
Г~\ ?
/ Wh-c-r
\ /
S T
Схема 3.211
Наиболее удобный и мягкий способ получения алкйлиден^
карбенов, опубликованный до настоящего времени, заключается
в реакции между (триметилсилил)винилтрифторметансуль;фо-
натами и фторид-ионом при температурах от —20 до 0°С в.
течение 1—2 ч. Количественные выходы аддуктов циклогексена
или этилвинилового эфира были получены [901] в системах
фторид калия/18-краун-6, безводный R^N+F" либо аликват 336/
/фторид калия путем следующей последовательности реакций:
Me2C=C(SiMe3)OSO2CFs + F~ *■ Ме2С=С: -f FSiMe3 + -OSO2CFg
Генерированный таким образом изопропилиденкарбен
может быть уловлен также изонитрилами; конечными продуктами
этого процесса являются замещенные акриламиды, получае?
мые с выходом 35—52% [1165]: ■
Ме2С=С: + :C=N—R >■ [Me2C=C=C=N—R] > Ме2С=СН—СО—NHR
а-Элиминирование в 1-бромвинильных соединениях обычно
приводит к перегруппировке, но ю-бромметиленадамантан
может быть превращен в адамантилиденкарбен при помощи
грег-бутилата калия и 18-крауна-6. Аддукты олефинов и этого
карбена получены с выходами вплоть до 97% [1166];
Алкенилиденкарбены
Аддукты U алкенилиденкарбенов были получены в МФК-
процессе из пропаргилгалогенидов V или аллилгалогенидов W
(схема 3.212).
Таблица 3.26. Превращения некоторых олефинов
в (днметилвннилидеи)циклопропаны
Олефва
Вкход.
Катализатор
Примечания
CI
НзС
с-с=сн
н,с
Стирол
Изобутоксивиниловый
эфир
Циклопентен
Циклогексен
3-Метилбутен-2-ол-1
2,3-Днметилбутен-2
а-Пинен
$-Пинев
1,5-Циклооктадиен
Коричный спирт
Изоцрен
Инден ,
$-Метилстирол
Дигидропиран
Норборнадиен
81
60
69
48
27
37
16
87
20
31
19
21
26
38
16
36
38
18
Аликват 336
ТЭБА
Дициклогексано-18-
крауи-6
Аликват 336
Аликват 336
Аликват 336
Аликват 336
Аликват 336
ТЭБА
ТЭБА
ТЭБА
ТЭБА
ТЭБА
Дициклогексано-18-
краун-6
ТЭБА
ТЭБА
Дициклогексано-18-
краун-6
Дициклогексано-18-
краун-6
NaOH/пентан
КОН/бензол
КОН/бензол
NaOH/пеитан
NaOH/пентан
NaOH/пеитан
NaOH/пентан
NaOH/пентан
КОН/бензол
КОН/бензол
КОН/бензол
КОН/бензол
КОН/бензол
КОН/бензол
КОН/бензол
КОН/бензол
КбН/бензол
КОН/бензол
827
828
829
827
827
827
827
827
828
828
828
828
828
82S
828
828
82S
82S
н3с.
НзС.
"\
Br
N,C
у
W
2,3-Диметилбутен-2
Циклогексен
Гексен-1
Норборнадиен
68
61
25
21
Аликват 336
Аликват 336
Аликват 336
Bu.NI
16 ч
2 сут
3 сут
6 сут
830
830
830
831
3.20. а-Элимшшрование 363
Большое количество олефинов было превращено в винили-
денциклопроланы. В табл. 3.26 приведены выходы, полученные
при использовании соединений V и W. Выходы превосходят
величины, достигнутые при помощи более ранних методик, и
в условиях МФК, по всей видимости, генерируется свободный
карбен [970].
До некоторой степени были изучены параметры процесса.
Так, прн использовании V в качестве исходного продукта и
ТЭБА как катализатора реакцию лучше вести при комнатной
температуре, чем при 45°С; применение концентрированного
гидроксида калия дает лучшие результаты, чем использование
разбавленных растворов; бензол — лучший растворитель, чем
гексан; оптимальное время реакции составляет 10 ч '[828].
В определенных условиях использование краун-эфиров
обеспечивает такие же или гораздо большие выходы [829]. Однако
было обнаружено '[829], что выходы в реакциях,
катализируемых 18-крауном-6, после достижения максимума при
дальнейшем увеличении времени реакции снижаются более или менее
резко. Этот эффект наиболее ярко выражен при температуре
45 °С, когда через 2 ч наблюдается максимальный выход 85%,
а через 10 ч — лишь 53%. Низкокипящие (например, бутадиен)
или гидрофильные субстраты могут реагировать с системой
твердый гидроксид калия/краун-эфир даже при таких низких
температурах, как —78 °С [829].
3 ^
=С=С. с N.OHU.UKOH
N-OH'""'KOH> \=С=С. с \, „-/
катализатор / •*" катализатор уЛ-==*-=С.
Н3С Н3С Вг
, W
Н3С ^
R4
U
Схема 3.212
Для исходного соединения W и NaOH в качестве
катализаторов применялись аликват 336 {830] и иодид тетрабутилам-
мония [831]. В реакции норборнадиена с последним
катализатором выходы продуктов и обработка реакционной массы
364 Глава 3. Практическое использование МФК
аналогичны получаемым при использовании других 'солей
аммония [831]. В данном случае для достижения максимального
выхода необходимо вести реакцию 6 сут при температуре 60 °С..
Взаимодействием метил (диметил)аллилсульфида V
получали [827] продукт алкилирования X вместо аддукта карбена.
Из азобензола образуется бензимидазольное производное V.
Возможный механизм этого превращения представлен на
схеме 3.213 [832].
.СНз
СНз Ph—N=M—I
НзС СНз
Схема 3.213
Даже более высоконенасыщенные карбены Z улавливаются
циклогексеном, хотя с довольно низким выходом [833].
R'
I
ВгСН—СН=С—С==СН
N»OH>
Bu*Nl
f '' 1
[RCH=CH—C=C=C:J
(R. R' = H, CH3)
Схема 3.214
Ниже представлены типичные методики.
Получение производных диметилвинилиденциклопропана из
З-хлор-З-метилбутина-1 (V) [828]. Смесь 30 ммолей олефина
и 4 мл бензола энергично перемешивают со 150 мг ТЭБА и
50 мл 50%-ного раствора гидроксида калия при комнатной
температуре в атмосфере азота. В течение 2,5 ч медленно
прибавляют раствор 10 мМблей V в 6 мл бензола. Когда
прибавление закончено, -массу перемешивают еще -10—г 13 ч. Смесь
разбавляют водой, экстрагируют эфиром и продукт выделяют
обычными способами. •. . .
3.21. Реакции восстановления 365
Получение производных диметилвинилиденциклопропана из
1-бром-3-метцлбутадиена-1,2 (W) [830]. Смесь 20 мл олефина,
7 мл концентрированного водного раствора гидроксида натрия,
1 г аликвата 3§6 и 3 г W энергично перемешивают при
температурах 0—60^ в течение 3 сут. Смесь обрабатывают, как
указано выше.
3.21. Реакции восстановления
3.21.1. Комплексные гидриды и диборан
Среди обычно используемых для восстановления гидридных
комплексов, пожалуй, только литийалюминийгидриД является
наиболее подходящим реагентом для усовершенствования
методики восстановления гидридами в системе твердая
фаза/жидкая фаза путем распространения на эту методику принципов
МФК- В самом деле, литийалюминийгидрид разлагается гид-
роксилсодержащими растворителями и плохо растворим во
многих апротонных растворителях (г/100 г при 25 °С): ТГФ 13,
диоксан 0,1, дибутиловый эфир-'■% "диэтиловый эфир 35—40
(только в случае упаривания более разбавленных растворов).
На первый взгляд можно добиться успеха, используя
замещение лития на аммонийную группу или комплексообразование
гидрида с краун-эфиром. Тетрабутиламмонийалюминийгидрид
может быть получен осаждением эфиром из раствора Bu4N+Bi—
и 1ЛАЩ4 в тетрагидрофуране. Аналогично получают BtuNGaHt-
Другие способы синтеза основаны на аланатах натрия или
калия [525]. Известны следующие аммонийные производные:
Me4N+AlH4- Et4N+AlH4-, (н-С3Н7) (h-CsHi7)<*N+A1H*- и
Me4N+GaH4~. Однако нет сведений о том, что эти соединения
использовались для восстановления; поэтому может оказаться,
что аланаты четвертичного аммония не могут применяться для
этой цели. Напротив, было показано, что для восстановления
совершенно необходимо присутствие катионов щелочного
металла. Пьзр и сотр. [526—528] изучили роль катиона при
восстановлений кетонов действием LiAlH4 и NaBH4, Они
сравнивали результаты восстановления циклогексанона этими двумя
комплексными гидридами в диглиме как в присутствии, так и
в отсутствие криптанда [2.1.1] (А), образующего комплекс с
Li+, или криптанда [2.2.1] (В), образующего комплекс с Na+.
При использовании LiAlH4 прибавление эквимолярного
количества А полностью подавляет восстановление. Добавление
иодида лития или натрия полностью возвращает системе
восстанавливающую способность. Из этого можно сделать вывод,
что роль ионов металла чрезвычайно велика. Однако вполне
366 Глава 3. Практическое использование МФК
NN/AorV\/N
Схема 3.215
достаточно каталитических количеств этих ионов. Был
предложен следующий механизм:
Li®
н
'О6 Li®
Схема 3.216
Аналогичное замедление реакции было обнаружено Лупи и
сотр. [529] при добавлении А в случае восстановления альде^
гидов и кетонов в эфире или ТГФ. Этот эффект был
использован для селективного 1,4-восстановления замещенных 2-цикло-
гексенонов, в результате которого образуется смесь циклогекса-
нонов и циклогексанолов. В то время как в стандартных
условиях основными продуктами реакции являются циклогекса-
нолы (соотношение атаки С1: С3 ж 86 : 14), в присутствии А
или 12-крауна-4 это соотношение изменяется до 14:86 [1629].
Отсюда вытекает, что вопреки высказанному ранее
предположению процесс восстановления алюмогидридом лития не
может быть существенно улучшен использованием межфазного
катализа, поскольку для этого процесса необходимо
присутствие свободных катионов металла, а в неполярных
растворителях катионы металлов всегда комплексуются. Единственным
возможным решением этой задачи может быть использование
добавок солей. Восстановление карбоксилатов калия с
помощью 1ЛАЧН4 возможно в диглиме в присутствии краун-эфи-
POBJ1620].
Перейдем теперь к боргидридам, которые более
перспективны для межфазного катализа. Однако необходимость
улучшения традиционных методик для NaBH4 не слишком
настоятельна, поскольку этот реагент разлагается гндроксйлсодержащими
растворителями очень медленно.
3J21. Реакции восстановления 367
Исследовано [4, 38, 39] восстановление октанона-2 водным
NaBH4 в 2 н. NaOH как в присутствии, так и в отсутствие
аликвата 336. Хотя каталитическая реакция проходит очень
быстро, она все же не так эффективна, как обычный
гомогенный процесс. Из данных работы [526] следует, что в
абсолютном диглиме циклогексанон не восстанавливается
непосредственно действием NaBH4, входящего в комплекс с 1 экв. В.
В этом случае добавление небольшого количества воды или
(что более вероятно) прибавление воды прн обработке
катализирует превращение.
Схема 3.217
Таким же образом катализируют восстановление добавки
NaC104 или LiOlO4.
В течение нескольких лет боргндриды тетраалкиламмония
предлагали в качестве восстанавливающих реагентов в
неполярных растворителях [530]. Растворимость и
восстанавливающая способность зависят отчасти от величины катиона. Так,
Me4N+BH4~ — белый устойчивый порошок, который по
восстанавливающей способности равноценен NaBH*, — обладает тем
преимуществом, что растворим даже в бензоле. Кроме того, он
в меньшей степени разлагается в таких растворителях, как
этанол и этиленгликоль. Высшие аналоги, такие, как триметил-
цетиламмонийборонат (белый гранулированный порошок) и
трикаприлметиламмонийборонат (воск), еще лучше растворимы
в гексане или минеральном масле [530]. Однако
восстанавливающая способность этих реагентов меньше, чем у NaBH4*, это
подтверждается тем, что Bu4N+BH4~ можно перекристаллизо-
вывать из этилацетата или даже ацетона, если работать
достаточно быстро [531].
Альдегиды восстанавливаются этими боронатами при
комнатной температуре достаточно быстро; кетоны в этих же
условиях восстанавливаются медленнее, а сложные эфиры
реагируют медленно даже при повышенной температуре. Нитрилы не
восстанавливаются совсем, а пероксиды и хлорангидриды
кислот реагируют быстро. Ароматические нитросоединения не
способны к восстановлению при низких температурах и лишь
частично восстанавливаются при 65 °С [530]. Истинная причина
такой селективности аммонийборонатов может быть связана с
тем,' что реагенты содержат различное количество остаточной
368 Глава 3. Практическое использование МФК
воды. Низшие аналоги боронатов связывают воды больше, чем
боронаты с высшими алкильными заместителями. Однако нет
сведений о том, что присутствие каталитических .количеств во^
ды или катионов щелочных металлов необходимо только при
восстановлении карбонильных групп или оно требуется и при
восстановлении других функциональных групп Кроме того, не--
которые функциональные группы, по-вщцййому, образуют с
реагентом рыхлые комплексы, которые с легкостью распадаются
при обработке. Некоторые аммонийборонаты имеются в
продаже, хотя и их цена относительно высока. Аммонийборонаты
получают традиционным способом: реакцией обмена и
выделением путем осаждения
R4N+X~ + NaBH4 »- R4N+BH4" + NaX
Метод экстракции ионных пар по Брендстрёму очень
упрощает их приготовление. Поэтому, прежде чем перейти к
истинным межфазным процессам, мы остановимся на реакциях
восстановления с помощью аммонийборонатов в гомогенной среде.
Тетра-н-бутиламмонийборонат [531]. Смесь 1 моля
Bu4>NHSO4, растворенного в 200 мл воды, и 250 мл 5 н. NaOH
добавляют к раствору 1,1 моля NaBH4 в 100 мл воды при
комнатной температуре. После экстракции СНгСЬ, высушивания и;
упаривания получают аммонийборонат с количественным
выходом. Полученный раствор можно также непосредственно
использовать для восстановления. Ниже приведен пример
экзотермической реакции, выход конечного продукта в которой 90%
{532].
СНЯ—СООН ^г^ ^СНЯ—СН2—ОН
5мол«п
НзСО
3,3 моля
Схема 3.218
Недавно описаны превращения алкалоидов, в которых
этот аммонийборонат был использован в системе СС14/СНаОН
для гидрогенолиза связи С—Н [1169]. В та время как в
работе [530]. использовали для создания гомогенной среды бензол,
к-гексан или минеральное масло, другие исследователи [533,
1521] предпочитали применять метиленхлорид* В этом
растворителе кетоны восстанавливаются довольно быстро даже при
комнатной температуре (24—48 ч при использовании 4 мол,
экв. Bu4NBH4); сложные эфиры реагируют весьма медленно, з
. Реакции восстановления 369
хлорангидриды кислот образуют спирты очень быстро [533] s.
Селективное восстановление альдегидов в присутствии кетонов.
возможно. осуществить действием Et4NBH4 в СНгС12 (только
1,2 мол. экв. реагента, 20 ч, комнатная температура, выход 53—
89%) [1521]. Индолы при этом восстанавливаются до индоли-
нов [1843].
Предложены два альтернативных механизма реакции:
(1) прямое гидридное восстановление карбонильного
соединения и (2) первоначальная реакция боргидридного иона с
CH2Gl2 с образованием истинного восстанавливающего агента
диборана. Можно предположить, что восстановление
альдегидов протекает в: большей степени по пути (1), тогда как путь
(2) более вероятен для восстановления кетонов.
NR«®BH4e R~CO~">
н
RCH2OH
(1)
NR4eBH4e
B,H.
" O—BH2
R—C—R
I
. H
RaCHOH (2)
Схема 3.219.
Восстановление 2-циклогексенона и изофорона действием
Bu4NBH4 в ТГФ протекает с образованием циклогексанонов и
циклогексанолов (1,4-атака) с выходом 85 и 96% соответствен^
но. Этот способ проще и дешевле, чем описанный выше метод
восстановления с помощью LiAlH4/A- Интересно, что лри
проведении этой реакции в системе толуол/вода образуется
значительное количество аллильиых спиртов [1744].
Превращение сложных эфиров в альдегиды с выходами
75—85 % [534] можно осуществить также следующим путем i
CHaCl*. -78 "С
R_CHo
Схема 3.220
Кроме того, в гомогенном растворе (растворитель метилеи-
хлорид) при —30 °С восстанавливается тритиокарбонат-З.Б-ди-
370 Глава 3. Практическое использование МФК
оксиды [535]:
2R._so2-c=s "30°CVB^,BHif > R1-so2-ch^-s-c=s
| CH2Cl2 |. I
SR2 SR2 SR2
Схема 3.221
Наконец, следует упомянуть восстановительное алкилиро-
вание с образованием диорганоселенидов и диорганотеллури:
дов в толуольном растворе [1396]:
>■ R—TeiSe)-R + 2R3N-BH,+ Hj
Более мягким восстанавливающим реагентом является циа-
нотригидридоборонат. Сильная электроакцепторная группа CN
способствует тому, что анион становится поставщиком гидрид-
ионов. Реагент можно легко приготовить методом экстракции
ионных пар.
Тетрабутиламмонийцианборонат [536]. Суспензию 0,1 моля
B114N+HSO4 в 50 мл воды обрабатывают 35 мл 5 н. NaOH.
Добавляют при комнатной температуре раствор 0,1 моля
NaBH3CN в 50 мл воды и спустя несколько минут
экстрагируют смесь метиленхлоридом:
Этот мягкий реагент восстанавливает в ГМФТА первичные
нодиды в соответствующие углеводороды; элиминирования га»-
логеиов с образованием олефинов при этом не наблюдается
(например, 1-иоддекан превращается в декан при комнатной
температуре за 21 ч с выходом 81%). Бромиды, хлориды и то-
зилаты реагируют медленнее. Гидридное замещение у
вторичных галогенидов проходит преимущественно с инверсией
конфигурации; скорость реакции меньше, чем у первичных
соединений [536, 537]. Первичные и вторичные бензилгалогениды
восстанавливаются гладко. В последнем случае не было
обнаружено возникновения двойных связей. В то время как
аллильные галогениды превращаются в алкены, винильные галогениды
инертны к восстановлению. Тогда как геминальные дигалоге-
ниды легко дают моногалогениды под действием Bu4NBH3CN,
1,2-дигалогениды вступают в реакцию менее активно [537].
Альдегиды, кетоны, сложные эфиры, амиды, цианиды и
ароматические нитросоединения инертны при комнатной температуре
[536]. Однако альдегиды можно восстановить в присутствии
кетонов, если добавить каталитическое количество 0,1 н. НС1
[536]. Увеличение концентрации кислоты увеличивает- также
и скорость восстановления кетонов. Другие заместители, такие,
3.21. Реакции восстановления 37Е
как цианоли нитрогруппы, а также сложнрэфирная.и амидная
группы, ииерт&ы в этих условиях. Несколько позже было
описано [538] восстановление ненасыщенных карбонильных
соединений при комнатной температуре с помощью NaBHsGN ила
Bu4NBHgGN в подкисленном метаноле (рН около 3).
Аммонийные соединения « этом случае не являются оптимальными ре-^
агентами. За 1—2 ч большинство ненасыщенных альдегидов и
кетонов образует аллильные спирты с примесью аллильных
метиловых эфиров в качестве побочных продуктов. Замена
растворителя на ГМФТА увеличивает скорость реакции. Кроме
того, в этом случае не обнаружено образования продуктов с
участием растворителя, однако существенно возрастает выход
аллильных углеводородов. Если а^-ненасыщенная
карбонильная группа дополнительно сопряжена с ароматическим
кольцом, цианидной, карбоксильной или сложноэфирной группой
либо если карбонильная группа входит в пяти- или шестичлен-
ный цикл [538], то основным направлением реакции
становится 1,4-присоединение, приводящее в конечном итоге к
образованию заметных количеств предельного спирта.
Восстановительное аминирование типа
»- R4R*CH—NHR»
можно осуществить в метиленхлориде, содержащем немного
метанольного раствора НС1, действием NaBHaCN/аликват 336
или Bu«NBH3GN [1842].
Другим мощным восстановительным1 реагентом является
В2Нб, который упоминался выше как возможный
промежуточный продукт. Он может быть легко получен в растворе при
добавлении алкилгалогенида (например, метилиодида) к
безводному раствору тетраалкиламмонийбороната в
метиленхлориде. Побочный продукт (R4N—галогенид) может быть легко
удален [531]:
2ВН4" + 2СН„1 * В2Нв + 2СН4 + 21"
По простоте и безопасности этот метод, несомненно,
превосходит традиционные методики.
Реакции с дибораном, полученным in situ. Раствор
ВифЫ+ВН4~ в сухом метиленхлориде (получен, как описано
выше) и субстрат перемешивают при охлаждении льдом в
атмосфере аргона. Медленно добавляют 2 экв. СН31 таким образом,
чтобы выделение метана не было слишком бурным.
Перемешивают в течение 30 мин при комнатной температуре, избыток
гидрида разлагают добавлением этанола и смесь гидролизуют
НС1.
Примерами реакций восстановления и гидроборирования,
проведенных аналогично, являются следующие превращения
372 Глава 3. Практическое использование МФК
[531, 640]: 3-нитробензойная кислота^З-нитро^ензиловый
епирт (90%), этал-(4-бензилоксифенил)ацетат-*2-|4/-бензшгок':
еифенил) этанол .(87%), бензонитрил-^бензилайин (95%),
«-хлорбензальдегид-»-л-хлорбензиловый спирт (94%), циклогек-
сен-мщклогексанол (98%), а также восстановление N-формильт
ной группы в N-метильную в гетероциклических соединениях
[912]. Более сложными примерами гидроборирования/окисления
являются превращения 8-циклододецен-1-она в смесь 1,3- и 1,4-
диклододекандиолов [541] и образование 3-(4-гидрокси-З-ме-
токсифенил) пропанола (96%) из эвгенола [1465]. .
В упомянутых в этой главе работах, как правило,
используются восстанавливающие реагенты в гомогенной среде. Мацуда
и Коида [539] восстанавливали кетоны в кипящем ксилоле или
толуоле в присутствии эквимольных количеств катализаторов,
что способствовало увеличению растворимости., В качестве
катализаторов использовали диглим, диметоксиэтан и дибензо-18-
краун-6. Последний из них приводит к лучшим результатам,
однако вследствие протекания побочных реакций конденсации
выходы целевых продуктов посредственные.
Можно предположить, что катион межфазного катализатора,
взаимодействуя с водным боргидридом натрия, наряду с
ВН4~ экстрагирует некоторое количество воды (сольватирую-
щие молекулы), которого вполне достаточно, чтобы
способствовать восстановлению. Это предположение вытекает из опытов
Ландини и сотр. [44, 45], которые на примере восстановления
•октанона-2 действием КВН4 или NaBH4 в системе бензол/вода
яри 80 °С проверили каталитическую активность следующих
краун-эфиров: 18-крауна-6, дибензо-18-крауна-6, бензо-15-крау-
на-5 и дициклогексаног18-крауна-6. Наиболее активным- оказал^
ся последний. Решающим фактором, очевидно, является
увеличение липофильности.
Шикк [120, 999] сообщил, что стереоселективность восстат
новления кетонов, приведенных ниже, в условиях межфазного
катализа и в гомогенной метанольной среде различна^
он
сн3
он
он
н он
главный продукт
главный продукт
Схема 3.222
Боргидрид натрия способен восстанавливать при 65 °С слож-
зше эфиры до спиртов в полиэтиленгликоле 400 с хорошими
3.2L Реакции восстановления 333
выходами |Д767]. Проведению этой реакции в гомогенной р
де для превращения галогенидов и сульфонатов в алканы благ
гоприятствует использование гексадецилтрибутилфосфонийброт
мида в присутствии избытка водного NaBH4. Гидрогенолизпер-г
вичных бршшдоь и.иодидов проходит экзотермически. Для гид-
^зогенолиза менее реакционноспособных. субстратов (первичные
хлориды, сульфрнаты и вторичные производные) требуется по-,
вышение температуры вплоть до 80 °С. Влнильные галогени-
ды дегалогенируются медленно; группы COOR, CONiRa. NOg н
•ON не изменяются. В мягких условиях алкены также не восста-.
лавливаются [1768]. . .
Частичное восстановление бензилхлоридов в системе толу,
•ол/вода в присутствии теллура и Bu4NX привело к получению
теллурбензойных эфнров [1536]:
2ArCOCl+Te+NaBH4—^-Ar—GO—Те^-€Н2—Аг
Восстановительное меркурирование, которое уже
упоминалось в связи с гомогенными фазовыми реакциями, в сиетецефод?
ный NaOH/NaBH4/T9BA в дихлорметановрм растворе предетавт
ляет собой быстрый процесс; при этом выходы конечных
продуктов намного выше, чем в "классических условиях [1223J, Та
же система используется для высокоэффективного восстадбвде-,
лия карбонилов металлов, приводящего к,следующим аниона»
лли гидридам [1669]:
ВгМп(СО)6 >-. [Мп(СО)6Г
BrMn(CO)4PPhi, >- [HMn(CO)4PPhg]-
Fe3(CO)la ^ [HFe3(CO)u]-
Со2(СО)8 —*■• [Со(СО)4Г
(C6H6)Ru(CO)2Br »- [(C5H6)Ru(CO)2H]
(С6НБ)Мо(СО)3С1 >- [(СБНБ)Мо(СО)зГ
2,3,4- и 2,4,5-тринитробензолы могут быть восстановлены до
2,4-динитробензола (выход около 90%) в системе дихлорм«-
тан/воднгЭДаВН4/катализатор [1294]. Восстановление ' Ййвййн*
ароматических карбоновых кислот до о-метйлфейолой «Wfgeal
промежуточное этоксикарбонильное производнёё) ;'в уаювия?
межфазного катализа не имеет преимуществ переД;Другйми ме-!
тодами [1443]. Однако новый синтез циангидриновых эфиров
без выделения промежуточных продуктов -(«в одном горшке»)
оказался выгодным при использовании МФК-условий £ЙШ]:
2ArCOCl-f-NaCN+NaBH4(+Bu4NBr) » ArCH(CN)—О—СО—Аг (92%)
В качестве исходных соединений для получения таких
продуктов можно использовать также ацилцианнды. ' '
О попытках и истинной асимметрической-индукции привое?
становлении карбонильных .соединений действием ВН4- ем.
374 Глава 3. Практическое использование МФК
в разд. 3.1.5. Вопреки предсказаниям правила Прелога реакции
боргидрида с (3$) -ментилбензоилформиатом в присутстии
катализатора, полученного на основе (IS,2S)-эфедрина, приводят
к (З^)-ментиловому эфиру (S)-миндальной кислоты [1171].
Отметим, что в этом случае молекула субстрата уже являлась
асимметрической. Кроме того, были определены константы
скорости реакций восстановления ацетофенона в присутствии соли
эфедрина в качестве катализатора [1172]. Добавки различных,
частично замещенных гидроксимоносахаридов солюбилизируют
до известной степени NaBH4 в бензоле и ТГФ и при
восстановлении кетонов повышают оптический выход продуктов до 39%
[1201].
Дарст и сотр. [546] разработали метод восстановления сульф-
оксидов до сульфидов, заключающийся в метилировании ме-
тилфторсульфонатом в метиленхлориде с последующим
восстановлением цианборогидридом натрия. Таким способом можно
защитить и другие чувствительные к восстановлению группы.
В стандартных условиях растворитель после метилирования
удаляют, а остаток разбавляют этанолом для последующего
восстановления. В улучшенном варианте межфазного катализа
18-краун-6 и NaBH3CN добавляют непосредственно к
охлажденной реакционной смеси и продолжают перемешивание в
течение нескольких часов.
U с R' FSO3OCH3 ® NaBH3CN
R~irR сн2с.2.<гсзч R-S—R > R-S-R
о °сн3
Схема 3.223
3.21.2. Другие восстанавливающие реагенты
Метод межфазного катализа был использован для модельной
системы NADP-H, в которой восстанавливающим реагентом
является Na2S2O4, катализатором — бензилтриметиламмоний-
хлорид, а роль редокс-модели играл бензилникотинамидхлорид
[1174]. Более широко межфазный катализ использовали для
восстановления кетонов дитионитом натрия в системе
бензол/водный бикарбонат натрия (20 ч, комнатная температура).
В присутствии адогена 464 выходы повышаются с 15 до 55%.
Стереоселективность реакции не отличается от наблюдаемой
при обычном гидридном восстановлении [1233]. Восстановление
растворами металлов можно осуществить в присутствии краун-
эфиров, которые солюбилизируют металлы. Так, сплав натрия
с калием восстанавливает алкины в смесь цис/транс-алкенов
(70—83%, 0,5—2 ч), бензойную кислоту в циклогексанкарбоно-
вую кислоту (86%, 18 ч) и антрацен в 9,10-дигидропроизводное
3.21. Реакции восстановления 375
(67%, 1 ч) в ТГФ в присутствии 18-крауна-6 [1598]. Деокси-
генирование первичных и вторичных спиртов (в том числе
стероидов) проходит при реакции соответствующих эфиров [1219,
1793] или, лучше, карбаматов с калием в присутствии 18-крау-
на-6 в системе грег-бутиламин/ТГФ [1220]:
RiRaCHOH ► IWCH—О—СО—R3 >- RiR*CH2 (~50%)
>- RiR*CH—О—CS—NHEt >- R^sffl,, (до 87%)
Описано также восстановительное дегалогенирование галоген-
бензолов действием КН/криптанд [2.2.2] в ТГФ [1600]:
СвНБ1 > СвНБВг> СвНБС1 > CeH5F
Имеются указания на то, что реакция осуществляется на
поверхности твердого КН. Каталитическое действие связано с
удалением с поверхности образующегося КХ.
В химии карбонилов металлов межфазный катализ был
впервые использован для восстановления нитросоединений
действием Fe3(CO)i2 в системе бензол/1 н. NaOH при комнатной
температуре в присутствии катализатора ТЭБА или 18-крауна-6
[547, 548]. Более эффективным катализатором оказался ТЭБА,
в присутствии которого выходы достигали 60—90%.
NaOH/ышалюотор
R
Схема 3.224
Раньше такое восстановление требовало кипячения в
течение 10—17 ч в бензоле, содержащем немного метанола. Авторы
предположили, что ОН~ реагирует с карбонилом железа с
образованием [Fe3(CO)i2]2~, который в свою очередь протониру-
ется, давая [HFe3(CO)i2]~. Ионная пара этого аниона,
по-видимому, является истинным восстанавливающим реагентом [547,
548]. В присутствии газообразного оксида углерода выход
анилина значительно снижается [1168]. Восстановление
ароматических нитросоединений также возможно в бензоле при комнатной
температуре и использовании системы Ru3(CO)i5/12 н. NaOH/
/ТЭБА. В этом случае в атмосфере оксида углерода выходы
существенно повышаются [1376].
о о / о о \
д-с-сн2вг + Со2(со)8 ТЗБА- SH-NaOH> r_c-ch3 + [r_c-ch2-ch2-c-rJ
G H
Схема 3.225
376 Глава 3. Практическое использование МФК
Обработка а-бромкетона G (1?=арил, адамантил) эквимо-
лярным количеством октакарбонилдикобальта, 5 н. гидрокси-
дом натрия, бензолом и ТЭБА, позволяет за 2 ч при комнатной,
температуре получить дегалогенированный монокетон с почти
количественным выходом. МФК-методика дает прекрасные
выходы при отношении G/соединение кобальта, равном 10: 1.
Иногда 1,4-дикетоны Н образуются в небольших количествах
как побочные продукты и редко в качестве основных продуктов
[549]. Возможная схема восстановления включает реакцию ОН~
с Со2(СО)в с образованием иона Со(СО)4~, который
экстрагируется в органический слой. Затем, как полагают, проходят
следующие превращения:
о О
NR,e[Co(CO),]e + R—С—СН2Вг > R—С—СН2' + Хо(СО)« + NR4eBr&
Н о Со2(СО)в
II
R—С—СН2—Со(СО),
ОНв,наО
О
R—С—СНз
Схема 3.226
Для реакции восстановления/окисления, приводящей к анио-
ну^ способному к экстракции, принята следующая схема [511]:
32ОН" + 11Со2(СО)в *■ 20Со(СО)4- + 6СО82- + 2СоСО8 + 16Н2О
Восстановление бензальацетона до 4-фенилбутанона-2 возт
можно с помощью Fe(CO)5 в системе бензол/вода в присутствии
первичных аминов и 18-крауна-6 или дициклогексано-18-крау-
на-6. Восстанавливающим агентом является, по-видимому,
[KpayH-RNH3]+[HFe(CO)4]- [1366].
Дешевым и простым в приготовлении является катализатор
гидрирования K»[Co(CN)5H]. В присутствии ТЭБА анион
(H2[Co(GN)5H]~??) способен экстрагироваться в бензол, где
он может при атмосферном давлении Действовать как
катализатор гидрирования. Этот анион оказался полезным для
восстановления главным образом а,р-ненасыщенных кетонов в
насыщенные кетоны. Так, карвон дает 2-метил-5-изопропенилцикло-
гексанон (транс/цис-6: 1) с выходом 93% [1175]. В
последующих работах диены были прогидрированы до смеси моноенов,
3.21. Реакции восстановления 377
в которой преобладали продукты 1,4-присрединения [1540].
Метиловый эфир сорбиновой кислоты дает в основном метиловый
эфир гранс-3-гексеновой кислоты (%) и гранс-2-гексеновой кис-
доты ОД), который является основным продуктом в отсутствие
межфазного катализатора [1724]. Межфазные катализаторы и
мицеллообразующие агенты увеличивают скорость реакции и
оказывают стабилизирующий эффект на те катализаторы,
которые обычно разлагаются на неактивные частицы за несколько
часов. В качестве катализаторов, в этих работах использовались
Me4NCl и ТЭБА, т. е. достаточно гидрофильные соединения.
Предварительные опыты в лаборатории авторов этой книги
показали, что более липофильный Hep4NBr вызывает еще
большее увеличение скорости реакции.
Восстановление ароматических (в том числе
гетероциклических) хлоридов или бромидов водными формиатами в
присутствии катализатора гидрирования и межфазного катализатора
описано в патенте [553]. Примером является восстановление
о-хлорнитробензола, который далее дегалогенируется до
анилина. Эта реакция осуществляется на поверхности раздела фаз,
о чем свидетельствует тот факт, что анионные
поверхностно-активные реагенты также оказывают каталитическое действие.
Другая группа исследователей [1616] использовала систему
муравьиная кислота/триэтиламин при 100°С для селективного
восстановления с помощью Pd/C одной из нитрогрупп до
аминогруппы в полинитробензблах. Примерами являются: 3-нитро-
анилин (77%), 2-амино-4-нитрофенол (57%), метил-З-амино-5-
нитробензоат (65%). Подобная же смесь реагентов была
использована: а) для восстановления фенила или двойной связи
в сопряженных алкинах с образованием цыс-алкенов и алканов
(48—84%) и б) для гидрогенолиза третичных алкиламинов
(61—93%) [1617]:
R2N—СН2—C(CHS)=CH—СНЙ—Ph *- (СН3)2С=СН—CHa—Ph (66%)
В другой методике были использованы водный формиат натрия,
липофильные аммонийные соли в качестве межфазных
катализаторов и RuCbfPPhsb как катализатор, гидрирования в
следующем соотношении:
Ки-кат:МФ-кат:субстрат: формиат = 1:2:100:1000
Растворителем в этих опытах был о-дихлорбензол.
Эффективность межфазных катализаторов уменьшалась в следующем
порядке [1747]: Нех^ШЭО^аликват 336>(Ci0H2i')4NBr>
>Ви4РВг>ТЭБА. Следует отметить, что дигидрофосфинат
(Н2РО2~) также экстрагируется в межфазных условиях с
помощью ТЭБА и используется как источник водорода при вое-
378 Глава 3. Практическое использование МФК
становлении алкинов до цмс-алкенов на Pd/C,
модифицированном Hg2+ [1853].
NO2
oj
NaOaCH,
ТЭБА.
Схема
Pd/C, NaOH
Зч, кипячение'
3227
NH2
(o)
Мощным восстанавливающим агентом, с которым легко
работать, является формамидинсульфиновая кислота (I),
получаемая при окислении тиомочевины пероксидом водорода:
h2n—c-so2h
II
NH
R-S-S-R l/NaOH> 2 R—S-H
като/шлшвр
Rl—S— Rg "NaOH) R-l^-S—R2
|| катализатор
N—Tos
Схема 3.228
Баргоньо и др. [550] показали, что при использовании
1,8 экв. I, 5,6 экв. 3,7 н. NaOH и небольших количеств трибу-
тилгексадецилфосфонийбромида восстановление дисульфидов
происходит с выходом 60—90% без растворителя при80°С.
Аналогично N-тозилсульфинимины восстанавливаются при 70 °С за
3—Б ч при использовании в качестве органической фазы диизо-
пропилового эфира. Результаты восстановления в условиях
МФК превосходят результаты других известных методов по
выходам, простоте проведения реакций и легкости обработки.
Для восстановления сульфонатов до дисульфидов была
использована комбинация полифосфорной кислоты, йодистого калия
и Bu4NI [1854].
3.22. Реакции окисления
Многие неорганические окислители могут быть перенесены
в органическую фазу с помощью катализаторов. Нужно
помнить, однако, что в зависимости от природы окислителя
реакция окисления требует присутствия ионов Н+ или ОН~. Извест-
3.22. Реакции окисления 379
но несколько случаев, когда использование метода межфазного
катализа не имеет преимуществ перед традиционными
методами окисления, например в случае сульфидов и сульфоксидов
[615].
3.22.1. Окисление перманганатом
Водный раствор КМ11О4 — хорошо известный окислитель для
многих соединений. Обычно используется довольно большой
избыток (более чем 100%) перманганата. Это объясняется тем,
что в обычных условиях КМпО4 частично разлагается до
диоксида марганца с выделением кислорода. Раствор КМпО4 в 0,04 н.
серной кислоте разлагается приблизительно в 20 раз быстрее,
чем нейтральный раствор. Однако щелочная среда, как и
присутствие диоксида марганца, также ускоряет распад. Этот
процесс имеет автокаталитический характер. Поскольку продукты
окисления часто прочно адсорбируются, то в большинстве
препаративных методик объемистый коричневый осадок
многократно промывают значительным количеством растворителя или
даже экстрагируют в аппарате Сокслета.
Метод межфазного каталитического окисления с помощью
КМпО4 свободен от этих недостатков. В качестве катализаторов
для перманганатного окисления в неводных растворителях с
одинаковым успехом используют четвертичные аммониевые и
арсониевые соли, а также краун-эфиры*. Твердый КМпО4
можно солюбилизировать, например, в бензоле при 25 °С при
перемешивании с эквимолярным количеством дициклогексано-18-кра-
уна-6. Раствор, приготовленный таким образом (его называют
«малиновым бензолом»**) можно довести упариванием до
концентрации 0,06 моль/л [557]. Среди различных краун-эфиров со-
любилизирующая способность увеличивается в следующем ряду:
18-краун-6<дибензо-18-краун-6<бензо-15-краун-5 <
дициклогексано-18-краун-6. Сначала предполагали, что для
приготовления органических растворов МпО4~ в неполярных средах при-
* В качестве альтернативных окислителей можно упомянуть тетраалкилам-
монийперманганаты. Их считали устойчивыми соединениями и использовали
как окислители в органических растворах в мягких условиях [974, 1182, 1183,
1646, 1651, 1667, 1668]. Однако позднее были сообщения [1176, 1283, 13071
о возможности их бурного самовозгорания и даже о взрывах.
** Способность давать малиновую окраску в органической фазе можно
использовать как быстрый диагностический метод проверки степени комплексо-
образования и экстрагируемости. Таким образом были испытаны в системе
хлороформ/вода новые пиклические лигаиды различного объема и гибкости
с пятью кислородными донорными атомами и одним внешним циклическим
гетероатомом (например, азотом пиридина, кислородом N-оксида пиридина илн
метиламина) [558].
Таблица 3.27. Окисление с помощью КМпО4 в условиях межфазного
катализа (органические соединения обычно растворены в бензоле)
Реакция
Катализатор
КМпО,
Выход, % Литература
Олефин—*-кислота, кетон или альдегид
Стильбен—«-бензойная кислота
Децен-1—>-ноиановая кислота
Октеи-1—>-гептановая кислота
Циклогексен—>-адипиновая кислота
а-Пинен—>-г|«с-пиноновая кислота
R—СН=СН2—->-R—COOH
R—C = CH—>-R—COOH
9-Октадецен—>-нонановая кислота
сн, сн3
сн,
СН=СН—СНз
Ph
Мео' ^ОМе
с.н,
Аг—СН—О(СН2).,—СН=С
Другие примеры
Дициклогексано-18-краун-6
Аликват 336
Аликват 336
Аликват 336
Дициклогексано-18-краун-6
Дициклогексано-18-краун-6
Дицнклогексано-18-краун-6
Аликват 336/бензол/НОАс
Алнкват 336/петролейный эфир/НОАс
Bu4NBr, Ci6HiiNMe3Br
Аликват 336
Bu4NBr
Твердый
Водн. раствор
Водн. раствор
Водн. раствор
Твердый
Твердый
Твердый
Водн. раствор
Водн. раствор
Водн. раствор
Водн. раствор
Водн. раствор
ОМе
2_._соон Аликват 336
Различные
Водн. раствор
Водн. раствор
100
53
95
91
81
78
100
100
100
100
80
557
38, 39
559
4
559
44, 50
557
557
960
960, 1184
1003
69
60
560
561
563
J626 '
Спирт, альдегид—^кислота
Ph—CHaOH—*Ph—COOH
«■октановая кислота
■>гептановая кислота
—>-пипероннловая кисло-
►кислота
Октанол-1—
Гептанол-1-
Пиперональ-
та
п-Хлорбензальдегид-
Ароматические боковые цепи
Аг—СНз^Аг—СООН
Мезитнлен—>-тримезнновая кислота
и аналогичные реакции
Реакции других типов
Ph—СН2—CN—>-бензойная кислота
Бензгидрол—>-бензофенон
,он
'он
Олецловый спирт—^эритро-
кантриол-1,9,10
Элаидиловый спирт—>-грео-октаде-
кантриол-1,9,10
Другие примеры
Циклогексадиен-1,4—>■ бензол
Триазолин—>-триазол
R—CsC^R'^R—CO—CO—R'
Аликват 336
Дициклогексано-18-кр ау н-6
Аликват 336
Дициклогексано-18-краун-6
Цетилтриметиламмонийбромид
Bu4NHSO<
Дициклогексано-18-краун-6
R4NX
18-Краун-6
Аликват 336
Дициклогексано- 18-краун-6
ТЭБА
Bu4PCl, BU4NCI
BU4PCI, BU4NCI
Bu4NBr, C16H33NMe3Br, ТЭБА
Водн. раствор
Твердый
Водн. раствор
Твердый
Водн. раствор
Водн. раствор
Твердый
Водн. раствор
Твердый
Водн. раствор
Твердый
Водн. раствор+
+4О°/о-ный водн.
NaOH
Водн. раствор/NaOH
Водн. раствор/NaOH
Водн. раствор/NaOH
Дициклогексано-18-крау н-6
Bu4NCl
Адоген 464, краун-эфнры, эфиры по- Твердый
лиэтиленглнколя
92
100
47
70
65
90
78—100
80-90
97
86
100
50
40—80
40—80
45-83
40—80
559
557
559
557
475
1178
564
559
557
567
1001
1001
382 Глава 3. Практическое использование МФК
годны только краун-эфиры*. Межфазный катализ в системе
жидкость/жидкость с водным раствором КМпО4 противопоставлялся
методике, используемой в системе твердая фаза/жидкость.
Считалось, что только в первой методике можно применять как ониевые
соли, так и краун-эфиры. Однако уже с помощью простой
качественной пробы в пробирке можно показать, что, например,
аликват 336 и твердый КМпО4 немедленно образуют
окрашенный бензольный раствор даже более интенсивного цвета, чем в
случае пары дибензо-18-краун-6/КМпО4. Таким образом,
принимая во внимание высокую стоимость краун-эфиров,
использование четвертичных аммониевых солей в качестве
катализаторов более приемлемо независимо от того, применяют ли
окислитель в форме твердой фазы или в виде водного раствора.
Вместо этих катализаторов в обеих системах можно использовать
тюлиэтиленгликоль [1177].
Вернемся теперь к экстракции МпО4~ из водного раствора;
известно, что среди аммонийных солей Me4NCl вообще не
используется для экстракции. Хорошими экстракционными
свойствами обладают ТЭБА и триметилцетиламмонийбромид;
наиболее оптимальными являются тетрабутиламмонийбромид, тет-
рабутиламмонийхлорид и аликват 336 [559]. Херриотт и Пик-
кер [559] показали, что прибавление 0,02 М Bu4NBr к равным
объемам бензола и 0,02 М водного КМпО4 вызывает
немедленное окрашивание органической фазы. Применение избытка этой
аммонийной соли позволяет достигнуть почти количественной
экстракции перманганата. Растворы, полученные таким
образом, достаточно устойчивы: в течение 45 мин титр МпО4-
уменьшается всего на 17%. Такими растворами осуществляют почти
количественное фотометрическое титрование некоторых
ненасыщенных углеводородов [910]. Реакции окисления можно
проводить при перемешивании гомогенного бензольного раствора
окисляемого вещества с малиновым бензолом при 25 СС. Быстрое
(в течение нескольких минут) обесцвечивание наблюдается при
добавлении олефинов; для окисления толуола требуется около
7$ ч [559]. Однако удобнее исключить предварительное
образование раствора реагента в каталитическом процессе. Некоторые
из реакций окисления олефинов и спиртов, приведенные в
табл. 3.27, являются сильноэкзотермическими. При добавлении
некоторого количества уксусной кислоты в органическую фазу
опасность переокисления становится минимальной [960]. Тем
не менее окисление алкильных групп в ароматических
соединениях требует температуры выше 60 °С и/или более
продолжительного времени реакции. Широко исследованы были следую-
* С помощью 18-крауна-6 можно солюбнлизнровать также и NaMnO4
19111
3.22. Реакции окисления 383
щие классы соединений (см. табл. 3.27):
R-CH=CH—R' v R-XOOH + HOOC—R'
R—СН2ОН v R—СООН
ArCHs »- Аг—СООН
Аг—СН2—R >- Аг—СО—R
Типичная методика приводится ниже [559]. См. также
«Синтезы органических препаратов» [1850]. Следует упомянуть, что
при использовании системы водный КМпО4/аликват 336
качественная проба на ненасыщенные жиры дает лучшие результаты,
чем в отсутствие катализатора [1271].
Окисление олефинов. К раствору 4,8 г (0,03 моля) КМпО4 в
50 мл Н2О при перемешивании и охлаждении водой добавляют
раствор 0,01 моля олефина в 30 мл бензола, содержащий 0,5 г
Bu4NBr. После перемешивания в течение 3 ч при комнатной
температуре реакционную смесь обрабатывают NaHSO3l
подкисляют, отделяют органический слой, высушивают его и
удаляют растворитель.
При использовании системы твердый КМпО4 или NaMnO«/
/краун [911] необходимо тонкое измельчение окислителя и
высокоэффективное перемешивание, чтобы предотвратить
торможение реакции образующимся МпОг. Некоторые автбры
предлагают проводить реакцию в шаровой мельнице.
Рассмотрим вкратце пример, иллюстрирующий, как можно-
с помощью экстракции выделить и перенести гидрофильные
катионы в органическую фазу с последующим окислением в ней.
Бензилонийбромид (антихолинергический препарат)
представляет собой четвертичную аммониевую соль, содержащую в
эфирной части остаток бензиловой кислоты. Для количественного-
определения его в плазме крови сначала необходимо
экстрагировать этот гидрофильный катион в органическую фазу. Это
достигается добавлением липофильного противоиона в виде 2-гид-
рокси-3,5-ди-трет-бутилбензолсульфоната натрия. Прибавление
раствора КМ11О4 приводит к межфазному окислению бензило-
ниевой соли до бензофенона, содержание которого оценивается;
методом газовой хроматографии [1737].
Некоторые исследователи использовали перманганатное-
окисление в условиях межфазного катализа для менее обычных
субстратов. Например, Димрот [561] превращал боковую цепь-
фосфорсодержащего гетероцикла, показанного на схеме 3.229,
в соответствующий альдегид. В этих условиях гетероцикл
неокисляется. Реакцию, приведенную на схеме 3.229, можно
осуществить и при использовании хромовой кислоты. Окисление
сульфидов (и особенно сернистых гетероциклов с малым
размером цикла) до сульфонов с использованием водного или
твердого КМпО4 при комнатной температуре в присутствии боль-
384 Глава 3. Практическое использование МФК
шинства межфазных катализаторов имеет, кажется, лишь orpat
ничейное применение. Полезно добавление небольших количеств
уксусной кислоты в CH2CI2, используемый в качестве
растворителя [1517].
нсн, Hv^° °
Беюоя,8оди.к.МпО«
BiijNBr
МеО
Схема 3.229
Несопряженные гексадиены количественно превращаются в
ароматические соединения при межфазном окислении пермангаг
натом в системе твердая фаза/жидкость, а их сопряженные
аналоги Ь этих условиях не реагируют [1179]. В системе бен-
зол/водиый KMnCVBtuNCl из Ш-4,5-дигидро-1,2,3-триазоловоб-
разуются ароматические гетероциклы [1180]. Однако, когда эти
триазолины' содержат 6-грег-аминогруппу, боковая цепь
окисляется легче, чем кольцо [1181]:
R
| сно |
Аг Аг
Схема 3.230
'• Авторы настоящей книги не видят особых преимуществ в ра?
<5оте с предварительно выделенными солями R4NMn04, особенно
если обращение с ними небезопасно (см. первое примечание
на с. 379), Тем не менее описаны реакции следующих типов,
Осуществленные в гомогенных растворах (пиридине, уксусной
кислоте илн дихлорметане) с использованием различных чет,
вертичных аммониевых перманганатов:
R—CH2—OR' *■ R—CO—OR' [1183]
R3CH *• RSC—ОН [1182, 1307]
RCHO v R—COOH [1667]
RCH2—NRa' > RCO—NR2' [1646]
RCHS—OH+ CHaCla »- RCO—O—CH2—O—COR [1651]
R—S—R' >■ R—SO2—R' [1668]
R—CH=CH—R' »- R—CHOH—CH—OH—R' [133P]
(реакция при —70 °C)
R—CHa—R' *■ R—CO—R' [1182, 1307, 1609]
(единичные случаи, низкие выходы) -
3.22. Реакции окисления 385
В присутствии минеральных кислот (например, полифосфорной
кислоты) третичные амины являются удовлетворительными
межфазными катализаторами для окисления с помощью КМ11О4.
Так, Старке и сотр. [600] окисляли тетрадецен-1 до тридекано-
вой кислоты, используя в двухфазной реакции тридодециламин
в качестве катализатора. Макк и Дарст [564, 565} получили
о-хинон при перемешивании в течение 30 мин при комнатной
температуре раствора 3,5-ди-грег-бутилпирокатехина в метилен-
хлориде в присутствии 18-крауна-6 и порошкообразного КМ1Ю4:
X
щелочной KMnO^ Y-OH
катализатор L_qH
Схема 3231
цис-Гидроксилирование олефинов щелочным КМ11О4
является обычной реакцией с низкими выходами продуктов, за
некоторыми исключениями, например при окислении длинноцепочеч-
ных мононенасыщенных жирных кислот. Поэтому до развития
межфазного катализа стандартные методики включали
использование тетраоксида осмия, обработку иодом или ацетатом
серебра во влажной уксусной кислоте по Вудворду. Вебер и Ше-
пард [567] осуществили цые-гидроксилирование олефинов в
условиях межфазного катализа* с выходом- около 50%- Низкие
выходы наблюдаются только в том случае, когда образующийся
гликоль хорошо растворим в воде (например, цые-дигидрокси-
циклогексан). Дополнительно образуется небольшое
количество кислоты. Приложение этого метода к более сложным
молекулам см. в работах [878, 1237, 1263, 1666]. Когда цис-гилр-
оксилирование катализируется применением хиральных солей, то
наблюдается невысокая оптическая индукция [1761].
Тщательный подбор условий реакций позволяет проводить их достаточно
селективно [1237, 1263]. Нейтральный перманганат дает смесь
двух продуктов (схема 3.232).
Окисление дизамещенных алкинов перманганатом в условиях
межфазного катализа может протекать в зависимости от
условий по двум направлениям. При использовании твердого
КМпО4 и адогена 464, дициклогексано-18-крауна-6 или диметил-
полиэтиленгликоля в кипящем дихлорметане, содержащем
немного уксусной кислоты, были получены а-дикетоны с выходом
40—80% [1184, 1308]:
R—C=C—R' > R—CO-CO-R'
* Субстрат, растворенный в СН2С12, ТЭБА, 40%-ный водный NaOH
и твердый КМ11О4 смешивают при О "С и интенсивно перемешивают до окон-
и о Шла noQrniira
чания реакции.
386 Глава 3. Практическое использование МФК
но«
83%
81%
сно
Схема 3.232
В тех же условиях, но при использовании водного перман-
ганата происходит окислительное расщепление. В этом случае
один из фрагментов имеет на один углеродный атом меньше, что
указывает на возможное образование енола и а-кетокарбоновой
кислоты в качестве промежуточных продуктов [1308]:
R—C=C—CH2—R' -—>■ R—CO—C(OH)=CHR' >■
R—CO—COOH + HOOC—R' > R—СООН + НООС—R'
В заключение следует отметить, что метилтрифениларсоний-
хлорид был использован в качестве межфазного катализатора
задолго до того, как была сформулирована концепция
межфазного катализа. Джибсон и Хоскинг [569], развивая ранние
наблюдения об экстракции МпО4~ в СНС13 и нитробензоле
[668], встряхивали водный КМ1Ю4 с органическими растворами
спиртов, олефинов, 1- и 2-нитропропанов и муравьиной кислотой
в присутствии (CH3)Ph3AsCl и наблюдали окисление. трет-Ъу-
танол, толуол, этилацетат, эфир и кетоны в этих условиях не
вза имодействуют.
Рассмотренные реакции представляли собой гомогенные или
межфазные процессы. Окисление октена перманганатом было
исследовано также с использованием катализаторов иного
типа [570]. В обычных условиях стеарат и лаурат натрия сильнее
увеличивали скорость реакции, чем некоторые межфазные
катализаторы. Это является убедительным доказательством того,
что окисление перманганатом может осуществляться на поверх-
дости и/или проходить как мицеллярная реакция. Подобный
эффект — сильное увеличение скорости реакции — был
обнаружен, когда к системе пиперональ/водный КМпО4 был добавлен
триметилцетиламмонийбромид [475]. В отличие от больших
симметричных четвертичных аммониевых солей триметилцетил-
аммоний даже частично не солюбилизирует твердый КМпО4
3.22. Реакции окисления 387
в бензоле и не экстрагирует его из водного раствора, так что
в этом случае механизм межфазного катализа, по-видимому, не
действует.
3.22.2. Роль пероксида водорода в межфазных реакциях
Как известно, экстракция ионов ОН~ из водной фазы в
органическую среду проходит с трудом. Поэтому можно ожидать,
что ион НОг~ также будет плохо экстрагироваться, или,
другими словами, анионы, входящие в состав катализатора, такие,
как хлориды или бромиды, будут иметь большую константу
экстракции, чем моноанион пероксида водорода. Однако было
найдено, что экстракция частиц с окислительными свойствами
из 35%-ного (~10 М) Н2О2 в метиленхлорид легко
осуществляется при использовании некоторых катализаторов [57]. Иодо-
метрическое титрование органической фазы после установления
равновесия с катализатором приводит к результатам,
представленным в табл. 3.28.
Таблица 3.28. Иодометрическое титрование Н2О2
в СН2С12 после установления равновесия
в присутствии катализатора (5 мл 35%-иого
20 мл CHgCia и 2 ммоля катализатора)
Катализатор
ТЭБА
(Bu4N)2SO4
BU4NHSO4
B114NCI
Bu4NBr
BiuN-P-нафталннсульфонат
Алнкват 336
Hex4NBr
Hep4NBr
Oct4NBr
Эквивалентное
соотношение НгОг/ка-
тализатор
0,013
0,09
0,10
0,30
0,68
0,92
0,88
0,99
1,00
1,00
Совершенно очевидно |(и этого следовало ожидать), что
гидрофильные катализаторы в этом случае мало приемлемы.
Кроме того, не может быть пригодным н катализатор,
находящийся преимущественно в водной фазе. Как анион, так и катион
катализатора влияют на его линофильность (гл. 1), и чем более
липофилен катализатор, тем сильнее его экстрагирующая спо-
388 Глава 3. Практическое использование МФК
собность. Небольшие добавки серной кислоты не вызывают
никакого изменения в экстракционных свойствах Н2О2, но
добавка 1,25 ммоль гидроксида натрия (меньше, чем молярное
количество катализатора и намного меньше количества Н2О2)
уменьшает наполовину экстрактивный окислительный
эквивалент. Поскольку Н2О2 является более сильной кислотой (рКа
11,6), чем вода (рКа 15,7), эти наблюдения свидетельствуют
против экстракции НО2~, но в пользу переноса пероксида
водорода в виде сольвата катализатора. Дополнительным
доказательством этого служит то, что в органической фазе при
экстракции Н2О2 с помощью тетрагексиламмонийбромида было
обнаружено около 100% теоретического количества бромида [57].
Следует отметить, что ранее были выделены кристаллические
комплексы [R4N+Br--H2O2] и [R4N+C1--H2O2] [1736]. Для
межфазного окисления пероксидом водорода некоторых
типичных олефинов описано применение сложного катализатора,
представляющего собой МФ-катализатор плюс соединения
тяжелого металла [39, 599]. Использовался тридецилметиламмо-
нийхлорид. Он выполнял две функции: перенос пероксида
водорода в органическую фазу и предотвращение его распада на
воду и кислород. Реакции осуществлялись при комнатной
температуре в течение нескольких часов. При использовании в
качестве катализаторов OsO4, МоО3 или H2W64 из циклогексена
образовывались преимущественно продукты Е и F:
ОН
н
Если же применялись SeOCl2, V2O5, Cr2O3, TiO2, CeSO4, NiO,
MnCl2, CoCls, PtO2, FeSO4, Pb(OAc)2 или PdCl2, то основными
продуктами были G и Н, а продукт Е образовывался в
небольшом количестве. Эпоксидирование олефинов в системе 90%-ный
водный Н2Ог/диоксан при 80 °С возможно с использованием в
качестве трехфазного катализатора мышьяковой кислоты,
нанесенной на полистирол [1282]. Как альтернатива
использовалась система 30%-ный Н2О2/хлороформ. Подобные растворитель
и температурные условия применяли для высокоэффективного
окисления кетонов по Байеру—Виллигеру [1281].
Изучено окисление с использованием системы твердый
КгОг/катализатор. Этим методом халкон был эпоксидирован в
органическом растворителе, однако эта методика не имеет ни-
3.22. Реакции окисления 389
каких явных преимуществ перед существующими методами
[604]. Впоследствии было установлено [1594], что «,р>оксид
цитраля — соединение, получаемое обычным путем лишь с
небольшим выходом, — может быть приготовлено с выходом 77%
при быстром добавлении 50%-ного раствора NaOH в 30%-ном
НгО2 к охлаждаемой смеси цитраля, бензола и небольших
количеств ТЭБА. Очень интересный метод окисления двойной
связи описан Винбергом и сотр. [603]: в условиях межфазного
катализа с использованием бензилхининийхлорида (М) или бен-
зилхинидинийхлорида в качестве катализаторов были
получены оптически активные эпоксиды с превосходным химическим
выходом и с энантиомерным избытком до 25%. Эпоксидирова-
ние соединений I, К и L проводили в системе 30%-ный Н2О2/
/разбавленный раствор гидроксида натрия в присутствии
каталитических количеств М при интенсивном перемешивании то-
луольных растворов в течение 24 ч при комнатной температуре.
Другие примеры этой реакции [1250, 1369, 1589] появились
позднее (см. дискуссию в разд. 3.1.5).
/ \ /- А,'
С—Аг'
МеО
Схема 3.234
390 Глава 3. Практическое использование МФК.
Механизм не обсуждался, однако поскольку реакция
катализируется основанием, а последнее не переносится в
органическую фазу, то возможно, что енон захватывает НО2~ на
границе фаз. Образующийся енолят-анион остается закрепленным на
межфазной поверхности до тех пор, пока катион катализатора,
обладающий хиральностью, не снимет его с поверхности.
Происходит мгновенная потеря ОН-, и образуется хиральный эпок-
сид. Такая модель не требует переноса окислителя в
органическую фазу. Продолжительность реакции скорее всего
определяется липофильным характером М.
Реакция между различными фенолами и системой твердый
КгОг/аликват 336 в метиленхлориде определяется, очевидно,
основным характером реагента, а не его окисляющей
способностью: атака фенолята на растворитель ведет к образованию
метилендифенольных эфиров [604, см. 234]:
KaOa/CHsa
АгОН »- АгО—СН.^ОАг
катализатор
Наконец, следует упомянуть о галогенировании олефинов в
условиях межфазного катализа. При этом галоген генерируется
и расходуется непосредственно в ионной форме без
возникновения побочных радикальных реакций. Этот метод удобно
применять в тех случаях, когда использовать элементарный хлор по
каким-либо причинам затруднительно или невозможно. К
раствору алкена в тетрахлориде углерода при охлаждении льдом
и перемешивании с концентрированной соляной кислотой и
небольшим количеством ТЭБА добавляют по каплям 30%-ный
Н2О2 [869]:
R1CH=CH~R!! + 2HX + Н2О2 >■ R1CH(X)—CH(X)—R
Твердая натриевая соль гидропероксида кумола
используется для окисления и гидратирования 2-арилциннамоилнитрилов
в бензоле в присутствии ТЭБА в качестве катализатора [602]:
Ph— С(СНз)а—OO-Na++Ar— CH=C(Ar')— CN >- Аг—СО—СН(Аг')—CONHa
3.22.3. Реакции с супероксидом калия*
Окисление органических субстратов с помощью супероксид-
иона представляет общий интерес, во-первых, по препаративным
соображениям, а во-вторых, из-за той функции, которую, как
оказалось, выполняет О2"" в биологических системах.
Супероксид-ион имеет достаточно сложную химическую природу,
потому что он может проявлять себя как окисляющий или восста-
Специальиые обзор»! по химии КО2 см. [913, 1339, 1844].
3.22. Реакции окисления 391
навливающий реагент в такой же мере, как и нуклеофил:
(1) R~ + O2*~ *-R--f-Oa*- окисление
(2) R-+O2-~ »-R~-J-Oat восстановление
(3) RX + <V~ »-R-7-O,-+ X" замещение
Реакция (1) требует протонов для стабилизации пероксид-
ного дианиона. Однако в протонной среде происходит распад:
(4) 2О2-—*(у
Таким образом, использование этого дешевого реагента
более или менее ограничено необходимостью применения апротон-
ных растворителей, но в отсутствие краун-эфиров он частично
растворим только в одном органическом растворителе — ДМСО.
Для приготовления бледно-желтого 0,15 М раствора КОг
может быть использован 0,3 М раствор дициклогексано-18-крау-
на-6 в ДМСО [576]. В большинстве случаев использование
бензола более целесообразно, чем ДМСО, потому что
применение бензола устраняет потенциально существующие сложности
при использовании ДМСО-аниона [677]. Комплекс КО2 с ди-
циклогексано-18-крауном-6 растворим в бензоле до
концентрации 0,05 моль/л £577]. В присутствии 18-крауна-6 могут быть
получены растворы КО2 в ДМФА, ДМЭ и даже эфире [578].
Стабильные растворы тетраэтиламмонийсупероксида в апротон-
ных растворителях были приготовлены путем
электрохимического генерирования [579, 587], а недавно показано, что
супероксид может быть активирован межфазным катализатором
аликватом 336 [1016]. Ряд исследователей-использовали нуклео-
фильные свойства супероксида. Сравнение реакционной
способности KI и КО2 (0,5 М) по отношению к 1-бромоктаиу (0,5 М
в ДМСО) в присутствии Я8-крауна-6 (0,05 М) показало, что
периоды полупревращения равны примерно 20 ч и 45 с
соответственно [580]. Таким образом, супероксид является «супериук-
леофилом>. Разные авторы сообщают о различном строении
продуктов реакции алкилгалогенидов и алкилсульфонатов в
зависимости от условий.
Джонсон и Найди [581] синтезировали диалкилсуперокснды,
используя стехиометрические количества алкилбромйдов, ди-
циклогексано-18-крауна-6 и КО2 в бензоле (3—6 ч при
комнатной температуре). Спирты и олефины образовывались в
незначительной степени. Если использовать только 0,1 экв: краун-эфи*
ра, выходы несколько снижаются, а время реакции
увеличивается. В противоположность этому при избытке КО2 в ДМСО
Сан Филиппо, Черн и Валентин [580] получили в качестве
основных продуктов спирты, а карбонильные соединения только как
примесь. Взрывоопасные беизо-1,2-диоксены были приготовлены
из о-быс-галогенометилароматических соединений в системе
392 Глава 3. Практическое использование МФК
КО2/дициклогексано-18-краун-6 {927]. Кори и др. [578, см.
также 861, 925] синтезировали спирты, проведя эту реакцию в ап-
ротонной среде, содержащей ДМСО. Интересно и важно с
препаративной точки зрения то, что первым актом всех этих
превращений должно быть Б^-замещение [578, 579, 580, 681, 861]:
СНз / СНз Л
|- СвН13СН—Вг ко'/кРа«н > \(S, S)- СвН,з—СН—О/г
свнв
СНз / СНз
[58Ц
СНз v СНэ
(S)- СвН,з—СН—OTos К<У.КГРп'|Н) (R)" СвН13—СН—ОН [5801
/7-гполуолс1)яьфона1Л холестерина Дм°о/Ьмэ/дмф^ 3-эпихолестерин 1578J
КОа/18-крсун-6
ДМСО/ДМЭ/ДМТА
MesO H
Схема 3.235
Сообщалось [1742] об очень быстром окислительном
дехлорировании СО4, СНС13, СН2С12 и ДДТ (только
алифатического С1) в ДМФА или ДМСО в присутствии Et4NC104 как
катализатора. Неудачными были попытки расширить этот тип
превращений на получение продуктов внедрения дигалокарбенов в
углеводороды с образованием, например, R3C—СНХ2 [1894].
Основные свойства КОг проявились в присутствии адогена 464 в
качестве катализатора: из СНС13 в бензоле как растворителе
с низким выходом был получен :СС12 [1895].
Последовательность отдельных стадий и взаимодействие
образующихся интермедиатов в предлагаемых механизмах
обсуждены также в работах [59, 582, 583, 587, 1188, 1189, 1191, 1350].
. В табл. 3.29 представлен подбор соответствующих работ.
Выходы первичных диалкилсупероксидов сравнимы или лучше,
чем в традиционных методах, а выходы вторичных субстратов
(кроме циклогексильного) выше. Достаточно интересен способ
приготовления днацилпероксидов из КО2 и соляной кислоты,
легко осуществляемый даже без использования краун-эфира в
бензоле [585]. Высокий выход инвертированных спиртов из
тозилатов или. мезилатов, приведенный в нижней части.
3.22. Реакции окисления
Таблица 3.29. Реакции алкнлгалогенидов и сульфонатов
с супероксидом калия
Субстрат
Условия8
Выход,
диалкил-
пероксид
спирт
фин(ы)
кетой
или
альдегид
H-C5-7Hl,_,s— ВГ
к-С18Нз7-Х
(X=Br, OTos, OMs)
Циклогексил—Br
Циклопеитил—Br
С6Н13СН(СНз)—X
(X=Br, OTos, OMs)
H-CeHi7X
(Х=С1, Br, I, OTos)
н-С6Н13СН(СН8)—X
(X=C1, Br, I, OTos)
Ph—CHjCl
СзН7С(СНз)2—Br
A
A
A
A
A
Б
Б
Б
Б
53-56
46—77
42
44—55
б
—
—
21—42
0—13
46—756
36—75
41
20
—
—
67 —
24 —
14—37 —
0—3 1—12
12—48 1
— 6
30 —
581
581
581
581
581
580
580
580
580
OTos
Tos
Br
В
В
он
-ОН
а
— 95
— 96
— 100
— — 578
— — 578
— — 578
а Условия А: эквимолярные количества КО], дициклогексаво-18-храува-б, субстрата
в бензоле; 3—6 ч при комнатной температуре. Условия Б: 3,33 иноля субстрата, 10 ммо-
лей КОа, 1 ымоль 18-крауна-6 в 20 мл сухого ДМСО; 1,5—3 ч при комнатной
температуре. Условия В: 4 экв. КО2 н 4 экв. 18-крауна-б в смеси ДМСО/ДМЭ при комнатной
температуре.
6 Другие авторы [11911 нашли, что в идентичных условиях главным продуктом
были перокснды.
табл. 3.29, имеет с препаративной точки зрения особую
значимость. В некоторых случаях, однако, реакция может
сопровождаться отщеплением [578]. трет-Бутилбромид дает
одновременно продукты замещения (трет-бутанол) и элиминирования (изо-
бутен) [578]. Аналогично ведут себя циклогексильные произвол-
594 Глава 3. Практическое использование МФК
ные, представленные на схеме 3-236. .N-Хлорпиперидин и N-хлор-
пирролидин образуют циклические имины при использовании
чжстемы КОг/краун-эфир в диэтиловом эфире [977, 1341].
С1
Схема 3.236
Фраймер и Розенталь [589] наблюдали нуклеофильное
замещение галогена анион-радикалом Оу" в ароматических циклах,
активированных электроноакцепторными заместителями,
такими/как нитрогруппа. Избытек как КО2 (4-кратныЙ), так и ди-
циклогексано-18-крауна-6 (2-кратный) приводит к быстрой
(несколько часов) реакции в бензоле при комнатной температуре:
NO2
NO2
NO2
NO2
NO2
NOa
OH
МрногалогендинитросЬединения реагируют в 100 раз
быстрее моногалогенмононитрозамещенных аналогов. Эксперименты
с.мейерым *Ю показали, что первой стадией этих реакций.явля-
8.22. Реакции окисления 395
ется электронный перенос от Оу^ к замещенному бензолу с
образованием анион-радикала, который впоследствии гасится
молекулярным кислородом. Таким образом, эта реакция не
является обычным нуклеофильным ароматическим замещением.
Исследуя совершенно различные типы реакций, Сан Филип-
по с сотр. [586, 1194] получили высокие выходы в реакции
расщепления сложноэфирных связей избытком КОг в присутствии
каталитических количеств (7з моля) 18-крауна-6 при энергичном
перемешивании в бензоле от 8 ч и редко до 140 ч и
последующей обработкой водой. Оказалось, что такое расщепление на
спирт и кислоту проходит со многими сложными эфирами
первичных, вторичных и третичных спиртов, а также фенолов и
тиолов. Также расщепляются фосфаты. При использовании в
качестве растворителя ДМСО время реакции сокращается.
Возможность разрыва связи кислород—алкил в результате
воздействия супернуклеофила рассматривалась, но была отвергнута,
по крайней мере для вторичных спиртов, так как наблюдалось
обращение конфигурации на 99% [586]. Простые амиды й тщт-
рилы не реагируют.
Ряд исследователей [588] проверили возможность
применения как окислителя системы избыток твердого КО$/ката:литйче-
ские количества 18*крауна-6/бензол на примере гидразинов,
гидразонов и родственных соединений. В большинстве случаев
реакцию ведут при перемешивании в течение 24 ч. Монозаме-
щенные гидразины, особенно арилгидразины, превращались в
продукты, не содержащие азот (частр в углеводороды),
вероятнее всего, в результате свободнорадикального процесса. 1,2-Ди-
арилгидразины дают соответствующие азосоединения;
окисление 1,1-дизамещенных гидразинов приводит к N-нитрозосоеди-
нениям. Гидразоны ароматических кетонов образуют азины.
Анилин и его производные превращаются в производные
азобензола, а о-фенилендиамин дает смесь о-нитрозоанилина,
о-нитроанилина и о,о'-диаминоазобензола [1218].
Тетрациклон (А) окисляется при действии избытка КОг и
молярного количества дициклогексано-18-крауна-6 в бензоле до
2-гидрокси-2,4,5-трифенилфуранона-3 (В) [577]. Предложенный
механизм этого превращения основывается на результатах
изучения халконовых систем с меченым 18О [590] (схема 3.238).
В аналогичных условиях халконы С расщепляются с
образованием замещенных бензойных кислот (D) и арилуксусных
кислот (Е) [590]. Краун-эфиры в реакции А-»-В выступают как
доноры протонов и необратимо разрушаются. Родственные'
соединения, такие, как флуоренон, тетрафенилэтилен и 2,5-дифе-
нилфуран, в аналогичных условиях образуют лишь минимальные
количества полярных соединений, а алкилзамещенные оле-
фины и ароматические углеводороды не реагируют: Однако суб-
396 Глава 3. Практическое использование МФК
страты, содержащие активные атомы водорода, например 9,10-
дигидроантрацен, 1,3- и 1,4-гексадиен, антрон и подобные
соединения окисляются [591] (отношение КО2/18-краун-6/субстрат
равно 2:2:1, в сухом ДМСО).
о
II
Аг—С— СН=СН—Аг'
С
КОа
АгСООН + НООС—СН2—Аг'
D Е
Схема 3.238
2,5-Ди-грег-бутилгидрохинон быстро превращается в хинон
[591], а 2,6-ди-грег-бутилфеноксильные свободные радикалы
восстанавливаются до феноксидов [592] (схема 3.239). Тем не
менее совершенно очевидно, что система КО2/краун способна
преподнести еще много неожиданностей при изучении ее
реакционной способности. В сухом бензоле при пиридине витамин
Ki и его производные образуют нафтохинон-2,3-оксиды (F) и
фталевый ангидрид [1334].
он
Схема 3.239
Обнаружено [593] также образование семихинонных анион-
радикалов при окислении 1,2- и 1,4-дигидроксиаренов и при
восстановлении 1,4-хинонов в системе К02/дициклогексано-18-кра-
ун-6/ТГФ. В случае о-дигидроксиаренов происходит дальнейшее
■окисление семихинонов до дикарбоновых кислот.
3.22. Реакции окисления 397
Несмотря на упоминавшееся выше образование
соединения F, 4,4,6,6-тетразамещенные циклогексеноны инертны к
супероксиду. Однако при действии водородсодержащих кислот
циклогексеноны подвергаются окислению [1260].
о о
+ „тример
Схема 3.240
Обедненные электронами олефины типа а-нитро- и а,а-ди-
цианостирола расщепляются до соответствующих бензойных
кислот или кетонов при перемешивании их в сухом бензоле с
системой избыток КОг/избыток 18-крауна-6 в темноте [898].
Предполагается первоначальное образование радикал-аниона,
который атакуется предпочтительно О"г~.
При действии системы КО2/18-краун-6 в эфире при
комнатной температуре (перемешивание в течение 4—6 ч) вторичные
N-хлорамины превращаются в имины и далее в карбонильные
соединения [977]:
н3о+
—ЩСЩ1 » R4?*C=N—R1 *- R1R2C=O
Бензилам иды гидроксилируются в п- и о-положения при
помощи КО2/краун в бензоле ;[1846]. В последних работах
сообщалось об окислении 3-гидроксифлавонов [1847], об
образовании серусодержащих пероксидов в процессе окисления сульф-
оксидов, об окислении фосфинов и олефинов [1848], о
расщеплении гидроксикумаринов и образовании лактолов из 2-гидрокси-
2,5-циклогексадиенонов [1849].
Модель а-токоферола была окислена действием системы
КО2/дициклогексано-18-каун-6 в ацетонитриле [1193]:
НО
он
Схема 3241
398 Глава 3. Практическое использование МФК
I
Новое окислительное расщепление в нейтральных условиях
было обнаружено в лаборатории авторов этой книги Лисселем:
кетоны, содержащие по крайней мере один а-водородный атом,
расщепляются с помощью КО2/аликват 336 в бензоле при
комнатной температуре [1016, 1780]:
RCOOH + HOOCR' 90-95 % [1016]
НООС—(CHiV-CO—R 80-90% [1780]
соон
93% [1016]
н соон
Схема 3.242
Некоторые циклические кетоны дают со-гидроксилактоны
[1845].
Предполагалось, что при действии КОг/18-крауна-б в
бензоле на дибензоил-или дилауроилпероксиды образуется синглет-
ньгй кислород в соответствии со следующим уравнением
[1190]:
"2КО2 + RCO—О—О—COR *■ 2К+ "(
Однако впоследствии в этом стали сомневаться [1350].
Совершенно очевидно, что реакции с участием пероксидов и
супероксидов требуют дальнейшего тщательного изучения, прежде чем
будет окончательно выяснен иХ механизм. Диспропорциониро-
взние системы урет-бутилгидропероксид/98%-ный Н2Оа в бен*.
золе с образованием суперокеида, -дающего Oj, проходит с
выделением протонов. Так же осуществляется диспропорциониро-
вацие Н2О2, катализируемое основанием. В ацетонитриле
перокси-анионы реагируют с растворителем, давая в конечном
счете ацетамид. Реакции супероксидов с диацилпероксидами, хлор-
ангидридами и ангидридами карбоновых кислот проходят, очень
(сложно, однако при этом в реакционной системе генерируются
промежуточные продукты, способные образовывать эпоксиды из
олефинов. Использование в качестве катализатора аликвата 336
вместо 18-крауна-6 увеличивает скорость реакции, однако
снижает выход эпоксидов. Выходы эпоксидов сильно зависят
также от природы используемого ангидрида или хлорангидрида
J1350, 1725]. Как альтернативу, для эпоксидирования можно ис-
S.22. Реакции окисления 399
пользовать хлорангидриды сульфокислот в системе КОг/18-кра-
ун-6 в бензоле или ацетокитриле [1470].
S—S-Связи дисульфидов, тиосульфинатов и тиосульфонатов
подвергаются окислительному расщеплению с образованием
сульфиновой и сульфоновой кислот при действии КОг/18-крау-
на-6 в пиридине [1192, 1653]:
R—S—S—R, R—SO—S—R, R—SO2—S—R * R—S<V + R—SO8~
3.22.4. Хроматы
Гибсон и сотр. [569] приготовили органические растворы би-
хромата в хлороформе или нитробензоле, используя метилентри-
фениларсонийхлорид как источник противоиона. Бихромат в
противоположность MePhsAs+MnO4~ не является активным
окислителем для спиртов. Причина этого кроется, возможно,
в том, что окисление спиртов хроматом протекает через стадию
образования эфиров хромовой кислоты, которые трудно
получаются в отсутствие кислот. Недавно Хатчинс и др. [890]
экстрагировали СггОт2" (переносимый ион неизвестен) в Ьензол, СС14,
СНС1з или СН2С12 с помощью адогена 464. Эти растворы спо*
собны окислять аллильные и бензильные спирты до. альдегидов
и кетонов, однако реакция идет достаточно медленно (15—
18 ч при 55 °С). Так, первичные алканолы в этих условиях
практически инертны, а вторичные реагируют крайне медленно
[890]. Однако при содержании в водной фазе 3—10 М H2SO4
реагент можно экстрагировать в CH2CI2 с помощью тетрабутил-
аммонийбисульфата, что позволяет окислять даже первичные
спирты в течение нескольких минут [991]. Наилучшие
результаты дает небольшой избыток К2СГ2О7 в водной фазе.
Возможно, что действующим агентом является НСг&О7~\ Первинные
спирты могут быть превращены в альдегиды только в том
случае, когда условия реакции тщательно контролируются и
избыточное окисление минимальнр [991, 1196], В оптимальных
условиях реакцию ведут при температуре от 5 до 0°С, прибавляя
по каплям раствор К2СГ2О7 в 30% -ной H2SO4 и используя
в качестве катализатора Bu4N+HSO4- [1196], а в качестве рась
творителей СНС13 или СН2С12.
Добавление тетрабутиламмонийхлорида к водному раствору
СгОз приводит к образованию осадка, представляющего собой
Bu4N+HCr04^. Хотя этот реагент, по-видимому, должен быть
устойчив при комнатной температуре, однако следует принимать
меры предосторожности на случай непредвиденного взрыва.
Этот реагент окисляет в кипящем хлороформе вторичные
спирты до кетонов (3—12 ч), а аллильные и бензильные спирты до
альдегидов (1—4 ч) [1198]. Другим эффективным окислителем
.400 Глава 3. Практическое использование МФК
спиртов до альдегидов или кетонов является реагент
R^N+XCrOa", получающийся при растворении СгО3 и R4NX в ди-
хлорметане [1597]. X может представлять собой HSO4, P, С1,
Вг или CF3CO2 [1596].
Образование эфиров хромовой кислоты обеспечивается в том
случае, когда аллил- или бензилгалогениды реагируют с
системой хромат/дициклогексано-18-краун-6 (или дибензо-18-кра-
ун-6) в ГМФТА [594]. Выходы целевых продуктов достигают
80% за 2 ч при 100 °С Соответствующие спирты в этих
условиях инертны, «-а л кил бромиды реагируют очень медленно
(выход 20%). Образование альдегидов и кетонов из бензил- и ал-
лилгалогенидов возможно также при реакции в хлороформе с
предварительно выделенной солью бихромата [1197]. Реагент
[по-видимому, (ВщЫ)2Сг207] получают экстракцией дихлорме-
таном из водного раствора K2CrO4/Bu4NHSO4. Следует
отметить, что отличить эту соль от описанной выше Bu^N+HCrO^""
с помощью элементного (С, Н, N) анализа или иодметрическо-
го титрования достаточно трудно. Так, недавно описанные ре^
акции в кипящем метиленхлориде (окисление бензиловых и ал-
лиловых спиртов при помощи предварительно выделенной соли)
могут в действительности осуществляться при действии
бихромата [1337]. Система адоген 4б4/КгСг2О7 (в молярном
соотношении 2:1)/бензол была использована для предположительно
нейтрального окисления о-аллилфенолов в хромены-3. При
кипячении реакционной смеси в течение 1 ч выходы целевых
продуктов 45—83% [1234].
3.22.5. Гипохлорит
Ли и Фридман [595, 998, 1195] установили, что арилкарби-
нолы гладко превращаются в альдегиды или кетоны при
перемешивании раствора субстрата в СН2С12, содержащем 5%
BU4NHSO4 с избытком 10%-ного водного NaOCl, в течение 1—
3 ч при комнатной температуре. В отсутствие катализатора
реакция проходит в небольшой степени или вообще не идет.
R' ГИ' 1 ГЯ' 1 R
СЫ*Н ^ ^)CHNHClj [/NHj С
Схема 3.243
Для этого окисления наблюдается интересный, но до сих пор
необъяснимый эффект растворителя: подходящими
растворителями являются бензол, тетрахлорид углерода, хлороформ и ме-
тиленхлорид, однако только этилацетат увеличивает скорость
3.22. Реакции окисления 401
реакции настолько.Ччто становится возможным окисление
алифатических вторичных спиртов. Первичные амины также
могут быть окислены гипохлорит-ионом ОС1~ в условиях
межфазного катализа. Однако важными в синтетическом отношении
являются только образующие кетоны первичные амины, у которых
а-углеродный атом несет два заместителя. Монозамещенньге по
«-углеродному атому первичные амины дают преимущественно
нитрилы с примесью альдегидов в качестве побочных
продуктов. Амиды реагируют с системой ОС1~/МФ-катализатор путем
последовательных расщепления по Гофману и окисления
полученного амина. Обычно наблюдается низкий общий выход
нитрилов. Результаты представлены в табл. 3.30. Гипохлоритное
окисление может быть осуществлено также в трехфазном
каталитическом варианте с использованием специально
приготовленного полимерсвязанного катализатора [62] и полиэтиленгли-
колей в качестве обычных межфазных катализаторов [1779].
На схеме 3.244 показаны другие реакции гипохлорита в
условиях межфазного катализа. В первой схеме, предложенной
Хори и сотр. [1240], представлен привлекательный
препаративный метод для превращения кетонов в нитросоединения. Табу-
ши и Кори [1358] наблюдали комбинацию каталитических
процессов двух типов — межфазного и электронного переноса,—
действующих в одном направлении, при гипохлоритном
окислении бензилового спирта и бензилового эфира до бензальдегида
и циклогексана в циклогексилхлорид.
NOH
a no
а no,
н ко,
11240]
O2N
NOj 11358]
NaOCyKOH/НдО
толуол, Bu4NBr
R
Схема 3.244
Исследованная систета содержала метилтриоктиламмонийхло-
рид и лезо-тетрафенюторфиринмарганец(Ш)хлорид в водно-
Глава 3. Практическое использование МФК
Таблица 3.30. Окисление с помощью системы
Реакция
Спирт—^-альдегид или кетон
Ph-rCftOH
о.СНзО—СвН4СН2ОН
п-СНзО—СбН4СН2рН
n-СНз—СбН4СН2ОН
«-С1—С6Н4СН2ОН
Ph2CHOH
9-Флуоренол
Цнклргептанол
2'Норборнеол
4-грег-Бутйяциклогексанол
Амин—*-кетон, амин—унитрил
Циклогексиламин—>-циклогексанон
Норборниламин—»-норборнанон
Бензгидриламин—>-бензофенои
а-Метилбён8иламин—»-ацетофенон
(Циклогексилметил)амии—>-циклогекснлнитрнл
1 -Октиламин—»-1 -цианогептан
Полициклические арены—*-ареноксиды [961}
Гидрохиноны—*-п-хиноны [1479]
З-трет-Бутилпирокатехины—*-о-хиноны [1479]
Метилкетон—*-карбоновая кислота
Мезитилоксид—иВ,0-диметилакриловая кислота+
+дихлорид [1575]
Халкон (гетероциклический)—t-халконоксид
MaQCI/Bu4NHSO4
Растворитель
СН2С12
CH2CI2
ЕЮАс
CtI2Cl2
ЕЮАс
CH2CI2
ЕЮАс
CH2CI2
СН2С18
СН2С12
ЕЮАс
ЕЮАс
ЕЮАс
ЕЮАс
ЕЮАс
ЕЮАс
ЕЮАс
ЕЮАс
ЕЮАс
НССЦ
HCCIS или
ЕЮАс
неси
сн^ь
[595]
Выход, %
76
47
94
79
92
78
100
82
82
92
89
36
49
98
84
94
98
76
60
10—95
14—91
91
Высокий
32—96
дихлорметановой среде; при комнатной температуре реакция
проходит очень быстро.
Эпоксидирование стирола и циклогексена с помощью гипо»
хлорита натрия также требует присутствия межфазного
катализатора и катализатора с переходным металлом. При
использовании бензилдиметилтетрадециламмонийхлорида и мезо-тетра*
3.22. Реакции окисления 40&
феншшорфиринмарганец (III) ацетата в системе Н2О/СН2С12
с 0,35 М NaOCl реакция проходит за несколько часов при ком-,
натной температуре. Были испытаны аналогичные комплексы
железа .и кобальта и различные комплексы основании ^Шиффа.
с Мп(Ш), но они оказались менее эффективными [1571].
Гипабромит
Известны реакции окисления в которых гипобромит
генерировался in situ путем электрохимического окисления бромида;
в качестве катализатора использовался Bu4NHSO4 в водно-
амилацетатной эмульсии [1002, 1639]. Этот метод особенно
привлекателен вследствие широких возможностей ' его
применения.
3.22.6. Калийгексацианоферрат(Ш)
(Bu4N)3[Fe(CN)6] может быть экстрагирован в хлороформ*.
При комнатной температуре он не окисляет фенол А. Однако
окисление начинается сразу же, если добавить 2 экв. л-толуол-
сульфокислоты. Образуется В, а собственно окислителем
является дигидрогексацианоферрат(Ш) [596]. Если проводить
окисление в подкисленном метаноле в присутствии гидрогексациано-
феррата, образуются два различных продукта (Си D),
соотношение которых отчасти зависит От кислотности среды. В менее
кислой среде образуется больше n-метоксиметилфенола D.
ОН
СНз
А
^-^снэон
H3F«CN).
Схема 3.245
* Ион [Fe(CN)6]3~ более липофнлен, чем бромид; fFe(CN)e]4- цо
экстрактивной способности располагается между хлоридом и бромидом [1185].
404 Глава 3. Практическое использование МФК
Эти результаты позволяют предположи1», что при
использовании хлороформенного раствора процесс окисления имеет
свободно-радикальный механизм. В метанольном растворе
первоначально возникающий О-радикал может либо подвергнуться
дальнейшему окислению до феноксойий иона
(преимущественный процесс в сильнокислом растворе), либо диспропорциони*
роваться до метилхинона и родственного фенола:
Б
снон
сан5
Схема 3.246
С=О
С2Н5
Подобное изменение в направлении реакции в зависимости
от кислотности и растворителя наблюдалось и для замещенного
n-гидроксибензилового спирта (Е) [596].
3.22.7. Периодат
Et4N+IO4"~— белый кристаллический порошок. Его получают
нейтрализацией Et4N+OH~ рассчитанным количеством йодной
кислоты. Соль устойчива в течение нескольких недель при
хранении в эксикаторе, однако требует осторожного обращения
(возможен взрыв! [1203]). Соль хорошо растворима в воде,
а также во многих органических растворителях. Квареши и
3.22. Реакции окисления 405
Скларц [597] использовали этот реагент для проведения
реакции окисления, представленной на схеме 3.247, при комнатной
температуре в хлороформе. Некоторые реакции проводят в
водно-спиртовой среде в присутствии NalO*. Подобным образом
при помощи (СзНг^Ы+Юа" была окислена беизгидроксамоваяз
кислота в бензоилнитрозойроизводное — весьма реакционноспо-
собный интермедиат [1200J., В других случаях в отсутствие
гидроксилсодержащего растворителя окисление не
происходило [1186].
он
Схема 3.247
Другие авторы использовали раствор предварительно
выделенного четвертичного аммониевого периодата. в хлороформе»
ТГФ или диоксане для расщепления гликолей [1385],
селективного окисления сульфидов до сульфоксидов [1402] и для
окислительного декарбоксилирования [1381, 1402] следующих типов
соединений (кипячение в течение 2—24 ч):
RCH(OH)—СООН >■ R—СНО
ArCR<OH)—COOH *• ArCOR
АгСОСН2Х >• АгСООН
Приведенные выше реакции проводили в гомогенной среде.
Наиболее близким к межфазному катализу и наиболее
интересным с препаративной точки зрения является расщепление
гликолей в двухфазной системе. Низшие гликоли весьма
гидрофильны и не нуждаются в применении двухфазной системы. Но ряд
неполностью защищенных производных Сахаров ,может быть
подвергнут расщеплению в органическом растворе; в присутствии
водной системы NaIO4/R4NX [1343, 1410]. Кроме того, липо-
фильные гликоли являются промежуточными продуктами в
хорошо известном методе Лемьё — Джонсона [598],
заключающемся в окислении олефинов действием системы периодат/тетра-
оксид осмия. В этом случае вариант с использованием
межфазного катализа может обладать преимуществами. Так, Старке и
сотр. [39, 599, ср. 1385] показали, что окисление октена-1 в
углеводородном растворителе с образованием к-гептанал5Г
(и небольшого количества гептановой кислоты) протекает как
слабо экзотермическая реакция при использовании 2 экв. вод-
■406 Глава 3. Практическое использование МФК
ной йодной кислоты в присутствии 0,2 мол.Я> OsC>4 и менее
1 мол.% аликвата 336:
H6IOe/OsO4 H6IOe/RiiO2
R—CHO -« R—СН=СН, —■ *~ R—COOH
аликват, ] ч * аликват, 2 ч
В той же реакции при использовании 4 экв. Н5Ю6 и
диоксида рутения вместо тетраоксида осмия образуется почти с
количественным выходом м-гептановая кислота [599]. В обоих
превращениях в качестве катализатора могут быть использованы
третичные амины (например, тридодециламин), так как ионная
пара типа R3HN+H4lO6~~ может экстрагироваться в
органическую среду [600]. Однако оказалось, что третичные амины —
намного менее эффективные катализаторы, чем четвертичные
аммониевые или фосфониевые соли (например, трибутилстеарил-
фосфонийбромид) [599]. В отсутствие катализаторов эти
реакции идут крайне медленно.
Дитиоацетали формальдегида окисляются до moho-S-оксидов
(ArSCHjSOAr) с частичной оптической индукцией при
взаимодействии с водным NaIO4 в присутствии альбумина бычьей сы*
воротки [1272].
.3.22.8. Надуксусная кислота
Имеются две близкие методики окисления гидразонов ди-
арилкетонов в диарилдиазометаны [601]; в классических
условиях для этого используется оксид ртути. В методе (а), не
являющемся межфазным, могут быть использованы в качестве
растворителей СН2С12, ЕЮН, СНС13 или С1СН2—СН2С1.
<а) надуксусная кислота, следы
1а. 1,1,3.3-тетраметилгуанидин
(Ar)2C=N—NH2 : *■ (Ar)2C=N2
у " * (б) надуксусная кислота, следы
12, NaOH (pH 10) МФ-катализатор
Существенным фактором является присутствие сокатализатора
(h) в оптимальной концентрации 10~4 моль/л. Если
использовать меньшее количество, окисление идет слишком медленно;
большее количество 12 ускоряет распад диазосоединения до
азина. Наилучшим основанием оказался 1,1,3,3-тетраметилгуа-
нидин; многие другие гуанидины подвергаются окислительному
разложению. Были получены с выходами 70—98,6% различные
диарилдиазометаны. В качестве окислителей помимо надуксус-
яой кислоты можно использовать ж-хлорнадбензойную кислоту
и хлорамин Т.
3.22. Реакции окисления 407
При использовании метода межфазного катализа для
приготовления дифенилдйазометана были опробованы различные
катализаторы:
Катализатор
Отсутствует
Bu4NOH
B114NCI
ВщШ
(C8H17)3C3H7NC1
Выход, %
17,5
78,6
65,5
48,0
84,9
Оказалось, что весьма гидрофильный надацетат требует
катализатора с липофильным катионом и нелипофильным
анионом. Среди других изученных окислителей только хлорамин Т
по своей эффективности приближается к надуксусной кислоте.
МФК-методика. 0,1 моль гидразона бензофенона растворяют
в 100 мл CH2CI2, добавляют 3 ммоля (C8Hi7)3C3H7NCl и 4 мл
10%-ного 1г. Смесь перемешивают при 0°С и добавляют
одновременно раствор 0,1 моля надуксусной кислоты и водный NaOH
таким образом, чтобы поддерживать рН около 10. Реакция
заканчивается через 15 мин.
3.22.9. Кислород как окислитель
Кислород — хорошо известный окислитель для некоторых
карбанионов. Возможность легкой генерации карбанионов в
условиях межфазного катализа позволяет усовершенствовать
методику проведения таких реакций. Дитрих и Лен [359]
сообщили о количественном превращении флуорен->-флуоренон,
осуществленном в ТГФ с использованием атмосферного
кислорода в присутствии твердого гидроксида калия и криптан-
да [2.2.2] (криптофикс [222]). Дарст [571] осуществил такую
же реакцию в бензоле, применив 18-краун-6. Дициклогексано-
18-краун-6 оказался не пригодным для этой цели. Алнери и
сотр. [572] смогли окислить ароматические соединения с
активными метиленовыми группами и частично гидрированные
ароматические соединения в присутствии 50%-ного водного
гидроксида натрия и четвертичного аммониевого катализатора.
Был найден следующий ряд активности катализаторов:
Et4N+OH-~PhCH2N+Me3OH-> (C16H33)2Et2N+Cl- > ТЭБА >
>Et4N+Cl->Et4N+Br-. Окисление проходит при 30—50 °С. Про-
408 Глава 3. Практическое использование МФК
ведено также широкое изучение условий реакций,
катализируемых краун-эфирами [1393]: /
О
[359, 571, 572, 1393]
[572]
н н н н н н
н н
[572]
НН НН Н Н
Кроме того, было показано, что анилин может подвергаться
окислительной димеризации до азобензола [573] и что окисЛе^
ние происходит быстро, если через раствор натриевых
производных замещенных бензилцианидов в диоксане или бензоле
пропускать кислород в присутствии межфазных катализаторов:
Ph—NH2
18-краун-б, КОН
О2
* Ph—N=N—Ph
Ph—С—CH2R
CN
Ph—С—CH2R + Nae NCO
О
Схема 3.249
При этом образуются ароматические кетоны [574, 1187,
1610]. Если реакцию осуществлять в системе 50%-ный NaOH/
/ДМСО/ТЭБА, конечные продукты получаются с выходами от
50 до 90%. Окислительное децианирование алифатических
нитрилов (без разветвления в а-положении) происходит в ТГФ,
при использовании системы трет-бутоксид калия/18-краун-6/воз-
дух. Время реакции 48—92 ч, выходы достигают 90% [1518]
3.22. Реакции окисления 409
Несомненно, что в подобных условиях можно использовать
ряд других окислителей. Как было недавно показано [973,
1625], соединения типа Рейссерта при взаимодействии в
бензольном растворе с системой 50%-ный NaOH/воздух дают
продукты, указанные на схеме 3.250 с выходами соответственно
37—80 и 65—97%:
CN
Бензольные растворы <х,р- или Р/у-ненасыщенных кетонов в
присутствии системы конц. NaOH/ТЭБА на воздухе окисляются
до одного и того же гидропероксида с выходом 15—20% [981].
.Л
6 ^ \
о
Схема 3251
Условия, подобные-межфазным, были использованы для солю-
билизации сенсибилизаторов при фотохимическом
генерировании синглетного кислорода. Авторы работали с анионными
красителями роза бенгальская и эозин-Y в CS2 или CH2CI2,
применяя 18-краун-6 или аликват 336 для перевода красителей
в растворенное состояние. Модельные реакции представлены
(4+2)-циклоприсоединением О2 к антрацену и еновой
реакцией с 2,3-диметилбутеном-2 [575, 1199].
3.22.10. Другие реагенты-окислители
Хорошо известное окисление альдегидов до карбоновых
кислот действием оксида серебра ускоряется в присутствии
межфазного катализатора. Это позволяет предположить, что экст-
410 Глава 3. Практическое использование МФК
рагируемой активной частицей является rR4N+Ag-(OH)2l
[1293].
Оба персульфата — производные серной кислоты — в
межфазных условиях действуют более эффективно. Пероксимоносуль-
фат (кароат) эпоксидирует водонерастворимый олефин в
бензол/водно-ацетон-фосфатном буфере (рН 7,5) при 60 °С в
присутствии небольших количеств 18-крауна-6 или Bu4NHSO±
[1644]. Пероксидисульфат калия является инициатором свобод-
норадикальных реакций, но широко не применяется из-за era
нерастворимости в большинстве органических растворителей.
Если использовать различные крауны или четвертичные
аммониевые соли, можно осуществить экстракцию аниона из водной
фазы и провести полимеризацию бутилакрилата во многих
органических растворителях. При использовании как инициатора
азо-б«с-изобутиронитрила (АИБН) в этих условиях
полимеризация практически не наблюдается, хотя АИБН имеет такую
же энергию активации разложения, как и K2S2O8 в 0,1 н.
водном основании [1743, 1776, 1851]. Окисление АгСН3->-АгСНО
осуществляется в бензольном растворе КгЗгОв/ТЭБА [1852].
Ионы церия (IV) могут быть перенесены в неполярные
органические среды типа циклогексана путем комплексообразова*
ния с 4,4'-диоктадекаокси-2,2'-бипиридин-1, Г-диоксидом А
(схема 3.252). Это является одним из примеров межфазного
процесса экстракции катиона, что встречается довольно редко.
Реагент был использован для некоторых реакций окисления,
приведенных ниже. Противоионом экстрагируемому Се-комплек-
су является нитрат-ион.
ОСНз
ОСНз О
АгСНз ► АгСНО + ArCH2ONO2
Схема 3.252
Интересна идея [1304, см. 1002, 1639] использования этой и
других реакций окисления для проведения весьма экономически
выгодного процесса без выделения промежуточных продуктов
(«в одном горшке»), в котором окислитель непрерывно
регенерируется путем электрохимического окисления как в водной
фазе, так и в органической, а межфазный катализатор служит
одновременно в качестве фонового электролита.
Литература*
К главе 1
Ц] J. Jarrousse, C.R. Acad. Sei. Paris, 232, 1424 (1951).
*[2] A. T. Babayan, N. Gambaryan, and N. P. Gambaryan. Zh. Obshch. Шт., 24, 1887
(1954); Chem. Abstr., 49, 10879 (1955); Chem. Zentralbl. Sonderb., 1950/54, 4532.
[3] G. Maerker, J. F. Carmichael, and W. Port, J. Org. Chem., 26, 2681 (1961).
£4] H. B. Copelin and G. B. Crane, U.S. Pat. 2,779,781 (1957), Chem. Abstr., 51, 10579
(1957); R. Kohler and H. Pietsch, Germ. Pat. 944,995 (1956), Chem. Abstr.,53, 1831
(1959); P. Edwards, U.S. Pat. 2,537,981 (1951), Chem. Abstr., 45, 5177 (1951); A.
Conix and U. L. Laridon, Belg. Pat. 563,173 (1958) and 565,478 (1958), Chem.
Abstr., 55,25356 (1961); E. Miiiler, O. Bayer, and H. Morschel, Germ. Pat. 959,497
(1957), Chem. Abstr., 53,13665(1959); Germ.Offenl.268621 {mS),Chem.ZentealbL,
1914, 310.
|5] M. Makosza and B. Serafinowa, Rocz. Chem., 39,1223 (1965), and subsequent papers.
{6] M. Mqkosza and W. Wawrzyniewicz, Tetrahedron Lett., 1969, 4659.
[7] A. Brandstrom and K. Gustavii, Ada. Chem. Scand., 23, 1215 (1969).
[8] A. Brandstrom, Kern. Tidskr., 1970, Nos. 5-6, 1.
[9] С M. Starks and D. R. Napier, Ital. Pat. 832,967 (1968); Brit. Pat. 1,227,144 (1971);
French Pat. 1,573,164 (1969) and further concordances; Chem. Abstr., 72, 115271
(1970).
Ц0] С M. Starks» J. Am, Chem. Soc, 95, 195 (1971).
Ill] A. Brandstrom, "Preparative Ion Pair Extraction, an Introduction to Theory and
Practice,". Apotekarsocieteten/Hassle LSkemedel, Stockholm, 1974, ■«
(12] E. V. Dehmlow, Angew. Chem., 86, J87 (1974); Angew. Chem. Ш. Ed. Etigl, 13, 170
(1974); reprinted in: New Synthetic Methods, Vol. 1, Verlag Chemie, Weinheim, 1975,
p. 1, and—in slightly shortened form—in Chem. Techno!., 1975, 210.
[13] E. V. Dehmlow, Angew. Chem. 89, 521 (1977), Angew. Chem. Int. Ed. EngL, 16, 493
(1977); "New Synthetic Methods," Vol. 6, Verlag Chemie, Weinheim, 1979, p. 205.
[14] -M. Makosza, Pure Appl. Chem., 43. 439 (1975).
[15] M. Makosza, Naked anions-^phase transfer, in "Modern Synthetic Methods 1976,"
R. Scheffold, Eds, Schweizerischer Chemike^Verband, Zurich, 1976» pi 7.
[16] J. Dockx, Synthesis, 1973, 441.
[17] R. A. Jones. Aldrichimica Acta, 9, 35 (1976).
[18] M. Makosza, Survey Progr. Chem. 9, 1 (1980).
[19] E. V. Dehmlow, Chimia, 34, 12 (1980).
[20] J. P. Antoine, I. de Aguirre, F. Janssens, and F. Thyrion, Bull. Soc. Chim. Fr,, 198», 207.
*[21] L. P. Hammett, "Physical Organic Chemistry—Reaction Rates, Equilibria, and
Mechanisms," McGraw-Hill, Inc., New York, 1970; "Physik'alische Organische
Chemie—Reaktionsgeschwindigkeiten, Gleichgewichte und Mechanismen," Verlag
Chemie, Weinheim, 1973.
*[22] M. Szwarc, Ed., "Ions and Ion Pairs in Organic Reactions," Vols. 1 and 2, Wiley-
Interscience, New York, 1972.
[23] G. A. Olah and P. v. R. Schieyer, Eds., "Carbonium Ions, Vol. 1, General Aspects and
Methods of Investigation," Interscience Publishers, New York, 1968.
124] C. Reichardt, "Losungsmittel-Effekte in der Organischen Chemie," Verlag Chemie,
Weinheim 1969; C. Reichardt," Solvent Effects in Organic "Chemistry," Monographs
in Modern Chemistry, Vol. 3, Verlag Chemie, Weinheim, 1979.
* Работы, которые отмечены звездочкой, стоящей впереди номера,
опубликованы на русском языке (см. с. 462). — Прим. ред.
[25] D. T. Copenhafer and С. A. Kraus, /. Am. Chem. Soc, 73,4557 (f 951).
(26] С R. Witschonke and C. A. Kraus, /. Am. Chem. Soc, 69, 2472 (1947).
[27] K. H. Wong, M. Bourgoin, and J. Smid, J. Chem. Soc. Chem. Commun., 1974, 715.
[28] J. Almy, D. C. Garword, and D. J. Cram, /. Am. Chem. Soc, 92,4321 (1970).
*[29] E. J. Fendler and J. H. Fendler, Micellar catalysis in organic reactions—kinetic and
mechanistic implications, in "Advances in Physical' Organic Chemistry," Vol. 8..
V. Gold, Ed., Academic Press, London and New York, 1970, p. 271.
[30] J. H. Fendler and E. J. Fendler, "Catalysis in Micellar and Macromolecular Systems,"
Academic Press, New York, 1975.
[31] J. H. Fendler, Ace Chem. Res., 9, 153 (1976).
[32] J. Smid, Angew. Chem., 84, 127 (1972); Angew. Chem. Int. Ed. Engl., 11, 112 (1972).
[33] R. A. Sneen, Ace. Chem. Res., 6, 46 (1973).
•[34] I. P. Beletskaya, Usp. Khim., 44, 2205 (1975); Engl. transl. p. 1067.
[35] D. J. McLennan, Ace. Chem. Res., 9, 281 (1976).
[36] D. J. Raber, J. M. Harris, and P. v. R. Schleyer, in ref. [22], Vol. 2, p. 247.
•[37] B. S. Krumgal'z, Zh. Obshch. Khim., 44, 1617 (1974), Engl. transl. p. 1585.
[38] R. M. Fuoss.and F. Accascina, "Electrolytic Conductance," Interscience Publishers,
New York, 1959.
1 [39] M. Szwarc, "Carbanions, Living Polymers, and Electron Transfer Processes,'*'
Interscience Publishers, New York, 1968, viz. Chapter V.
[40] H. F. Ebel, Fortschr. Chem. Forsch., 12, 387 (1969).
[41] H. F. Ebel, "Die Aciditat der CH-Sauren," G. Thieme, Verlag, Stuttgart, 1969.
[42] M. Schlosser, "Struktur und Reaktivitat polarer Organometalle," Springer-Verlag,
Berlin-Heidelberg, 1973.
•[43] D. J. Cram, "Fundamentals of Carbanion Chemistry," Academic Press, New York,
1965.
[44] R. Modin, Ada. Pharm. Suec, 9, 157 (1972), and papers cited therein.
[45] R. Modin and G. Schill, Acta. Pharm. Suec, 4, 301 (1967), and subsequent papers of
the same series.
[46] A. Brandstrom, ref. [Ш], p. 276.
[47] G. Schill, R. Modin; and B.-A. Persson, Acta. Pharm. Suec, 2, 119 (1965).
[48] B.-A. Persson and G. Schill, Acta. Pharm. Suec, 3, 281 (1966).
[49] K. Gustavii and G. Schill, Acta. Pharm. Suec, 3, 241 (1966).
[50] J. H. Blakeley and V. J. Zatka, Anal. Chim. Acta, 74, 139 (1975).
[51] P.-O. Lagerstrom, K. O. Borg, and D. Westerlund, Acta Pharm. Suec, 9, 53
(1972).
[52] K. Gustavii, A. Brandstrom, and S. Allanson, Acta Chem. Scand., 25, 77 (1971).
[53] K. Gustavii, Ada Pharm. Suec, 4, 233 (1967).
[54] S. O. Jannsson, R. Modin, and G. Schill, Talanta, 21, 905 (1974).
[55] R. Modin and A;!Tilly, Acta Pharm. Suec, 5, 311 (1968).
[56] T. Nordgren and R. Modin, Acta Pharm. Suec, 12, 407 (1975),
[57] S. Eksborg and fc. Schill, Anal. Chem., 45, 2092 (1973).
[58] K. Gustavii and G. Schill, Ada Pharm. Suec, 3, 259 (1966).
[59] B. Czapkiewicz-Tutaj and J. Czapkiewicz, Rocz. Chem., 49, 1353 (1975).
[60] R. Modin and M. Schroder-Nielsen, Acta Pharm. Suec, 8, 573 (1971).
[61] К. О. Borg and G. Schill, Acta Pharm. Suec, 5, 323 (1968).
[62] К. О. Borg and,D. Westerlund, Z. Anal. Chem. 252. 275 (1970).
[63] W. E. Clifford and H. Irving, Anal. Chim. Ada. 31, I (1964).
*[64] O. E. Zvyagintsev, N. M. Sinitsyn. and V. N. Pichkov, Zh. Neorg. Khim., 11, 198"
(1966); Engl. transl. p. .107; Chem. Abstr. 64, 8983 (1966).
*[65] 1. M. lvanov, L. M. Gindin, and G. N. Chichagova, Izv. Sib. Otd. Akad.Nauk SSSR,
Ser. Khim. Nauk., 1967, 100; Chem. Abstr., «9, !3377..(1968> and Chem. Abstr., 78,
8481 (1973).
412
[66] M. Makosza and E. Biaieeka, Synth. Commun., 6, 313 (1976).
[67] E. V. Dehmlow, M. Slopianka, and J. Heiderj Tetrahedron Lett., 1977, 2361.
*[68] Yu. G. Frolov, V. V. Sergievskii, A. V. Ochkin, and A. P. Zuev, Radiokhimiya, 14,
643 (1972); Engl. transl. p. 664; Chem. Abstr., 78, 8479 (1973). Solvent Extr., Proc.
Int. Solvent Extr. Conf.rl, 1229 (1971); Chem. Abstr., 83, 153505 (1975).
[69] D. Landini, A. M. Maia, F. Montanari, and F. M. Pirisi, /. Chem. Soc. Chem. Commun.,
1975, 950.
* [70] Yu. G. Frolov, V. V. Sergievskii, A. P. Zuev, and L. B. Fedyanina, Zh. Fiz. Шт., 47,
1956(1973), Engl. transl. p. 1101; Chem. Abstr.,79,140143 (1973).
[71] Y. Marcus and A. S. Kerts, "Ion Exchange and Solvent Extraction of Metal
Complexes," Wiley-Interscience, New York, 1969.
[72] "Recent Advances in Liquid-Liquid Extraction," C. Hanson, Ed., Pergamon Press,
Oxford, 1971.
[73] "Solvent Extraction Chemistry," D. Dyrrssen, J. O. Liljenzin, and J. Rydberg, Ed.,
North-Holland Co., Amsterdam, 1967.
[74] "Solvent Extraction Research, Proc. 5thInt. Conf. on Solvent Extraction Chemistry,"
A. S. Kertes and Y. Marcus. Eds., Wiley-Interscience, New York, 1969.
[75] Ref. [71], p. 796; R. Bock and G. M. Beilstein, Z. Anal. Chem., 192, 44 (1963);
R. Bock and J. Jainz, Z. Anal Chem., 198, 315 (1963).
*[76] M. J. Gugeshashvils, M. A. Manvelyan, and L. J. Boguslavskii, Elektrokhimiya 10,
819 (1974); Engl. transl. p. 782; Chem. Abstr., 81, 98514 (1974).
[77] С J. Pedersen and H. K. Frensdorff, Angew. Chem. 84, 16 (1972); Angew. Chem. Int.
Ed. Engl., 11, 16 (1972).
[78] D. J. Cram and J. M. Cram, Science, 183, 803 (1974).
[79] J. J. Christensen, D. J. Eatough, and R. M. Izatt, Chem. Rev., 74, 351 (1974).
[80] A. C. Knipe, /. Chem. Educ.,53, 618 (1976).
[81] G. W. Gokel and H. D. Durst, Synthesis, 1976, 168.
[82] J. J. Christensen, J. O. Hill, and R. M. Izatt, Science, 174, 459 (1971).
[83] B. Dietrich, J. M. Lehn, and J. P. Sauvage, Tetrahedron Lett., 1969, 2885, 2889; B.
Dietrich and J. M. Lehn, Tetrahedron Lett., 1973, 1225; review: Ace. Chem. Res., 11.
49 (1978).
[84] R. С Helgeson, K. Koga, J. M. Timko, and D. J. Cram, J. Am. Chem. Soc, 95, 3021
(1973); R. С Helgeson, J. M. Timko, P. Moreau, S. C. Peacock, J. M. Mayer, and
D. J. Cram, ibid, 96, 6762 (1974); M. Newcomb, R. С Helgeson, and D, J. Cram,
ibid., 96, 7367 (1974); L. R. Sousa, D. H. Hoffman, L. Kaplan, and D. J. Cram, ibid.,
96, 7100 (1974); M. Newcomb, G. W. Gokel, and D. J. Cram, ibid., 96, 6811 (1974);
G. Dotsevi, Y. Sogah, and D. J. Cram, ibid., 97, 1259 (1973), 98, 3038 (1976); S. С
Peacock, and D. J. Cram, /. Chem. Soc. Chem. Commun., 1976, 282; G. W. Gokel,
J. M. Timko, and D. J. Cram, /. Chem. Soc. Chem. Commun., 1975, 394, 444; cf.,
W. D. Curtis, R. M. King, J. F. Stoddart, and G. H. Jones, J. Chem. Soc. Chem.
Commun., 1976, 284, and further references,
[85] Recent references: F. Vogtle and B. Jansen, Tetrahedron Lett., 1976,4895; I. J. Burden,
A. C. Coxon, J. F. Stoddart, and С M. Wheatley, /. Chem. Soc. Perkin Trans. I,
1977, 220; M. P. Mack, R. R. Hendrixson, R. A. Palmer, and R. G. Ghirardelli,
/. Am. Chem. Soc, 98, 7830 (1976); R. C. Hayward, С H. Overton, and G. H.
Whitham, J. Chem. Soc. Perkin Trans. 1,1976,2413; D. A. Laidler and J. F. Stoddart,
J. Chem. Soc. Chem. Commun., 1977, 481; J. P. Behr, J. M. Lehn, and P. Vierling,
/. Chem. Soc. Chem. Commun., 1976, 621; J. M. Girodeau, J. M. Lehn, and J. P.
Sauvage, Angew. Chem., 87, 813 (1975), Angew. Chem. Int. Ed. Engl., 14, 764 (1975);
D. G. Parsons, J. Chem. Soc. Perkin Trans. 1, 1975, 245; D. A. Laidler, and J. F.
Stoddart, /. Chem. Soc. Chem. Commun., 1976, 979; D. Curtis, J. F. Stoddart, and
G. H. Jones, J. Chem. Soc. Perkin Trans. I, 1977, 1756; N. Ando, Y. Yamamoto,
J. Oda and Y. Inouye, Synthesis, 1978,688; P. DiCesare and B. Gross, Synthesis, 1979,458;
413
J.-P. Behr, J.-M. Girodeau, R. C. Hayward, J.-M. Lehn, and J.-P. Sauvage, Helv. Chim.
Ada, «3, 2096 (1980).
[86] J. D. Lamb, J. J. Christensen, 3. L. Oscarson, B. L. Nielsen, B. W. Asay, and R. M. Izatt, J.
Am. Chem. Soc, 192, 6820(1980).
[87] S. Shinkai, T. Nakaji, T. Ogawa, K. Shigematsu, and O. Manabe, J. Am. Chem. Soc, 103,
111 (1981).
[88] E. Graf and J. M. Lehn, /. Am. Chem. Soc, 98, 6403 (1976); F. P. Schmidtchen,
Angew. Chem., 89, 751 (1977); Angew. Chem. Int. Engl, 16, 720 (1977); B. Metz,
J. M. Rosalky, and R. Weiss,./. Chem. Soc. Chem. Commun., 1976, 533.
[89] J. L. Dye, M. G. De Backer, and V. A. Nicely, /. Am. Chem. Soc, 92, 5226 (1970).
[90] F. J. Tehan, B. L. Barnett, and J. L. Dye, /. Am. Chem. Soc, 96, 7203 (1974).-
[91] G. W. Gokel and D. J. Cram, J. Chem. Soc. Chem, Commun,, 1973, 481.
[92] E. V. Dehmlow, Tetrahedron Lett., 1976, 91; E. V. Dehmlow and TV Remmler, Л
Chem. Res., 1977 (S) 72, (M) 766.
[93] A. Knochel, J. Oehler, and G. Rudolph, Tetrahedron Lett., 1975, 3167.
[94] H. Normant, T. Cuvigny, and P. Savignac, Synthesis, 1975, 805.
[95] G. V. Nelson and A. von Zelewsky, /. Am. Chem. Soc, 97, 6279 (1975); M. A. Kor-
marynski and S. I. Weissman, ibid., 91,1589 (1975); B, Kaempf, S. Raynal, A. Collet,
F. Schne, S. Boileau, and J. M. Lehn, Angew. Chem., 86,670 (1974), Angew. Chem. Int.
Ed. Engl, 13, 611 (1974).
[96] B. R. Agarwal and R. M. Diamond, /. Phys. Chem., 57, 2785 (1963).
. [97] F, Vogtle and E. Weber, Angew. Chem., 86, 896 (1974), Angew. Chem. Int. Ed. Engl.,
13, 814 (1974); F. Vogtle and H.Sieger, Angew. Chem., 89, 410 (1977), Angew. Chem.
Int. Ed. Engl., 16, 396 (1977); B. Tummler, G. Maass, E. Weber, W. Wehner, and F.
VogUe,/. Am. Chem, Soc.t 99,4683 (1977); F. Vogtle, W. M. Miiller, W. Wehner, and
E. Buhleier, Angew. Chem., 89, 564 (1977); Angew. Chem. Int. Ed. Engl,, 16, 548
(1977).— For various PTC applications of open chain polyethers cf., Chapter 3 and
refs. [93,94].
[98] M. Kirch and J. M. Lehn, Angew. Chem., 87,542 П975), Angew. Chem. Int. Ed. Engl.,
14, 555 (1975); M. Newcomb, R. С Helgeson, and D. J. Cram, /. Am. Chem. Soc, 96,
7367 (1974); J. J. Christensen, J. D. Lamb, S. R. Izatt, S. E. Starr, G. С Weed,
M. S. Astin, B. D. Stitt, and R. M. Izatt, /. Am. Chem. Soc, 100, 3219 (1978).
[99] C. L. Liotta and E. E. Grisdale, Tetrahedron Lett., 1975, 420S.
[ИЮ] J. P. Tarn, W. P. Cunningham-Rundles, B. W. Erickson, and R. B. Merrifield,
* Tetrahedron Lett., »T7, 4001.
[101] M. J. Maskornick, Tetrahedron Lett.,1972, 1797.
[102] L. M. Thomassen, T. Ellingsen, and'j. Ugelstad, Ada Chem. Scand., 25, 3024
(1971).
[103] R. A. Bartsch, E. A. Mintz, and R. M. Parlman, /. Am. Chem. Soc, 96, 4249 (1974);
R. A. Bartsdi, Ace Chem. Res., 8,239 (1975).
[104] W. Dorn, A. Knochel, J. Oehler, and G. Rudolph.Z. Naturforsch, TeilB, 32. 776(1977).
[105] A. Knochel and G. Rudolph, Tetrahedron Lett., 1974, 3739.
[1061 Eg-, G. W- Gokel and B. J. Garcia, Tetrahedron Lett., 1977, 317.
[107] A. Knochel, J. Oehler, and G. Rudolph, Z. Naturforsch, Teil B, 32, 783 (1977).
[108] A. el Bosyony, J. Klimes, A. Knochel, J. Oehler, and G. Rudolph, Z. Naturforsch.,
TeilB, 31, 1192(1976).
[109] T. Iwachido, M, Kimura, and K. Toei, Chem. Lett., 1976, 1101.
[110] F. de Jong, D. N. Reinhoudt, and С J. Smit, Tetrahedron Lett., 1976, 1371, 1375.
[1111 B.g., R. A. Bartsch, H. Chen. N. F. Haddock, and P. N. Juri, /. Am. Chem. Soc, 98,
6753(1976).
[112] A. Brandstrom, Principles of phase transfer catalysis by quaternary ammonium salts,
■in "Advances in Physical Organic Chemistry," Vol. 15, V. Gold, Ed., Academic Press,
London and New York, 1977, p. 267.
*[113] W. P. Weber and G. W. Gokel, Phase transfer catalysis in organic synthesis, in
"Reactivity and Structure," Vol. 4, K. Hafner et aL, Eds., Sprlnger-Verlag, Berlin-
Heidelberg, 1977.
*[114J M. Makqsza, Uspekhi Khim., 46, 2174<J977); Engl. translp. 1151.
[115] J. M. Mclntosh, J. Chem. Educ, 55, 235 (J978); G. W. Gokel and W. P. Weber,
ibid., 55, 350, 429 (1978).
[116] C. M. Starks and C.. Liotta, "Phase Transfer Catalysis* Principles and Techniques,"
Academic Press, London and New York, 1978. :
[117] J. Czapkiewicz, private communication.
[118] N. A. Gibson and D. С Weatherburn, Anal. Chim. Acta, 58, 159 {1972).
[119] J. Czapkiewicz and B. Czapkiewicz-Tutaj, /. Chem. Soc. Faraday Tram. 1,1980, 1663.
1120] J. Czapkiewicz, B. Czapkiewicz-Tutaj, and D. Struk, Pol, J. Chem., 52, 2203 (1978).
[121] Y. Inoue and O. Tochiyama, Bull. Chem. Soc. Jpn., 53, 1618 (1980).
[122] ref. [116], p. 25.
[123] H. Brink, R. Modin, and J. Vessman, Acta Pharm. Suec, 16, 247 (1979).
[124] G. A. Harlow, Anal. Chem., 34, 1487 (1962).
[125] H. Brink, Dissertation, Uppsala University, 1980.
[126] A. Kndchel, Institut fur Anorg. Chemie, Universitat Hamburg, unpublished
[127] J. Hayami, N. Hihara, and A. Kaji, Chem. Lett., 1979,413.
*I128] V.V. Yakushin, V. M. Abashkin, and B,, N. Laskqrin, Doklady Akad. Nauk SSSR, Ser.
Khim., 252, 373 (1980); Engl. trans», p. 239.
[129J P.'Vfouf, J. Molecul. Catal., 10, 231 (1981).
[130] M. H. Abraham and J. Liszi, /. Inorg. Nucl. Chem., 43r 14^ (1981).
[131] E. V. Dehmlow arid R. Thieser, unpublished.
К главе 2
II] A. Brandstrom, Principles of phase transfer catalysis by quaternary ammonium salts,
in "Advances in Physical Organic Chemistry," Vol. 15, V. Gold, Ed., Academic Press,
London and New York, 1977, p. 267. '■ , rr
[2] С M. Starks,/. Am. Chem. Soc., 93,195(1971); С M. Starks andЖ. M. Oweiis,!>. Am.
Chem. Soc, 95, 3613 (1973).
{3] D. H. Picker, Ph.D. dissertation, Department of Chemistry, State University of New
York at Albany, 1974; Diss. Abstr. 35, 3229 (1975).
[4] A. W. Herriott and D. Picker, /. Am. Chem. Soc, 97, 2345 (1975).
(5] D. Landini, A. M- Maia.F. Montanari.and F. M. Krisi,/. Chem'.'Sttc Cheni. Commun.,
1975,950; D. Landini, A. Maia, and F. Montanari, /. Am. Chem. Soc, Ш, 2796 (1978).
[6] F, M. Menger, /. Am. Chem. Soc, 92, 5965 (1970).
[7] H. H. Freedman and R. A. Dubois, Tetrahedron Lett., 1975, 3251.
[8] A. Merz. Angew. Chem., 85, 868 (1973); Angew. Chem. Int. Ed. Engl. 1», 846 (1973).
[9] E. V. Dehmlow and M. Lissel, Tetrahedron Lett., 1976,1783.
[10] J. E. Gordon and R. Ё. Kutina, /. Am. Chem. Soc, 99, 3903 (1977).
[11] J. H. Fendler and E. J. Fendler, "Catalysis in Micellar and Macromolecular Systems,"
Academic Press, New York, 1975.
(12] A. Brandstrom, private communication; ref. [1], p. 307.
[13] D. Landini, A. Maia, and F. Montanari, /. Chem. Soc. Chem. Commun., 1977, 112.
[14] A. Brandstrom and H. Kolind-Andersen, Acta Chem Scand., 29B, 201 (1975).
[15] A. Brandstrom, Acta Chem. Scand., ЗОВ, 203 (1976).
[16] M. Tomoi, T. Takubo, M. Ikeda, and H. Kakiuchi, Chem. Lett., 1976, 473; M. Tomoi, T.
Hasegawa, M. Ikeda, and H. Kakiuchi, Bull. Chem. Soc. Jpn., 52, 1653 (1979).
[17] M. Mikolajczyk, S. Grzejszczak, A. Zatorski, F. Montanari, and" M. Cihquini,
Tetrahedron Lett., 1975, 3757. :
[18] T. Tanaka and T. Mukaiyama, Chem. Lett., 1976, 1259.
415
[19] S. L. Regen, J. Am. Chetn. Soc, 98, 6270 (1976).
[20] M. Cinquini, S. Colonna, H. Molinari, and F. Montanari, J. Chem. Soc. Chem.
Comrmn., 1976, 394.
[21] S. L. Regen and L. Dulak, J. Am. Chem. Soc, 99, 623 (1977); S. L. Regen, J. J. Besse,
and J. McLick, J. Am. Chem. Soc, 101, 116 (1979).
[22] S. L. Regen, /. Am. Chem. Soc, 99, 3838 (1977).
[23] A. W. Herriott and D. Picker, Tetrahedron Lett., \W1, 4521.
[24] H. F. Ebel, "Die Aciditat der CH-Sauren.," G. Thieme Verlag, Stuttgart, 1969.
[25] E. V. Dehmlow and M. Slopianka, unpublished results.
[26] M. Makosza, Pure Appl. Chem., 43, 439 (1975).
[27] M. Makosza, Naked anions—phase transfer, in "Modern Synthetic Methods," R.
Scheffold, Ed., Schweizerischer Chemiker-Verband, Zurich, 1976, p. 7.
[28] E. V. Dehmlow, Angew. Chem. 89, 521 (1977); Angew. Chem. Iht. Ed. Engl., 16, 493
(1977).
[29] M. Lissel, Ph.D. dissertation, Technische Universitat, Berlin, 1977; E. V. Dehmlow and M.
Lissel, /. Chem. Res., 1978 (S) 310; (M) 4163.
[30] E. V. Dehmlow, M. Slopianka, and J. Heider, Tetrahedron Lett., 1977, 2361 (1977).
[31] M. Makosza and E. Bialecka, Tetrahedron Lett., 1977, 183.
[32] E. V. Dehmlow and M. Lissel, unpublished results.
[33] E. V. Dehmlow and T. Remmler, /. Chem. Res. 1977, (S) 72, (M) 766.
[34] S. Barahona-Naranjo, M.S. Diploma thesis, Technische Universitat, Berlin, 1977.
[35] E. V. Dehmlow and S. Barahona-Naranjo, /. Chem. Res. 1979, (S) 238.
[36] S. Lindenbaum and G. E. Boyd, /. Phys. Chem., 68. 911 (1964).
[37] W.-Y. Wen and S. Saito, /. Phys. Chem., 68, 2639 (1964).
[38] J. E. Gordon. J. С Robertson, and R. L. Thorne, /. Phys. Chem., 74, 957 (1970).
[39] S. Shinkai and T. Kunitake, J. Chem. Soc. Perkin Trans. 2, 1976, 980.
[40] Y. Okahata, R. Ando, andT. Kunitake, J. Am. Chem. Soc, 99,3067 (1977); T. Kunitake, Y
Okahata, R. Ando, S. Shinkai, and S. Hirakawa, ibid., 102, 7877 (1980).
[41] A. Tomita, N. Ebina, and Y. Tamai, /. Am. Chem. Soc, 99, 5725 (1977).
[42] A. W. Herriott and D. Picker, Tetrahedron Lett., 1974, 1511. )
[43] S. Colonna and R. Fornasier, Synthesis, 1975, 531. '
[44] M. Makosza and B. Serafinowa, Rocz. Chem., 39, 1223 (1965).
[45] E. V. Dehmlow. M. Slopianka, and E. Menzel, unpublished results.
[46] E. V. Dehmlow and E. Menzel, unpublished results; E. Menzel, State Examination
Thesis, Technische Universitat, Berlin, 1977.
[47] J. Dockx, Synthesis, 1973, 441.
[48] P. A. Verbrugge and E. W. Uurbanus, Germ. Offenl. 2,324,390 (1973); Chem. Abstr.y
80, 70420 (1974).
[49] J. Kranz, State Examination Thesis, Technische Universitat, Berlin, 1977
[50] T Hiyama, M Tsukanaka, and H. Nozaki,/. Am. Chem. Soc, 96, 3713 (1974)
[51] T Hiyama, H Sawada, and M. Tsukanaka, Tetrahedron Lett., 1975, 3013
[52] E V. Dehmlow, M. Lissel, and J. Heider, Tetrahedron, 33. 363 (1977).
[53] M. Makosza, private communication, July 1977
[54] Y Kimura, К Isagawa, and Y Otsuji, Chem. Lett., 1977. 951
[55] K.-H. Wong, J. Chem. Soc. Chem. Commun.. 1978, 282
[56] S. L. Regen. Angew. Chem., 91,464(1979); Angew Chem. Int. Ed. Engl., 18. 421 (1979>,
IV' S. L. Regen, \. Nigam, and J. J. Besse, Tetrahedron Lett.. 1978. 2757 M Tomoi,
M. Ikeda, and H. Kakiuchi, ibid., 1978. 1757
[58] b. L. Regen and A. Nigam,/ Am. Chem. Soc, 100, 7773 (1978)
[59] S. Samaan and F. Rolla. Phosphorus Sulfur. 4, 145 П978).
[60] J. A. Hyatt, J. Org. Chem., 43, 1808 (1978).
[61] B. G. Zupancic and M. Sopcic, Synthesis, 1979, 123.
[62] S. Farhat, R Gallo. and I. Metzger, С R. 4cad. Sci. Ser С 287 581
416
[63] S, Julia, A. Ginebreda, and J. J. Marco, Afinidad, 35, 235 (1978); Chem. Abstr., 90,
22507 (1979).
[64] P. Savignac, T. Cuvigny, and H. Normant, C. R. Acad. Sci. Ser. C, 287, 35 (1978).
[65] M. С Vander Zwan and F. W. Hartner, /. Org. Chem., 43, 2655 (1978).
[66] L. Horner and J. Gerhard, Liebigs Am. Chem., 1980, 838.
[67] D. Landini, A. Maia, and F. Montanari, Nouv. J. Chim., 3, 575 (1979).
[68] J. P. Antoine, 1. de Aguirre, F. Janssens, and F. Thyrion, Bull. Soc. Chim. Fr., 1980, 207.
[69] E. V. Dehmlow and M. Lissel, Liebigs Ann. Chem., 1981, 28.
[70] R. Solaro, S. D'Antone, and E. Chiellini, /. Org. Chem., 45, 4179 (1980).
[71] H. Brink, R. Modin, and J. Vessman, Ada Pharm. Suec, 16, 247 (1979); H. Brink,
Dissertation, Uppsala University, 1980.
[72] A. M. Tivert and K. Gustavii, Ada Pharm. Suec, 16, 1 (1979).
[73] S. Quici and S. L. Regen, J. Org. Chem., 45, 1700 (1980).
[74] W. M. Mac Kenzie and D. С Sherrington, Polymer, 21, 791 (1980).
[75] A. Arbin, H. Brink, and J. Vessman, J. Chromat., 170, 25 (1979).
[76] M. Fedorynski, K. Wojciechowski, Z. Matacz, and M. Makosza, /. Org. Chem., 43, 4682
(1978).
[77] M. Lissel and E. V. Dehmlow, Chem. Ber.;\U, 1210 (1981).
[78] R. Le Goaller, H. Handel, M. A. Pasquini, and J. L. Pierre, Tetrahedron, 35,1437 (1979); R..
Le Goaller, M. A. Pasquini, and i. L. Pierre, ibid., 36, 237 (1980).
[79] E. Buncell and B. Menon, /. Am. Chem. Soc, 99, 4457 (1977).
[80] B. Dietrich and J. M. Lehn, Tetrahedron Lett.,1973, 1225.
[81 ] D. Landini, A. Maia, F. Montanari, and F. M. Pirisi, /. Chem. Soc. Perkin Trans., 2,1980,46,
[82] D. Landini, A. Maia, F. Montanari, and P. Tundo, J. Am. Chem. Soc, 101, 2526 (1979).
[83] P. E. Stott, J. S. Bradshaw, and W. W. Parish, /. Am. Chem. Soc, 102, 4810 (1980).
[84] K. Fukanaga, H. Shirai, S. Ide, and M. Kimura, Nippon Kagaku Kaishi, 1980,1148; Chem..
Abstr., 93, 238497 (1980).
[85] Y. Hayashi, Jap. Kokai 77, 111, 486 (1977); Chem. Abstr., 88, 104653 (1978).
[86] B. J. Garcia, A. Leopold, and G. W. Gokel, Tetrahedron Lett., 1980, 2115.
[87] D. W: Armstrong and H. Kornahrens, Tetrahedron Lett.,1979, 4525.
[88] K. Hiratanai, P. Reuter, and G. Manecke, /. Mol. Catal., 5, 241 (1979).
[89] E. Santaniello, A. Manzocchi, and P. Sozzani, Tetrahedron Lett., 1979, 4581.
[90] L. Токе, G. T. Szabo, and K. Somogyi-Werner, Ada Chim. Acad. Sci. Hung., 101,47 (1979)..
[91] L. Токе, G. T. Szabo, and K. Aranyosi, Ada Chim. Acad. Sci. Hung. 100, 257 (1979).
[92] D. Balasubramanian, P. Sukumar, and B. Chandani, Tetrahedron Lett., 1979, 3543.
[93] D. Landini, A. Maia, F. Montanari, and F. Rolla, /. C. S. Perkin 7>а/и.;1981, 821.
[94] G. D. Yadav and M. M. Sharma, Ind. Eng. Chem. Proc Des. Dev., 20, 385 (1981).
[95] H. Fujihara, K. lmaoka, N. Furukawa, and S. Oae, Chem. Lett., Л9Я1, 1293.
[96] N. Furukawa, K. Kishimoto, S. Ogawa, T. Kawai, H. Fujihara, and S. Oae, Tetrahedron
Lett., 1981, 4409.
[97] T. Hideshima, M. Moringa, and H. Kimizuka, Bull. Chem. Soc. Jpn., 54, 85 (1981).
[98] D. Landini, A. Maia, and G. Podda, /. Org. Chem., 47, 2265 (1982).
К главе 3
[1] A. Brandstrom and B. Lamm, Ada Chem.'Scand. Ser. B, 28, 590 (1974).
[2] E. V. Dehmlow, unpublished results.
[3] R. Ikan, A. Markus, and Z. Goldschmidt, Is. J. Chem., 11, 591 (1973).
[4] С. М. Starks, /. Am, Chem. Soc, 93, 195 (1971).
[5] A. Brandstrom, "Preparative Ion Pair Extraction. An Introduction to Theory and'
Practice," Apotekarsocieteten/Hassle Lakemedel, Stockholm, 1974, pp. 139-140.
[6] J. Goerdeler in Houben-Weyl, "Methoden der Organischen Chemie," Band 11,2, 4..
• Aufl., G. Thieme Verlag, Stuttgart, 1958, pp. 620-622.
[7J R. Kunin and F. X. McGarvey, Ind. Eng. Chem., 41, 1265 (1949).
417
18] С. Kaiser and J. Weinstqck, Org. Synth., 45, 3 (1976).
* [9] Ref. [5] pp. 82, 83, 143, 144.
110] Ref. [5] pp. 83-87, 142-144, 147-148.
[11] M. Makosza and E. Bialecka. Synth. Commun,, 6, 313 (1976).
[12] E. V. Dehmlow, M. Slopianka, and J. Heider, Tetrahedron Lett.; 1977, 2361.
[13] J. Dale and P. O. Kristiansen, Ada Chem. Scand, 26, 1471 (1972).
[14] R. N. Greene, Tetrahedron Lett., 1972, 1793.
[15] G. W. Gokel, D. J. Cram, C. L. Liotta, H. P. Harris, and F. L. Cook, /. Org. Client.,
39, 2445 (1974).
[161 C. J. Pedersen, Org. Synth., 52,66 (1972).
[17] J. Ashby, R. Hull, M. J. Cooper, and E. M. Ramage, Synth. Commun., 4, 113 (1974).
[18] I. J. Burden, А. С Coxon, J. F. Stoddart, and С M. Wheatley, /. Chem. Soc.
Perkin Trans. 1, 1977, 220.
(19] R. С Hayward, С H. Overton, and G. H. Whitham, /. Chem. Soc. Perkin Trans, t, ,
1976, 2413.
[20] G. Johns, C. J. Ransom, and С. В. Reese, Synthesis, 1976, 515.
;[21] F. L. Cook, T. C. Caruso, M. P. Byrne, С W. Bowers, D. H. Speak, and C. L.
Liotta, Tetrahedron Lett., 1974, 4029.
{22] J. Dale and K. Daasvatn, /. Chem. Soc. Chem. Commun., 1976, 295.
[23] B. Dietrich, J. M. Lehn, and J. P. Sauvage, Tetrahedron Lett., 1969, 2885.
[24] R. Noyori, K. Yokoyama, J. Skata, I. Kuwajima, E. Nakamura, and M. Shimizu,
/, Am. Chem. Soc, 99, 1265 (1977).
■'{25] I. Kuwajima, T. Murofushi, and E. Nakamura, Synthesis, 1976, 602.
[26] P. W. Kent and R. С Young, Tetrahedron, 27,4057 (1971).
[27] A. W. Herriot and D. Picker, J. Am. Chem. Soc, 97, 2345 (1975).
[28] A. W. Herriott and D. Picker, Tetrahedron, Lett., 1972, 4521.
[29] A. McKillop, I.-C. Fiaud, and R. P. Hug, Tetrahedron, 30, 1379 (1974).
[30] L. Dalgaard, L. Jensen, and S.-O. Lawessdn, Tetrahedron, 30, 93X1974).
Л31] E. V. Dehmlow and T. Remmler, J. Chem. Res., 1977, (S) 72, (M) 766.
[32] E. V. Dehmlow and M. Lissel, Tetrahedron Lett., 1976, 1783.
[33] Ref. [5], p. 141.
[34] J. v. Ъгшщ, Justus Liebigs.Ann. Chem., 382, 1 (1911).
[35] D. H. Hunter and R. A. Perry, Synthesis, 1977, 37.
[36] W. T. MiMer, Jr., J. H. Fried, and H. Goldwhite, /. Am. Chem. Soc, 82, 3091 (1960).
[37] L. H. Andersen, Finn. Pat. 40,462 (1968); Chem. Abstr., 70, 88036 (1969).
[38] С M. Starks and D. R. Napier,. Brit. Pat. 1,227,144 (1971) = ltal. Pat. 832,967
(1968) = French Pat. 1,573,164 (1969) and further concordances; Chem. Abstr., 72,
115271 (1970).
[39] D. R. Napier and С M. Starks, U.S. Pat. 3,992,432 (1976); Derwent Abstr., 90477
(1976).
[40] D. Landini and F. Rolla, Chem. Ind. (London), 1974, 533.
[41] D. Forster,/. Chem. Soc. Chem. Commun., 197S, 917.
:[42] E. V. Dehmlow and J. Heider, unpublished results.
[43] D. J. Sam and H. E. Simmons, J. Am. Chem. Soc, 96, 2252 (1974).
[44] D. Landini, F. Montanarr, and F. M. Pirisi.y. Chem. Soc. Chem. Commun., 1974,879.
i[451 D. Landini, A. M. Maia, F. Montanari, and F. NL Pirisi, Gazz. Chim. ltal., 105, 863
(1975).
J46] R. Fornasier, F. Montanari, G. Podda, and P. Tundo, Tetrahedron Lett., 1976,1381.
|[47] H. Lehmkuhl and F. Rabet, Germ. Offenl. 2,534,851 (1977).
[48] M. Cinquini, F. Montanari, and P. Tundo, (a)/. Chem. Soc. Chem. Commun., 1975,
, 393; (b) Gazz. Chim. ltal.,WI, 11 (1977).
,[49] M. S. Newman and H. M. Chung, /. Org. Chem., 39,1036 (1974).
418
(50] D Landim, A. M. Maia, R Montanari, and F: M. Pirisi, 3. Chem. Soc. Chem-
Common., 1975,930; D. Landini, A. Maia, and F. Montanari, /.'Am. Chem. Soc, 100,.
2796 (1978)
(51J D. Landini, S. Quid, and F. Rolla, Synthesis, 1975, 430.
[52] D Landini, F Montanari, and F. Rolla, Synthesis, 1974, 428.
[53] С L Liotta and H. P Harris, / Am. Chem. Soc, 96,2250 (1974).
[54] L. Fitjer, Synthesis, 1977, 189.
[55] P Ykman and H. К Hall, Jr., Tetrahedron Lett., 1975, 2429.
[56] M Gross and F Peter, Bull. Soc. Chim. Fr., 1975,871.
[57] E. V Dehmlow and M. Slopianka, Chem. Ber., 112, 2768 (1979).
[58] D. Landini, F Montanari, and F. Rolla, Synthesis, 1974, 37.
[59] D. Bethell, K. McDonald, and K. S. Rao, Tetrahedron Lett., 1977, 1447.
[60] F Jeanne and A. Trichet, L'Air Liquide, Centre d'Etudes Cryogeniques, F-38360*
Sassenage, France, "poster" Journees de Chimie Organique d'Orsay, Societe.
Chimique de France, Sept. 17-19, 1975.
[61] H. Lehmkuhl, F. Rabet, and K. Hauschild, Synthesis, 1977,184.
[62] S. L. Regen, /. Org. Chem., 42, 875 (1977).
[63] С M. Starks and R. M. Owens, J. Am. Chem. Soc, 95, 3613 (1973).
[64] S. L. Regen, J. Am. Chem. Soc, 98, 6270 (1976).
[65] J. W. Zubrick, B. 1. Dunbar, and H. D. Durst, Tetrahedron Lett., 1975, 71.
[66] M. E. Childs and W. P. Weber,/. Org. Chem., 41, 3486 (1976).
[67] G. Simchen and H. Kobler, Synthesis, 1975, 605; H. Kobler, K.-H. Schuster, and.
G. Simchen, JustusLiebigs Ann. Chem., 1978, 1946.
[68] S. L. Regen, J. Am. Chem. Soc, 97, 5956 (1975).
[69] H. B. Copelin and G. B. Crane, U.S. Pat. 2,779,781 (1957); Chem. Abstr., 51,10579-
(1957).
[70] H. Leuchs and K. H. Schmidt, Brit. Pat. 1,200,970 (1970); Chem. Abstr., 73, 87662.
(1970).
[71] H. Coates, R. L. Barker, R. Guest, and A. Kent, Brit. Pat. 1,336,883 (1973); Chem.
Abstr., 80, 70564 (1973).
[72] w: P. Reeves and M. R. White, Synth. Commun., 6, 193 (1976).
[73] N. Sugimoto, T. Fujita, N. Shigematsu, and A. Ayada, Chem. Pharm. Bull., 10,427 П962)..
[74] A. S. Kende and L. S. Liebeskind, /. Am. Chem. Soc, 98, 267(1976).
[75] M. S. Newman, T. G. Barbee, Jr., C.*N. Blakesley, Z. ud fifin, S. Gromelski, Jr.*.
V. K. Khanna, L.-F. Lee, J. Radhakrishnan, R. L. Robey, V. Sahkaran, S. K.
Sankarappa, and J. M. Springer, J. Org. Chem.; 40, 2863 (197i).
[76] K. E. Koenig and W. P. Weber, Tetrahedron Lett., 1914, 2275. ■ '
[77] F. L. Cook, С W. Bowers, and С L. Liotta, /. Org. Chem., 3», 3446 (1974).
[78] L. B. Engemyr, A. Martinsen, and J. Songstad, Acta Chem. Scand. Ser. A., 28, 25$
(1974); J. Songstad, L. J. Stangeland, and T. Austad, ibid., 24, 355 (1970).
[79] L. Eberson and B. Helgee, Chem. Scr., 5,47 (1974).
[80] G. W. Gekel, S. H. Korzeniowski, and L. Blum, Tetrahedron Lett., 1977, 1633.
[81] S. H. Korzeniowski and G. W. Gokel, Tetrahedron Lett., 1977, 1637.
[82] W. P. Reeves, M. R. White* R. G. Hilbrich, and L. L. Biegert, Synth. Commun., 6*.
509 (1976).
[83] С. L. Liotta, E. E. Grisdale, and H. P. Hopkins, Jr., Tetrahedron Lett., 1975, 4205..
[84] A. Knochel and G. Rudolph, Tetrahedron Lett., 1974, 3739.
[85] W. P. Reeves and M. L. Bahr, Synthesis, 1976, 823.
[86] A. Brandstrom, B. Lamm, and I. Palmertz, Acta Chem. Scand. Ser. B, 28, 699 (1974).
[87] K. J. MacNay, E. R. Rogier, and M. M. Kreevoy, Germ. Oflehlegungsschrift 2,245,.
611; Chem. Abstr., 81, 63387 (1974).
[88] M. Takeishi, Y. Naito, and M. Okawara, Angew. Makromol. Chem., 28, 111 (1973).
[89} M. Takeishi, R. Kawashima, and M. Okawara, Makromol. Chem., 167, 261 (1973)..'
[90] B. Graham, U.S. Pat. 2,866,802 (1958); Chem. Abstr., 53, 9146 (1959).
^[91] N. P. Smetankina and N. 1. Miryan, Zh. Obshch. Khim., 39, 2299 (1969) (Engl.
transl. p. 2239); N. P. Smetankina and N. N. Laskovenko, Zh. Obshch. Khim., 42,
617 (1972) (Engl. transl. p. 613).
[92] G. E. Vennstra and B. Zwaneburg, Synthesis, 1975, 519.
[93] G. D. Hartman and S. Ё. Biffer, J. Org. Chem., 42, 1468 (1977).
[94] S. H. Korzeniowski, L. Blum, and G. W. Gokel, J. Org. Chem., 42, 1469 (1977).
[95] V. D. Parker and L. Eberson, J. Chem. Soc. Chem. Commun., 1972, 441.
[961 H. E. Hennis, L. R. Thompson, and J. P. Long, Ind. Eng. Chem. Prod. Res. Dev.,
1968, 96; Chem. Abslr., 69, 18796 (1968).
, [97] J. H. Wagenknecht, M. M. Baizer, and J. L. Chruma, Synth. Commun., 2,215 (1972).
[98] R. H. Mills, M. W. Farrar, and O. J. Weinkauf, Chem. hid (London), 1962, 2144.
[99] R. L. Merker and M. J. Scott,/. Org. Chem., 26, 5180 (1961).
r{100] Y. YamashitaandT. Shimamura,KogyoRagakuZasshi.W,423(1957); Chem. Abstr.,
53, 9025 (1959).
[101] R. Kay, Brit. Pat. 916,772 (1963); Chem. Abstr., 59, 2728 (1963) and Brit. Pat.
966,266 (1964); Chem. Abstr., 61, 10629 (1964).
[102] T. Torn, S. Kurozumi, T. Tanaka, S. Miura, M. Kobayashi, and S. Ishimoto,
Synthesis, 1974, 867.
■1103] E. V. Dehmlow, unpublished results, 1974.
[104] K. Holmberg and B. Hansen, Tetrahedron Lett., 1975, 2303.
[105] Ref. 5, p. 155.
[106] Ref. 5, pp. 109-111.
[107] F. С V. Larsson and S.-O. Lawesson, Tetrahedron, 28, 5341 (1972).
;[108] H. Ehrsson, Acta Pharm. Suec, 8, 113 (1971); Chem. Abstr.,75,44,678 (1971); J. E.
Greving, J. H. G. Jonkman, and R. A. de Zeeuw, J. Chromatogr., 148, 389 (1978).
[109] A. Knochel, J. Oehler, and G. Rudolph, Tetrahedron Lett., 1975, 3167.
[110] C. L. Liotta, H. P. Harris, M. McDermott, T. Gonzalez, and K. Smith, Tetrahedron
Lett., 1974, 2417
[111] M. D. Durst, Tetrahedron Lett., 1974, 2421.
[112] "S. Akabori and M. Ohtomi, Bull. Chem. Soc. J/m., 48, 2991 (1975).
[113] E. Grushka, H. D. Durst, and E. J. Kikta, Jr.,/. Chromatogr., 112, 673 (1975).
[114] H. D. Durst, M. Milano, E. J. Kikta, Jr., S. H. Connelly, and E. Grushka, Anal.
Chem., 47, 1797 (1975).
[115] A. Padwa and D. Dehm, J. Org. Chem., 40, 3139 (1975).
[116] G. Gelbard and S. Colonna, Synthesis, 1977, 113.
[117] H. Normant, T Cuvigny, and P. Savignac, Synthesis, 1975, 805.
[118] H. Normant and С Laurenco, С R, Acad. Sci. Ser. C, 283, 483 (1976).
[119] P. i. Garegg, T Iversen, and S. Oscarson, Carbohydr. Res., 53, C5 (1977).
:[120] H. Schick, Pharmazie, 30, 817 (1975).
[121] S. Schwarz and G. Weber, Z. Chem., 15, 270(1975).
[122] S. Schwarz, G Weber, M. Wahren, and M. Herrmann, Z. Chem., 16, 439 (1976).
[123] H. J Fex, S K. Kristensson, and A. R. Stamvik, Germ. Offenleg, 2,629,657 (1977).
,[124] R. A. Bauman. Synthesis, 1974, 870. *
[125] R. W Ridgway. H. S Greenside, and H. H. Freedman, J. Am. Chem. Soc, 98,1979
(1976).
[126] A Zwierzak, Synthesis, 1975, 507.
[127] A Zwierzak and I Brylikowska, Synthesis, 1975, 712.
[128] A Zwierzak and A Sulewska, Synthesis, 1976, 835.
[129] A Zwierzak. Synthesis, 1976, 305.
1130] Bayer A G. Germ. Offenl. 2,442,883 (1976); Derwent Abstr., 22819 (1976).
4131] Bayer A G., Germ. Offenl. 2,512,498 (1976); Derwent Abstr., 38189 (1976).
[132] Wacker-Chemie GmbH., Belg. Pat. 795,616 (1972); Derwent Abstr., 50232 (1973).
■ШЗ] M A lohnson and К Yang, U.S. Pat. 3,641,172 (1972); Chem. Abstr., 7V99072
< 1972)
-420
[134] Hokko Chem. Ind., Jap. Kokai 75,14,607; Chem. Abstr., 83, 27529 (1975).
[135] W. Schoenlebcn, J. Daiow, H. Hoffmann, and S. Winder!, Germ. Offenl. 2,149,822
(1973); Chem. Ahstr.,19, 52768 (1973).
[136] R. P. Johnson, U.S. Pat. 3,941,827 (1976); Chem. Abstr., 84, 179 707 (1976).
[137] Ethyl Corp., Belg. Pal. 841,131 (1976); Derwenl Abstr., 83041 (1976).
[138] Dow Chemical Corp., U.S. Pat. 3,564,252 (1976); Derwenl Abstr., 20403 (1976).
[139] С M. Starks, U.S. Pat. 3,725,458 (1973); Chem. Abstr., 78, 158991 (J973).
[140] С M. Starks and R. D. Gordon, Germ. Offenl. 2 103,547 (1971); Chem. Abstr.,7ST
117 993(1971).
[141] С Kimura, K. Kashiwaya, and K. Murai, Chem. Abstr., 84, 121027 (1976); C.
Kimura, K. Murai, Y. Ishikawa, and K. Kashiwaya, Chem. Abstr., 87,133781 (1977).
[142] M. Hiraoka, T. Nakamura, and T. Ozeki, Jap. Kokai 76,06,928; Chem. Abstr.x
164159^1976).
[143] Toyfo Chemical, Jap. Kokai 76,06,927; Derwent Abstr., 17505 (1976).
[144] Uniroyal Inc., Germ. Offenl. 2,558,163 (1976); Derwent Abstr., 54478 (1976).
[145] S. Paul and N. Ranby, Macromolecules, 9, 337 (1976).
[146] A. J. Coary and D. D. Cozad, U.S. Pat. 3,873,589 (1975); Chem. Abstr., 82, 170037
(1975).
[147] K. D. MacKay, E. R. Rogier, and M. M. Kreevoy, U.S. Pat. 3,707,495 (1972); Chem.
Abstr,, 78091 (1973).
[148] K. F. Zeiiner and H. Appel, Germ. Offenl. 2,126,296 (1972); Chem. Abstr., 78,58473
(1973).
[149] Y. Nadachi and M. Kokura, Jap. Kokai 75,95,289; Chem. Abstr., 84, 164852
(1976).
[150] D. G. Brady, U.S. Pat. 3,674,750 {\912);.Chem. Abstr., 77, 89339 (1972).
[151] Y. Nadachi and M. Kokura, Jap. Kokai 73,84,529; Chem. Abstr., 84, 30650 (1976).
[152] P. Edwards, U.S. Pat. 2,537,981 (1951); Chem. Abstr., 45, 5177 (1951).
[153] W. J. Heilman, U.S. Pat. 3,661,938 (1972J; Chem. Abstr., 77, 49108 (1972).
[154] M. Yasuda, M. Sakai, H. Ono, and M. Tanaka, Jap. Pat. 72,41,884 (1972); Chem.
Abstr., 78, 44212 (1973).
[155] Gulf Res. and Dev. Co., Jap. Pat. 72,11,365; Derwent Abstr., 46931 (1972).
[156] M. Yoshino, S. Sato, H. Mochida, and M. Shibata, Jap. Pat. 71,37,326(1971); Chem.
Abstr., 76, 154695 (1972).
[157] N. Hashi and Y. Takase, Jap. Pat. 72,47,366; Chem. Abstr., 78, 125136 (1973).
•[158] F. N. Bodnaryuk, M. A. Korshunov, V. M. Melekhov, et al., U.S.S.R. Pat. 357,196
(1972); Chem. Abstr., 78, 160443 (1973).
[159] S. Sato, K. Kamezawa, and M. Shiba, Jap. Pat. 73,04,006; Chem. Abstr., 78,160320
(1973).
[160] T. Horii, K. Kawamata, T. Yagi, H. Sasaki, and S. Nose, Jap. Kokai 73,39,423;
Chem. Abstr., 80, 17641 (1974).
[161] Gulf Res. and Dev. Co., U.S. Pat. 3,957,831 (1976); Derwent Abstr., 41873 (1976).
[162] G. Maerke?, J. F. Carmichael, and W. Port, J. Org. Chem., 26, 2681 (1961).
[163] R. Kohler and H. Pietsch, Germ. Pat. 944,995 (1956); Chem. Abstr., 53, 1831 (1959).
[164] Ciba-Geigy A. G., Jap. Pat. 72,10,822; Derwent Abstr., 42486 (1972).
[165] Shell Int. Res. Mij NV, Belg. Pat. 793,432 (1973); Derwent Abstr., 39744 (1973);
Netherl. Pat. 7,105,426 (1972); Chem. Abstr., 78, 98456 (1971).
[166] Ciba, S. A., Belg. Pat. 739,526 (1970); Derwent Abstr., 21737 (1970).
[167] H. Matsuda, Y Tange, and F Yamauchi, Jap. Pat. 70,36,733; Chem. Abstr., 74,
126854 (1971).
[168] A. Heer apd W. Schaffner, Swiss Pat. 536,835 (1973); Chem. Abstr,, 79,79726 (1973).
[169] A. Suzui and K. Matsumoto. Jap. Pat. 70,04,975 (1970); Chem. Abstr., 100314 (1970).
[170] F. H. Sinnema and G. C. Vegter, Brit. Pat. 1,205,180 (1970); Chem. Abstr., 73.
130686 (1970).
421
[171] W. Hoffmann and K. v. Fraunberg, Germ. Offenl. 2,324,047(1974); Chem. Abstr.,S2,
73244(1975).
{172] D. G. Brady, U.S. Pat. 3,843,719 (1974); Chem. Abstr., 82, 16570(1975).
(173] A. Suzui and T. Horii, Jap. Pat. 70,36,737 (1970); Chem. Abstr., 74, 125 197 (1971).
i[!74] R. A. Gray and D. G. Brady, U.S. Pat. 3,879,445 (1975); Chem. Abstr., 84, 31962
(1976).
({175] R. Kay, French Pat. 1,391,727 (1965); Chem. Abstr., 63,10134 (1965).
1176] H. Trautmann, East Germ. Pat. 105,620 (1974); Dement Abstr., 56825 (1974).
1177] J. G. Scruggs andJD. G. Brady, U.S. Pat. 3,676,484 (1972); Chem. Abstr., 77,126249
(1972).
[178] J. Berthoux and G. Schwachhofer, Germ. Offenl. 2,338,552 (1974); Chem. Abstr., 80,
133063 (1974).
[179] Hodoga Chem. Ind., Jap. Kokai 76,065,725; Derwent Abstr., 56607 (1976).
[180] Union Camp. Corp., Netherl. Pat. Appl. 6,909,697 (1969).
[181] T. Hayashi and H. Hitoshi, Jap. Pat. 73,30,611; Chem. Abstr., 80,26771 (1974).
[182] T. Nishikubo, S. Ukai, H. Idemitsu, and M. Kishida, Jap. Kokai 72,17,715; Chem.
Abstr., 77, 139450 (1972).
*[183] M. A. Korshunov, R. G. Kuzovleva, and I. V. Furaeva, U.S.S.R. Pat. 432,128(1974);
, Chem. Abstr., 81, 77496 (1974).
{184] F. N. Bodnaryak, M. A. Korshunov, V. E. Lazaryants, V. M. Melekhov, A. I.
Ezrielev, A. V. Lebedev, A. B. Peiznev.and L. V. Utkinn, Brit. Pat. 1,242,980(1969);
Chem. Abstr., 76, 4320 (1976); Germ. Offenl. 1,960,716 (1971); Chem. Abstr., 75
77491 (1971). ,
[185] Y. Inamori, Y. Yanagawa, and I. Iwasa, Jap. Pat. 74,02,098; them. Abstr., 81,
77493 (1974).
[186] J. E. Masters, U.S. Pat. 3,351,674 (1967); Chem. Abstr., 68,22480 (1968).
[187] E. Muller, O. Bayer, and H. Morschel, Germ. Pat. 959,497 (1957); Chem. Abstr., 53,
13665 (1959).
[188] M. Tomikawa, H. Kaji, and K. Ueda, Chem. Abstr., 68,40263 (1968).
[189] J. F. Yanes and O. L. Castellanos, An. R. Soc. Esp. Fis. Quim. Ser. B, 57, 815 (1962);
Qiem. Abstr., 57, 7455 (1962).
[190] B. E. Jennings, Brit. Pat. 907,649 (1962); Chem. Abstr., 58, 5810 (1963).
[191] Z. Jedlinski and D. Sek, Polimer, 14,105 (1969); Chem. Abstr., 71, 39537 (1969).
[192] A. Conix and U. L. Laridon, Belg. Pat. 563,173 (1958) and 565,478 (1958); Chem.
Abstr., 55; 25356 (1961).
[193] A. J. Conix and L. M. Dohmen, Germ. Pat. 1,199,500 (1962); Chem. Abstr., 63,
16502 (1965).
[194] Politechnica Warszawska, East Germ. Pat. 101,687 (1974); Derwent Abstr., 13584
. ^ (1974).
[195] Gevaert Photo-Prod. N.V., Belg. Pat. 6Q0.053 (1961); Chem. Abstr., 58,11491 (1963);
Belg. Pat. 602,793 (1961); Chem. Abstr., 59, 15405 (1963).
[196] E. P. Goldberg and F. Scardiglia, U.S. Pat. 3,271,368 (1966); Chem. Abstr., 66, 3015
(1967).
{197] Dow Chemical Co., U.S. Pat. 3,972,887 (1976); Derwent Abstr., 63289 (1976).
1198] L. M. Kroposki, M. Yoshimine, and H. Freedman, U.S. Pat. 3,917,621 (1975);
Chem. Abstr., 84, 121436 (1976).
1199] L. M. Kroposki and M. Yosbimine, U.S. Pat. 3,907,815 (1975); Chem. Abstr., 84,
43864 (1976).
[200] Dow Chemical Co., Belg. Pat. 832,047 (1976); Derwent Abstr., 13078 (1976).
[201] A. W. Herriott and D. Picker, J. Am. Chem. Soc., 97, 2345 (1975).
{202] D. Landini and F. Rolla, Synthesis, 1974, 565.
{203] A. Tozzi and P. Cassandrini, Germ. Offenl. 2,513,805 (1975); Chem. Abstr., 84,4476
(1976).
482
[204] I. К. Kim and J. S. Noh, Chem. Abstr., 82, 124967 (1975).
[205] A. W. Herriott and D. Picker, Synthesis, 1975, 447.
[206] S. Cabiddu, A. Maccioni, and M. Secci, Synthesis, 1976, 797.
[207] N. H. Nilsson and A. Senning, Chem. Ber., 107, 2345 (1974).
[208] L. Dalgaard and S.-O. Lawesson, Ada Chem. Scand. Ser. B, 28, 1077 (1974).
[209] L. Dalgaard and S.-O. Lawesson, Tetrahedron, 28, 2051 (1972).
[210] T. Nakabayashi, S. Kawamura, T. Horii, and M. Hamada, Chem. Lett., 1976, 869.
*[211] A. T. Babayan and M. G. Indzhikyan, Dokl. Akad.Nauk. Arm. SSR, 28, 67 (1959);
Chem. Abstr., 64, 1368 (1960); Chem. Zentralbl., 1965, 14-0870.
[212] H. F. Ebel, " Die Aciditat der CH-Sauren," G. Thieme Verlag, Stuttgart, 1969.
*[213] A. J. Gordon and R. A. Ford, "The Chemist's Companion, Handbook of Practical
Data, Techniques, and References," Wiley-Interscience, New York, 1972, pp. 60-64.
[214] M. Makosza, Pure Appl. Chem., 43, 439 (1975).
[215] G. W. Gokel, S. A. Di Biase, and B..A. Lipisko, Tetrahedron Lett., 1976, 3495.
[216] D. J. Cram, C. A. Kingsburg, and B. Rickborn, J. Am. Chem. Soc., 83, 3688 (1961).
[217] A. Schrieschein and C. A. Rowe, Jr.,/. Am. Chem. Soc, 84, 3160 (1962).
[218] D. Bethell and A. F. Cockerill, J. Chem. Soc. B, 1966, 913.
[219] M. J. Maskornick, Tetrahedron Lett., 1972, 1797.
[220] R. A. Bartsch, G. M. Pruss, R. L. Buswell, and R. A. Bushaw, Tetrahedron Lett.t
1972, 2621.
[221] R. A. Bartsch and К. Е. Wiegers, Tetrahedron Lett., 1972, 3819.
[222] R. A. Bartsch, E. A. Mintz, and R. M. Parlman,/. Am. Chem. Soc., 96,4249 (1974).
♦[223] Survey: L. F. and M. Fieser, "Reagents for Organic Syntheses," Vol. I, J. Wiley &
Sons, New York, 1967, p. 1252.
[224] J. Jarrousse, С R. Acad. Sci. Ser. C, 232, 1424 (1951).
*[225] A. T. Babayan, M. G. Indzhikyan, and T. A. Azizyan, Dokl. Akad. Nauk Army.
SSR, 31, 79 (1960); Chem. Abstr., 55, 11342 (1961); Chem. Zentralbl., 1966, 5-0867.
•[226] A. T. Babayan and M. G. Indzhikyan, Zh. Obshch. Khim., 27, 1201 (1957), Engl.
transl. p. 1284; Chem. Abstr., 52, 3707 (1958); Chem. Zentralbl., 1961, 2246.
[227] S. Korzeniowski, L. Blum, and G. W. Gokel, Tetrahedron Lett., 1977, 1871.
[228] W. J. Belanger, U.S. Pat. 2,940,953 (1960); Chem. Abstr., 55,1072 (1961); U.S. Pat.
1 2,928,810 (1960); Chem. Abstr., 54, 13729 (1960).
[229] R. D. Gordon, U.S. Pat. 3,914,320 (1975); Chem. Abstr., 84, 30423 (1976); U.S. Pat.
3,824,295 (1974); Chem. Abstr., 81, 77464 (1974).
[230] С M. Starks, U.S. Pat. 3,931,238 (1976); Chem. Abstr., 84, 104988 (1976).
[231] H. Baw, J. Indian Chem. Soc, 3, 101 (1926).
1232] H. H. Freedman and R. A. Dubois, Tetrahedron Lett., 1975, 3251.
1233] A. Merz, Angew. Chem., 85, 868 (1973), Angew. Chem. Int. Ed. Ettgl, 11, 846 (1973).
[234] E. V. Dehmlow and J. Schmidt, Tetrahedron Lett., 1976, 95.
[235] A. Merz and R. Tomahogh, Chem. Ber., 110, 96 (1977).
1236] J. Villieras, С Baquet, and J, F. Normant, J. Organomet. Chem., 97, 355 (1975).
[237] B. Cazes and S. Julia, Tetrahedron Lett., 1974, 2077; Bull. Soc. Chim. Fr., 1977,
925.
[238] B. Cazes and S. Julia, Synth. Commun., 7, 113 (1977).
[239] P. J. Garegg, T. Ivensen, and S. Oscarson, Carbohydr. Res., 50, C12 (1976).
[240] P. di Cesare and B. Gross, Carbohydr. Res., 48, 271 (1976).
[241] G. J. H. Rail, M. E. Oberholzer, D. Ferreira and D. G. Roux, Tetrahedron Lett.,
1976, 1033.
[242] J. D. Daley, J. M. Rosenfeld, and E. V. Young-Lai, Steroids, 27, 481 (1976).
[243] K. Olsson, Ada Chem. Scand. Ser. B, 29, 405 (1975).
[244] A. P. Bashall and J. F. Collins, Tetrahedron Leu., 1975, 3489. ,
[245] Takasago Perfumery, Jap. Kokai 76,023,265, Derwent Abstr., 27219 (1976).
[246] D. Picker, Ph.D. Dissertation, State University of New York at Albany, 197J4;
Diss. Abstr., 35, 3229 (1975). !
423
[247] Dow Chemical Co., U.S. Pat. 3,969,360 (1976); Derwent Abstr., 57726 (1976).
[248J P- M. Quail and S. R. Korn, Germ. Offenl. 2,634,419 (1977).
[249] J. Ugelstad, T. Ellingsen, and A. Berge, Ada Chem. Scand., 20, 1S93 (1966); L. M.
Thomassen, T. Elligsen, and J. Ugelstad, ibid., 25, 3024 (1971).
[250] H. Brotell, H. Ehrsson, and O. Gyllenhaal, J. Chromatogr., 78, 293 (1973).
*t251] A. T. Babayan and A. A. Grigoryan, Izv. Akad. Nauk Arm. SSR, Fiz. Mai., Estestv.
Tekh. Nauki, 8, 81 (1955); Chem. Abstr., 50, 10023 (1956); Chem. Zentralbl. 1959,
4446.
[252] R. Kreher and K. J. Herd, Z. Naturforsch, Teil B, 29, 683 (1974).
[253] K. Berg-Nielsen and E. Bernatek, Ada. Chem. Scand., 26, 4130 (1972).
(254] A. Jonczyk, J. Wlostpwska, and M. Makosza, Synthesis, 1976, 795.
[255] (a) Ref. [5], p. 156. (b) ibid., p. 112.
[256] U. Junggren, Dissertation Goteborg, 1972, cited by Brandstrom, ref. [5].
[257] R. Brehme, Synthesis, 1976,113; East Germ. Pat.,117,446 (1976); Derwent Abstr., 24554
(1976); R. Brehme and P. Thelen, J. Prakt. Chem., 323, 299 (1981).
[258] D. Landini and F. Rolla, Synthesis, 1976, 389.
[259] M.-T. Maurette, A. Lopez, R. Martino, and A. Lattes, С R. Acad. Sci. Ser. C, 282,
599 (1976).
1260] J. Palecek and J. Kuthan, Synthesis, 1976, 550.
[261] A. Jonczyk and M. Makosza, Rocz. Chem., 49, 1203 (1975).
[262] V. Bocchi, G. Gasnati, A. Dossena, and F. Villani, Synthesis, 1976, 414.
[263] A. Barco, S. Benetti, G. P. Pollini, and P. G. Baraldi, Synthesis, 1976, 124.
•[264] N. N. Suvorov, Y. I. Smushkevich, V. S. Velezhevan, V. S. Rozhkov, and S. V,
Simakov, Khim. Ceterotsikl. Soedin., 12, 191 (1976); Engl. transl., p, 167.
[265] T. Greibrokk, Ada Chem. Scand., 26, 3305 (1972).
[266] H. Ehrsson and A. Tilly, Anal. Lett., 6, 197 (1973).
1267] M. Evrik and K. Gustavii, Anal. Chem. 46, 39 (1974).
[268] H. Ehrsson, Anal. Chem., 46, 922 (1974).
[269] С Hoppel, M. Garle, and M. Elander, J. Chromatogr., 116, 53 (1976).
(270] O. Gyllenhaal and H. Ehrsson, J. Chromatogrr/lVJ, 237 (1975); O. Gyllenhaal,
N. Tjarnlund, H. Ehrsson, and P. Hartvig, ibid., 156, 275 (1978).
[271] A. Arbin and.P.-O. Edlund, Ada Pharm. Suec, 12, 119 (1975).
[272] A. Zwierzak ind J. Brylikowska-Piotrowicz, Angew. Chem., 89, 109 (1977); Angew.
Chem. Int. Ed. Engl., 16, 107 (1977).
[273] S. Cacchi, F. La Torre, and D. Misiti, Synthesis, 1977, 301.
[274] H. J.-M. Dou and J. Metzger, Bull. Soc. Chim. Ft., 1976, 1861.
[275] J. Masse, Synthesis, 1977, 341.
[276] A, Suzui and T. Horii, Jap. Pat. 68,29,940; Chem. Abstr., 70, 68П0 (I960).,
[277] R. W. Miller and J. A. Schmitt, U.S. Pat. 3,193,528 (1965); Chem. Abstr., 63,11730
(1965).
[278] F. J. Ponder, Germ. Pat. 1,806,870 (1969); Chem. Abstr., 71, 101545 (1969).
1279] H. Wunderlich, D. Lugenheim, and A. Stark, East Germ. Pat. 51,642 (1966); Chem.
Zentrolbl, 1969, 14-1986.
[280] M. Makosza and A. Jonczyk, Org. Synth., 55, 91 (1976).
1281] M. Makosza and B. Serafinowa, Rocz. Chem., 39, 1223 (1965).
1282] M. Makosza and B. Serafinowa, Rocz. Chem., 39, 1401 (1965).
:[283] M. Makosza and B. Serafinowa, Rocz. Chem., 39, 1595 (1965).
|[284] M. Makosza and B. Serafinowa, Rocz. Chem., 39, 1799 (1965).
£285] M. Makosza and B. Serafinowa, Rocz. Chem., 39, 1805 (1965).
P86] M. Makosza and B. Serafinowa, Rocz. Chem., 40, 1647 (1966).
(287] M. Makosza and B. Serafinowa, Rocz. Chem., 40, 1839 (1966).
(288] J. Lange and M. Makosza, Rocz. Chem., 41, 1303 (1967).
[289] M. Makosza, 1. Kmiotek-Skarzyiiska, and M. Jawdosiuk, Synthesis, 1977, 56.
424
[290] P. Vittorelli, J. Peter-Katalinic, G. Mukherjee-Muller, H.-J. Hansen, and H. Schmid,
Helv. Chim. Acta, 58, 1379 (1975).
[291] M. Makosza, B. Serafinowa, and M. Jawdosiuk, Rocz. Chem., 41, 1037(1967),
[292] M. Makosza, Rocz. Chem., 43, 79 (1965).
[293] M. Makosza, B. Serafinowa, and T. Bolestawska, Rocz. Chem., 42,817 (1968).
[294] M. Makosza, Rocz. Chem., 43, 333 (1969).
[295] M. Makosza and T. Goetzen, Rocz. Chem., 46, 1239 (1972).
[296] M. Makosza, E. Bialecka, and M. Ludwikow, Tetrahedron Lett., 1972, 2391.
[297] B. Gutkowska, Acta Pol. Pharm., 30, 109 (1973); Chem. Abstr., 79, 78552 (1973).
[298] M. Makosza and E. Bialecka, Tetrahedron. Lett., 1977, 183.
[299] H. KomeLli-Zadeh, H. J.-M. Dou, and J. Metzger, С R. Acad. Sci. Ser. C, 283, 41
(1976).
[300] M. Makosza and M. Fedorynski, Synthesis, 1974, 274.
[301] M. Makosza, M. Ludwikow, and A. Urniaz, Rocz. Chem., 49, 297 (1975).
[302] M. Makosza and M. Ludwikow, Angew. Chem., 86, 744 (1974); Angew. Chem. Int,
Ed. Engl., 13, 665 (1974).
[303] M. Makosza, Tetrahedron Lett., 1969, 673.
[304] M. Makosza, J. M. Jagusztyn-Grochowska, and M. Jawdosiuk, Rocz. Chem., 45,
, 851 (1971).
[305] M. Makosza and M. Ludwikow, Bull. Acad. Pol. Sci. Ser. Sci. Chim., 19, 231 (1971).
[306] M. Makosza, J. M. Jagusztyn-Grochowska, and M. Jawdosiuk, Rocz. Chem., 50^
1841 (1976).
[307] M. Makosza, M. Jagusztyn-Grochowska, M. Ludwikow, and M. Jawdosiuk,
Tetrahedron, 30, 3723 (1974).
[308] M. Makosza and J. M. Jagusztyn-Grochowska, Rocz. Chem., 50,1859 (1976).
[309] M. Makosza and M. Jawdosiuk, Bull. Acad. Pol. Sci. Ser. Sci. Chim., 16, 597 (1968).
[310] M. Makosza, Tetrahedron Lett., 1969, 677.
[311] A. Jonczyk, Bull. Acad. Pol. Sci. Ser. Sci. Chim., 22, 849 (1974).
[312] T. Tanaka and T. Mukaiyama, Chem. Lett., 1976, 1259.
[313] M. Makosza and M. Ludwikow, Rocz. Chem., 51, 829 (1977).
[314] Sumitomo Chemical KK, Netherl. Pat. Appl. 7,513,627 (1976); Derwent Abstr.,
45249 (1976).
[315] Sigurta Farm. Spa., Belg. Pat. 837,624 (1976); Derwent Abstr., 46067 (1976).
[316] S. L. Regen and L. Dulak, J. Am. Chem. Soc, 99, 623 (1977).
[317] L. Rylski and F. Gajewski, Acta Pol. Pharm., 26, 115 (1969); Chem. Abstr., 71,
38741 (1969).
[318] G. Carenini, R. D'Ambrosio, M. Carissimi, E. Grumelli, E. Milla, and F. Ravenna,
Farmaco Ed. Sci., 28, 265 (1973); Chem. Abstr., 78, 159023 (1973).
[319] M. Barreau and M. Julia, Tetrahedron Lett., 1973, 1537.
[320] M. Makosza and T. Goetzen, Organ. Prep. Proced. Int., 5, 203 (1973).
[321] A. Brandstrom and U. Junggren, Tetrahedron Lett., 1972,473.
[322] Y. Masuyama, Y. Ueno, and Okawara, Tetraltedron Lett., 1976, 2967.
[323] E. D'Incan and J. Seyden-Penne, Synthesis, 1975, 516.
[324] J. Blanchard, N. Collignon, P. Savignac, and H. Normant, Tetrahedron, 32, 455
(1976).
[325] J. Blanchard, N. Collignon, P. Savignac, and H. Normant, Synthesis, 1975, 655.
[326] A. Brandstrom and H. Junggren, Acta Chem. Scand., 23, 2203 (1969).
[327] U. SchSHkopf, D. Hoppe, and R. Jentsch, Chem. Ber., 108, 1580(1975).
[328] A. M. van Leusen, R. J. Bouma, and O. Possel. Tetrahedron Lett., 1975, 3487.
[329] R. K. Singh and S. Danishefsky, J. Org. Chem., 40, 2969 (1975), Organ. Symh., 60, 66
(1981).
[330] A. Jonczyk, B. Serafinowa, and J. Czyzewski, Rocz. Chem., 47, 529 (1973).
[331] A. Jonczyk, B. Serafin, and M. Makosza, Rocz. Chem., 45, 1027 (1971).
[332] A. Jonczyk, B. Serafin, and M. Makosza, Rocz. Chem., 45, 2097 (1971).
425
1333] M. Makosza, A. Jonczyk, В. Serafinowa, and Z. Mroczek, Rocz. Chem., 47, 77
(1973).
[334] A. Jonczyk, B. Serafin, and M. Makosza, Tetrahedron Lett., 1971, 1351.
[335] A. Jonczyk and T. Pytlewski, Roscz. Chem., 49, 1425 (1975).
[336] A. Jonczyk, B. Serafin, and E. Skulimowska, Rocz. Chem., 45, 1259 (1971).
[337] A. Jonczyk, M. Fedorynski, and M. Makosza, Rocz. Chem., 48, 1713 (1974).
[338] A. Jonczyk, M. Ludwikow, and M. Makosza, Rocz. Chem., 47, 89 (1973).
[339] M. Makosza and M. Fedorynski, Rocz. Chem., 45, 1861 (1971).
*[340] A. T. Babayan, N. Gambaryan, and N. P. Gambaryan, Zh. Obshch. Khim., 24, 1887
(1954); Chem. Abstr., 49, 10879 (1955); Chem. Zentralbl. Sonderb., 1950/54, 4532.
[341] M. A. Boudville and H. des Abbayes, Tetrahedron Lett., 1975, 2727.
[342] B. Koutek, L. Pavlickova, and M. Soucek, Collect. Czech. Chem. Commun., 39, 192
(1974).
[343] A. Jonczyk, M. Ludwikow, arid M. Makosza, Rocz. Chem., 51, 175 (1977).
[344] A. Jonczyk, K. Banco, and M. Makosza, /. Org. Chem., 40, 266 (1975).
•[345] I. S. Aul'chenko, L. A. Kheifits, T. I. Konstantinova, and T. P. Cherkasova, Chem.
Abstr., 84, 73591 (1976).
[346] J. Itakura and H. Ito, Jap. Kokai 73,00,515; Chem. Abstr., 78, 71443 (1973).
[347] Y. Tamai, T. Nishida, Y. Ohmura, T. Hosogai, Y. Ninagawa, Y. Fujita, and K. Itoi,
Jap. Kokai 75,96,514; Chem. Abstr., 84, 4483 (1976).
1348] Y. Tamai, T. Nishida, F. Mori, Y. Ohmura, M. Tanomura, T. Hosogai, Y. Ninagawa,
and K. Itoi, Germ. Offenl. 2,356,866 (1974); Chem. Abstr., 81, 77475 (1974).
[349] H. K. Dietl and К. С Brannyck, Tetrahedron Lett., 1973, 1273.
[350] BASF, Germ. Offenl. 2,513,996 (1976); Derwent Abstr., 76050 (1976).
[351] H. des Abbayes and С Neveau, С R. Acad. Sci. Ser. C, 278, 805 (1974).
*[352] V. M. Andreev, A. I. Bibicheva, and M. I. Zhuravleva, Zh. Org. Khim., 10, 1470
(1974), Engl. transl., p. 1479.
'[353] H. D. Zook and W. L. Gumby, /. Am. Chem. Soc, 82, 1386 (1960).
[354] W. Beck and M. Girnth, Chem. Ber., 109, 965 (1976).
[355] B. Samuelsson and B. Lamm, Ada Chem. Scand., 25, 1535 (1971).
[356] U. JunggreruPh.D. Dissertation, Goteborg, 1972, as cited in ref. [5], p. 122.
[357] M. Makosza; Bull. Acad. Pol. Sci. Ser. Sci. Chem. 15, 165 (1967).
[358] M. Makosza, Tetrahedron Lett., 1966, 4621.
[359] B. Dietrich and J. M. Lehn, Tetrahedron Lett., 1973, 1225.
(360] M. Makosza, in: "Modern Synthetic Methods 1976," R. Scheffold, Ed., Schwei-
zerischer Chemikerverband, Zurich, 1976, p. 7.
[361] F. Guibe and G. Brain, Bull. Soc. Chim. Fr., 1975, 933.
[362] H. E. Zaugg and A. D. Schaefer, /. Am. Chem. Soc, 87, 1857 (1965).
[363] E. M. Arnett and V. M. De Palma, /. Am. Chem. Soc, 98, 2447 (1976).
[364] С Riche, C. Pascard-Billy, С Cambillau, and G. Bram, /. Chem. Soc. Chem.
Commun., 1977, 183.
[365] E. A. Noe and M. Raban, /. Chem. Soc. Chem. Commun., 1976, 165.
[366] E. A. Noe and M. Raban, /. Am. Chem. Soc, 96, 6184 (1974).
[367] R. Gelin, S. Gelin, and A. Galliaud, Bull. Soc. Chem. Fr., 1973, 3416.
[368] Summary in: W. J. le Noble, "Highlights of Organic Chemistry," Marcel Dekker
Inc., New York, 1974, p. 821 ff.
[369] A. Brandstrom and U. Junggren, Acta Chem. Scand., 25, 1469 (1971).
[370] H. Kolind-Andersen, R. Dyrnesli, and S.-O. Lawesson, Bull. Soc Chim. Belg., 84,
341 (1975).
[371] B. Miller, H. Margulies, T. Drabb, and R. Wayne, Tetrahedron Lett., 1970, 3805.
[37,2] F. Guibe, P. Sarthou, and G. Bram, Tetrahedron, 30, 3139 (1974).
[373] H. Kolind-Andersen and S. O. Lawesson, Acta Chem. Scand. Ser. B, 29,430 (1975).
[374] A. Brandstrom and U. Junggren, Acta Chem. Scand., 23, 2536 (1969).
[375] A. Brandstrom and U. Junggren, Acta Chem. Scand., 23, 3585 (1969).
426
[376] A. Brandstrom and U. Junggren, Acta Chem. Scand., 23, 2204 (1969).
[377] C. Cambillau, P.'Sartou, and G. Bram, Tetrahedron Lett., 1976, 281; С Cambillau,
G. Bram, J. Corset, С Riche, and С Pascard-Billy, Tetrahedron, 34, 2675 (1978).
*[378] A. L. Kurts, S. M. Sakembaeve, I. P. Beletskaya, and O. A. Reutov.ZA. Org. Khim.,
10, 1512 (1974); Engl. transl., p. 1588.
*[379] A. L. Kurts, P. 1. Derh'yanov, 1. P. Beletskaya, and O. A. Reutov.ZA. Org. Khim.,9,
1313 (1973): Engl. transl., p. 1341.
[380]" E. D'Incan and P. Viout, Tetrahedron, 31, 159 (1975).
[381] J. H. Clark and J. M. Miller,./. Chem. Soc. Chem. Commun., 1977,64;/. Chem. Soc.
Perkin Trans., 1, 1977, 1743.
[382] G. Nee and B. Tchoubar, С R. Acad. Sci. Ser. C, 283, 223 (1976).
[383] J.-C. Fiaud, Tetrahedron Lett., 1975, 3495.
[384] E. V. Dehmlow, M. Lissel, and J. Heider, Tetrahedron, 33, 363 (1977).
[385] G. Hata, Jap. Kokai 74,109,346; Chem. Abstr., 82, 124876 (1975).
[386] B. Cederlund and A.-B. Hornfeldt, Ada Client. Scand., 25,3546 (1971).
[387] B. Cederlund, A. Jesperson, and A.-B. Hornfeldt, Acta Chem. Scand, 25, 3656 (1971).
[388] B. Cederlund and A.-B. Hornfeldt, Chem. Scrip., 8, 140 (1975).
[389] S. Gronowitz and T. Frejd, Acta Chem. Scand. Ser. В 30,439 (1976).
1390] В. Cederlund and A.-B. Hornfeldt, Acta Chem. Scand. Ser. B, 30, 101 (1976).
[391 ] L. Dalgaard, H. Kolind-Andersen, and S.-O. Lawesson, Tetrahedron, 29,2077 (1973).
[392] L. Jensen, L. Dalgaard, and S.-O. Lawesson, Tetrahedron, 30, 2413 (1974).
[393] P. Hassanaly, H. J.-M. Dou, and J. Metzger, G. Assef, and J. Kister, Synthesis,
1977, 253.
[394] H. J.-M. Dou, P. Hassanaly, and J. Metzger, /. Heterocycl. Chem., 14, 321 (1977).
[395] N. O. Vesterager, E. B. Pedersen, and S.-O. Lawesson, Tetrahedron, 29, 321 (1973).
[396] T. Bianchi and L. A. Cate, /. Org. Chem., 41, 2031 (1977).
1397] H. D. Durst and L. Liebeskind, /. Org. Chem., 39, 3271 (1974).
1398] A. W. Burgstahler, M. E. Sanders, С G. Shaefer, and L. O. Weigel, Synthesis, 1977,
405.
1399] В. Т. Golding and P. V. loannou, Synthesis, 1977, 423.
[400] E. V. Dehmlow, unpublished results.
[401] Y. Jasor, M. Gaudry, and A. Marquet, Tetrahedron Lett., 1976, 53.
[402] K. Yamada, M. Kamai, Y. Nakano, H. Kasimura, and H. Iida, Tetrahedron Lett.,
1974, 1741.
[403] W. J. Spillane, H. J.-M. Dou, and J. Metzger, Tetrahedron Lett., 1976, 2269.
[404] W. J. Spillane, P. Hassanaly, and H. J.-M.Dou, С R. Acad. Sci. Ser. C, 283, 289
(1976).
[405] T. Higgin, W. J. Spillane, H. J.-M. Dou, and J. Merger* С R. Acad. Sci. Ser. C,
284, 929 (1977).
[406] Y. Yano, T. Okonogi, and W. Tagaki, /. Org. Chem., 38, 3912 (1973).
*[407] B. A. Trofimov, S. V. Amasova, and V. V. Nosyreva, Zh. Org. Khim., 12, 1366
(1976); Engl. transl., p. 1358.
[408] M. Makosza, J. Czyewski, and M. Jawdosiuk, Org. Synth., 55, 99 (1976).
[409] M. Makosza, Tetrahedron Lett., 1966, 5489.
[410] M. Makosza and M. Jawdosiuk, Bull. Acad. Pol. Sci. Ser. Sci. Chim., 16, 589 (1968).
*[411] L. F. and M. Fieser, "Reagents for Organic Synthesis," J. Wiley and Sons, Inc.,
New York, 1967, (a) p. 1252, (b) p. 933.
[412] H. A. Bruson, in: "Organic Reactions," R. Adams, Ed., Vol. 5, J. Wiley and Sons,
Inc., New York, 1949, p. 79.
[413] D. A. White and M. M. Baizer, J. Chem. Soc. Perkin Trans. 1,1973, 2230.
[414] S. Hoz, M. Albeck, and Z. Rappoport, Synthesis, 1975, 162.
[415] T. Yanami, M. Kato, and A. Yoshikoshi, /. Chem. Soc. Chem. Commun., 1975, 726.
[416] I. Belsky, /. Chem. Soc. Chem. Commun., 1977, 237.
[417] С L. Liotta, A. M. Dabdoub, and L. H. Zalkow, Tetrahedron Lett., 1977, 1117.
28* 427
[418] Т. Sakakibara and R. Sudoh, /. Org. Chem., 40, 2823 (1975).
[419] T. Sakakibara, M. Yamada, and R. Sudoh, J. Org. Chem., 41, 736 (1976).
[420] M. Fedorynski, I. Gorzkowska, and M. Makosza, Synthesis, 1977, 120.
[421] A. Joifczyk and M. Makosza, Synthesis, 1976, 387.
[422] I. Artaud, J. Seyden-Penne, and P. Viout, С R. Acad. Sci. Ser. C, 283, 503 (1976).
[423]'T. Wakabayashi and Y. Kato, Tetrahedron Lett., 1977, 1235.
[424] H. Wynberg and R. Helder, Tetrahedron Lett., 1975, 4057.
[425] T. Sakakibara and R. Sudoh, Carbohydr. Res., 50, 197 (1976).
[426] J. M. Mclntosh and H. Khalil,/. Org. Chem., 42, 2123 (1977).
[427] S. Kurozumi, T. Toru, T. Tanaka, S. Miura, M. Kobayashi, and S. I. Ishimoto,
Bull. Chem. Soc. Jpn., 50, 1357 (1977).
[428] A. Merz, Angew. Chem., 89, 484 (1977); Angew. Chem. Int. Ed. Engl., 16, 467 (1977).
[429] С Hansson and E. Rosengreen, Ada Chem. Scand. Ser. B, 30, 871 (1976).
[430] E. Buncel and B. Menon, J. Am. Chem. Soc, 99, 4457 (1977).
[431] A. M. van Leusen and J. Wildeman, Synthesis, 1977, 501.
• [432] W. P. Reeves and R. G. Hilbrich, Tetrahedron, 32, 2235 (1976).
[433] M. Makosza, A. Kacprowicz, and M. Fedorynski, Tetrahedron Lett., 1975, 2119.
[434] J. N. Roitman and D. J. Cram, J. Am. Chem. Soc, 93, 2231 (1971).
[435] J. Solodar, tetrahedron Lett., 1971, 287.
. [436] J. Solodar, Synth. Inorg. Met. Org. Chem., 1, 141 (1971).
[437] S. Akabori, M. Ohtomi, and K. Arai, Bull. Chem. Soc. Jpn., 49, 746 (1976).
, {438] V. Dryanska and C. Ivanov, Tetrahedron Lett., 1975, 3519.
[439] G. Cardillo, D. Savoia, and A. Umani-Ronchi, Synthesis, 1975, 453.
. [440] G. W. Gokel, H. M. Gerdes, and N. W. Rebert, Tetrahedron Lett., 1976, 653.
.•[441] I. N. Rozhkov, N. D. Kuleshova, and I. N. Knunyants, Izv. Akad. Nauk SSSR Ser.
Khim., 22, 128 (1973); Engl. transl., p. 124. ■
[442] E. Nakamura and I. Kuwajima, Angew. Chem., 88, 539 (1976); Angew. Chem. Int.
Ed. Engl., 15, 498 (1976).
[443] E. Nakamura, M. Shimizu, and I. Kuwajima, Tetrahedron Lett., 1976, 1699.
'[444] D. A. Evans and L. K. Truesdale, Tetrahedron Lett., 1973, 4929.
[445] D. A. Evans, J. M. Hoffman, and L. K. Truesdale, J. Am. Chem. Soc, 95,5822 (1973).
[446] H. des Abbayes and A. Buloup, J. Chem. Soc. Chem. Commun., 1978, 1090.
,.[447] E. V. Dehmlow, unpublished rg*ults.
[448] J. Jonczyk, M. Fedorynski, and M. Makosza, Tetrahedron Lett., 1972, 2395.
[449] T. Koizumi, K. Takeda, K. Yoshida, and E. Yoshii, Synthesis, 1977, 497.
i [450] F. Naso and L. Ronzini, /. Chem. Soc. Perkin Trans 1, 1974, 340.
[451] E. J. Corey and В. В. Snider, J. Am. Chem. Soc, 94, 2549 (1972).
;[452] Т. Н. Chan and W. Mychajlowskij, Tetrahedron Lett., 1974, 171.
[453] J. Hayami, N. Ono, and A. Kaji, Tetrahedron Lett., 1970, 2727.
[454] R. A. Bartsch, J. R. Allaway, and J. G. Lee, Tetrahedron Lett., 1977, 779.
[455] R. A. Bartsch, Ace. Chem. Res., 8, 239 (1975).
[456] D. Landini, S. Quici, and F. Rolla, Synthesis, 1975, 397.
[457] J. B. Campbell, U.S. Pat. 3,639,493 (1972); eq. to Fr. Demande 2,002,799 (1969);
Chem. Abstr., 72, 78366 (1970).
[458] J. B. Campbell and R. E. Tarney, French Pat. 1,525,661 (1968); Chem. Abstr., 71,
80648(1969).
, [459] J. B. Campbell and R. E. Tarney, U.S. Pat. 3,981,937 (1976); Derwent Abstr.,
75550(1976).
[460] du Pont de Nemours Co., Netherl. Pat. Appl. 6,707,711 (1967); Bayer-Patentschnell-
ber., 11-008(1968).
[461] du Pont de Nemours Co., Netherl. Pat. Appl. 6,903,151 (1969); Bayer-Patentschnell-
ber., 51-034(1969).
[462] R, G. Gordon, U.S. Pat. 3,664,966 (1972); Chem. Abstr., 77, 75756 (1972). i
428
[463] К. Sennewald, К. Gehrmann, and G, Viertel, Germ. Pat. 1,271,107 (1968); them.
Zentralbl, 1969, 18-1806.
[464] E. F. Lutz, J. T. Kelly, and D. W. Hall, Brit. Pat. 1,112,068 (1968); Chem. Abstr., 69,
10079 (1968).
[465] H. Baader, K. Sennewald, and H. Reis, Brit. Pat. 1,154,445 (1969); Chem. Abstr., 71,
30094 (1969).
[466] J. Dockx, Synthesis, 1973, 441.
[467] Unpublished results by A. Verstraelen, W. Helsen, and J. Dockx, cited in ref. [466],
p. 449.
[468] M. S. Newman, B. Dhawan, M. M. Hashem, V. K. Khanna, and J. M. Springer,
J. Org. Chem., 41, 3925 (1976).
[469] A. Gorgues, С R. Acad. Sci. Ser. C, 278, 287 (1974).
[470]. A. Gorgues, and A. LeCoq,,Bu//. Soc. Chim. Fr. 11,1976, 125.
[471] A. Gorgues and A. LeCojq, Tetrahedron Lett., 1976, 4723.
[472] E. T. McBee and R. O. Bolt, lnd. Eng. Chem., 39, 412 (1947).
[473] J. H. Fendler and E. J. Fendler," Catalysis in Micellar and Macromolecular Systems,"
Academic Press, New York, 1975.
.1474] J. H. Fendler, Ace. Chem. Res., 9, 153 (1976).
[475] F. M. Menger, J. U. Knee,and H. K. Rhee, J. Org. Chem.,40,3803 (1975).
.[476] E. Josefowicz and R. Soloniewicz, Mitteilungsbl. Chem. Ges. DDR, Sonderheft 1959,,
Katalyse, p. 215.
[477] E. V. Dehmlow and S. Barahona, unpublished results; S. Barahona, Diploma Thesis,
Technical University, Berlin 1977.
[478] H. J. Bestmann, in: "Neuere Methoden des praparativen organischenChemie," Vol.
5, W. Foerst, Ed., Verlag Chemie, Weinheim, 1967, p. 1.
[479] U. Schollkopf, Angew. Chem., 71, 260 (1959).
[480] R. J. Anderson and С A. Henrick, J. Am. Chem. Soc, 97, 4327 (1975).
[4811 M. Schlosser, "Topics in Stereochemistry," Vol 5, E. L. Eliel and N. L. Allinger,
Eds., Wiley-Interscience, New York, 1970, p. 1.
[482] W. P. Schneider, J. Chem. Soc. Chem. Commun., 1969, 7^5.
[483] G. Markl and A. Merz, Synthesis, 1973, 295.
[484] R. M. Boden, Synthesis, 1975, 784.
[485] W. Tagaki, I. Inoue, Y. Yano, and T. Okonogi, Tetrahedron Lett., 1974, 2587.
[486] R. Broos and M. Anteunis, Synth. Commun., 6, 53 (1976).
[487] S. Hiinig and I. Stemmler, Tetrahedron Lett., 1974, 3151.
[488] K. W. Lee and L. A. Singer, J. Org. Chem., 39, 3780 (1974).
[489] J. Boutagy and R. Thomas, Chem. Rev., 74, 87 (1974).
[490] T. H. Kinstle and B. Y. Mandanas, J. Chem. Soc. Chem. Commun., 196», 1699.
[491] G. Jones and R. F. Maisey, J. Chem. Soc. Chem. Commun., 1968, 543.
[492] M. Mikolajczyk, S. Grzejszczak, W. Midura, and A. Zatorski, Synthesis, 1976, 396.
[493] M. Mikolajczyk, S. Grzejszczak, W. Midura, and A. Zatorski, Synthesis, 1975, 278.
{494] С Piechucki, Synthesis, 1976, 187.
[495] С Piechucki, Synthesis, 1974, 869.
[496] E. D'Incan, Tetrahedron, 33, 951 (1977).
[497] Y. Yano, T. Okonogi, M. Sunaga, and W. Tagaki, J. Chem. Soc. Chem. Commun.,
1973, 527.
E498J A. Merz and G. Markl, Angew. Chem., 85, 867 (1973); Angew. Chem. Int. Ed. Engl.,
11, 845 (1973).
[499] M. J. Hatch, J. Org. Chem., 34, 2133 (1969).
[500] T. Hiyama, T. Mishima, H. Sawada, and H. Nozaki, J. Am. Chem. Soc, 97, 1626
(1975).
[501] T. Hiyama, T. Mishima, H. Sawada, and H. Nozaki, /. Am. Chem. Soc, 98, 641
(1976).
£94
[502] К. Kleveiand, L. Skatteb0l, and L. K. Sydnes, Ada Chem. Scand. Ser. B, 31, 463
(1977).
[503] E. d'Incan and J. Seyden-Penne, С R. Acad. Sci. Ser. C, 281, 1031 (1975).
[504] C. J. Pedersen and H. K. Frensdorff, Angew. Chem., 84,16 (1972); Angew. Chem. Int.
Ed. Engl, 11, 16 (1972).
[505] C. J. Pedersen, J. Am. Chem. Soc, 89, 7017 (1967).
[506] D. H. Hunter, W. Lee, and S. K. Sim,/. Chem. Soc. Chem. Commun., 1974, 1018.
[507] J. Smid, S. Shah, L. Wong, and J. Hurley, J. Am. Chem. Soc, 97, 5932 (1975).
[508] H. Ledon, Synthesis, 1974, 347.
[509] M. Nakajima and J.-P. Anselme, Tetrahedron Lett., 1976, 4421.
[510] H. Alper and D. Des Roches, J. Organomet. Chem., 117, C44 (1976).
[511] H. Alper, H. Des Abbayes, and D. Des Roches, J. Organomet. Chem., 121 C31
(1976).
[512] L. Cassar, M. Foa, and A. Gordano,/. Organomet. Chem., 121 C55 (1976).
[513] Dow Chemical Co., U.S. Pat. 3,974,199 (1976); Derwent Abstr., 64914 (1976).
[514] S. Yanagida, Y. Noji, and M. Okafiara, Tetrahedron Lett., 1977, 2337, 2893.
[515] A. C. Knipe, N. Sridhar, and A. Loughran,/. Chem. Soc. Chem. Commun., 1976,630.
[516] H. Iida, K. Takahashi, and K. Yamada, Nippon Kagaku Kaishi, 1974, 2127; Chem.
Abstr., 83, 27739 (1975); Chem. Informationsdienst, 1975, 11-248.
|517] К. С Chan, S. H. Goh, S. E. Teoh, and W. H. Wong, Aust. J. Chem., 27,421 (1974).
[518] J. R. Grunwell and S. L. Dye, Tetrahedron Lett., 1975, 1739.
[519] L. Rand and M. J. Albinak, J. Org. Chem., 25, 1837 (1960).
[520] J. Hayami, N. Ono, and A. Kaji, Tetrahedron Lett., 1968, 1385.
[521] M. Mitami, T. Tsuchida, and K. Koyama, Chem. Lett., 1974, 1209.
[522] M. Makosza, B. Serafinowa, and I. Gajos, Rocz. Chem., 43, 671 (1969).
[523] T Gajda and A. Zwierzak, Synthesis, 1976, 243.
[524] M. Guainazzi, G. Silvestri, and G. Serravalle, J. Chem. Soc. Chem. Commun., 1975,
200.
K[525] S. J. Bakum and S. F. Ereshko, lzv. Akad. Nauk SSSR Ser. Khim., 23, 2138 (1974),
Engl. transl., p. 2058.
(526] J. L. Pierre and H. Handel, Tetrahedron Lett., 1974, 2317.
[527] J. L. Pierre, H. Handel, and R. Perrand, Tetrahedron, 31, 2795 (1975).
[528] H. Handel and J. L. Pierre, Tetrahedron, 31, 2799 (1975).
[529] A. Loupy, J. Seiden-Penne, and B. Tschoubar, Tetrahedron Lett., 1976, 1677.
[530] E. A. Sullivan and A. A. Hinckley, J. Org. Chem., 27, 3731 (1962).
(531] A. Brandstrom, H. Junggren, and B. Lamm, Tetrahedron Lett., Wll, 3173.
(532] Ref. [5], p. 165.
(533] D. J. Raber and W C. Guida, J. Org. Chem., 41, 690 (1976).
<534] D. I. Raber and W С Guida, Synthesis, 1974, 808.
(535] N H. Nilson and A. Senning, Chem. Ber., 107, 2345 (1974).
(536] R. O. Hutchins and D. Kandasamy, J. Am. Спет. Soc, 9§, 6131 (1973).
15371 R О Hutchins, D. Kandasamy, С A. Maryanoff, D. Masilamani, and В. Е.
Maryanoff, J Org. Chem., 42, 82 (1977).
(538] l. О Hutchins and D. Kandasamy, J. Org. Chem., 40, 2530 (1975).
(539] i Matsuda and K. Koida, Bull. Chem. Soc. Jpn., 46, 2259 (1973).
(540] Ref. (5], pp. 164-166.
l>41 j 3. Bradamante, A. Merchesi, and U. M. Pagnoni, Ann. Chim. {Rome), 65, 131 (1975).
1542J Balcells, S. Colonna, and R. Fornasier, Synthesis, 1976, 266.
i>4J] H Masse and E. R Parayre,</. Chem. Soc. Chem. Commun., 1976, 438.
1.544] В Witkop and С. М. Foltz, J Am. Chem. Soc, 79, 197 (1957).
O45] t. V Dehmlow, J Heider, U. Brenner, unpublished results.
O46J H Durst, J W Zubrick, and G. R. Kieczykowski, Tetrahedron Lett., 1974, 1777
15471 H des Abbayes and H. Alper, / Am. Chem. Soc, 99, 98 (1977).
430
[548] H. Alper, D. Des Roches, and H. des Abbayes, Angew. Chem., 89. 43 (1977): Angew.
Chem. Int. Edit. Engl, 16, 41 (1977).
[549J- H. Alper, K. D. Logbo, and H. des Abbayes, Tetrahedron Lett., 1977. 2861
[550] G. Borgogno, S. Colonna, and R. Fornasier, Synthesis, 1975, 529.
[551] K. Y. Hui and B. L. Shaw, J. Organomet. Chem., 124, 262 (1977).
[552] R. Rebois, J. Chauveau, M. Deyris, and H. Condanne, C. R. Acad. Sci. Ser С 282.
947 (J976),
[553] Imp. Chem. Ind. Ltd., Belg. Pat. 833,241 (1976); Derwent Abstr., 22647 (1976).
[554] R. Helder, R. Arends, W. Bolt, H. Hiemstra, and H. Wynberg, Tetrahedron Lett..
1977, 2181.
[555] Kohjin, KK, Jap. Kokai 76,105,024 Derwent Abstr., 82123 (1976).
[556] P. D. Woodgate, H. H. Lee, P. S. Rutledge, and R. С Cambie, Synthesis, 1977, 462,
[557] D. J. Sam and H. E. Simmons, J. Am. Client. Soc., 94, 4024 (1972).
[558] E. Weber and F. Vegtle, Chem. Ber., 109, 1803 (1976).
[559] A. W. Herriott and D. Picker, Tetrahedron Lett., 1974, 1511.
[560] M. S. Newman, H. M. Dali, and W. M. Hung,/. Org. Chem., 40, 263 (1975).
[561] H. H. Pohl and K. Dimroth, Angew. Chem., 87, 135 (1975); Angew. Chem. Int. Ed.
Engl., 14, 111 (1975).
[562] F. Vegtle and W. Offermann, Chem. Exp. Didakt., 1, 147 (1975).
[563] S. G. Boot, M. R. Boots, K. E. Guyer, and P. E. Marecki, J. Pharnx. Sci., 65, 1374
(1976).
[564] G. W. Gokel and H. D. Durst, Synthesis, 1976, 168.
[565] M. Mack and H. D. Durst, unpublished results, 1975, cited in ref. [564] as ref. 155.
[566] Ref. [5], p. 161.
[567] W. P. Weber and J. P. Shepherd, Tetrahedron Lett., 1972, 4907.
[568] M. L. Richardson, Analyst {London), 87, 435 (1962); J. M. Matusek and Т. Т. Sugi-
hara, Anal. Chem., 33, 35 (1961).
[569] N. A. Gibson and J. W. Hosking, Aust. J. Chem., 18, 123 (1965).
[570] F. Yamashita, A. Atsumi, and H. Inoue, Nippon Kagaku Kaishi, 1975, 1102; Chem.
Abstr., 83, 113378 (1975); Chem. Informationsdienst, 1975, 48-162.
[571] H. D. Durst, unpublished results, cited in ref. [564] as ref. 181, 189.
[572] E. Alneri, G. Bottaccio, and V. Carletti, Tetrahedron Lett., 1977, 2117.
[573] B. J. Garcia, G. W. Gokel, and D. W. Tudor, unpublished results, cited in ref. [564]
as ref. 182.
[574] J. Jarrousse and J.-C. Raulin, С R. Acad. Sci. Ser. C, 284,'503 (1977).
1575] R. M. Boden, Synthesis, 1975, 783.
[576] J. S. Valentine and A. B. Curtis, J. Am. Chem. Soc, 97, 224 (1975).
1577] I. Rosenthal and A. Frimer, Tetrahedron Lett., 1975, 3731.
[578] E. J. Corey, К. С Nicolaou, M. Shibasaki, Y. Machida, and С S. Shiner, Tetrahedron
Lett., 1975, 3183..
[579] M. V. Merritt and D. T. Sawyer, J. Org. Chem., 35, 2157 (1970).
[580] J. San Filippo, Jr., C.-I. Chern, and J. S. Valentine,/. Org. Chem., 40, 1678 (1975).
[581] R. A. Johnson and E. G. Nidy, /. Org. Chem., 40, 1680 (1975).
[582] M. J. Gibian and T. Ungermann,/. Org. Chem., 41, 2500 (1976).
[583] W. С Danen and R. J. Warner, Tetrahedron, 11, 989 (1977).
[584] M. V. Meritt and R. A. Johnson, J. Am. Chem. Soc, 99, 3713 (1977).
[585].R. A. Johnson, Tetrahedron Lett., 1976, 331.
1586] J. San Filippo, Jr., L. J. Romano, C.-I. Chern, and J. S. Valentine,/. Org. Chem., 41,
586 (1976).
[587] J. W. Peters and С S. Foote, J. Am. Chem. Soc, 98, 873 (1976).
[588] C.-I. Chern and J. San Filippo, Jr.,/. Org. Chem., 42, 178 (1977).
[589] A. Frimer and I. Rosenthal, Tetrahedron Lett., 1976, 2809.
[590] I. Rosenthal and A. Primer, Tetrahedron Lett., 1976, 2805.
1591] Y. Мого-Oka, P. J. Chung, H. Arakawa, and T. Ikawa, Chem. Lett., 1976,1293.
431
. Nishinaga, I. Shimizu, and T Matsuura, Chem. Lett., 1977, 547.
[593] E. Lee-Ruff, A. B. P. Lever, and J. Rigaudy, Can. J. Chem., 54, 1837 (1976),
1594] G. Cardillo, M. Orena, and S. Sandri, J. Chem. Soc. Chem. Commwu, 1976,190.
[595] G. A. Lee and H. H. Freedman, Tetrahedron Lett., 1976,1641.
[596] G. Brunow and S. Sumelius, Ada Chem. Scan. Ser. B, 29.499 (1975).
1597] A. R. Qureshi and B. Sklarz, /. Chem. Soc, C, 1966, 412.
[598] R. Pappo, D. S. Alien, Jr., R. U. Lemieux, and W. S. Johnson, J. Org. Chem., 21.
478 (1956),
[599] С M. Starks and D. R. Napier, S. African. Pat. 7,101,495 (1971) s Brit, Pat.
1,324,763 (1973); Chem. Abstr., 76, 153191 (1972).
[600] С M. Starks and P. H. Washecheck, U.S. Pat. 3,547,962 (1970); Chenu Abstr., 74,
140895 (1971).
[601] J. R. Adamson, R. Bywood, D. T. Eastlick, G. Gallagher, D. Walker, and E. M.
Wilson, J. Chem. Soc. Perkin Trans. 1,1975, 2030; R. Bywood, G. Gallagher, G. K.
Sharma, and D. Walker, ibid., 1975, 2019. ^-.
[602] K. Takahashi and H. lida. Synthesis, 1979, 301.
[603] R. Helder, J. С Hummelen, R. W. P. iv£ Laane, J. S. Wiering, and H. Wynbcrg,
Tetrahedron Lett., 1976, 1831; r/, also H. Wynberg. Cliimia, 30,445 (1976).
[604] E. V. Dehmlow and J. Schmidt, unpublished results (1975).
[605] W. Kirmse, "Carbene Chemistry," Second Ed., Academic Press, New York and
London, 1971, Chapters 4, 8,.9, 10, and 11.
[606] W. von E. Doering and A. K. Hoffmann,/. Am. Chem. Soc, 76, 6162 (1954).
[607] G. С Robinson, Tetrahedron Lett., 1965, 1749.
[608] M. Ma,kosza and W. Wawryzniewicz, Tetrahedron Lett., 1969, 4659.
[609] P. S. Skell and M. S. Cholod, J. Am. Chem. Soc, 91, 6035, 7131 (1969).
[610] G. Kobrich, H. Buttner,and E. Wagner, Angew. Chem., 82,177(1970); Angew. Chem.
Int. Edit. Engl., 9, 169 (1970).
[611] D. Seyferth, M. E. Gordon, J. Y.-P. Mui, and J. M. Burlitch, J. Am. Chem. Soc, 89,
959, 4953 (1967).
[612] R. A. Moss and F. G. Pilkiewicz, J. Am. Chem. Soc, 96, 5632 (1974).
[613] E. V. Dehmlow and M. Lissel, J. Chem. Res, 191%, (S)310, (M)4163.
[614] W. M. Wagner, H. Kloosterziel, S. van <^r Ven, and A. F. Bickel, Лес Trav. Chim.
Pays-Bas; 81, 925, 933, 947 (1962).
[615] H. J.-M. Dou, H. Komeili-Zadeh, and M. Crozet, С R. Acad. Sci. Ser. C, 284, 685
(1977).
[616] P. A. Verbrugge and E. W. Uurbanus, Germ. Offenl. 2,324,390 (J973); s DutchPat.
7,306,662 (1973) as Fr. Pat. 2,184,793; Chem. Abstr., 80, 70420 (1974).
1617] K. Isagawa, Y. Kimura, and S. Kwon, J. Org. Chem., 39, 3171 (1974).
*I618] R. R. Kostikov and A. P. Molchanov, Zh. Org. Khim., 11,1767 (1975); Engl. transl.»
p. 1767.
[619] M. Mqkosza, private communication.
[620] Shell B.V., Brit. Pat. 1,436,854 (1976); Derwent Abstr., 40579 (1976).
[621] T. Hiyama, M. Tsukanaka, and H. Nozaki.y. Am. Chem. Soc, 96, 3713 (1974).
[622] T. Hiyama, H. Sawada, M. Tsukanaka, and H. Nozaki, Tetrahedron Lett., 1975,
3013.
[623] Y. Kimura, Y. Ogaki, K. Isagawa, and Y. Otsuji, Chem. Lett., 1976, 1149.
[624] E. V. Dehmlow; Angew. Chem., 89, 521 (1977); Angew. Chem. Int. Ed. Engl., 16, 493
(1977).
[625] E. V. Dehmlow, Angew. Chem., 86, 187 (1974); Angew. Chem. Int. Ed. Engl., 13, 170
(1974).
[626] E. V. Dehmlow and J. Schfinefeld, Justus Liebigs Ann. Chem., 744, 42 (1971).
[627] G. W. Gokel, J. P. Shepherd, W. P. Weber, H. G. Boettger, J. L. Holwick, and D. J.
• McAdoo, J. Org. Chem., 38,1913 (1973).
432
[628] M. J. Aroney, К. E. Calderbank, and H. J. S toot man, /. Chem. Soc. Perkin Trans. 1,
1973, 2060.
[629] E. Wada, S. Fujisaki, A. Nagashima, and S. Kajigaeshi, Bull. Chem. Soc. Jpn., 48,
739 (1975).
[630] F. Effenberger and W. Kurtz, Chem. Ber., 106, 511 (1973).
[631] K.-O. Henseling and P. Weyerstahl, Chem. Ber., 108,2803 (1975).
[632] S. Kajigaeshi, N. Kuroda, G. Matsumoto, E. Wada, and A. Nagashima, Tetrahedron
Lett., 1971, 4887.
[6331 K. Kobayashi and J. B. Lambert, J. Org. Chem., 42,1254 (1977).
[634] G. W. Gokel, J. P. Shepherd, and W. P. Weber, J. Org. Chem., 38,1913 (1973).
[635] M. Makosza and I. Gajos, Rocz. Chem., 48, 1883 (1974).
[636] G. C. Joshi, N. Singh, and L. M. Pande, Tetrahedron Lett., 1972, 1461.
[637] G. C. Joshi, N. Singh, and L. M. Pande, Synthesis, 1972, 317.
[638] S. Takano, K. Yuta, and K. Ogasawara,,Heterocycles, 4, 947 (1976).
[639] R. lkan, A. Markus, and Z. Goldschmidt, J. Chem. Sot. Perkin Trans. 1, 1972,
2423.
[640] Y. M. Sheikh, J. Leclercq, and C. Djerassi, J. Chem. Soc. Perkin Trans. 1,1974, 909.
[641] G. Hammen, T. Bassler, and M. Hanack, Chem. Ber., 107, 1676 (1974).
[642] S. S. Hixon, J. Am. Chem. Soc, 97, 1981 (1975).
[643] M. Nakazaki, K. Yamamoto, and J. Yanagi, J. Chem. Soc. Chem. Commun., 1977,
346.
[644] E. V. Dehmlow and G. С Ezimora, Z. Naturforsch. Teil B, 30, 825 (1975).
[645] K. Kawashima, T. Saraie, Y. Kawano, and I. Ishiguro, Germ. Offenl. 2,404,744
(1974); Chem. Abstr., 81, 135997 (1974); Belg. Pat. 809,816 (1973); Derwent Abstr.J
43495 (1974).
*[646] N. I. Yakushkina, L. F. Germanova, V. D. Klebanova, L. I. Leonova, and 1. G.
Bolesov, Zh. Org. Khim., 12, 2141 (1976); Engl. transl., p. 2082.
[647] E. Dunkelblum and B. Singer, Synthesis, 1975, 323.
[648] T Sasaki, S. Eguchi, and M. Mizutani, Org. Prep. Proc. Int., 6, 57 (1974).
[649] T Sasaki, S. Eguchi, and Y Hirako, Tetrahedron Lett., 1976, 541.
[650] R. B. Miller, Synth. Commun., 4, 341 (1974).
[651] R. A. Moss and D. J. Smudin, J. Org. Chem., 41, 611 (1976).
[652] R. F Boswell and R. G. Bass, J. Org. Chem., 40, 2419 (1975), 42, 2342 (1977).
[653] Y Gaoni, Tetrahedron Lett., 1976, 2167.
[654] L. Pizzala, J.-P Aycard, and H. Bodot, С R. Acad. Set. Ser. C, 285,9 (1977).
[655] D. Davalian and P. J. Garratt.y. Am. Chem. Soc, 97, 6883 (1975).
[656] T Hiyama, T Mishima, K. Kitatani, and H. Nozaki, Tetrahedron Lett., 1974, 3297.
*[6571 M. G. Voronkov, S. M. Shostakovskii, V G. Kozyrev, Y V Artst, L. N. Balabanova,
and O. B. Bannikova, U.S.S.R. Pat. 488,814 (1976); Chem. Abstr., 84, 43839 (9176).
*'{658] A A. Bredikhin and V V Plemenkov, Zft. Org: Khim., 12,1001 (1976); Engl. transl.,
p. 1011
*[6591 N. V Kuznetsov and I. I. Krasavtsev, Ukr. Khem. Zh., 42, 968 (1976); Chem.
Infdrmationsdienst. 7. 7650-293 (1976).
*[660) А. К Khusid, G V Kryshtal, V F. Kucherov, and L. A. Yanoskaya, Izv. Akad.
Nauk SSSR Ser Khim., 24, 2577 0975); Engl. transl., p. 2462.
Г661] А К Khusid, G V Kryshtal, V A. Dombrovsky, V F. Kucherov, L. A. Yanovs-
kaya, V. I Kadentsev, and O. S. Chizhov, Tetrahedron, 33, ?7 (1977).
"[6621 M F Shostakovskii, V I Erofeev, and V S. Aksenov, Izv. Akad: Nauk SSSR Ser
Khim., 24. 2577 (1975); Engl. transl., p. 2462.
f663) а К Khusid. G v Kryshtal. V F Kucherov, and L. A, Yanovskaya, Synthesis.
1977 428
(664J M Makosza anO л Kacprowicz. Bull. Acad. Pot. Set Ser. Set. Chem.. 11. Affi
(1974)
(665) Graefe. M Adlei and M Miihlstadt, Z. Chem., 15, 14 (1975).
433
[666] S. A. G. De Graaf and U. K. Pandit, Tetrahedron, 29, 4263 (1973).
[667] M. Makosza and A. Kacprowicz, Rocz. Chem., 48, 2129 (1974).
•[668] R. R. Kostikov, A. F. Khlebnikov, and K. A. Ogloblin, U.S.S.R Pat. 482,448 (1976);
Chem. Abstr., 83, 206087 (1975).
[669] J. Graefe, Z. Chem., 14, 469 (1974).
[670] D. J. Sikkema, E. Molenaar, and D. B. van Guldener, Rec. Trav. Chim. Pays-Bas,
95, 154 (1976).
[671] E. V. Dehmlow, Justus Liebigs Ann. Chem., 758, 148 (1972).
[672] D. F. Hayman, Germ. Offenl. 2,201,514 (1972); Chem. Abstr., 78, 3793 (1973).
[673] B.D.H. Pharmaceuticals Ltd., Belg. Pat. 777705 (1972); Derwent Abstr., 48539 (1972).
[674] R. Barlet, C. JR. Acad. Sci. Ser. C, 278, 621 (1974).
[675] E. V. Dehmlow, Tetrahedron Lett., 1976, 91.
[676] K. Idemori, M. Tagaki, and T. Matsuda, Bull. Chem. Soc. Jpn., 50,1355 (1977).
[677] M. Makosza and I. Gajos, Bull. Acad. Pol. Sci. Ser. Sci. Chim., 20, 33 (1972).
[678] E. V. Dehmlow, S. S. Dehmlow, and F. Marschner, Chem. Ber., 110, 154 (1977).
[679] E. V. Dehmlow, Chem. Ber., 100, 3829 (1967).
[680] E. V. Dehmlow and G. Hofle, Chem. Ber., 107, 2760 (1974).
•[681] G. V. Kryshtal, V. F. Kucherov, and L. A. Yanovskaya, Izv. Akad. Nauk SSSR Ser.
Khim., 25, 929 (1976), Engl. transl., p. 909.
•[682] G. V. Kryshtal, A. K. Khusid, V. F. Kucherov, and L. A. Yanovskaya, Izv. Akad.
Nauk SSSR Ser. Khim., 24, 424 (1976); Engl. transl., p. 424.
[683] S. S. Dehmlow and E. V. Dehmlow, Justus Liebigs Ann. Chem., 1973, 1753.
[684] F. Kasper and T. Beier, Z. Chem., 16, 435 (1976).
•[685] R. R. Kostikov and A. P. Molchanov, Zh. Org. Khim., 11,1861 (1975); Engl. transl.,
p. 1871.
[686] W. Kraus, W. Rothenwohrer, H. Sadlo, and G. Klein, Angew. Chem., 84,643 (1972);
Angew. Chem. Int. Ed. Engl, 11, 641 (1972).
[687J E. V. Dehmlow, Tetrahedron, 28, 175 (1972).
[688] H. D. Beckhaus, J. Schoch, and С Riichardt, Chem. Ber., 109, 1369 (1976).
[689] W. Kuhn, H. Marschall, and P. Weyerstahl, Chem. Ber., 110, 1564 (1977).
*[690J S. M. Shostakovskii, A. A. Retinskii, and A. V. Bobrov, Izv. Akad. Nauk SSSR Ser.
Khim., 23,1818 (1975); Engl. transl., p. 1736.
[691] C. B. Chapleo, C. E. Dahl, and A. S. Dreiding, Helv. Chim. Ada, 57, 1876 (1975).
[692] A. De Smet, M. Anteuris, and D. Tavernis, Bull. Soc. Chim. Belg., 84, 67 (1975).
[693] T. Sasaki, K. Kanematsu, and N. Okamura, J. Org. Chem., 40, 3322 (1975).
[694] A. Busch and H. M. R. Hoffmann^Tetrahedron Lett., 1976, 2379.
[695] G. Klein and L. A. Paquette, Tetrahedron Lett., 1976, 2419.
[696J Z. Goldschmidt and U. Gutman, Tetrahedron, 30, 3327 (1974).
[697] B. Cheminat and B. Mege, C. R. Acad. Sci. Ser. C, 280, 1003 (1975).
[698J P. F. Ranken, B. J. Harty, L. Kapicak, and M. A. Battiste, Synth. Commun., 3, 311
(1973).
[699] T. Sasaki, S. Eguchi, and T. Kiriyama, J. Org. Chem., 38,2230 (1973).
[700J W. Kraus, G. Klein, H- Sadlo, and W. Rothenwohrer, Synthesis, 1912, 485.
[701J C. W. Jefford, A. Sweeney, and F. Delay, Helv. Chim. Acta, 55, 2214 (1972),
[702] С W. Jefford, U. Burger, and F. Delay, Helv. Chim. Acta, 56,1083 (1973).
[703] С W. Jefford, W. D. Graham, and U. Burger, Tetrahedron Lett., 1975, 4717.
[704] С W. Jefford, V. de los Heros, and U. Burger, Tetrahedron Lett., 1976. 703.
[705] P. M. Kwantes and G. W. Klumpp, Tetrahedron Lett.. 1976, 707
[706] H. Hart and M. Nitta, Tetrahedron Lett., 1974, 2109
[707J E. V Dehmlow, Tetrahedron Lett., 1975, 203
[708] T. Greibrokk, Acta Chem. Scand., 27, 3207 (1973).
[709] J. С Jochims and G. Karich, Tetrahedron Lett., 1974, 4215.
[710] G. Karich and J. С Jochims, Chem. Ber., 110, 2680 (1977).
[711] M. R. Detty and L. A. Paquette, J. Am. Chem, Soc, 99, 821 (1977)
434
[712] Е. V. Dehmlow, H. Klabuhn, and E.-C. Hass, Justus Liebigs Ann. Chem., 1973,1063.
[713] T. Sasaki, K. Kanematsu, and Y. Yukimoto, J. Org. Chem., 39,455 (1974).
[714] T. Sasaki, K.Kanematsu, and Y. Yukimoto, Heterocycles, 1,1 (1973); Chem. Abstr.,
80, 82615 (1974).
[715J G. Andrews and D. A. Evans, Tetrahedron Lett., 1972, 5121.
[716] T. Sasaki, S. Eguchi, T. Kiriyama, and Y. Sakito, J. Org. Chem., 38,1648 (1973).
[717J G. Blume and P. Weyerstahl, Tetrahedron Lett., 1970, 3669.
[718] P. Weyerstahl and G. Blume, Tetrahedron, 28, 5281 (1972).
[719J G. Blume, T. Neumann, and P. Weyerstahl, Justus Liebigs Ann. Chem., 1975,201.
[720] B. Miiller and P. Weyerstahl, Tetrahedron, 32, 865 (1976).
[721] M. Sato, S. Ebine, and J. Tsunetsugu,./. Chem. Soc. Chem. Commun., 1974, 846.
[722] M. Sato, T. Tanaka, J. Tsunetsugu, and S. Ebine, Bull. Chem. Soc. Jpn., 48, 2395
(1975).
[723J M. Sato, J. Tsunetsugu, and S. Ebine, Bull. Chem. Soc. Jpn., 49, 2230 (1976).
[724] M. Sato, A. Uchida, J. Tsunetsugu, and S. Ebine, Tetrahedron Lett., 1977, 2151.
[725] W.-H. Giindel, Z. Naturforsch. Teil B, 30, 616 (1975).
[726J W.-H. Gundel, Z. Naturforsch. Teil B, 31, 807 (1976).
[727] W.-H. Gundel, Z. Naturforsch. Teil B, 32, 193 (1977).
[728J F. De Angelis, A. Gambacorta, and R. Nicoletti, Synthesis, 1976, 798.
[•729] S. Kwon, Y. Nishimura, M. Ikeda, and Y. Tamura, Synthesis, 1976, 249.
[730] E. V. Dehmlow and M. Lissel, Liebigs Ann. Chem., 1979, 181.
[731] E. V. Dehmlow and K.-H. Franke, unpublished results (1976-1977).
[732] R. A. Magadan and E. J. Benjamin, J. Pharm. Set, 64, 1626 (1975).
[733] T. T. Coburn and W. M. Jones, J. Am. Chem. Soc, 96, 5218 (1974).
[734] J. Tsunetsugu, M. Sato, and S. Ebine, J. Chem. Soc. Chem. Commun., 1973, 363.
[735] K. Berg-Nielsen, Acta Chem. Scand. Ser. B, 31, 224 (1977).
*[736J I. G. Tishchenko, O. G. Kulinkovich, and Y. V. Glazkov, Zh. Org. Khim., 11, 581
(1975); Engl. transl., p. 579.
*[737J I. G. Tishchenko, Y. V. Glazkov, and O. G. Kulinkovich, Zh. Org. Khim., 9, 2510
(1973); Engl. transl., p. 2530.
*[738] I. G. Tishchenko, O. G. Kulinkovich, Y. V. Glazkov, and M. K. Pirshtuk, Zh. Org.
Khim., 11, 576 (1975); Engl. transl., p. 574.
[739J E. V. Dehmlow, Tetrahedron, H, 4071 (1971).
[740] I. Tabushi, Z. Yoshida, and N. Takahashi, J. Am. Chem. Soc, 92,6670 (19'70):
[741] M. Makosza and M. Fedorynski, Rocz. Chem. 46, 311 (1972).
[742] S.-H. Goh, K.-C. Chan, T.-S. Kam, and H.-C. Chong, Aus'tr. J. Chem., 28, 381
(1975).
[743J S.-H. Goh, J. Chem. Educ, 52, 399 (1975).
[744J A. de Meijere, O. Schallner, and С Weitemeyer, Angew. Chem., 84, 63 (1972);
Angew. Chem. Int. Ed. Engl., 11, 56 (1972).
[745] A. de Meijere, С Weitemeyer, and O. Schallner, Chem. Ber., 110, 1504 (1977)?
1746] T. Greibrokk, Tetrahedron Lett., 1972, 1663.
[747] T. Tabushi, Y. Aoyama, and N. Takahashi, Tetrahedron Lett., 1973, 107.
1748] T. Saraie, T. Ishiguro, K. Kawashima, and K. Morita, Tetrahedron Lett., 1973, 2121.
I749J G. Hofle, Z. Naturforsch. Teil B, 28, 831 (1973).
[750] J. Graefe, Z. Chem., 15, 301 (1975).
[751] W. P. Weber and G. W. Gokel, Tetrahedron Lett., 1972, 1637.
[752J W. P. Weber, G. W. Gokel, and I. K. Ugi, Angew. Chem., 84, 587 (1972); Angew.
Chem. Int. Ed. Engl., 11, 530 (1972).
[753] T. Sasaki, S. Eguchi, and T. Katada, J. Org. Chem., 39, 1239 (1974).
[754] G. W. Gokel, R. P Widera, and W. P. Weber, Org. Synth., 55,96 (1976).
[755] G. Domschke, R. Beckert, and R. Mayer, Synthesis, 1977, 275; East Germ. Pat.,
128,531 (1977).
[756] D. T. Sepp, K. V. Scherer, and W. P Weber, Tetrahedron Lett., 1974,2983.
435
[757] J. Graefe, В. Striegler, and M. Miihistadt, Z. Chem., 16, 356 (1976/
•[758] G. V. Kryshtal, A. K. Khusid, V. F. Kucherov, and L. A. Yanovjfcaya, Izv. Akad.
Nauk SSSR Ser. Khim., 26, 709 (1977).
[759] M. Makosza and A. Kacprowicz, Rocz. Chem. 49, 1627 (1975),.
[760] J. Graefe, I. Frohlich, and M. Muhlstadt.Z. Chem., 14,434 (1974).
[761] T. Hiyama, Y. Ozaki, and H. Nozaki, Tetrahedron, 30, 2661 (19^4).
[762] M. Makosza, B. Jerzak, and M. Fedorynski, Rocz. Chem., 49, 1783 (1975).
[763] I. Tabushi, Z. Yoshida, and N. Takahashi, J. Am. Chem. Soc, 93,1820 (1971).
[764] I. Tabushi and Y. Aoyami, J. Org. Chem., 38, 3447 (1973).
[765] P. Stromquist, M. Radcliffe, and W. P. Weber, Tetrahedron Lett., 1973, 4523. N
[766] I."Tabushi, Y. Kuroda, and Z. Yoshida, Tetrahedron, 32, 997 (1976).
1767] M. Fedorynski and M. Mqkosza, J. Organomet. Chem., 51, 89 (1973).
[768] M. Makosza and M. Fedorynski, Rocz. Chem., 46, 533 (1972).
[769] M. Makosza and M. Fedorynski, Rocz. Chem., 49, 1779 (1975).
[770] A. Merz, Synthesis, 1974, 724.
[771] P. Kuhl, M. Miihlstadt, and J. Graefe, Synthesis, 1976, 825; 1977, 502.
[772] Nisshin Flour Mill KK, Jap. Kokai 76,05,028; Derwent Ahstr., 82125 (1976).
[773] S. Colonna and R. Fornasier, Synthesis, 1975, 531.
[774] J. Lange, Racz. Chem., 42, 1619 (1968).
[775] M. Cinquini, S. Colonna, H. Molinari, F. Montanari, and P. Tundo, J. Chem. Soc.
Chem. Commun., 1976, 394.
[776] M. Cinquini and P. Tundo, Synthesis, 1976, 516.
[777] G. Cainelli, F. Manescalchi, and M. Panunzio, Synthesis, 1976, 472.
[778] J. M. Saa and M. P. Cava, J. Org. Chem., 42, 347 (1977),
[779] H. Bieraugel, J. M. Akkerman, J. С, Lapierre-Armande, and U.K. Pandit, Rec.
Trav. Chim. Pays-Bas, 95, 266 (1976).
[780] R. A. Moss, M. A. Joyce, and J. K. Huselton, Tetrahedron Lett., 1975,4621.
[781] M. Makosza and M. Fedorynski, Bull. Acad. Pol. Sci. Ser. Sci. Chim., 19,105 (1971).
[782] J. Hine, "Divalent Carbon," The Ronald Press Co., New York, 1964. *
[783] E. V. Dehmlow, J. Heider, and U. Brenner, unpublished results.
[784] L. Skattebal, G. A. Abskharoun, and T. Greibrokk, Tetrahedron Lett., 1973,1367.
[785] M. Makosza and M. Fedorynski, Synth. Commun., 3, 305 (1973).
[786]M. Makosza and M. Fedorynski, Rocz. Chem., 50, 2223 (1976).
[787]E. V. Dehmlow and J. Kranz, unpublished results.
1788] E. Vogel and J. Ippen, Angew. Chem., 86, ?78 (1974); Angew. Chem. Int. Ed. Engl.,
13, 734 (1974).
[789] J.-L. Luche, J.-C. Damiano, and P. Crabbe, J. Chem. Res., 1977, (S) 32, (M) 443.
[790] J.-C. Damiano, J.-L. Luche, and P. Crabbe, Tetrahedron Lett., 1976, 779.
[791] H. Maskill,/. Chem. Soc. Perkin TYans. 2,1975, 197.
[792] EVV. Dehmlow and M. Lissel, unpublished results.
[793h"ft. R. Allan and M. S. Baird, J. Chem. Soc. Chem. Commun., 1975, 172.
[794] E. Vogel, M. Konigshofen, K. Miiiler, and J. F. M. Oth, Angew. Chem., 86, 229
(1974); Angew. Chem. Int. Ed. EngL, 13, 281 (1974).
[795] L. K. Sydnes, L. SkatteW, С. В. Capleo, D. G. Leppard, K. L. Svanholt, and A. Si
Dreiding, Helv. Chim. Ada, 58, 2061 (1975).
[796] L. Sydnes and L. Skalleb0\^Tetraheclron Lett., 1975, 4603.
1797] M. Braun, R. Dammann, and D. Seebach, Chem. Ber., 108, 2368 (1975).
[798] M. Braun and D. Seebach, Chem. Ber., 109, 669 (1975).
[799] i. J. Landheer, W. H. de Wolf, and F. Bickelhaupt, Tetrahedron Lett., 1974,2813.
1800] E. V. Dehmlow and G. С Ezimoft, Tetrahedron Lett., 1970,4047.
[801] D. Reinhard and P. Weyerstahl, Chem. Ber., 110, 138 (1977).
[802] R. Dammann, M. Braun, and D. Seebach, Helv. Chim. Acta, 59, 2821 (1976).
[803] W. Kirmse, "Carbene Chemistry," Second Ed., Academic Press, New York and
London, 1971.
43C
[804] D. Seyfehh, S. P. Hopper, and T, F. Jula, J. Organomet. Chem., 17,193 (1969)
[805] M. Schlossfei;, B. Spahic, С Tarchini,and L. V. Chau, Angew. Chem.,87, 346(1975);
Angew. Chen\lnt. Ed. Engl., 14, 365 (1975).
[806] M. Schlosser Aid L. V. Chau, Helv. Chim. Ada, 58, 2595 (1975).
(807] L. V. Chau andW Schlosser, Synthesis, 1973, 112.
[808] P. Weyerstahl, GABlume, and С Muller, Tetrahedron Lett., 1971, 3869.
[809] P. Weyerstahl, R. Mathias, and G. Blume, Tetrahedron Lett., 1973, 611.
[810] R. Mathias, Ph.D. Dissertation, Technical University, Berlin, 1977.
[8111 С Muller arid P. Weyerstahl, Tetrahedron, 31, 1787 (1975).
[812] С Muller, F. Stier, andKP. Weyerstahl, Chem. Ber., 110, 124 (1977).
[813] R. Mathias and P. Weyefttahl, Angew. Chem., 86, 42 (1974); Angew. Chem. Int. Ed.
Engl., 13, 134 (1974).
[814] M. S. Baird, J. Chem. Soc. terkin Trans. 1, 1976, 54.
t815J B. Giese, Angew. Chem., 89,162 (1977); Angew. Chem. Int. Ed. Engl., 16,125 (1977),
[816] E. V. Dehmlow, unpublished results.
[817] M. Makosza and E. Bialecka, Tetrahedron Lett., 1971,4517.
[818] G. Boche and D. R. Schneider, Tetrahedron Lett., 1975, 4247.
[819] R. A. Moss and F. G. Pilkiewicz, Synthesis, 1973, 209.
[820] M. S. Newman and T. B. Patrick, J. Am. Chem. Soc., 91, 6461 (1969) and previous
work cited therein.
[821] M. S. Newman and S. J. Gromelski, J. Org. Chem., 37, 3220 (1972).
[822] M. S. Newman and Z. ud Din, J. Org. Chem., 38, 547 (1973).
[823] P. S. Stang and M. G. Mangum, J. Am. Chem. Soc., 97, 6478 (1975).
[824] M. S. Newman and M. С Van der Zwan, J. Org, Chem., 39,1186 (1974).
[825] M, S. Newman and M. С Van der Zwan, J. Org. Chem., 39. 761 (1974).
[826] M. S. Newman and W. C. Liang, J. Org. Chem., 38, 2438 (1973).
[827] S. Julia, D. Micbelot, and G. Linstrumelle, С R. Acad. Sci. Ser. C.tlb, 1523(1974).
[828] T. Sasaki, S. Eguchi, and T. Qgawa, J. Org. Chem., 39,1927 (1974). j
'[«29] T. Sasaki, S. Eguchi, M. Ohno.and F. Nakato, J. Org. Chem., 41, 2Ш (1976). ?
[830] Т. В. Patrick, Tetrahedron Lett., 1974, 1407.
[831 ] D. Y. Aue and M. J. Meshishnek, J. Am, Chem. Sac, 99, 223 (1977).
[832] T. Sasaki, S. Eguchi, and T. Ogawa, Heterocycles, 3, 193 (1975).
[833] A. Doutheaw and J. Gore, Bull. Soc. Chim, Fr., 1976, 1189.
[834] M. Schlosser and Y. Bessiere, Heb. Chim. Ada, 60, 590 (1977).
[835] M. Fedorynski, Synthesis, 1977, 783.
[8^] M. Fedorynski, M. Poptawska, K. Nitschke, W. Kawalski, and M. Makosza,
Synth. Commun., 7, 287 (1977).
[837] R. A. Jones, S. Nokkeo, and S. Singh, Synth. Commun., 7, 195 (1977).
[838] B. L. Burt, D. J. Freeman, D. G. Gray, R. K. Norris, and D. Randies, Tetrahedron
Lett., 1977, 3063.
1839] S. E. Callander, Germ. Offenl. 2,656,062 (1977), Chem. Abstr., 87, 215436 U978).
[840] S. W. Tobey and R. West, J. Am. Chem. Soc, 86, 56 (1964).
[841] E. W. Meijer and H. Wynberg, private communication. • i
[842] D. С Duffey, R. С Gueldner, B. R. Layton, and J. P. Minyard, Jr., J. Org. Chem.i
42, 1082 (1977).
[843] J. Heider and E. V. Dehmlow, unpublished results.
[844] W. Kimpenhaus and J. Buddrus, Chem. Ber., 110, 1304 (1977).
[845] E. V. Dehmlow, unpublished results.
[846] R. A. Moss, M. A. Joyce, and F. G. Pilkiewicz, Tetrahedron Lett., 1975, 2425.
[847]. J. Palecek and J. Kuthan, Z. Chem., 17, 260 (1977). '
[848] R. O. Hutchins and F. J. Dux, J. Org. Chem., 38, 1961 <1973).
[84?) T.-L. Ho, Synlh. Commun., 3, 99 (1973).
[850] G. P,:FuHer, R. Greenhouse, and I. Itoh, Synth. Commun,, 4,183 (1974).
[851] M. Tomoi, T. Takubo, M. Ikeda, and H. Kakiuchi, Chem. Lett., 1976, 473.
437
1852] Е. V. Dehmlow and E. Menzel, unpublished results.
[853] E. V. Dehmlow and S. Barahona-Naranjo, 3. Chem. Res., 1982 (S),y
[854] E. V. Dehmlow and E. Timm, unpublished results.
[855] S. H. Korzeniowski and G. W. Gokel, Tetrahedron Lett., 197^3519.
[856] D. Landini and F. Rolla, Org. Synth., 58, 143 (1978).
[857] S. A. Di Biase and G. W. Gokel, Synthesis, 1977, 629.
[858] H. Molinari, F^Montanari, and P. Tundo,/. Chem. Soc. Chem. Contmun., 1977,639.
[859] P. Tundo, У. Chem. Soc. Chem. Commun., 1977, 641.
[860] J. M. Brown and J. A. Jenkins, J. Chem. Soc. Chem. dommun., 1976, 458.
[861] M. Shibasaki and S. Ikeyami, Tetrahedron Lett., 1977,* 4037.
[862] L. Eberson and B. Helgee, Ada Chem. Scand. Ser.,6, 32, 157 (1978).
[863] L. Eberson and B. Helgee, Acta Chem. Scand. Seri B, 31, 813 (1977).
[864] L. Eberson and B. Helgee, Acta Chem. Scand. Sir. B, 29, 451 (1975).
[865] Y. Nakajima, R. Kinishi, J. Oda, and Y. Inouye, Bull. Chem. Soc. Jpn., 50, 202$
(1977).
{866] Y. Kimura, K. Isagawa, and Y. Otsuji, Chem. Lett., 1977, 951.
1867] J. L. Ripoll, Tetrahedron Lett., 33, 389 (1977).
1868] S. Julia and A. Ginebreda, Synthesis, 1977, 682.
[869] T.-L. Ho, B. G. B. Gupta, and G. A. Olah, Synthesis, 1977, 676.
1870] W. Wilczynski, M. Jawdosiuk, and M. Ma.kosza, Rocz. Chem., 51, 1643 (1977).
[871] J. Renault, P. Mailliet, J. Berlot, and S. Renault, С R. Acad. Sci. Ser. C, 285, 199
(1977).
J872] CSIR, Germ. Pat. 2,653,189 (1977); Derwent Abstr., 42013 (1977).
1873] P. Maggioni and C. Montevechia, Germ. Offenl. 2,703,640 (1977).
[874] A. W. Burgstahler, L. O. Weigel, M. E. Sanders, and С G. Shaefer, /. Org. Chem.,
42, 566 (1977).
£875] S. C^Cchi, F. La Torre, and D. Misiti, Chem. Ind. {London), 1977, 691.
[876] Y. Masuyama, Y. Heno, and M. Okawara, Chem. Lett., 1977, 835.
J877] G. R. Kieczykowski, С S. Pogonowski, J. E.' Richman, and R. M. Schlessinger, Л'
Org. Chem., 42, 175 (1977).
£878] A. Chollet, С Mahaim, С Foetisch, M. Hardy, and P. Vogel, Helv. Chim. Acta, 60,
59 (1977).
[879] M. Muhammed, J. Szabon, and E. Hegfeldt, Chem. Scr., 6, 61 (1974).
[880] J. Cousseau, L. Gouin, L. V. Jones, G. Jugic, and J. A. S. Smith, /. Chem. Soc.
-Faraday Trans. 2, 1973, 1821.
[881] J. Cousseau and L. Gouin, J. Chem. Soc. Perkin Trans. 1, 1977, 1797.
[882] E. V. Dehmlow and M. Slopianka, unpublished results.
[883] R. Beugelmans, M.-T. Le Goff, J. Pusset, and G. Roussi, Tetrahedron Lett., 1976,
2305; J. Chem. Soc. Chem. Commun., 1976, 377.
[884] K. Kitatani, T. Hiyama, and H. Nozaki,7. Am. Chem. Soc, 98, 2362 (1976).
[885] A. Zwierzak and [. Padstawszyiiska, Angew. Chem., 89, 737 (1977); Angew. Chem.
Int. Ed. Engl., 16, 702 (1977).
[886] M. D. Rozwasowska, Can. J. Chem., 55, 164 (1977).
[887] R. A. Sheldon, P. Been, D. A. Wood, and R. F. Mason, Germ. Offenl. 2,708,590
(1977).
[888] N. Ono, R. Tamura, R. Tanikaga, and A. Kaji, Synthesis, 1977, 690.
[889] G. Entenmann, Tetrahedron Lett., 1975, 4241.
[890] R. O. Hutchins, N. R. Natale, and W. J. Cook, Tetrahedron Lett., 1911,4167.
[891] M. A. Pericas and F. Serratosa, Tetrahedron Lett., 1977, 4437.
(892] K. Hermann and H. Wynberg, Helv. Chim. Acta, 60, 2208 (1977).
[893] K. Yamamura and S. Murahashi, Tetrahedron Lett., 1977, 4429.
[894] A. Brandstrom, P. Berntsson, S. Carlsson, A. Djurhuus, K. Gustavi), U* Junggren,
B. Lamm, and B. Samuelsson, Acta Chem. Scand., 23, 2202 (1969).
438
[895] К. М. More and J. Wemple, Syntltesis, 1977, 791.
[896] L. M. JacRman and В. С Lange, Tetrahedron, 33, 2737 (1977).
[897] S. Gronowitk and T. Frejd, Synth. Commun., 6, 475 (1976).
[898] A. A. FrimeiyJ. Rosenthal, and S. Hoz, Tetrahedron Lett., 1911, 4631.
[899] A. BrandstromVid H. Kolind-Andersen, Ada Chem. Scand. Ser. B, 29, 201 (1975).
[900] A. Merz and R.\omahogh, J. Chem. Res., 1977, (S) 273 (M) 3070.
[901] P. J. Stang and dV. Fox, J. Org. Chem., 42, 1667 (1977).
[902] E. Chiellini and R. &>laro, J. Chem. Soc. Chem. Commun., 1977, 231.
[903] P. Cazeau and B. Mufckensturm, Tetrahedron Lett., 1977, 1493.
[904] R. Lantz and A.-B. Hornfeldt, Chem. Ser., 10, 126 (1976).
[905] С Innes and G. Lamaty\7Vow. J. Chim., 1, 503 (1977).
[906] G. Cardillo, M. Contento, lyl. Panunzio, and A. Umani-Ronchi, Chem. Ind. (London),
1977, 873.
[907] L. Jensen, I. Thomsen, and J>\O. Lawesson, Bull. Soc. Chim. Belg., 86, 309 (1977).
[908] A. Jonczyk, J. Wlostowska, and M. Majcosza, Bull. Soc. Chim. Belg., 86,739 (1977).
[909] J.-P. Hagenbuch and P. Vogel, Chimia, 31, 136 (1977).
[910] С Wyganowski, Talanta, 24, 190 (1977).
[911] K. H. Pannell and J. Mclntosh, Chem. Ind. (London), 1977, 873.
[912] G. Adembri, A. Camparini, F. Ponticelli, and P. Tedeschi, J. Chem. Soc. Perkin
Trans. 1, 1977, 971.
[913] E. Lee-Ruff, Chem. Soc. Rev., 6, 195 (1977).
[914] G. Markl, H. Baier, and R. Liebl, Synthesis, 1977, 842.
[915] I. Degani, R. Fochi, and M. Santi, Synthesis, 1977, 873.
[916] R. Beugelmans, H. Ginsburg, A. Lecas, M.-T. Le Goff, J. Pusset, and G. Roussi,
J. Chem. Soc. Chem. Commun., 1977, 885; Tetrahedron Lett., 1978, 3271.
[917] Allied Chem. Corp., Neth. Pat. Appl. 6,400,872 (1964) = U.S.Pat. 3,297,634(1967);
Chem. Abstr., 62, 6587 (1965).
[918] S. Alunni, E. Baciocchio, and P. Perucci, J. Org. Chem., 41, 2636 (1976).
[919] Z. K. Brzozowski, J. Kielkiewicz, anAz. Goclawski, Angew. Makromol. Chem., 44,1
(1975).
[920] L. Cassar and M. Foa, J. Organomet. Chem., 134, С 15 (1977).
[921] M. Chorev and Y. S. Klausner, J. Chem. Soc. Chem. Commun., 1976, 596; Y. S.
Klausner and M. Chorev, J. Chem. Soc. Perkin Trans. 1,1977, 627.
[922] J. L. Cihonski and R. A. Levenson, Inorg. Chem., 14, 1717 (1975).
[923] S. Colonna, R. Fornasier, and U. Pfeiffer, J. Chem. Soc. Perkin Trans. X, 1978, 8.
[924] S. Colonna, H. Hiemstra, and H. Wynberg, J. Chem. Soc. Chem. Commun., 1978,
238.
1925] E. J. Corey, К. С Nicolaou, and M. Shibasaki,/. Chem. Soc. Chem. Commun., 1975,
658.
[926] U. Curtius, V. Boellert, G. Fritz, and J. Nentwig, Belg. Pat. 616 919 (1962); Chem.
Abstr., 58, 3360 (1963).
[927] L. H. Dao, A. C. Hopkinson, E. Lee-Ruff, and J. Rigaudy, Can. J. Chem., 55, 3791
(1977).
[928] D. J. Darensbourg and J. A. Froelich,/. Am. Chem. Soc, 100, 338 (1978).
[929] J. K. Rasmussen, Chem. Lett., 1977, 1295.
I?30] H. des Abbayes and M.-A. Boudeville, J. Org. Chem., 42, 4104 (1977).
[931] S. A. DiBiase and G. W. Gokel,7. Org. Chem., 43, 447 (1978).
I?32] H. J.-M. Dou, P. Hassanaly, J. Kister, and J. Metzger, Phosphorus Sulfur, 3, 355
(1977).
[933] H. J.-M. Dou, R. Gallo, P. Hassanaly, and J. Metzger,J. Org. Chem., 42,4275 (1977).
[934] S. O. De Silva and V. Snieckus, Can. J. Chem., 56, 1621 (1978).
[935] R. Rucman, J. Stres, and M. Jurgcc, Chem. Abstr., 89, 215637 (1978).
[936] V. O. Illi, Synthesis, 1979, 136.
[937] R. Boehm, Pharmazie, 33, 83 (1978).
439
[938] N.-C. Wang, K.-E. Teo, and H. J. Anderson, Can. J. Chem., 55, 41/2 (1977).
[939] M. Garle and I. Petters, /. Chromatogr., 140, 165 (1977).
[940] S. Julia, A. Ginebreda, and J. Guixer, J. Chem. Sod Chem. Com/nun., 1978, 742.
[941] J. E. Nordlander, D. B. Catalane, T. H. Eberlein, L. V. FarkasfR. S. Howe, R. M.
Stevens, N. A. Tripoulas, R. E. Stansfield, J. L. Cox, M. J. Pa/ne, and A. Viehbeck,
Tetrahedron Lett., 1978, 4987.
{942] E. V Dehmlow, unpublished results.
[943] A. Lopez, M. T. Maurette, R. Martino, and A. Lattes, Tetrahedron Lett., 1978, 2013.
[944] M. E. Fakley and A. Pidcock, J. Chem. Soc. Dalton Tr/ns., WTJ, 1444.
[945] Y. Gaoni and N. Shoef, Bull. Soc. Chim. Fr., 1977,
[946] General Mills Inc., Jap. Kokai 74,66,634 (1974); Chim. Abstr.,94, 179663 (1976).
[947] R. С Hahn and R. P. Johnson, Tetrahedron Lett.AviS, 2149.
[948] В. Hanquet, R. Guilard, and P. Fournari, Bull. Sbc. Chim. Fr., 1977, 571.
[949] J. Heider, Doctoral Dissertation, T. U. Berlin, 1978.
[950] F. R. van Heerden, J. J. van Zyl, G. J. H. Rail, E. V. Brandt, and D. G. Roux,
Tetrahedron Lett., 1978, 661.
[951] J. С Hummelen and H. Wynberg, Tetrahedron Lett., 1978,1089.
[952] A. Jonczyk, A. Kwast, and M. Makosza, J. Chem. Soc. Chem. Commun., 1977,
902.
[953] A. Jonczyk, M. Ludwikow, and M. Makosza, Angew. Chem., 90, 58 (1978); Angew.
Chem. Int. Ed. Engl, 17, 62 (1978).
[954] K. S. Kim and W. A. Szarek, Synthesis, 1978, 48.
[955] G. Klein und W Kraus, Tetrahedron, 33, 3121 (1977).
[956] G. Klein and L. A. Paquette,/. Org. Chem., 43, 1293 (1978).
[957] M Kluba and A. Zwierzak, Synthesis, 1978,134.
[958] H. Komeili-Zadeh, H. J.-Ы- Dou, and J. Metzger, J. Org. Chem., 43, 156 (1978).
* (959] R. A Kostikov, A. P. Molchanov, G. V. Golovanova, and I. G. Zenkevich, Zh. Org,
Khim., 13, 1846 (1977), Engl. tranl., p. 1712.
[960] A. P Krapcho, J. R. Larson, and J. M. Eldridge, J. Org. Chem., 42, 3749 (1977).
[961] S. Krishnan, D. G. Kuhn, and G. A. Hamilton,/. Am. Chem. Soc, 99, 8121 (1977).
[962] B. Ly, HJ.-M.Dou, P, Hassanaly, and J. Verducci, /. Heterocycl. Chem., 14, 1275
(1977).
[963] I M Mclntosh, Can. J. Chem., 55, 4200 (1977).
Г964] M К Meilahn, D K. Olsen, W. J. Brittain, and R. T. Anders, J. Org. Chem., 43,
1346 (1978).
1965) А. С Mueller, Ш5. Pat. 2,772,296 (1956); Chem. Abstr., 51, 7416 (1957).
[966] D 1 Nelson and E. A. Uschak,/. Org. Chem., 42, 3308 (1977).
[967] M S Newman and 1. O. Landers, J. Org. Chem., 42, 2556 (1977).
[968] L я Paquette, D R. James, and G. Klein, J.'Org. Chem., 43, 1287 (1978).
(969] K. M Patel. H I. Pownall, J. D. Morrisett, and J. T. Sparrow, Tetrahedron Lett.*
1976. 4015.
[970] Г В Patrick and D I Schmidt, J. Org. Chem., 41, 3355 (1977).
[971] v Ratovelomanana and S. Julia, Synth. Comm., 8, 87 (1978).
[972] R W Roeske and P D. Gesellchen, Tetrahedron Lett., 1976, 3369.
[973] M D Rozwadowska and D. Br6zda, Tetrahedron Lett., 1978, 589.
[974J T Sala and M v Sargent. / Chem. Soc. Chem. Commun., 1978, 253.
(9751 W H -Saunders. li SD Bonadies, M Braunstein, J. K. Borchardt, and R. T
Hargreaves, Tetrahedron. S3, 1577 (1977).
(9761 S Schwar? and G Weber East Germ Pat. 114,806 (1975); Chem. Abstr., 85,63238
(.1976)
L977J F Ь Scully li and R С Davies, / Org. Chem., 43, 1467 (1978).
[978J I b Shaw Q v Hsin G S Parries, and T K. Swayer, J. Org. Chem.. 43, 1017
U978>
(979) H Singli ana и Singh Chem fnd. (London), 197*. 126.
440
[980] к. Steinbe&k, Tetrahedron Lett., 1978, 1103.
{981J L. K. SydneX .Acta Chem. Scand. Ser. B, 31, 903 (1977).
[982] В. С Uff, A.\l-kolla, K. E. Adamali, and V. Harutunian, Synth. Commun., 8,163>
(1978).
1983] J. Vessman, M. Johansson, P. Magnusson, and S. Strbmberg, Anal. Chem., 49,1545
(1977).
[984] E. J. Walsh, E. DerbV and J. Smegal, Inorg. Chim. Acta, 16, C9 (1976).
[98S] T. Wakabayashi and Ц Watanabe, Tetrahedron Lett., 1978, 361.
[986] I. Willner, M. Halpern\and M. Rabinovitz, J. Chem. Soc. Chem. Commun., 1978,.
155.
[987] E. V. Dehmlow and K.-H. ftranke, Liebigs Ann. Chem., 1979, 1456.
[988] E. V. Dehmlow and M. Lissek unpublished results.
[989J E. V. Dehmlow and M. SlopiaW Liebigs Ann. Chem., 1979, 1465.
[990J E. V. Dehmlow and A. EulenbergV Angew. Chem., 90,716 (1978); Angew. Chem. Int. Edr
Engl., 17, 674 (1978); Liebigs AnnSchem., 1979, 1112. ;
J991] D- Pktcher arid S. J..D. Tail, Tetrahedron Lett., 1978, 1601; J. Chem. Soc. РегШ.
Trans. 2,1979, 788.
[992] D. Landini, F. Montanari, and F. Rolla, Synthesis, 1978, 223.
J993] В. С Uff and R. S. Budhram, HeterocycUs, 6, 1789 (1977).
[994] С Goralski and G. A. Burk, U.S. Pat. 4,014,891 (1977); Chem. Abstr., 87, 22763-
(1977).
[995] D. Martinetz and A. Hiller, Z. Chem., 16, 320 (1976).
[996] H. Alper and H. des Abbayes, J. Organomet. Chem., 134, СП (1977).
(997J С. Kimura, К. Kashiwaya, and K. Koichi, Chem. Abstr., 86, 5112 (1977).
[998] G. A. Lee and H. H. Freedman, U.S. Pat. 3,996,259 (1976); Chem. Abstr., 86, 88453-
(1977).
[999] S. Schwarz, U. Eberhardt, and H. Schick, East German Pat. 120 649 (1976); Chem,
Abstr., 86, 72996 (1977).
[1000] Y. Hayashi, Jap. Kokai 77,111,486 (1976); Chem. Abstr., 88, 104653 (1978).
11001J T. Okimoto and D. Swern, J. Amer. Oil Chem. Soc., 54, 867 A (1977); Chem. Abstr.y
88,37180(1978).
[1002] D. Pletcher and N. Tomov,7. Appl. Electrochem., 7, 501 (1977).
[1003] T. A. Foglia, A. P. Barr, and A. J. Malley, J. Amer. Oil Chem. Soc, 54,858 A (1977);
Chem. Abstr., 88,17210 (1978).
[1004] L. Тбке and G. T. Szabo, Acta Chim. Acad. Set. Hung., 93,421 (1977); Chem. Abstr.,
88, 38298 (1978).
[1005] A. F. Rosenthal, L. A. Vargas, and J. F. Dixon, Chem. Phys. Lipids, 20, 205 (1977);
Chem. Abstr., 88, 121598 (1978).
[1006] G. W. Gokel, D. J. Cram, С L. Liotta, H. P, Harris, and F. L. Cook, Organ. Synth.*
57, 30 (1977).
[1007] M. Amouyal and H. Sekiguchi, С R. Acad. Sci. Ser. C, 286, 233 (1978).
[1008] P. Tundo, Synthesis, 1978, 315.
[1009] I. Degani and R. Fochii Synthesis, 1978, 365.
[1010] D. Martinetz and A. Hiller, Z. Chem., 1978, 61.
[1011] H. Wynberg and B. Greijdanus, J. Chem. Soc. Chem. Commun., 1978,427.
[1012] E. V. Dehmlow, U. Brenner, and M. Slopianka, unpublished results.
[1013] G. W. Gokel and B. J. Garcia, Tetrahedron Lett., 1978, 1743.
[1014] S. Colonna and R. Fornasier, J. Chem. Soc. Perkin Trans. 1,1978, 371.
[1015] T. Kitazume and N. Ishikawa, Chem. Lett., 1978, 283.
[1016] M. Lissel and E. V. Dehmlow, Tetrahedron Lett., 1978, 3689.
[1017] J. J. Kaminski, K. W. Knutson, and N. Bodor, Tetrahedron, 34, 2857 (1978).
[1018] H. Kobler, R. Munz, G. Al Gasser, and G. Simchen, Justus Liebigs Ann. Chem.,-
1978,1937.
[1019] J. H. Clark, J. Chem, Soc. Chem. Commun., 197$, 789.
441
41020] H. J.-M. Dou, P. Hassanaly, and J. Metzger, Nouv. J. Chim., 2, 445 (1978)?
11021] J. Elguero and M. Espada, С R. Acad. Sci. Ser. C, 287, 439 (p).
11022] W. M. McKenzie and D. С Sherrington, J. Chem. Soc. Chem/Commun., 1978, 541.
J1023] M. Tomoi, O. Abe, M. Ikeda, K. Kihara, and H. Kakindii./etrahedronLett., 1978,
3031.
{1024] S. Akabori, S. Miyamoto, and H. Tanabe, /. Polym. Sci/Polym. Lett. Ed., 16, 533
(1978).
11025] M. Tomoi, M. Ikeda, and H. Kakiuchi, Tetrahedron Lien., 1978, 3757; S. L. Regen,
A. Nigam, and J. J. Besse, ibid., 1978, 2757.
11026] F. Rolla, W. Roth, and L. Homer, Naturwissenschdften, 64, 337 (1977).
11027] H. J. Cristau, A. Long, and H. Christol, Tetrahe/ron Lett., 1979, 349.
[1028] P. Tundo, Tetrahedron Lett., 1978, 4693.
[1029] S. Samaan and F. Rolla, Phosphorus Sulfur, 4/145 (1978); Chem. Abstr., 89, 41853
(1978).
|1030] M. Ravey, L. M. Shorr, and I. Hertz, J. 0iem. Soc. Perkin Trans. 2, 1977, 1462.
|1031] R. L. Markezich, O. S. Zamek, P. E. Donahue, and F. J. Williams, J. Org. Chem*
42, 3435 (1977). .
[1032] D. Landini, F. Montanari, and F. Rolla, Synthesis, 1978, 771.
{1033] R. Lantzsch, A. Marhold, and K.-F. Lehment, Germ. Offenl. 2,545,644 (1977); Chem».
Abstr., 87, 38874 (1977).
[1034] L. Eberson and B. Helgee, Acta Chem. Scand. Ser. B, 32, 313 (1978).
[1.035] L. С Packman and D. J. H. Smith, FEBS Lett., 91, 178 (1978).
[1036] J. G. Smith and D. С Irwin, Synthesis, 1978, 894.
11037] K. Soga, S. Hosoda, and S. Ikeda, /. Polym. Sci. Polym. Lett. Ed., 15, 611 (1977).
{1038] B. Davis, Anal. Chem., 49, 832 (1977).
Ц039] K. Hermann aJfi H. Wynberg, /. Org. Chem., AA, 2239 (1979).
{1040] С. Е. Hatch III, J. Org. Chem., 43, 3953 (1978).
{1041] M. С Vander Zwan and F. W. Hartner, /. Org. Chem., 43, 2655 (1978).
[1042] L. G. Wade Jr., J. M. Gerdes, and R. P. Wirth, Tetrahedron Lett., 1978, 731.
11043] I. Degani, R. Fochi, and V. Regondi, Synthesis, 1979, 178.
[1044] M. Kluba and A. Zwierzak, Synthesis, 1978, 770.
[1045] W. P. Reeves, M. R. White, and D. Bier, /. Chem. Educ, 55, 56 (1978).
[1046] Y. Nakajima, J. Oda, and Y. Inouye, Tetrahedron Lett., 1978, 3107.
[1047] A. Jonczyk, Angew. Chem., 91, 228 (1979); Angew. Chem. Int. Ed. Engl., 18, 217
(1979).
41048] M. Fedoryiiski, K. Wojciechowski, Z. Matacz, and M. Makosza, /. Org. Chem., 43,
4682(1978).
*[1049] Y. A. Zhdanov, Y. E. Alekseev, S. S. Doroshenko, G. V. Bodganova, T. P. Sudareva,
and V. G. Akekseeva, Dokl. Akad. Nauk SSSR Ser. Khim., 238, 1102 (1978); Engl.
transl., p. 65.
[1050] T. D. N'Guyen, A. Deffieux, and S. Boileau, Polymer, 19, 423 (1978).
*[1051] V. M. Andreev, A. I. Bibicheva, and L. S. Lapikova, Zh. Vses. Khim. Ova., 23, 231
(1978); Chem. Abstr. 89, 23704 (1978).
[1052] J. D. Nicholson, Analyst, 103, 1, 13 (1978).
[1053] A. Arbin, /. Chromawgr., 144, 85 (1977).
[1054] P. Hartwig and С Fagerlund, J. Chromatogr., 140, 170 (1977).
{1055] J. M. Rosenfeld and J. L. Crocco, Anal. Chem., 50, 701 (1978).
{1056] A. Berlin-Wahlen and R. Sandberg, Acta Pharm. Suec, 14, 321 (1977).
[1057] W. S. Di Menna, С Piantadosi, and R. G. Lamb, /. Medic. Chem., 21, 1073!
(1978).
[1058] H. Segawa and M. Tanyo, Jap. Kokai, 76,75,084 (1976); Chem. Abstr., 86, 106661
(1977).
{1059] J. P. Ferris, S. Singh, andT. A. Newton, J. Org. Chem., 44,173 (1979).
442
[1060] £. V Dehmlow and M. Lissel, unpublished results.
[1061] B. Mlotkovuska and A. Zwierzak, Tetrahedron Lett., 1978, 4731.
[1062] G. Vernin and J. Metzger, Synthesis, 1978,^21.
[1063] D. Reuschling^H. Pietsch, and A. Linkies, Tetrahedron Lett., 1978, 615.
[1064] S. R. Fletcher a\d I. T. Kay, J. Chem. Soc. Chem. Commun., 1978, 903.
[1065] A. Jonczyk, Z. obhal, and M. MaJcosza, Synthesis, 1978, 882.
[1066] M. Jawdosiuk, A. Jpficzyk, A. Kwast, M. Msjkosza, I. Kmiotek-Skarzynska, and
K. Wojciechowski, /W/. Chem., 53, 191 (1979).
[1067] M. Jawdosiuk, M. M^osza, E. Malinowsky, and W. Wilczynski, Pol.J. Chem., S2r
2189 (1978).
[1068] J. W. Skiles and M. P. Ckva, Heterocycles, 9, 653 (1978).
[1069] E. Chiellini and R. SolaroV. Org. Chem., 43, 2550 (1978).
[1070] M. J. O'Donnell, J. M. Boniece, and S. E. Earp, Tetrahedron Lett., 1978, 2641; M. J-
O'Donnell and Т. М. EckrichV/Ш., 1978, 4625.
[1071] Y. Masuyama, Y. Ueno, and Mv Okawara, Bull. Chem. Soc. Jpn., 50, 3071 (1977).
[1072] J. M. J. Frechet, M. de Smet, and M. J. Farrall, Tetrahedron Lett., 1979, 137.
[1073] T. Shioiri and Y. Hamada, /. Org. Chem. 43, 3631 (1978).
[1074] M. Makosza, K. Wojciechowski, and M. Jawdosiuk, Pol. J. Chem., 52, 1173 (1978).
[1075] A. J. Fry and J. P. Bujanauskas, J. Org. Chem., 43, 3157 (1978). '-
[1076] E. Baxmann and E. Winterfeldt, Chem. Ber., Ill, 3403 (1978).
[1077] A. Jonczyk and T. Pytlewski, Synthesis, 1978, 883.
[1078] B. Lamm and K. Ankner, Acta Chem. Scand. Ser. B, 32, 193 (1978).
[1079] L. S. Hart, C. R. J. Killen, and K. D. Saunders, J. Chem. Soc. Chem. Commun., 1979»
24.
[1080] V. G. Purohit and R. Subramanian, Chem. Ind. (London), 1978, 731.
[1081] H. Kise, Y. Kaneko, T. Sato, and M. Seno, Yukagaku, 26, 474 (1977); Chem*
Abstr., 87, 133818 (1977).
[1082] S. Akabori and H. Tuji, Bull. Chem. Soc. Jpn., 51, 1197 (1978).
[1083] E. D'Incan and P. Viout, Tetrahedron, 34, 2469 (1978).
[1084]. U. Burckhardt, L, Werthemann, and R. J. Troxler, Germ. Offenl., 2, 738,588 (1978);
Chem. Abstr., 89, 6,114 (1978).
[1085] I. Willner and M. Halpern, Synthesis, 1979, 177.
[1086] H. J.-M. Dou, P. Hassanaly, J. Kister, G. Vernin, and J. Metzger, Helv. Chim. Actat
61, 3143 (1978).
Ц087] J. Golinski and M. Majcosza, Synthesis, 1978, 823.
11088] S. P. Markey and G. J. Shaw, /. Org. Chem., 43, 3414 (1978).
11089] R. Durand, P. Geneste, G. Lamaty, С Moreau, O. Pomares, and J. P. Roque, Reel,
Trav. Chim. Pays-Bas, 97, 42 (1978).
P090] V. Scherrer, M. Jackson-Mully, Z. Zsindely, and H. Schmid, Helv. Chim. Acta, 61,
716 (1978).
Ц091] Y. Ali and W. A. Szarek, Carbohydr. Res., 67, С 17 (1978).
[1092] J. H. Clark, J. M. Miller, and K.-M. So, /. Chem. Soc. Perkin Trans. 1, 1978, 941.
[1093] G. V. Kryshtal, V. V. Kulganek, V. F. Kucherov, and L. A. Yanovskaya, Synthesist
1979, 107.
[1094] G. A. Russel, M. MaJcosza, and J. Hershberger, /. Org. Chem., 44, 1195 (1979).
[1095] J. M. Mclntosh and H. Khalil, Can. J. Chem., 56, -2134 (1978).
[1096] A. Jonczyk, A. Kwast, and M. MaJcosza, Tetrahedron Lett., 1979, 541.
[1097] V. Dryanska, K. Popandova-Yambolieva, and C. Ivanov, Tetrahedron Lett., 1979,
443.
[1098] K. Takahashi, S. Kimura, Y. Ogawa, K. Yamada, and H. Iida, Synthesis, 1978, 892.
*[1099] Y. A. Zhdanov, Y. E. Alekseev, G. V. Zinchenko, and S. S. Dorosihenko, DokU
Akad. Nauk SSSR Ser. Khim., 231, 868 (1976); Engl. transl. p. 703.
[1100] b. Cassar, S. Panossian, and С Giordano, Synthesis,-1978, 917. - ';
[1101] D. H. Hunter, M- Hanity, У, Patpl.andR. A, Perry,Can. J. Chem., 56,104.(1978).!
443
{1102) a. Chollei. i Y Hagcnbuch ana P. Vogei, Helv Chim. Ada, 6lA\\ (1979).
Ц103) l T Scon arid» R JBfunsvola J Org. Chem.. 44. 641 (1979У
11104) E V Dehmlow and M Lissei, Synthesis. 1979, 372
11105) E V. Dehmloftand M Lissei Liebigs Ann .Шт.. 1980, l
[1106) P Hayden Germ Offenl 2,633,228 (1977): Chem Abstr., 8>S, 139395 (1977).
11107) V О Illi Synthesis 1979 387
[1108) W RaBhotei and F Vogtle. Tetrahedron Lett., 1979, 12t
[1109j E V Dehmlov, and S Barahona-Naranjo, J. Chem. R/s., 1979, (S) 238.
[1110J V Mancini О Morelli antl L Standoli, Gazz Chin/ltal., 107, 47 (1977).
[1111) G Wittigand L> Schoch-Grublei Justus Liebigs Afh. Chem., 1978, 362.
11112) C. Botteghi O. Caccia. and S Gladiali: Chim. In/. (Milan), 59, 839 (1977); Chenu
Abstr., Щ 152172(1978).
pi 13) E. V. Dehmlow and S. Barahona-Naranjo, unpublished work.
*[1114J А. К Khusid. G V Kryshtal, V F Kucher/W, and L. A. Yanovskaya, Izv. Akad.
Nauk SSSR Ser Khtm., 26, 692 (1977); En^l. transl., p. 628.
[1115) M J Farrall, T Durst, and J. M. J. Frtchet, Tetrahedron Lett., 1979, 203.
[1116] B. Stanovnik, M. TiSler, M. Kunaver, D. GabrijdCiC, and M. Kocevar, Tetrahedron
Lett., 1978, 3059.
[1117] P D. Klemmensen, H. Kolind-Andersen, M. B. Madsen, and A. Svendsen, J. Org*
Chem... 44, 416 (1979)
[1118] M. Seno, T. Namba, ana H. Kise,J. Org. Chem., 43, 3345 (1978).
[1119] A R. Butler and P. T. Shepherd, J. Chem. Res., 197g, (S) 339, (M) 4471.
{1120] A. Jonczyk, A. Kwast, and M. Makosza, /. Org. Chem., 44, 1192 (1979).
[1121] S. Yanagida, K, Takahashi, and M. Okahara, J. Org. Chem., 44, 1099 (1978).
[1122] H AlperWid H.-N. Paik.7. Am. Chem. Soc, 100, 508 (1978).
J1123] A. K. Shukla and W. Preetz, Angew. Chem., 91, 160 (1979); Angew. Chem. Int. Ed,
Engl., 18, 151 (1979).
[1124] P. S. Braterman, B. S. Walker, and Т. Н. Robertson, J. Chem. Soc. Chem. Common.,
1977, 651
{1125] H. Alper, J. K. Currie, and H. des Abbayes, /. Chem. Soc. Chem. Common., 1978,
311.
[1126] Y Gaoni, Tetrahedron Lett., 1978, 3277.
Г1127] T. Uyehara, A. Ichida, M. Funamizu, H. Nanbu, and Y. Kitahara, Bull. Chem. Soc.
Jpn., 52, 273 (1979).
41128] M. Seno, S. Shiraishi, Y. Suzuki, and T. Asahara, Bull. Chem. Soc. Jpn., 51, 1413
(1978).
[1129] L. Paryulescu and M. D. Gheorghiu, Rev. Roum. Chim., 22, 1089 (1977).
[1130] G. F Weber and S. S. Hall, J. Org. Chem., 44, 447 (1979).
•[1131] N. N. Labeish, E. M. Kharicheva, T.-V. Mandelshtam, and R. R. Kostkov, Zh. Org",
Khim., 14, 878 (1978); Engl. transl., p. 815.
[1132] P. Duchaussoy, P. DiCesare, and B. Gross, Synthesis, 1979, 198.
[ПЗЗ] K. Steinbeck and J. Klein, J. Chem. Res., 1978, (S) 396, (M) 4771.
Jl 134] K. Steinbeck, Chem. Ber., 112, 2402 (1979).
[1135] K. Steinbeck, private communication.
[1136] K. Kawashima, T. Saraie, Y. Kawano, and T. Ishiguro, Chem. Pharm. Bull., 26, 94Z
(1978).
[1137] U. K. Pandit, Heterocycles, 8, 609 (1977).
[1138] W. Ando, H. Higuchi, and T. Migata, /. Org. Chem., 42, 3365 (1977).
[1139] J. N. Shah, Y. P. Mehta, and G. M. Shah, J. Org. Chem., 4i, 2078 (1978).
[1140] A. R. Shamout, Diploma Thesis, Teclyiische Universitat Berlin, 1979.
[1141] D. Landini, F. Montanari, and F. Rolla, Synthesis, 1979, 26.
[1142] H. S. D. Soysa and W. P. Weber, Tetrahedron Lett., 1978,1969.
Ц143] £. V Dehmlow and M. Lissei, Chem. JBer., HI, 3873 (1978).
444
•(1144J R. R. Kostikov and A. P. Molchanov, Zh. Org. Khim,, 14, 879 (1978); Engl. transl.,
p. 816. \
i[1145] R. R. Kostikov, A. P. Molchanov, I. A. Vasil'eva, and Y. M. Slobodin, Zhur. Org.
Khim., 13, 25*1 (1977); Engl. transl., p. 2361.
II146] O. P. Vig, I. R-Yrehan, G. L. Kad, and A. L. Bedi, Indian J. Chem. Ser. B, 16,455
(1978). . \
[1147] O. P. Vig, G. L. СЦ, A. L. Bodin, and S. D. Kumar, Indian J. Chem. Ser. B, 16,452
(1978). \
[1148] R. F. Heldeweg and f\ Hogeveen, J. Org. Chem., 43, 1916 (1978).
[1149] W. Amman, R. A. PfunjJ, and.C. Ganter, Chimia, 31, 61 (1977).
J1150] G. I. Fray and R. G. Sa*jon, Tetrahedron, 34, 2663 (1978).
[1151] L. K. Sydnes, Ada Chem. Scand. Ser. B, 31, 823 (1977).
{1152] E. Piers and E. H. Ruediger,/. Chem. Soc. Chem. Commun., 1978,166.
[1153] H. Sadlo and W. Kraus, Tetrahedron, 34,1965 (1978).
[1154] M. S. Baird, A. G. W. Baxter, B. R. J. Devlin, and R. J. G. Searle, J. Chem. Soc.
Chem. Commun., 1979, 210.
[1155] E. Piers, I. Nagakura, and H. E. Morton, J. Org. Chem., 43, 3630 (1978).
[1156] E. Piers, I. Nagakura, and J. E. Shaw, J. Org. Chem., 43, 3431 (1978).
[1157] K. H. Holm, D. G. Lee, and L. Skattetol, Ada Chem. Scand. Ser. B, 32, 693 (1978).
[1158] J. B. Lambert, K. Kobayashf, and P. H. Mueller, Tetrahedron Lett., 1978, 4253;
M. Jones, Jr., V. J. Tortorelli, P. P. Gaspar, and J. B. Lambert, ibid., 1978, 4257.
[1159] M.S. Baird,/. Chem. Soc. Perkin Trans. 1, 1979, 1020.
[1160] B>Giese and J. Meister, Angew. Chem., 90, 636 (1978); Angew. Chem. Int. Ed. Engl,
17, 595 (1978).
[1161] K. P. Butin, A. N. Kashin, I. P. Beletskaya, L. S. German, and V. R. Polwchchuk,
J. Organomet. Chem., 25, 11 (1970).
[1162] Y. Bessiere, D. N. H. Savary, and M. Schlosser, Helv. Chim. Ada, 60, 1739 (1977>.
[1163] M. Christl, G. Freitag, and G. Bruntrup, Chem. Ber., Ill, 2307 (1978).
*[1164] V. S. Aksenov and G. A. Terenfeva, Izv. Akad. Nauk SSSR Ser. Khim., 26, 623
(1977); Engl. transl., p. 560.
[1165] P. J. Stang and J. A. Bjork, J. Chem. Soc. Chem. Commun., 1978,1057.
[1166] T. Sasaki, S. Eguchi, and F. Nakata, Tetrahedron Lett., 1978, 1999.
[1167] J. T. Martz, G. W. Gokel, and R. A. Olofson, Tetrahedron Lett., 1979, 1473.
[1168] H. Alper and H.-N. Paik, Nouv. J. Chim., 2, 245 (1978).
[1169] P. Mangeney and Y. Langlois, Tetrahedron Lett., 1978, 3015.
Ц170] L. Homer and W. Brkh, Justus Liebigs Am. Chem., 1978, 710.
[1171] R. Kinishi, Y. Nakajima, J. Oda, and Y. Inouye, Agric. Biol. Chem., 42, 869 (1978);
Chem. Abstr., 89, 106932 (1978).
[1172] J. Mass* and E. Parayre, Bull. Soc. Chim. Fr., 1978, II 395.
[1173] J. M. Mclntosh, Tetrahedron Lett., 1979, 403.
[1174] K. Nakamura, A. Ohno, S. Yasui, and S. Oka, Tetrahedron Lett., 1978, 4815.
[1175] D. L. Reger, M. M. Habib, and D. J. Fauth, Tetrahedron Lett., 1979, 115.
[1176] J. A. Morris and D. С Mills, Chem. Br., 14, 326 (1978).
[1177] D. G. Lee and V. S. Chang, J: Org. Chem., 43, 1532 (1978).
[1178] ref. [5], p. 161.
[1179] A. Poulose and R. Croteau, J. Chem. Soc. Chem. Commun., 1979, 243.
[1180] P. K. Kadaba, Synthesis, 1978, 694.
[1181] L. M. Rossi and P. Trimarco, Synthesis, 1978, 733.
[1182] H.-J. Schmidt and H. J. Schafer, Angew. Chem., 91, 77 (1979); Angew. Chem. Int.
Ed. Engl., 18, 77 (1979).
11183] H.-J. Schmidt and H. J. Schafer, Angew. Chem., 91, 78(1979); Angew. Chem. Int.
Ed. Engl., 18, 78 (1979).
[1184] D. G. Lee and V. S. Chang, Synthesis, 1978, 462.
445
*[1185] М. L. Navtanovich, L A Dzhanashvili, and V L. Kheifets, Zh.ubshch. Khim., 48,
1925 (1978); Engl. transl., p. 1755. /
[11861 D. R Bender, J Brennan, and H. Rapoport,/. Org. Chem.,fi, 3354 (1978).
[1187] Y. Masuyama, Y. Ueno, and M. Okawara, Chem. Lett., 1917, 1439.
[1188] M J. Gibian and T Ungermann, /. Am. Chem. Soc, 101,,/1291 (1979).
[1189] C.-l. Chern, R. DiCosimo, R. DeJesus, and J. S. San Filipi>o Jr., /. Am. Chem. Soc,
100, 7317 (1978). /
[1190] W С Danen and R. L. Arudi, /. Am. Chem. Soc., 100, 3944 (1978).
[1191] R. A. Johnson, E. G. Nidy, and M. V. Merritt,/. Am. Chem. Soc., 100, 7960 (1978).
[1192] T Takata, Y H. Kim, and S. Oae, Tetrahedron Lett., 1979, 821. i
[1193] M Matsuo, S. Matsumoto, Y Iitaka, A. Hanaki, and T. Ozawe, /. Chem. SoK
Chem. Commun., 1979, 105.
[1194] M. J. Gibian, D. T Sawyer, T. Ungermann, R. Tangpoonpholvivat, and M. M.
Morrison, J. Am. Chem. Soc, 101, 640 (1979).
[1195] G. A. Lee and H. H. Freedman, U.S. Pat. 4,079,075 (1978); Chem. Abstr., 89, 41578
(1978).
[1196] D. Landint, F. Montanan, and F. Rolla, Synthesis, 1979, 134.
[1197] D. Landini and F. Rolla, Chem. Irtd. {London), 1979, 213.
[H98] S. Cacchi, F. La Torre, and D. Misiti, Synthesis, 1979, 356.
[1199] G. Aksnes and B. H. Vagstad, Acta Chem. Scand. Ser. B, 33, 47 (1979).
[12( ч G. E. Keck and S. A. Fleming, Tetrahedron Lett., 1978, 4763.
[1201] M. Hirao, H. Mochizuki, S. Nakahama, and N. Yamazaki, /. Org. Chem., 44, 1720
(1979).
[1202] D. W. Armstrong and M. Godat, J. Am. Chem. Soc. 101, 2489 (1979).
[1203] R. C. Cookson and I. D. R. Stevens, Chem. Br., 15, 329 (1979).
[1204] Y. Yamoto, J. Oda%nd Y. Inouye, Tetrahedron Lett., 1979, 2411.
[1205] M. S. Baird and M. Mitra, J. Chem. Soc. Chem. Commun., 1979, 563.
[1206] M. F. Ahern and G. W. Gokel, J. Chem. Soc. Chem. Commun., 1979,1019. M. F. Ahem, A.
Leopold, J. R. Beadle, and G. W. Gokel, J. Am. Chem. Soc. 104, 548 (1982).
[1207] S. Akabori and M. Ohtomi, Synthesis, 1979, 616.
[1208] H. Alper and S. Amaratunga, Tetrahedron Lett., 1980, 1589.
[1209] H. Alper and J. K. Currie, Tetrahedron Lett., 1979, 2665.
[1210]. S. Ambasht, S. K. Chiu, P. E. Peterson, and J. Queen, Synthesis, 1980, 318.
[1211] R. Annunziata, M. Cinquini, and S. Colonna, Chem. Ind. (London), 1980, 238.
[1212] A. Arbin, H. Brink, and J. Vessman, /. Chromatogr., 170, 25 (1979).
•[1213] V. D. Novokreshchennykh, S. S. Mochalov, and Y. S. Shabarov, Zh. Org. Khim, 15,485
(1979); Engl. transl. p. 430.
[1214] I. Artaud, J. Seyden-Penne, and P. Viout, Synthesis, 1980, 34.
[1215] I. Artaud, J. Seyden-Penne, and P. Viout, Tetrahedron Lett., 1980, 613.
[1216], С A. Audeh, S. E. Fuller, R. J. Hutchinson, and J. R. L. Smith, /. Chem. Res., 1979, (S)
' 270; (M) 2985.
[1217] T. P. N'Guyen and S. Boileau, Tetrahedron Lett., 1979, 2651.
[1218] E. Balogh-Hergovich, G. Speier, and E. Winkelmann, Tetrahedron Lett., 1979, 3541.
[1219] A. G. M. Barrett, P. A. Prokopiou, D. H. R. Barton, R. B. Boar, and J. F. McGhie, Л
Chem. Soc. Chem. Commun., 1979, 1173.
[1220] A. G. M. Barrett, P. A. Prokopiou, and D. H. R. Barton, J. Chem. Soc. Chem. Commun.,
1979, 1175.
[1221] R. A. Bartsch, P. N.Juri, and M. A. Mills, Tetrahedron Lett.,1979,2499; R. A. Bartschand
1. W. Yang, ibid., 1979, 2503.
[1222] H. Oedigerand A. Schulze, Germ. Ausl. 2,757,483(28.6.1979); A. Schulze and H. Oediger,
» Liebigs Ann. Chem., 1981, 1725.
[1223] M. С Benhamou, G. Etemad-Moghadam, V. Speziale, and A. Lattes, Synthesis, 1979,89b
G. Etemad-Moghadam, M. C. Benhamou, V. Speziale, A. Lattes, and A. Bielawska, Nov.
J. Chim., 4, 727 (1980).
[1224] J. F. BieUmann, H. D'Orchymont, and M. P. Goeldner, Tetrahedron Lett., 1979, 4209.
446
[1225] V. Bocchft G. Casnati, A. Dossena, and R. Marchelh, Synthesis, 1979, 957, 961.
*[1226] A. V Bogatskii, N. G. Lukyanenko. and V N. Pastushok, Dokl. Akad. Nauk SSSR, 247,
1153 (1979); Engl. transl. p. 379.
[1227] T. Nishikubo, T Iizawa, and К Kobayashi, Makromol. Chem. Rapid Commun. 1. 765
(1980).
[1228] K. Brewster, J. M. Harrison, and T. D. Inch, Tetrahedron Lett., 1979, 5051
[1229] H. Brink, R. Modin, and J. Vessman, Ada Pharm. Suet. 16, 247 (1979).
{1230] E. Buschmann, and B. Zeeh, Liebigs Ann. Chem. 1979, 1585.
J1231] S. Cacchi, D. Misiti, and M. Felici, Synthesis, 1980, 147
|1232] S. Cacchi, D. Misiti, and F. La Torre, Synthesis, 1980, 243.
{1233] F. .Camps, J. Coll, and M. Riba, J. Chem. Soc. Chem. Commun., 1979, 1080.
J1234] G. Cardillo, M. Orena, G. Porzi, and S. Sandri, J. Chem. Soc. Chem. Commun. 1979, 836.
J1235] L. A. Carpino, and A. C. Sau, J. Chem. Soc. Chem. Commun. 1979, 514.
(1236] L. Cassar, M. Foa, F Montanari, and G. P. Marinelli, J. Organomet. Chem. 173, 335
(1979).
11237] T. Ogino, Tetrahedron Lett., 1980, 177.
(1238] M. Cieslak, E. Grochowski, J. Jurczak, K. Kurowska, and W. Szelejewski, Przem. Chem.,
58, 154 (1979); Chem. Abstr. 91, 20062 (1979).
{1239] S. D. Clarke, С R. Harrison, and P. Hodge, Tetrahedron Lett., 1980, 1375.
(1240J E. J. Corey and H. Estreicher, Tetrahedron Lett., 1980, 1117.
{1241] G. Coudert, G. Guillaumet, and B. Loubinoux, Synthesis, 1979, 541.
11242] H. J. Cristau, A. Long, and H. Christol, Tetrahedron Lett., 1979, 349.
[1243] W. Czuba, P. Kowalski, and K. Rutkowski, Polish J. Chem. 53, 1477 (1979).
11244] I. Degani, M. Dolci, and R. Fochi, Synth. Commun., 10, 161 (1980).
(1245] I. Degani, R. Fochi, and V. Regondi, Synthesis, 1980, 375.
{1246] H. Des Abbayes, and A. Bouloup, J. Organomet. Chem., 179, С 21 (1979).
[1247] R. R. Dewald, S. R. Jones, and B. S. Schwartz, J. Chem. Soc. Chem. Commun., 1980, 272.
[1248J S. A. Di Biase, B. A. Lipisko, A. Haag, R. A. Wolak, and G. W. Gokel, J. Org. Chem. 44,
4640 (1979).
[1249] P. Di Cesare, and B. Gross, Synthesis, 1979, 458.
,[1250] J. M. Domagala and R. D. Bach, J. Org. Chem. 44, 3168 (1979)..
[1251] H. J. M. Dou, J. Elguero, M. Espada, and P. Hassanaly, Ann. Quim., 74, 1137 (1978).
*{1252] E. A. Drygailova, R. R. Kostikov, and K. A. Ogloblin, Zh. Org. Khim, 14, 2008 (1978);
Engl. transl. p. 1863.
[1253] N. T. Dung and S. Boileau, Polym. Bull., 1, 817 (1979); Chem. Abstr. 92, 76974 (1980).
[1254] .1. Dyong, N. Jersch, and Q. Lam-Chi, Chem. Ber., 112, 1859 (1979).
[1255] J, Elguero, M. Espada, D. Mathieu, and R. Phan Tan Lun, Ann. Quim. 75, 729 (1979).
[1256] P. O. Ellingsen and K. Undheim, Ada Chem. Scand., Ser. В., 33, 528 (1979).
[1257] M. EUwood, J. Griffith, and P. Gregory, J. Chem. Soc. Chem. Commun., 1980, 181.
[1258] E. Flesia, R. Nouguier, and J. M. Surzur, Tetrahedron Lett., 1979, 197.
[1259] J. M. J. Frechet, M. D. de Smet, and M. J. Farrall, J. Org. Chem. 44, 1774 (1979).
[1260] A. A. Frimer and P. Gilinsky, Tetrahedron Lett., 1979, 4331.
[1261] T. Gajda, A. Koziara, St. Zawadzki, and A. Zwierzak, Synthesis, 1979, 549.
[1262] J. P. Galy, J. Elguero, E. J. Vincent, A. M. Galy, and J. Barbe, Synthesis 1979, 944; A.
Mahamoud, J. P. Galy, E. J. Vincent, and J. Barbe, ibid., 1981, 917.
[1263] T. Ogino and K. Mochizuki, -Chem. Lett., 1979, 443
[1264] T. Gibson, J. Org. Chem., 45, 1095 (1980).
[1265] D. H. Gibson, W.-L. Hsu, and D.-S. Lin, J. Organomet. Chem., 172, С 7 (1979).
[1266] J. Golinski, A. Jonczyk, and M. Makosza, Synthesis, 1979, 461.
[1267] H. Greuter, T. Winkler, and D. BelluS, Belt. Chim. Ada 62, 1275 (1979).
[1268] N. Hayashi, T. Toga, N. Tada, and T. Azuma, J. Label. Сотр. Radiopharm., 15,489 (1978).
11269] M. Hamana, F. Sato, Y. Kimura, N. Nishikawa, and H. Noda, Heterocycles, 11, 371
(19,78).
[1270] M. Hedayatullah, С R. Acad. Sci. Ser. C, 289, 365 (1979).
11271] J. W. Hill and Т. С Boyd, J. Chem. Educ., 56, 824 (1979).
[1272] K. Ogura, M. Fujita, and H. Iida, Tetrahedron Lett., 1980, 2233.
447
[1273] А. В. Holmes, С. L. D. Jennings-White, A. H. Schulthess, A. Akinde, and D. R. M. Walton,'
J. Chem. Soc. Chem. Commun., 1979, 840.'
[1274] T. Hosokawa, T. Ohta, Y. Okamoto, and S.-I. Murahashi, Tetrahedron Lett., 1980,12S9.
[1275] S. Hfinig, M. Oiler, and G. Wehner, LtMgs Ann. Chem. 1979,1925.
[1276] S. Cacchi, F. La Torre, arid D. Misiti, Tetrahedron Lett., 1979, 4591.
[1277] S. Hflnig and G. Wehner, Synthesis, 1979, 522; c/. also: J. K. Rasmussen and S. M.
Heilmann, Synthesis, 1979, 523.
[1278] V. O. Illi, Tetrahedron Lett. 1979, 2431.
[1279] Y. Imai, A. Kato, M. Ii, and M. Ueda, J. Polym. Sci. Polym. Lett. Ed. 17, 579 (1979).
[1280] Imp. Chem. Ind. Ltd., Belg. Pat. 854, 903 (1977); Chem. Abstr. 89, 112310 (1978).
[1281] S. E. Jacobsen, F. Mares, and P. M. Zambri, J. Am. Chem. Soc, 101, 6938 (1979).
[1282] S. E. Jacobsen, F. Mares, and P. M. Zambri, J. Am. Chem. Soc. 101, 6946 (1979).
[1283a] H. Jager, J. Lutolf, and M. W. Meyer, Angew. Chem., 91,852 (1979); Angew. Chem. Int. Ed.
Engl., 18, 786 (1979).
[1283b] H. J. Schmidt and H. J. SchSfer, Angew. Chem., 91,852 (1979); Angew. Chem. Int. Ed. Engl.%
18, 787 (1979).
[1284] A. Jarczewski, G. Schroeder, and K. T. Leffek, J. Chem. Soc. Perkin Trans. 2, 1979, 866.
[1285] M. Jawdosiuk and I. Kmiotek-Skaiiynska, Pol. J. Chem., 53, 2259 (1979).
[1286] M. Jawdosiuk, M. Ludwikow, and B. Bednarska, Pol. J, Chem. 53, 805 (1979).
[1287] M. Jawdosiuk, M. Makosza, E. Malinowska, and W. Wilczynski, Pol. J. Chem., 53, 617
(1979).
[1288] С W. Jefford, J.-C. E. Gehret, and V. de los Heros, Bull. Soc. Chim. Belg., 88, 973 (1979).
[1289] J.-I. Jin, Y.-M. Jung, K.-S. Lee, andK.-W. Chung, Taehan Hwahakhoe Chi., 23,259 (1979);
Chem. Abstr. 92, 129392 (1980).
[1290] G. C. Johnson and R. G. Bergman, Tetrahedron Lett., 1979, 2093.
[1291] S. Julia and A. Ginebreda, Arm. Quint., 75, 346 (1979).
[1292] S. Julia and A. Ginebreda, Tetrahedron Lett., 1979, 2171.
[1293] S. Julia, A. Ginebra, and J. Pallas, Aflnidad, 36,326 (1979); Chem. Abstr., 92,93483 (1980).
[1294] Y. Okamoto and S. T. Attarwala, J. Org. Chem., 44, 3269 (1979).
[1295] Y. Kimura, Y. Tomita, S. Nakanishi, and Y. Otsuji, Chem. Lett., 1979, 321.
[1296] S. J. Kirsch and H. Schellmg, J. Org. Chem., 44, 3970 (1979).
[1297] G. KoBmehl and R. Nuck, Chem. Ber., 112, 2342 (1979).
* [1298] R. R. Kostikov, E. N. Grishina, and Ya. M. Slobodin, Zh. Org. Khim., 15,33J (1979); Engl.
transl. p. 287.
[1299] E. Kunesch, Diploma Thesis, Technical University Berlin, 1980.
[1300] R. A. Olofson and J. Cuomo, Tetrahedron Lett., 1980, 819.
•[1301] G. V. Kryshtal, V. V. Kulganek, V. F. Kucherov, and!. A. Yanovskaya, Izv. Akad. Nauk
SSSR Ser. Khim., 27, 2808 (1978); Engl. transl. p. 2508.
• [1302] G. V. Kryshtal, V. F. Kucherov, and L. A. Yanovskaya, Izv. Akad. Nauk SSSR Ser. Khim.,
27, 2803^1978); Engl. transl. p. 2503.
11303], S. Kulstad and L. A, Malmsten, Tetrahedron Lett., 1980, 643.
[1304]' suggested by Prof. Hinze, Universitat Bielefeld.
[1305] J. T. Lai and J. С Westfall, J. Org. Chem., 45, 1513 (1980).
[1306] A. Le Coq and A. Gorgues, Org. Synth., 59, 10 (1980).
[1307] B. P. Leddy, M. A. McKervey, and P. McSweeny, Tetrahedron Lett., 1980, 2261.
[1308] D. G. Lee and V. S. Chang, J. Org. Chem. 44, 2726 (1979).
[1309] R. Le Goaller, H. Handel, M. A. Pasquini, and J. L. Pierre, Tetrahedron, 35,1437 (1979).
[1310] R. Le Goaller, M. A. Pasquini, and J. L. Pierre, Tetrahedron, 36, 237 (1980).
[1311] M. Lind and T. Winkler, Tetrahedron Lett., 1980, 119.
[1312] A. Jonczyk, M. Ludwikow, and M. Makosza, Organ. Prep. Proced. Int. 11, 275 (1979).
[1313] O. Markl, P. Hofmeister, and R. SchieBl, Tetrahedron Lett. 1979. 3503.
[1314] MakhteshjmChem. Works Ltd., Jap. Kokai 79,132,529 (1979); Chem. Abstr. 92, 111499
(1980).
[1315] M. Makosza and J. Winiarski, J. Org. Chem., 45, 1534 (1980).
[1316] L. Mandolini and B. Masci, Synth. Commun., 9, 851 (1979).
[1317] L. J. Mathias and D. Burkett, Tetrahedron Lett., 1979, 4709.
448
[1318] R. Mathias and P Weyerstahl, Chem. Ber., 112, 3041 (1979).
[1319] K. Mizuno, Y. Kimura, and Y. Otsuji, Synthesis, 1979, 688.
[1320] F. Montanari and P. Tundo, Tetrahedron Lett., 1979, 5055
[1321] G. Moore, T A Foglia, and T. J McGahan, /. Crg. Chem., 44, 2425 (1979).
*[1322] M. M. Movsumzade, A. L. Shabanov, and G. Kh. Mamedov, Izv. Vyss. Uchebn. Zaved,
Khim. Khim. Tekhnol., 22, 758 (1979); Chem. Abstr. 91, 157711 (1979).
[1323] R. Munz and G. Simchen, Liebigs Am. Chem., -1979, 628.
[1324] Y. Nakano end H. Hiura, Organ. Prep. Proced. Int., 11, 267 (1979).
• [1325] O. M. Nefedov, I. E. Dolgii, and E. V. Bulusheva, Izv.. Akad. Nauk. SSSR Ser. Khim., 27,
1454 (1978); Engl. transl. p. 1271.
• [1326] O. M. Nefedov, I.E. Dolgii, I. B. Shvedova, and E. A. Baidzhigitova, Izv. Akad. Nauk SSSR
Ser. Khim., 27, 1339 (1978); Engl. transl. p. 1164,
[1327] J. Ledon, Org. Synth., 59, 66 (1980).
. [1328] K. M. Patel and J. T. Sparrow, Synth. Common., 9, 251 (1979).
[1329] P. M. Quan and J. S. Hunter, Germ. Offenl. 2,905, 274 (1979); Chem. Abstr. 91, 194 616
(1979). D. B. Baird, R. D. McClelland, and P. M. Quan, Brit. Pat. 1,578,733 (1980); Chem
Abstr. 94, 210286 (1981).
. [1330] W. Reischl and E. Zbiral, Tetrahedron, 35, 1109 (1979).
[1331] G. Rosini, A. Salomoni, and F. Squarcia, Synthesis, 1979, 942.
[1332] J. E. Rowe, Synthesis, 1980, 114.
[1333] K. Saigo, H. Koda, and H. Nohira, Bull. Chem. Soc. Jp., 52, 3119 (1979).
[1334] I. Saito, T. Otsuki, and T. Matsuura, Tetrahedron Lett., 1979, 1693.
[1335] M. SaliSova and H. Alper, Angew. Chem., 91, 858 (1979); Angew. Chem. Int. Ed. Engl, 18,
792 (1979).
[1336] E. Santaniello, С Farachi, and F. Ponti, Synthesis, 1979, 617.
[1337] E. Santaniello and P. Ferraboschi, Synth. Commm.,10, 75 (198.0).
[1338] Y. Sasson and M. Yonovich, Tetrahedron Lett., 1979, 3753.
[1339] D. T. Sawyer and M. J. Gibian, Tetrahedron, 35, 1471 (1979).
[1340] Y. Watanabe and T. Mukaiyama, Chem. Letters, 1981, 443.
[1341] F. E. Scully Jr., J. Org. Chem., 45, 1515 (1980); 43, 1467 (1978).
[1342] F. Seela and D. Winkeler, Angew. Chem., 91,570 (1979); Angew. Chem. biter. Ed. Engl. 18,
536 (1979).
[1343] A. Sele, B. Baehler, and J. M. J. Tronchet, Helv. Chim. Acta, 62, 866 (1979).
[1344] Y. Shida, N. Ando, Y. Yamamoto, J. Oda, and Y. Inouye, Agric. Bid. Chem., 43, 1797
(1979).
[1345] S. Shinkai, T. Fukunaga, and O. Manabe, J. Org. Chem., 44, 4990 (1979).
11346] H. Singh and P. Singh, Chem. bid. (London), 1978, 807.
[1347] H. Singh and P. Singh, Indian J. Chem., 17b, 1 (1979).
. [1348] S. Snipes and A. W. Herriott, J. Am. Chem. Soc., 101,6441 (1979).
•^1349] V. A. Sokolenko, V. A. Markova, and В. Е. Kogai, Zh. Org. Khim., 14,1111 (1978); Engl.
transl. p. 1035.
11350] J. P. Stanley, J. Org. Chem., 45, 1413 (1980).
[1351] K. Steinbeck, Tetrahedron Lett., 1980, 2149.
[1352] K. Steinbeck, Liebigs Ann. Chem., 1979, 920.
[1353] K. Steinbeck and J. Klein, J. Chem. Res., 1980, (S) 94, (M) 1150; (S) 95, (M) 1163.
[1354] A. O. Sy and J. W. Raksis, Tetrahedron Lett., 1980, 2223.
[1355] W. Szeja, Synthesis, 1980, 402.
[1356] W. Szeja, Synthesis, 1979, 822.
[1357] W. Szeja, Synthesis, 1979, 821.
[1358] I. Tabushi and N. Koya, Tetrahedron Lett., 1У79, 3681.
[1359] K. Tanaka, N. Ono, Y. Kubo, and A. Kaji, Synthesis, 1979, 890.
[1360] F. Texier-Boullet and A. Foucaud, Synthesis, 1979, 884.
[1361] F. Tewer-Boullet and A. Foucaud, Tetrahedron Lett., 1980, 2161.
[1362] W. Chin-Hsien, Synthesis, 1981,622; H. lilies and P. E. Hoch, U.S. Pat. 4,147,715 (1979);
Chem. Abstr., 91, 19927 (1979).
449
[1363] L. M. Weinstock, A. M. De Marco, R. J. Tull, and I. Shinkai, Org. Prep. Prac. Intern., 13,103.
(1981); R. J. Tuli, L. M. Weinstock, and I. Shinkai, U. S.-Pat. 4,140,719 (1979); Chem. Abstr.,
90, 186558 (1979).
[1364] P. Tundo, J. Org. Chem., 44, 2048 (1979).
[1365] P. Tundo and P. Venturello, Synthesis, 1979, 952.
[1366] F. Wada, R. Ishihara, У. Kamohara, and T. Matsuda, Bull. Chem. Soc. Jpn., 52„2959 (1979).
[1367] J. D. Warren and J. B. Press, Synth. Commun., 10, 107 (1980).
[1368] J. WUdeman and A. M. van Leusen, Synthesis, 1979, 733.
[1369] H. Wynberg and B. Marsman, /. Org. Chem., 45, 158 (1980).
[1370] Y. Yano, A. Kurashima, and W. Tagaki, Bull. Chem. Soc. Chem. Jpn., 52,2739 (1979).
[1371] M. Yonovich and Y. Sasson, Tetrahedron Lett., 1980, 1875.
[1372] S. Yoshida, H. Kawara, and N. Shigeyuki, Asahi Garasu Keukya Нококы (Jpn.), 28, 14$
(1978); Chem. Abstr.92, 129817 (1980).
*[1373] S. S. Yufrt and I. A. Esikova, Izv. Akad. NaukSSSR Ser. Khim. 28,1706 (1979);. Engl. transl.
p. 1573.
[1374] A. Koziara, S. Zawadzki, and A. A. Zwierzak, Synthesis, 1979, 527.
*Ц375] Y. A. Zhdanov, Y. E. Alekseev, V. G. Alekseeva, E. L. Korol, V. А. Tyumcnev,.!- I. Popov,
and V. A. Polenov, Dokl. Akad. Nauk SSSR Ser. Khim., 244,1122 (1979); EngL transi. p.
63.
[1376] H. Alper and S. Amaratunga, Tetrahedron Lett., 1980, 2603.
[1377] M. G. Banwell and B. Halton, Austr. J. Chem., 32, 849 (1979).
[1378] M. Jay, W. J. Layton, and G. A. Digenis, Tetrahedron Lett., 1980, 2621.
[1379] M, A. Pasquini, R. Le Goaller, and J. L. Pierre, Tetrahedron, 36, 1223 (1980).
[1380] W. RaBhofer, G. Oepen, and F. Vogtle, Isr. J. Chem., 18, 249 (1979).
[1381] E. Santaniello, F. Ponti, and A. Manzocchi, Tetrahedron Lett., 1980, 2655.
•[1382] G. N. Suvorova and M. I. Komendantov, Zh. Org. Khim., 15,1435 (1979); Engl. transl. p.
1280.
•[1383] Y. A. Zhdanov, Y. E. Alekseev, I. N. Siilis, T. P. Sudareva, and G. V. Bogdanova, Dokl.
■Akad. Nauk SSSR Ser. Khim., 245, 846 (1979); Engl. transl. p. 157.
[1384] T. Hiyama, M. Shinoda, and M. Tsukanaka, Bull. Chem. Soc. Jpn., 53, 1010 (1980).
■11385] K. Inomata, Y. Nakayama, and H. Kotake, Bull. Chem. Soc. Jpn., 53, 565 (1930).
[1386] H.-D. Winkeler and F: Seela, Chem. Ber., 113, 2069 (1980).
[1387] G. Tarrago, A. Ramdani, J. Elguero, and M. Espada, /. Heterocycl. Chem., 17,137 (1980).
[1388] W. H. Daly and J. D. Caldwell, J. Polym. Sc. Polym. Lett. Ed., 17, 55 (1979).
[1389] F. J. Jaggi, P. Buchs, and С Ganter, Helv. Chim. Acta, 63, 872 (1980).
•[1390] O. M. Nefedov, I. E. Dolgii, E. V. Bulusheva, and A. Y. Shteinshneider, Izv. Akad. Nauk
SSSR Ser. Khim., 28, 1535 (1979); Engl. transl. p. 1422.
[1391] F. Vogtle and W. M. Miiller, Chem. Ber., 113, 2081 (1980).
*{1392] R. R. Kostikov, A. P. Molchanov, O. A. Tarasova, S. V. Amosova, and B. A. Trofimov, Izv.
Akad. Nauk SSSR Ser. Khim., 28, 1286 (1979); Engl. transl. p. 1201.
[1393] J. Yamashita, S. Ishikawa, and H. Hashimoto, Bull. Chem. Soc. Jpn., 53, 736 (1980).
[1394] J. S. Bradshaw and P. E. Stott, Tetrahedron, 36, 461 (1980).
[1395] L. Cassar, Ann. N. Y. Acad. Sci., 333, 208 (1980).
[1396] J. Bergman and L. Engman, Synthesis, 1980, 569
[1397] E. V. Dehmlow and M. Lissel, Liebigs Ann. Chem., 1981, 28.
[1398] E. V. Dehmlow, Chimia, 34, 12 (1980).
[1399] A. J. S. Duggan, E. J. J. Grabowski, and W. K. Russ, Synthesis, 1980, 573.
[1400] M. Foa and L. Cassar, Gazz. Chim. Ital. 109, 619 (1979).
• [1401] S. Gambarotta and H. Alper, J. Organomet. Chem., 194, С 19 (1980).
[1402] E. Santaniello, A. Manzocchi, and С Farachi, Synthesis, 1980, 563.
[1403] O-Santelli, Tetrahedron Lett., 1980, 2893.
[1404] M. Tanaka, Tetrahedron Lett., 1980, 2959.
[1405] H. Tappe, Synthesis, 1980, 577.
[1406] H. J.-M. Dou, B. Delfort, P. Hassanaly, R. Gallo, and J. Kister, Bull. Soc. Chim. Belg., 89,
■ 421 (1980).
[1407] W.-H. Gundel and H. Berenbold, Z. Natwforsch., Teil B, 34, 1593 (1979).
450
[1408] R. Luckenbach and N. Mffller, Z. Naturforsch., Teil B, 34, 464 (1979).
[1409] С Boche, К Buckl, D. Martens, D. R. Schneider, and H.-U. Wagner, Chem. Ber., 112,2961
(1979).
(1410] J. P. H. Verheyden, A. C. Richardson, R. S. Bhatt, B. D. Grant, W. L. Fitch, and J. G.
Moflat, Pure Appl. Chem., 50, 1363 (1978).
(14111 J. Cuomo and R. A. Oiofson, /. Org. Chem. 44, 1016 (1979).
[1412] С Boan and L. Skattetykl, /. Chem. Soc. Perkin Trans. 1, 1978, 1568.
[1413] M Maeda, /. Label. Сотр. Radiopharm., 16, 175 (1979).
[1414] D. Seebach, H. Neumann, and R. Dammann, Helv. Chim. Ada, 62, 1162 (1979).
[1415] H. Alsaidi, R. Galio, and J. Metzger, С R. Acad. Sci. Ser. C, 289, 203 (1979).
[1416] V. Pozsgay, Carbohydr. Res., 69, 284 (1979).
[1417] R. H. Ellison, /. Org. Chem., 45, 2509 (1980).
[1418] G. Bram, D. Cabaret, N. Maigrot, J.-P. Mazaleyrat, and Z. Welvart, J. Chem. Res., 1979
(S) 196, (M) 2301; G. Bram, D. Cabaret, E. D'Incan, N. Maigrot, and Z. Welvart, ibid.,
1982, (S) 86.
(1419] M. S. Baird and A. G. W. Baxter, J. Chem. Soc. Perkin Trans. 1,1979, 2317.
[И20] G. S. Ponticello, R. D. Hartman, W. С Lumma Jr., and J. J. Baldwin, J. Org. Chem., 44,3080
(1979).
[1421] J. Yamawaki and T. Ando, Chem. Lett., 1980, 533.
{1422] M. Yamada, Y. Watabe, T. Sakakibara, and R. Sudoh, J. Chem, Soc. Chem. Comrnun., 1979,
179.
(1423] D. J. Collins and J. Sjovall, Tetrahedron Lett., 1979, 629.
[1424] T. Ido, J. Label. Сотр. Radiopharm., 16, 153 (1979).
• [1425] N. E. Akhmetova, V. M. Vlasov, and G. G. Yakobson, Izv. Akad. Nauk SSSR Ser. Khim., 27,
949 (1978); Engl. transl. p. 823.
*[1426] N. M. Abramova and S. V. Zotova, Izv. Akad. Каик SSSR Ser. Khim. 28,697 (1979); Engl
transl. p. 653.
*[1427] N. I. Shtemenko, V. F. Kucherov, and L. A. Yanovsfcaya, Izv. Akad. Nauk SSSR Ser. Khim^
27, 1444 (1978); Engl. transl. p. 1261.
|1428] A. S. Wengel, T. Reflstrup, and P. M. Boll, Tetrahedron, 35, 2181 (1979).
[1429] B. Fuchs and A. EUencweig, J. Org. Chem., 44, 2274 (1979).
[1430] R. W. Hoffmann and N. Hauel, Tetrahedron Lett., 1979, 4959.
[1431] G. Stork and V. Nair, J. Am. Chem. Soc., 101, 1315 (1979).
[1432] R. Gray and V. Boekelheide, J. Am. Chem. Soc., 101. 2128 (1979).
[1433] С Tarchini, M. Rohmer, and С Djerassi, Helv. Chim. Ada, 62, 1210 (1979).
[1434] A. KrutoSikova, J. Kova£, M. Dandarova, and M. Veverka, Coll. Czech. Chem. Commun.,
44, 1805 (1979).
(1435] A. Pusino, V. Rosnati, A. Saba, and F. Soccolini, Gazz. Chim. ltd., 108, 557 (1978).
[1436] F. G. de las Heras, R.Alonso, and G. Alonso, J. tied. Chem., 12, 496 (1979).
*[1437] V. M. Vlasov, V. V. Aksenov, and G. G. Yakobson, Zh. Org. Khim., 15,2156 (1979); EngL
transl. p. 1953.
[1438] K. Clausen, B. S. Pedersen, S. Scheibye, and S.-O. Laweson, Org. Mass. Spectr. 14, 101
(1979).
[1439] F. Jung, D. J^adjama, and J. J. Riehl, Synthesis, 1979, 507.
[1440] M. Maeda, H. Shimoirisa, H. Komatsu, and M. Kojima, Int. J. Appl. Rod. Isotopes, 30,255
(1979).
[1441] J. A. Foulkes and J. Hutton, Synth. Commun., 9, 625 (1979).
[1442] L. Trogen and U. Edlund, Acta Chem. Scand. Ser. B, 33, 109 (1979).
[1443] N. Minami and S. Kijima, Chem. Pharm. Bull., 27, 816 (1979). '
{1444] G. M. Buchan and G. G. Cameron, Polymer, 19, 1089 (i978).
[1445] M. R. Thissen and W. P. Duncan, J. Label. Сотр. Radiopharm., 15; 59 (1978).
[1446] R. Asami, M. Gyi, M. Takaki, and I. Ikuta, Polym. Journ., 10, 301 (1978).
[1447] J. D. Henion and D. G. I. Kingston, Org. Mass. Spectr., 13,431 (1978).
[1448] Z. E. Golubski, Synthesis, 1980, 632.
[1449] K. Schulze, M. Rentsch, P. Kuhl, and M. Muhlstadt, Z. Chem., 20, 186 (1980).
[1450] B. Schwabe,*}. Westphal, and H.-G. Henning, Z. Chem., 20, 184 (1980).
451
•11451] E. A. Runova, L. N. Borodina, А. К. Shcherbakov. and E. H. Karakhanov, Vestn. Mask.
Univ. Ser. Khim., 20, 581 (1979); Chem. Abstr., 92, 198217 (1980).
[1452] J. T. F. KaoandK. E. Wiegang,Can. Pat. 1,064,507(1979); Chem.Abstr.,92,180992(1980)..
.[1453] Q.-P. Huang, S.-K. Tang, R.-Q. Lao, and K.-H. Long, Hua Hsueh Tung Pao, 1979, 524;.
Chem. Abslr., 92, 180736 (1980).
[1454] M. Kodomari, M. Sawamura, N. Naoya, and S. Yoshitomi, Nippon Kagaku Kaishi, 1980,.
58; Chem. Abstr., 92, 180760 (1980).
[1455] R. P. Johnson, Can. Pat. 1,064,052 (1979); Chem. Abstr., 92, 163594 (1980).
[1456] R. Hummerich, W. D. Balzer, H. Klug, W. Trimborn, and H. J. Krause, Germ. OffenL
2,831,613 (1980); Chem. Abstr., 92, 181 897 (1980).
[1457] J. Gillois, G. Guillerm, M. Savignac, E. Stephen, and L. Vo-Quang, J. Chem. Educ, 57,161
(1980).
[1058] G. R. Pettit, J. Polym. Sci. Polym. Chem. Ed., 18, 345 (1980).
[14591 R. F. Cunico and B. B. Chou, J, Organomet. Chem., 154, С 45 (1978).
[1460] С. M. Starks, Chem. Technol, 10, 110 (1980).
[1461] H. Alper and K. Hachem, J. Org. Chem., 45, 2269 (1980).
* [1462] L. K. Novikova, O. B. Skripnik, G. Sh. Bikbulatova, S. G, Vulfson, Z. G. Isaeva, and A.
Vereshchagin, Izv. Akad. Nauk SSSR Ser. Khim., П, 605 (1978); Engl. transl. p. 520.
•[1463] Y. A. Zhdanov, Y. E. Alekseev, S. S. Doroshenko, T. P. Sudareva, G. V. Bogdanova, V. Q.
Alekseeva, and V. A. Tynmenev, Zh. Obsh. Khim., 48, 2614 (1978); Engl. transl*.
p. 2373
[1464] D. J. Darensbourg, B. J. Baldwin, and J. A. Froelich, J. Am. Chem. Sac, 102,4688 (1980).
[1465] O. Lindeberg, Acta Chem. Scand. Ser. B, 34, 15 (1980).
11466] P.-L. Kuo, N. Kawamura, M. Miki, and M. Okahara, Bull. Chem. Soc. Jpn., 53, 1689"
(1980).
X1467] H. Shinozaki, N. Yoshida, and M. Tajima, Chem. Lett., 1980, 869. д ■•
[1468] G. Lhommet, M.-G. Richaud, and P. Maitte, С R. Acad. Sci. Ser. C, 290, 445 (1980)Г
11469] S. Julia, A. Ginebreda, J. Gutter, and A. Tomas, Tetrahedron Lett., 1980, 3709. >
[1470] S. Oae and T. Takata, Tetrahedron Lett., 1980, 3689.
[1471] G. J. Bird, D. J. Collins, F. W. Eastwood, R. H. Exner, M. L. Romanelli, and D. D. Small,
Austr. J.<:hem., 32, 783 (1979).
[1472] L. Cassar, S. Panossian, and С Giordano, Phosphorus Sulfur, 6, 363 (1979).
[1473] S. Gronowitz, R. Svenson, G. Bondesson, and O: Magnusson, Acta Pharm. Suec, 15,361
(1978). i
[1474] P. di Cesare and B. Gross, Synthesis, 1980, 714. j
11475] H. Brink, Dissertation, Uppsala, 1980; Acta Pharm. Suec, 17, 233 (1980). ;
[1476] A. Arbin, H. Brink, and J. Vessman, J. Chromatogr., 196, 255 (1980).
"-[1477] V. M. Andreev, A. I. Bibicheva, and I. E. Radionova, Maslo-Zhir. Prom-su, 1980, 27;
Chem. Abstr., 93, 7795 (1980).
11478] T. D. N'guyen and S. Boileau, Polym. Bull, 1, 817 (1979). :■
[1479] F. Ishii and K.-l. Kishi, Synthesis, 1980, 706.
[1480] E. Slusarska and A. Zwierzak, Synthesis, 1980, 717.
[1481] H. P. Soetens and U. K. Pandit, Rec Trav. Chim. Pays-Bas, 99, 271 (1980).
[1482] R. Perraud, H. Handel, and J. L. Pierre, Bull. Soc Chim. Fr., 1980, 283.
{1483] G. Manikumar and M. Shamma, J. Org. Chem., 46, 386 (1981).
•[1484] V. I. Boevand A. V. Dombrovski, Zh. Obsh. Khim., 50,121 (1980); Engl. transl. p. 103.
[1485] J. SoJoducho and A. Zabza, Bull. Acad. Pol. Sci. Ser. Sci. Chim., 27, 447 (1979).
[1486] J.-P. Battioni, D. Mansuy, and J.-C. Chottard, Inorg. Chem., 19, 791 (1980).
[1487] F. С Churchill II and D. N. Ku, J. Chromatogr., 189, 375 (1980).
[1488] H. J.-M. Dou, M. Ludwikow, P. Hassanaly, J. Kister, and J. Metzger, J. Heterocycl. Chem^
17, 393 (1980).
[1489] J. T. Lai, J. Org. (fhem., 45, 754 (1980).
[1490] B. Cazes and S. Julia, Tetrahedron, 35, 2655 (1979).
[1491] R. Hull, F. Hollywood, and H. Suschitzky, /. Chem. Soc. Perkin, 1, 1979, 3037.
[1492] O. Hoshino, S. Sawaki, N. Shimamura, A. Onodera, and B. Umezawa, Heterocycles, 10,61
(1978).
452
[1493] К Gurumurthy and К. Narasimhan, Chem. Ind. (London), 1980. 698.
[1494] B. Stanovnik. M. Tisler, D. Gabrijeltic, M. Kunaver, and J. Zmitek, J. Helerocycl. Chem'.,
16. 1567(1979).
•114951 A. Esikova and S. S Yufit, Izv. Akad. Nauk SSSR Ser. Khim., 29,507, 515 (1980); EngL
transl p. 328, 336
11496] I. P Galy, Г Elguero, h. J. Vincent, A. M. Galy, and J. Barbe, Heterocycles, 14,311 (1980)j:
Chem. Abstr 93, 26244 (1980).
11497] H Des Abbayes and A. Buloup. J. Organomet. Chem., 198, С 36 (1980).
[1498] В M Trost and D. P. Curran, J Am. Chem. Soc, 102, 5699 (1980).
[1499] L Rafecas and J. J Artus, Tetrahedron Lett., 1980, 977.
[1500] К Soga, S. Hosoda, and S. Ikeda. J. Polym. Sci. Polym. Chem. Ed., 17, 517 (1979).
[1501] M Beran, J Krepelka,and M. Semonsky, Coll. Czech. Chem. Commun., 44, 3385 (1980).
'[1502] > A. Esikova, V F. Kucherov, B. A. Rudenko. and S. S. Yufit, Izv. Akad. Nauk SSSR Ser.
Khim., 28, 1468 (1979); Engl. transl. p. 1367 :
[1503] J Lewis, M. К Naqvi, and G. S. Park, Makromol. Chem. Rap. Commun., 1, 119 (1980).
[1504] V S. Hawaldar and S. V Sunthankar, Indian J. Chem., 19b, 151 (1980).
[1505] B. Czech, Tetrahedron Lett., 1980, 4197 ;
[1506] D. E. Rosenberg, J. R. Beadle, S. H. Korzeniowski, and G. W. Gokel, Tetrahedron Lett.
1980, 4141
11507] С R. Harrison and P. Hodge, Synthesis, 1980; 299.
[1508] R. R. Whitney and D. A. Jaeger, Tetrahedron, 36, 769 (1980).
[1509] P Sarthon, G. Bram, F. Guibe, and J. Corset, Tetrahedron, 36, 1043 (1980).
[1510] A E. Feiring, /. Org. Chem., 45, 1958 (1980).
[1511] R. A. Olofson and J. Cuomo, J. Org. Chem., 45, 2538 (1980).
[1512] K. N. Slessor, A. C. Oehlschlager, B. D. Johnston, H. D. Pierce Jr., S. K. Grewal, and L.
G. Wickremesinghe, J. Org. Chem., 45, 2290 (1980).
[1513] J. Cousseau, Synthesis, 1980, 805.
- [1514] M Vlassa, M. Kedzi, and I. Goia, Synthesis, 1980, 850.
[1515] H. Alper and D. E. Laycock, Synthesis, 1980, 799.
[1516] H. J.-M. Dou, P. Hassanaly, and J. Metzger, Chim. Ada Turc, 7,291 (1979); Chem. Abstr.,
94, 1S639 (1981). :
[1517] G. W Gokel, H. M. Gerdes, and D. M. Dishong, J. Org. Chem., 45, 3634 (1980).
[1518] S. A. Di Biase, R. P. Wolak Jr., D. M. Dishong, and G. W. Gokel, J. Org. Chem., 45,363O
(1980).
[1519] R. Kinishi, N. Uchida, Y. Yamamoto, J. Oda, and Y. Inouye, Agric. Biol. Chem., 44,643*
(1980).
[1520] J. T. Lai, J. Org. Chem., 45, 3671 (1980).
[1521] T. N. Sorrell and P. S. Pearlman, Tetrahedron Lett., 1980, 3963.
[1522] A. Arnoldi, R. Galli, and A. Zagni, Heterocycles, 12, 1335 (1979).
[1523] Y. Imai, M. Ueda, and M. Ii, Kobunshi Ronbunshu, 35, 807 (1978) (Japan); Chem. Abstr.*
90, 87899 (1979).
[1524] E. Santaniello and P. Ponti, Synth. Commun., 10, 611 (1980).
[1525] W. P. Reeves, A. Simmons Jr., and K. Keller, Synth. Commun., 10, 633 (1980).
[1526] L. Garanti and G. Zecchi, J. Chem. Soc. Perkin Trans. 1, 1980, 116.
[1527] M. Michihiro, H. Mori, J. Nakanishi, and J. Kasai, Germ. Offenl. 2,838,030 (1980); Chem~
Abstr., 93, 7657 (1980).
?[1528] G. A. Artamkina, A. A. Grinfel'd, and I. P. Beletskaya, Zh. Org. Khim., 16, 698 (1980);:
Engl. transl. p. 612.
[1529] B. jenko, I. Langof, and J. Habjan, Brit. Pat. Appl. 2,019,842 (1979); Chem. Abstr., 93^
71768 (1980).
11530] Asahi Glass Co. Ltd., Japan, Jap. Kokai 80,52,323 (1980); Chem. Abstr., 93,96139 (1980)-
[1531] Asahi Glass Co. Ltd., Japan, Jap. Kokai 80,52,325 (1980); Chem. Abstr., 93,96137 (1980).-
[1532] E. Waldvogel, Brit. Pat. Appl. 2,025,930 (1980); Chem. Abstr., 93, 114048 (1980).
* [1533] S. S. Yufit and I. A. Esikova, Izv. Akad. Nauk SSSR Ser. Khim., 29,519 (1980); Engl. transl..
o. 340.
453
[1534] N. Uchida, Y. Nakajima, J. Oda, and Y. Inouye, Agric. ВЫ. Chem., 43,2169 (1979) CChem.
Abstr., 92, 93574 (1980).
=♦[1535] N. N. Suvorov, D. N. Plutitskii, and Y. I. Smushkevich, Zh. Org. Khim., 16, 872 (1980),
Ehgl. transl. p. 766.
{1536] J. Bergman and L. Engman, Z. Naturforsch. Teil B, 35, 217 (1980).
(15371 W. С Guida and D. J. Mathre, J. Org. Chem., 45, 3172 (1980).
[1538] J.-R. Dormoy, B. Castro, G. Chappuis, U.-S. Fritschi, and P. Grogg, Angew. Chem.,92,761
(1980); Angew. Chem. Int. Ed. Engl., 19, 742 (1980).
11539] J. M. Mclntosh, H. Khalil, and D. W. Pillon, J. Org. Chem., 45, 3436 (1980).
[1540] D. L. Reger, M. M. Habib, and D. J. Fauth, J. Org. Chem., 45, 3860 (1980).
[1541] Y. Kawakami and Y. Yamashita, J. Org. Chem., 45, 3930 (1980).
[1542] A. A. Jaxa-Chamiec, P. G. Sammes, and P. D. Kennewell, J. Chem. Soc. Perkin Trans. 1,
1980,170.
[1543] L. Lombardo and L. N. Mander, Synthesis, 1980, 368.
[1544] K. Eicken, W. Rohr, and F. Linhart, Germ. Offenl. 2,830,764 (1980); Chem. Abstr., 93,
46212 (1980).
[1545] H. Takahata, Y. Ohnishi, and T. Yamazaki, Heterocycles, 14,467 (1980); Chem. Abstr., 93,
46266 (1980).
11546] Sandoz, Netherl. Pat. Appl. 7,806,134 (1979); Chem. Abstr., 93, 46068 (1980).
"♦[1547] T. 1. Lozanskaya, I. A. Esikova, S. S. Yufit, and V. F. Kucherov, Izv. Akad. Naiik SSSR
Ser. Khim., 28, 794 (1980); Engl. transl. p. 548.
•11548] A. T. Malkhasyan, Z. L. Dzhandzhylyan, R. T. Grigoryan, S. M. Mirakyan, and G. T.
Martirosyan, Arm. Khim. Zh., 33, 152 (1980); Chem. Abstr., 93, 94949 (1980).
11549] B. Zupancic and M. Sopcic, Brit. Pat. Appl. 2,021,562 (1979); Chem. Abstr., 93, 94999
(1980).
11550] M. Rakoutz, Germ. Offenl. 2,925,763 (1980); Chem. Abstr., 93, 71272 (1980).
11551] H. Fah, U.S. Pat. 4,185,018 (1980); Chem. Abstr., 92, 146780 (1980).
[1552] J. E. Hallgren and G. M. Lucas, J. Organomet. Chem., 212,135 (1981); General Electric, Fr.
Demande 2,422,621 (1979); Chem. Abstr., 92, 146429 (1980) and J. E. Hallgren, Germ.
Offenl. 2,815,512 (1979); Chem. Abstr., 92, 163723 (1980).
: 11553] H. Takahata, T. Hashizume, and T. Yamazaki, Heterocycles, 12, 1449 (1979); Chem.
Abstr., 92, 146577 (1980); H. Takahata, Y. Ohnishi, H. Takahara, K. Tsuritani, and T.
Yamazaki, Chem. Pharm. Bull., 29, 1063 (1981).
[1554] S. Campolmi, M. G. Felicioli, V. Carletti, and R. Santi, Germ. Offenl. 2,919,919 (1979);
Chem. Abstr., 92,146491 (1980).
[1555] F. Mossini, M. R. Mingiardi, E. Gaetani, M. Nardelli, and G. Pelizzi, J. Chem. Soc. Perkin
Trans. 2, 1979, 1665.
; [1556] Y. Hamada and K. Shigemura, Yukagaku Zasshi,99,982 (1980); Chem. Abstr., 92,163869
(1980).
,.£1557] G. Meuhier, P. Hemery, J. P. Senet, and S. Boileau, Polym. Bull.,1, 809 (1979); Chem.
Abstr., 92, 94722 (1980).
[1558] R. Kuboszek, K. Slon, M. Makosza, W. Zulachowski, and R. Przybylik, Pol. Pat. 101,422
(1979); Chem. Abstr., 92, 76091 (1980).
[1559] M. Lissel, Synth. Commun., 11, 343 (1981).
[1560] M. Lissel and E. V. Dehmlow, Chem. Ber., 114, 1210 (1981).
[1561 ] R. S. Raman and M. A. Ashrof, Curr. Sci., 48,583 (1979); Chem. Abstr., 91,123526 (1979).
[1562] G. Chapelet, P. Lubin, and R. Nouguier, Germ. Offenl. 2,844,756 (1979); Chem. Abstr., 91,
107661 (1979).
[1563] S. Maeke and A. Bauer, perm. Offenl. 2,753,440 (1979); Chem. Abstr., 91, 91190 (1979).
, [1564] D. Jyotsna and A. V. S. Rao, Curr. Sci., 48,439 (1979); Chem. Abstr., 91, 56563 (1979).
[1565] L. de Vincentiis, Belg. Pat. 872,650 (1979); Chem. Abstr., 91, 56365 (1979).
[1566] R. Annunciata, Synth. Commun., 9, 171 (1979).
[1567] P. J. Me Mahon and F. Sylvester, Germ. Offenl. 2,855,230(1979); Chem. Abstr., 91,140519
(1979).
[1568] D. V. Banthorpe and P. N. Christou, Phytochem., 18, 666 (1979).
.[ [1569] L.-F. Tietze and U. Reichert, Angew. Chem., 92,832 (1980); Angew. Chem. Int. Ed. Engl.,
19, 830 (1980).
454
[1570] С. R. Johnson, К. Mori, and A. Nakanishi, J. Org. Chem., 44, 2065 (1979).
[1571] E. Guilmet and B. Meunier, Tetrahedron Lett., 1980, 4449.
[1572] H. des Abbayes and A. Buloup, Tetrahedron Lett., 1980, 4343.
[1573] Y. Nii, K. Okano, S. Kobayashi, and M. Ohno, Tetrahedron Lett., 1979, 2517.
[1574] M. Yoshifuji, F. Hanafusa, and N. Inamoto, Chem. Lett., 1979, 723.
[1575] F. Iordache,S. Badica, A. Ionescu, and F. Badea, Rev. Chim.,30,629(1979); Chem. Abstr.^
92, 58T81 (1980).
[1576] Y. lmai, M. Ueda, and M. Ii, J. Polym. Sci. Polym. Lett. Ed., 17,85 (1979); Chem. Abstr.,,
90, 187398 (1980).
[1577] A. Marhold, R. Lantzsch, E. Klauke, and E. Kysela, Germ. Oflenl. 2,814,860 (1979);
Chem. Abstr., 92, 76082 (1980).
[1578] S. A. Al-Jibori and B. L. Shaw, J. Organomet. Chem., 192, 83 (1980).
[1579] G. Gelbard, T. Brunelet, and С Jouitteau, Tetrahedron Lett., 1980, 4653.
[1580] Y. lmai, M. Ueda, and M. Ii, Makromol. Chem., 179, 2085 (1978).
[1581] S. Akabori, S. Miyamoto, and H. Tanabe, J. Polym. Sci. Polym. Chem. Ed., 17,3933 (1979).
[1582] Z. K. Brzozowski, J. Dubczynski, and J. Petrus, J. Macrom. Sc. Chem., A 13, 875 (1979).
[1583] J. Rosenfeld, J. Crocco, and T. L. Ling, Anal Lett., 1980, 283.
[1584] D. G. Brown, Germ. Oflenl., 3,012,918 (1980).
[1585] A. M. Kolodziejczyk and A. Arendt, Pol. J. Chem., 54, 1327 (1980).
[1586] W. Szeja, Pol. J. Chem., 54, 1301 (1980).
[1587] W. Szeja, Pol. J. Chem., 54, 1323 (1980).
* [1588] A. T. Malkhasyan, E. M. Nazaryan, S. M. Mirakyan, and G. T. Martirosyan, Arm. Khim.
Zh., 32, 952 (1979); Chem. Abstr., 93, 132318 (1980).
[1589] S. Julia, J. Masana, and J. С Vega, Angew. Chem., 92,968 (1980); Angew. Chem. Intern. Ed.
Engl., 19, 929 (1980).
* [1590] S. S. Yufit, T. I. Lozanskaya, and I. A. Esikova, lzv. Akad. Nauk SSSR Ser. Khim., 29,797
(1980); Engl. transl. p. 551.
*[1591) K. A. Kufginyan, I. M. Rostomyan, A. E. Kalaidzhyan, and G. A. Chukhadzyan, Arm..
Khim. Zh., 32, 945 (1979); Chem. Abstr., 93, 132000 (1980); A. E. Kalaidzhyan, I. M.
Rostomyan, K. A. Kurginyan, and G. A. Chukhadzyan, ibid., 33,57 (1980); Chem. Abstr.,
93, 132001 (1980).
[1592J Y. lmai, N. Sato, and M. Ueda, Makromol. Chem. Rap. Commun., 1, 419 (1980); Chem..
Abstr., 93, 132881 (1980).
[1593] E. Angeletti, P. Tundo, and P. Venturello, J. Chem. Soc. Chem. Commun., 1980, 1127.
^[1594] L. P. Glushko, V. N. Samsonova, M. S. Malinovskii, and L. A. Yanovskaya, lzv. Akad.-
Nauk SSSR Ser. Khim., 29, 1048<1980); Engl. transl. p. 754.
[1595] H. Alsaidi, R. Gallo, and J. Metzger, Synthesis, 1980, 921.
[1596] B. Badet, M. Julia, and M. Ramirez-Munoz, Synthesis, 1980, 926.
[1597] P. Di Cesare, P. Duchaussoy, and B. Cross, Synthesis, 1980, 953.
[1598] D. J. Mathre and W. С Guida, Tetrahedron Lett., 1980, 4773.
[1599] M. Delmas, Y. Le Bigot, and A. Gaset, Tetrahedron Lett., 1980, 4831.
[1600] H. Handel, M. A. Pasquini, and J. L. Pierre, Tetrahedron, 36, 3205 (1980).
[1601J J. M. Mclntosh, Can. J. Chem., 58, 2604 (1980).
[1602J J. B. Hall, J. J. Kryschuk, and M. A. Sprecker, U.S. Pat. 4,205,186 (1980); Chem. Abstr., 93,.
204064 (1980).
[1603J Asahi Glass Co., Jap. Kokai 80,65, 226 (1980); Chem. Abstr., 93, 150903 (1980).
* [1604] L. A. Kheifits, A. V. Gurevich, N. V. Lepikhina, and N. A. Novikov, Maslo-Zhir. Prom-st.,.
1980, 24; Chem. Abstr., 93, 167814 (1980).
[1605] N. Ishikawa,T. Kitazume,andM. Nakabayashi, Chem. Lett., 1980,1089;T. Kitazume and-
N. Ishikawa, ibid., 1980, 1327.
[1606] J. С W. Chien, T. Kohara, С P. Lillya, T. Sarubbi, B.-H. Su, and R. S. Miller, J. Polym,
Sci. Polym. Chem. Ed., 18, 2723 (1980).
[1607] A. Mizuno, Y. Hamada. and T. Shioiri, Synthesis, 1980, 1007.
[1608] J. Skarzewski, J. Chem. Res., 1980, (S) 410.
[1609] R. Sangaiah and G. S. K. Rao, Synthesis, 1980, 1018.
[1610] A. Donetti, O. Boniardi, and A. Ezhaya, Synthesis, 1980, 1009.
455
J1611] Е. V. Dehmlow and M. Lissel, Tetrahedron, 37, 1653 (1981).
11612] M. Tomoi and W. T. Ford, J. Amer. Chem. Soc, 102, 7140 (1980).
[1613] E. V. Dehmlow and A. R. Sharaout, J. Chem. Res., 1981 (S) 106; (M) 1178.
{1614] R. Annunziata, M. Cinquini, and S. Colonna, J. Chem. Soc Perkin Trans., 1, 1980, 2422.
11615] D. Davalian, P. J. Garrat, W. Roller, and M. M. Mansuri, J. Org. Chem., 45,4183 (1980).
11616] M. O. Terpko and R. F. Heck, J. Org. Chem., 45, 4992 (1980).
{1617] J. R. Weir, B. A. Patel, and R. F. Heck, J. Org. Chem., 45, 4926 (1980).
[1618] J.-P. Behr, J.-M. Girodeau, R. С Hayward, J.-M. Lehn, and J.-P. Sauvage, Helv. Chim.
Ada, 63, 2096(1980).
[1619] K. O. Knollmueller, U.S. Pat. 4,207,247 (1980); Chem. Abslr., 93, 185753 (1980).
[1620] S. Szakacs, S. Gobolos, and J. Szammer, Magy. Kern. Foly., 86, 273 (1980); Chem. Abslr.,
93, 185442 (1980); Monatsh. Chem., 112, 883 (1981).
[1621] A. Mihailovski, U.S. Pat. 4,206,141 (1980); Chem. Abslr., 93, 186000 (1980).
[1622] M. Sato, K. Inaba, S. Ebine, and J. Tsunetsugu, Bull. Chem. Soc. Jpn., 53, 2334 (1980).
:*[1623] V. P. Ivshin, M. S. Komelin, and N. P. Belik, Zh. Org. Khim., 16,980 (1980); Engl. transl. p.
858.
[1624] A. Arcoria, F. P. Ballistreri, A. Cantone, G. Musumarra, and M. Tripolone, Gazz. Chim.
Hal., 110, 267 (1980).
[1625] M. D. Rozwadowska and D. Brozda, Can. J. Chem., 58, 1239 (1980).
[1626] F. Turecek, Coll. Czech. Chem. Commun., 45, 1820 (1980).
[1627] "M. Lissel and J. Weiffen, Synth. Commun., 1981, 545.
[1628] H. Koinuma, K. Naito, and H. Hirai, Makromol. Chem. Rap. Commun., 1, 493 (1980).
[1629] A. Loupy and J. Seyden-Penne, Tetrahedron, 36, 1937 (1980).
[1630] F. Seela, U. Lupke, and D. Hasselmann, Chem. Ber., 113, 2808 (1980).
{1631] R. Baker and R. J. Sims, Tetrahedron Lett., 1981, 161
[1632] E. V. Dehmlow and S. Barahona-Naranjo, J. Chem. Res., 1981, (S) 142, (M) 1748.
[1633] E. V. Dehmlow and S. Barahona-Naranjo, J. Chem! Res., 1981, (S) 143, (M) 1763.
[1634] D. Landini and F. Rolla, J. Org. Chem., 45, 3527 (1980).
[1635] B. B. Snider and J. V. Duncia, J. Amer. Chem. Soc, 102, 5926 (1980).
[1636] N. A. Porter, D. H. Roberts, and С. В. Ziegler Jr., J. Amer. Chem. Soc, 102, 5912 (1980).
*[1637] B. A. Arbuzov, A; M. Salikhova, S. M. Shostakovskii, A. N. Vereshchagin, and N. S.
Nikolskii, Izv. Akad. Nauk SSSR Ser. Khim., 28, 2786 (1979); Engl. transl. p. 2594.
[1638] J. Leroy, J. Bensoam, M. Humiliere, С Wakselman, and F. Mathey, Tetrahedron, 36,1931
(1980).
[1639] R. E. W. Jansson and N. R. Tomov, J. Appl. Eleclrochem., 10, 583 (1980).
[1640] M. Rosenberger, W. Jackson, and G. Saucy, Helv. Chim. Ada, 63, 1665 (1980).
[1641] D. Landini, A. Maia, F. Montanari, M. Mikolajczyk, and A. Zatorski, Nouv. J. Chim., 4,
723 (1980).
[1642] M. Argentini, M. Weidmann, M. Zahner, K. Zimmermann, and P. A. Schubiger,
Radiochem. Radioanal. Lett., 44, 269 (1980).
[1643] H. Alper, K. Hachem, and S. Gambarotta, Can. J. Chem., 58, 1599 (1980).
[1644] R. Curci, M. Fiorentino, L. Troisi, J. O. Edwards, and R. H. Pater, J. Org. Chem., 45,4758
(1980).
[1645] A. Shinohara, N. Wada, Y. Tokunaga, and С Yazawa, Germ. Offenl. 2,944,030 (1980);
Chem. Abstr., 93, 239 025 (1980).
{1646] H.-J. Schmidt and H. J. Schafer, Angew. Chem., 93, r24 (1981); Angew. Chem. Intern. Ed.
Engl., 20, 109 (1981).
" [1647] M. Halpern, M. Yonowich-Weiss, Y. Sasson, and M. Rabinovitz, Tetrahedron Lett., 1981,
703.
[1648] S. Julia and A. Ginebreda, Afinidad, 1980, 194.
[1649] J. С Dyer and S. A. Evans Jr., J. Org. Chem., 45, 5350 (1980).
.[1650] R. Gurumurthy, P. Balasubramanian, and K. Narasimhan, Synth. Commun., 10, 833
/1980).
;[1651] H.-J. Schmidt and H. J. Schafer, Angew. Chem., 93,124(1981); Angew. Chem. Intern. Ed.
Engl., 20, 104 (1981).
456
[1652] M. Delmas and A. Gaset, Tetrahedron Lett., 1981, 723.
[1653] S. Oae, T. Takata, and Y. H. Kim, Tetrahedron, 37, 37 (1981).
[1654] D. Martens, E V. Dehralow, D. J. Hanlon, M. B. Hossain, and D. van der Helm, J. Am.
Chem. Soc, 103, 4940 (1981).
[1655] E. Hannig, R. Pech, and C. Dressier, Pharmazie, 34, 670 (1979).
[1656] I. Photaki, I. Samonilidis, S. Caranikas.and L. Zervas,/. Chem. Soc. Perkin Trans. 1,1979,
2599.
[1657] T. Kunitake, Y. Okahata, R. Ando, S. Shinkai, and S.-i. Hirakawa, J. Am. Chem. Soc, 102,
7877 (1980).
[1658] R. Baker and R. J. Sims, Synthesis, 1981, 117.
[1659] K. J. Kolonko, M. L. Deinzer, and T. L. Miller, Synthesis, 1981, 133.
[1660] S. Colonna, A. Re, and H. Wynberg, J. Chem. Soc. Perkin Trans. 1, 1981, 547.
[1661] S. Julia, A. Ginebreda, J. Guixer, J. Masana, A. Tomas, and S. Colonna, J. Chem. Soc.
Perkin Trans. 1, 1981, 574.
[1662] I. Degani, R. Fochi, and V. Regondi, Synthesis, 1981, 149.
[1663] E. Slusarska and A. Zwierzak, Synthesis, 1981, 155.
[1664] J. H. Clark, Chem. Rev., 80, 429 (1980).
[1665] G. W. Klumpp and P. M. Kwantes, Tetrahedron Lett., 1981, 831.
[1666] M. M. Campbell, R.C. Craig, A. C. Boyd, I. M. Gilbert, R. T. Logan, J. Redpath, R. G.
Roy, D. S. Savage, and T. Sleigh, J. Chem. Soc. Perkin Trans. 1, 1979, 2235.
[1667] D. Scholz, Monatsh. Chem., 110, 1471 (1979).
{1668] D. Scholz, Monatsh. Chem., 112, 241 (1981).
[1669] D. H. Gibson, F. U. Ahmed, and K. R. Phillips, J. Organomet. Chem., 206, C17 (1981).
[1670] M. S. Chiles and P. С Reeves, Tetrahedron Lett., 1979, 3367.
11671] V. Ragaini and G. Saed, Z. Phys. Chem. N. F, 119, 117 (1980).
[1672] M. S. Chiles, D. D. Jackson, and P. С Reeves, J. Org. Chem., 45, 2915 (1980).
J1673] H. Kise, K. Araki, and M. Seno, Tetrahedron Lett., 1981, 1017.
[1674] R.Maeda, Y. Hayashi, and K. Teramura, Chem. Lett., 1980, 677.
[1675] S. L. Regen, J. С. К. Heh, and J. McLick, J. Org. Chem., 44, 1961 (1979).
J1676] S. Tamagaki, K. Hotta, and S. Kazuka, Mem. Fac. Eng. Osaka City Univ., 20,93 (1979);
Chem. Abstr., 93, 204215 (1980).
[1677] T. Saegusa, S. Kobayashi, A. Yamada, and S. Kashimura, Polym. J., 11, 1 (1979).
[1678] P. Tundo and P. Venturello, J. Am. Chem. Soc., 101, 6606 (1979).
[1679] P. Tundo and P. Venturello, Tetrahedron Lett., 1980, 2581.
[1680] S. Quid and S. L. Regen, J. Org. Chem., 44, 3436 (1979).
[1681] M. Okahara, Y. Yanagida, and N. Shiokawa, Jap. Kokai. 79,63,196(1979); Chem. Abstr.,
91, 141630 (1979).
[1682] J. Kiji, T. Okano, H. Konishi, and Y. Nishio, Makromol. Chem., 180, 2241 (1979).
[1683] M. Molinari, F. Montanari, S. Quici, and P. Tundo, Am. Chem. Soc., 101, 3920 (1979).
[1684] M. Tomoi, K. Kihara, and H. Kakiuchi, Tetrahedron Lett., 1979, 3485.
[1685] K. Kimura, M. Sawada, and T. Shono, Anal. Lett., 12, 1095 (1979).
[1686] M. Ricard, D. Villemin, and A. Ricard, Tetrahedron Lett., 1980, 47.
[1687] S. L. Regen and J. J. Besse, J. Am. Chem. Soc, 101, 4059 (1979).
[1688] J. Kelly, W. M. Mac Kenzie, D. С Sherrington, and G. Reiss, Polymer, 20,1048 (1979).
[1689] S. Yanagida, K. Takahashi, and M. Okahara, Yukagaku, 28,14(1979); Chem. Abstr., 90,
169794 (1979).
[1690] H. Maeda, Y. Hayashi, and K. Teramura, Chem. Lett., 1980, 677.
[1691] W. M. MacKenzie and D. С Sherrington, Polymer, 21,791 (1980); ibid., 22,431 (1981); J.
G. Hefferman, W. M. MacKenzie, and D. С Sherrington, J. Chem. Soc. Perkin 2,1981,
514.
[1692] M. Gisler and H. Zollinger, Angew. Chem., 93,184 (1981); Angew. Chem. Intern. Ed. Engl.,
20.203(1981); J. F. Bunnett, M. Gisler, and H. Zollinger, Heh. Chim. Ada, 65,63(1982).
[1693] S. L. Regen and A. Nigan, J. Am. Chem. Soc, 100, 7773 (1978).
[1694] S. L. Regen, J. J. Besse, and J. McLick, J. Am. Chem. Soc, 101, 116 (1979).
[1695] S. Inokuma, E. Kameyama, T. Osawa, and t. Kuwamura, Yukagaku, 29, 354 (1980);
Chem. Absir., 93, 185710 (1980).
30—751 457
[1696] W. Szeja, J. Chem. Soc. Chem. Commun., 1981, 215.
[1697] E. V Dehmlow, P; Singh, and J. Heider, J. Chem. Res. 1981 (S) 292.
.[1698] К Kiec-Kononowicz. A. Zejc, M. MikoJajczyk, and A. Zatorski, Tetrahedron, 37, 409
(1981).
[1699] E. Stamm and R. Keese, Synthesis, 1981. 231.
[1700] W. Schroth, H. Kluge, M. Mathias, R. Krieg,and H.-D. Schadler, Z. Chemie,21,25(1981).
[1701] H. Hiemstra and H. Wynberg, J. Am. Chem. Soc, 103, 417 (1981).
J17O2] J. M. Photis. J. Qrg. Chem.»46, 182 (1981).
[1703] K. Matsunaga and T Yamashita, Kogyo Kayaku, 41, 3 (1980); Chem. Abstr., 94, 31679
(1981).
[1704] H. Hiller, H. Eilingsfeld,,H. Reinicke, and F. Traub, Germ. Ofienl. 2,910,716(1980); Chem.
Abstr., 94, 32179 (1981).
J1705] A. Weber, G. Sabbioni, R. Galli, and M, Neuenschwander, Chimia, 35, 57 (1981).
[1706] D. H. Buchanan and G. McComas, Tetrahedron Lett., 1980, 4317.
[1707] A. J. Bloodworth, D. J. Lapham, and R. A. Savva, J. Chem. Soc. Chem. Commun., 1980,
925.
[1708] M. Kimura, K. Satake, S. Yonemori, and S. Morosawa, Bull. Chem. Soc. Jpn., 53, 3232
(1980).
£1709] J. W. Hill and J. Corredor, J. Chem. Educ, 57, 822 (1980).
/1710] L. Colombo, C. Gennari, C. Scolastico, F. Aragozzini, and С Mercudi, J. Chem. Soc.
1 Perkin 1, 1980, 2549.
[1711] K. Idel, V. Serini, D. Freitag, and G. Fengler, Germ. Offenl. 2,901,665 (1980); Chem.
Abstr., 93,168 888 (1980).
[1712] F. Seela and D. Hasselmann, Chem. Ber., 113, 3389 (1980).
[1713] M. Cieslak, K. Katarzyna, J. Swietoslawski, W. Kulicki, S. Malina, and R. Hupert, East
Germ. Pat. 140,452 (1980); Chem. Abstr., 93, 185925 (1980).
[1714] M. L, Deem and К. С Stueben, US Pat. 4,215,076 (1980); Chem. Abstr., 93,238842 (1980).
[1715] T. Asai, T. Aoyama, and T. Shioiri, Synthesis, 1980, 811.
[1716] K. Sjoberg, Aldrichimica Ada, 13, 55 (1980).
[1717] Aktiebolag Leo, US-Pat. 4,081,461 (1978); Germ. Pat. 2,629,657 (1977); Chem. Abstr., Щ
189570(1977).
[17181 M. С Van der Zwan, US-Pat. 4,100,159 (1978); Chem. Abstr., 89, 215436 (1978).
[1719] F. Masuko, H. Yamachiku, K. Fujiyoshi, K. Shiota, Germ. Offenl. 2,820,710 (1978);
Chem. Abstr., 90, 203724 (1979).
[1720] J. H. Fendler, Texas A and M University, priv. commun. 1980
[1721] P. Bamberg, B. Ekstrom, and A. Bertil, Swed. Pat. 397,981 (1977); Germ. Pat. 2,345,236
(1974); Chem. Abstr., 81, 152251 (1974).
[1722] Y. Kawakami, T. Sugiura, Y. Mizutani, and Y. Yamashita, J. Polym. Sci., Polym. Chem.
Ed., 18, 3009 (1980).
[1723] M. Aglietto, G. Ruggeri, B. Tarquini, F. Ciardelli, and P. Gianni, Polymer, 21,541 (1980).
[1724] D. L. Reger and M. M. Habib, J. Molecul. Catal., 7, 365 (1980).
[1725] T. Nagano, K. Arakane, and M. Hirobe, Chem. Pharm. Bull, 28, 3719 (1980).
[1726] J. Smid, Ind. Eng. Chem. Prod. Res. Dev., 19, 364 (1980).
[1727] J. ApSimon and R. Seguin, Synth. Commun., 10, 897 (1980).
•[1728] R. R. Kostikov, A. P. Molchanov, S. V. Amosova, and B. A. Trofimov, Izv. Akad. NaUk-
SSSR Ser. Khim., 29, 1354 (1980); Engl. transl. p. 971.
[1729] M. Mizutani, A. Higo, N. Ohno, and H. Hirai, Germ. Ofienl. 2,737,299 (1978); Chem.
Abstr., 88, 169779 0978).
[1730] I. Akimoto and A. Suzuki, Synth. Commun., 11, 475 (1981).
[1731] F. Seela and H.-D. Winkeler, Angew. Chem., 93,105 (1981); Angew. Chem. Intern. Ed., 20,
97(1981). ;
[1732] G. A. Pinna, M. Loriga, G. Paglietti, and G. Cignarella, Farmaco Ed. Sci., 35,684 (1980).
[1733] H. С Walters, Canad. Pat. 1,019,681 (1977); Chem. Abstr., 88, 38309 (1978).
[1734] E Blasius, K.-P. Janzen, W. Klein, H. Klotz, N. B. Nguyen, T. Nguyen-Tien, R. Pfeifier, G.
Scholten, H. Simon, H. Stockemer, and A. Toussaint, J. Chromatogr., 201, 147 (1980).
Г1735] M. Gacek and K. Undheim, Ada Chem. Scand. Ser. B, 35, 69 (1981).
4Ьв
* [1736] N. A: SokoJov, G. Y. Perchugov, aiidO.S. Morozov, Zh. Obshch. Khim., 49,1856 (1979);
Engl. transl. p. 1633.
[1737] H. Dahlstrom, K. Leander, B. Lindstrom, and M. Molander, J. Chromatogi., 183, 511
(1980).
{1738] S. Julia and A. Ginebreda, Ann. Quint., 76, 136 (1980).
[1739] J. M. J. Frechet, J. Macromol. Sci. Chem., A 15, 877 (1981).
[1740] P. K. Jain,J. K. Makrandi, S. K. Grover,and M. Nogradi, Cwr. Set.,49,664(1980); Chem.
Abslr., 94, 47075(1981).
[1741] J. P. Behr, J. M. Lehn, D. Moras, and J. С Thierry, J. Am. Chem. Soc, 103, 701 (1981).
[1742] J. L. Roberts Jr. and D. T. Sawyer, J. Am. Chem. Soc., 103, 712 (1981).
[1743] J. K. Rasmussen and H. K. Smith II, J. Am. Chem. Soc, 103, 730 (1981).
[1744] E. D'Incan and A. Loupy, Tetrahedron, 37, 1171 (1981).
[1745] K. Fukunishi, B. Czech, and S. L. Regen, J. Org. Chem., 46, 1218 (1981).
[1746] M. Halpern, Y. Sasson, I. Willner, and M. Rabinovitz, Tetrahedron Leu., 1981, 1719.
[1747] R. Bar and Y. Sasson, Tetrahedron Lett., 1981, 1709.
[1748] P. Tundo and P. Venturello, J. Am, Chem. Soc, 103, 856 (1981).
[1749] I. Degani, R. Focbi, and V. Regondi, Tetrahedron Lett., 1981, 1821.
^ [1750] F. Bohlmann, J. Jakupovic, L. Muller, and A. Schuster, Angew. Chem,, 93, 280 (1981);
Angew. Chem. Inter. Ed., 20, 292 (1981).
*[1751] V. B. Vol'eva, I. S. Belostotskaya, I. A. Novikova, E. V. Dzhuaryan, and V. V. Ershov, /i».
Akad. Nauk SSSR Ser. Khim., 29, 2414 (1980) (Russ.); Chem. Abslr., 94, 83711 (1981).
[1752] P. Piccardi, P. Massardo, F. Bettarini, and A. Longoni, Pestic. Sci., 11, 423 (1980).
[1753] B. Loubinoux, G. Coudert, and G. Guillauraet, Tetrahedron Lett.,.19Sl, 1973.
[1754] T. Gajda and A. Zwierzak, Synthesis, 1981, 1005.
Ц755] M. Kluba and A. Zwierzak, Synthesis, 1981, 537.
[1756] A. Zwierzak, private-communication 1981.
[1757] R. Ljotta and G. Brons, /. Am. Chem. Soc, 103, 1735 (1981),
[1758] G. S. Bates and J. O'Doherty, J. Org. Chem.,- 46, 1745 (1981).
[1759] S. Akabori, M. Ohtomi, and S. Yatabe, Bull. Chem. Soc. Jpn., 53, 1463 (1980).
• [1760] A. Kh. Khusid and M. S. Lyubich, Zh. Org. Khim. SSSR, 16,2437(1980){Russ.]; Chem.
Abstr. 94, 102763 (1981).
[1761] K. Inoue, H. Noguchi, H. Masanobu, and Y. Uchida, Yukagaku, 29, 397 (1980); Chem.
Abstr. 94, 102469 (1981).
[1762] Y, Watanabe and T. Mukaiyama, Chem. Letters, 1981, 285.
[1763] T. Okawara, Y. Noguchi, T. Matsuda, and M. Furukawa, Chem. Letters, 1981, 185.
[1764] M. Jautelat, D. Aril, R. Lantzsch, R. A. Fuchs, H. Riebel, R. Schroeder, and H. Harnisch,
Germ. Offenl., 2,916,321 (1980); Chem. Abstr., 94, 121121 (1981).
[1765] H. J. Koch, Canad. Pat., 1,085,423 (1980); Chem. Abstr., 94, 120834 (1981).
[1766] E. V, Dehmlow and A. R. Shamout, Liebigs Am. Chem., 1982, 1753.
[1767] E. Santaniello, P. Ferraboshi, and P. Sozzani, ESOC-Conference II, June 1-5,1981, Stresa,
Italy, poster 56A.
[1768] F. Rolla, J. Org. Chem. 46, 3909 (1981).
[1769] F. Montanari and P. Tundo, J. Org. Chem., 46, 2125 (1981).
[1770] R. Annunziata, F. Montanari, S. Quid, and M.T. Vitali, J. Chem. Soc. Chem. Commun.,
1981, 777.
[1771] E. V. Dehmlow and J. Soufi, unpublished.
[1772] P. N. Juri and R. A. Bartsch, J. Org. Chem., 45, 2028 (1980); J. Krane and T. Skjetne,
Tetrahedron Lett., 1980,1775; T. Kuokkanen and P. O. I. Virtanen, A. Chem. Scand. Ser. B,
33, 725 (1979).
{1773] K. D. Cooper and H. M. Walborsky, J. Org. Chem., 46, 2110 (1981).
[1774] W. A. Feld, J. Macromol. Sci. Chem., A 15, 891 (1981).
[1775] A. M. Kolodziejczyk and M. Manning, J. Org. Chem., 46,1944 (1981).
[1776] J. K. Rasmussen and H. K. Smith II, Macromol. Chem. Phys., 182, 701 (1981).
[1777] R^Colman, E. F. V; Scriven, H. Suschitzky, and D. R. Thomas, Chem. bid. (London), 1981,
249.
[1778] E. V. Dehmlow and R. Meier, unpublished.
30* 459
- [1779] D. Balasubramanian, P. Suktunar, and B. Chandani, Tetrahedron Lett., 1979, 3543. . '
[1780] M. Lissel, Bielefeld University, unpublished.
. (1781] S. L. Regen, A. Mehrotra, and A. Singh, j. Org. Chem., 46, 2182 (1981).
[1782] M. Delmas, Y. Le Bigot, A. Gaset, and J. P. Gorrichon, Synth. Commun., 11,125 (1981).
[1783] S. H. Korzeniowski, A. Leopold, J. R. Beadle, M. F. Ahern, W. A. Sheppard, R. K. Kharaa,
and G. W. Gokel, J. Org. Chem., 46, 2153 (1981).
.:(1784] S. Gambarotta and H. Alper, J. Org. Chem., 46, 2142 (1981).
[1785] H. H. Hergott and G. Simchen, Synthesis, 1980, 626.
.f [1786] G. Vernin,H. Domloj.C. Siv, J. Metzger, H. Archavlis, and J. R. Llinas, Chem. Scr., 16,157
(1980).
[1787] G. Isele, J. A. Martinetz, and G. Schill, Synthesis, 1981, 455.
[1788] K. Steinbeck, T. Schenke, and J. Runsink, Chem. Ber., 114, 1836 (1981).
[1789] G. Mouzin, H. Cousse, and J.-M. Autin, Synthesis, 1981, 448.
[4790] L. Anke, D. Reinhard, and P. Weyerstahl, Liebigs Ann. Chem., 1981, 591.
[1791] ref. 5, pages 80, 167, 168.
[1792] F. Seela, Q. Tran-Thi, and H.-D. Winkeler, Chem. Ber., 114, 1217 (1980).
[1793] A. G. M. Barrett, С R. A. Godfrey, D: M. Holinshead, P. A. Prokopiou, D. H. R. Barton,
;. R. B. Boar, L. Jonkhadar, J. F. McGhie, and S. С Misra, J. C. S. Perkin Trans. 1,1981,
1501.
. [1794] G.PhilippossianandM.Enslen,Europ.Pat. Appl. 19,165(1980);Chem.Abstr.,94,156970
[1795] H. Kobayashi, T. Sonoda, and H. Iwamoto, Chem. Lett., »81, 579
[1796] H. Alper and D. Ё. Laycock, Tetrahedron Lett., 1981, 33
[1797] S. O. Jansson, R. Modin, and G. Schill, Talanla, 21, 905 (1974).
[1798] S.Oae, K. Iida, K. Shinhama, and T. Takahata, Bull. Chem. Soc. Jpn., 54, 2374(1981).
[1799] S. Paul and B. Raanby, J. Appl. Polym. Set., 26, 3927 (1981).
[1800] S. L. Regen, D. Bolikal, and C. Barcelon, J.Org. Chem., 46. 2511 (1981).
[1801] P. Monsef-Mirzai and W. R. McWhinnie, Inorg. Chim. Ada, 52, 211 (1981).
[1802] W. Tomoi arid W. T. Ford, J. Am. Chem. Soc, 103, 3821, 3828 (1981).
4l803] А,тап Zon, F. de Jong, and Y. Onwezen, Rec. Trav. Chim. Pays-Bas, 100, 429 (1981);
[1804] E. P. Woo, US Pat. 4,256,859 (1981); Chem. Abstr., 95, 81039 (1981).
[1805] T. Balakrishnan and W. T. Ford, Tetrahedron Lett., 1981, 4377.
[1806] M. Schneider, J.-V. Weber, and P. Faller, J. Org. Chem., 47, 364 (1982).
[1807] A. Cornells and P. La'szk), Synthesis, 1982, 162.
[1808] J. V. Comasseto and С A. Brandt, J. Chem. Res., 1982, (S) 56.
[1809] A. Jonczyk and J. Kacprzak, private communication 1982.
[1810] H. Wynberg, Rec Trav. Chim. Pays-Bas, 100, 393 (1981).
[1811] N. Kobayashi and<K. Iwai, Makrtmol. Chem. Rapid Commun., X, 105 (1981).
11812] S. Banfi, M. Cinquini, and S. Colonna, Bull. Chem. Soc. Jpn., 54,1841 (1981).
[1813] Y. Harigaya, H. Yamaguchi, and M. Onda, Chem. Pharm. Bull., 29, 1321 (1981).
[1814] D. J. Cram and G. D. Y. Sogah, J. Chem. Soc. Chem. Commun., 1981, 625.
[1815] T. Nishikubo, T. Iizawa, K. Kobayashi, and M. Okawara, Tetrahedron Lett., 1981, 3873.
[1816] J. L. Charlton, V. A. Sayeed, and G. N. Lypka, Synth. Commun., 11, 931 (1981).
[1817] A. Chatterjee, B. Sen, and S. K. Chatterjee, J. Chem. Soc. Perkin l,Л9Л1, 1707.
• [1818] G. A. Chukhadzyan, E. L. Sarkisyan, I. M. Rostomyan, and A. G. Israelyan, Arm. Khim.
Zh., 34, 519 (1981); Chem. Abstr., 95, 114684 (1981).
*[1819] K. A. Kurginyan, A..E. Kalaidzhyan, I. M. Rostomyan, and G. A. Chakhadzyan, Arm.
Khim. Zh., 34, 422 (1981); Chem. Abstr., 95, 186557 (1981).
♦[18201 A. E. Kalaidzhyan, K. A. Kurginyan, I. M. Rostomyan, and G. A. Chukhadzyan, Arm.
Khim. Zh., 33, 845 (1980); Chem. Abstr., 94, 191802 (1981).
[1821] S. L. J. Daren, D. Vofsi, and M. Asscher, US. Pat. 4,292,453 (1981); Chem. Abstr. 96,6345
(1982).
[1822] H. J. Pettelkau, Germ. Offenl. 3,007,634 (1981); Chem. Abstr., 96, 7208 (1982).
[1823] H. J. Krause, Germ. Offenl. 2,925,521 (1981); Chem. Abstr., 94, 174 341 (1981).
[1824] T. R. Bucke Jr. and L. R. Pohl, J. Label. Сотр. Rajiopharm., 18, 663 (1981).
460
[1825] W. J. Spillane. P. Kavanagh, F. Young, H. J.-M. Dou, and J. Metzger, J. Chem. Soc. Perkin
I, 1941,1763. .
[1826] M. Ossd, Liebigs Arm Chem, mi, 1589.
[1827] M. Lissd, J. Chem. Res, 1981, (S) 286, (M) 2946.
• [1828] I. Rico and С Wakselman, Tetrahedron Lett., 1981, 323.
[1829] D. Martinetz and A. Hiller, Z. Chem., 21. 263 (1981).
[1830] A. S. Gozdz,Makromol. Chem. Rap. Commun., 2, 595 (1981).
[1831] B. ZupanSit and M. Kokalj, Synthesis, 1981, 913.
[1832] A. Ricci, A. Degl'innocenti, M. Fiorenza, M. Taddei, M. A. Spartera, and D. R. M.
Walton, Tetrahedron Lett., 1982, 577. .
[1833] T. Livinghouse, Organ. Synth., 60, 126 (1981).
[1834] K. Takahashi, T. Nishizuka, and H. lida, Synth. Commun., 11, 757 (1981).
[1835] S. Gladiali, Chim. Ind. (Milano), 63, 544 (1981).
[1836] M. Govindan and H. W. Pinnick, J. Org. Chem., 46, 5011 (1981).
•[1837] E. A. Parfenov, A. R. Bekker, andG. F. Kostereva,Zh.Org.Khim.SSSRiYI, 1591 (1981),
Engl. transl. p. 1412.
[1838] E. V. Dehmlow and A. R. Shamout, Liebigs Ann. Chem., 1982, 1750.
[1839] E. V. Dehmlow and W. Broda, Chem. Ber., 115, (1982), in press.
[1840] A. Jonczyk and T. Pytlewski, private commun. 1982.
[1841] J. H. Poupaert, P. van der Jeugd, B. M. Gerardy, M. Claesen, and P. Dumont, J. Chem.
Res., 1981, (S) 192, (M) 2484.
[1842] R. O. Hutchins and M. Markowitz, J. Org. Chem., 46, 3571 (1981).
[1843] T. Wakamatsu, H. Inaki, A. Ogawa, M. Watanabe, and Y. Ban, Heterocycles, 14,1441
(1980).
[1844] D. T. Sawyer and J. S. Valentine, Ace. Chem. Res., 14, 393 (1981).
• {1845] E. Alvarez, C. Betancor, R. Freire, A. Martin, and E. Suarez, Tetrahedron Lett., 1981,4335.
[1846] G. Galliani and B. RiudoDe, Tetrahedron, 37, 2313 (1981). :
[1847] M. M. A. El-Sukkary and G. Speier, J. Chem. Soc. Chem. Commun., 1981,745. .
[1848} S. Oae, T. Takata, and Y. H. Kim, Bull. Chem. Soc: Jpn., 54, 2712 (1981). :
[1849] A. A. Frimer, P. Gilinsky-Sharon, and G. Aljadeff, Tetrahedron Lett., 1982,. 1301. j
[1850] D. G. Lee, S. E. Lamb, and V. S. Chang, Organ: Synth., 69,11 (1981):
[1851] M. Takeishi, H. Ohkawa, and S. Hayama, Makromol. Chem. Rap. Commun.,2,457 (1981).
[18S2] Seitetsu KagakuCo., Ltd., Jap. Kokai 81,20,539 (1981); Chem. Abstr., 9S. 24557 (1981).
.11853] R. A. W. Johnstone and A. H. Wilby, Tetrahedron, 37, 3667 (1981). >
[18S4] S. Oae and H. Togo, Synthesis, 1982, 152.
[1855] L. del Valle, S. Sandoval, and G. L. Larson, J. Organomet. Chem., 215, С 45 (1981).
[1856] I. Crossland, Org. Synth,, 60, 6 (1981).
[1857] D. Landini and F. Rolla, J. Org. Chem., 47,154 (1982).
[1858] G. Cardillo, M. Orena, G. Porzi, aad S. Sandri, Synthesis, 1981, 793.
[1859] L. Horner and H. W. Kappa, Phosphorus Sulfur, 11, 339 (1981).
[I860] A. Merz, M. Eichner, and R. Tomahogh; Liebigs Am. Chem., 1981, 1774.
[1861] J. Alais and A. Veyrieres, J. Chan. Soc. Perkin I, 1981, 377.
[1862] J. A, Bristol, I. Gross, and R. G. Lovey, Synthesis, 1981, 971.
[1863] T. Hosokawa, T. Ohta, and S. Murahashi, J. Chem. Soc. Chem. Commun., 1982, 7.
[1864] K.-W. Glombitza and G. Lentz, Tetrahedron,22, 3861 (1981).
[1865] D. Dess, H. P. Kletne, D. V. Weinberg, R. J. Kaufman, and R. S.Suibu»Synthesis, 1981,
883,< л •
[1866] В. Clement, Chemik. Zeit., 104, 368 (1980). v .
[Ш7] W. Kuran and J. Petrus, / Macromol. Sci. Chem., A 15, 381 (1981). ,, .
[1868] Y. Imai, Org. Coat Plast. Chem., 42, 257 (1980); Chem. Abstr., 96, 35771 <1982).
[1869J Y. Hirai, I. Kamide, and T. Yamazati, Heterocycles, 15,1101 (1981); Chem: Abstr., 94,
192023(1981). : '
{1870] S. A. Rao, A. Kumar, H. Ha, and H. Junjappa, Synthesis^ Щ%, $25. ^ ., , ,
{1871] F. Seela, Di Hasselmann, and H.-D. Winkeler, Liebigs Ann. Clum,4992,499 and F.Seela,
private commun. 1982.
461
462 Работы, опубликованные на русском языке
[1872] I. Shinkai, М. С. van der Zwan, F. W. Hartner, R. A. Reamer, R. J. Tull, and L. M.
Weinstock, J. Heterocycl. Chem., 18, 197 (1981).
[1873] Iwashiro Seiyaku Co., Ltd., Jap. Kokai 81,59,775#nd 81,53,680(1981); Chem. Abstr., 95,
115608 and 115609(1981).
.[1874] M. Hedayatullah, J. Heterocycl. Chem., 18, 339(1981).
[1875] G. Vernin, H. Domlog, С Siv, J. Metzger, and A. K. El-Shafei, /. HeterQgycl. Chem., 18,85
(1981).
[1876] J. P. Galy, E. J. Vincent, M. A. Galy, J. Barbe, and J. Elguero, Bull. Soc. Chim. Belg.,99,947
(1981).
[1877] H. Nishi, H. Kohno, and T. Kano, Mil. Chem. Soc. Jpn., 54, 1897 (1981).
,[1878] F. Seela and W. BuBmann, Chem. Ber., 114, 2056 (1981).
[1879] T. Yamada and M. Ohki, Synthesis, 1981, 631.
[1880] A. K. El-Shafei, G. Vernin; and J. Metzger, Gazz. Chim. hai, 111, 413 (1981).
[1881] F. de Jong, D. N. Reinhoudt, A. van Zon, G. J. Torny, E. M. van de Vondervoort, Rec.
Trav. Chim. Pays-Bas, 100, 449 (1981).
[1882] P. Bako, L. Fenichel, U Take, and M. Czugler, Liebigs Ann. Chem.. 1981, 1163.
[1883] S. E. Lauritzen, С Remming, and L. Skattebel, A. Chem. Scand. Ser. B, 35, 263 (1981).
[1884] D. H. Gibson, W.-L. Hsu, and F. U. Ahmed, /. Organomet. Chem., 215, 379 (1981).
[1885] J.-Л. Brunet, С Sidot, and P. Caubere, Tetrahedron Lett., 1981, 1013.
[1886] Y. Sasson, A. Zoran, and J. Blum, J. Molec. Catal., 11, 293 (1981).
[1887] H. Alper and K. Hachem, Transit. Met. Chem., 6, 219 (1981)
-11888] R: Rossi, A, Garpito, M. G. Quirici, and M. L. Gaudenzi, Tetrahedron, 38, 631 (1982).
[1889] S. Bhaduri and K. R. Sharma, /. Organomet. Chem., 218, С 37 (1981).
[1890] E. V. Dehmlow, Z. Naturf., 37b, 1216 (1982).
[1891] S. T. Jolley, J. S. Bradshaw, and R. M. lzatt, j. Heterocycl. Chem., 19, 3 (1982).
[1892] M. Nilssonvv4c«i Chem Scand. Ser. B, 36, 125 (1982).
[1893] G. D. Hartmann and R. D. Hartman, Synthesis, 1982, 504.
- [1894] E. V. Dehmlow and coworkers, unpublished.
• [1895] S. T. Purrington and G. B. Kenion, /. Chem. Soc. Chem. Comrmm., 1982, 731.
«[1896] T. V. Mandelshtham, E. M. Kharicheva, N. N. Labeich, and R. R. Kostikov, Zhur. Org.
Khim., 16, 2513 (1980); Engl. transl. p. 2143.
[1897] E. V. Dehmlow and coworkers, unpublished.
[1898] E. V. Dehmlow and M. Prashad, /. Chem. Res., 1982, submitted.
[1899] E. V. Dehmlow and A. R. Shamout, Liebigs Ann. Chem., 1982, in press.
[1900] M. A. Hashem and P. Weyerstahl, Tetrahedron, 37, 2473 (1981).
Работы, опубликованные на русском языке
К главе 1
[2] Бабаян А. Т., Гамбарян И., Гамбарян Н. Л. —Журя. общ. хим. 1954,
т. 24, с. 1887.
[21] Гаммет Л. Основы физической органической химии: Пер. с англ. — М.:
Мир, 1972.
[22] Шварц М., ред. Ионы н ионные пары в органических реакциях: Пер.
с англ.—М.: Мир, 1975.
[29] Фендлер Е., Фендлер Дж. Мицеллярный катализ в органических
реакциях: кинетика и катализ. — В кн.: Методы н достижения в физико-
органической химии: Пер. с англ.—М.: Мир, 1973
[34] Белецкая И. П. — Усп. хим., 1975, т. 44, с. 2205.
[37] Крумгольц Б. С— Журн. общ. хим., 1974, т. 44, с. 1617.
[43] Крам Д. Основы химии карбаннонов: Пер. с аигл. — М.: Мир, 1967.
Работы, опубликованные на русском языке 463
[641 Звягинцев О. Е., Синицын.Н. М„ Пинков В. #. —Журн.. неорган, хим.,
1966, т. 11, с. 198.
Г651 Иванов И. М., Гиндин Л. М., Чичагова Г.-Я.— Изв. Сиб. отд. АН
СССР, сер. хим., 1967, с. 100. . -. . .
[68] Фролов Ю. Г., Сергиевский В. В., Очкин А. В., Зуев А. Л.—-
Радиохимия, 1972, т. 14, с. 643.
[70] Фролов Ю. Г., Сергиевский В. В., Зуев А. П., Федянина Л. Б.—Жури,
физ. хик., 1972, т. 47, с. 1956.
[76] Гугешашвили М. И., Манвелян М. А., Богуславский Л. И. —
Электрохимия, 1974, т. 10, с 819.
[113] Вебер В., Гокель Г. Межфазный катализ в органической Химии: Пер. с
англ.—М.: Мир, 1980.
[1141 Макоша М. — Усп. хнм„ 1977, т. 46, с. 2174.
[128] Якушин В. В., Абашкин В. М., ЛаскОрин Б. Я. —Докл. АН СССР, сер.
хим., 1980, т. 252, с. 373.
К главе 3
[91] Сметанкина Я. /7., Мирян И. И. — Журн. общ. хим., 1969, т. 39, с. 2299;
Сметанкина Я. П., Ласковенко И. п. — Журн. общ. хим., 1972, т. 42,
е. 617.
[158] Бондарюк Ф. И., Коршунов М. А., Мелехов В„ М. и др. Авт. свид.
№ 357196 (1972). " '
[183] Коршунов М: А., КузовлёЩ Р. Г., ФурЬева И. В., Авт. свид. № 432128
(1974).
[211] Бабаян А. Т., Инджжян М. Г. — Докл. АН АрмССР, 1959, т. 28, с. 67.
[213] Гордон А., Форд Р. Спутник химика: Пер. с &нгл. — М.: Мир, 1976. ,
[223] Физер Л., Физер М. Реагенты для органического синтеза, В 7-мн томах.
Т. 1: Пер. с англ.—М:: Мир, 1970:
[225] Бабаян А. Т., Инджикян М. Г., Азизян Т. Л. —Докл. АН АрмССР.
I960, т. 31, с. 79.
[226] Бабаян А. Т., Инджикян М. Г.—Щрн. фбт. хим., 1957,'т. 27, с. 1201.
[251] Бабаян А. Т., Григорян А. А. ИзЬ. АрмССР, сер. физ. мат., естеств. тех.
науки, 1955, т. 8, с. 81. -
[264] Суворов Я. И., Смушкевич Ю. И., ВелеЖёва B.C., Рожков В. С,
Симаков С. В. — Хим. гетероцикл. сОед., 1976, т. 12, с, 191.
[340] Бабаян А. Т., Гамбарян Я, Гамбарян Н. Я. —Журн. общ. хим., 1954,
т. 24* с. 1887.
[345] Аульченко И. С, Хейфиц Л. А., Константинова Т. И.,
Черкасова Т. П.—Масло-жир, пром., 1975. с. 24. .
[352] Андреев В. М., Бибичева А. И„ Журавлева К. И. —Журн. орг. хим.,
т. 10, С. 1470, ■ ' "
[378] КурЦ А. Л., Закембаева С. М., Белецкая И. П., Реутов О. А — Журн.
орг. хим., 1974. т. 10, с. 1512. . . ■ .-•:
[379] КурЦ А. Л., Демьянов П. И., Белецкая И. П., Реутов О.'А, —Журя.
орг. хим., 1973, т. 9, с. 1313.
[407] Трофимов Б. А., Амосова С. В., Носырева В. В.—-Журн. орг. хим.,:
1976, т. 12, с. 1366.
[411] Физер Л., Физер М. Реагенты для органического' синтеза. В 7ми
томах. Т. 1: Пер. с англ. —М.: Мир, 1970.
[441] Рожков И. Я, Кулешова Я. Д., Кнунянц И. Я. —Изв. АН СССР, сер.
хнм;, 1973, т. 22, с. 128.
[525] Бакум С. Я., Ерешко С. Ф. — Изв. АН СССР, сер! хим., 1974, с. 2138.
[618] Костиков Р; Р., Молчанов А. Я. —Журн. орг. хим.,-1975, т. 11, с. 1767.
[646] Якушкина Я. И., Германова Л. Ф., Клебанова В. Д., Леонова Л. И.,
'■- Болесов И. Г. — Журн. орг. хим., 1976, т. 12, е.,2141. -
464 Работы, опубликованные, на русском языке
[657] Воронков М. Г., Шостаковский С. М., Козырев В: Г., Арц Ю. В.,
Балабанова Л. И., Банникова О. Б. Авт. свид. № 488814 (1976).
[6581 Бредихин А. А., Племенков В. В. — Жури. орг. хим., 1976, т. 12, с. 1001.
[659] Кузнецов И. В., Красавцев И. И.—Укр. хим. журн., 1976, т. 12,
с. 968. .
[660] Хусид А. К.. Крышталь Г. В., Кучеров В. Ф.; Яновская Л. Л. Изв. АН
СССР, сер. хим., 1975, т. 24, с. 2577.
[662] Шостаковский М. Ф., Ерофеев В. И., Аксенов В. С. — Изв. АН СССР,
сер. хим., 1975, т. 24, с. 2577.
[668] Костиков Р. Р., Хлебников А. Ф., ОглоблинК. А. — Авт. свид. № 482448
(1976).
[681] Крышталь Г. В., Кучеров В. Ф., Яновская Л. А.— Изв.'АН СССР, сер.
хим., 1976, т. 25, с. 929.
[682] Крышаль Г; В., Хусид А, К., Кучеров В. Ф„ Яновская Л. А. — Изв. АН
СССР, сер. хим., 1976, т. 24, с. 424. , .
[685] Костиков Р. Р., Молчанов А. Л. —Журн. орг. хим. 1975, т. 11, с. 1861.
[690] Шостаковский С. М., Ретинский А. А., Бобров А. В. —Изв. АН СССР,
сер. хим., 1975, т. 23, с. 1818. ...
[736] Тищенко И. Г., Кулинкович О. Г., Глазков Ю. В. —Журн. орг. хим.,
1975, т. .11. с. 581.
[737} 'Тъщенко И. Г., Глазков Ю. В., Кулинкович'О. Г. —Журн. орг. хим.,
1973, т. 9, с. 25ia.;
[738] Тищенко И. Г., Кулинкович О. Г., Глазков Ю. В., Пирштюк М. /С.—
■ Журн. орг. хим., 1975, т. 11, С 576.
[758]. Крышталь Г. В„ Хусид. А. К, Куперов В. Ф., Яновская Л. А. — Изв.
АН СССР/сер. хнн:, 1977, т. 26; с. 709.
[959] Костиков Р. Р„ Молчанов А. П., Голованова Г. Вн Зенкевич И. Г.—
Журн. орг. хим., 1977, т. 13, с. 1846.
[1049] Жданов ТО. А., Алексеев Ю. £., Дорошенко С. С, Богданова Г. В.,
Cjfdajpeea Т. П., Алексеева В. Г. —Докл. АН СССР, сер. хим., 1978,
т. 238, с. 1102.
Г105Г] Андреев В. М., Бибичева А. И., Лапикова Л. С. — ВХО нм. Менделеева,
. 1978. т. 23. с. 231. г
[1099] Жданов Ю. А., Алексеев Ю. Е., Зинченко Г. В., Дорошенко С. С.—
Докл. АН СССР, сер. хим., 1976, т. 231, с. 868.
[1114] Хусид А.. /С„ Крышталь Г, В., Кучеров В. Ф.. Яновская Л. А.— Изв:
АН СССР, сер: хим., 1977, т. 26; с. 692.
[1131] Лабич.Н. Н., Харичева Е. М, Мандельштам Т. В., Костиков Р. Р.~~
Журн. орг. хим., 1978, т. 14, с. 878.
[1144] Костикрв Р. Р., Молчанов А. Я,—*Журн. орг. хим.,-1978, т. 14, с. &Т9;
[1145] Костиков Р. Р., Молчанов А. .П., Васильева И. А.. Слободин Ю. М.—
Жури. орг. хим., Д977, т. 13. с 2541.
[Н64] Аксенов В. С, Терентьева Г. А:—Изв. АН СССР, сер. .хим., 197/, т. 26,
. . с. 623, . ..•■■"*.
[1185] Натанович М. Л., Джанашвили Л. А., Хейфец В. Л. — Журн. общ.
... хим., 1978, т, 48, с 1925, - .'.■--? ......
[1213] Новокрёщенник В. Д., Мочалов С. С, Шабаров Ю. С — Жури. орг.
хим., 1979,т. 15, с^485. . ,, - -
[1226] БогатскийАгЪ., Лукьяненко Н. Г., Пастушок В. Я. —Докл. АН СССР,
„1979, т. 247, с. 1153- .. . -
[1252J Дрыгайлова Ё. А., Костиков Р. В., Оглоблин К. Л. —Журн. орг. хим.,
I97i8, 7.14, 2008. , . •
[1298] Костиков Р: Р., Гришина Ё. Н., Слободин Я. Ж —Жури. орг. хим.,
^ 1979. .т. 15, с. 331. , - . . - .- -,
{130.1} Крышталь Т.- В., Кульганек В. В., Кучеров В. Ф., Яновская Л. А.—
Изв. АН СССР, сер. хим., 1978, т, 27, с. 2808,
{1302] Крышталь Г. В, Кучеров В. Ф., Яновская Л. А. — Изв. АН СССР,
сер. хим., 1978; т. 27, с. 2803.
Работы, опубликованные на русском, языке 465
[13221 Мавсумзаде М. М... Шабанов А. Л.» Мамвдве.Т. X *—Изв, высших.
учебн. завед. Химия я хим. технод., 1979, т, 22, с. 758.: . .
[1325J Нефедове М., Долгцй И, -£., Булушева £, В.-¥Изв..АН СССР, сер.
хим., 1978, т. 27, с. 1454.
[1326] Нефедов О. М., Долгий И. В., Шведова И: £., Байдокигитова Е.А.—
Изв. АН СССР, сер. хим., .1978, т. 27, с. 1339.. . . ' *
[1349] Соколенка В. А., Маркова В. А., Когай Б. £. — ЖУрн. орг; хнм.,г3978,
т. 14, с. 1111. .', •' , . ..-.:"
[1373] Юфит С. С, Есикова И. Л.—Изв. АН СССР, сер..хим., 1Й9, т.,28,
с. 1706. . j ...
[1375] Жданов Л. А.. Алексеев Ю. Е.. Алексеева В. Г., Король Е. Л., Тюме-
нев В. А^ Попов И. И., Поленов В. А. —Докл. АН СССР» сер. хим.,
1979, т. 244, с. 1122.
[1382] Суворова Г. И., Комендантов М, Я.— Журн. орг. хим.,. 1979, т. 15,
с. 1435. ... .
[1383] Жданов Ю. А., Алексеев Ю. Е., Сиилис И. П., Сухарева Т: П.,
Богданова Г. В. — Докл: АН СССР, сер. хим., 1979, т. 245, с. 8461
[1390] Нефедов О. М., Долгий И. Е.. БуАушева Е. В., Штейншнейдер А. Я. —
Изв. АН СССР, сер. хим., 1979. т. 28, с. 1535. .
[1392] Костиков Р. Р., Молчанов А. П., Тарасова <Х А., Амосова С. В., Тро->
фимов Б. А. — Изв. АН СССР, сер. хим., 1979, т. 28, с. 1286. . :
[14251 Ахметова Н. Е., Власов В. М., Якобсон Г. А^-г.Иай АН СССР,;сер,
хим., 1978, т. 27, с. 949. .- ■ ■
[1426] Абрамова Н. М., Зотова С. В.-^ Изв. АН СССР,\ сер. яим., 1979, т.28,
с. 697. ... : :.:-'■.
[1427] Штеменко Н. И., Кучеров В. Ф., Яновская Л; Я.—.Изв.--АН СССР, сер.
хим., 1978, т. 27, с. 1444.
[14.37] Власов В. Mv Аксенов В. В., Якобсон Г. Т,-й-Жури. орг. хтац 19Г9,:
т. 15, с. 2156. . ■} ... .: л " : ..--'. ■ .-.,,;: ч
[1451] Рунова Е. Л, Бородина Л. Н., Щербаков А. К, Кораханов Е. X.—
Вести. МГУ, сер. хим., 1979, т. 20, с. 581.
[1462] Новикова Л. К, Скрипник О. Б., Бикбулатова Г. Ш., Вулфсон С. Г.,
Исаева 3. Г.. Верещагин А. Н. — Изв. АН СССР, сер. хим., 1978, т. 27,
с. 605.
[1463] Жданов Ю. А., Алексеев Ю. Е., Дорошенко С. С, Сударева Т. П.,
Богданова Г. В., Алексеева В. Г., Тюменев В. А. — Жури. общ. хим., 1978,
т. 48, с. 2614.
[1477] Андреев В. А1, Бабичева А. И., Родионова И. Е. — Масло-жир, пром.,
1980. с. 27.
[1484] Боев В. И., Домбровский А. В.—Жури. общ. хим., 1980, т. 50, с. 121.
[1495] Есикова И. А., Юфит С. С —Изв. АН СССР, сер. хим., 1980, т. 29,
с. 507, 515.
[1502] Есикова И. А., Кучеров В. Ф., Руденко Б. А., Юфит С. С —Изв. АН
СССР, сер. хим., 1979, т. 28, с. 1468.
[1528] Артамкина Г. А., Гринфельд А. А., Белецкая И. П. — Журн. орг. хим.,
1980, т. 16, с. 698.
[1533] Юфит С. С, Есикова И. А. — Изв. АН СССР, сер. хим., 1980, т. 29,
с. 519.
[1535] Суворов Н. Н., Плутитский Д. Н., Смушкевич Ю. И. — Журн. орг. хим..
1980, т. 16, с 872.
[1547] Лоэанская Г. И., Есикова И. А., Юфит С. С, Кучеров В. Ф. — Изв. АН
СССР, сер. хим., 1980, т. 28, с. 794.
[1548] Малхасян А. Т., Джанджилян 3. Л., Григорян Р. Т., Миракян С. М.
Арм. хим. журн., 1980, т. 33, с. 152.
[1588] Малхасян А. Т., Назарян Е. М., Миракян С. М.. Мартиросян Г. Т. —
Арм. хим. журн., 1979, т. 32. с. 952.
[1590] Юфит С. С, Лозанская Т. И.. Есикова И. А. — Изв. АН СССР, сер.
хим., 1980, т. 29, с. 797.
466 Работы, опубликованные на русском языке
[1591] Кургинян К. А., Ростомян И. М., Калайджян А. Е., Чухаджян Г. А.—
Арм. хим. журн., 1979, т. 32, с. 945; Калайджян А. Е., Ростомян И. М.,
, Кургинян К. А., Чухаджян Г. А.—Арм. хим. журн., 1980, т. 33,
с. 57.
[1594]. Глушко Л. П., Самсонова В. Н., Малиновский М. С, Яновская Л. А.—
Изв. АН СССР, сер. хим., 1980, т. 29, с. 1048.
[1604]. Хейфиц Л. А., Гуревин А. В., Лепихина Н. В., Новиков Н. А. —Масло-
жир, пром., 1980, с. 24.
[1623] Иешин В. П., Комелин М. С, Велик И. Я, — Журн. орг. хим., 1980, т. 16,
с. 980.
[1637] Арбузов Б. А., Салихова А. М., Шостаковский С. М., Верещагин А. Я.,
Никольский Н. С. —Изв. АН СССР, сер. хим., 1979, т. 28, с. 2786.
[1728] Костиков Р. Р., Молчанов А. П., Амосова С. В., Трофимов Б. А.—
Лзв. АН СССР, сер, хим., 1980, т. 29, с. 1354.
[1736] Соколов Я. А., Перчугов Г. Ю., Морозов О. С. — Журн. общ. хим.,
1979, т. 49, с. 1856.
[1751] Вольева В. Б., Белостоцкая И. С, Новикова И. А., Джуарян Е. В., Ер?
шов В. В. — Изв. АН СССР, сер. хим., 1980, т. 29, с. 2414.
[1760] Хусид А. К., Любим М. С—Журн. орг. хим., 1980, т. 16, с. 2437.
[1818] Чухаджян Г. А., Саркисян Е. Л., Ростомян И. М., Исраелян А. Г.—
Арм. хим. журн., 1981, т. 34, с. 519.
[1819] Кургинян К. А., Калайджян А. Е., Ростомян И. М.у Чухаджян Г. А. —.
Арм. хим. жури., 1981, т. 34, с. 422.
{1820] Калайджян А. Е., Кургинян К. А., Ростомян И. М., Чухаджян Г. А. —
Арм. хим. журн., 1980, т. 33, с. 845.
[1837] Парфенов Е. А., Беккер А. Р., Костерова Г. Ф. — Жури. орг. хим., 1981,
т. 17, с. 1591.
[1886] Мандельштам Т. В., Харичева Е. М., Лабич Я. Я., Костиков Р. Р. —
Журн. орг. хим., 1980, т. 16, с. 2513.